
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 93
CHLOROPHENOLS OTHER THAN PENTACHLOROPHENOL
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Organization
Geneva, 1989
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organization, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Chlorophenols other than pentachlorophenol
(Environmental health criteria; 93)
1. Chlorophenols I. Series
ISBN 92 4 154293 4 (NLM Classification: QV 223)
ISSN 0250-863X
(c) World Health Organization 1989
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. For rights of reproduction or
translation of WHO publications, in part or in toto, application
should be made to the Office of Publications, World Health
Organization, Geneva, Switzerland. The World Health Organization
welcomes such applications.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city or area or
of its authorities, or concerning the delimitation of its frontiers or
boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
1. SUMMARY
1.1. Identity, physical and chemical properties,
analytical methods
1.2. Sources of human and environmental exposure
1.2.1. Production figures
1.2.2. Manufacturing processes
1.2.3. Uses
1.2.4. Waste disposal
1.2.5. Release of chlorophenols into the environment
1.2.6. Natural sources
1.3. Environmental transport, distribution, and transformation
1.3.1. Degradation
1.3.2. Bioaccumulation
1.3.3. Effects of physical chemical and biological
factors on degradation
1.4. Environmental levels and human exposure
1.4.1. Chlorophenol levels in the environment
1.4.2. Chlorophenol levels in food, drinking-water, and
treated wood
1.5. Kinetics and metabolism
1.6. Effects on organisms in the environment
1.7. Effects on experimental animals and in vitro systems
1.8. Effects on man
1.8.1. Non-occupational exposure
1.8.2. Occupational exposure
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sample collection and storage
2.4.2. Sample preparation and analysis
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Man-made sources
3.2.1. Production levels and processes
3.2.1.1 World production figures
3.2.1.2 Manufacturing processes
3.2.2. Uses
3.2.2.1 Wood treatment
3.2.2.2 Agriculture
3.2.2.3 Domestic
3.2.2.4 Water treatment
3.2.2.5 Additives
3.2.2.6 Intermediates in industrial syntheses
3.2.3. Other sources
3.3. Waste disposal
3.4. Losses of chlorophenols into the environment
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution
4.1.1. Atmospheric movement
4.1.1.1 Volatilization
4.1.2. Soil movement
4.1.2.1 Adsorption
4.1.2.2 Leaching
4.1.3. Transport in aquatic environments
4.2. Degradation and bioaccumulation
4.2.1. Degradation
4.2.1.1 Abiotic degradation
4.2.1.2 Degradation by microorganisms
4.2.2. Bioaccumulation
4.3. Effects of other physical, chemical, or biological factors
4.3.1. pH
4.3.2. Lack of oxygen
4.3.3. Inorganic nutrients
4.3.4. Organic matter
4.3.5. Temperature
4.4. Persistence
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.2.1 Sediments
5.1.3. Soil
5.1.4. Food and feed, drinking-water
5.1.4.1 Food
5.1.4.2 Livestock feed
5.1.4.3 Drinking-water
5.1.5. Treated wood
5.1.6. Terrestrial and aquatic organisms
5.1.6.1 Invertebrates
5.1.6.2 Fish
5.1.6.3 Other non-human vertebrates
5.2. General population exposure
5.3. Occupational exposure
6. KINETICS AND METABOLISM
6.1. Absorption
6.2. Distribution
6.2.1. Tissue distribution following chlorophenol
exposure
6.2.2. Tissue distribution following exposure to
chemicals metabolized to chlorophenols
6.3. Metabolic transformation
6.4. Elimination and excretion
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Laboratory toxicity studies
7.1.1. Acute toxicity
7.1.2. Long-term toxicity
7.1.3. Organoleptic effects
7.2. Toxicity studies under natural environment conditions
7.2.1. Bacteria
7.2.2. Phytoplankton
7.2.3. Zooplankton
7.2.4. Fish
7.2.5. Effects on physical and chemical variables
7.3. Treatment levels
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO SYSTEMS
8.1. Acute studies
8.2. Skin and eye irritation; sensitization
8.3. Short-term exposure
8.4. Long-term exposure
8.5. Reproduction, embryotoxicity, and teratogenicity
8.6. Mutagenicity and related end-points
8.7. Carcinogenicity
8.8. Factors modifying toxicity; metabolism
8.9. Mechanisms of toxicity, mode of action
9. EFFECTS ON MAN
9.1. Acute toxicity
9.2. Long-term exposure
9.2.1. Effects on skin and mucous membranes
9.2.2. Systemic effects
9.2.3. Psychological and neurological effects
9.2.4. Reproductive effects
9.2.5. Carcinogenicity
9.2.5.1 Case-control studies reviewed by IARC
9.2.5.2 Cohort studies reviewed by IAC
9.2.5.3 More recent studies
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health risks
10.1.1. Exposure levels
10.1.1.1 Non-occupational exposure
10.1.1.2 Occupational exposure
10.1.2. Toxic effects
10.1.3. Risk evaluation
10.2. Evaluation of effects on the environment
10.2.1. Levels of exposure
10.2.2. Transport
10.2.3. Degradation
10.2.4. Bioaccumulation
10.2.5. Persistence
10.2.6. Toxic effects on environmental organisms
10.2.7. Risk evaluation
11. RECOMMENDATIONS
11.1. Production
11.2. Disposal
11.3. Occupational exposure
11.4. General population exposure
11.5. Recommendations for future research
11.5.1. Environmental Aspects
11.5.2. Toxicology
11.5.3. Epidemiology
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR CHLOROPHENOLS
OTHER THAN PENTACHLOROPHENOL
Members
Dr U.G. Ahlborg, Unit of Toxicology, National Institute of
Environmental Medicine, Stockholm, Sweden
Dr L.A. Albert, Division of Studies on Environmental Pollution,
National Institute for Research on Biotic Resources, Vera Cruz,
Mexico (Vice-Chairman)
Dr F.A. Chandra, Toxicology and Environmental Health, Department of
Health and Social Security, London, United Kingdom
Dr A. Gilman, Industrial Chemicals and Product Safety Section, Bureau
of Chemical Hazards, Environmental Health Directorate, Department
of National Health and Welfare, Tunney's Pasture, Ottawa, Canada
Dr I. Gut, Biotransformation, Institute for Hygiene and Epidemiology,
Prague, Czechoslovakia (Chairman)
Dr R. Jones, Health and Safety Executive, Bootie, Merseyside, United
Kingdom
Dr J. Kangas, Kuopio Regional Institute of Occupational Health,
Kuopio, Finland
Dr E. Lynge, Danish Cancer Registry, Institute of Cancer Epidemiology,
Copenhagen, Denmark
Dr U.G. Oleru, Department of Community Health, College of Medicine,
University of Lagos, Lagos, Nigeria
Dr J.K. Selkirk, Division of Toxicology Research and Testing,
Carcinogenesis and Toxicological Evaluation Branch, National
Institute of Environmental Health Sciences, Research Triangle
Park, NC, USA
Dr A. van der Gen, Leiden University, Leiden, Netherlands
Observer
Dr S. Lambert (European Chemical Industry Ecology and Toxicology
Centre), Rhône Poulenc, Décines Charpieu, France
Secretariat
Dr G.C. Becking, Team Leader, International Programme on Chemical
Safety, Interregional Research Unit, World Health Organization,
Research Triangle Park, NC, USA (Secretary)
Dr T. Kauppinen, International Agency for Research on Cancer, Lyons,
France
Mr R. Newhook, Bureau of Chemical Hazards, Environmental Health
Directorate, Department of National Health and Welfare, Tunney's
Pasture, Ottawa, Canada (Temporary Adviser, Rapporteur)
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the criteria
documents as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria documents, readers are kindly requested to communicate any
errors that may have occurred to the Manager of the International
Programme on Chemical Safety, World Health Organization, Geneva,
Switzerland, in order that they may be included in corrigenda, which
will appear in subsequent volumes.
***
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Palais des
Nations, 1211 Geneva 10, Switzerland (Telephone no. 7988400-7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR CHLOROPHENOLS OTHER THAN
PENTACHLOROPHENOL
A WHO Task Group on Environmental Health Criteria for
Chlorophenols other than Pentachlorophenol met at the Monitoring and
Assessment Research Centre, London, United Kingdom, on 21-25 March,
1988. Dr M. Hutton opened the meeting and welcomed the members on
behalf of the host institute and on behalf of the United Kingdom
Department of Health and Social Security, who sponsored the meeting.
Dr G.C. Becking addressed the meeting on behalf of the three
Cooperating Organizations of the IPCS (UNEP, ILO, and WHO). The Task
Group reviewed and revised the draft criteria document and made an
evaluation of the risks for human health and the environment from
exposure to chlorophenols other than pentachlorophenol.
The drafts of this document were prepared by Mr R. NEWHOOK and
Dr A. GILMAN, Health Protection Branch, Ottawa, Canada. Dr G. BECKING,
IPCS Interregional Research Unit, was responsible for the overall
scientific content of the document and Mrs M.O. HEAD, Oxford, England,
for the editing.
The efforts of all who helped in the preparation and finalization
of the document are gratefully acknowledged.
***
Partial financial support for the publication of this criteria
document was kindly provided by the United States Department of Health
and Human Services, through a contract from the National Institute of
Environmental Health Sciences, Research Triangle Park, North Carolina,
USA -- a WHO Collaborating Centre for Environmental Health Effects.
The United Kingdom Department of Health and Social Security generously
supported the costs of printing.
1. SUMMARY
1.1 Identity, Physical and Chemical Properties, Analytical Methods
Chlorophenols (CPs) are organic chemicals formed from phenol
(1-hydroxybenzene) by substitution in the phenol ring with one or more
atoms of chlorine. Nineteen congeners are possible, ranging from
monochlorophenols to the fully chlorinated pentachlorophenol (PCB).
Chlorophenols, particularly trichlorophenols (T3CP), tetrachloro-
phenols (T4CP), and PCP, are also available as sodium or potassium
salts.
Chlorophenols are solids at room temperature, except for 2-MCP,
which is a liquid. The aqueous solubility of chlorophenols is low, but
the sodium or potassium salts of chlorophenols are up to four orders
of magnitude more soluble in water than the parent compounds. The
acidity of chlorophenols increases as the number of chlorine sub-
stitutions increases. The n-octanol/water partition coefficients
of chlorophenols increase with chlorination, indicating a propensity
for the higher chlorophenols to bioaccumulate. Taste and odour
thresholds are quite low.
Technical grade chlorophenol products are heterogeneous mixtures
of chlorophenols, unreacted precursors, and a variety of dimeric
microcontaminants. As a result of the semiquantitative nature of the
reaction of chlorine with molten phenol, commercial formulations of
chlorophenols contain substantial quantities of other chlorophenols.
When the alkaline hydrolysis of chlorobenzenes is used to manufacture
chlorophenols, the technical product can contain unreacted
chlorobenzene.
A number of other compounds are present as microcontaminants in
technical tri- and tetrachlorophenol preparations, as a result of the
elevated reaction temperatures used. These include the polychlorinated
dibenzo- p-dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs),
polychlorinated phenoxyphenols ("predioxins"), polychlorinated
diphenyl ethers, polychlorinated benzenes, and polychlorinated
biphenyls. Lower chlorophenol preparations do not contain detectable
levels of dioxins, presumably because their manufacture does not occur
at sufficiently high temperatures. Tri- and tetrachloro-dibenzo-
p-dioxins predominate in T3CP formulations, while the hexa, hepta,
and octa congeners are the major PCDD contaminants in technical T4CP
and PCP. 2,3,7,8-Tetra-chlorodibenzo- p-dioxin (2,3,7,8-TCDD) occurs
primarily as a contaminant of 2,4,5,-T3CP, though it is present at
low µg/litre concentrations in T4CP, PCP, and Na-PCP. Chlorophenol
formulations contain a similar array of PCDFs. Phenoxyphenols may
comprise as much as 1-5% of the formulation.
A large number of sampling and analytical methods have been
developed for the determination of chlorophenols in different media.
Sensitive methods, such as gas chromatography, high-performance liquid
chromatography, and mass spectrometry are increasingly used.
1.2 Sources of Human and Environmental Exposure
1.2.1 Production figures
Recent data on production levels of chlorophenols other than PCP
are not readily available. Around 1975, the combined global production
of all chlorophenols approached 200 million kg; slightly more than
half of this quantity consisted of non-PCP chlorophenols, primarily
2,4-dichlorophenol (2,4-DCP), 2,4,5-trichlorophenol (2,4,5-T3CP),
and 2,3,4,6-tetrachlorophenol (2,3,4,6-T4CP). Consumption has since
declined in some countries as a consequence of health-based concerns
(particularly for 2,4,5-T3CP), and the use of alternative wood
preservatives. Some European countries and the USA are major producers
and consumers of chlorophenols.
1.2.2 Manufacturing processes
The compounds 2-MCP, 4-MCP, 2,4-DCP, 2,3,4-T3CP, 2,4,6-T3CP,
2,3,4,6-T4CP, and PCP have been made by direct stepwise chlorination
of phenol or lower chlorinated phenols at a high temperature; a
catalyst is necessary if the last two chlorophenols are being
produced. Alternatively, some chlorophenols (2,5-DCP, 3,4-DCP,
2,4,5-T3CP, 2,3,4,5-T4CP and PCP) can be produced by the alkaline
hydrolysis of the appropriate chlorobenzene.
Both methods yield contaminants that are themselves potential
health hazards, including polychlorinated dibenzo- p-dioxins (PCDDs),
polychlorinated dibenzofurans (PCDFs), and 2-phenoxyphenols.
1.2.3 Uses
Chlorophenols are toxic for a wide range of organisms, a property
that accounts for many of their uses. Large quantities of higher
chlorophenols are used in pressure treatment in the wood preservation
industry; in addition, substantial amounts of the sodium salts of
T4CP, PCP, and T3CP are used to surface-treat fresh-cut logs and
lumber against sapstain fungi and surface mould. Large quantities of
lower chlorophenols serve as intermediates in the production of
pesticides, such as T4CP, PCP, 2,4-D, and 2,4,5-T. The use of 2,4,5-T
has been discontinued in a number of countries. Lesser amounts of
chlorophenols are used as wood preservatives in agricultural and
domestic applications, and as additives to inhibit microbial growth in
a wide array of products, such as adhesives, oils, textiles, and
pharmaceutical products.
1.2.4 Waste disposal
As a result of process design, the quantities of chlorophenolic
wastes generated are reportedly small. Available treatment methods for
such waste should prove satisfactory, if they are carefully applied.
Gravity separation is the primary treatment method most often used to
recover oil and the associated chlorophenol for recycling and
treatment. Organisms during secondary treatment degrade roughly 90% of
most chlorophenol waste, provided that they are acclimated to the
waste, and precautions are taken against shock loadings. Adsorption
onactivated carbon as a final clean-up step removes almost 100% of
remaining waste chlorophenols in waste-streams. Incineration appears
to be an effective means of disposal, if the temperatures are high
enough and residence times long enough to ensure complete combustion
and prevent the formation of PCDDs and PCDFs in the incinerator.
1.2.5 Release of chlorophenols into the environment
Patterns of losses to the environment appear similar in most
industrialized countries. The majority of chlorophenol wastes are
released in spills and leaching from treated lumber (PCP, NaPCP,
NaT4CP), and as contaminants or breakdown products of agricultural
pesticides (2,4-DCP, 2,4,5-T3CP). Substantial amounts of
chlorophenol wastes (NaT4CP, NaPCP) are released from sawmills,
planer mills, and the incineration of wood wastes. Significant amounts
of chlorophenols can be formed and subsequently released into the
environment from the chlorine bleaching process in pulp and
paper-mills, the chlorination of waste-water and drinking-water, and
the incineration of municipal waste. A significant amount of wastes is
discharged from manufacturing sites. Losses during storage and
transport are negligible. No estimates are available of the quantities
of chlorophenols released as a result of the disinfection of
waste-waters with chlorine, volatilization, or domestic uses of
products containing these compounds.
1.2.6 Natural sources
While some chlorophenols and related organohalogens occur
naturally, as metabolites of certain flora and fauna, these sources
are thought to make a negligible contribution to overall environmental
levels.
1.3 Environmental Transport, Distribution, and Transformation
Chlorophenols adsorb strongly on acidic soils, and those with a
high organic content. Leaching is more significant in basic and
mineral soils. Studies to date have not addressed the quantitative
contribution of these processes to the transport of chlorophenols
in situ.
Adsorption appears to play an important role in surface waters.
Chlorophenols that are not degraded in the water body are incorporated
into the sediments, most likely because they adsorb on sediment
particulates. They may persist in sediments for years. However, it is
not known how important this process is for lower chlorophenols, since
they should be adsorbed to a lesser extent than the T4CPs and PCP
studied to date.
While a large part of the chlorophenols entering natural waters is
probably degraded, they are nonetheless fairly persistent and, thus,
may be transported considerable distances by water.
Although chlorophenols are principally water and soil
contaminants, some atmospheric movement occurs, and low levels of PCP
have been found in rain, snow, and outdoor air. No corresponding
measurements have been made for other chlorophenols, but it is highly
probable that they too are transported in this manner.
1.3.1 Degradation
Chlorophenol residues are removed from the environment by both
biological and non-biological degradation. Laboratory studies have
shown that ultraviolet radiation can break down chlorophenols in a
matter of hours to days, and the shifts in the ratio of PCP to some of
its breakdown products in situ suggest that this process is
important in exposed habitats.
A large number of bacteria and fungi from different habitats are
able to degrade chlorophenols in the laboratory, sometimes eliminating
tens of mg/litre in a matter of hours or days. Degradation is
generally slowest for the higher chlorinated phenols, and for those
with a chlorine in the "meta" position. Previous exposure to a given
chlorophenol or a related compound enables a microorganism to
metabolize it immediately and/or at a faster rate, presumably by
inducing the necessary enzymes. In general, anaerobic biodegradation
of these compounds is much slower than aerobic metabolism.
Considerable overlap appears to exist in the rates of biodegradation
of the compounds in different habitats.
But chlorophenols should only persist in environments where the
rates of these transformations are minor. The persistence of
chlorophenols other than PCP has not been studied under controlled
conditions, but spills and applications of PCP as a herbicide
reportedly disappear in a matter of weeks or months.
1.3.2 Bioaccumulation
Bioaccumulation of chlorophenols appears to be moderate, and most
bioconcentration factors (BCFs) fall roughly between 100 and 1000. The
biocentration factor is usually a positive function of the chlorine
number, and there are no obvious relationships between it and the type
of organism (algae, plants, invertebrates, fish). Once exposure is
discontinued, chlorophenols clear rapidly from biota, indicating that
the bioaccumulation observed in field studies is the result of
long-term exposure rather than persistence.
1.3.3 Effects of physical, chemical, and biological factors on
degradation
Both the rate of evaporation and the extent of adsorption of PCP
(and undoubtedly other chlorophenols) are inversely related to pH. In
contrast, the rates of photolysis of 4-MCP and 2,4-DCP both increase
with pH, and shortage of oxygen, inorganic nutrients, or organic
matter may all influence the biodegradation rate of various lower
chlorophenols. Higher temperatures increase the rates of evaporation,
photolysis, and microbial degradation of chlorophenols, although the
last process obviously has an upper limit.
1.4 Environmental Levels and Human Exposure
1.4.1 Chlorophenol levels in the environment
Data on levels of chlorophenols other than PCP in the environment
are not available for air. Levels of PCP in outdoor air range from 1
to several ng/m3. Work-place air concentrations of chlorophenols are
much higher. Facilities in which chlorophenols are used, such as
sawmills, often have air levels of several tens of µg/m3, while in
manufacturing facilities, concentrations may be in the mg/m3 range.
Residues of all chlorophenol isomers have been found in fresh and
marine waters. In relatively undeveloped areas, levels are often
undetectable in receiving waters, and only occasionally exceed
1 µg/litre close to industrial sources of chlorophenols. In receiving
waters from heavily industrialized regions, ambient levels are
somewhat higher, but still median concentrations do not exceed
1 µg/litre, while the maximum concentrations in surface waters and
ground waters can reach several µg/litre. As a result of spills,
isolated levels as high as 61 000 µg/litre of chlorophenols (T4CP +
PCP) in ground water, and 18 090 µg/litre in surface waters have been
reported.
Levels of some chlorophenols in effluents from chemical and wood
preservation industries may reach several thousand µg/litre, though
typical levels are in the low µg/litre range, and dilution apparently
reduces these to the observed low ambient levels.
Chlorophenol concentrations in sediments are generally higher than
those in the overlying water. Levels in sediments from waters not
receiving large chlorophenol inputs generally contain less than 1 µg
of the individual chlorophenols/kg dry sediment. The maximum levels of
all chlorophenol isomers in fresh-water sediments in industrialized
regions seldom exceed 50 µg/kg. However, in some instances, thousands
of µg chlorophenols/kg have been detected in fresh-water sediments
adjacent to point sources (spillage sites and effluent discharges).
In waters receiving chlorophenolic wastes, invertebrates generally
contain from trace levels to 20 µg of chlorophenols from the
surrounding environments/kg wet tissue, though levels approaching
200 µg/kg have been observed in some instances. Fish can contain
similar whole-body levels of chlorophenols, usually concentrated in
the liver and viscera. For example, liver tissues from sculpins
inhabiting polluted waters contained up to 1600 µg/kg wet weight. In
birds, muscle tissues exhibited only trace to moderate (50 µg/kg wet
weight) levels of chlorophenols, however, higher concentrations have
been found in single samples of liver, brain, kidney, and eggs. For
instance, a level of 1017 µg 2,4-DCP/kg (fresh weight) was found in
the kidney of an eagle.
1.4.2 Chlorophenol levels in food, drinking-water, and treated wood
Quantities of T4CP range from trace to several µg/kg in carrots,
potatoes (also 2,4-DCP), turnips, cabbages, beets, and raw milk,
though contamination from treated wood storage containers can elevate
these levels considerably. Recent restrictions on the agricultural use
of chlorophenols have reduced this contamination. T4CP has been
detected in poultry, but no reports of residues in other meat have
been found.
Drinking-water supplies are characterized by relatively low
concentrations of chlorophenols. While a variety of congeners have
been detected, these are usually present in the range of 10-3 to
10-1 µg/litre.
Concentrations of PCP or T4CP in treated wood are predictably
high, and can reach several hundred mg/kg of wood dust or shavings.
1.5 Kinetics and Metabolism
The lower chlorophenols are readily absorbed across the skin of
both laboratory animals and human beings. The results of studies on
rats further suggest that absorption via the skin is greater for the
sodium salts than for the parent molecules (2,3,5,6-T4CP and its salt
were used). Ingested chlorophenols are also readily taken up from the
gastrointestinal tract. The absorption of inhaled lower chlorophenols
by experimental animals has not been studied.
Experimental animals accumulate chlorophenols mostly in the liver
and kidney, and to a lesser extent in the brain, muscle, and fat
tissues. The higher levels in the liver and kidney may reflect their
greater circulating blood volume, as well as the role these organs
play in the detoxification and elimination of these compounds. Related
compounds, such as trichlorophenyl acetate, 2,4-D, Nemacide, Silvex,
2,4,5-T, and lindane, yield similar tissue distributions of
chlorophenol metabolites.
In the animals studied to date, most chlorophenols were rapidly
conjugated to glucuronates or sulfates in the liver. This binding, and
also dechlorination and methylation, serve to detoxify these
compounds. At present, the only chlorinated phenol that is known to be
metabolized to a more toxic substance is 2,3,5,6,-T4CP, which gives
rise to tetrachloro- p-hydroquinone. The corresponding quinone has
been shown to bind covalently to protein and DNA.
Chlorophenols are eliminated by test mammals primarily through the
urine (roughly 80-90%), in both free and bound forms. Smaller amounts
are eliminated in faecal matter. A single dose of chlorophenols is
virtually eliminated within one to several days. Elimination rates
appear to be even more rapid for some tissues.
1.6 Effects on Organisms in the Environment
The available information on the effects of chlorophenols in the
environment centres primarily on aquatic organisms. Considerable
overlap exists in the concentrations that are toxic for bacteria,
phytoplankton, plants, invertebrates, and fish, most of the EC50 and
LC50 values falling in the several mg/litre range. Toxicity generally
increases with the degree of chlorination of the phenol ring. However,
chlorophenols with chlorine in the 3 and 5 positions ("meta"
chlorophenols) are often more toxic than expected solely on the basis
of their chlorine number. Species-specific sensitivity can override
these general patterns. Furthermore, particularly in the case of the
higher chlorophenols, acute toxicity is a strong inverse function of
pH, reflecting the degree of ionization of the chemical. In long-term
studies, sublethal levels of 2,4-DCP reduced both growth and survival
of fathead minnows. In one study, exposure to a concentration of only
0.5 µg 2,4,6-T3CP/litre was fetotoxic in trout.
Fish kills have resulted from PCP spills, some of which have also
involved T4CP. In controlled field studies, exposure to large
quantities (100-5000 µg/litre) of chlorophenols (4-MCP, 2,4-DCP,
2,4,6-T3CP) generally impaired algal primary production and
reproduction, altered algal species composition dramatically, and
reduced zooplankton biomass and production. These studies shed little
light on the hazard, if any, presented by the low-level contamination
observed in most environments. The low concentrations of several
chlorophenols typically found in moderately contaminated waters have
been reported to impair the flavour of fish.
1.7 Effects on Experimental Animals and In Vitro Systems
In rats, lethal doses of lower chlorinated phenols resulted in
tremors and convulsions (except for T4CP and some T3CPs), hypotonia,
and, after death, a rapid onset of rigor mortis. Acute LD50s for rats
for all lower chlorophenols and routes of administration ranged from
130 to 4000 mg/kg body weight. The range of toxicity of the compounds
generally occurred in the following order: T4CPs > MCP > DCPs >
T3CPs, when the toxicant was administered either orally or by
subcutaneous injection. When injected intraperitoneally, the
toxicities of MCP, DCPs, and T3CPs were similar, while T4CP was 2-3
times more toxic. In studies on dermal exposure, 2,3,5,6-T4CP was the
most toxic of the T4CP isomers. These variations according to route
of administration may reflect differences in the rate of absorption of
the compounds. Acute effects are attributable to the parent
chlorophenol itself rather than to the microcontaminants.
Some reports have indicated that lower chlorinated phenols cause
mild irritation of the eye in rats. This effect increases with the
number of chlorine atoms on the phenol ring. Skin sensitization has
not been shown for the chlorophenols.
Short-term exposures of rats and mice to 2,4-DCP at hundreds of
mg/kg have been consistently associated with increased spleen and
liver weights and, in some instances, with haematological or
immunological effects. The very few studies concerning exposure to
various tri- and tetrachlorophenols have also identified
exposure-related changes in the weight or histology of the liver and,
in some instances, of the spleen or kidney. In one study, combined
pre- and postnatal exposure to 2-MCP and 2,4-DCP resulted in
haematological changes in exposed rats, but only 2,4-DCP elicited
immune responses.
Several lower chlorophenols appear to be mildly fetotoxic, though
the data are inconsistent in this regard. While female rats exposed to
2-MCP, 2,4-DCP, or 2,4,6-T3CP in the drinking-water produced smaller
litters with an increased frequency of stillborn offspring in one
study, similar or higher exposures in other studies did not have any
effects on these and other reproductive parameters. A dose of 30 mg/kg
body weight per day of pure or technical 2,3,4,6-T4CP delayed
ossification of fetal skull bones, but was not embryolethal.
Birth defects did not arise as a result of daily exposure of rats
to concentrations of up to 500 mg 2-MCP/litre, 300 mg 2,4-DCP/litre
(both in the drinking-water), 1000 mg 2,4,6-T3CP/kg body weight and
30 mg 2,3,4,6-T4CP/kg body weight (both by gavage).
Limited information indicates that 2,4,6-T3CP (in yeast and
mammalian test systems) and 2,3,4,6-T4CP (Chinese hamster cell
cultures) elicited weak mutagenic responses, but were not clastogenic.
Most of the other chlorophenols that have been tested have been found
to be non-mutagenic in the few test systems used (primarily
bacterial).
Exposure of rats and mice (both sexes) to 2,4-DCP for 2 years at
doses as high as 440 and 1300 mg/kg body weight per day, respectively,
proved negative with respect to carcinogenicity. In a test with a
similar design, 2,4,6-T3CP at doses of up to 10 000 mg/kg body
weight per day caused cancer in mice (hepatocellular carcinomas or
adenomas) and male rats (lymphomas, leukaemia). The 2,4,6-T3CP used
was commercial grade and was not analysed for impurities, such as
PCDDs and PCDFs.
Studies on rats on the carcinogenicity of 2-MCP or 2,4-DCP
(500 µg/litre and 300 µg/litre, respectively, for 15-24 months) were
inadequate. Some chlorophenols appeared to be promoters (MCPs,
2,4-DCP, and 2,4,5-T3CP); others did not.
Exposure of female rats to 2,4-DCP in the drinking-water, at
0-300 mg/litre, altered the major immune function in offspring exposed
prenatally and postnatally, but not in rats exposed only in utero.
In contrast, in a similar study, a concentration of 2-MCP as high as
500 mg/litre did not have any adverse effects on the immune systems of
rats.
The major effects observed with lethal exposures to chlorophenols
indicated a general effect on the nervous system. Long-term studies
implicated the liver and kidney as organs that accumulate high
concentrations of chlorophenols and are often adversely affected by
exposure to chlorophenols, perhaps reflecting their roles in the
detoxification and elimination of xenobiotics. On the basis of the
suppression of cell-mediated immunity in rats exposed to 2,4-DCP, it
can be assumed that the thymus and spleen may be target organs.
The toxicology of chlorophenols is complicated by the presence of
PCDD and PCDF microcontaminants in technical grade products.
Assessment of toxicity studies with chlorophenols requires a knowledge
of the types, levels, and effects of the microcontaminants that are
present in the formulation studied, because some PCDDs and PCDFs are
extremely toxic.
The major mode of action in the acute toxicity of chlorophenols
involves the uncoupling of oxidative phosphorylation and the
inhibition of the electron transport system. These effects are related
to the number of chlorine atoms on the molecule and to a lesser extent
by their positions on the molecule. PCP is 40 times more potent than
2,4-DCP as an uncoupler. The chlorophenate ion is evidently
responsible for the uncoupling reaction, while the undissociated
molecule causes convulsions.
Other enzyme systems are also inhibited by exposure to
chlorophenols in vitro, though, in some instances, such inhibition
is not observed with in vivo exposures.
1.8 Effects on Man
1.8.1 Non-occupational exposure
Low (usually 10 mg/kg) levels of the lower chlorinated phenols are
found in the serum, urine, and adipose tissues of the general
population. The major identifiable sources of these chlorophenols are
food and drinking-water. Chlorophenol levels in the ambient atmosphere
have not been measured.
In the only instance of acute exposure of the general population
to chlorophenols, an explosion at a manufacturing plant contaminated
an area, with a population of 37 000 persons, with sodium hydroxide,
2,4,5-T3CP, and TCDD. However, the effects, if any, of the released
2,4,5-T3CP were masked by those of TCDD. Clinical symptoms
attributed to TCDD were recorded in the exposed individuals. No toxic
effects have been attributed to the low concentrations of
chlorophenols typical of most non-occupational exposures. However,
undesirable organoleptic effects are produced by chlorophenols at very
low concentrations.
1.8.2 Occupational exposure
Worker exposure is a major concern in industries in which
chlorophenols are used extensively, as respiratory and dermal
absorption of these compounds results in measurable levels in the
blood and urine of exposed workers. In the manufacture of
chlorophenols, clinical symptoms associated with exposure include eye,
nose, and airway irritation, dermatitis, chloracne, and porphyria.
Abnormal liver function tests, changes in brain wave activity, and
slowed visual reaction time have been reported in association with
high-level exposure.
In sawmill workers, Na-T4CP exposures have caused numerous cases
of dermatitis and respiratory irritation. Eye, nose, and airway
irritation from exposure to T3CP have been reported by gas mask
testers.
Conflicting results have come from epidemiological studies
relating cancer incidence and mortality to chlorophenol exposure in
the work place. Associations between soft-tissue sarcoma, malignant
lymphoma, and nasal and nasopharyngeal cancer, have been shown in some
epidemiological studies, but not in others. Exposure levels have not
been accurately determined in these studies, and the conflicting
results remain unresolved, at present.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Chlorophenols are organic chemicals formed from phenol
(1-hydroxybenzene) by substitution in the phenol ring with one or more
atoms of chlorine. Nineteen congeners are possible, ranging from
mono-chlorophenols to the fully substituted pentachlorophenol.a
However, this document does not deal with pentachlorophenol, which has
been evaluated previously (WHO, 1987b). The chlorophenols
(particularly trichlorophenols and tetrachlorophenols) are also used
in the form of sodium or potassium salts. The CAS number, name,
chemical (molecular) formula, commercial uses, and common synonyms and
trade names for each chlorophenol congener, are presented in Table 1.
The general chemical structure for the chlorophenol congeners is shown
below.
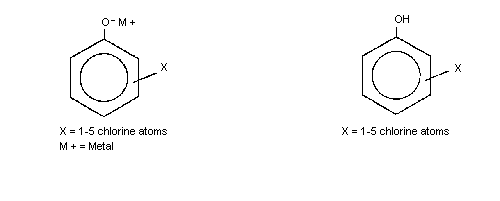 a The chlorophenol congeners are designated as follows:
monochlorophenols (MCP); dichlorophenols (DCP); trichlorophenols
(T3CP); tetrachlorophenols (T4CP); pentachlorophenol (PCP).
Chlorine substitution is also indicated: 2,4-dichlorophenol
(2,4-DCP); 2,4,6-trichlorophenol (2,4,6-T3CP), etc.
Table 1. Information on the identity of chlorophenol congenersa
CAS numberb Common name Abbreviation Molecular Common synonymsc Common trade
formula names
95-57-8 2-monochlorophenol 2-MCP C6H5ClO o-chlorophenol;
ortho-chlorophenol;
1-chloro-2-hydroxybenzene
108-43-0 3-monochlorophenol 3-MCP C6H5ClO m-chlorophenol;
meta-chlorophenol;
1-chloro-3-hydroxybenzene
106-48-9 4-monochlorophenol 4-MCP C6H5ClO p-chlorophenol;
para-chlorophenol;
1-chloro-4-hydroxybenzene
576-24-9 2,3-dichlorophenol 2,3-DCP C6H4Cl20
120-83-2 2,4-dichlorophenol 2,4-DCP C6H4Cl20 NCl-C55345
583-78-8 2,5-dichlorophenol 2,5-DCP C6H4Cl20
87-65-0 2,6-dichlorophenol 2,6-DCP C6H4Cl20
95-77-2 3,4-dichlorophenol 3,4.DCP C6H4Cl20
591-35-5 3,5-dichlorophenol 3,5-DCP C6H4Cl20
15950-66-0 2,3,4-trichlorophenol 2,3,4-T3CP C6H3Cl30
933-78-8 2,3,5-trichlorophenol 2,3,5-T3CP C6H3Cl30
933-75-5 2,3,6-trichlorophenol 2,3,6-T3CP C6H3Cl30
95-95-4 2,4,5-trichlorophenol 2,4,5-T3CP C6H3Cl30 NCl-C61187 Collunosol;
Dowicide 2;
Dowicide B;
Nurelle;
Preventol 1
Table 1. (contd).
CAS numberb Common name Abbreviation Molecular Common synonymsc Common trade
formula names
88-06-2 2,4,6-trichlorophenol 2,4,6-T3CP C6H3Cl30 NCl-C02904 Dowicide 2; Omal;
Phenachlor
609-19-8 3,4,5-trichlorophenol 3,4,5-T3CP C6H3Cl30
4901-51-3 2,3,4,5-tetrachlorophenol 2,3,4,5-T4CP C6H2Cl40
58-90-2 2,3,4,6-tetrachlorophenol 2,3,4,6-T4CP C6H2Cl40 Dowicide 6
935-95-5 2,3,5,6-tetrachlorophenol 2,3,5,6-T4CP C6H2Cl40
a From: Jones (1981).
b Chemical Abstracts Service Registry number.
c From: NIOSH (1983) and Verschueren (1983).
Note: Owing to the planar nature of the phenol ring, other congeners (e.g., 2,4,5,6-T4CP) are possible, but these are
identical in structure to the listed congeners.
Technical grade chlorophenols are heterogeneous mixtures of
chlorophenol congeners, unreacted precursors, and a variety of dimeric
microcontaminants. For example, Cochrane et al. (1983) found that
technical 2,4-DCP contained on average, 92.24% 2,4-DCP, 4.48% 2,6-DCP,
1.24% 2,4,6-T3CP, 1.09% 2-MCP, and 0.46% 4-MCP.
Similarly, Levin et al. (1976) examined the composition of 3
commercial chlorophenol formulations used to control fungi in Swedish
sawmills and found that Na-2,4,6-T3CP contained approximately 5%
T4CP, Na-2,3,4,6-T4CP included 5% T3CP and 10% PCP, and technical
NaPCP contained 5% T4CP.
Kleinman et al. (1986) determined that commercial Na-T4CP, used
in the USA, contained 3.1% PCP, 20.7% 2,3,4,6-T4CP, and less than
0.4% of other chlorophenol congeners. These results are typical,
showing that roughly 2-12% T4CP congeners occur in technical PCP
formulations, together with trace quantities of several lower
chlorophenols (Jones, 1981; Lanouette et al., 1984).
Contamination of technical chlorophenols varies according to the
production process used. Because of the elevated reaction temperatures
used to produce chlorophenols, a number of compounds are present as
microcontaminants in technical chlorophenol preparations prepared by
this procedure. These include the polychlorinated dibenzo- p-dioxins
(PCDDs) polychlorinated dibenzofurans (PCDFs), polychlorinated
diphenyl ethers, polychlorinated phenoxyphenols, polychlorinated
benzenes, and polychlorinated biphenyls. Where the alkaline hydrolysis
of chlorobenzenes is used to manufacture chlorophenols, the technical
product also contains the unreacted chlorobenzene. Technical
chlorophenol salts usually also contain an excess of sodium or
potassium hydroxide.
While commercial MCP and DCP contain little or no detectable PCDDs
and PCDFs, presumably because their manufacture does not involve high
enough temperatures, other chlorophenols may contain up to many mg/kg
of particular PCDDs, and PCDFs (Firestone et al., 1972; Woolson et
al., 1972; Levin et al., 1976; Levin & Nilsson, 1977; Rappe et al.,
1979; Cedar, 1984; Kleinman et al., 1986). Concentrations of PCDDs and
PCDFs in some American and European chlorophenols are provided in
Table 2. Tri- and tetrachloro-dibenzoxo-dioxins predominate in T3CP
formulations, while the hexa, hepta, and octa congeners are the major
PCDD contaminants in technical T4CP and PCP (Firestone et al., 1972;
Rappe et al., 1978a). 2,3,7,8-Tetrachloro-dibenzo- p-dioxin
(2,3,7,8-TCDD) occurs primarily as a contaminant of 2,4,5-T3CP
(Table 2), though it is present at low µg/kg concentrations in T4CP,
PCP, and NaPCP (Hagenmaier, 1986; Hagenmaier & Brunner, 1987).
Predioxins (chlorinated phenoxyphenols) may comprise as much as 5% of
technical CP preparations (Levin et al., 1976; Levin & Nilsson, 1977).
Most of the data in Table 2 concern chlorophenol formulations from
the 1970s. As a result of modifications in production chemistry, it is
likely that the levels of microcontaminants in current formulations
are somewhat lower. Indeed, all of the 1986 tetrachlorophenol products
assayed by Agriculture Canada (1987) contained levels of H6CDD that
were several times lower than those in the earlier reports (Table 2).
2.2 Physical and Chemical Properties
Data on some physical and chemical properties of chlorophenols are
summarized in Table 3. All of the CPs are solids at room temperature,
except for 2-MCP, which is a liquid. They have strong odours that have
been described as pungent or medicinal, particularly those of
2-monochlorophenol (2-MCP) and 2,4-dichlorophenol (2,4-DCP). Taste and
odour thresholds are so low that Maximum Acceptable Concentrations of
chlorophenols in drinking-water are based on organoleptic rather than
toxicological criteria (US EPA, 1980c; WHO, 1984).
Although the solubility in water of all chlorophenols is poor,
varying from 2.1 × 10-1 mol/litre for 2-MCP t o 7.9 × 10-4 mol/litre
for 2,3,4,6-T4CP (US EPA 1980c) they readily dissolve in a number of
organic solvents. In contrast, the sodium or potassium salts of
chloropenols (most commonly NaT3CP, NaT4CP, and NaPCP) are up to
four orders of magnitude more soluble in water than the parent
compounds. The acidity of chlorophenols increases as the number of
chlorine substitutions increases. Thus, ionization of the higher
chlorophenols begins at a lower pH than that of the lower
chlorophenols (pH approximately 3.5 versus 7 for PCP and 2-MCP,
respectively), with important implications for the interactions
between pH and chlorophenol sorption (section 4.1.2.1), or toxicity
(section 6.1.1). The n-octanol-water partition coefficient of
chlorophenols also increases with chlorination, indicating a
propensity on the part of the higher chlorophenols to bioaccumulate.
2.3 Conversion Factors
MCP 1 mg/m3 = 0.190 ppm; 1 ppm = 5.258 mg/m3
DCP 1 mg/m3 = 0.150 ppm; 1 ppm = 6.667 mg/m3
T3CP 1 mg/m3 = 0.124 ppm; 1 ppm = 8.076 mg/m3
T4CP 1 mg/m3 = 0.105 ppm; 1 ppm = 9.488 mg/m3
Table 2. Polychlorodibenzo-p-dioxins (PCDDs) and polychtorodibenzofurans (PCDFs) in some American and European
mono-, di-, tri-, and tetrachlorophenolsa
Formulation PCDD Concentration PCDF Concentration Year
(mg/kg) (mg/kg) sample
received
2-MCP ND T4CDF presentb 1967
2,4-DCP ND ND 1970
2,6-DCP ND ND 1970d
Na-2,4,5-T3CP ND ND 1967
Na-2,4,5-T3CP 2,7-D2CDD 0.72 ND 1969
2,3,7,8-T4CDDc 1.4
2,4,5-T3CP 1,3,6,8-T4CDD 0.30 ND 1969
2,3,7,8-T4CDc 6.2
2,4,5-T3CP P5CDD 1.5 ND 1970
2,4,5-T3CP ND T3CDF presentb 1970
2,4,5-T3CP 2,3,7,8-T4CDD 0.07 ND 1970
2,4,6-T3CPf 2,3,7-T3CDD 93 T4CDF 1.5 1970d
1,3,6,8-T4CDD 49 P5CDF 17.5
H6CDF 36
H7CDF 4.8
O8DF
Table 2. (contd.)
Formulation PCDD Concentration PCDF Concentration Year
(mg/kg) (mg/kg) sample
received
2,3,4,6-T4CP H6CDDc 15 H6CDF presentb 1970d
H6CDDc 14 H7CDF present
H6CDDc 5.1 O8CDF
O8CDD 0.17
2,3,4,6-T4CP H6CDc 4.1 T4CDF < 0.5 1967
P5CDF 10 (PCDDs);
H6CDF 70 1967d
H7CDF 70 (PCDFs)
O8CDF 10
2,3,4,6-T4CPf ND T4CDF presentb 1967d
H6CDF present
2,3,4,6-T4CPe T4CDD 0.7 T4CDF ca.10 1970d
P5CDD 5.2 P5CDF ca.10
H6CDD 9.5 H6CDF ca.60-70
H7CDD 5.6 H7CDF ca.60-70
O8CDD 0.7 O8CDF ca.10
2,3,4,6-T4CPe T4CDD 0.4 T4CDF ca.10 1970d
P5CDD 3.5 P5CDF ca.10
H6CDD 5.3 H6CDF ca.60-70
H7CDD 2.1 H7CDF ca.60-70
O8CDD 0.3 O8CDF ca.10
Table 2. (contd.)
Formulation PCDD Concentration PCDF Concentration Year
(mg/kg) (mg/kg) sample
received
TCP/PCPg H6CDD 1-4 (n = 6) not reported 1986
H7CDD 40-102 (n = 6) not reported
O8CDD 27-55 (n = 6) not reported
Na-T4CP/PCPg H6CDD N.D.-4 (n = 13) not reported 1986
H7CDD 10-119 (n = 13) not reported
O8CDD 5-330 (n = 13) not reported
a Reports of PCDDs from Firestone et al (1972), except where otherwise indicated; quantitative data on
PCDF concentrations from Rappe et al. (1978a).
b Unquantified. See Firestone et al. (1972).
c Confirmed by combined gas chromatography-mass spectrometry,
d Not reported.
e Rappe et al. (1979).
f Rappe et al. (1978b).
g Agriculture Canada (1987).
ND: No congener detected; limit of detection from Firestone et al. (1972) is approximately 0.02 ppm for PCDDs,
that from Rappe et al. (1978a) is roughly 0.01-0.04 mg/kg (Buser & Bosshardt, 1976).
Table 3. Physical and chemical properties of chlorophenols other than pentachlorophenola
Compound Relative Density Boiling point Melting point Flash Vapour log
molecular (°C at 760 mm) (°C at 760 mm) point pressure n-octanol/
mass (°C) (mm) water
(temperature) partition
coefficient
2-MCP 128.56 1.2634 (20/4) 174.9 9 63.9 1 (12.1 °C) 2.15b
3-MCP 128.56 1.268 (25/4) 214 33 1 (44.2 °C) 2.50b
4-MCP 128.56 1.2651 (30/4) 219.75 43.2-43.7 121.1 1 (49.8 °C) 2.39b
2,3-DCP 163 206 57-59
2,4-DCP 163 1.38 (60/7) 210 45 62 1 (76.5 °C) 3.06c
2,5-DCP 163 211 (744 mm) 59 3.20c
2,6-DCP 163 219-220 (740 mm) 68-69 1 (59.5 °C)d
3,4-DCP 163 253.5 (767 mm) 68
3,5-DCP 163 253 (757 mm) 68
2,3,4-T3CP 197.45 sublimes 83.5
2,3,5-T3CP 197.45 248.5-249.5 62
2,3,6-T3CP 197.45 272 58
2,4,5-T3CP 197.45 1.68 (25/25)e sublimes 68-70.5 1 (72 °C) 3.72f
(275 mm) 1 (53 °C) 3.62c
1 (76.5 °C)
Table 3. (contd).
Compound Relative Density Boiling point Melting point Flash Vapour log
molecular (°C at 760 mm) (°C at 760 mm) point pressure n-octanol/
mass (°C) (mm) water
(temperature) partition
coefficient
2,4,6-T3CP 197.45 1.49 (75/4)e 246 69.5 113.9
3,4,5-T3CP 197.45 271-277 (746 mm) 101
2,3,4,5-T4CP 231.98 1.67d sublimes 116-117
2,3,4,6-T4CP 231.98 1.6 (60/4)g 150 (15 mm) 70 1 (100 °C) 4.10c
2,3,5,6-T4CP 231.98 115
a Principal source: Jones (1981).
b From: Fujita et al. (1964).
c From: Stockdale & Selwyn (1971).
d From: US EPA (1980a).
e From: Kozak et al. (1979).
f From: Leo et al. (1971).
g From: Verschueren (1983).
2.4 Analytical Methods
2.4.1 Sample collection and storage
Proper sampling and sample storage are essential prerequisites for
residue determinations, particularly as picogram or nanogram
quantities are often encountered in environmental samples. It is,
therefore, important to minimize contamination, and to collect
representative samples.
Chlorophenols in the air have been collected by drawing air
through an absorbent liquid at a given rate for a given period, using
absorbents such as potassium carbonate (Dahms & Metzher, 1979) or
ethylene glycol (Wyllie et al., 1975). If a significant proportion of
the chlorophenols present is likely to bind to container walls, as
occurs with water samples, glass containers are preferable to plastic
ones (Kozak et al., 1979).
To avoid erroneous determinations, samples should be processed
immediately or appropriate steps taken to avoid losses through
degradation. If samples are to be stored for an extended length of
time after collection, major losses of chlorophenols may occur as a
result of photodecomposition, oxidation, biodegradation, or
evaporation (section 4). If it is necessary to store samples, changes
in residue levels can be reduced by refrigeration or freezing. The
American Public Health Association (Greenberg et al., 1985) recommends
preserving waste-water samples containing phenolic compounds by
acidification with phosphoric acid and treatment with copper sulfate,
prior to refrigeration.
2.4.2 Sample preparation and analysis
The early procedures used to analyse for chlorophenols were
reviewed by Bevenue & Beckman (1967). Most were colorimetric
techniques, the most popular being the 4-aminoantipyrine method; none
of the methods was either very specific or sensitive. They are no
longer widely used, and are not discussed here. Instead, more
sophisticated analytical techniques are being increasingly used,
including thin-layer chromatography (TLC), gas chromatography (GC),
high-performance liquid chromatography (HPLC), ion exchange
chromatography, infrared (IR) and ultraviolet (uv) spectroscopy, mass
spectrometry (MS), and mass fragmentography. Table 4 includes examples
of the techniques available for the sampling and determination of
chlorophenols other than pentachlorophenols. An indication of the
sensitivity of each method is given, when available.
Table 4. Analytical methods for chlorophenols other than PCPa
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Air T4CP Bubbler collection; Derivatization with 0.05 µg/m3 Dahms & Metzner
absorption in acetyl chloride; GC (1979)
potassium carbonate analysis, EC detector
solution; hexane
extraction
Air 3-MCP Polyether-type HPLC analysis, EC 5 ng US EPA (1980d)
4-MCP polyurethane foam; detector
2,4-DCP Soxhlet extraction
2,4,5-T3CP with diethyl ether/
2,4,6-T3CP hexane; evaporation;
extraction with NaOH,
buffered with phosphoric
acid
Water 2-MCP Adsorption on Florisil column Eichelberger
2,4-DCP activated carbon; with anhydrous et al. (1970)
2,4,5-T3CP adsorbates extracted sodium sulfate for
2,4,6-T3CP with chloroform then clean-up; GC analysis
sodium hydroxide
followed by ethyl ether
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Surface 2-MCP Adsorption on Form pentafluorobenzyl Kawahara (1971)
water 2,4-DCP activated carbon; ether derivatives;
adsorbates extracted GC analysis
with chloroform then
partitioned into acetone
Water 2-MCP Methylene chloride HPLC analysis, 4.2-12.6 ng; Realini (1981)
2,4-DCP extraction of acidified UV254 detector 93-97%
2,4,6-T3CP sample, followed
by ion-pair extraction
of basic sample with
acetonitrile
Air T4CP Collection in 0.1 N GC analysis, 0.5 µg/m3 Kleinman et al.
sodium hydroxide in EC detector (1986)
impingers, acidification,
extraction with toluene
Air T3CP Collection in Acetylation and 2-5 µg/m3 Kauppinen &
T4CP toluene in impingers, extraction with Lindroos (1985)
extraction into basic hexane; GC analysis,
borax solution EC detector
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Water 4-MCP Adsorb basic sample GC analysis, Fl 80-102% Chriswell et al.
2,4,6-T3CP on anion exchange detector; confirmation (1975)
resin; extraction by GC-MS
of hydrochloric acid
and acetone-water
eluates with methylene
chloride
Water T3CP Adsorb on XAD-4 Derivatization with T3CP: 1 µg/litre, Woodrow et al.
T4CP resin; extract with diazomethane; 74.8-77.7%; (1976)
acetone, hydrochloric dissolve in hexane; T4CP: 0.5 µg/litre,
acid; concentrate, HPLC clean-up on 46.7-61.4%;
then dilute with Partisil silica PCP: 0.5 µg/litre,
water; partition column; GC analysis 72.5-85.1%
with sodium sulfate, with EC detector
dry over
dichloromethane
Surface 2,4-DCP Addition of sodium Extract & derivatize 1-2 ng/litre, Abrahamsson & Xie
waste, or 2,6-DCP phosphate buffer by adding 98-105% (1983)
drinking- 2,4,6-T3CP solution, for acid hexane containing
water 2,3,4,6-T4CP waste-water pH internal standard
adjustment to 7 (2,6-dibromophenol)
with sodium and acetic anhydride
hydroxide directly to sample;
GC analysis, EC
detector
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Urine 2,4-DCP Partitioned into Purified by TLC; GC Kurihara & Nakajima
(mouse) 2,4,5-T3CP ethanol or benzene, analysis thermal (1974)
2,4,6-T3CP and water, both conductivity detector;
phases analysed; confirmation by MS
enzyme hydrolysis
of water soluble CP
conjugates
Urine T3CP Benzene extraction Der. with diazom 1 µg/litre, Edgerton et al.
(human, T4CP from acidified, ethane; separation on 89.3-97.0% (1979)
rat) hydrolysed solution acid alumina column;
GC analysis, EC detector;
GC-MS confirmation
Urine T4CP Acidic hydrolysis; LC analysis column: 23 µg/litre, Pekari & Aitio
(human) hexane/isopropanol Spherisorb ODS; 54.6% (1982)
extraction; mobile phase:
evaporation and methanol +
redistillation in ammonium carbonate;
methanol-water UV254 detector
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Urine 2-MCP Hydrolyse conjugates Der. with sodium Hargesheimer & Courts
(human) 4-MCP in acidified sample bicarbonate then (1983)
2,4-DCP by boiling; make acetic anhydride
2,6-DCP basic with sodium extraction methylene
2,4,5-T3CP hydroxide extract chloride; GC analysis,
2,4,6-T3CP with methylene EC and FI detectors;
chloride; neutralize, confirmation by MS
dry with sodium
sulfate
Blood 3-MCP Hydrolysis with HPLC analysis, 5 ng, US EPA (1980d)
(human) 4-MCP hydrochloric acid EC detector 80-100%
2,4-DCP extraction with hexane
2,4,5-T3CP ethyl ether;
extraction with sodium
hydroxide, buffered
with phosphoric acid
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Animal 2,4-DCP Optional alkaline Der. to trimethylsilyl 0.02 mg/kg Clark et al. (1975)
tissue 2,4,5-T3CP digestion; acid ether; GC analysis (2,4-DCP);
(sheep hydrolysis; 0.01 mg/kg, > 95%
cattle) distillation with water; (2,4,5-T3CP)
methylene chloride
Sediment T4CP Homogenization; toluene Derivatization 0.5-25 µg/kg, Butte et al. (1983)
and clams extraction from pyrolytic ethylation 76.7-98.8%
acidified sample with triethylsulfonium
2,4,6-tribromophenol iodide; GC analysis,
as internal standard EC detector;
Confirmation by MS
Fish 2-MCP Gel permeation Derivatization with 2-MCP-47% Stalling et al.
tissue 2,4-DCP chromatography to remove pentafluorobenzyl 2,4-DCP-78% (1979)
2,4,6-T3CP lipids, free fatty bromide; silica 2,4,6-T3CP-86%
acids; acid-base gel chromatography PCP-63%
extraction clean-up; GC analysis,
EC detector
Meat and 2,4-DCP Alkaline digestion; Derivatization 10 µg/kg, Sackmauerova-Veningerova
poultry 2,3,4-T3CP steam distillation of with methyl iodide; 92-98% et al. (1981)
livers 2,4,5-T3CP acidified sample; GC analysis;
2,4,6-T3CP toluene extraction EC detector
dry sodium sulfate,
evaporate
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Muscle 2,4-DCP Blended in GC analysis, Sherman et al. (1972)
(hen) hexane-sulfuric acid; EC detector
extraction with
NaOH, then hexane
Liver 2,4-DCP Ground; dried with Florisil column Sherman et al. (1972)
(hen) sodium sulfate eluted for clean-up; GC
with hexane; analysis, EC detector
extraction of eluate
with acetonitrile,
then hexane; dried
with sodium sulfate
Soil 2-MCP Steam distillation of GC analysis, EC < 0.1 mg/kg Narang et al. (1983)
2,4-DCP acidified sample; detector 2-MCP-59%;
2,4,6-T3CP extraction with 2,4-DCP-64%;
toluene dichloromethane 2,4,6-T3CP-70%
eluted through anhydrous
sodium sulfate
extraction with
hexane
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Wood 2,3,4,6-T4CP Extraction with diethyl Elution from TLC 200 mg/kg dust Levin & Nilsson (1977)
dust ether; evaporation; with n-hexane; 70%
dissolve in acetone derivatization
and TLC (silica gel) with diazomethane;
GC with Ni63
EC detector
Wood 2,4,6-T3CP Pumped through GC analysis, EC Kauppinen & Lindroos
dust 2,3,4,6-T4CP membrane filter; detector (1985)
Soxhlet extraction
with diethyl ether;
evaporate; dissolved
in hexane
a GC = gas chromatography.
TLC = thin-layer chromatography.
HPLC = high-performance liquid chromatography.
MS = mass spectrometry.
EC = electron capture detection.
FID = flame ionization detection.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural Occurrence
Some chlorophenols are present in the environment independent of
man-made input. Dichlorophenols have been detected in a variety of
organisms (Siuda, 1980). 2,4-Dichlorophenol occurs naturally in a
Penicillium sp., while 2,6-DCP serves as a sex pheromone for several
species of tick. A number of related organohalogens are also found in
flora and fauna (Arsenault, 1976; Siuda, 1980). However, these sources
cannot account for the significant amounts of chlorophenols,
particularly the higher chlorinated phenols, found in the environment.
3.2 Man-made Sources
3.2.1 Production levels and processes
3.2.1.1 World production figures
Reliable data on production levels of chlorophenols other than PCP
are not readily available. In 1975, the combined global production of
all chlorophenols approached 200 million kilograms (Table 5). Slightly
more than half consisted of chlorophenols other than PCP, with
2,4-DCP, 2,4,5-T3CP, and 2,3,4,6-T4CP predominating. Where
commercial use data are available, recent figures indicate that
consumption has declined (IARC, 1986). Chlorophenols are used in
countries other than those shown in Table 5, but the quantities used
are not known. Information on PCP production is presented in WHO
(1987b).
3.2.1.2 Manufacturing processes
While most chlorophenols can be produced by several different
procedures, only a few methods are actually used in commercial
manufacture (Doedens, 1963; Freiter, 1979). Most chlorophenols are
made by the direct stepwise chlorination of phenol or lower
chlorinated phenols at an elevated temperature. The compounds 2-MCP,
4-MCP, 2,4-DCP, 2,6-DCP, 2,4,6-T3CP, 2,3,4,6-T4CP, and PCP are
manufactured by this means. The manufacture of T4CP or PCP requires
the use of a catalyst, such as iodine, aluminium chloride (AlCl3),
ferric chloride (FeCl3), or antimony chloride (SbCl3). The process
is not quantitative, with the result that batches of one chlorophenol
will usually contain substantial amounts of other CPs (section 2.1).
Table 5. Production/consumption of chlorophenols other than PCP
Country Compound Year Production/ Reference
consumption
(kg/year)
Global total chlorophenols 1975 1.8 × 108 (P)a Levin & Nilsson
(1977)
non-PCP 1978 0.98 × 108 (P) Ahlborg &
chlorophenolsb Thunberg
(1980)
Canada total chlorophenols 1976 3.4 × 106 (C) Jones (1981)
non-PCP chlorophenols 1.5 × 106 (C)b
total chlorophenols 1981 > 5.266 × 106 (C) Jones (1984)
1981 4.000 × 106 (P)
non-PCP chlorophenols 1981 > 3.730 × 106 (C)
tetrachlorophenol 1981 7.86 × 105 (C) Jones (1984)
and Na-T4CP 12.44 × 105 (P)
Na-T3CP 1981 3.0 × 103 (C) Jones (1984)
1.0 × 103 (P)
2,4-dichlorophenol 1981 3.700 × 106 (C) Jones (1984)
1.850 × 106 (P)
total chlorophenols 1984 3.89 × 106 (S) Environment
non-PCP chlorophenols 1984 4.91 × 105 (S)b Canada
(1986)
tetrachlorophenol 1984 4.9 × 105 (S) Environment
and Na-T4CP Canada
(1986)
2,4,5-trichlorophenol 1984 < 1.0 × 103 (S) Environment
and Na-2,4,5-T3CP Canada
(1986)
Europe monochlorophenols 4.5 × 106 (P) Krijgsheld &
van der Gen
2,4-dichlorophenol 9.1 × 106 (P) (1986)
Table 5. (contd).
Country Compound Year Production/ Reference
consumption
(kg/year)
United total chlorophenols 1972 > 1.14 × 106 (C)d Ahlborg &
Kingdom Thunberg
(1980)
USA total chlorophenols 1976 > 2.421 × Buikema et al.
107 (S,P)c,d (1979)
non-PCP chlorophenols 1976 > 1.995 × Buikema et al.
106 (S,P)b (1979)
2,4-Dichlorophenol 1976 1.995 × 106 (S)
a P = production, C = consumption, S = sales volume.
b By difference, from data presented in reference.
c Sales approximate consumption, since most use is domestic (Jones, 1981).
d A conservative estimate, derived by adding figures for major chlorophenols.
Alternatively, some chlorophenols are produced by the alkaline
hydrolysis of hexachlorobenzene (HCB) or other chlorobenzenes in
methanol, ethylene glycol, and other solvents. The compounds 2,5-DCP,
3,4-DCP, 2,4,5-T3CP, 2,3,4,5-T4CP, 2,3,5,6-T4CP, and PCP can be
synthesized by this type of reaction (Doedens, 1963; Freiter, 1979).
Both methods may yield contaminants that are themselves potential
health hazards, specifically PCDDs, PCDFs, and 2-phenoxyphenols
(section 2.1), especially if optimum reaction conditions are not
maintained (particularly temperature and pressure) in the production
of higher chlorophenols. In addition, chlorophenols derived from the
hydrolysis of chlorobenzenes may include substantial amounts of the
initial isomer in the final product.
3.2.2 Uses
The uses of commercial chlorophenols are summarized in Table 6.
These compounds are biocides, a property that accounts for many of
their uses. Chlorophenols, particularly tetra-, and to a lesser
extent, trichlorophenols, have been used as bactericides, algicides,
molluscicides, acaricides, fungicities, and mould inhibitors, and for
less specific uses, such as general antiseptics and disinfectants.
Chlorophenols are also used as intermediates in the production of
certain herbicides, dyes, and drugs.
At present, use patterns are more restricted than is indicated in
Table 6. For example, revisions to Canadian standards for chlorophenol
in 1980 resulted in a sharp reduction in use in domestic interiors,
agriculture, the leather industry, and as slimicides in the pulp and
paper industries. Both the quantities and patterns of use are even
more restricted in some countries. For example, in Sweden and Finland,
chlorophenols are no longer used, or use is severely restricted in the
wood preservation or pulp and paper industries (Ahlborg & Thunberg,
1980; Lindroos et al., 1987).
Most chlorophenols are applied in the form of a chlorophenol-oil
mixture, but some are dissolved in a "clean" carrier that can be
recovered, such as methylene chloride (Jones, 1981). In contrast, the
sodium salts of higher chlorophenols (particularly T3CP, T4CP, and
PCP) are readily soluble in water.
3.2.2.1 Wood treatment
Large quantities of higher chlorophenols are used in wood
preservation (Table 6). In Canada in 1981, most chlorophenol-treated
wood was preserved by pressure treatment with pentachlorophenol
(Table 7). This compound has been evaluated previously (WHO, 1987b),
and will not be covered here.
Substantial amounts of the sodium salts of T4CP (ca. 13% of total
1981 chlorophenol consumption: Table 7), and lesser amounts of NaT3CP
and NaPCP have been used to protect fresh-cut logs and lumber. These
compounds, which are readily soluble in water, are used to surface-
treat lumber by dipping or spraying to protect against sapstain or
mould. Some plywood mills also use T4CP to reduce decay and mould,
and insect attack. The preservative is usually added to the glue.
3.2.2.2 Agriculture
At one time, chlorophenol-treatment was widely used in
agriculture, to prevent wood decay in buildings, food containers, and
horticultural timbers. Recently, such chlorophenol applications have
been considerably restricted in some countries (section 3.2.2), and as
a result, the quantities of non-PCP chlorophenols used in agriculture
are minor (Jones, 1981).
Table 6. Principal uses and reactions of selected chlorophenols other than PCPa
Compound Principal uses Other uses
2-chlorophenol Intermediate for Polymer intermediate for
further chlorination to fire-retardant varnishes;
2,4-dichlorophenol, cotton fabric treatment
2,4,6-trichlorophenol, to provide rot resistance;
and pentachlorophenol ingredient in coal
processing
4-chlorophenolb Intermediate for
higher chlorophenols;
intermediate dyes,
fungicides, and drugs
2,4-dichlorophenol Intermediate for Intermediate for
production of 2,4-D and production of Sesone,
other herbicides; Nitrofen, Nemacide,
ingredient of Genite-EM-923; raw
antiseptics; starting material for polyester
material for higher films; mothproofing;
chlorophenols miticide
2,4,5-trichlorophenol Intermediate in Germicides and
manufacture of 2,4,5-T ingredients of
and related herbicides; germicidal soaps
fungicide, bactericide,
algicide
2,4,6-trichloropenol Precursor for higher CPs;
germicide, particularly for
preservation of wood,
leather, glue, and textiles;
intermediate in preparation
of insecticides and soap
germicides
2,3,4,6- Fungicide and bactericide Preservative for latex
tetrachlorophenol, for wood preservation; and leather; preservative
and its sodium salt sodium salt is sapstain in glue for plywood
inhibitor; pesticide
a From: US EPA (1979).
b From: US EPA (1980c).
3.2.2.3 Domestic
T4CP is an active ingredient in formulations of PCP used as wood
preservatives for homes, and as an additive to paints and stains
(Table 6). Sales in Canada for these purposes contribute only a small
fraction to the total PCP market (Jones, 1981) (Table 7) as a result
of recent government restrictions on their use (section 3.2.2). T3CP
is used as a general-purpose home antiseptic and as the active
ingredient in some throat lozenges. At one time, 4-MCP was found in
disinfectants for home, farm, dental, hospital, and veterinary uses,
but has been largely replaced by other chemicals (Exon, 1984).
Table 7. Canadian use patterns for chlorophenols and their sodium
salts in 1981a
Use Product Consumptionb % of Total
(kg × 103/year)
Wood preservation PCP 1536 25.2
(pressure treatment)
Wood protection Na-PCP 32 0.5
(surface treatment) Na-T4CP 786 12.9
Na-T3CPc 1 0.02
Intermediates for 2,4-DCPd 3700 60.7
phenoxy herbicides
Additives in products NaPCP 38 0.6
listed in footnote e NaT3CPc 2 0.03
Total 6095
a From: Jones (1984).
b Includes chlorophenols present in exports (13.6% of total
consumption), principally treated wood products.
c Chlorophenols are no longer registered for use in Canada.
d 2,4-D is no longer produced domestically, though considerable
quantities continue to be imported.
e Adhesives, construction materials, fabrics, fibreboard
products, finished paper, leather, paper machine felts,
photographic solutions, pulp and paper process solutions,
rayon emulsions, rubber, rubber gaskets.
3.2.2.4 Water treatment
Information is lacking on the use of non-PCP chlorophenols in
water-treatment applications (Jones, 1981).
3.2.2.5 Additives
Sodium salts of T3CP and T4CP have been used to inhibit
microbial growth in a diverse array of products (Tables 6 & 7). These
applications make up only a small fraction of the total consumption of
chlorophenols (Jones, 1981).
3.2.2.6 Intermediates in industrial syntheses
Production of chlorophenols is stepwise and not quantitative,
hence lower chlorophenols are generally recycled within a reactor
system, or recycled from other manufacturing processes in the
production of the higher chlorinated phenols. The lower chlorophenols
also serve as intermediates in the production of other pesticides
(Table 6). Large amounts of 2,4-DCP are consumed in the manufacture of
the phenoxy herbicide 2,4-D (Table 7), and also as a precursor for the
production of the pesticides Sesone, Nitrofen, Nemacide, and
Genite-EM-923. 2,4,5-T3CP is used in the manufacture Ronnel(R),
2,4,5-T, and related herbicides, while 4-MCP is used in the production
of the germicide 4-chlorophenol- o-cresol. Small amounts of lower
chlorinated phenols have been used in the manufacture of some dyes and
drugs.
3.2.3 Other sources
Chlorophenols are also generated by human activity via several
indirect routes. They are formed as by-products of chlorine bleaching
in paper-mills, and subsequently released into the environment
(Ahlborg & Thunberg, 1980; Xie et al., 1986) (section 5.1.2.1). The
chlorination of municipal and industrial wastes, and municipal
drinking-water can give rise to mono-, di-, and trichlorophenols in
the µg/litre range (NRCC, 1978). At these levels, the taste and odour
of water may be affected locally, though the chlorophenol
concentrations are well below those that produce any observable toxic
effect in test organisms (section 6.1). The incomplete incineration of
chlorophenol wastes can release substantial quantities of these
compounds into the environment (section 3.4). The lower chlorinated
phenols are also formed as a result of the bioconversion of lower
chlorinated benzenes and related compounds (Ballschmiter & Scholz,
1980). The contributions of these sources to environmental release or
human exposures to chlorophenols are generally not well-defined, and
are not considered in subsequent sections.
3.3 Waste Disposal
Waste-waters containing chlorophenols arise from three sources,
i.e., the manufacture of chlorophenols, the manufacture of compounds
in which chlorophenols are used as intermediates, and wood-treatment
facilities. Both manufacturers and regulatory agencies have emphasized
appropriate process design, in order to minimize the volume of waste
generated, particularly in the treatment of lumber (Richardson, 1978).
Information on the handling of chlorophenol-containing wastes in
Canada is limited. In the past, some industries disposed of
2,3,4,6-T4CP and PCP-contaminated wastes as raw effluent into deep
wells, or into lagoons, prior to discharge into the North Saskatchewan
River (Jones, 1981). However, most Canadian wood-treatment plants
report that they do not have any discharge and are able to dispose of
their minimal wastes by incineration, or containment and evaporation
in lagoons. Data to confirm the adequacy of such treatments are
generally not collected (Richardson, 1978), but they are probably
adequate, if applied correctly.
While waste-water treatment plants have been used in only a few
large wood-preserving plants and by some chemical manufacturers (US
EPA, 1979), their use is increasing in response to environmental
concerns. Such methods and their efficiency have been described (US
EPA, 1979).
Usually primary treatment is applied only in instances where the
chlorophenol in question is dissolved in a carrier oil, when gravity
separation tanks are used to recover the oil and associated
chlorophenol for subsequent recycling or waste treatment. A few plants
also use hay or sand filtration to remove some oil droplets and wood
particles (Richardson, 1978). Flocculation is not widely used, because
flocculents have proved ineffective or inconsistent in removing
chlorophenols (US EPA, 1979).
Chlorophenols are effectively removed by secondary treatment under
favourable conditions. Roughly 90% of total phenols were removed from
waters containing wastes from the manufacture of phenoxy herbicides in
aerated lagoons (US EPA, 1971) or by trickling filter/activated sludge
treatments (Mills, 1959). Several laboratory and treatment-plant
studies have shown that PCP can be degraded by activated sludge (Dust
& Thompson, 1973; Kirsch & Etzel, 1973; Etzel & Kirsch, 1974; Moos et
al., 1983; Guthrie et al., 1984; Hickman & Novak, 1984), a fluidized
bed reactor (Hakulinen & Salkinoja-Salonen, 1982), and a biofilm
reactor (Salkinoja-Salonen et al., 1984). However, of 14 municipal
treatment plants surveyed by the US EPA, 8 did not remove any of the
PCP load, while the remainder were considered to remove PCP (6-87%)
primarily by adsorption on solids (Hickman & Novak, 1984).
Furthermore, degradation by microorganisms is sharply reduced, when
chlorophenol concentrations are excessive (Broecker & Zahn, 1977;
Reiner et al., 1978; El-Gohary & Nasr, 1984; Salkinoja-Salonen et al.,
1984). If secondary treatment facilities are to remove chlorophenols
reliably, they must include acclimated organisms, and chlorophenol
concentrations must be dilute and fairly stable (Hickman & Novak,
1984). These considerations suggest that such wastes are best handled
by a facility designed specifically to treat them, rather than being
treated at general-purpose sewage-treatment plants.
Chemical oxidation, using such chemicals as chlorine or potassium
permanganate, may also be effective in treating
chlorophenol-contaminated wastes. While chlorination of municipal
wastes can actually produce mono-, di-, and tri- chlorophenols, they
are subsequently oxidized together with higher chlorophenols to
compounds that are less toxic and/or more biodegradable (US EPA, 1979;
Sithole & Williams, 1986).
Adsorption of chlorophenols on activated carbon is sometimes used
as a final clean-up step for waste-waters, though this is feasible
only when waste treatment is handled in the same plant from start to
finish. Removal of 2,4-DCP (Aly & Faust, 1964) and PCP (Richardson,
1978) approaches 100% using this method.
Incineration has also been used to dispose of chlorophenol wastes,
but the available information deals mainly with PCP. A controlled air
incinerator destroyed more than 99.99% of PCP in treated wood at
combustion temperatures of between 916 and 1032°C, and yielded no
measurable T4CDD or T4CDF in the off-gas (Stretz & Vawuska, 1984).
However, incinerator temperatures must be high enough and residence
times long enough to ensure complete combustion. Rappe et al. (1978b)
demonstrated that burning technical T4CP at low temperatures
increased the content of PCDDs. Similarly, low-temperature destruction
in hog-fuel or "wigwam" burners fed chlorophenol-contaminated sawdust
and wood shavings can lead to the formation of PCDDs and PCDFs (Crosby
et al., 1981).
3.4 Losses of Chlorophenols into the Environment
In the absence of information from other countries, releases of
chlorophenols into the Canadian environment for 1981 (Jones, 1984) are
presented in Table 8 by way of an example. Of the 5.27 × 106 kg of
chlorophenols consumed in Canada in 1984, 1.37 × 106 kg (26%) were
eventually released into the environment. A large proportion (less
than 28%) of these releases would have been as PCP and NaPCP, but the
data compiled in the table do not distinguish these from T4CP and
Na-T4CP.
Table 8. Chlorophenol releases into the Canadian environment in 1981a
Source Quantity (kg × 103/year)
1. Releases in wastes from production sites
emissions 3
effluents 70
solids -
sub-total > 73
2. Releases in other wastes
(a) Industrial
(i) Wood preservation sites
liquid 2
solids -
incineration (hog-fuel) -
landfill 1 (PCP, T4CP)
sub-total > 3
(ii) Saw-mill/planer mill
liquid 21
solids -
(ii) Incineration (hog-fuel) 272
Pulp mills/landfill -
sub-total >293 (NaPCP, NaT4CP)
(b) Agricultural
solids (livestock litter)
landfill
(c) Domestic
solids
incineration (mun.)
landfill
3. Releases during storage and transport
(solids and liquids)
(a) Industrial 3.5
(b) Agricultural 3.4
(c) Domestic 0.1
sub-total 7.0
Table 8. (cont'd).
Source Quantity (kg × 103/year)
4. Releases in situ from treated products
(solids and liquids)
(a) Industrial 618
(b) Agricultural 370 (2,4-DCP)
(c) Domestic 1
sub-total 989
Grand total > 1365
a From: Jones (1984).
In 1981, a significant amount (ca. 5%) of the total releases of
chlorophenols in Canada occurred from production sites. Following this
estimate, production of all chlorophenols ceased in Canada. However,
this route could be a significant source of chlorophenol contamination
in countries where they are still manufactured. Releases from plants
will include a variety of chlorophenols from the manufacture of
chlorophenols and chlorophenol-derived products. A small proportion of
these materials reaches the environment after incineration (Table 8),
but the bulk of the chlorophenols released from production sites in
Canada is diluted and released as untreated effluent.
Losses into the environment during the storage and transport of
chlorophenols are small (Table 8), comprising less than 1% of the
total production.
The majority (>70%) of the chlorophenols released into the
Canadian environment arose from treated products (Table 8). About
two-thirds of these came from industrial sources, which were not
identified by Jones (1984). Petrochemical drilling fluids contain
large amounts of chlorophenols (from 700 to 1400 mg NaPCP/kg), to
prevent fermentation of polysaccharides, starch, and polymers (Jones,
1981). Once used, the drilling waste is stored on site, in sumps that
are often subject to flooding and washing out. In-service treatment
with wood preservatives, principally PCP and its salts, also results
in some spillage. Large spills have been responsible for fish kills in
waters contaminated in this fashion (Jones, 1981). In addition,
unknown, but presumably large, quantities of PCP and T4CP are leached
from treated lumber in storage or in service. The remaining third of
the environmental releases, which Jones (1984) identifies as primarily
2,4-DCP, is from agricultural sources. Commercial preparations of
pesticides, particularly 2,4-D, 2,4,5-T, and Lindane, contain
chlorophenols as contaminants. Furthermore, chlorophenols are among
the early degradation products of these widely-used chemicals.
Chlorophenols from these sources contaminate soils treated with the
pesticides, and runoff from these soils finds its way into adjacent
water bodies and ground water.
Much of the remaining input of chlorophenols into the environment
occurs in the form of industrial wastes. These comprise roughly 22% of
the total chlorophenol releases, primarily as NaT4CP and NaPCP. Most
of these are released in liquid wastes from pulp-mills (where they are
by-products of chlorine bleaching), untreated wastes from sawmills,
planer mills, and other facilities where they are used in wood
preservation, and during incineration of contaminated sawdust and wood
shavings. Losses of other chlorophenols from such commercial uses are
negligible (Table 8).
The quantities of chlorophenols lost to the atmosphere by
volatilization are not known. Nanogram-per-litre quantities of
chlorophenols have been detected in rainfall and snow (Paasivirta et
al., 1985), suggesting that significant quantities volatilize or are
adsorbed on airborne particulates.
The contributions to the environmental load of chlorophenols from
municipal and industrial chlorination processes, the metabolism of
other chlorinated compounds to chlorophenols, and the domestic use of
such products as cosmetics, drugs, home-care products, stains, wood
preservatives, and pesticides are not known.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and Distribution
4.1.1 Atmospheric movement
While chlorophenols are considered to be primarily water and soil
contaminants, atmospheric movement also occurs. Measurable quantities
of chlorophenols have been detected in air, rainfall, and snow,
sometimes far from obvious point sources (Paasivirta et al., 1985)
(section 5.1.1). Furthermore, considerable quantities of chlorophenols
are released as part of incinerator emissions. The relative
contributions of volatilization and adsorption on particulates to
these atmospheric levels are not known.
4.1.1.1 Volatilization
No estimates of the rate of volatilization of chlorophenols in the
environment have been published. It appears that losses through this
process are minimal in natural waters. If the half-times for
volatilization of 2-MCP and 4-MCP from 0.38 cm of still water measured
by Chiou et al. (1980) are extrapolated to 1 m depth, the estimated
half-times are 395 h and 3421 h, respectively (Krijgsheld & van der
Gert, 1986).
Diffusion, a process related to volatilization, does not
contribute significantly to the long-range transport of substances in
either the soil or aquatic habitats, though it is essential in the
local replacement of materials lost through volatilization or
breakdown.
4.1.2 Soil movement
4.1.2.1 Adsorption
Environmental transport of chlorophenols, particularly in soils,
can be affected by adsorption on particulates. Such deposition is
quite variable. Acidic soils bind chlorophenols strongly, while
adsorption is minimal under alkaline conditions. Chlorophenols also
adsorb on organic matter, with the result that adsorption is strong in
organic soils, but low in mineral soils.
Thus, Aly & Faust (1964) found that large amounts of three types
of clay were necessary to adsorb small quantities of 2,4-DCP in
aqueous suspensions, even under extremely acidic conditions
(pH 3.6-4.8). Seip et al. (1986) compared the migration rates of
tritiated water and of dilute solutions (12.5-25 µg/litre) of 2,4-DCP,
2,4,6-T3CP, 2,3,4,6-T4CP, and PCP through packed soil columns. All
of the chlorophenols migrated more slowly than water. Adsorption was
moderate in a weakly acid inorganic soil and a basic soil with a
higher organic content, while no chlorophenols were detected in the
eluate from a soil with both a low pH and a high content of organic
matter. In studies by Choi & Aomine (1972, 1974), strongly acidic
soils adsorbed PCP, while weakly acidic or neutral soils did not
adsorb it at all. Moreover, soils with a high organic content adsorbed
PCP strongly, regardless of their pH, while hydrogen peroxide
digestion of organic matter reduced apparent adsorption (Choi &
Aomine, 1974). Miller & Faust (1973) confirmed that sorption of a
number of phenolic compounds on organo-clay was pH-dependent.
It is difficult to assess the impact of adsorption on chlorophenol
transport in the environment from the results of the preceding
studies. Chlorophenol dynamics observed by Seip et al. (1986) suggest
that chlorophenols bound to soils are continually turned over, and
that binding sites may be saturated under the appropriate conditions,
leading to increased mobility and a decreased residence time of
chlorophenols in the soil body.
4.1.2.2 Leaching
In instances when adsorption is minimal, leaching will be an
important means of chlorophenol transport in the soil. Most
chlorophenols should be carried into ground- and surface-waters from
soils that are neutral or alkaline or have a low organic content, or
through which material can percolate rapidly. No information has been
found on the leaching of the lower chlorophenols, but Kuwatsuka (1972)
noted that much of the PCP applied to flooded rice paddies was carried
through the soil in solution, and it has been reported by Jones (1981)
that Na-PCP leaches readily from soils.
4.1.3 Transport in aquatic environments
While a large fraction of the chlorophenols entering waters is
probably degraded in situ (section 4.2), they are nonetheless
moderately soluble and fairly persistent, and so can be transported
considerable distances by water. For example, Fox & Joshi (1984)
detected elevated levels of T4CP and PCP in the surface waters of the
Bay of Quinte, Lake Ontario, as far away as 82 km from the
wood-preserving plant where they originated.
Chlorophenols that are not degraded are concentrated in the
sediments, perhaps through adsorption on sediment particulates.
Schellenberg et al. (1984) determined that sorption of chlorophenols
on natural sediments and aquifer materials was a combined function of
the pH, the organic carbon content of the potential sorbent, and the
partition coefficients (known also as soil adsorption coefficients).
Adsorption was quite strong on non-mineral sediments. Xie et al.
(1986) observed that the disappearance of chlorophenolic compounds
discharged from a sulfate pulp-mill was related to the partition
coefficients of the compounds, and that sediment concentrations were
quite high near to their source (section 5.1.2.1), suggesting that
adsorption strongly influenced the transport of chlorophenolic
compounds. It was reported by Eder & Weber (1980) that the
concentration factor (relative to water) of chlorophenols in the
sediments (38- to 680-fold) and suspended solids (6.3- to 240-fold) of
the Weser estuary was inversely related to the degree of ionization of
the chlorophenol. In contrast, Kuiper & Hantsveit (1984) reported that
both the water and the sediment on the bottom of marine enclosures
contained similar levels of 4-MCP and 2,4-DCP; this discrepancy is not
obviously attributable to differences in system pH, or the nature of
the particulates.
4.2 Degradation and Bioaccumulation
4.2.1 Degradation
As yet, there are few studies addressing the persistence of
chlorophenols in the environment.
4.2.1.1 Abiotic degradation
(a) Photodecomposition
Many, if not all, chlorophenol isomers are degraded to some extent
by exposure to ultraviolet radiation (UVR). The breakdown most often
involves an oxidation reaction that dechlorinates the molecule (Boule
et al., 1982), though a variety of reactions have been described.
2,4-DCP in aqueous solution was decomposed in a matter of minutes
by irradiation from a UV lamp (Aly & Faust, 1964; Crosby & Tutass,
1966; Nakagawa & Crosby, 1974). The major pathway involved the
degradation of 2,4-dichlorophenol to 4-chlorocatechol, which in turn
produced 1,2,4-benzenetriol, and finally a mixture of polyquinoid
humic acids (Crosby & Tutass, 1966). A similar sequential degradation
was reported for 2,4,5-T3CP (Crosby & Wong, 1973). Freitag et al.
(1982) reported that 65.8% of 14C-2,4,6-T3CP on silica gel was
degraded after 17 h of UVR exposure. No organic by-products were
detected, most radioactivity being recovered as carbon dioxide.
The breakdown of chlorophenols is markedly affected by the number
and position of chlorine substituents on the molecule. Under UVR,
2,4-DCP degrades to diameric products (Crosby & Tutass, 1966), while
2,5-DCP degrades to 4-resorcinol (Crosby & Wong, 1973). Omura &
Matsuura (1971) found that alkaline solutions of monochlorophenols
degraded as follows: 2-MCP (82.5% lost in 5 h at 40°C) to a complex
mixture with much resinous material, 3-MCP (70%) to resorcinol, and
4-MCP (55%) to hydroquinone, phenol, and three diphenyls. Aqueous
solutions of the three monochlorophenol isomers yielded similarly
varied products in studies by Boule et al. (1982). In later work,
Boule et al. (1984) determined that among the dichlorophenol
congeners, substitution at the ortho and meta positions made the
chemical more reactive than para substitution.
Only one study has been reported on whether photolysis
significantly reduces concentrations of chlorophenols other than PCP
in situ. Hwang et al. (1986) concluded that photolysis was the
principal degradative pathway (half-life of 3 h or less) for 2,4-DCP,
2,4,5-T3CP, and PCP, but not 4-MCP, in estuarine surface waters
(though they noted that mineralization by other mechanisms was
substantially photo-inhibited under the experimental conditions). Fox
& Joshi (1984) observed an increase in the ratio of T4CP/PCP in
surface waters with increasing distance from a wood-preserving plant
discharge, suggesting that substantial photolysis of PCP occurred, but
noted that the concentrations were remarkably stable, once the
chlorophenols were incorporated into the sediments.
(b) Chemical degradation
There is one report that indicates that chlorophenols may be
degraded in the environment by chemical processes. Baker & Mayfield
(1980) observed losses of 2-MCP, 4-MCP, and 2,4-DCP from sterile
washed silica sand, sterile aerobic soils, non-sterile anaerobic
soils, and sterile anaerobic soils. Microbial contamination,
photolysis, and volatilization were eliminated as causes. The authors
suggested that the chlorophenols were auto-oxidized, or broken down at
catalytic sites, but did not eliminate polymerization as a means of
loss. A number of other research workers, using a wide range of
chlorophenols, have not detected such abiotic losses (Alexander &
Aleem, 1961; Aly & Faust, 1964; Tabak et al., 1964; Boyd & Shelton,
1984).
4.2.1.2 Degradation by microorganisms
Although chlorophenols are quite toxic for microorganisms in
general, they are nonetheless readily metabolized by a large number
that occur in soils, natural waters, sediments, and sewage sludges.
This decomposition is often quite rapid, i.e., completed in a matter
of hours or days. For instance, of 206 isolates from a petroleum waste
lagoon, 46% were able to degrade chlorophenols as a sole source of
carbon after acclimation to the particular chlorophenol (Tabak et al.,
1964). Up to 95% of the added 3-MCP and 4-MCP (initially 150 and
300 mg/litre respectively) was consumed in 3-6 days, while the same
amount of 2,4-DCP (200 mg/litre) and 2,4,6-T3CP (initially
300 mg/litre) disappeared in 7-10 days. No breakdown of 2,6-DCP was
observed. Similarly, in batch cultures enriched with 50 mg
chlorophenol/litre and inoculated with soil, 2-MCP, 4-MCP, 2,4-DCP,
and 2,4,6-T3CP were readily biodegraded and were often removed
completely in less than 10 days, while 2,6-DCP was only metabolized in
some studies; 3-MCP, 2,5-DCP, 2,3-DCP, 3,4-DCP, 3,5-DCP, 2,4,5-T3CP,
2,3,4,6-T4CP and PCP were refractory (Alexander & Aleera, 1961).
Using an acclimated, activated sludge derived from soil, Ingols et al.
(1966) observed complete ring degradation of the following compounds
at 100 mg/litre: 2-MCP in 3 days, 3-MCP in 2 days, 4-MCP in 3 days,
2,4-DCP in 5 days, and 2,4,6-T3CP in 3 days. As much as 52% of
2,5-DCP disappeared in 4 days. No decomposition of sodium
pentachlorophenate occurred. More recently, aerobic microorganisms in
clay loam soils were able to degrade most of the 2-MCP, 4-MCP,
2,4-DCP, 2,6-DCP, or 2,4,6-T3CP present (100 mg/kg) within a few days
without a lag phase (Baker & Mayfield, 1980). More than 70% of added
3-MCP, 3,4-DCP, 2,4,5-T3CP, and PCP disappeared within 80-100 days,
while 3,4,5-T3CP and 2,3,4,5-T4CP levels were little changed after
160 days.
The results of many other studies have confirmed that most
chlorophenols can be metabolized by certain microorganisms in water
(Aly & Faust, 1964; Lee & Ryan, 1979; Baker et al., 1980;
Blades-Fillmore et al., 1982; Hwang et al., 1986), sediment (Lee &
Ryan, 1979; Baker et al., 1980), soil (Walker, 1954; Loos et al.,
1967; Spokes & Walker, 1974; Baker et al., 1980; Pal et al., 1980),
and activated sludge (Baird et al., 1974; Pitter, 1976; Pal et al.,
1980; Boyd & Shelton, 1984).
While bacteria are most frequently studied as the agents
responsible for chlorophenol biotransformation, they are not alone in
this capability. Fungi on wood shavings, used as litter for broiler
chickens, converted 2,3,4,6-T4CP to 2,3,4,6-tetrachloroanisole,
leading to a musty taint in the chicken flesh (Curtis et al, 1972; Gee
& Peel, 1974). The genera Aspergillus and Penicillium readily
degrade chlorophenols. Walker (1973) determined that a yeast isolated
from soil and grown on phenol could metabolize 2-MCP, 3-MCP, 4-MCP,
and 2,4-DCP, but not 2,6-DCP.
The relative rate of degradation of chlorophenols generally
decreases as the number of chlorine atoms on the phenolic ring
increases (Alexander & Aleem, 1961; Tabak et al., 1964; Ingols et al.,
1966; Baker & Mayfield, 1980). However, it is possible to obtain the
reverse result with organisms able to use PCP as the sole carbon
source: the KC3 bacterium studied by Chu & Kirsch (1973) grew on
2,3,4,6-T4CP and 2,4,6-T3CP, but metabolized the dichlorophenois
poorly; the monochlorophenols were not metabolized at all. Rates of
biodegradation are further affected by the relative position of the
chlorine atoms on the phenolic ring. Compounds with a chlorine in the
meta position are generally more stable than those without (Alexander
& Aleera, 1961; Chu & Kirsch, 1973; Etzel & Kirsch, 1974; Baker &
Mayfield, 1980). The chlorophenolic products of PCP degradation in
soils in vitro support this hypothesis (Ide et al., 1972).
Microorganisms that have been previously exposed to a compound are
usually able to metabolize it immediately when re-exposed, and at a
faster rate than unexposed organisms (Walker, 1954; Alexander &
Aleera, 1961; Tyler & Finn, 1974; Pal et al., 1980; Blades-Fillmore et
al., 1982), presumably because exposure induces the enzymes necessary
to metabolize the chlorophenol. Microorganisms not previously
acclimated often exhibit a lag time of as much as several days before
they begin to degrade the compound (Bollag et al., 1968; Spokes &
Walker, 1974; Lee & Ryan, 1979; de Kreuk & Hantsveit, 1981).
Similarly, prior exposure to a structurally related compound can
facilitate the metabolism of chlorophenols, indicating that the
enzymes induced by the original compound are somewhat nonspecific. As
noted earlier, PCP-adapted microorganisms utilize T3CPs and T4CPs
readily (Chu & Kirsch, 1973), while bacteria raised on phenol, lower
chlorophenols, or phenoxyacetic acids are able to metabolize various
other lower chlorophenols (Tabak et al., 1964; Loos et al., 1967;
Walker, 1973; Spokes & Walker, 1974; Boyd & Shelton, 1984).
Research workers have found little or no anaerobic biodegradation
of chlorophenols (Gee & Peel, 1974; Lee & Ryan, 1979; Baker &
Mayfield, 1980; Horowitz et al., 1982; Pignatello et al., 1986). The
persistence of PCP and T4CP in sediment cores, several decades old,
which were presumably anaerobic, supports these findings (Fox & Joshi,
1984). However, under the right conditions, anaerobic metabolism can
be substantial: acclimated anaerobic sludge from a municipal sewage
plant degraded 25 mg monochlorophenols/litre in a few days (Boyd &
Shelton, 1984).
Only a few studies can be used to compare chlorophenol
biodegradation between habitats under conditions that may be readily
extrapolated to a natural situation. The in vitro aerobic breakdown
of 2-MCP, 4-MCP, and 2,4-DCP (100 mg/kg) has been studied in clay loam
soil (Baker & Mayfield, 1980; Baker et al., 1980), freshwater
sediments (Baker et al., 1980), and streams (Baker et al., 1980) at
temperatures ranging from 0 to 23°C. In soil incubated at 23°C, at
least 70% of added 2-MCP disappeared in 0.5-1.0 days, 4-MCP in 1-2
days, and 2,4-DCP in 7-20 days (Baker & Mayfield, 1980). In contrast,
decomposition in sediments was slower: at 20°C, 2-MCP disappeared in
10-15 days, 4-MCP, in 30 days, and 73% of 2,4-DCP, in 15-30 days
(Baker et al., 1980). Virtually no biological degradation of
monochlorophenols occurred in the stream water at 20°C, but 2,4-DCP
levels were reduced by 74% in 10 days at 20°C. These differences may
be related to the favourable conditions for microorganisms that exist
in soils and sediments, in which levels of organic matter and
particulate surface area are high. Addition of sterile sediments or
several inert substances enhanced the degradation of 50 µg
2,4,6-T3CP/litre in river water (Blades-Fillmore et al., 1982).
In other reports, chlorophenol degradation in water has proceeded
more rapidly (eliminated in 1-3 weeks) (de Kreuk & Hantsveit, 1981;
Blades-Fillmore et al., 1982; Hwang et al., 1986). It is possible that
chlorophenols are generally degraded faster in soils and aerobic
sediments than in water but, wherever a suitable combination of
microflora and physical and chemical factors occurs, these general
differences can be overridden.
In summary, a number of microorganisms from a variety of habitats
can readily degrade chlorophenols, especially if previous exposure to
these compounds has induced the enzymes necessary for their
metabolism. This process is slowest with exposure to the higher
chlorophenols, particularly those that are meta-substituted. The
results of incubation studies in the laboratory suggest that
biodegradation is most rapid in aerobic soils and sediments, and is
reduced in anaerobic or nutrient-poor habitats.
4.2.2 Bioaccumulation
A number of field and laboratory studies have yielded information
on the bioaccumulation of the chlorophenols. Most of these have
involved aquatic organisms. Although organisms ranging from bacteria
to fish generally contain higher levels of chlorophenol residues than
the environment at large, the concentrations are not large compared
with those of some other chemicals. Most bioconcentration factors
(BCFs) fall between 1 × 102 and 1 × 103 (Table 9), and substantial
biomagnification is not evident. Ernst & Weber (1978) and Ernst (1979)
suggested that Lanice conchilega displayed exceptionally high BCFs
because of an unusual halogen metabolism (detectable levels of
bromophenols were noted, unlike the other invertebrates studied).
The results of most of the studies in which a range of
chlorophenols has been surveyed have indicated that bioconcentration
is a positive function of chlorine number (Kobayashi et al., 1979;
Hattula et al., 1981b). The higher BCF with increasing chlorine
substitution most likely results from the high partition coefficient
or the lower dissociation constant. Other experimental conditions,
such as length of exposure and exposure concentration, may also
contribute to the substantial range of BCF values shown in Table 9.
Clearance rates of chlorophenols from biota are rapid, indicating
that the bioaccumulation observed in field studies is the result of
long-term exposure rather than persistence. Landner et al. (1977)
reported that 2,4,6-T3CP was eliminated from rainbow trout livers,
three weeks after dosing was discontinued. Similarly, 84-92% of
2,4,5-T3CP was lost from fathead minnows in the first day after
exposure (Call et al., 1980), and the half-life for 2-MCP in bluegills
was less than 1 day (Barrows et al., 1980).
4.3 Effects of Other Physical, Chemical, or Biological Factors
4.3.1 pH
One of the major factors affecting the transport, breakdown, and
toxicity (section 6) of chlorophenols is pH. Because chlorophenols are
weak acids in aqueous solution, they exist primarily in the molecular
form under acidic conditions, while the anion predominates at neutral
or basic pH. Since the molecular and ionic forms of chlorophenols
react differently, pH affects a variety of processes that in turn
influence chlorophenol dynamics. Ionization is further affected by the
degree of chlorine saturation of the chlorophenol; in general, higher
chlorophenols are increasingly acid. Throughout the pH range
characterizing physiological and environmental situations,
monochlorophenols are present mainly in their molecular form while,
above pH 3.5, PCP is primarily dissociated. No information on the
interaction of pH and evaporation of lower chlorophenols was
available. In their studies on PCP volatilization, Kloppfer et al.
(1982) determined that the half-life for PCP disappearance, through
volatilization from their apparatus, was 167 h at pH 3.3 and 3120 h at
pH 6. No evaporation was detected at pH8.
Similarly, pH influences particulate sorption phenomena through
changes in the molecular form of the chlorophenol. As was discussed in
section 4.2, adsorption on soils, sediments, and suspended solids is
inversely related to pH.
The rate of the photolysis of chlorophenols is also altered by pH.
Aly & Faust (1964) determined that the breakdown of 2,4-DCP in aqueous
solution was extremely rapid under alkaline conditions and relatively
slow under acidic conditions: approximate half-lives for photolysis at
pH values of 4, 7, and 9 were 34, 15, and 2 min, respectively.
Similarly, Omura & Matsuura (1971) reported that the rate of
photolysis of 4-MCP increased as the pH increased. In addition, pH
exerts an influence on the biodegradation of chlorophenols. An
activated sludge culture grew well on 2-MCP and 3-MCP at neutral, but
not at alkaline pH (Ingols et al., 1966). Tyler & Finn (1974)
determined that a Pseudomonas species grew best on 2,4-DCP at a pH
range of 7.1-7.8.
Table 9. Bioconcentration estimates for various chlorophenols from field and laboratory data
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Plants
Oedogonium 2,4,6-T3CP 36 1720 Aquatic microcosm, 0.5 µg/litre, Virtanen & Hattula
long-term exposure (1982)
Echinodorus 2,4,6-T3CP 36 1000 Aquatic microcosm, 0.5 µg/litre, Virtanen & Hattula
long-term exposure (1982)
Elodea 2,4,6-T3CP 36 4460 Aquatic microcosm, 0.5 µg/litre, Virtanen & Hattula
long-term exposure (1982)
Chlorella 2,4,6-T3CP 1 51 50 µg/litre screening test, Freitag et al.
fusca var. as 14C (1982)
vacuolate 2,4,6-T3CP 1 580 49 µg/litre screening test, Korte et al.
as 14C (1978)
Invertebrates
Lymnae 2,4,6-T3CP 36 3020 Aquatic microcosm, 0.5 µg/litre Virtanen & Hattula
(adult) long-term exposure (1982)
Lanice 2,4,5-T3CP indefinite 24 088b Field data Ernst & Weber
conchilega 2,4,6-T3CP indefinite 20 269b Field data (1978)
2,3,4,S-T4CP indefinite 17 625b Field data
2,3,4,6 +
2,3,S,6-T4CP indefinite 11 163b Field data
Table 9. (cont'd).
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Invertebrates (contd).
Mytilus edulis 2,3,4,6-T4CP indefinite 45-60 Field data, receiving waters Folke et al.
for dump leachate (1984)
Fish
Roach 2,3,4,6-T4CP indefinite 200 Field data, pulp-mill inputs Paasivirta et al.
(Rutilus) (1985)
Pike 2,3,4,6-T4CP indefinite 150 Field data, pulp-mill inputs Paasivirta et al.
(Esox lucius) (1985)
Trout (Salmo 2,4-DCP 1 10 1.7 mg/litre (LC50) Hattula et al.
trutta) 2,3,5-T3CP 1 12 0.8 mg/litre (LC50) (1981b)
2,3,4,6-T4CP 1 450 0.5 mg/litre (LC50)
Poecilia young 2,4,6-T3CP 36 1 020 Aquatic microcosm, 0.5 µg/litre Virtanen & Hattula
female 2,4,6-T3CP 36 12 180 long-term exposure (1982)
male 2,4,6-T3CP 36 7 000
Fathead 2,4,5-T3CP 28 1900 Aquatic microcosm, 4.8 mg/litre, Call et al. (1980)
minnow 2,4,5-T3CP 28 1800 single addition, 49.3 µg/litre
(Pimephales as 14C
promelas)
Table 9. (cont'd).
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Sunfish 2,3,5,6-T4CP indefinite Muscle/field data, Mississippi Pierce & Victor
lake/spill in December, n = 2 (1978)
6 Januaryd 79 Muscle/spill in December, n = 2
27 Aprild 21
2,3,5,6-T4CP indefinite Liver/spill in December, n = 1 Pierce & Victor
6 Januaryd 962 Liver/spill in December, n = 2 (1978)
27 April 72
Bluegill 2-MCP 28 214 Continuous-flow aquarium, Barrows et al.
(Lepomis 2-MCP at 9.18 l&g/litre, (1980)
macrochirus) as 14C
Bass 2,3,5,6-T4CP indefinite Muscle/field data, Mississippi Pierce & Victor
lake (1978)
6 Januaryd 218 Spill in December, n = 2
27 Aprild 4962 Muscle, n = 2
Catfish 2,3,5,6-T4CP indefinited Muscle, n = 1
6 Januaryd 222 Muscle, n = 2
27 Aprild 53
2,3,5,6-T4CP indefinited Liver, n = 1
6 Januaryd 8608 Liver, n = 2
27 April 1005
Table 9. (cont'd).
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Fish (contd.)
Goldfish 2-MCP 25h 6.4 Static lab. assay, 16 mg/litrec Kobayashi et al.
4-MCP 25h 10.1 Static lab. assay, 9 mg/litrec (1979)
2,4-DCP 25h 34 Static lab. assay, 7.8 mg/litrec
2 4 5-T3CP 25h 62 Static lab. assay, 1.7 mg/litrec
2,4,6-T3CP 25h 20 Static lab. assay, 10.0 mg/litrec
2,3,4,6-T4CP 25h 93 Static lab. assay, 0.75 mg/litrec
Golden orfes 2,4,6-T3CP 3 310 50 µg/litre, screening test Freitag et al.
(Leuciscus idus (1982)
melanotus)
Golden orfes 2,4,6-T3CP 3 250 30 µg/litre, screening test Korte et al.
(Leuciscus idus (1978)
melanotus)
a Ratio of concentration in organism or tissue:water.
b Based on bioconcentration relative to PCP.
c Concentrations near LC50 values, but still sublethal.
d Sampling date.
4.3.2 Lack of oxygen
In general, higher chlorinated phenols are persistent in anaerobic
environments, because of the low microbial degradation of
chlorophenols under such conditions. (section 4.2.1.2).
4.3.3 Inorganic nutrients
Inorganic nutrients may restrict the rate of chlorophenol
biodegradation where a shortage of nutrients limits microbial
activity. Striking seasonal variations noted in the rate of 4-MCP
degradation in natural sea water incubated in the laboratory (de Kreuk
& Hantsveit, 1981) were not related to microbial biomass, but
paralleled the levels of phosphate and nitrate in the samples.
In vitro addition of nutrients to the sea water stimulated
biodegradation. Similar results were reported by Kuiper & Hantsveit
(1984).
4.3.4 Organic matter
Like inorganic nutrients, levels of organic matter can influence
the microbial breakdown of chlorophenols through the control of
microbial biomass and activity. For example, while chlorophenol
decomposition was apparent in most soils studied (section 4.2.1.2), it
was virtually absent from those containing little or no organic matter
(Kuwatsuka, 1972). Similarly, low heterotrophic activity, determined
by the low concentrations of organic substances in water (typically
mg/kg) relative to those found in soils or sediments (g/kg), may
account for differences observed in chlorophenol biodegradation
between these environmental compartments (section 4.2.1.2). Organic
matter bound to the surface of the soil or sediment particulates may
also absorb chlorophenols and thereby affect their transport (sections
4.1.2.1, 4.1.3).
4.3.5 Temperature
Volatilization is a direct function of temperature. In their
studies of PCP evaporation, Kloppfer et al. (1982) determined that the
half-life for volatilization of PCP was 653 h at 23°C, 328 h at 30°C,
and 211 h at 40°C. Photolysis can also be temperature-dependent. Omura
& Matsuura (1971) found that higher solution temperatures increased
the photodecomposition of 4-MCP. The microbial breakdown of lower
chlorophenols as a function of temperature was investigated in water,
soil, and sediment by Baker et al. (1980). As expected, biodegradation
was higher at 20°C or 4°C than at 0°C. However, high temperatures also
limit the microbial decomposition of chlorophenols; Tyler & Finn
(1974) found that growth of Pseudomonas on 2,4-DCP fell off sharply
above 25°C.
Under some conditions, exposure of chlorophenols to elevated
temperatures, such as those used in heating and burning, can lead to
the formation of chlorinated dibenzo- p-dioxins (PCDDs) and
dibenzofurans (PCDFs) (Rappe et al., 1978b). Many PCDDs and PCDFs are
extremely persistent in the environment and are toxic for living
systems (WHO, in press).
4.4 Persistence
As a consequence of chlorophenol decomposition through photolysis,
biodegradation, and perhaps chemical catalysis, virtually all
chlorophenol compounds will be eliminated from most environments.
Some, notably PCP and the lower chlorophenols with a chlorine in the
meta position, will persist for a longer time than others, but even
these should eventually be broken down, wherever suitable light
exposure or microorganisms occur.
No information is available on the persistence of chlorophenols in
air. Photodecomposition may be an important removal mechanism,
particularly for the higher chlorophenols (Callahan et al., 1979).
The long-term persistence of chlorophenol isomers is only expected
where there is a lack of degradative activity and/or outward
transport, allowing them to accumulate, as is illustrated by the range
of residence times for chlorophenols in aquatic environments.
Substantial quantities of chlorophenols were eliminated from in situ
marine pelagic enclosures (4-MCP and 2,4-DCP) (Kuiper & Hansveit,
1984) and from fresh waters in vitro: 2-MCP and 4-MCP (Ettinger &
Ruchhoft 1950; 2,4-DCP (Aly & Faust, 1964); 2,4,6-T3CP and PCP
(Schauerte et al., 1982; Sugiara et al., 1984) in roughly 1-3 weeks.
In contrast, it has been shown that T4CP and PCP in sediments, where
photolysis and apparently biodegradation are minimal, may persist for
years (Pierce & Victor, 1978; DeLaune et al., 1983; Fox & Joshi,
1984).
A similar range of persistences has been reported for soils. Most
of 2-MCP and 4-MCP was removed by microorganisms in soil after 10 and
20 days, respectively (Walker, 1954). Virtually all of 2,3,4,5-,
2,3,4,6-, and 2,3,5,6-T4CP (100 mg/kg dry soil) disappeared from
paddy soils after 4 weeks of incubation in vitro (Ide et al., 1972).
Concentrations of trichlorophenols and tetrachlorophenols derived from
PCP degradation in vitro were in turn, substantially reduced after
many days (Kuwatsuka & Igarashi, 1975). The disappearance of at least
90% of added PCP from soils in vitro took from 21 to 205 days,
proceeding most rapidly in soils with a moderate to high organic
content and acclimated microorganisms (Kozak et al., 1979).
Most studies of chlorophenol metabolism have only monitored the
disappearance of the parent compound, but a few others have indicated
that subsequent metabolism may completely mineralize CPs. Thus,
labelled 4-MCP, 2,4-DCP, and 2,4,5-T3CP were converted to 14CO2 in
the in vitro incubation of estuarine waters and sediments (Lee &
Ryan, 1979; Hwang et al., 1986). About one half of the radioactivity
added to aerobic artificial streams as 14C-PCP was recovered as
carbon dioxide after 21 days (Pignatello et al., 1983). Pure cultures
and activated sludges may also mineralize chlorophenols to carbon
dioxide (Teidje & Alexander, 1969; Duxbury et al., 1970; Chu & Kirsch,
1973; Moos et al., 1983). Methane may be produced under anaerobic
conditions (Boyd & Shelton, 1984), but more often little or no
mineralization occurs in anaerobic sediments and sludges (Lee & Ryan,
1979; Horowitz et al., 1982; Pignatello et al., 1983).
In most instances, aerobic metabolism involves dechlorination and
hydroxylation, which are usually followed by cleavage of the phenol
ring at the ortho position and subsequent complete degradation. The
products of ring cleavage at the meta position are more resistant to
degradation and tend to accumulate in the medium. Reductive
dechlorination is an initial step towards complete mineralization
under anaerobic conditions (Krijgsheld & van der Gen, 1986).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental Levels
A substantial amount of research has been carried out on the
concentration of pentachlorophenol in the environment; however,
relatively few studies have been concerned with the determination of
the levels of other chlorophenols. Nonetheless, enough information is
available to make a preliminary survey of the residues of these
chlorophenols in the environment.
5.1.1 Air
No information is available on the ambient levels of
chlorophenols, other than PCP, in the atmosphere. The data on PCP are
limited but may provide a useful indication of the potential for
atmospheric distribution of the other chlorophenols. Measurable
quantities of PCP are present in ambient air, and are surprisingly
ubiquitous: Cautreels et al. (1977) detected 0.93 and 0.25 ng PCP/m3
in the mountains high above La Paz, Bolivia, a presumably
uncontaminated environment. Concentrations at 4 sites in Antwerp,
Belgium, ranged from 5.7 to 7.8 ng PCP/m3. It is not known whether
the compound was present as a vapour, or adsorbed on airborne
particulates. Presumably as a result of such transport, chlorophenols
have been detected in rainwater, alpine lakes, and snow (Bevenue et
al., 1972; Paasivirta et al., 1985).
5.1.2 Water
Residues of all chlorophenol isomers have been detected in aquatic
systems. Generally, residues are present at measurable concentrations
in discharges from such sources as manufacturing plants,
wood-treatment facilities, municipal waste discharges, and in the
receiving waters adjacent to these sources. Concentrations in other
receiving waters are more sporadic and quite low. While the levels are
low, chlorophenols have been detected in some of the least polluted
waters in the world.
Most reports of chlorophenol levels in water are from sites in the
vicinity of wood-treatment facilities. For instance, Fox & Joshi
(1984) measured concentrations of PCP and tetrachlorophenols in water,
sediments, and selected biota from the Bay of Quinte, in an
investigation of contamination by a wood-treatment plant. Levels of
both T4CP (2,3,4,5 plus 2,3,5,6) and PCP generally declined with
increasing distance from the source. Adjacent subsurface water levels
of T4CP ranged from 0.005 to 0.086 µg/litre over the summer of 1978,
while at the furthest site, 100 km distant, the range was 0.005-
0.030 µg/litre. Surface film samples contained T4CP concentrations
from 2.3-200 times higher than those below the surface. Bacon (1978)
assayed chlorophenols in the effluent from a Kraft pulp-mill at St.
John, New Brunswick, and found 2,4-DCP and 2,4,6-T3CP in the samples
before and after chlorination and caustic extraction. No chlorophenols
were detected in the receiving waters, perhaps as a result of tidal
flushing. Similarly, in a Great Lakes survey conducted by the Ontario
Ministry of the Environment (Jones, 1981), chlorophenol congeners were
detected in receiving waters: samples from the St. Mary's River near a
pulp and paper-mill did not contain any detectable levels of
chlorophenols, while 1 of 10 samples taken from near another mill on
Thunder Bay, contained 4 µg DCP/litre, and 2 contained 3 and 23 µg
T3CP/litre, respectively.
An Environment Canada survey (1979) of British Columbia coastal
waters for chlorophenol contaminants in surface waters, effluents,
sediments (section 5.1.2.2), and biota (section 5.1.6) did not reveal
any DCP or T3CP residues in water samples, but low levels of T4CP
(and PCP) were present at almost all sites: tetrachlorophenol
concentrations ranged from trace levels to 1.0 µg/litre in fresh
waters and 5.2 µg/litre in sea water. Effluent concentrations of T4CP
were high at 2 out of 4 discharges sampled (530 and 8270 µg/litre),
even exceeding PCP levels. Garrett (1980) reported sources and levels
of chlorophenols in sediments (section 5.1.2.2), fish (section 5.1.6),
and a variety of discharges from industry, waste disposal systems, and
runoff from landfills in the lower Fraser River and estuary. The most
frequently detected chlorophenols were 2,4,6-T3CP, 2,3,4,6-T4CP, and
PCP. Levels in most discharges were less than 7 µg/litre. Discharges
from municipal sewage-treatment plants in the same study also
contained several chlorophenols, most frequently 2,4,6-T3CP
(trace-1.2 µg/litre), 2,3,4,6-T4CP (trace-28.3 µg/litre) and PCP.
As in the Canadian studies, levels in receiving waters in the USA
are typically low. Morgade and co-workers (1980) did not find any
detectable levels of chlorophenols, other than PCP, in drinking-waters
in Dade County, Florida. In vitro chlorination of secondary sewage
effluent and power plant cooling waters, using chlorine levels and
treatment times similar to those used in practice, yielded only
µg/litre quantities of 2-MCP, 3-MCP, and 4-MCP (Jolley et al., 1975).
Pierce & Victor (1978) measured the levels of PCP and some of its
degradation products (2,3,5,6-T4CP and PCP-OCH3) in a Mississippi
lake contaminated by an overflow from a wood pole-treatment plant.
Prior to the spill, levels of 2,3,5,6-T4CP in the water were low,
ranging from 0.07 to 0.21 µg/litre. Concentrations were higher after
the spill (0.25-2 µg/litre), and remained relatively stable for at
least 4 months. Sediment (section 5.1.2.2) and fish tissue (section
5.1.6) samples were also collected.
Dutch research workers have monitored chlorophenol levels in the
water and sediments of the major rivers in industrial areas in the
Netherlands since 1976 (Wegman & Hofstee, 1979; Wegman & van den
Broek, 1983). In both reports, maximum levels of all chlorophenols,
other than PCP, seldom exceeded 1 µg/litre. Medians for the most
frequent congeners for 6 rivers in 1976 and 1977 ranged as follows:
2,6-DCP, trace-0.15 µg/litre; 2,4,5-T3CP, trace-0.15 µg/litre;
2,4,6-T3CP, trace-0.19 µg/litre; 2,3,4,6-T4CP, trace-0.11 µg/litre
(Wegman & Hofstee, 1979). Likewise, Piet & de Grunt (1975) reported
that levels of monochlorophenols ranged from not detectable to
20 µg/litre, dichlorophenols from not detectable to 1.5 µg/litre, and
trichlorophenols from not detectable to 0.1 µg/litre in Netherland
rivers and coastal waters. These ranges include the levels reported by
Zoeteman (1975) for Rhine river water and drinking-water. Zoeteman et
al. (1981) compiled information on concentrations of a variety of
chemicals in Dutch ground waters. The highest concentrations of
chlorophenols reported were as follows: 2,3,6-T3CP, 1 µg/litre;
2,4,5-T3CP, 2 µg/litre; 2,3,4,6-T4CP, 3 µg/litre; 2,3,5,6-T4CP,
5 µg/litre; PCP, 1 µg/litre.
In the Glatt river in Switzerland, concentrations of 2,3,4,6-T4CP
over the year averaged about 0.04-0.05 µg/litre at each of several
stations along a 35-km stretch of the river (Ahel et al., 1984).
Paasivirta et al. (1985), assayed water, snow, ash, benthic
invertebrates, fish, and birds from relatively unpolluted Finnish
lakes for chlorophenol residues. (Levels in the biota are reported in
section 5.1.6). The compounds 2,6-DCP, 2,4-DCP, 2,4,6-T3CP,
2,4,5-T3CP, 2,3,4,6-T4CP, and PCP were widespread, and present at
µg/litre concentrations in pulp-bleaching liquors and ng/litre levels
in lake waters (Table 10). Chlorination of some waters elevated the
concentration of the total chlorophenols measured almost 6-fold, from
0.043 µg/litre to 0.243 µg/litre. Elevated levels have been associated
with specific discharges in Europe. Leachate from a Danish chemical
dump site used during 1953-71 contained, among other compounds, PCP
and T4CP, the latter ranging in concentration from 0.030 to
80 µg/litre (Folke et al., 1984). Folke (1984) analysed effluent from
a Danish sewage-treatment plant, which received a portion of its
wastes from the manufacture of phenoxy herbicides, for a number of
chlorophenols. The effluent contained 0.1 µg 2-MCP/litre, 0.03 µg
4-MCP/litre, 0.5 µg 2,4-DCP/ litre, 0.6 µg 2,6-DCP/litre, 8 µg
2,4,6-T3CP/litre, and 0.03 µg 2,3,4,6-T4CP/litre.
Table 10. Concentration (µg/litre) of chlorophenols in Finnish pulp mill waste
liquors and fresh watersa
Chlorophenol Type of sample
Waste liquorsb Fresh watersb Janakka water Jyvaskyla
raw tap tap
2,6-DCP ND - 12c ND - 0.073 0.010 0.062 0.272
2,4-DCP ND - 11 ND - 0.014 0.014 0.053 0.093
2,4,6-T3CP 15 - 28 ND - 0.011 ND 0.030 0.014
2,4,5-T3CP ND - 66 ND - 0.019 0.019 0.059 0.035
2,3,4,6-T4CP ND - 10 ND - 0.090 ND 0.016 0.009
PCP ND - 01 0.064 - 0.011 0.023 0.005
a From Paasivirta et al. (1985).
b Range reported.
c ND -- Not detectable.
A similar variety of congeners was detected in effluent from a
sewage-treatment plant that was processing paper-mill wastes.
Lindstrom & Nordin (1976) found 115 µg 2,4,6-T3CP/litre in spent
bleach liquors from kraft mill pulp chlorinated in vitro, and noted
that dichlorophenols were also present.
As in Canada, high concentrations of chlorophenols in European
fresh waters are associated with wood-treatment facilities. Waters on
the sites of 2 Finnish sawmills, in which a sodium chlorophenate
preservative (mostly Na-2,3,4,6- T4CP, with substantial quantities of
the 2,4,6-T3CP and PCP salts) was used to protect against sapstain
fungi, contained total chlorophenol concentrations ranging from 1.6 to
20 000 µg/litre. The highest concentration occurred in a blind drain
adjacent to the dip site (Valo et al., 1984). Off-site levels in
ground water and lake water were much lower, with a maximum of 1.17 µg
chlorophenols/litre. Concentrations of chlorophenols in the effluent
from a Swedish sawmill on two separate dates were, respectively: 8.3
and 24 µg 2,4-DCP/litre; 40 and 22 µg 2,4,6-T3CP/litre; 7.5 and
5.8 µg 2,3,4,6-T4CP/litre (Xie et al., 1986).
Chlorophenols are also widespread in European marine waters,
generally at lower concentrations than in fresh waters. Weber & Ernst
(1978) noted that coastal waters off the Federal Republic of Germany
yielded only trace quantities (1 ng/litre) of 2,4-/2,5-DCP, 2,6-DCP,
2,4,5-T3CP, 2,4,6- T3CP, 2,3,4,5-T4CP, and 2,3,4,6/2,3,5,6-T4CP.
Danish marine waters receiving chemical dump leachate including T4CP
(at 0.030-80 µg/litre) showed corresponding levels in water of
0.006-0.008 µg/litre (Folke et al., 1984). Chlorophenol levels in
coastal waters off Sweden fell off rapidly with increasing distance
from a sulfate pulp-mill, from maximum concentrations on one date of
0.123 µg 2,4-DCP/litre, 0.040 µg 2,6-DCP/litre, 0.370 µg
2,4,6-T3CP/litre, and 0.084 µg 2,3,4,6-T4CP/litre to undetectable
levels (Xie et al., 1986). For example, the half-distances for
2,4,6-T4CP and 2,3,4,6-T4CP disappearance were 1 and 0.8 km,
respectively (see also sections 4.1.3, 5.1.2.2).
5.1.2.1 Sediments
Chlorophenol concentrations in sediments are for the most part
much greater than those in the overlying water. This may reflect
adsorption of the chlorophenols on suspended particulates in the water
column, with subsequent sedimentation. For instance, Eder & Weber
(1980) reported higher levels of chlorophenols (di- to penta-) in both
sediments and suspended solids compared with those in water.
Fox & Joshi (1984) analysed sediment cores for T4CP and PCP in
their study of chlorophenol contamination from a wood-preservation
facility on the Bay of Quinte, Lake Ontario. For the upper sediments
(1/2-cm sections of the top 5 cm of the core) levels ranged from 1 to
48 µg/kg dry weight. In a similar study, Environment Canada (1979)
analysed sediments in British Columbia waters associated with
wood-preservation plants. T4CP was present at all 11 sites, and
ranged from a trace to 1600 mg/kg dry sediment. T3CP was found at 4
of the 11 sites, with as much as 170 µg/kg of dry sediment measured.
Although the kraft pulp-mill effluent studied by Bacon (1978)
contained 2,4-DCP and 2,4,6-T3CP, only PCP was detected in sediment
samples downstream.
As a result of a PCP spill from a pole-treatment plant, sediments
in a Mississippi lake were contaminated with PCP and its degradation
products, including 2,3,5,6-T4CP (Pierce & Victor, 1978). Four months
after the spill, the levels of 2,3,5,6-T4CP in surface sediment
ranged from 3.8 to 71 µg/kg dry sediment whereas, 1 month after the
spill, levels of between 12 and 97 µg/kg dry sediment had been
detected.
Interpretation of these results is confounded by their
variability, the residence time (several weeks) of PCP in the water
column, and a 1974 spill at the same site. T4CP was present in the
sediments of a nearby, reportedly uncontaminated, pond at 1 µg/kg.
High concentrations of DCP and T3CP were present in sediments
adjacent to hazardous waste dumps near the Niagara River, at maximum
levels of approximately 2000 and 500 µg/kg, respectively (Elder et
al., 1981).
In studies in progress on chlorophenols that are present in
surface and coastal waters and sediments in The Netherlands, the
compounds 2,5-DCP, 2,3,5-T3CP, 2,4,5-T3CP, 2,3,4,5-T4CP,
2,3,4,6-T4CP, and PCP are observed most frequently (Wegman & van den
Broek, 1983). In the industrial regions that have been the focus of
this research, moderate levels of contamination prevail (several µg/kg
of dry sediment for most isomers) (Table 11).
Table 11. Levels of chlorophenols (µg/kg) in sediments from Dutch
surface waters and the Weser estuary
Compound Lake Ketelmeera Other Netherlanda Weser
surface waters Estuaryb
(Range) (Mean)
Median Maximum
3-MCP - 43
2,3-DCP 1.9 2.2
2,4-DCP 4.4 10 ND - 3.6 1.170
2,5-DCP 6.3 11 ND - 3.8
2,6-DCP 1.8 31 ND - 2.5
3,4-DCP 9.8 49 ND - 4.1
3,5-DCP 6.6 12 ND - 9.3
2,3,4-T3CP 0.7 0.8 ND - 0.6
2,3,5-T3CP 2.4 11 ND - 1.5
2,4,5-T3CP 6.4 15 ND - 6.3 1.170
2,4,6-T3CP 1.9 3.7 ND - 0.9 0.300
3,4,5-T3CP 1.2 19 ND - 1.7 0.310
2,3,4,5-T4CP 0.9 8.9 ND - 0.9
2,3,4,6-T4CP 1.7 4.9 ND - 1.7 1.546
2,3,5,6-T4CP 1.4 2.8 ND - 0.4
a From:Wegman & van den Broek (1983) (dry sediment).
b From: Eder & Weber (1980) (wet sediment).
Paasivirta et al. (1980) analysed sediments and biota (section
5.1.6) from three Finnish lakes for several chlorophenols and related
compounds. In one lake, contaminated only from sawmills upstream,
concentrations of T3CP and T4CP were 4.68 and 33.4 µg/kg of
sediment; respectively (Table 12, section 5.1.6.1), while in another
lake downstream from a pulp-mill, the corresponding values were 27.7
and 50.1 µg/kg. The third lake, further downstream from pulp and paper
inputs, contained intermediate levels of both compounds.
Levels in estuarine and marine sediments overlap considerably with
those from fresh waters. Butte et al. (1985) determined chlorophenol
concentrations in sediments and clams (section 5.1.6) from a German
bight that had received untreated PCP waste from a pulp and paper-mill
for 13 or 14 years, until 2 years before the study. Sediments from one
site near the former discharge contained from 28.3 to 30.9 µg
2,3,4,5-T4CP/kg of dry sediment, while those from another site
contained 92.8 µg 2,3,4,5-T4CP/kg and 7.9-11.6 µg 2,3,4,6- plus
2,3,5,6-T4CP/kg. Sediments taken some distance from the discharge
contained only 1.2 µg 2,3,4,5-T4CP/kg or less. Eder & Weber (1980)
reported levels of chlorophenols (di- to penta-) that corresponded
with the lower end of the range for The Netherlands surface waters;
mean concentrations of chlorophenols other than pentachlorophenol
ranged from 0.300-1.546 µg/litre. Similar levels were found in Baltic
Sea sediments from a site 2 km distant from a sulfate pulp-mill; these
contained 0.9 µg/kg dry sediment of 2,4-DCP, 0.4 µg 2,4,6-T3CP/kg,
and 3.1 µg 2,3,4,6-T4CP/kg (Xie, 1983). In a subsequent study, higher
levels of the same congeners were found at this location (Xie et al.,
1986). While surface sediments from sites roughly 5-10 km from the
discharge contained levels of chlorophenol that were near or below the
limits of detection, sediments 2 km or less from the mill contained as
much as 16 µg 2,4-DCP/kg dry sediment, 19 µg 2,4,6-T3CP/kg, and 89 µg
2,3,4,6-T4CP/kg.
5.1.3 Soil
Information on ambient levels of chlorophenol residues in soils is
limited, perhaps reflecting limited use of these compounds on soils.
The processes of degradation and movement also combine to reduce soil
residues (section 4).
The sole report on levels of chlorophenols in Canadian soils is
undoubtedly atypical of the environment at large. Garrett (1980)
reported that soil samples from the former site of a pesticide plant
in Richmond, British Columbia contained 2 mg T4CP/kg dry soil,
0.18 mg T3CP/kg, as well as low levels of PCP.
Valo et al. (1984) assayed chlorophenols in the soil and water
(section 5.1.2) around two Finnish sawmills where lumber was treated
against sap-staining fungi. Soils at both facilities were heavily
contaminated with chlorophenols; up to 70 mg/kg were found near the
dipping site, and up to 6 mg/kg occurred in a storage area for the
preserved wood. Soil outside the storage area contained only
0.1 mg/kg. The most common chlorophenols in the preservative
formulation also predominated in the near-surface soils, but lower
chlorinated phenols (particularly dichlorophenols) became increasingly
important further down the soil horizon, presumably as a result of
decomposition of the preservative. In general, chlorophenol
concentrations declined in the progressively deeper layers.
Kitunen et al. (1987) determined the concentrations of
chlorophenols and their contaminants in soil near the preserving
facilities at 4 different sawmills. Concentrations of chlorophenols in
soil ranged from 500 to 3500 mg/kg, polychlorinated phenoxyphenols,
from 1-5 mg/kg, and polychlorinated dibenzofurans, from 0.2-5 mg/kg.
No clear decrease in the soil concentrations of these compounds was
seen during the first year after the mill stopped using technical
chlorophenol.
5.1.4 Food, feed, and drinking-water
5.1.4.1 Food
Little information is available on residues in food of
chlorophenols, other than PCP. Low-level contamination undoubtedly
occurs as a result of contact with treated wood storage and transport
containers, and from herbicide applications. However, both of these
uses are prohibited in a number of countries (section 3.2.2); hence,
the following cases may overestimate the extent of contamination.
Both T4CP and PCP were identified in agricultural products by the
Alberta Department of Agriculture (Jones, 1981). Trace levels of T4CP
(mainly 1 µg/kg, maximum 45 µg/kg) occurred in grab samples of
carrots, potatoes, turnips, and beets. T4CP concentrations as high as
472 µg/kg occurred as a result of contamination from treated wood.
No residues were detected in 45 samples of Southern Ontario milk
analysed for 2,4,5-T3CP, 2,4,6-T3CP, 2,3,4,6-T4CP, and PCP (Frank
et al., 1979).
In the USA, Bristol et al. (1982) determined 2,4-DCP
concentrations in 3 varieties of potatoes sprayed with 2,4-D as a
growth regulator at an application rate of 140 g/ha. Levels of
chlorophenol, which could arise as a contaminant or degradation
product of the herbicides, varied slightly in 3 varieties of potatoes,
but ranged between 3 and 8.8 µg/kg. Potatoes not sprayed with 2,4-D
did not contain any detectable 2,4-DCP.
Stijve (1981) determined chlorophenol residues in edible products
derived from the bones and hides industry, where PCP was used as a
preservative and disinfectant. Fleshing grease intended as an
ingredient in cow feed, contained 50-480 µg 2,4,5-T3CP/kg, 2060-13
400 µg 2,3,4,6-T4CP/kg, and 210-1090 µg PCP/kg. A survey of 50
samples of edible gelatins (worldwide distribution), which are
produced from the collagen in hides and bones, all contained PCP; 30%
also contained 2,3,4,6-T4CP and trace amounts of trichlorophenols. In
some countries, where chlorophenola are used for the disinfection of
hides, chiorophenol levels in some gelatins range from 1000 to
5000 µg/kg.
Chlorophenols have been found at very low levels in the tissues of
commercial livestock and poultry. Fartington & Munday (1976) found
2,3,4,6-T4CP (at 2-3 µg/kg) in chicken flesh from 3 out of 4 shops in
the United Kingdom.
5.1.4.2 Livestock feed
No data are available on levels of the lower chlorophenols in
animal feed. Jones (1981) cited a case in which a boxcar that had been
used to ship PCP was later filled with feed oats, contaminating the
oats with roughly 2000 mg PCP/kg. Presumably, the feed was
simultaneously contaminated with T4CP at roughly 10% of the PCP
levels, given the usual formulation for technical Na-PCP. Livestock
illness and mortality were associated with this incident.
5.1.4.3 Drinking-water
Data on chlorophenol residues in drinking-water are quite limited,
but suggest that levels vary considerably between locations. Sithole &
Williams (1986) reported that low levels of a number of lower
chlorophenols occurred infrequently in potable waters at 40 Canadian
treatment plants. Chlorination increased the concentrations of 2-MCP
(maximum observed: 65 ng/litre), 4-MCP (127 ng/litre), 2,4-DCP
(72 ng/litre), 2,6-DCP (33 ng/litre), and 2,4,6-T3CP (719 ng/litre),
but decreased those of 2,3,4,5-T4CP (reduced below the limit of
detection) and PCP (34 ng/litre). Lower chlorophenols were not
detected in drinking-water supplies from Dade County, Florida (Morgade
et al., 1980). Dietz & Traud (1978) found low concentrations of a
variety of CP congeners in drinking-water from the Ruhr area of the
Federal Republic of Germany, including: 3-6 ng 2,4-DCP/litre; 20 ng
2,6-DCP/litre; 1 ng 2,4,6-T3CP/litre; 1 ng 2,3,5-T3CP/litre; 3 ng
2,4,5-T3CP/litre; 1 ng 2,3,4,5-T3CP/litre; and 3 ng
2,3,4,6-T4CP/litre. For comparison, levels in the effluent from a
sewage plant were concurrently 2 orders of magnitude higher.
In contrast, Paasivirta et al. (1985) found a number of
chlorophenols in Finnish tap waters at levels roughly one order of
magnitude higher than those in the German study (Table 10). As these
data indicate, chlorophenol concentrations in drinking-water are
generally quite low; indeed, the low threshold concentrations
producing undesirable organoleptic (taste and odour) properties would
make higher levels in drinking-water unacceptable (WHO, 1984 and Table
21).
5.1.5 Treated wood
The treatment of wood continues to be an important use for
chlorophenols (section 3.2.2), with considerable potential for
environmental contamination, as well as general and occupational human
exposure. In 1978-79, levels of chlorophenols, principally PCP, were
measured in samples of wood shavings that were used as livestock
litter in Southern Ontario (Jones, 1981). No trichlorophenols were
detected, but T4CP, PCP, and related chlorinated anisoles were
quantified. T4CP levels were as high as 70 mg/kg. Daniels & Swan
(1979) determined that 15 lumber samples from a British Columbia
sawmill protected with a commercial formulation of T4CP and PCP salts
("sodium penta") contained on average 44 µg chlorophenols/cm2 (range,
29-86 µg/cm2), most of which was T4CP.
In the United Kingdom, Parr et al. (1974) found that wood shavings
from imported wood used as litter for hens were contaminated with the
sodium salts of T4CP and PCP. When lumber is planed, most of the
chlorophenates are removed in the shavings. As a result, levels of
2,3,4,6-T4CP and PCP in fresh litter are quite high. According to
Parr et al.(1974), T4CP concentrations averaged 54 mg/kg and ranged
from 4 to 310 mg/kg. When the litter is used, chlorophenol levels fall
off as they are converted to their corresponding chloroanisoles (Gee &
Peel, 1974): spent litter contained on average 0.7 mg T4CP/kg.
Similarly, Curtis et al.(1972) measured as much as 100 mg
2,3,4,6-T4CP/kg in fresh shavings and sawdust from the United
Kingdom.
Levin & Nilsson (1977) assayed for T4CP, PCP, and related
compounds in wood dust from a Swedish sawmill. The wood had been
treated with 2% Na-2,3,4,6-T4CP; T4CP levels in the dust ranged from
100 to 800 mg/kg.
5.1.6 Terrestrial and aquatic organisms
5.1.6.1 Invertebrates
Invertebrates contain levels of chlorophenols that are higher than
those found in the environment at large, reflecting moderate
bioconcentration by these organisms (section 4.2.2). Environment
Canada (1979) found tetrachlorophenol in invertebrates from the
receiving waters for wood-treatment plant effluents in British
Columbia. In fresh waters, crayfish (Pacifasticus sp.) pincer muscle
contained traces of T4CP. The same tissue from a marine crab (Cancer
magister) contained T4CP levels ranging from a trace to 20 µg/kg
wet weight. One clam (Macoma) sampled contained 12 µg T4CP/kg,
presumably in muscle tissue. T3CP was not detected in any of the
organisms. Similar chlorophenol burdens were reported by Bacon (1978),
who assayed for 2,4-DCP, 2,4,6-T3CP, and PCP in lipids from a clam
(Mya arenia) and sandshrimp (O'angon septemspinosa) from waters
receiving pulp-mill effluent. Trace quantities of 2,4-DCP were present
in both organisms, while 2,4,6-T3CP levels ranged from undetectable
to 7.2 µg/kg wet weight for the clam and 9.2 µg/kg wet weight for the
sandshrimp.
During the course of their survey of chlorophenol residues in the
Weser Estuary and German Bight, Ernst & Weber (1978) determined that a
polychaete (Lanice conchilega) contained on average 11.8 µg 2,4-DCP
and 2,5-DCP/kg, 19.3 µg 2,4,5-T3CP/kg, 26 µg 2,4,6-T3CP/kg, 7 µg
2,3,4,5-T4CP/kg, 66.9 µg 2,3,4,6-T4CP and 2,3,5,6-T4CP/kg, and
117.5 µg PCP/kg (all values wet weight).
Paasivirta et al. (1980) have surveyed levels of chlorophenols in
a variety of biota in 3 lakes with different chlorophenol inputs
(section 5.1.2.1 and Table 12). Plankton generally contained little or
no tri-chlorophenol, but relatively high concentrations of
tetrachlorophenol. Mussels (Anodonta piscinalis) and sponges
(Spongilla lacustris) contained moderate levels of both T3CP and
T4CP. Chlorophenol burdens in organisms from the different lakes
generally ranked in the same order as the perceived chlorophenol
inputs into the 3 lakes.
In more recent work (Paasivirta et al., 1985), mussels, chironomids,
sponges, and fly larvae from Lake Vatia, 5 km downstream from a
pulp-mill, were analysed for several chlorophenols. Low levels
(usually not detected to 20 µg/kg fresh weight) of chlorophenols were
found in all invertebrates except sponges, which inexplicably
contained 195 µg 2,4-DCP/kg fresh weight and 22 µg 2,4,6-T3CP/kg.
Table 12. Chlorophenol levels in various environmental compartments in three
Finnish Lakes (µg/kg wet weight, except for sediment, dry weight)a,b,c
Population Lake N Trichlorophenol Tetrachlorophenol
species
X S CV X S CV
Kd 8 0.79 1.6 2.03 20.2 40.0 1.98
Pike pd 6 17.3 18.1 1.05 11.1 14.9 1.34
Vd 10 13.6 19.1 1.40 19.0 12.4 0.65
Roach K 9 ND ND 2.19 1.82 0.83
P 10 4.67 5.29 1.13 6.41 3.42 0.53
V 10 55.9 53.4 0.96 11.5 8.26 0.72
Mussel K 10 ND ND 2.83 4.44 1.57
P 9 1.44 2.21 1.53 7.44 3.47 0.47
Sponge K 5 0.36 0.80 2.22 6.30 6.36 1.01
P 5 6.86 9.14 1.33 1.45 2.43 1.68
V 5 4.96 3.73 0.75 2.56 1.12 0.44
Plankton K 4 ND ND ND ND
(100 µm) P 4 ND ND 9.28 10.0 1.08
V 4 2.45 4.90 2.00 9.90 1.73 0.17
Plankton K 4 ND ND 7.95 15.6 1.96
(25 µm) P 4 NO ND 14.3 10.5 0.73
V 3 ND ND 23.1 4.08 0.18
Sediment K 5 4.68 10.4 2.22 33.4 38.6 1.16
(0-2 cm) P 5 10.7 15.1 1.42 37.5 29.3 0.78
V 5 27.7 17.2 0.62 50.1 17.3 0.35
a From: Paasivirta et al. (1980).
b N = number of samples analysed;
x = mean;
s = standard deviation;
cv = coefficient of variation.
c Wet weight of plankton was calculated from dry weight by multiplying by
13.69.
d Lake areas: K = Konnevesi; P = Paijanne; V = Vatia.
Butte et al. (1985) analysed clams from a German bight that had
received untreated PCP-contaminated discharge from a paper-mill for a
number of years (section 5.1.2.1). Clams near the discharge contained
0.7-55.6 µg 2,3,4,5-T4CP/kg dry tissue, while those further away
contained, at the most, 2.6 µg 2,3,4,5-T4CP/kg and 0.2 µg 2,3,4,6-
plus 2,3,5,6-T4CP/kg. Similar low concentrations of 2,3,4,6-T4CP and
PCP were found in blue mussels (Mytilus edulis) from Danish coastal
waters (Folke & Birklund, 1986). Tissue levels of T4CP averaged from
0.2 to 2.9 µg/kg fresh weight (1-23 µg/kg dry weight) for mussels from
various locations in 1985, with no obvious relation to a nearby
chemical dump from which chlorophenols were leaching.
5.1.6.2 Fish
In general, levels of chlorophenols in fish are similar to, or
slightly higher than, those in invertebrates. Bacon (1978) studied
chlorophenol levels in different tissues from several fish species
from the St. John River estuary, New Brunswick, which receives
pulp-mill effluents. Residues of 2,4-DCP and, 2,4,6-T3CP were
detected, usually at several tens of µg/kg wet weight and several
µg/kg wet weight respectively, in all tissues including muscle,
viscera, skin, and liver (calculated from per-lipid-weight data in
original publication). In some instances, levels in liver were much
higher than this; concentrations as high as 242.9 µg 2,4-DCP/kg and
128 µg T3CP/kg (wet weight) were measured. In surface and coastal
waters in British Columbia, Environment Canada (1979) detected T4CP
in marine and freshwater sculpins. Across all sites, skeletal muscle
burdens averaged 30 µg/kg wet weight, and ranged from a trace to
100 µg/kg. Levels were roughly an order of magnitude higher in liver.
Similarly, Garrett (1980) reported that marine sculpins (Leptocottus
armams) from the lower Fraser River were the principal fish with
detectable amounts of T4CP, averaging 24.9 µg/kg wet weight, and
ranging from a trace to 62 µg/kg. Significant levels of T4CP were
also found in squawfish, which averaged 10.5 µg/kg wet weight and
contained as much as 18 µg/kg. Chlorophenol levels were higher in fish
that were caught in the industrialized areas of the river. Spottail
shiners (Notropis hudsonius) from Lakes Erie and Ontario contained
2,4,5-T3CP and 2,4,6-T3CP at maximum concentrations of 22 and
33 µg/kg (wet weight, whole fish), respectively (Canada-Ontario Review
Board, 1981).
Similar tissue concentrations have been detected in fish from
European waters. In Finnish lakes spanning a gradient of chlorophenol
inputs (Paasivirta et al., 1980), skeletal muscle of roach contained
on average 0-55.9 µg T3CP/kg wet weight, and 2.19-11.5 µg T4CP/kg
(Table 12). Chlorophenol concentrations in roach muscle were related
to the chlorophenol inputs into the lake. In contrast, levels in pike
skeletal muscle (average ranges 0.79-17.3 µg T3CP/kg, and
11.1-20.2 µg T4CP/kg) bore no relation to chlorophenol inputs. In
subsequent studies (Paasivirta et al., 1981, 1983, 1985), average CP
levels in muscle of pike, burbot, ide, and roach taken from waters
receiving pulp-mill discharges also fell within the same range, except
in the case of heavily polluted waters.
In Lake Tiiranselka, which receives a large volume of pulp-mill
effluent, average concentrations of 2,4,6-T3CP and 2,3,4,6-T4CP in
pike muscle were 37.02 and 125.02 µg/kg wet weight, respectively. Two
species of Baltic salmon analysed for chlorophenols contained similar
levels of contamination to those found in fish from moderately
polluted lakes (Paasivirta et al., 1985). Muscle tissue of salmon from
2 rivers and a hatchery contained an average of 3 µg 2,4,6-T3CP/kg
fresh weight, 1.8 µg 2,4,5-T3CP/kg, and 12.5 µg 2,3,4,6-T4CP/kg.
Fish collected in the vicinity of a pulp-mill effluent in Sweden
contained 2,4,6-trichlorophenol and related compounds that were
present in the discharge (Landner et al., 1977). Perch (Perca
fluviatilis) contained levels of 2700 µg/kg in liver fat (62.1 µg/kg
fresh weight), while Northern Pike (Esox lucius) contained
400-500 µg/kg (27.5-40.4 µg/kg fresh weight).
Extremely high chlorophenol levels have occurred as a result of
accidental spills. Following contamination of a Mississippi lake by
PCP in December 1976 (section 5.1.2.1), levels of the degradation
product 2,3,5,6-T4CP in sunfish liver and muscle increased by 1-2
orders of magnitude (Pierce & Victor, 1978) (Table 13). Four to 5
months after the spill, liver levels in sunfish were approaching
pre-spill levels, while levels in muscle tissue apparently cleared
more slowly. Bass and catfish showed particularly high levels of T4CP
after the spill, but unfortunately no baseline data were provided.
5.1.6.3 Other non-human vertebrates
Data on chlorophenol concentrations in vertebrates other than fish
or human beings are quite limited. Purple martin fledglings analysed
for chlorophenol residues contained 2 µg T4CP/kg (Jones, 1981). The
tissue analysed was not specified. Levels of CP residues in eggs,
embryos, and chick tissues of ring-billed gulls on the Ottawa and St
Lawrence Rivers have been reported (NRCC, 1982). The compounds
2,4-DCP, 2,4,5-T3CP, 2,4,6-T3CP, and PCP were present in most
tissues, the highest concentrations occurring in liver and brain
(Table 14). Paasivirta et al. (1985) measured chlorophenol residues in
the muscle tissue of 45 juvenile starlings from southern Finland.
Detectable levels of residues were not common: 2 birds contained 1 µg
2,3,4,6-T4CP/kg fresh muscle and 2 others contained 1 and 2 µg
2,4,6-T4CP/kg, respectively.
Table 13. Levels of 2,3,5,6-T4CP in tissues of fish from a
Mississippi lake (USA) contaminated by PCP from a
wood-pole treatment facilitya
Date Fish Concentration (µg/kg wet weight)
Muscle Liver
October 11/76b Sunfish 1 <1 30
Sunfish 2 <1 50
January 6/77 Sunfish 1 95 950
Sunfish 2 60 NAc
Bass 2 300 1600
Bass 3 130 8200
Catfish 1 219 8500
April 27/77 Sunfish 1 27 25
Sunfish 2 22 150
Catfish 1 82 1400
Catfish 2 41 940
a From: Pierce & Victor (1978).
b Spill in December, 1976.
c NA = not analysed.
Table 14. Range of concentrations (µg/kg) of several chlorophenols
in ring-billed gull eggs, embryos, and chick tissuesa
Compound
2,4-DCP 2,4,5-T3CP 2,4,6-T3CP
Fresh eggs 0-176 0-26 12-87
Embryos 7 25
Chick liver 14-210 0 47-157
Chick brain 177-476 NDb 144-234
a From: NRCC (1982).
b ND = not detectable.
The same authors analysed osprey eggs, and the pectoral muscle,
brain, liver, eggs, and kidney of white-tailed eagles. 2,3,4,6-T4CP
levels in osprey eggs ranged from 0 to 17 µg/kg fresh weight; MCPs,
DCPs, and T3CPs were not detected. Similarly, only 2,3,4,6-T4CP
occurred (15-22 µg/kg fresh weight) in fresh eagle eggs. Fresh eagle
muscle tissue (2 samples) contained moderate levels of 2,4,6-T3CP (26
and 50 µg/kg respectively) and 2,3,4,6-T4CP (0 and 26 µg/kg). Single
samples of eagle brain, liver, and kidney revealed that all of these
tissues contained chlorophenol residues, some at high levels; kidney,
for example, contained 1017 µg 2,4-DCP/kg.
5.2 General Population Exposure
The general population is exposed to chlorinated phenols through
diverse sources and routes, which have been summarized by the NRCC
(1982). Chlorophenols can be ingested as contaminants in food
including produce sprayed with phenolic pesticides, flesh of livestock
given feed contaminated with these pesticides, and general food items,
usually at mg/kg levels (section 5.1.4).
In addition, sub-µg/litre quantities of chlorophenol congeners
have been detected in drinking-water (section 5.1.4). These 2 routes
of exposure are generally considered to be the major sources of
exposure of the general population to chlorophenols (US EPA, 1980c).
In addition, minor quantities may be taken up through the dermal and
respiratory routes. Sources include industrial discharges (solid,
liquid, atmospheric) of chlorophenolic wastes, exposure to treated
wood, exposure to general consumer products including adhesives,
textiles, wood-treatment products, mouth-washes and disinfectants, and
break down products of hexachlorobenzene and phenoxy acid herbicides.
Because of this diversity of sources of chlorophenols, there are
no comprehensive estimates of the chlorophenol levels to which the
general population is exposed. On the basis of preliminary estimates
from the literature of total chlorinated phenol residues in food,
water, air, and miscellaneous sources, the Canadian Department of
National Health and Welfare (NHW, 1988) estimated typical
non-occupational exposure to all chlorophenols to be:
6.0 µg/person per day in food
2.8 µg/person per day in water
1.9 µg/person per day in air
2.0 µg/person per day from other sources
12.7 µg/person per day in total (= 0.18 µg/kg body weight per day
for 70-kg adult).
Similarly, the NRCC (1982) estimated that the total chlorophenol
exposure per day in the general population in Canada was 10-30 µg/
person (0.17-0.50 µg/kg body weight per day for a 60-kg adult). This
estimate was based on the following assumptions: 6 µg/person per day
from food; 4 µg/person per day from water; and 20 µg/person per day
from air. The last figure is extremely high, based on monitoring data,
and was derived by assuming that indoor rooms were treated with a
chlorophenol preservative. This figure should be considered tentative
in view of the meagre data base available on environmental levels.
On the basis of approximate levels of several trichlorophenols in
drinking-water and fish flesh, SENES (1985) estimated the daily
general population intake of each of 2,4,5-T3CP, 2,4,6-T3CP, and
(2,3,5- + 2,3,6-) T3CP to be 0.44 µg/person per day. If it is assumed
that the uptake of each of the remaining 2 isomers is also
0.44 µg/person per day, the total T3CP intake would then be
2.20 µg/person per day.
These low estimated levels of exposure are confirmed by the few
studies in which the residue levels of lower chlorinated phenols have
been determined in the general population. Although contamination
generally appears to be widespread, the concentrations of
chlorophenols in the tissues and fluids of people, not occupationally
exposed, are extremely low.
Kutz et al. (1978) determined the levels of pesticide-related
phenolic residues in human urine samples from all over the USA with a
limit of detection of 5-30 µg/litre. In over 1.7% of 400 samples
collected from the general population, 2,4,5-T3CP was present at a
mean concentration of less than 5 µg/litre, and a maximum of
32.4 µg/litre.
In comparing different methods of detection of hexachlorobenzene
and 2,4,5-T3CP in human serum and urine, Yost et al. (1984) found
2,4,5-T3CP levels in the 2 fluids, in the USA, to be 0.25-
6.7 µg/litre and 0.25-1.9 µg/litre, respectively. Samples were pooled
from the general population, but neither the sample size nor the site
of origin was specified.
As part of the development of an analytical method for
chlorophenols, Edgerton et al. (1980) determined chlorophenol
concentrations in urine samples from the general population. The
origin of the samples was not specified, but was presumably the
southeastern USA. Chlorophenol concentrations ranged widely as
follows:
2,6-DCP, 1-112 µg/litre; 2,4-/2,5-DCP, 2-161 µg/litre (mean,
34.1); 3,5-DCP, 15-44 µg/litre; 2,4,5-T3CP, 1-9 µg/litre;
2,4,6-T3CP, 1-6 µg/litre; 2,3,4,6-T4CP, 2-15 µg/litre.
Similar levels of T4CP were found in urine samples from 25
members of the general population in Barcelona, Spain (Gomez-Catalan
et al., 1987). The mean urine concentration was 6.2 µg/litre (standard
error of mean = 1.6). No trichlorophenols were detected.
In Dade County, Florida, where large quantities of lindane
(gamma-HCH) and Bromophos ( O-(4-bromo-2,5-dichlorophenyl) O,O
dimethyl-phosphorothioate) are used in agriculture, Morgade et al.
(1980) measured the serum concentrations of 2,4-DCP, 2,3,5-, 2,4,5-,
2,4,6-T3CP, 2,3,4,5-, 2,3,4,6-T4CP, and PCP in 58 female residents.
In addition, 10 samples of human adipose tissue from autopsies were
analysed for the same series of compounds. No detectable levels of any
of the non-fully substituted chlorophenols were found in the serum or
adipose tissue of the study group, but traces of PCP were found in the
drinking-water and in both biological compartments.
Williams et al. (1984) analysed adipose tissue from autopsies of
male and female residents of Ottawa (n = 84) and Kingston (n = 91),
Ontario, for organochlorine residues. Levels of 2,3,4,5-T4CP were
typically 6 or 7 µg/kg tissue, and did not differ significantly
between locations or sexes. Tissues from Kingston contained roughly 3
times more of other T4CPs (2,3,4,6 plus 2,3,5,6; male 24 µg/kg;
female 20 µg/kg) than those from Ottawa (male 6 µg/kg; female
8 µg/kg), but this difference was not statistically significant.
These data support the hypothesis that the general population is
exposed to very low levels of the lower-chlorinated phenols. However,
estimates of this burden are highly speculative at present, as data
are lacking for most congeners. Quantitative analyses for these
compounds in meat, poultry, produce, and drinking-water are scarce.
Atmospheric measurements have not been documented at all, and the
extent of dermal absorption by the general population, assumed to be
low, is not known.
5.3 Occupational Exposure
The potential for both acute and long-term exposure to
chlorophenols may be heavy for workers from industries using these
compounds. The routes of exposure for Canadian workers have been
summarized by NRCC (1982); the same routes undoubtedly apply in most
other countries. Large numbers of workers are exposed to
chlorophenols, other than PCP, in the lumber industry, particularly in
instances where lumber is surface-treated with Na-T4CP, during the
dipping, sorting, handling, planing, trimming, or the grading of
lumber.
In-service treatment of wood by painters, wood preservation workers,
or telephone linemen could result in similar dermal and inhalation
exposure. Employees in the chemical industry, who are involved in the
manufacture of chlorophenols or their derivatives, may also be exposed
to high levels. The same is true of employees in manufacturing
industries that use chlorophenols as preservatives, such as the
photographic, paint, textile, rubber, construction, electrical,
pharmaceutical, and disinfectant industries. Finally, employees
working with products containing chlorophenols may be exposed, such as
commercial applicators and farmers using phenoxy herbicides, or those
exposed to treated wood in the fields of construction (carpentry), or
railways. For such occupational exposures, inhalation and dermal
absorption are the major routes of uptake.
Unfortunately, there is little quantitative information on
occupational exposure to low chlorine-substituted chlorophenols. As
might be expected of such moderately volatile compounds, high
atmospheric concentrations are found in work areas where they are in
use. In addition, the body fluids of persons working in such areas
contain elevated levels of chlorophenols. In general, concentrations
of chlorophenols in air at chemical manufacturing plants can reach
mg/m3 levels while much lower concentrations occur in facilities in
the lumber industry that use chlorophenols.
Ott et al. (1980) examined worker exposure to T3CP and 2,4,5-T at
a manufacturing plant in the USA. The time-weighted average
concentrations of T3CP in the air at work locations adjacent to the
reactor, salt wheel, acid wheel, and dryer, were 2.1, 2.1, 9.7, and
1.6 mg/m3 respectively.
In a factory manufacturing PCP in Japan, crude exhaust air vented
from a"drying room" contained 3.54 mg T4CP/m3 and 14.04 mg PCP/m3
(Akisada, 1964). Urine-T4CP concentrations of personnel in the
factory ranged from 0.07 to 0.37 mg/litre, compared with 0.01-
0.03 mg/litre for unexposed persons.
An industrial hygiene survey of worker exposure to chlorophenols
and hexachlorobenzene at a PCP-production facility revealed that
workers in different tasks were exposed to average concentrations of
2,3,4,6- plus 2,3,5,6-T4CP of 0.016-0.320 mg/m3 in conjunction with
several-times-higher exposures to PCP. The highest average exposures
were experienced by handymen and block-casting workers (Marlow, 1986).
Recent data from Kauppinen & Lindroos (1985) showed much lower
average atmospheric chlorophenol levels in 10 Finnish sawmills,
ranging from 24 to 75 µg/m3. The values given are the sum of the
three chlorophenols present, as the Na-2,3,4,6-T4CP formulation used
also contained 10-20% 2,4,6-T3CP and 5% PCP. The highest mean
concentrations in the general work place occurred at the site where
the solution was prepared and at the machine stacking the lumber. Much
higher levels were also detected inside the drying kilns, where
chlorophenol concentrations averaged 5800 µg/m3. Levels of
2,4,6-T3CP were measured separately; particularly high concentrations
were noted at the machine stacking site (58 µg/m3) and the outdoor
dipping site (44 µg/m3), while it could not be detected at the
preparation site. Average urine levels of T4CP and PCP (measured
together) ranged from 0.10 to 3.3 µmol/litre (approximately
25-825 µg/litre). The highest mean concentration occurred among the
loaders at the trough dipping area (mean air levels 55 µg/m3, dermal
uptake substantial), while the other urine values parallelled the
atmospheric readings in terms of relative concentration.
Kauppinen (1986) reported that air concentrations of chlorophenols
(T4CP and PCP combined) for a variety of tasks in Finnish plywood
plants usually ranged from < 1 to 6 µg/m3. The levels in air plus
wood dust, where plywood was sawed, ranged from 3 to 6 µg/m3 and were
usually higher than those in air at work sites where wood dust was
minimal.
A detailed study of the chlorophenol exposure of sawmill workers
in a pulp, paper, and sawmill complex in British Columbia was
conducted by Embree et al. (1984). They divided the workers into 3
groups: a control group of 351 workers in areas with no identifiable
air contaminants; a group of 31 workers in close proximity to recently
treated lumber, who did not have manual contact with it (airborne
exposure); and a group of 40 who handled recently treated lumber
(dermal plus airborne exposure). Air levels of chlorophenols were
determined using personal monitors. Tetrachlorophenol levels in the
plant air were elevated, and similar for the airborne group (3.3 ±
2.1 µg/m3; mean ± standard deviation), and the dermal-plus-airborne
group (3.0 ± 2.7 µg/m3). Serum levels were related to perceived
exposure in a dose-dependent manner; tetrachlorophenol concentrations
for the dermal-plus- airborne group (204 ± 92 µg/litre) were
approximately twice those in the airborne group (112 ± 136 µg/litre),
and 8 times those in the controls (26 ± 7 µg/litre). Urine levels for
the 2 exposed groups were also dose-dependent (airborne 93 ±
43 µg/litre; dermal-plus-airborne 125 ± 20 µg/litre). Urine levels in
the control group were not reported.
Similar urine concentrations were reported for American
wood-workers exposed to Permatox(R) (3% PCP, 21% 2,3,4,6- T4CP)
(Kalman & Hortsman, 1983). Of 47 workers, 28 showed urine levels of
more than 100 µg 2,3,4,6-T4CP/litre, 13, levels between 20 and
100 µg/litre, and 6 levels of less than 20 µg/litre. Air levels were
reportedly below 25 µg/m3. Because atmospheric concentrations were
this low, the authors suggested that the individuals with the highest
urine levels were taking up most of the dose through non-respiratory
routes, most likely dermal. Over a 2-week holiday period, T4CP levels
in the three groups declined by averages of 84%, 67%, and 34%,
respectively, a slower rate of elimination than that found in
experimental animals (section 6.4).
Kleinman et al. (1986) and Fenske et al. (1987) also evaluated the
extent and impact of occupational exposure to Permatox(R) in 100
workers from a lumber-mill in Washington State. Plant air
concentrations of T4CP ranged from 0.8 to 12.2 µg/m3, while no PCP
was detected (limit of detection 0.5 µg/m ). It was estimated that
dermal exposure accounted for 95% of the dose taken up by exposed
workers. Average chlorophenol concentrations in the urine were higher
for exposed workers than for controls (range of averages:
T4CP-exposed = 31.2-497.5 µg/litre, control = 6.3-28.7 µg/litre;
PCP-exposed = 57.4-102.8 µg/litre, control = 28.9-38.8 µg/litre).
In a recent report, 230 sawmill workers in Finland were examined
for urinary levels of chlorophenols (Lindroos et al., 1987). In
occupations where dermal exposure was greatest, workers (n = 112) had
a median urinary chlorophenol level of approximately 1.8 mg/litre
(range, 0.02-49 mg/litre, assuming all chlorophenols were T4CP)
whereas employees (n = 34) exposed mainly via the respiratory route
had a median urinary level of 0.2 mg chlorophenols/litre (range,
0.02-3.1 mg/litre). These results support the hypothesis of Kalman &
Hortsman (1983) regarding the importance of the dermal route of
exposure for chlorophenols.
The chlorophenol levels in urine among workers handling imported
lumber treated with 2,3,4,6-T4CP ranged from 0.13 to 2.2 µmol/litre
(30.2-510.4 µg/litre, assuming all chlorophenols were T4CP) with a
mean value of 0.86 µmol/litre (199.5 µg/litre) (Rappe et al., 1982).
These exposure data are static, and, as such, give no information
on the actual amount of chlorophenols taken up by a worker. In the
course of designating permissible levels of chlorophenol exposure for
regulatory purposes, the US EPA (1978) modelled the chlorophenol
exposure experienced by a worker performing various tasks (Table 15).
The exposure levels indicate that, as expected, occupational
exposure to chlorophenols is much higher than non-occupational; these
rates of uptake are 2 or more orders of magnitude higher than
estimates of exposure of the general population to all chlorophenols
summarized in section 5.2.
However, the estimates of chlorophenol burdens given in Table 15
are for 2,4,5-T3CP (the use of which has been discontinued in many
countries), and are based on speculative scenarios that exaggerate
worker exposure to this compound. Despite the longstanding concern for
the potential health hazards associated with occupational exposure to
chlorophenols, meaningful estimates of worker exposure to the
chlorophenols that are currently extensively used do not appear to
have been made to date. Exposures have been estimated qualitatively
or, at best, semi-quantitatively. Although occupational uptake of
chlorophenols is thought to be principally through inhalation and
dermal absorption, there are no data on the rates of such uptake. To
obtain such information, air monitoring should be continuous
throughout the shift, using personal monitors, and urine levels of
chlorophenols should be measured for consecutive 24-h periods.
Table 15. Estimates of occupational exposure to 2,4,5-T3CPa
Site Dermal exposure Inhalation exposure
(µg/kg body weight per day)
Cooling tower 3.7b 23c
Water Systems 14d 90.3e
Pulp and paper mill 2f 55g
Tannery 49h 87i
Hospital 70j 9k
a From: US EPA (1978).
b Exposure to 100 ml containing 22 mg Na-2,4,5-T3CP/litre; 10%
absorption for 60-kg female maintenance worker.
c 100% relative humidity, 20 °C; therefore, 1 m3 air contains
0.0173 litre H2O with 22 mg Na-2,4,5-T3CP/litre H2O;
breathing rate, 1.8 m3/litre for 2 h; 60-kg female worker.
d Exposure to 100 ml containing 87 mg Na-2,4,S-T3CP/litre; 10%
absorption; 60-kg female worker.
e As footnote c, but product concentration 87 mg
Na-2,4,5-T3CP/litre H2O.
f Exposure to 80 ml (13 × 10 ml; one hand) containing 15 mg
Na-2,4,5-T3CP/litre; 10% absorption; 60-kg female worker.
g As footnote c, but product concentration 15 mg
Na-2,4,5-T3CP/litre H20, 7-h exposure.
h Exposure to 1.4 litre containing 21 µg Na-2,4,5-T3CP/litre;
10% absorption;
i 60-kg female worker. As footnote c, but product concentration
21 µg Na-2,4,S-T3CP/litre H2O, 8-h exposure.
j Exposure to 1 cup (0.24 litre; 16 cups per gallon) containing
686 mg Na-2,4,S-T3CP per gallon; 10% absorption; 60-kg
female worker.
k Hospital volume, 1800 m3; recommended air ventilation rate,
60 m3/h per person; 60 persons; 8-h day; total circulated air,
28 800 m3; 100 gallons disinfectant used; 100 cups remain giving
a total of 4.3 g Na-2,4,5-T3CP; volatilization at 25 °C;
breathing rate, 1.8 m3/h; 8-h day; 60-kg worker.
6. KINETICS AND METABOLISM
6.1 Absorption
Hoben et al. (1976a) exposed male Sprague-Dawley rats to an
aerosol of sodium-PCP (repeated exposures to about 5.9 mg PCP/kg body
weight) and found very rapid absorption into the blood. Unfortunately,
no information is available on the absorption of the lower chlorinated
phenols via the mammalian lung during inhalation exposure.
In general, chlorophenols are readily absorbed through the skin.
Using the skin of the hairless mouse, Huq et al. (1986) found that
aqueous solutions of 2-MCP, 2,4-DCP, and 2,4,6- T3CP readily
penetrated the skin, provided that the compound was not ionized (i.e.,
pH pKa). In vitro studies on epidermal membranes from human skin
taken at autopsy showed penetration by 2-MCP, 4-MCP, 2,4-DCP, and
2,4,6-T3CP (Roberts et al., 1977, 1978). The lipophilic character of
the solutes and their hydrogen-bonding capacity are the 2 main
features determining this penetration. Shen et al. (1983) investigated
the dermal absorption of T4CP in Sprague-Dawley rats and found the
Na-2,3,5,6-T4CP was more toxic than 2,3,5,6-T4CP itself. Toxic
amounts of 2,3,4,6-T4CP in organic solvents can be absorbed through
the skin (Gosselin et al., 1976), and the use of 2,4,5-T3CP in
hospitals has been suggested as a potential problem because of its
absorption through the skin (US EPA, 1978). Similarly, on the basis of
data concerning urine levels of chlorophenols, absorption through the
skin has been reported to be a major route of exposure among workers
occupationally exposed to chlorophenols or to their salts (section
5.3).
The greater part of orally-administered tri- and
tetra-chlorophenols is recovered in the urine and faeces of test
animals (section 6.4), indicating that lower chlorophenols are readily
absorbed through the gastrointestinal tract. More than 90% of the oral
dose was excreted in the urine of volunteers after ingestion of PCP,
which indicates similarly effective absorption via the
gastrointestinal tract in human beings (Braun et al., 1979).
6.2 Distribution
6.2.1 Tissue distribution following chlorophenol exposure
No information is available on the distribution of
mono-chlorophenols in animal systems.
With respect to dichlorophenols, single intravenous injections of
2,4-DCP (10 mg/kg body weight) in Sprague-Dawley rats weighing
250-300 g resulted in a maximum concentration (17.7 mg/kg of tissue) in
the kidney, 10 min after injection (Somani & Khalique, 1982). Levels
in liver, brain, and fat peaked at 15 min at 10.5 mg/kg, 3.2 mg/kg,
and 4.1 mg/kg tissue, respectively. A level of 1.64 mg/litre was
recorded in plasma, 10 min after injection.
Following intraperitoneal administration of 25 mg 2,4,6-T3CP/kg
body weight to male Wistar rats (Pekari et al., 1986), concentrations
in all tissues assayed were maximal, 30 min after injection: kidney
levels peaked at 329 ± 117 nmol/g, while maximum concentrations were
progressively lower in blood, liver, fat, muscle, and brain.
Hattula et al. (1981a) reported that Wistar rats fed 2,3,4,6-T4CP
in olive oil at 100 mg/kg intragastrically for 55 days showed the
following tissue concentrations of 2,3,4,6-T4CP: kidney, 5.1 mg/kg;
spleen, 3.2 mg/kg; liver, 2.2 mg/kg; brain, 1.2 mg/kg; and muscle,
0.46 mg/kg tissue.
6.2.2 Tissue distribution following exposure to chemicals metabolized
to chlorophenols
The distribution of chlorophenols as metabolites following the
administration of other organochlorine compounds has been investigated
in several studies. Like the original chlorophenols, these metabolites
accumulate most often in the kidney and liver. Clark et al. (1975)
investigated the tissue distribution of 2,4-DCP in sheep and cattle
fed 2,4-D. Cattle were given a diet containing 2,4-D at 0, 300, 1000,
or 2000 mg/kg (9, 30, or 60 mg/kg body weight per day). Muscle, fat,
liver, and kidney were analysed for 2,4-DCP. Sheep were given a diet
containing 2,4-D at 2000 mg/kg for 28 days. At 2000 mg 2,4-D/kg in the
diet, 2,4-DCP concentrations in kidney and liver from sheep were
0.26 mg/kg tissue and 0.16 mg/kg tissue, respectively; in cattle,
levels were 1.06 mg/kg and 0.31 mg/kg tissue, respectively.
In laying hens fed VC-13 Nemacide(R) [ O-(2,4-dichlorophenyl)-
O,O-diethyl phosphorothioate) at a dose of 800 mg/kg for 55 days,
Sherman et al. (1972) reported similar levels of 2,4-DCP in both liver
tissue and egg yolk (average values ranged from 0.122 to 0.613 mg/kg).
2,4-DCP was not detected in the muscle and fat of these birds.
Levels of 2,4,5,-T3CP in the tissues of sheep and cattle fed
trichlorophenoxy acid herbicides for 28 days were determined by Clark
et al. (1975). In sheep fed Silvex (2-(2,4,5- trichlorophenoxy)-
propionic acid) at 2000 mg/kg, residues of 2,4,5-T3CP were 0.22 mg/kg
in liver and 0.17 mg/kg in kidney.
Cattle fed this compound at 9, 30, or 60 mg/kg body weight had
2,4,5-T3CP tissue concentrations ranging from 0.06 to 0.48 mg/kg in
the liver and from 0.05 to 0.10 mg/kg in kidney. No residues were
detected in samples of muscle and fat from sheep or cattle fed Silvex.
Sheep exposed to 2,4,5-T in the diet at 2000 mg/kg for 28 days
exhibited 2,4,5-T3CP levels of 6.1 mg/kg, 0.90 mg/kg, 0.13 mg/kg, and
0.05 mg/kg tissue, in the liver, kidney, muscle, and fat,
respectively.
Sheep dosed orally with Erbon(R) (2-(2,4,5-trichloro-phenoxy)
ethyl 2,2-dichloropropionate) metabolized it to 2,4,5-T3CP and
2-(2,4,5-trichlorophenoxy) ethanol in less than 7 h (Wright et al.,
1970). Most of these compounds were eliminated in the urine (section
6.4), but mg/kg quantities of 2,4,5-T3CP and the other metabolite
were found in the kidney, liver, omental fat, muscle, and brain of
sheep given 100 mg Erbon(R)/kg body weight daily for 10 days (5.54,
3.14, 2.06, 1.00, and 0.21 mg/kg, respectively).
Male Wistar rats dosed with 8 mg lindane (gamma-hexachlorocyclo-
hexane)/kg body weight by gavage for 19 days showed 2,4,6-T3CP and
2,3,4,6-T4CP in heart tissue, 2,3,4,6- T4CP and/or 2,3,5,6-T4CP in
the liver, and 2,4,6-T3CP and 2,3,4,6-T4CP in the kidney (Engst et
al., 1976), but no quantitative data were given.
6.3 Metabolic Transformation
The major metabolic transformation for the lower chlorinated
chlorophenols appears to be conjugation with sulfate or glucuronate,
prior to clearance in the urine. Perhaps, because of similarities
between the structure and lipophilicity of T4CP and PCP, a small
proportion of these congeners undergo the same dechlorination and/or
oxidation reactions that PCP does prior to conjugation (Renner &
Mücke, 1986).
As much as 84.7% of administered 2-MCP was reportedly excreted as
sulfate and glucuronate conjugates in dogs (Karpow, 1893). In the
rabbit, oral administration of monochlorobenzene resulted in surf ate
and glucuronide conjugates of 2-MCP in the urine (Lindsay-Smith et
al., 1972). It has been suggested that in mice o-methylation might
be a relevant mechanism for 2-MCP detoxification (Angel & Rogers,
1972). Similarly, conjugates were detected in the kidney, liver, fat,
brain, and plasma of rats after the iv injection of 2.5-3 mg 2,4-DCP
(10 mg/kg body weight) (Somani & Khalique, 1982). Of the total
conjugates determined, glucuronide conjugates were the major
metabolite in kidney (79.6%), liver (62.7%), brain (77.9%), and plasma
(79.5%). No glucuronide conjugates were found in fat. Free 2,4-DCP did
not accumulate in rat tissues and was rapidly metabolized to its
conjugates. In another study using 14C-2,4-DCP on isolated perfused
rat liver, Somani et al. (1984) demonstrated that the liver is capable
of the formation of glucuronide conjugates, and that 2 dichlorometho-
xyphenols are metabolites of 2,4-DCP when glucuronide formation is
blocked by galactosamine (section 8.8).
Bahig et al. (1981) have suggested that, in rats, 2,4,6-T3CP is
isomerized to 2,4,5- and 2,3,6-T3CP before being excreted as
glucuronide conjugates. However, in a similar study on rats given
25 mg 2,4,6-T3CP/kg body weight (ip), 83 ± 11% was present in the
blood as glucuronides rather than being converted to another isomer
(Pekari et al., 1986).
Concerning T4CP, Ahlborg & Larsson (1978) showed that, of the 3
isomers, only 2,3,5,6-T4CP was metabolized to a significant extent in
the rat. Thirty-five percent of the given dose (10 mg/kg body weight
by ip injection) was metabolized to tetrachloro- p-hydroquinone,
which, when also given ip, is more toxic than the parent compound
(section 8.9). Trichloro- p-hydroquinone was a minor metabolite of
the other isomers. Recently, it has been shown that the microsomal
metabolism of PCP yields tetrachloro-1,2-and tetrachloro-1, 4-hydro-
quinone (van Ommen et al. 1986). Covalent binding to protein and DNA
occurs via the corresponding tetrachloroquinones (van Oremen et al.,
1988). To what extent this kind of metabolic activation plays a role
in the toxicity of lower chlorinated phenols is not known, at present.
The metabolism of compounds that axe structurally related to
chlorophenols also yields conjugates of chlorophenols. Kurihara &
Nakajima (1974) studied the metabolism in mice of injected 14C-hexa-
chlorocyclohexane (14C-HCH). The major metabolites were conjugates of
2,4,6-T3CP with sulfate or glucuronide, as well as conjugates of
2,4-DCP. The proportion of 2,4,6-T3CP sulfate to glucuronide
conjugates varied from 80%:20% to 40%:60%, depending on whether
gamma-HCH or beta-HCH was used. Trace amounts of free 2,4,6-T3CP and
2,4,5-T3CH were also found in the urine. Koransky et al. (1975) also
found that injection of 14C-HCH into rats resulted in glucuronide
and sulfate urinary metabolites of 2,4,6- and 2,4,5-T3CP; small
amounts of the free phenols were also detected. The ratio of sulfate
to glucuronide conjugates was not determined. Engst et al. (1976)
found that lindane (gamma-HCH) administration in rats produced free
2,4,6-T3CP, 2,3,4,6- and/or 2,3,5,6-T4CP and PCP in the urine, as
well as glucuronide-bound 2,3,4-T3CP, 2,3,4,5-, 2,3,4,6- and/or
2,3,5,6-T4CP.
In terms of glucuronide formation of PCP, the rat is probably a
better model for human beings than the monkey, which does not
metabolize this compound (Braun et al., 1978, 1979). Whether the rat
model can be used to predict human responses to the lower chlorinated
phenols is not known at present, since no human data exist. However,
hydrolysis of human urine samples indicated that most of the T4CP in
human urine is conjugated (Dahms & Metzner, 1979; Butte, 1984; Currie
& McDonald, 1986).
6.4 Elimination and Excretion
In experimental mammals, chlorophenols are eliminated primarily in
the urine. For example, Freitag et al. (1982) administered 14C-2,4,6-
T3CP to rats orally for 3 days to examine retention, dispersion, and
excretion rates. Within 7 days, 82.3% of the label was excreted in the
urine and 22.2% in the faeces. At sacrifice on the 8th day, residues
in the liver, lung, and adipose tissues were below the level of
detection (i.e., less than 0.01% of the label), whereas the carcass
retained 7.8% of the label. Bahig et al. (1981) found that 92.5% of a
daily oral dose (25 µg by gavage) of 14C-2,4,6-T3CP was excreted by
rats in the urine, while 6.4% was found in the faeces. Thus, the
ingested 2,4,6-T3CP was largely eliminated within 24 h. Similarly,
Ahlborg & Thunberg (1980) reported that 2,4,5-T3CP given to rats was
excreted rapidly (within 24 h) with very little retention by the
animal, and Pekari et al. (1986) estimated the half-times for the
elimination of 2,4,6-T3CP from the blood, liver, muscle, fat, brain,
and kidney of rats at between 1.4 and 1.8 h, after dosing ip with
25 mg/kg body weight.
Excretion by rats of the different T4CP isomers injected
intraperitoneally was examined by Ahlborg & Larsson (1978). While
2,3,5,6-T4CP was eliminated in the urine within 24 h and
2,3,4,6-T4CP within 48 h, only 60% of the injected 2,3,4,5-T4CP was
collected in 72 h.
In a study on the elimination of 2,4,-DCP from various tissues in
the rat following intravenous administration of 10 mg/kg body weight
(Somani & Khalique, 1982), the compound was eliminated most rapidly
from brain tissue followed by plasma, fat, liver, and kidney.
Half-lives for 2,4-DCP were 6 min in the brain, 10 min in fat and
plasma, 15.1 min in the liver, and 30.1 min in the kidney.
Much of the information on the excretion of chlorophenols has come
from studies of the uptake and clearance of chlorophenols that have
been formed metabolically from other compounds. As in the studies
described previously, these chlorophenols are generally eliminated
rapidly in the urine. Thus, Lindsay-Smith et al. (1972) identified
free and conjugated forms of all monochlorophenol isomers in the urine
of rabbits dosed with 14C-monochlorobenzene. Similarly, 2,4-DCP was
eliminated in the urine of rats injected with Nemacide(R) (67% of
dose excreted as 2,4-DCP within 3 days) (Shafik et al., 1973). Shafik
et al. (1973) also found that rats cleared 53% of a dose of
Ronnel(R) ( O, O-dimethyl- O (2,4,5-trichloro-phenyl)phosphoro-
thioate) as 2,4,5-T3CP within 2 days. A sheep given 50 mg/kg body
weight of Erbon(R) [2-(2,4,5-trichloro-phenoxy)-ethyl 2,2-dichloro-
propionate] as an oral drench metabolized it to 2,4,5-T3CP and
2-(2,4,5-trichloro-phenoxy) ethanol in less than 7 h (Wright et al.,
1970). Within 96 h, 68.42% of the dose was eliminated in the urine and
1.74% in the faeces, approximately half of these amounts as
2,4,5-T3CP.
Karapally et al. (1973) identified chlorinated phenols derived
from lindane in rabbit urine. Of the 14 chlorophenols identified,
comprising at least 19.9% of the total dose, the most abundant were
(in decreasing order) 2,4,5-T3CP, 2,3,5-T3CP, 2,4,6-T3CP,
2,3,4,6-T4CP, 2,3-DCP, 2,4-DCP, and 2,3,4-T3CP. The results of a
similar study on the rat (Engst et al., 1976) showed that 2,4,6-T3CP,
2,3,4,6-T4CP and/or 2,3,5,6-T4CP, and 2,3,4,5-T4CP derived from
lindane were eliminated via the urine. Chadwick & Freal (1972)
observed that, following one week of dosing with lindane, rats
excreted 3,4-DCP, 2,4,5-T3CP, 2,3,5-T3CP, 2,4,6-T3CP,
2,3,4,5-T3CP, and 2,3,4,6-T4CP for at least 1 month.
The clearance from tissues of chlorophenols derived from other
compounds may be slower than their elimination via the urine. Sherman
et al. (1972) found that from 60 to 83% of the 2,4-DCP metabolized
from Nemacide(R) disappeared from the liver of chickens within 21
days of the cessation of dosing. 2,4-DCP found in the yolk of eggs
from these hens dropped to non-detectable levels in 10 days for the
high-dose (800 mg/kg diet) group, while at the lower dosages (50, 100,
200 mg/kg diet), a shorter time was required for complete clearance
(see also section 6.2). In sheep fed 2,4,5-T3CP at 2000 mg/kg diet
(Clark et al., 1975), liver and kidney 2,4,5-T3CP levels remained
relatively constant one week after exposure ceased, while muscle
concentrations dropped roughly 3-fold.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
There are few studies on the effects of chlorophenols, other than
PCP, on organisms in the environment. This lack of information may
stem, in part, from the fact that many wastes contain other
potentially toxic components in addition to chlorophenols. Moreover,
most laboratory studies on the toxicity of chlorophenols for
environmental organisms have involved much higher exposure levels than
those that are usually found in the environment.
The information that is available on the effects of chlorophenols
deals primarily with aquatic habitats, perhaps because many point
discharges of chlorophenols are released into water bodies.
7.1 Laboratory Studies
7.1.1 Acute toxicity
Recent laboratory studies on the acute toxicity of chlorophenols
for aquatic biota are summarized in Table 16. The data are derived
primarily from studies published since 1980; information from research
prior to this date is presented by Jones (1981). In general, the
patterns evident in the review by Jones (1981) are also seen in the
more recent data (Table 16).
Considerable overlap exists in the chlorophenol levels that
produce toxic effects in bacteria, phytoplankton, macrophytes,
invertebrates, and fish (Table 16). For instance, LeBlanc (1984)
(Table 16) reported that LC50 values compiled for 4-MCP toxicity in
algae (Selenastrum, capricornutum, Skeletonema costatum),
invertebrates (Daphnia magna), and fish (Lepomis macrochirus,
Cyprinidon variegatus) ranged from 3.27-5.35 mg/litre. Most of the
EC50/LC50 values for other organisms compiled in Table 16 also fall
within the several mg/litre range. However, there are isolated reports
of certain bacterial, fungal, and protozoan processes that are
insensitive to chlorophenol exposure (Table 16).
In general, chlorophenol toxicity for aquatic organisms increases
with the degree of chlorination of the phenol ring (Table 16),
presumably as a result of increasing lipophilicity (Table 3).
Table 16. Acute toxicity of chlorophenols for aquatic biota
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Phytoplankton
Scenedesmus SB, FW 3,5-DCP 5.32 (EC50) Growth rate, Weibull model Christensen & Nyholm
spicatus 5.87 (EC50) Growth rate, Probit model (1984)
6.10 (EC50) Growth rate, Logit model
Selenastrum SB, FW 4-MCP 5.01 (EC50) Growth LeBlanc (1984)
capricornutum
Skeletonema SB, FW 4-MCP 3.27 (EC50) Growth LeBlanc (1984)
costatum
Bacteria
Bacillus sp. SB, FW 2-MCP 700
3-MCP 450
4-MCP 400
2,3-DCP 130
2,4-DCP 75
2,5-DCP 85
2,6-DCP 550
3,4-DCP 52 IC50c reduction in Liu et al. (1982)
3,5-DCP 25 activity after 30 min
2,3,4-T3CP 13 incubation with toxicant
2,3,5-T3CP 10
2,3,6-T3CP 190
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Bacteria
(contd).
Bacillus sp. 2,4,5-T3CP 12
(contd). 2,4,6-T3CP 240
3,4,5-T3CP 5
2,3,4,5-T4CP 4
2,3,5,6-T4CP 54
Photobacterium SW 2-MCP 33.8 Ribo & Kaiser
phosphoreum 3-MCP 14.1 (1983)
(Microtox(R)) 4-MCP 8.30
2,3-DCP 4.92
2,4-DCP 5.52
2,5-DCP 9.38 EC50 for inhibition
2,6-DCP 13.2 of light emission with
3,4-DCP 1.63 30-min toxicant exposure
3,5-DCP 2.77
2,3,4-T3CP 1.25
2,3,5-T3CP 1.11
2,3,6-T3CP 12.7
2,4,5-T3CP 1.27
2,4,6-T3CP 7.68
3,4,5-T3CP 0.359
2,3,4,5-T4CP 0.176
2,3,4,6-T4CP 1.27
2,3,5,6-T4CP 2.22
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Activated SB, FW 3,5-DCP 30.2 IC50 for O2 Dutka & Kwan
sludge consumption, 3 h (1984)
Photobacterium SW 2-MCP 22.1 EC50 for inhibition Indorato et al.
phosphoreum 2,4-DCP 3.36 of light emission with (1984)
(Microtox(R))
toxicant exposure
Activated SB, FW 3,5-DCP 7 EC50 for O2 King (1984)
sludge consumption
Nitrifying 5 EC50 for nitrate and
activated nitrite production
sludge
Photobacterium SW 3.2 EC50 for light emission
phosphoreum
(Microtox(R))
Sewage 8 EC50 for growth inhibition,
microorganisms 6 h
Sewage 6 EC50 for growth inhibition,
microorganisms 16 h
Sewage effluent 15 EC50 for BOD, 5 days,
supplemented
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Bacteria
(contd).
Photobacterium SW 3,5-DCP 2.9 EC50 for light emission Dutka & Kwan (1984)
phosphoreum after 15 min
(Microtox(R))
Pseudomonas SB, FW 3,5-DCP 3.2 EC50 for growth Dutka & Kwan (1984)
fluorescens inhibition, 18 h
Activated FB, FW 2-MCP 104.4 Beltrame et al.
sludge 3-MCP 67.5 (1984)
4-MCP 71.0
2,3-DCP 55.1
2,4-DCP 47.6
2,5-DCP 50.2
2,6-DCP 65.2 IC50 for phenol
3,4-DCP 42.7 biodegradation
3,5-DCP 58.3
2,3,4-T3CP 27.4
2,3,5-T3CP 22.3
2,3,6-T3CP 39.3
2,4,5-T3CP 23.6
2,4,6-T3CP 42.0
3,4,5-T3CP 19.6
2,3,4,5-T4CP 20.4
2,3,4,6-T4CP 40.5
2,3,5,6-T4CP 44.3
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Bacteria
(contd).
Nitrobacter SB, FW 2-MCP 50 25 and 27% inhibition of Wang & Reed (1984)
nitrite uptake
3-MCP 50 0 and 15% inhibition of
nitrite intake
4-MCP 50 0 and 5% inhibition of
nitrite intake
2,3-DCP 30 96 and 73% inhibition of
nitrite intake
2,4-DCP 30 21 and 77% inhibition of
nitrite intake
2,4,6-T3CP 10 88 and 100% inhibition of
nitrite intake
Nitrosomonas SB, FW 2-MCP 100 24% loss in ATP Parker & Pribyl
europa 2,4,6-T3CP 150 17.6% loss in ATP (1984)
Escherichia SB, FW 2-MCP 100 6.5% loss in ATP
coil 2,4,6-T3CP 150 12.9% loss in ATP
Pseudomonas SB, FW 2,3,4,5-T4CP 10 0% reduction in CFUs, Trevors et al.
fluorescens 1-h exposure (1982)
25 86.6 and 87.2% reduction
in CFUs, 1-h exposure
35 99.4 and 99.9% reduction
in CFUs, 1-h exposure
23.2 LC50 of CFUs, 1-h exposure
Table 16. (cont'd).
Test organism Test Chlorphenol Concentration Criterion Reference
conditions (mg/litre)
Protozoa
Tetrahymena 2-MCP 67.97
4-MCP 36.68 IC50 for growth Schultz &
2,4-DCP 15.00 at 60 h Riggin (1985)
2,5-DCP 12.15
2,4,6-T3CP 3.99
2,3,5,6-T4CP 1.40
Fungi
16 fungal SB 3-MCP 257.1 Average minumum Ruckdeschel &
strains, 4-MCP 184.9 concentration for Renner (1986)
(14 genera) 2,3-DCP 60.8 complete inhibition
2,4-DCP 54.1 of growth
2,5-DCP 54.8
2,6-DCP 180.8
3,4-DCP 30.1
3,6-DCP 13.2
SB 2,3,4-T3CP 11.6
2,3,5-T3CP 140.4
2,4,5-T3CP 19.2
2,4,6-T3CP 4.1
2,3,4,5-T4CP 4.6
2,3,4,6-T4CP 72.8
2,3,5,-T4CP 119.7
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fungi (contd).
Pichia SB Na-4-MCP 145 IC50 for culture growth Kwasniewska &
(fermentative Na-2,4-DCP 42.5 Kaiser (1983)
yeast) Na-2,4,5-T3CP 4.3
Rhodoturula SB Na-4-MCP 62.5 IC50 for culture growth
rubra Na-2,4-DCP 16.5
(oxidative Na-2,4,5-T3CP 2.0
yeast)
Invertebrates
Daphnia magna SB, FW 2-MCP 22 24-h LC50 LeBlanc (1980)
(water flea) 2.6 48-h LC50
4-K4CP 8.8 24-h LC50
4.1 48-h LC50
2,4-DCP > 10 24-h LC50
2.6 48-h LC50
2,4,5-T3CP 3.8 24-h LC50
2.7 48-h LC50
2,4,6-T3CP 15 24-h LC50
6.0 48-h LC50
2,3,4,6-T4CP > 1.0 24-h LC50
0.29 48-h LC50
2,3,5,6-T4CP 2.5 24-h LC50
0.57 48-h LC50
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Invertebrates
(contd.)
Daphnia magna 2-MCP 17.95 IC50 for Devillers &
(water flea) 3-MCP 15.78 immobilization Chambon (1986)
4-MCP 8.07 after 24 h
2,3-DCP 5.19
2,4-DCP 2.68
2,6-DCP 9.38
3,4-DCP 2.77
3,5-DCP 2.09
2,3,4-T3CP 2.24
2,3,5-T3CP 2.28
2,3,6-T3CP 7.38
2,4,5-T3CP 2.08
2,4,6-T3CP 5.47
3,4,5-T3CP 0.88
2,3,4,5-T4CP 1.76
2,3,5,6-T4CP 2.27
Astacus SB, FW 2,3,6-T3CP 5.4 at pH 6.5 8-day LC50 Kaila &
fluviatilis 19.0 at pH 7.5 8-day LC50 Saarikoski (1977)
Mysidiopsis SB, SW 4-MCP 29.7 at pH 6 96-h LC50 LeBlanc (1984)
bahia
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Invertebrates (contd).
Palaemonetes SS, SW 2,4-DCP 2.55(I)a; 2.16 (M)b 96-h LC50 Rao et al. (1981)
pugio 2,4,6-T3CP 3.95(I); 1.21 (M) 96-h LC50
(grass shrimp) 2,4,5-T3CP 1.12(I); 0.64 (M) 96-h LC50
2,3,4,5-T4CP 0.86(I); 0.37 (M) 96-h LC50
2,3,4,6-T4CP 3.70(I); 0.81 (M) 96- LC50
2,3,5,6-T4CP 4.10(I); 1.17 (M) 96-h LC50
Palaemonetes SS, SW 2,3,4,5-T4CP 0.30 EC50 for intermolt Rao et al. (1981)
pugio limb regeneration
(grass shrimp) 2,3,4,6-T4CP 0.78 EC50 for intermolt
limb regeneration
Fish
Pimephales SB/FB, F-W 2-MCP 11.0-13.0 96-b LC50 flowthrough Phipps et al.
promelas 6.3 192-h LC50 flowthrough (1981)
(fathead 9.7 48-h LC50 static
minnow) 2,4-DCP 8.2-8.3 96-h LC50 flowthrough
6.5 192-h LC50 flowthrough
8.4 48-h LC50 Static
2,4,6-T3CP 8.6-9.7 96-h LC50 flowthrough
5.8-6.4 192-h LC50 flowthrough
7.7 48-h LC50 static
Cyprinidon SB, SW 4-MCP 5.7 24-h LC50 Heitmuller et al.
variegatus 5.4 48-h LC50 (1981)
(sheepshead 5.4 72-h LC50
minnow) 5.4 96-h LC50
3.2 NOECc
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fish (contd).
Cyprinidon 2,4,5-T3CP 2.4 24-h LC50
variegatus 1.7 48-h LC50
(contd). 1.7 72-h LC50
1.7 96-h LC50
1.0 NOECc
2,3,5,6-T4CP 2.0 24-h LC50
2.0 48-h LC50
2.0 72-h LC50
1.9 96-h LC50
1.0 NOECc
Salmo trutta SB, FW 2,4-DCP 1.7 24-h LC50 Hattula et al.
(trout) 2,6-DCP 4.0 (1981b)
2,3,5-T3CP 0.8
2,4,5-T3CP 0.9
2,3,4,6-T4CP 1.1
Poecilia SS, FW 4-MCP 49.0 at pH 5 96-h-LC50 Saarikoski &
reticulatus 61.0 at pH 6 96-h LC50 Viluksela (1981)
(guppy) 66.0 at pH 7 96-h LC50
2,4,5-T3CP 50.0 at pH 5 96-h LC50
6.3 at pH 7 96-h LC50
15.3 at pH 8 96-h LC50
2,4,6-T3CP 3.1 at pH 5 96-h LC50
4.5 at pH 6 96-h LC50
11.6 at pH 7 96-h LC50
39.8 at pH 8 96-h LC50
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Poecilia SB, FW 2-MCP 13.5 at pH 7.8 24-h LC50 Könemann &
reticulatus 7.1 at pH 6.1 24-h IC50 Musch (1981)
(guppy) 3-MCP 7.9 at pH 7.8 24-h LC50
6.4 at pH 6.1 24-h LC50
2,4-DCP 5.9 at pH 7.8 24-h LC50
3.3 at pH 6.1 24-h LC50
3,5-DCP 4.7 at pH 7.8 24-h LC50
2.6 at pH 6.1 24-h LC50
2,3,5-T3CP 4.7 at pH 7.8 24-h LC50
0.88 at pH 6.1 24-h LC50
2,3,6-T3CP 13.3 at pH 7.8 24-h LC50
0.94 at pH 6.1 24-h LC50
3,4,5-T3CP 2.4 at pH 7.8 24-h LC50
2,3,4,5-T4CP 1.1 at pH 6.1 24-h LC50
2,3,5,6-T4CP 2.3 at pH 7.8 24-h LC50
0.44 at pH 6.1 24-h LC50
3.9 at pH 7.8 24-h LC50
0.36 at pH 6.1 24-h LC50
Carassius SB, FW 2-MCP 16 25-h LC50 Kobayashi et al.
auratus 4-MCP 9.0 (1979)
(goldfish) 2,4-DCP 7.8
2,4,5-T3CP 1.7
2,4,6-T3CP 10.0
2,3,4,6-T4CP 0.75
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fish (contd).
Lebistes S8, FW 2-MCP 13.4 24-h LC50 Benoit-Guyod et al.
reticulatus 3-MCP 27.0 (1984)
(guppy) 4-MCP 9.0
2,3-DCP 18.0
2,4-DCP 6.8
2,5-DCP 11.0
2,6-DCP 8.9
Lebistes 3,4-DCP 7.4
reticulatus 3,5-DCP 6.1
(guppy) 2,3,6-T3CP 53.0
2,4,5-T3CP 2.7
2,4,6-T3CP 2.3
2,3,4,5-T4CP 1.70
2,3,5,6-T4CP 3.60
Lepomis SB, FW 2-MCP 7.2 24-h LC50 Buccafusco
macrochirus 6.6 96-h LC50 et al. (1981)
(bluegill) 4,-MCP 4.0 24-h LC50
3.8 96-h LC50
2,4-DCP 4.7 24-h LC50
2.0 96-h LC50
2,4,5-T3CP 0.61 24-h LC50
0.45 96-h LC50
2,4,6-T3CP 0.72 24-h LC50
0.32 96-h LC50
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fish (contd).
2,3,4,6-T4CP 0.19 24-h LC50
0.14 96-h LC50
2,3,5,6-T4CP 0.40 24-h LC50
0.17 96-h LC50
Plants
Lemna minor SB, FW 4-MCP 282.8 50% chlorosis of fronds Blackman et al.
(duckweed) 2,4-DCP 58.7 (1955)
2,4,6-T3CP 5.9
2,4,5-T3CP 1.7
2,3,4,6-T4CP 0.6
a SB = Static bioassay.
SS = Semistatic bioassay.
SW = Marine.
FB = Continuous flow bioassay.
FW = Fresh water.
M = Molting.
I = Intermolt.
b NOEC = No-observed-effect concentration.
c IC50 = Concentration resulting in 50% inhibition.
The position of the chlorines on the phenol ring also influences
chlorophenol toxicity. Chlorophenols with chlorines in the 2 and 6
positions are often relatively non-toxic (Kobayashi et al., 1979;
Hattula et al., 1981b; Liu et al., 1982; Ribo & Kaiser, 1983;
Devillers & Chambon, 1986; Ruckdeschel & Renner, 1986), perhaps,
because the chlorines shield the hydroxyl group. These patterns
parallel the biodegradability of the compounds, as ortho-substituted
chlorophenols are less stable than their meta-substituted isomers
(section 4.2.1.2). However, the effects of chlorine position on
toxicity are not evident in all of the studies included in Table 16,
suggesting that the toxicity of any particular chlorophenol is highly
species-specific.
In addition, pH affects the toxicity of chlorophenols (Table 16).
At low pH, a given chlorophenol is relatively toxic, because it is
mainly in the form of molecules that can readily cross biological
membranes. As the pH is increased, chlorophenol toxicity is reduced
because the ionic form becomes abundant. Under the range of conditions
in most natural habitats, this effect becomes more important as the
number of chlorines in the chlorophenol increases, because the pKa is
related to chlorine number. Thus, monochlorophenol toxicity is
relatively unchanged by environmental pH, whereas that of
pentachlorophenol, which is present in the molecular form only under
very acid conditions, is greatly affected.
Studies on the toxicity of chlorophenols for terrestrial organisms
in the environment are much more limited. Blackman et al. (1955)
determined that the EC50s of several chlorophenols for the inhibition
of radial growth of the mould Trichoderma viride were as follows:
4-MCP, 47.6 mg/litre agar; 2,4-DCP, 8.6 mg/litre; 2,4,6-T3CP,
5.7 mg/litre; and 2,3,4,6-T4CP, 0.8 mg/litre. A similar pattern of
increasing toxicity with increasing chlorination of chlorophenols was
also observed by Sund & Nomura (1963) in their investigation of the
inhibition of seed germination by a number of chlorophenols. In
addition, they noted that chlorination at the 3 or 5 position enhanced
chlorophenol toxicity for germinating seeds. In a survey on the
contact toxicity of chemicals for the earthworm Eisenia foetida
(Roberts & Dorough, 1984), 2,4-DCP and 2,4,5-T3CP were classified as
extremely toxic, on the basis that their 48-h LC50 values fell within
the range of 1-10 µg/cm2 of filter paper.
7.1.2 Long-term toxicity
There are very few studies on the long-term effects of
chlorophenols on environmental organisms. Holcombe et al. (1982)
exposed the embryo, larval, and early juvenile stages of fathead
minnows to a range of sublethal concentrations of 2,4-DCP and other
phenolic compounds, in 32-day flow-through tests using Lake Superior
water. Survival of larvae and juvenile minnows was significantly
reduced after exposure for 28 days to 2,4-DCP at 460 µg/litre. The
growth of larval and juvenile stages was reduced by 1240 µg
2,4-DCP/litre. Hatching success was unaffected by the maximum
concentration used (1240 µg/litre).
Survival, reproduction, and growth were all reduced in Daphnia
magna exposed to 2,4-DCP concentrations of 1.48 mg/litre in
long-term (21-day) static renewal tests (Gersich & Milazzo, 1988).
In a study more relevant to field conditions, Virtanen & Hattula
(1982) used a flow-through aquarium microcosm with levels of
2,4,6-T3CP of 0.5 µg/litre, in order to track its incorporation into
sediment, algae, invertebrates, and fish. Male and female Poecilia
reticulatus fish were included in the microcosm, and aspects of
their reproduction and histopathology were monitored. Over a 10-month
period following their 56-day exposure in the aquarium and subsequent
transfer to uncontaminated water, only 90 offspring were born to
exposed fish and 22 of these died. Control fish produced 180
offspring, only 8 of which died. In addition, several offspring from
exposed parents had abnormally curved spines. Thus, under the test
conditions, 2,4,6-T3CP appeared to be very fetotoxic, and perhaps
teratogenic. No histological changes were noted in the livers or
kidneys of P. reticulatus as a result of these exposures.
7.1.3 Organoleptic effects
Exposure to low levels of chlorophenols can also impair the
flavour of fish (see section 7.2.4). According to Boetius (1954), as
little as 0.1 Ul 2-MCP/litre (v/v) tainted the flesh of eels and
oysters after exposure for 11 and 4 days, respectively. Shumway &
Palerisky (1973) estimated the threshold concentrations of several
chlorophenols for the impairment of the flavour of rainbow trout to
be: 2-MCP, 60 µg/litre; 3-MCP, 25 µg/litre; 4-MCP, 45 µg/litre;
2,3-DCP, 84 µg/litre; 2,4-DCP, 1 µg/litre; 2,5-DCP, 23 µg/litre;
2,6-DCP, 35 µg/litre; and 2,4,6-T3CP, 52 µg/litre.
7.2 Toxicity Studies under Natural Environmental Conditions
7.2.1 Bacteria
During the course of studies in Dutch coastal waters, Kuiper &
Hantsveit (1984) examined the effects of the addition of 4-MCP and
2,4-DCP on plankton communities enclosed in 1500-litre plastic bags.
In the first of 3 studies, total bacterial densities (by direct count)
were prevented from increasing by 0.1 and 1 mg 2,4-DCP/litre but, in a
second study, 2,4-DCP at 1 mg/litre did not have any effect on total
bacterial densities, and, in the final study, 1 mg 2,4-DCP/litre was
necessary to inhibit bacterial population growth. No effects of 4-MCP
were detected, even at 1 mg/litre. As treatments were not replicated
and bacterial densities were quite variable, it is not clear whether
the effects observed were in fact responses to chlorophenol exposure.
In contrast, when 5 mg 2,4,6-T3CP/litre was added to enclosures
in a West German pond, the numbers of aerobic heterotrophic bacteria
(by plate count) in the water increased more than 10 times compared
with the controls, within 4 days (Schauerte et al, 1982). This
response coincided with the disappearance of Daphnia from the
CP-treated tubes, suggesting that the increase was the result of a
release from grazing by the Daphnia.
There has been some concern that chlorophenols in industrial
wastes may impair the efficiency of secondary waste treatment through
their toxic effects on bacteria. Using a bench-scale activated sludge
plant, Broecker & Zahn (1977) determined that the degradation of waste
water declined after exposure to 25 µg 3,5-DCP/litre. Similarly, in a
laboratory scale model of a trickling filter (El-Gohary & Nasr, 1984),
exposure of acclimated microbes to 50 mg 2,4-DCP/litre reduced
Biological Oxygen Demand (BOD), and Chemical Oxygen Demand (COD).
However, chlorophenols in industrial wastes are unlikely to pose a
serious hazard for organisms important for secondary treatment. Levels
of chlorophenol entering treatment facilities are far below those used
in the studies just described (Folke, 1984), and recovery from shock
loadings of chlorophenols is rapid (El-Gohary & Nasr, 1984).
7.2.2 Phytoplankton
Kuiper & Hantsveit (1984) monitored the response of enclosed
marine plankton to chlorophenol additions, and determined that
exposure to chlorophenols affected algal biomass, composition, and
activity. In the first of 3 studies, 1 mg 4-MCP or 2,4-DCP/litre
prevented the increase in algal biomass (as chlorophyll) that occurred
in control enclosures. Large flagellates made up a greater proportion
of the algal community in 1 mg/litre-treated enclosures compared with
controls, perhaps because grazing was reduced (section 7.3). Primary
productivity generally parallelled the dynamics of algal biomass, as
it was reduced by exposure to 1 mg 4-MCP/litre; however, the addition
of 1 mg 2,4-DCP/litre did not affect photosynthetic radiolabelled
dissolved inorganic carbon (DIC) uptake. Results were generally
similar during the 2 subsequent manipulations, though the magnitude
and timing of the effects varied.
In their studies on ponds, Schauerte et al. (1982) observed major
shifts in the species composition of the phytoplankton following the
addition of 5 mg 2,4,6-T3CP/litre. The large population of the
blue-green alga Chroococcus limneticus was sharply reduced, and the
diatom Nitzschia acicularis was eliminated, while the flagellated
algae Euglena and Trachelomonas appeared in large numbers
following exposure to 2,4,6-T3CP. The dynamics of other
phytoplankton, which were less abundant, were not discussed.
Chorophenols were among the toxicants used by Erickson & Hawkins
(1980), who measured the response of estuarine phytoplankton
communities to 15 compounds produced during the chlorination of sea
water. Natural phytoplankton assemblages, pumped from the estuary to
flow-through aquaria in the laboratory, were insensitive to
chlorophenol concentrations of 0.5-2 mg/litre. Photosynthetic
radiolabelled DIC uptake was not depressed by exposure to 2 mg
2,4,6-T3CP or 4-MCP/litre.
7.2.3 Zooplankton
Marine zooplankton were strongly affected by chlorophenol
additions during field studies in 1500-litre plastic enclosures
(Kuiper & Hantsveit, 1984). While the zooplankton communities in the
control enclosures and those treated with 0.1 mg 4-MCP/litre or 0.1 mg
2,4-DCP/litre displayed similar dynamics, total biomass and production
in enclosures treated with 1 mg 4-MCP/litre and 1 mg 2,4-DCP/litre
were reduced relative to controls throughout the first three-quarters
of the study. All life-history stages of several copepod species were
similarly affected. Results in subsequent studies were generally
similar, though the magnitude of the impact varied.
More severe effects for Daphnia exposed to 5 mg
2,4,6-T3CP/litre were reported by Schauerte et al. (1982) during
studies on a pond. From initial levels of more than 20 individuals per
100 ml before the toxicant was added, Daphnia was eliminated from
T3CP-treated enclosures in 3 days. The abundance of Daphnia in
control enclosures was high and stable during the 24 days of the
study.
7.2.4 Fish
There are no controlled field studies on the effects of
chlorophenols on fish, but fish kills have occurred as a result of
chlorophenol spills. Mackenzie et al. (1975) compiled information on
such incidents in British Columbia salmon waters during 1960-73. In
one instance, an over- flow from a lumber-treatment tank released both
T4CP and PCP into the Mamquam Channel in 1973, killing an estimated
500 adult and juvenile coho salmon.
Chlorophenols may also impair the flavour of fish, even when
present in the minute quantities detected in moderately-contaminated
natural waters (section 7.1.3). Chatterjee (1974) reported that kraft
and groundwood pulp discharges into Lakes Superior and Huron, which
included phenolic compounds, were apparently responsible for the
tainting of flesh from fish captured nearby.
7.2.5 Effects on physical and chemical variables
The only instance in which chlorophenols affected physical or
chemical factors in the environment apparently involved a secondary
effect. In studies in which 5 mg 2,4,6-T3CP/litre was added to
enclosures in a pond, oxygen levels declined from initial levels of
3-4 mg/litre to less than 1 mg/litre, within 6 days of treatment, as
the balance between heterotrophic and autotrophic metabolism shifted
(Schauerte et al., 1982). Apart from this secondary effect, physical
and chemical variables appear insensitive to CP additions. Schauerte
et al. (1982) did not find any significant differences in temperature,
pH, hardness, sulfide, carbonate, or chloride levels between control
and 2,4,6-T3CP-treated enclosures. Similarly, levels of phosphate,
ammonia, nitrate, nitrite, silicate, and pH were unaffected by
additions of as much as 1 mg 4-MCP or 2,4-DCP/litre (Kuiper &
Hantsveit, 1984).
7.3 Treatment Levels
Unfortunately, the effects of chlorophenols at the low (mg/litre)
levels that characterize the aquatic environment at large (section
5.1.2) were not examined in most of these studies. As a result, they
shed little light on the possible hazards presented by the widespread
low-level contamination observed in most environments. The microcosm
study by Virtanen & Hattula (1982) was an exception in this regard.
However, the mg/litre concentrations used by most research workers are
relevant to major accidental spills of chlorophenols in the
environment.
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO SYSTEMS
8.1 Acute Studies
In general, the toxicity of chlorophenols increases with an
increase in the chlorination of the phenol molecule. A convulsant
effect is associated with the less-chlorinated phenols and oxidative
phosphorylation uncoupling is more prominent with the highly
substituted compounds (Ahlborg & Thunberg, 1980; Jones, 1981; Exon,
1984).
Farquharson et al. (1958) studied the effects of a series of
chlorophenols on male albino rats (Table 17). Symptoms associated with
the lethal intraperitoneal injection of the monochlorophenols, 2,6-DCP
and 2,4,6-T3CP, as well as phenol, included an initial increase in
physical activity (rapid running, nose rubbing) followed by tremors,
convulsions, and loss of righting reflex. With 2,3,6-T3CP, rats
suffered convulsions only when handled, and otherwise lay prostrate
with hypotonia. Hypotonia, starting at the hind limbs, was observed
within 2-3 min of injection with 2,4-DCP, 2,3,6-, 3,4,5- and
2,4,5-T3CP, T4CP, and PCP. Body temperature was slightly reduced by
phenol, MCPs and DCPs, while T3CP caused a slight elevation and T4CP
and PCP a marked rise in temperature (4-4.5°C). Respiratory rate
increased initially then declined as coma developed, especially with
T4CP and PCP. The extremities were cyanosed and asphyxial spasms
occurred about 30 seconds before death. With T4CP and PCP,
respiration stopped usually one-half to 2 min before cessation of the
heart, whereas with the other chlorophenols, respiration ceased
concomitantly with the heart or just before.
Animal studies indicate that most mono-, di-, and tri-
chlorophenols are moderately toxic when administered orally, with
LD50 values ranging between 230 and 4000 mg/kg body weight
(Table 18). In general, the less-chlorinated phenols have an acute
oral toxicity very close to that of phenol. T4CP is considerably more
acutely toxic, with LD50 values of between 100 and 400 mg/kg body
weight (Ahlborg & Thunberg, 1980; Hattula et al., 1981a). Thus, the
data indicate that the general order of decreasing acute toxicity is:
T4CP, MCP, DCP, T3CP.
The subcutaneous and intraperitoneal routes of exposure have also
been investigated (Table 18). As with oral administration,
subcutaneous injections revealed a general order of decreasing acute
toxicity of: T4CP, MCP, DCP, T3CP.
Table 17. Effect of lethal chlorophenol doses given intraperitoneally to ratsa
Compound Convulsant Hypotonia Max. Respiration Rigor mortis
onset (min) change onset (min)
activity in temp.
(°C)
Ether 50
Monochlorophenols
2(o) + -b -2.0 -d > 5.2 < 50
3(m) + -b -2.5 -d > 5.2 < 50
4(p) + -b -2.5 -d > 5.2 < 50
Dichlorophenols
2,4- (occasional 2-3c -0.5 -d > 5.2 < 50
twitches)
2,6- + -b -0.7 -d > 5.2 < 50
Trichlorophenols
3,4,5- - 2-3 + 0.5 -d < 5
2,4,5- - 2-3 + 0.5 -d < 5
2,4,6- + + 0.5 -d < 5
2,3,6- (sometimes 2-3 + 0.5 < 5
convulsions
when
handled)
Table 17. (cont'd).
Compound Convulsant Hypotonia Max. Respiration Rigor mortis
onset (min) change onset (min)
activity in temp.
(°C)
Tetrachlorophenol
2,3,4,6- - 2-3 + 4.0 -e < 6
Pentachlorophenol
2,3,4,5,6- - 2-3 + 4.5 -e < 5
a From Farquharson et al. (1958).
b Apparent after convulsions have lessened.
c Muscle twitches evoked by auditory and mechanical stimuli.
d Initial increase, then decrease as coma developed; asphyxial spasms 30
seconds before death; ceases just before or simultaneously with cardiac arrest.
e Ceases 1/2 to 2 min before stopping of heart.
Table 18. Acute toxicity (LD50s) of phenols and chlorophenols for rats and micea
RAT MOUSE
Oral Subcutaneous Dermal IP Oral Subcutaneous IP
Phenol 530-650b 669 250 300 344 360 -
Monochlorophenol
-2(o) 670 950 - 230 347, 345c - -
-3(m) 570 1390 - 355 521, 530c - -
-4(p) 261 1030 - 281 1373, 1422c - -
Dichlorophenol
-2,3 - - - - 2585, 2376c - -
-2,4 580, 4000d 1730 - 430 1276, 1352c 1600h - -
-2,5 - - - - 1600, 946c - -
-2,6 2940 1730 - 390 2198, 2120c - -
-3,4 - - - - 1685, 2046c - -
-3,5 - - - - 2643, 2389c - -
Trichlorophenol
-2,3,6 - - - 308 - - -
-2,4,5 820e 2260 - 355 - - -
-2,4,6 820e - - 276 - - -
-3,4,5 - - - 372 - - -
Table 18 (contd).
RAT MOUSE
Oral Subcutaneous Dermal IP Oral Subcutaneous IP
Tetrachlorophenol
-2,3,4,5 - - > 2000h - 400g - 97g
-2,3,4,6 140, 360f 210g - 130 131g 120i 82g
-2,3,5,6 > 2000h 109g 48g
-Commercial 485-565h - - -
mix
Tetrachlorophenate-Na
-2,3,4,5 - - > 2000h - - - -
-2,3,5,6 - - 294-469h - - - -
a Principle database NIOSH (1983). LD50 values given as mg/kg body weight.
b Babich & Davis (1981).
c Borzelleca et al. (1985a). Data for males presented first, then females.
d Kobayashi et al. (1972).
e More recent values in US EPA (1979) are 4 times higher (2460-2960 mg/kg).
f Hattula et al. (1981a).
g Ahlborg & Larsson (1978).
h Shen et al. (1983) commercial mixture was primarily 2,3,4,6-T3CP.
i Kozak et al. (1979).
However, in ip injection studies, the toxicities of the mono-,
di-, and trichlorophenols were comparable while T4CP was 2-3 times
more toxic.
The only study available on the acute dermal toxicity of the less
chlorinated phenols was carried out by Shen et al. (1983). In this
investigation, Sprague-Dawley rats of both sexes were given 2,3,4,5-,
2,3,5,6-T4CP, their sodium salts, or a commercial T4CP preparation
containing over 90% of 2,3,4,6-T4CP and 5-10% of PCP. A very low
dermal toxicity was reported for 2,3,4,5-T4CP, its phenate salt, and
2,3,5,6-T4CP, while the sodium salt of 2,3,5,6-T4CP had the highest
toxicity of all, followed by the commercial T4CP (Table 18). The
relatively high toxicity of the commercial T4CP could result from its
content of PCP. The rapid death of the rats (usually 6 h) argues
against any role being played by the microcontaminants, because the
PCDDs and PCDFs are usually associated with delayed acute toxicity.
The effects on animals of these less-chlorinated phenols
administered via inhalation have not been investigated. However, it
can be inferred from the one report on the inhalation of sodium-PCP
(Hoben et al., 1976b) that the toxicity via inhalation would be
greater than that associated with the oral, intraperitoneal,
subcutaneous, or dermal routes.
In their study on the toxicity of 3 different T4CP isomers in
mice, Ahlborg & Larsson (1978) noted that the LD50 values for all 3
isomers were lower with ip injection than with oral (gavage)
administration. This effect of the mode of administration on toxicity
is apparent in the data on the other chlorophenols (Table 18) and may
be related to differences in the rates of absorption, metabolism, and
excretion of these compounds. The low toxicity values obtained with
subcutaneous injections may be due to a low rate of absorption. The
vehicle used for administration can also have a significant influence
on absorption.
8.2 Skin and Eye Irritation; Sensitization
Very little information is available on skin and eye irritation or
sensitization in experimental animals exposed to chlorophenols. Only
very slight irritation was noted in rabbits following the application
of dry 2,4,5-T3CP to their skin (McCollister et al., 1961). The
authors suggested that mild erythema might be caused by high
concentrations of the material in solution. In a study on the dermal
toxicity of the T4CP isomers, Shen et al. (1983) found that
dermatosis occurred in rats painted with 2,3,4,5-T4CP or its phenate
salt but not with the other 2 isomers or their salts.
8.3 Short-term Exposure
All the available information on short-term exposure has been
obtained by means of oral studies, except for one report concerning
dermal application. Information is lacking on the effects of
inhalation and other routes of exposure in experimental animals.
Exon & Koller (1985) investigated the possible immunological
effects of 3 chlorophenols on rats exposed both prenatally and
post-natally. Weanling female rats (3 weeks of age) were given 2-MCP
(98% pure) (0, 5, 50, or 500 mg/litre), 2,4-DCP (99% pure), or
2,4,6-T3CP (98% pure); (both at 0, 3, 30, or 300 mg/litre) in the
drinking-water, for 90 days from weaning through breeding and
pregnancy. A randomly chosen group of offspring was given the same
dose regime as the dams for an additional 12-15 weeks after weaning.
Both groups were observed for another 10 weeks. 2-MCP did not have any
adverse effects on humoral immunity, cell-mediated immunity, or
macrophage function in the exposed progeny at any of the exposure
levels. Exposure of progeny to the highest concentration of 2,4-DCP
significantly enhanced (P less than 0.05) humoral immune
responsiveness and decreased cell-mediated immunity in rats with both
prenatal and postnatal treatments, but not in rats exposed only
in utero. 2,4,6-T3CP did not have any effects on the immune
responses tested. Spleen and liver weights were increased in the
progeny receiving water containing 2,4-DCP or 2,4,6-T3CP at
300 mg/litre, but not 2-MCP. The authors suggested that this increase
in organ weight was due to hyperplasia, as no histological anomalies
were observed.
Exon & Koller (1985) also monitored haematological parameters and
organ weights in exposed rat progeny. Combined pre- and postnatal
(24 months) exposure to 500 mg 2-MCP/litre or 300 mg 2,4-DCP/litre
significantly elevated red blood cell counts, haemoglobin
concentrations, and packed cell volumes of offspring. Each of the 3
compounds was also fetotoxic (section 8.5).
In studies by Kobayashi et al. (1972), 2,4-DCP in the diet at the
maximum dose of 230 mg/kg body weight per day, over a 6-month period,
caused swelling of the hepatocytes in male mice but did not
substantially affect liver, kidney, spleen, or adrenal histology.
Furthermore, no significant exposure-related changes were observed in
organ weight, body weight, food consumption, serum concentrations of
liver enzymes, or numbers of red and white blood cells.
In a 90-day study, Borzelleca et al. (1985b) exposed mice of both
sexes to 2,4-DCP in their drinking-water at mean daily doses of 40,
114, or 383 mg/kg body weight for males, and 50,143, or 491 mg/kg body
weight for females. These dosages were calculated from daily water
consumption data and the concentrations added to the drinking-water.
At the end of the study period, there were no significant
treatment-related differences in organ weights, electrolyte levels,
haematological factors, or the activities of hepatic mixed-function
oxidases (MFO) or serum enzymes.
In short-term studies that were preliminary to a carcinogenicity
bioassay (section 8.6), rats and mice of both sexes were fed diets
containing 2500-40 000 mg 2,4-DCP/kg for 13 weeks (NTP, 1988). All
animals survived to the end of the study, except the mice receiving
the highest dose, all of which died. Male mice and rats of both sexes
receiving the 20 000 mg/kg diet showed reduced final mean body
weights. Dose-related effects were apparent in the form of bone-marrow
atrophy in rats and liver damage (necrosis, multi-nucleated
hepatocytes) in mice.
A study on the short-term exposure of rats and rabbits to
2,4,5-T3CP was carried out by McCollister et al. (1961). Rabbits,
given 20 oral doses of 500 mg/kg body weight over a 28-day period,
showed only "very slight kidney and liver changes". In rats receiving
18 doses of 1000 mg/kg body weight over a 24-day period, there was a
slight increase in kidney weight, while growth, mortality,
haematological parameters, and the histology of the lung, heart,
liver, kidney, spleen, adrenal, pancreas, and testis were unaffected.
In the same report, male and female rats given a diet containing
0, 0.1, 0.3, or 1.0 g 2,4,5-T3CP/kg for 98 days did not exhibit
behavioural changes, increased mortality, changes in food consumption,
growth, or histology (McCollister et al., 1961). At the 10 g/kg level,
an increase in the frequency of day-time urination was noted in both
males and females, as well as significant growth retardation in
females. Kidney and liver degeneration, which was judged reversible,
was also found at this exposure level.
During the course of a study on reproduction (section 8.5.1),
Blackburn et al. (1986) dosed rats for 5 days per week (males, 11
weeks; females, 2 weeks and through gestation) by gavage with 0, 100,
500 or 1000 mg 2,4,6-T3CP/kg body weight in corn oil. A number of
rats from the 1000 mg/kg group died as a result of treatment (males, 8
out of 25; females, 3 out of 40). Males and females in this group
exhibited significant but transitory weight loss compared with
controls. Male kidney, liver, lung, adrenal, spleen, heart, testis,
prostate, seminal vesicle, and epididymis weights were unaffected by
all levels of 2,4,6-T3CP exposure. Females dosed at 1000 mg/kg body
weight lost hair, were lethargic, and breathed irregularly.
In a range-finding study, rats and mice of both sexes were exposed
to 2,4,6-T3CP for 7 weeks to determine the maximum tolerated dose
(NCI, 1979). The compound was given in the feed up to a maximum level
of 46 000 mg/kg for rats and 31 500 mg/kg for mice. Weight gain was
reduced in both male and female rats at all exposure levels, but only
at the 2 highest dose levels (21 500 and 31 500 mg/kg) in mice. At the
highest dose, a moderate to marked increase in splenic haematopoeisis
was found in the rats and midzonal vacuolation of hepatocytes was seen
in 2 males. All tissues in the mice were normal at the end of 7 weeks.
Hattula et al. (1981a) dosed rats by garage daily with 0, 10, 50,
or 100 mg 2,3,4,6-T4CP/kg body weight in olive oil for 55 days.
Histological changes were found only in the liver, even though the
kidneys and spleen contained higher concentrations of 2,3,4,6-T4CP.
This finding suggests that 2,3,4,6-T4CP has a specific effect on the
liver of rats.
8.4 Long-Term Exposure
Investigations of the effects of long-term exposure to
chlorophenols have been designed primarily to test their carcinogenic
properties and are described in section 8.7.
8.5 Reproduction, Embryotoxicity, and Teratogenicity
Exon & Koller (1981, 1983) and Exon et al. (1984) examined the
effects of 2-MCP (98% pure; 0, 5, 50, or 500 mg/litre), 2,4-DCP (99%
pure; 0, 3, 30, or 300 mg/litre), and 2,4,6- T3CP (98% pure; 0, 3,
30, or 300 mg/litre) on reproductive parameters in mice exposed via
drinking-water (section 8.3). Females were exposed from 3 weeks of age
until breeding at 90 days, and through-out gestation to parturition.
Reanalysis of their original data (Exon & Koller, 1985) led the
authors to conclude that there was a weakly significant (P less than
0.1) effect of 2-MCP at 500 mg/litre and 2,4-DCP and 2,4,6-T3CP at
300 mg/litre, expressed as reduced litter size. The percentage of
stillborn offspring compared with controls tended to increase in all
exposed groups. However, the differences were not significant at P
less than 0.05.
In mice, sperm motility and penetration of ova were not affected
by acute (0.1 to 1 mmol/litre) and long-term exposure (90 days at
50-500 mg/kg body weight per day) to 2,4-DCP (Seyler et al., 1984).
In vitro penetration was depressed by 2,5-, 3,4-, or 3,5-DCP at
1 mmol/litre, and it appeared that this concentration of 3,4- or
3,5-DCP also disrupted the sperm acrosome.
The effects of oral dosing with 2,4,6-T3CP on rat reproduction
were investigated by Blackburn et al. (1986). Male rats were given 0,
100, 500, or 1000 mg 2,4,6-T3CP/kg body weight in corn oil, by
gavage, 5 days per week, for 11 weeks.
These exposures to T3CP did not significantly affect male sexual
behaviour (mount and ejaculation latencies, number of mounts and
intromissions), plasma-testosterone levels, or sperm counts, motility,
or morphology. Males from the control and 1000 mg/kg groups were then
mated with unexposed females, which were sacrificed on day 18 of
gestation. The exposure regime of the males did not significantly
affect litter size, sex ratio, mean pup weight by sex, number of dead
fetuses, or the numbers of resorptions or implantation sites. As part
of the same study, female rats were dosed in the same manner as the
males for 5 days per week over 2 weeks and then mated, after which
dosing continued until day 21 of gestation. No treatment-related
effects were evident in the breeding success, mean litter size, or
offspring survival of these females. Litter weights were initially
significantly reduced in the 500 and 1000 mg/kg body weight groups,
but this difference had disappeared at 4 days postpartum. This effect
was suggested to have been a secondary manifestation of female
toxicity (section 8.2), or of initial differences in litter size
between treatments.
In a teratogenicity study of 2,4,5-trichlorophenoxyacetic acid
(Neubert & Dillman, 1972), 2,4,5-T3CP was also tested, but only at
doses of 0.9 or 9 mg/kg body weight per day. A slight increase in
embryo mortality was observed at the higher dose, but its significance
is unclear. No teratogenic effects were observed.
Schwetz et al. (1974) studied the embryotoxic and fetotoxic
effects on rats of purified (99.6%) 2,3,4,6-T4CP and commercial grade
2,3,4,6-T4CP (73% T4CP + 27% PCP plus 100 ppm total of each of
dioxins and dibenzofurans). Doses of 10 and 30 mg/kg were given by
gavage to pregnant rats from day 6 to day 15 of gestation, after which
they were sacrificed on day 21. A delay in ossification of the skull
bones was found. Neither of the compounds was embryolethal (evidenced
by no increase in resorptions or abortions) even at the high dose of
30 mg/kg per day. No differences were observed in the toxicity of the
2 grades of T4CP.
Chlorinated phenols do not appear to be teratogenic for
experimental animals. Of the compounds tested, levels as high as
500 mg 2-MCP/litre in drinking-water (Exon & Koller, 1981), 300 mg
2,4-DCP/litre in drinking-water (Exon et al., 1984), 1000 mg
2,4,6-T3CP/kg body weight (Blackburn et al., 1986), and 30 mg
2,3,4,6-T4CP/kg body weight (Schwetz et al. 1974) did not produce any
teratogenic effects in rats.
8.6 Mutagenicity and Related End-Points
Rasanen et al. (1977) found that all dichlorophenol isomers, 4 out
of 6 trichlorophenols, and 2,3,4,6-T4CP were negative in Salmonella
typhimurium mutagenicity bioassays. Similarly, Haworth et al. (1983)
reported that 2,3-, 2,4-, 2,5-, 2,6-, 3,4-, 3,5-DCPs and 2,4,5- and
2,4,6-T3CP were negative in S. typhimurium TA 98, 100, 1535 and
1537 strains. Nestmann et al. (1980) reported similar findings for
2,6-DCP and 2,4,5-T3CP. It was reported by Kinae et al. (1981) that
2,4,6-T3CP was negative in S. typhimurium TA 98, 100, and 1537
strains, with and without exogenous metabolic activation, but positive
in a B. subtilis recombination assay. At a concentration of
400 mg/litre, purified 2,4,6-T3CP caused a weak but significant
increase in the frequency of forward mutations, but did not affect
intergenic or intragenic re-combinations in the yeast Saccharomyces
cervisiae MP-1 strain (Fahrig et al. 1978).
In mutagenicity studies conducted to supplement a carcinogenicity
bioassay (NTP, 1988), 2,4-DCP did not produce any revertant colonies
in S. typhimurium strains TA 98, 100, or 1537 and yielded equivocal
results with TA 1535 only in the presence of hamster S9 activation. In
the mouse L5178Y assay (without metabolic activation),
trifluorothymidine resistance was increased by 2,4-DCP exposure. In
the same study, in vitro exposure of Chinese hamster ovary cells to
2,4-DCP increased the frequency of sister chromatid exchanges, but did
not cause chromosomal aberrations.
Hattula & Knuutinen (1985) showed that purified 2,4,6- T3CP and
2,3,4,6-T4CP were weakly mutagenic in V-79 Chinese hamster cells
in vitro, in the absence of metabolic activation by hepatocyte
co-cultures. They were both non-mutagenic in the presence of metabolic
activation by hepatocyte co-cultures. 2,6-DCP and PCP were negative
regardless of the presence of metabolic activators, and 2,4,6-T3CP
did not induce chromosomal aberrations or sister chromatid exchanges
in cultures of Chinese hamster ovary cells (Galloway et al., 1987).
Only very few data are available from in vivo studies, and these
are limited to the mouse spot test (Fahrig et al., 1978). Pregnant
mice were injected intraperitoneally with 50 or 100 mg purified
2,4,6-T3CP/kg body weight on day 10 of gestation. Examination of
offspring revealed an increased frequency of coat spots (0.6% in the
50 mg/kg group versus 0.1% in controls), indicative of a weak
mutagenic response.
8.7 Carcinogenicity
Both 2,4,6-T3CP and 2,4-DCP have been tested for carcinogenicity
in a 2-year bioassay. The other lower chlorophenols have not been
adequately tested for their carcinogenic properties.
A 2-year carcinogenicity study on 2,4-DCP was recently completed
under the US National Toxicology Program (NTP, 1988). Test animals fed
diets containing 2,4-DCP (more than 99% pure) received the following
average calculated doses (mg/kg body weight):
F344/N rats B6C3F1 mice
Male Female Male Female
Low dose 210 120 800 430
High dose 440 250 1300 820
Mean body weights were reduced, usually by several percent, in all
of the high-dose groups, as well as the low-dose groups of female
mice. Mean food consumption was reduced in all treated groups, by
several percent in rats, and in a dose-related manner in mice (up to
22%). No significant differences in the survival of any treated group
occurred. There were dose-related increases in the incidence of
multinucleated hepatocytes in male mice. No compound-related increases
in the incidence of neoplastic lesions were observed; in fact, these
were reduced for mononuclear cell leukaemia in male rats (both doses)
and malignant lymphomas in female mice (high dose only).
Innes et al. (1969) tested 120 pesticides and industrial chemicals
for tumorigenicity in male and female mice. The individual compounds
were administered at a maximum tolerated dose, by stomach tube, from 7
days of age to 4 weeks old, and then in the diet at approximately the
same dosage. The 100 mg/kg daily dose of Omal or Dowicide 2S
(2,4,6-T3CP) increased tumour incidence in the treated animals at the
end of 72 weeks. The authors recommended additional statistical
evaluation and/or studies before a meaningful interpretation could be
made.
A long-term oral exposure study was carried out on rats and mice
to test the carcinogenicity of 2,4,6-T3CP (NCI, 1979). Male and
female F344 rats were given dose levels of 2,4,6-T3CP of 5000 and
10 000 mg/kg in the feed for 106 weeks. Male B6C3F1 mice were dosed at
the same levels as the rats for 105 weeks. Female mice were initially
given dietary levels of 10 000 and 20 000 mg/kg for 38 weeks, which
were then reduced to 2500 and 5000 mg/kg, respectively, for the
remaining 67 weeks of the study, because they showed a marked
reduction in weight gain. At the end of the study, the treated male
rats showed a significantly higher incidence of malignant lymphomas
and leukaemias. Leukocytosis and monocytosis of the peripheral blood
and hyperplasia of the bone marrow were found in those that did not
show lymphomas and leukaemias. No lymphomas and/or leukaemias were
detected in female rats, but leukocytosis and monocytosis of
peripheral blood and bone-marrow hyperplasia were evident. Both male
and female mice displayed a dose-related statistically significant
incidence of both hepatocellular carcinomas and adenomas. It was
concluded that 2,4,6-T3CP was carcinogenic for male Fisher rats and
both sexes of B6C3F1 mice under the assay conditions used.
Boutwell & Bosch (1959) studied the tumour-promoting action of 2-
and 3-MCP, 2,4-DCP, 2,4,5-T3CP, and 2,4,6- T3CP in mice following
the dermal application of 9,10- dimethyl-l,2-benzanthracene (DMBA)
(25 µl of 0.3% DMBA in benzene) as an initiator. One week after a
single exposure to DMBA, twice weekly applications of 25 µl of a 20%
solution of the test compound in benzene were made for 12-24 weeks.
The control group received the pre-treatment dose of DMBA only. The
authors reported that the monochlorophenols, 2,4- DCP, and 2,4,5-T3CP
all had a tumour-promoting action similar to that of phenol, while
2,4,6-T3CP did not have any effect on tumour promotion.
Exon & Koller (1981,1985) carried out 15 to 24-month studies on
the effects of pre- and postnatal exposure of rats to 2-MCP and
2,4-DCP. No evidence of tumour initiation was revealed with exposure
to 2-MCP (98% pure) at 500 mg/litre drinking-water, or to 2,4-DCP
(99% pure) at 300 mg/litre drinking-water (Exon & Koller, 1985).
However, 2-MCP acted as a promoter of the carcinogenic activity of
ethylnitrosoures (ENU), reducing tumour latency and increasing tumour
incidence in male rats exposed both pre- and postnatally, compared
with controls receiving only ENU.
There is sufficient evidence of the carcinogenicity for animals of
2,3,7,8-TCDD (IARC, 1987) and a mixture of two H6CDD isomers (NCI,
1979), which may be present as microimpurities in some technical
chlorophenols.
8.8 Factors Modifying Toxicity Metabolism
Any factor that can interfere with sulfate and/or glucuronide
conjugation would modify the toxicity of a chlorophenol by inhibiting
chlorophenol detoxification. Somani et al. (1984) demonstrated with an
isolated liver-perfusion system that the glucuronide conjugation of
14C-2,4-DCP can be blocked by galactosamine in the rat. In high
enough concentrations, galactosamine or similar compounds would
prolong the residence time in the body and thus the toxicity of the
chlorophenol molecule.
In some instances, metabolism of chlorophenol yields molecules
that are more, rather than less, toxic than the parent molecule. For
example, when injected into rats intraperitoneally, tetrachloro-
p-hydroquinone, a metabolite of 2,3,5,6-T4CP, is much more toxic
than the parent compound (Ahlborg & Larsson, 1978).
The toxicology of chlorophenols is further complicated by
micro-contaminants in technical grade products, particularly in the
higher chlorinated compounds. Some of these impurities are themselves
extremely toxic. Conversely, microcontaminants are known to induce
enzymes that can affect the rate of chlorophenol metabolism and
excretion (Ahlborg & Thunberg, 1978). For these reasons, assessment of
toxicity studies with chlorophenols requires a knowledge of the types,
levels, and toxicology of the contaminants present. This is
particularly true when extrapolating animal studies (which often
involve the use of purified compounds) to human beings, who are
generally exposed to technical formulations. The residue
concentrations and toxicology of the dibenzodioxin and dibenzofuran
microcontaminants have been reviewed by WHO (in press).
8.9 Mechanisms of Toxicity, Mode of Action
The major mode of action of chlorophenols appears to be the
uncoupling of oxidative phosphorylation. The strength of the
uncoupling effect is related to the degree of chlorination: PCP is the
strongest inhibitor of oxidative phosphorylation, MCP the weakest
(Farquharson et al., 1958; Mitsuda et al., 1963; Weinbach & Garbus,
1965; Carlson, 1978). To a lesser extent, inhibition of oxidative
phosphorylation is affected by the positions of the chlorine atoms on
the molecule. There appears to be a relationship between chlorination
and the toxicity of PCP and T4CP (Table 18), although there is no
clear-cut relationship between the degree of chlorination and
toxicityin MCP, DCP, and the T3CP series. PCP and to a lesser extent
other chlorophenols, depending on their degree of chlorination, have
been shown to bind strongly to mitochondrial protein (Weinbach &
Garbus, 1965). At low levels, PCP uncouples oxidative phosphorylation,
at intermediate levels, it inhibits the formation of high-energy
intermediates in the phosphorylation system and, at high
concentrations, it inhibits the electron transport system (Mitsuda et
al., 1963). PCP has been shown to bind directly to mitochondrial
ATPase (Stockdale & Selwyn, 1971). Inhibition of ATPase would prevent
the breakdown of ATP to ADP and energy release to the mitochondria. In
response to the reduced availability of energy, a compensatory
increase in catabolism would be expected. Increased catabolism would
result in higher rates of oxygen consumption and, under short-term
exposure conditions, depletion of metabolic stores. With oxidative
phosphorylation uncoupled, the energy provided by catabolism would be
released as heat. This process would underlie, respectively, the
elevated respiratory rate and body temperature, and the long-term
weight loss observed in intoxicated organisms (Weinbach, 1957;
Farquharson et al., 1958; Mitsuda et al., 1963; Wood et al., 1983).
Similarly, Exon & Koller (1985) suggested that the immune effects of
2,4-DCP might result from the uncoupling of oxidative phosphorylation,
thus impairing cellular energy production in immunocompetent cells, or
perhaps, from a direct toxic effect on subpopulations of cells
involved in immune responses. The uncoupling of phosphorylation
appears to be due to the chlorophenate ion, while the convulsant
action is caused by the undissociated molecule (Farquharson et al.,
1958).
Chlorophenol toxicity may also result from a more general
inhibition of enzyme activity by these compounds. Arrhenius et al.
(1977) demonstrated that, in rat liver microsomal preparations,
purified chlorophenols selectively inhibited cytochrome P-450 activity
at the terminal oxygenation step of the MFO enzyme system by
interfering with the coupling of flavin to this enzyme. 2,4-DCP,
2,4,6-T3CP, and 2,3,4,6-T4CP concentrations of 0.3 mmol-3 mmol/litre
showed a weaker inhibition of C-oxygenation than PCP. Kaneki & Tanaka
(1984) reported that the inhibition of porcine lipase activity and, to
a lesser extent, wheat germ lipase activity increased with the number
of chlorine atoms on the phenol ring. The inhibiting action of PCP was
greater than that of 2,3,4,6-T4CP, which in turn was stronger than
2,3,6- T3CP, while DCP and MCP did not have any effect. Carlson
(1978) found that certain T3CP isomers inhibited glucuronyl
transferase activity and EPN detoxification in vitro, but not
in vivo. The reason for these effects could be the absence from the
in vitro preparations of binding proteins or alternate metabolic
enzyme systems (Somani et al., 1984).
The mechanisms of carcinogenicity or co-carcinogenicity of
chlorophenols (section 8.7) remain unresolved at present. Exon &
Koller (1985) suggested that chlorophenols may play a role in
carcinogenicity by altering the toxicity of carcinogens (by inhibiting
detoxifying enzymes, damaging DNA, or altering DNA repair) or by
reducing immunosurveillance. It has been shown by Vizethun & Goerz
(1979) that 2,4,5-T3CP and PCP can induce different species of
cytochrome P-450 in nuclei and microsomes. The importance of this
finding in relation to carcinogenesis is as yet unclear, but nuclear
monoxygenases could play a critical role in cell alterations, because
of their proximity to DNA and their ability to induce binding of
electrophilic compounds.
9. EFFECTS ON MAN
As a result of the diverse range of applications of chlorophenols
(section 3.2.2), there is considerable potential for human exposure to
these compounds and their associated contaminants. Knowledge
concerning the toxic effects of chlorophenols on people is based
primarily on studies on persons employed in the chemical-manufacturing
industry, where mainly DCP and T3CP are involved, and the
wood-preservation/ protection industries, where T3CP, T4CP, and PCP
are the major forms used (see reviews by Behrbohm, 1959, Kozak et al,
1979, and Ahlborg & Thunberg, 1980). Reports of chlorophenol toxicity
in the general population are few in number, though chlorophenol
contamination of human tissues and fluids seems widespread (sections
5.2, 5.3).
9.1 Acute Toxicity
Accidental and suicidal poisonings with commercial chlorinated
phenols have been reported (WHO, 1987b), and a number of the most
heavy acute exposures have resulted in death. With the support of
animal studies, the signs and symptoms of acute exposure to
chlorinated phenols include: convulsions (especially with
less-chlorinated phenols), ataxia, mental and physical fatigue,
headache, dizziness, disorientation, tachycardia, body temperature
change (decreased with monochlorophenols, increased with,
particularly, tetrachlorophenols and pentachlorophenol), and increased
sweating. Cyanosis and asphyxia spasms shortly precede death. Death is
apparently due to cardiac arrest and is followed, at least in animals,
by rapid rigor mortis, especially with T3CP and T4CP poisoning.
In general, the acute toxicity of chlorophenols in animals
increases with the number of chlorine atoms in the molecule (section
7.1.1). In man, the only published estimate of a minimum lethal oral
dose (LDLo) for any chlorophenol is for PCP (29 mg/kg body weight,
approximately 2 g for an average person) (WHO, 1987b). The human LDLo
for unchlorinated phenol is estimated to be 140 mg/kg body weight.
Acute exposure of human beings to lower chlorophenols has also
occurred as a result of industrial accidents during the production of
2,4,5-T3CP and has been most consistently associated with chloracne
(a persistent form of acne with keratotic follicles associated with
exposure to chlorinated compounds) and symptoms of liver toxicity.
For example, in April 1968, an explosion in a reactor producing
2,4,5-T3CP at a manufacturing plant in England released a
considerable amount of 2,4,5-T3CP and TCDD, exposing 14 people inside
the plant (May, 1973). Abnormalities in liver function tests (elevated
thymol turbidity, zinc turbidity), serum-transaminases, and urine
analyses were detected immediately after the incident; 10 days later,
the same tests gave normal values. Plant activity resumed but, by
December, 79 cases of chloracne were reported, some of whom also
suffered from conjunctivitis. The entire building was then thoroughly
cleaned, the interior walls resurfaced, and contaminated equipment was
buried. In a follow-up study conducted 10 years after the accident,
the frequencies of chromosomal aberrations and sister chromatid
exchange rates in lymphocyte cultures from exposed subjects were
normal (Blank et al., 1983).
To date, acute exposure of the general population to lower
chlorinated chlorophenols has been documented only from the ICMESA
plant accident in Seveso, Italy. An over-heated chemical reactor
discharged a cloud containing sodium hydroxide, Na-T3CP, and TCDD
into the atmosphere, contaminating an area south of the factory
containing 37 000 people (Hay, 1976; Del Como et al., 1982). Within 2
weeks of the accident, toxic effects were being treated in some 500
people (Hay, 1976). The most prevalent signs of exposure were skin
burns and chloracne, which was evident in 193 of the inhabitants. The
highest soil concentrations of 2,3,7,8-TCDD were associated with the
most severe cases of chloracne (Caramaschi et al., 1982). An
international steering group formed by the Italian government stated
in their final 1984 document that, with the exception of chloracne, no
clear health effects remained in the 193 persons in Seveso who were
registered as having chloracne, 20 of whom still showed symptoms in
1984. Exposed children had indications of increased enzyme activities
(increased D-glucaric acid in the urine) up to 3 years after exposure
(WHO, in press).
9.2 Long-term Exposure
9.2.1 Effects on skin and mucous membranes
Workers may display a variety of overt symptoms of chlorophenol
exposure. Persons often complain of irritations of the skin, mucous
membranes and respiratory tract as a result of direct airborne
contact. In addition, chronic skin ailments, particularly chloracne,
but also other skin lesions, ulcerations, and porphyria cutanea tarda
have been reported, mainly from plants manufacturing chlorophenols for
phenoxy-acetic acid herbicides. Clinical indications of liver damage
and haematological and neurological effects have also been reported,
particularly in association with high exposures.
For instance, among workers in a 2,4-DCP and 2,4,5-T3CP
manufacturing plant in the USA, 29 cases of chloracne and 11 cases of
porphyria were detected (Bleiberg et al., 1964). In addition to the
two chlorophenols, PCDDs and PCDFs, acetic acid, phenol,
monochloroacetic acid, and sodium hydroxide may have contributed to
the symptoms of the workers. Poland et al. (1971) examined employees
from the same plant 6 years after the report of Bleiberg et al.
(1964). Of the 73 male workers examined, 48 (66%) had some degree of
acne, and chloracne was found in 13 workers (18%). The severity of the
chloracne was not correlated with job location within the plant or
duration of employment, suggesting that there is a large variation in
the susceptibility of individuals to exposure to chlorophenols or
their contaminants.
Workers exposed to 2,4,5-T3CP in a production plant in the USSR
also developed dermatitis, as reported by Kozak et al. (1979).
Similarly, an incident of acute dermatitis in Russian agricultural
workers exposed to copper trichlorophenate was reported (Kozak et al,
1979).
Ott et al. (1980) examined the effects of exposure to commercial
2,4,5-T3CP and 2,4,5-T at a plant in the USA. In unacclimated
personnel, levels of less than 4 mg T3CP/m3 and/or 0.1 mg
2,4,5-T/m3 caused nasal irritation, sneezing, and a bitter taste in
the mouth. TCDD concentrations in both preparations were less than
1 mg/kg, in 1966, and 0.1 mg/kg, after 1972. Medical records of 204
exposed employees over the period 1950-76, did not reveal any cases of
chloracne or porphyria. The mortality rate of employees was lower than
expected (6 versus 13.3 expected) in workers exposed for less than 1
year and close to expected in those exposed for 1 year or more
(5 versus 7 expected).
In clinical studies on workers at a 2,4,5-T-manufacturing plant
(presumably exposed to 2,4,5-T3CP and its microcontaminants),
exposure was strongly associated with the development of chloracne,
plus increased prevalences of actinic elastosis and hirsutism (Suskind
& Hertzberg, 1984).
Jirasek et al. (1974) and Pazelerova et al. (1974) reported the
cases of about 80 people with both acute (industrial accident) and
long-term (up to 6 years) exposure to Na-T3CP, tetrachlorobenzene,
the sodium salt of trlchlorophenoxyacetate, and their contaminants.
Symptoms developed as long as 18 months after exposure. Chloracne
appeared in 96% of 55 people examined.
In lumber-mill workers, lesions and ulcerations were found in skin
areas in direct contact with a Na-T4CP solution, usually through
soaked clothing (Stingily, 1940). On the basis of roughly 300-400
cases, it was concluded that Na-T4CP caused the dermatitis while the
subsequent chronic lesions and nicerations were caused by fungal
infections.
Kleinman et al. (1986) and Fenske et al. (1987) evaluated the
extent and impact of occupational exposure to Permatox 100 (20.7%
2,3,4,6-T4CP, 3.1% PCP, plus substantial quantities of PCDDs and
PCDFs) in workers from a lumber mill in Washington state. The results
of their monitoring of air and urine are presented in section 5.3. In
health effects questionnaires, exposed workers complained
significantly more frequently than controls of headaches, eye and
upper respiratory irritations, and unusual sweating, though there was
no significant correlation between urinary levels and the frequency of
these symptoms. The effects of exposure to a commercial Na-T4CP
solution (containing hexa-, hepta-, and oetachlorodibenzo- p-dioxins,
dibenzofurans, and probably PCP and T3CP) were investigated by
Sterling et al. (1982) in workers at 2 sawmills in British Columbia,
Canada. The study included 1014 men with from 1 to over 20 years of
known exposure, compared with 103 loggers and outdoor municipal
workers, who served as controls. In self-administered questionnaires,
exposed workers reported significantly increased incidences of various
dermatological, upper respiratory, and general respiratory symptoms,
as well as eye irritation. These disorders were significantly more
frequent in the high-exposure group (247 workers) than in the workers
considered to be in the low/moderate-exposure group (767 workers),
who, in turn, had a higher incidence than the controls.
A detailed study of chlorophenol exposure in a sawmill in the same
geographical area was carried out by Embree et al. (1984) (section
5.3). They divided the workers into a control group from areas with no
identifiable air contaminants, a group of workers who worked in close
proximity to recently treated lumber but who did not have manual
contact (airborne), and a group who were responsible for the manual
handling of recently treated lumber (dermal plus airborne). Serum and
urine levels of chlorophenols were related to exposure in a
dose-dependent manner (section 5.3). From health histories, the only
symptoms that occurred significantly more frequently in exposed
workers were a productive cough and a reduced rate of forced
exhalation in the "airborne" group. These symptoms could not be
attributed to chlorophenol exposure, as the "dermal-plus-airborne"
group were exposed to similar atmospheric chlorophenol levels and had
higher levels of overall exposure, yet recorded a significantly lower
incidence of productive coughing.
Alexandersson & Hedenstierna (1982) examined the effects of
long-term exposure to T3CP vapours in workers at a gas-mask factory.
Trichlorophenol vapour, because of its characteristic smell, was used
at the factory for checking leaks in gas masks. Complaints of eye,
nose, and airway irritation were voiced by 7 individuals who had been
employed in testing masks for from 2 to 10 or more years. Pulmonary
function tests revealed that exposed workers displayed reduced forced
expiratory flow and increased closing volume in the lungs compared
with controls.
9.2.2 Systemic effects
Effects on liver and kidney function and haematological parameters
have also been investigated in workers exposed to chlorophenols. The
findings have been generally negative. In studies on Canadian sawmill
workers (Enarson et al., 1986), serum levels of creatinine, bilirubin,
glutamic oxaloacetic transaminase, and alkaline phosphatase, and
patient histories of jaundice, liver, kidney, and heart disease did
not differ from those of the controls. Blood-leukocyte counts and
haematocrit decreased, and urine-erythrocyte levels increased
following chlorophenate exposure. These effects were significant only
for the haematocrit and haematuria, and only for workers handling
treated lumber.
Sterling et al. (1982) reported that chlorophenol-exposed sawmill
workers filling out self-administered questionnaires reported
significantly increased incidences of gastrointestinal,
musculoskeletal, acute systemic, liver, kidney, and neurological
symptoms.
In the study on industrial workers exposed to di- and
tri-chlorophenols, Bleiberg et al. (1964) reported elevated
coproporphyrin excretion in the maintenance men that could have been
due to more intense, though sporadic, exposure. Hepatotoxic effects
were not found in the study group.
Initially, more than one-third of Czechoslovakian 2,4,5-T3CP- and
2,4,5-T-manufacturing workers described by Jirasek et al. (1974) and
Pazelerova et al. (1974) showed indications of mild liver damage,
which, in some instances, was confirmed by needle biopsy. The workers
had increased serum levels of cholesterol (56%), total lipids (67%),
and lipid-phosphorus (42%). A small but significant decrease in
serum-albumin, and an increase in serum-globulin were also found.
9.2.3 Psychological and neurological effects
A range of psychological and neurological symptoms have also been
associated with exposure to chlorophenols, often in association with
other chemicals. Workers from a plant in the USSR who were
occupationally exposed to 4-MCP complained of "... sleep disorders
(usually sleepiness and sometimes insomnia), irritability, frequent
mood changes, and rapid fatigability" (Gurova, 1964).
Similarly, Kleu & Goltz (1971) reported that 10 persons suffering
from chloracne as a result of 15 years' exposure to a T3CP
formulation complained of "... decreased sexual activity, easy
fatigability, alcohol intolerance, and loss of interest ... reduced
vital psychic and intellectual capacities combined with neurasthenia
and mental depression". The actual occupations of these individuals
were not stated.
Gilioli et al. (1983) conducted electroencephalographic analyses
of workers exposed to T3CP and TCDD at the Seveso plant in Italy,
site of an accident in 1976. These workers had both long-term, and
possibly acute high-level exposure to TCDD and T3CP. Exposed workers
generally exhibited an increased incidence of abnormal EEG tracings,
which were particularly associated with increased proportions of theta
waves, and had a slower visual reaction time than a non-exposed group.
The 2,4,5-T3CP-and 2,4,5-T-manufacturing employees reported by
Jirasek et al. (1974) and Pazelerova et al. (1974) showed neurological
abnormalities, including myographic changes in 23% of those tested.
Neurasthenic symptoms were present in 60% of the workers, compared
with 30-40% of the general population. Some of the workers complained
of fatigue, loss of appetite, weight loss, and abdominal pain.
9.2.4 Reproductive effects
The effects of chlorophenols on reproduction have been
investigated in 3 studies. Corddry (1981) provided data on pregnancy
outcomes in women married to workers from a sawmill in British
Columbia using Na-T4CP and Na-PCP. Analysis of data from 43 women,
with a total of 100 pregnancies, did not reveal any significant
differences in the pregnancy outcomes of women living with exposed men
compared with those living with unexposed men. A slight trend towards
more adverse pregnancy outcomes in the exposed group disappeared when
corrected for alcohol consumption. Male fertility was not studied.
Suskind & Hertzberg (1984) reported pregnancy outcomes in the
families of male workers manufacturing 2,4,5-T (and with probable
exposure to 2,4,5-T3CP and 2,3,7,8-T4CDD) compared with those of
other males from the same plant. There were no significant differences
between the families of exposed and non-exposed workers, but the rates
for stillbirth and death during the first 4 weeks were higher in the
families of exposed workers.
The pregnancy outcomes were surveyed in wives of workers from a
chlorophenol-manufacturing plant in Michigan, USA, potentially exposed
to PCP, 2,4,5-T3CP, and their microcontaminants for the group as a
whole (Townsend et al., 1982). There was no significant association
between exposure to dioxins (these were the focus of the study) and
adverse pregnancy outcomes. When the conceptions were divided into
subgroups according to risk factors associated with the mother, a
subgroup of 9 TCDD-exposed conceptions was identified of which 3 were
spontaneous abortions.
9.2.5 Carcinogenicity
A large number of epidemiological studies have been published
concerning human cancer outcomes following occupational exposure to
chlorophenols, phenoxy herbicides (made from and contaminated with
chlorophenols), and chlorinated dibenzo- p-dioxins and dibenzofurans
(microcontaminants found in some chlorophenols and phenoxy
herbicides). Most of these studies have been described and reviewed in
several publications by the International Agency for Research on
Cancer (IARC, 1979, 1986, 1987), and readers are referred to the IARC
monographs for details of individual studies. Only the conclusions of
these studies are given in this publication with the emphasis on
studies that specifically concern exposure of populations to
chlorophenols. Studies that address the effects of exposure to phenoxy
herbicides in general and MCPA, and do not involve substantial
concomitant exposure to chlorophenols are not discussed. These
include: cohort studies on phenoxy herbicide sprayers (Axelson et al.,
1980; Hogsted & Westerlund, 1980); studies on Vietnam war veterans
(Royal Commission on the Use and Effects of Chemical Agents on
Australian Personnel in Vietnam, 1985; Lathrop et al., 1987); and
workers involved in the manufacture of MCPA (Coggon et al., 1986).
Case-control studies covering phenoxy herbicide exposure, but with no
mention of exposure to chlorophenols (Balarajan & Acheson, 1984;
Greenwald et al., 1984; Kogan & Clapp, 1985; Hoar et al., 1986; Kang
et al., 1986; Vineis et al., 1987) and data from occupational
mortality statistics in which exposure data are inferred only from job
titles are also not included (Milham, 1982, 1985; Callagher &
Threlfall, 1984).
9.2.5.1 Case-control studies reviewed by IARC
Soft tissue sarcoma (STS) was studied in individuals exposed to
chlorophenols and other chlorophenol-based chemicals in Sweden
(Hardell & Sandstrom, 1979; Eriksson et al., 1981), New Zealand (Smith
et al., 1984), and the USA (Woods et al., 1987). Relative risks (RR)
of STS for exposure to chlorophenols alone were elevated in both of
the Swedish studies (RR = 6.6; 96% CI, 2.1-20.9; and RR = 3.3; 95%
CI, 1.3-8.1) and for one potentially exposed subgroup in the New
Zealand study (RR = 7.2; 90% CI, 1-ND). They were not elevated in the
US study, unless the subgroup of cases with Scandinavian names
(RR = 7.2; 95% CI, 2.1-24.7) and the subgroup of lumber graders
(RR = 2.6, 95% CI, 1.1-6.4) were considered separately. Chlorophenol
exposures (2,4,6-T3CP, PCP, and others) were poorly described and not
quantified. Case-control studies of malignant lymphomas (Hodgkins
disease and non-Hodgkins lymphoma) have also been carried out in
Sweden (Hardell et al., 1981), New Zealand (Pearce et al., 1986), and
the USA (Woods et al., 1987). In Sweden, low-grade exposure (up to one
week continuous or 1 month intermittent) and high-grade exposure
(greater than the low-grade exposure criteria) to unspecified types of
chlorophenols resulted in relative risk values of 2.2 (95%
CI, 1.1-4.6) and 7.6 (95% CI, 3.2-17.7), respectively. In the New
Zealand study of 83 cases of non-Hodgkins lymphoma, exposures were not
classified into high or low, and were not quantified; however, some
cases were not likely to have been exposed to 2,4,6-T3CP and PCP.
The relative risk for non-Hodgkins lymphoma in pelt department
workers with potential exposure to 2,4,6- T3CP was 1.9 (90%
CI, 0.9-4.0); however, other meat workers without exposure to
chlorophenols had a relative risk of the same order. In the US study,
the relative risks for non-Hodgkins lymphoma were not elevated for
groups exposed to low, medium, or high levels of chlorophenols.
A statistically significant increase in nasal and nasopharyngeal
cancer was found among workers exposed to chlorophenols (mainly tri-,
tetra-, and pentachlorophenol) in a Swedish case-control study
(Hardell et al., 1982). In an inter-Nordic study, 2 cases of sinonasal
cancer out of 167 were identified as having been exposed to
chlorophenols (not further specified) compared with 0/167 colorectal
cancer controls (Hernberg et al., 1983). In a Danish study, no
association was found between sinonasal cancer and potential exposure
to chlorophenols, as inferred from employment records (Olsen &
Moller-Jensen, 1984).
The results of 2 Swedish studies did not show statistically
significant associations between primary liver cancer or colon cancer,
and exposure to chlorophenols (mainly tri-, tetra-, and
pentachlorophenol) (Hardell, 1981; Hardell et al., 1984).
No significant associations were found between multiple myeloma
and potential exposure to chlorophenols (probably mainly
penta-chlorophenol) in a case-control study carried out in New Zealand
(Pearce et al., 1986).
9.2.5.2 Cohort studies reviewed by IARC
Three follow-up studies have been undertaken on small groups of
workers exposed to 2,3,7,8-TCDD during accidents in the manufacture of
2,4,5-T3CP (Cook et al., 1980; Zack & Suskind, 1980; Thiess et al.,
1982). When combined, these cohort studies show 19 cancer deaths
observed versus 14.7 expected (RR = 1.29; 95% CI, 0.78-2.02). Two
studies (Ott et al., 1980; Zack & Galley, 1983) covered workers
employed in the manufacture of 2,4,5-T from 2,4,5-T3CP. The observed
cancer deaths versus expected cancer deaths for these cohorts were 1
versus 3.6 and 35 versus 30.9, respectively.
A total of 3 deaths from soft-tissue sarcoma (STS) were identified
in US cohorts studied by Honchar & Halperin (1981). This was
equivalent to 2.9% of the deaths in the 4 cohorts, where approximately
only 0.07% would be expected. Later, one additional STS case was
observed in one of these cohorts (Cook, 1981), and an additional 3
cases of STS were reported in workers in 2,4,5-T-manufacturing plants
(Johnson et al., 1981; Moses & Selikoff, 1981). Fingerhut et al.
(1984) reviewed the histological specimens for the 7 cases reported as
having STS. In the review, 5 of the 7 cases were diagnosed as having
STS. A review of the employment records for the 7 patients showed that
all of them had worked in 2,4,5-T-manufacturing plants; 4 of the
patients had specific assignments to 2,4,5-T3CP or 2,4,5-T
departments.
A cohort study on workers in 2 Danish chemical plants (Lynge,
1985), in which 2,4-D and 2,4-DP were produced together with MCPA and
MCPP, showed the overall cancer incidence to be close to that of the
Danish population (208 observed cases and 216.5 expected). In men, 5
cases of STS were observed, where 1.84 were expected. The number of
malignant lymphoma cases in men was 7, with 5.37 expected cases. In
the subgroup of men assigned to the phenoxy herbicide-manufacturing
department, 11 lung cancer cases were observed in men, when 5.33 cases
were expected.
9.2.5.3 More recent studies
A Swedish study on soft-tissue sarcoma and exposure to phenoxy
acid herbicides and chlorophenols was recently repeated using 55 new
cases diagnosed in 1978-83 (Hardell & Eriksson, 1988). In addition to
comparing the cases with controls from the general population, a
control group of patients with cancers other than malignant lymphoma
and nasopharyngeal cancer was selected. An elevated relative risk for
exposure to phenoxy herbicides was found, but no statistically
significant differences were observed between cases and controls with
regard exposure to chlorophenols. The relative risk for chlorophenols
was based on only 4 exposed cases.
In a case-control study nested within a cohort of Finnish
woodworkers, no association was found between respiratory cancer and
exposure to chlorophenols (mainly tetra- and trichlorophenols), but
this result was based on only 3 exposed cases (Kauppinen et al.,
1986).
In a more recent cohort study on 2192 employees at a plant
involved in the production of higher chlorophenols and phenoxy acids
(Cook et al., 1987; Ott et al., 1987), mortality during the period
1940-82 among workers with potential occupational exposure to
chlorophenols was similar to that of US white males for all causes and
for all cancers; there were 81 observed cancer deaths versus 79.3
expected deaths. No statistically significant excesses of mortality
were observed for the cancers of a prior interest (nasal and
nasopharyngeal, stomach, liver, connective and soft tissue,
lymphomas). Excesses in mortality that did not reach the 5% level of
probability were reported for stomach cancer (6/3.8) and non-Hodgkins
lymphoma (5/2.6).
A cohort of 878 persons, employed in the manufacture of 2,4-D at
the same plant between 1945 and 1983, was followed up for mortality
until 1983. Some of these employees may also have been exposed to
other chlorophenols at the plant site. There were 20 cancer deaths
against 16.9 expected. There were 5 deaths from lymphatic and
haematopoietic cancer against 2.5 expected. Two cases had
lymphosarcoma and reticuiosarcoma, and there was one case of Hodgkins
disease. Both workers who died from non-Hodgkins lymphoma had had
potential exposure to PCDDs. Death certificates were reviewed with
special attention directed to soft-tissue sarcoma, but no cases were
identified (Bond et al., 1988).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of Human Health Risks
10.1.1 Exposure levels
10.1.1.1 Non-occupational exposure
Exposure to chlorophenols other than pentachlorophenol may occur
via ingestion, inhalation, or dermal absorption (section 5.2). The
general population is thought to be exposed mainly through the
ingestion of food and drinking-water. These compounds have not been
quantified in the ambient atmosphere, but atmospheric levels are
likely to be of the same order of magnitude (ng/m3) as those of PCP.
Even with 100% absorption, uptake via this route would be much less
than 1 µg/day. Similarly, even though low levels of the lower
chlorinated phenols occur widespread throughout the environment,
direct dermal contact with these compounds will not be an important
route of exposure for the general population. Non-occupational
exposure (inhalation, dermal) to chlorophenol-treated lumber has not
been investigated, but may be significant, if chlorophenols are used
for extensive treatment of the interior of houses. Significant
exposures may also occur if consumers use chlorophenol-based products
without appropriate care and protection.
An estimate of non-occupational exposure through the ingestion of
drinking-water and food can be made using representative published
concentrations of the major commercial chlorophenols other than
pentachlorophenol. The daily exposure of a 60-kg person in Canada to
2,4-DCP, 2,4,5-T3CP, and 2,3,4,6-T4CP is estimated in Table 19, on
the basis of assumptions detailed in the table. These calculated
exposures may be overestimates in that residue determinations are
generally taken from contaminated areas, and because kinetic or
metabolic studies usually involve high chlorophenol concentrations. On
the other hand, estimates have not been made for all foodstuffs; for
example, no data were found for fruits, dairy products, or nuts.
Exposure to other chlorophenols, in particular 2,4,6-T3CP, cannot be
estimated, because there are insufficient data on the concentrations
present in water and food. Nevertheless, the values calculated should
suffice as a first approximation of exposure.
Table 19. Estimated daily per capita exposure to 2,4-DCP, 2,4,5-T3CP, and 2,3,4,6-T4CP from food and drinking-water in Canada
Source Daily 2,4-DCP 2,4,5-T3CP 2,3,4,6-T4CP
consumption
in Canada Concentration Exposure Concentration Exposure Concentration Exposure
(kg)a (µg/kg fresh (µg/kg (µg/kg fresh (µg/kg (µg/kg (µg/kg
weight) body weight weight) body weight fresh weight) body weight
per day)b per day) per day)
Tap 1.4 0.093c 0.002 0.035c 0.0008 0.009c 0.0002
water (litres)d
Vegetables
and
potatoes 0.36 6e 0.036 0.041f 0.0002 1.917g 0.011
Meat and
poultry 0.26 5h 0.022 0.034f 0.0002 3i 0.013
Fish and
seafood 0.019 13.75j 0.004 0.76k 0.0002 10.3k 0.003
Total
exposure 0.064 0.0014 0.027
Table 19. (contd).
Source Daily 2,4-DCP 2,4,5-T3CP 2,3,4,6-T4CP
consumption
in Canada Concentration Exposure Concentration Exposure Concentration Exposure
(kg)a (µg/kg fresh (µg/kg (µg/kg fresh (µg/kg (µg/kg (µg/kg
weight) body weight weight) body weight fresh weight) body weight
per day)b per day) per day)
Tolerable 200.0l 100.0m 10.0n
daily intake
(µg/kg body
weight per
day)
a Per capita per day in 1984 (Statistics Canada, 1986).
b Based on a 60-kg person.
c Jyvaskyla, Finland (Paasivirta et al., 1985).
d Water consumption for 60 kg man per day (NHW, 1983).
e Average for potatoes treated with 2,4-D (Bristol et al., 1982).
f Assuming that the ratio of 2,4-DCP: 2,4,5-T3CP is the same as for 1984 Canadian imports.
g Average from Alberta Government assay (Jones, 1981), assuming all T4CP is 2,3,4,6-T4CP.
h Muscle concentration from chickens fed 50 mg Nemacide/kg, 7 days post-treatment (Sherman et al., 1972).
i Upper concentration detected in chicken flesh from British shops (Farrington & Munday, 1976).
j Mean muscle concentration calculated as µg/kg wet weight Bacon, (1978).
k Mean muscle concentration (Paasivirta et al., 1985).
l NOEL taken from Kobayashi et al. (1972) using a 500-fold safety factor.
m NOEL taken from McCollister et al. (1961) using a 1000-fold safety factor.
n NOEL taken from Schweiz et al. (1974) using a 1000-fold safety factor.
The daily ingestion of these chlorophenols by a 60-kg person is
estimated to be 3.84 µg of 2,4-DCP; 0.084 µg of 2,4,5-T3CP, and
1.62 µg of 2,3,4,6-T4CP (on the basis of data in Table 19 and
calculated for a 60-kg person). These values are presented on a
per-unit-weight basis in Table 19. In Canada, the amount of PCP used
is approximately equal to the amounts of all other chlorophenols
combined, suggesting that PCP exposure is roughly equal to that of the
other chlorophenols. Thus, the total estimated exposure of Canadians
to all chlorophenols including PCP is about 10 µg/person per day. This
value agrees with the 10-30 µg/ person per day estimated by NRCC
(1982) and the 12.7 µg/person per day estimated by NHW (1988) (section
5.2) for the general population in Canada. The total estimated
exposure levels in other countries may differ from this value,
depending on product use patterns, food and water consumption, and
levels of environmental contamination.
Although the above estimate of exposure is based on limited
information on residue levels and uptake rates, it is supported by the
low levels of chlorophenols found in human tissues and fluids; in the
few studies available, µg/kg quantities have been detected in human
tissues and fluids (section 5.2).
10.1.1.2 Occupational exposure
The potential exposure to chlorophenols may be significant for
certain workers employed in the lumber industry, pesticide manufacture
and use, use of treated wood for construction, railroad ties, or
telephone poles, and a variety of other industries in which
chlorophenols are used as biocides. In the work place, exposure would
be mainly through dermal absorption and inhalation; ingestion of the
compounds is more likely to occur if eating, drinking, and smoking are
allowed in the work area, or if proper cleansing procedures are not
practised. The air in work areas where chlorophenols are used contains
elevated concentrations of chlorophenols. Concentrations of 14 mg/m3
have been reported in such work areas, but typical concentrations were
1-3 orders of magnitude below this in work places studied recently in
North America and Scandinavia (section 5.3). Persons working in high
exposure areas have elevated levels of chlorophenols in their body
fluids, particularly if their job combines dermal and inhalation
exposure to chlorophenols. Urine levels of up to 49 mg T4CP/litre
have been reported (section 5.3).
Few data are available with which to model uptake and excretion of
chlorophenols other than PCP. However, preliminary estimates of some
occupational exposures can be derived from recent reports of
concentrations in the urine of workers in the sawmill industry, where
handling of treated wood and the proximity of workers to the
open-treatment apparatus can result in relatively high uptake of
chlorophenols.
Urinary concentrations of employees at Finnish, American, and
Canadian sawmills have been compiled in Table 20, and have been used
to estimate worker exposure in this industry. These estimates are
based on two principal assumptions. First, it was assumed that urinary
concentrations of T4CP reach a sustained level following long-term
exposure, since Braun et al. (1979) calculated that PCP in the urine
of exposed persons reached a fairly constant level within one week.
Second, it was assumed that all T4CP is cleared in the urine, because
it is known that human beings clear 86% of administered PCP in urine
(Braun et al., 1979) and that rats clear more than 95% of single doses
of 2,3,4,6-T4CP and 2,3,5,6-T4CP (the major tetrachlorophenols in
commercial preparations) via the urine (Ahlborg & Larsson, 1978).
On the basis of these assumptions, the urinary concentrations in
Table 20, and a urine production of 1.4 litres daily, a 60-kg sawmill
worker is estimated to take up from 2 to 42 µg/kg per day on average
(Table 20). Estimates of the exposure of workers with the highest
urine concentrations from each study, who presumably had extensive
dermal uptake of chlorophenates, ranged from 53 to 1142 µg/kg per day.
A more comprehensive knowledge of chlorophenol levels and fluxes and
their dynamics would be necessary for a better estimate.
As expected, the estimated occupational exposure is much higher
(often 2 or more orders of magnitude) than that calculated for members
of the general population. This difference is confirmed by levels of
chlorophenols measured in the biological tissues and fluids of
workers, and the ambient atmosphere (sections 5.2, 5.3), as well as
the association of intoxications and adverse symptoms with
occupational exposure to chlorophenols (section 9).
Since the foregoing calculations are based on only 5 studies, all
involving sawmills, these estimates are not directly applicable to
other occupations and/or geographical regions.
10.1.2 Toxic effects
Acute lethal doses of lower chlorophenols in experimental mammals
(section 8.1) are associated with restlessness, hyperpyrexia, rapid
respiratory rates, at axia, and eventually dyspnoea, coma, and death.
MCPs, DCPs, and 2,4,6-T3CP are convulsive agents, while the other
trichlorophenols and tetrachlorophenols are not. All of the
chlorophenols are irritating or corrosive to the skin, eyes, and
mucuous membranes (section 8.2).
Table 20. Estimates of occupational exposure to tetrachlorophenols (based on urinary concentrations from recent studies
on exposed workers)
Situation Estimated intake Reference
Urinary concentrations (µg/litre) µg/kg body weight per day)a
Median/mean Maximum Averageb Maximum
Finnish sawmill; Dermal exposure primarily Dermal exposure Lindroos et al. (1987)
workers exposed to 1809.4c (median) 48 924.6 42 1142
KY-S (NaT4CP)
formulation; 1980-81 Respiratory exposure primarily Respiratory exposure
208.8c (median) 3085.3 5 72
Finnish sawmill; 2841.5d,e (mean) 39 436.6d,e 66 920 Kauppinen & Lindroos (1985)
workers exposed to (1985)
NaT4CP
Canadian (British 423f (mean) 1479f 10 35 Currie & McDonald (1986)
Columbia) planer (1986)
mill; workers
exposed to NaT4CP;
1985
Washington state (USA) 160.4g (mean) 2255 4 53 Kleinman et al. (1986)
sawmill; workers
exposed primarily to
NaT4CP; 1981-82
Table 20. (contd).
Situation Estimated intake Reference
Urinary concentrations (µg/litre) µg/kg body weight per day)a
Median/mean Maximum Averageb Maximum
Canadian (British Dermal + airborne exposure Dermal + airborne exposure Embree et al. (1984)
Columbia) sawmill; 1250e (mean) 29
workers; exposed to
Na-T4CP; 1978-79 Airborne only Airborne only
930e 22
a For a 60-kg worker.
b Based on mean concentration, except for estimate from Lindroos et al. (1987), which is based on the median.
c Assumes all chlorophenols are T4CP, which predominates in these formulations.
d Not clear if sample hydrolysed.
e Assumes free concentrations are 10% of total concentrations.
f From samples collected on final morning of a 5-day work week.
g Grand mean for exposed workers, all dates.
Note: No correction for density has been made in any table entries.
Short-term exposure of experimental animals to 2,4-DCP,
2,4,5-T3CP, 2,4,6-T3CP, or 2,3,4,6-T4CP produces moderate adverse
effects on the liver, kidney, and spleen, while 2,4-DCP is also
haematotoxic and immunotoxic (section 8.2). 2-MCP, 2,4-DCP,
2,4,5-T3CP, 2,4,6-T3CP, and 2,3,4,6-T4CP produce fetotoxic or
embryo-toxic effects, but none of the chlorophenols tested to date has
produced teratogenic effects. Isomers of DCP other than 2,4-DCP may
reduce male fertility (section 8.5).
2,4,6-T3CP and 2,3,4,6-T4CP appear to have some mutagenic
capability; however, chlorophenols do not appear to be potent mutagens
capable of exerting significant genotoxic effects (section 8.6). There
is sufficient evidence that 2,4,6-T3CP (commercial grade) is an
animal carcinogen. However, the trichlorophenol used in this study was
not analysed for PCDD and PCDF microcontaminants. 2,4-DCP (more than
99% pure) was found not to be carcinogenic in mice and rats. The other
lower chlorophenols have not been adequately tested for their
carcinogenic properties. Some chlorophenols appear to be tumour
promoters; others do not (section 8.6).
The microcontaminants in the commercial grades of the more highly
chlorinated phenols can lead to toxic effects even more severe than
those produced by the chlorophenol itself (sections 6.3, 8.9). Their
possible significance in terms of the carcinogenicity of 2,4,6-T3CP
should not be overlooked.
Toxic effects in man from acute exposure to high concentrations of
the lower chlorinated phenols include acute and chronic skin
irritation, chloracne, respiratory disorders, recurring headaches,
dizziness, nausea, vomiting, loss of coordination, tremor, weakness,
and lethargy (section 9.1).
The question of carcinogenicity in man from exposure to
chlorophenols is a matter of controversy. IARC concluded that there is
limited evidence of carcinogenicity from occupational exposure to
chlorophenols. Following a review of studies published since the IARC
evaluation, the Task Group still finds this conclusion appropriate.
10.1.3 Risk Evaluation
The information available to date indicates that the general
population is exposed to low levels of chlorinated phenols. As derived
in section 10.1, the estimated exposure to the major chlorophenols
other than PCP of a person who does not work with these compounds is
0.0833 µg/kg per day. This burden is made up of 0.058 µg 2,4-DCP/kg
per day, 0.0013 µg 2,4,5-T3CP/kg per day, and 0.024 µg
2,3,4,6-T4CP/kg per day. People who manufacture or apply
chlorophenols predictably experience much higher levels of exposure.
As an example, the total estimated average exposures to T4CP for
sawmill workers ranged from 2 to 42 µg/kg per day (section 10.1.1.2).
For comparison with these estimated exposures, Tolerable Daily
Intake (TDI) levels have been calculated (from no-observed-effect
levels determined in short- term studies) of 100 mg/kg body weight for
2,4-DCP (mice, in feed) (Kobayashi et al., 1972), 100 mg/kg body
weight for 2,4,5-T3CP (rats, oral) (McCollister et al., 1961), and
10 mg/kg body weight for 2,3,4,6-T4CP (rats, oral) (Schwetz et al.,
1974). Using an uncertainty factor of 1000 for 2,4,5- T3CP and
2,3,4,6-T4CP, because of the lack of long-term animal data, and an
uncertainty factor of 500 for 2,4-DCP, because of the availability of
long-term data (WHO, 1986), the TDI values for 2,4-DCP, 2,4,5-T3CP,
and 2,3,4,6-T4CP were estimated to be 200, 100, and 10 µg/kg per day,
respectively. The long-term carcinogenicity study available for
2,4-DCP (section 8.7) does not provide data that would alter this
estimate. The embryotoxicity data available for 2,4,5- T3CP (section
8.5) were found to be of too limited significance to be taken into
consideration.
Non-occupational exposure levels are usually well below these
values, indicating that the anticipated health hazards for the general
population from exposure to chlorophenols other than PCP are minimal.
The estimated occupational intakes are considerably higher, especially
when there is skin contact with chlorophenols (Table 20). Exposure
levels in sawmills may, in some cases, exceed the TDI values.
If tetra- or trichlorophenol preparations are used in wood
protection, workers can also be exposed to 2,4,6-T3CP. There is
sufficient evidence that 2,4,6-T3CP is carcinogenic for mice and
rats. Because of the arbitrary nature of assumptions inherent in
developing estimates of exposure, and extrapolating from effects on
animals to effects on human beings, risk assessments are never
precise. Nevertheless, it is prudent to ensure that human exposure to
2,4,6-T3CP is kept to a minimum.
Acute exposure to high concentrations of chlorophenol formulations
can be a significant hazard for the health of workers involved in the
production or use of chlorophenols. While no deaths have been reported
with exposure to chlorophenols other than PCP, fatalities due to high
exposure to the latter are well documented. Exposures to non-PCP
chlorophenols result in adverse signs and symptoms similar to those
caused by PCP. Exposure to commercial formulations of chlorinated
phenols has been associated with an increased relative risk of
soft-tissue sarcomas, lyrephotons, and nasal and nasopharyngeal
cancers in some studies; such associations have not been found in
other similar studies.
On the basis of toxicological effects and current exposure levels,
there does not appear to be any strong reason for eliminating all use
of chlorophenols. Furthermore, the need to reduce the levels of
exposure to chlorophenols appears minimal as long as the necessary
precautions to prevent high-level dermal and respiratory uptake are
observed. Exposure of the general population is much lower, and in the
absence of release from an industrial accident, the overall risk from
sustained non-occupational exposure is probably negligible.
Chlorophenols produce undesirable organoleptic effects at very low
concentrations. Contamination of the environment and of drinking-water
with chlorophenols above the threshold for organoleptic effects is
therefore unacceptable. Comprehensive monitoring of chlorophenol
levels, sources, and fluxes is essential to characterize both
occupational and non-occupational exposure to these compounds, and to
alert the responsible agency to potentially hazardous exposures where
they exist.
Microcontaminants, in particular PCDDs and PCDFs, found in
commercial formulations of tri-, tetra-, and pentachlorophenol, are
probably the causal agents for chloracne in human beings. PCDD and
PCDF levels in commercial preparations should be kept as low as
technically feasible. Care should also be taken to minimize their
formation during the incineration of wastes containing chlorophenols.
10.2 Evaluation of Effects on the Environment
10.2.1 Levels of exposure
Data on levels of chlorophenol residues other than PCP in the
environment are limited primarily to aquatic habitats, and indicate
that chlorophenol contamination is widespread in these systems. This
contamination may result from either the use or the formation of
chlorophenols, e.g., in the chlorine-bleaching process in pulp and
paper-mills (sections 3.2, 3.3, 3.4). Where data are not available on
levels of other chlorophenols in the environment and for evaluation
purposes, examples of monitoring data on PCP are used. On the basis of
relative rates of degradation and use patterns, it is likely that, in
general, environmental levels of other chlorophenols would be lower
than those found for PCP.
Ambient levels of PCP in air are less than 1 ng/m3 in
uncontaminated areas, while concentrations of several ng/m3 have been
detected in residential areas. Other chlorophenols may well be present
at comparable levels, but confirmatory data are lacking.
Residues of all chlorophenol congeners have been found in fresh
and marine waters (section 5.1.2). In Canada, concentrations are often
undetectable at the ng/litre level in receiving waters, and only
occasionally exceed 1 µg/litre; these higher levels are only observed
in close proximity to industrial sources of chlorophenols,
particularly pulp and paper-mills. Ambient levels are higher in waters
in the industrialized areas of Europe, but median concentrations still
do not exceed 1 µg/litre, and maximum concentrations in surface waters
and groundwaters only reach several µg/litre in heavily industrialized
regions. Levels of particular chlorophenols in industrial effluents
can reach several thousand µg/litre (section 5.1.2), but dilution
apparently reduces these to the low ambient levels observed.
Chlorophenol concentrations in sediments are usually higher than
those in the overlying water (section 5.1.2), as a result of
adsorption and low rates of anaerobic degradation. Water bodies not
receiving large chlorophenol inputs generally contain less than 1 µg
of chlorophenol congeners per kg dry sediment. Typical levels of all
chlorophenol congeners in fresh-water sediments of industrialized
regions are less than 50 µg/kg of dry sediment. In some instances,
hundreds or thousands of µg/kg have been detected from sites adjacent
to spills or discharges.
Soils may contain significant quantities of chlorophenols,
particularly at timber preservation facilities, or where phenoxy
herbicides have been applied. Levels as high as 70 mg chlorophenols/kg
were detected in soils from Finnish sawmills (section 5.1.3), but
ambient levels in soil were found to be much lower (< 0.1 mg/kg).
10.2.2 Transport
While chlorophenols are considered to be mainly water and soil
contaminants, some atmospheric movement also occurs. PCP has been
detected in rain, snow, and in the air (section 5.1.1), and presumably
other chlorophenols are also transported in this manner. Adsorption
controls chlorophenol transport in acidic or organic soils, but is
much less important in basic or mineral soils (section 4.1.2.1). In
surface waters, the fraction that is not degraded is incorporated into
the sediments, most likely through adsorption on sedimenting
particulates (section 4.1.3). While much of the chlorophenols entering
natural waters are probably degraded by photolysis or microorganisms,
they are moderately soluble and fairly persistent, and so can be
transported considerable distances by water (section 4.1.3).
10.2.3 Degradation
Both abiotic and biotic degradation eliminate chlorophenols from
the environment. Numerous in vitro studies have shown that
ultraviolet radiation can rapidly break down chlorophenols. Evidence
suggests that photolysis is important in natural surface waters
(section 4.2.1.1), and presumably in other exposed habitats. A large
number of microorganisms from different habitats are able to degrade
chlorophenols in laboratory cultures. In some instances, quantities as
high as tens of mg/litre are eliminated in a matter of hours or days
(section 4.2.1.2), though it is necessary to acclimate them first.
Degradation is generally slowest for the higher chlorinated congeners
or those with a chlorine atom in the meta position. In general,
anaerobic biodegradation of these compounds, if it occurs at all, is
much slower than aerobic metabolism. Some evidence suggests that
biodegradation is faster in soil than in sediments, and slower in
stream waters.
10.2.4 Bioaccumulation
Bioaccumulation of chlorophenols appears moderate, and most
bioconcentration factors (BCFs) fall between 100 and 1000.
Bioconcentration is usually a positive function of chlorine number.
There are no obvious patterns in BCF in relation to the type of
organism (for algae, plants, invertebrates, and fish). Once exposure
is discontinued, chlorophenols clear rapidly from biota, indicating
that the bioaccumulation observed in field studies is the result of
long-term exposure rather than persistence.
10.2.5 Persistence
Chlorophenols should only persist in the environment where the
rates of the various degradative processes are minor. Indeed, residues
in sediments, where photolysis and apparently microbial degradation
are minimal, have been estimated to be decades old (section 4.4).
Herbicidal applications and spills of PCP in soils reportedly
disappear in a matter of weeks or months.
10.2.6 Toxic effects on environmental organisms
Considerable overlap exists in the chlorophenol concentrations
that are toxic for bacteria, phytoplankton, plants, invertebrates, and
fish. Most of the LC50 and EC50 values for these organisms, which to
date have been primarily aquatic, fall within the several mg/litre
range (Table 16). Toxicity generally increases with the degree of
chlorination of the phenol ring, though chlorophenols with chlorines
in the 2 and 6 positions are often less toxic than expected on the
basis of the number of chlorines. Particularly in the case of the
higher chlorophenols, acute toxicity is a strong inverse function of
pH, as the phenol form of the compound is more toxic than the ionized
form. Exposure to chlorophenols affects a wide variety of processes in
environmental organisms (section 7) (Table 16).
In controlled field studies on aquatic ecosystems, exposure to
high concentrations (100-5000 mg/litre) of lower chlorophenols
generally impairs algal production and reproduction, alters the algal
species composition dramatically, and reduces zooplankton biomass and
production. The low levels of chlorophenols present in moderately
contaminated waters have been reported to impair the flavour of fish.
There is very little information on the toxic effects exerted by
concentrations similar to ambient levels.
10.2.7 Risk evaluation
The information available to assess the environmental hazards
presented by chlorophenols other than PCP is deficient in at least 2
respects: (a) knowledge of the quantities of chlorophenols entering
the environment, and of their subsequent dynamics, is insufficient for
all chlorophenols other than PCP; and (b) not enough toxicology
studies using concentrations characteristic of the environment have
been conducted. As a result, it is not yet possible to predict
quantitatively the environmental impact of the widespread low-level
contamination that has recently become apparent (section 5).
However, it is possible to get a first approximation of the hazard
presented by a given chlorophenol, by comparing the levels that
produce toxic effects on test organisms in vitro with the residue
concentrations that have been measured in the environment. The
information for such a comparison is presented in Table 21, using data
for aquatic organisms and environments. It is necessary to restrict
the data in this manner because the vast majority of studies on the
environmental toxicity of chlorophenols have been on aquatic test
systems.
Furthermore, this focus is appropriate because many producers and
users of chlorophenols still discharge them as wastes into water
bodies (section 3.2.3).
The environmental data in Table 21 include measured levels for
each chlorophenol in water, sediment, and effluent, since these are
the environmental sources most readily comparable with the results of
toxicity studies. The latter data are taken from the laboratory
toxicity studies outlined in section 5.1.2 and include the
concentrations that cause toxic effects, the no-observed-effect
levels, and those showing organoleptic effects in fish and
drinking-water. In order to ensure that the comparison of
environmental concentrations and toxic levels provides a margin of
safety, the environmental data used for evaluation are the maximum
values reported in highly-contaminated industrialized waters, while
the levels producing toxic effects are the minimum levels reported to
cause effects from a large number of studies compiled by Buikema et
al. (1979), and Jones (1981, 1984). Table 21 is designed so that
comparisons can be made across the rows; because of the diversity of
test conditions, organisms, and response variables, the data on the
toxicity of the individual compounds are not directly comparable.
In virtually all instances, the maximum ambient levels in water
and sediments are orders of magnitude below the lowest concentrations
that are toxic for aquatic organisms: effects typically occur in the
mg/litre range, while environmental levels are generally in the
µg/litre range. On this basis, it appears that the ambient
chlorophenol levels measured in aquatic environments are unlikely to
have adverse effects on the ecosystems receiving them, except in the
case of accidental spills, high-concentration, point-source
discharges, or in the immediate vicinity of manufacturers' undiluted
waste streams. However, the elevated concentrations found in some
industrialized regions, or in habitats adjacent to discharges, can
compromise the flavour and/or smell of drinking-water and fish.
It is advisable to control chlorophenol discharges into the
environment at levels that would not increase the present
environmental concentrations, in view of the taste and odour effects
of chlorophenols and the lack of data on the long-term effects on
ecosystems from the present low levels of chlorophenols detected.
Furthermore, the levels of such microcontaminants as PCDDs and PCDFs
in technical formulations of T3CP and T4CP should be reduced as much
as possible, in order to decrease the levels of such toxic chemicals
released into the environment.
Table 21. Maximum reported water, sediment, and effluent levels, contrasted with minimum levels producing toxic and organoleptic
effects and no-observed-effect levels
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2-MCP 2.3m 4p 2600 Daphnia 1000 Daphnia 60
magna magna
48-h LC50b survivalb
3-MCP 6.0m 43 6p 1800 Unidentified 10000 Chlorella 0.1 25
fish pyrenoidosa
24-h TLmd growthc
4-MCP 3.9m 150q 4100 Daphnia 500 Estuarine 0.1 45
magnab phytoplankton
48-h LC50 growthe
2,3-DCP 0.72m 2.2 0.04 84
Table 21. (contd).
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2,4-DCP 0.59m 10 3304 100 Crayfish 0.3-8.0 1.0
elevation of
blood-glucosef
2,5-DCP 0.29m 11 0.5 23
2,6-DCP 0.45m 31 2204 4000 Salmo trutta 0.2-2.0 35
24-h LC50g
3,4-DCP 0.23m 49 5000 Fishhdeath 0.3
3hh
3,5-DCP 0.52m 12 1500 Shrimp
96-h lethal
thresholdi
2,3,4-T3CP 0.04n 0.8 3.64 2000 Shrimp
96-h lethal
thresholdi
2,3,5-T3CP 0.28n 11 800 Salmo truttag
24-h LC50
Table 21. (contd).
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2,3,6-T3CP 0.36n 2700 Shrimp 0.5
96-h lethal
thresholdl
2,4,5-T3CP 0.66m 15 2400q 640 Palaemonetes 100 Rainbow trout 1.0 52
puglo
96-h LC50j
2,4,6-T3CP 2.5m 3.7 3120q .0.5 Guppy < 410 Daphnia 2.0
fecundity magna
offspring survival
survival
2,4,6-T3CP 2.5m 3.7 3120q > 100- Fathead minnow
< 1000 96-h TLm
Table 21. (contd).
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2,3,4,5-T4CP 0.02n 8.9 12q < 300 Grass shrimp
EC50-limb
regenerationj
2,3,4,6-T4CP 3r 4.9 2100q 290 Daphnia 10 Daphnia 1.0
magna magnab
48-h LC50b survival
2,3,5,6-T4CP 5r 2.8 35 570 Daphnia 10 Daphnia
magna magna
48-h LC50b survivalb
a Organoleptic threshold is for drinking-water (US EPA, 1980c) and ambient water that leads to the tainting of fish
flesh (Shumway & Palensky, 1973).
b LeBlanc (1980). j Rao et al. (1981).
c Huang & Gloyna (1968). k Virtanen & Hattula (1982).
d Ingols et al. (1966). l Barnhart & Campbell (1972).
e Erickson & Hawkins (1980). m Wegman & Hofstee (1979).
f Telford (1974). n Wegman & van den Broek (1983).
g Hattula et al. (1981b). p Jolley et al. (1975).
h Jones (1981). q Garrett (1980).
i McLeese et al. (1979). r Zoeteman et al. (1981).
11. RECOMMENDATIONS
11.1 Production
(a) The concentrations of microcontaminants in chlorophenols and
products derived from them should be determined and specified.
(b) Levels of PCDDs and PCDFs in chlorophenols and related products
should be kept as low as is technically possible.
(c) Since data on the quantities of chlorophenols produced and
consumed are not available for most countries, international agencies
should seek the assistance of industry to compile such data in
different countries.
11.2 Disposal
(a) Disposal of chlorophenols and chlorophenol-contaminated waste
should be carried out in a manner that minimizes their release into
the environment. Contaminated waste waters should undergo primary and
secondary treatment. Chlorophenols should only be incinerated at high
temperatures and under strictly controlled conditions.
(b) Contamination of surface and ground waters with chlorophenols
arising as a result of industrial chlorination processes or waste
treatments using chlorine should be avoided as far as is technically
feasible.
11.3 Occupational Exposure
(a) Work-place exposure to chlorophenols should be minimized, and
absorption of these compounds through the dermal and inhalation routes
prevented, by:
-- enclosure and automation of industrial processes that use
chlorophenols;
-- adequate ventilation of the work area;
-- provision of appropriate protective clothing for employees working
with chlorophenols;
-- provision of proper washing and laundry facilities;
-- instruction of workers in the safe use and handling of
chlorophenols, the importance of personal hygiene (washing before
eating or smoking, showering before leaving work, and daily
laundering of clothing), and the application of proper emergency
procedures;
-- provision of eating and rest areas in the work place that are
isolated from potential chlorophenol contamination.
(b) The effectiveness of measures to reduce occupational exposure
should be surveyed by monitoring both the work-place air and the urine
of the workers.
11.4 General Population Exposure
(a) The availability and use of consumer products containing
chlorophenols should be reduced wherever practicable.
(b) Products containing chlorophenols should be clearly labelled by
the manufacturer to alert the consumer to their toxicity and to
instruct consumers in the safe use and handling of these products.
(c) The use of tri- and tetrachlorophenols for wood preservation
should be avoided where such wood is to be used:
-- for shipping or storing food;
-- for the retention of soil on which food may be grown;
-- for animal housing or bedding on farms.
11.5 Recommendations for Future Research
11.5.1 Environmental Aspects
Given the continued release of chlorophenols into the environment,
research is needed to study:
(a) the transport and distribution of chlorophenols in the
environment;
(b) the effects of long-term exposure to chlorophenols on both
aquatic and terrestrial organisms at concentrations typical of the
environment;
(c) the suitability of controlled landfill sites for the disposal of
chlorophenols and related wastes;
(d) means of reducing the contribution of industrial and municipal
chlorination to the overall environmental releases of chlorophenols.
11.5.2 Toxicology
The toxicology database for chlorophenols other than PCP has major
deficiencies, particularly for tetrachlorophenols. A more accurate
estimate of the risk posed by these chemicals necessitates research
into:
(a) uptake, distribution, metabolism, and excretion of
chlorophenols, especially in man;
(b) further study of the effects of chlorophenols on reproduction;
(c) the in vivo genotoxic potential of chlorophenols;
(d) long-term carcinogenicity studies with pure and technical grade
2,4,5-T3CP, 2,4,6-T3CP, and 2,3,4,6-T4CP;
(e) the cancer-promoting potential of chlorophenols;
(f) the mechanism of toxicity of chlorophenols at the molecular
level;
(g) the extent to which the toxic effects exerted by technical grade
chlorophenols are attributable to microcontaminants;
(h) the contribution made by the biotransformation of other
chlorinated compounds (e.g., hexachlorobenzene) to the human body
burden of chlorophenols.
11.5.3 Epidemiology
(a) Epidemiological investigations of previously-studied cohorts
should be followed-up and updated.
(b) Studies on new groups of workers exposed specifically to
chlorophenols, i.e., sawmill employees working with these compounds,
should be conducted. End-points studied should not only be cancers,
but should also include pulmonary, reproductive, and other effects.
(c) The higher chlorinated PCDDs and PCDFs, which occur as
contaminants in several chlorophenols, have long half-lives in human
beings and can therefore be used as indicators of exposure to the
higher chlorophenols. Since the present contradictory results from
epidemiological studies may, in part, be because of inaccurate
information on exposure to CPs, the potential use of PCDD and PCDF
concentrations in human tissues and fluids as markers of previous
exposure to these chlorophenols should be investigated.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
A guideline value of 10 µg/litre was recommended by WHO (WHO,
1984) for 2,4,6-trichlorophenol in drinking-water, based on animal
carcinogenicity data using a conservative mathematical model. In the
supporting documentation for this guideline value (WHO, 1985), it was
noted that the taste threshold level for 2,4,6-T3CP was 1.0 µg/litre
and that, on the basis of aesthetic qualities, the level would be
0.1 µg/litre. Guideline values for other individual CPs were not set,
but the odour threshold concentration of 0.1 µg/litre was considered
appropriate for chlorophenols other than pentachlorophenol.
The carcinogenicity of 2,4,5- and 2,4,6-T3CP has been evaluated
by the International Agency for Research on Cancer (IARC, 1979, 1986).
It was concluded that: there was sufficient evidence for
carcinogenicity in animals for 2,4,6-T3CP and inadequate data for the
assessment of the carcinogenicity of 2,4,5-T3CP in animals. IARC
(1986) also concluded that there was limited evidence for the
carcinogenicity of occupational exposures to all chlorophenols for
human beings. Although not stated directly in the IARC monographs,
occupational exposures were primarily to T3CP and T4CP formulations.
Regulatory standards for chlorophenols established by national
bodies in different countries and the EEC are summarized in the Legal
File of the International Register of Potentially Toxic Chemicals
(IRPTC, 1986).
REFERENCES
ABRAHAMSSON, K. & XIE, T.M. (1983) Direct determination of trace
amounts of chlorophenols in fresh water, waste water and sea
water. J. Chromatogr., 279: 199-208.
AGRICULTURE CANADA (1987) Pentachlorophenol wood preservative,
Ottawa, Pesticides Directorate, Agriculture Canada (Discussion
Document No. 87-02).
AHEL, M., GIGER, W., MOLNAR-KUBICA, E., & SCHAFFNER, C. (1984) Organic
micropollutants in surface waters of the Glatt Valley,
Switzerland. In: Angeletti, G. & Bjorseth, A., ed. Analysis of
organic micropollutants in water. Proceedings of the 3rd European
Symposium, Oslo, 19-21 September 1983, Luxembourg, Commission of
the European Communities, pp. 280-288.
AHLBORG, U.G. & LARSSON, K. (1978) Metabolism of tetrachlorophenols in
rat. Arch. Toxicol., 40: 63-74.
AHLBORG, U.G. & THUNBERG, T. (1978) Effects of 2,3,7,8-tetra-
chlorodibenzo- p-dioxin on the in vivo and in vitro
dechlorination of pentachlorophenol. Arch. Toxicol., 40: 55-61.
AHLBORG, U.G. & THUNBERG, T.M. (1980) Chlorinated phenols: occurrence,
toxicity, metabolism, and environmental impact. CRC Crit. Rev.
Toxicol., 7(1): 1-35.
AKISADA, T. (1964) Simultaneous determination of pentachlorophenol and
tetrachlorophenol in air and urine. Jpn. Anal., 14: 101-105.
ALEXANDER, M. & ALEEM, M.I.H. (1961) Effect of chemical structure on
microbial degradation of aromatic herbicide. J. agric. food
Chem., 9(1): 44-47.
ALEXANDERSSON, R. & HEDENSTIERNA, G. (1982) Pulmonary function after
long-term exposure to trichlorophenol. Int. Arch. occup. environ.
Health, 49: 275-280.
ester derivatives in natural surface waters. J. agric. food Chem.,
12(6): 541-546.
ANGEL, A. & ROGERS, K.J. (1972) An analysis of the convulsant activity
of substituted benzenes in the mouse. Toxicol. appl. Pharmacol.,
21(2): 214-229.
APPLEGATE, V.L., HOWELL, J.H., HALL, A.E., Jr, & SMITH, M.A. (1957)
Toxicity of 4,346 chemicals to larval lampreys and fishes,
Washington, DC, US Department of the Interior, Fish and Wildlife
Service, 157 pp (Special Report, Fish No. 207).
ARRHENIUS, E., RENBERG, L., JOHANSSON, L., & ZETTERQVIST, M.-A. (1977)
Disturbance of microsomal detoxication mechanisms in liver by
chlorophenol pesticides. Chem.-biol. Interact., 18: 35-46.
ARSENAULT, R.D. (1976) Pentachlorophenol and contained chlorinated
dibenzodioxins in the environment. A study of environmental fate,
stability, and significance when used in wood preservation.
Am. Wood-Preserv. Assoc., 20: 122-148.
AXELSON, O., SUNDELL, L., ANDERSON, K., EDLING, C., & KLING, H. (1980)
Herbicide exposure and tumour mortality. Scand. J. Work Environ.
Health, 6: 73-79.
BABICH, H. & DAVIS, D.L. (1981) Phenol: a review of environmental and
health risks. Reg. Toxicol. Pharmacol., 1: 90-109.
BACON, G.B. (1978) Bioaccumulation of toxic compounds in pulpmill
effluents by aquatic organisms in receiving waters, Ottawa,
Environment Canada (Annual Report CPAR Project No. 675; draft
report No. M-79-76).
BAHIG, M.E., KRAUS, A., & KLEIN, W. (1981) Excretion and metabolism of
2,4,6-trichlorophenol-14C in rats. Chemosphere, 10(3): 323-327.
BAIRD, R.B., KUO, C.L., SHAPIRO, J.S., & YANKO, W.A. (1974) The fate
of phenolics in wastewater -- determination by direct-injection
GLC and Warburg respirometry. Arch. environ. Contam. Toxicol.,
2(2): 165-178.
BAKER, M.D. & MAYFIELD, C.I. (1980) Microbial and non-biological
decomposition of chlorophenols and phenol in soil. Water Air Soil
Pollut., 13(4): 411-424.
BAKER, M.D., MAYFIELD, C.I., & INNISS, W.E. (1980) Degradation of
chlorophenols in soil, sediment and water at low temperature.
Water Res., 14(12): 1765-1771.
BALARAJAN, R. & ACHESON, E.D. (1984) Soft tissue sarcomas in
agriculture and forestry workers. J. Epidemiol. community Health,
38: 113-116.
BALLSCHMITER, K. & SCHOLZ, C. (1980) Microbial decomposition of
chlorinated aromatic substances. VI. Formation of dichlorophenols
and dichloropyrocatechol from dichlorobenzenes in a micromolar
solution by Pseudomonas sp. Chemosphere, 9(7-8): 457-467.
BARNHART, E.L. & CAMPBELL, G.R. (1972) The effect of chlorination on
selected organic chemicals, Washington, DC, US Environmental
Protection Agency, 105 pp (Water Pollution Control Research Series
No. PB 211-160).
BARROWS, M.E., PETROCELLI, S.R., MACEK, K.J., & CARROLL, K.J. (1980)
Bioconcentration and elimination of selected water pollutants by
bluegill sunfish (Lepomis macrochirus). In: Haque, R., ed.
Dynamics exposure and hazard assessment of "toxic chemicals",
Ann Arbor, Michigan, Ann Arbor Science Publishers Inc.,
pp. 379-392.
BEHRBOHM, P. (1959) [On the hazards of exposure to chlorinated
phenols.] Dtsch. Gesundheitswes., 14: 614-619 (in German).
BELTRAME, P., BELTRAME, P.L., & CARNITI, P. (1984) Inhibiting action
of chloro- and nitrophenols on biodegradation of phenols: a
structure-toxicity relationship. Chemosphere, 13(1): 3-9.
BENOIT-GUYOD, J.-L., ANDRE, C., TAILLANDIER, G., ROCHAT, J., &
BOUCHERLE, A. (1984) Toxicity and QSAR of chlorophenols on
Lebistes reticulatus. Ecotoxical. environ. Saf., 8: 227-235.
BEVENUE, A. & BECKMAN, H. (1967) Pentachlorophenol: a discussion of
its properties and its occurrence as a residue in human and animal
tissues. Residue Rev., 19: 83-134.
BEVENUE, A., OGATA, J.N., & HYLIN, J.W. (1972) Organochlorine
pesticides in rainwater, Oahu, Hawaii, 1971-1972. Bull. environ.
Contam. Toxicol., 8(4): 238-241.
BLACKBURN, K., ZENICK, H., HOPE, E., MANSON, J.M., GEORGE, E.L., &
SMITH, M.K. (1986) Evaluation of the reproductive toxicology of
2,4,6-trichlorophenol in male and female rats. Fundam. appl.
Toxicol., 6(2): 233-239.
BLACKMAN, G.E., PARKE, M.A., & GARTON, F. (1955) The physiological
action of substituted phenols. I. Relationships between chemical
structure and physiological activity. Arch. Biochem. Biophys.,
54(1): 45-54.
BLADES-FILLMORE, L.A., CLEMENT, W.H., & FAUST, S.D. (1982) The effect
of sediment on the biodegradation of 2,4,6-trichlorophenol in
Delaware River water. J. environ. Sci. Health, A17(6): 797-818.
BLANK, C.E., COOKE, P., & POTTER, A.M. (1983) Investigations for
genotoxic effects after exposure to crude 2,4,5-trichlorophenol.
Br. J. ind. Med., 40: 87-91.
BLEIBERG, J., WALLEN, M., BRODKIN, R., & APPLEBAUM, I.L. (1964)
Industrially acquired porphyria. Arch. Dermatol., 89: 793-797.
BOETIUS, J. (1954) Foul taste of fish and oysters caused by
chlorophenol. Medd. Dan. Fisk. Havunders. N.S., 1(4): 3-8.
BOLLAG, J.-M., HELLING, C.S., & ALEXANDER, M. (1968) 2,4-D metabolism:
enzymatic hydroxylation of chlorinated phenols. J. agr. food
Client., 16(5): 826-828.
BOND, G.G., WETTERSTROEM, N.H., ROUSH, G.J., MCLAREN, E.A., LIPPS,
T.E., & COOK, R.R. (1988) Cause-specific mortality among employees
engaged in the manufacture, formulation, or packaging of
2,4-dichlorophenoxyacetic acid and related salts. Br. J. ind.
Med., 45(2): 98-105.
BORZELLECA, J.F., HAYES, J.R., CONDIE, L.W., & EGLE, J.L., Jr (1985a)
Acute toxicity of monochlorophenols, dichlorophenols and
pentachlorophenol in the mouse. Toxicol. Lett., 29(1): 39-42.
BORZELLECA, J.F., HAYES, J.R., CONDIE, L.W., & EGLE, J.L., Jr (1985b)
Acute and subchronic toxicity of 2,4-dichlorophenol in CD-1 mice.
Fundam. appl. Toxicol., 5(3): 478-486.
BOULE, P., GUYON, C., & LEMAIRE, J. (1982) Photochemistry and
environment. IV. Photochemical behaviour of monochlorophenols in
dilute aqueous solution. Chemosphere, 11(12): 1179-1188.
BOULE, P., GUYON, C., & LEMAIRE, J. (1984) Photochimie et
environnement. VIII. Comportement photochimique de dichlorophénols
en solution aqueuse diluée. Chemosphere, 13: 603-612.
BOUTWELL, R.K. & BOSCH, D.K. (1959) The tumour-promoting action of
phenol and related compounds for mouse skin. Cancer Res.,
19: 413-424.
BOYD, S.A. & SHELTON, D.R. (1984) Anaerobic biodegradation of
chlorophenols in fresh and acclimated sludge. Appl. environ.
Microbiol., 47(2): 272-277.
BRAUN, W.H., BLAU, G.E., & CHENOWETH, M.B. (1978) The
metabolism/pharmacokinetics of pentachlorophenol in man, and a
comparison with the rat and monkey model. Toxicol. appl.
Pharmacol., 45: 278.
BRAUN, W.H., BLAU, G.E., & CHENOWETH, M.B. (1979) The metabolism/
pharmacokinetics of pentachlorophenol in man, and a comparison
with the rat and monkey. In: Deichmann, W.B., ed. Toxicology and
occupational medicine, New York, Amsterdam, Oxford, Elsevier/
North-Holland, pp. 289-296.
BRISTOL, D.W., COOK, L.W., KOTERBA, M.T., & NELSON, D.C. (1982)
Determination of free and hydrolyzable residues of 2,4-dichloro-
phenoxyacetic acid and 2,4-dichlorophenol in potatoes. J. agric.
food Chem., 30(1): 137-144.
BROECKER, B. & ZAHN, R. (1977) The performance of activated sludge
plants compared with the results of various bacterial toxicity
tests a study with 3,5-dichlorophenol. Water Res., 11: 165-172.
BUCCAFUSCO, R.J., ELLS, S.J., & LEBLANC, G.A. (1981) Acute toxicity of
priority pollutants to bluegill (Lepomis macrochirus). Bull.
environ. Contam. Toxicol., 26: 446-452.
BUIKEMA, A.L., Jr, MCGINNISS, M.J., & CAIRNS, J., Jr (1979) Phenolics
in aquatic ecosystems: a selected review of recent literature.
Mar. environ. Res., 2: 87-181.
BUSER, H.-R. & BOSSHARDT, H.P. (1976) Determination of polychlorinated
dibenzo- p-dioxins and dibenzofurans in commercial
pentachlorophenols by combined gas chromatography-mass
spectrometry. J. Assoc. Off. Anal. Chem., 59(3): 562-569.
BUTTE, W. (1984) Determination of free and total pentachlorophenol in
urine. Fresenius Z. anal. Chem., 317: 659.
BUTTE, W., KIRSCH, M., & DENKER, J. (1983) The determination of
pentachlorophenol and tetrachlorophenols in Wadden sediment and
clams (Mya arenia) using triethylsulfonium hydroxide for
excretion and pyrolytic ethylation. Int. J. environ. anal. Chem.,
13: 141-153.
BUTTE, W., DENKER, J., KIRSCH, M., & HOPNER, Th. (1985)
Pentachlorophenol and tetrachlorophenols in Wadden sediment and
clams (Mya arenia) of the Jadebusen after a 14-year period of
wastewater discharge containing pentachlorophenol. Environ.
Pollut., B9: 29-39.
CALL, D.J., BROOKE, L.T., & LU, P.-Y. (1980) Uptake, elimination, and
metabolism of three phenols by fathead minnows. Arch. environ.
Contam. Toxicol., 9: 699-714.
CALLAGHER, R.P. & THRELFALL, W.J. (1984) Cancer and occupational
exposure to chlorophenols. Lancet, II: 48.
CALLAGHER, R.P. & THRELFALL, W.J. (1985) Cancer risk in wood and pulp
workers. In: Stich, H.F., ed. Carcinogens and mutagens in the
environment, Boca Raton, Florida, CRC Press, Vol. 5,
pp. 125-137.
CALLAHAN, M.A., SLIMAK, M.W., GABEL, N.W., MAY, I.P., FOWLER, C.F.,
FREED, J.R., JENNINGS, P., DURFEE, R.L., WHITMORE, F.C., MAESTRI,
B., MABEY, W.R., HOLT, B.R., & GOULD, C. (1979). Water-related
environmental fate of 129 priority pollutants, Washington, DC,
US Environmental Protection Agency, Vol. II (EPA-440/4-79-029b).
CANADA-ONTARIO REVIEW BOARD (1981) Environmental baseline report of
the Niagara River, Ottawa, Environment Canada (November 1981
Update).
CARAMASCHI, F., MONTESARCHIO, E., DEL CORNO, G., FAVARETTI, C.,
GIAMBELLUCA, S.E., BONETTI, F., & VOLPATO, C. (1982) Analysis of
exposure to environmental contamination by TCDD in individuals
affected by dermatological lesions. Ig. mod., 77: 681-706.
CARLSON, G.P. (1978) Effect of trichlorophenols on xenobiotic
metabolism in the rat. Toxicology, 11: 145-151.
CAUTREELS, W., VAN CAUWENBERGHE, K., & GUZOLAN, L.A. (1977) Comparison
between the organic fraction of suspended matter at a background
and an urban station. Sci. total Environ., 8: 79-88.
CEDAR, F.J. (1984) Historical and current developments of antisapstain
chemical use. In: Proceedings of a Symposium on Preservatives in
the Wood Industry, 16-17 May, 1984, Vancouver, Division of
Occupational and Environmental Health, Department of Health Care
and Epidemiology, University of British Columbia.
CHADWICK, R.W. & FREAL, J.J. (1972) The identification of five
unreported lindane metabolites recovered from rat urine. Bull.
environ. Contam. Toxicol., 7(2/3): 137-146.
CHATTERJEE, R.M. (1974) Fish tainting in the Upper Great Lakes,
Ottawa, Ontario Ministry of Environment, 31 pp (Water Resources
Branch Report).
CHIOU, C.T., FREED, V.H., PETERS, L.J., & KOHNERT, R.L. (1980)
Evaporation of solutes from water. Environ. Int., 3: 231-236.
CHOI, J. & AOMINE, S. (1972) Effects of the soil on the activity of
pentachlorophenol. Soil Sci. plant Nutr., 18(6): 255-260.
CHOI, J. & AOMINE, S. (1974) Adsorption of pentachlorophenol by soils.
Soil Sci. plant Nutr., 20(2): 135-144.
CHRISTENSEN, E.R. & NYHOLM, N. (1984) Ecotoxicological assays with
algae: Weibull dose-response curves. Environ. Sci. Technol.,
18(9): 713-718.
CHRISWELL, C.D, CHANG, R.C, & FRITZ, J.S. (1975) Chromatographic
determination of phenols in water. Anal. Chem.,
47(8): 1325-1329.
CHU, J. & KIRSCH, E.J. (1973) Utilization of halophenols by a
pentachlorophenol metabolizing bacterium. Der. bid. Microbiol,
14: 264-273.
CLARK, D.E., PALMER, J.S., RADELEFF, R.D., CROOKSHANK, H.R., & FARR,
F.M. (1975) Residues of chlorophenoxy acid herbicides and their
phenolic metabolites in tissues of sheep and cattle. J. agric.
food Chem., 23(3): 573-578.
COCHRANE, W.P., LANOUETTE, M, & SINGH, J. (1983) High pressure liquid
chromatographic determination of impurity phenols in technical
2,4-D acid and 2,4-dichlorophenol. J. Assoc. Off. Anal. Chem.,
66(3): 804-809.
COGGON, D., PANNETT, B., WINTER, P.D.., ACHESON, E.D, & BONSALL, J.
(1986) Mortality of workers exposed to 2 methyl-4-chloropheno-
xyacetic acid. Scand. J. Work Environ. Health, 12: 448-454.
COOK, R.R. (1981) Dioxin, chloracne and soft-tissue sarcoma. Lancet,
1: 618-619.
COOK, R.R., TOWNSEND, J.C., OTT, M.G., & SILVERSTEIN, L.G. (1980)
Mortality experience of employees exposed to 2,3,7,8-tetra-
chlorodibenzo- p-dioxin (TCDD). J. occup. Med., 22(8): 530-532.
COOK, R.R., BOND, G.G., OLSON, R.A., & OTT, M.G. (1987) Update of the
mortality experience of workers exposed to chlorinated dioxins.
Chemosphere, 16: 2111-2116.
CORDDRY, A.E. (1981) A pregnancy outcome study of the wives of
workers exposed to chlorophenate wood preservatives at a sawmill,
Seattle, University of Washington, Department of Environmental
Health, 191 pp (Thesis).
CROSBY, D.G. & TUTASS, H.O. (1966) Photodecomposition of
2,4-dichlorophenoxyacetic acid. J. agric. food Chem.,
14(6): 596-599.
CROSBY, D.G. & WONG, A.S. (1973) Photodecomposition of
2,4,5-trichlorophenoxyacetic acid (2,4,5-T) in water. J. agric.
food Chem., 21(6): 1052-1054.
CROSBY, D.G., BEYNON, K.I., GREVE, P.A., KORTE, F., STILL, G.G., &
VOUK, J.W. (1981) Environmental chemistry of chlorophenol. A
special report on pentachlorophenol in the environment. Pure.
appl. Chem., 53: 1051-1080.
CURRIE, K. & MCDONALD, E. (1986) Uptake/excretion of chlorophenols by
sawmill employees. Phase II, Ottawa, Health and Welfare Canada
(B.C. Research Report to Pesticides Division; Contract No. 1424).
CURTIS, R.F., LAND, D.G., GRIFFITHS, N.M., GEE, M., ROBINSON, D.,
PEEL, J.L., DENNIS, C., & GEE, J.M. (1972) 2,3,4,6-Tetrachloro-
anisole association with musty taint in chickens and
microbiological formation. Nature (Lond.), 235: 223-224.
DAHMS, A. & METZNER, W. (1979) [Analysis of pentachlorophenol and
tetrachlorophenol in air and urine.] Holz. Roh-Werkstoff,
37: 331-344 (in German).
DANIELS, C.R. & SWAN, E.P. (1979) Determination of chlorinated phenols
in surface-treated lumber by HPLC. J. chromatogr. Sci.,
17: 628-630.
DE KREUK, J.F. & HANTSVEIT, A.O. (1981) Determination of the
biodegradability of the organic fraction of chemical wastes.
Chemosphere, 10(6): 561-573.
DELAUNE, R.D., GAMBRELL, R.P., & REDDY, K.S. (1983) Fate of PCP in
estuarine sediment. Environ. Pollut., B6: 297-308.
DEL CORNO, G., FAVARETTI, C., CARAMASCHI, F., GIAMBELLUCA, S.E.,
MONTESARCHIO, E., BONETTI, F., & VOLPATO, C. (1982) Distribution
of chloracne cases in the Seveso area following contamination by
TCDD. Ig. mod., 77: 635-658.
DEVILLERS, J. & CHAMBON, P. (1986) Acute toxicity and QSAR of
chlorophenols on Daphnia magna. Bull. environ. Contam. Toxicol.,
37: 599-605.
DIETZ, F. & TRAUD, J. (1978) [Trace analysis of phenols, especially
chlorophenols in water, by gas chromatography: methods and
results.] Vom Wasser, 51: 235-237 (in German with English
summary).
DOEDENS, J.D. (1963) Chlorophenols. In: Kirk-Othmer Encyclopedia of
chemical technology, 2nd ed., New York, John Wiley and Sons,
Vol. 5, pp. 325-338.
DUST, J.V. & THOMPSON, W.S. (1973) Pollution control in the
wood-preserving industry. Part IV. Biological methods of treating
wastewater. Forest. Prod. J., 23(9): 59-66.
DUTKA, B.J. & KWAN, K.K. (1984) Studies in a synthetic activated
sludge toxicity screening procedure with comparison to three
microbial toxicity tests. In: Liu, D. & Dutka, B.J., ed. Toxicity
screening procedures using bacterial systems, New York, Basel,
Marcel Dekker Inc., pp. 125-138.
DUXBURY, J.M., TIEDJE, J.M., ALEXANDER, M., & DAWSON, J.E. (1970)
2,4-D metabolism: enzymatic conversion of chloromaleylacetic acid
to succinic acid. J. agric. food Chem., 18(2): 199-201.
EDER, G. & WEBER, K. (1980) Chlorinated phenols in sediments and
suspended matter of the Weser estuary. Chemosphere,
9(2): 111-118.
EDGERTON, T.R., MOSEMAN, R.F., LINDER, R.E., & WRIGHT, L.H. (1979)
Multi-residue method for the determination of chlorinated phenol
metabolites in urine. J. Chromatogr., 170: 331-342.
EDGERTON, T.R., MOSEMAN, R.F., LORES, E.M., & WRIGHT, L.H. (1980)
Determination of trace amounts of chlorinated phenols in human
urine by gas chromatography. Anal. Chem., 52: 1774-1777.
EICHELBERGER, J.W., DRESSMAN, R.C., & LONGBOTTOM, J.E. (1970)
Separation of phenolic compounds from carbon chloroform extract
for individual chromatographic identification and measurement.
Environ. Sci. Technol., 4(7): 576-578.
ELDER, V.A., PROCTOR, B.L., & HITES, R.A. (1981) Organic compounds
found near dumpsites in Niagara Falls, New York. Environ. Sci.
Technol., 15(10): 1237-1243.
EL-GOHARY, F.A. & NASR, F.A. (1984) Fate and effect of
2,4-dichlorophenol on biological treatment processes. Environ.
Int., 10: 211-215.
EMBREE, V, ENARSON, D.A., CHAN-YEUNG, M, DYBUNCIO, A., DENNIS, R., &
LEACH, J. (1984) Occupational exposure to chlorophenates:
toxicology and respiratory effects. Clin. Toxicol,
22(4): 317-329.
ENARSON, D.A., CHAN-YEUNG, M., EMBREE, V., WANG, R., & SCHULZER, M.
(1986) Occupational exposure to chlorophenates: Renal, hepatic and
other health effects. Scand. J. Work Environ. Health,
12: 144-148.
ENGST, R., MACHOLZ, R.M., KUJAWA, M., LEWERENZ, H.-J., & PLASS, R.
(1976) The metabolism of lindane and its metabolites
gamma-2,3,4,5,6-pentachlorocyclohexane, pentachlorobenzene, and
pentachlorophenol in rats and the pathways of lindane metabolism.
J. environ. Sci. Health, B11(2): 95-17.
ENVIRONMENT CANADA (1979) Monitoring environmental contamination from
chlorophenol contaminated wastes generated in the wood
preservation industry, Ottawa, Environmental Protection Branch,
Environmental Protection Service, Pacific and Yukon Region,
Environment Canada (Regional Program Report No. 79-24).
ENVIRONMENT CANADA (1986) Dioxin-containing chemicals: sales and
imports. 1985 Update, Ottawa, Commercial Chemicals Branch,
Environmental Protection Service, Environment Canada.
ERICKSON, S.J. & HAWKINS, C.E. (1980) Effects of halogenated organic
compounds on photosynthesis in estuarine phytoplankton. Bull.
environ. Contam. Toxicol., 24: 910-915.
ERIKSSON, M., HARDELL, L., BERG, N.O., MOLLER, T., & AXELSON, O.
(1981) Soft-tissue sarcomas and exposure to chemical substances: a
case-referent study. Br. J. ind. Med., 38: 27-33.
ERNST, W. (1979) Factors affecting the evaluation of chemicals in
laboratory experiments using marine organisms. Ecotoxicol.
environ. Saf, 3: 90-98.
ERNST, W. & WEBER, K. (1978) Chlorinated phenols in selected estuarine
bottom fauna. Chemosphere, 7(11): 867-872.
ETTINGER, M.B. & RUCHHOFT, C.C. (1950) Persistence of
monochlorophenols in polluted river water and sewage dilutions.
Presented at the Annual Meeting of the Central States Sewage Works
Association, Cincinnati, Environmental Health Centre, 11 pp
(PB 215-498).
ETZEL, J.E. & KIRSCH, E.J. (1974) Biological treatment of contrived
and industrial wastewater containing pentachlorophenol. Dev. ind.
Microbiol., 16: 287-295.
EXON, J.H. (1984) A review of chlorinated phenols. Vet. hum.
Toxicol., 26(6): 508-520.
EXON, J.H. & KOLLER, L.D. (1981) Alteration of transplacental
carcinogenesis by chlorinated phenols. In: Jolley, R.L., Brungs,
W.A., Cotruvo, J.A., Cumming, R.B., Mattice, J.S., & Jacobs, V.A,
ed. Water chlorination: Environmental impact and health effects,
Ann Arbor, Michigan, Ann Arbor Science Publishers, Vol. 4, Book 2,
pp. 1177-1188.
EXON, J.H. & KOLLER, L.D. (1983) Effects of chlorinated phenols on
immunity in rats. Int. J. Immunopharmacol., 5(2): 131-136.
EXON, J.H. & KOLLER, L.D. (1985) Toxicity of 2-chlorophenol,
2,4-dichlorophenol, and 2,4,6-trichlorophenol. In: Jolley, R.L,
Bull, R.J., Davis, W.P., Katz, S., Roberts, M.H., Jr, & Jacobs,
V.H., ed. Water chlorination: Environmental impact and health
effects, Chelsea, Michigan, Lewis Publishers Inc., Vol. 5,
pp. 307-330.
EXON, J.H., HENNINGSEN, G.M., OSBORNE, C.A., & KOLLER, L.D. (1984)
Toxicologic, pathologic, and immunotoxic effects of
2,4-dichlorophenol in rats. J. Toxicol. environ. Health,
14: 723-730.
FAHRIG, R., NILSSON, C.-A., & RAPPE, C. (1978) Genetic activity of
chlorophenols and chlorophenol impurities. In: Rao, K.R., ed.
Pentachlorophenol: Chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 325-338.
FARQUHARSON, M.E., CAGE, J.C., & NORTHOVER, J. (1958) The biological
action of chlorophenols. Br. J. Pharmacol., 13: 20-24.
FARRINGTON, D.S. & MUNDAY, J. W. (1976) Determination of trace amounts
of chlorophenols by gas-liquid chromatography. Analyst,
101: 639-643.
FENSKE, R.A., HORSTMAN, S.W., & BENTLEY, R.K. (1987) Assessment of
dermal exposure to chlorophenols in lumber mills. Appl. ind.
Hyg., 2(4): 143-147.
FINGERHUT, M.A., HALPERIN, W.E., HONCHAR, P.A., SMITH, A.B., GROTH,
D.H. & RUSSELL, W.O. (1984) An evaluation of reports of dioxin
exposure and soft-tissue sarcoma pathology among chemical workers.
Scand. J. Work Environ. Health, 10: 299-303.
FIRESTONE, D., RESS, J., BROWN, N.L., BARRON, R.P., & DAMICO, J.N.
(1972) Determination of polychlorodibenzo- p-dioxins and related
compounds in commercial chlorophenols. J. Assoc. Off. Anal.
Chem., 55(1): 85-92.
FOLKE, J. (1984) Organic micropollutants in the sewage from a
phenoxyalkanoic acid production plant after biological treatment
in a municipal trickling filters plant. J. high Resolut.
Chromatogr. Chromatogr. Commun., 7: 25-32.
FOLKE, J. & BIRKLUND, J. (1986) Danish coastal water levels of
2,3,4,6-tetrachlorophenol, pentachlorophenol, and total
organohalogens in blue mussels (Mytilus edulis). Chemosphere,
15(7): 895-900.
FOLKE, J., BIRKLUND, J., SORENSEN, A.K., & LUND, U. (1984) The impact
on the ecology of polychlorinated phenols and other organics
dumped at the bank of a small marine inlet. In: Angeletti, G. &
Bjorseth, A., ed. Analysis of organic micropollutants in water.
Proceedings of the 3rd European Symposium, Oslo, 19-21 September,
1983, Luxembourg, Commission of the European Communities,
pp. 242-254.
FOX, M.E. & JOSHI, S.R. (1984) The fate of pentachlorophenol in the
Bay of Quinte, Lake Ontario. J. Great Lakes Res.,
10(2): 190-196.
FRANK, R., BRAUN, H.E., HOLDRINET, M., SIRONS, G.J, SMITH, E.H., &
DIXON, D.W. (1979) Organochlorine insecticides and industrial
pollutants in the milk supply of Southern Ontario, Canada 1977.
J. food Prot., 42(1): 31-37.
FREITAG, D., GEYER, H., KRAUS, A., VISWANATHAN, R., KOTZIAS, D, ATTAR,
A., KLEIN, W., & KORTE, F. (1982) Ecotoxicological profile
analysis. VII. Screening chemicals for their environmental
behaviour by comparative evaluation. Ecotoxicol. environ. Saf.,
6(1): 60-81.
FREITER, E.R. (1979) Chlorophenols. In: Kirk-Othmer Encyclopedia of
chemical technology, 3rd ed., New York, John Wiley & Sons,
Vol. 5, pp. 864-872.
FUJITA, T., IWASA, J., & HANSCH, C. (1964) A new substituent constant,
pi, derived from partition coefficients. J. Am. Chem. Soc.,
86: 5175-5180.
GALLAGHER, R.P. & THRELFALL, W.J. (1984) Cancer and occupational
exposure to chlorophenols. Lancet, II(8393): 48.
GALLOWAY, S.M., ARMSTRONG, M.J., REUBEN, C., COLMAN, S., BROWN, B.,
CANNON, C., BLOOM, A.D., NAKAMURA, F., AHMED, M., DUK, S., RIMPO,
J., MARGOLIN, B.H., RESNICK, M.A., ANDERSEN, B., & ZEIGER, E.
(1987) Chromosome aberrations and sister chromatid exchanges in
Chinese hamster ovary cells: evaluations of 108 chemicals.
Environ. mol. Mutagenesis., 10(10): 1-175.
GARRETT, C.L. (1980) Fraser river estuary study: Toxic organic
contaminants, Ottawa, Environmental Protection Service, Pacific
and Yukon Region, Environment Canada, 123 pp (Water Quality
Series).
GEE, J.M. & PEEL, J.L. (1974) Metabolism of 2,3,4,6-tetrachlorophenol
by microorganisms from broiler house litter. J. gen. Microbiol.,
88: 237-243.
GERSICH, F.M. & MILAZZO, D.P. (1988) Chronic toxicity of aniline and
2,4-dichlorophenol to Daphnia magna Straus. Bull environ.
Contam. Toxicol., 40: 1-7.
GILIOLI, R., GENTA, P., COTRONEO, L., & CANNATELLI, P. (1983)
Electroencephalographic spectral analysis in chemical and
engineering workers. In: Gilioli, R., Cassitto, M.G., & Foa, V.,
ed. Neurobehavioral methods in occupational health: advances in
the bioscience, Oxford, Pergamon Press, pp. 219-229.
GOMEZ-CATALAN, J., TO-FIGUERAS, J., PLANAS, J., RODAMILANS, M., &
CORBELLA, J. (1987) Pentachlorophenol and hexachlorobenzene in
serum and urine of the population of Barcelona. Hum. Toxicol.,
6: 397-400.
GOSSELIN, R.E, HODGE, H.C., SMITH, R.P., & GLEASON, M.N. (1976)
Clinical toxicology of commercial products, 4th ed., Baltimore,
Maryland, Williams and Wilkins Co., pp. 130-132.
GREENBERG, A.T., TRUSSEL, R.R., & CLESCERI, L.S., ed. (1985) Standard
methods for the examination of water and wastewater, 16th ed.,
Washington, DC, American Public Health Association, pp. 556-570.
GREENWALD, P., KOVASNAY, B., COLLINS, D.N., & THERRIAULT, G. (1984)
Sarcomas of soft tissues after Vietnam service. J. Natl Cancer
Inst., 73: 1107-1109.
GUROVA, A.I. (1964) Hygienic characteristics of p-chlorophenol in the
aniline dye industry. Hyg. Sanit., 29(10): 46-51.
GUTHRIE, M.A., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C.P.L., Jr (1984)
Pentachlorophenol biodegradation. II. Anaerobic. Water Res.,
18(4): 451-461.
HAGENMAIER, H. (1986) Determination of 2,3,7,8-tetrachlorodibenzo-
p-dioxin in commercial chlorophenols and related products.
Fresenius Z. anal Chem., 325: 603-606.
HAGENMAIER, H. & BRUNNER, H. (1987) Isomers specific analysis of
pentachlorophenol and sodium pentachlorophenate for 2,3,7,8-
substituted PCDD and PCDF at sub-ppb levels. Chemosphere,
16: 1759-1764.
HAKULINEN, R. & SALKINOJA-SALONEN, M. (1982) Treatment of pulp and
paper industry waste waters in an anaerobic fluidized bed reactor.
Process Biochem., 314: 18-22.
HARDELL, L. & ERIKSSON, M. (1988) The association between soft tissue
sarcomas and exposure to phenoxyacetic acids: A new case-referent
study. Cancer, 62: 652-656.
HARDELL, L. & SANDSTROM, A. (1979) Case-control study: soft-tissue
sarcomas and exposure to phenoxyacetic acids or chlorophenols.
Br. J. Cancer, 39: 711-717.
HARDELL, L. (1981) Relation of soft-tissue sarcoma, malignant lymphoma
and colon cancer to phenoxy acids, chlorophenols and other agents.
Scand. J. Work Environ. Health, 7: 119-130.
HARDELL, L., ERIKSSON, M., LENNER, P., & LUNDGREN, E. (1981) Malignant
lymphoma and exposure to chemicals, especially organic solvents,
chlorophenols and phenoxy acids: a case-control study. Br. J.
Cancer, 43: 169-176.
HARDELL, L., JOHANSSON, B., & AXELSON, O. (1982) Epidemiological study
of nasal and nasopharyngeal cancer and their relation to phenoxy
acid or chlorophenol exposure. Am. J. ind. Med., 3: 247-257.
HARDELL, L., BENGTSSON, N.O., JONSSON, U., ERIKSSON, S., & LARSSON,
L.G. (1984) Aetiological aspects on primary liver cancer with
special regard to alcohol, organic solvents and acute intermittent
pophyria: an epidemiological investigation. Br. J. Cancer,
50: 389-397.
HARGESHEIMER, E.E. & COUTTS, R.T. (1983) Selected ion mass
spectrometric identification of chlorophenol residues in human
urine. J. Assoc. Off. Anal. Chem., 66(1): 13-21.
HATTULA, M.L., WASENIUS, V.-M., KREES, R, ARSTILA, A.U., & KIHLSTROM,
M. (1981a) Acute and short-term toxicity of
2,3,4,6-tetrachlorophenol in rats. Bull. environ. Contam.
Toxicol., 26: 795-800.
HATTULA, M.L., WASENIUS, V.-M., REUNANEN, H., & ARSTILA, A.U. (1981b)
Acute toxicity of some chlorinated phenols, catechols and cresols
to trout. Bull. environ. Contam. Toxicol., 26: 295-298.
HATTULA, M.L. & KNUUTINEN, J. (1985) Mutagenesis of mammalian cells in
culture by chlorophenols, chlorocatechols, and chloroguaiacols.
Chemosphere, 14: 1617-1625.
HAWORTH, S., LEWLOR, T., MORTELMANS, K., SPECK, W., & ZEIGER, E.
(1983) Salmonella mutagenicity test results for 250 chemicals.
Environ. Mutagen., 1 (Suppl.): 3-142.
HAY, A. (1976) Toxic cloud over Seveso. Nature (Lond.),
262: 636-638.
HEITMULLER, P.T., HOLLISTER, T.A., & PARRISH, P.R. (1981) Acute
toxicity of 54 industrial chemicals to sheepshead minnows
(Cyprinodon variegatus). Bull. environ. Contam. Toxicol.,
27: 596-604.
HERNBERG, S., WESTERHOLM, P., SCHULTZ-LARSEN, K., DEGERTH, R., KUOSMA,
E., ENGLUND, A., ENGZELL, U., HANSEN, H.S., & MUTANEN, P. (1983)
Nasal and sinonasal cancer: connection with occupational exposures
in Denmark, Finland, and Sweden. Scand. Z Work Environ. Health,
9: 315-326.
HICKMAN, G.T. & NOVAK, J.T. (1984) Acclimation of activated sludge to
pentachlorophenol. J. Water Pollut. Control Fed., 56: 364-369.
HOAR, S.K., BLAIR, A., HOLMES, F.F., BOYSEN, C.D., ROBEL, R.J.,
HOOVER, R., & FRAUMENI, J.F. (1986) Agricultural herbicide use and
risk of lymphoma and soft-tissue sarcoma. J. Am. Med. Assoc.,
256: 1141-1147.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976a) A study of
inhalation of pentachlorophenol by rats. IV. Distribution and
excretion of inhaled pentachlorophenol. Bull. environ. Contam.
Toxicol., 15(4): 466-474.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976b) A study of
inhalation of pentachlorophenol by rats. III. Inhalation toxicity
study. Bull. environ. Contam. Toxicol., 15(4): 463-465.
HOGSTED, C. & WESTERLUND, B. (1980) [Cohort study of mortality among
forestry workers with and without exposure for phenoxy
herbicides.] Läkartidningen, 77: 1828-1831 (in Swedish).
HOLCOMBE, G.W., PHIPPS, G.L., & FIANDT, J.T. (1982) Effects of phenol,
2,4-dimethylphenol, 2,4-dichlorophenol, and pentachlorophenol on
embryo, larval, and early-juvenile fathead minnows (Pimephales
promelas). Arch. environ. Contam. Toxicol., 11: 73-78.
HONCHAR, P.A. & HALPERIN, W.E. (1981) 2,4,5-T, trichlorophenol, and
soft tissue sarcoma. Lancet, 1: 268-269.
HOROWITZ, A., SHELTON, D.R., CORNELL, C.P., & TIEDJE, J.M. (1982)
Anaerobic degradation of aromatic compounds in sediments and
digested sludge. In: Proceedings of the 38th General Meeting of
the Society of Industrial Microbiology, Richmond, Virginia, 9-14
August, 1981, pp. 435-444.
HUANG, J. & GLOYNA, E.F. (1968) Effect of organic compounds on
photosynthetic oxygenation. I. Chlorophyll destruction and
suppression of photosynthetic oxygen production. Water Res.,
2: 347-366.
HUQ, A.S, HO, N.F.H., HUSARI, N., FLYNN, G.L., JETZER, W.E., & CONDIE,
L, Jr (1986) Permeation of water contaminative phenols through
hairless mouse skin. Arch. environ. Contam. Toxicol.,
15: 557-566.
HWANG, H.-M., HODSON, R.E., & LEE, R.F. (1986) Degradation of phenol
and chlorophenols by sunlight and microbes in estuarine waters.
Environ. Sci. Technol., 20(10): 1002-1007.
IARC (1979) Trichlorophenols. In: Some halogenated hydrocarbons,
Lyons, International Agency for Research on Cancer, pp. 349-367
(IARC Monographs on the Evaluation of the Carcinogenicity Risk of
Chemicals to Humans, Vol. 20).
IARC (1986) Occupational exposures to chlorophenols. In: Some
halogenated hydrocarbons and pesticide exposures, Lyons,
International Agency for Research on Cancer, pp. 319-356 (IARC
Monographs on the Evaluation of the Carcinogenicity Risk of
Chemicals to Humans, Vol. 41).
IARC (1987) Overall evaluations of carcinogenicity: an update of IARC
Monographs, Volumes 1 to 42, Lyons, International Agency for
Research on Cancer, pp. 154-156 (IARC Monographs, Supplement 7).
IDE, A., NIKI, Y., SAKAMOTO, F., WATANABE, I., & WATANABE, H. (1972)
Decomposition of pentachlorophenol in paddy soil. Agric. biol.
Chem., 36(11): 1937-1944.
INDORATO, A.M., SNYDER, K.B., & USINOWlCZ, P.J. (1984) Toxicity
screening using Microtox(R) analyzer. In: Liu, D. & Dutka, B.J.,
ed. Toxicity screening procedures using bacterial systems, New
York, Basel, Marcel Dekker Inc., pp. 37-53.
INGOLS, R.S., GAFFNEY, P.E., & STEVENSON, P.C. (1966) Biological
activity of halophenols. J. Water Pollut. Control Fed.,
38(4): 629-635.
INNES, J.R.M., ULLAND, B.M., VALERIO, M.G., PETRUCELLI, L., FISHBIEN,
L., HART, E.R., PALLOTTA, A.J., BATES, R.R., FALK, H.L., GART,
J.J., KLEIN, M., MITCHELL, I., & PETERS, J. (1969) Bioassay of
pesticides and industrial chemicals for tumorigenicity in mice: A
preliminary note. J. Natl Cancer Inst., 42: 1101-1114.
IRPTC (1986) IRPTC Legal file on chlorophenols, Geneva,
International Register of Potentially Toxic Chemicals, United
Nations Environment Programme.
JIRASEK, L, KALENSKY, J., KUBEC, K., PAZDEROVA, J., & LUKAS, E. (1974)
[Acne chlorina, porphyria cutanea tarda, and other manifestations
of general poisoning during the manufacture of herbicides.] Cesk.
Dermatol., 49: 145-157 (in Czechoslovakian with English
summary).
JOHNSON, F.E., KUGLER, M.A, & BROWN, S.M. (1981) Soft-tissue sarcomas
and chlorinated phenols. Lancet, 7: 40.
JOLLEY, R.L., JONES, G., PITT, W.W., & THOMPSON, J.E. (1975)
Chlorination of organics in cooling waters and process effluents.
In: Jolley, R.L, ed. The environmental impact of chlorination.
Proceedings of the Conference on Environmental Impact of Water
Chlorination, Oak Ridge, 22-24 October, 1975, Oak Ridge,
Tennessee, Oak Ridge National Laboratories, pp. 115-152.
JONES, P.A. (1981) Chlorophenols and their impurities in the Canadian
environment, Ottawa, Environmental Protection Service,
Environment Canada, 434 pp (Report No. EPS-3-EC-81-2).
JONES, P.A. (1984) Chlorophenols and their impurities in the Canadian
environment: 1983 Supplement, Ottawa, Environmental Protection
Service, Environment Canada, 93 pp (Report No. EPS-3-EP-84-3).
KAILA, K. & SAARIKOSKI, J. (1977) Toxicity of pentachlorophenol and
2,3,6-trichlorophenol to the crayfish (Astacus fluviatilis L.)
Environ. Pollut., 12: 119-123.
KALMAN, D.A. & HORSTMAN, S.W. (1983) Persistence of tetrachlorophenol
and pentachlorophenol in exposed woodworkers. J. Toxicol. clin.
Toxicol., 20(4): 343-352.
KANEKI, H. & TANAKA, M. (1984) Effects of some chlorinated pesticides
on porcine pancreatic and Rhizopus delemar lipase activities.
Eisei Kagaku, 30(4): 233-237.
KANG, H.K., WEATHERBEE, L., BRESLIN, P., & SHEPARD, B.M. (1986) Soft
tissue sarcoma and military service in Vietnam: A case comparison
group analysis of hospital patients. J. occup. Med.,
28: 1215-1218.
KARPALLY, J.C., SAHA, J.G., & LEE, Y.W. (1973) Metabolism of
lindane-14C in the rabbit: ether-soluble urinary metabolites.
J. agric. food Chem., 21(5): 811-818.
KARPOW, G. (1983) [On the antiseptic action of three isomeric
chlorophenols and of their salicylate esters and their fate in the
metabolism.] Arch. Sci. Biol. St. Petersburg, 2: 304-327
(in Russian and French).
KAUPPINEN, T. (1986) Occupational exposure to chemical agents in the
plywood industry. Ann. occup. Hyg., 30(1): 19-29.
KAUPPINEN, T.P., PARTANEN, T.J., NURMINEN, M.M., NICKELS, J.I.,
HERNBERG, S.G., HAKULINEN, T.R., PUKKALA, E.I., & SAVONEN, E.T,
(1986) Respiratory cancers and chemical exposures in the wood
industry: a nested case-control study. Br. J. ind. Med.,
43: 84-90.
KAUPPINEN, T.P. & LINDROOS, L. (1985) Chlorophenol exposure in
sawmills. Am. Ind. Hyg. Assoc. J., 46(1): 34-38.
KAWAHARA, F.K. (1971) Gas chromatographic analysis of mercaptous,
phenols, and organic acids in surface waters with use of
pentafluorobenzyl derivatives. Environ. Sci. Technol.,
5(3): 235-239.
KINAE, N., HASHIZUME, T., MAKITA, T., TOMITA, I., KIMURA, I., &
KANAMORI, H. (1981) Studies on the toxicity of pulp and paper mill
effluents. I. Mutagenicity of the sediment samples derived from
kraft paper mills. Water Res., 15: 17-24.
KING, E.F. (1984) A comparative study of methods for assessing the
toxicity to bacteria of single chemicals and mixtures. In: Liu, D.
& Dutka, B.J., ed. Toxicity screening procedures using bacterial
systems, New York, Basel, Marcel Dekker Inc., pp. 175-193.
KIRSCH, E.J. & ETZEL, J.E. (1973) Microbial decomposition of
pentachlorophenol. J. Water Pollut. Control Fed.,
45(2): 359-364.
KITUNEN, V.H., VALO, R.J., & SALKINOJA-SALONEN, M.S. (1987)
Contamination of soil around wood-preserving facilities by
polychlorinated aromatic compounds. Environ. Sci. Technol.,
21(1): 96-101.
KLEINMAN, G.D, HORSTMAN, S.W., KALMAN, D.A., MCKENZIE, J., & STANZEL,
D. (1986) Industrial hygiene, chemical and biological assessments
of exposures to a chlorinated phenolic sapstain control agent.
Am. Ind. Hyg. Assoc. J., 47(12): 731-741.
KLEU, G. & GOLTZ, R. (1971) [Late and long-term injuries following the
chronic occupational action of chlorophenol compounds.] Med.
Klin. (Munich), 66(2): 53-58 (in German with English abstract).
KLOPPFER, W., KAUFMANN, G., RIPPEN, G., & POREMSKI, H.-J. (1982) A
laboratory method for the volatility from aqueous solution: first
results and comparison with theory. Ecotoxicol. environ. Saf.,
6: 545-559.
KOBAYASHI, S., TODA, S., KAWAMURA, H., CHANG, H.S., FUKUDA, T., &
KAWAGUCHI, K. (1972) Chronic toxicity of 2,4-dichlorophenol in
mice: a simple design for the toxicity of residual metabolites of
pesticides. J. Med. Soc. Toho Univ. Jpn, 19: 356-362.
KOBAYASHI, K., AKITAKE, H., & MANABE, K. (1979) Relation between
toxicity and accumulation of various chlorophenols in goldfish.
Bull. Jpn. Soc. Sci. Fish., 45(2): 173-175.
KOGAN, M. & CLAPP, R. (1985) Mortality among Vietnam veterans in
Massachusetts, 1972-1983. Am. J. Epidemiol, 122: 523.
KONEMANN, H. & MUSCH, A. (1981) Quantitative structure-activity
relationships in fish toxicity studies. Part 2: The influence of
pH on the QSAR of chlorophenols. Toxicology, 19: 223-228.
KORANSKY, W., MUNCH, G., NOACK, G., PORTIG, J., SODOMANN, S., &
WIRSCHING, M. (1975) Biodegradation of hexachlorocyclohexane. V.
Characterization of the major urinary metabolites.
Naunyn-Schmiedeberg's Arch. Pharmacol., 288: 65-78.
KORTE, F., FREITAG, D., GEYER, H., KLEIN, W., KRAUS, A.G., &
LAHANIATIS, E. (1978) Ecotoxicologic profile analysis: a concept
for establishing ecotoxicologic priority lists for chemicals.
Chemosphere, 7(1): 79-102.
KOZAK, V.P., SIMSIMAN, G.V., CHESTERS, G., STENSBY, D., & HARKIN, J.
(1979) Reviews of the environmental effects of pollutants. XI.
Chlorophenols, Washington, DC, US Environmental Protection
Agency, 492 pp (EPA Report No. 600/1-79-O/Z).
KRIJGSHELD, K.R. & VAN DER GEN, A. (1986) Assessment of the impact of
the emission of certain organochlorine compounds on the aquatic
environment. Part I: Monochlorophenols and 2,4-dichlorophenol.
Chemosphere, 15(7): 825-860.
KUIPER, J. & HANTSVEIT, A.O. (1984) Fate and effects of 4-chlorophenol
and 2,4-dichlorophenol in marine plankton communities in
experimental enclosures. Ecotoxicol. environ. Saf., 8: 15-33.
KURIHARA, N. & NAKAJIMA, M. (1974) Studies on BHC isomers and related
compounds. VIII. Urinary metabolites produced from gamma-and ß -
BHC in the mouse: chlorophenol conjugates. Pestic. Biochem.
Physiol, 4(2): 220-231.
KUTZ, F.W., MURPHY, R.S., & STRASSMAN, S.C. (1978) Survey of pesticide
residues and their metabolites in urine from the general
population. In: Rao, K.R., ed. Pentachlorophenol: Chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 363-369.
KUWATSUKA, S. (1972) Degradation of several herbicides in soils under
different conditions. In: Matsuura, F., Boush, G.M., & Misato, T.,
ed. Environmental toxicology of pesticides, New York, London,
Academic Press, pp. 385-400.
KUWATSUKA, S. & IGARASHI, M. (1975) Degradation of PCP in soils. II.
The relationship between the degradation of soils, and the
identification of the degradation products of PCP. Soil Sci.
plant Nutr., 21: 405-414.
KWASNIEWSKA, K. & KAISER, K.L.E. (1983) Toxicities of selected phenols
to fermentative and oxidative yeasts. Bull. environ. Contam.
Toxicol., 31: 188-194.
LANDNER, L., LINDSTROM, K., KARLSSON, M., NORDIN, J., & SÖRENSEN, L.
(1977) Bioaccumulation in fish of chlorinated phenols from kraft
pulp mill bleachery effluents. Bull. environ. Contam. Toxicol.,
18(6): 663-673.
LANOUETTE, M., COCHRANE, W.P., & SINGH, J. (1984) Liquid
chromatographic determination of chlorinated phenol impurities in
technical pentachlorophenol using electrochemical detection
(Coulometric mode). J. Assoc. Off. Anal. Chem., 67(3): 494-497.
LATHROP, G.D., MACHADO, S.G., KARRISON, T.G., GRUBBS, W.D., THOMAS,
W.F., WOOLFE, W.H., MICHALEK, J.E., MINER, J.C., & PETERSON, M.R.
(1987) Air Force Health Study. First follow-up examination
results. An epidemiologic investigation of health effects in Air
Force personnel following exposure to herbicides, Texas, USAF
School of Aerospace Medicine Human Systems Division, 440 pp.
LEBLANC, G,A. (1980) Acute toxicity of priority pollutants to water
fleas (Daphnia magna). Bull. environ. Con tam. Toxicol.,
24: 684-691.
LEBLANC, G.A. (1984) Interspecies relationships in acute toxicity of
chemicals to aquatic organisms. Environ. Toxicol. Chem.,
3: 47-60.
LEE, R.F. & RYAN, C. (1979) Microbial degradation of organo-chlorine
compounds in estuarine waters and sediments. In: Bourquin, A.W. &
Pritchard, P.H., ed. Proceedings of the Workshop on Microbial
Degradation of Pollutants in Marine Environments, held at
Pennsicola Beach, Florida, 9-14 April 1978, Washington, DC, US
Environmental Protection Agency, pp. 443-450 (EPA Report
No. 60/9-79-012).
LEO, A., HANTSCH, C., & ELKINS, D. (1971) Partition coefficients and
their uses. Chem. Rev., 71(6): 525-616.
LEVIN, J.O. & NILSSON, C.A. (1977) Chromatographic determination of
polychlorinated phenols, phenoxyphenols, dibenzofurans and
dibenzodioxins in wood-dust from worker environments.
Chemosphere, 7: 443-448.
LEVIN, J.-O., RAPPE, C., & NILSSON., C.-A. (1976) Use of chlorophenols
as fungicides in sawmills. Scand. J. Work Environ. Health,
2: 71-81.
LINDSAY SMITH, J.R., SHAW, B.A.J., & FOULKES, D.M. (1972) Mechanisms
of mammalian hydroxylation: some novel metabolites of
chlorobenzene. Xenobiotica, 2(3): 215-226.
LINDROOS, L., KOSKINEN, H., MUTANEN, P., & JARVISALO, J. (1987)
Urinary chlorophenols in sawmill workers. Int. Arch. occup.
environ. Health, 59: 463-467.
LINDSTROM, K. & NORDIN, J. (1976) Gas chromatography -- mass
spectrometry of chlorophenols in spent bleach liquors.
J. Chromatogr., 128: 13-26.
LIU, D., THOMSON, K., & KAISER, K.L.E. (1982) Quantitative
structure-toxicity relationship of halogenated phenols on
bacteria. Bull. environ. Contam. Toxicol., 29: 130-136.
LOOS, M.A., ROBERTS, R.N., & ALEXANDER, M. (1967) Phenols as
intermediates in the decomposition of phenoxyacetates by an
Arthrobacter species. Can. J. Microbiol., 13: 679-690.
LYNGE, E. (1985) A follow-up study of cancer incidence among workers
in manufacture of phenoxy herbicides in Denmark. Br. J. Cancer,
52: 259-270.
MCCOLLISTER, D.D., LOCKWOOD, D.T., & ROWE, V.K. (1961) Toxicologic
information on 2,4,5-trichlorophenol. Toxicol. appl. Pharmacol.,
3: 63-70.
MACKENZIE, C.J.G., OLDHAM, W.K., & POWRIE, W.D. (1975) Effects
of pesticides on fish and wildlife in British Columbia.
Appendix RR, Vancouver, British Columbia Royal Commission
(Inquiry into the Use of Pesticides and Herbicides. Final report
of the Commissioners).
MCLEESE, D.W., ZITKO, V., & PETERSON, M.R. (1979) Structure-lethality
relationships for phenols, anilines and other aromatic compounds
in shrimp and clams. Chemosphere, 8(2): 53-57.
MARLOW, D.A. (1986) Hexachlorobenzene exposure in the production of
chlorophenols. In: Morris, C.R. & Cabral, J.R.P., ed.
Hexachlorobenzene. Proceedings of an International Symposium,
Lyons, International Agency for Research on Cancer, pp. 161-169
(IARC Scientific Publications No. 77).
MAY, G. (1973) Chloracne from the accidental production of
tetrachlorodibenzodioxin. Br. J. ind. Med., 30: 276-283.
MILHAM. S. (1982) Herbicides, occupation and cancer. Lancet,
I: 1464-1465.
MILHAM, S. (1985) Cancer in Washington State woodworkers. In: Stich,
H.F., ed. Carcinogens and mutagens in the environment, Boca
Raton, Florida, CRC Press, Vol. 5, pp. 117-123.
MILLER, R.W. & FAUST, S.D. (1973) Sorption from aqueous solutions by
organo-clay. III. The effect of pH on sorption of various phenols.
Environ. Lett, 4(3): 211-223.
MILLS, R.E. (1959) Development of design criteria for biological
treatment of an industrial effluent containing 2,4-D waste water.
In: Proceedings of the 14th Industrial Waste Conference,
Lafayette, Indiana, Purdue University, pp. 340-358.
MITSUDA, H., MURAKAMI, K., & KAWAI, F. (1963) Effect of chlorophenol
analogues on the oxidative phosphorylation in rat liver
mitochondria. Agric. biol. Chem., 27(5): 366-372.
MOOS, L.P., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C,P.L., Jr. (1983)
Pentachlorophenol biodegradation. I. Aerobic. Water Res.,
17(11): 1575-1584.
MORGADE, C., BARQUET, A., & PFAFFENBERGER, C.D. (1980) Determination
of polyhalogenated phenolic compounds in drinking water, human
blood serum and adipose tissue. Bull. environ. Contam. Toxicol.,
24: 257-264.
MOSES, M. & SELIKOFF, I.J. (1981) Soft-tissue sarcomas, phenoxy
herbicides, and chlorinated phenols. Lancet, 1: 1370.
NAKAGAWA, M. & CROSBY, D.G. (1974) Photodecomposition of nitrofen.
J. Agric. food Chem., 22(5): 849-853.
NARANG, A.S., VERNOY, C.A., & EADON, G.A. (1983) Evaluation of
Nielsen-Kryger steam distillation technique for recovery of
phenols from soil. J. Assoc. Off. Anal. Chem., 66: 1330-1334.
NCI (1968) Evaluation of carcinogenic, teratogenic, and mutagenic
activities of selected pesticides and industrial chemicals,
Bethesda, Maryland, National Cancer Institute, Vol. I (Report
prepared by Bionetics Research Laboratories Inc.) (NTIS
PB-223 160).
NCI (1979) Bioassay of 2, 4,6-trichlorophenol for possible
carcinogenicity (CAS No. 88-06-2), Bethesda, Maryland, National
Cancer Institute, 115 pp (NCI Carcinogenesis Technical Report
Series, No. 155).
NCI (1980) Bioassay of a mixture of 1,2,3,6,7,8-hexachlorodibenzo-p-
dioxin and 1,2, 3, 7,8, 9-hexachlorodibenzo-p-dioxin (gavage)
for possible carcinogenicity, Bethesda, Maryland, National
Cancer Institute (NCI Technical Report Series, No. 198)
(NIH 80-1758).
NESTMANN, E.R., LEE, E.G., MATULA, T.I., DOUGLAS, G.R., & MUELLER,
J.C. (1980) Mutagenicity of constituents identified in pulp and
paper mill effluents using the Salmonella/mammalian-microsome
assay. Mutat. Res., 79: 203-212.
NEUBERT, D. & DILLMAN, I. (1972) Embryotoxic effects in mice treated
with 2,4,5-trichlorophenoxyacetic acid and 2,3,7,8-tetra-
chlorodibenzo- p-dioxin. Naunyn-Schmiedeberg's Arch. Pharmacol.,
272: 243-264.
NHW (1983) Tap water consumption in Canada, Ottawa, Department of
National Health and Welfare, Environmental Health Directorate,
Health Protection Branch (Publication No. 82-EHD-80).
NHW (1988) Chlorophenols and their impurities: a health hazard
evaluation, Ottawa, Department of National Health and Welfare,
Environmental Health Directorate, Health Protection Branch
(Publication No. 84-EHD-110).
NIOSH (1983) Registry of toxic effects of chemical substances,
1981-1982, Cincinnati, Ohio, US National Institute for
Occupational Safety and Health.
NRCC (1978) The aqueous chlorination of organic compounds: chemical
reactivity and effects on environmental quality, Ottawa,
Associate Committee on Scientific Criteria for Environmental
Quality, National Research Council of Canada (NRCC Publication
No. 16450).
NRCC (1982) Chlorinated phenols: Criteria for environmental quality,
Ottawa, Associate Committee on Scientific Criteria for
Environmental Quality, National Research Council of Canada,
191 pp (NRCC Publication No. 18578).
NTP (1988) NTP Technical Report on the toxicology and carcinogenesis
studies of 2,4-dichlorophenols (CAS No. 120-83-2) in F344/N rats
and B6C3F1 mice (feed studies), Research Triangle Park, North
Carolina, National Toxicology Programme, US Department of Health
and Human Services (NTP TR 353) (NIH Publication No. 88-2808).
OLSEN, J.H. & MOLLER-JENSEN, O. (1984) Nasal cancer and chlorophenols.
Lancet, II (8393): 47-48.
OMURA, K. & MATSUURA, T. (1971) Photoinduced reactions -- L.
Photolysis of halogenophenols in aqueous alkali and in aqueous
cyanide. Tetrahedron Lett., 27: 3101-3109.
OTT, M.G., HOLDER, B.B., & OLSON, R.D. (1980) A mortality analysis of
employees engaged in the manufacture of 2,4,5-trichloro-
phenoxyacetic acid. J. occup. Med., 22(1): 47-50.
OTT, M.G., OLSON, R.A., COOK, R.R, & BOND, G.G. (1987) Cohort
mortality study of chemical workers with potential exposure to the
higher chlorinated dioxins. J. occup. Med., 29(5): 422-429.
PAASIVIRTA, J., SARKKA, J., LESKIJARVI, T., & ROOS, A. (1980)
Transportation and enrichment of chlorinated phenolic compounds in
different aquatic food chains. Chemosphere, 9: 441-456.
PAASIVIRTA, J., SARKKA, J., AHO, M., SURMA-AHO, K., TARHANEN, J, 8,:
ROOS, A. (1981) Recent trends of biocides in pikes of the Lake
Päijäne. Chemosphere, 10(4): 405-414.
PAASIVIRTA, J., SARKKA, J., SURMA-AHO, K., HUMPPI, T., KUOKKANEN, T.,
& MARTTINEN, M. (1983) Food chain enrichment of organochlorine
compounds and mercury in clean and polluted lakes of Finland.
Chemosphere, 12(2): 239-252.
PAASIVIRTA, J., HEINOLA, K., HUMPPI, T., KARJALAINEN, A., KNUUTINEN,
J., MANTYKOSKI, K., PAUKKU, R., PIILOLA, T., SURMA-AHO, K.,
TARHANEN, J., WELLING, L., & VIHONEN, H. (1985) Polychlorinated
phenols, guaiacols and catechols in environment. Chemosphere,
14(5): 469-491.
PAL, H.S., MURPHY, T., CARTER, A.C,, & DREW, S.W. (1980) Rapid assays
for microbial degradation of 2-chlorophenol, Blacksburg,
Virginia, Virginia Water Resources Research Center, Virginia
Polytechnic Institute and State University (Report No. 128).
PARKER, C.E. & PRIBYL, E.J., Jr (1984) Assessment of bacterial ATP
response as a measurement of aquatic toxicity. In: Liu, D. &
Dutka, B.J., ed. Toxicity screening procedures using bacterial
systems, New York, Basel, Marcel Dekker Inc., pp. 283-293.
PARR, L.J., GEE, M.G., LAND, D.G., ROBINSON, D., & CURTIS, R.F. (1974)
Chlorophenols from wood preservatives in broiler house litter.
J. Sci. Food Agric., 25(7): 835-841.
PAZELEROVA, J., LUKAS, E., NEMCOVA, M., SPACILOVA, M., JIRASEK, L.,
KALENSKY, J., JOHN, J., JIRASEK, A., & PICKOVA, J. (1974) [Chronic
poisoning by chlorinated hydrocarbons, formed in the production of
2,4,5-sodium trichlorophenoxyacetate.] Prac. Lek.,
26(9): 332-339 (in Czech, with English summary).
PEARCE, N.E., SMITH, A.H., HOWARD, J.K., SHEPPARD, R., GILES, H.J., &
TEAGUE, C.A. (1986) Non-Hodgkin's lymphoma and exposure to phenoxy
herbicides, chlorophenols, fencing work, and meat works
employment: a case study. Br. J. ind. Med., 43: 75-83.
PEKARI, K. & AITIO, A. (1982) A simple liquid chromatographic method
for the anaylsis of penta and tetrachlorophenols in urine of
exposed workers. J. Chromatogr., 232: 129-136.
PEKARI, K., BOUDENE, C., & AITIO, A. (1986) Kinetics of
2,4,6-trichlorophenol in different organs in the rat.
Arch. Toxicol., 59: 41-44.
PHIPPS, G.L., HOLCOMBE, G.W., & FIANDT, J.T. (1981) Acute toxicity of
phenol and substituted phenols to the fathead minnow. Bull.
environ. Contam. Toxicol., 26: 585-593.
PIERCE, R.H., Jr & VICTOR, D.M. (1978) The fate of pentachlorophenol
in an aquatic ecosystem. In: Rao, K.R., ed. Pentachlorophenol:
Chemistry, pharmacology, and environmental toxicology, New York,
London, Plenum Press, pp. 41-52.
PIET, G.J. & DE GRUNT, F. (1975) Organic chloro compounds in surface
and drinking water of the Netherlands. In: Problems raised by the
contamination of man and his environment by persistent pesticides
and organo-halogenated compounds. European Colloquium,
Luxembourg, Commission of the European Communities, pp. 81-92.
PIGNATELLO, J.J., MARTINSON, M.M., STEIERT, J.G., CARLSON, R.E., &
CRAWFORD, R.L. (1983) Biodegradation and photolysis of
pentachlorophenol in artificial freshwater streams. Appl.
environ. Microbiol., 46: 1024-1031.
PIGNATELLO, J.J., JOHNSON, L.K., MARTINSON, M.M., CARLSON, R.E., &
CRAWFORD, R.L. (1986) Response of the microflora in outdoor
experimental streams to pentachlorophenol: environmental factors.
Can. J. Microbiol., 32: 38-46.
PITTER, P. (1976) Determination of biological degradability of organic
substances. Water Res., 10: 231-235.
POLAND, A.P., SMITH, D., METTER, G., & POSSICK, P. (1971) A health
survey of workers in a 2,4-D and 2,4,5-T plant. Arch. environ.
Health, 22: 316-327.
RAO, K.R., FOX, F.R., CONKLIN, P.J., & CANTELMO, A.C. (1981)
Comparative toxicology and pharmacology of chlorophenols: studies
on the grass shrimp, Palaemonetespugio. In: Vernberg, F.J.,
Calabrese, A., Thurberg, F.P., & Vernberg, W.B., ed. Biological
monitoring of marine pollutants, New York, London, Academic
Press, pp. 37-72.
RAPPE, C, GARA, A., & BUSER, H.R. (1978a) Identification of
polychlorinated dibenzofurans (PCDFs) in commercial chlorophenol
formulations. Chemosphere, 12: 981-991.
RAPPE, C., MARKLUND, B., BUSER, H.-R., & BOSSHARDT, H.-P. (1978b)
Formation of polychlorinated dibenzo- p-dioxins (PCDDs) and
dibenzofurans (PCDFs) by burning or heating chlorophenates.
Chemosphere, 3: 269-281.
RAPPE, C., BUSER, H.-R., & BOSSHARDT, H.-P. (1979) Dioxins,
dibenzofurans, and other polyhalogenated aromatics: Production,
use, formation and destruction. Ann. N.Y. Acad. Sci., 320: 1-18.
RAPPE, C., NYGREN, M., BUSER, H.-R., & KAUPPINEN, T. (1982)
Occupational exposure to polychlorinated dioxins and
dibenzofurans. In: Hutzinger, O., Frei, R.W., Merian, E., &
Pocchiari, F., ed. Chlorinated dioxins and related compounds:
Impact on the environment, Oxford, Pergamon Press, pp. 495-514
(Pergamon Series on Environmental Science, Vol. 5).
RASANEN, L., HATTULA, M.L., & ARSTILA, A.U. (1977) The mutagenicity of
MCPA and its soil metabolites, chlorinated phenols, catechols and
some widely used slimicides in Finland. Bull. environ. Contam.
Toxicol., 18(5): 565-571.
REALINI, P.A. (1981) Determination of priority pollutant phenols in
water by HPLC. J. chromatogr. Sci., 19: 124-129.
REINER, E.A., CHU, J.P., & KIRSCH, E.J. (1978) Microbial metabolism of
pentachlorophenol. In: Rao, K.R., ed. Pentachlorophenol:
Chemistry, pharmacology, and environmental toxicology, New York,
London, Plenum Press, pp. 67-81.
RENNER, G. & MUCKE, W. (1986) Transformations of pentachlorophenol.
Part I: Metabolism in animals and man. Toxicol. environ. Chem.,
11: 9-29.
RIBO, J.M. & KAISER, K.L.E. (1983) Effects of selected chemicals to
photoluminescent bacteria and their correlations with acute and
sublethal effects on other organisms. Chemosphere,
12(11): 1421-1442.
RICHARDSON, N.G. (1978) Wood preserving effluents and their treatment.
In: The timber processing industry. Proceedings of a Seminar on
Technology Transfer, Toronto, Ontario, 10-11 March, 1977,
Ottawa, Environment Canada (Report No. EPS 3-WP).
ROBERTS, B.L. & DOROUGH, H.W. (1984) Relative toxicities of chemicals
to the earthworm, Eisenia foetida. Environ. Toxicol. Chem.,
3: 67-78.
ROBERTS, M.S., ANDERSON, R.A., & SWARBRICK, J. (1977) Permeability of
human epidermis to phenolic compounds. J. Pharm. Pharmacol.,
29: 677-683.
ROBERTS, M.S., ANDERSON, R.A., SWARBRICK, J., & MOORE, D.E. (1978) The
percutaneous absorption of phenolic compounds: the mechanism of
diffusion across the stratum corneum. J. Pharm. Pharmacol.,
30: 486-490.
ROYAL COMMISSION ON THE USE AND EFFECTS OF CHEMICAL AGENTS ON
AUSTRALIAN PERSONNEL IN VIETNAM (1985) Final report. Volume 4:
Cancer, Canberra, Australian Government Publishing Service,
pp. 71-74.
RUCKDESCHEL, G. & RENNER, G. (1986) Effects of pentachlorophenol and
some of its known and possible metabolites on fungi. Appl.
environ. Microbiol., 51(6): 1370-1372.
SAARIKOSKI, J. & VILUKSELA, M. (1981) Influence of pH on the toxicity
of substituted phenols to fish. Arch. environ. Contam. Toxicol.,
10: 747-753.
SACKMAUEROVA-VENINGEROVA, M., UHNAK, J., SZOKOLAY, A., & KOCAN, A.
(1981) Identification of chlorinated phenols as degradation
products of chlorinated pesticides in biological materials.
J. Chromatogr., 205: 194-198.
SALKINOJA-SALONEN, M.S., VALO, R., APAJALAHTI, J., HAKULINEN, R.,
SILAKOSKI, R., & JAAKKOLA, T. (1984) Biodegradation of
chlorophenolic compounds in wastes from wood-processing industry.
In: Klug, M. & Reddy, C., ed. Current perspectives in microbial
ecology, Washington, DC, American Society for Microbiology,
pp. 668-676.
SCHAUERTE, W., LAY, J.P., KLEIN, W., & KORTE, F. (1982) Influence of
2,4,6-trichlorophenol and pentachlorophenol on the biota of
aquatic systems. Chemosphere, 11(1): 71-79.
SCHELLENBERG, K., LEUENBERGER, C., & SCHWARZENBACH, R.P. (1984)
Sorption of chlorinated phenols by natural sediments and aquifer
materials. Environ. Sci. Technol., 18: 652-657.
SCHULTZ, T.W. & RIGGIN, G.W. (1985) Predictive correlations for the
toxicity of alkyl- and halogen-substituted phenols. Toxicol.
Lett., 25: 47-54.
SCHWETZ, B.A., KEELER, P.A., & GEHRING, P.J. (1974) Effect of purified
and commercial grade tetrachlorophenol on rat embryonal and fetal
development. Toxicol. appl. Pharmacol., 28: 146-150.
SEIP, H.M., ALSTAD, J., CARLBERG, G.E., MARTINSEN, K., & SKAANE, R.
(1986) Measurement of mobility of organic compounds in soils.
Sci. total Environ., 50: 87-101.
SENES (1985) Drinking water criteria reviews for 2, 4,5-trichloro-
phenol, 2, 4,6-trichlorophenol, 2,3,5-trichlorophenol, and
2,3,6-trichlorophenol, Willowdale, Ontario, Senes Consultants
Limited (Report prepared for the Ontario Ministry of the
Environment, Water Resources Branch, Drinking Water Section).
SEYLER, D.E., EAST, J.M., CONDIE, L.W., & BORZELLECA, J.F. (1984) The
use of in vitro methods for assessing reproductive toxicity.
Dichlorophenols. Toxicol. Lett., 20(3): 309-315.
SHAFIK, T.M., SULLIVAN, H.C., & ENOS, H.R. (1973) Multiresidue
procedure for halo- and nitrophenols: measurement of exposure to
biodegradable pesticides yielding these compounds as metabolites.
J. agric. food Chem., 21(2): 295-298.
SHEN, S.Y., VILLENEUVE, D.C., CHU, I., KELLY, J., & GILMAN, A.P.
(1983) Acute dermal toxicity of tetrachlorophenols in the rat.
Bull. environ. Contam. Toxicol., 31: 680-685.
SHERMAN, M., BECK, J., & HERRICK, R.B. (1972) Chronic toxicity and
residues from feeding Nemacide [ O-(2,4-dichlorophenyl)
O,O-diethyl phosphothionate] to laying hens. J. agric. food
Chem., 20(3): 617-624.
SHUMWAY, D.L. & PALENSKY, J.R. (1973) Impairment of the flavour of
fish by water pollutants, Corvallis, Oregon State University,
Department of Fish and Wildlife, 83 pp (Report No. W73-11322)
(EPA Report No. R3-73-010).
SITHOLE, B.B. & WILLIAMS, D.T. (1986) Halogenated phenols in water at
forty Canadian potable water treatment facilities. J. Assoc. Off.
Anal. Chem., 69(5): 807-810.
SIUDA, J.F. (1980) Natural production of organohalogens. In: Jolley,
R.L., Brungs, W.A., Cumming, R.B., & Jacobs, V.A., ed. Water
chlorination: Environmental impact and health effects, Ann
Arbor, Michigan, Ann Arbor Science Publishers Inc., Vol. 3,
pp. 63-72.
SMITH, A.H., PEARCE, N.E., FISHER, D.O., GILES, H.J., TEAGUE, C.A., &
HOWARD, J.K. (1984) Soft-tissue sarcoma and exposure to phenoxy
herbicides and chlorophenols in New Zealand. J. Natl Cancer
Inst., 73(5): 1111-1117.
SOMANI, S.M. & KHALIQUE, A. (1982) Distribution and metabolism of
2,4-dichlorophenol in rats. J. Toxicol. environ. Health,
9: 889-897.
SOMANI, S.M., SMART, T., & KHALIQUE, A. (1984) Metabolism of
2,4-dichlorophenol by isolated perfused rat liver. I. Toxicol.
environ. Health, 13: 787-798.
SPOKES, J.R. & WALKER, N. (1974) Chlorophenol and chlorobenzoic acid
co-metabolism by different genera of soil bacteria. Arch.
Microbiol., 96(2): 125-134.
STALLING, D.L., SMITH, L.M., & PETTY, J.D. (1979) Approaches to
comprehensive analyses of persistent halogenated environmental
contaminants. In: Van Hall, C.D., ed. Measurement of organic
pollutants in water and wastewater, Washington, DC, American
Society for Testing and Materials, pp. 302-323 (ASTM STP 686).
STATISTICS CANADA (1986) Apparent per capita food consumption in
Canada. Part I & II. 1984, Ottawa, Ministry of Supply and
Services Canada (Catalogue No. 32-230).
STERLING, T.D., STOFFMAN, L.D., STERLING, D.A., & MATE, G. (1982)
Health effects of chlorophenol wood preservatives on sawmill
workers. Int. J. Health Serv., 12(4): 559-571.
STIJVE, T. (1981) Determination of pentachlorophenol and
2,3,4,6-tetrachlorophenol in edible gelatins. Dtsch. Lebensm.
Rundschau, 77: 249-253.
STINGILY, K.O. (1940) A new industrial chemical dermatitis. Southern
med. J., 33(12): 1268-1272.
STOCKDALE, M. & SELWYN, M.J. (1971) Influence of ring substituents on
the action of phenols on some dehydrogenases, phosphokinases and
the soluble ATPase from mitochondria. Eur. J. Biochem.,
21: 416-423.
STRETZ, L.A. & VAVRUSKA, J.S. (1984) Controlled air incineration of
pentachlorophenol-treated wood, Washington, DC, US Environmental
Protection Agency, 110 pp (EPA Report No. 600/2-84-089).
SUGIARA, K., AOKI, M., KANEKO, S., DAISAKU, I., KOMATSU, Y., &
SHIBUYA, H. (1984) Fate of 2,4,6-trichlorophenol, penta-
chlorophenol, p-chlorobiphenyl, and hexachlorobenzene in an
outdoor experimental pond: comparison between observation and
predictions based on laboratory data. Arch. environ. Contam.
Toxicol., 13: 745-758.
SUSKIND, R.R. & HERTZBERG, V.S. (1984) Human health effects of 2,4,5-T
and its toxic contaminants. J. Am. Med. Assoc.,
251(18): 2372-2380.
SUND, K.A. & NOMURA, N. (1963) Laboratory evaluation of several
herbicides. Weed Res., 3: 35-43.
TABAK, H.H., CHAMBERS, C.W., & KABLER, P.W. (1964) Microbial
metabolism of aromatic compounds. I. Decomposition of phenolic
compounds and aromatic hydrocarbons by phenol-adapted bacteria.
J. Bacteriol., 87(4): 910-919.
TEIDJE, J.M. & ALEXANDER, M. (1969). Enzymatic cleavage of the ether
bond of 2,4-dichlorophenoxyacetate. J. agric. food Chem.,
17: 1080-1084.
TELFORD, M. (1974) Blood glucose in crayfish. II. Variations induced
by artificial stress. Comp. Biochem. Physiol., 48A: 555-560.
THIESS, A.M, FRENTZEL-BEYME, R., & LINK, R. (1982) Mortality study of
persons exposed to dioxin in a trichlorophenol-process accident
that occurred in the BASF AG on November 17, 1953. Am. J. ind.
Med., 3: 179-189.
TOWNSEND, J.C., BODNER, K.M., VAN PEENEN, P.F.D., OLSON, R.D., & COOK,
R.R. (1982) Survey of reproductive events of wives of employees
exposed to chlorinated dioxins. Am. J. Epidemiol.,
115(5): 695-713.
TREVORS, J.T., MAYFIELD, C.I., & INNISS, W.E. (1982) Effect of
sequence of exposure to chlorophenols in short-term bacterial
bioassays. Arch. environ. Contam. Toxicol., 11: 203-207.
TYLER, J.E. & FINN, R.K. (1974) Growth rates of a pseudomonad on
2,4-dichlorophenoxyacetic acid and 2,4-dichlorophenol. Appl.
Microbiol., 28: 181-184.
US EPA (1971) Biological treatment of chlorophenolic wastes,
Washington, DC, US Environmental Protection Agency (Water
Pollution Control Research Series).
US EPA (1978) Rebuttable presumption against registration and
continued registration of pesticide products containing
2,4,5-trichlorophenol and its salts. Fed. Reg.,
43(149): 34026-34054.
US EPA (1979) In: Kozak, V.P., Simsiman, G.V., Chesters, G., Stensby,
D., & Harkin, J. ed. Reviews of the environmental effects of
pollutants: XI. Chlorophenols, Washington, DC, US Environmental
Protection Agency, 492 pp (EPA Report No. 600/1-79-012).
US EPA (1980a) Ambient water quality criteria for 2-chlorophenol,
Washington, DC, US Environmental Protection Agency, Office of
Water Regulations and Standards Criteria (EPA Report
No. 440/5-80-034).
US EPA (1980b) Ambient water quality criteria for 2, 4-dichloro-
phenol, Washington, DC, US Environmental Protection Agency,
Office of Water Regulations and Standards Criteria (EPA Report
No. 440/5-80-042).
US EPA (1980c) Ambient water quality criteria of chlorinated
phenols, Washington, DC, US Environmental Protection Agency,
Office of Water Regulations and Standards Criteria (EPA Report
No. 440/5-80-032).
US EPA (1980d) Analytical methodology for determination of
chlorophenols in human and environmental media, Research
Triangle Park, North Carolina, US Environmental Protection Agency,
Health Effects Research Laboratory (EPA Report No. 600/2-80-181).
VALO, R., KITUNEN, V., SALKINOJA-SALONEN, M., & RAISANEN, S. (1984)
Chlorinated phenols as contaminants of soil and water in the
vicinity of two Finnish sawmills. Chemosphere, 13(8): 835-844.
VAN OMMEN, B., ADANG, A.E.P., BRADER, L., POSTHUMUS, M.A., MULLER, F.,
& VAN BLADEREN, P.J. (1966) The microsomal metabolism of
hexachlorobenzene. Origin of the covalent binding to protein.
Biochem. Pharmacol., 35(19): 3233-3238.
VAN OMMEN, B, ADANG, A., MULLER, F., & VAN BLADEREN, P.J. (1986) The
microsomal metabolism of pentachlorophenol and its covalent
binding to protein and DNA. Chem.-biol. Interact., 60: 1-11.
VAN OMMEN, B., VONCKEN, J.W., MULLER, F., & VAN BLADEREN, P.J. (1988)
The oxidation of tetrachloro-1,4-hydroquinone by microsomes and
purified cytochrome P-450s. Implications for covalent binding to
protein and involvement of reactive oxygen species. Chem.-biol.
Interact., 65(3): 247-259.
VERSCHUEREN, K. (1983) Handbook of environmental data on organic
chemicals, 2nd ed., New York, Van Nostrand Reinhold Co.
VINEIS, P., TERRACINI, B., CICCONE, G., CIGNETTI, A., COLOMBO, E.,
DONNA, A, MAFFI, L., PISA, R., RICCI, P., ZANINI, E., & COMBA, P.
(1987) Phenoxy herbicides and soft-tissue sarcomas in female rice
weeders: A population-based case-referent study. Scand. J. Work
Environ. Health, 13: 9-17.
VIRTANEN, M.T. & HATTULA, M.-L. (1982) The fate of 2,4,6-trichloro-
phenol in an aquatic continuous-flow system. Chemosphere,
11(7): 641-649.
VIZETHUM, W. & GOERZ, G. (1979) Induction of the hepatic microsomal
and nuclear cytochrome P-450 system by hexachloro-benzene,
pentachlorophenol and trichlorophenol. Chem.-biol. Interact.,
28: 291-299.
WALKER, N. (1954) Preliminary observations on the decomposition of
chlorophenols in soil. Plant Soil, 5(2): 194-204.
WALKER, N. (1973) Metabolism of chlorophenols by Rhodoturula
glutinis. Soil Biol. Biochem., 5(5): 525-530.
WANG, W. & REED, P. (1984) Nitrobacter bioassay for aquatic toxicity.
In: Liu, D. & Dutka, B.J., ed. Toxicity screening procedures
using bacterial systems, New York, Basel, Marcel Dekker Inc.,
pp. 309-325.
WEBER, K. & ERNST, W. (1978) Levels and pattern of chlorophenols in
water of the Weser Estuary and the German Bight. Chemosphere,
7(11): 873-879.
WEGMAN, R.C.C. & HOFSTEE, A.W.M. (1979) Chlorophenols in surface
waters of the Netherlands (1976-1977). Water Res., 13: 651-657.
WEGMAN, R.C.C. & VAN DEN BROEK, H.H. (1983) Chlorophenols in river
sediment in the Netherlands. Water Res., 17(2): 227-230.
WEINBACH, E.C. (1957) Biochemical basis for the toxicity of
pentachlorophenols. Proc. Natl Acad. Sci., 43: 393-397.
WEINBACH, E.C. & GARBUS, J. (1965) The interaction of uncoupling
phenols with mitochondria and with mitochondrial protein.
J. biol. Chem., 240(4): 1811-1819.
WHO (1984) Guidelines for drinking-water quality. Vol. 1:
Recommendations, Geneva, World Health Organization, 130 pp.
WHO (1985) Guidelines for drinking-water quality. Vol. 2: Health
Criteria and other supporting information, Geneva, World Health
Organization, 335 pp.
WHO (1987a) Environmental Health Criteria 70: Principles for the
safety assessment of food additives and contaminant in food,
Geneva, World Health Organization, 174 pp.
WHO (1987b) Environmental Health Criteria 71: Pentachlorophenol,
Geneva, World Health Organization, 236 pp.
WHO (in press) Environmental Health Criteria: Polychlorinated
dibenzo-p-clioxins and dibenzofurans. Geneva, World Health
Organization.
WILLIAMS, D.T., LEBEL, G.L., & JUNKINS, E. (1984) A comparison of
organochlorine residues in human adipose tissue autopsy samples
from two Ontario municipalities. J. Toxicol. environ. Health,
13: 19-29.
WOOD, S., ROM, W.N., WHITE, G.L., & LOGAN, D.C. (1983)
Pentachlorophenol poisoning. J. occup. Med., 25(7): 527-530.
WOODROW, J.E., MAJEWSKI, M.S., & SEIBER, J.N. (1976) Accumulative
sampling of trace pesticides and other organics in surface water
using XAD-4 resin. J. environ. Sci. Health, B21(2): 143-164.
WOODS, J.S., POLISSAR, L., SEVERSON, R.K., HEUSER, L.S., & KULANDER,
B.G. (1987) Soft tissue sarcoma and non-Hodgkin's lymphoma in
relation to phenoxy herbicide and chlorinated phenol exposure in
Western Washington. J. Natl Cancer Inst, 78(5): 899-910.
WOOLSON, E.A, THOMAS, R.F., & ENSOR, P.D.J. (1972) Survey of
polychlorodibenzo- p-dioxin content in selected pesticides.
J. agric. food Chem., 20: 351-354.
WRIGHT, F.C., RINER, J.C., PALMER, J.S., & SCHLINKE, J.C. (1970)
Metabolic and residue studies with 2-(2,4,5-trichlorophenoxy)-
ethyl-2,2-dichloropropionate (Erbon) herbicide in sheep.
J. agric. food Chem., 18(5): 845-847.
WYLLIE, J.A., GABICA, J., BENSON, W.W, & YODER, J. (1975) Exposure and
contamination of the air and employees of a pentachlorophenol
plant, Idaho -- 1972. Pestic. monit. J., 9(3): 150-153.
XIE, T.-M. (1983) Determination of trace amounts of chlorophenols and
chloroguaiacols in sediments. Chemosphere, 12: 1183-1191.
XIE, T.-M., ABRAHAMSSON, K., FOGELQVIST, E., & JOSEFSSON, B. (1986)
Distribution of chlorophenolics in a marine environment. Environ.
Sci. Technol., 20: 457-463.
YOST, R.A., FETTEROLF, D.D., HASS, J.R., HARVAN, D.J., WESTON, A.F.,
SKOTNICKI, P.A., & SIMON, N.M. (1984) Comparison of mass
spectrometric methods for trace level screening of
hexachlorobenzene and trichlorophenol in human blood serum and
urine. Anal. Chem., 56: 2223-2228.
ZACK, J.A. & SUSKIND, R.R. (1980) The mortality experience of workers
exposed to tetrachlorobenzodioxin in a trichlorophenol process
accident. J. occup. Med, 22(1): 11-14.
ZACK, J.A. & GAFFEY, W.R. (1983) A mortality study of workers employed
at the Monsanto plant in Nitro, West Virginia. Environ. Sci.
Res., 26: 575-591.
ZOETEMAN, B.C.J. (1975) Odour nuisance by organo-halogenated compounds
in water and its toxicological impact. In: Problems raised by the
contamination of man and his environment by persistent pesticides
and organo-halogenated compounds. European Colloquium,
Luxembourg, Commission of the European Communities, pp. 527-544
(EUR 5196).
ZOETEMAN, B.C.J., DE GREEF, E., & BRINKMAN, F.J.J. (1981) Persistency
of organic contaminants in groundwater, lessons from soil
pollution incidents in the Netherlands. Sci. total Environ.,
21: 187-202.
a The chlorophenol congeners are designated as follows:
monochlorophenols (MCP); dichlorophenols (DCP); trichlorophenols
(T3CP); tetrachlorophenols (T4CP); pentachlorophenol (PCP).
Chlorine substitution is also indicated: 2,4-dichlorophenol
(2,4-DCP); 2,4,6-trichlorophenol (2,4,6-T3CP), etc.
Table 1. Information on the identity of chlorophenol congenersa
CAS numberb Common name Abbreviation Molecular Common synonymsc Common trade
formula names
95-57-8 2-monochlorophenol 2-MCP C6H5ClO o-chlorophenol;
ortho-chlorophenol;
1-chloro-2-hydroxybenzene
108-43-0 3-monochlorophenol 3-MCP C6H5ClO m-chlorophenol;
meta-chlorophenol;
1-chloro-3-hydroxybenzene
106-48-9 4-monochlorophenol 4-MCP C6H5ClO p-chlorophenol;
para-chlorophenol;
1-chloro-4-hydroxybenzene
576-24-9 2,3-dichlorophenol 2,3-DCP C6H4Cl20
120-83-2 2,4-dichlorophenol 2,4-DCP C6H4Cl20 NCl-C55345
583-78-8 2,5-dichlorophenol 2,5-DCP C6H4Cl20
87-65-0 2,6-dichlorophenol 2,6-DCP C6H4Cl20
95-77-2 3,4-dichlorophenol 3,4.DCP C6H4Cl20
591-35-5 3,5-dichlorophenol 3,5-DCP C6H4Cl20
15950-66-0 2,3,4-trichlorophenol 2,3,4-T3CP C6H3Cl30
933-78-8 2,3,5-trichlorophenol 2,3,5-T3CP C6H3Cl30
933-75-5 2,3,6-trichlorophenol 2,3,6-T3CP C6H3Cl30
95-95-4 2,4,5-trichlorophenol 2,4,5-T3CP C6H3Cl30 NCl-C61187 Collunosol;
Dowicide 2;
Dowicide B;
Nurelle;
Preventol 1
Table 1. (contd).
CAS numberb Common name Abbreviation Molecular Common synonymsc Common trade
formula names
88-06-2 2,4,6-trichlorophenol 2,4,6-T3CP C6H3Cl30 NCl-C02904 Dowicide 2; Omal;
Phenachlor
609-19-8 3,4,5-trichlorophenol 3,4,5-T3CP C6H3Cl30
4901-51-3 2,3,4,5-tetrachlorophenol 2,3,4,5-T4CP C6H2Cl40
58-90-2 2,3,4,6-tetrachlorophenol 2,3,4,6-T4CP C6H2Cl40 Dowicide 6
935-95-5 2,3,5,6-tetrachlorophenol 2,3,5,6-T4CP C6H2Cl40
a From: Jones (1981).
b Chemical Abstracts Service Registry number.
c From: NIOSH (1983) and Verschueren (1983).
Note: Owing to the planar nature of the phenol ring, other congeners (e.g., 2,4,5,6-T4CP) are possible, but these are
identical in structure to the listed congeners.
Technical grade chlorophenols are heterogeneous mixtures of
chlorophenol congeners, unreacted precursors, and a variety of dimeric
microcontaminants. For example, Cochrane et al. (1983) found that
technical 2,4-DCP contained on average, 92.24% 2,4-DCP, 4.48% 2,6-DCP,
1.24% 2,4,6-T3CP, 1.09% 2-MCP, and 0.46% 4-MCP.
Similarly, Levin et al. (1976) examined the composition of 3
commercial chlorophenol formulations used to control fungi in Swedish
sawmills and found that Na-2,4,6-T3CP contained approximately 5%
T4CP, Na-2,3,4,6-T4CP included 5% T3CP and 10% PCP, and technical
NaPCP contained 5% T4CP.
Kleinman et al. (1986) determined that commercial Na-T4CP, used
in the USA, contained 3.1% PCP, 20.7% 2,3,4,6-T4CP, and less than
0.4% of other chlorophenol congeners. These results are typical,
showing that roughly 2-12% T4CP congeners occur in technical PCP
formulations, together with trace quantities of several lower
chlorophenols (Jones, 1981; Lanouette et al., 1984).
Contamination of technical chlorophenols varies according to the
production process used. Because of the elevated reaction temperatures
used to produce chlorophenols, a number of compounds are present as
microcontaminants in technical chlorophenol preparations prepared by
this procedure. These include the polychlorinated dibenzo- p-dioxins
(PCDDs) polychlorinated dibenzofurans (PCDFs), polychlorinated
diphenyl ethers, polychlorinated phenoxyphenols, polychlorinated
benzenes, and polychlorinated biphenyls. Where the alkaline hydrolysis
of chlorobenzenes is used to manufacture chlorophenols, the technical
product also contains the unreacted chlorobenzene. Technical
chlorophenol salts usually also contain an excess of sodium or
potassium hydroxide.
While commercial MCP and DCP contain little or no detectable PCDDs
and PCDFs, presumably because their manufacture does not involve high
enough temperatures, other chlorophenols may contain up to many mg/kg
of particular PCDDs, and PCDFs (Firestone et al., 1972; Woolson et
al., 1972; Levin et al., 1976; Levin & Nilsson, 1977; Rappe et al.,
1979; Cedar, 1984; Kleinman et al., 1986). Concentrations of PCDDs and
PCDFs in some American and European chlorophenols are provided in
Table 2. Tri- and tetrachloro-dibenzoxo-dioxins predominate in T3CP
formulations, while the hexa, hepta, and octa congeners are the major
PCDD contaminants in technical T4CP and PCP (Firestone et al., 1972;
Rappe et al., 1978a). 2,3,7,8-Tetrachloro-dibenzo- p-dioxin
(2,3,7,8-TCDD) occurs primarily as a contaminant of 2,4,5-T3CP
(Table 2), though it is present at low µg/kg concentrations in T4CP,
PCP, and NaPCP (Hagenmaier, 1986; Hagenmaier & Brunner, 1987).
Predioxins (chlorinated phenoxyphenols) may comprise as much as 5% of
technical CP preparations (Levin et al., 1976; Levin & Nilsson, 1977).
Most of the data in Table 2 concern chlorophenol formulations from
the 1970s. As a result of modifications in production chemistry, it is
likely that the levels of microcontaminants in current formulations
are somewhat lower. Indeed, all of the 1986 tetrachlorophenol products
assayed by Agriculture Canada (1987) contained levels of H6CDD that
were several times lower than those in the earlier reports (Table 2).
2.2 Physical and Chemical Properties
Data on some physical and chemical properties of chlorophenols are
summarized in Table 3. All of the CPs are solids at room temperature,
except for 2-MCP, which is a liquid. They have strong odours that have
been described as pungent or medicinal, particularly those of
2-monochlorophenol (2-MCP) and 2,4-dichlorophenol (2,4-DCP). Taste and
odour thresholds are so low that Maximum Acceptable Concentrations of
chlorophenols in drinking-water are based on organoleptic rather than
toxicological criteria (US EPA, 1980c; WHO, 1984).
Although the solubility in water of all chlorophenols is poor,
varying from 2.1 × 10-1 mol/litre for 2-MCP t o 7.9 × 10-4 mol/litre
for 2,3,4,6-T4CP (US EPA 1980c) they readily dissolve in a number of
organic solvents. In contrast, the sodium or potassium salts of
chloropenols (most commonly NaT3CP, NaT4CP, and NaPCP) are up to
four orders of magnitude more soluble in water than the parent
compounds. The acidity of chlorophenols increases as the number of
chlorine substitutions increases. Thus, ionization of the higher
chlorophenols begins at a lower pH than that of the lower
chlorophenols (pH approximately 3.5 versus 7 for PCP and 2-MCP,
respectively), with important implications for the interactions
between pH and chlorophenol sorption (section 4.1.2.1), or toxicity
(section 6.1.1). The n-octanol-water partition coefficient of
chlorophenols also increases with chlorination, indicating a
propensity on the part of the higher chlorophenols to bioaccumulate.
2.3 Conversion Factors
MCP 1 mg/m3 = 0.190 ppm; 1 ppm = 5.258 mg/m3
DCP 1 mg/m3 = 0.150 ppm; 1 ppm = 6.667 mg/m3
T3CP 1 mg/m3 = 0.124 ppm; 1 ppm = 8.076 mg/m3
T4CP 1 mg/m3 = 0.105 ppm; 1 ppm = 9.488 mg/m3
Table 2. Polychlorodibenzo-p-dioxins (PCDDs) and polychtorodibenzofurans (PCDFs) in some American and European
mono-, di-, tri-, and tetrachlorophenolsa
Formulation PCDD Concentration PCDF Concentration Year
(mg/kg) (mg/kg) sample
received
2-MCP ND T4CDF presentb 1967
2,4-DCP ND ND 1970
2,6-DCP ND ND 1970d
Na-2,4,5-T3CP ND ND 1967
Na-2,4,5-T3CP 2,7-D2CDD 0.72 ND 1969
2,3,7,8-T4CDDc 1.4
2,4,5-T3CP 1,3,6,8-T4CDD 0.30 ND 1969
2,3,7,8-T4CDc 6.2
2,4,5-T3CP P5CDD 1.5 ND 1970
2,4,5-T3CP ND T3CDF presentb 1970
2,4,5-T3CP 2,3,7,8-T4CDD 0.07 ND 1970
2,4,6-T3CPf 2,3,7-T3CDD 93 T4CDF 1.5 1970d
1,3,6,8-T4CDD 49 P5CDF 17.5
H6CDF 36
H7CDF 4.8
O8DF
Table 2. (contd.)
Formulation PCDD Concentration PCDF Concentration Year
(mg/kg) (mg/kg) sample
received
2,3,4,6-T4CP H6CDDc 15 H6CDF presentb 1970d
H6CDDc 14 H7CDF present
H6CDDc 5.1 O8CDF
O8CDD 0.17
2,3,4,6-T4CP H6CDc 4.1 T4CDF < 0.5 1967
P5CDF 10 (PCDDs);
H6CDF 70 1967d
H7CDF 70 (PCDFs)
O8CDF 10
2,3,4,6-T4CPf ND T4CDF presentb 1967d
H6CDF present
2,3,4,6-T4CPe T4CDD 0.7 T4CDF ca.10 1970d
P5CDD 5.2 P5CDF ca.10
H6CDD 9.5 H6CDF ca.60-70
H7CDD 5.6 H7CDF ca.60-70
O8CDD 0.7 O8CDF ca.10
2,3,4,6-T4CPe T4CDD 0.4 T4CDF ca.10 1970d
P5CDD 3.5 P5CDF ca.10
H6CDD 5.3 H6CDF ca.60-70
H7CDD 2.1 H7CDF ca.60-70
O8CDD 0.3 O8CDF ca.10
Table 2. (contd.)
Formulation PCDD Concentration PCDF Concentration Year
(mg/kg) (mg/kg) sample
received
TCP/PCPg H6CDD 1-4 (n = 6) not reported 1986
H7CDD 40-102 (n = 6) not reported
O8CDD 27-55 (n = 6) not reported
Na-T4CP/PCPg H6CDD N.D.-4 (n = 13) not reported 1986
H7CDD 10-119 (n = 13) not reported
O8CDD 5-330 (n = 13) not reported
a Reports of PCDDs from Firestone et al (1972), except where otherwise indicated; quantitative data on
PCDF concentrations from Rappe et al. (1978a).
b Unquantified. See Firestone et al. (1972).
c Confirmed by combined gas chromatography-mass spectrometry,
d Not reported.
e Rappe et al. (1979).
f Rappe et al. (1978b).
g Agriculture Canada (1987).
ND: No congener detected; limit of detection from Firestone et al. (1972) is approximately 0.02 ppm for PCDDs,
that from Rappe et al. (1978a) is roughly 0.01-0.04 mg/kg (Buser & Bosshardt, 1976).
Table 3. Physical and chemical properties of chlorophenols other than pentachlorophenola
Compound Relative Density Boiling point Melting point Flash Vapour log
molecular (°C at 760 mm) (°C at 760 mm) point pressure n-octanol/
mass (°C) (mm) water
(temperature) partition
coefficient
2-MCP 128.56 1.2634 (20/4) 174.9 9 63.9 1 (12.1 °C) 2.15b
3-MCP 128.56 1.268 (25/4) 214 33 1 (44.2 °C) 2.50b
4-MCP 128.56 1.2651 (30/4) 219.75 43.2-43.7 121.1 1 (49.8 °C) 2.39b
2,3-DCP 163 206 57-59
2,4-DCP 163 1.38 (60/7) 210 45 62 1 (76.5 °C) 3.06c
2,5-DCP 163 211 (744 mm) 59 3.20c
2,6-DCP 163 219-220 (740 mm) 68-69 1 (59.5 °C)d
3,4-DCP 163 253.5 (767 mm) 68
3,5-DCP 163 253 (757 mm) 68
2,3,4-T3CP 197.45 sublimes 83.5
2,3,5-T3CP 197.45 248.5-249.5 62
2,3,6-T3CP 197.45 272 58
2,4,5-T3CP 197.45 1.68 (25/25)e sublimes 68-70.5 1 (72 °C) 3.72f
(275 mm) 1 (53 °C) 3.62c
1 (76.5 °C)
Table 3. (contd).
Compound Relative Density Boiling point Melting point Flash Vapour log
molecular (°C at 760 mm) (°C at 760 mm) point pressure n-octanol/
mass (°C) (mm) water
(temperature) partition
coefficient
2,4,6-T3CP 197.45 1.49 (75/4)e 246 69.5 113.9
3,4,5-T3CP 197.45 271-277 (746 mm) 101
2,3,4,5-T4CP 231.98 1.67d sublimes 116-117
2,3,4,6-T4CP 231.98 1.6 (60/4)g 150 (15 mm) 70 1 (100 °C) 4.10c
2,3,5,6-T4CP 231.98 115
a Principal source: Jones (1981).
b From: Fujita et al. (1964).
c From: Stockdale & Selwyn (1971).
d From: US EPA (1980a).
e From: Kozak et al. (1979).
f From: Leo et al. (1971).
g From: Verschueren (1983).
2.4 Analytical Methods
2.4.1 Sample collection and storage
Proper sampling and sample storage are essential prerequisites for
residue determinations, particularly as picogram or nanogram
quantities are often encountered in environmental samples. It is,
therefore, important to minimize contamination, and to collect
representative samples.
Chlorophenols in the air have been collected by drawing air
through an absorbent liquid at a given rate for a given period, using
absorbents such as potassium carbonate (Dahms & Metzher, 1979) or
ethylene glycol (Wyllie et al., 1975). If a significant proportion of
the chlorophenols present is likely to bind to container walls, as
occurs with water samples, glass containers are preferable to plastic
ones (Kozak et al., 1979).
To avoid erroneous determinations, samples should be processed
immediately or appropriate steps taken to avoid losses through
degradation. If samples are to be stored for an extended length of
time after collection, major losses of chlorophenols may occur as a
result of photodecomposition, oxidation, biodegradation, or
evaporation (section 4). If it is necessary to store samples, changes
in residue levels can be reduced by refrigeration or freezing. The
American Public Health Association (Greenberg et al., 1985) recommends
preserving waste-water samples containing phenolic compounds by
acidification with phosphoric acid and treatment with copper sulfate,
prior to refrigeration.
2.4.2 Sample preparation and analysis
The early procedures used to analyse for chlorophenols were
reviewed by Bevenue & Beckman (1967). Most were colorimetric
techniques, the most popular being the 4-aminoantipyrine method; none
of the methods was either very specific or sensitive. They are no
longer widely used, and are not discussed here. Instead, more
sophisticated analytical techniques are being increasingly used,
including thin-layer chromatography (TLC), gas chromatography (GC),
high-performance liquid chromatography (HPLC), ion exchange
chromatography, infrared (IR) and ultraviolet (uv) spectroscopy, mass
spectrometry (MS), and mass fragmentography. Table 4 includes examples
of the techniques available for the sampling and determination of
chlorophenols other than pentachlorophenols. An indication of the
sensitivity of each method is given, when available.
Table 4. Analytical methods for chlorophenols other than PCPa
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Air T4CP Bubbler collection; Derivatization with 0.05 µg/m3 Dahms & Metzner
absorption in acetyl chloride; GC (1979)
potassium carbonate analysis, EC detector
solution; hexane
extraction
Air 3-MCP Polyether-type HPLC analysis, EC 5 ng US EPA (1980d)
4-MCP polyurethane foam; detector
2,4-DCP Soxhlet extraction
2,4,5-T3CP with diethyl ether/
2,4,6-T3CP hexane; evaporation;
extraction with NaOH,
buffered with phosphoric
acid
Water 2-MCP Adsorption on Florisil column Eichelberger
2,4-DCP activated carbon; with anhydrous et al. (1970)
2,4,5-T3CP adsorbates extracted sodium sulfate for
2,4,6-T3CP with chloroform then clean-up; GC analysis
sodium hydroxide
followed by ethyl ether
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Surface 2-MCP Adsorption on Form pentafluorobenzyl Kawahara (1971)
water 2,4-DCP activated carbon; ether derivatives;
adsorbates extracted GC analysis
with chloroform then
partitioned into acetone
Water 2-MCP Methylene chloride HPLC analysis, 4.2-12.6 ng; Realini (1981)
2,4-DCP extraction of acidified UV254 detector 93-97%
2,4,6-T3CP sample, followed
by ion-pair extraction
of basic sample with
acetonitrile
Air T4CP Collection in 0.1 N GC analysis, 0.5 µg/m3 Kleinman et al.
sodium hydroxide in EC detector (1986)
impingers, acidification,
extraction with toluene
Air T3CP Collection in Acetylation and 2-5 µg/m3 Kauppinen &
T4CP toluene in impingers, extraction with Lindroos (1985)
extraction into basic hexane; GC analysis,
borax solution EC detector
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Water 4-MCP Adsorb basic sample GC analysis, Fl 80-102% Chriswell et al.
2,4,6-T3CP on anion exchange detector; confirmation (1975)
resin; extraction by GC-MS
of hydrochloric acid
and acetone-water
eluates with methylene
chloride
Water T3CP Adsorb on XAD-4 Derivatization with T3CP: 1 µg/litre, Woodrow et al.
T4CP resin; extract with diazomethane; 74.8-77.7%; (1976)
acetone, hydrochloric dissolve in hexane; T4CP: 0.5 µg/litre,
acid; concentrate, HPLC clean-up on 46.7-61.4%;
then dilute with Partisil silica PCP: 0.5 µg/litre,
water; partition column; GC analysis 72.5-85.1%
with sodium sulfate, with EC detector
dry over
dichloromethane
Surface 2,4-DCP Addition of sodium Extract & derivatize 1-2 ng/litre, Abrahamsson & Xie
waste, or 2,6-DCP phosphate buffer by adding 98-105% (1983)
drinking- 2,4,6-T3CP solution, for acid hexane containing
water 2,3,4,6-T4CP waste-water pH internal standard
adjustment to 7 (2,6-dibromophenol)
with sodium and acetic anhydride
hydroxide directly to sample;
GC analysis, EC
detector
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Urine 2,4-DCP Partitioned into Purified by TLC; GC Kurihara & Nakajima
(mouse) 2,4,5-T3CP ethanol or benzene, analysis thermal (1974)
2,4,6-T3CP and water, both conductivity detector;
phases analysed; confirmation by MS
enzyme hydrolysis
of water soluble CP
conjugates
Urine T3CP Benzene extraction Der. with diazom 1 µg/litre, Edgerton et al.
(human, T4CP from acidified, ethane; separation on 89.3-97.0% (1979)
rat) hydrolysed solution acid alumina column;
GC analysis, EC detector;
GC-MS confirmation
Urine T4CP Acidic hydrolysis; LC analysis column: 23 µg/litre, Pekari & Aitio
(human) hexane/isopropanol Spherisorb ODS; 54.6% (1982)
extraction; mobile phase:
evaporation and methanol +
redistillation in ammonium carbonate;
methanol-water UV254 detector
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Urine 2-MCP Hydrolyse conjugates Der. with sodium Hargesheimer & Courts
(human) 4-MCP in acidified sample bicarbonate then (1983)
2,4-DCP by boiling; make acetic anhydride
2,6-DCP basic with sodium extraction methylene
2,4,5-T3CP hydroxide extract chloride; GC analysis,
2,4,6-T3CP with methylene EC and FI detectors;
chloride; neutralize, confirmation by MS
dry with sodium
sulfate
Blood 3-MCP Hydrolysis with HPLC analysis, 5 ng, US EPA (1980d)
(human) 4-MCP hydrochloric acid EC detector 80-100%
2,4-DCP extraction with hexane
2,4,5-T3CP ethyl ether;
extraction with sodium
hydroxide, buffered
with phosphoric acid
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Animal 2,4-DCP Optional alkaline Der. to trimethylsilyl 0.02 mg/kg Clark et al. (1975)
tissue 2,4,5-T3CP digestion; acid ether; GC analysis (2,4-DCP);
(sheep hydrolysis; 0.01 mg/kg, > 95%
cattle) distillation with water; (2,4,5-T3CP)
methylene chloride
Sediment T4CP Homogenization; toluene Derivatization 0.5-25 µg/kg, Butte et al. (1983)
and clams extraction from pyrolytic ethylation 76.7-98.8%
acidified sample with triethylsulfonium
2,4,6-tribromophenol iodide; GC analysis,
as internal standard EC detector;
Confirmation by MS
Fish 2-MCP Gel permeation Derivatization with 2-MCP-47% Stalling et al.
tissue 2,4-DCP chromatography to remove pentafluorobenzyl 2,4-DCP-78% (1979)
2,4,6-T3CP lipids, free fatty bromide; silica 2,4,6-T3CP-86%
acids; acid-base gel chromatography PCP-63%
extraction clean-up; GC analysis,
EC detector
Meat and 2,4-DCP Alkaline digestion; Derivatization 10 µg/kg, Sackmauerova-Veningerova
poultry 2,3,4-T3CP steam distillation of with methyl iodide; 92-98% et al. (1981)
livers 2,4,5-T3CP acidified sample; GC analysis;
2,4,6-T3CP toluene extraction EC detector
dry sodium sulfate,
evaporate
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Muscle 2,4-DCP Blended in GC analysis, Sherman et al. (1972)
(hen) hexane-sulfuric acid; EC detector
extraction with
NaOH, then hexane
Liver 2,4-DCP Ground; dried with Florisil column Sherman et al. (1972)
(hen) sodium sulfate eluted for clean-up; GC
with hexane; analysis, EC detector
extraction of eluate
with acetonitrile,
then hexane; dried
with sodium sulfate
Soil 2-MCP Steam distillation of GC analysis, EC < 0.1 mg/kg Narang et al. (1983)
2,4-DCP acidified sample; detector 2-MCP-59%;
2,4,6-T3CP extraction with 2,4-DCP-64%;
toluene dichloromethane 2,4,6-T3CP-70%
eluted through anhydrous
sodium sulfate
extraction with
hexane
Table 4. (cont'd).
Matrix Chlorophenol Sampling, extraction Analytical method Detection limit/ Reference
recovery
Wood 2,3,4,6-T4CP Extraction with diethyl Elution from TLC 200 mg/kg dust Levin & Nilsson (1977)
dust ether; evaporation; with n-hexane; 70%
dissolve in acetone derivatization
and TLC (silica gel) with diazomethane;
GC with Ni63
EC detector
Wood 2,4,6-T3CP Pumped through GC analysis, EC Kauppinen & Lindroos
dust 2,3,4,6-T4CP membrane filter; detector (1985)
Soxhlet extraction
with diethyl ether;
evaporate; dissolved
in hexane
a GC = gas chromatography.
TLC = thin-layer chromatography.
HPLC = high-performance liquid chromatography.
MS = mass spectrometry.
EC = electron capture detection.
FID = flame ionization detection.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural Occurrence
Some chlorophenols are present in the environment independent of
man-made input. Dichlorophenols have been detected in a variety of
organisms (Siuda, 1980). 2,4-Dichlorophenol occurs naturally in a
Penicillium sp., while 2,6-DCP serves as a sex pheromone for several
species of tick. A number of related organohalogens are also found in
flora and fauna (Arsenault, 1976; Siuda, 1980). However, these sources
cannot account for the significant amounts of chlorophenols,
particularly the higher chlorinated phenols, found in the environment.
3.2 Man-made Sources
3.2.1 Production levels and processes
3.2.1.1 World production figures
Reliable data on production levels of chlorophenols other than PCP
are not readily available. In 1975, the combined global production of
all chlorophenols approached 200 million kilograms (Table 5). Slightly
more than half consisted of chlorophenols other than PCP, with
2,4-DCP, 2,4,5-T3CP, and 2,3,4,6-T4CP predominating. Where
commercial use data are available, recent figures indicate that
consumption has declined (IARC, 1986). Chlorophenols are used in
countries other than those shown in Table 5, but the quantities used
are not known. Information on PCP production is presented in WHO
(1987b).
3.2.1.2 Manufacturing processes
While most chlorophenols can be produced by several different
procedures, only a few methods are actually used in commercial
manufacture (Doedens, 1963; Freiter, 1979). Most chlorophenols are
made by the direct stepwise chlorination of phenol or lower
chlorinated phenols at an elevated temperature. The compounds 2-MCP,
4-MCP, 2,4-DCP, 2,6-DCP, 2,4,6-T3CP, 2,3,4,6-T4CP, and PCP are
manufactured by this means. The manufacture of T4CP or PCP requires
the use of a catalyst, such as iodine, aluminium chloride (AlCl3),
ferric chloride (FeCl3), or antimony chloride (SbCl3). The process
is not quantitative, with the result that batches of one chlorophenol
will usually contain substantial amounts of other CPs (section 2.1).
Table 5. Production/consumption of chlorophenols other than PCP
Country Compound Year Production/ Reference
consumption
(kg/year)
Global total chlorophenols 1975 1.8 × 108 (P)a Levin & Nilsson
(1977)
non-PCP 1978 0.98 × 108 (P) Ahlborg &
chlorophenolsb Thunberg
(1980)
Canada total chlorophenols 1976 3.4 × 106 (C) Jones (1981)
non-PCP chlorophenols 1.5 × 106 (C)b
total chlorophenols 1981 > 5.266 × 106 (C) Jones (1984)
1981 4.000 × 106 (P)
non-PCP chlorophenols 1981 > 3.730 × 106 (C)
tetrachlorophenol 1981 7.86 × 105 (C) Jones (1984)
and Na-T4CP 12.44 × 105 (P)
Na-T3CP 1981 3.0 × 103 (C) Jones (1984)
1.0 × 103 (P)
2,4-dichlorophenol 1981 3.700 × 106 (C) Jones (1984)
1.850 × 106 (P)
total chlorophenols 1984 3.89 × 106 (S) Environment
non-PCP chlorophenols 1984 4.91 × 105 (S)b Canada
(1986)
tetrachlorophenol 1984 4.9 × 105 (S) Environment
and Na-T4CP Canada
(1986)
2,4,5-trichlorophenol 1984 < 1.0 × 103 (S) Environment
and Na-2,4,5-T3CP Canada
(1986)
Europe monochlorophenols 4.5 × 106 (P) Krijgsheld &
van der Gen
2,4-dichlorophenol 9.1 × 106 (P) (1986)
Table 5. (contd).
Country Compound Year Production/ Reference
consumption
(kg/year)
United total chlorophenols 1972 > 1.14 × 106 (C)d Ahlborg &
Kingdom Thunberg
(1980)
USA total chlorophenols 1976 > 2.421 × Buikema et al.
107 (S,P)c,d (1979)
non-PCP chlorophenols 1976 > 1.995 × Buikema et al.
106 (S,P)b (1979)
2,4-Dichlorophenol 1976 1.995 × 106 (S)
a P = production, C = consumption, S = sales volume.
b By difference, from data presented in reference.
c Sales approximate consumption, since most use is domestic (Jones, 1981).
d A conservative estimate, derived by adding figures for major chlorophenols.
Alternatively, some chlorophenols are produced by the alkaline
hydrolysis of hexachlorobenzene (HCB) or other chlorobenzenes in
methanol, ethylene glycol, and other solvents. The compounds 2,5-DCP,
3,4-DCP, 2,4,5-T3CP, 2,3,4,5-T4CP, 2,3,5,6-T4CP, and PCP can be
synthesized by this type of reaction (Doedens, 1963; Freiter, 1979).
Both methods may yield contaminants that are themselves potential
health hazards, specifically PCDDs, PCDFs, and 2-phenoxyphenols
(section 2.1), especially if optimum reaction conditions are not
maintained (particularly temperature and pressure) in the production
of higher chlorophenols. In addition, chlorophenols derived from the
hydrolysis of chlorobenzenes may include substantial amounts of the
initial isomer in the final product.
3.2.2 Uses
The uses of commercial chlorophenols are summarized in Table 6.
These compounds are biocides, a property that accounts for many of
their uses. Chlorophenols, particularly tetra-, and to a lesser
extent, trichlorophenols, have been used as bactericides, algicides,
molluscicides, acaricides, fungicities, and mould inhibitors, and for
less specific uses, such as general antiseptics and disinfectants.
Chlorophenols are also used as intermediates in the production of
certain herbicides, dyes, and drugs.
At present, use patterns are more restricted than is indicated in
Table 6. For example, revisions to Canadian standards for chlorophenol
in 1980 resulted in a sharp reduction in use in domestic interiors,
agriculture, the leather industry, and as slimicides in the pulp and
paper industries. Both the quantities and patterns of use are even
more restricted in some countries. For example, in Sweden and Finland,
chlorophenols are no longer used, or use is severely restricted in the
wood preservation or pulp and paper industries (Ahlborg & Thunberg,
1980; Lindroos et al., 1987).
Most chlorophenols are applied in the form of a chlorophenol-oil
mixture, but some are dissolved in a "clean" carrier that can be
recovered, such as methylene chloride (Jones, 1981). In contrast, the
sodium salts of higher chlorophenols (particularly T3CP, T4CP, and
PCP) are readily soluble in water.
3.2.2.1 Wood treatment
Large quantities of higher chlorophenols are used in wood
preservation (Table 6). In Canada in 1981, most chlorophenol-treated
wood was preserved by pressure treatment with pentachlorophenol
(Table 7). This compound has been evaluated previously (WHO, 1987b),
and will not be covered here.
Substantial amounts of the sodium salts of T4CP (ca. 13% of total
1981 chlorophenol consumption: Table 7), and lesser amounts of NaT3CP
and NaPCP have been used to protect fresh-cut logs and lumber. These
compounds, which are readily soluble in water, are used to surface-
treat lumber by dipping or spraying to protect against sapstain or
mould. Some plywood mills also use T4CP to reduce decay and mould,
and insect attack. The preservative is usually added to the glue.
3.2.2.2 Agriculture
At one time, chlorophenol-treatment was widely used in
agriculture, to prevent wood decay in buildings, food containers, and
horticultural timbers. Recently, such chlorophenol applications have
been considerably restricted in some countries (section 3.2.2), and as
a result, the quantities of non-PCP chlorophenols used in agriculture
are minor (Jones, 1981).
Table 6. Principal uses and reactions of selected chlorophenols other than PCPa
Compound Principal uses Other uses
2-chlorophenol Intermediate for Polymer intermediate for
further chlorination to fire-retardant varnishes;
2,4-dichlorophenol, cotton fabric treatment
2,4,6-trichlorophenol, to provide rot resistance;
and pentachlorophenol ingredient in coal
processing
4-chlorophenolb Intermediate for
higher chlorophenols;
intermediate dyes,
fungicides, and drugs
2,4-dichlorophenol Intermediate for Intermediate for
production of 2,4-D and production of Sesone,
other herbicides; Nitrofen, Nemacide,
ingredient of Genite-EM-923; raw
antiseptics; starting material for polyester
material for higher films; mothproofing;
chlorophenols miticide
2,4,5-trichlorophenol Intermediate in Germicides and
manufacture of 2,4,5-T ingredients of
and related herbicides; germicidal soaps
fungicide, bactericide,
algicide
2,4,6-trichloropenol Precursor for higher CPs;
germicide, particularly for
preservation of wood,
leather, glue, and textiles;
intermediate in preparation
of insecticides and soap
germicides
2,3,4,6- Fungicide and bactericide Preservative for latex
tetrachlorophenol, for wood preservation; and leather; preservative
and its sodium salt sodium salt is sapstain in glue for plywood
inhibitor; pesticide
a From: US EPA (1979).
b From: US EPA (1980c).
3.2.2.3 Domestic
T4CP is an active ingredient in formulations of PCP used as wood
preservatives for homes, and as an additive to paints and stains
(Table 6). Sales in Canada for these purposes contribute only a small
fraction to the total PCP market (Jones, 1981) (Table 7) as a result
of recent government restrictions on their use (section 3.2.2). T3CP
is used as a general-purpose home antiseptic and as the active
ingredient in some throat lozenges. At one time, 4-MCP was found in
disinfectants for home, farm, dental, hospital, and veterinary uses,
but has been largely replaced by other chemicals (Exon, 1984).
Table 7. Canadian use patterns for chlorophenols and their sodium
salts in 1981a
Use Product Consumptionb % of Total
(kg × 103/year)
Wood preservation PCP 1536 25.2
(pressure treatment)
Wood protection Na-PCP 32 0.5
(surface treatment) Na-T4CP 786 12.9
Na-T3CPc 1 0.02
Intermediates for 2,4-DCPd 3700 60.7
phenoxy herbicides
Additives in products NaPCP 38 0.6
listed in footnote e NaT3CPc 2 0.03
Total 6095
a From: Jones (1984).
b Includes chlorophenols present in exports (13.6% of total
consumption), principally treated wood products.
c Chlorophenols are no longer registered for use in Canada.
d 2,4-D is no longer produced domestically, though considerable
quantities continue to be imported.
e Adhesives, construction materials, fabrics, fibreboard
products, finished paper, leather, paper machine felts,
photographic solutions, pulp and paper process solutions,
rayon emulsions, rubber, rubber gaskets.
3.2.2.4 Water treatment
Information is lacking on the use of non-PCP chlorophenols in
water-treatment applications (Jones, 1981).
3.2.2.5 Additives
Sodium salts of T3CP and T4CP have been used to inhibit
microbial growth in a diverse array of products (Tables 6 & 7). These
applications make up only a small fraction of the total consumption of
chlorophenols (Jones, 1981).
3.2.2.6 Intermediates in industrial syntheses
Production of chlorophenols is stepwise and not quantitative,
hence lower chlorophenols are generally recycled within a reactor
system, or recycled from other manufacturing processes in the
production of the higher chlorinated phenols. The lower chlorophenols
also serve as intermediates in the production of other pesticides
(Table 6). Large amounts of 2,4-DCP are consumed in the manufacture of
the phenoxy herbicide 2,4-D (Table 7), and also as a precursor for the
production of the pesticides Sesone, Nitrofen, Nemacide, and
Genite-EM-923. 2,4,5-T3CP is used in the manufacture Ronnel(R),
2,4,5-T, and related herbicides, while 4-MCP is used in the production
of the germicide 4-chlorophenol- o-cresol. Small amounts of lower
chlorinated phenols have been used in the manufacture of some dyes and
drugs.
3.2.3 Other sources
Chlorophenols are also generated by human activity via several
indirect routes. They are formed as by-products of chlorine bleaching
in paper-mills, and subsequently released into the environment
(Ahlborg & Thunberg, 1980; Xie et al., 1986) (section 5.1.2.1). The
chlorination of municipal and industrial wastes, and municipal
drinking-water can give rise to mono-, di-, and trichlorophenols in
the µg/litre range (NRCC, 1978). At these levels, the taste and odour
of water may be affected locally, though the chlorophenol
concentrations are well below those that produce any observable toxic
effect in test organisms (section 6.1). The incomplete incineration of
chlorophenol wastes can release substantial quantities of these
compounds into the environment (section 3.4). The lower chlorinated
phenols are also formed as a result of the bioconversion of lower
chlorinated benzenes and related compounds (Ballschmiter & Scholz,
1980). The contributions of these sources to environmental release or
human exposures to chlorophenols are generally not well-defined, and
are not considered in subsequent sections.
3.3 Waste Disposal
Waste-waters containing chlorophenols arise from three sources,
i.e., the manufacture of chlorophenols, the manufacture of compounds
in which chlorophenols are used as intermediates, and wood-treatment
facilities. Both manufacturers and regulatory agencies have emphasized
appropriate process design, in order to minimize the volume of waste
generated, particularly in the treatment of lumber (Richardson, 1978).
Information on the handling of chlorophenol-containing wastes in
Canada is limited. In the past, some industries disposed of
2,3,4,6-T4CP and PCP-contaminated wastes as raw effluent into deep
wells, or into lagoons, prior to discharge into the North Saskatchewan
River (Jones, 1981). However, most Canadian wood-treatment plants
report that they do not have any discharge and are able to dispose of
their minimal wastes by incineration, or containment and evaporation
in lagoons. Data to confirm the adequacy of such treatments are
generally not collected (Richardson, 1978), but they are probably
adequate, if applied correctly.
While waste-water treatment plants have been used in only a few
large wood-preserving plants and by some chemical manufacturers (US
EPA, 1979), their use is increasing in response to environmental
concerns. Such methods and their efficiency have been described (US
EPA, 1979).
Usually primary treatment is applied only in instances where the
chlorophenol in question is dissolved in a carrier oil, when gravity
separation tanks are used to recover the oil and associated
chlorophenol for subsequent recycling or waste treatment. A few plants
also use hay or sand filtration to remove some oil droplets and wood
particles (Richardson, 1978). Flocculation is not widely used, because
flocculents have proved ineffective or inconsistent in removing
chlorophenols (US EPA, 1979).
Chlorophenols are effectively removed by secondary treatment under
favourable conditions. Roughly 90% of total phenols were removed from
waters containing wastes from the manufacture of phenoxy herbicides in
aerated lagoons (US EPA, 1971) or by trickling filter/activated sludge
treatments (Mills, 1959). Several laboratory and treatment-plant
studies have shown that PCP can be degraded by activated sludge (Dust
& Thompson, 1973; Kirsch & Etzel, 1973; Etzel & Kirsch, 1974; Moos et
al., 1983; Guthrie et al., 1984; Hickman & Novak, 1984), a fluidized
bed reactor (Hakulinen & Salkinoja-Salonen, 1982), and a biofilm
reactor (Salkinoja-Salonen et al., 1984). However, of 14 municipal
treatment plants surveyed by the US EPA, 8 did not remove any of the
PCP load, while the remainder were considered to remove PCP (6-87%)
primarily by adsorption on solids (Hickman & Novak, 1984).
Furthermore, degradation by microorganisms is sharply reduced, when
chlorophenol concentrations are excessive (Broecker & Zahn, 1977;
Reiner et al., 1978; El-Gohary & Nasr, 1984; Salkinoja-Salonen et al.,
1984). If secondary treatment facilities are to remove chlorophenols
reliably, they must include acclimated organisms, and chlorophenol
concentrations must be dilute and fairly stable (Hickman & Novak,
1984). These considerations suggest that such wastes are best handled
by a facility designed specifically to treat them, rather than being
treated at general-purpose sewage-treatment plants.
Chemical oxidation, using such chemicals as chlorine or potassium
permanganate, may also be effective in treating
chlorophenol-contaminated wastes. While chlorination of municipal
wastes can actually produce mono-, di-, and tri- chlorophenols, they
are subsequently oxidized together with higher chlorophenols to
compounds that are less toxic and/or more biodegradable (US EPA, 1979;
Sithole & Williams, 1986).
Adsorption of chlorophenols on activated carbon is sometimes used
as a final clean-up step for waste-waters, though this is feasible
only when waste treatment is handled in the same plant from start to
finish. Removal of 2,4-DCP (Aly & Faust, 1964) and PCP (Richardson,
1978) approaches 100% using this method.
Incineration has also been used to dispose of chlorophenol wastes,
but the available information deals mainly with PCP. A controlled air
incinerator destroyed more than 99.99% of PCP in treated wood at
combustion temperatures of between 916 and 1032°C, and yielded no
measurable T4CDD or T4CDF in the off-gas (Stretz & Vawuska, 1984).
However, incinerator temperatures must be high enough and residence
times long enough to ensure complete combustion. Rappe et al. (1978b)
demonstrated that burning technical T4CP at low temperatures
increased the content of PCDDs. Similarly, low-temperature destruction
in hog-fuel or "wigwam" burners fed chlorophenol-contaminated sawdust
and wood shavings can lead to the formation of PCDDs and PCDFs (Crosby
et al., 1981).
3.4 Losses of Chlorophenols into the Environment
In the absence of information from other countries, releases of
chlorophenols into the Canadian environment for 1981 (Jones, 1984) are
presented in Table 8 by way of an example. Of the 5.27 × 106 kg of
chlorophenols consumed in Canada in 1984, 1.37 × 106 kg (26%) were
eventually released into the environment. A large proportion (less
than 28%) of these releases would have been as PCP and NaPCP, but the
data compiled in the table do not distinguish these from T4CP and
Na-T4CP.
Table 8. Chlorophenol releases into the Canadian environment in 1981a
Source Quantity (kg × 103/year)
1. Releases in wastes from production sites
emissions 3
effluents 70
solids -
sub-total > 73
2. Releases in other wastes
(a) Industrial
(i) Wood preservation sites
liquid 2
solids -
incineration (hog-fuel) -
landfill 1 (PCP, T4CP)
sub-total > 3
(ii) Saw-mill/planer mill
liquid 21
solids -
(ii) Incineration (hog-fuel) 272
Pulp mills/landfill -
sub-total >293 (NaPCP, NaT4CP)
(b) Agricultural
solids (livestock litter)
landfill
(c) Domestic
solids
incineration (mun.)
landfill
3. Releases during storage and transport
(solids and liquids)
(a) Industrial 3.5
(b) Agricultural 3.4
(c) Domestic 0.1
sub-total 7.0
Table 8. (cont'd).
Source Quantity (kg × 103/year)
4. Releases in situ from treated products
(solids and liquids)
(a) Industrial 618
(b) Agricultural 370 (2,4-DCP)
(c) Domestic 1
sub-total 989
Grand total > 1365
a From: Jones (1984).
In 1981, a significant amount (ca. 5%) of the total releases of
chlorophenols in Canada occurred from production sites. Following this
estimate, production of all chlorophenols ceased in Canada. However,
this route could be a significant source of chlorophenol contamination
in countries where they are still manufactured. Releases from plants
will include a variety of chlorophenols from the manufacture of
chlorophenols and chlorophenol-derived products. A small proportion of
these materials reaches the environment after incineration (Table 8),
but the bulk of the chlorophenols released from production sites in
Canada is diluted and released as untreated effluent.
Losses into the environment during the storage and transport of
chlorophenols are small (Table 8), comprising less than 1% of the
total production.
The majority (>70%) of the chlorophenols released into the
Canadian environment arose from treated products (Table 8). About
two-thirds of these came from industrial sources, which were not
identified by Jones (1984). Petrochemical drilling fluids contain
large amounts of chlorophenols (from 700 to 1400 mg NaPCP/kg), to
prevent fermentation of polysaccharides, starch, and polymers (Jones,
1981). Once used, the drilling waste is stored on site, in sumps that
are often subject to flooding and washing out. In-service treatment
with wood preservatives, principally PCP and its salts, also results
in some spillage. Large spills have been responsible for fish kills in
waters contaminated in this fashion (Jones, 1981). In addition,
unknown, but presumably large, quantities of PCP and T4CP are leached
from treated lumber in storage or in service. The remaining third of
the environmental releases, which Jones (1984) identifies as primarily
2,4-DCP, is from agricultural sources. Commercial preparations of
pesticides, particularly 2,4-D, 2,4,5-T, and Lindane, contain
chlorophenols as contaminants. Furthermore, chlorophenols are among
the early degradation products of these widely-used chemicals.
Chlorophenols from these sources contaminate soils treated with the
pesticides, and runoff from these soils finds its way into adjacent
water bodies and ground water.
Much of the remaining input of chlorophenols into the environment
occurs in the form of industrial wastes. These comprise roughly 22% of
the total chlorophenol releases, primarily as NaT4CP and NaPCP. Most
of these are released in liquid wastes from pulp-mills (where they are
by-products of chlorine bleaching), untreated wastes from sawmills,
planer mills, and other facilities where they are used in wood
preservation, and during incineration of contaminated sawdust and wood
shavings. Losses of other chlorophenols from such commercial uses are
negligible (Table 8).
The quantities of chlorophenols lost to the atmosphere by
volatilization are not known. Nanogram-per-litre quantities of
chlorophenols have been detected in rainfall and snow (Paasivirta et
al., 1985), suggesting that significant quantities volatilize or are
adsorbed on airborne particulates.
The contributions to the environmental load of chlorophenols from
municipal and industrial chlorination processes, the metabolism of
other chlorinated compounds to chlorophenols, and the domestic use of
such products as cosmetics, drugs, home-care products, stains, wood
preservatives, and pesticides are not known.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and Distribution
4.1.1 Atmospheric movement
While chlorophenols are considered to be primarily water and soil
contaminants, atmospheric movement also occurs. Measurable quantities
of chlorophenols have been detected in air, rainfall, and snow,
sometimes far from obvious point sources (Paasivirta et al., 1985)
(section 5.1.1). Furthermore, considerable quantities of chlorophenols
are released as part of incinerator emissions. The relative
contributions of volatilization and adsorption on particulates to
these atmospheric levels are not known.
4.1.1.1 Volatilization
No estimates of the rate of volatilization of chlorophenols in the
environment have been published. It appears that losses through this
process are minimal in natural waters. If the half-times for
volatilization of 2-MCP and 4-MCP from 0.38 cm of still water measured
by Chiou et al. (1980) are extrapolated to 1 m depth, the estimated
half-times are 395 h and 3421 h, respectively (Krijgsheld & van der
Gert, 1986).
Diffusion, a process related to volatilization, does not
contribute significantly to the long-range transport of substances in
either the soil or aquatic habitats, though it is essential in the
local replacement of materials lost through volatilization or
breakdown.
4.1.2 Soil movement
4.1.2.1 Adsorption
Environmental transport of chlorophenols, particularly in soils,
can be affected by adsorption on particulates. Such deposition is
quite variable. Acidic soils bind chlorophenols strongly, while
adsorption is minimal under alkaline conditions. Chlorophenols also
adsorb on organic matter, with the result that adsorption is strong in
organic soils, but low in mineral soils.
Thus, Aly & Faust (1964) found that large amounts of three types
of clay were necessary to adsorb small quantities of 2,4-DCP in
aqueous suspensions, even under extremely acidic conditions
(pH 3.6-4.8). Seip et al. (1986) compared the migration rates of
tritiated water and of dilute solutions (12.5-25 µg/litre) of 2,4-DCP,
2,4,6-T3CP, 2,3,4,6-T4CP, and PCP through packed soil columns. All
of the chlorophenols migrated more slowly than water. Adsorption was
moderate in a weakly acid inorganic soil and a basic soil with a
higher organic content, while no chlorophenols were detected in the
eluate from a soil with both a low pH and a high content of organic
matter. In studies by Choi & Aomine (1972, 1974), strongly acidic
soils adsorbed PCP, while weakly acidic or neutral soils did not
adsorb it at all. Moreover, soils with a high organic content adsorbed
PCP strongly, regardless of their pH, while hydrogen peroxide
digestion of organic matter reduced apparent adsorption (Choi &
Aomine, 1974). Miller & Faust (1973) confirmed that sorption of a
number of phenolic compounds on organo-clay was pH-dependent.
It is difficult to assess the impact of adsorption on chlorophenol
transport in the environment from the results of the preceding
studies. Chlorophenol dynamics observed by Seip et al. (1986) suggest
that chlorophenols bound to soils are continually turned over, and
that binding sites may be saturated under the appropriate conditions,
leading to increased mobility and a decreased residence time of
chlorophenols in the soil body.
4.1.2.2 Leaching
In instances when adsorption is minimal, leaching will be an
important means of chlorophenol transport in the soil. Most
chlorophenols should be carried into ground- and surface-waters from
soils that are neutral or alkaline or have a low organic content, or
through which material can percolate rapidly. No information has been
found on the leaching of the lower chlorophenols, but Kuwatsuka (1972)
noted that much of the PCP applied to flooded rice paddies was carried
through the soil in solution, and it has been reported by Jones (1981)
that Na-PCP leaches readily from soils.
4.1.3 Transport in aquatic environments
While a large fraction of the chlorophenols entering waters is
probably degraded in situ (section 4.2), they are nonetheless
moderately soluble and fairly persistent, and so can be transported
considerable distances by water. For example, Fox & Joshi (1984)
detected elevated levels of T4CP and PCP in the surface waters of the
Bay of Quinte, Lake Ontario, as far away as 82 km from the
wood-preserving plant where they originated.
Chlorophenols that are not degraded are concentrated in the
sediments, perhaps through adsorption on sediment particulates.
Schellenberg et al. (1984) determined that sorption of chlorophenols
on natural sediments and aquifer materials was a combined function of
the pH, the organic carbon content of the potential sorbent, and the
partition coefficients (known also as soil adsorption coefficients).
Adsorption was quite strong on non-mineral sediments. Xie et al.
(1986) observed that the disappearance of chlorophenolic compounds
discharged from a sulfate pulp-mill was related to the partition
coefficients of the compounds, and that sediment concentrations were
quite high near to their source (section 5.1.2.1), suggesting that
adsorption strongly influenced the transport of chlorophenolic
compounds. It was reported by Eder & Weber (1980) that the
concentration factor (relative to water) of chlorophenols in the
sediments (38- to 680-fold) and suspended solids (6.3- to 240-fold) of
the Weser estuary was inversely related to the degree of ionization of
the chlorophenol. In contrast, Kuiper & Hantsveit (1984) reported that
both the water and the sediment on the bottom of marine enclosures
contained similar levels of 4-MCP and 2,4-DCP; this discrepancy is not
obviously attributable to differences in system pH, or the nature of
the particulates.
4.2 Degradation and Bioaccumulation
4.2.1 Degradation
As yet, there are few studies addressing the persistence of
chlorophenols in the environment.
4.2.1.1 Abiotic degradation
(a) Photodecomposition
Many, if not all, chlorophenol isomers are degraded to some extent
by exposure to ultraviolet radiation (UVR). The breakdown most often
involves an oxidation reaction that dechlorinates the molecule (Boule
et al., 1982), though a variety of reactions have been described.
2,4-DCP in aqueous solution was decomposed in a matter of minutes
by irradiation from a UV lamp (Aly & Faust, 1964; Crosby & Tutass,
1966; Nakagawa & Crosby, 1974). The major pathway involved the
degradation of 2,4-dichlorophenol to 4-chlorocatechol, which in turn
produced 1,2,4-benzenetriol, and finally a mixture of polyquinoid
humic acids (Crosby & Tutass, 1966). A similar sequential degradation
was reported for 2,4,5-T3CP (Crosby & Wong, 1973). Freitag et al.
(1982) reported that 65.8% of 14C-2,4,6-T3CP on silica gel was
degraded after 17 h of UVR exposure. No organic by-products were
detected, most radioactivity being recovered as carbon dioxide.
The breakdown of chlorophenols is markedly affected by the number
and position of chlorine substituents on the molecule. Under UVR,
2,4-DCP degrades to diameric products (Crosby & Tutass, 1966), while
2,5-DCP degrades to 4-resorcinol (Crosby & Wong, 1973). Omura &
Matsuura (1971) found that alkaline solutions of monochlorophenols
degraded as follows: 2-MCP (82.5% lost in 5 h at 40°C) to a complex
mixture with much resinous material, 3-MCP (70%) to resorcinol, and
4-MCP (55%) to hydroquinone, phenol, and three diphenyls. Aqueous
solutions of the three monochlorophenol isomers yielded similarly
varied products in studies by Boule et al. (1982). In later work,
Boule et al. (1984) determined that among the dichlorophenol
congeners, substitution at the ortho and meta positions made the
chemical more reactive than para substitution.
Only one study has been reported on whether photolysis
significantly reduces concentrations of chlorophenols other than PCP
in situ. Hwang et al. (1986) concluded that photolysis was the
principal degradative pathway (half-life of 3 h or less) for 2,4-DCP,
2,4,5-T3CP, and PCP, but not 4-MCP, in estuarine surface waters
(though they noted that mineralization by other mechanisms was
substantially photo-inhibited under the experimental conditions). Fox
& Joshi (1984) observed an increase in the ratio of T4CP/PCP in
surface waters with increasing distance from a wood-preserving plant
discharge, suggesting that substantial photolysis of PCP occurred, but
noted that the concentrations were remarkably stable, once the
chlorophenols were incorporated into the sediments.
(b) Chemical degradation
There is one report that indicates that chlorophenols may be
degraded in the environment by chemical processes. Baker & Mayfield
(1980) observed losses of 2-MCP, 4-MCP, and 2,4-DCP from sterile
washed silica sand, sterile aerobic soils, non-sterile anaerobic
soils, and sterile anaerobic soils. Microbial contamination,
photolysis, and volatilization were eliminated as causes. The authors
suggested that the chlorophenols were auto-oxidized, or broken down at
catalytic sites, but did not eliminate polymerization as a means of
loss. A number of other research workers, using a wide range of
chlorophenols, have not detected such abiotic losses (Alexander &
Aleem, 1961; Aly & Faust, 1964; Tabak et al., 1964; Boyd & Shelton,
1984).
4.2.1.2 Degradation by microorganisms
Although chlorophenols are quite toxic for microorganisms in
general, they are nonetheless readily metabolized by a large number
that occur in soils, natural waters, sediments, and sewage sludges.
This decomposition is often quite rapid, i.e., completed in a matter
of hours or days. For instance, of 206 isolates from a petroleum waste
lagoon, 46% were able to degrade chlorophenols as a sole source of
carbon after acclimation to the particular chlorophenol (Tabak et al.,
1964). Up to 95% of the added 3-MCP and 4-MCP (initially 150 and
300 mg/litre respectively) was consumed in 3-6 days, while the same
amount of 2,4-DCP (200 mg/litre) and 2,4,6-T3CP (initially
300 mg/litre) disappeared in 7-10 days. No breakdown of 2,6-DCP was
observed. Similarly, in batch cultures enriched with 50 mg
chlorophenol/litre and inoculated with soil, 2-MCP, 4-MCP, 2,4-DCP,
and 2,4,6-T3CP were readily biodegraded and were often removed
completely in less than 10 days, while 2,6-DCP was only metabolized in
some studies; 3-MCP, 2,5-DCP, 2,3-DCP, 3,4-DCP, 3,5-DCP, 2,4,5-T3CP,
2,3,4,6-T4CP and PCP were refractory (Alexander & Aleera, 1961).
Using an acclimated, activated sludge derived from soil, Ingols et al.
(1966) observed complete ring degradation of the following compounds
at 100 mg/litre: 2-MCP in 3 days, 3-MCP in 2 days, 4-MCP in 3 days,
2,4-DCP in 5 days, and 2,4,6-T3CP in 3 days. As much as 52% of
2,5-DCP disappeared in 4 days. No decomposition of sodium
pentachlorophenate occurred. More recently, aerobic microorganisms in
clay loam soils were able to degrade most of the 2-MCP, 4-MCP,
2,4-DCP, 2,6-DCP, or 2,4,6-T3CP present (100 mg/kg) within a few days
without a lag phase (Baker & Mayfield, 1980). More than 70% of added
3-MCP, 3,4-DCP, 2,4,5-T3CP, and PCP disappeared within 80-100 days,
while 3,4,5-T3CP and 2,3,4,5-T4CP levels were little changed after
160 days.
The results of many other studies have confirmed that most
chlorophenols can be metabolized by certain microorganisms in water
(Aly & Faust, 1964; Lee & Ryan, 1979; Baker et al., 1980;
Blades-Fillmore et al., 1982; Hwang et al., 1986), sediment (Lee &
Ryan, 1979; Baker et al., 1980), soil (Walker, 1954; Loos et al.,
1967; Spokes & Walker, 1974; Baker et al., 1980; Pal et al., 1980),
and activated sludge (Baird et al., 1974; Pitter, 1976; Pal et al.,
1980; Boyd & Shelton, 1984).
While bacteria are most frequently studied as the agents
responsible for chlorophenol biotransformation, they are not alone in
this capability. Fungi on wood shavings, used as litter for broiler
chickens, converted 2,3,4,6-T4CP to 2,3,4,6-tetrachloroanisole,
leading to a musty taint in the chicken flesh (Curtis et al, 1972; Gee
& Peel, 1974). The genera Aspergillus and Penicillium readily
degrade chlorophenols. Walker (1973) determined that a yeast isolated
from soil and grown on phenol could metabolize 2-MCP, 3-MCP, 4-MCP,
and 2,4-DCP, but not 2,6-DCP.
The relative rate of degradation of chlorophenols generally
decreases as the number of chlorine atoms on the phenolic ring
increases (Alexander & Aleem, 1961; Tabak et al., 1964; Ingols et al.,
1966; Baker & Mayfield, 1980). However, it is possible to obtain the
reverse result with organisms able to use PCP as the sole carbon
source: the KC3 bacterium studied by Chu & Kirsch (1973) grew on
2,3,4,6-T4CP and 2,4,6-T3CP, but metabolized the dichlorophenois
poorly; the monochlorophenols were not metabolized at all. Rates of
biodegradation are further affected by the relative position of the
chlorine atoms on the phenolic ring. Compounds with a chlorine in the
meta position are generally more stable than those without (Alexander
& Aleera, 1961; Chu & Kirsch, 1973; Etzel & Kirsch, 1974; Baker &
Mayfield, 1980). The chlorophenolic products of PCP degradation in
soils in vitro support this hypothesis (Ide et al., 1972).
Microorganisms that have been previously exposed to a compound are
usually able to metabolize it immediately when re-exposed, and at a
faster rate than unexposed organisms (Walker, 1954; Alexander &
Aleera, 1961; Tyler & Finn, 1974; Pal et al., 1980; Blades-Fillmore et
al., 1982), presumably because exposure induces the enzymes necessary
to metabolize the chlorophenol. Microorganisms not previously
acclimated often exhibit a lag time of as much as several days before
they begin to degrade the compound (Bollag et al., 1968; Spokes &
Walker, 1974; Lee & Ryan, 1979; de Kreuk & Hantsveit, 1981).
Similarly, prior exposure to a structurally related compound can
facilitate the metabolism of chlorophenols, indicating that the
enzymes induced by the original compound are somewhat nonspecific. As
noted earlier, PCP-adapted microorganisms utilize T3CPs and T4CPs
readily (Chu & Kirsch, 1973), while bacteria raised on phenol, lower
chlorophenols, or phenoxyacetic acids are able to metabolize various
other lower chlorophenols (Tabak et al., 1964; Loos et al., 1967;
Walker, 1973; Spokes & Walker, 1974; Boyd & Shelton, 1984).
Research workers have found little or no anaerobic biodegradation
of chlorophenols (Gee & Peel, 1974; Lee & Ryan, 1979; Baker &
Mayfield, 1980; Horowitz et al., 1982; Pignatello et al., 1986). The
persistence of PCP and T4CP in sediment cores, several decades old,
which were presumably anaerobic, supports these findings (Fox & Joshi,
1984). However, under the right conditions, anaerobic metabolism can
be substantial: acclimated anaerobic sludge from a municipal sewage
plant degraded 25 mg monochlorophenols/litre in a few days (Boyd &
Shelton, 1984).
Only a few studies can be used to compare chlorophenol
biodegradation between habitats under conditions that may be readily
extrapolated to a natural situation. The in vitro aerobic breakdown
of 2-MCP, 4-MCP, and 2,4-DCP (100 mg/kg) has been studied in clay loam
soil (Baker & Mayfield, 1980; Baker et al., 1980), freshwater
sediments (Baker et al., 1980), and streams (Baker et al., 1980) at
temperatures ranging from 0 to 23°C. In soil incubated at 23°C, at
least 70% of added 2-MCP disappeared in 0.5-1.0 days, 4-MCP in 1-2
days, and 2,4-DCP in 7-20 days (Baker & Mayfield, 1980). In contrast,
decomposition in sediments was slower: at 20°C, 2-MCP disappeared in
10-15 days, 4-MCP, in 30 days, and 73% of 2,4-DCP, in 15-30 days
(Baker et al., 1980). Virtually no biological degradation of
monochlorophenols occurred in the stream water at 20°C, but 2,4-DCP
levels were reduced by 74% in 10 days at 20°C. These differences may
be related to the favourable conditions for microorganisms that exist
in soils and sediments, in which levels of organic matter and
particulate surface area are high. Addition of sterile sediments or
several inert substances enhanced the degradation of 50 µg
2,4,6-T3CP/litre in river water (Blades-Fillmore et al., 1982).
In other reports, chlorophenol degradation in water has proceeded
more rapidly (eliminated in 1-3 weeks) (de Kreuk & Hantsveit, 1981;
Blades-Fillmore et al., 1982; Hwang et al., 1986). It is possible that
chlorophenols are generally degraded faster in soils and aerobic
sediments than in water but, wherever a suitable combination of
microflora and physical and chemical factors occurs, these general
differences can be overridden.
In summary, a number of microorganisms from a variety of habitats
can readily degrade chlorophenols, especially if previous exposure to
these compounds has induced the enzymes necessary for their
metabolism. This process is slowest with exposure to the higher
chlorophenols, particularly those that are meta-substituted. The
results of incubation studies in the laboratory suggest that
biodegradation is most rapid in aerobic soils and sediments, and is
reduced in anaerobic or nutrient-poor habitats.
4.2.2 Bioaccumulation
A number of field and laboratory studies have yielded information
on the bioaccumulation of the chlorophenols. Most of these have
involved aquatic organisms. Although organisms ranging from bacteria
to fish generally contain higher levels of chlorophenol residues than
the environment at large, the concentrations are not large compared
with those of some other chemicals. Most bioconcentration factors
(BCFs) fall between 1 × 102 and 1 × 103 (Table 9), and substantial
biomagnification is not evident. Ernst & Weber (1978) and Ernst (1979)
suggested that Lanice conchilega displayed exceptionally high BCFs
because of an unusual halogen metabolism (detectable levels of
bromophenols were noted, unlike the other invertebrates studied).
The results of most of the studies in which a range of
chlorophenols has been surveyed have indicated that bioconcentration
is a positive function of chlorine number (Kobayashi et al., 1979;
Hattula et al., 1981b). The higher BCF with increasing chlorine
substitution most likely results from the high partition coefficient
or the lower dissociation constant. Other experimental conditions,
such as length of exposure and exposure concentration, may also
contribute to the substantial range of BCF values shown in Table 9.
Clearance rates of chlorophenols from biota are rapid, indicating
that the bioaccumulation observed in field studies is the result of
long-term exposure rather than persistence. Landner et al. (1977)
reported that 2,4,6-T3CP was eliminated from rainbow trout livers,
three weeks after dosing was discontinued. Similarly, 84-92% of
2,4,5-T3CP was lost from fathead minnows in the first day after
exposure (Call et al., 1980), and the half-life for 2-MCP in bluegills
was less than 1 day (Barrows et al., 1980).
4.3 Effects of Other Physical, Chemical, or Biological Factors
4.3.1 pH
One of the major factors affecting the transport, breakdown, and
toxicity (section 6) of chlorophenols is pH. Because chlorophenols are
weak acids in aqueous solution, they exist primarily in the molecular
form under acidic conditions, while the anion predominates at neutral
or basic pH. Since the molecular and ionic forms of chlorophenols
react differently, pH affects a variety of processes that in turn
influence chlorophenol dynamics. Ionization is further affected by the
degree of chlorine saturation of the chlorophenol; in general, higher
chlorophenols are increasingly acid. Throughout the pH range
characterizing physiological and environmental situations,
monochlorophenols are present mainly in their molecular form while,
above pH 3.5, PCP is primarily dissociated. No information on the
interaction of pH and evaporation of lower chlorophenols was
available. In their studies on PCP volatilization, Kloppfer et al.
(1982) determined that the half-life for PCP disappearance, through
volatilization from their apparatus, was 167 h at pH 3.3 and 3120 h at
pH 6. No evaporation was detected at pH8.
Similarly, pH influences particulate sorption phenomena through
changes in the molecular form of the chlorophenol. As was discussed in
section 4.2, adsorption on soils, sediments, and suspended solids is
inversely related to pH.
The rate of the photolysis of chlorophenols is also altered by pH.
Aly & Faust (1964) determined that the breakdown of 2,4-DCP in aqueous
solution was extremely rapid under alkaline conditions and relatively
slow under acidic conditions: approximate half-lives for photolysis at
pH values of 4, 7, and 9 were 34, 15, and 2 min, respectively.
Similarly, Omura & Matsuura (1971) reported that the rate of
photolysis of 4-MCP increased as the pH increased. In addition, pH
exerts an influence on the biodegradation of chlorophenols. An
activated sludge culture grew well on 2-MCP and 3-MCP at neutral, but
not at alkaline pH (Ingols et al., 1966). Tyler & Finn (1974)
determined that a Pseudomonas species grew best on 2,4-DCP at a pH
range of 7.1-7.8.
Table 9. Bioconcentration estimates for various chlorophenols from field and laboratory data
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Plants
Oedogonium 2,4,6-T3CP 36 1720 Aquatic microcosm, 0.5 µg/litre, Virtanen & Hattula
long-term exposure (1982)
Echinodorus 2,4,6-T3CP 36 1000 Aquatic microcosm, 0.5 µg/litre, Virtanen & Hattula
long-term exposure (1982)
Elodea 2,4,6-T3CP 36 4460 Aquatic microcosm, 0.5 µg/litre, Virtanen & Hattula
long-term exposure (1982)
Chlorella 2,4,6-T3CP 1 51 50 µg/litre screening test, Freitag et al.
fusca var. as 14C (1982)
vacuolate 2,4,6-T3CP 1 580 49 µg/litre screening test, Korte et al.
as 14C (1978)
Invertebrates
Lymnae 2,4,6-T3CP 36 3020 Aquatic microcosm, 0.5 µg/litre Virtanen & Hattula
(adult) long-term exposure (1982)
Lanice 2,4,5-T3CP indefinite 24 088b Field data Ernst & Weber
conchilega 2,4,6-T3CP indefinite 20 269b Field data (1978)
2,3,4,S-T4CP indefinite 17 625b Field data
2,3,4,6 +
2,3,S,6-T4CP indefinite 11 163b Field data
Table 9. (cont'd).
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Invertebrates (contd).
Mytilus edulis 2,3,4,6-T4CP indefinite 45-60 Field data, receiving waters Folke et al.
for dump leachate (1984)
Fish
Roach 2,3,4,6-T4CP indefinite 200 Field data, pulp-mill inputs Paasivirta et al.
(Rutilus) (1985)
Pike 2,3,4,6-T4CP indefinite 150 Field data, pulp-mill inputs Paasivirta et al.
(Esox lucius) (1985)
Trout (Salmo 2,4-DCP 1 10 1.7 mg/litre (LC50) Hattula et al.
trutta) 2,3,5-T3CP 1 12 0.8 mg/litre (LC50) (1981b)
2,3,4,6-T4CP 1 450 0.5 mg/litre (LC50)
Poecilia young 2,4,6-T3CP 36 1 020 Aquatic microcosm, 0.5 µg/litre Virtanen & Hattula
female 2,4,6-T3CP 36 12 180 long-term exposure (1982)
male 2,4,6-T3CP 36 7 000
Fathead 2,4,5-T3CP 28 1900 Aquatic microcosm, 4.8 mg/litre, Call et al. (1980)
minnow 2,4,5-T3CP 28 1800 single addition, 49.3 µg/litre
(Pimephales as 14C
promelas)
Table 9. (cont'd).
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Sunfish 2,3,5,6-T4CP indefinite Muscle/field data, Mississippi Pierce & Victor
lake/spill in December, n = 2 (1978)
6 Januaryd 79 Muscle/spill in December, n = 2
27 Aprild 21
2,3,5,6-T4CP indefinite Liver/spill in December, n = 1 Pierce & Victor
6 Januaryd 962 Liver/spill in December, n = 2 (1978)
27 April 72
Bluegill 2-MCP 28 214 Continuous-flow aquarium, Barrows et al.
(Lepomis 2-MCP at 9.18 l&g/litre, (1980)
macrochirus) as 14C
Bass 2,3,5,6-T4CP indefinite Muscle/field data, Mississippi Pierce & Victor
lake (1978)
6 Januaryd 218 Spill in December, n = 2
27 Aprild 4962 Muscle, n = 2
Catfish 2,3,5,6-T4CP indefinited Muscle, n = 1
6 Januaryd 222 Muscle, n = 2
27 Aprild 53
2,3,5,6-T4CP indefinited Liver, n = 1
6 Januaryd 8608 Liver, n = 2
27 April 1005
Table 9. (cont'd).
Organism Compound Length of Bioconcentration Remarks Reference
exposure factora
(days)
Fish (contd.)
Goldfish 2-MCP 25h 6.4 Static lab. assay, 16 mg/litrec Kobayashi et al.
4-MCP 25h 10.1 Static lab. assay, 9 mg/litrec (1979)
2,4-DCP 25h 34 Static lab. assay, 7.8 mg/litrec
2 4 5-T3CP 25h 62 Static lab. assay, 1.7 mg/litrec
2,4,6-T3CP 25h 20 Static lab. assay, 10.0 mg/litrec
2,3,4,6-T4CP 25h 93 Static lab. assay, 0.75 mg/litrec
Golden orfes 2,4,6-T3CP 3 310 50 µg/litre, screening test Freitag et al.
(Leuciscus idus (1982)
melanotus)
Golden orfes 2,4,6-T3CP 3 250 30 µg/litre, screening test Korte et al.
(Leuciscus idus (1978)
melanotus)
a Ratio of concentration in organism or tissue:water.
b Based on bioconcentration relative to PCP.
c Concentrations near LC50 values, but still sublethal.
d Sampling date.
4.3.2 Lack of oxygen
In general, higher chlorinated phenols are persistent in anaerobic
environments, because of the low microbial degradation of
chlorophenols under such conditions. (section 4.2.1.2).
4.3.3 Inorganic nutrients
Inorganic nutrients may restrict the rate of chlorophenol
biodegradation where a shortage of nutrients limits microbial
activity. Striking seasonal variations noted in the rate of 4-MCP
degradation in natural sea water incubated in the laboratory (de Kreuk
& Hantsveit, 1981) were not related to microbial biomass, but
paralleled the levels of phosphate and nitrate in the samples.
In vitro addition of nutrients to the sea water stimulated
biodegradation. Similar results were reported by Kuiper & Hantsveit
(1984).
4.3.4 Organic matter
Like inorganic nutrients, levels of organic matter can influence
the microbial breakdown of chlorophenols through the control of
microbial biomass and activity. For example, while chlorophenol
decomposition was apparent in most soils studied (section 4.2.1.2), it
was virtually absent from those containing little or no organic matter
(Kuwatsuka, 1972). Similarly, low heterotrophic activity, determined
by the low concentrations of organic substances in water (typically
mg/kg) relative to those found in soils or sediments (g/kg), may
account for differences observed in chlorophenol biodegradation
between these environmental compartments (section 4.2.1.2). Organic
matter bound to the surface of the soil or sediment particulates may
also absorb chlorophenols and thereby affect their transport (sections
4.1.2.1, 4.1.3).
4.3.5 Temperature
Volatilization is a direct function of temperature. In their
studies of PCP evaporation, Kloppfer et al. (1982) determined that the
half-life for volatilization of PCP was 653 h at 23°C, 328 h at 30°C,
and 211 h at 40°C. Photolysis can also be temperature-dependent. Omura
& Matsuura (1971) found that higher solution temperatures increased
the photodecomposition of 4-MCP. The microbial breakdown of lower
chlorophenols as a function of temperature was investigated in water,
soil, and sediment by Baker et al. (1980). As expected, biodegradation
was higher at 20°C or 4°C than at 0°C. However, high temperatures also
limit the microbial decomposition of chlorophenols; Tyler & Finn
(1974) found that growth of Pseudomonas on 2,4-DCP fell off sharply
above 25°C.
Under some conditions, exposure of chlorophenols to elevated
temperatures, such as those used in heating and burning, can lead to
the formation of chlorinated dibenzo- p-dioxins (PCDDs) and
dibenzofurans (PCDFs) (Rappe et al., 1978b). Many PCDDs and PCDFs are
extremely persistent in the environment and are toxic for living
systems (WHO, in press).
4.4 Persistence
As a consequence of chlorophenol decomposition through photolysis,
biodegradation, and perhaps chemical catalysis, virtually all
chlorophenol compounds will be eliminated from most environments.
Some, notably PCP and the lower chlorophenols with a chlorine in the
meta position, will persist for a longer time than others, but even
these should eventually be broken down, wherever suitable light
exposure or microorganisms occur.
No information is available on the persistence of chlorophenols in
air. Photodecomposition may be an important removal mechanism,
particularly for the higher chlorophenols (Callahan et al., 1979).
The long-term persistence of chlorophenol isomers is only expected
where there is a lack of degradative activity and/or outward
transport, allowing them to accumulate, as is illustrated by the range
of residence times for chlorophenols in aquatic environments.
Substantial quantities of chlorophenols were eliminated from in situ
marine pelagic enclosures (4-MCP and 2,4-DCP) (Kuiper & Hansveit,
1984) and from fresh waters in vitro: 2-MCP and 4-MCP (Ettinger &
Ruchhoft 1950; 2,4-DCP (Aly & Faust, 1964); 2,4,6-T3CP and PCP
(Schauerte et al., 1982; Sugiara et al., 1984) in roughly 1-3 weeks.
In contrast, it has been shown that T4CP and PCP in sediments, where
photolysis and apparently biodegradation are minimal, may persist for
years (Pierce & Victor, 1978; DeLaune et al., 1983; Fox & Joshi,
1984).
A similar range of persistences has been reported for soils. Most
of 2-MCP and 4-MCP was removed by microorganisms in soil after 10 and
20 days, respectively (Walker, 1954). Virtually all of 2,3,4,5-,
2,3,4,6-, and 2,3,5,6-T4CP (100 mg/kg dry soil) disappeared from
paddy soils after 4 weeks of incubation in vitro (Ide et al., 1972).
Concentrations of trichlorophenols and tetrachlorophenols derived from
PCP degradation in vitro were in turn, substantially reduced after
many days (Kuwatsuka & Igarashi, 1975). The disappearance of at least
90% of added PCP from soils in vitro took from 21 to 205 days,
proceeding most rapidly in soils with a moderate to high organic
content and acclimated microorganisms (Kozak et al., 1979).
Most studies of chlorophenol metabolism have only monitored the
disappearance of the parent compound, but a few others have indicated
that subsequent metabolism may completely mineralize CPs. Thus,
labelled 4-MCP, 2,4-DCP, and 2,4,5-T3CP were converted to 14CO2 in
the in vitro incubation of estuarine waters and sediments (Lee &
Ryan, 1979; Hwang et al., 1986). About one half of the radioactivity
added to aerobic artificial streams as 14C-PCP was recovered as
carbon dioxide after 21 days (Pignatello et al., 1983). Pure cultures
and activated sludges may also mineralize chlorophenols to carbon
dioxide (Teidje & Alexander, 1969; Duxbury et al., 1970; Chu & Kirsch,
1973; Moos et al., 1983). Methane may be produced under anaerobic
conditions (Boyd & Shelton, 1984), but more often little or no
mineralization occurs in anaerobic sediments and sludges (Lee & Ryan,
1979; Horowitz et al., 1982; Pignatello et al., 1983).
In most instances, aerobic metabolism involves dechlorination and
hydroxylation, which are usually followed by cleavage of the phenol
ring at the ortho position and subsequent complete degradation. The
products of ring cleavage at the meta position are more resistant to
degradation and tend to accumulate in the medium. Reductive
dechlorination is an initial step towards complete mineralization
under anaerobic conditions (Krijgsheld & van der Gen, 1986).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental Levels
A substantial amount of research has been carried out on the
concentration of pentachlorophenol in the environment; however,
relatively few studies have been concerned with the determination of
the levels of other chlorophenols. Nonetheless, enough information is
available to make a preliminary survey of the residues of these
chlorophenols in the environment.
5.1.1 Air
No information is available on the ambient levels of
chlorophenols, other than PCP, in the atmosphere. The data on PCP are
limited but may provide a useful indication of the potential for
atmospheric distribution of the other chlorophenols. Measurable
quantities of PCP are present in ambient air, and are surprisingly
ubiquitous: Cautreels et al. (1977) detected 0.93 and 0.25 ng PCP/m3
in the mountains high above La Paz, Bolivia, a presumably
uncontaminated environment. Concentrations at 4 sites in Antwerp,
Belgium, ranged from 5.7 to 7.8 ng PCP/m3. It is not known whether
the compound was present as a vapour, or adsorbed on airborne
particulates. Presumably as a result of such transport, chlorophenols
have been detected in rainwater, alpine lakes, and snow (Bevenue et
al., 1972; Paasivirta et al., 1985).
5.1.2 Water
Residues of all chlorophenol isomers have been detected in aquatic
systems. Generally, residues are present at measurable concentrations
in discharges from such sources as manufacturing plants,
wood-treatment facilities, municipal waste discharges, and in the
receiving waters adjacent to these sources. Concentrations in other
receiving waters are more sporadic and quite low. While the levels are
low, chlorophenols have been detected in some of the least polluted
waters in the world.
Most reports of chlorophenol levels in water are from sites in the
vicinity of wood-treatment facilities. For instance, Fox & Joshi
(1984) measured concentrations of PCP and tetrachlorophenols in water,
sediments, and selected biota from the Bay of Quinte, in an
investigation of contamination by a wood-treatment plant. Levels of
both T4CP (2,3,4,5 plus 2,3,5,6) and PCP generally declined with
increasing distance from the source. Adjacent subsurface water levels
of T4CP ranged from 0.005 to 0.086 µg/litre over the summer of 1978,
while at the furthest site, 100 km distant, the range was 0.005-
0.030 µg/litre. Surface film samples contained T4CP concentrations
from 2.3-200 times higher than those below the surface. Bacon (1978)
assayed chlorophenols in the effluent from a Kraft pulp-mill at St.
John, New Brunswick, and found 2,4-DCP and 2,4,6-T3CP in the samples
before and after chlorination and caustic extraction. No chlorophenols
were detected in the receiving waters, perhaps as a result of tidal
flushing. Similarly, in a Great Lakes survey conducted by the Ontario
Ministry of the Environment (Jones, 1981), chlorophenol congeners were
detected in receiving waters: samples from the St. Mary's River near a
pulp and paper-mill did not contain any detectable levels of
chlorophenols, while 1 of 10 samples taken from near another mill on
Thunder Bay, contained 4 µg DCP/litre, and 2 contained 3 and 23 µg
T3CP/litre, respectively.
An Environment Canada survey (1979) of British Columbia coastal
waters for chlorophenol contaminants in surface waters, effluents,
sediments (section 5.1.2.2), and biota (section 5.1.6) did not reveal
any DCP or T3CP residues in water samples, but low levels of T4CP
(and PCP) were present at almost all sites: tetrachlorophenol
concentrations ranged from trace levels to 1.0 µg/litre in fresh
waters and 5.2 µg/litre in sea water. Effluent concentrations of T4CP
were high at 2 out of 4 discharges sampled (530 and 8270 µg/litre),
even exceeding PCP levels. Garrett (1980) reported sources and levels
of chlorophenols in sediments (section 5.1.2.2), fish (section 5.1.6),
and a variety of discharges from industry, waste disposal systems, and
runoff from landfills in the lower Fraser River and estuary. The most
frequently detected chlorophenols were 2,4,6-T3CP, 2,3,4,6-T4CP, and
PCP. Levels in most discharges were less than 7 µg/litre. Discharges
from municipal sewage-treatment plants in the same study also
contained several chlorophenols, most frequently 2,4,6-T3CP
(trace-1.2 µg/litre), 2,3,4,6-T4CP (trace-28.3 µg/litre) and PCP.
As in the Canadian studies, levels in receiving waters in the USA
are typically low. Morgade and co-workers (1980) did not find any
detectable levels of chlorophenols, other than PCP, in drinking-waters
in Dade County, Florida. In vitro chlorination of secondary sewage
effluent and power plant cooling waters, using chlorine levels and
treatment times similar to those used in practice, yielded only
µg/litre quantities of 2-MCP, 3-MCP, and 4-MCP (Jolley et al., 1975).
Pierce & Victor (1978) measured the levels of PCP and some of its
degradation products (2,3,5,6-T4CP and PCP-OCH3) in a Mississippi
lake contaminated by an overflow from a wood pole-treatment plant.
Prior to the spill, levels of 2,3,5,6-T4CP in the water were low,
ranging from 0.07 to 0.21 µg/litre. Concentrations were higher after
the spill (0.25-2 µg/litre), and remained relatively stable for at
least 4 months. Sediment (section 5.1.2.2) and fish tissue (section
5.1.6) samples were also collected.
Dutch research workers have monitored chlorophenol levels in the
water and sediments of the major rivers in industrial areas in the
Netherlands since 1976 (Wegman & Hofstee, 1979; Wegman & van den
Broek, 1983). In both reports, maximum levels of all chlorophenols,
other than PCP, seldom exceeded 1 µg/litre. Medians for the most
frequent congeners for 6 rivers in 1976 and 1977 ranged as follows:
2,6-DCP, trace-0.15 µg/litre; 2,4,5-T3CP, trace-0.15 µg/litre;
2,4,6-T3CP, trace-0.19 µg/litre; 2,3,4,6-T4CP, trace-0.11 µg/litre
(Wegman & Hofstee, 1979). Likewise, Piet & de Grunt (1975) reported
that levels of monochlorophenols ranged from not detectable to
20 µg/litre, dichlorophenols from not detectable to 1.5 µg/litre, and
trichlorophenols from not detectable to 0.1 µg/litre in Netherland
rivers and coastal waters. These ranges include the levels reported by
Zoeteman (1975) for Rhine river water and drinking-water. Zoeteman et
al. (1981) compiled information on concentrations of a variety of
chemicals in Dutch ground waters. The highest concentrations of
chlorophenols reported were as follows: 2,3,6-T3CP, 1 µg/litre;
2,4,5-T3CP, 2 µg/litre; 2,3,4,6-T4CP, 3 µg/litre; 2,3,5,6-T4CP,
5 µg/litre; PCP, 1 µg/litre.
In the Glatt river in Switzerland, concentrations of 2,3,4,6-T4CP
over the year averaged about 0.04-0.05 µg/litre at each of several
stations along a 35-km stretch of the river (Ahel et al., 1984).
Paasivirta et al. (1985), assayed water, snow, ash, benthic
invertebrates, fish, and birds from relatively unpolluted Finnish
lakes for chlorophenol residues. (Levels in the biota are reported in
section 5.1.6). The compounds 2,6-DCP, 2,4-DCP, 2,4,6-T3CP,
2,4,5-T3CP, 2,3,4,6-T4CP, and PCP were widespread, and present at
µg/litre concentrations in pulp-bleaching liquors and ng/litre levels
in lake waters (Table 10). Chlorination of some waters elevated the
concentration of the total chlorophenols measured almost 6-fold, from
0.043 µg/litre to 0.243 µg/litre. Elevated levels have been associated
with specific discharges in Europe. Leachate from a Danish chemical
dump site used during 1953-71 contained, among other compounds, PCP
and T4CP, the latter ranging in concentration from 0.030 to
80 µg/litre (Folke et al., 1984). Folke (1984) analysed effluent from
a Danish sewage-treatment plant, which received a portion of its
wastes from the manufacture of phenoxy herbicides, for a number of
chlorophenols. The effluent contained 0.1 µg 2-MCP/litre, 0.03 µg
4-MCP/litre, 0.5 µg 2,4-DCP/ litre, 0.6 µg 2,6-DCP/litre, 8 µg
2,4,6-T3CP/litre, and 0.03 µg 2,3,4,6-T4CP/litre.
Table 10. Concentration (µg/litre) of chlorophenols in Finnish pulp mill waste
liquors and fresh watersa
Chlorophenol Type of sample
Waste liquorsb Fresh watersb Janakka water Jyvaskyla
raw tap tap
2,6-DCP ND - 12c ND - 0.073 0.010 0.062 0.272
2,4-DCP ND - 11 ND - 0.014 0.014 0.053 0.093
2,4,6-T3CP 15 - 28 ND - 0.011 ND 0.030 0.014
2,4,5-T3CP ND - 66 ND - 0.019 0.019 0.059 0.035
2,3,4,6-T4CP ND - 10 ND - 0.090 ND 0.016 0.009
PCP ND - 01 0.064 - 0.011 0.023 0.005
a From Paasivirta et al. (1985).
b Range reported.
c ND -- Not detectable.
A similar variety of congeners was detected in effluent from a
sewage-treatment plant that was processing paper-mill wastes.
Lindstrom & Nordin (1976) found 115 µg 2,4,6-T3CP/litre in spent
bleach liquors from kraft mill pulp chlorinated in vitro, and noted
that dichlorophenols were also present.
As in Canada, high concentrations of chlorophenols in European
fresh waters are associated with wood-treatment facilities. Waters on
the sites of 2 Finnish sawmills, in which a sodium chlorophenate
preservative (mostly Na-2,3,4,6- T4CP, with substantial quantities of
the 2,4,6-T3CP and PCP salts) was used to protect against sapstain
fungi, contained total chlorophenol concentrations ranging from 1.6 to
20 000 µg/litre. The highest concentration occurred in a blind drain
adjacent to the dip site (Valo et al., 1984). Off-site levels in
ground water and lake water were much lower, with a maximum of 1.17 µg
chlorophenols/litre. Concentrations of chlorophenols in the effluent
from a Swedish sawmill on two separate dates were, respectively: 8.3
and 24 µg 2,4-DCP/litre; 40 and 22 µg 2,4,6-T3CP/litre; 7.5 and
5.8 µg 2,3,4,6-T4CP/litre (Xie et al., 1986).
Chlorophenols are also widespread in European marine waters,
generally at lower concentrations than in fresh waters. Weber & Ernst
(1978) noted that coastal waters off the Federal Republic of Germany
yielded only trace quantities (1 ng/litre) of 2,4-/2,5-DCP, 2,6-DCP,
2,4,5-T3CP, 2,4,6- T3CP, 2,3,4,5-T4CP, and 2,3,4,6/2,3,5,6-T4CP.
Danish marine waters receiving chemical dump leachate including T4CP
(at 0.030-80 µg/litre) showed corresponding levels in water of
0.006-0.008 µg/litre (Folke et al., 1984). Chlorophenol levels in
coastal waters off Sweden fell off rapidly with increasing distance
from a sulfate pulp-mill, from maximum concentrations on one date of
0.123 µg 2,4-DCP/litre, 0.040 µg 2,6-DCP/litre, 0.370 µg
2,4,6-T3CP/litre, and 0.084 µg 2,3,4,6-T4CP/litre to undetectable
levels (Xie et al., 1986). For example, the half-distances for
2,4,6-T4CP and 2,3,4,6-T4CP disappearance were 1 and 0.8 km,
respectively (see also sections 4.1.3, 5.1.2.2).
5.1.2.1 Sediments
Chlorophenol concentrations in sediments are for the most part
much greater than those in the overlying water. This may reflect
adsorption of the chlorophenols on suspended particulates in the water
column, with subsequent sedimentation. For instance, Eder & Weber
(1980) reported higher levels of chlorophenols (di- to penta-) in both
sediments and suspended solids compared with those in water.
Fox & Joshi (1984) analysed sediment cores for T4CP and PCP in
their study of chlorophenol contamination from a wood-preservation
facility on the Bay of Quinte, Lake Ontario. For the upper sediments
(1/2-cm sections of the top 5 cm of the core) levels ranged from 1 to
48 µg/kg dry weight. In a similar study, Environment Canada (1979)
analysed sediments in British Columbia waters associated with
wood-preservation plants. T4CP was present at all 11 sites, and
ranged from a trace to 1600 mg/kg dry sediment. T3CP was found at 4
of the 11 sites, with as much as 170 µg/kg of dry sediment measured.
Although the kraft pulp-mill effluent studied by Bacon (1978)
contained 2,4-DCP and 2,4,6-T3CP, only PCP was detected in sediment
samples downstream.
As a result of a PCP spill from a pole-treatment plant, sediments
in a Mississippi lake were contaminated with PCP and its degradation
products, including 2,3,5,6-T4CP (Pierce & Victor, 1978). Four months
after the spill, the levels of 2,3,5,6-T4CP in surface sediment
ranged from 3.8 to 71 µg/kg dry sediment whereas, 1 month after the
spill, levels of between 12 and 97 µg/kg dry sediment had been
detected.
Interpretation of these results is confounded by their
variability, the residence time (several weeks) of PCP in the water
column, and a 1974 spill at the same site. T4CP was present in the
sediments of a nearby, reportedly uncontaminated, pond at 1 µg/kg.
High concentrations of DCP and T3CP were present in sediments
adjacent to hazardous waste dumps near the Niagara River, at maximum
levels of approximately 2000 and 500 µg/kg, respectively (Elder et
al., 1981).
In studies in progress on chlorophenols that are present in
surface and coastal waters and sediments in The Netherlands, the
compounds 2,5-DCP, 2,3,5-T3CP, 2,4,5-T3CP, 2,3,4,5-T4CP,
2,3,4,6-T4CP, and PCP are observed most frequently (Wegman & van den
Broek, 1983). In the industrial regions that have been the focus of
this research, moderate levels of contamination prevail (several µg/kg
of dry sediment for most isomers) (Table 11).
Table 11. Levels of chlorophenols (µg/kg) in sediments from Dutch
surface waters and the Weser estuary
Compound Lake Ketelmeera Other Netherlanda Weser
surface waters Estuaryb
(Range) (Mean)
Median Maximum
3-MCP - 43
2,3-DCP 1.9 2.2
2,4-DCP 4.4 10 ND - 3.6 1.170
2,5-DCP 6.3 11 ND - 3.8
2,6-DCP 1.8 31 ND - 2.5
3,4-DCP 9.8 49 ND - 4.1
3,5-DCP 6.6 12 ND - 9.3
2,3,4-T3CP 0.7 0.8 ND - 0.6
2,3,5-T3CP 2.4 11 ND - 1.5
2,4,5-T3CP 6.4 15 ND - 6.3 1.170
2,4,6-T3CP 1.9 3.7 ND - 0.9 0.300
3,4,5-T3CP 1.2 19 ND - 1.7 0.310
2,3,4,5-T4CP 0.9 8.9 ND - 0.9
2,3,4,6-T4CP 1.7 4.9 ND - 1.7 1.546
2,3,5,6-T4CP 1.4 2.8 ND - 0.4
a From:Wegman & van den Broek (1983) (dry sediment).
b From: Eder & Weber (1980) (wet sediment).
Paasivirta et al. (1980) analysed sediments and biota (section
5.1.6) from three Finnish lakes for several chlorophenols and related
compounds. In one lake, contaminated only from sawmills upstream,
concentrations of T3CP and T4CP were 4.68 and 33.4 µg/kg of
sediment; respectively (Table 12, section 5.1.6.1), while in another
lake downstream from a pulp-mill, the corresponding values were 27.7
and 50.1 µg/kg. The third lake, further downstream from pulp and paper
inputs, contained intermediate levels of both compounds.
Levels in estuarine and marine sediments overlap considerably with
those from fresh waters. Butte et al. (1985) determined chlorophenol
concentrations in sediments and clams (section 5.1.6) from a German
bight that had received untreated PCP waste from a pulp and paper-mill
for 13 or 14 years, until 2 years before the study. Sediments from one
site near the former discharge contained from 28.3 to 30.9 µg
2,3,4,5-T4CP/kg of dry sediment, while those from another site
contained 92.8 µg 2,3,4,5-T4CP/kg and 7.9-11.6 µg 2,3,4,6- plus
2,3,5,6-T4CP/kg. Sediments taken some distance from the discharge
contained only 1.2 µg 2,3,4,5-T4CP/kg or less. Eder & Weber (1980)
reported levels of chlorophenols (di- to penta-) that corresponded
with the lower end of the range for The Netherlands surface waters;
mean concentrations of chlorophenols other than pentachlorophenol
ranged from 0.300-1.546 µg/litre. Similar levels were found in Baltic
Sea sediments from a site 2 km distant from a sulfate pulp-mill; these
contained 0.9 µg/kg dry sediment of 2,4-DCP, 0.4 µg 2,4,6-T3CP/kg,
and 3.1 µg 2,3,4,6-T4CP/kg (Xie, 1983). In a subsequent study, higher
levels of the same congeners were found at this location (Xie et al.,
1986). While surface sediments from sites roughly 5-10 km from the
discharge contained levels of chlorophenol that were near or below the
limits of detection, sediments 2 km or less from the mill contained as
much as 16 µg 2,4-DCP/kg dry sediment, 19 µg 2,4,6-T3CP/kg, and 89 µg
2,3,4,6-T4CP/kg.
5.1.3 Soil
Information on ambient levels of chlorophenol residues in soils is
limited, perhaps reflecting limited use of these compounds on soils.
The processes of degradation and movement also combine to reduce soil
residues (section 4).
The sole report on levels of chlorophenols in Canadian soils is
undoubtedly atypical of the environment at large. Garrett (1980)
reported that soil samples from the former site of a pesticide plant
in Richmond, British Columbia contained 2 mg T4CP/kg dry soil,
0.18 mg T3CP/kg, as well as low levels of PCP.
Valo et al. (1984) assayed chlorophenols in the soil and water
(section 5.1.2) around two Finnish sawmills where lumber was treated
against sap-staining fungi. Soils at both facilities were heavily
contaminated with chlorophenols; up to 70 mg/kg were found near the
dipping site, and up to 6 mg/kg occurred in a storage area for the
preserved wood. Soil outside the storage area contained only
0.1 mg/kg. The most common chlorophenols in the preservative
formulation also predominated in the near-surface soils, but lower
chlorinated phenols (particularly dichlorophenols) became increasingly
important further down the soil horizon, presumably as a result of
decomposition of the preservative. In general, chlorophenol
concentrations declined in the progressively deeper layers.
Kitunen et al. (1987) determined the concentrations of
chlorophenols and their contaminants in soil near the preserving
facilities at 4 different sawmills. Concentrations of chlorophenols in
soil ranged from 500 to 3500 mg/kg, polychlorinated phenoxyphenols,
from 1-5 mg/kg, and polychlorinated dibenzofurans, from 0.2-5 mg/kg.
No clear decrease in the soil concentrations of these compounds was
seen during the first year after the mill stopped using technical
chlorophenol.
5.1.4 Food, feed, and drinking-water
5.1.4.1 Food
Little information is available on residues in food of
chlorophenols, other than PCP. Low-level contamination undoubtedly
occurs as a result of contact with treated wood storage and transport
containers, and from herbicide applications. However, both of these
uses are prohibited in a number of countries (section 3.2.2); hence,
the following cases may overestimate the extent of contamination.
Both T4CP and PCP were identified in agricultural products by the
Alberta Department of Agriculture (Jones, 1981). Trace levels of T4CP
(mainly 1 µg/kg, maximum 45 µg/kg) occurred in grab samples of
carrots, potatoes, turnips, and beets. T4CP concentrations as high as
472 µg/kg occurred as a result of contamination from treated wood.
No residues were detected in 45 samples of Southern Ontario milk
analysed for 2,4,5-T3CP, 2,4,6-T3CP, 2,3,4,6-T4CP, and PCP (Frank
et al., 1979).
In the USA, Bristol et al. (1982) determined 2,4-DCP
concentrations in 3 varieties of potatoes sprayed with 2,4-D as a
growth regulator at an application rate of 140 g/ha. Levels of
chlorophenol, which could arise as a contaminant or degradation
product of the herbicides, varied slightly in 3 varieties of potatoes,
but ranged between 3 and 8.8 µg/kg. Potatoes not sprayed with 2,4-D
did not contain any detectable 2,4-DCP.
Stijve (1981) determined chlorophenol residues in edible products
derived from the bones and hides industry, where PCP was used as a
preservative and disinfectant. Fleshing grease intended as an
ingredient in cow feed, contained 50-480 µg 2,4,5-T3CP/kg, 2060-13
400 µg 2,3,4,6-T4CP/kg, and 210-1090 µg PCP/kg. A survey of 50
samples of edible gelatins (worldwide distribution), which are
produced from the collagen in hides and bones, all contained PCP; 30%
also contained 2,3,4,6-T4CP and trace amounts of trichlorophenols. In
some countries, where chlorophenola are used for the disinfection of
hides, chiorophenol levels in some gelatins range from 1000 to
5000 µg/kg.
Chlorophenols have been found at very low levels in the tissues of
commercial livestock and poultry. Fartington & Munday (1976) found
2,3,4,6-T4CP (at 2-3 µg/kg) in chicken flesh from 3 out of 4 shops in
the United Kingdom.
5.1.4.2 Livestock feed
No data are available on levels of the lower chlorophenols in
animal feed. Jones (1981) cited a case in which a boxcar that had been
used to ship PCP was later filled with feed oats, contaminating the
oats with roughly 2000 mg PCP/kg. Presumably, the feed was
simultaneously contaminated with T4CP at roughly 10% of the PCP
levels, given the usual formulation for technical Na-PCP. Livestock
illness and mortality were associated with this incident.
5.1.4.3 Drinking-water
Data on chlorophenol residues in drinking-water are quite limited,
but suggest that levels vary considerably between locations. Sithole &
Williams (1986) reported that low levels of a number of lower
chlorophenols occurred infrequently in potable waters at 40 Canadian
treatment plants. Chlorination increased the concentrations of 2-MCP
(maximum observed: 65 ng/litre), 4-MCP (127 ng/litre), 2,4-DCP
(72 ng/litre), 2,6-DCP (33 ng/litre), and 2,4,6-T3CP (719 ng/litre),
but decreased those of 2,3,4,5-T4CP (reduced below the limit of
detection) and PCP (34 ng/litre). Lower chlorophenols were not
detected in drinking-water supplies from Dade County, Florida (Morgade
et al., 1980). Dietz & Traud (1978) found low concentrations of a
variety of CP congeners in drinking-water from the Ruhr area of the
Federal Republic of Germany, including: 3-6 ng 2,4-DCP/litre; 20 ng
2,6-DCP/litre; 1 ng 2,4,6-T3CP/litre; 1 ng 2,3,5-T3CP/litre; 3 ng
2,4,5-T3CP/litre; 1 ng 2,3,4,5-T3CP/litre; and 3 ng
2,3,4,6-T4CP/litre. For comparison, levels in the effluent from a
sewage plant were concurrently 2 orders of magnitude higher.
In contrast, Paasivirta et al. (1985) found a number of
chlorophenols in Finnish tap waters at levels roughly one order of
magnitude higher than those in the German study (Table 10). As these
data indicate, chlorophenol concentrations in drinking-water are
generally quite low; indeed, the low threshold concentrations
producing undesirable organoleptic (taste and odour) properties would
make higher levels in drinking-water unacceptable (WHO, 1984 and Table
21).
5.1.5 Treated wood
The treatment of wood continues to be an important use for
chlorophenols (section 3.2.2), with considerable potential for
environmental contamination, as well as general and occupational human
exposure. In 1978-79, levels of chlorophenols, principally PCP, were
measured in samples of wood shavings that were used as livestock
litter in Southern Ontario (Jones, 1981). No trichlorophenols were
detected, but T4CP, PCP, and related chlorinated anisoles were
quantified. T4CP levels were as high as 70 mg/kg. Daniels & Swan
(1979) determined that 15 lumber samples from a British Columbia
sawmill protected with a commercial formulation of T4CP and PCP salts
("sodium penta") contained on average 44 µg chlorophenols/cm2 (range,
29-86 µg/cm2), most of which was T4CP.
In the United Kingdom, Parr et al. (1974) found that wood shavings
from imported wood used as litter for hens were contaminated with the
sodium salts of T4CP and PCP. When lumber is planed, most of the
chlorophenates are removed in the shavings. As a result, levels of
2,3,4,6-T4CP and PCP in fresh litter are quite high. According to
Parr et al.(1974), T4CP concentrations averaged 54 mg/kg and ranged
from 4 to 310 mg/kg. When the litter is used, chlorophenol levels fall
off as they are converted to their corresponding chloroanisoles (Gee &
Peel, 1974): spent litter contained on average 0.7 mg T4CP/kg.
Similarly, Curtis et al.(1972) measured as much as 100 mg
2,3,4,6-T4CP/kg in fresh shavings and sawdust from the United
Kingdom.
Levin & Nilsson (1977) assayed for T4CP, PCP, and related
compounds in wood dust from a Swedish sawmill. The wood had been
treated with 2% Na-2,3,4,6-T4CP; T4CP levels in the dust ranged from
100 to 800 mg/kg.
5.1.6 Terrestrial and aquatic organisms
5.1.6.1 Invertebrates
Invertebrates contain levels of chlorophenols that are higher than
those found in the environment at large, reflecting moderate
bioconcentration by these organisms (section 4.2.2). Environment
Canada (1979) found tetrachlorophenol in invertebrates from the
receiving waters for wood-treatment plant effluents in British
Columbia. In fresh waters, crayfish (Pacifasticus sp.) pincer muscle
contained traces of T4CP. The same tissue from a marine crab (Cancer
magister) contained T4CP levels ranging from a trace to 20 µg/kg
wet weight. One clam (Macoma) sampled contained 12 µg T4CP/kg,
presumably in muscle tissue. T3CP was not detected in any of the
organisms. Similar chlorophenol burdens were reported by Bacon (1978),
who assayed for 2,4-DCP, 2,4,6-T3CP, and PCP in lipids from a clam
(Mya arenia) and sandshrimp (O'angon septemspinosa) from waters
receiving pulp-mill effluent. Trace quantities of 2,4-DCP were present
in both organisms, while 2,4,6-T3CP levels ranged from undetectable
to 7.2 µg/kg wet weight for the clam and 9.2 µg/kg wet weight for the
sandshrimp.
During the course of their survey of chlorophenol residues in the
Weser Estuary and German Bight, Ernst & Weber (1978) determined that a
polychaete (Lanice conchilega) contained on average 11.8 µg 2,4-DCP
and 2,5-DCP/kg, 19.3 µg 2,4,5-T3CP/kg, 26 µg 2,4,6-T3CP/kg, 7 µg
2,3,4,5-T4CP/kg, 66.9 µg 2,3,4,6-T4CP and 2,3,5,6-T4CP/kg, and
117.5 µg PCP/kg (all values wet weight).
Paasivirta et al. (1980) have surveyed levels of chlorophenols in
a variety of biota in 3 lakes with different chlorophenol inputs
(section 5.1.2.1 and Table 12). Plankton generally contained little or
no tri-chlorophenol, but relatively high concentrations of
tetrachlorophenol. Mussels (Anodonta piscinalis) and sponges
(Spongilla lacustris) contained moderate levels of both T3CP and
T4CP. Chlorophenol burdens in organisms from the different lakes
generally ranked in the same order as the perceived chlorophenol
inputs into the 3 lakes.
In more recent work (Paasivirta et al., 1985), mussels, chironomids,
sponges, and fly larvae from Lake Vatia, 5 km downstream from a
pulp-mill, were analysed for several chlorophenols. Low levels
(usually not detected to 20 µg/kg fresh weight) of chlorophenols were
found in all invertebrates except sponges, which inexplicably
contained 195 µg 2,4-DCP/kg fresh weight and 22 µg 2,4,6-T3CP/kg.
Table 12. Chlorophenol levels in various environmental compartments in three
Finnish Lakes (µg/kg wet weight, except for sediment, dry weight)a,b,c
Population Lake N Trichlorophenol Tetrachlorophenol
species
X S CV X S CV
Kd 8 0.79 1.6 2.03 20.2 40.0 1.98
Pike pd 6 17.3 18.1 1.05 11.1 14.9 1.34
Vd 10 13.6 19.1 1.40 19.0 12.4 0.65
Roach K 9 ND ND 2.19 1.82 0.83
P 10 4.67 5.29 1.13 6.41 3.42 0.53
V 10 55.9 53.4 0.96 11.5 8.26 0.72
Mussel K 10 ND ND 2.83 4.44 1.57
P 9 1.44 2.21 1.53 7.44 3.47 0.47
Sponge K 5 0.36 0.80 2.22 6.30 6.36 1.01
P 5 6.86 9.14 1.33 1.45 2.43 1.68
V 5 4.96 3.73 0.75 2.56 1.12 0.44
Plankton K 4 ND ND ND ND
(100 µm) P 4 ND ND 9.28 10.0 1.08
V 4 2.45 4.90 2.00 9.90 1.73 0.17
Plankton K 4 ND ND 7.95 15.6 1.96
(25 µm) P 4 NO ND 14.3 10.5 0.73
V 3 ND ND 23.1 4.08 0.18
Sediment K 5 4.68 10.4 2.22 33.4 38.6 1.16
(0-2 cm) P 5 10.7 15.1 1.42 37.5 29.3 0.78
V 5 27.7 17.2 0.62 50.1 17.3 0.35
a From: Paasivirta et al. (1980).
b N = number of samples analysed;
x = mean;
s = standard deviation;
cv = coefficient of variation.
c Wet weight of plankton was calculated from dry weight by multiplying by
13.69.
d Lake areas: K = Konnevesi; P = Paijanne; V = Vatia.
Butte et al. (1985) analysed clams from a German bight that had
received untreated PCP-contaminated discharge from a paper-mill for a
number of years (section 5.1.2.1). Clams near the discharge contained
0.7-55.6 µg 2,3,4,5-T4CP/kg dry tissue, while those further away
contained, at the most, 2.6 µg 2,3,4,5-T4CP/kg and 0.2 µg 2,3,4,6-
plus 2,3,5,6-T4CP/kg. Similar low concentrations of 2,3,4,6-T4CP and
PCP were found in blue mussels (Mytilus edulis) from Danish coastal
waters (Folke & Birklund, 1986). Tissue levels of T4CP averaged from
0.2 to 2.9 µg/kg fresh weight (1-23 µg/kg dry weight) for mussels from
various locations in 1985, with no obvious relation to a nearby
chemical dump from which chlorophenols were leaching.
5.1.6.2 Fish
In general, levels of chlorophenols in fish are similar to, or
slightly higher than, those in invertebrates. Bacon (1978) studied
chlorophenol levels in different tissues from several fish species
from the St. John River estuary, New Brunswick, which receives
pulp-mill effluents. Residues of 2,4-DCP and, 2,4,6-T3CP were
detected, usually at several tens of µg/kg wet weight and several
µg/kg wet weight respectively, in all tissues including muscle,
viscera, skin, and liver (calculated from per-lipid-weight data in
original publication). In some instances, levels in liver were much
higher than this; concentrations as high as 242.9 µg 2,4-DCP/kg and
128 µg T3CP/kg (wet weight) were measured. In surface and coastal
waters in British Columbia, Environment Canada (1979) detected T4CP
in marine and freshwater sculpins. Across all sites, skeletal muscle
burdens averaged 30 µg/kg wet weight, and ranged from a trace to
100 µg/kg. Levels were roughly an order of magnitude higher in liver.
Similarly, Garrett (1980) reported that marine sculpins (Leptocottus
armams) from the lower Fraser River were the principal fish with
detectable amounts of T4CP, averaging 24.9 µg/kg wet weight, and
ranging from a trace to 62 µg/kg. Significant levels of T4CP were
also found in squawfish, which averaged 10.5 µg/kg wet weight and
contained as much as 18 µg/kg. Chlorophenol levels were higher in fish
that were caught in the industrialized areas of the river. Spottail
shiners (Notropis hudsonius) from Lakes Erie and Ontario contained
2,4,5-T3CP and 2,4,6-T3CP at maximum concentrations of 22 and
33 µg/kg (wet weight, whole fish), respectively (Canada-Ontario Review
Board, 1981).
Similar tissue concentrations have been detected in fish from
European waters. In Finnish lakes spanning a gradient of chlorophenol
inputs (Paasivirta et al., 1980), skeletal muscle of roach contained
on average 0-55.9 µg T3CP/kg wet weight, and 2.19-11.5 µg T4CP/kg
(Table 12). Chlorophenol concentrations in roach muscle were related
to the chlorophenol inputs into the lake. In contrast, levels in pike
skeletal muscle (average ranges 0.79-17.3 µg T3CP/kg, and
11.1-20.2 µg T4CP/kg) bore no relation to chlorophenol inputs. In
subsequent studies (Paasivirta et al., 1981, 1983, 1985), average CP
levels in muscle of pike, burbot, ide, and roach taken from waters
receiving pulp-mill discharges also fell within the same range, except
in the case of heavily polluted waters.
In Lake Tiiranselka, which receives a large volume of pulp-mill
effluent, average concentrations of 2,4,6-T3CP and 2,3,4,6-T4CP in
pike muscle were 37.02 and 125.02 µg/kg wet weight, respectively. Two
species of Baltic salmon analysed for chlorophenols contained similar
levels of contamination to those found in fish from moderately
polluted lakes (Paasivirta et al., 1985). Muscle tissue of salmon from
2 rivers and a hatchery contained an average of 3 µg 2,4,6-T3CP/kg
fresh weight, 1.8 µg 2,4,5-T3CP/kg, and 12.5 µg 2,3,4,6-T4CP/kg.
Fish collected in the vicinity of a pulp-mill effluent in Sweden
contained 2,4,6-trichlorophenol and related compounds that were
present in the discharge (Landner et al., 1977). Perch (Perca
fluviatilis) contained levels of 2700 µg/kg in liver fat (62.1 µg/kg
fresh weight), while Northern Pike (Esox lucius) contained
400-500 µg/kg (27.5-40.4 µg/kg fresh weight).
Extremely high chlorophenol levels have occurred as a result of
accidental spills. Following contamination of a Mississippi lake by
PCP in December 1976 (section 5.1.2.1), levels of the degradation
product 2,3,5,6-T4CP in sunfish liver and muscle increased by 1-2
orders of magnitude (Pierce & Victor, 1978) (Table 13). Four to 5
months after the spill, liver levels in sunfish were approaching
pre-spill levels, while levels in muscle tissue apparently cleared
more slowly. Bass and catfish showed particularly high levels of T4CP
after the spill, but unfortunately no baseline data were provided.
5.1.6.3 Other non-human vertebrates
Data on chlorophenol concentrations in vertebrates other than fish
or human beings are quite limited. Purple martin fledglings analysed
for chlorophenol residues contained 2 µg T4CP/kg (Jones, 1981). The
tissue analysed was not specified. Levels of CP residues in eggs,
embryos, and chick tissues of ring-billed gulls on the Ottawa and St
Lawrence Rivers have been reported (NRCC, 1982). The compounds
2,4-DCP, 2,4,5-T3CP, 2,4,6-T3CP, and PCP were present in most
tissues, the highest concentrations occurring in liver and brain
(Table 14). Paasivirta et al. (1985) measured chlorophenol residues in
the muscle tissue of 45 juvenile starlings from southern Finland.
Detectable levels of residues were not common: 2 birds contained 1 µg
2,3,4,6-T4CP/kg fresh muscle and 2 others contained 1 and 2 µg
2,4,6-T4CP/kg, respectively.
Table 13. Levels of 2,3,5,6-T4CP in tissues of fish from a
Mississippi lake (USA) contaminated by PCP from a
wood-pole treatment facilitya
Date Fish Concentration (µg/kg wet weight)
Muscle Liver
October 11/76b Sunfish 1 <1 30
Sunfish 2 <1 50
January 6/77 Sunfish 1 95 950
Sunfish 2 60 NAc
Bass 2 300 1600
Bass 3 130 8200
Catfish 1 219 8500
April 27/77 Sunfish 1 27 25
Sunfish 2 22 150
Catfish 1 82 1400
Catfish 2 41 940
a From: Pierce & Victor (1978).
b Spill in December, 1976.
c NA = not analysed.
Table 14. Range of concentrations (µg/kg) of several chlorophenols
in ring-billed gull eggs, embryos, and chick tissuesa
Compound
2,4-DCP 2,4,5-T3CP 2,4,6-T3CP
Fresh eggs 0-176 0-26 12-87
Embryos 7 25
Chick liver 14-210 0 47-157
Chick brain 177-476 NDb 144-234
a From: NRCC (1982).
b ND = not detectable.
The same authors analysed osprey eggs, and the pectoral muscle,
brain, liver, eggs, and kidney of white-tailed eagles. 2,3,4,6-T4CP
levels in osprey eggs ranged from 0 to 17 µg/kg fresh weight; MCPs,
DCPs, and T3CPs were not detected. Similarly, only 2,3,4,6-T4CP
occurred (15-22 µg/kg fresh weight) in fresh eagle eggs. Fresh eagle
muscle tissue (2 samples) contained moderate levels of 2,4,6-T3CP (26
and 50 µg/kg respectively) and 2,3,4,6-T4CP (0 and 26 µg/kg). Single
samples of eagle brain, liver, and kidney revealed that all of these
tissues contained chlorophenol residues, some at high levels; kidney,
for example, contained 1017 µg 2,4-DCP/kg.
5.2 General Population Exposure
The general population is exposed to chlorinated phenols through
diverse sources and routes, which have been summarized by the NRCC
(1982). Chlorophenols can be ingested as contaminants in food
including produce sprayed with phenolic pesticides, flesh of livestock
given feed contaminated with these pesticides, and general food items,
usually at mg/kg levels (section 5.1.4).
In addition, sub-µg/litre quantities of chlorophenol congeners
have been detected in drinking-water (section 5.1.4). These 2 routes
of exposure are generally considered to be the major sources of
exposure of the general population to chlorophenols (US EPA, 1980c).
In addition, minor quantities may be taken up through the dermal and
respiratory routes. Sources include industrial discharges (solid,
liquid, atmospheric) of chlorophenolic wastes, exposure to treated
wood, exposure to general consumer products including adhesives,
textiles, wood-treatment products, mouth-washes and disinfectants, and
break down products of hexachlorobenzene and phenoxy acid herbicides.
Because of this diversity of sources of chlorophenols, there are
no comprehensive estimates of the chlorophenol levels to which the
general population is exposed. On the basis of preliminary estimates
from the literature of total chlorinated phenol residues in food,
water, air, and miscellaneous sources, the Canadian Department of
National Health and Welfare (NHW, 1988) estimated typical
non-occupational exposure to all chlorophenols to be:
6.0 µg/person per day in food
2.8 µg/person per day in water
1.9 µg/person per day in air
2.0 µg/person per day from other sources
12.7 µg/person per day in total (= 0.18 µg/kg body weight per day
for 70-kg adult).
Similarly, the NRCC (1982) estimated that the total chlorophenol
exposure per day in the general population in Canada was 10-30 µg/
person (0.17-0.50 µg/kg body weight per day for a 60-kg adult). This
estimate was based on the following assumptions: 6 µg/person per day
from food; 4 µg/person per day from water; and 20 µg/person per day
from air. The last figure is extremely high, based on monitoring data,
and was derived by assuming that indoor rooms were treated with a
chlorophenol preservative. This figure should be considered tentative
in view of the meagre data base available on environmental levels.
On the basis of approximate levels of several trichlorophenols in
drinking-water and fish flesh, SENES (1985) estimated the daily
general population intake of each of 2,4,5-T3CP, 2,4,6-T3CP, and
(2,3,5- + 2,3,6-) T3CP to be 0.44 µg/person per day. If it is assumed
that the uptake of each of the remaining 2 isomers is also
0.44 µg/person per day, the total T3CP intake would then be
2.20 µg/person per day.
These low estimated levels of exposure are confirmed by the few
studies in which the residue levels of lower chlorinated phenols have
been determined in the general population. Although contamination
generally appears to be widespread, the concentrations of
chlorophenols in the tissues and fluids of people, not occupationally
exposed, are extremely low.
Kutz et al. (1978) determined the levels of pesticide-related
phenolic residues in human urine samples from all over the USA with a
limit of detection of 5-30 µg/litre. In over 1.7% of 400 samples
collected from the general population, 2,4,5-T3CP was present at a
mean concentration of less than 5 µg/litre, and a maximum of
32.4 µg/litre.
In comparing different methods of detection of hexachlorobenzene
and 2,4,5-T3CP in human serum and urine, Yost et al. (1984) found
2,4,5-T3CP levels in the 2 fluids, in the USA, to be 0.25-
6.7 µg/litre and 0.25-1.9 µg/litre, respectively. Samples were pooled
from the general population, but neither the sample size nor the site
of origin was specified.
As part of the development of an analytical method for
chlorophenols, Edgerton et al. (1980) determined chlorophenol
concentrations in urine samples from the general population. The
origin of the samples was not specified, but was presumably the
southeastern USA. Chlorophenol concentrations ranged widely as
follows:
2,6-DCP, 1-112 µg/litre; 2,4-/2,5-DCP, 2-161 µg/litre (mean,
34.1); 3,5-DCP, 15-44 µg/litre; 2,4,5-T3CP, 1-9 µg/litre;
2,4,6-T3CP, 1-6 µg/litre; 2,3,4,6-T4CP, 2-15 µg/litre.
Similar levels of T4CP were found in urine samples from 25
members of the general population in Barcelona, Spain (Gomez-Catalan
et al., 1987). The mean urine concentration was 6.2 µg/litre (standard
error of mean = 1.6). No trichlorophenols were detected.
In Dade County, Florida, where large quantities of lindane
(gamma-HCH) and Bromophos ( O-(4-bromo-2,5-dichlorophenyl) O,O
dimethyl-phosphorothioate) are used in agriculture, Morgade et al.
(1980) measured the serum concentrations of 2,4-DCP, 2,3,5-, 2,4,5-,
2,4,6-T3CP, 2,3,4,5-, 2,3,4,6-T4CP, and PCP in 58 female residents.
In addition, 10 samples of human adipose tissue from autopsies were
analysed for the same series of compounds. No detectable levels of any
of the non-fully substituted chlorophenols were found in the serum or
adipose tissue of the study group, but traces of PCP were found in the
drinking-water and in both biological compartments.
Williams et al. (1984) analysed adipose tissue from autopsies of
male and female residents of Ottawa (n = 84) and Kingston (n = 91),
Ontario, for organochlorine residues. Levels of 2,3,4,5-T4CP were
typically 6 or 7 µg/kg tissue, and did not differ significantly
between locations or sexes. Tissues from Kingston contained roughly 3
times more of other T4CPs (2,3,4,6 plus 2,3,5,6; male 24 µg/kg;
female 20 µg/kg) than those from Ottawa (male 6 µg/kg; female
8 µg/kg), but this difference was not statistically significant.
These data support the hypothesis that the general population is
exposed to very low levels of the lower-chlorinated phenols. However,
estimates of this burden are highly speculative at present, as data
are lacking for most congeners. Quantitative analyses for these
compounds in meat, poultry, produce, and drinking-water are scarce.
Atmospheric measurements have not been documented at all, and the
extent of dermal absorption by the general population, assumed to be
low, is not known.
5.3 Occupational Exposure
The potential for both acute and long-term exposure to
chlorophenols may be heavy for workers from industries using these
compounds. The routes of exposure for Canadian workers have been
summarized by NRCC (1982); the same routes undoubtedly apply in most
other countries. Large numbers of workers are exposed to
chlorophenols, other than PCP, in the lumber industry, particularly in
instances where lumber is surface-treated with Na-T4CP, during the
dipping, sorting, handling, planing, trimming, or the grading of
lumber.
In-service treatment of wood by painters, wood preservation workers,
or telephone linemen could result in similar dermal and inhalation
exposure. Employees in the chemical industry, who are involved in the
manufacture of chlorophenols or their derivatives, may also be exposed
to high levels. The same is true of employees in manufacturing
industries that use chlorophenols as preservatives, such as the
photographic, paint, textile, rubber, construction, electrical,
pharmaceutical, and disinfectant industries. Finally, employees
working with products containing chlorophenols may be exposed, such as
commercial applicators and farmers using phenoxy herbicides, or those
exposed to treated wood in the fields of construction (carpentry), or
railways. For such occupational exposures, inhalation and dermal
absorption are the major routes of uptake.
Unfortunately, there is little quantitative information on
occupational exposure to low chlorine-substituted chlorophenols. As
might be expected of such moderately volatile compounds, high
atmospheric concentrations are found in work areas where they are in
use. In addition, the body fluids of persons working in such areas
contain elevated levels of chlorophenols. In general, concentrations
of chlorophenols in air at chemical manufacturing plants can reach
mg/m3 levels while much lower concentrations occur in facilities in
the lumber industry that use chlorophenols.
Ott et al. (1980) examined worker exposure to T3CP and 2,4,5-T at
a manufacturing plant in the USA. The time-weighted average
concentrations of T3CP in the air at work locations adjacent to the
reactor, salt wheel, acid wheel, and dryer, were 2.1, 2.1, 9.7, and
1.6 mg/m3 respectively.
In a factory manufacturing PCP in Japan, crude exhaust air vented
from a"drying room" contained 3.54 mg T4CP/m3 and 14.04 mg PCP/m3
(Akisada, 1964). Urine-T4CP concentrations of personnel in the
factory ranged from 0.07 to 0.37 mg/litre, compared with 0.01-
0.03 mg/litre for unexposed persons.
An industrial hygiene survey of worker exposure to chlorophenols
and hexachlorobenzene at a PCP-production facility revealed that
workers in different tasks were exposed to average concentrations of
2,3,4,6- plus 2,3,5,6-T4CP of 0.016-0.320 mg/m3 in conjunction with
several-times-higher exposures to PCP. The highest average exposures
were experienced by handymen and block-casting workers (Marlow, 1986).
Recent data from Kauppinen & Lindroos (1985) showed much lower
average atmospheric chlorophenol levels in 10 Finnish sawmills,
ranging from 24 to 75 µg/m3. The values given are the sum of the
three chlorophenols present, as the Na-2,3,4,6-T4CP formulation used
also contained 10-20% 2,4,6-T3CP and 5% PCP. The highest mean
concentrations in the general work place occurred at the site where
the solution was prepared and at the machine stacking the lumber. Much
higher levels were also detected inside the drying kilns, where
chlorophenol concentrations averaged 5800 µg/m3. Levels of
2,4,6-T3CP were measured separately; particularly high concentrations
were noted at the machine stacking site (58 µg/m3) and the outdoor
dipping site (44 µg/m3), while it could not be detected at the
preparation site. Average urine levels of T4CP and PCP (measured
together) ranged from 0.10 to 3.3 µmol/litre (approximately
25-825 µg/litre). The highest mean concentration occurred among the
loaders at the trough dipping area (mean air levels 55 µg/m3, dermal
uptake substantial), while the other urine values parallelled the
atmospheric readings in terms of relative concentration.
Kauppinen (1986) reported that air concentrations of chlorophenols
(T4CP and PCP combined) for a variety of tasks in Finnish plywood
plants usually ranged from < 1 to 6 µg/m3. The levels in air plus
wood dust, where plywood was sawed, ranged from 3 to 6 µg/m3 and were
usually higher than those in air at work sites where wood dust was
minimal.
A detailed study of the chlorophenol exposure of sawmill workers
in a pulp, paper, and sawmill complex in British Columbia was
conducted by Embree et al. (1984). They divided the workers into 3
groups: a control group of 351 workers in areas with no identifiable
air contaminants; a group of 31 workers in close proximity to recently
treated lumber, who did not have manual contact with it (airborne
exposure); and a group of 40 who handled recently treated lumber
(dermal plus airborne exposure). Air levels of chlorophenols were
determined using personal monitors. Tetrachlorophenol levels in the
plant air were elevated, and similar for the airborne group (3.3 ±
2.1 µg/m3; mean ± standard deviation), and the dermal-plus-airborne
group (3.0 ± 2.7 µg/m3). Serum levels were related to perceived
exposure in a dose-dependent manner; tetrachlorophenol concentrations
for the dermal-plus- airborne group (204 ± 92 µg/litre) were
approximately twice those in the airborne group (112 ± 136 µg/litre),
and 8 times those in the controls (26 ± 7 µg/litre). Urine levels for
the 2 exposed groups were also dose-dependent (airborne 93 ±
43 µg/litre; dermal-plus-airborne 125 ± 20 µg/litre). Urine levels in
the control group were not reported.
Similar urine concentrations were reported for American
wood-workers exposed to Permatox(R) (3% PCP, 21% 2,3,4,6- T4CP)
(Kalman & Hortsman, 1983). Of 47 workers, 28 showed urine levels of
more than 100 µg 2,3,4,6-T4CP/litre, 13, levels between 20 and
100 µg/litre, and 6 levels of less than 20 µg/litre. Air levels were
reportedly below 25 µg/m3. Because atmospheric concentrations were
this low, the authors suggested that the individuals with the highest
urine levels were taking up most of the dose through non-respiratory
routes, most likely dermal. Over a 2-week holiday period, T4CP levels
in the three groups declined by averages of 84%, 67%, and 34%,
respectively, a slower rate of elimination than that found in
experimental animals (section 6.4).
Kleinman et al. (1986) and Fenske et al. (1987) also evaluated the
extent and impact of occupational exposure to Permatox(R) in 100
workers from a lumber-mill in Washington State. Plant air
concentrations of T4CP ranged from 0.8 to 12.2 µg/m3, while no PCP
was detected (limit of detection 0.5 µg/m ). It was estimated that
dermal exposure accounted for 95% of the dose taken up by exposed
workers. Average chlorophenol concentrations in the urine were higher
for exposed workers than for controls (range of averages:
T4CP-exposed = 31.2-497.5 µg/litre, control = 6.3-28.7 µg/litre;
PCP-exposed = 57.4-102.8 µg/litre, control = 28.9-38.8 µg/litre).
In a recent report, 230 sawmill workers in Finland were examined
for urinary levels of chlorophenols (Lindroos et al., 1987). In
occupations where dermal exposure was greatest, workers (n = 112) had
a median urinary chlorophenol level of approximately 1.8 mg/litre
(range, 0.02-49 mg/litre, assuming all chlorophenols were T4CP)
whereas employees (n = 34) exposed mainly via the respiratory route
had a median urinary level of 0.2 mg chlorophenols/litre (range,
0.02-3.1 mg/litre). These results support the hypothesis of Kalman &
Hortsman (1983) regarding the importance of the dermal route of
exposure for chlorophenols.
The chlorophenol levels in urine among workers handling imported
lumber treated with 2,3,4,6-T4CP ranged from 0.13 to 2.2 µmol/litre
(30.2-510.4 µg/litre, assuming all chlorophenols were T4CP) with a
mean value of 0.86 µmol/litre (199.5 µg/litre) (Rappe et al., 1982).
These exposure data are static, and, as such, give no information
on the actual amount of chlorophenols taken up by a worker. In the
course of designating permissible levels of chlorophenol exposure for
regulatory purposes, the US EPA (1978) modelled the chlorophenol
exposure experienced by a worker performing various tasks (Table 15).
The exposure levels indicate that, as expected, occupational
exposure to chlorophenols is much higher than non-occupational; these
rates of uptake are 2 or more orders of magnitude higher than
estimates of exposure of the general population to all chlorophenols
summarized in section 5.2.
However, the estimates of chlorophenol burdens given in Table 15
are for 2,4,5-T3CP (the use of which has been discontinued in many
countries), and are based on speculative scenarios that exaggerate
worker exposure to this compound. Despite the longstanding concern for
the potential health hazards associated with occupational exposure to
chlorophenols, meaningful estimates of worker exposure to the
chlorophenols that are currently extensively used do not appear to
have been made to date. Exposures have been estimated qualitatively
or, at best, semi-quantitatively. Although occupational uptake of
chlorophenols is thought to be principally through inhalation and
dermal absorption, there are no data on the rates of such uptake. To
obtain such information, air monitoring should be continuous
throughout the shift, using personal monitors, and urine levels of
chlorophenols should be measured for consecutive 24-h periods.
Table 15. Estimates of occupational exposure to 2,4,5-T3CPa
Site Dermal exposure Inhalation exposure
(µg/kg body weight per day)
Cooling tower 3.7b 23c
Water Systems 14d 90.3e
Pulp and paper mill 2f 55g
Tannery 49h 87i
Hospital 70j 9k
a From: US EPA (1978).
b Exposure to 100 ml containing 22 mg Na-2,4,5-T3CP/litre; 10%
absorption for 60-kg female maintenance worker.
c 100% relative humidity, 20 °C; therefore, 1 m3 air contains
0.0173 litre H2O with 22 mg Na-2,4,5-T3CP/litre H2O;
breathing rate, 1.8 m3/litre for 2 h; 60-kg female worker.
d Exposure to 100 ml containing 87 mg Na-2,4,S-T3CP/litre; 10%
absorption; 60-kg female worker.
e As footnote c, but product concentration 87 mg
Na-2,4,5-T3CP/litre H2O.
f Exposure to 80 ml (13 × 10 ml; one hand) containing 15 mg
Na-2,4,5-T3CP/litre; 10% absorption; 60-kg female worker.
g As footnote c, but product concentration 15 mg
Na-2,4,5-T3CP/litre H20, 7-h exposure.
h Exposure to 1.4 litre containing 21 µg Na-2,4,5-T3CP/litre;
10% absorption;
i 60-kg female worker. As footnote c, but product concentration
21 µg Na-2,4,S-T3CP/litre H2O, 8-h exposure.
j Exposure to 1 cup (0.24 litre; 16 cups per gallon) containing
686 mg Na-2,4,S-T3CP per gallon; 10% absorption; 60-kg
female worker.
k Hospital volume, 1800 m3; recommended air ventilation rate,
60 m3/h per person; 60 persons; 8-h day; total circulated air,
28 800 m3; 100 gallons disinfectant used; 100 cups remain giving
a total of 4.3 g Na-2,4,5-T3CP; volatilization at 25 °C;
breathing rate, 1.8 m3/h; 8-h day; 60-kg worker.
6. KINETICS AND METABOLISM
6.1 Absorption
Hoben et al. (1976a) exposed male Sprague-Dawley rats to an
aerosol of sodium-PCP (repeated exposures to about 5.9 mg PCP/kg body
weight) and found very rapid absorption into the blood. Unfortunately,
no information is available on the absorption of the lower chlorinated
phenols via the mammalian lung during inhalation exposure.
In general, chlorophenols are readily absorbed through the skin.
Using the skin of the hairless mouse, Huq et al. (1986) found that
aqueous solutions of 2-MCP, 2,4-DCP, and 2,4,6- T3CP readily
penetrated the skin, provided that the compound was not ionized (i.e.,
pH pKa). In vitro studies on epidermal membranes from human skin
taken at autopsy showed penetration by 2-MCP, 4-MCP, 2,4-DCP, and
2,4,6-T3CP (Roberts et al., 1977, 1978). The lipophilic character of
the solutes and their hydrogen-bonding capacity are the 2 main
features determining this penetration. Shen et al. (1983) investigated
the dermal absorption of T4CP in Sprague-Dawley rats and found the
Na-2,3,5,6-T4CP was more toxic than 2,3,5,6-T4CP itself. Toxic
amounts of 2,3,4,6-T4CP in organic solvents can be absorbed through
the skin (Gosselin et al., 1976), and the use of 2,4,5-T3CP in
hospitals has been suggested as a potential problem because of its
absorption through the skin (US EPA, 1978). Similarly, on the basis of
data concerning urine levels of chlorophenols, absorption through the
skin has been reported to be a major route of exposure among workers
occupationally exposed to chlorophenols or to their salts (section
5.3).
The greater part of orally-administered tri- and
tetra-chlorophenols is recovered in the urine and faeces of test
animals (section 6.4), indicating that lower chlorophenols are readily
absorbed through the gastrointestinal tract. More than 90% of the oral
dose was excreted in the urine of volunteers after ingestion of PCP,
which indicates similarly effective absorption via the
gastrointestinal tract in human beings (Braun et al., 1979).
6.2 Distribution
6.2.1 Tissue distribution following chlorophenol exposure
No information is available on the distribution of
mono-chlorophenols in animal systems.
With respect to dichlorophenols, single intravenous injections of
2,4-DCP (10 mg/kg body weight) in Sprague-Dawley rats weighing
250-300 g resulted in a maximum concentration (17.7 mg/kg of tissue) in
the kidney, 10 min after injection (Somani & Khalique, 1982). Levels
in liver, brain, and fat peaked at 15 min at 10.5 mg/kg, 3.2 mg/kg,
and 4.1 mg/kg tissue, respectively. A level of 1.64 mg/litre was
recorded in plasma, 10 min after injection.
Following intraperitoneal administration of 25 mg 2,4,6-T3CP/kg
body weight to male Wistar rats (Pekari et al., 1986), concentrations
in all tissues assayed were maximal, 30 min after injection: kidney
levels peaked at 329 ± 117 nmol/g, while maximum concentrations were
progressively lower in blood, liver, fat, muscle, and brain.
Hattula et al. (1981a) reported that Wistar rats fed 2,3,4,6-T4CP
in olive oil at 100 mg/kg intragastrically for 55 days showed the
following tissue concentrations of 2,3,4,6-T4CP: kidney, 5.1 mg/kg;
spleen, 3.2 mg/kg; liver, 2.2 mg/kg; brain, 1.2 mg/kg; and muscle,
0.46 mg/kg tissue.
6.2.2 Tissue distribution following exposure to chemicals metabolized
to chlorophenols
The distribution of chlorophenols as metabolites following the
administration of other organochlorine compounds has been investigated
in several studies. Like the original chlorophenols, these metabolites
accumulate most often in the kidney and liver. Clark et al. (1975)
investigated the tissue distribution of 2,4-DCP in sheep and cattle
fed 2,4-D. Cattle were given a diet containing 2,4-D at 0, 300, 1000,
or 2000 mg/kg (9, 30, or 60 mg/kg body weight per day). Muscle, fat,
liver, and kidney were analysed for 2,4-DCP. Sheep were given a diet
containing 2,4-D at 2000 mg/kg for 28 days. At 2000 mg 2,4-D/kg in the
diet, 2,4-DCP concentrations in kidney and liver from sheep were
0.26 mg/kg tissue and 0.16 mg/kg tissue, respectively; in cattle,
levels were 1.06 mg/kg and 0.31 mg/kg tissue, respectively.
In laying hens fed VC-13 Nemacide(R) [ O-(2,4-dichlorophenyl)-
O,O-diethyl phosphorothioate) at a dose of 800 mg/kg for 55 days,
Sherman et al. (1972) reported similar levels of 2,4-DCP in both liver
tissue and egg yolk (average values ranged from 0.122 to 0.613 mg/kg).
2,4-DCP was not detected in the muscle and fat of these birds.
Levels of 2,4,5,-T3CP in the tissues of sheep and cattle fed
trichlorophenoxy acid herbicides for 28 days were determined by Clark
et al. (1975). In sheep fed Silvex (2-(2,4,5- trichlorophenoxy)-
propionic acid) at 2000 mg/kg, residues of 2,4,5-T3CP were 0.22 mg/kg
in liver and 0.17 mg/kg in kidney.
Cattle fed this compound at 9, 30, or 60 mg/kg body weight had
2,4,5-T3CP tissue concentrations ranging from 0.06 to 0.48 mg/kg in
the liver and from 0.05 to 0.10 mg/kg in kidney. No residues were
detected in samples of muscle and fat from sheep or cattle fed Silvex.
Sheep exposed to 2,4,5-T in the diet at 2000 mg/kg for 28 days
exhibited 2,4,5-T3CP levels of 6.1 mg/kg, 0.90 mg/kg, 0.13 mg/kg, and
0.05 mg/kg tissue, in the liver, kidney, muscle, and fat,
respectively.
Sheep dosed orally with Erbon(R) (2-(2,4,5-trichloro-phenoxy)
ethyl 2,2-dichloropropionate) metabolized it to 2,4,5-T3CP and
2-(2,4,5-trichlorophenoxy) ethanol in less than 7 h (Wright et al.,
1970). Most of these compounds were eliminated in the urine (section
6.4), but mg/kg quantities of 2,4,5-T3CP and the other metabolite
were found in the kidney, liver, omental fat, muscle, and brain of
sheep given 100 mg Erbon(R)/kg body weight daily for 10 days (5.54,
3.14, 2.06, 1.00, and 0.21 mg/kg, respectively).
Male Wistar rats dosed with 8 mg lindane (gamma-hexachlorocyclo-
hexane)/kg body weight by gavage for 19 days showed 2,4,6-T3CP and
2,3,4,6-T4CP in heart tissue, 2,3,4,6- T4CP and/or 2,3,5,6-T4CP in
the liver, and 2,4,6-T3CP and 2,3,4,6-T4CP in the kidney (Engst et
al., 1976), but no quantitative data were given.
6.3 Metabolic Transformation
The major metabolic transformation for the lower chlorinated
chlorophenols appears to be conjugation with sulfate or glucuronate,
prior to clearance in the urine. Perhaps, because of similarities
between the structure and lipophilicity of T4CP and PCP, a small
proportion of these congeners undergo the same dechlorination and/or
oxidation reactions that PCP does prior to conjugation (Renner &
Mücke, 1986).
As much as 84.7% of administered 2-MCP was reportedly excreted as
sulfate and glucuronate conjugates in dogs (Karpow, 1893). In the
rabbit, oral administration of monochlorobenzene resulted in surf ate
and glucuronide conjugates of 2-MCP in the urine (Lindsay-Smith et
al., 1972). It has been suggested that in mice o-methylation might
be a relevant mechanism for 2-MCP detoxification (Angel & Rogers,
1972). Similarly, conjugates were detected in the kidney, liver, fat,
brain, and plasma of rats after the iv injection of 2.5-3 mg 2,4-DCP
(10 mg/kg body weight) (Somani & Khalique, 1982). Of the total
conjugates determined, glucuronide conjugates were the major
metabolite in kidney (79.6%), liver (62.7%), brain (77.9%), and plasma
(79.5%). No glucuronide conjugates were found in fat. Free 2,4-DCP did
not accumulate in rat tissues and was rapidly metabolized to its
conjugates. In another study using 14C-2,4-DCP on isolated perfused
rat liver, Somani et al. (1984) demonstrated that the liver is capable
of the formation of glucuronide conjugates, and that 2 dichlorometho-
xyphenols are metabolites of 2,4-DCP when glucuronide formation is
blocked by galactosamine (section 8.8).
Bahig et al. (1981) have suggested that, in rats, 2,4,6-T3CP is
isomerized to 2,4,5- and 2,3,6-T3CP before being excreted as
glucuronide conjugates. However, in a similar study on rats given
25 mg 2,4,6-T3CP/kg body weight (ip), 83 ± 11% was present in the
blood as glucuronides rather than being converted to another isomer
(Pekari et al., 1986).
Concerning T4CP, Ahlborg & Larsson (1978) showed that, of the 3
isomers, only 2,3,5,6-T4CP was metabolized to a significant extent in
the rat. Thirty-five percent of the given dose (10 mg/kg body weight
by ip injection) was metabolized to tetrachloro- p-hydroquinone,
which, when also given ip, is more toxic than the parent compound
(section 8.9). Trichloro- p-hydroquinone was a minor metabolite of
the other isomers. Recently, it has been shown that the microsomal
metabolism of PCP yields tetrachloro-1,2-and tetrachloro-1, 4-hydro-
quinone (van Ommen et al. 1986). Covalent binding to protein and DNA
occurs via the corresponding tetrachloroquinones (van Oremen et al.,
1988). To what extent this kind of metabolic activation plays a role
in the toxicity of lower chlorinated phenols is not known, at present.
The metabolism of compounds that axe structurally related to
chlorophenols also yields conjugates of chlorophenols. Kurihara &
Nakajima (1974) studied the metabolism in mice of injected 14C-hexa-
chlorocyclohexane (14C-HCH). The major metabolites were conjugates of
2,4,6-T3CP with sulfate or glucuronide, as well as conjugates of
2,4-DCP. The proportion of 2,4,6-T3CP sulfate to glucuronide
conjugates varied from 80%:20% to 40%:60%, depending on whether
gamma-HCH or beta-HCH was used. Trace amounts of free 2,4,6-T3CP and
2,4,5-T3CH were also found in the urine. Koransky et al. (1975) also
found that injection of 14C-HCH into rats resulted in glucuronide
and sulfate urinary metabolites of 2,4,6- and 2,4,5-T3CP; small
amounts of the free phenols were also detected. The ratio of sulfate
to glucuronide conjugates was not determined. Engst et al. (1976)
found that lindane (gamma-HCH) administration in rats produced free
2,4,6-T3CP, 2,3,4,6- and/or 2,3,5,6-T4CP and PCP in the urine, as
well as glucuronide-bound 2,3,4-T3CP, 2,3,4,5-, 2,3,4,6- and/or
2,3,5,6-T4CP.
In terms of glucuronide formation of PCP, the rat is probably a
better model for human beings than the monkey, which does not
metabolize this compound (Braun et al., 1978, 1979). Whether the rat
model can be used to predict human responses to the lower chlorinated
phenols is not known at present, since no human data exist. However,
hydrolysis of human urine samples indicated that most of the T4CP in
human urine is conjugated (Dahms & Metzner, 1979; Butte, 1984; Currie
& McDonald, 1986).
6.4 Elimination and Excretion
In experimental mammals, chlorophenols are eliminated primarily in
the urine. For example, Freitag et al. (1982) administered 14C-2,4,6-
T3CP to rats orally for 3 days to examine retention, dispersion, and
excretion rates. Within 7 days, 82.3% of the label was excreted in the
urine and 22.2% in the faeces. At sacrifice on the 8th day, residues
in the liver, lung, and adipose tissues were below the level of
detection (i.e., less than 0.01% of the label), whereas the carcass
retained 7.8% of the label. Bahig et al. (1981) found that 92.5% of a
daily oral dose (25 µg by gavage) of 14C-2,4,6-T3CP was excreted by
rats in the urine, while 6.4% was found in the faeces. Thus, the
ingested 2,4,6-T3CP was largely eliminated within 24 h. Similarly,
Ahlborg & Thunberg (1980) reported that 2,4,5-T3CP given to rats was
excreted rapidly (within 24 h) with very little retention by the
animal, and Pekari et al. (1986) estimated the half-times for the
elimination of 2,4,6-T3CP from the blood, liver, muscle, fat, brain,
and kidney of rats at between 1.4 and 1.8 h, after dosing ip with
25 mg/kg body weight.
Excretion by rats of the different T4CP isomers injected
intraperitoneally was examined by Ahlborg & Larsson (1978). While
2,3,5,6-T4CP was eliminated in the urine within 24 h and
2,3,4,6-T4CP within 48 h, only 60% of the injected 2,3,4,5-T4CP was
collected in 72 h.
In a study on the elimination of 2,4,-DCP from various tissues in
the rat following intravenous administration of 10 mg/kg body weight
(Somani & Khalique, 1982), the compound was eliminated most rapidly
from brain tissue followed by plasma, fat, liver, and kidney.
Half-lives for 2,4-DCP were 6 min in the brain, 10 min in fat and
plasma, 15.1 min in the liver, and 30.1 min in the kidney.
Much of the information on the excretion of chlorophenols has come
from studies of the uptake and clearance of chlorophenols that have
been formed metabolically from other compounds. As in the studies
described previously, these chlorophenols are generally eliminated
rapidly in the urine. Thus, Lindsay-Smith et al. (1972) identified
free and conjugated forms of all monochlorophenol isomers in the urine
of rabbits dosed with 14C-monochlorobenzene. Similarly, 2,4-DCP was
eliminated in the urine of rats injected with Nemacide(R) (67% of
dose excreted as 2,4-DCP within 3 days) (Shafik et al., 1973). Shafik
et al. (1973) also found that rats cleared 53% of a dose of
Ronnel(R) ( O, O-dimethyl- O (2,4,5-trichloro-phenyl)phosphoro-
thioate) as 2,4,5-T3CP within 2 days. A sheep given 50 mg/kg body
weight of Erbon(R) [2-(2,4,5-trichloro-phenoxy)-ethyl 2,2-dichloro-
propionate] as an oral drench metabolized it to 2,4,5-T3CP and
2-(2,4,5-trichloro-phenoxy) ethanol in less than 7 h (Wright et al.,
1970). Within 96 h, 68.42% of the dose was eliminated in the urine and
1.74% in the faeces, approximately half of these amounts as
2,4,5-T3CP.
Karapally et al. (1973) identified chlorinated phenols derived
from lindane in rabbit urine. Of the 14 chlorophenols identified,
comprising at least 19.9% of the total dose, the most abundant were
(in decreasing order) 2,4,5-T3CP, 2,3,5-T3CP, 2,4,6-T3CP,
2,3,4,6-T4CP, 2,3-DCP, 2,4-DCP, and 2,3,4-T3CP. The results of a
similar study on the rat (Engst et al., 1976) showed that 2,4,6-T3CP,
2,3,4,6-T4CP and/or 2,3,5,6-T4CP, and 2,3,4,5-T4CP derived from
lindane were eliminated via the urine. Chadwick & Freal (1972)
observed that, following one week of dosing with lindane, rats
excreted 3,4-DCP, 2,4,5-T3CP, 2,3,5-T3CP, 2,4,6-T3CP,
2,3,4,5-T3CP, and 2,3,4,6-T4CP for at least 1 month.
The clearance from tissues of chlorophenols derived from other
compounds may be slower than their elimination via the urine. Sherman
et al. (1972) found that from 60 to 83% of the 2,4-DCP metabolized
from Nemacide(R) disappeared from the liver of chickens within 21
days of the cessation of dosing. 2,4-DCP found in the yolk of eggs
from these hens dropped to non-detectable levels in 10 days for the
high-dose (800 mg/kg diet) group, while at the lower dosages (50, 100,
200 mg/kg diet), a shorter time was required for complete clearance
(see also section 6.2). In sheep fed 2,4,5-T3CP at 2000 mg/kg diet
(Clark et al., 1975), liver and kidney 2,4,5-T3CP levels remained
relatively constant one week after exposure ceased, while muscle
concentrations dropped roughly 3-fold.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
There are few studies on the effects of chlorophenols, other than
PCP, on organisms in the environment. This lack of information may
stem, in part, from the fact that many wastes contain other
potentially toxic components in addition to chlorophenols. Moreover,
most laboratory studies on the toxicity of chlorophenols for
environmental organisms have involved much higher exposure levels than
those that are usually found in the environment.
The information that is available on the effects of chlorophenols
deals primarily with aquatic habitats, perhaps because many point
discharges of chlorophenols are released into water bodies.
7.1 Laboratory Studies
7.1.1 Acute toxicity
Recent laboratory studies on the acute toxicity of chlorophenols
for aquatic biota are summarized in Table 16. The data are derived
primarily from studies published since 1980; information from research
prior to this date is presented by Jones (1981). In general, the
patterns evident in the review by Jones (1981) are also seen in the
more recent data (Table 16).
Considerable overlap exists in the chlorophenol levels that
produce toxic effects in bacteria, phytoplankton, macrophytes,
invertebrates, and fish (Table 16). For instance, LeBlanc (1984)
(Table 16) reported that LC50 values compiled for 4-MCP toxicity in
algae (Selenastrum, capricornutum, Skeletonema costatum),
invertebrates (Daphnia magna), and fish (Lepomis macrochirus,
Cyprinidon variegatus) ranged from 3.27-5.35 mg/litre. Most of the
EC50/LC50 values for other organisms compiled in Table 16 also fall
within the several mg/litre range. However, there are isolated reports
of certain bacterial, fungal, and protozoan processes that are
insensitive to chlorophenol exposure (Table 16).
In general, chlorophenol toxicity for aquatic organisms increases
with the degree of chlorination of the phenol ring (Table 16),
presumably as a result of increasing lipophilicity (Table 3).
Table 16. Acute toxicity of chlorophenols for aquatic biota
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Phytoplankton
Scenedesmus SB, FW 3,5-DCP 5.32 (EC50) Growth rate, Weibull model Christensen & Nyholm
spicatus 5.87 (EC50) Growth rate, Probit model (1984)
6.10 (EC50) Growth rate, Logit model
Selenastrum SB, FW 4-MCP 5.01 (EC50) Growth LeBlanc (1984)
capricornutum
Skeletonema SB, FW 4-MCP 3.27 (EC50) Growth LeBlanc (1984)
costatum
Bacteria
Bacillus sp. SB, FW 2-MCP 700
3-MCP 450
4-MCP 400
2,3-DCP 130
2,4-DCP 75
2,5-DCP 85
2,6-DCP 550
3,4-DCP 52 IC50c reduction in Liu et al. (1982)
3,5-DCP 25 activity after 30 min
2,3,4-T3CP 13 incubation with toxicant
2,3,5-T3CP 10
2,3,6-T3CP 190
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Bacteria
(contd).
Bacillus sp. 2,4,5-T3CP 12
(contd). 2,4,6-T3CP 240
3,4,5-T3CP 5
2,3,4,5-T4CP 4
2,3,5,6-T4CP 54
Photobacterium SW 2-MCP 33.8 Ribo & Kaiser
phosphoreum 3-MCP 14.1 (1983)
(Microtox(R)) 4-MCP 8.30
2,3-DCP 4.92
2,4-DCP 5.52
2,5-DCP 9.38 EC50 for inhibition
2,6-DCP 13.2 of light emission with
3,4-DCP 1.63 30-min toxicant exposure
3,5-DCP 2.77
2,3,4-T3CP 1.25
2,3,5-T3CP 1.11
2,3,6-T3CP 12.7
2,4,5-T3CP 1.27
2,4,6-T3CP 7.68
3,4,5-T3CP 0.359
2,3,4,5-T4CP 0.176
2,3,4,6-T4CP 1.27
2,3,5,6-T4CP 2.22
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Activated SB, FW 3,5-DCP 30.2 IC50 for O2 Dutka & Kwan
sludge consumption, 3 h (1984)
Photobacterium SW 2-MCP 22.1 EC50 for inhibition Indorato et al.
phosphoreum 2,4-DCP 3.36 of light emission with (1984)
(Microtox(R))
toxicant exposure
Activated SB, FW 3,5-DCP 7 EC50 for O2 King (1984)
sludge consumption
Nitrifying 5 EC50 for nitrate and
activated nitrite production
sludge
Photobacterium SW 3.2 EC50 for light emission
phosphoreum
(Microtox(R))
Sewage 8 EC50 for growth inhibition,
microorganisms 6 h
Sewage 6 EC50 for growth inhibition,
microorganisms 16 h
Sewage effluent 15 EC50 for BOD, 5 days,
supplemented
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Bacteria
(contd).
Photobacterium SW 3,5-DCP 2.9 EC50 for light emission Dutka & Kwan (1984)
phosphoreum after 15 min
(Microtox(R))
Pseudomonas SB, FW 3,5-DCP 3.2 EC50 for growth Dutka & Kwan (1984)
fluorescens inhibition, 18 h
Activated FB, FW 2-MCP 104.4 Beltrame et al.
sludge 3-MCP 67.5 (1984)
4-MCP 71.0
2,3-DCP 55.1
2,4-DCP 47.6
2,5-DCP 50.2
2,6-DCP 65.2 IC50 for phenol
3,4-DCP 42.7 biodegradation
3,5-DCP 58.3
2,3,4-T3CP 27.4
2,3,5-T3CP 22.3
2,3,6-T3CP 39.3
2,4,5-T3CP 23.6
2,4,6-T3CP 42.0
3,4,5-T3CP 19.6
2,3,4,5-T4CP 20.4
2,3,4,6-T4CP 40.5
2,3,5,6-T4CP 44.3
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Bacteria
(contd).
Nitrobacter SB, FW 2-MCP 50 25 and 27% inhibition of Wang & Reed (1984)
nitrite uptake
3-MCP 50 0 and 15% inhibition of
nitrite intake
4-MCP 50 0 and 5% inhibition of
nitrite intake
2,3-DCP 30 96 and 73% inhibition of
nitrite intake
2,4-DCP 30 21 and 77% inhibition of
nitrite intake
2,4,6-T3CP 10 88 and 100% inhibition of
nitrite intake
Nitrosomonas SB, FW 2-MCP 100 24% loss in ATP Parker & Pribyl
europa 2,4,6-T3CP 150 17.6% loss in ATP (1984)
Escherichia SB, FW 2-MCP 100 6.5% loss in ATP
coil 2,4,6-T3CP 150 12.9% loss in ATP
Pseudomonas SB, FW 2,3,4,5-T4CP 10 0% reduction in CFUs, Trevors et al.
fluorescens 1-h exposure (1982)
25 86.6 and 87.2% reduction
in CFUs, 1-h exposure
35 99.4 and 99.9% reduction
in CFUs, 1-h exposure
23.2 LC50 of CFUs, 1-h exposure
Table 16. (cont'd).
Test organism Test Chlorphenol Concentration Criterion Reference
conditions (mg/litre)
Protozoa
Tetrahymena 2-MCP 67.97
4-MCP 36.68 IC50 for growth Schultz &
2,4-DCP 15.00 at 60 h Riggin (1985)
2,5-DCP 12.15
2,4,6-T3CP 3.99
2,3,5,6-T4CP 1.40
Fungi
16 fungal SB 3-MCP 257.1 Average minumum Ruckdeschel &
strains, 4-MCP 184.9 concentration for Renner (1986)
(14 genera) 2,3-DCP 60.8 complete inhibition
2,4-DCP 54.1 of growth
2,5-DCP 54.8
2,6-DCP 180.8
3,4-DCP 30.1
3,6-DCP 13.2
SB 2,3,4-T3CP 11.6
2,3,5-T3CP 140.4
2,4,5-T3CP 19.2
2,4,6-T3CP 4.1
2,3,4,5-T4CP 4.6
2,3,4,6-T4CP 72.8
2,3,5,-T4CP 119.7
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fungi (contd).
Pichia SB Na-4-MCP 145 IC50 for culture growth Kwasniewska &
(fermentative Na-2,4-DCP 42.5 Kaiser (1983)
yeast) Na-2,4,5-T3CP 4.3
Rhodoturula SB Na-4-MCP 62.5 IC50 for culture growth
rubra Na-2,4-DCP 16.5
(oxidative Na-2,4,5-T3CP 2.0
yeast)
Invertebrates
Daphnia magna SB, FW 2-MCP 22 24-h LC50 LeBlanc (1980)
(water flea) 2.6 48-h LC50
4-K4CP 8.8 24-h LC50
4.1 48-h LC50
2,4-DCP > 10 24-h LC50
2.6 48-h LC50
2,4,5-T3CP 3.8 24-h LC50
2.7 48-h LC50
2,4,6-T3CP 15 24-h LC50
6.0 48-h LC50
2,3,4,6-T4CP > 1.0 24-h LC50
0.29 48-h LC50
2,3,5,6-T4CP 2.5 24-h LC50
0.57 48-h LC50
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Invertebrates
(contd.)
Daphnia magna 2-MCP 17.95 IC50 for Devillers &
(water flea) 3-MCP 15.78 immobilization Chambon (1986)
4-MCP 8.07 after 24 h
2,3-DCP 5.19
2,4-DCP 2.68
2,6-DCP 9.38
3,4-DCP 2.77
3,5-DCP 2.09
2,3,4-T3CP 2.24
2,3,5-T3CP 2.28
2,3,6-T3CP 7.38
2,4,5-T3CP 2.08
2,4,6-T3CP 5.47
3,4,5-T3CP 0.88
2,3,4,5-T4CP 1.76
2,3,5,6-T4CP 2.27
Astacus SB, FW 2,3,6-T3CP 5.4 at pH 6.5 8-day LC50 Kaila &
fluviatilis 19.0 at pH 7.5 8-day LC50 Saarikoski (1977)
Mysidiopsis SB, SW 4-MCP 29.7 at pH 6 96-h LC50 LeBlanc (1984)
bahia
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Invertebrates (contd).
Palaemonetes SS, SW 2,4-DCP 2.55(I)a; 2.16 (M)b 96-h LC50 Rao et al. (1981)
pugio 2,4,6-T3CP 3.95(I); 1.21 (M) 96-h LC50
(grass shrimp) 2,4,5-T3CP 1.12(I); 0.64 (M) 96-h LC50
2,3,4,5-T4CP 0.86(I); 0.37 (M) 96-h LC50
2,3,4,6-T4CP 3.70(I); 0.81 (M) 96- LC50
2,3,5,6-T4CP 4.10(I); 1.17 (M) 96-h LC50
Palaemonetes SS, SW 2,3,4,5-T4CP 0.30 EC50 for intermolt Rao et al. (1981)
pugio limb regeneration
(grass shrimp) 2,3,4,6-T4CP 0.78 EC50 for intermolt
limb regeneration
Fish
Pimephales SB/FB, F-W 2-MCP 11.0-13.0 96-b LC50 flowthrough Phipps et al.
promelas 6.3 192-h LC50 flowthrough (1981)
(fathead 9.7 48-h LC50 static
minnow) 2,4-DCP 8.2-8.3 96-h LC50 flowthrough
6.5 192-h LC50 flowthrough
8.4 48-h LC50 Static
2,4,6-T3CP 8.6-9.7 96-h LC50 flowthrough
5.8-6.4 192-h LC50 flowthrough
7.7 48-h LC50 static
Cyprinidon SB, SW 4-MCP 5.7 24-h LC50 Heitmuller et al.
variegatus 5.4 48-h LC50 (1981)
(sheepshead 5.4 72-h LC50
minnow) 5.4 96-h LC50
3.2 NOECc
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fish (contd).
Cyprinidon 2,4,5-T3CP 2.4 24-h LC50
variegatus 1.7 48-h LC50
(contd). 1.7 72-h LC50
1.7 96-h LC50
1.0 NOECc
2,3,5,6-T4CP 2.0 24-h LC50
2.0 48-h LC50
2.0 72-h LC50
1.9 96-h LC50
1.0 NOECc
Salmo trutta SB, FW 2,4-DCP 1.7 24-h LC50 Hattula et al.
(trout) 2,6-DCP 4.0 (1981b)
2,3,5-T3CP 0.8
2,4,5-T3CP 0.9
2,3,4,6-T4CP 1.1
Poecilia SS, FW 4-MCP 49.0 at pH 5 96-h-LC50 Saarikoski &
reticulatus 61.0 at pH 6 96-h LC50 Viluksela (1981)
(guppy) 66.0 at pH 7 96-h LC50
2,4,5-T3CP 50.0 at pH 5 96-h LC50
6.3 at pH 7 96-h LC50
15.3 at pH 8 96-h LC50
2,4,6-T3CP 3.1 at pH 5 96-h LC50
4.5 at pH 6 96-h LC50
11.6 at pH 7 96-h LC50
39.8 at pH 8 96-h LC50
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Poecilia SB, FW 2-MCP 13.5 at pH 7.8 24-h LC50 Könemann &
reticulatus 7.1 at pH 6.1 24-h IC50 Musch (1981)
(guppy) 3-MCP 7.9 at pH 7.8 24-h LC50
6.4 at pH 6.1 24-h LC50
2,4-DCP 5.9 at pH 7.8 24-h LC50
3.3 at pH 6.1 24-h LC50
3,5-DCP 4.7 at pH 7.8 24-h LC50
2.6 at pH 6.1 24-h LC50
2,3,5-T3CP 4.7 at pH 7.8 24-h LC50
0.88 at pH 6.1 24-h LC50
2,3,6-T3CP 13.3 at pH 7.8 24-h LC50
0.94 at pH 6.1 24-h LC50
3,4,5-T3CP 2.4 at pH 7.8 24-h LC50
2,3,4,5-T4CP 1.1 at pH 6.1 24-h LC50
2,3,5,6-T4CP 2.3 at pH 7.8 24-h LC50
0.44 at pH 6.1 24-h LC50
3.9 at pH 7.8 24-h LC50
0.36 at pH 6.1 24-h LC50
Carassius SB, FW 2-MCP 16 25-h LC50 Kobayashi et al.
auratus 4-MCP 9.0 (1979)
(goldfish) 2,4-DCP 7.8
2,4,5-T3CP 1.7
2,4,6-T3CP 10.0
2,3,4,6-T4CP 0.75
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fish (contd).
Lebistes S8, FW 2-MCP 13.4 24-h LC50 Benoit-Guyod et al.
reticulatus 3-MCP 27.0 (1984)
(guppy) 4-MCP 9.0
2,3-DCP 18.0
2,4-DCP 6.8
2,5-DCP 11.0
2,6-DCP 8.9
Lebistes 3,4-DCP 7.4
reticulatus 3,5-DCP 6.1
(guppy) 2,3,6-T3CP 53.0
2,4,5-T3CP 2.7
2,4,6-T3CP 2.3
2,3,4,5-T4CP 1.70
2,3,5,6-T4CP 3.60
Lepomis SB, FW 2-MCP 7.2 24-h LC50 Buccafusco
macrochirus 6.6 96-h LC50 et al. (1981)
(bluegill) 4,-MCP 4.0 24-h LC50
3.8 96-h LC50
2,4-DCP 4.7 24-h LC50
2.0 96-h LC50
2,4,5-T3CP 0.61 24-h LC50
0.45 96-h LC50
2,4,6-T3CP 0.72 24-h LC50
0.32 96-h LC50
Table 16. (cont'd).
Test organism Test Chlorophenol Concentration Criterion Reference
conditions (mg/litre)
Fish (contd).
2,3,4,6-T4CP 0.19 24-h LC50
0.14 96-h LC50
2,3,5,6-T4CP 0.40 24-h LC50
0.17 96-h LC50
Plants
Lemna minor SB, FW 4-MCP 282.8 50% chlorosis of fronds Blackman et al.
(duckweed) 2,4-DCP 58.7 (1955)
2,4,6-T3CP 5.9
2,4,5-T3CP 1.7
2,3,4,6-T4CP 0.6
a SB = Static bioassay.
SS = Semistatic bioassay.
SW = Marine.
FB = Continuous flow bioassay.
FW = Fresh water.
M = Molting.
I = Intermolt.
b NOEC = No-observed-effect concentration.
c IC50 = Concentration resulting in 50% inhibition.
The position of the chlorines on the phenol ring also influences
chlorophenol toxicity. Chlorophenols with chlorines in the 2 and 6
positions are often relatively non-toxic (Kobayashi et al., 1979;
Hattula et al., 1981b; Liu et al., 1982; Ribo & Kaiser, 1983;
Devillers & Chambon, 1986; Ruckdeschel & Renner, 1986), perhaps,
because the chlorines shield the hydroxyl group. These patterns
parallel the biodegradability of the compounds, as ortho-substituted
chlorophenols are less stable than their meta-substituted isomers
(section 4.2.1.2). However, the effects of chlorine position on
toxicity are not evident in all of the studies included in Table 16,
suggesting that the toxicity of any particular chlorophenol is highly
species-specific.
In addition, pH affects the toxicity of chlorophenols (Table 16).
At low pH, a given chlorophenol is relatively toxic, because it is
mainly in the form of molecules that can readily cross biological
membranes. As the pH is increased, chlorophenol toxicity is reduced
because the ionic form becomes abundant. Under the range of conditions
in most natural habitats, this effect becomes more important as the
number of chlorines in the chlorophenol increases, because the pKa is
related to chlorine number. Thus, monochlorophenol toxicity is
relatively unchanged by environmental pH, whereas that of
pentachlorophenol, which is present in the molecular form only under
very acid conditions, is greatly affected.
Studies on the toxicity of chlorophenols for terrestrial organisms
in the environment are much more limited. Blackman et al. (1955)
determined that the EC50s of several chlorophenols for the inhibition
of radial growth of the mould Trichoderma viride were as follows:
4-MCP, 47.6 mg/litre agar; 2,4-DCP, 8.6 mg/litre; 2,4,6-T3CP,
5.7 mg/litre; and 2,3,4,6-T4CP, 0.8 mg/litre. A similar pattern of
increasing toxicity with increasing chlorination of chlorophenols was
also observed by Sund & Nomura (1963) in their investigation of the
inhibition of seed germination by a number of chlorophenols. In
addition, they noted that chlorination at the 3 or 5 position enhanced
chlorophenol toxicity for germinating seeds. In a survey on the
contact toxicity of chemicals for the earthworm Eisenia foetida
(Roberts & Dorough, 1984), 2,4-DCP and 2,4,5-T3CP were classified as
extremely toxic, on the basis that their 48-h LC50 values fell within
the range of 1-10 µg/cm2 of filter paper.
7.1.2 Long-term toxicity
There are very few studies on the long-term effects of
chlorophenols on environmental organisms. Holcombe et al. (1982)
exposed the embryo, larval, and early juvenile stages of fathead
minnows to a range of sublethal concentrations of 2,4-DCP and other
phenolic compounds, in 32-day flow-through tests using Lake Superior
water. Survival of larvae and juvenile minnows was significantly
reduced after exposure for 28 days to 2,4-DCP at 460 µg/litre. The
growth of larval and juvenile stages was reduced by 1240 µg
2,4-DCP/litre. Hatching success was unaffected by the maximum
concentration used (1240 µg/litre).
Survival, reproduction, and growth were all reduced in Daphnia
magna exposed to 2,4-DCP concentrations of 1.48 mg/litre in
long-term (21-day) static renewal tests (Gersich & Milazzo, 1988).
In a study more relevant to field conditions, Virtanen & Hattula
(1982) used a flow-through aquarium microcosm with levels of
2,4,6-T3CP of 0.5 µg/litre, in order to track its incorporation into
sediment, algae, invertebrates, and fish. Male and female Poecilia
reticulatus fish were included in the microcosm, and aspects of
their reproduction and histopathology were monitored. Over a 10-month
period following their 56-day exposure in the aquarium and subsequent
transfer to uncontaminated water, only 90 offspring were born to
exposed fish and 22 of these died. Control fish produced 180
offspring, only 8 of which died. In addition, several offspring from
exposed parents had abnormally curved spines. Thus, under the test
conditions, 2,4,6-T3CP appeared to be very fetotoxic, and perhaps
teratogenic. No histological changes were noted in the livers or
kidneys of P. reticulatus as a result of these exposures.
7.1.3 Organoleptic effects
Exposure to low levels of chlorophenols can also impair the
flavour of fish (see section 7.2.4). According to Boetius (1954), as
little as 0.1 Ul 2-MCP/litre (v/v) tainted the flesh of eels and
oysters after exposure for 11 and 4 days, respectively. Shumway &
Palerisky (1973) estimated the threshold concentrations of several
chlorophenols for the impairment of the flavour of rainbow trout to
be: 2-MCP, 60 µg/litre; 3-MCP, 25 µg/litre; 4-MCP, 45 µg/litre;
2,3-DCP, 84 µg/litre; 2,4-DCP, 1 µg/litre; 2,5-DCP, 23 µg/litre;
2,6-DCP, 35 µg/litre; and 2,4,6-T3CP, 52 µg/litre.
7.2 Toxicity Studies under Natural Environmental Conditions
7.2.1 Bacteria
During the course of studies in Dutch coastal waters, Kuiper &
Hantsveit (1984) examined the effects of the addition of 4-MCP and
2,4-DCP on plankton communities enclosed in 1500-litre plastic bags.
In the first of 3 studies, total bacterial densities (by direct count)
were prevented from increasing by 0.1 and 1 mg 2,4-DCP/litre but, in a
second study, 2,4-DCP at 1 mg/litre did not have any effect on total
bacterial densities, and, in the final study, 1 mg 2,4-DCP/litre was
necessary to inhibit bacterial population growth. No effects of 4-MCP
were detected, even at 1 mg/litre. As treatments were not replicated
and bacterial densities were quite variable, it is not clear whether
the effects observed were in fact responses to chlorophenol exposure.
In contrast, when 5 mg 2,4,6-T3CP/litre was added to enclosures
in a West German pond, the numbers of aerobic heterotrophic bacteria
(by plate count) in the water increased more than 10 times compared
with the controls, within 4 days (Schauerte et al, 1982). This
response coincided with the disappearance of Daphnia from the
CP-treated tubes, suggesting that the increase was the result of a
release from grazing by the Daphnia.
There has been some concern that chlorophenols in industrial
wastes may impair the efficiency of secondary waste treatment through
their toxic effects on bacteria. Using a bench-scale activated sludge
plant, Broecker & Zahn (1977) determined that the degradation of waste
water declined after exposure to 25 µg 3,5-DCP/litre. Similarly, in a
laboratory scale model of a trickling filter (El-Gohary & Nasr, 1984),
exposure of acclimated microbes to 50 mg 2,4-DCP/litre reduced
Biological Oxygen Demand (BOD), and Chemical Oxygen Demand (COD).
However, chlorophenols in industrial wastes are unlikely to pose a
serious hazard for organisms important for secondary treatment. Levels
of chlorophenol entering treatment facilities are far below those used
in the studies just described (Folke, 1984), and recovery from shock
loadings of chlorophenols is rapid (El-Gohary & Nasr, 1984).
7.2.2 Phytoplankton
Kuiper & Hantsveit (1984) monitored the response of enclosed
marine plankton to chlorophenol additions, and determined that
exposure to chlorophenols affected algal biomass, composition, and
activity. In the first of 3 studies, 1 mg 4-MCP or 2,4-DCP/litre
prevented the increase in algal biomass (as chlorophyll) that occurred
in control enclosures. Large flagellates made up a greater proportion
of the algal community in 1 mg/litre-treated enclosures compared with
controls, perhaps because grazing was reduced (section 7.3). Primary
productivity generally parallelled the dynamics of algal biomass, as
it was reduced by exposure to 1 mg 4-MCP/litre; however, the addition
of 1 mg 2,4-DCP/litre did not affect photosynthetic radiolabelled
dissolved inorganic carbon (DIC) uptake. Results were generally
similar during the 2 subsequent manipulations, though the magnitude
and timing of the effects varied.
In their studies on ponds, Schauerte et al. (1982) observed major
shifts in the species composition of the phytoplankton following the
addition of 5 mg 2,4,6-T3CP/litre. The large population of the
blue-green alga Chroococcus limneticus was sharply reduced, and the
diatom Nitzschia acicularis was eliminated, while the flagellated
algae Euglena and Trachelomonas appeared in large numbers
following exposure to 2,4,6-T3CP. The dynamics of other
phytoplankton, which were less abundant, were not discussed.
Chorophenols were among the toxicants used by Erickson & Hawkins
(1980), who measured the response of estuarine phytoplankton
communities to 15 compounds produced during the chlorination of sea
water. Natural phytoplankton assemblages, pumped from the estuary to
flow-through aquaria in the laboratory, were insensitive to
chlorophenol concentrations of 0.5-2 mg/litre. Photosynthetic
radiolabelled DIC uptake was not depressed by exposure to 2 mg
2,4,6-T3CP or 4-MCP/litre.
7.2.3 Zooplankton
Marine zooplankton were strongly affected by chlorophenol
additions during field studies in 1500-litre plastic enclosures
(Kuiper & Hantsveit, 1984). While the zooplankton communities in the
control enclosures and those treated with 0.1 mg 4-MCP/litre or 0.1 mg
2,4-DCP/litre displayed similar dynamics, total biomass and production
in enclosures treated with 1 mg 4-MCP/litre and 1 mg 2,4-DCP/litre
were reduced relative to controls throughout the first three-quarters
of the study. All life-history stages of several copepod species were
similarly affected. Results in subsequent studies were generally
similar, though the magnitude of the impact varied.
More severe effects for Daphnia exposed to 5 mg
2,4,6-T3CP/litre were reported by Schauerte et al. (1982) during
studies on a pond. From initial levels of more than 20 individuals per
100 ml before the toxicant was added, Daphnia was eliminated from
T3CP-treated enclosures in 3 days. The abundance of Daphnia in
control enclosures was high and stable during the 24 days of the
study.
7.2.4 Fish
There are no controlled field studies on the effects of
chlorophenols on fish, but fish kills have occurred as a result of
chlorophenol spills. Mackenzie et al. (1975) compiled information on
such incidents in British Columbia salmon waters during 1960-73. In
one instance, an over- flow from a lumber-treatment tank released both
T4CP and PCP into the Mamquam Channel in 1973, killing an estimated
500 adult and juvenile coho salmon.
Chlorophenols may also impair the flavour of fish, even when
present in the minute quantities detected in moderately-contaminated
natural waters (section 7.1.3). Chatterjee (1974) reported that kraft
and groundwood pulp discharges into Lakes Superior and Huron, which
included phenolic compounds, were apparently responsible for the
tainting of flesh from fish captured nearby.
7.2.5 Effects on physical and chemical variables
The only instance in which chlorophenols affected physical or
chemical factors in the environment apparently involved a secondary
effect. In studies in which 5 mg 2,4,6-T3CP/litre was added to
enclosures in a pond, oxygen levels declined from initial levels of
3-4 mg/litre to less than 1 mg/litre, within 6 days of treatment, as
the balance between heterotrophic and autotrophic metabolism shifted
(Schauerte et al., 1982). Apart from this secondary effect, physical
and chemical variables appear insensitive to CP additions. Schauerte
et al. (1982) did not find any significant differences in temperature,
pH, hardness, sulfide, carbonate, or chloride levels between control
and 2,4,6-T3CP-treated enclosures. Similarly, levels of phosphate,
ammonia, nitrate, nitrite, silicate, and pH were unaffected by
additions of as much as 1 mg 4-MCP or 2,4-DCP/litre (Kuiper &
Hantsveit, 1984).
7.3 Treatment Levels
Unfortunately, the effects of chlorophenols at the low (mg/litre)
levels that characterize the aquatic environment at large (section
5.1.2) were not examined in most of these studies. As a result, they
shed little light on the possible hazards presented by the widespread
low-level contamination observed in most environments. The microcosm
study by Virtanen & Hattula (1982) was an exception in this regard.
However, the mg/litre concentrations used by most research workers are
relevant to major accidental spills of chlorophenols in the
environment.
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO SYSTEMS
8.1 Acute Studies
In general, the toxicity of chlorophenols increases with an
increase in the chlorination of the phenol molecule. A convulsant
effect is associated with the less-chlorinated phenols and oxidative
phosphorylation uncoupling is more prominent with the highly
substituted compounds (Ahlborg & Thunberg, 1980; Jones, 1981; Exon,
1984).
Farquharson et al. (1958) studied the effects of a series of
chlorophenols on male albino rats (Table 17). Symptoms associated with
the lethal intraperitoneal injection of the monochlorophenols, 2,6-DCP
and 2,4,6-T3CP, as well as phenol, included an initial increase in
physical activity (rapid running, nose rubbing) followed by tremors,
convulsions, and loss of righting reflex. With 2,3,6-T3CP, rats
suffered convulsions only when handled, and otherwise lay prostrate
with hypotonia. Hypotonia, starting at the hind limbs, was observed
within 2-3 min of injection with 2,4-DCP, 2,3,6-, 3,4,5- and
2,4,5-T3CP, T4CP, and PCP. Body temperature was slightly reduced by
phenol, MCPs and DCPs, while T3CP caused a slight elevation and T4CP
and PCP a marked rise in temperature (4-4.5°C). Respiratory rate
increased initially then declined as coma developed, especially with
T4CP and PCP. The extremities were cyanosed and asphyxial spasms
occurred about 30 seconds before death. With T4CP and PCP,
respiration stopped usually one-half to 2 min before cessation of the
heart, whereas with the other chlorophenols, respiration ceased
concomitantly with the heart or just before.
Animal studies indicate that most mono-, di-, and tri-
chlorophenols are moderately toxic when administered orally, with
LD50 values ranging between 230 and 4000 mg/kg body weight
(Table 18). In general, the less-chlorinated phenols have an acute
oral toxicity very close to that of phenol. T4CP is considerably more
acutely toxic, with LD50 values of between 100 and 400 mg/kg body
weight (Ahlborg & Thunberg, 1980; Hattula et al., 1981a). Thus, the
data indicate that the general order of decreasing acute toxicity is:
T4CP, MCP, DCP, T3CP.
The subcutaneous and intraperitoneal routes of exposure have also
been investigated (Table 18). As with oral administration,
subcutaneous injections revealed a general order of decreasing acute
toxicity of: T4CP, MCP, DCP, T3CP.
Table 17. Effect of lethal chlorophenol doses given intraperitoneally to ratsa
Compound Convulsant Hypotonia Max. Respiration Rigor mortis
onset (min) change onset (min)
activity in temp.
(°C)
Ether 50
Monochlorophenols
2(o) + -b -2.0 -d > 5.2 < 50
3(m) + -b -2.5 -d > 5.2 < 50
4(p) + -b -2.5 -d > 5.2 < 50
Dichlorophenols
2,4- (occasional 2-3c -0.5 -d > 5.2 < 50
twitches)
2,6- + -b -0.7 -d > 5.2 < 50
Trichlorophenols
3,4,5- - 2-3 + 0.5 -d < 5
2,4,5- - 2-3 + 0.5 -d < 5
2,4,6- + + 0.5 -d < 5
2,3,6- (sometimes 2-3 + 0.5 < 5
convulsions
when
handled)
Table 17. (cont'd).
Compound Convulsant Hypotonia Max. Respiration Rigor mortis
onset (min) change onset (min)
activity in temp.
(°C)
Tetrachlorophenol
2,3,4,6- - 2-3 + 4.0 -e < 6
Pentachlorophenol
2,3,4,5,6- - 2-3 + 4.5 -e < 5
a From Farquharson et al. (1958).
b Apparent after convulsions have lessened.
c Muscle twitches evoked by auditory and mechanical stimuli.
d Initial increase, then decrease as coma developed; asphyxial spasms 30
seconds before death; ceases just before or simultaneously with cardiac arrest.
e Ceases 1/2 to 2 min before stopping of heart.
Table 18. Acute toxicity (LD50s) of phenols and chlorophenols for rats and micea
RAT MOUSE
Oral Subcutaneous Dermal IP Oral Subcutaneous IP
Phenol 530-650b 669 250 300 344 360 -
Monochlorophenol
-2(o) 670 950 - 230 347, 345c - -
-3(m) 570 1390 - 355 521, 530c - -
-4(p) 261 1030 - 281 1373, 1422c - -
Dichlorophenol
-2,3 - - - - 2585, 2376c - -
-2,4 580, 4000d 1730 - 430 1276, 1352c 1600h - -
-2,5 - - - - 1600, 946c - -
-2,6 2940 1730 - 390 2198, 2120c - -
-3,4 - - - - 1685, 2046c - -
-3,5 - - - - 2643, 2389c - -
Trichlorophenol
-2,3,6 - - - 308 - - -
-2,4,5 820e 2260 - 355 - - -
-2,4,6 820e - - 276 - - -
-3,4,5 - - - 372 - - -
Table 18 (contd).
RAT MOUSE
Oral Subcutaneous Dermal IP Oral Subcutaneous IP
Tetrachlorophenol
-2,3,4,5 - - > 2000h - 400g - 97g
-2,3,4,6 140, 360f 210g - 130 131g 120i 82g
-2,3,5,6 > 2000h 109g 48g
-Commercial 485-565h - - -
mix
Tetrachlorophenate-Na
-2,3,4,5 - - > 2000h - - - -
-2,3,5,6 - - 294-469h - - - -
a Principle database NIOSH (1983). LD50 values given as mg/kg body weight.
b Babich & Davis (1981).
c Borzelleca et al. (1985a). Data for males presented first, then females.
d Kobayashi et al. (1972).
e More recent values in US EPA (1979) are 4 times higher (2460-2960 mg/kg).
f Hattula et al. (1981a).
g Ahlborg & Larsson (1978).
h Shen et al. (1983) commercial mixture was primarily 2,3,4,6-T3CP.
i Kozak et al. (1979).
However, in ip injection studies, the toxicities of the mono-,
di-, and trichlorophenols were comparable while T4CP was 2-3 times
more toxic.
The only study available on the acute dermal toxicity of the less
chlorinated phenols was carried out by Shen et al. (1983). In this
investigation, Sprague-Dawley rats of both sexes were given 2,3,4,5-,
2,3,5,6-T4CP, their sodium salts, or a commercial T4CP preparation
containing over 90% of 2,3,4,6-T4CP and 5-10% of PCP. A very low
dermal toxicity was reported for 2,3,4,5-T4CP, its phenate salt, and
2,3,5,6-T4CP, while the sodium salt of 2,3,5,6-T4CP had the highest
toxicity of all, followed by the commercial T4CP (Table 18). The
relatively high toxicity of the commercial T4CP could result from its
content of PCP. The rapid death of the rats (usually 6 h) argues
against any role being played by the microcontaminants, because the
PCDDs and PCDFs are usually associated with delayed acute toxicity.
The effects on animals of these less-chlorinated phenols
administered via inhalation have not been investigated. However, it
can be inferred from the one report on the inhalation of sodium-PCP
(Hoben et al., 1976b) that the toxicity via inhalation would be
greater than that associated with the oral, intraperitoneal,
subcutaneous, or dermal routes.
In their study on the toxicity of 3 different T4CP isomers in
mice, Ahlborg & Larsson (1978) noted that the LD50 values for all 3
isomers were lower with ip injection than with oral (gavage)
administration. This effect of the mode of administration on toxicity
is apparent in the data on the other chlorophenols (Table 18) and may
be related to differences in the rates of absorption, metabolism, and
excretion of these compounds. The low toxicity values obtained with
subcutaneous injections may be due to a low rate of absorption. The
vehicle used for administration can also have a significant influence
on absorption.
8.2 Skin and Eye Irritation; Sensitization
Very little information is available on skin and eye irritation or
sensitization in experimental animals exposed to chlorophenols. Only
very slight irritation was noted in rabbits following the application
of dry 2,4,5-T3CP to their skin (McCollister et al., 1961). The
authors suggested that mild erythema might be caused by high
concentrations of the material in solution. In a study on the dermal
toxicity of the T4CP isomers, Shen et al. (1983) found that
dermatosis occurred in rats painted with 2,3,4,5-T4CP or its phenate
salt but not with the other 2 isomers or their salts.
8.3 Short-term Exposure
All the available information on short-term exposure has been
obtained by means of oral studies, except for one report concerning
dermal application. Information is lacking on the effects of
inhalation and other routes of exposure in experimental animals.
Exon & Koller (1985) investigated the possible immunological
effects of 3 chlorophenols on rats exposed both prenatally and
post-natally. Weanling female rats (3 weeks of age) were given 2-MCP
(98% pure) (0, 5, 50, or 500 mg/litre), 2,4-DCP (99% pure), or
2,4,6-T3CP (98% pure); (both at 0, 3, 30, or 300 mg/litre) in the
drinking-water, for 90 days from weaning through breeding and
pregnancy. A randomly chosen group of offspring was given the same
dose regime as the dams for an additional 12-15 weeks after weaning.
Both groups were observed for another 10 weeks. 2-MCP did not have any
adverse effects on humoral immunity, cell-mediated immunity, or
macrophage function in the exposed progeny at any of the exposure
levels. Exposure of progeny to the highest concentration of 2,4-DCP
significantly enhanced (P less than 0.05) humoral immune
responsiveness and decreased cell-mediated immunity in rats with both
prenatal and postnatal treatments, but not in rats exposed only
in utero. 2,4,6-T3CP did not have any effects on the immune
responses tested. Spleen and liver weights were increased in the
progeny receiving water containing 2,4-DCP or 2,4,6-T3CP at
300 mg/litre, but not 2-MCP. The authors suggested that this increase
in organ weight was due to hyperplasia, as no histological anomalies
were observed.
Exon & Koller (1985) also monitored haematological parameters and
organ weights in exposed rat progeny. Combined pre- and postnatal
(24 months) exposure to 500 mg 2-MCP/litre or 300 mg 2,4-DCP/litre
significantly elevated red blood cell counts, haemoglobin
concentrations, and packed cell volumes of offspring. Each of the 3
compounds was also fetotoxic (section 8.5).
In studies by Kobayashi et al. (1972), 2,4-DCP in the diet at the
maximum dose of 230 mg/kg body weight per day, over a 6-month period,
caused swelling of the hepatocytes in male mice but did not
substantially affect liver, kidney, spleen, or adrenal histology.
Furthermore, no significant exposure-related changes were observed in
organ weight, body weight, food consumption, serum concentrations of
liver enzymes, or numbers of red and white blood cells.
In a 90-day study, Borzelleca et al. (1985b) exposed mice of both
sexes to 2,4-DCP in their drinking-water at mean daily doses of 40,
114, or 383 mg/kg body weight for males, and 50,143, or 491 mg/kg body
weight for females. These dosages were calculated from daily water
consumption data and the concentrations added to the drinking-water.
At the end of the study period, there were no significant
treatment-related differences in organ weights, electrolyte levels,
haematological factors, or the activities of hepatic mixed-function
oxidases (MFO) or serum enzymes.
In short-term studies that were preliminary to a carcinogenicity
bioassay (section 8.6), rats and mice of both sexes were fed diets
containing 2500-40 000 mg 2,4-DCP/kg for 13 weeks (NTP, 1988). All
animals survived to the end of the study, except the mice receiving
the highest dose, all of which died. Male mice and rats of both sexes
receiving the 20 000 mg/kg diet showed reduced final mean body
weights. Dose-related effects were apparent in the form of bone-marrow
atrophy in rats and liver damage (necrosis, multi-nucleated
hepatocytes) in mice.
A study on the short-term exposure of rats and rabbits to
2,4,5-T3CP was carried out by McCollister et al. (1961). Rabbits,
given 20 oral doses of 500 mg/kg body weight over a 28-day period,
showed only "very slight kidney and liver changes". In rats receiving
18 doses of 1000 mg/kg body weight over a 24-day period, there was a
slight increase in kidney weight, while growth, mortality,
haematological parameters, and the histology of the lung, heart,
liver, kidney, spleen, adrenal, pancreas, and testis were unaffected.
In the same report, male and female rats given a diet containing
0, 0.1, 0.3, or 1.0 g 2,4,5-T3CP/kg for 98 days did not exhibit
behavioural changes, increased mortality, changes in food consumption,
growth, or histology (McCollister et al., 1961). At the 10 g/kg level,
an increase in the frequency of day-time urination was noted in both
males and females, as well as significant growth retardation in
females. Kidney and liver degeneration, which was judged reversible,
was also found at this exposure level.
During the course of a study on reproduction (section 8.5.1),
Blackburn et al. (1986) dosed rats for 5 days per week (males, 11
weeks; females, 2 weeks and through gestation) by gavage with 0, 100,
500 or 1000 mg 2,4,6-T3CP/kg body weight in corn oil. A number of
rats from the 1000 mg/kg group died as a result of treatment (males, 8
out of 25; females, 3 out of 40). Males and females in this group
exhibited significant but transitory weight loss compared with
controls. Male kidney, liver, lung, adrenal, spleen, heart, testis,
prostate, seminal vesicle, and epididymis weights were unaffected by
all levels of 2,4,6-T3CP exposure. Females dosed at 1000 mg/kg body
weight lost hair, were lethargic, and breathed irregularly.
In a range-finding study, rats and mice of both sexes were exposed
to 2,4,6-T3CP for 7 weeks to determine the maximum tolerated dose
(NCI, 1979). The compound was given in the feed up to a maximum level
of 46 000 mg/kg for rats and 31 500 mg/kg for mice. Weight gain was
reduced in both male and female rats at all exposure levels, but only
at the 2 highest dose levels (21 500 and 31 500 mg/kg) in mice. At the
highest dose, a moderate to marked increase in splenic haematopoeisis
was found in the rats and midzonal vacuolation of hepatocytes was seen
in 2 males. All tissues in the mice were normal at the end of 7 weeks.
Hattula et al. (1981a) dosed rats by garage daily with 0, 10, 50,
or 100 mg 2,3,4,6-T4CP/kg body weight in olive oil for 55 days.
Histological changes were found only in the liver, even though the
kidneys and spleen contained higher concentrations of 2,3,4,6-T4CP.
This finding suggests that 2,3,4,6-T4CP has a specific effect on the
liver of rats.
8.4 Long-Term Exposure
Investigations of the effects of long-term exposure to
chlorophenols have been designed primarily to test their carcinogenic
properties and are described in section 8.7.
8.5 Reproduction, Embryotoxicity, and Teratogenicity
Exon & Koller (1981, 1983) and Exon et al. (1984) examined the
effects of 2-MCP (98% pure; 0, 5, 50, or 500 mg/litre), 2,4-DCP (99%
pure; 0, 3, 30, or 300 mg/litre), and 2,4,6- T3CP (98% pure; 0, 3,
30, or 300 mg/litre) on reproductive parameters in mice exposed via
drinking-water (section 8.3). Females were exposed from 3 weeks of age
until breeding at 90 days, and through-out gestation to parturition.
Reanalysis of their original data (Exon & Koller, 1985) led the
authors to conclude that there was a weakly significant (P less than
0.1) effect of 2-MCP at 500 mg/litre and 2,4-DCP and 2,4,6-T3CP at
300 mg/litre, expressed as reduced litter size. The percentage of
stillborn offspring compared with controls tended to increase in all
exposed groups. However, the differences were not significant at P
less than 0.05.
In mice, sperm motility and penetration of ova were not affected
by acute (0.1 to 1 mmol/litre) and long-term exposure (90 days at
50-500 mg/kg body weight per day) to 2,4-DCP (Seyler et al., 1984).
In vitro penetration was depressed by 2,5-, 3,4-, or 3,5-DCP at
1 mmol/litre, and it appeared that this concentration of 3,4- or
3,5-DCP also disrupted the sperm acrosome.
The effects of oral dosing with 2,4,6-T3CP on rat reproduction
were investigated by Blackburn et al. (1986). Male rats were given 0,
100, 500, or 1000 mg 2,4,6-T3CP/kg body weight in corn oil, by
gavage, 5 days per week, for 11 weeks.
These exposures to T3CP did not significantly affect male sexual
behaviour (mount and ejaculation latencies, number of mounts and
intromissions), plasma-testosterone levels, or sperm counts, motility,
or morphology. Males from the control and 1000 mg/kg groups were then
mated with unexposed females, which were sacrificed on day 18 of
gestation. The exposure regime of the males did not significantly
affect litter size, sex ratio, mean pup weight by sex, number of dead
fetuses, or the numbers of resorptions or implantation sites. As part
of the same study, female rats were dosed in the same manner as the
males for 5 days per week over 2 weeks and then mated, after which
dosing continued until day 21 of gestation. No treatment-related
effects were evident in the breeding success, mean litter size, or
offspring survival of these females. Litter weights were initially
significantly reduced in the 500 and 1000 mg/kg body weight groups,
but this difference had disappeared at 4 days postpartum. This effect
was suggested to have been a secondary manifestation of female
toxicity (section 8.2), or of initial differences in litter size
between treatments.
In a teratogenicity study of 2,4,5-trichlorophenoxyacetic acid
(Neubert & Dillman, 1972), 2,4,5-T3CP was also tested, but only at
doses of 0.9 or 9 mg/kg body weight per day. A slight increase in
embryo mortality was observed at the higher dose, but its significance
is unclear. No teratogenic effects were observed.
Schwetz et al. (1974) studied the embryotoxic and fetotoxic
effects on rats of purified (99.6%) 2,3,4,6-T4CP and commercial grade
2,3,4,6-T4CP (73% T4CP + 27% PCP plus 100 ppm total of each of
dioxins and dibenzofurans). Doses of 10 and 30 mg/kg were given by
gavage to pregnant rats from day 6 to day 15 of gestation, after which
they were sacrificed on day 21. A delay in ossification of the skull
bones was found. Neither of the compounds was embryolethal (evidenced
by no increase in resorptions or abortions) even at the high dose of
30 mg/kg per day. No differences were observed in the toxicity of the
2 grades of T4CP.
Chlorinated phenols do not appear to be teratogenic for
experimental animals. Of the compounds tested, levels as high as
500 mg 2-MCP/litre in drinking-water (Exon & Koller, 1981), 300 mg
2,4-DCP/litre in drinking-water (Exon et al., 1984), 1000 mg
2,4,6-T3CP/kg body weight (Blackburn et al., 1986), and 30 mg
2,3,4,6-T4CP/kg body weight (Schwetz et al. 1974) did not produce any
teratogenic effects in rats.
8.6 Mutagenicity and Related End-Points
Rasanen et al. (1977) found that all dichlorophenol isomers, 4 out
of 6 trichlorophenols, and 2,3,4,6-T4CP were negative in Salmonella
typhimurium mutagenicity bioassays. Similarly, Haworth et al. (1983)
reported that 2,3-, 2,4-, 2,5-, 2,6-, 3,4-, 3,5-DCPs and 2,4,5- and
2,4,6-T3CP were negative in S. typhimurium TA 98, 100, 1535 and
1537 strains. Nestmann et al. (1980) reported similar findings for
2,6-DCP and 2,4,5-T3CP. It was reported by Kinae et al. (1981) that
2,4,6-T3CP was negative in S. typhimurium TA 98, 100, and 1537
strains, with and without exogenous metabolic activation, but positive
in a B. subtilis recombination assay. At a concentration of
400 mg/litre, purified 2,4,6-T3CP caused a weak but significant
increase in the frequency of forward mutations, but did not affect
intergenic or intragenic re-combinations in the yeast Saccharomyces
cervisiae MP-1 strain (Fahrig et al. 1978).
In mutagenicity studies conducted to supplement a carcinogenicity
bioassay (NTP, 1988), 2,4-DCP did not produce any revertant colonies
in S. typhimurium strains TA 98, 100, or 1537 and yielded equivocal
results with TA 1535 only in the presence of hamster S9 activation. In
the mouse L5178Y assay (without metabolic activation),
trifluorothymidine resistance was increased by 2,4-DCP exposure. In
the same study, in vitro exposure of Chinese hamster ovary cells to
2,4-DCP increased the frequency of sister chromatid exchanges, but did
not cause chromosomal aberrations.
Hattula & Knuutinen (1985) showed that purified 2,4,6- T3CP and
2,3,4,6-T4CP were weakly mutagenic in V-79 Chinese hamster cells
in vitro, in the absence of metabolic activation by hepatocyte
co-cultures. They were both non-mutagenic in the presence of metabolic
activation by hepatocyte co-cultures. 2,6-DCP and PCP were negative
regardless of the presence of metabolic activators, and 2,4,6-T3CP
did not induce chromosomal aberrations or sister chromatid exchanges
in cultures of Chinese hamster ovary cells (Galloway et al., 1987).
Only very few data are available from in vivo studies, and these
are limited to the mouse spot test (Fahrig et al., 1978). Pregnant
mice were injected intraperitoneally with 50 or 100 mg purified
2,4,6-T3CP/kg body weight on day 10 of gestation. Examination of
offspring revealed an increased frequency of coat spots (0.6% in the
50 mg/kg group versus 0.1% in controls), indicative of a weak
mutagenic response.
8.7 Carcinogenicity
Both 2,4,6-T3CP and 2,4-DCP have been tested for carcinogenicity
in a 2-year bioassay. The other lower chlorophenols have not been
adequately tested for their carcinogenic properties.
A 2-year carcinogenicity study on 2,4-DCP was recently completed
under the US National Toxicology Program (NTP, 1988). Test animals fed
diets containing 2,4-DCP (more than 99% pure) received the following
average calculated doses (mg/kg body weight):
F344/N rats B6C3F1 mice
Male Female Male Female
Low dose 210 120 800 430
High dose 440 250 1300 820
Mean body weights were reduced, usually by several percent, in all
of the high-dose groups, as well as the low-dose groups of female
mice. Mean food consumption was reduced in all treated groups, by
several percent in rats, and in a dose-related manner in mice (up to
22%). No significant differences in the survival of any treated group
occurred. There were dose-related increases in the incidence of
multinucleated hepatocytes in male mice. No compound-related increases
in the incidence of neoplastic lesions were observed; in fact, these
were reduced for mononuclear cell leukaemia in male rats (both doses)
and malignant lymphomas in female mice (high dose only).
Innes et al. (1969) tested 120 pesticides and industrial chemicals
for tumorigenicity in male and female mice. The individual compounds
were administered at a maximum tolerated dose, by stomach tube, from 7
days of age to 4 weeks old, and then in the diet at approximately the
same dosage. The 100 mg/kg daily dose of Omal or Dowicide 2S
(2,4,6-T3CP) increased tumour incidence in the treated animals at the
end of 72 weeks. The authors recommended additional statistical
evaluation and/or studies before a meaningful interpretation could be
made.
A long-term oral exposure study was carried out on rats and mice
to test the carcinogenicity of 2,4,6-T3CP (NCI, 1979). Male and
female F344 rats were given dose levels of 2,4,6-T3CP of 5000 and
10 000 mg/kg in the feed for 106 weeks. Male B6C3F1 mice were dosed at
the same levels as the rats for 105 weeks. Female mice were initially
given dietary levels of 10 000 and 20 000 mg/kg for 38 weeks, which
were then reduced to 2500 and 5000 mg/kg, respectively, for the
remaining 67 weeks of the study, because they showed a marked
reduction in weight gain. At the end of the study, the treated male
rats showed a significantly higher incidence of malignant lymphomas
and leukaemias. Leukocytosis and monocytosis of the peripheral blood
and hyperplasia of the bone marrow were found in those that did not
show lymphomas and leukaemias. No lymphomas and/or leukaemias were
detected in female rats, but leukocytosis and monocytosis of
peripheral blood and bone-marrow hyperplasia were evident. Both male
and female mice displayed a dose-related statistically significant
incidence of both hepatocellular carcinomas and adenomas. It was
concluded that 2,4,6-T3CP was carcinogenic for male Fisher rats and
both sexes of B6C3F1 mice under the assay conditions used.
Boutwell & Bosch (1959) studied the tumour-promoting action of 2-
and 3-MCP, 2,4-DCP, 2,4,5-T3CP, and 2,4,6- T3CP in mice following
the dermal application of 9,10- dimethyl-l,2-benzanthracene (DMBA)
(25 µl of 0.3% DMBA in benzene) as an initiator. One week after a
single exposure to DMBA, twice weekly applications of 25 µl of a 20%
solution of the test compound in benzene were made for 12-24 weeks.
The control group received the pre-treatment dose of DMBA only. The
authors reported that the monochlorophenols, 2,4- DCP, and 2,4,5-T3CP
all had a tumour-promoting action similar to that of phenol, while
2,4,6-T3CP did not have any effect on tumour promotion.
Exon & Koller (1981,1985) carried out 15 to 24-month studies on
the effects of pre- and postnatal exposure of rats to 2-MCP and
2,4-DCP. No evidence of tumour initiation was revealed with exposure
to 2-MCP (98% pure) at 500 mg/litre drinking-water, or to 2,4-DCP
(99% pure) at 300 mg/litre drinking-water (Exon & Koller, 1985).
However, 2-MCP acted as a promoter of the carcinogenic activity of
ethylnitrosoures (ENU), reducing tumour latency and increasing tumour
incidence in male rats exposed both pre- and postnatally, compared
with controls receiving only ENU.
There is sufficient evidence of the carcinogenicity for animals of
2,3,7,8-TCDD (IARC, 1987) and a mixture of two H6CDD isomers (NCI,
1979), which may be present as microimpurities in some technical
chlorophenols.
8.8 Factors Modifying Toxicity Metabolism
Any factor that can interfere with sulfate and/or glucuronide
conjugation would modify the toxicity of a chlorophenol by inhibiting
chlorophenol detoxification. Somani et al. (1984) demonstrated with an
isolated liver-perfusion system that the glucuronide conjugation of
14C-2,4-DCP can be blocked by galactosamine in the rat. In high
enough concentrations, galactosamine or similar compounds would
prolong the residence time in the body and thus the toxicity of the
chlorophenol molecule.
In some instances, metabolism of chlorophenol yields molecules
that are more, rather than less, toxic than the parent molecule. For
example, when injected into rats intraperitoneally, tetrachloro-
p-hydroquinone, a metabolite of 2,3,5,6-T4CP, is much more toxic
than the parent compound (Ahlborg & Larsson, 1978).
The toxicology of chlorophenols is further complicated by
micro-contaminants in technical grade products, particularly in the
higher chlorinated compounds. Some of these impurities are themselves
extremely toxic. Conversely, microcontaminants are known to induce
enzymes that can affect the rate of chlorophenol metabolism and
excretion (Ahlborg & Thunberg, 1978). For these reasons, assessment of
toxicity studies with chlorophenols requires a knowledge of the types,
levels, and toxicology of the contaminants present. This is
particularly true when extrapolating animal studies (which often
involve the use of purified compounds) to human beings, who are
generally exposed to technical formulations. The residue
concentrations and toxicology of the dibenzodioxin and dibenzofuran
microcontaminants have been reviewed by WHO (in press).
8.9 Mechanisms of Toxicity, Mode of Action
The major mode of action of chlorophenols appears to be the
uncoupling of oxidative phosphorylation. The strength of the
uncoupling effect is related to the degree of chlorination: PCP is the
strongest inhibitor of oxidative phosphorylation, MCP the weakest
(Farquharson et al., 1958; Mitsuda et al., 1963; Weinbach & Garbus,
1965; Carlson, 1978). To a lesser extent, inhibition of oxidative
phosphorylation is affected by the positions of the chlorine atoms on
the molecule. There appears to be a relationship between chlorination
and the toxicity of PCP and T4CP (Table 18), although there is no
clear-cut relationship between the degree of chlorination and
toxicityin MCP, DCP, and the T3CP series. PCP and to a lesser extent
other chlorophenols, depending on their degree of chlorination, have
been shown to bind strongly to mitochondrial protein (Weinbach &
Garbus, 1965). At low levels, PCP uncouples oxidative phosphorylation,
at intermediate levels, it inhibits the formation of high-energy
intermediates in the phosphorylation system and, at high
concentrations, it inhibits the electron transport system (Mitsuda et
al., 1963). PCP has been shown to bind directly to mitochondrial
ATPase (Stockdale & Selwyn, 1971). Inhibition of ATPase would prevent
the breakdown of ATP to ADP and energy release to the mitochondria. In
response to the reduced availability of energy, a compensatory
increase in catabolism would be expected. Increased catabolism would
result in higher rates of oxygen consumption and, under short-term
exposure conditions, depletion of metabolic stores. With oxidative
phosphorylation uncoupled, the energy provided by catabolism would be
released as heat. This process would underlie, respectively, the
elevated respiratory rate and body temperature, and the long-term
weight loss observed in intoxicated organisms (Weinbach, 1957;
Farquharson et al., 1958; Mitsuda et al., 1963; Wood et al., 1983).
Similarly, Exon & Koller (1985) suggested that the immune effects of
2,4-DCP might result from the uncoupling of oxidative phosphorylation,
thus impairing cellular energy production in immunocompetent cells, or
perhaps, from a direct toxic effect on subpopulations of cells
involved in immune responses. The uncoupling of phosphorylation
appears to be due to the chlorophenate ion, while the convulsant
action is caused by the undissociated molecule (Farquharson et al.,
1958).
Chlorophenol toxicity may also result from a more general
inhibition of enzyme activity by these compounds. Arrhenius et al.
(1977) demonstrated that, in rat liver microsomal preparations,
purified chlorophenols selectively inhibited cytochrome P-450 activity
at the terminal oxygenation step of the MFO enzyme system by
interfering with the coupling of flavin to this enzyme. 2,4-DCP,
2,4,6-T3CP, and 2,3,4,6-T4CP concentrations of 0.3 mmol-3 mmol/litre
showed a weaker inhibition of C-oxygenation than PCP. Kaneki & Tanaka
(1984) reported that the inhibition of porcine lipase activity and, to
a lesser extent, wheat germ lipase activity increased with the number
of chlorine atoms on the phenol ring. The inhibiting action of PCP was
greater than that of 2,3,4,6-T4CP, which in turn was stronger than
2,3,6- T3CP, while DCP and MCP did not have any effect. Carlson
(1978) found that certain T3CP isomers inhibited glucuronyl
transferase activity and EPN detoxification in vitro, but not
in vivo. The reason for these effects could be the absence from the
in vitro preparations of binding proteins or alternate metabolic
enzyme systems (Somani et al., 1984).
The mechanisms of carcinogenicity or co-carcinogenicity of
chlorophenols (section 8.7) remain unresolved at present. Exon &
Koller (1985) suggested that chlorophenols may play a role in
carcinogenicity by altering the toxicity of carcinogens (by inhibiting
detoxifying enzymes, damaging DNA, or altering DNA repair) or by
reducing immunosurveillance. It has been shown by Vizethun & Goerz
(1979) that 2,4,5-T3CP and PCP can induce different species of
cytochrome P-450 in nuclei and microsomes. The importance of this
finding in relation to carcinogenesis is as yet unclear, but nuclear
monoxygenases could play a critical role in cell alterations, because
of their proximity to DNA and their ability to induce binding of
electrophilic compounds.
9. EFFECTS ON MAN
As a result of the diverse range of applications of chlorophenols
(section 3.2.2), there is considerable potential for human exposure to
these compounds and their associated contaminants. Knowledge
concerning the toxic effects of chlorophenols on people is based
primarily on studies on persons employed in the chemical-manufacturing
industry, where mainly DCP and T3CP are involved, and the
wood-preservation/ protection industries, where T3CP, T4CP, and PCP
are the major forms used (see reviews by Behrbohm, 1959, Kozak et al,
1979, and Ahlborg & Thunberg, 1980). Reports of chlorophenol toxicity
in the general population are few in number, though chlorophenol
contamination of human tissues and fluids seems widespread (sections
5.2, 5.3).
9.1 Acute Toxicity
Accidental and suicidal poisonings with commercial chlorinated
phenols have been reported (WHO, 1987b), and a number of the most
heavy acute exposures have resulted in death. With the support of
animal studies, the signs and symptoms of acute exposure to
chlorinated phenols include: convulsions (especially with
less-chlorinated phenols), ataxia, mental and physical fatigue,
headache, dizziness, disorientation, tachycardia, body temperature
change (decreased with monochlorophenols, increased with,
particularly, tetrachlorophenols and pentachlorophenol), and increased
sweating. Cyanosis and asphyxia spasms shortly precede death. Death is
apparently due to cardiac arrest and is followed, at least in animals,
by rapid rigor mortis, especially with T3CP and T4CP poisoning.
In general, the acute toxicity of chlorophenols in animals
increases with the number of chlorine atoms in the molecule (section
7.1.1). In man, the only published estimate of a minimum lethal oral
dose (LDLo) for any chlorophenol is for PCP (29 mg/kg body weight,
approximately 2 g for an average person) (WHO, 1987b). The human LDLo
for unchlorinated phenol is estimated to be 140 mg/kg body weight.
Acute exposure of human beings to lower chlorophenols has also
occurred as a result of industrial accidents during the production of
2,4,5-T3CP and has been most consistently associated with chloracne
(a persistent form of acne with keratotic follicles associated with
exposure to chlorinated compounds) and symptoms of liver toxicity.
For example, in April 1968, an explosion in a reactor producing
2,4,5-T3CP at a manufacturing plant in England released a
considerable amount of 2,4,5-T3CP and TCDD, exposing 14 people inside
the plant (May, 1973). Abnormalities in liver function tests (elevated
thymol turbidity, zinc turbidity), serum-transaminases, and urine
analyses were detected immediately after the incident; 10 days later,
the same tests gave normal values. Plant activity resumed but, by
December, 79 cases of chloracne were reported, some of whom also
suffered from conjunctivitis. The entire building was then thoroughly
cleaned, the interior walls resurfaced, and contaminated equipment was
buried. In a follow-up study conducted 10 years after the accident,
the frequencies of chromosomal aberrations and sister chromatid
exchange rates in lymphocyte cultures from exposed subjects were
normal (Blank et al., 1983).
To date, acute exposure of the general population to lower
chlorinated chlorophenols has been documented only from the ICMESA
plant accident in Seveso, Italy. An over-heated chemical reactor
discharged a cloud containing sodium hydroxide, Na-T3CP, and TCDD
into the atmosphere, contaminating an area south of the factory
containing 37 000 people (Hay, 1976; Del Como et al., 1982). Within 2
weeks of the accident, toxic effects were being treated in some 500
people (Hay, 1976). The most prevalent signs of exposure were skin
burns and chloracne, which was evident in 193 of the inhabitants. The
highest soil concentrations of 2,3,7,8-TCDD were associated with the
most severe cases of chloracne (Caramaschi et al., 1982). An
international steering group formed by the Italian government stated
in their final 1984 document that, with the exception of chloracne, no
clear health effects remained in the 193 persons in Seveso who were
registered as having chloracne, 20 of whom still showed symptoms in
1984. Exposed children had indications of increased enzyme activities
(increased D-glucaric acid in the urine) up to 3 years after exposure
(WHO, in press).
9.2 Long-term Exposure
9.2.1 Effects on skin and mucous membranes
Workers may display a variety of overt symptoms of chlorophenol
exposure. Persons often complain of irritations of the skin, mucous
membranes and respiratory tract as a result of direct airborne
contact. In addition, chronic skin ailments, particularly chloracne,
but also other skin lesions, ulcerations, and porphyria cutanea tarda
have been reported, mainly from plants manufacturing chlorophenols for
phenoxy-acetic acid herbicides. Clinical indications of liver damage
and haematological and neurological effects have also been reported,
particularly in association with high exposures.
For instance, among workers in a 2,4-DCP and 2,4,5-T3CP
manufacturing plant in the USA, 29 cases of chloracne and 11 cases of
porphyria were detected (Bleiberg et al., 1964). In addition to the
two chlorophenols, PCDDs and PCDFs, acetic acid, phenol,
monochloroacetic acid, and sodium hydroxide may have contributed to
the symptoms of the workers. Poland et al. (1971) examined employees
from the same plant 6 years after the report of Bleiberg et al.
(1964). Of the 73 male workers examined, 48 (66%) had some degree of
acne, and chloracne was found in 13 workers (18%). The severity of the
chloracne was not correlated with job location within the plant or
duration of employment, suggesting that there is a large variation in
the susceptibility of individuals to exposure to chlorophenols or
their contaminants.
Workers exposed to 2,4,5-T3CP in a production plant in the USSR
also developed dermatitis, as reported by Kozak et al. (1979).
Similarly, an incident of acute dermatitis in Russian agricultural
workers exposed to copper trichlorophenate was reported (Kozak et al,
1979).
Ott et al. (1980) examined the effects of exposure to commercial
2,4,5-T3CP and 2,4,5-T at a plant in the USA. In unacclimated
personnel, levels of less than 4 mg T3CP/m3 and/or 0.1 mg
2,4,5-T/m3 caused nasal irritation, sneezing, and a bitter taste in
the mouth. TCDD concentrations in both preparations were less than
1 mg/kg, in 1966, and 0.1 mg/kg, after 1972. Medical records of 204
exposed employees over the period 1950-76, did not reveal any cases of
chloracne or porphyria. The mortality rate of employees was lower than
expected (6 versus 13.3 expected) in workers exposed for less than 1
year and close to expected in those exposed for 1 year or more
(5 versus 7 expected).
In clinical studies on workers at a 2,4,5-T-manufacturing plant
(presumably exposed to 2,4,5-T3CP and its microcontaminants),
exposure was strongly associated with the development of chloracne,
plus increased prevalences of actinic elastosis and hirsutism (Suskind
& Hertzberg, 1984).
Jirasek et al. (1974) and Pazelerova et al. (1974) reported the
cases of about 80 people with both acute (industrial accident) and
long-term (up to 6 years) exposure to Na-T3CP, tetrachlorobenzene,
the sodium salt of trlchlorophenoxyacetate, and their contaminants.
Symptoms developed as long as 18 months after exposure. Chloracne
appeared in 96% of 55 people examined.
In lumber-mill workers, lesions and ulcerations were found in skin
areas in direct contact with a Na-T4CP solution, usually through
soaked clothing (Stingily, 1940). On the basis of roughly 300-400
cases, it was concluded that Na-T4CP caused the dermatitis while the
subsequent chronic lesions and nicerations were caused by fungal
infections.
Kleinman et al. (1986) and Fenske et al. (1987) evaluated the
extent and impact of occupational exposure to Permatox 100 (20.7%
2,3,4,6-T4CP, 3.1% PCP, plus substantial quantities of PCDDs and
PCDFs) in workers from a lumber mill in Washington state. The results
of their monitoring of air and urine are presented in section 5.3. In
health effects questionnaires, exposed workers complained
significantly more frequently than controls of headaches, eye and
upper respiratory irritations, and unusual sweating, though there was
no significant correlation between urinary levels and the frequency of
these symptoms. The effects of exposure to a commercial Na-T4CP
solution (containing hexa-, hepta-, and oetachlorodibenzo- p-dioxins,
dibenzofurans, and probably PCP and T3CP) were investigated by
Sterling et al. (1982) in workers at 2 sawmills in British Columbia,
Canada. The study included 1014 men with from 1 to over 20 years of
known exposure, compared with 103 loggers and outdoor municipal
workers, who served as controls. In self-administered questionnaires,
exposed workers reported significantly increased incidences of various
dermatological, upper respiratory, and general respiratory symptoms,
as well as eye irritation. These disorders were significantly more
frequent in the high-exposure group (247 workers) than in the workers
considered to be in the low/moderate-exposure group (767 workers),
who, in turn, had a higher incidence than the controls.
A detailed study of chlorophenol exposure in a sawmill in the same
geographical area was carried out by Embree et al. (1984) (section
5.3). They divided the workers into a control group from areas with no
identifiable air contaminants, a group of workers who worked in close
proximity to recently treated lumber but who did not have manual
contact (airborne), and a group who were responsible for the manual
handling of recently treated lumber (dermal plus airborne). Serum and
urine levels of chlorophenols were related to exposure in a
dose-dependent manner (section 5.3). From health histories, the only
symptoms that occurred significantly more frequently in exposed
workers were a productive cough and a reduced rate of forced
exhalation in the "airborne" group. These symptoms could not be
attributed to chlorophenol exposure, as the "dermal-plus-airborne"
group were exposed to similar atmospheric chlorophenol levels and had
higher levels of overall exposure, yet recorded a significantly lower
incidence of productive coughing.
Alexandersson & Hedenstierna (1982) examined the effects of
long-term exposure to T3CP vapours in workers at a gas-mask factory.
Trichlorophenol vapour, because of its characteristic smell, was used
at the factory for checking leaks in gas masks. Complaints of eye,
nose, and airway irritation were voiced by 7 individuals who had been
employed in testing masks for from 2 to 10 or more years. Pulmonary
function tests revealed that exposed workers displayed reduced forced
expiratory flow and increased closing volume in the lungs compared
with controls.
9.2.2 Systemic effects
Effects on liver and kidney function and haematological parameters
have also been investigated in workers exposed to chlorophenols. The
findings have been generally negative. In studies on Canadian sawmill
workers (Enarson et al., 1986), serum levels of creatinine, bilirubin,
glutamic oxaloacetic transaminase, and alkaline phosphatase, and
patient histories of jaundice, liver, kidney, and heart disease did
not differ from those of the controls. Blood-leukocyte counts and
haematocrit decreased, and urine-erythrocyte levels increased
following chlorophenate exposure. These effects were significant only
for the haematocrit and haematuria, and only for workers handling
treated lumber.
Sterling et al. (1982) reported that chlorophenol-exposed sawmill
workers filling out self-administered questionnaires reported
significantly increased incidences of gastrointestinal,
musculoskeletal, acute systemic, liver, kidney, and neurological
symptoms.
In the study on industrial workers exposed to di- and
tri-chlorophenols, Bleiberg et al. (1964) reported elevated
coproporphyrin excretion in the maintenance men that could have been
due to more intense, though sporadic, exposure. Hepatotoxic effects
were not found in the study group.
Initially, more than one-third of Czechoslovakian 2,4,5-T3CP- and
2,4,5-T-manufacturing workers described by Jirasek et al. (1974) and
Pazelerova et al. (1974) showed indications of mild liver damage,
which, in some instances, was confirmed by needle biopsy. The workers
had increased serum levels of cholesterol (56%), total lipids (67%),
and lipid-phosphorus (42%). A small but significant decrease in
serum-albumin, and an increase in serum-globulin were also found.
9.2.3 Psychological and neurological effects
A range of psychological and neurological symptoms have also been
associated with exposure to chlorophenols, often in association with
other chemicals. Workers from a plant in the USSR who were
occupationally exposed to 4-MCP complained of "... sleep disorders
(usually sleepiness and sometimes insomnia), irritability, frequent
mood changes, and rapid fatigability" (Gurova, 1964).
Similarly, Kleu & Goltz (1971) reported that 10 persons suffering
from chloracne as a result of 15 years' exposure to a T3CP
formulation complained of "... decreased sexual activity, easy
fatigability, alcohol intolerance, and loss of interest ... reduced
vital psychic and intellectual capacities combined with neurasthenia
and mental depression". The actual occupations of these individuals
were not stated.
Gilioli et al. (1983) conducted electroencephalographic analyses
of workers exposed to T3CP and TCDD at the Seveso plant in Italy,
site of an accident in 1976. These workers had both long-term, and
possibly acute high-level exposure to TCDD and T3CP. Exposed workers
generally exhibited an increased incidence of abnormal EEG tracings,
which were particularly associated with increased proportions of theta
waves, and had a slower visual reaction time than a non-exposed group.
The 2,4,5-T3CP-and 2,4,5-T-manufacturing employees reported by
Jirasek et al. (1974) and Pazelerova et al. (1974) showed neurological
abnormalities, including myographic changes in 23% of those tested.
Neurasthenic symptoms were present in 60% of the workers, compared
with 30-40% of the general population. Some of the workers complained
of fatigue, loss of appetite, weight loss, and abdominal pain.
9.2.4 Reproductive effects
The effects of chlorophenols on reproduction have been
investigated in 3 studies. Corddry (1981) provided data on pregnancy
outcomes in women married to workers from a sawmill in British
Columbia using Na-T4CP and Na-PCP. Analysis of data from 43 women,
with a total of 100 pregnancies, did not reveal any significant
differences in the pregnancy outcomes of women living with exposed men
compared with those living with unexposed men. A slight trend towards
more adverse pregnancy outcomes in the exposed group disappeared when
corrected for alcohol consumption. Male fertility was not studied.
Suskind & Hertzberg (1984) reported pregnancy outcomes in the
families of male workers manufacturing 2,4,5-T (and with probable
exposure to 2,4,5-T3CP and 2,3,7,8-T4CDD) compared with those of
other males from the same plant. There were no significant differences
between the families of exposed and non-exposed workers, but the rates
for stillbirth and death during the first 4 weeks were higher in the
families of exposed workers.
The pregnancy outcomes were surveyed in wives of workers from a
chlorophenol-manufacturing plant in Michigan, USA, potentially exposed
to PCP, 2,4,5-T3CP, and their microcontaminants for the group as a
whole (Townsend et al., 1982). There was no significant association
between exposure to dioxins (these were the focus of the study) and
adverse pregnancy outcomes. When the conceptions were divided into
subgroups according to risk factors associated with the mother, a
subgroup of 9 TCDD-exposed conceptions was identified of which 3 were
spontaneous abortions.
9.2.5 Carcinogenicity
A large number of epidemiological studies have been published
concerning human cancer outcomes following occupational exposure to
chlorophenols, phenoxy herbicides (made from and contaminated with
chlorophenols), and chlorinated dibenzo- p-dioxins and dibenzofurans
(microcontaminants found in some chlorophenols and phenoxy
herbicides). Most of these studies have been described and reviewed in
several publications by the International Agency for Research on
Cancer (IARC, 1979, 1986, 1987), and readers are referred to the IARC
monographs for details of individual studies. Only the conclusions of
these studies are given in this publication with the emphasis on
studies that specifically concern exposure of populations to
chlorophenols. Studies that address the effects of exposure to phenoxy
herbicides in general and MCPA, and do not involve substantial
concomitant exposure to chlorophenols are not discussed. These
include: cohort studies on phenoxy herbicide sprayers (Axelson et al.,
1980; Hogsted & Westerlund, 1980); studies on Vietnam war veterans
(Royal Commission on the Use and Effects of Chemical Agents on
Australian Personnel in Vietnam, 1985; Lathrop et al., 1987); and
workers involved in the manufacture of MCPA (Coggon et al., 1986).
Case-control studies covering phenoxy herbicide exposure, but with no
mention of exposure to chlorophenols (Balarajan & Acheson, 1984;
Greenwald et al., 1984; Kogan & Clapp, 1985; Hoar et al., 1986; Kang
et al., 1986; Vineis et al., 1987) and data from occupational
mortality statistics in which exposure data are inferred only from job
titles are also not included (Milham, 1982, 1985; Callagher &
Threlfall, 1984).
9.2.5.1 Case-control studies reviewed by IARC
Soft tissue sarcoma (STS) was studied in individuals exposed to
chlorophenols and other chlorophenol-based chemicals in Sweden
(Hardell & Sandstrom, 1979; Eriksson et al., 1981), New Zealand (Smith
et al., 1984), and the USA (Woods et al., 1987). Relative risks (RR)
of STS for exposure to chlorophenols alone were elevated in both of
the Swedish studies (RR = 6.6; 96% CI, 2.1-20.9; and RR = 3.3; 95%
CI, 1.3-8.1) and for one potentially exposed subgroup in the New
Zealand study (RR = 7.2; 90% CI, 1-ND). They were not elevated in the
US study, unless the subgroup of cases with Scandinavian names
(RR = 7.2; 95% CI, 2.1-24.7) and the subgroup of lumber graders
(RR = 2.6, 95% CI, 1.1-6.4) were considered separately. Chlorophenol
exposures (2,4,6-T3CP, PCP, and others) were poorly described and not
quantified. Case-control studies of malignant lymphomas (Hodgkins
disease and non-Hodgkins lymphoma) have also been carried out in
Sweden (Hardell et al., 1981), New Zealand (Pearce et al., 1986), and
the USA (Woods et al., 1987). In Sweden, low-grade exposure (up to one
week continuous or 1 month intermittent) and high-grade exposure
(greater than the low-grade exposure criteria) to unspecified types of
chlorophenols resulted in relative risk values of 2.2 (95%
CI, 1.1-4.6) and 7.6 (95% CI, 3.2-17.7), respectively. In the New
Zealand study of 83 cases of non-Hodgkins lymphoma, exposures were not
classified into high or low, and were not quantified; however, some
cases were not likely to have been exposed to 2,4,6-T3CP and PCP.
The relative risk for non-Hodgkins lymphoma in pelt department
workers with potential exposure to 2,4,6- T3CP was 1.9 (90%
CI, 0.9-4.0); however, other meat workers without exposure to
chlorophenols had a relative risk of the same order. In the US study,
the relative risks for non-Hodgkins lymphoma were not elevated for
groups exposed to low, medium, or high levels of chlorophenols.
A statistically significant increase in nasal and nasopharyngeal
cancer was found among workers exposed to chlorophenols (mainly tri-,
tetra-, and pentachlorophenol) in a Swedish case-control study
(Hardell et al., 1982). In an inter-Nordic study, 2 cases of sinonasal
cancer out of 167 were identified as having been exposed to
chlorophenols (not further specified) compared with 0/167 colorectal
cancer controls (Hernberg et al., 1983). In a Danish study, no
association was found between sinonasal cancer and potential exposure
to chlorophenols, as inferred from employment records (Olsen &
Moller-Jensen, 1984).
The results of 2 Swedish studies did not show statistically
significant associations between primary liver cancer or colon cancer,
and exposure to chlorophenols (mainly tri-, tetra-, and
pentachlorophenol) (Hardell, 1981; Hardell et al., 1984).
No significant associations were found between multiple myeloma
and potential exposure to chlorophenols (probably mainly
penta-chlorophenol) in a case-control study carried out in New Zealand
(Pearce et al., 1986).
9.2.5.2 Cohort studies reviewed by IARC
Three follow-up studies have been undertaken on small groups of
workers exposed to 2,3,7,8-TCDD during accidents in the manufacture of
2,4,5-T3CP (Cook et al., 1980; Zack & Suskind, 1980; Thiess et al.,
1982). When combined, these cohort studies show 19 cancer deaths
observed versus 14.7 expected (RR = 1.29; 95% CI, 0.78-2.02). Two
studies (Ott et al., 1980; Zack & Galley, 1983) covered workers
employed in the manufacture of 2,4,5-T from 2,4,5-T3CP. The observed
cancer deaths versus expected cancer deaths for these cohorts were 1
versus 3.6 and 35 versus 30.9, respectively.
A total of 3 deaths from soft-tissue sarcoma (STS) were identified
in US cohorts studied by Honchar & Halperin (1981). This was
equivalent to 2.9% of the deaths in the 4 cohorts, where approximately
only 0.07% would be expected. Later, one additional STS case was
observed in one of these cohorts (Cook, 1981), and an additional 3
cases of STS were reported in workers in 2,4,5-T-manufacturing plants
(Johnson et al., 1981; Moses & Selikoff, 1981). Fingerhut et al.
(1984) reviewed the histological specimens for the 7 cases reported as
having STS. In the review, 5 of the 7 cases were diagnosed as having
STS. A review of the employment records for the 7 patients showed that
all of them had worked in 2,4,5-T-manufacturing plants; 4 of the
patients had specific assignments to 2,4,5-T3CP or 2,4,5-T
departments.
A cohort study on workers in 2 Danish chemical plants (Lynge,
1985), in which 2,4-D and 2,4-DP were produced together with MCPA and
MCPP, showed the overall cancer incidence to be close to that of the
Danish population (208 observed cases and 216.5 expected). In men, 5
cases of STS were observed, where 1.84 were expected. The number of
malignant lymphoma cases in men was 7, with 5.37 expected cases. In
the subgroup of men assigned to the phenoxy herbicide-manufacturing
department, 11 lung cancer cases were observed in men, when 5.33 cases
were expected.
9.2.5.3 More recent studies
A Swedish study on soft-tissue sarcoma and exposure to phenoxy
acid herbicides and chlorophenols was recently repeated using 55 new
cases diagnosed in 1978-83 (Hardell & Eriksson, 1988). In addition to
comparing the cases with controls from the general population, a
control group of patients with cancers other than malignant lymphoma
and nasopharyngeal cancer was selected. An elevated relative risk for
exposure to phenoxy herbicides was found, but no statistically
significant differences were observed between cases and controls with
regard exposure to chlorophenols. The relative risk for chlorophenols
was based on only 4 exposed cases.
In a case-control study nested within a cohort of Finnish
woodworkers, no association was found between respiratory cancer and
exposure to chlorophenols (mainly tetra- and trichlorophenols), but
this result was based on only 3 exposed cases (Kauppinen et al.,
1986).
In a more recent cohort study on 2192 employees at a plant
involved in the production of higher chlorophenols and phenoxy acids
(Cook et al., 1987; Ott et al., 1987), mortality during the period
1940-82 among workers with potential occupational exposure to
chlorophenols was similar to that of US white males for all causes and
for all cancers; there were 81 observed cancer deaths versus 79.3
expected deaths. No statistically significant excesses of mortality
were observed for the cancers of a prior interest (nasal and
nasopharyngeal, stomach, liver, connective and soft tissue,
lymphomas). Excesses in mortality that did not reach the 5% level of
probability were reported for stomach cancer (6/3.8) and non-Hodgkins
lymphoma (5/2.6).
A cohort of 878 persons, employed in the manufacture of 2,4-D at
the same plant between 1945 and 1983, was followed up for mortality
until 1983. Some of these employees may also have been exposed to
other chlorophenols at the plant site. There were 20 cancer deaths
against 16.9 expected. There were 5 deaths from lymphatic and
haematopoietic cancer against 2.5 expected. Two cases had
lymphosarcoma and reticuiosarcoma, and there was one case of Hodgkins
disease. Both workers who died from non-Hodgkins lymphoma had had
potential exposure to PCDDs. Death certificates were reviewed with
special attention directed to soft-tissue sarcoma, but no cases were
identified (Bond et al., 1988).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of Human Health Risks
10.1.1 Exposure levels
10.1.1.1 Non-occupational exposure
Exposure to chlorophenols other than pentachlorophenol may occur
via ingestion, inhalation, or dermal absorption (section 5.2). The
general population is thought to be exposed mainly through the
ingestion of food and drinking-water. These compounds have not been
quantified in the ambient atmosphere, but atmospheric levels are
likely to be of the same order of magnitude (ng/m3) as those of PCP.
Even with 100% absorption, uptake via this route would be much less
than 1 µg/day. Similarly, even though low levels of the lower
chlorinated phenols occur widespread throughout the environment,
direct dermal contact with these compounds will not be an important
route of exposure for the general population. Non-occupational
exposure (inhalation, dermal) to chlorophenol-treated lumber has not
been investigated, but may be significant, if chlorophenols are used
for extensive treatment of the interior of houses. Significant
exposures may also occur if consumers use chlorophenol-based products
without appropriate care and protection.
An estimate of non-occupational exposure through the ingestion of
drinking-water and food can be made using representative published
concentrations of the major commercial chlorophenols other than
pentachlorophenol. The daily exposure of a 60-kg person in Canada to
2,4-DCP, 2,4,5-T3CP, and 2,3,4,6-T4CP is estimated in Table 19, on
the basis of assumptions detailed in the table. These calculated
exposures may be overestimates in that residue determinations are
generally taken from contaminated areas, and because kinetic or
metabolic studies usually involve high chlorophenol concentrations. On
the other hand, estimates have not been made for all foodstuffs; for
example, no data were found for fruits, dairy products, or nuts.
Exposure to other chlorophenols, in particular 2,4,6-T3CP, cannot be
estimated, because there are insufficient data on the concentrations
present in water and food. Nevertheless, the values calculated should
suffice as a first approximation of exposure.
Table 19. Estimated daily per capita exposure to 2,4-DCP, 2,4,5-T3CP, and 2,3,4,6-T4CP from food and drinking-water in Canada
Source Daily 2,4-DCP 2,4,5-T3CP 2,3,4,6-T4CP
consumption
in Canada Concentration Exposure Concentration Exposure Concentration Exposure
(kg)a (µg/kg fresh (µg/kg (µg/kg fresh (µg/kg (µg/kg (µg/kg
weight) body weight weight) body weight fresh weight) body weight
per day)b per day) per day)
Tap 1.4 0.093c 0.002 0.035c 0.0008 0.009c 0.0002
water (litres)d
Vegetables
and
potatoes 0.36 6e 0.036 0.041f 0.0002 1.917g 0.011
Meat and
poultry 0.26 5h 0.022 0.034f 0.0002 3i 0.013
Fish and
seafood 0.019 13.75j 0.004 0.76k 0.0002 10.3k 0.003
Total
exposure 0.064 0.0014 0.027
Table 19. (contd).
Source Daily 2,4-DCP 2,4,5-T3CP 2,3,4,6-T4CP
consumption
in Canada Concentration Exposure Concentration Exposure Concentration Exposure
(kg)a (µg/kg fresh (µg/kg (µg/kg fresh (µg/kg (µg/kg (µg/kg
weight) body weight weight) body weight fresh weight) body weight
per day)b per day) per day)
Tolerable 200.0l 100.0m 10.0n
daily intake
(µg/kg body
weight per
day)
a Per capita per day in 1984 (Statistics Canada, 1986).
b Based on a 60-kg person.
c Jyvaskyla, Finland (Paasivirta et al., 1985).
d Water consumption for 60 kg man per day (NHW, 1983).
e Average for potatoes treated with 2,4-D (Bristol et al., 1982).
f Assuming that the ratio of 2,4-DCP: 2,4,5-T3CP is the same as for 1984 Canadian imports.
g Average from Alberta Government assay (Jones, 1981), assuming all T4CP is 2,3,4,6-T4CP.
h Muscle concentration from chickens fed 50 mg Nemacide/kg, 7 days post-treatment (Sherman et al., 1972).
i Upper concentration detected in chicken flesh from British shops (Farrington & Munday, 1976).
j Mean muscle concentration calculated as µg/kg wet weight Bacon, (1978).
k Mean muscle concentration (Paasivirta et al., 1985).
l NOEL taken from Kobayashi et al. (1972) using a 500-fold safety factor.
m NOEL taken from McCollister et al. (1961) using a 1000-fold safety factor.
n NOEL taken from Schweiz et al. (1974) using a 1000-fold safety factor.
The daily ingestion of these chlorophenols by a 60-kg person is
estimated to be 3.84 µg of 2,4-DCP; 0.084 µg of 2,4,5-T3CP, and
1.62 µg of 2,3,4,6-T4CP (on the basis of data in Table 19 and
calculated for a 60-kg person). These values are presented on a
per-unit-weight basis in Table 19. In Canada, the amount of PCP used
is approximately equal to the amounts of all other chlorophenols
combined, suggesting that PCP exposure is roughly equal to that of the
other chlorophenols. Thus, the total estimated exposure of Canadians
to all chlorophenols including PCP is about 10 µg/person per day. This
value agrees with the 10-30 µg/ person per day estimated by NRCC
(1982) and the 12.7 µg/person per day estimated by NHW (1988) (section
5.2) for the general population in Canada. The total estimated
exposure levels in other countries may differ from this value,
depending on product use patterns, food and water consumption, and
levels of environmental contamination.
Although the above estimate of exposure is based on limited
information on residue levels and uptake rates, it is supported by the
low levels of chlorophenols found in human tissues and fluids; in the
few studies available, µg/kg quantities have been detected in human
tissues and fluids (section 5.2).
10.1.1.2 Occupational exposure
The potential exposure to chlorophenols may be significant for
certain workers employed in the lumber industry, pesticide manufacture
and use, use of treated wood for construction, railroad ties, or
telephone poles, and a variety of other industries in which
chlorophenols are used as biocides. In the work place, exposure would
be mainly through dermal absorption and inhalation; ingestion of the
compounds is more likely to occur if eating, drinking, and smoking are
allowed in the work area, or if proper cleansing procedures are not
practised. The air in work areas where chlorophenols are used contains
elevated concentrations of chlorophenols. Concentrations of 14 mg/m3
have been reported in such work areas, but typical concentrations were
1-3 orders of magnitude below this in work places studied recently in
North America and Scandinavia (section 5.3). Persons working in high
exposure areas have elevated levels of chlorophenols in their body
fluids, particularly if their job combines dermal and inhalation
exposure to chlorophenols. Urine levels of up to 49 mg T4CP/litre
have been reported (section 5.3).
Few data are available with which to model uptake and excretion of
chlorophenols other than PCP. However, preliminary estimates of some
occupational exposures can be derived from recent reports of
concentrations in the urine of workers in the sawmill industry, where
handling of treated wood and the proximity of workers to the
open-treatment apparatus can result in relatively high uptake of
chlorophenols.
Urinary concentrations of employees at Finnish, American, and
Canadian sawmills have been compiled in Table 20, and have been used
to estimate worker exposure in this industry. These estimates are
based on two principal assumptions. First, it was assumed that urinary
concentrations of T4CP reach a sustained level following long-term
exposure, since Braun et al. (1979) calculated that PCP in the urine
of exposed persons reached a fairly constant level within one week.
Second, it was assumed that all T4CP is cleared in the urine, because
it is known that human beings clear 86% of administered PCP in urine
(Braun et al., 1979) and that rats clear more than 95% of single doses
of 2,3,4,6-T4CP and 2,3,5,6-T4CP (the major tetrachlorophenols in
commercial preparations) via the urine (Ahlborg & Larsson, 1978).
On the basis of these assumptions, the urinary concentrations in
Table 20, and a urine production of 1.4 litres daily, a 60-kg sawmill
worker is estimated to take up from 2 to 42 µg/kg per day on average
(Table 20). Estimates of the exposure of workers with the highest
urine concentrations from each study, who presumably had extensive
dermal uptake of chlorophenates, ranged from 53 to 1142 µg/kg per day.
A more comprehensive knowledge of chlorophenol levels and fluxes and
their dynamics would be necessary for a better estimate.
As expected, the estimated occupational exposure is much higher
(often 2 or more orders of magnitude) than that calculated for members
of the general population. This difference is confirmed by levels of
chlorophenols measured in the biological tissues and fluids of
workers, and the ambient atmosphere (sections 5.2, 5.3), as well as
the association of intoxications and adverse symptoms with
occupational exposure to chlorophenols (section 9).
Since the foregoing calculations are based on only 5 studies, all
involving sawmills, these estimates are not directly applicable to
other occupations and/or geographical regions.
10.1.2 Toxic effects
Acute lethal doses of lower chlorophenols in experimental mammals
(section 8.1) are associated with restlessness, hyperpyrexia, rapid
respiratory rates, at axia, and eventually dyspnoea, coma, and death.
MCPs, DCPs, and 2,4,6-T3CP are convulsive agents, while the other
trichlorophenols and tetrachlorophenols are not. All of the
chlorophenols are irritating or corrosive to the skin, eyes, and
mucuous membranes (section 8.2).
Table 20. Estimates of occupational exposure to tetrachlorophenols (based on urinary concentrations from recent studies
on exposed workers)
Situation Estimated intake Reference
Urinary concentrations (µg/litre) µg/kg body weight per day)a
Median/mean Maximum Averageb Maximum
Finnish sawmill; Dermal exposure primarily Dermal exposure Lindroos et al. (1987)
workers exposed to 1809.4c (median) 48 924.6 42 1142
KY-S (NaT4CP)
formulation; 1980-81 Respiratory exposure primarily Respiratory exposure
208.8c (median) 3085.3 5 72
Finnish sawmill; 2841.5d,e (mean) 39 436.6d,e 66 920 Kauppinen & Lindroos (1985)
workers exposed to (1985)
NaT4CP
Canadian (British 423f (mean) 1479f 10 35 Currie & McDonald (1986)
Columbia) planer (1986)
mill; workers
exposed to NaT4CP;
1985
Washington state (USA) 160.4g (mean) 2255 4 53 Kleinman et al. (1986)
sawmill; workers
exposed primarily to
NaT4CP; 1981-82
Table 20. (contd).
Situation Estimated intake Reference
Urinary concentrations (µg/litre) µg/kg body weight per day)a
Median/mean Maximum Averageb Maximum
Canadian (British Dermal + airborne exposure Dermal + airborne exposure Embree et al. (1984)
Columbia) sawmill; 1250e (mean) 29
workers; exposed to
Na-T4CP; 1978-79 Airborne only Airborne only
930e 22
a For a 60-kg worker.
b Based on mean concentration, except for estimate from Lindroos et al. (1987), which is based on the median.
c Assumes all chlorophenols are T4CP, which predominates in these formulations.
d Not clear if sample hydrolysed.
e Assumes free concentrations are 10% of total concentrations.
f From samples collected on final morning of a 5-day work week.
g Grand mean for exposed workers, all dates.
Note: No correction for density has been made in any table entries.
Short-term exposure of experimental animals to 2,4-DCP,
2,4,5-T3CP, 2,4,6-T3CP, or 2,3,4,6-T4CP produces moderate adverse
effects on the liver, kidney, and spleen, while 2,4-DCP is also
haematotoxic and immunotoxic (section 8.2). 2-MCP, 2,4-DCP,
2,4,5-T3CP, 2,4,6-T3CP, and 2,3,4,6-T4CP produce fetotoxic or
embryo-toxic effects, but none of the chlorophenols tested to date has
produced teratogenic effects. Isomers of DCP other than 2,4-DCP may
reduce male fertility (section 8.5).
2,4,6-T3CP and 2,3,4,6-T4CP appear to have some mutagenic
capability; however, chlorophenols do not appear to be potent mutagens
capable of exerting significant genotoxic effects (section 8.6). There
is sufficient evidence that 2,4,6-T3CP (commercial grade) is an
animal carcinogen. However, the trichlorophenol used in this study was
not analysed for PCDD and PCDF microcontaminants. 2,4-DCP (more than
99% pure) was found not to be carcinogenic in mice and rats. The other
lower chlorophenols have not been adequately tested for their
carcinogenic properties. Some chlorophenols appear to be tumour
promoters; others do not (section 8.6).
The microcontaminants in the commercial grades of the more highly
chlorinated phenols can lead to toxic effects even more severe than
those produced by the chlorophenol itself (sections 6.3, 8.9). Their
possible significance in terms of the carcinogenicity of 2,4,6-T3CP
should not be overlooked.
Toxic effects in man from acute exposure to high concentrations of
the lower chlorinated phenols include acute and chronic skin
irritation, chloracne, respiratory disorders, recurring headaches,
dizziness, nausea, vomiting, loss of coordination, tremor, weakness,
and lethargy (section 9.1).
The question of carcinogenicity in man from exposure to
chlorophenols is a matter of controversy. IARC concluded that there is
limited evidence of carcinogenicity from occupational exposure to
chlorophenols. Following a review of studies published since the IARC
evaluation, the Task Group still finds this conclusion appropriate.
10.1.3 Risk Evaluation
The information available to date indicates that the general
population is exposed to low levels of chlorinated phenols. As derived
in section 10.1, the estimated exposure to the major chlorophenols
other than PCP of a person who does not work with these compounds is
0.0833 µg/kg per day. This burden is made up of 0.058 µg 2,4-DCP/kg
per day, 0.0013 µg 2,4,5-T3CP/kg per day, and 0.024 µg
2,3,4,6-T4CP/kg per day. People who manufacture or apply
chlorophenols predictably experience much higher levels of exposure.
As an example, the total estimated average exposures to T4CP for
sawmill workers ranged from 2 to 42 µg/kg per day (section 10.1.1.2).
For comparison with these estimated exposures, Tolerable Daily
Intake (TDI) levels have been calculated (from no-observed-effect
levels determined in short- term studies) of 100 mg/kg body weight for
2,4-DCP (mice, in feed) (Kobayashi et al., 1972), 100 mg/kg body
weight for 2,4,5-T3CP (rats, oral) (McCollister et al., 1961), and
10 mg/kg body weight for 2,3,4,6-T4CP (rats, oral) (Schwetz et al.,
1974). Using an uncertainty factor of 1000 for 2,4,5- T3CP and
2,3,4,6-T4CP, because of the lack of long-term animal data, and an
uncertainty factor of 500 for 2,4-DCP, because of the availability of
long-term data (WHO, 1986), the TDI values for 2,4-DCP, 2,4,5-T3CP,
and 2,3,4,6-T4CP were estimated to be 200, 100, and 10 µg/kg per day,
respectively. The long-term carcinogenicity study available for
2,4-DCP (section 8.7) does not provide data that would alter this
estimate. The embryotoxicity data available for 2,4,5- T3CP (section
8.5) were found to be of too limited significance to be taken into
consideration.
Non-occupational exposure levels are usually well below these
values, indicating that the anticipated health hazards for the general
population from exposure to chlorophenols other than PCP are minimal.
The estimated occupational intakes are considerably higher, especially
when there is skin contact with chlorophenols (Table 20). Exposure
levels in sawmills may, in some cases, exceed the TDI values.
If tetra- or trichlorophenol preparations are used in wood
protection, workers can also be exposed to 2,4,6-T3CP. There is
sufficient evidence that 2,4,6-T3CP is carcinogenic for mice and
rats. Because of the arbitrary nature of assumptions inherent in
developing estimates of exposure, and extrapolating from effects on
animals to effects on human beings, risk assessments are never
precise. Nevertheless, it is prudent to ensure that human exposure to
2,4,6-T3CP is kept to a minimum.
Acute exposure to high concentrations of chlorophenol formulations
can be a significant hazard for the health of workers involved in the
production or use of chlorophenols. While no deaths have been reported
with exposure to chlorophenols other than PCP, fatalities due to high
exposure to the latter are well documented. Exposures to non-PCP
chlorophenols result in adverse signs and symptoms similar to those
caused by PCP. Exposure to commercial formulations of chlorinated
phenols has been associated with an increased relative risk of
soft-tissue sarcomas, lyrephotons, and nasal and nasopharyngeal
cancers in some studies; such associations have not been found in
other similar studies.
On the basis of toxicological effects and current exposure levels,
there does not appear to be any strong reason for eliminating all use
of chlorophenols. Furthermore, the need to reduce the levels of
exposure to chlorophenols appears minimal as long as the necessary
precautions to prevent high-level dermal and respiratory uptake are
observed. Exposure of the general population is much lower, and in the
absence of release from an industrial accident, the overall risk from
sustained non-occupational exposure is probably negligible.
Chlorophenols produce undesirable organoleptic effects at very low
concentrations. Contamination of the environment and of drinking-water
with chlorophenols above the threshold for organoleptic effects is
therefore unacceptable. Comprehensive monitoring of chlorophenol
levels, sources, and fluxes is essential to characterize both
occupational and non-occupational exposure to these compounds, and to
alert the responsible agency to potentially hazardous exposures where
they exist.
Microcontaminants, in particular PCDDs and PCDFs, found in
commercial formulations of tri-, tetra-, and pentachlorophenol, are
probably the causal agents for chloracne in human beings. PCDD and
PCDF levels in commercial preparations should be kept as low as
technically feasible. Care should also be taken to minimize their
formation during the incineration of wastes containing chlorophenols.
10.2 Evaluation of Effects on the Environment
10.2.1 Levels of exposure
Data on levels of chlorophenol residues other than PCP in the
environment are limited primarily to aquatic habitats, and indicate
that chlorophenol contamination is widespread in these systems. This
contamination may result from either the use or the formation of
chlorophenols, e.g., in the chlorine-bleaching process in pulp and
paper-mills (sections 3.2, 3.3, 3.4). Where data are not available on
levels of other chlorophenols in the environment and for evaluation
purposes, examples of monitoring data on PCP are used. On the basis of
relative rates of degradation and use patterns, it is likely that, in
general, environmental levels of other chlorophenols would be lower
than those found for PCP.
Ambient levels of PCP in air are less than 1 ng/m3 in
uncontaminated areas, while concentrations of several ng/m3 have been
detected in residential areas. Other chlorophenols may well be present
at comparable levels, but confirmatory data are lacking.
Residues of all chlorophenol congeners have been found in fresh
and marine waters (section 5.1.2). In Canada, concentrations are often
undetectable at the ng/litre level in receiving waters, and only
occasionally exceed 1 µg/litre; these higher levels are only observed
in close proximity to industrial sources of chlorophenols,
particularly pulp and paper-mills. Ambient levels are higher in waters
in the industrialized areas of Europe, but median concentrations still
do not exceed 1 µg/litre, and maximum concentrations in surface waters
and groundwaters only reach several µg/litre in heavily industrialized
regions. Levels of particular chlorophenols in industrial effluents
can reach several thousand µg/litre (section 5.1.2), but dilution
apparently reduces these to the low ambient levels observed.
Chlorophenol concentrations in sediments are usually higher than
those in the overlying water (section 5.1.2), as a result of
adsorption and low rates of anaerobic degradation. Water bodies not
receiving large chlorophenol inputs generally contain less than 1 µg
of chlorophenol congeners per kg dry sediment. Typical levels of all
chlorophenol congeners in fresh-water sediments of industrialized
regions are less than 50 µg/kg of dry sediment. In some instances,
hundreds or thousands of µg/kg have been detected from sites adjacent
to spills or discharges.
Soils may contain significant quantities of chlorophenols,
particularly at timber preservation facilities, or where phenoxy
herbicides have been applied. Levels as high as 70 mg chlorophenols/kg
were detected in soils from Finnish sawmills (section 5.1.3), but
ambient levels in soil were found to be much lower (< 0.1 mg/kg).
10.2.2 Transport
While chlorophenols are considered to be mainly water and soil
contaminants, some atmospheric movement also occurs. PCP has been
detected in rain, snow, and in the air (section 5.1.1), and presumably
other chlorophenols are also transported in this manner. Adsorption
controls chlorophenol transport in acidic or organic soils, but is
much less important in basic or mineral soils (section 4.1.2.1). In
surface waters, the fraction that is not degraded is incorporated into
the sediments, most likely through adsorption on sedimenting
particulates (section 4.1.3). While much of the chlorophenols entering
natural waters are probably degraded by photolysis or microorganisms,
they are moderately soluble and fairly persistent, and so can be
transported considerable distances by water (section 4.1.3).
10.2.3 Degradation
Both abiotic and biotic degradation eliminate chlorophenols from
the environment. Numerous in vitro studies have shown that
ultraviolet radiation can rapidly break down chlorophenols. Evidence
suggests that photolysis is important in natural surface waters
(section 4.2.1.1), and presumably in other exposed habitats. A large
number of microorganisms from different habitats are able to degrade
chlorophenols in laboratory cultures. In some instances, quantities as
high as tens of mg/litre are eliminated in a matter of hours or days
(section 4.2.1.2), though it is necessary to acclimate them first.
Degradation is generally slowest for the higher chlorinated congeners
or those with a chlorine atom in the meta position. In general,
anaerobic biodegradation of these compounds, if it occurs at all, is
much slower than aerobic metabolism. Some evidence suggests that
biodegradation is faster in soil than in sediments, and slower in
stream waters.
10.2.4 Bioaccumulation
Bioaccumulation of chlorophenols appears moderate, and most
bioconcentration factors (BCFs) fall between 100 and 1000.
Bioconcentration is usually a positive function of chlorine number.
There are no obvious patterns in BCF in relation to the type of
organism (for algae, plants, invertebrates, and fish). Once exposure
is discontinued, chlorophenols clear rapidly from biota, indicating
that the bioaccumulation observed in field studies is the result of
long-term exposure rather than persistence.
10.2.5 Persistence
Chlorophenols should only persist in the environment where the
rates of the various degradative processes are minor. Indeed, residues
in sediments, where photolysis and apparently microbial degradation
are minimal, have been estimated to be decades old (section 4.4).
Herbicidal applications and spills of PCP in soils reportedly
disappear in a matter of weeks or months.
10.2.6 Toxic effects on environmental organisms
Considerable overlap exists in the chlorophenol concentrations
that are toxic for bacteria, phytoplankton, plants, invertebrates, and
fish. Most of the LC50 and EC50 values for these organisms, which to
date have been primarily aquatic, fall within the several mg/litre
range (Table 16). Toxicity generally increases with the degree of
chlorination of the phenol ring, though chlorophenols with chlorines
in the 2 and 6 positions are often less toxic than expected on the
basis of the number of chlorines. Particularly in the case of the
higher chlorophenols, acute toxicity is a strong inverse function of
pH, as the phenol form of the compound is more toxic than the ionized
form. Exposure to chlorophenols affects a wide variety of processes in
environmental organisms (section 7) (Table 16).
In controlled field studies on aquatic ecosystems, exposure to
high concentrations (100-5000 mg/litre) of lower chlorophenols
generally impairs algal production and reproduction, alters the algal
species composition dramatically, and reduces zooplankton biomass and
production. The low levels of chlorophenols present in moderately
contaminated waters have been reported to impair the flavour of fish.
There is very little information on the toxic effects exerted by
concentrations similar to ambient levels.
10.2.7 Risk evaluation
The information available to assess the environmental hazards
presented by chlorophenols other than PCP is deficient in at least 2
respects: (a) knowledge of the quantities of chlorophenols entering
the environment, and of their subsequent dynamics, is insufficient for
all chlorophenols other than PCP; and (b) not enough toxicology
studies using concentrations characteristic of the environment have
been conducted. As a result, it is not yet possible to predict
quantitatively the environmental impact of the widespread low-level
contamination that has recently become apparent (section 5).
However, it is possible to get a first approximation of the hazard
presented by a given chlorophenol, by comparing the levels that
produce toxic effects on test organisms in vitro with the residue
concentrations that have been measured in the environment. The
information for such a comparison is presented in Table 21, using data
for aquatic organisms and environments. It is necessary to restrict
the data in this manner because the vast majority of studies on the
environmental toxicity of chlorophenols have been on aquatic test
systems.
Furthermore, this focus is appropriate because many producers and
users of chlorophenols still discharge them as wastes into water
bodies (section 3.2.3).
The environmental data in Table 21 include measured levels for
each chlorophenol in water, sediment, and effluent, since these are
the environmental sources most readily comparable with the results of
toxicity studies. The latter data are taken from the laboratory
toxicity studies outlined in section 5.1.2 and include the
concentrations that cause toxic effects, the no-observed-effect
levels, and those showing organoleptic effects in fish and
drinking-water. In order to ensure that the comparison of
environmental concentrations and toxic levels provides a margin of
safety, the environmental data used for evaluation are the maximum
values reported in highly-contaminated industrialized waters, while
the levels producing toxic effects are the minimum levels reported to
cause effects from a large number of studies compiled by Buikema et
al. (1979), and Jones (1981, 1984). Table 21 is designed so that
comparisons can be made across the rows; because of the diversity of
test conditions, organisms, and response variables, the data on the
toxicity of the individual compounds are not directly comparable.
In virtually all instances, the maximum ambient levels in water
and sediments are orders of magnitude below the lowest concentrations
that are toxic for aquatic organisms: effects typically occur in the
mg/litre range, while environmental levels are generally in the
µg/litre range. On this basis, it appears that the ambient
chlorophenol levels measured in aquatic environments are unlikely to
have adverse effects on the ecosystems receiving them, except in the
case of accidental spills, high-concentration, point-source
discharges, or in the immediate vicinity of manufacturers' undiluted
waste streams. However, the elevated concentrations found in some
industrialized regions, or in habitats adjacent to discharges, can
compromise the flavour and/or smell of drinking-water and fish.
It is advisable to control chlorophenol discharges into the
environment at levels that would not increase the present
environmental concentrations, in view of the taste and odour effects
of chlorophenols and the lack of data on the long-term effects on
ecosystems from the present low levels of chlorophenols detected.
Furthermore, the levels of such microcontaminants as PCDDs and PCDFs
in technical formulations of T3CP and T4CP should be reduced as much
as possible, in order to decrease the levels of such toxic chemicals
released into the environment.
Table 21. Maximum reported water, sediment, and effluent levels, contrasted with minimum levels producing toxic and organoleptic
effects and no-observed-effect levels
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2-MCP 2.3m 4p 2600 Daphnia 1000 Daphnia 60
magna magna
48-h LC50b survivalb
3-MCP 6.0m 43 6p 1800 Unidentified 10000 Chlorella 0.1 25
fish pyrenoidosa
24-h TLmd growthc
4-MCP 3.9m 150q 4100 Daphnia 500 Estuarine 0.1 45
magnab phytoplankton
48-h LC50 growthe
2,3-DCP 0.72m 2.2 0.04 84
Table 21. (contd).
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2,4-DCP 0.59m 10 3304 100 Crayfish 0.3-8.0 1.0
elevation of
blood-glucosef
2,5-DCP 0.29m 11 0.5 23
2,6-DCP 0.45m 31 2204 4000 Salmo trutta 0.2-2.0 35
24-h LC50g
3,4-DCP 0.23m 49 5000 Fishhdeath 0.3
3hh
3,5-DCP 0.52m 12 1500 Shrimp
96-h lethal
thresholdi
2,3,4-T3CP 0.04n 0.8 3.64 2000 Shrimp
96-h lethal
thresholdi
2,3,5-T3CP 0.28n 11 800 Salmo truttag
24-h LC50
Table 21. (contd).
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2,3,6-T3CP 0.36n 2700 Shrimp 0.5
96-h lethal
thresholdl
2,4,5-T3CP 0.66m 15 2400q 640 Palaemonetes 100 Rainbow trout 1.0 52
puglo
96-h LC50j
2,4,6-T3CP 2.5m 3.7 3120q .0.5 Guppy < 410 Daphnia 2.0
fecundity magna
offspring survival
survival
2,4,6-T3CP 2.5m 3.7 3120q > 100- Fathead minnow
< 1000 96-h TLm
Table 21. (contd).
Compound Maximum reported Lowest reported levels No-observed-effect Organoleptic
environmental levels causing toxic effects level (NOEL) threshold in water
(µg/litre)
Water Sedimentn Effluents Level Effect Level Parameter Drinking- Fish
(µg/ (µg/kg) (µg/litre) (µg/litre) (µg/litre) watera flesha
litre)
2,3,4,5-T4CP 0.02n 8.9 12q < 300 Grass shrimp
EC50-limb
regenerationj
2,3,4,6-T4CP 3r 4.9 2100q 290 Daphnia 10 Daphnia 1.0
magna magnab
48-h LC50b survival
2,3,5,6-T4CP 5r 2.8 35 570 Daphnia 10 Daphnia
magna magna
48-h LC50b survivalb
a Organoleptic threshold is for drinking-water (US EPA, 1980c) and ambient water that leads to the tainting of fish
flesh (Shumway & Palensky, 1973).
b LeBlanc (1980). j Rao et al. (1981).
c Huang & Gloyna (1968). k Virtanen & Hattula (1982).
d Ingols et al. (1966). l Barnhart & Campbell (1972).
e Erickson & Hawkins (1980). m Wegman & Hofstee (1979).
f Telford (1974). n Wegman & van den Broek (1983).
g Hattula et al. (1981b). p Jolley et al. (1975).
h Jones (1981). q Garrett (1980).
i McLeese et al. (1979). r Zoeteman et al. (1981).
11. RECOMMENDATIONS
11.1 Production
(a) The concentrations of microcontaminants in chlorophenols and
products derived from them should be determined and specified.
(b) Levels of PCDDs and PCDFs in chlorophenols and related products
should be kept as low as is technically possible.
(c) Since data on the quantities of chlorophenols produced and
consumed are not available for most countries, international agencies
should seek the assistance of industry to compile such data in
different countries.
11.2 Disposal
(a) Disposal of chlorophenols and chlorophenol-contaminated waste
should be carried out in a manner that minimizes their release into
the environment. Contaminated waste waters should undergo primary and
secondary treatment. Chlorophenols should only be incinerated at high
temperatures and under strictly controlled conditions.
(b) Contamination of surface and ground waters with chlorophenols
arising as a result of industrial chlorination processes or waste
treatments using chlorine should be avoided as far as is technically
feasible.
11.3 Occupational Exposure
(a) Work-place exposure to chlorophenols should be minimized, and
absorption of these compounds through the dermal and inhalation routes
prevented, by:
-- enclosure and automation of industrial processes that use
chlorophenols;
-- adequate ventilation of the work area;
-- provision of appropriate protective clothing for employees working
with chlorophenols;
-- provision of proper washing and laundry facilities;
-- instruction of workers in the safe use and handling of
chlorophenols, the importance of personal hygiene (washing before
eating or smoking, showering before leaving work, and daily
laundering of clothing), and the application of proper emergency
procedures;
-- provision of eating and rest areas in the work place that are
isolated from potential chlorophenol contamination.
(b) The effectiveness of measures to reduce occupational exposure
should be surveyed by monitoring both the work-place air and the urine
of the workers.
11.4 General Population Exposure
(a) The availability and use of consumer products containing
chlorophenols should be reduced wherever practicable.
(b) Products containing chlorophenols should be clearly labelled by
the manufacturer to alert the consumer to their toxicity and to
instruct consumers in the safe use and handling of these products.
(c) The use of tri- and tetrachlorophenols for wood preservation
should be avoided where such wood is to be used:
-- for shipping or storing food;
-- for the retention of soil on which food may be grown;
-- for animal housing or bedding on farms.
11.5 Recommendations for Future Research
11.5.1 Environmental Aspects
Given the continued release of chlorophenols into the environment,
research is needed to study:
(a) the transport and distribution of chlorophenols in the
environment;
(b) the effects of long-term exposure to chlorophenols on both
aquatic and terrestrial organisms at concentrations typical of the
environment;
(c) the suitability of controlled landfill sites for the disposal of
chlorophenols and related wastes;
(d) means of reducing the contribution of industrial and municipal
chlorination to the overall environmental releases of chlorophenols.
11.5.2 Toxicology
The toxicology database for chlorophenols other than PCP has major
deficiencies, particularly for tetrachlorophenols. A more accurate
estimate of the risk posed by these chemicals necessitates research
into:
(a) uptake, distribution, metabolism, and excretion of
chlorophenols, especially in man;
(b) further study of the effects of chlorophenols on reproduction;
(c) the in vivo genotoxic potential of chlorophenols;
(d) long-term carcinogenicity studies with pure and technical grade
2,4,5-T3CP, 2,4,6-T3CP, and 2,3,4,6-T4CP;
(e) the cancer-promoting potential of chlorophenols;
(f) the mechanism of toxicity of chlorophenols at the molecular
level;
(g) the extent to which the toxic effects exerted by technical grade
chlorophenols are attributable to microcontaminants;
(h) the contribution made by the biotransformation of other
chlorinated compounds (e.g., hexachlorobenzene) to the human body
burden of chlorophenols.
11.5.3 Epidemiology
(a) Epidemiological investigations of previously-studied cohorts
should be followed-up and updated.
(b) Studies on new groups of workers exposed specifically to
chlorophenols, i.e., sawmill employees working with these compounds,
should be conducted. End-points studied should not only be cancers,
but should also include pulmonary, reproductive, and other effects.
(c) The higher chlorinated PCDDs and PCDFs, which occur as
contaminants in several chlorophenols, have long half-lives in human
beings and can therefore be used as indicators of exposure to the
higher chlorophenols. Since the present contradictory results from
epidemiological studies may, in part, be because of inaccurate
information on exposure to CPs, the potential use of PCDD and PCDF
concentrations in human tissues and fluids as markers of previous
exposure to these chlorophenols should be investigated.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
A guideline value of 10 µg/litre was recommended by WHO (WHO,
1984) for 2,4,6-trichlorophenol in drinking-water, based on animal
carcinogenicity data using a conservative mathematical model. In the
supporting documentation for this guideline value (WHO, 1985), it was
noted that the taste threshold level for 2,4,6-T3CP was 1.0 µg/litre
and that, on the basis of aesthetic qualities, the level would be
0.1 µg/litre. Guideline values for other individual CPs were not set,
but the odour threshold concentration of 0.1 µg/litre was considered
appropriate for chlorophenols other than pentachlorophenol.
The carcinogenicity of 2,4,5- and 2,4,6-T3CP has been evaluated
by the International Agency for Research on Cancer (IARC, 1979, 1986).
It was concluded that: there was sufficient evidence for
carcinogenicity in animals for 2,4,6-T3CP and inadequate data for the
assessment of the carcinogenicity of 2,4,5-T3CP in animals. IARC
(1986) also concluded that there was limited evidence for the
carcinogenicity of occupational exposures to all chlorophenols for
human beings. Although not stated directly in the IARC monographs,
occupational exposures were primarily to T3CP and T4CP formulations.
Regulatory standards for chlorophenols established by national
bodies in different countries and the EEC are summarized in the Legal
File of the International Register of Potentially Toxic Chemicals
(IRPTC, 1986).
REFERENCES
ABRAHAMSSON, K. & XIE, T.M. (1983) Direct determination of trace
amounts of chlorophenols in fresh water, waste water and sea
water. J. Chromatogr., 279: 199-208.
AGRICULTURE CANADA (1987) Pentachlorophenol wood preservative,
Ottawa, Pesticides Directorate, Agriculture Canada (Discussion
Document No. 87-02).
AHEL, M., GIGER, W., MOLNAR-KUBICA, E., & SCHAFFNER, C. (1984) Organic
micropollutants in surface waters of the Glatt Valley,
Switzerland. In: Angeletti, G. & Bjorseth, A., ed. Analysis of
organic micropollutants in water. Proceedings of the 3rd European
Symposium, Oslo, 19-21 September 1983, Luxembourg, Commission of
the European Communities, pp. 280-288.
AHLBORG, U.G. & LARSSON, K. (1978) Metabolism of tetrachlorophenols in
rat. Arch. Toxicol., 40: 63-74.
AHLBORG, U.G. & THUNBERG, T. (1978) Effects of 2,3,7,8-tetra-
chlorodibenzo- p-dioxin on the in vivo and in vitro
dechlorination of pentachlorophenol. Arch. Toxicol., 40: 55-61.
AHLBORG, U.G. & THUNBERG, T.M. (1980) Chlorinated phenols: occurrence,
toxicity, metabolism, and environmental impact. CRC Crit. Rev.
Toxicol., 7(1): 1-35.
AKISADA, T. (1964) Simultaneous determination of pentachlorophenol and
tetrachlorophenol in air and urine. Jpn. Anal., 14: 101-105.
ALEXANDER, M. & ALEEM, M.I.H. (1961) Effect of chemical structure on
microbial degradation of aromatic herbicide. J. agric. food
Chem., 9(1): 44-47.
ALEXANDERSSON, R. & HEDENSTIERNA, G. (1982) Pulmonary function after
long-term exposure to trichlorophenol. Int. Arch. occup. environ.
Health, 49: 275-280.
ester derivatives in natural surface waters. J. agric. food Chem.,
12(6): 541-546.
ANGEL, A. & ROGERS, K.J. (1972) An analysis of the convulsant activity
of substituted benzenes in the mouse. Toxicol. appl. Pharmacol.,
21(2): 214-229.
APPLEGATE, V.L., HOWELL, J.H., HALL, A.E., Jr, & SMITH, M.A. (1957)
Toxicity of 4,346 chemicals to larval lampreys and fishes,
Washington, DC, US Department of the Interior, Fish and Wildlife
Service, 157 pp (Special Report, Fish No. 207).
ARRHENIUS, E., RENBERG, L., JOHANSSON, L., & ZETTERQVIST, M.-A. (1977)
Disturbance of microsomal detoxication mechanisms in liver by
chlorophenol pesticides. Chem.-biol. Interact., 18: 35-46.
ARSENAULT, R.D. (1976) Pentachlorophenol and contained chlorinated
dibenzodioxins in the environment. A study of environmental fate,
stability, and significance when used in wood preservation.
Am. Wood-Preserv. Assoc., 20: 122-148.
AXELSON, O., SUNDELL, L., ANDERSON, K., EDLING, C., & KLING, H. (1980)
Herbicide exposure and tumour mortality. Scand. J. Work Environ.
Health, 6: 73-79.
BABICH, H. & DAVIS, D.L. (1981) Phenol: a review of environmental and
health risks. Reg. Toxicol. Pharmacol., 1: 90-109.
BACON, G.B. (1978) Bioaccumulation of toxic compounds in pulpmill
effluents by aquatic organisms in receiving waters, Ottawa,
Environment Canada (Annual Report CPAR Project No. 675; draft
report No. M-79-76).
BAHIG, M.E., KRAUS, A., & KLEIN, W. (1981) Excretion and metabolism of
2,4,6-trichlorophenol-14C in rats. Chemosphere, 10(3): 323-327.
BAIRD, R.B., KUO, C.L., SHAPIRO, J.S., & YANKO, W.A. (1974) The fate
of phenolics in wastewater -- determination by direct-injection
GLC and Warburg respirometry. Arch. environ. Contam. Toxicol.,
2(2): 165-178.
BAKER, M.D. & MAYFIELD, C.I. (1980) Microbial and non-biological
decomposition of chlorophenols and phenol in soil. Water Air Soil
Pollut., 13(4): 411-424.
BAKER, M.D., MAYFIELD, C.I., & INNISS, W.E. (1980) Degradation of
chlorophenols in soil, sediment and water at low temperature.
Water Res., 14(12): 1765-1771.
BALARAJAN, R. & ACHESON, E.D. (1984) Soft tissue sarcomas in
agriculture and forestry workers. J. Epidemiol. community Health,
38: 113-116.
BALLSCHMITER, K. & SCHOLZ, C. (1980) Microbial decomposition of
chlorinated aromatic substances. VI. Formation of dichlorophenols
and dichloropyrocatechol from dichlorobenzenes in a micromolar
solution by Pseudomonas sp. Chemosphere, 9(7-8): 457-467.
BARNHART, E.L. & CAMPBELL, G.R. (1972) The effect of chlorination on
selected organic chemicals, Washington, DC, US Environmental
Protection Agency, 105 pp (Water Pollution Control Research Series
No. PB 211-160).
BARROWS, M.E., PETROCELLI, S.R., MACEK, K.J., & CARROLL, K.J. (1980)
Bioconcentration and elimination of selected water pollutants by
bluegill sunfish (Lepomis macrochirus). In: Haque, R., ed.
Dynamics exposure and hazard assessment of "toxic chemicals",
Ann Arbor, Michigan, Ann Arbor Science Publishers Inc.,
pp. 379-392.
BEHRBOHM, P. (1959) [On the hazards of exposure to chlorinated
phenols.] Dtsch. Gesundheitswes., 14: 614-619 (in German).
BELTRAME, P., BELTRAME, P.L., & CARNITI, P. (1984) Inhibiting action
of chloro- and nitrophenols on biodegradation of phenols: a
structure-toxicity relationship. Chemosphere, 13(1): 3-9.
BENOIT-GUYOD, J.-L., ANDRE, C., TAILLANDIER, G., ROCHAT, J., &
BOUCHERLE, A. (1984) Toxicity and QSAR of chlorophenols on
Lebistes reticulatus. Ecotoxical. environ. Saf., 8: 227-235.
BEVENUE, A. & BECKMAN, H. (1967) Pentachlorophenol: a discussion of
its properties and its occurrence as a residue in human and animal
tissues. Residue Rev., 19: 83-134.
BEVENUE, A., OGATA, J.N., & HYLIN, J.W. (1972) Organochlorine
pesticides in rainwater, Oahu, Hawaii, 1971-1972. Bull. environ.
Contam. Toxicol., 8(4): 238-241.
BLACKBURN, K., ZENICK, H., HOPE, E., MANSON, J.M., GEORGE, E.L., &
SMITH, M.K. (1986) Evaluation of the reproductive toxicology of
2,4,6-trichlorophenol in male and female rats. Fundam. appl.
Toxicol., 6(2): 233-239.
BLACKMAN, G.E., PARKE, M.A., & GARTON, F. (1955) The physiological
action of substituted phenols. I. Relationships between chemical
structure and physiological activity. Arch. Biochem. Biophys.,
54(1): 45-54.
BLADES-FILLMORE, L.A., CLEMENT, W.H., & FAUST, S.D. (1982) The effect
of sediment on the biodegradation of 2,4,6-trichlorophenol in
Delaware River water. J. environ. Sci. Health, A17(6): 797-818.
BLANK, C.E., COOKE, P., & POTTER, A.M. (1983) Investigations for
genotoxic effects after exposure to crude 2,4,5-trichlorophenol.
Br. J. ind. Med., 40: 87-91.
BLEIBERG, J., WALLEN, M., BRODKIN, R., & APPLEBAUM, I.L. (1964)
Industrially acquired porphyria. Arch. Dermatol., 89: 793-797.
BOETIUS, J. (1954) Foul taste of fish and oysters caused by
chlorophenol. Medd. Dan. Fisk. Havunders. N.S., 1(4): 3-8.
BOLLAG, J.-M., HELLING, C.S., & ALEXANDER, M. (1968) 2,4-D metabolism:
enzymatic hydroxylation of chlorinated phenols. J. agr. food
Client., 16(5): 826-828.
BOND, G.G., WETTERSTROEM, N.H., ROUSH, G.J., MCLAREN, E.A., LIPPS,
T.E., & COOK, R.R. (1988) Cause-specific mortality among employees
engaged in the manufacture, formulation, or packaging of
2,4-dichlorophenoxyacetic acid and related salts. Br. J. ind.
Med., 45(2): 98-105.
BORZELLECA, J.F., HAYES, J.R., CONDIE, L.W., & EGLE, J.L., Jr (1985a)
Acute toxicity of monochlorophenols, dichlorophenols and
pentachlorophenol in the mouse. Toxicol. Lett., 29(1): 39-42.
BORZELLECA, J.F., HAYES, J.R., CONDIE, L.W., & EGLE, J.L., Jr (1985b)
Acute and subchronic toxicity of 2,4-dichlorophenol in CD-1 mice.
Fundam. appl. Toxicol., 5(3): 478-486.
BOULE, P., GUYON, C., & LEMAIRE, J. (1982) Photochemistry and
environment. IV. Photochemical behaviour of monochlorophenols in
dilute aqueous solution. Chemosphere, 11(12): 1179-1188.
BOULE, P., GUYON, C., & LEMAIRE, J. (1984) Photochimie et
environnement. VIII. Comportement photochimique de dichlorophénols
en solution aqueuse diluée. Chemosphere, 13: 603-612.
BOUTWELL, R.K. & BOSCH, D.K. (1959) The tumour-promoting action of
phenol and related compounds for mouse skin. Cancer Res.,
19: 413-424.
BOYD, S.A. & SHELTON, D.R. (1984) Anaerobic biodegradation of
chlorophenols in fresh and acclimated sludge. Appl. environ.
Microbiol., 47(2): 272-277.
BRAUN, W.H., BLAU, G.E., & CHENOWETH, M.B. (1978) The
metabolism/pharmacokinetics of pentachlorophenol in man, and a
comparison with the rat and monkey model. Toxicol. appl.
Pharmacol., 45: 278.
BRAUN, W.H., BLAU, G.E., & CHENOWETH, M.B. (1979) The metabolism/
pharmacokinetics of pentachlorophenol in man, and a comparison
with the rat and monkey. In: Deichmann, W.B., ed. Toxicology and
occupational medicine, New York, Amsterdam, Oxford, Elsevier/
North-Holland, pp. 289-296.
BRISTOL, D.W., COOK, L.W., KOTERBA, M.T., & NELSON, D.C. (1982)
Determination of free and hydrolyzable residues of 2,4-dichloro-
phenoxyacetic acid and 2,4-dichlorophenol in potatoes. J. agric.
food Chem., 30(1): 137-144.
BROECKER, B. & ZAHN, R. (1977) The performance of activated sludge
plants compared with the results of various bacterial toxicity
tests a study with 3,5-dichlorophenol. Water Res., 11: 165-172.
BUCCAFUSCO, R.J., ELLS, S.J., & LEBLANC, G.A. (1981) Acute toxicity of
priority pollutants to bluegill (Lepomis macrochirus). Bull.
environ. Contam. Toxicol., 26: 446-452.
BUIKEMA, A.L., Jr, MCGINNISS, M.J., & CAIRNS, J., Jr (1979) Phenolics
in aquatic ecosystems: a selected review of recent literature.
Mar. environ. Res., 2: 87-181.
BUSER, H.-R. & BOSSHARDT, H.P. (1976) Determination of polychlorinated
dibenzo- p-dioxins and dibenzofurans in commercial
pentachlorophenols by combined gas chromatography-mass
spectrometry. J. Assoc. Off. Anal. Chem., 59(3): 562-569.
BUTTE, W. (1984) Determination of free and total pentachlorophenol in
urine. Fresenius Z. anal. Chem., 317: 659.
BUTTE, W., KIRSCH, M., & DENKER, J. (1983) The determination of
pentachlorophenol and tetrachlorophenols in Wadden sediment and
clams (Mya arenia) using triethylsulfonium hydroxide for
excretion and pyrolytic ethylation. Int. J. environ. anal. Chem.,
13: 141-153.
BUTTE, W., DENKER, J., KIRSCH, M., & HOPNER, Th. (1985)
Pentachlorophenol and tetrachlorophenols in Wadden sediment and
clams (Mya arenia) of the Jadebusen after a 14-year period of
wastewater discharge containing pentachlorophenol. Environ.
Pollut., B9: 29-39.
CALL, D.J., BROOKE, L.T., & LU, P.-Y. (1980) Uptake, elimination, and
metabolism of three phenols by fathead minnows. Arch. environ.
Contam. Toxicol., 9: 699-714.
CALLAGHER, R.P. & THRELFALL, W.J. (1984) Cancer and occupational
exposure to chlorophenols. Lancet, II: 48.
CALLAGHER, R.P. & THRELFALL, W.J. (1985) Cancer risk in wood and pulp
workers. In: Stich, H.F., ed. Carcinogens and mutagens in the
environment, Boca Raton, Florida, CRC Press, Vol. 5,
pp. 125-137.
CALLAHAN, M.A., SLIMAK, M.W., GABEL, N.W., MAY, I.P., FOWLER, C.F.,
FREED, J.R., JENNINGS, P., DURFEE, R.L., WHITMORE, F.C., MAESTRI,
B., MABEY, W.R., HOLT, B.R., & GOULD, C. (1979). Water-related
environmental fate of 129 priority pollutants, Washington, DC,
US Environmental Protection Agency, Vol. II (EPA-440/4-79-029b).
CANADA-ONTARIO REVIEW BOARD (1981) Environmental baseline report of
the Niagara River, Ottawa, Environment Canada (November 1981
Update).
CARAMASCHI, F., MONTESARCHIO, E., DEL CORNO, G., FAVARETTI, C.,
GIAMBELLUCA, S.E., BONETTI, F., & VOLPATO, C. (1982) Analysis of
exposure to environmental contamination by TCDD in individuals
affected by dermatological lesions. Ig. mod., 77: 681-706.
CARLSON, G.P. (1978) Effect of trichlorophenols on xenobiotic
metabolism in the rat. Toxicology, 11: 145-151.
CAUTREELS, W., VAN CAUWENBERGHE, K., & GUZOLAN, L.A. (1977) Comparison
between the organic fraction of suspended matter at a background
and an urban station. Sci. total Environ., 8: 79-88.
CEDAR, F.J. (1984) Historical and current developments of antisapstain
chemical use. In: Proceedings of a Symposium on Preservatives in
the Wood Industry, 16-17 May, 1984, Vancouver, Division of
Occupational and Environmental Health, Department of Health Care
and Epidemiology, University of British Columbia.
CHADWICK, R.W. & FREAL, J.J. (1972) The identification of five
unreported lindane metabolites recovered from rat urine. Bull.
environ. Contam. Toxicol., 7(2/3): 137-146.
CHATTERJEE, R.M. (1974) Fish tainting in the Upper Great Lakes,
Ottawa, Ontario Ministry of Environment, 31 pp (Water Resources
Branch Report).
CHIOU, C.T., FREED, V.H., PETERS, L.J., & KOHNERT, R.L. (1980)
Evaporation of solutes from water. Environ. Int., 3: 231-236.
CHOI, J. & AOMINE, S. (1972) Effects of the soil on the activity of
pentachlorophenol. Soil Sci. plant Nutr., 18(6): 255-260.
CHOI, J. & AOMINE, S. (1974) Adsorption of pentachlorophenol by soils.
Soil Sci. plant Nutr., 20(2): 135-144.
CHRISTENSEN, E.R. & NYHOLM, N. (1984) Ecotoxicological assays with
algae: Weibull dose-response curves. Environ. Sci. Technol.,
18(9): 713-718.
CHRISWELL, C.D, CHANG, R.C, & FRITZ, J.S. (1975) Chromatographic
determination of phenols in water. Anal. Chem.,
47(8): 1325-1329.
CHU, J. & KIRSCH, E.J. (1973) Utilization of halophenols by a
pentachlorophenol metabolizing bacterium. Der. bid. Microbiol,
14: 264-273.
CLARK, D.E., PALMER, J.S., RADELEFF, R.D., CROOKSHANK, H.R., & FARR,
F.M. (1975) Residues of chlorophenoxy acid herbicides and their
phenolic metabolites in tissues of sheep and cattle. J. agric.
food Chem., 23(3): 573-578.
COCHRANE, W.P., LANOUETTE, M, & SINGH, J. (1983) High pressure liquid
chromatographic determination of impurity phenols in technical
2,4-D acid and 2,4-dichlorophenol. J. Assoc. Off. Anal. Chem.,
66(3): 804-809.
COGGON, D., PANNETT, B., WINTER, P.D.., ACHESON, E.D, & BONSALL, J.
(1986) Mortality of workers exposed to 2 methyl-4-chloropheno-
xyacetic acid. Scand. J. Work Environ. Health, 12: 448-454.
COOK, R.R. (1981) Dioxin, chloracne and soft-tissue sarcoma. Lancet,
1: 618-619.
COOK, R.R., TOWNSEND, J.C., OTT, M.G., & SILVERSTEIN, L.G. (1980)
Mortality experience of employees exposed to 2,3,7,8-tetra-
chlorodibenzo- p-dioxin (TCDD). J. occup. Med., 22(8): 530-532.
COOK, R.R., BOND, G.G., OLSON, R.A., & OTT, M.G. (1987) Update of the
mortality experience of workers exposed to chlorinated dioxins.
Chemosphere, 16: 2111-2116.
CORDDRY, A.E. (1981) A pregnancy outcome study of the wives of
workers exposed to chlorophenate wood preservatives at a sawmill,
Seattle, University of Washington, Department of Environmental
Health, 191 pp (Thesis).
CROSBY, D.G. & TUTASS, H.O. (1966) Photodecomposition of
2,4-dichlorophenoxyacetic acid. J. agric. food Chem.,
14(6): 596-599.
CROSBY, D.G. & WONG, A.S. (1973) Photodecomposition of
2,4,5-trichlorophenoxyacetic acid (2,4,5-T) in water. J. agric.
food Chem., 21(6): 1052-1054.
CROSBY, D.G., BEYNON, K.I., GREVE, P.A., KORTE, F., STILL, G.G., &
VOUK, J.W. (1981) Environmental chemistry of chlorophenol. A
special report on pentachlorophenol in the environment. Pure.
appl. Chem., 53: 1051-1080.
CURRIE, K. & MCDONALD, E. (1986) Uptake/excretion of chlorophenols by
sawmill employees. Phase II, Ottawa, Health and Welfare Canada
(B.C. Research Report to Pesticides Division; Contract No. 1424).
CURTIS, R.F., LAND, D.G., GRIFFITHS, N.M., GEE, M., ROBINSON, D.,
PEEL, J.L., DENNIS, C., & GEE, J.M. (1972) 2,3,4,6-Tetrachloro-
anisole association with musty taint in chickens and
microbiological formation. Nature (Lond.), 235: 223-224.
DAHMS, A. & METZNER, W. (1979) [Analysis of pentachlorophenol and
tetrachlorophenol in air and urine.] Holz. Roh-Werkstoff,
37: 331-344 (in German).
DANIELS, C.R. & SWAN, E.P. (1979) Determination of chlorinated phenols
in surface-treated lumber by HPLC. J. chromatogr. Sci.,
17: 628-630.
DE KREUK, J.F. & HANTSVEIT, A.O. (1981) Determination of the
biodegradability of the organic fraction of chemical wastes.
Chemosphere, 10(6): 561-573.
DELAUNE, R.D., GAMBRELL, R.P., & REDDY, K.S. (1983) Fate of PCP in
estuarine sediment. Environ. Pollut., B6: 297-308.
DEL CORNO, G., FAVARETTI, C., CARAMASCHI, F., GIAMBELLUCA, S.E.,
MONTESARCHIO, E., BONETTI, F., & VOLPATO, C. (1982) Distribution
of chloracne cases in the Seveso area following contamination by
TCDD. Ig. mod., 77: 635-658.
DEVILLERS, J. & CHAMBON, P. (1986) Acute toxicity and QSAR of
chlorophenols on Daphnia magna. Bull. environ. Contam. Toxicol.,
37: 599-605.
DIETZ, F. & TRAUD, J. (1978) [Trace analysis of phenols, especially
chlorophenols in water, by gas chromatography: methods and
results.] Vom Wasser, 51: 235-237 (in German with English
summary).
DOEDENS, J.D. (1963) Chlorophenols. In: Kirk-Othmer Encyclopedia of
chemical technology, 2nd ed., New York, John Wiley and Sons,
Vol. 5, pp. 325-338.
DUST, J.V. & THOMPSON, W.S. (1973) Pollution control in the
wood-preserving industry. Part IV. Biological methods of treating
wastewater. Forest. Prod. J., 23(9): 59-66.
DUTKA, B.J. & KWAN, K.K. (1984) Studies in a synthetic activated
sludge toxicity screening procedure with comparison to three
microbial toxicity tests. In: Liu, D. & Dutka, B.J., ed. Toxicity
screening procedures using bacterial systems, New York, Basel,
Marcel Dekker Inc., pp. 125-138.
DUXBURY, J.M., TIEDJE, J.M., ALEXANDER, M., & DAWSON, J.E. (1970)
2,4-D metabolism: enzymatic conversion of chloromaleylacetic acid
to succinic acid. J. agric. food Chem., 18(2): 199-201.
EDER, G. & WEBER, K. (1980) Chlorinated phenols in sediments and
suspended matter of the Weser estuary. Chemosphere,
9(2): 111-118.
EDGERTON, T.R., MOSEMAN, R.F., LINDER, R.E., & WRIGHT, L.H. (1979)
Multi-residue method for the determination of chlorinated phenol
metabolites in urine. J. Chromatogr., 170: 331-342.
EDGERTON, T.R., MOSEMAN, R.F., LORES, E.M., & WRIGHT, L.H. (1980)
Determination of trace amounts of chlorinated phenols in human
urine by gas chromatography. Anal. Chem., 52: 1774-1777.
EICHELBERGER, J.W., DRESSMAN, R.C., & LONGBOTTOM, J.E. (1970)
Separation of phenolic compounds from carbon chloroform extract
for individual chromatographic identification and measurement.
Environ. Sci. Technol., 4(7): 576-578.
ELDER, V.A., PROCTOR, B.L., & HITES, R.A. (1981) Organic compounds
found near dumpsites in Niagara Falls, New York. Environ. Sci.
Technol., 15(10): 1237-1243.
EL-GOHARY, F.A. & NASR, F.A. (1984) Fate and effect of
2,4-dichlorophenol on biological treatment processes. Environ.
Int., 10: 211-215.
EMBREE, V, ENARSON, D.A., CHAN-YEUNG, M, DYBUNCIO, A., DENNIS, R., &
LEACH, J. (1984) Occupational exposure to chlorophenates:
toxicology and respiratory effects. Clin. Toxicol,
22(4): 317-329.
ENARSON, D.A., CHAN-YEUNG, M., EMBREE, V., WANG, R., & SCHULZER, M.
(1986) Occupational exposure to chlorophenates: Renal, hepatic and
other health effects. Scand. J. Work Environ. Health,
12: 144-148.
ENGST, R., MACHOLZ, R.M., KUJAWA, M., LEWERENZ, H.-J., & PLASS, R.
(1976) The metabolism of lindane and its metabolites
gamma-2,3,4,5,6-pentachlorocyclohexane, pentachlorobenzene, and
pentachlorophenol in rats and the pathways of lindane metabolism.
J. environ. Sci. Health, B11(2): 95-17.
ENVIRONMENT CANADA (1979) Monitoring environmental contamination from
chlorophenol contaminated wastes generated in the wood
preservation industry, Ottawa, Environmental Protection Branch,
Environmental Protection Service, Pacific and Yukon Region,
Environment Canada (Regional Program Report No. 79-24).
ENVIRONMENT CANADA (1986) Dioxin-containing chemicals: sales and
imports. 1985 Update, Ottawa, Commercial Chemicals Branch,
Environmental Protection Service, Environment Canada.
ERICKSON, S.J. & HAWKINS, C.E. (1980) Effects of halogenated organic
compounds on photosynthesis in estuarine phytoplankton. Bull.
environ. Contam. Toxicol., 24: 910-915.
ERIKSSON, M., HARDELL, L., BERG, N.O., MOLLER, T., & AXELSON, O.
(1981) Soft-tissue sarcomas and exposure to chemical substances: a
case-referent study. Br. J. ind. Med., 38: 27-33.
ERNST, W. (1979) Factors affecting the evaluation of chemicals in
laboratory experiments using marine organisms. Ecotoxicol.
environ. Saf, 3: 90-98.
ERNST, W. & WEBER, K. (1978) Chlorinated phenols in selected estuarine
bottom fauna. Chemosphere, 7(11): 867-872.
ETTINGER, M.B. & RUCHHOFT, C.C. (1950) Persistence of
monochlorophenols in polluted river water and sewage dilutions.
Presented at the Annual Meeting of the Central States Sewage Works
Association, Cincinnati, Environmental Health Centre, 11 pp
(PB 215-498).
ETZEL, J.E. & KIRSCH, E.J. (1974) Biological treatment of contrived
and industrial wastewater containing pentachlorophenol. Dev. ind.
Microbiol., 16: 287-295.
EXON, J.H. (1984) A review of chlorinated phenols. Vet. hum.
Toxicol., 26(6): 508-520.
EXON, J.H. & KOLLER, L.D. (1981) Alteration of transplacental
carcinogenesis by chlorinated phenols. In: Jolley, R.L., Brungs,
W.A., Cotruvo, J.A., Cumming, R.B., Mattice, J.S., & Jacobs, V.A,
ed. Water chlorination: Environmental impact and health effects,
Ann Arbor, Michigan, Ann Arbor Science Publishers, Vol. 4, Book 2,
pp. 1177-1188.
EXON, J.H. & KOLLER, L.D. (1983) Effects of chlorinated phenols on
immunity in rats. Int. J. Immunopharmacol., 5(2): 131-136.
EXON, J.H. & KOLLER, L.D. (1985) Toxicity of 2-chlorophenol,
2,4-dichlorophenol, and 2,4,6-trichlorophenol. In: Jolley, R.L,
Bull, R.J., Davis, W.P., Katz, S., Roberts, M.H., Jr, & Jacobs,
V.H., ed. Water chlorination: Environmental impact and health
effects, Chelsea, Michigan, Lewis Publishers Inc., Vol. 5,
pp. 307-330.
EXON, J.H., HENNINGSEN, G.M., OSBORNE, C.A., & KOLLER, L.D. (1984)
Toxicologic, pathologic, and immunotoxic effects of
2,4-dichlorophenol in rats. J. Toxicol. environ. Health,
14: 723-730.
FAHRIG, R., NILSSON, C.-A., & RAPPE, C. (1978) Genetic activity of
chlorophenols and chlorophenol impurities. In: Rao, K.R., ed.
Pentachlorophenol: Chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 325-338.
FARQUHARSON, M.E., CAGE, J.C., & NORTHOVER, J. (1958) The biological
action of chlorophenols. Br. J. Pharmacol., 13: 20-24.
FARRINGTON, D.S. & MUNDAY, J. W. (1976) Determination of trace amounts
of chlorophenols by gas-liquid chromatography. Analyst,
101: 639-643.
FENSKE, R.A., HORSTMAN, S.W., & BENTLEY, R.K. (1987) Assessment of
dermal exposure to chlorophenols in lumber mills. Appl. ind.
Hyg., 2(4): 143-147.
FINGERHUT, M.A., HALPERIN, W.E., HONCHAR, P.A., SMITH, A.B., GROTH,
D.H. & RUSSELL, W.O. (1984) An evaluation of reports of dioxin
exposure and soft-tissue sarcoma pathology among chemical workers.
Scand. J. Work Environ. Health, 10: 299-303.
FIRESTONE, D., RESS, J., BROWN, N.L., BARRON, R.P., & DAMICO, J.N.
(1972) Determination of polychlorodibenzo- p-dioxins and related
compounds in commercial chlorophenols. J. Assoc. Off. Anal.
Chem., 55(1): 85-92.
FOLKE, J. (1984) Organic micropollutants in the sewage from a
phenoxyalkanoic acid production plant after biological treatment
in a municipal trickling filters plant. J. high Resolut.
Chromatogr. Chromatogr. Commun., 7: 25-32.
FOLKE, J. & BIRKLUND, J. (1986) Danish coastal water levels of
2,3,4,6-tetrachlorophenol, pentachlorophenol, and total
organohalogens in blue mussels (Mytilus edulis). Chemosphere,
15(7): 895-900.
FOLKE, J., BIRKLUND, J., SORENSEN, A.K., & LUND, U. (1984) The impact
on the ecology of polychlorinated phenols and other organics
dumped at the bank of a small marine inlet. In: Angeletti, G. &
Bjorseth, A., ed. Analysis of organic micropollutants in water.
Proceedings of the 3rd European Symposium, Oslo, 19-21 September,
1983, Luxembourg, Commission of the European Communities,
pp. 242-254.
FOX, M.E. & JOSHI, S.R. (1984) The fate of pentachlorophenol in the
Bay of Quinte, Lake Ontario. J. Great Lakes Res.,
10(2): 190-196.
FRANK, R., BRAUN, H.E., HOLDRINET, M., SIRONS, G.J, SMITH, E.H., &
DIXON, D.W. (1979) Organochlorine insecticides and industrial
pollutants in the milk supply of Southern Ontario, Canada 1977.
J. food Prot., 42(1): 31-37.
FREITAG, D., GEYER, H., KRAUS, A., VISWANATHAN, R., KOTZIAS, D, ATTAR,
A., KLEIN, W., & KORTE, F. (1982) Ecotoxicological profile
analysis. VII. Screening chemicals for their environmental
behaviour by comparative evaluation. Ecotoxicol. environ. Saf.,
6(1): 60-81.
FREITER, E.R. (1979) Chlorophenols. In: Kirk-Othmer Encyclopedia of
chemical technology, 3rd ed., New York, John Wiley & Sons,
Vol. 5, pp. 864-872.
FUJITA, T., IWASA, J., & HANSCH, C. (1964) A new substituent constant,
pi, derived from partition coefficients. J. Am. Chem. Soc.,
86: 5175-5180.
GALLAGHER, R.P. & THRELFALL, W.J. (1984) Cancer and occupational
exposure to chlorophenols. Lancet, II(8393): 48.
GALLOWAY, S.M., ARMSTRONG, M.J., REUBEN, C., COLMAN, S., BROWN, B.,
CANNON, C., BLOOM, A.D., NAKAMURA, F., AHMED, M., DUK, S., RIMPO,
J., MARGOLIN, B.H., RESNICK, M.A., ANDERSEN, B., & ZEIGER, E.
(1987) Chromosome aberrations and sister chromatid exchanges in
Chinese hamster ovary cells: evaluations of 108 chemicals.
Environ. mol. Mutagenesis., 10(10): 1-175.
GARRETT, C.L. (1980) Fraser river estuary study: Toxic organic
contaminants, Ottawa, Environmental Protection Service, Pacific
and Yukon Region, Environment Canada, 123 pp (Water Quality
Series).
GEE, J.M. & PEEL, J.L. (1974) Metabolism of 2,3,4,6-tetrachlorophenol
by microorganisms from broiler house litter. J. gen. Microbiol.,
88: 237-243.
GERSICH, F.M. & MILAZZO, D.P. (1988) Chronic toxicity of aniline and
2,4-dichlorophenol to Daphnia magna Straus. Bull environ.
Contam. Toxicol., 40: 1-7.
GILIOLI, R., GENTA, P., COTRONEO, L., & CANNATELLI, P. (1983)
Electroencephalographic spectral analysis in chemical and
engineering workers. In: Gilioli, R., Cassitto, M.G., & Foa, V.,
ed. Neurobehavioral methods in occupational health: advances in
the bioscience, Oxford, Pergamon Press, pp. 219-229.
GOMEZ-CATALAN, J., TO-FIGUERAS, J., PLANAS, J., RODAMILANS, M., &
CORBELLA, J. (1987) Pentachlorophenol and hexachlorobenzene in
serum and urine of the population of Barcelona. Hum. Toxicol.,
6: 397-400.
GOSSELIN, R.E, HODGE, H.C., SMITH, R.P., & GLEASON, M.N. (1976)
Clinical toxicology of commercial products, 4th ed., Baltimore,
Maryland, Williams and Wilkins Co., pp. 130-132.
GREENBERG, A.T., TRUSSEL, R.R., & CLESCERI, L.S., ed. (1985) Standard
methods for the examination of water and wastewater, 16th ed.,
Washington, DC, American Public Health Association, pp. 556-570.
GREENWALD, P., KOVASNAY, B., COLLINS, D.N., & THERRIAULT, G. (1984)
Sarcomas of soft tissues after Vietnam service. J. Natl Cancer
Inst., 73: 1107-1109.
GUROVA, A.I. (1964) Hygienic characteristics of p-chlorophenol in the
aniline dye industry. Hyg. Sanit., 29(10): 46-51.
GUTHRIE, M.A., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C.P.L., Jr (1984)
Pentachlorophenol biodegradation. II. Anaerobic. Water Res.,
18(4): 451-461.
HAGENMAIER, H. (1986) Determination of 2,3,7,8-tetrachlorodibenzo-
p-dioxin in commercial chlorophenols and related products.
Fresenius Z. anal Chem., 325: 603-606.
HAGENMAIER, H. & BRUNNER, H. (1987) Isomers specific analysis of
pentachlorophenol and sodium pentachlorophenate for 2,3,7,8-
substituted PCDD and PCDF at sub-ppb levels. Chemosphere,
16: 1759-1764.
HAKULINEN, R. & SALKINOJA-SALONEN, M. (1982) Treatment of pulp and
paper industry waste waters in an anaerobic fluidized bed reactor.
Process Biochem., 314: 18-22.
HARDELL, L. & ERIKSSON, M. (1988) The association between soft tissue
sarcomas and exposure to phenoxyacetic acids: A new case-referent
study. Cancer, 62: 652-656.
HARDELL, L. & SANDSTROM, A. (1979) Case-control study: soft-tissue
sarcomas and exposure to phenoxyacetic acids or chlorophenols.
Br. J. Cancer, 39: 711-717.
HARDELL, L. (1981) Relation of soft-tissue sarcoma, malignant lymphoma
and colon cancer to phenoxy acids, chlorophenols and other agents.
Scand. J. Work Environ. Health, 7: 119-130.
HARDELL, L., ERIKSSON, M., LENNER, P., & LUNDGREN, E. (1981) Malignant
lymphoma and exposure to chemicals, especially organic solvents,
chlorophenols and phenoxy acids: a case-control study. Br. J.
Cancer, 43: 169-176.
HARDELL, L., JOHANSSON, B., & AXELSON, O. (1982) Epidemiological study
of nasal and nasopharyngeal cancer and their relation to phenoxy
acid or chlorophenol exposure. Am. J. ind. Med., 3: 247-257.
HARDELL, L., BENGTSSON, N.O., JONSSON, U., ERIKSSON, S., & LARSSON,
L.G. (1984) Aetiological aspects on primary liver cancer with
special regard to alcohol, organic solvents and acute intermittent
pophyria: an epidemiological investigation. Br. J. Cancer,
50: 389-397.
HARGESHEIMER, E.E. & COUTTS, R.T. (1983) Selected ion mass
spectrometric identification of chlorophenol residues in human
urine. J. Assoc. Off. Anal. Chem., 66(1): 13-21.
HATTULA, M.L., WASENIUS, V.-M., KREES, R, ARSTILA, A.U., & KIHLSTROM,
M. (1981a) Acute and short-term toxicity of
2,3,4,6-tetrachlorophenol in rats. Bull. environ. Contam.
Toxicol., 26: 795-800.
HATTULA, M.L., WASENIUS, V.-M., REUNANEN, H., & ARSTILA, A.U. (1981b)
Acute toxicity of some chlorinated phenols, catechols and cresols
to trout. Bull. environ. Contam. Toxicol., 26: 295-298.
HATTULA, M.L. & KNUUTINEN, J. (1985) Mutagenesis of mammalian cells in
culture by chlorophenols, chlorocatechols, and chloroguaiacols.
Chemosphere, 14: 1617-1625.
HAWORTH, S., LEWLOR, T., MORTELMANS, K., SPECK, W., & ZEIGER, E.
(1983) Salmonella mutagenicity test results for 250 chemicals.
Environ. Mutagen., 1 (Suppl.): 3-142.
HAY, A. (1976) Toxic cloud over Seveso. Nature (Lond.),
262: 636-638.
HEITMULLER, P.T., HOLLISTER, T.A., & PARRISH, P.R. (1981) Acute
toxicity of 54 industrial chemicals to sheepshead minnows
(Cyprinodon variegatus). Bull. environ. Contam. Toxicol.,
27: 596-604.
HERNBERG, S., WESTERHOLM, P., SCHULTZ-LARSEN, K., DEGERTH, R., KUOSMA,
E., ENGLUND, A., ENGZELL, U., HANSEN, H.S., & MUTANEN, P. (1983)
Nasal and sinonasal cancer: connection with occupational exposures
in Denmark, Finland, and Sweden. Scand. Z Work Environ. Health,
9: 315-326.
HICKMAN, G.T. & NOVAK, J.T. (1984) Acclimation of activated sludge to
pentachlorophenol. J. Water Pollut. Control Fed., 56: 364-369.
HOAR, S.K., BLAIR, A., HOLMES, F.F., BOYSEN, C.D., ROBEL, R.J.,
HOOVER, R., & FRAUMENI, J.F. (1986) Agricultural herbicide use and
risk of lymphoma and soft-tissue sarcoma. J. Am. Med. Assoc.,
256: 1141-1147.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976a) A study of
inhalation of pentachlorophenol by rats. IV. Distribution and
excretion of inhaled pentachlorophenol. Bull. environ. Contam.
Toxicol., 15(4): 466-474.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976b) A study of
inhalation of pentachlorophenol by rats. III. Inhalation toxicity
study. Bull. environ. Contam. Toxicol., 15(4): 463-465.
HOGSTED, C. & WESTERLUND, B. (1980) [Cohort study of mortality among
forestry workers with and without exposure for phenoxy
herbicides.] Läkartidningen, 77: 1828-1831 (in Swedish).
HOLCOMBE, G.W., PHIPPS, G.L., & FIANDT, J.T. (1982) Effects of phenol,
2,4-dimethylphenol, 2,4-dichlorophenol, and pentachlorophenol on
embryo, larval, and early-juvenile fathead minnows (Pimephales
promelas). Arch. environ. Contam. Toxicol., 11: 73-78.
HONCHAR, P.A. & HALPERIN, W.E. (1981) 2,4,5-T, trichlorophenol, and
soft tissue sarcoma. Lancet, 1: 268-269.
HOROWITZ, A., SHELTON, D.R., CORNELL, C.P., & TIEDJE, J.M. (1982)
Anaerobic degradation of aromatic compounds in sediments and
digested sludge. In: Proceedings of the 38th General Meeting of
the Society of Industrial Microbiology, Richmond, Virginia, 9-14
August, 1981, pp. 435-444.
HUANG, J. & GLOYNA, E.F. (1968) Effect of organic compounds on
photosynthetic oxygenation. I. Chlorophyll destruction and
suppression of photosynthetic oxygen production. Water Res.,
2: 347-366.
HUQ, A.S, HO, N.F.H., HUSARI, N., FLYNN, G.L., JETZER, W.E., & CONDIE,
L, Jr (1986) Permeation of water contaminative phenols through
hairless mouse skin. Arch. environ. Contam. Toxicol.,
15: 557-566.
HWANG, H.-M., HODSON, R.E., & LEE, R.F. (1986) Degradation of phenol
and chlorophenols by sunlight and microbes in estuarine waters.
Environ. Sci. Technol., 20(10): 1002-1007.
IARC (1979) Trichlorophenols. In: Some halogenated hydrocarbons,
Lyons, International Agency for Research on Cancer, pp. 349-367
(IARC Monographs on the Evaluation of the Carcinogenicity Risk of
Chemicals to Humans, Vol. 20).
IARC (1986) Occupational exposures to chlorophenols. In: Some
halogenated hydrocarbons and pesticide exposures, Lyons,
International Agency for Research on Cancer, pp. 319-356 (IARC
Monographs on the Evaluation of the Carcinogenicity Risk of
Chemicals to Humans, Vol. 41).
IARC (1987) Overall evaluations of carcinogenicity: an update of IARC
Monographs, Volumes 1 to 42, Lyons, International Agency for
Research on Cancer, pp. 154-156 (IARC Monographs, Supplement 7).
IDE, A., NIKI, Y., SAKAMOTO, F., WATANABE, I., & WATANABE, H. (1972)
Decomposition of pentachlorophenol in paddy soil. Agric. biol.
Chem., 36(11): 1937-1944.
INDORATO, A.M., SNYDER, K.B., & USINOWlCZ, P.J. (1984) Toxicity
screening using Microtox(R) analyzer. In: Liu, D. & Dutka, B.J.,
ed. Toxicity screening procedures using bacterial systems, New
York, Basel, Marcel Dekker Inc., pp. 37-53.
INGOLS, R.S., GAFFNEY, P.E., & STEVENSON, P.C. (1966) Biological
activity of halophenols. J. Water Pollut. Control Fed.,
38(4): 629-635.
INNES, J.R.M., ULLAND, B.M., VALERIO, M.G., PETRUCELLI, L., FISHBIEN,
L., HART, E.R., PALLOTTA, A.J., BATES, R.R., FALK, H.L., GART,
J.J., KLEIN, M., MITCHELL, I., & PETERS, J. (1969) Bioassay of
pesticides and industrial chemicals for tumorigenicity in mice: A
preliminary note. J. Natl Cancer Inst., 42: 1101-1114.
IRPTC (1986) IRPTC Legal file on chlorophenols, Geneva,
International Register of Potentially Toxic Chemicals, United
Nations Environment Programme.
JIRASEK, L, KALENSKY, J., KUBEC, K., PAZDEROVA, J., & LUKAS, E. (1974)
[Acne chlorina, porphyria cutanea tarda, and other manifestations
of general poisoning during the manufacture of herbicides.] Cesk.
Dermatol., 49: 145-157 (in Czechoslovakian with English
summary).
JOHNSON, F.E., KUGLER, M.A, & BROWN, S.M. (1981) Soft-tissue sarcomas
and chlorinated phenols. Lancet, 7: 40.
JOLLEY, R.L., JONES, G., PITT, W.W., & THOMPSON, J.E. (1975)
Chlorination of organics in cooling waters and process effluents.
In: Jolley, R.L, ed. The environmental impact of chlorination.
Proceedings of the Conference on Environmental Impact of Water
Chlorination, Oak Ridge, 22-24 October, 1975, Oak Ridge,
Tennessee, Oak Ridge National Laboratories, pp. 115-152.
JONES, P.A. (1981) Chlorophenols and their impurities in the Canadian
environment, Ottawa, Environmental Protection Service,
Environment Canada, 434 pp (Report No. EPS-3-EC-81-2).
JONES, P.A. (1984) Chlorophenols and their impurities in the Canadian
environment: 1983 Supplement, Ottawa, Environmental Protection
Service, Environment Canada, 93 pp (Report No. EPS-3-EP-84-3).
KAILA, K. & SAARIKOSKI, J. (1977) Toxicity of pentachlorophenol and
2,3,6-trichlorophenol to the crayfish (Astacus fluviatilis L.)
Environ. Pollut., 12: 119-123.
KALMAN, D.A. & HORSTMAN, S.W. (1983) Persistence of tetrachlorophenol
and pentachlorophenol in exposed woodworkers. J. Toxicol. clin.
Toxicol., 20(4): 343-352.
KANEKI, H. & TANAKA, M. (1984) Effects of some chlorinated pesticides
on porcine pancreatic and Rhizopus delemar lipase activities.
Eisei Kagaku, 30(4): 233-237.
KANG, H.K., WEATHERBEE, L., BRESLIN, P., & SHEPARD, B.M. (1986) Soft
tissue sarcoma and military service in Vietnam: A case comparison
group analysis of hospital patients. J. occup. Med.,
28: 1215-1218.
KARPALLY, J.C., SAHA, J.G., & LEE, Y.W. (1973) Metabolism of
lindane-14C in the rabbit: ether-soluble urinary metabolites.
J. agric. food Chem., 21(5): 811-818.
KARPOW, G. (1983) [On the antiseptic action of three isomeric
chlorophenols and of their salicylate esters and their fate in the
metabolism.] Arch. Sci. Biol. St. Petersburg, 2: 304-327
(in Russian and French).
KAUPPINEN, T. (1986) Occupational exposure to chemical agents in the
plywood industry. Ann. occup. Hyg., 30(1): 19-29.
KAUPPINEN, T.P., PARTANEN, T.J., NURMINEN, M.M., NICKELS, J.I.,
HERNBERG, S.G., HAKULINEN, T.R., PUKKALA, E.I., & SAVONEN, E.T,
(1986) Respiratory cancers and chemical exposures in the wood
industry: a nested case-control study. Br. J. ind. Med.,
43: 84-90.
KAUPPINEN, T.P. & LINDROOS, L. (1985) Chlorophenol exposure in
sawmills. Am. Ind. Hyg. Assoc. J., 46(1): 34-38.
KAWAHARA, F.K. (1971) Gas chromatographic analysis of mercaptous,
phenols, and organic acids in surface waters with use of
pentafluorobenzyl derivatives. Environ. Sci. Technol.,
5(3): 235-239.
KINAE, N., HASHIZUME, T., MAKITA, T., TOMITA, I., KIMURA, I., &
KANAMORI, H. (1981) Studies on the toxicity of pulp and paper mill
effluents. I. Mutagenicity of the sediment samples derived from
kraft paper mills. Water Res., 15: 17-24.
KING, E.F. (1984) A comparative study of methods for assessing the
toxicity to bacteria of single chemicals and mixtures. In: Liu, D.
& Dutka, B.J., ed. Toxicity screening procedures using bacterial
systems, New York, Basel, Marcel Dekker Inc., pp. 175-193.
KIRSCH, E.J. & ETZEL, J.E. (1973) Microbial decomposition of
pentachlorophenol. J. Water Pollut. Control Fed.,
45(2): 359-364.
KITUNEN, V.H., VALO, R.J., & SALKINOJA-SALONEN, M.S. (1987)
Contamination of soil around wood-preserving facilities by
polychlorinated aromatic compounds. Environ. Sci. Technol.,
21(1): 96-101.
KLEINMAN, G.D, HORSTMAN, S.W., KALMAN, D.A., MCKENZIE, J., & STANZEL,
D. (1986) Industrial hygiene, chemical and biological assessments
of exposures to a chlorinated phenolic sapstain control agent.
Am. Ind. Hyg. Assoc. J., 47(12): 731-741.
KLEU, G. & GOLTZ, R. (1971) [Late and long-term injuries following the
chronic occupational action of chlorophenol compounds.] Med.
Klin. (Munich), 66(2): 53-58 (in German with English abstract).
KLOPPFER, W., KAUFMANN, G., RIPPEN, G., & POREMSKI, H.-J. (1982) A
laboratory method for the volatility from aqueous solution: first
results and comparison with theory. Ecotoxicol. environ. Saf.,
6: 545-559.
KOBAYASHI, S., TODA, S., KAWAMURA, H., CHANG, H.S., FUKUDA, T., &
KAWAGUCHI, K. (1972) Chronic toxicity of 2,4-dichlorophenol in
mice: a simple design for the toxicity of residual metabolites of
pesticides. J. Med. Soc. Toho Univ. Jpn, 19: 356-362.
KOBAYASHI, K., AKITAKE, H., & MANABE, K. (1979) Relation between
toxicity and accumulation of various chlorophenols in goldfish.
Bull. Jpn. Soc. Sci. Fish., 45(2): 173-175.
KOGAN, M. & CLAPP, R. (1985) Mortality among Vietnam veterans in
Massachusetts, 1972-1983. Am. J. Epidemiol, 122: 523.
KONEMANN, H. & MUSCH, A. (1981) Quantitative structure-activity
relationships in fish toxicity studies. Part 2: The influence of
pH on the QSAR of chlorophenols. Toxicology, 19: 223-228.
KORANSKY, W., MUNCH, G., NOACK, G., PORTIG, J., SODOMANN, S., &
WIRSCHING, M. (1975) Biodegradation of hexachlorocyclohexane. V.
Characterization of the major urinary metabolites.
Naunyn-Schmiedeberg's Arch. Pharmacol., 288: 65-78.
KORTE, F., FREITAG, D., GEYER, H., KLEIN, W., KRAUS, A.G., &
LAHANIATIS, E. (1978) Ecotoxicologic profile analysis: a concept
for establishing ecotoxicologic priority lists for chemicals.
Chemosphere, 7(1): 79-102.
KOZAK, V.P., SIMSIMAN, G.V., CHESTERS, G., STENSBY, D., & HARKIN, J.
(1979) Reviews of the environmental effects of pollutants. XI.
Chlorophenols, Washington, DC, US Environmental Protection
Agency, 492 pp (EPA Report No. 600/1-79-O/Z).
KRIJGSHELD, K.R. & VAN DER GEN, A. (1986) Assessment of the impact of
the emission of certain organochlorine compounds on the aquatic
environment. Part I: Monochlorophenols and 2,4-dichlorophenol.
Chemosphere, 15(7): 825-860.
KUIPER, J. & HANTSVEIT, A.O. (1984) Fate and effects of 4-chlorophenol
and 2,4-dichlorophenol in marine plankton communities in
experimental enclosures. Ecotoxicol. environ. Saf., 8: 15-33.
KURIHARA, N. & NAKAJIMA, M. (1974) Studies on BHC isomers and related
compounds. VIII. Urinary metabolites produced from gamma-and ß -
BHC in the mouse: chlorophenol conjugates. Pestic. Biochem.
Physiol, 4(2): 220-231.
KUTZ, F.W., MURPHY, R.S., & STRASSMAN, S.C. (1978) Survey of pesticide
residues and their metabolites in urine from the general
population. In: Rao, K.R., ed. Pentachlorophenol: Chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 363-369.
KUWATSUKA, S. (1972) Degradation of several herbicides in soils under
different conditions. In: Matsuura, F., Boush, G.M., & Misato, T.,
ed. Environmental toxicology of pesticides, New York, London,
Academic Press, pp. 385-400.
KUWATSUKA, S. & IGARASHI, M. (1975) Degradation of PCP in soils. II.
The relationship between the degradation of soils, and the
identification of the degradation products of PCP. Soil Sci.
plant Nutr., 21: 405-414.
KWASNIEWSKA, K. & KAISER, K.L.E. (1983) Toxicities of selected phenols
to fermentative and oxidative yeasts. Bull. environ. Contam.
Toxicol., 31: 188-194.
LANDNER, L., LINDSTROM, K., KARLSSON, M., NORDIN, J., & SÖRENSEN, L.
(1977) Bioaccumulation in fish of chlorinated phenols from kraft
pulp mill bleachery effluents. Bull. environ. Contam. Toxicol.,
18(6): 663-673.
LANOUETTE, M., COCHRANE, W.P., & SINGH, J. (1984) Liquid
chromatographic determination of chlorinated phenol impurities in
technical pentachlorophenol using electrochemical detection
(Coulometric mode). J. Assoc. Off. Anal. Chem., 67(3): 494-497.
LATHROP, G.D., MACHADO, S.G., KARRISON, T.G., GRUBBS, W.D., THOMAS,
W.F., WOOLFE, W.H., MICHALEK, J.E., MINER, J.C., & PETERSON, M.R.
(1987) Air Force Health Study. First follow-up examination
results. An epidemiologic investigation of health effects in Air
Force personnel following exposure to herbicides, Texas, USAF
School of Aerospace Medicine Human Systems Division, 440 pp.
LEBLANC, G,A. (1980) Acute toxicity of priority pollutants to water
fleas (Daphnia magna). Bull. environ. Con tam. Toxicol.,
24: 684-691.
LEBLANC, G.A. (1984) Interspecies relationships in acute toxicity of
chemicals to aquatic organisms. Environ. Toxicol. Chem.,
3: 47-60.
LEE, R.F. & RYAN, C. (1979) Microbial degradation of organo-chlorine
compounds in estuarine waters and sediments. In: Bourquin, A.W. &
Pritchard, P.H., ed. Proceedings of the Workshop on Microbial
Degradation of Pollutants in Marine Environments, held at
Pennsicola Beach, Florida, 9-14 April 1978, Washington, DC, US
Environmental Protection Agency, pp. 443-450 (EPA Report
No. 60/9-79-012).
LEO, A., HANTSCH, C., & ELKINS, D. (1971) Partition coefficients and
their uses. Chem. Rev., 71(6): 525-616.
LEVIN, J.O. & NILSSON, C.A. (1977) Chromatographic determination of
polychlorinated phenols, phenoxyphenols, dibenzofurans and
dibenzodioxins in wood-dust from worker environments.
Chemosphere, 7: 443-448.
LEVIN, J.-O., RAPPE, C., & NILSSON., C.-A. (1976) Use of chlorophenols
as fungicides in sawmills. Scand. J. Work Environ. Health,
2: 71-81.
LINDSAY SMITH, J.R., SHAW, B.A.J., & FOULKES, D.M. (1972) Mechanisms
of mammalian hydroxylation: some novel metabolites of
chlorobenzene. Xenobiotica, 2(3): 215-226.
LINDROOS, L., KOSKINEN, H., MUTANEN, P., & JARVISALO, J. (1987)
Urinary chlorophenols in sawmill workers. Int. Arch. occup.
environ. Health, 59: 463-467.
LINDSTROM, K. & NORDIN, J. (1976) Gas chromatography -- mass
spectrometry of chlorophenols in spent bleach liquors.
J. Chromatogr., 128: 13-26.
LIU, D., THOMSON, K., & KAISER, K.L.E. (1982) Quantitative
structure-toxicity relationship of halogenated phenols on
bacteria. Bull. environ. Contam. Toxicol., 29: 130-136.
LOOS, M.A., ROBERTS, R.N., & ALEXANDER, M. (1967) Phenols as
intermediates in the decomposition of phenoxyacetates by an
Arthrobacter species. Can. J. Microbiol., 13: 679-690.
LYNGE, E. (1985) A follow-up study of cancer incidence among workers
in manufacture of phenoxy herbicides in Denmark. Br. J. Cancer,
52: 259-270.
MCCOLLISTER, D.D., LOCKWOOD, D.T., & ROWE, V.K. (1961) Toxicologic
information on 2,4,5-trichlorophenol. Toxicol. appl. Pharmacol.,
3: 63-70.
MACKENZIE, C.J.G., OLDHAM, W.K., & POWRIE, W.D. (1975) Effects
of pesticides on fish and wildlife in British Columbia.
Appendix RR, Vancouver, British Columbia Royal Commission
(Inquiry into the Use of Pesticides and Herbicides. Final report
of the Commissioners).
MCLEESE, D.W., ZITKO, V., & PETERSON, M.R. (1979) Structure-lethality
relationships for phenols, anilines and other aromatic compounds
in shrimp and clams. Chemosphere, 8(2): 53-57.
MARLOW, D.A. (1986) Hexachlorobenzene exposure in the production of
chlorophenols. In: Morris, C.R. & Cabral, J.R.P., ed.
Hexachlorobenzene. Proceedings of an International Symposium,
Lyons, International Agency for Research on Cancer, pp. 161-169
(IARC Scientific Publications No. 77).
MAY, G. (1973) Chloracne from the accidental production of
tetrachlorodibenzodioxin. Br. J. ind. Med., 30: 276-283.
MILHAM. S. (1982) Herbicides, occupation and cancer. Lancet,
I: 1464-1465.
MILHAM, S. (1985) Cancer in Washington State woodworkers. In: Stich,
H.F., ed. Carcinogens and mutagens in the environment, Boca
Raton, Florida, CRC Press, Vol. 5, pp. 117-123.
MILLER, R.W. & FAUST, S.D. (1973) Sorption from aqueous solutions by
organo-clay. III. The effect of pH on sorption of various phenols.
Environ. Lett, 4(3): 211-223.
MILLS, R.E. (1959) Development of design criteria for biological
treatment of an industrial effluent containing 2,4-D waste water.
In: Proceedings of the 14th Industrial Waste Conference,
Lafayette, Indiana, Purdue University, pp. 340-358.
MITSUDA, H., MURAKAMI, K., & KAWAI, F. (1963) Effect of chlorophenol
analogues on the oxidative phosphorylation in rat liver
mitochondria. Agric. biol. Chem., 27(5): 366-372.
MOOS, L.P., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C,P.L., Jr. (1983)
Pentachlorophenol biodegradation. I. Aerobic. Water Res.,
17(11): 1575-1584.
MORGADE, C., BARQUET, A., & PFAFFENBERGER, C.D. (1980) Determination
of polyhalogenated phenolic compounds in drinking water, human
blood serum and adipose tissue. Bull. environ. Contam. Toxicol.,
24: 257-264.
MOSES, M. & SELIKOFF, I.J. (1981) Soft-tissue sarcomas, phenoxy
herbicides, and chlorinated phenols. Lancet, 1: 1370.
NAKAGAWA, M. & CROSBY, D.G. (1974) Photodecomposition of nitrofen.
J. Agric. food Chem., 22(5): 849-853.
NARANG, A.S., VERNOY, C.A., & EADON, G.A. (1983) Evaluation of
Nielsen-Kryger steam distillation technique for recovery of
phenols from soil. J. Assoc. Off. Anal. Chem., 66: 1330-1334.
NCI (1968) Evaluation of carcinogenic, teratogenic, and mutagenic
activities of selected pesticides and industrial chemicals,
Bethesda, Maryland, National Cancer Institute, Vol. I (Report
prepared by Bionetics Research Laboratories Inc.) (NTIS
PB-223 160).
NCI (1979) Bioassay of 2, 4,6-trichlorophenol for possible
carcinogenicity (CAS No. 88-06-2), Bethesda, Maryland, National
Cancer Institute, 115 pp (NCI Carcinogenesis Technical Report
Series, No. 155).
NCI (1980) Bioassay of a mixture of 1,2,3,6,7,8-hexachlorodibenzo-p-
dioxin and 1,2, 3, 7,8, 9-hexachlorodibenzo-p-dioxin (gavage)
for possible carcinogenicity, Bethesda, Maryland, National
Cancer Institute (NCI Technical Report Series, No. 198)
(NIH 80-1758).
NESTMANN, E.R., LEE, E.G., MATULA, T.I., DOUGLAS, G.R., & MUELLER,
J.C. (1980) Mutagenicity of constituents identified in pulp and
paper mill effluents using the Salmonella/mammalian-microsome
assay. Mutat. Res., 79: 203-212.
NEUBERT, D. & DILLMAN, I. (1972) Embryotoxic effects in mice treated
with 2,4,5-trichlorophenoxyacetic acid and 2,3,7,8-tetra-
chlorodibenzo- p-dioxin. Naunyn-Schmiedeberg's Arch. Pharmacol.,
272: 243-264.
NHW (1983) Tap water consumption in Canada, Ottawa, Department of
National Health and Welfare, Environmental Health Directorate,
Health Protection Branch (Publication No. 82-EHD-80).
NHW (1988) Chlorophenols and their impurities: a health hazard
evaluation, Ottawa, Department of National Health and Welfare,
Environmental Health Directorate, Health Protection Branch
(Publication No. 84-EHD-110).
NIOSH (1983) Registry of toxic effects of chemical substances,
1981-1982, Cincinnati, Ohio, US National Institute for
Occupational Safety and Health.
NRCC (1978) The aqueous chlorination of organic compounds: chemical
reactivity and effects on environmental quality, Ottawa,
Associate Committee on Scientific Criteria for Environmental
Quality, National Research Council of Canada (NRCC Publication
No. 16450).
NRCC (1982) Chlorinated phenols: Criteria for environmental quality,
Ottawa, Associate Committee on Scientific Criteria for
Environmental Quality, National Research Council of Canada,
191 pp (NRCC Publication No. 18578).
NTP (1988) NTP Technical Report on the toxicology and carcinogenesis
studies of 2,4-dichlorophenols (CAS No. 120-83-2) in F344/N rats
and B6C3F1 mice (feed studies), Research Triangle Park, North
Carolina, National Toxicology Programme, US Department of Health
and Human Services (NTP TR 353) (NIH Publication No. 88-2808).
OLSEN, J.H. & MOLLER-JENSEN, O. (1984) Nasal cancer and chlorophenols.
Lancet, II (8393): 47-48.
OMURA, K. & MATSUURA, T. (1971) Photoinduced reactions -- L.
Photolysis of halogenophenols in aqueous alkali and in aqueous
cyanide. Tetrahedron Lett., 27: 3101-3109.
OTT, M.G., HOLDER, B.B., & OLSON, R.D. (1980) A mortality analysis of
employees engaged in the manufacture of 2,4,5-trichloro-
phenoxyacetic acid. J. occup. Med., 22(1): 47-50.
OTT, M.G., OLSON, R.A., COOK, R.R, & BOND, G.G. (1987) Cohort
mortality study of chemical workers with potential exposure to the
higher chlorinated dioxins. J. occup. Med., 29(5): 422-429.
PAASIVIRTA, J., SARKKA, J., LESKIJARVI, T., & ROOS, A. (1980)
Transportation and enrichment of chlorinated phenolic compounds in
different aquatic food chains. Chemosphere, 9: 441-456.
PAASIVIRTA, J., SARKKA, J., AHO, M., SURMA-AHO, K., TARHANEN, J, 8,:
ROOS, A. (1981) Recent trends of biocides in pikes of the Lake
Päijäne. Chemosphere, 10(4): 405-414.
PAASIVIRTA, J., SARKKA, J., SURMA-AHO, K., HUMPPI, T., KUOKKANEN, T.,
& MARTTINEN, M. (1983) Food chain enrichment of organochlorine
compounds and mercury in clean and polluted lakes of Finland.
Chemosphere, 12(2): 239-252.
PAASIVIRTA, J., HEINOLA, K., HUMPPI, T., KARJALAINEN, A., KNUUTINEN,
J., MANTYKOSKI, K., PAUKKU, R., PIILOLA, T., SURMA-AHO, K.,
TARHANEN, J., WELLING, L., & VIHONEN, H. (1985) Polychlorinated
phenols, guaiacols and catechols in environment. Chemosphere,
14(5): 469-491.
PAL, H.S., MURPHY, T., CARTER, A.C,, & DREW, S.W. (1980) Rapid assays
for microbial degradation of 2-chlorophenol, Blacksburg,
Virginia, Virginia Water Resources Research Center, Virginia
Polytechnic Institute and State University (Report No. 128).
PARKER, C.E. & PRIBYL, E.J., Jr (1984) Assessment of bacterial ATP
response as a measurement of aquatic toxicity. In: Liu, D. &
Dutka, B.J., ed. Toxicity screening procedures using bacterial
systems, New York, Basel, Marcel Dekker Inc., pp. 283-293.
PARR, L.J., GEE, M.G., LAND, D.G., ROBINSON, D., & CURTIS, R.F. (1974)
Chlorophenols from wood preservatives in broiler house litter.
J. Sci. Food Agric., 25(7): 835-841.
PAZELEROVA, J., LUKAS, E., NEMCOVA, M., SPACILOVA, M., JIRASEK, L.,
KALENSKY, J., JOHN, J., JIRASEK, A., & PICKOVA, J. (1974) [Chronic
poisoning by chlorinated hydrocarbons, formed in the production of
2,4,5-sodium trichlorophenoxyacetate.] Prac. Lek.,
26(9): 332-339 (in Czech, with English summary).
PEARCE, N.E., SMITH, A.H., HOWARD, J.K., SHEPPARD, R., GILES, H.J., &
TEAGUE, C.A. (1986) Non-Hodgkin's lymphoma and exposure to phenoxy
herbicides, chlorophenols, fencing work, and meat works
employment: a case study. Br. J. ind. Med., 43: 75-83.
PEKARI, K. & AITIO, A. (1982) A simple liquid chromatographic method
for the anaylsis of penta and tetrachlorophenols in urine of
exposed workers. J. Chromatogr., 232: 129-136.
PEKARI, K., BOUDENE, C., & AITIO, A. (1986) Kinetics of
2,4,6-trichlorophenol in different organs in the rat.
Arch. Toxicol., 59: 41-44.
PHIPPS, G.L., HOLCOMBE, G.W., & FIANDT, J.T. (1981) Acute toxicity of
phenol and substituted phenols to the fathead minnow. Bull.
environ. Contam. Toxicol., 26: 585-593.
PIERCE, R.H., Jr & VICTOR, D.M. (1978) The fate of pentachlorophenol
in an aquatic ecosystem. In: Rao, K.R., ed. Pentachlorophenol:
Chemistry, pharmacology, and environmental toxicology, New York,
London, Plenum Press, pp. 41-52.
PIET, G.J. & DE GRUNT, F. (1975) Organic chloro compounds in surface
and drinking water of the Netherlands. In: Problems raised by the
contamination of man and his environment by persistent pesticides
and organo-halogenated compounds. European Colloquium,
Luxembourg, Commission of the European Communities, pp. 81-92.
PIGNATELLO, J.J., MARTINSON, M.M., STEIERT, J.G., CARLSON, R.E., &
CRAWFORD, R.L. (1983) Biodegradation and photolysis of
pentachlorophenol in artificial freshwater streams. Appl.
environ. Microbiol., 46: 1024-1031.
PIGNATELLO, J.J., JOHNSON, L.K., MARTINSON, M.M., CARLSON, R.E., &
CRAWFORD, R.L. (1986) Response of the microflora in outdoor
experimental streams to pentachlorophenol: environmental factors.
Can. J. Microbiol., 32: 38-46.
PITTER, P. (1976) Determination of biological degradability of organic
substances. Water Res., 10: 231-235.
POLAND, A.P., SMITH, D., METTER, G., & POSSICK, P. (1971) A health
survey of workers in a 2,4-D and 2,4,5-T plant. Arch. environ.
Health, 22: 316-327.
RAO, K.R., FOX, F.R., CONKLIN, P.J., & CANTELMO, A.C. (1981)
Comparative toxicology and pharmacology of chlorophenols: studies
on the grass shrimp, Palaemonetespugio. In: Vernberg, F.J.,
Calabrese, A., Thurberg, F.P., & Vernberg, W.B., ed. Biological
monitoring of marine pollutants, New York, London, Academic
Press, pp. 37-72.
RAPPE, C, GARA, A., & BUSER, H.R. (1978a) Identification of
polychlorinated dibenzofurans (PCDFs) in commercial chlorophenol
formulations. Chemosphere, 12: 981-991.
RAPPE, C., MARKLUND, B., BUSER, H.-R., & BOSSHARDT, H.-P. (1978b)
Formation of polychlorinated dibenzo- p-dioxins (PCDDs) and
dibenzofurans (PCDFs) by burning or heating chlorophenates.
Chemosphere, 3: 269-281.
RAPPE, C., BUSER, H.-R., & BOSSHARDT, H.-P. (1979) Dioxins,
dibenzofurans, and other polyhalogenated aromatics: Production,
use, formation and destruction. Ann. N.Y. Acad. Sci., 320: 1-18.
RAPPE, C., NYGREN, M., BUSER, H.-R., & KAUPPINEN, T. (1982)
Occupational exposure to polychlorinated dioxins and
dibenzofurans. In: Hutzinger, O., Frei, R.W., Merian, E., &
Pocchiari, F., ed. Chlorinated dioxins and related compounds:
Impact on the environment, Oxford, Pergamon Press, pp. 495-514
(Pergamon Series on Environmental Science, Vol. 5).
RASANEN, L., HATTULA, M.L., & ARSTILA, A.U. (1977) The mutagenicity of
MCPA and its soil metabolites, chlorinated phenols, catechols and
some widely used slimicides in Finland. Bull. environ. Contam.
Toxicol., 18(5): 565-571.
REALINI, P.A. (1981) Determination of priority pollutant phenols in
water by HPLC. J. chromatogr. Sci., 19: 124-129.
REINER, E.A., CHU, J.P., & KIRSCH, E.J. (1978) Microbial metabolism of
pentachlorophenol. In: Rao, K.R., ed. Pentachlorophenol:
Chemistry, pharmacology, and environmental toxicology, New York,
London, Plenum Press, pp. 67-81.
RENNER, G. & MUCKE, W. (1986) Transformations of pentachlorophenol.
Part I: Metabolism in animals and man. Toxicol. environ. Chem.,
11: 9-29.
RIBO, J.M. & KAISER, K.L.E. (1983) Effects of selected chemicals to
photoluminescent bacteria and their correlations with acute and
sublethal effects on other organisms. Chemosphere,
12(11): 1421-1442.
RICHARDSON, N.G. (1978) Wood preserving effluents and their treatment.
In: The timber processing industry. Proceedings of a Seminar on
Technology Transfer, Toronto, Ontario, 10-11 March, 1977,
Ottawa, Environment Canada (Report No. EPS 3-WP).
ROBERTS, B.L. & DOROUGH, H.W. (1984) Relative toxicities of chemicals
to the earthworm, Eisenia foetida. Environ. Toxicol. Chem.,
3: 67-78.
ROBERTS, M.S., ANDERSON, R.A., & SWARBRICK, J. (1977) Permeability of
human epidermis to phenolic compounds. J. Pharm. Pharmacol.,
29: 677-683.
ROBERTS, M.S., ANDERSON, R.A., SWARBRICK, J., & MOORE, D.E. (1978) The
percutaneous absorption of phenolic compounds: the mechanism of
diffusion across the stratum corneum. J. Pharm. Pharmacol.,
30: 486-490.
ROYAL COMMISSION ON THE USE AND EFFECTS OF CHEMICAL AGENTS ON
AUSTRALIAN PERSONNEL IN VIETNAM (1985) Final report. Volume 4:
Cancer, Canberra, Australian Government Publishing Service,
pp. 71-74.
RUCKDESCHEL, G. & RENNER, G. (1986) Effects of pentachlorophenol and
some of its known and possible metabolites on fungi. Appl.
environ. Microbiol., 51(6): 1370-1372.
SAARIKOSKI, J. & VILUKSELA, M. (1981) Influence of pH on the toxicity
of substituted phenols to fish. Arch. environ. Contam. Toxicol.,
10: 747-753.
SACKMAUEROVA-VENINGEROVA, M., UHNAK, J., SZOKOLAY, A., & KOCAN, A.
(1981) Identification of chlorinated phenols as degradation
products of chlorinated pesticides in biological materials.
J. Chromatogr., 205: 194-198.
SALKINOJA-SALONEN, M.S., VALO, R., APAJALAHTI, J., HAKULINEN, R.,
SILAKOSKI, R., & JAAKKOLA, T. (1984) Biodegradation of
chlorophenolic compounds in wastes from wood-processing industry.
In: Klug, M. & Reddy, C., ed. Current perspectives in microbial
ecology, Washington, DC, American Society for Microbiology,
pp. 668-676.
SCHAUERTE, W., LAY, J.P., KLEIN, W., & KORTE, F. (1982) Influence of
2,4,6-trichlorophenol and pentachlorophenol on the biota of
aquatic systems. Chemosphere, 11(1): 71-79.
SCHELLENBERG, K., LEUENBERGER, C., & SCHWARZENBACH, R.P. (1984)
Sorption of chlorinated phenols by natural sediments and aquifer
materials. Environ. Sci. Technol., 18: 652-657.
SCHULTZ, T.W. & RIGGIN, G.W. (1985) Predictive correlations for the
toxicity of alkyl- and halogen-substituted phenols. Toxicol.
Lett., 25: 47-54.
SCHWETZ, B.A., KEELER, P.A., & GEHRING, P.J. (1974) Effect of purified
and commercial grade tetrachlorophenol on rat embryonal and fetal
development. Toxicol. appl. Pharmacol., 28: 146-150.
SEIP, H.M., ALSTAD, J., CARLBERG, G.E., MARTINSEN, K., & SKAANE, R.
(1986) Measurement of mobility of organic compounds in soils.
Sci. total Environ., 50: 87-101.
SENES (1985) Drinking water criteria reviews for 2, 4,5-trichloro-
phenol, 2, 4,6-trichlorophenol, 2,3,5-trichlorophenol, and
2,3,6-trichlorophenol, Willowdale, Ontario, Senes Consultants
Limited (Report prepared for the Ontario Ministry of the
Environment, Water Resources Branch, Drinking Water Section).
SEYLER, D.E., EAST, J.M., CONDIE, L.W., & BORZELLECA, J.F. (1984) The
use of in vitro methods for assessing reproductive toxicity.
Dichlorophenols. Toxicol. Lett., 20(3): 309-315.
SHAFIK, T.M., SULLIVAN, H.C., & ENOS, H.R. (1973) Multiresidue
procedure for halo- and nitrophenols: measurement of exposure to
biodegradable pesticides yielding these compounds as metabolites.
J. agric. food Chem., 21(2): 295-298.
SHEN, S.Y., VILLENEUVE, D.C., CHU, I., KELLY, J., & GILMAN, A.P.
(1983) Acute dermal toxicity of tetrachlorophenols in the rat.
Bull. environ. Contam. Toxicol., 31: 680-685.
SHERMAN, M., BECK, J., & HERRICK, R.B. (1972) Chronic toxicity and
residues from feeding Nemacide [ O-(2,4-dichlorophenyl)
O,O-diethyl phosphothionate] to laying hens. J. agric. food
Chem., 20(3): 617-624.
SHUMWAY, D.L. & PALENSKY, J.R. (1973) Impairment of the flavour of
fish by water pollutants, Corvallis, Oregon State University,
Department of Fish and Wildlife, 83 pp (Report No. W73-11322)
(EPA Report No. R3-73-010).
SITHOLE, B.B. & WILLIAMS, D.T. (1986) Halogenated phenols in water at
forty Canadian potable water treatment facilities. J. Assoc. Off.
Anal. Chem., 69(5): 807-810.
SIUDA, J.F. (1980) Natural production of organohalogens. In: Jolley,
R.L., Brungs, W.A., Cumming, R.B., & Jacobs, V.A., ed. Water
chlorination: Environmental impact and health effects, Ann
Arbor, Michigan, Ann Arbor Science Publishers Inc., Vol. 3,
pp. 63-72.
SMITH, A.H., PEARCE, N.E., FISHER, D.O., GILES, H.J., TEAGUE, C.A., &
HOWARD, J.K. (1984) Soft-tissue sarcoma and exposure to phenoxy
herbicides and chlorophenols in New Zealand. J. Natl Cancer
Inst., 73(5): 1111-1117.
SOMANI, S.M. & KHALIQUE, A. (1982) Distribution and metabolism of
2,4-dichlorophenol in rats. J. Toxicol. environ. Health,
9: 889-897.
SOMANI, S.M., SMART, T., & KHALIQUE, A. (1984) Metabolism of
2,4-dichlorophenol by isolated perfused rat liver. I. Toxicol.
environ. Health, 13: 787-798.
SPOKES, J.R. & WALKER, N. (1974) Chlorophenol and chlorobenzoic acid
co-metabolism by different genera of soil bacteria. Arch.
Microbiol., 96(2): 125-134.
STALLING, D.L., SMITH, L.M., & PETTY, J.D. (1979) Approaches to
comprehensive analyses of persistent halogenated environmental
contaminants. In: Van Hall, C.D., ed. Measurement of organic
pollutants in water and wastewater, Washington, DC, American
Society for Testing and Materials, pp. 302-323 (ASTM STP 686).
STATISTICS CANADA (1986) Apparent per capita food consumption in
Canada. Part I & II. 1984, Ottawa, Ministry of Supply and
Services Canada (Catalogue No. 32-230).
STERLING, T.D., STOFFMAN, L.D., STERLING, D.A., & MATE, G. (1982)
Health effects of chlorophenol wood preservatives on sawmill
workers. Int. J. Health Serv., 12(4): 559-571.
STIJVE, T. (1981) Determination of pentachlorophenol and
2,3,4,6-tetrachlorophenol in edible gelatins. Dtsch. Lebensm.
Rundschau, 77: 249-253.
STINGILY, K.O. (1940) A new industrial chemical dermatitis. Southern
med. J., 33(12): 1268-1272.
STOCKDALE, M. & SELWYN, M.J. (1971) Influence of ring substituents on
the action of phenols on some dehydrogenases, phosphokinases and
the soluble ATPase from mitochondria. Eur. J. Biochem.,
21: 416-423.
STRETZ, L.A. & VAVRUSKA, J.S. (1984) Controlled air incineration of
pentachlorophenol-treated wood, Washington, DC, US Environmental
Protection Agency, 110 pp (EPA Report No. 600/2-84-089).
SUGIARA, K., AOKI, M., KANEKO, S., DAISAKU, I., KOMATSU, Y., &
SHIBUYA, H. (1984) Fate of 2,4,6-trichlorophenol, penta-
chlorophenol, p-chlorobiphenyl, and hexachlorobenzene in an
outdoor experimental pond: comparison between observation and
predictions based on laboratory data. Arch. environ. Contam.
Toxicol., 13: 745-758.
SUSKIND, R.R. & HERTZBERG, V.S. (1984) Human health effects of 2,4,5-T
and its toxic contaminants. J. Am. Med. Assoc.,
251(18): 2372-2380.
SUND, K.A. & NOMURA, N. (1963) Laboratory evaluation of several
herbicides. Weed Res., 3: 35-43.
TABAK, H.H., CHAMBERS, C.W., & KABLER, P.W. (1964) Microbial
metabolism of aromatic compounds. I. Decomposition of phenolic
compounds and aromatic hydrocarbons by phenol-adapted bacteria.
J. Bacteriol., 87(4): 910-919.
TEIDJE, J.M. & ALEXANDER, M. (1969). Enzymatic cleavage of the ether
bond of 2,4-dichlorophenoxyacetate. J. agric. food Chem.,
17: 1080-1084.
TELFORD, M. (1974) Blood glucose in crayfish. II. Variations induced
by artificial stress. Comp. Biochem. Physiol., 48A: 555-560.
THIESS, A.M, FRENTZEL-BEYME, R., & LINK, R. (1982) Mortality study of
persons exposed to dioxin in a trichlorophenol-process accident
that occurred in the BASF AG on November 17, 1953. Am. J. ind.
Med., 3: 179-189.
TOWNSEND, J.C., BODNER, K.M., VAN PEENEN, P.F.D., OLSON, R.D., & COOK,
R.R. (1982) Survey of reproductive events of wives of employees
exposed to chlorinated dioxins. Am. J. Epidemiol.,
115(5): 695-713.
TREVORS, J.T., MAYFIELD, C.I., & INNISS, W.E. (1982) Effect of
sequence of exposure to chlorophenols in short-term bacterial
bioassays. Arch. environ. Contam. Toxicol., 11: 203-207.
TYLER, J.E. & FINN, R.K. (1974) Growth rates of a pseudomonad on
2,4-dichlorophenoxyacetic acid and 2,4-dichlorophenol. Appl.
Microbiol., 28: 181-184.
US EPA (1971) Biological treatment of chlorophenolic wastes,
Washington, DC, US Environmental Protection Agency (Water
Pollution Control Research Series).
US EPA (1978) Rebuttable presumption against registration and
continued registration of pesticide products containing
2,4,5-trichlorophenol and its salts. Fed. Reg.,
43(149): 34026-34054.
US EPA (1979) In: Kozak, V.P., Simsiman, G.V., Chesters, G., Stensby,
D., & Harkin, J. ed. Reviews of the environmental effects of
pollutants: XI. Chlorophenols, Washington, DC, US Environmental
Protection Agency, 492 pp (EPA Report No. 600/1-79-012).
US EPA (1980a) Ambient water quality criteria for 2-chlorophenol,
Washington, DC, US Environmental Protection Agency, Office of
Water Regulations and Standards Criteria (EPA Report
No. 440/5-80-034).
US EPA (1980b) Ambient water quality criteria for 2, 4-dichloro-
phenol, Washington, DC, US Environmental Protection Agency,
Office of Water Regulations and Standards Criteria (EPA Report
No. 440/5-80-042).
US EPA (1980c) Ambient water quality criteria of chlorinated
phenols, Washington, DC, US Environmental Protection Agency,
Office of Water Regulations and Standards Criteria (EPA Report
No. 440/5-80-032).
US EPA (1980d) Analytical methodology for determination of
chlorophenols in human and environmental media, Research
Triangle Park, North Carolina, US Environmental Protection Agency,
Health Effects Research Laboratory (EPA Report No. 600/2-80-181).
VALO, R., KITUNEN, V., SALKINOJA-SALONEN, M., & RAISANEN, S. (1984)
Chlorinated phenols as contaminants of soil and water in the
vicinity of two Finnish sawmills. Chemosphere, 13(8): 835-844.
VAN OMMEN, B., ADANG, A.E.P., BRADER, L., POSTHUMUS, M.A., MULLER, F.,
& VAN BLADEREN, P.J. (1966) The microsomal metabolism of
hexachlorobenzene. Origin of the covalent binding to protein.
Biochem. Pharmacol., 35(19): 3233-3238.
VAN OMMEN, B, ADANG, A., MULLER, F., & VAN BLADEREN, P.J. (1986) The
microsomal metabolism of pentachlorophenol and its covalent
binding to protein and DNA. Chem.-biol. Interact., 60: 1-11.
VAN OMMEN, B., VONCKEN, J.W., MULLER, F., & VAN BLADEREN, P.J. (1988)
The oxidation of tetrachloro-1,4-hydroquinone by microsomes and
purified cytochrome P-450s. Implications for covalent binding to
protein and involvement of reactive oxygen species. Chem.-biol.
Interact., 65(3): 247-259.
VERSCHUEREN, K. (1983) Handbook of environmental data on organic
chemicals, 2nd ed., New York, Van Nostrand Reinhold Co.
VINEIS, P., TERRACINI, B., CICCONE, G., CIGNETTI, A., COLOMBO, E.,
DONNA, A, MAFFI, L., PISA, R., RICCI, P., ZANINI, E., & COMBA, P.
(1987) Phenoxy herbicides and soft-tissue sarcomas in female rice
weeders: A population-based case-referent study. Scand. J. Work
Environ. Health, 13: 9-17.
VIRTANEN, M.T. & HATTULA, M.-L. (1982) The fate of 2,4,6-trichloro-
phenol in an aquatic continuous-flow system. Chemosphere,
11(7): 641-649.
VIZETHUM, W. & GOERZ, G. (1979) Induction of the hepatic microsomal
and nuclear cytochrome P-450 system by hexachloro-benzene,
pentachlorophenol and trichlorophenol. Chem.-biol. Interact.,
28: 291-299.
WALKER, N. (1954) Preliminary observations on the decomposition of
chlorophenols in soil. Plant Soil, 5(2): 194-204.
WALKER, N. (1973) Metabolism of chlorophenols by Rhodoturula
glutinis. Soil Biol. Biochem., 5(5): 525-530.
WANG, W. & REED, P. (1984) Nitrobacter bioassay for aquatic toxicity.
In: Liu, D. & Dutka, B.J., ed. Toxicity screening procedures
using bacterial systems, New York, Basel, Marcel Dekker Inc.,
pp. 309-325.
WEBER, K. & ERNST, W. (1978) Levels and pattern of chlorophenols in
water of the Weser Estuary and the German Bight. Chemosphere,
7(11): 873-879.
WEGMAN, R.C.C. & HOFSTEE, A.W.M. (1979) Chlorophenols in surface
waters of the Netherlands (1976-1977). Water Res., 13: 651-657.
WEGMAN, R.C.C. & VAN DEN BROEK, H.H. (1983) Chlorophenols in river
sediment in the Netherlands. Water Res., 17(2): 227-230.
WEINBACH, E.C. (1957) Biochemical basis for the toxicity of
pentachlorophenols. Proc. Natl Acad. Sci., 43: 393-397.
WEINBACH, E.C. & GARBUS, J. (1965) The interaction of uncoupling
phenols with mitochondria and with mitochondrial protein.
J. biol. Chem., 240(4): 1811-1819.
WHO (1984) Guidelines for drinking-water quality. Vol. 1:
Recommendations, Geneva, World Health Organization, 130 pp.
WHO (1985) Guidelines for drinking-water quality. Vol. 2: Health
Criteria and other supporting information, Geneva, World Health
Organization, 335 pp.
WHO (1987a) Environmental Health Criteria 70: Principles for the
safety assessment of food additives and contaminant in food,
Geneva, World Health Organization, 174 pp.
WHO (1987b) Environmental Health Criteria 71: Pentachlorophenol,
Geneva, World Health Organization, 236 pp.
WHO (in press) Environmental Health Criteria: Polychlorinated
dibenzo-p-clioxins and dibenzofurans. Geneva, World Health
Organization.
WILLIAMS, D.T., LEBEL, G.L., & JUNKINS, E. (1984) A comparison of
organochlorine residues in human adipose tissue autopsy samples
from two Ontario municipalities. J. Toxicol. environ. Health,
13: 19-29.
WOOD, S., ROM, W.N., WHITE, G.L., & LOGAN, D.C. (1983)
Pentachlorophenol poisoning. J. occup. Med., 25(7): 527-530.
WOODROW, J.E., MAJEWSKI, M.S., & SEIBER, J.N. (1976) Accumulative
sampling of trace pesticides and other organics in surface water
using XAD-4 resin. J. environ. Sci. Health, B21(2): 143-164.
WOODS, J.S., POLISSAR, L., SEVERSON, R.K., HEUSER, L.S., & KULANDER,
B.G. (1987) Soft tissue sarcoma and non-Hodgkin's lymphoma in
relation to phenoxy herbicide and chlorinated phenol exposure in
Western Washington. J. Natl Cancer Inst, 78(5): 899-910.
WOOLSON, E.A, THOMAS, R.F., & ENSOR, P.D.J. (1972) Survey of
polychlorodibenzo- p-dioxin content in selected pesticides.
J. agric. food Chem., 20: 351-354.
WRIGHT, F.C., RINER, J.C., PALMER, J.S., & SCHLINKE, J.C. (1970)
Metabolic and residue studies with 2-(2,4,5-trichlorophenoxy)-
ethyl-2,2-dichloropropionate (Erbon) herbicide in sheep.
J. agric. food Chem., 18(5): 845-847.
WYLLIE, J.A., GABICA, J., BENSON, W.W, & YODER, J. (1975) Exposure and
contamination of the air and employees of a pentachlorophenol
plant, Idaho -- 1972. Pestic. monit. J., 9(3): 150-153.
XIE, T.-M. (1983) Determination of trace amounts of chlorophenols and
chloroguaiacols in sediments. Chemosphere, 12: 1183-1191.
XIE, T.-M., ABRAHAMSSON, K., FOGELQVIST, E., & JOSEFSSON, B. (1986)
Distribution of chlorophenolics in a marine environment. Environ.
Sci. Technol., 20: 457-463.
YOST, R.A., FETTEROLF, D.D., HASS, J.R., HARVAN, D.J., WESTON, A.F.,
SKOTNICKI, P.A., & SIMON, N.M. (1984) Comparison of mass
spectrometric methods for trace level screening of
hexachlorobenzene and trichlorophenol in human blood serum and
urine. Anal. Chem., 56: 2223-2228.
ZACK, J.A. & SUSKIND, R.R. (1980) The mortality experience of workers
exposed to tetrachlorobenzodioxin in a trichlorophenol process
accident. J. occup. Med, 22(1): 11-14.
ZACK, J.A. & GAFFEY, W.R. (1983) A mortality study of workers employed
at the Monsanto plant in Nitro, West Virginia. Environ. Sci.
Res., 26: 575-591.
ZOETEMAN, B.C.J. (1975) Odour nuisance by organo-halogenated compounds
in water and its toxicological impact. In: Problems raised by the
contamination of man and his environment by persistent pesticides
and organo-halogenated compounds. European Colloquium,
Luxembourg, Commission of the European Communities, pp. 527-544
(EUR 5196).
ZOETEMAN, B.C.J., DE GREEF, E., & BRINKMAN, F.J.J. (1981) Persistency
of organic contaminants in groundwater, lessons from soil
pollution incidents in the Netherlands. Sci. total Environ.,
21: 187-202.