
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 177
1,2-Dibromoethame
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First draft prepared by Dr J. Sekizawa,
National Institute of Health Science, Japan
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization
World Health Organization
Geneva, 1996
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
1,2-Dibromoethame.
(Environmental health criteria ; 177)
1.Ethylene dibromide - adverse effects 2.Solvents
3.Environmental exposure I.Series
ISBN 92 4 157177 2 (NLM Classification: QV 633)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1996
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved. The designations
employed and the presentation of the material in this publication do
not imply the expression of any opinion whatsoever on the part of the
Secretariat of the World Health Organization concerning the legal
status of any country, territory, city or area or of its authorities,
or concerning the delimitation of its frontiers or boundaries. The
mention of specific companies or of certain manufacturers' products
does not imply that they are endorsed or recommended by the World
Health Organization in preference to others of a similar nature that
are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR 1,2-DIBROMOETHANE
Preamble
1. SUMMARY
1.1. Identity, physical and chemical properties, and analytical
methods
1.2. Sources of human and environmental exposure
1.3. Environmental levels and degradation
1.4. Kinetics and metabolism in laboratory animals
1.5. Effects on laboratory mammals and in vitro test systems
1.6. Effects on humans
1.7. Effects on organisms in the environment
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Air
2.4.2. Water
2.4.3. Soils and sediment
2.4.4. Food
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.1.1 World production figures
3.2.1.2 Manufacturing processes
3.2.2. Uses
3.2.2.1 Petrol additive
3.2.2.2 Fumigant
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Air
4.1.2. Soil
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.3. Food
5.2. Occupational exposure
6. KINETICS AND METABOLISM
6.1. Absorption
6.2. Distribution
6.3. Metabolic transformation
6.4. Elimination and excretion in expired air, faeces and urine
6.5. Retention and turnover
6.6. Reaction with body components
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Oral
7.1.1.1 Rat
7.1.1.2 Chicken
7.1.2. Inhalation
7.1.2.1 Rat
7.1.2.2 Guinea-pig
7.1.3. Intraperitoneal injection
7.1.3.1 Mouse
7.1.3.2 Rat
7.2. Short-term exposure
7.2.1. Oral
7.2.1.1 Chicken
7.2.2. Inhalation
7.2.2.1 Mouse
7.2.2.2 Rat
7.2.2.3 Guinea-pig
7.2.2.4 Rabbit
7.2.2.5 Monkey
7.3. Eye and skin irritation
7.3.1. Rabbit
7.4. Long-term exposure
7.4.1. Oral
7.4.1.1 Mouse
7.4.1.2 Rat
7.4.2. Inhalation
7.4.2.1 Mouse
7.4.2.2 Rat
7.5. Developmental toxicity
7.5.1. Reproduction
7.5.1.1 Effects on sperm
7.5.1.2 Effects on ova
7.5.2. Teratogenicity
7.5.2.1 Effects on neonatal behaviour
7.6. Mutagenicity and related end-points
7.6.1. In vitro assays
7.6.2. In vivo assays
7.6.3. Other studies
7.7. Carcinogenicity
7.7.1. Administration by gavage
7.7.1.1 Mouse
7.7.1.2 Rat
7.7.2. Administration in drinking-water
7.7.2.1 Mouse
7.7.3. Inhalation
7.7.3.1 Mouse
7.7.3.2 Rat
7.7.4. Dermal application
7.7.4.1 Mouse
7.7.5. Cell transformation
7.8. Biochemical studies and species specificity
8. EFFECTS ON HUMANS
8.1. Acute toxicity
8.2. Occupational exposure
8.2.1. Cancer incidence
8.2.2. Reproductive effects
9. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
9.1. Aquatic organisms
9.1.1. Invertebrates
9.1.2. Fish
9.2. Terrestrial biota
9.3. Microorganisms
9.4. Plants
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health risks
10.2. Evaluation of effects on the environment
11. CONCLUSIONS AND RECOMMENDATIONS
12. FURTHER RESEARCH
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (Telephone No. 9799111).
* * *
This publication was made possible by grant number 5 U01
ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial support
from the European Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and
in vitro studies provide support and are used mainly to supply
evidence missing from human studies. It is mandatory that research on
human subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary - a review of the salient facts and the risk evaluation
of the chemical
* Identity - physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph. Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson.
Observers do not participate in the final evaluation of the
chemical; this is the sole responsibility of the Task Group members.
When the Task Group considers it to be appropriate, it may meet
in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
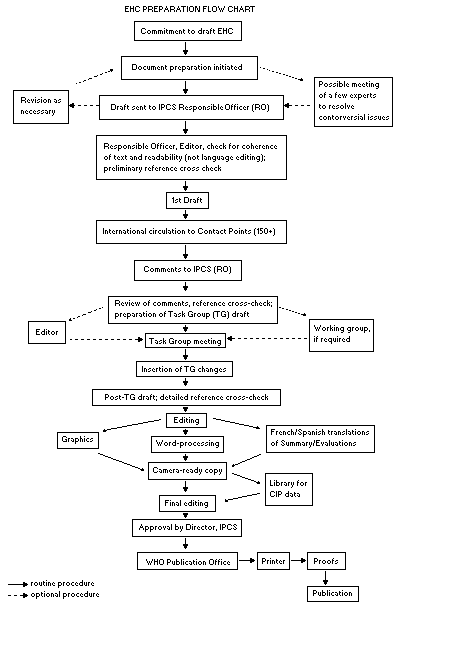 All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR 1,2-DIBROMOETHANE
Members
Dr T. Bailey, US Environmental Protection Agency, Washington DC, USA
Dr A.L. Black, Department of Human Services and Health, Canberra,
Australia
Mr D.J. Clegg, Carp, Ontario, Canada
Dr S. Dobson, Institute of Terrestrial Ecology, Monks Wood, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom
(Vice-Chairman)
Dr P.E.T. Douben, Her Majesty's Inspectorate of Pollution, London,
United Kingdom (EHC Joint Rapporteur)
Dr P. Fenner-Crisp, US Environmental Protection Agency, Washington DC,
USA
Dr R. Hailey, National Institute of Environmental Health Sciences,
National Institutes of Health, Research Triangle Park, USA
Ms K. Hughes, Environmental Health Directorate, Health Canada, Ottawa,
Ontario, Canada (EHC Joint Rapporteur)
Dr D. Kanungo, Central Insecticides Laboratory, Government of India,
Ministry of Agriculture & Cooperation, Directorate of Plant
Protection, Quarantine & Storage, Faridabad, Haryana, India
Dr L. Landner, MFG, European Environmental Research Group Ltd,
Stockholm, Sweden
Dr M.H. Litchfield, Melrose Consultancy, Denmans Lane, Fontwell,
Arundel, West Sussex, United Kingdom (CAG Joint Rapporteur)
Professor M. Lotti, Institute of Occupational Medicine, University of
Padua, Padua, Italy (Chairman)
Professor D.R. Mattison, University of Pittsburgh, Graduate School of
Public Health, Pittsburgh, Pennsylvania, USA
Dr J. Sekizawa, National Institute of Health Sciences, Tokyo, Japan
Dr P. Sinhaseni, Chulalongkorn University, Bangkok, Thailand
Dr S.A. Soliman, King Saud University, Bureidah, Saudi Arabia
Dr M. Tasheva, National Centre of Hygiene, Medical Ecology and
Nutrition, Sofia, Bulgaria (CAG Joint Rapporteur)
Mr J.R. Taylor, Pesticides Safety Directorate, Ministry of
Agriculture, Fisheries and Food, York, United Kingdom
Dr H.M. Temmink, Wageningen Agricultural University, Wageningen, The
Netherlands
Dr M.I. Willems, TNO Nutrition and Food Research Institute, Zeist, The
Netherlands
Secretariat
Ms A. Sundén Byléhn, International Register of Potentially Toxic
Chemicals, United Nations Environment Programme, Châtelaine,
Switzerland
Dr P. Chamberlain, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr J. Herrman, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr K. Jager, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
Dr P. Jenkins, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr W. Kreisel, World Health Organization, Geneva, Switzerland
Dr M. Mercier, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr M.I. Mikheev, Occupational Health, World Health Organization,
Geneva, Switzerland
Dr G. Moy, Food Safety, World Health Organization, Geneva, Switzerland
Mr I. Obadia, International Labour Organisation, Geneva,
Switzerland
Dr R. Plestina, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland (EHC Secretary)
Mr J. Wilbourn, International Agency for Research on Cancer, Lyon,
France
ENVIRONMENTAL HEALTH CRITERIA FOR 1,2-DIBROMOETHANE
The Core Assessment Group (CAG) of the Joint Meeting on
Pesticides (JMP) met at the World Health Organization, Geneva from
25 October to 3 November 1994. Dr W. Kreisel, Executive Director,
welcomed the participants on behalf of WHO, and Dr M. Mercier,
Director, IPCS, on behalf of the IPCS and its cooperating
organizations (UNEP/ILO/WHO). The Core Assessment Group reviewed and
revised the draft monograph and made an evaluation of the risks for
human health and the environment from exposure to 1,2-dibromoethane
(ethylene dibromide).
The preparation of the first draft of the monograph was
coordinated by Dr J. Sekizawa, National Institute of Health Sciences,
Japan. The second draft, revised in the light of international
comment, was prepared under the coordination of Dr Sekizawa.
Dr E. Smith and Dr P.G. Jenkins, both members of the IPCS Central
Unit, were responsible for the scientific content and technical
editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
The authors who contributed to the first draft were:
Dr C. Hashida The Jikei University School of Medicine, Japan
Dr Y. Hayashi National Institute of Health Sciences, Japan
Dr E. Kamata National Institute of Health Sciences, Japan
Dr Y. Kurokawa National Institute of Health Sciences, Japan
Dr A. Matsuoka National Institute of Health Sciences, Japan
Dr T. Matsushima The Japan Industrial Safety and Health
Association
Dr K. Morimoto National Institute of Health Sciences, Japan
Dr M. Nakadate National Institute of Health Sciences, Japan
Dr G. Ohmori The Jikei University School of Medicine, Japan
Dr Y. Saito National Institute of Health Sciences, Japan
Dr J. Sekizawa National Institute of Health Sciences, Japan
Dr T. Sohuni National Institute of Health Sciences, Japan
Dr M. Takeda National Institute of Health Sciences, Japan
Dr M. Takemura Ashiya University, Japan
Dr Y. Takenaka National Institute of Health Sciences, Japan
Dr S. Tanaka National Institute of Health Sciences, Japan
ABBREVIATIONS
BCF bioconcentration factor
BUN blood urea nitrogen
ECD electron capture detector
EDB 1,2-dibromoethane (ethylene dibromide)
FID flame ionization detector
GC gas chromatography
GSH glutathione
gamma-GT gamma-glutamyltranspeptidase
HECD Hall electron capture detector
LOEL lowest-observed-effect level
MS mass spectrometry
NADPH reduced nicotinamide adenine dinucleotide phosphate
NOEL no-observed-effect level
PIB piperonyl butoxide
SGOT serum glutamic-oxalic transaminase
SGPT serum glutamic-pyruvic transaminase
TEAM total exposure assessment methodology
TWA time-weighted average
UDS unscheduled DNA synthesis
VHH volatile halogenated hydrocarbon
VOC volatile organic carbon compound
1. Summary
1.1 Identity, physical and chemical properties, and analytical
methods
1,2-Dibromoethane (ethylene dibromide) is a colourless liquid
(melting point 9.9°C, boiling point 131.4°C) with a chloroform-like
odour. It is quite volatile, with a vapour pressure of 1.47 kPa
(11 mmHg) at 25°C and a vapour density compared to air of 6.1.
1,2-Dibromoethane is miscible with most organic solvents. Its
solubility in water is 4.3 g/litre at 30°C.
1,2-Dibromoethane in ambient air is analysed by GC after
absorption to porous polymers followed by rapid thermal desorption. A
purge-trap method is used for water samples. 1,2-Dibromoethane
residues in foods and other media can either be extracted by solvents
or be subjected to automated headspace analysis under cryogenic
conditions followed by analysis by GC and HPLC after derivatization.
1.2 Sources of human and environmental exposure
1,2-Dibromoethane is used as a scavenger of lead antiknock agents
in gasoline. It is also used as a soil fumigant and for fumigation of
grains and fruits. Reduced use of leaded gasoline in some countries
and cancellations of registrations for the use of 1,2-dibromoethane
for agricultural applications has reduced human exposure to
1,2-dibromoethane. However, it is still used as a lead scavenger in
gasoline in some countries, as a fumigant, for quarantine purposes, as
a solvent and as an intermediate for industrial chemicals.
1.3 Environmental levels and degradation
Concentrations of 1,2-dibromoethane measured in air range from
undetectable to the order of ng/m3 in urban areas. 1,2-Dibromoethane
has been found in ground water at up to 0.2 µg/litre and in surface
water at up to 50 µg/litre in areas of extensive agricultural use.
Although 1,2-dibromoethane leaches through soil, some is retained in
the soil matrix and may later contaminate irrigation wells. There is
a lack of information on microbial breakdown in soils.
The high volatility of 1,2-dibromoethane means that the major
environmental sink is the atmosphere. Stratospheric photolysis may
lead to the formation of breakdown products with ozone-depleting
potential.
1.4 Kinetics and metabolism in laboratory animals
1,2-Dibromoethane is rapidly absorbed orally, dermally and by
inhalation. Metabolites are thought to play an important role in
1,2-dibromoethane toxicity for humans. It can be metabolized by an
oxidative pathway (cytochrome P-450 system) and a conjugation pathway
(glutathione S-transferase system). Two reactive metabolites,
bromacetaldehyde formed via the oxidation pathway and thiiranium ion
formed via the conjugation pathway, are thought to interact with
cellular macromolecules (proteins, DNA) to form covalently bound
products.
1.5 Effects on laboratory mammals and in vitro test systems
1,2-Dibromoethane is acutely toxic to animals (oral LD50 for
rats of 146-417 mg/kg body weight, inhalation LC50 for rats of
3080 mg/m3 after a 2-h exposure, mortality observed following dermal
application of 210 mg/kg to rabbits). Toxic effects of
1,2-dibromoethane were mainly observed in the liver and kidneys.
Inhaled 1,2-dibromoethane vapour produced nasal irritation and
depression of the central nervous system. In rats exposed to
concentrations between 1540 and 77 000 mg/m3 (200-10 000 ppm) for
exposure durations between 0.1 and 16.0 h, deaths occurred in all
groups and were related to concentration and time. 1,2-Dibromoethane
(1.0% solution) caused irritation of shaved abdominal skin and eye
irritation in rabbits.
After oral subchronic exposure, mortality and decreases in weight
gain were observed in rats and mice at 100 mg/kg body weight per day.
Decreases in weight gain and nasal pathological effects were noted in
rats exposed to 1,2-dibromoethane at 115 mg/m3 (578 ppm) for
6 h/day, 5 days/week, for 13 weeks. The NOEL for histopathological
alterations of the nasal cavity was 23 mg/m3 (3 ppm) in this study.
In a similar study in mice, the same pathological changes were
observed, also with a NOEL of 23 mg/m3 (3 ppm).
After mice or rats were administered 1,2-dibromoethane by gavage
at 37-107 mg/kg body weight per day (TWA) for 49-90 weeks or mice were
administered 103-117 mg/kg body weight per day in drinking-water for
15-17 months, non-carcinogenic changes such as liver degeneration,
testicular atrophy, and forestomach acanthosis and hyperkeratosis in
addition to mortality were observed. After inhalation exposure (mice
or rats exposed to 77-388 mg/m3 for 6-18 months), inflammation of
the trachea and nasal cavity, testicular degeneration and hepatic
necrosis were observed.
1,2-Dibromoethane was not teratogenic in rats or mice following
inhalational exposure. Developmental toxicity (impairment of
development of motor coordination) was observed in the offspring of
male rats treated intraperitoneally with 1.25 mg/kg body weight per
day and in the offspring of female rats treated by inhalation
509 mg/m3, 4 h/day, 3 days/week from day 3 to day 20 of gestation.
1,2-Dibromoethane affected the reproductive performance of rats (in
males at the exposure level of 684 mg/m3, 7 h/day, 5 days/week, for
10 weeks, and in females at the exposure level of 614 mg/m3,
7 h/day, 7 days/week, for 3 weeks). The NOEL for this parameter was
300 mg/m3 in both sexes. The NOEL for reproductive performance of
male rats in a feeding study was 50 mg/kg per day after a 90-day
exposure. Spermatogenesis was affected in bulls following oral dosing
with 2 mg/kg per day for less than 21 days and in rabbits following
subcutaneous injection of 15 mg/kg for 5 days. Feeding of
1,2-dibromoethane caused diminution of egg size in hens after exposure
to 12.5 mg/kg per day for 12 weeks.
1,2-Dibromoethane did not induce dominant lethal mutations in
mice or rats, and did not produce chromosomal aberrations or
micronuclei in the bone-marrow cells of mice treated in vivo.
However, it was mutagenic in bacterial assays and caused single-strand
DNA breaks. Metabolites of 1,2-dibromoethane were covalently bound to
DNA, in vivo and in vitro. Sister chromatid exchange, mutations
and unscheduled DNA synthesis were observed in human cells in vitro.
Carcinogenicity studies involving oral administration (mice and
rats exposed by gavage to 37-107 mg/kg body weight per day (TWA) for
49-90 weeks; mice given 1,2-dibromoethane in drinking-water at
103-117 mg/kg body weight per day for 15-17 months), inhalational
exposure (mice and rats exposed at 10-40 ppm for 6-18 months) or skin
administration (25-50 mg/mice, 3 times/week for 400-594 days) showed
that 1,2-dibromoethane is carcinogenic to rats and mice, causing
tumours in a variety of organs (both at the application site and
distant sites, including the nasal cavity, lung, stomach, liver, skin,
circulatory system and mammary glands). In many cases it reduced the
latency period in developing tumours.
1.6 Effects on humans
1,2-Dibromoethane may produce adverse effects on the respiratory,
nervous and renal systems.
Acute (single) inhalation exposure to 1,2-dibromoethane at
215 mg/m3 (28 ppm) for 30 min or more has been shown to be fatal for
humans. Ingestion of 140 mg/kg body weight was fatal. Long-term
exposure to 1,2-dibromoethane (5 y) at a concentration of 0.68 mg/m3
in the breathing zone significantly decreased sperm counts and
fertility in occupationally exposed workers.
1.7 Effects on organisms in the environment
Few aquatic ecotoxicity studies have been performed with
1,2-dibromoethane. The LC50s for aquatic organisms are greater than
5 mg/litre. No information is available on terrestrial organisms.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Chemical name 1,2-dibromoethane
(IUPAC):
Chemical structure: Br - CH2 - CH2 - Br
Molecular formula: C2H4Br2
Relative molecular 187.9
mass:
CAS chemical name: ethylene dibromide
CAS registry number: 106-93-4
Synonyms: sym-dibromothane, DBE, dibromo, bromure
d'ethylene, 1,2-ethylene dibromide, ethylene
bromide
Major trade names: Nematron, Nemafume, Bromofume, Dowfume W-85,
Aadibrom, Iscobrome D
Formulations: kerosene (30 and 97%),
emulsifiable concentrate (40 and 48%)
in combination with other pesticides
2.2 Physical and chemical properties
Appearance: colourless liquid with chloroform-like odour
Melting point: 9.9°C (Stenger, 1983)
Boiling point: 131.4°C (Stenger, 1983)
Vapour pressure: 1.47 kPa (11.0 mmHg)
(at 25°C) (Verschueren, 1983)
Vapour density: 6.1
Specific gravity: 2.172 (Stenger, 1983)
(at 25°C)
Refractive index (n20): 1.5379
Solubility in water: 4.3 g/litre at 30°C (Verschueren, 1983)
soluble in ether, etanol, benzene, acetone
(Weast et al., 1988)
Saturating concentration 113 g/m3 (at 20°C), 168 g/m3 (at 30°C)
in air:
Log Pow 1.76 or 1.93
Stability: decomposes gradually when exposed to light
1,2-Dibromoethane is flammable. Chemically, 1,2-dibromoethane is
a bifunctional alkylating agent.
2.3 Conversion factors
1 ppm = 7.68 mg/m3 (at 25°C);
1 mg/m3 = 0.13 ppm
2.4 Analytical methods
Analytical methods for volatile halogenated hydrocarbons (VHH)
are applicable to 1,2-dibromoethane. Determination of
1,2-dibromoethane is usually carried out by gas chromatography with
electron capture detection (GC-ECD). High resolution GC capillary
columns can be used for multiple analysis in high-resolution gas
chromatography (HR-GC) or high-resolution gas chromatography - mass
spectrometry (HR-GC-MS). A sensitive photoionization detector (Dumas
& Bond, 1982; Collins & Barker, 1983), a Hall electroconductivity
detector (Cairns et al., 1984) or mass spectrometry can also be used
for determination and confirmation of 1,2-dibromoethane. GC-ECD is
the most sensitive method.
The preconcentration of trace 1,2-dibromoethane in samples is
usually carried out through collection by cryogenic trapping or by
absorption on solid absorbents. The former is the preferred
preconcentration technique. Ice formation in the trap-tube during
sampling can be a problem, especially with ambient water and
homogenized food samples. Co-collected water can alter sample or
column flow rates in separation techniques that require subfreezing at
initial GC oven temperature (Pleil et al., 1987).
2.4.1 Air
A convenient analytical method for trace levels of
1,2-dibromoethane in ambient air is a combination of preconcentration
by absorption on porous polymers, such as Tenax, Porapack, Florisil,
silica-gel or charcoal, followed by rapid thermal desorption and
direct application for GC. Tenax GC resin is widely used for
1,2-dibromoethane sampling in ambient air (Barkley et al., 1980; Clark
et al., 1982, 1984a,b; Krost et al., 1982; Harkov et al., 1984),
although Porapack, Chromosorb, silica gel and charcoal have also been
used extensively (Kojima & Seo, 1976; Jagielski et al., 1978; Mann et
al., 1980). 1,2-Dibromoethane is absorbed by passing air samples
through the columns followed by thermal desorption and direct
application to GC. Alternatively, 1,2-dibromoethane in air is
collected by cryogenic cooling in capillary trap-tubes and then
thermally desorbed for GC analysis using trap-ovens with carrier gases
(Barkley et al., 1980; Harkov et al., 1984; McClenny et al., 1984;
Ballschmiter et al., 1986).
The relatively high concentrations of 1,2-dibromoethane in or
near fumigation chambers for foods and in automobile exhaust gases can
be directly determined by sampling with a gas-tight syringe followed
by GC analysis (Hasanen et al., 1979; Dumas & Bond, 1982; Morris et
al., 1982; Collins & Barker, 1983).
Analytical methods for measuring 1,2-dibromoethane in ambient air
are summarized in Table 1.
2.4.2 Water
A purge-trap method using absorbents such as Tenax GC and
Amberlite XAD-4 resin is the most effective concentration technique
for recovering 1,2-dibromoethane from water samples before GC
analysis. The GC test solution is prepared by eluting the absorbent
columns with a small volume of hexane (Spingarn et al., 1982;
Stottmeister et al., 1986). Another method for 1,2-dibromoethane
concentration is direct absorption on organic resins like Amberlite
XAD-1, 2, 4, 7 and 8, and XE-340 (Libbey, 1986). Direct absorption of
water samples on, for example, Amberlite XAD resins can be used for
the concentration of 1,2-dibromoethane in aquatic media (Libbey, 1986;
Woodrow et al., 1986).
Solvent extraction and headspace collection are simple methods
for recovering 1,2-dibromoethane from water samples (Saito et al.,
1978; Keough et al., 1984; Koida et al., 1986).
Analytical methods for measuring 1,2-dibromoethane in water are
summarized in Table 2.
2.4.3 Soils and sediment
1,2-Dibromoethane in sediments can be concentrated by a purge-
trap procedure, either after dilution of sediment samples with water
or after vacuum extraction from sediment samples into a cryogenically
cooled trap (Amin & Narang, 1985).
Analytical methods for measuring 1,2-dibromoethane in soils are
summarized in Table 3.
Table 1. Analytical methods for 1,2-dibromoethane in air
Collection from air Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected on Chromosorb extracted with hexane 1.5% OV-17+1.95% OV-210; 100 pg ECD Mann et al.
101 cartridge (20 ml); extracted with column temp: 75°C; in 70% FSRD (1980)
(10 mm i.d. × 10 cm) 1% MeOH in benzene, gas flow: N2
(ambient air) extract kept in a 70 ml/min
screw-capped test tube
and injected into GC
Collected directly with a applied directly with 5.5% DC-200+11% GF-1/Ga 0.1 ng ECD Morris et al.
gas-tight syringe gas-tight syringe Chrim Q (0.3 mm o.d. × 150 cm, (1982); Morris
stainless steel); column temp: & Rippon (1985)
90°C; gas flow: 40 ml/min
Collected directly with a applied directly with 5% Carbowax 20M/Chromosorb 2 µg PID Dumas &
gas-tight syringe gas-tight syringe W (3 mm i.d. × 200 cm, stainless FSRD Bond (1982)
steel); column temp: 120°C;
gas flow: N2 30 ml/min (portable
gas chromatograph)
Collected with a gas-tight applied by direct CPS-20 M (1/8- × 4 tefron tube); 1 ppb PID Cairns et al.
syringe (ambient air) injection column temp: ambient temp; (1984)
Collected on Tenax GC applied by rapidly Fused silica SP-2000 FSOT not given ECD Harkov et al.
cartridge at a flow rate of raising trap oven GC/MS (1984)
approx.300-1000 ml/min for temperature to 140°C
24 h; desorbed from the with purge of high
cartridge by heating purity N2 (50 ml/min)
rapidly at 250°C and
collected in an evacuated
stainless steel cylinder
under cryogenic conditions
using vacuum distillation
for 30 min (ambient air)
Table 1 (cont'd)
Collection from air Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected on a double applied by rapidly OV-1 fused silica capillary 1 ppb ECD McClenny et
loop of 0.32 mm (o.d.) heating the tube (-150 column (0.32 mm i.d. × 50 m); al. (1984)
nickel tubing packed with to 100°C in 55 sec) and column temp: -50°C (3 min)
60-80 mesh Pyrex beads cooling quickly (120 8°C/min 150°C (7 min)
under cryogenic conditions to -150°C in 225 sec) -50°C (10 min); gas flow:
(-150°C) (ambient air) H2 4 ml/min
a ECD = electron caputure detector; PID = photoionization detector; GC = gas chromatography; MS = mass spectrometry;
FSRD = full-scale recorder deflection
Table 2. Analytical methods for 1,2-dibromoethane in water
Collection Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Headspace method applied on a fused 1.J & W FSOT (0.326 mm i.d. × 30 m); 1.8 pg GC/MS Keough et al.
silica line Temp: column 70°C, ion source 299°C; (1984)
(0.25 mm i.d. × 100 cm) split ratio: 1:10; gas flow:
at 250-275°C with a 1.5 ml/min; ion dwell time:
gas-tight syringe 100 msec;
2.OV-1 FSOT (0.32 mm i.d. × 50 m); not given ECD
column temp: 60-90°C; split ratio:
1:1; gas flow: He 10 ml/min
N2 100 ml/min
Extracted with applied directly with a 1.5% OV-17 on Chromosorb W 0.05 mg/litre GC/MS Koida et al.
hexane micro-syringe (3 mm i.d. × 150 cm); temp: column (CI-NID) (1986)
200°C; separator 120°C; ion source
200°C; reaction gas: isotutane;
gas flow: H2 20 ml/min
Collected by purging applied by rapidly 0.2% Carbowax 1500 on Carbopack C 0.3 mg/litre FID Stottmeister et
into purge-trap thermal desorption (3 mm i.d. × 200 cm); column temp: al. (1986)
tubing (Tenax GC (200°C) 35°C (4 min), (8°C/min) 170°C
155 mg) at flow (20 min); gas flow: N2 4 ml/min
rate of 50 ml/min
for 30 min
Table 2 (cont'd)
Collection Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected by absorption extracted with ether 1.DB-1701 FSOT(0.25 mm i.d. × 30 m); 1 ppt ECD Woodrow et
on Amberlite and concentrated in column temp: 230°C; split ratio: al. (1986)
XAD-4 cartridge Kudernadanish 1:10; gas flow: H2 19 ml/sec at
column ball concentrator with 3 230°C; make-up gas Ar/CH4 20 ml/sec;
(4.7 mm o.d. × 12 cm) at Snyder column; applied 2.SE-54 FSOT (0.25 mm i.d. × 30 m);
flow rate of 10 ml/min with a microsyringe column temp: 100°C (5°C/min)
for 18-24 h 250°C (other conditions described
above)
a ECD = electron capture detector; GC = gas chromatography; MS = mass spectrometry; FID = flame ionization detector;
CI = chemical ionization; NID = negative ion detector
Table 3. Analytical methods for 1,2-dibromoethane in soil
Collection Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected by steam fortify 50 ml of the not given ECD Abdel-Kader
distillation of an extract to folder paper et al. (1979)
aqueous slurry of soil and then measure with
into dry-ice-cooled molecular emission cavity
solvent (acetone: analyser; 4 mm × 4 mm
isooctane = 1:1) deep stainless steel
under N2 stream at cavity; gas flow:
60°C and drying H2 2.5 litre/min;
over Na2SO4 N2 4.0 litre/min;
wave length: 376 nm;
split: 1.4 nm
Collected on Prapack apply by thermal 4% OV-11 & 6% SP 2100 Supelcopor 7 ppb PID Amin &
N by purge of desorption of the (2.0 mm × 2.7 m); column temp: 40°C Narang (1985)
an aqueous slurry absorbent spiked with (5 min), (3°C/min) 70°C; gas flow:
of soil for 30 min fluorobenzene as an N2 30 ml/min, 15% SF-95 & 6% OV-225 1 ppb ECD
internal standard on Chromosorb W (2 mm i.d. × 3.6 m);
column temp: 60°C (10 min),
(3°C/min) 80°C (10 min) 65°C-75°C
(isothermal)
DB-5 FSOT(0.25 mm i.d. × 60 m) PID
column temp: 50°C (15 min) (4°C/min) ECD
170°C (14 min); gas flow:
He 60 cm/sec make-up gas He 8 ml/min
a ECD = electron caputure detector; PID = photoionization detector; GC = gas chromatography; FID = flame ionization detector
2.4.4 Food
Continuous extraction with hexane in a Dean-Stark apparatus for
one hour or soaking in a solvent solution of ethanol or acetone and
water (1 : 5) for 2 or 3 days is used for the analysis of
1,2-dibromoethane in agricultural crops and their products. A highly
sensitive method for the analysis of 1,2-dibromoethane in flour and
biscuits was developed by Rains & Holder (1981). Continuous
extraction with hexane is used for fruit, vegetables and grains
(Sekita et al., 1981, 1983; Kato et al., 1982; Iwata et al., 1983;
Konishi et al., 1985; De Vries et al., 1985; Alleman et al., 1986;
Nakamura, 1986).
Soaking in aqueous ethanol or acetone and water solution is used
for grains and their products (Clower, 1980; Daft, 1983, 1985, 1987;
Cairns et al., 1984; Barry & Petzinger, 1985; Sawyer & Walters, 1986;
Clower et al., 1986). For fruit (papaya and lemon), hexane, hexane-
water and acetonitrile are used. In the case of grain, intermediate
products, ready-to-eat products, corn bread mix, baby cereal and
bread, 1,2-dibromoethane can be extracted with an acetone-water (5+1)
solution, 0.1N HCl or light petroleum. Where necessary, Florisil
cleanup is useful for the removal of materials interfering with GC
analysis.
The purge-trap method on layers of Tenax TA (Heikes, 1985a,b),
Tenax resins (GC and TA) or Amberlite XAD-4 (Heikes & Hopper, 1986;
Daft, 1988) under a nitrogen gas stream is also used for the
collection of 1,2-dibromoethane from grains and their products. The
resins are eluted with hexane.
Automated headspace analysis is employed for measurement of
1,2-dibromoethane in fumigated crops in combination with GC-ECD and
GC-MS (Mestres et al., 1980; Entz & Hollifield, 1982; Gilbert et al.,
1985; Pranoto-Soetardhi et al., 1986). Equilibrium partitioning
between the samples and the gaseous headspace can be accelerated by
warming the vials. For detection, the gas chromatographic method with
an electron capture detector is used in all the above-mentioned
methods. Gas chromatography-mass spectrometry is also used. The
detection limit ranges from 0.1 µg/kg to a few µg/kg according to the
method used and the food being tested.
1,2-Dibromoethane can be analysed in animal feed by continuous
extraction with hexane in a Dean-Stark apparatus, followed by cleanup
on a Florisil column (Ishikuro, 1986).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
1,2-Dibromoethane is not a naturally occurring substance.
3.2 Anthropogenic sources
3.2.1 Production levels and processes
3.2.1.1 World production figures
In 1975, 1400 tonnes was produced in Japan. Production in
Belgium, France, Italy, the Netherlands, Spain, Switzerland and the
United Kingdom, was estimated to be between 3000 and 30 000 tonnes
(IARC, 1977).
3.2.1.2 Manufacturing processes
1,2-Dibromoethane is made by direct bromination of ethylene or
reacting hydrobromic acid with acetylene (Roskill, 1992).
3.2.2 Uses
Major uses of 1,2-dibromoethane are as a lead scavenger in
tetraalkyllead petrol and antiknock preparations, as a soil and grain
fumigant, as an intermediate in the synthesis of dyes and
pharmaceuticals, and as a solvent for resins, gums and waxes (IARC,
1977).
Reduction in the use of leaded gasoline from the late-1970s in
developed countries and of 1,2-dibromoethane for agricultural
applications in the 1980s, owing to its carcinogenicity in animals,
reduced human exposure to 1,2-dibromoethane. However, it is still used
in large amounts for many industrial purposes in developed countries,
and as a petrol additive in developing countries.
3.2.2.1 Petrol additive
1,2-Dibromoethane has been added to scavenge the inorganic lead
compounds (e.g., lead oxide and sulfate) remaining after fuel
combustion. Lead accumulation is prevented by the reaction of
1,2-dibromoethane with lead oxide to form volatile lead bromide, which
can pass from the combustion chamber to the atmosphere (IARC, 1977).
In 1981, use as a lead scavenger represented 83% of the
1,2-dibromoethane consumed (SRI International, 1982).
In 1972, 122 000 tonnes 1,2-dibromoethane was added to petrol
formulations in the USA; this figure declined to 73 000 tonnes in 1980
and to 24 000 tonnes in 1992 (Roskill, 1992). In 1992, sales of
unleaded petrol accounted for more than 90% of petrol in the USA. In
the European Community, all new vehicles must be fitted with
three-way convertors that can only use unleaded petrol by the
mid-1990s. This is also true of Japan, where almost all cars run on
unleaded petrol (Roskill, 1992).
In 1992, demand for 1,2-dibromoethane as a gasoline additive in
the USA was 24 000 tonnes and consumption outside the USA, principally
in Europe, was 25 000 to 30 000 tonnes, giving an estimated world
demand of 49 000 to 54 000 tonnes. The amount of 1,2-dibromoethane
used in Germany in 1989 was 980 tonnes, calculated on the basis of the
petrol consumed in the Federal Republic of Germany in 1989 (BUA,
1991). Legislation banning the use of lead in gasoline and controlling
the agricultural use of 1,2-dibromoethane has reduced world demand for
1,2-dibromoethane by at least 75% (Roskill, 1992).
3.2.2.2 Fumigant
The volatility of 1,2-dibromoethane allows it to be distributed
as a gas through substances such as soil in sufficiently high
concentrations to kill target pests. Its chemical and biocidal
properties allowed it to be effectively utilized in a wide range of
applications. Its primary pesticidal use has been as a soil
nematocide (Pignatello & Cohen, 1989).
1,2-Dibromoethane has been used in the spot fumigation of grain
milling machinery, post-harvest fumigation of grain, and in the
control and prevention of infestations in produce. Additional minor
uses have been the control of bark beetles in felled logs, moths in
stored furniture and clothing, termites under concrete slab
foundations and porches, Japanese beetles in balled ornamental trees
and grass sod, and wax moths in stored honeycombs and beehive
superstructures.
In post-harvest grain fumigation of barley, maize, oats, rice,
rye, sorghum and wheat, 1,2-dibromoethane has often been used in
conjunction with 1,2-dichloroethane (ethylene dichloride) or carbon
tetrachloride.
Residues of 1,2-dibromoethane in tropical fruits, imported wheat
and beans have been prohibited in Japan (MHW, 1985, 1987, 1988). Use
of 1,2-dibromoethane for agricultural purposes has been prohibited in
Egypt, Kenya, the Netherlands, Sweden, the United Kingdom and the USA
(BUA, 1991; IRPTC, 1993). However, it is still used for quarantine
purpose in some countries.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
The use of 1,2-dibromoethane on a field contaminated both the
field and crops for 2 years (Yuita, 1984). About 10% of bromine-
containing pesticides was retained, in the form of bromine, in the
soil and crops. The remaining 90% seemed to have moved to underground
water and rivers.
4.1.1 Air
The atmospheric chemistry of bromine compounds has received
attention because of the role that they play in the depletion of the
stratospheric ozone layer. Wofsy et al. (1975) suggested that bromine
atoms can be more effective than chlorine atoms in the catalytic
destruction of ozone. A major uncertainty is the absolute
concentration of bromine compounds in both the troposphere and the
stratosphere.
4.1.2 Soil
Injection of 1,2-dibromoethane as a soil fumigant at 70 kg/ha
into fine sandy loam resulted in a concentration of 130 µg/kg nearly
one year later (Steinberg et al., 1987).
The disappearance with time of 1,2-dibromoethane was measured in
a sediment-water mixture (ratio 0.075) and a half-life of 55 h was
calculated (Jafvert & Wolfe, 1987).
Important factors influencing the movement of soil fumigants
include their physical and chemical characteristics, temperature,
moisture, presence of organic matter, soil texture and soil profile
variability (Munnecke & Van Gundy, 1979).
1,2-Dibromoethane is moderately hydrophillic, having a calculated
octanol-water partition coefficient of 58 (Lyman, 1982). At
environmental levels (10-1000 ppb), 1,2-dibromoethane has a soil
organic carbon partition coefficient of 66 ml/g (Rogers & McFarlane,
1981).
1,2-Dibromoethane has a low vapour pressure and moves slowly in
the vapour phase. Little, if any, mass flow occurs except in
extremely warm soil or when water is applied. Soil temperature is
important and may affect 1,2-dibromoethane movement in several ways.
A rise in temperature increases the vapour pressure and decreases the
solubility. This alters the phase distribution and results in an
increase in the rate of diffusion of 1,2-dibromoethane through soils.
Fumigation of warm soils (25°C) results in a faster rate and greater
distance of nematicide diffusion. In colder soils (5°C), the rate of
diffusion is slower and the persistence of the chemical is longer, but
the total distance of diffusion of an effective dosage is decreased.
The approximate movement and fate of 1,2-dibromoethane in two soils
were predicted using extrapolations from laboratory experiments and
soil-vapour phase concentrations obtained from simulated field
experiments. The most far-reaching diffusion patterns in mineral
soils are those obtained in soils whose moisture content is nearest
the wilting point of plants (15 bars moisture tension). As the
moisture content of the soil is increased, the diffusion pattern
gradually becomes more limited. The soil texture and type determine
to a large extent the amount of soil moisture present and the size of
the connecting air spaces. Soil air space and the size of pores are
important because these chemicals move primarily in the vapour phase
and smaller pores are most easily blocked when water is present. A
material balance for 1,2-dibromoethane was surveyed when
1,2-dibromoethane (equivalent to 47 litres/ha) was applied under
various conditions to several soils using a soil fumigation technique
in both field and laboratory experiments. Most of the
1,2-dibromoethane was accounted for; the remainder was mostly
irreversibly adsorbed or lost during sampling. The 1,2-dibromoethane
not accounted for represented between 10 and 40%. After 3 days at
15°C, about 40% of the 1,2-dibromoethane was absorbed in the
soil-particle phase, 25% was in the soil-water phase, and 20% remained
in the liquid state (McKenry & Thomason, 1974).
1,2-Dibromoethane soil fumigation is used for the control of
plant parasitic nematodes on high value crops. In Ontario, Canada,
soil types fumigated varied from loamy sand to muck. Three soils
differing in texture (Fox loam sand, Vineland silt loam and Lincoln
clay) were studied for penetration of 1,2-dibromoethane (Townshend et
al., 1980). Fox loam sand (highest content of sand and lowest of
organic matter) showed the most rapid penetration; moisture level,
temperature and their interactions had the greatest effects on
movement of 1,2-dibromoethane. On Vineland silt loam (medium-textured
soil) the degree of penetration was dependent on moisture, temperature
and bulk density, and there were relatively small interaction effects.
On Lincoln clay (high content of organic matter and fine-textured
soil) 1,2-dibromoethane did not move in the soil, regardless of
edaphic factors, thus explaining the difficulty of using
1,2-dibromoethane fumigation to control nematodes in clay.
1,2-Dibromoethane persists in top soil at µg/kg levels for at
least 20 years, despite its predicted lability in the environment
(high water solubility and low soil-water partition coefficient).
Misleading results were obtained when studies of microbial
degradation, sorption, desorption and analytical recovery were
conducted with freshly spiked soils or sediments (Pignatello, 1986).
1,2-Dibromoethane can serve as a C1 unit and energy source for some
soil aerobic or anaerobic microorganisms. However, residual
1,2-dibromoethane is strongly bound to soils and can only be extracted
from them by warming with polar solvents. Surfactants showed no
enhanced extraction ability. Thermal desorption at temperatures as
high as 200°C in an N2 stream resulted in more decomposition than
desorption, while a fresh spike of 14C-labelled 1,2-dibromoethane
was recovered quantitatively.
Diffusion of residual 1,2-dibromoethane from soil to water is
very slow and highly temperature-dependent (diffusion coefficient:
10-16 cm2/sec) (Pignatello et al., 1987). 1,2-Dibromoethane, when
present as a groundwater contaminant in areas where it had been used
as a soil fumigant, was degraded anaerobically by microorganisms in
two types of soils from 1,2-dibromoethane-contaminated groundwater
discharge areas. At initial concentrations of 6 to 8 µg/litre,
1,2-dibromoethane was degraded in a few days to near or below the
detection limit (0.02 µg/litre). At 15 to 18 µg/litre degradation was
slow. Bromide ion released at the higher concentration was 1.4 ± 0.3
and 23.1 ± 0.2 molar equivalents for the two soil types. A study
using 14C-1,2-dibromoethane showed that 1,2-dibromoethane was
converted to approximately equal amounts of CO2 and cellular carbon;
only small amounts of 14C were not attributable to these products.
However, 1,2-dibromoethane was not degraded in autoclaved soil water
samples. The results suggested that, initially, microbial degradation
of 1,2-dibromoethane in the topsoil was too slow to prevent leaching
of large quantities to groundwater. With continued application the
microbial community may have adapted to the higher levels and
degradation rates increased; this has been observed with other
agricultural chemicals. The results of acetate incorporation studies
suggested that the highest application rates of 1,2-dibromoethane are
definitely toxic to topsoil microbial communities.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
1,2-Dibromoethane enters the atmosphere from its use as a petrol
additive to scavenge the lead oxide resulting from the combustion of
alkyllead antiknock additive, and from its use in agriculture as an
insecticidal and fungicidal fumigant.
Nilsson et al. (1987) reported that the exhaust gas of chain saws
fuelled with petrol contained a mean 1,2-dibromoethane level of
0.0008 (0.0001-0.001) mg/m3 under snow-free conditions and
0.002 (0.0001-0.005) mg/m3 with snow on the ground during the
winter. 1,2-Dibromoethane levels are around 45% higher in cold start
than in hot start conditions and have a tendency to decrease with
increasing vehicle speed (see Tables 4 and 5). Concentrations of
1,2-dibromoethane in raw, undiluted exhaust from vehicles using leaded
petrol are in the range of 55-146 µg/m3 (7.2-19 ppb), 46-122 µg/m3
(6.0-16 ppb) and 38-115 mg/m3 (5.0-15 ppb) under the conditions of
USA Federal test driving, idle, and a steady speed of 30 mph,
respectively (Jacobs, 1980). Based on these levels, 1,2-dibromoethane
concentrations in air alongside roads due to vehicle exhaust emissions
may range from 0.04 to 122 µg/m3 (0.005-0.19 ppb). These results
are similar to the observations of Leinster et al. (1978).
Table 4. 1,2-Dibromoethane produced by motor vehicles (petrol engine)
under constant speed test conditions
Vehicle speed km/h Concentration (µg/m3)
Engine not Vehicle with Vehicle with
defineda 3 litre engineb 0.85 litre engineb
Cold start (idle) 70 878 332
10 78
30 62-70 618 165
40 61
50 2 669 155
64 180 139
80 98 135
a Leinster et al. (1978)
b Tsani-Bazaca et al. (1981)
Table 5. 1,2-Dibromoethane in the exhaust emission of motor vehicles
(petrol engine) (µg/m3)a
Conditions 3 litre engine 0.85 letre engine
ECEb
cold start 292-560 733-538
hot start 200-234 533-538
ECD/CVSc 14-25 26-34
USA Federald
cold start 48 29
hot start 22
a From: Tsani-Bazaca et al. (1981)
b standard European driving cycle
c ECE driving cycle under constant volume sampling condition
d 1973 driving cycle
1,2-Dibromoethane levels in air have been measured at several
sites around the world (Table 6). Leinster et al. (1978) concluded
that the lower levels during the autumn were the result of a reduction
in evaporative loss particularly from parked vehicles (the calculated
evaporation rate for 1,2-dibromoethane at 5°C is less than one third
of that at 30°C). An indication of the magnitude of evaporative loss
from parked vehicles was provided by levels of 0.02-0.05 µg/m3
measured in a car park. It was also probable that an opposite trend
would be produced by a change in driving conditions. For example,
cold starts and driving speeds of vehicles have a marked influence on
the 1,2-dibromoethane content of exhaust emissions.
The 1,2-dibromoethane added to leaded petrol contributes to a
large amount of methyl bromide in urban atmospheres. IPCS (1995)
estimated that per annum between 7000 and 18 000 tonnes of methyl
bromide could be emitted from car exhausts. Reactions in the lower
troposphere with hydroxyl radicals and other chemical species are the
most important of the possible removal mechanisms within the
atmosphere (UNEP, 1992). The end-products of both photodissociation
of methyl bromide and reactions with hydroxyl radicals in the
atmosphere include bromide species (BUA, 1987). Active bromine
species react with ozone mainly in the lower stratosphere and are
thought to be partly responsible for the destruction of the ozone
layer. However, 1,2-dibromoethane was not included as a controlled
substance in the "Montreal Protocol on Substances that Deplete the
Ozone Layer".
Table 6. Environmental concentrations of 1,2-dibromoethane
Location Measuring period Concentration (µg/m3) Reference
London August 1976a 0.08-0.09 µg/m3 Leinster et al. (1978)
December 1976b 0.001-0.01 µg/m3
12 Canadian cities 1989-1992 mean 0.05 ± 0.05 µg/m3 Environment Canada
range n.d.-1.73 µg/m3 (1994)
Busy streets at 2 m height 0.07-1.26 µg/m3 Tsani-Bazaca et al.
and 5 m from kerbside (1981)
Los Angeles, California, USA 9-21 April 1979 0.25 ± 0.20 ng/m3 (33.2 ± 26.2 ppt) Singh et al. (1981)
range 0.041-1.4 ng/m3 (5.4-187.2 ppt)
Oakland, California, USA 28 June-10 July 1979 0.12 ± 0.10 ng/m3 (15.8 ± 12.5 ppt) Singh et al. (1981)
range 0.018-0.65 ng/m3 (2.4-84.5 ppt)
Phoenix, Arizona, USA 23 April-6 May 1979 0.31 ± 0.29 ng/m3 (40.3 ± 38.3 ppt) Singh et al. (1981)
range 0.018-1.6 ng/m3 (2.4-204.4 ppt)
Background 38 ng/m3
Various cities (7) 1-2 weeks in 1980 0.122-0.453 ng/m3 (0.016-0.059 ppt)c Singh et al. (1982)
2.826 ng/m3 (0.368 ppt)d
Denver, Colorado, USA 1-2 weeks n.d.-2.304 µg/m3 (0.3 ppb) Going & Spigarelli (1976);
Leinster et al. (1978)
Sites in New Jersey, USA late 1983-Spring 1984 0.077-5.4 µg/m3 Harkov et al. (1985)
Summer 1981 < 0.038 µg/m3 Harkov et al. (1983)
Winter 1982 < 0.038 µg/m3 Harkov et al. (1983)
Table 6 (cont'd)
Location Measuring period Concentration (µg/m3) Reference
Central London Summer 1982 0.23 µg/m3 (0.03 ppb)e,f Clark et al. (1984a,b)
Exhibition Road May-August 1983 0.23 µg/m3 (0.03 ppb)e
Rural site Summer 1982 0.12 µg/m3 (0.015 ppb)e,g Clark et al. (1984a,b)
Silwood Park, United Kingdom May-August 1983 0.15 µg/m3 (0.019 ppb)e
Motorway outside London, Summer 1982 0.39 µg/m3 (0.05 ppb)e,h Clark et al. (1984a,b)
Toddington May-August 1983 0.31 µg/m3 (0.04 ppb)e
Central London Summer 1982 1.0 µg/m3 (0.13 ppb)i Clark et al. (1984a,b)
May-August 1983 0.62 µg/m3 (0.08 ppb)i
Motorway site Summer 1982 2.0 µg/m3 (0.26 ppb)i Clark et al. (1984a,b)
May-August 1983 1.2 µg/m3 (0.15 ppb)i
Anchorage (Alaska) March 1983 31-177 ng/m3 (4-23 ppt) Berg et al. (1984)
Barrow (Alaska) March 1983 n.d.-177 ng/m3 (n.d.-28 ppt) Berg et al. (1984)
Mould Bay Coast (Alaska) March 1983 38-284 ng/m3 (5-37 ppt) Berg et al. (1984)
Thule (Greenland) March 1983 15-246 ng/m3 (2-32 ppt) Berg et al. (1984)
North Pole March 1983 92.2 ng/m3 (12 ppt) Berg et al. (1984)
Ny-Alesund (Norway) March-April 1983 31-150 ng/m3 (4-20 ppt) Berg et al. (1984)
Table 6 (cont'd)
Location Measuring period Concentration (µg/m3) Reference
Bear Island (Norway) March-April 1983 23-100 ng/m3 (3-13 ppt) Berg et al. (1984)
Bodo (Norway) March-April 1983 38-110 ng/m3 (5-14 ppt) Berg et al. (1984)
a temperature range 28-30°C
b temperature range 4-8°C
c average level for each city
d maximum concentration found in Houston, Texas, USA
e mean hourly concentrations
f range 0.078-1 µg/m3 (0.01-0.13 ppb)
g range ND-0.78 µg/m3 (ND-0.01 ppb)
h range 0.07-2.0 µg/m3 (0.009-0.26 ppb)
i maximum hourly concentrations
1,2-Dibromoethane levels of 0.07-1.26 µg/m3 have been found in
busy streets. Higher levels were found in a road tunnel and were
associated with poor ventilation (Tsani-Bazaca et al., 1981). Three
field studies on the measurement of selected potentially hazardous
organic compounds in urban environments were conducted in the USA in
1979 (Los Angeles, California, 9-21 April; Oakland, California,
28 June-10 July; and Phoenix, Arizona, 23 April-6 May). These studies
were performed to characterize the atmospheric abundance, fate and
human exposure to these compounds (Table 6). The background
concentration of 1,2-dibromoethane was 38 ng/m3 (5 ppt). Assuming
an average respiratory volume of 23 m3 at 25°C and 1 atm for a 70-kg
male, the average daily dose (µg/day) of 1,2-dibromoethane at these
locations can be calculated as 6.0 ± 2.7 for Los Angeles, 2.9 ± 0.8
for Oakland and 7.0 ± 2.7 for Phoenix. The ratios of
1,2-dibromoethane to total haloethane and VHH (volatile halogenated
hydrocarbons) in the average daily doses were 2.7% and 0.57%, 5.6% and
1.14%, and 2.8% and 1.03%, respectively. The chemical loss rate of
1,2-dibromoethane was 2.8% per day (sunlight = 12 h). There was
diurnal variation in 1,2-dibromoethane levels at the three locations.
The afternoon minimum at Phoenix was attributed to deep vertical
mixing associated with hot and dry weather. The afternoon maximum at
Oakland was most likely a result of transport from upwind sources
(Singh et al., 1981).
Other studies measuring 1,2-dibromoethane in the ambient
atmosphere of urban and rural areas have been performed (Going & Long,
1975; Going & Spigarelli, 1976). Sources of 1,2-dibromoethane in air
were considered to be emissions from stations dispensing leaded petrol
and evaporative emissions from motor vehicles using leaded petrol.
Atmospheric levels of 1,2-dibromoethane were low (0.046 to
3.5 µg/m3) (0.006 to 0.45 ppb) in worst case conditions near petrol
stations and with heavy traffic in cities. These levels are 10 to
10 000 times less than the occupational exposure level of 1 mg/m3
(0.13 ppm) for 15 min recommended by the US National Institute of
Occupational Safety and Health (Jacobs, 1980).
Tsani-Bazaca et al. (1981) monitored the concentrations of VHH
collected in 1979 at several locations and utilizing vehicles
operating under various conditions on a busy road in central London
(2000 vehicles/h), a poorly ventilated tunnel (1600 vehicles/h at peak
traffic flow), and a semi-rural industrialized area. The
concentration of 1,2-dibromoethane varied between 0.07 and
1.26 µg/m3. There was a good correlation between 1,2-dibromoethane
and benzene concentrations (correlation coefficient : 0.93) at the
three locations and a higher correlation between 1,2-dibromoethane and
1,2-dichloroethane (correlation coefficient : 0.94).
In 1983, 54 air samples at 6 urban sites and 54 air samples at
6 mountainous sites were collected in Japan and were analysed for the
presence of 1,2-dibromoethane. A total of 35 samples from 5 urban
sites contained 1,2-dibromoethane at concentrations of
0.008-0.322 µg/m3 (0.001-0.042 ppb). The detection limit was
0.005-0.008 µg/m3 (0.0007-0.001 ppb). A total of 36 samples from
5 mountainous sites contained 1,2-dibromoethane at concentrations of
0.008-0.515 µg/m3 (0.001-0.067 ppb). The detection limit was
0.002-0.008 µg/m3 (0.0003-0.001 ppb) (Environment Agency Japan,
1985).
Urban 1,2-dibromoethane levels at seven sites in selected cities
in the USA in 1980, using on-site and real-time measurement instrument
following a 24-h measurement schedule for a period of 1-2 weeks, were
0.12-0.45 µg/m3 (16-59 ppt) (Singh et al., 1982). The average
concentration of 1,2-dibromoethane did not exceed 0.015-0.46 µg/m3
(0.06 ppb) (average range 0.002-0.06 ppb) at any study site and
average levels ranged from 0.122 µg/m3 (0.016 ppb) at St. Louis,
Missouri, to 0.46 µg/m3 (0.059 ppb) at Houston, Texas. The maximum
concentration of 2.83 µg/m3 (0.368 ppb) was found at Houston. In
general, the highest average levels were found during the night and
early morning. In the case of Denver, Colorado, typical ambient
concentration data suggested a range of not detectable to 2.3 µg/m3
(0.300 ppb) (Going & Spigarelli, 1976; Leinster et al., 1978).
The Office of Science and Research (USA) monitored VHH in ambient
air at listed abandoned hazardous waste sites and sanitary landfills
in New Jersey (Harkov et al., 1985). 1,2-Dibromoethane was found at
mean levels of 2.1 µg/m3 (0.27 ppb), 2.2 µg/m3 (0.288 ppb),
3.6 µg/m3 (0.47 ppb), 5.4 µg/m3 (0.7 ppb), 0.38 µg/m3
(0.05 ppb), 0.077 µg/m3 (0.01 ppb) and 0.15 µg/m3 (0.02 ppb) at
different sites during late 1983 and early 1984. It was below the
detection limit 0.038 µg/m3 (0.005 ppb) at three sites during the
summer of 1981 (Harkov et al., 1983) and the winter of 1982 (Harkov et
al., 1984).
Ambient air monitoring survey of VHH at a busy road in central
London, a rural site and a motorway location near London showed mean
hourly 1,2-dibromoethane concentrations of 0.23, 1.2 and 0.39 µg/m3
(0.03, 0.15 and 0.05 ppb), respectively, in summer 1982 and 0.23, 0.15
and 0.31 µg/m3 (0.03, 0.019 and 0.04 ppb) between May and August
1983 (Clark et al., 1984a,b). The maximum hourly concentrations of
1,2-dibromoethane at the urban and motorway sites were 1.0 and
2.0 µg/m3 (0.13 and 0.26 ppb) in 1982, and 0.61 and 1.2 µg/m3
(0.08 and 0.15 ppb) in 1983, respectively. 1,2-Dibromoethane
concentrations at the urban site were in the same ranges
0.07-0.31 ng/m3 (0.01-0.04 ppt) as those measured by other workers
(Leinster et al., 1978; Tsani-Bazaca et al., 1981; Singh et al.,
1982). The low concentrations found at the rural site were primarily
related to the low incidence of vehicular pollutant sources in the
area. However, the site was near the urban fringe of London and near
several small towns and this may explain occasional elevated
concentrations.
1,2-Dibromoethane concentrations were measured at Point Arena,
California between 1979 and 1981; the background level of
1,2-dibromoethane in the troposphere was found to be less than
0.023 µg/m3 (3 ppt) (Singh et al., 1983).
Berg et al. (1984) measured atmospheric 1,2-dibromoethane
concentrations at eight arctic sites in 1983. Concentrations at three
sites in Alaska (Anchorage, Barrow, Mould Bay Coast) in March were
0.031-0.177 µg/m3 (4-23 ppt), not detectable to 0.22 µg/m3
(29 ppt) and 0.038-0.284 µg/m3 (5-37 ppt), respectively.
1,2-Dibromoethane levels in Greenland (Thule) and at the North Pole in
March were 0.015-0.246 µg/m3 (2-32 ppt) and 0.096 µg/m3 (12 ppt),
respectively. Those at Norwegian sites (Ny-Alesund, Bear Island,
Bodo) during March-April were 0.031-0.15 µg/m3 (4-20 ppt),
0.023-0.10 µg/m3 (3-13 ppt) and 0.038-0.11 µg/m3 (5-14 ppt),
respectively. The mean concentration ± standard deviation was
0.084-0.77 µg/m3 (11 ± 10 ppt). Other organobromine compounds, such
as methyl bromide, methylene dibromide and bromoform, were detected at
similar concentrations.
Monthly monitoring of the atmosphere of Barrow, Alaska (72 °N),
showed that the 1,2-dibromoethane concentration was higher in winter
than in other seasons, although the monthly average concentrations did
not differ greatly (7.68-10.7 ng/m3) (1.0-1.4 ppt) except in
January. From the results of atmospheric VHH monitoring, Rasmussen &
Khalil (1984) suggested that VHH in arctic air might be an indicator
of polluted air transported from industrial mid-latitude sources.
5.1.2 Water
Widespread use of 1,2-dibromoethane as a soil fumigant in the USA
resulted in its detection in both groundwater and surface water in
California, Florida, Georgia, and Hawaii (Sun, 1984), Connecticut
(Isaacson et al., 1984) and New Jersey (Page, 1981), and in wells used
for irrigation in Georgia (Martl et al., 1984). 1,2-Dibromoethane
was reported in groundwater in Georgia, California, Florida, and
Hawaii by US EPA (1986).
Laboratory studies have shown that 1,2-dibromoethane
photohydrolyses rapidly in aqueous solutions when irradiated. The
degradation is a two-stage process in which 1,2-dibromoethane is
converted to bromoethanol (half-life, 7.6 min) and then to ethylene
oxide (half-life, 64 min). Further degradation to ethylene glycol was
less influenced by light, as shown by a half-life of 10 days (Castro &
Belser, 1978). While the above study provides some understanding of
aqueous degradation, Logan (1988) cautions that the efficiency of the
photo-reactions were not reported in terms of quantum yield.
1,2-Dibromoethane was found in ground- and surface water in New
Jersey (over 1000 different wells and 600 different sites) during
1977-1979; the highest levels were 0.2 µg/litre in surface water and
48.8 µg/litre in groundwater (Page, 1981).
Analyses of 350 well water samples from Connecticut in 1984
revealed concentrations of up to 2 µg/litre. 1,2-Dibromoethane was
rapidly lost from water samples exposed to the atmosphere or boiled
for few minutes. It could not be detected in water samples purged
with nitrogen for 10 min (Isaacson et al., 1984).
In southwest Georgia, USA, agricultural practices involve
intensive use of groundwater for irrigation and pesticides for control
of plant and insect pests. 1,2-Dibromoethane was found at levels of
between 1 and 90 µg/litre in water samples from three irrigation wells
collected between 1981 and 1983. Application at ratios of
14-19 litres/ha) near wells showed that 1,2-dibromoethane
concentrations in the aquifer did not appear to be directly related to
the application rate of the compound to the surface. The
concentrations in the wells may reflect application of the compound at
sites some distance from the wells (Martl et al., 1984).
In 1982, 27 water samples and 27 bottom sediment samples were
collected at nine sites in Japan and were analysed for the presence of
1,2-dibromoethane. None of the water or bottom sediment samples
contained 1,2-dibromoethane. The detection limit was 0.3-2 µg/litre
for water and 0.0016-0.01 µg/kg for bottom sediment (Environment
Agency Japan, 1985).
In 1983, 1,2-dibromoethane surveillance of the water of six rural
wells in Ibaraki prefecture, Japan, where 1,2-dibromoethane was used
for soil fumigation or as a pesticide on pine tree, showed no
1,2-dibromoethane contamination (detection limit, 5 µg/litre) (Nemoto
et al., 1984).
Groundwater samples from nine sites in and around vegetable-
growing areas in Gifu Prefecture, Japan, were collected twelve times
between July 1983 and December 1984. 1,2-Dibromoethane levels ranged
from 0.06 to 0.55 µg/litre at seven sites and the mean values of
1,2-dibromoethane at each site varied between 0.15 and 0.28 µg/litre.
1,2-Dibromoethane levels in groundwater around the vegetable-growing
areas did not differ from those within the areas, where
1,2-dibromoethane application was limited to once a year in the first
two weeks of July. Sites where 1,2-dibromoethane were detected around
these areas overlapped completely the stream of groundwater coming
from these areas (Terao et al., 1985). The annual variation of
concentrations in the groundwater was small. 1,2-Dibromoethane
concentration showed good correlation with bromine ion concentration
and bromine ion/chlorine ion ratio at each site (Terao et al., 1984).
Mayer et al. (1991) studied 1,2-dibromoethane concentrations in
detail in water from a domestic well, approximately 10 m deep, in a
fruit growing area of Whatcom County, Washington, USA where
1,2-dibromoethane had been used extensively prior to its 1983 ban.
Additional wells (n = 107) were also sampled over a 4-year period; no
details of well depths were given. Correlation analysis showed no
relationship between 1,2-dibromoethane concentration in water and
temperature but significant negative correlation between precipitation
and 1,2-dibromoethane. The analysis allowed lag times of between
0 and 12 months; a 3-month lag was found to give the best relationship
between precipitation and 1,2-dibromoethane in the water. The
dilution effect of precipitation was followed by slow
1,2-dibromoethane infiltration from overlying soils which tended to
re-establish prior concentrations over about 3 months. The authors
stated that water contamination can result from such continuing
infiltration of soil-matrix-derived 1,2-dibromoethane long after
agricultural use has ceased.
5.1.3 Food
Beckman et al. (1967) reported that part of the inorganic bromine
in foods and raw agricultural commodities comes from the soil.
1,2-Dibromoethane was applied annually at 54 kg/ha and samples from
40 crops grown in soil treated with 1,2-dibromoethane were analysed
over a 3-year period. In general, leafy portions of plants contained
the highest levels of bromide on the basis of weight. Residue levels
were calculated as inorganic bromide ion present in the crop after
harvest. Levels in crop samples from untreated soil were less than
1.6 mg/m3 (0.2 ppm), and the highest level in crops from treated
soil was 137 mg/kg (17.8 ppm) in sugar beet tops. Most of the crops
were harvested about 100 days after soil treatment but time from
treatment to harvest ranged from 55 days for strawberries to 10 years
for walnuts.
1,2-Dibromoethane was absorbed strongly by cereal, grains, cereal
products and other produce during the fumigation period. Even when
normal ventilation procedures were followed, residues of
1,2-dibromoethane disappeared very slowly. Nearly all the
1,2-dibromoethane was physically sorbed and at normal temperatures
there was little formation of inorganic bromide. However,
occasionally in produce at higher temperatures and moisture content
there was rapid breakdown to inorganic bromide (Heuser & Scudamore,
1967).
Levels of 1,2-dibromoethane in wheat were between 10 and
20 mg/kg, and, for its products, between 2 and 4 mg/kg in flour,
0.002-0.04 in white bread and 0.006-0.16 in wholemeal bread. When
flour was treated directly with 1,2-dibromoethane, ventilated
thoroughly, and then baked into loaves, there were residues of
20-24 mg/kg in the flour and 0.33-0.47 mg/kg in the bread (FAO/WHO,
1972). Desorption of 1,2-dibromoethane occurred at low (14-17°C)
rather than high (30-37°C) temperatures, and was abolished by grinding
the grain (Bielorai & Alumot, 1975).
Rappaport et al. (1984) reported that the decay of the outgassing
rate over time from fumigated oranges was approximately first order.
Outgassing was significantly slowed by reducing either the temperature
or the ventilation rate. In laboratory trials, ventilation at
0.6 air changes/h removed 1,2-dibromoethane vapours from the surface
of oranges, and prevented reabsorption onto the fruits.
A pesticide formulation, consisting of carbon tetrachloride (CT),
1,2-dichloroethane (EDC), 1,2-dibromoethane in 63 : 30 : 7 w/w
proportions, was applied to 27.3 tonnes of wheat stored in a paper
laminate bin (Berck, 1974). The CT-EDC-1,2-dibromoethane distribution-
persistence patterns were monitored at 16 bin locations over a 14-day
period by GC. Fumigant residues in the wheat, in flour, bran, and
middlings derived from the wheat, and in bread baked from the flour
were determined over a 7-week period. 1,2-Dibromoethane residues in
the wheat varied, depending on the bin location and contact time, and
ranged from 0 to 3.3 mg/kg. Residues in bran and middlings were
greater than those in flour, and ranged from 0 to 0.4 mg/kg. No
1,2-dibromoethane residues were found in any of the bread tested
(detection limit, 10 ng/kg).
1,2-Dibromoethane levels were studied in biscuits (22 samples)
and flour (22 samples), the biscuits being baked from each of the
flour samples for 12 min at 268°C. After baking, the samples were
sealed in plastic bags and frozen to prevent any further loss of
1,2-dibromoethane. Flour samples were also sealed in plastic bags and
frozen. Levels of 1,2-dibromoethane in flour and biscuits ranged from
non-detectable to 4.2 mg/kg and to 0.26 mg/kg, respectively. There
was poor correlation between the levels of 1,2-dibromoethane in flour
and biscuits.
5.2 Occupational exposure
Air concentrations of 1,2-dibromoethane in ventilated containers
dropped from several ppm immediately after fumigation to a few ppb
after 5-10 days; levels remained between 15 and 23 mg/kg (2 and 3 ppm)
for 15-20 days during unventilated, refrigerated storage. Results of
experiments on a laboratory scale (0.25 carton) and a large scale
(400 cartons) suggested that workers transporting and distributing
fumigated citrus fruit could routinely be exposed to airborne
1,2-dibromoethane at concen trations greater than 998 µg/kg (130 ppb)
(OSHA, 1983).
The US National Institute for Occupational Safety and Health
(NIOSH) estimated that approximately 108 000 workers in the USA were
potentially exposed to 1,2-dibromoethane in their workplaces (Table 7)
and that another 875 000 workers handling leaded petrol were exposed
to very low levels (NIOSH, 1981). There is no estimate of the number
of motorists exposed to 1,2-dibromoethane during self-service
operations at filling stations.
Table 7. Occupations with potenial exposure to 1,2-dibromoethanea
Antiknock compound makers Motor fuel workers
Cabbage growers Oil processors
Corn growers Organic chemical synthesizers
1,2-Dibromoethane workers Petrol blenders
Drug makers Resin makers
Fat processors Seed corn maggot controllers
Fire-extinguisher makers Soil fumigators
Fruit fumigators Termite controllers
Fumigant workers Tetraethyllead makers
Grain elevator workers Waterproofing makers
Grain fumigators Waxmakers
Gum processors Wood insect controllers
Lead scavenger makers Wool reclaimers
a From: NIOSH (1977)
In the 1970s, US EPA examined the exposure of professional
pesticide applicators involved with 1,2-dibromoethane soil fumigation.
It was estimated that applicators applying 1,2-dibromoethane for
30-40 days/year would receive a total annual inhalation dose of
3-40 mg/kg and farmer-applicators applying 1,2-dibromoethane for
7-10 days/year would receive a total annual inhalation dose of
0.7-10 mg/kg (US EPA, 1977).
1,2-Dibromoethane exposures were measured in a plant where lead
antiknock blends for petrol were prepared (Jacobs, 1980). The
antiknock blend constituents were mixed in tanks under enclosed-system
conditions and the only manual operations were connecting and
disconnecting hoses while loading and unloading tank cars, taking
quality control samples, and processing and loading drums. The levels
of worker exposure to 1,2-dibromoethane in antiknock blending and
storage areas were 0.77 µg/m3 (0.1 ppb) (laboratory technician) to
6.3 µg/m3 (0.82 ppb) (raw maternal blender). In addition to
long-term personal sampling, some short-term monitoring of specific
tasks was conducted. The results are shown in Table 8.
Table 8. Short-term air levels of 1,2-dibromoethane in antiknock
blending plant tank cars (Jacobs, 1980)
Taska Sampling time Concentration
mg/m3 ppm
Quality control sample 13 min, 10 sec 5.38 0.7
Loading tank car 7 min 1.07 0.14
a Respirator worn during these tasks
Personal air monitoring during vehicle refuelling at a petroleum
laboratory 10 m downwind of the fuel pump and fuel-handling facilities
was performed at a USA plant in July, 1975 (Table 9). The average
exposure of filling station attendants to 1,2-dibromoethane during
refuelling was 1.8 µg/m3 (0.24 ppb). Measurements at the car fuel
tank filler pipe showed maximum instantaneous 1,2-dibromoethane
concentrations of 105 µg/m3 (13.7 ppb), with an average for four
samples of 10.0 µg/m3 (1.3 ppb). This represented the maximum for a
short-term exposure. The concentration of 1,2-dibromoethane in air at
the fuel pump island was similar to values measured at upwind and
downwind sites. Overall, the very low 1,2-dibromoethane air levels
measured in this study indicated that the potential for filling
station attendant exposure to 1,2-dibromoethane while refuelling cars
was low and less than the current or proposed USA occupational air
standard for 1,2-dibromoethane exposure (Jacobs, 1980).
Exhaust emissions from various types of internal combustion
engines, including four-stroke Otto engines and diesel engines, are a
major source of environmental and occupational exposure to
1,2-dibromoethane (Hasanen et al., 1981). There are few data on the
composition of and exposure to exhaust emissions from two-stroke
engines.
Table 9. Petrol station attendant exposure to 1,2-dibromoethane
during vehicle refuelling (Jacobs, 1980)
Sample Concentration
mg/m3 ppb
Upwind background < 0.77 < 0.1
Downwind background < 0.77 < 0.1
Fuel pump island 0.99 0.13
Near vehicle fuel pipe 9.98 1.3a
during refueling 105.2 13.7b
Personal air sampler 2.15 0.28
a Average
b Maximum
Seven chain saws fuelled with 93-octane standard petrol-
containing tetramethyllead (lead content 0.15 g/litre) and
1,2-dibromoethane as a scavenger were tested on a test-bench
permitting a variable load to be applied by an electric power brake
(Nilsson et al., 1987). 1,2-Dibromoethane emissions were low
(2.5 mg/m3). Exposure to chain saw exhaust during logging was
studied under snowy and snow-free conditions. The time-weighted
average exposure to 1,2-dibromoethane was lower in the snow-free
conditions (0.0008 (0.0004-0.001) mg/m3) than in the snowy
conditions (0.002 (0.0001-0.005) mg/m3).
1,2-Dibromoethane is mainly used as a scavenger in tetraalkyllead
petrol and antiknock preparations, as a soil and grain fumigant, as
an intermediate in the synthesis of dyes and pharmaceuticals and as a
solvent for resins, gums and waxes (Alexeeff et al., 1990).
Rumsey & Tanita (1978) performed an industrial hygiene survey of
two manufacturing and two user facilities involving 1,2-dibromoethane.
Samples were taken from more than 69 potentially-exposed workers in
17 job classifications. Median 1,2-dibromoethane exposure (by similar
job types) in the manufacturing process ranged from 0.076 to
3.8 mg/m3 (0.010 to 0.5 ppm) (35 TWA personal samples). General
area samples collected at breathing zone heights had median TWA levels
of 1.5 mg/m3 (0.2 ppm) for 10 samples at process sites, and
3.8 mg/m3 (0.5 ppm) for 3 samples at laboratory sites.
Papaya workers in Hawaii were exposed to a geometric mean of
676 mg/m3 (88 ppb), and peaks up to 2.01 mg/m3 (262 ppb) were
measured (Steenland et al., 1986).
6. KINETICS AND METABOLISM
6.1 Absorption
1,2-Dibromoethane was found in the blood of rodents almost
immediately after dermal and oral exposure. Jakobson et al. (1982)
reported that during a 6-h dermal exposure of guinea-pigs of both
sexes (weighing between 600 and 1000 g) with undiluted
1,2-dibromoethane applied to 3.1 cm2 of shaved skin on the back
(1.0 ml/animal), the blood concentration of 1,2-dibromoethane
increased rapidly during 1 h to a level of 2 mg/litre and then slowly
decreased. The influx of 1,2-dibromoethane into the blood after 1 h
was largely in equilibrium with its disappearance.
In male Sprague-Dawley rats given 15 mg/kg body weight of
[14C-1,2] 1,2-dibromoethane in corn oil by gavage, the blood levels
at 24 h and 48 h were 0.90 and 0.64 mg/litre, respectively (Plotnick
et al., 1979). The excretion of radioactivity in faeces within 24 h
was 1.7% of the dose. The remainder was recovered either in the urine
(72%) or in the tissues (2.8%) (Table 10). The results indicated
rapid 1,2-dibromoethane absorption from the gastrointestinal tract.
No absorption information regarding inhalation exposure exists.
Table 10. The distribution of 14C in selected tissues and body
fluids of male rats 24 h after a single oral dose of
14C-1,2-dibromoethane (15 mg/kg)a
Tissue Tissue concentrationsb Percentage of dose
(mg equivalent/kg or mg/litre) (%)
Liver 4.78 ± 0.24 1.7 ± 0.07
Kidneys 3.32 ± 0.42 0.21 ± 0.02
Spleen 1.00 ± 0.03 0.22 ± < 0.01
Testes 0.49 ± 0.05 0.04 ± < 0.01
Brain 0.41 ± 0.04 0.02 ± < 0.01
Fat 0.35 ± 0.04 0.15 ± 0.02
Blood 0.90 ± 0.05 0.59 ± 0.03
Plasma 0.46 ± 0.04
Urinec 72.38 ± 0.98
Faecesc 1.65 ± 0.28
a From: Plotnick et al. (1979)
b Values represent mean concentrations (expressed as parent
compound) ± S.E.M. of duplicate determinations for six animals.
c n = 12
6.2 Distribution
Plotnick et al. (1979) compared levels of 14C in selected
tissues of male Sprague-Dawley rats following oral administration of
14C-1,2-dibromoethane. One day after the administration, the
highest levels of radioactivity were found in the liver and kidneys
(Table 10).
Distribution of 14C-1,2-dibromoethane (30 mg/kg body weight)
after intraperitoneal administration to male guinea-pigs was studied
by Plotnick & Conner (1976). The liver and kidneys contained the
highest levels of radioactivity followed by the adrenal glands
(Table 11).
Table 11. Distribution of 14C-1,2-dibromoethane in selected tissues
of male guinea-pigs at various time intervals following
intraperitoneal administrationa
Tissues/organs 4 h 8 h 24 h 72 h
Liver 129.0 104.9 38.0 15.6
Kidneys 286.6b 236.5 3.5 10.5
Adrenals 60.7 60.8 28.6 10.4
Pancreas 35.0 36.8 18.7 6.0
Spleen 15.8 14.0 14.9 7.0
Heart 14.0 15.6 9.5 3.3
Lungs 20.9 19.0 15.4 5.8
Testes 10.7 10.7 8.3 4.0
Brain 6.2 7.6 6.5 2.5
Fatc 21.4 7.9 3.2 2.1
Muscle 5.5 5.0 4.2 2.2
Blood 10.0 3.4 5.0 2.8
a From: Plotnick & Conner (1976)
b Values represent mean levels in mg equivalent/kg of tissue or
litre of fluid for three animals at each time interval
c Suprarenal fat
Kowalski et al. (1985) reported epithelial binding of
1,2-dibromoethane in the respiratory and upper alimentary tracts of
C57BL mice, Sprague-Dawley rats and Fischer-344 rats after intravenous
and intraperitoneal injection of 14C-1,2-dibromoethane. In C57BL
mice, there was a high level of radioactivity in the nasal and
bronchial mucosa and liver 5 min after intravenous injection of
14C-1,2-dibromoethane. In the nose, the highest labelling was
present in a spotty band beneath the epithelium of the
ethmoturbinates. The radioactive labelling of the mucosa of the
respiratory tract was persistent, and 10 days after injection a
selectively bound radioactivity remained. High labelling was also
present in the mucosa of the forestomach, whereas there was no
selective uptake of radioactivity in the glandular stomach or
intestine. Similar distribution patterns were observed in the
intraperitoneally injected mice killed after 30 min or subsequently.
6.3 Metabolic transformation
The metabolism of 1,2-dibromoethane has been extensively studied
and metabolites have been identified in in vivo and in vitro
studies (Table 12, Fig. 1).
Table 12. Metabolites of 1,2-dibromoethane
(a) In vivo
Metabolite Animal; route; substrate Reference
Bromide Swiss-Webster mice; White et al.
intraperitoneal; plasma (1983)
N-acetyl-S-(2-hydroxy male Wistar rat; oral; Van Bladeren et
ethyl)-L-cysteine urine al. (1981b)
GS-CH2-CH2SG female white rat; oral; Nachtomi (1970)
liver
GSCH2CH2OH sulfoxide liver Nachtomi (1970)
GSCH2CH2OH liver and kidney Nachtomi (1970)
S-(2-hydroxyethyl) kidney Nachtomi (1970)
mercapturic acid
(b) In vitro
Metabolite Tissue Reference
GSCH2CH2SG rat liver and kidney extract Nachtomi (1970)
GSCH2CH2OH rat liver and kidney extract Nachtomi (1970)
Bromide (1984) rat liver cytosols White et al.
Bromide (1983) mouse liver cytosols White et al.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR 1,2-DIBROMOETHANE
Members
Dr T. Bailey, US Environmental Protection Agency, Washington DC, USA
Dr A.L. Black, Department of Human Services and Health, Canberra,
Australia
Mr D.J. Clegg, Carp, Ontario, Canada
Dr S. Dobson, Institute of Terrestrial Ecology, Monks Wood, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom
(Vice-Chairman)
Dr P.E.T. Douben, Her Majesty's Inspectorate of Pollution, London,
United Kingdom (EHC Joint Rapporteur)
Dr P. Fenner-Crisp, US Environmental Protection Agency, Washington DC,
USA
Dr R. Hailey, National Institute of Environmental Health Sciences,
National Institutes of Health, Research Triangle Park, USA
Ms K. Hughes, Environmental Health Directorate, Health Canada, Ottawa,
Ontario, Canada (EHC Joint Rapporteur)
Dr D. Kanungo, Central Insecticides Laboratory, Government of India,
Ministry of Agriculture & Cooperation, Directorate of Plant
Protection, Quarantine & Storage, Faridabad, Haryana, India
Dr L. Landner, MFG, European Environmental Research Group Ltd,
Stockholm, Sweden
Dr M.H. Litchfield, Melrose Consultancy, Denmans Lane, Fontwell,
Arundel, West Sussex, United Kingdom (CAG Joint Rapporteur)
Professor M. Lotti, Institute of Occupational Medicine, University of
Padua, Padua, Italy (Chairman)
Professor D.R. Mattison, University of Pittsburgh, Graduate School of
Public Health, Pittsburgh, Pennsylvania, USA
Dr J. Sekizawa, National Institute of Health Sciences, Tokyo, Japan
Dr P. Sinhaseni, Chulalongkorn University, Bangkok, Thailand
Dr S.A. Soliman, King Saud University, Bureidah, Saudi Arabia
Dr M. Tasheva, National Centre of Hygiene, Medical Ecology and
Nutrition, Sofia, Bulgaria (CAG Joint Rapporteur)
Mr J.R. Taylor, Pesticides Safety Directorate, Ministry of
Agriculture, Fisheries and Food, York, United Kingdom
Dr H.M. Temmink, Wageningen Agricultural University, Wageningen, The
Netherlands
Dr M.I. Willems, TNO Nutrition and Food Research Institute, Zeist, The
Netherlands
Secretariat
Ms A. Sundén Byléhn, International Register of Potentially Toxic
Chemicals, United Nations Environment Programme, Châtelaine,
Switzerland
Dr P. Chamberlain, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr J. Herrman, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr K. Jager, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
Dr P. Jenkins, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr W. Kreisel, World Health Organization, Geneva, Switzerland
Dr M. Mercier, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr M.I. Mikheev, Occupational Health, World Health Organization,
Geneva, Switzerland
Dr G. Moy, Food Safety, World Health Organization, Geneva, Switzerland
Mr I. Obadia, International Labour Organisation, Geneva,
Switzerland
Dr R. Plestina, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland (EHC Secretary)
Mr J. Wilbourn, International Agency for Research on Cancer, Lyon,
France
ENVIRONMENTAL HEALTH CRITERIA FOR 1,2-DIBROMOETHANE
The Core Assessment Group (CAG) of the Joint Meeting on
Pesticides (JMP) met at the World Health Organization, Geneva from
25 October to 3 November 1994. Dr W. Kreisel, Executive Director,
welcomed the participants on behalf of WHO, and Dr M. Mercier,
Director, IPCS, on behalf of the IPCS and its cooperating
organizations (UNEP/ILO/WHO). The Core Assessment Group reviewed and
revised the draft monograph and made an evaluation of the risks for
human health and the environment from exposure to 1,2-dibromoethane
(ethylene dibromide).
The preparation of the first draft of the monograph was
coordinated by Dr J. Sekizawa, National Institute of Health Sciences,
Japan. The second draft, revised in the light of international
comment, was prepared under the coordination of Dr Sekizawa.
Dr E. Smith and Dr P.G. Jenkins, both members of the IPCS Central
Unit, were responsible for the scientific content and technical
editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
The authors who contributed to the first draft were:
Dr C. Hashida The Jikei University School of Medicine, Japan
Dr Y. Hayashi National Institute of Health Sciences, Japan
Dr E. Kamata National Institute of Health Sciences, Japan
Dr Y. Kurokawa National Institute of Health Sciences, Japan
Dr A. Matsuoka National Institute of Health Sciences, Japan
Dr T. Matsushima The Japan Industrial Safety and Health
Association
Dr K. Morimoto National Institute of Health Sciences, Japan
Dr M. Nakadate National Institute of Health Sciences, Japan
Dr G. Ohmori The Jikei University School of Medicine, Japan
Dr Y. Saito National Institute of Health Sciences, Japan
Dr J. Sekizawa National Institute of Health Sciences, Japan
Dr T. Sohuni National Institute of Health Sciences, Japan
Dr M. Takeda National Institute of Health Sciences, Japan
Dr M. Takemura Ashiya University, Japan
Dr Y. Takenaka National Institute of Health Sciences, Japan
Dr S. Tanaka National Institute of Health Sciences, Japan
ABBREVIATIONS
BCF bioconcentration factor
BUN blood urea nitrogen
ECD electron capture detector
EDB 1,2-dibromoethane (ethylene dibromide)
FID flame ionization detector
GC gas chromatography
GSH glutathione
gamma-GT gamma-glutamyltranspeptidase
HECD Hall electron capture detector
LOEL lowest-observed-effect level
MS mass spectrometry
NADPH reduced nicotinamide adenine dinucleotide phosphate
NOEL no-observed-effect level
PIB piperonyl butoxide
SGOT serum glutamic-oxalic transaminase
SGPT serum glutamic-pyruvic transaminase
TEAM total exposure assessment methodology
TWA time-weighted average
UDS unscheduled DNA synthesis
VHH volatile halogenated hydrocarbon
VOC volatile organic carbon compound
1. Summary
1.1 Identity, physical and chemical properties, and analytical
methods
1,2-Dibromoethane (ethylene dibromide) is a colourless liquid
(melting point 9.9°C, boiling point 131.4°C) with a chloroform-like
odour. It is quite volatile, with a vapour pressure of 1.47 kPa
(11 mmHg) at 25°C and a vapour density compared to air of 6.1.
1,2-Dibromoethane is miscible with most organic solvents. Its
solubility in water is 4.3 g/litre at 30°C.
1,2-Dibromoethane in ambient air is analysed by GC after
absorption to porous polymers followed by rapid thermal desorption. A
purge-trap method is used for water samples. 1,2-Dibromoethane
residues in foods and other media can either be extracted by solvents
or be subjected to automated headspace analysis under cryogenic
conditions followed by analysis by GC and HPLC after derivatization.
1.2 Sources of human and environmental exposure
1,2-Dibromoethane is used as a scavenger of lead antiknock agents
in gasoline. It is also used as a soil fumigant and for fumigation of
grains and fruits. Reduced use of leaded gasoline in some countries
and cancellations of registrations for the use of 1,2-dibromoethane
for agricultural applications has reduced human exposure to
1,2-dibromoethane. However, it is still used as a lead scavenger in
gasoline in some countries, as a fumigant, for quarantine purposes, as
a solvent and as an intermediate for industrial chemicals.
1.3 Environmental levels and degradation
Concentrations of 1,2-dibromoethane measured in air range from
undetectable to the order of ng/m3 in urban areas. 1,2-Dibromoethane
has been found in ground water at up to 0.2 µg/litre and in surface
water at up to 50 µg/litre in areas of extensive agricultural use.
Although 1,2-dibromoethane leaches through soil, some is retained in
the soil matrix and may later contaminate irrigation wells. There is
a lack of information on microbial breakdown in soils.
The high volatility of 1,2-dibromoethane means that the major
environmental sink is the atmosphere. Stratospheric photolysis may
lead to the formation of breakdown products with ozone-depleting
potential.
1.4 Kinetics and metabolism in laboratory animals
1,2-Dibromoethane is rapidly absorbed orally, dermally and by
inhalation. Metabolites are thought to play an important role in
1,2-dibromoethane toxicity for humans. It can be metabolized by an
oxidative pathway (cytochrome P-450 system) and a conjugation pathway
(glutathione S-transferase system). Two reactive metabolites,
bromacetaldehyde formed via the oxidation pathway and thiiranium ion
formed via the conjugation pathway, are thought to interact with
cellular macromolecules (proteins, DNA) to form covalently bound
products.
1.5 Effects on laboratory mammals and in vitro test systems
1,2-Dibromoethane is acutely toxic to animals (oral LD50 for
rats of 146-417 mg/kg body weight, inhalation LC50 for rats of
3080 mg/m3 after a 2-h exposure, mortality observed following dermal
application of 210 mg/kg to rabbits). Toxic effects of
1,2-dibromoethane were mainly observed in the liver and kidneys.
Inhaled 1,2-dibromoethane vapour produced nasal irritation and
depression of the central nervous system. In rats exposed to
concentrations between 1540 and 77 000 mg/m3 (200-10 000 ppm) for
exposure durations between 0.1 and 16.0 h, deaths occurred in all
groups and were related to concentration and time. 1,2-Dibromoethane
(1.0% solution) caused irritation of shaved abdominal skin and eye
irritation in rabbits.
After oral subchronic exposure, mortality and decreases in weight
gain were observed in rats and mice at 100 mg/kg body weight per day.
Decreases in weight gain and nasal pathological effects were noted in
rats exposed to 1,2-dibromoethane at 115 mg/m3 (578 ppm) for
6 h/day, 5 days/week, for 13 weeks. The NOEL for histopathological
alterations of the nasal cavity was 23 mg/m3 (3 ppm) in this study.
In a similar study in mice, the same pathological changes were
observed, also with a NOEL of 23 mg/m3 (3 ppm).
After mice or rats were administered 1,2-dibromoethane by gavage
at 37-107 mg/kg body weight per day (TWA) for 49-90 weeks or mice were
administered 103-117 mg/kg body weight per day in drinking-water for
15-17 months, non-carcinogenic changes such as liver degeneration,
testicular atrophy, and forestomach acanthosis and hyperkeratosis in
addition to mortality were observed. After inhalation exposure (mice
or rats exposed to 77-388 mg/m3 for 6-18 months), inflammation of
the trachea and nasal cavity, testicular degeneration and hepatic
necrosis were observed.
1,2-Dibromoethane was not teratogenic in rats or mice following
inhalational exposure. Developmental toxicity (impairment of
development of motor coordination) was observed in the offspring of
male rats treated intraperitoneally with 1.25 mg/kg body weight per
day and in the offspring of female rats treated by inhalation
509 mg/m3, 4 h/day, 3 days/week from day 3 to day 20 of gestation.
1,2-Dibromoethane affected the reproductive performance of rats (in
males at the exposure level of 684 mg/m3, 7 h/day, 5 days/week, for
10 weeks, and in females at the exposure level of 614 mg/m3,
7 h/day, 7 days/week, for 3 weeks). The NOEL for this parameter was
300 mg/m3 in both sexes. The NOEL for reproductive performance of
male rats in a feeding study was 50 mg/kg per day after a 90-day
exposure. Spermatogenesis was affected in bulls following oral dosing
with 2 mg/kg per day for less than 21 days and in rabbits following
subcutaneous injection of 15 mg/kg for 5 days. Feeding of
1,2-dibromoethane caused diminution of egg size in hens after exposure
to 12.5 mg/kg per day for 12 weeks.
1,2-Dibromoethane did not induce dominant lethal mutations in
mice or rats, and did not produce chromosomal aberrations or
micronuclei in the bone-marrow cells of mice treated in vivo.
However, it was mutagenic in bacterial assays and caused single-strand
DNA breaks. Metabolites of 1,2-dibromoethane were covalently bound to
DNA, in vivo and in vitro. Sister chromatid exchange, mutations
and unscheduled DNA synthesis were observed in human cells in vitro.
Carcinogenicity studies involving oral administration (mice and
rats exposed by gavage to 37-107 mg/kg body weight per day (TWA) for
49-90 weeks; mice given 1,2-dibromoethane in drinking-water at
103-117 mg/kg body weight per day for 15-17 months), inhalational
exposure (mice and rats exposed at 10-40 ppm for 6-18 months) or skin
administration (25-50 mg/mice, 3 times/week for 400-594 days) showed
that 1,2-dibromoethane is carcinogenic to rats and mice, causing
tumours in a variety of organs (both at the application site and
distant sites, including the nasal cavity, lung, stomach, liver, skin,
circulatory system and mammary glands). In many cases it reduced the
latency period in developing tumours.
1.6 Effects on humans
1,2-Dibromoethane may produce adverse effects on the respiratory,
nervous and renal systems.
Acute (single) inhalation exposure to 1,2-dibromoethane at
215 mg/m3 (28 ppm) for 30 min or more has been shown to be fatal for
humans. Ingestion of 140 mg/kg body weight was fatal. Long-term
exposure to 1,2-dibromoethane (5 y) at a concentration of 0.68 mg/m3
in the breathing zone significantly decreased sperm counts and
fertility in occupationally exposed workers.
1.7 Effects on organisms in the environment
Few aquatic ecotoxicity studies have been performed with
1,2-dibromoethane. The LC50s for aquatic organisms are greater than
5 mg/litre. No information is available on terrestrial organisms.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
Chemical name 1,2-dibromoethane
(IUPAC):
Chemical structure: Br - CH2 - CH2 - Br
Molecular formula: C2H4Br2
Relative molecular 187.9
mass:
CAS chemical name: ethylene dibromide
CAS registry number: 106-93-4
Synonyms: sym-dibromothane, DBE, dibromo, bromure
d'ethylene, 1,2-ethylene dibromide, ethylene
bromide
Major trade names: Nematron, Nemafume, Bromofume, Dowfume W-85,
Aadibrom, Iscobrome D
Formulations: kerosene (30 and 97%),
emulsifiable concentrate (40 and 48%)
in combination with other pesticides
2.2 Physical and chemical properties
Appearance: colourless liquid with chloroform-like odour
Melting point: 9.9°C (Stenger, 1983)
Boiling point: 131.4°C (Stenger, 1983)
Vapour pressure: 1.47 kPa (11.0 mmHg)
(at 25°C) (Verschueren, 1983)
Vapour density: 6.1
Specific gravity: 2.172 (Stenger, 1983)
(at 25°C)
Refractive index (n20): 1.5379
Solubility in water: 4.3 g/litre at 30°C (Verschueren, 1983)
soluble in ether, etanol, benzene, acetone
(Weast et al., 1988)
Saturating concentration 113 g/m3 (at 20°C), 168 g/m3 (at 30°C)
in air:
Log Pow 1.76 or 1.93
Stability: decomposes gradually when exposed to light
1,2-Dibromoethane is flammable. Chemically, 1,2-dibromoethane is
a bifunctional alkylating agent.
2.3 Conversion factors
1 ppm = 7.68 mg/m3 (at 25°C);
1 mg/m3 = 0.13 ppm
2.4 Analytical methods
Analytical methods for volatile halogenated hydrocarbons (VHH)
are applicable to 1,2-dibromoethane. Determination of
1,2-dibromoethane is usually carried out by gas chromatography with
electron capture detection (GC-ECD). High resolution GC capillary
columns can be used for multiple analysis in high-resolution gas
chromatography (HR-GC) or high-resolution gas chromatography - mass
spectrometry (HR-GC-MS). A sensitive photoionization detector (Dumas
& Bond, 1982; Collins & Barker, 1983), a Hall electroconductivity
detector (Cairns et al., 1984) or mass spectrometry can also be used
for determination and confirmation of 1,2-dibromoethane. GC-ECD is
the most sensitive method.
The preconcentration of trace 1,2-dibromoethane in samples is
usually carried out through collection by cryogenic trapping or by
absorption on solid absorbents. The former is the preferred
preconcentration technique. Ice formation in the trap-tube during
sampling can be a problem, especially with ambient water and
homogenized food samples. Co-collected water can alter sample or
column flow rates in separation techniques that require subfreezing at
initial GC oven temperature (Pleil et al., 1987).
2.4.1 Air
A convenient analytical method for trace levels of
1,2-dibromoethane in ambient air is a combination of preconcentration
by absorption on porous polymers, such as Tenax, Porapack, Florisil,
silica-gel or charcoal, followed by rapid thermal desorption and
direct application for GC. Tenax GC resin is widely used for
1,2-dibromoethane sampling in ambient air (Barkley et al., 1980; Clark
et al., 1982, 1984a,b; Krost et al., 1982; Harkov et al., 1984),
although Porapack, Chromosorb, silica gel and charcoal have also been
used extensively (Kojima & Seo, 1976; Jagielski et al., 1978; Mann et
al., 1980). 1,2-Dibromoethane is absorbed by passing air samples
through the columns followed by thermal desorption and direct
application to GC. Alternatively, 1,2-dibromoethane in air is
collected by cryogenic cooling in capillary trap-tubes and then
thermally desorbed for GC analysis using trap-ovens with carrier gases
(Barkley et al., 1980; Harkov et al., 1984; McClenny et al., 1984;
Ballschmiter et al., 1986).
The relatively high concentrations of 1,2-dibromoethane in or
near fumigation chambers for foods and in automobile exhaust gases can
be directly determined by sampling with a gas-tight syringe followed
by GC analysis (Hasanen et al., 1979; Dumas & Bond, 1982; Morris et
al., 1982; Collins & Barker, 1983).
Analytical methods for measuring 1,2-dibromoethane in ambient air
are summarized in Table 1.
2.4.2 Water
A purge-trap method using absorbents such as Tenax GC and
Amberlite XAD-4 resin is the most effective concentration technique
for recovering 1,2-dibromoethane from water samples before GC
analysis. The GC test solution is prepared by eluting the absorbent
columns with a small volume of hexane (Spingarn et al., 1982;
Stottmeister et al., 1986). Another method for 1,2-dibromoethane
concentration is direct absorption on organic resins like Amberlite
XAD-1, 2, 4, 7 and 8, and XE-340 (Libbey, 1986). Direct absorption of
water samples on, for example, Amberlite XAD resins can be used for
the concentration of 1,2-dibromoethane in aquatic media (Libbey, 1986;
Woodrow et al., 1986).
Solvent extraction and headspace collection are simple methods
for recovering 1,2-dibromoethane from water samples (Saito et al.,
1978; Keough et al., 1984; Koida et al., 1986).
Analytical methods for measuring 1,2-dibromoethane in water are
summarized in Table 2.
2.4.3 Soils and sediment
1,2-Dibromoethane in sediments can be concentrated by a purge-
trap procedure, either after dilution of sediment samples with water
or after vacuum extraction from sediment samples into a cryogenically
cooled trap (Amin & Narang, 1985).
Analytical methods for measuring 1,2-dibromoethane in soils are
summarized in Table 3.
Table 1. Analytical methods for 1,2-dibromoethane in air
Collection from air Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected on Chromosorb extracted with hexane 1.5% OV-17+1.95% OV-210; 100 pg ECD Mann et al.
101 cartridge (20 ml); extracted with column temp: 75°C; in 70% FSRD (1980)
(10 mm i.d. × 10 cm) 1% MeOH in benzene, gas flow: N2
(ambient air) extract kept in a 70 ml/min
screw-capped test tube
and injected into GC
Collected directly with a applied directly with 5.5% DC-200+11% GF-1/Ga 0.1 ng ECD Morris et al.
gas-tight syringe gas-tight syringe Chrim Q (0.3 mm o.d. × 150 cm, (1982); Morris
stainless steel); column temp: & Rippon (1985)
90°C; gas flow: 40 ml/min
Collected directly with a applied directly with 5% Carbowax 20M/Chromosorb 2 µg PID Dumas &
gas-tight syringe gas-tight syringe W (3 mm i.d. × 200 cm, stainless FSRD Bond (1982)
steel); column temp: 120°C;
gas flow: N2 30 ml/min (portable
gas chromatograph)
Collected with a gas-tight applied by direct CPS-20 M (1/8- × 4 tefron tube); 1 ppb PID Cairns et al.
syringe (ambient air) injection column temp: ambient temp; (1984)
Collected on Tenax GC applied by rapidly Fused silica SP-2000 FSOT not given ECD Harkov et al.
cartridge at a flow rate of raising trap oven GC/MS (1984)
approx.300-1000 ml/min for temperature to 140°C
24 h; desorbed from the with purge of high
cartridge by heating purity N2 (50 ml/min)
rapidly at 250°C and
collected in an evacuated
stainless steel cylinder
under cryogenic conditions
using vacuum distillation
for 30 min (ambient air)
Table 1 (cont'd)
Collection from air Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected on a double applied by rapidly OV-1 fused silica capillary 1 ppb ECD McClenny et
loop of 0.32 mm (o.d.) heating the tube (-150 column (0.32 mm i.d. × 50 m); al. (1984)
nickel tubing packed with to 100°C in 55 sec) and column temp: -50°C (3 min)
60-80 mesh Pyrex beads cooling quickly (120 8°C/min 150°C (7 min)
under cryogenic conditions to -150°C in 225 sec) -50°C (10 min); gas flow:
(-150°C) (ambient air) H2 4 ml/min
a ECD = electron caputure detector; PID = photoionization detector; GC = gas chromatography; MS = mass spectrometry;
FSRD = full-scale recorder deflection
Table 2. Analytical methods for 1,2-dibromoethane in water
Collection Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Headspace method applied on a fused 1.J & W FSOT (0.326 mm i.d. × 30 m); 1.8 pg GC/MS Keough et al.
silica line Temp: column 70°C, ion source 299°C; (1984)
(0.25 mm i.d. × 100 cm) split ratio: 1:10; gas flow:
at 250-275°C with a 1.5 ml/min; ion dwell time:
gas-tight syringe 100 msec;
2.OV-1 FSOT (0.32 mm i.d. × 50 m); not given ECD
column temp: 60-90°C; split ratio:
1:1; gas flow: He 10 ml/min
N2 100 ml/min
Extracted with applied directly with a 1.5% OV-17 on Chromosorb W 0.05 mg/litre GC/MS Koida et al.
hexane micro-syringe (3 mm i.d. × 150 cm); temp: column (CI-NID) (1986)
200°C; separator 120°C; ion source
200°C; reaction gas: isotutane;
gas flow: H2 20 ml/min
Collected by purging applied by rapidly 0.2% Carbowax 1500 on Carbopack C 0.3 mg/litre FID Stottmeister et
into purge-trap thermal desorption (3 mm i.d. × 200 cm); column temp: al. (1986)
tubing (Tenax GC (200°C) 35°C (4 min), (8°C/min) 170°C
155 mg) at flow (20 min); gas flow: N2 4 ml/min
rate of 50 ml/min
for 30 min
Table 2 (cont'd)
Collection Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected by absorption extracted with ether 1.DB-1701 FSOT(0.25 mm i.d. × 30 m); 1 ppt ECD Woodrow et
on Amberlite and concentrated in column temp: 230°C; split ratio: al. (1986)
XAD-4 cartridge Kudernadanish 1:10; gas flow: H2 19 ml/sec at
column ball concentrator with 3 230°C; make-up gas Ar/CH4 20 ml/sec;
(4.7 mm o.d. × 12 cm) at Snyder column; applied 2.SE-54 FSOT (0.25 mm i.d. × 30 m);
flow rate of 10 ml/min with a microsyringe column temp: 100°C (5°C/min)
for 18-24 h 250°C (other conditions described
above)
a ECD = electron capture detector; GC = gas chromatography; MS = mass spectrometry; FID = flame ionization detector;
CI = chemical ionization; NID = negative ion detector
Table 3. Analytical methods for 1,2-dibromoethane in soil
Collection Preparation for GC GC conditions Minimum detection Detectora Reference
limit (amount)
Collected by steam fortify 50 ml of the not given ECD Abdel-Kader
distillation of an extract to folder paper et al. (1979)
aqueous slurry of soil and then measure with
into dry-ice-cooled molecular emission cavity
solvent (acetone: analyser; 4 mm × 4 mm
isooctane = 1:1) deep stainless steel
under N2 stream at cavity; gas flow:
60°C and drying H2 2.5 litre/min;
over Na2SO4 N2 4.0 litre/min;
wave length: 376 nm;
split: 1.4 nm
Collected on Prapack apply by thermal 4% OV-11 & 6% SP 2100 Supelcopor 7 ppb PID Amin &
N by purge of desorption of the (2.0 mm × 2.7 m); column temp: 40°C Narang (1985)
an aqueous slurry absorbent spiked with (5 min), (3°C/min) 70°C; gas flow:
of soil for 30 min fluorobenzene as an N2 30 ml/min, 15% SF-95 & 6% OV-225 1 ppb ECD
internal standard on Chromosorb W (2 mm i.d. × 3.6 m);
column temp: 60°C (10 min),
(3°C/min) 80°C (10 min) 65°C-75°C
(isothermal)
DB-5 FSOT(0.25 mm i.d. × 60 m) PID
column temp: 50°C (15 min) (4°C/min) ECD
170°C (14 min); gas flow:
He 60 cm/sec make-up gas He 8 ml/min
a ECD = electron caputure detector; PID = photoionization detector; GC = gas chromatography; FID = flame ionization detector
2.4.4 Food
Continuous extraction with hexane in a Dean-Stark apparatus for
one hour or soaking in a solvent solution of ethanol or acetone and
water (1 : 5) for 2 or 3 days is used for the analysis of
1,2-dibromoethane in agricultural crops and their products. A highly
sensitive method for the analysis of 1,2-dibromoethane in flour and
biscuits was developed by Rains & Holder (1981). Continuous
extraction with hexane is used for fruit, vegetables and grains
(Sekita et al., 1981, 1983; Kato et al., 1982; Iwata et al., 1983;
Konishi et al., 1985; De Vries et al., 1985; Alleman et al., 1986;
Nakamura, 1986).
Soaking in aqueous ethanol or acetone and water solution is used
for grains and their products (Clower, 1980; Daft, 1983, 1985, 1987;
Cairns et al., 1984; Barry & Petzinger, 1985; Sawyer & Walters, 1986;
Clower et al., 1986). For fruit (papaya and lemon), hexane, hexane-
water and acetonitrile are used. In the case of grain, intermediate
products, ready-to-eat products, corn bread mix, baby cereal and
bread, 1,2-dibromoethane can be extracted with an acetone-water (5+1)
solution, 0.1N HCl or light petroleum. Where necessary, Florisil
cleanup is useful for the removal of materials interfering with GC
analysis.
The purge-trap method on layers of Tenax TA (Heikes, 1985a,b),
Tenax resins (GC and TA) or Amberlite XAD-4 (Heikes & Hopper, 1986;
Daft, 1988) under a nitrogen gas stream is also used for the
collection of 1,2-dibromoethane from grains and their products. The
resins are eluted with hexane.
Automated headspace analysis is employed for measurement of
1,2-dibromoethane in fumigated crops in combination with GC-ECD and
GC-MS (Mestres et al., 1980; Entz & Hollifield, 1982; Gilbert et al.,
1985; Pranoto-Soetardhi et al., 1986). Equilibrium partitioning
between the samples and the gaseous headspace can be accelerated by
warming the vials. For detection, the gas chromatographic method with
an electron capture detector is used in all the above-mentioned
methods. Gas chromatography-mass spectrometry is also used. The
detection limit ranges from 0.1 µg/kg to a few µg/kg according to the
method used and the food being tested.
1,2-Dibromoethane can be analysed in animal feed by continuous
extraction with hexane in a Dean-Stark apparatus, followed by cleanup
on a Florisil column (Ishikuro, 1986).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
1,2-Dibromoethane is not a naturally occurring substance.
3.2 Anthropogenic sources
3.2.1 Production levels and processes
3.2.1.1 World production figures
In 1975, 1400 tonnes was produced in Japan. Production in
Belgium, France, Italy, the Netherlands, Spain, Switzerland and the
United Kingdom, was estimated to be between 3000 and 30 000 tonnes
(IARC, 1977).
3.2.1.2 Manufacturing processes
1,2-Dibromoethane is made by direct bromination of ethylene or
reacting hydrobromic acid with acetylene (Roskill, 1992).
3.2.2 Uses
Major uses of 1,2-dibromoethane are as a lead scavenger in
tetraalkyllead petrol and antiknock preparations, as a soil and grain
fumigant, as an intermediate in the synthesis of dyes and
pharmaceuticals, and as a solvent for resins, gums and waxes (IARC,
1977).
Reduction in the use of leaded gasoline from the late-1970s in
developed countries and of 1,2-dibromoethane for agricultural
applications in the 1980s, owing to its carcinogenicity in animals,
reduced human exposure to 1,2-dibromoethane. However, it is still used
in large amounts for many industrial purposes in developed countries,
and as a petrol additive in developing countries.
3.2.2.1 Petrol additive
1,2-Dibromoethane has been added to scavenge the inorganic lead
compounds (e.g., lead oxide and sulfate) remaining after fuel
combustion. Lead accumulation is prevented by the reaction of
1,2-dibromoethane with lead oxide to form volatile lead bromide, which
can pass from the combustion chamber to the atmosphere (IARC, 1977).
In 1981, use as a lead scavenger represented 83% of the
1,2-dibromoethane consumed (SRI International, 1982).
In 1972, 122 000 tonnes 1,2-dibromoethane was added to petrol
formulations in the USA; this figure declined to 73 000 tonnes in 1980
and to 24 000 tonnes in 1992 (Roskill, 1992). In 1992, sales of
unleaded petrol accounted for more than 90% of petrol in the USA. In
the European Community, all new vehicles must be fitted with
three-way convertors that can only use unleaded petrol by the
mid-1990s. This is also true of Japan, where almost all cars run on
unleaded petrol (Roskill, 1992).
In 1992, demand for 1,2-dibromoethane as a gasoline additive in
the USA was 24 000 tonnes and consumption outside the USA, principally
in Europe, was 25 000 to 30 000 tonnes, giving an estimated world
demand of 49 000 to 54 000 tonnes. The amount of 1,2-dibromoethane
used in Germany in 1989 was 980 tonnes, calculated on the basis of the
petrol consumed in the Federal Republic of Germany in 1989 (BUA,
1991). Legislation banning the use of lead in gasoline and controlling
the agricultural use of 1,2-dibromoethane has reduced world demand for
1,2-dibromoethane by at least 75% (Roskill, 1992).
3.2.2.2 Fumigant
The volatility of 1,2-dibromoethane allows it to be distributed
as a gas through substances such as soil in sufficiently high
concentrations to kill target pests. Its chemical and biocidal
properties allowed it to be effectively utilized in a wide range of
applications. Its primary pesticidal use has been as a soil
nematocide (Pignatello & Cohen, 1989).
1,2-Dibromoethane has been used in the spot fumigation of grain
milling machinery, post-harvest fumigation of grain, and in the
control and prevention of infestations in produce. Additional minor
uses have been the control of bark beetles in felled logs, moths in
stored furniture and clothing, termites under concrete slab
foundations and porches, Japanese beetles in balled ornamental trees
and grass sod, and wax moths in stored honeycombs and beehive
superstructures.
In post-harvest grain fumigation of barley, maize, oats, rice,
rye, sorghum and wheat, 1,2-dibromoethane has often been used in
conjunction with 1,2-dichloroethane (ethylene dichloride) or carbon
tetrachloride.
Residues of 1,2-dibromoethane in tropical fruits, imported wheat
and beans have been prohibited in Japan (MHW, 1985, 1987, 1988). Use
of 1,2-dibromoethane for agricultural purposes has been prohibited in
Egypt, Kenya, the Netherlands, Sweden, the United Kingdom and the USA
(BUA, 1991; IRPTC, 1993). However, it is still used for quarantine
purpose in some countries.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
The use of 1,2-dibromoethane on a field contaminated both the
field and crops for 2 years (Yuita, 1984). About 10% of bromine-
containing pesticides was retained, in the form of bromine, in the
soil and crops. The remaining 90% seemed to have moved to underground
water and rivers.
4.1.1 Air
The atmospheric chemistry of bromine compounds has received
attention because of the role that they play in the depletion of the
stratospheric ozone layer. Wofsy et al. (1975) suggested that bromine
atoms can be more effective than chlorine atoms in the catalytic
destruction of ozone. A major uncertainty is the absolute
concentration of bromine compounds in both the troposphere and the
stratosphere.
4.1.2 Soil
Injection of 1,2-dibromoethane as a soil fumigant at 70 kg/ha
into fine sandy loam resulted in a concentration of 130 µg/kg nearly
one year later (Steinberg et al., 1987).
The disappearance with time of 1,2-dibromoethane was measured in
a sediment-water mixture (ratio 0.075) and a half-life of 55 h was
calculated (Jafvert & Wolfe, 1987).
Important factors influencing the movement of soil fumigants
include their physical and chemical characteristics, temperature,
moisture, presence of organic matter, soil texture and soil profile
variability (Munnecke & Van Gundy, 1979).
1,2-Dibromoethane is moderately hydrophillic, having a calculated
octanol-water partition coefficient of 58 (Lyman, 1982). At
environmental levels (10-1000 ppb), 1,2-dibromoethane has a soil
organic carbon partition coefficient of 66 ml/g (Rogers & McFarlane,
1981).
1,2-Dibromoethane has a low vapour pressure and moves slowly in
the vapour phase. Little, if any, mass flow occurs except in
extremely warm soil or when water is applied. Soil temperature is
important and may affect 1,2-dibromoethane movement in several ways.
A rise in temperature increases the vapour pressure and decreases the
solubility. This alters the phase distribution and results in an
increase in the rate of diffusion of 1,2-dibromoethane through soils.
Fumigation of warm soils (25°C) results in a faster rate and greater
distance of nematicide diffusion. In colder soils (5°C), the rate of
diffusion is slower and the persistence of the chemical is longer, but
the total distance of diffusion of an effective dosage is decreased.
The approximate movement and fate of 1,2-dibromoethane in two soils
were predicted using extrapolations from laboratory experiments and
soil-vapour phase concentrations obtained from simulated field
experiments. The most far-reaching diffusion patterns in mineral
soils are those obtained in soils whose moisture content is nearest
the wilting point of plants (15 bars moisture tension). As the
moisture content of the soil is increased, the diffusion pattern
gradually becomes more limited. The soil texture and type determine
to a large extent the amount of soil moisture present and the size of
the connecting air spaces. Soil air space and the size of pores are
important because these chemicals move primarily in the vapour phase
and smaller pores are most easily blocked when water is present. A
material balance for 1,2-dibromoethane was surveyed when
1,2-dibromoethane (equivalent to 47 litres/ha) was applied under
various conditions to several soils using a soil fumigation technique
in both field and laboratory experiments. Most of the
1,2-dibromoethane was accounted for; the remainder was mostly
irreversibly adsorbed or lost during sampling. The 1,2-dibromoethane
not accounted for represented between 10 and 40%. After 3 days at
15°C, about 40% of the 1,2-dibromoethane was absorbed in the
soil-particle phase, 25% was in the soil-water phase, and 20% remained
in the liquid state (McKenry & Thomason, 1974).
1,2-Dibromoethane soil fumigation is used for the control of
plant parasitic nematodes on high value crops. In Ontario, Canada,
soil types fumigated varied from loamy sand to muck. Three soils
differing in texture (Fox loam sand, Vineland silt loam and Lincoln
clay) were studied for penetration of 1,2-dibromoethane (Townshend et
al., 1980). Fox loam sand (highest content of sand and lowest of
organic matter) showed the most rapid penetration; moisture level,
temperature and their interactions had the greatest effects on
movement of 1,2-dibromoethane. On Vineland silt loam (medium-textured
soil) the degree of penetration was dependent on moisture, temperature
and bulk density, and there were relatively small interaction effects.
On Lincoln clay (high content of organic matter and fine-textured
soil) 1,2-dibromoethane did not move in the soil, regardless of
edaphic factors, thus explaining the difficulty of using
1,2-dibromoethane fumigation to control nematodes in clay.
1,2-Dibromoethane persists in top soil at µg/kg levels for at
least 20 years, despite its predicted lability in the environment
(high water solubility and low soil-water partition coefficient).
Misleading results were obtained when studies of microbial
degradation, sorption, desorption and analytical recovery were
conducted with freshly spiked soils or sediments (Pignatello, 1986).
1,2-Dibromoethane can serve as a C1 unit and energy source for some
soil aerobic or anaerobic microorganisms. However, residual
1,2-dibromoethane is strongly bound to soils and can only be extracted
from them by warming with polar solvents. Surfactants showed no
enhanced extraction ability. Thermal desorption at temperatures as
high as 200°C in an N2 stream resulted in more decomposition than
desorption, while a fresh spike of 14C-labelled 1,2-dibromoethane
was recovered quantitatively.
Diffusion of residual 1,2-dibromoethane from soil to water is
very slow and highly temperature-dependent (diffusion coefficient:
10-16 cm2/sec) (Pignatello et al., 1987). 1,2-Dibromoethane, when
present as a groundwater contaminant in areas where it had been used
as a soil fumigant, was degraded anaerobically by microorganisms in
two types of soils from 1,2-dibromoethane-contaminated groundwater
discharge areas. At initial concentrations of 6 to 8 µg/litre,
1,2-dibromoethane was degraded in a few days to near or below the
detection limit (0.02 µg/litre). At 15 to 18 µg/litre degradation was
slow. Bromide ion released at the higher concentration was 1.4 ± 0.3
and 23.1 ± 0.2 molar equivalents for the two soil types. A study
using 14C-1,2-dibromoethane showed that 1,2-dibromoethane was
converted to approximately equal amounts of CO2 and cellular carbon;
only small amounts of 14C were not attributable to these products.
However, 1,2-dibromoethane was not degraded in autoclaved soil water
samples. The results suggested that, initially, microbial degradation
of 1,2-dibromoethane in the topsoil was too slow to prevent leaching
of large quantities to groundwater. With continued application the
microbial community may have adapted to the higher levels and
degradation rates increased; this has been observed with other
agricultural chemicals. The results of acetate incorporation studies
suggested that the highest application rates of 1,2-dibromoethane are
definitely toxic to topsoil microbial communities.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
1,2-Dibromoethane enters the atmosphere from its use as a petrol
additive to scavenge the lead oxide resulting from the combustion of
alkyllead antiknock additive, and from its use in agriculture as an
insecticidal and fungicidal fumigant.
Nilsson et al. (1987) reported that the exhaust gas of chain saws
fuelled with petrol contained a mean 1,2-dibromoethane level of
0.0008 (0.0001-0.001) mg/m3 under snow-free conditions and
0.002 (0.0001-0.005) mg/m3 with snow on the ground during the
winter. 1,2-Dibromoethane levels are around 45% higher in cold start
than in hot start conditions and have a tendency to decrease with
increasing vehicle speed (see Tables 4 and 5). Concentrations of
1,2-dibromoethane in raw, undiluted exhaust from vehicles using leaded
petrol are in the range of 55-146 µg/m3 (7.2-19 ppb), 46-122 µg/m3
(6.0-16 ppb) and 38-115 mg/m3 (5.0-15 ppb) under the conditions of
USA Federal test driving, idle, and a steady speed of 30 mph,
respectively (Jacobs, 1980). Based on these levels, 1,2-dibromoethane
concentrations in air alongside roads due to vehicle exhaust emissions
may range from 0.04 to 122 µg/m3 (0.005-0.19 ppb). These results
are similar to the observations of Leinster et al. (1978).
Table 4. 1,2-Dibromoethane produced by motor vehicles (petrol engine)
under constant speed test conditions
Vehicle speed km/h Concentration (µg/m3)
Engine not Vehicle with Vehicle with
defineda 3 litre engineb 0.85 litre engineb
Cold start (idle) 70 878 332
10 78
30 62-70 618 165
40 61
50 2 669 155
64 180 139
80 98 135
a Leinster et al. (1978)
b Tsani-Bazaca et al. (1981)
Table 5. 1,2-Dibromoethane in the exhaust emission of motor vehicles
(petrol engine) (µg/m3)a
Conditions 3 litre engine 0.85 letre engine
ECEb
cold start 292-560 733-538
hot start 200-234 533-538
ECD/CVSc 14-25 26-34
USA Federald
cold start 48 29
hot start 22
a From: Tsani-Bazaca et al. (1981)
b standard European driving cycle
c ECE driving cycle under constant volume sampling condition
d 1973 driving cycle
1,2-Dibromoethane levels in air have been measured at several
sites around the world (Table 6). Leinster et al. (1978) concluded
that the lower levels during the autumn were the result of a reduction
in evaporative loss particularly from parked vehicles (the calculated
evaporation rate for 1,2-dibromoethane at 5°C is less than one third
of that at 30°C). An indication of the magnitude of evaporative loss
from parked vehicles was provided by levels of 0.02-0.05 µg/m3
measured in a car park. It was also probable that an opposite trend
would be produced by a change in driving conditions. For example,
cold starts and driving speeds of vehicles have a marked influence on
the 1,2-dibromoethane content of exhaust emissions.
The 1,2-dibromoethane added to leaded petrol contributes to a
large amount of methyl bromide in urban atmospheres. IPCS (1995)
estimated that per annum between 7000 and 18 000 tonnes of methyl
bromide could be emitted from car exhausts. Reactions in the lower
troposphere with hydroxyl radicals and other chemical species are the
most important of the possible removal mechanisms within the
atmosphere (UNEP, 1992). The end-products of both photodissociation
of methyl bromide and reactions with hydroxyl radicals in the
atmosphere include bromide species (BUA, 1987). Active bromine
species react with ozone mainly in the lower stratosphere and are
thought to be partly responsible for the destruction of the ozone
layer. However, 1,2-dibromoethane was not included as a controlled
substance in the "Montreal Protocol on Substances that Deplete the
Ozone Layer".
Table 6. Environmental concentrations of 1,2-dibromoethane
Location Measuring period Concentration (µg/m3) Reference
London August 1976a 0.08-0.09 µg/m3 Leinster et al. (1978)
December 1976b 0.001-0.01 µg/m3
12 Canadian cities 1989-1992 mean 0.05 ± 0.05 µg/m3 Environment Canada
range n.d.-1.73 µg/m3 (1994)
Busy streets at 2 m height 0.07-1.26 µg/m3 Tsani-Bazaca et al.
and 5 m from kerbside (1981)
Los Angeles, California, USA 9-21 April 1979 0.25 ± 0.20 ng/m3 (33.2 ± 26.2 ppt) Singh et al. (1981)
range 0.041-1.4 ng/m3 (5.4-187.2 ppt)
Oakland, California, USA 28 June-10 July 1979 0.12 ± 0.10 ng/m3 (15.8 ± 12.5 ppt) Singh et al. (1981)
range 0.018-0.65 ng/m3 (2.4-84.5 ppt)
Phoenix, Arizona, USA 23 April-6 May 1979 0.31 ± 0.29 ng/m3 (40.3 ± 38.3 ppt) Singh et al. (1981)
range 0.018-1.6 ng/m3 (2.4-204.4 ppt)
Background 38 ng/m3
Various cities (7) 1-2 weeks in 1980 0.122-0.453 ng/m3 (0.016-0.059 ppt)c Singh et al. (1982)
2.826 ng/m3 (0.368 ppt)d
Denver, Colorado, USA 1-2 weeks n.d.-2.304 µg/m3 (0.3 ppb) Going & Spigarelli (1976);
Leinster et al. (1978)
Sites in New Jersey, USA late 1983-Spring 1984 0.077-5.4 µg/m3 Harkov et al. (1985)
Summer 1981 < 0.038 µg/m3 Harkov et al. (1983)
Winter 1982 < 0.038 µg/m3 Harkov et al. (1983)
Table 6 (cont'd)
Location Measuring period Concentration (µg/m3) Reference
Central London Summer 1982 0.23 µg/m3 (0.03 ppb)e,f Clark et al. (1984a,b)
Exhibition Road May-August 1983 0.23 µg/m3 (0.03 ppb)e
Rural site Summer 1982 0.12 µg/m3 (0.015 ppb)e,g Clark et al. (1984a,b)
Silwood Park, United Kingdom May-August 1983 0.15 µg/m3 (0.019 ppb)e
Motorway outside London, Summer 1982 0.39 µg/m3 (0.05 ppb)e,h Clark et al. (1984a,b)
Toddington May-August 1983 0.31 µg/m3 (0.04 ppb)e
Central London Summer 1982 1.0 µg/m3 (0.13 ppb)i Clark et al. (1984a,b)
May-August 1983 0.62 µg/m3 (0.08 ppb)i
Motorway site Summer 1982 2.0 µg/m3 (0.26 ppb)i Clark et al. (1984a,b)
May-August 1983 1.2 µg/m3 (0.15 ppb)i
Anchorage (Alaska) March 1983 31-177 ng/m3 (4-23 ppt) Berg et al. (1984)
Barrow (Alaska) March 1983 n.d.-177 ng/m3 (n.d.-28 ppt) Berg et al. (1984)
Mould Bay Coast (Alaska) March 1983 38-284 ng/m3 (5-37 ppt) Berg et al. (1984)
Thule (Greenland) March 1983 15-246 ng/m3 (2-32 ppt) Berg et al. (1984)
North Pole March 1983 92.2 ng/m3 (12 ppt) Berg et al. (1984)
Ny-Alesund (Norway) March-April 1983 31-150 ng/m3 (4-20 ppt) Berg et al. (1984)
Table 6 (cont'd)
Location Measuring period Concentration (µg/m3) Reference
Bear Island (Norway) March-April 1983 23-100 ng/m3 (3-13 ppt) Berg et al. (1984)
Bodo (Norway) March-April 1983 38-110 ng/m3 (5-14 ppt) Berg et al. (1984)
a temperature range 28-30°C
b temperature range 4-8°C
c average level for each city
d maximum concentration found in Houston, Texas, USA
e mean hourly concentrations
f range 0.078-1 µg/m3 (0.01-0.13 ppb)
g range ND-0.78 µg/m3 (ND-0.01 ppb)
h range 0.07-2.0 µg/m3 (0.009-0.26 ppb)
i maximum hourly concentrations
1,2-Dibromoethane levels of 0.07-1.26 µg/m3 have been found in
busy streets. Higher levels were found in a road tunnel and were
associated with poor ventilation (Tsani-Bazaca et al., 1981). Three
field studies on the measurement of selected potentially hazardous
organic compounds in urban environments were conducted in the USA in
1979 (Los Angeles, California, 9-21 April; Oakland, California,
28 June-10 July; and Phoenix, Arizona, 23 April-6 May). These studies
were performed to characterize the atmospheric abundance, fate and
human exposure to these compounds (Table 6). The background
concentration of 1,2-dibromoethane was 38 ng/m3 (5 ppt). Assuming
an average respiratory volume of 23 m3 at 25°C and 1 atm for a 70-kg
male, the average daily dose (µg/day) of 1,2-dibromoethane at these
locations can be calculated as 6.0 ± 2.7 for Los Angeles, 2.9 ± 0.8
for Oakland and 7.0 ± 2.7 for Phoenix. The ratios of
1,2-dibromoethane to total haloethane and VHH (volatile halogenated
hydrocarbons) in the average daily doses were 2.7% and 0.57%, 5.6% and
1.14%, and 2.8% and 1.03%, respectively. The chemical loss rate of
1,2-dibromoethane was 2.8% per day (sunlight = 12 h). There was
diurnal variation in 1,2-dibromoethane levels at the three locations.
The afternoon minimum at Phoenix was attributed to deep vertical
mixing associated with hot and dry weather. The afternoon maximum at
Oakland was most likely a result of transport from upwind sources
(Singh et al., 1981).
Other studies measuring 1,2-dibromoethane in the ambient
atmosphere of urban and rural areas have been performed (Going & Long,
1975; Going & Spigarelli, 1976). Sources of 1,2-dibromoethane in air
were considered to be emissions from stations dispensing leaded petrol
and evaporative emissions from motor vehicles using leaded petrol.
Atmospheric levels of 1,2-dibromoethane were low (0.046 to
3.5 µg/m3) (0.006 to 0.45 ppb) in worst case conditions near petrol
stations and with heavy traffic in cities. These levels are 10 to
10 000 times less than the occupational exposure level of 1 mg/m3
(0.13 ppm) for 15 min recommended by the US National Institute of
Occupational Safety and Health (Jacobs, 1980).
Tsani-Bazaca et al. (1981) monitored the concentrations of VHH
collected in 1979 at several locations and utilizing vehicles
operating under various conditions on a busy road in central London
(2000 vehicles/h), a poorly ventilated tunnel (1600 vehicles/h at peak
traffic flow), and a semi-rural industrialized area. The
concentration of 1,2-dibromoethane varied between 0.07 and
1.26 µg/m3. There was a good correlation between 1,2-dibromoethane
and benzene concentrations (correlation coefficient : 0.93) at the
three locations and a higher correlation between 1,2-dibromoethane and
1,2-dichloroethane (correlation coefficient : 0.94).
In 1983, 54 air samples at 6 urban sites and 54 air samples at
6 mountainous sites were collected in Japan and were analysed for the
presence of 1,2-dibromoethane. A total of 35 samples from 5 urban
sites contained 1,2-dibromoethane at concentrations of
0.008-0.322 µg/m3 (0.001-0.042 ppb). The detection limit was
0.005-0.008 µg/m3 (0.0007-0.001 ppb). A total of 36 samples from
5 mountainous sites contained 1,2-dibromoethane at concentrations of
0.008-0.515 µg/m3 (0.001-0.067 ppb). The detection limit was
0.002-0.008 µg/m3 (0.0003-0.001 ppb) (Environment Agency Japan,
1985).
Urban 1,2-dibromoethane levels at seven sites in selected cities
in the USA in 1980, using on-site and real-time measurement instrument
following a 24-h measurement schedule for a period of 1-2 weeks, were
0.12-0.45 µg/m3 (16-59 ppt) (Singh et al., 1982). The average
concentration of 1,2-dibromoethane did not exceed 0.015-0.46 µg/m3
(0.06 ppb) (average range 0.002-0.06 ppb) at any study site and
average levels ranged from 0.122 µg/m3 (0.016 ppb) at St. Louis,
Missouri, to 0.46 µg/m3 (0.059 ppb) at Houston, Texas. The maximum
concentration of 2.83 µg/m3 (0.368 ppb) was found at Houston. In
general, the highest average levels were found during the night and
early morning. In the case of Denver, Colorado, typical ambient
concentration data suggested a range of not detectable to 2.3 µg/m3
(0.300 ppb) (Going & Spigarelli, 1976; Leinster et al., 1978).
The Office of Science and Research (USA) monitored VHH in ambient
air at listed abandoned hazardous waste sites and sanitary landfills
in New Jersey (Harkov et al., 1985). 1,2-Dibromoethane was found at
mean levels of 2.1 µg/m3 (0.27 ppb), 2.2 µg/m3 (0.288 ppb),
3.6 µg/m3 (0.47 ppb), 5.4 µg/m3 (0.7 ppb), 0.38 µg/m3
(0.05 ppb), 0.077 µg/m3 (0.01 ppb) and 0.15 µg/m3 (0.02 ppb) at
different sites during late 1983 and early 1984. It was below the
detection limit 0.038 µg/m3 (0.005 ppb) at three sites during the
summer of 1981 (Harkov et al., 1983) and the winter of 1982 (Harkov et
al., 1984).
Ambient air monitoring survey of VHH at a busy road in central
London, a rural site and a motorway location near London showed mean
hourly 1,2-dibromoethane concentrations of 0.23, 1.2 and 0.39 µg/m3
(0.03, 0.15 and 0.05 ppb), respectively, in summer 1982 and 0.23, 0.15
and 0.31 µg/m3 (0.03, 0.019 and 0.04 ppb) between May and August
1983 (Clark et al., 1984a,b). The maximum hourly concentrations of
1,2-dibromoethane at the urban and motorway sites were 1.0 and
2.0 µg/m3 (0.13 and 0.26 ppb) in 1982, and 0.61 and 1.2 µg/m3
(0.08 and 0.15 ppb) in 1983, respectively. 1,2-Dibromoethane
concentrations at the urban site were in the same ranges
0.07-0.31 ng/m3 (0.01-0.04 ppt) as those measured by other workers
(Leinster et al., 1978; Tsani-Bazaca et al., 1981; Singh et al.,
1982). The low concentrations found at the rural site were primarily
related to the low incidence of vehicular pollutant sources in the
area. However, the site was near the urban fringe of London and near
several small towns and this may explain occasional elevated
concentrations.
1,2-Dibromoethane concentrations were measured at Point Arena,
California between 1979 and 1981; the background level of
1,2-dibromoethane in the troposphere was found to be less than
0.023 µg/m3 (3 ppt) (Singh et al., 1983).
Berg et al. (1984) measured atmospheric 1,2-dibromoethane
concentrations at eight arctic sites in 1983. Concentrations at three
sites in Alaska (Anchorage, Barrow, Mould Bay Coast) in March were
0.031-0.177 µg/m3 (4-23 ppt), not detectable to 0.22 µg/m3
(29 ppt) and 0.038-0.284 µg/m3 (5-37 ppt), respectively.
1,2-Dibromoethane levels in Greenland (Thule) and at the North Pole in
March were 0.015-0.246 µg/m3 (2-32 ppt) and 0.096 µg/m3 (12 ppt),
respectively. Those at Norwegian sites (Ny-Alesund, Bear Island,
Bodo) during March-April were 0.031-0.15 µg/m3 (4-20 ppt),
0.023-0.10 µg/m3 (3-13 ppt) and 0.038-0.11 µg/m3 (5-14 ppt),
respectively. The mean concentration ± standard deviation was
0.084-0.77 µg/m3 (11 ± 10 ppt). Other organobromine compounds, such
as methyl bromide, methylene dibromide and bromoform, were detected at
similar concentrations.
Monthly monitoring of the atmosphere of Barrow, Alaska (72 °N),
showed that the 1,2-dibromoethane concentration was higher in winter
than in other seasons, although the monthly average concentrations did
not differ greatly (7.68-10.7 ng/m3) (1.0-1.4 ppt) except in
January. From the results of atmospheric VHH monitoring, Rasmussen &
Khalil (1984) suggested that VHH in arctic air might be an indicator
of polluted air transported from industrial mid-latitude sources.
5.1.2 Water
Widespread use of 1,2-dibromoethane as a soil fumigant in the USA
resulted in its detection in both groundwater and surface water in
California, Florida, Georgia, and Hawaii (Sun, 1984), Connecticut
(Isaacson et al., 1984) and New Jersey (Page, 1981), and in wells used
for irrigation in Georgia (Martl et al., 1984). 1,2-Dibromoethane
was reported in groundwater in Georgia, California, Florida, and
Hawaii by US EPA (1986).
Laboratory studies have shown that 1,2-dibromoethane
photohydrolyses rapidly in aqueous solutions when irradiated. The
degradation is a two-stage process in which 1,2-dibromoethane is
converted to bromoethanol (half-life, 7.6 min) and then to ethylene
oxide (half-life, 64 min). Further degradation to ethylene glycol was
less influenced by light, as shown by a half-life of 10 days (Castro &
Belser, 1978). While the above study provides some understanding of
aqueous degradation, Logan (1988) cautions that the efficiency of the
photo-reactions were not reported in terms of quantum yield.
1,2-Dibromoethane was found in ground- and surface water in New
Jersey (over 1000 different wells and 600 different sites) during
1977-1979; the highest levels were 0.2 µg/litre in surface water and
48.8 µg/litre in groundwater (Page, 1981).
Analyses of 350 well water samples from Connecticut in 1984
revealed concentrations of up to 2 µg/litre. 1,2-Dibromoethane was
rapidly lost from water samples exposed to the atmosphere or boiled
for few minutes. It could not be detected in water samples purged
with nitrogen for 10 min (Isaacson et al., 1984).
In southwest Georgia, USA, agricultural practices involve
intensive use of groundwater for irrigation and pesticides for control
of plant and insect pests. 1,2-Dibromoethane was found at levels of
between 1 and 90 µg/litre in water samples from three irrigation wells
collected between 1981 and 1983. Application at ratios of
14-19 litres/ha) near wells showed that 1,2-dibromoethane
concentrations in the aquifer did not appear to be directly related to
the application rate of the compound to the surface. The
concentrations in the wells may reflect application of the compound at
sites some distance from the wells (Martl et al., 1984).
In 1982, 27 water samples and 27 bottom sediment samples were
collected at nine sites in Japan and were analysed for the presence of
1,2-dibromoethane. None of the water or bottom sediment samples
contained 1,2-dibromoethane. The detection limit was 0.3-2 µg/litre
for water and 0.0016-0.01 µg/kg for bottom sediment (Environment
Agency Japan, 1985).
In 1983, 1,2-dibromoethane surveillance of the water of six rural
wells in Ibaraki prefecture, Japan, where 1,2-dibromoethane was used
for soil fumigation or as a pesticide on pine tree, showed no
1,2-dibromoethane contamination (detection limit, 5 µg/litre) (Nemoto
et al., 1984).
Groundwater samples from nine sites in and around vegetable-
growing areas in Gifu Prefecture, Japan, were collected twelve times
between July 1983 and December 1984. 1,2-Dibromoethane levels ranged
from 0.06 to 0.55 µg/litre at seven sites and the mean values of
1,2-dibromoethane at each site varied between 0.15 and 0.28 µg/litre.
1,2-Dibromoethane levels in groundwater around the vegetable-growing
areas did not differ from those within the areas, where
1,2-dibromoethane application was limited to once a year in the first
two weeks of July. Sites where 1,2-dibromoethane were detected around
these areas overlapped completely the stream of groundwater coming
from these areas (Terao et al., 1985). The annual variation of
concentrations in the groundwater was small. 1,2-Dibromoethane
concentration showed good correlation with bromine ion concentration
and bromine ion/chlorine ion ratio at each site (Terao et al., 1984).
Mayer et al. (1991) studied 1,2-dibromoethane concentrations in
detail in water from a domestic well, approximately 10 m deep, in a
fruit growing area of Whatcom County, Washington, USA where
1,2-dibromoethane had been used extensively prior to its 1983 ban.
Additional wells (n = 107) were also sampled over a 4-year period; no
details of well depths were given. Correlation analysis showed no
relationship between 1,2-dibromoethane concentration in water and
temperature but significant negative correlation between precipitation
and 1,2-dibromoethane. The analysis allowed lag times of between
0 and 12 months; a 3-month lag was found to give the best relationship
between precipitation and 1,2-dibromoethane in the water. The
dilution effect of precipitation was followed by slow
1,2-dibromoethane infiltration from overlying soils which tended to
re-establish prior concentrations over about 3 months. The authors
stated that water contamination can result from such continuing
infiltration of soil-matrix-derived 1,2-dibromoethane long after
agricultural use has ceased.
5.1.3 Food
Beckman et al. (1967) reported that part of the inorganic bromine
in foods and raw agricultural commodities comes from the soil.
1,2-Dibromoethane was applied annually at 54 kg/ha and samples from
40 crops grown in soil treated with 1,2-dibromoethane were analysed
over a 3-year period. In general, leafy portions of plants contained
the highest levels of bromide on the basis of weight. Residue levels
were calculated as inorganic bromide ion present in the crop after
harvest. Levels in crop samples from untreated soil were less than
1.6 mg/m3 (0.2 ppm), and the highest level in crops from treated
soil was 137 mg/kg (17.8 ppm) in sugar beet tops. Most of the crops
were harvested about 100 days after soil treatment but time from
treatment to harvest ranged from 55 days for strawberries to 10 years
for walnuts.
1,2-Dibromoethane was absorbed strongly by cereal, grains, cereal
products and other produce during the fumigation period. Even when
normal ventilation procedures were followed, residues of
1,2-dibromoethane disappeared very slowly. Nearly all the
1,2-dibromoethane was physically sorbed and at normal temperatures
there was little formation of inorganic bromide. However,
occasionally in produce at higher temperatures and moisture content
there was rapid breakdown to inorganic bromide (Heuser & Scudamore,
1967).
Levels of 1,2-dibromoethane in wheat were between 10 and
20 mg/kg, and, for its products, between 2 and 4 mg/kg in flour,
0.002-0.04 in white bread and 0.006-0.16 in wholemeal bread. When
flour was treated directly with 1,2-dibromoethane, ventilated
thoroughly, and then baked into loaves, there were residues of
20-24 mg/kg in the flour and 0.33-0.47 mg/kg in the bread (FAO/WHO,
1972). Desorption of 1,2-dibromoethane occurred at low (14-17°C)
rather than high (30-37°C) temperatures, and was abolished by grinding
the grain (Bielorai & Alumot, 1975).
Rappaport et al. (1984) reported that the decay of the outgassing
rate over time from fumigated oranges was approximately first order.
Outgassing was significantly slowed by reducing either the temperature
or the ventilation rate. In laboratory trials, ventilation at
0.6 air changes/h removed 1,2-dibromoethane vapours from the surface
of oranges, and prevented reabsorption onto the fruits.
A pesticide formulation, consisting of carbon tetrachloride (CT),
1,2-dichloroethane (EDC), 1,2-dibromoethane in 63 : 30 : 7 w/w
proportions, was applied to 27.3 tonnes of wheat stored in a paper
laminate bin (Berck, 1974). The CT-EDC-1,2-dibromoethane distribution-
persistence patterns were monitored at 16 bin locations over a 14-day
period by GC. Fumigant residues in the wheat, in flour, bran, and
middlings derived from the wheat, and in bread baked from the flour
were determined over a 7-week period. 1,2-Dibromoethane residues in
the wheat varied, depending on the bin location and contact time, and
ranged from 0 to 3.3 mg/kg. Residues in bran and middlings were
greater than those in flour, and ranged from 0 to 0.4 mg/kg. No
1,2-dibromoethane residues were found in any of the bread tested
(detection limit, 10 ng/kg).
1,2-Dibromoethane levels were studied in biscuits (22 samples)
and flour (22 samples), the biscuits being baked from each of the
flour samples for 12 min at 268°C. After baking, the samples were
sealed in plastic bags and frozen to prevent any further loss of
1,2-dibromoethane. Flour samples were also sealed in plastic bags and
frozen. Levels of 1,2-dibromoethane in flour and biscuits ranged from
non-detectable to 4.2 mg/kg and to 0.26 mg/kg, respectively. There
was poor correlation between the levels of 1,2-dibromoethane in flour
and biscuits.
5.2 Occupational exposure
Air concentrations of 1,2-dibromoethane in ventilated containers
dropped from several ppm immediately after fumigation to a few ppb
after 5-10 days; levels remained between 15 and 23 mg/kg (2 and 3 ppm)
for 15-20 days during unventilated, refrigerated storage. Results of
experiments on a laboratory scale (0.25 carton) and a large scale
(400 cartons) suggested that workers transporting and distributing
fumigated citrus fruit could routinely be exposed to airborne
1,2-dibromoethane at concen trations greater than 998 µg/kg (130 ppb)
(OSHA, 1983).
The US National Institute for Occupational Safety and Health
(NIOSH) estimated that approximately 108 000 workers in the USA were
potentially exposed to 1,2-dibromoethane in their workplaces (Table 7)
and that another 875 000 workers handling leaded petrol were exposed
to very low levels (NIOSH, 1981). There is no estimate of the number
of motorists exposed to 1,2-dibromoethane during self-service
operations at filling stations.
Table 7. Occupations with potenial exposure to 1,2-dibromoethanea
Antiknock compound makers Motor fuel workers
Cabbage growers Oil processors
Corn growers Organic chemical synthesizers
1,2-Dibromoethane workers Petrol blenders
Drug makers Resin makers
Fat processors Seed corn maggot controllers
Fire-extinguisher makers Soil fumigators
Fruit fumigators Termite controllers
Fumigant workers Tetraethyllead makers
Grain elevator workers Waterproofing makers
Grain fumigators Waxmakers
Gum processors Wood insect controllers
Lead scavenger makers Wool reclaimers
a From: NIOSH (1977)
In the 1970s, US EPA examined the exposure of professional
pesticide applicators involved with 1,2-dibromoethane soil fumigation.
It was estimated that applicators applying 1,2-dibromoethane for
30-40 days/year would receive a total annual inhalation dose of
3-40 mg/kg and farmer-applicators applying 1,2-dibromoethane for
7-10 days/year would receive a total annual inhalation dose of
0.7-10 mg/kg (US EPA, 1977).
1,2-Dibromoethane exposures were measured in a plant where lead
antiknock blends for petrol were prepared (Jacobs, 1980). The
antiknock blend constituents were mixed in tanks under enclosed-system
conditions and the only manual operations were connecting and
disconnecting hoses while loading and unloading tank cars, taking
quality control samples, and processing and loading drums. The levels
of worker exposure to 1,2-dibromoethane in antiknock blending and
storage areas were 0.77 µg/m3 (0.1 ppb) (laboratory technician) to
6.3 µg/m3 (0.82 ppb) (raw maternal blender). In addition to
long-term personal sampling, some short-term monitoring of specific
tasks was conducted. The results are shown in Table 8.
Table 8. Short-term air levels of 1,2-dibromoethane in antiknock
blending plant tank cars (Jacobs, 1980)
Taska Sampling time Concentration
mg/m3 ppm
Quality control sample 13 min, 10 sec 5.38 0.7
Loading tank car 7 min 1.07 0.14
a Respirator worn during these tasks
Personal air monitoring during vehicle refuelling at a petroleum
laboratory 10 m downwind of the fuel pump and fuel-handling facilities
was performed at a USA plant in July, 1975 (Table 9). The average
exposure of filling station attendants to 1,2-dibromoethane during
refuelling was 1.8 µg/m3 (0.24 ppb). Measurements at the car fuel
tank filler pipe showed maximum instantaneous 1,2-dibromoethane
concentrations of 105 µg/m3 (13.7 ppb), with an average for four
samples of 10.0 µg/m3 (1.3 ppb). This represented the maximum for a
short-term exposure. The concentration of 1,2-dibromoethane in air at
the fuel pump island was similar to values measured at upwind and
downwind sites. Overall, the very low 1,2-dibromoethane air levels
measured in this study indicated that the potential for filling
station attendant exposure to 1,2-dibromoethane while refuelling cars
was low and less than the current or proposed USA occupational air
standard for 1,2-dibromoethane exposure (Jacobs, 1980).
Exhaust emissions from various types of internal combustion
engines, including four-stroke Otto engines and diesel engines, are a
major source of environmental and occupational exposure to
1,2-dibromoethane (Hasanen et al., 1981). There are few data on the
composition of and exposure to exhaust emissions from two-stroke
engines.
Table 9. Petrol station attendant exposure to 1,2-dibromoethane
during vehicle refuelling (Jacobs, 1980)
Sample Concentration
mg/m3 ppb
Upwind background < 0.77 < 0.1
Downwind background < 0.77 < 0.1
Fuel pump island 0.99 0.13
Near vehicle fuel pipe 9.98 1.3a
during refueling 105.2 13.7b
Personal air sampler 2.15 0.28
a Average
b Maximum
Seven chain saws fuelled with 93-octane standard petrol-
containing tetramethyllead (lead content 0.15 g/litre) and
1,2-dibromoethane as a scavenger were tested on a test-bench
permitting a variable load to be applied by an electric power brake
(Nilsson et al., 1987). 1,2-Dibromoethane emissions were low
(2.5 mg/m3). Exposure to chain saw exhaust during logging was
studied under snowy and snow-free conditions. The time-weighted
average exposure to 1,2-dibromoethane was lower in the snow-free
conditions (0.0008 (0.0004-0.001) mg/m3) than in the snowy
conditions (0.002 (0.0001-0.005) mg/m3).
1,2-Dibromoethane is mainly used as a scavenger in tetraalkyllead
petrol and antiknock preparations, as a soil and grain fumigant, as
an intermediate in the synthesis of dyes and pharmaceuticals and as a
solvent for resins, gums and waxes (Alexeeff et al., 1990).
Rumsey & Tanita (1978) performed an industrial hygiene survey of
two manufacturing and two user facilities involving 1,2-dibromoethane.
Samples were taken from more than 69 potentially-exposed workers in
17 job classifications. Median 1,2-dibromoethane exposure (by similar
job types) in the manufacturing process ranged from 0.076 to
3.8 mg/m3 (0.010 to 0.5 ppm) (35 TWA personal samples). General
area samples collected at breathing zone heights had median TWA levels
of 1.5 mg/m3 (0.2 ppm) for 10 samples at process sites, and
3.8 mg/m3 (0.5 ppm) for 3 samples at laboratory sites.
Papaya workers in Hawaii were exposed to a geometric mean of
676 mg/m3 (88 ppb), and peaks up to 2.01 mg/m3 (262 ppb) were
measured (Steenland et al., 1986).
6. KINETICS AND METABOLISM
6.1 Absorption
1,2-Dibromoethane was found in the blood of rodents almost
immediately after dermal and oral exposure. Jakobson et al. (1982)
reported that during a 6-h dermal exposure of guinea-pigs of both
sexes (weighing between 600 and 1000 g) with undiluted
1,2-dibromoethane applied to 3.1 cm2 of shaved skin on the back
(1.0 ml/animal), the blood concentration of 1,2-dibromoethane
increased rapidly during 1 h to a level of 2 mg/litre and then slowly
decreased. The influx of 1,2-dibromoethane into the blood after 1 h
was largely in equilibrium with its disappearance.
In male Sprague-Dawley rats given 15 mg/kg body weight of
[14C-1,2] 1,2-dibromoethane in corn oil by gavage, the blood levels
at 24 h and 48 h were 0.90 and 0.64 mg/litre, respectively (Plotnick
et al., 1979). The excretion of radioactivity in faeces within 24 h
was 1.7% of the dose. The remainder was recovered either in the urine
(72%) or in the tissues (2.8%) (Table 10). The results indicated
rapid 1,2-dibromoethane absorption from the gastrointestinal tract.
No absorption information regarding inhalation exposure exists.
Table 10. The distribution of 14C in selected tissues and body
fluids of male rats 24 h after a single oral dose of
14C-1,2-dibromoethane (15 mg/kg)a
Tissue Tissue concentrationsb Percentage of dose
(mg equivalent/kg or mg/litre) (%)
Liver 4.78 ± 0.24 1.7 ± 0.07
Kidneys 3.32 ± 0.42 0.21 ± 0.02
Spleen 1.00 ± 0.03 0.22 ± < 0.01
Testes 0.49 ± 0.05 0.04 ± < 0.01
Brain 0.41 ± 0.04 0.02 ± < 0.01
Fat 0.35 ± 0.04 0.15 ± 0.02
Blood 0.90 ± 0.05 0.59 ± 0.03
Plasma 0.46 ± 0.04
Urinec 72.38 ± 0.98
Faecesc 1.65 ± 0.28
a From: Plotnick et al. (1979)
b Values represent mean concentrations (expressed as parent
compound) ± S.E.M. of duplicate determinations for six animals.
c n = 12
6.2 Distribution
Plotnick et al. (1979) compared levels of 14C in selected
tissues of male Sprague-Dawley rats following oral administration of
14C-1,2-dibromoethane. One day after the administration, the
highest levels of radioactivity were found in the liver and kidneys
(Table 10).
Distribution of 14C-1,2-dibromoethane (30 mg/kg body weight)
after intraperitoneal administration to male guinea-pigs was studied
by Plotnick & Conner (1976). The liver and kidneys contained the
highest levels of radioactivity followed by the adrenal glands
(Table 11).
Table 11. Distribution of 14C-1,2-dibromoethane in selected tissues
of male guinea-pigs at various time intervals following
intraperitoneal administrationa
Tissues/organs 4 h 8 h 24 h 72 h
Liver 129.0 104.9 38.0 15.6
Kidneys 286.6b 236.5 3.5 10.5
Adrenals 60.7 60.8 28.6 10.4
Pancreas 35.0 36.8 18.7 6.0
Spleen 15.8 14.0 14.9 7.0
Heart 14.0 15.6 9.5 3.3
Lungs 20.9 19.0 15.4 5.8
Testes 10.7 10.7 8.3 4.0
Brain 6.2 7.6 6.5 2.5
Fatc 21.4 7.9 3.2 2.1
Muscle 5.5 5.0 4.2 2.2
Blood 10.0 3.4 5.0 2.8
a From: Plotnick & Conner (1976)
b Values represent mean levels in mg equivalent/kg of tissue or
litre of fluid for three animals at each time interval
c Suprarenal fat
Kowalski et al. (1985) reported epithelial binding of
1,2-dibromoethane in the respiratory and upper alimentary tracts of
C57BL mice, Sprague-Dawley rats and Fischer-344 rats after intravenous
and intraperitoneal injection of 14C-1,2-dibromoethane. In C57BL
mice, there was a high level of radioactivity in the nasal and
bronchial mucosa and liver 5 min after intravenous injection of
14C-1,2-dibromoethane. In the nose, the highest labelling was
present in a spotty band beneath the epithelium of the
ethmoturbinates. The radioactive labelling of the mucosa of the
respiratory tract was persistent, and 10 days after injection a
selectively bound radioactivity remained. High labelling was also
present in the mucosa of the forestomach, whereas there was no
selective uptake of radioactivity in the glandular stomach or
intestine. Similar distribution patterns were observed in the
intraperitoneally injected mice killed after 30 min or subsequently.
6.3 Metabolic transformation
The metabolism of 1,2-dibromoethane has been extensively studied
and metabolites have been identified in in vivo and in vitro
studies (Table 12, Fig. 1).
Table 12. Metabolites of 1,2-dibromoethane
(a) In vivo
Metabolite Animal; route; substrate Reference
Bromide Swiss-Webster mice; White et al.
intraperitoneal; plasma (1983)
N-acetyl-S-(2-hydroxy male Wistar rat; oral; Van Bladeren et
ethyl)-L-cysteine urine al. (1981b)
GS-CH2-CH2SG female white rat; oral; Nachtomi (1970)
liver
GSCH2CH2OH sulfoxide liver Nachtomi (1970)
GSCH2CH2OH liver and kidney Nachtomi (1970)
S-(2-hydroxyethyl) kidney Nachtomi (1970)
mercapturic acid
(b) In vitro
Metabolite Tissue Reference
GSCH2CH2SG rat liver and kidney extract Nachtomi (1970)
GSCH2CH2OH rat liver and kidney extract Nachtomi (1970)
Bromide (1984) rat liver cytosols White et al.
Bromide (1983) mouse liver cytosols White et al.
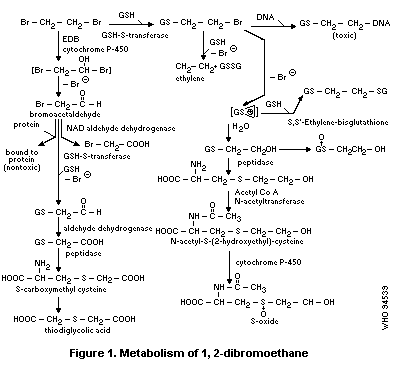 Inorganic bromide may be formed as a consequence of attack by GSH
or oxidative catabolism. In the first case, the expected intermediate
would be S-(2-bromoethyl)-GSH, which can be converted to bis-GSH or
S-(2-hydroxyethyl)-GSH. Sulfoxidation of S-(2-hydroxyethyl)-GSH
would yield S-(2-hydroxyethyl)-GSH- S-oxide or further metabolism
would produce S-(2-hydroxyethyl)-cysteine, which in turn may undergo
sulfoxidation to yield N-acetyl- S-(2-hydroxyethyl)-cysteine-
S-oxide. The oxidative metabolism of 1,2-dibromoethane by
cytochrome P-450-dependent mixed function oxidases would be expected
to yield 2-bromoacetaldehyde as the initial product. This may be
converted by dehydrogenase to 2-bromoacetic acid or undergo attack by
GSH and subsequent dehydrogenase activity to give rise to
S-carboxymethyl-GSH. S-carboxymethyl-cysteine may be further
metabolized to thioglycolic acid. A reactive intermediate binds
mainly to DNA guanyl remnants and may be responsible for the
genotoxicity.
White et al. (1983) reported a deuterium isotope effect on the
metabolism of 1,2-dibromoethane. The metabolism of 1,2-dibromoethane
and tetradeutero-1,2-dibromoethane (d4-1,2-dibromoethane) was compared
in male Swiss-Webster mice. Three hours after intraperitoneal
administration of 1,2-dibromoethane or d4-1,2-dibromoethane
(50 mg/kg), there was 42% less bromide in the plasma of
d4-1,2-dibromoethane-treated mice than in the plasma of
1,2-dibromoethane-treated mice. This difference demonstrated a
significant deuterium isotope effect on the metabolism of
1,2-dibromoethane in vivo. In in vitro studies, which measured
bromide ion released from the substrate to monitor the rate of
metabolism, hepatic glutathione- S-transferase was unaffected. Since
the decreased metabolism of d4-1,2-dibromoethane was apparently due to
a reduced rate of microsomal oxidation, these data supported the
hypothesis that conjugation with GSH is responsible for the genotoxic
effect of 1,2-dibromoethane.
White et al. (1984) studied metabolism in isolated rat
hepatocytes. Cytosolic metabolism of 1,2-dibromoethane was not
affected by deuterium substitution. Both compounds caused DNA
single-strand breaks, as measured by the alkaline elution technique,
when incubated at a concentration of 0.1 mM with hepatocytes. No
difference in the degree of DNA damage was demonstrated between
hepatocytes incubated with 1,2-dibromoethane and those incubated with
d4-1,2-dibromoethane.
1,2-Dibromoethane can be metabolized by freshly isolated rat
hepatocytes to S-(2-hydroxyethyl)glutathione, S-(carboxymethyl)
glutathione and S,S'-(1,2-ethanediyl)bis(glutathione). These three
metabolites account for 84% of the total intracellular glutathione
depletion (Jean & Reed, 1992). These reactions were negligible in the
presence of rat glutathione- S-transferase, but conjugation was
catalysed by the rat alpha class enzyme 2-2 and, to a lesser extent,
the rat µ class enzyme 3-3. Of the three classes of human cytosolic
glutathione- S-transferases, 1,2-dibromoethane conjugation was
catalysed by the alpha class enzymes (Cmarik et al., 1990).
Human fetal liver appears to be especially active (several times
higher specific activity of glutathione- S-transferase, compared to
adult liver, as reported by Wiesma et al., 1986) in metabolizing
1,2-dibromoethane in vitro (Kulkarni et al., 1992).
1,2-Dibromoethane-induced lipid peroxidation and cytotoxicity
were increased upon concomitant exposure to carbon tetrachloride.
Similarly, the amount of 1,2-dibromoethane metabolites bound
covalently to proteins was enhanced. The effect of carbon
tetrachloride has been related to a shift in the 1,2-dibromoethane
metabolism from GSH-dependent to P-450-dependent (Chiarpotto et al.,
1993).
Oral administration of large doses of 1,2-dibromoethane
(37.6 mg/animal) to male Wistar rats (weighing around 200 g),
following a single dose of disulfiram (12 mg/kg), led to decreased
excretion of the mercapturic acid metabolite, a phenomenon associated
with a decrease in cytochrome P-450 levels (van Bladeren et al.,
1981a). In an additional reaction, 1,2-dibromoethane is debrominated
by an oxidative process catalysed by an enzyme in hepatic microsomes.
This system requires NADPH and oxygen and is inducible by
phenobarbital but not by methyl-cholanthrene (Hill et al., 1978).
Simula et al. (1993) reported an increased mutagenicity of
1,2-dibromoethane in the Salmonella typhimurium strain TA100
expressing human glutathione- S-transferase A1-1, indicating that
human glutathione- S-transferases are able to metabolize
1,2-dibromoethane to reactive intermediates.
In a study with isolated human hepatocytes (Cmarik et al., 1990),
it was found that concurrent treatment with diethylmaleate reduced the
intracellular glutathione level and inhibited 1,2-dibromoethane
concentration-dependent unscheduled DNA synthesis.
6.4 Elimination and excretion in expired air, faeces and urine
When 14C-1,2-dibromoethane (30 mg/kg body weight) was given
intraperitoneally to male guinea-pigs of the Hartley strain, 66% of
the radioactivity was excreted in the urine within 72 h of
administration (Plotnick & Conner, 1976). Faecal excretion was
relatively insignificant, representing less than 3% of the dose. The
excretion of unchanged 1,2-dibromoethane in the expired air was
significant (10-12% of dose).
Plotnick et al. (1979) reported that urinary extraction of
radioactivity from male rats of the Sprague-Dawley strain, 24 h after
a single oral dose of 14C-1,2-dibromoethane (15 mg/kg), was 72.4%.
Faecal radioactivity was 1.7%. Concomitant exposure to dietary
disulfiram significantly depressed urinary excretion of
1,2-dibromoethane.
6.5 Retention and turnover
Following intraperitoneal administration of [1,2-14C]-
1,2-dibromoethane (40 mg/kg) to RF/Hiraki mice, the circulating
radiolabel was mainly accounted for by S-(2-hydroxyethyl)
cysteine- N-acetate. Less than 1% of the dose was present in the
blood as a volatile component (Edwards et al., 1970).
Jakobson et al. (1982) reported that the elimination curve for
1,2-dibromoethane from blood after a 4-h dermal exposure of
guinea-pigs was non-linear and corresponded to a kinetic model
involving at least two compartments.
6.6 Reaction with body components
Radioactivity from [1,2-14C]-1,2-dibromoethane is bound
irreversibly to macromolecules in rat tissues after intraperitoneal
injection. For protein, DNA, and RNA, the largest amounts of bound
radioactivity were found to be present in the liver and kidney. Lung,
testis, stomach and the large and small intestines showed less
radioactivity (Hill et al., 1978).
Ozawa & Guengerich (1983) reported the formation of an
S-[2-(N7-guanyl)ethyl]glutathione adduct. 1,2-Dibromoethane and
GSH were irreversibly bound to calf thymus DNA in equimolar amounts
when in vitro incubation was carried out in the presence of
glutathione- S-transferase. The labelled DNA was enzymatically
digested to deoxyribonucleosides and separated by HPLC. The level of
adducts in DNA isolated from human hepatocytes incubated with 0.5 mM
1,2-dibromoethane was about 40% of the value obtained for rat
hepatocytes (Cmarik et al., 1990).
S-[2-(N7-guanyl)ethyl]glutathione was the only major DNA adduct
formed in vivo in rat (male Sprague-Dawley) liver (1.3 nmol/mg DNA)
or kidney (0.95 nmol/mg DNA) 8 h after intraperitoneal administration
of 37 mg 1,2-dibromoethane/kg body weight. The in vivo half-life of
S-[2-(N7-guanyl)ethyl] glutathione in rat liver, kidney, stomach
and lung was estimated to be between 70 and 100 h (Inskeep et al.,
1986).
Following intraperitoneal injection of 1,2-dibromoethane
(37 mg/kg) in rats and mice of several strains, it was found that more
of the S-[2-(N7-guanyl)ethyl]glutathione adduct of DNA was formed
in the livers of rats than in those of mice (Kim & Guengerich, 1990).
Incubation of 1,2-dibromoethane with calf thymus DNA and cytosol from
rats or mice did not result in different adduct levels, whereas the
level of adduct formation by human liver cytosol was about half of
those values for rats or mice. Induction of glutathione-
S-transferase in rat liver by phenobarbital or ß-naphthoflavone did
not increase DNA adduct levels, whereas cytochrome P-450 inhibition by
disulfiram did increase DNA adducts without altering the transferase
activity. Depletion of reduced glutathione in vivo by
diethylmaleate correlated with a reduction in DNA adduct levels.
Bromine atoms generated upon reductive degradation of
1,2-dibromoethane have been shown to react with polyunsaturated fatty
acids via both abstraction of bisallylic hydrogen and addition to the
double bond. Bromine atoms may be a potential initiator for lipid
peroxidation and provide a chemical basis for the toxic action of
1,2-dibromoethane (Guha et al., 1993).
The cytotoxic action of 1,2-dibromoethane has been studied by
Khan et al. (1993). They found that both cytochrome P-450- and
GSH-dependent metabolism of 1,2-dibromoethane contributed to its
cytotoxic effect in hepatocytes. Antioxidants or removal of oxygen
delayed the cytotoxicity. Furthermore, cytotoxicity could be
increased markedly if aldehyde dehydrogenase was inhibited with
disulfiram. In addition, cytotoxicity could be reduced if the
hepatocytes were depleted of GSH before the addition of
1,2-dibromoethane.
1,2-Dibromoethane has also been shown to cause cytotoxicity in
rabbit pulmonary cells (Nichols et al., 1992), being cytotoxic to
Clara cells, type II cells and alveolar macrophages.
The irreversible binding of radioactivity from [1,2-14C]-
1,2-dibromoethane to protein, DNA and RNA in rats was measured 24 h
after an intraperitoneal injection of 14C-1,2-dibromoethane
(2.6-3.2 mg/kg) (Hill et al., 1978). For each of these classes of
macromolecules, the largest amounts of bound radioactivity were found
in the liver and kidneys.
Botti et al. (1982) evaluated the effect of 1,2-dibromoethane on
GSH levels and cytosolic glutathione- S-transferase activity
following administration by gavage to male rats. Doses of either 75
or 150 mg/kg were found to decrease GSH and glutathione- S-
transferase activities. Maximal effects were observed 2 h following
exposure, although a significant decrease in GSH levels was observed
within 15 min. Mann & Darby (1985) also noted GSH depletion in both
male and female rats, the maximum effect occurring 2 h after an
intraperitoneal 1,2-dibromoethane dose of 80 mg/kg. A greater effect
was observed in males.
Brandt et al. (1987) used autoradiography and 14C-labelled
1,2-dibromoethane to study the tissue distribution of
1,2-dibromoethane following an intraperitoneal injection to a
cynomolgus monkey. 1,2-Dibromoethane was found to bind preferentially
to the liver and renal tubules, particularly the adrenal zona
reticularis.
Kaphalia & Ansari (1992) measured the rate of incorporation of
label into albumin and other large plasma proteins following exposure
to [14C]-1,2-dibromoethane either in vivo or in vitro. About
37% of the total label, administered by gavage (25 mg/kg body weight
in mineral oil over a 12-day period), was estimated to be bound to
plasma proteins, the majority (72%) being bound to albumin. In the
case of in vitro exposure, the incorporation of label to human
albumin or plasma after incubation with radioactive 1,2-dibromoethane
could be increased by the addition of either microsomal enzymes or an
NADPH-generating system.
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
Toxic effects of 1,2-dibromoethane have been mainly observed in
the liver and kidneys. Inhaled 1,2-dibromoethane vapour produces
nasal irritation and depression of the central nervous system. In
solution, 1,2-dibromoethane causes skin irritation on the shaved
abdomen, and eye irritation.
7.1.1 Oral
7.1.1.1 Rat
1,2-Dibromoethane (99% pure) in olive or corn oil was given by
gavage to albino rats, guinea-pigs, rabbits, mice and chickens. The
difference in LD50 of 1,2-dibromoethane in male and female rats was
statistically significant; rabbits appeared to be the most sensitive
and mice the least sensitive (Rowe et al., 1952) (Table 13).
Adult male albino rats (weight 140-160 g) were given 110 mg
1,2-dibromoethane/kg in olive oil by gavage and were killed 2, 4, 8,
12, 17 or 22 h later. During the first 4 h, no changes in the liver
were detectable by light microscopy. From 8 h after administration,
1,2-dibromoethane induced sinusoidal dilatation and centrilobular
necrosis in the liver (Broda et al., 1976).
7.1.1.2 Chicken
Leghorn chickens of both sexes were given 1,2-dibromoethane
(110 mg/kg body weight) in soybean oil by gavage. There was an
increase in liver weight and NAD concentration, and a decrease in
liver and blood alkaline phosphatase activity (Nachtomi et al., 1968).
Five-week-old male Leghorn chicks (weight 580-620 g) were given
1,2-dibromoethane (110 mg/kg body weight) in olive oil by gavage and
were killed 8, 12 or 22 h later. The central areas of the liver were
not changed, but portal areas were affected by 1,2-dibromoethane. The
concentration of eosinophilic granulocytes was much greater in
1,2-dibromoethane-treated livers than in controls (Broda et al.,
1976).
7.1.2 Inhalation
7.1.2.1 Rat
When rats were exposed to concentrations of 770, 1540, 3080,
6610, 12 300, 23 100, 38 500 or 77 000 mg/m3 (20 males and females
per group in most concentrations, and no control group) for durations
Table 13. Acute toxicity of 1,2-dibromoethane
Species Route Vehicle Parameter Value Reference
Rat (M) oral olive oil LD50 146 mg/kg Rowe et al. (1952)
Rat (F) oral olive oil LD50 117 mg/kg Rowe et al. (1952)
Rat (M,F) oral corn oil LD50 140 mg/kg McCollister
et al. (1956)
Mouse (F) oral olive oil LD50 420 mg/kg Rowe et al. (1952)
Rabbit (F) oral olive oil LD50 55 mg/kg Rowe et al. (1952)
Guinea-pig oral olive oil LD50 110 mg/kg Rowe et al. (1952)
(M,F)
Chicken (M,F) oral olive oil LD50 79 mg/kg Rowe et al. (1952)
Rabbit skin LD50 450 mg/kg Rowe et al. (1952)
Rat inhalation vapour by LC50 4620 mg/m3 Rowe et al. (1952)
aeration 1 h
Rat (M,F) inhalation vapour by LC50 2304 mg/m3 McCollister
aeration 4 h et al. (1956)
Mouse (ICR) intraperitoneal corn oil LD50 205 mg/kg Kluwe et al.
(1981)
ranging from 0.01 to 16.0 h (exposure time varied at different
concentrations), slight anaesthetic actions and depression of the
central nervous system were observed in rats exposed to 1540 mg/m3
or more. Deaths occurred within 24 h at concentrations between 1540
and 3080 mg/m3, related to exposure duration, due to respiratory or
cardiac failure. The LC50 concentration for a 2-h exposure was
3080 mg/m3. Deaths occurring from exposures at lower concentrations
were almost always delayed, sometimes as long as 12 days after
exposure. The majority of these deaths were due to pneumonia. The
animals usually lost weight, appeared rough and unkempt, became
irritable, had a bloodstained nasal discharge, and died. Animals
surviving the exposure at the lower concentrations exhibited a typical
progression of symptoms for several days before recovery took place.
Rats exposed to concentrations producing mortality, which were
sacrificed and autopsied 16-24 h after exposure, showed an increased
weight of lungs, liver and kidneys. The lungs showed congestion,
oedema, haemorrhages and inflammation; the liver cells had cloudy
swelling, centrilobular fatty degeneration and necrosis; the kidneys
showed slight interstitial congestion and oedema, with slight cloudy
swelling of the tubular epithelium in some cases (Rowe et al., 1952).
7.1.2.2 Guinea-pig
All guinea-pigs (20 males and females per group) exposed to 1540
or 3080 mg/m3 (no control group) for 2 to 7 h died, whereas all
those exposed to 770 mg/m3 for 7 h or 1540 mg/m3 for 2 h survived
(Rowe et al., 1952).
7.1.3 Intraperitoneal injection
7.1.3.1 Mouse
Intraperitoneal injection (46.8 and 93.7 mg/kg; 0.25 and
0.5 mmol/kg) of 1,2-dibromoethane (> 99.9%) in corn oil in male
B6C3F1 mice (weight 20-26 g) produced hepatic damage. The mice were
killed 4 h later and in vivo genotoxicity was determined by a
sensitive in vivo/in vitro alkaline DNA unwinding assay for the
presence of single-strand breaks and/or alkali-labile sites in hepatic
DNA. Significant hepatic DNA damage was found with a dose of
0.5 mmol/kg. In an assessment of the acute hepatotoxicity and
nephrotoxicity of 1,2-dibromoethane, male B6C3F1 mice were given
intraperitoneal injections (93.7, 140, 187.7 or 281.6 mg/kg; 0.5,
0.75, 1.0 or 1.5 mmol/kg) of 1,2-dibromoethane in corn oil and
sacrificed 24 h later. Serum L-iditol dehydrogenase (IDH), alanine
aminotransferase (ATT), and blood urea nitrogen were determined. At a
dose of 187.7 mg/kg 1,2-dibromoethane produced statistically
significant increases in relative liver and kidney weights, serum IDH
and ATT levels, and blood urea nitrogen levels. Four out of five
animals given a dose of 281.6 mg/kg died (Storer & Conolly, 1983).
7.1.3.2 Rat
Adult male Fischer-344 rats were given a single intraperitoneal
injection of 99% 1,2-dibromoethane (50 mg/kg; 0.27 mmol/kg) in corn
oil; they were sacrificed 2, 12, 24, 48 or 96 h later and the kidneys
were removed. Histopathological alterations in the kidney were most
prominent 48 h after 1,2-dibromoethane injection, and consisted of
acute proximal tubular degeneration (proximal tubular swelling and
vacuolation) (Kluwe et al., 1982).
7.2 Short-term exposure
7.2.1 Oral
7.2.1.1 Chicken
In a study by Schlinke (1970), 1,2-dibromoethane (formulation
which contains 83% of active ingredient) was administered orally at
doses of 50, 100 or 200 mg/kg per day to groups of five unsexed SPF
White Leghorn chickens (6-7 weeks old) for 10 days. There was an
untreated control group. All five chickens given 200 mg/kg per day
showed lack of appetite and depression, and died after the third dose.
Chickens which died had inflamed crops, excess pericardial fluid, and
congestion of the liver. Chickens given 50 or 100 mg/kg per day
showed no toxic effects.
7.2.2 Inhalation
7.2.2.1 Mouse
B6C3F1 mice (10 males and 10 females per group) were exposed by
inhalation to 23.1, 115.5 and 577.5 mg/m3 (3, 15 and 75 ppm) of
1,2-dibromoethane (6 h/day, 5 days per week) for 13 weeks. Four male
mice in the low dose group died before the end of the exposure period.
At 13 weeks, mice showed severe necrosis and atrophy of the olfactory
epithelium in the nasal cavity after inhaling the highest
concentration. Lower concentrations induced squamous cell metaplasia,
hyperplasia and cytomegaly of the epithelium of the respiratory nasal
turbinals. Squamous metaplasia, hyperplasia and cytomegaly of the
epithelium were also seen in larynx, trachea, bronchi and bronchioles.
The NOEL, based on histopathological alterations in the nasal cavity,
was 23.1 mg/m3 (3 ppm) (Reznik et al., 1980).
7.2.2.2 Rat
A group of 10 female rats (strain unknown) exposed to a
concentration of 768 mg/m3 (100 ppm) of 1,2-dibromoethane vapour for
7 h/day, lost weight steadily and three died after 1, 5 and
7 exposures, respectively. Surviving rats were thin and unkempt after
7 exposures in 9 days. At autopsy the stomachs were full of food, and
the contents were bloodstained. Lung, liver and kidney weights were
significantly elevated. Microscopic examination revealed some
thickening of the alveolar walls, with slight leukocytic infiltration
of the lungs, widespread cloudy swelling of the liver (but no fatty
degeneration), and slight congestion and haemosiderosis of the spleen
(Rowe et al., 1952).
F-344 rats (five males and five females per group) were exposed
by inhalation to 23.1, 115.5 and 577.5 mg/m3 (3, 15 and 75 ppm) of
1,2-dibromoethane (6 h/day, 5 days per week) for 13 weeks. At 13
weeks, they showed severe necrosis and atrophy of the olfactory
epithelium in the nasal cavity after inhalation of 577.5 mg/m3.
Lower concentrations induced squamous cell metaplasia, hyperplasia and
cytomegaly of the epithelium of the respiratory nasal turbinals.
Squamous metaplasia, hyperplasia and cytomegaly of the epithelium were
also seen in the larynx, trachea, bronchi and bronchioles. Other
compound-related toxic lesions in rats were seen in the liver, kidney
and testes. At 115.5 mg/m3, 1,2-dibromoethane induced only minor
changes in the nasal cavity. No lesions were seen in other tissues.
The NOEL based on histopathological alterations in the nasal cavity
was 23.1 mg/m3 (3 ppm) (Reznik et al., 1980).
Nitschke et al. (1981) conducted a 13-week inhalation study on
1,2-dibromoethane in rats. Male and female F-344 rats were exposed to
0, 23, 77 or 307 mg/m3 (0, 3, 10 or 40 ppm) (6 h/day, 5 days per
week) for 13 weeks. Those exposed to 307 mg/m3 (40 ppm) of
1,2-dibromoethane exhibited a decrease in body weight gain, an
increase in liver and kidney weight, and hyperplasia and metaplasia of
the respiratory epithelium of the nasal turbinates. A slight
epithelial hyperplasia of the nasal tubinates was also noted at
77 mg/m3 (10 ppm). A recovery period of 88 days resulted in
regression of the lesions in all but one animal.
7.2.2.3 Guinea-pig
Guinea-pigs (8 of each sex per group) that were administered
385 mg/m3 (50 ppm) of 1,2-dibromoethane for up to 7 h, 57 times in
80 days, had decreased final body weight and increased lung, liver and
kidney weights. Microscopic examination of the tissues showed slight
central fatty degeneration in the liver, and slight interstitial
congestion and oedema, with some parenchymatous degeneration of the
tubular epithelium in the kidney. Blood urea nitrogen values were
normal. In females, the total lipid content in the liver revealed no
significant variation. Guinea-pigs (4-8 of each sex per group)
tolerated without toxic effects 145 exposures (7 h each) in 205 days
at a 1,2-dibromoethane concentration of 193 mg/m3 (25 ppm).
However, four out of eight males and two out of eight females died of
pulmonary infection during the experiment (Rowe et al., 1952).
7.2.2.4 Rabbit
When four female rabbits were exposed by inhalation to
770 mg/m3 (7 h/day) for 4 days, two of the rabbits died after the
second exposure. Microscopic examination of tissues revealed
widespread central fatty degeneration of the liver with areas of
necrosis. Rabbits given 59 exposures each lasting 7 h (385 mg/m3)
in 84 days showed no evidence of adverse effects except for a slight
increase in liver and kidney weights; those given 152 exposures
(each 7 h) (192.5 mg/m3) in 214 days showed no adverse effects
(Rowe et al., 1952).
7.2.2.5 Monkey
When one male and one female monkey were given 7-h exposures
(385 mg/m3) 49 times in 70 days, they appeared ill, nervous and
unkempt throughout the experiment. Liver weights were increased with
slight central fatty degeneration and increased total lipid values.
No significant changes were observed in other organs except for a
slight increase in kidney weight. Another pair (one male and one
female) of monkeys received 7-h exposures (193 mg/m3) 156 times in
214 days. This group showed no evidence of adverse effects and the
NOEL was established as 193 mg/m3 (25 ppm) (Rowe et al., 1952).
7.3 Eye and skin irritation
7.3.1 Rabbit
1,2-Dibromoethane (undiluted, 1% and 10% in propylene glycol,
quantity not stated) was introduced into both eyes of a rabbit and
after 30 seconds one eye was flushed for 3 min with copious amounts of
running water. Conjunctival irritation occurred in both eyes, and
there was slight superficial necrosis of the cornea. However, healing
was prompt and complete 12 days after exposure; there was no corneal
scarring and no apparent injury to the iris or the lens. A 1%
1,2-dibromoethane solution in propylene glycol elicited a response
very similar to undiluted 1,2-dibromoethane (Rowe et al., 1952).
A 1.0% solution of 1,2-dibromoethane in butyl carbitol acetate
was applied 10 times in 14 days to the rabbit ear and also to the
shaved abdomen where it was then protected by a bandage. On the ear,
it caused slight irritation (erythema and exfoliation), whereas on the
shaved abdomen there was marked irritation with erythema and oedema
progressing to necrosis and sloughing of the superficial layers of the
skin. Healing was complete without scarring within 7 days after
termination of exposure (Rowe et al., 1952).
7.4 Long-term exposure
Designs of long-term/carcinogenicity studies are given in Table
14 and tumorigenic effects seen in these studies are summarized in
Table 15.
7.4.1 Oral
7.4.1.1 Mouse
Groups of 50 male and 50 female B6C3F1 mice (5 weeks old) were
given technical-grade 1,2-dibromoethane (99.6% pure) in corn oil by
gavage on 5 consecutive days per week. The time-weighted average high
and low doses of 1,2-dibromoethane were 107 and 62 mg/kg per day for
Table 14. Carcinogenicity studies on 1,2-dibromoethane
Route Species Number of animals Sexa Dose and dosing scheduleb Durationc Reference
(strain) per group
Gavage
Mouse 50 M 62 mg/kg body weight TWAd 78 weeks(53) NCI (1978)
(B6C3F1) (treated) 107 mg/kg body weight TWAd 77 weeks(53)
F 62 mg/kg body weight TWAd 90 weeks(53)
107 mg/kg body weight TWAd 78 weeks(53)
20 M,F
(control)e
Rat 50 M 38 mg/kg body weight TWAd 49 weeks(47) NCI (1978)
(Osborn- (treated) 41 mg/kg body weight TWAd 49 weeks(34)
Mendel) F 37 mg/kg body weight TWAd 61 weeks(57)
39 mg/kg body weight TWAd 61 weeks(44)
20 M,F
(control)e
Drinking-water
Mouse 30 M 117 mg/kg body weight per day 15 months Van Duuren et
(B6C3F1) (treated) F 103 mg/kg body weight per day 17 months al. (1985)
50 M,F
(control)
Inhalation
Mouse 60 F 154 mg/m3 6 h/day, 5 days/week 6 months Adkins et al.
(A/J) (treated) 384 mg/m3 6 h/day, 5 days/week 6 months (1986)
60 F 154 mg/m3 6 h/day, 5 days/week 6 months
(control)e 384 mg/m3 6 h/day, 5 days/week 6 months
90 F
(treatead)
60
(control)
Table 14 (cont'd)
Route Species Number of animals Sexa Dose and dosing scheduleb Durationc Reference
(strain) per group
Mouse 50 M 77 mg/m3 6 h/day, 5 days/week 78 weeks NTP (1982)
(B6C3F1) (treated) 307 mg/m3 6 h/day, 5 days/week 78 weeks
50 F 77 mg/m3 6 h/day, 5 days/week 103 weeks
(treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks
50 M,F
(control)
Mouse 50 M 77 mg/m3 6 h/day, 5 days/week 103 weeks Stinson et
(B6C3F1) (treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks al. (1981)
50 F 77 mg/m3 6 h/day, 5 days/week 103 weeks
(treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks
50 M,F 104 weeks
(control)
Rat 50 M 77 mg/m3 6 h/day, 5 days/week 103 weeks NTP (1982)
(Fisher-344) (treated) 307 mg/m3 6 h/day, 5 days/week 88 weeks
50 F 77 mg/m3 6 h/day, 5 days/week 103 weeks
(treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks
50 M,F
(control)
Rat 48 M,F disulfiramf (0.05% in diet) 18 months Wong et
(Sprague- (treated) ± EDB (119-167 mg/m3 TWAb) al. (1982)
Dawley) 7 h/day, 5 days/week
48 M,F
(control)
Table 14 (cont'd)
Route Species Number of animals Sexa Dose and dosing scheduleb Durationc Reference
(strain) per group
Dermal
Mouse 30 F 25 mg/mouse, 3 times/week 400-594 days Van Duuren et
(Ha:ICR (treated) 50 mg/mouse, 3 times/week al. (1979)
Swiss) 100 F
(control)g
a M = male, F = female
b TWA = time-weighted average
c observation period: treated and untreated weeks (exposed weeks)
d doses changed in the course of the experiment with intermitting untreated weeks
e the controls consisted of vehicle (corn oil) & untreated controls
f This combination was chosen to examine effects of disulfiram used for alcoholism control programmes on workers
who were exposed to 1,2-dibromoethane occupationally.
g controls consisted of vehicle (acetone) & untreated controls
Table 15. Summaries of results of carcinogenicity studies on 1,2-dibromoethane
Study Route Animal Statistically significant effects dose (numbers of animals with
effects/total numbers of animals)
NCI (1978) gavage mouse squamous cell carcinoma of the forestomach
control (M: 0/20, F: 0/20), 62 mg/kg (M: 45/50, F: 46/49)
107 mg/kg (M: 29/49, F: 28/50)
alveolar/bronchiolar adenoma
control (M: 0/20, F: 0/20), 62 mg/kg (M: 4/45, F: 11/43)
107 mg/kg (M: 10/47, F: 6/46)
rat squamous cell carcinoma of the forestomach
control (M: 0/20, F: 0/20), 38 mg/kg (M: 45/50), 37 mg/kg (F: 40/50)
41 mg/kg (M: 33/50), 39 mg/kg (F: 29/50)
hepatocellular carcinoma
control (M: 0/20, F: 0/20), 39 mg/kg (F: 6/48)
haemangiosarcoma
control (M: 0/20, F: 0/20), 38 mg/kg (M: 11/50), 41 mg/kg (M: 4/50)
Van Duuren et al. (1985) drinking- mouse squamous cell carcinoma
water 117 mg/kg (M: 26/30), 103 mg/kg (F: 22/30)
oesophageal papilloma
103 mg/kg (F: 3/30)
squamous cell papillomaa
115 mg/kg (M: 9/30, F: 10/30)
Adkins et al. (1986) inhalation mouse (A/J) i) pulmonary adenoma
(all females) control (F: 0/60), 154 mg/m3 (F: 60/60), 384 mg/m3 (F: 60/60)
ii) pulmonary adenoma
control (F: 0/60), 154 mg/m3 (F: 75/90), 384 mg/m3 (F: 90/90)
Table 15 (cont'd)
Study Route Animal Statistically significant effects dose (numbers of animals with
effects/total numbers of animals)
NTP (1982) inhalation mouse alveolar/bronchiolar carcinoma
control (M: 0/41, F: 1/49), 77 mg/m3 (M: 3/48, F: 5/49),
307 mg/m3 (M: 19/46, F: 37/50)
alveolar/bronchiolar adenoma
control (M: 0/41, F: 3/49), 77 mg/m3 (F: 7/49),
307 mg/m3 (M: 11/46, F:13/50)
haemangiosarcoma of the circulatory system
control (F: 0/50), 77 mg/m3 (F: 11/50), 307 mg/m3 (F: 23/50)
subcutaneous fibrosarcoma
control (F: 0/50), 77 mg/m3 (F: 5/50), 307 mg/m3 (F: 11/50)
nasal cavity carcinoma, or adenoma
control (F: 0/50), 307 mg/m3 (F: 6/50, 8/50)
mammary gland adenocarcinoma
control (F: 2/50), 77 mg/m3 (F: 14/50), 307 mg/m3 (F: 8/50)
inhalation rat nasal cavity carcinoma, adenocarcinoma, adenoma
control (M: 0/50, F: 0/50), 77 mg/m3 (M: 1/50, 20/50, 11/50, F: 0/50,
20/50, 11/50), 307 mg/m3 (M: 21/50, 0/50, 28/50, F: 25/50, 29/50, 3/50)
haemangiosarcoma of the circulatory system
control (M: 0/50, F: 0/50), 307 mg/m3 (M: 15/50, F: 5/50)
tunica vaginalis mesothelioma
control (M: 0/50), 77 mg/m3 (M: 7/50), 307 mg/m3 (M: 25/50)
alveolar/bronchiolar adenoma, carcinoma (combined)
control (F: 0/50), 307 mg/m3 (F: 5/47)
nasal cavity adenomatous polyps
control (M: 0/50), 77 mg/m3 (M: 18/50), 307 mg/m3 (M: 5/50)
mammary gland fibroadenoma
control (F: 4/50), 77 mg/m3 (F: 29/50), 307 mg/m3 (F: 24/50)
Table 15 (cont'd)
Study Route Animal Statistically significant effects dose (numbers of animals with
effects/total numbers of animals)
Stinson et al. (1981) inhalation mouse nasal cavity carcinoma (squamous carcinoma, adenocarcinoma)
control (M: 0/45, F: 0/50), 77 mg/m3 (M: 0/44, F: 0/49)
307 mg/m3 (M: 0/46, F: 7/49)
sarcoma
control (M: 0/45, F: 0/50), 77 mg/m3 (M: 0/44, F: 1/49)
307 mg/m3 (M: 0/46, F: 2/49)
benign neoplasms (squamous papilloma, adenoma)
control (M: 0/45, F: 0/50), 77 mg/m3 (M: 0/44, F: 0/49)
307 mg/m3 (M: 3/46, F: 7/49)
Wong et al. (1982) inhalation rat hepatocellular tumour
control (M: 0/48, F: 0/48), EDB (M: 3/46, F: 1/48)
EDB+DSb (M: 36/48, F: 32/48)
kidney (adenoma and adenocarcinoma)
control (M: 0/48, F: 0/48), EDB (M: 3/46, F: 1/48)
EDB+DSb (M: 17/48, F: 7/48)
Van Duuren et al. (1979) skin mouse lung tumours
control untreated (F: 30/100)
control acetone (F: 11/100)
25 mg/mouse (F: 24/30), 50 mg/mouse (F: 26/30)
skin carcinoma
control untreated (F: 0/100)
control acetone (F: 0/100)
25 mg/mouse (F: 2/30), 50 mg/mouse (F: 8/30)
a carcinogenicity study on bromoethanol
b DS: disulfirum given in diet
both sexes. All surviving male mice and high-dose female mice were
killed on week 78, and low-dose females were killed on week 90. In
low-dose females, body weight depression was apparent after the first
10 weeks. Soft faeces, alopecia and body sores were observed in all
surviving animals at week 14. In mice receiving the high dose,
acanthosis of the forestomach was observed in 5/49 males and 9/50
females, compared with 1/50 in low-dose females. Hyperkeratosis
occurred in the stomachs of 13/49 high-dose males, 12/50 high-dose
females, and 1/48 low-dose females. There was testicular atrophy
(10/47) related to compound administration in males receiving the high
dose (NCI, 1978).
7.4.1.2 Rat
Groups (50 of each sex per group) of Osborne-Mendel rats (8 weeks
old) were administered technical-grade 1,2-dibromoethane in corn oil
by gavage on five consecutive days per week. The time-weighted
average high and low doses of 1,2-dibromoethane in treated groups were
41 and 38 mg/kg per day for male rats, and 39 and 37 mg/kg per day for
females. Body weight depression was apparent in the treated rats
after the first 10 weeks. Reddened ears, a hunched appearance, firm
distended abdomens and abdominal urine stains were observed in treated
groups. There was high mortality in both the high- and low-dose
groups. All surviving treated male (24 out of 100) and female (3 out
of 100) rats were sacrificed on weeks 49 and 61, respectively. In
rats given the high dose, hyperkeratosis and acanthosis of the
forestomach were observed in 12/50 males and 18/50 females, and in
low-dose females the value was 4/50. In the treated rats,
degenerative changes in the liver and adrenal gland and early
development of testicular atrophy were reported (NCI, 1978). However,
total development of testicular atrophy in controls, low-dose group
and high-dose group were 11/20, 14/49 and 18/5, respectively.
7.4.2 Inhalation
7.4.2.1 Mouse
In an NTP carcinogenicity study (NTP, 1982), groups of 50 male
and 50 female B6C3F1 mice were exposed to 1,2-dibromoethane
(> 99% pure) concentrations of 0, 77 or 308 mg/m3 (0, 10, or
40 ppm) for 6 h/day, 5 days per week, for 78 to 103 weeks, throughout
the study. Mean body weights of high-dose male and female mice were
lower than those of the untreated controls. Survival of high-dose
female mice was significantly reduced compared with controls.
Survival was also reduced in both control and treated male mice, the
principal cause of death being an ascending suppurative urinary tract
infection unrelated to compound administration. Surviving male mice
were killed at 79 weeks, while female mice were killed at 104-106
weeks, except for the high-dose group (killed at 91 weeks). The NTP
also reported inflammation of the nasal cavity and epithelial
hyperplasia of the respiratory system in both sexes (NTP, 1982).
Stinson et al. (1981) reported similar findings in mice using
data from a 2-year study. Epithelial hyperplasia of the urinary
bladder and inflammation of the prostate gland were also observed in
dosed male mice.
7.4.2.2 Rat
Groups of 50 male and 50 female inbred Fischer-344 rats were
exposed to 1,2-dibromoethane (> 99% pure) concentrations of 0, 77 or
308 mg/m3 (0, 10 or 40 ppm) for 6 h/day, 5 days per week, for 78 to
103 weeks, throughout the study. Mean body weights of high-dose rats
were lower than those of untreated controls. Survival of high-dose
male and female rats was significantly reduced compared with the
controls. Surviving female and male rats were killed at 104-106
weeks, except for the male high-dose group (5 out of 50 alive, killed
at 89 weeks) and female high-dose group (8 out of 50 alive, killed at
91 weeks). Among the observed compound-related non-neoplastic lesions
were hepatic necrosis and toxic nephropathy in both sexes, testicular
degeneration and atrophy in males, and retinal degeneration in females
(NTP, 1982).
Four groups of 48 male and 48 female Sprague-Dawley rats were
exposed to either control air, 154 mg 1,2-dibromoethane/m3, 0.05%
disulfiram in the diet, or 20 mg 1,2-dibromoethane/kg and 0.05%
disulfiram in the diet for 18 months. Disulfiram which is used in
alcohol control programmes is known to inhibit acetaldehyde
dehydrogenase. It may therefore alter the biotransformation of
1,2-dibromoethane and cause the accumulation of 2-bromoacetaldehyde,
one of the possible toxic metabolites of 1,2-dibromoethane. The
average starting weights ranged from 131 to 134 g for male rats and
118 and 124 g for females. Between inhalation exposures (7 h/day,
5 days/week), rats were permitted free access to water and food. Rats
receiving 0.05% disulfiram showed reduced body weight gain compared
with control rats or those given 20 mg 1,2-dibromoethane/kg or the
control diet. Rats exposed to 154 mg/m3 of 1,2-dibromoethane alone
and those receiving a combination of 20 mg 1,2-dibromoethane/kg and
0.05% disulfiram had high mortality compared with control and
disulfiram-treated rats. In rats exposed by inhalation to 154 mg
1,2-dibromoethane/m3 alone, mortality was 90% in males and 77% in
females at 18 months. Haematological parameters were within the
normal range for moribund rats in this group. Male rats receiving
20 mg 1,2-dibromoethane/kg with 0.05% disulfiram had a high incidence
of testicular atrophy and there was atrophy of the spleen in 30/48
males and 19/48 females (Wong et al., 1982).
7.5 Developmental toxicity
1,2-Dibromoethane causes testicular effects and mating failure in
rats at high inhalation levels that result in mortality. It also
causes temporary malformation of sperm cells in bulls and rams, but
not in chickens. In laying hens, it causes decreased egg size. In
rats and mice there is no evidence of embryotoxicity and
teratogenicity. In rats, paternal exposure causes behavioural changes
in F1 progeny.
7.5.1 Reproduction
Male Sprague-Dawley rats (3-4 per group) were exposed by
inhalation to average daily concentrations of 146, 300 or 684 mg/m3
(19, 39 or 89 ppm) of 1,2-dibromoethane for 7 h/day, 5 days/week, for
10 weeks. There was reduced weight gain in the 300 and 684 mg/m3
groups with mortality (21%) and morbidity in the 684 mg/m3 group.
Animals in this group had reduced testicular weights, reduced serum
testosterone levels, and atrophy of the testes (10/10), epididymis
(10/10), prostate (10/10), and seminal vesicles (9/9). When mated
with untreated females none of the males exposed to 684 mg/m3
impregnated any females, while 90% of those exposed to the low and
intermediate concentrations impregnated females who produced litters
that were normal in terms of total implants, viable implants and
resorptions. In female Sprague-Dawley rats, inhaling average daily
concentrations of 154, 300 or 614 mg/m3 (20, 39 or 80 ppm) of
1,2-dibromoethane for 7 h/day, 7 days/week, for 3 weeks, there was
reduced weight gain, morbidity and mortality (20%) in the 614 mg/m3
group. At the end of the 3-week exposure, females were mated with
untreated males. Females in the 614 mg/m3 group were in diestrus
and did not cycle normally until 3-4 days later; consequently fewer
females mated during a 10-day mating period than in the case of the
other two groups. The vaginal smears from females exposed to 154 or
300 mg/m3 were normal. In all three groups the reproductive
performance (total implants/dam, viable implants/dam and resorptions/
dam) was unimpaired. Histopathological examination of the ovaries and
uterus did not reveal any significant lesions (Short et al., 1979).
It was considered that the NOEL for reproductive performance was
300 mg/m3 for male and female rats.
Weanling male albino rats (10 rats per group) were fed
1,2-dibromoethane in the diet at levels of 100 or 500 mg/kg
(equivalent to 10 or 50 mg/kg body weight per day) for 90 days. There
was no evidence of toxicity; serum enzyme activities were unchanged.
Five rats from each group were mated with untreated virgin females.
There was no impairment of reproductive performance in the male rats.
At the end of the 2-week mating period the males were sacrificed.
Histology of the testis was normal. The pregnant females were allowed
to go to term, and the mean number of litters per group, mean pup
weight at birth, and sex ratio were found to be similar to the values
for a control group mated with untreated males (Shivanandappa et al.,
1987). The NOEL for male rat reproductive performance was considered
to be 50 mg/kg body weight per day.
In a study of sperm quality and fertility, mature (12 months old)
male New Zealand White rabbits (8-10 group) were injected
subcutaneously with 1,2-dibromoethane in corn oil at dose levels of
15, 30 or 45 mg/kg body weight per day for 5 days. There were also
untreated and vehicle control groups. Male fertility was assessed
before exposure, and at 4 and 12 weeks after injection, by artificial
insemination of three females/male per time point with one million
motile sperm. The percentage of pregnant females, litter size, fetal
body weights and structural development were assessed. In the highest
dose group there was 30% mortality and liver damage in 43% of the
survivors, indicated by increased levels of serum enzymes. There were
also changes in some sperm parameters (see 7.5.1.1). The percentage
of pregnant females and mean litter sizes were similar to those
produced by sperm from vehicle control animals, demonstrating that
fertilizing capacity and gestational outcome were unaffected (Williams
et al., 1991).
7.5.1.1 Effects on sperm
Four bull calves of the Israel-Friesian breed were administered
1,2-dibromoethane orally at a dose of 2 mg/kg body weight from the age
of 4 days by adding 1,2-dibromoethane to milk or feed concentrates.
When the calves reached an age of about 12 months, 1,2-dibromoethane
was administered in gelatin capsules. The treatment did not affect
the growth or health of the treated animals, and their libido was
similar to that of untreated animals. However, sperm density in
treated bulls was low, and sperm motility was poor. Semen showed
abnormally shaped spermatozoa (tailless, coiled tail, pyriform head).
Recovery after discontinuation of treatment varied from 10 days to
about 3 months in different animals. In a further study,
1,2-dibromoethane (4 mg/kg body weight) was administered orally to a
previously untreated bull. Two weeks after the start of the treatment
the semen exhibited abnormalities (Amir & Volcani, 1965). Other
studies also showed that 1,2-dibromoethane caused reversible
abnormalities of sperm cells in bulls (Amir, 1973, 1975; Amir et al.,
1979; Courtens et al., 1980) and in rams (ElJack & Hrudka, 1979), but
not in chickens (Alumot et al., 1968).
In a study by Williams et al. (1991), male New Zealand White
rabbits (8-10/group) were injected subcutaneously with
1,2-dibromoethane in corn oil (0, 15, 30 or 45 mg/kg body weight for
5 days). Weekly semen samples (for 6 weeks before exposure, during
treatment and 12 weeks after dosing) were analysed for sperm
concentration, number, morphology, viability and motion parameters
(velocity, linearity, beat cross-frequency, amplitude of lateral head
displacement (ALH) and circularity), and for semen pH, osmolality,
volume, and levels of fructose, citric acid, carnitine, protein and
acid phosphatase (AP). In the 45-mg/kg dose group, 1,2-dibromoethane
produced significant decreases in sperm velocity, percentage motility
and ALH (up to 25% at various times after dosing). There were also
dose-related decreases in semen pH (up to 2%) and total ejaculate
volume (up to 23%, 15 and 30 mg/kg groups only). Acid phosphatase
activities were significantly elevated (up to 116%) 2 weeks after
dosing in the 45 mg/kg dose group. All other semen parameters
evaluated were unaffected. Rabbits appear less sensitive than humans
to the reproductive effects of 1,2-dibromoethane, since semen
parameters were affected only at doses close to the LD50 and some
parameters (sperm numbers, viability and morphology) were unaffected.
A NOEL was not obtained in this study.
7.5.1.2 Effects on ova
Feeding studies with laying hens showed that 1,2-dibromoethane
absorbed by grain adversely affected egg production. When hens were
fed grain containing 200 mg/kg (corresponding to 25 mg/kg per day) for
56 days or grain containing 300 mg/kg (corresponding to 38 mg/kg per
day) for 46 days, the hens ceased laying completely. Feeding of grain
containing 10 mg/kg (corresponding to 12.5 mg/kg per day) caused a
diminution of egg size after 12 weeks (Bondi et al., 1955).
7.5.2 Teratogenicity
Pregnant Sprague-Dawley rats and CD-1 mice inhaled
1,2-dibromoethane concentrations of 146, 292 or 614 mg/m3 (20, 38 or
80 ppm) (23 h/day for 10 days) from day 6 to day 15 of gestation.
Adverse effects on maternal animals, measured by body weight gain and
food consumption, were observed in both species at all doses tested.
A marked increase in maternal mortality occurred in rats exposed to
614 mg/m3 and in mice exposed to 292 or 614 mg/m3. Some
morphological changes, such as haematomas, exencephaly and skeletal
variations, were observed in the fetuses of rats and mice. However,
these changes occurred only at high concentrations that caused
maternal toxicity (Short et al., 1978).
The embryotoxic effects of 1,2-dibromoethane bioactivation,
mediated by purified rat liver glutathione- S-transferases (GST),
were investigated using rat embryos in culture (Mitra et al., 1992).
Significant 1,2-dibromoethane metabolism was observed with rat liver
GST purified by affinity chromatography. 1,2-Dibromoethane activation
caused a significant reduction in general development as measured by
crown-rump length, yolk sac diameter, somite number, and the composite
score for different morphological parameters. Structures most
significantly affected were the central nervous and olfactory systems
as well as the yolk sac circulation and allantois. The results of
this study clearly indicate that under in vitro conditions,
bioactivation of 1,2-dibromoethane by GST can lead to embryotoxicity.
GST isozymes from human fetal liver were purified and used to
investigate the toxicity of 1,2-dibromoethane in an in vitro model
of rat embryos in culture as passive targets (Mitra et al., 1992).
1,2-Dibromoethane bioactivation by the GST isozyme P-3 resulted in
toxicity to cultured rat embryos. Significant reductions in crown
rump length, yolk sac diameter, and the composite score of
morphological parameters were observed. The central nervous, optic
and olfactory systems, and the hind limb were most significantly
affected.
When pregnant Sprague-Dawley rats were injected intraperitoneally
with 1,2-dibromoethane at a dose of 50 mg/kg body weight from day 1 to
day 15 of gestation, there was no evidence of embryotoxicity or
teratogenicity, although there were maternal toxic effects (change in
organ weights) (Hardin et al., 1981).
7.5.2.1 Effects on neonatal behaviour
Pregnant Long-Evans rats (16 animals/group, litter size 8-10)
were exposed to 3.3, 51.2 or 512 mg/m3 by inhalation (4 h/day,
3 days/week) from day 3 to day 20 of gestation. The highest
concentration produced enhanced rotorod performance and T-maze
brightness discrimination acquisition in the offspring. Similar
behavioural changes were noted in the offspring of mothers exposed to
51.2 mg/m3, but the magnitude of the effect was reduced. Exposure
to 3.3 mg/m3 produced no effects. DRL-20 acquisition (differential
reinforcement of low rates), straight alley running speed, and passive
avoidance were not affected at any dose level (Smith & Goldman, 1983).
In a study by Fanini et al. (1984), adult male Fischer-344 rats
were injected intraperitoneally with 1,2-dibromoethane at daily doses
of 0, 1.25, 2.5, 5 or 10 mg/kg body weight for 5 days. The treated
males were then mated with untreated female rats 4 or 9 weeks after
treatment. A total of 19 litters composed of 172 animals, 84 males
and 88 females were obtained from breeding the exposed males with
untreated females. Behavioural assessments of all F1 progeny were
carried out up to 21 days of age. Assessment of behavioural
development was made by means of an extensive testing battery.
Pre-weaning behavioural assessment included simple reflexes (surface
righting, cliff avoidance and negative geotaxis), motor coordination
(e.g., swimming and open field activity) and locomotor activity.
Significant impairment in the development of motor coordination and
motor activity was observed in the F1 progeny of males of all
treated groups. A NOEL was not found in this study.
The effects of 1,2-dibromoethane exposure on several
neurotransmitter enzymes in male rats were examined in various brain
regions of the F1 progeny (from 7 to 90 days of age) (Hsu et al.,
1985). Significant increase of choline acetyltransferase in the
cerebellum, corpus striatum, hippocampus and hypothalamus, alterations
of acetylcholinesterase in various brain regions, and an increase of
glutamic acid decarboxylase activity in the corpus striatum may be
related to early-development behavioural abnormalities.
7.6 Mutagenicity and related end-points
Mutagenicity assays are summarized in Table 16.
Table 16. Mutagenicity studies on 1,2-dibromoethane
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
In vitro
Gene mutation Salmonella E503 - Alper & Ames (1975)
typhimurium G46(-S9) + Ames & Yanofsky (1971); Von Buselmaier et al. (1972)
TA1530(-S9) + Ames & Yanofsky (1971); Brem et al. (1974); Rosenkranz
(1977); Buijs et al. (1984)
TA1535(+/-S9) +l Brem et al. (1974); McCann et al. (1975); Rannug & Beije (1979);
Shiau et al. (1980); Barber et al. (1981); Principe et al. (1981);
Moriya et al. (1983); Buijs et al. (1984); Dunkel et al. (1985);
Tennant et al. (1986, 1987); Zoetemelk et al. (1987); Barber &
Donish (1982)
TA98(+/-S9) +l Barber et al. (1981); Moriya et al. (1983);Dunkel et al. (1985);
Tennant et al. (1986)
TA100(+/-S9) +l McCann et al. (1975); Barber et al. (1981); Stolzenberg & Hine
(1980); van Bladeren et al. (1980, 1981b); Principe et al. (1981);
Moriya et al. (1983); Buijs et al. (1984); Dunkel et al. (1985);
Kerklaan et al. (1985); Guobaitis et al. (1986);
Tennant et al. (1986);
Hughes et al. (1987); Zoetemelk et al. (1987); Barber & Donish (1982)
TA1535(GSH-) + Kerklann et al. (1983)
(-S9,+ GSH) Zoetemelk et al. (1987)
TA100(GSH-) + Kerklaan et al. (1985)
(-S9,+GSH) Zoetemelk et al. (1987)
TA100W(Str',8AGr')(-S9) +g Ong et al. (1989)
TA1535 (bile of rats) + Rannug & Beije (1979)
TA1537(+/-S9) - Principe et al. (1981); Moriya et al. (1983); Dunkel et al. (1985);
Tennant et al. (1986)
TA1538(+/-S9) - Brem et al. (1974); Principe et al. (1981); Moriya et al. (1983);
Dunkel et al. (1985)
TA98 (+/-S9) - Principe et al. (1981); Wildeman & Nazar (1982)
TA100(+/-S9) - Shiau et al. (1980); Wildeman & Nazar (1982)
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Serratia a21 (-S9) - Von Buselmaier et al. (1972)
marcescens
Escherichia coli WP2 (+/-S9) + Scott et al. (1978); Hemminki et al. (1980); Moriya et al. (1983);
Dunkel et al. (1985)
CHY832 (-S9) + Hayes et al. (1984)
343/286 (+/-S9) + Mohn et al. (1984)
KI201, KI211 (-S9) + Izutani et al. (1980)
uvrB5 + Foster et al. (1988)
343/113 (-S9) - Mohn et al. (1984)
Bacillus subtilis TKJ5211, TKJ6321 (+S9) + Shiau et al. (1980)
Streptomyces (-S9, spot test) + Principe et al. (1981)
coelicolor (-S9, plate method) - Principe et al. (1981)
Aspergillus methG1BiA1 + Scott et al. (1978)
nidulans (+/-plant extract)
haploid strain 35 (-S9) + Principe et al. (1981)
Neurospora crassa ad-3 (forward mutation) + de Serres & Malling (1983)
Tradescantia clone 02, 0106, 4430 +g Sparrow et al. (1974); Nauman et al. (1976);
Vant'Hof & Schairer (1982)
Mouse L5178Y (+/-S9) + Clive et al. (1979); Tennant et al. (1986, 1987)
Chinese hamster CHO-K1 (+/-S9) + Tan & Hsie (1981); Brimer et al. (1982)
Human cell line AHH-1, TK6 + Crespi et al. (1985)
(-S9)
Human cell line EUE (-S9) + Ferreri et al. (1983)
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Unscheduled Rat hepatocytes + Williams et al. (1982); Tennant et al. (1986)
DNA synthesis
Opossum lymphocytes + Meneghini (1974)
Human lymphocytes (+/-S9) + Perocco & Prodi (1981)
Mouse (C3Hfx101)F1, + Sega & Rene (1980)
germ cells
Sister-chromatid Fish lymphocytes (- S9) + Ellingham et al. (1986)
exchange Chinese hamster V79 cl-15 (-S9) + Tezuka et al. (1980)
Chinese hamster CHO (+/-S9) + Tennant et al. (1987)
Human lymphocytes (-S9) +g Ong et al. (1989); Tucker et al. (1984)
Chromosome Fish lymphocytes (- S9) + Ellingham et al. (1986)
aberrations Chinese hamster V79 cl-15 (-S9) + Tezuka et al. (1980)
CHO (+/-S9) + Tennant et al. (1987)
Micronuclei Tradescantia clone 03, 4430 +l Ma et al. (1978, 1984)
DNA damage E. coli polA1-/polA+ (-S9) +w Brem et al. (1974)
B. subtilis TKJ5211, TKJ6321 - Shiau et al. (1980)
(+/-S9)
SOS induction S. typhimurium TA1535/pSK1002 +g Ong et al. (1987)
(+/-S9)
E. coli PQ37 (-S9) + Ohta et al. (1984); Quillardet et al. (1985)
Mitotic gene Saccharomyces ade2, trp5 + Fahrig (1974)
conversion cerevisiae
Somatic A. nidulans diploid 35 x 17 (-S9) +g Crebelli et al. (1984)
segregation
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
DNA binding E. coli Q13 (+/-S9) - Kubinski et al. (1981)
Mouse Ehrlich ascites (+/-S9) - Kubinski et al. (1981)
Cell Human lymphocyte + Channarayappa et al. (1992)
proliferation
DNA strand Rat hepatocytes + Sina et al. (1983)
breaks
Cell Mouse Balb/c 3T3 (-S9) - Tennant et al. (1986); Perocco et al. (1991)
transformation
In vivo
Gene mutation S. typhimurium G46 (host-mediated) + Von Buselmaier et al. (1972)
Serratia a21 (host-mediated) - Von Buselmaier et al. (1972)
marcescens
Barley + Ehrenberg et al. (1974)
Silkworm (egg color mutation) - Sugiyama (1980)
Drosophila (wing spot) +g Graf et al. (1984)
melanogaster
Recombination D. melanogaster (wing spot) +g Graf et al. (1984)
Sex-linked D. melanogaster +l Kale & Baum (1979a,b, 1981, 1982, 1983); Yoshida & Inagaki
recessive lethal (1986); Vogel & Chandler (1974)
mutations D.melanogaster spermatozoa + Ballering et al. (1993)
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Specific Mouse - Russell (1986)
locus test Mouse DBA/2J - Barnett et al. (1992)
Chromosome Barley root tips + Ehrenberg et al. (1974)
aberration Mouse (ip) CD1, (bone marrow) - Krishna et al. (1985)
Sister-chromatid Mouse (ip) CD1, bone marrow - Krishna et al. (1985)
exchange
Dominant lethal Rat (ih, po) CD, SD - Short et al. (1979); Teramoto et al. (1980)
Mouse (po) BDF1 - Teramoto et al. (1980)
Mouse (ip, po) ICR/Ha - Epstein et al. (1972)
Mouse DBA/2J - Barnett et al. (1992)
DNA strand Rat (po) hepatocytes + Nachtomi & Sarma (1977)
breaks Mouse (ip) hepatocytes + Storer & Conolly (1983)
breaks Mouse (ip) hepatocytes + White (1982)
Micronuclei Amphibians: erythrocytes Fernandez et al. (1993)
Pleurodeles waltl +
(newt)
Ambystoma +
mexicanum (axolotl)
Anuran: Xenopus +
laevis (toad)
Mouse ddY(peripheral blood - Asita et al. (1992)
reticulocytes)
Mouse(ip) CD1, bone marrow - Krishna et al. (1985)
Tradescantia tetrads of + Ma et al. (1978)
microsporogenesis
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Sperm Bull Friesian + Amir et al. (1977)
abnormality
a ip = intraperitoneal administration; ih = inhalation exposure; iv = intravenous administration; po = oral administration
b g = test in gaseous phase; l = test in liquid or gaseous phase; w = weak positive results
7.6.1 In vitro assays
1,2-Dibromoethane was mutagenic in reverse mutation assays
(Ames test) using Salmonella typhimurium strains G46, TA1530,
TA1535, TA98, TA100 and TA100W, and in the host-mediated assay using
strain G46. Bile from mice and rats injected intraperitoneally with
1,2-dibromoethane or following liver perfusion was mutagenic in a test
using strain TA1535. Gene mutation assays using Escherichia coli,
strain WP2 and several other strains, were positive. A positive
response was reported in assays using Bacillus subtilis, Streptomyces
coelicolor, Aspergillus nidulans and Neurospora crassa.
A requirement for intracellular activation of 1,2-dibromoethane
was observed for a mutagenic response in S. typhimurium TA1535.
Glutathione- S-transferase 5-5 transfected into the bacteria allowed
the induction of base-pair mutations on exposure to 1,2-dibromoethane,
whereas a 20-fold extracellular excess of the enzyme did not permit
1,2-dibromoethane-induced mutations in non-transfected bacteria in the
presence of reduced glutathione (Thier et al., 1993).
1,2-dibromoethane failed to induce reverse mutations in a number
of assays using S. typhimurium E503, TA1537 and TA1538, with or
without S9 mix. A negative response was reported in several assays
using E. coli, Serratia marcescens and silkworms, and in the mouse
specific locus test.
Chromosomal aberrations were induced by 1,2-dibromoethane in
cultured fish lymphocytes (Ellingham et al., 1986), Chinese hamster
V79 and CHO cell lines (Tezuka et al., 1980; Tennant et al., 1987).
1,2-Dibromoethane was negative in a test for dominant lethal and
specific locus mutations in germ cells from DBA/2J male mice (Barnett
et al., 1992).
1,2-Dibromoethane increased significantly the frequency of sister
chromatid exchanges in cultured fish lymphocytes (Ellingham et al.,
l986), Chinese hamster cell line (V79) and cultured human lymphocytes
(Tucker et al., 1984; Ong et al., 1989), but not in bone marrow cells
of mice injected intraperitoneally (Krishna et al., 1985).
In isolated human mononucleated and binucleated peripheral
lymphocytes, 1,2-dibromoethane caused an increased frequency of
micronuclei in vitro (Channarayappa et al., 1992).
A positive response was obtained in the unscheduled DNA synthesis
assay using cultured rat hepatocytes (Williams et al., 1982; Tennant
et al., 1986), cultured opossum lymphocytes (Meneghini, 1974),
cultured human lymphocytes (Perocco & Prodi, 1981) and mouse germ
cells (Sega & Rene, 1980), and in the DNA strand break assay using rat
or mouse hepatocytes (Nachtomi & Sarma, 1977; White, 1982; Sina et
al., l983). Tests for DNA damage using E. coli (Brem et al., 1974),
SOS induction using S. typhimurium and E. coli (Ohta et al., 1984;
Quillardet et al., 1985; Ong et al., 1987), mitotic gene conversion
using Saccharomyces cerevisiae (Fahrig, 1974), somatic segregation
using A. nidulans (Crebelli et al., 1984), recombinant DNA synthesis
using D. melanogaster (Graf et al., 1984), and sperm abnormality in
bulls (Amir et al., 1977) were positive. However, there was a
negative response in a test for DNA damage using B. subtilis (Shiau
et al., 1980), in DNA-binding tests using E. coli and Ehrlich
ascites tumour cells (Kubinski et al., 1981), and in the cell
transformation assay using mouse Balb/c 3T3 cells (Tennant et al.,
1986).
7.6.2 In vivo assays
1,2-Dibromoethane did not induce chromosomal aberrations or
sister chromatid exchanges in occupationally exposed papaya fruit
packers (Steenland et al., 1986).
1,2-Dibromoethane was negative in tests for specific locus
mutations in germ cells in male mice (Russell, 1986; Barnett et al.,
1992). It did not induce dominant lethal mutations in mice (Epstein
et al., 1972; Teramoto et al., 1980; Barnett et al., 1992) or rats
(Short et al., 1979; Teramoto et al., 1980), chromosomal aberrations
in mice (Krishna et al., 1985) or micronuclei in bone-marrow cells of
mice (Krishna et al., 1985); there was a weak sister chromatid
exchange response that was not dose-related (Krishna et al., 1985).
Tests for recombinant DNA synthesis using D. melanogaster (Graf
et al., 1984) and sperm abnormality using bulls (Amir et al., 1977),
and a sex-linked recessive lethal test using D. melanogaster with
exposure to 1,2-dibromoethane in the gaseous phase (Kale & Baum, 1982)
were all positive.
The frequency of micronuclei in pollen mother cells of
Tradescantia was significantly increased by exposure to
1,2-dibromoethane in the liquid or gaseous phase (Ma et al., 1978,
1984). Gene mutation was demonstrated in barley (Ehrenberg et al.,
1974) and Drosophila melanogaster (wing spot) (Graf et al., 1984).
It induced DNA strand breaks in rat (Nachtomi & Sarma, 1977) and mouse
(White, 1982) hepatic cells. 1,2-Dibromoethane was negative in a gene
mutation assay (egg colour mutation) in silkworms (Sugiyama, 1980).
7.6.3 Other studies
Two doses of 1,2-dibromoethane with dose levels ranging between
10 and 300 µmol/kg body weight (1.8-56 mg/kg body weight) were given
by gavage to 90-day-old female Sprague-Dawley rats, 21 and 4 h before
sacrifice. 1,2-Dibromoethane caused marked DNA damage. The fraction
of DNA eluted from samples of blood, bone marrow, liver, kidney,
spleen or thymus of the rats given 1,2-dibromoethane doses of 100 µmol
body weight (18 mg/kg body weight) 21 and 4 h before sacrifice was
higher than in controls. However, the difference was statistically
significant only for kidney and liver (Kitchin & Brown, 1986).
Intraperitoneal injection (0.25 and 0.5 mmol/kg) of
1,2-dibromoethane (> 99 9%) in corn oil in B6C3F1 mice (weight
20-26 g) produced hepatic damage. The mice were sacrificed 4 h later
and in vivo genotoxicity was determined by a sensitive in vivo/
in vitro alkaline DNA-unwinding assay for the presence of
single-strand breaks and/or alkali-labile sites in hepatic DNA.
Significant hepatic DNA damage was found with a dose of 0.5 mmol/kg.
Although 1,2-dibromoethane is a direct-acting mutagen, its
mutagenicity is generally enhanced by metabolic activation. Two
different pathways have been postulated for its activation to the
ultimate mutagenic form. One is mediated by the mixed-function
oxygenases of liver microsomes, in which 1,2-dibromoethane is
converted to bromoacetaldehyde and 2-bromoethanol, both of which are
potential DNA-damaging agents (Hill et al., 1978; Banerjee et al.,
1979). The other pathway is mediated by enzymes present in liver
cytosol, in which glutathione conjugation can yield a half-sulfur-
mustard or an episulfonium ion as reaction products (Rannug, 1980; Van
Bladeren et al., 1980).
Glutathione conjugation also contributes to the binding of
1,2-dibromoethane to DNA, and an S-[2-(N7-guanyl)ethyl]
glutathione adduct has been identified (Ozawa & Guengerich, 1983;
Inskeep & Guengerich, 1984). Foster et al. (1988) reported that the
majority of mutations induced by 1,2-dibromoethane consist of GC to AT
and AT to GC base changes, suggesting that it acts like an alkylating
agent.
This glutathione conjugate accounts for > 95% of the total DNA
adducts formed by 1,2-dibromoethane. S. typhimurium TA100 and
sequence analysis were used to determine the type, site and frequency
of mutations in a portion of the lacZ gene resulting from in vitro
modification of bacteriophage M13mp18 DNA with S-(2-chloroethyl)
glutathione, an analogue of the 1,2-dibromoethane-glutathione
conjugate. An adduct level of approx.8 nmol per mg DNA resulted in a
10-fold increase in mutation frequency. The mutations were mainly
base substitutions in which GC to AT transitions accounted for 75%
(70% of the total mutations) (Cmarik et al., 1992).
The steady-state levels of c-fos, c-jum, and c-myc mRNA were
investigated in male Wistar rat liver following oral dosing with
100 mg 1,2-dibromoethane/kg body weight. This dose induced
hyperplasia. Increases in the expression of c-myc and c-jum genes
were observed in the absence of c-fos expression (Coni et al.,
1993).
Sundheimer et al. (1982) examined the relationship between
glutathione metabolism and the rate of DNA alkylation by
1,2-dibromoethane in cultured hepatocytes. The rate of alkylation was
decreased by the addition of diethyl maleate and increased by the
addition of cytosolic microsomal enzymes. While high concentrations
of 1,2-dibromoethane were capable of depleting glutathione in the
hepatocytes, depletion did not appear to be necessary for binding to
occur. On the basis of these results, the authors concluded that
glutathione- S-transferases are involved in the bioactivation of
1,2-dibromoethane to an alkylating species.
Working et al. (1986) assessed the ability of 1,2-dibromoethane
to cause DNA damage by quantifying the rate of unscheduled DNA
synthesis (UDS) in F-344 rat hepatocytes and spermatocytes exposed to
1,2-dibromoethane in vivo and in vitro. Pretreatment of cells
with inhibitors of cytochrome-P450-mediated oxidation had no effect on
the induction of UDS by 1,2-dibromoethane (10-100 µmol/litre)
in vitro, whereas depletion of cellular glutathione strongly
inhibited UDS induction in both cell types. Pretreatment of rats with
metyrapone (an inhibitor of hepatic mixed-function oxidases) in vivo
had no effect on 1,2-dibromoethane-induced UDS in hepatocytes, but
produced a positive UDS response in spermatocytes. This suggests that
the mixed-function oxidase pathway in metabolism is the primary route
of clearance of 1,2-dibromoethane and the inhibition of this enzyme
system leads to more extensive tissue distribution of the parent
compound. The data also suggest that the pathway which produces
genotoxic metabolites from 1,2-dibromoethane in hepatocytes and
spermatocytes, in vivo and in vitro, involves the conjugation of
1,2-dibromoethane to glutathione and its subsequent metabolism.
In studies of DNA adducts, Kim & Guengerich (1989) measured
urinary excretion of S-[2-( N7-guanyl)ethyl]- N-acetyl-cysteine,
derived from the nucleic acid adduct, S-[2-( N7-guanyl)
ethyl]glutathione, in rats treated with 1,2-dibromoethane. A good
correlation was found between the excretion of this mercapturic acid
and the in vivo formation of the DNA adduct in liver and kidney DNA.
Inskeep et al. (1986) determined that the major DNA adduct formed upon
exposure to 1,2-dibromoethane, S-[2-( N7-guanyl)ethyl]glutathione,
had a half-life in rat liver, kidney, stomach and lung between 70 and
100 h.
Inskeep & Guengerich (1984) measured the rate of formation of
1,2-dibromoethane adducts to calf thymus DNA in vitro. Adduct
formation was dependent on the presence of both glutathione and
glutathione- S-transferase.
7.7 Carcinogenicity
1,2-Dibromoethane has been tested for carcinogenicity by oral
administration and inhalation in mice and rats, and by skin
application in mice (IARC, 1987) (see Table 14).
It has been shown to cause tumours in various organs, by several
dosage routes and, in some cases, with a latency of less than
12 months (Table 15).
The following tumours have been observed:
a) By oral administration
hepatocellular carcinomas and neoplastic nodules in female rats
haemangiosarcomas in various circulatory system organs in male
rats
alveolar/bronchiolar adenomas in male and female mice
b) By inhalation exposure
nasal cavity carcinomas and adenocarcinomas in male and female
rats
alveolar/bronchiolar carcinomas in female rats
alveolar/bronchiolar carcinomas in male and female mice
haemangiosarcomas in the circulatory system of male and female
rats
mesotheliomas in male rats
mammary fibroadenomas in female mice
subcutaneous fibrosarcomas in female mice
c) By skin administration
skin papillomas and lung papillomas in female mice
7.7.1 Administration by gavage
7.7.1.1 Mouse
When technical grade 1,2-dibromoethane (99.1% pure) in corn oil
was administered on 5 consecutive days per week by gavage to B6C3F1
mice (groups of 50 males and 50 females in the treated groups, and 20
of each sex in the untreated and vehicle control groups) at time-
weighted average dose levels of 62 and 107 mg/kg per day, early
development of squamous cell carcinomas of the forestomach was
observed in both sexes. The incidence of alveolar/bronchiolar
adenomas was significantly higher in treated mice of both sexes than
in controls (NCI, 1978).
7.7.1.2 Rat
Osborne-Mendel rats (50 animals of each sex in the treated groups
and 120 animals of each sex in the untreated and vehicle control
groups) were given on 5 consecutive days per week, by gavage,
technical grade 1,2-dibromoethane (99.1% pure) in corn oil at time-
weighted average dose levels of 38 or 41 mg/kg per day for males, and
37 or 39 mg/kg per day for females. Squamous cell carcinomas of the
forestomach were observed in more than half the male and female rats
at both dose levels, while none were observed in controls. The
lesions, seen as early as week 12, were locally invasive and
eventually metastasized. Significantly higher incidences of
hepatocellular carcinomas and haemangiosarcomas were observed in
treated males and females, respectively (NCI, 1978).
Ledda-Columbano et al. (1987b) examined the interaction of an
intragastric dose of either 1,2-dibromoethane or carbon tetrachloride
(CCl4) with diethylnitrosomine (DENA). The administration of either
1,2-dibromoethane or CCl4 resulted in similar increases in cell
proliferation. However, while pretreatment with CCl4 caused an
increase in the incidence of hepatic foci resulting from subsequent
DENA administration, pretreatment with 1,2-dibromoethane did not.
This difference in the ability of the two compounds to act as a
promoter was attributed to the nature of cell proliferation response.
The authors concluded that the cell proliferation induced by
1,2-dibromoethane, unlike the compensatory cell proliferation induced
by CCl4, is not an effective process for increasing the rate of
tumour initiation.
7.7.2 Administration in drinking-water
7.7.2.1 Mouse
1,2-Dibromoethane (> 99% purity) and its metabolites,
bromoethanol or bromoacetaldehyde, were administered to B6C3F1 mice
(groups of 30 males and 30 females) at a concentration of 4 mmol/litre
in distilled drinking-water (equivalent to 116 mg/kg body weight for
males and 103 mg/kg body weight for females), for 450 days in the case
of 1,2-dibromoethane and for 560 days in the case of the metabolites.
A control group (60 males and 60 females) was given distilled
drinking-water. 1,2-Dibromoethane induced squamous cell carcinomas of
the forestomach in 26/30 males and 27/30 females, and squamous cell
papillomas of the oesophagus in 3/30 females. Bromoethanol in
drinking-water at a concentration of 4 mmol/litre (equivalent to
76 mg/kg body weight for males and 71 mg/kg body weight for females)
induced squamous cell papillomas of the forestomach in 9/29 males and
10/29 females, but bromoacetaldehyde at the same concentration
(equivalent to 62 mg/kg body weight for females and 62 mg/kg body
weight for males) did not induce a significant incidence of
forestomach tumours. The incidence of tumours in the control group
was not significant. Bromoethanol and bromoacetaldehyde were not
considered likely to be active intermediates of 1,2-dibromoethane
carcinogenicity (Van Duuren et al., 1985).
7.7.3 Inhalation
7.7.3.1 Mouse
Groups of 50 male and 50 female B6C3F1 mice were exposed in
inhalation chambers to air containing 77 or 308 mg/m3 (10 or 40 ppm)
of 1,2-dibromoethane (99.3-99.4% pure) for 78-106 weeks. The
incidences of alveolar/bronchiolar carcinomas and alveolar/bronchiolar
adenomas were significantly higher in exposed male and female mice
than in controls. Haemangiosarcomas of the circulatory system,
fibrosarcomas in subcutaneous tissue, carcinomas of the nasal cavity,
and adenocarcinomas of the mammary gland were significantly increased
in females. Exposure to 1,2-dibromoethane was also associated with
epithelial hyperplasia of the respiratory system (NTP, 1982).
7.7.3.2 Rat
Groups of 50 male and 50 female F-344 rats were exposed in
inhalation chambers to air containing 77 or 308 mg/m3 (10 or 40 ppm)
of 1,2-dibromoethane (99.3-99.4%) for 88-106 weeks. Carcinomas,
adenocarcinomas and adenomas of the nasal cavity, and
haemangiosarcomas of the circulatory system were significantly
increased in exposed male and female rats. The incidences of
mesotheliomas of the tunica vaginalis and adenomatous polyps of the
nasal cavity in males, and of fibroadenomas of the mammary gland and
alveolar/bronchiolar adenomas and carcinomas (combined) in females
were significantly increased (NTP, 1982).
Wong et al. (1982) reported that the coadministration of dietary
disulfiram increases the rate of tumour incidence in rats exposed to
1,2-dibromoethane (153.6 mg/m3, 20 ppm) by inhalation. There is
evidence to suggest that the synergistic effect of disulfiram may be
the result of increased liver glutathione- S-transferase activity in
animals treated with this drug (Elliot & Ashby, 1980).
7.7.4 Dermal application
7.7.4.1 Mouse
Doses (25 mg or 50 mg) of 1,2-dibromoethane (> 99% pure)
dissolved in 0.2 ml acetone were applied 3 times/week to the shaved
dorsal skin of female Ha:ICR Swiss mice (groups of 30 animals). There
were an acetone only and untreated control groups. The times to first
appearance of skin tumour (papilloma) were 434 days for the 25-mg
group and 395 days for 50-mg group. By comparison with controls, both
groups showed a statistically significant increase in skin papillomas,
and in the 50-mg group there was also a significant increase in lung
papillomas. Both groups also had dermal squamous carcinomas and
stomach, tumours but these were not statistically significant (Van
Duuren et al., 1979).
7.7.5 Cell transformation
1,2-Dibromoethane caused transformation of BALB/C 3T3 cells both
in the presence and absence of an exogenous metabolism system (Perocco
et al., 1991).
7.8 Biochemical studies and species specificity
1,2-Dibromoethane (75-100 mg/kg body weight) given by gavage to
non-fasted Wistar rats induced DNA synthesis and cell division in the
liver. The peak of DNA synthesis, as measured by 3H-methyl
thymidine incorporation, was attained at or shortly after 24 h. The
mitotic waves measured with the aid of colchicine occurred at 24-30 h
and 48 to 54 h after 1,2-dibromoethane treatment. Increase in DNA
synthesis was confirmed by autoradiography. The stimulation of liver
cell mitosis occurred in non-fasted animals without any apparent cell
necrosis. 1,2-Dibromoethane was an effective mitogen for liver under
these experimental conditions (Nachtomi & Farber, 1978).
In a study by Ledda-Columbano et al. (1987a) 1,2-dibromoethane in
corn oil was administered by gavage at a dose of 100 mg/kg body weight
to male Wistar rats (weight 250-280 g). The rats were given an
intraperitoneal injection of 3H-thymidine 1 h prior to sacrifice,
10, 20, 30, 36 and 48 h after the treatment. No mortality
attributable to 1,2-dibromoethane was observed in the treated rats.
The body weights of 1,2-dibromoethane-treated rats were similar to
those of controls. No changes in the specific activity of DNA were
observed in kidney 10 h after treatment. There was an increase in
labelled thymidine incorporation into DNA and this was maximal after
20-30 h. At 48 h the extent of incorporation of labelled thymidine
decreased rapidly even though it was still higher than in controls.
When effects on the kidneys were investigated, mitotic activity was
noted predominantly in the proximal tubular epithelium of the renal
cortex. Histological examination of the kidney did not reveal any
signs of necrosis.
Of possible significance for the prediction of species
differences in response to 1,2-dibromoethane are the observations
(Dibiasio et al., 1991) that hepatic cytosolic glutathione-
S-transferase activities are similar in rats and mice, but about 40%
of these values in rhesus monkeys. In addition, cytosolic
glutathione- S-transferase activities in rhesus monkey and human
testis are only about 5% of the activities in rat and mouse testes.
8. EFFECTS ON HUMANS
8.1 Acute toxicity
1,2-Dibromoethane is strongly irritant to the eyes, skin, and
respiratory tract (Peoples et al., 1978; Letz et al., 1984). Deaths
from acute exposure to high concentrations of 1,2-dibromoethane are
usually due to pneumonia following damage to the lungs. In addition,
acute inhalation exposure may lead to liver and kidney damage.
Six people who attempted suicide by ingesting 1,2-dibromoethane
suffered from vomiting, nausea and burning throat; death followed in
two cases. The characteristic pathological lesions were present in
liver, lungs and kidneys. Intense jaundice was observed and was due
to massive necrosis of the liver (Sarawat et al., 1986).
It is estimated that 200 mg/kg is lethal to humans, based on the
observation that 12 g caused the death of a woman weighing about 60 kg
(Alexeef et al., 1990).
8.2 Occupational exposure
In cases of poisoning following occupational exposure, headache,
severe vomiting, diarrhoea, respiratory tract irritation and death
have been reported. Exposure to 1,2-dibromoethane in air at
concentrations above 384 mg/m3 (50 ppm) caused nasal and throat
irritation. Two deaths were reported after exposure by inhalation to
a mean concentration of 215 mg/m3 (28 ppm) for 30 and 45 min,
respectively, during the cleaning of a storage tank containing
residues of 1,2-dibromoethane (Letz et al., 1984, Jacobs, 1985).
There was also absorption from dermal exposure to the 0.1-0.3%
solution in the tank (Letz et al., 1984). The first worker collapsed
while working inside the tank and died 12 h later with metabolic
acidosis, depression of the CNS, and laboratory evidence of liver
damage. A supervisor attempting to rescue the worker also collapsed
inside the tank and died 64 h later with intractable metabolic
acidosis, hepatic and renal failure, and necrosis of skeletal muscle
and other organs. Coughing, vomiting, diarrhoea, eye, skin and
respiratory irritation, coma, metabolic acidosis, delirium, confusion,
nausea, low urine output, renal failure, tachycardia and asystole
were noted. Autopsy revealed pulmonary oedema, liver damage and
extensive autolysis in the kidney.
Inhalation exposure to concentrations over 154 mg/m3 (20 ppm)
for more than 30 min is considered fatal to humans.
8.2.1 Cancer incidence
Mortality in employees exposed to 1,2-dibromoethane in two
production units operated from 1942 to 1969 and from the mid-1920s to
1976 was investigated (Ott et al., 1980). The study population was
161 employees. In the first production unit two deaths from malignant
neoplasms were observed against 3.6 expected, and in the second unit,
where there was potential exposure to various organic bromide
products, there were five deaths from malignant neoplasms against
2.2 expected (p < 0.072). However, no statistically significant
increase in total deaths or malignant neoplasms relatives to duration
of exposure was observed.
Epidemiological studies of four worker populations did not show
any increase in cancer that could be attributed to 1,2-dibromoethane
(Ter Haar, 1980)
8.2.2 Reproductive effects
In an investigation of possible sterility from exposure to
1,2-dibromoethane, sperm levels in workers exposed to
1,2-dibromoethane were not affected, and there was no evidence of
effects on offspring (Ter Haar, 1980).
Ratcliffe et al. (1987) and Schrader et al. (1987) conducted a
cross-sectional study of semen quality in 46 men employed in the
papaya fumigation industry in Hawaii, with an average duration of
exposure of 5 years and a geometric mean breathing zone exposure to
airborne 1,2-dibromoethane of 0.68 mg/m3 (88 ppb) (8-h time-weighted
average). The control group consisted of 43 unexposed men from a
nearby sugar refinery. Statistically significant decreases in sperm
count per ejaculate and percentage of viable and motile sperm,
together with increases in the proportion of sperm with specific
morphological abnormalities, were observed among exposed men, compared
to controls, after consideration of smoking, caffeine and alcohol
consumption, subject's age, abstinence, history of urogenital
disorders, and other potentially confounding variables. The data
indicated that 1,2-dibromoethane could cause reproductive inpairment
in males exposed to this concentration. Schrader et al. (1988)
conducted a short-term longitudinal study on the effect of
1,2-dibromoethane exposure on male reproductive potential in ten
forestry workers and six unexposed men in Colorado. The time-weighted
average inhalation exposure over 6 weeks was 0.46 mg/m3 (peak
exposure of 16 mg/m3) and there was extensive skin exposure. Sperm
velocity and semen volume were decreased significantly in the exposed
workers. Both studies suggested that 1,2-dibromoethane has multiple
sites of action on male accessory sex glands and testes.
In five studies on the reproductive effects of occupational
exposure to 1,2-dibromoethane, four showed potential reproductive
impairment but this was not large enough to be statistically
significant. The power of all of the studies was low and they were
considered inconclusive for assessing reproductive risk (Dobbins,
1987).
A retrospective study of four plants in the United Kingdom where
male workers were exposed to 1,2-dibromoethane revealed statistically
marginally reduced fertility rates (i.e., live births to their wives).
The average exposure was probably below 38.5 mg/m3 (5 ppm), although
actual concentrations were not measured (Wong et al., 1985).
9. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
9.1 Aquatic organisms
9.1.1 Invertebrates
Nishiuchi (1980, 1981) studied the acute toxicity of
1,2-dibromoethane in aquatic larvae of insects and aquatic
invertebrates, such as a mayfly (Cloeon dipterum), two dragonflies
(Sympetrum frequens and Orthetrum albistylum speciosum), a
Japanese diving beetle (Eretes sticticus) and a water boatman
(Micronecta sedula) (Table 17). The toxicity threshold (48-h LC50
values) of 1,2-dibromoethane machine oil formulation for the above
aquatic invertebrates and that of 1,2-dibromoethane for mayfly were
greater than 40 mg/m3. In mayfly the reported 48-h toxicity
thresholds for two mixed formulations, which consisted of
1,2-dibromoethane, malathion, diazinon and machine oil (3:1:1:80), and
1,2-dibromoethane, diazinon and O-sec-butylphenyl methylcarbamate
(25:5:3) were 28 and 65 mg/m3, respectively (Nishiuchi & Asano,
1979). There was no apparent difference in toxicity of technical
1,2-dibromoethane and an oil-based formulation at least up to
40 mg/litre.
Table 17. Acute toxicity of 1,2-dibromoethane to aquatic
invertebrates (From: Nishiuchi, 1980)
Species Test material Temperature 48-h LC50
(°C)
Mayfly Technical material 25 > 40
(Cloeon dipterum) (in acetone)
Dragonfly 1,2-dibromoethane (oil) 25 > 40
(Sympetrum frequens)
Dragonfly 1,2-dibromoethane (oil) 25 > 40
(Orthetrum albistylum
speciosum)
Japanese diving beetle 1,2-dibromoethane (oil) 25 > 40
(Eretes sticticus)
Water boatman 1,2-dibromoethane (oil) 25 > 40
(Micronecta sedula)
Herring et al. (1988) evaluated the toxicity of 1,2-dibromoethane
to Hydra oligactis in a series of three experiments. Study 1
evaluated lethality, feeding behaviour and mobility in a series of
concentrations ranging from 7.5 to 75 mg/litre. In Study 2, adult
Hydra were pre-treated for 14 days with a sublethal dose of
1,2-dibromoethane (5 mg/litre) prior to exposure to a range of
1,2-dibromoethane concentrations (25-300 mg/litre) for a total of
72 h. In the third study, the F1 offspring of pre-treated adult
Hydra were also exposed to a series of 1,2-dibromoethane
concentrations. The 48-h LC50 for Hydra was determined to be
70 mg/litre. When adult Hydra were pre-treated with sublethal
concentrations of 1,2-dibromoethane, the 48-h LC50 was increased to
200 mg/litre. Furthermore, the F1 offspring of pre-treated adult
exhibited mortalities of only 10% and 20%, respectively, after 24-h
and 48-h exposures to 200 mg 1,2-dibromoethane/litre. These results
suggest that Hydra and first-generation offspring are capable of
developing tolerance to 1,2-dibromoethane.
Adams & Kennedy (1992) exposed first-stage budding Hydra
oligactis to 1,2-dibromoethane at 5 mg/litre. The 1,2-dibromoethane
was dissolved using acetone at 15 mg/litre, and an acetone control was
used. Exposure was for 24, 48 or 72 h. Following exposure, the
animals were washed several times with the medium, sectioned through
the gastric region, and the base/apical sections were grafted.
Regeneration was significantly affected by all exposures to
1,2-dibromoethane but not by acetone. The severity of the effect
increased with increasing exposure.
Adams et al. (1989) reported dose-sensitive relationships for the
loss and recovery of locomotor response, chromatophore expansion and
lethality in three species of laboratory-reared octopus. Three
species of octopus (Octopus bimaculoides, O. joubini, and O. maya)
were exposed to 25, 50, 75 and 100 mg 1,2-dibromoethane/litre for
either one hour, followed by transfer to chemical-free water, or
continuously for a period of 72 h. Generally, responses by the
octopuses were evident after only 1 min of exposure. Chromatophore
expansion and loss of locomotor response occurred at 25 mg/litre after
30 min, but recovery was noted 6 h after transfer to chemical-free
water. Lethality occurred in all three species at 25 mg/litre after
48 h of exposure. O. maya was the most sensitive species,
exhibiting 100% mortality after 3 h of exposure. The acute LC50
values for O. bimaculoides, O. joubini and O. maya were 42.7, 35.3
and 30.6 mg/litre respectively (Table 18). Although the authors
reported a chronic LC50 of 100 mg/litre occurring within 12 h, the
Task Group considered this to be an acute exposure.
Table 18. Acute LC50 values approximated for lethality data
(Adams et al., 1989) using either the moving average,
binomial or probit methods
Test species Estimated 48-h LC50 95% confidence
(mg/litre) limits
Octopus bimaculoides 42.7 28.1-58.8a
Octopus joubini 35.3 -b
Octopus maya 30.6 0-52.02c
a moving average method
b confidence limits exceeded 95%, therefore the limits would
range from 0 to infinity
c probit method used
9.1.2 Fish
There are few data for the acute toxicity of 1,2-dibromoethane in
fish. A study of the effect of pH on the acute toxicity of
1,2-dibromoethane in killifish (Oryzias latipes) was carried out in
open static systems. Altering the pH of the breeding water between pH
5.0 and pH 10.0 had no effect on 48-h LC50 values (Nishiuchi, 1982).
Goldfish quickly absorbed 1,2-dibromoethane from water (at 1 mg/litre)
and quickly eliminated it. The concentration in the goldfish
(1.75 ± 0.041 mg/kg) was in equilibrium with the concentration in the
water 1.5 h after the initiation of exposure. From the results of the
elimination study, the biological half-life was calculated to be less
than 30 min (Ogino, 1978).
Landau & Tucker (1984) found the 48-h LC50 values for
sheepshead minnow Cyprinodon variegautus and snook Centropomus
undecimatis, both estuarine fish, to be 4.8 and 6.2 mg/litre,
respectively.
Nishiuchi & Asano (1979) measured toxicity thresholds for carp
(Cyprinus carpio) exposed to pesticide mixtures containing various
concentrations of 1,2-dibromoethane (Table 19). After 24 h of
exposure at 24°C, no difference in the toxicity threshold (> 40)
could be detected at unit pH increases between 5 and 9.
Table 19. Toxicity threshold after 48 h exposure of Cyprinus carpio
to mixtures containing 1,2-dibromoethane at 25°C
(Nishiuchi & Asano, 1979)
Mixture constituents Concentrations Toxicity threshold
1,2-Dibromoethane 2.5% oil > 100
Fenitrothion 0.5% oil
1,2-Dibromoethane 2.5% oil 86
Fenitrothion 0.5% oil
1,2-Dibromoethane 1.5% emulsion
Fenitrothion 10% emulsion 45
Carbaryl 5% emulsion
Diazinon 20% emulsion 28
1,2-Dibromoethane 10% emulsion
Cyanophos 10% emulsion 32
1,2-Dibromoethane 10% emulsion
1,2-Dibromoethane 25% oil
Diazinon 5% oil 65
O-sec-butylphenyl 3% oil
methylcarbamate
9.2 Terrestrial biota
Data on the effects of 1,2-dibromoethane on terrestrial biota
(other than mammals) are limited. In a study on the effects of
different foods on the susceptibility of the adzuki bean weevil
(Callosobruchus chinensis Linn.) to 1,2-dibromoethane, the 24-h
LC50 at 29°C was 4.40, 7.232, 6.130, 5.249 and 4.951 mg/litre for
chickpea, pea, green gram, black gram, and pigeon pea, respectively
(Mundhe & Pandey, 1980).
Exposure of the nematode Aphelenchus avenae to low
concentrations of 1,2-dibromoethane resulted in a very small
conversion of the halide just before death. A study with
14C-labelled 1,2-dibromoethane showed that two primary products were
ethylene (5%) and O-acetylserine (> 95%). These transformations
were indicative of two primary modes of intoxication of the nematodes,
postulated to be a direct reaction of 1,2-dibromoethane with an iron
centre in the respiratory sequence and the substitution of a serine at
the active site of an esterase or protease (Castro & Belser, 1978).
9.3 Microorganisms
The toxicity of 1,2-dibromoethane to microsclerotia of
Verticillium dahliae in air and in soil was determined in a sealed
container. At concentrations of 470 mg/litre in air or 12.5 mg/kg in
soil, 1,2-dibromoethane killed 97% of the microsclerotia, after
incubation for 16 days in both cases. The toxicity of
1,2-dibromoethane increased with increasing temperature and with
increase in soil moisture (0-80%) (Ben-Yephet et al., 1981).
Pignatello (1986) investigated 1,2-dibromoethane effects on
microorganisms from two soils/sediments taken from a
1,2-dibromoethane-contaminated groundwater discharge area. Labelled
potassium acetate was added to slurries of soil, which had been shaken
with added 1,2-dibromoethane (up to 1000 mg/litre), for 3 or 12 h.
Incorporation of acetate into microbial lipids was used as the
end-point. The EC50 for inhibition of acetate incorporation
following 12-h incubation with 1,2-dibromoethane was 50 and
100 mg/litre for the two soils; soil 1 showed an EC50 of 100
following 3 h of incubation. Both soils showed "slight" inhibition at
10 mg 1,2-dibromoethane/litre and > 94% at 1000 mg/litre. Soil 1 was
a muddy soil with a total organic carbon (TOC) content of 14%, whereas
soil 2 was a stream-bed soil with a TOC of 0.24%.
9.4 Plants
1,2-Dibromoethane was phytotoxic to fruit after they had been
fumigated against several species of fruit flies. Fumigation with
1,2-dibromoethane at a concentration of 4 g/m3 stimulated fruit
respiration and ethylene evolution. A higher concentration
(32 g/m3) increased respiration rate and ethylene evolution in the
fruits and increased tissue leakage. Fruits stored at 1°C after
fumigation with 32 g/m3 suffered more severe damage than those
stored at 20°C. Storage at 1°C abolished the increases in gas
exchange observed in fumigated fruits at 20°C. The injurious effect
of cooling might be ascribed to a higher residue of unchanged fumigant
persisting in the cooled fruits or to a decreased capacity of the
fruits to repair cellular damage at low temperatures (Wade & Rigney,
1979).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
1,2-Dibromoethane is carcinogenic for rats and mice causing
tumours (adenomas and carcinomas) in a variety of organs including the
nasal cavity, lung, stomach, liver, skin, and mammary gland, as well
as haemangiosarcomas. In many cases, a reduced latency period for
developing tumours was observed. 1,2-Dibromoethane has been shown to
be mutagenic in various in vivo and in vitro assays and to cause
single-strand DNA breaks in vitro. Some metabolites have been shown
to covalently bind to DNA. Based on these data, 1,2-dibromoethane is
thought to be a genotoxic carcinogen to rodents. Although adequate
studies in humans are not available, the extensive evidence for
carcino genicity in animal studies indicates that 1,2-dibromoethane
is a potential human carcinogen.
10.2 Evaluation of effects on the environment
The high volatility of 1,2-dibromoethane makes the atmosphere the
predominant environmental sink. Consequently, measured concentrations
in surface waters are low (< 0.2 µg/litre). Air concentrations of
< 0.2 µg/litre have been measured in cities, while concentrations of
up to 90 µg/litre in irrigation wells reflect the mobility of the
compound in soil. Persistent contamination of irrigation wells may
result from the slow release of 1,2-dibromoethane from the soil matrix
many years after its use as a soil fumigant.
There is a lack of information on the degradation of
1,2-dibromoethane in the aquatic and soil environments.
Stratospheric photodegradation occurs and potentially leads to
breakdown products with ozone-depleting capacity. However,
1,2-dibromoethane is not listed in the Montreal Convention.
Few aquatic ecotoxicity tests have been conducted with
1,2-dibromoethane. Those reported show LC50s greater than
5 mg/litre. There is a difference of at least 4 orders of magnitude
between measured water concentrations and these toxic concentrations,
indicating that 1,2-dibromoethane poses no risk to aquatic organisms.
11. CONCLUSIONS AND RECOMMENDATIONS
Considering the toxicological characteristics of
1,2-dibromoethane, both qualitatively and quantitatively, it was
concluded that an exposure that would not cause adverse effects in
humans after any route of exposure could not be estimated.
Consequently, all appropriate measures should be taken to eliminate or
minimize human exposure to 1,2-dibromoethane.
12. FURTHER RESEARCH
Further information from epidemiology studies on
1,2-dibromoethane would be useful.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
IARC (1977, 1982, 1987) concluded, from biological data relevant
to the evaluation of carcinogenic risk, that 1,2-dibromoethane is
carcinogenic in mice and rats after oral administration, and by
inhalation, producing squamous cell carcinomas of the forestomach.
1,2-Dibromoethane given orally or by intraperitoneal injection did not
produce dominant lethal mutations in mice. Prolonged contact with
1,2-dibromoethane causes skin irritation. However, no case reports or
epidemiological studies were available to the Working Group. IARC has
classified 1,2-dibromoethane as a Group 2A carcinogen (probably
carcinogenic to humans).
WHO has not established a drinking-water quality guideline for
1,2-dibromoethane. This is because 1,2-dibromoethane appears to be a
genotoxic carcinogen and the studies are inadequate for mathematical
extrapolation (WHO, 1993).
1,2-Dibromoethane was evaluated by the Joint FAO/WHO Expert
Committee on Pesticide Residues in 1965 (FAO/WHO, 1965) and 1966
(FAO/WHO, 1967). The 1965 evaluation concluded that 1,2-dibromoethane
should be used for fumigation of food only on the condition that no
residue of the unchanged compound reached the consumer. In the 1966
evaluation an Acceptable Daily Intake (ADI) of 1 mg/kg body weight as
bromide was established. In 1991 the Codex Alimentarius Commission
deleted the Guideline Levels for 1,2-dibromoethane in food commodities
(FAO/WHO, 1992).
REFERENCES
Abdel-Kader MHK, Peach ME, & Stiles DA (1979) Determination of
ethylene dibromide in fortified soils by molecular emission cavity
analysis using a modified extraction process. J Assoc Off Anal Chem,
62(1): 114-118.
Adams JA & Kennedy AA (1992) Sublethal effects of ethylene dibromide
on wound healing and morphogenesis in Hydra oligactis. Arch Environ
Contam Toxicol, 22: 272-277.
Adams PM, Hanlon RT, & Forsythe JW (1989) Toxic exposure to ethylene
dibromide and mercuric chloride: Effects on laboratory-reared
octopuses. Neurotoxicol Teratol, 10: 519-523.
Adkins B, Van Stee EW, Simmons JE, & Eustis SL (1986) Oncogenic
response of strain A/J mice to inhaled chemicals. J Toxicol Environ
Health, 17: 311-322.
Alexeeff GV, Kilgore WW, & Li MY (1990) Ethylene dibromide: Toxicology
and risk assessment. Berlin, Heidelberg, New York, Spinger Verlag
Inc., pp 49-122.
Alleman TG, Sanders RA, & Madison BL (1986) Simultaneous analysis of
grain and grain-based products for ethylene dibromide, carbon
tetrachloride, and ethylene dichloride. J Assoc Off Anal Chem,
69(4): 575-580.
Alper MD & Ames BN (1975) Positive selection of mutants with deletions
of the gal-ch1 region of the salmonella chromosome as a screening
procedure for mutagens that cause deletions. J Bacteriol,
121(1): 259-266.
Alumot E, Nachtomi E, Kempenich-Pinto O, Mandel E, & Schindler H
(1968) The effect of ethylene dibromide in feed on the growth, sexual
development and fertility of chickens. Poult Sci, 47: 1979-1985.
Ames BN & Yanofsky C (1971) The detection of chemical mutagens with
enteric bacteria. In: Hollaender A ed. Chemical mutagens: Principles
and methods for their detection. New York, London, Plenum Press,
vol 1, pp 267-282.
Amin TA & Narang RS (1985) Determination of volatile organics in
sediment at nanogram-per-gram concentrations by gas chromatography.
Anal Chem, 57: 648-651.
Amir D (1973) The sites of the spermicidal action of ethylene
dibromide in bulls. J Reprod Fertil, 35: 519-525.
Amir D (1975) Individual and age differences in the spermicidal effect
of ethylene dibromide in bulls. J Reprod Fertil, 44: 561-565.
Amir D & Volcani R (1965) Effect of dietary ethylene dibromide on bull
semen. Nature (Lond), 206(4979): 99-100.
Amir D, Esnault C, Nicolle JC, & Courot M (1977) DNA and protein
changes in the spermatozoa of bulls treated orally with ethylene
dibromide. J Reprod Fertil, 51: 453-456.
Amir D, Nicolle JC, & Courot M (1979) Changes induced to bull
spermatids and testicular spermatozoa by a single peritesticular
injection of ethylene dibromide. Int J Androl, 2: 162-170.
Asita AO, Hayashi M, Kodama Y, Matsuoka A, Suzuki T, & Sofuni T (1992)
Micronucleated reticulocyte induction by ethylating agents in mice.
Mutat Res, 271: 29-37.
Ballering LAP, Nivard MJM, & Vogel EW (1993) Characterization of the
genotoxic action of three structurally related 1,2-dihaloalkanes in
Drosophila melanogaster. Mutat Res, 285: 209-217.
Ballschmiter K, Mayer P, & Class Th (1986) Chemistry of organic traces
in air. IV. Analysis of C1- and C2-halocarbons in ambient air by
cold trap injection and wide bore glass capillary gas chromatography.
Fresenius Z Anal Chem, 323: 334-339.
Banerjee S, Van Duuren BL, & Kline SA (1979) Interaction of potential
metabolites of the carcinogen ethylene dibromide with protein and DNA
in vitro. Biochem Biophys Res Commun, 90(4): 1214-1220.
Barber ED & Donish WH (1982) An exposure system for quantitative
measurements of the microbial mutagenicity of volatile liquids.
Measurements of microbial mutagenicity. In: Tice RR, Costa DL, &
Schaich KM ed. Genotoxic effects of airborne agents. New York, London,
Plenum Press, pp 3-18.
Barber ED, Donish WH, & Mueller KR (1981) A procedure for the
quantitative measurement of the mutagenicity of volatile liquids in
the Ames Salmonella/microsome assay. Mutat Res, 90: 31-48.
Barkley J, Bunch J, Bursey JT, Castillo N, Cooper SD, Davis JM,
Erickson MD, Harris BSH III, Kirkpatrick M, Michael LC, Parks SP,
Pellizzari ED, Ray M, Smith D, Tomer KB, Wagner R, & Zweidinger RA
(1980) Gas chromatography mass spectrometry computer analysis of
volatile halogenated hydrocarbons in man and his environment - a
multimedia environmental study. Biomed Mass Spectrom, 7(4): 139-147.
Barnett LB, Lovell DP, Felton CF, Gibson BJ, Cobb RR, Sharpe DS,
Shelby MD, & Lewis SE (1992) Ethylene dibromide: Negative results with
the mouse dominant lethal assay and the electrophoretic specific-locus
test. Mutat Res, 282: 127-133.
Barry TL & Petzinger G (1985) GC/MS confirmation of ethylene dibromide
residue in foods. J Food Saf, 7: 171-176.
Beckman H, Crosby DG, Allen PT, & Mourer C (1967) The inorganic
bromide content of foodstuffs due to soil treatment with fumigants.
J Food Sci, 32: 138-140.
Ben-Yephet Y, Letham D, & Evans G (1981) Toxicity of 1,2-dibromoethane
and 1,3-dichloropropene to microsclerotia of Verticillium dahliae.
Pestic Sci, 12: 170-174.
Berck B (1974) Fumigant residues of carbon tetrachloride, ethylene
dichloride, and ethylene dibromide in wheat, flour, bran, middlings,
and bread. J Agric Food Chem, 22(6): 977-984.
Berg WW, Heidt LE, Pollock W, Sperry PD, & Cicerone RJ (1984)
Brominated organic species in the arctic atmosphere. Geophys Res Lett,
11(5): 429-432.
Bielorai R & Alumot E (1975) The temperature effect on fumigant
desorption from cereal grain. J Agric Food Chem, 23(3): 426-429.
Bondi A, Olomucki E, & Calderon M (1955) Problems connected with
ethylene dibromide fumigation of cereals. II. Feeding experiments with
laying hens. J Sci Food Agric, 6: 600-602.
Botti B, Moslen MT, Trieff NM, & Reynolds ES (1982) Transient decrease
of liver cytosolic glutathione s-transferase activities in rats given
1,2-dibromoethane or CCL. Chem-Biol Interact, 42: 259-270.
Brandt I, Brittebo EB, Kowalski B, & Lund B-O (1987) Tissue binding
of 1,2-dibromoethane in the cynomolgus monkey (Macaca fascicularis).
Carcinogenesis, 8(9): 1359-1361.
Brem H, Stein AB, & Rosenkranz HS (1974) The mutagenicity and
DNA-modifying effect of haloalkanes. Cancer Res, 34: 2576-2579.
Brimer PA, Tan E-L, & Hsie AW (1982) Effect of metabolic activation on
the cytotoxicity and mutagenicity of 1,2-dibromoethane in the
CHO/HGPRT system. Mutat Res, 95: 377-388.
Broda C, Nachtomi E, & Alumot E (1976) Differences in liver morphology
between rats and chicks treated with ethylene dibromide. Gen
Pharmacol, 7: 345-348.
BUA (Society of German Chemists-Advisory Committee on Existing
Chemicals of Environmental Relevance) (1987) [Methyl bromide.]
Weinheim, VCH Publishers, 64 pp (BUA Report 14) (in German).
BUA (Society of German Chemists-Advisory Committee on Existing
Chemicals of Environmental Relevance) (1991) [1,2-Dibromoethane.]
Weinheim, VCH Publishers, 290 pp (BUA Report 66) (in German).
Buijs W, Van Der Gen A, Mohn GR, & Breimer DD (1984) The direct
mutagenic activity of alpha,omega-dihalogenoalkanes in Salmonella
typhimurium. Mutat Res, 141: 11-14.
Cairns T, Siegmund EG, Doose GM, Hundley HK, Barry T, & Petzinger G
(1984) Confirmation of ethylene dibromide in fruits and grains by mass
spectrometry. Anal Chem, 56: 2138-2141.
Castro CE & Belser NO (1978) Intoxication of Aphelenchus avenae by
ethylene dibromide. Nematologica, 24: 37-44.
Channarayappa, Ong T, & Nath J (1992) Cytogenetic effects of
vincristine sulfate and ethylene dibromide in human peripheral
lymphocytes: Micronucleus analysis. Environ Mol Mutagen, 20: 117-126.
Chiarpotto E, Biasi F, Aragno M, Scavazza A, Danni O, Albano E, &
Poli G (1993) Change of liver metabolism of 1,2-dibromoethane during
simultaneous treatment with carbon tetrachloride. Cell Biochem Funct,
11: 71-75.
Clark AI, McIntyre AE, Lester JN, & Perry R (1982) Evaluation of a
tenax GC sampling procedure for collection and analysis of vehicle-
related aromatic and halogenated hydrocarbons in ambient air.
J Chromatogr, 252: 147-157.
Clark AI, McIntyre AE, Lester JN, & Perry R (1984a) Ambient air
measurements of aromatic and halogenated hydrocarbons at urban, rural
and motorway locations. Sci Total Environ, 39: 265-279.
Clark AI, McIntyre AE, Perry R, & Lester JN (1984b) Monitoring and
assessment of ambient atmospheric concentrations of aromatic and
halogenated hydrocarbons at urban, rural and motorway locations.
Environ Pollut, B7: 141-158.
Clive D, Johnson KO, Spector JFS, Batson AG, & Brown MMM (1979)
Validation and characterization of the L5178Y/TK+/-mouse lymphoma
mutagen assay system. Mutat Res, 59: 61-108.
Clower M (1980) Modification of the AOAC method for determination of
fumigants in wheat. J Assoc Off Anal Chem, 63(3): 539-545.
Clower M, McCarthy JP, & Carson LJ (1986) Comparison of methodology
for determination of ethylene dibromide in grains and grain-based
foods. J Assoc Off Anal Chem, 69(1): 87-90.
Cmarik JL, Inskeep PB, Meredith MJ, Meyer DJ, Ketterer B, & Guengerich
FP (1990) Selectivity of rat and human glutathione S-transferases in
activation of ethylene dibromide by glutathione conjugation and DNA
binding and induction of unscheduled DNA synthesis in human
hepatocytes. Cancer Res, 50(9): 2747-2752.
Cmarik JL, Humphreys WG, Bruner KL, Lloyd RS, Tibbetts C, & Guengerich
FP (1992) Mutation spectrum and sequence alkylation selectivity
resulting from modification of bacteriophage M13mp18 DNA with
s-(2-chloroethyl)glutathione. Evidence for a role of s-[2-(N-guanyl)
ethyl]glutathione as a mutagenic lesion formed from ethylene
dibromide. J Biol Chem, 267(10): 6672-6679.
Collins M & Barker NJ (1983) Direct monitoring of ambient air for
ethylene oxide and ethylene dibromide. Am Lab (Fairfield, Conn),
15: 72, 74-76, 78-81.
Coni P, Simbula G, De Prati AC, Menegazzi M, Suzuki H, Sarma DSR,
Ledda-Columbano GM, & Columbano A (1993) Differences in the
steady-state levels of c-fos, c-jun and c-myc messenger RNA during
mitogen-induced liver growth and compensatory regeneration.
Hepatology, 17: 1109-1116.
Courtens JL, Amir D, & Durand J (1980) Abnormal spermiogenesis in
bulls treated with ethylene dibromide: An ultrastructural and
ultracytochemical study. J Ultrastruct Res, 71: 103-115.
Crebelli R, Conti G, Conti L, & Carere A (1984) Induction of somatic
segregation by halogenated aliphatic hydrocarbons in Aspergillus
nidulans. Mutat Res, 138: 33-38.
Crespi CL, Seixas GM, Turner TR, Ryan CG, & Penman BW (1985)
Mutagenicity of 1,2-dichloroethane and 1,2-dibromoethane in two human
lymphoblastoid cell lines. Mutat Res, 142: 133-140.
Daft JL (1983) Gas chromatographic determination of fumigant residues
in stored grains, using isooctane partitioning and dual column
packings. J Assoc Off Anal Chem, 66(2): 228-233.
Daft JL (1985) Preparation and use of mixed fumigant standards for
multiresidue level determination by gas chromatography. J Agric Food
Chem, 33: 563-566.
Daft JL (1987) Determining multifumigants in whole grains and legumes,
milled and low-fat grain products, spices, citrus fruit, and
beverages. J Assoc Off Anal Chem, 70(4): 734-760.
Daft JL (1988) Rapid determination of fumigant and industrial chemical
residues in food. J Assoc Off Anal Chem, 71(4): 748-760.
De Serres FJ & Malling HV (1983) The role of Neurospora in
evaluating environmental chemicals for mutagenic activity. Ann NY Acad
Sci, 407: 177-185.
De Vries JW, Larson PA, Bowers RH, Keating AM, Broge JM, Wehling PS,
Patel HH, & Zurawski JW (1985) Improved codistillation method for
determination of carbon tetrachloride, ethylene dichloride and
ethylene dibromide in grain and grain-based products. J Assoc Off Anal
Chem, 68(4): 759-762.
Dibiasio KW, Silva MH, Shull LR, Overstreet JW, Hammock BD, & Miller
MG (1991) Xenobiotic metabolizing enzyme activities in rat, mouse,
monkey, and human testes. Drug Metab Dispos, 19(1): 227-232.
Dobbins JG (1987) Regulation and the use of "Negative" results from
human reproductive studies: The case of ethylene dibromide. Am J Ind
Med, 12: 33-45.
Dumas T & Bond EJ (1982) Microdetermination of ethylene dibromide in
air by gas chromatography. J Assoc Off Anal Chem, 65(6): 1379-1381.
Dunkel VC, Zeiger E, Brusick D, McCoy E, McGregor D, Mortelmans K,
Rosenkranz HS, & Simmon VF (1985) Reproducibility of microbial
mutagenicity assays: Testing of carcinogens and noncarcinogens in
Salmonella typhimurium and Escherichia coli. Environ Mutagen,
7(5): 1-248.
Edwards K, Jackson H, & Jones AR (1970) Studies with alkylating
esters-II: A chemical interpretation through metabolic studies of the
antifertility effects of ethylene dimethanesulphonate and ethylene
dibromide. Biochem Pharmacol, 19: 1783-1789.
Ehrenberg L, Osterman-Golkar S, Singh D, & Lundqvist U (1974) On the
reaction kinetics and mutagenic activity of methylating and
beta-halogenoethylating gasoline additives. Radiat Bot, 15: 185-194.
ElJack AH & Hrudka F (1979) Pattern and dynamics of teratospermia
induced in rams by parenteral treatment with ethylene dibromide.
J Ultrastruct Res, 67: 124-134.
Ellingham TJ, Christensen EA, & Maddock MB (1986) In vitro induction
of sister chromatid exchanges and chromosomal aberrations in
peripheral lymphocytes of the oyster toadfish and American eel.
Environ Mutagen, 8: 555-569.
Elliott BM & Ashby J (1980) Ethylene dibromide and disulfiram: Studies
in vivo and in vitro on the mechanism of the observed synergistic
carcinogenic response. Carcinogenesis, 1(12): 1049-1057.
Entz RC & Hollifield HC (1982) Headspace gas chromatographic analysis
of foods for volatile halocarbons. J Agric Food Chem, 30: 84-88.
Environment Agency Japan (1985) Chemicals in the environment: Annual
report - Annex. Tokyo, Environment Agency, pp 16-28.
Environment Canada (1994) Summary of EDB concentration at Canadian
sites. Ottawa, Environment Canada, Environment Protection Service,
Pollution Measurement Division (Unpublished report).
Epstein SS, Arnold E, Andrea J, Bass W, & Bishop Y (1972) Detection of
chemical mutagens by the dominant lethal assay in the mouse. Toxicol
Appl Pharmacol, 23: 288-325.
Fahrig R (1974) Comparative mutagenicity studies with pesticides.
In: Chemical carcinogenesis essays. Lyon, International Agency for
Research on Cancer, pp 161-181 (IARC Scientific Publications No. 10).
Fanini D, Legator MS, & Adams PM (1984) Effects of paternal ethylene
dibromide exposure on F1 generation behavior in the rat. Mutat Res,
139: 133-138.
FAO/WHO (1965) Evaluation of the hazards to consumers resulting from
the use of fumigants in the protection of food. Report of the 2nd
Meeting of the FAO/WHO Joint Expert Committee on Pesticide Residues.
Rome, Food and Agriculture Organization of the United Nations,
pp 36-42 (FAO Meeting Report No. PL/1965/10/2).
FAO/WHO (1967) Pesticide residues in food. Joint report of the FAO
Working Party on Pesticide Residues and the WHO Expert Committee on
Pesticide Residues. Geneva, World Health Organization, 19 pp (WHO
Technical Report Series 370).
FAO/WHO (1972) 1971 Evaluations of some pesticide residues in food.
Geneva, World Health Organization, p 267 (WHO Pesticide Residues
Series, No. 1).
FAO/WHO (1992) Report of the Twenty-Fourth Session of the Codex
Committee on Pesticide Residues. Rome, Food and Agriculture
Organization of the United Nations, Codex Alimentarius Commission,
82 pp (Alinorm 93/24).
Fernandez M, L'Haridon J, Gauthier L, & Zoll-Moreux C (1993) Amphibian
micronucleus test(s): A simple and reliable method for evaluating
in vivo genotoxic effects of freshwater pollutants and radiations.
Initial assessment. Mutat Res, 292: 83-99.
Ferreri AM, Rocchi P, Capucci A, & Prodi G (1983) Induction of
diphtheria toxin-resistant mutants in human cells by halogenated
compounds. J Cancer Res Clin Oncol, 105: 111-112.
Foster PL, Wilkinson WG, Miller JK, Sullivan AD, & Barnes WM (1988) An
analysis of the mutagenicity of 1,2-dibromoethane to Escherichia
coli: influence of DNA repair activities and metabolic pathways.
Mutat Res, 194: 171-181.
Gilbert J, Startin JR, & Grew C (1985) Automated headspace GC-MS
analysis of ethylene dibromide fumigant residues in fresh fruits. Food
Addit Contam, 2(1): 55-61.
Going JE & Long S (1975) Sampling and analysis of selected toxic
substances: Task II - Ethylene dibromide. Washington, DC, US
Environmental Protection Agency, Office of Toxic Substances
(EPA-560/6-75-001).
Going JE & Spigarelli JE (1976) Sampling and analysis of selected
toxic substances: Task IV - Ethylene dibromide. Washington, DC, US
Environmental Protection Agency, Office of Toxic Substances
(EPA-560/6-76-0211).
Graf U, Wurgler FE, Katz AJ, Frei H, Juon H, Hall CB, & Kale PG (1984)
Somatic mutation and recombination test in Drosophila melanogaster.
Environ Mutagen, 6: 153-188.
Guha SN, Schoneich C, & Asmus KD (1993) Free radical reductive
degradation of vic-dibromoalkanes and reaction of bromine atoms with
polyunsaturated fatty acids: possible involvement of Br* in the
1,2-dibromoethane-induced lipid peroxidation. Arch Biochem Biophys,
305(1): 132-140.
Guobaitis RJ, Ellingham TJ, & Maddock MB (1986) The effects of
pretreatment with cytochrome P-450 inducers and preincubation with a
cytochrome P-450 effector on the mutagenicity of genotoxic carcinogens
mediated by hepatic and renal S9 from two species of marine fish.
Mutat Res, 164: 59-70.
Hardin BD, Bond GP, Sikov MR, Andrew FD, Beliles RP, & Niemeier RW
(1981) Testing of selected workplace chemicals for teratogenic
potential. Scand J Work Environ Health, 7(4): 66-75.
Harkov R, Kebbekus B, Bozzelli J, & Lioy PJ (1983) Measurement of
selected volatile organic compounds at three locations in New
Jersey during the summer season. J Air Pollut Control Assoc,
33(12): 1177-1183.
Harkov R, Kebbekus B, Bozzelli JW, Lioy P, & Daisy J (1984) Comparison
of selected volatile organic compounds during the summer and winter at
urban sites in New Jersey. Sci Total Environ, 38: 259-274.
Harkov R, Gianti SJ, Bozzelli JW, & LaRegina JE (1985) Monitoring
volatile organic compounds at hazardous and sanitary landfills in New
Jersey. J Environ Sci Health, A20(5): 491-501.
Hasanen E, Soininen V, Pyysalo H, & Leppamaki E (1979) On the
occurrence of aliphatic chlorine and bromine compounds in automobile
exhaust. Atmos Environ, 13: 1217-1219.
Hasanen E, Karlsson V, Leppamaki E, & Juhala M (1981) Benzene, toluene
and xylene concentrations in car exhausts and in city air. Atmos
Environ, 15(9): 1755-1757.
Hayes S, Gordon A, Sadowski I,& Hayes C (1984) RK bacterial test for
independently measuring chemical toxicity and mutagenicity: Short-term
forward selection assay. Mutat Res, 130: 97-106.
Heikes DL (1985a) Purge and trap method for determination of ethylene
dibromide in table ready foods. J Assoc Off Anal Chem, 68(3): 431-436.
Heikes DL (1985b) Purge and trap method for determination of ethylene
dibromide in whole grains, milled grain products, intermediate
grain-based foods and animal feeds. J Assoc Off Anal Chem,
68(6): 1108-1111.
Heikes DL & Hopper ML (1986) Purge and trap method for determination
of fumigants in whole grains, milled grain products and intermediate
grain-based foods. J Assoc Off Anal Chem, 69(6): 990-998.
Hemminki K, Falck K, & Vainio H (1980) Comparison of alkylation rates
and mutagenicity of directly acting industrial and laboratory
chemicals. Arch Toxicol, 46: 277-285.
Herring CO, Adams JA, Wilson BA, & Pollard S (1988) Dose-response
studies using ethylene dibromide(EDB) in Hydra oligactis. Bull
Environ Contam Toxicol, 40: 35-40.
Heuser SG & Scudamore KA (1967) Determination of ethylene
chlorohydrin, ethylene dibromide and other volatile fumigant residues
in flour and whole wheat. Chem Ind, 37: 1557-1560.
Hill DL, Shih TW, Johnston TP, & Struck RF (1978) Macromolecular
binding and metabolism of the carcinogen 1,2-dibromoethane. Cancer
Res, 38: 2438-2442.
Hsu LL, Adams PM, Fanini D, & Legator MS (1985) Ethylene dibromide:
Effects of paternal exposure on the neurotransmitter enzymes in the
developing brain of progeny. Mutat Res, 147: 197-203.
Hughes TJ, Simmons DM, Monteith LG, & Claxton LD (1987) Vaporization
technique to measure mutagenic activity of volatile organic chemicals
in the Ames/Salmonella assay. Environ Mutagen, 9: 421-441.
IARC (1977) Ethylene dibromide. In: Some fumigants, the herbicides
2,4-D and 2,4,5-T, chlorinated dibenzodioxins and miscellaneous
industrial chemicals. Lyon, International Agency for Research on
Cancer, pp 195-209 (IARC Monographs on the Evaluation of Carcinogenic
Risks to Humans, Volume 15).
IARC (1982) Ethylene dibromide. In: Chemicals, industrial processes
and industries associated with cancer in humans - IARC monographs,
volumes 1 to 29. Lyon, International Agency for Research on Cancer,
pp 124-126 (IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, Supplement 4).
IARC (1987) Ethylene dibromide. In: Overall evaluations of
carcinogenicity: An updating of IARC monographs, volumes 1 to 42.
Lyon, International Agency for Research on Cancer, pp 204-205 (IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans,
Supplement 7).
Inskeep PB & Guengerich FP (1984) Glutathione-mediated binding of
dibromoalkanes to DNA: Specificity of rat glutathione-S-transferases
and dibromoalkane structure. Carcinogenesis, 5(6): 805-808.
Inskeep PB, Koga N, Cmarik JL, & Guengerich FP (1986) Covalent binding
of 1,2-dihaloalkanes to DNA and stability of the major DNA adduct,
S-[2-(N7-guanyl)ethyl]glutathione1. Cancer Res, 46: 2839-2844.
IPCS (International Programme on Chemical Safety) (1995) Environmental
health criteria 166: Methyl bromide. Geneva, World Health
Organization, 324 pp.
IRPTC (1993) Legal file. Geneva, International Register of Potentially
Toxic Chemicals, United Nations Environment Programme.
Isaacson PJ, Hankin L, & Frink CR (1984) Boiling drinking water
removes ethylene dibromide. Science, 225: 672.
Ishikuro E (1986) [Quantitative analysis of ethylene dibromide (EDB)
in feed ingredients and mixed feed.] Shiryo Kenkyo Hokoku, 11: 1-7
(in Japanese).
Iwata Y, Dusch ME, & Gunther FA (1983) Use of sulfuric acid cleanup
step in the determination of 1,2-dibromoethane residues in lemons,
oranges, and grapefruits. J Agric Food Chem, 31: 171-174.
Izutani K, Nakata A, Shinagawa H, & Kawamata J (1980) Forward mutation
assay for screening carcinogens by alkaline phosphatase constitutive
mutations in Escherichia coli K-12. J Res Inst Microb Dis (Biken J),
23: 69-75.
Jacobs ES (1980) Use and air quality impact of EDC and EDB scavengers
in leaded gasoline. In: Ames B, Infante P, & Peirtz R ed. Ethylene
dichloride: A potential health risk? Cold Spring Harbor, New York,
Cold Spring Harbor Laboratory, pp 239-255 (Banbury Report No. 5).
Jacobs RS (1985) Ethylene dibromide poisoning. J Am Med Assoc,
253: 2961.
Jafvert CT & Wolfe NL (1987) Degradation of selected halogenated
ethanes in anoxic sediment-water systems. Environ Toxicol Chem,
6: 827-837.
Jagielski J, Scudamore KA, & Heuser SG (1978) Residues of carbon
tetrachloride and 1,2-dibromoethane in cereals and processed foods
after liquid fumigant grain treatment for pest control. Pestic Sci,
9: 117-126.
Jakobson I, Wahlberg JE, Holmberg B, & Johansson G (1982) Uptake via
the blood and elimination of 10 organic solvents following
epicutaneous exposure of anesthetized guinea pigs. Toxicol Appl
Pharmacol, 63: 181-187.
Jean PA & Reed DJ (1992) Utilization of glutathione during
1,2-dihaloethane metabolism in rat hepatocytes. Chem Res Toxicol,
5: 386-391.
Kale PG & Baum JW (1979a) Sensitivity of Drosophila melanogaster to
low concentrations of the gaseous 1,2-dibromoethane: I. Acute
exposures. Environ Mutagen, 1: 15-18.
Kale PG & Baum JW (1979b) Sensitivity of Drosophila melanogaster to
low concentrations of gaseous mutagens: II. Chronic exposures. Mutat
Res, 68: 59-68.
Kale PG & Baum JW (1981) Sensitivity of Drosophila melanogaster to
low concentrations of gaseous mutagens: III. Dose-rate effects.
Environ Mutagen, 3: 65-70.
Kale PG & Baum JW (1982) Genetic effects of ethylene dibromide in
Drosophila melanogaster. In: Tice RR, Costa DL, & Schaich KM ed.
Genotoxic effects of airborne agents. New York, London, Plenum Press,
pp 291-300.
Kale PG & Baum JW (1983) Sensitivity of Drosophila melanogaster to
low concentrations of gaseous mutagens: IV. Mutations in embryonic
spermatogonia. Mutat Res, 113: 135-143.
Kaphalia BS & Ansari GAS (1992) Covalent binding of ethylene dibromide
and its metabolites to albumin. Toxicol Lett, 62: 221-230.
Kato K, Nakamura M, Nakaoka T, & Ito K (1982) [Determination of
ethylene bromide residues in fruits.] Kanagawa-ken Eisei Kenkyosho
Hokoku, 12: 33-34 (in Japanese).
Keough T, Strife RJ, Rodriguez PA, & Sanders RA (1984) Separation and
use of the perdeuterated analogue as an internal standard for the
analysis of ethylene dibromide. J Chromatogr, 312: 450-455.
Kerklaan P, Bouter S, & Mohn G (1983) Isolation of a mutant of
Salmonella typhimurium strain TA1535 with decreased levels of
glutathione (GSH-): Primary characterization and chemical mutagenesis
studies. Mutat Res, 122: 257-266.
Kerklaan P, Zoetemelk CEM, & Mohn GR (1985) Mutagenic activity of
various chemicals in Salmonella strain TA100 and glutathione-deficient
derivatives: On the role of glutathione in the detoxification or
activation of mutagens inside bacterial cells. Biochem Pharmacol,
34(12): 2151-2156.
Khan S, Sood C, & O'Brien PJ (1993) Molecular mechanisms of
dibromoalkane cytotoxicity in isolated rat hepatocytes. Biochem
Pharmacol, 45(2): 439-447.
Kim DH & Guengerich FP (1989) Excretion of the mercapturic acid
S-[2-(N7-guanyl) ethyl]-N-acetylcysteine in urine following
administration of ethylene dibromide to rats. Cancer Res,
49: 5843-5847.
Kim DH & Guengerich FP (1990) Formation of the DNA adduct
S-[2-(N7-guanyl)ethyl] glutathione as an adduct formed in RNA and DNA
from 1,2-dibromoethane. Chem Res Toxicol, 3(6): 587-594.
Kitchin KT & Brown JL (1986) 1,2-dibromoethane causes rat hepatic DNA
damage at low doses. Biochem Biophys Res Commun, 141(2): 723-727.
Kluwe WM, McNish R, Smithson K, & Hook JB (1981) Depletion by
1,2-dibromoethane, 1,2-dibromo-3-chloropropane, tris(2,3-
dibromopropyl)phosphate, and hexachloro-1,3-butadiene of reduced
non-protein sulfhydryl groups in target and non-target organs. Biochem
Pharmacol, 30(16): 2265-2271.
Kluwe WM, Harrington FW, & Cooper SE (1982) Toxic effects of
organohalide compounds on renal tubular cells in vivo and in vitro.
J Pharmacol Exp Ther, 220: 597-603.
Koida K, Tokumori Y, Saimatsu J, Sekigawa K, Kohno U, & Oka A (1986)
[Determination of ethylene dibromide and dibromochloropropane in
groundwater by gas chromatography-negative ion chemical ionization
mass spectrometry.] Hiroshima-shi Eisei Kenkyusho Nenpo, 5: 34-36
(in Japanese).
Kojima T & Seo Y (1976) [Selective detection of alkyl nitrites, alkyl
nitrates, nitroparaffins and halogen compounds by gas chromatography.]
Bunseki Kagaku, 25: 855-858 (in Japanese).
Konishi Y, Yoshida S, & Nakamura A (1985) [Determination of EDB on
capillary ECD-GC.] Osaka-furitsu Koshu Eisei Kenkyusho Kenkyu Hokoku,
Shokuhin Eisei Hen, 16: 63-67 (in Japanese).
Kowalski B, Brittebo EB, & Brandt I (1985) Epithelial binding of
1,2-dibromoethane in the respiratory and upper alimentary tracts of
mice and rats. Cancer Res, 45: 2616-2625.
Krishna G, Xu J, Nath J, Petersen M, & Ong T (1985) In vivo
cytogenetic studies on mice exposed to ethylene dibromide. Mutat Res,
158: 81-87.
Krost KJ, Pellizzari ED, Walburn SG, & Hubbard SA (1982) Collection
and analysis of hazardous organic emissions. Anal Chem, 54: 810-817.
Kubinski H, Gutzke GE, & Kubinski ZO (1981) DNA-cell-binding (DCB)
assay for suspected carcinogens and mutagens. Mutat Res, 89: 95-136.
Kulkarni AP, Edwards J, & Richards IS (1992) Metabolism of
1,2-dibromoethane in the human fetal liver. Gen Pharmacol, 23(1): 1-5.
Landau M & Tucker JW (1984) Acute toxicity of EDB and aldicarb to
young of two estuarine fish species. Bull Environ Contam Toxicol,
33: 127-132.
Ledda-Columbano GM, Columbano A, Coni P, Curto M, Faa G, & Pani P
(1987a) Cell proliferation in rat kidney induced by 1,2-dibromoethane.
Toxicol Lett, 37: 85-90.
Ledda-Columbano GM, Columbano A, Coni P, Liguori C, & Pani P (1987b)
Liver cell proliferation induced by the mitogen ethylene dibromide
unlike compensatory cell proliferation, does not achieve initiation of
rat liver carcinogenesis by diethylnitrosamine. Cancer Lett,
36: 247-252.
Leinster P, Perry R, & Young RJ (1978) Ethylene dibromide in urban
air. Atmos Environ, 12: 2383-2387.
Letz GA, Pond SM, Osterloh JD, Wade RL, & Becker CE (1984) Two
fatalities after acute occupational exposure to ethylene dibromide.
J Am Med Assoc, 252(17): 2428-2431.
Libbey AJ (1986) Determination of ethylene dibromide in aquatic
environments. Analyst, 111: 1221-1222.
Logan SR (1988) Comments on photohydrolysis of ethylene dibromide.
J Agric Food Chem, 36: 872-873.
Lyman WJ (1982) Octanol/water partition coefficient. In: Lyman WJ &
Peehl WF ed. Handbook of chemical property estimation methods -
Environmental behaviour of organic compounds. New York, McGraw-Hill
Publishers, pp 1/1-1/54.
Ma T-H, Sparrow AH, Schairer LA, & Nauman AF (1978) Effect of 1,2
dibromoethane (EDB) on meiotic chromosomes of Tradescantia. Mutat Res,
58: 251-258.
Ma T-H, Harris MM, Anderson VA, Ahmed I, Mohammad K, Bare JL, & Lin G
(1984) Tradescantia-micronucleus (Trad-MCN) tests on 140 health-
related agents. Mutat Res, 138: 157-167.
McCann J, Choi E, Yamasaki E, & Ames BN (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc Natl Acad Sci (USA), 72(12): 5135-5139.
McClenny WA, Pleil JD, Holdren MW, & Smith RN (1984) Automated
cryogenic preconcentration and gas chromatographic determination of
volatile organic compounds in air. Anal Chem, 56: 2947-2951.
McCollister DD, Hollingsworth RL, Oyen F, & Rowe VK (1956) Comparative
inhalation toxicity of fumigant mixtures. Am Med Assoc Arch Ind
Health, 13: 1-7.
McKenry MV & Thomason IJ (1974) 1,3-Dichloropropene and
1,2-dibromoethane compounds: I. Movement and fate as affected by
various conditions in several soils: II. Organism-dosage-response
studies in the laboratory with several nematode species. Hilgardia,
42(11): 393-438.
Mann AM & Darby FJ (1985) Effects of 1,2-dibromoethane on glutathione
metabolism in rat liver and kidney. Biochem Pharmacol,
34(16): 2827-2830.
Mann JB, Freal JJ, Enos HF, & Danauskas JX (1980) Evaluation of
methodology for determining 1,2-dibromoethane (EDB) in ambient air. J
Environ Sci Health, B15(5): 507-518.
Martl LR, de Kanel J, & Dougherty RC (1984) Screening for organic
contamination of groundwater: ethylene dibromide in Georgia irrigation
wells. Environ Sci Technol, 18: 973-974.
Mayer JR, Lacher TE, Elkins NR, & Thorn CJ (1991) Temporal variation
of ethylene dibromide (EDB) in an unconfined aquifer, Whatcom County,
Washington, USA: a twenty-seven month study. Bull Environ Contam
Toxicol, 47: 368-373.
Meneghini R (1974) Repair replication of opossum lymphocyte DNA:
effect of compounds that bind to DNA. Chem-Biol Interact, 8: 113-126.
Mestres R, Atmawijaya S, & Francois CL (1980) XXXV. Méthode de
recherche et de dosage des résidus de pesticides dans les produits
céréaliers. Ann Fals Exp Chim, 73(788): 407-420.
MHW (1985) Prohibition of EDB residues in imported wheat, tropical
fruit etc. Official notice of the Ministry of Health and Welfare.
Tokyo, Ministry of Health and Welfare.
MHW (1987) Prohibition of EDB residues in imported wheat, tropical
fruit etc. Official notice of the Ministry of Health and Welfare.
Tokyo, Ministry of Health and Welfare.
MHW (1988) Prohibition of EDB residues in imported wheat, tropical
fruit etc. Official notice of the Ministry of Health and Welfare.
Tokyo, Ministry of Health and Welfare.
Mitra A, Hilbelink DR, Dwornik JJ, & Kulkarni A (1992) A novel model
to assess developmental toxicity of dihaloalkanes in humans:
Bioactivation of 1,2-dibromoethane by the isozymes of human fetal
liver glutathione S-transferase. Teratog Carcinog Mutagen,
12(3): 113-127.
Mohn GR, Kerklaan PRM, Van Zeeland AA, Ellenberger J, Baan RA, Lohman
PHM, & Pons F-W (1984) Methodologies for the determination of various
genetic effects in permeable strains of E. coli K-12 differing in
DNA repair capacity: Quantification of DNA adduct formation,
experiments with organ homogenates and hepatocytes, and animal-
mediated assays. Mutat Res, 125: 153-184.
Moriya M, Ohta T, Watanabe K, Miyazawa T, Kato K, & Shirasu Y (1983)
Further mutagenicity studies on pesticides in bacterial reversion
assay systems. Mutat Res, 116: 185-216.
Morris SC & Rippon LE (1985) Distribution of ethylene dibromide within
a fumigation chamber during fumigation of citrus fruit. J Agric Food
Chem, 33: 801-803.
Morris SC, Rippon LE, & Halamek R (1982) Rapid analysis and gas
sampling of ethylene dibromide in the gaseous phase and as residues in
citrus fruit. J Chromatogr, 246: 136-140.
Mundhe DR & Pandey ND (1980) Effect of different foods on the
susceptibility of Callosobruchus chinensis Linn, to some fumigants.
Indian J Entomol, 42: 787-790.
Munnecke DE & Van Gundy SD (1979) Movement of fumigants in soil,
dosage responses, and differential effects. Annu Rev Phytopathol,
17: 405-429.
Nachtomi E (1970) The metabolism of ethylene dibromide in the rat: the
enzymic reaction with glutathione in vitro and in vivo. Biochem
Pharmacol, 19: 2853-2860.
Nachtomi E & Farber E (1978) Ethylene dibromide as a mitogen for
liver. Lab Invest, 38(3): 279-283.
Nachtomi E & Sarma DSR (1977) Repair of rat liver DNA in vivo
damaged by ethylene dibromide. Biochem Pharmacol, 26: 1941-1945.
Nachtomi E, Alumot E, & Bondi A (1968) Biochemical changes in organs
of chicks and rats poisoned with ethylene dibromide and carbon
tetrachloride. Israel J Chem, 6: 803-811.
Nakamura M (1986) [Analytical method for ethylene dibromide in wheat
products using capillary column GC-mass spectrometry.] Fukuoka-shi
Eisei Shikenshiho, 11: 47-53 (in Japanese).
Nauman CH, Sparrow AH, & Schairer LA (1976) Comparative effects of
ionizing radiation and two gaseous chemical mutagens on somatic
mutation induction in one mutable and two non-mutable clones of
Tradescantia. Mutat Res, 38: 53-70.
NCI (1978) Bioassay of 1,2-dibromoethane for possible carcinogenicity.
Bethesda, Maryland, National Cancer Institute, Division of Cancer
Cause and Prevention, Carcinogenesis Testing Program, 64 pp (Technical
Report Series No 86; DHEW Publication No. (NIH) 78-1336).
Nemoto Y, Sasamoto T, Nakata M, Suzuki Y, Kubota K, & Kurokawa K
(1984) [Survey of EDB groundwaters in Ibaraki Prefecture.] Ibaraki-ken
Eisei Kenkyujo Nenpo, 22: 80-81 (in Japanese).
Nichols WK, Covington MO, Seiders CD, Safiullah S, & Yost GS (1992)
Bioactivation of halogenated hydrocarbons by rabbit pulmonary cells.
Pharmacol Toxicol, 71(5): 335-339.
Nilsson CA, Lindahl R, & Norstrom A (1987) Occupational exposure to
chain saw exhausts in logging operations. Am Ind Hyg Assoc J,
48(2): 99-105.
NIOSH (1977) Criteria for a recommended standard: occupational
exposure to ethylene dibromide. Cincinnati, Ohio, National Institute
for Occupational Safety and Health, 208 pp (DHEW (NIOSH) Publication
No. 77-221).
NIOSH (1981) Current intelligence bulletin No. 37: Ethylene bromide
(revised). Cincinnati, Ohio, National Institute for Occupational
Safety and Health, 13 pp (Publication No. 82-105).
Nishiuchi Y (1980) [Toxicity of pesticide formulas to some fresh water
organisms - LXX.] Suisan Zoushoku, 27(4): 225-237 (in Japanese).
Nishiuchi Y (1981) [Toxicity of pesticides to some aquatic organisms:
I. Toxicity of pesticides to some aquatic insects.] Seitai Kagaku,
4(2): 31-46 (in Japanese).
Nishiuchi Y (1982) [Toxicity of pesticide formulas to some fresh water
organisms: effects of pH on toxicity revelation.] Suisan Zoushoku,
30(3): 172-177 (in Japanese).
Nishiuchi Y & Asano K (1979) [Toxicity of pesticide formulas to some
fresh water organisms - LXVI.] Suisan Zoushoku, 27(3): 172-177 (in
Japanese).
Nitschke KD, Kociba RJ, Keyes DG, & Mckenna MJ (1981) A thirteen week
repeated inhalation study of ethylene dibromide in rats. Fundam Appl
Toxicol, 1: 437-442.
NTP (1982) Carcinogenesis bioassay of 1,2-dibromoethane in F344 rats
and B6C3F1 mice (inhalation study). Research Triangle Park, North
Carolina, US Department of Health and Human Services, National
Toxicology Program, 163 pp (NTP Technical Report Series No. 210).
Ogino Y (1978) [Study of the effect of the environment - polluting
substances on ecological systems: I. Translocation of
bromohydrocarbons to the fish body.] Okayamo Igakkai Zasshi,
90: 1451 -1455 (in Japanese).
Ohta T, Nakamura N, Moriya M, Shirasu Y, & Kada T (1984) The
SOS-function-inducing activity of chemical mutagens in Escherichia
coli. Mutat Res, 131: 101-109.
Ong T-M, Stewart JD, Wen Y-F, & Whong W-Z (1987) Application of SOS
umu-test for the detection of genotoxic volatile chemicals and air
pollutants. Environ Mutagen, 9: 171-176.
Ong T-M, Stewart JD, Tucker JD, & Whong W-Z (1989) Development of an
in situ test system for detection of mutagens in the workplace. In:
Waters MD, Sandhu SS, Lewtas J, Claxton L, Strauss G, & Nesnow S, ed.
Short-term bioassays in the analysis of complex environmental
mixtures, IV. New York, Plenum Publishing Corporation, pp 25-36.
OSHA (Occupational Safety and Health Agency) (1983) Occupational
exposure to ethylene dibromide. Fed Reg, 48(196): 45956-46003.
Ott MG, Scharnweber HC, & Langner RR (1980) Mortality experience of
161 employees exposed to ethylene dibromide in two production units.
Br J Ind Med, 37: 163-168.
Ozawa N & Guengerich FP (1983) Evidence for formation of an
S-[2-(N7-guanyl)ethyl] glutathione adduct in glutathione-mediated
binding of the carcinogen 1,2-dibromoethane to DNA. Proc Natl Acad Sci
(USA), 80: 5266-5270.
Page GW (1981) Comparison of groundwater and surface water for
patterns and levels of contamination by toxic substances. Environ Sci
Technol, 15(12): 1475-1481.
Peoples SA, Maddy KT, & Riddle LC (1978) Human occupational health
problems resulting from exposure to ethylene dibromide in California
in 1975 and 1976. Vet Hum Toxicol, 20: 241-244.
Perocco P & Prodi G (1981) DNA damage by haloalkanes in human
lymphocytes cultured in vitro. Cancer Lett, 13: 213-218.
Perocco P, Colacci A, Santucci MA, Vaccari M, & Grilli S (1991)
Transforming activity of ethylene dibromide in BALB/c 3T3 cells. Res
Commun Chem Pathol Pharmacol, 73(2): 159-172.
Pignatello JJ (1986) Ethylene dibromide mineralization in soils under
aerobic conditions. Appl Environ Microbiol, 51(3): 588-592.
Pignatello JJ & Cohen SZ (1989) Environmental chemistry of ethylene
dibromide in soil and ground water. Rev Environ Contam Toxicol,
112: 1-47.
Pignatello JJ, Sawhney BL, & Frink CR (1987) EDB: Persistence in soil.
Science, 236: 898.
Pleil JD, Oliver KD, & McClenny WA (1987) Enhanced performance of
nation dryers in removing water from air samples prior to gas
chromatographic analysis. JAPCA, 37(30): 244-248.
Plotnick HB & Conner WL (1976) Tissue distribution of 14C-labeled
ethylene dibromide in the guinea pig. Res Commun Chem Pathol
Pharmacol, 13(2): 251-259.
Plotnick HB, Weigel WW, Richards DE, & Cheever KL (1979) The effect of
dietary disulfiram upon the tissue distribution and excretion of
14C-1,2-dibromoethane in the rat. Res Commun Chem Pathol Pharmacol,
26(3): 535-545.
Pranoto-Soetardhi LA, Rijk MAH, De Kruijf N, & De Vos RH (1986)
Automatic headspace sampling for determination of ethylene dibromide
residues in cereals. Int J Environ Anal Chem, 25: 151-159.
Principe P, Dogliotti E, Bignami M, Crebelli R, Falcone E, Fabrizi M,
Conti G, & Comba P (1981) Mutagenicity of chemicals of industrial and
agricultural relevance in Salmonella, Streptomyces and Aspergillus. J
Sci Food Agric, 32: 826-832.
Quillardet P, De Bellecombe C, & Hofnung M (1985) The SOS chromotest,
a colorimetric bacterial assay for genotoxins: validation study with
83 compounds. Mutat Res, 147: 79-95.
Rains DM & Holder JW (1981) Technical Communications: Ethylene
dibromide residues in biscuits and commercial flour. J Assoc Off Anal
Chem, 64(5): 1252.
Rannug U (1980) Genotoxic effects of 1,2-dibromoethane and
1,2-dichloroethane. Mutat Res, 76: 269-295.
Rannug U & Beije B (1979) The mutagenic effect of 1,2-dichloroethane
on Salmonella typhimurium. II. Activation by the isolated perfused
rat liver. Chem-Biol Interact, 24: 265-285.
Rappaport SM, Cameron W, & McAllister J (1984) Outgassing of ethylene
dibromide from fumigated oranges. J Agric Food Chem, 32(5): 1112-1116.
Rasmussen RA & Khalil MAK (1984) Gaseous bromine in the arctic and
arctic haze. Geophys Res Lett, 11(5): 433-436.
Ratcliffe JM, Schrader SM, Steenland K, Clapp DE, Turner T, & Hornung
RW (1987) Semen quality in papaya workers with long term exposure to
ethylene dibromide. Br J Ind Med, 44: 317-326.
Reznik G, Stinson SF, & Ward JM (1980) Respiratory pathology in rats
and mice after inhalation of 1,2-dibromo-3-chloropropane or 1,2
dibromoethane for 13 weeks. Arch Toxicol, 46: 233-240.
Rogers RD & McFarlane JC (1981) Sorption of carbon tetrachloride,
ethylene dibromide, and trichloroethylene in soil and clay. Environ
Monit Assess, 1: 155-162.
Rosenkranz HS (1977) Mutagenicity of halogenated alkanes and their
derivatives. Environ Health Perspect, 21: 79-84.
Roskill (1992) The economics of bromine, 1992. London, Roskill
Information Services, pp 57-58.
Rowe VK, Spencer HC, McCollister DD, Hollingsworth RL, & Adams EM
(1952) Toxicity of ethylene dibromide determined on experimental
animals. Am Med Assoc Arch Ind Hyg Occupat Med, 6: 158-173.
Rumsey DW & Tanita RK (1978) NIOSH industrial hygiene survey.
Cincinnati, Ohio, National Institute for Occupational Safety and
Health, 20 pp (Publication No. 79-112).
Russell WL(1986) Positive genetic hazard predictions from short-term
tests have proved false for results in mammalian spermatogonia with
all environmental chemicals so far tested. In: Genetic toxicology of
environmental chemicals - Part B: Genetic effects and applied
mutagenesis. New York, Alan R. Liss Inc., pp 67-74.
Saito N, Ogino Y, Katayama Y, Matsunaga K, Nagao M, & Ishida T (1978)
[Determination of aliphatic brominated hydrocarbons in environmental
samples.] Kogaito Taisaku, 14(3): 316-320 (in Japanese).
Sarawat PK, Kandara M, Dhurva AK, Malhotra VK, & Jhanwar RS (1986)
Poisoning by ethylene dibromide - six cases: A clinicopathological and
toxicological study. Indian J Med Sci, 40(5): 121-123.
Sawyer LD & Walters SM (1986) Gas chromatographic method for ethylene
dibromide in grains and grain-based products: Collaborative study. J
Assoc Off Anal Chem, 69(5): 847-851.
Schlinke JC (1970) Toxicologic effects of five soil nematocides in
chickens. Am J Vet Res, 31(6): 1119-1121.
Schrader SM, Ratcliffe JM, Turner T, & Hornung RW (1987) The use of
new field methods of semen analysis in the study of occupational
hazards to reproduction the example of ethylene dibromide. J Occup
Med, 29: 963-966.
Schrader SM, Turner TW, & Ratclife JM (1988) The effects of ethylene
dibromide on semen quality: A comparison of short-term and chronic
exposure. Reprod Toxicol, 2: 191-198.
Schultz K, Ghosh L, & Banerjee S (1992) Neoplastic expression in
murine cells induced by halogenated hydrocarbons. In vitro Cell Dev
Biol, 28A: 267-272.
Scott BR, Sparrow AH, Schwemmer SS, & Schairer LA (1978) Plant
metabolic activation of 1,2-dibromoethane (EDB) to a mutagen of
greater potency. Mutat Res, 49: 203-212.
Sega GA & Rene E (1980) Chemical dosimetry and unscheduled DNA
synthesis studies of ethylene dibromide in the germ cells of male
mice. Environ Mutagen, 5: 274.
Sekita H, Takeda M, & Uchiyama M (1981) [Studies on analysis of
pesticide residues in foods. XXXIII. Determination of ethylene
dibromide residues in litchi (lychee) fruits imported from Formosa.]
Bull Natl Inst Hyg Sci (Tokyo), 99: 130-132 (in Japanese).
Sekita H, Takeda M, & Uchiyama M (1983) [Ethylene dibromide (EDB)
residues in fresh fruits imported after fumigation with EDB and
decrease of the residues with time.] Shokuhin Eisei Gaku Zasshi,
24(1): 57-63 (in Japanese).
Shiau SY, Huff RA, Wells BC, & Felkner IC (1980) Mutagenicity and
DNA-damaging activity for several pesticides tested with Bacillus
subtilis mutants. Mutat Res, 71: 169-179.
Shivanandappa T, Krishnakumari MK, & Majumder SK (1987) Reproductive
potential of male rats fed dietary ethylene dibromide. J Food Saf,
8: 147-155.
Short RD, Minor JL, Winston JM, Seifter J, & Lee CC (1978) Inhalation
of ethylene dibromide during gestation by rats and mice. Toxicol Appl
Pharmacol, 46: 173-182.
Short RD, Winston JM, Hong CB, Minor JL, Lee CC, & Seifter J (1979)
Effects of ethylene dibromide on reproduction in male and female rats.
Toxicol Appl Pharmacol, 49: 97-105.
Simula TP, Glancey MJ, & Wolf CR (1993) Human glutathione
S-transferase-expressing Salmonella typhimurium tester strains to
study the activation/detoxification of mutagenic compounds: studies
with halogenated compounds, aromatic amines and aflatoxin B1.
Carcinogenesis, 14: 1371-1376.
Sina JF, Bean CL, Dysart GR, Taylor VI, & Bradley MO (1983) Evaluation
of the alkaline elution/rat hepatocyte assay as a predictor of
carcinogenic/mutagenic potential. Mutat Res, 113: 357-391.
Singh HB, Salas LJ, Smith AJ, & Shigeishi H (1981) Measurements of
some potentially hazardous organic chemicals in urban environments.
Atmos Environ, 15: 601-612.
Singh HB, Salas LJ, & Stiles RE (1982) Distribution of selected
gaseous organic mutagens and suspect carcinogens in ambient air.
Environ Sci Technol, 16(12): 872-880.
Singh HB, Salas LJ, & Stiles RE (1983) Selected man-made halogenated
chemicals in the air and oceanic environment. J Geophys Res,
88(C6): 3675-3683.
Sipes IG, Wiersma DA, & Armstrong DJ (1986) The role of glutathion in
the toxicity of xenobiotic compounds: Metabolic activation of
1,2-dibromoethane by glutathione. Adv Exp Med Biol, 197: 457-467.
Smith RF & Goldman L (1983) Behavioral effects of prenatal exposure to
ethylene dibromide. Neurobehav Toxicol Teratol, 5: 579-585.
Sparrow AH, Schairer LA, & Villalobos-Pietrini R (1974) Comparison of
somatic mutation rates induced in Tradescantia by chemical and
physical mutagens. Mutat Res, 26: 265-276.
Springarn NE, Northington DJ, & Pressely T (1982) Analysis of volatile
hazardous substances by GC/MS. J Chromatogr Sci, 20: 286-288.
SRI International (1982) Ethylene dibromide: US salient statistics -
Chemical economics handbook. Menlo Park, California, Stanford Research
International.
Steenland K, Carrano A, Ratcliffe J, Clapp D, Ashworth L, & Meinhardt
T (1986) A cytogenetic study of papaya workers exposed to ethylene
dibromide. Mutat Res, 170: 151-160.
Steinberg SM, Pignatello JJ, & Sawhney BL (1987) Persistence of
1,2-dibromomethane in soils: entrapment in intraparticle micropores.
Environ Sci Technol, 21: 1202-1208.
Stenger VA (1978) Bromine compounds. In: Kirk-Othmer encyclopedia of
chemical technology, 3rd ed. New York, John Wiley, vol 4, pp 243-263.
Stinson SF, Reznik G, & Ward JM (1981) Characteristics of
proliferative lesions in the nasal cavities of mice following chronic
inhalation of 1,2-dibromoethane. Cancer Lett, 12: 121-129.
Stolzenberg SJ & Hine CH (1980) Mutagenicity of 2-and 3-carbon
halogenated compounds in the Salmonella/mammalian-microsome test.
Environ Mutagen, 2: 59-66.
Storer RD & Conolly RB (1983) Comparative in vivo genotoxicity and
acute hepatotoxicity of three 1,2-dihaloethanes. Carcinogenesis,
4(11): 1491-1494.
Stottmeister E, Hendel P, & Engewald W (1986) [Gas-chromatographic
trace analysis of highly volatile halogenated hydrocarbons in water
with FID detection.] Acta Hydrochim Hydrobiol, 14(6): 573-580 (in
German).
Sugiyama H (1980) Effects of EDB (1,2-dibromoethane) on the silkworm
(Bombyx mori L.). J Pestic Sci, 5: 599-602.
Sun M (1984) EDB contamination kindles federal action. Science,
233: 464-466.
Sundheimer DW, White RD, & Sipes IG (1982) The bioactivation of
1,2-dibromoethane in rat hepatocytes: Covalent binding to nucleic
acids. Carcinogenesis, 3(10): 1129-1133.
Tan E-L & Hsie AW (1981) Mutagenicity and cytotoxicity of haloethanes
as studied in the CHO/HGPRT system. Mutat Res, 90: 183-191.
Tennant RW, Stasiewicz S, & Spalding JW (1986) Comparison of multiple
parameters of rodent carcinogenicity and in vitro genetic toxicity.
Environ Mutagen, 8: 205-227.
Tennant RW, Margolin BH, Shelby MD, Zeiger E, Haseman JK, Spalding J,
Caspary W, Resnick M, Stasiewicz S, Anderson B, & Minor R (1987)
Prediction of chemical carcinogenicity in rodents from in vitro
genetic toxicity assays. Science, 236: 933-941.
Teramoto S, Saito R, Aoyama H, & Shirasu Y (1980) Dominant lethal
mutation induced in male rats by 1,2-dibromo-3-chloropropane (DBCP).
Mutat Res, 77: 71-78.
Terao H, Kajikawa M, Morishita Y, & Kato K (1984) [Bromide ion
concentration and Br/Cl ratio of ground water in the southwest region
of Gifu Prefecture.] Chikyu Kagaku, 18: 21-28 (in Japanese).
Terao H, Kajikawa M, Morishita K, & Kato K (1985) [Influence of
pesticide and fertilizer on the ground water in vegetable field zone.]
Chikyu Kagaku, 19: 31-38 (in Japanese).
Ter Haar G (1980) An investigation of possible sterility and health
effects from exposure to ethylene dibromide. In: Ames B, Infante O, &
Peirtz R ed. Ethylene dichloride: A potential health risk? Cold Spring
Harbor, New York, Cold Spring Harbor Laboratory, pp 167-188 (Banbury
Report No. 5).
Tezuka H, Ando N, Suzuki R, Terahata M, Moriya M, & Shirasu Y (1980)
Sister-chromatid exchanges and chromosomal aberrations in cultured
Chinese hamster cells treated with pesticides positive in microbial
reversion assays. Mutat Res, 78: 177-191.
Thier R, Taylor JB, Pemble SE, Humphreys WG, Persmark M, Ketterer B, &
Guengerich FP (1993) Expression of mammalian glutathione S-transferase
5-5 in Salmonella typhimurium TA1535 leads to base-pair mutations
upon exposure to dihalomethanes. Proc Natl Acad Sci (USA),
90: 8576-8580.
Townshend JL, Dirks VA, & Marks CF (1980) Temperature, moisture and
compaction and their effects on the diffusion of ethylene dibromide in
three Ontario soils. Can J Soil Sci, 60: 177-184.
Tsani-Bazaca E, McIntyre AE, Lester JN, & Perry R (1981)
Concentrations and correlations of 1,2-dibromoethane,
1,2-dichloroethane, benzene and toluene in vehicle exhaust and ambient
air. Environ Technol Lett, 2: 303-316.
Tucker JD, Xu J, Stewart J, & Ong T-M (1984) Detection of sister-
chromatid exchanges in human peripheral lymphocytes induced by
ethylene dibromide vapor. Mutat Res, 138: 93-98.
UNEP (1992) Methyl bromide and the ozone layer: a summary of current
understanding. Montreal Protocol assessment supplement: Synthesis
report of the methyl bromide interim scientific assessment and methyl
bromide interim technology and economic assessment requested by the
United Nations Environment Programme on behalf of the Contracting
Parties to the Montreal Protocol. Nairobi, United Nations Environment
Programme.
US EPA (1977) EPA notice of rebuttable presumption against
registration and continued registration of pesticide products
containing ethylene dibromide. Fed Reg, 42: 63134.
US EPA (1986) US EPA reports results of study of pesticides in
groundwater. J Am Water Works Assoc, 1986: 128-129.
Van Bladeren PJ, Breimer DD, Rotteveel-Smijs GMT, De Jong RAW, Buus W,
Van Der Gen A, & Mohn GR (1980) The role of glutathione conjugation in
the mutagenicity of 1,2-dibromoethane. Biochem Pharmacol,
29: 2975-2982.
Van Bladeren PJ, Hoogeterp JJ, Breimer DD, & Van Der Gen A (1981a) The
influence of disulfiram and other inhibitors of oxidative metabolism
on the formation of 2-hydroxyethyl-mercapturic acid from
1,2-dibromoethane by the rat. Biochem Pharmacol, 30(21): 2983-2987.
Van Bladeren PJ, Breimer DD, Rotteveel-Smijs GMT, De Knijff P, Mohn
GT, Van Meeteren-Walchli B, Buijs W, & Van der Gen A (1981b) The
relation between the structure of vicinal dihalogen compounds and
their mutagenic activation via conjugation to glutathione.
Carcinogenesis, 2(6): 499-505.
Van Duuren BL, Goldschmidt BM, Loewengart G, Smith AC, Melchlonne S,
Seldman I, & Roth D (1979) Carcinogenicity of halogenated olefinic and
aliphatic hydrocarbons in mice. J Natl Cancer Inst, 63(6): 1433-1439.
Van Duuren BL, Seidman I, Melchionne S, & Kline SA (1985)
Carcinogenicity bioassays of bromoacetaldehyde and bromoethanol-
potential metabolites of dibromoethane. Teratogen Carcinogen Mutagen,
5: 393-403.
Vant'Hof J & Schairer LA (1982) Tradescantia assay system for gaseous
mutagens: A report of the US Environmental Protection Agency Gene-Tox
Program. Mutat Res, 99: 303-315.
Verschueren K (1983) Handbook of environmental data on organic
chemicals, 2nd ed. New York, Van Nostrand Reinhold, pp 635-636.
Vogel E & Chandler JLR (1974) Mutagenicity testing of cyclamate and
some pesticides in Drosophila melanogaster. Experientia (Basel),
30: 621-623.
Von Buselmaier W, Rohrborn G, & Propping P (1972) [Mutagenicity
investigations with pesticides in the host mediated assay and the
dominant lethal test in mice.] Biol Zent.bl, 91: 311-325 (in German).
Wade NL & Rigney CJ (1979) Phytotoxicity of ethylene dibromide to
cherry and banana fruit. J Am Soc Hortic Sci, 104(6): 900-903.
Weast RC, Astle MJ, & Beyer WH (1988) CRC Handbook of chemistry and
physics, 68th ed. Boca Raton, Florida, CRC Press, p C-264.
White RD (1982) Chemical induction of genetic injury: The
bioactivation of 1,2-dibromoethane. Diss Abstr Int, 43(03): 696B-697B.
White RD, Gandolfi AJ, Bowden GT, & Sipes IG (1983) Deuterium isotope
effect on the metabolism and toxicity of 1,2-dibromoethane. Toxicol
Appl Pharmacol, 69: 170-178.
White RD, Petry TW, & Sipes IG (1984) The bioactivation of
1,2-dibromoethane in rat hepatocytes: deuterium isotope effect.
Chem-Biol Interact, 49: 225-233.
WHO (1993) Guidelines for drinking-water quality: Volume 1 - WHO
Recommendations, 2nd ed. Geneva, World Health Organization, 130 pp.
Wildeman AG & Nazar RN (1982) Significance of plant metabolism in the
mutagenicity and toxicity of pesticides. Can J Genet Cytol,
24: 437-449.
Williams GM, Laspia MF, & Dunkel VC (1982) Reliability of the
hepatocyte primary culture/DNA repair test in testing of coded
carcinogens and noncarcinogens. Mutat Res, 97: 359-370.
Williams J, Gladen BC, Turner TW, Schrader SM, & Chapin RE (1991) The
effects of ethylene dibromide on semen quality and fertility in the
rabbit: Evaluation of a model for human seminal characteristics.
Fundam Appl Toxicol, 16: 687-700.
Wofsy SC, McElroy MB, & Yung YL (1975) The chemistry of atmospheric
bromine. Geophys Res Lett, 2(6): 215-218.
Wong LCK, Winston JM, Hong CB, & Plotnick H (1982) Carcinogenicity and
toxicity of 1,2-dibromoethane in the rat. Toxicol Appl Pharmacol,
63: 155-165.
Wong O, Morgan RW, & Whorton MD (1985) An epidemiologic surveillance
program for evaluating occupational reproductive hazards. Am J Ind
Med, 7: 295-306.
Woodrow JE, Majewski MS, & Seiber JN (1986) Accumulative sampling of
trace pesticides and other organics in surface water using XAD-4
resin. J Environ Sci Health, B21(2): 143-164.
Working PK, Smith-Oliver T, White RD, & Butterworth BE (1986)
Induction of DNA repair in rat spermatocytes and hepatocytes by
1,2-dibromoethane: the role of glutathione conjugation.
Carcinogenesis, 7(3): 467-472.
Yoshida YH & Inagaki E (1986) [Mutagenicity of ethylene dibromide in
Drosophila melanogaster]. Tachikawa Tandai Kiyo, 19: 49-50
(in Japanese).
Yuita K (1984) [Residues and pollutions of bromine derived from soil
fumigants and disinfectants crops, soils and ground water.] Seitai
Kogaku, 7(2): 3-12 (in Japanese).
Zoetemelk CEM, Mohn GR, Van Der Gen A, & Breimer DD (1987)
Mutagenicity in Salmonella strains differing in glutathione content
and their alkylating potential. Biochem Pharmacol, 36(11): 1829-1835.
Résumé
1. Identité, propriétés physiques et chimiques, et méthodes d'analyse
Le 1,2-dibromoéthane (dibromure d'éthylène) est un liquide
incolore d'odeur chloroformique dont le point de fusion est de 9,9°C
et le point d'ébullition de 131,4°C. Il est assez volatil, avec une
tension de vapeur de 1,47 kPa (11 mmHg) à 25°C et une densité de
vapeur par rapport à l'air de 6,1. Le 1,2-dibromoéthane est miscible
à la plupart des solvants organiques. Sa solubilité dans l'eau est de
4,3 g/litre à 30°C.
Dans l'air ambiant, l'analyse s'effectue par chromatographie en
phase gazeuse après absorption sur polymères poreux puis désorption
thermique rapide. Pour les échantillons d'eau, on utilise un système
avec piège et purgeur. Les résidus de 1,2-dibromoéthane présents dans
les denrées alimentaires et d'autres milieux peuvent être soit
extraits par solvent, soit soumis à une analyse par la technique de
l'espace de tête dans des conditions cryogéniques, après quoi on
prépare un dérivé et on poursuit par chromatographie en phase gazeuse
ou chromatographie liquide à haute performance.
2. Sources d'exposition humaine et environnementale
Le 1,2-dibromoéthane est utilisé comme agent d'épuration des
agents antidétonnants à base de plomb ajoutés à l'essence. On
l'emploie aussi pour la fumigation du sol ou de certains fruits ou
céréales. L'essence additionnée de plomb étant moins utilisée dans
certains pays et les homologations pour les usages agricoles ayant été
annulées, l'exposition humaine à ce produit a diminué. Il reste
cependant encore en usage dans certains pays comme épurateur de
l'essence au plomb, comme fumigant, au fins de quarantaine, comme
solvent et comme intermédiaire dans l'industrie chimique.
3. Concentrations et dégradation dans l'environnement
Dans l'air, on mesure des concentrations qui vont de zéro à des
teneurs de l'ordre du ng/m3 en zone urbaine. On a trouvé du
1,2-dibromoéthane dans des eaux souterraines à des concentrations
allant jusqu'à 0,2 µg/litre, la teneur pouvant atteindre 50 µg/litre
dans les eaux superficielles des zones d'exploitation agricole
intensive. Bien qu'il y ait lessivage du 1,2-dibromoéthane à travers
le sol, il en reste une certaine quantité dans la matrice
édaphique,d'où risque de contamination ultérieure de la nappe
phréatique. On connaît mal les conditions de dégradation microbienne
du 1,2-dibromoéthane dans le sol.
Le composé étant très volatil, c'est l'atmosphère qui en est le
principal réceptacle. Par photolyse dans la stratosphère, il peut se
former des produits de décomposition susceptibles d'attaquer la couche
d'ozone.
4. Cinétique et métabolisme chez les animaux de laboratoire
Le 1,2-dibromoéthane est rapidement absorbé par la voie orale,
percutanée et respiratoire. On pense que la toxicité du composé est
largement due à ses métabolites. La métabolisation s'effectue soit
par une voie oxydative (cytochrome P-450), soit par conjugaison
(glutathion- S-transférase). Deux métabolites réactifs, le
bromoacétaldéhyde formé par la voie oxydative, et l'ion thiiranium,
formé par conjugaison, interagissent avec les macromolécules
cellulaires (protéines, ADN), pour donner naissance à divers produits
par l'établissement de liaisons covalentes.
5. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
Le 1,2-dibromoéthane est fortement toxique pour les animaux
(DL50 par voie orale pour le rat égale à 146-417 mg/kg de poids
corporel; CL50 inhalatoire pour le rat égale à 3080 mg/m3 après
2 h d'exposition; mortalité observée chez des lapins à la suite d'une
application cutanée à raison de 210 mg/kg). Les effets toxiques
observés se sont produits principalement au niveau des reins et du
foie. L'inhalation de vapeurs provoque une irritation de la muqueuse
nasale et une dépression du système nerveux central. Chez des groupes
de rats exposés à des concentrations comprises entre 1540 et
77 000 mg/m3 (200-10 000 ppm) pendant 0,1 à 16,0 h, on a observé
dans tous les groupes une mortalité qui était fonction de la durée
d'exposition et de la concentration. En solution à 1,0%, le
1,2-dibromoéthane a provoqué une irritation sur la peau abdominale de
lapins après rasage ainsi qu'une irritation oculaire.
Chez des rats et des souris qui avaient reçu du 1,2-dibromoéthane
par voie orale de manière subchronique, on a observé une certaine
mortalité et une baisse du gain de poids à la dose quotidienne de
100 mg/kg de poids corporel. Chez des rats exposés au composé à la
dose de 115 mg/m3 (578 ppm), 6 h par jour, 5 jours par semaine et
ce, pendant 13 semaines, on a noté un moindre gain de poids et des
effets pathologiques au niveau du nez. Cette étude a permis d'établir
que la dose sans effets histopathologiques observables au niveau de la
cavité nasale était égale à 23 mg/m3 (3 ppm). Lors d'une étude
analogue sur des souris, on a observé le même genre d'effets
histopathologiques avec la même dose sans effets observables
(23 mg/m3, 3 ppm).
On a administré du 1,2-dibromoéthane par gavage à des rats selon
les modalités suivantes: 37-107 mg/kg de poids corporel (moyenne
pondérée par rapport au temps) tous les jours pendant 49-90 semaines;
de même des souris en ont reçu pendant 15-17 mois dans leur eau de
boisson à raison de 103-117 mg/kg de poids corporel. A la suite de
cela, on a observé des anomalies non malignes telles qu'une
dégénérescence hépatique, une atrophie testiculaire, ainsi qu'une
acanthose et une hyperkératose au niveau de la portion cardiaque de
l'estomac. Après exposition par la voie respiratoire de rats et de
souris à des doses de 77-388 mg/m3 pendant 6 à 18 mois, on a observé
une inflammation de la trachée et de la cavité nasale, une
dégénérescence testiculaire et une nécrose hépatique.
Après exposition par la voie respiratoire, le 1,2-dibromoéthane
ne se révèle pas tératogène pour le rat ou la souris. Chez des rats
qui en avaient reçu par la voie intrapéritonéale une dose quotidienne
de 1,25 mg/kg de poids corporel (mâles) ou de 509 mg/m3 (voie
respiratoire, femelles, 4 h/jour, 3 jours par semaine, du jour 3 au
jour 20 de la gestation), on a constaté une action toxique sur le
développement (anomalies de la coordination motrice). Le
1,2-dibromoéthane a également eu une action délétère sur la fonction
de reproduction de rats (chez les mâles, dans les conditions
d'exposition suivantes: 684 mg/m3, 7 h/jour, 5 jours par semaine,
pendant 10 semaines; chez les femelles, aux doses et pendant les
durées suivantes: 614 mg/m3, 7 h/jour, 7 jours par semaine, pendant
3 semaines). La dose sans effet observable pour ce paramètre était
égale à 300 mg/m3 chez les deux sexes. Lors d'une étude où des rats
mâles ont reçu pendant 90 jours une alimentation contenant le
composé, on a constaté que la dose sans effet observable sur la
capacité de reproduction était égale à 50 mg/kg et par jour. On a
observé une atteinte de la spermatogénèse chez des taureaux après
administration du composé par voie orale à la dose quotidienne de
2 mg/kg pendant moins de 21 jours et chez des lapins après injection
sous-cutanée du produit à raison de 15 mg/kg pendant 5 jours. Chez
des poules qui avaient reçu pendant 12 semaines une nourriture
contenant du 1,2-dibromoéthane à la dose de 12,5 mg/kg, on a constaté
une diminution du calibre des oeufs.
Le composé n'a pas entraîné de mutations léthales dominantes chez
des souris ou des rats, ni produit d'aberrations chromosomiques ou de
micronoyaux dans les cellules de la moëlle osseuse de souris traitées
in vivo. Toutefois, il s'est révélé mutagène dans les épreuves sur
bactéries et a provoqué des ruptures de l'ADN monocaténaire. On a
constaté in vivo comme in vitro, que les métabolites du
1,2-dibromoéthane étaient fixés à l'ADN par des liaisons covalentes.
Des échanges entre chromatides soeurs, des mutations et une synthèse
non programmée de l'ADN, ont été observées dans des cellules humaines
in vitro.
Des études de cancérogénicité ont été effectuées selon le schéma
suivant: souris et rats ayant reçu par gavage une dose quotidienne de
1,2-dibromoéthane égale à 37-107 mg/kg de poids corporel (en moyenne
pondérée par rapport au temps), pendant 49-90 semaines; souris ayant
reçu dans leur eau de boisson une dose de 1,2-dibromoéthane de
103-117 mg/kg de poids corporel, quotidiennement pendant 15-17 mois;
souris et rats exposés à une dose de 10-40 ppm pendant 6-18 mois par
voie respiratoire ou encore, souris badigeonnées au 1,2-dibromoéthane
pendant 400-594 jours, 3 fois par semaine à raison de 25-50 mg/souris.
Ces études ont montré que le 1,2-dibromoéthane était cancérogène pour
les rats et les souris et provoquait l'apparition de tumeurs au niveau
de divers organes, soit au point d'application, soit à distance de ce
point: cavité nasale, poumons, estomac, foie, peau, système
circulatoire et glandes mammaires. Dans de nombreux cas, il y avait
réduction du temps de latence des tumeurs.
6. Effets sur l'homme
Le 1,2-dibromoéthane peut avoir des effets nocifs sur le système
respiratoire, le système nerveux et les reins.
Ainsi, une seule exposition, par la voie respiratoire, à ce
composé (215 mg/m3, soit 28 ppm) pendant 30 min ou davantage, s'est
révélée mortelle pour l'homme. L'ingestion d'une dose de 140 mg/kg de
poids corporel s'est également révélée mortelle. Chez des
travailleurs exposés de par leur profession, une exposition de longue
durée au 1,2-dibromoéthane (5 ans), à la concentration de 0,68 mg/m3
dans la zone de respiration, a provoqué une diminution sensible du
nombre de spermatozoïdes et une baisse de la fécondité.
7. Effets sur les êtres vivants dans leur milieu naturel
Peu d'études d'ecotoxicité aquatique ont été consacrées au
1,2-dibromoéthane. Les valeurs de la CL50 pour les organismes
aquatiques sont supérieures à 5 mg/litre. On ne possède aucune donnée
au sujet des organismes terrestres.
RESUMEN
1. Identidad, propiedades físicas y químicas y métodos analíticos
El 1,2-dibromoetano (dibromuro de etileno) es un líquido incoloro
(punto de fusión: 9,9°C; punto de ebullición: 131,4°C) con olor
similar al del cloroformo. Es muy volátil, con una presión de vapor
de 1,47 kPa (11 mmHg) a 25°C y una densidad de vapor, en comparación
con el aire, de 6,1. El 1,2-dibromoetano es invisible en la mayor
parte de los disolventes orgánicos. Su solubilidad en el agua es de
4,3 g/litro a 30°C.
El 1,2-dibromoetano existente en el aire ambiental se analiza por
cromatografía de gases tras su absorción por polímeros porosos,
seguida por una rápida desasorpción térmica. Para las muestras de
agua se utiliza un método de «purge-and-trap». Los residuos de
1,2-dibromoetano presentes en los alimentos y en otros medios pueden
extraerse mediante disolventes o ser sometidos a un análisis
automatizado de la fase gaseosa superior en condiciones criogénicas,
seguido de análisis por cromatografía de gases y cromatografía líquida
de alta resolución, previa derivación.
2. Fuentes de exposición humana y ambiental
El 1,2-dibromoetano se utiliza para eliminar las sustancias
antidetonantes derivadas del plomo presentes en la gasolina.
Asimismo, se utiliza como fumigante de suelos y para la fumigación de
granos y frutas. El consumo reducido de gasolina con plomo en algunos
países y la anulación de inscripciones para la utilización de
1,2-dibromoetano con fines agrícolas ha reducido la exposición humana
a esa sustancia. Sin embargo, aún se utiliza para eliminar el plomo
de la gasolina en algunos países, como fumigante, para fines de
cuarentena, como disolvente y como producto intermedio en las
sustancias químicas industriales.
3. Niveles ambientales y degradación
Las concentraciones de 1,2-dibromoetano medidas en el aire
abarcan desde niveles indetectables hasta otros expresados en ng/m3
en las zonas urbanas. En zonas de explotación agrícola extensiva se
han detectado concentraciones de 1,2-dibromoetano superiores a
0,2 µg/litro en aguas subterráneas y a 50 µg/litro en aguas
superficiales. Aunque el 1,2-dibromoetano se filtra a través de la
tierra, parte de él queda retenido en la matriz del suelo y puede
contaminar posteriormente los pozos de riego. Se carece de
información suficiente sobre la descomposición microbiana en los
suelos.
La alta volatilidad del 1,2-dibromoetano determina que el
principal receptor ambiental sea la atmósfera. La fotólisis
estratosférica puede dar lugar a la formación de productos de
descomposición potencialmente destructores del ozono.
4. Cinética y metabolismo en animales de laboratorio
El 1,2-dibromoetano se absorbe rápidamente por vía oral y cutánea
y por inhalación. Se cree que los metabolitos desempeñan una función
importante en la toxicidad de esa sustancia para los seres humanos.
El 1,2-dibromoetano se puede metabolizar por vía oxidativa (sistema
del citocromo P-450) y por vía conjugada (sistema de la glutatión
S-transferasa). Según parece, dos metabolitos reactivos, el
bromacetaldehído formado por oxidación y el ion de tiranio formado por
conjugación interactúan con las macromoléculas celulares (proteínas,
ADN) para formar productos de enlace covalente.
5. Efectos en mamíferos de laboratorio y en sistemas de pruebas
in vitro
El 1,2-dibromoetano tiene una toxicidad aguda para los animales
(DL50 por vía oral en ratas de 146-417 mg/kg de peso corporal,
CL50 por inhalación en ratas de 3080 mg/m3 tras una exposición de
2 h, y una mortalidad observada tras la aplicación cutánea de
210 mg/kg a conejos). Los efectos tóxicos del 1,2-dibromoetano se
observaron principalmente en el hígado y en los riñones. La
inhalación de vapor de 1,2-dibromoetano produjo irritación nasal y
depresión del sistema nervioso central. En ratas expuestas a
concentraciones de 1540 mg/m3 a 77 000 mg/m3 (200 a 10 000 partes
por millón), con una duración de la exposición de 0,1 a 16,0 h, se
produjeron muertes en todos los grupos, en función de la concentración
y del tiempo. El 1,2-dibromoetano (solución al 1,0%) causó irritación
de la piel abdominal afeitada e irritación ocular en conejos.
Tras la exposición oral subcrónica, se observaron efectos
mortales y menor adquisición de peso en ratas y ratones con dosis de
100 mg/kg de peso corporal al día. Asimismo, se observaron reducciones
en la adquisición de peso y efectos patológicos nasales en ratas
expuestas al 1,2-dibromoetano en una proporción de 115 mg/m3
(578 partes por millón) durante 6 h al día y 5 días por semana a lo
largo de 13 semanas. El NOEL relativo a las alteraciones
histopatológicas de la cavidad nasal fue de 23 mg/m3 (3 ppm) en ese
estudio. En otro similar realizado en ratones se observaron los
mismos cambios patológicos, también con un NOEL de 23 mg/m3 (3 ppm).
Tras la administración por sonda a ratones o ratas de
1,2-dibromoetano en dosis de 37 a 107 mg/kg de peso corporal al día
(promedio ponderado por el tiempo) durante un periodo de 49 a
90 semanas, o la administración a ratones en dosis de 103 a 117 mg/kg
de peso corporal al día en el agua de beber durante un periodo de 15 a
17 meses, se observaron cambios no carcinogénicos tales como
degeneración hepática, atrofia testicular, y acantosis e
hiperqueratosis del preestómago, además de mortalidad. Tras la
exposición por inhalación (ratones o ratas expuestos a dosis de 77 a
388 mg/m3 durante un periodo de 6 a 18 meses), se observaron
inflamación de la tráquea y de la cavidad nasal, degeneración
testicular y necrosis hepática.
El 1,2-dibromoetano no resultó teratogénico en ratas o ratones
tras la exposición por inhalación. Se observó toxicidad para el
desarrollo (daños en el desarrollo de la coordinación motora) en la
descendencia de ratas macho tratadas por vía intraperitoneal con
1,25 mg/kg de peso corporal al día y en la descendencia de ratas
hembra tratadas mediante la inhalación de 509 mg/m3 durante 4 h al
día y 3 días por semana desde el día 3 al día 20 de la gestación. El
1,2-dibromoetano influyó en el comportamiento reproductivo de las
ratas (en los machos, con un nivel de exposición de 684 mg/m3
durante 7 h al día y 5 días/semana a los largo de 10 semanas, y en las
hembras con un nivel de exposición de 614 mg/m3 durante 7 h al día y
7 días por semana a lo largo de 3 semanas). El NOEL para ese
parámetro fue de 300 mg/m3 en ambos sexos. El NOEL para el
comportamiento reproductivo de las ratas macho en un estudio de
alimentación fue de 50 mg/kg al día tras una exposición de 90 días.
La espermatogénesis resultó afectada en toros tras la administración
de dosis orales de 2 mg/kg al día durante menos de 21 días, y en
conejos tras la inyección subcutánea de 15 mg/kg durante 5 días. La
administración de 1,2-dibromoetano a gallinas a través de la
alimentación causó una disminución del tamaño de los huevos tras la
exposición a 12,5 mg/kg al día durante 12 semanas.
El 1,2-dibromoetano no indujo mutaciones dominantes letales en
los ratones o las ratas, ni produjo aberraciones cromosómicas o
micronúcleos en las células de médula ósea de ratones tratados
in vivo. Sin embargo, resultó mutagénico en análisis bacterianos y
causó roturas del ADN de una sola hebra. Los metabolitos del
1,2-dibromoetano se fijaron al ADN mediante enlaces covalentes,
in vivo e in vitro. En células humanas in vitro se observó
intercambio de cromátides hermanas, mutaciones y síntesis de ADN no
programada.
Los estudios de carcinogenicidad en los que se administró la
sustancia por vía oral (ratones y ratas sometidas mediante sonda a
dosis de 37 a 107 mg/kg de peso corporal al día (promedio ponderado
por el tiempo) durante un periodo de 49 a 90 semanas; y ratones a los
que se administró 1,2-dibromoetano en el agua de beber en dosis de
103 a 117 mg/kg de peso corporal al día durante un periodo de 15 a
17 meses), mediante exposición inhalacional (ratones y ratas expuestos
a dosis de 10 a 40 partes por millón durante un periodo de 6 a
18 meses) o por vía cutánea de 25 a 50 mg/ratón, 3 veces por semana
durante un periodo 400 a 594 días) mostraron que el 1,2-dibromoetano
es carcinogénico para las ratas y los ratones y causa tumores en
diversos órganos (tanto en la zona de aplicación como en zonas
distantes, entre ellas, la cavidad nasal, los pulmones, el estómago,
el hígado, la piel, el sistema circulatorio y las glándulas mamarias).
En muchos casos reduce el periodo de latencia de tumores en
desarrollo.
6. Efectos en el ser humano
El 1,2-dibromoetano puede producir efectos adversos en los
sistemas respiratorio, nervioso y renal.
La exposición aguda (única) a la inhalación de 1,2-dibromoetano
en dosis de 215 mg/m3 (28 ppm) durante 30 minutos o más ha resultado
mortal para el ser humano. La ingestión de 140 mg/kg de peso corporal
resultó asimismo mortal. La exposición duradera (5 años) de la zona
respiratoria al 1,2-dibromoetano a una concentración de 0,68 mg/m3
redujo notablemente la densidad de espermatozoides y la fecundidad en
los trabajadores expuestos en su entorno laboral.
7. Efectos en otros organismos en el medio ambiente
Se han realizado pocos estudios sobre la ecotoxicidad acuática
del 1,2-dibromoetano. La CL50 para los organismos acuáticos es
superior a 5 mg/litro. No se dispone de información acerca de los
organismos terrestres.
Inorganic bromide may be formed as a consequence of attack by GSH
or oxidative catabolism. In the first case, the expected intermediate
would be S-(2-bromoethyl)-GSH, which can be converted to bis-GSH or
S-(2-hydroxyethyl)-GSH. Sulfoxidation of S-(2-hydroxyethyl)-GSH
would yield S-(2-hydroxyethyl)-GSH- S-oxide or further metabolism
would produce S-(2-hydroxyethyl)-cysteine, which in turn may undergo
sulfoxidation to yield N-acetyl- S-(2-hydroxyethyl)-cysteine-
S-oxide. The oxidative metabolism of 1,2-dibromoethane by
cytochrome P-450-dependent mixed function oxidases would be expected
to yield 2-bromoacetaldehyde as the initial product. This may be
converted by dehydrogenase to 2-bromoacetic acid or undergo attack by
GSH and subsequent dehydrogenase activity to give rise to
S-carboxymethyl-GSH. S-carboxymethyl-cysteine may be further
metabolized to thioglycolic acid. A reactive intermediate binds
mainly to DNA guanyl remnants and may be responsible for the
genotoxicity.
White et al. (1983) reported a deuterium isotope effect on the
metabolism of 1,2-dibromoethane. The metabolism of 1,2-dibromoethane
and tetradeutero-1,2-dibromoethane (d4-1,2-dibromoethane) was compared
in male Swiss-Webster mice. Three hours after intraperitoneal
administration of 1,2-dibromoethane or d4-1,2-dibromoethane
(50 mg/kg), there was 42% less bromide in the plasma of
d4-1,2-dibromoethane-treated mice than in the plasma of
1,2-dibromoethane-treated mice. This difference demonstrated a
significant deuterium isotope effect on the metabolism of
1,2-dibromoethane in vivo. In in vitro studies, which measured
bromide ion released from the substrate to monitor the rate of
metabolism, hepatic glutathione- S-transferase was unaffected. Since
the decreased metabolism of d4-1,2-dibromoethane was apparently due to
a reduced rate of microsomal oxidation, these data supported the
hypothesis that conjugation with GSH is responsible for the genotoxic
effect of 1,2-dibromoethane.
White et al. (1984) studied metabolism in isolated rat
hepatocytes. Cytosolic metabolism of 1,2-dibromoethane was not
affected by deuterium substitution. Both compounds caused DNA
single-strand breaks, as measured by the alkaline elution technique,
when incubated at a concentration of 0.1 mM with hepatocytes. No
difference in the degree of DNA damage was demonstrated between
hepatocytes incubated with 1,2-dibromoethane and those incubated with
d4-1,2-dibromoethane.
1,2-Dibromoethane can be metabolized by freshly isolated rat
hepatocytes to S-(2-hydroxyethyl)glutathione, S-(carboxymethyl)
glutathione and S,S'-(1,2-ethanediyl)bis(glutathione). These three
metabolites account for 84% of the total intracellular glutathione
depletion (Jean & Reed, 1992). These reactions were negligible in the
presence of rat glutathione- S-transferase, but conjugation was
catalysed by the rat alpha class enzyme 2-2 and, to a lesser extent,
the rat µ class enzyme 3-3. Of the three classes of human cytosolic
glutathione- S-transferases, 1,2-dibromoethane conjugation was
catalysed by the alpha class enzymes (Cmarik et al., 1990).
Human fetal liver appears to be especially active (several times
higher specific activity of glutathione- S-transferase, compared to
adult liver, as reported by Wiesma et al., 1986) in metabolizing
1,2-dibromoethane in vitro (Kulkarni et al., 1992).
1,2-Dibromoethane-induced lipid peroxidation and cytotoxicity
were increased upon concomitant exposure to carbon tetrachloride.
Similarly, the amount of 1,2-dibromoethane metabolites bound
covalently to proteins was enhanced. The effect of carbon
tetrachloride has been related to a shift in the 1,2-dibromoethane
metabolism from GSH-dependent to P-450-dependent (Chiarpotto et al.,
1993).
Oral administration of large doses of 1,2-dibromoethane
(37.6 mg/animal) to male Wistar rats (weighing around 200 g),
following a single dose of disulfiram (12 mg/kg), led to decreased
excretion of the mercapturic acid metabolite, a phenomenon associated
with a decrease in cytochrome P-450 levels (van Bladeren et al.,
1981a). In an additional reaction, 1,2-dibromoethane is debrominated
by an oxidative process catalysed by an enzyme in hepatic microsomes.
This system requires NADPH and oxygen and is inducible by
phenobarbital but not by methyl-cholanthrene (Hill et al., 1978).
Simula et al. (1993) reported an increased mutagenicity of
1,2-dibromoethane in the Salmonella typhimurium strain TA100
expressing human glutathione- S-transferase A1-1, indicating that
human glutathione- S-transferases are able to metabolize
1,2-dibromoethane to reactive intermediates.
In a study with isolated human hepatocytes (Cmarik et al., 1990),
it was found that concurrent treatment with diethylmaleate reduced the
intracellular glutathione level and inhibited 1,2-dibromoethane
concentration-dependent unscheduled DNA synthesis.
6.4 Elimination and excretion in expired air, faeces and urine
When 14C-1,2-dibromoethane (30 mg/kg body weight) was given
intraperitoneally to male guinea-pigs of the Hartley strain, 66% of
the radioactivity was excreted in the urine within 72 h of
administration (Plotnick & Conner, 1976). Faecal excretion was
relatively insignificant, representing less than 3% of the dose. The
excretion of unchanged 1,2-dibromoethane in the expired air was
significant (10-12% of dose).
Plotnick et al. (1979) reported that urinary extraction of
radioactivity from male rats of the Sprague-Dawley strain, 24 h after
a single oral dose of 14C-1,2-dibromoethane (15 mg/kg), was 72.4%.
Faecal radioactivity was 1.7%. Concomitant exposure to dietary
disulfiram significantly depressed urinary excretion of
1,2-dibromoethane.
6.5 Retention and turnover
Following intraperitoneal administration of [1,2-14C]-
1,2-dibromoethane (40 mg/kg) to RF/Hiraki mice, the circulating
radiolabel was mainly accounted for by S-(2-hydroxyethyl)
cysteine- N-acetate. Less than 1% of the dose was present in the
blood as a volatile component (Edwards et al., 1970).
Jakobson et al. (1982) reported that the elimination curve for
1,2-dibromoethane from blood after a 4-h dermal exposure of
guinea-pigs was non-linear and corresponded to a kinetic model
involving at least two compartments.
6.6 Reaction with body components
Radioactivity from [1,2-14C]-1,2-dibromoethane is bound
irreversibly to macromolecules in rat tissues after intraperitoneal
injection. For protein, DNA, and RNA, the largest amounts of bound
radioactivity were found to be present in the liver and kidney. Lung,
testis, stomach and the large and small intestines showed less
radioactivity (Hill et al., 1978).
Ozawa & Guengerich (1983) reported the formation of an
S-[2-(N7-guanyl)ethyl]glutathione adduct. 1,2-Dibromoethane and
GSH were irreversibly bound to calf thymus DNA in equimolar amounts
when in vitro incubation was carried out in the presence of
glutathione- S-transferase. The labelled DNA was enzymatically
digested to deoxyribonucleosides and separated by HPLC. The level of
adducts in DNA isolated from human hepatocytes incubated with 0.5 mM
1,2-dibromoethane was about 40% of the value obtained for rat
hepatocytes (Cmarik et al., 1990).
S-[2-(N7-guanyl)ethyl]glutathione was the only major DNA adduct
formed in vivo in rat (male Sprague-Dawley) liver (1.3 nmol/mg DNA)
or kidney (0.95 nmol/mg DNA) 8 h after intraperitoneal administration
of 37 mg 1,2-dibromoethane/kg body weight. The in vivo half-life of
S-[2-(N7-guanyl)ethyl] glutathione in rat liver, kidney, stomach
and lung was estimated to be between 70 and 100 h (Inskeep et al.,
1986).
Following intraperitoneal injection of 1,2-dibromoethane
(37 mg/kg) in rats and mice of several strains, it was found that more
of the S-[2-(N7-guanyl)ethyl]glutathione adduct of DNA was formed
in the livers of rats than in those of mice (Kim & Guengerich, 1990).
Incubation of 1,2-dibromoethane with calf thymus DNA and cytosol from
rats or mice did not result in different adduct levels, whereas the
level of adduct formation by human liver cytosol was about half of
those values for rats or mice. Induction of glutathione-
S-transferase in rat liver by phenobarbital or ß-naphthoflavone did
not increase DNA adduct levels, whereas cytochrome P-450 inhibition by
disulfiram did increase DNA adducts without altering the transferase
activity. Depletion of reduced glutathione in vivo by
diethylmaleate correlated with a reduction in DNA adduct levels.
Bromine atoms generated upon reductive degradation of
1,2-dibromoethane have been shown to react with polyunsaturated fatty
acids via both abstraction of bisallylic hydrogen and addition to the
double bond. Bromine atoms may be a potential initiator for lipid
peroxidation and provide a chemical basis for the toxic action of
1,2-dibromoethane (Guha et al., 1993).
The cytotoxic action of 1,2-dibromoethane has been studied by
Khan et al. (1993). They found that both cytochrome P-450- and
GSH-dependent metabolism of 1,2-dibromoethane contributed to its
cytotoxic effect in hepatocytes. Antioxidants or removal of oxygen
delayed the cytotoxicity. Furthermore, cytotoxicity could be
increased markedly if aldehyde dehydrogenase was inhibited with
disulfiram. In addition, cytotoxicity could be reduced if the
hepatocytes were depleted of GSH before the addition of
1,2-dibromoethane.
1,2-Dibromoethane has also been shown to cause cytotoxicity in
rabbit pulmonary cells (Nichols et al., 1992), being cytotoxic to
Clara cells, type II cells and alveolar macrophages.
The irreversible binding of radioactivity from [1,2-14C]-
1,2-dibromoethane to protein, DNA and RNA in rats was measured 24 h
after an intraperitoneal injection of 14C-1,2-dibromoethane
(2.6-3.2 mg/kg) (Hill et al., 1978). For each of these classes of
macromolecules, the largest amounts of bound radioactivity were found
in the liver and kidneys.
Botti et al. (1982) evaluated the effect of 1,2-dibromoethane on
GSH levels and cytosolic glutathione- S-transferase activity
following administration by gavage to male rats. Doses of either 75
or 150 mg/kg were found to decrease GSH and glutathione- S-
transferase activities. Maximal effects were observed 2 h following
exposure, although a significant decrease in GSH levels was observed
within 15 min. Mann & Darby (1985) also noted GSH depletion in both
male and female rats, the maximum effect occurring 2 h after an
intraperitoneal 1,2-dibromoethane dose of 80 mg/kg. A greater effect
was observed in males.
Brandt et al. (1987) used autoradiography and 14C-labelled
1,2-dibromoethane to study the tissue distribution of
1,2-dibromoethane following an intraperitoneal injection to a
cynomolgus monkey. 1,2-Dibromoethane was found to bind preferentially
to the liver and renal tubules, particularly the adrenal zona
reticularis.
Kaphalia & Ansari (1992) measured the rate of incorporation of
label into albumin and other large plasma proteins following exposure
to [14C]-1,2-dibromoethane either in vivo or in vitro. About
37% of the total label, administered by gavage (25 mg/kg body weight
in mineral oil over a 12-day period), was estimated to be bound to
plasma proteins, the majority (72%) being bound to albumin. In the
case of in vitro exposure, the incorporation of label to human
albumin or plasma after incubation with radioactive 1,2-dibromoethane
could be increased by the addition of either microsomal enzymes or an
NADPH-generating system.
7. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
Toxic effects of 1,2-dibromoethane have been mainly observed in
the liver and kidneys. Inhaled 1,2-dibromoethane vapour produces
nasal irritation and depression of the central nervous system. In
solution, 1,2-dibromoethane causes skin irritation on the shaved
abdomen, and eye irritation.
7.1.1 Oral
7.1.1.1 Rat
1,2-Dibromoethane (99% pure) in olive or corn oil was given by
gavage to albino rats, guinea-pigs, rabbits, mice and chickens. The
difference in LD50 of 1,2-dibromoethane in male and female rats was
statistically significant; rabbits appeared to be the most sensitive
and mice the least sensitive (Rowe et al., 1952) (Table 13).
Adult male albino rats (weight 140-160 g) were given 110 mg
1,2-dibromoethane/kg in olive oil by gavage and were killed 2, 4, 8,
12, 17 or 22 h later. During the first 4 h, no changes in the liver
were detectable by light microscopy. From 8 h after administration,
1,2-dibromoethane induced sinusoidal dilatation and centrilobular
necrosis in the liver (Broda et al., 1976).
7.1.1.2 Chicken
Leghorn chickens of both sexes were given 1,2-dibromoethane
(110 mg/kg body weight) in soybean oil by gavage. There was an
increase in liver weight and NAD concentration, and a decrease in
liver and blood alkaline phosphatase activity (Nachtomi et al., 1968).
Five-week-old male Leghorn chicks (weight 580-620 g) were given
1,2-dibromoethane (110 mg/kg body weight) in olive oil by gavage and
were killed 8, 12 or 22 h later. The central areas of the liver were
not changed, but portal areas were affected by 1,2-dibromoethane. The
concentration of eosinophilic granulocytes was much greater in
1,2-dibromoethane-treated livers than in controls (Broda et al.,
1976).
7.1.2 Inhalation
7.1.2.1 Rat
When rats were exposed to concentrations of 770, 1540, 3080,
6610, 12 300, 23 100, 38 500 or 77 000 mg/m3 (20 males and females
per group in most concentrations, and no control group) for durations
Table 13. Acute toxicity of 1,2-dibromoethane
Species Route Vehicle Parameter Value Reference
Rat (M) oral olive oil LD50 146 mg/kg Rowe et al. (1952)
Rat (F) oral olive oil LD50 117 mg/kg Rowe et al. (1952)
Rat (M,F) oral corn oil LD50 140 mg/kg McCollister
et al. (1956)
Mouse (F) oral olive oil LD50 420 mg/kg Rowe et al. (1952)
Rabbit (F) oral olive oil LD50 55 mg/kg Rowe et al. (1952)
Guinea-pig oral olive oil LD50 110 mg/kg Rowe et al. (1952)
(M,F)
Chicken (M,F) oral olive oil LD50 79 mg/kg Rowe et al. (1952)
Rabbit skin LD50 450 mg/kg Rowe et al. (1952)
Rat inhalation vapour by LC50 4620 mg/m3 Rowe et al. (1952)
aeration 1 h
Rat (M,F) inhalation vapour by LC50 2304 mg/m3 McCollister
aeration 4 h et al. (1956)
Mouse (ICR) intraperitoneal corn oil LD50 205 mg/kg Kluwe et al.
(1981)
ranging from 0.01 to 16.0 h (exposure time varied at different
concentrations), slight anaesthetic actions and depression of the
central nervous system were observed in rats exposed to 1540 mg/m3
or more. Deaths occurred within 24 h at concentrations between 1540
and 3080 mg/m3, related to exposure duration, due to respiratory or
cardiac failure. The LC50 concentration for a 2-h exposure was
3080 mg/m3. Deaths occurring from exposures at lower concentrations
were almost always delayed, sometimes as long as 12 days after
exposure. The majority of these deaths were due to pneumonia. The
animals usually lost weight, appeared rough and unkempt, became
irritable, had a bloodstained nasal discharge, and died. Animals
surviving the exposure at the lower concentrations exhibited a typical
progression of symptoms for several days before recovery took place.
Rats exposed to concentrations producing mortality, which were
sacrificed and autopsied 16-24 h after exposure, showed an increased
weight of lungs, liver and kidneys. The lungs showed congestion,
oedema, haemorrhages and inflammation; the liver cells had cloudy
swelling, centrilobular fatty degeneration and necrosis; the kidneys
showed slight interstitial congestion and oedema, with slight cloudy
swelling of the tubular epithelium in some cases (Rowe et al., 1952).
7.1.2.2 Guinea-pig
All guinea-pigs (20 males and females per group) exposed to 1540
or 3080 mg/m3 (no control group) for 2 to 7 h died, whereas all
those exposed to 770 mg/m3 for 7 h or 1540 mg/m3 for 2 h survived
(Rowe et al., 1952).
7.1.3 Intraperitoneal injection
7.1.3.1 Mouse
Intraperitoneal injection (46.8 and 93.7 mg/kg; 0.25 and
0.5 mmol/kg) of 1,2-dibromoethane (> 99.9%) in corn oil in male
B6C3F1 mice (weight 20-26 g) produced hepatic damage. The mice were
killed 4 h later and in vivo genotoxicity was determined by a
sensitive in vivo/in vitro alkaline DNA unwinding assay for the
presence of single-strand breaks and/or alkali-labile sites in hepatic
DNA. Significant hepatic DNA damage was found with a dose of
0.5 mmol/kg. In an assessment of the acute hepatotoxicity and
nephrotoxicity of 1,2-dibromoethane, male B6C3F1 mice were given
intraperitoneal injections (93.7, 140, 187.7 or 281.6 mg/kg; 0.5,
0.75, 1.0 or 1.5 mmol/kg) of 1,2-dibromoethane in corn oil and
sacrificed 24 h later. Serum L-iditol dehydrogenase (IDH), alanine
aminotransferase (ATT), and blood urea nitrogen were determined. At a
dose of 187.7 mg/kg 1,2-dibromoethane produced statistically
significant increases in relative liver and kidney weights, serum IDH
and ATT levels, and blood urea nitrogen levels. Four out of five
animals given a dose of 281.6 mg/kg died (Storer & Conolly, 1983).
7.1.3.2 Rat
Adult male Fischer-344 rats were given a single intraperitoneal
injection of 99% 1,2-dibromoethane (50 mg/kg; 0.27 mmol/kg) in corn
oil; they were sacrificed 2, 12, 24, 48 or 96 h later and the kidneys
were removed. Histopathological alterations in the kidney were most
prominent 48 h after 1,2-dibromoethane injection, and consisted of
acute proximal tubular degeneration (proximal tubular swelling and
vacuolation) (Kluwe et al., 1982).
7.2 Short-term exposure
7.2.1 Oral
7.2.1.1 Chicken
In a study by Schlinke (1970), 1,2-dibromoethane (formulation
which contains 83% of active ingredient) was administered orally at
doses of 50, 100 or 200 mg/kg per day to groups of five unsexed SPF
White Leghorn chickens (6-7 weeks old) for 10 days. There was an
untreated control group. All five chickens given 200 mg/kg per day
showed lack of appetite and depression, and died after the third dose.
Chickens which died had inflamed crops, excess pericardial fluid, and
congestion of the liver. Chickens given 50 or 100 mg/kg per day
showed no toxic effects.
7.2.2 Inhalation
7.2.2.1 Mouse
B6C3F1 mice (10 males and 10 females per group) were exposed by
inhalation to 23.1, 115.5 and 577.5 mg/m3 (3, 15 and 75 ppm) of
1,2-dibromoethane (6 h/day, 5 days per week) for 13 weeks. Four male
mice in the low dose group died before the end of the exposure period.
At 13 weeks, mice showed severe necrosis and atrophy of the olfactory
epithelium in the nasal cavity after inhaling the highest
concentration. Lower concentrations induced squamous cell metaplasia,
hyperplasia and cytomegaly of the epithelium of the respiratory nasal
turbinals. Squamous metaplasia, hyperplasia and cytomegaly of the
epithelium were also seen in larynx, trachea, bronchi and bronchioles.
The NOEL, based on histopathological alterations in the nasal cavity,
was 23.1 mg/m3 (3 ppm) (Reznik et al., 1980).
7.2.2.2 Rat
A group of 10 female rats (strain unknown) exposed to a
concentration of 768 mg/m3 (100 ppm) of 1,2-dibromoethane vapour for
7 h/day, lost weight steadily and three died after 1, 5 and
7 exposures, respectively. Surviving rats were thin and unkempt after
7 exposures in 9 days. At autopsy the stomachs were full of food, and
the contents were bloodstained. Lung, liver and kidney weights were
significantly elevated. Microscopic examination revealed some
thickening of the alveolar walls, with slight leukocytic infiltration
of the lungs, widespread cloudy swelling of the liver (but no fatty
degeneration), and slight congestion and haemosiderosis of the spleen
(Rowe et al., 1952).
F-344 rats (five males and five females per group) were exposed
by inhalation to 23.1, 115.5 and 577.5 mg/m3 (3, 15 and 75 ppm) of
1,2-dibromoethane (6 h/day, 5 days per week) for 13 weeks. At 13
weeks, they showed severe necrosis and atrophy of the olfactory
epithelium in the nasal cavity after inhalation of 577.5 mg/m3.
Lower concentrations induced squamous cell metaplasia, hyperplasia and
cytomegaly of the epithelium of the respiratory nasal turbinals.
Squamous metaplasia, hyperplasia and cytomegaly of the epithelium were
also seen in the larynx, trachea, bronchi and bronchioles. Other
compound-related toxic lesions in rats were seen in the liver, kidney
and testes. At 115.5 mg/m3, 1,2-dibromoethane induced only minor
changes in the nasal cavity. No lesions were seen in other tissues.
The NOEL based on histopathological alterations in the nasal cavity
was 23.1 mg/m3 (3 ppm) (Reznik et al., 1980).
Nitschke et al. (1981) conducted a 13-week inhalation study on
1,2-dibromoethane in rats. Male and female F-344 rats were exposed to
0, 23, 77 or 307 mg/m3 (0, 3, 10 or 40 ppm) (6 h/day, 5 days per
week) for 13 weeks. Those exposed to 307 mg/m3 (40 ppm) of
1,2-dibromoethane exhibited a decrease in body weight gain, an
increase in liver and kidney weight, and hyperplasia and metaplasia of
the respiratory epithelium of the nasal turbinates. A slight
epithelial hyperplasia of the nasal tubinates was also noted at
77 mg/m3 (10 ppm). A recovery period of 88 days resulted in
regression of the lesions in all but one animal.
7.2.2.3 Guinea-pig
Guinea-pigs (8 of each sex per group) that were administered
385 mg/m3 (50 ppm) of 1,2-dibromoethane for up to 7 h, 57 times in
80 days, had decreased final body weight and increased lung, liver and
kidney weights. Microscopic examination of the tissues showed slight
central fatty degeneration in the liver, and slight interstitial
congestion and oedema, with some parenchymatous degeneration of the
tubular epithelium in the kidney. Blood urea nitrogen values were
normal. In females, the total lipid content in the liver revealed no
significant variation. Guinea-pigs (4-8 of each sex per group)
tolerated without toxic effects 145 exposures (7 h each) in 205 days
at a 1,2-dibromoethane concentration of 193 mg/m3 (25 ppm).
However, four out of eight males and two out of eight females died of
pulmonary infection during the experiment (Rowe et al., 1952).
7.2.2.4 Rabbit
When four female rabbits were exposed by inhalation to
770 mg/m3 (7 h/day) for 4 days, two of the rabbits died after the
second exposure. Microscopic examination of tissues revealed
widespread central fatty degeneration of the liver with areas of
necrosis. Rabbits given 59 exposures each lasting 7 h (385 mg/m3)
in 84 days showed no evidence of adverse effects except for a slight
increase in liver and kidney weights; those given 152 exposures
(each 7 h) (192.5 mg/m3) in 214 days showed no adverse effects
(Rowe et al., 1952).
7.2.2.5 Monkey
When one male and one female monkey were given 7-h exposures
(385 mg/m3) 49 times in 70 days, they appeared ill, nervous and
unkempt throughout the experiment. Liver weights were increased with
slight central fatty degeneration and increased total lipid values.
No significant changes were observed in other organs except for a
slight increase in kidney weight. Another pair (one male and one
female) of monkeys received 7-h exposures (193 mg/m3) 156 times in
214 days. This group showed no evidence of adverse effects and the
NOEL was established as 193 mg/m3 (25 ppm) (Rowe et al., 1952).
7.3 Eye and skin irritation
7.3.1 Rabbit
1,2-Dibromoethane (undiluted, 1% and 10% in propylene glycol,
quantity not stated) was introduced into both eyes of a rabbit and
after 30 seconds one eye was flushed for 3 min with copious amounts of
running water. Conjunctival irritation occurred in both eyes, and
there was slight superficial necrosis of the cornea. However, healing
was prompt and complete 12 days after exposure; there was no corneal
scarring and no apparent injury to the iris or the lens. A 1%
1,2-dibromoethane solution in propylene glycol elicited a response
very similar to undiluted 1,2-dibromoethane (Rowe et al., 1952).
A 1.0% solution of 1,2-dibromoethane in butyl carbitol acetate
was applied 10 times in 14 days to the rabbit ear and also to the
shaved abdomen where it was then protected by a bandage. On the ear,
it caused slight irritation (erythema and exfoliation), whereas on the
shaved abdomen there was marked irritation with erythema and oedema
progressing to necrosis and sloughing of the superficial layers of the
skin. Healing was complete without scarring within 7 days after
termination of exposure (Rowe et al., 1952).
7.4 Long-term exposure
Designs of long-term/carcinogenicity studies are given in Table
14 and tumorigenic effects seen in these studies are summarized in
Table 15.
7.4.1 Oral
7.4.1.1 Mouse
Groups of 50 male and 50 female B6C3F1 mice (5 weeks old) were
given technical-grade 1,2-dibromoethane (99.6% pure) in corn oil by
gavage on 5 consecutive days per week. The time-weighted average high
and low doses of 1,2-dibromoethane were 107 and 62 mg/kg per day for
Table 14. Carcinogenicity studies on 1,2-dibromoethane
Route Species Number of animals Sexa Dose and dosing scheduleb Durationc Reference
(strain) per group
Gavage
Mouse 50 M 62 mg/kg body weight TWAd 78 weeks(53) NCI (1978)
(B6C3F1) (treated) 107 mg/kg body weight TWAd 77 weeks(53)
F 62 mg/kg body weight TWAd 90 weeks(53)
107 mg/kg body weight TWAd 78 weeks(53)
20 M,F
(control)e
Rat 50 M 38 mg/kg body weight TWAd 49 weeks(47) NCI (1978)
(Osborn- (treated) 41 mg/kg body weight TWAd 49 weeks(34)
Mendel) F 37 mg/kg body weight TWAd 61 weeks(57)
39 mg/kg body weight TWAd 61 weeks(44)
20 M,F
(control)e
Drinking-water
Mouse 30 M 117 mg/kg body weight per day 15 months Van Duuren et
(B6C3F1) (treated) F 103 mg/kg body weight per day 17 months al. (1985)
50 M,F
(control)
Inhalation
Mouse 60 F 154 mg/m3 6 h/day, 5 days/week 6 months Adkins et al.
(A/J) (treated) 384 mg/m3 6 h/day, 5 days/week 6 months (1986)
60 F 154 mg/m3 6 h/day, 5 days/week 6 months
(control)e 384 mg/m3 6 h/day, 5 days/week 6 months
90 F
(treatead)
60
(control)
Table 14 (cont'd)
Route Species Number of animals Sexa Dose and dosing scheduleb Durationc Reference
(strain) per group
Mouse 50 M 77 mg/m3 6 h/day, 5 days/week 78 weeks NTP (1982)
(B6C3F1) (treated) 307 mg/m3 6 h/day, 5 days/week 78 weeks
50 F 77 mg/m3 6 h/day, 5 days/week 103 weeks
(treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks
50 M,F
(control)
Mouse 50 M 77 mg/m3 6 h/day, 5 days/week 103 weeks Stinson et
(B6C3F1) (treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks al. (1981)
50 F 77 mg/m3 6 h/day, 5 days/week 103 weeks
(treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks
50 M,F 104 weeks
(control)
Rat 50 M 77 mg/m3 6 h/day, 5 days/week 103 weeks NTP (1982)
(Fisher-344) (treated) 307 mg/m3 6 h/day, 5 days/week 88 weeks
50 F 77 mg/m3 6 h/day, 5 days/week 103 weeks
(treated) 307 mg/m3 6 h/day, 5 days/week 90 weeks
50 M,F
(control)
Rat 48 M,F disulfiramf (0.05% in diet) 18 months Wong et
(Sprague- (treated) ± EDB (119-167 mg/m3 TWAb) al. (1982)
Dawley) 7 h/day, 5 days/week
48 M,F
(control)
Table 14 (cont'd)
Route Species Number of animals Sexa Dose and dosing scheduleb Durationc Reference
(strain) per group
Dermal
Mouse 30 F 25 mg/mouse, 3 times/week 400-594 days Van Duuren et
(Ha:ICR (treated) 50 mg/mouse, 3 times/week al. (1979)
Swiss) 100 F
(control)g
a M = male, F = female
b TWA = time-weighted average
c observation period: treated and untreated weeks (exposed weeks)
d doses changed in the course of the experiment with intermitting untreated weeks
e the controls consisted of vehicle (corn oil) & untreated controls
f This combination was chosen to examine effects of disulfiram used for alcoholism control programmes on workers
who were exposed to 1,2-dibromoethane occupationally.
g controls consisted of vehicle (acetone) & untreated controls
Table 15. Summaries of results of carcinogenicity studies on 1,2-dibromoethane
Study Route Animal Statistically significant effects dose (numbers of animals with
effects/total numbers of animals)
NCI (1978) gavage mouse squamous cell carcinoma of the forestomach
control (M: 0/20, F: 0/20), 62 mg/kg (M: 45/50, F: 46/49)
107 mg/kg (M: 29/49, F: 28/50)
alveolar/bronchiolar adenoma
control (M: 0/20, F: 0/20), 62 mg/kg (M: 4/45, F: 11/43)
107 mg/kg (M: 10/47, F: 6/46)
rat squamous cell carcinoma of the forestomach
control (M: 0/20, F: 0/20), 38 mg/kg (M: 45/50), 37 mg/kg (F: 40/50)
41 mg/kg (M: 33/50), 39 mg/kg (F: 29/50)
hepatocellular carcinoma
control (M: 0/20, F: 0/20), 39 mg/kg (F: 6/48)
haemangiosarcoma
control (M: 0/20, F: 0/20), 38 mg/kg (M: 11/50), 41 mg/kg (M: 4/50)
Van Duuren et al. (1985) drinking- mouse squamous cell carcinoma
water 117 mg/kg (M: 26/30), 103 mg/kg (F: 22/30)
oesophageal papilloma
103 mg/kg (F: 3/30)
squamous cell papillomaa
115 mg/kg (M: 9/30, F: 10/30)
Adkins et al. (1986) inhalation mouse (A/J) i) pulmonary adenoma
(all females) control (F: 0/60), 154 mg/m3 (F: 60/60), 384 mg/m3 (F: 60/60)
ii) pulmonary adenoma
control (F: 0/60), 154 mg/m3 (F: 75/90), 384 mg/m3 (F: 90/90)
Table 15 (cont'd)
Study Route Animal Statistically significant effects dose (numbers of animals with
effects/total numbers of animals)
NTP (1982) inhalation mouse alveolar/bronchiolar carcinoma
control (M: 0/41, F: 1/49), 77 mg/m3 (M: 3/48, F: 5/49),
307 mg/m3 (M: 19/46, F: 37/50)
alveolar/bronchiolar adenoma
control (M: 0/41, F: 3/49), 77 mg/m3 (F: 7/49),
307 mg/m3 (M: 11/46, F:13/50)
haemangiosarcoma of the circulatory system
control (F: 0/50), 77 mg/m3 (F: 11/50), 307 mg/m3 (F: 23/50)
subcutaneous fibrosarcoma
control (F: 0/50), 77 mg/m3 (F: 5/50), 307 mg/m3 (F: 11/50)
nasal cavity carcinoma, or adenoma
control (F: 0/50), 307 mg/m3 (F: 6/50, 8/50)
mammary gland adenocarcinoma
control (F: 2/50), 77 mg/m3 (F: 14/50), 307 mg/m3 (F: 8/50)
inhalation rat nasal cavity carcinoma, adenocarcinoma, adenoma
control (M: 0/50, F: 0/50), 77 mg/m3 (M: 1/50, 20/50, 11/50, F: 0/50,
20/50, 11/50), 307 mg/m3 (M: 21/50, 0/50, 28/50, F: 25/50, 29/50, 3/50)
haemangiosarcoma of the circulatory system
control (M: 0/50, F: 0/50), 307 mg/m3 (M: 15/50, F: 5/50)
tunica vaginalis mesothelioma
control (M: 0/50), 77 mg/m3 (M: 7/50), 307 mg/m3 (M: 25/50)
alveolar/bronchiolar adenoma, carcinoma (combined)
control (F: 0/50), 307 mg/m3 (F: 5/47)
nasal cavity adenomatous polyps
control (M: 0/50), 77 mg/m3 (M: 18/50), 307 mg/m3 (M: 5/50)
mammary gland fibroadenoma
control (F: 4/50), 77 mg/m3 (F: 29/50), 307 mg/m3 (F: 24/50)
Table 15 (cont'd)
Study Route Animal Statistically significant effects dose (numbers of animals with
effects/total numbers of animals)
Stinson et al. (1981) inhalation mouse nasal cavity carcinoma (squamous carcinoma, adenocarcinoma)
control (M: 0/45, F: 0/50), 77 mg/m3 (M: 0/44, F: 0/49)
307 mg/m3 (M: 0/46, F: 7/49)
sarcoma
control (M: 0/45, F: 0/50), 77 mg/m3 (M: 0/44, F: 1/49)
307 mg/m3 (M: 0/46, F: 2/49)
benign neoplasms (squamous papilloma, adenoma)
control (M: 0/45, F: 0/50), 77 mg/m3 (M: 0/44, F: 0/49)
307 mg/m3 (M: 3/46, F: 7/49)
Wong et al. (1982) inhalation rat hepatocellular tumour
control (M: 0/48, F: 0/48), EDB (M: 3/46, F: 1/48)
EDB+DSb (M: 36/48, F: 32/48)
kidney (adenoma and adenocarcinoma)
control (M: 0/48, F: 0/48), EDB (M: 3/46, F: 1/48)
EDB+DSb (M: 17/48, F: 7/48)
Van Duuren et al. (1979) skin mouse lung tumours
control untreated (F: 30/100)
control acetone (F: 11/100)
25 mg/mouse (F: 24/30), 50 mg/mouse (F: 26/30)
skin carcinoma
control untreated (F: 0/100)
control acetone (F: 0/100)
25 mg/mouse (F: 2/30), 50 mg/mouse (F: 8/30)
a carcinogenicity study on bromoethanol
b DS: disulfirum given in diet
both sexes. All surviving male mice and high-dose female mice were
killed on week 78, and low-dose females were killed on week 90. In
low-dose females, body weight depression was apparent after the first
10 weeks. Soft faeces, alopecia and body sores were observed in all
surviving animals at week 14. In mice receiving the high dose,
acanthosis of the forestomach was observed in 5/49 males and 9/50
females, compared with 1/50 in low-dose females. Hyperkeratosis
occurred in the stomachs of 13/49 high-dose males, 12/50 high-dose
females, and 1/48 low-dose females. There was testicular atrophy
(10/47) related to compound administration in males receiving the high
dose (NCI, 1978).
7.4.1.2 Rat
Groups (50 of each sex per group) of Osborne-Mendel rats (8 weeks
old) were administered technical-grade 1,2-dibromoethane in corn oil
by gavage on five consecutive days per week. The time-weighted
average high and low doses of 1,2-dibromoethane in treated groups were
41 and 38 mg/kg per day for male rats, and 39 and 37 mg/kg per day for
females. Body weight depression was apparent in the treated rats
after the first 10 weeks. Reddened ears, a hunched appearance, firm
distended abdomens and abdominal urine stains were observed in treated
groups. There was high mortality in both the high- and low-dose
groups. All surviving treated male (24 out of 100) and female (3 out
of 100) rats were sacrificed on weeks 49 and 61, respectively. In
rats given the high dose, hyperkeratosis and acanthosis of the
forestomach were observed in 12/50 males and 18/50 females, and in
low-dose females the value was 4/50. In the treated rats,
degenerative changes in the liver and adrenal gland and early
development of testicular atrophy were reported (NCI, 1978). However,
total development of testicular atrophy in controls, low-dose group
and high-dose group were 11/20, 14/49 and 18/5, respectively.
7.4.2 Inhalation
7.4.2.1 Mouse
In an NTP carcinogenicity study (NTP, 1982), groups of 50 male
and 50 female B6C3F1 mice were exposed to 1,2-dibromoethane
(> 99% pure) concentrations of 0, 77 or 308 mg/m3 (0, 10, or
40 ppm) for 6 h/day, 5 days per week, for 78 to 103 weeks, throughout
the study. Mean body weights of high-dose male and female mice were
lower than those of the untreated controls. Survival of high-dose
female mice was significantly reduced compared with controls.
Survival was also reduced in both control and treated male mice, the
principal cause of death being an ascending suppurative urinary tract
infection unrelated to compound administration. Surviving male mice
were killed at 79 weeks, while female mice were killed at 104-106
weeks, except for the high-dose group (killed at 91 weeks). The NTP
also reported inflammation of the nasal cavity and epithelial
hyperplasia of the respiratory system in both sexes (NTP, 1982).
Stinson et al. (1981) reported similar findings in mice using
data from a 2-year study. Epithelial hyperplasia of the urinary
bladder and inflammation of the prostate gland were also observed in
dosed male mice.
7.4.2.2 Rat
Groups of 50 male and 50 female inbred Fischer-344 rats were
exposed to 1,2-dibromoethane (> 99% pure) concentrations of 0, 77 or
308 mg/m3 (0, 10 or 40 ppm) for 6 h/day, 5 days per week, for 78 to
103 weeks, throughout the study. Mean body weights of high-dose rats
were lower than those of untreated controls. Survival of high-dose
male and female rats was significantly reduced compared with the
controls. Surviving female and male rats were killed at 104-106
weeks, except for the male high-dose group (5 out of 50 alive, killed
at 89 weeks) and female high-dose group (8 out of 50 alive, killed at
91 weeks). Among the observed compound-related non-neoplastic lesions
were hepatic necrosis and toxic nephropathy in both sexes, testicular
degeneration and atrophy in males, and retinal degeneration in females
(NTP, 1982).
Four groups of 48 male and 48 female Sprague-Dawley rats were
exposed to either control air, 154 mg 1,2-dibromoethane/m3, 0.05%
disulfiram in the diet, or 20 mg 1,2-dibromoethane/kg and 0.05%
disulfiram in the diet for 18 months. Disulfiram which is used in
alcohol control programmes is known to inhibit acetaldehyde
dehydrogenase. It may therefore alter the biotransformation of
1,2-dibromoethane and cause the accumulation of 2-bromoacetaldehyde,
one of the possible toxic metabolites of 1,2-dibromoethane. The
average starting weights ranged from 131 to 134 g for male rats and
118 and 124 g for females. Between inhalation exposures (7 h/day,
5 days/week), rats were permitted free access to water and food. Rats
receiving 0.05% disulfiram showed reduced body weight gain compared
with control rats or those given 20 mg 1,2-dibromoethane/kg or the
control diet. Rats exposed to 154 mg/m3 of 1,2-dibromoethane alone
and those receiving a combination of 20 mg 1,2-dibromoethane/kg and
0.05% disulfiram had high mortality compared with control and
disulfiram-treated rats. In rats exposed by inhalation to 154 mg
1,2-dibromoethane/m3 alone, mortality was 90% in males and 77% in
females at 18 months. Haematological parameters were within the
normal range for moribund rats in this group. Male rats receiving
20 mg 1,2-dibromoethane/kg with 0.05% disulfiram had a high incidence
of testicular atrophy and there was atrophy of the spleen in 30/48
males and 19/48 females (Wong et al., 1982).
7.5 Developmental toxicity
1,2-Dibromoethane causes testicular effects and mating failure in
rats at high inhalation levels that result in mortality. It also
causes temporary malformation of sperm cells in bulls and rams, but
not in chickens. In laying hens, it causes decreased egg size. In
rats and mice there is no evidence of embryotoxicity and
teratogenicity. In rats, paternal exposure causes behavioural changes
in F1 progeny.
7.5.1 Reproduction
Male Sprague-Dawley rats (3-4 per group) were exposed by
inhalation to average daily concentrations of 146, 300 or 684 mg/m3
(19, 39 or 89 ppm) of 1,2-dibromoethane for 7 h/day, 5 days/week, for
10 weeks. There was reduced weight gain in the 300 and 684 mg/m3
groups with mortality (21%) and morbidity in the 684 mg/m3 group.
Animals in this group had reduced testicular weights, reduced serum
testosterone levels, and atrophy of the testes (10/10), epididymis
(10/10), prostate (10/10), and seminal vesicles (9/9). When mated
with untreated females none of the males exposed to 684 mg/m3
impregnated any females, while 90% of those exposed to the low and
intermediate concentrations impregnated females who produced litters
that were normal in terms of total implants, viable implants and
resorptions. In female Sprague-Dawley rats, inhaling average daily
concentrations of 154, 300 or 614 mg/m3 (20, 39 or 80 ppm) of
1,2-dibromoethane for 7 h/day, 7 days/week, for 3 weeks, there was
reduced weight gain, morbidity and mortality (20%) in the 614 mg/m3
group. At the end of the 3-week exposure, females were mated with
untreated males. Females in the 614 mg/m3 group were in diestrus
and did not cycle normally until 3-4 days later; consequently fewer
females mated during a 10-day mating period than in the case of the
other two groups. The vaginal smears from females exposed to 154 or
300 mg/m3 were normal. In all three groups the reproductive
performance (total implants/dam, viable implants/dam and resorptions/
dam) was unimpaired. Histopathological examination of the ovaries and
uterus did not reveal any significant lesions (Short et al., 1979).
It was considered that the NOEL for reproductive performance was
300 mg/m3 for male and female rats.
Weanling male albino rats (10 rats per group) were fed
1,2-dibromoethane in the diet at levels of 100 or 500 mg/kg
(equivalent to 10 or 50 mg/kg body weight per day) for 90 days. There
was no evidence of toxicity; serum enzyme activities were unchanged.
Five rats from each group were mated with untreated virgin females.
There was no impairment of reproductive performance in the male rats.
At the end of the 2-week mating period the males were sacrificed.
Histology of the testis was normal. The pregnant females were allowed
to go to term, and the mean number of litters per group, mean pup
weight at birth, and sex ratio were found to be similar to the values
for a control group mated with untreated males (Shivanandappa et al.,
1987). The NOEL for male rat reproductive performance was considered
to be 50 mg/kg body weight per day.
In a study of sperm quality and fertility, mature (12 months old)
male New Zealand White rabbits (8-10 group) were injected
subcutaneously with 1,2-dibromoethane in corn oil at dose levels of
15, 30 or 45 mg/kg body weight per day for 5 days. There were also
untreated and vehicle control groups. Male fertility was assessed
before exposure, and at 4 and 12 weeks after injection, by artificial
insemination of three females/male per time point with one million
motile sperm. The percentage of pregnant females, litter size, fetal
body weights and structural development were assessed. In the highest
dose group there was 30% mortality and liver damage in 43% of the
survivors, indicated by increased levels of serum enzymes. There were
also changes in some sperm parameters (see 7.5.1.1). The percentage
of pregnant females and mean litter sizes were similar to those
produced by sperm from vehicle control animals, demonstrating that
fertilizing capacity and gestational outcome were unaffected (Williams
et al., 1991).
7.5.1.1 Effects on sperm
Four bull calves of the Israel-Friesian breed were administered
1,2-dibromoethane orally at a dose of 2 mg/kg body weight from the age
of 4 days by adding 1,2-dibromoethane to milk or feed concentrates.
When the calves reached an age of about 12 months, 1,2-dibromoethane
was administered in gelatin capsules. The treatment did not affect
the growth or health of the treated animals, and their libido was
similar to that of untreated animals. However, sperm density in
treated bulls was low, and sperm motility was poor. Semen showed
abnormally shaped spermatozoa (tailless, coiled tail, pyriform head).
Recovery after discontinuation of treatment varied from 10 days to
about 3 months in different animals. In a further study,
1,2-dibromoethane (4 mg/kg body weight) was administered orally to a
previously untreated bull. Two weeks after the start of the treatment
the semen exhibited abnormalities (Amir & Volcani, 1965). Other
studies also showed that 1,2-dibromoethane caused reversible
abnormalities of sperm cells in bulls (Amir, 1973, 1975; Amir et al.,
1979; Courtens et al., 1980) and in rams (ElJack & Hrudka, 1979), but
not in chickens (Alumot et al., 1968).
In a study by Williams et al. (1991), male New Zealand White
rabbits (8-10/group) were injected subcutaneously with
1,2-dibromoethane in corn oil (0, 15, 30 or 45 mg/kg body weight for
5 days). Weekly semen samples (for 6 weeks before exposure, during
treatment and 12 weeks after dosing) were analysed for sperm
concentration, number, morphology, viability and motion parameters
(velocity, linearity, beat cross-frequency, amplitude of lateral head
displacement (ALH) and circularity), and for semen pH, osmolality,
volume, and levels of fructose, citric acid, carnitine, protein and
acid phosphatase (AP). In the 45-mg/kg dose group, 1,2-dibromoethane
produced significant decreases in sperm velocity, percentage motility
and ALH (up to 25% at various times after dosing). There were also
dose-related decreases in semen pH (up to 2%) and total ejaculate
volume (up to 23%, 15 and 30 mg/kg groups only). Acid phosphatase
activities were significantly elevated (up to 116%) 2 weeks after
dosing in the 45 mg/kg dose group. All other semen parameters
evaluated were unaffected. Rabbits appear less sensitive than humans
to the reproductive effects of 1,2-dibromoethane, since semen
parameters were affected only at doses close to the LD50 and some
parameters (sperm numbers, viability and morphology) were unaffected.
A NOEL was not obtained in this study.
7.5.1.2 Effects on ova
Feeding studies with laying hens showed that 1,2-dibromoethane
absorbed by grain adversely affected egg production. When hens were
fed grain containing 200 mg/kg (corresponding to 25 mg/kg per day) for
56 days or grain containing 300 mg/kg (corresponding to 38 mg/kg per
day) for 46 days, the hens ceased laying completely. Feeding of grain
containing 10 mg/kg (corresponding to 12.5 mg/kg per day) caused a
diminution of egg size after 12 weeks (Bondi et al., 1955).
7.5.2 Teratogenicity
Pregnant Sprague-Dawley rats and CD-1 mice inhaled
1,2-dibromoethane concentrations of 146, 292 or 614 mg/m3 (20, 38 or
80 ppm) (23 h/day for 10 days) from day 6 to day 15 of gestation.
Adverse effects on maternal animals, measured by body weight gain and
food consumption, were observed in both species at all doses tested.
A marked increase in maternal mortality occurred in rats exposed to
614 mg/m3 and in mice exposed to 292 or 614 mg/m3. Some
morphological changes, such as haematomas, exencephaly and skeletal
variations, were observed in the fetuses of rats and mice. However,
these changes occurred only at high concentrations that caused
maternal toxicity (Short et al., 1978).
The embryotoxic effects of 1,2-dibromoethane bioactivation,
mediated by purified rat liver glutathione- S-transferases (GST),
were investigated using rat embryos in culture (Mitra et al., 1992).
Significant 1,2-dibromoethane metabolism was observed with rat liver
GST purified by affinity chromatography. 1,2-Dibromoethane activation
caused a significant reduction in general development as measured by
crown-rump length, yolk sac diameter, somite number, and the composite
score for different morphological parameters. Structures most
significantly affected were the central nervous and olfactory systems
as well as the yolk sac circulation and allantois. The results of
this study clearly indicate that under in vitro conditions,
bioactivation of 1,2-dibromoethane by GST can lead to embryotoxicity.
GST isozymes from human fetal liver were purified and used to
investigate the toxicity of 1,2-dibromoethane in an in vitro model
of rat embryos in culture as passive targets (Mitra et al., 1992).
1,2-Dibromoethane bioactivation by the GST isozyme P-3 resulted in
toxicity to cultured rat embryos. Significant reductions in crown
rump length, yolk sac diameter, and the composite score of
morphological parameters were observed. The central nervous, optic
and olfactory systems, and the hind limb were most significantly
affected.
When pregnant Sprague-Dawley rats were injected intraperitoneally
with 1,2-dibromoethane at a dose of 50 mg/kg body weight from day 1 to
day 15 of gestation, there was no evidence of embryotoxicity or
teratogenicity, although there were maternal toxic effects (change in
organ weights) (Hardin et al., 1981).
7.5.2.1 Effects on neonatal behaviour
Pregnant Long-Evans rats (16 animals/group, litter size 8-10)
were exposed to 3.3, 51.2 or 512 mg/m3 by inhalation (4 h/day,
3 days/week) from day 3 to day 20 of gestation. The highest
concentration produced enhanced rotorod performance and T-maze
brightness discrimination acquisition in the offspring. Similar
behavioural changes were noted in the offspring of mothers exposed to
51.2 mg/m3, but the magnitude of the effect was reduced. Exposure
to 3.3 mg/m3 produced no effects. DRL-20 acquisition (differential
reinforcement of low rates), straight alley running speed, and passive
avoidance were not affected at any dose level (Smith & Goldman, 1983).
In a study by Fanini et al. (1984), adult male Fischer-344 rats
were injected intraperitoneally with 1,2-dibromoethane at daily doses
of 0, 1.25, 2.5, 5 or 10 mg/kg body weight for 5 days. The treated
males were then mated with untreated female rats 4 or 9 weeks after
treatment. A total of 19 litters composed of 172 animals, 84 males
and 88 females were obtained from breeding the exposed males with
untreated females. Behavioural assessments of all F1 progeny were
carried out up to 21 days of age. Assessment of behavioural
development was made by means of an extensive testing battery.
Pre-weaning behavioural assessment included simple reflexes (surface
righting, cliff avoidance and negative geotaxis), motor coordination
(e.g., swimming and open field activity) and locomotor activity.
Significant impairment in the development of motor coordination and
motor activity was observed in the F1 progeny of males of all
treated groups. A NOEL was not found in this study.
The effects of 1,2-dibromoethane exposure on several
neurotransmitter enzymes in male rats were examined in various brain
regions of the F1 progeny (from 7 to 90 days of age) (Hsu et al.,
1985). Significant increase of choline acetyltransferase in the
cerebellum, corpus striatum, hippocampus and hypothalamus, alterations
of acetylcholinesterase in various brain regions, and an increase of
glutamic acid decarboxylase activity in the corpus striatum may be
related to early-development behavioural abnormalities.
7.6 Mutagenicity and related end-points
Mutagenicity assays are summarized in Table 16.
Table 16. Mutagenicity studies on 1,2-dibromoethane
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
In vitro
Gene mutation Salmonella E503 - Alper & Ames (1975)
typhimurium G46(-S9) + Ames & Yanofsky (1971); Von Buselmaier et al. (1972)
TA1530(-S9) + Ames & Yanofsky (1971); Brem et al. (1974); Rosenkranz
(1977); Buijs et al. (1984)
TA1535(+/-S9) +l Brem et al. (1974); McCann et al. (1975); Rannug & Beije (1979);
Shiau et al. (1980); Barber et al. (1981); Principe et al. (1981);
Moriya et al. (1983); Buijs et al. (1984); Dunkel et al. (1985);
Tennant et al. (1986, 1987); Zoetemelk et al. (1987); Barber &
Donish (1982)
TA98(+/-S9) +l Barber et al. (1981); Moriya et al. (1983);Dunkel et al. (1985);
Tennant et al. (1986)
TA100(+/-S9) +l McCann et al. (1975); Barber et al. (1981); Stolzenberg & Hine
(1980); van Bladeren et al. (1980, 1981b); Principe et al. (1981);
Moriya et al. (1983); Buijs et al. (1984); Dunkel et al. (1985);
Kerklaan et al. (1985); Guobaitis et al. (1986);
Tennant et al. (1986);
Hughes et al. (1987); Zoetemelk et al. (1987); Barber & Donish (1982)
TA1535(GSH-) + Kerklann et al. (1983)
(-S9,+ GSH) Zoetemelk et al. (1987)
TA100(GSH-) + Kerklaan et al. (1985)
(-S9,+GSH) Zoetemelk et al. (1987)
TA100W(Str',8AGr')(-S9) +g Ong et al. (1989)
TA1535 (bile of rats) + Rannug & Beije (1979)
TA1537(+/-S9) - Principe et al. (1981); Moriya et al. (1983); Dunkel et al. (1985);
Tennant et al. (1986)
TA1538(+/-S9) - Brem et al. (1974); Principe et al. (1981); Moriya et al. (1983);
Dunkel et al. (1985)
TA98 (+/-S9) - Principe et al. (1981); Wildeman & Nazar (1982)
TA100(+/-S9) - Shiau et al. (1980); Wildeman & Nazar (1982)
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Serratia a21 (-S9) - Von Buselmaier et al. (1972)
marcescens
Escherichia coli WP2 (+/-S9) + Scott et al. (1978); Hemminki et al. (1980); Moriya et al. (1983);
Dunkel et al. (1985)
CHY832 (-S9) + Hayes et al. (1984)
343/286 (+/-S9) + Mohn et al. (1984)
KI201, KI211 (-S9) + Izutani et al. (1980)
uvrB5 + Foster et al. (1988)
343/113 (-S9) - Mohn et al. (1984)
Bacillus subtilis TKJ5211, TKJ6321 (+S9) + Shiau et al. (1980)
Streptomyces (-S9, spot test) + Principe et al. (1981)
coelicolor (-S9, plate method) - Principe et al. (1981)
Aspergillus methG1BiA1 + Scott et al. (1978)
nidulans (+/-plant extract)
haploid strain 35 (-S9) + Principe et al. (1981)
Neurospora crassa ad-3 (forward mutation) + de Serres & Malling (1983)
Tradescantia clone 02, 0106, 4430 +g Sparrow et al. (1974); Nauman et al. (1976);
Vant'Hof & Schairer (1982)
Mouse L5178Y (+/-S9) + Clive et al. (1979); Tennant et al. (1986, 1987)
Chinese hamster CHO-K1 (+/-S9) + Tan & Hsie (1981); Brimer et al. (1982)
Human cell line AHH-1, TK6 + Crespi et al. (1985)
(-S9)
Human cell line EUE (-S9) + Ferreri et al. (1983)
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Unscheduled Rat hepatocytes + Williams et al. (1982); Tennant et al. (1986)
DNA synthesis
Opossum lymphocytes + Meneghini (1974)
Human lymphocytes (+/-S9) + Perocco & Prodi (1981)
Mouse (C3Hfx101)F1, + Sega & Rene (1980)
germ cells
Sister-chromatid Fish lymphocytes (- S9) + Ellingham et al. (1986)
exchange Chinese hamster V79 cl-15 (-S9) + Tezuka et al. (1980)
Chinese hamster CHO (+/-S9) + Tennant et al. (1987)
Human lymphocytes (-S9) +g Ong et al. (1989); Tucker et al. (1984)
Chromosome Fish lymphocytes (- S9) + Ellingham et al. (1986)
aberrations Chinese hamster V79 cl-15 (-S9) + Tezuka et al. (1980)
CHO (+/-S9) + Tennant et al. (1987)
Micronuclei Tradescantia clone 03, 4430 +l Ma et al. (1978, 1984)
DNA damage E. coli polA1-/polA+ (-S9) +w Brem et al. (1974)
B. subtilis TKJ5211, TKJ6321 - Shiau et al. (1980)
(+/-S9)
SOS induction S. typhimurium TA1535/pSK1002 +g Ong et al. (1987)
(+/-S9)
E. coli PQ37 (-S9) + Ohta et al. (1984); Quillardet et al. (1985)
Mitotic gene Saccharomyces ade2, trp5 + Fahrig (1974)
conversion cerevisiae
Somatic A. nidulans diploid 35 x 17 (-S9) +g Crebelli et al. (1984)
segregation
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
DNA binding E. coli Q13 (+/-S9) - Kubinski et al. (1981)
Mouse Ehrlich ascites (+/-S9) - Kubinski et al. (1981)
Cell Human lymphocyte + Channarayappa et al. (1992)
proliferation
DNA strand Rat hepatocytes + Sina et al. (1983)
breaks
Cell Mouse Balb/c 3T3 (-S9) - Tennant et al. (1986); Perocco et al. (1991)
transformation
In vivo
Gene mutation S. typhimurium G46 (host-mediated) + Von Buselmaier et al. (1972)
Serratia a21 (host-mediated) - Von Buselmaier et al. (1972)
marcescens
Barley + Ehrenberg et al. (1974)
Silkworm (egg color mutation) - Sugiyama (1980)
Drosophila (wing spot) +g Graf et al. (1984)
melanogaster
Recombination D. melanogaster (wing spot) +g Graf et al. (1984)
Sex-linked D. melanogaster +l Kale & Baum (1979a,b, 1981, 1982, 1983); Yoshida & Inagaki
recessive lethal (1986); Vogel & Chandler (1974)
mutations D.melanogaster spermatozoa + Ballering et al. (1993)
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Specific Mouse - Russell (1986)
locus test Mouse DBA/2J - Barnett et al. (1992)
Chromosome Barley root tips + Ehrenberg et al. (1974)
aberration Mouse (ip) CD1, (bone marrow) - Krishna et al. (1985)
Sister-chromatid Mouse (ip) CD1, bone marrow - Krishna et al. (1985)
exchange
Dominant lethal Rat (ih, po) CD, SD - Short et al. (1979); Teramoto et al. (1980)
Mouse (po) BDF1 - Teramoto et al. (1980)
Mouse (ip, po) ICR/Ha - Epstein et al. (1972)
Mouse DBA/2J - Barnett et al. (1992)
DNA strand Rat (po) hepatocytes + Nachtomi & Sarma (1977)
breaks Mouse (ip) hepatocytes + Storer & Conolly (1983)
breaks Mouse (ip) hepatocytes + White (1982)
Micronuclei Amphibians: erythrocytes Fernandez et al. (1993)
Pleurodeles waltl +
(newt)
Ambystoma +
mexicanum (axolotl)
Anuran: Xenopus +
laevis (toad)
Mouse ddY(peripheral blood - Asita et al. (1992)
reticulocytes)
Mouse(ip) CD1, bone marrow - Krishna et al. (1985)
Tradescantia tetrads of + Ma et al. (1978)
microsporogenesis
Table 16 (cont'd)
Test method Species (route Strain/cell type Resultsb Reference
of administration)a
Sperm Bull Friesian + Amir et al. (1977)
abnormality
a ip = intraperitoneal administration; ih = inhalation exposure; iv = intravenous administration; po = oral administration
b g = test in gaseous phase; l = test in liquid or gaseous phase; w = weak positive results
7.6.1 In vitro assays
1,2-Dibromoethane was mutagenic in reverse mutation assays
(Ames test) using Salmonella typhimurium strains G46, TA1530,
TA1535, TA98, TA100 and TA100W, and in the host-mediated assay using
strain G46. Bile from mice and rats injected intraperitoneally with
1,2-dibromoethane or following liver perfusion was mutagenic in a test
using strain TA1535. Gene mutation assays using Escherichia coli,
strain WP2 and several other strains, were positive. A positive
response was reported in assays using Bacillus subtilis, Streptomyces
coelicolor, Aspergillus nidulans and Neurospora crassa.
A requirement for intracellular activation of 1,2-dibromoethane
was observed for a mutagenic response in S. typhimurium TA1535.
Glutathione- S-transferase 5-5 transfected into the bacteria allowed
the induction of base-pair mutations on exposure to 1,2-dibromoethane,
whereas a 20-fold extracellular excess of the enzyme did not permit
1,2-dibromoethane-induced mutations in non-transfected bacteria in the
presence of reduced glutathione (Thier et al., 1993).
1,2-dibromoethane failed to induce reverse mutations in a number
of assays using S. typhimurium E503, TA1537 and TA1538, with or
without S9 mix. A negative response was reported in several assays
using E. coli, Serratia marcescens and silkworms, and in the mouse
specific locus test.
Chromosomal aberrations were induced by 1,2-dibromoethane in
cultured fish lymphocytes (Ellingham et al., 1986), Chinese hamster
V79 and CHO cell lines (Tezuka et al., 1980; Tennant et al., 1987).
1,2-Dibromoethane was negative in a test for dominant lethal and
specific locus mutations in germ cells from DBA/2J male mice (Barnett
et al., 1992).
1,2-Dibromoethane increased significantly the frequency of sister
chromatid exchanges in cultured fish lymphocytes (Ellingham et al.,
l986), Chinese hamster cell line (V79) and cultured human lymphocytes
(Tucker et al., 1984; Ong et al., 1989), but not in bone marrow cells
of mice injected intraperitoneally (Krishna et al., 1985).
In isolated human mononucleated and binucleated peripheral
lymphocytes, 1,2-dibromoethane caused an increased frequency of
micronuclei in vitro (Channarayappa et al., 1992).
A positive response was obtained in the unscheduled DNA synthesis
assay using cultured rat hepatocytes (Williams et al., 1982; Tennant
et al., 1986), cultured opossum lymphocytes (Meneghini, 1974),
cultured human lymphocytes (Perocco & Prodi, 1981) and mouse germ
cells (Sega & Rene, 1980), and in the DNA strand break assay using rat
or mouse hepatocytes (Nachtomi & Sarma, 1977; White, 1982; Sina et
al., l983). Tests for DNA damage using E. coli (Brem et al., 1974),
SOS induction using S. typhimurium and E. coli (Ohta et al., 1984;
Quillardet et al., 1985; Ong et al., 1987), mitotic gene conversion
using Saccharomyces cerevisiae (Fahrig, 1974), somatic segregation
using A. nidulans (Crebelli et al., 1984), recombinant DNA synthesis
using D. melanogaster (Graf et al., 1984), and sperm abnormality in
bulls (Amir et al., 1977) were positive. However, there was a
negative response in a test for DNA damage using B. subtilis (Shiau
et al., 1980), in DNA-binding tests using E. coli and Ehrlich
ascites tumour cells (Kubinski et al., 1981), and in the cell
transformation assay using mouse Balb/c 3T3 cells (Tennant et al.,
1986).
7.6.2 In vivo assays
1,2-Dibromoethane did not induce chromosomal aberrations or
sister chromatid exchanges in occupationally exposed papaya fruit
packers (Steenland et al., 1986).
1,2-Dibromoethane was negative in tests for specific locus
mutations in germ cells in male mice (Russell, 1986; Barnett et al.,
1992). It did not induce dominant lethal mutations in mice (Epstein
et al., 1972; Teramoto et al., 1980; Barnett et al., 1992) or rats
(Short et al., 1979; Teramoto et al., 1980), chromosomal aberrations
in mice (Krishna et al., 1985) or micronuclei in bone-marrow cells of
mice (Krishna et al., 1985); there was a weak sister chromatid
exchange response that was not dose-related (Krishna et al., 1985).
Tests for recombinant DNA synthesis using D. melanogaster (Graf
et al., 1984) and sperm abnormality using bulls (Amir et al., 1977),
and a sex-linked recessive lethal test using D. melanogaster with
exposure to 1,2-dibromoethane in the gaseous phase (Kale & Baum, 1982)
were all positive.
The frequency of micronuclei in pollen mother cells of
Tradescantia was significantly increased by exposure to
1,2-dibromoethane in the liquid or gaseous phase (Ma et al., 1978,
1984). Gene mutation was demonstrated in barley (Ehrenberg et al.,
1974) and Drosophila melanogaster (wing spot) (Graf et al., 1984).
It induced DNA strand breaks in rat (Nachtomi & Sarma, 1977) and mouse
(White, 1982) hepatic cells. 1,2-Dibromoethane was negative in a gene
mutation assay (egg colour mutation) in silkworms (Sugiyama, 1980).
7.6.3 Other studies
Two doses of 1,2-dibromoethane with dose levels ranging between
10 and 300 µmol/kg body weight (1.8-56 mg/kg body weight) were given
by gavage to 90-day-old female Sprague-Dawley rats, 21 and 4 h before
sacrifice. 1,2-Dibromoethane caused marked DNA damage. The fraction
of DNA eluted from samples of blood, bone marrow, liver, kidney,
spleen or thymus of the rats given 1,2-dibromoethane doses of 100 µmol
body weight (18 mg/kg body weight) 21 and 4 h before sacrifice was
higher than in controls. However, the difference was statistically
significant only for kidney and liver (Kitchin & Brown, 1986).
Intraperitoneal injection (0.25 and 0.5 mmol/kg) of
1,2-dibromoethane (> 99 9%) in corn oil in B6C3F1 mice (weight
20-26 g) produced hepatic damage. The mice were sacrificed 4 h later
and in vivo genotoxicity was determined by a sensitive in vivo/
in vitro alkaline DNA-unwinding assay for the presence of
single-strand breaks and/or alkali-labile sites in hepatic DNA.
Significant hepatic DNA damage was found with a dose of 0.5 mmol/kg.
Although 1,2-dibromoethane is a direct-acting mutagen, its
mutagenicity is generally enhanced by metabolic activation. Two
different pathways have been postulated for its activation to the
ultimate mutagenic form. One is mediated by the mixed-function
oxygenases of liver microsomes, in which 1,2-dibromoethane is
converted to bromoacetaldehyde and 2-bromoethanol, both of which are
potential DNA-damaging agents (Hill et al., 1978; Banerjee et al.,
1979). The other pathway is mediated by enzymes present in liver
cytosol, in which glutathione conjugation can yield a half-sulfur-
mustard or an episulfonium ion as reaction products (Rannug, 1980; Van
Bladeren et al., 1980).
Glutathione conjugation also contributes to the binding of
1,2-dibromoethane to DNA, and an S-[2-(N7-guanyl)ethyl]
glutathione adduct has been identified (Ozawa & Guengerich, 1983;
Inskeep & Guengerich, 1984). Foster et al. (1988) reported that the
majority of mutations induced by 1,2-dibromoethane consist of GC to AT
and AT to GC base changes, suggesting that it acts like an alkylating
agent.
This glutathione conjugate accounts for > 95% of the total DNA
adducts formed by 1,2-dibromoethane. S. typhimurium TA100 and
sequence analysis were used to determine the type, site and frequency
of mutations in a portion of the lacZ gene resulting from in vitro
modification of bacteriophage M13mp18 DNA with S-(2-chloroethyl)
glutathione, an analogue of the 1,2-dibromoethane-glutathione
conjugate. An adduct level of approx.8 nmol per mg DNA resulted in a
10-fold increase in mutation frequency. The mutations were mainly
base substitutions in which GC to AT transitions accounted for 75%
(70% of the total mutations) (Cmarik et al., 1992).
The steady-state levels of c-fos, c-jum, and c-myc mRNA were
investigated in male Wistar rat liver following oral dosing with
100 mg 1,2-dibromoethane/kg body weight. This dose induced
hyperplasia. Increases in the expression of c-myc and c-jum genes
were observed in the absence of c-fos expression (Coni et al.,
1993).
Sundheimer et al. (1982) examined the relationship between
glutathione metabolism and the rate of DNA alkylation by
1,2-dibromoethane in cultured hepatocytes. The rate of alkylation was
decreased by the addition of diethyl maleate and increased by the
addition of cytosolic microsomal enzymes. While high concentrations
of 1,2-dibromoethane were capable of depleting glutathione in the
hepatocytes, depletion did not appear to be necessary for binding to
occur. On the basis of these results, the authors concluded that
glutathione- S-transferases are involved in the bioactivation of
1,2-dibromoethane to an alkylating species.
Working et al. (1986) assessed the ability of 1,2-dibromoethane
to cause DNA damage by quantifying the rate of unscheduled DNA
synthesis (UDS) in F-344 rat hepatocytes and spermatocytes exposed to
1,2-dibromoethane in vivo and in vitro. Pretreatment of cells
with inhibitors of cytochrome-P450-mediated oxidation had no effect on
the induction of UDS by 1,2-dibromoethane (10-100 µmol/litre)
in vitro, whereas depletion of cellular glutathione strongly
inhibited UDS induction in both cell types. Pretreatment of rats with
metyrapone (an inhibitor of hepatic mixed-function oxidases) in vivo
had no effect on 1,2-dibromoethane-induced UDS in hepatocytes, but
produced a positive UDS response in spermatocytes. This suggests that
the mixed-function oxidase pathway in metabolism is the primary route
of clearance of 1,2-dibromoethane and the inhibition of this enzyme
system leads to more extensive tissue distribution of the parent
compound. The data also suggest that the pathway which produces
genotoxic metabolites from 1,2-dibromoethane in hepatocytes and
spermatocytes, in vivo and in vitro, involves the conjugation of
1,2-dibromoethane to glutathione and its subsequent metabolism.
In studies of DNA adducts, Kim & Guengerich (1989) measured
urinary excretion of S-[2-( N7-guanyl)ethyl]- N-acetyl-cysteine,
derived from the nucleic acid adduct, S-[2-( N7-guanyl)
ethyl]glutathione, in rats treated with 1,2-dibromoethane. A good
correlation was found between the excretion of this mercapturic acid
and the in vivo formation of the DNA adduct in liver and kidney DNA.
Inskeep et al. (1986) determined that the major DNA adduct formed upon
exposure to 1,2-dibromoethane, S-[2-( N7-guanyl)ethyl]glutathione,
had a half-life in rat liver, kidney, stomach and lung between 70 and
100 h.
Inskeep & Guengerich (1984) measured the rate of formation of
1,2-dibromoethane adducts to calf thymus DNA in vitro. Adduct
formation was dependent on the presence of both glutathione and
glutathione- S-transferase.
7.7 Carcinogenicity
1,2-Dibromoethane has been tested for carcinogenicity by oral
administration and inhalation in mice and rats, and by skin
application in mice (IARC, 1987) (see Table 14).
It has been shown to cause tumours in various organs, by several
dosage routes and, in some cases, with a latency of less than
12 months (Table 15).
The following tumours have been observed:
a) By oral administration
hepatocellular carcinomas and neoplastic nodules in female rats
haemangiosarcomas in various circulatory system organs in male
rats
alveolar/bronchiolar adenomas in male and female mice
b) By inhalation exposure
nasal cavity carcinomas and adenocarcinomas in male and female
rats
alveolar/bronchiolar carcinomas in female rats
alveolar/bronchiolar carcinomas in male and female mice
haemangiosarcomas in the circulatory system of male and female
rats
mesotheliomas in male rats
mammary fibroadenomas in female mice
subcutaneous fibrosarcomas in female mice
c) By skin administration
skin papillomas and lung papillomas in female mice
7.7.1 Administration by gavage
7.7.1.1 Mouse
When technical grade 1,2-dibromoethane (99.1% pure) in corn oil
was administered on 5 consecutive days per week by gavage to B6C3F1
mice (groups of 50 males and 50 females in the treated groups, and 20
of each sex in the untreated and vehicle control groups) at time-
weighted average dose levels of 62 and 107 mg/kg per day, early
development of squamous cell carcinomas of the forestomach was
observed in both sexes. The incidence of alveolar/bronchiolar
adenomas was significantly higher in treated mice of both sexes than
in controls (NCI, 1978).
7.7.1.2 Rat
Osborne-Mendel rats (50 animals of each sex in the treated groups
and 120 animals of each sex in the untreated and vehicle control
groups) were given on 5 consecutive days per week, by gavage,
technical grade 1,2-dibromoethane (99.1% pure) in corn oil at time-
weighted average dose levels of 38 or 41 mg/kg per day for males, and
37 or 39 mg/kg per day for females. Squamous cell carcinomas of the
forestomach were observed in more than half the male and female rats
at both dose levels, while none were observed in controls. The
lesions, seen as early as week 12, were locally invasive and
eventually metastasized. Significantly higher incidences of
hepatocellular carcinomas and haemangiosarcomas were observed in
treated males and females, respectively (NCI, 1978).
Ledda-Columbano et al. (1987b) examined the interaction of an
intragastric dose of either 1,2-dibromoethane or carbon tetrachloride
(CCl4) with diethylnitrosomine (DENA). The administration of either
1,2-dibromoethane or CCl4 resulted in similar increases in cell
proliferation. However, while pretreatment with CCl4 caused an
increase in the incidence of hepatic foci resulting from subsequent
DENA administration, pretreatment with 1,2-dibromoethane did not.
This difference in the ability of the two compounds to act as a
promoter was attributed to the nature of cell proliferation response.
The authors concluded that the cell proliferation induced by
1,2-dibromoethane, unlike the compensatory cell proliferation induced
by CCl4, is not an effective process for increasing the rate of
tumour initiation.
7.7.2 Administration in drinking-water
7.7.2.1 Mouse
1,2-Dibromoethane (> 99% purity) and its metabolites,
bromoethanol or bromoacetaldehyde, were administered to B6C3F1 mice
(groups of 30 males and 30 females) at a concentration of 4 mmol/litre
in distilled drinking-water (equivalent to 116 mg/kg body weight for
males and 103 mg/kg body weight for females), for 450 days in the case
of 1,2-dibromoethane and for 560 days in the case of the metabolites.
A control group (60 males and 60 females) was given distilled
drinking-water. 1,2-Dibromoethane induced squamous cell carcinomas of
the forestomach in 26/30 males and 27/30 females, and squamous cell
papillomas of the oesophagus in 3/30 females. Bromoethanol in
drinking-water at a concentration of 4 mmol/litre (equivalent to
76 mg/kg body weight for males and 71 mg/kg body weight for females)
induced squamous cell papillomas of the forestomach in 9/29 males and
10/29 females, but bromoacetaldehyde at the same concentration
(equivalent to 62 mg/kg body weight for females and 62 mg/kg body
weight for males) did not induce a significant incidence of
forestomach tumours. The incidence of tumours in the control group
was not significant. Bromoethanol and bromoacetaldehyde were not
considered likely to be active intermediates of 1,2-dibromoethane
carcinogenicity (Van Duuren et al., 1985).
7.7.3 Inhalation
7.7.3.1 Mouse
Groups of 50 male and 50 female B6C3F1 mice were exposed in
inhalation chambers to air containing 77 or 308 mg/m3 (10 or 40 ppm)
of 1,2-dibromoethane (99.3-99.4% pure) for 78-106 weeks. The
incidences of alveolar/bronchiolar carcinomas and alveolar/bronchiolar
adenomas were significantly higher in exposed male and female mice
than in controls. Haemangiosarcomas of the circulatory system,
fibrosarcomas in subcutaneous tissue, carcinomas of the nasal cavity,
and adenocarcinomas of the mammary gland were significantly increased
in females. Exposure to 1,2-dibromoethane was also associated with
epithelial hyperplasia of the respiratory system (NTP, 1982).
7.7.3.2 Rat
Groups of 50 male and 50 female F-344 rats were exposed in
inhalation chambers to air containing 77 or 308 mg/m3 (10 or 40 ppm)
of 1,2-dibromoethane (99.3-99.4%) for 88-106 weeks. Carcinomas,
adenocarcinomas and adenomas of the nasal cavity, and
haemangiosarcomas of the circulatory system were significantly
increased in exposed male and female rats. The incidences of
mesotheliomas of the tunica vaginalis and adenomatous polyps of the
nasal cavity in males, and of fibroadenomas of the mammary gland and
alveolar/bronchiolar adenomas and carcinomas (combined) in females
were significantly increased (NTP, 1982).
Wong et al. (1982) reported that the coadministration of dietary
disulfiram increases the rate of tumour incidence in rats exposed to
1,2-dibromoethane (153.6 mg/m3, 20 ppm) by inhalation. There is
evidence to suggest that the synergistic effect of disulfiram may be
the result of increased liver glutathione- S-transferase activity in
animals treated with this drug (Elliot & Ashby, 1980).
7.7.4 Dermal application
7.7.4.1 Mouse
Doses (25 mg or 50 mg) of 1,2-dibromoethane (> 99% pure)
dissolved in 0.2 ml acetone were applied 3 times/week to the shaved
dorsal skin of female Ha:ICR Swiss mice (groups of 30 animals). There
were an acetone only and untreated control groups. The times to first
appearance of skin tumour (papilloma) were 434 days for the 25-mg
group and 395 days for 50-mg group. By comparison with controls, both
groups showed a statistically significant increase in skin papillomas,
and in the 50-mg group there was also a significant increase in lung
papillomas. Both groups also had dermal squamous carcinomas and
stomach, tumours but these were not statistically significant (Van
Duuren et al., 1979).
7.7.5 Cell transformation
1,2-Dibromoethane caused transformation of BALB/C 3T3 cells both
in the presence and absence of an exogenous metabolism system (Perocco
et al., 1991).
7.8 Biochemical studies and species specificity
1,2-Dibromoethane (75-100 mg/kg body weight) given by gavage to
non-fasted Wistar rats induced DNA synthesis and cell division in the
liver. The peak of DNA synthesis, as measured by 3H-methyl
thymidine incorporation, was attained at or shortly after 24 h. The
mitotic waves measured with the aid of colchicine occurred at 24-30 h
and 48 to 54 h after 1,2-dibromoethane treatment. Increase in DNA
synthesis was confirmed by autoradiography. The stimulation of liver
cell mitosis occurred in non-fasted animals without any apparent cell
necrosis. 1,2-Dibromoethane was an effective mitogen for liver under
these experimental conditions (Nachtomi & Farber, 1978).
In a study by Ledda-Columbano et al. (1987a) 1,2-dibromoethane in
corn oil was administered by gavage at a dose of 100 mg/kg body weight
to male Wistar rats (weight 250-280 g). The rats were given an
intraperitoneal injection of 3H-thymidine 1 h prior to sacrifice,
10, 20, 30, 36 and 48 h after the treatment. No mortality
attributable to 1,2-dibromoethane was observed in the treated rats.
The body weights of 1,2-dibromoethane-treated rats were similar to
those of controls. No changes in the specific activity of DNA were
observed in kidney 10 h after treatment. There was an increase in
labelled thymidine incorporation into DNA and this was maximal after
20-30 h. At 48 h the extent of incorporation of labelled thymidine
decreased rapidly even though it was still higher than in controls.
When effects on the kidneys were investigated, mitotic activity was
noted predominantly in the proximal tubular epithelium of the renal
cortex. Histological examination of the kidney did not reveal any
signs of necrosis.
Of possible significance for the prediction of species
differences in response to 1,2-dibromoethane are the observations
(Dibiasio et al., 1991) that hepatic cytosolic glutathione-
S-transferase activities are similar in rats and mice, but about 40%
of these values in rhesus monkeys. In addition, cytosolic
glutathione- S-transferase activities in rhesus monkey and human
testis are only about 5% of the activities in rat and mouse testes.
8. EFFECTS ON HUMANS
8.1 Acute toxicity
1,2-Dibromoethane is strongly irritant to the eyes, skin, and
respiratory tract (Peoples et al., 1978; Letz et al., 1984). Deaths
from acute exposure to high concentrations of 1,2-dibromoethane are
usually due to pneumonia following damage to the lungs. In addition,
acute inhalation exposure may lead to liver and kidney damage.
Six people who attempted suicide by ingesting 1,2-dibromoethane
suffered from vomiting, nausea and burning throat; death followed in
two cases. The characteristic pathological lesions were present in
liver, lungs and kidneys. Intense jaundice was observed and was due
to massive necrosis of the liver (Sarawat et al., 1986).
It is estimated that 200 mg/kg is lethal to humans, based on the
observation that 12 g caused the death of a woman weighing about 60 kg
(Alexeef et al., 1990).
8.2 Occupational exposure
In cases of poisoning following occupational exposure, headache,
severe vomiting, diarrhoea, respiratory tract irritation and death
have been reported. Exposure to 1,2-dibromoethane in air at
concentrations above 384 mg/m3 (50 ppm) caused nasal and throat
irritation. Two deaths were reported after exposure by inhalation to
a mean concentration of 215 mg/m3 (28 ppm) for 30 and 45 min,
respectively, during the cleaning of a storage tank containing
residues of 1,2-dibromoethane (Letz et al., 1984, Jacobs, 1985).
There was also absorption from dermal exposure to the 0.1-0.3%
solution in the tank (Letz et al., 1984). The first worker collapsed
while working inside the tank and died 12 h later with metabolic
acidosis, depression of the CNS, and laboratory evidence of liver
damage. A supervisor attempting to rescue the worker also collapsed
inside the tank and died 64 h later with intractable metabolic
acidosis, hepatic and renal failure, and necrosis of skeletal muscle
and other organs. Coughing, vomiting, diarrhoea, eye, skin and
respiratory irritation, coma, metabolic acidosis, delirium, confusion,
nausea, low urine output, renal failure, tachycardia and asystole
were noted. Autopsy revealed pulmonary oedema, liver damage and
extensive autolysis in the kidney.
Inhalation exposure to concentrations over 154 mg/m3 (20 ppm)
for more than 30 min is considered fatal to humans.
8.2.1 Cancer incidence
Mortality in employees exposed to 1,2-dibromoethane in two
production units operated from 1942 to 1969 and from the mid-1920s to
1976 was investigated (Ott et al., 1980). The study population was
161 employees. In the first production unit two deaths from malignant
neoplasms were observed against 3.6 expected, and in the second unit,
where there was potential exposure to various organic bromide
products, there were five deaths from malignant neoplasms against
2.2 expected (p < 0.072). However, no statistically significant
increase in total deaths or malignant neoplasms relatives to duration
of exposure was observed.
Epidemiological studies of four worker populations did not show
any increase in cancer that could be attributed to 1,2-dibromoethane
(Ter Haar, 1980)
8.2.2 Reproductive effects
In an investigation of possible sterility from exposure to
1,2-dibromoethane, sperm levels in workers exposed to
1,2-dibromoethane were not affected, and there was no evidence of
effects on offspring (Ter Haar, 1980).
Ratcliffe et al. (1987) and Schrader et al. (1987) conducted a
cross-sectional study of semen quality in 46 men employed in the
papaya fumigation industry in Hawaii, with an average duration of
exposure of 5 years and a geometric mean breathing zone exposure to
airborne 1,2-dibromoethane of 0.68 mg/m3 (88 ppb) (8-h time-weighted
average). The control group consisted of 43 unexposed men from a
nearby sugar refinery. Statistically significant decreases in sperm
count per ejaculate and percentage of viable and motile sperm,
together with increases in the proportion of sperm with specific
morphological abnormalities, were observed among exposed men, compared
to controls, after consideration of smoking, caffeine and alcohol
consumption, subject's age, abstinence, history of urogenital
disorders, and other potentially confounding variables. The data
indicated that 1,2-dibromoethane could cause reproductive inpairment
in males exposed to this concentration. Schrader et al. (1988)
conducted a short-term longitudinal study on the effect of
1,2-dibromoethane exposure on male reproductive potential in ten
forestry workers and six unexposed men in Colorado. The time-weighted
average inhalation exposure over 6 weeks was 0.46 mg/m3 (peak
exposure of 16 mg/m3) and there was extensive skin exposure. Sperm
velocity and semen volume were decreased significantly in the exposed
workers. Both studies suggested that 1,2-dibromoethane has multiple
sites of action on male accessory sex glands and testes.
In five studies on the reproductive effects of occupational
exposure to 1,2-dibromoethane, four showed potential reproductive
impairment but this was not large enough to be statistically
significant. The power of all of the studies was low and they were
considered inconclusive for assessing reproductive risk (Dobbins,
1987).
A retrospective study of four plants in the United Kingdom where
male workers were exposed to 1,2-dibromoethane revealed statistically
marginally reduced fertility rates (i.e., live births to their wives).
The average exposure was probably below 38.5 mg/m3 (5 ppm), although
actual concentrations were not measured (Wong et al., 1985).
9. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
9.1 Aquatic organisms
9.1.1 Invertebrates
Nishiuchi (1980, 1981) studied the acute toxicity of
1,2-dibromoethane in aquatic larvae of insects and aquatic
invertebrates, such as a mayfly (Cloeon dipterum), two dragonflies
(Sympetrum frequens and Orthetrum albistylum speciosum), a
Japanese diving beetle (Eretes sticticus) and a water boatman
(Micronecta sedula) (Table 17). The toxicity threshold (48-h LC50
values) of 1,2-dibromoethane machine oil formulation for the above
aquatic invertebrates and that of 1,2-dibromoethane for mayfly were
greater than 40 mg/m3. In mayfly the reported 48-h toxicity
thresholds for two mixed formulations, which consisted of
1,2-dibromoethane, malathion, diazinon and machine oil (3:1:1:80), and
1,2-dibromoethane, diazinon and O-sec-butylphenyl methylcarbamate
(25:5:3) were 28 and 65 mg/m3, respectively (Nishiuchi & Asano,
1979). There was no apparent difference in toxicity of technical
1,2-dibromoethane and an oil-based formulation at least up to
40 mg/litre.
Table 17. Acute toxicity of 1,2-dibromoethane to aquatic
invertebrates (From: Nishiuchi, 1980)
Species Test material Temperature 48-h LC50
(°C)
Mayfly Technical material 25 > 40
(Cloeon dipterum) (in acetone)
Dragonfly 1,2-dibromoethane (oil) 25 > 40
(Sympetrum frequens)
Dragonfly 1,2-dibromoethane (oil) 25 > 40
(Orthetrum albistylum
speciosum)
Japanese diving beetle 1,2-dibromoethane (oil) 25 > 40
(Eretes sticticus)
Water boatman 1,2-dibromoethane (oil) 25 > 40
(Micronecta sedula)
Herring et al. (1988) evaluated the toxicity of 1,2-dibromoethane
to Hydra oligactis in a series of three experiments. Study 1
evaluated lethality, feeding behaviour and mobility in a series of
concentrations ranging from 7.5 to 75 mg/litre. In Study 2, adult
Hydra were pre-treated for 14 days with a sublethal dose of
1,2-dibromoethane (5 mg/litre) prior to exposure to a range of
1,2-dibromoethane concentrations (25-300 mg/litre) for a total of
72 h. In the third study, the F1 offspring of pre-treated adult
Hydra were also exposed to a series of 1,2-dibromoethane
concentrations. The 48-h LC50 for Hydra was determined to be
70 mg/litre. When adult Hydra were pre-treated with sublethal
concentrations of 1,2-dibromoethane, the 48-h LC50 was increased to
200 mg/litre. Furthermore, the F1 offspring of pre-treated adult
exhibited mortalities of only 10% and 20%, respectively, after 24-h
and 48-h exposures to 200 mg 1,2-dibromoethane/litre. These results
suggest that Hydra and first-generation offspring are capable of
developing tolerance to 1,2-dibromoethane.
Adams & Kennedy (1992) exposed first-stage budding Hydra
oligactis to 1,2-dibromoethane at 5 mg/litre. The 1,2-dibromoethane
was dissolved using acetone at 15 mg/litre, and an acetone control was
used. Exposure was for 24, 48 or 72 h. Following exposure, the
animals were washed several times with the medium, sectioned through
the gastric region, and the base/apical sections were grafted.
Regeneration was significantly affected by all exposures to
1,2-dibromoethane but not by acetone. The severity of the effect
increased with increasing exposure.
Adams et al. (1989) reported dose-sensitive relationships for the
loss and recovery of locomotor response, chromatophore expansion and
lethality in three species of laboratory-reared octopus. Three
species of octopus (Octopus bimaculoides, O. joubini, and O. maya)
were exposed to 25, 50, 75 and 100 mg 1,2-dibromoethane/litre for
either one hour, followed by transfer to chemical-free water, or
continuously for a period of 72 h. Generally, responses by the
octopuses were evident after only 1 min of exposure. Chromatophore
expansion and loss of locomotor response occurred at 25 mg/litre after
30 min, but recovery was noted 6 h after transfer to chemical-free
water. Lethality occurred in all three species at 25 mg/litre after
48 h of exposure. O. maya was the most sensitive species,
exhibiting 100% mortality after 3 h of exposure. The acute LC50
values for O. bimaculoides, O. joubini and O. maya were 42.7, 35.3
and 30.6 mg/litre respectively (Table 18). Although the authors
reported a chronic LC50 of 100 mg/litre occurring within 12 h, the
Task Group considered this to be an acute exposure.
Table 18. Acute LC50 values approximated for lethality data
(Adams et al., 1989) using either the moving average,
binomial or probit methods
Test species Estimated 48-h LC50 95% confidence
(mg/litre) limits
Octopus bimaculoides 42.7 28.1-58.8a
Octopus joubini 35.3 -b
Octopus maya 30.6 0-52.02c
a moving average method
b confidence limits exceeded 95%, therefore the limits would
range from 0 to infinity
c probit method used
9.1.2 Fish
There are few data for the acute toxicity of 1,2-dibromoethane in
fish. A study of the effect of pH on the acute toxicity of
1,2-dibromoethane in killifish (Oryzias latipes) was carried out in
open static systems. Altering the pH of the breeding water between pH
5.0 and pH 10.0 had no effect on 48-h LC50 values (Nishiuchi, 1982).
Goldfish quickly absorbed 1,2-dibromoethane from water (at 1 mg/litre)
and quickly eliminated it. The concentration in the goldfish
(1.75 ± 0.041 mg/kg) was in equilibrium with the concentration in the
water 1.5 h after the initiation of exposure. From the results of the
elimination study, the biological half-life was calculated to be less
than 30 min (Ogino, 1978).
Landau & Tucker (1984) found the 48-h LC50 values for
sheepshead minnow Cyprinodon variegautus and snook Centropomus
undecimatis, both estuarine fish, to be 4.8 and 6.2 mg/litre,
respectively.
Nishiuchi & Asano (1979) measured toxicity thresholds for carp
(Cyprinus carpio) exposed to pesticide mixtures containing various
concentrations of 1,2-dibromoethane (Table 19). After 24 h of
exposure at 24°C, no difference in the toxicity threshold (> 40)
could be detected at unit pH increases between 5 and 9.
Table 19. Toxicity threshold after 48 h exposure of Cyprinus carpio
to mixtures containing 1,2-dibromoethane at 25°C
(Nishiuchi & Asano, 1979)
Mixture constituents Concentrations Toxicity threshold
1,2-Dibromoethane 2.5% oil > 100
Fenitrothion 0.5% oil
1,2-Dibromoethane 2.5% oil 86
Fenitrothion 0.5% oil
1,2-Dibromoethane 1.5% emulsion
Fenitrothion 10% emulsion 45
Carbaryl 5% emulsion
Diazinon 20% emulsion 28
1,2-Dibromoethane 10% emulsion
Cyanophos 10% emulsion 32
1,2-Dibromoethane 10% emulsion
1,2-Dibromoethane 25% oil
Diazinon 5% oil 65
O-sec-butylphenyl 3% oil
methylcarbamate
9.2 Terrestrial biota
Data on the effects of 1,2-dibromoethane on terrestrial biota
(other than mammals) are limited. In a study on the effects of
different foods on the susceptibility of the adzuki bean weevil
(Callosobruchus chinensis Linn.) to 1,2-dibromoethane, the 24-h
LC50 at 29°C was 4.40, 7.232, 6.130, 5.249 and 4.951 mg/litre for
chickpea, pea, green gram, black gram, and pigeon pea, respectively
(Mundhe & Pandey, 1980).
Exposure of the nematode Aphelenchus avenae to low
concentrations of 1,2-dibromoethane resulted in a very small
conversion of the halide just before death. A study with
14C-labelled 1,2-dibromoethane showed that two primary products were
ethylene (5%) and O-acetylserine (> 95%). These transformations
were indicative of two primary modes of intoxication of the nematodes,
postulated to be a direct reaction of 1,2-dibromoethane with an iron
centre in the respiratory sequence and the substitution of a serine at
the active site of an esterase or protease (Castro & Belser, 1978).
9.3 Microorganisms
The toxicity of 1,2-dibromoethane to microsclerotia of
Verticillium dahliae in air and in soil was determined in a sealed
container. At concentrations of 470 mg/litre in air or 12.5 mg/kg in
soil, 1,2-dibromoethane killed 97% of the microsclerotia, after
incubation for 16 days in both cases. The toxicity of
1,2-dibromoethane increased with increasing temperature and with
increase in soil moisture (0-80%) (Ben-Yephet et al., 1981).
Pignatello (1986) investigated 1,2-dibromoethane effects on
microorganisms from two soils/sediments taken from a
1,2-dibromoethane-contaminated groundwater discharge area. Labelled
potassium acetate was added to slurries of soil, which had been shaken
with added 1,2-dibromoethane (up to 1000 mg/litre), for 3 or 12 h.
Incorporation of acetate into microbial lipids was used as the
end-point. The EC50 for inhibition of acetate incorporation
following 12-h incubation with 1,2-dibromoethane was 50 and
100 mg/litre for the two soils; soil 1 showed an EC50 of 100
following 3 h of incubation. Both soils showed "slight" inhibition at
10 mg 1,2-dibromoethane/litre and > 94% at 1000 mg/litre. Soil 1 was
a muddy soil with a total organic carbon (TOC) content of 14%, whereas
soil 2 was a stream-bed soil with a TOC of 0.24%.
9.4 Plants
1,2-Dibromoethane was phytotoxic to fruit after they had been
fumigated against several species of fruit flies. Fumigation with
1,2-dibromoethane at a concentration of 4 g/m3 stimulated fruit
respiration and ethylene evolution. A higher concentration
(32 g/m3) increased respiration rate and ethylene evolution in the
fruits and increased tissue leakage. Fruits stored at 1°C after
fumigation with 32 g/m3 suffered more severe damage than those
stored at 20°C. Storage at 1°C abolished the increases in gas
exchange observed in fumigated fruits at 20°C. The injurious effect
of cooling might be ascribed to a higher residue of unchanged fumigant
persisting in the cooled fruits or to a decreased capacity of the
fruits to repair cellular damage at low temperatures (Wade & Rigney,
1979).
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
1,2-Dibromoethane is carcinogenic for rats and mice causing
tumours (adenomas and carcinomas) in a variety of organs including the
nasal cavity, lung, stomach, liver, skin, and mammary gland, as well
as haemangiosarcomas. In many cases, a reduced latency period for
developing tumours was observed. 1,2-Dibromoethane has been shown to
be mutagenic in various in vivo and in vitro assays and to cause
single-strand DNA breaks in vitro. Some metabolites have been shown
to covalently bind to DNA. Based on these data, 1,2-dibromoethane is
thought to be a genotoxic carcinogen to rodents. Although adequate
studies in humans are not available, the extensive evidence for
carcino genicity in animal studies indicates that 1,2-dibromoethane
is a potential human carcinogen.
10.2 Evaluation of effects on the environment
The high volatility of 1,2-dibromoethane makes the atmosphere the
predominant environmental sink. Consequently, measured concentrations
in surface waters are low (< 0.2 µg/litre). Air concentrations of
< 0.2 µg/litre have been measured in cities, while concentrations of
up to 90 µg/litre in irrigation wells reflect the mobility of the
compound in soil. Persistent contamination of irrigation wells may
result from the slow release of 1,2-dibromoethane from the soil matrix
many years after its use as a soil fumigant.
There is a lack of information on the degradation of
1,2-dibromoethane in the aquatic and soil environments.
Stratospheric photodegradation occurs and potentially leads to
breakdown products with ozone-depleting capacity. However,
1,2-dibromoethane is not listed in the Montreal Convention.
Few aquatic ecotoxicity tests have been conducted with
1,2-dibromoethane. Those reported show LC50s greater than
5 mg/litre. There is a difference of at least 4 orders of magnitude
between measured water concentrations and these toxic concentrations,
indicating that 1,2-dibromoethane poses no risk to aquatic organisms.
11. CONCLUSIONS AND RECOMMENDATIONS
Considering the toxicological characteristics of
1,2-dibromoethane, both qualitatively and quantitatively, it was
concluded that an exposure that would not cause adverse effects in
humans after any route of exposure could not be estimated.
Consequently, all appropriate measures should be taken to eliminate or
minimize human exposure to 1,2-dibromoethane.
12. FURTHER RESEARCH
Further information from epidemiology studies on
1,2-dibromoethane would be useful.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
IARC (1977, 1982, 1987) concluded, from biological data relevant
to the evaluation of carcinogenic risk, that 1,2-dibromoethane is
carcinogenic in mice and rats after oral administration, and by
inhalation, producing squamous cell carcinomas of the forestomach.
1,2-Dibromoethane given orally or by intraperitoneal injection did not
produce dominant lethal mutations in mice. Prolonged contact with
1,2-dibromoethane causes skin irritation. However, no case reports or
epidemiological studies were available to the Working Group. IARC has
classified 1,2-dibromoethane as a Group 2A carcinogen (probably
carcinogenic to humans).
WHO has not established a drinking-water quality guideline for
1,2-dibromoethane. This is because 1,2-dibromoethane appears to be a
genotoxic carcinogen and the studies are inadequate for mathematical
extrapolation (WHO, 1993).
1,2-Dibromoethane was evaluated by the Joint FAO/WHO Expert
Committee on Pesticide Residues in 1965 (FAO/WHO, 1965) and 1966
(FAO/WHO, 1967). The 1965 evaluation concluded that 1,2-dibromoethane
should be used for fumigation of food only on the condition that no
residue of the unchanged compound reached the consumer. In the 1966
evaluation an Acceptable Daily Intake (ADI) of 1 mg/kg body weight as
bromide was established. In 1991 the Codex Alimentarius Commission
deleted the Guideline Levels for 1,2-dibromoethane in food commodities
(FAO/WHO, 1992).
REFERENCES
Abdel-Kader MHK, Peach ME, & Stiles DA (1979) Determination of
ethylene dibromide in fortified soils by molecular emission cavity
analysis using a modified extraction process. J Assoc Off Anal Chem,
62(1): 114-118.
Adams JA & Kennedy AA (1992) Sublethal effects of ethylene dibromide
on wound healing and morphogenesis in Hydra oligactis. Arch Environ
Contam Toxicol, 22: 272-277.
Adams PM, Hanlon RT, & Forsythe JW (1989) Toxic exposure to ethylene
dibromide and mercuric chloride: Effects on laboratory-reared
octopuses. Neurotoxicol Teratol, 10: 519-523.
Adkins B, Van Stee EW, Simmons JE, & Eustis SL (1986) Oncogenic
response of strain A/J mice to inhaled chemicals. J Toxicol Environ
Health, 17: 311-322.
Alexeeff GV, Kilgore WW, & Li MY (1990) Ethylene dibromide: Toxicology
and risk assessment. Berlin, Heidelberg, New York, Spinger Verlag
Inc., pp 49-122.
Alleman TG, Sanders RA, & Madison BL (1986) Simultaneous analysis of
grain and grain-based products for ethylene dibromide, carbon
tetrachloride, and ethylene dichloride. J Assoc Off Anal Chem,
69(4): 575-580.
Alper MD & Ames BN (1975) Positive selection of mutants with deletions
of the gal-ch1 region of the salmonella chromosome as a screening
procedure for mutagens that cause deletions. J Bacteriol,
121(1): 259-266.
Alumot E, Nachtomi E, Kempenich-Pinto O, Mandel E, & Schindler H
(1968) The effect of ethylene dibromide in feed on the growth, sexual
development and fertility of chickens. Poult Sci, 47: 1979-1985.
Ames BN & Yanofsky C (1971) The detection of chemical mutagens with
enteric bacteria. In: Hollaender A ed. Chemical mutagens: Principles
and methods for their detection. New York, London, Plenum Press,
vol 1, pp 267-282.
Amin TA & Narang RS (1985) Determination of volatile organics in
sediment at nanogram-per-gram concentrations by gas chromatography.
Anal Chem, 57: 648-651.
Amir D (1973) The sites of the spermicidal action of ethylene
dibromide in bulls. J Reprod Fertil, 35: 519-525.
Amir D (1975) Individual and age differences in the spermicidal effect
of ethylene dibromide in bulls. J Reprod Fertil, 44: 561-565.
Amir D & Volcani R (1965) Effect of dietary ethylene dibromide on bull
semen. Nature (Lond), 206(4979): 99-100.
Amir D, Esnault C, Nicolle JC, & Courot M (1977) DNA and protein
changes in the spermatozoa of bulls treated orally with ethylene
dibromide. J Reprod Fertil, 51: 453-456.
Amir D, Nicolle JC, & Courot M (1979) Changes induced to bull
spermatids and testicular spermatozoa by a single peritesticular
injection of ethylene dibromide. Int J Androl, 2: 162-170.
Asita AO, Hayashi M, Kodama Y, Matsuoka A, Suzuki T, & Sofuni T (1992)
Micronucleated reticulocyte induction by ethylating agents in mice.
Mutat Res, 271: 29-37.
Ballering LAP, Nivard MJM, & Vogel EW (1993) Characterization of the
genotoxic action of three structurally related 1,2-dihaloalkanes in
Drosophila melanogaster. Mutat Res, 285: 209-217.
Ballschmiter K, Mayer P, & Class Th (1986) Chemistry of organic traces
in air. IV. Analysis of C1- and C2-halocarbons in ambient air by
cold trap injection and wide bore glass capillary gas chromatography.
Fresenius Z Anal Chem, 323: 334-339.
Banerjee S, Van Duuren BL, & Kline SA (1979) Interaction of potential
metabolites of the carcinogen ethylene dibromide with protein and DNA
in vitro. Biochem Biophys Res Commun, 90(4): 1214-1220.
Barber ED & Donish WH (1982) An exposure system for quantitative
measurements of the microbial mutagenicity of volatile liquids.
Measurements of microbial mutagenicity. In: Tice RR, Costa DL, &
Schaich KM ed. Genotoxic effects of airborne agents. New York, London,
Plenum Press, pp 3-18.
Barber ED, Donish WH, & Mueller KR (1981) A procedure for the
quantitative measurement of the mutagenicity of volatile liquids in
the Ames Salmonella/microsome assay. Mutat Res, 90: 31-48.
Barkley J, Bunch J, Bursey JT, Castillo N, Cooper SD, Davis JM,
Erickson MD, Harris BSH III, Kirkpatrick M, Michael LC, Parks SP,
Pellizzari ED, Ray M, Smith D, Tomer KB, Wagner R, & Zweidinger RA
(1980) Gas chromatography mass spectrometry computer analysis of
volatile halogenated hydrocarbons in man and his environment - a
multimedia environmental study. Biomed Mass Spectrom, 7(4): 139-147.
Barnett LB, Lovell DP, Felton CF, Gibson BJ, Cobb RR, Sharpe DS,
Shelby MD, & Lewis SE (1992) Ethylene dibromide: Negative results with
the mouse dominant lethal assay and the electrophoretic specific-locus
test. Mutat Res, 282: 127-133.
Barry TL & Petzinger G (1985) GC/MS confirmation of ethylene dibromide
residue in foods. J Food Saf, 7: 171-176.
Beckman H, Crosby DG, Allen PT, & Mourer C (1967) The inorganic
bromide content of foodstuffs due to soil treatment with fumigants.
J Food Sci, 32: 138-140.
Ben-Yephet Y, Letham D, & Evans G (1981) Toxicity of 1,2-dibromoethane
and 1,3-dichloropropene to microsclerotia of Verticillium dahliae.
Pestic Sci, 12: 170-174.
Berck B (1974) Fumigant residues of carbon tetrachloride, ethylene
dichloride, and ethylene dibromide in wheat, flour, bran, middlings,
and bread. J Agric Food Chem, 22(6): 977-984.
Berg WW, Heidt LE, Pollock W, Sperry PD, & Cicerone RJ (1984)
Brominated organic species in the arctic atmosphere. Geophys Res Lett,
11(5): 429-432.
Bielorai R & Alumot E (1975) The temperature effect on fumigant
desorption from cereal grain. J Agric Food Chem, 23(3): 426-429.
Bondi A, Olomucki E, & Calderon M (1955) Problems connected with
ethylene dibromide fumigation of cereals. II. Feeding experiments with
laying hens. J Sci Food Agric, 6: 600-602.
Botti B, Moslen MT, Trieff NM, & Reynolds ES (1982) Transient decrease
of liver cytosolic glutathione s-transferase activities in rats given
1,2-dibromoethane or CCL. Chem-Biol Interact, 42: 259-270.
Brandt I, Brittebo EB, Kowalski B, & Lund B-O (1987) Tissue binding
of 1,2-dibromoethane in the cynomolgus monkey (Macaca fascicularis).
Carcinogenesis, 8(9): 1359-1361.
Brem H, Stein AB, & Rosenkranz HS (1974) The mutagenicity and
DNA-modifying effect of haloalkanes. Cancer Res, 34: 2576-2579.
Brimer PA, Tan E-L, & Hsie AW (1982) Effect of metabolic activation on
the cytotoxicity and mutagenicity of 1,2-dibromoethane in the
CHO/HGPRT system. Mutat Res, 95: 377-388.
Broda C, Nachtomi E, & Alumot E (1976) Differences in liver morphology
between rats and chicks treated with ethylene dibromide. Gen
Pharmacol, 7: 345-348.
BUA (Society of German Chemists-Advisory Committee on Existing
Chemicals of Environmental Relevance) (1987) [Methyl bromide.]
Weinheim, VCH Publishers, 64 pp (BUA Report 14) (in German).
BUA (Society of German Chemists-Advisory Committee on Existing
Chemicals of Environmental Relevance) (1991) [1,2-Dibromoethane.]
Weinheim, VCH Publishers, 290 pp (BUA Report 66) (in German).
Buijs W, Van Der Gen A, Mohn GR, & Breimer DD (1984) The direct
mutagenic activity of alpha,omega-dihalogenoalkanes in Salmonella
typhimurium. Mutat Res, 141: 11-14.
Cairns T, Siegmund EG, Doose GM, Hundley HK, Barry T, & Petzinger G
(1984) Confirmation of ethylene dibromide in fruits and grains by mass
spectrometry. Anal Chem, 56: 2138-2141.
Castro CE & Belser NO (1978) Intoxication of Aphelenchus avenae by
ethylene dibromide. Nematologica, 24: 37-44.
Channarayappa, Ong T, & Nath J (1992) Cytogenetic effects of
vincristine sulfate and ethylene dibromide in human peripheral
lymphocytes: Micronucleus analysis. Environ Mol Mutagen, 20: 117-126.
Chiarpotto E, Biasi F, Aragno M, Scavazza A, Danni O, Albano E, &
Poli G (1993) Change of liver metabolism of 1,2-dibromoethane during
simultaneous treatment with carbon tetrachloride. Cell Biochem Funct,
11: 71-75.
Clark AI, McIntyre AE, Lester JN, & Perry R (1982) Evaluation of a
tenax GC sampling procedure for collection and analysis of vehicle-
related aromatic and halogenated hydrocarbons in ambient air.
J Chromatogr, 252: 147-157.
Clark AI, McIntyre AE, Lester JN, & Perry R (1984a) Ambient air
measurements of aromatic and halogenated hydrocarbons at urban, rural
and motorway locations. Sci Total Environ, 39: 265-279.
Clark AI, McIntyre AE, Perry R, & Lester JN (1984b) Monitoring and
assessment of ambient atmospheric concentrations of aromatic and
halogenated hydrocarbons at urban, rural and motorway locations.
Environ Pollut, B7: 141-158.
Clive D, Johnson KO, Spector JFS, Batson AG, & Brown MMM (1979)
Validation and characterization of the L5178Y/TK+/-mouse lymphoma
mutagen assay system. Mutat Res, 59: 61-108.
Clower M (1980) Modification of the AOAC method for determination of
fumigants in wheat. J Assoc Off Anal Chem, 63(3): 539-545.
Clower M, McCarthy JP, & Carson LJ (1986) Comparison of methodology
for determination of ethylene dibromide in grains and grain-based
foods. J Assoc Off Anal Chem, 69(1): 87-90.
Cmarik JL, Inskeep PB, Meredith MJ, Meyer DJ, Ketterer B, & Guengerich
FP (1990) Selectivity of rat and human glutathione S-transferases in
activation of ethylene dibromide by glutathione conjugation and DNA
binding and induction of unscheduled DNA synthesis in human
hepatocytes. Cancer Res, 50(9): 2747-2752.
Cmarik JL, Humphreys WG, Bruner KL, Lloyd RS, Tibbetts C, & Guengerich
FP (1992) Mutation spectrum and sequence alkylation selectivity
resulting from modification of bacteriophage M13mp18 DNA with
s-(2-chloroethyl)glutathione. Evidence for a role of s-[2-(N-guanyl)
ethyl]glutathione as a mutagenic lesion formed from ethylene
dibromide. J Biol Chem, 267(10): 6672-6679.
Collins M & Barker NJ (1983) Direct monitoring of ambient air for
ethylene oxide and ethylene dibromide. Am Lab (Fairfield, Conn),
15: 72, 74-76, 78-81.
Coni P, Simbula G, De Prati AC, Menegazzi M, Suzuki H, Sarma DSR,
Ledda-Columbano GM, & Columbano A (1993) Differences in the
steady-state levels of c-fos, c-jun and c-myc messenger RNA during
mitogen-induced liver growth and compensatory regeneration.
Hepatology, 17: 1109-1116.
Courtens JL, Amir D, & Durand J (1980) Abnormal spermiogenesis in
bulls treated with ethylene dibromide: An ultrastructural and
ultracytochemical study. J Ultrastruct Res, 71: 103-115.
Crebelli R, Conti G, Conti L, & Carere A (1984) Induction of somatic
segregation by halogenated aliphatic hydrocarbons in Aspergillus
nidulans. Mutat Res, 138: 33-38.
Crespi CL, Seixas GM, Turner TR, Ryan CG, & Penman BW (1985)
Mutagenicity of 1,2-dichloroethane and 1,2-dibromoethane in two human
lymphoblastoid cell lines. Mutat Res, 142: 133-140.
Daft JL (1983) Gas chromatographic determination of fumigant residues
in stored grains, using isooctane partitioning and dual column
packings. J Assoc Off Anal Chem, 66(2): 228-233.
Daft JL (1985) Preparation and use of mixed fumigant standards for
multiresidue level determination by gas chromatography. J Agric Food
Chem, 33: 563-566.
Daft JL (1987) Determining multifumigants in whole grains and legumes,
milled and low-fat grain products, spices, citrus fruit, and
beverages. J Assoc Off Anal Chem, 70(4): 734-760.
Daft JL (1988) Rapid determination of fumigant and industrial chemical
residues in food. J Assoc Off Anal Chem, 71(4): 748-760.
De Serres FJ & Malling HV (1983) The role of Neurospora in
evaluating environmental chemicals for mutagenic activity. Ann NY Acad
Sci, 407: 177-185.
De Vries JW, Larson PA, Bowers RH, Keating AM, Broge JM, Wehling PS,
Patel HH, & Zurawski JW (1985) Improved codistillation method for
determination of carbon tetrachloride, ethylene dichloride and
ethylene dibromide in grain and grain-based products. J Assoc Off Anal
Chem, 68(4): 759-762.
Dibiasio KW, Silva MH, Shull LR, Overstreet JW, Hammock BD, & Miller
MG (1991) Xenobiotic metabolizing enzyme activities in rat, mouse,
monkey, and human testes. Drug Metab Dispos, 19(1): 227-232.
Dobbins JG (1987) Regulation and the use of "Negative" results from
human reproductive studies: The case of ethylene dibromide. Am J Ind
Med, 12: 33-45.
Dumas T & Bond EJ (1982) Microdetermination of ethylene dibromide in
air by gas chromatography. J Assoc Off Anal Chem, 65(6): 1379-1381.
Dunkel VC, Zeiger E, Brusick D, McCoy E, McGregor D, Mortelmans K,
Rosenkranz HS, & Simmon VF (1985) Reproducibility of microbial
mutagenicity assays: Testing of carcinogens and noncarcinogens in
Salmonella typhimurium and Escherichia coli. Environ Mutagen,
7(5): 1-248.
Edwards K, Jackson H, & Jones AR (1970) Studies with alkylating
esters-II: A chemical interpretation through metabolic studies of the
antifertility effects of ethylene dimethanesulphonate and ethylene
dibromide. Biochem Pharmacol, 19: 1783-1789.
Ehrenberg L, Osterman-Golkar S, Singh D, & Lundqvist U (1974) On the
reaction kinetics and mutagenic activity of methylating and
beta-halogenoethylating gasoline additives. Radiat Bot, 15: 185-194.
ElJack AH & Hrudka F (1979) Pattern and dynamics of teratospermia
induced in rams by parenteral treatment with ethylene dibromide.
J Ultrastruct Res, 67: 124-134.
Ellingham TJ, Christensen EA, & Maddock MB (1986) In vitro induction
of sister chromatid exchanges and chromosomal aberrations in
peripheral lymphocytes of the oyster toadfish and American eel.
Environ Mutagen, 8: 555-569.
Elliott BM & Ashby J (1980) Ethylene dibromide and disulfiram: Studies
in vivo and in vitro on the mechanism of the observed synergistic
carcinogenic response. Carcinogenesis, 1(12): 1049-1057.
Entz RC & Hollifield HC (1982) Headspace gas chromatographic analysis
of foods for volatile halocarbons. J Agric Food Chem, 30: 84-88.
Environment Agency Japan (1985) Chemicals in the environment: Annual
report - Annex. Tokyo, Environment Agency, pp 16-28.
Environment Canada (1994) Summary of EDB concentration at Canadian
sites. Ottawa, Environment Canada, Environment Protection Service,
Pollution Measurement Division (Unpublished report).
Epstein SS, Arnold E, Andrea J, Bass W, & Bishop Y (1972) Detection of
chemical mutagens by the dominant lethal assay in the mouse. Toxicol
Appl Pharmacol, 23: 288-325.
Fahrig R (1974) Comparative mutagenicity studies with pesticides.
In: Chemical carcinogenesis essays. Lyon, International Agency for
Research on Cancer, pp 161-181 (IARC Scientific Publications No. 10).
Fanini D, Legator MS, & Adams PM (1984) Effects of paternal ethylene
dibromide exposure on F1 generation behavior in the rat. Mutat Res,
139: 133-138.
FAO/WHO (1965) Evaluation of the hazards to consumers resulting from
the use of fumigants in the protection of food. Report of the 2nd
Meeting of the FAO/WHO Joint Expert Committee on Pesticide Residues.
Rome, Food and Agriculture Organization of the United Nations,
pp 36-42 (FAO Meeting Report No. PL/1965/10/2).
FAO/WHO (1967) Pesticide residues in food. Joint report of the FAO
Working Party on Pesticide Residues and the WHO Expert Committee on
Pesticide Residues. Geneva, World Health Organization, 19 pp (WHO
Technical Report Series 370).
FAO/WHO (1972) 1971 Evaluations of some pesticide residues in food.
Geneva, World Health Organization, p 267 (WHO Pesticide Residues
Series, No. 1).
FAO/WHO (1992) Report of the Twenty-Fourth Session of the Codex
Committee on Pesticide Residues. Rome, Food and Agriculture
Organization of the United Nations, Codex Alimentarius Commission,
82 pp (Alinorm 93/24).
Fernandez M, L'Haridon J, Gauthier L, & Zoll-Moreux C (1993) Amphibian
micronucleus test(s): A simple and reliable method for evaluating
in vivo genotoxic effects of freshwater pollutants and radiations.
Initial assessment. Mutat Res, 292: 83-99.
Ferreri AM, Rocchi P, Capucci A, & Prodi G (1983) Induction of
diphtheria toxin-resistant mutants in human cells by halogenated
compounds. J Cancer Res Clin Oncol, 105: 111-112.
Foster PL, Wilkinson WG, Miller JK, Sullivan AD, & Barnes WM (1988) An
analysis of the mutagenicity of 1,2-dibromoethane to Escherichia
coli: influence of DNA repair activities and metabolic pathways.
Mutat Res, 194: 171-181.
Gilbert J, Startin JR, & Grew C (1985) Automated headspace GC-MS
analysis of ethylene dibromide fumigant residues in fresh fruits. Food
Addit Contam, 2(1): 55-61.
Going JE & Long S (1975) Sampling and analysis of selected toxic
substances: Task II - Ethylene dibromide. Washington, DC, US
Environmental Protection Agency, Office of Toxic Substances
(EPA-560/6-75-001).
Going JE & Spigarelli JE (1976) Sampling and analysis of selected
toxic substances: Task IV - Ethylene dibromide. Washington, DC, US
Environmental Protection Agency, Office of Toxic Substances
(EPA-560/6-76-0211).
Graf U, Wurgler FE, Katz AJ, Frei H, Juon H, Hall CB, & Kale PG (1984)
Somatic mutation and recombination test in Drosophila melanogaster.
Environ Mutagen, 6: 153-188.
Guha SN, Schoneich C, & Asmus KD (1993) Free radical reductive
degradation of vic-dibromoalkanes and reaction of bromine atoms with
polyunsaturated fatty acids: possible involvement of Br* in the
1,2-dibromoethane-induced lipid peroxidation. Arch Biochem Biophys,
305(1): 132-140.
Guobaitis RJ, Ellingham TJ, & Maddock MB (1986) The effects of
pretreatment with cytochrome P-450 inducers and preincubation with a
cytochrome P-450 effector on the mutagenicity of genotoxic carcinogens
mediated by hepatic and renal S9 from two species of marine fish.
Mutat Res, 164: 59-70.
Hardin BD, Bond GP, Sikov MR, Andrew FD, Beliles RP, & Niemeier RW
(1981) Testing of selected workplace chemicals for teratogenic
potential. Scand J Work Environ Health, 7(4): 66-75.
Harkov R, Kebbekus B, Bozzelli J, & Lioy PJ (1983) Measurement of
selected volatile organic compounds at three locations in New
Jersey during the summer season. J Air Pollut Control Assoc,
33(12): 1177-1183.
Harkov R, Kebbekus B, Bozzelli JW, Lioy P, & Daisy J (1984) Comparison
of selected volatile organic compounds during the summer and winter at
urban sites in New Jersey. Sci Total Environ, 38: 259-274.
Harkov R, Gianti SJ, Bozzelli JW, & LaRegina JE (1985) Monitoring
volatile organic compounds at hazardous and sanitary landfills in New
Jersey. J Environ Sci Health, A20(5): 491-501.
Hasanen E, Soininen V, Pyysalo H, & Leppamaki E (1979) On the
occurrence of aliphatic chlorine and bromine compounds in automobile
exhaust. Atmos Environ, 13: 1217-1219.
Hasanen E, Karlsson V, Leppamaki E, & Juhala M (1981) Benzene, toluene
and xylene concentrations in car exhausts and in city air. Atmos
Environ, 15(9): 1755-1757.
Hayes S, Gordon A, Sadowski I,& Hayes C (1984) RK bacterial test for
independently measuring chemical toxicity and mutagenicity: Short-term
forward selection assay. Mutat Res, 130: 97-106.
Heikes DL (1985a) Purge and trap method for determination of ethylene
dibromide in table ready foods. J Assoc Off Anal Chem, 68(3): 431-436.
Heikes DL (1985b) Purge and trap method for determination of ethylene
dibromide in whole grains, milled grain products, intermediate
grain-based foods and animal feeds. J Assoc Off Anal Chem,
68(6): 1108-1111.
Heikes DL & Hopper ML (1986) Purge and trap method for determination
of fumigants in whole grains, milled grain products and intermediate
grain-based foods. J Assoc Off Anal Chem, 69(6): 990-998.
Hemminki K, Falck K, & Vainio H (1980) Comparison of alkylation rates
and mutagenicity of directly acting industrial and laboratory
chemicals. Arch Toxicol, 46: 277-285.
Herring CO, Adams JA, Wilson BA, & Pollard S (1988) Dose-response
studies using ethylene dibromide(EDB) in Hydra oligactis. Bull
Environ Contam Toxicol, 40: 35-40.
Heuser SG & Scudamore KA (1967) Determination of ethylene
chlorohydrin, ethylene dibromide and other volatile fumigant residues
in flour and whole wheat. Chem Ind, 37: 1557-1560.
Hill DL, Shih TW, Johnston TP, & Struck RF (1978) Macromolecular
binding and metabolism of the carcinogen 1,2-dibromoethane. Cancer
Res, 38: 2438-2442.
Hsu LL, Adams PM, Fanini D, & Legator MS (1985) Ethylene dibromide:
Effects of paternal exposure on the neurotransmitter enzymes in the
developing brain of progeny. Mutat Res, 147: 197-203.
Hughes TJ, Simmons DM, Monteith LG, & Claxton LD (1987) Vaporization
technique to measure mutagenic activity of volatile organic chemicals
in the Ames/Salmonella assay. Environ Mutagen, 9: 421-441.
IARC (1977) Ethylene dibromide. In: Some fumigants, the herbicides
2,4-D and 2,4,5-T, chlorinated dibenzodioxins and miscellaneous
industrial chemicals. Lyon, International Agency for Research on
Cancer, pp 195-209 (IARC Monographs on the Evaluation of Carcinogenic
Risks to Humans, Volume 15).
IARC (1982) Ethylene dibromide. In: Chemicals, industrial processes
and industries associated with cancer in humans - IARC monographs,
volumes 1 to 29. Lyon, International Agency for Research on Cancer,
pp 124-126 (IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, Supplement 4).
IARC (1987) Ethylene dibromide. In: Overall evaluations of
carcinogenicity: An updating of IARC monographs, volumes 1 to 42.
Lyon, International Agency for Research on Cancer, pp 204-205 (IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans,
Supplement 7).
Inskeep PB & Guengerich FP (1984) Glutathione-mediated binding of
dibromoalkanes to DNA: Specificity of rat glutathione-S-transferases
and dibromoalkane structure. Carcinogenesis, 5(6): 805-808.
Inskeep PB, Koga N, Cmarik JL, & Guengerich FP (1986) Covalent binding
of 1,2-dihaloalkanes to DNA and stability of the major DNA adduct,
S-[2-(N7-guanyl)ethyl]glutathione1. Cancer Res, 46: 2839-2844.
IPCS (International Programme on Chemical Safety) (1995) Environmental
health criteria 166: Methyl bromide. Geneva, World Health
Organization, 324 pp.
IRPTC (1993) Legal file. Geneva, International Register of Potentially
Toxic Chemicals, United Nations Environment Programme.
Isaacson PJ, Hankin L, & Frink CR (1984) Boiling drinking water
removes ethylene dibromide. Science, 225: 672.
Ishikuro E (1986) [Quantitative analysis of ethylene dibromide (EDB)
in feed ingredients and mixed feed.] Shiryo Kenkyo Hokoku, 11: 1-7
(in Japanese).
Iwata Y, Dusch ME, & Gunther FA (1983) Use of sulfuric acid cleanup
step in the determination of 1,2-dibromoethane residues in lemons,
oranges, and grapefruits. J Agric Food Chem, 31: 171-174.
Izutani K, Nakata A, Shinagawa H, & Kawamata J (1980) Forward mutation
assay for screening carcinogens by alkaline phosphatase constitutive
mutations in Escherichia coli K-12. J Res Inst Microb Dis (Biken J),
23: 69-75.
Jacobs ES (1980) Use and air quality impact of EDC and EDB scavengers
in leaded gasoline. In: Ames B, Infante P, & Peirtz R ed. Ethylene
dichloride: A potential health risk? Cold Spring Harbor, New York,
Cold Spring Harbor Laboratory, pp 239-255 (Banbury Report No. 5).
Jacobs RS (1985) Ethylene dibromide poisoning. J Am Med Assoc,
253: 2961.
Jafvert CT & Wolfe NL (1987) Degradation of selected halogenated
ethanes in anoxic sediment-water systems. Environ Toxicol Chem,
6: 827-837.
Jagielski J, Scudamore KA, & Heuser SG (1978) Residues of carbon
tetrachloride and 1,2-dibromoethane in cereals and processed foods
after liquid fumigant grain treatment for pest control. Pestic Sci,
9: 117-126.
Jakobson I, Wahlberg JE, Holmberg B, & Johansson G (1982) Uptake via
the blood and elimination of 10 organic solvents following
epicutaneous exposure of anesthetized guinea pigs. Toxicol Appl
Pharmacol, 63: 181-187.
Jean PA & Reed DJ (1992) Utilization of glutathione during
1,2-dihaloethane metabolism in rat hepatocytes. Chem Res Toxicol,
5: 386-391.
Kale PG & Baum JW (1979a) Sensitivity of Drosophila melanogaster to
low concentrations of the gaseous 1,2-dibromoethane: I. Acute
exposures. Environ Mutagen, 1: 15-18.
Kale PG & Baum JW (1979b) Sensitivity of Drosophila melanogaster to
low concentrations of gaseous mutagens: II. Chronic exposures. Mutat
Res, 68: 59-68.
Kale PG & Baum JW (1981) Sensitivity of Drosophila melanogaster to
low concentrations of gaseous mutagens: III. Dose-rate effects.
Environ Mutagen, 3: 65-70.
Kale PG & Baum JW (1982) Genetic effects of ethylene dibromide in
Drosophila melanogaster. In: Tice RR, Costa DL, & Schaich KM ed.
Genotoxic effects of airborne agents. New York, London, Plenum Press,
pp 291-300.
Kale PG & Baum JW (1983) Sensitivity of Drosophila melanogaster to
low concentrations of gaseous mutagens: IV. Mutations in embryonic
spermatogonia. Mutat Res, 113: 135-143.
Kaphalia BS & Ansari GAS (1992) Covalent binding of ethylene dibromide
and its metabolites to albumin. Toxicol Lett, 62: 221-230.
Kato K, Nakamura M, Nakaoka T, & Ito K (1982) [Determination of
ethylene bromide residues in fruits.] Kanagawa-ken Eisei Kenkyosho
Hokoku, 12: 33-34 (in Japanese).
Keough T, Strife RJ, Rodriguez PA, & Sanders RA (1984) Separation and
use of the perdeuterated analogue as an internal standard for the
analysis of ethylene dibromide. J Chromatogr, 312: 450-455.
Kerklaan P, Bouter S, & Mohn G (1983) Isolation of a mutant of
Salmonella typhimurium strain TA1535 with decreased levels of
glutathione (GSH-): Primary characterization and chemical mutagenesis
studies. Mutat Res, 122: 257-266.
Kerklaan P, Zoetemelk CEM, & Mohn GR (1985) Mutagenic activity of
various chemicals in Salmonella strain TA100 and glutathione-deficient
derivatives: On the role of glutathione in the detoxification or
activation of mutagens inside bacterial cells. Biochem Pharmacol,
34(12): 2151-2156.
Khan S, Sood C, & O'Brien PJ (1993) Molecular mechanisms of
dibromoalkane cytotoxicity in isolated rat hepatocytes. Biochem
Pharmacol, 45(2): 439-447.
Kim DH & Guengerich FP (1989) Excretion of the mercapturic acid
S-[2-(N7-guanyl) ethyl]-N-acetylcysteine in urine following
administration of ethylene dibromide to rats. Cancer Res,
49: 5843-5847.
Kim DH & Guengerich FP (1990) Formation of the DNA adduct
S-[2-(N7-guanyl)ethyl] glutathione as an adduct formed in RNA and DNA
from 1,2-dibromoethane. Chem Res Toxicol, 3(6): 587-594.
Kitchin KT & Brown JL (1986) 1,2-dibromoethane causes rat hepatic DNA
damage at low doses. Biochem Biophys Res Commun, 141(2): 723-727.
Kluwe WM, McNish R, Smithson K, & Hook JB (1981) Depletion by
1,2-dibromoethane, 1,2-dibromo-3-chloropropane, tris(2,3-
dibromopropyl)phosphate, and hexachloro-1,3-butadiene of reduced
non-protein sulfhydryl groups in target and non-target organs. Biochem
Pharmacol, 30(16): 2265-2271.
Kluwe WM, Harrington FW, & Cooper SE (1982) Toxic effects of
organohalide compounds on renal tubular cells in vivo and in vitro.
J Pharmacol Exp Ther, 220: 597-603.
Koida K, Tokumori Y, Saimatsu J, Sekigawa K, Kohno U, & Oka A (1986)
[Determination of ethylene dibromide and dibromochloropropane in
groundwater by gas chromatography-negative ion chemical ionization
mass spectrometry.] Hiroshima-shi Eisei Kenkyusho Nenpo, 5: 34-36
(in Japanese).
Kojima T & Seo Y (1976) [Selective detection of alkyl nitrites, alkyl
nitrates, nitroparaffins and halogen compounds by gas chromatography.]
Bunseki Kagaku, 25: 855-858 (in Japanese).
Konishi Y, Yoshida S, & Nakamura A (1985) [Determination of EDB on
capillary ECD-GC.] Osaka-furitsu Koshu Eisei Kenkyusho Kenkyu Hokoku,
Shokuhin Eisei Hen, 16: 63-67 (in Japanese).
Kowalski B, Brittebo EB, & Brandt I (1985) Epithelial binding of
1,2-dibromoethane in the respiratory and upper alimentary tracts of
mice and rats. Cancer Res, 45: 2616-2625.
Krishna G, Xu J, Nath J, Petersen M, & Ong T (1985) In vivo
cytogenetic studies on mice exposed to ethylene dibromide. Mutat Res,
158: 81-87.
Krost KJ, Pellizzari ED, Walburn SG, & Hubbard SA (1982) Collection
and analysis of hazardous organic emissions. Anal Chem, 54: 810-817.
Kubinski H, Gutzke GE, & Kubinski ZO (1981) DNA-cell-binding (DCB)
assay for suspected carcinogens and mutagens. Mutat Res, 89: 95-136.
Kulkarni AP, Edwards J, & Richards IS (1992) Metabolism of
1,2-dibromoethane in the human fetal liver. Gen Pharmacol, 23(1): 1-5.
Landau M & Tucker JW (1984) Acute toxicity of EDB and aldicarb to
young of two estuarine fish species. Bull Environ Contam Toxicol,
33: 127-132.
Ledda-Columbano GM, Columbano A, Coni P, Curto M, Faa G, & Pani P
(1987a) Cell proliferation in rat kidney induced by 1,2-dibromoethane.
Toxicol Lett, 37: 85-90.
Ledda-Columbano GM, Columbano A, Coni P, Liguori C, & Pani P (1987b)
Liver cell proliferation induced by the mitogen ethylene dibromide
unlike compensatory cell proliferation, does not achieve initiation of
rat liver carcinogenesis by diethylnitrosamine. Cancer Lett,
36: 247-252.
Leinster P, Perry R, & Young RJ (1978) Ethylene dibromide in urban
air. Atmos Environ, 12: 2383-2387.
Letz GA, Pond SM, Osterloh JD, Wade RL, & Becker CE (1984) Two
fatalities after acute occupational exposure to ethylene dibromide.
J Am Med Assoc, 252(17): 2428-2431.
Libbey AJ (1986) Determination of ethylene dibromide in aquatic
environments. Analyst, 111: 1221-1222.
Logan SR (1988) Comments on photohydrolysis of ethylene dibromide.
J Agric Food Chem, 36: 872-873.
Lyman WJ (1982) Octanol/water partition coefficient. In: Lyman WJ &
Peehl WF ed. Handbook of chemical property estimation methods -
Environmental behaviour of organic compounds. New York, McGraw-Hill
Publishers, pp 1/1-1/54.
Ma T-H, Sparrow AH, Schairer LA, & Nauman AF (1978) Effect of 1,2
dibromoethane (EDB) on meiotic chromosomes of Tradescantia. Mutat Res,
58: 251-258.
Ma T-H, Harris MM, Anderson VA, Ahmed I, Mohammad K, Bare JL, & Lin G
(1984) Tradescantia-micronucleus (Trad-MCN) tests on 140 health-
related agents. Mutat Res, 138: 157-167.
McCann J, Choi E, Yamasaki E, & Ames BN (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc Natl Acad Sci (USA), 72(12): 5135-5139.
McClenny WA, Pleil JD, Holdren MW, & Smith RN (1984) Automated
cryogenic preconcentration and gas chromatographic determination of
volatile organic compounds in air. Anal Chem, 56: 2947-2951.
McCollister DD, Hollingsworth RL, Oyen F, & Rowe VK (1956) Comparative
inhalation toxicity of fumigant mixtures. Am Med Assoc Arch Ind
Health, 13: 1-7.
McKenry MV & Thomason IJ (1974) 1,3-Dichloropropene and
1,2-dibromoethane compounds: I. Movement and fate as affected by
various conditions in several soils: II. Organism-dosage-response
studies in the laboratory with several nematode species. Hilgardia,
42(11): 393-438.
Mann AM & Darby FJ (1985) Effects of 1,2-dibromoethane on glutathione
metabolism in rat liver and kidney. Biochem Pharmacol,
34(16): 2827-2830.
Mann JB, Freal JJ, Enos HF, & Danauskas JX (1980) Evaluation of
methodology for determining 1,2-dibromoethane (EDB) in ambient air. J
Environ Sci Health, B15(5): 507-518.
Martl LR, de Kanel J, & Dougherty RC (1984) Screening for organic
contamination of groundwater: ethylene dibromide in Georgia irrigation
wells. Environ Sci Technol, 18: 973-974.
Mayer JR, Lacher TE, Elkins NR, & Thorn CJ (1991) Temporal variation
of ethylene dibromide (EDB) in an unconfined aquifer, Whatcom County,
Washington, USA: a twenty-seven month study. Bull Environ Contam
Toxicol, 47: 368-373.
Meneghini R (1974) Repair replication of opossum lymphocyte DNA:
effect of compounds that bind to DNA. Chem-Biol Interact, 8: 113-126.
Mestres R, Atmawijaya S, & Francois CL (1980) XXXV. Méthode de
recherche et de dosage des résidus de pesticides dans les produits
céréaliers. Ann Fals Exp Chim, 73(788): 407-420.
MHW (1985) Prohibition of EDB residues in imported wheat, tropical
fruit etc. Official notice of the Ministry of Health and Welfare.
Tokyo, Ministry of Health and Welfare.
MHW (1987) Prohibition of EDB residues in imported wheat, tropical
fruit etc. Official notice of the Ministry of Health and Welfare.
Tokyo, Ministry of Health and Welfare.
MHW (1988) Prohibition of EDB residues in imported wheat, tropical
fruit etc. Official notice of the Ministry of Health and Welfare.
Tokyo, Ministry of Health and Welfare.
Mitra A, Hilbelink DR, Dwornik JJ, & Kulkarni A (1992) A novel model
to assess developmental toxicity of dihaloalkanes in humans:
Bioactivation of 1,2-dibromoethane by the isozymes of human fetal
liver glutathione S-transferase. Teratog Carcinog Mutagen,
12(3): 113-127.
Mohn GR, Kerklaan PRM, Van Zeeland AA, Ellenberger J, Baan RA, Lohman
PHM, & Pons F-W (1984) Methodologies for the determination of various
genetic effects in permeable strains of E. coli K-12 differing in
DNA repair capacity: Quantification of DNA adduct formation,
experiments with organ homogenates and hepatocytes, and animal-
mediated assays. Mutat Res, 125: 153-184.
Moriya M, Ohta T, Watanabe K, Miyazawa T, Kato K, & Shirasu Y (1983)
Further mutagenicity studies on pesticides in bacterial reversion
assay systems. Mutat Res, 116: 185-216.
Morris SC & Rippon LE (1985) Distribution of ethylene dibromide within
a fumigation chamber during fumigation of citrus fruit. J Agric Food
Chem, 33: 801-803.
Morris SC, Rippon LE, & Halamek R (1982) Rapid analysis and gas
sampling of ethylene dibromide in the gaseous phase and as residues in
citrus fruit. J Chromatogr, 246: 136-140.
Mundhe DR & Pandey ND (1980) Effect of different foods on the
susceptibility of Callosobruchus chinensis Linn, to some fumigants.
Indian J Entomol, 42: 787-790.
Munnecke DE & Van Gundy SD (1979) Movement of fumigants in soil,
dosage responses, and differential effects. Annu Rev Phytopathol,
17: 405-429.
Nachtomi E (1970) The metabolism of ethylene dibromide in the rat: the
enzymic reaction with glutathione in vitro and in vivo. Biochem
Pharmacol, 19: 2853-2860.
Nachtomi E & Farber E (1978) Ethylene dibromide as a mitogen for
liver. Lab Invest, 38(3): 279-283.
Nachtomi E & Sarma DSR (1977) Repair of rat liver DNA in vivo
damaged by ethylene dibromide. Biochem Pharmacol, 26: 1941-1945.
Nachtomi E, Alumot E, & Bondi A (1968) Biochemical changes in organs
of chicks and rats poisoned with ethylene dibromide and carbon
tetrachloride. Israel J Chem, 6: 803-811.
Nakamura M (1986) [Analytical method for ethylene dibromide in wheat
products using capillary column GC-mass spectrometry.] Fukuoka-shi
Eisei Shikenshiho, 11: 47-53 (in Japanese).
Nauman CH, Sparrow AH, & Schairer LA (1976) Comparative effects of
ionizing radiation and two gaseous chemical mutagens on somatic
mutation induction in one mutable and two non-mutable clones of
Tradescantia. Mutat Res, 38: 53-70.
NCI (1978) Bioassay of 1,2-dibromoethane for possible carcinogenicity.
Bethesda, Maryland, National Cancer Institute, Division of Cancer
Cause and Prevention, Carcinogenesis Testing Program, 64 pp (Technical
Report Series No 86; DHEW Publication No. (NIH) 78-1336).
Nemoto Y, Sasamoto T, Nakata M, Suzuki Y, Kubota K, & Kurokawa K
(1984) [Survey of EDB groundwaters in Ibaraki Prefecture.] Ibaraki-ken
Eisei Kenkyujo Nenpo, 22: 80-81 (in Japanese).
Nichols WK, Covington MO, Seiders CD, Safiullah S, & Yost GS (1992)
Bioactivation of halogenated hydrocarbons by rabbit pulmonary cells.
Pharmacol Toxicol, 71(5): 335-339.
Nilsson CA, Lindahl R, & Norstrom A (1987) Occupational exposure to
chain saw exhausts in logging operations. Am Ind Hyg Assoc J,
48(2): 99-105.
NIOSH (1977) Criteria for a recommended standard: occupational
exposure to ethylene dibromide. Cincinnati, Ohio, National Institute
for Occupational Safety and Health, 208 pp (DHEW (NIOSH) Publication
No. 77-221).
NIOSH (1981) Current intelligence bulletin No. 37: Ethylene bromide
(revised). Cincinnati, Ohio, National Institute for Occupational
Safety and Health, 13 pp (Publication No. 82-105).
Nishiuchi Y (1980) [Toxicity of pesticide formulas to some fresh water
organisms - LXX.] Suisan Zoushoku, 27(4): 225-237 (in Japanese).
Nishiuchi Y (1981) [Toxicity of pesticides to some aquatic organisms:
I. Toxicity of pesticides to some aquatic insects.] Seitai Kagaku,
4(2): 31-46 (in Japanese).
Nishiuchi Y (1982) [Toxicity of pesticide formulas to some fresh water
organisms: effects of pH on toxicity revelation.] Suisan Zoushoku,
30(3): 172-177 (in Japanese).
Nishiuchi Y & Asano K (1979) [Toxicity of pesticide formulas to some
fresh water organisms - LXVI.] Suisan Zoushoku, 27(3): 172-177 (in
Japanese).
Nitschke KD, Kociba RJ, Keyes DG, & Mckenna MJ (1981) A thirteen week
repeated inhalation study of ethylene dibromide in rats. Fundam Appl
Toxicol, 1: 437-442.
NTP (1982) Carcinogenesis bioassay of 1,2-dibromoethane in F344 rats
and B6C3F1 mice (inhalation study). Research Triangle Park, North
Carolina, US Department of Health and Human Services, National
Toxicology Program, 163 pp (NTP Technical Report Series No. 210).
Ogino Y (1978) [Study of the effect of the environment - polluting
substances on ecological systems: I. Translocation of
bromohydrocarbons to the fish body.] Okayamo Igakkai Zasshi,
90: 1451 -1455 (in Japanese).
Ohta T, Nakamura N, Moriya M, Shirasu Y, & Kada T (1984) The
SOS-function-inducing activity of chemical mutagens in Escherichia
coli. Mutat Res, 131: 101-109.
Ong T-M, Stewart JD, Wen Y-F, & Whong W-Z (1987) Application of SOS
umu-test for the detection of genotoxic volatile chemicals and air
pollutants. Environ Mutagen, 9: 171-176.
Ong T-M, Stewart JD, Tucker JD, & Whong W-Z (1989) Development of an
in situ test system for detection of mutagens in the workplace. In:
Waters MD, Sandhu SS, Lewtas J, Claxton L, Strauss G, & Nesnow S, ed.
Short-term bioassays in the analysis of complex environmental
mixtures, IV. New York, Plenum Publishing Corporation, pp 25-36.
OSHA (Occupational Safety and Health Agency) (1983) Occupational
exposure to ethylene dibromide. Fed Reg, 48(196): 45956-46003.
Ott MG, Scharnweber HC, & Langner RR (1980) Mortality experience of
161 employees exposed to ethylene dibromide in two production units.
Br J Ind Med, 37: 163-168.
Ozawa N & Guengerich FP (1983) Evidence for formation of an
S-[2-(N7-guanyl)ethyl] glutathione adduct in glutathione-mediated
binding of the carcinogen 1,2-dibromoethane to DNA. Proc Natl Acad Sci
(USA), 80: 5266-5270.
Page GW (1981) Comparison of groundwater and surface water for
patterns and levels of contamination by toxic substances. Environ Sci
Technol, 15(12): 1475-1481.
Peoples SA, Maddy KT, & Riddle LC (1978) Human occupational health
problems resulting from exposure to ethylene dibromide in California
in 1975 and 1976. Vet Hum Toxicol, 20: 241-244.
Perocco P & Prodi G (1981) DNA damage by haloalkanes in human
lymphocytes cultured in vitro. Cancer Lett, 13: 213-218.
Perocco P, Colacci A, Santucci MA, Vaccari M, & Grilli S (1991)
Transforming activity of ethylene dibromide in BALB/c 3T3 cells. Res
Commun Chem Pathol Pharmacol, 73(2): 159-172.
Pignatello JJ (1986) Ethylene dibromide mineralization in soils under
aerobic conditions. Appl Environ Microbiol, 51(3): 588-592.
Pignatello JJ & Cohen SZ (1989) Environmental chemistry of ethylene
dibromide in soil and ground water. Rev Environ Contam Toxicol,
112: 1-47.
Pignatello JJ, Sawhney BL, & Frink CR (1987) EDB: Persistence in soil.
Science, 236: 898.
Pleil JD, Oliver KD, & McClenny WA (1987) Enhanced performance of
nation dryers in removing water from air samples prior to gas
chromatographic analysis. JAPCA, 37(30): 244-248.
Plotnick HB & Conner WL (1976) Tissue distribution of 14C-labeled
ethylene dibromide in the guinea pig. Res Commun Chem Pathol
Pharmacol, 13(2): 251-259.
Plotnick HB, Weigel WW, Richards DE, & Cheever KL (1979) The effect of
dietary disulfiram upon the tissue distribution and excretion of
14C-1,2-dibromoethane in the rat. Res Commun Chem Pathol Pharmacol,
26(3): 535-545.
Pranoto-Soetardhi LA, Rijk MAH, De Kruijf N, & De Vos RH (1986)
Automatic headspace sampling for determination of ethylene dibromide
residues in cereals. Int J Environ Anal Chem, 25: 151-159.
Principe P, Dogliotti E, Bignami M, Crebelli R, Falcone E, Fabrizi M,
Conti G, & Comba P (1981) Mutagenicity of chemicals of industrial and
agricultural relevance in Salmonella, Streptomyces and Aspergillus. J
Sci Food Agric, 32: 826-832.
Quillardet P, De Bellecombe C, & Hofnung M (1985) The SOS chromotest,
a colorimetric bacterial assay for genotoxins: validation study with
83 compounds. Mutat Res, 147: 79-95.
Rains DM & Holder JW (1981) Technical Communications: Ethylene
dibromide residues in biscuits and commercial flour. J Assoc Off Anal
Chem, 64(5): 1252.
Rannug U (1980) Genotoxic effects of 1,2-dibromoethane and
1,2-dichloroethane. Mutat Res, 76: 269-295.
Rannug U & Beije B (1979) The mutagenic effect of 1,2-dichloroethane
on Salmonella typhimurium. II. Activation by the isolated perfused
rat liver. Chem-Biol Interact, 24: 265-285.
Rappaport SM, Cameron W, & McAllister J (1984) Outgassing of ethylene
dibromide from fumigated oranges. J Agric Food Chem, 32(5): 1112-1116.
Rasmussen RA & Khalil MAK (1984) Gaseous bromine in the arctic and
arctic haze. Geophys Res Lett, 11(5): 433-436.
Ratcliffe JM, Schrader SM, Steenland K, Clapp DE, Turner T, & Hornung
RW (1987) Semen quality in papaya workers with long term exposure to
ethylene dibromide. Br J Ind Med, 44: 317-326.
Reznik G, Stinson SF, & Ward JM (1980) Respiratory pathology in rats
and mice after inhalation of 1,2-dibromo-3-chloropropane or 1,2
dibromoethane for 13 weeks. Arch Toxicol, 46: 233-240.
Rogers RD & McFarlane JC (1981) Sorption of carbon tetrachloride,
ethylene dibromide, and trichloroethylene in soil and clay. Environ
Monit Assess, 1: 155-162.
Rosenkranz HS (1977) Mutagenicity of halogenated alkanes and their
derivatives. Environ Health Perspect, 21: 79-84.
Roskill (1992) The economics of bromine, 1992. London, Roskill
Information Services, pp 57-58.
Rowe VK, Spencer HC, McCollister DD, Hollingsworth RL, & Adams EM
(1952) Toxicity of ethylene dibromide determined on experimental
animals. Am Med Assoc Arch Ind Hyg Occupat Med, 6: 158-173.
Rumsey DW & Tanita RK (1978) NIOSH industrial hygiene survey.
Cincinnati, Ohio, National Institute for Occupational Safety and
Health, 20 pp (Publication No. 79-112).
Russell WL(1986) Positive genetic hazard predictions from short-term
tests have proved false for results in mammalian spermatogonia with
all environmental chemicals so far tested. In: Genetic toxicology of
environmental chemicals - Part B: Genetic effects and applied
mutagenesis. New York, Alan R. Liss Inc., pp 67-74.
Saito N, Ogino Y, Katayama Y, Matsunaga K, Nagao M, & Ishida T (1978)
[Determination of aliphatic brominated hydrocarbons in environmental
samples.] Kogaito Taisaku, 14(3): 316-320 (in Japanese).
Sarawat PK, Kandara M, Dhurva AK, Malhotra VK, & Jhanwar RS (1986)
Poisoning by ethylene dibromide - six cases: A clinicopathological and
toxicological study. Indian J Med Sci, 40(5): 121-123.
Sawyer LD & Walters SM (1986) Gas chromatographic method for ethylene
dibromide in grains and grain-based products: Collaborative study. J
Assoc Off Anal Chem, 69(5): 847-851.
Schlinke JC (1970) Toxicologic effects of five soil nematocides in
chickens. Am J Vet Res, 31(6): 1119-1121.
Schrader SM, Ratcliffe JM, Turner T, & Hornung RW (1987) The use of
new field methods of semen analysis in the study of occupational
hazards to reproduction the example of ethylene dibromide. J Occup
Med, 29: 963-966.
Schrader SM, Turner TW, & Ratclife JM (1988) The effects of ethylene
dibromide on semen quality: A comparison of short-term and chronic
exposure. Reprod Toxicol, 2: 191-198.
Schultz K, Ghosh L, & Banerjee S (1992) Neoplastic expression in
murine cells induced by halogenated hydrocarbons. In vitro Cell Dev
Biol, 28A: 267-272.
Scott BR, Sparrow AH, Schwemmer SS, & Schairer LA (1978) Plant
metabolic activation of 1,2-dibromoethane (EDB) to a mutagen of
greater potency. Mutat Res, 49: 203-212.
Sega GA & Rene E (1980) Chemical dosimetry and unscheduled DNA
synthesis studies of ethylene dibromide in the germ cells of male
mice. Environ Mutagen, 5: 274.
Sekita H, Takeda M, & Uchiyama M (1981) [Studies on analysis of
pesticide residues in foods. XXXIII. Determination of ethylene
dibromide residues in litchi (lychee) fruits imported from Formosa.]
Bull Natl Inst Hyg Sci (Tokyo), 99: 130-132 (in Japanese).
Sekita H, Takeda M, & Uchiyama M (1983) [Ethylene dibromide (EDB)
residues in fresh fruits imported after fumigation with EDB and
decrease of the residues with time.] Shokuhin Eisei Gaku Zasshi,
24(1): 57-63 (in Japanese).
Shiau SY, Huff RA, Wells BC, & Felkner IC (1980) Mutagenicity and
DNA-damaging activity for several pesticides tested with Bacillus
subtilis mutants. Mutat Res, 71: 169-179.
Shivanandappa T, Krishnakumari MK, & Majumder SK (1987) Reproductive
potential of male rats fed dietary ethylene dibromide. J Food Saf,
8: 147-155.
Short RD, Minor JL, Winston JM, Seifter J, & Lee CC (1978) Inhalation
of ethylene dibromide during gestation by rats and mice. Toxicol Appl
Pharmacol, 46: 173-182.
Short RD, Winston JM, Hong CB, Minor JL, Lee CC, & Seifter J (1979)
Effects of ethylene dibromide on reproduction in male and female rats.
Toxicol Appl Pharmacol, 49: 97-105.
Simula TP, Glancey MJ, & Wolf CR (1993) Human glutathione
S-transferase-expressing Salmonella typhimurium tester strains to
study the activation/detoxification of mutagenic compounds: studies
with halogenated compounds, aromatic amines and aflatoxin B1.
Carcinogenesis, 14: 1371-1376.
Sina JF, Bean CL, Dysart GR, Taylor VI, & Bradley MO (1983) Evaluation
of the alkaline elution/rat hepatocyte assay as a predictor of
carcinogenic/mutagenic potential. Mutat Res, 113: 357-391.
Singh HB, Salas LJ, Smith AJ, & Shigeishi H (1981) Measurements of
some potentially hazardous organic chemicals in urban environments.
Atmos Environ, 15: 601-612.
Singh HB, Salas LJ, & Stiles RE (1982) Distribution of selected
gaseous organic mutagens and suspect carcinogens in ambient air.
Environ Sci Technol, 16(12): 872-880.
Singh HB, Salas LJ, & Stiles RE (1983) Selected man-made halogenated
chemicals in the air and oceanic environment. J Geophys Res,
88(C6): 3675-3683.
Sipes IG, Wiersma DA, & Armstrong DJ (1986) The role of glutathion in
the toxicity of xenobiotic compounds: Metabolic activation of
1,2-dibromoethane by glutathione. Adv Exp Med Biol, 197: 457-467.
Smith RF & Goldman L (1983) Behavioral effects of prenatal exposure to
ethylene dibromide. Neurobehav Toxicol Teratol, 5: 579-585.
Sparrow AH, Schairer LA, & Villalobos-Pietrini R (1974) Comparison of
somatic mutation rates induced in Tradescantia by chemical and
physical mutagens. Mutat Res, 26: 265-276.
Springarn NE, Northington DJ, & Pressely T (1982) Analysis of volatile
hazardous substances by GC/MS. J Chromatogr Sci, 20: 286-288.
SRI International (1982) Ethylene dibromide: US salient statistics -
Chemical economics handbook. Menlo Park, California, Stanford Research
International.
Steenland K, Carrano A, Ratcliffe J, Clapp D, Ashworth L, & Meinhardt
T (1986) A cytogenetic study of papaya workers exposed to ethylene
dibromide. Mutat Res, 170: 151-160.
Steinberg SM, Pignatello JJ, & Sawhney BL (1987) Persistence of
1,2-dibromomethane in soils: entrapment in intraparticle micropores.
Environ Sci Technol, 21: 1202-1208.
Stenger VA (1978) Bromine compounds. In: Kirk-Othmer encyclopedia of
chemical technology, 3rd ed. New York, John Wiley, vol 4, pp 243-263.
Stinson SF, Reznik G, & Ward JM (1981) Characteristics of
proliferative lesions in the nasal cavities of mice following chronic
inhalation of 1,2-dibromoethane. Cancer Lett, 12: 121-129.
Stolzenberg SJ & Hine CH (1980) Mutagenicity of 2-and 3-carbon
halogenated compounds in the Salmonella/mammalian-microsome test.
Environ Mutagen, 2: 59-66.
Storer RD & Conolly RB (1983) Comparative in vivo genotoxicity and
acute hepatotoxicity of three 1,2-dihaloethanes. Carcinogenesis,
4(11): 1491-1494.
Stottmeister E, Hendel P, & Engewald W (1986) [Gas-chromatographic
trace analysis of highly volatile halogenated hydrocarbons in water
with FID detection.] Acta Hydrochim Hydrobiol, 14(6): 573-580 (in
German).
Sugiyama H (1980) Effects of EDB (1,2-dibromoethane) on the silkworm
(Bombyx mori L.). J Pestic Sci, 5: 599-602.
Sun M (1984) EDB contamination kindles federal action. Science,
233: 464-466.
Sundheimer DW, White RD, & Sipes IG (1982) The bioactivation of
1,2-dibromoethane in rat hepatocytes: Covalent binding to nucleic
acids. Carcinogenesis, 3(10): 1129-1133.
Tan E-L & Hsie AW (1981) Mutagenicity and cytotoxicity of haloethanes
as studied in the CHO/HGPRT system. Mutat Res, 90: 183-191.
Tennant RW, Stasiewicz S, & Spalding JW (1986) Comparison of multiple
parameters of rodent carcinogenicity and in vitro genetic toxicity.
Environ Mutagen, 8: 205-227.
Tennant RW, Margolin BH, Shelby MD, Zeiger E, Haseman JK, Spalding J,
Caspary W, Resnick M, Stasiewicz S, Anderson B, & Minor R (1987)
Prediction of chemical carcinogenicity in rodents from in vitro
genetic toxicity assays. Science, 236: 933-941.
Teramoto S, Saito R, Aoyama H, & Shirasu Y (1980) Dominant lethal
mutation induced in male rats by 1,2-dibromo-3-chloropropane (DBCP).
Mutat Res, 77: 71-78.
Terao H, Kajikawa M, Morishita Y, & Kato K (1984) [Bromide ion
concentration and Br/Cl ratio of ground water in the southwest region
of Gifu Prefecture.] Chikyu Kagaku, 18: 21-28 (in Japanese).
Terao H, Kajikawa M, Morishita K, & Kato K (1985) [Influence of
pesticide and fertilizer on the ground water in vegetable field zone.]
Chikyu Kagaku, 19: 31-38 (in Japanese).
Ter Haar G (1980) An investigation of possible sterility and health
effects from exposure to ethylene dibromide. In: Ames B, Infante O, &
Peirtz R ed. Ethylene dichloride: A potential health risk? Cold Spring
Harbor, New York, Cold Spring Harbor Laboratory, pp 167-188 (Banbury
Report No. 5).
Tezuka H, Ando N, Suzuki R, Terahata M, Moriya M, & Shirasu Y (1980)
Sister-chromatid exchanges and chromosomal aberrations in cultured
Chinese hamster cells treated with pesticides positive in microbial
reversion assays. Mutat Res, 78: 177-191.
Thier R, Taylor JB, Pemble SE, Humphreys WG, Persmark M, Ketterer B, &
Guengerich FP (1993) Expression of mammalian glutathione S-transferase
5-5 in Salmonella typhimurium TA1535 leads to base-pair mutations
upon exposure to dihalomethanes. Proc Natl Acad Sci (USA),
90: 8576-8580.
Townshend JL, Dirks VA, & Marks CF (1980) Temperature, moisture and
compaction and their effects on the diffusion of ethylene dibromide in
three Ontario soils. Can J Soil Sci, 60: 177-184.
Tsani-Bazaca E, McIntyre AE, Lester JN, & Perry R (1981)
Concentrations and correlations of 1,2-dibromoethane,
1,2-dichloroethane, benzene and toluene in vehicle exhaust and ambient
air. Environ Technol Lett, 2: 303-316.
Tucker JD, Xu J, Stewart J, & Ong T-M (1984) Detection of sister-
chromatid exchanges in human peripheral lymphocytes induced by
ethylene dibromide vapor. Mutat Res, 138: 93-98.
UNEP (1992) Methyl bromide and the ozone layer: a summary of current
understanding. Montreal Protocol assessment supplement: Synthesis
report of the methyl bromide interim scientific assessment and methyl
bromide interim technology and economic assessment requested by the
United Nations Environment Programme on behalf of the Contracting
Parties to the Montreal Protocol. Nairobi, United Nations Environment
Programme.
US EPA (1977) EPA notice of rebuttable presumption against
registration and continued registration of pesticide products
containing ethylene dibromide. Fed Reg, 42: 63134.
US EPA (1986) US EPA reports results of study of pesticides in
groundwater. J Am Water Works Assoc, 1986: 128-129.
Van Bladeren PJ, Breimer DD, Rotteveel-Smijs GMT, De Jong RAW, Buus W,
Van Der Gen A, & Mohn GR (1980) The role of glutathione conjugation in
the mutagenicity of 1,2-dibromoethane. Biochem Pharmacol,
29: 2975-2982.
Van Bladeren PJ, Hoogeterp JJ, Breimer DD, & Van Der Gen A (1981a) The
influence of disulfiram and other inhibitors of oxidative metabolism
on the formation of 2-hydroxyethyl-mercapturic acid from
1,2-dibromoethane by the rat. Biochem Pharmacol, 30(21): 2983-2987.
Van Bladeren PJ, Breimer DD, Rotteveel-Smijs GMT, De Knijff P, Mohn
GT, Van Meeteren-Walchli B, Buijs W, & Van der Gen A (1981b) The
relation between the structure of vicinal dihalogen compounds and
their mutagenic activation via conjugation to glutathione.
Carcinogenesis, 2(6): 499-505.
Van Duuren BL, Goldschmidt BM, Loewengart G, Smith AC, Melchlonne S,
Seldman I, & Roth D (1979) Carcinogenicity of halogenated olefinic and
aliphatic hydrocarbons in mice. J Natl Cancer Inst, 63(6): 1433-1439.
Van Duuren BL, Seidman I, Melchionne S, & Kline SA (1985)
Carcinogenicity bioassays of bromoacetaldehyde and bromoethanol-
potential metabolites of dibromoethane. Teratogen Carcinogen Mutagen,
5: 393-403.
Vant'Hof J & Schairer LA (1982) Tradescantia assay system for gaseous
mutagens: A report of the US Environmental Protection Agency Gene-Tox
Program. Mutat Res, 99: 303-315.
Verschueren K (1983) Handbook of environmental data on organic
chemicals, 2nd ed. New York, Van Nostrand Reinhold, pp 635-636.
Vogel E & Chandler JLR (1974) Mutagenicity testing of cyclamate and
some pesticides in Drosophila melanogaster. Experientia (Basel),
30: 621-623.
Von Buselmaier W, Rohrborn G, & Propping P (1972) [Mutagenicity
investigations with pesticides in the host mediated assay and the
dominant lethal test in mice.] Biol Zent.bl, 91: 311-325 (in German).
Wade NL & Rigney CJ (1979) Phytotoxicity of ethylene dibromide to
cherry and banana fruit. J Am Soc Hortic Sci, 104(6): 900-903.
Weast RC, Astle MJ, & Beyer WH (1988) CRC Handbook of chemistry and
physics, 68th ed. Boca Raton, Florida, CRC Press, p C-264.
White RD (1982) Chemical induction of genetic injury: The
bioactivation of 1,2-dibromoethane. Diss Abstr Int, 43(03): 696B-697B.
White RD, Gandolfi AJ, Bowden GT, & Sipes IG (1983) Deuterium isotope
effect on the metabolism and toxicity of 1,2-dibromoethane. Toxicol
Appl Pharmacol, 69: 170-178.
White RD, Petry TW, & Sipes IG (1984) The bioactivation of
1,2-dibromoethane in rat hepatocytes: deuterium isotope effect.
Chem-Biol Interact, 49: 225-233.
WHO (1993) Guidelines for drinking-water quality: Volume 1 - WHO
Recommendations, 2nd ed. Geneva, World Health Organization, 130 pp.
Wildeman AG & Nazar RN (1982) Significance of plant metabolism in the
mutagenicity and toxicity of pesticides. Can J Genet Cytol,
24: 437-449.
Williams GM, Laspia MF, & Dunkel VC (1982) Reliability of the
hepatocyte primary culture/DNA repair test in testing of coded
carcinogens and noncarcinogens. Mutat Res, 97: 359-370.
Williams J, Gladen BC, Turner TW, Schrader SM, & Chapin RE (1991) The
effects of ethylene dibromide on semen quality and fertility in the
rabbit: Evaluation of a model for human seminal characteristics.
Fundam Appl Toxicol, 16: 687-700.
Wofsy SC, McElroy MB, & Yung YL (1975) The chemistry of atmospheric
bromine. Geophys Res Lett, 2(6): 215-218.
Wong LCK, Winston JM, Hong CB, & Plotnick H (1982) Carcinogenicity and
toxicity of 1,2-dibromoethane in the rat. Toxicol Appl Pharmacol,
63: 155-165.
Wong O, Morgan RW, & Whorton MD (1985) An epidemiologic surveillance
program for evaluating occupational reproductive hazards. Am J Ind
Med, 7: 295-306.
Woodrow JE, Majewski MS, & Seiber JN (1986) Accumulative sampling of
trace pesticides and other organics in surface water using XAD-4
resin. J Environ Sci Health, B21(2): 143-164.
Working PK, Smith-Oliver T, White RD, & Butterworth BE (1986)
Induction of DNA repair in rat spermatocytes and hepatocytes by
1,2-dibromoethane: the role of glutathione conjugation.
Carcinogenesis, 7(3): 467-472.
Yoshida YH & Inagaki E (1986) [Mutagenicity of ethylene dibromide in
Drosophila melanogaster]. Tachikawa Tandai Kiyo, 19: 49-50
(in Japanese).
Yuita K (1984) [Residues and pollutions of bromine derived from soil
fumigants and disinfectants crops, soils and ground water.] Seitai
Kogaku, 7(2): 3-12 (in Japanese).
Zoetemelk CEM, Mohn GR, Van Der Gen A, & Breimer DD (1987)
Mutagenicity in Salmonella strains differing in glutathione content
and their alkylating potential. Biochem Pharmacol, 36(11): 1829-1835.
Résumé
1. Identité, propriétés physiques et chimiques, et méthodes d'analyse
Le 1,2-dibromoéthane (dibromure d'éthylène) est un liquide
incolore d'odeur chloroformique dont le point de fusion est de 9,9°C
et le point d'ébullition de 131,4°C. Il est assez volatil, avec une
tension de vapeur de 1,47 kPa (11 mmHg) à 25°C et une densité de
vapeur par rapport à l'air de 6,1. Le 1,2-dibromoéthane est miscible
à la plupart des solvants organiques. Sa solubilité dans l'eau est de
4,3 g/litre à 30°C.
Dans l'air ambiant, l'analyse s'effectue par chromatographie en
phase gazeuse après absorption sur polymères poreux puis désorption
thermique rapide. Pour les échantillons d'eau, on utilise un système
avec piège et purgeur. Les résidus de 1,2-dibromoéthane présents dans
les denrées alimentaires et d'autres milieux peuvent être soit
extraits par solvent, soit soumis à une analyse par la technique de
l'espace de tête dans des conditions cryogéniques, après quoi on
prépare un dérivé et on poursuit par chromatographie en phase gazeuse
ou chromatographie liquide à haute performance.
2. Sources d'exposition humaine et environnementale
Le 1,2-dibromoéthane est utilisé comme agent d'épuration des
agents antidétonnants à base de plomb ajoutés à l'essence. On
l'emploie aussi pour la fumigation du sol ou de certains fruits ou
céréales. L'essence additionnée de plomb étant moins utilisée dans
certains pays et les homologations pour les usages agricoles ayant été
annulées, l'exposition humaine à ce produit a diminué. Il reste
cependant encore en usage dans certains pays comme épurateur de
l'essence au plomb, comme fumigant, au fins de quarantaine, comme
solvent et comme intermédiaire dans l'industrie chimique.
3. Concentrations et dégradation dans l'environnement
Dans l'air, on mesure des concentrations qui vont de zéro à des
teneurs de l'ordre du ng/m3 en zone urbaine. On a trouvé du
1,2-dibromoéthane dans des eaux souterraines à des concentrations
allant jusqu'à 0,2 µg/litre, la teneur pouvant atteindre 50 µg/litre
dans les eaux superficielles des zones d'exploitation agricole
intensive. Bien qu'il y ait lessivage du 1,2-dibromoéthane à travers
le sol, il en reste une certaine quantité dans la matrice
édaphique,d'où risque de contamination ultérieure de la nappe
phréatique. On connaît mal les conditions de dégradation microbienne
du 1,2-dibromoéthane dans le sol.
Le composé étant très volatil, c'est l'atmosphère qui en est le
principal réceptacle. Par photolyse dans la stratosphère, il peut se
former des produits de décomposition susceptibles d'attaquer la couche
d'ozone.
4. Cinétique et métabolisme chez les animaux de laboratoire
Le 1,2-dibromoéthane est rapidement absorbé par la voie orale,
percutanée et respiratoire. On pense que la toxicité du composé est
largement due à ses métabolites. La métabolisation s'effectue soit
par une voie oxydative (cytochrome P-450), soit par conjugaison
(glutathion- S-transférase). Deux métabolites réactifs, le
bromoacétaldéhyde formé par la voie oxydative, et l'ion thiiranium,
formé par conjugaison, interagissent avec les macromolécules
cellulaires (protéines, ADN), pour donner naissance à divers produits
par l'établissement de liaisons covalentes.
5. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
Le 1,2-dibromoéthane est fortement toxique pour les animaux
(DL50 par voie orale pour le rat égale à 146-417 mg/kg de poids
corporel; CL50 inhalatoire pour le rat égale à 3080 mg/m3 après
2 h d'exposition; mortalité observée chez des lapins à la suite d'une
application cutanée à raison de 210 mg/kg). Les effets toxiques
observés se sont produits principalement au niveau des reins et du
foie. L'inhalation de vapeurs provoque une irritation de la muqueuse
nasale et une dépression du système nerveux central. Chez des groupes
de rats exposés à des concentrations comprises entre 1540 et
77 000 mg/m3 (200-10 000 ppm) pendant 0,1 à 16,0 h, on a observé
dans tous les groupes une mortalité qui était fonction de la durée
d'exposition et de la concentration. En solution à 1,0%, le
1,2-dibromoéthane a provoqué une irritation sur la peau abdominale de
lapins après rasage ainsi qu'une irritation oculaire.
Chez des rats et des souris qui avaient reçu du 1,2-dibromoéthane
par voie orale de manière subchronique, on a observé une certaine
mortalité et une baisse du gain de poids à la dose quotidienne de
100 mg/kg de poids corporel. Chez des rats exposés au composé à la
dose de 115 mg/m3 (578 ppm), 6 h par jour, 5 jours par semaine et
ce, pendant 13 semaines, on a noté un moindre gain de poids et des
effets pathologiques au niveau du nez. Cette étude a permis d'établir
que la dose sans effets histopathologiques observables au niveau de la
cavité nasale était égale à 23 mg/m3 (3 ppm). Lors d'une étude
analogue sur des souris, on a observé le même genre d'effets
histopathologiques avec la même dose sans effets observables
(23 mg/m3, 3 ppm).
On a administré du 1,2-dibromoéthane par gavage à des rats selon
les modalités suivantes: 37-107 mg/kg de poids corporel (moyenne
pondérée par rapport au temps) tous les jours pendant 49-90 semaines;
de même des souris en ont reçu pendant 15-17 mois dans leur eau de
boisson à raison de 103-117 mg/kg de poids corporel. A la suite de
cela, on a observé des anomalies non malignes telles qu'une
dégénérescence hépatique, une atrophie testiculaire, ainsi qu'une
acanthose et une hyperkératose au niveau de la portion cardiaque de
l'estomac. Après exposition par la voie respiratoire de rats et de
souris à des doses de 77-388 mg/m3 pendant 6 à 18 mois, on a observé
une inflammation de la trachée et de la cavité nasale, une
dégénérescence testiculaire et une nécrose hépatique.
Après exposition par la voie respiratoire, le 1,2-dibromoéthane
ne se révèle pas tératogène pour le rat ou la souris. Chez des rats
qui en avaient reçu par la voie intrapéritonéale une dose quotidienne
de 1,25 mg/kg de poids corporel (mâles) ou de 509 mg/m3 (voie
respiratoire, femelles, 4 h/jour, 3 jours par semaine, du jour 3 au
jour 20 de la gestation), on a constaté une action toxique sur le
développement (anomalies de la coordination motrice). Le
1,2-dibromoéthane a également eu une action délétère sur la fonction
de reproduction de rats (chez les mâles, dans les conditions
d'exposition suivantes: 684 mg/m3, 7 h/jour, 5 jours par semaine,
pendant 10 semaines; chez les femelles, aux doses et pendant les
durées suivantes: 614 mg/m3, 7 h/jour, 7 jours par semaine, pendant
3 semaines). La dose sans effet observable pour ce paramètre était
égale à 300 mg/m3 chez les deux sexes. Lors d'une étude où des rats
mâles ont reçu pendant 90 jours une alimentation contenant le
composé, on a constaté que la dose sans effet observable sur la
capacité de reproduction était égale à 50 mg/kg et par jour. On a
observé une atteinte de la spermatogénèse chez des taureaux après
administration du composé par voie orale à la dose quotidienne de
2 mg/kg pendant moins de 21 jours et chez des lapins après injection
sous-cutanée du produit à raison de 15 mg/kg pendant 5 jours. Chez
des poules qui avaient reçu pendant 12 semaines une nourriture
contenant du 1,2-dibromoéthane à la dose de 12,5 mg/kg, on a constaté
une diminution du calibre des oeufs.
Le composé n'a pas entraîné de mutations léthales dominantes chez
des souris ou des rats, ni produit d'aberrations chromosomiques ou de
micronoyaux dans les cellules de la moëlle osseuse de souris traitées
in vivo. Toutefois, il s'est révélé mutagène dans les épreuves sur
bactéries et a provoqué des ruptures de l'ADN monocaténaire. On a
constaté in vivo comme in vitro, que les métabolites du
1,2-dibromoéthane étaient fixés à l'ADN par des liaisons covalentes.
Des échanges entre chromatides soeurs, des mutations et une synthèse
non programmée de l'ADN, ont été observées dans des cellules humaines
in vitro.
Des études de cancérogénicité ont été effectuées selon le schéma
suivant: souris et rats ayant reçu par gavage une dose quotidienne de
1,2-dibromoéthane égale à 37-107 mg/kg de poids corporel (en moyenne
pondérée par rapport au temps), pendant 49-90 semaines; souris ayant
reçu dans leur eau de boisson une dose de 1,2-dibromoéthane de
103-117 mg/kg de poids corporel, quotidiennement pendant 15-17 mois;
souris et rats exposés à une dose de 10-40 ppm pendant 6-18 mois par
voie respiratoire ou encore, souris badigeonnées au 1,2-dibromoéthane
pendant 400-594 jours, 3 fois par semaine à raison de 25-50 mg/souris.
Ces études ont montré que le 1,2-dibromoéthane était cancérogène pour
les rats et les souris et provoquait l'apparition de tumeurs au niveau
de divers organes, soit au point d'application, soit à distance de ce
point: cavité nasale, poumons, estomac, foie, peau, système
circulatoire et glandes mammaires. Dans de nombreux cas, il y avait
réduction du temps de latence des tumeurs.
6. Effets sur l'homme
Le 1,2-dibromoéthane peut avoir des effets nocifs sur le système
respiratoire, le système nerveux et les reins.
Ainsi, une seule exposition, par la voie respiratoire, à ce
composé (215 mg/m3, soit 28 ppm) pendant 30 min ou davantage, s'est
révélée mortelle pour l'homme. L'ingestion d'une dose de 140 mg/kg de
poids corporel s'est également révélée mortelle. Chez des
travailleurs exposés de par leur profession, une exposition de longue
durée au 1,2-dibromoéthane (5 ans), à la concentration de 0,68 mg/m3
dans la zone de respiration, a provoqué une diminution sensible du
nombre de spermatozoïdes et une baisse de la fécondité.
7. Effets sur les êtres vivants dans leur milieu naturel
Peu d'études d'ecotoxicité aquatique ont été consacrées au
1,2-dibromoéthane. Les valeurs de la CL50 pour les organismes
aquatiques sont supérieures à 5 mg/litre. On ne possède aucune donnée
au sujet des organismes terrestres.
RESUMEN
1. Identidad, propiedades físicas y químicas y métodos analíticos
El 1,2-dibromoetano (dibromuro de etileno) es un líquido incoloro
(punto de fusión: 9,9°C; punto de ebullición: 131,4°C) con olor
similar al del cloroformo. Es muy volátil, con una presión de vapor
de 1,47 kPa (11 mmHg) a 25°C y una densidad de vapor, en comparación
con el aire, de 6,1. El 1,2-dibromoetano es invisible en la mayor
parte de los disolventes orgánicos. Su solubilidad en el agua es de
4,3 g/litro a 30°C.
El 1,2-dibromoetano existente en el aire ambiental se analiza por
cromatografía de gases tras su absorción por polímeros porosos,
seguida por una rápida desasorpción térmica. Para las muestras de
agua se utiliza un método de «purge-and-trap». Los residuos de
1,2-dibromoetano presentes en los alimentos y en otros medios pueden
extraerse mediante disolventes o ser sometidos a un análisis
automatizado de la fase gaseosa superior en condiciones criogénicas,
seguido de análisis por cromatografía de gases y cromatografía líquida
de alta resolución, previa derivación.
2. Fuentes de exposición humana y ambiental
El 1,2-dibromoetano se utiliza para eliminar las sustancias
antidetonantes derivadas del plomo presentes en la gasolina.
Asimismo, se utiliza como fumigante de suelos y para la fumigación de
granos y frutas. El consumo reducido de gasolina con plomo en algunos
países y la anulación de inscripciones para la utilización de
1,2-dibromoetano con fines agrícolas ha reducido la exposición humana
a esa sustancia. Sin embargo, aún se utiliza para eliminar el plomo
de la gasolina en algunos países, como fumigante, para fines de
cuarentena, como disolvente y como producto intermedio en las
sustancias químicas industriales.
3. Niveles ambientales y degradación
Las concentraciones de 1,2-dibromoetano medidas en el aire
abarcan desde niveles indetectables hasta otros expresados en ng/m3
en las zonas urbanas. En zonas de explotación agrícola extensiva se
han detectado concentraciones de 1,2-dibromoetano superiores a
0,2 µg/litro en aguas subterráneas y a 50 µg/litro en aguas
superficiales. Aunque el 1,2-dibromoetano se filtra a través de la
tierra, parte de él queda retenido en la matriz del suelo y puede
contaminar posteriormente los pozos de riego. Se carece de
información suficiente sobre la descomposición microbiana en los
suelos.
La alta volatilidad del 1,2-dibromoetano determina que el
principal receptor ambiental sea la atmósfera. La fotólisis
estratosférica puede dar lugar a la formación de productos de
descomposición potencialmente destructores del ozono.
4. Cinética y metabolismo en animales de laboratorio
El 1,2-dibromoetano se absorbe rápidamente por vía oral y cutánea
y por inhalación. Se cree que los metabolitos desempeñan una función
importante en la toxicidad de esa sustancia para los seres humanos.
El 1,2-dibromoetano se puede metabolizar por vía oxidativa (sistema
del citocromo P-450) y por vía conjugada (sistema de la glutatión
S-transferasa). Según parece, dos metabolitos reactivos, el
bromacetaldehído formado por oxidación y el ion de tiranio formado por
conjugación interactúan con las macromoléculas celulares (proteínas,
ADN) para formar productos de enlace covalente.
5. Efectos en mamíferos de laboratorio y en sistemas de pruebas
in vitro
El 1,2-dibromoetano tiene una toxicidad aguda para los animales
(DL50 por vía oral en ratas de 146-417 mg/kg de peso corporal,
CL50 por inhalación en ratas de 3080 mg/m3 tras una exposición de
2 h, y una mortalidad observada tras la aplicación cutánea de
210 mg/kg a conejos). Los efectos tóxicos del 1,2-dibromoetano se
observaron principalmente en el hígado y en los riñones. La
inhalación de vapor de 1,2-dibromoetano produjo irritación nasal y
depresión del sistema nervioso central. En ratas expuestas a
concentraciones de 1540 mg/m3 a 77 000 mg/m3 (200 a 10 000 partes
por millón), con una duración de la exposición de 0,1 a 16,0 h, se
produjeron muertes en todos los grupos, en función de la concentración
y del tiempo. El 1,2-dibromoetano (solución al 1,0%) causó irritación
de la piel abdominal afeitada e irritación ocular en conejos.
Tras la exposición oral subcrónica, se observaron efectos
mortales y menor adquisición de peso en ratas y ratones con dosis de
100 mg/kg de peso corporal al día. Asimismo, se observaron reducciones
en la adquisición de peso y efectos patológicos nasales en ratas
expuestas al 1,2-dibromoetano en una proporción de 115 mg/m3
(578 partes por millón) durante 6 h al día y 5 días por semana a lo
largo de 13 semanas. El NOEL relativo a las alteraciones
histopatológicas de la cavidad nasal fue de 23 mg/m3 (3 ppm) en ese
estudio. En otro similar realizado en ratones se observaron los
mismos cambios patológicos, también con un NOEL de 23 mg/m3 (3 ppm).
Tras la administración por sonda a ratones o ratas de
1,2-dibromoetano en dosis de 37 a 107 mg/kg de peso corporal al día
(promedio ponderado por el tiempo) durante un periodo de 49 a
90 semanas, o la administración a ratones en dosis de 103 a 117 mg/kg
de peso corporal al día en el agua de beber durante un periodo de 15 a
17 meses, se observaron cambios no carcinogénicos tales como
degeneración hepática, atrofia testicular, y acantosis e
hiperqueratosis del preestómago, además de mortalidad. Tras la
exposición por inhalación (ratones o ratas expuestos a dosis de 77 a
388 mg/m3 durante un periodo de 6 a 18 meses), se observaron
inflamación de la tráquea y de la cavidad nasal, degeneración
testicular y necrosis hepática.
El 1,2-dibromoetano no resultó teratogénico en ratas o ratones
tras la exposición por inhalación. Se observó toxicidad para el
desarrollo (daños en el desarrollo de la coordinación motora) en la
descendencia de ratas macho tratadas por vía intraperitoneal con
1,25 mg/kg de peso corporal al día y en la descendencia de ratas
hembra tratadas mediante la inhalación de 509 mg/m3 durante 4 h al
día y 3 días por semana desde el día 3 al día 20 de la gestación. El
1,2-dibromoetano influyó en el comportamiento reproductivo de las
ratas (en los machos, con un nivel de exposición de 684 mg/m3
durante 7 h al día y 5 días/semana a los largo de 10 semanas, y en las
hembras con un nivel de exposición de 614 mg/m3 durante 7 h al día y
7 días por semana a lo largo de 3 semanas). El NOEL para ese
parámetro fue de 300 mg/m3 en ambos sexos. El NOEL para el
comportamiento reproductivo de las ratas macho en un estudio de
alimentación fue de 50 mg/kg al día tras una exposición de 90 días.
La espermatogénesis resultó afectada en toros tras la administración
de dosis orales de 2 mg/kg al día durante menos de 21 días, y en
conejos tras la inyección subcutánea de 15 mg/kg durante 5 días. La
administración de 1,2-dibromoetano a gallinas a través de la
alimentación causó una disminución del tamaño de los huevos tras la
exposición a 12,5 mg/kg al día durante 12 semanas.
El 1,2-dibromoetano no indujo mutaciones dominantes letales en
los ratones o las ratas, ni produjo aberraciones cromosómicas o
micronúcleos en las células de médula ósea de ratones tratados
in vivo. Sin embargo, resultó mutagénico en análisis bacterianos y
causó roturas del ADN de una sola hebra. Los metabolitos del
1,2-dibromoetano se fijaron al ADN mediante enlaces covalentes,
in vivo e in vitro. En células humanas in vitro se observó
intercambio de cromátides hermanas, mutaciones y síntesis de ADN no
programada.
Los estudios de carcinogenicidad en los que se administró la
sustancia por vía oral (ratones y ratas sometidas mediante sonda a
dosis de 37 a 107 mg/kg de peso corporal al día (promedio ponderado
por el tiempo) durante un periodo de 49 a 90 semanas; y ratones a los
que se administró 1,2-dibromoetano en el agua de beber en dosis de
103 a 117 mg/kg de peso corporal al día durante un periodo de 15 a
17 meses), mediante exposición inhalacional (ratones y ratas expuestos
a dosis de 10 a 40 partes por millón durante un periodo de 6 a
18 meses) o por vía cutánea de 25 a 50 mg/ratón, 3 veces por semana
durante un periodo 400 a 594 días) mostraron que el 1,2-dibromoetano
es carcinogénico para las ratas y los ratones y causa tumores en
diversos órganos (tanto en la zona de aplicación como en zonas
distantes, entre ellas, la cavidad nasal, los pulmones, el estómago,
el hígado, la piel, el sistema circulatorio y las glándulas mamarias).
En muchos casos reduce el periodo de latencia de tumores en
desarrollo.
6. Efectos en el ser humano
El 1,2-dibromoetano puede producir efectos adversos en los
sistemas respiratorio, nervioso y renal.
La exposición aguda (única) a la inhalación de 1,2-dibromoetano
en dosis de 215 mg/m3 (28 ppm) durante 30 minutos o más ha resultado
mortal para el ser humano. La ingestión de 140 mg/kg de peso corporal
resultó asimismo mortal. La exposición duradera (5 años) de la zona
respiratoria al 1,2-dibromoetano a una concentración de 0,68 mg/m3
redujo notablemente la densidad de espermatozoides y la fecundidad en
los trabajadores expuestos en su entorno laboral.
7. Efectos en otros organismos en el medio ambiente
Se han realizado pocos estudios sobre la ecotoxicidad acuática
del 1,2-dibromoetano. La CL50 para los organismos acuáticos es
superior a 5 mg/litro. No se dispone de información acerca de los
organismos terrestres.