
IVERMECTIN
1. EXPLANATION
Ivermectin is a broad spectrum antiparasitic drug which is
registered in over 60 countries. It is currently registered for use
in cattle, sheep, horses, goats, swine, camels, reindeer, bison, and
dogs (Di Netta, 1989). Ivermectin is active against two major phyla
of animal parasites, the Nemathelminthes and the Arthropoda (Campbell
et al., 1983). Ivermectin has not been evaluated previously by
the Joint FAO/WHO Expert Committee on Food Additives.
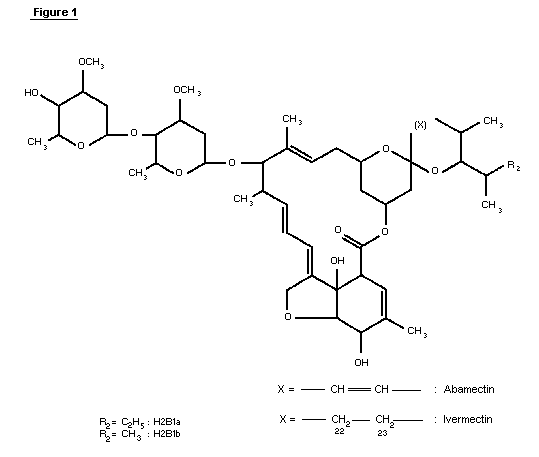
1.1 Chemical identity of ivermectin
Ivermectin (CAS-7-288-86-7) is a mixture of two compounds
belonging to a class of substances known as avermectins ( See Figure
1). The chemical names are 5-0-demethyl-22,23-dihydroavermectin A1a
and 5-0-demethyl-22,23-dihydroavermectin A1b. These are also known
as 22,23-dihydroavermectin B1a and 22,23-dihydroavermectin B1b.
Ivermectin contains at least 80% of 22,23-dihydroavermectin B1a and
less than 20% of 22,23-dihydroavermectin B1b.
The avermectins are derivatives of pentacyclic sixteen-membered
lactones. Within the family of the avermectins, there exist two
series, A and B, within which are two structural subsets, designated
1 and 2, consisting of two homologs a and b. Members of the A-series
are methoxylated at the carbon atom in position five, whereas the
B-series compounds have an underivatized hydroxyl-group at this
position. Compounds of the 1-subset possess an olefinic bond between
the two carbon atoms C22 and C23; this double bond is hydrated in the
2-subset, resulting in a hydroxyl group at position 23. This
difference has a profound effect on the conformation of the ring
bearing these functionalities and causes subtle changes in bioactivity
(Chabala et al., 1980).
The a- and b-homologs differ by their substituents at position
25, with a-homologs having an isopropyl group, derived from L-valine,
and b-homologs possessing a sec-butyl group derived from L-iso-leucine
during biosynthesis. Avermectins are glycosides with a disaccharide
attached to the hydroxyl group at C13. The two identical sugars have
been identified as L-oleandrose, a dideoxy-methyl-aldohexose.
 2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Absorption, distribution, and excretion
2.1.1.1 Rats
Radio-labeled ivermectin (mixture of 92.2% 3H-H2B1a and 7.8%
3H-H2B1b was administered to Sprague-Dawley rats, approximately 8
weeks old, once orally as a solution in sesame oil by gavage at 0.3
mg/kg b.w. (based on an average body weight of 294 grams for the males
and 218 grams for the females), or topically, after shaving, as a
solution in topical vehicle at 0.5 mg/kg b.w. (males only, average
body weight 279 grams). The animals were subdivided into groups of
three. Three treatment groups (sacrifice at days 1, 3, and 5
post-dose) and one control group (sacrifice at day 5 post-dose) of
each sex were used in the oral study. In the topical study, three
treatment groups (sacrifice on days 1, 3, and 6 post-dose) and one
control group (sacrifice on day 6 post-dose) of males were used.
Urine and faeces were collected daily. At sacrifice, blood was
collected by heart puncture; liver, kidney, muscle (from the hind
legs), and fat (males: testicular fat pad; females: peripheral fat)
were also harvested. Samples from the gastrointestinal tract were
also taken at the time of sacrifice from the animals dosed orally.
For each tissue, the individual samples from animals of the same group
were composited. Radioactivity was measured and calculated as drug
equivalents.
Residue levels in all samples were generally much higher
following oral application compared with the topical application. The
times of peak concentrations were delayed following topical
application. The main route of excretion was via the faeces.
Concentrations in faeces, however, were higher in females compared
with males; concentrations in urine were lower in females than in
males. This was consistently observed at all sampling times. In the
oral study 57.4% (males) and 58.4% (females) of the administered drug
had been eliminated one day after administration. These figures
increased to 83% (males) and 91.7% (females) five days after
administration. In the oral study, the highest residue levels in body
tissues were observed in fat, followed by liver, kidney, and muscle.
The approximate ratios for the residue levels in these tissues were
100:55:40:15 and were similar for both sexes. For the topical study
the situation was less clear due to the smaller data base and the
delay in the appearance of the residues in the tissues. At day 3,
when maximum residue levels were observed, concentrations in the above
tissues ranged in the same order, i.e., fat, liver, kidney, and muscle
(Merck & Co., Inc., 1987).
A residue study with labeled H2B1a (3H-labeled in the
22,23-position) was conducted in CRCD-rats.
Group 1: Six female rats aged approximately 8 weeks, and weighing
189-240 grams at initiation were administered labeled ivermectin for
61 days, then throughout mating, gestation, and until day 9
post-partum. One animal failed to mate; another animal was determined
not to be pregnant.
Group 2: Six female rats, aged approximately 12 weeks and weighing
248-298 grams, received radiolabeled ivermectin at the same dose and
specific activity from days 1 to 9 post-partum.
The dose, administered as an oral solution in sesame oil and by
metal catheter to both groups, was 2.5 mg/kg b.w./day (specific
activity 0.2 mCi/mg). Litter sizes were standardized to 5 males and
5 females on day 1 post-partum in both groups. At the time of
sacrifice, kidney, liver, brain, and carcass were collected. Milk
samples were obtained from 2 dams in each group on days 4, 6, and 10
post-partum. Blood, liver, brain, and carcass samples were obtained
from selected offspring (2 from each litter) of both groups on days 1,
4, 6, and 10 post-partum. Radioactivity was measured by
liquid-scintillation counting following oxidation of the tissues.
Dams: The concentration of ivermectin equivalents in plasma
increased to reach a plateau at approximately day 10 of treatment in
group 1. On day 1 post-partum, a three to four times higher
concentration was observed, possibly due to an increased mobilization
of body fat (ivermectin is highly lipophilic). Plasma levels then
decreased gradually and reached values comparable to those seen during
the pre-mating dosing period by day 10 post-partum. In group 2,
plasma levels increased gradually throughout the lactation period. On
day 10 post-partum, the individual values in both groups were
comparable. Concentrations in milk were at least three to four times
higher than corresponding concentrations in plasma. Tissue residues
in brain were very low relative to residues in kidney, liver and
carcass.
Offspring: From the concentrations in milk and based on daily milk
consumption by neonatal rats, a daily intake of 0.5 to 0.6 mg/kg body
weight on days 4-5 post-partum has been estimated. This corresponds
to approximately one half of the acute oral LD50. Under these
conditions concentrations in plasma increased dramatically between
days 1 and 6 post-partum, and were finally three times higher than the
concentrations found in maternal plasma. Similarly, residue
concentrations in livers were two times higher than those seen in the
corresponding dams. In contrast to the dams, the residue
concentrations in brain from offspring were comparable to plasma
concentrations on days 1 and 4 post-partum in group 1. On days 6 and
10, however brain levels were approximately three times lower than
plasma levels. The results of this study suggest that the transfer of
drug via the milk is probably responsible for the increase in neonatal
mortality associated with repeated administration in multigeneration
studies with rats when the blood-brain barrier to the drug is still
incomplete (Merck & Co., Inc., 1980a).
2.1.1.2 Dogs
To determine whether plasma and/or brain tissue levels of
ivermectin are proportional to dose, groups of 4 female beagle dogs,
approximately 34-39 weeks old at initiation of the study, weighing 7.6
to 9.6 kg, received ivermectin orally (solution in sesame oil, gavage)
at dose levels of 0.5 mg/kg body weight or 2.0 mg/kg b.w., once per
day over a period of 35 days (except one animal in the high dose group
which received 24 doses prior to sacrifice on day 24). Surviving
animals were not dosed on the day of sacrifice. On days 2, 8, 15, 22,
and 29 (6 hours post-dose), and on day 36, blood was withdrawn.
Following withdrawal of the last blood sample, cerebrospinal fluid was
collected. Samples were assayed for H2B1a. Beginning in week 2,
mydriasis was observed in two animals of the high dose group. This
effect was also noted in the remaining two animals of this group
during week 3 and continuing until termination. On day 21, one animal
of the high dose group exhibited ataxia, and fine whole-body tremors.
This animal's condition deteriorated rapidly and by approximately 4
hours post-dose on day 22 the animal was recumbent with salivation and
marked tremors. On arousal it exhibited marked ataxia. It had to be
sacrificed on day 24. With the exception of mydriasis, the remaining
three animals of the high dose group exhibited no other
treatment-related effect. No treatment-related physical signs were
observed in the low dose animals. Slight increases in body weight
were found in all animals receiving 0.1 mg/kg b.w./day. Slight to
marked weight losses occurred among the high dose animals with the
largest loss (2.1 kg) found in the dog which was sacrificed on drug
day 24. Plasma concentration of H2B1a increased dramatically in both
groups between days 2 and 8 of treatment. Thereafter, gradual
increases occurred, reaching approximate steady-state levels at day 22
in both groups. The animal which had to be sacrificed on day 24
achieved the highest plasma concentrations. Unexplained decreases in
plasma concentrations occurred in weeks 4 and 5 in the high dose group
and in week 4 in the low dose group. The mean of the ratios of the
plasma concentrations in the 2.0 mg/kg b.w./day group compared to the
0.5 mg/kg b.w./day group was 8.4. Plasma concentrations were thus
not linear to dose, since a 4-fold increase in dose led to a 8-fold
increase in plasma concentrations. Concentrations in cerebrospinal
fluid remained below the limit of detection (1 ng/ml), except in the
animal which was sacrificed on day 24 in which severe signs of CNS
depression were evident. In this animal 3 ng/ml was determined in the
cerebrospinal fluid (Merck & Co., Inc., 1982c).
2.1.1.3 Monkeys
Concentrations of 22,23-dihydroavermectin-B1a and avermectin-B1a
were measured in plasma at three dose levels (2.0, 8.0, and 24.0 mg/kg
body weight) in a combined oral toxicity and plasma level study of
ivermectin and abamectin with immature rhesus monkeys. The time
post-administration when blood levels reached a maximum could not be
precisely determined from the data. However, the concentrations
observed following treatment with ivermectin were higher than those
measured following the administration of abamectin. For both
substances, the concentrations in plasma were proportional to the
administered dose but this proportionality was apparently not linear
(Merck & Co., Inc., 1985).
2.1.1.4 Humans
The pharmacokinetics in plasma of ivermectin were studied in
randomized three-period crossover studies with 12 healthy adult male
volunteers in each study. Peak plasma concentrations were reached
approximately 4 hours after administration. Bioavailability (area
under the curve) was highest following administration of ivermectin as
a solution. No significant differences between the bioavailabilities
of the capsule and the tablet formulations were observed (Merck & Co..
Inc., 1988b).
2.1.2 Biotransformation
Because of the extremely low levels of residues in tissues and
the many difficulties associated with purification, the following
approach was taken:
* steer and rat liver microsomes were incubated in vitro
with ivermectin components. Metabolites were isolated,
purified and identified;
* major steer-liver metabolites were isolated from large
quantities of this tissue and were compared to the products
of in vitro incubations;
* fractions obtained from smaller amounts of liver were
chromatographically separated and cochromatographed with
in vitro products, where possible;
* chromatographic profiles of rat hydrolyzed isolates were
compared in the same way to those obtained from liver.
Based on this approach it was possible to identify the major
metabolites obtained from the various steer liver, rat liver and steer
fat isolates. (Merck & Co. Inc., 1980b).
2.1.2.1 In vitro studies of metabolism
Rat liver microsomes were incubated in vitro with either
avermectin-B1a, 22,23-dihydroavermectin-B1a, or
22,23-dihydroaver-mectin-B1b. The samples were extracted and
metabolites were purified by solvent extraction and chromatographic
procedures. The structures of the isolated metabolites were
determined by mass spectrometry and nuclear magnetic resonance
spectrometry.
With each substrate >70% of the radioactivity was associated
with the respective parent compounds. Two major polar metabolites
(total amount 2-11%) were formed. One metabolite has been identified
as the C24-methyl alcohol of the parent compound which had been used
as the substrate for incubation. A smaller quantity was identified as
the monosaccharide of the C24-alcohol. These two metabolites
represented the major fraction of metabolites which were more polar
than the respective parent compounds (>50% with the substrate H2B1a
and 80% with the substrate H2B1b). The metabolites retained
antiparasitic activities which quantitatively depend on test species
and conditions of testing (e.g., in vitro / in vivo) (Merck & Co.,
Inc., 1980b).
2.1.2.2 Rats
An experiment was designed to provide composite samples of
tissues and excreta from 24 male CRCD rats (weighing 295 to 329
grams), dosed orally by gavage at 0.3 mg/kg body weight with tritiated
ivermectin. The animals were sacrificed at days 1 and 3 post-dose.
The cumulatively measured radioactive drug equivalents in the
gastrointestinal tract, urine, and faeces accounted for 6.4, 0.8, and
84.9%, respectively, of the administered dose at day 3. Tables 1 and
2 summarize the amount of "total residue" and the percentage of
unchanged drug in tissues, and the proportions of metabolite fractions
isolated from the liver of rats dosed with tritium-labeled ivermectin
(Merck & Co., Inc., 1980c).
A group of nonpolar metabolites has been detected in fat tissue.
Upon hydrolysis, these metabolites gave rise to polar products that
were similar to the ivermectin metabolites present in liver. From
residue profiles (reversed-phase HPLC) of extracts purified from the
fat obtained on day 3 post-dose from rats treated with 3H-ivermectin,
it was estimated that about 17% of the total residue consisted of the
nonpolar metabolites and about 49% was the parent drug. It was
suggested that polar ivermectin metabolites produced in the liver were
esterified with fatty acids and stored in the fat as nonpolar entities
(Chiu et al., 1988).
2.1.2.3 Humans
Four healthy male volunteers received 14 mg of 3H-labeled
ivermectin. Blood, urine, and faeces were assayed for radioactivity
by liquid scintillation counting and/or following HPLC separation.
Approximately 0.6% of the radioactive dose was excreted in the urine
(over four days). An average of 49% was recovered from faeces (over
five days). Table 3 summarizes the kinetic parameters of the study.
Mean plasma concentrations of radiolabeled metabolites were about
twice that of the parent drug. Peak plasma concentrations occurred
seven hours post-treatment for radioactivity and six hours post-dose
for the parent drug. Radioactivity was eliminated from the blood more
slowly than the non-metabolized component. Discontinuities in plasma
profiles of the parent drug were observed. This study suggested the
possibility of enterohepatic recycling (Merck & Co., Inc., 1988b).
2.1.2.4 Mechanism of action
Most studies on the mode of action have been performed with
avermectin B1. However it is assumed that all active members and
derivatives of the avermectin family share a common mechanism of
action. Avermectin B1 apparently affects interneuron-motorneuron
transmission in nematodes and neuromuscular transmission in
arthropods. In both cases receptors for gamma - aminobutyric acid
(GABA) are involved. The low amount of GABA-ergic synapses in
helminths and arthropods hindered complete elucidation of the
mechanism (Fritz et al. 1979; Kass et al., 1980).
2.2 Toxicological Studies
2.2.1 Acute toxicity studies
2.2.1.1 Mice
Ivermectin was tested in adult male and female mice. The
components of ivermectin, H2B1a and H2B1b, were also studied in the
female mouse. In an additional study, the acute toxicities of
tetrahydroavermectin B1, the major contaminant (up to 4%) of
ivermectin, and of ivermectin itself were compared in the female
mouse. Either ten or twenty albino mice (Careworth CF-1 strain)
weighing 19-24 grams, and approximately 7 weeks of age, received the
compounds tested as a solution in sesame oil by gastric intubation.
All mice were observed at the day of drug administration and
thereafter for 14 days. Calculation of LD50 values were based on 14-
day mortality response.
Table 1: Equivalents of total radioactive residues [ng/g] and percent of unaltered drug in rat tissues
Tissue: fat liver kidney muscle plasma
Days post dose 1 3 1 3 1 3 1 3 1 3
Total residue 232 137 47 40 40 46 44 18 12 5
H2B1a [%] 50 66 66 56 28 34 53 51
H2B1b [%] 13 12 5.8 9 21 27 9.6 11
Table 2: Classification of the total radioactive residue in rat liver
at day 1 post-dose
Metabolite Isolation Percent of total
Group polarity fraction radio-activity
I very polar aqueous buffer 0.06
II very polar Sep-Pak, eluate 0.4
with methanol
II-A polar (at least HPLC 0.3
two compounds)
III-B polar (not HPLC 3
identified)
IV polar (identfied HPLC 8
metabolites)
V polar (at least 10
four compounds)
VI unaltered drug HPLC 71
VII non-polar HPLC 7.3
VIII non-polar isooctane 1.9
Table 3: Kinetic parameters in plasma of radioactivity and H2B1a
following administration of 3H-ivermectin to healthy
human volunteers.
Cmax tmax t1/2
[ng/ml] [hours] [hours]
radioactivity 54.2 7 70
equivalents
H2B1a 21.7 6 11.8
component
H2B1a: Two lots were studied at different times. Dose levels
were 2.5, 5, 10, 20, 40, 80 and 160 mg/kg body weight with the first
lot: with the second lot the same dose levels were tested, except that
the dose level of 2.5 mg/kg body weight was omitted. Signs of
toxicity were seen within 30 to 90 minutes at a dose at or above 5
mg/kg body weight which consisted of ataxia, tremors, bradypnoea,
decreased activity and loss of righting. The majority of deaths
occurred from 45 minutes to 3 days with four later deaths from the 4th
to 7th days. Most of the survivors appeared normal by the fourth day.
H2B1b: Two lots were tested at different times at dose levels
of 5, 10, 20, 40, 80, and 160 mg/kg body weight. Signs of toxicity,
which were generally similar to those observed following
administration of H2B1a, (ataxia, tremor, bradypnoea, and loss of
righting) were seen within 90 minutes, and were scattered through all
doses. Most deaths occurred from 26 minutes to the fourth day with
one death on the sixth day.
Ivermectin: Two different lots (one 80% H2B1a/20% H2B1b, and
the other 84% H2B1a/16% H2B1a) were tested at dose levels of 5, 10,
20, 40, 80 and 160 mg/kg body weight. Ivermectin was found to be
significantly more toxic orally in the male mouse than in the female
mouse. Signs of drug effects, however, were generally similar in both
sexes. When these compounds were tested concurrently in the female
mouse, there was no significant difference in the toxicity (Merck &
Co., Inc., 1979a).
Tetrahydroavermectin-B1: This substance was also tested at dose
levels of 5, 10, 20, 40, 80 and 160 mg/kg body weight in direct
comparison with ivermectin. The results indicated that this compound
was significantly less toxic than ivermectin in the female mouse.
Signs of drug effects were seen at 80 and 160 mg/kg body weight and
consisted of ptosis, salivation, ataxia, and loss of righting.
Eighteen hours after dosing, tremors and ataxia were seen at dose
levels of 10 mg/kg body weight and above. The duration of signs was
approximately 36 hours (Merck & Co., Inc., 1980d).
2.2.1.2 Rats
Ivermectin was studied in young adult male and female rats and
infant rats. Young adult rats of two different strains were used:
Sprague-Dawley Camm rats weighing 150 to 175 grams which were 7 to 9
weeks of age were treated at dose levels of 2.5, 5, 10, 20, 40, and 80
mg/kg body weight. Charles River-CD strain rats approximately 7 weeks
old and weighing 125 to 175 grams were treated with 25, 35, 49, 68.6,
and 96 mg/kg body weight ivermectin. Ten rats of each sex were used at
each dose. The infant rats (Charles River-CD strain) were 24 to 48
hours old and weighed on a littermate group average 7.3 to 9.4 grams.
Ten infant rats of undiscriminated sex were used at each dose level of
1, 2, 4, 8, 16 and 32 mg/kg body weight. Signs of drug effects were
similar in both strains of adult rats (decreased activity, salivation,
bradypnoea, and ataxia, depending on the dose). Deaths occurred from
overnight to the second day. There was no significant sex-related
difference in toxicity.
No signs were observed following the oral administration to
infant rats. The majority of deaths occurred in 131 minutes to
overnight with one death on the fifth and one death on the sixth day
(Merck & Co., Inc., 1979a).
The acute oral toxicity of ivermectin was also studied in 13 week
old Charles River CD rats obtained from a cross-fostering study in
order to determine if prenatal or postnatal exposure increased the
toxicity of subsequent exposure. The rats had been exposed to
ivermectin or a vehicle control, prenatally, postnatally, or both pre-
and postnatally, and were grouped (30 males, weighing 224-497 grams
and 30 females, weighing 151-272 grams, per group) as follows:
Group 1: F0 treated x F1 treated
Group 2: F0 treated x F1 control
Group 3: F0 control x F1 control
Group 4: F0 control x F1 treated
The rats (six animals of each sex per dose level) were given a
single dose by gastric intubation of a 0.8% solution of ivermectin at
dose levels of 25.0, 32.5, 42.3, 55, or 71.5 mg/kg body weight. All
rats were observed on the day of drug administration and daily
thereafter for 14 days.
Signs of drug effects were generally similar in all four groups
of rats tested and in both sexes. On the second day, decreased
activity, bradypnoea, and a reddish-brown discharge around the nose
and mouth were seen at all dose levels. In the male rats, loss of
righting was observed at 42.3 mg/kg body weight and higher. In the
females this effect was seen at the 55 and 71.5 mg/kg body weight dose
levels. In the surviving animals these effects persisted until the
seventh day. The majority of deaths occurred from overnight to day 4,
but a few deaths occurred on days 5, 6, 9, and 10. The number of
deaths was too small to allow the calculation of the LD50. The
results of the study indicated no significant differences in toxicity
between controls (group 3) and the other groups (Merck & Co., Inc.,
1979c).
The acute percutaneous toxicity of ivermectin was studied in 10
male and 10 female rats (Charles River CD strain) which weighed 224 to
408 grams and were 12 to 13 weeks of age. Five rats of each sex were
used at each of two dose levels, 330 and 660 mg/kg body weight. The
doses were applied to occluded unabraded skin. The animals were
observed daily (except on weekends) for 14 days.
Rats at both dosage levels began to show signs of systemic
toxicity two days after treatment. At the 330 mg/kg body weight dose
level, one male died seven days after treatment. At the 660 mg/kg
body weight dose level one male (on day five) and one female (on day
three) died. The relevant effects were bradypnoea and tremors (Merck
& Co., Inc., 1979d).
2.2.1.3 Rabbits
Three groups of three male and three female albino rabbits each,
weighing 2.61 to 3.47 kg and 18 to 20 weeks of age, were used to
determine the acute percutaneous toxicity of ivermectin. The hair was
removed and in each group three animals were abraded. The test
compound was applied as a dry powder at doses of 165, 330, and 660
mg/kg body weight. Animals were examined during a 14 day post dosing
period. The signs of systemic toxicity (bradypnoea, tremor, and
anorexia) were similar for both abraded and unabraded rabbits. The
percutaneous LD50 was estimated as 406 mg/kg body weight (Merck & Co.,
Inc., 1979d).
No toxic effects (except mucosal irritations) developed in an
acute inhalation toxicity study with ivermectin. Five male and 5
female Sprague-Dawley rats were exposed for 60 minutes to the maximum
attainable concentration of 5.11 mg/1 overall nominal air
concentration. With 0.37% of the particles showing sizes of 15
microns or less, the corresponding dose was estimated as < 0.4
mg/kg body weight (Merck & Co., Inc., 1979e).
Only slight irritation developed in an acute ocular irritation
study in 2 male and 2 female New Zealand rabbits when 100 mg
ivermectin powder was placed in the conjunctival sac of the left eye
(Merck & Co., Inc., 1979d).
2.2.1.4 Dogs
The acute oral toxicity of ivermectin was studied in eight male
and 8 female beagle dogs, 10 to 14 months of age and weighing between
8.1 and 15.2 kg. Three groups, each containing two male and two
female dogs, received doses of 2.5, 5.0, or 10.0 mg/kg body weight as
a 1.6% solution in sesame oil by gastric intubation. A fourth group
received sesame oil (0.625 mg/kg body weight) in the same way. The
animals were observed throughout a 14-day test period.
Mydriasis and the absence of pupil response were seen in two dogs
at the lowest dose, and in all dogs at the two higher doses. Within
75 minutes of drug administration, one dog at the high dose vomited.
This same dog exhibited emesis and salivation two additional times
within four hours. At both the high and the intermediate doses one
additional dog had emesis following drug administration. Tremors
which were seen in five animals (2 dogs at 5 mg/kg body weight and 3
dogs at 10 mg/kg body weight) were first observed about six hours
following drug administration and were still present on the third day
in some animals.
One dog in the high dose group became ataxic and heavily sedated.
This dog recovered from the sedation within 48 hours and from ataxia,
tremors, and salivation within 72 hours (Merck & Co., Inc., 1979f).
In a further oral toxicity study with dogs, three groups of two
male and 2 female beagle dogs, 6-9 months of age and weighing 6.3 to
10.1 kg, were given ivermectin 1.6% solution in sesame oil at doses of
5, 10, and 20 mg/kg body weight by gastric intubation. Because no
deaths occurred, two additional groups of two males and two females
each were dosed with a 6.4% suspension/solution in sesame oil at doses
of 40 and 80 mg/kg body weight. Signs included emesis (all dose
levels, except 40 mg/kg body weight), mydriasis (all dose levels),
ataxia and tremors (doses >10 mg/kg body weight), salivation (in the
40 and 80 mg/kg body weight dose-groups) and death preceded by a
comatose like state (in the 40 and 80 mg/kg body weight dose-groups)
(Merck & Co., Inc., 1981a).
Sixteen rough-coated collies ranging in age from 7 months to 9
years were used in an oral toxicity study with ivermectin. Treatment
groups (two males, two females, half of the animals of each sex having
collie eye anomaly) received 0.05, 0.2 or 0.6 mg/kg body weight of
ivermectin once orally (plastic syringe) as a solution in fractionated
coconut oil with 2% benzyl alcohol. At the end of the trial (7 days)
three previously untreated control dogs also received the lowest dose
(0.05 mg/kg body weight). Samples of plasma, cerebrospinal fluid,
brain, spinal cord, and liver were assayed for H2B1a. No drug-
related effects were seen in the 7 animals treated at the lowest dose
level. Two dogs given 0.2 mg/kg body weight and two dogs given 0.6
mg/kg body weight showed signs of toxicity which were mild and
transitory in one dog of each group. One animal per group
progressively developed similar severe clinical signs: Ataxia with
increasing hypermetria which progressed to paresis and, finally,
paralysis. In both dogs segmental reflexes remained strong. Both
salivated excessively and had predominantly diaphragmatic respiration.
The dog from the 0.6 mg/kg body weight group was killed at 28 hours
post-dose; the dog from the 0.2 mg/kg body weight group died 51 hours
post-dose. These two severely affected dogs showed significantly
increased brain drug levels (Pulliam et al., 1985).
The acute subcutaneous toxicity of ivermectin injectable micelle
solution and its vehicle were studied in 11 to 12 week old beagle
dogs. Five groups of three males and three females received 4.7, 9.4,
18.8, 37.5, and 75.0 mg/kg body weight ivermectin. A sixth group of
three males and two females received an identical volume (1 ml/kg body
weight) of vehicle. Signs were seen in all ivermectin dose groups.
There were no sex related significant differences in toxicity. No
deaths occurred the 4.7 mg/kg body weight dose. Three of the six dogs
dosed at 9.4 mg/kg body weight, and all of the dogs administered doses
>9.4 mg/kg body weight, died. Dogs receiving the vehicle appeared
normal throughout the fifteen-day observation period. Mydriasis and
negative pupil response were observed in all ivermectin treated groups
with time of onset and duration (in the surviving dogs) depending on
the dose (e.g., onset was three hours after dosing at 75 mg/kg body
weight and approximately 24 hours after dosing at 47 mg/kg body
weight). Other signs included tremors, ataxia, salivation, and
decreased activity. At necropsy, treatment-related slight to very
slight changes were present only in dogs that did not survive to
termination (thymus-hemorrhage, lung-congestion, lung-edema, lung
acute suppurative pneumonia, skin edema). LD50 values were estimated
as 8.4 mg/kg body weight in males and 10.5 mg/kg body weight in
females (Merck & Co., Inc., 1981b).
2.2.1.5 Pigs
Male and female Yorkshire swine were given subcutaneously 0.3, 3,
15, or 30 mg/kg body weight of ivermectin. Signs of toxicity
(decreased food and water intake, lethargy, ataxia, mydriasis, tremor,
labored breathing and lateral recumbency) were seen at the highest
dose level (Merck & Co., Inc., 1982d).
2.2.1.6 Sheep
Ten male and ten female lambs weighing 28 to 39 kg, allocated to
20 individual pens and fed restrictively once daily (with a diet
containing ethoxyquin; drinking water ad libitum), were assigned to
five treatment groups of four each. Treatment-sex combinations were
randomly allocated. There were five treatment groups, control
(distilled water), 0.3, 2.0, 4.0, and 8.0 mg/kg body weight. Two
additional animals were dosed with propylene glycol two days after the
other animals were treated. The animals were sampled 6 and 4 days
prior to treatment as well as on days 1, 2, 4, 7, 10, 14, and 18.
Surviving animals were killed on days 21 to 23 after treatment. At
8.0 mg/kg body weight, all sheep were ataxic within three hours after
dosing. They were depressed (head and ear drooping). One animal went
into lateral recumbency in a shock-like condition. Reflexes were
present but delayed. The animal was up after 24 hours and appeared
clinically normal by day 3 after dosing. After 24 hours all animals
were still mildly depressed and slightly incoordinated. At 4.0 mg/kg
body weight, the sheep were mildly incoordinated and depressed and had
initially delayed feed consumption although the 24-hour feed
consumption was normal. All animals were clinically normal at 24
hours. Since two additional sheep given vehicle (propylene glycol)
only showed the same physical signs as those given 8 mg/kg body
weight (the female fell into lateral recumbency in a shock-like
condition and was found dead 24 hours after dosing), it was suggested
that the observed signs were due to the propylene glycol vehicle
(Merck & Co., Inc., 1981c).
2.2.1.7 Cattle
Approximately 6 month old male and female Holstein Friesian
calves ranging in weight between 95 and 143 kg were injected once
subcutaneously with 0.3, 2, or 8 mg/kg body weight ivermectin. The
observed signs of toxicity at the highest dose level were increased
respiratory rate, muscular tremors, and rigidity of the extremities
and death (Merck & Co., Inc., 1979g).
2.2.1.8 Horses
In a target animal safety study, horses were injected
intramuscularly with ivermectin at dose levels of 3, 6, or 12 mg/kg
body weight. Signs of toxicity were seen at all dose levels (Egerton
et al., 1984).
No effects were observed when ivermectin was given as an oral
paste at 0.4 mg/kg body weight to 26 male and female miniature and
farabella pure and crossbred horses aged 5 months to 13 years and
weighing 40-180 kg. Ivermectin was administered once as a micelle
solution to groups of four horses (264-431 kg body weight) at 3, 6, or
12 mg/kg body weight. At 3 mg/kg body weight one group was injected
with ivermectin concentrate; a second group and all other treatment
groups were injected with a ready-to-use micelle solution. Mydriasis
was seen at all dose levels. All horses dosed at 12 mg/kg body weight
showed additional signs of drug toxicity including depression and
ataxia. One horse was found in lateral recumbency 24 hours after
dosing and was killed 72 hours after dosing (Merck & Co., Inc.,
1981d).
2.2.1.9 Rhesus monkeys
An oral toxicity study was conducted in order to determine the
minimum toxic dose of ivermectin and abamectin in rhesus monkeys and
to determine the plasma levels of the drug at that dose. Immature
rhesus monkeys, aged 2 to 3 years at initiation, weighing 2.6 to 3.1
kg (males) and 2.4 to 3.2 kg (females), were given single increasing
oral doses (0.2, 0.5, 1, 2, 4, 6, 8, 12, 24 mg/kg body weight, in
chronological order) of ivermectin and abamectin in sesame oil, by
gavage, at intervals of 2 to 3 weeks before the administration of the
next higher dose to same group of four animals (two of each sex). The
0.2 mg/kg body weight dose was repeated twice due to uncertainties as
to whether mydriasis was occurring in two treated monkeys and because
two monkeys regurgitated part of their dose. The second repetition
was a cross-over study in which the monkeys formerly treated with
ivermectin were given abamectin and vice versa. The 2 and 8 mg/kg
body weight doses were repeated to measure plasma levels. Therefore,
each animal received a total of 13 doses.
The following treatment-related physical signs were observed: (a)
Doses of 2.0 mg/kg body weight and higher caused emesis. The time of
onset tended to decrease as the dose increased. (b) Pupil dilation
and/or decreased constriction was observed following doses of 6.0
mg/kg body weight of abamectin and above and after doses of 12.0 or
24.0 mg/kg body weight of ivermectin. Most of the observations were
slight and difficult to assess and were considered equivocal, except
those seen following the 24 mg/kg body weight dose. No relationship
appeared to exist between the dose levels and the time of onset or
duration. (c) Several animals displayed decreased levels of activity
or slight to moderate sedation at 24 mg/kg body weight of both
compounds. All animals recovered within 48 hours. No tremors or
convulsions occurred. A difference in potency between the two test
substances was not discernible. It was unlikely that the lack of more
profound toxicity was due to regurgitation, since emesis did not
generally occur before four hours post-dose. Plasma concentrations
increased with dose but not in a linear manner. Signs of toxicity did
not correlate with plasma levels over the investigated dose-range.
The most sensitive indicator of toxicity was emesis with a NOEL of 1.0
mg/kg body weight and a dose-related increase over the range of 2.0 to
24.0 mg/kg body weight (Merck & Co., Inc., 1985).
2.2.1.10 Summary of the results obtained from acute toxicity studies
The following main symptoms of central-nervous disorders were
observed within one hour and up to seven days following a single oral
dose of ivermectin depending on the test species and the applied dose:
tremor, depression, ataxia, paresis, paralysis and death. LD50 values
for experimental animals are given in Table 4.
Mice, particularly males, were found to be more sensitive than
rats. LD50 values ranged from 11.6 mg/kg bw in LD50 values male mice
to 40 mg/kg body weight in females. The reason for the rather high
variability of the results remains unclear. In studies with female
mice when either ivermectin or one of its individual main components
H2B1a and H2B1b was applied, no significant differences in acute
toxicity were observed. Tetrahydroavermectin B1, however, the most
abundant potential impurity (up to 4%), was of significantly lower
acute oral toxicity.
In the rat, a higher sensitivity of neonatal rats (LD50 = 2.3
mg/kg body weight) was evident if compared with 42.8-52.8 mg/kg body
weight LD50s reported for adult animals (male and female). The
increased toxicity of ivermectin in neonatal rats is likely due to a
combination of excessive plasma levels resulting from exposure via
maternal milk and the increased permeability of the blood-brain
barrier during the early postnatal period in this species (Lankas et
al., 1989).
Great differences in sensitivities were observed among various
other species including target animals. Large variation has been
observed between breeds of dogs and individual dogs within the same
breed.
Table 4: Tabular representation of acute toxicity data in laboratory
animals
LD50 Reference
Species Sex Route (mg/kg bw) (Merck & Co., Inc.)
Mouse M oral 11.61A 1979a
(CF-1) F oral 24.61A 1979a
27.11A 1979a
41.61B 1979a
40.01A 1979a
30.01A 1980d
F oral 31.72 1979a
87.22 1979a
F oral 27.63 1979a
56.63 1979a
F oral 1604 1980d
Rat MA oral 42.81A 1979a
(young) FA 44.31A 1979a
MB 42.81A 1979a
FB 52.81A 1979a
M+F percutan. >660 1979d
(infant) M+F oral 2.31C 1979a
Rabbit percutan. 406 1979d
Dog
(beagle) F oral >10.0 1979f
M+F oral approx. 80.0 1981a
M subcutaneous 8.4 1981b
F 10.5 1981b
1A) test substance: ivermectin; 80:20 mixture;
1B) test substance: ivermectin; 84:16 mixture;
1C) test substance: ivermectin; ratio not indicated
2) test substance: H2B1a;
3) test substance; H2B1b;
4) test substance: Tetrahydroavermectin B1;
A) Charles River, CD;
B) Sprague-Dawley, Camm
2.2.2 Short-term studies
2.2.2.1 Rats
A fourteen-week toxicity study following in utero exposure was
reported. Twenty rat pups of each sex between 3 and 4 weeks of age
weighing 49 to 86 grams (males) or 43 to 77 grams (females) were
treated at dosage levels of 0.4, 0.8, and 1.6 mg/kg b.w./day. No
changes due to treatment occurred at 0.4 mg/kg b.w./day. The
following effects could not be excluded as being treatment-related in
the two other dosage level groups: spleen-enlargement and reactive
bone-marrow hyperplasia, which occurred in 1 animal at 0.8 mg/kg
b.w./day and in 3 animals at 1.6 mg/kg b.w/day (Merck & Co., Inc.,
1979b).
2.2.2 Dogs
Twenty male and 20 female beagle dogs, 39-43 weeks of age,
weighing initially 8.2 - 12.1 kg (males) and 6.2 - 9.2 kg (females)
were selected for oral treatment (gastric intubation) in five groups
of four males and four females at doses of 0.5, 1.0, 2.0 mg/kg
b.w./day. Controls received water or vehicle (sesame oil). At 2.0
mg/kg b.w./day, three males and one female developed tremors, ataxia,
anorexia, and dehydration. All of these animals exhibited ptyalism
and mydriasis followed by slight tremors, characterized by
intermittent or constant shaking of all limbs, which generally
increased in severity over 3 to 6 days. These animals were frequently
found laterally recumbent and were ataxic when standing. They were
sacrificed between weeks 4 and 12. Mydriasis was observed in all dogs
at this level (beginning in week 1 and continuing until week 12 when
it decreased in incidence). The four dogs sacrificed showed weight
losses between 1.0 and 1.6 kg. At 1.0 mg/kg b.w./day, mydriasis was
occasionally seen, particularly in week 3. Weight gain was retarded.
At 0.5 mg/kg b.w./day, only slight retardation of weight gain was
observed. No significant drug-related changes were observed for the
following parameters: ocular abnormalities, electrocardiograms,
haematologic parameters, urine-analysis, and pathological changes
(Merck & Co., Inc., 1978a).
2.2.2.3 Rhesus monkeys
A 16-day oral toxicity study with ivermectin was conducted to
determine its toxicity in immature rhesus monkeys (13 - 21 months old,
weighing 2.1 to 3.2 kg (males) and 1.9 to 2.7 kg (females) at
initiation). Each of the treatment groups (4 females, 4 males per
group) were dosed daily by nasogastric intubation with ivermectin in
sesame oil at dose levels of 0.3, 0.6, and 1.2 mg/kg body weight.
These dose levels were chosen to provide an appropriate 6-fold safety
margin relative to the human clinical dose, and based on the acute
toxicity in rhesus monkeys. All animals were treated for at least 14
days and then sacrificed on days 15, 16 or (one animal) 17. No drug-
related effects (physical signs, body weight, ocular lesions,
haematology, serum biochemical parameters, or necropsy findings) were
noted in any of the treated animals (Merck & Co., Inc., 1986a).
To assess the potential significance of neonatal exposure to
ivermectin, a study in neonatal rhesus monkeys (7 to 13 days old; 400
to 600 g body weight) was conducted. The animals (3 females, 5 males
per group) received ivermectin as a solution in sesame oil once daily
via nasogastric intubation at dose levels of either 0.04 or 0.1 mg/kg
body weight for 14 days. A control group of the same size received
the vehicle. Approximately four hours post-dose, animals were
examined for mydriasis and pupillary light response, and for adverse
reactions. The results of the examinations (physical, ophthalmic,
haematologic, serum biochemical examination, body weight, and
necropsy) indicated no treatment-related effects (Merck & Co., Inc.,
1986b).
2.2.3 Long-term/carcinogenicity studies
2.2.3.1 Mice
No specific study of ivermectin per se beyond 14 weeks has
been reported. Carcinogenicity was studied in mice (Crl:Cd-1(ICR)BR)
and rats (Crl:CD(SD)BR) with abamectin, a close chemical analog of
ivermectin differing only by being unsaturated between the carbon
atoms at positions C22 and C23.
In a 94 week dietary carcinogenicity study in Crl:CD-1 mice
abamectin was given at doses of 2, 4, or 8 mg/kg b.w./day. Seventy-
four mice of each sex were assigned to each group and to control
groups I and II. At the start of the study the males weighed 15.5-
33.2 grams and the females weighed 15.8-22.1 grams. Twelve
mice/sex/group were killed for bleeding at weeks 25 or 26 and 52.
The examinations included: daily observation; weekly detailed
examination including palpation for masses, body weight, and food
consumption; eye examination (pretest, weeks 51 or 53 and 91, control
and high-dose group only); haematology and serum biochemistry (weeks
25 or 26 and 52; in moribund mice after week 69; in all surviving mice
at scheduled sacrifice); and complete necropsy (gross necropsy on
animals killed for bleeding; histopathology on all mice assigned to
the carcinogenic segment of the study).
Treatment-related tremors were seen among females in all dosage
groups. Seven females of the 8 mg/kg b.w./day group and 3 females of
the 4 mg/kg b.w./day group died the day after the beginning of the
treatment for unexplained reasons (dietary concentrations were checked
and found correct). The females were killed and discarded. The males
were continued in the study. About a month later a new group of
females without adverse signs was started. Treatment-related tremors
were seen in drug weeks 89 and 91 in two females of the high-dose
group.
Increased mortality was seen among the high-dose males but not
the females (common causes: amyloidosis or lymphoma). Dosing of this
group was stopped in week 90 when there was 40% survival. All other
mice were treated until sacrifice in week 94. Treatment-related
decreases in body weight gain (males 7%; females 21%) were seen in the
high-dose groups. A dose-related increase in feed consumption and
decrease in feed efficiency (20%) was seen among the females of the
high-dose group. There were neither treatment-related ophthalmic
changes nor haematological nor serum biochemical effects. No
treatment-related changes in organ weights or gross lesions were seen
at necropsy. There were no neoplastic or non-neoplastic changes seen
in any tissue at necropsy examinations. Abamectin was not
carcinogenic to mice when given in the diet for 94 weeks at the above
dosage levels (Merck & Co., Inc., 1983).
2.2.3.2 Rats
Abamectin was tested in Crl:Cd(SD)BR rats (size of treatment
groups and controls I and II: 65 males [115-191 grams] and 65 females
[93-154 grams]) in a 105 week dietary study at doses of 0.75, 15, or
2.0 mg/kg b.w./day. Fifteen males and 15 females per group were
randomly selected at the start of the study for interim necropsy. The
examinations included: daily observation; weekly detailed examination
including palpation for masses, body weight, and food consumption; eye
examination (pretest and about every six months [controls and high-
dose group only]); haematology, serum biochemistry and urine analysis
(10 rats/sex/group in weeks 12, 25, 38, and 51 [from animals selected
for interim necropsy]), in week 78 (10 replacement rats from the
carcinogenic segment), in week 105 (sacrifice; haematology and serum
biochemistry) and in moribund animals (begining in week 89); complete
necropsy (all rats that died or were sacrificed before scheduled
termination, all rats sacrificed at scheduled time); and
histopathology (including histological examination of all gross
lesions).
Treatment-related increases in body weight gain were seen during
the first year on study (males and females of all dosage groups); by
the end of the study, body weight gain was statistically significant
only in the males. Tremors occurred in some rats receiving the
highest dose and one rat of the mid dose group which had exceedingly
high feed consumption. There were no gross or microscopic changes in
the nervous or muscular systems of rats which died with tremors.
There was no treatment-related increase in deaths. There were no
treatment-related changes in ophthalmic abnormalities, nor treatment-
related effects seen in haematology, serum biochemistry and
urinalysis. No treatment-related changes in organ weights or gross
lesions were seen at necropsy. There was no statistically significant
increase in tumour incidence, and no non-neoplastic changes were seen
in any tissue at necropsy examinations. Abamectin was not
carcinogenic to rats in this study (Merck & Co., Inc., 1982e).
2.2.4 Reproduction studies
2.2.4.1 Rats
Ivermectin was administered orally once daily to three groups of
15 female rats at dose levels of 0.4, 0.8, and 1.6 mg/kg body weight
from 15 days prior to mating until 20 days post-partum. Two vehicle
control groups received sesame oil in the same dosing regimen as the
treated animals.
There was no mortality or clinical evidence of toxicity in the
females. Average body weight was significantly increased among
females at 0.8 and 1.6 mg/kg b.w./day during the prebreeding period
and at all dose levels during gestation.
Ivermectin had no effect on mating, reproductive status, average
length of gestation or post implantation survival rate. Statistically
significant treatment-related increases in mortality among pups in the
1.6 mg/kg b.w./day group were observed on day 1 and from days 7-14
post-partum. Prior to death, several pups were observed to be
hypothermic and to have no externally observable milk in the
epigastric region. Throughout the lactation period, average pup
weights were slightly higher than controls in the 0.4 mg/kg b.w./day
group and significantly higher in the two other dose-level groups.
Development (eye opening, ear opening, incisor eruption, and hair
growth) was also slightly accelerated (Merck & Co., Inc., 1979a,b).
A series of three multigeneration studies was initiated in rats,
the first two of which were halted prior to scheduled termination
because neonatal toxicity was apparent at all dose levels tested.
Dose rates of 0.4, 1.2, and 3.6 mg/kg b.w./day were used in the first
study. It was necessary, however, to terminate this study before
mating of the F1b-generation because it became apparent from toxic
symptoms observed in the F1a-, F1b-, and F2a-generations that a NOEL
could not be derived from this study.
Effects on the F0-generation were a significant increase in the
average length of gestation and a significantly decreased maternal
weight gain during lactation in females in the 3.6 mg/kg b.w./day
group. Following the production of the F1b-litter, the average
maternal weight gain during lactation was significantly decreased
compared to that of the control.
Effects on the F1a-generation were a high (92%) mortality during
the lactation period of F1a-offspring in the 3.6 mg/kg b.w./day group
with the majority dying between days 5 and 10 post-partum. The most
common clinical sign of toxicity in pups that died was an absence of
milk in the stomach one to several days prior to death. Average pup
weights on day 1 post-partum and subsequent weight gain in surviving
pups were also significantly reduced in this group. During postnatal
development there was a significant decrease in the time to occurrence
of incisor eruption in the 3.6 mg/kg b.w./day group, an effect which
was believed to be secondary to a lower body weight in these
offspring.
Effects on the F1b-generation were evidence of treatment-related
toxicity among F1b-offspring in both remaining dose-level groups (1.2
and 0.4 mg/kg b.w./day). An increased pup mortality occurred between
days 2 and 7 post-partum. Slight decreases in average live pup weight
per litter were also observed during the lactation period. The time
to occurrence of the auditory startle reflex was delayed and earlier
incisor eruption was also observed compared to the corresponding
control. At 1.2 mg/kg b.w./day, vaginal opening was significantly
delayed and there was a nonsignificant but treatment-related delay in
testes descent. It is likely that these effects reflected slightly
lower average body weights. During the lactation period females in
the 1.2 mg/kg b.w./day group showed a significant decrease in average
maternal body weight gain.
Effects on the F2a-generation were significant increases in pup
mortality between days 2 and 7 post-partum in both dosage groups. The
average live pup weight per litter was decreased (statistically
significant only in the 1.2 mg/kg b.w./day group) on days 7, 14, and
21 post-partum. During postnatal development, there were significant
delays in the appearance of the righting reflex and the auditory
startle reflex and significantly earlier incisor eruptions in the 1.2
mg/kg b.w./day group (Merck & Co., Inc., 1980e).
A second multigeneration study was initiated at a dose of 2.0
mg/kg b.w./day in order to provide clear evidence of toxicity while
allowing sufficient surviving offspring to permit continuous dosing
throughout the production of two litters in each of three generations.
This study was terminated prior to the production of the F1b-litter
when it became apparent that there was treatment-related neonatal
toxicity present in the above concurrent multigeneration study at dose
levels 1.2 and 0.4 mg/kg b.w./day (Merck & Co., Inc., 1981e).
In a final multi-generation study the following dose groups were
included: 0.05, 0.1, 0.2, and 0.4 mg/kg b.w./day. A vehicle control
group received sesame oil daily in the same volume as drug-treated
rats. The animals were 28 days old at the onset of the daily
treatment and were mated 71 days later. Exposure was continued for
the entire life-span.
The F1a-litter was sacrificed on day 21 post-partum.
Approximately three weeks later the F0-rats were mated again to
produce the F1b-litter. On day 21 post partum of the F1b-offspring,
the F0-generation was sacrificed. After 71 days of treatment, the
F1b-rats were mated to produce the F2a-offspring which were also
sacrificed on day 21 post-partum. Approximately three weeks later the
F1b-rats were again mated to produce the F2b-offspring. Twenty-one
days post partum of this offspring, the F1b-generation was sacrificed.
After 71 days of drug treatment, F2b-rats were mated to produce the
F3a-offspring which were sacrificed on day 21 postpartum.
Approximately three weeks later the F2b-rats were again mated to
produce the F3b-litter. The parents were sacrificed after weaning of
the F3b-litter. Twenty males and 20 females from each F3b-offspring
group were randomly selected for necropsy at 28 to 43 days of age.
There was no treatment-related mortality or physical signs of
toxicity among parents or offspring in any dosage group throughout the
production of two litters in each of the F0-, F1-, and F2-
generations. Ivermectin had no treatment-related effects on the
reproductive performance of male or female rats in any dosage group.
Treatment-related effects on body weight gain were limited to a
slight but statistically significant decrease during the postweaning
period in mean body weight gain among F1b-females in the 0.4 mg/kg
b.w./day group and among F2b-males from the 0.2 and 0.4 mg/kg b.w./day
groups. External, visceral, and skeletal examination of both the F3a-
and F3b-offspring revealed no evidence of teratogenicity. Doses of
less than or equal to 0.2 mg/kg b.w./day had no adverse effects on
parents or progeny (Merck & Co., Inc., 1980e, 1981e).
2.2.4.2 Target animal species
No adverse effects on reproduction have been observed in target
animal species (Campbell & Benz, 1984; Egerton et al., 1984;
Schroder et al., 1986).
2.2.5 Special studies on embryotoxicity
Ivermectin has been tested in the mouse, the rat, the rabbit, and
the dog. Additionally, the effects of H2B1a and H2B1b have been
tested separately in mice.
2.2.5.1 Mice
22,23-Dihydroavermectin-B1a: In a teratogenic study groups of
20 pregnant CF1-mice each received H2B1a at dosage levels of 0.2,
0.4, 0.8, or 1.6 mg/kg b.w./day from days 6 through 15 of gestation
once daily by gavage as a solution in sesame oil. An additional
control group received the vehicle.
There were treatment-related maternal deaths. Whole body tremors
and coma developed in 2 mice after the first dose at 1.6 mg/kg
b.w./day. Dosing was suspended, but the coma persisted. One of these
mice was found dead on day 8; the second mouse was sacrificed while
aborting on day 9. At 0.8 mg/kg b.w./day, two mice developed tremor
and coma after the second dose. One of these mice died on day 9; the
second mouse was sacrificed on day 11 while aborting. At 0.4 mg/kg
b.w./day two mice developed slight to moderate tremors after the
second dose which became more pronounced after the third dose. Dosing
was then suspended. One mouse was sacrificed while aborting. The
second became comatose on day 9 and was sacrificed moribund on day 11.
Average maternal weight gain and reproductive status of surviving mice
was unaffected.
The average fetal weight per litter was unaffected by treatment
at any dosage level. There was a teratogenic effect as evidenced by
cleft palate (10 fetuses from five litters at 1.6 mg/kg b.w./day).
22,23-Dihydroavermectin-B1b: H2B1b was administered orally as
a solution in sesame oil by metal catheter to three groups of pregnant
CF1-mice from days 6 to 15 of gestation at dosage levels of 0.4, 0.8,
or 1.6 mg/kg bw/day. A control group of 35 mice received the vehicle.
At 1.6 mg/kg b.w./day one mouse was found comatose 24 hours after
the first dose, did not recover and was sacrificed 48 hours later. At
0.8 mg/kg b.w./day one mouse became prostrate and hypothermic after
five doses, and was sacrificed on day 11 of gestation. No other signs
of toxicity were observed at any other dosage level. The average
fetal weight was unaffected by treatment. Teratogenicity was
evidenced by a dose-related increased incidence of cleft palate (ten
fetuses from four litters at 1.6 mg/kg b.w./day; four fetuses from
four litters at 0.8 mg/kg b.w./day) (Merck & Co., Inc., 1979a).
Ivermectin: Groups of 25 mated female CF1-mice were
administered ivermectin orally as a solution in sesame oil at dose
levels of 0.1, 0.2, 0.4, or 0.8 mg/kg b.w./day from days 6 through 15
of gestation. The 0.4 mg/kg body weight day group had only 24 mice
because one mistakenly assigned male had to be discarded. A control
group of 25 mice received the vehicle.
There was treatment-related mortality in each of the three
highest dose level groups (0.8 mg/kg b.w./day: 3 females sacrificed
moribund after 14 doses; 0.4 mg/kg b.w./day: 1 female found dead after
the 3rd dose, two others sacrificed in poor physical condition after
1 and 8 doses, respectively; 0.2 mg/kg b.w./day: 1 female sacrificed
moribund after 4 doses). Physical signs were confined to those mice
which died or were sacrificed. Neither the reproductive status of
females (as measured by the number of implants, resorptions, and live
and dead fetuses per litter) nor average maternal body weight was
influenced by the treatment. Teratogenicity was evidenced by an
increased incidence of cleft palate (3 fetuses from 3 litters at 0.8
mg/kg b.w./day; 5 fetuses from 4 litter at 0.4 mg/kg b.w./day). There
was no evidence of a teratogenic effect at 0.1 or 0.2 mg/kg b.w./day
(Merck & Co., Inc., 1980g).
2.2.5.2 Rats
A summary of a teratogenic study in rats was available.
Ivermectin was administered as a solution in sesame oil to groups of
25 mated CRCD rats from days 6 through 17 of gestation at dose levels
of 2.5, 5, or 10 mg/kg b.w./day. A control group of the same size
received the vehicle. In the 10 mg/kg b.w./day group, three females
were sacrificed in poor physical condition after receiving 7-9 doses.
There were no treatment-related toxicity signs observed in the two
other groups. Teratogenicity, as evidenced by cleft palate in 4
fetuses from 2 litters was seen at 10.0 mg/kg b.w./day. No other
treatment-related external malformations were observed at the other
dosage levels. Visceral and skeletal examination produced no further
evidence of teratogenicity in any dosage group (Merck & Co., Inc.,
1980g).
2.2.5.3 Rabbits
A summary of a teratogenic study in rabbits was available.
Ivermectin was administered as a solution in sesame oil to groups of
16 pregnant rabbits from days 6 through 18 of gestation at dose levels
of 1.5, 3, or 6 mg/kg b.w./day. A control group of the same size
received the vehicle. In the 6 mg/kg b.w./day group, slight to marked
sedation was observed 24 hours after the 7th dose and persisted in
some females up to eight days after cessation of dosing. There was
also a significant decrease in mean maternal body weight during the
period of drug administration in this group. Six females of this
group aborted between days 22 and 27 of gestation, possibly due to
embryo-/feto-toxicity (increase in fetal deaths). No treatment-
related maternal effects were seen in the two other groups.
Teratogenicity was indicated by a dose-related increased incidence of
cleft palate and clubbed forepaws. At 3 mg/kg b.w./day 1 fetus had
cleft palate and 5 fetuses from 1 litter had clubbed forepaws. At 6
mg/kg b.w./day, 8 fetuses from 3 litters had cleft palate and 6
fetuses from 3 litters had clubbed forepaws (Merck & Co., Inc.,
1980g).
2.2.5.4 Dogs
An oral teratogenic study in beagle dogs was conducted. The
mated bitches weighed 8.2-17.1 kg at initiation of the treatment.
Seventeen mated bitches received 0.5 mg/kg body weight of ivermectin
in sesame oil on days 5, 15, 25, and 35 of gestation. A second group
of 19 mated bitches received the same dose on days 10, 20, 30, and 40
of gestation. A third group of 17 mated bitches served as the control
and received vehicle on days 5, 10, 15, 20, 25, 30, 35, and 40 of
gestation. On day 48 of gestation the females were hysterectomized.
At hysterectomy it was determined that 14, 15, and 12 bitches were
pregnant in groups 1, 2 and the control, respectively. There was no
maternal mortality and there were no maternal signs of toxicity in the
course of the study. There was no apparent effect on the average
fetal weight per litter. There was no evidence of a teratogenic
effect at external, visceral, and skeletal examination (Merck & Co.,
Inc., 1981f).
Summary of teratogenicity studies
Table 5 summarizes the results of the above teratogenicity
studies.
2.2.6 Special study on cross-fostering
Results of the initial multigeneration study in rats (Section
2.2.4.1) suggested that at doses of 0.4 and 1.2 mg/kg b.w./day the
F2a-progeny may have been more sensitive to the toxic effects of
ivermectin than the F1a- or F1b-progeny. A cross-fostering study was
conducted in order to determine whether this was due to biological
variation or to a real increase in sensitivity following prenatal,
postnatal or a combination of pre- and postnatal exposure to the drug.
One group of 40 female Charles River CD rats, approximately 8 weeks
old and weighing 169-256 grams, was administered 2.4 mg/kg b.w./day of
ivermectin in sesame oil for 61 days. A vehicle group of the same
size was administered sesame oil according to the same regimen. They
were mated with untreated males. On day 1 post-partum litter sizes
were standardized to 4 males and 4 females by random selection.
Litters were then cross-fostered to one of the following groups:
group treatment of prenatal exposure number of
F0-dams of F1-litters litters/group
1 + + 12
2 + - 12
3 - - 15
4 - + 12
Table 5: Teratogenicity in laboratory animals (oral administration)
Species Dams/ Dose Maternal toxicity Teratogenicity
(strain group [mg/kg
or breed) bw/day]
a) 22,23-dihydro-avermectin-B1a:
Mouse 20 0.2 no observed effects no observed
effects
(CF-1) 0.4 2 sacrificed no observed
effects
0.8 2 sacrificed/dead no observed
effects
1.6 2 sacrificed/dead cleft palate
(10/151)
b) 22,23-dihydro-avermectin-B1b:
Mouse 20 0.4 no observed effects no observed
effects
(CF-1) 0.8 1 sacrificed cleft palate
4/243)
1.6 1 sacrificed cleft palate
(10/205)
Mouse 25 0.1 no observed effects no observed
effects
(CRCF) 0.2 1 sacrificed no observed
effects
0.4 3 sacrificed/dead cleft palate
(5/244)
0.8 3 sacrificed cleft palate
3/254)
Rat 25 2.5 no observed effects no observed
effects
(CRDC) 5.0 no observed effects no observed
effects
10.0 3 sacrificed cleft palate
(4/264)
Table 5 (contd)
Species Dams/ Dose Maternal toxicity Teratogenicity
(strain group [mg/kg
or breed) bw/day]
Rabbit 16 1.5 no observed effects no observed
effects
3.0 no observed effects reduced
litter weight
cleft palate
(1/136)
clubbed forepaw
(5/136)
6.0 sedation reduced litter
decreased body weight
weight increased fetal
aborts death
cleft palates
(1(7*)/50
(18*))
clubbed forepaw
(6/50(18*))
Dog 17 ** no observed effects no observed
effects
(beagle) 19 ** no observed effects no observed
effects
* Numbers in parenthesis: dead fetuses
** These animals were dosed with 0.5 mg/kg b.w. every ten days.
Offspring were examined for weight gain, clinical signs of
toxicity, and postnatal development (occurrence of surface righting
reflex and time of eye opening). Thirteen weeks post-partum offspring
from all four groups were selected for continuation on an acute oral
toxicity study. The average live pup weight per litter on days 7, 14,
and 21 post-partum among offspring fostered within the treated group
(group 1) was significantly decreased. Among control litters
cross-fostered to treated females (group 2) there was a significant
decrease in average live pup weight on days 14 and 21 only. The
average live pup weight among litters prenatally exposed and
cross-fostered to control dams (group 4) was comparable to that
of litters cross-fostered within the control group. There was a
significant decrease in average postweaning body gain in groups 1,
2, and 4 compared to the control group 3. The magnitude of the
decrease was significantly greater in groups 1 and 2 than that of
group 4. There was a significant increase in pup mortality in litters
cross-fostered to F0-dams administered ivermectin (groups 1 and
2). Pup mortality among litters from treated F0-dams cross-fostered
to control dams (group 4) was comparable to that of pups
cross-fostered within the control group (group 3). There was no
significant variation in the time to occurrence of the righting reflex
among all four groups. The time to occurrence of eye opening
was significantly retarded in group 1 only. The results of this study
indicated that the neonatal toxicity of ivermectin in rats was
primarily a function of postnatal exposure. It also appeared that
in utero exposure did not increase the toxicity of subsequent
exposure via milk during the lactation period (Merck & Co., Inc.,
1980f).
2.2.7 Special studies on genotoxicity
2.2.7.1 Bacterial systems
Ivermectin and each component of ivermectin were tested in the
Ames test. Tests were done with and without rat liver metabolic
activation systems. None of the agents studied produced any noteworthy
increase in revertants to histidine prototrophy. The positive controls
(either 1-methyl-2(3a,4,5,6,7a-hexahydro-1,2-benzisoxazolol-3-yl)-5-
nitroimidazole or 2-amino-anthracene) produced significant increases
in revertants, particularly after metabolic activation with all the
tester strains used (Merck & Co., Inc., 1978b).
2.2.7.2 Mouse lymphoma cells
The ability of ivermectin to produce forward mutation at the
thymidine kinase locus (TK+/- to TK-/-) of mouse lymphoma cells (Fischer
L5178Y) was studied. Two cytotoxicity studies revealed that ivermectin
was detoxified in the presence of rat liver S-9 fraction. The mutagenic
assays were done by exposing cells to ivermectin for four hours; they
were then washed, fed and cultured for three days. TK-/- mutants were
detected by cloning the cells in selection medium containing
bromodeoxyuridine and plating diluted aliquots in nonselective medium.
Dose levels of 40, 60, and 80 µg/ml were used with and without S-9.
However the cells died in this initial study. A second assay was done
with 20, 40 and 60 µg/ml in the presence of S-9 only. Without S-9 the
dose levels were 5, 10, and 20 µg/ml. This study was replicated with
an identical protocol. The results of both tests were negative when
compared with appropriate negative controls. The positive control,
3-methylcholanthrene with S-9 produced significant increases in mutation
frequency (Merck & Co., Inc., 1980h).
2.2.7.3 Human Fibroblasts
Effects of ivermectin on unscheduled DNA synthesis were studied in
IMR-90 normal human embryonic lung fibroblasts in the presence and absence
of rat liver microsomal activation systems. The drug concentration ranged
from 10 to 1000 µg/ml. Ivermectin did not produce any significant
increase in background thymidine incorporation. In contrast it produced
an unexplained decrease at 10, 100, 300, and 1000 µg/ml but not at 30
µg/ml. The positive controls, methylmethane sulfonate and aflatoxin B-1,
both produced significant increases in UDS (Merck & Co., Inc., 1980i).
2.2.7.4 Summary of studies on genotoxicity
Table 6 summarizes the genotoxicity studies that have been performed
on ivermectin and its components.
2.3 Observations in humans
One 15 year old male was accidentally injected with an unknown
quantity of IVOMECTM 1% in a needle fingerprick accident. His arm
later became paralysed, due to coincidental viral polyneuritis which was
probably unrelated to ivermectin. An adult female injected herself
accidentally with a small quantity (estimated to be 200 micrograms/kg
body weight) of IVOMECTM 1%. Twelve hours later she experienced
colicky pain with nausea, but recovered within 12 hours. A 16 month old
boy weighing about 15 kg accidentally drank an estimated 10-13 ml of
IVOMECTM 1%. Mydriasis was noted in one pupil, along with vomiting,
pallor, 35°C body temperature, tachycardia, somnolence, and variable
blood pressure. The next morning urticaria occurred. He was normal
after three days. Therapy in hospital included calcium-gluconate,
caffeine, and an antihistamine. A woman, 8 months pregnant, sprayed
EQVALANTM into her eye. The eye was rinsed. Stinging at the
application side was the only adverse effect described (Merck & Co.,
Inc., 1988a).
2.3.1 Clinical use of ivermectin
Ivermectin was first administered to humans in 1981. Initial
studies were performed in Senegalese patients with onchocerciasis (Aziz
et al., 1982, Diallo et al., 1984). Since the first clinical
experience numerous multiclinic double-blind studies have been conducted
in endemic areas of onchocerciasis in different countries (Lariviere
et al., 1985; Awadzie et al., 1986; Diallo et al., 1986;
Dadzie et al., 1987; Albiez et al., 1988; Vingtain et al., 1988).
In 1987 MECTIZANTM (ivermectin) of Merck, Sharp and Dohme has been
approved in France for the treatment of onchocerciasis. The Onchocerciasis
Control Programme (OCP) of WHO has elaborated for 1989 and 1990 a dual
strategy of vector control and mass population drug treatment with
MECTIZANTM. It is expected that over 250,000 individuals in the OCP
area of Central Africa will be treated (Bradshaw, 1989). In addition to
its use for the treatment of onchocerciasis, ivermectin has been used
successfully in the treatment of Wuchereria bancrofti filariasis
(Kumaraswami et al., 1988), loiasis (Richard-Lenoble et al.,
1988), strongyloidiasis and enterobiasis (Naquira et al., 1989).
Table 6: Results of genotoxicity assays on ivermectin and its components
Test system Test object Concentration Results Reference
[ug/plate]
Ames test* TA1535, TA1537, 400, 1000, 2000 negative Merck & Co.,
(ivermectin) TA98, TA100 400, 1000, 2000 negative Inc. (1978b)
TA 1535, TA100 500, 1000, 2000 negative
Ames test* TA100 1000 negative Merck & Co.,
(H2B1a) TA1537, TA98, 100, 500, 1000 negative Inc. (1978b)
TA100,TA92 100, 500, 1000 negative
Ames test* TA1535, TA1537, 20, 200, 2000 negative Merck & Co.,
(H2B1b) TA98, TA100 20, 200, 2000 negative Inc. (1978b)
[ug/ml]
mouse lymphoma L5178Y 5, 10, 201) negative Merck & Co.,
cells L5178Y 20, 40, 602) negative Inc. (1980h)
[ug/ml]
unscheduled* human IMR-90 10, 30, 100 negative Merck & Co.,
DNA synthesis fibroblasts 300, 1000 negative Inc. (1980i)
(ivermectin)
*with and without S-9; 1)with S-9; 2)without S-9.
2.3.2 Studies in healthy subjects
Ivermectin was assessed for tolerability in several clinical
pharmacology studies. A total of 54 healthy individuals received
single oral doses of MECTIZANTM. Adverse experiences reported were
headaches in three persons who received a 6 mg dose. These
experiences were assessed as either probably or definitely not drug
related. Decreases in white blood cell counts occurred in one subject
after single oral doses of 12 mg as a solution and as tablets. Both
were assessed as being possibly drug related (Merck & Co., Inc.,
1988b).
2.3.3 Studies on tolerability in patients
Treatment of onchocerciasis with ivermectin requires a single
oral dose of 0.15 - 0.2 mg/kg body weight every 12 months. The
tolerability of MECTIZANTM has been closely examined in a number of
clinical trails. The observed side effects in some patients were
mostly mild and transient. These side effects can probably be
attributed to hypersensitivity reactions resulting from death of
microfilariae (the symptoms most frequently reported include pruritus,
arthralgia, dizziness, myalgia, fever, edema, lymphadenitis, nausea,
vomiting, diarrhoea, postural hypotension, tachycardia, weakness,
rash, and headache) (Merck & Co., Inc., 1988b).
3. COMMENTS
The Committee reviewed toxicological data from studies on
pharmacokinetics, biotransformation, acute and short-term toxicity,
effects on reproduction and development, genotoxicity, and
observations in humans. Aspects of the comparative toxicities of
ivermectin and abamectin were also considered.
Pharmacokinetic data were available from studies in mice, rats,
dogs, rhesus monkeys, and human volunteers. In mice, peak plasma
levels were reached approximately four hours after a single oral dose
of 51 mg per kg of body weight. The average plasma to brain ratio of
the concentrations of the drug was approximately 11:1. When
ivermectin was administered at 0.1 to 0.5 mg per kg of body weight per
day for 35 days, steady-state concentrations were observed from day
21. The concentration in the plasma and brain was proportional to the
dose.
In a study in rats in which H2B1a was administered orally at
0.06 to 0.75 mg per kg of body weight the dose and residue levels in
plasma and tissues were also shown to be well correlated. In a study
in which [3H] ivermectin was given orally at 0.3 mg per kg of body
weight the residue concentrations were highest in fat, followed by the
liver, kidney and muscle. The main route of excretion was via the
faeces.
In female rats aged eight weeks at initiation of dosing and
receiving daily oral doses of 2.5 mg/kg b.w. for 61 days and then
throughout mating, gestation, and until day 9 postpartum, steady-state
plasma concentrations were reached on day 10 of treatment. On day 1
postpartum, however, the plasma concentration was three to four times
that of the steady-state concentration, probably due to an increased
mobilization of body fat. When treatment was restricted to days 1 to
9 postpartum, the concentration of ivermectin in the plasma increased
gradually throughout the lactation period, and concentrations in milk
were at least three to four times the corresponding concentrations in
plasma. Under these conditions, the concentrations in the plasma of
the offspring increased dramatically between days 1 and 6 postpartum,
and on day 10 they were up to three times the concentrations found in
maternal plasma. On days 1 and 4 postpartum; residue levels in the
brain tissue from offspring were similar to their plasma
concentrations. The results of this study suggested that the transfer
of the drug via the milk was probably responsible for the increase in
neonatal mortality observed in multigeneration studies.
In a 36-day study in the beagle dog in which ivermectin was
administered orally at 0.5 and 2.0 mg/kg b.w. per day, the
concentrations of H2B1a in the plasma increased dramatically between
days 2 and 8 of treatment and reached steady state after approximately
three weeks. A fourfold increase in the dose resulted in an average
eightfold increase in plasma levels. In a comparative study with
abamectin and ivermectin in immature rhesus monkeys, higher plasma
concentrations were reached with ivermectin at all the dose levels
investigated (2, 8, and 24 mg per kg of body weight). For both
substances the concentrations in plasma were related to the dose, but
the relationship was not linear.
In a study with human volunteers in which various formulations of
ivermectin were administered orally, peak plasma concentrations were
reached within approximately four hours. Administration of [3H]
ivermectin showed that approximately 49% of the dose was eliminated in
the faeces within five days. In a clinical study in lactating women
treated with a single dose of ivermectin, a maximum concentration of
23 µg of the drug was found in milk on the day after treatment. This
level decreased to less than 0.1 µg/1 approximately one week after
treatment. Although plasma levels were not reported for this
particular study and the data determined from other studies were not
directly comparable, it appears that concentrations in human milk are
similar to or slightly less than in plasma.
Most of the studies on biotransformation were conducted using
[3H] ivermectin. When rat liver microsomes were incubated in vitro
with the individual components and a NADPH-regenerating system, more
than 70% of the radioactivity was associated with the corresponding
parent compound. The major polar metabolite was identified as the 24-
desmethyl-24-hydroxymethyl alcohol. The corresponding monosaccharide
was also detected. These findings correlated well with the results of
in vivo liver metabolism studies. In addition, a group of nonpolar
metabolites was detected in fat, which secreted polar products on
hydrolysis that were similar to the ivermectin metabolites present in
liver.
Acute toxicity studies were conducted in mice, rats, rabbits,
dogs, rhesus monkeys and a variety of target species (pigs, sheep,
cattle and horses). The typical signs of acute toxicity of ivermectin
were attributed to its effects on the central nervous system. These
were most severe in CF1 mice, which exhibited ataxia, bradypnoea and
tremors. Deaths occurred from approximately one hour to six days
after dosing. Ivermectin was more toxic in neonatal rats than in
young adult rats. This was believed to be due to postnatal completion
of the blood-brain barrier in this species.
In beagle dogs, mydriasis was the most sensitive indicator of
toxicity. More severe signs included ataxia and tremors. Deaths were
preceded by a comatose-like state. Approximately 30% of collies
tested were highly sensitive to ivermectin (as estimated from reports
from non-approved use of the drug). In immature rhesus monkeys no
tremors or convulsions occurred. The most sensitive indicator was
vomiting, which occurred in one of four monkeys given ivermectin at
2.0 mg per kg of body weight. The steep dose response curve in
rodents for the toxicity of ivermectin was not reproduced in monkeys.
Short-term studies were considered in rats, dogs, and monkeys.
In a 14-week study in rats in which ivermectin was administered orally
to pregnant dams, splenic enlargement and bone-marrow hyperplasia were
noted in the offspring of dams dosed at 0.8 and 1.6 mg per kg of body
weight per day. The NOEL was 0.4 mg per kg of body weight per day.
These changes did not occur in other species which received
ivermectin.
In a 14-week study in beagle dogs in which the compound was given
orally, mydriasis and loss of body weight were observed at 1.0 and 2.0
mg per kg of body weight per day (each group consisted of four females
and four males). Four dogs in the group receiving ivermectin at 2.0
mg per kg of body weight per day developed tremors, ataxia, anorexia,
and dehydration, and were killed prior to scheduled necropsy. No
other treatment-related effects were found. The NOEL was 0.5 mg per
kg of body weight per day.
In a two-week study in which ivermectin was administered orally
to neonatal monkeys at 0.04 and 0.1 mg per kg of body weight per day,
and to immature monkeys at 0.3, 0.6 and 1.2 mg per kg of body weight
per day, no drug treatment-related effects were observed.
Three multigeneration studies were initiated in rats, but the
first two were halted prior to scheduled termination because neonatal
toxicity was apparent at all dose-levels tested. In the final (three-
generation) study, the highest dose level was 0.4 mg per kg of body
weight per day. The results indicated that ivermectin was toxic to
neonatal rats at doses of 0.4 mg per kg of body weight per day or
above (administered to adult females) as evidenced by increased
neonatal mortality up to approximately ten days postpartum, and by the
decreased weights of surviving offspring. The results of a cross-
fostering study indicated that the neonatal toxicity was not related
to in utero exposure but to postnatal exposure via maternal milk.
The developmental toxicity of ivermectin has been investigated in
mice, rats, rabbits, and dogs. The results demonstrated that
teratogenic effects (cleft palates in mice, rats, and rabbits; clubbed
fore-paws without skeletal alterations in rabbits) were produced only
at dose levels similar to those causing severe toxic effects in
pregnant animals. The no-observed-effect level for teratogencity in
the most sensitive species and strain, the CF1 mouse, was 0.2 mg/kg
b.w./day, while for maternal toxicity it was 0.1 mg/kg b.w./day.
Ivermectin was negative in three in vitro assays for
genotoxicity. The Committee noted that no test of clastogenicity had
been performed.
There were no carcinogenicity studies available on ivermectin.
The Committee noted the very close structrual similarities of
ivermectin and abamectin. Extensive toxicological tests had been
conducted on both compounds by one particular manufacturer, using the
same strains of test animals over the same period of time. The
Committee, therefore, reviewed several aspects of comparative
toxicology of the two products. The compounds were indistinguishable
at the level of receptor binding. Clinical signs of the toxicity of
both compounds included mydriasis in dogs, vomiting in monkeys, and
tremors, convulsions, and coma at higher doses in most species. CF1
mice were most sensitive to the compounds. In general, ivermectin was
slightly less toxic than abamectin in laboratory animals (2-4-fold
higher threshold). In 14-week studies in rats in which ivermectin and
abamectin were administered orally at 0.4 mg per kg of body weight per
day, no adverse effects were observed. In a 14-week study with
ivermectin in dogs, mydriasis was seen at 1.0 mg per kg of body weight
per day and above, and tremors, ataxia and anorexia at 2.0 mg/kg of
body weight per day. In a 12-week study with abamectin in dogs,
mydriasis occurred at 1.0 mg per kg of body weight per day and above
and extreme weight loss at 2.0 mg per kg of body weight per day and
above. In multigeneration studies, toxicity in pups was the most
sensitive indicator, and occurred at 0.4 mg per kg of body weight per
day. The no-observed-effect levels for the formation of cleft palates
in CF1 mice were the same. Both compounds were negative in a number
of in vitro tests for genotoxicity. Abamectin was also negative in
in vivo tests, including clastogenicity. Carcinogenicity studies
with abamectin were negative at maximum tolerated doses in mice and
rats. The Committee therefore concluded that it was unnecessary to
request data from long-term toxicity and carcinogenicity studies on
ivermectin.
Ivermectin is widely used in humans for the treatment of
onchocerciasis at single doses of 0.2 mg per kg of body weight.
Tolerance to the compound has been assessed in healthy volunteers and
in patients; adverse effects are usually mild and transient. In
particular, no effects on the central nervous system were observed in
patients.
4. EVALUATION
The Committee concluded that the most relevant effect for the
safety evaluation of residues of ivermectin was its effect on the
mammalian nervous system. An ADI of 0-0.0002 mg per kg of body weight
was established based on a no-observed-effect level of 0.1 mg per kg
of body weight per day for maternal toxicity in CF1 mouse. A safety
factor of 500 was selected on the basis of the absence of neurological
effects in patients. This also provided a 1000-fold margin of safety
for the developmental toxicity of ivermectin.
5. REFERENCES
ABALIS, I.M., ELDEFRANI, M.E., & ELDEFRANI, A.T. (1986). Effects of
insecticides on GABA-induced chloride influx into rat brain microsacs.
J. Toxicol. Environ. Health, 18, 13-23.
ABLIEZ, E.J, NEWLAND, H.S., WHITE, A.T., KAISER, A., GREENE, B.M.,
TAYLOR, H.R., & BUETTNER, D.W. (1988). Chemotherapy of onchoceriasis
with high doses of diethylcarbamazine or a single dose of ivermectin:
microfilaria levels and side effects. Trop. Med. Parasitol., 39,
19-24.
AWADZI, K., DADZIE, K.Y., SCHULZ-KEY, H., GILLES, H.M, FULFORD, A.J.,
& AZIA, M.A. (1986). The chemotherapy of onchocerciasis. XI. A
double-blind comparative study of ivermectin, diethylcarbamazine and
placebo in human onchocerciasis in northern Ghana. Ann. Trop. Med.
Parasitol., 80, 433-442.
AZIZ, M.A., DIALLO, S., DIOP, I.M. LARIVIERE, M., & PORTA, M. (1982).
Efficacy and tolerance of ivermectin in human onchocerciasis.
Lancet, 2, 171-173.
BRADSHAW, H. (1989). Onchocerciasis and the Mectizan programme.
Parasitol. Today, 5, 63-64.
BURG, R.W., MILLER, B.M., BAKER, E.E., BIRNBAUM, J., CURRIE, S.A.,
HARTMAN, R., & KONG, Y.L. (1979). Avermectins, a new family of potent
antihelmintic agents: producing organism and fermentation.
Antimicrob. Agents Chemother., 15, 361-367.
CAMPBELL, W.C., FISHER, M.H., STAPLEY,E.O., ALBERS-SCHONBERG, G., &
JACOB, T.A., (1983). Ivermectin: a potent new antiparasitic agent.
Science, 221, 823-828.
CAMPBELL, W.C., & BENZ, G.W. (1984). Ivermectin: a review of
efficacy and safety. J. Vet. Phamacol. Ther., 23, 1134-1136.
CHABALA, J.C., MROZIK, H., TOLMAN, R.L., ESKOLA, P., LUSI, A.,
PETERSON, L.H., WOODS, M.F., & FISHER, M.H. (1980). Ivermectin, a new
broad-spectrum antiparasitic agent. J. Med. Chem., 23, 1934-1136.
CHIU, S.H.L., CARLIN; J.R., TAUB, R., SESTOKAS, E., ZWEIG, J.,
VANDENHEUVEL, W.J.A., & JACOB, T.A. (1988). Comparative metabolic
disposition of ivermectin in fat tissues of cattle, sheep and rats.
Drug Metab. and Dispos., 16, 728-736.
DADZIE, K.Y., BIRD, A.C., AWADIZ, K., SCHULZ-KEY, H., GILLES, H.M., &
AZIZ, M.A. (1987). Ocular findings in a double-blind study of
ivermectin versus diethylcarbamazine versus placebo in the treatment
of onchocerciasis. Br. J. Ophtalmol., 71, 78-85.
DIALLO, S., AZIZ, M.A., LARIVIERE, M., DIALLO, J.S., DIOP-MAR, I.,
N'DIR, O., BADIANE, S., PY, D., SCHULZ-KEY, H., & GAXOTTE, P. (1986).
A double-blind comparison of the efficacy and safety of invermectin
and diethylcarbamazine in a placebo controlled study of Senegalese
patients with onchocerciasis. Trans. Rog., Soc. Trop. Med. Hyg., 80,
927-934.
DIALLO, S., LARIVIERE, M., DIOP-MAR, I., N'DIR, O., N'DIAYE, R.,
BADIANE, S., PORTA, M., & AZIZ, M. (1984). Conduite au Senegal des
premiers etudes d'efficacite et de tolerance de l'ivermectine (MK 933)
dans l'onchocercose humaine. Bull. soc. Pathol. Exot. Filiales., 77,
196-205.
DI NETTA, J. (1989). List of registrations. Appendix III to
Campbell, W.C., (ed.) Ivermectin and Abamectin. Springer-Verlag, New
York pp. 344-366.
DREXLER, G. & SIEGHART, W. (1984). Properties of high affinity
binding site for 3H-Avermectin B1a. Eur. J. Pharmacol., 99, 269-
277.
EGERTON, J.R., SEWARD, R.L., & ROBIN, B. (1984). L'ivermectine: un
agent antiparasitaire pour les chevaux. Rec. Med. Vet., 6, 595-599.
ELDEFRANI, A.T.. & ELDEFRANI, M.E. (1987). Receptors for gamma-
aminobutyric acid and voltage-dependant chloride channels as targets
for drugs and toxicants. FASEB J., 1, 262-271.
FRITZ, L.C., WANG, C.C., & GORIO, A. (1979). Avermectin B1a
irreversibly blocks postsynaptic potentials at the lobster
neuromuscular junction by reducing muscle membrane resistance. Proc.
Natl. Acad. Sci. USA, 76, 2062-2066.
KASS, I.S., WANG, C.C., WALROND, J.P., & STRETTON, A.O.W. (1980).
Avermectin B1a, a paralyzing anthelmintic that effects interneurons
and inhibitory motoneurons in ascaris. Proc. Natl. Acad. Sci. USA.,
77/10, 6211-6215.
KUMARASWAMI, V., OTTESEN, E.A., VIJAYASEKARAN, V, DEVI, U.,
SWAMINATHAN, M., AZIZ, M.A., SARMA, G.R., PRABHAKAR, R, & TRIPATHY,
S.P. (1988). Ivermectin for the treatment of wuchereria bancrofti
filariasis. JAMA, 259, 3105-3153.
LANKAS, G.R, MINSKER, D.H., & ROBERTSON, R.T. (1989). Effects of
ivermectin on reproduction and neonatal toxicity in rats. Fd. Chem.
Toxic., 27, 523-529.
LARIVIERE, M., VINGTAIN, P., AZIZ, M., BEAUVAIS, B., WEIMANN,D.,
DEROUIN F., GINOUX, J., SCHULZ-KEY, H., GAXOTE, P., & BASSET, D.,
(1985). Double-blind study of ivermectin and diethylcarbamazine in
African onchcerciasis patients with ocular involvement. Lancet,
2, 147-177.
MERCK & CO., INC. (1978a). Fourteen week oral toxicity study in dogs.
TT #78-038-00. Unpublished study; submitted to WHO by Merck & Co.,
Inc.
MERCK & CO., INC. (1978b). MK-0933: microbial mutagen test with and
without rat liver enzyme activation. TT #77-8068. Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979a). MK-0933: Toxicological evaluation (April
30, 1979). Reports of unpublished studies; submitted to WHO by Merck
& Co., Inc.
MERCK & CO., INC. (1979b). MK-933: Summary of toxicity studies.
Summary of unpublished studies; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979c). Acute oral toxicity study in rats. TT
#78-3087. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979d). MK-933: Acute dermal toxicity studies in
rabbits and rats. Acute ocular toxicity studies in rabbits. Acute
oral toxicity in dogs. Unpublished studies; submitted to WHO by Merck
& Co., Inc.
MERCK & CO., INC. (1979e). Acute inhalation toxicity study in rats.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979f). MK-933 (L-640,471-00W51): Acute oral
toxicity in dogs. TT #79-2869. Unpublished study; submitted to WHO
by Merck & Co., Inc.
MERCK & CO., INC. (1979g). MK-933/cattle/safety/ toxicity/clinical
pathology/N.O.T. 4480. Unpublished study; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1980a). MK-932: metabolism study in the rat. TT #
79-711-0. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980b). In vitro metabolism studies of
MK-0932/0933. Unpublished study; submitted to WHO by Merck & Co.,
Inc.
MERCK & CO., INC. (1980c). Tissue residues and radioactive balances
in rats dosed with 3H-labeled MK-0933 (OM-45). Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980d). L-640,497-OOH: Acute oral toxicity in
female mice. Unpublished study: submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980e). MK-933: Multigeneration studies in rats.
Unpublished studies; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980f). MK-933: Cross fostering study in rats. TT
#79-710-0. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980g). MK-0933: Teratogenic evaluation.
Unpublished report: submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980h). MK-0933: Mouse lymphoma cytotoxicity
study. TT #79-8034. Unpublished study; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1980i). MK 933: Unscheduled DNA synthesis in human
IMR90-fibroblasts. TT #80-8205. Unpublished study; submitted to WHO
by Merck & Co., Inc.
MERCK & CO., INC. (1981a). MK-933 (L-640,471-OOW72): Acute oral
toxicity in dogs. TT #81-2500. Unpublished study; submitted to WHO by
Merck & Co., Inc.
MERCK & CO., INC. (1981b). MK-933 Ivermectin injectable micelle
solution: acute subcutaneous toxicity study in young dogs. TT
#81-025-0. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981c). MK-933: Acute toxicity in the ovine.
N.O.T. 7000. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981d).MK-933/horses/safety/toxicity/clinical
pathology/N.O.T. 8292, protocol 554. Unpublished study; submitted to
WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981e). MK-933: Multigeneration study in rats.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981f). MK-933: oral teratogenic study in dogs.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981g). Metabolism of ivermectin (MK-0933) in
cattle and rats. Unpublished studies; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1982a). MK-0933: Three-day study of plasma and
brain drug levels in mice. TT #82-088-0. Unpublished study; submited
to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1982b). MK-0933: Thirty-six day study of plasma
and brain levels in mice. TT #82-071-0. Unpublished study: submitted
to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1982c). MK-0933: Thirty-six day study of plasma
and brain levels in dogs. TT #82-071-0. Unpublished study; submitted
by Merck & Co., Inc.
MERCK & CO., INC. (1982d). MK-0933 / Swine / Safety / Toxicity
clinical pathology / Anatomic pathology. N.O.T. 9739. Unpublished
study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1982e). MK-936: One-hundred-five week dietary
carcinogenicity and toxicity study in rats with a fifty-three week
interim necropsy. Final report. TT #82-099-0. Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1983). MK936: Ninety-four week dietary
carcinogenicity and toxicity study in mice. TT #83-002,-0, -1, -2, -3.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1985). MK-0933 and MK-0936: oral toxicity and
plasma level study in monkeys. Unpublished study; submitted WHO by
Merck & Co., Inc.
MERCK & CO., INC. (1986a). MK-0933: 16-day oral toxicity study of MK-
0933 in immature rhesus monkeys. TT #85-9033. Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1986b). MK-0933: Fifteen-day toxicity study of
orally administered MK-0933 (ivermectin) in neonatal rhesus monkeys.
TT #869005, GLP-13. Unpublished study; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1987). The metabolism, distribution, and excretion
of 22,23-3H-MKO933 in rats dosed orally and percutaneously (Expt.
ADM-66). Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC (1988a). Ivermectin: Poison Control Monograph.
Submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1988b). MECTIZANTM (IVERMECTIN, MSD). Product
Monograph 7-89 MCT 88-R-1152M. Submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1989). Identity of Ivermectin. Structural Formula.
Unpublished study; submitted to WHO by Merck & Co., Inc.
NAQUIRA, C., JIMENEZ, G., GUERRA, J.G., BERNAL, R., NALIN, D.R., NEU,
D., & AZIZ, M. (1989). Ivermectin for human strongyloidiasis and
other intestinal helminths. Am. J. Trop. Med. Hyg., 40, 304-309.
OLSEN, R.W, & SNOWMAN, A.M. (1985). Avermectin B,, modulation of
gamma aminobutyric acid/benzodiazepine receptor binding in mammalian
brain. J. Neurochem., 44/4, 1074-1082.
PAUL, S.M., SKOLNICK, P., & ZATZ, M. (1980). Avermectin B1a: An
irreversible activator of the GABA-Benzodiazepine-chloride-ionophore
receptor complex. Biochem. Biophys. Res. Communs., 96, 632-638.
PONG, S.S., DEHAVEN, R., & WANG, C.C. (1981). Stimulation of
benzo-diazepine binding to rat brain membranes and solubilized
receptor complex by avermectin B1a and GABA. Biochim. Biophys.
Acta., 646, 143-150.
PONG, S.S., DEHAVEN, R., & WANG, C.C. (1982). A comparative study of
avermectin B1a and other modulators of the gamma-aminobutyric acid
receptor-chloride ion channel complex. J. Neurosci, 2/7, 966-971.
PONG, S.S. & WANG, C.C., (1980). The specificity of high affinity
binding of avermectin B1a to mammalian brain membranes.
Neuropharmacology, 19, 311-312.
PONG, S.S., & WANG, C.C., (1982). Avermectin B1a modulation of gamma
aminobutyric acid receptors in rat brain membranes. J. Neurochem.,
38/2, 375-379.
PONG, S.S., WANG, C.C., & FRITZ, L.C., (1980). Studies on the
mechanism of action of avermectin B1a; stimulation of release of
gamma-aminobutyric acid from brain synaptosomes. J. Neurochem.,
34/2, 351-358.
PULLIAM, J.D., SEWARD, R.L., HENRY, R.T., & STEINBERG, S.A., (1985).
Investigation of ivermectin toxicity in collies. Vet. Med., 6, 33-
40.
RICHARD-LENOBLE, D., KOMBILA, M., RUPP, E.A., PAPPAYLIOU, E.S.,
GAXOTTE, P., NGUIRI, C., & AZIZ, M.A. (1988). Ivermectin in loiasis
and concomitant O. volvulus and M. perstans infections. Am.
J. Trop. Med. Hyg., 39, 480-483.
SCHOFIELD, P.R., DARLISON, M.G., FUJITA, N., BURT, D.R., STEPHENSON,
F.A., RODRIGUEZ, H., RHEE, L.M, RAMCHANDRAN, J., REALE, V., GLENCORSE,
T.A., SEEBURG, P.H., & BARNARD, E.A. (1987). Sequence and functional
expression of the GABAa receptor shows a ligand-gated receptor super-
family. Nature, 328, 221-227.
SCHRODER, J., SWAN, G.E., BARRICK, R.A., & PULLIAM, J.D., (1986).
Effect of ivermectin on the reproductive potential of breeding rams.
J. S. Afr. Vet. Assoc., 57/4, 211-213.
SUPAVILAI, P. & KAROBATH, M. (1981). In vitro modulation by
avermectin B1a of the GABA/benzodiazepine receptor complex of rat
cerebellum. J. Neurochem., 36/3, 798-803.
VINGTAIN, P., PICHARD, E., GINOUX, J., COULIBALY, S.M., BISSAN, Y.,
RANQUE, P., & THILAYE, B. (1988). Ivermectine et onchocercose
humaine. A propos d'une etude portant sur 234 onchocerquiens en
Republique du Mali. Bull. Soc. Pathol. Exot. Fialiales., 81, 260-
270.
WILLIAMS, M., & YARBROUGH, G.G. (1979). Enhancement of in vitro
binding and some of the pharmacological properties of diazepam by
novel anthelmintic agent avermectin B1a. Eur. J. Pharmacol., 56,
273-276.
WILLIAMS, M. & RISLEY, E.A. (1982). Interactions of avermectins with
3H-B-carboline-3-carboxylate-ethylester and 3H-diazepam binding
sites in rat brain cortical membrane. Eur. J. Pharmacol., 77, 307-
312.
WILLIAMS, M. & RISLEY, E.A., (1984). Ivermectin interactions with
benzodiazepine receptors in rat cortex and cerebellum in vitro.
J. Neurochem., 42/3, 745-753.
2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Absorption, distribution, and excretion
2.1.1.1 Rats
Radio-labeled ivermectin (mixture of 92.2% 3H-H2B1a and 7.8%
3H-H2B1b was administered to Sprague-Dawley rats, approximately 8
weeks old, once orally as a solution in sesame oil by gavage at 0.3
mg/kg b.w. (based on an average body weight of 294 grams for the males
and 218 grams for the females), or topically, after shaving, as a
solution in topical vehicle at 0.5 mg/kg b.w. (males only, average
body weight 279 grams). The animals were subdivided into groups of
three. Three treatment groups (sacrifice at days 1, 3, and 5
post-dose) and one control group (sacrifice at day 5 post-dose) of
each sex were used in the oral study. In the topical study, three
treatment groups (sacrifice on days 1, 3, and 6 post-dose) and one
control group (sacrifice on day 6 post-dose) of males were used.
Urine and faeces were collected daily. At sacrifice, blood was
collected by heart puncture; liver, kidney, muscle (from the hind
legs), and fat (males: testicular fat pad; females: peripheral fat)
were also harvested. Samples from the gastrointestinal tract were
also taken at the time of sacrifice from the animals dosed orally.
For each tissue, the individual samples from animals of the same group
were composited. Radioactivity was measured and calculated as drug
equivalents.
Residue levels in all samples were generally much higher
following oral application compared with the topical application. The
times of peak concentrations were delayed following topical
application. The main route of excretion was via the faeces.
Concentrations in faeces, however, were higher in females compared
with males; concentrations in urine were lower in females than in
males. This was consistently observed at all sampling times. In the
oral study 57.4% (males) and 58.4% (females) of the administered drug
had been eliminated one day after administration. These figures
increased to 83% (males) and 91.7% (females) five days after
administration. In the oral study, the highest residue levels in body
tissues were observed in fat, followed by liver, kidney, and muscle.
The approximate ratios for the residue levels in these tissues were
100:55:40:15 and were similar for both sexes. For the topical study
the situation was less clear due to the smaller data base and the
delay in the appearance of the residues in the tissues. At day 3,
when maximum residue levels were observed, concentrations in the above
tissues ranged in the same order, i.e., fat, liver, kidney, and muscle
(Merck & Co., Inc., 1987).
A residue study with labeled H2B1a (3H-labeled in the
22,23-position) was conducted in CRCD-rats.
Group 1: Six female rats aged approximately 8 weeks, and weighing
189-240 grams at initiation were administered labeled ivermectin for
61 days, then throughout mating, gestation, and until day 9
post-partum. One animal failed to mate; another animal was determined
not to be pregnant.
Group 2: Six female rats, aged approximately 12 weeks and weighing
248-298 grams, received radiolabeled ivermectin at the same dose and
specific activity from days 1 to 9 post-partum.
The dose, administered as an oral solution in sesame oil and by
metal catheter to both groups, was 2.5 mg/kg b.w./day (specific
activity 0.2 mCi/mg). Litter sizes were standardized to 5 males and
5 females on day 1 post-partum in both groups. At the time of
sacrifice, kidney, liver, brain, and carcass were collected. Milk
samples were obtained from 2 dams in each group on days 4, 6, and 10
post-partum. Blood, liver, brain, and carcass samples were obtained
from selected offspring (2 from each litter) of both groups on days 1,
4, 6, and 10 post-partum. Radioactivity was measured by
liquid-scintillation counting following oxidation of the tissues.
Dams: The concentration of ivermectin equivalents in plasma
increased to reach a plateau at approximately day 10 of treatment in
group 1. On day 1 post-partum, a three to four times higher
concentration was observed, possibly due to an increased mobilization
of body fat (ivermectin is highly lipophilic). Plasma levels then
decreased gradually and reached values comparable to those seen during
the pre-mating dosing period by day 10 post-partum. In group 2,
plasma levels increased gradually throughout the lactation period. On
day 10 post-partum, the individual values in both groups were
comparable. Concentrations in milk were at least three to four times
higher than corresponding concentrations in plasma. Tissue residues
in brain were very low relative to residues in kidney, liver and
carcass.
Offspring: From the concentrations in milk and based on daily milk
consumption by neonatal rats, a daily intake of 0.5 to 0.6 mg/kg body
weight on days 4-5 post-partum has been estimated. This corresponds
to approximately one half of the acute oral LD50. Under these
conditions concentrations in plasma increased dramatically between
days 1 and 6 post-partum, and were finally three times higher than the
concentrations found in maternal plasma. Similarly, residue
concentrations in livers were two times higher than those seen in the
corresponding dams. In contrast to the dams, the residue
concentrations in brain from offspring were comparable to plasma
concentrations on days 1 and 4 post-partum in group 1. On days 6 and
10, however brain levels were approximately three times lower than
plasma levels. The results of this study suggest that the transfer of
drug via the milk is probably responsible for the increase in neonatal
mortality associated with repeated administration in multigeneration
studies with rats when the blood-brain barrier to the drug is still
incomplete (Merck & Co., Inc., 1980a).
2.1.1.2 Dogs
To determine whether plasma and/or brain tissue levels of
ivermectin are proportional to dose, groups of 4 female beagle dogs,
approximately 34-39 weeks old at initiation of the study, weighing 7.6
to 9.6 kg, received ivermectin orally (solution in sesame oil, gavage)
at dose levels of 0.5 mg/kg body weight or 2.0 mg/kg b.w., once per
day over a period of 35 days (except one animal in the high dose group
which received 24 doses prior to sacrifice on day 24). Surviving
animals were not dosed on the day of sacrifice. On days 2, 8, 15, 22,
and 29 (6 hours post-dose), and on day 36, blood was withdrawn.
Following withdrawal of the last blood sample, cerebrospinal fluid was
collected. Samples were assayed for H2B1a. Beginning in week 2,
mydriasis was observed in two animals of the high dose group. This
effect was also noted in the remaining two animals of this group
during week 3 and continuing until termination. On day 21, one animal
of the high dose group exhibited ataxia, and fine whole-body tremors.
This animal's condition deteriorated rapidly and by approximately 4
hours post-dose on day 22 the animal was recumbent with salivation and
marked tremors. On arousal it exhibited marked ataxia. It had to be
sacrificed on day 24. With the exception of mydriasis, the remaining
three animals of the high dose group exhibited no other
treatment-related effect. No treatment-related physical signs were
observed in the low dose animals. Slight increases in body weight
were found in all animals receiving 0.1 mg/kg b.w./day. Slight to
marked weight losses occurred among the high dose animals with the
largest loss (2.1 kg) found in the dog which was sacrificed on drug
day 24. Plasma concentration of H2B1a increased dramatically in both
groups between days 2 and 8 of treatment. Thereafter, gradual
increases occurred, reaching approximate steady-state levels at day 22
in both groups. The animal which had to be sacrificed on day 24
achieved the highest plasma concentrations. Unexplained decreases in
plasma concentrations occurred in weeks 4 and 5 in the high dose group
and in week 4 in the low dose group. The mean of the ratios of the
plasma concentrations in the 2.0 mg/kg b.w./day group compared to the
0.5 mg/kg b.w./day group was 8.4. Plasma concentrations were thus
not linear to dose, since a 4-fold increase in dose led to a 8-fold
increase in plasma concentrations. Concentrations in cerebrospinal
fluid remained below the limit of detection (1 ng/ml), except in the
animal which was sacrificed on day 24 in which severe signs of CNS
depression were evident. In this animal 3 ng/ml was determined in the
cerebrospinal fluid (Merck & Co., Inc., 1982c).
2.1.1.3 Monkeys
Concentrations of 22,23-dihydroavermectin-B1a and avermectin-B1a
were measured in plasma at three dose levels (2.0, 8.0, and 24.0 mg/kg
body weight) in a combined oral toxicity and plasma level study of
ivermectin and abamectin with immature rhesus monkeys. The time
post-administration when blood levels reached a maximum could not be
precisely determined from the data. However, the concentrations
observed following treatment with ivermectin were higher than those
measured following the administration of abamectin. For both
substances, the concentrations in plasma were proportional to the
administered dose but this proportionality was apparently not linear
(Merck & Co., Inc., 1985).
2.1.1.4 Humans
The pharmacokinetics in plasma of ivermectin were studied in
randomized three-period crossover studies with 12 healthy adult male
volunteers in each study. Peak plasma concentrations were reached
approximately 4 hours after administration. Bioavailability (area
under the curve) was highest following administration of ivermectin as
a solution. No significant differences between the bioavailabilities
of the capsule and the tablet formulations were observed (Merck & Co..
Inc., 1988b).
2.1.2 Biotransformation
Because of the extremely low levels of residues in tissues and
the many difficulties associated with purification, the following
approach was taken:
* steer and rat liver microsomes were incubated in vitro
with ivermectin components. Metabolites were isolated,
purified and identified;
* major steer-liver metabolites were isolated from large
quantities of this tissue and were compared to the products
of in vitro incubations;
* fractions obtained from smaller amounts of liver were
chromatographically separated and cochromatographed with
in vitro products, where possible;
* chromatographic profiles of rat hydrolyzed isolates were
compared in the same way to those obtained from liver.
Based on this approach it was possible to identify the major
metabolites obtained from the various steer liver, rat liver and steer
fat isolates. (Merck & Co. Inc., 1980b).
2.1.2.1 In vitro studies of metabolism
Rat liver microsomes were incubated in vitro with either
avermectin-B1a, 22,23-dihydroavermectin-B1a, or
22,23-dihydroaver-mectin-B1b. The samples were extracted and
metabolites were purified by solvent extraction and chromatographic
procedures. The structures of the isolated metabolites were
determined by mass spectrometry and nuclear magnetic resonance
spectrometry.
With each substrate >70% of the radioactivity was associated
with the respective parent compounds. Two major polar metabolites
(total amount 2-11%) were formed. One metabolite has been identified
as the C24-methyl alcohol of the parent compound which had been used
as the substrate for incubation. A smaller quantity was identified as
the monosaccharide of the C24-alcohol. These two metabolites
represented the major fraction of metabolites which were more polar
than the respective parent compounds (>50% with the substrate H2B1a
and 80% with the substrate H2B1b). The metabolites retained
antiparasitic activities which quantitatively depend on test species
and conditions of testing (e.g., in vitro / in vivo) (Merck & Co.,
Inc., 1980b).
2.1.2.2 Rats
An experiment was designed to provide composite samples of
tissues and excreta from 24 male CRCD rats (weighing 295 to 329
grams), dosed orally by gavage at 0.3 mg/kg body weight with tritiated
ivermectin. The animals were sacrificed at days 1 and 3 post-dose.
The cumulatively measured radioactive drug equivalents in the
gastrointestinal tract, urine, and faeces accounted for 6.4, 0.8, and
84.9%, respectively, of the administered dose at day 3. Tables 1 and
2 summarize the amount of "total residue" and the percentage of
unchanged drug in tissues, and the proportions of metabolite fractions
isolated from the liver of rats dosed with tritium-labeled ivermectin
(Merck & Co., Inc., 1980c).
A group of nonpolar metabolites has been detected in fat tissue.
Upon hydrolysis, these metabolites gave rise to polar products that
were similar to the ivermectin metabolites present in liver. From
residue profiles (reversed-phase HPLC) of extracts purified from the
fat obtained on day 3 post-dose from rats treated with 3H-ivermectin,
it was estimated that about 17% of the total residue consisted of the
nonpolar metabolites and about 49% was the parent drug. It was
suggested that polar ivermectin metabolites produced in the liver were
esterified with fatty acids and stored in the fat as nonpolar entities
(Chiu et al., 1988).
2.1.2.3 Humans
Four healthy male volunteers received 14 mg of 3H-labeled
ivermectin. Blood, urine, and faeces were assayed for radioactivity
by liquid scintillation counting and/or following HPLC separation.
Approximately 0.6% of the radioactive dose was excreted in the urine
(over four days). An average of 49% was recovered from faeces (over
five days). Table 3 summarizes the kinetic parameters of the study.
Mean plasma concentrations of radiolabeled metabolites were about
twice that of the parent drug. Peak plasma concentrations occurred
seven hours post-treatment for radioactivity and six hours post-dose
for the parent drug. Radioactivity was eliminated from the blood more
slowly than the non-metabolized component. Discontinuities in plasma
profiles of the parent drug were observed. This study suggested the
possibility of enterohepatic recycling (Merck & Co., Inc., 1988b).
2.1.2.4 Mechanism of action
Most studies on the mode of action have been performed with
avermectin B1. However it is assumed that all active members and
derivatives of the avermectin family share a common mechanism of
action. Avermectin B1 apparently affects interneuron-motorneuron
transmission in nematodes and neuromuscular transmission in
arthropods. In both cases receptors for gamma - aminobutyric acid
(GABA) are involved. The low amount of GABA-ergic synapses in
helminths and arthropods hindered complete elucidation of the
mechanism (Fritz et al. 1979; Kass et al., 1980).
2.2 Toxicological Studies
2.2.1 Acute toxicity studies
2.2.1.1 Mice
Ivermectin was tested in adult male and female mice. The
components of ivermectin, H2B1a and H2B1b, were also studied in the
female mouse. In an additional study, the acute toxicities of
tetrahydroavermectin B1, the major contaminant (up to 4%) of
ivermectin, and of ivermectin itself were compared in the female
mouse. Either ten or twenty albino mice (Careworth CF-1 strain)
weighing 19-24 grams, and approximately 7 weeks of age, received the
compounds tested as a solution in sesame oil by gastric intubation.
All mice were observed at the day of drug administration and
thereafter for 14 days. Calculation of LD50 values were based on 14-
day mortality response.
Table 1: Equivalents of total radioactive residues [ng/g] and percent of unaltered drug in rat tissues
Tissue: fat liver kidney muscle plasma
Days post dose 1 3 1 3 1 3 1 3 1 3
Total residue 232 137 47 40 40 46 44 18 12 5
H2B1a [%] 50 66 66 56 28 34 53 51
H2B1b [%] 13 12 5.8 9 21 27 9.6 11
Table 2: Classification of the total radioactive residue in rat liver
at day 1 post-dose
Metabolite Isolation Percent of total
Group polarity fraction radio-activity
I very polar aqueous buffer 0.06
II very polar Sep-Pak, eluate 0.4
with methanol
II-A polar (at least HPLC 0.3
two compounds)
III-B polar (not HPLC 3
identified)
IV polar (identfied HPLC 8
metabolites)
V polar (at least 10
four compounds)
VI unaltered drug HPLC 71
VII non-polar HPLC 7.3
VIII non-polar isooctane 1.9
Table 3: Kinetic parameters in plasma of radioactivity and H2B1a
following administration of 3H-ivermectin to healthy
human volunteers.
Cmax tmax t1/2
[ng/ml] [hours] [hours]
radioactivity 54.2 7 70
equivalents
H2B1a 21.7 6 11.8
component
H2B1a: Two lots were studied at different times. Dose levels
were 2.5, 5, 10, 20, 40, 80 and 160 mg/kg body weight with the first
lot: with the second lot the same dose levels were tested, except that
the dose level of 2.5 mg/kg body weight was omitted. Signs of
toxicity were seen within 30 to 90 minutes at a dose at or above 5
mg/kg body weight which consisted of ataxia, tremors, bradypnoea,
decreased activity and loss of righting. The majority of deaths
occurred from 45 minutes to 3 days with four later deaths from the 4th
to 7th days. Most of the survivors appeared normal by the fourth day.
H2B1b: Two lots were tested at different times at dose levels
of 5, 10, 20, 40, 80, and 160 mg/kg body weight. Signs of toxicity,
which were generally similar to those observed following
administration of H2B1a, (ataxia, tremor, bradypnoea, and loss of
righting) were seen within 90 minutes, and were scattered through all
doses. Most deaths occurred from 26 minutes to the fourth day with
one death on the sixth day.
Ivermectin: Two different lots (one 80% H2B1a/20% H2B1b, and
the other 84% H2B1a/16% H2B1a) were tested at dose levels of 5, 10,
20, 40, 80 and 160 mg/kg body weight. Ivermectin was found to be
significantly more toxic orally in the male mouse than in the female
mouse. Signs of drug effects, however, were generally similar in both
sexes. When these compounds were tested concurrently in the female
mouse, there was no significant difference in the toxicity (Merck &
Co., Inc., 1979a).
Tetrahydroavermectin-B1: This substance was also tested at dose
levels of 5, 10, 20, 40, 80 and 160 mg/kg body weight in direct
comparison with ivermectin. The results indicated that this compound
was significantly less toxic than ivermectin in the female mouse.
Signs of drug effects were seen at 80 and 160 mg/kg body weight and
consisted of ptosis, salivation, ataxia, and loss of righting.
Eighteen hours after dosing, tremors and ataxia were seen at dose
levels of 10 mg/kg body weight and above. The duration of signs was
approximately 36 hours (Merck & Co., Inc., 1980d).
2.2.1.2 Rats
Ivermectin was studied in young adult male and female rats and
infant rats. Young adult rats of two different strains were used:
Sprague-Dawley Camm rats weighing 150 to 175 grams which were 7 to 9
weeks of age were treated at dose levels of 2.5, 5, 10, 20, 40, and 80
mg/kg body weight. Charles River-CD strain rats approximately 7 weeks
old and weighing 125 to 175 grams were treated with 25, 35, 49, 68.6,
and 96 mg/kg body weight ivermectin. Ten rats of each sex were used at
each dose. The infant rats (Charles River-CD strain) were 24 to 48
hours old and weighed on a littermate group average 7.3 to 9.4 grams.
Ten infant rats of undiscriminated sex were used at each dose level of
1, 2, 4, 8, 16 and 32 mg/kg body weight. Signs of drug effects were
similar in both strains of adult rats (decreased activity, salivation,
bradypnoea, and ataxia, depending on the dose). Deaths occurred from
overnight to the second day. There was no significant sex-related
difference in toxicity.
No signs were observed following the oral administration to
infant rats. The majority of deaths occurred in 131 minutes to
overnight with one death on the fifth and one death on the sixth day
(Merck & Co., Inc., 1979a).
The acute oral toxicity of ivermectin was also studied in 13 week
old Charles River CD rats obtained from a cross-fostering study in
order to determine if prenatal or postnatal exposure increased the
toxicity of subsequent exposure. The rats had been exposed to
ivermectin or a vehicle control, prenatally, postnatally, or both pre-
and postnatally, and were grouped (30 males, weighing 224-497 grams
and 30 females, weighing 151-272 grams, per group) as follows:
Group 1: F0 treated x F1 treated
Group 2: F0 treated x F1 control
Group 3: F0 control x F1 control
Group 4: F0 control x F1 treated
The rats (six animals of each sex per dose level) were given a
single dose by gastric intubation of a 0.8% solution of ivermectin at
dose levels of 25.0, 32.5, 42.3, 55, or 71.5 mg/kg body weight. All
rats were observed on the day of drug administration and daily
thereafter for 14 days.
Signs of drug effects were generally similar in all four groups
of rats tested and in both sexes. On the second day, decreased
activity, bradypnoea, and a reddish-brown discharge around the nose
and mouth were seen at all dose levels. In the male rats, loss of
righting was observed at 42.3 mg/kg body weight and higher. In the
females this effect was seen at the 55 and 71.5 mg/kg body weight dose
levels. In the surviving animals these effects persisted until the
seventh day. The majority of deaths occurred from overnight to day 4,
but a few deaths occurred on days 5, 6, 9, and 10. The number of
deaths was too small to allow the calculation of the LD50. The
results of the study indicated no significant differences in toxicity
between controls (group 3) and the other groups (Merck & Co., Inc.,
1979c).
The acute percutaneous toxicity of ivermectin was studied in 10
male and 10 female rats (Charles River CD strain) which weighed 224 to
408 grams and were 12 to 13 weeks of age. Five rats of each sex were
used at each of two dose levels, 330 and 660 mg/kg body weight. The
doses were applied to occluded unabraded skin. The animals were
observed daily (except on weekends) for 14 days.
Rats at both dosage levels began to show signs of systemic
toxicity two days after treatment. At the 330 mg/kg body weight dose
level, one male died seven days after treatment. At the 660 mg/kg
body weight dose level one male (on day five) and one female (on day
three) died. The relevant effects were bradypnoea and tremors (Merck
& Co., Inc., 1979d).
2.2.1.3 Rabbits
Three groups of three male and three female albino rabbits each,
weighing 2.61 to 3.47 kg and 18 to 20 weeks of age, were used to
determine the acute percutaneous toxicity of ivermectin. The hair was
removed and in each group three animals were abraded. The test
compound was applied as a dry powder at doses of 165, 330, and 660
mg/kg body weight. Animals were examined during a 14 day post dosing
period. The signs of systemic toxicity (bradypnoea, tremor, and
anorexia) were similar for both abraded and unabraded rabbits. The
percutaneous LD50 was estimated as 406 mg/kg body weight (Merck & Co.,
Inc., 1979d).
No toxic effects (except mucosal irritations) developed in an
acute inhalation toxicity study with ivermectin. Five male and 5
female Sprague-Dawley rats were exposed for 60 minutes to the maximum
attainable concentration of 5.11 mg/1 overall nominal air
concentration. With 0.37% of the particles showing sizes of 15
microns or less, the corresponding dose was estimated as < 0.4
mg/kg body weight (Merck & Co., Inc., 1979e).
Only slight irritation developed in an acute ocular irritation
study in 2 male and 2 female New Zealand rabbits when 100 mg
ivermectin powder was placed in the conjunctival sac of the left eye
(Merck & Co., Inc., 1979d).
2.2.1.4 Dogs
The acute oral toxicity of ivermectin was studied in eight male
and 8 female beagle dogs, 10 to 14 months of age and weighing between
8.1 and 15.2 kg. Three groups, each containing two male and two
female dogs, received doses of 2.5, 5.0, or 10.0 mg/kg body weight as
a 1.6% solution in sesame oil by gastric intubation. A fourth group
received sesame oil (0.625 mg/kg body weight) in the same way. The
animals were observed throughout a 14-day test period.
Mydriasis and the absence of pupil response were seen in two dogs
at the lowest dose, and in all dogs at the two higher doses. Within
75 minutes of drug administration, one dog at the high dose vomited.
This same dog exhibited emesis and salivation two additional times
within four hours. At both the high and the intermediate doses one
additional dog had emesis following drug administration. Tremors
which were seen in five animals (2 dogs at 5 mg/kg body weight and 3
dogs at 10 mg/kg body weight) were first observed about six hours
following drug administration and were still present on the third day
in some animals.
One dog in the high dose group became ataxic and heavily sedated.
This dog recovered from the sedation within 48 hours and from ataxia,
tremors, and salivation within 72 hours (Merck & Co., Inc., 1979f).
In a further oral toxicity study with dogs, three groups of two
male and 2 female beagle dogs, 6-9 months of age and weighing 6.3 to
10.1 kg, were given ivermectin 1.6% solution in sesame oil at doses of
5, 10, and 20 mg/kg body weight by gastric intubation. Because no
deaths occurred, two additional groups of two males and two females
each were dosed with a 6.4% suspension/solution in sesame oil at doses
of 40 and 80 mg/kg body weight. Signs included emesis (all dose
levels, except 40 mg/kg body weight), mydriasis (all dose levels),
ataxia and tremors (doses >10 mg/kg body weight), salivation (in the
40 and 80 mg/kg body weight dose-groups) and death preceded by a
comatose like state (in the 40 and 80 mg/kg body weight dose-groups)
(Merck & Co., Inc., 1981a).
Sixteen rough-coated collies ranging in age from 7 months to 9
years were used in an oral toxicity study with ivermectin. Treatment
groups (two males, two females, half of the animals of each sex having
collie eye anomaly) received 0.05, 0.2 or 0.6 mg/kg body weight of
ivermectin once orally (plastic syringe) as a solution in fractionated
coconut oil with 2% benzyl alcohol. At the end of the trial (7 days)
three previously untreated control dogs also received the lowest dose
(0.05 mg/kg body weight). Samples of plasma, cerebrospinal fluid,
brain, spinal cord, and liver were assayed for H2B1a. No drug-
related effects were seen in the 7 animals treated at the lowest dose
level. Two dogs given 0.2 mg/kg body weight and two dogs given 0.6
mg/kg body weight showed signs of toxicity which were mild and
transitory in one dog of each group. One animal per group
progressively developed similar severe clinical signs: Ataxia with
increasing hypermetria which progressed to paresis and, finally,
paralysis. In both dogs segmental reflexes remained strong. Both
salivated excessively and had predominantly diaphragmatic respiration.
The dog from the 0.6 mg/kg body weight group was killed at 28 hours
post-dose; the dog from the 0.2 mg/kg body weight group died 51 hours
post-dose. These two severely affected dogs showed significantly
increased brain drug levels (Pulliam et al., 1985).
The acute subcutaneous toxicity of ivermectin injectable micelle
solution and its vehicle were studied in 11 to 12 week old beagle
dogs. Five groups of three males and three females received 4.7, 9.4,
18.8, 37.5, and 75.0 mg/kg body weight ivermectin. A sixth group of
three males and two females received an identical volume (1 ml/kg body
weight) of vehicle. Signs were seen in all ivermectin dose groups.
There were no sex related significant differences in toxicity. No
deaths occurred the 4.7 mg/kg body weight dose. Three of the six dogs
dosed at 9.4 mg/kg body weight, and all of the dogs administered doses
>9.4 mg/kg body weight, died. Dogs receiving the vehicle appeared
normal throughout the fifteen-day observation period. Mydriasis and
negative pupil response were observed in all ivermectin treated groups
with time of onset and duration (in the surviving dogs) depending on
the dose (e.g., onset was three hours after dosing at 75 mg/kg body
weight and approximately 24 hours after dosing at 47 mg/kg body
weight). Other signs included tremors, ataxia, salivation, and
decreased activity. At necropsy, treatment-related slight to very
slight changes were present only in dogs that did not survive to
termination (thymus-hemorrhage, lung-congestion, lung-edema, lung
acute suppurative pneumonia, skin edema). LD50 values were estimated
as 8.4 mg/kg body weight in males and 10.5 mg/kg body weight in
females (Merck & Co., Inc., 1981b).
2.2.1.5 Pigs
Male and female Yorkshire swine were given subcutaneously 0.3, 3,
15, or 30 mg/kg body weight of ivermectin. Signs of toxicity
(decreased food and water intake, lethargy, ataxia, mydriasis, tremor,
labored breathing and lateral recumbency) were seen at the highest
dose level (Merck & Co., Inc., 1982d).
2.2.1.6 Sheep
Ten male and ten female lambs weighing 28 to 39 kg, allocated to
20 individual pens and fed restrictively once daily (with a diet
containing ethoxyquin; drinking water ad libitum), were assigned to
five treatment groups of four each. Treatment-sex combinations were
randomly allocated. There were five treatment groups, control
(distilled water), 0.3, 2.0, 4.0, and 8.0 mg/kg body weight. Two
additional animals were dosed with propylene glycol two days after the
other animals were treated. The animals were sampled 6 and 4 days
prior to treatment as well as on days 1, 2, 4, 7, 10, 14, and 18.
Surviving animals were killed on days 21 to 23 after treatment. At
8.0 mg/kg body weight, all sheep were ataxic within three hours after
dosing. They were depressed (head and ear drooping). One animal went
into lateral recumbency in a shock-like condition. Reflexes were
present but delayed. The animal was up after 24 hours and appeared
clinically normal by day 3 after dosing. After 24 hours all animals
were still mildly depressed and slightly incoordinated. At 4.0 mg/kg
body weight, the sheep were mildly incoordinated and depressed and had
initially delayed feed consumption although the 24-hour feed
consumption was normal. All animals were clinically normal at 24
hours. Since two additional sheep given vehicle (propylene glycol)
only showed the same physical signs as those given 8 mg/kg body
weight (the female fell into lateral recumbency in a shock-like
condition and was found dead 24 hours after dosing), it was suggested
that the observed signs were due to the propylene glycol vehicle
(Merck & Co., Inc., 1981c).
2.2.1.7 Cattle
Approximately 6 month old male and female Holstein Friesian
calves ranging in weight between 95 and 143 kg were injected once
subcutaneously with 0.3, 2, or 8 mg/kg body weight ivermectin. The
observed signs of toxicity at the highest dose level were increased
respiratory rate, muscular tremors, and rigidity of the extremities
and death (Merck & Co., Inc., 1979g).
2.2.1.8 Horses
In a target animal safety study, horses were injected
intramuscularly with ivermectin at dose levels of 3, 6, or 12 mg/kg
body weight. Signs of toxicity were seen at all dose levels (Egerton
et al., 1984).
No effects were observed when ivermectin was given as an oral
paste at 0.4 mg/kg body weight to 26 male and female miniature and
farabella pure and crossbred horses aged 5 months to 13 years and
weighing 40-180 kg. Ivermectin was administered once as a micelle
solution to groups of four horses (264-431 kg body weight) at 3, 6, or
12 mg/kg body weight. At 3 mg/kg body weight one group was injected
with ivermectin concentrate; a second group and all other treatment
groups were injected with a ready-to-use micelle solution. Mydriasis
was seen at all dose levels. All horses dosed at 12 mg/kg body weight
showed additional signs of drug toxicity including depression and
ataxia. One horse was found in lateral recumbency 24 hours after
dosing and was killed 72 hours after dosing (Merck & Co., Inc.,
1981d).
2.2.1.9 Rhesus monkeys
An oral toxicity study was conducted in order to determine the
minimum toxic dose of ivermectin and abamectin in rhesus monkeys and
to determine the plasma levels of the drug at that dose. Immature
rhesus monkeys, aged 2 to 3 years at initiation, weighing 2.6 to 3.1
kg (males) and 2.4 to 3.2 kg (females), were given single increasing
oral doses (0.2, 0.5, 1, 2, 4, 6, 8, 12, 24 mg/kg body weight, in
chronological order) of ivermectin and abamectin in sesame oil, by
gavage, at intervals of 2 to 3 weeks before the administration of the
next higher dose to same group of four animals (two of each sex). The
0.2 mg/kg body weight dose was repeated twice due to uncertainties as
to whether mydriasis was occurring in two treated monkeys and because
two monkeys regurgitated part of their dose. The second repetition
was a cross-over study in which the monkeys formerly treated with
ivermectin were given abamectin and vice versa. The 2 and 8 mg/kg
body weight doses were repeated to measure plasma levels. Therefore,
each animal received a total of 13 doses.
The following treatment-related physical signs were observed: (a)
Doses of 2.0 mg/kg body weight and higher caused emesis. The time of
onset tended to decrease as the dose increased. (b) Pupil dilation
and/or decreased constriction was observed following doses of 6.0
mg/kg body weight of abamectin and above and after doses of 12.0 or
24.0 mg/kg body weight of ivermectin. Most of the observations were
slight and difficult to assess and were considered equivocal, except
those seen following the 24 mg/kg body weight dose. No relationship
appeared to exist between the dose levels and the time of onset or
duration. (c) Several animals displayed decreased levels of activity
or slight to moderate sedation at 24 mg/kg body weight of both
compounds. All animals recovered within 48 hours. No tremors or
convulsions occurred. A difference in potency between the two test
substances was not discernible. It was unlikely that the lack of more
profound toxicity was due to regurgitation, since emesis did not
generally occur before four hours post-dose. Plasma concentrations
increased with dose but not in a linear manner. Signs of toxicity did
not correlate with plasma levels over the investigated dose-range.
The most sensitive indicator of toxicity was emesis with a NOEL of 1.0
mg/kg body weight and a dose-related increase over the range of 2.0 to
24.0 mg/kg body weight (Merck & Co., Inc., 1985).
2.2.1.10 Summary of the results obtained from acute toxicity studies
The following main symptoms of central-nervous disorders were
observed within one hour and up to seven days following a single oral
dose of ivermectin depending on the test species and the applied dose:
tremor, depression, ataxia, paresis, paralysis and death. LD50 values
for experimental animals are given in Table 4.
Mice, particularly males, were found to be more sensitive than
rats. LD50 values ranged from 11.6 mg/kg bw in LD50 values male mice
to 40 mg/kg body weight in females. The reason for the rather high
variability of the results remains unclear. In studies with female
mice when either ivermectin or one of its individual main components
H2B1a and H2B1b was applied, no significant differences in acute
toxicity were observed. Tetrahydroavermectin B1, however, the most
abundant potential impurity (up to 4%), was of significantly lower
acute oral toxicity.
In the rat, a higher sensitivity of neonatal rats (LD50 = 2.3
mg/kg body weight) was evident if compared with 42.8-52.8 mg/kg body
weight LD50s reported for adult animals (male and female). The
increased toxicity of ivermectin in neonatal rats is likely due to a
combination of excessive plasma levels resulting from exposure via
maternal milk and the increased permeability of the blood-brain
barrier during the early postnatal period in this species (Lankas et
al., 1989).
Great differences in sensitivities were observed among various
other species including target animals. Large variation has been
observed between breeds of dogs and individual dogs within the same
breed.
Table 4: Tabular representation of acute toxicity data in laboratory
animals
LD50 Reference
Species Sex Route (mg/kg bw) (Merck & Co., Inc.)
Mouse M oral 11.61A 1979a
(CF-1) F oral 24.61A 1979a
27.11A 1979a
41.61B 1979a
40.01A 1979a
30.01A 1980d
F oral 31.72 1979a
87.22 1979a
F oral 27.63 1979a
56.63 1979a
F oral 1604 1980d
Rat MA oral 42.81A 1979a
(young) FA 44.31A 1979a
MB 42.81A 1979a
FB 52.81A 1979a
M+F percutan. >660 1979d
(infant) M+F oral 2.31C 1979a
Rabbit percutan. 406 1979d
Dog
(beagle) F oral >10.0 1979f
M+F oral approx. 80.0 1981a
M subcutaneous 8.4 1981b
F 10.5 1981b
1A) test substance: ivermectin; 80:20 mixture;
1B) test substance: ivermectin; 84:16 mixture;
1C) test substance: ivermectin; ratio not indicated
2) test substance: H2B1a;
3) test substance; H2B1b;
4) test substance: Tetrahydroavermectin B1;
A) Charles River, CD;
B) Sprague-Dawley, Camm
2.2.2 Short-term studies
2.2.2.1 Rats
A fourteen-week toxicity study following in utero exposure was
reported. Twenty rat pups of each sex between 3 and 4 weeks of age
weighing 49 to 86 grams (males) or 43 to 77 grams (females) were
treated at dosage levels of 0.4, 0.8, and 1.6 mg/kg b.w./day. No
changes due to treatment occurred at 0.4 mg/kg b.w./day. The
following effects could not be excluded as being treatment-related in
the two other dosage level groups: spleen-enlargement and reactive
bone-marrow hyperplasia, which occurred in 1 animal at 0.8 mg/kg
b.w./day and in 3 animals at 1.6 mg/kg b.w/day (Merck & Co., Inc.,
1979b).
2.2.2 Dogs
Twenty male and 20 female beagle dogs, 39-43 weeks of age,
weighing initially 8.2 - 12.1 kg (males) and 6.2 - 9.2 kg (females)
were selected for oral treatment (gastric intubation) in five groups
of four males and four females at doses of 0.5, 1.0, 2.0 mg/kg
b.w./day. Controls received water or vehicle (sesame oil). At 2.0
mg/kg b.w./day, three males and one female developed tremors, ataxia,
anorexia, and dehydration. All of these animals exhibited ptyalism
and mydriasis followed by slight tremors, characterized by
intermittent or constant shaking of all limbs, which generally
increased in severity over 3 to 6 days. These animals were frequently
found laterally recumbent and were ataxic when standing. They were
sacrificed between weeks 4 and 12. Mydriasis was observed in all dogs
at this level (beginning in week 1 and continuing until week 12 when
it decreased in incidence). The four dogs sacrificed showed weight
losses between 1.0 and 1.6 kg. At 1.0 mg/kg b.w./day, mydriasis was
occasionally seen, particularly in week 3. Weight gain was retarded.
At 0.5 mg/kg b.w./day, only slight retardation of weight gain was
observed. No significant drug-related changes were observed for the
following parameters: ocular abnormalities, electrocardiograms,
haematologic parameters, urine-analysis, and pathological changes
(Merck & Co., Inc., 1978a).
2.2.2.3 Rhesus monkeys
A 16-day oral toxicity study with ivermectin was conducted to
determine its toxicity in immature rhesus monkeys (13 - 21 months old,
weighing 2.1 to 3.2 kg (males) and 1.9 to 2.7 kg (females) at
initiation). Each of the treatment groups (4 females, 4 males per
group) were dosed daily by nasogastric intubation with ivermectin in
sesame oil at dose levels of 0.3, 0.6, and 1.2 mg/kg body weight.
These dose levels were chosen to provide an appropriate 6-fold safety
margin relative to the human clinical dose, and based on the acute
toxicity in rhesus monkeys. All animals were treated for at least 14
days and then sacrificed on days 15, 16 or (one animal) 17. No drug-
related effects (physical signs, body weight, ocular lesions,
haematology, serum biochemical parameters, or necropsy findings) were
noted in any of the treated animals (Merck & Co., Inc., 1986a).
To assess the potential significance of neonatal exposure to
ivermectin, a study in neonatal rhesus monkeys (7 to 13 days old; 400
to 600 g body weight) was conducted. The animals (3 females, 5 males
per group) received ivermectin as a solution in sesame oil once daily
via nasogastric intubation at dose levels of either 0.04 or 0.1 mg/kg
body weight for 14 days. A control group of the same size received
the vehicle. Approximately four hours post-dose, animals were
examined for mydriasis and pupillary light response, and for adverse
reactions. The results of the examinations (physical, ophthalmic,
haematologic, serum biochemical examination, body weight, and
necropsy) indicated no treatment-related effects (Merck & Co., Inc.,
1986b).
2.2.3 Long-term/carcinogenicity studies
2.2.3.1 Mice
No specific study of ivermectin per se beyond 14 weeks has
been reported. Carcinogenicity was studied in mice (Crl:Cd-1(ICR)BR)
and rats (Crl:CD(SD)BR) with abamectin, a close chemical analog of
ivermectin differing only by being unsaturated between the carbon
atoms at positions C22 and C23.
In a 94 week dietary carcinogenicity study in Crl:CD-1 mice
abamectin was given at doses of 2, 4, or 8 mg/kg b.w./day. Seventy-
four mice of each sex were assigned to each group and to control
groups I and II. At the start of the study the males weighed 15.5-
33.2 grams and the females weighed 15.8-22.1 grams. Twelve
mice/sex/group were killed for bleeding at weeks 25 or 26 and 52.
The examinations included: daily observation; weekly detailed
examination including palpation for masses, body weight, and food
consumption; eye examination (pretest, weeks 51 or 53 and 91, control
and high-dose group only); haematology and serum biochemistry (weeks
25 or 26 and 52; in moribund mice after week 69; in all surviving mice
at scheduled sacrifice); and complete necropsy (gross necropsy on
animals killed for bleeding; histopathology on all mice assigned to
the carcinogenic segment of the study).
Treatment-related tremors were seen among females in all dosage
groups. Seven females of the 8 mg/kg b.w./day group and 3 females of
the 4 mg/kg b.w./day group died the day after the beginning of the
treatment for unexplained reasons (dietary concentrations were checked
and found correct). The females were killed and discarded. The males
were continued in the study. About a month later a new group of
females without adverse signs was started. Treatment-related tremors
were seen in drug weeks 89 and 91 in two females of the high-dose
group.
Increased mortality was seen among the high-dose males but not
the females (common causes: amyloidosis or lymphoma). Dosing of this
group was stopped in week 90 when there was 40% survival. All other
mice were treated until sacrifice in week 94. Treatment-related
decreases in body weight gain (males 7%; females 21%) were seen in the
high-dose groups. A dose-related increase in feed consumption and
decrease in feed efficiency (20%) was seen among the females of the
high-dose group. There were neither treatment-related ophthalmic
changes nor haematological nor serum biochemical effects. No
treatment-related changes in organ weights or gross lesions were seen
at necropsy. There were no neoplastic or non-neoplastic changes seen
in any tissue at necropsy examinations. Abamectin was not
carcinogenic to mice when given in the diet for 94 weeks at the above
dosage levels (Merck & Co., Inc., 1983).
2.2.3.2 Rats
Abamectin was tested in Crl:Cd(SD)BR rats (size of treatment
groups and controls I and II: 65 males [115-191 grams] and 65 females
[93-154 grams]) in a 105 week dietary study at doses of 0.75, 15, or
2.0 mg/kg b.w./day. Fifteen males and 15 females per group were
randomly selected at the start of the study for interim necropsy. The
examinations included: daily observation; weekly detailed examination
including palpation for masses, body weight, and food consumption; eye
examination (pretest and about every six months [controls and high-
dose group only]); haematology, serum biochemistry and urine analysis
(10 rats/sex/group in weeks 12, 25, 38, and 51 [from animals selected
for interim necropsy]), in week 78 (10 replacement rats from the
carcinogenic segment), in week 105 (sacrifice; haematology and serum
biochemistry) and in moribund animals (begining in week 89); complete
necropsy (all rats that died or were sacrificed before scheduled
termination, all rats sacrificed at scheduled time); and
histopathology (including histological examination of all gross
lesions).
Treatment-related increases in body weight gain were seen during
the first year on study (males and females of all dosage groups); by
the end of the study, body weight gain was statistically significant
only in the males. Tremors occurred in some rats receiving the
highest dose and one rat of the mid dose group which had exceedingly
high feed consumption. There were no gross or microscopic changes in
the nervous or muscular systems of rats which died with tremors.
There was no treatment-related increase in deaths. There were no
treatment-related changes in ophthalmic abnormalities, nor treatment-
related effects seen in haematology, serum biochemistry and
urinalysis. No treatment-related changes in organ weights or gross
lesions were seen at necropsy. There was no statistically significant
increase in tumour incidence, and no non-neoplastic changes were seen
in any tissue at necropsy examinations. Abamectin was not
carcinogenic to rats in this study (Merck & Co., Inc., 1982e).
2.2.4 Reproduction studies
2.2.4.1 Rats
Ivermectin was administered orally once daily to three groups of
15 female rats at dose levels of 0.4, 0.8, and 1.6 mg/kg body weight
from 15 days prior to mating until 20 days post-partum. Two vehicle
control groups received sesame oil in the same dosing regimen as the
treated animals.
There was no mortality or clinical evidence of toxicity in the
females. Average body weight was significantly increased among
females at 0.8 and 1.6 mg/kg b.w./day during the prebreeding period
and at all dose levels during gestation.
Ivermectin had no effect on mating, reproductive status, average
length of gestation or post implantation survival rate. Statistically
significant treatment-related increases in mortality among pups in the
1.6 mg/kg b.w./day group were observed on day 1 and from days 7-14
post-partum. Prior to death, several pups were observed to be
hypothermic and to have no externally observable milk in the
epigastric region. Throughout the lactation period, average pup
weights were slightly higher than controls in the 0.4 mg/kg b.w./day
group and significantly higher in the two other dose-level groups.
Development (eye opening, ear opening, incisor eruption, and hair
growth) was also slightly accelerated (Merck & Co., Inc., 1979a,b).
A series of three multigeneration studies was initiated in rats,
the first two of which were halted prior to scheduled termination
because neonatal toxicity was apparent at all dose levels tested.
Dose rates of 0.4, 1.2, and 3.6 mg/kg b.w./day were used in the first
study. It was necessary, however, to terminate this study before
mating of the F1b-generation because it became apparent from toxic
symptoms observed in the F1a-, F1b-, and F2a-generations that a NOEL
could not be derived from this study.
Effects on the F0-generation were a significant increase in the
average length of gestation and a significantly decreased maternal
weight gain during lactation in females in the 3.6 mg/kg b.w./day
group. Following the production of the F1b-litter, the average
maternal weight gain during lactation was significantly decreased
compared to that of the control.
Effects on the F1a-generation were a high (92%) mortality during
the lactation period of F1a-offspring in the 3.6 mg/kg b.w./day group
with the majority dying between days 5 and 10 post-partum. The most
common clinical sign of toxicity in pups that died was an absence of
milk in the stomach one to several days prior to death. Average pup
weights on day 1 post-partum and subsequent weight gain in surviving
pups were also significantly reduced in this group. During postnatal
development there was a significant decrease in the time to occurrence
of incisor eruption in the 3.6 mg/kg b.w./day group, an effect which
was believed to be secondary to a lower body weight in these
offspring.
Effects on the F1b-generation were evidence of treatment-related
toxicity among F1b-offspring in both remaining dose-level groups (1.2
and 0.4 mg/kg b.w./day). An increased pup mortality occurred between
days 2 and 7 post-partum. Slight decreases in average live pup weight
per litter were also observed during the lactation period. The time
to occurrence of the auditory startle reflex was delayed and earlier
incisor eruption was also observed compared to the corresponding
control. At 1.2 mg/kg b.w./day, vaginal opening was significantly
delayed and there was a nonsignificant but treatment-related delay in
testes descent. It is likely that these effects reflected slightly
lower average body weights. During the lactation period females in
the 1.2 mg/kg b.w./day group showed a significant decrease in average
maternal body weight gain.
Effects on the F2a-generation were significant increases in pup
mortality between days 2 and 7 post-partum in both dosage groups. The
average live pup weight per litter was decreased (statistically
significant only in the 1.2 mg/kg b.w./day group) on days 7, 14, and
21 post-partum. During postnatal development, there were significant
delays in the appearance of the righting reflex and the auditory
startle reflex and significantly earlier incisor eruptions in the 1.2
mg/kg b.w./day group (Merck & Co., Inc., 1980e).
A second multigeneration study was initiated at a dose of 2.0
mg/kg b.w./day in order to provide clear evidence of toxicity while
allowing sufficient surviving offspring to permit continuous dosing
throughout the production of two litters in each of three generations.
This study was terminated prior to the production of the F1b-litter
when it became apparent that there was treatment-related neonatal
toxicity present in the above concurrent multigeneration study at dose
levels 1.2 and 0.4 mg/kg b.w./day (Merck & Co., Inc., 1981e).
In a final multi-generation study the following dose groups were
included: 0.05, 0.1, 0.2, and 0.4 mg/kg b.w./day. A vehicle control
group received sesame oil daily in the same volume as drug-treated
rats. The animals were 28 days old at the onset of the daily
treatment and were mated 71 days later. Exposure was continued for
the entire life-span.
The F1a-litter was sacrificed on day 21 post-partum.
Approximately three weeks later the F0-rats were mated again to
produce the F1b-litter. On day 21 post partum of the F1b-offspring,
the F0-generation was sacrificed. After 71 days of treatment, the
F1b-rats were mated to produce the F2a-offspring which were also
sacrificed on day 21 post-partum. Approximately three weeks later the
F1b-rats were again mated to produce the F2b-offspring. Twenty-one
days post partum of this offspring, the F1b-generation was sacrificed.
After 71 days of drug treatment, F2b-rats were mated to produce the
F3a-offspring which were sacrificed on day 21 postpartum.
Approximately three weeks later the F2b-rats were again mated to
produce the F3b-litter. The parents were sacrificed after weaning of
the F3b-litter. Twenty males and 20 females from each F3b-offspring
group were randomly selected for necropsy at 28 to 43 days of age.
There was no treatment-related mortality or physical signs of
toxicity among parents or offspring in any dosage group throughout the
production of two litters in each of the F0-, F1-, and F2-
generations. Ivermectin had no treatment-related effects on the
reproductive performance of male or female rats in any dosage group.
Treatment-related effects on body weight gain were limited to a
slight but statistically significant decrease during the postweaning
period in mean body weight gain among F1b-females in the 0.4 mg/kg
b.w./day group and among F2b-males from the 0.2 and 0.4 mg/kg b.w./day
groups. External, visceral, and skeletal examination of both the F3a-
and F3b-offspring revealed no evidence of teratogenicity. Doses of
less than or equal to 0.2 mg/kg b.w./day had no adverse effects on
parents or progeny (Merck & Co., Inc., 1980e, 1981e).
2.2.4.2 Target animal species
No adverse effects on reproduction have been observed in target
animal species (Campbell & Benz, 1984; Egerton et al., 1984;
Schroder et al., 1986).
2.2.5 Special studies on embryotoxicity
Ivermectin has been tested in the mouse, the rat, the rabbit, and
the dog. Additionally, the effects of H2B1a and H2B1b have been
tested separately in mice.
2.2.5.1 Mice
22,23-Dihydroavermectin-B1a: In a teratogenic study groups of
20 pregnant CF1-mice each received H2B1a at dosage levels of 0.2,
0.4, 0.8, or 1.6 mg/kg b.w./day from days 6 through 15 of gestation
once daily by gavage as a solution in sesame oil. An additional
control group received the vehicle.
There were treatment-related maternal deaths. Whole body tremors
and coma developed in 2 mice after the first dose at 1.6 mg/kg
b.w./day. Dosing was suspended, but the coma persisted. One of these
mice was found dead on day 8; the second mouse was sacrificed while
aborting on day 9. At 0.8 mg/kg b.w./day, two mice developed tremor
and coma after the second dose. One of these mice died on day 9; the
second mouse was sacrificed on day 11 while aborting. At 0.4 mg/kg
b.w./day two mice developed slight to moderate tremors after the
second dose which became more pronounced after the third dose. Dosing
was then suspended. One mouse was sacrificed while aborting. The
second became comatose on day 9 and was sacrificed moribund on day 11.
Average maternal weight gain and reproductive status of surviving mice
was unaffected.
The average fetal weight per litter was unaffected by treatment
at any dosage level. There was a teratogenic effect as evidenced by
cleft palate (10 fetuses from five litters at 1.6 mg/kg b.w./day).
22,23-Dihydroavermectin-B1b: H2B1b was administered orally as
a solution in sesame oil by metal catheter to three groups of pregnant
CF1-mice from days 6 to 15 of gestation at dosage levels of 0.4, 0.8,
or 1.6 mg/kg bw/day. A control group of 35 mice received the vehicle.
At 1.6 mg/kg b.w./day one mouse was found comatose 24 hours after
the first dose, did not recover and was sacrificed 48 hours later. At
0.8 mg/kg b.w./day one mouse became prostrate and hypothermic after
five doses, and was sacrificed on day 11 of gestation. No other signs
of toxicity were observed at any other dosage level. The average
fetal weight was unaffected by treatment. Teratogenicity was
evidenced by a dose-related increased incidence of cleft palate (ten
fetuses from four litters at 1.6 mg/kg b.w./day; four fetuses from
four litters at 0.8 mg/kg b.w./day) (Merck & Co., Inc., 1979a).
Ivermectin: Groups of 25 mated female CF1-mice were
administered ivermectin orally as a solution in sesame oil at dose
levels of 0.1, 0.2, 0.4, or 0.8 mg/kg b.w./day from days 6 through 15
of gestation. The 0.4 mg/kg body weight day group had only 24 mice
because one mistakenly assigned male had to be discarded. A control
group of 25 mice received the vehicle.
There was treatment-related mortality in each of the three
highest dose level groups (0.8 mg/kg b.w./day: 3 females sacrificed
moribund after 14 doses; 0.4 mg/kg b.w./day: 1 female found dead after
the 3rd dose, two others sacrificed in poor physical condition after
1 and 8 doses, respectively; 0.2 mg/kg b.w./day: 1 female sacrificed
moribund after 4 doses). Physical signs were confined to those mice
which died or were sacrificed. Neither the reproductive status of
females (as measured by the number of implants, resorptions, and live
and dead fetuses per litter) nor average maternal body weight was
influenced by the treatment. Teratogenicity was evidenced by an
increased incidence of cleft palate (3 fetuses from 3 litters at 0.8
mg/kg b.w./day; 5 fetuses from 4 litter at 0.4 mg/kg b.w./day). There
was no evidence of a teratogenic effect at 0.1 or 0.2 mg/kg b.w./day
(Merck & Co., Inc., 1980g).
2.2.5.2 Rats
A summary of a teratogenic study in rats was available.
Ivermectin was administered as a solution in sesame oil to groups of
25 mated CRCD rats from days 6 through 17 of gestation at dose levels
of 2.5, 5, or 10 mg/kg b.w./day. A control group of the same size
received the vehicle. In the 10 mg/kg b.w./day group, three females
were sacrificed in poor physical condition after receiving 7-9 doses.
There were no treatment-related toxicity signs observed in the two
other groups. Teratogenicity, as evidenced by cleft palate in 4
fetuses from 2 litters was seen at 10.0 mg/kg b.w./day. No other
treatment-related external malformations were observed at the other
dosage levels. Visceral and skeletal examination produced no further
evidence of teratogenicity in any dosage group (Merck & Co., Inc.,
1980g).
2.2.5.3 Rabbits
A summary of a teratogenic study in rabbits was available.
Ivermectin was administered as a solution in sesame oil to groups of
16 pregnant rabbits from days 6 through 18 of gestation at dose levels
of 1.5, 3, or 6 mg/kg b.w./day. A control group of the same size
received the vehicle. In the 6 mg/kg b.w./day group, slight to marked
sedation was observed 24 hours after the 7th dose and persisted in
some females up to eight days after cessation of dosing. There was
also a significant decrease in mean maternal body weight during the
period of drug administration in this group. Six females of this
group aborted between days 22 and 27 of gestation, possibly due to
embryo-/feto-toxicity (increase in fetal deaths). No treatment-
related maternal effects were seen in the two other groups.
Teratogenicity was indicated by a dose-related increased incidence of
cleft palate and clubbed forepaws. At 3 mg/kg b.w./day 1 fetus had
cleft palate and 5 fetuses from 1 litter had clubbed forepaws. At 6
mg/kg b.w./day, 8 fetuses from 3 litters had cleft palate and 6
fetuses from 3 litters had clubbed forepaws (Merck & Co., Inc.,
1980g).
2.2.5.4 Dogs
An oral teratogenic study in beagle dogs was conducted. The
mated bitches weighed 8.2-17.1 kg at initiation of the treatment.
Seventeen mated bitches received 0.5 mg/kg body weight of ivermectin
in sesame oil on days 5, 15, 25, and 35 of gestation. A second group
of 19 mated bitches received the same dose on days 10, 20, 30, and 40
of gestation. A third group of 17 mated bitches served as the control
and received vehicle on days 5, 10, 15, 20, 25, 30, 35, and 40 of
gestation. On day 48 of gestation the females were hysterectomized.
At hysterectomy it was determined that 14, 15, and 12 bitches were
pregnant in groups 1, 2 and the control, respectively. There was no
maternal mortality and there were no maternal signs of toxicity in the
course of the study. There was no apparent effect on the average
fetal weight per litter. There was no evidence of a teratogenic
effect at external, visceral, and skeletal examination (Merck & Co.,
Inc., 1981f).
Summary of teratogenicity studies
Table 5 summarizes the results of the above teratogenicity
studies.
2.2.6 Special study on cross-fostering
Results of the initial multigeneration study in rats (Section
2.2.4.1) suggested that at doses of 0.4 and 1.2 mg/kg b.w./day the
F2a-progeny may have been more sensitive to the toxic effects of
ivermectin than the F1a- or F1b-progeny. A cross-fostering study was
conducted in order to determine whether this was due to biological
variation or to a real increase in sensitivity following prenatal,
postnatal or a combination of pre- and postnatal exposure to the drug.
One group of 40 female Charles River CD rats, approximately 8 weeks
old and weighing 169-256 grams, was administered 2.4 mg/kg b.w./day of
ivermectin in sesame oil for 61 days. A vehicle group of the same
size was administered sesame oil according to the same regimen. They
were mated with untreated males. On day 1 post-partum litter sizes
were standardized to 4 males and 4 females by random selection.
Litters were then cross-fostered to one of the following groups:
group treatment of prenatal exposure number of
F0-dams of F1-litters litters/group
1 + + 12
2 + - 12
3 - - 15
4 - + 12
Table 5: Teratogenicity in laboratory animals (oral administration)
Species Dams/ Dose Maternal toxicity Teratogenicity
(strain group [mg/kg
or breed) bw/day]
a) 22,23-dihydro-avermectin-B1a:
Mouse 20 0.2 no observed effects no observed
effects
(CF-1) 0.4 2 sacrificed no observed
effects
0.8 2 sacrificed/dead no observed
effects
1.6 2 sacrificed/dead cleft palate
(10/151)
b) 22,23-dihydro-avermectin-B1b:
Mouse 20 0.4 no observed effects no observed
effects
(CF-1) 0.8 1 sacrificed cleft palate
4/243)
1.6 1 sacrificed cleft palate
(10/205)
Mouse 25 0.1 no observed effects no observed
effects
(CRCF) 0.2 1 sacrificed no observed
effects
0.4 3 sacrificed/dead cleft palate
(5/244)
0.8 3 sacrificed cleft palate
3/254)
Rat 25 2.5 no observed effects no observed
effects
(CRDC) 5.0 no observed effects no observed
effects
10.0 3 sacrificed cleft palate
(4/264)
Table 5 (contd)
Species Dams/ Dose Maternal toxicity Teratogenicity
(strain group [mg/kg
or breed) bw/day]
Rabbit 16 1.5 no observed effects no observed
effects
3.0 no observed effects reduced
litter weight
cleft palate
(1/136)
clubbed forepaw
(5/136)
6.0 sedation reduced litter
decreased body weight
weight increased fetal
aborts death
cleft palates
(1(7*)/50
(18*))
clubbed forepaw
(6/50(18*))
Dog 17 ** no observed effects no observed
effects
(beagle) 19 ** no observed effects no observed
effects
* Numbers in parenthesis: dead fetuses
** These animals were dosed with 0.5 mg/kg b.w. every ten days.
Offspring were examined for weight gain, clinical signs of
toxicity, and postnatal development (occurrence of surface righting
reflex and time of eye opening). Thirteen weeks post-partum offspring
from all four groups were selected for continuation on an acute oral
toxicity study. The average live pup weight per litter on days 7, 14,
and 21 post-partum among offspring fostered within the treated group
(group 1) was significantly decreased. Among control litters
cross-fostered to treated females (group 2) there was a significant
decrease in average live pup weight on days 14 and 21 only. The
average live pup weight among litters prenatally exposed and
cross-fostered to control dams (group 4) was comparable to that
of litters cross-fostered within the control group. There was a
significant decrease in average postweaning body gain in groups 1,
2, and 4 compared to the control group 3. The magnitude of the
decrease was significantly greater in groups 1 and 2 than that of
group 4. There was a significant increase in pup mortality in litters
cross-fostered to F0-dams administered ivermectin (groups 1 and
2). Pup mortality among litters from treated F0-dams cross-fostered
to control dams (group 4) was comparable to that of pups
cross-fostered within the control group (group 3). There was no
significant variation in the time to occurrence of the righting reflex
among all four groups. The time to occurrence of eye opening
was significantly retarded in group 1 only. The results of this study
indicated that the neonatal toxicity of ivermectin in rats was
primarily a function of postnatal exposure. It also appeared that
in utero exposure did not increase the toxicity of subsequent
exposure via milk during the lactation period (Merck & Co., Inc.,
1980f).
2.2.7 Special studies on genotoxicity
2.2.7.1 Bacterial systems
Ivermectin and each component of ivermectin were tested in the
Ames test. Tests were done with and without rat liver metabolic
activation systems. None of the agents studied produced any noteworthy
increase in revertants to histidine prototrophy. The positive controls
(either 1-methyl-2(3a,4,5,6,7a-hexahydro-1,2-benzisoxazolol-3-yl)-5-
nitroimidazole or 2-amino-anthracene) produced significant increases
in revertants, particularly after metabolic activation with all the
tester strains used (Merck & Co., Inc., 1978b).
2.2.7.2 Mouse lymphoma cells
The ability of ivermectin to produce forward mutation at the
thymidine kinase locus (TK+/- to TK-/-) of mouse lymphoma cells (Fischer
L5178Y) was studied. Two cytotoxicity studies revealed that ivermectin
was detoxified in the presence of rat liver S-9 fraction. The mutagenic
assays were done by exposing cells to ivermectin for four hours; they
were then washed, fed and cultured for three days. TK-/- mutants were
detected by cloning the cells in selection medium containing
bromodeoxyuridine and plating diluted aliquots in nonselective medium.
Dose levels of 40, 60, and 80 µg/ml were used with and without S-9.
However the cells died in this initial study. A second assay was done
with 20, 40 and 60 µg/ml in the presence of S-9 only. Without S-9 the
dose levels were 5, 10, and 20 µg/ml. This study was replicated with
an identical protocol. The results of both tests were negative when
compared with appropriate negative controls. The positive control,
3-methylcholanthrene with S-9 produced significant increases in mutation
frequency (Merck & Co., Inc., 1980h).
2.2.7.3 Human Fibroblasts
Effects of ivermectin on unscheduled DNA synthesis were studied in
IMR-90 normal human embryonic lung fibroblasts in the presence and absence
of rat liver microsomal activation systems. The drug concentration ranged
from 10 to 1000 µg/ml. Ivermectin did not produce any significant
increase in background thymidine incorporation. In contrast it produced
an unexplained decrease at 10, 100, 300, and 1000 µg/ml but not at 30
µg/ml. The positive controls, methylmethane sulfonate and aflatoxin B-1,
both produced significant increases in UDS (Merck & Co., Inc., 1980i).
2.2.7.4 Summary of studies on genotoxicity
Table 6 summarizes the genotoxicity studies that have been performed
on ivermectin and its components.
2.3 Observations in humans
One 15 year old male was accidentally injected with an unknown
quantity of IVOMECTM 1% in a needle fingerprick accident. His arm
later became paralysed, due to coincidental viral polyneuritis which was
probably unrelated to ivermectin. An adult female injected herself
accidentally with a small quantity (estimated to be 200 micrograms/kg
body weight) of IVOMECTM 1%. Twelve hours later she experienced
colicky pain with nausea, but recovered within 12 hours. A 16 month old
boy weighing about 15 kg accidentally drank an estimated 10-13 ml of
IVOMECTM 1%. Mydriasis was noted in one pupil, along with vomiting,
pallor, 35°C body temperature, tachycardia, somnolence, and variable
blood pressure. The next morning urticaria occurred. He was normal
after three days. Therapy in hospital included calcium-gluconate,
caffeine, and an antihistamine. A woman, 8 months pregnant, sprayed
EQVALANTM into her eye. The eye was rinsed. Stinging at the
application side was the only adverse effect described (Merck & Co.,
Inc., 1988a).
2.3.1 Clinical use of ivermectin
Ivermectin was first administered to humans in 1981. Initial
studies were performed in Senegalese patients with onchocerciasis (Aziz
et al., 1982, Diallo et al., 1984). Since the first clinical
experience numerous multiclinic double-blind studies have been conducted
in endemic areas of onchocerciasis in different countries (Lariviere
et al., 1985; Awadzie et al., 1986; Diallo et al., 1986;
Dadzie et al., 1987; Albiez et al., 1988; Vingtain et al., 1988).
In 1987 MECTIZANTM (ivermectin) of Merck, Sharp and Dohme has been
approved in France for the treatment of onchocerciasis. The Onchocerciasis
Control Programme (OCP) of WHO has elaborated for 1989 and 1990 a dual
strategy of vector control and mass population drug treatment with
MECTIZANTM. It is expected that over 250,000 individuals in the OCP
area of Central Africa will be treated (Bradshaw, 1989). In addition to
its use for the treatment of onchocerciasis, ivermectin has been used
successfully in the treatment of Wuchereria bancrofti filariasis
(Kumaraswami et al., 1988), loiasis (Richard-Lenoble et al.,
1988), strongyloidiasis and enterobiasis (Naquira et al., 1989).
Table 6: Results of genotoxicity assays on ivermectin and its components
Test system Test object Concentration Results Reference
[ug/plate]
Ames test* TA1535, TA1537, 400, 1000, 2000 negative Merck & Co.,
(ivermectin) TA98, TA100 400, 1000, 2000 negative Inc. (1978b)
TA 1535, TA100 500, 1000, 2000 negative
Ames test* TA100 1000 negative Merck & Co.,
(H2B1a) TA1537, TA98, 100, 500, 1000 negative Inc. (1978b)
TA100,TA92 100, 500, 1000 negative
Ames test* TA1535, TA1537, 20, 200, 2000 negative Merck & Co.,
(H2B1b) TA98, TA100 20, 200, 2000 negative Inc. (1978b)
[ug/ml]
mouse lymphoma L5178Y 5, 10, 201) negative Merck & Co.,
cells L5178Y 20, 40, 602) negative Inc. (1980h)
[ug/ml]
unscheduled* human IMR-90 10, 30, 100 negative Merck & Co.,
DNA synthesis fibroblasts 300, 1000 negative Inc. (1980i)
(ivermectin)
*with and without S-9; 1)with S-9; 2)without S-9.
2.3.2 Studies in healthy subjects
Ivermectin was assessed for tolerability in several clinical
pharmacology studies. A total of 54 healthy individuals received
single oral doses of MECTIZANTM. Adverse experiences reported were
headaches in three persons who received a 6 mg dose. These
experiences were assessed as either probably or definitely not drug
related. Decreases in white blood cell counts occurred in one subject
after single oral doses of 12 mg as a solution and as tablets. Both
were assessed as being possibly drug related (Merck & Co., Inc.,
1988b).
2.3.3 Studies on tolerability in patients
Treatment of onchocerciasis with ivermectin requires a single
oral dose of 0.15 - 0.2 mg/kg body weight every 12 months. The
tolerability of MECTIZANTM has been closely examined in a number of
clinical trails. The observed side effects in some patients were
mostly mild and transient. These side effects can probably be
attributed to hypersensitivity reactions resulting from death of
microfilariae (the symptoms most frequently reported include pruritus,
arthralgia, dizziness, myalgia, fever, edema, lymphadenitis, nausea,
vomiting, diarrhoea, postural hypotension, tachycardia, weakness,
rash, and headache) (Merck & Co., Inc., 1988b).
3. COMMENTS
The Committee reviewed toxicological data from studies on
pharmacokinetics, biotransformation, acute and short-term toxicity,
effects on reproduction and development, genotoxicity, and
observations in humans. Aspects of the comparative toxicities of
ivermectin and abamectin were also considered.
Pharmacokinetic data were available from studies in mice, rats,
dogs, rhesus monkeys, and human volunteers. In mice, peak plasma
levels were reached approximately four hours after a single oral dose
of 51 mg per kg of body weight. The average plasma to brain ratio of
the concentrations of the drug was approximately 11:1. When
ivermectin was administered at 0.1 to 0.5 mg per kg of body weight per
day for 35 days, steady-state concentrations were observed from day
21. The concentration in the plasma and brain was proportional to the
dose.
In a study in rats in which H2B1a was administered orally at
0.06 to 0.75 mg per kg of body weight the dose and residue levels in
plasma and tissues were also shown to be well correlated. In a study
in which [3H] ivermectin was given orally at 0.3 mg per kg of body
weight the residue concentrations were highest in fat, followed by the
liver, kidney and muscle. The main route of excretion was via the
faeces.
In female rats aged eight weeks at initiation of dosing and
receiving daily oral doses of 2.5 mg/kg b.w. for 61 days and then
throughout mating, gestation, and until day 9 postpartum, steady-state
plasma concentrations were reached on day 10 of treatment. On day 1
postpartum, however, the plasma concentration was three to four times
that of the steady-state concentration, probably due to an increased
mobilization of body fat. When treatment was restricted to days 1 to
9 postpartum, the concentration of ivermectin in the plasma increased
gradually throughout the lactation period, and concentrations in milk
were at least three to four times the corresponding concentrations in
plasma. Under these conditions, the concentrations in the plasma of
the offspring increased dramatically between days 1 and 6 postpartum,
and on day 10 they were up to three times the concentrations found in
maternal plasma. On days 1 and 4 postpartum; residue levels in the
brain tissue from offspring were similar to their plasma
concentrations. The results of this study suggested that the transfer
of the drug via the milk was probably responsible for the increase in
neonatal mortality observed in multigeneration studies.
In a 36-day study in the beagle dog in which ivermectin was
administered orally at 0.5 and 2.0 mg/kg b.w. per day, the
concentrations of H2B1a in the plasma increased dramatically between
days 2 and 8 of treatment and reached steady state after approximately
three weeks. A fourfold increase in the dose resulted in an average
eightfold increase in plasma levels. In a comparative study with
abamectin and ivermectin in immature rhesus monkeys, higher plasma
concentrations were reached with ivermectin at all the dose levels
investigated (2, 8, and 24 mg per kg of body weight). For both
substances the concentrations in plasma were related to the dose, but
the relationship was not linear.
In a study with human volunteers in which various formulations of
ivermectin were administered orally, peak plasma concentrations were
reached within approximately four hours. Administration of [3H]
ivermectin showed that approximately 49% of the dose was eliminated in
the faeces within five days. In a clinical study in lactating women
treated with a single dose of ivermectin, a maximum concentration of
23 µg of the drug was found in milk on the day after treatment. This
level decreased to less than 0.1 µg/1 approximately one week after
treatment. Although plasma levels were not reported for this
particular study and the data determined from other studies were not
directly comparable, it appears that concentrations in human milk are
similar to or slightly less than in plasma.
Most of the studies on biotransformation were conducted using
[3H] ivermectin. When rat liver microsomes were incubated in vitro
with the individual components and a NADPH-regenerating system, more
than 70% of the radioactivity was associated with the corresponding
parent compound. The major polar metabolite was identified as the 24-
desmethyl-24-hydroxymethyl alcohol. The corresponding monosaccharide
was also detected. These findings correlated well with the results of
in vivo liver metabolism studies. In addition, a group of nonpolar
metabolites was detected in fat, which secreted polar products on
hydrolysis that were similar to the ivermectin metabolites present in
liver.
Acute toxicity studies were conducted in mice, rats, rabbits,
dogs, rhesus monkeys and a variety of target species (pigs, sheep,
cattle and horses). The typical signs of acute toxicity of ivermectin
were attributed to its effects on the central nervous system. These
were most severe in CF1 mice, which exhibited ataxia, bradypnoea and
tremors. Deaths occurred from approximately one hour to six days
after dosing. Ivermectin was more toxic in neonatal rats than in
young adult rats. This was believed to be due to postnatal completion
of the blood-brain barrier in this species.
In beagle dogs, mydriasis was the most sensitive indicator of
toxicity. More severe signs included ataxia and tremors. Deaths were
preceded by a comatose-like state. Approximately 30% of collies
tested were highly sensitive to ivermectin (as estimated from reports
from non-approved use of the drug). In immature rhesus monkeys no
tremors or convulsions occurred. The most sensitive indicator was
vomiting, which occurred in one of four monkeys given ivermectin at
2.0 mg per kg of body weight. The steep dose response curve in
rodents for the toxicity of ivermectin was not reproduced in monkeys.
Short-term studies were considered in rats, dogs, and monkeys.
In a 14-week study in rats in which ivermectin was administered orally
to pregnant dams, splenic enlargement and bone-marrow hyperplasia were
noted in the offspring of dams dosed at 0.8 and 1.6 mg per kg of body
weight per day. The NOEL was 0.4 mg per kg of body weight per day.
These changes did not occur in other species which received
ivermectin.
In a 14-week study in beagle dogs in which the compound was given
orally, mydriasis and loss of body weight were observed at 1.0 and 2.0
mg per kg of body weight per day (each group consisted of four females
and four males). Four dogs in the group receiving ivermectin at 2.0
mg per kg of body weight per day developed tremors, ataxia, anorexia,
and dehydration, and were killed prior to scheduled necropsy. No
other treatment-related effects were found. The NOEL was 0.5 mg per
kg of body weight per day.
In a two-week study in which ivermectin was administered orally
to neonatal monkeys at 0.04 and 0.1 mg per kg of body weight per day,
and to immature monkeys at 0.3, 0.6 and 1.2 mg per kg of body weight
per day, no drug treatment-related effects were observed.
Three multigeneration studies were initiated in rats, but the
first two were halted prior to scheduled termination because neonatal
toxicity was apparent at all dose-levels tested. In the final (three-
generation) study, the highest dose level was 0.4 mg per kg of body
weight per day. The results indicated that ivermectin was toxic to
neonatal rats at doses of 0.4 mg per kg of body weight per day or
above (administered to adult females) as evidenced by increased
neonatal mortality up to approximately ten days postpartum, and by the
decreased weights of surviving offspring. The results of a cross-
fostering study indicated that the neonatal toxicity was not related
to in utero exposure but to postnatal exposure via maternal milk.
The developmental toxicity of ivermectin has been investigated in
mice, rats, rabbits, and dogs. The results demonstrated that
teratogenic effects (cleft palates in mice, rats, and rabbits; clubbed
fore-paws without skeletal alterations in rabbits) were produced only
at dose levels similar to those causing severe toxic effects in
pregnant animals. The no-observed-effect level for teratogencity in
the most sensitive species and strain, the CF1 mouse, was 0.2 mg/kg
b.w./day, while for maternal toxicity it was 0.1 mg/kg b.w./day.
Ivermectin was negative in three in vitro assays for
genotoxicity. The Committee noted that no test of clastogenicity had
been performed.
There were no carcinogenicity studies available on ivermectin.
The Committee noted the very close structrual similarities of
ivermectin and abamectin. Extensive toxicological tests had been
conducted on both compounds by one particular manufacturer, using the
same strains of test animals over the same period of time. The
Committee, therefore, reviewed several aspects of comparative
toxicology of the two products. The compounds were indistinguishable
at the level of receptor binding. Clinical signs of the toxicity of
both compounds included mydriasis in dogs, vomiting in monkeys, and
tremors, convulsions, and coma at higher doses in most species. CF1
mice were most sensitive to the compounds. In general, ivermectin was
slightly less toxic than abamectin in laboratory animals (2-4-fold
higher threshold). In 14-week studies in rats in which ivermectin and
abamectin were administered orally at 0.4 mg per kg of body weight per
day, no adverse effects were observed. In a 14-week study with
ivermectin in dogs, mydriasis was seen at 1.0 mg per kg of body weight
per day and above, and tremors, ataxia and anorexia at 2.0 mg/kg of
body weight per day. In a 12-week study with abamectin in dogs,
mydriasis occurred at 1.0 mg per kg of body weight per day and above
and extreme weight loss at 2.0 mg per kg of body weight per day and
above. In multigeneration studies, toxicity in pups was the most
sensitive indicator, and occurred at 0.4 mg per kg of body weight per
day. The no-observed-effect levels for the formation of cleft palates
in CF1 mice were the same. Both compounds were negative in a number
of in vitro tests for genotoxicity. Abamectin was also negative in
in vivo tests, including clastogenicity. Carcinogenicity studies
with abamectin were negative at maximum tolerated doses in mice and
rats. The Committee therefore concluded that it was unnecessary to
request data from long-term toxicity and carcinogenicity studies on
ivermectin.
Ivermectin is widely used in humans for the treatment of
onchocerciasis at single doses of 0.2 mg per kg of body weight.
Tolerance to the compound has been assessed in healthy volunteers and
in patients; adverse effects are usually mild and transient. In
particular, no effects on the central nervous system were observed in
patients.
4. EVALUATION
The Committee concluded that the most relevant effect for the
safety evaluation of residues of ivermectin was its effect on the
mammalian nervous system. An ADI of 0-0.0002 mg per kg of body weight
was established based on a no-observed-effect level of 0.1 mg per kg
of body weight per day for maternal toxicity in CF1 mouse. A safety
factor of 500 was selected on the basis of the absence of neurological
effects in patients. This also provided a 1000-fold margin of safety
for the developmental toxicity of ivermectin.
5. REFERENCES
ABALIS, I.M., ELDEFRANI, M.E., & ELDEFRANI, A.T. (1986). Effects of
insecticides on GABA-induced chloride influx into rat brain microsacs.
J. Toxicol. Environ. Health, 18, 13-23.
ABLIEZ, E.J, NEWLAND, H.S., WHITE, A.T., KAISER, A., GREENE, B.M.,
TAYLOR, H.R., & BUETTNER, D.W. (1988). Chemotherapy of onchoceriasis
with high doses of diethylcarbamazine or a single dose of ivermectin:
microfilaria levels and side effects. Trop. Med. Parasitol., 39,
19-24.
AWADZI, K., DADZIE, K.Y., SCHULZ-KEY, H., GILLES, H.M, FULFORD, A.J.,
& AZIA, M.A. (1986). The chemotherapy of onchocerciasis. XI. A
double-blind comparative study of ivermectin, diethylcarbamazine and
placebo in human onchocerciasis in northern Ghana. Ann. Trop. Med.
Parasitol., 80, 433-442.
AZIZ, M.A., DIALLO, S., DIOP, I.M. LARIVIERE, M., & PORTA, M. (1982).
Efficacy and tolerance of ivermectin in human onchocerciasis.
Lancet, 2, 171-173.
BRADSHAW, H. (1989). Onchocerciasis and the Mectizan programme.
Parasitol. Today, 5, 63-64.
BURG, R.W., MILLER, B.M., BAKER, E.E., BIRNBAUM, J., CURRIE, S.A.,
HARTMAN, R., & KONG, Y.L. (1979). Avermectins, a new family of potent
antihelmintic agents: producing organism and fermentation.
Antimicrob. Agents Chemother., 15, 361-367.
CAMPBELL, W.C., FISHER, M.H., STAPLEY,E.O., ALBERS-SCHONBERG, G., &
JACOB, T.A., (1983). Ivermectin: a potent new antiparasitic agent.
Science, 221, 823-828.
CAMPBELL, W.C., & BENZ, G.W. (1984). Ivermectin: a review of
efficacy and safety. J. Vet. Phamacol. Ther., 23, 1134-1136.
CHABALA, J.C., MROZIK, H., TOLMAN, R.L., ESKOLA, P., LUSI, A.,
PETERSON, L.H., WOODS, M.F., & FISHER, M.H. (1980). Ivermectin, a new
broad-spectrum antiparasitic agent. J. Med. Chem., 23, 1934-1136.
CHIU, S.H.L., CARLIN; J.R., TAUB, R., SESTOKAS, E., ZWEIG, J.,
VANDENHEUVEL, W.J.A., & JACOB, T.A. (1988). Comparative metabolic
disposition of ivermectin in fat tissues of cattle, sheep and rats.
Drug Metab. and Dispos., 16, 728-736.
DADZIE, K.Y., BIRD, A.C., AWADIZ, K., SCHULZ-KEY, H., GILLES, H.M., &
AZIZ, M.A. (1987). Ocular findings in a double-blind study of
ivermectin versus diethylcarbamazine versus placebo in the treatment
of onchocerciasis. Br. J. Ophtalmol., 71, 78-85.
DIALLO, S., AZIZ, M.A., LARIVIERE, M., DIALLO, J.S., DIOP-MAR, I.,
N'DIR, O., BADIANE, S., PY, D., SCHULZ-KEY, H., & GAXOTTE, P. (1986).
A double-blind comparison of the efficacy and safety of invermectin
and diethylcarbamazine in a placebo controlled study of Senegalese
patients with onchocerciasis. Trans. Rog., Soc. Trop. Med. Hyg., 80,
927-934.
DIALLO, S., LARIVIERE, M., DIOP-MAR, I., N'DIR, O., N'DIAYE, R.,
BADIANE, S., PORTA, M., & AZIZ, M. (1984). Conduite au Senegal des
premiers etudes d'efficacite et de tolerance de l'ivermectine (MK 933)
dans l'onchocercose humaine. Bull. soc. Pathol. Exot. Filiales., 77,
196-205.
DI NETTA, J. (1989). List of registrations. Appendix III to
Campbell, W.C., (ed.) Ivermectin and Abamectin. Springer-Verlag, New
York pp. 344-366.
DREXLER, G. & SIEGHART, W. (1984). Properties of high affinity
binding site for 3H-Avermectin B1a. Eur. J. Pharmacol., 99, 269-
277.
EGERTON, J.R., SEWARD, R.L., & ROBIN, B. (1984). L'ivermectine: un
agent antiparasitaire pour les chevaux. Rec. Med. Vet., 6, 595-599.
ELDEFRANI, A.T.. & ELDEFRANI, M.E. (1987). Receptors for gamma-
aminobutyric acid and voltage-dependant chloride channels as targets
for drugs and toxicants. FASEB J., 1, 262-271.
FRITZ, L.C., WANG, C.C., & GORIO, A. (1979). Avermectin B1a
irreversibly blocks postsynaptic potentials at the lobster
neuromuscular junction by reducing muscle membrane resistance. Proc.
Natl. Acad. Sci. USA, 76, 2062-2066.
KASS, I.S., WANG, C.C., WALROND, J.P., & STRETTON, A.O.W. (1980).
Avermectin B1a, a paralyzing anthelmintic that effects interneurons
and inhibitory motoneurons in ascaris. Proc. Natl. Acad. Sci. USA.,
77/10, 6211-6215.
KUMARASWAMI, V., OTTESEN, E.A., VIJAYASEKARAN, V, DEVI, U.,
SWAMINATHAN, M., AZIZ, M.A., SARMA, G.R., PRABHAKAR, R, & TRIPATHY,
S.P. (1988). Ivermectin for the treatment of wuchereria bancrofti
filariasis. JAMA, 259, 3105-3153.
LANKAS, G.R, MINSKER, D.H., & ROBERTSON, R.T. (1989). Effects of
ivermectin on reproduction and neonatal toxicity in rats. Fd. Chem.
Toxic., 27, 523-529.
LARIVIERE, M., VINGTAIN, P., AZIZ, M., BEAUVAIS, B., WEIMANN,D.,
DEROUIN F., GINOUX, J., SCHULZ-KEY, H., GAXOTE, P., & BASSET, D.,
(1985). Double-blind study of ivermectin and diethylcarbamazine in
African onchcerciasis patients with ocular involvement. Lancet,
2, 147-177.
MERCK & CO., INC. (1978a). Fourteen week oral toxicity study in dogs.
TT #78-038-00. Unpublished study; submitted to WHO by Merck & Co.,
Inc.
MERCK & CO., INC. (1978b). MK-0933: microbial mutagen test with and
without rat liver enzyme activation. TT #77-8068. Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979a). MK-0933: Toxicological evaluation (April
30, 1979). Reports of unpublished studies; submitted to WHO by Merck
& Co., Inc.
MERCK & CO., INC. (1979b). MK-933: Summary of toxicity studies.
Summary of unpublished studies; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979c). Acute oral toxicity study in rats. TT
#78-3087. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979d). MK-933: Acute dermal toxicity studies in
rabbits and rats. Acute ocular toxicity studies in rabbits. Acute
oral toxicity in dogs. Unpublished studies; submitted to WHO by Merck
& Co., Inc.
MERCK & CO., INC. (1979e). Acute inhalation toxicity study in rats.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1979f). MK-933 (L-640,471-00W51): Acute oral
toxicity in dogs. TT #79-2869. Unpublished study; submitted to WHO
by Merck & Co., Inc.
MERCK & CO., INC. (1979g). MK-933/cattle/safety/ toxicity/clinical
pathology/N.O.T. 4480. Unpublished study; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1980a). MK-932: metabolism study in the rat. TT #
79-711-0. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980b). In vitro metabolism studies of
MK-0932/0933. Unpublished study; submitted to WHO by Merck & Co.,
Inc.
MERCK & CO., INC. (1980c). Tissue residues and radioactive balances
in rats dosed with 3H-labeled MK-0933 (OM-45). Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980d). L-640,497-OOH: Acute oral toxicity in
female mice. Unpublished study: submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980e). MK-933: Multigeneration studies in rats.
Unpublished studies; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980f). MK-933: Cross fostering study in rats. TT
#79-710-0. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980g). MK-0933: Teratogenic evaluation.
Unpublished report: submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1980h). MK-0933: Mouse lymphoma cytotoxicity
study. TT #79-8034. Unpublished study; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1980i). MK 933: Unscheduled DNA synthesis in human
IMR90-fibroblasts. TT #80-8205. Unpublished study; submitted to WHO
by Merck & Co., Inc.
MERCK & CO., INC. (1981a). MK-933 (L-640,471-OOW72): Acute oral
toxicity in dogs. TT #81-2500. Unpublished study; submitted to WHO by
Merck & Co., Inc.
MERCK & CO., INC. (1981b). MK-933 Ivermectin injectable micelle
solution: acute subcutaneous toxicity study in young dogs. TT
#81-025-0. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981c). MK-933: Acute toxicity in the ovine.
N.O.T. 7000. Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981d).MK-933/horses/safety/toxicity/clinical
pathology/N.O.T. 8292, protocol 554. Unpublished study; submitted to
WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981e). MK-933: Multigeneration study in rats.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981f). MK-933: oral teratogenic study in dogs.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1981g). Metabolism of ivermectin (MK-0933) in
cattle and rats. Unpublished studies; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1982a). MK-0933: Three-day study of plasma and
brain drug levels in mice. TT #82-088-0. Unpublished study; submited
to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1982b). MK-0933: Thirty-six day study of plasma
and brain levels in mice. TT #82-071-0. Unpublished study: submitted
to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1982c). MK-0933: Thirty-six day study of plasma
and brain levels in dogs. TT #82-071-0. Unpublished study; submitted
by Merck & Co., Inc.
MERCK & CO., INC. (1982d). MK-0933 / Swine / Safety / Toxicity
clinical pathology / Anatomic pathology. N.O.T. 9739. Unpublished
study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1982e). MK-936: One-hundred-five week dietary
carcinogenicity and toxicity study in rats with a fifty-three week
interim necropsy. Final report. TT #82-099-0. Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1983). MK936: Ninety-four week dietary
carcinogenicity and toxicity study in mice. TT #83-002,-0, -1, -2, -3.
Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1985). MK-0933 and MK-0936: oral toxicity and
plasma level study in monkeys. Unpublished study; submitted WHO by
Merck & Co., Inc.
MERCK & CO., INC. (1986a). MK-0933: 16-day oral toxicity study of MK-
0933 in immature rhesus monkeys. TT #85-9033. Unpublished study;
submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1986b). MK-0933: Fifteen-day toxicity study of
orally administered MK-0933 (ivermectin) in neonatal rhesus monkeys.
TT #869005, GLP-13. Unpublished study; submitted to WHO by Merck &
Co., Inc.
MERCK & CO., INC. (1987). The metabolism, distribution, and excretion
of 22,23-3H-MKO933 in rats dosed orally and percutaneously (Expt.
ADM-66). Unpublished study; submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC (1988a). Ivermectin: Poison Control Monograph.
Submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1988b). MECTIZANTM (IVERMECTIN, MSD). Product
Monograph 7-89 MCT 88-R-1152M. Submitted to WHO by Merck & Co., Inc.
MERCK & CO., INC. (1989). Identity of Ivermectin. Structural Formula.
Unpublished study; submitted to WHO by Merck & Co., Inc.
NAQUIRA, C., JIMENEZ, G., GUERRA, J.G., BERNAL, R., NALIN, D.R., NEU,
D., & AZIZ, M. (1989). Ivermectin for human strongyloidiasis and
other intestinal helminths. Am. J. Trop. Med. Hyg., 40, 304-309.
OLSEN, R.W, & SNOWMAN, A.M. (1985). Avermectin B,, modulation of
gamma aminobutyric acid/benzodiazepine receptor binding in mammalian
brain. J. Neurochem., 44/4, 1074-1082.
PAUL, S.M., SKOLNICK, P., & ZATZ, M. (1980). Avermectin B1a: An
irreversible activator of the GABA-Benzodiazepine-chloride-ionophore
receptor complex. Biochem. Biophys. Res. Communs., 96, 632-638.
PONG, S.S., DEHAVEN, R., & WANG, C.C. (1981). Stimulation of
benzo-diazepine binding to rat brain membranes and solubilized
receptor complex by avermectin B1a and GABA. Biochim. Biophys.
Acta., 646, 143-150.
PONG, S.S., DEHAVEN, R., & WANG, C.C. (1982). A comparative study of
avermectin B1a and other modulators of the gamma-aminobutyric acid
receptor-chloride ion channel complex. J. Neurosci, 2/7, 966-971.
PONG, S.S. & WANG, C.C., (1980). The specificity of high affinity
binding of avermectin B1a to mammalian brain membranes.
Neuropharmacology, 19, 311-312.
PONG, S.S., & WANG, C.C., (1982). Avermectin B1a modulation of gamma
aminobutyric acid receptors in rat brain membranes. J. Neurochem.,
38/2, 375-379.
PONG, S.S., WANG, C.C., & FRITZ, L.C., (1980). Studies on the
mechanism of action of avermectin B1a; stimulation of release of
gamma-aminobutyric acid from brain synaptosomes. J. Neurochem.,
34/2, 351-358.
PULLIAM, J.D., SEWARD, R.L., HENRY, R.T., & STEINBERG, S.A., (1985).
Investigation of ivermectin toxicity in collies. Vet. Med., 6, 33-
40.
RICHARD-LENOBLE, D., KOMBILA, M., RUPP, E.A., PAPPAYLIOU, E.S.,
GAXOTTE, P., NGUIRI, C., & AZIZ, M.A. (1988). Ivermectin in loiasis
and concomitant O. volvulus and M. perstans infections. Am.
J. Trop. Med. Hyg., 39, 480-483.
SCHOFIELD, P.R., DARLISON, M.G., FUJITA, N., BURT, D.R., STEPHENSON,
F.A., RODRIGUEZ, H., RHEE, L.M, RAMCHANDRAN, J., REALE, V., GLENCORSE,
T.A., SEEBURG, P.H., & BARNARD, E.A. (1987). Sequence and functional
expression of the GABAa receptor shows a ligand-gated receptor super-
family. Nature, 328, 221-227.
SCHRODER, J., SWAN, G.E., BARRICK, R.A., & PULLIAM, J.D., (1986).
Effect of ivermectin on the reproductive potential of breeding rams.
J. S. Afr. Vet. Assoc., 57/4, 211-213.
SUPAVILAI, P. & KAROBATH, M. (1981). In vitro modulation by
avermectin B1a of the GABA/benzodiazepine receptor complex of rat
cerebellum. J. Neurochem., 36/3, 798-803.
VINGTAIN, P., PICHARD, E., GINOUX, J., COULIBALY, S.M., BISSAN, Y.,
RANQUE, P., & THILAYE, B. (1988). Ivermectine et onchocercose
humaine. A propos d'une etude portant sur 234 onchocerquiens en
Republique du Mali. Bull. Soc. Pathol. Exot. Fialiales., 81, 260-
270.
WILLIAMS, M., & YARBROUGH, G.G. (1979). Enhancement of in vitro
binding and some of the pharmacological properties of diazepam by
novel anthelmintic agent avermectin B1a. Eur. J. Pharmacol., 56,
273-276.
WILLIAMS, M. & RISLEY, E.A. (1982). Interactions of avermectins with
3H-B-carboline-3-carboxylate-ethylester and 3H-diazepam binding
sites in rat brain cortical membrane. Eur. J. Pharmacol., 77, 307-
312.
WILLIAMS, M. & RISLEY, E.A., (1984). Ivermectin interactions with
benzodiazepine receptors in rat cortex and cerebellum in vitro.
J. Neurochem., 42/3, 745-753.