
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization.
Concise International Chemical Assessment Document 69
First draft prepared by Dr James H. Kim and Dr Herman J. Gibb, Sciences International Inc., Alexandria, Virginia, USA; and Mr Paul D. Howe, Centre for Ecology and Hydrology, Monks Wood, Huntingdon, Cambridgeshire, United Kingdom
Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals.
The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
Kim, James H.
Cobalt and inorganic cobalt compounds/prepared by James H. Kim, Herman J. Gibb, Paul D. Howe
(Concise international chemical assessment document ; 69)
1. Cobalt - adverse effects. 2.Cobalt - toxicity. 3. Environmental exposure. 4 Risk
assessment. I.Gibb, Herman J. II. Howe, Paul D. III. World Health Organization
IV. International Programme on Chemical Safety. V.Title. VI. Series
ISBN 92 4 153069 3 (NLM Classification: QV 290)
ISSN 978 92 4 153069 9
©World Health Organization 2006
All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 3264; fax: +41 22 791 4857; email: bookorders@who.int). Requests for permission to reproduce or translate WHO publications — whether for sale or for noncommercial distribution — should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; email: permissions@who.int).
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
All reasonable precautions have been taken by WHO to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either express or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use.
Risk assessment activities of the International Programme on Chemical Safety, including the production of Concise International Chemical Assessment Documents, are supported financially by the Department of Health and Department for Environment, Food & Rural Affairs, UK, Environmental Protection Agency, Food and Drug Administration, and National Institute of Environmental Health Sciences, USA, European Commission, German Federal Ministry of Environment, Nature Conservation and Nuclear Safety, Health Canada, Japanese Ministry of Health, Labour and Welfare, and Swiss Agency for Environment, Forests and Landscape.
Concise International Chemical Assessment Documents (CICADs) are published by the International Programme on Chemical Safety (IPCS) — a cooperative programme of the World Health Organization (WHO), the International Labour Organization (ILO), and the United Nations Environment Programme (UNEP). CICADs have been developed from the Environmental Health Criteria documents (EHCs), more than 200 of which have been published since 1976 as authoritative documents on the risk assessment of chemicals.
International Chemical Safety Cards on the relevant chemical(s) are attached at the end of the CICAD, to provide the reader with concise information on the protection of human health and on emergency action. They are produced in a separate peer-reviewed procedure at IPCS. They may be complemented by information from IPCS Poison Information Monographs (PIM), similarly produced separately from the CICAD process.
CICADs are concise documents that provide summaries of the relevant scientific information concerning the potential effects of chemicals upon human health and/or the environment. They are usually based on selected national or regional evaluation documents or on existing EHCs. Before acceptance for publication as CICADs by IPCS, these documents undergo extensive peer review by internationally selected experts to ensure their completeness, accuracy in the way in which the original data are represented, and the validity of the conclusions drawn.
The primary objective of CICADs is characterization of hazard and dose–response from exposure to a chemical. CICADs are not a summary of all available data on a particular chemical; rather, they include only that information considered critical for characterization of the risk posed by the chemical. The critical studies are, however, presented in sufficient detail to support the conclusions drawn. For additional information, the reader should consult the identified source documents upon which the CICAD has been based.
Risks to human health and the environment will vary considerably depending upon the type and extent of exposure. Responsible authorities are strongly encouraged to characterize risk on the basis of locally measured or predicted exposure scenarios. To assist the reader, examples of exposure estimation and risk characterization are provided in CICADs, whenever possible. These examples cannot be considered as representing all possible exposure situations, but are provided as guidance only. The reader is referred to EHC 170.1
While every effort is made to ensure that CICADs represent the current status of knowledge, new information is being developed constantly. Unless otherwise stated, CICADs are based on a search of the scientific literature to the date shown in the executive summary. In the event that a reader becomes aware of new information that would change the conclusions drawn in a CICAD, the reader is requested to contact IPCS to inform it of the new information.
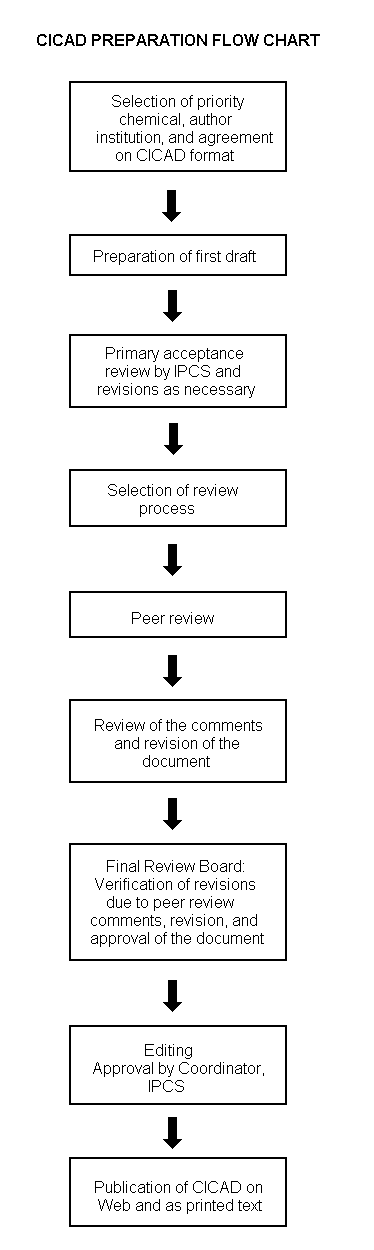
Procedures
The flow chart shows the procedures followed to produce a CICAD. These procedures are designed to take advantage of the expertise that exists around the world — expertise that is required to produce the high-quality evaluations of toxicological, exposure, and other data that are necessary for assessing risks to human health and/or the environment. The IPCS Risk Assessment Steering Group advises the Coordinator, IPCS, on the selection of chemicals for an IPCS risk assessment based on the following criteria:
Thus, it is typical of a priority chemical that:
|
Advice from Risk Assessment Steering Group Criteria of priority:
Thus, it is typical of a priority chemical that
Special emphasis is placed on avoiding duplication of effort by WHO and other international organizations. A prerequisite of the production of a CICAD is the availability of a recent high-quality national/regional risk assessment document = source document. The source document and the CICAD may be produced in parallel. If the source document does not contain an environmental section, this may be produced de novo, provided it is not controversial. If no source document is available, IPCS may produce a de novo risk assessment document if the cost is justified. Depending on the complexity and extent of controversy of the issues involved, the steering group may advise on different levels of peer review:
|
The Steering Group will also advise IPCS on the appropriate form of the document (i.e. a standard CICAD or a de novo CICAD) and which institution bears the responsibility of the document production, as well as on the type and extent of the international peer review.
The first draft is usually based on an existing national, regional, or international review. When no appropriate source document is available, a CICAD may be produced de novo. Authors of the first draft are usually, but not necessarily, from the institution that developed the original review. A standard outline has been developed to encourage consistency in form. The first draft undergoes primary review by IPCS to ensure that it meets the specified criteria for CICADs.
The second stage involves international peer review by scientists known for their particular expertise and by scientists selected from an international roster compiled by IPCS through recommendations from IPCS national Contact Points and from IPCS Participating Institutions. Adequate time is allowed for the selected experts to undertake a thorough review. Authors are required to take reviewers’ comments into account and revise their draft, if necessary. The resulting second draft is submitted to a Final Review Board together with the reviewers’ comments. At any stage in the international review process, a consultative group may be necessary to address specific areas of the science. When a CICAD is prepared de novo, a consultative group is normally convened.
The CICAD Final Review Board has several important functions:
Board members serve in their personal capacity, not as representatives of any organization, government, or industry. They are selected because of their expertise in human and environmental toxicology or because of their experience in the regulation of chemicals. Boards are chosen according to the range of expertise required for a meeting and the need for balanced geographic representation.
Board members, authors, reviewers, consultants, and advisers who participate in the preparation of a CICAD are required to declare any real or potential conflict of interest in relation to the subjects under discussion at any stage of the process. Representatives of nongovernmental organizations may be invited to observe the proceedings of the Final Review Board. Observers may participate in Board discussions only at the invitation of the Chairperson, and they may not participate in the final decision-making process.
This CICAD2 on cobalt and inorganic cobalt compounds was prepared by Sciences International, Inc. in the United States and the Centre for Ecology and Hydrology in the United Kingdom and was based on reviews prepared by the Agency for Toxic Substances and Disease Registry (ATSDR, 2004) and the International Agency for Research on Cancer (IARC, 2005). To address literature citations not included in either of these reviews, a comprehensive literature search of several online databases was conducted in April 2005. Information on the source documents and their peer reviews is presented in Appendix 2. Information on the peer review of this CICAD is presented in Appendix 3. This CICAD was considered and approved as an international assessment at a meeting of the Final Review Board held in Nagpur, India, on 31 October – 3 November 2005. Participants at the Final Review Board meeting are presented in Appendix 4. International Chemical Safety Cards for cobalt, cobalt(II) oxide, cobalt(III) oxide, cobalt(II) sulfide, cobalt(II) chloride, cobalt(II) sulfate, cobalt(II) sulfate heptahydrate, cobalt(II) nitrate, cobalt(II) nitrate hexahydrate, cobalt(II) acetate tetrahydrate, cobalt naphthenate, and cobalt carbonyl, produced by the International Programme on Chemical Safety (IPCS, 2000, 2001a–e, 2004a–f) in a separate, peer-reviewed process, have also been presented in this CICAD.
Cobalt (atomic number 27) is a naturally occurring element with one stable isotope (59Co) and 26 known radioactive isotopes. There are three valence states of cobalt (0, +2, and +3). Because cobalt may occur as a radioactive isotope, it can produce ionizing radiation. This document focuses primarily on stable cobalt. The reader should consult other sources, such as ATSDR (2004), for information on the effects of ionizing radiation from radioactive cobalt isotopes.
Cobalt (CAS No.
Cobalt and inorganic cobalt compounds are non-volatile and released into the atmosphere in particulate form. Anthropogenic cobalt from combustion sources is assumed to be primarily in the form of oxides. Sulfide and arsenide forms are also released into the atmosphere during ore extraction and refining processes.
Cobalt released into the atmosphere is deposited on soil, and cobalt released to water may sorb to particles and settle into sediment or sorb directly to sediment. The distribution coefficient of cobalt (e.g. from water to sediment) varies due to pH, redox conditions, ionic strength, and dissolved organic matter concentrations. Factors affecting the speciation and fate of cobalt in water, sediments, and soil include organic ligands such as humic acids, anions, pH, and redox potential. The soil mobility of cobalt is inversely related to the strength of adsorption by soil constituents. Although plants may take up cobalt from the soil, the translocation of cobalt from the roots to other parts of the plant is not significant.
Measured atmospheric concentrations of cobalt are about 1 ng/m3 or less in non-source areas and generally less than 10 ng/m3 in source areas, although higher concentrations in source areas have been reported. Surface water and groundwater concentrations of cobalt are low, below 1 µg/l in pristine areas and 1–10 µg/l in populated areas. Surface water and groundwater concentrations can be much higher in mining and agricultural areas — as much as several hundred milligrams per litre. Mean cobalt concentrations in seawater have been reported to be less than 1 µg/l. Cobalt concentrations in drinking-water are generally <1–2 µg/l. In rainwater, mean concentrations are 0.3–1.7 µg/l. The earth’s crust contains an average cobalt concentration of 20–25 mg/kg. Near some anthropogenic sources, the concentration of cobalt in soil may be several hundred milligrams per kilogram.
The largest source of exposure to cobalt for the general population is the food supply. The estimated intake from food is 5–40 µg/day, most of which is inorganic cobalt. Occupational exposure to cobalt occurs in several industries. Levels of cobalt in tobacco range from <0.3 to 2.3 µg/g dry weight, and approximately 0.5% of this cobalt is present in mainstream smoke. Cobalt concentrations in coal, crude oil, fuel oil, and gasoline in the United States were found to be 5 mg/kg, 0.001–10 mg/kg, 0.03–0.3 mg/kg, and <0.1 mg/kg, respectively.
Inhalation of cobalt particles results in deposition in the upper and lower respiratory tract, where they can be retained or absorbed into the blood after dissolution or mechanically transferred to the gastrointestinal tract by mucociliary action and swallowing. Approximately 50% of the cobalt that enters the gastrointestinal tract will be absorbed. Cobalt absorption is increased among individuals who are iron deficient. Water-soluble forms are better absorbed than insoluble forms. Cobalt is essential as a component of vitamin B12; therefore, it is found in most tissues. Total body burden is estimated as 1.1–1.5 mg, with 0.11 mg in the liver. After inhalation exposure, higher levels of cobalt have been found in the lung. No studies describe the distribution of cobalt in humans following ingestion, but animal studies indicate that cobalt is retained primarily in the liver. In a controlled human aerosol exposure study, 40% of the initial lung burden of cobalt oxide was retained at 6 months after exposure. Urinary excretion increases with time following inhalation exposure. Particle size affects elimination of inhaled cobalt, since more cobalt is mechanically cleared to the gastrointestinal tract when particles are larger. Faecal elimination is the primary route of excretion following oral exposure in humans.
The inhalation LC50 for cobalt hydrocarbonyl in rats was found to be 165 mg/m3 for a 30-min exposure. Oral LD50s for soluble cobalt compounds have been reported to range from 42.4 to 317 mg/kg body weight, depending on the compound and species tested. Tricobalt tetraoxide, an insoluble cobalt compound, is reported to have an LD50 of 3672 mg of cobalt per kilogram body weight in rats.
Rats and mice exposed short term (16 days) to cobalt sulfate by inhalation at cobalt concentrations of 19 mg/m3 and 1.9 mg/m3, respectively, exhibited necrosis and inflammation of the respiratory tract epithelium. Rats also developed thymus necrosis and testicular atrophy. Male rats exposed orally to cobalt chloride at a cobalt concentration of 12.4 mg/kg body weight per day for 3 weeks exhibited cardiac damage. Rats, rabbits, and mice exposed by inhalation to cobalt compounds at concentrations of >0.3 mg/m3 (cobalt concentrations of >0.11 mg/m3) for 3–4 months exhibited lesions of the respiratory tract. Rats exposed for 2–3 months to cobalt sulfate in the diet or to cobalt chloride in the drinking-water at cobalt doses of 26–30.2 mg/kg body weight per day exhibited increased heart weight and degenerative heart lesions. Rats exposed to cobalt sulfate at a cobalt dose of 8.4 mg/kg body weight per day in the diet for 24 weeks had significant reductions in heart enzyme activity levels. Rats exposed to cobalt chloride for 4–5 months at cobalt doses of 10–18 mg/kg body weight per day exhibited kidney damage.
Hamsters exposed by inhalation to cobalt oxide for a lifetime developed emphysema. Mice and rats exposed to cobalt sulfate by inhalation for 105 weeks developed lung tumours in a dose-related manner. Cobalt (as cobalt metal powder) produces tumours such as sarcomas in rats when injected intramuscularly.
Many cobalt compounds are genotoxic in mammals and in mammalian and bacterial test systems. Cobalt(III) compounds are positive in bacterial test systems. Cobalt(II) compounds were positive for genetic conversions in Saccharomyces cerevisiae but otherwise demonstrated little genotoxic activity.
Cobalt has been found to have reproductive and developmental effects in animals. Rats exposed to cobalt (as cobalt chloride) at 13.3–58.9 mg/kg body weight per day for 2–3 months and mice exposed to cobalt (as cobalt chloride) at 43.4 mg/kg body weight per day for 13 weeks exhibited testicular degeneration and atrophy. Male mice exposed to cobalt chloride at doses of 46.9 or 93.0 mg/kg body weight per day and mated with unexposed female mice displayed decreased epididymal weight, sperm count, testes weight, and fertility, as measured by the number of successful matings. In developmental studies, pregnant rats exposed to maternally toxic doses of cobalt chloride (5.4 or 21.8 mg of cobalt per kilogram body weight per day) produced newborn pups with stunted growth and decreased survival, but no teratogenic effects were observed. Rabbits exposed to cobalt (as cobalt sulfate) at 7.6 mg/kg body weight per day had increased fetal resorption and an increased number of fetuses with retarded body weight.
Inhalation and dermal exposure to cobalt in humans can result in sensitization. Bronchial asthma has been described in workers exposed to various forms of cobalt.
Humans ingesting cobalt chloride at 150 mg/day for 22 days experienced polycythaemia and an increase in haemoglobin. Studies have also reported cardiomyopathy in humans who had consumed large quantities of beer that contained cobalt sulfate.
Interstitial lung disease caused by metallic cobalt-containing particles is an occupational lung disease generally referred to as hard metal lung disease.
Mortality studies of the hard metal industry suggest an increase in lung cancer mortality. Cobalt is used as a binder in this industry, and exposures to other compounds, including tungsten carbide and other metallic compounds, such as titanium carbide, tantalium carbide, and niobium carbide, also occur.
A cross-sectional study of diamond polishers exposed to cobalt was used to derive an inhalation tolerable concentration of 1 × 10−4 mg/m3 based on lung function decrement. The difference between the tolerable concentration and the cobalt concentrations found in the ambient air near anthropogenic sources is generally about 10-fold.
A 96-h EC50 for cobalt based on growth of the freshwater green alga Chlorella vulgaris was reported as 0.6 mg/l, whereas EC50s for aquatic vascular plants were 0.1 and 0.2 mg/l. The 5-day EC50 for cobalt based on growth of the marine diatom Ditylum brightwellii was 0.3 mg/l. For freshwater invertebrates, acute LC50s (24–96 h) range from 1.1 to 239 mg/l. Several studies on Daphnia magna reproduction were reported, with a 21-day EC50 at 0.01 mg/l and a 28-day NOEC of 0.003 mg/l; however, later studies found 21-day NOECs ranging from 0.03 to 0.05 mg/l for varying levels of calcium carbonate. The lowest reported NOEC for aquatic organisms was for the water flea Ceriodaphnia dubia in a 7-day test, at <0.003 mg/l. The most sensitive marine invertebrates were lobster larvae, with 96-h LC50s ranging from 4.5 to 22.7 mg/l. Ninety-six-hour LC50s for freshwater fish range from 1.4 to 333 mg/l. A 16-day NOEC based on survival was reported at 0.06 mg/l. Test results for marine fish suggest that at least the species tested are relatively insensitive to cobalt, with 96-h LC50s ranging from 52.5 to >1000 mg/l. Ca2+ competition and dissolved organic matter complexation were the most important factors preventing Co2+ from binding at the gills in natural water tests. However, the effect of Ca2+ ions on the uptake and potential toxicity of cobalt occurs at very low Ca2+ concentrations, probably lower than those used in any of the reported toxicity tests.
Moderate-reliability guidance values were determined for the marine environment at 20 µg/l (for the protection of 99% of marine species with 50% confidence) and for the freshwater environment at 8 µg/l (for the protection of 95% of freshwater species with 50% confidence). A comparison of the guidance values with environmental concentrations would suggest that effects are likely only in the vicinity of major anthropogenic releases. There is some evidence that under freshwater conditions of extremely low Ca2+ there is less competition for cobalt at fish gill binding sites and therefore greater uptake of cobalt. Therefore, the greatest risk to aquatic organisms might be in very soft water areas (where the Ca2+ ion concentration is extremely low) close to sources of anthropogenic release.
Data regarding the toxicity of cobalt to soil microorganisms are limited. There is little evidence of cobalt toxicity to plants due to elevated concentrations in soil. Cobalt tolerance, along with tolerance to other metals, has been found in plant populations growing on soils high in particular metals. Exclusion of the metal has been demonstrated in the cobalt tolerance of some species, whereas others growing on cobalt-rich copper clearings are hyperaccumulators of cobalt. Adverse effects on earthworm growth and springtail reproduction have been reported at 300–400 mg/kg dry weight. In the terrestrial environment, adverse effects of cobalt on birds and wild mammals would appear unlikely, with cobalt deficiency in ruminants more likely than cobalt toxicosis.
Cobalt (CAS No. 7440-48-4) is a naturally occurring element (atomic number 27) in the first transition series of Group 9 of the periodic chart of elements. 59Co is the only stable isotope. There are 26 known radioactive isotopes, of which only 57Co and 60Co are commercially important.
Cobalt occurs in the 0, +2, and +3 valence states. Cobalt(II) is more stable than cobalt(III), which is a powerful oxidizing agent that can oxidize water and liberate oxygen. Metallic cobalt(0) occurs in two allotropic forms, hexagonal and cubic, which are stable at room temperature. Cobalt has a relative molecular mass of 58.93 and is a silvery grey solid at room temperature. Its melting point is 1493 °C. At room temperature (20 °C), the density of cobalt is 8.9 g/cm3. Cobalt is soluble in dilute acids, and ultrafine metal cobalt powder is soluble in water at 1.1 mg/l.
Selected chemical and physical properties of cobalt and several inorganic cobalt compounds are presented in Table 1, with further details contained in the International Chemical Safety Cards reproduced at the end of this document.
Table 1: Physical and chemical properties of selected cobalt compounds.
|
Species |
CAS No. |
Relative molecular mass |
Molecular formula |
Melting point |
Solubility |
|
Cobalt |
7440-48-4 |
58.93 |
Co |
1493 °C |
Insoluble in water |
|
Cobalt(II) acetate |
71-48-7 |
177.03 |
Co(C2H4O2)2 |
No data |
Soluble in water, 2.1 g/100 g methanol |
|
Cobalt(II) acetate tetrahydrate |
6147-53-1 |
249.1 |
Co(C2H4O2)2·4H20 |
140 °C |
Very soluble in water |
|
Cobalt(III) acetate |
917-69-1 |
236.07 |
Co(C2H4O2)3 |
Decomposes at 100 °C |
Soluble in water, alcohol, acetic acid |
|
Cobalt(II) carbonate |
513-79-1 |
118.94 |
CoCO3 |
Decomposes |
0.18 g/100 g water |
|
Cobalt carbonyl |
10210-68-1 |
341.9 |
Co2(CO)8 |
51 °C |
Insoluble in water; soluble in ether |
|
Cobalt(II) chloride |
7646-79-9 |
129.84 |
CoCl2 |
724 °C |
450 g/l water, 544 g/l ethanol, 86 g/l acetone |
|
Cobalt(II) hydroxide |
21041-93-0 |
92.95 |
Co(OH)2 |
No data |
0.0032 g/l water |
|
Cobalt(II) mesoporphyrin |
21158-51-0 |
621.2 |
C34H34CoN4O4 |
No data |
No data |
|
Cobalt(II) naphthenate |
61789-51-3 |
407.0 |
Co(C11H10O2)2 |
140 °C |
Insoluble in water |
|
Cobalt(II) nitrate |
10141-05-6 |
182.96 |
Co(NO3)2 |
Decomposes at 100–105 °C |
Soluble in water (133.8 g/l), ethanol, acetone |
|
Cobalt(II) nitrate hexahydrate |
10026-22-9 |
291.03 |
Co(NO3)2·6H2O |
55 °C |
133.8 g/100 ml water at 0 °C |
|
Cobalt(II) oxide |
1307-96-6 |
74.93 |
CoO |
1935 °C |
Insoluble in water |
|
Cobalt(III) oxide |
1308-04-9 |
165.86 |
Co2O3 |
Decomposes at 895 °C |
Insoluble |
|
Cobalt(II,III) oxide |
1308-06-1 |
250.80 |
Co3O4 |
−O2 at 900–950 °C |
Insoluble |
|
Cobalt(II) sulfate |
10124-43-3 |
154.99 |
CoSO4 |
Decomposes at 735 °C |
36.2 g/100 ml water at 20 °C |
|
Cobalt(II) sulfate heptahydrate |
10026-24-1 |
281.1 |
CoSO4·7H2O |
96.8 °C |
60.4 g/100 ml water at 3 °C |
|
Cobalt sulfide |
1317-42-6 |
91.0 |
CoS |
>1116 °C |
Insoluble in water |
Cobalt can be analysed in human biological samples, such as urine, blood, serum, and tissues. Analysis of cobalt in urine usually involves sample chelation and/or acid digestion, followed by GF-AAS (Heinrick & Angerer, 1984; Ichikawa et al., 1985; Bouman et al., 1986; Kimberly et al., 1987; Alexandersson, 1988; Sunderman et al., 1989; Templeton, 1996). Detection limits range from 0.1 to 2.4 μg/l. Analysis in whole blood can be done by GF-AAS, by acid digestion, chelation, preconcentration, and extraction followed by differential pulse cathodic stripping voltammetry, or by a colorimetric method (Heinrick & Angerer, 1984; Afeworki & Chandravanshi, 1987). GF-AAS and differential pulse cathodic stripping voltammetry have detection limits of 2 µg/l and 0.8 μg/l, respectively. The colorimetric method has a detection limit of 150 μg/l. Analysis of cobalt in serum also uses the GF-AAS method, with a detection limit of 0.02 μg/l (Sunderman et al., 1989). NIOSH method 8005 utilizes ICP-AES, with detection limits of 10 µg/l for blood and 0.2 μg/g for tissue (NIOSH, 1994b). ICP-MS is more widely available since the 1990s and is used for multi-elemental analysis of human blood, serum, and urine.
Environmental samples are analysed by atomic absorption spectrometry, instrumental neutron activation analysis, and mass spectrometry (USEPA, 1982, 1986; Haddad & Zikovsky, 1985; Nojiri et al., 1985; Fishman et al., 1986; Hansson et al., 1988; Nakashima et al., 1988; NIOSH, 1994a). Using these methods, the detection limits for cobalt in air range from 0.17 to 0.5 μg/m3. A more recent NIOSH method for the analysis of cobalt in workplace air utilizes sample collection on cellulose or PVC membrane and analysis by ICP-AES; the limit of detection for a 2-m3 sample is 6 ng/m3 (NIOSH, 2003). Detection limits for cobalt in water range from 0.004 μg/l (from lake water using ICP-AES) to 0.05 mg/l (using flame atomic absorption spectrometry).
Cobalt comprises 0.0025% of the weight of the earth’s crust and is the 33rd most abundant element (Smith & Carson, 1981; Merian, 1985; Abbasi et al., 1989). Cobalt does not occur naturally as a base metal, but is a component of more than 70 naturally occurring minerals, including various sulfides, arsenides, sulfoarsenides, hydrates, and oxides. The most common cobalt minerals are the arsenide CoAs2–3 (smeltite), the arsenosulfide CoAsS (cobaltine), and the sulfide Co3S4 (linneite) (IARC, 1991). Identified world cobalt resources are about 14 million tonnes. The vast majority of these resources are in nickel-bearing laterite deposits, with most of the rest occurring in nickel–copper sulfide deposits hosted in mafic and ultramafic rocks in Australia, Canada, and the Russian Federation and in the sedimentary copper deposits in Kinshasha, Democratic Republic of Congo, and Zambia (USGS, 2005). Significant resources of cobalt are also present in the deep-sea nodules and crusts that occur in the mid-Pacific Ocean and are estimated to contain anywhere from 2.5 to 10 million tonnes of cobalt (Cobalt Development Institute, undated a). Encrustation deposits ("cobalt-rich crusts") in shallow waters close to the Hawaiian Islands are believed to contain up to 2.5% cobalt and constitute an important potential source of cobalt (Cobalt Development Institute, 2004).
Sources of environmental cobalt are both natural and anthropogenic (Barceloux, 1999). Natural sources include erosion (wind-blown continental dusts), weathering of rocks and soil, seawater spray, volcanoes, forest fires, extraction by plants, and continental and marine biogenic emissions. The worldwide estimate for atmospheric cobalt emissions is 5350–6170 tonnes per year (Lantzy & Mackenzie, 1979; Nriagu, 1989). Cobalt compounds have been found to occur naturally in seawater, surface water, spring water, and groundwater (Smith & Carson, 1981).
Cobalt is normally associated with copper or nickel; mined ore often contains only 0.1% elemental cobalt. About 44% of world production of cobalt comes from nickel ores. Cobalt is extracted from the metals in the ore by both flotation (sulfide ores) and gravity (arsenide ores); roasting or acid leaching is necessary to concentrate the cobalt (Barceloux, 1999). Cobalt is also extracted from the ore and concentrated by pyrometallurgical, hydrometallurgical, and electrolytic processes alone or in combination (Donaldson et al., 1986). Cobalt is currently mined in 12 countries and refined in 23 countries. The global mine production of cobalt in 2003 totalled 46 900 tonnes, with the principal nine producing countries as follows (production in tonnes): Democratic Republic of Congo, 11 000; Zambia, 9000; Australia, 7000; Canada, 5200; Russian Federation, 4800; Cuba, 3400; New Caledonia, 1500; Brazil, 1300; Morocco, 1300; and other counties, 2400 (USGS, 2005). The approximate refined quantity of cobalt in 2004 was 43 000 tonnes, with the largest amounts (in tonnes) produced in Finland (8000), Zambia (6500), Canada, China, Russian Federation, and Norway (4500 each), Australia (3900), Belgium, Morocco, New Caledonia, and Democratic Republic of Congo (1200 each) (Cobalt Development Institute, 2004). A significant source of cobalt is the recycling of scrap metal. In 1998, an estimated 32% of cobalt supply in the United States was derived from scrap, and the ratio of cobalt derived from new scrap to that derived from old scrap was estimated to be 50:50. Of all the cobalt in old scrap available for recycling, an estimated 68% was either consumed in the United States or exported to be recycled (Shedd, 2004). In 2003, 2200 tonnes of cobalt were recycled in the United States (USGS, 2004).
In 2002, consumption of cobalt metal, organic and inorganic cobalt compounds, and cobalt scrap in the United States was 3870, 1270, and 2800 tonnes, respectively (Shedd, 2002). The use pattern (end use: tonnes) was as follows: superalloys: 3700; steel alloys: 555; other alloys including magnetic alloys: 1050; cemented carbides: 617; chemical and ceramic use: 1950; and miscellaneous: 63 (Shedd, 2002). Cobalt metal is used in alloys with iron, nickel, and other metals to make Alnico, an alloy of unusual magnetic strength; and in Stellite alloys, which contain cobalt, chromium, and tungsten and are used for high-speed, heavy-duty, high-temperature cutting tools (Cobalt Development Institute, 2004). Cobalt metal has three major uses in the petrochemical and plastic industries as both heterogeneous and homogeneous catalysts: (1) hydro-treating and desulfurization catalysts for oil and gas; these catalysts are typically 3–5% cobalt oxide (Co3O4), 14% manganese trioxide (MnO3), and the balance aluminium oxide (Al2O3); (2) mixed cobalt acetate/manganese–sodium bromide homogeneous catalyst for the production of terephthalic acid and dimethyl terephthalate; and (3) cobalt catalyst in the oxo synthesis (hydroformylation) for the production of alcohols and aldehydes for plastic and detergent production, employing freshly reduced cobalt metal, carbonyls, or cobalt salts (transformed in situ to carbonyl) (Cobalt Development Institute, undated b; USGS, 2005).
The major anthropogenic sources of environmental cobalt include mining and processing (smelting) of cobalt-bearing ores, the use of cobalt-containing sludge or phosphate fertilizers on soil, the disposal of cobalt-containing waste, and atmospheric deposition from activities such as the burning of fossil fuels and smelting and refining of metals (Smith & Carson, 1981). Cobalt-containing sewage sludge, phosphate fertilizers, processing of cobalt alloys, and industries that use or process cobalt compounds are estimated to emit an estimated 4000 tonnes per year of atmospheric cobalt (Lantzy & Mackenzie, 1979). More than 2000 tonnes of cobalt are released annually from mining and mineral processing in the United States, including 480 tonnes of cobalt in coal produced; losses generated during cobalt chemical and powder processing were estimated at 50–80 tonnes annually, whereas losses from alloy processing and manufacture of parts and products were estimated to be 360 tonnes and 120 tonnes, respectively (Donaldson, 1986; Donaldson et al., 1986; Shedd, 1993). The total environmental release of cobalt by industrial sources in the United States that was reportable to the Toxics Release Inventory for 2000 was approximately 228 400 kg, which included air release (16 150 kg), water release (1633 kg), and land release (210 600 kg). Additionally, the total off-site waste transfer of cobalt was 2 967 000 kg (USEPA, 2002).
Cobalt and inorganic cobalt compounds are non-volatile. Therefore, they are released into the atmosphere in particulate form. Atmospheric transport depends on particle size and density and meteorological conditions. Coarse particles with diameters >2 μm may deposit within 10 km from the point of emission, while smaller particles may travel longer distances. The mass median diameter of atmospheric cobalt was found to be 2.6 μm in one study (Milford & Davidson, 1985). Data on the transformations of cobalt in the atmosphere are limited. Anthropogenic cobalt from combustion processes is assumed to be primarily oxides (Schroeder et al., 1987). Arsenide and sulfide forms are also released into the atmosphere during ore extraction processes. It is unclear whether these forms of cobalt are transformed in the atmosphere. If oxides are transformed into more soluble species such as sulfates, then these may be washed out of the atmosphere in rain.
Ultimately, the final repository for cobalt is soil and sediment. Released into water, cobalt may sorb to particles and settle into sediment or sorb directly to sediment. Complexation of cobalt to dissolved organic substances can reduce sediment sorption (Albrecht, 2003). Interparticle migration of cobalt can affect the transport of metal ions in sediments (Jackman et al., 2001). In addition, cobalt can be transported in dissolved form or as suspended sediment by rivers and by sea and ocean currents. Concentration profiles of cobalt in deep water suggest that dissolved amounts decrease with increasing depth and that dissolved cobalt is precipitated in the adsorbed state with oxides of iron and manganese and with crystalline sediments such as aluminosilicate and goethite. In the deep sea, formation of manganese nodules removes cobalt by interaction with manganese oxide (MnO) (Barceloux, 1999). Polluted water with higher concentrations of organic pollutants may result in higher concentrations of soluble organic cobalt complexes (Nriagu & Coker, 1980; Glooschenko et al., 1981; Smith & Carson, 1981; Knauer et al., 1982; Brügmann, 1988; Finney & Huh, 1989; Windom et al., 1989; Shine et al., 1995; Szefer et al., 1996; Bargagli, 2000). Humic substances/humic acids are naturally present in aquatic environments and bind strongly to cobalt (Burba et al., 1994). Over time, these complexes may transform into stronger complexes where cobalt is less readily disassociated (Zhang et al., 1990).
The distribution coefficient of cobalt in water varies due to pH, redox conditions, ionic strength, and dissolved organic matter concentrations (Mahara & Kudo, 1981). For example, as pH is increased from 5 to 7.5, the uptake of 60Co from the water to sediment increased rapidly (Benes et al., 1989a, 1989b). Liquid-to-solids ratio and ionic strength did not affect 60Co uptake by sediment. 60Co has also been found to be more mobile in anaerobic aquatic environments than in aerobic freshwater environments (Mahara & Kudo, 1981). For example, in anaerobic seawater–sediment systems, 60Co was 250 times more mobile than in aerobic freshwater–sediment systems. In anaerobic conditions, 30% of 60Co added to a freshwater–sediment system was mobile, whereas in aerobic conditions, 98% was permanently fixed. In anaerobic seawater systems, mobile 60Co consisted of non-ionic forms associated with low molecular weight organic substances that were stable as pH changed. Mobile 60Co was mostly ionic.
Factors that affect the speciation and fate of cobalt in water and sediments include organic ligands such as humic acids and EDTA, anions such as Cl−, OH−, CO32−, HCO3−, and SO42−, pH, and redox potential. The mole percentages of cobalt species in a Welsh lake were 76% free Co2+, 9.8% CoCO3, 9.6% CoHCO3+, 4.0% humate complexes, and 0.5% CoSO4 based on stability constant data used in conjunction with the HALTAFALL program (Mantoura et al., 1978). Similarly, Smith & Carson (1981) reported the rank concentrations of cobalt species in fresh water as free Co2+ > CoCO3 > CoSO4. In the Rhone River in France, where organic wastes are present in high levels, cobalt is almost completely complexed. The distribution of 60Co in the Rhone River at Arles, France, was 45% particulate phase, 30% dissolved phase, and 25% colloidal phase (Eyrolle & Charmasson, 2001). As pH decreases, adsorption of cobalt by particulate matter also decreases, since increasing H+ concentrations compete with metal binding sites. Therefore, levels of dissolved cobalt will be increased at low pH (ATSDR, 2004). In a study of riverine, estuarine, and marine surface water in England, cobalt carbonate complexes (HCO3− and CO32−) constituted 70% of dissolved cobalt, whereas free Co2+ was a major species at 25% (Tipping et al., 1998). As water alkalinity increases, the proportion of cobalt carbonate complexes increases as free Co2+ decreases. In seawater, the proportion of carbonate and free cobalt species is similar. Sulfate complexes are estimated to make up 20% of cobalt in seawater (Tipping et al., 1998). Smith & Carson (1981) estimated the rank concentrations of cobalt species in seawater to be CoCl+ > free Co2+ > CoCO3 > CoSO4, whereas Mantoura et al. (1978) reported the rank concentrations of cobalt species in seawater (35‰) as CoCO3 > free Co2+ > CoSO4 > CoHCO3+ > CoCl+ > CoOH+. Redox potential can also affect speciation of cobalt. For example, the concentration of dissolved cobalt has been found to increase by several orders of magnitude with increasing depth in Baltic waters. This is because of the formation of soluble bisulfide and polysulfide complexes in anoxic zones (ATSDR, 2004).
Soil mobility of cobalt is inversely related to the strength of adsorption by soil constituents. The adsorption of cobalt to soil occurs rapidly, within 1–2 h. Mineral oxides such as iron and manganese oxide, crystalline materials such as aluminosilicate and goethite, and organic substances can retain cobalt. Soil oxides adsorb larger levels of cobalt than do other materials. Clay minerals adsorb relatively smaller amounts of cobalt (McLaren et al., 1986). Desorption of cobalt from soil oxides is low, although humic acids and montmorillonite desorb substantial amounts. Adsorption in clay soils is most likely due to ion exchange at cationic sites of clay with simple ionic cobalt or hydrolysed ionic species such as CoOH+. Adsorption of cobalt with iron or manganese increases with pH (Brooks et al., 1998). As pH increases, insoluble hydroxides and carbonates may form that also reduce cobalt mobility. In contrast, adsorption to mobile colloids would enhance cobalt mobility. Typically, cobalt is more mobile than other metals, such as lead, chromium(II), zinc, and nickel, in soil, but less mobile than cadmium (Mahara & Kudo, 1981; Smith & Carson, 1981; Baes & Sharp, 1983; King, 1988). The partition coefficient, KD, of cobalt ranged from 0.2 to 3800 l/kg in a wide variety of soils. In 36 Japanese agricultural soils, the mean KD was 1840 l/kg (minimum 130 l/kg, maximum 104 000 l/kg, median 1735 l/kg) (Yasuda et al., 1995). Soil properties that exhibited the highest correlation with KD were exchangeable calcium, pH, water content, and cation exchange capacity. The mean Freundlich adsorption constant, KF, and isotherm exponent, n, values in 11 soils in the United States were 37 l/kg and 0.754, respectively (Buchter et al., 1989). The KF values ranged from 2.6 to 363 l/kg and correlated with soil pH and cation exchange capacity. In another study, 13 soils from the south-eastern United States had soil pH values that ranged from 3.9 to 6.5, and cobalt sorption ranged from 15% to 93% (King, 1988). Soil pH accounted for 84–95% of sorption variation.
Decontamination at nuclear facilities involves the use of organic complexing agents such as EDTA, which greatly enhances cobalt mobility in soil (Killey et al., 1984; Toste et al., 1984; McLaren et al., 1986). Cobalt has been found to leach from municipal and low-level radioactive waste sites (Czyscinski et al., 1982; Cyr et al., 1987; Friedman & Kelmers, 1988). In soils from two sites in Nevada, USA, cobalt was sorbed at >90% when the pH was above 7 and the solids concentration was 20 g/l (USDOE, 1996). Only under extreme conditions, such as pH <4 or high ionic strength soil (0.1 mol/l), would cobalt be capable of migrating.
Factors that affect cobalt speciation in soil and sediment include the nature of the soil and sediment, the concentration of chelating and complexing agents, pH, and redox potential. Dissolved cobalt may form complexes with fulvic acid, humic acid, or other organic ligands, or it may be absorbed by ion exchange mechanisms. However, humic and fulvic cobalt complexes are not as stable as those of copper, lead, iron, and nickel. Sediment from nine sites in the Red Sea was assessed for cobalt speciation using a sequential extraction technique: 5.5% exchangeable, 5% carbonate, 24% iron/manganese oxides, 30.4% organic, 13% sulfides, and 22% lithogenous (Hanna, 1992). The Red Sea is unique, since no permanent streams flow into it. Mean cobalt concentrations increased from 3 mg/kg in 1934 to 6 mg/kg in 1984, although the cobalt distribution was not altered. A reduction of soil redox potential may occur when soil is flooded or in deeper oxygen-depleted layers. This may result in the reduction of iron and manganese and the release of adsorbed cobalt from mineral oxides. A decrease in soil pH may also cause a solubilization of precipitated cobalt, desorption of cobalt, and an increase in cobalt mobility (Smith & Carson, 1981).
Although plants may take up cobalt from the soil, the translocation of cobalt from the roots to other parts of the plants is not significant (Smith & Carson, 1981; Mermut et al., 1996). The transfer coefficient, defined as the ratio of the plant concentration to the soil cobalt concentration, for cobalt is 0.01–0.3 (Mascanzoni, 1989). Higher amounts of cobalt translocation are observed in highly acidic soils (pH 3.3) and in some higher plants (Tolle et al., 1983; Kloke et al., 1984; Watabe et al., 1984; Boikat et al., 1985; Francis et al., 1985; Mejstrik & Svacha, 1988; Palko & Yli-Halla, 1988).
60Co is taken up by unicellular algae with reported concentration factors (dry weight) of 40 000 for Scenedesmus obliquus and 18 000 for Selenastrum capricornutum (Nucho et al., 1988; Corisco & Carreiro, 1999). Freshwater molluscs have concentration factors of 100–14 000 (~1–300 in soft tissue). Much of the cobalt taken up by molluscs and crustaceans from water or sediment is adsorbed to the shell or exoskeleton; very little cobalt is generally accumulated in the edible parts (Amiard & Amiard-Triquet, 1979; Smith & Carson, 1981). Similarly, in laboratory studies with Daphnia magna, adsorption to the exoskeleton was the major contamination process (Adam et al., 2001). In studies with starfish (Asterias rubens), accumulation of 57Co was found to be predominately from seawater rather than from food (Warnau et al., 1999). Bioaccumulation factors for marine fish and freshwater fish are 100–4000 and <10–1000, respectively (Smith & Carson, 1981). However, accumulation is mostly in the viscera and skin of the fish, not the edible parts of the fish (Smith & Carson, 1981). In carp (Cyprinus carpio), accumulation from water accounted for 75% of 60Co accumulated from both water and food; accumulation from water and food was additive (Baudin & Fritsch, 1989). Depuration half-lives were 53 and 87 days for fish contaminated from food and water, respectively (Bandin & Fritsch, 1989). Biomagnification of cobalt up the food-chain does not occur (Smith & Carson, 1981).
Atmospheric cobalt is associated with particulate matter principally to the extent to which particles of soil are dispersed by the wind. At unpolluted sites, mean cobalt levels are typically <1–2 ng/m3 (Smith & Carson, 1981; Hamilton, 1994). At the South Pole, the concentration of cobalt was 0.000 49 ± 0.000 15 ng/m3 in 1974–1975 (Maenhaut et al., 1979). In open-ocean environments, mean cobalt concentrations ranged from 0.0004 to 0.08 ng/m3 (Chester et al., 1991). As examples of cobalt concentrations in urban areas, the annual average cobalt concentration at Nahant, Massachusetts (near Boston), USA, in 1992–1993 was 1.7 ng/m3 (Golomb et al., 1997), whereas in Seville, Spain, during 1996 it was 0.5 ng/m3 (Espinosa et al., 2001). In southern Norway, the mean cobalt level was 0.10 ng/m3 in 1985–1986 (Amundsen et al., 1992). In source areas, cobalt concentrations may exceed 10 ng/m3. The highest average atmospheric cobalt concentration was recorded near a nickel refinery in Wales, at 48 ng/m3 (Smith & Carson, 1981).
Surface water and groundwater concentrations of stable cobalt are low: <1 μg/l in pristine areas and 1–10 μg/l in populated areas (Smith & Carson, 1981; Hamilton, 1994). In 1962–1967, cobalt was detected in 2.8% of 1577 raw surface waters in the United States, with a detection limit of 1 μg/l and a maximum level of 48 μg/l (NAS, 1977). United States Geological Survey data for 6805 ambient surface water stations reported mean and median cobalt levels of 2.9 and 2.0 μg/l, respectively (Eckel & Jacob, 1988). Mean dissolved cobalt concentrations ranging from 0.1 to 1.1 µg/l were reported for rivers in the United Kingdom sampled between 1993 and 1998 (Neal et al., 1996, 1998, 2000). Water concentrations can be much higher in mining and agricultural areas. For example, surface water and groundwater samples collected near the Blackbird Mine in Idaho, USA, where lead and silver mining was conducted from the 1880s to 1982, exhibited cobalt concentrations that ranged from <1 to 625 000 μg/l and from not detected to 315 000 μg/l, respectively (ATSDR, 1995). Levels in Mineral Creek, Arizona, USA (which is near a copper mine and smelter), were recorded at 4500 μg/l, and levels in the Little St. Francis River, Missouri, USA (which receives cobalt mining and milling effluent), were 6500 μg/l (Smith & Carson, 1981).
Mean cobalt levels in seawater were reported as 0.078 μg/l in the Caribbean Sea and 0.17–0.39 μg/l in the Indian Ocean (Hamilton, 1994).
Cobalt is rarely detected in drinking-water. The concentration of cobalt in drinking-water is low and ranges from 0.1 to 5 µg/l (Barceloux, 1999). Only 0.5% of 380 finished drinking-waters in the United States were found to contain cobalt at concentrations above 1 μg/l, with a maximum concentration of 29 μg/l (NAS, 1977). In finished drinking-water in Canada, the median and maximum cobalt concentrations were <2.0 μg/l and 6.0 μg/l, respectively (Meranger et al., 1981). Household tap water in the United States from 35 geographical areas had cobalt concentrations ranging from 2.6 to 107 µg/l in 9.8% of 3834 grab samples (Greathouse & Craun, 1978). In the National Community Water Supply Study in the United States, 62% of 2500 samples contained <1 μg/l, whereas the average and maximum cobalt concentrations were 2.2 and 19 μg/l, respectively (Smith & Carson, 1981).
In rainwater, mean cobalt concentrations are 0.3–1.7 μg/l, with ranges from 0.002 μg/l at Enewetak Atoll to 2.9 μg/l at Swansea Valley, Wales (Smith & Carson, 1981; Arimoto et al., 1985; Hansson et al., 1988; Dasch & Wolff, 1989; Heaton et al., 1990; Nimmo & Chester, 1993; Helmers & Schrems, 1995; Nimmo & Fones, 1997). The highest recorded concentration was 68.9 μg/l in the vicinity of a nickel smelter at Monchegorsk in the Russian Arctic (Reimann et al., 1997). Data on rain from the Mediterranean and the United Kingdom demonstrated that 33–44% of the cobalt occurred as stable organic complexes (Nimmo & Chester, 1993; Nimmo & Fones, 1997).
The earth’s crust contains an average cobalt concentration of 20–25 mg/kg (Smith & Carson, 1981; Merian, 1985; Abbasi et al., 1989). The average concentration of cobalt in soil in the United States is 7.2 mg/kg, with a range of 1–40 mg/kg (Smith & Carson, 1981). Soils that contain cobalt at <0.5–3 mg/kg are considered deficient, since vegetation growing on such soils has insufficient cobalt (<0.08–0.1 mg/kg) to meet the dietary requirements of cattle and sheep. Generally, concentrations of up to 800 mg/kg have been reported in soils near ore deposits, phosphate rocks, ore smelting facilities, and soils contaminated by airport traffic, highway traffic, or other industrial pollution (Smith & Carson, 1981; Kloke et al., 1984). However, soils near the aforementioned Blackbird Mine in Idaho, USA, had cobalt concentrations ranging from 26.5 to 7410 mg/kg (ATSDR, 1995). Cobalt levels in surface soils from two active volcano islands of Sicily ranged from 5.1 to 59.0 mg/kg (Bargagli et al., 1991). Soils surrounding large copper–nickel smelters in Sudbury, Ontario, Canada, illustrate the increasing concentrations of cobalt with closer proximity: 42–154 mg/kg between 0.8 and 1.3 km from the smelter, 33 mg/kg at 10 km, 48 mg/kg at 19 km, and 19 mg/kg at 50 km (Smith & Carson, 1981). Soils surrounding a tungsten carbide tool grinding factory had cobalt levels as high as 12 700 mg/kg; however, neighbourhood soils located 30 and 160 m from the factory had 12–18 mg/kg (Abraham & Hunt, 1995).
Unpolluted freshwater sediment contains about the same levels of cobalt as does cobalt-sufficient soil, generally <20 mg/kg. Cobalt concentrations in polluted lake and river sediment ranged from 0.16 to 133 mg/kg (Smith & Carson, 1981). Knutson et al. (1987) reported cobalt concentrations of up to 700 mg/kg in surficial sediment (Hudson River, New York, USA) near a disused nickel–cadmium battery plant (4 years after closure). In the Hudson River estuary, cobalt levels were an order of magnitude higher in suspended sediment than in bottom sediment (Gibbs, 1994). This can be attributed to the finer grain size of suspended sediment or local sources. Cobalt levels in core samples (surface to 42 cm deep) from the Upper St. Lawrence River estuary in Canada were independent of depth, indicating the lack of any recent significant anthropogenic releases (Coakley et al., 1993).
The cobalt content of living plants depends on the species, the cobalt content of the soil, and numerous environmental factors. The mean cobalt concentration reported for terrestrial plants was 0.48 µg/g (Bowen, 1966). Median cobalt concentrations in freshwater vascular plants of 0.32 and 0.37 µg/g dry weight were reported for unpolluted and polluted environments, respectively (Outridge & Noller, 1991). Grasses normally contain cobalt concentrations of 0.2–0.35 µg/g, but grasses from cobalt-deficient regions contain only 0.02–0.06 µg/g (Hamilton, 1994). Cobalt tolerance, along with tolerance to other metals, has been found in plant populations growing on soils high in particular metals. For example, some plants growing on cobalt-rich soils in Zaire are hyperaccumulators of cobalt, with the plant Haumaniastrum robertii containing a mean concentration of 4304 mg/kg dry weight (1368–10 222 mg/kg) (Brooks, 1977).
Cobalt concentrations have been reported in various aquatic animals. Fish from three Dutch polder lakes contained cobalt at 2.5–25 mg/kg wet weight (Badsha & Goldspink, 1988). Muscle tissue of ocean fish and rock crabs caught near dump sites off New York City, New Haven, Connecticut, and Delaware Bay, USA, contained 10–40 µg/kg and 16.0 µg/kg, respectively (Greig & Jones, 1976). Cobalt has also been detected at remote sites; mean cobalt levels in fish and amphipods in Antarctica were 0.11–0.14 µg/g dry weight and 1.01 µg/g dry weight, respectively (Szefer et al., 1993). The concentration of cobalt in the tissue of 14 bluefin tuna (Thunnus thynnus) caught by various commercial fishing vessels off Newfoundland, Canada, was essentially the same, 0.01 ± 0.004 µg/g (Hellou et al., 1992). In a broad survey of contaminant levels in nine species of fish and fiddler crabs from 11 sites in the lower Savannah River, Georgia, and the Savannah National Wildlife Refuge, USA, mean cobalt levels (0.1–2.5 mg/kg wet weight) among different species and sites were statistically indistinguishable (Winger et al., 1990). These studies suggest that cobalt does not biomagnify up the food-chain (Smith & Carson, 1981).
In a study of the levels and distribution of 14 elements in oceanic seabirds, the concentration of cobalt, an essential element, appeared to be highly regulated, with over 80% of the body burden residing in the skeleton. The mean cobalt concentration in the livers of 11 seabird species ranged from 0.048 to 0.078 µg/g dry weight; of the elements studied, cobalt had the lowest coefficient of variation in the different species (Kim et al., 1998). Mean cobalt levels in the tissues of penguin and other Antarctic seabirds ranged from 0.09 to 0.11 µg/g (Szefer et al., 1993). The geometric mean concentrations of cobalt in tern eggs collected from coastal New Jersey, USA, in 1971 and 1982 were 0.48 mg/kg and 0.50 mg/kg, respectively. Unlike the levels of many other metals, the level of cobalt showed no decline over the 11-year period (Burger & Gochfeld, 1988).
Cobalt concentrations in coal, crude oil, fuel oil, and gasoline in the United States are 5 mg/kg, 0.001–10 mg/kg, 0.03–0.3 mg/kg, and <0.1 mg/kg, respectively (Smith & Carson, 1981).
The largest potential source of cobalt exposure for the general population is food. Most of the cobalt that is ingested is inorganic. Vitamin B12 contains cobalt but occurs in foods of animal origin and represents only a small fraction of cobalt intake. Green vegetables and fresh cereals are the richest sources of cobalt (0.2–0.6 µg/g dry mass), whereas dairy products, refined cereals, and sugar contain the least cobalt (0.01–0.03 µg/g dry mass) (IARC, 1991; Cobalt Development Institute, 2003). Plant products have been estimated to contribute up to 88% of the total cobalt in the Japanese diet (Yamagata et al., 1963; IARC, 1991). The Total Diet Study in the United Kingdom in 1994 estimated the population average intake of cobalt to be 0.12 mg/day (MAFF, 1997; EVM, 2002). Cobalt intake in the United States has been estimated to be 5–40 μg/day (Jenkins, 1980), with relatively high concentrations of cobalt occurring in fish and vegetables (Barceloux, 1999). In Canada, the estimated average daily intake is 11 μg/day (Dabeka & McKenzie, 1995). Bakery goods/cereals and vegetables contributed most to this daily intake, at 29.8% and 21.9%, respectively. The cobalt intake of Canadian children (age 1–19 years) has been estimated to range from 7 to 14 μg/day (Dabeka & McKenzie, 1995). In France, the estimated average daily intake is 29 μg/day (Biego et al., 1998). Foodstuffs that contributed most to this intake were milk and dairy products (32%), fish/ crustaceans (20%), and condiments/sugar/oil (16%). A study in Sweden from 1983 to 1990 evaluated cobalt levels in various foodstuffs (Jorhem & Sundström, 1993). Cobalt levels were highest in seeds (alfalfa seeds, 0.86 μg/g fresh weight; linseed, 0.56 μg/g), beef liver (0.043 μg/g), and milk chocolate (0.34 μg/g), whereas fish, fruit, and leafy vegetables contained <0.01 μg/g fresh weight. In Spain, cobalt concentrations in 20 brands of beer ranged from 0.16 to 0.56 μg/l, with a median concentration of 0.39 μg/l (Cameαn et al., 1998). The cobalt content of five brewed teas averaged 0.2 µg/g (range 0.16–0.34 µg/g), and that of seven brewed coffees, 0.75 µg/g (range 0.42–2.0 µg/g) (Horwitz & van der Linden, 1974).
Tobacco contains cobalt at <0.3–2.3 μg/g dry weight, and 0.5% of the cobalt is present in mainstream smoke (Munita & Mazzilli, 1986; Ostapczuk et al., 1987; Stebbins et al., 1992; Barceloux, 1999).
Occupational exposure to cobalt occurs in several industries, including hard metal manufacturing, welding, and grinding. Air concentrations of cobalt in occupational settings generally range from 1.0 × 104 to 1.7 × 106 ng/m3 (IARC, 1991; Barceloux, 1999).
Inhalation of cobalt particles results in deposition in the upper and lower respiratory tract (Casarett & Doull, 1986). Particle size is the primary factor determining deposition patterns. Large particles (diameter >2 μm) deposit in the upper respiratory tract. High airstream velocities promote the inertial impaction of these large particles. Smaller particles tend to escape this inertial impaction and deposit in the lower respiratory tract, where sedimentation and diffusion can occur. Fractional deposition varies due to particle size and the age and breathing patterns of the exposed individual. Fractional deposition of cobalt oxide in humans varied from approximately 50% of the inhaled dose for particles with a geometric mean diameter of 0.8 μm to approximately 75% for particles with a geometric mean diameter of 1.7 μm (Foster et al., 1989). Studies in hamsters suggest that the lungs absorb approximately 30% of an inhaled dose of cobalt oxide (Wehner et al., 1977).
Transfer pathways in humans and laboratory animals have been studied using 57Co in the form of cobalt oxide (Bailey et al., 1989). Cobalt particles deposited in the respiratory tract can be absorbed into the blood after dissolution or mechanically transferred to the gastrointestinal tract by mucociliary action and swallowing. Approximately 50% of the cobalt that enters the gastrointestinal tract will be absorbed. Large particles (>2 μm) tend to deposit in the upper respiratory tract, where mechanical clearance processes occur more readily than translocation. Smaller particles that deposit in the lower respiratory tract will usually remain dissolved or be phagocytosed by macrophages and then translocated. The ratio of translocation to mechanical clearance in humans is 5:1 for particle sizes ranging from 0.8 to 1.7 μm (Foster et al., 1989).
Cobalt oxide (using 57Co tracer) was found to persist in the respiratory tracts of humans at half the original lung burden after 6 months of exposure (Bailey et al., 1989). In contrast, rats exhibited nearly complete clearance after 6 months. Since cobalt may bind to cellular components in human lung, the elimination half-time in human lung increases with increasing times after exposure (Sedlet et al., 1958; Foster et al., 1989).
Gastrointestinal absorption of cobalt in humans has been found to vary from 18% to 97% of the administered dose, depending on the type and dose of the cobalt compound and the nutritional status of the individual (Harp & Scoular, 1952; Valberg et al., 1969; Sorbie et al., 1971; Smith et al., 1972). Studies of the absorption of cobalt chloride in volunteers indicate that the absorption rate from the gastrointestinal tract ranges from 5% to >20% between doses of <1 μg and 1.2 mg cobalt (Smith et al., 1972). Cobalt absorption was increased among individuals who were iron deficient (31–71% absorption in iron-deficient subjects, 18–44% in controls) (Valberg et al., 1969; Sorbie et al., 1971). The absorption of vitamin B12 occurs by a complex, yet specific pathway that involves the interaction of the molecule with factors in the stomach and intestine that facilitate absorption (Russell-Jones & Alpers, 1999).
Data on absorption via the gastrointestinal tract are available from animal experiments. Several rat studies have found that soluble cobalt chloride was 13–34% absorbed, whereas insoluble cobalt oxides were only 1–3% absorbed (Taylor, 1962; Barnaby et al., 1968; Schade et al., 1970; Hollins & McCullough, 1971; Bailey et al., 1989; Collier et al., 1989; Patrick et al., 1989; Kirchgessner et al., 1994; Ayala-Fierro et al., 1999). Particle size did not affect gastrointestinal absorption in baboons, guinea-pigs, HMT rats, F-344 rats, hamsters, or CBA/H mice (Bailey et al., 1989). In rats, cobalt chloride (with 58Co tracer) that was complexed with histidine, lysine, glycylglycine, EDTA, casein, or glycine was absorbed less than free cobalt chloride (Taylor, 1962). Cobalt chloride administered in conjunction with cow’s milk resulted in significantly greater gastrointestinal absorption (~40%) (Taylor, 1962). Water-soluble cobalt compounds have been found to exhibit greater absorption than non-water-soluble forms (Kinoshita & Fujita, 1972; Inaba et al., 1980; Deka et al., 1981; Firriolo et al., 1999). As in humans, iron deficiency in animals increased cobalt absorption, while simultaneous administration of cobalt and iron resulted in less cobalt absorption (Schade et al., 1970; Reuber et al., 1994). As oral cobalt doses increase, fractional absorption decreases (Houk et al., 1946; Taylor, 1962; Kirchgessner et al., 1994). Rats and guinea-pigs aged 1–60 days have 3- to 15-fold greater absorption than adult animals aged 200 days or more (Naylor & Harrison, 1995). Species differences in absorption rates have not been observed; however, absorption of soluble cobalt compounds is greater in rats (13–34%) than in cows (1–2%) or guinea-pigs (4–5%) (Taylor, 1962; Barnaby et al., 1968; Schade et al., 1970; Hollins & McCullough, 1971; van Bruwaene et al., 1984; Bailey et al., 1989; Kirchgessner et al., 1994; Naylor & Harrison, 1995; Ayala-Fierro et al., 1999).
Since cobalt is an essential metal and a component of vitamin B12, it has been found in most tissues, such as muscle, lung, lymph nodes, heart, skin, bone, hair, stomach, brain, pancreatic juice, kidneys, plasma, urinary bladder, and liver (highest levels), of non-occupationally exposed subjects (Forbes et al., 1954; Yamagata et al., 1962; Yukawa et al., 1980; Teraoka, 1981; Collecchi et al., 1986; Ishihara et al., 1987; Hewitt, 1988; Muramatsu & Parr, 1988). These tissue levels reflect exposure from all routes and all sources. Total body burden in humans has been estimated as 1.1–1.5 mg, with 0.11 mg in the liver (Yamagata et al., 1962; ICRP, 1979).
Workers exposed occupationally to airborne cobalt had higher tissue levels of cobalt when examined at death. Lung concentrations of cobalt are significantly higher in copper smelter workers, metal workers, and coal miners who were occupationally exposed compared with non-occupationally exposed workers (Teraoka, 1981; Hillerdal & Hartung, 1983; Gerhardsson et al., 1984; Hewitt, 1988). In copper smelter workers, no increases in liver or kidney cobalt levels were observed compared with controls (Gerhardsson et al., 1984). However, metal workers had increased cobalt levels in lymph nodes, liver, spleen, and kidneys (Teraoka, 1981; Hillerdal & Hartung, 1983).
Tissue distribution of cobalt in laboratory animals is similar to that in humans. After inhalation exposure, marked increases of cobalt have been found in the lung (Barnes et al., 1976; Brune et al., 1980; Kreyling et al., 1986; Patrick et al., 1989; Talbot & Morgan, 1989; Collier et al., 1991; Kyono et al., 1992). Histological analysis revealed that cobalt particles were localized to macrophages within the bronchial wall or in the interstitium close to the terminal bronchioli (Brune et al., 1980). Cobalt has been found in significant amounts in the liver, kidney, trachea, spleen, bones, and heart, with the highest levels in the liver and kidney (Wehner & Craig, 1972; Kerfoot, 1975; Barnes et al., 1976; Brune et al., 1980; Kreyling et al., 1986).
Although no studies describe the distribution of cobalt after oral exposure in humans, laboratory animal studies indicate that cobalt absorbed in the gastrointestinal tract is primarily retained in the liver (Simesen, 1939; Greenberg et al., 1943; Ayala-Fierro et al., 1999). Cobalt was also found in the kidneys, heart, stomach, and intestines (Simesen, 1939; Persson et al., 1992; Ayala-Fierro et al., 1999). In pregnant rats, oral exposure to cobalt caused a dose-dependent increase in fetal blood and amniotic fluid (Szakmary et al., 2001). Long-term oral exposure of rats caused significantly increased cobalt levels in the liver, kidney, muscle, brain, and testes (Barnaby et al., 1968; Thomas et al., 1976; Bourg et al., 1985).
Cobalt (as 55CoCl2 and 56CoCl2) administered to two human volunteers by intravenous injection was found to be distributed primarily to the liver and kidneys (Jansen et al., 1996). In rats, intravenous injection of 57CoCl2 resulted in cobalt accumulation in the liver (22.8%), kidneys (10.2%), and intestines (3.16%) 2 h after exposure (Gregus & Klaassen, 1986). When rats were intracardially injected with cobalt nitrate, similar results were observed: 29% accumulation in the liver, 10% in the kidneys, and 4.6% in the intestines (Patrick et al., 1989). In a rat study in which tissue cobalt levels were determined 100 days after intravenous injection of 60CoCl2, the highest levels were found in spleen, followed by heart and then bone (Thomas et al., 1976). Liver and kidney had the highest initial cobalt concentrations, but concentrations were comparatively low at 100 days. Intramuscular injection of cobalt mesoporphyrin in rats resulted in the highest levels in liver and blood, followed by kidneys, lung, spleen, adrenal glands, and heart, at 7 days after exposure (Feng et al., 1998). Subcutaneous injection of cobalt protoporphyrin resulted in the highest levels in kidney, followed by spleen, liver, lung, thymus, and gonads, at 4 weeks after exposure (Rosenberg, 1993).
In humans, there are no available data on the elimination of soluble cobalt particles after inhalation exposure. Elimination of insoluble cobalt particles after inhalation exposure appears to follow three-phase kinetics. The first phase is the mucociliary clearance of particles deposited in the tracheobronchial region and has a half-time of 2–44 h (Apostoli et al., 1994; Mosconi et al., 1994). The second phase is the macrophage-mediated clearance of lung cobalt particles and has a half-time of 10–78 days (Beleznay & Osvay, 1994; Mosconi et al., 1994). The third phase represents long-term lung clearance and has a half-time on the order of years (Newton & Rundo, 1971; Bailey et al., 1989; Beleznay & Osvay, 1994; Mosconi et al., 1994). In a controlled aerosol exposure study in humans, 40% of the initial lung burden of cobalt oxide (using 57Co as a tracer) was retained at 6 months after exposure (Foster et al., 1989). In the first week after exposure, 17% of the initial lung burden was eliminated, of which 90% was mechanically cleared to the gastrointestinal tract and excreted in faeces. By 6 months after exposure, 33% of the initial lung burden was cumulatively eliminated in urine and 28% was cumulatively eliminated in faeces. The ratio of peak absorption rate and mechanical clearance rate is 5:1. Urinary excretion increases with time following exposure. Particle size affects elimination, since more cobalt is mechanically cleared to the gastrointestinal tract when particles are larger (Bailey et al., 1989; Foster et al., 1989). Smokers with no occupational exposure to cobalt had a significantly higher mean concentration in urine (0.6 μg/l; SD 0.6) than non-smokers (0.3 μg/l; SD 0.1). There was no difference between smokers and non-smokers in the cobalt levels in blood (Alexandersson, 1988).
Animal data on cobalt elimination indicate that the solubility of the cobalt compound greatly affects its long-term clearance. For example, the more soluble cobalt(II) oxide is cleared from the lungs at a faster rate than the less soluble cobalt(II,III) oxide (Barnes et al., 1976; Kreyling, 1984). Soluble cobalt compounds are absorbed into the blood at a faster rate than less soluble compounds and excreted in the urine and faeces (Barnes et al., 1976). Urinary excretion rates seem to correlate with the translocation rate of cobalt from the lungs to blood, whereas faecal excretion rates seem to correlate with mechanical clearance rates of cobalt from the lungs to the gastrointestinal tract (Kreyling et al., 1986, 1989; Andre et al., 1989; Bailey et al., 1989; Collier et al., 1989; Patrick et al., 1989; Talbot & Morgan, 1989). Following an initial high rate of faecal clearance, urinary excretion is the primary route of cobalt elimination after a single inhalation exposure or 3 months of exposure (Palmes et al., 1959; Kerfoot, 1975).
Faecal elimination is the primary route of excretion following oral exposure in humans. Faecal elimination has been found to vary (3–99% of the dose) depending on the amount and type of cobalt administered and the nutritional status of the subject (Harp & Scoular, 1952; Paley et al., 1958; Valberg et al., 1969; Sorbie et al., 1971; Smith et al., 1972). Several days after an oral exposure, 10 times more cobalt was excreted in faeces than in urine (Paley et al., 1958). In subjects with an iron deficiency, less cobalt was eliminated in the faeces, and more was absorbed (Valberg et al., 1969; Sorbie et al., 1971).
Faecal elimination is also the primary route of excretion in animals following oral exposure. Faecal clearance has been noted to decrease as cobalt particle solubility increases. In several species, oral exposure to cobalt(II,III) oxide (with 57Co tracer) resulted in little gastrointestinal absorption and a rapid elimination in faeces (>96%) (Bailey et al., 1989). No significant differences in cobalt(II,III) oxide elimination were observed among species (Andre et al., 1989; Bailey et al., 1989; Collier et al., 1989; Patrick et al., 1989; Talbot & Morgan, 1989). Cobalt(II) chloride, which is more soluble, was excreted primarily via faeces (70–83% of the administered dose) in rats, with urinary excretion accounting for the remainder of the dose (Barnaby et al., 1968; Hollins & McCullough, 1971; Ayala-Fierro et al., 1999). In lactating dairy cows, 97% of an oral dose of cobalt chloride was recovered in the faeces by 70 days after exposure, whereas urine and milk contained 0.26% and 0.012% of the dose, respectively (van Bruwaene et al., 1984). Single exposures in beagle dogs demonstrated that insoluble cobalt(II,III) oxide was eliminated in the faeces and urine at 90% and 5%, respectively, while the more soluble cobalt nitrate was eliminated at 70% in the faeces and 25% in the urine (Kreyling et al., 1986). Similar to humans, iron deficiency in rats also caused less elimination in the faeces, whereas co-administration of iron caused an increase in faecal elimination (Schade et al., 1970; Reuber et al., 1994).
After an intravenous injection of cobalt chloride in humans, 30% of the dose was excreted in urine within 24 h, 56–73% within 48 h, and 57% within 2 weeks (Kent & McCance, 1941; Paley et al., 1958; Smith et al., 1972). In various animal species, urinary excretion has also been shown to be the primary elimination route following intravenous injection of cobalt nitrate (Andre et al., 1989; Bailey et al., 1989; Collier et al., 1989; Patrick et al., 1989; Talbot & Morgan, 1989). In animals, 80% of the dose was excreted via urine within 21 days. Most of the remaining dose (5–30% of the total dose) was excreted in the faeces, with little long-term retention. Biliary excretion has also been reported in animals, at 2–7% of the injected dose (Sheline et al., 1945; Cikrt & Tich, 1981; Gregus & Klaassen, 1986).
The ICRP has developed two physiologically based pharmacokinetic/pharmacodynamic models that are applicable to cobalt: a human respiratory tract model for radiological protection (ICRP, 1994) and a biokinetic model of ingested cobalt in humans (ICRP, 1979, 1994). The PBPK model for the human respiratory tract was developed for a wide variety of radionuclides and their chemical forms. It models the behaviour of aerosols and vapours in the respiratory tract. It provides inhalation dose coefficients for estimating the committed dose equivalents and the effective doses to organs and tissues based on a unit intake of radioactive material, the distribution and retention of the material, the radioactive decay, and the energy of radiation emitted from the material and absorbed by tissues. This model applies to various particle sizes (0.0005–100 μm in diameter) and can be adjusted for population characteristics such as sex, age, and level of physical activity. Inhaled particles may be redistributed either upwards into the respiratory tract or to the lymph and blood by particle removal mechanisms. Deposition of vapours and gases is modelled as a partitioning process and is based on physiological parameters and the solubility and reactivity of the compound of interest. The solubility and reactivity of compounds are classified into three categories: SR-0, insoluble and non-reactive gases; SR-1, soluble and reactive gases and vapours that are expected to be taken up and deposited on respiratory tract tissues; and SR-2, soluble and reactive gases and vapours that are completely retained in the extrathoracic regions of the respiratory tract. This model also accounts for mechanical particle clearance and is based primarily on human data, although particle retention in airway walls is based on data from experimental animals. This model assumes that blood absorption occurs at equivalent rates in all areas of the respiratory tract, except for the anterior nasal passages, where absorption does not occur. Particles undergo dissociation, after which the dissolved molecules diffuse across capillary walls into the blood. Absorption is classified into four types: Type V (complete and instantaneous absorption), Type F (fast, 100% absorption within 10 min), Type M (medium, 70% absorption within 10 min), and Type S (slow, 0.1% absorption within 10 min). Cobalt compounds have been classified as follows: Type F, cobalt chloride and nitrate; Type M or S, cobalt oxides, cobalt metal, and metal alloys; Type M, cobalt in mineral dusts, such as fly ash and volcano ash, and all cobalt aerosols in the absence of specific information; and Type S, cobalt-infused aluminosilicate or polystyrene.
The ICRP’s cobalt biokinetics model (ICRP, 1994) is a three-compartment model for ingested cobalt that is applicable to infants, children, adolescents, and adults. Absorption of ingested cobalt is assumed to be 60% in infants up to 3 months of age, 30% from 3 months to 15 years of age, and 10% after 15 years of age. The distribution of cobalt is assumed to be 50% excreted in the urine and faeces at a 6:1 ratio, 5% in the liver, and 45% in other tissues. Elimination from tissues is assumed to follow three first-order rate constants that represent slow, medium, and fast, with half-times of 6, 80, and 600 days, respectively. These half-times are assumed to be independent of age. The validation of this model is not described by the ICRP, but the model has been used to establish radiation dose equivalents (Sv/Bq) of ingested 57Co, 58Co, and 60Co for ages 3 months to 70 years (ICRP, 1994). This model is designed for human dosimetry and would need modification for other species. Radiation doses from cobalt radionuclides to all major organs can be estimated using this model and can be used to assess environmental and occupational exposures to radioactive cobalt.
Analysis of urinary cobalt has been recommended for biological monitoring of exposure to cobalt at work (Templeton, 1996), and different institutions have proposed biomonitoring action limits for acceptable exposure (ACGIH, 1999; FIOH, 1999). The concentration of cobalt in a urine specimen collected at the end of the last work shift of the week reflects exposure over the preceding work week, and that collected on Monday morning reflects chronic occupational exposure in comparison with the reference population (Templeton, 1996).
Urine and blood cobalt levels correlate positively with occupational exposure to cobalt. Unexposed humans have blood cobalt levels ranging from 0.05 to 0.19 μg/dl and urine cobalt levels ranging from 0.04 to 2 μg/l (Ichikawa et al., 1985; Alexandersson, 1988). Workers exposed to 0.1 mg/m3 reported blood levels ranging from 0.57 to 0.79 μg/dl (95% CI), compared with 0.19 μg/dl in unexposed subjects (Ichikawa et al., 1985). This study also reported urine cobalt levels of 59–78 μg/l in the workers compared with 2 μg/l in unexposed subjects (Ichikawa et al., 1985).
The LC50 for a 30-min inhalation exposure of rats was 165 mg/m3 as cobalt hydrocarbonyl (Palmes et al., 1959). One of 14 Syrian golden hamsters exposed by inhalation to cobalt oxide at 106 mg/m3 for 3 h died within 24 h; after a 6-h exposure, the 24-h mortality rate was 2/44 (Wehner & Craig, 1972). Oral LD50 values are dependent on the type of cobalt compound tested and the test species. Wistar rats and Sprague-Dawley rats had LD50 values ranging from 42.4 mg of cobalt (as cobalt chloride) per kilogram body weight to 317 mg of cobalt (as cobalt carbonate) per kilogram body weight (FDRL, 1984a, 1984b, 1984c; Singh & Junnarkar, 1991). Tricobalt tetraoxide, an insoluble compound, had an LD50 in Sprague-Dawley rats of 3672 mg of cobalt per kilogram body weight (FDRL, 1984c). Speijers et al. (1982) reported an LD50 of 418 mg/kg body weight for cobalt chloride in Wistar rats. In male Swiss mice, LD50 values ranged from 89.3 mg of cobalt (as cobalt chloride) per kilogram body weight to 123 mg of cobalt (as cobalt sulfate) per kilogram body weight (Singh & Junnarkar, 1991).
Rats and mice exposed by inhalation to cobalt sulfate heptahydrate at cobalt concentrations of 19 and 1.9 mg/m3, respectively, for 16 days exhibited necrosis and inflammation of the respiratory tract epithelium. Rats also developed thymus necrosis and testicular atrophy (Bucher et al., 1990; NTP, 1991). Male CFY rats exposed orally to cobalt chloride at 50 mg/kg body weight per day (equivalent to 12.4 mg of cobalt per kilogram body weight per day) for 3 weeks and co-exposed to drinking-water that contained 10% ethanol and 5% sugar exhibited cardiac damage that presented as incipient, multifocal myocytolysis with degeneration of myofibrils (Morvai et al., 1993). Rats exposed to ultrafine cobalt particles (diameter 20 nm) at concentrations of 2.72 mg/m3 for 5 h or 2.12 mg/m3 for 5 h/day for 4 days displayed focal hypertrophy or proliferation of lower airway epithelium, macrophage damage, intracellular oedema of type I alveolar epithelium, interstitial oedema, and proliferation of type II alveolar epithelium (Kyono et al., 1992).
Rats (strain not specified), guinea-pigs (strain not specified), and beagle dogs exposed to cobalt (as cobalt hydrocarbonyl) at 9 mg/m3 for 6 h/day, 5 days/week, for 3 months displayed foam cell aggregates (Palmes et al., 1959). These foam cell aggregates were composed of nodules of large macrophages with foamy cytoplasm, accompanied by moderate interstitial and peribronchial fibrosis, mild emphysema, and moderate peribronchial lymphoid hyperplasia. These aggregates were not present when animals were sacrificed and evaluated at 3 months or 6 months post-exposure. Rabbits exposed by inhalation for 1–4 months to cobalt chloride (0.4–2 mg/m3) exhibited lesions of the alveolar region of the respiratory tract that were characterized by nodular accumulation of Type II epithelial cells and interstitial inflammation (Johansson et al., 1984, 1987, 1991, 1992).
F344/N rats and B6C3F1 mice exposed by inhalation to cobalt sulfate heptahydrate (0, 0.3, 1, 3, 10, and 30 mg/m3; equivalent to cobalt concentrations of 0, 0.11, 0.38, 1.14, 3.80, and 11.38 mg/m3) for 6 h/day, 5 days/ week, for 13 weeks developed adverse effects throughout the respiratory tract (Bucher et al., 1990; NTP, 1991). At concentrations >0.3 mg/m3 (cobalt concentrations >0.11 mg/m3), both rats and mice developed squamous metaplasia of the larynx (the most sensitive tissue), such that a NOAEC could not be determined. Rats developed chronic inflammation of the larynx at >1 mg/m3 and more severe effects in the nose, larynx, and lung at higher exposures. Mice exhibited acute inflammation of the nose at >3 mg/m3 and more severe effects in the nose, larynx, and lung at higher exposures. At 30 mg/m3, mice exhibited hyperplasia of the mediastinal lymph nodes and testicular atrophy and increased estrous cycle length in females. Both rats and mice exhibited histiocytic infiltrates of the lung at similar exposure levels. Sperm motility was decreased in mice exposed to 3 mg/m3 or higher (lower exposures not assessed), and increased abnormal sperm and decreased testis and epididymal weights were observed in mice exposed to 30 mg/m3.
Rats exposed for 2–3 months to cobalt at 26–30.2 mg/kg body weight per day in the diet (as cobalt sulfate) or in drinking-water (as cobalt chloride) exhibited increased heart weight and degenerative heart lesions (Grice et al., 1969; Domingo et al., 1984). Rats exposed to cobalt (as cobalt sulfate) in the diet at 8.4 mg/kg body weight per day for 24 weeks experienced significant reductions in cardiac enzyme activity levels, such as manganese superoxide dismutase, succinate cytochrome c oxidase, NADH cytochrome c reductase, and cytochrome c oxidase, and a reduction in mitochondrial ATP production (Clyne et al., 2001). Rats exposed to cobalt (as cobalt chloride) at 10–18 mg/kg body weight per day for 4–5 months exhibited renal injury, such as histological alteration of proximal tubules (Holly, 1955; Murdock, 1959).
A study by the NTP examined the carcinogenicity of cobalt by inhalation in mice (NTP, 1998; Bucher et al., 1999). Groups of 50 male and 50 female B6C3F1 mice were exposed to cobalt sulfate heptahydrate at 0, 0.3, 1, or 3 mg/m3 for 6 h/day, 5 days/week, for 105 weeks. Cobalt concentrations in this study were 0, 0.11, 0.38, 1.14, and 3.80 mg/m3. Mean body weights were increased in all treated females and decreased only in the high-dose males. Survival was not adversely affected by treatment. The incidences of benign and malignant alveolar/bronchiolar neoplasms were increased in a concentration-dependent manner: males, 11/50, 14/50, 19/50, and 28/50 for 0, 0.3, 1, and 3 mg/m3, respectively; females, 4/50, 7/50, 13/50, and 18/50 for 0, 0.3, 1, and 3 mg/m3, respectively. There were no increased incidences of neoplasms in other tissues. The NTP concluded that there was clear evidence of carcinogenic activity.
Another study by the NTP examined the carcinogenicity of cobalt by inhalation in rats (NTP, 1998; Bucher et al., 1999). Groups of 50 male and 50 female Fischer 344/N rats were exposed to cobalt sulfate heptahydrate at 0, 0.3, 1, and 3 mg/m3 (equivalent to cobalt concentrations of 0, 0.11, 0.38, 1.14, and 3.80 mg/m3) for 6 h/day, 5 days/week, for 105 weeks. Mean body weights and survival were unaffected by treatment. Rats exhibited a concentration-related increase in the incidence of benign and malignant alveolar/bronchiolar neoplasms in male and female rats and benign and malignant pheochromocytomas in female rats. The incidences of benign and malignant alveolar/bronchiolar neoplasms were 1/50, 4/50, 4/48, and 7/50 for 0, 0.3, 1, and 3 mg/m3, respectively, in males and 0/50, 3/49, 16/50, and 16/50 for 0, 0.3, 1, and 3 mg/m3, respectively, in females. Although many of the alveolar/bronchiolar lesions were morphologically similar to those that arise spontaneously, the lesions in rats, unlike those in mice, were predominantly fibrotic, squamous, or mixtures of alveolar/bronchiolar epithelium and squamous or fibrous components. Squamous metaplasia of alveolar/bronchiolar epithelium, which is a common response to pulmonary injury, was observed in a number of rats. In females, incidences of benign and malignant pheochromocytomas of the adrenal medulla were 2/48, 1/49, 4/50, and 10/48 for 0, 0.3, 1, and 3 mg/m3, respectively. In males, the incidences of benign and malignant pheochromocytomas of the adrenal medulla were 15/50, 19/50, 25/50, and 20/50 for 0, 0.3, 1, and 3 mg/m3, respectively. Pheochromocytomas are common spontaneous neoplasms in male Fischer F344/N rats, but have a lower spontaneous occurrence in females. There were no increased incidences of neoplasms in other tissues. The NTP concluded that there was some evidence of carcinogenic activity in male rats, but clear evidence in female rats.
Syrian golden hamsters (51 per group) exposed by inhalation to cobalt (as cobalt oxide) at 10.0 mg/m3 for 7 h/day, 5 days/week, for a lifetime developed emphysema, but the incidence of pulmonary tumours was not different from controls. While tobacco smoke exposure induced pulmonary tumours in 14/51 animals, the incidence in animals exposed to both tobacco smoke and cobalt oxide was 11/51 (Wehner et al., 1977).
Steinhoff & Mohr (1991) conducted a study of rats exposed to a cobalt–aluminium–chromium spinel, with an empirical formula Co(II) 0.66, Al 0.7, Cr(III) 0.3, and O 3.66 (80% of the particles < 1.5 μm), or to cobalt(II) oxide. Groups of 50 male and 50 female Sprague-Dawley rats were exposed by intratracheal instillations of the spinel in saline at 10 mg/kg body weight every 2 weeks for 18 treatments, followed by every 4 weeks from the 19th to the 30th treatments for a total of 2 years. The rats were allowed to live until a natural death, or they were sacrificed when moribund. Alveolar/bronchiolar proliferation was not observed in 100 untreated controls and 100 saline controls; however, 61/100 rats exhibited this effect in the spinel treatment group. Likewise, no pulmonary tumours were observed in the untreated or saline controls. In the spinel-treated groups, one male and two female rats exhibited squamous cell carcinomas. When cobalt(II) oxide was administered by intratracheal instillation at doses of 2 mg/kg body weight (total dose 78 mg/kg body weight) or 10 mg/kg body weight (total dose 390 mg/kg body weight), there were two benign pulmonary tumours among the 100 rats in the low-dose group and two benign and four malignant pulmonary tumours among the 100 rats in the high-dose group. Steinhoff & Mohr (1991) also administered subcutaneous doses of 5 × 2 and 1 × 10 mg of cobalt(II) oxide per kilogram body weight per week, and 5/10 and 4/10 rats, respectively, developed local malignant tumours in a lifetime study. In a related study (Steinhoff & Mohr, 1991), groups of 10 male and 10 female rats were administered three intraperitoneal injections of saline or cobalt–aluminium–chromium spinel powder at 2-month intervals for a total dose of 600 mg/kg body weight. The rats were observed for their natural life span or were sacrificed when moribund. Malignant peritoneal tumours occurred in 1/20 controls (histiocytoma) and 2/20 spinel-treated rats (one histiocytoma and one sarcoma). After intraperitoneal administration of 3 × 200 mg of cobalt(II) oxide per kilogram body weight, 14/20 rats developed malignant intraperitoneal tumours.
Heath (1954, 1956, 1960) treated groups of 10 male and 10 female hooded rats with a single intramuscular injection of 28 mg of cobalt metal powder. The cobalt metal particles ranged in size from 3.5 μm Χ 3.5 μm to 17 μm Χ 12 μm, with large numbers of long narrow particles 10 μm Χ 4 μm. The rats were injected in the thigh. The observation period was 122 weeks, during which 4/10 male and 5/10 female rats developed sarcomas, mostly rhabdomyosarcomas, at the injection site. In a related study, 80 female hooded rats (divided into three groups of 16, 14, and 50) were intramuscularly injected with 28 mg of wear particles (ground artificial hip or knee prostheses composed of a cobalt–chromium–molybdenum alloy) (Heath et al., 1971; Swanson et al., 1973). No control group was reported. Animals were observed for up to 29 months. The incidences of sarcomas at the injection site were 3/16, 4/14, and 16/50. Half the tumours were rhabdomyosarcomas, and the remainder were fibrosarcomas. In a related study, Heath & Daniel (1962) injected two groups of 10 female hooded rats with 28 mg cobalt metal powder (3.5 μm Χ 3.5 μm to 17 μm Χ 12 μm, with large numbers of long narrow particles 10 μm Χ 4 μm) through the right dome of the diaphragm (first group) or through the fourth left intercostal space (second group). Animals were observed for up to 28 months. Of the diaphragm-treated rats, 6/10 died within 3 days, and in the rats injected through the intercostal space, 2/10 died within 3 days. Of the 12 rats that survived the injection, 4 developed intrathoracic sarcomas. Three of these sarcomas were of mixed origin and included rhabdomyosarcomatous elements, while the fourth rhabdomyosarcoma arose in the intercostal muscle.
Meachim et al. (1982) conducted a follow-up study to Heath et al. (1971) and Swanson et al. (1973). Female Wistar rats (n = 51) received intramuscular implants of 28 mg coarse particles (100–250 μm diameter) of ground cobalt–chromium–molybdenum alloy, and 61 Wistar and 53 hooded rats received implants of fine particles (0.5–50 μm). The rats were observed for life. Survival at 2 years was 11/41 for Wistar rats receiving coarse particles, 7/61 for Wistar rats receiving fine particles, 0/53 for hooded rats receiving fine particles, and 5/50 for Wistar controls. No tumours were observed at the implantation sites. Meachim et al. (1982) conducted a similar study in a group of 46 female Dunkin Hartley guinea-pigs that received intramuscular implants of 28 mg of fine particles of ground cobalt–chromium–molybdenum alloy. At 3 years, 12/46 animals were alive. No tumours were reported; however, nodular fibroblastic hyperplasia was observed in eight animals at the implantation site.
Mitchell et al. (1960) implanted a cobalt–chromium–molybdenum pellet (Vitallium alloy) subcutaneously into five male and five female Wistar rats. Rats were observed for up to 27 months, and no sarcomas were reported.
Memoli et al. (1986) implanted seven different test materials containing cobalt alloyed with chromium and nickel, molybdenum, tungsten, and/or zirconium into the femoral bone of groups of 10–17 male and 8–15 female Sprague-Dawley rats. The test materials were small rods (1.6 mm diameter and 4 mm length), powders, or porous compacted wire. The rats were observed for up to 30 months. Untreated and sham-operated controls, consisting of groups of 13 male and 13 female rats, were also studied. Sarcomas at the site of implantation were observed in 1/18 rats given a cobalt alloy powder (41% cobalt) and 3/26 rats given a nickel–cobalt-based powder (51% cobalt). No tumours were observed in two groups of 25 rats given rods with 69% or 47% cobalt, in two groups of 26 rats given rods with 0.11% or 33% cobalt, or in the untreated and sham-operated controls.
Vollmann (1938) implanted metallic cobalt dust into the femoral cavity of two groups of 15–20 rabbits. No tumours were observed at 3 years post-implantation. A follow-up study of these survivors at 6 years revealed sarcomas at the site of implantation in two cobalt-treated rabbits (Schinz & Uehlinger, 1942).
Jasmin & Riopelle (1976) injected groups of 20 and 18 female Sprague-Dawley rats with 5 mg of metallic cobalt powder or cobalt sulfide powder, respectively, into each pole of the right kidney. After 12 months, necropsies were conducted, and no tumours were observed in the kidneys of treated or control rats.
There are no available studies on genotoxic effects in animals exposed by inhalation. Male Swiss mice administered a single oral dose of cobalt (as cobalt chloride) at 0, 4.96, 9.92, or 19.8 mg/kg body weight exhibited a dose–response increase in percentages of chromosomal breaks and chromosomal aberrations in bone marrow cells (Palit et al., 1991a, 1991b, 1991c, 1991d). A single intraperitoneal injection of cobalt (as cobalt(II) chloride) at 12.4 or 22.3 but not 6.19 mg/kg body weight in BALB/c mice caused an increase in micronucleus formation after 30 h (Suzuki et al., 1993). F344 rats injected intraperitoneally with cobalt at 3 or 6 mg/kg body weight exhibited increased levels of oxidatively damaged DNA bases in the liver, kidney, and lung at 2 and 10 days following injection (Kasprzak et al., 1994).
Cobalt, in compounds with a valence state of +2, was mostly negative in mutagenicity tests conducted in Salmonella typhimurium, Escherichia coli, and yeast, but weakly positive in Bacillus subtilis (Kanematsu et al., 1980; Tso & Fung, 1981; Fukunaga et al., 1982; Singh, 1983; Arlauskas et al., 1985; Kharab & Singh, 1985; Ogawa et al., 1986). The only positive report for cobalt(II) is from S. typhimurium TA100 both with and without liver S9 metabolic enzymes (NTP, 1998). S. typhimurium strains TA98 and TA1535 were negative. Cobalt(II) compounds caused genetic conversions in S. cerevisiae (Fukunaga et al., 1982; Singh, 1983; Kharab & Singh, 1985). The reasons for this dichotomy in yeast are unknown. Cobalt in compounds with a valence state of +3 was positive in S. typhimurium and E. coli (Schultz et al., 1982).
In mammalian test systems, many cobalt compounds and metals are genotoxic. Cobalt compounds and cobalt metals have been reported to cause clastogenic effects in mammalian cells such as human lymphocytes (Painter & Howard, 1982; Hamilton-Koch et al., 1986; Anard et al., 1997), transformation in hamster cells (Costa et al., 1982), sister chromatid exchanges in human lymphocytes (Andersen, 1983), and micronucleus formation in mouse bone marrow cells (Suzuki et al., 1993), human lymphocytes (Capomazza & Botta, 1991; Olivero et al., 1995; van Goethem et al., 1997), and rat type II epithelial lung cells (DeBoeck et al., 2003). Cobalt particles are genotoxic in vitro in human peripheral blood mononucleated cells (Anard et al., 1997; van Goethem et al., 1997; De Boeck et al., 1998, 2003). In general, hard cobalt metal is more genotoxic than other cobalt compounds in in vitro test systems.
A study by the NTP that examined the carcinogenicity of cobalt sulfate heptahydrate by inhalation in B6C3F1 mice (NTP, 1998; Bucher et al., 1999) (see section 8.4) also evaluated K-ras mutation frequency and spectra in lung tumours. A higher frequency (5/9; 55%) of G to T transversions was detected in codon 12 of K-ras compared with chamber controls (0/1) or historical controls (1/24). G to T transversions are common DNA changes associated with active oxygen species. This provides supportive evidence that cobalt sulfate heptahydrate may indirectly damage DNA by oxidative stress.
Both rats exposed to cobalt (as cobalt chloride) at 13.3–58.9 mg/kg body weight per day for 2–3 months in drinking-water or diet (Nation et al., 1983; Domingo et al., 1984; Corrier et al., 1985; Mollenhauer et al., 1985; Pedigo et al., 1988; Pedigo & Vernon, 1993) and mice exposed to cobalt (as cobalt chloride) at 43.4 mg/kg body weight per day for 13 weeks in drinking-water exhibited testicular degeneration and atrophy (Anderson et al., 1992, 1993).
In an abstract reported by Elbetieha et al. (2004), sexually mature male mice exposed to cobalt(II) chloride at 200, 400, or 800 mg/l in their drinking-water for 12 weeks were assessed for effects on fertility by breeding these exposed males to unexposed females. Fertility, as measured by successful matings, was reduced in mice exposed to cobalt chloride at 400 and 800 mg/l (internal doses of 46.91 ± 4.78 and 93.01 ± 6.76 mg/kg body weight per day, respectively). The number of implantation sites was significantly reduced in females mated with exposed males at 400 and 800 mg/l. The number of viable fetuses was decreased in females mated with males at all three exposure levels. In the 800 mg/l males, absolute epididymal weight was significantly decreased, whereas relative and absolute testes weights were decreased in males exposed to both 400 and 800 mg/l. Epididymal sperm count was decreased in males of all three exposure levels. At 400 and 800 mg/l, males also exhibited reduced testicular sperm counts and daily sperm production. The testes displayed severe abnormalities, including hypertrophy of the interstitial Leydig cells, congested blood vessels, degeneration of the spermatogonial cells, and necrosis of seminiferous tubules and interstitial tissue.
In a study in which B6C3F1 mice were exposed by inhalation to cobalt sulfate heptahydrate (0, 0.3, 1, 3, 10, and 30 mg/m3; equivalent to cobalt concentrations of 0, 0.11, 0.38, 1.14, 3.80, and 11.38 mg/m3) for 6 h/day, 5 days/week, for 13 weeks, testicular atrophy in males and increased estrous cycle length in females were observed at 30 mg/m3. Sperm motility was decreased in mice exposed to 3 mg/m3 or higher (lower exposures not assessed), and increased abnormal sperm and decreased testis and epididymal weights were observed in mice exposed to 30 mg/m3 (Bucher et al., 1990; NTP, 1991) (see also section 8.3).
Oral exposure of female rats to cobalt (as cobalt chloride) at doses of 5.4 or 21.8 mg/kg body weight per day from gestation day 14 to lactation day 21 caused newborn pups to exhibit stunted growth and decreased survival. However, these effects occurred at exposures that also caused maternal toxicity, such as reduced body weight, reduced food consumption, and altered haematological measurements. No teratogenic effects were observed (Domingo et al., 1985). Another study reported that exposure of pregnant rats to cobalt (as cobalt sulfate) at 0–38 mg/kg body weight per day did not affect fetal death rates, maternal body weight gain, average litter size, or average fetal and placental weights. However, a dose-related increase was noted in the percentage of fetuses with retarded body weights (Szakmary et al., 2001). In contrast, Paternain et al. (1988) found no effects on fetal growth or survival after exposing rats to cobalt (as cobalt chloride) at 24.8 mg/kg body weight per day during gestation days 6–15. Exposure of pregnant mice to cobalt (as cobalt sulfate) at 19 mg/kg body weight per day also did not affect litter size, postimplantation loss, or average fetal and placental weights (Szakmary et al., 2001). Rabbits exposed to cobalt (as cobalt sulfate) at doses of >38 mg/kg body weight per day exhibited complete maternal lethality and fetal loss. At 7.6 mg/kg body weight per day, rabbits had increased mortality, fetal resorption, and number of fetuses with retarded body weight (Szakmary et al., 2001).
Dermal exposures on 3 consecutive days to cobalt(II) chloride (in dimethylsulfoxide) caused an increase in cellular proliferation in the lymph node assay in mice (10.8, 27, or 54.1 mg of cobalt per kilogram body weight per day), rats (9.6 or 19.2 mg of cobalt per kilogram body weight per day), and guinea-pigs (14.7 mg of cobalt per kilogram body weight per day) (Ikarashi et al., 1992a, 1992b).
Several studies have demonstrated that a hard metal alloy of tungsten carbide and cobalt matrix is more toxic than either tungsten carbide or cobalt alone. In a proposed mechanism, tungsten carbide facilitates the oxidation of cobalt metal to ionic cobalt (Co2+) by transferring electrons from the cobalt atom to molecular oxygen (Lison et al., 1995, 1996). This causes an increase in the solubility of cobalt, relative to cobalt metal, and the generation of reactive oxygen species. The ionic cobalt may be transported by blood throughout the body, causing adverse effects by the generation of reactive oxygen species. In vitro evidence consists of the ability of hard metal particles to generate substantial levels of oxidant species and cause lipid peroxidation (Lison et al., 1995; Zanetti & Fubini, 1997), which does not occur by cobalt or tungsten carbide alone. In addition, hard metal particles have been shown to increase inducible nitric oxide synthase levels, which is responsive to oxidant stress (Rengasamy et al., 1999).
Cobalt toxicity may also be caused through oxidant-based and free radical-based processes. Exposure to soluble cobalt leads to increased indices of oxidative stress, diminished levels of reduced glutathione, increased levels of oxidized glutathione, activation of the hexose monophosphate shunt, and free radical-induced DNA damage (Lewis et al., 1991; Kasprzak et al., 1994; Zhang et al., 1998; Hoet et al., 2002). In the presence of hydrogen peroxide, cobalt(II) stimulates in vitro formation of 8-hydroxy-2’-deoxyguanosine (Ivancsits et al., 2002). A Fenton-type mechanism causes cobalt to generate oxygen radicals, such as superoxide, in both in vitro and in vivo studies (Moorhouse et al., 1985; Kadiiska et al., 1989; Kawanishi et al., 1994; Lloyd et al., 1997). Exposure of rats and guinea-pigs to cobalt results in liver lipid peroxidation and reduced levels of glutathione, superoxide dismutase, catalase, haem oxygenase, and glutathione peroxidase (Sunderman & Zaharia, 1988; Christova et al., 2001, 2002). Cobalt accumulation in cardiac tissues is believed to stimulate carotid body chemoreceptors, which mimics the action of hypoxia (Di Giulio et al., 1990, 1991; Hatori et al., 1993; Morelli et al., 1994). Cobalt exposure also affects genes that are sensitive to oxidant status, such as hypoxia-inducible factor 1, erythropoietin, vascular endothelial growth factor, catalase, and monooxygenase enzymes (Yasukochi et al., 1974; Dalvi & Robbins, 1978; Legrum et al., 1979; Goldberg et al., 1988; Di Giulio et al., 1991; Goldberg & Schneider, 1994; Ladoux & Frelin, 1994; Semenza et al., 1994; Ho & Bunn, 1996; Bunn et al., 1998; Daghman et al., 1999; Hoet et al., 2002). These effects may also lead to the induction of apoptosis, through either these genes or other pathways (Zou et al., 2001).
Soluble cobalt has been shown to block inorganic calcium channels (Henquin et al., 1983; Moger, 1983; Yamatani et al., 1998). This has been shown to reduce steroidogenesis in isolated mouse Leydig cells (Moger, 1983). Calcium influx in liver cells, pancreatic beta cells, and isolated rat islets is altered by soluble cobalt (Henquin & Lambert, 1975; Henquin et al., 1983; Yamatani et al., 1998). By antagonizing calcium, cobalt may also affect neuromuscular transmissions (Weakly, 1973).
In the past, cobalt used to be added to beer as a defoaming agent. Cobalt was found to accumulate in the hearts of heavy beer drinkers and result in cardiomyopathy (see section 9 below). Microscopic analysis found fragmentation and degeneration of myofibres and aggregates of abnormal mitochondria (Ferrans et al., 1964). Mitochondrial effects result in disturbances in energy production and utilization and may be related to the irreversible chelation of lipoic acids under aerobic conditions by cobalt (Webb, 1962). Lipoic acid is a cofactor for the oxidative decarboxylation of pyruvate to acetyl CoA and of alpha-ketoglutarate to succinate (Lehninger, 1982). In rats treated with cobalt, the myocardium exhibits an impairment of pyruvate and fatty acid oxidation (Wiberg, 1968).
Cobalt ions, in the presence of oxidants such as UV radiation or hydrogen peroxide, can cause increased levels of DNA damage in vitro (Hartwig et al., 1991; Nackerdien et al., 1991; De Boeck et al., 1998). Cobalt is hypothesized to inhibit DNA repair, particularly the steps of incision and polymerization, by interacting with zinc finger DNA repair proteins (Sarkar, 1995; Kasten et al., 1997; Asmuß et al., 2000).
Cobalt is hypothesized to affect haem and haem-containing enzymes. Two sites of the biosynthetic pathway are thought to be the target for cobalt: synthesis of 5-aminolevulinate and conversion of 5-aminolevulinate to haem (de Matteis & Gibbs, 1977). This could result in the formation of cobalt protoporphyrin instead of haem (Sinclair et al., 1979). Cobalt may also act by inducing haem oxygenase and causing haem oxidation in organs (Sunderman, 1987). Haem-containing proteins that would be affected include monooxygenase enzymes (cytochrome P450) and catalase (Yasukochi et al., 1974; Legrum et al., 1979). Cobalt may also increase erythropoietin, which results in the increased production of red blood cells (Smith & Fisher, 1973; Goldberg et al., 1988; Di Giulio et al., 1991).
Glucose metabolism has also been demonstrated to be affected by cobalt. Animals treated with cobalt exhibit depressed serum and tissue glucose levels (Wiberg, 1968; Eaton & Pommer, 1973; Ybarra et al., 1997). Cobalt-induced glucose depression was persistent in diabetic rats (by pretreatment with streptozotocin) but transient in normal rats (Ybarra et al., 1997). Cobalt may alter the expression of the GLUT family of glucose transport proteins, which are Na+-independent proteins that mediate non-insulin-dependent glucose transport. Soluble cobalt has been found to increase the expression of these genes, particularly GLUT-1, in the liver, kidney cortex, myocardium, skeletal muscle, and cerebrum (Behrooz & Ismail-Beigi, 1997; Ybarra et al., 1997). Cobalt has also been found to reduce the amount of glucose produced in liver cells that were stimulated by glucagons and reduce insulin release in isolated rat islets (Eaton & Pommer, 1973; Henquin & Lambert, 1975; Yamatani et al., 1998).
During the early to mid-1960s, breweries in the United States, Canada, and Europe added cobalt sulfate to beer as a foam stabilizer. Several studies reported lethal cardiomyopathy in people who consumed large quantities of beer with cobalt sulfate (Morin & Daniel, 1967; Kesteloot et al., 1968; Alexander, 1969, 1972; Bonenfant et al., 1969; Sullivan et al., 1969; Morin et al., 1971). Cobalt exposures leading to death ranged from 0.04 to 0.14 mg/kg body weight per day for several years (approximately 8–30 pints per day). Acute mortality occurred in 18% of these deaths (Alexander, 1972). Approximately 40–50% of the patients admitted to the hospital for cardiomyopathy died within several years of diagnosis (Alexander, 1972). Possible confounders for cobalt-induced cardiomyopathy include the protein-poor diets of heavy beer drinkers and cardiac damage from alcohol abuse. Cardiomyopathy induced by beer cobalt was similar to alcoholic cardiomyopathy and beriberi, except that the onset was abrupt. These patients also exhibited liver injury characterized by central hepatic necrosis and increased levels of serum bilirubin and serum enzymes.
A study by Davis & Fields (1958) demonstrated that six normal men aged 20–47 exposed to a daily oral dose of cobalt chloride (150 mg/day) for up to 22 days experienced polycythaemia. Red blood cell numbers increased by 0.5–1.19 million over initial values, approximately a 16–20% increase over pretreatment levels. Haemoglobin levels were also increased, by 6–11% over pretreatment levels.
In humans, inhalation and dermal exposure have been observed to result in sensitization to cobalt (Marcussen, 1963; Valer et al., 1967; Dooms-Goossens et al., 1980; Bencko et al., 1983; Fischer & Rystedt, 1983; Alomar et al., 1985; Goh et al., 1986; Kanerva et al., 1988; Shirakawa et al., 1988, 1989). Contact allergy was reported in 22 of 223 (9.9%) nurses who were tested with a patch test of 1.0% cobalt chloride (Kieć-Świerczyńska & Kręcisz, 2000), as well as 16 of 79 (20.3%) of examined dentists (Kieć-Świerczyńska & Kręcisz, 2002). Nielsen et al. (2000) demonstrated that daily repeated exposure to aqueous cobalt salts did not result in hand eczema in patients known to have cobalt allergy, suggesting that the allergic properties of cobalt result mainly from exposure to the metal itself, rather than to a cobalt salt. Shirakawa et al. (1989) reported that inhalation of cobalt chloride aerosols can precipitate an asthmatic attack in sensitized individuals. Sensitization has been observed in hard metal workers with work-related asthma and exposures ranging from 0.007 to 0.893 mg/m3 for 3 years or more (Shirakawa et al., 1988, 1989). Bronchial asthma has been described in workers exposed to various forms of cobalt — i.e. not only in workers exposed to hard metal dust, but also in those exposed to "pure" cobalt particles (Swennen et al., 1993; Linna et al., 2003). Cobalt-specific IgE and IgA antibodies have been reported in humans (Bencko et al., 1983; Shirakawa et al., 1988, 1989).
A cross-sectional study of 82 workers in a cobalt refinery examined cobalt in blood and urine, erythropoietic variables, thyroid metabolism, pulmonary function, skin lesions, and several serum enzymes (Swennen et al., 1993). Cobalt concentrations in blood and urine were significantly correlated with airborne cobalt levels. Workers exposed to cobalt metal, salts, or oxides (mean concentration 0.125 mg/m3 in air, range 0.001–7.7 mg/m3) displayed a statistically significant increase in the prevalence of dyspnoea and wheezing and also had significantly more skin lesions, such as eczema and erythema, compared with controls. A dose–response relationship between decreased FEV1 and cobalt exposure assessed by blood, urine, or air cobalt levels was observed. Verougstraete et al. (2004) examined lung function among 122 workers in a cobalt-producing plant in a 13-year (1988–2001) follow-up study. The FEV1 was found to decrease over time, but only in association with smoking.
A cross-sectional study of 194 diamond polishers from 10 diamond-polishing workshops in Belgium and 59 workers from three other workshops in the diamond industry (control subjects) examined cobalt exposure and respiratory effects (Nemery et al., 1992). Cobalt exposure of the diamond polishers was a result of the generation of airborne cobalt from use of the cobalt-containing polishing discs. Analysis of air samples showed the presence of cobalt and no tungsten. Occasionally there were traces of other metals. Questionnaires inquiring about work history, working conditions, medical history, respiratory symptoms, and smoking habits were administered to the workers. Urine samples were collected from the workers and analysed for cobalt. Both area and personal air samples were collected. There was a good correlation between the results of area and personal samples at all workshops with the exception of one. When this workshop was excluded, a good correlation was also found between urinary cobalt and cobalt in the air. The workers were divided into three exposure categories: control (mean personal sample concentration of 0.0004 ± 0.0006 mg/m3), low (mean personal sample concentration of 0.0053 ± 0.0032 mg/m3), and high (mean personal sample concentration of 0.0151 ± 0.0117 mg/m3). The high exposure group was more likely to complain about respiratory symptoms and had significantly higher prevalence of eye, nose, and throat irritation and cough. The prevalence of some symptoms (e.g. cough, phlegm) was elevated in the low exposure group compared with the control group, but they were not significantly (P < 0.05) elevated. Lung function, assessed by FVC, FEV1, MMEF (forced expiratory flow between 25% and 75% of the FVC), and mean PEFR, was significantly reduced in workers in the high exposure group compared with workers in the lower exposure and control groups. The effect on women was greater than that on men, although the interaction of gender and exposure was not statistically significant. Lung function was not decreased in the low exposure group compared with the control group. Smoking habits were similar in the high exposure, low exposure, and control groups. The average concentration in the low exposure group was determined to be the NOAEC (0.0053 mg/m3).
A group of female workers occupationally exposed to a semisoluble cobalt glaze (cobalt–zinc silicate, estimated cobalt concentration of 0.05 mg/m3) showed significantly elevated levels of serum thyroxine and free thyroxine, but no change in triiodothyronine levels (Prescott et al., 1992). In contrast, Swennen et al. (1993) reported no significant change in serum thyroxine levels but a significant reduction in serum triiodothyronine in workers occupationally exposed to cobalt oxides, cobalt salts, and cobalt metal.
Interstitial lung disease caused by metallic cobalt-containing particles is a rare occupational lung disease. Several reviews are available on this fibrosing alveolitis, which is generally called hard metal lung disease (Bech et al., 1962; Anthoine et al., 1982; Hartung, 1986; Balmes, 1987; Van Den Eeckhout et al., 1988; Cugell, 1992; Seghizzi et al., 1994; Lison, 1996; Newman et al., 1998; Nemery et al., 2001a, 2001b). Potolicchio et al. (1997, 1999) suggested that individuals with a polymorphism in the HLA-DP gene (presence of glutamate 69 in the beta chain) may be more susceptible to hard metal lung disease. Individuals with ongoing respiratory illness may also be more susceptible to the effects of inhaled cobalt.
Hard metals are manufactured by a process of powder metallurgy from tungsten and carbon (tungsten carbide) and small amounts of a few other metallic compounds (titanium carbide, tantalium carbide, niobium carbide, etc.) using cobalt as a binder. Four mortality studies of the hard metal industry have been conducted in Sweden and France. Hogstedt & Alexandersson (1990) reported on 3163 male workers, each with at least 1 year of occupational exposure at hard metal manufacturing plants in Sweden during 1940–1982 and followed from 1951 to 1982. Exposures included a number of other substances used in the production of hard metal, such as tungsten carbide. The lung cancer SMR was 1.34 (95% CI = 0.77–2.13); the all-cause mortality SMR was slightly less than unity. Among workers with more than 10 years of employment and more than 20 years since first exposure, a significant excess of lung cancer mortality was observed (SMR = 2.78, 95% CI = 1.11–5.72). Smoking habits among hard metal workers were reported to be similar to those of the male Swedish population.
Lasfargues et al. (1994) conducted a cohort mortality study of 709 male workers employed for >1 year at a hard metal manufacturing plant (including two workshops) in central France. Follow-up was from 1956 to 1989. Categories of exposure were defined based on dust and urinary measurements of cobalt taken in 1983. Workers who had been employed in jobs with different degrees of exposure were categorized according to their highest exposure. Job histories were obtained from company records; before 1970, however, the records were often missing. The overall mortality did not differ from expected (SMR = 1.05, 95% CI = 0.82–1.31). Mortality due to lung cancer was in excess (SMR = 2.13, 95% CI = 1.02–3.93), and the excess was highest among workers in the areas with highest exposures to cobalt (SMR = 5.03, 95% CI = 1.85–10.95).
An industry-wide cohort mortality study of the French hard metal industry was conducted by Moulin et al. (1998) to further evaluate the potential association of lung cancer risk with occupational exposure to cobalt and tungsten carbide. The cohort included 5777 men and 1682 women (total = 7459 workers) from 10 factories (most of which were in eastern France), including the factory studied by Lasfargues et al. (1994). Workers were included in the cohort if they had 3 months of employment in nine factories or 1 year of employment in the factory studied by Lasfargues et al. (1994) and were first employed between the date each factory opened (1945–1965) and December 31, 1991. The follow-up period was 1968–1991. There were 1131 workers lost to follow-up (15%). The all-cause mortality SMR was 0.93; the lung cancer SMR was 1.30 (95% CI = 1.00–1.66). Sixty-one of the 63 lung cancer deaths in the cohort were included in a nested case–control study. Three controls that were alive on the date the case died were matched to each case based on gender and age. Occupational exposure of the cases and controls was evaluated based on a job–exposure matrix involving 320 job periods and exposure intensity scores from 0 to 9. Data on smoking were available for 80% of the cases and controls. The odds ratio for workers exposed to cobalt and tungsten carbide was 1.93 (95% CI = 1.03–3.62) for exposure levels 2–9 versus levels 0–1. The odds ratio for cobalt with tungsten carbide increased with duration of exposure and cumulative dose, but less so for level of exposure. Adjustments for exposure to known or suspected carcinogens and smoking did not change the results.
A study of the largest plant in the multicentre cohort of Moulin et al. (1998) was conducted by Wild et al. (2000). The authors used the same job–exposure matrix of Moulin et al. (1998) but made use of the more detailed job histories available. Follow-up was from 1968 to 1992. The SMR for the all-cause mortality was 1.02 (95% CI = 0.92–1.13). The SMR for lung cancer among men was increased (SMR = 1.70, 95% CI = 1.24–2.26). The lung cancer SMR for exposure to hard metal dust at an intensity score of >2 was 2.02 (95% CI = 1.32–2.96). In a Poisson regression model including terms for smoking and other occupational carcinogens, the risk for lung cancer increased with duration of exposure to cobalt with tungsten before sintering; there was no evidence of risk from exposure to sintered hard metal dust.
Moulin et al. (1993) studied the mortality of a cohort of 1148 workers in a cobalt electrochemical plant in France that produced cobalt and sodium by electrochemistry, extending the follow-up of an earlier study by Mur et al. (1987). The cohort included all men who had worked at the plant for a minimum of 1 year between 1950 and 1980. Follow-up was to the end of 1988 and was obtained for 99% of French-born workers. Because of difficulty in follow-up of non-French workers, results were presented only for the 870 French-born (i.e. a loss to follow-up of 24%). The SMR for all causes of death was 0.95 (95% CI = 0.78–1.26). The SMR for lung cancer was 1.16 (95% CI = 0.24–3.40) among workers exclusively employed in cobalt production and 1.18 (95% CI = 0.32–3.03) for workers ever employed in cobalt production.
Tüchsen et al. (1996) did not find evidence of an increased risk of lung cancer among a cohort of 874 women occupationally exposed to poorly soluble cobalt–aluminate spinel in two porcelain production factories in Denmark compared with that expected based on national rates for Danish women.
There are no available studies on genotoxic effects in humans exposed to cobalt by the oral and dermal routes of exposure. A cohort of 26 male workers who were occupationally exposed to cobalt, chromium, nickel, and iron exhibited increased sister chromatid exchange rank values (by analysis of variance) that were related to metal exposures and smoking habits (Gennart et al., 1993). De Boeck et al. (2000) performed a comet assay on lymphocytes from non-smoking workers occupationally exposed to cobalt or hard metal dusts and reported no significant effects. This study reported a positive association between hard metal exposure and increased micronucleus formation in smokers only. Hengstler et al. (2003) determined DNA single-strand break induction from mononuclear blood cells of 78 workers occupationally exposed to cadmium (range of concentrations in air, 0.05–138 μg/m3), cobalt (range, 0–10 μg/m3), and lead (range, 0–125 μg/m3), compared with 22 unexposed control workers. Non-parametric correlation analysis demonstrated significant correlations between DNA single-strand breaks and cobalt (P < 0.001; r = 0.401) and cadmium (P < 0.001; r = 0.371), but not lead.
Cobalt is essential for nitrogen fixation by free-living bacteria, blue-green algae, and symbiotic systems (e.g. Rhizobium in the root nodules of legumes) (Adriano, 1986). Cobalt is an essential element for the growth of many marine algal species, including diatoms, chrysophytes, and dinoflagellates (McLachlan, 1973; Bruland et al., 1991). In higher plants, cobalt has been shown to be an essential element for legumes, which have nodules containing nitrogen-fixing bacteria (Ozanne et al., 1963; Gladstones et al., 1977). In non-leguminous plants, cobalt is reported to be beneficial rather than essential. Smith & Carson (1981) reported inconclusive evidence of low concentrations of cobalt being beneficial to non-leguminous plants, whereas cobalt supplements have been reported to increase growth of rubber plants and tomatoes and length of pea stem sections (Adriano, 1986).
Studies with earthworms (Eisenia foetida) involving the addition of cobalt chloride supplements to a food source low in cobalt indicated that total cobalt concentrations of 17.6 and 25.9 mg/kg dry weight resulted in significantly increased maximum weights and numbers of cocoons produced compared with control worms exposed to 9.4 mg/kg dry weight (Neuhauser et al., 1984). Although cobalt is essential for animal nutrition, the metal is not required by animals in ionic form. It is, however, a dietary essential element for ruminants and horses, in which it is incorporated into vitamin B12 molecules by gastrointestinal microbes (Smith, 1987). Low levels of cobalt in feedstuff can cause nutritional diseases in ruminants — e.g. "bush sickness" in cattle or sheep or "pining" in sheep (Adriano, 1986). In the natural environment, NAS (1980) noted that cobalt deficiency in ruminants is more likely than cobalt toxicosis. Suttle et al. (2003), using acetic acid-extractable cobalt as a predictor of "plant availability" and therefore of potentially deficient soils, found that 29% of 103 soils analysed in the United Kingdom were deficient (<0.4 mg of acetic acid-extractable cobalt per kilogram dry weight) for grazing livestock. Frank et al. (2004) suggested that a wasting, debilitating disease affecting a wild population of moose (Alces alces americana) in eastern North America might be due to cobalt/vitamin B12 deficiency.
Acute exposure (<96 h) to cobalt concentrations in the range of 5–20 mg/l has been shown to result in a reduction in growth of the cyanobacterium Anabaena variabilis (Ahluwalia & Kaur, 1988). A delay in the onset of the log phase of growth of Anacystis nidulans has been reported following a 17-day exposure to cobalt concentrations of 15 mg/l; concentrations of 30 mg/l caused complete cessation of growth (Lee et al., 1992). Toxicity of cobalt to aquatic organisms is summarized in Table 2. A 96-h EC50 based on growth of the freshwater green alga Chlorella vulgaris was reported at 0.6 mg/l (Rachlin & Grosso, 1993), whereas EC50s for aquatic vascular plants were 0.1 and 0.2 mg/l (Gaur et al., 1994). The 5-day EC50 based on growth of the marine diatom Ditylum brightwellii was reported at 0.3 mg/l (Canterford & Canterford, 1980).
Table 2: Toxicity of cobalt to aquatic organisms.
|
Organism |
End-point |
Salt |
Cobalt concentration (mg/l) |
Reference |
|
Microorganisms |
||||
|
Freshwater |
||||
|
Blue-green alga (Spirulina platensis) |
96-h EC50 (biomass) |
Chloride |
10.8 |
Sharma et al. (1987) |
|
Green alga (Chlorella vulgaris) |
96-h EC50 (growth) |
Chloride |
0.6 |
Rachlin & Grosso (1993) |
|
21-day NOEC (growth) |
Nitrate |
0.6 |
Coleman et al. (1971) |
|
|
21-day LOEC (growth) |
Nitrate |
1.6 |
Coleman et al. (1971) |
|
|
Green alga (Euglena viridis) |
21-day LOEC (growth) |
Nitrate |
0.6 |
Coleman et al. (1971) |
|
Green alga (Pediastrum tetras) |
21-day LOEC (growth) |
Nitrate |
0.6 |
Coleman et al. (1971) |
|
Protozoan (Tetrahymena pyriformis) |
9-h IC50 (growth) |
Chloride |
56 |
Sauvant et al. (1995b) |
|
36-h IC50 (growth) |
Chloride |
24 |
Sauvant et al. (1995a) |
|
|
Ciliated protozoan (Spirostomum ambiguum) |
24-h LC50 |
Nitrate |
11.8 |
Nalecz-Jawecki & Sawicki (1998) |
|
Marine |
||||
|
Diatom (Ditylum brightwellii) |
5-day EC50 (growth) |
Chloride |
0.3 |
Canterford & Canterford (1980) |
|
Diatom (Nitzschia closterium) |
96-h EC50 (growth) |
Not given |
10.2 |
Rosko & Rachlin (1975) |
|
Vascular plants |
||||
|
Greater duckweed (Spirodela polyrhiza) |
96-h EC50 (growth) |
Chloride |
0.1 |
Gaur et al. (1994) |
|
Water velvet (Azolla pinnata) |
96-h EC50 (growth) |
Chloride |
0.2 |
Gaur et al. (1994) |
|
Invertebrates |
||||
|
Freshwater |
||||
|
Water flea (Daphnia magna) |
48-h LC50 |
Chloride |
1.5 |
Khangarot & Ray (1989a) |
|
48-h LC50 |
Chloride |
1.1 |
Biesinger & Christensen (1972) |
|
|
48-h LC50 |
Sulfate |
6 |
Kimball (1978) |
|
|
96-h LC50 |
Chloride |
1.5 |
Ewell et al. (1986) |
|
|
21-day LC50 |
Chloride |
0.02 |
Biesinger & Christensen (1972) |
|
|
21-day EC50 (reproduction) |
Chloride |
0.01 |
Biesinger & Christensen (1972) |
|
|
21-day NOEC (reproduction and survival) |
Not given |
0.03–0.05a |
Nagpal (2004) |
|
|
28-day LC50 |
Sulfate |
0.03 |
Kimball (1978) |
|
|
28-day NOEC (reproduction) |
Sulfate |
0.003 |
Kimball (1978) |
|
|
Water flea (Daphnia hyalina) |
48-h LC50 |
Chloride |
1.3 |
Baudouin & Scoppa (1974) |
|
Water flea (Ceriodaphnia dubia) |
24-h LC50 |
Chloride |
2.4–>5.3b |
Diamond et al. (1992) |
|
7-day NOEC (reproduction) |
Not given |
<0.003–0.013a |
Nagpal (2004) |
|
|
Rotifer (Philodina acuticornis) |
24-h LC50 |
Chloride |
27.8 |
Buikema et al. (1984) |
|
Copepod (Diaptomus forbesi) |
96-h LC50 |
Chloride |
3.4 |
Das & Kaviraj (1994) |
|
Copepod (Cyclops abyssorum) |
48-h LC50 |
Chloride |
15.5 |
Baudouin & Scoppa (1974) |
|
Copepod (Eudiaptomus padanus) |
48-h LC50 |
Chloride |
4 |
Baudouin & Scoppa (1974) |
|
Crayfish (Austropotamobius pallipes) |
96-h LC50 |
Chloride |
8.8 |
Boutet & Chaisemartin (1973) |
|
Crayfish (Orconectes limosus) |
96-h LC50 |
Chloride |
10.2 |
Boutet & Chaisemartin (1973) |
|
Amphipod (Crangonyx pseudogracilis) |
96-h LC50 |
Chloride |
39.2 |
Martin & Holdich (1986) |
|
Flatworm (Dugesia tigrina) |
96-h LC50 |
Chloride |
11.3 |
Ewell et al. (1986) |
|
Snail (Helisoma trivolvis) |
96-h LC50 |
Chloride |
>45 |
Ewell et al. (1986) |
|
Sideswimmer (Gammarus fasciatus) |
96-h LC50 |
Chloride |
>45 |
Ewell et al. (1986) |
|
Pillbug (Asellus intermedius) |
96-h LC50 |
Chloride |
>45 |
Ewell et al. (1986) |
|
Segmented worm (Lumbriculus variegatus) |
96-h LC50 |
Chloride |
>45 |
Ewell et al. (1986) |
|
Tubificid worm (Tubifex tubifex) |
96-h LC50 |
Chloride |
95.4–239c |
Rathore & Khangarot (2002) |
|
Oligochaete (Branchiura sowerbyi) |
96-h LC50 |
Chloride |
133 |
Das & Kaviraj (1994) |
|
Midge (Chironomus tentans) |
48-h LC50 |
Chloride |
57 |
Khangarot & Ray (1989b) |
|
Mayfly (Ephemerella subvaria) |
96-h LC50 |
Sulfate |
16 |
Warnick & Bell (1969) |
|
Marine |
||||
|
Nematode (Monhystera disjuncta) |
96-h LC50 |
Not given |
94 |
Vranken et al. (1991) |
|
Brown mussel (Perna perna) |
1-h EC50 (filtering rate) |
Chloride |
1.7 |
Watling & Watling (1982) |
|
Brine shrimp (Artemia salina) |
48-h LC50 |
Nitrate |
172 |
Kissa et al. (1984) |
|
48-h EC50 (hatching rate) |
Nitrate |
10.3 |
Kissa et al. (1984) |
|
|
Common prawn (Palaemon serratus) |
96-h LC50 |
Chloride |
227–454 (adult) |
Amiard (1976) |
|
96-h LC50 |
Chloride |
22.7–45.4 (larva) |
Amiard (1976) |
|
|
Shore crab (Carcinus maenus) |
96-h LC50 |
Chloride |
227–454 (adult) |
Amiard (1976) |
|
96-h LC50 |
Chloride |
22.7 (larva) |
Amiard (1976) |
|
|
Lobster (Homarus vulgaris) |
96-h LC50 |
Chloride |
4.5–22.7 (larva) |
Amiard (1976) |
|
Isopod (Idotea baltica) |
52-day LC50 |
Chloride |
10 |
El-Nady & Atta (1996) |
|
Fish |
||||
|
Freshwater |
||||
|
Rainbow trout (Oncorhynchus mykiss) |
96-h LC50 |
Chloride |
1.4 |
Marr et al. (1998) |
|
14-day NOEC (growth) |
Chloride |
0.1 |
Marr et al. (1998) |
|
|
28-day LC50d |
Nitrate |
0.49 |
Birge et al. (1980) |
|
|
Fathead minnow (Pimephales promelas) |
96-h LC50 |
Formate |
12.8 |
Curtis & Ward (1981) |
|
96-h LC50 |
Bromide |
24.8 |
Curtis & Ward (1981) |
|
|
96-h LC50 |
Sulfate |
3.6 |
Kimball (1978) |
|
|
96-h LC50 |
Chloride |
21.8 |
Ewell et al. (1986) |
|
|
7-day NOECe (survival) |
Chloride |
1.2–3.8b |
Diamond et al. (1992) |
|
|
Goldfish (Carassius auratus) |
96-h LC50 |
Chloride |
333 |
Das & Kaviraj (1994) |
|
7-day LC50d |
Nitrate |
0.8 |
Birge et al. (1979) |
|
|
Zebrafish (Danio rerio) |
16-day NOECd (hatching rate) |
Chloride |
3.8 |
Dave & Xiu (1991) |
|
16-day NOECd (survival) |
Chloride |
0.06 |
Dave & Xiu (1991) |
|
|
Giant gourami (Colisa fasciata) |
96-h LC50 |
Chloride |
102 |
Srivastava & Agrawal (1979) |
|
Marine |
||||
|
Plaice (Pleuronectes platessa) |
96-h LC50 |
Chloride |
454–681 |
Amiard (1976) |
|
Shanny (Blennius pholis) |
96-h LC50 |
Chloride |
454–681 |
Amiard (1976) |
|
Mummichog (Fundulus heteroclitus) |
96-h LC50 |
Chloride |
275f |
Dorfman (1977) |
|
96-h LC50 |
Carbonic acid |
>1000g |
Dorfman (1977) |
|
|
Crescent perch (Therapon jarbua) |
96-h LC50 |
Sulfate |
52.5 |
Krishnakumari et al. (1983) |
|
Amphibians |
||||
|
Freshwater |
||||
|
Frog (Rana hexadactyla) |
96-h LC50 |
Chloride |
18 |
Khangarot et al. (1985) |
|
Narrow-mouthed toad (Gastrophryne carolinensis) |
7-day LC50d |
Nitrate |
0.05 |
Birge et al. (1979) |
|
a |
Hardness ranging from 50 to 200 mg of calcium carbonate per litre. |
|
b |
Hardness ranging from 50 to 800 mg of calcium carbonate per litre. |
|
c |
Temperature ranging from 30 °C to 15 °C. |
|
d |
Embryo–larval toxicity test. |
|
e |
Early life stage toxicity test (larvae <24 h old). |
|
f |
Salinity 5–25‰. |
|
g |
Salinity 8–19‰. |
For freshwater invertebrates, acute LC50s (24–96 h) range from 1.1 mg/l (water flea Daphnia magna) to 239 mg/l (tubificid worm Tubifex tubifex). Several studies on D. magna reproduction were reported, with a 21-day EC50 at 0.01 mg/l and a 28-day NOEC at 0.003 mg/l (Biesinger & Christensen, 1972; Kimball, 1978); later studies found 21-day NOECs ranging from 0.03 to 0.05 mg/l for varying levels of calcium carbonate (Nagpal, 2004). The lowest reported NOEC for aquatic organisms was for the water flea Ceriodaphnia dubia in a 7-day test, at <0.003 mg/l (Nagpal, 2004). The most sensitive marine invertebrates were lobster larvae (Homarus vulgaris), with 96-h LC50s ranging from 4.5 to 22.7 mg/l (Amiard, 1976). Ninety-six-hour LC50s for freshwater fish range from 1.4 to 333 mg/l. A 16-day NOEC based on survival of zebrafish (Danio rerio) was reported at 0.06 mg/l (Dave & Xiu, 1991). Test results for marine fish suggest that at least the species tested are relatively insensitive to cobalt, with 96-h LC50s ranging from 52.5 to >1000 mg/l.
Marr et al. (1998) reported a temporal pattern to cobalt toxicity in rainbow trout (Oncorhynchus mykiss). Cobalt concentrations that would eventually cause 100% lethality caused no lethality until at least 72 h of exposure. A one-compartment uptake–depuration model was used to estimate the incipient lethal level for 50% mortality (time-independent concentration); the authors noted that the majority of the lethality occurred between 72 and 192 h, suggesting that the standard short-term 96-h LC50 could underpredict cobalt toxicity substantially. It should be noted that the 96-h LC50 was 1.4 mg/l and the incipient lethal level for 50% mortality was 0.4 mg/l.
Under most environmental conditions, including both fresh water and marine water, much of cobalt is dissolved either as cobalt carbonate or as Co2+ ions (Tipping et al., 1998). However, the actual bioavailability appears to depend on the water chemistry and particularly the concentration of Ca2+ ions and dissolved organic matter complexation. Diamond et al. (1992) suggested that there might be an effect of water hardness on aquatic toxicity. They reported that the 24-h LC50 for Ceriodaphnia dubia varied from 2.4 mg/l to greater than 5.3 mg/l in water with hardness ranging from 50 to 800 mg/l as calcium carbonate. The 7-day NOECs, based on survival, for C. dubia were <0.05 mg/l at a water hardness of 50 mg of calcium carbonate per litre and 0.6 mg/l at 800 mg of calcium carbonate per litre. The 48-h NOECs, based on survival, for fathead minnow (Pimephales promelas) varied from 1.3 mg/l at a water hardness of 50 mg of calcium carbonate per litre to 13.7 mg/l at 400 mg of calcium carbonate per litre, whereas 7-day NOECs ranged from 1.2 to 3.8 mg/l (Diamond et al., 1992). However, the results of a 7-day C. dubia partial life cycle toxicity test and a 21-day D. magna partial life cycle toxicity test reported by Nagpal (2004) did not support the cobalt toxicity–water hardness relationship suggested by Diamond et al. (1992). The 95% confidence limits for test end-points were overlapping for each of the three water hardnesses tested (50, 100, and 200 mg of calcium carbonate per litre).
Further studies on the interaction between Ca2+ ions and cobalt uptake suggest that cobalt ions compete with Ca2+ ions at the fish gill–water interface (Comhaire et al., 1994; Richards & Playle, 1998). Comhaire et al. (1994) found a clear decrease in the uptake of Co2+ by common carp (Cyprinus carpio) at Ca2+ concentrations of 0.4 and 14 mg/l; however, there was no further effect at >40 mg/l. The rate of Ca2+ uptake in gills and blood did not depend on the amount of calcium present in the water, and the results suggested that the effect of calcium on Co2+ uptake involves a direct interaction with the systems involved in the translocation of these metal ions across the gill epithelium. Richards & Playle (1998) found significant uptake of cobalt in artificial soft water (<1 mg of Ca2+ per litre); however, there was no significant uptake of cobalt in natural soft waters with Ca2+ concentrations ranging from 20 to 100 mg/l. Using a gill–cobalt binding model, they were able to predict an absence of gill cobalt accumulation in natural waters from nine different water bodies across Ontario, Canada, spanning a wide range of Ca2+ and Na+, pH (4.2–7.6), and dissolved organic matter levels. Overall, the model analysis indicated that Ca2+ competition and dissolved organic matter complexation were the most important factors preventing Co2+ from binding at the gills in these natural water tests. However, the effect of Ca2+ ions on the uptake and potential toxicity of cobalt occurs at very low Ca2+ concentrations, probably lower than those used in any of the reported toxicity tests. The data indicate that Co2+ binds to gill sites >1000 times more weakly than Cd2+, 10 times more weakly than Pb2+, and about 6 times more weakly than Zn2+ (Niyogi & Wood, 2004).
Rathore & Khangarot (2002) reported effects of temperature on the sensitivity of the sludge worm Tubifex tubifex to cobalt. In 96-h tests, worms were more sensitive at 30 °C than at 15 °C; overall, however, there was no clear relationship between temperature and acute toxicity — i.e. LC50s were 239, 180, 247, and 95.4 mg/l at 15 °C, 20 °C, 25 °C, and 30°C, respectively. Furthermore, at shorter exposure times, the pattern of sensitivity with temperature varied.
Behavioural avoidance of cobalt in soft water differed greatly between rainbow trout (Oncorhynchus mykiss) and chinook salmon (O. tshawytscha). Chinook salmon avoided cobalt concentrations of at least 0.02 mg/l, whereas rainbow trout avoided at least 0.2 mg/l (Hansen et al., 1999).
Data regarding the toxicity of cobalt to soil microorganisms are limited. Lighthart et al. (1977) studied the effects of several metals, including cobalt, at single concentrations on respiration of native soil microflora in soil/litter microcosms. A 1362 mg/l solution of cobalt mixed into the soil and litter in the microcosm resulted in a reduction in respiration of 23%.
There is little evidence of cobalt toxicity to plants due to elevated concentrations in soil. Vanselow (1966) reported that concentrations of cobalt in soil of up to 100 mg/kg have little effect on citrus crops. USEPA (2005) reported mean EC20 values, based on growth of alfalfa (Medicago sativa), barley (Hordeum vulgare), and radish (Raphanus sativus), ranging from 0.6 to 45.2 mg/kg dry weight.
Data from a number of nutrient solution studies were used to evaluate the potential for toxicity to plants from irrigation water containing cobalt. Wallace et al. (1977) reported reduction of leaf dry weight in bush beans (Phaseolus vulgaris) grown in nutrient solution containing a cobalt concentration of 0.06 mg/l for 21 days. A reduction of chysanthemum (Chrysanthemum morifolium) seedling root weight after 21 days of growth in nutrient solution containing cobalt at 0.06 mg/l was reported by Patel et al. (1976). Inhibition of mung bean (Vigna radiata) seedling growth occurred at 295 mg/l and was associated with chlorosis of the younger leaves (Liu et al., 2000). Misra et al. (1994) studied the effects of heavy metals, including cobalt, alone and in combination, on germination and root elongation of broad bean (Vicia faba). They found that seed germination was not affected by exposure to cobalt; however, root elongation was reduced, but not significantly, at concentrations of 8000 and 10 000 mg/l. Further, an increase in root elongation was observed at lower cobalt concentrations, with a significant increase at the lowest concentration studied (2000 mg/l). Patterson & Olsen (1983) assessed the toxicity of cobalt in solution to seedlings of white spruce (Picea glauca), black spruce (Picea mariana), paper birch (Betula papyrifera), jack pine (Pinus banksiana), white pine (Pinus strobus), red pine (Pinus resinosa), and honeysuckle (Lonicera tatarica). Toxic concentrations ranged from 5 mg/l for honeysuckle and paper birch to 100 mg/l for white pine. NAS/NAE (1973) reported that toxicity to a variety of food crops has been observed due to the application of nutrient solution containing cobalt at concentrations of approximately 0.1–5 mg/l.
Cobalt tolerance, along with tolerance to other metals, has been found in plant populations growing on soils high in particular metals: for example, populations of bladder campion (Silene vulgaris) and redtop (Agrostis gigantea) on mine tailings in Ontario, Canada (Hogan & Rauser, 1979; Paliouris & Hutchinson, 1991), tufted hair grass (Deschampsia cespitosa) around the Sudbury smelters, Ontario, Canada (Cox & Hutchinson, 1979), and Silene cobalticola (a campion species) in Zaire (Baker et al., 1983). Exclusion of the metal has been demonstrated in the cobalt tolerance of Silene cobalticola (Baker et al., 1983), whereas other species growing on cobalt-rich copper clearings are hyperaccumulators of cobalt (Brooks, 1977; Malaisse et al., 1979; Morrison et al., 1979).
Hartenstein et al. (1981) exposed the earthworm Eisenia foetida in a silt loam covered with activated sludge spiked at different cobalt concentrations and observed a significant effect on growth after 8 weeks of exposure at 300 mg/kg but not at 30 mg/kg. Neuhauser et al. (1984) exposed E. foetida to soil covered with horse manure spiked at different cobalt concentrations and found no significant effect on growth after 4 weeks of exposure at concentrations up to 91.9 mg/kg dry weight. Fischer & Molnár (1997) exposed E. foetida to a mixture of peaty marshland soil and horse manure spiked at different cobalt concentrations for 10 weeks. They found a total inhibition of reproduction and ultimately 77% mortality at 4720 mg/kg dry weight. The 28-day EC50, based on reproduction, of the springtail Folsomia candida was 1480 mg/kg dry weight in standard OECD artificial soil and 409 mg/kg in standard field soil. The difference in toxicity was reported to be due to the pH and cation exchange capacity of the two soils (Lock et al., 2004). Tatara et al. (1998) reported the 24-h LC50 for total cobalt at 1274 mg/l and for the free ion at 1210 mg/l for the free-living soil nematode Caenorhabditis elegans exposed to cobalt nitrate.
Dietary levels of 125, 250, and 500 mg of cobalt per kilogram of feed were given to 1-day-old broiler chicks for 14 days. All levels of cobalt reduced feed intake, body weight gain, and gain:feed ratio and caused a dose-dependent increase in mortality (Diaz et al., 1994). Hill (1974) reported a significant adverse effect on growth of 2-week-old chickens at a cobalt (as cobalt chloride) concentration of 100 mg/kg diet; no effect was found at 50 mg/kg. After 5 weeks, there was significant mortality at 200 mg/kg. Van Vleet et al. (1981) exposed white peking ducklings (Anas sp.) to dietary cobalt (as cobalt chloride) concentrations of 200 or 500 mg/kg for 15–28 days. Ducklings fed cobalt at 200 mg/kg for 15 days developed lesions characteristic of selenium–vitamin E deficiency, such as necrosis of skeletal and cardiac muscle and of smooth muscle of the gizzard and intestine; no significant mortality was reported. Significant mortality was reported at 500 mg/kg during a 28-day exposure.
Inhalation of cobalt metal is associated with respiratory effects in humans, including respiratory symptoms and effects on lung function as measured by FVC, FEV1, MMEF, and mean PEFR in a cross-sectional study of diamond polishers (Nemery et al., 1992). The NOAEC was determined to be 0.0053 mg/m3.
Inhalation and dermal exposure to cobalt are known to result in sensitization. Bronchial asthma has been described in workers exposed to various forms of cobalt.
Interstitial lung disease caused by metallic cobalt-containing particles is an occupational lung disease generally referred to as hard metal lung disease.
Four mortality studies, one in Sweden and three in France, of hard metal industry workers exposed to tungsten carbide, cobalt, and small amounts of other metals found an increased risk of death from lung cancer. The three French studies were not independent. Mortality studies of cobalt production workers and of workers exposed to cobalt during porcelain production found no increased lung cancer risk. Rats and mice exposed to cobalt sulfate heptahydrate by inhalation developed a dose-related lung tumour response, and cobalt metal administered by injection was found to produce injection-site sarcomas.
In the early to mid-1960s, cobalt sulfate was added to beer as a foam stabilizer. Ingestion of cobalt sulfate at 0.04–0.14 mg/kg body weight per day (8–30 pints per day) over several years was found to be associated with cardiomyopathy in humans. This effect may have been confounded by poor diet and high alcohol consumption. Rats exposed to cobalt sulfate in the diet at higher doses also experienced adverse cardiac effects.
No long-term feeding studies have been conducted with cobalt, and there are no long-term studies of humans ingesting cobalt. Short-term (22 days) ingestion of cobalt at 150 mg/day in human volunteers produced polycythaemia and an increase in haemoglobin.
Cobalt has been shown to be mutagenic in somatic and germ cells in in vivo and in vitro experiments. Increased sister chromatid exchange was observed in male workers exposed to cobalt and other metals. Clastogenic effects in bone marrow cells were observed in mice orally exposed to cobalt. Intraperitoneal injection of cobalt produced an increase in micronuclei in mice and oxidatively damaged DNA in rats. Cobalt has been found to cause genotoxic effects in mammalian test systems. Cobalt(III) was positive, but cobalt(II) produced mixed responses, in bacterial mutagenicity tests. Cobalt ions in the presence of oxidants can cause increased levels of DNA damage in vitro.
Mice and rats exposed to high oral doses of cobalt chloride for 2–3 months experienced testicular degeneration and atrophy. Stunted growth and decreased survival were observed among newborn rats at doses that caused maternal toxicity in one study. Similar doses did not produce such effects in another study of rats or in a study of mice. Rabbits exposed at high doses were found to have increased mortality, fetal resorption, and number of fetuses with decreased body weight. No teratogenic effects were reported in any of the studies.
Cobalt has been found to decrease glucose metabolism in animals.
Cobalt toxicity is hypothesized to be the result of oxidant-based and free radical-based processes. Cobalt exposure affects genes that are sensitive to oxidant status, potentially leading to apoptosis. Soluble cobalt has also been shown to block inorganic calcium channels, which can affect neuromuscular transmissions. Cobalt is hypothesized to affect haem synthesis.
The study of diamond polishers by Nemery et al. (1992) provides an adequate basis for setting a tolerable concentration for inhaled cobalt. The NOAEC in the study was 0.0053 mg/m3. Assuming an 8-h workday and a 5 days/week exposure, the NOAEC in the study is adjusted to derive a NOAEC for the general population of 0.0013 mg/m3 (0.0053 mg/m3 × 8/24 × 5/7). This NOAEC is divided by an uncertainty factor of 10 for human variability to give a tolerable concentration of 0.000 13 mg/m3, which is rounded to 1 × 10−4 mg/m3, for the general population.
No previous peer-reviewed documents have done a quantitative cancer risk estimate for cobalt. The tolerable concentration is based on a non-cancer end-point. To provide some assurance, at least with respect to cancer, that the tolerable concentration is protective, a BMC approach (USEPA, 2003) was used to estimate the lung cancer risk at the tolerable concentration of 1 × 10−4 mg/m3. Using this approach,3 the lifetime cancer risk at the tolerable concentration derived from the Nemery et al. (1992) study was estimated to be 3 × 10−5.
There are no suitable data with which to derive a tolerable intake for chronic ingestion of cobalt.4
At unpolluted sites, mean cobalt air concentrations are typically <1–2 ng/m3. The cobalt concentration in the air near Boston, Massachusetts, USA, in 1992–1993 was 1.7 ng/m3; in southern Norway, the mean cobalt concentration was 0.10 ng/m3 in 1985–1986. In source areas, cobalt concentrations may exceed 10 ng/m3.
The general population is exposed to cobalt primarily through the food supply, with estimated intake of 5–40 µg/day through the diet. Most of the ingested cobalt is inorganic.
The margin of exposure between ambient cobalt concentrations near anthropogenic sources of cobalt and the tolerable concentration of 1 × 10−4 mg/m3 is a factor of about 10-fold.
The study used to derive an inhalation tolerable concentration is a cross-sectional study of lung function and respiratory symptoms. The effects may be a reflection of recent, not chronic, exposure, and thus it is unknown if the tolerable concentration is protective for chronic effects. Studies of cohorts of hard metal workers exposed to cobalt and tungsten carbide have consistently found an increased risk of lung cancer. Cobalt sulfate has been found to induce pulmonary tumours in rats and mice. Cobalt is also known to be a sensitizing agent and genotoxic. Given that cobalt is an animal carcinogen, a sensitizing agent, and genotoxic, health risks may be present at exposures lower than the tolerable concentration.
The inhalation tolerable concentration was based on a cohort of 192 workers (among many thousands worldwide), in one industry (of many), which uses cobalt in a specific form, and may not be reflective of the form of cobalt to which workers are exposed in all industries that use cobalt.
Cobalt and inorganic cobalt compounds are non-volatile and released into the atmosphere in particulate form. Anthropogenic cobalt from combustion sources is assumed to be primarily in the form of oxides. Arsenide and sulfide forms are also released into the atmosphere during ore extraction and refining processes.
Cobalt released into the atmosphere is deposited on soil, and cobalt released into water may sorb to particles and settle into sediment or sorb directly to sediment. The distribution coefficient of cobalt (e.g. from water to sediment) varies due to pH, redox conditions, ionic strength, and dissolved organic matter concentrations. Factors affecting the speciation and fate of cobalt in water, sediments, and soil include organic ligands such as humic acids, anions, pH, and redox potential. In fresh water, cobalt complexed with carbonate (HCO3− and CO32−) constituted about 70% of dissolved cobalt, whereas a further 25% was present as the free Co2+ ion. The proportion of cobalt complexed with carbonate increases at the expense of free Co2+ as the alkalinity of the water increases. The proportions, but not the concentrations, of cobalt that exist as the free ion and as carbonate complexes in river water are independent of the level of fulvic acid in the water. In seawater, the carbonate species and the free aqua species assume roughly equal importance. The proportion of dissolved cobalt complexed with fulvic acid decreases with increasing salinity. About 20% of cobalt in seawater was estimated to be present as sulfate complexes. Soil mobility of cobalt is inversely related to the strength of adsorption by soil constituents. Although plants may take up cobalt from the soil, the translocation of cobalt from the roots to other parts of the plant is not significant.
Measured atmospheric concentrations of cobalt are approximately 1 ng/m3 or less in non-source areas and approximately 10 ng/m3 in source areas. Surface water and groundwater concentrations of stable cobalt are less than 1 µg/l in pristine areas and 1–10 µg/l in populated areas. Surface water and groundwater concentrations can be much higher in mining and agricultural areas, with values of up to several hundred milligrams per litre. Mean cobalt concentrations in seawater have been reported to be less than 1 µg/l. In rainwater, mean concentrations are 0.3–1.7 µg/l. The earth’s crust contains an average cobalt concentration of 20–25 mg/kg. Near some anthropogenic sources, the concentration of cobalt in soil may be several hundred milligrams per kilogram.
Cobalt is essential for nitrogen fixation by free-living bacteria, blue-green algae, and symbiotic systems (e.g. Rhizobium in the root nodules of legumes). Although cobalt is essential for animal nutrition, the metal is not required by animals in the ionic form. It is, however, a dietary essential element for ruminants and horses, in which it is incorporated into vitamin B12 molecules by gastrointestinal microbes.
A 96-h EC50 based on growth of the freshwater green alga Chlorella vulgaris was reported at 0.6 mg/l, whereas EC50s for vascular plants were 0.1 and 0.2 mg/l. The 5-day EC50 based on growth of the marine diatom Ditylum brightwellii was 0.3 mg/l. For freshwater invertebrates, acute LC50s (24–96 h) range from 1.1 to 239 mg/l. Several studies on Daphnia magna reproduction were reported, with a 21-day EC50 of 0.01 mg/l and a 28-day NOEC of 0.003 mg/l; however, later studies found 21-day NOECs ranging from 0.03 to 0.05 mg/l for varying levels of calcium carbonate. The lowest reported NOEC for aquatic organisms was for the water flea Ceriodaphnia dubia in a 7-day test, at <0.003 mg/l. The most sensitive marine invertebrates were lobster larvae, with 96-h LC50s ranging from 4.5 to 22.7 mg/l. Ninety-six-hour LC50s for freshwater fish range from 1.4 to 333 mg/l. A 16-day NOEC based on survival was reported at 0.06 mg/l. Test results for marine fish suggest that at least the species tested are relatively insensitive to cobalt, with 96-h LC50s ranging from 52.5 mg/l (as sulfate) to >1000 mg/l (as carbonate).
Ca2+ competition and dissolved organic matter complexation were the most important factors preventing Co2+ from binding at the gills in natural water tests. However, the effect of Ca2+ ions on the uptake and potential toxicity of cobalt occurs at very low Ca2+ concentrations, probably lower than those used in any of the reported toxicity tests. While the gill–cobalt binding model developed by Richards & Playle (1998) provides a basic framework, Niyogi & Wood (2004) suggest that a biotic ligand model should be developed to enhance the understanding of cobalt uptake by aquatic organisms. They recommended that future focus should be on correlating the model-simulated influence of water chemistry on gill cobalt accumulations with measured acute toxicity (96-h LC50) in fish under well defined water chemistry so as to quantify the critical gill cobalt burdens. Additional water chemistry variables such as alkalinity and Mg2+ should be investigated in this context. The approach could then be extended to model aquatic invertebrates such as daphnids.
Guidance values for cobalt toxicity in the marine and freshwater environments can be derived using a probabilistic approach, since the data set is sufficiently large to warrant it. Appendix 5 details the methodology used as an example.
For the marine environment, 12 toxicity values were chosen to derive a guidance value. The criteria for choosing the toxicity values and the values are presented in Appendix 5. These acute values have been converted to chronic estimates (see Table A5-1, Appendix 5). A moderate-reliability guidance value for the protection of 99% of marine species with 50% confidence was derived at 0.02 mg/l (20 µg/l) (see Figure A5-2, Appendix 5). A comparison of this value with environmental concentrations would suggest that effects are likely only in the vicinity of major anthropogenic releases.
For the freshwater environment, 28 data points were used in the derivation in the same way. For further details, the reader should refer to Appendix 5 and Table A5-2. A moderate-reliability guidance value for the protection of 95% of freshwater species with 50% confidence was derived at 0.008 mg/l (8 µg/l) (see Figure A5-3, Appendix 5). This value is based on mean values for studies carried out on the same species over the same test period but under differing test conditions. A comparison of this value with environmental concentrations would suggest that effects are likely only in the vicinity of major anthropogenic releases. There is some evidence that under conditions of extremely low Ca2+ ion concentrations, there is less competition for cobalt at binding sites and therefore greater uptake. Therefore, the greatest risk to aquatic organisms would be in very soft water areas (where the Ca2+ ion concentration is <10 mg/l) close to sources of anthropogenic release.
Data regarding the toxicity of cobalt to soil microorganisms are limited. There is little evidence of cobalt toxicity to plants due to elevated concentrations in soil. Cobalt tolerance, along with tolerance to other metals, has been found in plant populations growing on soils high in particular metals. Exclusion of the metal has been demonstrated in the cobalt tolerance of some species, whereas others growing on cobalt-rich copper clearings are hyperaccumulators of cobalt.
A significant adverse effect on the growth of earthworms (Eisenia foetida) was reported at 300 mg/kg; however, no effects were found at concentrations up to 91.9 mg/kg dry weight. Total inhibition of reproduction and ultimately 77% mortality were reported at 4720 mg/kg dry weight. The 28-day EC50, based on reproduction, for the springtail Folsomia candida was 409 mg/kg in standard field soil, whereas the 24-h LC50 for the free-living soil nematode Caenorhabditis elegans was 1210 mg/l based on the free cobalt ion.
In the terrestrial environment, adverse effects of cobalt on birds and wild mammals would appear unlikely, with cobalt deficiency in ruminants more likely than cobalt toxicosis.
The Australian protocol was used as an example of a probabilistic approach. Alternative probabilistic and deterministic approaches are available, such as those in the OECD guidelines, which might give different guidance values.
IARC (2005) evaluated the carcinogenic hazards of cobalt and cobalt compounds and concluded that:
The overall evaluation was that:
Abbasi SA, Nipaney PC, Soni R (1989) Environmental status of cobalt and its micro determination with 7-nitroso-8-hydroxyquinoline-5-sulfonic acid in waters, aquatic weeds and animal tissues. Analytical Letters, 22(1):225–235.
Abraham JL, Hunt A (1995) Environmental contamination by cobalt in the vicinity of a cemented tungsten carbide tool grinding plant. Environmental Research, 69:67–74.
ACGIH (1999) 1999 TLVs and BEIs. Threshold limit values for chemical substances and physical agents. Biological exposure indices. Cincinnati, OH, American Conference of Governmental Industrial Hygienists, 184 pp.
Adam C, Baudin JP, Garnier-Laplace J (2001) Kinetics of 110mAg, 60Co, 137Cs and 54Mn bioaccumulation from water and depuration by the crustacean Daphnia magna. Water, Air, and Soil Pollution, 125:171–188.
Adriano DC (1986) Trace elements in the terrestrial environment. New York, NY, Springer-Verlag.
Afeworki S, Chandravanshi BS (1987) Simultaneous determination of iron(III) and cobalt(II) with N-phenylcinnamohydroxamic acid and thiocyanate by extraction and spectrophotometry. Mikrochimica Acta, 92:143–152.
Ahluwalia AS, Kaur M (1988) Effect of some heavy metal compounds on growth and differentiation in a blue-green and a green alga. Microbios, 53:37–45.
Albrecht A (2003) Validating riverine transport and speciation models using nuclear reactor-derived radiocobalt. Journal of Environmental Radioactivity, 66:295–307.
Aldenberg T, Slob W (1993) Confidence limits for hazardous concentrations based on logistically distributed NOEC toxicity data. Ecotoxicology and Environmental Safety, 25:48–63.
Alexander CS (1969) Cobalt and the heart. Annals of Internal Medicine, 70:411–413.
Alexander CS (1972) Cobalt–beer cardiomyopathy: A clinical and pathological study of twenty-eight cases. American Journal of Medicine, 53:395–417.
Alexandersson R (1988) Blood and urinary concentrations as estimators of cobalt exposure. Archives of Environmental Health, 43(4):299–303.
Alomar A, Conde-Salazar L, Romaguera C (1985) Occupational dermatosis from cutting oils. Contact Dermatitis, 12:129–138.
Amiard JC (1976) Experimental study on the acute toxicity of cobalt, antimony, strontium and silver salts in some crustacea and their larvae and some teleostei. Revue Internationale d’Océanographie Médicale, 43:79–95.
Amiard JC, Amiard-Triquet C (1979) Distribution of cobalt 60 in a mollusc, a crustacean and freshwater teleost: Variations as a function of the source of pollution and during elimination. Environmental Pollution, 20(3):199–213.
Amundsen CE, Hanssen JE, Semb A, Steinnes E (1992) Long-range atmospheric transport of trace elements to southern Norway. Atmospheric Environment, 26A(7):1309–1324.
Anard D, Kirsch-Volders M, Elhajouji A, Belpaeme K, Lison D (1997) In vitro genotoxic effects of hard metal particles assessed by alkaline single cell gel and elution assays. Carcinogenesis, 18(1):177–184.
Andersen O (1983) Effects of coal combustion products and metal compounds on sister chromatid exchange (SCE) in a macrophage like cell line. Environmental Health Perspectives, 47:239–253.
Anderson MB, Pedigo NG, Katz RP, George WJ (1992) Histopathology of testes from mice chronically treated with cobalt. Reproductive Toxicology, 6:41–50.
Anderson MB, Lepak K, Farinas V, George WJ (1993) Protective action of zinc against cobalt-induced testicular damage in the mouse. Reproductive Toxicology, 7:49–54.
Andre S, Metivier H, Masse R (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part III: Lung clearance of inhaled cobalt oxide particles in baboons. Journal of Aerosol Science, 20(2):205–217.
Anthoine D, Petiet G, Wurtz MC, Simon B, Stefani F, François MC (1982) [Hard metal pulmonary fibroses and their distribution in France.] Médecine et Hygiene, 40:4280–4286 (in French).
ANZECC/ARMCANZ (2000) Australian and New Zealand guidelines for fresh and marine water quality. Canberra, Australian and New Zealand Environment Conservation Council, Agriculture and Resource Management Council of Australia and New Zealand, National Water Quality Management Strategy (http://www.deh.gov.au/water/publications/index.html).
Apostoli P, Porru S, Alessio L (1994) Urinary cobalt excretion in short time occupational exposure to cobalt powders. Science of the Total Environment, 150:129–132.
Arimoto R, Duce RA, Ray BJ, Uni CK (1985) Atmospheric trace elements at Enewetak Atoll: 2. Transport to the ocean by wet and dry deposition. Journal of Geophysical Research, 90(D1):2391–2408.
Arlauskas A, Baker RS, Bonin AM, Tandon RK, Crisp PT, Ellis J (1985) Mutagenicity of metal ions in bacteria. Environmental Research, 36:379–388.
Asmuß M, Mullenders LH, Hartwig A (2000) Interference by toxic metal compounds with isolated zinc finger DNA repair proteins. Toxicology Letters, 112–113:227–231.
ATSDR (1995) Public health assessment, Blackbird Mine, Cobalt, Lemhi County, Idaho. CERCLIS No. IDD980725832. Atlanta, GA, United States Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry, 12 January (http://www.atsdr.cdc.gov/HAC/PHA/blackbird/bla_toc.html).
ATSDR (2004) Toxicological profile for cobalt. Atlanta, GA, United States Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry.
Ayala-Fierro F, Firriolo JM, Carter DE (1999) Disposition, toxicity, and intestinal absorption of cobaltous chloride in male Fischer 344 rats. Journal of Toxicology and Environmental Health, Part A, 56:571–591.
Badsha KS, Goldspink CR (1988) Heavy metal levels in three species of fish in Tjeukemeer, a Dutch polder lake. Chemosphere, 17(2):459–463.
Baes CF, Sharp RD (1983) A proposal for estimation of soil leaching and leaching constants for use in assessment models. Journal of Environmental Quality, 12(1):17–28.
Bailey MR, Kreyling WG, Andre S, Batchelor A, Collier CG, Drosselmeyer E, Ferron GA, Foster P, Haider B, Hodgson A, Masse R, Métivier H, Morgan A, Müller H-L, Patrick G, Pearman I, Pickering S, Ramsden D, Stirling C, Talbot RJ (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part 1: Objectives and summary of results. Journal of Aerosol Science, 20(2):169–188.
Baker AJM, Brooks RR, Pease AJ, Malaisse F (1983) Studies on copper and cobalt tolerance in three closely related taxa within the genus Silene L. (Caryophyllaceae) from Zaire. Plant and Soil, 73:377–385.
Balmes JR (1987) Respiratory effects of hard-metal dust exposure. Occupational Medicine, 2:327–344.
Barceloux DG (1999) Cobalt. Clinical Toxicology, 37(2):201–216.
Bargagli R (2000) Trace metals in Antarctica related to climate changes and increasing human impact. Reviews of Environmental Contamination and Toxicology, 166:129–173.
Bargagli R, Barghigiani C, Siegel BZ, Siegel SM (1991) Trace metal anomalies in surface soils and vegetation on two active island volcanos: Stromboli and Vulcano (Italy). Science of the Total Environment, 102:209–222.
Barnaby CF, Smith T, Thompson BD (1968) Dosimetry of the radioisotopes of cobalt. Physics in Medicine and Biology, 13(3):421–433.
Barnes JE, Kanapilly GM, Newton GJ (1976) Cobalt-60 oxide aerosols: Methods of production and short-term retention and distribution kinetics in the beagle dog. Health Physics, 30:391–398.
Baudin JP, Fritsch AF (1989) Relative contributions of food and water in the accumulation of 60Co by a freshwater fish. Water Research, 23(7):817–823.
Baudouin MF, Scoppa P (1974) Acute toxicity of various metals to freshwater zooplankton. Bulletin of Environmental Contamination and Toxicology, 12(6):745–751.
Bech AO, Kipling MD, Heather JC (1962) Hard metal disease. British Journal of Industrial Medicine, 19:239–252.
Behrooz A, Ismail-Beigi F (1997) Dual control of glut1 glucose transporter gene expression by hypoxia and by inhibition of oxidative phosphorylation. Journal of Biological Chemistry, 272(9):5555–5562.
Beleznay E, Osvay M (1994) Long-term clearance of accidentally inhaled 60Co aerosols in humans. Health Physics, 66:392–399.
Bencko V, Wagner V, Wagnerova M, Reichrtova E (1983) Immuno-biochemical findings in groups of individuals occupationally and non-occupationally exposed to emissions containing nickel and cobalt. Journal of Hygiene, Epidemiology, Microbiology and Immunology, 27(4):387–394.
Benes P, Jurak M, Crenik M (1989a) Factors affecting interaction of radiocobalt with river sediments: II. Composition and concentration of sediment temperature. Journal of Radioanalytical and Nuclear Chemistry Letters, 132(2):225–239.
Benes P, Jurak M, Kunkova M (1989b) Factors affecting interaction of radiocobalt with river sediments: I. pH and composition of water and contact time. Journal of Radioanalytical and Nuclear Chemistry Letters, 132(2):209–223.
Biego GH, Joyeux M, Hartemann P, Debry G (1998) Daily intake of essential minerals and metallic micropollutants from foods in France. Science of the Total Environment, 217:27–36.
Biesinger KE, Christensen GM (1972) Effects of various metals on survival, growth, reproduction, and metabolism of Daphnia magna. Journal of the Fisheries Research Board of Canada, 29:1691–1700.
Birge WJ, Black JA, Westerman AG (1979) Evaluation of aquatic pollutants using fish and amphibian eggs as bioassay organisms. In: Nielsen SW, Migaki G, Scarpelli DG, eds. Animals as monitors of environmental pollutants. Washington, DC, National Academy of Sciences, pp. 108–118.
Birge WJ, Black JA, Westerman AG, Hudson JE (1980) Aquatic toxicity tests on inorganic elements occurring in oil shale. In: Gale C, ed. Oil shale symposium. Sampling, analysis and quality assurance. Cincinnati, OH, United States Environmental Protection Agency, pp. 519–534 (EPA-600/9-80-022; NTIS PB80 221435).
Boikat U, Fink A, Bleck-Neuhaus J (1985) Cesium and cobalt transfer from soil to vegetation on permanent pastures. Radiation and Environmental Biophysics, 24:287–301.
Bonenfant JL, Auger C, Miller G, Chenard J, Roy PE (1969) Quebec beer-drinkers’ myocardosis: pathological aspects. Annals of the New York Academy of Sciences, 156(1):577–582.
Bouman AA, Platenkamp AJ, Posma FD (1986) Determination of cobalt in urine by flameless atomic absorption spectrometry. Comparison of direct analysis using Zeeman background correction and indirect analysis using extraction in organic solution. Annals of Clinical Biochemistry, 23:346–350.
Bourg WJ, Nation JR, Clark DE (1985) The effects of chronic cobalt exposure on passive-avoidance performance in the adult rat. Bulletin of the Psychonomic Society, 23(6):527–530.
Boutet C, Chaisemartin C (1973) Specific toxic properties of metallic salts in Austropotamobius pallipes pallipes and Orconectes limosus. Compte Rendu des Séances de la Société de Biologie (Paris), 167(12):1933–1938.
Bowen HJM (1966) Trace elements in biochemistry. New York, NY, Academic Press.
Brooks RR (1977) Copper and cobalt uptake by Haumaniastrum species. Plant and Soil, 48:541–544.
Brooks SC, Herman JS, Hornberger GM, Mills AL (1998) Biodegradation of cobalt–citrate complexes: Implications for cobalt mobility in groundwater. Journal of Contaminant Hydrology, 32:99–115.
Brügmann L (1988) Some peculiarities of the trace-metal distribution in Baltic waters and sediments. Marine Chemistry, 23:425–440.
Bruland KW, Donat JR, Hutchins DA (1991) Interactive influences of bioactive trace metals on biological production in oceanic waters. Limnology and Oceanography, 36(8):1555–1577.
Brune D, Kjaerheim A, Paulsen G, Beltesbrekke H (1980) Pulmonary deposition following inhalation of chromium–cobalt grinding dust in rats and distribution in other tissues. Scandinavian Journal of Dental Research, 88:543–551.
Bucher JR, Elwell MR, Thompson MB, Chou BJ, Renne R, Ragan HA (1990) Inhalation toxicity studies of cobalt sulfate in F344/N rats and B6C3F1 mice. Fundamental and Applied Toxicology, 15:357–372.
Bucher JR, Hailey JR, Roycroft JR, Haseman JK, Sills RC, Grumbein SL, Mellick PW, Chou BJ (1999) Inhalation toxicity and carcinogenicity studies of cobalt sulfate. Toxicological Sciences, 49:56–67.
Buchter B, Davidoff B, Amacher MC, Hinz C, Iskandar IK, Selim HM (1989) Correlation of Freundlich Kd and n retention parameters with soils and elements. Soil Science, 148(5):370–379.
Buikema AL, Cairns J, Sullivan GW (1984) Evaluation of Philodina acuticornis (Rotifera) as bioassay organisms for heavy metals. Water Resources Bulletin, 10:648–661.
Bunn HF, Gu J, Huang LE, Park JW, Zhu H (1998) Erythropoietin: A model system for studying oxygen-dependent gene regulation. Journal of Experimental Biology, 201:1197–1201.
Burba P, Rocha J, Klockow D (1994) Labile complexes of trace metals in aquatic humic substances: Investigations by means of an ion exchange-based flow procedure. Fresenius Journal of Analytical Chemistry, 349:800–807.
Burger J, Gochfeld M (1988) Metals in tern eggs in a New Jersey estuary: A decade of change. Environmental Monitoring and Assessment, 11:127–135.
Cameán A, Lopez-Artiguez M, Roca I, Herce-Pagliai C, Menendez M, Repetto M (1998) Determination of cobalt, manganese, and alcohol content in beers. Journal of Food Protection, 61(1):129–131.
Canterford GS, Canterford DR (1980) Toxicity of heavy metals to the marine diatom Ditylum brightwellii (West) Grunow: Correlation between toxicity and metal speciation. Journal of the Marine Biological Association of the United Kingdom, 60:227–242.
Capomazza C, Botta A (1991) Cobalt chloride induces micronuclei in human lymphocytes. Medical Science Research, 19:219–220.
Casarett LJ, Doull J (1986) Toxicology: The basic science of poisons, 3rd ed. New York, NY, Macmillan Publishing Company, pp. 56–57.
Chapman PM, Fairbrother A, Brown D (1998) A critical evaluation of safety (uncertainty) factors for ecological risk assessment. Environmental Toxicology and Chemistry, 17:99–108.
Chester R, Berry AS, Murphy KJT (1991) The distributions of particulate atmospheric trace metals and mineral aerosols over the Indian Ocean. Marine Chemistry, 34:261–290.
Christova T, Duridanova D, Braykova A, Setchenska M, Bolton T (2001) Heme oxygenase is the main protective enzyme in rat liver upon 6-day administration of cobalt chloride. Archives of Toxicology, 75(8):445–451.
Christova TY, Duridanova DB, Setchenska MS (2002) Enhanced heme oxygenase activity increases the antioxidant defense capacity of guinea pig liver upon acute cobalt chloride loading: comparison with rat liver. Comparative Biochemistry and Physiology C — Pharmacology, Toxicology & Endocrinology, 131(2):177–184.
Cikrt M, Tich M (1981) Biliary excretion of cobalt in rats. Journal of Hygiene, Epidemiology, Microbiology, and Immunology, 25(4):364–368.
Clyne N, Hofman-Bang C, Haga Y, Hatori N, Marklund SL, Pehrsson SK, Wibom R (2001) Chronic cobalt exposure affects antioxidants and ATP production in rat myocardium. Scandinavian Journal of Clinical and Laboratory Investigation, 61(8):609–614.
Coakley JP, Nagy E, Serodes JB (1993) Spatial and vertical trends in sediment-phase contaminants in the upper estuary of the St. Lawrence River. Estuaries, 16(3B):653–669.
Cobalt Development Institute (undated a) About cobalt. Guildford, Surrey, Cobalt Development Institute (http://www.thecdi.com/general.php?r=aboutcobalt).
Cobalt Development Institute (undated b) Cobalt facts. Guildford, Surrey, Cobalt Development Institute (http://www.thecdi.com/cobaltfacts.php).
Cobalt Development Institute (2003) Cobalt occurrence, supply and demand. Guildford, Surrey, Cobalt Development Institute.
Cobalt Development Institute (2004) Cobalt supply & demand 2004. Guildford, Surrey, Cobalt Development Institute (http://www.thecdi.com/cdi/images/documents/facts/Cobalt_Facts_Supply-Demand_000.pdf).
Coleman RD, Coleman RL, Rice EL (1971) Zinc and cobalt bioconcentration and toxicity in selected algal species. Botanical Gazette, 132(2):102–109.
Collecchi P, Esposito M, Brera S, Mora E, Mazzucotelli A, Oddone M (1986) The distribution of arsenic and cobalt in patients with laryngeal carcinoma. Journal of Applied Toxicology, 6(4):287–289.
Collier CG, Bailey MR, Hodgson A (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part V: Lung clearance of inhaled cobalt oxide particles in hamsters, rats and guinea-pigs. Journal of Aerosol Science, 20(2):233–247.
Collier CG, Hodgson A, Gray SA, Moody JC, Ball A (1991) The lung clearance kinetics of 57Co3O4. Journal of Aerosol Science, 22(4):537–549.
Comhaire S, Blust R, Vanginneken L, Vanderborght OLJ (1994) Cobalt uptake across the gills of the common carp, Cyprinus carpio, as a function of calcium concentration in the water of acclimation and exposure. Comparative Biochemistry and Physiology C — Pharmacology, Toxicology & Endocrinology, 109(1):63–76.
Corisco JAG, Carreiro MCV (1999) Co-60 transfer from water to the freshwater planktonic algae Selenastrum capricornutum Prinz. In: Anagnostoopoulos P, Brebbia CA, eds. Water pollution V: Modeling, measuring, and prediction. Boston, MA, WIT Press, pp. 427–436 (Progress in Water Resources, Vol. 1).
Corrier DE, Mollenhauer HH, Clark DE, Hare MF, Elissalde MH (1985) Testicular degeneration and necrosis induced by dietary cobalt. Veterinary Pathology, 22:610–616.
Costa M, Heck JD, Robison S (1982) Selective phagocytosis of crystalline metal sulfide particles and DNA strand breaks as a mechanism for the induction of cellular transformation. Cancer Research, 42:2757–2763.
Cox RM, Hutchinson TC (1979) Metal co-tolerances in the grass Deschampsia cespitosa. Nature, 297:231–233.
Cugell DW (1992) The hard metal diseases. Clinical Chest Medicine, 13:269–279.
Curtis MW, Ward CH (1981) Aquatic toxicity of forty industrial chemicals: testing in support of hazardous substance spill prevention regulation. Journal of Hydrology, 51:359–367.
Cyr F, Mehra MC, Mallet VN (1987) Leaching of chemical contaminants from a municipal landfill site. Bulletin of Environmental Contamination and Toxicology, 38:775–782.
Czyscinski KS, Pietrzak RF, Weiss AJ (1982) Evaluation of isotope migration — land burial: Water chemistry at commercially operated low-level radioactive waste disposal sites. Washington, DC, Nuclear Regulatory Commission, Office of Nuclear Regulatory Research (NTIS/NUREG/CR-2124).
Dabeka RW, McKenzie AD (1995) Survey of lead, cadmium, fluoride, nickel, and cobalt in food composites and estimation of dietary intakes of these elements by Canadians in 1986–1988. Journal of AOAC International, 78(4):897–909.
Daghman NA, Elder GE, Savage GA, Winter PC, Maxwell AP, Lappin TR (1999) Erythropoietin production: evidence for multiple oxygen sensing pathways. Annals of Hematology, 78:275–278.
Dalvi RR, Robbins TJ (1978) Comparative studies on the effect of cadmium, cobalt, lead, and selenium on hepatic microsomal monooxygenase enzymes and glutathione levels in mice. Journal of Environmental Pathology and Toxicology, 1:601–607.
Das BK, Kaviraj A (1994) Individual and interactive lethal toxicity of cadmium, potassium permanganate and cobalt chloride to fish, worm and plankton. Geobios, 21(4):223–227.
Dasch JM, Wolff GT (1989) Trace inorganic species in precipitation and their potential use in source apportionment studies. Water, Air, and Soil Pollution, 43:401–412.
Dave G, Xiu RQ (1991) Toxicity of mercury, copper, nickel, lead, and cobalt to embryos and larvae of zebrafish, Brachydanio rerio. Archives of Environmental Contamination and Toxicology, 21(1):126–134.
Davis JE, Fields JP (1958) Experimental production of polycythemia in humans by administration of cobalt chloride. Proceedings of the Society for Experimental Biology and Medicine, 99:493–495.
De Boeck M, Lison D, Kirsh-Volders M (1998) Evaluation of the in vitro direct and indirect genotoxic effects of cobalt compounds using the alkaline comet assay. Influence of interdonor and interexperimental variability. Carcinogenesis, 19:2021–2129.
De Boeck M, Lardau S, Buchet JP, Kirsch-Volders M, Lison D (2000) Absence of significant genotoxicity in lymphocytes and urine from workers exposed to moderate levels of cobalt-containing dust: a cross-sectional study. Environmental and Molecular Mutagenesis, 36(2):151–160.
De Boeck M, Hoet P, Lombaert N, Nemery B, Kirsch-Volders M, Lison D (2003) In vivo genotoxicity of hard metal dust: Induction of micronuclei in rat type II epithelial lung cells. Carcinogenesis, 24:1793–1800.
Deka NC, Sehgal AK, Chhuttani PN (1981) Absorption and transport of radioactive 57cobalt vitamin B12 in experimental giardiasis in rats. Indian Journal of Medical Research, 74:675–679.
de Matteis F, Gibbs AH (1977) Inhibition of haem synthesis caused by cobalt in rat liver. Biochemical Journal, 162:213–216.
Diamond JM, Winchester EL, Mackler DG, Rasnake WJ, Fanelli JK, Gruber D (1992) Toxicity of cobalt to fresh-water indicator species as a function of water hardness. Aquatic Toxicology, 22(3):163–179.
Diaz GJ, Julian RJ, Squires EJ (1994) Lesions in broiler chickens following experimental intoxication with cobalt. Avian Diseases, 38(2):308–316.
Di Guilio C, Huang WX, Lahiri S, Mokashi A, Buerk DG (1990) Cobalt stimulates carotid body chemoreceptors. Journal of Applied Physiology, 68(5):1844–1849.
Di Giulio C, Data PG, Lahiri S (1991) Chronic cobalt causes hypertrophy of glomus cells in the rat carotid body. American Journal of Physiology, Cell Physiology, 261:102–105.
Domingo JL, Llobet JM, Bernat R (1984) A study of the effects of cobalt administered orally to rats. Archivos de Farmacologia y Toxicologia, 10:13–20.
Domingo JL, Paternain JL, Llobet JM, Corbella J (1985) Effects of cobalt on postnatal development and late gestation in rats upon oral administration. Revista Espanola de Fisiologia, 41:293–298.
Donaldson JD (1986) Cobalt in the environment. In: Proceedings of the first congress on cobalt and the environment, Toronto, Ontario, 2–3 April 1986. Guildford, Surrey, Cobalt Development Institute, pp.1–21.
Donaldson JD, Clark SJ, Grimes SM (1986) Cobalt in medicine, agriculture and environment. Guildford, Surrey, Cobalt Development Institute.
Dooms-Goossens A, Ceuterick A, Vanmaele N, Degreef H (1980) Follow-up study of patients with contact dermatitis caused by chromates, nickel, and cobalt. Dermatologica, 160:249–260.
Dorfman D (1977) Tolerance of Fundulus heteroclitus to different metals in salt waters. Bulletin of the New Jersey Academy of Sciences, 22(2):21–23.
Eaton RP, Pommer I (1973) Glucagon secretion and activity in the cobalt chloride-treated rat. American Journal of Physiology, 225:67–72.
Eckel WP, Jacob TA (1988) Ambient levels of 24 dissolved metals in U.S. surface and ground waters. In: Proceedings of the 196th meeting of the American Chemical Society, Division of Environmental Chemistry. New York, NY, American Chemical Society, pp. 317–372.
Elbetieha A, Al-Thani AS, Al-Thani RK, Darmani H, Owais W (2004) Chronic exposure to cobaltous chloride caused adverse effects on fertility of male mice. Toxicology and Applied Pharmacology, 197(3):351 (abstract).
El-Nady FE, Atta MM (1996) Toxicity and bioaccumulation of heavy metals to some marine biota from the Egyptian coastal waters. Journal of Environmental Science and Health, Part A, Environmental Science and Engineering & Toxic and Hazardous Substance Control, 31(7):1529–1545.
Espinosa AJF, Ternero-Rodriguez M, Barragan de la Rosa FJ, Jimenez-Sanchez JC (2001) Size distribution of metals in urban aerosols in Seville (Spain). Atmospheric Environment, 35(14):2595–2601.
EVM (2002) Review of cobalt. Expert Group on Vitamins and Minerals Secretariat, revised August (EVM/00/07; http://www.foodstandards.gov.uk/multimedia/pdfs/evm0007p.pdf).
Ewell WS, Gorsuch JW, Kringle RO, Robillard KA, Spiegel RC (1986) Simultaneous evaluation of the acute effects of chemicals on seven aquatic species. Environmental Toxicology and Chemistry, 5(9):831–840.
Eyrolle F, Charmasson S (2001) Distribution of organic carbon, selected stable elements and artificial radionuclides among dissolved, colloidal and particulate phases in the Rhone River (France): preliminary results. Journal of Environmental Radioactivity, 55(2):145–155.
FDRL (1984a) Acute oral LD50 study of cobalt sulphate lot no. S88336/A in Sprague-Dawley rats. Waverly, NY, Food and Drug Research Laboratories, Inc., 11 April 1984 (FDRL Study No. 8005D).
FDRL (1984b) Study of cobalt (II) carbonate tech gr. CoCO3, lot #030383 in Sprague-Dawley rats. Waverly, NY, Food and Drug Research Laboratories, Inc., 12 April 1984.
FDRL (1984c) Acute oral toxicity study of cobalt oxide tricobalt tetraoxide in Sprague-Dawley rats. Waverly, NY, Food and Drug Research Laboratories, Inc., 5 April 1984.
Feng MR, Rossi DT, Strenkoski C, Black A, Dehart P, Lovdahl M, McNally W (1998) Disposition kinetics of cobalt mesoporphyrin in mouse, rat, monkey and dog. Xenobiotica, 28(4):413–426.
Ferrans VJ, Hibbs RG, Weilbaecher DG (1964) Alcoholic cardiomyopathy: a histochemical and electron microscopic study. American Journal of Cardiology, 13:106–107.
Finney BP, Huh C-A (1989) History of metal pollution in the southern California bight: An update. Environmental Science & Technology, 23:294–303.
FIOH (1999) [Biomonitoring of exposure to chemicals 2000. Guidelines for sample collection.] Helsinki, Finnish Institute of Occupational Health, 128 pp. (in Finnish).
Firriolo JM, Ayala-Fierro F, Sipes IG, Carter DE (1999) Absorption and disposition of cobalt naphthenate in rats after a single oral dose. Journal of Toxicology and Environmental Health A, 58:383–395.
Fischer E, Molnár L (1997) Growth and reproduction of Eisenia fetida (Oligochaeta, Lumbricidae) in semi-natural soil containing various metal chlorides. Soil Biology and Biochemistry, 29:667–670.
Fischer T, Rystedt I (1983) Cobalt allergy in hard metal workers. Contact Dermatitis, 9:115–121.
Fishman MJ, Perryman GR, Schroder LJ, Matthews EW (1986) Determination of trace metals in low ionic strength waters using Zeeman and deuterium background correction for graphite furnace absorption spectrometry. Journal of the Association of Official Analytical Chemists, 69(4):704–708.
Forbes RM, Cooper AR, Mitchell HH (1954) On the occurrence of beryllium, boron, cobalt, and mercury in human tissues. Journal of Biological Chemistry, 209:857–865.
Foster PP, Pearman I, Ramsden D (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part II: Lung clearance of inhaled cobalt oxide in man. Journal of Aerosol Science, 20(2):189–204.
Francis CW, Davis EC, Goyert JC (1985) Plant uptake of trace elements from coal gasification ashes. Journal of Environmental Quality, 14(4):561–569.
Frank A, McPartlin J, Danielsson R (2004) Nova Scotia moose mystery — a moose sickness related to cobalt and vitamin B12 deficiency. Science of the Total Environment, 318:89–100.
Friedman HA, Kelmers AD (1988) Investigation of leaching of radionuclides and hazardous materials from low-level wastes at Oak Ridge National Laboratory. Washington, DC, United States Department of Energy (NTIS/DE87013363).
Fukunaga M, Kurachi Y, Mizuguchi Y (1982) Action of some metal ions on yeast chromosomes. Chemical & Pharmaceutical Bulletin, 30(8):3017–3019.
Gaur JP, Noraho N, Cauhan YS (1994) Relationship between heavy metal accumulation and toxicity in Spirodella polyrhiza and Azolla pinnata. Aquatic Botany, 49:183–192.
Gennart JP, Baleux C, Verellen-Dumoulin C, Buchet JP, De Meyer R, Lauwerys R (1993) Increased sister chromatid exchanges and tumor markers in workers exposed to elemental chromium-, cobalt- and nickel-containing dusts. Mutation Research, 299:55–61.
Gerhardsson L, Wester PO, Nordberg GF, Brune D (1984) Chromium, cobalt and lanthanum in lung, liver and kidney tissue from deceased workers. Science of the Total Environment, 37:233–246.
Gibbs RJ (1994) Metals in the sediments along the Hudson River estuary. Environment International, 20(4):507–516.
Gladstones JS, Loneragan JF, Goodchild NA (1977) Field responses to cobalt and molybdenum by different legume species, with inferences on the role of cobalt in legume growth. Australian Journal of Agricultural Research, 28(4):619–628.
Glooschenko WA, Capocianco J, Coburn J, Glooschenko V (1981) Geochemical distribution of trace metals and organochlorine contaminants of a Lake Ontario shoreline marsh. Water, Air, and Soil Pollution, 15:197–213.
Goh CL, Gan SL, Ngui SJ (1986) Occupational dermatitis in a prefabrication construction factory. Contact Dermatitis, 15:235–240.
Goldberg MA, Schneider TJ (1994) Similarities between the oxygen-sensing mechanisms regulating the expression of vascular endothelial growth factor and erythropoietin. Journal of Biological Chemistry, 269(6):4355–4359.
Goldberg MA, Dunning SP, Bunn HF (1988) Regulation of the erythropoietin gene: Evidence that the oxygen sensor is a heme protein. Science, 242:1412–1415.
Golomb D, Ryan D, Eby N, Underhill J, Zemba S (1997) Atmospheric deposition of toxics onto Massachusetts Bay — I. Metals. Atmospheric Environment, 31(9):1349–1359.
Greathouse DG, Craun GF (1978) Cardiovascular disease study — occurrence of inorganics in household tap water and relationships to cardiovascular mortality rates. In: Proceedings of the 12th annual conference on trace substances in environmental health. Columbia, MO, University of Missouri, pp. 31–39.
Greenberg DM, Copp DH, Cuthbertson EM (1943) Studies in mineral metabolism with the aid of artificial radioactive isotopes: VII. The distribution and excretion, particularly by way of the bile, of iron, cobalt, and manganese. Journal of Biological Chemistry, 147:749–756.
Gregus Z, Klaassen CD (1986) Disposition of metals in rats: A comparative study of fecal, urinary, and biliary excretion and tissue distribution of eighteen metals. Toxicology and Applied Pharmacology, 85:24–38.
Greig RA, Jones J (1976) Nondestructive neutron activation analysis of marine organisms collected from ocean dump sites of the middle eastern United States. Archives of Environmental Contamination and Toxicology, 4(4):420–434.
Grice HC, Goodman T, Munro IC, Wiberg GS, Morrison AB (1969) Myocardial toxicity of cobalt in the rat. Annals of the New York Academy of Sciences, 156:189–194.
Haddad E, Zikovsky L (1985) Determination of Al, As, Cr, Cs, Fe, Mn, Sb, Sc, W and Zn in the workroom air by instrumental neutron activation analysis. Journal of Radioanalytical and Nuclear Chemistry Letters, 93(6):371–378.
Hamilton EI (1994) The geobiochemistry of cobalt. Science of the Total Environment, 150:7–39.
Hamilton-Koch W, Snyder RD, Lavelle JM (1986) Metal-induced DNA damage and repair in human diploid fibroblasts and Chinese hamster ovary cells. Chemico-Biological Interactions, 59:17–28.
Hanna RGM (1992) The level of heavy metals in the Red Sea after 50 years. Science of the Total Environment, 125:417–448.
Hansen JA, Marr JCA, Lipton J, Cacela D, Bergman HL (1999) Differences in neurobehavioral responses of chinook salmon (Oncorhynchus tshawytscha) and rainbow trout (Oncorhynchus mykiss) exposed to copper and cobalt: Behavioral avoidance. Environmental Toxicology and Chemistry, 18(9):1972–1978.
Hansson H-C, Ekholm A-KP, Ross HB (1988) Rainwater analysis: A comparison between proton-induced x-ray emission and graphite furnace atomic absorption spectroscopy. Environmental Science & Technology, 22:527–531.
Harp MJ, Scoular FI (1952) Cobalt metabolism of young college women on self-selected diets. Journal of Nutrition, 47:67–72.
Hartenstein R, Neuhauser EF, Narahara A (1981) Effects of heavy metal and other elemental additives to activated sludge on growth of Eisenia foetida. Journal of Environmental Quality, 10:372–376.
Hartung M (1986) Lungenfibrosen bei Hartmetallschleifern. Bedeutung der Cobalteinwirkung. Sankt Augustin, Hauptverband der gewerblichen Berufsgenossenschaften.
Hartwig A, Snyder RD, Schlepegrell R, Beyersmann D (1991) Modulation by Co(II) of UV-induced DNA repair, mutagenesis and sister-chromatid exchanges in mammalian cells. Mutation Research, 248:177–185.
Hatori N, Pehrsson SK, Clyne N, Hansson G, Hofman-Bang C, Marklund SL, Ryden L, Sjoqvist PO, Svensson L (1993) Acute exposure and oxygen radical scavengers in the rat myocardium. Biochimica et Biophysica Acta, 1181:257–260.
Heath JC (1954) Cobalt as a carcinogen. Nature, 173:822–823.
Heath JC (1956) The production of malignant tumours by cobalt in the rat. British Journal of Cancer, 10:668–673.
Heath JC (1960) The histogenesis of malignant tumors induced by cobalt in the rat. British Journal of Cancer, 15:478–482.
Heath JC, Daniel MR (1962) The production of malignant tumors by cobalt in the rat: Intrathoracic tumors. British Journal of Cancer, 16:473–478.
Heath JC, Freeman MA, Swanson SA (1971) Carcinogenic properties of wear particles from prostheses made in cobalt–chromium alloy. Lancet, 1(7699):564–566.
Heaton RW, Rahn KA, Lowenthal DH (1990) Determination of trace elements, including regional tracers, in Rhode Island precipitation. Atmospheric Environment, 24A(1):147–153.
Heinrich R, Angerer J (1984) Determination of cobalt in biological materials by voltammetry and electrothermal atomic absorption spectrometry. International Journal of Environmental Analytical Chemistry, 16:305–314.
Hellou J, Fancey LL, Payne JF (1992) Concentrations of twenty-four elements in bluefin tuna, Thunnus thynnus from the northwest Atlantic. Chemosphere, 24(2):211–218.
Helmers E, Schrems O (1995) Wet deposition of metals to the tropical north and the south Atlantic Ocean. Atmospheric Environment, 29(18):2475–2484.
Hengstler JG, Bolm-Audorff U, Faldum A, Janssen K, Reifenrath M, Gotte W, Jung D, Mayer-Popken O, Fuchs J, Gebhard S, Bienfait HG, Schlink K, Dietrich C, Faust D, Epe B, Oesch F (2003) Occupational exposure to heavy metals: DNA damage induction and DNA repair inhibition prove co-exposures to cadmium, cobalt and lead as more dangerous than hitherto expected. Carcinogenesis, 24:63–73.
Henquin J-C, Lambert AE (1975) Cobalt inhibition of insulin secretion and calcium uptake by isolated rat islets. American Journal of Physiology, 228(6):1669–1677.
Henquin J-C, Schmeer W, Meissner HP (1983) Forskolin, an activator of adenylate cyclase, increases Ca2+-dependent electrical activity induced by glucose in mouse pancreatic B cells. Endocrinology, 112(6):2218–2220.
Hewitt PJ (1988) Accumulation of metals in the tissues of occupationally exposed workers. Environmental Geochemistry and Health, 10(3–4):113–116.
Hill CH (1974) Influence of high levels of minerals on the susceptibility of chicks to Salmonella gallinarum. Journal of Nutrition, 104(10):1221–1226.
Hillerdal G, Hartung M (1983) Short communication on cobalt in tissues from hard metal workers. International Archives of Occupational and Environmental Health, 53:89–90.
Ho VT, Bunn HF (1996) Effects of transition metals on the expression of the erythropoietin gene: further evidence that the oxygen sensor is a heme protein. Biochemical and Biophysical Research Communications, 223:175–180.
Hoet PMH, Roesems G, Demedts MG, Nemery B (2002) Activation of the hexose monophosphate shunt in rat type II pneumocytes as an early marker of oxidative stress caused by cobalt particles. Archives of Toxicology, 76(1):1–7.
Hogan GD, Rauser WE (1979) Tolerance and toxicity of cobalt, copper, nickel and zinc in clones of Agrostis gigantes. New Phytologist, 83:665–670.
Hogstedt C, Alexandersson R (1990) [Mortality among hard metal workers.] Solna, National Institute of Occupational Health, pp. 1–16 (Arbete och Hälsa 21) (in Swedish with English abstract).
Hollins JG, McCullough RS (1971) Radiation dosimetry of internal contamination by inorganic compounds of cobalt: An analysis of cobalt metabolism in rats. Health Physics, 21:233–246.
Holly RG (1955) Studies on iron and cobalt metabolism. Journal of the American Medical Association, 158:1349–1352.
Horwitz C, van der Linden SE (1974) Cadmium and cobalt in tea and coffee and their relationship to cardiovascular disease. South African Medical Journal, 48:230–233.
Houk AEH, Thomas AW, Sherman HC (1946) Some interrelationships of dietary iron, copper and cobalt in metabolism. Journal of Nutrition, 31:609–620.
IARC (1991) Chlorinated drinking-water; chlorination by-products; some other halogenated compounds; cobalt and cobalt compounds. Lyon, International Agency for Research on Cancer (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 52).
IARC (2005) Metallic cobalt particles. In: Cobalt in hard-metals and cobalt sulfate, gallium arsenide, indium phosphide and vanadium pentoxide (draft). Lyon, International Agency for Research on Cancer (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 86).
Ichikawa Y, Kusaka Y, Goto S (1985) Biological monitoring of cobalt concentrations in blood and urine. International Archives of Occupational and Environmental Health, 55:269–276.
ICRP (1979) Limits for intakes of radionuclides by workers. New York, NY, Pergamon Press on behalf of the International Commission on Radiological Protection (ICRP Publication 30; Annals of the ICRP, Vol. 2, No. 3/4).
ICRP (1994) Age-dependent doses to members of the public from intake of radionuclides: Part 2. Ingestion dose coefficients. New York, NY, Pergamon Press on behalf of the International Commission on Radiological Protection (ICRP Publication 67; Annals of the ICRP, Vol. 23, No. 3–4).
Ikarashi Y, Ohno K, Tsuchiya T, Nakamura A (1992a) Differences of draining lymph node cell proliferation among mice, rats and guinea pigs following exposure to metal allergens. Toxicology, 76:283–292.
Ikarashi Y, Tsuchiya T, Nakamura A (1992b) Detection of contact sensitivity of metal salts using the murine local lymph node assay. Toxicology Letters, 62:53–61.
Inaba J, Nishimura Y, Ichikawa R (1980) Comparative metabolism of 54Mn, 59Fe, 60Co and 65Zn incorporated into Chlorella and in inorganic form in rats. Health Physics, 39:611–617.
IPCS (2000) International Chemical Safety Card — Cobalt naphthenate. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1093; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc10/icsc1093.htm).
IPCS (2001a) International Chemical Safety Card — Cobalt(II) nitrate hexahydrate. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 0784; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc07/icsc0784.htm).
IPCS (2001b) International Chemical Safety Card — Cobalt sulfate. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1127; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc11/icsc1127.htm).
IPCS (2001c) International Chemical Safety Card — Cobalt(II) acetate tetrahydrate. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1128; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc11/icsc1128.htm).
IPCS (2001d) International Chemical Safety Card — Cobalt(II) sulfate heptahydrate. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1396; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc13/icsc1396.htm).
IPCS (2001e) International Chemical Safety Card — Cobalt(II) nitrate. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1397; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc13/icsc1397.htm).
IPCS (2004a) International Chemical Safety Card — Cobalt. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 0782; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc07/icsc0782.htm).
IPCS (2004b) International Chemical Safety Card — Cobalt(II) chloride. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 0783; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc07/icsc0783.htm).
IPCS (2004c) International Chemical Safety Card — Cobalt(III) oxide. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 0785; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc07/icsc0785.htm).
IPCS (2004d) International Chemical Safety Card — Cobalt carbonyl. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 0976; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc09/icsc0976.htm).
IPCS (2004e) International Chemical Safety Card — Cobalt sulfide. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1529; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc15/icsc1529.htm).
IPCS (2004f) International Chemical Safety Card — Cobalt(II) oxide. Geneva, World Health Organization, International Programme on Chemical Safety (ICSC 1551; http://www.ilo.org/public/english/protection/safework/cis/products/icsc/dtasht/_icsc15/icsc1551.htm).
Ishihara N, Koizumi M, Yoshida A (1987) Metal concentrations in human pancreatic juice. Archives of Environmental Health, 42(6):356–360.
Ivancsits S, Diem E, Pilger A, Rudiger HW (2002) Induction of 8-hydroxy-2’-deoxyguanosine by cobalt(II) and hydrogen peroxide in vitro. Journal of Toxicology and Environmental Health, Part A, 65(9):665–676.
Jackman AP, Kennedy VC, Bhatia N (2001) Interparticle migration of metal cations in stream sediments as a factor in toxics transport. Journal of Hazardous Materials, B82:27–41.
Jansen HML, Knollema S, van der Duin LV, Willemsen AT, Wiersma A, Franssen EJ, Russel FG, Korf J, Paans AM (1996) Pharmacokinetics and dosimetry of cobalt-55 and cobalt-57. Journal of Nuclear Medicine, 37(12):2082–2086.
Jasmin G, Riopelle JL (1976) Renal carcinomas and erythrocytosis in rats following intrarenal injection of nickel subsulfide. Laboratory Investigations, 35:71–78.
Jenkins DW (1980) Biological monitoring of toxic trace metals: Vol. 1. Biological monitoring and surveillance. Washington, DC, United States Environmental Protection Agency, September (NTIS PB81-103475).
Johansson A, Curstedt T, Robertson B, Camner P (1984) Lung morphology and phospholipids after experimental inhalation of soluble cadmium, copper, and cobalt. Environmental Research, 34:295–309.
Johansson A, Robertson B, Camner P (1987) Nodular accumulation of type II cells and inflammatory lesions caused by inhalation of low cobalt concentrations. Environmental Research, 43:227–243.
Johansson A, Curstedt T, Camner P (1991) Lung lesions after combined inhalation of cobalt and nickel. Environmental Research, 54:24–38.
Johansson A, Curstedt T, Rasool O, Jarstrand C, Camner P (1992) Rabbit lung after combined exposure to soluble cobalt and trivalent chromium. Environmental Research, 58:80–96.
Jorhem L, Sundström B (1993) Levels of lead, cadmium, zinc, copper, nickel, chromium, manganese, and cobalt in foods on the Swedish market, 1983–1990. Journal of Food Composition and Analysis, 6:223–241.
Kadiiska MB, Maples KR, Mason RP (1989) A comparison of cobalt(II) and iron(II) hydroxyl and superoxide free radical formation. Archives of Biochemistry and Biophysics, 275(1):98–111.
Kanematsu N, Hara M, Kada T (1980) Rec assay and mutagenicity studies on metal compounds. Mutation Research, 77:109–116.
Kanerva L, Estlander T, Jolanki R (1988) Occupational skin disease in Finland. International Archives of Occupational and Environmental Health, 60:89–94.
Kasprzak KS, Zastawny TH, North SL, Riggs CW, Diwan BA, Rice JM, Dizdaroglu M (1994) Oxidative DNA base damage in renal, hepatic, and pulmonary chromatin of rats after intraperitoneal injection of cobalt(II) acetate. Chemical Research in Toxicology, 7:329–335.
Kasten U, Mullenders LH, Hartwig A (1997) Cobalt(II) inhibits the incision and the polymerization step of nucleotide excision repair in human fibroblasts. Mutation Research, 383:81–90.
Kawanishi S, Inoue S, Yamamoto K (1994) Active oxygen species in DNA damage induced by carcinogenic metal compounds. Environmental Health Perspectives, 102(Suppl. 3):17–20.
Kent NL, McCance RA (1941) The absorption and excretion of "minor" elements by man. Biochemical Journal, 35:877–883.
Kerfoot EJ (1975) Semi-chronic inhalation study on cobalt. Dissertation Abstracts International B, 35:6054–6055.
Kesteloot H, Roelandt J, Willems J, Claes JH, Joosens JV (1968) An inquiry into the role of cobalt in the heart disease of chronic beer drinkers. Circulation, 37:854–864.
Khangarot BS, Ray PK (1989a) Investigation of correlation between physicochemical properties of metals and their toxicity to the water flea Daphnia magna Straus. Ecotoxicology and Environmental Safety, 18(2):109–120.
Khangarot BS, Ray PK (1989b) Sensitivity of midge larvae of Chironomus tentans Fabricius (Diptera Chironomidae) to heavy metals. Bulletin of Environmental Contamination and Toxicology, 42:325–330.
Khangarot BS, Sehgal A, Bhasin MK (1985) "Man and biosphere" — Studies on the Sikkim Himalayas. Part 5: Acute toxicity of selected heavy metals on the tadpoles of Rana hexadactyla. Acta Hydrochimica et Hydrobiologica, 13(2):259–263.
Kharab P, Singh I (1985) Genotoxic effects of potassium dichromate, sodium arsenite, cobalt chloride and lead nitrate in diploid yeast. Mutation Research, 155:117–120.
Kieć-Swierczyńska M, Kręcisz B (2000) Occupational skin diseases among the nurses in the region of Lodz.
International Journal of Occupational Medicine and Environmental Health, 13(3):179–184.Kieć-Swierczyńska M, Kręcisz B (2002) Allergic contact dermatit
is in dentists and dental nurses. Exogenous Dermatology, 1(1):27–31.Killey RWD, McHugh JO, Champ DR, Cooper EL, Young JL (1984) Subsurface cobalt-60 migration from a low-level waste disposal site. Environmental Science & Technology, 18(3):148–157.
Kim EY, Goto R, Tanabe S, Tanaka H, Tatsukawa R (1998) Distribution of 14 elements in tissues and organs of oceanic seabirds. Archives of Environmental Contamination and Toxicology, 35(4):638–645.
Kimball G (1978) The effects of lesser known metals and one organic to fathead minnows (Pimephales promelas) and Daphnia magna. Minneapolis, MN, University of Minnesota, Department of Entomology, Fisheries and Wildlife (Report No. N: 88).
Kimberly MM, Bailey GG, Paschal DC (1987) Determination of urinary cobalt using matrix modification and graphite furnace atomic absorption spectrometry with Zeeman-effect background correction. Analyst, 112:287–290.
King LD (1988) Retention of metals by several soils of the southeastern United States. Journal of Environmental Quality, 17(2):239–246.
Kinoshita K, Fujita T (1972) Metabolism of 57Co-methylcobalamin in rat and guinea pig. Chemical and Pharmaceutical Bulletin, 20(12):2561–2569.
Kirchgessner M, Reuber S, Kreuzer M (1994) Endogenous excretion and true absorption of cobalt as affected by the oral supply of cobalt. Biological Trace Element Research, 41:175–189.
Kissa E, Moraitou-Apostolopoulou M, Kiortsis V (1984) Effects of four heavy metals on survival and hatching rate of Artemia salina (L.). Archiv für Hydrobiologie, 102(2):255–264.
Kloke A, Sauerbeck DR, Vetter H (1984) The contamination of plants and soils with heavy metals and the transport of metals in terrestrial food chains. In: Nriagu JO, ed. Changing metal cycles and human health. Berlin, Springer-Verlag, pp. 113–141.
Knauer GA, Martin JH, Gordon RM (1982) Cobalt in north-east Pacific waters. Nature, 297:49–51.
Knutson AB, Klerks PL, Levinton JS (1987) The fate of metal contaminated sediments in Foundry Cove, New York. Environmental Pollution, 45:291–304.
Kreyling WG, Ferron GA, Haider B (1984) The dependency of the lung retention on cobalt aerosol parameters. Journal of Aerosol Science, 15(3):229–232.
Kreyling WG, Ferron GA, Haider B (1986) Metabolic fate of inhaled Co aerosols in beagle dogs. Health Physics, 51(6):773–795.
Kreyling WG, Ferron GA, Haider B (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part IV: Lung clearance of inhaled cobalt oxide particles in beagle dogs. Journal of Aerosol Science, 20(2):219–232.
Krishnakumari L, Varshney PK, Gajbhiye SN, Govindan K, Nair VR (1983) Toxicity of some metals on the fish Therapon jarbua (Forsskal, 1775). Indian Journal of Marine Sciences, 12:64–66.
Kyono H, Kusaka Y, Homma K, Kubota H, Endo-Ichikawa Y (1992) Reversible lung lesions in rats due to short-term exposure to ultrafine cobalt particles. Industrial Health, 30:103–118.
Ladoux A, Frelin C (1994) Cobalt stimulates the expression of vascular endothelial growth factor and mRNA in rat cardiac cells. Biochemical and Biophysical Research Communications, 204(2):794–798.
Lantzy RJ, Mackenzie FT (1979) Atmospheric trace metals: Global cycles and assessment of man’s impact. Geochimica et Cosmochimica Acta, 43:511–525.
Lasfargues G, Wild P, Moulin JJ, Hammon B, Rosmorduc B, Rondeau du Noyer C, Lavandier M, Moline J (1994) Lung cancer mortality in a French cohort of hard-metal workers. American Journal of Industrial Medicine, 26:585–595.
Lee LH, Lustigman B, Chu IY, Hsu S (1992) Effect of lead and cobalt on the growth of Anacystis nidulans. Bulletin of Environmental Contamination and Toxicology, 48:230–236.
Legrum W, Stuehmeier G, Netter KJ (1979) Cobalt as a modifier of microsomal monooxygenases in mice. Toxicology and Applied Pharmacology, 48:195–204.
Lehninger AL (1982) Principles of biochemistry. New York, NY, Worth Publishers, pp. 361–466.
Lewis CPL, Demedts M, Nemery B (1991) Indices of oxidative stress in hamster lung following exposure to cobalt(II) ions: In vivo and in vitro studies. American Journal of Respiratory Cell and Molecular Biology, 5:163–169.
Lighthart B, Bond H, Ricard M (1977) Trace elements research using coniferous forest soil/litter microcosms. Washington, DC, United States Environmental Protection Agency (Report No. EPA-600/3-77-091).
Linna A, Oksa P, Palmroos P, Roto P, Laippala P, Uitti J (2003) Respiratory health of cobalt production workers. American Journal of Industrial Medicine, 44:124–132.
Lison D (1996) Human toxicity of cobalt-containing dust and experimental studies on the mechanism of interstitial lung disease (hard metal disease). Critical Reviews in Toxicology, 26:585–616.
Lison D, Carbonnelle P, Mollo L, Lauwerys R, Fubini B (1995) Physicochemical mechanism of the interaction between cobalt metal and carbide particles to generate toxic activated oxygen species. Chemical Research in Toxicology, 8:600–606.
Lison D, Lauwerys R, Demedts M, Nemery B (1996) Experimental research into the pathogenesis of cobalt/hard metal lung disease. European Respiratory Journal, 9:1024–1028.
Liu J, Reid RJ, Smith FA (2000) The mechanism of cobalt toxicity in mung beans. Physiologia Plantarum, 110(1):104–110.
Lloyd DR, Phillips DH, Carmichael PL (1997) Generation of putative intrastrand cross-links and strand breaks in DNA by transition metal ion-mediated oxygen radical attack. Chemical Research in Toxicology, 10:393–400.
Lock K, Because S, Criel P, Van Eeckhout H, Janssen CR (2004) Ecotoxicity of cobalt to the springtail Folsomia candida. Comparative Biochemistry and Physiology, Part C, Pharmacology, Toxicology & Endocrinology, 139:195–199.
Maenhaut W, Zoller WH, Duce RA, Hoffman GL (1979) Concentration and size distribution of particulate trace elements in the south polar atmosphere. Journal of Geophysical Research, 84:2421–2431.
MAFF (1997) 1994 Total Diet Study: Metals and other elements. London, United Kingdom Ministry of Agriculture, Food and Fisheries, Food Standards Agency (Food Surveillance Information Sheet No. 131; http://archive.food.gov.uk/maff/archive/food/infsheet/1997/no131/131tds.htm).
Mahara Y, Kudo A (1981) Interaction and mobility of cobalt-60 between water and sediments in marine environments possible effects by acid rain. Water Research, 15(4):413–419.
Malaisse F, Gregoire J, Morrison RS, Brooks RR, Reeves RD (1979) Copper and cobalt in vegetation of Fungurume, Shaba Province, Zaire. Oikos, 33:472–478.
Mantoura RFC, Dickson A, Riley JP (1978) The complexation of metals with humic materials in natural waters. Estuarine Coastal Shelf Science, 6:387–408.
Marcussen PV (1963) Cobalt dermatitis. Clinical picture. Acta Dermato-Venereologica, 43:231–234.
Marr JCA, Hansen JA, Meyer JS, Cacela D, Podrabsky T, Lipton J, Bergman HL (1998) Toxicity of cobalt and copper to rainbow trout: application of a mechanistic model for predicting survival. Aquatic Toxicology, 43(4):225–238.
Martin TR, Holdich DM (1986) The acute lethal toxicity of heavy metals to peracarid crustaceans (with particular reference to fresh-water asellids and gammarids). Water Research, 20(9):1137–1147.
Mascanzoni D (1989) Long-term transfer from soil to plant of radioactive corrosion products. Environmental Pollution, 57:49–62.
McLachlan J (1973) Growth media — marine. In: Stein JR, ed. Handbook of phycological methods, culture methods and growth measurements. Cambridge, Cambridge University Press, pp. 25–51.
McLaren RG, Lawson DM, Swift RS (1986) Sorption and desorption of cobalt by soils and soil components. Soil Sciences, 37:413–426.
Meachim G, Pedley RB, Williams DF (1982) A study of sarcogenicity associated with Co–Cr–Mo particles implanted in animal muscle. Journal of Biomedical Materials Research, 16:407–416.
Mejstrik V, Svacha J (1988) Concentrations of Co, Cd, Ni, and Zn in crop plants cultivated in the vicinity of coal-fired power plants. Science of the Total Environment, 72:57–67.
Memoli VA, Urban RM, Alroy J, Galante JO (1986) Malignant neoplasms associated with orthopedic implant materials in rats. Journal of Orthopaedic Research, 4: 346–355.
Meranger JC, Subramanian KS, Chalifoux C (1981) Metals and other elements: Survey for cadmium, cobalt, chromium, copper, nickel, lead, zinc, calcium, and magnesium in Canadian drinking water supplies. Journal of the Association of Official Analytical Chemists, 64(1):44–53.
Merian E (1985) Introduction on environmental chemistry and global cycles of chromium, nickel, cobalt, beryllium, arsenic, cadmium and selenium, and their derivatives. Current Topics in Environmental Toxicology and Chemistry, 8:3–32.
Mermut AR, Jain JC, Song L, Kerrich R, Kozak L, Jana S (1996) Trace element concentrations of selected soils and fertilizers in Saskatchewan, Canada. Journal of Environmental Quality, 25:845–853.
Milford JB, Davidson CI (1985) The size of particulate trace elements in the atmosphere — a review. Journal of the Air Pollution Control Association, 35(12):1249–1260.
Misra J, Pandey V, Singh N (1994) Effects of some heavy metals on root growth of germinating seeds of Vicia faba. Journal of Environmental Science and Health, Part A, Environmental Science and Engineering & Toxic and Hazardous Substance Control, 29(10):2229–2234.
Mitchell DF, Shankwalker GB, Shazer S (1960) Determining the tumorigenicity of dental materials. Journal of Dental Research, 39:1023–1028.
Moger WH (1983) Effects of the calcium-channel blockers cobalt, verapamil, and D600 on Leydig cell steroidogenesis. Biology of Reproduction, 28:528–535.
Mollenhauer HH, Corrier DE, Clark DE, Hare MF, Elissalde MH (1985) Effects of dietary cobalt on testicular structure. Virchows Archiv B: Cell Pathology Including Molecular Pathology, 49:241–248.
Moorhouse CP, Halliwell B, Grootveld M, Gutteridge JM (1985) Cobalt(II) ion as a promoter of hydroxyl radical and possible "crypto-hydroxyl" radical formation under physiological conditions. Differential effects of hydroxyl radical scavengers. Biochimica et Biophysica Acta, 843(3):261–268.
Morelli L, Di Giulio C, Iezzi M, Data PG (1994) Effect of acute and chronic cobalt administration on carotid body chemoreceptors responses. Science of the Total Environment, 150:215–216.
Morin Y, Daniel P (1967) Quebec beer-drinkers’ cardiomyopathy: etiological considerations. Canadian Medical Association Journal, 97:926–928.
Morin Y, Tetu A, Mercier G (1971) Cobalt cardiomyopathy: Clinical aspects. British Heart Journal, 33:175–178.
Morrison RS, Brooks RR, Reeves RDM, Malaisse F (1979) Copper and cobalt uptake by metallophytes from Zaire [short communication]. Plant and Soil, 53:535–539.
Morvai V, Szakmary E, Tatrai E, Ungvary G, Folly G (1993) The effects of simultaneous alcohol and cobalt chloride administration on the cardiovascular system of rats. Acta Physiologica Hungarica, 81(3):253–261.
Mosconi G, Bacis M, Vitali MT, Leghissa P, Sabbioni E (1994) Cobalt excretion in urine: Results of a study on workers producing diamond grinding tools and on a control group. Science of the Total Environment, 150:133–139.
Moulin JJ, Wild P, Mur JM, Fournier-Betz M, Mercier-Gallay M (1993) A mortality study of cobalt production workers: An extension of the follow-up. American Journal of Industrial Medicine, 23:281–288.
Moulin JJ, Wild P, Romazini S, Lasfargues G, Peltier A, Bozec C, Deguerry P, Pellet F, Perdrix A (1998) Lung cancer risk in hard-metal workers. American Journal of Epidemiology, 148(3):241–248.
Munita CS, Mazzilli BP (1986) Determination of trace elements in Brazilian cigarette tobacco by neutron activation analysis. Journal of Radioanalytical and Nuclear Chemistry Letters, 108(4):217–227.
Mur JM, Moulin JJ, Charruyer-Seinerra MP, Lafitte J (1987) A cohort mortality study among cobalt and sodium workers in an electrochemical plant. American Journal of Industrial Medicine, 11:75–81.
Muramatsu Y, Parr RM (1988) Concentrations of some trace elements in hair, liver and kidney from autopsy subjects — relationship between hair and internal organs. Science of the Total Environment, 76:29–40.
Murdock HR (1959) Studies on the pharmacology of cobalt chloride. Journal of the American Pharmaceutical Association, 48:140–142.
Nackerdien Z, Kasprak KS, Rao G, Halliwell B, Dizdaroglu M (1991) Nickle(II)- and cobalt(II)-dependent damage by hydrogen peroxide to the DNA bases in isolated human chromatin. Cancer Research, 51:5837–5842.
Nagpal NK (2004) Technical report — water quality guidelines for cobalt. Victoria, British Columbia, Ministry of Water, Land, and Air Protection, Water, Air and Climate Change Branch, Water Protection Section (http://www.env.gov.bc.ca/wat/wq/BCguidelines/cobalt/cobalt_tech.pdf).
Nakashima S, Sturgeon RE, Willie SN, Berman SS (1988) Determination of trace metals in seawater by graphite furnace atomic absorption spectrometry with preconcentration on silica-immobilized 8-hydroxyquinoline in a flow-system. Fresenius Journal of Analytical Chemistry, 330(7):592–595.
Nalecz-Jawecki G, Sawicki J (1998) Toxicity of inorganic compounds in the spirotox test: A miniaturized version of the Spirostomum ambiguum test. Archives of Environmental Contamination and Toxicology, 34(1):1–5.
NAS (1977) Drinking water and health. Washington, DC, National Academy of Sciences, pp. 209–211, 247.
NAS (1980) Mineral tolerance of domestic animals. A publication by the National Reseach Council’s Subcommittee on Mineral Toxicity in Animals. Washington, DC, National Academy of Sciences.
NAS/NAE (1973) Water quality criteria 1972. Washington, DC, National Academy of Sciences and National Academy of Engineering; funded by the United States Environmental Protection Agency (Report No. EPA-R3-73-033).
Nation JR, Bourgeois AE, Clark DE, Hare MF (1983) The effects of chronic cobalt exposure on behavior and metallothionein levels in the adult rat. Neurobehavioral Toxicology and Teratology, 5:9–15.
Naylor GPL, Harrison JD (1995) Gastrointestinal iron and cobalt absorption and iron status in young rats and guinea pigs. Human and Experimental Toxicology, 14:949–954.
Neal C, Smith CJ, Jeffery HA, Jarvie HP, Robson AJ (1996) Trace element concentrations in the major rivers entering the Humber estuary, NE England. Journal of Hydrology, 182:37–64.
Neal C, Robson AJ, Wass P, Wade AJ, Ryland GP, Leach DV, Leeks GJL (1998) Major, minor, trace element and suspended sediment variations in the River Derwent. Science of the Total Environment, 210/211:163–172.
Neal C, Jarvie HP, Whitton BA, Gemmell J (2000) The water quality of the River Wear, north-east England. Science of the Total Environment, 251/252:153–172.
Nemery B, Casier P, Roosels D, Lahaye D, Demedts M (1992) Survey of cobalt exposure and respiratory health in diamond polishers. American Review of Respiratory Disease, 145:610–616.
Nemery B, Bast A, Behr J, Borm PJA, Bourke SJ, Camus P, De Vuyst P, Jansen HM, Kinnula VL, Lison D, Pelkonen O, Saltini C (2001a) Interstitial lung disease induced by exogenous agents: factors governing susceptibility. European Respiratory Journal, 32(Suppl.):30S–42S.
Nemery B, Verbeken EK, Demedts M (2001b) Giant cell interstitial pneumonia (hard metal lung disease, cobalt lung). Seminars in Respiratory and Critical Care Medicine, 22:435–447.
Neuhauser EF, Meyer JA, Malecki MR, Thomas JM (1984) Dietary cobalt supplements and the growth and reproduction of the earthworm Eisenia foetida. Soil Biology and Biochemistry, 16:521–523.
Newman LS, Maier LA, Nemery B (1998) Interstitial lung disorders due to beryllium and cobalt. In: Schwartz MI, King TE Jr, eds. Interstitial lung disease. St Louis, MO, Mosby, pp. 367–392.
Newton D, Rundo J (1971) The long term retention of inhaled cobalt-60. Health Physics, 21(3):377–384.
Nielsen NH, Kristiansen J, Borg L, Christensen JM, Poulsen LK, Menne T (2000) Repeated exposures to cobalt or chromate on the hands of patients with hand eczema and contact allergy to that metal. Contact Dermatitis, 43(4):212–215.
Nimmo M, Chester R (1993) The chemical speciation of dissolved nickel and cobalt in Mediterranean rainwaters. Science of the Total Environment, 135:153–160.
Nimmo M, Fones GR (1997) The potential pool of Co, Ni, Cu, Pb and Cd organic complexing ligands in coastal and urban rain waters. Atmospheric Environment, 31(5):693–702.
NIOSH (1994a) Method 7027: Cobalt and cobalt compounds, as Co. In: NIOSH manual of analytical methods, 4th ed. Cincinnati, OH, National Institute for Occupational Safety and Health (http://www.cdc.gov/niosh/nmam/pdfs/7027.pdf).
NIOSH (1994b) Method 8005: Elements in blood or tissue. In: NIOSH manual of analytical methods, 4th ed. Cincinnati, OH, National Institute for Occupational Safety and Health (http://www.cdc.gov/niosh/nmam/pdfs/8005.pdf).
NIOSH (2003) Method 7300: Elements by ICP. In: NIOSH manual of analytical methods, 4th ed. Cincinnati, OH, National Institute for Occupational Safety and Health (http://www.cdc.gov/niosh/nmam/pdfs/7300.pdf).
Niyogi S, Wood CM (2004) Biotic ligand model, a flexible tool for developing site-specific water quality guidelines for metals. Environmental Science & Technology, 38(23):6177–6192.
Nojiri Y, Kawai T, Otsuki A, Fuwa K (1985) Simultaneous multielement determinations of trace metals in lake waters by ICP emission spectrometry with preconcentration and their background levels in Japan. Water Research, 19(4):503–509.
Nriagu JO (1989) A global assessment of natural sources of atmospheric trace metals. Nature, 338:47–49.
Nriagu JO, Coker RD (1980) Trace metals in humic and fulvic acids from Lake Ontario sediments. Environmental Science & Technology, 14:443–446.
NTP (1991) Toxicity studies of cobalt sulfate heptahydrate (CAS No. 10026-24-1) in F344/N rats and B6C3F1 mice (inhalation studies). Research Triangle Park, NC, United States Department of Health and Human Services, National Institutes of Health, National Toxicology Program (NIH Publication No. 91-3124).
NTP (1998) Report on the toxicology and carcinogenesis studies of cobalt sulfate heptahydrate (CAS No. 10026-24-1) in F344/N rats and B6C3F1 mice (inhalation studies). Research Triangle Park, NC, United States Department of Health and Human Services, National Institutes of Health, National Toxicology Program (NIH Publication No. 471).
Nucho R, Rambaud A, Foulquier L, Baudin JP (1988) Bioaccumulation du 60Co par une algue planctonique, Scenedesmus obliquus. Influence du stade de développement de la culture sur la fixation du radionucléide. Acta Oecologica Oecologia Applicata, 9(2):111–125.
OECD (1992) Report of the OECD workshop on extrapolation of laboratory aquatic toxicity data to the real environment. Paris, Organisation for Economic Co-operation and Development (OECD Environment Monograph No. 59).
OECD (1995) Guidance document for aquatic effects assessment. Paris, Organisation for Economic Co-operation and Development (OECD Environment Monograph No. 92).
Ogawa HI, Sakata K, Inouye T, Jyosui S, Niyitani Y, Kakimoto K, Morishita M, Tsuruta S, Kato Y (1986) Combined mutagenicity of cobalt(II) salt and heteroaromatic compounds in Salmonella typhimurium. Mutation Research, 172:97–104.
Olivero S, Villani P, Botta A (1995) Genotoxic effects of cobalt chloride, sulfate and nitrate on cultured human lymphocytes. Medical Science Research, 23:339–341.
Ostapczuk P, Valenta P, Rutzel H, Nürnberg HW (1987) Application of differential pulse anodic stripping voltammetry to the determination of heavy metals in environmental samples. Science of the Total Environment, 60:1–16.
Outridge PM, Noller BN (1991) Accumulation of toxic trace elements by freshwater vascular plants. Reviews of Environmental Contamination and Toxicology, 121:1–63.
Ozanne PG, Greenwood EAN, Shaw TC (1963) The cobalt requirement of subterranean clover in the field. Australian Journal of Agricultural Research, 14(1):39–50.
Painter RB, Howard R (1982) The hela DNA-synthesis inhibition test as a rapid screen for mutagenic carcinogens. Mutation Research, 92:427–437.
Paley KR, Sobel ES, Yalow RS (1958) Effect of oral and intravenous cobaltous chloride on thyroid function. Journal of Clinical Endocrinology and Metabolism, 18:850–859.
Paliouris G, Hutchinson TC (1991) Arsenic, cobalt and nickel tolerances in two populations of Silene vulgaris (Moench) Garcke from Ontario, Canada. New Phytologist, 117:449–459.
Palit S, Ghosh AK, Sharma A, Talukder G (1991a) Modification of the clastogenic effects of cobalt by calcium in bone marrow cells of mice in vivo. Cytologia, 56:373–377.
Palit S, Sharma A, Talukder G (1991b) Chromosomal aberrations induced by cobaltous chloride in mice in vivo. Biological Trace Element Research, 29:139–145.
Palit S, Sharma A, Talukder G (1991c) Cytotoxic effects of cobalt chloride on mouse bone marrow cells in vivo. Cytobios, 65:85–89.
Palit S, Sharma A, Talukder G (1991d) Protection by chlorophyllin against induction of chromosomal aberrations by cobalt in bone marrow cells of mice in vivo. Fitoterapia, 62(5):425–428.
Palko J, Yli-Halla M (1988) Solubility of Co, Ni, and Mn in some extractants in a Finnish acid sulphate soil area. Acta Agriculturae Scandinavica, 38:153–158.
Palmes ED, Nelson N, Laskin S, Kuschner M (1959) Inhalation toxicity of cobalt hydrocarbonyl. American Industrial Hygiene Association Journal, 20:453–468.
Patel PM, Wallace A, Mueller RT (1976) Some effects of copper, cobalt, cadmium, zinc, nickel, and chromium on growth and mineral element concentration in chrysanthemum. Journal of the American Society of Horticultural Science, 101(5):553–556.
Paternain JL, Domingo JL, Corbella J (1988) Developmental toxicity of cobalt in the rat. Journal of Toxicology and Environmental Health, 24:193–200.
Patrick G, Batchelor AL, Stirling C (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part VI: Lung clearance of inhaled cobalt oxide particles in SPF Fischer rats. Journal of Aerosol Science, 20(2):249–255.
Patterson WA, Olsen JJ (1983) Effects of heavy metals on radicle growth of selected woody species germinated on filter paper, mineral and organic substrates. Canadian Journal of Forest Research, 13:233–238.
Pedigo NG, Vernon MW (1993) Embryonic losses after 10-week administration of cobalt to male mice. Reproductive Toxicology, 7:111–116.
Pedigo NG, George WJ, Anderson MB (1988) Effects of acute and chronic exposure to cobalt in male reproduction in mice. Reproductive Toxicology, 2:45–53.
Persson B, Carlenor E, Clyne N, Hultman E, Lins LE, Pehrsson SK, Rydstrom J (1992) Binding of dietary cobalt to sarcoplasmic reticulum proteins. Scandinavian Journal of Clinical and Laboratory Investigation, 52:137–140.
Potolicchio I, Mosconi G, Forni A, Nemery B, Seghizzi P, Sorrentino R (1997) Susceptibility to hard metal lung disease is strongly associated with the presence of glutamate 69 in HLA-DP beta chain. European Journal of Immunology, 27:2741–2743.
Potolicchio I, Festucci A, Hausler P, Sorrentino R (1999) HLA-DP molecules bind cobalt: a possible explanation for the genetic association with hard metal disease. European Journal of Immunology, 29:2140–2147.
Prescott E, Netterstrom B, Faber J, Hegedus L, Suadicani P, Christensen JM (1992) Effect of occupational exposure to cobalt blue dyes on the thyroid volume and function of female plate painters. Scandinavian Journal of Work, Environment and Health, 18:101–104.
Rachlin JW, Grosso A (1993) The growth response of the green alga Chlorella vulgaris to combined divalent cation exposure. Archives of Environmental Contamination and Toxicology, 24:16–20.
Rathore RS, Khangarot BS (2002) Effects of temperature on the sensitivity of sludge worm Tubifex tubifex Muller to selected heavy metals. Ecotoxicology and Environmental Safety, 53(1):27–36.
Reimann C, De Caritat P, Halleraker JH, Volden T, Äyräs M, Niskavaara H, Chekushin VA, Pavlov VA (1997) Rainwater composition in eight Arctic catchments in northern Europe (Finland, Norway and Russia). Atmospheric Environment, 31(2):159–170.
Rengasamy A, Kommineni C, Jones JA, Fedan JS (1999) Effects of hard metal on nitric oxide pathways and airway reactivity to methacholine in rat lungs. Toxicology and Applied Pharmacology, 157:178–191.
Reuber S, Krcuzer M, Kirchgessner M (1994) Interactions of cobalt and iron in absorption and retention. Journal of Trace Elements and Electrolytes in Health and Disease, 8:151–158.
Richards JG, Playle RC (1998) Cobalt binding to gills of rainbow trout (Oncorhynchus mykiss): An equilibrium model. Comparative Biochemistry and Physiology, Part C, Pharmacology, Toxicology & Endocrinology, 119(2):185–197.
Rosenberg DW (1993) Pharmacokinetics of cobalt chloride and cobalt-protoporphyrin. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 21(5):846–849.
Rosko JJ, Rachlin JW (1975) The effect of copper, zinc, cobalt and manganese on the growth of the marine diatom Nitzschia closterium. Bulletin of the Torrey Botanical Club, 102(3):100–106.
Russell-Jones GJ, Alpers DH (1999) Vitamin B12 transporters. In: Amidon GL, Sadee W, eds. Pharmaceutical biotechnology. New York, NY, Kluwer Academic/Plenum Publishers, pp. 493–520.
Sarkar B (1995) Metal replacement in DNA-binding zinc finger proteins and its relevance to mutagenicity and carcinogenicity through free radical generation. Nutrition, 11(5):646–649.
Sauvant MP, Pepin D, Bohatier J, Groliere CA (1995a) Microplate technique for screening and assessing cytotoxicity of xenobiotics with Tetrahymena pyriformis. Ecotoxicology and Environmental Safety, 32(2):159–165.
Sauvant MP, Pepin D, Groliere CA, Bohatier J (1995b) Effects of organic and inorganic substances on the cell proliferation of L-929 fibroblasts and Tetrahymena pyriformis GL protozoa used for toxicological bioassays. Bulletin of Environmental Contamination and Toxicology, 55(2):171–178.
Schade SG, Felsher BF, Bernier GM, Conrad ME (1970) Interrelationship of cobalt and iron absorption. Journal of Laboratory and Clinical Medicine, 75:435–441.
Schinz HR, Uehlinger E (1942) Metals: A new principle of carcinogenesis. Zeitschrift für Krebsforschung, 52:425–437.
Schroeder WH, Dobson M, Kane DM, Johnson ND (1987) Toxic trace elements associated with airborne particulate matter: A review. Journal of the Air Pollution Control Association, 37(11):1267–1285.
Schultz PN, Warren G, Kosso C, Rogers S (1982) Mutagenicity of a series of hexacoordinate cobalt(III) compounds. Mutation Research, 102:393–400.
Sedlet J, Robinson J, Fairman W (1958) A cobalt and a tritium incident at Argonne National Laboratory. In: Proceedings of the fourth annual meeting on bioassay and analytical chemistry. Washington, DC, Atomic Energy Commission, Office of Technical Services, pp. 101–106 (AEC Report No. WASH-1023).
Seghizzi P, D’Adda F, Borleri D, Barbic F, Mosconi G (1994) Cobalt myocardiopathy. A critical review of literature. Science of the Total Environment, 150:105–109.
Semenza GL, Roth PH, Fang H-M, Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. Journal of Biological Chemistry, 269(38):23757–23763.
Sharma RM, Panigrahi S, Azeez PA (1987) Effect of cobalt on the primary productivity of Spirulina platensis. Bulletin of Environmental Contamination and Toxicology, 39(4):716–720.
Shedd KB (1993) The materials flow of cobalt in the United States. Reston, VA, United States Geological Survey, Bureau of Mines (Circular 9350).
Shedd KB (2002) Cobalt. In: USGS minerals yearbook. Vol. 1. Metals and minerals, 2002. Reston, VA, United States Geological Survey (http://minerals.usgs.gov/minerals/pubs/commodity/cobalt/cobalmyb02.pdf).
Shedd KB (2004) Cobalt recyling in the United States in 1998. In: Shipley SF, ed. Flow studies for recycling metal commodities in the United States. Reston, VA, United States Geological Survey (Circular 1196-A-M; http://pubs.usgs.gov/circ/2004/1196am/c1196a-m.pdf).
Sheline GE, Chaikoff IL, Montgomery ML (1945) The elimination of administered cobalt in pancreatic juice and bile of the dog, as measured with its radioactive isotopes. American Journal of Physiology, 145:285–290.
Shine JP, Ika RV, Ford TE (1995) Multivariate statistical examination of spatial and temporal patterns of heavy metal equipment in New Bedford Harbor marine sediments. Environmental Science & Technology, 29:1781–1788.
Shirakawa T, Kusaka Y, Fujimura N, Goto S, Morimoto K (1988) The existence of specific antibodies to cobalt in hard metal asthma. Clinical Allergy, 18:451–460.
Shirakawa T, Kusaka Y, Fujimura N, Goto S, Kato M, Heki S, Morimoto K (1989) Occupational asthma from cobalt sensitivity in workers exposed to hard metal dust. Chest, 95(1):29–37.
Simesen M (1939) The fate of cobalt after oral administration of metallic cobalt and subcutaneous injection of carbonatotetraminecobalt chloride, with remarks on the quantitative estimation of cobalt in organic materials. Archives Internationales de Pharmacodynamie et de Thérapie, 62:347–356.
Sinclair P, Gibbs AH, Sinclair JF, de Matteis F (1979) Formation of cobalt protoporphyrin in the liver of rats. Biochemical Journal, 178:529–538.
Singh I (1983) Induction of reverse mutation and mitotic gene conversion by some metal compounds in Saccharomyces cerevisiae. Mutation Research, 117:149–152.
Singh PP, Junnarkar AY (1991) Behavioral and toxic profile of some essential trace metal salts in mice and rats. Indian Journal of Pharmacology, 23:153–159.
Smith IC, Carson BL (1981) Trace metals in the environment. Ann Arbor, MI, Ann Arbor Science Publishers.
Smith RJ, Fisher JW (1973) Effects of cobalt on the renal erythropoietic factor kidney hydrolase activity in the rat. Blood, 42(2):893–905.
Smith RM (1987) Cobalt. In: Mertz W, ed. Trace elements in human and animal nutrition. San Diego, CA, Academic Press, pp. 143–183.
Smith T, Edmonds CJ, Barnaby CF (1972) Absorption and retention of cobalt in man by whole-body counting. Health Physics, 22:359–367.
Sorbie J, Olatunbosun D, Corbett WE, Valberg LS (1971) Cobalt excretion test for the assessment of body iron stores. Canadian Medical Association Journal, 104(9):777–782.
Speijers GJA, Krajnc EI, Berkvens JM, van Logten MJ (1982) Acute oral toxicity of inorganic cobalt compounds in rats. Food and Chemical Toxicology, 20:311–314.
Srivastava RN, Agrawal SJ (1979) Haematological anomalies in a fresh water teleost, Colisa fasciatus, on acute exposure to cobalt. Acta Pharmacologica et Toxicologica, 44(3):197–199.
Stebbins AI, Horstman SW, Daniell WE, Atallah R (1992) Cobalt exposure in a carbide tip grinding process. American Industrial Hygiene Association Journal, 53(3):186–192.
Steinhoff D, Mohr U (1991) On the question of a carcinogenic action of cobalt-containing compounds. Experimental Pathology, 41:169–174.
Sullivan JF, Egan JD, George RP (1969) A distinctive myocardiopathy occurring in Omaha, Nebraska: Clinical aspects. Annals of the New York Academy of Sciences, 156:526–543.
Sunderman FW (1987) Metal induction of heme oxygenase. Annals of the New York Academy of Sciences, 514:65–80.
Sunderman FW, Zaharia O (1988) Hepatic lipid peroxidation in CoCl2-treated rats, evidenced by elevated concentrations of thiobarbituric acid chromogens. Research Communications in Chemical Pathology and Pharmacology, 59(1):69–78.
Sunderman FW, Hopfer SM, Swift T, Rezuke WN, Ziebka L, Highman P, Edwards B, Folcik M, Gossling HR (1989) Cobalt, chromium, and nickel concentrations in body fluids of patients with porous-coated knee or hip prostheses. Journal of Orthopaedic Research, 7(3):307–315.
Suttle NF, Bell J, Thornton I, Agyriaki A (2003) Predicting the risk of cobalt deprivation in grazing livestock from soil composition data. Environmental Geochemistry and Health, 25:33–39.
Suzuki Y, Shimizu H, Nagae Y, Fukumoto M, Okonogi H, Kadokura M (1993) Micronucleus test and erythropoiesis: Effect of cobalt on the induction of micronuclei by mutagens. Environmental and Molecular Mutagenesis, 22:101–106.
Swanson SAV, Freeman MAR, Heath JC (1973) Laboratory tests on total joint replacement prostheses. Journal of Bone and Joint Surgery, 55B:759–773.
Swennen B, Buchet J-P, Stanescu D, Lison D, Lauwerys R (1993) Epidemiological survey of workers exposed to cobalt oxides, cobalt salts, and cobalt metal. British Journal of Industrial Medicine, 50:835–842.
Szakmary E, Ungvary G, Hudak A, Tatrai E, Naray M, Morvai V (2001) Effects of cobalt sulfate on prenatal development of mice, rats, and rabbits, and on early postnatal development of rats. Journal of Toxicology and Environmental Health A, 62:367–386.
Szefer P, Pempkowiak J, Skwarzec B, Bojanowski R, Holm E (1993) Concentration of selected metals in penguins and other representative fauna of the Antarctica. Science of the Total Environment, 138:281–288.
Szefer P, Szefer K, Glasby GP, Pempkowiak J, Kaliszan R
(1996) Heavy-metal pollution in surficial sediments from the southern Baltic Sea off Poland. Journal of Environmental Science and Health, Part A, Environmental Science and Engineering & Toxic and Hazardous Substance Control, 31(10):2723–2754.
Talbot RJ, Morgan A (1989) An interspecies comparison of the lung clearance of inhaled monodisperse cobalt oxide particles — Part VIII: Lung clearance of inhaled cobalt oxide particles in mice. Journal of Aerosol Science, 20(2):261–265.
Tatara CP, Newman MC, McCloskey JT, Williams PL (1998) Use of ion characteristics to predict relative toxicity of mono-, di- and trivalent metal ions: Caenorhabditis elegans LC50. Aquatic Toxicology, 42(4):255–269.
Taylor DM (1962) The absorption of cobalt from the gastro-intestinal tract of the rat. Physics in Medicine and Biology, 6:445–451.
Templeton D (1996) Cobalt. In: Biological monitoring of chemical exposure in the workplace. Vol. 2. Geneva, World Health Organization, pp. 35–50.
Teraoka H (1981) Distribution of 24 elements in the internal organs of normal males and the metallic workers in Japan. Archives of Environmental Health, 36(4):155–165.
Thomas RG, Furchner JE, London JE, Drake GA, Wilson JS, Richmond CR (1976) Comparative metabolism of radionuclides in mammals — X. Retention of tracer-level cobalt in the mouse, rat, monkey, and dog. Health Physics, 31:323–333.
Tipping E, Lofts S, Lawlor AJ (1998) Modelling the chemical speciation of trace metals in the surface waters of the Humber system. Science of the Total Environment, 210/211:63–77.
Tolle DA, Arthur MF, Van Voris P (1983) Microcosm/field comparison of trace element uptake in crops grown in fly ash-amended soil. Science of the Total Environment, 31:243–261.
Toste AP, Kirby LJ, Pahl TR (1984) Role of organics in the subsurface migration of radionuclides in groundwater. In: Barney GS, Navratil JD, Schulz WW, eds. Geochemical behavior of disposed radioactive waste. Washington, DC, American Chemical Society, pp. 251–270.
Tso W-W, Fung W-P (1981) Mutagenicity of metallic cations. Toxicology Letters, 8:195–200.
Tüchsen F, Jensen MV, Villadsen E, Lynge E (1996) Incidence of lung cancer among cobalt-exposed women. Scandinavian Journal of Work, Environment and Health, 22:444–450.
USDOE (1996) Evaluation of cobalt mobility in soils from the Nevada test site. Reno, NV, United States Department of Energy (DOE/NV/10845-58).
USEPA (1982) Methods for chemical analysis of water and wastes: Method 219.1. Washington, DC, United States Environmental Protection Agency, December (EPA-600/4-82-055).
USEPA (1986) Method 7200: Cobalt (atomic absorption, direct aspiration). In: Test methods for evaluating solid waste, physical/chemical methods. Washington, DC, United States Environmental Protection Agency (EPA Publication SW-846; http://www.epa.gov/SW-846/pdfs/7200.pdf).
USEPA (2002) Toxics Release Inventory report (TRI Explorer database). Washington, DC, United States Environmental Protection Agency (http://www.epa.gov/tri/tridata).
USEPA (2003) Benchmark dose software. Washington, DC, United States Environmental Protection Agency, National Center for Environmental Assessment (http://cfpub2.epa.gov/ncea/cfm/recordisplay.cfm?deid=20167).
USEPA (2005) Ecological soil screening levels for cobalt. Interim final report. Washington, DC, United States Environmental Protection Agency, Office of Solid Waste and Emergency Response, 57 pp. (OSWER Directive 9285.7-67; http://www.epa.gov/ecotox/ecossl/pdf/eco-ssl_cobalt.pdf).
USGS (2004) Mineral commodity summaries 2004. Reston, VA, United States Geological Survey (http://minerals.usgs.gov/minerals/pubs/mcs/2004/mcs2004.pdf).
USGS (2005) Mineral industry surveys. Reston, VA, United States Geological Survey.
Valberg LS, Ludwig J, Olatunbosun D (1969) Alteration in cobalt absorption in patients with disorders of iron metabolism. Gastroenterology, 56(2):241–251.
Valer M, Somogyi Z, Racz I (1967) Studies concerning the sensitizing effect of cobalt. Dermatologica, 134:36–50.
van Bruwaene R, Gerber GB, Kirchmann R, Colard J, Van Kerkom J (1984) Metabolism of 51Cr, 54Mn, 59Fe and 60Co in lactating dairy cows. Health Physics, 46(5):1069–1082.
van den Eeckhout A, Verbeken E, Demedts M (1988) [Pulmonary pathology due to cobalt and heavy metals.] Revue des Maladies Respiratoires, 5:201–207 (in French).
van Goethem F, Lison D, Kirsch-Volders M (1997) Comparative evaluation of the in vitro micronucleus test and the alkaline single cell gel electrophoresis assay for the detection of DNA damaging agents: Genotoxic effects of cobalt powder, tungsten carbide and cobalt–tungsten carbide. Mutation Research, 392:31–43.
Vanselow AP (1966) Cobalt. In: Chaptam HD, ed. Diagnostic criteria for plants and soils. Berkeley, CA, University of Berkeley, Division of Agricultural Sciences, pp. 141–156.
Van Vleet JF, Boon GD, Ferrans VJ (1981) Induction of lesions of selenium–vitamin E deficiency in ducklings fed silver, copper, cobalt, tellurium, cadmium, or zinc: protection by selenium or vitamin E supplements. American Journal of Veterinary Research, 42(7):1206–1217.
Verougstraete V, Mallants A, Buchet J, Swennen B, Lison D (2004) Lung function changes in workers exposed to cobalt compounds — a 13-year followup. American Journal of Respiratory and Critical Care Medicine, 170:162–166.
Vollmann J (1938) [Animal experiments with intraosseous arsenic, chromium and cobalt implants.] Schweizerische Zeitschrift für Pathologie und Bakteriologie, 1:440–443 (in German).
Vranken G, Vanderhaeghen R, Heip C (1991) Effects of pollutants on life-history parameters of the marine nematode Monhystera disjuncta. ICES Journal of Marine Science, 48(3):325–334.
Wallace A, Alexander G, Chaudhry FM (1977) Phytotoxicity of cobalt, vanadium, titanium, silver and chromium. Communications in Soil Science, 8(9):751–756.
Warnau M, Fowler SW, Teyssie J-L (1999) Biokinetics of radiocobalt in the asteroid Asterias rubens (Echinodermata): Sea water and food exposures. Marine Pollution Bulletin, 39(1–12):159–164.
Warne MS (1998) Critical review of methods to derive water quality guidelines for toxicants and a proposal for a new framework. Canberra, Environment Australia (Supervising Scientist Report 135).
Warnick S, Bell H (1969) The acute toxicity of some heavy metals to different species of aquatic insects. Journal of the Water Pollution Control Federation, 41(2):280–284.
Watabe T, Uchida S, Kamada H (1984) Transfer of radionuclides through soil–plant pathway. Journal of Radiation Research, 25:274–282.
Watling HR, Watling RJ (1982) Comparative effects of metals on the filtering rate of the brown mussel (Perna perna). Bulletin of Environmental Contamination and Toxicology, 29:651–657.
Weakly JN (1973) The action of cobalt ions on neuromuscular transmission in the frog. Journal of Physiology, 234:597–612.
Webb M (1962) The biological action of cobalt and other metals. III. Chelation of cations by dihydrolipoic acid. Biochimica et Biophysica Acta, 65:47–65.
Wehner AP, Craig DK (1972) Toxicology of inhaled NiO and CoO in Syrian golden hamsters. American Industrial Hygiene Association Journal, 33:146–155.
Wehner AP, Busch RH, Olson RJ, Craig DK (1977) Chronic inhalation of cobalt oxide and cigarette smoke by hamsters. American Industrial Hygiene Association Journal, 38:338–346.
Wiberg GS (1968) The effect of cobalt ions on energy metabolism in the rat. Canadian Journal of Biochemistry, 46:549–554.
Wild P, Perdrix A, Romazini S, Moulin JJ, Pellet F (2000) Lung cancer mortality in a site producing hard metals. Occupational and Environmental Medicine, 57:568–573.
Windom HL, Schropp SJ, Calder FD, Ryan JD, Smith RG, Burney LC, Lewis FG, Rawlinson CH (1989) Natural trace metal concentrations in estuarine and coastal marine sediments of the southeastern United States. Environmental Science & Technology, 23(3):314–320.
Winger PV, Schultz DP, Johnson WW (1990) Environmental contamination concentrations in biota from the lower Savannah River, Georgia and South Carolina. Archives of Environmental Contamination and Toxicology, 19:101–117.
Yamagata N, Murata S, Torii T (1962) The cobalt content of human body. Journal of Radiation Research, 5:4–8.
Yamagata N, Kurioka W, Shimizu T (1963) Balance of cobalt in Japanese people and diet. Journal of Radiation Research, 4:8–15.
Yamatani K, Saito K, Ikezawa Y, Ohnuma H, Sugiyama K, Manaka H, Takahashi K, Sasaki H (1998) Relative contribution of Ca2+-dependent mechanism in glucagon-induced glucose output from the liver. Archives of Biochemistry and Biophysics, 355(2):175–180.
Yasuda H, Uchida S, Muramatsu Y, Yoshida S (1995) Sorption of manganese, cobalt, zinc, strontium, and cesium onto agricultural soils: Statistical analysis on effects of soil properties. Water, Air, and Soil Pollution, 83:85–96.
Yasukochi Y, Nakamura M, Minakami S (1974) Effect of cobalt on the synthesis and degradation of hepatic catalase in vivo. Biochemical Journal, 144:455–464.
Ybarra J, Behrooz A, Gabriel A, Koseoglu MH, Ismail-Beigi F (1997) Glycemia-lowering effect of cobalt chloride in the diabetic rat: increased GLUT1 mRNA expression. Molecular and Cellular Endocrinology, 133:151–160.
Yukawa M, Amano K, Suzuki-Yasumoto M, Terai M (1980) Distribution of trace elements in the human body determined by neutron activation analysis. Archives of Environmental Health, 35:36–44.
Zanetti G, Fubini B (1997) Surface interaction between metallic cobalt and tungsten carbide particles as a primary cause of hard metal lung disease. Chemistry of Materials, 7(8):1647–1654.
Zhang C, Cai W, Li Y, Huang WQ, Su HC (1998) Quantitative analysis of calcitonin gene-related peptide- and neuropeptide Y-immunoreactive nerve fibers in mesenteric blood vessels of rats irradiated with cobalt-60 gamma rays. Radiation Research, 149:19–26.
Zhang H, Van Den Berg CMG, Wollast R (1990) The determination of interactions of cobalt (II) with organic compounds in seawater using cathodic stripping voltammetry. Marine Chemistry, 28:285–300.
Zou W, Yan M, Xu W, Huo H, Sun L, Zheng Z, Liu X (2001) Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation. Journal of Neuroscience Research, 64(6):646–653.
|
ATP |
adenosine triphosphate |
|
ATSDR |
Agency for Toxic Substances and Disease Registry (USA) |
|
BMC |
benchmark concentration |
|
BMCL10 |
lower limit of the benchmark concentration associated with a 10% incidence of an effect |
|
CAS |
Chemical Abstracts Service |
|
CI |
confidence interval |
|
CICAD |
Concise International Chemical Assessment Document |
|
CoA |
coenzyme A |
|
DNA |
deoxyribonucleic acid |
|
EC50 |
median effective concentration |
|
EDTA |
ethylenediaminetetraacetic acid |
|
FEV1 |
forced expiratory volume in 1 second |
|
FVC |
forced vital capacity |
|
GF-AAS |
graphite furnace atomic absorption spectrometry |
|
HCp |
hazardous concentration for p% of the species |
|
HCp(50) |
hazardous concentration for p% of the species with a 50% confidence level |
|
IARC |
International Agency for Research on Cancer |
|
IC50 |
median inhibitory concentration |
|
ICP-AES |
inductively coupled plasma atomic emission spectrometry |
|
ICP-MS |
inductively coupled plasma mass spectrometry |
|
ICRP |
International Commission on Radiological Protection |
|
Ig |
immunoglobulin |
|
IOMC |
Inter-Organization Programme for the Sound Management of Chemicals |
|
IPCS |
International Programme on Chemical Safety |
|
KD |
partition coefficient in soil |
|
KF |
Freundlich adsorption constant |
|
LC50 |
median lethal concentration |
|
LD50 |
median lethal dose |
|
LOEC |
lowest-observed-effect concentration |
|
MMEF |
maximal mid-expiratory flow rate |
|
NADH |
reduced nicotinamide adenine dinucleotide |
|
NIOSH |
National Institute for Occupational Safety and Health (USA) |
|
NOAEC |
no-observed-adverse-effect concentration |
|
NOEC |
no-observed-effect concentration |
|
NTP |
National Toxicology Program (USA) |
|
OECD |
Organisation for Economic Co-operation and Development |
|
PBPK |
physiologically based pharmacokinetic |
|
PEFR |
peak expiratory flow rate |
|
PVC |
polyvinyl chloride |
|
SD |
standard deviation |
|
SMR |
standardized mortality ratio |
|
UV |
ultraviolet |
Agency for Toxic Substances and Disease Registry
Copies of the ATSDR toxicological profile for cobalt (ATSDR, 2004) can be obtained from:
Agency for Toxic Substances and Disease Registry
Division of Toxicology/Toxicology Information Branch
1600 Clifton Road NE
Mailstop F-32
Atlanta, Georgia 30333
USA
The document is also available on the web at:
http://www.atsdr.cdc.gov/toxprofiles/tp33.html
The profile has undergone the following ATSDR internal reviews: Health Effects Review, Minimal Risk Level Review, and Data Needs Review. In addition, a peer review panel, which included Dr Herman Cember (Purdue University, USA), Dr James Hansen (United States Fish and Wildlife Service), Dr Dominique Lison (Catholic University of Louvain, Belgium), and Dr Nancy Pedigo (University of Kentucky Medical Center, USA), was assembled.
International Agency for Research on Cancer
Copies of the IARC (2005) monograph for cobalt particles may be obtained from:
IARC Press
150 Cours Albert Thomas
69008 Lyon, France
The draft CICAD on cobalt and inorganic cobalt compounds was sent for review to institutions and organizations identified by IPCS after contact with IPCS national Contact Points and Participating Institutions, as well as to identified experts. Comments were received from:
H. Ahlers, National Institute for Occupational Safety and Health, Morgantown, WV, USA
L. Alessio, Institute of Occupational Health, University of Brescia, Brescia, Italy
M. Baril, Institut de recherche Robert Sauvé en santé et en sécurité du travail, Montreal, Canada
R. Benson, United States Environmental Protection Agency Region 8, Denver, CO, USA
T. Brock, Cobalt Development Institute, Surrey, United Kingdom
J. Caley, Environment Agency, Wallingford, United Kingdom
J. Chapman, Department of Environment & Conservation, Lidcombe, New South Wales, Australia
R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
L. Fishbein, Fairfax, VA, USA
V. Foà, University of Milan, Milan, Italy
E. Frantik, Institute of Public Health, Prague, Czech Republic
G. Harvey, National Industrial Chemicals Notification and Assessment Scheme (NICNAS), Sydney, New South Wales, Australia
J. Högberg, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
M. Hoover, National Institute for Occupational Safety and Health, Morgantown, WV, USA
G. Hsu, United States Environmental Protection Agency, Washington, DC, USA
M. Keane, National Institute for Occupational Safety and Health, Morgantown, WV, USA
J. Kielhorn, Fraunhofer Institute of Toxicology & Experimental Medicine, Hanover, Germany
D. Lison, Catholic University of Louvain, Brussels, Belgium
I. Mangelsdorf, Fraunhofer Institute of Toxicology & Experimental Medicine, Hanover, Germany
M. Nordberg, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
N. Sahakian, National Institute for Occupational Safety and Health, Washington, DC, USA
H. Savolainen, Ministry of Social Affairs & Health, Tampere, Finland
J. Stauber, CSIRO Energy Technology, Menai, New South Wales, Australia
T. Stedeford, United States Environmental Protection Agency, Washington, DC, USA
A. Stefaniak, National Institute for Occupational Safety and Health, Morgantown, WV, USA
U. Stenius, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
M.H. Sweeney, United States Department of Health and Human Services, Hanoi, Viet Nam
S. Tao, Center for Food Safety and Applied Nutrition, Food and Drug Administration, College Park, MD, USA
G. Ungvary, Fodor József National Center for Public Health, Budapest, Hungary
R. Welton, Cobalt Development Institute, Surrey, United Kingdom
K. Ziegler-Skylakakis, Secretariat of the Commission for the Investigation of Health Hazards of Chemical Compounds in the Workplace Area (MAK Commission), Freising-Weihenstephan, Germany
Nagpur, India
31 October – 3 November 2005
Members
Dr T. Chakrabarti, National Environmental Engineering Research Institute, Nagpur, India
Dr R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
Mr P. Copestake, Toxicology Advice & Consulting Ltd, Surrey, United Kingdom
Dr C. De Rosa, Agency for Toxic Substances and Disease Registry, Atlanta, GA, USA
Dr S. Dobson, Centre for Ecology and Hydrology, Monks Wood, United Kingdom
Dr L. Fishbein, Fairfax, VA, USA
Dr L. Fruchtengarten, Poison Control Center of São Paulo, São Paulo, Brazil
Dr H. Gibb, Sciences International Inc., Alexandria, VA, USA
Dr R.F. Hertel, Federal Institute for Risk Assessment (BfR), Berlin, Germany
Mr P. Howe, Centre for Ecology and Hydrology, Monks Wood, United Kingdom
Ms K. Hughes, Health Canada, Ottawa, Ontario, Canada
Dr D. Kanungo, Directorate General of Health Services, New Delhi, India
Dr J. Kielhorn, Fraunhofer Institute of Toxicology and Experimental Medicine, Hanover, Germany
Dr G. Kong, Hanyang University, Seoul, Republic of Korea
Dr J. Rischer, Agency for Toxic Substances and Disease Registry, Chamblee, GA, USA
Dr O. Sabzevari, Tehran University of Medical Sciences, Tehran, Islamic Republic of Iran
Dr R. Sonawane, National Center for Environmental Assessment, Environmental Protection Agency, Washington, DC, USA
Dr J. Stauber, CSIRO Energy Technology, Menai, New South Wales, Australia
Dr M.H. Sweeney, United States Embassy, Hanoi, Viet Nam
Ms D. Willcocks, National Industrial Chemicals Notification and Assessment Scheme, Sydney, New South Wales, Australia
Dr Y. Zheng, National Institute for Occupational Health & Poison Control, Beijing, People’s Republic of China
Dr K. Ziegler-Skylakakis, Secretariat of the Commission for the Investigation of Health Hazards of Chemical Compounds in the Workplace Area (MAK Commission), Freising-Weihenstephan, Germany
Observer
Mr P. Ashford, Resorcinol Task Force, Wotton-under-edge, Gloucestershire, United Kingdom
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Ms L. Onyon, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr M. Shibatsuji, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
(DUTCH STATISTICAL EXTRAPOLATION METHOD) USED TO DERIVE
GUIDANCE VALUES FOR COBALT FOR THE
PROTECTION OF AQUATIC SPECIES
Introduction
The traditional approach to using single-species toxicity data to protect field ecosystems has been to apply standardized assessment factors, safety factors, or application factors to the lowest toxicity figure for a particular chemical. The magnitude of these safety factors depends on whether acute or chronic toxicity figures are available and the degree of confidence that one has in whether the figures reflect the field situation. Most of the factors are multiples of 10, and larger factors are applied where there is less certainty in the data. For example, a factor of 1000 is generally used for acute data, except for essential elements, including cobalt, where a factor of 200 is applied. This factor of 200 includes a factor of 10 for extrapolating from laboratory to field, a further factor of 10 for a limited data set, and a factor of 2 for conversion of an acute end-point to a chronic end-point for an essential metal.
Concerns have often been raised as to the arbitrary nature of assessment factors (Chapman et al., 1998) and the fact that they do not conform to risk assessment principles. OECD (1992) recommended that assessment factors be used only when there are inadequate data to allow statistical extrapolation methods to be used.
The following sections briefly outline the statistical extrapolation method used to derive the cobalt guidance values for the protection of marine and freshwater aquatic organisms for this CICAD. Much of the text is taken directly from the Australian and New Zealand guidelines for fresh and marine water quality (ANZECC/ARMCANZ, 2000).
Use of statistical extrapolation methods
New methods using statistical risk-based approaches have been developed over the last decade for deriving guideline (trigger) values. These are based on calculations of a statistical distribution of laboratory ecotoxicity data and attempt to offer a predetermined level of protection, usually 95%. The approach of Aldenberg & Slob (1993) has been adopted in the Netherlands, Australia, and New Zealand for guideline derivation and is recommended for use by the OECD. It was chosen because of its theoretical basis, its ease of use, and the fact that it has been extensively evaluated. Warne (1998) compared in detail the risk-based and assessment factor approaches used in various countries.
The Aldenberg & Slob (1993) method uses a statistical approach to protect 95% of species with a predetermined level of confidence, provided there is an adequate data set. This approach uses available data from all tested species (not just the most sensitive species) and considers these data to be a subsample of the range of concentrations at which effects would occur in all species in the environment. The method may be applied if toxicity data, usually chronic NOEC values, are available for at least five different species from at least four taxonomic groups. Data are entered into a computer program and generally fitted to a log-logistic distribution. A hazardous concentration for p per cent of the species (HCp) is derived. HCp is a value such that the probability of selecting a species from the community with a NOEC lower than HCp is equal to p (e.g. 5%, HC5). HC5 is the estimated concentration that should protect 95% of species. A level of uncertainty is associated with this derived value, and so values with a given confidence level (e.g. 50% or 95%) are computed in the program by attaching a distribution to the error in the tail (Figure A5-1). The ANZECC/ ARMCANZ (2000) guidelines use the median of 50% confidence.
Fig. A5-1: The Dutch statistical approach for the derivation
of trigger values (from Aldenberg & Slob, 1993)
HC5 is estimated by dividing the geometric mean of the NOEC values for m species by an extrapolation factor K (OECD, 1995), where:
K = exp(sm x k)
and where:
The Aldenberg & Slob (1993) extrapolation method is based on several critical assumptions, outlined below. Many of these are common to other statistical distribution methods:
Modification of the Aldenberg & Slob (1993) approach
The Aldenberg & Slob (1993) approach assumes that the data are best fitted to a log-logistic distribution. For some data sets, however, a better fit is obtained with other models. By using a program developed by CSIRO Biometrics, the data are compared with a range of statistical distributions called the Burr family of distributions, of which the log-logistic distribution is one case. The program determines the distribution that best fits the available toxicity data and calculates the HC5 with 50% confidence (ANZECC/ARMCANZ, 2000); this method has been used to calculate the HC5 for cobalt.
Table A5-1: Toxicity end-points and calculated chronic NOECs used in the derivation of a marine guidance value.
|
Organism |
End-point |
Cobalt concentration (mg/l) |
Calculated chronic NOEC (mg/l) |
|
Algae |
|||
|
Diatom (Ditylum brightwellii) |
5-day EC50 |
0.3 |
0.06 |
|
Diatom (Nitzschia closterium) |
96-h EC50 |
10.2 |
2 |
|
Invertebrates |
|||
|
Nematode (Monhystera disjuncta) |
96-h LC50 |
94 |
9.4 |
|
Brine shrimp (Artemia salina) |
48-h EC50 |
10.3 |
1 |
|
Common prawn (Palaemon serratus) |
96-h LC50 (larvae) |
22.7 |
2.3 |
|
Shore crab (Carcinus maenus) |
96-h LC50 (larvae) |
22.7 |
2.3 |
|
Lobster (Homarus vulgaris) |
96-h LC50 |
4.5 |
0.5 |
|
Isopod (Idotea baltica) |
52-day LC50 |
10 |
2 |
|
Fish |
|||
|
Plaice (Pleuronectes platessa) |
96-h LC50 |
454 |
45.4 |
|
Shanny (Blennius pholis) |
96-h LC50 |
454 |
45.4 |
|
Mummichog (Fundulus heteroclitus) |
96-h LC50 |
275 |
27.5 |
|
Crescent perch (Therapon jarbua) |
96-h LC50 |
52.5 |
5.3 |
Application to the data set for cobalt
For both the marine and freshwater risk assessments, acute LC50 values were converted to chronic values using a default acute to chronic ratio of 10. In cases where chronic values were reported as EC50s, these were then converted to chronic NOECs by applying a factor of 5, according to ANZECC/ ARMCANZ (2000) guidelines, prior to the species sensitivity distribution being undertaken. It would be better to use experimentally derived acute to chronic conversion factors, but these were not available for cobalt.
Marine guidance value
Twelve marine data were used from Table 2 (see section 10.2), and from these data were calculated chronic NOECs (see Table A5-1). Non-standard test end-points or end-points of uncertain significance were not included.
Using the calculated chronic NOECs, the HC5(50), i.e. the hazardous concentration to protect 95% of species with 50% confidence, was 0.14 mg/l. However, a guidance value of 0.14 mg/l is not sufficiently protective of the most sensitive marine species. To account for this, the HC1 50% value has been used to recalculate a moderate-reliability guidance value. Using the calculated chronic NOECs, the HC1(50) — i.e. the hazardous concentration to protect 99% of species with 50% confidence — was 0.02 mg/l. This is a "safe" value to ensure protection against chronic toxicity for most species (see Figure A5-2).
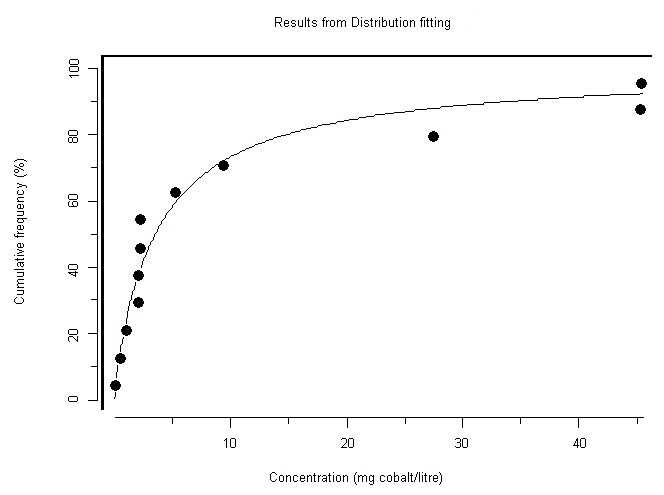
Fig. A5-2: Probability curve for cobalt in the marine environment
using actual and derived data from Table A5-1
Table A5-2: Toxicity end-points and calculated chronic NOECs used in the derivation of a freshwater guidance value.
|
Organism |
End-point |
Cobalt concentration (mg/l) |
Calculated chronic NOEC (mg/l) |
|
Microalgae |
|||
|
Green alga (Chlorella vulgaris) |
21-day NOEC |
0.6 |
0.6 |
|
Protozoa |
|||
|
Ciliated protozoan (Spirostomum ambiguum) |
24-h LC50 |
11.8 |
1.2 |
|
Invertebrates |
|||
|
Water flea (Daphnia magna) |
28-day NOEC |
0.003 |
0.003 |
|
Water flea (Daphnia hyalina) |
48-h LC50 |
1.3 |
0.1 |
|
Water flea (Ceriodaphnia dubia) |
7-day NOEC |
0.007a |
0.007 |
|
Rotifer (Philodina acuticornis) |
24-h LC50 |
27.8 |
2.8 |
|
Copepod (Diaptomus forbesi) |
96-h LC50 |
3.4 |
0.3 |
|
Copepod (Cyclops abyssorum) |
48-h LC50 |
15.5 |
1.6 |
|
Copepod (Eudiaptomus padanus) |
48-h LC50 |
4 |
0.4 |
|
Crayfish (Austropotamobius pallipes) |
96-h LC50 |
8.8 |
0.9 |
|
Crayfish (Orconectes limosus) |
96-h LC50 |
10.2 |
1 |
|
Amphipod (Crangonyx pseudogracilis) |
96-h LC50 |
39.2 |
3.9 |
|
Flatworm (Dugesia tigrina) |
96-h LC50 |
11.3 |
1.1 |
|
Snail (Helisoma trivolvis) |
96-h LC50 |
45 |
4.5 |
|
Pillbug (Asellus intermedius) |
96-h LC50 |
45 |
4.5 |
|
Sideswimmer (Gammarus fasciatus) |
96-h LC50 |
45 |
4.5 |
|
Segmented worm (Lumbriculus variegatus) |
96-h LC50 |
45 |
4.5 |
|
Tubificid worm (Tubifex tubifex) |
96-h LC50 |
178.5b |
17.9 |
|
Oligochaete (Branchiura sowerbyi) |
96-h LC50 |
133 |
13.3 |
|
Midge (Chironomus tentans) |
48-h LC50 |
57 |
5.7 |
|
Mayfly (Ephemerella subvaria) |
96-h LC50 |
16 |
1.6 |
|
Fish |
|||
|
Rainbow trout (Oncorhynchus mykiss) |
14-day NOEC |
0.1 |
0.1 |
|
Fathead minnow (Pimephales promelas) |
7-day NOEC |
2a |
2 |
|
Goldfish (Carassius auratus) |
7-day LC50 |
0.8 |
0.2 |
|
Zebrafish (Danio rerio) |
16-day NOEC (survival) |
0.06 |
0.06 |
|
Giant gourami (Colisa fasciata) |
96-h LC50 |
102 |
10.2 |
|
Amphibians |
|||
|
Frog (Rana hexadactyla) |
96-h LC50 |
18 |
1.8 |
|
Narrow-mouthed toad (Gastrophryne carolinensis) |
7-day LC50 |
0.05 |
0.01 |
a Geometric mean of NOECs for this species for the same time period.
b Geometric mean of LC50 values for this species for the same time period.
Freshwater guidance value
Twenty-eight freshwater data were used from Table 2 (see section 10.2), and from these data, chronic NOECs were calculated (see Table A5-2). Non-standard test end-points or end-points of uncertain significance were not included. Geometric means of multiple test results from the same species over the same time period were calculated.
Using the calculated chronic NOECs, the HC5(50), i.e. the hazardous concentration to protect 95% of species with 50% confidence — a "safe" value to ensure protection against chronic toxicity for most freshwater species — was 0.008 mg/l (see Figure A5-3).
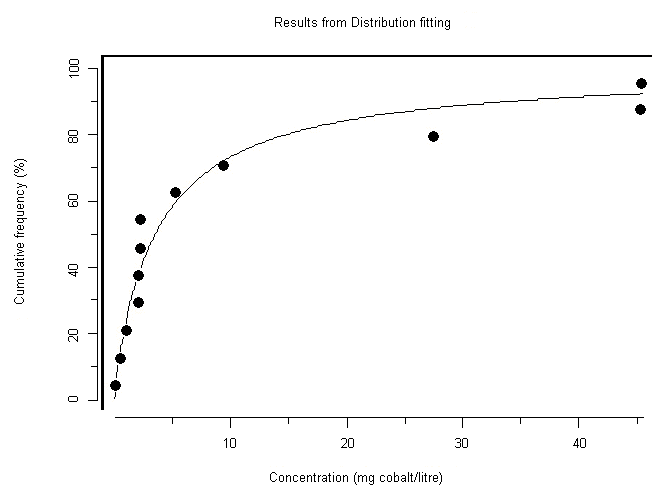
Fig. A5-3: Probability curve for cobalt in the freshwater environment
using actual and derived data from Table A5-2
COBALT(II) NITRATE HEXAHYDRATE ICSC:0784
COBALT(II) ACETATE TETRAHYDRATE ICSC:1128
COBALT(II) SULFATE HEPTAHYDRATE ICSC:1396
Le présent CICAD5 relatif au cobalt et à ses dérivés minéraux a été préparé aux Etats-Unis par Science International Inc. et au Royaume-Uni par le Centre d’écologie et d’hydrologie. Il s’inspire de mises au point rédigées par l’Agency for Toxic Substances and Disease Registry (ATSDR, 2004) et par le Centre international de recherches sur le cancer (IARC, 2005). Afin de prendre en compte les références qui ne figurent dans aucune de ces mises au point, une recherche bibliographique exhaustive a été effectuée en avril 2005 sur plusieurs bases de données en ligne. Des renseignements sur la disponibilité des documents de base et sur la nature de leur examen par des pairs sont donnés à l’appendice 2. L’appendice 3 donne des indications sur l’examen par des pairs du présent CICAD. Ce CICAD a été examiné et approuvé en tant qu’évaluation internationale lors d’une réunion du Comité d’évaluation finale qui s’est tenue à Nagpur (Inde) du 31 octobre au 3 novembre 2005. La liste des participants à cette réunion figure à l’appendice 4. Les fiches internationales sur la sécurité chimique du cobalt, de l’oxyde de cobalt (II), de l’oxyde de cobalt (III), du sulfure de cobalt (II), du chlorure de cobalt (II), du sulfate de cobalt (II), du sulfate de cobalt (II) à sept molécules d’eau, du nitrate de cobalt (II), du nitrate de cobalt (II) à six molécules d’eau, de l’acétate de cobalt (II) à quatre molécules d’eau, du naphténate de cobalt et du cobalt carbonyle, établies par le Programme international sur la sécurité chimique (IPCS, 2000, 2001a-e, 2004a-f) sont également reproduites dans le présent document.
Le cobalt (numéro atomique 27) est un élément présent naturellement dont on connaît un isotope stable (59Co) et 26 isotopes radioactifs. Il existe à trois degrés d’oxydation (0, +2 et +3). Etant donné qu’il peut se trouver sous la forme d’un isotope radioactif, cet élément est susceptible d’émettre un rayonnement ionisant. Le présent document porte essentiellement sur l’isotope stable du cobalt. Pour plus de renseignements sur les effets du rayonnement ionisant émis par les isotopes radioactifs du cobalt, on pourra consulter d’autres sources d’information et notamment la référence ATSDR (2004).
Le cobalt (No CAS 7440-48-4) se présente à la température ambiante sous l’aspect d’un solide gris argent. Il vient au 33ème rang par ordre d’abondance et il est présent dans divers milieux tels que l’air, les eaux superficielles, les produits de lessivage de zones de décharge classées comme dangereuses, les eaux souterraines, le sol et les sédiments. Les sources d’exposition au cobalt et à ses dérivés minéraux peuvent être naturelles ou anthropogéniques. Parmi les sources naturelles figurent les poussières soulevées par le vent, les embruns, les volcans, les feux de forêt ainsi que émissions continentales et marines d’origine biologique. Au nombre des sources anthropogéniques de cobalt on peut citer la combustion de combustibles fossiles, les boues d’égout, les engrais phosphatés, l’extraction minière et la fonte des minerais de cobalt, la préparation des alliages à base de cobalt et les industries qui utilisent ou transforment des dérivés du cobalt.
Le cobalt et ses dérivés minéraux ne sont pas volatils et passent dans l’atmosphère sous la forme de particules. On estime que le cobalt anthropogénique issu de la combustion se trouve principalement sous forme d’oxydes. Au cours de l’extraction des minerais et du raffinage, du cobalt est également libéré dans l’atmosphère sous forme de sulfure et d’arséniure.
Le cobalt libéré dans l’atmosphère vient se déposer sur le sol; lorsqu’il est déchargé dans l’eau, il peut subir une sorption sur des particules puis se déposer dans les sédiments ou se fixer directement à eux par sorption. Le coefficient de distribution du cobalt (par exemple de l’eau aux sédiments) varie en fonction du pH, des conditions rédox, de la force ionique et de la concentration en matières organiques dissoutes. Les facteurs qui influent sur la spéciation et le devenir du cobalt dans l’eau, les sédiments et le sol sont, entre autres, la présence de ligands organiques tels que les acides humiques, les anions, le pH et le potentiel rédox. La mobilité du cobalt dans le sol est inversement proportionnelle à l’intensité de son adsorption par les constituants du sol. Les végétaux peuvent capter le cobalt présent dans le sol, mais il n’est guère transporté de l’appareil racinaire vers d’autres parties de la plante.
Dans l’atmosphère, la concentration mesurée est d’environ un 1 ng/m3 ou moins dans les zones dépourvues de sources de cobalt et généralement inférieure à 10 ng/m3 dans les zones où de telles sources sont présentes, encore que des teneurs plus élevées aient été signalées dans ces zones. La teneur en cobalt des eaux superficielles ou souterraines est faible, inférieure à 1 µg/l dans les territoires vierges et comprise entre 1 et 10 µg/l dans les zones de peuplement. La teneur en cobalt des eaux superficielles ou souterraines peut être beaucoup plus élevée en zone minière ou agricole et atteindre plusieurs centaines de milligrammes par litre. Dans l’eau de mer, les concentrations relevées sont inférieures à 1 µg/l. Dans l’eau destinée à la boisson, la concentration est généralement inférieure à 1-2 µg/l. Des concentrations comprises entre 0,3 et 1,7 µg/l ont été mesurées dans l’eau de pluie. La concentration moyenne du cobalt dans l’écorce terrestre est de 20 à 25 mg/kg. A proximité d’un certain nombre de sources anthropogéniques, la concentration du cobalt dans le sol peut atteindre plusieurs centaines de milligrammes par kg.
Pour la population dans son ensemble, la principale source d’exposition au cobalt est constituée par la nourriture. L’apport de cobalt par la voie alimentaire est estimé à 5-40 m g par jour, en majorité sous forme minérale. Il y a exposition professionnelle au cobalt dans un certain nombre d’industries. Dans le tabac, la concentration du cobalt va de moins de 0,3 à 2,3 m g/g de poids sec et ce cobalt est présent à environ 0,5 % dans le courant principal de la fumée de tabac. Aux Etats-Unis, on a relevé des concentrations en cobalt dans le charbon, le pétrole brut, le mazout et l’essence respectivement égales à 5 mg/kg, 0,001-10 mg/kg, 0,03-0,3 mg/kg et moins de 0,1 mg/kg.
Une fois inhalées, les particules de cobalt se déposent dans les voies respiratoires supérieures et inférieures où elles peuvent rester prisonnières ou au contraire passer de là dans le sang après dissolution ou transport mécanique vers les voies digestives par l’action de l’ascenseur mucociliaire ou par déglutition. Le cobalt qui pénètre dans les voies digestives est absorbé dans la proportion d’environ 50 %. L’absorption est plus importante chez les sujets qui présentent une carence martiale. Les formes solubles dans l’eau sont mieux absorbées que les formes insolubles. Le cobalt est un constituant essentiel de la vitamine B12, c’est pourquoi on le retrouve dans la plupart des tissus. La quantité totale de cobalt présente dans l’organisme est estimée à 1,1-1,5 mg, dont 0,11 mg dans le foie. Après exposition au cobalt par la voie respiratoire, on en trouve des quantités plus élevées au niveau du poumon. Il n’existe pas d’études consacrées à la répartition du cobalt dans l’organisme humain après ingestion, mais l’expérimentation animale montre que le cobalt s’accumule principalement dans le foie. Lors d’une étude contrôlée sur l’exposition de sujets humains à des aérosols, on a constaté que 40 % de la quantité totale d’oxyde de cobalt présente dans les poumons était encore retenue à ce niveau 6 mois après l’exposition. Après inhalation, l’excrétion urinaire augmente au cours du temps. La granulométrie des particules inhalées influe sur l’élimination du cobalt car l’élimination mécanique par la voie digestive est plus importante lorsque les particules sont de plus grande taille. L’élimination dans les matières fécales est la principale voie d’excrétion chez l’Homme après exposition par voie orale.
Chez le rat, la CL50 par inhalation de cobalt hydrocarbonyle s’est révélée égale à 165 mg/m3 pour une durée d’exposition de 30 minutes. En ce qui concerne la DL50 par voie orale de divers dérivés solubles du cobalt, on a trouvé des valeurs allant de 42,4 à 317 mg/kg de poids corporel selon la nature du composé et l’espèce animale soumise à l’expérimentation. Dans le cas de l’oxyde salin de cobalt (tétroxyde de tricobalt), un composé insoluble, on a trouvé pour le rat une DL50 de 3672 mg de cobalt par kg de poids corporel.
Des rats et des souris exposés par voie respiratoire pendant une courte période (16 jours) à des doses de cobalt respectivement égales à 19 mg/m3 et 1,9 mg/m3 sous la forme de sulfate de cobalt ont présenté une nécrose et une inflammation de l’épithélium respiratoire. Chez les rats, on a également observé une nécrose du thymus et une atrophie des testicules. Des rats mâles exposés par voie orale pendant 3 semaines à une concentration quotidienne de cobalt de 12,4 mg/kg de poids corporel sous la forme de chlorure de cobalt ont présenté des lésions cardiaques. Exposés par la voie respiratoire à des dérivés du cobalt dont la concentration était supérieure ou égale à 0,3 mg/m3 (soit une concentration de cobalt > 0,11 mg/m3) pendant une durée de 3 à 4 mois, des rats, des lapins et des souris ont présenté des lésions au niveau des voies respiratoires. Chez des rats exposés pendant 2 à 3 mois à du sulfate de cobalt ajouté à leur alimentation ou à du chlorure de cobalt ajouté à leur eau de boisson, les doses journalières de cobalt correspondantes étant égales à 26-30,2 mg/kg de poids corporel, on a observé une augmentation du poids du myocarde et des lésions cardiaques dégénératives. Une réduction sensible de l’activité des enzymes cardiaques a été observée chez des rats exposés pendant 24 semaines à une dose journalière de cobalt égale à 8,4 mg/kg de poids corporel sous la forme de sulfate de cobalt ajouté à leur alimentation. Des rats exposés quotidiennement pendant 4 à 5 mois à des doses de cobalt égales à 10-18 mg/kg de poids corporel sous la forme de chlorure de cobalt, on présenté des lésions rénales.
Un emphysème a été observé chez des hamsters exposés pendant toute leur vie à de l’oxyde de cobalt par la voie respiratoire. Chez des souris et des rats exposés pendant 105 semaines à du sulfate de cobalt par la voie respiratoire, on a constaté des tumeurs pulmonaires dont la formation était liée à la dose. Sous la forme de poudre métallique injectée par voie intramusculaire, le cobalt provoque l’apparition de tumeurs sarcomateuses chez le rat.
De nombreux composés du cobalt sont génotoxiques chez les mammifères, cette génotoxicité se manifestant également dans des systèmes d’épreuve mammaliens ou bactériens. Les dérivés du cobalt (III) se révèlent positifs à cet égard dans les systèmes bactériens. Les dérivés du cobalt (II) provoquent des conversions géniques chez Saccharomyces cerevisiae, mais se révèlent peu génotoxiques par ailleurs.
On a constaté que le cobalt avait des effets sur la reproduction et le développement de l’animal. Des rats exposés à cet élément sous la forme de chlorure de cobalt aux doses quotidiennes de 13,3 à 58,9 mg/kg de poids corporel pendant 2 à 3 mois et des souris également exposés à ce composé à raison de 43,4 mg/kg de poids corporel par jour pendant 13 semaines, on présenté une dégénérescence et une atrophie testiculaires. Egalement exposés à du chlorure de cobalt à des doses égales à 46,9 ou 93,0 mg/kg de poids corporel par jour, puis accouplés avec des femelles non exposées, des souris mâles ont présenté une diminution du poids de l’épididyme, du nombre de spermatozoïdes, du poids des testicules et de la fécondité, cette dernière étant évaluée par le nombre d’accouplements entraînant la gravidité des femelles. Lors d’études sur le développement, des rattes gravides ont été exposées à des doses de chlorure de cobalt toxiques pour elles (soit 5,4 ou 21,8 mg de cobalt par kg de poids corporel et par jour); ces rattes ont mis bas des ratons présentant un retard de croissance et une moindre survie mais aucun effet tératogène n’a été relevé. Après exposition de lapines à du sulfate de cobalt à la dose de 7,6 mg/kg de poids corporel par jour, on a observé une augmentation des résorptions fœtales et un nombre plus élevé de fœtus présentant un retard pondéral.
Chez l’Homme, l’inhalation de dérivés du cobalt et l’exposition cutanée à ces produits peut provoquer une sensibilisation. Des cas d’asthme bronchique ont été décrits chez des ouvriers exposés à du cobalt sous diverses formes.
Des sujets humains qui avaient ingéré du chlorure de cobalt à raison de 150 mg par jour pendant 22 jours ont présenté une polycythémie en une augmentation du taux d’hémoglobine. Selon certaines études, des cas de myocardiopathies ont été observés chez des sujets humains qui avaient consommé de grandes quantités de bière contenant du sulfate de cobalt.
Les particules contenant du cobalt métallique sont à l’origine d’une maladie professionnelle appelée fibrose pulmonaire interstitielle, qui touche les travailleurs employés à la fabrication des carbures métalliques.
Les études de mortalité effectuées dans les industries produisant des carbures métalliques font état d’une augmentation de la mortalité par cancer du poumon. Le cobalt est utilisé comme liant dans ces industries et ce type de fabrication entraîne également une exposition à d’autres substances comme le carbure de tungstène et d’autres composés métalliques tels que les carbures de titane, de tantale et de niobium.
A partir des résultats d’une étude transversale effectuée chez des polisseurs de diamants exposés au cobalt on a évalué à 1 × 10−4 mg/m3 la concentration tolérable par inhalation, le critère retenu étant la diminution de la fonction respiratoire. Il y a généralement une différence d’un facteur 10 entre la concentration tolérable et les concentrations de cobalt mesurées dans l’air ambiant à proximité de sources anthropogéniques.
En prenant comme critère la croissance d’une algue verte dulçaquicole, Chlorella vulgaris, on a trouvé la valeur de 0,6 mg/l pour la CE50 à 96 heures du cobalt, alors que pour des plantes aquatiques vasculaires, ces valeurs étaient égales à 0,1 et 0,2 mg/l. La CE50 à cinq jours basée sur la croissance de la diatomée marine Ditylum brightwellii a été trouvée égale à 0,3 mg/l. Dans le cas d’invertébrés d’eau douce, les valeurs de la CL50 (24-96 h) allaient de 1,1 à 239 mg/l. Plusieurs études relatives à la reproduction de Daphnia magna ont été publiées; elles font état d’une CE50 à 21 jours égale à 0,01 mg/l et d’une NOEC (concentration sans effet observable) à 28 jours de 0,003 mg/l; cependant, selon des travaux ultérieurs, la valeur de la NOEC à 21 jours irait de 0,03 à 0,05 mg/l pour diverses valeurs de la teneur en carbonate de calcium. Dans le cas des organismes aquatiques, la valeur la plus faible trouvée pour la NOEC est inférieure à 0,003 mg/l. Elle a été mesurée lors d’un essai de 7 jours sur la puce d’eau Ceriodaphnia dubia. Chez les invertébrés marins, c’est dans le cas des larves de homard que la sensibilité la plus forte a été relevée, avec une valeur de la CL50 à 96 h allant de 4,5 à 22,7 mg/l. Pour les poissons d’eau douce, la CL50 à 96 h va de 1,4 à 333 mg/l. En prenant la survie comme critère, on a trouvé une valeur de 0,06 mg/l pour la NOEC à 16 jours. D’après les résultats des tests effectués sur des poissons de mer, il semblerait que les diverses espèces - tout au moins celles qui ont été étudiées - soient relativement insensibles au cobalt, avec des valeurs de la CL50 à 96 h allant de 52,5 à plus de 1000 mg/l. Les facteurs les plus importants qui empêchent la fixation des ions Co2+ aux branchies sont la compétition avec les ions calcium (Ca2+) et la complexation du cobalt par les matières organiques dissoutes. Toutefois, l’effet des ions calcium sur la fixation et la toxicité potentielle du cobalt s’exerce à de très faibles concentration en Ca2+, probablement plus faibles que celles qui ont été utilisées dans les épreuves toxicologiques rapportées.
En ce qui concerne le milieu marin, on a proposé une valeur-guide de fiabilité moyenne égale à 20 m g/l (protection de 99 % des espèces marines avec une confiance de 50 %); pour les eaux douces, la valeur est fixée à 8 m g/l (protection de 95 % des espèces dulçaquicoles avec une confiance de 50 %). Si l’on compare ces valeurs-guides aux concentrations relevées dans l’environnement, il semblerait que c’est uniquement à proximité des sites importants de pollution anthropogénique que des effets seraient susceptibles de se produire. On possède quelques données selon lesquelles, dans les eaux douces extrêmement pauvres en ions calcium, le calcium est moins à même d’entrer en compétition avec le cobalt pour se fixer sur les récepteurs branchiaux, ce qui conduit à une fixation plus importante du cobalt. C’est par conséquent dans les eaux particulièrement douces (à très faible teneur en ions calcium) proches des sources de pollution anthropogéniques que les espèces aquatiques sont les plus menacées.
Les données relatives à la toxicité du cobalt pour les microorganismes terricoles sont limitées. On ne possède guère de preuves d’une toxicité du cobalt pour les végétaux poussant sur des sols à forte teneur en cet élément. Une tolérance au cobalt et à d’autres métaux a été constatée chez des plantes poussant sur des sols où ces métaux sont présent à forte concentration. Chez certaines espèces, on a pu montrer que la tolérance était due à une exclusion du cobalt mais chez d’autres, présentes dans des clairières cuprifères au sol riche en cobalt, il y a suraccumulation de ce métal. Des effets indésirables sur la croissance des lombrics et sur la reproduction du collembole nivicole ont été observés à des concentrations de 300 à 400 mg/kg de poids sec. Dans l’environnement terrestre, il semblerait peu probable que le cobalt ait des effets nocifs sur les oiseaux et les mammifères sauvages, et chez les ruminants, la carence est plus vraisemblable que l’intoxication.
Este CICAD6 sobre el cobalto y sus compuestos inorgánicos, preparado por Sciences International, Inc. de los Estados Unidos y el Centro de Ecología e Hidrología del Reino Unido, se basó en los exámenes realizados por la Agencia para el Registro de Sustancias Tóxicas y Enfermedades (ATSDR, 2004) y el Centro Internacional de Investigaciones sobre el Cáncer (IARC, 2005). Para incluir las citas bibliográficas que no figuraban en ninguno de estos exámenes, se realizó una búsqueda bibliográfica amplia en diversas bases de datos en línea en abril de 2005. La información sobre los documentos originales y su examen colegiado se presenta en el apéndice 2. La información sobre el examen colegiado de este CICAD aparece en el apéndice 3. Este CICAD se examinó y aprobó como evaluación internacional en una reunión de la Junta de Evaluación Final, celebrada en Nagpur (India) del 31 de octubre al 3 de noviembre de 2005. La lista de participantes en esta reunión figura en el apéndice 4. También se reproducen en el presente documento las Fichas internacionales de seguridad química para el cobalto, el óxido de cobalto (II), el óxido de cobalto (III), el sulfuro de cobalto (II), el cloruro de cobalto (II), el sulfato de cobalto (II), el sulfato de cobalto (II) heptahidrato, el nitrato de cobalto (II), el nitrato de cobalto (II) hexahidrato, el acetato de cobalto (II) tetrahidrato, el naftalenato de cobalto y el carbonilo de cobalto, preparadas por el Programa Internacional de Seguridad de las Sustancias Químicas (IPCS, 2000, 2001a–e, 2004a–f) en un proceso separado de examen colegiado.
El cobalto (número atómico 27) es un elemento presente en la naturaleza con un isótopo estable (59Co) y 26 isótopos radiactivos conocidos. Tiene tres valencias (0, +2 y +3). Debido a su posible condición de isótopo radiactivo, puede producir radiaciones ionizantes. Este documento se concentra fundamentalmente en el cobalto estable. El lector debe consultar otras fuentes, por ejemplo ATSDR (2004), para buscar información sobre los efectos de las radiaciones ionizantes de los isótopos radiactivos de cobalto.
El cobalto (CAS Nº 7440-48-4) es una sustancia sólida de color gris plateado a temperatura ambiente. Es el 33er elemento más abundante y se ha encontrado en diversos medios, como el aire, el agua superficial, las filtraciones de vertederos de desechos peligrosos, el agua freática, el suelo y los sedimentos. Las fuentes de exposición al cobalto y a sus compuestos inorgánicos son tanto naturales como antropogénicas. Son fuentes naturales el polvo arrastrado por el viento, el agua marina pulverizada, los volcanes, los incendios forestales y las emisiones biogénicas continentales y marinas. Entre las fuentes antropogénicas cabe mencionar la quema de combustibles fósiles, los fangos de alcantarillado, los fertilizantes fosfatados, la extracción y fusión de menas de cobalto, la preparación de aleaciones de cobalto y las industrias que utilizan o elaboran compuestos de cobalto.
El cobalto y sus compuestos inorgánicos no son volátiles y se liberan en la atmósfera como partículas. Se supone que el cobalto antropogénico procedente de la combustión se encuentra fundamentalmente en forma de óxidos. Durante los procesos de extracción y refinado también se liberan en la atmósfera las formas de sulfuro y arseniuro.
El cobalto liberado en la atmósfera se deposita en el suelo y el liberado en el agua se puede adsorber en partículas y pasar al sedimento o adsorberse directamente en él. Su coeficiente de distribución (por ejemplo, del agua al sedimento) varía en función del pH, las condiciones redox, la fuerza iónica y las concentraciones de materia orgánica disuelta. Los factores que afectan a la especiación y el destino del cobalto en el agua, los sedimentos y el suelo incluyen ligandos orgánicos como los ácidos húmicos, los aniones, el pH y el potencial redox. Su movilidad en el suelo es inversamente proporcional a su grado de adsorción en los componentes del suelo. Aunque las plantas pueden absorber cobalto del suelo, su translocación desde las raíces hasta otras partes de la planta no es significativa.
Las concentraciones de cobalto medidas en la atmósfera son de alrededor de 1 ng/m3 o menos en zonas que carecen de fuentes de este elemento y en general son de menos de 10 ng/m3 en las zonas que sí las tienen, aunque también se han notificado concentraciones más altas en estas últimas. Las concentraciones de cobalto en las aguas superficiales y freáticas son bajas, inferiores a 1 µg/l en zonas vírgenes y de 1–10 µg/l en zonas pobladas, pudiendo alcanzar valores mucho más elevados en zonas mineras y agrícolas, de hasta varios cientos de mg por litro. Las concentraciones medias de cobalto notificadas en el agua marina son inferiores a 1 µg/l, en el agua de bebida suelen ser de <1–2 µg/l y en el agua de lluvia de 0,3–1,7 µg/l. La corteza terrestre contiene como promedio una concentración de cobalto de 20–25 mg/kg. Cerca de fuentes antropogénicas, sus concentraciones en el suelo pueden ser de varios cientos de mg por kg.
La fuente más importante de exposición al cobalto para la población general es el suministro de alimentos. La ingesta estimada a partir de los alimentos es de 5–40 µg/día, en su mayor parte como cobalto inorgánico. Son diversas las industrias en las que hay exposición profesional a este elemento. Sus niveles en el tabaco varían entre <0,3 y 2,3 µg/g de peso seco y alrededor del 0,5% de esa cantidad está presente en la corriente principal de humo. Se encontró que sus concentraciones en el carbón, el petróleo bruto, el combustible y la gasolina en los Estados Unidos eran de 5 mg/kg, 0,001–10 mg/kg, 0,03–0,3 mg/kg y <0,1 mg/kg, respectivamente.
La inhalación de partículas de cobalto da lugar a su deposición en las vías respiratorias superiores e inferiores, donde pueden quedar retenidas o pasar a la sangre tras la disolución, o bien transferirse de forma mecánica al tracto gastrointestinal por acción mucociliar y deglución. Se absorbe alrededor del 50% del cobalto que entra en el sistema gastrointestinal. La absorción es mayor en las personas con deficiencia de hierro. Las formas solubles en agua se absorben mejor que las insolubles. El cobalto es esencial como componente de la vitamina B12; por consiguiente, se encuentra en la mayoría de los tejidos. Su acumulación total en el organismo se estima en 1,1–1,5 mg, con 0,11 mg en el hígado. Se han observado concentraciones más altas de cobalto en los pulmones tras la exposición por inhalación. No hay estudios que describan su distribución en las personas tras la ingestión, pero diversos estudios en animales indican que se retiene fundamentalmente en el hígado. En un estudio de exposición humana controlada a aerosoles, el 40% de la acumulación inicial de óxido de cobalto en los pulmones se retuvo seis meses después de la exposición. La excreción urinaria aumenta con el tiempo tras la exposición por inhalación. El tamaño de las partículas influye en la eliminación del cobalto inhalado, puesto que cuanto más grandes son las partículas más cobalto pasa mecánicamente al tracto gastrointestinal. La eliminación fecal es la principal vía de excreción en las personas tras la exposición oral.
La CL50 del hidrocarbonilo de cobalto por inhalación en ratas fue de 165 mg/m3 para una exposición de 30 minutos. Se ha señalado que la DL50 de los compuestos solubles de cobalto por vía oral es del orden de 42,4 a 317 mg/kg de peso corporal, en función del compuesto y la especie sometida a prueba. Se ha notificado que el tetraóxido de tricobalto, un compuesto insoluble de cobalto, tiene una DL50 de 3672 mg de cobalto por kg de peso corporal en ratas.
En ratas y ratones expuestos durante un periodo breve (16 días) a sulfato de cobalto por inhalación en concentraciones de 19 mg/m3 y 1,9 mg/m3, respectivamente, se observó necrosis e inflamación del epitelio del tracto respiratorio. En ratas se detectó asimismo necrosis del timo y atrofia testicular. Las ratas macho expuestas a cloruro de cobalto por vía oral con una concentración de cobalto de 12,4 mg/kg de peso corporal al día durante tres semanas mostraron lesiones cardíacas. Las ratas, conejos y ratones expuestos a compuestos de cobalto por inhalación en concentraciones de >0,3 mg/m3 (concentraciones de cobalto de >0,11 mg/m3) durante 3–4 meses mostraron lesiones del tracto respiratorio. En ratas expuestas durante 2–3 meses a sulfato de cobalto en la alimentación o a cloruro de cobalto en el agua de bebida con dosis de cobalto de 26–30,2 mg/kg de peso corporal al día se observó un aumento de peso del corazón y lesiones cardíacas degenerativas. En ratas expuestas a sulfato de cobalto con una concentración de cobalto de 8,4 mg/kg de peso corporal al día en la alimentación durante 24 semanas se registró una reducción significativa de los niveles de actividad enzimática en el corazón. Las ratas expuestas a cloruro de cobalto durante 4–5 meses con dosis de cobalto de 10–18 mg/kg de peso corporal al día mostraron daños renales.
En hámsteres expuestos a óxido de cobalto por inhalación durante toda la vida se desarrolló enfisema. En ratones y ratas expuestos a sulfato de cobalto por inhalación durante 105 semanas aparecieron tumores pulmonares de una manera que estaba relacionada con la dosis. Cuando se inyecta cobalto a ratas (en forma de polvo metálico de cobalto) por vía intramuscular, produce tumores del tipo de sarcomas.
Muchos compuestos de cobalto son genotóxicos en mamíferos y en sistemas de prueba de mamíferos y bacterianos. Los compuestos de cobalto(III) dan resultado positivo en los sistemas de prueba bacterianos. Los compuestos de cobalto(II) fueron positivos para las conversiones genéticas en Saccharomyces cerevisiae, pero por lo demás demostraron escasa actividad genotóxica.
Se ha comprobado que el cobalto tiene efectos en la reproducción y el desarrollo de los animales. Las ratas expuestas a cobalto (en forma de cloruro de cobalto) en concentraciones de 13,3–58,9 mg/kg de peso corporal día durante 2–3 meses y los ratones expuestos a cobalto (en forma de cloruro de cobalto) en concentraciones de 43,4 mg/kg de peso corporal al día durante 13 semanas mostraron degeneración y atrofia testiculares. En los ratones macho expuestos a cloruro de cobalto con dosis de 46,9 ó 93,0 mg/kg de peso corporal al día y apareados con ratones hembra no expuestos se registró una disminución del peso del epidídimo, el recuento de espermatozoides, el peso de los testículos y la fecundidad, valores medidos por el número de apareamientos con éxito. En estudios de desarrollo, las ratas preñadas expuestas a dosis de cloruro de cobalto con toxicidad materna (5,4 ó 21,8 mg de cobalto por kg de peso corporal al día) produjeron crías recién nacidas con crecimiento retardado y disminución de la supervivencia, pero no se observó ningún efecto teratogénico. En los conejos expuestos a cobalto (en forma de sulfato de cobalto) con dosis de 7,6 mg/kg de peso corporal al día se produjo mayor resorción fetal y un aumento del número de fetos con peso corporal retardado.
La inhalación y la exposición cutánea al cobalto en personas puede dar lugar a sensibilización. En trabajadores expuestos a diversas formas de cobalto se ha descrito asma bronquial.
Las personas que ingirieron cloruro de cobalto en concentraciones de 150 mg/día durante 22 días sufrieron policitemia y un aumento de la hemoglobina. También hay estudios en los que se ha descrito cardiomiopatía en personas que habían consumido grandes cantidades de cerveza que contenía sulfato de cobalto.
La neumopatía intersticial ocasionada por partículas metálicas con cobalto es una enfermedad pulmonar ocupacional que recibe también el nombre de enfermedad pulmonar por metales duros.
Los estudios de mortalidad en la industria de los metales duros parecen indicar un aumento de la mortalidad por cáncer de pulmón. El cobalto se utiliza como aglutinante en esta industria y también se produce exposición a otras sustancias, entre ellas el carburo de tungsteno y otros compuestos metálicos, como el carburo de titanio, el carburo de tantalio y el carburo de niobio.
Se utilizó un estudio transversal de pulidores de diamantes expuestos al cobalto para derivar una concentración tolerable por inhalación de 1 × 10−4 mg/m3, basada en la disminución de la función pulmonar. La concentración tolerable suele ser 10 veces superior a las concentraciones de cobalto que se encuentran en el aire ambiente cerca de fuentes antropogénicas.
La CE50 notificada para el cobalto a las 96 horas basada en el crecimiento del alga de agua dulce Chlorella vulgaris fue de 0,6 mg/l, mientras que la CE50 para las plantas vasculares acuáticas fue de 0,1 y 0,2 mg/l. La CE50 del cobalto a los cinco días basada en el crecimiento de la diatomea marina Ditylum brightwellii fue de 0,3 mg/l. Para los invertebrados de agua dulce, la CL50 (24–96 horas) aguda oscila entre 1,1 y 239 mg/l. Se describieron varios estudios sobre la reproducción de Daphnia magna, con una CE50 a los 21 días de 0,01 mg/l y una NOEC a los 28 días de 0,003 mg/l; sin embargo, en estudios posteriores se encontró una NOEC a los 21 días que iba de 0,03 a 0,05 mg/l para diversos niveles de carbonato cálcico. La NOEC más baja notificada para organismos acuáticos fue la de la pulga de agua Ceriodaphnia dubia en una prueba de siete días, con <0,003 mg/l. Los invertebrados marinos más sensibles fueron las larvas de langosta, con una CL50 a las 96 horas comprendida entre 4,5 y 22,7 mg/l. La CL50 a las 96 horas para los peces de agua dulce oscila entre 1,4 y 333 mg/l. Se notificó una NOEC a los 16 días basada en la supervivencia de 0,06 mg/l. Los resultados de las pruebas realizadas con peces marinos parecen indicar que por lo menos las especies sometidas a prueba son relativamente insensibles al cobalto, con una CL50 a las 96 horas que va de 52,5 a >1000 mg/l. En pruebas realizadas en aguas naturales, la competencia del Ca2+ y la formación de complejos con materia orgánica disuelta fueron los factores más importantes que impidieron la unión del Co2+ a las agallas. Sin embargo, el efecto de los iones Ca2+ en la absorción y la posible toxicidad del cobalto se produce con concentraciones muy bajas de Ca2+, probablemente inferiores a las utilizadas en cualquiera de las pruebas de toxicidad descritas.
Los valores de orientación de una fiabilidad moderada fueron para el medio marino de 20 µg/l (para la protección del 99% de las especies marinas con una confianza del 50%) y para el medio de agua dulce de 8 µg/l (para la protección del 95% de las especies de agua dulce con una confianza del 50%). La comparación de los valores de orientación con las concentraciones en el medio ambiente parece indicar que probablemente sólo se producirán efectos en las proximidades de zonas con una liberación antropogénica importante. Hay algunas pruebas de que, en condiciones de agua dulce con una concentración extraordinariamente baja de Ca2+, el cobalto tiene menos competencia por los lugares de unión en las agallas de los peces y, en consecuencia, hay mayor absorción de cobalto. Por consiguiente, el mayor riesgo para los organismos acuáticos podría estar en las zonas de aguas muy blandas (con una concentración extraordinariamente baja de ión Ca2+) cercanas a fuentes de liberación antropogénica.
Los datos relativos a la toxicidad del cobalto para los microorganismos del suelo son limitados. Son escasas las pruebas de toxicidad del cobalto para las plantas debida a concentraciones elevadas en el suelo. Se ha encontrado tolerancia al cobalto, junto con tolerancia a otros metales, en poblaciones de plantas que crecen en suelos con una concentración elevada de metales concretos. Se ha demostrado que el metal no interviene en la tolerancia al cobalto de algunas especies, mientras que en otras que crecen en zonas de explotación de cobre ricas en cobalto se produce una hiperacumulación de éste. Se han notificado efectos adversos en el crecimiento de las lombrices de tierra y la reproducción de los colémbolos con 300–400 mg/kg de peso seco. En el medio terrestre, parecen poco probables los efectos adversos del cobalto en las aves y los mamíferos silvestres, siendo más probable en los rumiantes la deficiencia de cobalto que su toxicosis.
ENDNOTES:
See Also:
Toxicological Abbreviations