
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization.
Concise International Chemical Assessment Document 51
First draft prepared by Dr Bob Benson, US Environmental Protection Agency, Denver, Colorado, USA
Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 2003
The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
1,1-Dichloroethene (vinylidene chloride).
(Concise international chemical assessment document ; 51)
1.Dichloroethylenes - adverse effects 2.Dichloroethylenes - toxicity 3.Risk assessment
4.Environmental exposure I.International Programme on Chemical Safety II.Series
ISBN 92 4 153051 0 (NLM Classification: QV 633)
ISSN 1020-6167
©World Health Organization 2003
All rights reserved. Publications of the World Health Organization can be obtained from Marketing and Dissemination, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 2476; fax: +41 22 791 4857; email: ). Requests for permission to reproduce or translate WHO publications — whether for sale or for noncommercial distribution — should be addressed to Publications, at the above address (fax: +41 22 791 4806; email: permissions@who.int).
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
The World Health Organization does not warrant that the information contained in this publication is complete and correct and shall not be liable for any damages incurred as a result of its use.
The Federal Ministry for the Environment, Nature Conservation and Nuclear Safety, Germany, provided financial support for the printing of this publication.
Concise International Chemical Assessment Documents (CICADs) are the latest in a family of publications from the International Programme on Chemical Safety (IPCS) — a cooperative programme of the World Health Organization (WHO), the International Labour Organization (ILO), and the United Nations Environment Programme (UNEP). CICADs join the Environmental Health Criteria documents (EHCs) as authoritative documents on the risk assessment of chemicals.
International Chemical Safety Cards on the relevant chemical(s) are attached at the end of the CICAD, to provide the reader with concise information on the protection of human health and on emergency action. They are produced in a separate peer-reviewed procedure at IPCS. They may be complemented by information from IPCS Poison Information Monographs (PIM), similarly produced separately from the CICAD process.
CICADs are concise documents that provide summaries of the relevant scientific information concerning the potential effects of chemicals upon human health and/or the environment. They are based on selected national or regional evaluation documents or on existing EHCs. Before acceptance for publication as CICADs by IPCS, these documents undergo extensive peer review by internationally selected experts to ensure their completeness, accuracy in the way in which the original data are represented, and the validity of the conclusions drawn.
The primary objective of CICADs is characterization of hazard and dose–response from exposure to a chemical. CICADs are not a summary of all available data on a particular chemical; rather, they include only that information considered critical for characterization of the risk posed by the chemical. The critical studies are, however, presented in sufficient detail to support the conclusions drawn. For additional information, the reader should consult the identified source documents upon which the CICAD has been based.
Risks to human health and the environment will vary considerably depending upon the type and extent of exposure. Responsible authorities are strongly encouraged to characterize risk on the basis of locally measured or predicted exposure scenarios. To assist the reader, examples of exposure estimation and risk characterization are provided in CICADs, whenever possible. These examples cannot be considered as representing all possible exposure situations, but are provided as guidance only. The reader is referred to EHC 170.1
While every effort is made to ensure that CICADs represent the current status of knowledge, new information is being developed constantly. Unless otherwise stated, CICADs are based on a search of the scientific literature to the date shown in the executive summary. In the event that a reader becomes aware of new information that would change the conclusions drawn in a CICAD, the reader is requested to contact IPCS to inform it of the new information.
Procedures
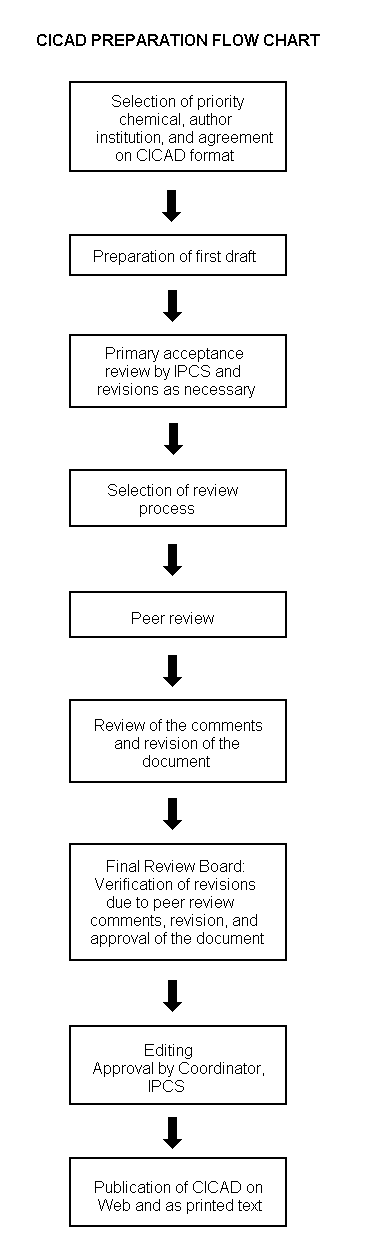
The flow chart on page 2 shows the procedures followed to produce a CICAD. These procedures are designed to take advantage of the expertise that exists around the world — expertise that is required to produce the high-quality evaluations of toxicological, exposure, and other data that are necessary for assessing risks to human health and/or the environment. The IPCS Risk Assessment Steering Group advises the Coordinator, IPCS, on the selection of chemicals for an IPCS risk assessment based on the following criteria:
Thus, it is typical of a priority chemical that
The Steering Group will also advise IPCS on the appropriate form of the document (i.e., EHC or CICAD) and which institution bears the responsibility of the document production, as well as on the type and extent of the international peer review.
The first draft is based on an existing national, regional, or international review. Authors of the first draft are usually, but not necessarily, from the institution that developed the original review. A standard outline has been developed to encourage consistency in form. The first draft undergoes primary review by IPCS to ensure that it meets the specified criteria for CICADs.
|
Advice from Risk Assessment Steering Group Criteria of priority:
Thus, it is typical of a priority chemical that
Special emphasis is placed on avoiding duplication of effort by WHO and other international organizations. A prerequisite of the production of a CICAD is the availability of a recent high-quality national/regional risk assessment document = source document. The source document and the CICAD may be produced in parallel. If the source document does not contain an environmental section, this may be produced de novo, provided it is not controversial. If no source document is available, IPCS may produce a de novo risk assessment document if the cost is justified. Depending on the complexity and extent of controversy of the issues involved, the steering group may advise on different levels of peer review:
|
The second stage involves international peer review by scientists known for their particular expertise and by scientists selected from an international roster compiled by IPCS through recommendations from IPCS national Contact Points and from IPCS Participating Institutions. Adequate time is allowed for the selected experts to undertake a thorough review. Authors are required to take reviewers’ comments into account and revise their draft, if necessary. The resulting second draft is submitted to a Final Review Board together with the reviewers’ comments. At any stage in the international review process, a consultative group may be necessary to address specific areas of the science.
The CICAD Final Review Board has several important functions:
Board members serve in their personal capacity, not as representatives of any organization, government, or industry. They are selected because of their expertise in human and environmental toxicology or because of their experience in the regulation of chemicals. Boards are chosen according to the range of expertise required for a meeting and the need for balanced geographic representation.
Board members, authors, reviewers, consultants, and advisers who participate in the preparation of a CICAD are required to declare any real or potential conflict of interest in relation to the subjects under discussion at any stage of the process. Representatives of nongovernmental organizations may be invited to observe the proceedings of the Final Review Board. Observers may participate in Board discussions only at the invitation of the Chairperson, and they may not participate in the final decision-making process.
This CICAD on 1,1-dichloroethene (vinylidene chloride) was prepared by the US Environmental Protection Agency (EPA). The US EPA considered data identified as of April 2001 in the national assessment document (US EPA, 2002d). Information on the nature of the peer review and availability of this national assessment document is presented in Appendix 1. The literature search for this CICAD was updated in August 2002. Information on the peer review of this CICAD is presented in Appendix 2. This CICAD was approved as an international assessment at a meeting of the Final Review Board, held in Monks Wood, United Kingdom, on 16–19 September 2002. Participants at the Final Review Board meeting are presented in Appendix 3. The International Chemical Safety Card for 1,1-dichloroethene (ICSC 0083), produced by the International Programme on Chemical Safety (IPCS, 2000), has also been reproduced in this document.
1,1-Dichloroethene (CAS No.
1,1-DCE can be found in the environment from release during its manufacture and use, from the breakdown of polyvinylidene (PVDC) products, and from the biotic or abiotic breakdown of 1,1,1-trichloroethane, tetrachloroethene, 1,1,2-trichloroethene, and 1,1-dichloroethane. The principal sources of environmental exposure for humans are ambient air and contaminated drinking-water.
In groundwater, biotransformation of 1,1-DCE can form vinyl chloride through reductive dechlorination.
The high vapour pressure and low water solubility of 1,1-DCE favour relatively high concentrations in the atmosphere compared with other environmental compartments. Atmospheric hydroxyl radicals play a major role in the degradation of 1,1-DCE. The atmospheric half-life is estimated at 16 h. Volatilization is the major transport process from water, soil, and sediment. Bioaccumulation is expected to be low based on the chemical’s octanol/water partition coefficient and water solubility.
1,1-DCE is rapidly absorbed following inhalation and oral exposure. Because of its low relative molecular mass and hydrophobic nature, dermal absorption is also likely, but there are no relevant published data. Although 1,1-DCE is rapidly distributed to all tissues, most of the free 1,1-DCE, its metabolites, and covalently bound derivatives are found in the liver and kidney. 1,1-DCE is rapidly oxidized by cytochrome P450-dependent monooxygenase 2E1 (CYP2E1) to 1,1-dichloroethene oxide (DCE-epoxide), 2-chloroacetyl chloride, and 2,2-dichloroacetaldehyde. The major metabolites, DCE-epoxide and 2-chloroacetyl chloride, can react with glutathione (GSH), water, or tissue macromolecules. It is not known if the metabolism of 1,1-DCE is the same in humans, although in vitro microsomal preparations from human liver and lung form the same initial products.
The only existing epidemiological study is inadequate to assess the cancer or non-cancer health effects of 1,1-DCE.
Following high-dose exposure by the oral or inhalation route, the target organs in experimental animals are the liver, the kidney, and the Clara cells of the lung. Following low-dose, long-term exposure, the liver is the major target organ in rats following oral or inhalation exposure, but the kidney is the major target organ in mice following inhalation exposure.
Bioassays for cancer by the oral route of exposure have been conducted in rats, mice, and trout. Although these bioassays have protocol limitations, none provides any significant evidence that 1,1-DCE is a carcinogen by the oral route of exposure. Bioassays for cancer by the inhalation route of exposure have been conducted in rats, mice, and hamsters. Most of these bioassays also have protocol limitations. One bioassay in male mice showed an increase in the incidence of kidney adenocarcinomas at one exposure level. There is evidence that the induction of kidney adenocarcinomas is a sex- and species-specific response related to the expression of CYP2E1 in the kidney of male mice. The results of the one bioassay showing an increase in tumours in one sex and at one exposure level in a single rodent species are not sufficient to justify an exposure–response assessment.
1,1-DCE causes gene mutations in microorganisms in the presence of an exogenous activation system. Most tests with mammalian cells in vitro or in vivo show no evidence of genotoxicity.
There is no evidence that reproductive toxicity or teratogenicity is a critical effect for 1,1-DCE. No reproductive or developmental toxicity was observed at an oral exposure that caused minimal toxicity in the liver of the dams. There is some evidence of developmental variations in the heart after oral exposure, but it is not clear if these effects are directly caused by exposure to 1,1-DCE. There is evidence of fetal toxicity (delayed ossification) following inhalation exposure in the absence of maternal toxicity.
One study shows no evidence that 1,1-DCE causes skin sensitization.
The toxicity of 1,1-DCE is associated with cytochrome P450-catalysed metabolism of 1,1-DCE to reactive intermediates that bind covalently to cellular macromolecules. The extent of binding is inversely related to loss of GSH, so that severities of tissue damage parallel the decline in GSH. Thus, the responses to 1,1-DCE at low doses with little depletion of GSH are expected to be very different from the responses at high doses causing substantial GSH depletion.
The critical effect from oral exposure is minimal hepatocellular mid-zonal fatty change in female Sprague-Dawley rats. Based on a BMDL10 (the lower 95% confidence limit on the benchmark dose [BMD] for a 10% response) of 4.6 mg/kg body weight per day and a total uncertainty factor of 100, the tolerable intake is 0.05 mg/kg body weight per day.
The critical effect from inhalation exposure is minimal hepatocellular mid-zonal fatty change in female Sprague-Dawley rats. Based on a BMCL10 (the lower 95% confidence limit on the benchmark concentration [BMC] for a 10% response) of 6.9 mg/m3 and a total uncertainty factor of 30, the tolerable concentration is 0.2 mg/m3.
Human exposure to 1,1-DCE is likely to be highly variable due to site-specific contamination. However, data suggest that the mean exposure from drinking-water will not exceed 6–9 × 10–5 mg/kg body weight per day for a 70-kg individual consuming 2 litres per day. The oral exposure from food and soil is most likely negligible. Data suggest that the upper end of the range for the mean concentration of 1,1-DCE in air will not exceed 0.004 mg/m3. Thus, human exposure is expected to be far below the tolerable intake of 0.05 mg/kg body weight per day and the tolerable concentration of 0.2 mg/m3.
There are only limited data on the effects of 1,1-DCE in the aquatic and terrestrial environments. In studies conducted in closed systems, the EC20 for inhibition of the growth of a mixed methanotrophic culture was 0.05 mg/litre; the 72-h EC50 for inhibition of growth of green alga Chlamydomonas reinhardtii was 9.12 mg/litre; and the 96-h LC50 for bluegill (Lepomis macrochirus) was 74 mg/litre. The limited data on occurrence of 1,1-DCE in surface water suggest that concentrations are in the microgram per litre range, indicating that acute toxic risks from 1,1-DCE for the aquatic environment are minimal. There are no long-term toxicity data with which to assess sublethal effects of 1,1-DCE on any organisms. However, because of the rapid volatilization of 1,1-DCE from the aquatic and terrestrial environments, no significant risk is expected.
Synonyms for 1,1-dichloroethene (CAS No.
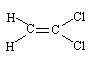
1,1-Dichloroethene
The chemical and physical properties of 1,1-DCE are summarized in the International Chemical Safety Card (ICSC 0083), which is included in this document.
The conversion factors2 for 1,1-dichloroethene in air (at 20 °C and 101.3 kPa) are as follows:
1 ppm = 4.0 mg/m3
1 mg/m3 = 0.25 ppm
Because of its volatility, 1,1-DCE is well suited to determination by gas chromatography (GC) using a variety of detectors, including flame ionization (FID), electron capture, electrolytic conductivity (ECD), and mass spectrometry (MS). The major analytical limitation is interference by other constituents of the media analysed.
Methods are available for quantifying 1,1-DCE in environmental samples (air, water, soil, sediment). Quantification of 1,1-DCE in air with a detection limit in the 1 µg/m3 range typically uses cold or absorbent column trapping followed by GC/FID (Foerst, 1979; Sidhu, 1980). A method for whole air samples using SUMMA canisters with a comparable method of detection is available (US EPA, 1988; Brymer et al., 1996). A method with a working range of 2–20 mg/m3 for a 5-litre air sample is available for monitoring in the workplace (NIOSH, 1994). This method uses GC/FID. Quantification of 1,1-DCE in water with a detection limit in the 0.1–0.5 µg/litre range uses headspace analysis and GC/FID or GC/ECD (Piet et al., 1978; Otson & Williams, 1982). Other methods with a comparable detection limit include purge and trap with GC/ion trap detector (Eichelberger et al., 1990; US EPA, 1995) and purge and trap with GC/MS (US EPA, 1998). Some newer techniques for sample collection include solid-phase microextraction (Arthur et al., 1992; Shirey, 1995) and sample introduction using membrane technology (Bauer & Solyom, 1994; Wong et al., 1995). Quantification of 1,1-DCE in soils and sediments with a detection limit in the 5–10 µg/kg range uses extraction with an organic solvent or purging with an inert gas, trapping, and GC/MS (DeLeon et al., 1980; Speis, 1980; Amaral et al., 1994; US EPA, 1998).
Methods are also available for quantifying 1,1-DCE in biological samples (breath, food, body tissues). Quantification of 1,1-DCE in human breath with a detection limit of 0.16 µg/m3 uses spirometry for sample collection, cryogenic or Tenax trapping, and GC/MS (Wallace et al., 1982, 1984). Quantification of 1,1-DCE in food (Gilbert et al., 1980) and body tissues (Lin et al., 1982) with a detection limit in the 5–10 µg/kg range uses a headspace technique (Gilbert et al., 1980), purge and trap, and GC/ECD (Lin et al., 1982) or GC/MS (Easley et al., 1981; Hiatt, 1983).
1,1-DCE is not known to occur as a natural product. It is produced commercially by the dehydrochlorination of 1,1,2-trichloroethane in the presence of excess base or by thermal decomposition of methyl chloroform (1,1,1-trichloroethane). 1,1-DCE is used as a captive intermediate in the production of hydrochlorofluorocarbons (HCFC-141b and HCFC-142b), in the production of chloroacetyl chloride, and in the production of homo-, co-, and terpolymers (latex and resin) (also known as PVDC polymers) (W. Stott, personal communication, 2002). These polymers are produced as emulsion polymers, as solvent-soluble powders for coating applications, and as resins for extrusion and co-extrusion. PVDC copolymers containing 79–90% 1,1-DCE are used to form moisture and vapour barrier coatings and films, with applications as food packaging products. PVDC copolymers containing 10–70% 1,1-DCE are used to improve flame and ignition resistance properties in the final product. Residual 1,1-DCE in PVDC used for food packaging products typically ranges from 5 to <1 mg/kg, the limit of detection of the method. Other consumer products containing PVDC include PVDC-latex for carpet backing (<2 mg/kg residual 1,1-DCE), PVDC-latex for Foil Scrim Kraft (<3 mg/kg residual 1,1-DCE), PVDC-latex for photographic film coating (<100 mg/kg residual 1,1-DCE), PVDC for flame retardant fibres for clothing and outdoor awnings (<100 mg/kg residual 1,1-DCE), and PVDC-fluorinated copolymers for application on textiles (<100 mg/kg residual 1,1-DCE). Further processing decreases the residual 1,1-DCE in the final consumer product.
1,1-DCE can be found in the environment from release during its manufacture and use; from the breakdown of PVDC products; and from the biotic or abiotic breakdown of 1,1,1-trichloroethane, tetrachloroethene, 1,1,2-trichloroethene, and 1,1-dichloroethane. 1,1-DCE is frequently found at hazardous waste sites.
In the early 1980s, annual world production was estimated at 306 000 tonnes (IPCS, 1990). IPCS (1990) estimated that 1%, or 3000 tonnes, was released to ambient air. The US EPA (2002c) reported that 74 tonnes were released to ambient air and 0.06 tonne was released to surface water in 1999 in the USA. For the period 1988–1999, the average annual release to ambient air was 99 tonnes and the average annual release to surface water was 0.39 tonne in the USA (US EPA, 2002c).
The high vapour pressure and low water solubility of 1,1-DCE favour relatively high concentrations in the atmosphere compared with other environmental compartments. Atmospheric hydroxyl radicals play a major role in the degradation of 1,1-DCE. The rate constant for the oxidation reaction ranges from 0.8 × 10–11 to 2.6 × 10–11 cm3/molecule per second, with an atmospheric half-life estimated at 16 h (Grosjean, 1991). The major reaction products are formaldehyde, phosgene, and hydroxyacetyl chloride. Removal of 1,1-DCE from the atmosphere by reactions with chlorine atoms, peroxy radicals, ozone, and nitrate are quantitatively insignificant, as is removal by rain droplets or by adsorption on particulate matter in the atmosphere (IPCS, 1990; Grosjean, 1991).
Consideration of the physical and chemical properties of 1,1-DCE indicates that volatilization is the major transport process from water. The half-life for evaporation of 1,1-DCE (1 g/litre) from a stirred aqueous solution with a depth of 6.5 cm was 27.2 min at 20 °C (Dilling, 1977). The calculated half-life of 1,1-DCE was 6 days in static pond water and 1 day in mobile river water (IPCS, 1990). Photolysis and hydrolysis are not likely to be significant pathways for removal of 1,1-DCE from aqueous solutions.
Volatilization is expected to be the major process for the removal of 1,1-DCE from soils and sediments. The chemical’s octanol/water partition coefficient and solubility in water also suggest that leaching from soils to water can occur.
MITI (1992) reported no significant biodegradation in a closed bottle test using activated sludge. This test was conducted according to Organisation for Economic Co-operation and Development (OECD) Guideline 302C. However, Tabak et al. (1981) measured microbial degradation of 78% of 1,1-DCE (5 mg/litre) following 7 days of incubation at 25 °C in a static culture flask, in the dark, with settled domestic wastewater as microbial inoculum. With subsequent incubations (after adaptation), 100% loss of compound occurred. At 10 mg/litre, 45% loss was found in the first 7 days of incubation. Volatilization losses over 7 days at 25 °C were 24% and 15% at 5 and 10 mg/litre, respectively.
The biotransformation of 1,1-DCE has been studied in anaerobic microcosms (Barrio-Lage et al., 1986; Vogel & McCarty, 1987). The biotransformation first produces vinyl chloride by reductive dehalogenation, which is subsequently mineralized to carbon dioxide. Barrio-Lage et al. (1986) obtained sediment and water from two different locations in the Florida Everglades. This material was spiked with 1,1-DCE to 5 mg/litre and incubated anaerobically at 25 °C in the dark for up to 6 months. In this experiment, 1,1-DCE was consumed and vinyl chloride was produced. The first-order rate constant for depletion of 1,1-DCE was 1.67 × 10–4/h in one microcosm and 3.57 × 10–4/h in the other, indicating a very slow rate of dehalogenation. Not all of the 1,1-DCE depleted appeared as vinyl chloride, indicating that mechanisms of biotransformation other than reductive dechlorination occur. Glod et al. (1997) presented evidence for an alternative pathway of biotransformation involving cobalamin. Cobalamin is involved in the enzymatic reduction of halogenated ethenes by a variety of anaerobic bacteria. Glod et al. (1997) investigated the mechanism of the reduction of 1,1-DCE in homogeneous solutions containing titanium(III) citrate as the bulk electron donor. Cob(I)alamin reduced 1,1-DCE to ethene and ethane in pH-dependent reactions. At pH 7, acetylene was detected as the major intermediate, and only a minor amount of vinyl chloride was formed. A possible mechanism involves addition of cob(I)alamin to 1,1-DCE. The deprotonated form of the β-dichloroethyl cobalamin then loses a chloride to form a carbene intermediate, which is transformed into a β-chlorovinyl cobalamin by transfer of an α-hydrogen. The β-chlorovinyl cobalamin is converted to acetylene by β-elimination through a cobalt (III)-pi complex. At pH 9, where the overall reaction was much slower than at pH 7, about equal amounts of vinyl chloride and ethene were formed simultaneously. The formation of vinyl chloride at pH 9 possibly involves two different pathways. The first pathway involves β-elimination of a chloride from β-dichloroethyl cobalamin with a cobalt (III)-pi complex as an intermediate. The second pathway involves a dissociative electron transfer yielding a chlorovinyl radical as the intermediate.
Dolan & McCarty (1995) studied the aerobic biotransformation of 1,1-DCE using a mixed methanotrophic culture isolated from a US Superfund site. The initial oxidation of 1,1-DCE is believed to be catalysed by methane monooxygenase, which has a broad substrate specificity. The degradation of 1,1-DCE stopped after the first few hours due to the toxicity of the metabolites of 1,1-DCE, most likely the epoxide or an acyl chloride.
Bioaccumulation is expected to be low based on the octanol/water partition coefficient and water solubility of 1,1-DCE. A bioconcentration factor of 4 and a bioaccumulation factor of 6.9 were reported for fish (Atri, 1985). A bioaccumulation factor of less than 13 was reported for common carp (Cyprinus carpio) (MITI, 1992).
Singh et al. (1981) and Brodzinski & Singh (1983) reported data on 1,1-DCE in ambient air in 30 US locations, and Guicherit & Schulting (1985) measured 1,1-DCE in ambient air at three sites in the Netherlands. All three data sets are consistent and indicate typical mean values of 20–120 µg/m3 and maxima of 40–560 µg/m3. Measurements taken in industrial areas near sources of 1,1-DCE in these studies yielded a much higher mean value of 120 × 103 µg/m3 and a maximum of 270 × 103 µg/m3. US EPA (1985) reported mean values of 0.02 µg/m3 in non-industrial areas and 8.7 µg/m3 in industrial areas. US EPA (2002a) compiled ambient air quality data from 1982 to 2001 for 1,1-DCE at locations throughout the USA. The data for 2001 provide the arithmetic mean of the samples collected from 87 individual locations. The range in the arithmetic mean values for these locations was 0.004–4 μg/m3.3
Of the 439 hazardous waste sites in the USA with detections of 1,1-DCE, 186 sites reported quantitative information on the concentration of 1,1-DCE in water (US EPA, 2002b). The mean maximum detected concentration in water was 2 mg/litre.
Hallbourg et al. (1992) investigated the occurrence of 1,1-DCE in groundwater and surface water near three municipal landfills in Florida, USA, using a method with a detection limit of 1 µg/litre. 1,1-DCE was not detected in any surface water or in any well near two of the landfills. At the third landfill, 1,1-DCE was not detected in two wells, but was detected in a third well at 24.4 µg/litre.
Yamamoto et al. (1997) determined the concentration of 1,1-DCE in surface water from 30 sites in Osaka, Japan, using a method with a detection limit of 0.4 µg/litre. 1,1-DCE was detected in only 3 of 136 samples. The maximum concentration found was 1 µg/litre.
Stangroom et al. (1998) reported on the occurrence of 1,1-DCE in 1995 from data collected by the National Centre for Toxic and Persistent Substances in the United Kingdom. 1,1-DCE was detected in 4% of industrial effluents in England and Wales at a mean concentration of 0.67 µg/litre.
Wilson et al. (1994) reported on the occurrence of 1,1-DCE in digested sewage sludge from 12 locations (rural, urban, and industrial) in northwest England. The mean concentration of 1,1-DCE was 7.97 mg/kg dry weight (range 1.92–16.6 mg/kg dry weight).
Of the 439 hazardous waste sites in the USA with detections of 1,1-DCE, 45 reported quantitative information on the concentration of 1,1-DCE in soil (US EPA, 2002b). The mean maximum detected concentration in soil was 90 mg/kg.
For the general population, potential sources of exposure to 1,1-DCE include breathing contaminated air, drinking contaminated water, dermal contact with contaminated water, and eating contaminated food.
Wallace et al. (1986) reported a 5-year US EPA study in urban populations of personal exposures to 1,1-DCE from air. Among the 400 participants, 1,1-DCE was detected only occasionally in personal air samples at levels exceeding 1 µg/m3 based on a 24-h average. No recent data were located.
Storm (1994) summarized the results of monitoring drinking-water sources in California, USA, from 1984 to 1992 using a method with a detection limit of 0.1 µg/litre. Of 11 686 sources sampled, 120 contained 1,1-DCE with a mean concentration of 2.45 µg/litre (based on 1437 individual samples from the 120 sources).4
Biziuk et al. (1996) investigated the occurrence of 1,1-DCE in the drinking-water of the Gdansk district of Poland using a method with a detection limit of 0.01 µg/litre. 1,1-DCE was not detected in the 22 samples analysed.
Chung et al. (1997) reported on the occurrence of 1,1-DCE in raw water, treated water, and tap water from 1993 to 1995 in water supplies from six cities in Korea (Seoul, Pusan, Taegu, Taejon, Kwangju, and Inchon) using a method with a detection limit of 0.012 µg/litre. The mean concentration of 1,1-DCE in raw water and treated water at the treatment plants and in tap water from individual households was 0.012, 0.022, and 0.019 µg/litre, respectively.
No reliable data are available to estimate the exposure from food. However, this source is expected to be negligible.
In the workplace, Ott (1976) reported a range of 20–280 mg/m3 for 1,1-DCE in a fibre production facility. These data were collected in 1960 and 1965. US EPA (1985) reported levels of 1,1-DCE in monomer and polymer plants of 90–100 µg/m3 and 25–50 µg/m3, respectively. No recent data were located.
1,1-DCE is rapidly absorbed following inhalation and oral exposures. Because of its low relative molecular mass and hydrophobic nature, dermal absorption is also likely, but there are no relevant published data. In rats treated with 1,1-DCE by gavage in corn oil, complete gastrointestinal absorption occurs at <350 mg/kg body weight (Jones & Hathway, 1978a,b; Putcha et al., 1986). 1,1-DCE is easily transported across the alveolar membrane. At a concentration of 600 mg/m3 and below in inspired air, equilibrium or near steady state is reached in the blood in rats in approximately 45 min (Dallas et al., 1983). Continued uptake in rats reflects to some extent continuing deposition in fatty tissues, but is primarily a result of metabolism of 1,1-DCE.
The major route of excretion for unchanged 1,1-DCE is through the lungs (Jones & Hathway, 1978a). The majority of the 1,1-DCE, however, is rapidly metabolized to non-volatile compounds and covalently bound derivatives (McKenna et al., 1978a,b). In both animal and human tissue, CYP2E1 catalyses the initial oxidation of 1,1-DCE (Dowsley et al., 1996). The covalent binding and cellular damage in kidney, lung, and liver correlate with the high concentration of CYP2E1 in certain cell populations in these tissues (Forkert, 2001). Mice metabolize more 1,1-DCE than rats. For example, when given 50 mg/kg body weight by oral gavage in corn oil, mice excrete 6% and rats excrete 28% of the dose as unchanged 1,1-DCE through the lungs (Jones & Hathway, 1978b). When exposed to 40 mg/m3 by inhalation for a single 6-h period, mice excrete 0.65% and rats excrete 1.63% of the absorbed dose as unchanged 1,1-DCE through the lungs (McKenna et al., 1977). Intraperitoneal administration of [14C]1,1-DCE at 125 mg/kg body weight to mice resulted in the highest concentrations of covalent binding (based on protein content) in the kidney, lung, and liver (Okine et al., 1985; Okine & Gram, 1986a,b).
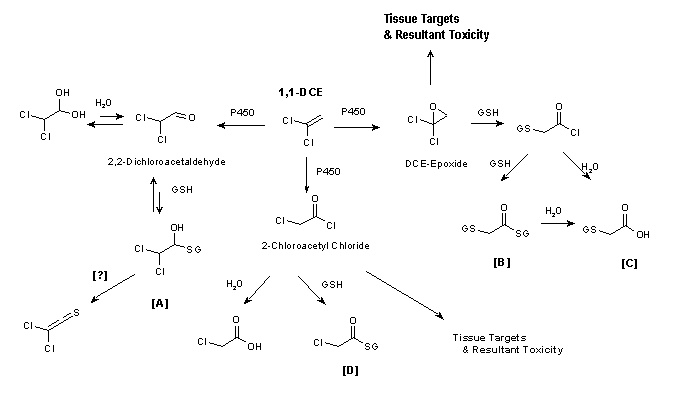
Fig. 1: Pathways for 1,1-dichloroethene metabolism and toxicity (see text for details).
The primary metabolites of 1,1-DCE formed in rat hepatic microsomal incubations are DCE-epoxide, 2,2-dichloroacetaldehyde, and 2-chloroacetyl chloride (Costa & Ivanetich, 1982, 1984; Liebler et al., 1985, 1988). These metabolites were also identified from mouse microsomal incubations (Dowsley et al., 1995). All these electrophilic metabolites undergo secondary reactions, including oxidation, conjugation with GSH, and hydrolysis. The major products formed are GSH conjugates, 2-(S-glutathionyl)acetyl glutathione [B] and 2-S-glutathionyl acetate [C], that are believed to be derived from the DCE-epoxide (Fig. 1). S-(2,2-Dichloro-1-hydroxy)ethyl glutathione [A], the GSH conjugate formed from reaction of GSH with 2,2-dichloroacetaldehyde, was not observed in liver microsomal incubations containing GSH (Dowsley et al., 1995). The acetal, together with chloroacetic acid and S-(2-chloroacetyl)-glutathione [D], the hydrolysis and GSH-conjugated products of 2-chloroacetyl chloride, respectively, were detected at levels much lower than those of the DCE-epoxide-derived conjugates [B] and [C]. In human liver and lung microsomal incubations (Dowsley et al., 1999), the DCE-epoxide-derived GSH conjugates [B] and [C] were the major metabolites detected. 2,2-Dichloroacetaldehyde was detected at low levels. Liver microsomes from three out of five human samples metabolized 1,1-DCE to the epoxide-derived GSH conjugates at levels that were 2.5- to 3-fold higher than those in mouse liver microsomes, based on GSH conjugate formed per milligram of microsomal protein. These GSH conjugates were also the major products formed in lung microsomes from eight human samples; only low levels of 2,2-dichloroacetaldehyde were formed. The mean level of GSH conjugates formed by lung microsomes from humans was about 50% of the amount formed by lung microsomes from mice.
The significance of the metabolic pathway in the liver involving 2,2-dichloroacetaldehyde is unclear. Existing evidence, however, suggests that this pathway is of minor toxicological importance. In addition to 2,2-dichloroacetaldehyde and the GSH conjugate, other potential metabolites include the acetal (the hydration product of the aldehyde), dichloroacetic acid, and dichloroethanol. An initial study with rat liver microsomes found a trace level of 2,2-dichloroacetaldehyde but no detectable dichloroacetic acid (Costa & Ivanetich, 1982). A later report using isolated rat hepatocytes detected dichloroacetic acid and trace levels of 2,2-dichloroacetaldehyde, 2,2-dichloroethanol, and chloroacetic acid (Costa & Ivanetich, 1984). Forkert (1999a) and Forkert & Boyd (2001), using intact mice, found no acetal in liver cytosol. However, the acetal was detected in the bile in one study (Forkert, 1999a) but not mentioned as being found in the bile in the other study (Forkert & Boyd, 2001). In early studies on the metabolism of 1,1-DCE, none of the potential metabolites from this pathway was reported as being found in the urine of rodents using techniques that readily identified chloroacetic acid (McKenna et al., 1977, 1978a,b; Jones & Hathway, 1978a,b). A pharmacokinetic analysis showed that any dichloroacetic acid formed in the liver is rapidly metabolized in the liver to two-carbon, non-chlorinated chemicals and carbon dioxide (Merdink et al., 1998).
The oxidative metabolism of 1,1-DCE reaches saturation in rats at an oral exposure of 10–50 mg/kg body weight and an inhalation exposure of 790 mg/m3 (McKenna et al., 1977; Andersen et al., 1979; Dallas et al., 1983; D’Souza & Andersen, 1988).
Because 1,1-DCE is lipophilic and has a blood-to-air partition coefficient of 5 in rats (D’Souza & Andersen, 1988), any 1,1-DCE not metabolized following oral or inhalation exposure is rapidly exhaled unchanged when exposure is terminated. Based on its low octanol/water partition coefficient, 1,1-DCE will not bioaccumulate in tissues to any significant extent.
D’Souza & Andersen (1988) developed physiologically based pharmacokinetic (PBPK) models for 1,1-DCE in the rat for both oral and inhalation exposure. No validated model is available for humans. D’Souza & Andersen (1988) used allometric scaling to estimate comparative amounts of epoxide formed (mg/kg body weight) in rats and humans. Cardiac output and pulmonary ventilation were scaled by (body weight)0.7, Vmax was scaled by (body weight)0.74, and body fat was estimated at 7% in the 200-g rat and 20% in the 70-kg human. When the oral exposure was less than 5 mg/kg body weight, the estimated amount of epoxide formed was about the same in rats and humans. When the inhalation exposure was less than 400 mg/m3, the estimated amount of epoxide formed was 5-fold lower in humans than in rats.
El-Masri et al. (1996a,b) used a combination of gas uptake experiments in Sprague-Dawley rats and PBPK modelling to assess the potential for interaction between 1,1-DCE and trichloroethene. Both substrates are activated by CYP2E1. Thus, there is a potential for competitive inhibition when simultaneous exposure to both substrates occurs. The results of the gas uptake experiments confirmed a model based on competitive inhibition. There was, however, no evidence of competitive inhibition when exposure to both substrates was 400 mg/m3 or less. As environmental exposures to these chemicals are expected to be below 400 mg/m3, there is little potential for reduced toxicity from 1,1-DCE when individuals are also exposed to trichloroethene.
Mice are more sensitive than rats to the acute toxicity of 1,1-DCE. Toxicity is enhanced by fasting (Jaeger et al., 1974, 1975, 1977a,b; Andersen & Jenkins, 1977; McKenna et al., 1978a,b; Chieco et al., 1981; Moslen et al., 1985), GSH depletion (Jaeger et al., 1974, 1977b; Andersen et al., 1980; Kanz et al., 1988; Moussa and Forkert, 1992), or vehicles that promote rapid oral absorption — for example, aqueous Tween versus mineral or corn oil (Chieco et al., 1981). Toxicity is decreased by agents that decrease metabolism by the cytochrome P450 system (Andersen et al., 1978; Moslen et al., 1989) or hypothyroidism, which increases intracellular GSH (Kanz et al., 1991).
The National Toxicology Program (NTP, 1982) conducted studies to determine lethality in five male and five female F344 rats (9 weeks old) and in five male and five female B6C3F1 mice (9 weeks old) after a single exposure to 1,1-DCE by gavage in corn oil at 0, 10, 50, 100, 500, or 1000 mg/kg body weight. Animals were observed for 14 days. Mortality was 0/10, 1/10, 0/10, 0/10, 1/10, and 2/10 in rats and 0/10, 0/10, 1/10, 0/10, 8/10, and 10/10 in mice at 0, 10, 50, 100, 500, and 1000 mg/kg body weight, respectively. NTP did not calculate an LD50 for either species or report clinical signs of toxicity.
Representative oral LD50s and inhalation LC50s are summarized in Table 1. Clinical signs of toxicity were not reported in these studies.
Table 1: Summary of oral LD50s and inhalation LC50s for exposure of rats and mice to 1,1-dichloroethene.
|
Species |
Dose/exposure |
Effect |
Reference |
|
Oral |
|||
|
Rat (male) |
1550 mg/kg body weight per day |
LD50 |
Jenkins et al., 1972 |
|
Rat (male) |
1800 mg/kg body weight per day |
LD50 |
Ponomarkov & Tomatis, 1980 |
|
Rat (female) |
1500 mg/kg body weight per day |
LD50 |
Ponomarkov & Tomatis, 1980 |
|
Mouse (male) |
217 mg/kg body weight per day |
LD50 |
Jones & Hathway, 1978b |
|
Mouse (female) |
194 mg/kg body weight per day |
LD50 |
Jones & Hathway, 1978b |
|
Inhalation |
|
|
|
|
Rat (male, fed) |
25 000 mg/m3 / 4 h |
LC50 |
Siegel et al., 1971 |
|
Rat (male, fasted) |
800 mg/m3 / 4.1 h |
LT50a |
Andersen et al., 1978 |
|
Mouse (male) |
390 mg/m3 / 22–23 h |
LC50 |
Short et al., 1977c |
|
Mouse (female) |
420 mg/m3 / 22–23 h |
LC50 |
Short et al., 1977c |
|
a |
LT50 values are times taken for 50% mortality following exposure at the concentrations indicated. |
The target organs for toxicity after acute oral or inhalation exposure are the liver, the kidney, and the Clara cells of the lung. The effects in the liver include an increase in liver enzymes in the serum (Jenkins et al., 1972; Jaeger, 1977; Jenkins & Andersen, 1978; Reynolds et al., 1980), severe histopathological damage, including disruption of bile canaliculi, cytoplasmic vacuolization, and haemorrhagic necrosis (Reynolds et al., 1984; Kanz & Reynolds, 1986), an increase in covalent binding of 1,1-DCE (Jaeger et al., 1977a,b; Forkert & Moussa, 1991, 1993), and a decrease in GSH (Reichert et al., 1978, 1979; Kanz et al., 1988; Forkert & Moussa, 1991, 1993) mediated by CYP2E1 metabolism of 1,1-DCE to intermediates that react with GSH (Kainz et al., 1993; Lee & Forkert, 1995). Details of these studies are reported in US EPA (2002d).
Toxic effects of 1,1-DCE exposure in the kidney include increased kidney weight, increased blood urea nitrogen and creatinine (Jenkins & Andersen, 1978; Jackson & Conolly, 1985), and histopathological changes, including vacuolization, tubular dilatation, and necrosis of the proximal tubules (Jenkins & Andersen, 1978; Jackson & Conolly, 1985). These changes are correlated with metabolic activation of 1,1-DCE by CYP2E1 in the proximal tubules, decreased GSH concentration, increased covalent binding of 1,1-DCE, and the presence of a relatively high concentration of β-lyase activity in kidney tissue (Dekant et al., 1989; Brittebo et al., 1993; Dekant, 1996). In addition, renal toxicity can be inhibited by pretreatment of animals with aminooxyacetic acid, an inhibitor of renal cysteinyl-β-lyase (Ban et al., 1995; Cavelier et al., 1996).
The effects in the Clara cells of the lung include extensive histopathological changes (Forkert & Reynolds, 1982; Forkert et al., 1985, 1990), repair of damage through cell proliferation (Forkert et al., 1985), depletion of GSH, and covalent binding of 1,1-DCE mediated through the formation of DCE-epoxide by CYP2E1 (Forkert & Moussa, 1991; Moussa & Forkert, 1992; Lee & Forkert, 1995; Dowsley et al., 1996; Forkert, 1999b). Details of these studies are reported in US EPA (2002d).
Warbrick et al. (2001) tested the ability of 1,1-DCE to cause skin sensitization using the local lymph node assay. 1,1-DCE was dissolved in acetone:olive oil (4:1 v/v) to give a 0%, 10%, 25%, or 50% concentration. Groups of mice (n = 4) were exposed topically on the dorsum of both ears to 25 µl of the test solution daily for 3 consecutive days. Five hours after the injection of [3H]methyl thymidine, the mice were sacrificed. The draining auricular lymph nodes were excised and tested for incorporation of [3H]thymidine. 1,1-DCE failed to elicit a positive response at any concentration tested.
NTP (1982) conducted a 14-day study in male and female F344 rats (five animals of each sex, 9 weeks old) administered 1,1-DCE by gavage in corn oil. Survival was 10/10, 10/10, 10/10, 10/10, 7/10, and 3/10 at 0, 10, 50, 100, 500, and 1000 mg/kg body weight per day, respectively. Mean body weight was significantly depressed at 500 mg/kg body weight per day and above. Haemorrhagic necrosis in the liver was observed in all of the rats that died at 500 and 1000 mg/kg body weight per day. NTP did not report clinical signs of toxicity.
NTP (1982) conducted a 14-day study in male and female B6C3F1 mice (five of each sex, 9 weeks old) administered 1,1-DCE by gavage in corn oil. Survival was 10/10, 10/10, 10/10, 10/10, 10/10, and 0/10 at 0, 10, 50, 100, 500, and 1000 mg/kg body weight per day, respectively. Haemorrhagic necrosis in the liver was observed in all mice at 1000 mg/kg body weight per day. NTP did not report clinical signs of toxicity.
Prendergast et al. (1967) evaluated the toxicity of 1,1-DCE in Long-Evans and Sprague-Dawley rats, Hartley guinea-pigs, beagle dogs, New Zealand albino rabbits, and squirrel monkeys. The test animals (15 rats per group, 15 guinea-pigs per group, 3 rabbits per group, 2 dogs per group, and 3 monkeys per group) were exposed to 1,1-DCE vapours for 8 h/day, 5 days/week, for a total of 30 exposures at 395 ± 32 mg/m3. The age of the animals was not specified. The exposed animals were evaluated for visible signs of toxicity, mortality, and haematological, pathological, and body weight changes. In this study, there were no deaths, no visible signs of toxicity, and no haematological or histopathological changes attributed to exposure to 1,1-DCE. Rabbits and monkeys lost weight (3.6% and 5.9%, respectively). The no-observed-adverse-effect level (NOAEL) in this study is 395 mg/m3 (the highest exposure tested), equivalent to an adjusted NOAEL based on continuous exposure of 94 mg/m3.
Plummer et al. (1990) exposed black hooded Wistar rats to 1,1-DCE at 1000 mg/m3 for 6 h/day, 5 days/week, for 4 weeks (six males and six females, age not specified) or continuously at 200 mg/m3 for 4 weeks (except for two 1.5-h periods per week) (18 males and 18 females, age not specified). The total exposure (concentration × time) was nearly the same for the two profiles (132 000 mg/m3·h for the continuous exposure and 120 000 mg/m3·h for the intermittent exposure). Animals in the intermittent exposure group showed signs of early coagulative necrosis in the liver (incidence not reported). Animals (11/12) in the continuous exposure group showed less severe injury, including fatty changes in variable numbers of hepatocytes and only very occasional focal liver cell necrosis. The lowest-observed-adverse-effect level (LOAEL) in this study is 200 mg/m3.
NTP (1982) conducted a study in male and female F344 rats (10 of each sex, 9 weeks old) administered 1,1-DCE by gavage in corn oil at 0, 5, 15, 40, 100, or 250 mg/kg body weight per day. Animals were exposed 5 times per week for 13 weeks. Representative tissues (skin, lung and bronchi, trachea, bone and bone marrow, spleen, lymph nodes, heart, salivary gland, liver, pancreas, stomach, small intestine, large intestine, kidneys, urinary bladder, pituitary, adrenal, thyroid, parathyroid, mammary gland, prostate and seminal vesicles or uterus, testis or ovary, brain, thymus, larynx, and oesophagus) from animals receiving 250 mg/kg body weight per day and from control animals were examined microscopically. Livers from all groups were examined. Three female rats receiving 250 mg/kg body weight per day died during the first week of the study. No other rats died. The mean body weight was depressed 13% for male rats receiving 250 mg/kg body weight per day compared with controls. Mean body weight in other groups was comparable. Only the liver showed effects attributed to 1,1-DCE. At 250 mg/kg body weight per day, the three female rats that died showed severe centrilobular necrosis. Minimal to moderate hepatocytomegaly was seen in the rest of the rats at 250 mg/kg body weight per day. Minimal to mild hepatocytomegaly was seen in 6/10 male rats and 3/10 female rats that received 100 mg/kg body weight per day. No biologically significant changes were observed in rats that received 40 mg/kg body weight per day or below. The NOAEL in this study is 40 mg/kg body weight per day (equivalent to 28.6 mg/kg body weight per day after adjusting for 5 days/week exposure); the LOAEL is 100 mg/kg body weight per day (equivalent to 71.4 mg/kg body weight per day after adjusting for 5 days/week exposure).
NTP (1982) conducted a study in male and female B6C3F1 mice (10 of each sex, 9 weeks old) administered 1,1-DCE by gavage in corn oil at 0, 5, 15, 40, 100, or 250 mg/kg body weight per day. Animals were exposed 5 times per week for 13 weeks. Representative tissues (see above paragraph) from mice receiving 100 and 250 mg/kg body weight per day and from control animals were examined microscopically. Livers from all groups were also examined. Survival was 10/10, 10/10, 10/10, 9/10, 8/10, and 0/10 in males and 10/10, 9/10, 9/10, 10/10, 7/10, and 1/10 in females at 0, 5, 15, 40, 100, and 250 mg/kg body weight per day, respectively. At 100 mg/kg body weight per day, there was a decrease in mean body weight in males (14%) but not females. No biologically significant change in mean body weight was observed at lower exposures. Only the liver showed effects attributed to 1,1-DCE. Centrilobular necrosis of the liver was observed in 5/10 males and 5/10 females that received 250 mg/kg body weight per day and 2/10 males and 2/10 females that received 100 mg/kg body weight per day. No biologically significant changes in the liver occurred in mice receiving 40 mg/kg body weight per day or below. The NOAEL in this study is 40 mg/kg body weight per day (equivalent to 28.6 mg/kg body weight per day after adjusting for 5 days/week exposure); the LOAEL is 100 mg/kg body weight per day (equivalent to 71.4 mg/kg body weight per day after adjusting for 5 days/week exposure).
Quast et al. (1983) conducted a study in beagle dogs (four per group, 8 months old) administered 1,1-DCE by gavage in peanut oil at 0, 6.25, 12.5, or 25 mg/kg body weight per day for 97 days. There were no significant differences among any groups in appearance and demeanour, mortality, body weight, food consumption, haematology, urinalysis, clinical chemistry determinations, organ weights, and organ-to-body-weight ratios. No exposure-related gross or histopathological changes were present in tissues. There was no depletion of the non-protein sulfhydryl levels in the liver or kidneys. The no-observed-effect level (NOEL) in this study is 25 mg/kg body weight per day (the highest exposure tested).
Prendergast et al. (1967) evaluated the toxicity of 1,1-DCE in Long-Evans or Sprague-Dawley rats, Hartley guinea-pigs, beagle dogs, New Zealand albino rabbits, and squirrel monkeys. The test animals (15 rats per group, 15 guinea-pigs per group, 3 rabbits per group, 2 dogs per group, and 3 or 9 monkeys per group) were exposed continuously for 90 days to 1,1-DCE vapours at 20 ± 2.1, 61 ± 5.7, 101 ± 4.4, or 189 ± 6.2 mg/m3. The concurrent controls included 304 rats, 314 guinea-pigs, 48 rabbits, 34 dogs, and 57 monkeys. The age of the animals was not specified. The exposed animals were evaluated for visible signs of toxicity, mortality, and haematological, biochemical, pathological, and body weight changes. There was apparent exposure-related mortality in guinea-pigs and monkeys. Mortality was 2/314, 2/45, 3/15, 3/15, and 7/15 in guinea-pigs and 1/57, 1/21, 0/9, 2/3, and 3/9 in monkeys in the 0, 20, 61, 101, or 189 mg/m3 exposure groups, respectively. The guinea-pigs died on the 3rd and 4th days of exposure, between the 3rd and 6th days of exposure, and between the 4th and 9th days of exposure in the 61, 101, and 189 mg/m3 groups, respectively. The monkeys died on days 39 and 47 and on days 26, 60, and 64 in the 101 and 189 mg/m3 groups, respectively. There were no visible signs of toxicity in any surviving animals. Because visible signs of toxicity were not observed and only non-life-threatening liver damage is apparent in this study (see below), the mortality data in guinea-pigs and monkeys are given no weight. Varying degrees of growth depression were found in all exposures, but growth depression was significant in all species only at 189 mg/m3. The test animals exhibited no significant haematological alterations, and serum urea nitrogen levels were within control limits in all exposures in which determinations were made. Significant elevations of serum glutamic–pyruvic transaminase and liver alkaline phosphatase activities were found in rats (a 3-fold and 1.75-fold increase, respectively) and guinea-pigs (a 7-fold and 2.4-fold increase, respectively) exposed to 189 mg/m3 (other species not tested), but not at 20 mg/m3 (enzyme levels at intermediate exposures not tested). Histopathological examination of liver from dogs, monkeys, and rats revealed damage at 189 mg/m3 (other species not examined). The effects observed included fatty metamorphosis, focal necrosis, haemosiderosis deposition, lymphocytic infiltration, bile duct proliferation, and fibrosis. The changes were most severe in dogs. Sections of kidney from all rats showed nuclear hypertrophy of the tubular epithelium. No detectable liver or kidney damage was observed in any species exposed to 101 mg/m3 or less. The NOAEL in this study is 101 mg/m3; the LOAEL is 189 mg/m3.
Bioassays for cancer by the oral route of exposure have been conducted in rats (Ponomarkov & Tomatis, 1980; NTP, 1982; Quast et al., 1983; Maltoni et al., 1985), mice (NTP, 1982), and trout (Hendricks et al., 1995). Some of these bioassays were conducted at an exposure below the maximum tolerated dose. The bioassay conducted by Maltoni et al. (1985) exposed the animals for only 1 year. The bioassay in rats (Quast et al., 1983) and the bioassay in mice (NTP, 1982) were well conducted, and both showed some toxicity in the liver at the highest exposure. Neither of these bioassays provides any significant evidence that 1,1-DCE is a carcinogen by the oral route of exposure.
Quast et al. (1983) summarized the results of a 2-year chronic toxicity and carcinogenicity study of 1,1-DCE conducted in Sprague-Dawley rats (6–7 weeks old). Humiston et al. (1978) reported detailed data from the study. There were 80 rats of each sex in the control group and 48 rats of each sex in each exposed group. The 1,1-DCE was incorporated in the drinking-water of the rats at nominal concentrations of 0, 50, 100, or 200 mg/litre. The time-weighted average exposure over the 2-year period was 7, 10, and 20 mg/kg body weight per day for males and 9, 14, and 30 mg/kg body weight per day for females. There were no significant differences among the groups in appearance and demeanour, mortality, body weight, food consumption, water consumption, haematology, urinalysis, clinical chemistry determinations, organ weights, or organ-to-body-weight ratios. After 1 year on study, there was no depletion of the non-protein sulfhydryl levels in the liver or the kidneys (Rampy et al., 1977). The only treatment-related effect observed in rats was minimal hepatocellular mid-zonal fatty change and hepatocellular swelling. In the male rats at the termination of the study, there was an increased incidence of minimal hepatocellular fatty change (control, 14/80; 50 mg/litre, 5/48; 100 mg/litre, 13/48; 200 mg/litre, 19/47) and minimal hepatocellular swelling (control, 0/80; 50 mg/litre, 1/48; 100 mg/litre, 2/48; 200 mg/litre, 3/47). The changes were statistically significant (P < 0.05) only in the 200 mg/litre group. In female rats at the termination of the study, there was an increased incidence of minimal hepatocellular fatty change (control, 10/80; 50 mg/litre, 12/48; 100 mg/litre, 14/48; 200 mg/litre, 22/48; statistically significant [P < 0.05] at 100 and 200 mg/litre) and minimal hepatocellular swelling (control, 3/80; 50 mg/litre, 7/48; 100 mg/litre, 11/48; 200 mg/litre, 20/48; statistically significant [P < 0.05] in all groups). No hepatocellular necrosis was evident at any exposure. In addition, there was no change in liver weight, no change in clinical chemistry measurements diagnostic for liver damage, and no other indication of abnormal liver function. Based on the minimal nature of the hepatocellular swelling reported by the authors, this effect is not considered biologically significant and is not an adverse effect in this study. The statistically significant hepatocellular mid-zonal fatty change, however, is considered a minimal adverse effect in this study. Accordingly, the NOAEL in male rats is 10 mg/kg body weight per day and the LOAEL is 20 mg/kg body weight per day; the NOAEL in female rats is 9 mg/kg body weight per day and the LOAEL is 14 mg/kg body weight per day. The US EPA conducted a BMD analysis for the results in female rats (Appendix 4). In female rats, the BMD10 (BMD for a 10% response) is 6.6 mg/kg body weight per day, and the BMDL10 is 4.6 mg/kg body weight per day.
Bioassays for cancer by the inhalation route of exposure have been conducted in rats (Lee et al., 1977, 1978; Viola & Caputo, 1977; Hong et al., 1981; Maltoni et al., 1985; Quast et al., 1986; Cotti et al., 1988), mice (Lee et al., 1977, 1978; Hong et al., 1981; Maltoni et al., 1985), and hamsters (Maltoni et al., 1985). None of these bioassays was conducted by a protocol that meets current standards. The major defects in most of these bioassays include exposure of the animals for 1 year and exposure at less than the maximum tolerated dose. The only bioassay showing some evidence of carcinogenicity was the study in Swiss-Webster mice (Maltoni et al., 1985). This study was conducted at or near the maximum tolerated dose, as animals exposed at 200 mg/m3 died after a few exposures.
Quast et al. (1986) and Rampy et al. (1977) reported results from studies that exposed male and female Sprague-Dawley rats (Spartan substrain, 86 animals per sex per group) to 1,1-DCE by inhalation 6 h/day, 5 days/week, for up to 18 months. Interim sacrifices
(4–5 animals per sex per group) occurred at 1, 6, and 12 months. Rats were exposed to 1,1-DCE concentrations of 40 or 160 mg/m3 for the first 5 weeks of the study. Based upon the absence of observable treatment-related effects among rats sacrificed after 1 month of exposure, the concentrations were increased to 100 and 300 mg/m3. Exposures were continued at these concentrations through the 18th month of the study. The surviving animals were then held without exposure to 1,1-DCE until 24 months. Cytogenetic evaluations were performed on a separate group of animals (four per sex) exposed to 0, 100, or 300 mg/m3 for 6 months. There were no exposure-related changes in mortality, appearance and demeanour, body weight, clinical chemistry determinations, haematological evaluations, urinalysis, or cytogenetic evaluation of bone marrow preparations. Minimal hepatocellular fatty change in the mid-zonal region of the hepatic lobule was observed in both male and female rats in the 100 and 300 mg/m3 groups at the 6-month interim sacrifice (male: control, 0/5; 100 mg/m3, 1/5; 300 mg/m3, 4/5; female: control, 0/5; 100 mg/m3, 2/5; 300 mg/m3, 4/5). The fatty change was also observed at the 12-month sacrifice, but there was no indication of progression of severity (male: control, 0/5; 100 mg/m3, 3/5; 300 mg/m3, 5/5; female: control, 0/5; 100 mg/m3, 5/5; 300 mg/m3, 5/5). At the 18-month sacrifice, the incidence of this change was no longer increased in male rats (control, 0/27; 100 mg/m3, 0/25; 300 mg/m3, 1/27). However, the change persisted in female rats (control, 0/16; 100 mg/m3, 6/29; 300 mg/m3, 7/20). In female rats, the fatty change was statistically significant (P < 0.05) only at the higher exposures. During the last 6 months of the study, after exposure had been discontinued, this effect was no longer discernible (male: control, 0/46; 100 mg/m3, 1/47; 300 mg/m3, 0/51; female: control, 0/49; 100 mg/m3, 0/46; 300 mg/m3, 1/48). Although the minimal hepatocellular mid-zonal fatty change is reversible and did not result in altered organ weight, clinical chemistry changes diagnostic for liver damage, or any obvious decrement in liver function, the fatty change in liver is considered a minimal adverse effect. Accordingly, the NOAEL in male rats in this study is 300 mg/m3 (the highest exposure tested). The NOAEL in female rats in this study is 100 mg/m3; the LOAEL is 300 mg/m3. The US EPA conducted a BMC analysis (Appendix 4). In female rats, using a conversion factor of 3.97, the BMC10 (the BMC for a 10% response) is 59.9 mg/m3 and the BMCL10 is 38.9 mg/m3, equivalent to 6.9 mg/m3 adjusted for continuous exposure (38.9 mg/m3 × 6/24 × 5/7).
Maltoni et al. (1985) conducted a carcinogenicity and toxicity study of 1,1-DCE in Swiss mice. Animals (9 or 16 weeks old) were exposed by inhalation to 0, 40, or 100 mg/m3 for 4 h/day, 4–5 days/week, for 52 weeks. Groups of animals exposed to 200 mg/m3 and higher in this study showed extreme toxicity after only a few exposures, causing termination of this portion of the bioassay. There were two control groups, one with 180 animals (90 of each sex) and the other with 200 animals (100 of each sex). The 40 mg/m3 group had 60 animals (30 of each sex). There were two groups exposed to 100 mg/m3, one with 60 animals (30 of each sex) and the other with 240 animals (120 of each sex). Following the 52-week exposure, animals were observed until spontaneous death (total duration 126 weeks). Body weight was measured every 2 weeks during the 52-week exposure and every 8 weeks thereafter. Full necropsy and histopathological examination were performed. There were no biologically significant changes in body weight. The exposed animals had a somewhat higher survival than controls. There was a statistically signifi-cant increase (P < 0.01) compared with controls in kidney adenocarcinomas in male mice at 100 mg/m3, but not in male mice at 40 mg/m3 or in female mice at either exposure. As reported in US EPA (1985), the incidence was 0/126 (0%), 0/25 (0%), and 28/119 (23.5%) in male mice in the combined control, 40 mg/m3, and combined 100 mg/m3 groups, respectively. Because of the study design, it is impossible to assess tumour development in relation to histopathological changes associated with typical age-related changes in the kidney. There was a statistically significant increase (P < 0.01) compared with control in mammary carcinomas in female mice at both exposures, but there was no clear exposure–response relationship. As reported in US EPA (1985), the incidence was 3/185 (1.6%), 6/30 (20%), and 16/148 (11%) in females in the combined control, 40 mg/m3, and combined 100 mg/m3 groups, respectively. There was also a statistically significant increase (P < 0.01) compared with control in pulmonary adenomas in both exposed groups, but there was no clear exposure–response relationship. As reported in US EPA (1985), the incidence was 6/153 (3.9%), 11/28 (39.3%), and 23/141 (16.3%) in males and 6/178 (3.4%), 3/30 (10%), and 18/147 (12.2%) in females in the combined controls, 40 mg/m3, and combined 100 mg/m3 groups, respectively. There were no pulmonary carcinomas in any mice. The incidence data are reported as the number of tumour-bearing animals compared with the number of animals alive when the first tumour was observed in that organ (kidney adenocarcinoma, 55 weeks; mammary tumour, 27 weeks; pulmonary adenoma, 36 weeks). The researchers discounted the significance of the mammary and pulmonary tumours.
Van Duuren et al. (1979) evaluated the carcinogenicity of 1,1-DCE in male and female non-inbred Ha:ICR Swiss mice. Carcinogenicity was assessed in three types of tests: a dermal initiation-promotion assay; a repeated dermal application assay; and a subcutaneous injection assay. Vehicle, no-treatment, and positive control groups were included in the tests. In the initiation-promotion assay, 1,1-DCE was tested as a tumour-initiating agent with phorbol myristate acetate as the promoter. Thirty female mice were treated with 121 mg 1,1-DCE each. A significant increase (P < 0.005) was observed in skin papillomas (nine in eight mice). In the repeated dermal application assay, exposures of 40 and 121 mg per mouse were used. 1,1-DCE was applied to the back of the shaved animals (30 females per dose). No sarcomas were observed at the treatment site. Although 19 mice in the high-dose group and 12 in the low-dose group had lung tumours and 2 mice in the high-dose group had stomach tumours, the tumour incidence at both sites was not significantly different from controls (30 lung tumours and 5 stomach tumours). In the subcutaneous injection assay, the test animals were given weekly injections of 2 mg of 1,1-DCE. After 548 days on test, none of the animals injected with 1,1-DCE developed sarcomas at the injection site. 1,1-DCE showed initiating activity in the two-stage carcinogenesis experiments but was inactive as a whole-mouse dermal carcinogen and after subcutaneous injection.
There is a fairly extensive database on the genotoxicity of 1,1-DCE. A complete summary of the results of genetic toxicity testing is provided in Table 2. 1,1-DCE induced mutations in Salmonella typhimurium and Escherichia coli in the presence of an exogenous metabolic activation system. 1,1-DCE was also weakly mutagenic in Salmonella typhimurium TA 100 in the absence of a metabolic activation system. In Saccharomyces cerevisiae, 1,1-DCE induced reverse mutation and mitotic gene conversion in vitro and in a host-mediated assay in mice. In a single study in Saccharomyces cerevisiae, it induced aneuploidy in the presence and absence of metabolic activation. In vitro, gene mutations were increased in mouse lymphoma cells but not in Chinese hamster lung cells with or without an exogenous metabolic system. In a single study, 1,1-DCE induced sister chromatid exchanges in Chinese hamster lung cells in the presence of an exogenous metabolic activation system, but not in its absence. In single studies in vivo, 1,1-DCE did not induce micronuclei or chromosomal aberrations in bone marrow or in fetal erythrocytes of mice or in bone marrow of rats, and it did not induce dominant lethal mutations in mice or rats. A test for chromosomal damage in the mouse lymphoma system has not been conducted.
Table 2: Genetic and related effects of 1,1-dichloroethene.
|
Test system |
Resulta |
Doseb |
Reference |
|
|
Without MA |
With MA |
|||
|
S. typhimurium BA13/BAL13, forward mutation |
– |
+ |
500 |
Roldan-Arjona et al., 1991 |
|
S. typhimurium TA 100, reverse mutation |
NT |
+ |
2% in air |
Malaveille et al., 1997 |
|
S. typhimurium TA 100, reverse mutation |
NT |
+ |
5% in air |
Jones & Hathway, 1978c |
|
S. typhimurium TA 100, reverse mutation |
– |
+ |
5% in air |
Simmon & Tardiff, 1978 |
|
S. typhimurium TA 100, reverse mutation |
+ |
+ |
5% in air |
Waskell, 1978 |
|
S. typhimurium TA 100, reverse mutation |
NT |
+ |
2% in air |
Bartsch et al., 1979 |
|
S. typhimurium TA 100, reverse mutation |
– |
+ |
1500 mg/m3 in air |
Oesch et al., 1983 |
|
S. typhimurium TA 100, reverse mutation |
(+) |
+ |
125 |
Strobel & Grummt, 1987 |
|
S. typhimurium TA 104, reverse mutation |
– |
– |
500 |
Strobel & Grummt, 1987 |
|
S. typhimurium TA 1535, reverse mutation |
– |
+ |
3% in air |
Baden et al., 1977 |
|
S. typhimurium TA 1535, reverse mutation |
NT |
+ |
5% in air |
Jones & Hathway, 1978c |
|
S. typhimurium TA 1535, reverse mutation |
– |
+ |
1500 mg/m3 in air |
Oesch et al., 1983 |
|
S. typhimurium TA 1537, reverse mutation |
– |
(+) |
1500 mg/m3 in air |
Oesch et al., 1983 |
|
S. typhimurium TA 98, reverse mutation |
– |
+ |
1500 mg/m3 in air |
Oesch et al., 1983 |
|
S. typhimurium TA 98, reverse mutation |
– |
(+) |
125 |
Strobel & Grummt, 1987 |
|
S. typhimurium TA 92, reverse mutation |
– |
(+) |
1500 mg/m3 in air |
Oesch et al., 1983 |
|
S. typhimurium TA 97, reverse mutation |
– |
+ |
5 |
Strobel & Grummt, 1987 |
|
E. coli K12, forward or reverse mutation |
– |
(+) |
242 |
Oesch et al., 1983 |
|
E. coli WP2 uvrA, reverse mutation |
– |
+ |
1500 mg/m3 in air |
Oesch et al., 1983 |
|
S. cerevisiae D7, gene conversion |
– |
+ |
2910 |
Bronzetti et al., 1983 |
|
S. cerevisiae D7, mitotic gene conversion |
+c |
– |
7300 |
Koch et al., 1988 |
|
S. cerevisiae D7, reverse mutation |
– |
+ |
2910 |
Bronzetti et al., 1983 |
|
S. cerevisiae D7, reverse mutation |
+c |
+ |
4876 |
Koch et al., 1988 |
|
S. cerevisiae D61.M, aneuploidy |
+ |
+ |
2435 |
Koch et al., 1988 |
|
Gene mutation, Chinese hamster lung V79 cells, hprt locus in vitro |
– |
– |
10% in air |
Drevon & Kuroki, 1979 |
|
Gene mutation, Chinese hamster lung V79 cells, ouabain resistance in vitro |
– |
– |
10% in air |
Drevon & Kuroki, 1979 |
|
Gene mutation, mouse lymphoma L5178Y cells, tk locus in vitro |
? |
+ |
0.16% in air |
McGregor et al., 1991 |
|
Sister chromatid exchange, Chinese hamster lung in vitro |
– |
+ |
75 |
Sawada et al., 1987 |
|
Chromosomal aberrations, Chinese hamster DON-6 cells in vitro |
– |
NT |
2910 |
Sasaki et al., 1980 |
|
Chromosomal aberrations, Chinese hamster fibroblast CHL cells in vitro |
– |
NT |
2000 |
Ishidate, 1983 |
|
Chromosomal aberrations, Chinese hamster lung cells in vitro |
– |
+ |
250 |
Sawada et al., 1987 |
|
Host-mediated assay, S. cerevisiae D7 in CD mouse hosts |
+ |
NT |
100 po × 23 |
Bronzetti et al., 1981 |
|
Host-mediated assay, S. cerevisiae D7 in CD mouse hosts |
+ |
NT |
400 po × 1 |
Bronzetti et al., 1981 |
|
Micronucleus test, mouse bone marrow in vivo |
– |
200 po × 1 |
Sawada et al., 1987 |
|
|
Micronucleus test, mouse fetal erythrocytes in vivo |
– |
100 po × 1 |
Sawada et al., 1987 |
|
|
Chromosomal aberrations, Sprague-Dawley rat bone marrow in vivo |
– |
300 mg/m3 inh, 6 h/day, 5 days/week, 6 months |
Rampy et al., 1977 |
|
|
Dominant lethal test, male CD-1 mice |
– |
200 mg/m3 inh, 6 h/day, 5 days |
Anderson et al., 1977 |
|
|
Dominant lethal test, CD rats |
– |
220 mg/m3 inh, 6 h/day, 5 days/week, 11 weeks |
Short et al., 1977b |
|
|
a |
MA, metabolic activation; +, positive; (+), weak positive; –, negative; NT, not tested; ?, inconclusive. |
|
b |
LED, lowest effective dose; HID, highest ineffective dose. In vitro tests, µg/ml; in vivo tests, mg/kg body weight; po, orally; inh, inhalation. |
|
c |
Positive in cells grown in logarithmic phase. |
Reitz et al. (1980) investigated the ability of 1,1-DCE to cause DNA alkylation, DNA repair, and DNA replication in liver and kidney of rats and mice. Male Sprague-Dawley rats (body weight 200–250 g) and male CD-1 mice (body weight 18–20 g) were exposed by inhalation for 6 h to 40 or 200 mg/m3. There was only a minimal increase in DNA alkylation in both rats and mice at 200 mg/m3. Similarly, DNA repair in kidney of mice was only minimally increased at 200 mg/m3. However, tissue damage (kidney nephrosis at 200 mg/m3, minimal effect at 40 mg/m3), an increase in DNA replication (7-fold increase in [3H]thymidine incorporation at 40 mg/m3, 25-fold increase at 200 mg/m3), and an increase in mitotic figures occurred. There was no observed histopathological damage or increased DNA replication in the liver of mice at 40 or 200 mg/m3. In rats, there was a small increase in DNA replication (2-fold increase in [3H]thymidine incorporation) in the kidney but no increase in liver at 40 mg/m3. This assay was not conducted in rats exposed to 200 mg/m3.
Nitschke et al. (1983) evaluated the reproductive and developmental toxicity of 1,1-DCE in Sprague-Dawley rats. Three generations of test animals were exposed to drinking-water containing nominal 1,1-DCE concentrations of 0 (initially 15 males and 30 females), 50, 100, or 200 mg/litre (initially 10 males and 20 females at each exposure). The authors provided no information on water consumption. This study was a companion study to Quast et al. (1983), using the same concentrations of 1,1-DCE in drinking-water. In Quast et al. (1983), the average exposure for females was 9, 14, and 30 mg/kg body weight per day. After 100 days of exposure, the rats were mated. In this three-generation study, there were no biologically significant changes in fertility index, average number of pups per litter, average body weight of pups, or pup survival at any exposure. Neonatal survival was decreased from concurrent control values in the F2 and F3a litters of dams ingesting 1,1-DCE from drinking-water. The survival indices, however, were within the range of control values for this strain of rats in this laboratory. The authors attributed the decreased survival index in F2 to increased litter size at birth in dams exposed to 1,1-DCE. The apparent effect seen in the F3a litters was not repeated in subsequent matings of the same adults to produce either the F3b or the F3c litters. The authors attributed the decreased survival in the F3a litters as being due to chance. Histopathological examination of tissues of rats exposed to 1,1-DCE in the drinking-water in utero, during lactation, and post-weaning revealed slight hepatocellular fatty change and an accentuated hepatic lobular pattern of a reversible nature in the adult rats (data not reported, but the observation is consistent with that reported by Quast et al. [1983] in a chronic bioassay). These effects were observed in the 100 and 200 mg/litre groups in the F1 generation and in all groups of the F2 generation. The authors did not present incidence data or report statistical analysis. Exposure to 1,1-DCE in drinking-water at concentrations causing mild, dose-related changes in the liver did not affect the reproductive capacity of rats through three generations, which produced six sets of litters. The NOAEL for reproductive and developmental toxicity in this study is 200 mg/litre for exposure to 1,1-DCE in drinking-water (the highest exposure tested and about 30 mg/kg body weight per day).
Murray et al. (1979) evaluated the developmental toxicity of 1,1-DCE administered in drinking-water at 0 (27 animals) or 200 mg/litre (26 animals) to pregnant Sprague-Dawley rats (body weight 250 g). Rats were exposed on gestation days 6–15 at 40 mg/kg body weight per day. No teratogenic effects were seen in the embryos using standard techniques for soft and hard tissue examination, and there was no evidence of toxicity to the dams or their offspring. The NOEL for developmental toxicity in this study is 40 mg/kg body weight per day (the only exposure tested).
Dawson et al. (1993) evaluated the ability of 1,1-DCE administered in drinking-water at 0.15 or 110 mg/litre to female Sprague-Dawley rats (body weight 250 g) to induce fetal cardiac changes. Rats were administered 110 mg 1,1-DCE/litre for 61 days before mating or for 48 days before mating and for 20 days during gestation. Other rats were administered 0.15 mg 1,1-DCE/litre for 82 days before mating or for 56 days before mating and for 20 days during gestation. The dams were killed on gestational day 22, and the gravid uterus was removed and examined. There was no effect on maternal weight gain, average resorption sites (sites where development began but resorption later occurred), or average implantation sites (sites that did not appear to develop beyond implantation and contained a metrial gland only). There was no increase in the incidence of cardiac changes when dams were exposed only before mating. There was, however, a statistically significant increase (P < 0.01) in the percentage of fetuses with cardiac changes (atrial septal, mitral valve, and aortic valve changes) when the dams were exposed before mating and during gestation. The incidences were control, 7/232 (3%); 0.15 mg/litre, 14/121 (11.6%); and 110 mg/litre, 24/184 (13%). This statistical analysis was based on total occurrence of affected fetuses. Because the dam, not individual fetuses, was exposed, a nested statistical analysis is preferred. Such an analysis takes into account the correlation among fetuses within a litter and the possible nesting of effects within litters. This analysis has not been conducted, because all the necessary data are not available. The study author (B. Dawson, personal communication, 2001) provided additional data to resolve typographical errors in the exposure information for each group and to clarify the number of affected litters and number of fetuses per litter affected. The exposure doses for dams before and during pregnancy were 0, 0.02, and 18 mg/kg body weight per day in the control, 0.15 mg/litre, and 110 mg/litre groups, respectively. The numbers of affected litters were 5/21 (24%), 8/11 (73%), and 13/17 (76%), respectively. The mean numbers of affected fetuses per litter, only affected litters considered, were 1.40 (13% of the fetuses in the litter), 1.75 (16% of the fetuses in the litter), and 1.85 (17% of the fetuses in the litter), respectively. The mean numbers of affected fetuses per litter, all litters considered, were 0.33 (3% of the fetuses in the litter), 1.27 (12% of the fetuses in the litter), and 1.41 (13% of the fetuses in the litter), respectively.
Dawson et al. (1993) did a much more thorough evaluation of alterations in cardiac development than is done in standard developmental toxicity testing protocols. There is no experience with the background rates or the functional significance of such alterations from other studies or laboratories. The incidence of alterations in control fetuses (3% of all fetuses, 24% of all litters, and 1.40 affected fetuses per litter) suggests a high background incidence. The authors report that examinations were done blind to the treatment group, so the data are presumed not to be affected by observer bias.
There is no demonstrated exposure–response relationship in Dawson et al. (1993). A 900-fold increase in exposure did not produce a significant increase in response in any measure of effect. The cardiac changes are of questionable biological significance, as no biologically significant effects on growth or survival were reported in the three-generation study (Nitschke et al., 1983). No cardiac effects were reported in a prenatal developmental study (Murray et al., 1979); however, in this study, exposure to 1,1-DCE did not occur throughout pregnancy. The pharmacokinetics of 1,1-DCE make it biologically implausible that the cardiac changes are causally associated with exposure. The exposures used in Dawson et al. (1993) are below the level of saturation of CYP2E1 in the rat liver. Essentially all of the 1,1-DCE administered to the dams will be metabolized in the liver and will react with GSH or macromolecules in the liver (see the discussion and references in section 7). Therefore, it is extremely unlikely that any significant amount of 1,1-DCE or any toxic metabolite will be in the fetal compartment. CYP2E1 is not expressed in fetal liver, but begins to be expressed shortly after birth (Cresteil, 1998). There is no information on the expression of CYP2E1 in fetal cardiac tissue. Cardiac tissue, however, is not generally considered to be a tissue with significant potential for metabolism of xenobiotics. For these reasons, it cannot be concluded that the cardiac changes are caused by exposure to 1,1-DCE.
Short et al. (1977a) evaluated the developmental toxicity of 1,1-DCE administered by inhalation to pregnant CD rats (Charles River). Animals were exposed to 0 mg/m3 (58 animals), 60 mg/m3 (18 animals), 230 mg/m3 (20 animals), 1200 mg/m3 (18 animals), or 1800 mg/m3 (18 animals) for 22–23 h/day on gestation days 6–16. Dams were sacrificed on gestation day 20. There was maternal toxicity, as shown by severe maternal weight loss (>28 g per dam) at 60 mg/m3 and higher and by statistically significant maternal mortality at 230 mg/m3 and higher. The mortality was 0/58, 2/18, 3/20, 4/18, and 7/8 in the control, 60 mg/m3, 230 mg/m3, 1200 mg/m3, and 1800 mg/m3 groups, respectively. There was a statistically significant increase in the mean number of fetuses per litter with hydrocephalus at 60 mg/m3 and 230 mg/m3, with malaligned sternebrae at 60 mg/m3, and with unossified sternebrae at 230 mg/m3. Because of the severe maternal toxicity at 60 mg/m3 and higher, this study is not useful for evaluating developmental toxicity.
Short et al. (1977a) evaluated the developmental toxicity of 1,1-DCE administered by inhalation to pregnant CD-1 mice (Charles River). Animals were exposed to 0 mg/m3 (65 animals), 60 mg/m3 (23 animals), 120 mg/m3 (19 animals), 230 mg/m3 (21 animals), 580 mg/m3 (18 animals), or 1200 mg/m3 (15 animals) for 22–23 h/day on gestation days 6–16. Dams were sacrificed on gestation day 17. At 120 mg/m3 and higher, there was severe fetal toxicity with complete early resorption of the litters. At 60 mg/m3, there was no evidence of maternal toxicity, no decrease in fetal body weight, and no decrease in the percentage of viable fetuses. At 60 mg/m3, there was an increase in the mean number of fetuses per litter with hydrocephalus, occluded nasal passages, microphthalmia, cleft palate, small liver, and hydronephrosis. None of these changes, however, was statistically significant when compared with controls. Also at 60 mg/m3, there was a statistically significant increase in the mean number of fetuses with an unossified incus and with incompletely ossified sternebrae. This study provides evidence of fetal toxicity at 60 mg/m3. In this study, the LOAEL for developmental toxicity is 60 mg/m3, the lowest exposure tested.
Short et al. (1977a) evaluated the developmental neurotoxicity of 1,1-DCE administered by inhalation to CD rats (Charles River). Pregnant rats were exposed to 0 mg/m3 (24 animals), 220 mg/m3 (20 animals), or 1100 mg/m3 (19 animals) for 22–23 h/day on gestation days 8–20. There was maternal toxicity at both exposures, as shown by weight loss of 7 g per dam at 220 mg/m3 and 15 g per dam at 1100 mg/m3, but no maternal mortality at either exposure. There was complete resorption of three litters at 1100 mg/m3. After normal delivery, there was a statistically significant decrease in average pup weight compared with control at both exposures on postnatal day 1. The difference in pup weight between control and exposed groups decreased with time and disappeared by postnatal day 21. There was no evidence of developmental neurotoxicity at either exposure in pups tested at various times from postnatal day 1 to day 21 in a battery of behavioural tasks, including surface righting, pivoting, auditory startle, bar holding, righting in air, visual placing, swimming ability, physical maturation, and activity. This study shows evidence of maternal and fetal toxicity at both exposures, but no evidence of developmental neurotoxicity at either exposure. Accordingly, the NOAEL for developmental neurotoxicity in this study is 1100 mg/m3, the highest exposure tested.
Murray et al. (1979) evaluated the developmental toxicity of 1,1-DCE administered by inhalation to pregnant Sprague-Dawley rats (body weight 250 g). Animals were exposed to 0 mg/m3 (20 or 47 animals), 80 mg/m3 (44 animals), 320 mg/m3 (30 animals), or 640 mg/m3 (30 animals) for 7 h/day on gestation days 6–15. At 80 mg/m3, there was no maternal toxicity and no effect on embryonal or fetal development. At 320 and 640 mg/m3, there was toxicity to the dams (statistically significant depression in weight gain at gestation days 6–9: 45% at 320 mg/m3 and 86% at 640 mg/m3). At 320 and 640 mg/m3, there was also a statistically significant increased incidence of wavy ribs and delayed ossification of the skull, regarded as an embryotoxic effect. Both effects were more severe at 640 mg/m3. No teratogenic effects were seen at any exposure. The NOAEL for developmental toxicity in this study is 80 mg/m3.
Murray et al. (1979) evaluated the developmental toxicity of 1,1-DCE administered by inhalation to New Zealand White rabbits (body weight 3.4–4.7 kg). Animals were exposed to 0 mg/m3 (16 animals), 320 mg/m3 (22 animals), or 640 mg/m3 (18 animals) for 7 h/day on gestation days 6–18. At 320 mg/m3, there was no maternal toxicity and no effect on embryonal or fetal development. Toxicity to both the dams and their developing embryos was observed at 640 mg/m3. There was a marked increase in the incidence of resorptions per litter (0.3 ± 0.6 versus 2.7 ± 3.9). There was also a significant change in the incidence of several minor skeletal variations in their offspring, including an increase in the occurrence of 13 pairs of ribs and an increased incidence of delayed ossification of the fifth sternebra (data not reported). No teratogenic effects were seen at any exposure. The NOAEL for developmental toxicity in this study is 320 mg/m3.
Siletchnik & Carlson (1974) investigated the effects of epinephrine on the cardiac sensitization of 1,1-DCE in male albino rats. The test animals (body weight 250–400 g) were exposed to 1,1-DCE at 0 or 100 000 ± 2400 mg/m3, and the dose of epinephrine was titrated to determine the minimum concentration needed to produce arrhythmias. A dose of 4 µg epinephrine/kg body weight failed to induce cardiac arrhythmias in air-exposed animals. However, the dose necessary to produce life-threatening arrhythmias was 2.0 µg/kg body weight following 58–61 min of exposure to 1,1-DCE, 1.0 µg/kg body weight following 64 min of exposure to 1,1-DCE, and 0.5 µg/kg body weight following 67–80 min of exposure to 1,1-DCE. The cardiac sensitization was found to be completely reversible upon discontinuation of the exposure to 1,1-DCE.
The various observations on the toxicity and metabolism of 1,1-DCE indicate that cytotoxicity is associated with cytochrome P450-catalysed metabolism of 1,1-DCE to reactive intermediates that bind covalently to cellular macromolecules. The extent of binding is inversely related to loss of GSH, so that severities of tissue damage parallel the decline in GSH (Forkert & Moussa, 1991; Moussa & Forkert, 1992). Hepatotoxicity is also exacerbated by treatments that diminish GSH (Jaeger et al., 1974, 1977b; McKenna et al., 1978b; Andersen et al., 1980). Thus, the responses to 1,1-DCE at low doses with little depletion of GSH are expected to be very different from the responses at high doses causing substantial GSH depletion. The targets of toxicity are centrilobular hepatocytes and bronchiolar Clara cells (Forkert et al., 1985), cell types that are rich in CYP2E1 (Forkert et al., 1991; Forkert, 1995). Immunohistochemical studies showed formation of DCE-epoxide–cysteine protein adducts within the centrilobular hepatocytes and Clara cells (Forkert, 1999a,b). Following short-term exposure of mice to high concentrations of 1,1-DCE, the degree of cellular damage in Clara cells and hepatocytes in various strains of mice correlates with the extent of formation of DCE-epoxide and the level of CYP2E1 in the tissue (Forkert, 2001; Forkert & Boyd, 2001; Forkert et al., 2001). In combination, these findings indicate that DCE-induced toxicity is associated with the formation and reactivity of the DCE-epoxide within the target centrilobular hepatocytes and Clara cells (Forkert, 2001).
Speerschneider & Dekant (1995) investigated the metabolic basis for the species- and sex-specific nephrotoxicity and tumorigenicity of 1,1-DCE. In kidney microsomes from Swiss-Webster male mice, the rate of oxidation of 1,1-DCE depended on the hormonal status of the animals. Oxidation of 1,1-DCE was decreased by castration and restored when the castrate was supplemented with exogenous testosterone. In kidney microsomes from naive female mice, the rate of oxidation of 1,1-DCE was significantly lower than in males, but could be increased by administration of exogenous testosterone. Using an antibody to rat liver CYP2E1, the researchers showed expression of a cross-reacting protein in male mouse kidney microsomes that was regulated by testosterone and correlated with the ability to oxidize 1,1-DCE and other substrates for CYP2E1 (e.g., p-nitrophenol and chlorozoxazone). The researchers also showed that different strains of mice express different levels of CYP2E1. The strains most sensitive to the effects of 1,1-DCE express greater levels of CYP2E1. Nephrotoxicity in Swiss-Webster mice after inhalation of 1,1-DCE was observed in males and in females treated with exogenous testosterone, but not in naive females. In kidney microsomes obtained from both sexes of rats and in six samples of human kidney from male donors, no p-nitrophenol oxidase activity was detected. Other research groups have also reported the absence of detectable CYP2E1 in human kidney tissue (Amet et al., 1997; Cummings et al., 2000).
Ott et al. (1976) investigated the health records of 138 employees occupationally exposed to 1,1-DCE in processes not involving vinyl chloride. The individuals included in the study had worked in experimental or pilot plant polymerization operations, in a monomer production process as tankcar loaders, or in a production plant manufacturing a monofilament fibre. Overall, there were no significant differences in haematology, clinical chemistry, or mortality between the exposed cohort and the controls. This study is too limited to derive useful information on the toxicity of 1,1-DCE to humans, because there was an inadequate number of subjects and only a limited number of end-points examined.
There are only limited data on the effects of 1,1-DCE on organisms in aquatic and terrestrial environments.
The data on the effects of 1,1-DCE on aquatic species are summarized in Table 3. A major limitation in most of these studies is the lack of prevention of the volatilization of 1,1-DCE. In addition, increase in biomass, not growth rate, was measured in the studies with aquatic plants.
Table 3: Aquatic toxicity of 1,1-dichloroethene.
|
Species |
Methoda |
Effect |
Value (µg/litre) |
Reference |
|
Freshwater species |
||||
|
Alga Chlamydomonas reinhardtii |
S, M, closed system |
72-h EC10 |
3 940 |
Brack & Rottler, 1994 |
|
Alga Selenastrum capricornutum |
S, N |
96-h EC50 |
>798 000 |
US EPA, 1978 |
|
Alga Scenedesmus subspicatus |
S, N |
96-h EC10 |
240 000 |
Geyer et al., 1985 |
|
Cladoceran Daphnia magna |
S, N |
24- and 48-h LC50 |
11 600 |
Dill et al., 1980 |
|
S, N, capped jar |
24-h LC50 |
98 000 |
Leblanc, 1980 |
|
|
S, N |
48-h LC50 |
79 000 |
US EPA, 1978 |
|
|
Fathead minnow Pimephales promelas |
S, N |
96-h LC50 |
169 000 |
Dill et al., 1980 |
|
FT, M |
96-h LC50 |
108 000 |
Dill et al., 1980 |
|
|
Bluegill Lepomis macrochirus |
S, N |
96-h LC50 |
73 900 |
US EPA, 1978 |
|
S, N |
96-h LC50 |
220 000 |
Dawson et al., 1977 |
|
|
S, N, capped jar |
96-h LC50 |
74 000 |
Buccafusco et al., 1981 |
|
|
Fathead minnow Pimephales promelas |
FT, M |
13-day LC50 |
29 000 |
Dill et al., 1980 |
|
E-L |
NOEC |
>2 800 |
US EPA, 1978 |
|
|
Saltwater species |
||||
|
Alga Skeletonema costatum |
S, N |
96-h EC50 |
>712 000 |
US EPA, 1978 |
|
Mysid shrimp Mysidopsis bahia |
S, N |
96-h LC50 |
224 000 |
US EPA, 1978 |
|
Sheepshead minnow Cyprinodon variegatus |
S, N |
96-h NOEC |
80 000 |
Heitmuller et al., 1981 |
|
S, N |
96-h LC50 |
249 000 |
US EPA, 1978 |
|
|
S, N |
96-h LC50 |
250 000 |
Dawson et al., 1977 |
|
|
Tidewater silversides Menidia beryllina |
S, N |
96-h LC50 |
250 000 |
Dawson et al., 1977 |
|
a |
S = static, FT = flow-through, N = nominal, M = measured, E-L = embryo-larval. |
Anderson & McCarty (1996) measured the effect of 1,1-DCE on the growth rate of a methanotrophic mixed culture. The growth rate of the culture on 460 µg methane/litre was reduced approximately 20% in the presence of 0.05 mg 1,1-DCE/litre.
Hendricks et al. (1995) conducted an 18-month carcinogenicity study of 1,1-DCE in rainbow trout (Oncorhynchus mykiss) (8 weeks old) at 4 mg/kg body weight per day. 1,1-DCE was incorporated in the feed. Tissues examined for neoplasms included liver, kidney, spleen, gill, gonads, thymus, thyroid, heart, stomach, pyloric caeca, duodenum, rectum, pancreas, and swimbladder. 1,1-DCE produced no neoplasms at the exposure levels used and no increase in liver weight. There was no evidence of any other chronic toxic effects.
Scheunert (1984) examined the toxicity of 1,1-DCE to terrestrial plants in a study conducted according to the OECD draft (1981) guideline "Growth test with higher plants." No effects on fresh weight of growth shoots were observed after a 14-day exposure of oats (Brassica rapa) and turnip (Avena sativa) to 1000 kg 1,1-DCE/kg soil dry weight (the highest concentration tested). Pestemer & Auspurg (1986) confirmed these results using a comparable test procedure with 14 more cultivated plants (Sinapis alba, Brassica napus, Brassica rapa, Brassica chinenesis, Raphanus sativus, Vicia sativa, Phaseolus aureus, Trifolium pratense, Trigonella meliotus-coerulea, Lolium perenne, Triticum aestivum, Sorghum vulgare, Lepidum sativum, and Lactuca sativa).
Viswanathan (1984) examined the toxicity of 1,1-DCE to earthworms (Eisenia fetida) in a study conducted according to OECD guidelines. After exposure in an artificial soil mixture, a 28-day LC50 value of >1000 mg/kg dry soil was established. Exposure to 1,1-DCE in the range of 100–1000 mg/kg soil resulted in a significant weight reduction of tested worms. The validity of the study seems limited. The minimum weight of test animals as prescribed by the guideline was not reached in 32% of the tests. Furthermore, the environmental relevance of the earthworm test seems questionable, as the factors determining the bioavailability of the applied chemical remain completely unconsidered.
The only existing epidemiological study is inadequate to assess the cancer or non-cancer health effects of 1,1-DCE.
In laboratory animals, 1,1-DCE is rapidly absorbed following oral and inhalation exposure. Although 1,1-DCE is rapidly distributed to all tissues, most of the free 1,1-DCE, its metabolites, and covalently bound derivatives are found in the liver and kidney. 1,1-DCE is rapidly oxidized by CYP2E1 (Figure 1). It is not known if the metabolism of 1,1-DCE is the same in humans, although in vitro microsomal preparations from human liver and lung form the same initial products.
Following high-dose exposure by the oral or inhalation route, the target organs are the liver, the kidney, and the Clara cells of the lung. Following longer-term continuous exposure at less than an acutely toxic exposure, the liver is the major target in rats following oral or inhalation exposure (Quast et al., 1983, 1986). The minimal fatty change observed in the liver of rats following long-term exposure occurs primarily in mid-zonal hepatocytes, but the change is not restricted to the centrilobular region. The minimal fatty change in the liver also occurs in the absence of significant depletion of cellular GSH. Although the minimal fatty change might not be considered adverse, as there is no evidence of a functional change in the liver of rats exposed at this level and GSH levels are not reduced, it is defined as the critical effect from both oral and inhalation exposure, as limiting exposure to this level will protect the liver from more serious damage that could compromise liver function.
The kidney is the major target organ in mice following inhalation exposure. This latter effect appears related to increased delivery of 1,1-DCE to the kidneys of mice following inhalation exposure relative to oral exposure, a gender-specific expression of CYP2E1 in male mice, and the presence of higher amounts of β-lyase in kidney tissue of mice relative to other species.
There is no evidence that toxicity occurs in the respiratory tract following exposure to 1,1-DCE at levels that cause minimal toxicity in the liver of rats and in the kidney of mice. However, regional responses in olfactory epithelium or bronchiolar changes in Clara cells might have been missed by the methods used to evaluate these regions in the toxicological studies.
As shown in a three-generation study, there is no evidence that reproductive toxicity is a critical effect for 1,1-DCE. No reproductive or developmental toxicity was observed at an exposure that caused minimal toxicity in the liver of the dams. There is also no evidence that teratogenicity is a critical effect. There is some evidence of developmental variations in the heart following ingestion of 1,1-DCE by pregnant rats from drinking-water (Dawson et al., 1993), but it is not clear if these effects are directly caused by exposure to 1,1-DCE. The biological significance of these cardiac structural variations is unclear. There is no indication that the structural variations have functional consequences in animals. However, animals known to have the structural variations have not been tested under conditions of stress.
There are no focused studies on neurotoxicity, but there is no indication from chronic, reproductive, or developmental bioassays in rats and mice by oral or inhalation exposure that neurotoxicity is an important toxic end-point. There are no long-term studies that have evaluated immunotoxicity in laboratory animals by any route of exposure. There is no indication from the chronic bioassays that immunotoxicity is likely to be a critical effect.
Bioassays for cancer by the oral route of exposure have been conducted in rats (Ponomarkov & Tomatis, 1980; NTP, 1982; Quast et al., 1983; Maltoni et al., 1985), mice (NTP, 1982), and trout (Hendricks et al., 1995). Some of these bioassays were conducted at an exposure below the maximum tolerated dose. The bioassay conducted by Maltoni et al. (1985) exposed the animals for only 1 year. The bioassay in rats (Quast et al., 1983) and the bioassay in mice (NTP, 1982) are well conducted, and both showed some toxicity in the liver at the highest exposure. Neither of these bioassays provides any significant evidence that 1,1-DCE is a carcinogen by the oral route of exposure.
Bioassays for cancer by the inhalation route of exposure have been conducted in rats (Lee et al., 1977, 1978; Viola & Caputo, 1977; Hong et al., 1981; Maltoni et al., 1985; Quast et al., 1986; Cotti et al., 1988), mice (Lee et al., 1977, 1978; Hong et al., 1981; Maltoni et al., 1985), and hamsters (Maltoni et al., 1985). None of these bioassays was conducted by a protocol that meets current standards. The major defects in most of these bioassays include exposure of the animals for 1 year and exposure at less than the maximum tolerated dose. The only bioassay showing some evidence of carcinogenicity was the study in Swiss-Webster mice (Maltoni et al., 1985). This study was conducted at or near the maximum tolerated dose, as animals exposed at 200 mg/m3 died after a few exposures. Although the animals were exposed for only 1 year and then observed until natural death, this study showed an increased incidence of kidney adenocarcinomas in male mice at 100 mg/m3, but not at 40 mg/m3. The incidence of mammary carcinomas in female mice and pulmonary adenomas in male and female mice did not increase with increased exposure. The responses were actually lower at 100 mg/m3 than at 40 mg/m3, but survival and other toxicities were comparable. There is evidence that the induction of kidney adenocarcinomas is a sex- and species-specific response related to the expression of CYP2E1 in the kidney of male mice (Speerschneider & Dekant, 1995; Amet et al., 1997; Cummings et al., 2000). The data presented by these researchers, however, are not sufficient to justify a conclusion that the kidney tumours in male mice have no relevance for a human health risk assessment. This recommendation is made with the knowledge that compounds similar in structure to 1,1-DCE (e.g., tetrachloroethene, trichloroethene, and 1,2-dichloroethene) produce varying degrees of kidney tumours in animal bioassays. The genotoxicity studies are incomplete, but most studies in mammalian cells indicate a lack of genotoxicity. Accordingly, the increased incidence of kidney adenocarcinomas in male mice (Maltoni et al., 1985) provides suggestive evidence of carcinogenicity by the inhalation route of exposure. The results of this bioassay showing a sex- and species-specific response are not sufficient to justify an exposure–response assessment.
1,1-DCE causes gene mutations in microorganisms in the presence of an exogenous activation system. Although most tests with mammalian cells show no evidence of genetic toxicity, the test battery is incomplete because it lacks a test for chromosomal damage in the mouse lymphoma system.
In the absence of a suitable PBPK model, a default procedure using an uncertainty factor for interspecies extrapolation is used to determine the tolerable intake for oral exposure and the tolerable concentration for inhalation exposure. The point of departure for the calculation of the tolerable intake for oral exposure and of the tolerable concentration for inhalation exposure is the lower 95% confidence limit on the BMD or the BMC for a 10% response (i.e., BMDL10, BMCL10), as described in Appendix 4.
The critical effect from oral exposure is minimal hepatocellular mid-zonal fatty change in female Sprague-Dawley rats (Quast et al., 1983). The BMDL10 for this effect is 4.6 mg/kg body weight per day (Appendix 4). The tolerable intake of 0.05 mg/kg body weight per day is calculated from the BMDL10 using a total uncertainty factor of 100 (4.6 mg/kg body weight per day ÷ 100 = 0.046, rounded to 0.05 mg/kg body weight per day). Individual uncertainty factors of 10 each were used for interspecies extrapolation and intraspecies variability, because there were no applicable data to justify a departure from the default values.
The critical effect from inhalation exposure is minimal hepatocellular mid-zonal fatty change in female Sprague-Dawley rats (Quast et al., 1986). The BMCLHEC (the BMCL10 adjusted to a human equivalent concentration) for this effect is 6.9 mg/m3 (Appendix 4). The tolerable concentration of 0.2 mg/m3 is calculated from the BMCLHEC of 6.9 mg/m3 using a total uncertainty factor of 30 (6.9 mg/m3 ÷ 30 = 0.23, rounded to 0.2 mg/m3). An uncertainty factor of 3 is used for interspecies extrapolation. Because a dosimetric adjustment was used, the default value of 3 for toxicokinetic differences was reduced to 1; the default value of 3 for toxicodynamic differences was retained. An uncertainty factor of 10 was used for intraspecies variability, because there were no applicable data to justify a departure from the default value.
None of the bioassays by the oral route of exposure provides any evidence that 1,1-DCE is a carcinogen. Accordingly, an oral slope factor is not derived. One bioassay by the inhalation route of exposure shows suggestive evidence of carcinogenicity. This study, however, does not provide sufficient weight of evidence to justify deriving an inhalation unit risk.
Human exposure to 1,1-DCE is likely to be highly variable due to site-specific contamination. However, data suggest that the mean concentration in drinking-water will not exceed 0.002–0.003 mg/litre, equivalent to 6–9 × 10–5 mg/kg body weight per day for a 70-kg individual consuming 2 litres per day. The oral exposure from food and soil is most likely negligible. Data suggest that the upper end of the range of the mean concentration of 1,1-DCE in air will not exceed 0.004 mg/m3. Thus, human exposure is expected to be far below the tolerable intake of 0.05 mg/kg body weight per day and the tolerable concentration of 0.2 mg/m3.
The quantitative estimates of 1,1-DCE in air, surface water, groundwater, and drinking-water are likely to be highly variable, depending on site-specific conditions.
Because the one epidemiological study showed no effects, there is some uncertainty that the laboratory animal data demonstrating the liver, lung, and kidney as target tissues have demonstrated the correct target tissues for humans. However, as CYP2E1 has been demonstrated to occur in liver and lung tissue from humans, it is likely that these tissues will be target tissues in humans. Data suggest that human kidney tissue does not contain CYP2E1, suggesting that human kidney will not be a target tissue for 1,1-DCE. Although there is also no evidence that teratogenicity is a critical effect, there is some evidence of developmental variations in the heart following ingestion of 1,1-DCE by pregnant rats from drinking-water. However, it is not clear if these effects are directly caused by exposure to 1,1-DCE. The biological significance of these cardiac structural variations is unclear.
There are a number of uncertainties in the assessment of the carcinogenicity of 1,1-DCE. Many of the bioassays by the inhalation route of exposure were not conducted at the maximum tolerated dose or for the full lifetime of the animals. In addition, our knowledge of the metabolic pathways for 1,1-DCE in humans is incomplete. While it is likely that the initial oxidation of 1,1-DCE in humans occurs via CYP2E1, there could be other CYP isoforms that could activate 1,1-DCE. Thus, there is some potential for a species-specific carcinogenic response in humans similar to the apparent species- and sex-specific response observed by Maltoni et al. (1985) in the kidney of male mice.
As there are no useful data in humans, the exposure–response assessment for humans is uncertain. The interspecies extrapolation was conducted using values of 10 for oral exposure and 3 for inhalation exposure. There is also uncertainty as to whether the uncertainty factor of 10 adequately accounts for human variability in expression of CYP2E1.
Studies on the effects of 1,1-DCE on aquatic species are limited to acute toxicity studies with algae, invertebrates, and fish and one carcinogenicity and chronic toxicity study in trout. A major limitation of these studies is the failure to prevent volatilization of 1,1-DCE. Only three species were tested using measured initial concentrations of 1,1-DCE. Of these studies, only the algal growth inhibition test with Chlamydomonas reinhardtii was carried out under closed test conditions. Algal growth inhibition with this species was also the most sensitive test end-point (72-h EC50 of 9.12 mg/litre). This result has been used to estimate the predicted no-effect concentration (PNEC), together with an uncertainty factor of 1000 (EC, 1996):
PNEC = 9.12 mg/litre / 1000 = 0.009 mg/litre
There were no available long-term effects data with which to assess the chronic toxicity of 1,1-DCE. However, the PNEC derived above included an uncertainty factor that should be sufficient to be protective for chronic toxicity.
The limited data on the occurrence of 1,1-DCE in surface waters suggest that concentrations are in the microgram per litre range. Using a maximum concentration of 0.001 mg/litre reported for surface waters in Osaka, Japan, as the predicted exposure concentration (PEC) gives a PEC/PNEC hazard quotient of 0.11. Because this is less than 1, no further information, testing, or risk reduction measures are required for freshwater species.
Data are inadequate to evaluate the effects of 1,1-DCE on the terrestrial environment. However, because of the rapid volatilization of 1,1-DEC, no significant risk is expected.
There were insufficient marine exposure or effects data to conduct a risk characterization specific for the marine environment. There is a need for further testing with a range of marine/estuarine species from different trophic levels using closed experimental systems with measurement of 1,1-DCE concentrations throughout the tests.
IPCS (1990) evaluated the human health and environmental effects of 1,1-DCE in 1990. This assessment updates that evaluation.
WHO established a drinking-water quality guideline for 1,1-DCE of 0.03 mg/litre in 1993. WHO has advised that this guideline is under review.5
IARC (1999) evaluated the carcinogenicity and genetic toxicity data for 1,1-DCE and concluded that there is inadequate evidence in humans for its carcinogenicity; that there is limited evidence in experimental animals for its carcinogenicity; and that it is not classifiable as to its carcinogenicity to humans (Group 3).
Amaral OC, Olivella L, Grimalt J, Albaiges J (1994) Combined solvent extraction–purge and trap method for the determination of volatile organic compounds in sediments. Journal of Chromatography, 675:177–187.
Amet Y, Berthou F, Fournier G, Dreano Y, Bardou L, Cledes J, Menez JF (1997) Cytochrome P450 4A and 2E1 expression in human kidney microsomes. Biochemical Pharmacology, 53:765–771.
Andersen ME, Jenkins LJ Jr (1977) Oral toxicity of 1,1-dichloroethylene in the rat: effects of sex, age, and fasting. Environmental Health Perspectives, 21:157–163.
Andersen ME, Jones RA, Jenkins LJ Jr (1978) The acute toxicity of single, oral doses of 1,1-dichloroethylene in the fasted, male rat: effect of induction and inhibition of microsomal enzyme activities on mortality. Toxicology and Applied Pharmacology, 46:227–234.
Andersen ME, French JE, Gargas ML, Jones RA, Jenkins LJ Jr (1979) Saturable metabolism and the acute toxicity of 1,1-dichloroethylene. Toxicology and Applied Pharmacology, 47:385–394.
Andersen ME, Thomas OE, Gargas ML, Jones RA, Jenkins LJ Jr (1980) The significance of multiple detoxification pathways for reactive metabolites in the toxicity of 1,1-dichloroethylene. Toxicology and Applied Pharmacology, 52:422–432.
Anderson D, Hodge MCE, Purchase IFH (1977) Dominant lethal studies with the halogenated olefins vinyl chloride and vinylidene chloride in male CD-1 mice. Environmental Health Perspectives, 21:71–78.
Anderson JE, McCarty PL (1996) Effect of three chlorinated ethenes on growth rates for a methanotrophic mixed culture. Environmental Science and Technology, 30:3517–3524.
Arthur CL, Pratt K, Motlagh S, Pawliszyn J, Belardi RP (1992) Environmental analysis of organic compounds in water using solid phase microextraction. Journal of High Resolution Chromatography, 15:741–744.
Atri FR (1985) [Chlorinated compounds in the environment.] Schriftenreihe des Vereins für Wasser-, Boden-, und Lufthygiene, Berlin-Dahlem, 60:309–317 (in German) [cited in IPCS, 1990].
Baden JM, Kelley M, Wharton RS, Hitt BA, Simmon VF, Mazze RI (1977) Mutagenicity of halogenated ether anesthetics. Anesthesiology, 46:346–350.
Ban M, Hettich D, Cavelier L (1995) Nephrotoxicity mechanism of 1,1-dichloroethylene in mice. Toxicology Letters, 78:87–92.
Barrio-Lage G, Parsons FZ, Nassar RS, Lorenzo PA (1986) Sequential dehalogenation of chlorinated ethenes. Environmental Science and Technology, 20:96–99.
Bartsch H, Malaveille C, Barbin A, Planche G (1979) Mutagenic and alkylating metabolites of halo-ethylenes, chlorobutadienes, and dichlorobutenes produced by rodent or human liver tissues. Evidence for oxirane formation by P450-linked microsomal mono-oxygenases. Archives of Toxicology, 41:249–277.
Bauer S, Solyom D (1994) Determination of volatile organic compounds at parts per trillion level in complex aqueous matrices using membrane introduction mass spectrometry. Analytical Chemistry, 66:4422–4431.
Biziuk M, Namiesnik J, Czerwinski J, Gorlo D, Makuch B, Janicki W, Polkowska Z, Wolska L (1996) Occurrence and determination of organic pollutants in tap and surface waters of the Gdansk district. Journal of Chromatography, 733:171–183.
Brack W, Rottler H (1994) Toxicity testing of highly volatile chemicals with green algae: a new assay. Environmental Science and Pollution Research International, 1:223–228.
Brittebo EB, Darnerud PO, Eriksson C, Brandt I (1993) Nephrotoxicity and covalent binding of 1,1-dichloroethylene in buthionine sulphoximine-treated mice. Archives of Toxicology, 67:605–612.
Brodzinski R, Singh HB (1983) Volatile organic chemicals in the atmosphere: an assessment of available data. Research Triangle Park, NC, US Environmental Protection Agency, Environmental Sciences Research Laboratory (EPA/600/3-83-027(a)).
Bronzetti G, Bauer C, Corsi C, Leporini C, Nieri R, del Carratore R (1981) Genetic activity of vinylidene chloride in yeast. Mutation Research, 89:179–185.
Bronzetti G, Bauer C, Corsi C, del Carratore R, Nieri R, Paolini M, Galli A, Giagoni P (1983) Comparison of genetic and biochemical effects of halogenated olefins. Mutation Research, 113:236–237 (Abstract No. 24).
Brymer DA, Ogle LD, Jones CJ, Lewis DL (1996) Viability of using SUMMA polished canisters for the collection and storage of parts per billion by volume level volatile organics. Environmental Science and Technology, 30:188–198.
Buccafusco RJ, Ells SJ, Leblanc GA (1981) Acute toxicity of priority pollutants to bluegill (Lepomis macrochirus). Bulletin of Environmental Contamination and Toxicology, 26:446–452.
Cavelier L, Bonnet P, Morel G, de Ceaurriz J (1996) Role of cysteine conjugation in vinylidene choride-induced nephrotoxicity and hepatotoxicity in fasted rats. Journal of Applied Toxicology, 16:109–113.
Chieco P, Moslen MT, Reynolds ES (1981) Effect of administration vehicle on oral 1,1-dichloroethylene toxicity. Toxicology and Applied Pharmacology, 57:146–155.
Chung Y, Shin D, Park S, Lim Y, Choi Y, Cho S, Yang J, Hwang M, Park Y, Lee H (1997) Risk assessment and management of drinking water pollutants in Korea. Water Science and Technology, 36:309–323.
Costa AK, Ivanetich KM (1982) Vinylidene chloride: its metabolism by hepatic microsomal cytochrome P-450 in vitro. Biochemical Pharmacology, 31:2083–2092.
Costa AK, Ivanetich KM (1984) Chlorinated ethylenes: their metabolism and effect on DNA repair in rat hepatocytes. Carcinogenesis, 5:1629–1636.
Cotti G, Maltoni C, Lefemine G (1988) Long-term carcinogenicity bioassay on vinylidene chloride administered by inhalation to Sprague-Dawley rats. New results. Annals of the New York Academy of Sciences, 534:160–168.
Cresteil T (1998) Onset of xenobiotic metabolism in children: toxicological implications. Food Additives and Contaminants, 15(Supplement): 45–51.
Cummings BS, Lasker JM, Lash LH (2000) Expression of glutathione-dependent enzymes and cytochrome P450s in freshly isolated and primary cultures of proximal tubular cells from human kidney. Journal of Pharmacology and Experimental Therapeutics, 293:677–685.
Dallas CE, Weir FW, Feldman S (1983) The uptake and disposition of 1,1-dichloroethylene in rats during inhalation exposure. Toxicology and Applied Pharmacology, 68:140–151.
Dawson GW, Jennings AL, Drozddowski D, Rider E (1977) The acute toxicity of 47 industrial chemicals to fresh and saltwater fishes. Journal of Hazardous Materials, 1:303–318.
Dawson BV, Johnson PD, Goldberg SJ, Ulreich JB (1993) Cardiac teratogenesis of halogenated hydrocarbon-contaminated drinking water. Journal of the American College of Cardiology, 21:1466–1472.
Dekant W (1996) Biotransformation and renal processing of nephrotoxic agents. Archives of Toxicology, Supplement 18:163–172.
Dekant W, Vamvakas S, Anders MW (1989) Bioactivation of nephrotoxic haloalkenes by glutathione conjugation: formation of toxic and mutagenic intermediates by cysteine conjugate β-lyase. Drug Metabolism Reviews, 20:43–83.
DeLeon IR, Maberry MA, Overton EB, Raschke CK, Remele PC, Steele CF, Laseter JL (1980) Rapid gas chromatographic method for the determination of volatile and semivolatile organochloride compounds in soil and chemical waste disposal site samples. Journal of Chromatographic Science, 18:85–88.
Dill DC, McCarty WM, Alexander HC, Bartlett EA (1980) Toxicity of 1,1-dichloroethylene (vinylidene chloride) to aquatic organisms. Midland, MI, Dow Chemical Company (PB 81-111098) [cited in US EPA, 1980].
Dilling WL (1977) Interphase transfer processes. II. Evaporation rates of chloromethanes, ethanes, ethylenes, propanes and propylenes from dilute aqueous solution. Comparison with theoretical predictions. Environmental Science and Technology, 11:405–409.
Dolan ME, McCarty PL (1995) Methanotropic chloroethene transformation capacities and 1,1-dichloroethene transformation product toxicity. Environmental Science and Technology, 29:2741–2747.
Dowsley TF, Forkert PG, Benesch LA, Bolton JL (1995) Reaction of glutathione with the electrophilic metabolities of 1,1-dichloroethylene. Chemico-Biological Interactions, 95:227–244.
Dowsley TF, Ulreich JB, Bolton JL, Park SS, Forkert PG (1996) CYP2E1-dependent bioactivation of 1,1-dichloroethylene in murine lung: formation of reactive intermediates and glutathione conjugates. Toxicology and Applied Pharmacology, 139:42–48.
Dowsley TF, Reid K, Petsikas D, Ulreich JB, Fisher RL, Forkert PG (1999) Cytochrome P-450-dependent bioactivation of 1,1-dichloroethylene to a reactive epoxide in human lung and liver microsomes. Journal of Pharmacology and Experimental Therapeutics, 289:641–648.
Drevon C, Kuroki T (1979) Mutagenicity of vinyl chloride, vinylidene chloride and chloroprene in V79 Chinese hamster cells. Mutation Research, 67:173–182.
D’Souza RW, Andersen ME (1988) Physiologically based pharmacokinetic model for vinylidene chloride. Toxicology and Applied Pharmacology, 95:230–240.
Easley DM, Kleopfer RD, Carasea AM (1981) Gas chromatographic–mass spectrometric determination of volatile organic compounds in fish. Journal of the Association of Official Analytical Chemists, 64:653–656.
EC (1996) Technical guidance document in support of the Commission Directive 93/EEC on risk assessment for new notified substances and the Commission Regulation (EC) 1488/94 on risk assessment for existing substances. Brussels, European Commission.
Eichelberger JW, Bellar TA, Donnelly JP, Budde WL (1990) Determination of volatile organics in drinking water using USEPA method 524.2 and the ion trap detector. Journal of Chromatographic Science, 28:460–467.
el-Masri HA, Constan AA, Ramsdell HS, Yang RS (1996a) Physiologically based pharmacodynamic modeling of an interaction threshold between trichloroethylene and 1,1-dichloroethylene in Fischer 344 rats. Toxicology and Applied Pharmacology, 141:124–132.
el-Masri HA, Tessari JD, Yang RS (1996b) Exploration of an interaction threshold for the joint toxicity of trichloroethylene and 1,1-dichloroethylene: utilization of a PBPK model. Archives of Toxicology, 70:527–539.
Foerst D (1979) A sampling and analytical method for vinylidene chloride in air. American Industrial Hygiene Association Journal, 40:888–893.
Forkert P-G (1995) CYP2E1 is preferentially expressed in Clara cells of murine lung: localization by in situ and immunohistochemical methods. American Journal of Respiratory Cell and Molecular Biology, 12:589–596.
Forkert P-G (1999a) In vivo formation and localization of 1,1-dichloroethylene epoxide in murine liver: identification of its glutathione conjugate 2-S-glutathionyl acetate. Journal of Pharmacology and Experimental Therapeutics, 290:1299–1306.
Forkert P-G (1999b) 1,1-Dichloroethylene-induced Clara cell damage is associated with in situ formation of the reactive epoxide. Immunohistochemical detection of its glutathione conjugate. American Journal of Respiratory Cell and Molecular Biology, 20:1310–1318.
Forkert P-G (2001) Mechanisms of 1,1-dichloroethylene-induced cytotoxicity in lung and liver. Drug Metabolism Reviews, 33:49–80.
Forkert P-G, Boyd SM (2001) Differential metabolism of 1,1-dichloroethylene in livers of A/J, CD-1, and C57BL/6 mice. Drug Metabolism and Disposition, 29:1396–1402.
Forkert P-G, Moussa M (1991) 1,1-Dichloroethylene elicits dose-dependent alterations in covalent binding and glutathione in murine liver. Drug Metabolism and Disposition, 19:580–586.
Forkert P-G, Moussa M (1993) Temporal effects of 1,1-dichloroethylene on nonprotein sulfhydryl content in murine lung and liver. Drug Metabolism and Disposition, 21:770–776.
Forkert P-G, Reynolds ES (1982) 1,1-Dichloroethylene-induced pulmonary injury. Experimental Lung Research, 3:57–68.
Forkert P-G, Forkert L, Farooqui M, Reynolds ES (1985) Lung injury and repair: DNA synthesis following 1,1-dichloroethylene. Toxicology, 36:199–214.
Forkert P-G, Geddes BA, Birch DW, Massey TE (1990) Morphologic changes and covalent binding of 1,1-dichloroethylene in Clara and alveolar type II cells isolated from lungs of mice following in vivo administration. Drug Metabolism and Disposition, 18:534–539.
Forkert P-G, Massey TE, Park SS, Gelboin HV, Anderson LM (1991) Distribution of cytochrome P450IIE1 in murine liver after ethanol and acetone administration. Carcinogenesis, 12:2259–2268.
Forkert P-G, Boyd SM, Ulreich JB (2001) Pulmonary bioactivation of 1,1-dichloroethylene is associated with CYP2E1 levels in A/J, CD-1, and C57Bl/6 mice. Journal of Pharmacology and Experimental Therapeutics, 297:1193–1200.
Geyer H, Scheunert I, Korte F (1985) The effects of organic environmental chemicals on the growth of the alga Scenedesmus subspicatus: a contribution to environmental biology. Chemosphere, 14:1355–1370.
Gilbert J, Shepherd MJ, Startin JR, McWeeny D (1980) Gas chromatographic determination of vinylidene chloride monomer in packing films and foods. Journal of Chromatography, 197:71–78.
Glod G, Brodmann U, Angst W, Hollinger C, Schwarzenbach RP (1997) Cobalamin-mediated reduction of cis- and trans-dichloroethene, 1,1-dichloroethene, and vinyl chloride in homogeneous aqueous solution: reaction kinetics and mechanistic considerations. Environmental Science and Technology, 31:3154–3160.
Grosjean D (1991) Atmospheric chemistry of toxic contaminants. 5. Unsaturated halogenated aliphatics: allyl chloride, chloroprene, hexachloropentadiene, vinylidene chloride. Journal of the Air and Waste Management Association, 41:182–189.
Guicherit R, Schulting FL (1985) The occurrence of organic chemicals in the atmosphere of the Netherlands. The Science of the Total Environment, 43(3):193–219.
Hallbourg RR, Delfina JJ, Miller WL (1992) Organic priority pollutants in groundwater and surface water at three landfills in north central Florida. Water, Air, and Soil Pollution, 65:307–322.
Heitmuller PT, Hollister TA, Parrish PR (1981) Acute toxicity of 54 industrial chemicals to sheepshead minnows (Cyprinodon variegatus). Bulletin of Environmental Contamination and Toxicology, 27:596–604.
Hendricks JD, Shelton DW, Loveland PM, Pereira CB, Bailey GS (1995) Carcinogenicity of dietary dimethylnitrosomorpholine, N-methyl-N'-nitro-N-nitrosoguanidine, and dibromoethane in rainbow trout. Toxicologic Pathology, 23:447–457.
Hiatt MH (1983) Determination of volatile organic compounds in fish samples by vacuum distillation and fused silica capillary gas chromatography/mass spectrometry. Analytical Chemistry, 55:506–516.
Hong CB, Winston JM, Thornburg LP, Lee CC, Woods JS (1981) Follow-up study on the carcinogenicity of vinyl chloride and vinylidene chloride in rats and mice; tumor incidence and mortality subsequent to exposure. Journal of Toxicology and Environmental Health, 7:909–924.
Humiston CG, Quast JF, Wade CE, Ballard JJ, Beyer JE, Lisowe RW (1978) Results of a two-year toxicity and oncogenicity study with vinylidene chloride incorporated in the drinking water of rats. Midland, MI, Dow Chemical USA, Health and Environmental Research, Toxicology Research Laboratory.
IARC (1999) Re-evaluation of some organic chemicals, hydrazine, and hydrogen peroxide (part 3). Lyon, International Agency for Research on Cancer, pp. 1163–1180 (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Volume 71).
IPCS (1990) Vinylidene chloride. Geneva, World Health Organization, International Programme on Chemical Safety, 187 pp. (Environmental Health Criteria 100).
IPCS (2000) International Chemical Safety Card — Vinylidene chloride. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 0083), at website http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc00/icsc0083.htm.
Ishidate M, ed. (1983) The data book of chromosomal aberration tests in vitro on 587 chemical substances using a Chinese hamster fibroblast cell line (CHL Cell). Tokyo, Realize Inc.
Jackson NM, Conolly RB (1985) Acute nephrotoxicity of 1,1-dichloroethylene in the rat after inhalation exposure. Toxicology Letters, 29:191–200.
Jaeger RJ (1977) Effect of 1,1-dichloroethylene exposure on hepatic mitochondria. Research Communications in Chemical Pathology and Pharmacology, 18:83–94.
Jaeger RJ, Conolly RB, Murphy SD (1974) Effect of 18-hr fast and glutathione depletion of 1,1-dichloroethylene-induced hepatotoxicity and lethality in rats. Experimental and Molecular Pathology, 20:187–198.
Jaeger RJ, Conolly RB, Murphy SD (1975) Short-term inhalation toxicity of halogenated hydrocarbons: effects on fasting rats. Archives of Environmental Health, 30:26–31.
Jaeger RJ, Shoner LG, Coffman L (1977a) 1,1-Dichloroethylene hepatotoxicity: proposed mechanism of action and distribution and binding of carbon-14 radioactivity following inhalation exposure in rats. Environmental Health Perspectives, 21:113–119.
Jaeger RJ, Szabo S, Coffman LJ (1977b) 1,1-Dichloroethylene hepatotoxicity: effect of altered thyroid function and evidence for the subcellular site of injury. Journal of Toxicology and Environmental Health, 3:545–556.
Jenkins LJ Jr, Andersen ME (1978) 1,1-Dichloroethylene nephrotoxicity in the rat. Toxicology and Applied Pharmacology, 46:131–142.
Jenkins LJ Jr, Trabulus MJ, Murphy SD (1972) Biochemical effects of 1,1-dichloroethylene in rats: comparison with carbon tetrachloride and 1,2-dichloroethylene. Toxicology and Applied Pharmacology, 23:501–510.
Jones BK, Hathway DE (1978a) The biological fate of vinylidene chloride in rats. Chemico-Biological Interactions, 20:27–41.
Jones BK, Hathway DE (1978b) Differences in metabolism of vinylidene chloride between mice and rats. British Journal of Cancer, 37:411–417.
Jones BK, Hathway DE (1978c) Tissue-mediated mutagenicity of vinylidene chloride in Salmonella typhimurium TA 1535. Cancer Letters, 5(1):1–6.
Kainz A, Cross H, Freeman S, Gescher A, Chipman JK (1993) Effects of 1,1-dichloroethene and of some of its metabolites on the functional viability of mouse hepatocytes. Fundamental and Applied Toxicology, 21(2):140–148.
Kanz MF, Reynolds ES (1986) Early effects of 1,1-dichloroethylene on canalicular and plasma membranes: ultrastructure and stereology. Experimental and Molecular Pathology, 44:93–110.
Kanz MF, Whitehead RF, Ferguson AE, Moslen MT (1988) Potentiation of 1,1-dichloroethylene hepatotoxicity: comparative effects of hyperthyroidism and fasting. Toxicology and Applied Pharmacology, 95:93–103.
Kanz MF, Taj Z, Moslen MT (1991) 1,1-Dichloroethylene hepatotoxicity: hyperthyroidism decreases metabolism and covalent binding but not injury in the rat. Toxicology, 70:213–229.
Koch R, Schlegelmilch R, Wolf HU (1988) Genetic effects of chlorinated ethylenes in the yeast Saccharomyces cerevisiae. Mutation Research, 206:209–216.
Leblanc GA (1980) Acute toxicity of priority pollutants to water flea (Daphnia magna). Bulletin of Environmental Contamination and Toxicology, 24:684–691.
Lee CC, Bhandari JC, Winston JM, House WB, Peters PJ, Dixon RL, Woods JS (1977) Inhalation toxicity of vinyl chloride and vinylidene chloride. Environmental Health Perspectives, 21:25–32.
Lee CC, Bhandari JC, Winston JM, House WB, Dixon RL, Woods JS (1978) Carcinogenicity of vinyl chloride and vinylidene chloride. Journal of Toxicology and Environmental Health, 24:15–30.
Lee RP, Forkert PG (1995) In vitro biotransformation of 1,1-dichloroethylene by hepatic cytochrome P4502E1 in mice. Journal of Pharmacology and Experimental Therapeutics, 270:371–376.
Liebler DC, Meredith TM, Guengerich FP (1985) Formation of glutathione conjugates by reactive metabolites of vinylidene chloride in microsomes and isolated hepatocytes. Cancer Research, 45:186–193.
Liebler DC, Latwesen DG, Reeder TC (1988) S-(2-chloroacetyl) glutathione, a reactive glutathione thio ester and a putative metabolite of 1,1-dichloroethylene. Biochemistry, 27:3652–3657.
Lin SN, Fu FWY, Bruckner JV, Feldman S (1982) Quantitation of 1,1- and 1,2-dichloroethylene in body tissues by purge-and-trap gas chromatography. Journal of Chromatography, 244:311–320.
Malaveille C, Planche G, Bartsch H (1997) Factors for efficiency of the Salmonella/microsome mutagenicity assay. Chemico-Biological Interactions, 17:129–136.
Maltoni C, Lefemine G, Cotti G, Chieco P, Patella V (1985) Experimental research on vinylidene chloride carcinogenesis. In: Maltoni C, Mehlman MA, eds. Archives of research on industrial carcinogenesis. Volume III. Princeton, NJ, Princeton Scientific Publishers, Inc., 229 pp.
McGregor D, Brown AG, Cattanach P, Edwards I, McBride D, Riach C, Shepherd W, Caspary WJ (1991) Responses of the L5178Y mouse lymphoma forward mutation assay: V. Gases and vapors. Environmental and Molecular Mutagenesis, 17:122–129.
McKenna MJ, Watanabe PG, Gehring PJ (1977) Pharmacokinetics of vinylidene chloride in the rat. Environmental Health Perspectives, 21:99–105.
McKenna MJ, Zempel JA, Madrid EO, Gehring PJ (1978a) The pharmacokinetics of [14C]vinylidene chloride in rats following inhalation exposures. Toxicology and Applied Pharmacology, 45:599–610.
McKenna MJ, Zempel JA, Madrid EO, Braun WH, Gehring PJ (1978b) Metabolism and pharmacokinetic profile of vinylidene chloride in rats following oral administration. Toxicology and Applied Pharmacology, 45:821–835.
Merdink JL, Gonzalez-Leon A, Bull RJ, Schultz IR (1998) The extent of dichloroacetate formation from trichloroethylene, chloral hydrate, trichloracetate, and trichloroethanol in B6C3F1 mice. Toxicological Science, 45:33–41.
MITI (1992) Biodegradation and bioaccumulation data of existing chemicals based on the CSCL Japan. Tokyo, Ministry of International Trade and Industry.
Moslen MT, Poisson LR, Reynolds ES (1985) Cholestasis and increased biliary excretion of inulin in rats given 1,1-dichloroethylene. Toxicology, 34:201–209.
Moslen MT, Whitehead RF, Ferguson AE, Kanz MF (1989) Protection by L-2-oxothiazolidine-4-carboxylate, a cysteine prodrug, against 1,1-dichloroethylene hepatotoxicity in rats is associated with decreases in toxin metabolism and cytochrome P-450. Journal of Pharmacology and Experimental Therapeutics, 248:157–163.
Moussa MT, Forkert PG (1992) 1,1-Dichloroethylene-induced alterations in glutathione and covalent binding in murine lung: morphological, histochemical, and biochemical studies. Journal of Pathology, 166:199–207.
Murray FJ, Nitschke KD, Rampy LW, Schwetz BA (1979) Embryotoxicity and fetotoxicity of inhaled or ingested vinylidene chloride in rats and rabbits. Toxicology and Applied Pharmacology, 49:189–202.
NIOSH (1994) NIOSH manual of analytical methods, 4th ed. Cincinnati, OH, National Institute of Occupational Safety and Health, at website http://www.cdc.gov/niosh/nmam/nmammenu.html.
Nitschke KD, Smith FA, Quast JF, Norris JM, Schwetz BA (1983) A three-generation rat reproductive toxicity study of vinylidene chloride in the drinking water. Fundamental and Applied Toxicology, 3:75–79.
NTP (1982) Carcinogenesis bioassay of vinylidene chloride in F344 rats and B6C3F1 mice (gavage study). Research Triangle Park, NC, US Department of Health and Human Services, National Toxicology Program (NTP Technical Report Series No. 228).
Oesch F, Protic-Sabljic M, Friedberg T, Klimisch HJ, Glatt HR (1983) Vinylidene chloride: changes in drug-metabolizing enzymes, mutagenicity and relation to its targets for carcinogenesis. Carcinogenesis, 4:1031–1038.
Okine LK, Gram TE (1986a) In vitro studies on the metabolism and covalent binding of [14C]1,1-dichloroethylene by mouse liver, kidney, and lung. Biochemical Pharmacology, 35:2789–2795.
Okine LK, Gram TE (1986b) Tissue distribution and covalent binding of [14C]1,1-dichloroethylene in mice: in vivo and in vitro studies. Advances in Experimental Medicine and Biology, 197:903–910.
Okine LK, Gochee JM, Gram TE (1985) Studies on the distribution and covalent binding of [14C]1,1-dichloroethylene in the mouse. Biochemical Pharmacology, 34:4051–4057.
Otson R, Williams DT (1982) Headspace chromatographic determination of water pollutants. Analytical Chemistry, 54:942–946.
Ott MG, Fishbeck WA, Townsend JC, Schneider EJ (1976) A health study of employees exposed to vinylidene chloride. Journal of Occupational Medicine, 18:735–738.
Pestemer W, Auspurg B (1986) Eignung eines Testpflanzensortiments zur Risikoabschätzung von Stoffwirkungen auf höhere Pflanzen im Rahmen des Chemikaliengesetzes. Nachrichtenblatt des Deutschen Pflanzenschutzdienstes (Braunschweig), 38:120–125.
Piet GJ, Slingerland P, DeGrunt FE, Van Den Heuvel MPM, Zoeteman BCL (1978) Determination of very volatile halogenated organic compounds in water by means of direct head-space analysis. Analytical Letters, A11:437–448.
Plummer JL, Hall PM, Ilsley AH, Jenner MA, Cousins MJ (1990) Influence of enzyme induction and exposure profile on liver injury due to chlorinated hydrocarbon inhalation. Pharmacology and Toxicology, 67:329–335.
Ponomarkov V, Tomatis L (1980) Long-term testing of vinylidene chloride and chloroprene for carcinogenicity in rats. Oncology, 37:136–141.
Prendergast JA, Jones RA, Jenkins LJ Jr, Siegel J (1967) Effects on experimental animals of long-term inhalation of trichloroethylene, carbon tetrachloride, 1,1,1-trichloroethane, dichlorodifluoromethane, and 1,1-dichloroethylene. Toxicology and Applied Pharmacology, 10:270–289.
Putcha L, Bruckner JV, D’Souza R (1986) Toxicokinetics and bioavailability of oral and intravenous 1,1-dichloroethylene. Fundamental and Applied Toxicology, 6:240–250.
Quast JF, Humiston CG, Wade CE, Ballard J, Beyer JE, Schwetz RW, Norris JM (1983) A chronic toxicity and oncogenicity study in rats and subchronic toxicity study in dogs on ingested vinylidene chloride. Fundamental and Applied Toxicology, 3:55–62.
Quast JF, McKenna MJ, Rampy LW, Norris JM (1986) Chronic toxicity and oncogenicity study on inhaled vinylidene chloride in rats. Fundamental and Applied Toxicology, 6:105–144.
Rampy LW, Quast JF, Humiston CG, Balmer MF, Schwetz BA (1977) Interim results of two-year toxicological studies in rats of vinylidene chloride incorporated in the drinking water or administered by repeated inhalation. Environmental Health Perspectives, 21:33–43.
Reichert D, Werner HW, Henschler D (1978) Role of liver glutathione in 1,1-dichloroethylene metabolism and hepatotoxicity in intact rats and isolated perfused rat liver. Archives of Toxicology, 41:169–178.
Reichert D, Werner HW, Metzler M, Henschler D (1979) Molecular mechanism of 1,1-dichloroethylene toxicity: excreted metabolites reveal different pathways of reactive intermediates. Archives of Toxicology, 42:159–169.
Reitz RH, Watanabe PG, McKenna MJ, Quast JF, Gehring PJ (1980) Effects of vinylidene chloride and DNA synthesis and DNA repair in the rat and mouse: a comparative study with dimethylnitrosamine. Toxicology and Applied Pharmacology, 52:357–370.
Reynolds ES, Moslen MT, Boor PJ, Jaeger RJ (1980) 1,1-Dichloroethylene hepatoxicity. Time course of GSH changes and biochemical aberrations. American Journal of Pathology, 101:331–342.
Reynolds ES, Kanz MF, Chieco P, Moslen MT (1984) 1,1-Dichloroethylene: an apoptotic hepatotoxin? Environmental Health Perspectives, 57:313–320.
Roldan-Arjona T, Garcia-Pedrajas MD, Luque-Romero FL, Hera C, Pueyo C (1991) An association between mutagenicity of the Ara test of Salmonella typhimurium and carcinogenicity in rodents for 16 halogenated aliphatic hydrocarbons. Mutagenesis, 6:199–205.
Sasaki M, Sugimura K, Yoshida MA, Abe S (1980) Cytogenic effects of 60 chemicals on cultured human and Chinese hamster cells. Kromosomo II, 20:574–584.
Sawada M, Sofuni T, Ishidate M Jr (1987) Cytogenic studies on 1,1-dichloroethylene and its two isomers in mammalian cells in vitro and in vivo. Mutation Research, 187:157–163.
Scheunert I (1984) Wachstumstest mit höheren Pflanzen. In: Ballhorn L, Freitag D, eds. Überprüfung der Durchführbarkeit von Prüfungsvorschriften und der Aussagekraft der Stufe I und II des E. Chem. G. Neuherberg, Gesellschaft für Strahlen- und Umweltforschung München mbH, pp. 113–123.
Shirey RE (1995) Rapid analysis of environmental samples using solid-phase microextractions (SPME) and narrow bore capillary columns. Journal of High Resolution Chromatography, 18:495–499.
Short RD, Minor JL, Peters P, Winston JM, Ferguson P, Unger T, Sawyer M, Lee CC (1977a) The developmental toxicity of vinylidene chloride inhaled by rats and mice during gestation. Report prepared by Midwest Research International, Kansas City, MO, for US Environmental Protection Agency, Washington, DC (EPA 560/6-77-022; PB-281-713).
Short RD, Minor JL, Winston JM, Lee CC (1977b) A dominant lethal study in male rats after repeated exposures to vinyl chloride or vinylidene chloride. Journal of Toxicology and Environmental Health, 3:965–968.
Short RD, Winston JM, Minor JL, Hong CB, Seifter J, Lee CC (1977c) Toxicity of vinylidene chloride in mice and rats and its alteration by various treatments. Journal of Toxicology and Environmental Health, 3:913–921.
Sidhu KS (1980) A gas-chromatographic method for the determination of vinylidene chloride in air. Journal of Analytical Toxicology, 4:266–268.
Siegel J, Jones RA, Coon A (1971) Effects on experimental animals of acute, repeated and continuous inhalation exposures to dichloroacetylene mixtures. Toxicology and Applied Pharmacology, 18:168–174.
Siletchnik LM, Carlson GP (1974) Cardiac sensitizing effects of 1,1-dichloroethylene: enhancement by phenobarbital pretreatment. Archives Internationales de Pharmacodynamie et de Therapie, 210:359–364.
Simmon VF, Tardiff RG (1978) The mutagenic activity of halogenated compounds found in chlorinated drinking water. In: Jolley RL, Gorchev H, Hamilton DH Jr, eds. Water chlorination. Environmental impact and health effects. Volume 2. Ann Arbor, MI, Ann Arbor Science, pp. 417–431.
Singh HB, Salas LJ, Smith AJ, Shigeishi M (1981) Measurements of some potentially hazardous organic chemicals in urban environments. Atmospheric Environment, 210:601–612.
Speerschneider P, Dekant W (1995) Renal tumorigenicity of 1,1-dichloroethene in mice: the role of male-specific expression of cytochrome P450 2E1 in the renal bioactivation of 1,1-dichloroethene. Toxicology and Applied Pharmacology, 130:48–56.
Speis DN (1980) Determination of purgeable organics in sediments. Environmental Science and Technology, 16:201–206.
Stangroom SJ, Collins CD, Lester JN (1998) Sources of organic micropollutants to lowland rivers. Environmental Technology, 19:643–666.
Storm DL (1994) Chemical monitoring of California’s public drinking water sources: public exposures and health impacts. In: Wang RGM, ed. Water contamination and health: Integration of exposure assessment, toxicology, and risk assessment. New York, NY, Marcel Dekker, pp. 67–124 (Environmental Science and Pollution Control Series 9).
Strobel K, Grummt T (1987) Aliphatic and aromatic hydrocarbons as potential mutagens in drinking water. III. Halogenated ethanes and ethenes. Toxicology and Environmental Chemistry, 15:101–128.
Tabak H, Quave SA, Mashni CI, Barth EF (1981) Biodegradability studies with organic priority pollutant compounds. Journal of the Water Pollution Control Federation, 53:1503–1518.
US EPA (1978) In-depth studies on health and environmental impacts of selected water pollutants. Washington, DC, US Environmental Protection Agency (Contract No. 68-01-4646) [cited in US EPA, 1980].
US EPA (1980) Ambient water quality criteria for dichloroethylenes. Washington, DC, US Environmental Protection Agency, Office of Water, Regulations and Standards Division (EPA 440/5-80-041).
US EPA (1985) Health assessment document for vinylidene chloride. Research Triangle Park, NC, US Environmental Protection Agency, Office of Health and Environmental Assessment, Environmental Criteria and Assessment Office (EPA/600/8-83-031F).
US EPA (1988) Compendium of methods for the determination of toxic organic compounds in ambient air. Research Triangle Park, NC, US Environmental Protection Agency, Office of Research and Development (EPA/600/4-84-041).
US EPA (1994) Methods for derivation of inhalation reference concentrations and application of inhalation dosimetry. Washington, DC, US Environmental Protection Agency (EPA/600/8-90/066F).
US EPA (1995) Method 502.2 and 524.2. Methods for the determination of organic compounds in drinking water — Supplement III. Washington, DC, US Environmental Protection Agency (EPA/600/R-95-131).
US EPA (1998) Method 8260B. SW-846 manual. Washington, DC, US Environmental Protection Agency, at website http://www.epa.gov/epaoswer/hazwaste/test/SW846.htm.
US EPA (2002a) AirData. Washington, DC, US Environmental Protection Agency, at website http://www.epa.gov/aqspubl1/annual_summary.html.
US EPA (2002b) CERCLIS database. Washington, DC, US Environmental Protection Agency.
US EPA (2002c) Toxics Release Inventory (TRI) Program. Washington, DC, US Environmental Protection Agency, at website http://www.epa.gov/tri.
US EPA (2002d) Toxicological review of 1,1-dichloroethylene (CAS No.
Van Duuren BL, Goldschmidt BM, Loewengart G, Smith AC, Melchionne S, Seldman I, Roth D (1979) Carcinogenicity of halogenated olefinic and aliphatic hydrocarbons in mice. Journal of the National Cancer Institute, 63:1433–1439.
Viola PL, Caputo A (1977) Carcinogenicity studies on vinylidene chloride. Environmental Health Perspectives, 21:45–47.
Viswanathan R (1984) Regenwurmtest. In: Ballhorn L, Freitag D, eds. Überprüfung der Durchführbarkeit von Prüfungsworschriften und der Aussagekraft der Stufe I und II des E. Chem. G. Neuherberg, Gesellschaft für Strahlen- und Umweltforschung München mbH, pp. 124–131.
Vogel TM, McCarty PL (1987) Abiotic and biotic transformations of 1,1,1-trichloroethane under methanogenic conditions. Environmental Science and Technology, 21:1208–1213.
Wallace L, Zweidinger R, Erickson M, Cooper S, Whitaker D, Pellizzari E (1982) Monitoring individual exposure. Measurements of volatile organic compounds in breathing-zone air, drinking water, food, and exhaled breath. Environment International, 8:269–282.
Wallace L, Pellizzari E, Hartwell T, Rozensweig M, Erickson M, Sparacino C, Zelon H (1984) Personal exposure to volatile organic compounds. I. Direct measurements in breathing-zone air, drinking water, food, and exhaled breath. Environmental Research, 35:293–319.
Wallace LA, Pellizzari E, Sheldon S, Hartwell T, Sparacino C, Zelon H (1986) The total exposure assessment methodology (TEAM) study: direct measurements of personal exposures through air and water for 600 residents of several US cities. In: Cohen Y, ed. Pollutants in a multimedia environment. New York, NY, Plenum Publishing Corporation, pp. 289–315.
Warbrick EV, Dearman RJ, Ashby J, Schmezer P, Kimber I (2001) Preliminary assessment of the skin sensitizing activity of selected rodent carcinogens using the local lymph node assay. Toxicology, 163:63–69.
Waskell L (1978) A study of the mutagenicity of anesthetics and their metabolism. Mutation Research, 57:141–153.
WHO (1993) Guidelines for drinking-water quality, 2nd ed. Volume 1. Recommendations. Geneva, World Health Organization.
Wilson SC, Burnett V, Waterhouse KS, Jones KC (1994) Volatile organic compounds in digested United Kingdom sewage sludges. Environmental Science and Technology, 28:259–266.
Wong PS, Cooks RG, Cisper ME, Hemberger PH (1995) On-line, in situ analysis with membrane introduction MS. Environmental Science and Technology, 29:215A–218A.
Yamamoto K, Fukushima M, Kakutani N, Kuroda K (1997) Volatile organic compounds in urban rivers and their estuaries in Osaka, Japan. Environmental Pollution, 95:135–143.
US EPA (2002d): Toxicological review of 1,1-dichloroethylene (CAS No.
Copies of the document may be obtained from:
EPA Risk Assessment Hotline
513-569-7254 (telephone)
513-569-7159 (fax)
rih.iris@epa.gov (e-mail address)
(Internet pdf file)
This document and summary information on IRIS have received peer review both by Environmental Protection Agency (EPA) scientists and by independent scientists external to EPA. Subsequent to external review and incorporation of comments, this assessment has undergone an Agency-wide review process whereby the IRIS Program Manager has achieved a consensus approval among the Office of Research and Development; Office of Air and Radiation; Office of Prevention, Pesticides, and Toxic Substances; Office of Solid Waste and Emergency Response; Office of Water; Office of Policy, Planning, and Evaluation; and the Regional Offices.
Internal EPA reviewers:
J. Cogliano, C. Kimmel, L. Flowers, K. Hogan, National Center for Environmental Assessment, Washington, DC
J. Strickland, National Center for Environmental Assessment, Research Triangle Park, NC
D. Niedzwiecki, Colorado Department of Public Health and Environment
External peer reviewers:
Melvin E. Andersen, Colorado State University, Ft. Collins, CO
James V. Bruckner, University of Georgia, Athens, GA
Poh-Gek Forkert, Queen’s University, Kingston, Ontario, Canada
Sam Kacew, University of Ottawa, Ottawa, Ontario, Canada
Kannan Krishnan, University of Montreal, Montreal, Quebec, Canada
US EPA (1980): Ambient water quality criteria for dichloroethylenes (EPA 440/5-80-041)
This report was prepared by the Office of Water Regulations and Standards, the Office of Research and Development, the Carcinogen Assessment Group, and the Environmental Research Laboratories of the US Environmental Protection Agency (EPA). It was reviewed by the Environmental Criteria and Assessment Office, US EPA, and approved for publication. It is available to the public from the National Technical Information Service in Springfield, Virginia, or on the Internet at: http://www.epa.gov/ost/pc/ambientwqc/dichloroethylenes80.pdf.
IPCS (1990): Vinylidene chloride (Environmental Health Criteria 100)
The first draft of this Environmental Health Criteria document was prepared by Dr J.K. Chipman, University of Birmingham, England. The draft was sent for peer review to IPCS national Contact Points and Collaborating Centres and was reviewed and approved as an international assessment document at a Task Group Meeting held in October 1988 in Rome, Italy.
This report is available from Marketing and Dissemination, World Health Organization, 1211 Geneva 27, Switzerland (fax + 41 22 791 4857) or at bookorders@who.int; it is also available on the Internet at http://www.inchem.org/documents/ehc/ehc/ehc100.htm.
The draft CICAD on 1,1-DCE was sent for review to institutions and organizations identified by IPCS after contact with IPCS national Contact Points and Participating Institutions, as well as to identified experts. Comments were received from:
Å. Andreassen, Department of Chemical Toxicology, Norwegian Institute of Public Health, Norway
R.M. Bruce, US Environmental Protection Agency, USA
R.J. Chhabra, National Institute of Environmental Health Sciences, USA
C. Cooke, Health and Safety Executive, United Kingdom
C. Cowles, Health and Safety Executive, United Kingdom
C.L. Deford, The Dow Chemical Company, USA
I. Desi, University of Szeged, Hungary
G. Dura, Fodor József National Public Health Centre, Hungary
C. Elliott-Minty, Health and Safety Executive, United Kingdom
L. Fishbein, Private Consultant, USA
E. Frantik, National Institute of Public Health, Czech Republic
M. Gwinn, National Institute of Occupational Safety and Health, USA
K. Hattefield, National Institute of Occupational Safety and Health, USA
R.F. Hertel, Federal Institute for Health Protection of Consumers and Veterinary Medicine, Germany
C. Hiremath, US Environmental Protection Agency, USA
A. Hirose, National Institute of Health Sciences, Japan
A. Hirvonen, Institute of Occupational Health, Finland
P. Howe, Centre for Ecology and Hydrology, United Kingdom
J. Kielhorn, Fraunhofer Institute of Toxicology and Aerosol Research, Germany
G. Koennecker, Fraunhofer Institute of Toxicology and Aerosol Research, Germany
S.H. Lee, Catholic University, Korea
J. Rantanen, Institute of Occupational Health, Finland
J. Stauber, CSIRO Energy Technology, Australia
J.H.M. Temmink, Wageningen Agricultural University, Netherlands
D. van Wijk, European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC) / Euro Chlor, Belgium
K. Victorin, Karolinska Institutet, Sweden
I. Zastenskaya, Scientific Institute of Sanitary and Hygiene, Belarus
Monks Wood, United Kingdom,
16–19 September 2002
Members
Dr R. Benson, US Environmental Protection Agency, Region VIII, Denver, CO, USA
Mr R. Cary, Health and Safety Executive, Bootle, Merseyside, United Kingdom
Dr R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
Dr S. Chou, Agency for Toxic Substances and Disease Registry (ATSDR), Atlanta, GA, USA
Dr S. Czerczak, Nofer Institute of Occupational Medicine, Lodz, Poland
Dr S. Dobson, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr G. Dura, National Institute of Environmental Health, Jozsef Fodor Public Health Centre, Budapest, Hungary
Dr L. Fishbein, Private Consultant, Fairfax, VA, USA
Dr H. Gibb, National Center for Environmental Assessment, US Environmental Protection Agency, Washington, DC, USA
Dr Y. Hayashi, Division of Chem-Bio Informatics, National Institute of Health Sciences, Ministry of Health, Labour and Welfare, Tokyo, Japan
Dr R.F. Hertel, Federal Institute for Health Protection of Consumers and Veterinary Medicine, Berlin, Germany
Dr A. Hirose, Division of Risk Assessment, National Institute of Health Sciences, Tokyo, Japan
Mr P. Howe, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Prof. J. Jeyaratnam, Colombo, Sri Lanka
Dr J. Kielhorn, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany
Prof. Y.-X. Liang, School of Public Health, Fudan University, Shanghai Medical College, Shanghai, People’s Republic of China
Dr R. Liteplo, Existing Substances Division, Environmental Contaminants Bureau, Health Canada, Ottawa, Ontario, Canada
Ms M.E. Meek, Existing Substances Division, Safe Environments Programme, Health Canada, Ottawa, Ontario, Canada
Mr F.K. Muchiri, Directorate of Occupational Health and Safety Services, Nairobi, Kenya
Dr O. Sabzevari, Department of Toxicology & Pharmacology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
Dr J. Sekizawa, Division of Chem-Bio Informatics, National Institute of Health Sciences, Tokyo, Japan
Dr F.P. Simeonova, Sofia, Bulgaria
Dr J. Stauber, CSIRO Energy Technology, Centre for Advanced Analytical Chemistry, Bangor, Australia
Dr M.H. Sweeney, Document Development Branch, Education and Information Division, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
Dr K. Ziegler-Skylakakis, European Commission, DG Employment & Social Affairs, Luxembourg
Resource Persons
Dr C. Cowles, Health and Safety Executive, Industrial Chemicals Unit HD, Bootle, Merseyside, United Kingdom
Dr C. Elliott-Minty, Health and Safety Executive, Industrial Chemicals Unit HD, Bootle, Merseyside, United Kingdom
Dr K. Fuller, Health and Safety Executive, Industrial Chemicals Unit HD, Bootle, Merseyside, United Kingdom
Observers
Mr A.G. Berends, Solvay S.A., Brussels, Belgium; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr W. Gulledge, American Chemistry Council, Arlington, VA, USA
Mr C. Newsome, Dow Chemical Company Limited, West Drayton, Middlesex, United Kingdom; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr M.A. Pemberton, Wilmslow, United Kingdom; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr W. Stott, Dow Chemical Company, Midland, MI, USA; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr J.M. Waechter, Jr, The Dow Chemical Company, Midland, MI, USA; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr T. Ehara, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr H. Malcolm, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Ms C. Vickers, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Oral
Data on fatty change in the liver from Quast et al. (1983) were analysed using EPA’s benchmark dose software. Each of the seven models gave an adequate fit (P > 0.2). The gamma, logistic, multistage, quantal-linear, and Weibull models showed the best visual fit to the data points. The gamma, multistage, quantal-linear, and Weibull models showed identical Akaike’s Information Criterion (AIC) values and identical BMDs and BMDLs. The results from the gamma model are presented.
The form of the probability function is:
P[response] = background + (1 − background) * CumGamma [slope * dose, power]
where CumGamma(.) is the cumulative Gamma distribution function.
Default initial (and specified) parameter values
|
Background = |
0.12963 |
|
Slope = |
0.0192007 |
|
Power = |
1.12817 |
Asymptotic correlation matrix of parameter estimates
(*** The model parameter Power has been estimated at a boundary point or has been specified by the user and does not appear in the correlation matrix)
|
Background |
Slope |
|
|
Background |
1 |
–0.54 |
|
Slope |
–0.54 |
1 |
Parameter estimates
|
Variable |
Estimate |
Standard error |
|
Background |
0.125627 |
0.0350171 |
|
Slope |
0.0158781 |
0.00405428 |
|
Power |
1 |
NA |
NA - Indicates that this parameter has hit a bound implied by some inequality constraint and thus has no standard error.
Analysis of deviance table
|
Model |
Log |
Deviance |
Test DF |
P-value |
|
Full model |
–119.212 |
|||
|
Fitted model |
–119.229 |
0.0326243 |
2 |
0.9838 |
|
Reduced model |
–128.113 |
17.8011 |
3 |
0.0004834 |
|
AIC = 242.458 |
||||
Goodness of fit
|
Dose |
Est. prob. |
Exp. |
Obs. |
Size |
Scaled residual |
|
0 |
0.1256 |
10.050 |
10 |
80 |
–0.01693 |
|
9 |
0.2421 |
11.619 |
12 |
48 |
0.1284 |
|
14 |
0.2999 |
14.396 |
14 |
48 |
–0.1246 |
|
30 |
0.4570 |
21.935 |
22 |
48 |
0.01895 |
Chi-square = 0.03; DF = 2; P-value = 0.9838
Benchmark dose computation
|
Specified effect = |
0.1 |
|
Risk type = |
Extra risk |
|
Confidence level = |
0.95 |
|
BMD = |
6.63557 mg/kg body weight per day |
|
BMDL = |
4.61215 mg/kg body weight per day |
Inhalation
Data on fatty change in the liver from Quast et al. (1986) were analysed using EPA’s benchmark dose software. The gamma, multistage, quantal-linear models gave an adequate fit (P > 0.2). These models also gave an adequate visual fit to the data points. The quantal-linear model gave the lowest AIC value. The results from this model are presented.
The form of the probability function is:
P[response] = background + (1 − background) * [1 − EXP(−slope * dose)]
Default initial (and specified) parameter values
|
Background = |
0.0294118 |
|
Slope = |
0.00549306 |
|
Power = |
1 Specified |
Asymptotic correlation matrix of parameter estimates
(*** The model parameters Background and Power have been estimated at a boundary point or have been specified by the user and do not appear in the correlation matrix)
Slope = 1
Parameter estimates
|
Variable |
Estimate |
Standard error |
|
Background |
0 |
NA |
|
Slope |
0.00697979 |
0.00194885 |
NA - Indicates that this parameter has hit a bound implied by some inequality constraint and thus has no standard error.
Analysis of deviance table
|
Model |
Log |
Deviance test |
DF |
P-value |
|
Full model |
−27.7336 |
|||
|
Fitted model |
−28.0929 |
0.718624 |
2 |
0.6982 |
|
Reduced model |
−32.5262 |
9.58514 |
2 |
0.008291 |
|
AIC = 58.1858 |
||||
Goodness of fit
|
Dose |
Est. prob. |
Exp. |
Obs. |
Size |
Scaled residual |
|
0 |
0.0000 |
0.000 |
0 |
16 |
0 |
|
25 |
0.1601 |
4.643 |
6 |
29 |
0.6869 |
|
75 |
0.4075 |
8.151 |
7 |
20 |
−0.5237 |
Chi-square = 0.75; DF = 2; P-value = 0.6886
Benchmark dose computation
|
Specified effect = |
0.1 |
|
Risk type = |
Extra risk |
|
Confidence level = |
0.95 |
|
BMC = |
15.0951 ppm |
|
BMCL = |
9.84365 ppm |
|
BMCLadj = |
BMCL x 6/24 x 5/7 x 3.97 mg/m3/ppm = 6.9 mg/m3 |
|
BMCLHEC = |
BMCLadj x (Hb/g)A/(Hb/g)H = 6.9 mg/m3 |
The blood:air partition coefficient in rats ((Hb/g)A) is 5 (D’Souza & Andersen, 1988). No published data are available to determine the blood:air partition coefficient in humans ((Hb/g)H). Therefore, the default value of 1 is used for (Hb/g)A/(Hb/g)H (US EPA, 1994). The BMCLHEC is 6.9 mg/m3.
Oral*
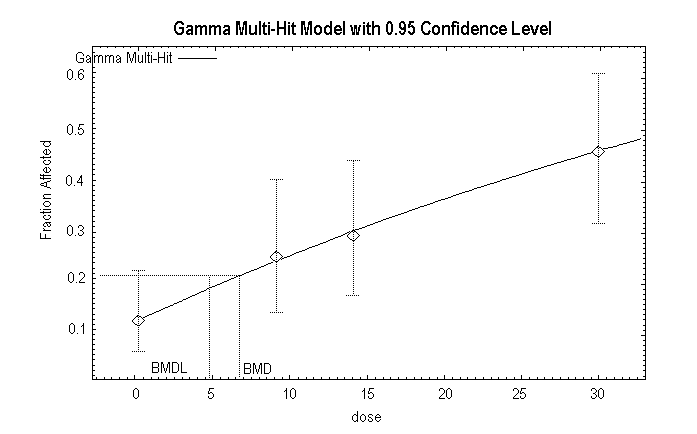
* Note: Dose units are mg/kg body weight per day.
Inhalation*
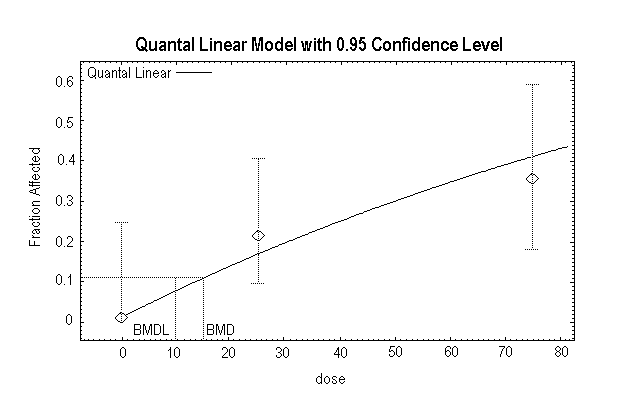
* Note: Dose units are ppm. BMD = BMC; BMDL = BMCL.
|
AIC |
Akaike’s Information Criterion |
|
BMC10 |
benchmark concentration for a 10% response |
|
BMCL10 |
lower 95% confidence limit on the benchmark concentration for a 10% response |
|
BMCLadj |
lower 95% confidence limit on the benchmark concentration for a 10% response adjusted to continuous exposure |
|
BMCLHEC |
lower 95% confidence limit on the benchmark concentration for a 10% response adjusted to a human equivalent concentration |
|
BMD |
benchmark dose |
|
BMD10 |
benchmark dose for a 10% response |
|
BMDL |
lower 95% confidence limit on the benchmark dose |
|
BMDL10 |
lower 95% confidence limit on the benchmark dose for a 10% response |
|
CYP2E1 |
cytochrome P450-dependent monooxygenase 2E1 |
|
1,1-DCE |
1,1-dichloroethene |
|
DCE-epoxide |
1,1-dichloroethene oxide |
|
EC10 |
effective concentration for a 10% response |
|
EC20 |
effective concentration for a 20% response |
|
EC50 |
effective concentration for a 50% response |
|
ECD |
electrolytic conductivity detector |
|
EPA |
Environmental Protection Agency (US) |
|
FID |
flame ionization detector |
|
GC |
gas chromatography |
|
GSH |
glutathione |
|
(Hb/g)A |
blood:air partition coefficient for laboratory animal species |
|
(Hb/g)H |
blood:air partition coefficient for human |
|
LC50 |
concentration for 50% lethality |
|
LD50 |
dose for 50% lethality |
|
LOAEL |
lowest-observed-adverse-effect level |
|
LT50 |
time for 50% lethality |
|
MS |
mass spectrometry |
|
NOAEL |
no-observed-adverse-effect level |
|
NOEC |
no-observed-effect concentration |
|
NOEL |
no-observed-effect level |
|
NTP |
National Toxicology Program (US) |
|
PBPK |
physiologically based pharmacokinetic |
|
PEC |
predicted exposure concentration |
|
PNEC |
predicted no-effect concentration |
|
PVDC |
polyvinylidene chloride |
Ce CICAD relatif au 1,1-dichloroéthylène (chlorure de vinylidène) a été préparé par l’Agence de protection de l’environnement des Etats-Unis (Environmental Protection Agency - EPA). L’EPA s’est servie des données prises en compte jusqu’à avril 2001 dans le document national d’évaluation. Des renseignements sur la nature de l’examen par des pairs et sur la disponibilité de ce document national d’évaluation sont donnés à l’appendice 1. Les références bibliographiques utilisées pour ce CICAD ont été mises à jour en août 2002. Les informations relatives à l’examen par des pairs du présent CICAD figurent à l’appendice 2. Ce CICAD a été approuvé en tant qu’évaluation internationale lors d’une réunion du Comité d’évaluation finale qui s’est tenue à Monks Wood (Royaume-Uni) du 16 au 19 septembre 2002. La liste des participants à cette réunion est donnée à l’appendice 3. La fiche internationale sur la sécurité chimique du 1,1-dichloroéthylène (ICSC 0083) établie par le Programme international sur la sécurité chimique (IPCS, 2000), est également reproduite dans le présent document.
Le 1,1-dichloroéthylène (No CAS
Le 1,1-DCE présent dans l’environnement provient d’émissions survenues lors de la production ou de l’utilisation de ce produit, de la décomposition de matériaux à base de chlorure de polyvinylidène (PVDC) ou encore de la dégradation biotique ou abiotique du 1,1,1-trichloroéthane, du tétrachloréthylène, du 1,1,2-trichloroéthylène et du 1,1-dichloroéthane. Les principales sources environnementales d’exposition humaine sont l’air ambiant et l’eau de boisson contaminée.
Dans les eaux souterraines, la biotransformation du 1,1-DCE peut conduite au chlorure de vinyle par déchloration réductrice.
Du fait de sa forte tension de vapeur et de sa faible solubilité dans l’eau, le 1,1-DCE est présent à concentration relativement élevée dans l’atmosphère, comparativement aux autres compartiments de l’environnement. Les radicaux hydroxyles atmosphériques jouent un rôle important dans la décomposition du 1,1-DCE. Sa demi-vie atmosphérique est estimée à 16 heures. Le 1,1-DCE présent dans l’eau, le sol et les sédiments est principalement transporté hors de ces compartiments par volatilisation. Sa bioaccumulation devrait être faible compte tenu de son coefficient de partage octanol/eau et de sa solubilité dans l’eau.
Le composé est rapidement résorbé après inhalation ou ingestion. En raison de sa faible masse moléculaire relative et de son caractère hydrophobe, il est probable qu’il puisse également être résorbé par voie percutanée mais rien n’a été publié à ce sujet. Bien qu’il se répartisse rapidement dans tous les tissus, on retrouve le 1,1-DCE ainsi que ses métabolites et un certain nombre de dérivés formés par liaison covalente en majeure partie dans le foie et le rein. La monooxygénase 2E1 dépendante du cytochrome P-450 (CYP2E1) catalyse l’oxydation rapide du 1,1-DCE en oxyde de 1,1-dichloroéthylène (DCE-époxyde), chlorure de 2-chloroacétyle et 2,2-dichloroacétaldéhyde. Ses principaux métabolites, le DCE-époxyde et le 2-chloroacétaldéhyde peuvent réagir avec le glutathion (GSH), l’eau ou encore les macromolécules tissulaires. On ignore si le métabolisme du 1,1-DCE est identique chez l’Homme, mais in vitro, en présence de préparations de microsomes obtenues à partir de tissus hépatiques et pulmonaires humains, il y a formation des mêmes produits initiaux.
La seule étude épidémiologique existante ne se prête pas à une évaluation des effets néoplasiques ou non néoplasiques du 1,1-DCE.
L’expérimentation animale montre qu’après exposition à une forte dose de 1,1-DCE par la voie orale ou respiratoire, les principaux organes cibles sont le foie, le rein et le poumon au niveau des cellules de Clara. Après exposition de longue durée à une faible dose par voie orale ou respiratoire, c’est le foie qui est le principal organe cible chez le rat; en revanche, c’est le rein chez des souris exposées par la voie respiratoire.
Des épreuves biologiques utilisant la voie orale ont été pratiquées sur des rats, des souris et des truites à la recherche d’éventuels effets cancérogènes. S’il est vrai que les protocoles expérimentaux utilisés présentent des insuffisances, aucun des résultats obtenus ne fournit la moindre preuve que le 1,1-DCE soit cancérogène après exposition par voie orale. D’autres épreuves de cancérogénicité utilisant cette fois la voie respiratoire ont été pratiquées sur des rats, des souris et des hamsters. Là encore, les protocoles expérimentaux présentent un certain nombre d’insuffisances. L’un des tests effectués sur des souris mâles a permis de constater une augmentation de l’incidence des adénocarcinomes du rein pour l’une des valeurs de l’exposition. On est fondé à penser que la formation d’adénocarcinomes du rein constitue une réaction spécifique à la fois de l’espèce et du sexe qui est liée à l’expression de la CYP2E1 dans le rein de la souris mâle. Toutefois, le fait de constater, dans une épreuve biologique, une augmentation des tumeurs chez les animaux d’une seule espèce, d’un sexe donné et pour une valeur donnée de l’exposition, ne suffit pas pour affirmer qu’il existe une relation exposition-réponse.
En présence d’un système d’activation exogène, le 1,1-DCE provoque des mutations géniques chez des microorganismes. En revanche, plupart des tests effectués in vitro et in vivo sur des cellules mammaliennes sont négatifs quant à la génotoxicité du composé.
Rien n’indique que le 1,1-DCE soit doté d’une toxicité génésique ou d’une tératogénicité importantes. En effet, après une exposition par voie orale qui n’a provoqué que des effets hépatotoxiques minimes sur les mères, aucun effet toxique n’a été constaté sur la reproduction des animaux ni sur le développement de leur progéniture. Il existe bien quelques éléments de preuve de certaines anomalies du développement cardiaque après exposition par voie orale au 1,1-DCE, mais on n’est pas sûr que ces effets soient la conséquence directe de l’exposition à ce composé. Par contre, après une exposition par la voie respiratoire, on a noté des signes de toxicité foetale (retard dans l’ossification) en l’absence de toxicité maternelle.
Selon une étude, le 1,1-DCE ne provoque pas de sensibilisation cutanée.
La toxicité du 1,1-DCE est liée à sa métabolisation, catalysée par le cytochrome P450, en intermédiaires réactifs qui se fixent par liaison covalente aux macromolécules cellulaires. L’ampleur de cette fixation est inversement proportionnelle à la diminution de la teneur en GSH. La réaction au 1,1-DCE devrait donc être sensiblement différente selon qu’il y a exposition à une faible dose de composé avec une petite déplétion en GSH ou exposition à une forte dose entraînant une importante déplétion en GSH.
L’effet essentiel constaté après exposition par voie orale est une dégénérescence graisseuse minime des hépatocytes avec présence de gouttelettes graisseuses en situation médiane chez des rattes Sprague-Dawley. Si l’on se base sur une BMDL10 (limite inférieure de l’intervalle de confiance à 95 % relatif à la dose de référence [BMD] pour une réponse de 10 %) égale à 4,6 mg/kg de poids corporel par jour et en prenant un facteur d’incertitude total de 100, on obtient une valeur de 0,05 mg/kg p.c. par jour pour la dose tolérable par ingestion.
Dans le cas d’une exposition par voie respiratoire, l’effet essentiel est également une dégénérescence graisseuse minime des hépatocytes avec présence de gouttelettes graisseuses en situation médiane chez des rattes Sprague-Dawley. En se basant sur une BMCL10 (limite inférieure de l’intervalle de confiance à 95 % relatif à la concentration de référence [BMC] pour une réponse de 10 %) égale à 6,9 mg/m3 et en prenant un facteur d’incertitude total de 30, on obtient une concentration tolérable de 0,2 mg/m3 par jour.
L’exposition humaine au 1,1-DCE est vraisemblablement très variable compte tenu du fait que la contamination dépend du site. Quoi qu’il en soit, les données indiquent que l’exposition moyenne due à l’eau de boisson ne devrait pas dépasser 6-9 × 10–5 mg/kg p.c. par jour pour un sujet de 70 kg consommant quotidiennement 2 litres d’eau. L’exposition par voie orale ayant pour origine les aliments ou le sol est très probablement négligeable. Selon les données disponibles, la limite supérieure de concentration du 1,1-DCE dans l’air ne devrait pas excéder 0,004 mg/m3. On est donc en droit de penser que l’exposition humaine est très inférieure à la dose journalière tolérable par ingestion de 0,05 mg/kg p.c. et à la concentration tolérable de 0,2 mg/m3.
On ne possède que des données limitées concernant les effets du 1,1-DCE sur l’environnement terrestre et aquatique. Des études effectuées dans des systèmes clos ont montré que la CE20 relative à l’inhibition de la croissance d’une culture méthanotrophe mixte était égale à 0,05 mg/litre. De même, on a obtenu une valeur de 9,12 mg/litre pour la CE50 à 72 h relative à l’inhibition de la croissance de l’algue verte Chlamydomonas reinhardtii, la CL50 à 96 h étant de 74 mg/litre pour la perche soleil Lepomis macrochirus. Selon les données limitées dont on dispose sur la présence du 1,1-DCE dans les eaux de surface, sa concentration serait de l’ordre du microgramme par litre, ce qui donne à penser que le risque de toxicité aiguë est minime dans l’environnement aquatique. On ne possède pas de données toxicologiques à long terme qui permettraient d’évaluer les effets sublétaux du composé sur les êtres vivants. Toutefois, en raison de la volatilisation rapide du 1,1-DCE à partir de l’environnement terrestre ou aquatique, il n’y a pas lieu de craindre un risque important.
Este CICAD sobre el 1,1-dicloroeteno (cloruro de vinilideno) fue preparado por la Agencia para la Protección del Medio Ambiente de los Estados Unidos (EPA). La EPA examinó los datos identificados hasta abril de 2001 en el documento de evaluación nacional (US EPA, 2002d). La información relativa al carácter del examen colegiado y a la disponibilidad de este documento de evaluación nacional figura en el apéndice 1. La búsqueda bibliográfica para este CICAD se actualizó en agosto de 2002. La información sobre el examen colegiado de este CICAD aparece en el apéndice 2. Este CICAD se aprobó como evaluación internacional en una reunión de la Junta de Evaluación Final, celebrada en Monks Wood (Reino Unido) del 16 al 19 de septiembre de 2002. La lista de participantes en esta reunión figura en el apéndice 3. La Ficha internacional de seguridad química (ICSC 0083) para el 1,1-dicloroeteno, preparada por el Programa Internacional de Seguridad de las Sustancias Químicas (IPCS, 2000), también se reproduce en este documento.
El 1,1-dicloroeteno (CAS Nº
El 1,1-DCE se puede encontrar en el medio ambiente procedente de las emisiones que se producen durante su fabricación y uso, de la desintegración de productos de polivinilideno y de la descomposición del 1,1,1-tricloroetano, el tetracloroeteno, el 1,1,2-tricloroeteno y el 1,1-dicloroetano. Las fuentes principales de exposición ambiental para las personas son el aire ambiente y el agua de bebida contaminada.
En el agua freática, la biotransformación del 1,1-DCE puede producir, mediante descloración reductiva, cloruro de vinilo.
La alta presión de vapor y la baja solubilidad en agua del 1,1-DCE favorecen la presencia de concentraciones relativamente altas en la atmósfera en comparación con otros compartimentos del medio ambiente. Los radicales hidroxilo atmosféricos desempeñan una función importante en la degradación del 1,1-DCE. Se estima que la semivida atmosférica es de 16 horas. La volatilización es el principal proceso de transporte a partir del agua, el suelo y los sedimentos. Basándose en el coeficiente de reparto octanol/agua y la solubilidad en agua de los productos químicos, se prevé una bioacumulación baja.
El 1,1-DCE se absorbe con rapidez tras la exposición por inhalación o por vía oral. También es probable la absorción cutánea, debido a su masa molecular relativamente baja y a su carácter hidrófobo, aunque no hay datos de interés publicados al respecto. Si bien el 1,1-DCE pasa con rapidez a todos los tejidos, la mayor parte del 1,1-DCE libre, sus metabolitos y los derivados de enlace covalente se encuentran en el hígado y el riñón. El 1,1-DCE se oxida con rapidez por acción de la monooxigenasa 2E1 dependiente del citocromo P450 (CYP2E1) a óxido de 1,1-dicloroeteno (epóxido-DCE), cloruro de 2-cloroacetilo y 2,2-dicloroacetaldehido. Los metabolitos principales, el epóxido-DCE y el cloruro de 2-cloroacetilo pueden reaccionar con el glutatión, el agua o las macromoléculas tisulares. No se sabe si el metabolismo del 1,1-DCE es el mismo en las personas, aunque se ha observado que en preparaciones microsomales in vitro de hígado y pulmón humanos se forman los mismos productos iniciales.
El único estudio epidemiológico existente es inadecuado para evaluar los efectos carcinogénicos del 1,1-DCE.
Tras la exposición a dosis altas por vía oral o por inhalación, los órganos destinatarios en animales de experimentación son el hígado, el riñón y las células Clara del pulmón. Tras la exposición prolongada de ratas a dosis bajas por vía oral o por inhalación se observó que el principal órgano destinatario es el hígado, sin embargo en los ratones expuestos por inhalación el órgano destinatario más importante es el riñón.
Se han realizado biovaloraciones para el cáncer mediante la exposición por vía oral de ratas, ratones y truchas. Si bien estas biovaloraciones tienen limitaciones de protocolo, ninguna proporciona pruebas significativas de que el 1,1-DCE sea carcinógeno por vía oral. Se han realizado biovaloraciones para el cáncer mediante la exposición de ratas, ratones y hámsteres por inhalación. La mayoría de estas biovaloraciones también tienen limitaciones de protocolo. En una biovaloración con ratones macho se puso de manifiesto un aumento de la incidencia de adenocarcinomas de riñón con un solo nivel de exposición. Hay pruebas de que la inducción de adenocarcinomas de riñón es una respuesta específica en función del sexo y de la especie, relacionada con la expresión del CYP2E1 en el riñón de los ratones macho. Los resultados de una sola biovaloración en la que se puso de manifiesto un aumento de tumores en un solo sexo y con un solo nivel de exposición en una única especie de roedores no son suficientes para justificar una evaluación de la exposición-respuesta.
El 1,1-DCE provoca mutaciones genéticas en microorganismos en presencia de un sistema de activación exógeno. La mayor parte de las pruebas con células de mamíferos in vitro o in vivo no ponen de manifiesto signos de genotoxicidad.
No hay pruebas de que la toxicidad reproductiva o la teratogenicidad sea un efecto crítico del 1,1-DCE. No se observó toxicidad reproductiva o del desarrollo en una exposición oral que provocó una toxicidad mínima en el hígado de las crías. Hay algunas pruebas de variaciones en el desarrollo del corazón tras la exposición oral, pero no está claro si estos efectos se deben a la exposición directa al 1,1-DCE. Hay pruebas de toxicidad fetal (osificación retardada) tras la exposición por inhalación en ausencia de toxicidad materna.
En un solo estudio no hay pruebas que demuestren que el 1,1-DCE provoca sensibilización cutánea.
La toxicidad del 1,1-DCE está asociada con la catalización de su metabolismo por el citocromo P-450 para formar intermediarios reactivos que se unen por enlace covalente a macromoléculas celulares. La magnitud de esta unión es inversamente proporcional a la pérdida de glutatión, de manera que la gravedad del daño tisular es paralela a la disminución de glutatión. Por consiguiente, cabe suponer que las respuestas al 1,1-DCE en dosis bajas con una escasa disminución del glutatión serán muy diferentes de las registradas con dosis altas, que provocan una reducción sustancial de dicho compuesto.
El efecto crítico de la exposición oral es un cambio mínimo de la grasa hepatocelular de la zona media en ratas Sprague-Dawley hembra. Basándose en un BMDL10 (límite de confianza mínimo del 95% con respecto a la dosis de referencia [BMD] para una respuesta del 10%) de 4,6 mg/kg de peso corporal al día y un factor de incertidumbre total de 100, la ingesta tolerable es de 0,05 mg/kg de peso corporal al día.
El efecto crítico de la exposición por inhalación es un cambio mínimo de la grasa hepatocelular de la zona media en ratas Sprague-Dawley hembra. Basándose en un BMCL10 (límite de confianza mínimo del 95% con respecto a la concentración de referencia [BMC] para una respuesta del 10%) de 6,9 mg/m3 y un factor de incertidumbre total de 30, la concentración tolerable es de 0,2 mg/m3.
La exposición humana al 1,1-DCE probablemente es muy variable, debido a la contaminación específica de determinados lugares. Sin embargo, los datos disponibles parecen indicar que la exposición media a partir del agua de bebida no es superior a 6-9 × 10–5 mg/kg de peso corporal al día para una persona de 70 kg que consuma dos litros de agua diarios. Es muy probable que la exposición oral a partir de los alimentos y el suelo sea insignificante. Los datos disponibles parecen indicar que el límite superior de la gama para la concentración media de 1,1-DCE en el aire no rebasa el valor de 0,004 mg/m3. Así pues, cabe prever una exposición humana muy inferior a la ingesta tolerable de 0,05 mg/kg de peso corporal al día y a la concentración tolerable de 0,2 mg/m3.
Sólo hay datos limitados sobre los efectos del 1,1-DCE en los compartimentos acuático y terrestre. En estudios realizados en sistemas cerrados, la CE20 para la inhibición del crecimiento de un cultivo metanotrófico mixto fue de 0,05 mg/l; la CE50 a las 72 horas para la inhibición del crecimiento del alga verde Chlamydomonas reinhardtii fue de 9,12 mg/l; y la CL50 a las 96 horas para Lepomis macrochirus fue de 74 mg/l. Hay datos limitados sobre la presencia de 1,1-DCE en el agua superficial que parecen indicar concentraciones del orden de µg/l, por lo que el riesgo de toxicidad aguda para el compartimento acuático debido al 1,1-DCE es mínimo. No hay datos sobre la toxicidad a largo plazo que permitan evaluar los efectos subletales del 1,1-DCE en ningún organismo. Sin embargo, debido a su volatilización rápida de los compartimentos acuático y terrestre no cabe esperar un riesgo significativo.
ENDNOTES:
|
International Programme on Chemical Safety (1994) Assessing human health risks of chemicals: derivation of guidance values for health-based exposure limits. Geneva, World Health Organization (Environmental Health Criteria 170) (also available at http://www.who.int/pcs/). |
|
|
In keeping with WHO policy, which is to provide measurements in SI units, all concentrations of gaseous chemicals in air will be given in SI units in the CICAD series. Where the original study or source document has provided concentrations in SI units, these will be cited here. Where the original study or source document has provided concentrations in volumetric units, conversions will be done using the conversion factors given here, assuming a temperature of 20 °C and a pressure of 101.3 kPa. Conversions are to no more than two significant digits. |
|
|
The upper end of the range of the mean concentration was used in the sample risk characterization in section 11. |
|
|
The results of this study were used in the sample risk characterization in section 11. |
|
|
New information relevant to a revised guideline should become available on the WHO website http://www.who.int/water_sanitation_health/GDWQ/Chemicals/orgconstitindex.htm. |
See Also:
Toxicological Abbreviations