
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 133
FENITROTHION
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First draft prepared by Dr. J. Sekizawa
(National Institute of Hygienic Sciences, Japan)
and Dr. M. Eto (Kyushu University, Japan) with
the assistance of Dr. J. Miyamoto and
Dr. M. Matsuo (Sumitomo Chemical Company)
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1992
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Fenitrothion.
(Environmental health criteria ; 133)
1.Fenitrothion - adverse effects 2.Fenitrothion - toxicity
3.Environmental exposure I.Series
ISBN 92 4 157133 0 (NLM Classification: WA 240)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1992
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR FENITROTHION
1. SUMMARY AND EVALUATION, CONCLUSIONS AND RECOMMENDATIONS
1.1. Summary and evaluation
1.1.1. Exposure
1.1.2. Uptake, metabolism, and excretion
1.1.3. Effects on organisms in the environment
1.1.4. Effects on experimental animals and
in vitro test systems
1.1.5. Effects on human beings
1.2. Conclusions
1.3. Recommendations
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Man-made sources
3.2.1. Production
3.2.2. Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.2. Abiotic and biotic transformation
4.2.1. Abiotic transformation
4.2.1.1 Thermal degradation
4.2.1.2 Photolysis in air
4.2.1.3 Hydrolysis and photolysis
in water
4.2.1.4 Photolysis on soil
4.2.2. Biotransformation
4.2.2.1 Biodegradation in soil
4.2.2.2 Biodegradation and bioaccumulation
in organisms
4.2.2.3 Abiotic and biological
degradation in/on plants
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.3. Soil
5.1.4. Food
5.2. Human exposure
5.2.1. Food
6. KINETICS AND METABOLISM
6.1. Absorption, distribution, metabolic
transformation, elimination, and excretion
6.2. Retention and turnover
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.2. Skin and eye irritation; skin sensitization
7.2.1. Skin and eye irritation
7.2.2. Skin sensitization
7.3. Short-term studies
7.3.1. Rat
7.3.2. Dog
7.3.3. Rabbit
7.3.4. Guinea-pig
7.4. Long-term and carinogenicity studies
7.5. Reproductive effects, embryotoxicity, and
teratogenicity
7.5.1. Reproductive effects
7.5.2. Embryotoxicity and teratogenicity
7.6. Mutagenicity
7.7. Neurotoxicity
7.8. Effects on hepatic enzymes
7.9. Effects on hormonal balance
7.10. Toxicity of metabolites and the S-isomer
7.11. Factors modifying toxicity
7.12. Mechanism of toxicity - mode of action
7.12.1. Mode of action
7.12.2. Selective toxicity
7.12.3. Potentiation of toxicity of
other chemicals
8. EFFECTS ON MAN
8.1. General population exposure
8.1.1. Acute toxicity
8.1.2. Poisoning incidents
8.1.3. Contact dermatitis
8.1.4. Possible links with Reye's syndrome
8.2. Occupational exposure
9. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
9.1. Microorganisms and algae
9.2. Aquatic organisms
9.2.1. Fish
9.2.2. Invertebrates
9.2.3. Amphibians and arthropods
9.3. Terrestrial organisms
9.3.1. Terrestrial invertebrates
9.3.2. Birds
9.3.3. Mammals
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
ANNEX I. TREATMENT OF ORGANOPHOSPHATE INSECTICIDE POISONING IN MAN
ANNEX II. NO-OBSERVED-EFFECT LEVELS IN PLASMA, RED BLOOD CELLS, AND
BRAIN ChE, IN ANIMALS TREATED WITH FENITROTHION
RESUME ET EVALUATION, CONCLUSIONS ET RECOMMANDATIONS
RESUMEN Y EVALUACION, CONCLUSIONES ET RECOMENDACIONES
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR TRICHLORFON
AND FENITROTHION
Members
Dr V. Benes, Department of Toxicology and Reference Laboratory,
Institute of Hygiene and Epidemiology, Prague, Czech and Slovak
Federal Republic
Dr C. Carrington, Division of Toxicological Review and Evaluation,
Food and Drug Administration, Washington, DC, USA (Joint
Rapporteur)
Dr W. Dedek, Department of Chemical Toxicology, Academy of
Sciences, Leipzig, Germany
Dr S. Dobson, Institute of Terrestrial Ecology, Monks Wood
Experimental Station, Huntingdon, United Kingdom
Dr D.J. Ecobichon, Department of Pharmacology and Therapeutics,
McGill University, Montreal, Canada
Dr M. Eto, Department of Agricultural Chemistry, Kyushu
University, Fukuoka-shi, Japan (Vice-Chairman)
Dr Bo Holmstedt, Department of Toxicology, Karolinska Institute,
Stockholm, Sweden
Dr S.K. Kashyap, National Institute of Occupational Health,
Ahmedabad, India
Dr J. Miyamoto, Takarazuka Research Centre, Hyogo, Japan
Dr H. Spencer, United States Environmental Protection Agency,
Washington, DC, USA (Chairman)
Dr M. Takeda, National Institute of Hygienic Sciences, Tokyo,
Japan
Observers
Dr M. Matsuo, Biochemistry and Toxicology Laboratory, Sumitomo
Chemical Co. Ltd, Osaka-shi, Japan (representing GIFAP)
Secretariat
Dr K.W. Jager, IPCS, World Health Organization, Geneva,
Switzerland (Secretary)
Dr J. Sekizawa, National Institute of Hygienic Sciences, Tokyo,
Japan (Joint Rapporteur)
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the
criteria documents as accurately as possible without unduly delaying
their publication. In the interest of all users of the Environmental
Health Criteria documents, readers are kindly requested to
communicate any errors that may have occurred to the Director of the
International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland, in order that they may be
included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Palais
des Nations, 1211 Geneva 10, Switzerland (Telephone No. 7988400 or
7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR FENITROTHION
A WHO Task Group on Environmental Health Criteria for
Trichlorfon and Fenitrothion met at the World Health Organization,
Geneva, from 10 to 14 December 1990. Dr K.W. Jager, IPCS, welcomed
the participants on behalf of Dr M. Mercier, Manager of the IPCS,
and the three IPCS cooperating organizations (UNEP/ILO/WHO). The
Group reviewed and revised the draft and made an evaluation of the
risks for human health and the environment from exposure to
fenitrothion.
The first draft was prepared by Dr J. Sekizawa of the National
Institute of Hygienic Sciences of Japan in collaboration with Dr J.
Miyamoto and Dr M. Matsuo of Sumitomo Chemical Company, and Dr M.
Eto of Kyushu University. Dr J. Sekizawa also prepared the second
draft, incorporating comments received following circulation of the
first drafts to the IPCS contact points for Environmental Health
Criteria.
Dr K.W. Jager of the IPCS Central Unit was responsible for the
scientific content and Mrs M.O. Head of Oxford for the editing.
The fact that Sumitomo Chemical Company Limited, Japan
(trichlorfon and fenitrothion) and Bayer AG, Germany (trichlorfon)
made available to the IPCS and the Task Group their proprietary
toxicological information on the products under discussion is
gratefully acknowledged. This allowed the Task Group to make its
evaluation on the basis of more complete data.
The efforts of all who helped in the preparation and
finalization of the document are gratefully acknowledged.
1. SUMMARY AND EVALUATION, CONCLUSIONS AND RECOMMENDATIONS
1.1 Summary and evaluation
1.1.1 Exposure
Fenitrothion is an organophosphorus insecticide that has been
in use since 1959. It is used in agriculture to control insects on
rice, cereals, fruits, vegetables, stored grains, and cotton. It is
also used to control insects in forests and for fly, mosquito, and
cockroach control in public health programmes. It is formulated as
emulsifiable concentrates, ultra-low-volume concentrates, powders,
granules, dusts, oil-based sprays, and in combination with other
pesticides. Between 15 000 and 20 000 tons of fenitrothion are
produced per year.
Fenitrothion enters the air by volatilization from contaminated
surfaces and may drift beyond the intended target area during
spraying. It leaches very slowly from most soils, but some run-off
can be expected.
Fenitrothion is degraded by photolysis and hydrolysis. In the
presence of ultraviolet radiation (UVR) or sunlight, the half-life
of fenitrothion in water is less than 24 h. The presence of
micro-flora may also accelerate degradation. In the absence of
sunlight or microbial contamination, fenitrothion is stable in
water. In soil, biodegradation is the primary route of degradation,
though photolysis may also play a role.
Airborne concentrations of fenitrothion may be as high as 5
µg/m3 directly after spraying, but may decrease markedly with time
and distance from the application site. Levels in water may be as
high as 20 µg/litre, but decrease rapidly.
Bioconcentration factors for fenitrothion with continuing
exposure have been estimated to range from 20 to 450 for a number of
different aquatic species.
Levels of fenitrothion residues in fruits, vegetables, and
cereal grains may range from 0.001 to 9.5 mg/kg immediately after
treatment, but decline rapidly, with a half-life of 1-2 days.
1.1.2 Uptake, metabolism, and excretion
Fenitrothion is rapidly absorbed from the intestinal tract of
experimental animals and distributed to various body tissues. The
half-life for the dermal absorption of fenitrothion in the monkey
was 15-17 h. Fenitrothion has been shown to be metabolized through
the major pathways of O-demethylation and by cleavage of the
P-O-aryl bond. The nitro group of fenitrothion is reduced by
intestinal microorganisms, in ruminants only. The major route of
elimination is via the urine, most of the metabolites being
eliminated within 2-4 days in the rat, guinea-pig, mouse, and dog.
The major metabolites reported are demethyl fenitrothion, demethyl
fenitrooxon, dimethylphosphorothioic acid and dimethyl phosphoric
acid, and 3-methyl-4-nitrophenol and its conjugates. Differences in
the composition of metabolites found among most laboratory test
animals and between sexes of the same species appear to be mainly
quantitative in nature. Only rabbits appear to excrete fenitrooxon
and aminofenitrooxon in small, though quantifiable, amounts in the
urine.
Evidence from studies on rabbits and dogs showed preferential
deposition of fenitrothion in the adipose tissue.
Residues found in the milk of cows following exposure to
fenitrothion were not detected two days later.
Though fenitrothion is readily absorbed via the oral route, it
is rapidly metabolized and excreted and is unlikely to remain in the
body for any prolonged period.
1.1.3 Effects on organisms in the environment
The concentrations of fenitrothion that are likely to be found
in the environment do not have any effects on microorganisms in soil
or water.
Fenitrothion is highly toxic for aquatic invertebrates in both
freshwater and seawater with LC50 values of a few µg/litre for
most species tested. The no-observed-effect level (NOEL) for
Daphnia, in 48-h tests, was < 2 µg/litre; in life-cycle tests, a
maximum acceptable toxicant concentration (MATC) of 0.14 µg per
litre was established. Field observations and studies on
experimental ponds have shown effects on populations of
invertebrates. However, most of the changes observed were temporary,
even at concentrations considerably higher than those likely to
occur after recommended usage.
Fish are less sensitive to fenitrothion than invertebrates and
show 96-h LC50 values in the range of 1.7-10 mg/litre. The most
sensitive life stage is the early larva. Long-term studies have
established a MATC at, or above, 0.1 mg/litre for 2 species of
freshwater fish. Field studies after application of fenitrothion to
forests showed no effects on wild populations of fish or on the
survival of caged test fish with measured water concentrations of
fenitrothion of up to 0.019 mg/litre. Repeated application of
fenitrothion to forests had no effect on fish populations.
In laboratory tests, freshwater molluscs showed LC50 values
in the range 1.2 to 15 mg/litre. No field effects were seen after
forest spraying at 140 g/ha.
Fenitrothion is highly toxic for bees (topical LD50,
0.03-0.04 µg/bee). Field effects have been reported with high
numbers of honey bees and other species killed locally. However, the
total numbers killed represented only a small percentage of the hive
population.
Acute oral LD50 values for birds range between 25 and 1190
mg/kg body weight and most 8-day dietary LC50s exceeded 5000 mg/kg
diet. NOEL values for reproduction were 10 mg/kg body weight for the
quail and 100 mg/kg body weight for the mallard. Song-bird deaths
occurred soon after application of fenitrothion at a rate of 280
g/ha and were markedly increased at 560 g/ha for species living in
the forest canopy. After spraying at 420 g/ha followed by 210 g/ha a
few days later, the reproductive success of White-throated Sparrows
was reduced. In many studies, song-birds showed inhibition of ChE
soon after the fenitrothion spraying of forests.
Field observations have not revealed any effects of
fenitrothion on populations of wild small mammals.
1.1.4 Effects on experimental animals and in vitro test systems
Fenitrothion is an organophosphate and causes cholinesterase
activity depression in plasma, red blood cells, and brain and liver
tissues. It is metabolized to fenitrooxon, which is more acutely
toxic. Its toxicity may be potentiated by some other organophosphate
compounds.
Fenitrothion is an insecticide of moderate toxicity with oral
LD50 values in rats and mice ranging from 330 to 1416 mg/kg body
weight. Acute dermal toxicity in rodents ranged from 890 mg/kg body
weight to more than 2500 mg/kg body weight.
Fenitrothion is only minimally irritating to the eyes and is
nonirritating to the skin. The chemical showed dermal sensitizing
potential in one of two studies on guinea-pigs.
Fenitrothion has been tested in short-term studies on rats,
dogs, guinea-pigs, and rabbits and in long-term carcinogenicity
studies on rats and mice. In short-term studies on rats and dogs,
the no-observed-adverse-effect levels (NOAELs), based on brain-ChE
activity, were, respectively, 10 mg/kg diet and 50 mg/kg diet.
Long-term studies on rats and mice indicated a NOAEL (based on
brain ChE activity) of 10 mg/kg diet.
No carcinogenic effects were found in any of the long-term
studies reported.
Fenitrothion was not mutagenic in in vitro and in vivo
studies.
Fenitrothion has not been found to be teratogenic at doses of
up to 30 mg/kg body weight in rabbits and up to 25 mg/kg body weight
in rats. Dose levels exceeding 8 mg/kg body weight were maternally
toxic.
Developing young rats exhibited behavioural deficits
post-natally following in utero exposure. A NOEL for this effect
was established at 5 mg/kg body weight per day.
Multigeneration reproduction studies on rats did not indicate
any morphological effects. A NOAEL of 120 mg/kg diet, based on
reproductive parameters, was demonstrated in these studies.
Delayed neurotoxicity has not been reported as a result of
exposure to fenitrothion.
1.1.5 Effects on human beings
Administration of fenitrothion as a single oral dose of 0.042-
0.33 mg/kg body weight and in repeated doses of 0.04-0.08 mg/kg body
weight to human volunteers did not cause inhibition in plasma and
erythrocyte ChE. The urinary excretion of a metabolite,
3-methyl-4-nitrophenol, was complete within 24 h.
Several cases of poisoning have occurred. The signs and
symptoms of poisoning were those of parasympathic stimulation. In a
few cases, the toxic manifestations were delayed in onset and
recurred for up to a few months. It has been suggested that the slow
release of the insecticide from adipose tissue can give rise to a
protracted clinical course or late symptoms of intoxication. In some
cases, contact dermatitis has been attributed to exposure to this
insecticide. There is no evidence of delayed neurotoxicity or of an
association with Reye's syndrome following exposure to fenitrothion.
Within WHO programmes, fenitrothion has been used in a few
countries for indoor residual spraying for malaria control
(application dose: 2.0 g of active ingredient/m2). No evidence of
toxicity was noted in thousands of inhabitants observed, with the
exception of one study in which less than 2% inhabitants reported
mild complaints. However, approximately 25% of spray operators
showed up to 50% inhibition of whole blood ChE activity. Following
aerial application of a 50% EC formulation, some workers developed
symptoms of poisoning and decreased whole blood ChE activity within
48 h. Occupational exposure for a period of over 5 years of male
workers in a production plant and female workers in the packaging
unit produced clinical signs and symptoms of poisoning in 15% of
male and 33% of female workers. The measured air concentration of
fenitrothion in the workplace ranged between 0.028 and 0.118
mg/m3.
1.2 Conclusions
* Fenitrothion is a moderately toxic organophosphorus ester
insecticide. However, over-exposure from handling during
manufacture or use and accidental or intentional ingestion may
cause serious poisoning.
* Exposure of the general population, resulting mainly from
agricultural and forestry practices and public health
programmes, should not constitute a health hazard.
* With good work practices, hygienic measures, and safety
precautions, fenitrothion is unlikely to present a hazard for
those occupationally exposed.
* Despite its high toxicity for non-target arthropods,
fenitrothion has been extensively used for pest control with
few, or no, adverse effects on populations in the environment.
1.3 Recommendations
* For the health and welfare of workers and the general
population, the handling and application of fenitrothion should
only be entrusted to competently supervised and well-trained
operators who will follow adequate safety measures and use
fenitrothion according to good application practices.
* The manufacture, formulation, use, and disposal of fenitrothion
should be carefully managed to minimize contamination of the
environment, particularly surface waters.
* Regularly exposed workers should receive periodic health
evaluations.
* Application rates of fenitrothion should be limited, to avoid
effects on non-target arthropods. The insecticide should never
be sprayed over water bodies or streams.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
Fenitrothion was first prepared in Czechoslovakia in 1956
(Drabek, Truchlik, 1957). Later, it was prepared independently by
Sumitomo Chemical Co. and by Bayer A.G. in 1959 and later by
American Cyanamid Co.
Its basic insecticidal activity was described by Nishizawa et
al. (1961).
2.1 Identity
Primary constituent
Chemical formula: C9H12NO5PS
Chemical structure:
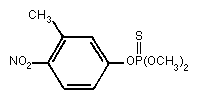 Relative molecular mass: 277.25
Common name: fenitrothion
CAS chemical name: O,O-dimethyl O-(3-methyl
-4-nitro-phenyl) phosphorothioate
IUPAC name: O,O-dimethyl O-(4-nitro-m-tolyl)
phosphorothioate
RTECS Registry number: TG0350000
CAS Registry number: 122-14-5
Synonyms: Accothion, Agrothion, Bayer 41831, Bayer S
5660, Cytel, Dybar, Fenitox, MEP,
Novathion, Nuvanol, Cyfen, Sumitomo 1102A
Technical product (FAO/WHO, 1988b)
Major trade names: Metathion, Novathion, Sumithion, Folithion
Purity: > 93% (Sumithion)
Impurities: O,O-dimethyl O-3-nitro- m-tolyl-
phosphorothioate < 1.5%
O-methyl O,O-bis(4-nitro- m-tolyl)
phosphorothioate < 2.5%
O-methyl S-methyl
O-(4-nitro- m-tolyl)
phosphorothioate(S-isomer) < 0.8%
O,O-dimethyl O-2-nitro- m-tolyl
phosphorothioate < 3.0%
O,O-dimethyl O-6-nitro- m-tolyl
phosphorothioate < 2.5%
O,O-dimethyl O-2,4-dinitro- m-tolyl
phosphorothioate < 2.0%
O,O-dimethyl O-4,6-dinitro- m-tolyl
phosphorothioate < 1.5%
3-methyl-4-nitrophenol < 0.5%
Isomeric composition: S-isomer, < 0.8%
2.2 Physical and chemical properties
Some physical and chemical properties of Fenitrothion are given
in Table 1.
2.3 Conversion factors
1 ppm = 11.5 mg/m3 (at 20 °C)
1 mg/m3 = 0.087 ppm.
2.4 Analytical methods
Methods for the determination of fenitrothion in foods,
environmental samples, technical products, and formulations are
summarized in Tables 2 and 3. The common procedure for determining
residues in foods and environmental media consists of (1)
extraction, (2) partition, (3) chromatographic separation
(clean-up), and (4) qualitative and quantitative analysis using
analytical instruments.
Fenitrothion levels in technical products and formulations are
usually determined by the diazo method, the colorimetric method, or
gas-liquid chromatography. The common procedure consists of: (1)
dissolution or extraction, (2) separation of impurities and (3)
determination. Granules should be pulverized before analysis.
The joint FAO/WHO Codex Alimentarius Commission has given
recommendations for the methods of analysis to be used for the
determination of fenitrothion residues (FAO/WHO, 1989d).
Table 1. Some physical and chemical properties of fenitrothiona
Physical state liquid
Colour yellow-brown
Odour chemical odour
Melting point 0.3 °C
Boiling point 140-145 °C (decomp.)/0.1 mmHg
Flash point 157 °C
Vapour pressure 18 mPa at 20 °C; 6 x 10-6 mmHg at 20 °C
25 25
Density d 1.32-1.34; d 1.3227
25 4
n-Octanol/water partition
coefficient (log P) 3.16
Solubility in water 14 mg/litre at 30 °C
Solubility in organic freely soluble in alcohols, esters,
solvents ketones, and aromatic hydrocarbons;
> 1000 g/kg dichloromethane, methanol,
xylene; 193 g/kg propan-2-ol; 42 g/kg
hexane at 20-25 °C
Stability hydrolysed by alkali: half-life
4.5 h in 0.01 N NaOH at 30 °C
decomposed by heat: 145 °C
a From: Martin & Worthing (1981); Worthing & Walker (1983);
Meister et al. (1985); Moody et al. (1987a).
Table 2. Analytical methods for the determination of fenitrothion in food and environmental samples
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
Residue analysis
apple, orange acetone ESd florisil acetone/ GC: FTD, NPD, FPD, 2% 0.1 95-96 (0.1) Ferreira &
peach, grape /2%Na2SO4 hexane DC-200 + 3% OF-1, 1.5% 86-97 (0.2) Fernandes
tomato, /n-hexane (4/96) OV-17 + 1.95% OF-1, 10% 88-102 (0.5) (1980)
cabbage DC-200 86-98 (1.0)
94-95 (2.0)
chinese methanol/ benzene GC: FPD, 3% OV-1, 89 (0.5) Talekar
cabbage acetone/ 10% DC-200 et al.
benzene (1977)
(Soxhlet)
onion acetonitrile Amberlite methanol GC: FPD, 4% OV-1, 200 °C 99 (0.5) Iwata
CH2Cl2/ XAD-8 benzene et al.
benzene (1:4) charcoal (1981)
apple,lettuce acetone ESd florisil benzene GC: ECD 0.1 70-102 Möllhoff
carrot,onion /CH3Cl3 5% SE-30, 190 °C, 6.3 min 64-89 (0.5) (1967)
tomato,potato 5% OF-1, 190 °C, 5.6 min 88-114 (1.0)
5% DC-200, 190 °C, 3.4 min
5% E-301, 190 °C, 2.9 min
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
orange,potato ethyl acetonitrile/ florisil petroleum sp/ GC:NPD, 0.001 Mestres
acetate petroleum ethyl et al.
CH2Cl2 spirit ether or (1974)
(4:1) ethyl
acetate
peach acetone water/ charcoal+ CH2Cl2 GC:NPD Ambrus et al.
potato CH2Cl2 MgO 3% OV-22; 3% OV-101; (1981)
1.95% SP-2401/1.5%
acetone alumina N hexane SP-2250; 3% NPGS;
3% SE-30 on 100-120
mesh Gas-Chrom Q
acetone silica gel benzene
5% water
apple acetonitrile ESd HPLC: UV (280 nm) 0.03 81-87 (0.5) Funch (1981)
salad 2% Nacl Radpak A with ODS
/CH2Cl2
unpolished n-hexane I. n-hexane charcoal/ acetone/ GC: FPD Aoki et al.
rice (Soxhlet) /CH3CN avicel n-hexane 10% DC-200 + 15% 76 (0.4) (1975)
(1/10) (50/50)
wheat OF-1, 180 °C, 7.9 min
buck wheat II. CH3CN 2% DEGS + 0.5% H3PO4, 0.0007 92 (0.4)
string bean /5% NaCl/ 180 °C, 10.2 min
soybean benzene 10% DC-200 + 15% 0.0008 79-101 (0.4)
pear OV-17, 190 °C, 9.4 min
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
water melon acetone ESd GC-MS 0.1 91-102 (0.5) Kobayashi
tomato NaCl/ 5% OV-1, 150 °C et al.
n-hexane (1979)
wheat grain methanol GC: AFI, FPD, 5% 0.1 86 (0.5) Smart
OV-17 + Epikote 1001 94 (1.0) (1980)
apple methanol/ GC: FTD 0.005 80-97 (0.1) Takimoto &
strawberry CH3CN/CHCl3 10% DC-200 + 20% OF-1, 97 (0.2) Miyamoto
pear, tomato 210 °C (1976b)
cucumber
potato methanol/ florisil benzene/ GC: FTD, 10% DC-200 0.005 96 (0.01) Takimoto &
CH3CN/CHCl3 ethyl + 20% OF-1, 210 °C Miyamoto
acetate (1976b)
(10/1)
soybean methanol/ CH3CN/ silica benzene 0.005 98 (0.2, 0.02) Takimoto &
(fresh) CH3CN/ n-hexane gel OF-1, 210 °C Miyamoto
CH3Cl3 (1976b)
green tea CH3CN/ CH3CN/ florisil benzene/ 0.005 90-93 (0.02) Takimoto &
rice grain benzene n-hexane ethyl 92-98 (0.5) Miyamoto
acetate/ (1976b)
(10/1)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
milk methanol/ CH3CN/ 0.005 89 (0.1) Takimoto &
CH3CN/ n-hexane Miyamoto
CHCl3 (1976b)
butter hexane I. hexane/ GC: ECD, 5% DC-200, 0.02 95 Gajduskova
CH3CN 180 °C (1974)
II. aq.
Na2SO4
CH3CN/
CH2Cl2
milk acetone I. acetone silica benzene GC: FPD, 10% DC-200, 0.001 96 (0.5), Bowman &
CH2Cl2 gel Bcraza
II. hexane/ (20% H2O) 180 °C 2.9 min 94 (0.05) (1969)
CH3CN
meat ethanol/ CH3CN/ TLC benzene/ GC: FTD, 10% DC-200 0.005 95 (0.1), Takimoto &
benzene n-hexane ethyl + 20% OF-1 86 (0.2) Miyamoto
acetate (1976b)
(4/1)
lettuce, petroleum- petroleum- TLC methanol Spectrophotometry 0.05- 89-115 Kovac &
apple ether ether (A12O3) 400 nm, after hydrolysis 0.1 (0.25-1) Sohler
cherries +CH3CN(1:1) (NaOH+ (1965)
plums H2O2) Cerna &
kohlrabi Benes
cauliflower (1972)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
cabbage
citrus, acetone ESd silica hexane/ TLC 3% OV-225 ng Gardner
potato CH3CCl3 gel acetone 170-240 °C 0.05 94 (0.5) (1971)
(9/1) Martindale
(1988)
Environmental analysis
water amberlite ethyl HPLC: UV (245 nm) RP-8 0.001 94 (0.05- Volpe &
XAD-4 acetate CH3CN/H2O (50/50) Mallet
(1981)
water amberlite CH2Cl2 or GC: FPD, 3.6% OV-101 90 (0.05- Volpe &
XAD-4 or 7 ethyl + 5% OV-210 0.005) Mallet
acetate (1980)
drinking- amberlite acetone/ GC: NPD, -MS, 3% 0.001 104 (0.1), LeBel
water XAD-2 hexane OV-17 96 (0.01) et al.
(15/85) (1979)
water amberlite ethyl GC: FPD, 4% OV-101 + 0.001 91.5 (0.01) Mallet
acetate et al.
XAD-2 6% OV-210, 195 °C (1978)
water uBondapak HPLC: UV (280 0.005 95.6 (0.737) Takaku
Phenyl uBondapak Phenyl H2O/ et al.
CH3CN (100/0-0/100) (1979a)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
water petroleum GC: FPD, 5% SE-30, 180 °C 98-102 Grift &
ether 5% DC-200, 180 °C (0.005-0.0005) Lockhart
(1974)
pasture CH3CN/ CH3CN/ florisil benzene/ GC: FTD, 10% DC-200 + 0.005 95 (0.1) Takimoto &
ethyl
grass benzene n-hexane acetate 20% OF-1, 210 °C Miyamoto
(1976b)
corn methanol/ silica benzene GC: FPD, 10% DC-200, 0.002 99-100 (5) Bowman &
gel Bcraza
grass CHCl3 (20% H2O) 180 °C, 2.9 min 98-99 (0.5) (1969)
97-98 (0.05)
jack pine ethyl I.carbon/ benzene GC: NPD, 6% SE-30 + 96-100 (0.2) McNeil
foliage acetate celite 4% OF-1, 225 °C et al.
(1979)
(1/6) II. 60%
florisil benzene
Si600 in hexane
water n-hexane GC: FPD, 11% OV-17 + 0.00001 100-108 Ripley
OF-1 3.6% OV-101, 225 °C (0.01-0.0001) et al.
(1974)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
fish ethyl ESd florisil benzene/ GC: FPD 0.05 93-110 Grift &
ethyl Lockhart
acetate CH3CN acetate 5% DC-200, 180 °C (1974)
sediment /hexane 5% SE-30, 180 °C 0.05 88-102 (5-0.1)
bivalve ethyl bio- CH2Cl2/ GC: FPD, -MS, 3% OV- 0.0009 94.4 (10-0.01) Sergeant
acetate beads, cyclohexane 101, 180 °C, 11% OV-17/ et al.
(Soxhlet) SX-3 OF-1, 200 °C (1979)
(50/50)
soil acetonitrile Amberlite ethyl GC: FPD, 3.6% OV-17 +0.05 98-112 (0.5) Hache
XAD-2 acetate et al.
chicken liver 5% OV-210, 210 °C (1981)
wine, clam
pine needle
soil acetone acetone florisil benzene GC: ECD 0.1 58-100 (0.1) Möllhoff
/CHCl3 5% SE-30, 190 °C, 6.3 min 76-98 (0.5) (1967)
5% QF-1, 190 °C, 5.6 min 98-122 (1.0)
5% DC-200, 190 °C, 3.4 min
5% E-301, 190 °C, 2.9 min
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
Ambient air
vapour trapped on 10% OV-101 on chromosorb GC: FPD, 3% OV-1, 5 x Krzymien
aerosol W packed in a glass tube and 205 °C 10-12 (1979)
thermally released and carried to GC column
aerosol consecutive plates of cascade 5 x 100 (30) Krzymien
impactor, plates washed 10-12 (1979)
with n-hexane, n-hexane solution
injected into GC
a Detectors for GC (FPD = flame photometric detector; FTD = flame thermionic detector; ECD = electron capture detector;
NPD = NP specific detector; AFI = alkali flame ionization detector), MS = mass spectrometry.
b RT = Retention time.
c MDC = minimum detectable concentration.
d ES = extraction solvent.
Table 3. Analytical methods for fenitrothion in technical products and formulationsa
Sample Sample preparation Determination
Diazo method
TG and EC dissolution (ether) reduction (Zn-acetic acid)
partition (ether/1% Na2CO3) titration (NaNO2)
end-point (potentiometer or
iodide-starch paper)
Colorimetric method
TG and EC dissolution (methanol) addition (1% Na2CO3)
determination (free NMC);
WP and dust extraction (methanol) 400 nm hydrolysis (5N KOH)
determination (total NMC);
Granule pulverization extraction (methanol) 400 nm
TLC-UV method
TG and EC dissolution (CHCl3) determination; 271 nm
TLC (benzene/diethyl ether=19/1)
WP extraction (methanol)
TLC (benzene/diethyl ether=19/1)
Dust extraction (CHCl3)
TLC (benzene/diethyl ether=19/1)
Granule pulverization extraction (CHCl3)
TLC (benzene/diethyl ether=19/1)
TLC-phosphorus method
TG and EC dissolution (CHCl3) TLC digestion (H2SO4 and HNO3)
colouring (ammonium metavanadate
WP extraction (methanol) TLC and ammonium molybdate)
determination;
Dust extraction (CHCl3) TLC 420 nm
Table 3. (cont'd).
Sample Sample preparation Determination
Granule pulverization extraction (CHCl3) TLC
GC method
TG and EC dissolution (IS solution) GC: FID
2% DC-QF-1, 170 °C
WP and dust extraction (IS solution) centrifuge
Granule pulverization extraction (IS solution)
centrifuge
a From: Takimoto et al. (1975).
TG = technical grade; EC = emulsifiable concentrate; WP = water-dispersible
powder; NMC = 3-methyl-4-nitrophenol; IS = internal standard (dibutyl
sebacate);
GC = gas-liquid chromatography.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Fenitrothion is not a natural product.
3.2 Man-made sources
3.2.1 Production
The global production volume is not available. However, the
global manufacturing capacity has been estimated to be between 15
000 and 20 000 tonnes. Production figures in Japan (a major
manufacturing country) are 5346 tonnes in 1982 increasing up to
about 10 000 tonnes in 1988 (Japan Plant Protection Association,
1984, 1986, 1988, 1989). Production in India was reported to be 400
tonnes in 1978, 350 tonnes in 1979, and 100 tonnes in 1980
(Battelle, 1982). Production in Czechoslovakia in 1989 was 964
tonnes (Benes, personal communication).
Fenitrothion is formulated as an emulsifiable concentrate
(50%), an ultra-low-volume concentrate, flowable (20%), a wettable
powder (40%), granules (3%), dust (3%), an oil-based liquid spray
alone or in combination with other pesticides, e.g., trichlorfon,
malathion; BPMC; fenvalerate (insecticide), tetramethrin (house-hold
insecticide); IBP, phthalide, thiophanate-methyl (fungicide).
3.2.2 Uses
Fenitrothion is mainly used in agriculture for controlling
chewing and sucking insects on rice, cereals, fruits, vegetables,
stored grains, cotton, and in forest areas. It is also used for the
control of flies, mosquitos, and cockroaches in public health
programmes and/or indoor use.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
Several studies were performed to elucidate the mechanism of
the apparent rapid disappearance of fenitrothion from the water
phase after the field spraying of fenitrothion formulation. The
processes most likely to explain the phenomena include sorption by
the sediments, photolysis, microbial degradation, hydrolysis, and
volatilization.
Marshall & Roberts (1977) discounted volatilization as a major
pathway for the disappearance on the basis of the calculated
half-life of 93 days obtained in a 1-m water column, designed as a
simple model for small, well-mixed lentic systems with compartments
representing the major pools, i.e., the water, the hydrosol, and the
suspended solids, which include both biotic and abiotic material.
Maguire & Hale (1980) studied the kinetics of fenitrothion
distribution and transformation in water and sediment, both
experimentally and after field spraying (see section 5.1.2 for
results after field spraying). Laboratory experiments demonstrated
that volatilization of fenitrothion from true solutions (5 mg/litre)
in distilled water followed first-order kinetics and that the
half-life of disappearance at 20 °C was estimated to be 64 ± 5 days.
The fact that the half-life was considerably longer (> 180 days) in
the presence of 5 mg fulvic acid/litre indicated that the rate would
be considerably reduced in natural waters too. In contrast with
this, the volatilization of fenitrothion that had been sprayed on
the surface of water appeared to be a very fast process in the
laboratory (half-life = 18 min for volatilization from the surface
of water). Surface volatilization was suggested to play a
significant role in the dissipation of fenitrothion from a small
pond after spraying the formulation.
Metcalf et al. (1980) also reported the significance of
volatilization in the disappearance of fenitrothion in a lake by
taking account of the effects of winds and water currents on natural
water bodies in experiments using various rates of aeration.
Observed half-lives of fenitrothion in Palfrey Lake and Brook in
Southwestern New Brunswick (water temperature: 11 °C, average pH
value in the lake: 6.7) were 6.3 days (bottom) - 7.2 days (surface)
and 0.9 days, respectively.
In a laboratory leaching study, 14C fenitrothion and its
degradation products hardly moved with water in 3 loam soils,
whereas, in Muko sand containing 0.2% clay and less than 0.1%
organic matter, about 15% of the applied 14C was eluted from the
soil column. Preincubation of the fenitrothion in sandy soil for 60
days before leaching decreased the degree of mobility. In the
effluent from sandy soil, a trace amount of fenitrothion (0-0.1%)
together with water-soluble products, such as
3-methyl-4-nitro-phenol [9]1 (0.6%) and amino-fenitrothion [13]
(11.3%), were detected (Takimoto et al., 1976, see Fig. 3).
Baarschers et al. (1983) examined the adsorption of
fenitrothion and 3-methyl-4-nitrophenol [9] in water-soil suspension
systems, using 4 different soils and 1 sediment as adsorbents. The
Freundlich k values were 15.5-354.8 for fenitrothion and 2.1-147.8
for 3-methyl-4-nitrophenol [9]. Both of the k values increased when
the organic matter content increased from 0.9 to 33.1%. The
adsorption characteristics of fenitrothion may be correlated with
the lower degree of mobility in the leaching study.
4.2 Abiotic and biotic transformation
4.2.1 Abiotic transformation
4.2.1.1 Thermal degradation
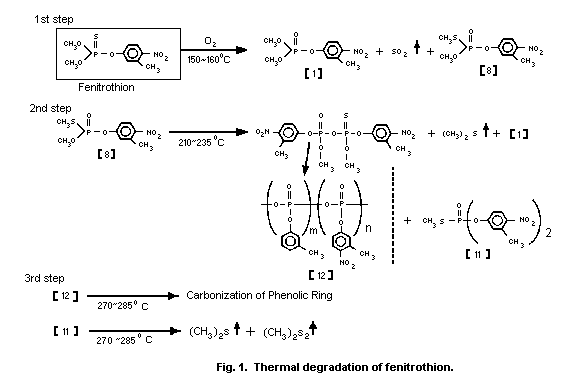
Tsuji et al. (1980) examined the mechanisms of thermal
degradation of fenitrothion in air and found that 3 major exothermic
steps were involved (Fig. 1). The first step was formation of
fenitrooxon [1], and S-methyl fenitrothion [8] with evolution of
sulfur dioxide at 150-160 °C.
The second step was formation of fenitrooxon [1], S-methyl
O,O-bis(3-methyl-4-nitrophenyl) phosphorothioate [11] and
polymetaphosphate [12] with evolution of dimethyl sulfide from
S-methyl fenitrothion [8] at 210-235 °C. The third step was
carbonization of the phenolic ring of [12] and gas evolution from
[11] at 270-285 °C. In a nitrogen atmosphere, the first step did not
take place and only the other two steps were involved. S-methyl
fenitrothion was produced by heating fenitrothion up to 193 °C.
The thermal degradation of fenitrothion in a closed system was
more rapid than that in an open system. Maeda et al. (1982) proposed
that dimethyl sulfide, which was evolved during thermal degradation
of S-methyl fenitrothion [8], catalysed isomerization of
fenitrothion to [8]. In fact, addition of 0.4-1.6% of dimethyl
sulfide in a closed system enhanced the degradation of fenitrothion.
Although metal salts, such as zinc, aluminum, ferric chloride, and
stannic chloride, also accelerated isomerization of fenitrothion,
calcium dodecylbenzene sulfonate (surfactant) showed no effect.
1 Chemical structures in Fig. 1-7 are referred to giving the
numbers in brackets.
Relative molecular mass: 277.25
Common name: fenitrothion
CAS chemical name: O,O-dimethyl O-(3-methyl
-4-nitro-phenyl) phosphorothioate
IUPAC name: O,O-dimethyl O-(4-nitro-m-tolyl)
phosphorothioate
RTECS Registry number: TG0350000
CAS Registry number: 122-14-5
Synonyms: Accothion, Agrothion, Bayer 41831, Bayer S
5660, Cytel, Dybar, Fenitox, MEP,
Novathion, Nuvanol, Cyfen, Sumitomo 1102A
Technical product (FAO/WHO, 1988b)
Major trade names: Metathion, Novathion, Sumithion, Folithion
Purity: > 93% (Sumithion)
Impurities: O,O-dimethyl O-3-nitro- m-tolyl-
phosphorothioate < 1.5%
O-methyl O,O-bis(4-nitro- m-tolyl)
phosphorothioate < 2.5%
O-methyl S-methyl
O-(4-nitro- m-tolyl)
phosphorothioate(S-isomer) < 0.8%
O,O-dimethyl O-2-nitro- m-tolyl
phosphorothioate < 3.0%
O,O-dimethyl O-6-nitro- m-tolyl
phosphorothioate < 2.5%
O,O-dimethyl O-2,4-dinitro- m-tolyl
phosphorothioate < 2.0%
O,O-dimethyl O-4,6-dinitro- m-tolyl
phosphorothioate < 1.5%
3-methyl-4-nitrophenol < 0.5%
Isomeric composition: S-isomer, < 0.8%
2.2 Physical and chemical properties
Some physical and chemical properties of Fenitrothion are given
in Table 1.
2.3 Conversion factors
1 ppm = 11.5 mg/m3 (at 20 °C)
1 mg/m3 = 0.087 ppm.
2.4 Analytical methods
Methods for the determination of fenitrothion in foods,
environmental samples, technical products, and formulations are
summarized in Tables 2 and 3. The common procedure for determining
residues in foods and environmental media consists of (1)
extraction, (2) partition, (3) chromatographic separation
(clean-up), and (4) qualitative and quantitative analysis using
analytical instruments.
Fenitrothion levels in technical products and formulations are
usually determined by the diazo method, the colorimetric method, or
gas-liquid chromatography. The common procedure consists of: (1)
dissolution or extraction, (2) separation of impurities and (3)
determination. Granules should be pulverized before analysis.
The joint FAO/WHO Codex Alimentarius Commission has given
recommendations for the methods of analysis to be used for the
determination of fenitrothion residues (FAO/WHO, 1989d).
Table 1. Some physical and chemical properties of fenitrothiona
Physical state liquid
Colour yellow-brown
Odour chemical odour
Melting point 0.3 °C
Boiling point 140-145 °C (decomp.)/0.1 mmHg
Flash point 157 °C
Vapour pressure 18 mPa at 20 °C; 6 x 10-6 mmHg at 20 °C
25 25
Density d 1.32-1.34; d 1.3227
25 4
n-Octanol/water partition
coefficient (log P) 3.16
Solubility in water 14 mg/litre at 30 °C
Solubility in organic freely soluble in alcohols, esters,
solvents ketones, and aromatic hydrocarbons;
> 1000 g/kg dichloromethane, methanol,
xylene; 193 g/kg propan-2-ol; 42 g/kg
hexane at 20-25 °C
Stability hydrolysed by alkali: half-life
4.5 h in 0.01 N NaOH at 30 °C
decomposed by heat: 145 °C
a From: Martin & Worthing (1981); Worthing & Walker (1983);
Meister et al. (1985); Moody et al. (1987a).
Table 2. Analytical methods for the determination of fenitrothion in food and environmental samples
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
Residue analysis
apple, orange acetone ESd florisil acetone/ GC: FTD, NPD, FPD, 2% 0.1 95-96 (0.1) Ferreira &
peach, grape /2%Na2SO4 hexane DC-200 + 3% OF-1, 1.5% 86-97 (0.2) Fernandes
tomato, /n-hexane (4/96) OV-17 + 1.95% OF-1, 10% 88-102 (0.5) (1980)
cabbage DC-200 86-98 (1.0)
94-95 (2.0)
chinese methanol/ benzene GC: FPD, 3% OV-1, 89 (0.5) Talekar
cabbage acetone/ 10% DC-200 et al.
benzene (1977)
(Soxhlet)
onion acetonitrile Amberlite methanol GC: FPD, 4% OV-1, 200 °C 99 (0.5) Iwata
CH2Cl2/ XAD-8 benzene et al.
benzene (1:4) charcoal (1981)
apple,lettuce acetone ESd florisil benzene GC: ECD 0.1 70-102 Möllhoff
carrot,onion /CH3Cl3 5% SE-30, 190 °C, 6.3 min 64-89 (0.5) (1967)
tomato,potato 5% OF-1, 190 °C, 5.6 min 88-114 (1.0)
5% DC-200, 190 °C, 3.4 min
5% E-301, 190 °C, 2.9 min
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
orange,potato ethyl acetonitrile/ florisil petroleum sp/ GC:NPD, 0.001 Mestres
acetate petroleum ethyl et al.
CH2Cl2 spirit ether or (1974)
(4:1) ethyl
acetate
peach acetone water/ charcoal+ CH2Cl2 GC:NPD Ambrus et al.
potato CH2Cl2 MgO 3% OV-22; 3% OV-101; (1981)
1.95% SP-2401/1.5%
acetone alumina N hexane SP-2250; 3% NPGS;
3% SE-30 on 100-120
mesh Gas-Chrom Q
acetone silica gel benzene
5% water
apple acetonitrile ESd HPLC: UV (280 nm) 0.03 81-87 (0.5) Funch (1981)
salad 2% Nacl Radpak A with ODS
/CH2Cl2
unpolished n-hexane I. n-hexane charcoal/ acetone/ GC: FPD Aoki et al.
rice (Soxhlet) /CH3CN avicel n-hexane 10% DC-200 + 15% 76 (0.4) (1975)
(1/10) (50/50)
wheat OF-1, 180 °C, 7.9 min
buck wheat II. CH3CN 2% DEGS + 0.5% H3PO4, 0.0007 92 (0.4)
string bean /5% NaCl/ 180 °C, 10.2 min
soybean benzene 10% DC-200 + 15% 0.0008 79-101 (0.4)
pear OV-17, 190 °C, 9.4 min
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
water melon acetone ESd GC-MS 0.1 91-102 (0.5) Kobayashi
tomato NaCl/ 5% OV-1, 150 °C et al.
n-hexane (1979)
wheat grain methanol GC: AFI, FPD, 5% 0.1 86 (0.5) Smart
OV-17 + Epikote 1001 94 (1.0) (1980)
apple methanol/ GC: FTD 0.005 80-97 (0.1) Takimoto &
strawberry CH3CN/CHCl3 10% DC-200 + 20% OF-1, 97 (0.2) Miyamoto
pear, tomato 210 °C (1976b)
cucumber
potato methanol/ florisil benzene/ GC: FTD, 10% DC-200 0.005 96 (0.01) Takimoto &
CH3CN/CHCl3 ethyl + 20% OF-1, 210 °C Miyamoto
acetate (1976b)
(10/1)
soybean methanol/ CH3CN/ silica benzene 0.005 98 (0.2, 0.02) Takimoto &
(fresh) CH3CN/ n-hexane gel OF-1, 210 °C Miyamoto
CH3Cl3 (1976b)
green tea CH3CN/ CH3CN/ florisil benzene/ 0.005 90-93 (0.02) Takimoto &
rice grain benzene n-hexane ethyl 92-98 (0.5) Miyamoto
acetate/ (1976b)
(10/1)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
milk methanol/ CH3CN/ 0.005 89 (0.1) Takimoto &
CH3CN/ n-hexane Miyamoto
CHCl3 (1976b)
butter hexane I. hexane/ GC: ECD, 5% DC-200, 0.02 95 Gajduskova
CH3CN 180 °C (1974)
II. aq.
Na2SO4
CH3CN/
CH2Cl2
milk acetone I. acetone silica benzene GC: FPD, 10% DC-200, 0.001 96 (0.5), Bowman &
CH2Cl2 gel Bcraza
II. hexane/ (20% H2O) 180 °C 2.9 min 94 (0.05) (1969)
CH3CN
meat ethanol/ CH3CN/ TLC benzene/ GC: FTD, 10% DC-200 0.005 95 (0.1), Takimoto &
benzene n-hexane ethyl + 20% OF-1 86 (0.2) Miyamoto
acetate (1976b)
(4/1)
lettuce, petroleum- petroleum- TLC methanol Spectrophotometry 0.05- 89-115 Kovac &
apple ether ether (A12O3) 400 nm, after hydrolysis 0.1 (0.25-1) Sohler
cherries +CH3CN(1:1) (NaOH+ (1965)
plums H2O2) Cerna &
kohlrabi Benes
cauliflower (1972)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
cabbage
citrus, acetone ESd silica hexane/ TLC 3% OV-225 ng Gardner
potato CH3CCl3 gel acetone 170-240 °C 0.05 94 (0.5) (1971)
(9/1) Martindale
(1988)
Environmental analysis
water amberlite ethyl HPLC: UV (245 nm) RP-8 0.001 94 (0.05- Volpe &
XAD-4 acetate CH3CN/H2O (50/50) Mallet
(1981)
water amberlite CH2Cl2 or GC: FPD, 3.6% OV-101 90 (0.05- Volpe &
XAD-4 or 7 ethyl + 5% OV-210 0.005) Mallet
acetate (1980)
drinking- amberlite acetone/ GC: NPD, -MS, 3% 0.001 104 (0.1), LeBel
water XAD-2 hexane OV-17 96 (0.01) et al.
(15/85) (1979)
water amberlite ethyl GC: FPD, 4% OV-101 + 0.001 91.5 (0.01) Mallet
acetate et al.
XAD-2 6% OV-210, 195 °C (1978)
water uBondapak HPLC: UV (280 0.005 95.6 (0.737) Takaku
Phenyl uBondapak Phenyl H2O/ et al.
CH3CN (100/0-0/100) (1979a)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
water petroleum GC: FPD, 5% SE-30, 180 °C 98-102 Grift &
ether 5% DC-200, 180 °C (0.005-0.0005) Lockhart
(1974)
pasture CH3CN/ CH3CN/ florisil benzene/ GC: FTD, 10% DC-200 + 0.005 95 (0.1) Takimoto &
ethyl
grass benzene n-hexane acetate 20% OF-1, 210 °C Miyamoto
(1976b)
corn methanol/ silica benzene GC: FPD, 10% DC-200, 0.002 99-100 (5) Bowman &
gel Bcraza
grass CHCl3 (20% H2O) 180 °C, 2.9 min 98-99 (0.5) (1969)
97-98 (0.05)
jack pine ethyl I.carbon/ benzene GC: NPD, 6% SE-30 + 96-100 (0.2) McNeil
foliage acetate celite 4% OF-1, 225 °C et al.
(1979)
(1/6) II. 60%
florisil benzene
Si600 in hexane
water n-hexane GC: FPD, 11% OV-17 + 0.00001 100-108 Ripley
OF-1 3.6% OV-101, 225 °C (0.01-0.0001) et al.
(1974)
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
fish ethyl ESd florisil benzene/ GC: FPD 0.05 93-110 Grift &
ethyl Lockhart
acetate CH3CN acetate 5% DC-200, 180 °C (1974)
sediment /hexane 5% SE-30, 180 °C 0.05 88-102 (5-0.1)
bivalve ethyl bio- CH2Cl2/ GC: FPD, -MS, 3% OV- 0.0009 94.4 (10-0.01) Sergeant
acetate beads, cyclohexane 101, 180 °C, 11% OV-17/ et al.
(Soxhlet) SX-3 OF-1, 200 °C (1979)
(50/50)
soil acetonitrile Amberlite ethyl GC: FPD, 3.6% OV-17 +0.05 98-112 (0.5) Hache
XAD-2 acetate et al.
chicken liver 5% OV-210, 210 °C (1981)
wine, clam
pine needle
soil acetone acetone florisil benzene GC: ECD 0.1 58-100 (0.1) Möllhoff
/CHCl3 5% SE-30, 190 °C, 6.3 min 76-98 (0.5) (1967)
5% QF-1, 190 °C, 5.6 min 98-122 (1.0)
5% DC-200, 190 °C, 3.4 min
5% E-301, 190 °C, 2.9 min
Table 2 (contd).
Sample Sample preparation Determination GLC MDCc Recovery (%), Reference
Extraction Partition Clean-up Elution or HPLC conditions; (mg/kg) (fortification
solvent column detectora, column, level, mg/kg)
temperature, RTb
Ambient air
vapour trapped on 10% OV-101 on chromosorb GC: FPD, 3% OV-1, 5 x Krzymien
aerosol W packed in a glass tube and 205 °C 10-12 (1979)
thermally released and carried to GC column
aerosol consecutive plates of cascade 5 x 100 (30) Krzymien
impactor, plates washed 10-12 (1979)
with n-hexane, n-hexane solution
injected into GC
a Detectors for GC (FPD = flame photometric detector; FTD = flame thermionic detector; ECD = electron capture detector;
NPD = NP specific detector; AFI = alkali flame ionization detector), MS = mass spectrometry.
b RT = Retention time.
c MDC = minimum detectable concentration.
d ES = extraction solvent.
Table 3. Analytical methods for fenitrothion in technical products and formulationsa
Sample Sample preparation Determination
Diazo method
TG and EC dissolution (ether) reduction (Zn-acetic acid)
partition (ether/1% Na2CO3) titration (NaNO2)
end-point (potentiometer or
iodide-starch paper)
Colorimetric method
TG and EC dissolution (methanol) addition (1% Na2CO3)
determination (free NMC);
WP and dust extraction (methanol) 400 nm hydrolysis (5N KOH)
determination (total NMC);
Granule pulverization extraction (methanol) 400 nm
TLC-UV method
TG and EC dissolution (CHCl3) determination; 271 nm
TLC (benzene/diethyl ether=19/1)
WP extraction (methanol)
TLC (benzene/diethyl ether=19/1)
Dust extraction (CHCl3)
TLC (benzene/diethyl ether=19/1)
Granule pulverization extraction (CHCl3)
TLC (benzene/diethyl ether=19/1)
TLC-phosphorus method
TG and EC dissolution (CHCl3) TLC digestion (H2SO4 and HNO3)
colouring (ammonium metavanadate
WP extraction (methanol) TLC and ammonium molybdate)
determination;
Dust extraction (CHCl3) TLC 420 nm
Table 3. (cont'd).
Sample Sample preparation Determination
Granule pulverization extraction (CHCl3) TLC
GC method
TG and EC dissolution (IS solution) GC: FID
2% DC-QF-1, 170 °C
WP and dust extraction (IS solution) centrifuge
Granule pulverization extraction (IS solution)
centrifuge
a From: Takimoto et al. (1975).
TG = technical grade; EC = emulsifiable concentrate; WP = water-dispersible
powder; NMC = 3-methyl-4-nitrophenol; IS = internal standard (dibutyl
sebacate);
GC = gas-liquid chromatography.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Fenitrothion is not a natural product.
3.2 Man-made sources
3.2.1 Production
The global production volume is not available. However, the
global manufacturing capacity has been estimated to be between 15
000 and 20 000 tonnes. Production figures in Japan (a major
manufacturing country) are 5346 tonnes in 1982 increasing up to
about 10 000 tonnes in 1988 (Japan Plant Protection Association,
1984, 1986, 1988, 1989). Production in India was reported to be 400
tonnes in 1978, 350 tonnes in 1979, and 100 tonnes in 1980
(Battelle, 1982). Production in Czechoslovakia in 1989 was 964
tonnes (Benes, personal communication).
Fenitrothion is formulated as an emulsifiable concentrate
(50%), an ultra-low-volume concentrate, flowable (20%), a wettable
powder (40%), granules (3%), dust (3%), an oil-based liquid spray
alone or in combination with other pesticides, e.g., trichlorfon,
malathion; BPMC; fenvalerate (insecticide), tetramethrin (house-hold
insecticide); IBP, phthalide, thiophanate-methyl (fungicide).
3.2.2 Uses
Fenitrothion is mainly used in agriculture for controlling
chewing and sucking insects on rice, cereals, fruits, vegetables,
stored grains, cotton, and in forest areas. It is also used for the
control of flies, mosquitos, and cockroaches in public health
programmes and/or indoor use.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
Several studies were performed to elucidate the mechanism of
the apparent rapid disappearance of fenitrothion from the water
phase after the field spraying of fenitrothion formulation. The
processes most likely to explain the phenomena include sorption by
the sediments, photolysis, microbial degradation, hydrolysis, and
volatilization.
Marshall & Roberts (1977) discounted volatilization as a major
pathway for the disappearance on the basis of the calculated
half-life of 93 days obtained in a 1-m water column, designed as a
simple model for small, well-mixed lentic systems with compartments
representing the major pools, i.e., the water, the hydrosol, and the
suspended solids, which include both biotic and abiotic material.
Maguire & Hale (1980) studied the kinetics of fenitrothion
distribution and transformation in water and sediment, both
experimentally and after field spraying (see section 5.1.2 for
results after field spraying). Laboratory experiments demonstrated
that volatilization of fenitrothion from true solutions (5 mg/litre)
in distilled water followed first-order kinetics and that the
half-life of disappearance at 20 °C was estimated to be 64 ± 5 days.
The fact that the half-life was considerably longer (> 180 days) in
the presence of 5 mg fulvic acid/litre indicated that the rate would
be considerably reduced in natural waters too. In contrast with
this, the volatilization of fenitrothion that had been sprayed on
the surface of water appeared to be a very fast process in the
laboratory (half-life = 18 min for volatilization from the surface
of water). Surface volatilization was suggested to play a
significant role in the dissipation of fenitrothion from a small
pond after spraying the formulation.
Metcalf et al. (1980) also reported the significance of
volatilization in the disappearance of fenitrothion in a lake by
taking account of the effects of winds and water currents on natural
water bodies in experiments using various rates of aeration.
Observed half-lives of fenitrothion in Palfrey Lake and Brook in
Southwestern New Brunswick (water temperature: 11 °C, average pH
value in the lake: 6.7) were 6.3 days (bottom) - 7.2 days (surface)
and 0.9 days, respectively.
In a laboratory leaching study, 14C fenitrothion and its
degradation products hardly moved with water in 3 loam soils,
whereas, in Muko sand containing 0.2% clay and less than 0.1%
organic matter, about 15% of the applied 14C was eluted from the
soil column. Preincubation of the fenitrothion in sandy soil for 60
days before leaching decreased the degree of mobility. In the
effluent from sandy soil, a trace amount of fenitrothion (0-0.1%)
together with water-soluble products, such as
3-methyl-4-nitro-phenol [9]1 (0.6%) and amino-fenitrothion [13]
(11.3%), were detected (Takimoto et al., 1976, see Fig. 3).
Baarschers et al. (1983) examined the adsorption of
fenitrothion and 3-methyl-4-nitrophenol [9] in water-soil suspension
systems, using 4 different soils and 1 sediment as adsorbents. The
Freundlich k values were 15.5-354.8 for fenitrothion and 2.1-147.8
for 3-methyl-4-nitrophenol [9]. Both of the k values increased when
the organic matter content increased from 0.9 to 33.1%. The
adsorption characteristics of fenitrothion may be correlated with
the lower degree of mobility in the leaching study.
4.2 Abiotic and biotic transformation
4.2.1 Abiotic transformation
4.2.1.1 Thermal degradation
Tsuji et al. (1980) examined the mechanisms of thermal
degradation of fenitrothion in air and found that 3 major exothermic
steps were involved (Fig. 1). The first step was formation of
fenitrooxon [1], and S-methyl fenitrothion [8] with evolution of
sulfur dioxide at 150-160 °C.
The second step was formation of fenitrooxon [1], S-methyl
O,O-bis(3-methyl-4-nitrophenyl) phosphorothioate [11] and
polymetaphosphate [12] with evolution of dimethyl sulfide from
S-methyl fenitrothion [8] at 210-235 °C. The third step was
carbonization of the phenolic ring of [12] and gas evolution from
[11] at 270-285 °C. In a nitrogen atmosphere, the first step did not
take place and only the other two steps were involved. S-methyl
fenitrothion was produced by heating fenitrothion up to 193 °C.
The thermal degradation of fenitrothion in a closed system was
more rapid than that in an open system. Maeda et al. (1982) proposed
that dimethyl sulfide, which was evolved during thermal degradation
of S-methyl fenitrothion [8], catalysed isomerization of
fenitrothion to [8]. In fact, addition of 0.4-1.6% of dimethyl
sulfide in a closed system enhanced the degradation of fenitrothion.
Although metal salts, such as zinc, aluminum, ferric chloride, and
stannic chloride, also accelerated isomerization of fenitrothion,
calcium dodecylbenzene sulfonate (surfactant) showed no effect.
1 Chemical structures in Fig. 1-7 are referred to giving the
numbers in brackets.
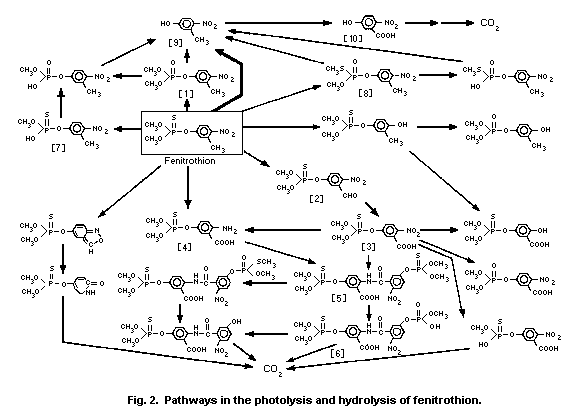
 4.2.1.2 Photolysis in air
Addison (1981) examined the photolysis of fenitrothion by UV
radiation (200-400 nm) from a xenon lamp in the vapour phase
(10-15 mg/50-1 reaction chamber) at about 85-90 °C, and computed the
half-life of disappearance to be 61 ± 11 min and 24 ± 3 min in the
absence or presence of ozone (0.7-0.9 ± 1 mg/m3), respectively.
Brewer et al. (1974) also studied the vapour phase photolysis
of fenitrothion at 313 nm UV radiation, and detected
3-methyl-4-nitrophenol [9] and an unidentified product as primary
photo-products (Fig. 2).
4.2.1.3 Hydrolysis and photolysis in water
Fenitrothion underwent hydrolysis in the absence of light
through a pH-independent process below pH 7 and a base-catalysed
process above pH 10, while both processes occurred between pH 7 and
pH 10. The half-lives of fenitrothion within the pH range of 5-9
(normally found in natural water) were about 200-630 days at 15 °C,
17-61 days at 30 °C, and 4-8 days at 45 °C. The predominant
hydrolysis products were 3-methyl-4-nitrophe-nol [9] above pH 10 and
demethylated fenitrothion [7] below pH 8 (Fig. 2; Mikami et al.,
1985a).
[Phenyl-14C]-fenitrothion was dissolved at 1.0 mg/litre in
aqueous buffer solutions at pH values of 5, 7, and 9 and kept at 25
± 1 °C in the dark for 30 days, free from microbial contamination.
Fenitrothion was less stable at pH 9 with a half-life of 100-101
days, compared with those of 180-186 days and 191-200 days at pH 7
and pH 5, respectively. Cleavage of the P-O-methyl linkage to form
demethylated fenitrothion [7] was predominant at pH 5 and pH 7,
while at pH 9 cleavage of the P-O-aryl linkage to form
3-methyl-4-nitrophenol [9] was the major hydrolytic path-way (Ito
et al., 1988).
Greenhalgh et al. (1980) and Aly & Badawy (1982) demonstrated
that hydrolysis of fenitrothion follows pseudo-first-order kinetics,
yielding mainly the phenol [9] at alkaline pH and the demethylated
form [7] under acidic conditions.
The rate of hydrolysis of fenitrothion may be accelerated
through the addition of peroxide ion (1.7 x 10-4 mol/litre),
particularly under alkaline conditions, since energies of activation
were reduced to 7.8 kcal/mol for peroxide hydrolysis from
16.3 kcal/mol for alkaline hydrolysis (Desmarchelier, 1987).
4.2.1.2 Photolysis in air
Addison (1981) examined the photolysis of fenitrothion by UV
radiation (200-400 nm) from a xenon lamp in the vapour phase
(10-15 mg/50-1 reaction chamber) at about 85-90 °C, and computed the
half-life of disappearance to be 61 ± 11 min and 24 ± 3 min in the
absence or presence of ozone (0.7-0.9 ± 1 mg/m3), respectively.
Brewer et al. (1974) also studied the vapour phase photolysis
of fenitrothion at 313 nm UV radiation, and detected
3-methyl-4-nitrophenol [9] and an unidentified product as primary
photo-products (Fig. 2).
4.2.1.3 Hydrolysis and photolysis in water
Fenitrothion underwent hydrolysis in the absence of light
through a pH-independent process below pH 7 and a base-catalysed
process above pH 10, while both processes occurred between pH 7 and
pH 10. The half-lives of fenitrothion within the pH range of 5-9
(normally found in natural water) were about 200-630 days at 15 °C,
17-61 days at 30 °C, and 4-8 days at 45 °C. The predominant
hydrolysis products were 3-methyl-4-nitrophe-nol [9] above pH 10 and
demethylated fenitrothion [7] below pH 8 (Fig. 2; Mikami et al.,
1985a).
[Phenyl-14C]-fenitrothion was dissolved at 1.0 mg/litre in
aqueous buffer solutions at pH values of 5, 7, and 9 and kept at 25
± 1 °C in the dark for 30 days, free from microbial contamination.
Fenitrothion was less stable at pH 9 with a half-life of 100-101
days, compared with those of 180-186 days and 191-200 days at pH 7
and pH 5, respectively. Cleavage of the P-O-methyl linkage to form
demethylated fenitrothion [7] was predominant at pH 5 and pH 7,
while at pH 9 cleavage of the P-O-aryl linkage to form
3-methyl-4-nitrophenol [9] was the major hydrolytic path-way (Ito
et al., 1988).
Greenhalgh et al. (1980) and Aly & Badawy (1982) demonstrated
that hydrolysis of fenitrothion follows pseudo-first-order kinetics,
yielding mainly the phenol [9] at alkaline pH and the demethylated
form [7] under acidic conditions.
The rate of hydrolysis of fenitrothion may be accelerated
through the addition of peroxide ion (1.7 x 10-4 mol/litre),
particularly under alkaline conditions, since energies of activation
were reduced to 7.8 kcal/mol for peroxide hydrolysis from
16.3 kcal/mol for alkaline hydrolysis (Desmarchelier, 1987).
 The photostability of fenitrothion in water is dependent on
both pH and energy of UVR or sunlight (Miyamoto, 1977a).
Fenitrothion rapidly decomposed in distilled water under sunlight
and in pH 7 and pH 9 solutions at ambient temperatures, but was
considerably more stable at pH 3. The half-life of fenitrothion was
10, 50, 20, and 6 h, respectively, in distilled water and in
solutions at pH 3, pH 7, and pH 9. Fenitrothion decomposed nearly 8
times faster at pH 9 than at pH 3.
Mikami et al. (1985a) determined the quantum yield of the
photodecomposition reaction of fenitrothion in distilled water (8.0
x 10-4) and calculated that the half-life of photolysis by
sunlight at the 40° north latitude was 7.6, 6.8, 11.3, and 17.0 h in
spring, summer, autumn and winter, respectively. The actual
half-life in autumn in Takarazuka, Japan, at a latitude of 35°
north, was 12 h, agreeing with the above calculation.
Photode-composition of fenitrothion in sterile lake water and sea
water was also rapid and the half-life in both these solutions was
less than 1.1 days.
Fenitrothion degraded fairly rapidly under sunlight to form
CO2; 14C ring-labelled fenitrothion released 39.4, 40.4, and
45.0% CO2 in 32 days in distilled water, in a buffer solution (pH
7), and in sterilized sea water (pH 7.8), respectively (Mikami et
al., 1985a).
[Phenyl-14C]-fenitrothion was dissolved at 1.0 mg/litre in
aqueous acetate buffer (pH 5.0), free from microbial contamination
and irradiated with artificial sunlight (wave length > 290 nm,
xenon arc lamp), for 30 days (10 h/day irradiation) at 25 ± 1 °C.
Fenitrothion was degraded rather rapidly with a half-life of
3.33-3.65 days (70.8-140.9 days under dark conditions).
Photo-degradation reactions were oxidation of the aryl methyl group
to a carboxyl group to form compound [3] (a main product, 8.0-12.4%
in 14 days), oxidation of the P=S group to P=O group, cleavage of
the P-O-CH3 or P-O-aryl linkage, and further decomposition to
14CO2 (41.2-42.0%) during 30 days (Katagi et al., 1988).
Kanazawa (1977) demonstrated that fenitrothion (20 µg/litre)
degraded to 8% of the original concentration under greenhouse
conditions and only to 55% in the absence of light, in sea water,
over 2 weeks.
Fenitrothion (about 0.1 mg/litre) degraded in sea waters
collected from various coasts of Japan to 44-62% and 63-94%, with
and without sediments, respectively, after 2 weeks in the absence of
sunlight and aeration (Environment Agency of Japan, 1978).
The effects of several factors on the persistence of
fenitrothion in sea water were examined using the experimental
design with an L16 orthogonal layout (Kodama & Kuwatsuka, 1980).
The persistence of fenitrothion was affected mainly by water quality
(river water or sea water) and sunlight (exposed or unexposed), but
also partially by temperature; it was not affected by the presence
of suspended solid or vaporization. After 72 h, the persistence of
fenitrothion in sea water was 56-97% of the original concentration
under varying conditions, while that in river water was 1-28%. When
river water was boiled, the rate of disappearance of fenitrothion
was the same as that in sea water, indicating that microbial
degradation was one of the most important contributing factors.
The photodegradation of fenitrothion (1 mg/litre) in sea water
collected along 3 different coastlines of Japan was very rapid with
a half-life of about 3 h, twice as fast as that in distilled water
(Takimoto et al., 1980).
A microcosm study using distilled, estuarine, and lake water
revealed that the ionic complement and/or microflora content of
estuarine water contributed more to the degradation of fenitrothion
than the pH. Sunlight irradiation in the static lake/bay models
decomposed 80% of fenitrothion to polar products within 6 h
(Weinberger et al., 1982a).
Twenty-one out of at least 50 radioactive photoproducts of
14C ring-labelled fenitrothion in water at various pHs were
identified. The major photoproducts were O,O-dimethyl
O-(3-carboxy-4-nitrophenyl) phosphorothioate [3] in distilled
water and in buffer solutions at pH 3 and 7. A dimeric compound [5]
composed of [3] and the corresponding amino analogue [4] was more
predominantly formed in buffer solutions at pH 7 and 9, in natural
river (pH 7.4) and sea (pH 7.8) water. On prolonged irradiation with
sunlight, these photoproducts decreased to less than 4% of the
initial concentration with concomitant increases in carbon dioxide
(21.5-45%) and the unextractable residues (29.3-51.4%) consisting of
polymeric humic acids. Demethylated products [6,7] and hydrolysis
products at the P-O-aryl linkage, such as 3-methyl-4-nitrophenol
[9], were of minor importance, independent of pH values. S-Methyl
fenitrothion [8] was occasionally detected in trace amounts (Mikami
et al., 1985a).
The UV irradiation of fenitrothion in oxygenated hexane
solution produced fenitrooxon [1] and O,O-dimethyl
O-(3-formyl-4-nitrophenyl)phosphorothioate [2] (Greenhalgh &
Marshall, 1976). However, these photochemical reactions might not
play a major role in the environmental photochemistry of
fenitrothion in water.
4.2.1.4 Photolysis on soil
When fenitrothion was applied to thin-layer plates with a 2 mm
thickness of 7 different types of soil and exposed to sunlight, it
took 50-150 days for the 90% disappearance of fenitrothion from the
soils (Miyamoto, 1977a). No clear correlation existed between the
rate of disappearance of fenitrothion and the physical and chemical
parameters of the soils. S-Methyl fenitrothion and
aminofenitrothion similarly applied to the soil decreased much more
rapidly than fenitrothion. The order of stability was fenitrothion
> S-methyl fenitrothion > aminofenitrothion. Under dark
conditions, decomposition of these 3 compounds proceeded more
slowly, the range of stability among them being the same as that
observed under irradiated conditions. At most, 10% of the applied
chemical was lost, probably by evaporation, from the soil after 14
days. Fenitrothion was degraded on the soil surface mainly by
oxidation of the P = S to the P = O group and cleavage of the P-O-
aryl linkage.
The rapid photodecomposition of fenitrothion on soil surfaces
was also demonstrated by Mikami et al. (1985a). Unlike photolysis in
water, the principal products were fenitrooxon [1] and
3-methyl-4-nitrophenol [9], amounting to 3.6-9.4% and 20.4-23.1% of
the applied 14C, respectively, after 12 days.
A photolysis study was conducted with
[phenyl- 14C]-fenitrothion applied on the surface of soil at a rate
of 23.4 µg/cm2. The samples were continuously irradiated (290 nm)
using an artificial light source (xenon arc lamp) over 30 days, the
soil temperature being maintained at 25 ± 1 °C throughout the
experiment. The rate constant and the half-life of photolysis were
determined to be 0.00814/day and 85 days, respectively, while under
dark conditions these were 0.0038/day and 182 days. Degradation
products identified included fenitrooxon [1] (2.0% at day 30),
demethyl fenitrothion [7] (2.1%) and 3-methyl-4-nitrophenol [9]
(3.0%) (Dykes & Carpenter, 1988).
Sunlight irradiation of fenitrooxon [1], 3-methyl-4-nitrophenol
[9], or carboxy-fenitrothion [3] on silica gel TLC plates resulted
in degradation and polymerization to humic acids, with half-lives of
3.9, 4.3, and 1.8 days, respectively (Ohkawa et al., 1974).
4.2.2 Biotransformation
4.2.2.1 Biodegradation in soil
The degradation pathways of fenitrothion in soils are shown in
Fig. 3.
When fenitrothion was incorporated at 10 mg/kg (on a dry-weight
basis) in soils with various physical and chemical properties, and
kept at 25 °C in the dark, under upland or submerged conditions, the
adsorption and decomposition of fenitrothion were quite variable,
depending on the properties of the soils and on the incubation
conditions (Takimoto et al., 1976; Miyamoto, 1977a). The half-life
of fenitrothion was 12-28 days under upland conditions, and 4-20
days under submerged conditions. However, no direct relationship was
observed between the decomposition of fenitrothion in soil and any
of the physical or chemical properties measured, namely clay
content, organic matter content, ion exchange capacity, and pH.
Under upland conditions, 3-methyl-4-nitrophenol [9] was formed at an
early stage of incubation, amounting to 10-20% of the applied
radioactivity ( m-methyl position). Levels of
3-methyl-4-nitrophenol decreased with longer incubation. Another
major decomposition product was radioactive carbon dioxide, which
amounted to approximately 40% of the initial fenitrothion after 60
days. No aminofenitrothion [13] was detected.
On the other hand, under submerged conditions,
3-methyl-4-nitrophenol [9] and carbon dioxide were minor products.
The major decomposition product was aminofenitrothion [13], its
formation being parallel to the decrease in fenitrothion. The
maximum amounts of aminofenitrothion were 18-66% of the initial
fenitrothion. Aminofenitrothion tended to disappear slowly on longer
incubation.
In soils, 14C-ring-labelled fenitrothion (10 mg/kg) degraded
at 25 °C in the dark with a half-life of 2-5 days under upland and
submerged conditions; after 8-26 weeks, levels declined to less than
0.1 mg/kg. After one year, the carbon dioxide evolved amounted to
60-70% of the initial radiocarbon under upland conditions and to
23-40% under submerged conditions, while the remaining radiocarbon
was mostly incorporated into the organic matter fractions of the
soil. When the soils containing the bound residues of the
radiocarbon were mixed with fresh soil, the release of radioactive
carbon dioxide was accelerated. Under upland conditions, degradation
of 3-methyl-4-nitrophenol [9] was more rapid than that of
fenitrothion (Mikami et al., 1985b).
Adhya et al. (1981a) also found that fenitrothion was degraded
primarily by reduction of the nitro group to aminofenitrothion in
flooded soil with lower redox potentials. Sterilization of the
prereduced flooded soil samples by autoclaving prevented the rapid
decomposition of fenitrothion in soil.
The photostability of fenitrothion in water is dependent on
both pH and energy of UVR or sunlight (Miyamoto, 1977a).
Fenitrothion rapidly decomposed in distilled water under sunlight
and in pH 7 and pH 9 solutions at ambient temperatures, but was
considerably more stable at pH 3. The half-life of fenitrothion was
10, 50, 20, and 6 h, respectively, in distilled water and in
solutions at pH 3, pH 7, and pH 9. Fenitrothion decomposed nearly 8
times faster at pH 9 than at pH 3.
Mikami et al. (1985a) determined the quantum yield of the
photodecomposition reaction of fenitrothion in distilled water (8.0
x 10-4) and calculated that the half-life of photolysis by
sunlight at the 40° north latitude was 7.6, 6.8, 11.3, and 17.0 h in
spring, summer, autumn and winter, respectively. The actual
half-life in autumn in Takarazuka, Japan, at a latitude of 35°
north, was 12 h, agreeing with the above calculation.
Photode-composition of fenitrothion in sterile lake water and sea
water was also rapid and the half-life in both these solutions was
less than 1.1 days.
Fenitrothion degraded fairly rapidly under sunlight to form
CO2; 14C ring-labelled fenitrothion released 39.4, 40.4, and
45.0% CO2 in 32 days in distilled water, in a buffer solution (pH
7), and in sterilized sea water (pH 7.8), respectively (Mikami et
al., 1985a).
[Phenyl-14C]-fenitrothion was dissolved at 1.0 mg/litre in
aqueous acetate buffer (pH 5.0), free from microbial contamination
and irradiated with artificial sunlight (wave length > 290 nm,
xenon arc lamp), for 30 days (10 h/day irradiation) at 25 ± 1 °C.
Fenitrothion was degraded rather rapidly with a half-life of
3.33-3.65 days (70.8-140.9 days under dark conditions).
Photo-degradation reactions were oxidation of the aryl methyl group
to a carboxyl group to form compound [3] (a main product, 8.0-12.4%
in 14 days), oxidation of the P=S group to P=O group, cleavage of
the P-O-CH3 or P-O-aryl linkage, and further decomposition to
14CO2 (41.2-42.0%) during 30 days (Katagi et al., 1988).
Kanazawa (1977) demonstrated that fenitrothion (20 µg/litre)
degraded to 8% of the original concentration under greenhouse
conditions and only to 55% in the absence of light, in sea water,
over 2 weeks.
Fenitrothion (about 0.1 mg/litre) degraded in sea waters
collected from various coasts of Japan to 44-62% and 63-94%, with
and without sediments, respectively, after 2 weeks in the absence of
sunlight and aeration (Environment Agency of Japan, 1978).
The effects of several factors on the persistence of
fenitrothion in sea water were examined using the experimental
design with an L16 orthogonal layout (Kodama & Kuwatsuka, 1980).
The persistence of fenitrothion was affected mainly by water quality
(river water or sea water) and sunlight (exposed or unexposed), but
also partially by temperature; it was not affected by the presence
of suspended solid or vaporization. After 72 h, the persistence of
fenitrothion in sea water was 56-97% of the original concentration
under varying conditions, while that in river water was 1-28%. When
river water was boiled, the rate of disappearance of fenitrothion
was the same as that in sea water, indicating that microbial
degradation was one of the most important contributing factors.
The photodegradation of fenitrothion (1 mg/litre) in sea water
collected along 3 different coastlines of Japan was very rapid with
a half-life of about 3 h, twice as fast as that in distilled water
(Takimoto et al., 1980).
A microcosm study using distilled, estuarine, and lake water
revealed that the ionic complement and/or microflora content of
estuarine water contributed more to the degradation of fenitrothion
than the pH. Sunlight irradiation in the static lake/bay models
decomposed 80% of fenitrothion to polar products within 6 h
(Weinberger et al., 1982a).
Twenty-one out of at least 50 radioactive photoproducts of
14C ring-labelled fenitrothion in water at various pHs were
identified. The major photoproducts were O,O-dimethyl
O-(3-carboxy-4-nitrophenyl) phosphorothioate [3] in distilled
water and in buffer solutions at pH 3 and 7. A dimeric compound [5]
composed of [3] and the corresponding amino analogue [4] was more
predominantly formed in buffer solutions at pH 7 and 9, in natural
river (pH 7.4) and sea (pH 7.8) water. On prolonged irradiation with
sunlight, these photoproducts decreased to less than 4% of the
initial concentration with concomitant increases in carbon dioxide
(21.5-45%) and the unextractable residues (29.3-51.4%) consisting of
polymeric humic acids. Demethylated products [6,7] and hydrolysis
products at the P-O-aryl linkage, such as 3-methyl-4-nitrophenol
[9], were of minor importance, independent of pH values. S-Methyl
fenitrothion [8] was occasionally detected in trace amounts (Mikami
et al., 1985a).
The UV irradiation of fenitrothion in oxygenated hexane
solution produced fenitrooxon [1] and O,O-dimethyl
O-(3-formyl-4-nitrophenyl)phosphorothioate [2] (Greenhalgh &
Marshall, 1976). However, these photochemical reactions might not
play a major role in the environmental photochemistry of
fenitrothion in water.
4.2.1.4 Photolysis on soil
When fenitrothion was applied to thin-layer plates with a 2 mm
thickness of 7 different types of soil and exposed to sunlight, it
took 50-150 days for the 90% disappearance of fenitrothion from the
soils (Miyamoto, 1977a). No clear correlation existed between the
rate of disappearance of fenitrothion and the physical and chemical
parameters of the soils. S-Methyl fenitrothion and
aminofenitrothion similarly applied to the soil decreased much more
rapidly than fenitrothion. The order of stability was fenitrothion
> S-methyl fenitrothion > aminofenitrothion. Under dark
conditions, decomposition of these 3 compounds proceeded more
slowly, the range of stability among them being the same as that
observed under irradiated conditions. At most, 10% of the applied
chemical was lost, probably by evaporation, from the soil after 14
days. Fenitrothion was degraded on the soil surface mainly by
oxidation of the P = S to the P = O group and cleavage of the P-O-
aryl linkage.
The rapid photodecomposition of fenitrothion on soil surfaces
was also demonstrated by Mikami et al. (1985a). Unlike photolysis in
water, the principal products were fenitrooxon [1] and
3-methyl-4-nitrophenol [9], amounting to 3.6-9.4% and 20.4-23.1% of
the applied 14C, respectively, after 12 days.
A photolysis study was conducted with
[phenyl- 14C]-fenitrothion applied on the surface of soil at a rate
of 23.4 µg/cm2. The samples were continuously irradiated (290 nm)
using an artificial light source (xenon arc lamp) over 30 days, the
soil temperature being maintained at 25 ± 1 °C throughout the
experiment. The rate constant and the half-life of photolysis were
determined to be 0.00814/day and 85 days, respectively, while under
dark conditions these were 0.0038/day and 182 days. Degradation
products identified included fenitrooxon [1] (2.0% at day 30),
demethyl fenitrothion [7] (2.1%) and 3-methyl-4-nitrophenol [9]
(3.0%) (Dykes & Carpenter, 1988).
Sunlight irradiation of fenitrooxon [1], 3-methyl-4-nitrophenol
[9], or carboxy-fenitrothion [3] on silica gel TLC plates resulted
in degradation and polymerization to humic acids, with half-lives of
3.9, 4.3, and 1.8 days, respectively (Ohkawa et al., 1974).
4.2.2 Biotransformation
4.2.2.1 Biodegradation in soil
The degradation pathways of fenitrothion in soils are shown in
Fig. 3.
When fenitrothion was incorporated at 10 mg/kg (on a dry-weight
basis) in soils with various physical and chemical properties, and
kept at 25 °C in the dark, under upland or submerged conditions, the
adsorption and decomposition of fenitrothion were quite variable,
depending on the properties of the soils and on the incubation
conditions (Takimoto et al., 1976; Miyamoto, 1977a). The half-life
of fenitrothion was 12-28 days under upland conditions, and 4-20
days under submerged conditions. However, no direct relationship was
observed between the decomposition of fenitrothion in soil and any
of the physical or chemical properties measured, namely clay
content, organic matter content, ion exchange capacity, and pH.
Under upland conditions, 3-methyl-4-nitrophenol [9] was formed at an
early stage of incubation, amounting to 10-20% of the applied
radioactivity ( m-methyl position). Levels of
3-methyl-4-nitrophenol decreased with longer incubation. Another
major decomposition product was radioactive carbon dioxide, which
amounted to approximately 40% of the initial fenitrothion after 60
days. No aminofenitrothion [13] was detected.
On the other hand, under submerged conditions,
3-methyl-4-nitrophenol [9] and carbon dioxide were minor products.
The major decomposition product was aminofenitrothion [13], its
formation being parallel to the decrease in fenitrothion. The
maximum amounts of aminofenitrothion were 18-66% of the initial
fenitrothion. Aminofenitrothion tended to disappear slowly on longer
incubation.
In soils, 14C-ring-labelled fenitrothion (10 mg/kg) degraded
at 25 °C in the dark with a half-life of 2-5 days under upland and
submerged conditions; after 8-26 weeks, levels declined to less than
0.1 mg/kg. After one year, the carbon dioxide evolved amounted to
60-70% of the initial radiocarbon under upland conditions and to
23-40% under submerged conditions, while the remaining radiocarbon
was mostly incorporated into the organic matter fractions of the
soil. When the soils containing the bound residues of the
radiocarbon were mixed with fresh soil, the release of radioactive
carbon dioxide was accelerated. Under upland conditions, degradation
of 3-methyl-4-nitrophenol [9] was more rapid than that of
fenitrothion (Mikami et al., 1985b).
Adhya et al. (1981a) also found that fenitrothion was degraded
primarily by reduction of the nitro group to aminofenitrothion in
flooded soil with lower redox potentials. Sterilization of the
prereduced flooded soil samples by autoclaving prevented the rapid
decomposition of fenitrothion in soil.
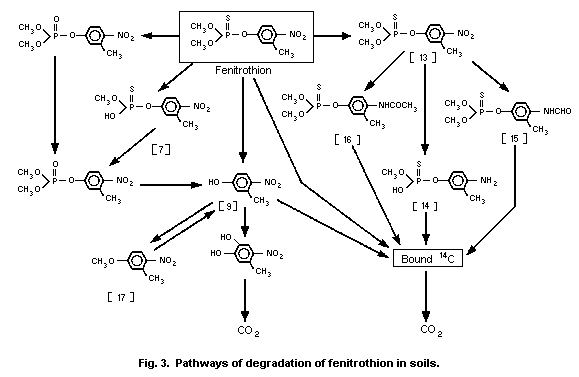 Aminofenitrothion was further degraded to demethyl
amino-fenitrothion [14] in typical flooded acid sulfate soils from
Kerala, India. Dealkylation also occurred in low sulfate soils under
submerged conditions, when supplemented with extraneous sulfate
(e.g., ammonium sulfate or ferrous sulfate). Hydrogen sulfide,
evolved as an end product of the anaerobic metabolism of sulfate,
catalysed the dealkylation of aminofenitrothion (Adhya et al.,
1981b).
Fenitrothion was stable when incubated in soil suspensions
containing streptomycin, cycloheximide, and mineral salts. However,
it decomposed rapidly when the soil suspension was added to a
culture medium suitable for fungal or bacterial growth (Takimoto et
al., 1976). The major decomposition product in the culture was
aminofenitrothion [13], the content of which reached 40-65%, and a
maximum 60-80% when formylamino-[15] and acetylamino- fenitrothion
[16] were combined. Demethyl fenitrothion [7] and
3-methyl-4-nitrophenol [9] were detected among other products. The
dominant species of microorganisms (Fusarium and Bacillus species),
isolated from the above soils, metabolized fenitrothion well.
When ring-labelled fenitrothion (7.4 mg/kg) was incubated with
two kinds of forest soils collected from the State of Maine, USA,
the half-life of fenitrothion at 30 °C was about 3 days. In 50 days,
94-97% of the fenitrothion had been decomposed yielding 35%
radioactive carbon dioxide, 5-7% 3-methyl-4-nitrophenol [9], 4%
3-methyl-4-nitroanisole [17], and approximately 50% of soil- bound
radioactivity (Spillner et al., 1979a).
It has been reported (National Research Council of Canada,
1975) that several species of soil and water bacteria, including
Bacillus subtilis, Escherichia coli, E. freundii, Pseudomonas
reptilovora, and P. aeruginosa, can metabolize or inactivate
fenitrothion.
The fungus Trichoderma viride can also hydrolyse fenitrothion
and fenitrooxon, and the hydrolysed product,
3-methyl-4-nitro-phenol, is co-metabolized by this fungus
(Baarschers & Heitland, 1986).
Flavobacterium sp. ATCC 27551, isolated from paddy field,
hydrolysed fenitrothion to yield 3-methyl-4-nitrophenol [9] in
culture solutions containing mineral salts (Adhya et al., 1981c).
A crude cell extract from a mixed bacterial culture growing on
parathion also hydrolysed fenitrothion to yield
3-methyl-4-nitro-phenol [9]. The chemical hydrolysis of fenitrothion
was 3-5 times slower than that of parathion. However, the rate of
enzymatic hydrolysis was 24-205 times faster than that of chemical
hydroly-sis by 0.1 N sodium hydroxide at 40 °C (Munnecke, 1976).
Liu et al. (1981) studied the biodegradability of fenitrothion
using a mixed-culture of microorganisms from activated sludge, soil,
and sediments, under aerobic and anaerobic conditions. Fenitrothion
was more readily degraded under anaerobic co-metabolic conditions
(half-time = 1.0 day) than under aerobic conditions (half-time = 5.5
days). When fenitrothion was applied to the medium as the sole
source of carbon, its stability was greater under aerobic
conditions, 77% of the initial dose being recovered after 164 h
incubation.
4.2.2.2 Biodegradation and bioaccumulation in organisms
a) Aquatic organisms
The accumulation and partitioning of fenitrothion residues
among different tissues and organs in wild trout were investigated
following 2 applications of the compound to a lake (280 g a.i./ha
with a 9-day interval). Fenitrothion residues accumulated fairly
rapidly in the tissues of both brook trout and lake trout. Peak
levels of fenitrothion were reached 2-4 days after the first
application (in lake trout, fat: 280 µg/kg, muscle: 96.8 µg/kg,
intestine: 96.3 µg/kg, liver: 16.1 µg/kg, ovary: 48.2 µg/kg) and 8
days after the second application (in lake trout, fat: 665 µg/kg,
muscle: 133 µg/kg, intestine: 114 µg/kg, liver: 39.6 µg/kg). These
levels were many times higher than those in the surrounding waters
of Lac Ste-Anne. Fenitrothion residues continued to persist in lake
trout tissues (but not in the tissues of brook trout) up to at least
8 days after the second application, even though the residues in the
water had declined to non-detectable levels 4 days earlier (Holmes
et al., 1984).
When exposed to running water containing 0.1 or 0.02 mg
fenitrothion/litre, both underyearling rainbow trout ( Salmo
gairdneri) and southern top-mouthed minnow ( Pseudorasbora parva)
rapidly absorbed the chemical (Takimoto & Miyamoto, 1976a). The
fenitrothion concentration in the fish reached a maximum after 1-3
days of exposure, and then remained virtually constant. The
bioaccumulation ratio did not increase on longer exposure and was
more or less independent of the fenitrothion concentration in water.
The ratio was similar in the 2 fish species, being approximately
250, 230, and 200 (at its maximum) in underyearling trout, yearling
trout, and minnow, respectively. Once the fish were transferred
from fenitrothion-containing water to fresh water, the levels of
fenitrothion in the fish decreased rapidly to about 0.01 mg/kg in 5
days. None of the fish species exhibited noticeable signs of
intoxication during the exposure period. In another study, the
bioaccumulation ratio for the fresh-water species, top-mouthed
minnow, was 246 (Kanazawa, 1981).
Killifish (Oryzias latipes) took up fenitrothion in a flow
system with bioaccumulation ratios at different developmental stages
of: 115 in embryos, 173 in yolk sac fry, 88 in postlarval stages,
441 in juveniles, 520 in adult males, 540 in adult females, and 224
in eggs produced from fenitrothion-exposed adults (Takimoto et al.,
1984a). However, the half-lives of disappearance of the compound in
clean water were less than 2 days, independent of fat content.
Similar results were reported with southern top-mouthed minnows
in a static aquarium test; the fenitrothion concentration in water
at 23 °C decreased from 0.81 mg/litre to 0.002 mg/litre in 28 days,
while the observed maximum concentration of fenitrothion in the fish
(162 mg/kg on the 4th day) decreased to 4.9 mg/kg after 28 days
(Kanazawa, 1975).
With respect to the distribution and metabolism of
fenitrothion, autoradiograms of rainbow trout exposed to labelled
fenitrothion under static water conditions for 6 h revealed that the
concentration of radiocarbon was highest in the gall bladder and
intestines; after 24 h, the radiocarbon was present in every tissue,
except the brain and heart. Twenty-four hours after the transfer of
the fish to fresh water, most of the radioactivity in the tissues
had disappeared. Only the gall bladder, intestines, and pyloric
caeca still contained an appreciable amount of the radiocarbon.
Intact fenitrothion accounted for 90% of the absorbed radioactivity
in fish. The remaining 10% comprised fenitrooxon [1], demethyl
fenitrothion [7], 3-methyl-4-nitrophenol [9], and its glucuronide
[27]. In water, the percentage of these degradation products
increased with time and amounted to 25% of the radioactivity.
Because fenitrothion is stable in water under the experimental
conditions, the degradation products are presumably derived from
fish metabolism (Takimoto & Miyamoto, 1976a) (see Fig. 4).
All developmental stages of killifish metabolized fenitrothion
mainly to 3-methyl-4-nitrophenyl-ß-glucuronide [27] (comprising
20-40% of 14C), except the embryo, which had the lowest metabolic
activity. Yolk sac fry contained the highest concentration (28%) of
demethyl fenitrothion [7]. Fenitrooxon [1] and demethyl fenitrooxon
[20] were present in small amounts, at the most, 0.5%. These
metabolites and the intact fenitrothion were eliminated into the
surrounding water, when the fish were transferred to fresh water
(Takimoto et al., 1984a).
Absorption of [methyl-14C]-fenitrothion at 0.1 mg/litre in
running water to a similar plateau level in the killifish (Oryzias
latipes) was more rapid at 25 °C than at 15 °C; the bioaccumulation
ratios of fenitrothion were 235 and 339, respectively. Water of
higher salinity (2.3%) resulted in slightly higher accumulation
ratios of fenitrothion in both killifish (303) and mullet, Mugil
cephalus (179), than fresh water (235 and 30, respectively), but the
half-lives were independent of temperature and salinity, with values
of 0.24-0.36 day. Fenitrothion was metabolized, primarily through
hydrolysis, to [9] by the killifish, demethylation to demethyl
fenitrothion [7] by the mullet, and conjugation of the liberated
phenol with glucuronic acid [27] by both species. Although the
metabolism of the compound in both fish was not affected by the
different salinities and temperatures, the glucuronide conjugate was
more directly excreted into water under conditions of lower
temperature and higher salinity. 14C-labelled compounds were
distributed primarily to the gall bladder, as shown by whole-body
radioautography (Takimoto et al., 1987a).
Bluegill sunfish (Lepomis macrochirus) was exposed to
[phenyl-14C] - fenitrothion or non-radiolabelled fenitrothion in a
flow-through system at concentrations of 0.049 mg/litre and 0.043
mg/litre, respectively, for a 28-day exposure period. The
concentrations of labelled and non-labelled fenitrothion in whole
fish reached an equilibrium on days 1-3 of exposure at levels of 5.4
and 1.3 mg/litre, respectively. Mean bioconcentration factors for
labelled and non-labelled fenitrothion during the uptake period were
respectively 111 and 29 for whole fish, 26 and 10 for the edible
portion, and 279 and 36 for the non-edible portion. When the exposed
fish were transferred to running fresh water, the concentrations of
labelled and non-labelled fenitrothion in the fish decreased
rapidly, with biological half-lives of less than 1 day in both the
edible and non-edible portions of the fish. A non-linear
2-compartment, kinetic modelling computer programme estimated 81.9
and 111 as the uptake rate constants (K1), 0.69 and 3.72 as the
depuration rate constants (K2), and 118 and 30 as the
bioconcentration factors (BCF) for labelled and non-labelled
fenitrothion, respectively. Fenitrothion was metabolized through the
oxidation of P=S to P=O, demethylation of the P-O-alkyl linkage,
cleavage of the P-O-aryl linkage, and conjugation of the phenol with
glucuronic acid. The major metabolites were demethylfenitrothion [7]
and 3-methyl-4-nitrophenyl-ß-glucuronide [27], amounting to 29-40%
and 11-15% of the recovered 14C from the whole fish, respectively
(Ohshima et al., 1988).
Freshwater teleosts ( Tilapia mossambica; body weight, 5-9 g;
length, 5-7 cm) were exposed to 200 mg fenitrothion/kg body weight
for 24 h. Fenitrothion and its metabolites, extracted from the
liver, kidney, and brain, were separated and identified using HPLC
and preparative silica gel TLC. The metabolites extracted from the
liver were identiied as fenitrooxon, N-acetylaminofenitrothion
[16], and fenitrothion. The metabolites from the kidney were
identified as demethyl- N-acetylaminofenitrooxon [37]. The
metabolites from the brain were identified as 3-methyl-4-nitrophenol
[9] and demethyl- N-acetylamino-fenitrooxon [37] (Anjum & Qadri,
1986).
Aminofenitrothion was further degraded to demethyl
amino-fenitrothion [14] in typical flooded acid sulfate soils from
Kerala, India. Dealkylation also occurred in low sulfate soils under
submerged conditions, when supplemented with extraneous sulfate
(e.g., ammonium sulfate or ferrous sulfate). Hydrogen sulfide,
evolved as an end product of the anaerobic metabolism of sulfate,
catalysed the dealkylation of aminofenitrothion (Adhya et al.,
1981b).
Fenitrothion was stable when incubated in soil suspensions
containing streptomycin, cycloheximide, and mineral salts. However,
it decomposed rapidly when the soil suspension was added to a
culture medium suitable for fungal or bacterial growth (Takimoto et
al., 1976). The major decomposition product in the culture was
aminofenitrothion [13], the content of which reached 40-65%, and a
maximum 60-80% when formylamino-[15] and acetylamino- fenitrothion
[16] were combined. Demethyl fenitrothion [7] and
3-methyl-4-nitrophenol [9] were detected among other products. The
dominant species of microorganisms (Fusarium and Bacillus species),
isolated from the above soils, metabolized fenitrothion well.
When ring-labelled fenitrothion (7.4 mg/kg) was incubated with
two kinds of forest soils collected from the State of Maine, USA,
the half-life of fenitrothion at 30 °C was about 3 days. In 50 days,
94-97% of the fenitrothion had been decomposed yielding 35%
radioactive carbon dioxide, 5-7% 3-methyl-4-nitrophenol [9], 4%
3-methyl-4-nitroanisole [17], and approximately 50% of soil- bound
radioactivity (Spillner et al., 1979a).
It has been reported (National Research Council of Canada,
1975) that several species of soil and water bacteria, including
Bacillus subtilis, Escherichia coli, E. freundii, Pseudomonas
reptilovora, and P. aeruginosa, can metabolize or inactivate
fenitrothion.
The fungus Trichoderma viride can also hydrolyse fenitrothion
and fenitrooxon, and the hydrolysed product,
3-methyl-4-nitro-phenol, is co-metabolized by this fungus
(Baarschers & Heitland, 1986).
Flavobacterium sp. ATCC 27551, isolated from paddy field,
hydrolysed fenitrothion to yield 3-methyl-4-nitrophenol [9] in
culture solutions containing mineral salts (Adhya et al., 1981c).
A crude cell extract from a mixed bacterial culture growing on
parathion also hydrolysed fenitrothion to yield
3-methyl-4-nitro-phenol [9]. The chemical hydrolysis of fenitrothion
was 3-5 times slower than that of parathion. However, the rate of
enzymatic hydrolysis was 24-205 times faster than that of chemical
hydroly-sis by 0.1 N sodium hydroxide at 40 °C (Munnecke, 1976).
Liu et al. (1981) studied the biodegradability of fenitrothion
using a mixed-culture of microorganisms from activated sludge, soil,
and sediments, under aerobic and anaerobic conditions. Fenitrothion
was more readily degraded under anaerobic co-metabolic conditions
(half-time = 1.0 day) than under aerobic conditions (half-time = 5.5
days). When fenitrothion was applied to the medium as the sole
source of carbon, its stability was greater under aerobic
conditions, 77% of the initial dose being recovered after 164 h
incubation.
4.2.2.2 Biodegradation and bioaccumulation in organisms
a) Aquatic organisms
The accumulation and partitioning of fenitrothion residues
among different tissues and organs in wild trout were investigated
following 2 applications of the compound to a lake (280 g a.i./ha
with a 9-day interval). Fenitrothion residues accumulated fairly
rapidly in the tissues of both brook trout and lake trout. Peak
levels of fenitrothion were reached 2-4 days after the first
application (in lake trout, fat: 280 µg/kg, muscle: 96.8 µg/kg,
intestine: 96.3 µg/kg, liver: 16.1 µg/kg, ovary: 48.2 µg/kg) and 8
days after the second application (in lake trout, fat: 665 µg/kg,
muscle: 133 µg/kg, intestine: 114 µg/kg, liver: 39.6 µg/kg). These
levels were many times higher than those in the surrounding waters
of Lac Ste-Anne. Fenitrothion residues continued to persist in lake
trout tissues (but not in the tissues of brook trout) up to at least
8 days after the second application, even though the residues in the
water had declined to non-detectable levels 4 days earlier (Holmes
et al., 1984).
When exposed to running water containing 0.1 or 0.02 mg
fenitrothion/litre, both underyearling rainbow trout ( Salmo
gairdneri) and southern top-mouthed minnow ( Pseudorasbora parva)
rapidly absorbed the chemical (Takimoto & Miyamoto, 1976a). The
fenitrothion concentration in the fish reached a maximum after 1-3
days of exposure, and then remained virtually constant. The
bioaccumulation ratio did not increase on longer exposure and was
more or less independent of the fenitrothion concentration in water.
The ratio was similar in the 2 fish species, being approximately
250, 230, and 200 (at its maximum) in underyearling trout, yearling
trout, and minnow, respectively. Once the fish were transferred
from fenitrothion-containing water to fresh water, the levels of
fenitrothion in the fish decreased rapidly to about 0.01 mg/kg in 5
days. None of the fish species exhibited noticeable signs of
intoxication during the exposure period. In another study, the
bioaccumulation ratio for the fresh-water species, top-mouthed
minnow, was 246 (Kanazawa, 1981).
Killifish (Oryzias latipes) took up fenitrothion in a flow
system with bioaccumulation ratios at different developmental stages
of: 115 in embryos, 173 in yolk sac fry, 88 in postlarval stages,
441 in juveniles, 520 in adult males, 540 in adult females, and 224
in eggs produced from fenitrothion-exposed adults (Takimoto et al.,
1984a). However, the half-lives of disappearance of the compound in
clean water were less than 2 days, independent of fat content.
Similar results were reported with southern top-mouthed minnows
in a static aquarium test; the fenitrothion concentration in water
at 23 °C decreased from 0.81 mg/litre to 0.002 mg/litre in 28 days,
while the observed maximum concentration of fenitrothion in the fish
(162 mg/kg on the 4th day) decreased to 4.9 mg/kg after 28 days
(Kanazawa, 1975).
With respect to the distribution and metabolism of
fenitrothion, autoradiograms of rainbow trout exposed to labelled
fenitrothion under static water conditions for 6 h revealed that the
concentration of radiocarbon was highest in the gall bladder and
intestines; after 24 h, the radiocarbon was present in every tissue,
except the brain and heart. Twenty-four hours after the transfer of
the fish to fresh water, most of the radioactivity in the tissues
had disappeared. Only the gall bladder, intestines, and pyloric
caeca still contained an appreciable amount of the radiocarbon.
Intact fenitrothion accounted for 90% of the absorbed radioactivity
in fish. The remaining 10% comprised fenitrooxon [1], demethyl
fenitrothion [7], 3-methyl-4-nitrophenol [9], and its glucuronide
[27]. In water, the percentage of these degradation products
increased with time and amounted to 25% of the radioactivity.
Because fenitrothion is stable in water under the experimental
conditions, the degradation products are presumably derived from
fish metabolism (Takimoto & Miyamoto, 1976a) (see Fig. 4).
All developmental stages of killifish metabolized fenitrothion
mainly to 3-methyl-4-nitrophenyl-ß-glucuronide [27] (comprising
20-40% of 14C), except the embryo, which had the lowest metabolic
activity. Yolk sac fry contained the highest concentration (28%) of
demethyl fenitrothion [7]. Fenitrooxon [1] and demethyl fenitrooxon
[20] were present in small amounts, at the most, 0.5%. These
metabolites and the intact fenitrothion were eliminated into the
surrounding water, when the fish were transferred to fresh water
(Takimoto et al., 1984a).
Absorption of [methyl-14C]-fenitrothion at 0.1 mg/litre in
running water to a similar plateau level in the killifish (Oryzias
latipes) was more rapid at 25 °C than at 15 °C; the bioaccumulation
ratios of fenitrothion were 235 and 339, respectively. Water of
higher salinity (2.3%) resulted in slightly higher accumulation
ratios of fenitrothion in both killifish (303) and mullet, Mugil
cephalus (179), than fresh water (235 and 30, respectively), but the
half-lives were independent of temperature and salinity, with values
of 0.24-0.36 day. Fenitrothion was metabolized, primarily through
hydrolysis, to [9] by the killifish, demethylation to demethyl
fenitrothion [7] by the mullet, and conjugation of the liberated
phenol with glucuronic acid [27] by both species. Although the
metabolism of the compound in both fish was not affected by the
different salinities and temperatures, the glucuronide conjugate was
more directly excreted into water under conditions of lower
temperature and higher salinity. 14C-labelled compounds were
distributed primarily to the gall bladder, as shown by whole-body
radioautography (Takimoto et al., 1987a).
Bluegill sunfish (Lepomis macrochirus) was exposed to
[phenyl-14C] - fenitrothion or non-radiolabelled fenitrothion in a
flow-through system at concentrations of 0.049 mg/litre and 0.043
mg/litre, respectively, for a 28-day exposure period. The
concentrations of labelled and non-labelled fenitrothion in whole
fish reached an equilibrium on days 1-3 of exposure at levels of 5.4
and 1.3 mg/litre, respectively. Mean bioconcentration factors for
labelled and non-labelled fenitrothion during the uptake period were
respectively 111 and 29 for whole fish, 26 and 10 for the edible
portion, and 279 and 36 for the non-edible portion. When the exposed
fish were transferred to running fresh water, the concentrations of
labelled and non-labelled fenitrothion in the fish decreased
rapidly, with biological half-lives of less than 1 day in both the
edible and non-edible portions of the fish. A non-linear
2-compartment, kinetic modelling computer programme estimated 81.9
and 111 as the uptake rate constants (K1), 0.69 and 3.72 as the
depuration rate constants (K2), and 118 and 30 as the
bioconcentration factors (BCF) for labelled and non-labelled
fenitrothion, respectively. Fenitrothion was metabolized through the
oxidation of P=S to P=O, demethylation of the P-O-alkyl linkage,
cleavage of the P-O-aryl linkage, and conjugation of the phenol with
glucuronic acid. The major metabolites were demethylfenitrothion [7]
and 3-methyl-4-nitrophenyl-ß-glucuronide [27], amounting to 29-40%
and 11-15% of the recovered 14C from the whole fish, respectively
(Ohshima et al., 1988).
Freshwater teleosts ( Tilapia mossambica; body weight, 5-9 g;
length, 5-7 cm) were exposed to 200 mg fenitrothion/kg body weight
for 24 h. Fenitrothion and its metabolites, extracted from the
liver, kidney, and brain, were separated and identified using HPLC
and preparative silica gel TLC. The metabolites extracted from the
liver were identiied as fenitrooxon, N-acetylaminofenitrothion
[16], and fenitrothion. The metabolites from the kidney were
identified as demethyl- N-acetylaminofenitrooxon [37]. The
metabolites from the brain were identified as 3-methyl-4-nitrophenol
[9] and demethyl- N-acetylamino-fenitrooxon [37] (Anjum & Qadri,
1986).
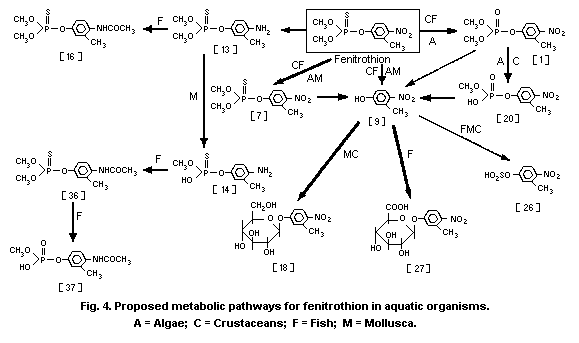 McLeese et al. (1979) reported that accumulation ratios of
fenitrothion were independent of the exposure levels, and were
19-35, 78-130, and 9, in marine clam (Mya arenaria), mussel (Mytilus
edulis), and freshwater clam (Anodonta cataractae), respectively.
When freshwater snails ( Cipangopaludina japonica and Physa
acuta) were exposed to 0.1 mg [methyl-14C]-fenitrothion/litre in
a dynamic flow system, the concentrations of fenitrothion and
14C-label in the body reached equilibrium on day one of exposure.
The maximum bioaccumulation ratios of fenitrothion were 18 and 53 in
C. japonica and P. acuta, respectively. These snails metabolized
the compound primarily by demethylation to [7], hydrolysis to [9],
and reduction to [13], and [14]. The liberated phenol moiety was
conjugated with sulfate [26] in C. japonica and mainly with
glucose [18] in P. acuta. When the snails were transferred to a
freshwater stream, fenitrothion and its metabolites were rapidly
eliminated, and the half-life of the parent compound was less than
0.5 days in both snails. In P. acute, fenitrothion and its
decomposition products were mainly distributed in the liver as shown
by whole-body radioautography (Takimoto et al., 1987b).
When the waterflea Daphnia pulex and the shrimp Palaemon
paucidens were exposed to 1.0 µg [methyl-14C]-fenitrothion/litre
in a flow-through system, the concentrations of fenitrothion and
14C-label in the body reached equilibrium (on day one of exposure)
and the maximum bioaccumulation ratios of fenitrothion were 71 and 6
in the daphnia and shrimp, respectively. These crustaceans primarily
metabolized the compound through oxidation of P = S to P = O to form
compound [1], hydrolysis of P-O-aryl linkage to form compound [9],
and demethylation to give compounds [7] and [20]. The liberated
phenol was conjugated with sulfate to form compound [26] in D.
pulex and with glucose to form compound [18] in the shrimp. When
the organisms were transferred to a freshwater stream, fenitrothion
and its metabolites were rapidly eliminated from their bodies, and
the half-life of the parent compound was less than 0.2 day in both
species (Takimoto et al., 1987c) (see Fig. 4).
The uptake rate of radiolabelled fenitrothion by the blue crab
(Callinectes sapides) increased with temperature and salinity. The
highest concentrations of radioactivity were found in the
hepato-pancreas and stomach. The blue crab can metabolize
fenitrothion to produce fenitrooxon, aminofenitrothion,
3-methyl-4-nitrophe-nol, 3-methyl-4-aminophenol, demethyl
fenitrothion, demethyl fenitrooxon, and glycoside and sulfate
conjugates of the phenols (Johnston & Corbett, 1986).
Aquatic plants were collected and analysed after the aerial
application of fenitrothion (280 g/ha) in Manitoba, Canada, (Moody
et al., 1978); the fenitrothion residues present in surface-
dwelling duckweed, obtained from stagnant water, disappeared rapidly
from 1.44 mg/kg after 1 h to 0.03 mg/kg after 192 h, while the
submerged hornwort, also from stagnant water, contained rather
persistent, but low, residues of fenitrothion ranging from 0.12 to
0.15 mg/kg after 192 h. No fenitrothion was detected in the
submerged flowering rush in running water.
Chlorella pyrenoidosa exposed to 10 mg radioactive
fenitrothion per litre rapidly took up the compound and the 14C
level reached equilibrium after 4 h with a bioaccumulation ratio of
417. On transfer to fresh water, the fenitrothion in the chlorella
was rapidly desorbed (Weinberger et al., 1982b). Other algae
( Chlamydomonas reinhardii and Euglena gracilis) showed less
bioaccumulation of fenitrothion. The aquatic macrophyte, Elodea
densa, showed a bioaccumulation ratio of 24-76 (Weinberger et al.,
1982b).
Three types of algae, Chlorella vulgaris, Nitzschia
closterium, and Anabaena flos-aquae, also rapidly absorbed
fenitrothion with maximum bioaccumulation ratios of 44, 105, and 53,
respectively (Kikuchi et al., 1984). Only A. flos-aquae (blue-green
algae) actively degraded fenitrothion. When transferred to a
fenitrothion-free medium, these algae released fenitrothion, as well
as its metabolites, with half-lives of the compounds of less than 1
day, except in the case of A. flos-aquae when the half-life was 2.6
days (see Fig. 4).
Bioaccumulation ratios for fenitrothion in 2 species of
blue-green algae (Anabaena sp. and Aulosira fertilissima) were
reported to be 42-347 and 136-784, respectively, when exposed to 1,
5, or 10 mg/litre (Lal et al., 1987).
In a field test in which fenitrothion was sprayed twice at 210
g/ha, to give a maximum concentration of 0.9 µg/litre in surface
water and 0.42 µg/litre in subsurface water (3 m depth),
phyto-planktons and zooplanktons contained maximum levels of 0.05
and 0.014 µg fenitrothion/litre, respectively, the concentrations
decreasing with time (Lakshminarayama & Bouque, 1980).
b) Birds
When male Hubbert chickens were intubated with fenitrothion at a
dose of 10 mg/kg, twice every other week, for 2-8 weeks, the residue
levels in the brain, blood, liver, and adipose tissue were less than
0.071 mg fenitrothion equivalent/kg wet tissue. None of the tissues
retained any significant amounts of fenitrothion or its metabolites,
and no tendency towards bioaccumulation was observed (Trottier &
Jankowska, 1980).
White Leghorn hens, dosed with 2 mg fenitrothion/kg body weight
for 7 consecutive days, discharged 95% of the radioactivity in the
excreta within 6 h following the last administration. The
radioactivity in the hen egg-white decreased sharply after the last
dosage, with the highest concentration (0.02 mg/kg) recorded on the
third day of administration. The egg yolk showed a maximum
radiocarbon level of 0.10 mg/kg (fenitrothion, 0.006 mg/kg), 1 day
after the last dose, followed by a decline to 0.02 mg/kg after 1
week (Mihara et al., 1979).
After oral administration of ring-labelled 14C fenitrothion
at 5 mg/kg body weight to female Japanese quails, 99% of the
radio-carbon was eliminated during the first 24 h (Miyamoto, 1977a).
When [phenyl-14C]-fenitrothion was administered orally to
Japanese quails in a single dose of 5 mg/kg body weight or to White
Leghorn hens at a daily dose of 2 mg/kg body weight for 7 days,
97-99% of the radiocarbon was eliminated in the mixture of urine and
faeces within one day. The radioactivity in the eggs was, at most,
0.2% of the parent compound (0.055 mg/kg). More than 18 metabolites
were found in the excreta. The major metabolites were
3-methyl-4-nitrophenol [9] and its sulfate conjugate [26], which
accounted for 70.5% of the dose in quails and 50.8% in hens.
Demethylfenitrothion [7] and demethylfenitrooxon [20] were found as
minor metabolites; several m-methyl oxidation products were also
detected. In vitro studies revealed that the oxidation activity of
hen, quail, pheasant, and duck liver enzymes at the m-methyl group
of fenitrooxon was higher than that of mammalian liver enzymes,
though the avian enzymes had extremely low O-demethylase activity
(Mihara et al., 1979) (see Fig. 5).
c) Terrestrial organisms
Lactating Japanese Sannen goats were treated orally with
[phenyl-14C]-fenitrothion at 0.5 mg/kg body weight per day for 7
days. One day after treatment, no residues of intact fenitrothion
were found in the organs and tissues, but a small amount of
aminofenitrothion [13] was detected in the digestive tracts (rumen,
omasum, and large intestine). The administered radiocarbon was
essentially quantitatively eliminated during the week following
treatment; 50% of the dose was recovered in the urine, 44%, in
faeces, and 0.1%, in the milk with a maximum concentration of 0.011
mg/litre. The major metabolites in the urine, faeces, and milk were
aminofenitrothion [13] and O-methyl O-hydrogen
O-(3-methyl-4-acetyl-aminophenyl) phosphate [37], and
O,O-dimethyl O-(3-methyl-4-sulfo-aminophenyl) phosphorothioate
(N-sulfo-aminofenitrothion) [38], respectively. No intact
fenitrothion or fenitrooxon was found in the milk, urine, or faeces
(Mihara et al., 1978) (see Fig. 6).
McLeese et al. (1979) reported that accumulation ratios of
fenitrothion were independent of the exposure levels, and were
19-35, 78-130, and 9, in marine clam (Mya arenaria), mussel (Mytilus
edulis), and freshwater clam (Anodonta cataractae), respectively.
When freshwater snails ( Cipangopaludina japonica and Physa
acuta) were exposed to 0.1 mg [methyl-14C]-fenitrothion/litre in
a dynamic flow system, the concentrations of fenitrothion and
14C-label in the body reached equilibrium on day one of exposure.
The maximum bioaccumulation ratios of fenitrothion were 18 and 53 in
C. japonica and P. acuta, respectively. These snails metabolized
the compound primarily by demethylation to [7], hydrolysis to [9],
and reduction to [13], and [14]. The liberated phenol moiety was
conjugated with sulfate [26] in C. japonica and mainly with
glucose [18] in P. acuta. When the snails were transferred to a
freshwater stream, fenitrothion and its metabolites were rapidly
eliminated, and the half-life of the parent compound was less than
0.5 days in both snails. In P. acute, fenitrothion and its
decomposition products were mainly distributed in the liver as shown
by whole-body radioautography (Takimoto et al., 1987b).
When the waterflea Daphnia pulex and the shrimp Palaemon
paucidens were exposed to 1.0 µg [methyl-14C]-fenitrothion/litre
in a flow-through system, the concentrations of fenitrothion and
14C-label in the body reached equilibrium (on day one of exposure)
and the maximum bioaccumulation ratios of fenitrothion were 71 and 6
in the daphnia and shrimp, respectively. These crustaceans primarily
metabolized the compound through oxidation of P = S to P = O to form
compound [1], hydrolysis of P-O-aryl linkage to form compound [9],
and demethylation to give compounds [7] and [20]. The liberated
phenol was conjugated with sulfate to form compound [26] in D.
pulex and with glucose to form compound [18] in the shrimp. When
the organisms were transferred to a freshwater stream, fenitrothion
and its metabolites were rapidly eliminated from their bodies, and
the half-life of the parent compound was less than 0.2 day in both
species (Takimoto et al., 1987c) (see Fig. 4).
The uptake rate of radiolabelled fenitrothion by the blue crab
(Callinectes sapides) increased with temperature and salinity. The
highest concentrations of radioactivity were found in the
hepato-pancreas and stomach. The blue crab can metabolize
fenitrothion to produce fenitrooxon, aminofenitrothion,
3-methyl-4-nitrophe-nol, 3-methyl-4-aminophenol, demethyl
fenitrothion, demethyl fenitrooxon, and glycoside and sulfate
conjugates of the phenols (Johnston & Corbett, 1986).
Aquatic plants were collected and analysed after the aerial
application of fenitrothion (280 g/ha) in Manitoba, Canada, (Moody
et al., 1978); the fenitrothion residues present in surface-
dwelling duckweed, obtained from stagnant water, disappeared rapidly
from 1.44 mg/kg after 1 h to 0.03 mg/kg after 192 h, while the
submerged hornwort, also from stagnant water, contained rather
persistent, but low, residues of fenitrothion ranging from 0.12 to
0.15 mg/kg after 192 h. No fenitrothion was detected in the
submerged flowering rush in running water.
Chlorella pyrenoidosa exposed to 10 mg radioactive
fenitrothion per litre rapidly took up the compound and the 14C
level reached equilibrium after 4 h with a bioaccumulation ratio of
417. On transfer to fresh water, the fenitrothion in the chlorella
was rapidly desorbed (Weinberger et al., 1982b). Other algae
( Chlamydomonas reinhardii and Euglena gracilis) showed less
bioaccumulation of fenitrothion. The aquatic macrophyte, Elodea
densa, showed a bioaccumulation ratio of 24-76 (Weinberger et al.,
1982b).
Three types of algae, Chlorella vulgaris, Nitzschia
closterium, and Anabaena flos-aquae, also rapidly absorbed
fenitrothion with maximum bioaccumulation ratios of 44, 105, and 53,
respectively (Kikuchi et al., 1984). Only A. flos-aquae (blue-green
algae) actively degraded fenitrothion. When transferred to a
fenitrothion-free medium, these algae released fenitrothion, as well
as its metabolites, with half-lives of the compounds of less than 1
day, except in the case of A. flos-aquae when the half-life was 2.6
days (see Fig. 4).
Bioaccumulation ratios for fenitrothion in 2 species of
blue-green algae (Anabaena sp. and Aulosira fertilissima) were
reported to be 42-347 and 136-784, respectively, when exposed to 1,
5, or 10 mg/litre (Lal et al., 1987).
In a field test in which fenitrothion was sprayed twice at 210
g/ha, to give a maximum concentration of 0.9 µg/litre in surface
water and 0.42 µg/litre in subsurface water (3 m depth),
phyto-planktons and zooplanktons contained maximum levels of 0.05
and 0.014 µg fenitrothion/litre, respectively, the concentrations
decreasing with time (Lakshminarayama & Bouque, 1980).
b) Birds
When male Hubbert chickens were intubated with fenitrothion at a
dose of 10 mg/kg, twice every other week, for 2-8 weeks, the residue
levels in the brain, blood, liver, and adipose tissue were less than
0.071 mg fenitrothion equivalent/kg wet tissue. None of the tissues
retained any significant amounts of fenitrothion or its metabolites,
and no tendency towards bioaccumulation was observed (Trottier &
Jankowska, 1980).
White Leghorn hens, dosed with 2 mg fenitrothion/kg body weight
for 7 consecutive days, discharged 95% of the radioactivity in the
excreta within 6 h following the last administration. The
radioactivity in the hen egg-white decreased sharply after the last
dosage, with the highest concentration (0.02 mg/kg) recorded on the
third day of administration. The egg yolk showed a maximum
radiocarbon level of 0.10 mg/kg (fenitrothion, 0.006 mg/kg), 1 day
after the last dose, followed by a decline to 0.02 mg/kg after 1
week (Mihara et al., 1979).
After oral administration of ring-labelled 14C fenitrothion
at 5 mg/kg body weight to female Japanese quails, 99% of the
radio-carbon was eliminated during the first 24 h (Miyamoto, 1977a).
When [phenyl-14C]-fenitrothion was administered orally to
Japanese quails in a single dose of 5 mg/kg body weight or to White
Leghorn hens at a daily dose of 2 mg/kg body weight for 7 days,
97-99% of the radiocarbon was eliminated in the mixture of urine and
faeces within one day. The radioactivity in the eggs was, at most,
0.2% of the parent compound (0.055 mg/kg). More than 18 metabolites
were found in the excreta. The major metabolites were
3-methyl-4-nitrophenol [9] and its sulfate conjugate [26], which
accounted for 70.5% of the dose in quails and 50.8% in hens.
Demethylfenitrothion [7] and demethylfenitrooxon [20] were found as
minor metabolites; several m-methyl oxidation products were also
detected. In vitro studies revealed that the oxidation activity of
hen, quail, pheasant, and duck liver enzymes at the m-methyl group
of fenitrooxon was higher than that of mammalian liver enzymes,
though the avian enzymes had extremely low O-demethylase activity
(Mihara et al., 1979) (see Fig. 5).
c) Terrestrial organisms
Lactating Japanese Sannen goats were treated orally with
[phenyl-14C]-fenitrothion at 0.5 mg/kg body weight per day for 7
days. One day after treatment, no residues of intact fenitrothion
were found in the organs and tissues, but a small amount of
aminofenitrothion [13] was detected in the digestive tracts (rumen,
omasum, and large intestine). The administered radiocarbon was
essentially quantitatively eliminated during the week following
treatment; 50% of the dose was recovered in the urine, 44%, in
faeces, and 0.1%, in the milk with a maximum concentration of 0.011
mg/litre. The major metabolites in the urine, faeces, and milk were
aminofenitrothion [13] and O-methyl O-hydrogen
O-(3-methyl-4-acetyl-aminophenyl) phosphate [37], and
O,O-dimethyl O-(3-methyl-4-sulfo-aminophenyl) phosphorothioate
(N-sulfo-aminofenitrothion) [38], respectively. No intact
fenitrothion or fenitrooxon was found in the milk, urine, or faeces
(Mihara et al., 1978) (see Fig. 6).
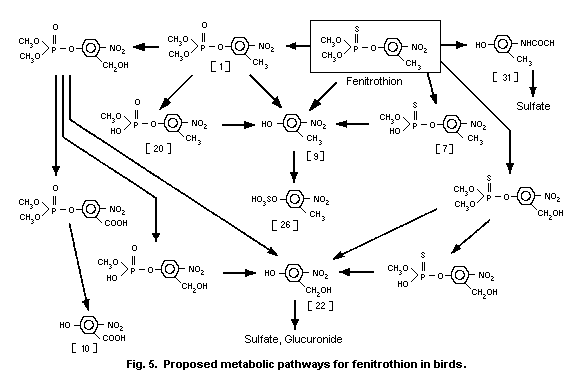 When 30 calves (1-1.5 years old, average weight, 243 kg) were
confined on a pasture sprayed with 378 g fenitrothion/ha (initial
residue on grass, 11.8 mg/kg), the meat and fat contained about 0.01
mg fenitrothion residues/kg on the first day. No fenitrothion
residues were found in the meat from the third day on, and only
0.004-0.007 mg fenitrothion/kg was found in the fat on the third
day; these amounts decreased to almost control levels by the seventh
day (Miyamoto & Sato, 1969).
Silage prepared from corn treated with 1.1, 2.2, or 3.4 kg
fenitrothion/ha was fed to lactating Jersey cattle for 8 weeks.
Although traces (0.001-0.005 mg/kg) of aminofenitrothion [13] were
found in the milk of cows fed 3.4 kg fenitrothion/ha silage, no
residues (< 0.001 mg/kg) were found in the milk of cows that had
consumed the silage treated at lower levels (Leuck et al., 1971).
Jersey cows, administered 3 mg fenitrothion/kg body weight for
7 consecutive days, produced milk containing fenitrothion and
aminofenitrothion [13] levels of up to 0.002 and 0.003 mg/kg,
respectively. However, levels were undetectable within 2 days of the
last dose (Miyamoto et al., 1967).
Johnson & Bowman (1972) reported that neither fenitrothion nor
its metabolites were detected in the milk of lactating Jersey cows,
7 days after being fed (for 28 days) a diet containing the pesticide
at a concentration of 1.84 mg/kg.
Topical application of a lethal dose of fenitrothion to spruce
budworm ( Choristoneura fumiferana) resulted in the formation of
3-methyl-4-nitrophenol (2-17%) and desmethyl fenitrothion (2-4%).
Trace levels (1-2% of the applied dose) of fenitrooxon were also
detected (Sundaram, 1988).
4.2.2.3 Abiotic and biological degradation in/on plants
The photolysis and metabolic pathways of fenitrothion in plants
are illustrated in Fig. 7.
One half the amount of fenitrothion applied at 12 mg/kg to rice
plants at the preheading stage was lost by evaporation and only 10%
was left on the plant surface after 24 h, 50% penetrating into
tissues (Miyamoto & Sato, 1969). Although fenitrooxon [1] was
detected at 0.01-0.86 mg/kg in leaf sheaths and blades (not in
harvested grains), it disappeared faster than fenitrothion.
The half-lives of fenitrothion were 1-3 days on, and in,
fenitrothion-treated apples (approx. 4.5 mg/kg) growing on the tree
under natural conditions, with fenitrooxon [1] (0.005 mg/kg) and
S-methyl fenitrothion [8] (approx. 0.005 mg/kg), on the fruit and
demethyl fenitrothion [7] (0.012 mg/kg), 3-methyl-4-nitro-phenol
[9] (0.024 mg/kg), and its glucose conjugate [18] in the fruit after
21 days (Hosokawa & Miyamoto, 1974). It was concluded that the fruit
metabolized the penetrating fenitrothion gradually to
3-methyl-4-nitrophenol [9], and further to the glucose conjugate
(e.g., [18]) in the tissues; this was combined with the
disappearance of fenitrothion on the fruit surface through
photodecomposition and volatilization.
A number of residue data are available on various feed plants
(Sumitomo, 1969; Takimoto & Miyamoto, 1976b). Coastal Bermuda grass
and corn treated with fenitrothion at 1, 2, and 3 kg/ha were
analysed for residues of the parent compound and some metabolites
(Leuck & Bowman, 1969); the residues of fenitrothion diminished
rapidly to approximately one hundredth of the initial levels after
14 days. The fenitrooxon [1] contents were low, declining more
rapidly, and none was detected after 21 days. While the amounts of
3-methyl-4-nitrophenol [9] were highest in the 1- and 7-day samples,
the total residues on both crops diminished to less than 1 mg/kg in
28 days.
Under operational spraying (280 g/ha) for the control of
budworm, fenitrothion deposits (2-4 mg/kg, wet weight) on the
foliage of red and white spruce and balsam fir decreased by about
50% within 4 days, and 70-85% within two weeks. In some cases, about
10% of the initial deposit (0.05-0.5 mg/kg) persisted for most of
the year following spraying (Yule & Duffy, 1972).
To monitor the persistence of fenitrothion in the Canadian
forest, LaPierre (1985) measured residues in leaves. Immediately
following application (15 min) of fenitrothion to poplar ( Populus
tremuloidus) and gray birch trees ( Betula populifolia),
fenitrothion levels of 22 and 18 mg/kg, respectively, were detected.
Residue levels decreased to less than 1 mg/kg and 0.1 mg/kg,
respectively, within 30 and 120 days. No fenitrooxon was detected in
any of the samples. The observation, made by McNeil & McLeod (1977),
indicating that sawfly populations were depressed by persistent
fenitrothion residues in jack pine foliage apparently supported the
persistence of fenitrothion within leaf tissues. However, it may be
that, in this case, the fenitrothion was rather persistent because
of the special circumstances (in micro sink).
A complete disappearance of fenitrothion from spruce foliage
was observed, within 45 days, following operational spraying with
280 g/ha. The hardwood species within mixed forests, such as red
maple, appeared to collect 3-4 times higher deposits on their
foliage when exposed to the same operational spraying (National
Research Council of Canada, 1975), but the residues decreased
rapidly.
When 30 calves (1-1.5 years old, average weight, 243 kg) were
confined on a pasture sprayed with 378 g fenitrothion/ha (initial
residue on grass, 11.8 mg/kg), the meat and fat contained about 0.01
mg fenitrothion residues/kg on the first day. No fenitrothion
residues were found in the meat from the third day on, and only
0.004-0.007 mg fenitrothion/kg was found in the fat on the third
day; these amounts decreased to almost control levels by the seventh
day (Miyamoto & Sato, 1969).
Silage prepared from corn treated with 1.1, 2.2, or 3.4 kg
fenitrothion/ha was fed to lactating Jersey cattle for 8 weeks.
Although traces (0.001-0.005 mg/kg) of aminofenitrothion [13] were
found in the milk of cows fed 3.4 kg fenitrothion/ha silage, no
residues (< 0.001 mg/kg) were found in the milk of cows that had
consumed the silage treated at lower levels (Leuck et al., 1971).
Jersey cows, administered 3 mg fenitrothion/kg body weight for
7 consecutive days, produced milk containing fenitrothion and
aminofenitrothion [13] levels of up to 0.002 and 0.003 mg/kg,
respectively. However, levels were undetectable within 2 days of the
last dose (Miyamoto et al., 1967).
Johnson & Bowman (1972) reported that neither fenitrothion nor
its metabolites were detected in the milk of lactating Jersey cows,
7 days after being fed (for 28 days) a diet containing the pesticide
at a concentration of 1.84 mg/kg.
Topical application of a lethal dose of fenitrothion to spruce
budworm ( Choristoneura fumiferana) resulted in the formation of
3-methyl-4-nitrophenol (2-17%) and desmethyl fenitrothion (2-4%).
Trace levels (1-2% of the applied dose) of fenitrooxon were also
detected (Sundaram, 1988).
4.2.2.3 Abiotic and biological degradation in/on plants
The photolysis and metabolic pathways of fenitrothion in plants
are illustrated in Fig. 7.
One half the amount of fenitrothion applied at 12 mg/kg to rice
plants at the preheading stage was lost by evaporation and only 10%
was left on the plant surface after 24 h, 50% penetrating into
tissues (Miyamoto & Sato, 1969). Although fenitrooxon [1] was
detected at 0.01-0.86 mg/kg in leaf sheaths and blades (not in
harvested grains), it disappeared faster than fenitrothion.
The half-lives of fenitrothion were 1-3 days on, and in,
fenitrothion-treated apples (approx. 4.5 mg/kg) growing on the tree
under natural conditions, with fenitrooxon [1] (0.005 mg/kg) and
S-methyl fenitrothion [8] (approx. 0.005 mg/kg), on the fruit and
demethyl fenitrothion [7] (0.012 mg/kg), 3-methyl-4-nitro-phenol
[9] (0.024 mg/kg), and its glucose conjugate [18] in the fruit after
21 days (Hosokawa & Miyamoto, 1974). It was concluded that the fruit
metabolized the penetrating fenitrothion gradually to
3-methyl-4-nitrophenol [9], and further to the glucose conjugate
(e.g., [18]) in the tissues; this was combined with the
disappearance of fenitrothion on the fruit surface through
photodecomposition and volatilization.
A number of residue data are available on various feed plants
(Sumitomo, 1969; Takimoto & Miyamoto, 1976b). Coastal Bermuda grass
and corn treated with fenitrothion at 1, 2, and 3 kg/ha were
analysed for residues of the parent compound and some metabolites
(Leuck & Bowman, 1969); the residues of fenitrothion diminished
rapidly to approximately one hundredth of the initial levels after
14 days. The fenitrooxon [1] contents were low, declining more
rapidly, and none was detected after 21 days. While the amounts of
3-methyl-4-nitrophenol [9] were highest in the 1- and 7-day samples,
the total residues on both crops diminished to less than 1 mg/kg in
28 days.
Under operational spraying (280 g/ha) for the control of
budworm, fenitrothion deposits (2-4 mg/kg, wet weight) on the
foliage of red and white spruce and balsam fir decreased by about
50% within 4 days, and 70-85% within two weeks. In some cases, about
10% of the initial deposit (0.05-0.5 mg/kg) persisted for most of
the year following spraying (Yule & Duffy, 1972).
To monitor the persistence of fenitrothion in the Canadian
forest, LaPierre (1985) measured residues in leaves. Immediately
following application (15 min) of fenitrothion to poplar ( Populus
tremuloidus) and gray birch trees ( Betula populifolia),
fenitrothion levels of 22 and 18 mg/kg, respectively, were detected.
Residue levels decreased to less than 1 mg/kg and 0.1 mg/kg,
respectively, within 30 and 120 days. No fenitrooxon was detected in
any of the samples. The observation, made by McNeil & McLeod (1977),
indicating that sawfly populations were depressed by persistent
fenitrothion residues in jack pine foliage apparently supported the
persistence of fenitrothion within leaf tissues. However, it may be
that, in this case, the fenitrothion was rather persistent because
of the special circumstances (in micro sink).
A complete disappearance of fenitrothion from spruce foliage
was observed, within 45 days, following operational spraying with
280 g/ha. The hardwood species within mixed forests, such as red
maple, appeared to collect 3-4 times higher deposits on their
foliage when exposed to the same operational spraying (National
Research Council of Canada, 1975), but the residues decreased
rapidly.
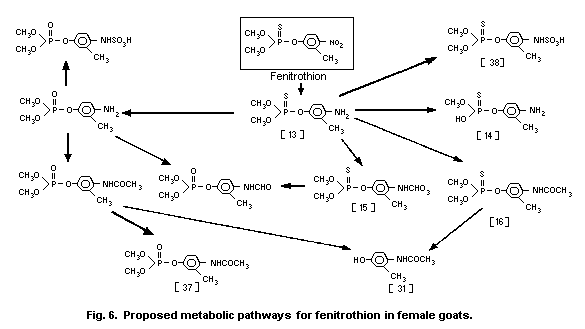
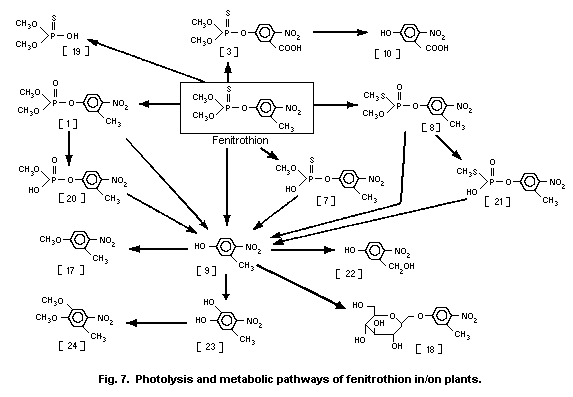 During environmental surveillance of aerial spraying of
fenitrothion, carried out from 1979 to 1982 in Quebec, the
insecticide concentrations were measured in foliage samples taken
from 1 to 4 h after spraying, when residue levels were likely to
have peaked. Over the 4 years studied, the median residue level of
fenitrothion found in balsam fir foliage was 3.81 mg/kg (dry
weight), with a maximum concentration of 111 mg/kg. On conifer
foliage, feni-trothion had a half-life of 2-4 days; 70-95% of the
residue dissipated in less than 2 weeks (Morin et al., 1986).
Takimoto et al. (1978) examined the stability of fenitrothion
(6 and 15 mg/kg) in stored rice grains. The insecticide decomposed
after 12 months to 22.0-26.3% and 64.7-65% of the initial dose at 30
and 15 °C, respectively. The major metabolites in rice grains were
demethyl fenitrothion [7] and 3-methyl-4-nitrophenol [9], which
amounted to 10.0-19.2% and 16.0-38.0% of the dose, respectively. In
addition, trace amounts of S-methyl fenitrothion [8], S-methyl
demethyl fenitrothion [21], fenitrooxon [1], demethyl fenitrooxon
[20], 3-hydroxymethyl-4-nitrophenol [22], 3-methyl-4-nitroanisole
[17], 1,2-dihydroxy-4-methyl-5-nitro-benzene [23], and
1,2-dimethoxy-4-methyl-5-nitrobenzene [24] were detected (see Fig.
7). Fenitrothion and its degradation products were distributed in
the outer portions of the endosperm and at 100 µm in depth from the
surface of rice grains stored for 12 months, as determined by
whole-body autoradiography. On cooking, the unpolished rice grains
treated with fenitrothion yielded 3-methyl-4-nitrophenol [9] and
demethyl fenitrothion [7] as primary degradation products.
Abdel-Kader & Webster (1980) and Abdel-Kader et al. (1982)
studied the stability of fenitrothion (8 mg/kg) in stored wheat.
Very little (< 3%) breakdown of the insecticide residue occurred on
wheat stored at -35 or -20 °C for 72 weeks. However, fenitrothion
residues decreased as the temperature increased. After 72 weeks, 18,
35, 56, 90, 96% of the initial deposit had degraded in wheat stored
at -5, 5, 10, 20, at 20 °C respectively. The major metabolites in
wheat stored at 20 °C for 12 months were demethyl fenitrothion [7],
3-methyl-4-nitrophenol [9], and dimethyl phosphorothioic acid [19]
(Fig. 7), as determined by GLC. Concentrations of demethyl
fenitrothion [7] and dimethyl phosphorothioic acid [19], which were
highest (2.01 and 0.55 mg/kg, respectively) after 6 months storage,
decreased to 0.98 and 0.21 mg/kg, respectively, at the end of
storage. The residue level of 3-methyl-4-nitrophenol [9] gradually
increased to 0.96 mg/kg after 12 months. No fenitrooxon [1] or
S-methyl fenitrothion [8] was detected throughout the experimental
period (Abdel-Kader & Webster, 1982).
When labelled fenitrothion was applied to bean leaves at a rate
of 84.5 µg/12.5 cm2, 26 and 64% of the radioactivity was lost by
volatilization after 1 and 3 days, respectively. The decrease of the
parent compound was rapid, both on and in the leaf.
After 12 days, the major products remaining on the bean leaves were
fenitrooxon [1] (0.1%), carboxy-fenitrothion [3] (0.1%), and
3-carboxy-4-nitrophenol [10] (0.1%) (Ohkawa et al., 1974).
The residues of fenitrothion in coastal Bermudagrass and corn
diminished to less than 0.13 mg/kg in 28 days, with half-lives of
less than 1 day, following a spray of the emulsifiable concentrate
at 1, 2, or 3 kg a.i./ha. The major metabolite was
3-methyl-4-nitrophenol [9], with smaller amounts of fenitrooxon [1].
After 28 days, the residues had declined to less than 1 mg/kg for
all 3 rates of application (Leuck & Bowman, 1969).
In leaves of shrubbery ( Maesa japonica) sprayed twice with
fenitrothion at 735 g/ha, fenitrothion was detected at 78.3 mg/kg on
the day of application, but 99% had disappeared within a week.
Although fenitrooxon [1] was detected at levels of 0.1-0.3% of
fenitrothion in the leaves, it disappeared after strong rainfall,
35-37 days after application. Fenitrothion was detected in grasses,
and the upper and lower layers of the soil, at levels of 0.1, 0.01,
and 0.001 mg/kg, respectively, 144 days after application (Ohmae et
al., 1981).
Hallett et al. (1973) observed the transport of fenitrothion to
the embryo, and its metabolism in seed tissues, when pine seeds were
germinated for up to 54 days in an aqueous solution or suspension of
fenitrothion (10 or 1000 mg/litre). Laboratory studies of
fenitrothion on seeds of eastern white pine demonstrated penetration
and accumulation of the parent compound, its oxon, and the
S-methyl metabolites in the embryo and perisperm. This appeared to
alter the amino acid metabolism in the seed but did not affect the
later growth of seedlings (Hallett et al., 1974). No significant
differences in germination and growth were reported between the
seeds of white pine from areas sprayed at 140-280 g/ha and from
unsprayed (control) areas (Pomber et al., 1974).
Similarly, the seeds of white pine, white spruce, and yellow
birch readily absorbed fenitrothion when germinated for up to 21
days in an aqueous solution or suspension of fenitrothion (10 or
1000 mg/litre). Fenitrooxon [1], demethyl fenitrothion [7], and
S-methyl fenitrothion [8] were detected as primary metabolites in
all 3 species. The highest concentrations of [1], [7], and [8] were
1.4-75 mg/kg, 10-37 mg/kg, and 1-8 mg/kg, respectively. Hallett et
al. (1977) proposed that the formation of S-methyl fenitrothion [8]
resulted from the alkylation of demethyl fenitrothion by excess
fenitrothion in the conifer seeds. It is probable that [8] will be
formed in plants via the non-enzymatic alkylation reaction, if the
concentrations of fenitrothion and [7] are extremely high. However,
formation of [7] will be extremely low or negligible, when
fenitrothion is used at the recommended dosage.
Sundaram & Prasad (1975) established the uptake and
transportation of fenitrothion in young spruce trees by growing the
plants in a nutrient solution containing the insecticide. However,
when fenitrothion was applied to the needles of spruce and fir
trees, which are the part of the tree most exposed to the
insecticide during spraying, the foliar penetration of fenitrothion
was found to be extremely small. Furthermore, less than 0.1% of the
fenitrothion that had penetrated was translocated laterally and
upward to the untreated parts of the foliage. The amounts found in
the stems and roots were also negligible (Sundaram et al., 1975).
On the other hand, Moody et al. (1977) using an
autoradiographic technique, suggested the systemic potential of
fenitrothion applied to 4-year-old seedlings of balsam fir and, to a
lesser extent, white spruce, and jackpine.
Prasad & Moody (1976) also found that, mainly because of
volatilization, 70% of the applied fenitrothion was lost one day
after treatment, though low levels of the insecticide (0.48 mg/kg
after 21 days) persisted in conifer tissues.
When labelled fenitrothion was applied to the leaves of
Japanese cypress at about 300 µg per leaf, approximately 70-80% of
the applied dose disappeared, mainly through evaporation, within 24
h. The major metabolites in the treated leaves were demethyl
fenitrothion [7], 3-methyl-4-nitrophenol [9] (approx. 2-10 µg), and
its glucoside conjugate [18] (Tabata & Okubo, 1980).
Immediately following the application of fenitrothion to poplar
( Populus tremuloidus) and birch trees ( Betula populifolia),
levels of 22 and 18 mg/kg, respectively, were detected. Residue
levels dropped to under 1 mg/kg within 30 days of treatment, and
under 0.1 mg/kg by 120 days. The oxygen analogue, fenitrooxon, was
not detected in any of the samples (La Pierre, 1985).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
In the 1980 spray programme in Canada, fenitrothion was
detected occasionally in air, the maximum concentration being 12
ng/m3 (Mallet, 1980), when the compound was sprayed at 140-280
g/ha.
Collaborative field studies were initiated in 1978 in Canada to
obtain relevant information concerning long-range pesticide drift
during the aerial spraying of fenitrothion in forest pest control,
using a surrogate (tris(2-ethylhexyl)phosphate) (Crabbe et al.,
1980a). At approximately 7.5 km downwind of the spray, the peak
treetop concentration of the drifting cloud varied from 1.4 ng/litre
in relatively neutral conditions to 5.0 ng/litre in the most stable
conditions. The fraction of the tracer material still airborne, 7.5
km from the spray line, was 6 and 16% for the neutral and most
stable environmental tests, respectively.
In 1980, a study was conducted to measure the atmospheric
fenitrothion exposure levels at breathing height near aerial
forestry spray operations (Crabbe et al., 1980b). The results
revealed an aerial concentration (or dosage) of 40 ng/min per litre1
at the spray line (flight path of aircraft) falling to an average of
8 ng/min per litre, 700 m from the spray line, in the first hour
after delivery. During the first hour after spraying, 30% of the
material collected in the samplers was vapour and the rest, aerosol.
Droplets of approx. 4.0 µm diameter contributed most to the mass of
drifting spray cloud at 500 m, about 15% of the airborne aerosol
mass consisting of droplets larger than 25 µm in diameter. The peak
aerosol concentration at chest height underneath the sprayed swath
of forest was approximately 15 ng/litre. One result of considerable
interest was the degree of volatilization of fenitrothion from an
early morning spray, which resulted in a sustained vapour plume
during the afternoon with a concentration
1 The value nanograms/min per litre represents the time
integrated aerial concentration of fenitrothion consisting of
the mean aerosol cloud concentration of agent (nanograms per
litre) divided by the residence time (minutes) of the cloud.
of 25 ng/litre at the spray line, falling to approximately 1.0
ng/litre 100 m downwind of the swath at an ambient temperature of
20-25 °C. The vapour plume persisted throughout the day and, in a
10-h period following spraying, was estimated to contribute up to
0.4 µg/person of respirable fenitrothion.
A pesticide residue survey was conducted in relation to the
1980 spruce budworm spray programme (210 g a.i./ha applied twice
with a 3-day interval) in New Brunswick, Canada. Fenitrothion was
detected occasionally in the air, the maximum level being 1.2
µg/m3. Amino-fenitrothion was sometimes present in the air at a
maximum level of 12.0 µg/m3 (Mallet & Volpe, 1982).
A chemical residue survey was undertaken to determine the
extent of the deposition and persistence of fenitrothion in the
environment in relation to the 1981 spruce budworm spray programme
(210 g a.i./ha applied twice with a 7-day interval) in New
Brunswick, Canada. Fenitrothion was only detected twice in the air
at levels of 0.08 and 0.04 µg/m3 (Mallet & Cassista, 1984).
When a 1.0% emulsion of fenitrothion was applied to a 61.2 m3
room (3.7 ± 0.3 g a.i./room) at 23.5-25.4 °C, using a compressed air
sprayer, the airborne concentrations of fenitrothion on days 0 and 3
after application were 3.3 µg/m3 and 0.5 µg/m3, respectively
(Wright et al., 1981).
5.1.2 Water
In actual field spraying in Canada, concentrations of
fenitrothion in stream waters varied greatly, depending on the spray
history and the weather. Spraying at 210 g/ha resulted in measurable
concentrations (> 0.03 µg/litre) in streams as far as 4 km from the
sprayed area. However, at 140-210 g/ha, most peak concentrations
were lower than 15 µg/litre and diminished very rapidly in
fast-flowing streams. Disappearance was still faster when a rain
storm followed spray application (Eidt & Sundaram, 1975).
High peak concentrations of fenitrothion were recorded in a few
cases, e.g., 64 µg/litre in flowing stream water approximately 1 h
after completion of spraying, and 75.5 µg/litre in stagnant water
some 17 h after spraying at 140-280 g/ha (Lockhart et al., 1977).
Fenitrothion concentrations usually dropped to less than 1 µg/litre
within a few days in forest streams and beaver ponds after
experimental and operational applications of 140-280 g/ha by
aircraft (Flannagan, 1973; Peterson & Zitko, 1974; Sundaram, 1974;
Eidt & Sundaram, 1975), and no measurable traces of fenitrothion
have been found in water longer than 40 days after spraying.
According to Symons (1977), the fenitrothion concentration in water
immediately after aerial spray at 140-280 g/ha in Canada seldom
exceeded 15 µg/litre.
One hour after the first application of fenitrothion (280 g
a.i./ha, applied twice with a 9-day interval) to a lake,
fenitrothion residues in the water were concentrated near the
surface (0.80-0.90 µg/litre), with small amounts (0.06 µg/litre) at
the bottom. Six hours later, residues were fairly evenly distributed
throughout the water column (0.91-1.49 µg/litre). Residue levels, 97
h after treatment, were similar at all depths at 0.41-0.46 µg per
litre, but declined rapidly in the next 48 h to < 0.01-0.06
µg/litre. Similar results were obtained after the second application
(Holmes et al., 1984).
Sundaram (1973) also reported that concentrations of
fenitrothion in aqueous systems diminished rapidly by dilution and
by physicochemical and microbial degradation to low levels (0.03
µg/litre) within a period of 40 days. The half-lives were found to
be short, ranging from 0.25 to 3.5 days.
In the 1980 spruce budworm spray programme in Canada,
fenitrothion was usually detected in water in the immediate vicinity
of the sprayed sites. The maximum level was 20.0 µg/litre and
persistence was usually limited to a few days. Aminofenitrothion
[13] was also found at a maximum of 8.0 µg/litre (Mallet, 1980;
Mallet & Volpe, 1982).
Water, suspended solids, and sediment samples were collected
from a small pond in a spruce-fir forest in New Brunswick, Canada,
before and after spraying with a fenitrothion formulation. When an
11% fenitrothion formulation was sprayed on a pond at 1500 g/ha, the
initial fenitrothion concentrations in the surface microlayer,
subsurface water, and suspended solids (0.25-0.67 h after spraying)
were 1.5, 0.015, and 1.9 µg/litre, respectively (Maguire & Hale,
1980). Thereafter, the fenitrothion disappeared exponentially with
half-lives of 0.34, 9.8, 1.8, and 3.2 h in the surface microlayer,
subsurface water, sediment, and suspended solids, respectively.
Residue levels declined rapidly to below detectable levels, 2 days
after spraying. The degradation products, 3-methyl-4-nitrophenol [9]
in water and aminofenitrothion [13] in sediment persisted for less
than 2 and 4 days, respectively. The concentration of fenitrothion
in rainwater during spraying (140-280 g/ha) in Canada was 77
µg/litre in the sprayed area, 17 out of 43 samples in the
neighbouring areas contained levels ranging from a trace to 1.1
µg/litre, while levels were less than 0.01 µg/litre in other regions
(Pearce et al., 1979a). No residues of fenitrooxon [1],
aminofenitrothion [13], or S-methyl fenitrothion [8] were
detected, even in the sprayed area (less than 0.05 µg/litre).
In the 1981 spruce budworm spray programme in Canada, the
amounts of fenitrothion residues detected in water were low (maximum
1.30 µg/litre) and post-spray samples did not contain detectable
amounts (less than 0.01 µg/litre) of fenitrothion. Small amounts of
aminofenitrothion [13] (0.1 µg/litre) were detected in some samples
(Mallet & Cassista, 1984).
In Quebec, environmental surveillance was performed in the
sprayed areas after aerial spraying, carried out from 1979 to 1982.
The insecticide concentrations were measured in foliage and water
samples taken from 1 to 4 h after spraying, when residue levels were
likely to peak. In lentic water, the median residue level was 5.82
µg fenitrothion/litre, with a maximum level of 1114 µg/litre. In
lotic water, the median concentration was 2.84 µg fenitrothion per
litre, with a maximum level of 127 µg/litre. In aquatic
environments, fenitrothion residues disappeared rapidly. Levels of
less than 1.0 µg/litre were found 2-8 days after spraying at 140-280
g a.i./ha (Morin et al., 1986).
Fenitrothion levels in a mountain stream increased to 20 µg per
litre, 6-9 h after spraying, and then decreased to below 1 µg/litre
within 24 h (Hatakeyama et al., 1990).
5.1.3 Soil
When operationally sprayed twice at 735 g/ha (Japan),
fenitrothion levels reached a maximum of 0.13-0.23 mg/kg in soil
under shrubbery after 1-8 days and then diminished with half-lives
of 2-4 days. Aminofenitrothion [13] was detected at maximum levels
of 1.5-6.5 µg/kg, but only shortly after spraying. Little
contamination with fenitrothion due to drift was observed (a maximum
of 0.01 µg/m2 per min, 1 h later) in areas that were 1.5 or 3.0 km
from the application site (Ohmae et al., 1981).
Yule & Duffy (1972) found that an operationally applied dosage
of fenitrothion (280 g/ha) produced a deposit of up to 0.04 mg/kg
(average depth 15 cm) in forest soil, and that the initial deposit
was too small to investigate degradation pathways. Sundaram (1974)
found a similar pattern of loss of fenitrothion from the soil after
5 years of repeated annual applications at a rate of 210-350 g/ha in
Canada; the insecticide content in the soil samples ranged from
traces to 0.1 mg/kg and disappeared within 45 days. No measurable
amounts of the breakdown products were found. Despite consecutive
annual spraying for up to 5 years at 210-350 g/ha in New Brunswick,
Canada, there was no evidence of build-up (< 0.005 mg/kg) in the
soil (Yule, 1974).
In the 1981 sprucce budworm spray programme in Canada, foilage
and soil litter collected after the second spray application
contained fenitrothion at levels of 0.2-0.5 and 0.06-0.14 mg/kg,
respectively; post-spray samples collected 2 weeks later contained
similar levels of fenitrothion. No residual fenitrothion was
detected in sediment (Mallet & Cassista, 1984).
5.1.4 Food
Fenitrothion is used for the pre-harvest treatment of a variety
of crops, such as fruits, vegetables, rice, cereals, soybeans and
coffee, at the rate of 0.5-2.0 kg a.i./ha. The initial
residue levels on fruits, vegetables, rice, and cereals were
relatively high (0.001-9.5 mg/kg), but declined rapidly with a
half-life of 1-2 days (FAO/WHO, 1970, 1975b).
A survey of pesticide residues in crops (total number of
samples: 697) collected in the Tokyo Metropolitan Central Vegetable
and Fruit Market was carried out from April 1984 to March 1989. Low
fenitrothion residues (0.01-0.06 mg/kg) were found in only a few
samples (Nagayama et al., 1986, 1987, 1988, 1989).
Fenitrothion (EC) was applied 3 times to peach at a rate of 2.0
kg a.i./ha in Japan. The residue levels in peach (pulp) were
0.03-0.07 mg/kg, 7 days after application and decreased to
0.005-0.015 mg/kg after 13-15 days. Peach peel contained most of the
residues (1.1-1.6 mg/kg), 7 days after application, but the levels
decreased to 0.3-0.6 mg/kg in 15 days.
Lettuce, plums, and apples were treated once with 0.2%
fenitrothion (EC) at a rate of 0.8 kg. a.i./ha. No increase in
3-methyl-4-nitrophenol indicating the hydrolysis of fenitrothion
residues was observed during 8 days following treatment. Levels of
3-methyl-4-nitrophenol did not exceed 10% of the fenitrothion
residues (Cerna & Benes, 1972).
Strawberries were treated twice with fenitrothion (EC) at a
rate of 1.0 kg a.i./ha; the residue levels were 0.02-0.03 mg/kg and
0.005-0.007 mg/kg, 7 and 15 days after treatment, respectively
(FAO/WHO, 1975b). Following 3 applications of fenitrothion to
tomatoes, a residue of 1.32 mg/kg was detected on the same day as
the final application (Kannan & Jayarnama, 1981). The residue level
dropped to 0.07 mg/kg, one day later, but there was very little
further reduction in fenitrothion levels over a further 2-week
period.
Fenitrothion (as EC, dust, or WP) was applied to rice plants in
Japan at rates of 0.5-1 kg a.i./ha at various time intervals (3-5
times/season) before harvest. Over 120 samples of harvested rice
were analysed for residues of the parent compound. The average
residue levels ranged from negligible (0.001 mg/kg) to 0.1 mg/kg
with a maximum level of 0.3 mg/kg, 11-20 days after the final
application, and 0.001 mg/kg, 41-67 days after treatment (Sumitomo
Chemical Co. Ltd, 1969).
A variety of fenitrothion formulations were applied 1-7 times
to rice crops pre-harvest at intervals ranging from 14 to 120 days.
Most of the samples of hulled rice did not contain any residues at
harvest (limit of determination 0.001 mg/kg), but a few contained
low levels (maximum level 0.025 mg/kg) (FAO/WHO, 1978b).
Fenitrothion is also used in the post-harvest treatment of
stored grain. When grains with a moisture content of 12-13% were
treated with fenitrothion at 15 mg/kg (18 mg/kg for brown rice), the
residues decreased after 3 and 6 month at 25 °C to 6.8 mg/kg and 3.1
mg/kg, respectively, in barley, 6.9 mg/kg and 4.2 mg/kg in oats, 6.2
mg/kg and 3.5 mg/kg in rice, 11.5 mg/kg and 8.0 mg/kg in brown rice,
and to 10 mg/kg and 7.5 mg/kg in polished rice. The rates of
decrease of fenitrothion in/on grains in storage can be predicted
accurately from the temperature, relative humidity, and dosage level
(FAO/WHO, 1978b).
When grains were subjected to milling, processing, preparation,
and cooking, there was a significant loss of fenitrothion.
Fenitrothion was deposited on the epidermis and removed with bran
during the milling process, so that the residue level on bran was
from 2 to 2.5 times higher than that on whole wheat and about 7
times higher than that on husked (brown) rice. After milling, the
residue of fenitrothion in white flour was about 10% or less of the
residue in the raw wheat. The residue in the white bread prepared
from the white flour was reduced to 1-2% of the residue in the raw
wheat. In case of wholemeal bread, however, 20-25% of the initial
concentration remained after baking. The residue of fenitrothion in
rice was decreased to 4.1 and 1.8% of the initial residue during
husking and milling, respectively. The residue in polished rice was
also decreased to 30% of original concentration. The concentration
of fenitrothion in oats, rice in husk, husked rice, and polished
rice decreased to 30-60% of the initial concentration after boiling.
The malting process removed substantially all of the fenitrothion
residue on raw barley. During the processing of oats for the
production of rolled oats and groats, more than 95% of the residues
on the raw oats were removed (FAO/WHO, 1977).
In the United Kingdom, fenitrothion was found in home-produced
and imported wheat at levels of up to 0.2-0.9 mg/kg in 1982 (MAFF,
1986). During the period of 1984-88, residues of fenitrothion were
found in haricot and mung beans at levels of up to 0.1 mg/kg, but
residues in several other pulses did not exceed 0.05 mg/kg. In
retail wheat products, fenitrothion was found at levels of up to
0.05 mg/kg in white bread, 0.3 mg/kg in brown bread, 0.5 mg/kg in
bran, and 0.02 mg/kg in cereal-based infant foods, but was not
detected in breakfast cereals, wheatgerm products, brown rice, rye
products, and processed oats, the level of detection being 0.01-0.1
mg/kg, depending on the commodity and the year of survey (MAFF,
1989).
5.2 Human exposure
5.2.1 Food
In the USA, average fenitrothion residues of 0.0001 mg/kg were
detected in grains and cereal products in 1979-80, and the average
intake for an adult from this source was calculated to be 0.0608
µg/day (or 0.001 µg/kg body weight per day) in 1980 (Gartrell et
al., 1985a). For the infant and toddler, maximum daily intakes were
estimated to be < 0.001 µg/kg body weight per day and 0.002 µg/kg
body weight per day, respectively, during 1977-80 (Gartrell et al.,
1985b).
6. KINETICS AND METABOLISM
The metabolic pathways of fenitrothion in mammals are shown in
Fig. 8.1
6.1 Absorption, distribution, metabolic transformation,
elimination, and excretion
32P-fenitrothion administered orally at doses of 15 and 500
mg/kg to rats and guinea-pigs, respectively, was readily absorbed
from the gastrointestinal tract and distributed among various
tissues. The concentrations of 32P in the blood of rats reached a
maximum (15.5 µg/g) 1-3 h after treatment, and diminished to below
detectable levels within 4 days (Miyamoto et al., 1963a).
Following intravenous injection of 32P-fenitrothion in rats
or guinea-pigs at a dose of 15 or 40 mg/kg, fenitrothion disappeared
equally rapidly from the blood and tissues (Miyamoto, 1964a, 1969).
The radioactivity (32P) was mostly excreted within 2-4 days into
the urine (85-97%), up to 10% being eliminated in the faeces,
accounting for nearly 100% recovery of the dose. The metabolites in
excreta were demethyl fenitrothion [7], demethyl fenitrooxon [20],
dimethylphosphorothioic acid [19], and dimethylphosphoric acid [25].
Fenitrooxon [1] was detected only after intravenous administration
of a large amount of fenitrothion and not after oral administration
(Miyamoto et al., 1963a; Miyamoto, 1964a).
Hollingworth et al. (1967a) studied the metabolism of
fenitrothion in white mice. 32P-fenitrothion administered orally
to mice at a dose of 3, 17, 200, or 850 mg/kg was rapidly eliminated
in the urine and faeces with a recovery of > 90% 72 h after
treatment. The isolated metabolites indicated that both the
P-O-alkyl and P-O-aryl bonds of fenitrothion and fenitrooxon [1]
were cleaved. No evidence was obtained indicating that the nitro
group was reduced to form aminofenitrothion [13] or that the ring
methyl group was oxidized. At the lower dose (17 mg/kg), demethyl
fenitrothion [7], demethyl fenitrooxon [20], dimethylphosphorothioic
acid [19], and dimethylphosphoric acid [25] were the major
metabolites. At the 200 mg/kg body weight dose, the amounts of
dimethylphosphorothioic acid [19] and dimethylphosphoic acid [25]
decreased, and demethyl fenitrothion [7] increased.
1 Chemical structures in Fig. 1 to 8 are referred to according to
numbers in the brackets.
During environmental surveillance of aerial spraying of
fenitrothion, carried out from 1979 to 1982 in Quebec, the
insecticide concentrations were measured in foliage samples taken
from 1 to 4 h after spraying, when residue levels were likely to
have peaked. Over the 4 years studied, the median residue level of
fenitrothion found in balsam fir foliage was 3.81 mg/kg (dry
weight), with a maximum concentration of 111 mg/kg. On conifer
foliage, feni-trothion had a half-life of 2-4 days; 70-95% of the
residue dissipated in less than 2 weeks (Morin et al., 1986).
Takimoto et al. (1978) examined the stability of fenitrothion
(6 and 15 mg/kg) in stored rice grains. The insecticide decomposed
after 12 months to 22.0-26.3% and 64.7-65% of the initial dose at 30
and 15 °C, respectively. The major metabolites in rice grains were
demethyl fenitrothion [7] and 3-methyl-4-nitrophenol [9], which
amounted to 10.0-19.2% and 16.0-38.0% of the dose, respectively. In
addition, trace amounts of S-methyl fenitrothion [8], S-methyl
demethyl fenitrothion [21], fenitrooxon [1], demethyl fenitrooxon
[20], 3-hydroxymethyl-4-nitrophenol [22], 3-methyl-4-nitroanisole
[17], 1,2-dihydroxy-4-methyl-5-nitro-benzene [23], and
1,2-dimethoxy-4-methyl-5-nitrobenzene [24] were detected (see Fig.
7). Fenitrothion and its degradation products were distributed in
the outer portions of the endosperm and at 100 µm in depth from the
surface of rice grains stored for 12 months, as determined by
whole-body autoradiography. On cooking, the unpolished rice grains
treated with fenitrothion yielded 3-methyl-4-nitrophenol [9] and
demethyl fenitrothion [7] as primary degradation products.
Abdel-Kader & Webster (1980) and Abdel-Kader et al. (1982)
studied the stability of fenitrothion (8 mg/kg) in stored wheat.
Very little (< 3%) breakdown of the insecticide residue occurred on
wheat stored at -35 or -20 °C for 72 weeks. However, fenitrothion
residues decreased as the temperature increased. After 72 weeks, 18,
35, 56, 90, 96% of the initial deposit had degraded in wheat stored
at -5, 5, 10, 20, at 20 °C respectively. The major metabolites in
wheat stored at 20 °C for 12 months were demethyl fenitrothion [7],
3-methyl-4-nitrophenol [9], and dimethyl phosphorothioic acid [19]
(Fig. 7), as determined by GLC. Concentrations of demethyl
fenitrothion [7] and dimethyl phosphorothioic acid [19], which were
highest (2.01 and 0.55 mg/kg, respectively) after 6 months storage,
decreased to 0.98 and 0.21 mg/kg, respectively, at the end of
storage. The residue level of 3-methyl-4-nitrophenol [9] gradually
increased to 0.96 mg/kg after 12 months. No fenitrooxon [1] or
S-methyl fenitrothion [8] was detected throughout the experimental
period (Abdel-Kader & Webster, 1982).
When labelled fenitrothion was applied to bean leaves at a rate
of 84.5 µg/12.5 cm2, 26 and 64% of the radioactivity was lost by
volatilization after 1 and 3 days, respectively. The decrease of the
parent compound was rapid, both on and in the leaf.
After 12 days, the major products remaining on the bean leaves were
fenitrooxon [1] (0.1%), carboxy-fenitrothion [3] (0.1%), and
3-carboxy-4-nitrophenol [10] (0.1%) (Ohkawa et al., 1974).
The residues of fenitrothion in coastal Bermudagrass and corn
diminished to less than 0.13 mg/kg in 28 days, with half-lives of
less than 1 day, following a spray of the emulsifiable concentrate
at 1, 2, or 3 kg a.i./ha. The major metabolite was
3-methyl-4-nitrophenol [9], with smaller amounts of fenitrooxon [1].
After 28 days, the residues had declined to less than 1 mg/kg for
all 3 rates of application (Leuck & Bowman, 1969).
In leaves of shrubbery ( Maesa japonica) sprayed twice with
fenitrothion at 735 g/ha, fenitrothion was detected at 78.3 mg/kg on
the day of application, but 99% had disappeared within a week.
Although fenitrooxon [1] was detected at levels of 0.1-0.3% of
fenitrothion in the leaves, it disappeared after strong rainfall,
35-37 days after application. Fenitrothion was detected in grasses,
and the upper and lower layers of the soil, at levels of 0.1, 0.01,
and 0.001 mg/kg, respectively, 144 days after application (Ohmae et
al., 1981).
Hallett et al. (1973) observed the transport of fenitrothion to
the embryo, and its metabolism in seed tissues, when pine seeds were
germinated for up to 54 days in an aqueous solution or suspension of
fenitrothion (10 or 1000 mg/litre). Laboratory studies of
fenitrothion on seeds of eastern white pine demonstrated penetration
and accumulation of the parent compound, its oxon, and the
S-methyl metabolites in the embryo and perisperm. This appeared to
alter the amino acid metabolism in the seed but did not affect the
later growth of seedlings (Hallett et al., 1974). No significant
differences in germination and growth were reported between the
seeds of white pine from areas sprayed at 140-280 g/ha and from
unsprayed (control) areas (Pomber et al., 1974).
Similarly, the seeds of white pine, white spruce, and yellow
birch readily absorbed fenitrothion when germinated for up to 21
days in an aqueous solution or suspension of fenitrothion (10 or
1000 mg/litre). Fenitrooxon [1], demethyl fenitrothion [7], and
S-methyl fenitrothion [8] were detected as primary metabolites in
all 3 species. The highest concentrations of [1], [7], and [8] were
1.4-75 mg/kg, 10-37 mg/kg, and 1-8 mg/kg, respectively. Hallett et
al. (1977) proposed that the formation of S-methyl fenitrothion [8]
resulted from the alkylation of demethyl fenitrothion by excess
fenitrothion in the conifer seeds. It is probable that [8] will be
formed in plants via the non-enzymatic alkylation reaction, if the
concentrations of fenitrothion and [7] are extremely high. However,
formation of [7] will be extremely low or negligible, when
fenitrothion is used at the recommended dosage.
Sundaram & Prasad (1975) established the uptake and
transportation of fenitrothion in young spruce trees by growing the
plants in a nutrient solution containing the insecticide. However,
when fenitrothion was applied to the needles of spruce and fir
trees, which are the part of the tree most exposed to the
insecticide during spraying, the foliar penetration of fenitrothion
was found to be extremely small. Furthermore, less than 0.1% of the
fenitrothion that had penetrated was translocated laterally and
upward to the untreated parts of the foliage. The amounts found in
the stems and roots were also negligible (Sundaram et al., 1975).
On the other hand, Moody et al. (1977) using an
autoradiographic technique, suggested the systemic potential of
fenitrothion applied to 4-year-old seedlings of balsam fir and, to a
lesser extent, white spruce, and jackpine.
Prasad & Moody (1976) also found that, mainly because of
volatilization, 70% of the applied fenitrothion was lost one day
after treatment, though low levels of the insecticide (0.48 mg/kg
after 21 days) persisted in conifer tissues.
When labelled fenitrothion was applied to the leaves of
Japanese cypress at about 300 µg per leaf, approximately 70-80% of
the applied dose disappeared, mainly through evaporation, within 24
h. The major metabolites in the treated leaves were demethyl
fenitrothion [7], 3-methyl-4-nitrophenol [9] (approx. 2-10 µg), and
its glucoside conjugate [18] (Tabata & Okubo, 1980).
Immediately following the application of fenitrothion to poplar
( Populus tremuloidus) and birch trees ( Betula populifolia),
levels of 22 and 18 mg/kg, respectively, were detected. Residue
levels dropped to under 1 mg/kg within 30 days of treatment, and
under 0.1 mg/kg by 120 days. The oxygen analogue, fenitrooxon, was
not detected in any of the samples (La Pierre, 1985).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
In the 1980 spray programme in Canada, fenitrothion was
detected occasionally in air, the maximum concentration being 12
ng/m3 (Mallet, 1980), when the compound was sprayed at 140-280
g/ha.
Collaborative field studies were initiated in 1978 in Canada to
obtain relevant information concerning long-range pesticide drift
during the aerial spraying of fenitrothion in forest pest control,
using a surrogate (tris(2-ethylhexyl)phosphate) (Crabbe et al.,
1980a). At approximately 7.5 km downwind of the spray, the peak
treetop concentration of the drifting cloud varied from 1.4 ng/litre
in relatively neutral conditions to 5.0 ng/litre in the most stable
conditions. The fraction of the tracer material still airborne, 7.5
km from the spray line, was 6 and 16% for the neutral and most
stable environmental tests, respectively.
In 1980, a study was conducted to measure the atmospheric
fenitrothion exposure levels at breathing height near aerial
forestry spray operations (Crabbe et al., 1980b). The results
revealed an aerial concentration (or dosage) of 40 ng/min per litre1
at the spray line (flight path of aircraft) falling to an average of
8 ng/min per litre, 700 m from the spray line, in the first hour
after delivery. During the first hour after spraying, 30% of the
material collected in the samplers was vapour and the rest, aerosol.
Droplets of approx. 4.0 µm diameter contributed most to the mass of
drifting spray cloud at 500 m, about 15% of the airborne aerosol
mass consisting of droplets larger than 25 µm in diameter. The peak
aerosol concentration at chest height underneath the sprayed swath
of forest was approximately 15 ng/litre. One result of considerable
interest was the degree of volatilization of fenitrothion from an
early morning spray, which resulted in a sustained vapour plume
during the afternoon with a concentration
1 The value nanograms/min per litre represents the time
integrated aerial concentration of fenitrothion consisting of
the mean aerosol cloud concentration of agent (nanograms per
litre) divided by the residence time (minutes) of the cloud.
of 25 ng/litre at the spray line, falling to approximately 1.0
ng/litre 100 m downwind of the swath at an ambient temperature of
20-25 °C. The vapour plume persisted throughout the day and, in a
10-h period following spraying, was estimated to contribute up to
0.4 µg/person of respirable fenitrothion.
A pesticide residue survey was conducted in relation to the
1980 spruce budworm spray programme (210 g a.i./ha applied twice
with a 3-day interval) in New Brunswick, Canada. Fenitrothion was
detected occasionally in the air, the maximum level being 1.2
µg/m3. Amino-fenitrothion was sometimes present in the air at a
maximum level of 12.0 µg/m3 (Mallet & Volpe, 1982).
A chemical residue survey was undertaken to determine the
extent of the deposition and persistence of fenitrothion in the
environment in relation to the 1981 spruce budworm spray programme
(210 g a.i./ha applied twice with a 7-day interval) in New
Brunswick, Canada. Fenitrothion was only detected twice in the air
at levels of 0.08 and 0.04 µg/m3 (Mallet & Cassista, 1984).
When a 1.0% emulsion of fenitrothion was applied to a 61.2 m3
room (3.7 ± 0.3 g a.i./room) at 23.5-25.4 °C, using a compressed air
sprayer, the airborne concentrations of fenitrothion on days 0 and 3
after application were 3.3 µg/m3 and 0.5 µg/m3, respectively
(Wright et al., 1981).
5.1.2 Water
In actual field spraying in Canada, concentrations of
fenitrothion in stream waters varied greatly, depending on the spray
history and the weather. Spraying at 210 g/ha resulted in measurable
concentrations (> 0.03 µg/litre) in streams as far as 4 km from the
sprayed area. However, at 140-210 g/ha, most peak concentrations
were lower than 15 µg/litre and diminished very rapidly in
fast-flowing streams. Disappearance was still faster when a rain
storm followed spray application (Eidt & Sundaram, 1975).
High peak concentrations of fenitrothion were recorded in a few
cases, e.g., 64 µg/litre in flowing stream water approximately 1 h
after completion of spraying, and 75.5 µg/litre in stagnant water
some 17 h after spraying at 140-280 g/ha (Lockhart et al., 1977).
Fenitrothion concentrations usually dropped to less than 1 µg/litre
within a few days in forest streams and beaver ponds after
experimental and operational applications of 140-280 g/ha by
aircraft (Flannagan, 1973; Peterson & Zitko, 1974; Sundaram, 1974;
Eidt & Sundaram, 1975), and no measurable traces of fenitrothion
have been found in water longer than 40 days after spraying.
According to Symons (1977), the fenitrothion concentration in water
immediately after aerial spray at 140-280 g/ha in Canada seldom
exceeded 15 µg/litre.
One hour after the first application of fenitrothion (280 g
a.i./ha, applied twice with a 9-day interval) to a lake,
fenitrothion residues in the water were concentrated near the
surface (0.80-0.90 µg/litre), with small amounts (0.06 µg/litre) at
the bottom. Six hours later, residues were fairly evenly distributed
throughout the water column (0.91-1.49 µg/litre). Residue levels, 97
h after treatment, were similar at all depths at 0.41-0.46 µg per
litre, but declined rapidly in the next 48 h to < 0.01-0.06
µg/litre. Similar results were obtained after the second application
(Holmes et al., 1984).
Sundaram (1973) also reported that concentrations of
fenitrothion in aqueous systems diminished rapidly by dilution and
by physicochemical and microbial degradation to low levels (0.03
µg/litre) within a period of 40 days. The half-lives were found to
be short, ranging from 0.25 to 3.5 days.
In the 1980 spruce budworm spray programme in Canada,
fenitrothion was usually detected in water in the immediate vicinity
of the sprayed sites. The maximum level was 20.0 µg/litre and
persistence was usually limited to a few days. Aminofenitrothion
[13] was also found at a maximum of 8.0 µg/litre (Mallet, 1980;
Mallet & Volpe, 1982).
Water, suspended solids, and sediment samples were collected
from a small pond in a spruce-fir forest in New Brunswick, Canada,
before and after spraying with a fenitrothion formulation. When an
11% fenitrothion formulation was sprayed on a pond at 1500 g/ha, the
initial fenitrothion concentrations in the surface microlayer,
subsurface water, and suspended solids (0.25-0.67 h after spraying)
were 1.5, 0.015, and 1.9 µg/litre, respectively (Maguire & Hale,
1980). Thereafter, the fenitrothion disappeared exponentially with
half-lives of 0.34, 9.8, 1.8, and 3.2 h in the surface microlayer,
subsurface water, sediment, and suspended solids, respectively.
Residue levels declined rapidly to below detectable levels, 2 days
after spraying. The degradation products, 3-methyl-4-nitrophenol [9]
in water and aminofenitrothion [13] in sediment persisted for less
than 2 and 4 days, respectively. The concentration of fenitrothion
in rainwater during spraying (140-280 g/ha) in Canada was 77
µg/litre in the sprayed area, 17 out of 43 samples in the
neighbouring areas contained levels ranging from a trace to 1.1
µg/litre, while levels were less than 0.01 µg/litre in other regions
(Pearce et al., 1979a). No residues of fenitrooxon [1],
aminofenitrothion [13], or S-methyl fenitrothion [8] were
detected, even in the sprayed area (less than 0.05 µg/litre).
In the 1981 spruce budworm spray programme in Canada, the
amounts of fenitrothion residues detected in water were low (maximum
1.30 µg/litre) and post-spray samples did not contain detectable
amounts (less than 0.01 µg/litre) of fenitrothion. Small amounts of
aminofenitrothion [13] (0.1 µg/litre) were detected in some samples
(Mallet & Cassista, 1984).
In Quebec, environmental surveillance was performed in the
sprayed areas after aerial spraying, carried out from 1979 to 1982.
The insecticide concentrations were measured in foliage and water
samples taken from 1 to 4 h after spraying, when residue levels were
likely to peak. In lentic water, the median residue level was 5.82
µg fenitrothion/litre, with a maximum level of 1114 µg/litre. In
lotic water, the median concentration was 2.84 µg fenitrothion per
litre, with a maximum level of 127 µg/litre. In aquatic
environments, fenitrothion residues disappeared rapidly. Levels of
less than 1.0 µg/litre were found 2-8 days after spraying at 140-280
g a.i./ha (Morin et al., 1986).
Fenitrothion levels in a mountain stream increased to 20 µg per
litre, 6-9 h after spraying, and then decreased to below 1 µg/litre
within 24 h (Hatakeyama et al., 1990).
5.1.3 Soil
When operationally sprayed twice at 735 g/ha (Japan),
fenitrothion levels reached a maximum of 0.13-0.23 mg/kg in soil
under shrubbery after 1-8 days and then diminished with half-lives
of 2-4 days. Aminofenitrothion [13] was detected at maximum levels
of 1.5-6.5 µg/kg, but only shortly after spraying. Little
contamination with fenitrothion due to drift was observed (a maximum
of 0.01 µg/m2 per min, 1 h later) in areas that were 1.5 or 3.0 km
from the application site (Ohmae et al., 1981).
Yule & Duffy (1972) found that an operationally applied dosage
of fenitrothion (280 g/ha) produced a deposit of up to 0.04 mg/kg
(average depth 15 cm) in forest soil, and that the initial deposit
was too small to investigate degradation pathways. Sundaram (1974)
found a similar pattern of loss of fenitrothion from the soil after
5 years of repeated annual applications at a rate of 210-350 g/ha in
Canada; the insecticide content in the soil samples ranged from
traces to 0.1 mg/kg and disappeared within 45 days. No measurable
amounts of the breakdown products were found. Despite consecutive
annual spraying for up to 5 years at 210-350 g/ha in New Brunswick,
Canada, there was no evidence of build-up (< 0.005 mg/kg) in the
soil (Yule, 1974).
In the 1981 sprucce budworm spray programme in Canada, foilage
and soil litter collected after the second spray application
contained fenitrothion at levels of 0.2-0.5 and 0.06-0.14 mg/kg,
respectively; post-spray samples collected 2 weeks later contained
similar levels of fenitrothion. No residual fenitrothion was
detected in sediment (Mallet & Cassista, 1984).
5.1.4 Food
Fenitrothion is used for the pre-harvest treatment of a variety
of crops, such as fruits, vegetables, rice, cereals, soybeans and
coffee, at the rate of 0.5-2.0 kg a.i./ha. The initial
residue levels on fruits, vegetables, rice, and cereals were
relatively high (0.001-9.5 mg/kg), but declined rapidly with a
half-life of 1-2 days (FAO/WHO, 1970, 1975b).
A survey of pesticide residues in crops (total number of
samples: 697) collected in the Tokyo Metropolitan Central Vegetable
and Fruit Market was carried out from April 1984 to March 1989. Low
fenitrothion residues (0.01-0.06 mg/kg) were found in only a few
samples (Nagayama et al., 1986, 1987, 1988, 1989).
Fenitrothion (EC) was applied 3 times to peach at a rate of 2.0
kg a.i./ha in Japan. The residue levels in peach (pulp) were
0.03-0.07 mg/kg, 7 days after application and decreased to
0.005-0.015 mg/kg after 13-15 days. Peach peel contained most of the
residues (1.1-1.6 mg/kg), 7 days after application, but the levels
decreased to 0.3-0.6 mg/kg in 15 days.
Lettuce, plums, and apples were treated once with 0.2%
fenitrothion (EC) at a rate of 0.8 kg. a.i./ha. No increase in
3-methyl-4-nitrophenol indicating the hydrolysis of fenitrothion
residues was observed during 8 days following treatment. Levels of
3-methyl-4-nitrophenol did not exceed 10% of the fenitrothion
residues (Cerna & Benes, 1972).
Strawberries were treated twice with fenitrothion (EC) at a
rate of 1.0 kg a.i./ha; the residue levels were 0.02-0.03 mg/kg and
0.005-0.007 mg/kg, 7 and 15 days after treatment, respectively
(FAO/WHO, 1975b). Following 3 applications of fenitrothion to
tomatoes, a residue of 1.32 mg/kg was detected on the same day as
the final application (Kannan & Jayarnama, 1981). The residue level
dropped to 0.07 mg/kg, one day later, but there was very little
further reduction in fenitrothion levels over a further 2-week
period.
Fenitrothion (as EC, dust, or WP) was applied to rice plants in
Japan at rates of 0.5-1 kg a.i./ha at various time intervals (3-5
times/season) before harvest. Over 120 samples of harvested rice
were analysed for residues of the parent compound. The average
residue levels ranged from negligible (0.001 mg/kg) to 0.1 mg/kg
with a maximum level of 0.3 mg/kg, 11-20 days after the final
application, and 0.001 mg/kg, 41-67 days after treatment (Sumitomo
Chemical Co. Ltd, 1969).
A variety of fenitrothion formulations were applied 1-7 times
to rice crops pre-harvest at intervals ranging from 14 to 120 days.
Most of the samples of hulled rice did not contain any residues at
harvest (limit of determination 0.001 mg/kg), but a few contained
low levels (maximum level 0.025 mg/kg) (FAO/WHO, 1978b).
Fenitrothion is also used in the post-harvest treatment of
stored grain. When grains with a moisture content of 12-13% were
treated with fenitrothion at 15 mg/kg (18 mg/kg for brown rice), the
residues decreased after 3 and 6 month at 25 °C to 6.8 mg/kg and 3.1
mg/kg, respectively, in barley, 6.9 mg/kg and 4.2 mg/kg in oats, 6.2
mg/kg and 3.5 mg/kg in rice, 11.5 mg/kg and 8.0 mg/kg in brown rice,
and to 10 mg/kg and 7.5 mg/kg in polished rice. The rates of
decrease of fenitrothion in/on grains in storage can be predicted
accurately from the temperature, relative humidity, and dosage level
(FAO/WHO, 1978b).
When grains were subjected to milling, processing, preparation,
and cooking, there was a significant loss of fenitrothion.
Fenitrothion was deposited on the epidermis and removed with bran
during the milling process, so that the residue level on bran was
from 2 to 2.5 times higher than that on whole wheat and about 7
times higher than that on husked (brown) rice. After milling, the
residue of fenitrothion in white flour was about 10% or less of the
residue in the raw wheat. The residue in the white bread prepared
from the white flour was reduced to 1-2% of the residue in the raw
wheat. In case of wholemeal bread, however, 20-25% of the initial
concentration remained after baking. The residue of fenitrothion in
rice was decreased to 4.1 and 1.8% of the initial residue during
husking and milling, respectively. The residue in polished rice was
also decreased to 30% of original concentration. The concentration
of fenitrothion in oats, rice in husk, husked rice, and polished
rice decreased to 30-60% of the initial concentration after boiling.
The malting process removed substantially all of the fenitrothion
residue on raw barley. During the processing of oats for the
production of rolled oats and groats, more than 95% of the residues
on the raw oats were removed (FAO/WHO, 1977).
In the United Kingdom, fenitrothion was found in home-produced
and imported wheat at levels of up to 0.2-0.9 mg/kg in 1982 (MAFF,
1986). During the period of 1984-88, residues of fenitrothion were
found in haricot and mung beans at levels of up to 0.1 mg/kg, but
residues in several other pulses did not exceed 0.05 mg/kg. In
retail wheat products, fenitrothion was found at levels of up to
0.05 mg/kg in white bread, 0.3 mg/kg in brown bread, 0.5 mg/kg in
bran, and 0.02 mg/kg in cereal-based infant foods, but was not
detected in breakfast cereals, wheatgerm products, brown rice, rye
products, and processed oats, the level of detection being 0.01-0.1
mg/kg, depending on the commodity and the year of survey (MAFF,
1989).
5.2 Human exposure
5.2.1 Food
In the USA, average fenitrothion residues of 0.0001 mg/kg were
detected in grains and cereal products in 1979-80, and the average
intake for an adult from this source was calculated to be 0.0608
µg/day (or 0.001 µg/kg body weight per day) in 1980 (Gartrell et
al., 1985a). For the infant and toddler, maximum daily intakes were
estimated to be < 0.001 µg/kg body weight per day and 0.002 µg/kg
body weight per day, respectively, during 1977-80 (Gartrell et al.,
1985b).
6. KINETICS AND METABOLISM
The metabolic pathways of fenitrothion in mammals are shown in
Fig. 8.1
6.1 Absorption, distribution, metabolic transformation,
elimination, and excretion
32P-fenitrothion administered orally at doses of 15 and 500
mg/kg to rats and guinea-pigs, respectively, was readily absorbed
from the gastrointestinal tract and distributed among various
tissues. The concentrations of 32P in the blood of rats reached a
maximum (15.5 µg/g) 1-3 h after treatment, and diminished to below
detectable levels within 4 days (Miyamoto et al., 1963a).
Following intravenous injection of 32P-fenitrothion in rats
or guinea-pigs at a dose of 15 or 40 mg/kg, fenitrothion disappeared
equally rapidly from the blood and tissues (Miyamoto, 1964a, 1969).
The radioactivity (32P) was mostly excreted within 2-4 days into
the urine (85-97%), up to 10% being eliminated in the faeces,
accounting for nearly 100% recovery of the dose. The metabolites in
excreta were demethyl fenitrothion [7], demethyl fenitrooxon [20],
dimethylphosphorothioic acid [19], and dimethylphosphoric acid [25].
Fenitrooxon [1] was detected only after intravenous administration
of a large amount of fenitrothion and not after oral administration
(Miyamoto et al., 1963a; Miyamoto, 1964a).
Hollingworth et al. (1967a) studied the metabolism of
fenitrothion in white mice. 32P-fenitrothion administered orally
to mice at a dose of 3, 17, 200, or 850 mg/kg was rapidly eliminated
in the urine and faeces with a recovery of > 90% 72 h after
treatment. The isolated metabolites indicated that both the
P-O-alkyl and P-O-aryl bonds of fenitrothion and fenitrooxon [1]
were cleaved. No evidence was obtained indicating that the nitro
group was reduced to form aminofenitrothion [13] or that the ring
methyl group was oxidized. At the lower dose (17 mg/kg), demethyl
fenitrothion [7], demethyl fenitrooxon [20], dimethylphosphorothioic
acid [19], and dimethylphosphoric acid [25] were the major
metabolites. At the 200 mg/kg body weight dose, the amounts of
dimethylphosphorothioic acid [19] and dimethylphosphoic acid [25]
decreased, and demethyl fenitrothion [7] increased.
1 Chemical structures in Fig. 1 to 8 are referred to according to
numbers in the brackets.
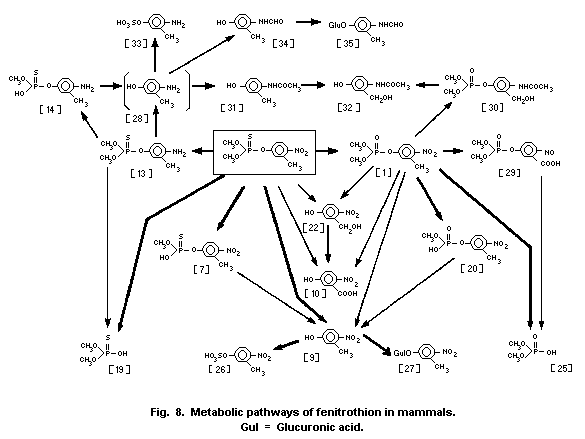 m-Methyl-14C-fenitrothion, administered orally at a dose of
15 mg/kg to Wistar rats, ICR mice, native Japanese rabbits, and
Beagle dogs, was readily absorbed from the gastrointestinal tract
and distributed to various tissues with maximum concentrations of
0.093 mg/kg (rats) - 0.144 mg/kg (dogs) after 1-3 h of the
treatment, and the radiocarbon was rapidly and completely
eliminated, mainly in the urine of rats (89-95%), mice (> 90%),
rabbits (86-94%), and dogs (88%) (Miyamoto et al., 1976b).
Examination of 11 rat tissues, including fat and muscle, revealed
that the concentrations of fenitrothion ranged from 0.004 mg/kg to
less than 0.001 mg/kg (except in fat-0.034 mg/kg), 24 h after
administration. Whole body autoradiography also indicated the rapid
disappearance of the radiocarbon from the tissues of mice. The
cumulative patterns of elimination were essentially the same among
these animals when radioactive fenitrothion was given at 15 mg/kg
body weight. Similar patterns were obtained in rats when
fenitrothion was administered at 105 mg/kg body weight or 15 mg/kg
body weight after a total of 5 pretreatments with non-radioactive
fenitrothion at 15 mg/kg body weight every other day. Thin-layer
chromatographic analysis of the urinary metabolites showed the
absence of intact fenitrothion and the presence of as many as 18
metabolites, of which 17 (92-99% of radioactivity) were identified.
The differences in the composition of the metabolites among these
animal species and between males and females of the same species
were mostly quantitative. The percentage of the metabolites
retaining the P-O-aryl linkage varied with animal species, ranging
from 8.6% (rabbits) to more than 56.9% (dogs). Most of these
metabolites were demethylated products [7,20] at the O-methyl
position. Rats and mice tended to eliminate greater amounts (15-26%)
of demethyl fenitrooxon [20] than rabbits and dogs (2-6%).
3-Methyl-4-nitrophenol [9], free or as conjugates with sulfuric acid
[26] or glucuronic acid [27], constituted another group of major
metabolites. Approximately 50-75% of the urinary radioactivity was
accounted for by these three metabolites ([9] [26] [27]), except in
dogs (36%). The urinary activity in rabbits (64-75%) exceeded that
in other animal species (50-60%). A trace amount of the oxidized
phenols (0.5-2%) [10,22] was present in rats and rabbits. Rabbits
were exceptional in excreting fenitrooxon [1] and animofenitrothion
[13], though in small amounts (0.1-3.7%). The urine of rabbits and,
to a lesser extent, of rats contained several minor metabolites
derived from aminofenitrothion [13] or from 3-methyl-4-aminophenol
[28]. The minor products totalled approximately 15% of the excreted
radiocarbon in rabbits and about 12% in rats. A few metabolites had
unique structures resulting from the reduction of the nitro group
and oxidation of the m-methyl group [29]. An appreciable amount of
the excreted metabolites from the 4 animal species was in the form
of conjugates. It appeared that rabbits were most active in this
regard, and dogs least active. In the 4 species treated, higher
amounts of sulfate conjugates than glucuronides were excreted. Thus,
fenitrothion was
metabolized through two major pathways, via O-demethylation and via
cleavage of the P-O-aryl bond. Neither S-(3-methyl-4-nitrophenyl)
glutathione mediated by glutathione S-aryltransferase nor ring
hydroxylation products have been positively demonstrated (Miyamoto
et al., 1976b).
Although reduction of the nitro group in the fenitrothion
molecule, presumably by intestinal microorganisms, was a minor
metabolic pathway in the above 4 animal species, it was the major
metabolic pathway in ruminants. In fact, a study of female goats
revealed that most of the urinary and faecal metabolites of
fenitrothion were amino derivatives [13,14,30] and were formed most
probably in rumen fluid (Mihara et al., 1978).
The metabolism of fenitrothion in Wistar rats with hepatic
lesions induced experimentally by dietary administration of
diaminodiphenyl-methane (DDM), by feeding a low protein-high fat
diet (LPHF), or by intramuscular treatment with carbon
tetrachloride, was also investigated. The in vitro rates of
degradation of fenitrothion were significantly reduced in all
preparations from livers with induced hepatic lesions, with the
greatest reduction produced by LPHF followed by carbon
tetra-chloride and DDM. The rate of degradation of fenitrothion and
fenitrooxon [1] was less severely affected than the rate of
activation of fenitrothion, because of the greater activity of
glutathione S-alkyltransferase (Miyamoto et al., 1977a). However,
the cumulative excretion patterns of radiocarbon were not altered by
these three hepatic lesions after oral administration to the injured
rats of either 15 or 50 mg 14C-fenitrothion/kg body weight, or 15
mg/kg body weight of the compound after pretreatment with approx.
2.8 mg/kg body weight per day of nonactive fenitrothion for 4 weeks.
Moreover, the injured rats metabolized fenitrothion equally as well
in vivo, as the control animals, even though the pathway yielding
demethyl fenitrothion [7] predominated. Higher or short-term doses
of fenitrothion tended to reduce the metabolic differences observed
between the injured and the control animals (Miyamoto et al.,
1977b). These results indicated that even very severe hepatic
lesions, such as those described here, hardly influenced the in vivo
detoxification of fenitrothion and the excretion of its metabolites.
The results also indicated that there was not much possibility of
retention or storage of the parent compound and/or its toxic
metabolites in the mammalian body under such unfavourable
conditions.
The dermal penetration of 14C-ring-labelled fenitrothion and
aminocarb was determined in male, rhesus monkeys and male,
Sprague-Dawley rats. In monkeys, 49 ± 4% of the fenitrothion
(urinary excretion half-life = 14 h) was absorbed from the
forehead, while 21 ± 10% of the fenitrothion (half-life = 17 h) was
absorbed from the ventral forearm. Monkey forehead was 2.3 times
more permeable than the forearm. In rats, 84 ± 12% of the
fenitrothion (half-life = 20 h) was absorbed from the middorsal
region (Moody & Franklin, 1987).
The presence of N,N-diethyl-m-toluamide (DEET), used as an
insect repellent by workers spraying fenitrothion, increased the
absorption of 14C fenitrothion through rat skin (32% DEET; 15%
control with acetone) and monkey skin (9.7% DEET; 3.2% control)
(Moody et al., 1987b).
6.2 Retention and turnover
To evaluate the possible retention of fenitrothion residues in
animal tissues, methyl-14C-fenitrothion was administered orally to
male Wistar rats at 15 mg/kg body weight per day for 7 days, and
then at 30 mg/kg body weight per day for a further 3 days. A
measurable amount (0.09 mg/kg) of fenitrothion was found in
abdominal fat and athe level increased (2.4 mg/kg) at the later
staes of administration. This amount, however, tended to disappear
quite rapidly on cessation of the administration (Miyamoto, 1977c).
Following administration of 10 or 3 mg fenitrothion/kg body
weight per day to male native Japanese rabbits for 6 months, blood,
skeletal muscle, and abdominal fat were analysed by gas
chromatography for fenitrothion and fenitrooxon. In most cases,
blood and muscle did not contain any detectable amounts of either
compound (detection limit for fenitrothion 0.005 or 0.002 mg/kg, and
that of fenitrooxon, 0.01 mg/kg). Averages of 0.131 mg
fenitrothion/kg (0.243 mg/kg maximum) and of 0.045 mg/kg were
measured in the fat of rabbits dosed at 10 and 3 mg/kg body weight
per day, respectively, while muscle contained a maximum of 0.006 mg
fenitrothion/kg. No fenitrooxon was detected (Miyamoto et al.,
1976a).
Three male beagle dogs were treated orally with 5 mg
fenitrothion/kg body weight per day, 6 days per week, for 10 months.
After termination of the treatment, only the fat contained trace
amounts of fenitrothion (maximum of 0.160 mg/kg) (Tomita et al.,
1974).
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term
effects on the nervous system, can be found in Environmental Health
Criteria 63: Organophosphorus insecticides - A general introduction
(WHO, 1986).
No-observed-effect levels on the ChE in the plasma,
erythrocytes, and brain of animals treated with fenitrothion under
various conditions are summarized in Annex II.
7.1 Single exposure
As with other organophosphorus insecticides, a large dose of
fenitrothion produces toxic signs and symptoms in various animals by
the inhibition of acetylcholinesterase (AChE) in the nervous system,
with the consequent accumulation of excessive levels of
acetylcholine. The major toxic signs in experimental animals are
salivation, tremor, exophthalmos, urinary incontinence,
piloerection, and dyspnoea, which are similar to the toxic signs
elicited by other organophosphorus insecticides (Namba, 1971).
The acute toxicity of fenitrothion varies according to the
species of the test animal, its sex, the route and method of
administration and/or the solvent used, as shown in Table 4. The
acute oral LD50s in mammals range from 330 mg/kg in the rat to
1850 mg/kg in the guinea-pig, the dermal LD50s range from
890 mg/kg in the rat to > 2500 mg/kg in the mouse.
Groups of 8 male and 8 female Sprague-Dawley rats were exposed
to fenitrothion mists dissolved in a mixture of deodorized kerosene
and xylene (particles mostly less than 3 µ in diameter) to assess
inhalation toxicity at aerial concentrations of 10, 70, and 186
mg/m3 for 2, 4, or 8 h. No significant toxic signs were observed
in the rats exposed to an aerial concentration of 10 mg/m3 for 2
and 4 h. With longer exposure and/or higher fenitrothion
concentrations, toxic symptoms developed including salivation,
urinary incontinence, tremor, lacrimation, muscular fibrillation,
and dyspnoea, and, after 8 h exposure to 186 mg fenitrothion/m3, 2
out of 8 males died. A transient body weight decrease was observed
after 8 h exposure to 10 and 70 mg fenitrothion/m3. A marked body
weight decrease was observed at a concentration of 186 mg/m3. The
LC50 value for fenitrothion in rats exposed for 8 h, was estimated
to be more than 186 mg/m3 (Kohda & Kadota, 1979).
Table 4. Acute toxicity of fenitrothion
Animal Strain Sex Route LD50 (mg/kg) Vehicle Reference
Mouse male oral 1336 Carshalton (1964)
female oral 1416
dd male oral 1030 10%Tween 80 aq. Miyamoto & Kadota (1972)
dd female oral 1040 10%Tween 80 aq.
dd male dermal > 2500 Undiluted Kadota et al. (1972)
female dermal > 2500 Undiluted
Rat Sherman male oral 740 Peanut oil Gaines (1969)
Sherman female oral 570 Peanut oil
Sprague-Dawley male oral 330 10%Tween 80 aq. Miyamoto & Kadota (1972)
Sprague-Dawley female oral 800 10%Tween 80 aq.
Wistar male oral 940 Benes & Cerna (1970)
Wistar female oral 600
Sprague-Dawley male dermal 890 Undiluted Kadota et al. (1972)
Sprague-Dawley female dermal 1200 Undiluted
Guinea-pig male oral 500 DuBois & Puchala (1960)
oral 1850 Sorpol 2001 Miyamoto et al. (1963b)
Dog Beagle male/ oral > 681 Gelatin capsule Mastalski (1971b)
female
Hen White Leghorn oral 500 10%Tween 80 aq. Kadota et al. (1975b)
The pulmonary toxicity of fenitrothion was evaluated following
a single exposure of caged rats (n = 160) placed in a field
oversprayed by aircraft during an operational forest treatment.
Depending on the position of the cages, the air concentrations of
fenitrothion measured in the first 30 min after spraying, were 6.19
and 33.2 µg/m3. Plasma pseudocholinesterase activity and pulmonary
alveoli ultrastructure were used as indices of fenitrothion
exposure. Although a few signs of toxic lung injury were observed on
days 3 and 7, there were no cholinergic signs or effects on
pseudocholinesterase activity within 12 h in exposed animals,
compared with controls. The alveolar toxic reaction was limited to
small and discrete foci, and was entirely reversible within a period
of 2 months (Coulombe et al., 1986).
Rat lungs were examined with light and electron microscopes,
3-60 days after exposure to fenitrothion. Groups of 9 male,
Sprague-Dawley rats were exposed by a "nose-only" apparatus for 1 h
to an aerosol of fenitrothion (15%) mixed with non aromatic
hydrocarbon solvent and oil diluent at 2 or 500 mg/m3. Only minor
modifications of lung alveolar tissue were observed after exposure
to the higher concentration. At 3 days, discrete foci of mild
inflammation were detected, including interstitial oedema, cellular
infiltration, and an increased number of alveolar macrophages. At 7
days, signs of irritation were diminished, and, at 21 and 60 days,
alveolar tissues were essentially normal. Exposure to the lower
concentration induced more limited changes at 3 days; no
modification was seen at later examinations. It was considered that
a single exposure to this fenitrothion mixture at 500 mg/m3 did
not present a serious hazard to the lung (Chevalier et al., 1984).
Alveolar irritation was also seen in the solvent- exposed control
animals.
When Holstein milk cows and sheep (male) were administered
fenitrothion orally at 500 and 770 mg/kg body weight, respectively,
they showed signs of poisoning, such as paralysis of the hindlegs
and salivation, but gradually recovered 4 h after treatment (Namba
et al., 1966). Pigs given a single oral dose of 310 mg/kg body
weight showed typical signs of ChE inhibition, but the toxic signs
disappeared within 48 h. When cows and sheep were given fenitrothion
at 3 mg/kg body weight per day, for 90 and 60 days, respectively,
the ChE activity in the plasma decreased in both species early in
the test period, but recovered fully after 30 days.
7.2 Skin and eye irritation; skin sensitization
7.2.1 Skin and eye irritation
One tenth ml of fenitrothion (technical) was applied to one eye
of 9 male, New Zealand White rabbits. The untreated eye served as a
control. The treated eyes of 6 animals remained unwashed. The
treated eyes of the other 3 animals were flushed for 1 min with
about 300 ml lukewarm water, 30 seconds after application. Slight
hyperaemia in the conjunctiva was observed 1 h after application in
the unwashed eyes. This change had disappeared 48 h after
application. No irritating reactions were induced in any washed
eyes. The irritating potency of fenitrothion in eyes was judged to
be minimal for the unwashed group, and negative for the washed group
(Hara & Suzuki, 1981).
A half ml of fenitrothion (technical) on a 1 x 1 inch lint
patch was applied to abraded and intact parts of the back skin of 6
New Zealand White rabbits. No irritating reactions, such as erythema
and oedema, were noticed in the animals. The irritating potency of
the material was estimated to be negative (Hara & Suzuki, 1981).
7.2.2 Skin sensitization
The Landsteiner-Draize test with fenitrothion (technical) was
conducted on 6 male, Hartley guinea-pigs using
2,4-dinitrochloro-benzene (DNCB) as a positive control, to examine
possible skin sensitization activity. Fenitrothion dissolved in corn
oil at concentrations of 1 or 5% was applied intradermally every
other day, 10 times. The animals were treated for a challenge with
intradermal injection and dermal application 2 weeks after the last
sensitizing treatment. Allergic reactions were not observed in any
animals treated with fenitrothion, while reactions, such as swelling
and hyperaemia, were clearly noticed in the animals treated with
DNCB. It was concluded that fenitrothion (technical) was not a skin
sensitizer (Kohda et al., 1972).
The maximization test was undertaken to study the allergenicity
of fenitrothion. Ten young adult, female, Hartley guinea-pigs
weighing from 300 to 500 g were studied, induction and challenge
being undertaken according to the original method. The
concentrations of the compounds for the induction were 5 and 25% for
intradermal and topical treatment, respectively. Challenge
concentrations of 0.05% showed the potential for allergenicity in
this test (Matsushita et al., 1985).
7.3 Short-term studies
7.3.1 Rat
Male rats were given daily oral doses of 13 mg fenitrothion/kg
body weight for 28 days. Erythrocyte ChE activity was severely
depressed, but recovered 30 days after withdrawal of fenitrothion
(Kimmerle, 1962a).
Oral administration of fenitrothion at 7.25 or 14.5 mg/kg body
weight per day to male, Wistar rats (5/group) for 28 days resulted
in increases in plasma corticosterone and glucose levels, 7 days
after the start of treatment, and in the relative adrenal weight, 2
weeks after the start of treatment (Yamamoto et al., 1982b).
Groups of 36 male, CD rats received technical fenitrothion by
oral gavage (2.5, 5.0, 10.0, or 20.0 mg/kg per day) for 30
consecutive days. No significant treatment-related changes were
observed by serum biochemistry or by light microscopy of liver.
However, a dose-dependent decrease was noted in the brain, plasma,
and erythrocyte ChEs as well as tissue esterase activities. Eight
out of 36 animals died at the 20.0 mg/kg level (Trottier et al.,
1980).
Male rats were fed 5, 10, or 20 mg fenitrothion/kg diet for 5
weeks. The activity of brain and erythrocyte ChE was normal in the 5
mg/kg group, whereas the 10 mg/kg group showed a slight depression
of erythrocyte ChE activity after 5 weeks and recovery 2 weeks after
withdrawal. The 20 mg/kg group showed some depression of activity in
both erythrocyte and brain ChE and the recovery in brain ChE
remained incomplete 2 weeks after withdrawal (Carshalton, 1964).
The neurobehavioural effects were examined in male,
Sprague-Dawley rats (6/group) of fenitrothion administered orally at
doses of 10, 20, or 40 mg/kg body weight, daily, for a period of 40
days. At the 2 higher doses, mortality was more than 50%, while
only one animal died at a dose of 10 mg/kg. Toxic signs, such as
cholinergic signs, loss of reflexes, and changes in motor activity
and indices of ataxia, were observed markedly at the 2 higher doses,
and slightly at the lowest dose. However, these effects tended to
recover gradually later in the study (Rondeau et al., 1981).
Groups of male, Wistar rats (16 or 17 in number) were fed 0,
32, 63, 125, 250, or 500 mg fenitrothion/kg diet for 90 days.
Mortality, food intake, growth, general behaviour, urinalysis,
average organ weights, and histopathology in the groups fed 32, 63,
125, and 250 mg/kg diet were comparable to those in the controls.
All animals fed 500 mg/kg diet showed clinical signs of
anticholinesterase poisoning and there were minimal toxic signs in 4
animals in the 250 mg/kg diet group. In the 500 mg/kg group, the
average weights of the testes and brain were increased in comparison
with those of the control group. On monthly interim sacrifice of 4
rats from each group, the cholinesterase (ChE) activity of plasma,
erythrocyte, brain cortex, liver, and kidney showed a dose-dependent
depression, the lowest activity being in the brain. The ChE activity
in the 32 and 63 mg/kg diet groups generally increased by day 60 of
feeding to a level within the normal limits (Misu et al., 1966).
Fenitrothion (60% water miscible concentrate) was given to
groups of 12 Wistar rats/sex at dose levels of 0, 20, 92.8, 430.7,
or 2000 mg/kg diet in the feed or 0, 10, 46.4, 215, or 1000 mg/litre
in drinking-water, for 90 days. The levels causing no appreciable
effect on the ChE activity of plasma, erythrocyte, and whole blood
were 20 mg/kg diet and 10 mg/litre drinking-water. The above
mentioned levels and 92.8 mg/kg diet and 46.4 mg/litre and 215
mg/litre in drinking-water did not cause any effects on food or
water intake, weight gain, average organ weights, haemogram, and
blood biochemistry. However, 92.8 mg fenitrothion/kg diet and 46.4
mg/litre in the drinking-water moderately depressed the activities
of whole blood and erythrocyte ChE and had a more marked depressive
effect on plasma ChE. The ChE activity recovered between 30-40 days
after withdrawal of fenitrothion. The levels of 430 mg/kg diet and
1000 mg/litre drinking-water caused a depression in body weight gain
(Cooper, 1966).
Two groups of male rats (20 each) were dosed by stomach tube
with fenitrothion (50% formulation) at 10 mg/kg or 11 mg/kg body
weight, for 6 days per week, over 6 months. During the first weeks,
the rats showed a temporary deterioration in general condition and
loss of weight. Haematology and urinalysis during the study and
gross and microscopic pathology at its termination did not reveal
any abnormalities (Klimmer, 1961).
Groups of rats (15 males and 15 females) were fed fenitrothion
at 0, 10, 30, or 150 mg/kg diet for 6 months. In all groups, growth,
food and water consumption, mortality, blood and urinalysis, and
blood biochemistry were comparable with those of the controls. The
ChE activities in the brain, erythrocytes, and plasma were depressed
in both sexes in the 150 mg/kg diet group and in females in the 30
mg/kg diet group. In the 10 mg/kg diet group, only the plasma ChE of
females was depressed. Absolute and relative organ weights were
within normal limits. No histopathological changes were found in the
organs examined. Based on brain acetylcholinesterase inhibition, a
NOAEL of 10 mg/kg diet was concluded (Kadota et al., 1975a).
Groups of 8 male and 8 female rats were fed diets containing 0,
10, 50, or 250 mg fenitrothion/kg for 34 weeks. In another test,
groups of 16 rats of each sex were fed fenitrothion at 0, 5, 25, or
125 mg/kg diet over the same period. The feeding of fenitrothion at
250 mg/kg diet resulted in a decrease in body weight gain in
females. In the 125 mg/kg diet group, a lower relative weight of
spleen was found in both sexes. The ChE activities in both the
plasma and erythrocytes, measured at intervals, showed a
dose-dependent decrease in all test groups, except for males at 5
mg/kg diet whose plasma ChE activity was not suppressed. In females,
the depression was slight. The ChE activity in the brain was
decreased only in the 250 mg/kg diet group (Benes & Cerna, 1970).
Groups of 9 and 10 male Sprague-Dawley rats were fed 0, 25,
100, or 400 mg fenitrothion/kg diet for 63 weeks. A positive control
group was fed 800 mg malathion/kg. At the 400 mg/kg level of
fenitrothion, body weight gain was decreased and only a few animals
survived the 63-week period. At this level, there was also a 100%
depression of erythrocyte ChE. At the 100 mg/kg level, a slight ChE
depression (10-30%) occurred in the brain and a moderate depression
(30-65%) in the erythrocyte and plasma. At 25 mg/kg diet,
fenitrothion had a slight effect on plasma ChE activity, but did not
cause any effects on other parameters (food intake, body weight
gain, brain and erythrocyte ChE) evaluated (Ueda & Nishimura, 1966).
Groups of male, Wistar rats (60/group with 120 controls) were
administered fenitrothion, by gavage, at 0, 0.5, 1, 5, or 10 mg/kg
body weight per day, for 12 months. Each month, 10 controls and 5
rats from each group were sacrificed. After 12 months,
administration was stopped and survivors were held for an additional
2 months to monitor recovery from the biological effects of the
treatment. No changes attributable to treatment with fenitrothion
were observed in haematology or blood biochemistry, except in
esterase activities. Marked reductions in hepatic ChE, and brain and
erythrocyte AchE activities, observed at doses of 5 and 10 mg/kg
body weight per day, within 1 month of treatment, persisted for the
duration of the treatment. Two months after treatment was
terminated, the esterase activities in all treated rats were
comparable to those in the controls. Little change was measured at
0.5 and 1 mg/kg per day (Ecobichon et al., 1980).
Groups of Wistar rats (26 males and 27 females) were fed a diet
containing 0, 1, 5, or 25 mg fenitrothion/kg for one year. No
significant changes were observed in body weight, food and water
intake, organ weights, or histopathology. The ChE activity in the
brain, liver, and erythrocytes was reduced at 5 and 25 mg/kg diet
during the first month but then recovered (Kanoh et al., 1982).
A nose-only inhalation toxicity study of fenitrothion was
conducted on Sprague-Dawley rats (Breckenridge et al., 1982). Groups
of 10 male and 10 female rats (20/sex for the controls) were exposed
to fenitrothion 11% emulsifier mixture at chamber concentrations of
0, 6.7, 20, or 60 mg/m3, for 2 h/day, for 30 consecutive days. ChE
activity in the plasma, erythrocytes, and brain was inhibited in the
60 mg/m3 males and in all the treated females, except those in the
6.7 and 20 mg/m3 groups, in which the erythrocyte ChE activity was
not affected. Hepatic and renal carboxyesterases were inhibited in
the 60 mg/m3 male group and the 20 and 60 mg/m3 female groups.
These enzyme activities had returned to normal values 30 days after
treatment. Suppression of the brain ChE activity in the 60 mg/m3
female group persisted. Haematological, urinary, blood biochemical,
and histopathological studies did not reveal any deleterious effects
of fenitrothion.
Durham et al. (1982) reported a short-term study of
fenitrothion (11% emulsifiable concentrate) in the absence or
presence of Atlox 3409F (emulsifier) and Dowanol TPM (cosolvent).
Forty-five male, Sprague-Dawley rats were assigned to each of the
treated and control groups. No changes in the haematological, serum
biochemical parameters or in tissue morphology were observed in rats
treated with fenitrothion at doses of 1, 5, 10, or 20 mg a.i./kg.
The emulsifier and co-solvent did not substantially enhance or
reduce the toxicity of fenitrothion.
7.3.2 Dog
Groups of dogs (1 male and 1 female) were given daily oral
doses (by capsule) of 0, 2, 9, or 40 mg fenitrothion/kg body weight
for 98 days. Body weights, blood biochemistry, ChE levels, and
haematology were checked at intervals. At the 2 mg/kg level,
fenitrothion did not produce any effects on the parameters examined.
At 9 mg/kg, a slight depression after 60 days and, at 40 mg/kg, a
moderate depression after 29 days occurred in whole blood, plasma,
and erythrocyte ChE activity. At 40 mg/kg, there were also marked
toxic signs typical of cholinergic stimulation, and the dogs in this
group were sacrificed before the end of 98-day period (Cooper,
1966).
Groups of dogs (6 males and 6 females) were fed fenitrothion at
levels of 0, 30, 100, or 200 mg/kg diet for two years. Body weights,
blood biochemistry, and plasma, erythrocyte, and brain ChE
activities were monitored, together with histopathological
examination. The only adverse effect was a reduction in the ChE
activities. Depression of plasma ChE activity was apparent in all
groups, while erythrocyte enzyme activity was unaffected in the 30
mg/kg group, when the treated and control animals were compared. The
brain ChE activity was decreased only after ingestion of 200 mg/kg
(Mastalski et al., 1973).
Groups of Beagle dogs (6 males and 6 females) were fed
fenitrothion at dose levels of 0, 5, 10, or 50 mg/kg diet for one
year. The only treatment-associated change was a depression of ChE
activity. At 50 mg/kg diet, plasma ChE was depressed in both sexes,
and erythrocyte ChE was slightly depressed in the males only.
However, brain ChE was not depressed at any levels. The
no-observed-effect level was 10 mg/kg or 50 mg/kg diet, based on the
effects on erythrocyte or brain ChE, respectively (Griggs et al.,
1984).
A group of 3 male Beagle dogs was given fenitrothion in
capsules at 5 mg/kg body weight per day, for 6 days a week, over 10
months. A marked depression of plasma and erythrocyte ChE activities
was observed compared with the control group (2 animals). However,
no appreciable accumulation of the compound was noted in the blood,
fat tissue, or liver (Tomita et al., 1974).
Groups of Beagle dogs (2 females) were treated orally with
fenitrothion at dose levels of 0 or 2 mg/kg body weight per day (by
capsule) for one year. A marked depression of plasma and erythrocyte
ChE activities was observed 2 weeks after initiation of treatment.
No changes attributable to treatment with fenitrothion were found in
haematology, blood biochemistry, or ophthalmology (Ogata, 1972).
7.3.3 Rabbit
When groups of male Japanese albino rabbits (15 animals per
group) were administered fenitrothion at 0, 3, or 10 mg/kg body
weight per day in the diet for 6 months, a significant depression of
the plasma and erythrocyte ChE activities was observed in both
treated groups. At the higher dosage, depression of the brain ChE
was also found. However, no noteworthy changes, either biochemical
or histochemical, were observed in musculi rectus medialis ChE, and
the fine structure of the neuromuscular junctions of the tissue was
essentially normal when examined using an electron microscope. No
other adverse effects were observed in behaviour, mortality,
haematology, clinical biochemistry, organ weights, or histopathology
of major organs and tissues (Miyamoto et al., 1976a).
7.3.4 Guinea-pig
Treatment of male guinea-pigs with 5 or 10 mg fenitrothion/kg
ip for 30 days resulted in a dose-dependent increase in
acetylcholine levels in the heart, decreased heart rate, and
hypocalcaemia (Anand et al., 1989).
Active sensitization toxicity by inhalation of fenitrothion
(technical) or fenitrothion 22% emulsifiable concentrate (EC) was
examined in 10-15 male and female Hartley guinea-pigs using
bacterial alpha amylase as a positive control material. Fenitrothion
(technical) suspended in 10% Tween 80 aqueous solution and the
undiluted EC formulation were sprayed daily for 120 min for 7 days.
The air concentrations of fenitrothion were 226 and 628 mg a.i./m3
in the chamber where the technical sample was sprayed, and 360 and
1048 mg a.i./m3 in the chamber where the EC formulation was
sprayed. Animals inhaling the material at the higher air
concentration of the EC formulation showed salivation from the third
day. The cholinesterase activities in the brain, packed red cells,
and plasma were severely inhibited in treated animals at the
completion of the sensitization period. The animals were challenged
7 days after the last sensitizing inhalation, but signs of allergic
asthma were not observed in any animals treated with fenitrothion.
In contrast, animals treated with bacterial alpha amylase showed
severe allergic signs of asthma. It was concluded that fenitrothion
did not cause allergic asthma (Okuno et al., 1977).
7.4 Long-term and carcinogenicity studies
Groups of Wistar rats (15 males and 15 females) were fed diets
containing fenitrothion at levels of 0, 2.5, 5, or 10 mg/kg for 92
weeks, to investigate the effects of fenitrothion on ChE activities.
The ChE activity in the blood was measured after 2, 4, 6, 8, 12, 16,
20, 24, 42, 68, and 92 weeks. In the 5 mg/kg group (equivalent to
0.27 mg/kg body weight per day), a 20-25% decrease in plasma ChE
activity in the males during the first 16 weeks and a 20-35%
decrease in the females during 12 weeks were recorded. The activity,
however, recovered during the remaining test period. In the 10 mg/kg
group, the plasma activity decreased during the first 8 weeks by
30-40% in males and 40-50% in females. The activity, however,
gradually returned to normal during the next 8 weeks. The activity
of erythrocyte ChE was decreased by 20-30% in both sexes during the
first 6 weeks at 10 mg/kg and then recovered fully. The brain ChE
activity, determined at the end of the test period, was not affected
by fenitrothion at any dose level. The no-observed-effect level in
this study was 5 mg/kg diet (0.27 mg/kg body weight per day in males
or 0.28 mg/kg body weight per day in females) (Kadota et al.,
1975a).
Three groups of Charles-River albino rats (50 males and 50
females) were administered fenitrothion at levels of 10, 30, or 100
mg/kg diet (corresponding to 0.5, 1.5, and 5 mg/kg body weight) for
a period of 104 weeks, with 60 females and 60 males as controls.
These animals were derived from the F1a generation of a
reproduction study (see section 7.5.1). Ten rats of each sex and
group were sacrificed after one year and all the surviving animals
at 104 weeks. The body weights of males and females at 100 mg/kg
were lower than those of the controls from the start of the test and
remained so in males until 52 weeks, but at the end of the test no
significant differences were seen. Food consumption at 52 weeks was
lower for the middle- and high-level males, but normal for low-level
males and all the females. The mortality rate in the treated females
was comparable with that in the control females, while, in males,
the mortality rate was significantly higher than the controls only
in the lowest-dose group. Blood and urine analyses were normal while
ChE activity showed a dose-dependent decrease among the test rats.
Significant depressions in ChE activities in the plasma were
recorded at all 3 dosage levels (F1a being affected from the
beginning), but erythrocyte ChE depression and brain ChE depression
occurred only at the 30 and 100 mg/kg levels in both sexes.
Statistical analysis of the probabilities of tumour incidence did
not reveal any differences between the controls and the animals
treated with 10 or 100 mg/kg. There was a decrease in the
probability of benign tumours in males treated with 30 mg/kg and an
increase in pituitary adenoma incidence in the 30
mg/kg females; however, since this was not observed at the 100 mg/kg
level, it was not considered related to treatment. Absolute and
relative organ weights and gross and histopathology did not reveal
any dose-dependent changes (Rutter & Nelson, 1974).
In a study by Rutter & Banas (1975), groups of ICR Swiss mice
(200 animals/sex) were fed a diet containing 0, 30, 100, or 200 mg
fenitrothion/kg for 78 weeks (18 months). No observable effects
attributable to the compound were detected on body weight gain, food
intake, mortality, or ophthalmological and gross/histopathological
findings. It was concluded that fenitrothion is not carcinogenic in
mice fed up to 200 mg/kg diet for 18 months.
Groups of B6C3F1 mice (100 males and 100 females) were
administered fenitrothion at levels of 0, 3, 10, 100, or 1000 mg/kg
diet. Each group of mice was divided into 2 subgroups (main and
satellite groups) comprising 50 mice of each sex. The main groups
were maintained on the control or test diets until death, or for a
maximum of 104 weeks. The satellite groups were used for interim
sacrifices. Throughout the administration period, no
treatment-related abnormal symptoms were observed, nor were there
any significant differences in the survival rates between control
and treated groups. Decreases in body weight gain, food consumption,
and water intake were observed at the 1000 mg/kg level in both
sexes. Urinalysis, ophthalmology, and haematology did not reveal any
treatment-related effects. Significant depression of ChE activity
occurred in plasma at the 3 highest levels, while, in erythrocytes
and brain, the depression occurred at 100 and 1000 mg/kg levels in
both sexes. The blood biochemical examination revealed an elevation
of total cholesterol values in both sexes in the 100 and 1000 mg/kg
groups and a decrease in glucose levels in both sexes in the 1000
mg/kg group. Some other biochemical changes were observed
transiently in the 1000 mg/kg group. The incidence of alopoecia was
decreased at the final sacrifice of 1000 mg/kg females. Brain
weights were increased in both sexes in the 1000 mg/kg group.
Histopathological examination revealed that calcification of brain
parenchyma in both sexes in the 1000 mg/kg group, and hair
follicular atrophy in 1000 mg/kg females were less frequent than in
the controls. No treatment-related increase was observed in the
incidence of any neoplastic changes. The level causing no
toxicological effect was concluded to be 10 mg/kg diet, based on the
depression of brain ChE (Tamano et al., 1990).
7.5 Reproductive effects, embryotoxicity, and teratogenicity
7.5.1 Reproductive effects
A 2-litter, 3-generation, reproduction study on Charles River
albino rats was carried out with 15 males and 30 females per test
group (20 males and 40 females for control) administered dietary
levels of 0, 10, 30, and 150 mg fenitrothion/kg. After the first
filial generation (F1a), the highest dose level was reduced to 100
mg/kg. Fertility, gestation, lactation, and live birth indices were
compared. Administration of 150 or 100 mg fenitrothion/kg in the
diet caused weight reduction in the parental animals in the P0 and
P1 generations and suppressed lactation indices through all
generations. The highest dose group also showed a higher incidence
of cannibalism and smallness at weaning, whereas all litters seemed
normal at birth. No dose-dependent malformations or
histopathological changes were seen (Rutter & Voelker, 1974).
Crl: CD(R)(SD)BR male and female rats (30 animals/group) were
exposed to technical fenitrothion (94.6% pure) in the feed for 2
generations (2 litters in the first generation and 1 litter in the
second generation) at dietary levels of 0, 10, 40, or 120 mg/kg. A
dose-dependent, statistically significant decrease in body weight
gain and absolute and relative feed consumption occurred in males
and females given 120 mg/kg diet. Body weights and body weight gains
were significantly affected at 40 mg/kg diet in the P1 generation
female rats during lactation, and in the F1a generation male rats
during post-cohabitation. Absolute feed consumption values were
significantly affected at 40 mg/kg diet in the F1a generation
female rats. There was no compound-related effect on reproductive
performance in either the P1 or F1a generations at levels of up
to 120 mg/kg diet. The body weights of pups were significantly
reduced in litters of both generations at 120 mg/kg diet and
mortality was significantly increased in the P1 generation, F1a
and F1b litters, and the F1a generation and F2 litters, at 120
mg/kg diet. There were no treatment-related histomorphological
changes in the male and female F0 and F1 generation rats at up to
120 mg/kg diet (Hoberman, 1990).
Groups of 10 male and 20 female rats were fed a diet containing
0, 10, 40, or 80 mg technical fenitrothion/kg in a 4-generation,
2-litter, reproduction study. The following parameters were
monitored: body weight and food consumption of parent animals and
indices of fertility, gestation, live birth, 24-h survival, 5-day
survival, and lactation; gross pathology of all pups, organ weights,
and histopathological examination of F4b weanlings; cholinesterase
activity in whole blood in males of F2a (aged 15 weeks) and in all
weanlings of F4b (aged 4 weeks). Fertility, gestation, and live
birth indices were normal in all groups, whereas the 24-h and 5-day
survival indices were reduced in one or both litters of the 80 mg/kg
group in almost all generations. The lactation index was reduced in
all generations in the 40 and 80 mg/kg groups. The mean litter size
was smaller in all but 5 test litters, but this was without any
clear dose-dependence. However, the lowest number of pups was found
in 6 out of 8 litters in the 80
mg/kg group. The mean weight of the pups at birth and at 21 days of
age was normal, whereas the growth of the parent animals was
slightly decreased in the 80 mg/kg group. Plasma and erythrocyte
cholinesterase activity was decreased in relation to the dose and
length of exposure; in the 10 mg/kg group, the decrease was not
significant. Organ weights and gross pathological examination did
not reveal any abnormalities (Benes et al., 1974).
Fenitrothion fed to 10 female rats and male rats at 200 mg/kg
diet before mating, and during the gestation period, in a
one-generation reproduction study did not affect the reproductive
indices, such as fertility, gestation, and live birth indices.
Moreover, it was revealed that fenitrothion did not have any
estrogenic activity, according to the mouse uterine weight assay
(Gowda & Sastry, 1979).
7.5.2 Embryotoxicity and teratogenicity
Groups of female albino rabbits were inseminated (gestation day
0) and during gestation days 6-18 were dosed with 0, 0.3, or 1 mg
fenitrothion/kg per day in gelatine capsules. A positive control
group given 37.5 mg thalidomide/kg per day was included. The
compound did not induce any effects on the does or on the number of
implantation sites, early or late resorption sites, number of dead
or live young, or aborted fetuses. In the thalidomide group,
approximately 10% of the fetuses showed external malformations,
while none were seen in the other groups. No effects related to the
administration of fenitrothion were seen on examination for internal
or skeletal deformities (Ladd et al., 1971).
Fenitrothion (technical grade, 96.6% purity) was dissolved in
corn oil and administered orally to 14-16 pregnant rabbits (New
Zealand White) during the period of organogenesis (gestation days
7-19) at dose levels of 0, 3, 10, or 30 mg/kg body weight per day.
There were no treatment-related effects on pregnancy rate, fetal
viability, fetal sex ratio, or mean fetal body weight values.
Implantation efficiency was slightly lower at 30 mg/kg body weight
per day compared with the controls. There were no dose-related
increases in the incidences of external, visceral, or skeletal
malformations at any dose level (Morseth et al., 1986).
Groups of 25 or 27 pregnant mice (ICR) were administered
fenitrothion orally at dose levels of 0, 20, 70, or 200 mg/kg per
day during gestation days 7-12. Groups of 23-26 pregnant SD rats
were also dosed orally with fenitrothion at levels of 0, 2, 7, or 20
mg/kg per day during gestation days 9-14. No embryotoxic or
teratogenic effects were observed in mice or rats at any of these
dose levels (Miyamoto et al., 1975).
Fenitrothion (technical grade, 96.9% purity) was dissolved in
corn oil and administered orally to 20-24 pregnant Sprague-Dawley
rats during the period of organogenesis (gestation days 6-15) at
dose levels of 0, 3, 8, or 25 mg/kg body weight per day. There were
no adverse effects on pregnancy rates, mean implantation efficiency,
fetal viability, fetal sex ratio, or mean fetal body weights. The
frequency and the distribution of fetal malformation findings did
not indicate a teratogenic response at any dose level. Maternal
toxicity, as evidenced by body weight decrease and clinical
observations (tremors), was indicated at a dose of 25 mg/kg body
weight per day (Morseth et al., 1987).
Prenatal administration of fenitrothion (50% EC) to CFY rats at
5, 10, or 15 mg/kg body weight by oral gavage from days 7 to 15 of
gestation resulted in dose-related decreases in open field activity
and motor coordination in the offspring of animals treated at the
two highest dose levels. Long-lasting alterations in the acquisition
and extinction of a conditioned escape response, as well as
increased social interactions were observed in the adult offspring.
The results indicated a no-observed-effect level of 5 mg/kg
(Lehotzky et al., 1989).
Fenitrothion (97.6% purity, 0.1% of 3-methyl-4-nitrophenol) was
administered to groups of 20 pregnant Wistar rats in sunflower oil,
by gavage, at single daily doses of 0, 2, 8, 16, or 24 mg/kg body
weight during days 6-15 of gestation. On day 20 of gestation, the
rats were sacrificed and the numbers of viable and dead fetuses,
resorptions, implantations, and corpora lutea were recorded. Fetuses
were subjected to external and internal (skeletal and soft tissue)
examination. Fenitrothion was not found to be teratogenic in this
study at any dose. At doses exceeding 8 mg/kg per day, fenitrothion
was maternally toxic (Benes & Tejnorova, 1986).
7.6 Mutagenicity
Fenitrothion was examined using a variety of mutagenicity tests
including in vitro and in vivo gene mutation, DNA damage/repair,
and chromosomal aberration assays (see Table 5). Most of the tests
showed that fenitrothion was not mutagenic, but some of the tests
gave weakly positive results: Ames tests with S. typhimurium TA100
(Kawachi, 1978; Moriya et al., 1983; Hara et al., 1989), sister
chromatid exchange (SCE) assays (Kawachi, 1978) and chromosomal
aberration tests (Kawachi, 1978). However, more recent studies
showed that fenitrothion was negative in gene mutation assays with
S. typhimurium TA100 NR, a nitro-reductase-deficient strain of
TA100, and in mammalian cells, V79 Chinese hamster lung cells.
Mutagenicity in S. typhimurium TA100 was considered attributable
to the nitro-reductase inherent to bacteria (Hara et al., 1989). An
SCE test in cultured mouse embryo cells showed a negative result for
clastogenic potential
(Suzuki & Miyamoto, 1980); in vitro and in vivo chromosomal
aberration tests in the rat, mouse, and Chinese hamster test systems
also gave negative results (Hara & Suzuki, 1982a,b; Hara et al.,
1988).
When CFLP mice were given 3-methyl-4-nitrophenol (25 mg/kg),
intraperitoneally, once a week for 10 weeks, the numbers of
chromosome gaps in bone marrow were significantly increased (Nehez
et al., 1985).
7.7 Neurotoxicity
Seven hens protected against acute anti-cholinesterase effects
with atropine and 2-pyridine aldoxime methiodide (2-PAM) were given
an oral dose of 250 mg fenitrothion/kg body weight and 3 other hens
were given 500 mg/kg. Two of the 500 mg/kg group died within 1-2
days, while the remaining 8 hens did not show any signs of paralysis
during the observation period of 6 weeks (Kimmerle, 1962b).
Groups of 6 adult White Leghorn hens were given single oral
doses of fenitrothion at 250, 500, or 1000 mg/kg body weight.
Tri- O-cresyl phosphate (TOCP, 300 mg/kg) was used as a positive
control. Toxic signs, which lasted 4-10 days, occurred in all
groups. One half of the hens in the middle dosage group and all
those in the highest fenitrothion group died within 24-48 h of
treatment. No delayed paralysis of the legs occurred in the
survivors in any dose group or at any time during the 5-week
observation period, while all TOCP dosed animals developed paralysis
within 3 weeks. The sciatic nerve in all the surviving hens given
fenitrothion was normal (Kadota et al., 1975b).
Sixteen adult White Leghorn hens received 500 mg
fenitrothion/kg orally and were protected against intoxication by
atropine and 2-PAM; this treatment was repeated 3 weeks later. TOCP
was used as a positive control. Five hens died 2 days after the
first treatment with fenitrothion and none after the second; the
survivors showed toxic signs of cholinesterase inhibition. No
paralysis was observed and histopathological findings in the sciatic
nerves were normal (Kadota et al., 1975b).
Groups of 8 adult, White Leghorn hens were given 16.7 or 33.7
mg fenitrothion/kg per day, 6 days/week, over 4 weeks and then
observed for another 3 weeks. Slight toxic signs were seen in both
groups during the administration period. One hen in the higher
dosage group died on the 5th day of dosing. Body weights were
decreased in both groups, but the decrease in the lower dosage group
was transient. No delayed leg paralysis or histopathological changes
in the sciatic nerve or spinal cord were recorded (Kadota et al.,
1975b).
Table 5. Mutagenicity tests on fenitrothion
Tests Strains Dose levels Metabolic Results Reference
activation
Gene mutation tests
Microorganism test E. coli K12 13.2, 132 µg/ml - Suzuki et al. (1974)
Coli-phage lambda 13.2, 132 µg/ml -
1847 sus E-h+
S. typhimurium
TA1535 10, 100, 1000, + - Suzuki & Miyamoto (1976)
10 000 µg/plate - -
TA1537 + -
- -
TA1538 + -
- -
S. typhimurium
TA98 500,750,1000, + - Kawachi (1978)
2500 µg/plate - -
TA100 + + (weakly)
- + (weakly)
TA1537 + -
- -
S. typhimurium
TA100 < 5000 µg/plate + + Moriya et al. (1983)
- +
TA98 + -
- -
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
TA1535 + -
- -
TA1537 + -
- -
TA1538 + -
- -
E. coli + -
WP2 hcr - -
S. typhimurium
TA98 100, 200, 500, 1000, + - Hara et al. (1989)
2000, 5000 µg/plate - -
TA1535 + -
- -
TA1537 + -
- -
TA100 + + (weakly)
- + (weakly)
TA100NR + -
- -
TA100 1,8-DNP6 + + (weakly)
- -
E. coli
WP2uvrA + -
- -
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
Host-mediated assay ICR mouse
S. typhimurium G46 100, 200 mg/kg body - Suzuki & Miyamoto (1976)
weight; oral, im/ip
(mouse)
Mammalian cell test Chinese hamster 0.01, 0.03, 0.1, + - Hara et al. (1989)
lung cells V79 0.3 mmol/litre - -
DNA-Damage and repair tests
Rec-assay B. subtilis up to 20 µlitre + - Shirasu et al. (1979)
H17/M45 - -
Sister chromatid Human embryonic cells 5 x 10-4 (mol/litre) 2.6 times Kawachi (1978)
higher
exchange (SCE) HE2144 SCE than
control
Chinese hamster cells 1 x 10-3 (mol/litre)
Don-6
ICR mouse embryo cells 10-5, 5 x 10-5, + - Suzuki & Miyamoto (1980)
10-4 (mol/litre) - -
Unscheduled DNA SD rat hepatocyte 300 mg/kg body - Kawamoto et al. (1989)
synthesis (UDS) weight, oral)
in vivo/in vitro
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
Chromosomal aberration tests
Chromosomal aberration Wistar rat F2b (male) 80 mg/kg diet - Benes et al. (1975)
F3b (male) 80 mg/kg diet -
Chinese hamster lung 0.1 mg/ml 21% Kawachi (1978)
cells aberration
Chinese hamster ovary 0.075, 0.15, 0.3 mg/ml + - Hara et al. (1988)
cells-K1 0.003, 0.01, 0.03 mg/ml - -
Long-Evans rat 400, 800, 1000 mg/kg + (weakly) Kawachi (1978)
body weight, oral;
20, 40 mg/kg body + (weakly)
weight, oral, x 5 days
ICR mouse (male) 200, 400, 800 mg/kg - Hara & Suzuki (1982a)
body weight, ip
SD rat (male) 100, 200, 400 mg/kg, - Hara & Suzuki (1982b)
oral, 20, 40, 80 mg/kg, -
oral, x 5 days
Wistar rat (male) 15, 30, 60 mg/kg body - Malhi & Grover (1987)
weight, ip, x 5 days
Q mouse (male) 1000 mg/kg body - Degraeve et al. (1984)
weight, ip
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
Micronucleus test
ICR mouse (male) 200, 400, 800 mg/kg - Hara & Suzuki (1982c)
body weight, ip
ddy mouse (male) 200, 400, 800 mg/kg - Hayashi et al. (1988)
body weight, ip
100, 300, 600 mg/kg body -
weight, ip, x 4 days
Wistar rat (male) 75, 165, 330 mg/kg - Grover & Malhi (1985)
body weight, ip
Dominant lethal test Wistar rat F2b 10, 40, 80 mg/kg diet - Benes et al. (1975)
F4b 10, 40, 80 mg/kg diet -
ICR mouse 20, 200 mg/kg body - Kohda & Kadota (1975)
weight, oral, x 5 days
SD rat 2, 7, 20 mg/kg body -
weight, oral, x 5 days
Q mouse 1000 mg/kg body - Degraeve et al. (1984)
weight, ip
Others S. cerevisiae 0.3% in DMSO per plate - Yadav et al. (1982)
D. melanogaster 50, 150 mg/kg diet - Velazquez et al. (1987)
The inhibitory activities of fenitrothion against neuropathy
target esterase (neurotoxic esterase, NTE) and acetylcholinesterase
(AChE) in the hen (White Leghorn) brain were examined. Oral
treatment with 500 mg fenitrothion/kg resulted in significant (80%)
inhibition of AChE, but not of NTE (less than 10%) (Ohkawa et al.,
1980).
The potential of a single, toxic dose of fenitrothion to elicit
delayed neurotoxicity in adult White Leghorn hens (5 per group) was
compared with the effects produced following similar treatment with
the known neurotoxin, tri-O-cresyl phosphate (TOCP). Hens receiving
single oral doses of either fenitrothion (500 mg/kg) or TOCP (500
mg/kg) were assessed for toxicity by measuring biochemical (brain
and spinal cord AChE and NTE), physiological (motor function) and
morphological parameters of the brain, spinal cord, and sciatic
nerve 24 h, and 7, 14, 28, 42, and 56 days after treatment. At 24 h,
fenitrothion caused a marked inhibition of neuronal AChE. No
alteration in NTE activity was found in any fenitrothion-treated
hens. A characteristic, central-peripheral, distal axonopathy was
observed following treatment with TOCP, mild signs appeared 7-14
days after treatment and increased in severity up to 28 days after
treatment, concomitant with morphological changes primarily in the
sciatic nerve and spinal cord. Minimal morphological changes were
elicited by fenitrothion at this dosage, but the tissues appeared
morphologically similar to those seen in vehicle-treated control
hens. The results demonstrated that fenitrothion was not neuropathic
in the classic manner of TOCP (Durham & Ecobichon, 1986).
7.8 Effects on hepatic enzymes
Groups of male and female Wistar rats (4 animals each) were
kept on a diet containing fenitrothion at 150 mg/kg or
3-methyl-4-nitrophenol at 500 or 1500 mg/kg, and the effects on
hepatic oxidative phosphorylation and mixed function oxidases were
studied. Respiratory control ratios of mitochondria were normal,
adenosine triphosphatase (ATPase) activity was normal, and no
differences were found between the treated and control groups. A
slight decrease in the adenosine diphosphate:oxygen ratio was
observed in females fed 1500 mg nitrophenol/kg. However, the
difference was not regarded as statistically significant. Thus, rat
hepatic mitochondrial respiration systems do not seem to be affected
by administration of fenitrothion or 3-methyl-4-nitrophenol.
Similarly, these chemicals did not affect hepatic microsomal mixed
function oxygenase activities (Hosokawa & Miyamoto, 1975).
Uchiyama et al. (1975) reported that intraperitoneal injection
of 25 mg fenitrothion/kg to male ddY mice inhibited aminopyrine
N-demethylation and aniline hydroxylation activities in the liver
by about 50%.
Yamamoto et al. (1982a) reported that a single oral
administration of about 13 mg fenitrothion/kg (5 µmol/rat) or
repeated oral administration of about 1.3 mg/kg per day (0.5
µmol/rat per day) for 10 days to 4 or 5 Wistar rats did not affect
aminopyrine N-demethylase activity and cytochrome P-450 content,
or inhibit the dearylation or desulfuration of fenitrothion.
Groups of 5-7 weanling male Wistar rats received 50 mg
fenitrothion/kg body weight per day dissolved in peanut oil, by
gavage, for 5 consecutive days. Overt signs of toxicity were
observed within 30 min of receiving the third dose. Severely
affected animals were sacrificed for analysis of tissue esterase
activities and insecticide residues and to assess the influence of
the insecticide on hepatic microsomal mono-oxygenase activity. Daily
treatment was terminated after 5 days and the survivors were
sacrificed at intervals to assess the recovery of activity of
esterases in the tissues. Fenitrothion caused a significant
reduction in liver and body weights, a marked increase in
pentobarbital sleeping time, and a marked reduction in hepatic
microsomal mono-oxygenase ( p-nitroanisole O-demethylase, aniline
hydroxylase) activities. Marked inhibition of plasma
pseudocholinesterase, hepatic and renal non-specific
carboxyesterase, and erythrocyte and brain acetylcholinesterase
occurred. Recovery from 5 such daily doses was slow for all tissue
esterases with the exception of plasma cholinesterase, which
returned to 78% of control activity values within 5 days. Activities
of the other tissue esterases were 20-50% of normal at 5 days, and
only 70-90% of normal 16 days after the final dose (Ecobichon &
Zelt, 1979).
A single oral dose of 250 mg fenitrothion/kg resulted in a
slight decrease in a number of biochemical indices of liver function
in rats, including mitochondrial ATPase activity, cytochrome P-450
content, aniline hydroxylase activity, and aminopyrine
N-demethylase activity (Mihara et al., 1981). A dose of 25 mg/kg
also had a slight effect on P-450 content and xenobiotic metabolism,
while 5 mg/kg did not have any significant effects. The magnitude of
the effects was greater in females than in males.
Intraperitoneal doses of 50 or 500 mg fenitrothion/kg,
administered to mice, depleted hepatic glutathione levels and
inhibited microsomal aniline hydroxylase and paranitroanisole
O-demethylase activities (Ginsberg et al., 1982).
7.9 Effects on hormonal balance
Osicka-Koprowska et al. (1987) reported a significant elevation
in plasma corticosterone levels in rats given a single oral dose or
260 mg fenitrothion/kg. Levels returned to control values within 12
h. Adrenal ascorbic acid content was also
significantly decreased between 1 and 5 h after dosing. The authors
attributed the changes to hypersecretion of ACTH. After daily dosing
with fenitrothion at 13 mg/kg for 14 days, there was no significant
difference in circulating corticoseterone levels between the
controls and treated animals. There was a significant decrease in
14C derived from injected (ip) 14C-corticosterone in the
hypothalamus, adrenals, blood, liver, and muscle, but not in the
pituitary gland, at the end of the dosing period and 30 min after
injection of the label.
7.10 Toxicity of metabolites and the S-isomer
The acute toxicity of fenitrooxon, a metabolite of
fenitrothion, is greater than that of the parent compound.
3-Methyl-4-nitro-phenol, another metabolite, is less toxic
(Table 6).
The intraperitoneal toxicity of fenitrothion metabolites and
their related compounds was tested in mice. 3-Hydroxymethyl and
3-formyl derivatives of fenitrothion and fenitrooxon were more toxic
than fenitrothion and fenitrooxon, respectively. Fifteen other
compounds tested, including aminofenitrothion, demethyl
fenitrothion, and demethyl fenitrooxon, were much less toxic than
the parent compound (Miyamoto et al., 1978).
The acute oral toxicity of the S-methyl-isomer ( O-methyl
S-methyl O-(3-methyl-4-nitrophenyl) phosphorothioate) in rats
and mice is approximately twice that of fenitrothion (e.g., LD50
in mice: 550 and 1400 mg/kg, respectively) and the signs of
poisoning are typical of the muscarinic and nicotinic actions of
acetylcholine, as seen with fenitrothion (Kovacicova et al., 1973;
Rosival et al., 1974). The S-isomer, when injected
intraperitoneally, is 7-9 times more toxic in mice than fenitrothion
(Miyamoto, 1977a).
The bimolecular inhibition constant of S-methyl fenitrothion
on rat brain acetylcholinesterase is about 1000 times greater than
that of fenitrothion (Thompson et al., 1989).
7.11 Factors modifying toxicity
The best therapy in rats against severe poisoning with a 100%
lethal dose of fenitrothion was confirmed to be repeated and
combined treatment with atropine and 2-PAM, resulting in a 90%
survival ratio and considerable alleviation of toxic signs
(Matsubara & Horikoshi, 1983).
Table 6. Acute toxicity of metabolites
Compound Animal Route LD50 Reference
(strain) (mg/kg)
Fenitrooxon
Mouse oral 90 Miyamoto et al. (1963b);
Miyamoto (1969)
Mouse oral 120 Hollingworth et al. (1967b)
(Swiss White)
Rat oral 24 Miyamoto et al. (1963b);
Miyamoto (1969)
iv 3.3 Miyamoto et al. (1963b);
Miyamoto (1969)
Guinea-pig oral 221 Miyamoto et al. (1963b);
Miyamoto (1969)
iv 32 Miyamoto et al. (1963b);
Miyamoto (1969)
Dog (Beagle) oral > 68.1 Mastalski et al. (1971)
Hen oral 35 Kadota et al. (1975b)
(White Leghorn)
3-Methyl-4-
nitrophenol Rat
(Male) (Wistar) oral 2300 Sumitomo (1971)
(Female) (Wistar) oral 1200 Sumitomo (1971)
Dog (Beagle) oral > 680 Mastalski (1971a)
The effects of adrenalectomy (Adx), SKF 525-A, phenobarbital
(PB), and diethyl maleate (DEM) on the acute toxicity of
fenitrothion were investigated in male Wistar rats. PB administered
(ip) at 60 mg/kg per day for 3 days, did not exert any effect on the
toxicity of fenitrothion (100 mg/kg) given orally 24 h after the
last injection of PB. In adrenalectomized and SKF 525-A-pretreated
rats, the toxicity of fenitrothion was lower than in the controls.
Fenitrothion toxicity was increased by administration of DEM at (1
ml/kg), which depletes hepatic glutathione (GSH) levels. In
in vitro experiments, the rates of fenitrothion decomposition
and fenitrooxon formation by microsomes were markedly affected by
PB, SKF 525-A, and Adx. The decomposition of fenitrooxon by the
microsomal fraction and GSH-dependent decomposition of fenitrooxon
by the soluble fraction were not affected by PB, SKF 525-A, and
Adx pretreatment. The GSH-dependent decomposition of fenitrothion
and fenitrooxon was increased by the addition of GSH to the incubation
mixture. It was considered that the GSH-dependent metabolic pathway
plays an important role in the detoxification of fenitrothion
(Yamamoto et al., 1983a).
7.12 Mechanism of toxicity - mode of action
7.12.1 Mode of action
Fenitrothion is not a strong inhibitor of AChE in vitro, but
is much more so in vivo. The compound is converted in the animal
body to the active esterase inhibitor, fenitrooxon [ O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphate], by the action of microsomal
mixed function monooxygenase in liver and other tissues (Miyamoto et
al., 1963b; Miyamoto, 1964a).
Plasma ChE is the enzyme most susceptible to acute and
short-term administration of fenitrothion in rats, guinea-pigs,
dogs, rabbits, and humans (Miyamoto et al., 1963b; FAO/WHO, 1975b).
Brain AChE in rabbits is less inhibited by fenitrothion (Miyamoto et
al., 1976a).
7.12.2 Selective toxicity
Hollingworth et al. (1967a,b) and Hollingworth (1969) concluded
that dealkylation was an important factor among the many that
contribute to the lower mammalian toxicity of fenitrothion compared
with that of methylparathion. For example, the cholinesterase
inhibition of fenitrooxon is less, the activation by conversion of
P=S to P=O, slower, the translocation of fenitrothion more rapid,
and the detoxification rate of fenitrothion, higher. Comparison of
metabolites, at equitoxic doses in white mice (i.e., 17 mg
methylparathion/kg and 850 mg fenitrothion/kg) showed that
demethylation is the major detoxification path for fenitrothion at
high doses (200-850 mg/kg), but not for methylparathion. However,
inherently, demethylation of both compounds proceeds in a similar
way (Miyamoto et al., 1968) and both fenitrothion and
methylparathion are metabolized at a similar rate in vivo
(Miyamoto, 1964a, 1969). On the basis of these findings, Miyamoto
(1969) concluded that the relatively poorer penetration of
fenitrooxon into the brain (Miyamoto, 1964b) explained the
selectively lower toxicity of fenitrothion rather than the
dealkylation detoxification mechanism. Similar conclusions were
reached by Kuroiwa & Yamamoto (1977).
The mechanisms for the lower toxicity of fenitrothion compared
with methylparathion were investigated in male Wistar rats. The
difference in acute toxicity between fenitrothion and
methylparathion could be because of the more rapid decomposition of
fenitrothion and fenitrooxon in rat liver compared with that of
methylparathion and methylparaoxon. In particular, the decomposition
of fenitrothion by hepatic microsomes was accelerated by increasing
the insecticide concentration. The oxygen analogues of both
insecticides, fenitrooxon and methylparaxon, were not detected in
the brain after administration of the parent compounds. It was
considered that the lower toxicity of fenitrothion compared with
that of methylparathion could be due to the greater rate of
decomposition of fenitrothion to its less toxic metabolites rather
than to the different rates of penetration of the oxygen analogues
into the brain (Yamamoto et al., 1983b).
7.12.3 Potentiation of toxicity of other chemicals
Female rats were administered ip a combination of fenitrothion
and parathion, malathion, diazinon, or carbaryl. No potentiation was
demonstrated. However, a marked potentiation (increased mortality)
was observed when male and female Sprague-Dawley rats were given
single oral doses of fenitrothion and phosphamidon. In female rats,
potentiation occurred only with mixtures containing relatively low
concentrations of fenitrothion (Dubois & Kinoshita, 1963; Braid &
Nix, 1968).
The effects of a combination of fenitrothion with malathion in
male rats were more than additive. The potentiation was most
pronounced (half of the expected LD50) with a combination rate of
1:1. No potentiation was observed with other tested
organophosphates, i.e., bromophos, amidithion, and trichlorfon
(Benes & Cerna, 1970).
Hladka et al. (1974) noted that a single dose of a mixture of
fenitrothion and malathion caused a significant increase in
fenitrooxon but not fenitrothion levels in the blood and muscles of
female Wistar rats.
Fenitrothion at a subtoxic oral dose (100 mg/kg body weight; 4
h pretreatment or 1000 mg/kg diet for one week) potentiated the
acute oral toxicity of 2-sec-butylphenyl methylcarbamate (BPMC) in
male ICR mice. Through several in vivo and in vitro studies, it
was suggested that competitive inhibition of BPMC metabolism in the
liver by fenitrothion played, in part, a role in the inhibition of
BPMC detoxication, resulting in the potentiation of its toxicity
(Takahashi et al., 1984, 1987; Tsuda et al., 1984).
The same effects were investigated in female Beagle dogs. Oral
co-administration of fenitrothion (100 mg/kg) doubled the duration
of symptoms of BPMC (50 mg/kg) (5 h). The plasma level of BPMC in
the eliminating phase was increased by the co-administration and
began to decrease after 6 h, while ChE inhibition continued for 8 h.
Pretreatment with fenitrothion at 5 mg/kg per day, for 7 days,
caused one death in 3 dogs after the administration of BPMC (100
mg/kg), while the same dose of BPMC without pretreatment did not
cause any deaths. The pretreatment with fenitrothion increased the
duration of the toxic symptoms of BPMC 2.5-fold. The time course of
the toxic symptoms was correlated with the plasma concentration of
BPMC (Miyaoka et al., 1983).
8. EFFECTS ON MAN
8.1 General population exposure
8.1.1 Acute toxicity
Fenitrothion was given to a total of 24 human volunteers in
single oral doses of 0.042-0.33 mg/kg body weight or 2.5-20 mg per
person. The excretion of a metabolite, 3-methyl-4-nitrophenol, in
the urine was almost complete within 24 h, and ranged from about 70%
of the dose (0.042 mg/kg) to about 50% (0.33 mg/kg). Neither plasma
nor erythrocyte cholinesterase (ChE) activities were depressed below
normal, except in one person given 0.33 mg/kg, whose plasma ChE
activity was about 65% of the pretest level after 6 and 24 h. When
repeated doses of 0.04-0.08 mg/kg were given to 5 individuals, 4
times at 24-h intervals, most of the metabolites appeared in the
urine within 12 h of administration. After receiving the third and
fourth doses, there was a trend towards a rise in erythrocyte ChE
activity (Nosal & Hladka, 1968; Hladka et al., 1977).
8.1.2 Poisoning incidents
Several cases of acute fenitrothion poisoning, a few of them
lethal, have been described in the literature. These were either
accidental, intentional (suicide), or due to gross neglect of safety
precautions. A review of these cases is given by Hayes & Lawes
(1991).
In all cases, the onset of poisoning was rapid, early signs and
symptoms being exhaustion, headache, weakness, confusion, vomiting,
abdominal pain, excessive sweating, and salivation. The pupils were
small. Difficulty in breathing may be experienced, due to either
congestion of the lungs or weakness of the respiratory muscles. In
severe cases of poisoning, muscle spasms, unconsciousness, and
convulsions may develop and death may result from respiratory
failure.
In atypical cases, symptoms of poisoning may be observed for a
more prolonged period and up to 8 months after exposure. It has been
suggested that fenitrothion can be stored in human fat tissue and
then released under stress conditions (Ecobichon et al., 1977).
An attempted suicide using a large quantity of fenitrothion was
treated successfully by 2-PAM, atropine, and glutathione with
artificial respiration. The case was characterized by delay in the
onset of severe signs of intoxication (the 3rd day after
hospitalization) and relatively prolonged poisoning (the 70th day
after hospitalization), as evidenced by electroencephalogram and
inhibition of ChE. It was reported that the patient (56-year-old
female) had been suffering from diabetes, which presumably caused
delayed biotransformation of fenitrothion to fenitrooxon or delayed
absorption of the compound from the intestines, resulting in the
delay in the onset of symptoms and prolonged poisoning. However, no
evidence leading to this presumption was seen in the study
(Tsukimoto et al., 1981).
A case of delayed neurotoxicity of late onset has been reported
in a 70-year-old female who ingested 40 ml of 50% Fenitrothion EC.
At first, no toxic symptoms were apparent. However, 48 h after
ingestion, certain signs became apparent. An impediment in
consciousness was observed. Fasciculation and muscular weakness were
noted, while plasma and urinary levels of 3-methyl-4-nitro-phenol
reached a maximum. Neither atropine sulfate nor 2-PAM was effective.
For 3 weeks, the patient required ventilatory support, and
consequently her muscle strength and neurological status gradually
recovered with falling of the phenol level (Sakamoto et al., 1984).
The late symptoms reported by Sakamoto et al. (1984) are not those
of the organophosphorus ester-induced delayed neurotoxicity (OPIDN)
reported for some other organo-phosphorus compounds (Martinez
Chuecos & Sole Violan, 1985).
A 56-year-old male attempted suicide by the ingestion of about
60 ml of 50% fenitrothion emulsion. Five hours later, combined
haemoperfusion and haemodialysis (HP-HD) treatment was performed for
60 min and, subsequently, the symptoms gradually improved. Four days
after ingestion, cholinergic symptoms recurred. Immediate HP-HD
treatment was of no use and the patient died 6 days after ingestion
of fenitrothion. Analysis of the organ and tissue distribution of
fenitrothion revealed that the highest concentration of fenitrothion
was found in fat (59.0 mg/kg wet weight, more than 10 times the
concentrations in other organs). It was suggested that a slow
release of the pesticide from adipose tissue can give rise to a
protracted clinical course or late symptoms of intoxication (Yoshida
et al., 1987).
8.1.3 Contact dermatitis
An analysis of 202 patients with contact dermatitis caused by
organophosphorus insecticides was undertaken. The organophosphorus
insecticides presumably attributing to the dermatitis were
dichlorvos, salithion, fenitrothion, leptophos, cyanophos,
amidothion, diazinon, and malathion. The dermatitis was located on
the fingers, face, forearms, neck and nape. About one quarter
(25.2%) of the cases with dermatitis had complications with symptoms
of acute poisoning by these compounds. The prognosis of the
dermatitis was relatively good; 44.1% of the cases healed but 23.8%
of the patients were incompletely cured (Matsushita et al., 1985).
8.1.4 Possible links with Reye's syndrome
Concerns have been raised about the possible association
between the aerial spraying of forests with fenitrothion
formulations and the incidence of Reye's Syndrome, an encephalopathy
complicated by hepatic damage, occurring primarily in children up to
18 years of age and linked to viral infections (Reye et al., 1963;
Ruben et al., 1976; Corey et al., 1976).
Crocker et al. (1976a) reported that patients from a
fenitrothion-sprayed area had reduced plasma ChE and erythrocyte
AChE activities compared with those in children living in an
unsprayed region. However, Pollack et al. (1977) confirmed that,
while the serum ChE of Reye's patients was lower, the difference was
not significant. No correlation between aerial spraying and the
incidence of Reye's Syndrome was established in epidemio-logical
studies conducted in New Brunswick, Canada, and Maine, USA
(Schneider, 1976; Spitzer, 1982; Wood & Bogdan, 1986). Attention
was focused on the potential of various emulsifiers and co-solvents
used in oil- and water-based formulations of fenitrothion to enhance
virus-induced lethality in neonatal mice or to promote viral growth
in cell cultures (Crocker et al., 1974, 1976b). However, in a
neonatal animal model, the infection of neonatal mice by influenza
virus was not potentiated by any emulsifiers in fenitrothion
formulations (Menna, 1985). Similarly, in an in vitro cell culture
system, the growth of the virus was not enhanced by co-incubation
with various concentrations of fenitrothion, in the presence or
absence of a wide range of emulsifiers, co-solvents and diluents,
commonly used in aerial spraying formulations (Brookman et al.,
1984).
8.2 Occupational exposure
In a WHO (1973) programme, 3 different water-dispersible powder
formulations of fenitrothion were applied indoors as a residual
spray with the aim of depositing 2 g active ingredient/m2 on the
wall. The first three rounds of spraying lasted for approximately
six, 5-day, working weeks and the fourth round lasted for 8 weeks. A
slight to moderate depression of cholinesterase (ChE) activity was
observed in most of the spraymen towards the end of the spraying
rounds. Except for one case of headache, no other complaints
attributable to insecticide exposure were recorded during a total of
2000 man-days of spraying. No complaints were received from the 20
000 inhabitants whose houses had been sprayed repeatedly. However,
no ChE activity determinations were performed.
During 30-days indoor spraying of fenitrothion (2 g/m2) for
malaria control in Southern Iran in August 1971, a group of 28 pest
control operators and 925 inhabitants were monitored with respect to
their health and ChE activity. Clinical investigations and ChE
tests on 840 workers showed 42 cases of clinical symptoms, most of
which were very slight and subsided after the workers had showered
and rested (2-3 h). Out of 20 spraymen, 8 showed decreased ChE
levels, and one individual preparing the spray mixture showed a
significant depression of the enzyme activity, which was reactivated
after an appropriate treatment. Among 925 inhabitants, only 15 cases
of very mild complaints, namely dizziness and nausea, were reported.
While fenitrothion was characterized as a pesticide that is safe for
the inhabitants in a subtropical region during dry and hot seasons,
further investigations were recommended on the toxic effects on
operators under tropical conditions (Motabar et al., 1972).
In a field spraying operation in a village in southern Nigeria
using a 5% spray of fenitrothion, 18 villagers examined one week
later did not show any clinical symptoms of toxicity or plasma ChE
depression. The ChE levels in the 3 spraymen examined on the first,
second, and sixth days after spraying were also not depressed
compared with pre-spraying levels (Vandekar, 1965).
In another field spraying operation in northern Nigeria, 10 000
huts, in which about 16 500 people lived, were sprayed. Field test
ChE determinations on whole blood did not show any appreciable
differences in the ChE levels of 535 villagers tested before
spraying and 299 villagers tested 5-30 days after spraying. After
one week of intensive spraying, 5 out of 20 spraymen developed a 50%
depression of ChE, which returned to a stable level after a period
of rest. One sprayman developed symptoms of intoxication that lasted
only a few hours and disappeared without treatment (Wilford et al.,
1965).
The health was monitored of workers employed in forest spraying
operations using fenitrothion, in New Brunswick, and plasma and
erythrocyte ChE activities, measured. There were no instances of ChE
activity fluctuating beyond the accepted range or of suspected
poisoning resulting from occupational exposure to this insecticide
(Braid & McCarthy, 1977).
Moderate poisoning of 25 workers was reported in Czechoslovakia
(Kalas, 1978; Hayes, 1982), where a formulation containing 50%
fenitrothion was applied by aircraft during a strong wind. Onset of
poisoning developed 2.5-6 h after inhalation and the symptoms were
typical. Whole blood ChE activity was decreased by 48%. Recovery
required 3 days of treatment with atropine.
In the Haitian malaria control programme, depressed whole blood
cholinesterase activity (< 50% of normal) was detected rapidly
prior to the development of serious symptoms. Evidence of
fenitrothion over-exposure appeared in spraymen early in the first
spray cycle, and was associated with faulty protective clothing and
failure to follow strictly the recommended safety measures at work.
After these deficiencies were corrected, insecticide application
continued without serious incidents or interruption of the
programme. It was recommended that training and monitoring
programmes should be instituted whenever organophosphate pesticides
are used as residual sprays for malaria control (Warren et al.,
1985a). Similar observations were made in another study undertaken
in Pakistan and Haiti (Miller & Shah, 1982).
Cholinesterase activity was significantly reduced at the end of
the working week in 3 out of 28 fenitrothion workers in Haiti.
Urinary levels of 3-methyl-4-nitrophenol in the spraymen ranged from
2.2 to 25.2 mg/litre. In fenitrothion workers who had no direct
contact with spraying (weighers and supervisors), the cholinesterase
activity remained > 75% of the normal control value, and the
urinary 3-methyl-4-nitrophenol levels were relatively low. The
cholinesterase levels improved and the urinary excretion of
metabolites decreased after 2 days of rest from the spraying
operations. In the residents of the sprayed houses, low
concentrations of 3-methyl-4-nitrophenol were detected in the urine,
1 day after spraying, and measurable, but reduced, levels were still
present after 7 days. In all these cases, the cholinesterase
activity remained > 75% of the normal control value (Warren et
al., 1985b).
In an epidemiological study on the effects of fenitrothion on
occupationally exposed male workers in the production department and
female workers in the packing room during a 5-year period, the
results of clinical examinations pointed to parasympathetic
stimulation and disposition to hypotonia. Neurological and
psychiatric findings revealed a low-grade pseudoneurasthenic
syndrome in 33% of females and in 15% of males. The results of
psychodiagnostic tests after exposure to fenitrothion and its
intermediate products showed partial deterioration of retention,
impairment of visuomotor coordination of movements in tapping,
impaired orientation readiness, prolonged average time in
decision-making, and prolonged average reaction time for complex
sensorimotor responses. The following effects on biochemical
parameters after exposure to fenitrothion should be mentioned:
inhibition of cholinesterase in the blood, increase of glutamate
pyruvate transaminase, increase of isoenzyme lactate dehydro-genase
(LDH-5), and changes in protein fractions, all of which were
statistically significant (P < 0.001). The values of
3-methyl-4-nitrophenol excreted in the urine of males after the
exposure to fenitrothion averaged 5.61 mg/litre, compared with an
average value of 1.54 mg 3-methyl-4-nitrophenol/litre before
exposure. The average values of 3-methyl-4-nitrophenol excreted in
the urine of females involved in bottling fenitrothion were 4.0 mg
3-methyl-4-nitrophenol before exposure and 6.58 mg
3-methyl-4-nitrophenol/litre of urine after exposure. The
concentrations of fenitrothion in the air of the workplace ranged
from 0.028 to 0.118 mg/m3. From the values of the
3-methyl-4-nitrophenol excreted in the urine of the exposed workers
and of volunteers, to whom fenitrothion was administered in doses of
2.5-20 mg, it could be judged that the exposed male workers received
approxi-mately 15 mg of fenitrothion per capita a day and the
exposed female workers received 20 mg or more per capita a day
(Liska et al., 1982).
The introduction of the ultra-low-volume application of the
organophosphate pesticide fenitrothion in grain terminals presented
a risk to workers of skin contact with a high concentration. Blood
testing, by the Ellman method, of a group of 5 grain terminal
workers engaged in grain treatment showed a lowering of mean
erythrocyte cholinesterase activity to 23 units/g Hb (normal value
28-40) with a range of 16-29. The probable cause was identified as
percutaneous absorption of fenitrothion through ungloved hands
exposed to clean blocked drip feed nozzles. Modification of work
practices was followed by a rise of mean erythrocyte ChE activity to
33.6 units/g Hb (range 32-36) during the following grain treatment
season. Erythrocyte ChE activity measured during the intervening
winter season, i.e., during a non-exposure period, showed a mean of
33.3 units/g Hb (range 23-40) (Gun et al., 1988).
9. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
9.1 Microorganisms and algae
Salonius (1972) reported that a 12-month incubation of forest
soils with massive doses of fenitrothion emulsion at the rate of 48
mg/pot, equivalent to approximately 112 kg/ha, based on surface area
of the soil, did not alter the population or respiration of the soil
microflora.
No effects of fenitrothion were observed on the growth of 6
species of fungi responsible for the decomposition of organic
detritus in streams and swamp water containing 15, 150, or 1500 µg
fenitrothion/litre for up to 3 months (Eidt, 1978).
In Maine (USA), viable populations of bacteria, yeasts, fungi,
and actinomycetes were monitored in forest leaf litter treated with
fenitrothion at 1 mg/kg (Spillner et al., 1979a); fenitrothion
treatment did not appear to affect microbial populations or
respiration in the forest litter. Furthermore, fenitrothion (1 or 5
mg/kg) did not have any effects on cellulose-degrading organisms
(Spillner et al., 1979b).
Total numbers of bacteria and the nitrification activity in the
soil were not affected by a 5-year application of fenitrothion 50%
EC diluted 1000 times at 125 ml/m2 (Nishio & Kusano, 1978). No
effects on urease and glucanase were observed when fenitrothion was
incorporated into silt loam soil at field rates (0.70-2.11 kg
a.i./ha) (Burns & Lethbridge, 1981).
Mandoul et al. (1968) reported that a concentration of 5 mg
fenitrothion/litre did not affect microplankton in a freshwater
environment. They stated that 2 applications of fenitrothion at 140
g/ha would not approach 5 mg/litre, even assuming that all of the
material reached the lakes or streams.
Growth inhibition tests were carried out using 3 species of
algae, and EC50 values were determined to be 4-9 mg/litre for diatom
and blue-green algae, and more than 100 mg/litre for green algae
(Kikuchi et al., 1984) (Table 7).
9.2 Aquatic organisms
9.2.1 Fish
Toxicity data of fenitrothion on aquatic non-target organisms
are summarized in Table 7. Fenitrothion is moderately toxic for fish
species with LC50 values of more than 1 mg/litre.
Table 7. Toxicity of fenitrothion for aquatic non-target organismsa
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Microorganisms
Activated sewage sludge 4-h growth EC50 450 22 ± 2 Kwasniewska et al. (1980)
sediment soil
Algae
(Chlorella vulgaris) F growth EC50 100 T 23 ± 2 Kikuchi et al. (1984)
(Nitzschia closterium) F growth EC50 3.9 T 23 ± 2 Kikuchi et al. (1984)
(Anabaena flos-aquae) F growth EC50 8.6 T 23 ± 2 Kikuchi et al. (1984)
Mollusca
(Pila globosa) F 30 ± 2 g 72-h LC50 1.2 T 29 ± 2 Madhu et al. (1982)
(Phya acuta) F 48-h LC50 15 EC Hashimoto & Nishiuchi (1981)
Red snail F 48-h LC50 8.5 EC Hashimoto & Nishiuchi (1981)
(Indoplanorbis exustus)
Marsh snail F 48-h LC50 6.0 EC Hashimoto & Nishiuchi (1981)
(Semisulcospira libertina)
Eastern oyster M juvenile 96-h EC50 0.450 T 27 Mayer (1987)
(Crassostrea virginica) growth
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Crustacean
Lobster M larva 96-h approx. T 15 Mcleese (1974)
0.001
(Homarus americanus) adult 96-h LC50 approx. T 15 Mcleese (1974)
0.001
Prawn M nauplius 24-h LC50 1.9 T 23-25 Hirayama & Tamanoi (1980)
(Penaeus japonicus) postlarva 24-h LC50 0.0005- T 23-25
0.0009
Crab M zoea 24-h LC50 0.005-0.008 T 23-26 Hirayama & Tamanoi (1980)
(Portunus trituberculatus) megalopa 24-h LC50 0.0002- T 23-26
0.0005
young 24-h LC50 0.003 T 23-26
Blue crab M 8.5- 96-h LC50 0.0086 T 22 Johnston & Corbett (1985)
(Callinectes sapidus) 11.0 cm
Brown shrimp M juvenile 96-h LC50 0.0015 T 29 Mayer (1987)
(Panaeus aztecus)
Water fleas
(Daphnia pulex) F adult 3-h LC50 0.050 T 24-26 Hashimoto & Nishiuchi (1981)
(Daphnia magna) adult 48-h LC50 0.0086 T 20 ± 2 Forbis (1987)
adult/young 21-day MATC 0.00014 T 20 ± 2 Burgess (1988)
(Moina macrocopa) F adult 3-h LC50 0.050 T 24-26 Hashimoto & Nishiuchi (1981)
Mite
(Hydrachna trilobata viets) F 48-h LC50 0.074 T 28 ± 3 Nair (1981)
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Arthropod Insects
Caddisfly larva F 96-h LC50 11 T 5 Symons & Metcalf (1978)
(Brachycentrus numberosus)
Scud M 96-h LC50 0.01 T 18 Swain & Ivanikiw (1976)
(Gammarus pseudolimnaeus) 96-h LC50 0.0043- T 12 Woodward & Mauck (1980)
0.0088
Stonefly naiad
(Peleronarcys californica) F 96-h LC50 0.004 T 16 Swain
& Ivanikiw (1976)
(Pteronarcella badia) 96-h LC50 0.0051- T 12 Woodward & Mauck (1980)
0.0072
Mayfly larva F 9.3 mm, 48-h LC50 0.0032 EC 25 Nishiuchi (1981)
(Cloeon dipterum) 5.6 mg
Dragon fly larva F 2.3 cm, 48-h LC50 0.055 EC 25 Nishiuchi (1981)
(Orthetrum albistylum 0.62 g
speciosum)
(Sigara substriata) F 5.9 mm, 48-h LC50 0.023 EC 25 Nishiuchi (1981)
6.1 mg
(Micronecta sedula) F 3.2 mm, 48-h LC50 0.058 EC 25 Nishiuchi (1981)
1.8 mg
(Sympetrum frequens) F 2.1 mm, 48-h LC50 0.050 EC 25 Nishiuchi (1981)
(larva) 0.56 g
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
(Eretes sticticus) 1.5 cm, 48-h LC50 0.062 EC 25 Nishiuchi (1981)
0.20 g
Amphibian
Clawed toad embryo 24-h LC50 10 T 18 Elliott-Feeley &
(Xenopus laevis) 24-h EC50 4.2 T 18 Armstrong (1982)
24-h LC50 10 T 25
24-h EC50 0.37 T 25
24-h LC50 0.33 T 30
24-h EC50 0.17 T 30
(Bufo b. japonics) tadpole 48-h LC50 9.0 EC Hashimoto & Nishiuchi (1981)
(Microhyla ornatas) embryo 96-h LC50 3.21 T Pawar & Katdare (1984)
tadpole 96-h LC50 1.14 T
Freshwater fish
Carp 5.1 cm 48-h LC50 8.2 T 25 ± 1 Hashimoto & Nishiuchi (1981)
(Cyprinus carpio) 7-9 cm 72-h LC50 2.3 50% EC 28- 32 Toor & Kaur (1974)
eyed egg 24-h LC50 3.5 EC 25 ± 2 Hashimoto et al. (1982)
sac fry 24-h LC50 1.5 EC 25 ± 2 Hashimoto et al. (1982)
floating fry 24-h LC50 1.7-4.0 EC 25 ± 2 Hashimoto et al. (1982)
6.0 cm, oral LD50 10 mg/kg T 20 ± 22 Hashimoto & Fukami (1969)
2.5 g
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Killifish 48-h LC50 7.0 T 25 ± 1 Hashimoto & Nishiuchi (1981)
(Oryzias latipes) embryo 96-h LC50 10 T 25 ± 1 Takimoto et al. (1984b)
yolk sac 96-h LC50 6.94 T 25 ± 1 Takimoto et al. (1984b)
fray
postlarva 96-h LC50 2.36 T 25 ± 1 Takimoto et al. (1984b)
juvenile 96-h LC50 3.54-4.66 T 25 ± 1 Takimoto et al. (1984b)
adult 96-h LC50 3.70 T 25 ± 1 Takimoto et al. (1984b)
Rainbow trout fingerling 96-h LC50 2.0 T 15 Klaverkamp et al. (1977)
(Salmo gairdneri) 9.2 ±
0.1 cm,
10.35 ±
0.78 g
egg/fry 60-days post- 0.12 T 10 ± 1 Cohle (1988)
hatch MATC
Brook trout 96-h LC50 1.7 T 12 Swain & Ivanikiw (1976)
(Salvelinus fontinalis) 96-h LC50 2.1 40% EC 7 Swain & Ivanikiw (1976)
96-h LC50 2.2 40% EC 12 Swain & Ivanikiw (1976)
96-h LC50 1.8 40% EC 17 Swain & Ivanikiw (1976)
15-21 cm No effect on 10 mg/kg T 15 ± 2 Wildish & Lister (1977)
(42-120 g) growth food
8.3-12.6 cm Effect on 0.50 T 15 Peterson (1974)
critical
swimming
velocity
Cutthroat 118 mm, 20 g 96-h LC50 2.57-2.88 T 12 Woodward & Mauck (1980)
(Salmo clarki) 96-h LC50 2.88 87% EC 12
96-h LC50 2.70 87% EC 7
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Gold fish 2-3 g, 4-5 24-h LC50 4.5 pure 25 ± 2 Gras (1966)
(Carassius auratus) product
Goldfish 9-11 cm 48-h LC50 3.4 T 25 Hashimoto & Nishiuchi (1981)
(Carassius auratus) threshhold for approx. 0.01 T 15 Scherer (1975)
avoidance
Eel (Anguilla anguilla) 2 g, 10 cm 24-h LC50 3.2 pure 25 ± Gras (1966)
product
Gambusia 0.25-0.60 g 24-h LC50 2.6 pure 25 ± Gras (1966)
(Gambusia affinis) product
Fathead minnow embryo-larva 31 days 0.13-0.30 T 23.8 ± 1 Kleiner et al. (1984)
(Pimephales promelas) MATC
Pond loach 48-h LC50 4.8 T 25 ± 1 Hashimoto & Nishiuchi (1981)
(Misqurnus anquilicaudatus)
(Acheilognathus moriokae 5.5-6.0 cm, 72-h LC50 5.0 T 25 Nishiuchi (1977)
1.2-1.4 g
(Channa gachua) 116 mm, 18 g 96-h LC50 12.2 50% EC 24 ± 2 Verma et al. (1978a)
(Saccobranchus fossilis) 135 mm, 25 g 96-h LC50 12.5 50% EC Verma et al. (1978b)
50-75 mm, 96-h LC50 12.6 50% EC 18 ± 2 Verma et al. (1982)
5-10 g
(Mystus cavasius) 6-8 cm 96-h LC50 3.3 T 28 ± 2 Murty et al. (1983)
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
(Labeo rohita) 118 mm, 20 g 96-h LC50 4.63 50% EC Verma et al. (1977)
3-4.4 cm 96-h LC50 2.8 T 28 ± 2 Murty et al. (1983)
4.5-5.9 cm 96-h LC50 4.1 T 28 ± 2 Murty et al. (1983)
6-8 cm 96-h LC50 4.6 T 28 ± 2 Murty et al. (1983)
(Aplocheilus latipes) 24-h LC50 4.5 T 25 Shim & Self (1973)
(Zacco platypus) 24-h LC50 7.4 T 25 Shim & Self (1973)
(Puntius ticto) 50-68 mm, 96-h LC50 5.8 50% EC 11-29 Bhatia (1971)
1.6-5.1 g
Seawater fish
Japanese striped knifejaw egg-prelarva 24-h LC50 2.4-4.2 50% EC 22 ± 0.1 Seikai (1982)
(Oplegnathus fasciatus) postlarva 24-h LC50 0.145 50% EC 22 ± 0.1 Seikai (1982)
postlarva- 24-h LC50 1.25-2.35 50% EC 22 ± 0.1 Seikai (1982)
juvenile
Agohaze (Chasmichthus 0.02-0.06 g 96-h LC50 1.1 50% EC 15 Hirose et al. (1979)
dolichognathus d.) 96-h LC50 1.7 50% EC 20 Hirose et al. (1979)
Sheepshead minnow juvenile 48-h LC50 > 1.0 T 9 Mayer (1987)
(Cyprinodon variegatus)
24-h EC50 0.17 T 30
a EC = Emulsifiable concentrate; F = Freshwater; M = Marine; MATC = Maximum Acceptable Toxicant
Concentration; NEC = No effect concentration; T = Technical ingredient.
In studies on the relative toxicity of fenitrothion in
different developmental stages of fish, postlarvae of killifish
(Takimoto et al., 1984b), and Japanese striped knifejaw (Seikai,
1982) and the sac fry of carp (Hashimoto et al., 1982) were most
susceptible with LC50 values of 2.36, 0.145, and 1.5 mg/litre,
respectively.
Oral administration of fenitrothion to carp showed a high
LD50 value (more than 10 mg/kg) (Hashimoto & Fukami, 1969),
whereas the 4-week, no-effect, dietary concentration was 1 mg/kg
food (Wildish & Lister, 1973).
Ingestion of contaminated, aquatic or terrestrial insects by
salmonids is extremely unlikely to cause lethal or sublethal effects
in the fish. Behavioural changes did not occur in laboratory studies
until brook trout ingested 3000 times more fenitrothion than the
3.19 mg/kg found in poisoned insects in treatment areas (Wildish &
Lister, 1973).
Exposure of young Atlantic salmon to 0.1 or 1 mg fenitrothion
per litre for 15-16 h caused a 20 or 50% reduction in numbers of
fish holding territories, respectively. Recovery of the parr from
the above effects appeared to be complete in 3 weeks. Feeding and
swimming behaviours returned to normal within 24-48 h (Symons,
1973). After exposure to 1.0 mg fenitrothion/litre for 24 h,
Atlantic salmon parr were more vulnerable than unexposed fish to
predation by large brook trout; fenitrothion at 0.1 mg/litre did not
have any noticeable effects on vulnerability (Hatfield & Anderson,
1972).
Juvenile Atlantic salmon exposed to 6.7 µg fenitrothion/litre
for 7 days showed a decrease in the reaction distance to prey, but
no significant decrease in the efficiency of the salmon's attack
sequence (Morgan & Kiceniuk, 1990).
The movement of goldfish was dependent on the fenitrothion
concentration in water, the threshold level being about 0.01
mg/litre (Scherer, 1975).
Long-term studies revealed that the no-observed-effect level or
maximum acceptable toxicant concentration (MATC) was at 0.1 mg/litre
and above 0.1 mg/litre, respectively, in fathead minnows in a 31-day
early life stage test (Kleiner et al., 1984) and in guppies in a
2-month reproduction test (Yasuno et al., 1980).
Exposure of freshwater murrel ( Channa punctatus) to a
subtoxic concentration of 1.5 mg fenitrothion 50% EC/litre resulted
in a reduction in protein-bound iodine levels after 60, but not 30,
days exposure (Saxena & Mani, 1985a). On day 120 of exposure,
effects on male and female reproductive function were observed, such
as reductions in testicular and ovarian weight, growth rates
of spermatocytes and oocytes, and the numbers of sperm and ova (Mani
& Saxena, 1985; Saxena & Mani, 1985b, 1987). However, it is not
clear whether such effects were derived from fenitrothion or other
EC components.
Pregnant female guppies were exposed to 10 mg
fenitrothion/litre for 4 h, 5, 10, or 15 days before the next
parturition. Half of the females gave premature birth when exposed 5
or 10 days before parturition, and only 32 or 63%, respectively, of
the eggs were delivered alive. The females exposed to the
fenitrothion 15 days before parturition had normal births and only
9.4% of the offspring were stillborn. The body lengths of the young
produced by the females after exposure were significantly shorter
than those produced before exposure in all the studies (Yasuno et
al., 1980). In a study by Miyashita (1984), exposure of pregnant
guppies ( Poecilia reticulata) to 10 mg/litre for 4 h at various
stages of gestation resulted in a reduction in the body lengths of
the offspring at birth.
Rainbow trout ( Salmo gairdneri) were exposed to fenitrothion
in a flow-through, early life stage, toxicity study at
concentrations of 0, 0.025, 0.046, 0.088, 0.17, or 0.35 mg/litre,
for 60-days after hatching at 10 ± 1 °C. Survival and growth of fry
showed that the maximum acceptable toxicant concentration (MATC)
limits were 0.088-0.17 mg/litre and that the point estimate MATC
value was 0.12 mg fenitrothion/litre (Cohle, 1988).
There were no direct lethal effects on wild and caged salmonid
fish after forest spraying with fenitrothion at dosages varying from
double sprays of 140 g/ha to single sprays of up to 280 g/ha in
Newfoundland, Canada (Hatfield & Riche, 1970).
Studies of the effects on wild salmon of fenitrothion sprays in
New Brunswick, Canada, from 1966 to 1969 revealed no mortality in
streams at spray levels of up to 560 g/ha (MacDonald & Penney,
1969). Studies in Manitoba, Canada, on stream-caged rainbow trout
exposed to a spray of 280 g fenitrothion/ha did not show any
significant biochemical changes, including changes in brain and
serum ChE activities. In the 24-h period after spraying,
fenitrothion residues in whole fish averaged 0.5 mg/kg wet weight.
Peak residue values in the fish were as high as 1.84 mg/kg wet
weight but declined to less than 0.02 mg/kg wet weight in 4 days.
Lockhart et al. (1973) concluded that no long-term toxic effects
would be expected among the caged fish.
In the 1979 aerial spraying programme in Canada,
concentrations of fenitrothion of up to 0.64 µg/litre were detected
in stream water in the spray block within 40 h; no
insecticide-related deaths of caged fish or abnormal fish behaviour
occurred (Gillis, 1980).
In 1979, fenitrothion was sprayed on a Scottish forest at a
rate of 300 g/ha. The maximum concentration in a river was 18.8 µg
per litre, 1-2 h after application. There was no evidence that the
resident fish population was disturbed, and no short-term effects
were noticeable in caged fish (Morrison & Wells, 1981). Gillis
(1978) demonstrated, in a 1977 study, that, in an area with a long
history of perennial treatment with fenitrothion (8 years since
1969), there were consistently higher numbers of salmon per 100 m of
stream, but that the density of trout was similar to that in the
control area. It was concluded that the long history of fenitrothion
usage in Canadian forests was unlikely to have resulted in depletion
of the population or biomass of salmon parr trout.
9.2.2 Invertebrates
The acute toxicity of fenitrothion for Daphnia magna was
assessed using the 48-h static method at 20 ± 2 °C. The 48-h LC50
value was 8.6 µg/litre (6.8-11 µg/litre) and the no-observed-effect
level was estimated to be < 2.0 µg/litre after 48 h (Forbis, 1987).
Daphnia magna was exposed to fenitrothion in a dynamic
21-day, life cycle, toxicity study at concentrations of 0, 0.029,
0.042, 0.087, 0.23, or 0.44 µg/litre, at 20 ± 2 °C. Statistical
analyses of survival, adult mean length, and length in days to first
brood, showed that the maximum acceptable toxicant concentration
(MATC) limits were estimated to be 0.087 and 0.23 µg/litre and the
point estimate MATC value was 0.14 µg/litre (Burgess, 1988).
Fenitrothion is highly toxic for arthropods, including insect
larvae, shrimps, crabs, and daphnids. LC50 values for these
invertebrates are generally at µg/litre levels, except that for
caddis fly larvae with a value of 11 mg/litre (Table 7).
Exposure of the freshwater rice-field crab ( Oziotelphusa
senex senex) to sublethal concentrations of fenitrothion showed a
number of effects: limb regeneration was completely inhibited at 0.1
mg/litre and partially inhibited at 0.01-0.05 mg/litre, during
continuous 60-days exposure (Reddy et al., 1983a); exposure to 0.04
mg/litre for 30 days reduced glycolysis and increased
gluco-neogenesis in the hepatopancreas and muscle (Reddy et al.,
1982a, 1983b); exposure to 0.1 mg/litre induced inhibition of
molting and ovarian growth, perhaps by triggering the release of
molt-inhibiting hormone and gonad-inhibiting hormone (Reddy et al.,
1982b, 1983c); exposure for 1 day to 0.1 mg/litre produced an
increase in muscle protease activity and a decrease in the protein
content, while exposure for 20 days resulted in increases in both
parameters (Bhagyalakshmi et al., 1983a); continuous exposure for
20-30 days to higher fenitrothion concentrations of 0.5, 1, or 2
mg/litre resulted in a decrease in oxygen consumption, an increase
in haemolymph glucose, and a reduction in carbohydrate metabolism
rate in the hepatopancreas, with the maximal effect occurring after
3-7 days of exposure. During the exposure, levels of haemolymph
glucose and oxygen consumption returned to the control levels,
accompanied by an increase in carbohydrate metabolism (Bhagyalakshmi
et al., 1983b, 1984a). Exposure to 1, 2, or 4 mg/litre for 48 h
resulted in a dose-dependent inhibition of acetylcholinesterase
activity of the thoracic ganglionic mass, where mean activity levels
in the treated groups ranged from 30 to 60% of the control values.
On cessation of exposure, activity levels returned to normal within
10-15 days (Bhagyalakshmi et al., 1984b).
Fenitrothion was experimentally added to a stream at a
concentration of 10 mg/litre, when the predominant and prevalent
species in the drift samples were shrimps ( Anisogammarus sp.),
mayfly ( Epeorus ikanonis, and Baetis sp.), stonefly ( Nemovra
sp.), and blackfly ( Simulium sp.) larvae. In the course of the
study, Simulium sp. increased in density, but Anisogammarus sp.
did not recover after 4 months (Yasuno et al., 1981). When
fenitrothion was applied at 1 mg/litre to a stream, drifting aquatic
invertebrates including shrimp ( Anisogammarus annandalei), Baetis
sp., Nemouridae, Epeorus sp., Perla quadrata, and Simulium
sp., but not caddis fly larvae (Arctopsyche sp.) or crab
( Geothelephusa dehaanii), were affected by the insecticide
(Hasegawa et al., 1982).
After treatment of a small lake with 140 g fenitrothion/ha,
surface populations of zooplankton and phantom midge larvae
(Chaoborus sp.) were depressed for a short period. No substantial
impact was found on benthic invertebrates, emerging insects, and
amphibians in the lake (Kingsbury, 1978).
9.2.3 Amphibians and arthropods
Acute toxicity tests revealed that fenitrothion at 0.1 mg/litre
did not induce any significant effects in amphibian embryos
( Xenopus laevis) (Elliott-Feeley & Armstrong, 1982).
When the frog embryo (Microhyla ornata), at the yolk-plug
stage, was exposed to 1 mg fenitrothion/litre for 96 h, no effects
were observed on development. Exposure at 3 mg/litre resulted in
blistering of the body surface and exposure at 5 mg/litre or more
resulted in abnormal behaviour, curvature of the spine, diminished
pigmentation, and retarded growth (Pawar & Katdare, 1983, 1984).
The LC50 value for fenitrothion in tadpoles of Bufo buto
japonicus was 9.0 mg/litre (Hashimoto & Nishiuchi, 1981). The
toxicity of fenitrothion in mollusca was low with LC50 values of
1.2-15 mg/litre in freshwater molluscs and 0.45 mg/litre in eastern
oyster (Table 7). Fenitrothion residues in frogs, collected near
stagnant water 1 day after spraying (280 g/ha), ranged from
0.03-0.17 mg/kg wet weight (Lockhart et al., 1977).
No effects were observed on amphibians and small mammals in
Northern Maine, USA, where the forest had been treated twice with
fenitrothion at the rate of 140 g/ha (USDA, 1976). Large numbers of
hatching salamander eggs were present throughout the treatment
period in the silty bottomed pond located in a treatment plot. No
deaths were observed up to a week after the second spray. When the
pond was examined approximately 2 months after treatment, it still
contained many larval salamanders and aquatic invertebrates.
A stream was treated with fenitrothion at a calculated
concentration of 73 µg/litre for 2.5 h. The standing crop of benthic
arthropods decreased, most of the kill consisting of stonefly larvae
(Leucta sp.). Benthos, including the stonefly larvae, completely
recovered 50 days after the treatment (Eidt, 1981).
MacDonald & Penney (1969) studying the effects on aquatic
insects of fenitrothion, applied twice at a rate of 140 g/ha, found
that the aquatic insect population remained stable. The results of
other studies suggested that populations of aquatic insects may be
reduced following operational spraying with fenitrothion at 140-280
g/ha (National Research Council of Canada, 1975). Stonefly nymphs,
in particular, and mayfly nymphs, to a lesser extent, appeared to be
susceptible to fenitrothion, along with caddis fly larvae.
Flannagan (1973) reported that within 24 h of spraying at 280
g/ha in Canada, the drift of chironomid larvae increased by 700-800%
(total numbers/net). In some instances, Eidt (1975) and Peterson &
Zitko (1974) observed that the drift returned to normal shortly
after the pulse of fenitrothion had cleared.
Fenitrothion was sprayed on a Scottish forest at a rate of 300
g/ha. The maximum concentration in a river within the sprayed
region, which was 18.8 µg/litre, fell to 0.5 µg/litre after 24 h.
Invertebrate drift increased 12-16 h after spraying, but decreased
to pre-spray levels within 48 h. Caged insects remained alive during
the 5-day post-spray period (Morrison & Wells, 1981).
Levels of fenitrothion residues in stream insects would not be
sufficiently high to cause mortality in fish; 3.19 mg fenitrothion
per kg was detected in a sample of mayfly nymphs one week after
spraying with fenitrothion at 280 g/ha, but no residues of
fenitrothion were found after this date (Kingsbury, 1976).
An application of fenitrothion at 210 g/ha in Quebec, Canada,
in 1978, had few or no adverse effects on aquatic invertebrates or
fish, though a small increase in the drift of mayfly nymphs was
observed after application (Holmes, 1979).
In an area with a long history of perennial fenitrothion
spraying (8 years since 1969), a light depletion of the invertebrate
population was observed on both drift and benthos in streams after
each spray, but the invertebrate density stabilized within 1 month
and was comparable with the control (Gillis, 1978).
When fenitrothion was dispersed by a helicopter in Japan at a
rate of 884 g a.i./ha on a paddy field, a peak concentration of 554
µg/litre was reached immediately after spraying, and the
concentration of fenitrothion in the paddy field decreased to less
than 5 µg/litre, 3 days after spray. A crustacean Moina sp., which
was prevalent in the field, disappeared after fenitrothion spraying,
but the population recovered to pre-spray levels within 10 days
(Takaku et al., 1979b).
9.3 Terrestrial organisms
The acute and short-term toxicities of fenitrothion for
terrestrial, non-target organisms are summarized in Table 8.
9.3.1 Terrestrial invertebrates
Studies using drop cloths (Leonard, 1971; Kettela & Varty,
1972; Miller et al., 1973) indicated that large numbers of
Lepidoptera, sawfly larvae, balsam twig aphids, and perching flies
(nematocerous Dipterans, etc.) were killed directly after the
spraying operation (140 or 210 g/ha), usually within the first 4
days. Generally, other defoliating insects were reduced in about the
same proportion as the target species; the larval sawflies were
affected more severely.
Various preliminary measurements have been made on the effects
of fenitrothion on invertebrate fauna (National Research Council of
Canada, 1975). Populations of ground-inhabiting invertebrates
declined after two operational applications of 140 and 210 g
fenitrothion/ha (Carter & Brown, 1973). The densities of predatory
invertebrates were higher in the 2 years before and the year
following the application. Thus, while the invertebrate populations
appeared to be depressed during the years of application, they were
generally able to recover to the normal levels.
On the basis of field observations on the long-term effects of
fenitrothion on non-target arthropods in Canada, Varty (1977)
concluded that:
- The non-target arthropod community on balsam fir was not
perceptibly destabilized by perennial or intermittent
applications at conventional rates (140-210 g/ha) and timing.
- A few fir-dwelling insects had become scarcer during the 1970s,
but the cause-effect was undetermined, and natural fluctuation
could account for the population decline.
- The abundance of predators was related primarily to cycles of
preabundance on fir and spruce, and to budworm modification of
habits.
- Herbivore populations tended to be depressed by budworm
competition, but fungivores might be favoured.
- The abundance of the two most important species parasitizing
spruce budworm larvae persisted, in spite of perennial
spraying.
- Population densities of some species-predatory arthropods in
the soil community might suffer some setback, but in short-term
studies recuperation seemed adequate.
- Minor-pest eruptions in the 1970s did not appear to be
triggered by the local disruption of biocontrol mechanisms
following spraying.
Fenitrothion is highly toxic for bees (Anderson & Atkins, 1968;
Okada & Hoshiba, 1970), the LD50 is approximately 0.03-0.13 µg per
bee (Anderson & Atkins, 1968) (Table 8). However, studies on the
effects of fenitrothion on colonies of honey-bees indicated that an
application of 280 g/ha had few long-term effects (National Research
Council of Canada, 1975). Initially, mortality of adult foraging
bees was evident. Within 4 days of treatment, the daily adult
mortality of foraging bees had apparently returned to normal. The
total detected mortality in excess of controls was about 500 adult
bees, which was estimated to be about 1% of the total hive
population. Buckner (National Research Council of Canada, 1975)
reported that hive activity was unaffected compared with controls,
and that hive weight and eventual honey yield were almost identical
in all experimental and control hives. The observed mortality was
probably restricted to actively foraging worker bees that were
actually exposed to the spray.
Table 8. Toxicity of fenitrothion for terrestrial non-target organismsa
Species Size Toxicity Formulation Condition Reference
Birds
Bobwhite quail 14 days old LC50 5000 mg/kg diet T 8-day dietary Hill et al. (1975)
(Colinus virginianus)
Japanese quail 14 days old LC50 5000 mg/kg T 8-day dietary Hill et al. (1975)
(Coturnix c japonica) diet (no mortality)
5 weeks old (M) LD50 84.85 mg/kg 50% EC oral Hattori et al. (1974)
5 weeks old (F) LD50 73.87 mg/kg 50% EC oral Hattori et al. (1974)
3-6 weeks old (M) LD50 110 mg/kg T oral Kadota & Miyamoto (1975)
3-6 weeks old (F) LD50 140 mg/kg T oral Kadota & Miyamoto (1975)
3-6 weeks old NEL approx. 5 mg/kg diet T 4-week dietary Kadota & Miyamoto (1975)
Ring-necked pheasant 10 days old LC50 5000 mg/kg T 8-day dietary Hill et al. (1975)
(Phasianus colchicus) diet (no mortality)
Mallard duck
(Anas platyrhynchos) 10 days old LC50 5000 mg/kg T 8-day dietary Hill et al. (1975)
diet (no mortality)
13-16 weeks old (M) LD50 1190 mg/kg T oral Hudson et al. (1979)
55-61 weeks old (M) LD50 504 mg/kg T percutaneous Hudson et al. (1979)
Redwinged blackbird
(Agelaius phoeniceus) LD50 25 mg/kg oral Schafer (1972)
Table 8 (contd).
Species Size Toxicity Formulation Condition Reference
Pigeon 250-380 g LD50 42.24 mg/kg 50% EC oral Hattori (1974)
(Columba livia)
Grackle
(Quiscalus quiscula) LC50 78 mg/kg diet T 5-day dietary Grue (1982)
Insect
Honeybee 7 days old 24-h LD50 0.13 µg/bee T topical Takeuchi et al. (1980)
(Apis mellifera) 32 ± 1 °C
24-h LD50 0.03 µg/bee 50% EC topical Okada & Hoshiba (1970)
32 ± 1 °C
LD90 0.25 µg/bee 60% EC oral Clinch & Ross (1970)
30 °C
Silkworm II-V instar 24-h LD50 T topical Kashi (1972)
(Bombyx mori) 4.88-18.7 µg/g
a M = Male; F = Female; T = Technical ingredient; EC = Emulsifiable concentrate.
Plowright (1977) demonstrated that fenitrothion, sprayed at a
dose level of 210 g/ha, was capable of inducing direct mortality in
caged bumble-bees. Aerially sprayed fenitrothion at 210 g/ha in
Canada caused 100% mortality in exposed habitats and 47% under dense
forest canopy, among caged bees (Bombus sp). Bumble bees foraging
in sprayed areas suffered significantly higher mortality than in
unsprayed areas. The abundance of bee species was 3 times less in
sprayed areas compared with unsprayed areas. Population recovery
appeared to be complete within a few years (Plowright et al., 1978).
Plowright & Rodd (1980) further examined the effects of
fenitrothion in wild bees, and found that fenitrothion sprayed at
210 g/ha caused high mortality among solitary bees and vespid wasps,
similar to that in bumble bees (Plowright et al., 1978). Wood (1979)
found that spraying of fenitrothion over nearby woodland resulted in
very low counts of native bees; however, the population returned to
normal levels within 3 years of spraying, with satisfactory
pollination of blueberry.
Wild Bombus sp. queen territories were identified prior to
treatment and a search was made for these queens after the
application of fenitrothion (280 g/ha) in Larose Forest, Canada.
Queens identified prior to a morning spray were again identified 1,
2, and 3 days after treatment. In this single study, no significant
effects were indicated (National Research Council of Canada, 1975).
The low yield of low-bush blueberry was reported to be
attributable to the mortality of native bees (Kevan, 1975) and lack
of pollination following aerial fenitrothion spraying to control
spruce budworm in nearby forests in New Brunswick. However, field
experiments in 1980 showed that there was no evidence of any harmful
effects on the blueberry as a result of fenitrothion spraying and
that the concentration of fenitrothion aerosols found in blueberry
fields during the 1980 budworm spray programme was estimated to be
well below the toxic levels for all bee species (Wood, 1980).
Bee, pollen, and wax samples were collected from forest areas
that had been sprayed with fenitrothion for budworm control at an
operational dosage of 140-280 g/ha, over a 3-year period from 1972.
The level of fenitrothion in bees was low (maximum 2.08 mg/kg)
initially, and decreased rapidly to trace levels within a week.
Fenitrothion levels in pollen were very low initially, but declined
less rapidly than in bees. Accumulations of the insecticide in honey
and wax samples were negligibly small, but traces seemed to persist
in the wax for some time (Sundaram, 1975).
Fenitrothion is toxic for one of the beneficial insects,
silkworm ( Bombyx mori) larvae. Silkworms were fed mulberry leaves
that had been treated with fenitrothion at the rate of 100 ml of
0.0025 or 0.005% fenitrothion solution /kg leaves (equivalent to 2.5
and 5 mg/kg). The administration during the 5 instar larva stage
resulted in a reduction in the number of eggs laid, fertility, and
hatching (Kuribayashi, 1981). At a lower concentration (1 mg/kg), no
effect was observed on mortality, egg laying, and hatching (Yamanoi,
1980). Growth after hatching was not affected by fenitrothion
treatment (Yamanoi, 1981).
The populations of nematodes, rotifers, and tardigrades in soil
were not affected after fenitrothion was sprayed on the surface of a
pasture at a rate of 2.24 kg a.i./ha (Martin & Yeates, 1975).
9.3.2 Birds
The acute oral LD50 values of fenitrothion in birds are shown
in Table 8. The values range from 25 mg/kg (Redwinged blackbird) to
1190 mg/kg (Mallard duck).
Except for the grackle with a dietary LD50 of 78 mg/kg,
values were generally more than 5000 mg/kg diet and, at this dose
level, no mortality was observed (Hill et al., 1975).
The effects of fenitrothion on the brain cholinergic system
were investigated in male Japanese quail (8-14 weeks old).
Cholinergic signs, such as salivation and convulsions in legs and
wings, were seen 6-120 min after the administration of fenitrothion
(250-350 mg/kg). Sixty minutes after an oral dose of fenitrothion
(300 mg/kg), free acetylcholine increased and acetylcholinesterase
(AChE) activity decreased to 20% of the control value. In vitro,
fenitrothion inhibited AChE activity in brain homogenate with an
I50 of 10-5 mol/litre (Kobayashi et al., 1983).
Japanese quail (60 per sex; 20 per sex in the 1.5 mg/kg group)
received 0, 1.5, 5, 15, or 50 mg fenitrothion/kg in the diet for 4
weeks. No animals died and no toxic signs were seen; no
abnormalities in body weight or food consumption were observed. A
dose-related decrease in blood ChE activity was observed in females
in the 5 mg/kg group (25%), and in males and females in the 15 mg/kg
(60%) and 50 mg/kg (85%) groups during the treat-ment period. When
these animals were fed the control diet again, partial recovery of
ChE activity occurred within 2 weeks, while the brain ChE activity
was reduced in females in the 15 mg/kg group (25%) and males and
females in the 50 mg/kg group (65%). Full recovery of ChE activity
occurred after 4 weeks. In the 50 mg/kg group, the egg-laying rate
was decreased, but returned to normal after 3 weeks (Kadota &
Miyamoto, 1975).
Reproduction studies on bobwhite quails and mallard ducks
revealed that short-term feeding of fenitrothion at rates of up to
10 mg/kg diet (quails) or 100 mg/kg diet (ducks) did not adversely
affect parental growth and reactions, egg production, egg weight,
hatchability, and the growth and viability of the young (Miyamoto,
1977b).
Adverse effects on bird species were observed occasionally when
fenitrothion was applied at dosages higher than those commonly used
for operational applications (National Research Council of Canada,
1975); above 280 g/ha, mortality was observed in adults that
inhabited the crown canopy, and, at rates of 560 g/ha or more, adult
mortality increased markedly. At rates of 210 or 280 g/ha,
behavioural changes were observed in adults as well as some juvenile
mortality.
When fenitrothion was sprayed at 1.7 kg a.i./ha, twice a year,
for three consecutive years in Japan, Takano & Hijikata (1981) were
unable to detect impacts on bird diversity, abundance, and
reproductive success, when 9 species (including the long-tailed tit,
which is most sensitive to fenitrothion) out of 48 were monitored in
the forests and fields (55 ha).
The various effects of fenitrothion on White-throated Sparrows
in New Brunswick, Canada, were investigated, with the following
results (Pearce & Busby, 1980):
- The parent compound fenitrothion was detected at levels as low
as 0.02 to 0.41 mg/kg in the male birds from the spray zone,
but none of its metabolites were found. There were no
correlations between fenitrothion residue levels and brain ChE
activity in sparrows.
- Singing frequency, song structure, clutch size, hatching
success, and fledging rates were similar in the control and
experimental area.
- Growth parameters, such as body weight, tarsus length, bill
length and width, and wing length, of the nestling birds
appeared to be lower in the exposed nestlings than in the
controls after fenitrothion spraying at a rate of 210 g
a.i./ha.
- The reproductive success was reduced in the exposed area after
repeated spraying of fenitrothion, first at 420 g/ha and
several days later at 210 g/ha. The young White-throated
Sparrows from 11.6% of the eggs laid fledged in the sprayed
area, whereas the young from 58.3% of the eggs laid fledged in
the control area.
- The main causes of reduced productivity in the sprayed area
were the high rate of nest desertion, the occasional death or
incapacitation of incubating females, and the high rate of
nestling disappearance from nests.
Eight hours after being exposed to an operational spray of
fenitrothion at 280 g/ha, the crops and carcasses of caged Japanese
quail contained 0.045-0.459 mg fenitrothion/kg (wet tissue basis),
when cages were placed in open locations, and the crops, up to 1.89
mg/kg, when the cages were placed below the tree canopy (Lockhart et
al., 1977). However, fenitrothion residues in all samples declined
sharply to an undetectable level (< 0.02 mg/kg) after 5 days.
Although these birds suffered a drop in serum ChE activity, they
were not killed and recovery of enzyme activity was observed 5 days
after spraying (Lockhart et al., 1977).
Experimental fenitrothion spraying at the rate of 210-280 g
a.i./ha was conducted using 2 types of spray hardware (boom and
nozzle and rotary atomizer) in southwest New Brunswick, Canada. The
brain ChE activity of the 5 avian species monitored (Tennessee
Warbler ( Vermivora peregrina), Magnolia Warbler ( Dendroica
magnolia), Blackburnian Warbler ( D. fusca), Bay-breasted Warbler
( D. castanea), and White-throated Sparrow ( Zonotrichia
albicollis)) was significantly inhibited compared with that in the
control birds, and 16-30% of the captive birds showed more than 20%
inhibition compared with the controls. However, no dead birds or
abnormal avian behaviour was observed (Busby et al., 1981).
Brain cholinesterase (ChE) inhibition and fenitrothion residues
were determined in White-throated Sparrows ( Zonotrichia
albicollis), exposed to aerial applications of fenitrothion (210 g
a.i./ha applied twice with a 5-8 day interval) during the breeding
seasons in 1978 and 1979, in New Brunswick (Canada). Brain ChE
activity was significantly reduced in birds exposed to the sprays
(50-66% inhibition) and fenitrothion and metabolite residues were
detected in all exposed birds (0.08-1.4 mg/kg); however, they did
not show any consistent correlation with brain ChE activity. An
acute brain ChE response, manifested as sudden ChE reduction
followed by gradual recovery, was noted in birds collected after
spraying with 420 g a.i./ha (Busby et al., 1983).
A study was conducted to assess the response of selected forest
songbird species, including the Tennessee Warbler ( Vermivora
peregrina), Bay-breasted Warbler ( Dendroica castanea), Magnolia
Warbler ( D. magnolia), and White-throated Sparrow ( Zonotrichia
albicollis), to aerial ultra ULV fenitrothion (40% content)
spraying (210 g a.i./ha applied twice with a 6-day interval) through
measurements of brain cholinesterase (ChE) depression. A slight
reduction (6-17% less brain ChE activity than in the controls) was
observed in birds collected on day 2
after the second spray; birds collected on day 1 of the first spray
were least affected (0-5% less activity). As a group, the Tennessee
Warbler, the upper canopy forager and singer, exhibited the highest
degree of ChE inhibition (6-17% less activity) and the
White-throated Sparrow, the ground-to-low crown dweller, showed the
least (0-8%) inhibition (Busby et al., 1987).
Brain AchE activity in songbirds exposed through experimental
fenitrothion spraying was monitored in Scotland in 1979 and 1980
(Hamilton et al., 1981). The brain ChE activity of songbirds was
significantly inhibited in the first 2 days after spraying of
fenitrothion at dosages of 300 g/ha in 1979 and 280 g/ha in 1980.
However, the enzyme activity in willow warblers ( Phylloscopus
trochilus) and chaffinches ( Fringilla coelebs) returned to the
normal levels, 7 and 21 days after spraying, respectively. The
residues of fenitrothion found in skin and plumage samples taken
from birds during the first few days after spraying were relatively
high, but declined rapidly to a low level. The fenitrothion residues
in viscera samples were very small initially and were not detectable
a few days after spraying (Hamilton et al., 1981).
Several publications concerning the possible effects of
fenitrothion on avian species including: Rushmore (1971), Hill et
al. (1975), National Research Council of Canada (1975), Paul &
Vadlamudi (1976), USDA (1976), Buckner & McLeod (1977), Germain
(1977), Pearce & Peakall (1977), Germain & Morin (1979), and Pearce
et al. (1979b) are not cited in extenso, because they report
findings similar to those above.
9.3.3 Mammals
Bailey & Swift (1968) classified fenitrothion as moderately
toxic for mammals.
Buckner reported that, at operational application rates below
420 g/ha, fenitrothion spraying did not produce any measurable
effects on small mammals in Maine (USA). Spraying of fenitrothion at
recommended rates would not be expected to cause mortality or
interrupt the breeding cycle of small mammal populations in forests
(Buckner & Sarrazin, 1975; Varty, 1976; Buckner et al., 1977)
though, at very high rates, some ill effects have been observed on
shrew and rodent populations (USDA, 1976). In addition, the
discriminatory behaviour of mature animals suggested that
fenitrothion-contaminated food material was rejected and that it was
a learned process (Buckner et al., 1977).
When fenitrothion was sprayed twice a year, at the rate of 1200
g/ha, by helicopter, there was no decrease in either plasma or
erythrocyte ChE activity in the Japanese wood mouse ( Apodemus
speciosus) inhabiting the sprayed area (Tabata & Kitahara, 1980).
The metabolism of fenitrothion in red-backed voles, which
inhabit the coniferous forests of Canada, did not differ
substantially from that demonstrated for laboratory strains of mice,
rats and guinea-pigs. When fenitrothion was administered ip at doses
of 48.4-2040 mg/kg, it was rapidly metabolized and excreted. No
accumulation of fenitrothion residues in the body was indicated. The
metabolic detoxification mechanism in red-backed voles involves the
cleavage of both the P-O-aryl and P-O-alkyl bonds, the latter being
more prominent at high dose levels (above 1250 mg/kg) (Tschaplinski
& Gardner, 1981).
Thus, the standard programme of forest spraying with the
insecticide does not appear to have serious ecological consequences
(Symons, 1977).
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Fenitrothion was evaluated by the Joint FAO/WHO Expert
Committee on Pesticide Residues (JMPR) in 1969, 1974, 1976, 1977,
1979, 1982, 1983, 1984, 1986, 1987, 1988, and 1989, (FAO/WHO, 1970,
1975a,b, 1977, 1978a,b, 1980a,b, 1983a,b, 1984a,b, 1985a,b, 1986a,b,
1987a,b, 1988a,b, 1989a,b,c). In 1988 the JMPR established an
Acceptable Daily Intake (ADI) for man of 0-0.005 mg/kg body weight.
This was based on the following levels causing no toxicological
effects:
- Rat: 10 mg/kg in the diet, equivalent to 0.5 mg/kg body
weight per day (based on brain acetylcholinesterase
inhibition and reproduction).
- Dog: 50 mg/kg in the diet, equivalent to 1.25 mg/kg body
weight per day.
- Man: 0.08 mg/kg body weight per day (highest dose tested).
The FAO/WHO Codex Committee advised maximum residue limits
(MRLs) in specified food commodities (FAO/WHO, 1986c; 1990) as
follows:
Milks 0.002 mg/kg
Cucumbers, meat, onions, potatoes 0.05 mg/kg
Cauliflower, cocoa beans, egg plants,
peppers, soybeans (dry) 0.1 mg/kg
Bread (white), leeks, radishes 0.2 mg/kg
Apples, cabbage, cabbage red, cherries,
grapes, lettuce, pear, peas, strawberries
tea (dried, green), tomatoes 0.5 mg/kg
Peach, rice (polished) 1 mg/kg
Citrus fruits, wheat flour (white)
processed wheat bran 2 mg/kg
Wheat flour (whole meal) 5 mg/kg
Cereal grains 10 mg/kg
Raw wheat bran, rice bran unprocessed 20 mg/kg
WHO (1990) classified technical fenitrothion as "moderately
hazardous" (Class II). WHO issued a data sheet on Fenitrothion (No.
30) (WHO, 1977).
REFERENCES
ABDEL-KADER, M.H.K. & WEBSTER, G.R.B. (1980) Low temperature
degradation and translocation of malathion and fenitrothion in
stored wheat. Can. Plant. Proc., 9: 143-153.
ABDEL-KADER, M.H.K. & WEBSTER, G.R.B. (1982) Analysis of
fenitrothion and metabolites in stored wheat. Int. J. environ. anal.
Chem., 11: 153-165.
ABDEL-KADER, M.H.K., WEBSTER, G.R.B., & LOSCHIAVO, S.R. (1982)
Effects of storage temperature on rate of degradation of
fenitrothion in stored wheat. J. econ. Entomol., 75: 422-424.
ADDISON, J.B. (1981) Vapour phase photochemistry of fenitrothion and
aminocarb. Bull. environ. Contam. Toxicol., 27: 250-255.
ADHYA, T.K., BARIK, S., & SETHUNATHAN, N. (1981a) Stability of
commercial formulations of fenitrothion, methyl parathion, and
parathion in anaerobic soils. J. agric. food Chem., 29: 90-93.
ADHYA, T.K., BARIK, S., & SETHUNATHAN, N. (1981b) Fate of
fenitrothion, methyl parathion, and parathion in anoxic
sulfur-containing soil systems. Pestic. Biochem. Physiol., 16:
14-20.
ADHYA, T.K., BARIK, S., & SETHUNATHAN, N. (1981c) Hydrolysis of
selected organophosphorus insecticides by two bacteria isolated from
flooded soil. J. appl. Bacteriol., 50: 167-172.
ALY, O.A. & BADAWY, M.I. (1982) Hydrolysis of organophosphate
insecticides in aqueous media. Environ. Int., 7: 373-377.
AMBRUS, A., LANTOS, J., VISI, E., CSATLOS, I., & SARVARI, L. (1981)
General method for determination of pesticide residues in samples of
plant origin, soil and water. I. Extraction and clean-up. J. Assoc.
Off. Anal. Chem., 64: 733-742.
ANAND, M., GULATI, A., GOPAL, K., KHANNA, R.N., & RAY, P.K. (1989)
Changes in cardiac rhythm and calcium levels in guinea pigs treated
chronically with fenitrothion. Toxicol. environ. Chem., 24: 199-205.
ANDERSON, L.D. & ATKINS, E.J., Jr (1968) Pesticide usage in relation
to bee-keeping. Annu. Rev. Entomol., 13: 213.
ANJUM, F. & QADRI, S.S.H. (1986) In vivo metabolism of fenitrothion
( O,O-dimethyl O-(4-nitro-m-tolyl)phosphorothioate) in fresh
water teleost ( Tilapia mossambica). Bull. environ. Contam.
Toxicol., 36: 140-145.
AOKI, Y., TAKEDA, M., & UCHIYAMA, M. (1975) Comparative study of
methods for the extraction of eleven organophosphorus pesticide
residues in rice. J. Assoc. Off. Anal. Chem., 58: 1286-1293.
BAARSCHERS, W.H., ELVISH, J., & RYAN, S.P. (1983) Adsorption of
fenitrothion and 3-methyl-4-nitrophenol on soil and sediment. Bull.
environ. Contam. Toxicol., 30: 621-627.
BAARSCHERS, W.H. & HEITLAND, H.S. (1986) Biodegradation of
fenitrothion and fenitrooxon by the fungus Trichoderma viride. J.
agric. food Chem., 34: 707-709.
BAILEY, J.B. & SWIFT, J.E. (1968) Pesticide information and safety
manual, Berkeley, University of California.
BATTELLE (1982) Pesticide programme of research and market planning.
Part II. Insecticides, Geneva, Switzerland, Battelle Research
Centres, 40 pp.
BATTELLE (1988) World pesticide programme: Insecticide III, Geneva,
Switzerland, Battelle Research Centres, Vol. 1.
BENES, V. & CERNA, V. (1970) Contribution to the toxicological
evaluation of fenitrothion
( O,O-dimethyl- O-(3-methyl-4-nitrophenyl)thiophosphate) and its
residues. In: Deichmann, W.B., Radomski, J.L., & Penalver, R.A., ed.
Proceedings of Pesticide Symposia. Inter-American Conferences on
Toxicology and Occupational Medicine, Miami, Florida, Halas and
Ass., Inc., pp. 219-222.
BENES, V. & TEJNOROVA, T. (1986) Fenitrothion: Teratogenicity study
in rats, Prague, Czechoslovakia, Institute of Hygiene and
Epidemiology (Unpublished report).
BENES, V., SRAM, R.J., & TUSCANY, R. (1974) Reproduction study in
rats, Prague, Czechoslovakia, Institute of Hygiene and Epidemiology
(Unpublished report).
BENES, V., SRAM, R.J., & TUSCANY, R. (1975) Fenitrothion I.: Study
of mutagenic activity in rats. J. Hyg. Epidemiol. Microbiol.
Immunol., 19(2): 163-172.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R. (1983a) Muscle
nitrogen metabolism of freshwater crab, Oziotelphusa senex senex
Fabricium, during acute and chronic sumithion intoxication. Toxicol.
Lett., 17: 89-93.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R. (1983b) Changes in
hemolymph glucose, hepatopancreas glycogen, total carbohydrates,
phosphorylase and amino transferases of sumition-stressed freshwater
rice-field crab. (Oziotelphusa senex senex). Toxicol. Lett., 18:
277-284.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R., (1984a) Subacute
stress induced by sumition on certain biochemical parameters in
Oziotelphusa senex senex, the freshwater rice field crab. Toxicol.
Lett., 21: 127-134.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R. (1984b) The in
vivo inhibition and recovery of acetylcholinesterase in the
thoracic ganglionic mass of the freshwater rice field crab
( Oziotelphusa senex senex) during and after exposure to
sumition. Toxicol. Lett., 21: 135-139.
BHATIA, H.L. (1971) Toxicity of some pesticides to Puntius ticto
(Hamilton). Sci. Cult., 37: 160-161.
BOWMAN, M.C. & BCRAZA, M. (1969) Determination of accothion, its
oxygen analog and its cresol in corn, grass, and milk by gas
chromatography. J. agric. food Chem., 17: 271-276.
BRAID, P.E. & MCCARTHY, T.F. (1977) Human exposure to fenitrothion
during forest spraying in eastern Canada. In: Proceedings of a
Symposium on Fenitrothion, Ottawa, National Research Council of
Canada, p. 451 (Publication NRCC No. 16073). BRAID, P.E. & NIX, M.
(1968) Potentiation of toxicity of Sumithion by phosphamidon in the
rat. Can. J. Physiol. Pharmacol., 46: 133-137.
BRECKENRIDGE, C., PESANT, M., DURHAM, H.D., & ECOBICHON, D.J. (1982)
A 30-day toxicity study of inhaled fenitrothion in the albino rat.
Toxicol. appl. Pharmacol., 62: 32-43.
BREWER, D.G., WOOD, G., & UNGER, I. (1974) The photodecomposition of
fenitrothion ( O,O-dimethyl- O-(3-methyl-4-nitrophenyl)-
phosphorothioate). Chemosphere, 3: 91-95.
BROOKMAN, D.H., CHOPRA, C., ECOBICHON, D.J., KANG, C.Y., RITTER, L.,
& THORSEN, J. (1984) Assessment of the potential of insecticides,
emulsifiers and solvent mixtures to enhance viral infection in
cultured mammalian cells. Appl. environ. Microbiol., 47: 80-83.
BUCKNER, C.H. & MCLEOD, B.B. (1977) Ecological impact studies of
experimental and operational spruce budworm (Choristoneura
fumiferana Clemens) control programs on selected non-target
organisms in Quebec, 1976, Ottawa, Environment Canada, Forestry
Service, Department of Fisheries and the Environment (Information
Report No. CC-X-137).
BUCKNER, C.H. & SARRAZIN, R., ed. (1975) Studies of the
environmental impact of the 1974 spruce budworm control operation in
Quebec, Ottawa, Canada, Chemical Control Research Institute
(Information Report No. CC-X-93).
BUCKNER, C.H., SARRAZIN, R., & MCLEOD, B.B. (1977) The effects of
the fenitrothion spray program on small mammals. In: Proceedings of
a Symposium on Fenitrothion, Ottawa, National Research Council of
Canada, pp. 377-390 (Publication NRCC No. 16073).
BURGESS, D. (1988) Chronic toxicity of fenitrothion technical to
Daphnia magna under flow-through test conditions, Osaka, Japan,
Sumitomo Chemical Co., Ltd (Technical Report. No. HW-81-0326).
BURNS, R.G. & LETHBRIDGE, G. (1981) The effect of pesticides and
fertilisers on enzyme activities in soil. J. Sci. Food Agric., 32:
626-627.
BUSBY, D.G., PEARCE, P.A., & GARRITY, N.R. (1981) Brain
cholinesterase response in songbirds exposed to experimental
fenitrothion spraying in New Brunswick, Canada. Bull. environ.
Contam. Toxicol., 26: 401-406.
BUSBY, D.G., PEARCE, P.A., GARRITY, N.R., & REYNOLDS, L.M. (1983)
Effect of an organophosphorus insecticide on brain cholinesterase
activity in White-throated Sparrows exposed to aerial forest
spraying. J. appl. Ecol., 20: 255-263.
BUSBY, D.G., PEARCE, P.A., & GARRITY, N.R. (1987) Effect of ULV
ultra fenitrothion spraying on brain cholinesterase activity in
forest songbirds. Bull. environ. Contam. Toxicol., 39: 304-311.
CARSHALTON LABORATORY (1964) OMS 43. Summary of mammalian toxicity,
Carshalton, United Kingdom, Carshalton Laboratory, Toxicology
Research Unit (Unpublished report submitted to WHO by Farbenfabriken
Bayer AG, Germany).
CARTER, N.E. & BROWN, N.R. (1973) Seasonal abundance of certain soil
arthropods in fenitrothion-treated red spruce stand. Can. Entomol.,
105: 1065-1073.
CERNA, V. & BENES, V. (1972) [Fenitrothion residues on fruits and
vegetables.] Nahrung, 16: 179-186 (in German).
CHEVALIER, G., HENIN, J.P., VANNIER, H., CANEVET, C., COTE, M.G., &
LE BOUFFANT, L. (1984) Pulmonary toxicity of aerosolized
oil-formulated fenitrothion in rat. Toxicol. appl. Pharmacol., 76:
349-355.
CLINCH, P.G. & ROSS, G.M. (1970) Laboratory assessment of the speed
of action on honey bees of orally dosed insecticides. N.Z. J. agric.
Res., 13: 717-725.
COHLE, P. (1988) Early life stage toxicity of fenitrothion technical
to rainbow trout (Salmo gairdneri) in a flow-through system,
Osaka, Japan, Sumitomo Chemical Co., Ltd (Technical Report No.
HW-81-0331).
COOPER TECHNICAL BUREAU (1966) Toxicity of sumithion (Unpublished
report submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
COREY, L., RUBIN, R.J., HATTWICK, M.A.W., NOBLE, G.R., & CASSIDY, E.
(1976) A nationwide outbreak of Reye's Syndrome. Its
epidemiological relationship to Influenza B. Am. J. Med., 61: 615.
COULOMBE, P.A., LORTIE, S., & COTE, M.G. (1986) Pulmonary toxicity
of the insecticide fenitrothion in the rat following a single field
exposure. J. appl. Toxicol., 6: 317-323.
CRABBE, R., ELIAS, L., KRZYMIEN, M., & DAVIE, S. (1980a) New
Brunswick forestry spray operations: field study of the effect of
atmospheric stability on long-range pesticide drift, Ottawa,
National Research Council of Canada, National Aeronautical
Establishment Laboratory (Report No. LTR-UA-52).
CRABBE, R., KRZYMIEN, M., ELIAS, L., & DAVIE, S. (1980b) New
Brunswick spray operations: measurement of atmospheric fenitrothion
concentrations near the spray area, Ottawa, National Research
Council of Canada, National Aeronautical Establishment Laboratory
(Technical Report No. LTR-UA-56).
CROCKER, J.F.S., OZERE, R.L., ROZEE, K.R., DIGOUT, S.C., &
HUTZINGER, O. (1974) Insecticide and viral infection as a cause of
fatty visceral changes and encephalopathy in the mouse. Lancet, II:
22-24.
CROCKER, J.F.S., OZERE, R.L., BERGH, R.M., MCDONALD, R.G., &
ECOBICHON, D.J. (1976a) Depressed red cell cholinesterase associated
with Reye's syndrome, Vancouver, Canadian Pediatric Society (Meeting
abstract).
CROCKER, J.F.S., OZERE, R.L., SAFE, S.N., DIGOUT, S.C., ROZEE, K.R.,
& HUTZINGER, O. (1976b) Lethal interaction of ubiquitous insecticide
carers with virus. Science, 192: 1351-1353.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984) Genetic and
cytogenetic effects of fenitrothion. Arch. environ. Health, 39:
25-26.
DESMARCHELIER, J.M. (1987) Kinetics of alkaline and
peroxide-catalyzed hydrolysis of fenitrothion. J. environ. Sci.
Health, B22: 403-411.
DRABEK, J. & TRUCHLIK, S. (1957) [Manufacture of O,O-dimethyl
O-(3-methyl-4-nitrophenyl) thiophosphate and its use in insect
control.] Czechoslovak Patent No. 89124 (in Slovak).
DUBOIS, K.P. & KINOSHITA, F. (1963) The acute toxicity of Bayer
41831 in combination with other anticholinesterase insecticides,
University of Chicago, Department of Pharmacology (Unpublished
report submitted to WHO by Farbenfabriken Bayer AG, Germany).
DUBOIS, K.P. & PUCHALA, E. (1960) The acute toxicity and
anticholinesterase action of Bayer 41831, University of Chicago,
Department of Pharmacology (Unpublished report submitted to WHO by
Farbenfabriken Bayer AG, Germany).
DURHAM, H.D. & ECOBICHON, D.J. (1986) An assessment of the
neurotoxic potential of fenitrothion in hen. Toxicology, 41:
319-332.
DURHAM, H.D., COMEAU, A.M., CAMERON, P.H., & ECOBICHON, D.J. (1982)
Subacute toxicity in rats of orally administered fenitrothion alone
and in a selected formulation. Toxicol. appl. Pharmacol., 62:
455-464.
DYKES, J. & CARPENTER, M. (1988) Photodegradation study of
14C-fenitrothion on soil surface, Osaka, Japan, Sumitomo Chemical
Co., Ltd (Technical Report No. HM-81-0098).
ECOBICHON, D.J. & ZELT, D. (1979) The acute toxicity of fenitrothion
in weanling rats and effects on tissue esterases and
mono-oxygenases. Toxicology, 13: 287-296.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S. (1977)
Acute fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
ECOBICHON, D.J., COMEAU, A.M., & CAMERON, P.H. (1980) Chronic
toxicity of fenitrothion in male rats. Toxicol. appl. Pharmacol.,
56: 409-417.
EIDT, D.C. (1975) The effect of fenitrothion from large-scale forest
spraying on benthos in New Brunswick headwater streams. Can.
Entomol., 107: 743-760.
EIDT, D.C. (1978) Effects of insecticide contamination on stream
litter decomposition processes, Ottawa, Environment Canada, Forestry
Service, Department of Fisheries and the Environment, pp. 9-11
(Report No. M-X-87).
EIDT, D.C. (1981) Recovery of aquatic arthropod populations in a
woodland stream after depletion by fenitrothion treatment. Can.
Entomol., 113: 303-313.
EIDT, D.C. & SUNDARAM, K.M.S. (1975) The insecticide fenitrothion in
headwater streams from large-scale forest spraying. Can. Entomol.,
107: 735-742.
ELLIOTT-FEELEY, E. & ARMSTRONG, J.B. (1982) Effects of fenitrothion
and carbaryl on Xenopus laevis development. Toxicology, 22:
319-335.
ENVIRONMENT AGENCY OF JAPAN (1978) The studies on persistence of the
pesticides in water, Tokyo, Environment Agency of Japan, Water
Quality Bureau.
FAO (1982) Second Government Consultation on International
Harmonization of Pesticide Registration Requirements, Rome, 11-15
October 1982, Rome, Food and Agriculture Organization of the United
Nations.
FAO/WHO (1970) Fenitrothion. In: 1969 Evaluations of some pesticide
residues in food: The monographs, Rome, Food and Agriculture
Organization of the United Nations, World Health Organization, pp.
117-135 (FAO/PL:1969/M/17/1; WHO/FOOD/ADD./70.38).
FAO/WHO (1975a) Fenitrothion. In: Pesticide residues in food. Report
of the 1974 Joint Meeting of the FAO Working Party of Experts on
Pesticide Residues and the WHO Expert Committee on Pesticide
Residues, Rome, 2-11 December 1974, Rome, Food and Agriculture
Organization of the United Nations, p. 17 (FAO Agricultural Studies
No. 97; WHO Technical Report Series No. 574).
FAO/WHO (1975b) Fenitrothion. In: 1974 Evaluations of some pesticide
residues in food: The monographs, Geneva, World Health Organization,
pp. 335-381 (WHO Pesticide Residues Series, No. 4).
FAO/WHO (1977) Fenitrothion. In: 1976 Evaluation of some pesticide
residues in food: The monographs, Rome, Food and Agriculture
Organization of the United Nations, pp. 361-376 (AGP:1976/M/14).
FAO/WHO (1978a) Fenitrothion. In: Pesticide residues in food -
Report 1977, Rome, Food and Agriculture Organization of the United
Nations, p. 35 (FAO Plant Production and Protection Paper 10 Rev.).
FAO/WHO (1978b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1977: The monographs, Rome, Food and Agriculture
Organization of the Untied Nations, pp. 263-272 (FAO Plant
Production and Protection Paper 10 Sup.).
FAO/WHO (1980a) Fenitrothion. In: Pesticide residues in food -
Report 1979, Rome, Food and Agriculture Organization of the United
Nations, pp. 45-46 (FAO Plant Production and Protection Paper 20).
FAO/WHO (1980b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1979: The monographs, Rome, Food and Agriculture
Organization of the United Nations, pp. 289-295 (FAO Plant
Production and Protection Paper 20 Sup.).
FAO/WHO (1983a) Fenitrothion. In: Pesticide residues in food -
Report 1982, Rome, Food and Agriculture Organization of the United
Nations, p. 27 (FAO Plant Production and Protection Paper 46).
FAO/WHO (1983b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1982: The monographs, Rome, Food and Agriculture
Organization of the United Nations, pp. 237-238 (FAO Plant
Production and Protection Paper 49).
FAO/WHO (1984a) Fenitrothion. In: Pesticide residues in food -
Report 1982, Rome, Food and Agriculture Organization of United
Nations, pp. 29-30 (FAO Plant Production and Protection Paper 56).
FAO/WHO (1984b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1983: The monographs, Rome, Food and Agriculture
Organization of United Nations, pp. 219-223 (FAO Plant Production
and Protection Paper 61).
FAO/WHO (1985a) Fenitrothion. In: Pesticide residues in food -
Report 1984, Rome, Food and Agriculture Organization of United
Nations, pp. 45-46 (FAO Plant Production and Protection Paper 62).
FAO/WHO (1985b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1984: The monographs, Rome, Food and Agriculture
Organization of United Nations, pp. 339-341, 645-648 (FAO Plant
Production and Protection Paper 67).
FAO/WHO (1986a) Fenitrothion. In: Pesticide residues in food -
Report 1986, Rome, Food and Agriculture Organization of United
Nations, p. 27, 61 (FAO Plant Production and Protection Paper 77).
FAO/WHO (1986b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1986: Part I. Residues, Rome, Food and Agriculture
Organization of United Nations, pp. 183-186 (FAO Plant Production
and Protection Paper 78).
FAO/WHO (1986c) Codex maximum limits for pesticide residues, 2nd
ed., Rome, Codex Alimentarius Commission, Food and Agriculture
Organization of the United Nations (CAC/Vol.XIII-Ed. 2).
FAO/WHO (1987a) Fenitrothion. In: Pesticide residues in food -
Evaluations 1986: Part II. Toxicology, Rome, Food and Agriculture
Organizations of United Nations, pp. 49-50 (FAO Plant Production and
Protection Paper 78/2).
FAO/WHO (1987b) Fenitrothion. In: Pesticide residues in food -
Report 1987, Rome, Food and Agriculture Organization of United
Nations, p. 26, 67 (FAO Plant Production and Protection Paper 84).
FAO/WHO (1988a) Fenitrothion. In: Pesticide residues in food.
Evaluations 1987: Part I. Residues, Rome, Food and Agriculture
Organization of United Nations, pp. 55-56 (FAO Plant Production and
Protection Paper, 86/1).
FAO/WHO (1988b) Fenitrothion. In: Pesticide residues in food -
Report 1988, Rome, Food and Agriculture Organization of United
Nations, p. 24 (FAO Plant Production and Protection Paper 92).
FAO/WHO (1989a) Fenitrothion. In: Pesticide residues in food -
Evaluations 1989: Part II. Toxicology, Rome, Food and Agriculture
Organization of the United Nations, pp. 31-34 (FAO Plant Production
and Protection Paper 93/2).
FAO/WHO (1989b) Fenitrothion. In: Pesticide residues in food -
Report 1989, Rome, Food and Agriculture Organization of the United
Nations, p. 28 (FAO Plant Production and Protection Paper 99).
FAO/WHO (1989c) Fenitrothion. In: Pesticide residues in food -
Evaluations 1989: Part I. Residues, Rome, Food and Agriculture
Organization of the United Nations, pp. 123-125 (FAO Plant
Production and Protection Paper 100).
FAO/WHO (1989d) Guide to Codex recommendations concerning pesticide
residues. Part 8. Recommendations for methods of analysis of
pesticide residues, 3rd ed., Rome, Codex Committee on Pesticide
Residues, Food and Agriculture Organization of the United Nations.
FAO/WHO (1990) In: Codex maximum limits for pesticide residues,
CAC/Supplement 2-1989 to FAO/WHO 1986c, Rome, Codex Alimentarius
Commission, Food and Agriculture Organization of the United Nations,
p. 1.
FERREIRA, J.R. & FERNANDES, A.M.S.S. (1980) Gas-liquid
chromatographic determination of organophosphorus insecticide
residues in fruits and vegetables. J. Assoc. Off. Anal. Chem., 63:
517-522.
FLANNAGAN, J.F. (1973) Field and laboratory studies of the effect of
exposure to fenitrothion on freshwater aquatic invertebrates.
Manitoba Entomol., 7: 15-25.
FORBIS, A.D. (1987) Acute toxicity of fenitrothion to Daphnia
magna, Osaka, Japan, Sumitomo Chemical Co., Ltd (Technical Report
No. HW-70-0236).
FUNCH, F.H. (1981) Analysis of residues of seven pesticides in some
fruits and vegetables by means of high pressure liquid
chromatography. Z. Lebensm. Unters. Forsch., 173: 95-98.
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol. appl.
Pharmacol., 14: 515-534.
GAJDUSKOVA, V. (1974) Dynamics of organophosphorus pesticide
residues in butter during manufacture and storage.
Milchwissenschaft, 29: 278-280.
GARDNER, A.M. (1971) Confirmation of organophosphorous pesticide
residues at nanogram levels by two dimensional thin layer
chromatography. J. Assoc. Off. Anal. Chem., 54: 517-524.
GARTRELL, M.J., CRAUN, J.C., PODREBARAC, D.S., & GUNDERSON, E.L.
(1985a) Pesticides selected elements, and other chemicals in adult
total diet samples, October 1979-September 1980. J. Assoc. Off.
Anal. Chem., 68: 1184-1195.
GARTRELL, M.J., CRAUN, J.C., PODREBARAC, D.S., & GUNDERSON, E.L.
(1985b) Pesticides, selected elements, and other chemicals in
infant and toddler total diet samples, October 1979-September 1980.
J. Assoc. Off. Anal. Chem., 68: 1163-1183.
GERMAIN, P. (1977) Songbird census in fenitrothion regime and 1977
forest spray program in New Brunswick. A final report to Forest
Protection Ltd and New Brunswick Department of Natural Resources,
Moncton, Canada, University of Moncton (Unpublished report).
GERMAIN, P. & MORIN, G. (1979) Forest songbird population, 1978
monitoring program in relation to aerial spray of insecticides
against spruce budworm. A final report to Forest Protection Ltd and
New Brunswick Department of Natural Resources, Moncton, Canada,
Avifauna Ltd.
GILLIS, G.F. (1978) Fish-growth rates. Benthic drift. Ottawa,
Environment Canada, Forestry Service, Department of Fisheries and
the Environment, 8 pp (Report M-X-87).
GILLIS, G.F. (1980) Assessment of the effects of insecticide
contamination in streams on the behaviour and growth of fish, 1979.
In: Environmental surveillance in New Brunswick 1978-1979, Moncton,
University of New Brunswick, pp. 49-50.
GINSBERG, G.L., PLACKE, M.E., WYAND, D.S., & COHEN, S.D. (1982)
Protection against acetaminophen-induced hepatotoxicity by prior
treatment with fenitrothion. Toxicol. appl. Pharmacol., 66: 383-399.
GOWDA, H. & SASTRY, M.S. (1979) Effect of O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphorothioate (Sumithion) on
reproductive performance in rats and oestrogenic activity in mice.
Indian J. Pharmacol., 11(4): 287.
GRAS, G. (1966) Toxicité du fénitrothion. Bull. Soc. Pharm.
Montpellier, 26: 375-404.
GREENHALGH, R. & MARSHALL, W.D. (1976) Ultraviolet irradiation of
fenitrothion and the synthesis of the photolytic oxidation products.
J. agric. food Chem., 24: 708-713.
GREENHALGH, R., DHAWAN, K.L., & WEINBERGER, P. (1980) Hydrolysis of
fenitrothion in model and natural aquatic systems. J. agric. food
Chem., 28: 102-105.
GRIGGS, L.M.P., JEFFERSON, N.D., BLAIR, N., KOPPLIN, J.R., RICHTER,
W.S., & SPICER, E.J.F. (1984) One-year dietary toxicity study in
dogs, Mattawan, Michigan, International Research and Development
Corporation (Unpublished report submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
GROVER, I.S. & MALHI, P.K. (1985) Genotoxic effects of some
organophosphorous pesticides I. Induction of micronuclei in bone
marrow cells in rat. Mutat. Res., 155: 131-134.
GRUE, C.E. (1982) Response of common grackles to dietary
concentrations of four organophosphate pesticides. Arch. environ.
Contam. Toxicol., 11: 617-626.
GUN, R.T., GRYCORCEWICZ, C., ESTERMAN, A.J., & EDWARDS, J.B. (1988)
Ultralow volume application of organophosphate concentrate in grain
terminals: A new occupational health hazard. Br. J. ind. Med., 45:
834-837.
HACHE, P., MARQUETTE, R., VOLPE, G., & MALLET, V.N. (1981) Fast
cleanup of difficult substrates for determination of fenitrothion
and some derivatives. J. Assoc. Off. Anal. Chem., 64: 1470-1473.
HALLETT, D.J., WEINBERG, P., & PRASAD, R. (1973) A preliminary study
on the fate of fenitrothion in forest seeds. I. Determination of
residues in eastern white pine (Pinus strobus L. Fr) and their
effects on amino acid metabolism, Ottawa, Environment Canada,
Forestry Service, Chemical Control Research Institute (Information
Report CC-X-50).
HALLETT, D.J., GREEENHALGH, R., WEINBERG, P., & PRASAD, R. (1974)
Fate of fenitrothion in forest trees. V. The formation of
metabolites in Pinus strobus L. and their detection by gas
chromatography and mass spectroscopy, Ottawa, Environment Canada,
Forestry Service, Chemical Control Research Institute (Information
Report CC-X-78).
HALLETT, D.J., GREENHALGH, R., WEINBERGER, P., & PRASAD, R. (1977)
The uptake and metabolism of fenitrothion by germinating white pine
spruce and yellow birch seeds. J. environ. Sci. Health, B12(1):
53-69.
HAMILTON, G.A., HUNTER, K., & RUTHVEN, A.D. (1981) Inhibition of
brain acetylcholinesterase activity in songbirds exposed to
fenitrothion during aerial spraying of forests. Bull. environ.
Contam. Toxicol., 27: 856-863.
HARA, S. & SUZUKI, T. (1981) Primary eye and skin irritation tests
of Sumithion Technical in rabbits (Unpublished Technical Report No.
HT-10-0201, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
HARA, M. & SUZUKI, H. (1982a) In vivo chromosomal aberration test
of Sumithion on bone marrow cells of mice (Unpublished Technical
Report No. HT-20-0235, submitted to WHO by Sumitomo Chemical Co.,
Ltd, Osaka, Japan).
HARA, M. & SUZUKI, H. (1982b) In vivo chromosomal aberration test
of sumithion on bone marrow cells of rats (Unpublished Technical
Report No. HT-30-0258, submitted to WHO by Sumitomo Chemical Co.,
Ltd, Osaka, Japan).
HARA, M. & SUZUKI, H. (1982c) The micronucleus test of Sumithion
(Unpublished Technical Report No. HT-20-0234, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
HARA, M., YAMAMOTO, K., KOGISO, S., & YOSHITAKE, A. (1988) In vivo
chromosomal aberration test of Sumithion in Chinese hamster ovary
cells (CHO-K1) in culture (Unpublished Technical Report No.
HT-80-0420, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
HARA, M., KOGISO, S., YAMADA, F., KAWAMOTO, M., YOSHITAKA, A., &
MIYAMOTO, J. (1989) Mutagenicity studies on fenitrothion in bacteria
and mammalian cells. Mutat. Res., 222: 53-61.
HASEGAWA, J., YASUNO, M., SAITO, K., NAKAMURA, Y., HATAKEYAMA, Y., &
SATO, H. (1982) Impact of temephos and fenitrothion on aquatic
invertebrates in a stream of Mt. Tsukuba. Jpn. J. Sanit. Zool., 33:
363-368.
HASHIMOTO, Y. & FUKAMI, J. (1969) Toxicity of orally and topically
applied pesticide ingredients to carp, Cyprimus carpio Linne.
Botyu-Kagaku, 34: 63-66.
HASHIMOTO, Y. & NISHIUCHI, Y. (1981) Establishment of bioassay
methods for evaluation of acute toxicity of pesticides to aquatic
organisms. J. pestic. Sci., 6: 257-264.
HASHIMOTO, Y., OKUBO, E., ITO, T., YAMAGUCHI, M., & TANAKA, S.
(1982) Changes in susceptibility of carp to several pesticides with
growth. J. pestic Sci., 7: 457-461.
HATAKEYAMA, S., SHIRAISHI, H., & KOBAYASHI, N. (1990) Effects of
aerial spraying of insecticides on nontarget macrobenthos in a
mountain stream. Ecotoxicol. environ. Saf., 19: 254-270.
HATFIELD, C.T. & ANDERSON, J.M. (1972) Effects of two insecticides
on the vulnerability of Atlantic salmon (Salmo salar) parr to
brook trout (Salvelinus fontinalis) predation. J. Fish Res. Board
Can., 29: 27-29.
HATFIELD, C.T. & RICHE, L.G. (1970) Effects of aerial Sumithion
spraying on juvenile Atlantic salmon (Salmo salar L.) and brook
trout (Salvelinus fontinalis Mitchill) in Newfoundland. Bull.
environ. Contam. Toxicol., 5: 440-442.
HATTORI, K. (1974) [Toxicological studies on the influence of
chemicals to the birds (part 2). Oral acute toxicity of three
organophosphate insecticides in common pigeon.] Hokkaidoritsu
Eiseikenkyuushoho, 24: 154 (in Japanese).
HATTORI, K., SATO, H., TSUCHIYA, K., YAMAMOTO, N., & OGAWA, E.
(1974) [Toxicological studies on influences of chemicals to the
birds (Part 1). Oral acute toxicity and cholinesterase inhibition of
three organophosphate insecticides in Japanese quail.] Hokkaidoritsu
Eiseikenkyuushoho, 24: 35-38 (in Japanese).
HAYASHI, M., KISHI, M., SOFUNI, T., & ISHIDATE, M. (1988)
Micronucleus test in mice on 39 food additives and eight miscellaneous
chemicals. Food chem. Toxicol., 26: 487-500.
HAYES, W.J., Jr (1982) Fenitrothion. In: Pesticides studies in man,
Baltimore, Maryland, Williams & Wilkins Company, pp. 367-372.
HAYES, W.J., Jr & LAWS, E.R., Jr (1991) Fenitrothion. In: Handbook
of pesticide toxicology, New York, London, Academic Press, pp.
1020-1023.
HILL, E.F., HEALTH, R.G., SPANN, J.W., & WILLIAMS, J.D. (1975)
Lethal dietary toxicities of environmental pollutants to birds,
Washington, DC, US Department of the Interior, Fish and Wildlife
Service, 61 pp (Special Scientific Report, Wildlife No. 191).
HIRAYAMA, K. & TAMANOI, S. (1980) Acute toxicity of MEP and diazinon
(pesticide) to larvae of kuruma prawn Penaeus japonicus and of
swimming crab Portunus trituberculatus. Bull. Jpn. Soc. Sci.
Fish., 46: 117-123.
HIROSE, K., YAMAZAKI, M., & ISHIKAWA, A. (1979) Effects of water
temperature on median lethal concentration (LC50) of a few
pesticides to seawater teleost. Bull. Tokai Reg. Fish. Res. Lab.,
98: 45-53.
HLADKA, A., KRAMPL, V., & KOVAC, J. (1974) Effect of malathion on
the content of fenitrothion and fenitrooxon in the rat. Bull.
environ. Contam. Toxicol., 12: 38-45.
HLADKA, A., BATORA, V., KOVACICOVA, J., & ROSIVAL, L. (1977) Effets
sur la santé professionnelle et importance toxicologique du
fénitrothion et de son contaminant (S-méthyl-fénitrothion) dans la
toxicologie de ses formulations. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, p. 416
(Publication NRCC No. 16073).
HOBERMAN, A.M. (1990) Reproductive effects of Sumithion administered
orally in feed to Crl: CDR(SD)BR rats for two generations, Argus
Research Laboratories, Inc. (Unpublished Technical Report No.
HT-10-0452 submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
HOLLINGWORTH, R.M. (1969) Dealkylation of organophosphorus esters by
mouse liver enzymes in vitro and in vivo. J. agric. food Chem., 17:
987-996.
HOLLINGWORTH, R.M., METCALF, R.M., & FUKUTO, T.R. (1967a) The
selectivity of Sumithion compared with methylparathion. The
metabolism in the white mouse. J. agric. food Chem., 15: 242-249.
HOLLINGWORTH, R.M., FUKUTO, T.R., & METCALF, R.L. (1967b)
Selectivity of Sumithion compared with methylparathion. Influence of
structure on anticholinesterase activity. J. agric. food Chem., 15:
235-241.
HOLMES, S.B. (1979) Aquatic impact studies of a spruce budworm
Choristoneura fumiferana clemens, control program in the lower St.
Lawrence region of Quebec in 1978, Ottawa, Forest Pest Management
Institute (Report No. FPM-X-26).
HOLMES, S.B., KINGSBURY, P.D., MAMARBACHI, G., & MATHIEU, P. (1984)
Distribution of fenitrothion residues in brook trout (Salvelinus
fontinalis) and lake trout (Salvelinus namaycush) tissues
following aerial applications to Lac Ste-Anne, Quebec. Bull.
environ. Contam. Toxicol., 33: 468-475.
HOSOKAWA, S. & MIYAMOTO, J. (1974) Metabolism of C-14 labelled
sumithion, O,O-dimethyl O-(3-methyl-4-nitrophenyl)-
phosphorothioate in apple. Botyu-Kagaku, 39: 49-53.
HOSOKAWA, S. & MIYAMOTO, J. (1975) Effects of subacute feeding of
Sumithion, Sumioxon and p-nitrocresol on rat hepatic oxidative
phosphorylation and mixed function oxidase. Botyu-Kagaku, 40: 33-38.
HUDSON, R.H., HAEGELE, M.A., & TUCKER, R.K. (1979) Acute oral and
percutaneous toxicity of pesticides to mallards: correlation with
mammalian toxicity data. Toxicol. appl. Pharmacol., 47: 451-460.
ITO, M., TAKAHASHI, M., & MIKAMI, N. (1988) Hydrolysis of
fenitrothion in water as a function of pH at 25 °C, Osaka, Japan,
Sumitomo Chemical Co., Ltd (Unpublished Technical Report No.
HM-80-0094).
IWATA, Y., SUGITANI, A., & YAMADA, F. (1981) An analytical method
for organophosphorus pesticide in onion. Shokuhin Eisei Zasshi, 22:
484-489.
JAPAN PLANT PROTECTION ASSOCIATION (1984) [Production statistics of
major pesticides.] In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 49 (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1986) [Production statistics of
major pesticides]. In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 55 (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1988) [Production statistics of
major pesticides.] In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 57 (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1989) [Production statistics of
major pesticides.] In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 60 (in Japanese).
JOHNSON, J.C., Jr & BOWMAN, M.C. (1972) Responses from cows fed
diets containing fenthion or fenitrothion. J. dairy Sci., 55:
777-782.
JOHNSTON, J.J. & CORBETT, M.D. (1985) The effects of temperature,
salinity and simulated tidal cycle on the toxicity of fenitrothion
to Callinectes sapidus. Comp. Biochem. Physiol., 80C: 145-149.
JOHNSTON, J.J. & CORBETT, M.D. (1986) The uptake and in vivo
metabolism of the organophosphate insecticide fenitrothion by the
blue crab, Callinectes sapidus. Toxicol. appl. Pharmacol., 85:
181-188.
KADOTA, T. & MIYAMOTO, J. (1975) Acute and sub-acute toxicity of
sumithion in Japanese quails. Botyu-Kagaku, 40: 54-58.
KADOTA, T., KAGOSHIMA, M., YAMAZAKI, H., & MIYAMOTO, J. (1972) Acute
oral, subcutaneous and dermal toxicities of Sumithion technical in
mice and rats (Unpublished Technical Report No. HT-20-0187,
submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
KADOTA, T., KOHDA, H., & MIYAMOTO, J. (1975a) Subchronic toxicity
studies of Sumithion, Sumioxon and p-nitrocresol in rats and
92-week feeding study of Sumithion with special reference to change
of cholinesterase activity. Botyu-Kagaku, 40: 38-48.
KADOTA, T., OKUNO, Y., & MIYAMOTO, J. (1975b) Acute oral toxicity
and delayed neurotoxicity of 5 organophosphorus compounds,
Salithion, Cyanox, Surecide, Sumithion an Sumioxon in adult hens.
Botyu-Kagaku, 40: 49-53.
KALAS, D. (1978) [Light group poisoning by metathion E-50 inhalation
when spraying by means of aircraft.] Prac. Lek., 30: 145-147 (in
Czech).
KANAZAWA, J. (1975) Uptake and excretion of organophosphorus and
carbamate insecticides by fresh water fish, Motsugo. Bull. environ.
Contam. Toxicol., 14: 346-352.
KANAZAWA, J. (1977) [Analytical chemistry related to evaluation of
the effects of pesticides on aquatic organisms.] In: [Proceeding of
a Symposium on the Effects of Pesticides on Aquatic Organisms and
the Methods for their Evaluation, 29-30 November, 1977], Scientist
Co., pp. 4/1-4/34 (in Japanese).
KANAZAWA, J. (1981) Measurement of the bioaccumulation factors of
pesticides by freshwater fish and their correlation with
physicochemical properties or acute toxicities. Pestic. Sci., 12:
417-424.
KANNAN, N. & JAYARNAMA, J. (1981) Impact monitoring of residues in
tomato. Int. J. environ. anal. Chem., 9: 145-151.
KANOH, S., KAWASAKI, H., YOSHIDA, M., & NISHIO, A. (1982) Studies on
chronic toxicity of low levels of O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphorothionate (Sumithion) in the rat.
J. toxicol. Sci., 7: 43-50.
KASHI, K.P. (1972) Tolerance of silkworm (Bombyx mori L.) larvae
to organophosphorous insecticides. J. Seric. Sci. Jpn, 41: 301-304.
KATAGI, T., TAKAHASHI, N., & MIKAMI, N. (1988) Photodegradation of
fenitrothion in water (Unpublished Technical Report No. HM-80-0093,
submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
KAWACHI, T. (1978) [Development of screening method for carcinogen
including mutagenicity tests.] In: [Annual Report of Cancer
Research], Tokyo, Ministry of Health and Welfare, pp. 803-820 (in
Japanese).
KAWAMOTO, M., HARA, M., KOGISO, S., & YOSHITAKE, A. (1989) In
vivo/in vitro unscheduled DNA synthesis (UDS) test of Sumithion
in rat hepatocytes (Unpublished Study No. 1487, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
KETTELA, E.G. & VARTY, I.W. (1972) Assessment of the 1971 spruce
budworm aerial spraying program in New Brunswick and forecast for
1972, Fredericton, New Brunswick, Canada, Maritimes Forest Research
Centre (Information Report M-X-29).
KEVAN, P.G. (1975) Forest application of the insecticide
fenitrothion and its effect on wild bee pollinators ( Hymenoptera:
Apoidea) of lowbush blueberries (Vaccinium SPP) in southern New
Brunswick, Canada. Biol. Conserv., 7: 301-309.
KIKUCHI, R., YASUTANIYA, T., TAKIMOTO, Y., YAMADA, H., & MIYAMOTO,
J. (1984) Accumulation and metabolism of fenitrothion in three
species of algae. J. pestic. Sci., 9: 331-337.
KIMMERLE, G. (1962a) [Toxicological study on additive S-5660],
Toxicology and Occupational Health Laboratory (Unpublished report
submitted to WHO by Bayer AG, Germany) (in German).
KIMMERLE, G. (1962b) [Neurotoxic effect of S-5660 on hens],
Toxicology and Occupational Health Laboratory (Unpublished report
submitted to WHO by Bayer AG, Germany) (in German).
KINGSBURY, P.D. (1976) A history of the effects of aerial forest
spraying in Canada on aquatic fauna, Ottawa, Chemical Contamination
Research Institute (Information Report No. CC-X-117).
KINGSBURY, P.D. (1978) A study of the distribution, persistence and
biological effects of fenitrothion applied to a small lake in and
oil formulation, Ottawa, Forest Pest Management Institute (Report
No. FPM-X-13).
KLAVERKAMP, J.F., DUANGSAWASDI, M., MACDONALD, W.A., & MAJEWSKI,
H.S. (1977) An evaluation of fenitrothion toxicity in four life
stages of rainbow trout, Salmo gairdneri, Philadelphia, American
Society for Testing and Materials, pp. 231-240 (Special Technical
Publication No. 77).
KLEINER, C.F., ANDERSON, R.L., & TANNER, D.K. (1984) Toxicity of
fenitrothion to fathead minnows (Pimephales promelas) and
alternative exposure duration studies with fenitrothion and
endosulfan. Arch. environ. Contam. Toxicol., 13: 573-578.
KLIMMER, O.R. (1961) Opinion on the toxicity of the compound ``S
5660'' of Farbenfabriken Bayer AC., Leverkusen, Bonn, Germany,
Pharmakologisches Institut der Rheinischen
Friedrich-Wilhelms-Universitat (Unpublished report submitted to WHO
by Farbenfabriken Bayer AG, Germany).
KOBAYASHI, H., KOJIMA, M., MATANO, O., & GOTO, S. (1979) Simple
multi-residue analysis of organophosphorus pesticides by mass
chromatography. J. pestic. Sci., 4: 463-472.
KOBAYASHI, H., YUYAMA, A., KUDO, M., & MATSUSAKA, N. (1983) Effects
of organophosphorus compounds, O,O-dimethyl
O-(2,2-dichlorovinyl)phosphate (DDVP) and O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphorothioate (fenitrothion), on
brain acetylcholine content and acetylcholinesterase activity in
Japanese quail. Toxicology, 28: 219-227.
KODAMA, T. & KUWATSUKA, S. (1980) Factors for the persistence of
parathion, methyl parathion, and fenitrothion in sea water. J.
pestic. Sci., 5: 351-355.
KOHDA, H. & KADOTA, T. (1975) Dominant lethal test of Sumithion in
rats and mice, Osaka, Japan, Sumitomo Chemical Co., Ltd (Technical
Report No. HT-60-0210).
KOHDA, H. & KADOTA, T. (1979) Acute inhalation toxicity study of
Sumithion in rats (Unpublished Technical Report No. HT-90-0085A,
submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
KOHDA, H., KAGOSHIMA, M., KADOTA, T., & MIYAMOTO, J. (1972) Skin
sensitization test of Sumithion Technical in guinea-pigs
(Unpublished Technical Report No. HT-00-0181, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
KOVAC, J. & SOHLER, E. (1965) [Determination of Fenitrothion
residues in fruits and vegetables after separation of coloured
co-extractives by thin-layer chromatography.] Z. anal. Chem., 208:
201-204 (in German).
KOVACICOVA, J., BATORA, V., & TRUCHLIK, S. (1973) Hydrolysis rate
and in vitro anticholinesterase activity of fenitrothion and
S-methyl Fenitrothion. Pestic. Sci., 4: 759-763.
KRZYMIEN, M. (1979) Determination of fenitrothion concentration in
ambient air, Ottawa, National Research Council of Canada, National
Aeronautical Establishment, 18 pp (Report LTR-UA-49).
KURIBAYASHI, S. (1981) Studies on the effect of pesticides on the
reproduction of the silkworm, Bombyx mori L. I. Effects of
chemicals administered during the larval stage on egg-laying and
hatching. J. toxicol. Sci., 16: 169-176.
KUROIWA, Y. & YAMAMOTO, T. (1977) [Toxicological study of
fenitrothion to rat. Comparative study with methylparathion.] In:
[Abstract papers of 9th Symposium on Drug Metabolism and Action],
Kumamoto, Pharmaceutical Society of Japan, pp. 137-140 (in
Japanese).
KWASNIEWSKA, K., LIU, D., & STRACHAN, W.M.J. (1980) The relative
biological toxicity effectiveness of chemical toward microorganisms.
Trace Subst. environ. Health, 14: 470-477.
LADD, R., JENKINS, D.H., WRIGHT, P.L., & KEPLINGER, M.L. (1971)
Teratogenic study with Sumithion technique in albino rabbits,
Northbrook, Illinois, Industrial Bio-Test Laboratories (Unpublished
report No. J9996, submitted to WHO by Sumitomo Chemical Co., Ltd,
Osaka, Japan).
LAKSHMINARAYAMA, J.S.S. & BOUQUE, H. (1980) Absorption of
fenitrothion by plankton and benthic algae. Bull. environ. Contam.
Toxicol., 24: 389-396.
LAL, S., LAL, R., & SAXENA, D.M. (1987) Bioconcentration and
metabolism of DDT, fenitrothion and chlorpyrifos by the blue-green
algae Anabaena sp. and Aulosira fertilissima. Environ. Pollut.,
46: 187-196.
LAPIERRE, L.E. (1985) Persistence of fenitrothion insecticide in
poplar Populus tremuloidus and gray birch Betula populifolia.
Bull. environ. Contam. Toxicol., 35: 471-475.
LEBEL, G.L., WILLIAMS, D.T., GRIFFITH, G., & BENOIT, F.M. (1979)
Isolation and concentration of organophosphorus pesticides from
drinking water at the ng/litre level, using macro-reticular resin.
J. Assoc. Off. Anal. Chem., 62: 241-249.
LEHOTZKY, K., SZEBERENYI, M.J., & KISS, A. (1989) Behavioral
consequences of prenatal exposure to the organophosphate insecticide
sumithion. Neurotoxicol. Teratol., 11: 321-324.
LEONARD, D.E. (1971) Effects of accothion on parasites and
predators. In: Nash, R.W., Peterson, J.W., & Chansler, J.F., ed.
Environmental studies of accothion for spruce budworm control,
Washington, DC, US Department of Agriculture and US Department of
Interior, p. 46.
LEUCK, D.B. & BOWMAN, M.C. (1969) Persistence of O,O-dimethyl
O-4-nitro- m-tolyl phosphorothioate, its oxygen analogue, and its
cresol in corn and grass forage. J. econ. Entomol., 62: 1282-1285.
LEUCK, D.B., JOHNSON, J.C., Jr, BOWMAN, M.C., KNOX, F.E., & BEROZA,
M. (1971) Fenitrothion residues in corn silage and their effects on
dairy cows. J. econ. Entomol., 64: 1394-1399.
LISKA, D., KOLESAR, D., HLADKA, A., VALKYOVA, L., & RAISKUP, J. CH.
(1982) Clinical and laboratory findings in Metation EK-50. Czech.
Med., 5: 146-154.
LIU, D., STRACHAN, W.M., THOMSON, K., & KWASNIEWSKA, K. (1981)
Determination of the biodegradability of organic compounds. Environ.
Sci. Technol., 15: 788-793.
LOCKHART, W.L., FLANNAGAN, J.F., MOODY, R.P., WEINBERGER, P., &
GREENHALGH, R. (1977) Fenitrothion monitoring in southern Manitoba.
In: Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 233-252 (Publication No. NRCC No.
16073).
LOCKHART, W.L., METNER, D.A., & GRIFT, T. (1973) Biochemical and
residue studies on rainbow trout (Salmo gairdneri) following field
and laboratory exposure to fenitrothion. Manitoba Entomol., 7:
26-36.
MACDONALD, J.R. & PENNEY, G.H. (1969) Preliminary report on the
effects of the 1969 New Brunswick forest spraying on juvenile salmon
and their food organisms, Halifax, Department of Fisheries and
Forestry of Canada, Resource Development Branch, 17 pp (Manuscript
report No. 69-4).
MCLEESE, D.W. (1974) Olfactory response and fenitrothion toxicity in
American lobsters (Homarus americanus). J. Fish. Res. Board Can.,
31: 1127-1131.
MCLEESE, D.W., ZITKO, V., & SERGEANT, D.B. (1979) Uptake and
excretion of fenitrothion by clams and mussels. Bull. environ.
Contam. Toxicol., 22: 800-806.
MCNEIL, J.N. & MCLEOD, J.M. (1977) Apparent impact of fenitrothion
on the swaine Jack-Pine. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, p. 203
(Publication NRCC No. 16073).
MCNEIL, J.N., GREENHALGH, R., & MCLEOD, J.M. (1979) Persistence and
accumulation of fenitrothion residues in jack pine foliage and their
effect on the swaine jack pine sawfly, Neodiprion Swainei
(Hymenoptera: Diprionidae). Entomol. Soc. Am., 8: 752-755.
MADHU, C.H., RAO, K.J., RAO, K.S., & RAO, K.V.R. (1982) Comparative
evaluation of the toxicity of lindane and sumithion on freshwater
snail, Pila globosa (Swainson). Geobios, 9: 181-183.
MAEDA, T., KAWASHIMA, M., OGASAWARA, Y., & TSUJI, K. (1982) Thermal
decomposition of O,O-dimethyl
O-(3-methyl-4-nitrophenol)-phosphorothioate-Autocatalytic
decomposition mechanism. J. pestic. Sci., 7: 341-347.
MAFF (MINISTRY OF AGRICULTURE, FISHERIES AND FOOD, UK) (1986) Food
surveillance paper No. 16, London, Her Majesty's Stationery Office
(HMSO), pp. 19-20.
MAFF (MINISTRY OF AGRICULTURE, FISHERIES AND FOOD, UK) (1989) Food
surveillance paper No. 25, London, Her Majesty's Stationery Office
(HMSO), pp. 12-34.
MAGUIRE, R.J. & HALE, E.J. (1980) Fenitrothion sprayed on a pond:
Kinetics of its distribution and transformation in water and
sediment. J. agric. food Chem., 28: 372-378.
MALHI, P.K. & GROVER, I.S. (1987) Genotoxic effects of some
organophosphorus pesticides II. In vivo chromosomal aberration
bioassay in bone marrow cells in rat. Mutat. Res., 188: 45-51.
MALLET, V.N. (1980) A chemical residue survey in relation to the
1980 spruce budworm spray program in New Brunswick, Moncton, Canada,
University of Moncton, Chemistry Department, 54 pp (Report No.
80-CP-18).
MALLET, V.N. & CASSISTA, A. (1984) Fenitrothion residue survey in
relation to the 1981 spruce budworm spray program in New Brunswick,
Canada. Bull. environ. Contam. Toxicol., 32: 65-74.
MALLET, V.N. & VOLPE, G. (1982) A chemical residue survey in
relation to the 1980 spruce budworm spray program in New Brunswick
(Canada). J. environ. Sci. Health, B17: 715-736.
MALLET, V.N., BRUN, G.L., MACDONALD, R.N., & BERKANE, K. (1978) A
comparative study of the use of XAD-2 resin and the conventional
serial solvent extraction procedure for the analysis of fenitrothion
and some derivatives in water preservation techniques. J.
Chromatogr., 160: 81-88.
MANDOUL, R., DUBOS, M., DECOMMAND, M., & MOULINIER, C.L. (1968)
Effets in vitro d'organo-phosphorés sur l'huître portugaise,
quelques mollusques dulçaquicoles et le micro-plancton d'eau douce.
Bull. Soc. Pathol. exot., 60: 568-580.
MANI, K. & SAXENA, P.K. (1985) Effect of safe concentrations of some
pesticides on ovarian recrudescence in the freshwater murrel,
Channa punctatus (BI): A quantitative study. Ecotoxicol.
environ. Saf., 9: 241-249.
MARSHALL, W.K. & ROBERTS, J.R. (1977) Simulation modelling of the
distribution of pesticides in ponds. In: Proceedings of a Symposium
on Fenitrothion, Ottawa, National Research Council of Canada, pp.
253-278 (Publication NRCC No. 16073).
MARTIN, H. & WORTHING, C.R. (1981) The pesticide manual, 5th ed.,
Croydon, The British Crop Protection Council, p. 267.
MARTIN, N.A. & YEATES, G.W. (1975) Effect of four insecticides on
the pasture ecosystem., III. Nematodes, rotifers, and tardigrades.
N.Z. J. agric. Res., 18: 307-312.
MARTINDALE, R.W. (1988) Determination of residues of a range of
fungicides, anti-sprouting agents and (organochlorine and
organophosphorus) insecticides. Analyst, 113: 1229-1233.
MARTINEZ CHUECOS, J. & SOLE VIOLAN, J. (1985) Letter to editor
concerning the article "Delayed neurotoxicity produced by an
organophosphorus compound (Sumithion)" by Sakamoto et al. Arch.
Toxicol., 58: 123-124.
MASTALSKI, K. (1971a) Acute oral toxicity study with p-nitro
metacresol in beagle dogs, Northbrook, Illinois, Industrial Bio-Test
Laboratories (Unpublished report No. J9994, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
MASTALSKI, K. (1971b) Acute oral toxicity study with Sumithion
technical grade in beagle dogs, Northbrook, Illinois, Industrial
Bio-Test Laboratories (Unpublished report No. J9994, submitted to
WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
MATSUBARA, T. & HORIKOSHI, I. (1983) Possible antidotes in severe
intoxication with fenitrothion in rats. J. Pharm. Dyn., 6: 699-707.
MATSUSHITA, T., AOYAMA, K., YOSHIMI, K., FUJITA, Y., & UEDA, A.
(1985) Allergic contact dermatitis from organophosphorus
insecticides. Ind. Health, 23: 145-153.
MAYER, F.L., Jr (1987) Acute toxicity handbook of chemicals to
estuarine organisms, Gulf Breeze, Florida, US Environmental
Protection Agency, Gulf Breeze Laboratory (EPA 600/8-87/017).
MEISTER, R.T., BERG, G.L., SINE, C., POPLYK, J., & SCHILLING, G.
(1985) Fenitrothion. In: Farm chemicals handbook, Willoughby,
Delaware, Meister Publishing Co., pp. C103-C104.
MENNA, J.H. (1985) Effect of emulsifiers on influenza type A virus
infection; in vivo and in vitro studies. Toxicol. environ.
Health, 16: 441-448.
MESTRES, R., TOURTE, J., CAMPO, M., & ILLES, S. (1974) Résidus de
pesticides. XXV-Méthode polyvalente de recherche et de dosage des
résidus de pesticides divers dans les matières alimentaires. Ann.
Fals. Exp. Chim., 1974: 513-526, 721-722.
METCALF, C.D., MCLEESE, D.W., & ZITKO, V. (1980) Rate of
volatilization of fenitrothion from fresh water. Chemosphere, 9:
151-155.
MIHARA, K., OKUNO, Y., MISAKI, Y., & MIYAMOTO, J. (1978) Metabolism
of fenitrothion in goats. J. pestic. Sci., 3: 233-242.
MIHARA, K., MISAKI, Y., & MIYAMOTO, J. (1979) Metabolism of
fenitrothion in birds. J. pestic. Sci., 4: 175-185.
MIHARA, K., ISOBE, N., OHKAWA, H., & MIYAMOTO, J. (1981) Effects of
organophosphorus insecticides on mitochondrial and microsomal
functions in the liver of rat with special emphasis on fenitrothion.
J. pestic. Sci., 6: 307-316.
MIKAMI, N., IMANISHI, K., YAMADA, H., & MIYAMOTO, J. (1985a)
Photodegradation of fenitrothion in water and soil surface, and its
hydrolysis in water. J. pestic. Sci., 10: 263-272.
MIKAMI, N., SAKATA, S., YAMADA, H., & MIYAMOTO, J. (1985b) Further
studies on degradation of fenitrothion in soils. J. pestic. Sci.,
10: 491-500.
MILLER, C.A., STEWART, J.F., ELGEE, D.E., SHAW, D.D., MORGAN, M.G.,
KETTELA, E.G., GREENBANK, D.O., GRESNER, G.N., & VARTY, I.W. (1973)
Aerial spraying against spruce budworm adults in New Brunswick. A
compendium of reports on the 1972 test program, Fredericton, Canada,
Maritimes Forest Research Centre (Information Report M-X-38).
MILLER, S. & SHAH, M.A. (1982) Cholinesterase activities of workers
exposed to organophosphorus insecticides in Pakistan and Haiti and
an evaluation of the tintometric method. J. environ. Sci. Health,
B17(2): 125-142.
MISU, Y., SEGAWA, T., KURUMA, I., KOJIMA, M., & TAKAGI, H. (1966)
Subacute toxicity of O,O-dimethyl- O-(3-methyl-4-nitrophenyl)
phosphorothioate (Sumithion) in the rat. Toxicol. appl. Pharmacol.,
9: 17-26.
MIYAMOTO, J. (1964a) Studies on the mode of action of
organophosphorus compounds. Part III. Activation and degradation of
Sumithion and methylparathion in vivo. Agric. biol. Chem., 28:
411-421.
MIYAMOTO, J. (1964b) Studies on the mode of action of
organophosphorus compounds. IV. Penetration of Sumithion,
methylparathion and their oxygen analogs into guinea-pig brain and
inhibition of cholinesterase in vivo. Agric. biol. Chem., 28:
422-430.
MIYAMOTO, J. (1969) Mechanism of low toxicity of Sumithion towards
mammals. Residue Rev., 25: 251-264.
MIYAMOTO, J. (1977a) Degradation of fenitrothion in terrestrial and
aquatic environments including photolytic and microbial reactions.
In: Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 105-109 (Publication NRCC No.
16073).
MIYAMOTO, J. (1977b) The implication of recent long-term
toxicological studies of fenitrothion on birds. In: Proceedings of a
Symposium on Fenitrothion, Ottawa, National Research Council of
Canada, pp. 307-319 (Publication NRCC No. 16073).
MIYAMOTO, J. (1977c) Long-term toxicological effects of fenitrothion
in mammals including carcinogenicity and mutagenicity. In:
Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 459-496 (Publication NRCC No.
16073).
MIYAMOTO, J. & KADOTA, T. (1972) Toxicological studies with
Sumithion (Unpublished report submitted to WHO by Sumitomo Chemical
Co., Ltd, Osaka, Japan).
MIYAMOTO, J. & SATO, Y. (1969) Determination of insecticide residue
in animal and plant tissues. VI. Determination of sumithion residue
in cattle tissues. Botyu-Kagaku, 34: 3-6.
MIYAMOTO, J., SATO, Y., KADOTA, T., FUJINAMI, A., & ENDO, M. (1963a)
Studies on the mode of action of organophosphorus compounds. Part I.
Metabolic fate of 32P-labelled Sumithion and methylparathion in
guinea-pigs and white rats. Agric. biol. Chem., 27: 381-389.
MIYAMOTO, J., SATO, Y., KADOTA, T., & FUJINAMI, A. (1963b) Studies
on the mode of action of organophosphorus compounds. Part II.
Inhibition of mammalian cholinesterase in vivo following
administration of Sumithion and methylparathion. Agric. biol. Chem.,
27: 669-676.
MIYAMOTO, J., SATO, Y., & SUZUKI, S. (1967) Determination of
insecticide residue in animal and plant tissue. IV. Determination of
residual amount of sumithion and some of its metabolites in fresh
milk. Botyu-Kagaku, 32: 95-100.
MIYAMOTO, J., SATO, Y., YAMAMOTO, K., & SUZUKI, S. (1968) Activation
and degradation of Sumithion, methylparathion and their oxygen
analogs by mammalian enzymes in vitro. Botyu-Kagaku, 33: 1-6.
MIYAMOTO, J., KOHDA, H., & KADOTA, T. (1975) Teratogenic studies
with Sumithion in ICR-JCL mice and Sprague-Dawley rats (Unpublished
report submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
MIYAMOTO, J., HOSOKAWA, S., KADOTA, T., KOHDA, H., ARAI, M.,
SUGIHARA, S., & HIRAO, K. (1976a) Studies on cholinesterase
inhibition and structural changes at neuromuscular junctions in
rabbits by subacute administration of Sumithion. J. pestic. Sci., 1:
171-178.
MIYAMOTO, J., MIHARA, K., & HOSOKAWA, S. (1976b) Comparative
metabolism of m-methyl-carbon-14-Sumithion in several species of
mammals in vivo. J. pestic. Sci., 1: 9-21.
MIYAMOTO, J., MIHARA, K., KADOTA, T., & OKUNO, Y. (1977a) Toxicity
and metabolism in vivo of fenitrothion in rats with experimental
hepatic lesion. J. pestic. Sci., 2: 271-277.
MIYAMOTO, J., OKUNO, Y., KADOTA, T., & MIHARA, K. (1977b)
Experimental hepatic lesions and drug metabolizing enzymes in rats.
J. pestic. Sci., 2: 257-269.
MIYAMOTO, J., MIKAMI, N., MIHARA, K., TAKIMOTO, Y., KOHDA, H., &
SUZUKI, H. (1978) Biological activity of fenitrothion and its
degradation products. J. pestic. Sci., 3: 35-41.
MIYAOKA, T., TAKAHASHI, H., TSUDA, S., & SHIRASU, Y. (1983) Effect
of O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate
(Fenitrothion) treatment on acute toxicity of 2-sec-butylphenyl
methylcarbamate (BPMC) in dogs. Jpn. J. vet. Sci., 45: 463-470.
MIYASHITA, M. (1984) Effects of a short period exposure of
fenitrothion on the reproduction of pregnant females of the guppy,
Poecilia reticulata. Jpn. J. Hyg., 39: 647-650.
MOLLHOFF, E. (1967) Gas-chromatographic determination of residues of
E 605 products and Agritox in plants and soil samples.
Pflanzenschutz-Nachr. "Bayer", 20: 557-574.
MOODY, R.P. & FRANKLIN, C.A. (1987) Percutaneous absorption of the
insecticides fenitrothion and aminocarb in rats and monkeys. J.
Toxicol. environ. Health, 20: 209-218.
MOODY, R.P., PRASAD, R., GREENHALGH, R., & WEINBERGER, P. (1977)
Translocation of ring labelled 14C-fenitrothion in conifers. In:
Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 217-231 (Publication NRCC No.
16073).
MOODY, R.P., GREENHALGH, R., LOCKHART, L., & WEINBERGER, P. (1978)
The fate of fenitrothion in an aquatic ecosystem. Bull. environ.
Contam. Toxicol., 19: 8-14.
MOODY, R.P., CARROLL, J.M., & KRESTA, A.M.E. (1987a) Automated high
performance liquid chromatography and liquid scintillation counting
determination of pesticide mixture octanol/water partition rates.
Toxicol. ind. Health, 3: 479-490.
MOODY, R.P., RIEDEL, D., RITTER, L., & FRANKLIN, C.A. (1987b) The
effect of DEET ( N,N-Diethyl- m-toluamide) on dermal persistence
and absorption of the insecticide fenitrothion in rats and monkeys.
J. Toxicol. environ. Health, 22: 471-479.
MORGAN M.J. & KICENIUK, J.W. (1990) Effect of fenitrothion on
foraging behavior of juvenile atlantic salmon. Environ. Toxicol.
Chem., 9: 489-495.
MORIN, R., GABCURY, G., & MANARBACHI, G. (1986) Fenitrothion and
aminocarb residues in water and balsam fir foliage following spruce
budworm spraying programs in Quebec, 1979-1982. Bull. environ.
Contam. Toxicol., 36: 622-628.
MORIYA, M., OHTA, T., WATANABE, K., MIYAZAWA, T., KATO, K., &
SHIRASU, Y. (1983) Further mutagenicity studies on pesticides in
bacterial reverse assay systems. Mutat. Res., 116: 185-216.
MORRISON, B.R.S. & WELLS, D.E. (1981) The fate of fenitrothion in a
stream environment and its effect on the fauna, following aerial
spraying of a Scottish forest. Sci. total Environ., 19: 233-252.
MORSETH, S.L., SERABIAN, M.A., LICHTENBERGER, J.M., VARGAS, K.J.,
THAKUR, A.K., & BURLEW, P.L. (1986) Teratology study in rabbits.
Fenitrothion T.G. (Sumithion): Final report, Vienna, Virginia,
Hazleton Laboratories (Technical Report No. HT-71-0382 (Project No.
343-914), submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
MORSETH, S.L., LANDES, S.G., LEWIS, S.A., THAKUR, A.K., &
LICHTENBERGER, J.M. (1987) Teratology study in rats with Sumithion,
Vienna, Virginia, Hazleton Laboratories (Technical Report No.
HT-61-0367 (Project No. 343-182), submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
MOTABAR, M., SANAI, G.H., & HEIDAI, A.A. (1972) Toxicological
evaluation of Sumithion on operators and inhabitants in the Mamasani
area, southern Iran. Iran. J. Pharm. Health, 2: 40-49.
MUNNECKE, D.M. (1976) Enzymatic hydrolysis of organophosphate
insecticides, a possible pesticide disposal method. Appl. environ.
Microbiol., 32: 7-13.
MURTY, A.S., RAJABHUSHANAM, B.R., RAMANI, A.V., & CHRISTOPHER, K.
(1983) Toxicity of fenitrothion to the fish Mystus cavasius and
Labeo rohita. Environ. Pollut., 30A: 225-232.
NAGAYAMA, T., MAKI, T., KIN, K., MAKI, M., & NISHIMA, T. (1986)
Survey of pesticide residues in vegetables and fruits. Annu. Rep.
Tokyo Metrop. Res. Lab. P.H., 37: 173-183.
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., & NISHIMA, T. (1987)
Survey of pesticide residues in vegetables and fruits. Annu. Rep.
Tokyo Metrop. Res. Lab. P.H., 38: 222-228.
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1988) Survey of pesticide residues in vegetables and fruits. Annu.
Rep. Tokyo Metrop. Res. Lab. P.H., 39: 130-138.
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1989) Survey of pesticide residues in vegetables and fruits. Annu.
Rep. Tokyo Metrop. Res. Lab. P.H., 40: 155-162.
NAIR, G.A. (1981) Toxic effects of certain biocides on a fresh water
mite, Hydrachna trilobata viets (Arachnida; Hydrachnoidae
Hydrachnidae) J. environ. Biol., 2: 91-96.
NAMBA, N., IWAMOTO, T., & SATOH, T. (1966) Oral toxicity and
metabolism of sumition on cattle, sheep and pigs. Res. Bull.
Hokkaido Natl Agric. Exp. Stn, 89: 82-90.
NAMBA, T. (1971) Cholinesterase inhibition by organophosphorus
compounds and its clinical effects. Bull. World Health Organization,
44: 289-307.
NATIONAL RESEARCH COUNCIL OF CANADA (1975) Fenitrothion: The effects
of its use on environmental quality and its chemistry, Ottawa,
National Research Council of Canada, pp. 22-32 (Publication NRCC No.
14104).
NEHEZ, M., HUSZTA, E., MAZZAG, E., & BERENCSI, G. (1985) A study of
the mutagenic effect of 3-methyl-4-nitrophenol on the somatic cells
of the mouse. Ecotoxicol. environ. Saf., 9: 47-52.
NISHIO, M. & KUSANO, S. (1978) [Effect of consecutive use of
organophosphorus insecticides to population and nitrification
activity of soil microorganisms.] Nojishikenjo-kenkyuuhokoku, 28:
39-48 (in Japanese).
NISHIUCHI, Y. (1977) Toxicity of pesticide formulations to some
fresh water organisms XXXXI. Aquiculture, 24: 146-150.
NISHIUCHI, Y. (1981) [Toxicity of pesticides to some aquatic
organisms I; Toxicity of pesticides to some aquatic insects.] Seitai
Kagaku, 4: 31-46 (in Japanese).
NISHIZAWA, Y., FUJII, K., KADOTA, T., MIYAMOTO, J., & SAKAMOTO, H.
(1961) Studies on the organophosphorus insecticides. Part VII.
Chemical and biological properties of new low toxic organophosphorus
insecticide. O,O-dimethyl- O-(3-methyl-4-nitrophenyl)
phosphorothioate. Agric. biol. Chem., 25: 605-610.
NOSAL, N. & HLADKA, A. (1968) Determination of the exposure to
fenitrothion ( O,O-dimethyl-0/3-methyl-4-nitrophenyl/thiophosphate)
on the basis of the excretion of p-nitro- m-cresol by the urine
of the persons tested. Int. Arch. Gewerbepathol. Gewerbehyg., 25:
28-38.
OGATA, S. (1972) Effects of organophosphate pesticide on the beagle
dogs in chronic toxicity studies experiments (Report 1). Acta Soc.
Ophthalmol. Jpn, 76: 1153-1150.
OHKAWA, H., MIKAMI, N., & MIYAMOTO, J. (1974) Photodecomposition of
sumithion, O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate.
Agric. biol. Chem., 38: 2247-2255.
OHKAWA, H., OSHITA, H., & MIYAMOTO, J. (1980) Comparison of
inhibitory activity of various organophosphorus compounds against
acetylcholinesterase and neurotoxic esterase of hen with respect to
delayed neurotoxicity. Biochem. Pharmacol., 29: 2721-2727.
OHMAE, T., UNO, M., OKADA, S., KAGECHI, Y., TERADA, I., & TANIGAWA,
K. (1981) Environmental behavior of fenitrothion and its
decomposition products after aerial application. J. pestic. Sci., 6:
437-446.
OHSHIMA, M., TAKAHASHI, N., MIKAMI, N., & MATSUDA, T. (1988)
Accumulation and metabolism of 14-C fenitrothion in bluegill
sunfish (Lepomis macrochirus) (Unpublished Technical Report No.
HM-80-0095, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
OKADA, I. & HOSHIBA, H. (1970) A laboratory experiment on some
insecticides to honeybee. Bull. Fac. Agric., Tamagawa Univ., 10:
79-85.
OKUNO, Y., HARA, S., KADOTA, T., & MIYAMOTO, J. (1977) Possible
allergic asthma by inhalation of Sumithion 22% emulsifiable
concentrate in guinea-pigs (Unpublished Technical Report No.
HT-70-0113, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
OSICKA-KOPROWSKA, A., GRADOWSKA-OLZEWSKA, I., & WYSOCKA-PARUSZEWSKA,
B. (1987) Adrenocortical changes and uptake of
4-14C-corticosterone in rats intoxicated with fenitrothion. Arch.
Toxicol., 61: 76-78.
PAUL, B.S. & VADLAMUDI, V.P. (1976) Teratogenic studies of
fenitrothion on White Leghorn chick embryos. Bull. environ. Contam.
Toxicol., 15: 223-229.
PAWAR, K.R. & KATDARE, M. (1983) Effect of the insecticide Sumithion
(fenitrothion) on embryonic development in a frog. Experientia
(Basel), 39: 297-298.
PAWAR, K.R. & KATDARE, M. (1984) Toxic and teratogenic effects of
fenitrothion, BHC and carbofuran on embryonic development of the
frog Microhyla ornatas. Toxicol. Lett., 22: 7-13.
PEARCE, P.A. & BUSBY, D.G. (1980) Research on the effects of
fenitrothion on the White-throated Sparrow. In: Research on the
effects of spray operations for forest protection against spruce
budworm. Environmental surveillance in New Brunswick 1978-79,
Fredericton, Canada, Maritimes Forest Research Centre, pp. 24-28.
PEARCE, P.A. & PEAKALL D.B. (1977) The impact of fenitrothion on
bird populations in New Brunswick. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, pp.
299-305 (Publication NRCC No. 16073).
PEARCE, P.A., BRUN, G.L., & WITTEMAN, J. (1979a) Off-target fallout
of fenitrothion during 1978 forest spraying operations in New
Brunswick. Bull. environ. Contam. Toxicol., 23: 503-508.
PEARCE, P.A., PEAKALL, D.B., & ERSKINE, A.J. (1979b) Impact on
forest birds of the 1976 spruce budworm spray operation in New
Brunswick, Ottawa, Environment Canada, Canadian Wildlife Service,
pp. 1-15 (Progress Note No. 97).
PETERSON, R.H. (1974) Influence of fenitrothion on swimming
velocities of brook trout (Salvelinus fontinalis). J. Fish Res.
Board Can., 31: 1757-1762.
PETERSON, R.H. & ZITKO, V. (1974) Variations in stream insect drift
associated with operational and experimental contamination with
fenitrothion in New Brunswick, St. Andrews, New Brunswick, Fisheries
Research Board of Canada, Biological Station, 23 pp (Technical
Report No. 439).
PLOWRIGHT, R.C. (1977) The effects of fenitrothion on forest
pollinators in New Brunswick. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, pp.
335-341 (Publication NRCC No. 16073).
PLOWRIGHT, R.C. & ROOD, F.H. (1980) The effect of aerial insecticide
spraying on hymenopterous pollinators in New Brunswick. Can.
Entomol., 112: 259-269.
PLOWRIGHT, R.C., PENDRER, B.A., & MCLAREN, I.A. (1978) The impact of
aerial fenitrothion spraying upon the population biology of bumble
bees ( Bombus LATR.: HYM.) in south-western New Brunswick. Can.
Entomol., 110: 1145-1156.
POLLACK, J.D., BURECH, D., & HAMPARIN, V.V. (1977) The role of
chemicals in potentiating viral infections and Reye's syndrome. In:
Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, p. 519 (Publication NRCC No. 16073).
POMBER, L., WEINBERGER, P., & PRASAD. R. (1974) Some physiological
and phytotoxic effects of fenitrothion on germination and seedling
growth of Pinus strobus L., Ottawa, Environment Canada, Forestry
Service, Chemical Control Research Institute, 25 pp (Information
Report CC-X-80).
PRASAD, R. & MOODY, R.P. (1976) Fate of fenitrothion in forest
trees. VIII. Persistence, translocation and metabolism of
C14-fenitrothion in Jack Pine ( Pinus banksiana Lamb.) in
relation to sawfly mortally, Ottawa, Environment Canada, Forestry
Service, Chemical Control Research Institute, 22 pp (Information
Report CC-X-126).
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1982a) In vivo
sub-acute physiological stress induced by sumithion on carbohydrate
metabolism in hepatopancreas of Oziotelphusa senex senex
Fabricius. Toxicol. Lett., 13: 179-182.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1982b) Effect of
sumithion on molting the freshwater rice-field crab ( Oziotelphusa
senex senex Fabricius). Toxicol. Lett., 14: 243-246.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1983a) Effect of
sublethal concentrations of sumithion on limb regeneration of
freshwater field crab Oziotelphusa senex senex. Experientia
(Basel), 39: 1380-1381.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1983b) Chronic
sumithion toxicity: Effect on carbohydrate metabolism in crab
muscle. Toxicol. Lett., 15: 113-117.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1983c) Effect of
sumithion on ovarian growth of a freshwater rice field crab
(Oziotelphusa senex senex Fabricius). Toxicol. Lett., 18: 273-276.
REYE, R.D.K., MORGAN, G., & BARAL, J. (1963) Encephalopathy and
fatty degeneration of the viscera. A disease entity in childfood.
Lancet, 7311: 749-752.
RIPLEY, B.D., HALL, J.A., & CHAU, A.S.Y. (1974) Determination of
fenitrothion, phosphamidon and dimethoate in natural water. Environ.
Lett., 7: 97-118.
RONDEAU, D.B., YOUNG, L., HEBERT, D., & TROTIER, B.L. (1981)
Behavioral toxicity of chronic administration of fenitrothion in
rats. Neurobehav. Toxicol. Teratol., 3: 313-319.
ROSIVAL, L., VARGOVA, M., & HLADKA, H., BATORA, V., KOVACICOVA, J.,
SZOKOLAYOVA, J., & CEREY, K. (1974) On the toxic effect of the
fenitrothion S-methyl isomer, Bratislava, Czechoslovakia, Research
Institute of Hygiene, Research Institute of Industrial Hygiene and
Professional Diseases, and Research Institute of Agrochemical
Technology (Unpublished report submitted to WHO by the authors).
RUBEN, F.L., STREIFF, E.J., NEAL, M., & MICHAELIS, R.H. (1976)
Epidemiologic study of Reye's Syndrome: cases seen in Pittsburgh,
October 1973-April 1975. Am. J. public Health, 66: 1096-1098.
RUSHMORE, F.M. (1971) Effects of accothion on birds. (Intensive
studies on sapsuckers). In: Nash, R.W., Peterson, J.W., & Chansler,
J.F., ed. Environmental studies of accothion for spruce budworm
control, Washington, DC, US Department of Agriculture and US
Department of Interior, p. 88.
RUTTER, H.A. & NELSON, L.W. (1974) Two-year dietary administration
in the rat, Vienna, Virginia, Hazleton Laboratories (Unpublished
Technical Report No. HT-41-0006, submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
RUTTER, H.A. & VOELKER, R.W. (1974) Three-generation reproduction
study: Rats, Vienna, Virginia, Hazleton Laboratories (Unpublished
Technical Report No. HT-41-0005, submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
RUTTER, H.A. & BANAS, D.A. (1975) Seventy eight week tumorigenic
study in the ICR Swiss mice with Sumithion, Vienna, Virginia,
Hazleton Laboratories (Technical Report No. HT-51-0002, submitted to
WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
SAKAMOTO, T., SAWADA, Y., NISHIDE, K., SADAMITSU, D., YOSHIOKA, T.,
SUGIMOTO, T., NISHII, S., & KISHII, H. (1984) Delayed neurotoxicity
produced by an organophosphorus compound (Sumithion). A case report.
Arch. Toxicol., 56: 136-138.
SALONIUS, P.O. (1972) Effect of DDT and fenitrothion on forest-soil
microflora. J. econ. Entomol., 65: 1089-1090.
SAXENA, P.K. & MANI, K. (1985a) Protein-bound iodine levels in the
blood plasma of freshwater teleost, Channa punctatus (BI) exposed
to subtoxic pesticides concentrations. Toxicol. Lett., 24: 33-36.
SAXENA, P.K. & MANI, K. (1985b) Quantitative study of testicular
recrudescence in the freshwater teleost, Channa punctatus (BI)
exposed to pesticides. Bull. environ. Contam. Toxicol., 34: 597-607.
SAXENA, P.K. & MANI, K. (1987) Effect of safe concentrations of some
pesticides on testicular recrudescence in the freshwater murrel,
Channa punctatus (BI): A morphological study. Ecotoxicol. environ.
Saf., 14: 56-63.
SCHAFER, E.W. (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol. appl.
Pharmacol., 21: 315-330.
SCHERER, E. (1975) Avoidance of fenitrothion by gold fish
(Carassius auratus). Bull. environ. Contam. Toxicol., 13:
492-496.
SCHNEIDER, W.G. (1976) Forest spray program and Reye's syndrome.
Report of the Panel convened by the Government of New Brunswick,
Fredericton, Canada, Government of New Brunswick, 22 pp.
SEIKAI, T. (1982) Acute toxicity of organophosphorous insecticides
on the developmental stages of eggs, larvae and juveniles of
Japanese striped knifejaw, Oplegnathus fasciatus. Bull. Jpn. Soc.
Sci. Fish., 48: 599.
SERGEANT, D.B., ZITKO, V., & BURRIDGE, L.E. (1979) The determination
of fenitrothion in bivalves, St. Andrews, Fisheries and Marine
Service, Biological Station, 27 pp (Technical Report No. 906).
SHIM, J.C. & SELF, L.S. (1973) Toxicity of agricultural chemicals to
larvivorous fish in Korean rice fields. Trop. Med., 15: 123-130.
SHIRASU, Y., MORIYA, M., & WATANABE, K. (1979) Mutagenicity studies
on Sumithion in bacterial systems. (Unpublished Technical Report No.
HT-91-0140, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
SMART, N.A. (1980) Determination of a range of organophosphorus
pesticide residues in grain. Report by a Working Group of the
Committee for Analytical Methods for Residues of Pesticides and
Veterinary Products in Foodstuffs and the Working Party on Pesticide
Residues of the Ministry of Agriculture, Fisheries and Food.
Analyst, 105: 515-517.
SPILLNER, C.J., DEBAUN, J.R., & MENN, J.J. (1979a) Degradation of
fenitrothion in forest soil and effects on forest soil microbes. J.
agric. food Chem., 27: 1054-1060.
SPILLNER, C.J., THOMAS, V.H., & DEBAUN, J.R. (1979b) Effect of
fenitrothion on microorganisms which degrade leaf-litter and
cellulose in forest soils. Bull. environ. Contam. Toxicol., 23:
601-606.
SPITZER, W.O. (1982) Report of the New Brunswick Task Force on the
Environment and Reye's Syndrome. Clin. invest. Med., 5: 203-214.
SUMITOMO CHEMICAL CO., LTD (1969) Residual amounts of Sumithion in
foods. Reports on trials conducted by the Japanese Ministry of
Agriculture and Forestry, 1968, Osaka, Japan, Sumitomo Chemical Co.,
Ltd (Unpublished report).
SUMITOMO CHEMICAL CO., LTD (1971) Acute oral toxicity study with
3-methyl-4-nitrophenol in rat, Osaka, Japan, Sumitomo Chemical Co.,
Ltd, Takarazuka Research Laboratory (Unpublished report).
SUMITOMO CHEMICAL CO., LTD. (1972) Six-month feeding study of
Sumithion in rats, Osaka, Japan, Sumitomo Chemical Co., Ltd,
Pesticide Research Department (Unpublished report).
SUNDARAM, K.M.S. (1973) Degradation dynamics of fenithrothion in
aqueous systems, Ottawa, Environment Canada, Forestry Service,
Chemical Control Research Institute, 19 pp (Information Report
CC-X-44).
SUNDARAM, K.M.S. (1974) Distribution and persistence of fenitrothion
residues in foliage, soil and water in larose forest, Ottawa,
Environment Canada, Forestry Service, Chemical Control Research
Institute, 43 pp (Information Report CC-X-64).
SUNDARAM, K.M.S. (1975) Fenitrothion residues in honey bees and
their products collected from forest areas sprayed with the
insecticide for budworm control, Ottawa, Environment Canada,
Forestry Service, Chemical Control Research Institute, 27 pp
(Information Report CC-X-89).
SUNDARAM, K.M.S. (1988) Cuticular penetration and in vivo metabolism
of fenitrothion in spruce budworm. J. environ. Sci. Health, B23:
643-659.
SUNDARAM, K.M.S. & PRASAD, R. (1975) Persistence studies of
insecticides: VI. Root penetration, translocation and metabolism of
14C-labelled fenitrothion in young spruce trees. Ottawa,
Environment Canada, Forestry Service, Chemical Control Research
Institute, 15 pp (Information Report CC-X-86).
SUNDARAM, K.M.S., YULE, W.M., & PRASAD, R. (1975) Studies of foliar
penetration, movement and persistence of 14C-labelled fenitrothion
in spruce and fir trees, Ottawa, Environment Canada, Forestry
Service, Chemical Control Research Institute, 17 pp (Information
Report CC-X-85).
SUZUKI, H. & MIYAMOTO, J. (1976) Studies on mutagenicity of
Sumithion, Cremart and Denmart. J. pestic. Sci., 1: 253-259.
SUZUKI, H. & MIYAMOTO, J. (1980) Effects of fenitrothion on sister
chromatid exchanges (SCE) in cultured mouse embryo cells
(Unpublished Technical Report No. HT-00-0280, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
SUZUKI, H., MIYAMOTO, J., & OKA, A. (1974) Mutagenicity and
radiomimeticity screening of Sumithion (Unpublished report submitted
to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
SWAIN, W.R. & IVANIKIW, N.K., ed. (1976) Proceedings of the Second
USA-USSR Symposium on the Effects of Pollutants upon Aquatic
Ecosystems, Vol. II.
SYMONS, P.E.K. (1973) Behavior of young Atlantic salmon (Salmo
salar) exposed or force-fed fenitrothion, an organophosphate
insecticide. J. Fish. Res. Board Can., 30: 651-655.
SYMONS, P.E.K. (1977) Disposal and toxicology of the insecticide
fenitrothion: Predicting hazards of forest spraying. Residue Rev.,
68: 1-36.
SYMONS, P.E.K. & METCALF, J.L. (1978) Mortality, recovery and
survival of larval Brochycentrus numerosus (Trichoptera) after
exposure to the insecticide fenitrothion. Can. J. Zool., 56:
1284-1290.
TABATA, K. & KITAHARA, E. (1980) Effect of fenitrothion (Sumithion)
spraying on the population density and blood cholinesterase activity
of Japanese woodmouse, Apodemus speciosus Temmink. Appl. Entomol.
Zool., 15: 242-248.
TABATA, K. & OKUBO, R. (1980) [Mechanism of the abnormal leaf
abscission phenomenon in Japanese cypress caused by fenitrothion
(Sumithion) (II) Metabolism of 14C-fenitrothion
( O,O-dimethyl- O-(3-methyl-4-nitrophenyl) phosphorothioate) in
Japanese cypress leaves.] J. Jpn. Forensic Soc., 62: 350-353 (in
Japanese, with English summary).
TAKAHASHI, H., MIYAOKA, T., TSUDA, S., & SHIRASU, Y. (1984)
Potentiated toxicity of 2-sec-butylphenyl methylcarbamate (BPMC) by
O,O-dimethyl O-(3-methyl-4-nitrophenyl)phosphorothioate
(fenitrothion) in mice; Relationship between acute toxicity and
metabolism of BPMC. Fundam. appl. Toxicol., 4: 718-723.
TAKAHASHI, H., KATO, A., YAMASHITA, E., NAITO, Y., TSUDA, S., &
SHIRASU, Y. (1987) Potentiations of N-methylcarbamate toxicities
by organophosphorus insecticides in male mice. Fundam. appl.
Toxicol., 8: 139-146.
TAKAKU, T., OTSUKI, A., & TAKAHASHI, M. (1979a) Simple and rapid
determination of fenitrothion in water by reversed phase adsorption
liquid chromatography. Bunseki Kagaku, 28: 702-704.
TAKAKU, T., TAKAHASHI, M., & OTSUKI, A. (1979b) Dispersion of an
organophosphorus insecticide, fenitrothion, in paddy fields and its
effects on the microorganisms. Jpn. J. Limnol., 40: 137-144.
TAKANO, H. & HIJIKATA, Y. (1981) [Environmental impact of
fenitrothion on forest - Effect on wild birds.] Forest Chem., 75:
15-18 (in Japanese).
TAKEUCHI, K., HIGO, M., & SAKAI, T. (1980) Susceptibility of the
honey bees to several insecticides. Bull. Fac. Agric. Tamagawa
Univ., 20: 40-46.
TAKIMOTO, Y. & MIYAMOTO, J. (1976a) Studies on accumulation and
metabolism of sumithion in fish. J. pestic. Sci., 1: 261-271.
TAKIMOTO, Y. & MIYAMOTO, J. (1976b) Residue analysis of Sumithion.
Residue Rev., 60: 83-101.
TAKIMOTO, Y., MURANO, A., & MIYAMOTO, J. (1975) Analytical methods
for Sumithion in technical products and formulated materials.
Residue Rev., 60: 11-28.
TAKIMOTO, Y., HIROTA, M., INUI, H., & MIYAMOTO, J. (1976)
Decomposition and leaching of radioactive sumithion of 4 different
soils under laboratory conditions. J. pestic. Sci., 1: 131-143.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1978) Degradation and
fate of the fenitrothion applied to harvested rice grains. J.
pestic. Sci., 3: 277-290.
TAKIMOTO, Y., OHSHIMA, M., KATO., T., & MIYAMOTO, J. (1980)
[Photodegradation of fenitrothion in sea water.] Abstract of papers
presented at the Annual Meeting of Pesticide Science Society of
Japan, Chiba, No. 143 (in Japanese).
TAKIMOTO, Y., OHSHIMA, M., YAMADA, H., & MIYAMOTO, J. (1984a) Fate
of fenitrothion in several developmental stages of the killifish
(Oryzias latipes). Arch. environ. Contam. Toxicol., 13: 579-587.
TAKIMOTO, Y., HAGINO, S., YAMADA, H., & MIYAMOTO, J. (1984b) The
acute toxicity of fenitrothion to killifish (Oryzias latipes) at
twelve different stages of its life history. J. pestic. Sci., 9:
463-470.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1987a) Comparative
metabolism of fenitrothion in aquatic organisms I. Metabolism in the
euryhaline fish, Oryzias latipes and Mugil cephalus. Ecotoxicol.
environ. Saf., 13: 104-117.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1987b) Comparative
metabolism of fenitrothion in aquatic organisms. II. Metabolism in
the freshwater snail, Cipangopaludina japonica and Physa acuta.
Ecotoxicol. environ. Saf., 13: 118-125.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1987c) Comparative
metabolism of fenitrothion in aquatic organisms. III. Metabolism in
the crustaceans, Daphnia pulex and Palaemon paucidens.
Ecotoxicol. environ. Saf., 13: 126-134.
TALEKAR, N.S., SUN, L.T., LEE, E.M., CHEN, J.S., LEE, T.M., & LU, S.
(1977) Residual behavior of several insecticides on chinese
cabbage. J. econ. Entomol., 70: 689-692.
TAMANO, S. HAGIWARA, A., SHIBATA, M., & SUZUKI, M. (1990) Chronic
dietary toxicity and carcinogenicity test of Sumithion technical in
mice, Aichi, Japan, Daiyu-Kai Medical Science Research Institute
(Unpublished Technical Report No. HT-01-0445, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
THOMPSON, C.M., FRICK, J.A., NATKE, B.C., & HANSEN, L.K. (1989)
Preparation, analysis and anticholinesterase properties of
O,O-dimethyl phosphorothioate isomerides. Chem. Res. Toxicol., 2:
386-391.
TOMITA, M., SOMEYA, N., NISHIMURA, M., & UEDA, K. (1974) [Studies on
the retention deposit of organophosphorus pesticides in animal and
human bodies.] Jpn. J. ind. Health, 16: 547-556 (in Japanese).
TOOR, H.S. & KAUR, K. (1974) Toxicity of pesticides to the fish,
Cyprinus carpio communis Linn. Indian J. exp. Biol., 12: 334-336.
TROTTIER, B.L. & JANKOWSKA, I. (1980) In vivo study on the storage
of fenitrothion in chicken tissues after long-term exposure to a
small dose. Bull. environ. Contam. Toxicol., 24: 606-610.
TROTTIER, B.L., FRASER, A.R., PLANET, G., & ECOBICHON, D.J. (1980)
Subacute toxicity of technical fenitrothion in male rats.
Toxicology, 17: 29-38.
TSCHAPLINSKI, P.J. & GARDNER, D.R. (1981) Metabolism of fenitrothion
in red-backed voles Clethrionomys gapperi. Pestic. Biochem.
Physiol., 16: 47-62.
TSUDA, S., MIYAOKA, T., IWASAKI, M., & SHIRASU, Y. (1984)
Pharmacokinetic analysis of increased toxicity of
2- sec-butylphenyl methyl carbamate (BPMC) by fenitrothion
pretreatment in mice. Fundam. appl. Toxicol., 4: 724-730.
TSUJI, K., HORIDE, F., MINOBE, M., SASAKI, M., SHIRAGA, N., &
HIROAKI, O. (1980) Thermal decomposition of O,O-dimethyl
O-(3-methyl-4-nitrophenyl) phosphorothioate (Sumithion). J.
pestic. Sci., 5: 371-384.
TSUKIMOTO, K., HAYASHI, H., NONOGUCHI, H., BABA, M., TAZAWA, J.,
IWAMOTO, H., ABE, T., & SHIMADA, Y. (1981) [A case of delayed onset
of symptoms in seriously-poisoned female with Sumithion.] J. Jpn.
rural Med., 30: 598-559 (in Japanese).
UCHIYAMA, M., YOSHIDA, T., HOMMA, K., & HONGO, T. (1975) Inhibition
of hepatic drug-metabolizing enzymes by triphosphate insecticides
and its drug toxicological implications. Biochem. Pharmacol., 24:
1221-1225.
UEDA, K. & NISHIMURA, M. (1966) Interim report. Chronic toxicity of
Sumithion: two-year feeding study in rats, Tokyo Dental College,
Department of Hygiene and Toxicology, (Unpublished report submitted
to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
USDA (1976) Final environmental statement cooperative spruce budworm
suppression project in Maine, Washington, DC, US Department of
Agriculture, Forest Service (Environmental Statement of April 19,
1976).
VANDEKAR, M. (1965) Observations on the toxicity of carbaryl,
folithion and 3-isopropylphenyl n-methylcarbamate in a
village-scale trial in southern Nigeria. Bull. World Health Organ.,
33: 107-115.
VARTY, I.W. (1976) Environmental effects of the spruce budworm spray
program in New Brunswick 1976, Fredericton, Canada, Maritimes Forest
Research Centre (Information Report No. M-X-67).
VARTY, I.W. (1977) Long-term effects of fenitrothion spray programs
on non-target terrestrial arthropods. In: Proceedings of a Symposium
on Fenitrothion, Ottawa, National Research Council of Canada, pp.
344-375 (Publication NRCC No. 16073).
VELAZQUEZ, A., XAMENA, N., CREUS, A., & MARCOS, R. (1987)
Mutagenicity studies on fenitrothion in Drosophila. Mutagenesis, 2:
333-336.
VERMA, S.R., BANSAL, S.K., & DALELA, R.C. (1977) Bioassay trials
with a few organic biocides on fresh water fish Laveo rohita. Indian
J. environ. Health, 19: 107-115.
VERMA, S.R., BHATNAGAR, M.C., & DALELA, R.C. (1978a) Biocides in
relation to water pollution. Part 2: Bioassay studies of few
biocides to a fresh water fish Channa gachua. Acta hydrochim.
hydrobiol., 6: 137-144.
VERMA, S.R., BANSAL, S.K., & DALELA, R.C. (1978b) Toxicity of
selected organic pesticides to a fresh water teleost fish,
Saccobranchus fossilis and its application in controlling water
pollution. Arch. environ. Contam. Toxicol., 7: 317-323.
VERMA, S.R., BANSAL, S.K., GUPTA, A.K., PAL, N., TYAGI, A.K.,
BHATNAGER, M.C., KUMAR, V., & DALELA, R.C. (1982) Bioassay trials
with twenty three pesticides to a fresh water teleost,
Saccobranchus fossilis. Water Res., 16: 525-529.
VOLPE, G.G. & MALLET, V.N. (1980) Development of an analytical
method for fenitrothion and five derivatives in water using XAD
resins and gas-liquid chromatography (GLC). Int. J. environ. anal.
Chem., 8: 291-301.
VOLPE, G.G. & MALLET, V.N. (1981) High-performance liquid
chromatography of fenitrothion and seven derivatives - a study of
their recovery from water using XAD resins as compared with organic
solvents. Chromatographia, 14: 333-336.
WARREN, M.W., RUEBUSH, T.K., II, HOBBS, J.H., HIPPOLYTE, R., & MILLER,
S. (1985a) Safety measures associated with the use of organophosphate
insecticides in the Haitian malaria control programme. Bull. World
Health Organ., 63(2): 345-351.
WARREN, M.W., SPENCER, H.C., CHURCHILL, F.C., FRANCOIS, V.J.,
HIPPOLYTE, R., & STAIGER, M.A. (1985b) Assessment of exposure to
organophosphate insecticides during spraying in Haiti: monitoring of
urinary metabolites and blood cholinesterase levels. Bull. World
Health Organ., 63(2): 353-360.
WEINBERGER, P., GREENHALGH, R., SHER, D., & OUELLETTE, M. (1982a)
Persistence of formulated fenitrothion in distilled, estuarine, and
lake water microcosmos in dynamic and static systems. Bull. environ.
Contam. Toxicol., 28: 484-489.
WEINBERGER, P., GREENHALGH, R., MOODY, R.P., & BOULTON, B. (1982b)
Fate of fenitrothion in aquatic microcosmos and the role of aquatic
plants. Environ. Sci. Technol., 16: 470-473.
WHO(1973) Fenitrothion. In: Safe use of pesticides. Twentieth report
of the WHO Expert Committee on Insecticides, Geneva, World Health
Organization, pp. 18-19 (WHO Technical Report Series No. 513).
WHO/FAO (1977) Data sheets on pesticides No. 30: Fenitrothion,
Geneva, World Health Organization, 9 pp (VBC/DS/77.30).
WHO (1986) Environmental Health Criteria 63: Organophosphorus
insecticides: A general introduction, Geneva, World Health
Organization, 181 pp.
WHO (1990) The WHO recommended classification of pesticides by
hazard and Guidelines to classification 1990-1991, Geneva, World
Health Organization (WHO/PCS/90.1).
WILDISH, D.J. & LISTER, N.A. (1973) Biological effects of
fenitrothion in the diet of brook trout. Bull. environ. Contam.
Toxicol., 10: u333-339.
WILDISH, D.J. & LISTER, N.A. (1977) Effects of dietary fenitrothion
on growth and hierarchial position in brook trout (Salvelinus
fontinalis). Prog. Fish-Cult., 39: 3-9.
WILFORD, K., LIETEART, P.E.A., & FOLL, C.V. (1965) Fenitrothion:
Working paper prepared for an informal meeting of toxicology of
insecticides, Geneva, World Health Organization, pp. 18-23
(65/TOX/1).
WOOD, G.W. (1979) Recuperation of native bee. Population in
blueberry fields exposed to drift of fenitrothion from forest spray
operations in New Brunswick. J. econ. Entomol., 72: 36-39.
WOOD, G.W. (1980) Bee toxicology from fenitrothion aerosol,
Fredericton, Agriculture Canada Research Station, 60 pp.
WOOD, R.B., Jr & BOGDAN, G.F. (1986) Reye's syndrome and spruce
budworm insecticide spraying in Maine, 1978-1982. Am. J. Epidemiol.,
124: 671-677.
WOODWARD, D.F. & MAUCK, W.L. (1980) Toxicity of five forest
insecticides to cutthroat trout and two species of aquatic
invertebrates. Bull. environ. Contam. Toxicol., 25: 846-853.
WORTHING, C.R. & WALKER, S.B., ed. (1983) Fenitrothion. In: The
pesticide manual 7th ed., Croydon, The British Crop Protection
Council, p. 261.
WRIGHT, C.G., LEIDY, R.B., & DUPREE, H.E., Jr (1981) Insecticides in
the ambient air of room following their application for control of
pest. Bull. environ. Contam. Toxicol., 26: 548-553.
YADAV, A.S., VASHISHAT, R.K., & KAKAR, S.N. (1982) Testing of
endosulfan and fenitrothion for genotoxicity in Saccharomyces
cerevisiae. Mutat. Res., 105: 403-407.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1982a)
Comparison of the effect of an equimolar and low dose of
fenitrothion and methylparathion on their own metabolism in rat
liver. J. toxicol. Sci., 7: 35-41.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1982b)
Increase of adrenal weight in rats by the repeated administration of
fenitrothion. Toxicol. Lett., 11: 187-191.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1983a)
Effect of adrenalectomy, pretreatment with SKF 525-A, phenobarbital
and diethyl-maleate on the acute toxicity of fenitrothion in male
rats. Arch. Toxicol., 52: 233-242.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1983b)
Comparative metabolism of fenitrothion and methylparathion in male
rats. Acta pharmacol. toxicol., 53: 96-102.
YAMANOI, F. (1980) [Effect of insecticides on the progeny in the
silkworm, Bombyx mori. I. Effect of organophosphorus insecticides
on egg laying and their hatching.] J. Sericult. Sci. Jpn, 49:
434-439 (in Japanese).
YAMANOI, F. (1981) [Effect of insecticides on the progeny in the
silkworm, Bombyx mori. II. Effect of organophosphorus insecticides
on the next generation.] J. Sericult. Sci. Jpn, 50: 83-87 (in
Japanese).
YASUNO, M., HATAKEYAMA, S., & MIYASHITA, M. (1980) Effect on
reproduction in Guppy (Poecilia reticulata) under chronic exposure
to temephos and fenitrothion. Bull. environ. Contam. Toxicol., 25:
29-33.
YASUNO, M., OKITA, J., SAITO, K., NAKAMURA, Y., HATAKEYAMA, S., &
KASUGA, S. (1981) Effect of fenitrothion on benethic fauna in small
stream of Mt. Tsukuba, Japan. Jpn. J. Ecol., 31: 237-245.
YOSHIDA, M., SHIMADA, E., YAMANAKA, H., AOYAMA, Y., YAMAMURA, Y., &
OWADA, S. (1987) A case of acute poisoning with fenitrothion
(Sumithion). Hum. Toxicol., 6: 403-406.
YULE, W.N. (1974) The persistence and fate of fenitrothion
insecticide in a forest environment II. Accumulation of residues in
balsam fir foliage. Bull. environ. Contam. Toxicol., 12: 249-252.
YULE, W.N. & DUFFY, J.R. (1972) The persistence and fate of
fenitrothion insecticide in a forest environment. Bull. environ.
Contam. Toxicol., 8: 10-18.
ANNEX I
TREATMENT OF ORGANOPHOSPHATE INSECTICIDE POISONING IN MAN
(From EHC 63: Organophosphorus Insecticides - A General
Introduction)
All cases of organophosphorus poisoning should be dealt with as
an emergency and the patient sent to hospital as quickly as
possible. Although symptoms may develop rapidly, delay in onset or a
steady increase in severity may be seen up to 48 h after ingestion
of some formulated organophosphorus insecticides.
Extensive descriptions of treatment of poisoning by
organophosphorus insecticides are given in several major references
(Kagan, 1977; Taylor, 1980; UK DHSS, 1983; Plestina, 1984) and will
also be included in the IPCS Health and Safety Guides to be prepared
for selected organophosphorus insecticides.
The treatment is based on:
(a) minimizing the absorption;
(b) general supportive treatment; and
(c) specific pharmacological treatment.
1.1 Minimizing the absorption
When dermal exposure occurs, decontamination procedures include
removal of contaminated clothes and washing of the skin with
alkaline soap or with a sodium bicarbonate solution. Particular care
should be taken in cleaning the skin area where venepuncture is
performed. Blood might be contaminated with direct-acting
organophosphorus esters and, therefore, inaccurate measures of ChE
inhibition might result. Extensive eye irrigation with water or
saline should also be performed. In the case of ingestion, vomiting
might be induced, if the patient is conscious, by the administration
of ipecacuanha syrup (10-30 ml) followed by 200 ml water. This
treatment is, however, contraindicated in the case of pesticides
dissolved in hydrocarbon solvents. Gastric lavage (with addition of
bicarbonate solution or activated charcoal) can also be performed,
particularly in unconscious patients, taking care to prevent
aspiration of fluids into the lungs (i.e., only after a tracheal
tube has been put into place).
The volume of fluid introduced into the stomach should be
recorded and samples of gastric lavage frozen and stored for
subsequent chemical analysis. If the formulation of the pesticide
involved is available, it should also be stored for further analysis
(i.e., detection of toxicologically relevant impurities). A
purgative can be administered to remove the ingested compound.
1.2 General supportive treatment
Artificial respiration (via a tracheal tube) should be started
at the first sign of respiratory failure and maintained for as long
as necessary.
Cautious administration of fluids is advised, as well as
general supportive and symptomatic pharmacological treatment and
absolute rest.
1.3 Specific pharmacological treatment
1.3.1 Atropine
Atropine should be given, beginning with 2 mg iv and given at
15-30-min intervals. The dose and the frequency of atropine
treatment varies from case to case, but should maintain the patient
fully atropinized (dilated pupils, dry mouth, skin flushing, etc.).
Continuous infusion of atropine may be necessary in extreme cases
and total daily doses up to several hundred mg may be necessary
during the first few days of treatment.
1.3.2 Oxime reactivators
Cholinesterase reactivators (e.g., pralidoxime, obidoxime)
specifically restore AChE activity inhibited by organophosphates.
This is not the case with enzymes inhibited by carbamates. The
treatment should begin as soon as possible, because oximes are not
effective on "aged" phosphorylated ChEs. However, if absorption,
distribution, and metabolism are thought to be delayed for any
reasons, oximes can be administered for several days after
intoxication. Effective treatment with oximes reduces the required
dose of atropine. Pralidoxime is the most widely available oxime. A
dose of 1 g pralidoxime can be given either im or iv and repeated
2-3 times per day or, in extreme cases, more often. If possible,
blood samples should be taken for AChE determinations before and
during treatment. Skin should be carefully cleansed before sampling.
Results of the assays should influence the decision whether to
continue oxime therapy after the first 2 days.
There are indications that oxime therapy may possibly have
beneficial effects on CNS-derived symptoms.
1.3.3 Diazepam
Diazepam should be included in the therapy of all but the
mildest cases. Besides relieving anxiety, it appears to counteract
some aspects of CNS-derived symptoms that are not affected by
atropine. Doses of 10 mg sc or iv are appropriate and may be
repeated as required (Vale & Scott, 1974). Other centrally acting
drugs and drugs that may depress respiration are not recommended in
the absence of artificial respiration procedures.
1.3.4 Notes on the recommended treatment
1.3.4.1 Effects of atropine and oxime
The combined effect far exceeds the benefit of either drug
singly.
1.3.4.2 Response to atropine
The response of the eye pupil may be unreliable in cases of
organophosphorus poisoning. A flushed skin and drying of secretions
are the best guide to the effectiveness of atropinization. Although
repeated dosing may well be necessary, excessive doses at any one
time may cause toxic side-effects. Pulse-rate should not exceed
120/min.
1.3.4.3 Persistence of treatment
Some organophosphorus pesticides are very lipophilic and may be
taken into, and then released from, fat depots over a period of many
days. It is therefore quite incorrect to abandon oxime treatment
after 1-2 days on the supposition that all inhibited enzyme will be
aged. Ecobichon et al. (1977) noted prompt improvement in both
condition and blood-ChEs in response to pralidoxime given on the
11th-15th days after major symptoms of poisoning appeared due to
extended exposure to fenitrothion (a dimethyl phosphate with a short
half-life for aging of inhibited AChE).
1.3.4.4 Dosage of atropine and oxime
The recommended doses above pertain to exposures, usual for an
occupational setting, but, in the case of very severe exposure or
massive ingestion (accidental or deliberate), the therapeutic doses
may be extended considerably. Warriner et al. (1977) reported the
case of a patient who drank a large quantity of dicrotophos, in
error, while drunk. Therapeutic dosages were progressively increased
up to 6 mg atropine iv every 15 min together with continuous iv
infusion of pralidoxime chloride at 0.5 g/h for 72 h, from days 3 to
6 after intoxication. After considerable improvement, the patient
relapsed and further aggressive therapy was given at a declining
rate from days 10 to 16 (atropine) and to day 23 (oxime),
respectively. In total, 92 g of pralidoxime chloride and 3912 mg of
atropine were given and the patient was discharged on the
thirty-third day with no apparent sequelae.
REFERENCES TO ANNEX I
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S (1977)
Acute fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
KAGAN, JU.S. (1977) [Toxicology of organophosphorus pesticides],
Moscow, Meditsina, pp. 111-121, 219-233, 260-269 (in Russian).
PLESTINA, R. (1984) Prevention, diagnosis, and treatment of
insecticide poisoning, Geneva, World Health Organization
(Unpublished document VBC/84.889).
TAYLOR, P. (1980) Anticholinesterase agents. In: Goodman, L.S. &
Gilman, A., ed. The pharmacological basis of therapeutics, 6th ed.,
New York, Macmillan Publishing Company, pp. 100-119.
UK DHSS (1983) Pesticide poisoning: notes for the guidance of
medical practitioners, London, United Kingdom Department of Health
and Social Security, pp. 41-47.
VALE, J.A. & SCOTT, G.W. (1974) Organophosphorus poisoning. Guy's
Hosp. Rep., 123: 13-25.
WARRINER, R.A., III, NIES, A.S., & HAYES, W.J., Jr (1977) Severe
organophosphate poisoning complicated by alcohol and terpentine
ingestion. Arch. environ. Health, 32: 203-205.
ANNEX II. No-observed-effect levels in plasma, red blood cells, and brain ChE, in animals treated with fenitrothion
ANNEX II. No-observed-effect levels in plasma, red blood cells, and brain ChE, in animals treated with fenitrothion
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Rat 30 days male 2.5 2.5 2.5 2.5 < 2.5 < 2.5 Trottier et al. (1980)
Gavage 0, 2.5, 5.0, 10.0, female 2.5 2.5 2.5 2.5 < 2.5 < 2.5
20.0 (mg/kg body weight)
Rat 5 weeksb male - - - 0.25 - 0.5 Carshalton (1964)
0, 5, 10, 20 mg/kg diet (5) (10)
Rat 90 daysb male 3.2 3.2 < 1.6 3.2 3.2 12.5 Misu et al. (1966)
0, 32, 63, 125, 250, 500 (63) (63) (< 32) (63) (63) (250)
mg/kg diet
Rat 90 days male - < 1.7 - < 1.7 - - Cooper (1966)
0, 20, 92.8, 430.7, 2000 (< 20) (< 20)
mg/kg diet female < 1.9 < 1.9 - -
(< 20) - (< 20)
Rat 90 days male - < 1.2 - 1.2 - - Cooper (1966)
0, 10, 46.4, 215, 1000 (< 10) (< 10)
(mg/litre water) female - < 1.3 - 1.3 - -
(< 10) (< 10)
Rat 6 months male - 1.8 - 1.8 - 1.8 Sumitomo Chem. Co.
0, 10, 30, 150 mg/kg diet (30) (30) (30) (1972)
female - < 0.64 - 0.64 - 0.64
(< 10) (< 10) (10)
Annex II (contd).
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Rat 34 weeksb male 0.25 0.25 < 0.25 < 0.25 - 6.3 Benes & Cerna (1970)
0, 10, 50, 150 (5) (5) (< 5) (< 5) (125)
0, 5, 25, 125 mg/kg diet female < 0.25 < 0.25 < 0.25 < 0.25 - 6.3
(< 5) (< 5) (< 5) (< 5) (125)
Rat 63 weeksb male - < 1.25 - 1.25 - 1.25 Ueda & Nishimura (1966)
0, 25, 100, 400 mg/kg diet (< 25) (25) (25)
Rat 1 year male 1.0 1.0 1.0 1.0 1.0 1.0 Ecobichon et al. (1980)
Gavage 0, 0.5, 1, 5, 10
(mg/kg body weight)
Rat 1 yearb male > 1.25 > 1.25 0.25 > 1.25 0.05 > 1.25 Kanoh et al. (1982)
0, 1, 5, 25 mg/kg diet (> 25) (> 25) (5) (> 25) (1) (> 25)
female > 1.25 > 1.25 0.25 > 1.25 0.25 > 1.25
(> 25) (> 25) (5) (> 25) (5) (> 25)
Rat 30 days male - 20 - 20 - 20 Breckenridge et al. (1982)
by inhalation 0, 6.7, female - < 6.7 - 20 - < 6.7
20, 60 (mg/m3)
Rat 92 weeksb male 0.13 > 0.5 0.25 > 0.5 - > 0.5 Kadota et al. (1975a)
0, 2.5, 5, 10 mg/kg diet (2.5) (> 10) (5) (> 10) (> 10)
female 0.13 > 0.5 0.25 > 0.5 - > 0.5
(2.5) (> 10) (5) (> 10) (> 10)
Annex II (contd).
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Rat 2 yearsb male < 0.5 < 0.5 0.5 0.5 0.5 1.5 Rutter & Nelson (1974)
0, 10, 30, 100 mg/kg diet (< 10) (< 10) (10) (10) (10) (30)
female < 0.5 < 0.5 0.5 0.5 0.5 1.5
(< 10) (< 10) (10) (10) (10) (30)
Mouse 2 yearsb male 0.43 0.43 1.43 1.43 1.43 1.43 Tamano et al. (1990)
0, 3, 10, 100, 1000 (3) (3) (10) (10) (10) (10)
mg/kg diet female 0.43 0.43 1.43 1.43 1.43 1.43
(3) (3) (10) (10) (10) (10)
Dog 98 days oral by male/ 2 - 2 - - - Cooper (1966)
capsule 0, 2, 9, 40
(mg/kg body weight)
Dog 1 yearb male 0.25 0.25 0.25 0.25 > 1.25 > 1.25 Griggs et al. (1984)
0.5, 10, 50 mg/kg diet (10) (10) (10) (10) (> 50) (> 50)
female 0.25 0.25 > 1.25 > 1.25 > 1.25 > 1.25
(10) (10) (> 50) (> 50) (> 50) (> 50)
Dog 10 months male - < 5 - < 5 - - Tomita et al. (1974)
oral by capsule
0, 5 (mg/kg body weight)
Dog 1 year female - < 2 - < 2 - - Ogata (1972)
oral by capsule
0, 2 (mg/kg body weight)
Annex II (contd).
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Dog 2 yearb male < 0.75 < 0.75 0.75 0.75 - 2.5 Mastalski et al. (1973)
0, 30, 100, 200 mg/kg diet (< 30) (< 30) (30) (30) (100)
female < 0.75 < 0.75 0.75 0.75 - 2.5
(< 30) (< 30) (30) (30) (100)
Rabbit 6 months male - <3 - < 3 - 3 Miyamoto et al. (1976a)
in diet, 0, 3, 10
(mg/kg body weight)
a I = Interim sacrifice; F = Final sacrifice.
b Calculated by dietary concentration providing that 1 mg/kg body weight per day equals 20, 7, and 40 mg/kg in
rats, mice, and dogs, respectively.
RESUME ET EVALUATION, CONCLUSIONS ET RECOMMANDATIONS
1. Résumé et évaluation
1.1 Exposition
Le fénitrothion est un insecticide organophosphoré utilisé
depuis 1959. On l'emploie en agriculture pour détruire les insectes
qui s'attaquent au riz, aux céréales, aux fruits, aux légumes, aux
céréales ensilées et au coton. On l'emploie également pour la
désinsectisation des forêts ainsi que pour la destruction des
mouches, des moustiques et des blattes dans le cadre des programmes
de santé publique. Il se présente sous la forme de concentrés
émulsionnables, de concentrés à très bas volume, de poudres, de
granulés, de poudres pour poudrage, de bouillies huileuses ainsi
qu'en association avec d'autres pesticides. La production de
fénitrothion oscille entre 15 000 et 20 000 tonnes par an.
Le fénitrothion pénètre dans l'air par volatilisation à partir
des surfaces contaminées et peut être entraîné pendant l'épandage
au-delà de la zone à traiter. Dans la plupart des terrains, il n'est
éliminé que très lentement par lessivage, néanmoins une certaine
quantité peut être entraînée par les eaux de ruissellement.
Le fénitrothion est décomposé par photolyse et hydrolyse. En
présence de rayonnement UV ou de lumière solaire, la demi-vie du
fénitrothion dans l'eau est inférieure à 24 heures. La présence
d'une microflore peut également accélérer la décomposition. En
l'absence de lumière solaire ou de contamination microbienne, le
fénitrothion est stable dans l'eau. Dans le sol, il est
essentiellement dégradé par voie biologique, encore que la photolyse
puisse jouer un certain rôle.
La concentration du fénitrothion dans l'air peut atteindre 5
µg/m3 immédiatement après l'épandage mais elle est susceptible de
diminuer fortement au cours du temps et en fonction de la distance
au lieu d'épandage. Dans l'eau, la concentration peut atteindre 20
µg/litre mais elle diminue rapidement.
En cas d'exposition continue, on observe des facteurs de
bioconcentration qui se situent entre 20 à 450 chez un certain
nombre d'espèces aquatiques.
Les résidus de fénitrothion dans les fruits, les légumes et les
céréales peuvent aller de 0,001 à 9,5 mg/kg immédiatement après le
traitement mais ils diminuent rapidement, leur demi-vie étant de 1 à
2 jours.
1.2 Fixation, métabolisme et excrétion
Le fénitrothion est rapidement résorbé dans les voies
digestives et se répartit ensuite dans les divers tissus. La
demi-vie du fénitrothion après absorption percutanée chez le singe a
été estimée à 15-17 heures. Sa métabolisation s'effectue selon les
grandes voies d' O-déméthylation ainsi que par clivage de la
liaison P-O-aryle. Le groupement NO2 est réduit par la microflore
intestinale, mais seulement chez les ruminants. La principale voie
d'élimination est la voie urinaire, la plupart des métabolites étant
excrétés en l'espace de 2 à 4 jours chez le rat, le cobaye, la
souris et le chien. Les principaux métabolites observés sont le
déméthyl- fénitrothion, le déméthyl-fénitrooxon, l'acide
diméthylphosphoro-thioïque et l'acide diméthylphosphorique ainsi que
le 3-méthyl-4-nitrophénol et ses conjugués. Les différences qui ont
été observées dans la composition en métabolites entre la plupart
des animaux de laboratoire et entre les sexes, chez une même espèce,
semblent être essentiellement d'ordre quantitatif. Seul le lapin
semble excréter du fénitrooxon et de l'aminofénitrooxon en quantités
faibles mais néanmoins mesurables, par la voie urinaire.
Des études portant sur des lapins et des chiens montrent que le
fénitrothion se dépose préférentiellement dans les tissus adipeux.
Des résidus qui avaient été décelés dans le lait de vache après
exposition à du fénitrothion ont disparu dans les deux jours.
Même s'il est rapidement résorbé après administration orale, le
fénitrothion est métabolisé et excrété sans délai et il est peu
probable qu'il demeure dans l'organisme pendant une longue période.
1.3 Effets sur les êtres vivants dans leur milieu naturel
A la concentration où on le rencontre vraisemblablement dans
l'environnement, le fénitrothion n'a aucun effet sur les
microorganismes présents dans le sol ou dans les eaux.
Il est extrêmement toxique pour les invertébrés aquatiques
d'eau douce ou d'eau de mer, la CL50 étant de l'ordre de quelques
µg/litre pour la plupart des espèces étudiées. La dose sans effet
observable pour la daphnie, lors d'épreuves à 48 heures, s'est
révélée < 2 µg/litre; lors de tests portant sur la totalité du
cycle évolutif, on a fixé à 0,14 µg/litre la concentration maximum
acceptable de produit toxique. Les observations et études effectuées
sur le terrain dans des étangs expérimentaux ont révélé l'existence
d'effets sur les populations d'invertébrés. Toutefois, la plupart
d'entre eux étaient passagers, même à des concentrations beaucoup
plus élevées que celles auxquelles donne vraisemblablement lieu
l'emploi de fénitrothion conformément aux recommandations.
Les poissons sont moins sensibles au fénitrothion que les
invertébrés, les valeurs de la CL50 à 96 heures dans leur cas
allant de 1,7 à 10 mg/litre. Ce sont les jeunes larves qui
constituent le stade le plus sensible. Des études à long terme ont
fixé à 0,1 mg/litre ou plus la concentration maximale admissible de
produit toxique pour deux espèces de poissons d'eau douce. Une étude
écologique effectuée après l'épandage de fénitrothion sur des forêts
a montré qu'il n'en résultait aucun effet sur les populations
sauvages de poissons ou sur la survie des poissons d'expérience
placés dans des nasses, les concentrations de fénitrothion dans
l'eau allant jusqu'à 0,019 mg/litre. Des épandages répétés de
fénitrothion sur des forêts n'ont eu aucun effet sur les poissons.
Lors d'essais en laboratoire, on a constaté que la CL50 pour
les mollusques dulçaquicoles allait de 1,2 à 15 mg/litre. Aucun
effet écologique n'a été constaté après épandage sur des forêts à la
dose de 140 g/hectare.
Le fénitrothion est extrêmement toxique pour les abeilles
(DL50 topique comprise entre 0,03 et 0,04 µg/abeille). Des effets
écologiques ont été observés qui consistaient essentiellement en une
mortalité localement élevée chez les abeilles et d'autres espèces.
Toutefois cette mortalité ne représentait qu'un faible pourcentage
de la population totale des ruches.
Pour les oiseaux, les valeurs de la DL50 aiguë par voie orale
vont de 25 à 1190 mg/kg de poids corporel et, dans la plupart des
cas, la CL50 alimentaire à huit jours dépasse 5000 mg/kg de
nourriture. Les valeurs de la dose sans effet observable sur la
reproduction sont de 10 mg/kg de poids corporel pour la caille et de
100 mg/kg pour le malard. Après épandage de fénitrothion à la dose
de 280 g/hectare, on a observé une mortalité parmi les oiseaux
chanteurs, mortalité qui augmentait sensiblement chez les espèces
peuplant la canopée lorsque la dose était portée à 500 g/hectare.
Après épandage à des doses de 420 g/hectare puis de 210 g/hectare
quelques jours plus tard, le nombre de nichées de pinsons à gorge
blanche (Zonotrichia albicollis) a diminué. Dans de nombreuses
études, on a constaté, chez les oiseaux chanteurs, une inhibition de
la cholinestérase peu après l'épandage de fénitrothion dans les
forêts.
L'observation sur le terrain n'a pas révélé d'effets sur les
populations de petits mammifères sauvages.
1.4 Effets sur les animaux d'expérience et les systèmes d'épreuve
in vitro
Le fénitrothion est un organophosphoré qui réduit l'activité de
la cholinestérase plasmatique, érythrocytaire, cérébrale et
hépatique. Il est métabolisé en fénitrooxon, composé dont la
toxicité aiguë est encore plus élevée. La toxicité du fénitrothion
peut être potentialisée par un certain nombre d'organophosphorés.
Le fénitrothion est un insecticide modérément toxique dont la
DL50 par voie orale chez le rat et la souris va de 330 à 1416
mg/kg de poids corporel. Sa toxicité aiguë par voie dermique chez
les rongeurs varie de 890 mg/kg de poids corporel à plus de 2500
mg/kg.
Le fénitrothion n'est que très peu irritant pour les yeux et
n'irrite pas la peau. Deux études portant sur des cobayes ont fait
ressortir une certaine tendance à la sensibilisation cutanée.
Le fénitrothion a fait l'objet d'études à court terme sur des
rats, des chiens, des cobayes, des lapins ainsi que d'études de
cancérogénicité à long terme chez des rats et des souris. Les études
à court terme sur les rats et les chiens ont permis de fixer
respectivement à 10 mg/kg de nourriture et 50 mg/kg de nourriture la
dose sans effet nocif observable, d'après l'activité de la
cholinestérase cérébrale.
Les études à long terme sur le rat et la souris ont donné une
dose sans effet nocif observable, basée sur l'activité de la
cholinestérase cérébrale, de 10 mg/kg de nourriture.
Aucune des études à long terme signalées n'a mis en évidence
d'effets cancérogènes.
Le fénitrothion ne s'est pas révélé mutagène dans les études
in vitro et in vivo.
A des doses allant jusqu'à 30 mg/kg de poids corporel chez le
lapin et jusqu'à 25 mg/kg de poids corporel chez le rat, le
fénitrothion n'a pas produit d'effets tératogènes. Lorsque la dose
dépassait 8 mg/kg de poids corporel, le fénitrothion était toxique
pour la mère.
Après exposition in utero, on a observé des déficits
comportementaux après la naissance chez des ratons en cours de
développement. La dose sans effet observable sur le comportement a
été fixée à 5 mg/kg de poids corporel et par jour.
Des études de reproduction portant sur plusieurs générations de
rats n'ont pas fait ressortir d'effets morphologiques. Ces études
ont permis de fixer à 120 mg/kg de nourriture, d'après les
paramètres génésiques, la dose sans effet nocif observable.
On a fait état d'une neurotoxicité retardée après exposition au
fénitrothion.
1.5 Effets sur l'homme
Administré à des volontaires humains en dose orale unique de
0,042 à 0,33 mg/kg de poids corporel puis en doses répétées de 0,04
à 0,08 mg/kg, le fénitrothion n'a pas entraîné d'inhibition de la
cholinestérase plasmatique ou érythrocytaire. Un des métabolites, le
3-méthyl-4-nitrophénol, a été totalement excrété par voie urinaire
dans les 24 heures.
Plusieurs cas d'intoxication se sont produits. La
symptomatologie de l'intoxication par le fénitrothion est celle
d'une stimulation du parasympathique. Il est arrivé que les
manifestations toxiques n'apparaissent pas immédiatement mais
récidivent pendant quelques mois. On a avancé que l'allongement de
la durée de l'évolution clinique de l'intoxication et l'apparition
de symptômes tardifs pouvaient s'expliquer par une libération lente
de l'insecticide à partir du tissu adipeux. Certaines dermatites de
contact ont été attribuées à une exposition à l'insecticide. Rien
n'indique qu'une exposition au fénitrothion puisse entraîner une
neurotoxicité retardée ou l'apparition d'un syndrome de Reye.
Dans le cadre des programmes de l'OMS, on utilise du
fénitrothion dans quelques pays pour la lutte antipaludique en
pulvérisation à effet rémanent à l'intérieur des habitations (dose
d'emploi: 2,0 gr de matière active par m2). Des observations
portant sur plusieurs milliers de résidents n'ont fait ressortir
aucun signe de toxicité à l'exception d'une enquête au cours de
laquelle 2% des personnes enquêtées se sont plaintes de légers
symptômes. Toutefois, chez environ 25% des ouvriers pulvériseurs, on
a observé une inhibition à 50% de l'activité de la cholinestérase du
sang total. Après épandage par voie aérienne d'un concentré
émulsionnable à 50%, on a constaté chez un certain nombre de
travailleurs des symptômes d'intoxication accompagnés d'une
réduction de l'activité cholinestérasique du sang total en l'espace
de 48 heures. Des ouvriers et des ouvrières exposés de par leur
profession pendant cinq ans à du fénitrothion respectivement dans un
atelier de production et dans une unité de conditionnement ont
présenté, pour 15% d'entre eux et 33% d'entre elles, des symptômes
cliniques d'intoxication. La concentration de fénitrothion dans
l'air des lieux de travail variait entre 0,028 et 0,118 mg/m3.
2. Conclusions
* Le fénitrothion est un insecticide organophosphoré modéré- ment
toxique. Toutefois, en cas d'exposition excessive résultant de
manipulations au cours de la production ou de l'épandage ou
encore par suite d'une ingestion accidentelle ou
intentionnelle, il peut s'ensuivre une grave intoxication.
* L'exposition de la population générale, qui résulte
principalement de l'emploi du fénitrothion en agriculture, en
foresterie et dans les programmes de santé publique, ne devrait
pas présenter de danger pour la santé.
* Moyennant de bonnes pratiques de fabrication et le respect des
mesures d'hygiène et de sécurité, le fénitrothion ne devrait
pas présenter de danger pour les personnes qui y sont exposées
de par leur profession.
* Malgré sa forte toxicité pour les arthropodes non visés, le
fénitrothion est utilisé très largement comme pesticide avec
des effets indésirables minimaux sur les populations animales
présentes dans l'environnement.
3. Recommandations
* Afin de préserver la santé et le bien-être des travailleurs et
de la population générale, il importe de ne confier la
manipulation et l'épandage du fénitrothion qu'à des personnes
expérimentées qui prendront les mesures de sécurité nécessaires
et procéderont à l'épandage en respectant les règles de bonne
pratique.
* Les opérations de production, de formulation, d'épandage et
d'élimination du fénitrothion doivent être effectuées avec tout
le soin nécessaire pour réduire au minimum la contamination de
l'environnement, et notamment des eaux de surface.
* Les travailleurs habituellement exposés doivent subir un examen
médical périodique.
* La dose d'emploi du fénitrothion doit être limitée afin
d'éviter tout effet sur les arthropodes non visés.
L'insecticide ne doit jamais être épandu sur des étendues ou
cours d'eau.
RESUMEN Y EVALUACION, CONCLUSIONES Y RECOMENDACIONES
1. Resumen y evaluación
1.1 Exposición
El fenitrotión es un insecticida organofosforado que se viene
utilizando desde 1959. Se emplea en la agricultura para combatir los
insectos del arroz, los cereales, las frutas, las hortalizas, el
grano almacenado y el algodón. Se usa también para luchar contra
insectos en los bosques, y en programas de salud pública para
combatir moscas, mosquitos y cucarachas. Se ha formulado como
concentrado emulsionable, concentrados de volumen muy bajo,
microgránulos, gránulos, polvo, pulverizadores oleosos y en
combinación con otros plaguicidas. Cada año se fabrican entre 15 000
y 20 000 toneladas.
El fenitrotión se incorpora al aire por volatilización a partir
de superficies contaminadas y puede ser arrastrado fuera de la zona
de tratamiento durante el rociamiento. Su lixiviación es muy lenta
en la mayoría de los suelos, pero es previsible que se produzca
algún arrastre en el agua.
Se degrada por fotólisis e hidrólisis. En presencia de la
radiación ultravioleta o de la luz solar, la semivida del
fenitrotión en el agua es inferior a 24 horas. La presencia de
microflora también puede acelerar la degradación. En ausencia de luz
solar o de contaminación microbiana, es estable en el agua. En el
suelo, la vía principal de eliminación es la biodegradación, aunque
la fotólisis también puede influir.
Las concentraciones de fenitrotión en el aire pueden ser de
hasta 5 µg/m3 inmediatamente después del rociamiento, pero
decrecen de manera considerable con el tiempo y la distancia del
lugar de aplicación. Los niveles en el agua pueden alcanzar valores
de hasta 20 µg/litro, pero disminuyen con rapidez.
Los factores de bioconcentración calculados en varias especies
acuáticas sometidas a una exposición constante al fenitrotión varían
entre 20 y 450.
Sus niveles residuales en frutas, hortalizas y grano de
cereales oscilan entre 0,001 y 9,5 mg/kg inmediatamente después del
tratamiento, pero decrecen rápidamente, con una semivida de 1 a 2
días.
1.2 Ingestión, metabolismo y excreción
El fenitrotión se absorbe con rapidez del tracto intestinal de
los animales de experimentación y se distribuye a diversos tejidos
corporales. La semivida en el caso de la absorción cutánea en el
mono fue de 15 a 17 horas. Se ha demostrado que su metabolismo se
realiza a través de las principales vías de demetilación oxidativa y
por rotura del enlace P-O-arilo. Los microorganismos intestinales
reducen, sólo en los rumiantes, el grupo nitrogenado del
fenitrotión. La principal vía de excreción es la orina; la mayor
parte de los metabolitos se eliminan en un periodo de 2 a 4 días en
el caso de la rata, el cobayo, el ratón y el perro. Los principales
metabolitos detectados son el demetilfenitrotión, el
demetilfenitrooxón, el ácido dimetilfosforotioico, el ácido
dimetilfosfórico y el 3-metil-4-nitrofenol y sus conjugados. Las
diferencias en la composición de los metabolitos que se detectan en
la mayor parte de los animales de laboratorio y entre los dos sexos
de la misma especie parecen ser principalmente de carácter
cuantitativo. Sólo los conejos parecen excretar fenitrooxón y
aminofenitrooxón en cantidades pequeñas, pero cuantificables, con la
orina.
En estudios realizados en conejos y perros se ha puesto de
manifiesto que el fenitrotión se deposita preferentemente en el
tejido adiposo.
Los residuos encontrados en la leche de vaca tras la exposición
no se detectaban dos días más tarde.
Aunque el fenitrotión se absorbe rápidamente por vía oral, se
metaboliza y excreta con rapidez y no es probable que permanezca en
el organismo durante un tiempo prolongado.
1.3 Efectos en los seres vivos del medio ambiente
La concentraciones normales de fenitrotión en el medio ambiente
no tienen efecto alguno en los microorganismos del suelo o del agua.
El fenitrotión es muy tóxico para los invertebrados acuáticos
de agua dulce y salada; los valores de la CL50 son de pocos
µg/litro en la mayoría de las especies analizadas. El nivel sin
efectos observados (NOEL) determinado en Daphnia en pruebas de 48
horas fue < 2 µg/litro; en pruebas del ciclo biológico se
estableció una concentración tóxica aceptable máxima (MATC) de 0,14
µg/litro. Las observaciones y los estudios sobre el terreno en
estanques experimentales han puesto de manifiesto ciertos efectos en
las poblaciones de invertebrados, si bien la mayor parte de los
cambios observados fueron transitorios, incluso con concentraciones
muy superiores a las que cabe esperar tras el uso recomendado.
Los peces son menos sensibles al fenitrotión que los
invertebrados y muestran valores de la CL50 a las 96 horas que
oscilan entre 1,7 y 10 mg/litro. La fase más sensible del ciclo
biológico es la larva joven. En estudios prolongados se ha
establecido una MATC igual o superior a 0,1 mg/litro para dos
especies de peces de agua dulce. Los estudios sobre el terreno tras
la aplicación de fenitrotión a bosques no demostraron efecto alguno
en las poblaciones silvestres de peces ni en la supervivencia de
peces de experimentación en vivero, con concentraciones medidas en
el agua de 0,019 mg/litro. La aplicación repetida en bosques no tuvo
efectos en las poblaciones de peces.
En pruebas de laboratorio, los valores de la CL50 para
moluscos de agua dulce oscilaban entre 1,2 y 15 mg/litro. No se
observaron efectos sobre el terreno tras rociar bosques con 140
g/ha.
El fenitrotión es muy tóxico para las abejas (DL50 tópica,
0,03-0,04 µg/abeja). Se han comunicado efectos sobre el terreno, con
un elevado número de abejas de la miel y de otras especies muertas
en la zona de aplicación. Sin embargo, el número total de individuos
muertos representaba solamente un pequeño porcentaje de la población
de las colmenas.
Los valores de la DL50 en la toxicidad aguda por vía oral en
las aves oscilan entre 25 y 1190 mg/kg de peso corporal, y en la
mayor parte de las dietas de ocho días las CL50 excedieron de 5000
mg/kg de la dieta. Los valores del NOEL para la reproducción fueron
de 10 mg/kg de peso corporal en la codorniz y de 100 mg/kg de peso
corporal en el pato silvestre. Poco después de aplicar fenitrotión
en una concentración de 280 g/ha, se observaron muertes de pájaros
cantores y la mortalidad aumentó considerablemente con 560 g/ha para
las especies que vivían en las copas de los árboles del bosque.
Después de rociar con una concentración de 420 g/ha y unos días más
tarde con 210 g/ha, se redujo la reproducción de la especie
zonotrichia albicollis. En muchos estudios, los pájaros cantores
mostraron inhibición de la colinesterasa inmediatamente después de
rociar los bosques con fenitrotión.
Las observaciones sobre el terreno no han puesto de manifiesto
efecto alguno del fenitrotión en las poblaciones de pequeños
mamíferos silvestres.
1.4 Efectos en los animales de experimentación y en sistemas
de prueba in vitro
El fenitrotión es un organofosfato y reduce la actividad de la
colinesterasa en el plasma, los eritrocitos y los tejidos cerebral y
hepático. Se metaboliza a fenitrooxón, con mayor toxicidad aguda.
Otros compuestos organofosfatados pueden potenciar aún más la
toxicidad del fenitrotión.
El fenitrotión es un insecticida de toxicidad moderada, con
valores de la DL50 por vía oral que oscilan entre 330 y 1416 mg/kg
de peso corporal en ratas y ratones. La toxicidad aguda cutánea en
roedores varió desde 890 mg/kg hasta más de 2500 mg/kg de peso
corporal.
El fenitrotión apenas irrita los ojos y no irrita la piel. En
uno de los dos estudios realizados en cobayos, el producto mostró
cierto potencial de sensibilización dérmica.
Se ha ensayado el fenitrotión en estudios de corta duración en
ratas, perros, cobayos y conejos y en estudios prolongados de
carcinogenicidad en ratas y ratones. En los estudios de corta
duración realizados en ratas y perros los niveles sin efectos
adversos observados (NOAEL), basados en la actividad de la
colinesterasa cerebral, fueron, respectivamente, de 10 mg/kg y 50
mg/kg de la dieta.
Los estudios de larga duración en ratas y ratones pusieron de
manifiesto un NOAEL (basado en la actividad de la colinesterasa
cerebral) de 10 mg/kg de la dieta.
No se han encontrado efectos carcinogénicos en ninguno de los
estudios de larga duración que se han publicado.
En los estudios in vitro e in vivo, el fenitrotión no
mostró efectos mutagénicos.
No se han detectado efectos teratogénicos con dosis de
fenitrotión de hasta 30 mg/kg de peso corporal en conejos y de hasta
25 mg/kg de peso corporal en ratas. Las dosis superiores a 8 mg/kg
de peso corporal produjeron toxicidad materna.
Las ratas jóvenes en fase de crecimiento presentaron problemas
de comportamiento postnatal tras la exposición in utero. Para este
efecto se estableció un NOEL de 5 mg/kg de peso corporal al día.
En estudios multigeneracionales de reproducción en ratas no se
detectó ningún efecto morfológico. En estos estudios se puso de
manifiesto un NOAEL de 120 mg/kg de la dieta, basado en parámetros
de la reproducción.
No se ha informado de neurotoxicidad retardada como
consecuencia de la exposición al fenitrotión.
1.5 Efectos en el ser humano
La administración de fenitrotión en una dosis oral única de
0,042 a 0,33 mg/kg de peso corporal y en dosis repetidas de 0,04 a
0,08 mg/kg de peso corporal en voluntarios humanos no causó la
inhibición de la colinesterasa del plasma y los eritrocitos. La
excreción urinaria del metabolito 3-metil-4-nitrofenol fue completa
en 24 horas.
Se han registrado varios casos de intoxicación, con los signos
y síntomas característicos de la estimulación parasimpática. En
algunos casos, las manifestaciones tóxicas tardaron en aparecer y se
repitieron durante unos meses. Se ha sugerido que la liberación
lenta del insecticida a partir del tejido adiposo puede dar lugar a
un curso clínico prolongado o a síntomas tardíos de intoxicación. En
algunos casos, la dermatitis de contacto se ha atribuido a la
exposición al insecticida. No hay pruebas de neurotoxicidad tardía
ni de relación con el síndrome de Reye.
El fenitrotión se ha utilizado en algunos países en programas
de la OMS para el rociamiento residual de interiores en la lucha
contra el paludismo (dosis de aplicación: 2,0 g de principio
activo/m2). No se apreciaron indicios de toxicidad en los miles de
habitantes observados, a excepción de un estudio en el que se
presentaron trastornos leves en menos del 2% de los habitantes. Sin
embargo, alrededor del 25% de los encargados del rociamiento
mostraron una inhibición de hasta el 50% de la actividad de la
colinesterasa en sangre total. En las 48 horas posteriores a la
aplicación aérea de una fórmula de concentrado emulsionable al 50%,
algunos trabajadores mostraron síntomas de intoxicación y una
disminución de la actividad de la colinesterasa en sangre total. En
una fábrica de producción, la exposición profesional durante más de
5 años de los trabajadores varones y de las mujeres del departamento
de envasado produjo signos clínicos y síntomas de intoxicación en el
15% de los hombres y en el 33% de las mujeres. La concentración de
fenitrotión medida en el aire del lugar de trabajo oscilaba entre
0,028 y 0,118 mg/m3.
2. Conclusiones
* El insecticida fenitrotión es un éster organofosforado
moderadamente tóxico. Sin embargo, la exposición excesiva
durante su fabricación o uso y la ingestión accidental o
intencional pueden ocasionar una intoxicación grave.
* La exposición de la población general, debida fundamentalmente
a las prácticas agrícolas y forestales y a los programas de
salud pública, no representa en principio amenaza alguna para
la salud.
* Con buenas prácticas de trabajo, medidas higiénicas y
precauciones de seguridad no es probable que el fenitrotión
represente un riesgo para las personas sujetas a exposición
profesional.
* A pesar de su elevada toxicidad para los artrópodos no
destinatarios, el fenitrotión se ha utilizado ampliamente en la
lucha contra las plagas, con pocos efectos adversos o ninguno
en las poblaciones presentes en el medio ambiente.
3. Recomendaciones
* Para salvaguardar la salud y el bienestar de los trabajadores y
de la población general, el manejo y la aplicación del
fenitrotión sólo se deben encomendar bajo una atenta
supervisión a personas bien capacitadas que se ajusten a las
medidas de seguridad adecuadas y utilicen el fenitrotión
correctamente.
* Se cuidarán especialmente la fabricación, la formulación, el
uso y la eliminación del compuesto, a fin de reducir al mínimo
la contaminación del medio ambiente, en particular de las aguas
superficiales.
* Los trabajadores regularmente expuestos deben someterse a
revisiones médicas periódicas.
* Se ha de limitar el número de aplicaciones de fenitrotión, para
evitar los efectos en artrópodos no destinatarios. No se deben
rociar jamás con este insecticida masas o corrientes de agua.
m-Methyl-14C-fenitrothion, administered orally at a dose of
15 mg/kg to Wistar rats, ICR mice, native Japanese rabbits, and
Beagle dogs, was readily absorbed from the gastrointestinal tract
and distributed to various tissues with maximum concentrations of
0.093 mg/kg (rats) - 0.144 mg/kg (dogs) after 1-3 h of the
treatment, and the radiocarbon was rapidly and completely
eliminated, mainly in the urine of rats (89-95%), mice (> 90%),
rabbits (86-94%), and dogs (88%) (Miyamoto et al., 1976b).
Examination of 11 rat tissues, including fat and muscle, revealed
that the concentrations of fenitrothion ranged from 0.004 mg/kg to
less than 0.001 mg/kg (except in fat-0.034 mg/kg), 24 h after
administration. Whole body autoradiography also indicated the rapid
disappearance of the radiocarbon from the tissues of mice. The
cumulative patterns of elimination were essentially the same among
these animals when radioactive fenitrothion was given at 15 mg/kg
body weight. Similar patterns were obtained in rats when
fenitrothion was administered at 105 mg/kg body weight or 15 mg/kg
body weight after a total of 5 pretreatments with non-radioactive
fenitrothion at 15 mg/kg body weight every other day. Thin-layer
chromatographic analysis of the urinary metabolites showed the
absence of intact fenitrothion and the presence of as many as 18
metabolites, of which 17 (92-99% of radioactivity) were identified.
The differences in the composition of the metabolites among these
animal species and between males and females of the same species
were mostly quantitative. The percentage of the metabolites
retaining the P-O-aryl linkage varied with animal species, ranging
from 8.6% (rabbits) to more than 56.9% (dogs). Most of these
metabolites were demethylated products [7,20] at the O-methyl
position. Rats and mice tended to eliminate greater amounts (15-26%)
of demethyl fenitrooxon [20] than rabbits and dogs (2-6%).
3-Methyl-4-nitrophenol [9], free or as conjugates with sulfuric acid
[26] or glucuronic acid [27], constituted another group of major
metabolites. Approximately 50-75% of the urinary radioactivity was
accounted for by these three metabolites ([9] [26] [27]), except in
dogs (36%). The urinary activity in rabbits (64-75%) exceeded that
in other animal species (50-60%). A trace amount of the oxidized
phenols (0.5-2%) [10,22] was present in rats and rabbits. Rabbits
were exceptional in excreting fenitrooxon [1] and animofenitrothion
[13], though in small amounts (0.1-3.7%). The urine of rabbits and,
to a lesser extent, of rats contained several minor metabolites
derived from aminofenitrothion [13] or from 3-methyl-4-aminophenol
[28]. The minor products totalled approximately 15% of the excreted
radiocarbon in rabbits and about 12% in rats. A few metabolites had
unique structures resulting from the reduction of the nitro group
and oxidation of the m-methyl group [29]. An appreciable amount of
the excreted metabolites from the 4 animal species was in the form
of conjugates. It appeared that rabbits were most active in this
regard, and dogs least active. In the 4 species treated, higher
amounts of sulfate conjugates than glucuronides were excreted. Thus,
fenitrothion was
metabolized through two major pathways, via O-demethylation and via
cleavage of the P-O-aryl bond. Neither S-(3-methyl-4-nitrophenyl)
glutathione mediated by glutathione S-aryltransferase nor ring
hydroxylation products have been positively demonstrated (Miyamoto
et al., 1976b).
Although reduction of the nitro group in the fenitrothion
molecule, presumably by intestinal microorganisms, was a minor
metabolic pathway in the above 4 animal species, it was the major
metabolic pathway in ruminants. In fact, a study of female goats
revealed that most of the urinary and faecal metabolites of
fenitrothion were amino derivatives [13,14,30] and were formed most
probably in rumen fluid (Mihara et al., 1978).
The metabolism of fenitrothion in Wistar rats with hepatic
lesions induced experimentally by dietary administration of
diaminodiphenyl-methane (DDM), by feeding a low protein-high fat
diet (LPHF), or by intramuscular treatment with carbon
tetrachloride, was also investigated. The in vitro rates of
degradation of fenitrothion were significantly reduced in all
preparations from livers with induced hepatic lesions, with the
greatest reduction produced by LPHF followed by carbon
tetra-chloride and DDM. The rate of degradation of fenitrothion and
fenitrooxon [1] was less severely affected than the rate of
activation of fenitrothion, because of the greater activity of
glutathione S-alkyltransferase (Miyamoto et al., 1977a). However,
the cumulative excretion patterns of radiocarbon were not altered by
these three hepatic lesions after oral administration to the injured
rats of either 15 or 50 mg 14C-fenitrothion/kg body weight, or 15
mg/kg body weight of the compound after pretreatment with approx.
2.8 mg/kg body weight per day of nonactive fenitrothion for 4 weeks.
Moreover, the injured rats metabolized fenitrothion equally as well
in vivo, as the control animals, even though the pathway yielding
demethyl fenitrothion [7] predominated. Higher or short-term doses
of fenitrothion tended to reduce the metabolic differences observed
between the injured and the control animals (Miyamoto et al.,
1977b). These results indicated that even very severe hepatic
lesions, such as those described here, hardly influenced the in vivo
detoxification of fenitrothion and the excretion of its metabolites.
The results also indicated that there was not much possibility of
retention or storage of the parent compound and/or its toxic
metabolites in the mammalian body under such unfavourable
conditions.
The dermal penetration of 14C-ring-labelled fenitrothion and
aminocarb was determined in male, rhesus monkeys and male,
Sprague-Dawley rats. In monkeys, 49 ± 4% of the fenitrothion
(urinary excretion half-life = 14 h) was absorbed from the
forehead, while 21 ± 10% of the fenitrothion (half-life = 17 h) was
absorbed from the ventral forearm. Monkey forehead was 2.3 times
more permeable than the forearm. In rats, 84 ± 12% of the
fenitrothion (half-life = 20 h) was absorbed from the middorsal
region (Moody & Franklin, 1987).
The presence of N,N-diethyl-m-toluamide (DEET), used as an
insect repellent by workers spraying fenitrothion, increased the
absorption of 14C fenitrothion through rat skin (32% DEET; 15%
control with acetone) and monkey skin (9.7% DEET; 3.2% control)
(Moody et al., 1987b).
6.2 Retention and turnover
To evaluate the possible retention of fenitrothion residues in
animal tissues, methyl-14C-fenitrothion was administered orally to
male Wistar rats at 15 mg/kg body weight per day for 7 days, and
then at 30 mg/kg body weight per day for a further 3 days. A
measurable amount (0.09 mg/kg) of fenitrothion was found in
abdominal fat and athe level increased (2.4 mg/kg) at the later
staes of administration. This amount, however, tended to disappear
quite rapidly on cessation of the administration (Miyamoto, 1977c).
Following administration of 10 or 3 mg fenitrothion/kg body
weight per day to male native Japanese rabbits for 6 months, blood,
skeletal muscle, and abdominal fat were analysed by gas
chromatography for fenitrothion and fenitrooxon. In most cases,
blood and muscle did not contain any detectable amounts of either
compound (detection limit for fenitrothion 0.005 or 0.002 mg/kg, and
that of fenitrooxon, 0.01 mg/kg). Averages of 0.131 mg
fenitrothion/kg (0.243 mg/kg maximum) and of 0.045 mg/kg were
measured in the fat of rabbits dosed at 10 and 3 mg/kg body weight
per day, respectively, while muscle contained a maximum of 0.006 mg
fenitrothion/kg. No fenitrooxon was detected (Miyamoto et al.,
1976a).
Three male beagle dogs were treated orally with 5 mg
fenitrothion/kg body weight per day, 6 days per week, for 10 months.
After termination of the treatment, only the fat contained trace
amounts of fenitrothion (maximum of 0.160 mg/kg) (Tomita et al.,
1974).
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
A more complete treatise on the effects of organophosphorus
insecticides in general, especially their short- and long-term
effects on the nervous system, can be found in Environmental Health
Criteria 63: Organophosphorus insecticides - A general introduction
(WHO, 1986).
No-observed-effect levels on the ChE in the plasma,
erythrocytes, and brain of animals treated with fenitrothion under
various conditions are summarized in Annex II.
7.1 Single exposure
As with other organophosphorus insecticides, a large dose of
fenitrothion produces toxic signs and symptoms in various animals by
the inhibition of acetylcholinesterase (AChE) in the nervous system,
with the consequent accumulation of excessive levels of
acetylcholine. The major toxic signs in experimental animals are
salivation, tremor, exophthalmos, urinary incontinence,
piloerection, and dyspnoea, which are similar to the toxic signs
elicited by other organophosphorus insecticides (Namba, 1971).
The acute toxicity of fenitrothion varies according to the
species of the test animal, its sex, the route and method of
administration and/or the solvent used, as shown in Table 4. The
acute oral LD50s in mammals range from 330 mg/kg in the rat to
1850 mg/kg in the guinea-pig, the dermal LD50s range from
890 mg/kg in the rat to > 2500 mg/kg in the mouse.
Groups of 8 male and 8 female Sprague-Dawley rats were exposed
to fenitrothion mists dissolved in a mixture of deodorized kerosene
and xylene (particles mostly less than 3 µ in diameter) to assess
inhalation toxicity at aerial concentrations of 10, 70, and 186
mg/m3 for 2, 4, or 8 h. No significant toxic signs were observed
in the rats exposed to an aerial concentration of 10 mg/m3 for 2
and 4 h. With longer exposure and/or higher fenitrothion
concentrations, toxic symptoms developed including salivation,
urinary incontinence, tremor, lacrimation, muscular fibrillation,
and dyspnoea, and, after 8 h exposure to 186 mg fenitrothion/m3, 2
out of 8 males died. A transient body weight decrease was observed
after 8 h exposure to 10 and 70 mg fenitrothion/m3. A marked body
weight decrease was observed at a concentration of 186 mg/m3. The
LC50 value for fenitrothion in rats exposed for 8 h, was estimated
to be more than 186 mg/m3 (Kohda & Kadota, 1979).
Table 4. Acute toxicity of fenitrothion
Animal Strain Sex Route LD50 (mg/kg) Vehicle Reference
Mouse male oral 1336 Carshalton (1964)
female oral 1416
dd male oral 1030 10%Tween 80 aq. Miyamoto & Kadota (1972)
dd female oral 1040 10%Tween 80 aq.
dd male dermal > 2500 Undiluted Kadota et al. (1972)
female dermal > 2500 Undiluted
Rat Sherman male oral 740 Peanut oil Gaines (1969)
Sherman female oral 570 Peanut oil
Sprague-Dawley male oral 330 10%Tween 80 aq. Miyamoto & Kadota (1972)
Sprague-Dawley female oral 800 10%Tween 80 aq.
Wistar male oral 940 Benes & Cerna (1970)
Wistar female oral 600
Sprague-Dawley male dermal 890 Undiluted Kadota et al. (1972)
Sprague-Dawley female dermal 1200 Undiluted
Guinea-pig male oral 500 DuBois & Puchala (1960)
oral 1850 Sorpol 2001 Miyamoto et al. (1963b)
Dog Beagle male/ oral > 681 Gelatin capsule Mastalski (1971b)
female
Hen White Leghorn oral 500 10%Tween 80 aq. Kadota et al. (1975b)
The pulmonary toxicity of fenitrothion was evaluated following
a single exposure of caged rats (n = 160) placed in a field
oversprayed by aircraft during an operational forest treatment.
Depending on the position of the cages, the air concentrations of
fenitrothion measured in the first 30 min after spraying, were 6.19
and 33.2 µg/m3. Plasma pseudocholinesterase activity and pulmonary
alveoli ultrastructure were used as indices of fenitrothion
exposure. Although a few signs of toxic lung injury were observed on
days 3 and 7, there were no cholinergic signs or effects on
pseudocholinesterase activity within 12 h in exposed animals,
compared with controls. The alveolar toxic reaction was limited to
small and discrete foci, and was entirely reversible within a period
of 2 months (Coulombe et al., 1986).
Rat lungs were examined with light and electron microscopes,
3-60 days after exposure to fenitrothion. Groups of 9 male,
Sprague-Dawley rats were exposed by a "nose-only" apparatus for 1 h
to an aerosol of fenitrothion (15%) mixed with non aromatic
hydrocarbon solvent and oil diluent at 2 or 500 mg/m3. Only minor
modifications of lung alveolar tissue were observed after exposure
to the higher concentration. At 3 days, discrete foci of mild
inflammation were detected, including interstitial oedema, cellular
infiltration, and an increased number of alveolar macrophages. At 7
days, signs of irritation were diminished, and, at 21 and 60 days,
alveolar tissues were essentially normal. Exposure to the lower
concentration induced more limited changes at 3 days; no
modification was seen at later examinations. It was considered that
a single exposure to this fenitrothion mixture at 500 mg/m3 did
not present a serious hazard to the lung (Chevalier et al., 1984).
Alveolar irritation was also seen in the solvent- exposed control
animals.
When Holstein milk cows and sheep (male) were administered
fenitrothion orally at 500 and 770 mg/kg body weight, respectively,
they showed signs of poisoning, such as paralysis of the hindlegs
and salivation, but gradually recovered 4 h after treatment (Namba
et al., 1966). Pigs given a single oral dose of 310 mg/kg body
weight showed typical signs of ChE inhibition, but the toxic signs
disappeared within 48 h. When cows and sheep were given fenitrothion
at 3 mg/kg body weight per day, for 90 and 60 days, respectively,
the ChE activity in the plasma decreased in both species early in
the test period, but recovered fully after 30 days.
7.2 Skin and eye irritation; skin sensitization
7.2.1 Skin and eye irritation
One tenth ml of fenitrothion (technical) was applied to one eye
of 9 male, New Zealand White rabbits. The untreated eye served as a
control. The treated eyes of 6 animals remained unwashed. The
treated eyes of the other 3 animals were flushed for 1 min with
about 300 ml lukewarm water, 30 seconds after application. Slight
hyperaemia in the conjunctiva was observed 1 h after application in
the unwashed eyes. This change had disappeared 48 h after
application. No irritating reactions were induced in any washed
eyes. The irritating potency of fenitrothion in eyes was judged to
be minimal for the unwashed group, and negative for the washed group
(Hara & Suzuki, 1981).
A half ml of fenitrothion (technical) on a 1 x 1 inch lint
patch was applied to abraded and intact parts of the back skin of 6
New Zealand White rabbits. No irritating reactions, such as erythema
and oedema, were noticed in the animals. The irritating potency of
the material was estimated to be negative (Hara & Suzuki, 1981).
7.2.2 Skin sensitization
The Landsteiner-Draize test with fenitrothion (technical) was
conducted on 6 male, Hartley guinea-pigs using
2,4-dinitrochloro-benzene (DNCB) as a positive control, to examine
possible skin sensitization activity. Fenitrothion dissolved in corn
oil at concentrations of 1 or 5% was applied intradermally every
other day, 10 times. The animals were treated for a challenge with
intradermal injection and dermal application 2 weeks after the last
sensitizing treatment. Allergic reactions were not observed in any
animals treated with fenitrothion, while reactions, such as swelling
and hyperaemia, were clearly noticed in the animals treated with
DNCB. It was concluded that fenitrothion (technical) was not a skin
sensitizer (Kohda et al., 1972).
The maximization test was undertaken to study the allergenicity
of fenitrothion. Ten young adult, female, Hartley guinea-pigs
weighing from 300 to 500 g were studied, induction and challenge
being undertaken according to the original method. The
concentrations of the compounds for the induction were 5 and 25% for
intradermal and topical treatment, respectively. Challenge
concentrations of 0.05% showed the potential for allergenicity in
this test (Matsushita et al., 1985).
7.3 Short-term studies
7.3.1 Rat
Male rats were given daily oral doses of 13 mg fenitrothion/kg
body weight for 28 days. Erythrocyte ChE activity was severely
depressed, but recovered 30 days after withdrawal of fenitrothion
(Kimmerle, 1962a).
Oral administration of fenitrothion at 7.25 or 14.5 mg/kg body
weight per day to male, Wistar rats (5/group) for 28 days resulted
in increases in plasma corticosterone and glucose levels, 7 days
after the start of treatment, and in the relative adrenal weight, 2
weeks after the start of treatment (Yamamoto et al., 1982b).
Groups of 36 male, CD rats received technical fenitrothion by
oral gavage (2.5, 5.0, 10.0, or 20.0 mg/kg per day) for 30
consecutive days. No significant treatment-related changes were
observed by serum biochemistry or by light microscopy of liver.
However, a dose-dependent decrease was noted in the brain, plasma,
and erythrocyte ChEs as well as tissue esterase activities. Eight
out of 36 animals died at the 20.0 mg/kg level (Trottier et al.,
1980).
Male rats were fed 5, 10, or 20 mg fenitrothion/kg diet for 5
weeks. The activity of brain and erythrocyte ChE was normal in the 5
mg/kg group, whereas the 10 mg/kg group showed a slight depression
of erythrocyte ChE activity after 5 weeks and recovery 2 weeks after
withdrawal. The 20 mg/kg group showed some depression of activity in
both erythrocyte and brain ChE and the recovery in brain ChE
remained incomplete 2 weeks after withdrawal (Carshalton, 1964).
The neurobehavioural effects were examined in male,
Sprague-Dawley rats (6/group) of fenitrothion administered orally at
doses of 10, 20, or 40 mg/kg body weight, daily, for a period of 40
days. At the 2 higher doses, mortality was more than 50%, while
only one animal died at a dose of 10 mg/kg. Toxic signs, such as
cholinergic signs, loss of reflexes, and changes in motor activity
and indices of ataxia, were observed markedly at the 2 higher doses,
and slightly at the lowest dose. However, these effects tended to
recover gradually later in the study (Rondeau et al., 1981).
Groups of male, Wistar rats (16 or 17 in number) were fed 0,
32, 63, 125, 250, or 500 mg fenitrothion/kg diet for 90 days.
Mortality, food intake, growth, general behaviour, urinalysis,
average organ weights, and histopathology in the groups fed 32, 63,
125, and 250 mg/kg diet were comparable to those in the controls.
All animals fed 500 mg/kg diet showed clinical signs of
anticholinesterase poisoning and there were minimal toxic signs in 4
animals in the 250 mg/kg diet group. In the 500 mg/kg group, the
average weights of the testes and brain were increased in comparison
with those of the control group. On monthly interim sacrifice of 4
rats from each group, the cholinesterase (ChE) activity of plasma,
erythrocyte, brain cortex, liver, and kidney showed a dose-dependent
depression, the lowest activity being in the brain. The ChE activity
in the 32 and 63 mg/kg diet groups generally increased by day 60 of
feeding to a level within the normal limits (Misu et al., 1966).
Fenitrothion (60% water miscible concentrate) was given to
groups of 12 Wistar rats/sex at dose levels of 0, 20, 92.8, 430.7,
or 2000 mg/kg diet in the feed or 0, 10, 46.4, 215, or 1000 mg/litre
in drinking-water, for 90 days. The levels causing no appreciable
effect on the ChE activity of plasma, erythrocyte, and whole blood
were 20 mg/kg diet and 10 mg/litre drinking-water. The above
mentioned levels and 92.8 mg/kg diet and 46.4 mg/litre and 215
mg/litre in drinking-water did not cause any effects on food or
water intake, weight gain, average organ weights, haemogram, and
blood biochemistry. However, 92.8 mg fenitrothion/kg diet and 46.4
mg/litre in the drinking-water moderately depressed the activities
of whole blood and erythrocyte ChE and had a more marked depressive
effect on plasma ChE. The ChE activity recovered between 30-40 days
after withdrawal of fenitrothion. The levels of 430 mg/kg diet and
1000 mg/litre drinking-water caused a depression in body weight gain
(Cooper, 1966).
Two groups of male rats (20 each) were dosed by stomach tube
with fenitrothion (50% formulation) at 10 mg/kg or 11 mg/kg body
weight, for 6 days per week, over 6 months. During the first weeks,
the rats showed a temporary deterioration in general condition and
loss of weight. Haematology and urinalysis during the study and
gross and microscopic pathology at its termination did not reveal
any abnormalities (Klimmer, 1961).
Groups of rats (15 males and 15 females) were fed fenitrothion
at 0, 10, 30, or 150 mg/kg diet for 6 months. In all groups, growth,
food and water consumption, mortality, blood and urinalysis, and
blood biochemistry were comparable with those of the controls. The
ChE activities in the brain, erythrocytes, and plasma were depressed
in both sexes in the 150 mg/kg diet group and in females in the 30
mg/kg diet group. In the 10 mg/kg diet group, only the plasma ChE of
females was depressed. Absolute and relative organ weights were
within normal limits. No histopathological changes were found in the
organs examined. Based on brain acetylcholinesterase inhibition, a
NOAEL of 10 mg/kg diet was concluded (Kadota et al., 1975a).
Groups of 8 male and 8 female rats were fed diets containing 0,
10, 50, or 250 mg fenitrothion/kg for 34 weeks. In another test,
groups of 16 rats of each sex were fed fenitrothion at 0, 5, 25, or
125 mg/kg diet over the same period. The feeding of fenitrothion at
250 mg/kg diet resulted in a decrease in body weight gain in
females. In the 125 mg/kg diet group, a lower relative weight of
spleen was found in both sexes. The ChE activities in both the
plasma and erythrocytes, measured at intervals, showed a
dose-dependent decrease in all test groups, except for males at 5
mg/kg diet whose plasma ChE activity was not suppressed. In females,
the depression was slight. The ChE activity in the brain was
decreased only in the 250 mg/kg diet group (Benes & Cerna, 1970).
Groups of 9 and 10 male Sprague-Dawley rats were fed 0, 25,
100, or 400 mg fenitrothion/kg diet for 63 weeks. A positive control
group was fed 800 mg malathion/kg. At the 400 mg/kg level of
fenitrothion, body weight gain was decreased and only a few animals
survived the 63-week period. At this level, there was also a 100%
depression of erythrocyte ChE. At the 100 mg/kg level, a slight ChE
depression (10-30%) occurred in the brain and a moderate depression
(30-65%) in the erythrocyte and plasma. At 25 mg/kg diet,
fenitrothion had a slight effect on plasma ChE activity, but did not
cause any effects on other parameters (food intake, body weight
gain, brain and erythrocyte ChE) evaluated (Ueda & Nishimura, 1966).
Groups of male, Wistar rats (60/group with 120 controls) were
administered fenitrothion, by gavage, at 0, 0.5, 1, 5, or 10 mg/kg
body weight per day, for 12 months. Each month, 10 controls and 5
rats from each group were sacrificed. After 12 months,
administration was stopped and survivors were held for an additional
2 months to monitor recovery from the biological effects of the
treatment. No changes attributable to treatment with fenitrothion
were observed in haematology or blood biochemistry, except in
esterase activities. Marked reductions in hepatic ChE, and brain and
erythrocyte AchE activities, observed at doses of 5 and 10 mg/kg
body weight per day, within 1 month of treatment, persisted for the
duration of the treatment. Two months after treatment was
terminated, the esterase activities in all treated rats were
comparable to those in the controls. Little change was measured at
0.5 and 1 mg/kg per day (Ecobichon et al., 1980).
Groups of Wistar rats (26 males and 27 females) were fed a diet
containing 0, 1, 5, or 25 mg fenitrothion/kg for one year. No
significant changes were observed in body weight, food and water
intake, organ weights, or histopathology. The ChE activity in the
brain, liver, and erythrocytes was reduced at 5 and 25 mg/kg diet
during the first month but then recovered (Kanoh et al., 1982).
A nose-only inhalation toxicity study of fenitrothion was
conducted on Sprague-Dawley rats (Breckenridge et al., 1982). Groups
of 10 male and 10 female rats (20/sex for the controls) were exposed
to fenitrothion 11% emulsifier mixture at chamber concentrations of
0, 6.7, 20, or 60 mg/m3, for 2 h/day, for 30 consecutive days. ChE
activity in the plasma, erythrocytes, and brain was inhibited in the
60 mg/m3 males and in all the treated females, except those in the
6.7 and 20 mg/m3 groups, in which the erythrocyte ChE activity was
not affected. Hepatic and renal carboxyesterases were inhibited in
the 60 mg/m3 male group and the 20 and 60 mg/m3 female groups.
These enzyme activities had returned to normal values 30 days after
treatment. Suppression of the brain ChE activity in the 60 mg/m3
female group persisted. Haematological, urinary, blood biochemical,
and histopathological studies did not reveal any deleterious effects
of fenitrothion.
Durham et al. (1982) reported a short-term study of
fenitrothion (11% emulsifiable concentrate) in the absence or
presence of Atlox 3409F (emulsifier) and Dowanol TPM (cosolvent).
Forty-five male, Sprague-Dawley rats were assigned to each of the
treated and control groups. No changes in the haematological, serum
biochemical parameters or in tissue morphology were observed in rats
treated with fenitrothion at doses of 1, 5, 10, or 20 mg a.i./kg.
The emulsifier and co-solvent did not substantially enhance or
reduce the toxicity of fenitrothion.
7.3.2 Dog
Groups of dogs (1 male and 1 female) were given daily oral
doses (by capsule) of 0, 2, 9, or 40 mg fenitrothion/kg body weight
for 98 days. Body weights, blood biochemistry, ChE levels, and
haematology were checked at intervals. At the 2 mg/kg level,
fenitrothion did not produce any effects on the parameters examined.
At 9 mg/kg, a slight depression after 60 days and, at 40 mg/kg, a
moderate depression after 29 days occurred in whole blood, plasma,
and erythrocyte ChE activity. At 40 mg/kg, there were also marked
toxic signs typical of cholinergic stimulation, and the dogs in this
group were sacrificed before the end of 98-day period (Cooper,
1966).
Groups of dogs (6 males and 6 females) were fed fenitrothion at
levels of 0, 30, 100, or 200 mg/kg diet for two years. Body weights,
blood biochemistry, and plasma, erythrocyte, and brain ChE
activities were monitored, together with histopathological
examination. The only adverse effect was a reduction in the ChE
activities. Depression of plasma ChE activity was apparent in all
groups, while erythrocyte enzyme activity was unaffected in the 30
mg/kg group, when the treated and control animals were compared. The
brain ChE activity was decreased only after ingestion of 200 mg/kg
(Mastalski et al., 1973).
Groups of Beagle dogs (6 males and 6 females) were fed
fenitrothion at dose levels of 0, 5, 10, or 50 mg/kg diet for one
year. The only treatment-associated change was a depression of ChE
activity. At 50 mg/kg diet, plasma ChE was depressed in both sexes,
and erythrocyte ChE was slightly depressed in the males only.
However, brain ChE was not depressed at any levels. The
no-observed-effect level was 10 mg/kg or 50 mg/kg diet, based on the
effects on erythrocyte or brain ChE, respectively (Griggs et al.,
1984).
A group of 3 male Beagle dogs was given fenitrothion in
capsules at 5 mg/kg body weight per day, for 6 days a week, over 10
months. A marked depression of plasma and erythrocyte ChE activities
was observed compared with the control group (2 animals). However,
no appreciable accumulation of the compound was noted in the blood,
fat tissue, or liver (Tomita et al., 1974).
Groups of Beagle dogs (2 females) were treated orally with
fenitrothion at dose levels of 0 or 2 mg/kg body weight per day (by
capsule) for one year. A marked depression of plasma and erythrocyte
ChE activities was observed 2 weeks after initiation of treatment.
No changes attributable to treatment with fenitrothion were found in
haematology, blood biochemistry, or ophthalmology (Ogata, 1972).
7.3.3 Rabbit
When groups of male Japanese albino rabbits (15 animals per
group) were administered fenitrothion at 0, 3, or 10 mg/kg body
weight per day in the diet for 6 months, a significant depression of
the plasma and erythrocyte ChE activities was observed in both
treated groups. At the higher dosage, depression of the brain ChE
was also found. However, no noteworthy changes, either biochemical
or histochemical, were observed in musculi rectus medialis ChE, and
the fine structure of the neuromuscular junctions of the tissue was
essentially normal when examined using an electron microscope. No
other adverse effects were observed in behaviour, mortality,
haematology, clinical biochemistry, organ weights, or histopathology
of major organs and tissues (Miyamoto et al., 1976a).
7.3.4 Guinea-pig
Treatment of male guinea-pigs with 5 or 10 mg fenitrothion/kg
ip for 30 days resulted in a dose-dependent increase in
acetylcholine levels in the heart, decreased heart rate, and
hypocalcaemia (Anand et al., 1989).
Active sensitization toxicity by inhalation of fenitrothion
(technical) or fenitrothion 22% emulsifiable concentrate (EC) was
examined in 10-15 male and female Hartley guinea-pigs using
bacterial alpha amylase as a positive control material. Fenitrothion
(technical) suspended in 10% Tween 80 aqueous solution and the
undiluted EC formulation were sprayed daily for 120 min for 7 days.
The air concentrations of fenitrothion were 226 and 628 mg a.i./m3
in the chamber where the technical sample was sprayed, and 360 and
1048 mg a.i./m3 in the chamber where the EC formulation was
sprayed. Animals inhaling the material at the higher air
concentration of the EC formulation showed salivation from the third
day. The cholinesterase activities in the brain, packed red cells,
and plasma were severely inhibited in treated animals at the
completion of the sensitization period. The animals were challenged
7 days after the last sensitizing inhalation, but signs of allergic
asthma were not observed in any animals treated with fenitrothion.
In contrast, animals treated with bacterial alpha amylase showed
severe allergic signs of asthma. It was concluded that fenitrothion
did not cause allergic asthma (Okuno et al., 1977).
7.4 Long-term and carcinogenicity studies
Groups of Wistar rats (15 males and 15 females) were fed diets
containing fenitrothion at levels of 0, 2.5, 5, or 10 mg/kg for 92
weeks, to investigate the effects of fenitrothion on ChE activities.
The ChE activity in the blood was measured after 2, 4, 6, 8, 12, 16,
20, 24, 42, 68, and 92 weeks. In the 5 mg/kg group (equivalent to
0.27 mg/kg body weight per day), a 20-25% decrease in plasma ChE
activity in the males during the first 16 weeks and a 20-35%
decrease in the females during 12 weeks were recorded. The activity,
however, recovered during the remaining test period. In the 10 mg/kg
group, the plasma activity decreased during the first 8 weeks by
30-40% in males and 40-50% in females. The activity, however,
gradually returned to normal during the next 8 weeks. The activity
of erythrocyte ChE was decreased by 20-30% in both sexes during the
first 6 weeks at 10 mg/kg and then recovered fully. The brain ChE
activity, determined at the end of the test period, was not affected
by fenitrothion at any dose level. The no-observed-effect level in
this study was 5 mg/kg diet (0.27 mg/kg body weight per day in males
or 0.28 mg/kg body weight per day in females) (Kadota et al.,
1975a).
Three groups of Charles-River albino rats (50 males and 50
females) were administered fenitrothion at levels of 10, 30, or 100
mg/kg diet (corresponding to 0.5, 1.5, and 5 mg/kg body weight) for
a period of 104 weeks, with 60 females and 60 males as controls.
These animals were derived from the F1a generation of a
reproduction study (see section 7.5.1). Ten rats of each sex and
group were sacrificed after one year and all the surviving animals
at 104 weeks. The body weights of males and females at 100 mg/kg
were lower than those of the controls from the start of the test and
remained so in males until 52 weeks, but at the end of the test no
significant differences were seen. Food consumption at 52 weeks was
lower for the middle- and high-level males, but normal for low-level
males and all the females. The mortality rate in the treated females
was comparable with that in the control females, while, in males,
the mortality rate was significantly higher than the controls only
in the lowest-dose group. Blood and urine analyses were normal while
ChE activity showed a dose-dependent decrease among the test rats.
Significant depressions in ChE activities in the plasma were
recorded at all 3 dosage levels (F1a being affected from the
beginning), but erythrocyte ChE depression and brain ChE depression
occurred only at the 30 and 100 mg/kg levels in both sexes.
Statistical analysis of the probabilities of tumour incidence did
not reveal any differences between the controls and the animals
treated with 10 or 100 mg/kg. There was a decrease in the
probability of benign tumours in males treated with 30 mg/kg and an
increase in pituitary adenoma incidence in the 30
mg/kg females; however, since this was not observed at the 100 mg/kg
level, it was not considered related to treatment. Absolute and
relative organ weights and gross and histopathology did not reveal
any dose-dependent changes (Rutter & Nelson, 1974).
In a study by Rutter & Banas (1975), groups of ICR Swiss mice
(200 animals/sex) were fed a diet containing 0, 30, 100, or 200 mg
fenitrothion/kg for 78 weeks (18 months). No observable effects
attributable to the compound were detected on body weight gain, food
intake, mortality, or ophthalmological and gross/histopathological
findings. It was concluded that fenitrothion is not carcinogenic in
mice fed up to 200 mg/kg diet for 18 months.
Groups of B6C3F1 mice (100 males and 100 females) were
administered fenitrothion at levels of 0, 3, 10, 100, or 1000 mg/kg
diet. Each group of mice was divided into 2 subgroups (main and
satellite groups) comprising 50 mice of each sex. The main groups
were maintained on the control or test diets until death, or for a
maximum of 104 weeks. The satellite groups were used for interim
sacrifices. Throughout the administration period, no
treatment-related abnormal symptoms were observed, nor were there
any significant differences in the survival rates between control
and treated groups. Decreases in body weight gain, food consumption,
and water intake were observed at the 1000 mg/kg level in both
sexes. Urinalysis, ophthalmology, and haematology did not reveal any
treatment-related effects. Significant depression of ChE activity
occurred in plasma at the 3 highest levels, while, in erythrocytes
and brain, the depression occurred at 100 and 1000 mg/kg levels in
both sexes. The blood biochemical examination revealed an elevation
of total cholesterol values in both sexes in the 100 and 1000 mg/kg
groups and a decrease in glucose levels in both sexes in the 1000
mg/kg group. Some other biochemical changes were observed
transiently in the 1000 mg/kg group. The incidence of alopoecia was
decreased at the final sacrifice of 1000 mg/kg females. Brain
weights were increased in both sexes in the 1000 mg/kg group.
Histopathological examination revealed that calcification of brain
parenchyma in both sexes in the 1000 mg/kg group, and hair
follicular atrophy in 1000 mg/kg females were less frequent than in
the controls. No treatment-related increase was observed in the
incidence of any neoplastic changes. The level causing no
toxicological effect was concluded to be 10 mg/kg diet, based on the
depression of brain ChE (Tamano et al., 1990).
7.5 Reproductive effects, embryotoxicity, and teratogenicity
7.5.1 Reproductive effects
A 2-litter, 3-generation, reproduction study on Charles River
albino rats was carried out with 15 males and 30 females per test
group (20 males and 40 females for control) administered dietary
levels of 0, 10, 30, and 150 mg fenitrothion/kg. After the first
filial generation (F1a), the highest dose level was reduced to 100
mg/kg. Fertility, gestation, lactation, and live birth indices were
compared. Administration of 150 or 100 mg fenitrothion/kg in the
diet caused weight reduction in the parental animals in the P0 and
P1 generations and suppressed lactation indices through all
generations. The highest dose group also showed a higher incidence
of cannibalism and smallness at weaning, whereas all litters seemed
normal at birth. No dose-dependent malformations or
histopathological changes were seen (Rutter & Voelker, 1974).
Crl: CD(R)(SD)BR male and female rats (30 animals/group) were
exposed to technical fenitrothion (94.6% pure) in the feed for 2
generations (2 litters in the first generation and 1 litter in the
second generation) at dietary levels of 0, 10, 40, or 120 mg/kg. A
dose-dependent, statistically significant decrease in body weight
gain and absolute and relative feed consumption occurred in males
and females given 120 mg/kg diet. Body weights and body weight gains
were significantly affected at 40 mg/kg diet in the P1 generation
female rats during lactation, and in the F1a generation male rats
during post-cohabitation. Absolute feed consumption values were
significantly affected at 40 mg/kg diet in the F1a generation
female rats. There was no compound-related effect on reproductive
performance in either the P1 or F1a generations at levels of up
to 120 mg/kg diet. The body weights of pups were significantly
reduced in litters of both generations at 120 mg/kg diet and
mortality was significantly increased in the P1 generation, F1a
and F1b litters, and the F1a generation and F2 litters, at 120
mg/kg diet. There were no treatment-related histomorphological
changes in the male and female F0 and F1 generation rats at up to
120 mg/kg diet (Hoberman, 1990).
Groups of 10 male and 20 female rats were fed a diet containing
0, 10, 40, or 80 mg technical fenitrothion/kg in a 4-generation,
2-litter, reproduction study. The following parameters were
monitored: body weight and food consumption of parent animals and
indices of fertility, gestation, live birth, 24-h survival, 5-day
survival, and lactation; gross pathology of all pups, organ weights,
and histopathological examination of F4b weanlings; cholinesterase
activity in whole blood in males of F2a (aged 15 weeks) and in all
weanlings of F4b (aged 4 weeks). Fertility, gestation, and live
birth indices were normal in all groups, whereas the 24-h and 5-day
survival indices were reduced in one or both litters of the 80 mg/kg
group in almost all generations. The lactation index was reduced in
all generations in the 40 and 80 mg/kg groups. The mean litter size
was smaller in all but 5 test litters, but this was without any
clear dose-dependence. However, the lowest number of pups was found
in 6 out of 8 litters in the 80
mg/kg group. The mean weight of the pups at birth and at 21 days of
age was normal, whereas the growth of the parent animals was
slightly decreased in the 80 mg/kg group. Plasma and erythrocyte
cholinesterase activity was decreased in relation to the dose and
length of exposure; in the 10 mg/kg group, the decrease was not
significant. Organ weights and gross pathological examination did
not reveal any abnormalities (Benes et al., 1974).
Fenitrothion fed to 10 female rats and male rats at 200 mg/kg
diet before mating, and during the gestation period, in a
one-generation reproduction study did not affect the reproductive
indices, such as fertility, gestation, and live birth indices.
Moreover, it was revealed that fenitrothion did not have any
estrogenic activity, according to the mouse uterine weight assay
(Gowda & Sastry, 1979).
7.5.2 Embryotoxicity and teratogenicity
Groups of female albino rabbits were inseminated (gestation day
0) and during gestation days 6-18 were dosed with 0, 0.3, or 1 mg
fenitrothion/kg per day in gelatine capsules. A positive control
group given 37.5 mg thalidomide/kg per day was included. The
compound did not induce any effects on the does or on the number of
implantation sites, early or late resorption sites, number of dead
or live young, or aborted fetuses. In the thalidomide group,
approximately 10% of the fetuses showed external malformations,
while none were seen in the other groups. No effects related to the
administration of fenitrothion were seen on examination for internal
or skeletal deformities (Ladd et al., 1971).
Fenitrothion (technical grade, 96.6% purity) was dissolved in
corn oil and administered orally to 14-16 pregnant rabbits (New
Zealand White) during the period of organogenesis (gestation days
7-19) at dose levels of 0, 3, 10, or 30 mg/kg body weight per day.
There were no treatment-related effects on pregnancy rate, fetal
viability, fetal sex ratio, or mean fetal body weight values.
Implantation efficiency was slightly lower at 30 mg/kg body weight
per day compared with the controls. There were no dose-related
increases in the incidences of external, visceral, or skeletal
malformations at any dose level (Morseth et al., 1986).
Groups of 25 or 27 pregnant mice (ICR) were administered
fenitrothion orally at dose levels of 0, 20, 70, or 200 mg/kg per
day during gestation days 7-12. Groups of 23-26 pregnant SD rats
were also dosed orally with fenitrothion at levels of 0, 2, 7, or 20
mg/kg per day during gestation days 9-14. No embryotoxic or
teratogenic effects were observed in mice or rats at any of these
dose levels (Miyamoto et al., 1975).
Fenitrothion (technical grade, 96.9% purity) was dissolved in
corn oil and administered orally to 20-24 pregnant Sprague-Dawley
rats during the period of organogenesis (gestation days 6-15) at
dose levels of 0, 3, 8, or 25 mg/kg body weight per day. There were
no adverse effects on pregnancy rates, mean implantation efficiency,
fetal viability, fetal sex ratio, or mean fetal body weights. The
frequency and the distribution of fetal malformation findings did
not indicate a teratogenic response at any dose level. Maternal
toxicity, as evidenced by body weight decrease and clinical
observations (tremors), was indicated at a dose of 25 mg/kg body
weight per day (Morseth et al., 1987).
Prenatal administration of fenitrothion (50% EC) to CFY rats at
5, 10, or 15 mg/kg body weight by oral gavage from days 7 to 15 of
gestation resulted in dose-related decreases in open field activity
and motor coordination in the offspring of animals treated at the
two highest dose levels. Long-lasting alterations in the acquisition
and extinction of a conditioned escape response, as well as
increased social interactions were observed in the adult offspring.
The results indicated a no-observed-effect level of 5 mg/kg
(Lehotzky et al., 1989).
Fenitrothion (97.6% purity, 0.1% of 3-methyl-4-nitrophenol) was
administered to groups of 20 pregnant Wistar rats in sunflower oil,
by gavage, at single daily doses of 0, 2, 8, 16, or 24 mg/kg body
weight during days 6-15 of gestation. On day 20 of gestation, the
rats were sacrificed and the numbers of viable and dead fetuses,
resorptions, implantations, and corpora lutea were recorded. Fetuses
were subjected to external and internal (skeletal and soft tissue)
examination. Fenitrothion was not found to be teratogenic in this
study at any dose. At doses exceeding 8 mg/kg per day, fenitrothion
was maternally toxic (Benes & Tejnorova, 1986).
7.6 Mutagenicity
Fenitrothion was examined using a variety of mutagenicity tests
including in vitro and in vivo gene mutation, DNA damage/repair,
and chromosomal aberration assays (see Table 5). Most of the tests
showed that fenitrothion was not mutagenic, but some of the tests
gave weakly positive results: Ames tests with S. typhimurium TA100
(Kawachi, 1978; Moriya et al., 1983; Hara et al., 1989), sister
chromatid exchange (SCE) assays (Kawachi, 1978) and chromosomal
aberration tests (Kawachi, 1978). However, more recent studies
showed that fenitrothion was negative in gene mutation assays with
S. typhimurium TA100 NR, a nitro-reductase-deficient strain of
TA100, and in mammalian cells, V79 Chinese hamster lung cells.
Mutagenicity in S. typhimurium TA100 was considered attributable
to the nitro-reductase inherent to bacteria (Hara et al., 1989). An
SCE test in cultured mouse embryo cells showed a negative result for
clastogenic potential
(Suzuki & Miyamoto, 1980); in vitro and in vivo chromosomal
aberration tests in the rat, mouse, and Chinese hamster test systems
also gave negative results (Hara & Suzuki, 1982a,b; Hara et al.,
1988).
When CFLP mice were given 3-methyl-4-nitrophenol (25 mg/kg),
intraperitoneally, once a week for 10 weeks, the numbers of
chromosome gaps in bone marrow were significantly increased (Nehez
et al., 1985).
7.7 Neurotoxicity
Seven hens protected against acute anti-cholinesterase effects
with atropine and 2-pyridine aldoxime methiodide (2-PAM) were given
an oral dose of 250 mg fenitrothion/kg body weight and 3 other hens
were given 500 mg/kg. Two of the 500 mg/kg group died within 1-2
days, while the remaining 8 hens did not show any signs of paralysis
during the observation period of 6 weeks (Kimmerle, 1962b).
Groups of 6 adult White Leghorn hens were given single oral
doses of fenitrothion at 250, 500, or 1000 mg/kg body weight.
Tri- O-cresyl phosphate (TOCP, 300 mg/kg) was used as a positive
control. Toxic signs, which lasted 4-10 days, occurred in all
groups. One half of the hens in the middle dosage group and all
those in the highest fenitrothion group died within 24-48 h of
treatment. No delayed paralysis of the legs occurred in the
survivors in any dose group or at any time during the 5-week
observation period, while all TOCP dosed animals developed paralysis
within 3 weeks. The sciatic nerve in all the surviving hens given
fenitrothion was normal (Kadota et al., 1975b).
Sixteen adult White Leghorn hens received 500 mg
fenitrothion/kg orally and were protected against intoxication by
atropine and 2-PAM; this treatment was repeated 3 weeks later. TOCP
was used as a positive control. Five hens died 2 days after the
first treatment with fenitrothion and none after the second; the
survivors showed toxic signs of cholinesterase inhibition. No
paralysis was observed and histopathological findings in the sciatic
nerves were normal (Kadota et al., 1975b).
Groups of 8 adult, White Leghorn hens were given 16.7 or 33.7
mg fenitrothion/kg per day, 6 days/week, over 4 weeks and then
observed for another 3 weeks. Slight toxic signs were seen in both
groups during the administration period. One hen in the higher
dosage group died on the 5th day of dosing. Body weights were
decreased in both groups, but the decrease in the lower dosage group
was transient. No delayed leg paralysis or histopathological changes
in the sciatic nerve or spinal cord were recorded (Kadota et al.,
1975b).
Table 5. Mutagenicity tests on fenitrothion
Tests Strains Dose levels Metabolic Results Reference
activation
Gene mutation tests
Microorganism test E. coli K12 13.2, 132 µg/ml - Suzuki et al. (1974)
Coli-phage lambda 13.2, 132 µg/ml -
1847 sus E-h+
S. typhimurium
TA1535 10, 100, 1000, + - Suzuki & Miyamoto (1976)
10 000 µg/plate - -
TA1537 + -
- -
TA1538 + -
- -
S. typhimurium
TA98 500,750,1000, + - Kawachi (1978)
2500 µg/plate - -
TA100 + + (weakly)
- + (weakly)
TA1537 + -
- -
S. typhimurium
TA100 < 5000 µg/plate + + Moriya et al. (1983)
- +
TA98 + -
- -
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
TA1535 + -
- -
TA1537 + -
- -
TA1538 + -
- -
E. coli + -
WP2 hcr - -
S. typhimurium
TA98 100, 200, 500, 1000, + - Hara et al. (1989)
2000, 5000 µg/plate - -
TA1535 + -
- -
TA1537 + -
- -
TA100 + + (weakly)
- + (weakly)
TA100NR + -
- -
TA100 1,8-DNP6 + + (weakly)
- -
E. coli
WP2uvrA + -
- -
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
Host-mediated assay ICR mouse
S. typhimurium G46 100, 200 mg/kg body - Suzuki & Miyamoto (1976)
weight; oral, im/ip
(mouse)
Mammalian cell test Chinese hamster 0.01, 0.03, 0.1, + - Hara et al. (1989)
lung cells V79 0.3 mmol/litre - -
DNA-Damage and repair tests
Rec-assay B. subtilis up to 20 µlitre + - Shirasu et al. (1979)
H17/M45 - -
Sister chromatid Human embryonic cells 5 x 10-4 (mol/litre) 2.6 times Kawachi (1978)
higher
exchange (SCE) HE2144 SCE than
control
Chinese hamster cells 1 x 10-3 (mol/litre)
Don-6
ICR mouse embryo cells 10-5, 5 x 10-5, + - Suzuki & Miyamoto (1980)
10-4 (mol/litre) - -
Unscheduled DNA SD rat hepatocyte 300 mg/kg body - Kawamoto et al. (1989)
synthesis (UDS) weight, oral)
in vivo/in vitro
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
Chromosomal aberration tests
Chromosomal aberration Wistar rat F2b (male) 80 mg/kg diet - Benes et al. (1975)
F3b (male) 80 mg/kg diet -
Chinese hamster lung 0.1 mg/ml 21% Kawachi (1978)
cells aberration
Chinese hamster ovary 0.075, 0.15, 0.3 mg/ml + - Hara et al. (1988)
cells-K1 0.003, 0.01, 0.03 mg/ml - -
Long-Evans rat 400, 800, 1000 mg/kg + (weakly) Kawachi (1978)
body weight, oral;
20, 40 mg/kg body + (weakly)
weight, oral, x 5 days
ICR mouse (male) 200, 400, 800 mg/kg - Hara & Suzuki (1982a)
body weight, ip
SD rat (male) 100, 200, 400 mg/kg, - Hara & Suzuki (1982b)
oral, 20, 40, 80 mg/kg, -
oral, x 5 days
Wistar rat (male) 15, 30, 60 mg/kg body - Malhi & Grover (1987)
weight, ip, x 5 days
Q mouse (male) 1000 mg/kg body - Degraeve et al. (1984)
weight, ip
Table 5 (contd).
Tests Strains Dose levels Metabolic Results Reference
activation
Micronucleus test
ICR mouse (male) 200, 400, 800 mg/kg - Hara & Suzuki (1982c)
body weight, ip
ddy mouse (male) 200, 400, 800 mg/kg - Hayashi et al. (1988)
body weight, ip
100, 300, 600 mg/kg body -
weight, ip, x 4 days
Wistar rat (male) 75, 165, 330 mg/kg - Grover & Malhi (1985)
body weight, ip
Dominant lethal test Wistar rat F2b 10, 40, 80 mg/kg diet - Benes et al. (1975)
F4b 10, 40, 80 mg/kg diet -
ICR mouse 20, 200 mg/kg body - Kohda & Kadota (1975)
weight, oral, x 5 days
SD rat 2, 7, 20 mg/kg body -
weight, oral, x 5 days
Q mouse 1000 mg/kg body - Degraeve et al. (1984)
weight, ip
Others S. cerevisiae 0.3% in DMSO per plate - Yadav et al. (1982)
D. melanogaster 50, 150 mg/kg diet - Velazquez et al. (1987)
The inhibitory activities of fenitrothion against neuropathy
target esterase (neurotoxic esterase, NTE) and acetylcholinesterase
(AChE) in the hen (White Leghorn) brain were examined. Oral
treatment with 500 mg fenitrothion/kg resulted in significant (80%)
inhibition of AChE, but not of NTE (less than 10%) (Ohkawa et al.,
1980).
The potential of a single, toxic dose of fenitrothion to elicit
delayed neurotoxicity in adult White Leghorn hens (5 per group) was
compared with the effects produced following similar treatment with
the known neurotoxin, tri-O-cresyl phosphate (TOCP). Hens receiving
single oral doses of either fenitrothion (500 mg/kg) or TOCP (500
mg/kg) were assessed for toxicity by measuring biochemical (brain
and spinal cord AChE and NTE), physiological (motor function) and
morphological parameters of the brain, spinal cord, and sciatic
nerve 24 h, and 7, 14, 28, 42, and 56 days after treatment. At 24 h,
fenitrothion caused a marked inhibition of neuronal AChE. No
alteration in NTE activity was found in any fenitrothion-treated
hens. A characteristic, central-peripheral, distal axonopathy was
observed following treatment with TOCP, mild signs appeared 7-14
days after treatment and increased in severity up to 28 days after
treatment, concomitant with morphological changes primarily in the
sciatic nerve and spinal cord. Minimal morphological changes were
elicited by fenitrothion at this dosage, but the tissues appeared
morphologically similar to those seen in vehicle-treated control
hens. The results demonstrated that fenitrothion was not neuropathic
in the classic manner of TOCP (Durham & Ecobichon, 1986).
7.8 Effects on hepatic enzymes
Groups of male and female Wistar rats (4 animals each) were
kept on a diet containing fenitrothion at 150 mg/kg or
3-methyl-4-nitrophenol at 500 or 1500 mg/kg, and the effects on
hepatic oxidative phosphorylation and mixed function oxidases were
studied. Respiratory control ratios of mitochondria were normal,
adenosine triphosphatase (ATPase) activity was normal, and no
differences were found between the treated and control groups. A
slight decrease in the adenosine diphosphate:oxygen ratio was
observed in females fed 1500 mg nitrophenol/kg. However, the
difference was not regarded as statistically significant. Thus, rat
hepatic mitochondrial respiration systems do not seem to be affected
by administration of fenitrothion or 3-methyl-4-nitrophenol.
Similarly, these chemicals did not affect hepatic microsomal mixed
function oxygenase activities (Hosokawa & Miyamoto, 1975).
Uchiyama et al. (1975) reported that intraperitoneal injection
of 25 mg fenitrothion/kg to male ddY mice inhibited aminopyrine
N-demethylation and aniline hydroxylation activities in the liver
by about 50%.
Yamamoto et al. (1982a) reported that a single oral
administration of about 13 mg fenitrothion/kg (5 µmol/rat) or
repeated oral administration of about 1.3 mg/kg per day (0.5
µmol/rat per day) for 10 days to 4 or 5 Wistar rats did not affect
aminopyrine N-demethylase activity and cytochrome P-450 content,
or inhibit the dearylation or desulfuration of fenitrothion.
Groups of 5-7 weanling male Wistar rats received 50 mg
fenitrothion/kg body weight per day dissolved in peanut oil, by
gavage, for 5 consecutive days. Overt signs of toxicity were
observed within 30 min of receiving the third dose. Severely
affected animals were sacrificed for analysis of tissue esterase
activities and insecticide residues and to assess the influence of
the insecticide on hepatic microsomal mono-oxygenase activity. Daily
treatment was terminated after 5 days and the survivors were
sacrificed at intervals to assess the recovery of activity of
esterases in the tissues. Fenitrothion caused a significant
reduction in liver and body weights, a marked increase in
pentobarbital sleeping time, and a marked reduction in hepatic
microsomal mono-oxygenase ( p-nitroanisole O-demethylase, aniline
hydroxylase) activities. Marked inhibition of plasma
pseudocholinesterase, hepatic and renal non-specific
carboxyesterase, and erythrocyte and brain acetylcholinesterase
occurred. Recovery from 5 such daily doses was slow for all tissue
esterases with the exception of plasma cholinesterase, which
returned to 78% of control activity values within 5 days. Activities
of the other tissue esterases were 20-50% of normal at 5 days, and
only 70-90% of normal 16 days after the final dose (Ecobichon &
Zelt, 1979).
A single oral dose of 250 mg fenitrothion/kg resulted in a
slight decrease in a number of biochemical indices of liver function
in rats, including mitochondrial ATPase activity, cytochrome P-450
content, aniline hydroxylase activity, and aminopyrine
N-demethylase activity (Mihara et al., 1981). A dose of 25 mg/kg
also had a slight effect on P-450 content and xenobiotic metabolism,
while 5 mg/kg did not have any significant effects. The magnitude of
the effects was greater in females than in males.
Intraperitoneal doses of 50 or 500 mg fenitrothion/kg,
administered to mice, depleted hepatic glutathione levels and
inhibited microsomal aniline hydroxylase and paranitroanisole
O-demethylase activities (Ginsberg et al., 1982).
7.9 Effects on hormonal balance
Osicka-Koprowska et al. (1987) reported a significant elevation
in plasma corticosterone levels in rats given a single oral dose or
260 mg fenitrothion/kg. Levels returned to control values within 12
h. Adrenal ascorbic acid content was also
significantly decreased between 1 and 5 h after dosing. The authors
attributed the changes to hypersecretion of ACTH. After daily dosing
with fenitrothion at 13 mg/kg for 14 days, there was no significant
difference in circulating corticoseterone levels between the
controls and treated animals. There was a significant decrease in
14C derived from injected (ip) 14C-corticosterone in the
hypothalamus, adrenals, blood, liver, and muscle, but not in the
pituitary gland, at the end of the dosing period and 30 min after
injection of the label.
7.10 Toxicity of metabolites and the S-isomer
The acute toxicity of fenitrooxon, a metabolite of
fenitrothion, is greater than that of the parent compound.
3-Methyl-4-nitro-phenol, another metabolite, is less toxic
(Table 6).
The intraperitoneal toxicity of fenitrothion metabolites and
their related compounds was tested in mice. 3-Hydroxymethyl and
3-formyl derivatives of fenitrothion and fenitrooxon were more toxic
than fenitrothion and fenitrooxon, respectively. Fifteen other
compounds tested, including aminofenitrothion, demethyl
fenitrothion, and demethyl fenitrooxon, were much less toxic than
the parent compound (Miyamoto et al., 1978).
The acute oral toxicity of the S-methyl-isomer ( O-methyl
S-methyl O-(3-methyl-4-nitrophenyl) phosphorothioate) in rats
and mice is approximately twice that of fenitrothion (e.g., LD50
in mice: 550 and 1400 mg/kg, respectively) and the signs of
poisoning are typical of the muscarinic and nicotinic actions of
acetylcholine, as seen with fenitrothion (Kovacicova et al., 1973;
Rosival et al., 1974). The S-isomer, when injected
intraperitoneally, is 7-9 times more toxic in mice than fenitrothion
(Miyamoto, 1977a).
The bimolecular inhibition constant of S-methyl fenitrothion
on rat brain acetylcholinesterase is about 1000 times greater than
that of fenitrothion (Thompson et al., 1989).
7.11 Factors modifying toxicity
The best therapy in rats against severe poisoning with a 100%
lethal dose of fenitrothion was confirmed to be repeated and
combined treatment with atropine and 2-PAM, resulting in a 90%
survival ratio and considerable alleviation of toxic signs
(Matsubara & Horikoshi, 1983).
Table 6. Acute toxicity of metabolites
Compound Animal Route LD50 Reference
(strain) (mg/kg)
Fenitrooxon
Mouse oral 90 Miyamoto et al. (1963b);
Miyamoto (1969)
Mouse oral 120 Hollingworth et al. (1967b)
(Swiss White)
Rat oral 24 Miyamoto et al. (1963b);
Miyamoto (1969)
iv 3.3 Miyamoto et al. (1963b);
Miyamoto (1969)
Guinea-pig oral 221 Miyamoto et al. (1963b);
Miyamoto (1969)
iv 32 Miyamoto et al. (1963b);
Miyamoto (1969)
Dog (Beagle) oral > 68.1 Mastalski et al. (1971)
Hen oral 35 Kadota et al. (1975b)
(White Leghorn)
3-Methyl-4-
nitrophenol Rat
(Male) (Wistar) oral 2300 Sumitomo (1971)
(Female) (Wistar) oral 1200 Sumitomo (1971)
Dog (Beagle) oral > 680 Mastalski (1971a)
The effects of adrenalectomy (Adx), SKF 525-A, phenobarbital
(PB), and diethyl maleate (DEM) on the acute toxicity of
fenitrothion were investigated in male Wistar rats. PB administered
(ip) at 60 mg/kg per day for 3 days, did not exert any effect on the
toxicity of fenitrothion (100 mg/kg) given orally 24 h after the
last injection of PB. In adrenalectomized and SKF 525-A-pretreated
rats, the toxicity of fenitrothion was lower than in the controls.
Fenitrothion toxicity was increased by administration of DEM at (1
ml/kg), which depletes hepatic glutathione (GSH) levels. In
in vitro experiments, the rates of fenitrothion decomposition
and fenitrooxon formation by microsomes were markedly affected by
PB, SKF 525-A, and Adx. The decomposition of fenitrooxon by the
microsomal fraction and GSH-dependent decomposition of fenitrooxon
by the soluble fraction were not affected by PB, SKF 525-A, and
Adx pretreatment. The GSH-dependent decomposition of fenitrothion
and fenitrooxon was increased by the addition of GSH to the incubation
mixture. It was considered that the GSH-dependent metabolic pathway
plays an important role in the detoxification of fenitrothion
(Yamamoto et al., 1983a).
7.12 Mechanism of toxicity - mode of action
7.12.1 Mode of action
Fenitrothion is not a strong inhibitor of AChE in vitro, but
is much more so in vivo. The compound is converted in the animal
body to the active esterase inhibitor, fenitrooxon [ O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphate], by the action of microsomal
mixed function monooxygenase in liver and other tissues (Miyamoto et
al., 1963b; Miyamoto, 1964a).
Plasma ChE is the enzyme most susceptible to acute and
short-term administration of fenitrothion in rats, guinea-pigs,
dogs, rabbits, and humans (Miyamoto et al., 1963b; FAO/WHO, 1975b).
Brain AChE in rabbits is less inhibited by fenitrothion (Miyamoto et
al., 1976a).
7.12.2 Selective toxicity
Hollingworth et al. (1967a,b) and Hollingworth (1969) concluded
that dealkylation was an important factor among the many that
contribute to the lower mammalian toxicity of fenitrothion compared
with that of methylparathion. For example, the cholinesterase
inhibition of fenitrooxon is less, the activation by conversion of
P=S to P=O, slower, the translocation of fenitrothion more rapid,
and the detoxification rate of fenitrothion, higher. Comparison of
metabolites, at equitoxic doses in white mice (i.e., 17 mg
methylparathion/kg and 850 mg fenitrothion/kg) showed that
demethylation is the major detoxification path for fenitrothion at
high doses (200-850 mg/kg), but not for methylparathion. However,
inherently, demethylation of both compounds proceeds in a similar
way (Miyamoto et al., 1968) and both fenitrothion and
methylparathion are metabolized at a similar rate in vivo
(Miyamoto, 1964a, 1969). On the basis of these findings, Miyamoto
(1969) concluded that the relatively poorer penetration of
fenitrooxon into the brain (Miyamoto, 1964b) explained the
selectively lower toxicity of fenitrothion rather than the
dealkylation detoxification mechanism. Similar conclusions were
reached by Kuroiwa & Yamamoto (1977).
The mechanisms for the lower toxicity of fenitrothion compared
with methylparathion were investigated in male Wistar rats. The
difference in acute toxicity between fenitrothion and
methylparathion could be because of the more rapid decomposition of
fenitrothion and fenitrooxon in rat liver compared with that of
methylparathion and methylparaoxon. In particular, the decomposition
of fenitrothion by hepatic microsomes was accelerated by increasing
the insecticide concentration. The oxygen analogues of both
insecticides, fenitrooxon and methylparaxon, were not detected in
the brain after administration of the parent compounds. It was
considered that the lower toxicity of fenitrothion compared with
that of methylparathion could be due to the greater rate of
decomposition of fenitrothion to its less toxic metabolites rather
than to the different rates of penetration of the oxygen analogues
into the brain (Yamamoto et al., 1983b).
7.12.3 Potentiation of toxicity of other chemicals
Female rats were administered ip a combination of fenitrothion
and parathion, malathion, diazinon, or carbaryl. No potentiation was
demonstrated. However, a marked potentiation (increased mortality)
was observed when male and female Sprague-Dawley rats were given
single oral doses of fenitrothion and phosphamidon. In female rats,
potentiation occurred only with mixtures containing relatively low
concentrations of fenitrothion (Dubois & Kinoshita, 1963; Braid &
Nix, 1968).
The effects of a combination of fenitrothion with malathion in
male rats were more than additive. The potentiation was most
pronounced (half of the expected LD50) with a combination rate of
1:1. No potentiation was observed with other tested
organophosphates, i.e., bromophos, amidithion, and trichlorfon
(Benes & Cerna, 1970).
Hladka et al. (1974) noted that a single dose of a mixture of
fenitrothion and malathion caused a significant increase in
fenitrooxon but not fenitrothion levels in the blood and muscles of
female Wistar rats.
Fenitrothion at a subtoxic oral dose (100 mg/kg body weight; 4
h pretreatment or 1000 mg/kg diet for one week) potentiated the
acute oral toxicity of 2-sec-butylphenyl methylcarbamate (BPMC) in
male ICR mice. Through several in vivo and in vitro studies, it
was suggested that competitive inhibition of BPMC metabolism in the
liver by fenitrothion played, in part, a role in the inhibition of
BPMC detoxication, resulting in the potentiation of its toxicity
(Takahashi et al., 1984, 1987; Tsuda et al., 1984).
The same effects were investigated in female Beagle dogs. Oral
co-administration of fenitrothion (100 mg/kg) doubled the duration
of symptoms of BPMC (50 mg/kg) (5 h). The plasma level of BPMC in
the eliminating phase was increased by the co-administration and
began to decrease after 6 h, while ChE inhibition continued for 8 h.
Pretreatment with fenitrothion at 5 mg/kg per day, for 7 days,
caused one death in 3 dogs after the administration of BPMC (100
mg/kg), while the same dose of BPMC without pretreatment did not
cause any deaths. The pretreatment with fenitrothion increased the
duration of the toxic symptoms of BPMC 2.5-fold. The time course of
the toxic symptoms was correlated with the plasma concentration of
BPMC (Miyaoka et al., 1983).
8. EFFECTS ON MAN
8.1 General population exposure
8.1.1 Acute toxicity
Fenitrothion was given to a total of 24 human volunteers in
single oral doses of 0.042-0.33 mg/kg body weight or 2.5-20 mg per
person. The excretion of a metabolite, 3-methyl-4-nitrophenol, in
the urine was almost complete within 24 h, and ranged from about 70%
of the dose (0.042 mg/kg) to about 50% (0.33 mg/kg). Neither plasma
nor erythrocyte cholinesterase (ChE) activities were depressed below
normal, except in one person given 0.33 mg/kg, whose plasma ChE
activity was about 65% of the pretest level after 6 and 24 h. When
repeated doses of 0.04-0.08 mg/kg were given to 5 individuals, 4
times at 24-h intervals, most of the metabolites appeared in the
urine within 12 h of administration. After receiving the third and
fourth doses, there was a trend towards a rise in erythrocyte ChE
activity (Nosal & Hladka, 1968; Hladka et al., 1977).
8.1.2 Poisoning incidents
Several cases of acute fenitrothion poisoning, a few of them
lethal, have been described in the literature. These were either
accidental, intentional (suicide), or due to gross neglect of safety
precautions. A review of these cases is given by Hayes & Lawes
(1991).
In all cases, the onset of poisoning was rapid, early signs and
symptoms being exhaustion, headache, weakness, confusion, vomiting,
abdominal pain, excessive sweating, and salivation. The pupils were
small. Difficulty in breathing may be experienced, due to either
congestion of the lungs or weakness of the respiratory muscles. In
severe cases of poisoning, muscle spasms, unconsciousness, and
convulsions may develop and death may result from respiratory
failure.
In atypical cases, symptoms of poisoning may be observed for a
more prolonged period and up to 8 months after exposure. It has been
suggested that fenitrothion can be stored in human fat tissue and
then released under stress conditions (Ecobichon et al., 1977).
An attempted suicide using a large quantity of fenitrothion was
treated successfully by 2-PAM, atropine, and glutathione with
artificial respiration. The case was characterized by delay in the
onset of severe signs of intoxication (the 3rd day after
hospitalization) and relatively prolonged poisoning (the 70th day
after hospitalization), as evidenced by electroencephalogram and
inhibition of ChE. It was reported that the patient (56-year-old
female) had been suffering from diabetes, which presumably caused
delayed biotransformation of fenitrothion to fenitrooxon or delayed
absorption of the compound from the intestines, resulting in the
delay in the onset of symptoms and prolonged poisoning. However, no
evidence leading to this presumption was seen in the study
(Tsukimoto et al., 1981).
A case of delayed neurotoxicity of late onset has been reported
in a 70-year-old female who ingested 40 ml of 50% Fenitrothion EC.
At first, no toxic symptoms were apparent. However, 48 h after
ingestion, certain signs became apparent. An impediment in
consciousness was observed. Fasciculation and muscular weakness were
noted, while plasma and urinary levels of 3-methyl-4-nitro-phenol
reached a maximum. Neither atropine sulfate nor 2-PAM was effective.
For 3 weeks, the patient required ventilatory support, and
consequently her muscle strength and neurological status gradually
recovered with falling of the phenol level (Sakamoto et al., 1984).
The late symptoms reported by Sakamoto et al. (1984) are not those
of the organophosphorus ester-induced delayed neurotoxicity (OPIDN)
reported for some other organo-phosphorus compounds (Martinez
Chuecos & Sole Violan, 1985).
A 56-year-old male attempted suicide by the ingestion of about
60 ml of 50% fenitrothion emulsion. Five hours later, combined
haemoperfusion and haemodialysis (HP-HD) treatment was performed for
60 min and, subsequently, the symptoms gradually improved. Four days
after ingestion, cholinergic symptoms recurred. Immediate HP-HD
treatment was of no use and the patient died 6 days after ingestion
of fenitrothion. Analysis of the organ and tissue distribution of
fenitrothion revealed that the highest concentration of fenitrothion
was found in fat (59.0 mg/kg wet weight, more than 10 times the
concentrations in other organs). It was suggested that a slow
release of the pesticide from adipose tissue can give rise to a
protracted clinical course or late symptoms of intoxication (Yoshida
et al., 1987).
8.1.3 Contact dermatitis
An analysis of 202 patients with contact dermatitis caused by
organophosphorus insecticides was undertaken. The organophosphorus
insecticides presumably attributing to the dermatitis were
dichlorvos, salithion, fenitrothion, leptophos, cyanophos,
amidothion, diazinon, and malathion. The dermatitis was located on
the fingers, face, forearms, neck and nape. About one quarter
(25.2%) of the cases with dermatitis had complications with symptoms
of acute poisoning by these compounds. The prognosis of the
dermatitis was relatively good; 44.1% of the cases healed but 23.8%
of the patients were incompletely cured (Matsushita et al., 1985).
8.1.4 Possible links with Reye's syndrome
Concerns have been raised about the possible association
between the aerial spraying of forests with fenitrothion
formulations and the incidence of Reye's Syndrome, an encephalopathy
complicated by hepatic damage, occurring primarily in children up to
18 years of age and linked to viral infections (Reye et al., 1963;
Ruben et al., 1976; Corey et al., 1976).
Crocker et al. (1976a) reported that patients from a
fenitrothion-sprayed area had reduced plasma ChE and erythrocyte
AChE activities compared with those in children living in an
unsprayed region. However, Pollack et al. (1977) confirmed that,
while the serum ChE of Reye's patients was lower, the difference was
not significant. No correlation between aerial spraying and the
incidence of Reye's Syndrome was established in epidemio-logical
studies conducted in New Brunswick, Canada, and Maine, USA
(Schneider, 1976; Spitzer, 1982; Wood & Bogdan, 1986). Attention
was focused on the potential of various emulsifiers and co-solvents
used in oil- and water-based formulations of fenitrothion to enhance
virus-induced lethality in neonatal mice or to promote viral growth
in cell cultures (Crocker et al., 1974, 1976b). However, in a
neonatal animal model, the infection of neonatal mice by influenza
virus was not potentiated by any emulsifiers in fenitrothion
formulations (Menna, 1985). Similarly, in an in vitro cell culture
system, the growth of the virus was not enhanced by co-incubation
with various concentrations of fenitrothion, in the presence or
absence of a wide range of emulsifiers, co-solvents and diluents,
commonly used in aerial spraying formulations (Brookman et al.,
1984).
8.2 Occupational exposure
In a WHO (1973) programme, 3 different water-dispersible powder
formulations of fenitrothion were applied indoors as a residual
spray with the aim of depositing 2 g active ingredient/m2 on the
wall. The first three rounds of spraying lasted for approximately
six, 5-day, working weeks and the fourth round lasted for 8 weeks. A
slight to moderate depression of cholinesterase (ChE) activity was
observed in most of the spraymen towards the end of the spraying
rounds. Except for one case of headache, no other complaints
attributable to insecticide exposure were recorded during a total of
2000 man-days of spraying. No complaints were received from the 20
000 inhabitants whose houses had been sprayed repeatedly. However,
no ChE activity determinations were performed.
During 30-days indoor spraying of fenitrothion (2 g/m2) for
malaria control in Southern Iran in August 1971, a group of 28 pest
control operators and 925 inhabitants were monitored with respect to
their health and ChE activity. Clinical investigations and ChE
tests on 840 workers showed 42 cases of clinical symptoms, most of
which were very slight and subsided after the workers had showered
and rested (2-3 h). Out of 20 spraymen, 8 showed decreased ChE
levels, and one individual preparing the spray mixture showed a
significant depression of the enzyme activity, which was reactivated
after an appropriate treatment. Among 925 inhabitants, only 15 cases
of very mild complaints, namely dizziness and nausea, were reported.
While fenitrothion was characterized as a pesticide that is safe for
the inhabitants in a subtropical region during dry and hot seasons,
further investigations were recommended on the toxic effects on
operators under tropical conditions (Motabar et al., 1972).
In a field spraying operation in a village in southern Nigeria
using a 5% spray of fenitrothion, 18 villagers examined one week
later did not show any clinical symptoms of toxicity or plasma ChE
depression. The ChE levels in the 3 spraymen examined on the first,
second, and sixth days after spraying were also not depressed
compared with pre-spraying levels (Vandekar, 1965).
In another field spraying operation in northern Nigeria, 10 000
huts, in which about 16 500 people lived, were sprayed. Field test
ChE determinations on whole blood did not show any appreciable
differences in the ChE levels of 535 villagers tested before
spraying and 299 villagers tested 5-30 days after spraying. After
one week of intensive spraying, 5 out of 20 spraymen developed a 50%
depression of ChE, which returned to a stable level after a period
of rest. One sprayman developed symptoms of intoxication that lasted
only a few hours and disappeared without treatment (Wilford et al.,
1965).
The health was monitored of workers employed in forest spraying
operations using fenitrothion, in New Brunswick, and plasma and
erythrocyte ChE activities, measured. There were no instances of ChE
activity fluctuating beyond the accepted range or of suspected
poisoning resulting from occupational exposure to this insecticide
(Braid & McCarthy, 1977).
Moderate poisoning of 25 workers was reported in Czechoslovakia
(Kalas, 1978; Hayes, 1982), where a formulation containing 50%
fenitrothion was applied by aircraft during a strong wind. Onset of
poisoning developed 2.5-6 h after inhalation and the symptoms were
typical. Whole blood ChE activity was decreased by 48%. Recovery
required 3 days of treatment with atropine.
In the Haitian malaria control programme, depressed whole blood
cholinesterase activity (< 50% of normal) was detected rapidly
prior to the development of serious symptoms. Evidence of
fenitrothion over-exposure appeared in spraymen early in the first
spray cycle, and was associated with faulty protective clothing and
failure to follow strictly the recommended safety measures at work.
After these deficiencies were corrected, insecticide application
continued without serious incidents or interruption of the
programme. It was recommended that training and monitoring
programmes should be instituted whenever organophosphate pesticides
are used as residual sprays for malaria control (Warren et al.,
1985a). Similar observations were made in another study undertaken
in Pakistan and Haiti (Miller & Shah, 1982).
Cholinesterase activity was significantly reduced at the end of
the working week in 3 out of 28 fenitrothion workers in Haiti.
Urinary levels of 3-methyl-4-nitrophenol in the spraymen ranged from
2.2 to 25.2 mg/litre. In fenitrothion workers who had no direct
contact with spraying (weighers and supervisors), the cholinesterase
activity remained > 75% of the normal control value, and the
urinary 3-methyl-4-nitrophenol levels were relatively low. The
cholinesterase levels improved and the urinary excretion of
metabolites decreased after 2 days of rest from the spraying
operations. In the residents of the sprayed houses, low
concentrations of 3-methyl-4-nitrophenol were detected in the urine,
1 day after spraying, and measurable, but reduced, levels were still
present after 7 days. In all these cases, the cholinesterase
activity remained > 75% of the normal control value (Warren et
al., 1985b).
In an epidemiological study on the effects of fenitrothion on
occupationally exposed male workers in the production department and
female workers in the packing room during a 5-year period, the
results of clinical examinations pointed to parasympathetic
stimulation and disposition to hypotonia. Neurological and
psychiatric findings revealed a low-grade pseudoneurasthenic
syndrome in 33% of females and in 15% of males. The results of
psychodiagnostic tests after exposure to fenitrothion and its
intermediate products showed partial deterioration of retention,
impairment of visuomotor coordination of movements in tapping,
impaired orientation readiness, prolonged average time in
decision-making, and prolonged average reaction time for complex
sensorimotor responses. The following effects on biochemical
parameters after exposure to fenitrothion should be mentioned:
inhibition of cholinesterase in the blood, increase of glutamate
pyruvate transaminase, increase of isoenzyme lactate dehydro-genase
(LDH-5), and changes in protein fractions, all of which were
statistically significant (P < 0.001). The values of
3-methyl-4-nitrophenol excreted in the urine of males after the
exposure to fenitrothion averaged 5.61 mg/litre, compared with an
average value of 1.54 mg 3-methyl-4-nitrophenol/litre before
exposure. The average values of 3-methyl-4-nitrophenol excreted in
the urine of females involved in bottling fenitrothion were 4.0 mg
3-methyl-4-nitrophenol before exposure and 6.58 mg
3-methyl-4-nitrophenol/litre of urine after exposure. The
concentrations of fenitrothion in the air of the workplace ranged
from 0.028 to 0.118 mg/m3. From the values of the
3-methyl-4-nitrophenol excreted in the urine of the exposed workers
and of volunteers, to whom fenitrothion was administered in doses of
2.5-20 mg, it could be judged that the exposed male workers received
approxi-mately 15 mg of fenitrothion per capita a day and the
exposed female workers received 20 mg or more per capita a day
(Liska et al., 1982).
The introduction of the ultra-low-volume application of the
organophosphate pesticide fenitrothion in grain terminals presented
a risk to workers of skin contact with a high concentration. Blood
testing, by the Ellman method, of a group of 5 grain terminal
workers engaged in grain treatment showed a lowering of mean
erythrocyte cholinesterase activity to 23 units/g Hb (normal value
28-40) with a range of 16-29. The probable cause was identified as
percutaneous absorption of fenitrothion through ungloved hands
exposed to clean blocked drip feed nozzles. Modification of work
practices was followed by a rise of mean erythrocyte ChE activity to
33.6 units/g Hb (range 32-36) during the following grain treatment
season. Erythrocyte ChE activity measured during the intervening
winter season, i.e., during a non-exposure period, showed a mean of
33.3 units/g Hb (range 23-40) (Gun et al., 1988).
9. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
9.1 Microorganisms and algae
Salonius (1972) reported that a 12-month incubation of forest
soils with massive doses of fenitrothion emulsion at the rate of 48
mg/pot, equivalent to approximately 112 kg/ha, based on surface area
of the soil, did not alter the population or respiration of the soil
microflora.
No effects of fenitrothion were observed on the growth of 6
species of fungi responsible for the decomposition of organic
detritus in streams and swamp water containing 15, 150, or 1500 µg
fenitrothion/litre for up to 3 months (Eidt, 1978).
In Maine (USA), viable populations of bacteria, yeasts, fungi,
and actinomycetes were monitored in forest leaf litter treated with
fenitrothion at 1 mg/kg (Spillner et al., 1979a); fenitrothion
treatment did not appear to affect microbial populations or
respiration in the forest litter. Furthermore, fenitrothion (1 or 5
mg/kg) did not have any effects on cellulose-degrading organisms
(Spillner et al., 1979b).
Total numbers of bacteria and the nitrification activity in the
soil were not affected by a 5-year application of fenitrothion 50%
EC diluted 1000 times at 125 ml/m2 (Nishio & Kusano, 1978). No
effects on urease and glucanase were observed when fenitrothion was
incorporated into silt loam soil at field rates (0.70-2.11 kg
a.i./ha) (Burns & Lethbridge, 1981).
Mandoul et al. (1968) reported that a concentration of 5 mg
fenitrothion/litre did not affect microplankton in a freshwater
environment. They stated that 2 applications of fenitrothion at 140
g/ha would not approach 5 mg/litre, even assuming that all of the
material reached the lakes or streams.
Growth inhibition tests were carried out using 3 species of
algae, and EC50 values were determined to be 4-9 mg/litre for diatom
and blue-green algae, and more than 100 mg/litre for green algae
(Kikuchi et al., 1984) (Table 7).
9.2 Aquatic organisms
9.2.1 Fish
Toxicity data of fenitrothion on aquatic non-target organisms
are summarized in Table 7. Fenitrothion is moderately toxic for fish
species with LC50 values of more than 1 mg/litre.
Table 7. Toxicity of fenitrothion for aquatic non-target organismsa
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Microorganisms
Activated sewage sludge 4-h growth EC50 450 22 ± 2 Kwasniewska et al. (1980)
sediment soil
Algae
(Chlorella vulgaris) F growth EC50 100 T 23 ± 2 Kikuchi et al. (1984)
(Nitzschia closterium) F growth EC50 3.9 T 23 ± 2 Kikuchi et al. (1984)
(Anabaena flos-aquae) F growth EC50 8.6 T 23 ± 2 Kikuchi et al. (1984)
Mollusca
(Pila globosa) F 30 ± 2 g 72-h LC50 1.2 T 29 ± 2 Madhu et al. (1982)
(Phya acuta) F 48-h LC50 15 EC Hashimoto & Nishiuchi (1981)
Red snail F 48-h LC50 8.5 EC Hashimoto & Nishiuchi (1981)
(Indoplanorbis exustus)
Marsh snail F 48-h LC50 6.0 EC Hashimoto & Nishiuchi (1981)
(Semisulcospira libertina)
Eastern oyster M juvenile 96-h EC50 0.450 T 27 Mayer (1987)
(Crassostrea virginica) growth
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Crustacean
Lobster M larva 96-h approx. T 15 Mcleese (1974)
0.001
(Homarus americanus) adult 96-h LC50 approx. T 15 Mcleese (1974)
0.001
Prawn M nauplius 24-h LC50 1.9 T 23-25 Hirayama & Tamanoi (1980)
(Penaeus japonicus) postlarva 24-h LC50 0.0005- T 23-25
0.0009
Crab M zoea 24-h LC50 0.005-0.008 T 23-26 Hirayama & Tamanoi (1980)
(Portunus trituberculatus) megalopa 24-h LC50 0.0002- T 23-26
0.0005
young 24-h LC50 0.003 T 23-26
Blue crab M 8.5- 96-h LC50 0.0086 T 22 Johnston & Corbett (1985)
(Callinectes sapidus) 11.0 cm
Brown shrimp M juvenile 96-h LC50 0.0015 T 29 Mayer (1987)
(Panaeus aztecus)
Water fleas
(Daphnia pulex) F adult 3-h LC50 0.050 T 24-26 Hashimoto & Nishiuchi (1981)
(Daphnia magna) adult 48-h LC50 0.0086 T 20 ± 2 Forbis (1987)
adult/young 21-day MATC 0.00014 T 20 ± 2 Burgess (1988)
(Moina macrocopa) F adult 3-h LC50 0.050 T 24-26 Hashimoto & Nishiuchi (1981)
Mite
(Hydrachna trilobata viets) F 48-h LC50 0.074 T 28 ± 3 Nair (1981)
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Arthropod Insects
Caddisfly larva F 96-h LC50 11 T 5 Symons & Metcalf (1978)
(Brachycentrus numberosus)
Scud M 96-h LC50 0.01 T 18 Swain & Ivanikiw (1976)
(Gammarus pseudolimnaeus) 96-h LC50 0.0043- T 12 Woodward & Mauck (1980)
0.0088
Stonefly naiad
(Peleronarcys californica) F 96-h LC50 0.004 T 16 Swain
& Ivanikiw (1976)
(Pteronarcella badia) 96-h LC50 0.0051- T 12 Woodward & Mauck (1980)
0.0072
Mayfly larva F 9.3 mm, 48-h LC50 0.0032 EC 25 Nishiuchi (1981)
(Cloeon dipterum) 5.6 mg
Dragon fly larva F 2.3 cm, 48-h LC50 0.055 EC 25 Nishiuchi (1981)
(Orthetrum albistylum 0.62 g
speciosum)
(Sigara substriata) F 5.9 mm, 48-h LC50 0.023 EC 25 Nishiuchi (1981)
6.1 mg
(Micronecta sedula) F 3.2 mm, 48-h LC50 0.058 EC 25 Nishiuchi (1981)
1.8 mg
(Sympetrum frequens) F 2.1 mm, 48-h LC50 0.050 EC 25 Nishiuchi (1981)
(larva) 0.56 g
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
(Eretes sticticus) 1.5 cm, 48-h LC50 0.062 EC 25 Nishiuchi (1981)
0.20 g
Amphibian
Clawed toad embryo 24-h LC50 10 T 18 Elliott-Feeley &
(Xenopus laevis) 24-h EC50 4.2 T 18 Armstrong (1982)
24-h LC50 10 T 25
24-h EC50 0.37 T 25
24-h LC50 0.33 T 30
24-h EC50 0.17 T 30
(Bufo b. japonics) tadpole 48-h LC50 9.0 EC Hashimoto & Nishiuchi (1981)
(Microhyla ornatas) embryo 96-h LC50 3.21 T Pawar & Katdare (1984)
tadpole 96-h LC50 1.14 T
Freshwater fish
Carp 5.1 cm 48-h LC50 8.2 T 25 ± 1 Hashimoto & Nishiuchi (1981)
(Cyprinus carpio) 7-9 cm 72-h LC50 2.3 50% EC 28- 32 Toor & Kaur (1974)
eyed egg 24-h LC50 3.5 EC 25 ± 2 Hashimoto et al. (1982)
sac fry 24-h LC50 1.5 EC 25 ± 2 Hashimoto et al. (1982)
floating fry 24-h LC50 1.7-4.0 EC 25 ± 2 Hashimoto et al. (1982)
6.0 cm, oral LD50 10 mg/kg T 20 ± 22 Hashimoto & Fukami (1969)
2.5 g
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Killifish 48-h LC50 7.0 T 25 ± 1 Hashimoto & Nishiuchi (1981)
(Oryzias latipes) embryo 96-h LC50 10 T 25 ± 1 Takimoto et al. (1984b)
yolk sac 96-h LC50 6.94 T 25 ± 1 Takimoto et al. (1984b)
fray
postlarva 96-h LC50 2.36 T 25 ± 1 Takimoto et al. (1984b)
juvenile 96-h LC50 3.54-4.66 T 25 ± 1 Takimoto et al. (1984b)
adult 96-h LC50 3.70 T 25 ± 1 Takimoto et al. (1984b)
Rainbow trout fingerling 96-h LC50 2.0 T 15 Klaverkamp et al. (1977)
(Salmo gairdneri) 9.2 ±
0.1 cm,
10.35 ±
0.78 g
egg/fry 60-days post- 0.12 T 10 ± 1 Cohle (1988)
hatch MATC
Brook trout 96-h LC50 1.7 T 12 Swain & Ivanikiw (1976)
(Salvelinus fontinalis) 96-h LC50 2.1 40% EC 7 Swain & Ivanikiw (1976)
96-h LC50 2.2 40% EC 12 Swain & Ivanikiw (1976)
96-h LC50 1.8 40% EC 17 Swain & Ivanikiw (1976)
15-21 cm No effect on 10 mg/kg T 15 ± 2 Wildish & Lister (1977)
(42-120 g) growth food
8.3-12.6 cm Effect on 0.50 T 15 Peterson (1974)
critical
swimming
velocity
Cutthroat 118 mm, 20 g 96-h LC50 2.57-2.88 T 12 Woodward & Mauck (1980)
(Salmo clarki) 96-h LC50 2.88 87% EC 12
96-h LC50 2.70 87% EC 7
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
Gold fish 2-3 g, 4-5 24-h LC50 4.5 pure 25 ± 2 Gras (1966)
(Carassius auratus) product
Goldfish 9-11 cm 48-h LC50 3.4 T 25 Hashimoto & Nishiuchi (1981)
(Carassius auratus) threshhold for approx. 0.01 T 15 Scherer (1975)
avoidance
Eel (Anguilla anguilla) 2 g, 10 cm 24-h LC50 3.2 pure 25 ± Gras (1966)
product
Gambusia 0.25-0.60 g 24-h LC50 2.6 pure 25 ± Gras (1966)
(Gambusia affinis) product
Fathead minnow embryo-larva 31 days 0.13-0.30 T 23.8 ± 1 Kleiner et al. (1984)
(Pimephales promelas) MATC
Pond loach 48-h LC50 4.8 T 25 ± 1 Hashimoto & Nishiuchi (1981)
(Misqurnus anquilicaudatus)
(Acheilognathus moriokae 5.5-6.0 cm, 72-h LC50 5.0 T 25 Nishiuchi (1977)
1.2-1.4 g
(Channa gachua) 116 mm, 18 g 96-h LC50 12.2 50% EC 24 ± 2 Verma et al. (1978a)
(Saccobranchus fossilis) 135 mm, 25 g 96-h LC50 12.5 50% EC Verma et al. (1978b)
50-75 mm, 96-h LC50 12.6 50% EC 18 ± 2 Verma et al. (1982)
5-10 g
(Mystus cavasius) 6-8 cm 96-h LC50 3.3 T 28 ± 2 Murty et al. (1983)
Table 7 (contd).
Species Size Parameters Toxicity Formulation Temperature Reference
(mg/litre) (°C)
(Labeo rohita) 118 mm, 20 g 96-h LC50 4.63 50% EC Verma et al. (1977)
3-4.4 cm 96-h LC50 2.8 T 28 ± 2 Murty et al. (1983)
4.5-5.9 cm 96-h LC50 4.1 T 28 ± 2 Murty et al. (1983)
6-8 cm 96-h LC50 4.6 T 28 ± 2 Murty et al. (1983)
(Aplocheilus latipes) 24-h LC50 4.5 T 25 Shim & Self (1973)
(Zacco platypus) 24-h LC50 7.4 T 25 Shim & Self (1973)
(Puntius ticto) 50-68 mm, 96-h LC50 5.8 50% EC 11-29 Bhatia (1971)
1.6-5.1 g
Seawater fish
Japanese striped knifejaw egg-prelarva 24-h LC50 2.4-4.2 50% EC 22 ± 0.1 Seikai (1982)
(Oplegnathus fasciatus) postlarva 24-h LC50 0.145 50% EC 22 ± 0.1 Seikai (1982)
postlarva- 24-h LC50 1.25-2.35 50% EC 22 ± 0.1 Seikai (1982)
juvenile
Agohaze (Chasmichthus 0.02-0.06 g 96-h LC50 1.1 50% EC 15 Hirose et al. (1979)
dolichognathus d.) 96-h LC50 1.7 50% EC 20 Hirose et al. (1979)
Sheepshead minnow juvenile 48-h LC50 > 1.0 T 9 Mayer (1987)
(Cyprinodon variegatus)
24-h EC50 0.17 T 30
a EC = Emulsifiable concentrate; F = Freshwater; M = Marine; MATC = Maximum Acceptable Toxicant
Concentration; NEC = No effect concentration; T = Technical ingredient.
In studies on the relative toxicity of fenitrothion in
different developmental stages of fish, postlarvae of killifish
(Takimoto et al., 1984b), and Japanese striped knifejaw (Seikai,
1982) and the sac fry of carp (Hashimoto et al., 1982) were most
susceptible with LC50 values of 2.36, 0.145, and 1.5 mg/litre,
respectively.
Oral administration of fenitrothion to carp showed a high
LD50 value (more than 10 mg/kg) (Hashimoto & Fukami, 1969),
whereas the 4-week, no-effect, dietary concentration was 1 mg/kg
food (Wildish & Lister, 1973).
Ingestion of contaminated, aquatic or terrestrial insects by
salmonids is extremely unlikely to cause lethal or sublethal effects
in the fish. Behavioural changes did not occur in laboratory studies
until brook trout ingested 3000 times more fenitrothion than the
3.19 mg/kg found in poisoned insects in treatment areas (Wildish &
Lister, 1973).
Exposure of young Atlantic salmon to 0.1 or 1 mg fenitrothion
per litre for 15-16 h caused a 20 or 50% reduction in numbers of
fish holding territories, respectively. Recovery of the parr from
the above effects appeared to be complete in 3 weeks. Feeding and
swimming behaviours returned to normal within 24-48 h (Symons,
1973). After exposure to 1.0 mg fenitrothion/litre for 24 h,
Atlantic salmon parr were more vulnerable than unexposed fish to
predation by large brook trout; fenitrothion at 0.1 mg/litre did not
have any noticeable effects on vulnerability (Hatfield & Anderson,
1972).
Juvenile Atlantic salmon exposed to 6.7 µg fenitrothion/litre
for 7 days showed a decrease in the reaction distance to prey, but
no significant decrease in the efficiency of the salmon's attack
sequence (Morgan & Kiceniuk, 1990).
The movement of goldfish was dependent on the fenitrothion
concentration in water, the threshold level being about 0.01
mg/litre (Scherer, 1975).
Long-term studies revealed that the no-observed-effect level or
maximum acceptable toxicant concentration (MATC) was at 0.1 mg/litre
and above 0.1 mg/litre, respectively, in fathead minnows in a 31-day
early life stage test (Kleiner et al., 1984) and in guppies in a
2-month reproduction test (Yasuno et al., 1980).
Exposure of freshwater murrel ( Channa punctatus) to a
subtoxic concentration of 1.5 mg fenitrothion 50% EC/litre resulted
in a reduction in protein-bound iodine levels after 60, but not 30,
days exposure (Saxena & Mani, 1985a). On day 120 of exposure,
effects on male and female reproductive function were observed, such
as reductions in testicular and ovarian weight, growth rates
of spermatocytes and oocytes, and the numbers of sperm and ova (Mani
& Saxena, 1985; Saxena & Mani, 1985b, 1987). However, it is not
clear whether such effects were derived from fenitrothion or other
EC components.
Pregnant female guppies were exposed to 10 mg
fenitrothion/litre for 4 h, 5, 10, or 15 days before the next
parturition. Half of the females gave premature birth when exposed 5
or 10 days before parturition, and only 32 or 63%, respectively, of
the eggs were delivered alive. The females exposed to the
fenitrothion 15 days before parturition had normal births and only
9.4% of the offspring were stillborn. The body lengths of the young
produced by the females after exposure were significantly shorter
than those produced before exposure in all the studies (Yasuno et
al., 1980). In a study by Miyashita (1984), exposure of pregnant
guppies ( Poecilia reticulata) to 10 mg/litre for 4 h at various
stages of gestation resulted in a reduction in the body lengths of
the offspring at birth.
Rainbow trout ( Salmo gairdneri) were exposed to fenitrothion
in a flow-through, early life stage, toxicity study at
concentrations of 0, 0.025, 0.046, 0.088, 0.17, or 0.35 mg/litre,
for 60-days after hatching at 10 ± 1 °C. Survival and growth of fry
showed that the maximum acceptable toxicant concentration (MATC)
limits were 0.088-0.17 mg/litre and that the point estimate MATC
value was 0.12 mg fenitrothion/litre (Cohle, 1988).
There were no direct lethal effects on wild and caged salmonid
fish after forest spraying with fenitrothion at dosages varying from
double sprays of 140 g/ha to single sprays of up to 280 g/ha in
Newfoundland, Canada (Hatfield & Riche, 1970).
Studies of the effects on wild salmon of fenitrothion sprays in
New Brunswick, Canada, from 1966 to 1969 revealed no mortality in
streams at spray levels of up to 560 g/ha (MacDonald & Penney,
1969). Studies in Manitoba, Canada, on stream-caged rainbow trout
exposed to a spray of 280 g fenitrothion/ha did not show any
significant biochemical changes, including changes in brain and
serum ChE activities. In the 24-h period after spraying,
fenitrothion residues in whole fish averaged 0.5 mg/kg wet weight.
Peak residue values in the fish were as high as 1.84 mg/kg wet
weight but declined to less than 0.02 mg/kg wet weight in 4 days.
Lockhart et al. (1973) concluded that no long-term toxic effects
would be expected among the caged fish.
In the 1979 aerial spraying programme in Canada,
concentrations of fenitrothion of up to 0.64 µg/litre were detected
in stream water in the spray block within 40 h; no
insecticide-related deaths of caged fish or abnormal fish behaviour
occurred (Gillis, 1980).
In 1979, fenitrothion was sprayed on a Scottish forest at a
rate of 300 g/ha. The maximum concentration in a river was 18.8 µg
per litre, 1-2 h after application. There was no evidence that the
resident fish population was disturbed, and no short-term effects
were noticeable in caged fish (Morrison & Wells, 1981). Gillis
(1978) demonstrated, in a 1977 study, that, in an area with a long
history of perennial treatment with fenitrothion (8 years since
1969), there were consistently higher numbers of salmon per 100 m of
stream, but that the density of trout was similar to that in the
control area. It was concluded that the long history of fenitrothion
usage in Canadian forests was unlikely to have resulted in depletion
of the population or biomass of salmon parr trout.
9.2.2 Invertebrates
The acute toxicity of fenitrothion for Daphnia magna was
assessed using the 48-h static method at 20 ± 2 °C. The 48-h LC50
value was 8.6 µg/litre (6.8-11 µg/litre) and the no-observed-effect
level was estimated to be < 2.0 µg/litre after 48 h (Forbis, 1987).
Daphnia magna was exposed to fenitrothion in a dynamic
21-day, life cycle, toxicity study at concentrations of 0, 0.029,
0.042, 0.087, 0.23, or 0.44 µg/litre, at 20 ± 2 °C. Statistical
analyses of survival, adult mean length, and length in days to first
brood, showed that the maximum acceptable toxicant concentration
(MATC) limits were estimated to be 0.087 and 0.23 µg/litre and the
point estimate MATC value was 0.14 µg/litre (Burgess, 1988).
Fenitrothion is highly toxic for arthropods, including insect
larvae, shrimps, crabs, and daphnids. LC50 values for these
invertebrates are generally at µg/litre levels, except that for
caddis fly larvae with a value of 11 mg/litre (Table 7).
Exposure of the freshwater rice-field crab ( Oziotelphusa
senex senex) to sublethal concentrations of fenitrothion showed a
number of effects: limb regeneration was completely inhibited at 0.1
mg/litre and partially inhibited at 0.01-0.05 mg/litre, during
continuous 60-days exposure (Reddy et al., 1983a); exposure to 0.04
mg/litre for 30 days reduced glycolysis and increased
gluco-neogenesis in the hepatopancreas and muscle (Reddy et al.,
1982a, 1983b); exposure to 0.1 mg/litre induced inhibition of
molting and ovarian growth, perhaps by triggering the release of
molt-inhibiting hormone and gonad-inhibiting hormone (Reddy et al.,
1982b, 1983c); exposure for 1 day to 0.1 mg/litre produced an
increase in muscle protease activity and a decrease in the protein
content, while exposure for 20 days resulted in increases in both
parameters (Bhagyalakshmi et al., 1983a); continuous exposure for
20-30 days to higher fenitrothion concentrations of 0.5, 1, or 2
mg/litre resulted in a decrease in oxygen consumption, an increase
in haemolymph glucose, and a reduction in carbohydrate metabolism
rate in the hepatopancreas, with the maximal effect occurring after
3-7 days of exposure. During the exposure, levels of haemolymph
glucose and oxygen consumption returned to the control levels,
accompanied by an increase in carbohydrate metabolism (Bhagyalakshmi
et al., 1983b, 1984a). Exposure to 1, 2, or 4 mg/litre for 48 h
resulted in a dose-dependent inhibition of acetylcholinesterase
activity of the thoracic ganglionic mass, where mean activity levels
in the treated groups ranged from 30 to 60% of the control values.
On cessation of exposure, activity levels returned to normal within
10-15 days (Bhagyalakshmi et al., 1984b).
Fenitrothion was experimentally added to a stream at a
concentration of 10 mg/litre, when the predominant and prevalent
species in the drift samples were shrimps ( Anisogammarus sp.),
mayfly ( Epeorus ikanonis, and Baetis sp.), stonefly ( Nemovra
sp.), and blackfly ( Simulium sp.) larvae. In the course of the
study, Simulium sp. increased in density, but Anisogammarus sp.
did not recover after 4 months (Yasuno et al., 1981). When
fenitrothion was applied at 1 mg/litre to a stream, drifting aquatic
invertebrates including shrimp ( Anisogammarus annandalei), Baetis
sp., Nemouridae, Epeorus sp., Perla quadrata, and Simulium
sp., but not caddis fly larvae (Arctopsyche sp.) or crab
( Geothelephusa dehaanii), were affected by the insecticide
(Hasegawa et al., 1982).
After treatment of a small lake with 140 g fenitrothion/ha,
surface populations of zooplankton and phantom midge larvae
(Chaoborus sp.) were depressed for a short period. No substantial
impact was found on benthic invertebrates, emerging insects, and
amphibians in the lake (Kingsbury, 1978).
9.2.3 Amphibians and arthropods
Acute toxicity tests revealed that fenitrothion at 0.1 mg/litre
did not induce any significant effects in amphibian embryos
( Xenopus laevis) (Elliott-Feeley & Armstrong, 1982).
When the frog embryo (Microhyla ornata), at the yolk-plug
stage, was exposed to 1 mg fenitrothion/litre for 96 h, no effects
were observed on development. Exposure at 3 mg/litre resulted in
blistering of the body surface and exposure at 5 mg/litre or more
resulted in abnormal behaviour, curvature of the spine, diminished
pigmentation, and retarded growth (Pawar & Katdare, 1983, 1984).
The LC50 value for fenitrothion in tadpoles of Bufo buto
japonicus was 9.0 mg/litre (Hashimoto & Nishiuchi, 1981). The
toxicity of fenitrothion in mollusca was low with LC50 values of
1.2-15 mg/litre in freshwater molluscs and 0.45 mg/litre in eastern
oyster (Table 7). Fenitrothion residues in frogs, collected near
stagnant water 1 day after spraying (280 g/ha), ranged from
0.03-0.17 mg/kg wet weight (Lockhart et al., 1977).
No effects were observed on amphibians and small mammals in
Northern Maine, USA, where the forest had been treated twice with
fenitrothion at the rate of 140 g/ha (USDA, 1976). Large numbers of
hatching salamander eggs were present throughout the treatment
period in the silty bottomed pond located in a treatment plot. No
deaths were observed up to a week after the second spray. When the
pond was examined approximately 2 months after treatment, it still
contained many larval salamanders and aquatic invertebrates.
A stream was treated with fenitrothion at a calculated
concentration of 73 µg/litre for 2.5 h. The standing crop of benthic
arthropods decreased, most of the kill consisting of stonefly larvae
(Leucta sp.). Benthos, including the stonefly larvae, completely
recovered 50 days after the treatment (Eidt, 1981).
MacDonald & Penney (1969) studying the effects on aquatic
insects of fenitrothion, applied twice at a rate of 140 g/ha, found
that the aquatic insect population remained stable. The results of
other studies suggested that populations of aquatic insects may be
reduced following operational spraying with fenitrothion at 140-280
g/ha (National Research Council of Canada, 1975). Stonefly nymphs,
in particular, and mayfly nymphs, to a lesser extent, appeared to be
susceptible to fenitrothion, along with caddis fly larvae.
Flannagan (1973) reported that within 24 h of spraying at 280
g/ha in Canada, the drift of chironomid larvae increased by 700-800%
(total numbers/net). In some instances, Eidt (1975) and Peterson &
Zitko (1974) observed that the drift returned to normal shortly
after the pulse of fenitrothion had cleared.
Fenitrothion was sprayed on a Scottish forest at a rate of 300
g/ha. The maximum concentration in a river within the sprayed
region, which was 18.8 µg/litre, fell to 0.5 µg/litre after 24 h.
Invertebrate drift increased 12-16 h after spraying, but decreased
to pre-spray levels within 48 h. Caged insects remained alive during
the 5-day post-spray period (Morrison & Wells, 1981).
Levels of fenitrothion residues in stream insects would not be
sufficiently high to cause mortality in fish; 3.19 mg fenitrothion
per kg was detected in a sample of mayfly nymphs one week after
spraying with fenitrothion at 280 g/ha, but no residues of
fenitrothion were found after this date (Kingsbury, 1976).
An application of fenitrothion at 210 g/ha in Quebec, Canada,
in 1978, had few or no adverse effects on aquatic invertebrates or
fish, though a small increase in the drift of mayfly nymphs was
observed after application (Holmes, 1979).
In an area with a long history of perennial fenitrothion
spraying (8 years since 1969), a light depletion of the invertebrate
population was observed on both drift and benthos in streams after
each spray, but the invertebrate density stabilized within 1 month
and was comparable with the control (Gillis, 1978).
When fenitrothion was dispersed by a helicopter in Japan at a
rate of 884 g a.i./ha on a paddy field, a peak concentration of 554
µg/litre was reached immediately after spraying, and the
concentration of fenitrothion in the paddy field decreased to less
than 5 µg/litre, 3 days after spray. A crustacean Moina sp., which
was prevalent in the field, disappeared after fenitrothion spraying,
but the population recovered to pre-spray levels within 10 days
(Takaku et al., 1979b).
9.3 Terrestrial organisms
The acute and short-term toxicities of fenitrothion for
terrestrial, non-target organisms are summarized in Table 8.
9.3.1 Terrestrial invertebrates
Studies using drop cloths (Leonard, 1971; Kettela & Varty,
1972; Miller et al., 1973) indicated that large numbers of
Lepidoptera, sawfly larvae, balsam twig aphids, and perching flies
(nematocerous Dipterans, etc.) were killed directly after the
spraying operation (140 or 210 g/ha), usually within the first 4
days. Generally, other defoliating insects were reduced in about the
same proportion as the target species; the larval sawflies were
affected more severely.
Various preliminary measurements have been made on the effects
of fenitrothion on invertebrate fauna (National Research Council of
Canada, 1975). Populations of ground-inhabiting invertebrates
declined after two operational applications of 140 and 210 g
fenitrothion/ha (Carter & Brown, 1973). The densities of predatory
invertebrates were higher in the 2 years before and the year
following the application. Thus, while the invertebrate populations
appeared to be depressed during the years of application, they were
generally able to recover to the normal levels.
On the basis of field observations on the long-term effects of
fenitrothion on non-target arthropods in Canada, Varty (1977)
concluded that:
- The non-target arthropod community on balsam fir was not
perceptibly destabilized by perennial or intermittent
applications at conventional rates (140-210 g/ha) and timing.
- A few fir-dwelling insects had become scarcer during the 1970s,
but the cause-effect was undetermined, and natural fluctuation
could account for the population decline.
- The abundance of predators was related primarily to cycles of
preabundance on fir and spruce, and to budworm modification of
habits.
- Herbivore populations tended to be depressed by budworm
competition, but fungivores might be favoured.
- The abundance of the two most important species parasitizing
spruce budworm larvae persisted, in spite of perennial
spraying.
- Population densities of some species-predatory arthropods in
the soil community might suffer some setback, but in short-term
studies recuperation seemed adequate.
- Minor-pest eruptions in the 1970s did not appear to be
triggered by the local disruption of biocontrol mechanisms
following spraying.
Fenitrothion is highly toxic for bees (Anderson & Atkins, 1968;
Okada & Hoshiba, 1970), the LD50 is approximately 0.03-0.13 µg per
bee (Anderson & Atkins, 1968) (Table 8). However, studies on the
effects of fenitrothion on colonies of honey-bees indicated that an
application of 280 g/ha had few long-term effects (National Research
Council of Canada, 1975). Initially, mortality of adult foraging
bees was evident. Within 4 days of treatment, the daily adult
mortality of foraging bees had apparently returned to normal. The
total detected mortality in excess of controls was about 500 adult
bees, which was estimated to be about 1% of the total hive
population. Buckner (National Research Council of Canada, 1975)
reported that hive activity was unaffected compared with controls,
and that hive weight and eventual honey yield were almost identical
in all experimental and control hives. The observed mortality was
probably restricted to actively foraging worker bees that were
actually exposed to the spray.
Table 8. Toxicity of fenitrothion for terrestrial non-target organismsa
Species Size Toxicity Formulation Condition Reference
Birds
Bobwhite quail 14 days old LC50 5000 mg/kg diet T 8-day dietary Hill et al. (1975)
(Colinus virginianus)
Japanese quail 14 days old LC50 5000 mg/kg T 8-day dietary Hill et al. (1975)
(Coturnix c japonica) diet (no mortality)
5 weeks old (M) LD50 84.85 mg/kg 50% EC oral Hattori et al. (1974)
5 weeks old (F) LD50 73.87 mg/kg 50% EC oral Hattori et al. (1974)
3-6 weeks old (M) LD50 110 mg/kg T oral Kadota & Miyamoto (1975)
3-6 weeks old (F) LD50 140 mg/kg T oral Kadota & Miyamoto (1975)
3-6 weeks old NEL approx. 5 mg/kg diet T 4-week dietary Kadota & Miyamoto (1975)
Ring-necked pheasant 10 days old LC50 5000 mg/kg T 8-day dietary Hill et al. (1975)
(Phasianus colchicus) diet (no mortality)
Mallard duck
(Anas platyrhynchos) 10 days old LC50 5000 mg/kg T 8-day dietary Hill et al. (1975)
diet (no mortality)
13-16 weeks old (M) LD50 1190 mg/kg T oral Hudson et al. (1979)
55-61 weeks old (M) LD50 504 mg/kg T percutaneous Hudson et al. (1979)
Redwinged blackbird
(Agelaius phoeniceus) LD50 25 mg/kg oral Schafer (1972)
Table 8 (contd).
Species Size Toxicity Formulation Condition Reference
Pigeon 250-380 g LD50 42.24 mg/kg 50% EC oral Hattori (1974)
(Columba livia)
Grackle
(Quiscalus quiscula) LC50 78 mg/kg diet T 5-day dietary Grue (1982)
Insect
Honeybee 7 days old 24-h LD50 0.13 µg/bee T topical Takeuchi et al. (1980)
(Apis mellifera) 32 ± 1 °C
24-h LD50 0.03 µg/bee 50% EC topical Okada & Hoshiba (1970)
32 ± 1 °C
LD90 0.25 µg/bee 60% EC oral Clinch & Ross (1970)
30 °C
Silkworm II-V instar 24-h LD50 T topical Kashi (1972)
(Bombyx mori) 4.88-18.7 µg/g
a M = Male; F = Female; T = Technical ingredient; EC = Emulsifiable concentrate.
Plowright (1977) demonstrated that fenitrothion, sprayed at a
dose level of 210 g/ha, was capable of inducing direct mortality in
caged bumble-bees. Aerially sprayed fenitrothion at 210 g/ha in
Canada caused 100% mortality in exposed habitats and 47% under dense
forest canopy, among caged bees (Bombus sp). Bumble bees foraging
in sprayed areas suffered significantly higher mortality than in
unsprayed areas. The abundance of bee species was 3 times less in
sprayed areas compared with unsprayed areas. Population recovery
appeared to be complete within a few years (Plowright et al., 1978).
Plowright & Rodd (1980) further examined the effects of
fenitrothion in wild bees, and found that fenitrothion sprayed at
210 g/ha caused high mortality among solitary bees and vespid wasps,
similar to that in bumble bees (Plowright et al., 1978). Wood (1979)
found that spraying of fenitrothion over nearby woodland resulted in
very low counts of native bees; however, the population returned to
normal levels within 3 years of spraying, with satisfactory
pollination of blueberry.
Wild Bombus sp. queen territories were identified prior to
treatment and a search was made for these queens after the
application of fenitrothion (280 g/ha) in Larose Forest, Canada.
Queens identified prior to a morning spray were again identified 1,
2, and 3 days after treatment. In this single study, no significant
effects were indicated (National Research Council of Canada, 1975).
The low yield of low-bush blueberry was reported to be
attributable to the mortality of native bees (Kevan, 1975) and lack
of pollination following aerial fenitrothion spraying to control
spruce budworm in nearby forests in New Brunswick. However, field
experiments in 1980 showed that there was no evidence of any harmful
effects on the blueberry as a result of fenitrothion spraying and
that the concentration of fenitrothion aerosols found in blueberry
fields during the 1980 budworm spray programme was estimated to be
well below the toxic levels for all bee species (Wood, 1980).
Bee, pollen, and wax samples were collected from forest areas
that had been sprayed with fenitrothion for budworm control at an
operational dosage of 140-280 g/ha, over a 3-year period from 1972.
The level of fenitrothion in bees was low (maximum 2.08 mg/kg)
initially, and decreased rapidly to trace levels within a week.
Fenitrothion levels in pollen were very low initially, but declined
less rapidly than in bees. Accumulations of the insecticide in honey
and wax samples were negligibly small, but traces seemed to persist
in the wax for some time (Sundaram, 1975).
Fenitrothion is toxic for one of the beneficial insects,
silkworm ( Bombyx mori) larvae. Silkworms were fed mulberry leaves
that had been treated with fenitrothion at the rate of 100 ml of
0.0025 or 0.005% fenitrothion solution /kg leaves (equivalent to 2.5
and 5 mg/kg). The administration during the 5 instar larva stage
resulted in a reduction in the number of eggs laid, fertility, and
hatching (Kuribayashi, 1981). At a lower concentration (1 mg/kg), no
effect was observed on mortality, egg laying, and hatching (Yamanoi,
1980). Growth after hatching was not affected by fenitrothion
treatment (Yamanoi, 1981).
The populations of nematodes, rotifers, and tardigrades in soil
were not affected after fenitrothion was sprayed on the surface of a
pasture at a rate of 2.24 kg a.i./ha (Martin & Yeates, 1975).
9.3.2 Birds
The acute oral LD50 values of fenitrothion in birds are shown
in Table 8. The values range from 25 mg/kg (Redwinged blackbird) to
1190 mg/kg (Mallard duck).
Except for the grackle with a dietary LD50 of 78 mg/kg,
values were generally more than 5000 mg/kg diet and, at this dose
level, no mortality was observed (Hill et al., 1975).
The effects of fenitrothion on the brain cholinergic system
were investigated in male Japanese quail (8-14 weeks old).
Cholinergic signs, such as salivation and convulsions in legs and
wings, were seen 6-120 min after the administration of fenitrothion
(250-350 mg/kg). Sixty minutes after an oral dose of fenitrothion
(300 mg/kg), free acetylcholine increased and acetylcholinesterase
(AChE) activity decreased to 20% of the control value. In vitro,
fenitrothion inhibited AChE activity in brain homogenate with an
I50 of 10-5 mol/litre (Kobayashi et al., 1983).
Japanese quail (60 per sex; 20 per sex in the 1.5 mg/kg group)
received 0, 1.5, 5, 15, or 50 mg fenitrothion/kg in the diet for 4
weeks. No animals died and no toxic signs were seen; no
abnormalities in body weight or food consumption were observed. A
dose-related decrease in blood ChE activity was observed in females
in the 5 mg/kg group (25%), and in males and females in the 15 mg/kg
(60%) and 50 mg/kg (85%) groups during the treat-ment period. When
these animals were fed the control diet again, partial recovery of
ChE activity occurred within 2 weeks, while the brain ChE activity
was reduced in females in the 15 mg/kg group (25%) and males and
females in the 50 mg/kg group (65%). Full recovery of ChE activity
occurred after 4 weeks. In the 50 mg/kg group, the egg-laying rate
was decreased, but returned to normal after 3 weeks (Kadota &
Miyamoto, 1975).
Reproduction studies on bobwhite quails and mallard ducks
revealed that short-term feeding of fenitrothion at rates of up to
10 mg/kg diet (quails) or 100 mg/kg diet (ducks) did not adversely
affect parental growth and reactions, egg production, egg weight,
hatchability, and the growth and viability of the young (Miyamoto,
1977b).
Adverse effects on bird species were observed occasionally when
fenitrothion was applied at dosages higher than those commonly used
for operational applications (National Research Council of Canada,
1975); above 280 g/ha, mortality was observed in adults that
inhabited the crown canopy, and, at rates of 560 g/ha or more, adult
mortality increased markedly. At rates of 210 or 280 g/ha,
behavioural changes were observed in adults as well as some juvenile
mortality.
When fenitrothion was sprayed at 1.7 kg a.i./ha, twice a year,
for three consecutive years in Japan, Takano & Hijikata (1981) were
unable to detect impacts on bird diversity, abundance, and
reproductive success, when 9 species (including the long-tailed tit,
which is most sensitive to fenitrothion) out of 48 were monitored in
the forests and fields (55 ha).
The various effects of fenitrothion on White-throated Sparrows
in New Brunswick, Canada, were investigated, with the following
results (Pearce & Busby, 1980):
- The parent compound fenitrothion was detected at levels as low
as 0.02 to 0.41 mg/kg in the male birds from the spray zone,
but none of its metabolites were found. There were no
correlations between fenitrothion residue levels and brain ChE
activity in sparrows.
- Singing frequency, song structure, clutch size, hatching
success, and fledging rates were similar in the control and
experimental area.
- Growth parameters, such as body weight, tarsus length, bill
length and width, and wing length, of the nestling birds
appeared to be lower in the exposed nestlings than in the
controls after fenitrothion spraying at a rate of 210 g
a.i./ha.
- The reproductive success was reduced in the exposed area after
repeated spraying of fenitrothion, first at 420 g/ha and
several days later at 210 g/ha. The young White-throated
Sparrows from 11.6% of the eggs laid fledged in the sprayed
area, whereas the young from 58.3% of the eggs laid fledged in
the control area.
- The main causes of reduced productivity in the sprayed area
were the high rate of nest desertion, the occasional death or
incapacitation of incubating females, and the high rate of
nestling disappearance from nests.
Eight hours after being exposed to an operational spray of
fenitrothion at 280 g/ha, the crops and carcasses of caged Japanese
quail contained 0.045-0.459 mg fenitrothion/kg (wet tissue basis),
when cages were placed in open locations, and the crops, up to 1.89
mg/kg, when the cages were placed below the tree canopy (Lockhart et
al., 1977). However, fenitrothion residues in all samples declined
sharply to an undetectable level (< 0.02 mg/kg) after 5 days.
Although these birds suffered a drop in serum ChE activity, they
were not killed and recovery of enzyme activity was observed 5 days
after spraying (Lockhart et al., 1977).
Experimental fenitrothion spraying at the rate of 210-280 g
a.i./ha was conducted using 2 types of spray hardware (boom and
nozzle and rotary atomizer) in southwest New Brunswick, Canada. The
brain ChE activity of the 5 avian species monitored (Tennessee
Warbler ( Vermivora peregrina), Magnolia Warbler ( Dendroica
magnolia), Blackburnian Warbler ( D. fusca), Bay-breasted Warbler
( D. castanea), and White-throated Sparrow ( Zonotrichia
albicollis)) was significantly inhibited compared with that in the
control birds, and 16-30% of the captive birds showed more than 20%
inhibition compared with the controls. However, no dead birds or
abnormal avian behaviour was observed (Busby et al., 1981).
Brain cholinesterase (ChE) inhibition and fenitrothion residues
were determined in White-throated Sparrows ( Zonotrichia
albicollis), exposed to aerial applications of fenitrothion (210 g
a.i./ha applied twice with a 5-8 day interval) during the breeding
seasons in 1978 and 1979, in New Brunswick (Canada). Brain ChE
activity was significantly reduced in birds exposed to the sprays
(50-66% inhibition) and fenitrothion and metabolite residues were
detected in all exposed birds (0.08-1.4 mg/kg); however, they did
not show any consistent correlation with brain ChE activity. An
acute brain ChE response, manifested as sudden ChE reduction
followed by gradual recovery, was noted in birds collected after
spraying with 420 g a.i./ha (Busby et al., 1983).
A study was conducted to assess the response of selected forest
songbird species, including the Tennessee Warbler ( Vermivora
peregrina), Bay-breasted Warbler ( Dendroica castanea), Magnolia
Warbler ( D. magnolia), and White-throated Sparrow ( Zonotrichia
albicollis), to aerial ultra ULV fenitrothion (40% content)
spraying (210 g a.i./ha applied twice with a 6-day interval) through
measurements of brain cholinesterase (ChE) depression. A slight
reduction (6-17% less brain ChE activity than in the controls) was
observed in birds collected on day 2
after the second spray; birds collected on day 1 of the first spray
were least affected (0-5% less activity). As a group, the Tennessee
Warbler, the upper canopy forager and singer, exhibited the highest
degree of ChE inhibition (6-17% less activity) and the
White-throated Sparrow, the ground-to-low crown dweller, showed the
least (0-8%) inhibition (Busby et al., 1987).
Brain AchE activity in songbirds exposed through experimental
fenitrothion spraying was monitored in Scotland in 1979 and 1980
(Hamilton et al., 1981). The brain ChE activity of songbirds was
significantly inhibited in the first 2 days after spraying of
fenitrothion at dosages of 300 g/ha in 1979 and 280 g/ha in 1980.
However, the enzyme activity in willow warblers ( Phylloscopus
trochilus) and chaffinches ( Fringilla coelebs) returned to the
normal levels, 7 and 21 days after spraying, respectively. The
residues of fenitrothion found in skin and plumage samples taken
from birds during the first few days after spraying were relatively
high, but declined rapidly to a low level. The fenitrothion residues
in viscera samples were very small initially and were not detectable
a few days after spraying (Hamilton et al., 1981).
Several publications concerning the possible effects of
fenitrothion on avian species including: Rushmore (1971), Hill et
al. (1975), National Research Council of Canada (1975), Paul &
Vadlamudi (1976), USDA (1976), Buckner & McLeod (1977), Germain
(1977), Pearce & Peakall (1977), Germain & Morin (1979), and Pearce
et al. (1979b) are not cited in extenso, because they report
findings similar to those above.
9.3.3 Mammals
Bailey & Swift (1968) classified fenitrothion as moderately
toxic for mammals.
Buckner reported that, at operational application rates below
420 g/ha, fenitrothion spraying did not produce any measurable
effects on small mammals in Maine (USA). Spraying of fenitrothion at
recommended rates would not be expected to cause mortality or
interrupt the breeding cycle of small mammal populations in forests
(Buckner & Sarrazin, 1975; Varty, 1976; Buckner et al., 1977)
though, at very high rates, some ill effects have been observed on
shrew and rodent populations (USDA, 1976). In addition, the
discriminatory behaviour of mature animals suggested that
fenitrothion-contaminated food material was rejected and that it was
a learned process (Buckner et al., 1977).
When fenitrothion was sprayed twice a year, at the rate of 1200
g/ha, by helicopter, there was no decrease in either plasma or
erythrocyte ChE activity in the Japanese wood mouse ( Apodemus
speciosus) inhabiting the sprayed area (Tabata & Kitahara, 1980).
The metabolism of fenitrothion in red-backed voles, which
inhabit the coniferous forests of Canada, did not differ
substantially from that demonstrated for laboratory strains of mice,
rats and guinea-pigs. When fenitrothion was administered ip at doses
of 48.4-2040 mg/kg, it was rapidly metabolized and excreted. No
accumulation of fenitrothion residues in the body was indicated. The
metabolic detoxification mechanism in red-backed voles involves the
cleavage of both the P-O-aryl and P-O-alkyl bonds, the latter being
more prominent at high dose levels (above 1250 mg/kg) (Tschaplinski
& Gardner, 1981).
Thus, the standard programme of forest spraying with the
insecticide does not appear to have serious ecological consequences
(Symons, 1977).
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Fenitrothion was evaluated by the Joint FAO/WHO Expert
Committee on Pesticide Residues (JMPR) in 1969, 1974, 1976, 1977,
1979, 1982, 1983, 1984, 1986, 1987, 1988, and 1989, (FAO/WHO, 1970,
1975a,b, 1977, 1978a,b, 1980a,b, 1983a,b, 1984a,b, 1985a,b, 1986a,b,
1987a,b, 1988a,b, 1989a,b,c). In 1988 the JMPR established an
Acceptable Daily Intake (ADI) for man of 0-0.005 mg/kg body weight.
This was based on the following levels causing no toxicological
effects:
- Rat: 10 mg/kg in the diet, equivalent to 0.5 mg/kg body
weight per day (based on brain acetylcholinesterase
inhibition and reproduction).
- Dog: 50 mg/kg in the diet, equivalent to 1.25 mg/kg body
weight per day.
- Man: 0.08 mg/kg body weight per day (highest dose tested).
The FAO/WHO Codex Committee advised maximum residue limits
(MRLs) in specified food commodities (FAO/WHO, 1986c; 1990) as
follows:
Milks 0.002 mg/kg
Cucumbers, meat, onions, potatoes 0.05 mg/kg
Cauliflower, cocoa beans, egg plants,
peppers, soybeans (dry) 0.1 mg/kg
Bread (white), leeks, radishes 0.2 mg/kg
Apples, cabbage, cabbage red, cherries,
grapes, lettuce, pear, peas, strawberries
tea (dried, green), tomatoes 0.5 mg/kg
Peach, rice (polished) 1 mg/kg
Citrus fruits, wheat flour (white)
processed wheat bran 2 mg/kg
Wheat flour (whole meal) 5 mg/kg
Cereal grains 10 mg/kg
Raw wheat bran, rice bran unprocessed 20 mg/kg
WHO (1990) classified technical fenitrothion as "moderately
hazardous" (Class II). WHO issued a data sheet on Fenitrothion (No.
30) (WHO, 1977).
REFERENCES
ABDEL-KADER, M.H.K. & WEBSTER, G.R.B. (1980) Low temperature
degradation and translocation of malathion and fenitrothion in
stored wheat. Can. Plant. Proc., 9: 143-153.
ABDEL-KADER, M.H.K. & WEBSTER, G.R.B. (1982) Analysis of
fenitrothion and metabolites in stored wheat. Int. J. environ. anal.
Chem., 11: 153-165.
ABDEL-KADER, M.H.K., WEBSTER, G.R.B., & LOSCHIAVO, S.R. (1982)
Effects of storage temperature on rate of degradation of
fenitrothion in stored wheat. J. econ. Entomol., 75: 422-424.
ADDISON, J.B. (1981) Vapour phase photochemistry of fenitrothion and
aminocarb. Bull. environ. Contam. Toxicol., 27: 250-255.
ADHYA, T.K., BARIK, S., & SETHUNATHAN, N. (1981a) Stability of
commercial formulations of fenitrothion, methyl parathion, and
parathion in anaerobic soils. J. agric. food Chem., 29: 90-93.
ADHYA, T.K., BARIK, S., & SETHUNATHAN, N. (1981b) Fate of
fenitrothion, methyl parathion, and parathion in anoxic
sulfur-containing soil systems. Pestic. Biochem. Physiol., 16:
14-20.
ADHYA, T.K., BARIK, S., & SETHUNATHAN, N. (1981c) Hydrolysis of
selected organophosphorus insecticides by two bacteria isolated from
flooded soil. J. appl. Bacteriol., 50: 167-172.
ALY, O.A. & BADAWY, M.I. (1982) Hydrolysis of organophosphate
insecticides in aqueous media. Environ. Int., 7: 373-377.
AMBRUS, A., LANTOS, J., VISI, E., CSATLOS, I., & SARVARI, L. (1981)
General method for determination of pesticide residues in samples of
plant origin, soil and water. I. Extraction and clean-up. J. Assoc.
Off. Anal. Chem., 64: 733-742.
ANAND, M., GULATI, A., GOPAL, K., KHANNA, R.N., & RAY, P.K. (1989)
Changes in cardiac rhythm and calcium levels in guinea pigs treated
chronically with fenitrothion. Toxicol. environ. Chem., 24: 199-205.
ANDERSON, L.D. & ATKINS, E.J., Jr (1968) Pesticide usage in relation
to bee-keeping. Annu. Rev. Entomol., 13: 213.
ANJUM, F. & QADRI, S.S.H. (1986) In vivo metabolism of fenitrothion
( O,O-dimethyl O-(4-nitro-m-tolyl)phosphorothioate) in fresh
water teleost ( Tilapia mossambica). Bull. environ. Contam.
Toxicol., 36: 140-145.
AOKI, Y., TAKEDA, M., & UCHIYAMA, M. (1975) Comparative study of
methods for the extraction of eleven organophosphorus pesticide
residues in rice. J. Assoc. Off. Anal. Chem., 58: 1286-1293.
BAARSCHERS, W.H., ELVISH, J., & RYAN, S.P. (1983) Adsorption of
fenitrothion and 3-methyl-4-nitrophenol on soil and sediment. Bull.
environ. Contam. Toxicol., 30: 621-627.
BAARSCHERS, W.H. & HEITLAND, H.S. (1986) Biodegradation of
fenitrothion and fenitrooxon by the fungus Trichoderma viride. J.
agric. food Chem., 34: 707-709.
BAILEY, J.B. & SWIFT, J.E. (1968) Pesticide information and safety
manual, Berkeley, University of California.
BATTELLE (1982) Pesticide programme of research and market planning.
Part II. Insecticides, Geneva, Switzerland, Battelle Research
Centres, 40 pp.
BATTELLE (1988) World pesticide programme: Insecticide III, Geneva,
Switzerland, Battelle Research Centres, Vol. 1.
BENES, V. & CERNA, V. (1970) Contribution to the toxicological
evaluation of fenitrothion
( O,O-dimethyl- O-(3-methyl-4-nitrophenyl)thiophosphate) and its
residues. In: Deichmann, W.B., Radomski, J.L., & Penalver, R.A., ed.
Proceedings of Pesticide Symposia. Inter-American Conferences on
Toxicology and Occupational Medicine, Miami, Florida, Halas and
Ass., Inc., pp. 219-222.
BENES, V. & TEJNOROVA, T. (1986) Fenitrothion: Teratogenicity study
in rats, Prague, Czechoslovakia, Institute of Hygiene and
Epidemiology (Unpublished report).
BENES, V., SRAM, R.J., & TUSCANY, R. (1974) Reproduction study in
rats, Prague, Czechoslovakia, Institute of Hygiene and Epidemiology
(Unpublished report).
BENES, V., SRAM, R.J., & TUSCANY, R. (1975) Fenitrothion I.: Study
of mutagenic activity in rats. J. Hyg. Epidemiol. Microbiol.
Immunol., 19(2): 163-172.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R. (1983a) Muscle
nitrogen metabolism of freshwater crab, Oziotelphusa senex senex
Fabricium, during acute and chronic sumithion intoxication. Toxicol.
Lett., 17: 89-93.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R. (1983b) Changes in
hemolymph glucose, hepatopancreas glycogen, total carbohydrates,
phosphorylase and amino transferases of sumition-stressed freshwater
rice-field crab. (Oziotelphusa senex senex). Toxicol. Lett., 18:
277-284.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R., (1984a) Subacute
stress induced by sumition on certain biochemical parameters in
Oziotelphusa senex senex, the freshwater rice field crab. Toxicol.
Lett., 21: 127-134.
BHAGYALAKSHMI, A., REDDY, P.S., & RAMAMURTHI, R. (1984b) The in
vivo inhibition and recovery of acetylcholinesterase in the
thoracic ganglionic mass of the freshwater rice field crab
( Oziotelphusa senex senex) during and after exposure to
sumition. Toxicol. Lett., 21: 135-139.
BHATIA, H.L. (1971) Toxicity of some pesticides to Puntius ticto
(Hamilton). Sci. Cult., 37: 160-161.
BOWMAN, M.C. & BCRAZA, M. (1969) Determination of accothion, its
oxygen analog and its cresol in corn, grass, and milk by gas
chromatography. J. agric. food Chem., 17: 271-276.
BRAID, P.E. & MCCARTHY, T.F. (1977) Human exposure to fenitrothion
during forest spraying in eastern Canada. In: Proceedings of a
Symposium on Fenitrothion, Ottawa, National Research Council of
Canada, p. 451 (Publication NRCC No. 16073). BRAID, P.E. & NIX, M.
(1968) Potentiation of toxicity of Sumithion by phosphamidon in the
rat. Can. J. Physiol. Pharmacol., 46: 133-137.
BRECKENRIDGE, C., PESANT, M., DURHAM, H.D., & ECOBICHON, D.J. (1982)
A 30-day toxicity study of inhaled fenitrothion in the albino rat.
Toxicol. appl. Pharmacol., 62: 32-43.
BREWER, D.G., WOOD, G., & UNGER, I. (1974) The photodecomposition of
fenitrothion ( O,O-dimethyl- O-(3-methyl-4-nitrophenyl)-
phosphorothioate). Chemosphere, 3: 91-95.
BROOKMAN, D.H., CHOPRA, C., ECOBICHON, D.J., KANG, C.Y., RITTER, L.,
& THORSEN, J. (1984) Assessment of the potential of insecticides,
emulsifiers and solvent mixtures to enhance viral infection in
cultured mammalian cells. Appl. environ. Microbiol., 47: 80-83.
BUCKNER, C.H. & MCLEOD, B.B. (1977) Ecological impact studies of
experimental and operational spruce budworm (Choristoneura
fumiferana Clemens) control programs on selected non-target
organisms in Quebec, 1976, Ottawa, Environment Canada, Forestry
Service, Department of Fisheries and the Environment (Information
Report No. CC-X-137).
BUCKNER, C.H. & SARRAZIN, R., ed. (1975) Studies of the
environmental impact of the 1974 spruce budworm control operation in
Quebec, Ottawa, Canada, Chemical Control Research Institute
(Information Report No. CC-X-93).
BUCKNER, C.H., SARRAZIN, R., & MCLEOD, B.B. (1977) The effects of
the fenitrothion spray program on small mammals. In: Proceedings of
a Symposium on Fenitrothion, Ottawa, National Research Council of
Canada, pp. 377-390 (Publication NRCC No. 16073).
BURGESS, D. (1988) Chronic toxicity of fenitrothion technical to
Daphnia magna under flow-through test conditions, Osaka, Japan,
Sumitomo Chemical Co., Ltd (Technical Report. No. HW-81-0326).
BURNS, R.G. & LETHBRIDGE, G. (1981) The effect of pesticides and
fertilisers on enzyme activities in soil. J. Sci. Food Agric., 32:
626-627.
BUSBY, D.G., PEARCE, P.A., & GARRITY, N.R. (1981) Brain
cholinesterase response in songbirds exposed to experimental
fenitrothion spraying in New Brunswick, Canada. Bull. environ.
Contam. Toxicol., 26: 401-406.
BUSBY, D.G., PEARCE, P.A., GARRITY, N.R., & REYNOLDS, L.M. (1983)
Effect of an organophosphorus insecticide on brain cholinesterase
activity in White-throated Sparrows exposed to aerial forest
spraying. J. appl. Ecol., 20: 255-263.
BUSBY, D.G., PEARCE, P.A., & GARRITY, N.R. (1987) Effect of ULV
ultra fenitrothion spraying on brain cholinesterase activity in
forest songbirds. Bull. environ. Contam. Toxicol., 39: 304-311.
CARSHALTON LABORATORY (1964) OMS 43. Summary of mammalian toxicity,
Carshalton, United Kingdom, Carshalton Laboratory, Toxicology
Research Unit (Unpublished report submitted to WHO by Farbenfabriken
Bayer AG, Germany).
CARTER, N.E. & BROWN, N.R. (1973) Seasonal abundance of certain soil
arthropods in fenitrothion-treated red spruce stand. Can. Entomol.,
105: 1065-1073.
CERNA, V. & BENES, V. (1972) [Fenitrothion residues on fruits and
vegetables.] Nahrung, 16: 179-186 (in German).
CHEVALIER, G., HENIN, J.P., VANNIER, H., CANEVET, C., COTE, M.G., &
LE BOUFFANT, L. (1984) Pulmonary toxicity of aerosolized
oil-formulated fenitrothion in rat. Toxicol. appl. Pharmacol., 76:
349-355.
CLINCH, P.G. & ROSS, G.M. (1970) Laboratory assessment of the speed
of action on honey bees of orally dosed insecticides. N.Z. J. agric.
Res., 13: 717-725.
COHLE, P. (1988) Early life stage toxicity of fenitrothion technical
to rainbow trout (Salmo gairdneri) in a flow-through system,
Osaka, Japan, Sumitomo Chemical Co., Ltd (Technical Report No.
HW-81-0331).
COOPER TECHNICAL BUREAU (1966) Toxicity of sumithion (Unpublished
report submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
COREY, L., RUBIN, R.J., HATTWICK, M.A.W., NOBLE, G.R., & CASSIDY, E.
(1976) A nationwide outbreak of Reye's Syndrome. Its
epidemiological relationship to Influenza B. Am. J. Med., 61: 615.
COULOMBE, P.A., LORTIE, S., & COTE, M.G. (1986) Pulmonary toxicity
of the insecticide fenitrothion in the rat following a single field
exposure. J. appl. Toxicol., 6: 317-323.
CRABBE, R., ELIAS, L., KRZYMIEN, M., & DAVIE, S. (1980a) New
Brunswick forestry spray operations: field study of the effect of
atmospheric stability on long-range pesticide drift, Ottawa,
National Research Council of Canada, National Aeronautical
Establishment Laboratory (Report No. LTR-UA-52).
CRABBE, R., KRZYMIEN, M., ELIAS, L., & DAVIE, S. (1980b) New
Brunswick spray operations: measurement of atmospheric fenitrothion
concentrations near the spray area, Ottawa, National Research
Council of Canada, National Aeronautical Establishment Laboratory
(Technical Report No. LTR-UA-56).
CROCKER, J.F.S., OZERE, R.L., ROZEE, K.R., DIGOUT, S.C., &
HUTZINGER, O. (1974) Insecticide and viral infection as a cause of
fatty visceral changes and encephalopathy in the mouse. Lancet, II:
22-24.
CROCKER, J.F.S., OZERE, R.L., BERGH, R.M., MCDONALD, R.G., &
ECOBICHON, D.J. (1976a) Depressed red cell cholinesterase associated
with Reye's syndrome, Vancouver, Canadian Pediatric Society (Meeting
abstract).
CROCKER, J.F.S., OZERE, R.L., SAFE, S.N., DIGOUT, S.C., ROZEE, K.R.,
& HUTZINGER, O. (1976b) Lethal interaction of ubiquitous insecticide
carers with virus. Science, 192: 1351-1353.
DEGRAEVE, N., CHOLLET, M.C., & MOUTSCHEN, J. (1984) Genetic and
cytogenetic effects of fenitrothion. Arch. environ. Health, 39:
25-26.
DESMARCHELIER, J.M. (1987) Kinetics of alkaline and
peroxide-catalyzed hydrolysis of fenitrothion. J. environ. Sci.
Health, B22: 403-411.
DRABEK, J. & TRUCHLIK, S. (1957) [Manufacture of O,O-dimethyl
O-(3-methyl-4-nitrophenyl) thiophosphate and its use in insect
control.] Czechoslovak Patent No. 89124 (in Slovak).
DUBOIS, K.P. & KINOSHITA, F. (1963) The acute toxicity of Bayer
41831 in combination with other anticholinesterase insecticides,
University of Chicago, Department of Pharmacology (Unpublished
report submitted to WHO by Farbenfabriken Bayer AG, Germany).
DUBOIS, K.P. & PUCHALA, E. (1960) The acute toxicity and
anticholinesterase action of Bayer 41831, University of Chicago,
Department of Pharmacology (Unpublished report submitted to WHO by
Farbenfabriken Bayer AG, Germany).
DURHAM, H.D. & ECOBICHON, D.J. (1986) An assessment of the
neurotoxic potential of fenitrothion in hen. Toxicology, 41:
319-332.
DURHAM, H.D., COMEAU, A.M., CAMERON, P.H., & ECOBICHON, D.J. (1982)
Subacute toxicity in rats of orally administered fenitrothion alone
and in a selected formulation. Toxicol. appl. Pharmacol., 62:
455-464.
DYKES, J. & CARPENTER, M. (1988) Photodegradation study of
14C-fenitrothion on soil surface, Osaka, Japan, Sumitomo Chemical
Co., Ltd (Technical Report No. HM-81-0098).
ECOBICHON, D.J. & ZELT, D. (1979) The acute toxicity of fenitrothion
in weanling rats and effects on tissue esterases and
mono-oxygenases. Toxicology, 13: 287-296.
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S. (1977)
Acute fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
ECOBICHON, D.J., COMEAU, A.M., & CAMERON, P.H. (1980) Chronic
toxicity of fenitrothion in male rats. Toxicol. appl. Pharmacol.,
56: 409-417.
EIDT, D.C. (1975) The effect of fenitrothion from large-scale forest
spraying on benthos in New Brunswick headwater streams. Can.
Entomol., 107: 743-760.
EIDT, D.C. (1978) Effects of insecticide contamination on stream
litter decomposition processes, Ottawa, Environment Canada, Forestry
Service, Department of Fisheries and the Environment, pp. 9-11
(Report No. M-X-87).
EIDT, D.C. (1981) Recovery of aquatic arthropod populations in a
woodland stream after depletion by fenitrothion treatment. Can.
Entomol., 113: 303-313.
EIDT, D.C. & SUNDARAM, K.M.S. (1975) The insecticide fenitrothion in
headwater streams from large-scale forest spraying. Can. Entomol.,
107: 735-742.
ELLIOTT-FEELEY, E. & ARMSTRONG, J.B. (1982) Effects of fenitrothion
and carbaryl on Xenopus laevis development. Toxicology, 22:
319-335.
ENVIRONMENT AGENCY OF JAPAN (1978) The studies on persistence of the
pesticides in water, Tokyo, Environment Agency of Japan, Water
Quality Bureau.
FAO (1982) Second Government Consultation on International
Harmonization of Pesticide Registration Requirements, Rome, 11-15
October 1982, Rome, Food and Agriculture Organization of the United
Nations.
FAO/WHO (1970) Fenitrothion. In: 1969 Evaluations of some pesticide
residues in food: The monographs, Rome, Food and Agriculture
Organization of the United Nations, World Health Organization, pp.
117-135 (FAO/PL:1969/M/17/1; WHO/FOOD/ADD./70.38).
FAO/WHO (1975a) Fenitrothion. In: Pesticide residues in food. Report
of the 1974 Joint Meeting of the FAO Working Party of Experts on
Pesticide Residues and the WHO Expert Committee on Pesticide
Residues, Rome, 2-11 December 1974, Rome, Food and Agriculture
Organization of the United Nations, p. 17 (FAO Agricultural Studies
No. 97; WHO Technical Report Series No. 574).
FAO/WHO (1975b) Fenitrothion. In: 1974 Evaluations of some pesticide
residues in food: The monographs, Geneva, World Health Organization,
pp. 335-381 (WHO Pesticide Residues Series, No. 4).
FAO/WHO (1977) Fenitrothion. In: 1976 Evaluation of some pesticide
residues in food: The monographs, Rome, Food and Agriculture
Organization of the United Nations, pp. 361-376 (AGP:1976/M/14).
FAO/WHO (1978a) Fenitrothion. In: Pesticide residues in food -
Report 1977, Rome, Food and Agriculture Organization of the United
Nations, p. 35 (FAO Plant Production and Protection Paper 10 Rev.).
FAO/WHO (1978b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1977: The monographs, Rome, Food and Agriculture
Organization of the Untied Nations, pp. 263-272 (FAO Plant
Production and Protection Paper 10 Sup.).
FAO/WHO (1980a) Fenitrothion. In: Pesticide residues in food -
Report 1979, Rome, Food and Agriculture Organization of the United
Nations, pp. 45-46 (FAO Plant Production and Protection Paper 20).
FAO/WHO (1980b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1979: The monographs, Rome, Food and Agriculture
Organization of the United Nations, pp. 289-295 (FAO Plant
Production and Protection Paper 20 Sup.).
FAO/WHO (1983a) Fenitrothion. In: Pesticide residues in food -
Report 1982, Rome, Food and Agriculture Organization of the United
Nations, p. 27 (FAO Plant Production and Protection Paper 46).
FAO/WHO (1983b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1982: The monographs, Rome, Food and Agriculture
Organization of the United Nations, pp. 237-238 (FAO Plant
Production and Protection Paper 49).
FAO/WHO (1984a) Fenitrothion. In: Pesticide residues in food -
Report 1982, Rome, Food and Agriculture Organization of United
Nations, pp. 29-30 (FAO Plant Production and Protection Paper 56).
FAO/WHO (1984b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1983: The monographs, Rome, Food and Agriculture
Organization of United Nations, pp. 219-223 (FAO Plant Production
and Protection Paper 61).
FAO/WHO (1985a) Fenitrothion. In: Pesticide residues in food -
Report 1984, Rome, Food and Agriculture Organization of United
Nations, pp. 45-46 (FAO Plant Production and Protection Paper 62).
FAO/WHO (1985b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1984: The monographs, Rome, Food and Agriculture
Organization of United Nations, pp. 339-341, 645-648 (FAO Plant
Production and Protection Paper 67).
FAO/WHO (1986a) Fenitrothion. In: Pesticide residues in food -
Report 1986, Rome, Food and Agriculture Organization of United
Nations, p. 27, 61 (FAO Plant Production and Protection Paper 77).
FAO/WHO (1986b) Fenitrothion. In: Pesticide residues in food -
Evaluations 1986: Part I. Residues, Rome, Food and Agriculture
Organization of United Nations, pp. 183-186 (FAO Plant Production
and Protection Paper 78).
FAO/WHO (1986c) Codex maximum limits for pesticide residues, 2nd
ed., Rome, Codex Alimentarius Commission, Food and Agriculture
Organization of the United Nations (CAC/Vol.XIII-Ed. 2).
FAO/WHO (1987a) Fenitrothion. In: Pesticide residues in food -
Evaluations 1986: Part II. Toxicology, Rome, Food and Agriculture
Organizations of United Nations, pp. 49-50 (FAO Plant Production and
Protection Paper 78/2).
FAO/WHO (1987b) Fenitrothion. In: Pesticide residues in food -
Report 1987, Rome, Food and Agriculture Organization of United
Nations, p. 26, 67 (FAO Plant Production and Protection Paper 84).
FAO/WHO (1988a) Fenitrothion. In: Pesticide residues in food.
Evaluations 1987: Part I. Residues, Rome, Food and Agriculture
Organization of United Nations, pp. 55-56 (FAO Plant Production and
Protection Paper, 86/1).
FAO/WHO (1988b) Fenitrothion. In: Pesticide residues in food -
Report 1988, Rome, Food and Agriculture Organization of United
Nations, p. 24 (FAO Plant Production and Protection Paper 92).
FAO/WHO (1989a) Fenitrothion. In: Pesticide residues in food -
Evaluations 1989: Part II. Toxicology, Rome, Food and Agriculture
Organization of the United Nations, pp. 31-34 (FAO Plant Production
and Protection Paper 93/2).
FAO/WHO (1989b) Fenitrothion. In: Pesticide residues in food -
Report 1989, Rome, Food and Agriculture Organization of the United
Nations, p. 28 (FAO Plant Production and Protection Paper 99).
FAO/WHO (1989c) Fenitrothion. In: Pesticide residues in food -
Evaluations 1989: Part I. Residues, Rome, Food and Agriculture
Organization of the United Nations, pp. 123-125 (FAO Plant
Production and Protection Paper 100).
FAO/WHO (1989d) Guide to Codex recommendations concerning pesticide
residues. Part 8. Recommendations for methods of analysis of
pesticide residues, 3rd ed., Rome, Codex Committee on Pesticide
Residues, Food and Agriculture Organization of the United Nations.
FAO/WHO (1990) In: Codex maximum limits for pesticide residues,
CAC/Supplement 2-1989 to FAO/WHO 1986c, Rome, Codex Alimentarius
Commission, Food and Agriculture Organization of the United Nations,
p. 1.
FERREIRA, J.R. & FERNANDES, A.M.S.S. (1980) Gas-liquid
chromatographic determination of organophosphorus insecticide
residues in fruits and vegetables. J. Assoc. Off. Anal. Chem., 63:
517-522.
FLANNAGAN, J.F. (1973) Field and laboratory studies of the effect of
exposure to fenitrothion on freshwater aquatic invertebrates.
Manitoba Entomol., 7: 15-25.
FORBIS, A.D. (1987) Acute toxicity of fenitrothion to Daphnia
magna, Osaka, Japan, Sumitomo Chemical Co., Ltd (Technical Report
No. HW-70-0236).
FUNCH, F.H. (1981) Analysis of residues of seven pesticides in some
fruits and vegetables by means of high pressure liquid
chromatography. Z. Lebensm. Unters. Forsch., 173: 95-98.
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol. appl.
Pharmacol., 14: 515-534.
GAJDUSKOVA, V. (1974) Dynamics of organophosphorus pesticide
residues in butter during manufacture and storage.
Milchwissenschaft, 29: 278-280.
GARDNER, A.M. (1971) Confirmation of organophosphorous pesticide
residues at nanogram levels by two dimensional thin layer
chromatography. J. Assoc. Off. Anal. Chem., 54: 517-524.
GARTRELL, M.J., CRAUN, J.C., PODREBARAC, D.S., & GUNDERSON, E.L.
(1985a) Pesticides selected elements, and other chemicals in adult
total diet samples, October 1979-September 1980. J. Assoc. Off.
Anal. Chem., 68: 1184-1195.
GARTRELL, M.J., CRAUN, J.C., PODREBARAC, D.S., & GUNDERSON, E.L.
(1985b) Pesticides, selected elements, and other chemicals in
infant and toddler total diet samples, October 1979-September 1980.
J. Assoc. Off. Anal. Chem., 68: 1163-1183.
GERMAIN, P. (1977) Songbird census in fenitrothion regime and 1977
forest spray program in New Brunswick. A final report to Forest
Protection Ltd and New Brunswick Department of Natural Resources,
Moncton, Canada, University of Moncton (Unpublished report).
GERMAIN, P. & MORIN, G. (1979) Forest songbird population, 1978
monitoring program in relation to aerial spray of insecticides
against spruce budworm. A final report to Forest Protection Ltd and
New Brunswick Department of Natural Resources, Moncton, Canada,
Avifauna Ltd.
GILLIS, G.F. (1978) Fish-growth rates. Benthic drift. Ottawa,
Environment Canada, Forestry Service, Department of Fisheries and
the Environment, 8 pp (Report M-X-87).
GILLIS, G.F. (1980) Assessment of the effects of insecticide
contamination in streams on the behaviour and growth of fish, 1979.
In: Environmental surveillance in New Brunswick 1978-1979, Moncton,
University of New Brunswick, pp. 49-50.
GINSBERG, G.L., PLACKE, M.E., WYAND, D.S., & COHEN, S.D. (1982)
Protection against acetaminophen-induced hepatotoxicity by prior
treatment with fenitrothion. Toxicol. appl. Pharmacol., 66: 383-399.
GOWDA, H. & SASTRY, M.S. (1979) Effect of O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphorothioate (Sumithion) on
reproductive performance in rats and oestrogenic activity in mice.
Indian J. Pharmacol., 11(4): 287.
GRAS, G. (1966) Toxicité du fénitrothion. Bull. Soc. Pharm.
Montpellier, 26: 375-404.
GREENHALGH, R. & MARSHALL, W.D. (1976) Ultraviolet irradiation of
fenitrothion and the synthesis of the photolytic oxidation products.
J. agric. food Chem., 24: 708-713.
GREENHALGH, R., DHAWAN, K.L., & WEINBERGER, P. (1980) Hydrolysis of
fenitrothion in model and natural aquatic systems. J. agric. food
Chem., 28: 102-105.
GRIGGS, L.M.P., JEFFERSON, N.D., BLAIR, N., KOPPLIN, J.R., RICHTER,
W.S., & SPICER, E.J.F. (1984) One-year dietary toxicity study in
dogs, Mattawan, Michigan, International Research and Development
Corporation (Unpublished report submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
GROVER, I.S. & MALHI, P.K. (1985) Genotoxic effects of some
organophosphorous pesticides I. Induction of micronuclei in bone
marrow cells in rat. Mutat. Res., 155: 131-134.
GRUE, C.E. (1982) Response of common grackles to dietary
concentrations of four organophosphate pesticides. Arch. environ.
Contam. Toxicol., 11: 617-626.
GUN, R.T., GRYCORCEWICZ, C., ESTERMAN, A.J., & EDWARDS, J.B. (1988)
Ultralow volume application of organophosphate concentrate in grain
terminals: A new occupational health hazard. Br. J. ind. Med., 45:
834-837.
HACHE, P., MARQUETTE, R., VOLPE, G., & MALLET, V.N. (1981) Fast
cleanup of difficult substrates for determination of fenitrothion
and some derivatives. J. Assoc. Off. Anal. Chem., 64: 1470-1473.
HALLETT, D.J., WEINBERG, P., & PRASAD, R. (1973) A preliminary study
on the fate of fenitrothion in forest seeds. I. Determination of
residues in eastern white pine (Pinus strobus L. Fr) and their
effects on amino acid metabolism, Ottawa, Environment Canada,
Forestry Service, Chemical Control Research Institute (Information
Report CC-X-50).
HALLETT, D.J., GREEENHALGH, R., WEINBERG, P., & PRASAD, R. (1974)
Fate of fenitrothion in forest trees. V. The formation of
metabolites in Pinus strobus L. and their detection by gas
chromatography and mass spectroscopy, Ottawa, Environment Canada,
Forestry Service, Chemical Control Research Institute (Information
Report CC-X-78).
HALLETT, D.J., GREENHALGH, R., WEINBERGER, P., & PRASAD, R. (1977)
The uptake and metabolism of fenitrothion by germinating white pine
spruce and yellow birch seeds. J. environ. Sci. Health, B12(1):
53-69.
HAMILTON, G.A., HUNTER, K., & RUTHVEN, A.D. (1981) Inhibition of
brain acetylcholinesterase activity in songbirds exposed to
fenitrothion during aerial spraying of forests. Bull. environ.
Contam. Toxicol., 27: 856-863.
HARA, S. & SUZUKI, T. (1981) Primary eye and skin irritation tests
of Sumithion Technical in rabbits (Unpublished Technical Report No.
HT-10-0201, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
HARA, M. & SUZUKI, H. (1982a) In vivo chromosomal aberration test
of Sumithion on bone marrow cells of mice (Unpublished Technical
Report No. HT-20-0235, submitted to WHO by Sumitomo Chemical Co.,
Ltd, Osaka, Japan).
HARA, M. & SUZUKI, H. (1982b) In vivo chromosomal aberration test
of sumithion on bone marrow cells of rats (Unpublished Technical
Report No. HT-30-0258, submitted to WHO by Sumitomo Chemical Co.,
Ltd, Osaka, Japan).
HARA, M. & SUZUKI, H. (1982c) The micronucleus test of Sumithion
(Unpublished Technical Report No. HT-20-0234, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
HARA, M., YAMAMOTO, K., KOGISO, S., & YOSHITAKE, A. (1988) In vivo
chromosomal aberration test of Sumithion in Chinese hamster ovary
cells (CHO-K1) in culture (Unpublished Technical Report No.
HT-80-0420, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
HARA, M., KOGISO, S., YAMADA, F., KAWAMOTO, M., YOSHITAKA, A., &
MIYAMOTO, J. (1989) Mutagenicity studies on fenitrothion in bacteria
and mammalian cells. Mutat. Res., 222: 53-61.
HASEGAWA, J., YASUNO, M., SAITO, K., NAKAMURA, Y., HATAKEYAMA, Y., &
SATO, H. (1982) Impact of temephos and fenitrothion on aquatic
invertebrates in a stream of Mt. Tsukuba. Jpn. J. Sanit. Zool., 33:
363-368.
HASHIMOTO, Y. & FUKAMI, J. (1969) Toxicity of orally and topically
applied pesticide ingredients to carp, Cyprimus carpio Linne.
Botyu-Kagaku, 34: 63-66.
HASHIMOTO, Y. & NISHIUCHI, Y. (1981) Establishment of bioassay
methods for evaluation of acute toxicity of pesticides to aquatic
organisms. J. pestic. Sci., 6: 257-264.
HASHIMOTO, Y., OKUBO, E., ITO, T., YAMAGUCHI, M., & TANAKA, S.
(1982) Changes in susceptibility of carp to several pesticides with
growth. J. pestic Sci., 7: 457-461.
HATAKEYAMA, S., SHIRAISHI, H., & KOBAYASHI, N. (1990) Effects of
aerial spraying of insecticides on nontarget macrobenthos in a
mountain stream. Ecotoxicol. environ. Saf., 19: 254-270.
HATFIELD, C.T. & ANDERSON, J.M. (1972) Effects of two insecticides
on the vulnerability of Atlantic salmon (Salmo salar) parr to
brook trout (Salvelinus fontinalis) predation. J. Fish Res. Board
Can., 29: 27-29.
HATFIELD, C.T. & RICHE, L.G. (1970) Effects of aerial Sumithion
spraying on juvenile Atlantic salmon (Salmo salar L.) and brook
trout (Salvelinus fontinalis Mitchill) in Newfoundland. Bull.
environ. Contam. Toxicol., 5: 440-442.
HATTORI, K. (1974) [Toxicological studies on the influence of
chemicals to the birds (part 2). Oral acute toxicity of three
organophosphate insecticides in common pigeon.] Hokkaidoritsu
Eiseikenkyuushoho, 24: 154 (in Japanese).
HATTORI, K., SATO, H., TSUCHIYA, K., YAMAMOTO, N., & OGAWA, E.
(1974) [Toxicological studies on influences of chemicals to the
birds (Part 1). Oral acute toxicity and cholinesterase inhibition of
three organophosphate insecticides in Japanese quail.] Hokkaidoritsu
Eiseikenkyuushoho, 24: 35-38 (in Japanese).
HAYASHI, M., KISHI, M., SOFUNI, T., & ISHIDATE, M. (1988)
Micronucleus test in mice on 39 food additives and eight miscellaneous
chemicals. Food chem. Toxicol., 26: 487-500.
HAYES, W.J., Jr (1982) Fenitrothion. In: Pesticides studies in man,
Baltimore, Maryland, Williams & Wilkins Company, pp. 367-372.
HAYES, W.J., Jr & LAWS, E.R., Jr (1991) Fenitrothion. In: Handbook
of pesticide toxicology, New York, London, Academic Press, pp.
1020-1023.
HILL, E.F., HEALTH, R.G., SPANN, J.W., & WILLIAMS, J.D. (1975)
Lethal dietary toxicities of environmental pollutants to birds,
Washington, DC, US Department of the Interior, Fish and Wildlife
Service, 61 pp (Special Scientific Report, Wildlife No. 191).
HIRAYAMA, K. & TAMANOI, S. (1980) Acute toxicity of MEP and diazinon
(pesticide) to larvae of kuruma prawn Penaeus japonicus and of
swimming crab Portunus trituberculatus. Bull. Jpn. Soc. Sci.
Fish., 46: 117-123.
HIROSE, K., YAMAZAKI, M., & ISHIKAWA, A. (1979) Effects of water
temperature on median lethal concentration (LC50) of a few
pesticides to seawater teleost. Bull. Tokai Reg. Fish. Res. Lab.,
98: 45-53.
HLADKA, A., KRAMPL, V., & KOVAC, J. (1974) Effect of malathion on
the content of fenitrothion and fenitrooxon in the rat. Bull.
environ. Contam. Toxicol., 12: 38-45.
HLADKA, A., BATORA, V., KOVACICOVA, J., & ROSIVAL, L. (1977) Effets
sur la santé professionnelle et importance toxicologique du
fénitrothion et de son contaminant (S-méthyl-fénitrothion) dans la
toxicologie de ses formulations. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, p. 416
(Publication NRCC No. 16073).
HOBERMAN, A.M. (1990) Reproductive effects of Sumithion administered
orally in feed to Crl: CDR(SD)BR rats for two generations, Argus
Research Laboratories, Inc. (Unpublished Technical Report No.
HT-10-0452 submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
HOLLINGWORTH, R.M. (1969) Dealkylation of organophosphorus esters by
mouse liver enzymes in vitro and in vivo. J. agric. food Chem., 17:
987-996.
HOLLINGWORTH, R.M., METCALF, R.M., & FUKUTO, T.R. (1967a) The
selectivity of Sumithion compared with methylparathion. The
metabolism in the white mouse. J. agric. food Chem., 15: 242-249.
HOLLINGWORTH, R.M., FUKUTO, T.R., & METCALF, R.L. (1967b)
Selectivity of Sumithion compared with methylparathion. Influence of
structure on anticholinesterase activity. J. agric. food Chem., 15:
235-241.
HOLMES, S.B. (1979) Aquatic impact studies of a spruce budworm
Choristoneura fumiferana clemens, control program in the lower St.
Lawrence region of Quebec in 1978, Ottawa, Forest Pest Management
Institute (Report No. FPM-X-26).
HOLMES, S.B., KINGSBURY, P.D., MAMARBACHI, G., & MATHIEU, P. (1984)
Distribution of fenitrothion residues in brook trout (Salvelinus
fontinalis) and lake trout (Salvelinus namaycush) tissues
following aerial applications to Lac Ste-Anne, Quebec. Bull.
environ. Contam. Toxicol., 33: 468-475.
HOSOKAWA, S. & MIYAMOTO, J. (1974) Metabolism of C-14 labelled
sumithion, O,O-dimethyl O-(3-methyl-4-nitrophenyl)-
phosphorothioate in apple. Botyu-Kagaku, 39: 49-53.
HOSOKAWA, S. & MIYAMOTO, J. (1975) Effects of subacute feeding of
Sumithion, Sumioxon and p-nitrocresol on rat hepatic oxidative
phosphorylation and mixed function oxidase. Botyu-Kagaku, 40: 33-38.
HUDSON, R.H., HAEGELE, M.A., & TUCKER, R.K. (1979) Acute oral and
percutaneous toxicity of pesticides to mallards: correlation with
mammalian toxicity data. Toxicol. appl. Pharmacol., 47: 451-460.
ITO, M., TAKAHASHI, M., & MIKAMI, N. (1988) Hydrolysis of
fenitrothion in water as a function of pH at 25 °C, Osaka, Japan,
Sumitomo Chemical Co., Ltd (Unpublished Technical Report No.
HM-80-0094).
IWATA, Y., SUGITANI, A., & YAMADA, F. (1981) An analytical method
for organophosphorus pesticide in onion. Shokuhin Eisei Zasshi, 22:
484-489.
JAPAN PLANT PROTECTION ASSOCIATION (1984) [Production statistics of
major pesticides.] In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 49 (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1986) [Production statistics of
major pesticides]. In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 55 (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1988) [Production statistics of
major pesticides.] In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 57 (in Japanese).
JAPAN PLANT PROTECTION ASSOCIATION (1989) [Production statistics of
major pesticides.] In: [Pesticide handbook], Tokyo, Japan Plant
Protection Association, p. 60 (in Japanese).
JOHNSON, J.C., Jr & BOWMAN, M.C. (1972) Responses from cows fed
diets containing fenthion or fenitrothion. J. dairy Sci., 55:
777-782.
JOHNSTON, J.J. & CORBETT, M.D. (1985) The effects of temperature,
salinity and simulated tidal cycle on the toxicity of fenitrothion
to Callinectes sapidus. Comp. Biochem. Physiol., 80C: 145-149.
JOHNSTON, J.J. & CORBETT, M.D. (1986) The uptake and in vivo
metabolism of the organophosphate insecticide fenitrothion by the
blue crab, Callinectes sapidus. Toxicol. appl. Pharmacol., 85:
181-188.
KADOTA, T. & MIYAMOTO, J. (1975) Acute and sub-acute toxicity of
sumithion in Japanese quails. Botyu-Kagaku, 40: 54-58.
KADOTA, T., KAGOSHIMA, M., YAMAZAKI, H., & MIYAMOTO, J. (1972) Acute
oral, subcutaneous and dermal toxicities of Sumithion technical in
mice and rats (Unpublished Technical Report No. HT-20-0187,
submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
KADOTA, T., KOHDA, H., & MIYAMOTO, J. (1975a) Subchronic toxicity
studies of Sumithion, Sumioxon and p-nitrocresol in rats and
92-week feeding study of Sumithion with special reference to change
of cholinesterase activity. Botyu-Kagaku, 40: 38-48.
KADOTA, T., OKUNO, Y., & MIYAMOTO, J. (1975b) Acute oral toxicity
and delayed neurotoxicity of 5 organophosphorus compounds,
Salithion, Cyanox, Surecide, Sumithion an Sumioxon in adult hens.
Botyu-Kagaku, 40: 49-53.
KALAS, D. (1978) [Light group poisoning by metathion E-50 inhalation
when spraying by means of aircraft.] Prac. Lek., 30: 145-147 (in
Czech).
KANAZAWA, J. (1975) Uptake and excretion of organophosphorus and
carbamate insecticides by fresh water fish, Motsugo. Bull. environ.
Contam. Toxicol., 14: 346-352.
KANAZAWA, J. (1977) [Analytical chemistry related to evaluation of
the effects of pesticides on aquatic organisms.] In: [Proceeding of
a Symposium on the Effects of Pesticides on Aquatic Organisms and
the Methods for their Evaluation, 29-30 November, 1977], Scientist
Co., pp. 4/1-4/34 (in Japanese).
KANAZAWA, J. (1981) Measurement of the bioaccumulation factors of
pesticides by freshwater fish and their correlation with
physicochemical properties or acute toxicities. Pestic. Sci., 12:
417-424.
KANNAN, N. & JAYARNAMA, J. (1981) Impact monitoring of residues in
tomato. Int. J. environ. anal. Chem., 9: 145-151.
KANOH, S., KAWASAKI, H., YOSHIDA, M., & NISHIO, A. (1982) Studies on
chronic toxicity of low levels of O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphorothionate (Sumithion) in the rat.
J. toxicol. Sci., 7: 43-50.
KASHI, K.P. (1972) Tolerance of silkworm (Bombyx mori L.) larvae
to organophosphorous insecticides. J. Seric. Sci. Jpn, 41: 301-304.
KATAGI, T., TAKAHASHI, N., & MIKAMI, N. (1988) Photodegradation of
fenitrothion in water (Unpublished Technical Report No. HM-80-0093,
submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
KAWACHI, T. (1978) [Development of screening method for carcinogen
including mutagenicity tests.] In: [Annual Report of Cancer
Research], Tokyo, Ministry of Health and Welfare, pp. 803-820 (in
Japanese).
KAWAMOTO, M., HARA, M., KOGISO, S., & YOSHITAKE, A. (1989) In
vivo/in vitro unscheduled DNA synthesis (UDS) test of Sumithion
in rat hepatocytes (Unpublished Study No. 1487, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
KETTELA, E.G. & VARTY, I.W. (1972) Assessment of the 1971 spruce
budworm aerial spraying program in New Brunswick and forecast for
1972, Fredericton, New Brunswick, Canada, Maritimes Forest Research
Centre (Information Report M-X-29).
KEVAN, P.G. (1975) Forest application of the insecticide
fenitrothion and its effect on wild bee pollinators ( Hymenoptera:
Apoidea) of lowbush blueberries (Vaccinium SPP) in southern New
Brunswick, Canada. Biol. Conserv., 7: 301-309.
KIKUCHI, R., YASUTANIYA, T., TAKIMOTO, Y., YAMADA, H., & MIYAMOTO,
J. (1984) Accumulation and metabolism of fenitrothion in three
species of algae. J. pestic. Sci., 9: 331-337.
KIMMERLE, G. (1962a) [Toxicological study on additive S-5660],
Toxicology and Occupational Health Laboratory (Unpublished report
submitted to WHO by Bayer AG, Germany) (in German).
KIMMERLE, G. (1962b) [Neurotoxic effect of S-5660 on hens],
Toxicology and Occupational Health Laboratory (Unpublished report
submitted to WHO by Bayer AG, Germany) (in German).
KINGSBURY, P.D. (1976) A history of the effects of aerial forest
spraying in Canada on aquatic fauna, Ottawa, Chemical Contamination
Research Institute (Information Report No. CC-X-117).
KINGSBURY, P.D. (1978) A study of the distribution, persistence and
biological effects of fenitrothion applied to a small lake in and
oil formulation, Ottawa, Forest Pest Management Institute (Report
No. FPM-X-13).
KLAVERKAMP, J.F., DUANGSAWASDI, M., MACDONALD, W.A., & MAJEWSKI,
H.S. (1977) An evaluation of fenitrothion toxicity in four life
stages of rainbow trout, Salmo gairdneri, Philadelphia, American
Society for Testing and Materials, pp. 231-240 (Special Technical
Publication No. 77).
KLEINER, C.F., ANDERSON, R.L., & TANNER, D.K. (1984) Toxicity of
fenitrothion to fathead minnows (Pimephales promelas) and
alternative exposure duration studies with fenitrothion and
endosulfan. Arch. environ. Contam. Toxicol., 13: 573-578.
KLIMMER, O.R. (1961) Opinion on the toxicity of the compound ``S
5660'' of Farbenfabriken Bayer AC., Leverkusen, Bonn, Germany,
Pharmakologisches Institut der Rheinischen
Friedrich-Wilhelms-Universitat (Unpublished report submitted to WHO
by Farbenfabriken Bayer AG, Germany).
KOBAYASHI, H., KOJIMA, M., MATANO, O., & GOTO, S. (1979) Simple
multi-residue analysis of organophosphorus pesticides by mass
chromatography. J. pestic. Sci., 4: 463-472.
KOBAYASHI, H., YUYAMA, A., KUDO, M., & MATSUSAKA, N. (1983) Effects
of organophosphorus compounds, O,O-dimethyl
O-(2,2-dichlorovinyl)phosphate (DDVP) and O,O-dimethyl
O-(3-methyl-4-nitrophenyl)phosphorothioate (fenitrothion), on
brain acetylcholine content and acetylcholinesterase activity in
Japanese quail. Toxicology, 28: 219-227.
KODAMA, T. & KUWATSUKA, S. (1980) Factors for the persistence of
parathion, methyl parathion, and fenitrothion in sea water. J.
pestic. Sci., 5: 351-355.
KOHDA, H. & KADOTA, T. (1975) Dominant lethal test of Sumithion in
rats and mice, Osaka, Japan, Sumitomo Chemical Co., Ltd (Technical
Report No. HT-60-0210).
KOHDA, H. & KADOTA, T. (1979) Acute inhalation toxicity study of
Sumithion in rats (Unpublished Technical Report No. HT-90-0085A,
submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
KOHDA, H., KAGOSHIMA, M., KADOTA, T., & MIYAMOTO, J. (1972) Skin
sensitization test of Sumithion Technical in guinea-pigs
(Unpublished Technical Report No. HT-00-0181, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
KOVAC, J. & SOHLER, E. (1965) [Determination of Fenitrothion
residues in fruits and vegetables after separation of coloured
co-extractives by thin-layer chromatography.] Z. anal. Chem., 208:
201-204 (in German).
KOVACICOVA, J., BATORA, V., & TRUCHLIK, S. (1973) Hydrolysis rate
and in vitro anticholinesterase activity of fenitrothion and
S-methyl Fenitrothion. Pestic. Sci., 4: 759-763.
KRZYMIEN, M. (1979) Determination of fenitrothion concentration in
ambient air, Ottawa, National Research Council of Canada, National
Aeronautical Establishment, 18 pp (Report LTR-UA-49).
KURIBAYASHI, S. (1981) Studies on the effect of pesticides on the
reproduction of the silkworm, Bombyx mori L. I. Effects of
chemicals administered during the larval stage on egg-laying and
hatching. J. toxicol. Sci., 16: 169-176.
KUROIWA, Y. & YAMAMOTO, T. (1977) [Toxicological study of
fenitrothion to rat. Comparative study with methylparathion.] In:
[Abstract papers of 9th Symposium on Drug Metabolism and Action],
Kumamoto, Pharmaceutical Society of Japan, pp. 137-140 (in
Japanese).
KWASNIEWSKA, K., LIU, D., & STRACHAN, W.M.J. (1980) The relative
biological toxicity effectiveness of chemical toward microorganisms.
Trace Subst. environ. Health, 14: 470-477.
LADD, R., JENKINS, D.H., WRIGHT, P.L., & KEPLINGER, M.L. (1971)
Teratogenic study with Sumithion technique in albino rabbits,
Northbrook, Illinois, Industrial Bio-Test Laboratories (Unpublished
report No. J9996, submitted to WHO by Sumitomo Chemical Co., Ltd,
Osaka, Japan).
LAKSHMINARAYAMA, J.S.S. & BOUQUE, H. (1980) Absorption of
fenitrothion by plankton and benthic algae. Bull. environ. Contam.
Toxicol., 24: 389-396.
LAL, S., LAL, R., & SAXENA, D.M. (1987) Bioconcentration and
metabolism of DDT, fenitrothion and chlorpyrifos by the blue-green
algae Anabaena sp. and Aulosira fertilissima. Environ. Pollut.,
46: 187-196.
LAPIERRE, L.E. (1985) Persistence of fenitrothion insecticide in
poplar Populus tremuloidus and gray birch Betula populifolia.
Bull. environ. Contam. Toxicol., 35: 471-475.
LEBEL, G.L., WILLIAMS, D.T., GRIFFITH, G., & BENOIT, F.M. (1979)
Isolation and concentration of organophosphorus pesticides from
drinking water at the ng/litre level, using macro-reticular resin.
J. Assoc. Off. Anal. Chem., 62: 241-249.
LEHOTZKY, K., SZEBERENYI, M.J., & KISS, A. (1989) Behavioral
consequences of prenatal exposure to the organophosphate insecticide
sumithion. Neurotoxicol. Teratol., 11: 321-324.
LEONARD, D.E. (1971) Effects of accothion on parasites and
predators. In: Nash, R.W., Peterson, J.W., & Chansler, J.F., ed.
Environmental studies of accothion for spruce budworm control,
Washington, DC, US Department of Agriculture and US Department of
Interior, p. 46.
LEUCK, D.B. & BOWMAN, M.C. (1969) Persistence of O,O-dimethyl
O-4-nitro- m-tolyl phosphorothioate, its oxygen analogue, and its
cresol in corn and grass forage. J. econ. Entomol., 62: 1282-1285.
LEUCK, D.B., JOHNSON, J.C., Jr, BOWMAN, M.C., KNOX, F.E., & BEROZA,
M. (1971) Fenitrothion residues in corn silage and their effects on
dairy cows. J. econ. Entomol., 64: 1394-1399.
LISKA, D., KOLESAR, D., HLADKA, A., VALKYOVA, L., & RAISKUP, J. CH.
(1982) Clinical and laboratory findings in Metation EK-50. Czech.
Med., 5: 146-154.
LIU, D., STRACHAN, W.M., THOMSON, K., & KWASNIEWSKA, K. (1981)
Determination of the biodegradability of organic compounds. Environ.
Sci. Technol., 15: 788-793.
LOCKHART, W.L., FLANNAGAN, J.F., MOODY, R.P., WEINBERGER, P., &
GREENHALGH, R. (1977) Fenitrothion monitoring in southern Manitoba.
In: Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 233-252 (Publication No. NRCC No.
16073).
LOCKHART, W.L., METNER, D.A., & GRIFT, T. (1973) Biochemical and
residue studies on rainbow trout (Salmo gairdneri) following field
and laboratory exposure to fenitrothion. Manitoba Entomol., 7:
26-36.
MACDONALD, J.R. & PENNEY, G.H. (1969) Preliminary report on the
effects of the 1969 New Brunswick forest spraying on juvenile salmon
and their food organisms, Halifax, Department of Fisheries and
Forestry of Canada, Resource Development Branch, 17 pp (Manuscript
report No. 69-4).
MCLEESE, D.W. (1974) Olfactory response and fenitrothion toxicity in
American lobsters (Homarus americanus). J. Fish. Res. Board Can.,
31: 1127-1131.
MCLEESE, D.W., ZITKO, V., & SERGEANT, D.B. (1979) Uptake and
excretion of fenitrothion by clams and mussels. Bull. environ.
Contam. Toxicol., 22: 800-806.
MCNEIL, J.N. & MCLEOD, J.M. (1977) Apparent impact of fenitrothion
on the swaine Jack-Pine. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, p. 203
(Publication NRCC No. 16073).
MCNEIL, J.N., GREENHALGH, R., & MCLEOD, J.M. (1979) Persistence and
accumulation of fenitrothion residues in jack pine foliage and their
effect on the swaine jack pine sawfly, Neodiprion Swainei
(Hymenoptera: Diprionidae). Entomol. Soc. Am., 8: 752-755.
MADHU, C.H., RAO, K.J., RAO, K.S., & RAO, K.V.R. (1982) Comparative
evaluation of the toxicity of lindane and sumithion on freshwater
snail, Pila globosa (Swainson). Geobios, 9: 181-183.
MAEDA, T., KAWASHIMA, M., OGASAWARA, Y., & TSUJI, K. (1982) Thermal
decomposition of O,O-dimethyl
O-(3-methyl-4-nitrophenol)-phosphorothioate-Autocatalytic
decomposition mechanism. J. pestic. Sci., 7: 341-347.
MAFF (MINISTRY OF AGRICULTURE, FISHERIES AND FOOD, UK) (1986) Food
surveillance paper No. 16, London, Her Majesty's Stationery Office
(HMSO), pp. 19-20.
MAFF (MINISTRY OF AGRICULTURE, FISHERIES AND FOOD, UK) (1989) Food
surveillance paper No. 25, London, Her Majesty's Stationery Office
(HMSO), pp. 12-34.
MAGUIRE, R.J. & HALE, E.J. (1980) Fenitrothion sprayed on a pond:
Kinetics of its distribution and transformation in water and
sediment. J. agric. food Chem., 28: 372-378.
MALHI, P.K. & GROVER, I.S. (1987) Genotoxic effects of some
organophosphorus pesticides II. In vivo chromosomal aberration
bioassay in bone marrow cells in rat. Mutat. Res., 188: 45-51.
MALLET, V.N. (1980) A chemical residue survey in relation to the
1980 spruce budworm spray program in New Brunswick, Moncton, Canada,
University of Moncton, Chemistry Department, 54 pp (Report No.
80-CP-18).
MALLET, V.N. & CASSISTA, A. (1984) Fenitrothion residue survey in
relation to the 1981 spruce budworm spray program in New Brunswick,
Canada. Bull. environ. Contam. Toxicol., 32: 65-74.
MALLET, V.N. & VOLPE, G. (1982) A chemical residue survey in
relation to the 1980 spruce budworm spray program in New Brunswick
(Canada). J. environ. Sci. Health, B17: 715-736.
MALLET, V.N., BRUN, G.L., MACDONALD, R.N., & BERKANE, K. (1978) A
comparative study of the use of XAD-2 resin and the conventional
serial solvent extraction procedure for the analysis of fenitrothion
and some derivatives in water preservation techniques. J.
Chromatogr., 160: 81-88.
MANDOUL, R., DUBOS, M., DECOMMAND, M., & MOULINIER, C.L. (1968)
Effets in vitro d'organo-phosphorés sur l'huître portugaise,
quelques mollusques dulçaquicoles et le micro-plancton d'eau douce.
Bull. Soc. Pathol. exot., 60: 568-580.
MANI, K. & SAXENA, P.K. (1985) Effect of safe concentrations of some
pesticides on ovarian recrudescence in the freshwater murrel,
Channa punctatus (BI): A quantitative study. Ecotoxicol.
environ. Saf., 9: 241-249.
MARSHALL, W.K. & ROBERTS, J.R. (1977) Simulation modelling of the
distribution of pesticides in ponds. In: Proceedings of a Symposium
on Fenitrothion, Ottawa, National Research Council of Canada, pp.
253-278 (Publication NRCC No. 16073).
MARTIN, H. & WORTHING, C.R. (1981) The pesticide manual, 5th ed.,
Croydon, The British Crop Protection Council, p. 267.
MARTIN, N.A. & YEATES, G.W. (1975) Effect of four insecticides on
the pasture ecosystem., III. Nematodes, rotifers, and tardigrades.
N.Z. J. agric. Res., 18: 307-312.
MARTINDALE, R.W. (1988) Determination of residues of a range of
fungicides, anti-sprouting agents and (organochlorine and
organophosphorus) insecticides. Analyst, 113: 1229-1233.
MARTINEZ CHUECOS, J. & SOLE VIOLAN, J. (1985) Letter to editor
concerning the article "Delayed neurotoxicity produced by an
organophosphorus compound (Sumithion)" by Sakamoto et al. Arch.
Toxicol., 58: 123-124.
MASTALSKI, K. (1971a) Acute oral toxicity study with p-nitro
metacresol in beagle dogs, Northbrook, Illinois, Industrial Bio-Test
Laboratories (Unpublished report No. J9994, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
MASTALSKI, K. (1971b) Acute oral toxicity study with Sumithion
technical grade in beagle dogs, Northbrook, Illinois, Industrial
Bio-Test Laboratories (Unpublished report No. J9994, submitted to
WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
MATSUBARA, T. & HORIKOSHI, I. (1983) Possible antidotes in severe
intoxication with fenitrothion in rats. J. Pharm. Dyn., 6: 699-707.
MATSUSHITA, T., AOYAMA, K., YOSHIMI, K., FUJITA, Y., & UEDA, A.
(1985) Allergic contact dermatitis from organophosphorus
insecticides. Ind. Health, 23: 145-153.
MAYER, F.L., Jr (1987) Acute toxicity handbook of chemicals to
estuarine organisms, Gulf Breeze, Florida, US Environmental
Protection Agency, Gulf Breeze Laboratory (EPA 600/8-87/017).
MEISTER, R.T., BERG, G.L., SINE, C., POPLYK, J., & SCHILLING, G.
(1985) Fenitrothion. In: Farm chemicals handbook, Willoughby,
Delaware, Meister Publishing Co., pp. C103-C104.
MENNA, J.H. (1985) Effect of emulsifiers on influenza type A virus
infection; in vivo and in vitro studies. Toxicol. environ.
Health, 16: 441-448.
MESTRES, R., TOURTE, J., CAMPO, M., & ILLES, S. (1974) Résidus de
pesticides. XXV-Méthode polyvalente de recherche et de dosage des
résidus de pesticides divers dans les matières alimentaires. Ann.
Fals. Exp. Chim., 1974: 513-526, 721-722.
METCALF, C.D., MCLEESE, D.W., & ZITKO, V. (1980) Rate of
volatilization of fenitrothion from fresh water. Chemosphere, 9:
151-155.
MIHARA, K., OKUNO, Y., MISAKI, Y., & MIYAMOTO, J. (1978) Metabolism
of fenitrothion in goats. J. pestic. Sci., 3: 233-242.
MIHARA, K., MISAKI, Y., & MIYAMOTO, J. (1979) Metabolism of
fenitrothion in birds. J. pestic. Sci., 4: 175-185.
MIHARA, K., ISOBE, N., OHKAWA, H., & MIYAMOTO, J. (1981) Effects of
organophosphorus insecticides on mitochondrial and microsomal
functions in the liver of rat with special emphasis on fenitrothion.
J. pestic. Sci., 6: 307-316.
MIKAMI, N., IMANISHI, K., YAMADA, H., & MIYAMOTO, J. (1985a)
Photodegradation of fenitrothion in water and soil surface, and its
hydrolysis in water. J. pestic. Sci., 10: 263-272.
MIKAMI, N., SAKATA, S., YAMADA, H., & MIYAMOTO, J. (1985b) Further
studies on degradation of fenitrothion in soils. J. pestic. Sci.,
10: 491-500.
MILLER, C.A., STEWART, J.F., ELGEE, D.E., SHAW, D.D., MORGAN, M.G.,
KETTELA, E.G., GREENBANK, D.O., GRESNER, G.N., & VARTY, I.W. (1973)
Aerial spraying against spruce budworm adults in New Brunswick. A
compendium of reports on the 1972 test program, Fredericton, Canada,
Maritimes Forest Research Centre (Information Report M-X-38).
MILLER, S. & SHAH, M.A. (1982) Cholinesterase activities of workers
exposed to organophosphorus insecticides in Pakistan and Haiti and
an evaluation of the tintometric method. J. environ. Sci. Health,
B17(2): 125-142.
MISU, Y., SEGAWA, T., KURUMA, I., KOJIMA, M., & TAKAGI, H. (1966)
Subacute toxicity of O,O-dimethyl- O-(3-methyl-4-nitrophenyl)
phosphorothioate (Sumithion) in the rat. Toxicol. appl. Pharmacol.,
9: 17-26.
MIYAMOTO, J. (1964a) Studies on the mode of action of
organophosphorus compounds. Part III. Activation and degradation of
Sumithion and methylparathion in vivo. Agric. biol. Chem., 28:
411-421.
MIYAMOTO, J. (1964b) Studies on the mode of action of
organophosphorus compounds. IV. Penetration of Sumithion,
methylparathion and their oxygen analogs into guinea-pig brain and
inhibition of cholinesterase in vivo. Agric. biol. Chem., 28:
422-430.
MIYAMOTO, J. (1969) Mechanism of low toxicity of Sumithion towards
mammals. Residue Rev., 25: 251-264.
MIYAMOTO, J. (1977a) Degradation of fenitrothion in terrestrial and
aquatic environments including photolytic and microbial reactions.
In: Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 105-109 (Publication NRCC No.
16073).
MIYAMOTO, J. (1977b) The implication of recent long-term
toxicological studies of fenitrothion on birds. In: Proceedings of a
Symposium on Fenitrothion, Ottawa, National Research Council of
Canada, pp. 307-319 (Publication NRCC No. 16073).
MIYAMOTO, J. (1977c) Long-term toxicological effects of fenitrothion
in mammals including carcinogenicity and mutagenicity. In:
Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 459-496 (Publication NRCC No.
16073).
MIYAMOTO, J. & KADOTA, T. (1972) Toxicological studies with
Sumithion (Unpublished report submitted to WHO by Sumitomo Chemical
Co., Ltd, Osaka, Japan).
MIYAMOTO, J. & SATO, Y. (1969) Determination of insecticide residue
in animal and plant tissues. VI. Determination of sumithion residue
in cattle tissues. Botyu-Kagaku, 34: 3-6.
MIYAMOTO, J., SATO, Y., KADOTA, T., FUJINAMI, A., & ENDO, M. (1963a)
Studies on the mode of action of organophosphorus compounds. Part I.
Metabolic fate of 32P-labelled Sumithion and methylparathion in
guinea-pigs and white rats. Agric. biol. Chem., 27: 381-389.
MIYAMOTO, J., SATO, Y., KADOTA, T., & FUJINAMI, A. (1963b) Studies
on the mode of action of organophosphorus compounds. Part II.
Inhibition of mammalian cholinesterase in vivo following
administration of Sumithion and methylparathion. Agric. biol. Chem.,
27: 669-676.
MIYAMOTO, J., SATO, Y., & SUZUKI, S. (1967) Determination of
insecticide residue in animal and plant tissue. IV. Determination of
residual amount of sumithion and some of its metabolites in fresh
milk. Botyu-Kagaku, 32: 95-100.
MIYAMOTO, J., SATO, Y., YAMAMOTO, K., & SUZUKI, S. (1968) Activation
and degradation of Sumithion, methylparathion and their oxygen
analogs by mammalian enzymes in vitro. Botyu-Kagaku, 33: 1-6.
MIYAMOTO, J., KOHDA, H., & KADOTA, T. (1975) Teratogenic studies
with Sumithion in ICR-JCL mice and Sprague-Dawley rats (Unpublished
report submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
MIYAMOTO, J., HOSOKAWA, S., KADOTA, T., KOHDA, H., ARAI, M.,
SUGIHARA, S., & HIRAO, K. (1976a) Studies on cholinesterase
inhibition and structural changes at neuromuscular junctions in
rabbits by subacute administration of Sumithion. J. pestic. Sci., 1:
171-178.
MIYAMOTO, J., MIHARA, K., & HOSOKAWA, S. (1976b) Comparative
metabolism of m-methyl-carbon-14-Sumithion in several species of
mammals in vivo. J. pestic. Sci., 1: 9-21.
MIYAMOTO, J., MIHARA, K., KADOTA, T., & OKUNO, Y. (1977a) Toxicity
and metabolism in vivo of fenitrothion in rats with experimental
hepatic lesion. J. pestic. Sci., 2: 271-277.
MIYAMOTO, J., OKUNO, Y., KADOTA, T., & MIHARA, K. (1977b)
Experimental hepatic lesions and drug metabolizing enzymes in rats.
J. pestic. Sci., 2: 257-269.
MIYAMOTO, J., MIKAMI, N., MIHARA, K., TAKIMOTO, Y., KOHDA, H., &
SUZUKI, H. (1978) Biological activity of fenitrothion and its
degradation products. J. pestic. Sci., 3: 35-41.
MIYAOKA, T., TAKAHASHI, H., TSUDA, S., & SHIRASU, Y. (1983) Effect
of O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate
(Fenitrothion) treatment on acute toxicity of 2-sec-butylphenyl
methylcarbamate (BPMC) in dogs. Jpn. J. vet. Sci., 45: 463-470.
MIYASHITA, M. (1984) Effects of a short period exposure of
fenitrothion on the reproduction of pregnant females of the guppy,
Poecilia reticulata. Jpn. J. Hyg., 39: 647-650.
MOLLHOFF, E. (1967) Gas-chromatographic determination of residues of
E 605 products and Agritox in plants and soil samples.
Pflanzenschutz-Nachr. "Bayer", 20: 557-574.
MOODY, R.P. & FRANKLIN, C.A. (1987) Percutaneous absorption of the
insecticides fenitrothion and aminocarb in rats and monkeys. J.
Toxicol. environ. Health, 20: 209-218.
MOODY, R.P., PRASAD, R., GREENHALGH, R., & WEINBERGER, P. (1977)
Translocation of ring labelled 14C-fenitrothion in conifers. In:
Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, pp. 217-231 (Publication NRCC No.
16073).
MOODY, R.P., GREENHALGH, R., LOCKHART, L., & WEINBERGER, P. (1978)
The fate of fenitrothion in an aquatic ecosystem. Bull. environ.
Contam. Toxicol., 19: 8-14.
MOODY, R.P., CARROLL, J.M., & KRESTA, A.M.E. (1987a) Automated high
performance liquid chromatography and liquid scintillation counting
determination of pesticide mixture octanol/water partition rates.
Toxicol. ind. Health, 3: 479-490.
MOODY, R.P., RIEDEL, D., RITTER, L., & FRANKLIN, C.A. (1987b) The
effect of DEET ( N,N-Diethyl- m-toluamide) on dermal persistence
and absorption of the insecticide fenitrothion in rats and monkeys.
J. Toxicol. environ. Health, 22: 471-479.
MORGAN M.J. & KICENIUK, J.W. (1990) Effect of fenitrothion on
foraging behavior of juvenile atlantic salmon. Environ. Toxicol.
Chem., 9: 489-495.
MORIN, R., GABCURY, G., & MANARBACHI, G. (1986) Fenitrothion and
aminocarb residues in water and balsam fir foliage following spruce
budworm spraying programs in Quebec, 1979-1982. Bull. environ.
Contam. Toxicol., 36: 622-628.
MORIYA, M., OHTA, T., WATANABE, K., MIYAZAWA, T., KATO, K., &
SHIRASU, Y. (1983) Further mutagenicity studies on pesticides in
bacterial reverse assay systems. Mutat. Res., 116: 185-216.
MORRISON, B.R.S. & WELLS, D.E. (1981) The fate of fenitrothion in a
stream environment and its effect on the fauna, following aerial
spraying of a Scottish forest. Sci. total Environ., 19: 233-252.
MORSETH, S.L., SERABIAN, M.A., LICHTENBERGER, J.M., VARGAS, K.J.,
THAKUR, A.K., & BURLEW, P.L. (1986) Teratology study in rabbits.
Fenitrothion T.G. (Sumithion): Final report, Vienna, Virginia,
Hazleton Laboratories (Technical Report No. HT-71-0382 (Project No.
343-914), submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
MORSETH, S.L., LANDES, S.G., LEWIS, S.A., THAKUR, A.K., &
LICHTENBERGER, J.M. (1987) Teratology study in rats with Sumithion,
Vienna, Virginia, Hazleton Laboratories (Technical Report No.
HT-61-0367 (Project No. 343-182), submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
MOTABAR, M., SANAI, G.H., & HEIDAI, A.A. (1972) Toxicological
evaluation of Sumithion on operators and inhabitants in the Mamasani
area, southern Iran. Iran. J. Pharm. Health, 2: 40-49.
MUNNECKE, D.M. (1976) Enzymatic hydrolysis of organophosphate
insecticides, a possible pesticide disposal method. Appl. environ.
Microbiol., 32: 7-13.
MURTY, A.S., RAJABHUSHANAM, B.R., RAMANI, A.V., & CHRISTOPHER, K.
(1983) Toxicity of fenitrothion to the fish Mystus cavasius and
Labeo rohita. Environ. Pollut., 30A: 225-232.
NAGAYAMA, T., MAKI, T., KIN, K., MAKI, M., & NISHIMA, T. (1986)
Survey of pesticide residues in vegetables and fruits. Annu. Rep.
Tokyo Metrop. Res. Lab. P.H., 37: 173-183.
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., & NISHIMA, T. (1987)
Survey of pesticide residues in vegetables and fruits. Annu. Rep.
Tokyo Metrop. Res. Lab. P.H., 38: 222-228.
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1988) Survey of pesticide residues in vegetables and fruits. Annu.
Rep. Tokyo Metrop. Res. Lab. P.H., 39: 130-138.
NAGAYAMA, T., MAKI, T., IIDA, M., KAN, K., KAWAI, Y., & NISHIMA, T.
(1989) Survey of pesticide residues in vegetables and fruits. Annu.
Rep. Tokyo Metrop. Res. Lab. P.H., 40: 155-162.
NAIR, G.A. (1981) Toxic effects of certain biocides on a fresh water
mite, Hydrachna trilobata viets (Arachnida; Hydrachnoidae
Hydrachnidae) J. environ. Biol., 2: 91-96.
NAMBA, N., IWAMOTO, T., & SATOH, T. (1966) Oral toxicity and
metabolism of sumition on cattle, sheep and pigs. Res. Bull.
Hokkaido Natl Agric. Exp. Stn, 89: 82-90.
NAMBA, T. (1971) Cholinesterase inhibition by organophosphorus
compounds and its clinical effects. Bull. World Health Organization,
44: 289-307.
NATIONAL RESEARCH COUNCIL OF CANADA (1975) Fenitrothion: The effects
of its use on environmental quality and its chemistry, Ottawa,
National Research Council of Canada, pp. 22-32 (Publication NRCC No.
14104).
NEHEZ, M., HUSZTA, E., MAZZAG, E., & BERENCSI, G. (1985) A study of
the mutagenic effect of 3-methyl-4-nitrophenol on the somatic cells
of the mouse. Ecotoxicol. environ. Saf., 9: 47-52.
NISHIO, M. & KUSANO, S. (1978) [Effect of consecutive use of
organophosphorus insecticides to population and nitrification
activity of soil microorganisms.] Nojishikenjo-kenkyuuhokoku, 28:
39-48 (in Japanese).
NISHIUCHI, Y. (1977) Toxicity of pesticide formulations to some
fresh water organisms XXXXI. Aquiculture, 24: 146-150.
NISHIUCHI, Y. (1981) [Toxicity of pesticides to some aquatic
organisms I; Toxicity of pesticides to some aquatic insects.] Seitai
Kagaku, 4: 31-46 (in Japanese).
NISHIZAWA, Y., FUJII, K., KADOTA, T., MIYAMOTO, J., & SAKAMOTO, H.
(1961) Studies on the organophosphorus insecticides. Part VII.
Chemical and biological properties of new low toxic organophosphorus
insecticide. O,O-dimethyl- O-(3-methyl-4-nitrophenyl)
phosphorothioate. Agric. biol. Chem., 25: 605-610.
NOSAL, N. & HLADKA, A. (1968) Determination of the exposure to
fenitrothion ( O,O-dimethyl-0/3-methyl-4-nitrophenyl/thiophosphate)
on the basis of the excretion of p-nitro- m-cresol by the urine
of the persons tested. Int. Arch. Gewerbepathol. Gewerbehyg., 25:
28-38.
OGATA, S. (1972) Effects of organophosphate pesticide on the beagle
dogs in chronic toxicity studies experiments (Report 1). Acta Soc.
Ophthalmol. Jpn, 76: 1153-1150.
OHKAWA, H., MIKAMI, N., & MIYAMOTO, J. (1974) Photodecomposition of
sumithion, O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate.
Agric. biol. Chem., 38: 2247-2255.
OHKAWA, H., OSHITA, H., & MIYAMOTO, J. (1980) Comparison of
inhibitory activity of various organophosphorus compounds against
acetylcholinesterase and neurotoxic esterase of hen with respect to
delayed neurotoxicity. Biochem. Pharmacol., 29: 2721-2727.
OHMAE, T., UNO, M., OKADA, S., KAGECHI, Y., TERADA, I., & TANIGAWA,
K. (1981) Environmental behavior of fenitrothion and its
decomposition products after aerial application. J. pestic. Sci., 6:
437-446.
OHSHIMA, M., TAKAHASHI, N., MIKAMI, N., & MATSUDA, T. (1988)
Accumulation and metabolism of 14-C fenitrothion in bluegill
sunfish (Lepomis macrochirus) (Unpublished Technical Report No.
HM-80-0095, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
OKADA, I. & HOSHIBA, H. (1970) A laboratory experiment on some
insecticides to honeybee. Bull. Fac. Agric., Tamagawa Univ., 10:
79-85.
OKUNO, Y., HARA, S., KADOTA, T., & MIYAMOTO, J. (1977) Possible
allergic asthma by inhalation of Sumithion 22% emulsifiable
concentrate in guinea-pigs (Unpublished Technical Report No.
HT-70-0113, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
OSICKA-KOPROWSKA, A., GRADOWSKA-OLZEWSKA, I., & WYSOCKA-PARUSZEWSKA,
B. (1987) Adrenocortical changes and uptake of
4-14C-corticosterone in rats intoxicated with fenitrothion. Arch.
Toxicol., 61: 76-78.
PAUL, B.S. & VADLAMUDI, V.P. (1976) Teratogenic studies of
fenitrothion on White Leghorn chick embryos. Bull. environ. Contam.
Toxicol., 15: 223-229.
PAWAR, K.R. & KATDARE, M. (1983) Effect of the insecticide Sumithion
(fenitrothion) on embryonic development in a frog. Experientia
(Basel), 39: 297-298.
PAWAR, K.R. & KATDARE, M. (1984) Toxic and teratogenic effects of
fenitrothion, BHC and carbofuran on embryonic development of the
frog Microhyla ornatas. Toxicol. Lett., 22: 7-13.
PEARCE, P.A. & BUSBY, D.G. (1980) Research on the effects of
fenitrothion on the White-throated Sparrow. In: Research on the
effects of spray operations for forest protection against spruce
budworm. Environmental surveillance in New Brunswick 1978-79,
Fredericton, Canada, Maritimes Forest Research Centre, pp. 24-28.
PEARCE, P.A. & PEAKALL D.B. (1977) The impact of fenitrothion on
bird populations in New Brunswick. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, pp.
299-305 (Publication NRCC No. 16073).
PEARCE, P.A., BRUN, G.L., & WITTEMAN, J. (1979a) Off-target fallout
of fenitrothion during 1978 forest spraying operations in New
Brunswick. Bull. environ. Contam. Toxicol., 23: 503-508.
PEARCE, P.A., PEAKALL, D.B., & ERSKINE, A.J. (1979b) Impact on
forest birds of the 1976 spruce budworm spray operation in New
Brunswick, Ottawa, Environment Canada, Canadian Wildlife Service,
pp. 1-15 (Progress Note No. 97).
PETERSON, R.H. (1974) Influence of fenitrothion on swimming
velocities of brook trout (Salvelinus fontinalis). J. Fish Res.
Board Can., 31: 1757-1762.
PETERSON, R.H. & ZITKO, V. (1974) Variations in stream insect drift
associated with operational and experimental contamination with
fenitrothion in New Brunswick, St. Andrews, New Brunswick, Fisheries
Research Board of Canada, Biological Station, 23 pp (Technical
Report No. 439).
PLOWRIGHT, R.C. (1977) The effects of fenitrothion on forest
pollinators in New Brunswick. In: Proceedings of a Symposium on
Fenitrothion, Ottawa, National Research Council of Canada, pp.
335-341 (Publication NRCC No. 16073).
PLOWRIGHT, R.C. & ROOD, F.H. (1980) The effect of aerial insecticide
spraying on hymenopterous pollinators in New Brunswick. Can.
Entomol., 112: 259-269.
PLOWRIGHT, R.C., PENDRER, B.A., & MCLAREN, I.A. (1978) The impact of
aerial fenitrothion spraying upon the population biology of bumble
bees ( Bombus LATR.: HYM.) in south-western New Brunswick. Can.
Entomol., 110: 1145-1156.
POLLACK, J.D., BURECH, D., & HAMPARIN, V.V. (1977) The role of
chemicals in potentiating viral infections and Reye's syndrome. In:
Proceedings of a Symposium on Fenitrothion, Ottawa, National
Research Council of Canada, p. 519 (Publication NRCC No. 16073).
POMBER, L., WEINBERGER, P., & PRASAD. R. (1974) Some physiological
and phytotoxic effects of fenitrothion on germination and seedling
growth of Pinus strobus L., Ottawa, Environment Canada, Forestry
Service, Chemical Control Research Institute, 25 pp (Information
Report CC-X-80).
PRASAD, R. & MOODY, R.P. (1976) Fate of fenitrothion in forest
trees. VIII. Persistence, translocation and metabolism of
C14-fenitrothion in Jack Pine ( Pinus banksiana Lamb.) in
relation to sawfly mortally, Ottawa, Environment Canada, Forestry
Service, Chemical Control Research Institute, 22 pp (Information
Report CC-X-126).
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1982a) In vivo
sub-acute physiological stress induced by sumithion on carbohydrate
metabolism in hepatopancreas of Oziotelphusa senex senex
Fabricius. Toxicol. Lett., 13: 179-182.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1982b) Effect of
sumithion on molting the freshwater rice-field crab ( Oziotelphusa
senex senex Fabricius). Toxicol. Lett., 14: 243-246.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1983a) Effect of
sublethal concentrations of sumithion on limb regeneration of
freshwater field crab Oziotelphusa senex senex. Experientia
(Basel), 39: 1380-1381.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1983b) Chronic
sumithion toxicity: Effect on carbohydrate metabolism in crab
muscle. Toxicol. Lett., 15: 113-117.
REDDY, P.S., BHAGYALAKSHMI, A., & RAMAMURTHI, R. (1983c) Effect of
sumithion on ovarian growth of a freshwater rice field crab
(Oziotelphusa senex senex Fabricius). Toxicol. Lett., 18: 273-276.
REYE, R.D.K., MORGAN, G., & BARAL, J. (1963) Encephalopathy and
fatty degeneration of the viscera. A disease entity in childfood.
Lancet, 7311: 749-752.
RIPLEY, B.D., HALL, J.A., & CHAU, A.S.Y. (1974) Determination of
fenitrothion, phosphamidon and dimethoate in natural water. Environ.
Lett., 7: 97-118.
RONDEAU, D.B., YOUNG, L., HEBERT, D., & TROTIER, B.L. (1981)
Behavioral toxicity of chronic administration of fenitrothion in
rats. Neurobehav. Toxicol. Teratol., 3: 313-319.
ROSIVAL, L., VARGOVA, M., & HLADKA, H., BATORA, V., KOVACICOVA, J.,
SZOKOLAYOVA, J., & CEREY, K. (1974) On the toxic effect of the
fenitrothion S-methyl isomer, Bratislava, Czechoslovakia, Research
Institute of Hygiene, Research Institute of Industrial Hygiene and
Professional Diseases, and Research Institute of Agrochemical
Technology (Unpublished report submitted to WHO by the authors).
RUBEN, F.L., STREIFF, E.J., NEAL, M., & MICHAELIS, R.H. (1976)
Epidemiologic study of Reye's Syndrome: cases seen in Pittsburgh,
October 1973-April 1975. Am. J. public Health, 66: 1096-1098.
RUSHMORE, F.M. (1971) Effects of accothion on birds. (Intensive
studies on sapsuckers). In: Nash, R.W., Peterson, J.W., & Chansler,
J.F., ed. Environmental studies of accothion for spruce budworm
control, Washington, DC, US Department of Agriculture and US
Department of Interior, p. 88.
RUTTER, H.A. & NELSON, L.W. (1974) Two-year dietary administration
in the rat, Vienna, Virginia, Hazleton Laboratories (Unpublished
Technical Report No. HT-41-0006, submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
RUTTER, H.A. & VOELKER, R.W. (1974) Three-generation reproduction
study: Rats, Vienna, Virginia, Hazleton Laboratories (Unpublished
Technical Report No. HT-41-0005, submitted to WHO by Sumitomo
Chemical Co., Ltd, Osaka, Japan).
RUTTER, H.A. & BANAS, D.A. (1975) Seventy eight week tumorigenic
study in the ICR Swiss mice with Sumithion, Vienna, Virginia,
Hazleton Laboratories (Technical Report No. HT-51-0002, submitted to
WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
SAKAMOTO, T., SAWADA, Y., NISHIDE, K., SADAMITSU, D., YOSHIOKA, T.,
SUGIMOTO, T., NISHII, S., & KISHII, H. (1984) Delayed neurotoxicity
produced by an organophosphorus compound (Sumithion). A case report.
Arch. Toxicol., 56: 136-138.
SALONIUS, P.O. (1972) Effect of DDT and fenitrothion on forest-soil
microflora. J. econ. Entomol., 65: 1089-1090.
SAXENA, P.K. & MANI, K. (1985a) Protein-bound iodine levels in the
blood plasma of freshwater teleost, Channa punctatus (BI) exposed
to subtoxic pesticides concentrations. Toxicol. Lett., 24: 33-36.
SAXENA, P.K. & MANI, K. (1985b) Quantitative study of testicular
recrudescence in the freshwater teleost, Channa punctatus (BI)
exposed to pesticides. Bull. environ. Contam. Toxicol., 34: 597-607.
SAXENA, P.K. & MANI, K. (1987) Effect of safe concentrations of some
pesticides on testicular recrudescence in the freshwater murrel,
Channa punctatus (BI): A morphological study. Ecotoxicol. environ.
Saf., 14: 56-63.
SCHAFER, E.W. (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol. appl.
Pharmacol., 21: 315-330.
SCHERER, E. (1975) Avoidance of fenitrothion by gold fish
(Carassius auratus). Bull. environ. Contam. Toxicol., 13:
492-496.
SCHNEIDER, W.G. (1976) Forest spray program and Reye's syndrome.
Report of the Panel convened by the Government of New Brunswick,
Fredericton, Canada, Government of New Brunswick, 22 pp.
SEIKAI, T. (1982) Acute toxicity of organophosphorous insecticides
on the developmental stages of eggs, larvae and juveniles of
Japanese striped knifejaw, Oplegnathus fasciatus. Bull. Jpn. Soc.
Sci. Fish., 48: 599.
SERGEANT, D.B., ZITKO, V., & BURRIDGE, L.E. (1979) The determination
of fenitrothion in bivalves, St. Andrews, Fisheries and Marine
Service, Biological Station, 27 pp (Technical Report No. 906).
SHIM, J.C. & SELF, L.S. (1973) Toxicity of agricultural chemicals to
larvivorous fish in Korean rice fields. Trop. Med., 15: 123-130.
SHIRASU, Y., MORIYA, M., & WATANABE, K. (1979) Mutagenicity studies
on Sumithion in bacterial systems. (Unpublished Technical Report No.
HT-91-0140, submitted to WHO by Sumitomo Chemical Co., Ltd, Osaka,
Japan).
SMART, N.A. (1980) Determination of a range of organophosphorus
pesticide residues in grain. Report by a Working Group of the
Committee for Analytical Methods for Residues of Pesticides and
Veterinary Products in Foodstuffs and the Working Party on Pesticide
Residues of the Ministry of Agriculture, Fisheries and Food.
Analyst, 105: 515-517.
SPILLNER, C.J., DEBAUN, J.R., & MENN, J.J. (1979a) Degradation of
fenitrothion in forest soil and effects on forest soil microbes. J.
agric. food Chem., 27: 1054-1060.
SPILLNER, C.J., THOMAS, V.H., & DEBAUN, J.R. (1979b) Effect of
fenitrothion on microorganisms which degrade leaf-litter and
cellulose in forest soils. Bull. environ. Contam. Toxicol., 23:
601-606.
SPITZER, W.O. (1982) Report of the New Brunswick Task Force on the
Environment and Reye's Syndrome. Clin. invest. Med., 5: 203-214.
SUMITOMO CHEMICAL CO., LTD (1969) Residual amounts of Sumithion in
foods. Reports on trials conducted by the Japanese Ministry of
Agriculture and Forestry, 1968, Osaka, Japan, Sumitomo Chemical Co.,
Ltd (Unpublished report).
SUMITOMO CHEMICAL CO., LTD (1971) Acute oral toxicity study with
3-methyl-4-nitrophenol in rat, Osaka, Japan, Sumitomo Chemical Co.,
Ltd, Takarazuka Research Laboratory (Unpublished report).
SUMITOMO CHEMICAL CO., LTD. (1972) Six-month feeding study of
Sumithion in rats, Osaka, Japan, Sumitomo Chemical Co., Ltd,
Pesticide Research Department (Unpublished report).
SUNDARAM, K.M.S. (1973) Degradation dynamics of fenithrothion in
aqueous systems, Ottawa, Environment Canada, Forestry Service,
Chemical Control Research Institute, 19 pp (Information Report
CC-X-44).
SUNDARAM, K.M.S. (1974) Distribution and persistence of fenitrothion
residues in foliage, soil and water in larose forest, Ottawa,
Environment Canada, Forestry Service, Chemical Control Research
Institute, 43 pp (Information Report CC-X-64).
SUNDARAM, K.M.S. (1975) Fenitrothion residues in honey bees and
their products collected from forest areas sprayed with the
insecticide for budworm control, Ottawa, Environment Canada,
Forestry Service, Chemical Control Research Institute, 27 pp
(Information Report CC-X-89).
SUNDARAM, K.M.S. (1988) Cuticular penetration and in vivo metabolism
of fenitrothion in spruce budworm. J. environ. Sci. Health, B23:
643-659.
SUNDARAM, K.M.S. & PRASAD, R. (1975) Persistence studies of
insecticides: VI. Root penetration, translocation and metabolism of
14C-labelled fenitrothion in young spruce trees. Ottawa,
Environment Canada, Forestry Service, Chemical Control Research
Institute, 15 pp (Information Report CC-X-86).
SUNDARAM, K.M.S., YULE, W.M., & PRASAD, R. (1975) Studies of foliar
penetration, movement and persistence of 14C-labelled fenitrothion
in spruce and fir trees, Ottawa, Environment Canada, Forestry
Service, Chemical Control Research Institute, 17 pp (Information
Report CC-X-85).
SUZUKI, H. & MIYAMOTO, J. (1976) Studies on mutagenicity of
Sumithion, Cremart and Denmart. J. pestic. Sci., 1: 253-259.
SUZUKI, H. & MIYAMOTO, J. (1980) Effects of fenitrothion on sister
chromatid exchanges (SCE) in cultured mouse embryo cells
(Unpublished Technical Report No. HT-00-0280, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
SUZUKI, H., MIYAMOTO, J., & OKA, A. (1974) Mutagenicity and
radiomimeticity screening of Sumithion (Unpublished report submitted
to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
SWAIN, W.R. & IVANIKIW, N.K., ed. (1976) Proceedings of the Second
USA-USSR Symposium on the Effects of Pollutants upon Aquatic
Ecosystems, Vol. II.
SYMONS, P.E.K. (1973) Behavior of young Atlantic salmon (Salmo
salar) exposed or force-fed fenitrothion, an organophosphate
insecticide. J. Fish. Res. Board Can., 30: 651-655.
SYMONS, P.E.K. (1977) Disposal and toxicology of the insecticide
fenitrothion: Predicting hazards of forest spraying. Residue Rev.,
68: 1-36.
SYMONS, P.E.K. & METCALF, J.L. (1978) Mortality, recovery and
survival of larval Brochycentrus numerosus (Trichoptera) after
exposure to the insecticide fenitrothion. Can. J. Zool., 56:
1284-1290.
TABATA, K. & KITAHARA, E. (1980) Effect of fenitrothion (Sumithion)
spraying on the population density and blood cholinesterase activity
of Japanese woodmouse, Apodemus speciosus Temmink. Appl. Entomol.
Zool., 15: 242-248.
TABATA, K. & OKUBO, R. (1980) [Mechanism of the abnormal leaf
abscission phenomenon in Japanese cypress caused by fenitrothion
(Sumithion) (II) Metabolism of 14C-fenitrothion
( O,O-dimethyl- O-(3-methyl-4-nitrophenyl) phosphorothioate) in
Japanese cypress leaves.] J. Jpn. Forensic Soc., 62: 350-353 (in
Japanese, with English summary).
TAKAHASHI, H., MIYAOKA, T., TSUDA, S., & SHIRASU, Y. (1984)
Potentiated toxicity of 2-sec-butylphenyl methylcarbamate (BPMC) by
O,O-dimethyl O-(3-methyl-4-nitrophenyl)phosphorothioate
(fenitrothion) in mice; Relationship between acute toxicity and
metabolism of BPMC. Fundam. appl. Toxicol., 4: 718-723.
TAKAHASHI, H., KATO, A., YAMASHITA, E., NAITO, Y., TSUDA, S., &
SHIRASU, Y. (1987) Potentiations of N-methylcarbamate toxicities
by organophosphorus insecticides in male mice. Fundam. appl.
Toxicol., 8: 139-146.
TAKAKU, T., OTSUKI, A., & TAKAHASHI, M. (1979a) Simple and rapid
determination of fenitrothion in water by reversed phase adsorption
liquid chromatography. Bunseki Kagaku, 28: 702-704.
TAKAKU, T., TAKAHASHI, M., & OTSUKI, A. (1979b) Dispersion of an
organophosphorus insecticide, fenitrothion, in paddy fields and its
effects on the microorganisms. Jpn. J. Limnol., 40: 137-144.
TAKANO, H. & HIJIKATA, Y. (1981) [Environmental impact of
fenitrothion on forest - Effect on wild birds.] Forest Chem., 75:
15-18 (in Japanese).
TAKEUCHI, K., HIGO, M., & SAKAI, T. (1980) Susceptibility of the
honey bees to several insecticides. Bull. Fac. Agric. Tamagawa
Univ., 20: 40-46.
TAKIMOTO, Y. & MIYAMOTO, J. (1976a) Studies on accumulation and
metabolism of sumithion in fish. J. pestic. Sci., 1: 261-271.
TAKIMOTO, Y. & MIYAMOTO, J. (1976b) Residue analysis of Sumithion.
Residue Rev., 60: 83-101.
TAKIMOTO, Y., MURANO, A., & MIYAMOTO, J. (1975) Analytical methods
for Sumithion in technical products and formulated materials.
Residue Rev., 60: 11-28.
TAKIMOTO, Y., HIROTA, M., INUI, H., & MIYAMOTO, J. (1976)
Decomposition and leaching of radioactive sumithion of 4 different
soils under laboratory conditions. J. pestic. Sci., 1: 131-143.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1978) Degradation and
fate of the fenitrothion applied to harvested rice grains. J.
pestic. Sci., 3: 277-290.
TAKIMOTO, Y., OHSHIMA, M., KATO., T., & MIYAMOTO, J. (1980)
[Photodegradation of fenitrothion in sea water.] Abstract of papers
presented at the Annual Meeting of Pesticide Science Society of
Japan, Chiba, No. 143 (in Japanese).
TAKIMOTO, Y., OHSHIMA, M., YAMADA, H., & MIYAMOTO, J. (1984a) Fate
of fenitrothion in several developmental stages of the killifish
(Oryzias latipes). Arch. environ. Contam. Toxicol., 13: 579-587.
TAKIMOTO, Y., HAGINO, S., YAMADA, H., & MIYAMOTO, J. (1984b) The
acute toxicity of fenitrothion to killifish (Oryzias latipes) at
twelve different stages of its life history. J. pestic. Sci., 9:
463-470.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1987a) Comparative
metabolism of fenitrothion in aquatic organisms I. Metabolism in the
euryhaline fish, Oryzias latipes and Mugil cephalus. Ecotoxicol.
environ. Saf., 13: 104-117.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1987b) Comparative
metabolism of fenitrothion in aquatic organisms. II. Metabolism in
the freshwater snail, Cipangopaludina japonica and Physa acuta.
Ecotoxicol. environ. Saf., 13: 118-125.
TAKIMOTO, Y., OHSHIMA, M., & MIYAMOTO, J. (1987c) Comparative
metabolism of fenitrothion in aquatic organisms. III. Metabolism in
the crustaceans, Daphnia pulex and Palaemon paucidens.
Ecotoxicol. environ. Saf., 13: 126-134.
TALEKAR, N.S., SUN, L.T., LEE, E.M., CHEN, J.S., LEE, T.M., & LU, S.
(1977) Residual behavior of several insecticides on chinese
cabbage. J. econ. Entomol., 70: 689-692.
TAMANO, S. HAGIWARA, A., SHIBATA, M., & SUZUKI, M. (1990) Chronic
dietary toxicity and carcinogenicity test of Sumithion technical in
mice, Aichi, Japan, Daiyu-Kai Medical Science Research Institute
(Unpublished Technical Report No. HT-01-0445, submitted to WHO by
Sumitomo Chemical Co., Ltd, Osaka, Japan).
THOMPSON, C.M., FRICK, J.A., NATKE, B.C., & HANSEN, L.K. (1989)
Preparation, analysis and anticholinesterase properties of
O,O-dimethyl phosphorothioate isomerides. Chem. Res. Toxicol., 2:
386-391.
TOMITA, M., SOMEYA, N., NISHIMURA, M., & UEDA, K. (1974) [Studies on
the retention deposit of organophosphorus pesticides in animal and
human bodies.] Jpn. J. ind. Health, 16: 547-556 (in Japanese).
TOOR, H.S. & KAUR, K. (1974) Toxicity of pesticides to the fish,
Cyprinus carpio communis Linn. Indian J. exp. Biol., 12: 334-336.
TROTTIER, B.L. & JANKOWSKA, I. (1980) In vivo study on the storage
of fenitrothion in chicken tissues after long-term exposure to a
small dose. Bull. environ. Contam. Toxicol., 24: 606-610.
TROTTIER, B.L., FRASER, A.R., PLANET, G., & ECOBICHON, D.J. (1980)
Subacute toxicity of technical fenitrothion in male rats.
Toxicology, 17: 29-38.
TSCHAPLINSKI, P.J. & GARDNER, D.R. (1981) Metabolism of fenitrothion
in red-backed voles Clethrionomys gapperi. Pestic. Biochem.
Physiol., 16: 47-62.
TSUDA, S., MIYAOKA, T., IWASAKI, M., & SHIRASU, Y. (1984)
Pharmacokinetic analysis of increased toxicity of
2- sec-butylphenyl methyl carbamate (BPMC) by fenitrothion
pretreatment in mice. Fundam. appl. Toxicol., 4: 724-730.
TSUJI, K., HORIDE, F., MINOBE, M., SASAKI, M., SHIRAGA, N., &
HIROAKI, O. (1980) Thermal decomposition of O,O-dimethyl
O-(3-methyl-4-nitrophenyl) phosphorothioate (Sumithion). J.
pestic. Sci., 5: 371-384.
TSUKIMOTO, K., HAYASHI, H., NONOGUCHI, H., BABA, M., TAZAWA, J.,
IWAMOTO, H., ABE, T., & SHIMADA, Y. (1981) [A case of delayed onset
of symptoms in seriously-poisoned female with Sumithion.] J. Jpn.
rural Med., 30: 598-559 (in Japanese).
UCHIYAMA, M., YOSHIDA, T., HOMMA, K., & HONGO, T. (1975) Inhibition
of hepatic drug-metabolizing enzymes by triphosphate insecticides
and its drug toxicological implications. Biochem. Pharmacol., 24:
1221-1225.
UEDA, K. & NISHIMURA, M. (1966) Interim report. Chronic toxicity of
Sumithion: two-year feeding study in rats, Tokyo Dental College,
Department of Hygiene and Toxicology, (Unpublished report submitted
to WHO by Sumitomo Chemical Co., Ltd, Osaka, Japan).
USDA (1976) Final environmental statement cooperative spruce budworm
suppression project in Maine, Washington, DC, US Department of
Agriculture, Forest Service (Environmental Statement of April 19,
1976).
VANDEKAR, M. (1965) Observations on the toxicity of carbaryl,
folithion and 3-isopropylphenyl n-methylcarbamate in a
village-scale trial in southern Nigeria. Bull. World Health Organ.,
33: 107-115.
VARTY, I.W. (1976) Environmental effects of the spruce budworm spray
program in New Brunswick 1976, Fredericton, Canada, Maritimes Forest
Research Centre (Information Report No. M-X-67).
VARTY, I.W. (1977) Long-term effects of fenitrothion spray programs
on non-target terrestrial arthropods. In: Proceedings of a Symposium
on Fenitrothion, Ottawa, National Research Council of Canada, pp.
344-375 (Publication NRCC No. 16073).
VELAZQUEZ, A., XAMENA, N., CREUS, A., & MARCOS, R. (1987)
Mutagenicity studies on fenitrothion in Drosophila. Mutagenesis, 2:
333-336.
VERMA, S.R., BANSAL, S.K., & DALELA, R.C. (1977) Bioassay trials
with a few organic biocides on fresh water fish Laveo rohita. Indian
J. environ. Health, 19: 107-115.
VERMA, S.R., BHATNAGAR, M.C., & DALELA, R.C. (1978a) Biocides in
relation to water pollution. Part 2: Bioassay studies of few
biocides to a fresh water fish Channa gachua. Acta hydrochim.
hydrobiol., 6: 137-144.
VERMA, S.R., BANSAL, S.K., & DALELA, R.C. (1978b) Toxicity of
selected organic pesticides to a fresh water teleost fish,
Saccobranchus fossilis and its application in controlling water
pollution. Arch. environ. Contam. Toxicol., 7: 317-323.
VERMA, S.R., BANSAL, S.K., GUPTA, A.K., PAL, N., TYAGI, A.K.,
BHATNAGER, M.C., KUMAR, V., & DALELA, R.C. (1982) Bioassay trials
with twenty three pesticides to a fresh water teleost,
Saccobranchus fossilis. Water Res., 16: 525-529.
VOLPE, G.G. & MALLET, V.N. (1980) Development of an analytical
method for fenitrothion and five derivatives in water using XAD
resins and gas-liquid chromatography (GLC). Int. J. environ. anal.
Chem., 8: 291-301.
VOLPE, G.G. & MALLET, V.N. (1981) High-performance liquid
chromatography of fenitrothion and seven derivatives - a study of
their recovery from water using XAD resins as compared with organic
solvents. Chromatographia, 14: 333-336.
WARREN, M.W., RUEBUSH, T.K., II, HOBBS, J.H., HIPPOLYTE, R., & MILLER,
S. (1985a) Safety measures associated with the use of organophosphate
insecticides in the Haitian malaria control programme. Bull. World
Health Organ., 63(2): 345-351.
WARREN, M.W., SPENCER, H.C., CHURCHILL, F.C., FRANCOIS, V.J.,
HIPPOLYTE, R., & STAIGER, M.A. (1985b) Assessment of exposure to
organophosphate insecticides during spraying in Haiti: monitoring of
urinary metabolites and blood cholinesterase levels. Bull. World
Health Organ., 63(2): 353-360.
WEINBERGER, P., GREENHALGH, R., SHER, D., & OUELLETTE, M. (1982a)
Persistence of formulated fenitrothion in distilled, estuarine, and
lake water microcosmos in dynamic and static systems. Bull. environ.
Contam. Toxicol., 28: 484-489.
WEINBERGER, P., GREENHALGH, R., MOODY, R.P., & BOULTON, B. (1982b)
Fate of fenitrothion in aquatic microcosmos and the role of aquatic
plants. Environ. Sci. Technol., 16: 470-473.
WHO(1973) Fenitrothion. In: Safe use of pesticides. Twentieth report
of the WHO Expert Committee on Insecticides, Geneva, World Health
Organization, pp. 18-19 (WHO Technical Report Series No. 513).
WHO/FAO (1977) Data sheets on pesticides No. 30: Fenitrothion,
Geneva, World Health Organization, 9 pp (VBC/DS/77.30).
WHO (1986) Environmental Health Criteria 63: Organophosphorus
insecticides: A general introduction, Geneva, World Health
Organization, 181 pp.
WHO (1990) The WHO recommended classification of pesticides by
hazard and Guidelines to classification 1990-1991, Geneva, World
Health Organization (WHO/PCS/90.1).
WILDISH, D.J. & LISTER, N.A. (1973) Biological effects of
fenitrothion in the diet of brook trout. Bull. environ. Contam.
Toxicol., 10: u333-339.
WILDISH, D.J. & LISTER, N.A. (1977) Effects of dietary fenitrothion
on growth and hierarchial position in brook trout (Salvelinus
fontinalis). Prog. Fish-Cult., 39: 3-9.
WILFORD, K., LIETEART, P.E.A., & FOLL, C.V. (1965) Fenitrothion:
Working paper prepared for an informal meeting of toxicology of
insecticides, Geneva, World Health Organization, pp. 18-23
(65/TOX/1).
WOOD, G.W. (1979) Recuperation of native bee. Population in
blueberry fields exposed to drift of fenitrothion from forest spray
operations in New Brunswick. J. econ. Entomol., 72: 36-39.
WOOD, G.W. (1980) Bee toxicology from fenitrothion aerosol,
Fredericton, Agriculture Canada Research Station, 60 pp.
WOOD, R.B., Jr & BOGDAN, G.F. (1986) Reye's syndrome and spruce
budworm insecticide spraying in Maine, 1978-1982. Am. J. Epidemiol.,
124: 671-677.
WOODWARD, D.F. & MAUCK, W.L. (1980) Toxicity of five forest
insecticides to cutthroat trout and two species of aquatic
invertebrates. Bull. environ. Contam. Toxicol., 25: 846-853.
WORTHING, C.R. & WALKER, S.B., ed. (1983) Fenitrothion. In: The
pesticide manual 7th ed., Croydon, The British Crop Protection
Council, p. 261.
WRIGHT, C.G., LEIDY, R.B., & DUPREE, H.E., Jr (1981) Insecticides in
the ambient air of room following their application for control of
pest. Bull. environ. Contam. Toxicol., 26: 548-553.
YADAV, A.S., VASHISHAT, R.K., & KAKAR, S.N. (1982) Testing of
endosulfan and fenitrothion for genotoxicity in Saccharomyces
cerevisiae. Mutat. Res., 105: 403-407.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1982a)
Comparison of the effect of an equimolar and low dose of
fenitrothion and methylparathion on their own metabolism in rat
liver. J. toxicol. Sci., 7: 35-41.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1982b)
Increase of adrenal weight in rats by the repeated administration of
fenitrothion. Toxicol. Lett., 11: 187-191.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1983a)
Effect of adrenalectomy, pretreatment with SKF 525-A, phenobarbital
and diethyl-maleate on the acute toxicity of fenitrothion in male
rats. Arch. Toxicol., 52: 233-242.
YAMAMOTO, T., EGASHIRA, T., YOSHIDA, T., & KUROIWA, Y. (1983b)
Comparative metabolism of fenitrothion and methylparathion in male
rats. Acta pharmacol. toxicol., 53: 96-102.
YAMANOI, F. (1980) [Effect of insecticides on the progeny in the
silkworm, Bombyx mori. I. Effect of organophosphorus insecticides
on egg laying and their hatching.] J. Sericult. Sci. Jpn, 49:
434-439 (in Japanese).
YAMANOI, F. (1981) [Effect of insecticides on the progeny in the
silkworm, Bombyx mori. II. Effect of organophosphorus insecticides
on the next generation.] J. Sericult. Sci. Jpn, 50: 83-87 (in
Japanese).
YASUNO, M., HATAKEYAMA, S., & MIYASHITA, M. (1980) Effect on
reproduction in Guppy (Poecilia reticulata) under chronic exposure
to temephos and fenitrothion. Bull. environ. Contam. Toxicol., 25:
29-33.
YASUNO, M., OKITA, J., SAITO, K., NAKAMURA, Y., HATAKEYAMA, S., &
KASUGA, S. (1981) Effect of fenitrothion on benethic fauna in small
stream of Mt. Tsukuba, Japan. Jpn. J. Ecol., 31: 237-245.
YOSHIDA, M., SHIMADA, E., YAMANAKA, H., AOYAMA, Y., YAMAMURA, Y., &
OWADA, S. (1987) A case of acute poisoning with fenitrothion
(Sumithion). Hum. Toxicol., 6: 403-406.
YULE, W.N. (1974) The persistence and fate of fenitrothion
insecticide in a forest environment II. Accumulation of residues in
balsam fir foliage. Bull. environ. Contam. Toxicol., 12: 249-252.
YULE, W.N. & DUFFY, J.R. (1972) The persistence and fate of
fenitrothion insecticide in a forest environment. Bull. environ.
Contam. Toxicol., 8: 10-18.
ANNEX I
TREATMENT OF ORGANOPHOSPHATE INSECTICIDE POISONING IN MAN
(From EHC 63: Organophosphorus Insecticides - A General
Introduction)
All cases of organophosphorus poisoning should be dealt with as
an emergency and the patient sent to hospital as quickly as
possible. Although symptoms may develop rapidly, delay in onset or a
steady increase in severity may be seen up to 48 h after ingestion
of some formulated organophosphorus insecticides.
Extensive descriptions of treatment of poisoning by
organophosphorus insecticides are given in several major references
(Kagan, 1977; Taylor, 1980; UK DHSS, 1983; Plestina, 1984) and will
also be included in the IPCS Health and Safety Guides to be prepared
for selected organophosphorus insecticides.
The treatment is based on:
(a) minimizing the absorption;
(b) general supportive treatment; and
(c) specific pharmacological treatment.
1.1 Minimizing the absorption
When dermal exposure occurs, decontamination procedures include
removal of contaminated clothes and washing of the skin with
alkaline soap or with a sodium bicarbonate solution. Particular care
should be taken in cleaning the skin area where venepuncture is
performed. Blood might be contaminated with direct-acting
organophosphorus esters and, therefore, inaccurate measures of ChE
inhibition might result. Extensive eye irrigation with water or
saline should also be performed. In the case of ingestion, vomiting
might be induced, if the patient is conscious, by the administration
of ipecacuanha syrup (10-30 ml) followed by 200 ml water. This
treatment is, however, contraindicated in the case of pesticides
dissolved in hydrocarbon solvents. Gastric lavage (with addition of
bicarbonate solution or activated charcoal) can also be performed,
particularly in unconscious patients, taking care to prevent
aspiration of fluids into the lungs (i.e., only after a tracheal
tube has been put into place).
The volume of fluid introduced into the stomach should be
recorded and samples of gastric lavage frozen and stored for
subsequent chemical analysis. If the formulation of the pesticide
involved is available, it should also be stored for further analysis
(i.e., detection of toxicologically relevant impurities). A
purgative can be administered to remove the ingested compound.
1.2 General supportive treatment
Artificial respiration (via a tracheal tube) should be started
at the first sign of respiratory failure and maintained for as long
as necessary.
Cautious administration of fluids is advised, as well as
general supportive and symptomatic pharmacological treatment and
absolute rest.
1.3 Specific pharmacological treatment
1.3.1 Atropine
Atropine should be given, beginning with 2 mg iv and given at
15-30-min intervals. The dose and the frequency of atropine
treatment varies from case to case, but should maintain the patient
fully atropinized (dilated pupils, dry mouth, skin flushing, etc.).
Continuous infusion of atropine may be necessary in extreme cases
and total daily doses up to several hundred mg may be necessary
during the first few days of treatment.
1.3.2 Oxime reactivators
Cholinesterase reactivators (e.g., pralidoxime, obidoxime)
specifically restore AChE activity inhibited by organophosphates.
This is not the case with enzymes inhibited by carbamates. The
treatment should begin as soon as possible, because oximes are not
effective on "aged" phosphorylated ChEs. However, if absorption,
distribution, and metabolism are thought to be delayed for any
reasons, oximes can be administered for several days after
intoxication. Effective treatment with oximes reduces the required
dose of atropine. Pralidoxime is the most widely available oxime. A
dose of 1 g pralidoxime can be given either im or iv and repeated
2-3 times per day or, in extreme cases, more often. If possible,
blood samples should be taken for AChE determinations before and
during treatment. Skin should be carefully cleansed before sampling.
Results of the assays should influence the decision whether to
continue oxime therapy after the first 2 days.
There are indications that oxime therapy may possibly have
beneficial effects on CNS-derived symptoms.
1.3.3 Diazepam
Diazepam should be included in the therapy of all but the
mildest cases. Besides relieving anxiety, it appears to counteract
some aspects of CNS-derived symptoms that are not affected by
atropine. Doses of 10 mg sc or iv are appropriate and may be
repeated as required (Vale & Scott, 1974). Other centrally acting
drugs and drugs that may depress respiration are not recommended in
the absence of artificial respiration procedures.
1.3.4 Notes on the recommended treatment
1.3.4.1 Effects of atropine and oxime
The combined effect far exceeds the benefit of either drug
singly.
1.3.4.2 Response to atropine
The response of the eye pupil may be unreliable in cases of
organophosphorus poisoning. A flushed skin and drying of secretions
are the best guide to the effectiveness of atropinization. Although
repeated dosing may well be necessary, excessive doses at any one
time may cause toxic side-effects. Pulse-rate should not exceed
120/min.
1.3.4.3 Persistence of treatment
Some organophosphorus pesticides are very lipophilic and may be
taken into, and then released from, fat depots over a period of many
days. It is therefore quite incorrect to abandon oxime treatment
after 1-2 days on the supposition that all inhibited enzyme will be
aged. Ecobichon et al. (1977) noted prompt improvement in both
condition and blood-ChEs in response to pralidoxime given on the
11th-15th days after major symptoms of poisoning appeared due to
extended exposure to fenitrothion (a dimethyl phosphate with a short
half-life for aging of inhibited AChE).
1.3.4.4 Dosage of atropine and oxime
The recommended doses above pertain to exposures, usual for an
occupational setting, but, in the case of very severe exposure or
massive ingestion (accidental or deliberate), the therapeutic doses
may be extended considerably. Warriner et al. (1977) reported the
case of a patient who drank a large quantity of dicrotophos, in
error, while drunk. Therapeutic dosages were progressively increased
up to 6 mg atropine iv every 15 min together with continuous iv
infusion of pralidoxime chloride at 0.5 g/h for 72 h, from days 3 to
6 after intoxication. After considerable improvement, the patient
relapsed and further aggressive therapy was given at a declining
rate from days 10 to 16 (atropine) and to day 23 (oxime),
respectively. In total, 92 g of pralidoxime chloride and 3912 mg of
atropine were given and the patient was discharged on the
thirty-third day with no apparent sequelae.
REFERENCES TO ANNEX I
ECOBICHON, D.J., OZERE, R.L., REID, E., & CROCKER, J.F.S (1977)
Acute fenitrothion poisoning. Can. Med. Assoc. J., 116: 377-379.
KAGAN, JU.S. (1977) [Toxicology of organophosphorus pesticides],
Moscow, Meditsina, pp. 111-121, 219-233, 260-269 (in Russian).
PLESTINA, R. (1984) Prevention, diagnosis, and treatment of
insecticide poisoning, Geneva, World Health Organization
(Unpublished document VBC/84.889).
TAYLOR, P. (1980) Anticholinesterase agents. In: Goodman, L.S. &
Gilman, A., ed. The pharmacological basis of therapeutics, 6th ed.,
New York, Macmillan Publishing Company, pp. 100-119.
UK DHSS (1983) Pesticide poisoning: notes for the guidance of
medical practitioners, London, United Kingdom Department of Health
and Social Security, pp. 41-47.
VALE, J.A. & SCOTT, G.W. (1974) Organophosphorus poisoning. Guy's
Hosp. Rep., 123: 13-25.
WARRINER, R.A., III, NIES, A.S., & HAYES, W.J., Jr (1977) Severe
organophosphate poisoning complicated by alcohol and terpentine
ingestion. Arch. environ. Health, 32: 203-205.
ANNEX II. No-observed-effect levels in plasma, red blood cells, and brain ChE, in animals treated with fenitrothion
ANNEX II. No-observed-effect levels in plasma, red blood cells, and brain ChE, in animals treated with fenitrothion
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Rat 30 days male 2.5 2.5 2.5 2.5 < 2.5 < 2.5 Trottier et al. (1980)
Gavage 0, 2.5, 5.0, 10.0, female 2.5 2.5 2.5 2.5 < 2.5 < 2.5
20.0 (mg/kg body weight)
Rat 5 weeksb male - - - 0.25 - 0.5 Carshalton (1964)
0, 5, 10, 20 mg/kg diet (5) (10)
Rat 90 daysb male 3.2 3.2 < 1.6 3.2 3.2 12.5 Misu et al. (1966)
0, 32, 63, 125, 250, 500 (63) (63) (< 32) (63) (63) (250)
mg/kg diet
Rat 90 days male - < 1.7 - < 1.7 - - Cooper (1966)
0, 20, 92.8, 430.7, 2000 (< 20) (< 20)
mg/kg diet female < 1.9 < 1.9 - -
(< 20) - (< 20)
Rat 90 days male - < 1.2 - 1.2 - - Cooper (1966)
0, 10, 46.4, 215, 1000 (< 10) (< 10)
(mg/litre water) female - < 1.3 - 1.3 - -
(< 10) (< 10)
Rat 6 months male - 1.8 - 1.8 - 1.8 Sumitomo Chem. Co.
0, 10, 30, 150 mg/kg diet (30) (30) (30) (1972)
female - < 0.64 - 0.64 - 0.64
(< 10) (< 10) (10)
Annex II (contd).
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Rat 34 weeksb male 0.25 0.25 < 0.25 < 0.25 - 6.3 Benes & Cerna (1970)
0, 10, 50, 150 (5) (5) (< 5) (< 5) (125)
0, 5, 25, 125 mg/kg diet female < 0.25 < 0.25 < 0.25 < 0.25 - 6.3
(< 5) (< 5) (< 5) (< 5) (125)
Rat 63 weeksb male - < 1.25 - 1.25 - 1.25 Ueda & Nishimura (1966)
0, 25, 100, 400 mg/kg diet (< 25) (25) (25)
Rat 1 year male 1.0 1.0 1.0 1.0 1.0 1.0 Ecobichon et al. (1980)
Gavage 0, 0.5, 1, 5, 10
(mg/kg body weight)
Rat 1 yearb male > 1.25 > 1.25 0.25 > 1.25 0.05 > 1.25 Kanoh et al. (1982)
0, 1, 5, 25 mg/kg diet (> 25) (> 25) (5) (> 25) (1) (> 25)
female > 1.25 > 1.25 0.25 > 1.25 0.25 > 1.25
(> 25) (> 25) (5) (> 25) (5) (> 25)
Rat 30 days male - 20 - 20 - 20 Breckenridge et al. (1982)
by inhalation 0, 6.7, female - < 6.7 - 20 - < 6.7
20, 60 (mg/m3)
Rat 92 weeksb male 0.13 > 0.5 0.25 > 0.5 - > 0.5 Kadota et al. (1975a)
0, 2.5, 5, 10 mg/kg diet (2.5) (> 10) (5) (> 10) (> 10)
female 0.13 > 0.5 0.25 > 0.5 - > 0.5
(2.5) (> 10) (5) (> 10) (> 10)
Annex II (contd).
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Rat 2 yearsb male < 0.5 < 0.5 0.5 0.5 0.5 1.5 Rutter & Nelson (1974)
0, 10, 30, 100 mg/kg diet (< 10) (< 10) (10) (10) (10) (30)
female < 0.5 < 0.5 0.5 0.5 0.5 1.5
(< 10) (< 10) (10) (10) (10) (30)
Mouse 2 yearsb male 0.43 0.43 1.43 1.43 1.43 1.43 Tamano et al. (1990)
0, 3, 10, 100, 1000 (3) (3) (10) (10) (10) (10)
mg/kg diet female 0.43 0.43 1.43 1.43 1.43 1.43
(3) (3) (10) (10) (10) (10)
Dog 98 days oral by male/ 2 - 2 - - - Cooper (1966)
capsule 0, 2, 9, 40
(mg/kg body weight)
Dog 1 yearb male 0.25 0.25 0.25 0.25 > 1.25 > 1.25 Griggs et al. (1984)
0.5, 10, 50 mg/kg diet (10) (10) (10) (10) (> 50) (> 50)
female 0.25 0.25 > 1.25 > 1.25 > 1.25 > 1.25
(10) (10) (> 50) (> 50) (> 50) (> 50)
Dog 10 months male - < 5 - < 5 - - Tomita et al. (1974)
oral by capsule
0, 5 (mg/kg body weight)
Dog 1 year female - < 2 - < 2 - - Ogata (1972)
oral by capsule
0, 2 (mg/kg body weight)
Annex II (contd).
Study/Dosage No-observed-effect levels in mg/kg body weight per day and in (mg/kg diet)a
Sex Plasma Red blood cells Brain Reference
I F I F I F
Dog 2 yearb male < 0.75 < 0.75 0.75 0.75 - 2.5 Mastalski et al. (1973)
0, 30, 100, 200 mg/kg diet (< 30) (< 30) (30) (30) (100)
female < 0.75 < 0.75 0.75 0.75 - 2.5
(< 30) (< 30) (30) (30) (100)
Rabbit 6 months male - <3 - < 3 - 3 Miyamoto et al. (1976a)
in diet, 0, 3, 10
(mg/kg body weight)
a I = Interim sacrifice; F = Final sacrifice.
b Calculated by dietary concentration providing that 1 mg/kg body weight per day equals 20, 7, and 40 mg/kg in
rats, mice, and dogs, respectively.
RESUME ET EVALUATION, CONCLUSIONS ET RECOMMANDATIONS
1. Résumé et évaluation
1.1 Exposition
Le fénitrothion est un insecticide organophosphoré utilisé
depuis 1959. On l'emploie en agriculture pour détruire les insectes
qui s'attaquent au riz, aux céréales, aux fruits, aux légumes, aux
céréales ensilées et au coton. On l'emploie également pour la
désinsectisation des forêts ainsi que pour la destruction des
mouches, des moustiques et des blattes dans le cadre des programmes
de santé publique. Il se présente sous la forme de concentrés
émulsionnables, de concentrés à très bas volume, de poudres, de
granulés, de poudres pour poudrage, de bouillies huileuses ainsi
qu'en association avec d'autres pesticides. La production de
fénitrothion oscille entre 15 000 et 20 000 tonnes par an.
Le fénitrothion pénètre dans l'air par volatilisation à partir
des surfaces contaminées et peut être entraîné pendant l'épandage
au-delà de la zone à traiter. Dans la plupart des terrains, il n'est
éliminé que très lentement par lessivage, néanmoins une certaine
quantité peut être entraînée par les eaux de ruissellement.
Le fénitrothion est décomposé par photolyse et hydrolyse. En
présence de rayonnement UV ou de lumière solaire, la demi-vie du
fénitrothion dans l'eau est inférieure à 24 heures. La présence
d'une microflore peut également accélérer la décomposition. En
l'absence de lumière solaire ou de contamination microbienne, le
fénitrothion est stable dans l'eau. Dans le sol, il est
essentiellement dégradé par voie biologique, encore que la photolyse
puisse jouer un certain rôle.
La concentration du fénitrothion dans l'air peut atteindre 5
µg/m3 immédiatement après l'épandage mais elle est susceptible de
diminuer fortement au cours du temps et en fonction de la distance
au lieu d'épandage. Dans l'eau, la concentration peut atteindre 20
µg/litre mais elle diminue rapidement.
En cas d'exposition continue, on observe des facteurs de
bioconcentration qui se situent entre 20 à 450 chez un certain
nombre d'espèces aquatiques.
Les résidus de fénitrothion dans les fruits, les légumes et les
céréales peuvent aller de 0,001 à 9,5 mg/kg immédiatement après le
traitement mais ils diminuent rapidement, leur demi-vie étant de 1 à
2 jours.
1.2 Fixation, métabolisme et excrétion
Le fénitrothion est rapidement résorbé dans les voies
digestives et se répartit ensuite dans les divers tissus. La
demi-vie du fénitrothion après absorption percutanée chez le singe a
été estimée à 15-17 heures. Sa métabolisation s'effectue selon les
grandes voies d' O-déméthylation ainsi que par clivage de la
liaison P-O-aryle. Le groupement NO2 est réduit par la microflore
intestinale, mais seulement chez les ruminants. La principale voie
d'élimination est la voie urinaire, la plupart des métabolites étant
excrétés en l'espace de 2 à 4 jours chez le rat, le cobaye, la
souris et le chien. Les principaux métabolites observés sont le
déméthyl- fénitrothion, le déméthyl-fénitrooxon, l'acide
diméthylphosphoro-thioïque et l'acide diméthylphosphorique ainsi que
le 3-méthyl-4-nitrophénol et ses conjugués. Les différences qui ont
été observées dans la composition en métabolites entre la plupart
des animaux de laboratoire et entre les sexes, chez une même espèce,
semblent être essentiellement d'ordre quantitatif. Seul le lapin
semble excréter du fénitrooxon et de l'aminofénitrooxon en quantités
faibles mais néanmoins mesurables, par la voie urinaire.
Des études portant sur des lapins et des chiens montrent que le
fénitrothion se dépose préférentiellement dans les tissus adipeux.
Des résidus qui avaient été décelés dans le lait de vache après
exposition à du fénitrothion ont disparu dans les deux jours.
Même s'il est rapidement résorbé après administration orale, le
fénitrothion est métabolisé et excrété sans délai et il est peu
probable qu'il demeure dans l'organisme pendant une longue période.
1.3 Effets sur les êtres vivants dans leur milieu naturel
A la concentration où on le rencontre vraisemblablement dans
l'environnement, le fénitrothion n'a aucun effet sur les
microorganismes présents dans le sol ou dans les eaux.
Il est extrêmement toxique pour les invertébrés aquatiques
d'eau douce ou d'eau de mer, la CL50 étant de l'ordre de quelques
µg/litre pour la plupart des espèces étudiées. La dose sans effet
observable pour la daphnie, lors d'épreuves à 48 heures, s'est
révélée < 2 µg/litre; lors de tests portant sur la totalité du
cycle évolutif, on a fixé à 0,14 µg/litre la concentration maximum
acceptable de produit toxique. Les observations et études effectuées
sur le terrain dans des étangs expérimentaux ont révélé l'existence
d'effets sur les populations d'invertébrés. Toutefois, la plupart
d'entre eux étaient passagers, même à des concentrations beaucoup
plus élevées que celles auxquelles donne vraisemblablement lieu
l'emploi de fénitrothion conformément aux recommandations.
Les poissons sont moins sensibles au fénitrothion que les
invertébrés, les valeurs de la CL50 à 96 heures dans leur cas
allant de 1,7 à 10 mg/litre. Ce sont les jeunes larves qui
constituent le stade le plus sensible. Des études à long terme ont
fixé à 0,1 mg/litre ou plus la concentration maximale admissible de
produit toxique pour deux espèces de poissons d'eau douce. Une étude
écologique effectuée après l'épandage de fénitrothion sur des forêts
a montré qu'il n'en résultait aucun effet sur les populations
sauvages de poissons ou sur la survie des poissons d'expérience
placés dans des nasses, les concentrations de fénitrothion dans
l'eau allant jusqu'à 0,019 mg/litre. Des épandages répétés de
fénitrothion sur des forêts n'ont eu aucun effet sur les poissons.
Lors d'essais en laboratoire, on a constaté que la CL50 pour
les mollusques dulçaquicoles allait de 1,2 à 15 mg/litre. Aucun
effet écologique n'a été constaté après épandage sur des forêts à la
dose de 140 g/hectare.
Le fénitrothion est extrêmement toxique pour les abeilles
(DL50 topique comprise entre 0,03 et 0,04 µg/abeille). Des effets
écologiques ont été observés qui consistaient essentiellement en une
mortalité localement élevée chez les abeilles et d'autres espèces.
Toutefois cette mortalité ne représentait qu'un faible pourcentage
de la population totale des ruches.
Pour les oiseaux, les valeurs de la DL50 aiguë par voie orale
vont de 25 à 1190 mg/kg de poids corporel et, dans la plupart des
cas, la CL50 alimentaire à huit jours dépasse 5000 mg/kg de
nourriture. Les valeurs de la dose sans effet observable sur la
reproduction sont de 10 mg/kg de poids corporel pour la caille et de
100 mg/kg pour le malard. Après épandage de fénitrothion à la dose
de 280 g/hectare, on a observé une mortalité parmi les oiseaux
chanteurs, mortalité qui augmentait sensiblement chez les espèces
peuplant la canopée lorsque la dose était portée à 500 g/hectare.
Après épandage à des doses de 420 g/hectare puis de 210 g/hectare
quelques jours plus tard, le nombre de nichées de pinsons à gorge
blanche (Zonotrichia albicollis) a diminué. Dans de nombreuses
études, on a constaté, chez les oiseaux chanteurs, une inhibition de
la cholinestérase peu après l'épandage de fénitrothion dans les
forêts.
L'observation sur le terrain n'a pas révélé d'effets sur les
populations de petits mammifères sauvages.
1.4 Effets sur les animaux d'expérience et les systèmes d'épreuve
in vitro
Le fénitrothion est un organophosphoré qui réduit l'activité de
la cholinestérase plasmatique, érythrocytaire, cérébrale et
hépatique. Il est métabolisé en fénitrooxon, composé dont la
toxicité aiguë est encore plus élevée. La toxicité du fénitrothion
peut être potentialisée par un certain nombre d'organophosphorés.
Le fénitrothion est un insecticide modérément toxique dont la
DL50 par voie orale chez le rat et la souris va de 330 à 1416
mg/kg de poids corporel. Sa toxicité aiguë par voie dermique chez
les rongeurs varie de 890 mg/kg de poids corporel à plus de 2500
mg/kg.
Le fénitrothion n'est que très peu irritant pour les yeux et
n'irrite pas la peau. Deux études portant sur des cobayes ont fait
ressortir une certaine tendance à la sensibilisation cutanée.
Le fénitrothion a fait l'objet d'études à court terme sur des
rats, des chiens, des cobayes, des lapins ainsi que d'études de
cancérogénicité à long terme chez des rats et des souris. Les études
à court terme sur les rats et les chiens ont permis de fixer
respectivement à 10 mg/kg de nourriture et 50 mg/kg de nourriture la
dose sans effet nocif observable, d'après l'activité de la
cholinestérase cérébrale.
Les études à long terme sur le rat et la souris ont donné une
dose sans effet nocif observable, basée sur l'activité de la
cholinestérase cérébrale, de 10 mg/kg de nourriture.
Aucune des études à long terme signalées n'a mis en évidence
d'effets cancérogènes.
Le fénitrothion ne s'est pas révélé mutagène dans les études
in vitro et in vivo.
A des doses allant jusqu'à 30 mg/kg de poids corporel chez le
lapin et jusqu'à 25 mg/kg de poids corporel chez le rat, le
fénitrothion n'a pas produit d'effets tératogènes. Lorsque la dose
dépassait 8 mg/kg de poids corporel, le fénitrothion était toxique
pour la mère.
Après exposition in utero, on a observé des déficits
comportementaux après la naissance chez des ratons en cours de
développement. La dose sans effet observable sur le comportement a
été fixée à 5 mg/kg de poids corporel et par jour.
Des études de reproduction portant sur plusieurs générations de
rats n'ont pas fait ressortir d'effets morphologiques. Ces études
ont permis de fixer à 120 mg/kg de nourriture, d'après les
paramètres génésiques, la dose sans effet nocif observable.
On a fait état d'une neurotoxicité retardée après exposition au
fénitrothion.
1.5 Effets sur l'homme
Administré à des volontaires humains en dose orale unique de
0,042 à 0,33 mg/kg de poids corporel puis en doses répétées de 0,04
à 0,08 mg/kg, le fénitrothion n'a pas entraîné d'inhibition de la
cholinestérase plasmatique ou érythrocytaire. Un des métabolites, le
3-méthyl-4-nitrophénol, a été totalement excrété par voie urinaire
dans les 24 heures.
Plusieurs cas d'intoxication se sont produits. La
symptomatologie de l'intoxication par le fénitrothion est celle
d'une stimulation du parasympathique. Il est arrivé que les
manifestations toxiques n'apparaissent pas immédiatement mais
récidivent pendant quelques mois. On a avancé que l'allongement de
la durée de l'évolution clinique de l'intoxication et l'apparition
de symptômes tardifs pouvaient s'expliquer par une libération lente
de l'insecticide à partir du tissu adipeux. Certaines dermatites de
contact ont été attribuées à une exposition à l'insecticide. Rien
n'indique qu'une exposition au fénitrothion puisse entraîner une
neurotoxicité retardée ou l'apparition d'un syndrome de Reye.
Dans le cadre des programmes de l'OMS, on utilise du
fénitrothion dans quelques pays pour la lutte antipaludique en
pulvérisation à effet rémanent à l'intérieur des habitations (dose
d'emploi: 2,0 gr de matière active par m2). Des observations
portant sur plusieurs milliers de résidents n'ont fait ressortir
aucun signe de toxicité à l'exception d'une enquête au cours de
laquelle 2% des personnes enquêtées se sont plaintes de légers
symptômes. Toutefois, chez environ 25% des ouvriers pulvériseurs, on
a observé une inhibition à 50% de l'activité de la cholinestérase du
sang total. Après épandage par voie aérienne d'un concentré
émulsionnable à 50%, on a constaté chez un certain nombre de
travailleurs des symptômes d'intoxication accompagnés d'une
réduction de l'activité cholinestérasique du sang total en l'espace
de 48 heures. Des ouvriers et des ouvrières exposés de par leur
profession pendant cinq ans à du fénitrothion respectivement dans un
atelier de production et dans une unité de conditionnement ont
présenté, pour 15% d'entre eux et 33% d'entre elles, des symptômes
cliniques d'intoxication. La concentration de fénitrothion dans
l'air des lieux de travail variait entre 0,028 et 0,118 mg/m3.
2. Conclusions
* Le fénitrothion est un insecticide organophosphoré modéré- ment
toxique. Toutefois, en cas d'exposition excessive résultant de
manipulations au cours de la production ou de l'épandage ou
encore par suite d'une ingestion accidentelle ou
intentionnelle, il peut s'ensuivre une grave intoxication.
* L'exposition de la population générale, qui résulte
principalement de l'emploi du fénitrothion en agriculture, en
foresterie et dans les programmes de santé publique, ne devrait
pas présenter de danger pour la santé.
* Moyennant de bonnes pratiques de fabrication et le respect des
mesures d'hygiène et de sécurité, le fénitrothion ne devrait
pas présenter de danger pour les personnes qui y sont exposées
de par leur profession.
* Malgré sa forte toxicité pour les arthropodes non visés, le
fénitrothion est utilisé très largement comme pesticide avec
des effets indésirables minimaux sur les populations animales
présentes dans l'environnement.
3. Recommandations
* Afin de préserver la santé et le bien-être des travailleurs et
de la population générale, il importe de ne confier la
manipulation et l'épandage du fénitrothion qu'à des personnes
expérimentées qui prendront les mesures de sécurité nécessaires
et procéderont à l'épandage en respectant les règles de bonne
pratique.
* Les opérations de production, de formulation, d'épandage et
d'élimination du fénitrothion doivent être effectuées avec tout
le soin nécessaire pour réduire au minimum la contamination de
l'environnement, et notamment des eaux de surface.
* Les travailleurs habituellement exposés doivent subir un examen
médical périodique.
* La dose d'emploi du fénitrothion doit être limitée afin
d'éviter tout effet sur les arthropodes non visés.
L'insecticide ne doit jamais être épandu sur des étendues ou
cours d'eau.
RESUMEN Y EVALUACION, CONCLUSIONES Y RECOMENDACIONES
1. Resumen y evaluación
1.1 Exposición
El fenitrotión es un insecticida organofosforado que se viene
utilizando desde 1959. Se emplea en la agricultura para combatir los
insectos del arroz, los cereales, las frutas, las hortalizas, el
grano almacenado y el algodón. Se usa también para luchar contra
insectos en los bosques, y en programas de salud pública para
combatir moscas, mosquitos y cucarachas. Se ha formulado como
concentrado emulsionable, concentrados de volumen muy bajo,
microgránulos, gránulos, polvo, pulverizadores oleosos y en
combinación con otros plaguicidas. Cada año se fabrican entre 15 000
y 20 000 toneladas.
El fenitrotión se incorpora al aire por volatilización a partir
de superficies contaminadas y puede ser arrastrado fuera de la zona
de tratamiento durante el rociamiento. Su lixiviación es muy lenta
en la mayoría de los suelos, pero es previsible que se produzca
algún arrastre en el agua.
Se degrada por fotólisis e hidrólisis. En presencia de la
radiación ultravioleta o de la luz solar, la semivida del
fenitrotión en el agua es inferior a 24 horas. La presencia de
microflora también puede acelerar la degradación. En ausencia de luz
solar o de contaminación microbiana, es estable en el agua. En el
suelo, la vía principal de eliminación es la biodegradación, aunque
la fotólisis también puede influir.
Las concentraciones de fenitrotión en el aire pueden ser de
hasta 5 µg/m3 inmediatamente después del rociamiento, pero
decrecen de manera considerable con el tiempo y la distancia del
lugar de aplicación. Los niveles en el agua pueden alcanzar valores
de hasta 20 µg/litro, pero disminuyen con rapidez.
Los factores de bioconcentración calculados en varias especies
acuáticas sometidas a una exposición constante al fenitrotión varían
entre 20 y 450.
Sus niveles residuales en frutas, hortalizas y grano de
cereales oscilan entre 0,001 y 9,5 mg/kg inmediatamente después del
tratamiento, pero decrecen rápidamente, con una semivida de 1 a 2
días.
1.2 Ingestión, metabolismo y excreción
El fenitrotión se absorbe con rapidez del tracto intestinal de
los animales de experimentación y se distribuye a diversos tejidos
corporales. La semivida en el caso de la absorción cutánea en el
mono fue de 15 a 17 horas. Se ha demostrado que su metabolismo se
realiza a través de las principales vías de demetilación oxidativa y
por rotura del enlace P-O-arilo. Los microorganismos intestinales
reducen, sólo en los rumiantes, el grupo nitrogenado del
fenitrotión. La principal vía de excreción es la orina; la mayor
parte de los metabolitos se eliminan en un periodo de 2 a 4 días en
el caso de la rata, el cobayo, el ratón y el perro. Los principales
metabolitos detectados son el demetilfenitrotión, el
demetilfenitrooxón, el ácido dimetilfosforotioico, el ácido
dimetilfosfórico y el 3-metil-4-nitrofenol y sus conjugados. Las
diferencias en la composición de los metabolitos que se detectan en
la mayor parte de los animales de laboratorio y entre los dos sexos
de la misma especie parecen ser principalmente de carácter
cuantitativo. Sólo los conejos parecen excretar fenitrooxón y
aminofenitrooxón en cantidades pequeñas, pero cuantificables, con la
orina.
En estudios realizados en conejos y perros se ha puesto de
manifiesto que el fenitrotión se deposita preferentemente en el
tejido adiposo.
Los residuos encontrados en la leche de vaca tras la exposición
no se detectaban dos días más tarde.
Aunque el fenitrotión se absorbe rápidamente por vía oral, se
metaboliza y excreta con rapidez y no es probable que permanezca en
el organismo durante un tiempo prolongado.
1.3 Efectos en los seres vivos del medio ambiente
La concentraciones normales de fenitrotión en el medio ambiente
no tienen efecto alguno en los microorganismos del suelo o del agua.
El fenitrotión es muy tóxico para los invertebrados acuáticos
de agua dulce y salada; los valores de la CL50 son de pocos
µg/litro en la mayoría de las especies analizadas. El nivel sin
efectos observados (NOEL) determinado en Daphnia en pruebas de 48
horas fue < 2 µg/litro; en pruebas del ciclo biológico se
estableció una concentración tóxica aceptable máxima (MATC) de 0,14
µg/litro. Las observaciones y los estudios sobre el terreno en
estanques experimentales han puesto de manifiesto ciertos efectos en
las poblaciones de invertebrados, si bien la mayor parte de los
cambios observados fueron transitorios, incluso con concentraciones
muy superiores a las que cabe esperar tras el uso recomendado.
Los peces son menos sensibles al fenitrotión que los
invertebrados y muestran valores de la CL50 a las 96 horas que
oscilan entre 1,7 y 10 mg/litro. La fase más sensible del ciclo
biológico es la larva joven. En estudios prolongados se ha
establecido una MATC igual o superior a 0,1 mg/litro para dos
especies de peces de agua dulce. Los estudios sobre el terreno tras
la aplicación de fenitrotión a bosques no demostraron efecto alguno
en las poblaciones silvestres de peces ni en la supervivencia de
peces de experimentación en vivero, con concentraciones medidas en
el agua de 0,019 mg/litro. La aplicación repetida en bosques no tuvo
efectos en las poblaciones de peces.
En pruebas de laboratorio, los valores de la CL50 para
moluscos de agua dulce oscilaban entre 1,2 y 15 mg/litro. No se
observaron efectos sobre el terreno tras rociar bosques con 140
g/ha.
El fenitrotión es muy tóxico para las abejas (DL50 tópica,
0,03-0,04 µg/abeja). Se han comunicado efectos sobre el terreno, con
un elevado número de abejas de la miel y de otras especies muertas
en la zona de aplicación. Sin embargo, el número total de individuos
muertos representaba solamente un pequeño porcentaje de la población
de las colmenas.
Los valores de la DL50 en la toxicidad aguda por vía oral en
las aves oscilan entre 25 y 1190 mg/kg de peso corporal, y en la
mayor parte de las dietas de ocho días las CL50 excedieron de 5000
mg/kg de la dieta. Los valores del NOEL para la reproducción fueron
de 10 mg/kg de peso corporal en la codorniz y de 100 mg/kg de peso
corporal en el pato silvestre. Poco después de aplicar fenitrotión
en una concentración de 280 g/ha, se observaron muertes de pájaros
cantores y la mortalidad aumentó considerablemente con 560 g/ha para
las especies que vivían en las copas de los árboles del bosque.
Después de rociar con una concentración de 420 g/ha y unos días más
tarde con 210 g/ha, se redujo la reproducción de la especie
zonotrichia albicollis. En muchos estudios, los pájaros cantores
mostraron inhibición de la colinesterasa inmediatamente después de
rociar los bosques con fenitrotión.
Las observaciones sobre el terreno no han puesto de manifiesto
efecto alguno del fenitrotión en las poblaciones de pequeños
mamíferos silvestres.
1.4 Efectos en los animales de experimentación y en sistemas
de prueba in vitro
El fenitrotión es un organofosfato y reduce la actividad de la
colinesterasa en el plasma, los eritrocitos y los tejidos cerebral y
hepático. Se metaboliza a fenitrooxón, con mayor toxicidad aguda.
Otros compuestos organofosfatados pueden potenciar aún más la
toxicidad del fenitrotión.
El fenitrotión es un insecticida de toxicidad moderada, con
valores de la DL50 por vía oral que oscilan entre 330 y 1416 mg/kg
de peso corporal en ratas y ratones. La toxicidad aguda cutánea en
roedores varió desde 890 mg/kg hasta más de 2500 mg/kg de peso
corporal.
El fenitrotión apenas irrita los ojos y no irrita la piel. En
uno de los dos estudios realizados en cobayos, el producto mostró
cierto potencial de sensibilización dérmica.
Se ha ensayado el fenitrotión en estudios de corta duración en
ratas, perros, cobayos y conejos y en estudios prolongados de
carcinogenicidad en ratas y ratones. En los estudios de corta
duración realizados en ratas y perros los niveles sin efectos
adversos observados (NOAEL), basados en la actividad de la
colinesterasa cerebral, fueron, respectivamente, de 10 mg/kg y 50
mg/kg de la dieta.
Los estudios de larga duración en ratas y ratones pusieron de
manifiesto un NOAEL (basado en la actividad de la colinesterasa
cerebral) de 10 mg/kg de la dieta.
No se han encontrado efectos carcinogénicos en ninguno de los
estudios de larga duración que se han publicado.
En los estudios in vitro e in vivo, el fenitrotión no
mostró efectos mutagénicos.
No se han detectado efectos teratogénicos con dosis de
fenitrotión de hasta 30 mg/kg de peso corporal en conejos y de hasta
25 mg/kg de peso corporal en ratas. Las dosis superiores a 8 mg/kg
de peso corporal produjeron toxicidad materna.
Las ratas jóvenes en fase de crecimiento presentaron problemas
de comportamiento postnatal tras la exposición in utero. Para este
efecto se estableció un NOEL de 5 mg/kg de peso corporal al día.
En estudios multigeneracionales de reproducción en ratas no se
detectó ningún efecto morfológico. En estos estudios se puso de
manifiesto un NOAEL de 120 mg/kg de la dieta, basado en parámetros
de la reproducción.
No se ha informado de neurotoxicidad retardada como
consecuencia de la exposición al fenitrotión.
1.5 Efectos en el ser humano
La administración de fenitrotión en una dosis oral única de
0,042 a 0,33 mg/kg de peso corporal y en dosis repetidas de 0,04 a
0,08 mg/kg de peso corporal en voluntarios humanos no causó la
inhibición de la colinesterasa del plasma y los eritrocitos. La
excreción urinaria del metabolito 3-metil-4-nitrofenol fue completa
en 24 horas.
Se han registrado varios casos de intoxicación, con los signos
y síntomas característicos de la estimulación parasimpática. En
algunos casos, las manifestaciones tóxicas tardaron en aparecer y se
repitieron durante unos meses. Se ha sugerido que la liberación
lenta del insecticida a partir del tejido adiposo puede dar lugar a
un curso clínico prolongado o a síntomas tardíos de intoxicación. En
algunos casos, la dermatitis de contacto se ha atribuido a la
exposición al insecticida. No hay pruebas de neurotoxicidad tardía
ni de relación con el síndrome de Reye.
El fenitrotión se ha utilizado en algunos países en programas
de la OMS para el rociamiento residual de interiores en la lucha
contra el paludismo (dosis de aplicación: 2,0 g de principio
activo/m2). No se apreciaron indicios de toxicidad en los miles de
habitantes observados, a excepción de un estudio en el que se
presentaron trastornos leves en menos del 2% de los habitantes. Sin
embargo, alrededor del 25% de los encargados del rociamiento
mostraron una inhibición de hasta el 50% de la actividad de la
colinesterasa en sangre total. En las 48 horas posteriores a la
aplicación aérea de una fórmula de concentrado emulsionable al 50%,
algunos trabajadores mostraron síntomas de intoxicación y una
disminución de la actividad de la colinesterasa en sangre total. En
una fábrica de producción, la exposición profesional durante más de
5 años de los trabajadores varones y de las mujeres del departamento
de envasado produjo signos clínicos y síntomas de intoxicación en el
15% de los hombres y en el 33% de las mujeres. La concentración de
fenitrotión medida en el aire del lugar de trabajo oscilaba entre
0,028 y 0,118 mg/m3.
2. Conclusiones
* El insecticida fenitrotión es un éster organofosforado
moderadamente tóxico. Sin embargo, la exposición excesiva
durante su fabricación o uso y la ingestión accidental o
intencional pueden ocasionar una intoxicación grave.
* La exposición de la población general, debida fundamentalmente
a las prácticas agrícolas y forestales y a los programas de
salud pública, no representa en principio amenaza alguna para
la salud.
* Con buenas prácticas de trabajo, medidas higiénicas y
precauciones de seguridad no es probable que el fenitrotión
represente un riesgo para las personas sujetas a exposición
profesional.
* A pesar de su elevada toxicidad para los artrópodos no
destinatarios, el fenitrotión se ha utilizado ampliamente en la
lucha contra las plagas, con pocos efectos adversos o ninguno
en las poblaciones presentes en el medio ambiente.
3. Recomendaciones
* Para salvaguardar la salud y el bienestar de los trabajadores y
de la población general, el manejo y la aplicación del
fenitrotión sólo se deben encomendar bajo una atenta
supervisión a personas bien capacitadas que se ajusten a las
medidas de seguridad adecuadas y utilicen el fenitrotión
correctamente.
* Se cuidarán especialmente la fabricación, la formulación, el
uso y la eliminación del compuesto, a fin de reducir al mínimo
la contaminación del medio ambiente, en particular de las aguas
superficiales.
* Los trabajadores regularmente expuestos deben someterse a
revisiones médicas periódicas.
* Se ha de limitar el número de aplicaciones de fenitrotión, para
evitar los efectos en artrópodos no destinatarios. No se deben
rociar jamás con este insecticida masas o corrientes de agua.