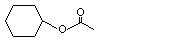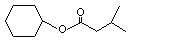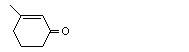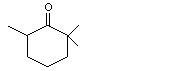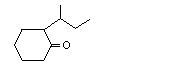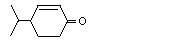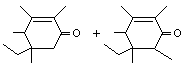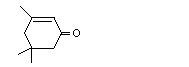First draft prepared by
Professor I.G. Sipes
Department of Pharmacology, College of Medicine, University of Arizona, Tucson, Arizona, USA
and Professor A.G. Renwick
Clinical Pharmacology Group, University of Southampton, Southampton, England
|
Application of the Procedure for the Safety Evaluation of Flavouring Agents |
|
Consideration of combined intake from use as flavouring agents |
The Committee evaluated a group of 25 flavouring agents consisting of alicyclic ketones, secondary alcohols and related esters (see Table 1). The evaluations were conducted according to the Procedure for the Safety Evaluation of Flavouring Agents (see Figure 1). None of these agents has been evaluated previously by the Committee.
Table 1. Summary of results of the safety evaluation of alicyclic ketones, secondary alcohols and related esters used as flavouring agentsa
|
Flavouring agent |
No. |
CAS No. and structure |
Step A3a,b |
Comments |
Conclusion based on current intake |
|
Structural class 1 |
|||||
|
Cyclohexyl acetate |
1093 |
622-45-7 |
No |
See note 1. |
No safety concern |
|
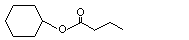
Cyclohexyl butyrate |
1094 |
1551-44-6 |
No |
See note 1. |
No safety concern |
|
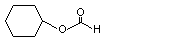
Cyclohexyl formate |
1095 |
4351-54-6 |
No |
See note 1. |
No safety concern |
|
Cyclohexyl isovalerate |
1096 |
7774-44-9 |
No |
See note 1. |
No safety concern |
|
Cyclohexyl propionate |
1097 |
6222-35-1 |
No |
See note 1. |
No safety concern |
|
3,3,5-Trimethyl cyclohexanol |
1099 |
116-02-9 |
No |
See note 2. |
No safety concern |
|
Structural class II |
|||||
|
cis- and trans-para-1(7),8-Menthadien-2-yl acetate |
1098 |
71660-03-2 |
No |
See note 1. |
No safety concern |
|
Cyclohexanone |
1100 |
108-94-1 |
No |
See note 2. |
No safety concern |
|
Cyclopentanone |
1101 |
120-92-3 |
No |
See note 2. |
No safety concern |
|
2-Methylcyclohexanone |
1102 |
583-60-8 |
No |
See note 2. |
No safety concern |
|
3-Methylcyclohexanone |
1103 |
591-24-2 |
No |
See note 2. |
No safety concern |
|
4-Methyl cyclohexanone |
1104 |
589-92-4 |
No |
See note 2. |
No safety concern |
|
1-Methyl-1-cyclopenten-3-one |
1105 |
2758-18-1 |
No |
See note 3. |
No safety concern |
|
Structural class II |
|||||
|
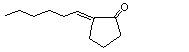
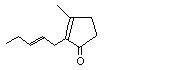
2-Hexylidene cyclopentanone |
1106 |
17373-89-6 |
No |
See notes 3 and 4. |
No safety concern |
|
3-Methyl-2-cyclohexen-1-one |
1107 |
1193-18-6 |
No |
See note 3. |
No safety concern |
|
2,2,6-Trimethylcyclohexanone |
1108 |
2408-37-9 |
No |
See note 2. |
No safety concern |
|
2-sec-Butylcyclohexanone |
1109 |
14765-30-1 |
No |
See note 4. |
No safety concern |
|
4-Isopropyl-2-cyclohexenone |
1110 |
500-02-7 |
No |
See note 3. |
No safety concern |
|
Tetramethyl ethyl cyclohexenone (mixture of isomers) |
1111 |
17369-60-7 |
No |
See note 3. |
No safety concern |
|
Isophorone |
1112 |
78-59-1 |
No |
See note 3. |
No safety concern |
|
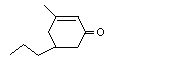
3-Methyl-5-propyl-2-cyclohexen-1-one |
1113 |
3720-16-9 |
No |
See note 3. |
No safety concern |
|
3-Methyl-2-(2-pentenyl)-2-cyclopenten-1-one |
1114 |
488-10-8 |
No |
See notes 3 and 4 . |
No safety concern |
|
Isojasmone |
1115 |
11050-62-7 |
No |
See notes 3 and 4. |
No safety concern |
|
(E)-2-(2-Octenyl) cyclopentanone |
1116 |
65737-52-2 |
No |
See note 4. |
No safety concern |
|
2-(3,7-Dimethyl-2,6-octadienyl) cyclo-pentanone |
1117 |
68133-79-9 |
No |
See note 4. |
No safety concern |
|
CAS: Chemical Abstracts Service; ND: No intake data reported |
|
|
a |
Step 2: All of the flavouring agents in this group were predicted to be metabolized to innocuous products. |
|
b |
The threshold for human intake is 1800 mg/person per day for class I and 540 mg/person per day for class II. All intake levels expressed in mg/person per day. The combined intake of flavouring agents in class I is 14 and 10 mg per person per day in Europe and the USA, respectively. The combined intake of flavouring agents in class II is 54 and 26 mg per person per day in Europe and the USA, respectively. |
|
Notes: |
|
|
1. |
Detoxicated by hydrolysis of ester and glucuronic acid conjugation of the resulting alicyclic alcohol and complete oxidation of the carboxylic acid |
|
2. |
Detoxicated by reduction of the ketone followed by glucuronic acid conjugation of the corresponding alcohol |
|
3. |
Detoxicated by reduction of the ketone functional group followed by glucuronic acid conjugation of the resulting alcohol and glutathione conjugation of the parent ketone |
|
4. |
Detoxicated by reduction of the ketone and alkyl side-chain oxidation and excretion |
Twelve of the 25 substances (Nos 1093, 1098, 1100–1104, 1107, 1108, 1110, 1112 and 1114) have been reported to occur naturally in foods. They have been detected in fruits, vegetables, cheese, meats, seafood, grains, alcoholic beverages, coffee and tea.
The total annual volume of production of the 25 alicyclic ketones, secondary alcohols and related esters in this group is approximately 520 kg in Europe (International Organization of the Flavor Industry, 1995) and 310 kg in the USA (Lucas et al., 1999). The reported annual production of only one agent, 3-methyl-2-(2-pentenyl)-2-cyclopenten-1-one (No. 1114), is greater than 100 kg (110 kg in Europe). The daily per capita intake of each of the agents in this group is < 15 µg in Europe and in the USA.
Esters (Nos 1093–1098) in this group are hydrolysed to their corresponding alcohols and carboxylic acids by carboxylesterases, which are found abundantly in hepatocytes. The resulting alicyclic secondary alcohols are conjugated with glucuronic acid and excreted mainly in the urine. Side-chain oxidation of methyl groups may also occur.
Alicyclic ketones (Nos 1100–1117) are reduced to the corresponding secondary alcohol and excreted primarily as glucuronic acid conjugates. If a double bond is present, the chemical may be reduced to the corresponding dihydro derivative. Reduction of the double-bond in metabolites excreted in the bile is probably associated with the action of gut microflora. Endocyclic double bonds (Nos 1105, 1107 and 1110–1115) are more prone to reduction than exocyclic double bonds (Nos 1098 and 1106). Alicyclic ketones containing an alkyl side-chain (Nos 1098, 1099 and 1102–1117) can not only follow reductive pathways but can undergo oxidation of the side-chain to form poly-oxygenated metabolites, which are excreted as the glucuronic acid or sulfate conjugates in the urine and, to a lesser extent, in the faeces. The nine chemicals that are alpha,beta-unsaturated ketones (Nos 1105–1107 and 1110–1116) are subject to glutathione conjugation, with subsequent elimination in the urine as mercapturic acids.
As the alpha,beta-unsaturated carbonyl group is a structural alert for toxicity, the Committee, at previous meetings, has devoted considerable attention to the safety of flavouring agents containing this reactive moiety. The Committee concluded at its fifty-seventh meeting (Annex 1, reference 154) that the presence of protective processes in cells provides adequate detoxication capacity at the low doses associated with use of such compounds as flavouring agents. With respect to alpha,beta-unsaturated ketones, these protective processes include reduction of the ketone to the corresponding alcohol (followed by conjugation of the alcohol with glucuronic acid) and conjugation with glutathione. These processes operate for the aliphatic ketones used as flavouring agents.
|
Step 1. |
In applying the Procedure for the Safety Evaluation of Flavouring Agents to this group of flavouring agents, the Committee assigned six (Nos 1093–1097 and 1099) of the 25 substances to structural class I (Cramer et al., 1978). These agents are either alicyclic secondary alcohols or are readily hydrolysed to secondary alcohols and simple short-chain carboxylic acids. They are simple aliphatic substances containing secondary hydroxyl groups, which have low toxicity. The remaining 19 compounds, which are monocyclic alkanones (Nos 1100–1117) or have a secondary alcohol attached to a vinyl group (No. 1098), were assigned to structural class II. |
|
Step 2. |
All the flavouring agents in this group were predicted to be metabolized to innocuous products. The evaluation of these agents therefore proceeded via the A (left-hand) side of the decision tree. |
|
Step A3. |
The estimated daily per capita intake of each of the six flavouring agents in structural class I and each of the 19 agents in structural class II is below the threshold for daily human intake of compounds in the respective structural class (i.e., 1800 µg per person for class I and 540 µg per person for class II). According to the Procedure, the safety of these 25 flavouring agents raises no concern when they are consumed at the currently estimated levels. |
Table 1 summarizes the evaluations of this group of acyclic ketones, secondary alcohols and related esters used as flavouring agents.
In the unlikely event that all the flavouring agents in structural class I or II were to be consumed concurrently on a daily basis, the estimated combined intake would not exceed the threshold for human intake of either class.
The Committee concluded that none of the flavouring agents in this group of alicyclic ketones, secondary alcohols and related esters would raise a safety concern at current estimated levels of intake. Other data on the toxicity of alicyclic ketones, secondary alcohols and related esters were consistent with the results of the safety evaluation.
This section summarizes the key data relevant to the safety evaluation of 25 alicyclic ketones, secondary alcohols and esters derived from secondary alcohols (see Table 1). The group includes both terpenoid and non-terpenoid substances and contains 18 saturated and unsaturated alicyclic ketones (Nos 1100–1117), one secondary alcohol (No. 1099) and six esters (Nos 1093–1098). Groups of structurally related menthol and carvone derivatives that are also alicyclic secondary alcohols and ketones were previously evaluated for use as flavouring agents (Annex 1, reference 138).
The flavouring agents in this group contain a ketone functional group or are esters of a related secondary alcohol. Before absorption, the esters are readily hydrolysed to an alicyclic secondary alcohol. Once they have been absorbed, secondary alcohols and ketones are readily interconverted in vivo (McMahon, 1982). Therefore, all the substances in this group are expected to participate in common routes of absorption, metabolism, distribution and excretion and show similar toxicological profiles.
Quantitative data on natural occurrence and consumption ratios have been reported for two substances, isophorone (No. 1112) and 3-methyl-2-(2-pentenyl)-2-cyclopenteny-1-one (No. 1114). These show that intake is predominantly from the consumption of traditional foods (i.e., consumption ratio > 1) (Stofberg & Kirschman, 1985; Stofberg & Grundschober, 1987; see Table 2).
Table 2. Annual usage volume of alicyclic ketones, secondary alcohols and related esters used as flavouring agents in Europe and the USA
|
Substance (No.) |
Most recent annual volume (kg)a |
Intake (‘eaters only’)b |
Annual volume in naturally occurring foods (kg)c |
Consumption ratiod |
|
|
µg/day |
µg/kg bw |
||||
|
Cyclohexyl acetate (1093) |
|||||
|
Europe |
99 |
14 |
0.24 |
||
|
USA |
77 |
10 |
0.17 |
+ |
NA |
|
Cyclohexyl butyrate (1094) |
|||||
|
Europe |
NR |
NA |
NA |
||
|
USA |
0.90 |
0.12 |
0.002 |
– |
NA |
|
Cyclohexyl formate (1095) |
|||||
|
Europe |
0.10 |
0.01 |
0.0002 |
||
|
USA |
1.4 |
0.18 |
0.003 |
– |
NA |
|
Cyclohexyl isovalerate (1096) |
|||||
|
Europe |
2.3 |
0.33 |
0.01 |
||
|
USA |
0.40 |
0.05 |
0.001 |
– |
NA |
|
Cyclohexyl propionate (1097) |
|||||
|
Europe |
0.10 |
0.01 |
0.0002 |
||
|
USA |
0.40 |
0.05 |
0.001 |
– |
NA |
|
cis- and trans-para-1(7),8-Menthadien-2-yl acetate (1098) |
|||||
|
Europe |
NR |
NA |
NA |
||
|
USA |
4.5 |
0.59 |
0.01 |
+ |
NA |
|
3,5,5-Trimethylcyclohexanol (1099) |
|||||
|
Europe |
1.0 |
0.14 |
0.002 |
||
|
USA |
1.0 |
0.13 |
0.002 |
– |
NA |
|
Cyclohexanone (1100) |
|||||
|
Europe |
1.0 |
0.14 |
0.002 |
||
|
USA |
1.0 |
0.13 |
0.002 |
+ |
NA |
|
Cyclopentanone (1101) |
|||||
|
Europe |
0.15 |
0.02 |
0.0003 |
||
|
USA |
0.15 |
0.02 |
0.0003 |
+ |
NA |
|
2-Methylcyclohexanone (1102)e |
|||||
|
Europe |
1.0 |
0.14 |
0.002 |
||
|
USA |
1.0 |
0.13 |
0.002 |
+ |
NA |
|
3-Methylcyclohexanone (1103)e |
|||||
|
Europe |
1.0 |
0.14 |
0.002 |
||
|
USA |
1.0 |
0.13 |
0.002 |
+ |
NA |
|
4-Methylcylcohexanone (1104)e |
|||||
|
Europe |
1.0 |
0.14 |
0.002 |
||
|
USA |
1.0 |
0.13 |
0.002 |
+ |
NA |
|
1-Methyl-1-cyclopenten-3-one (1105) |
|||||
|
Europe |
0.50 |
0.07 |
0.001 |
||
|
USA |
NR |
NA |
NA |
– |
NA |
|
2-Hexylidenecyclopentanone (1106) |
|||||
|
Europe |
2.0 |
0.29 |
0.005 |
||
|
USA |
0.05 |
0.01 |
0.0002 |
– |
NA |
|
3-Methyl-2-cyclohexen-1-one (1107) |
|||||
|
Europe |
0.10 |
0.01 |
0.0002 |
||
|
USA |
0.90 |
0.12 |
0.002 |
+ |
NA |
|
2,2,6-Trimethylcyclohexanone (1108) |
|||||
|
Europe |
17 |
2.4 |
0.04 |
||
|
USA |
0.30 |
0.04 |
0.001 |
+ |
NA |
|
2-sec-Butylcyclohexanone (1109) |
|||||
|
Europe |
42 |
6.0 |
0.10 |
||
|
USA |
NR |
NA |
NA |
– |
NA |
|
4-Isopropyl-2-cyclohexenone (1110)e |
|||||
|
Europe |
0.01 |
0.001 |
0.00002 |
||
|
USA |
0.01 |
0.001 |
0.00002 |
+ |
NA |
|
Tetramethylethylcyclohexenone (mixture of isomers) (1111) |
|||||
|
Europe |
64 |
9.1 |
0.15 |
||
|
USA |
1.4 |
0.18 |
0.003 |
– |
NA |
|
Isophorone (1112) |
|||||
|
Europe |
38 |
5.4 |
0.09 |
||
|
USA |
0.90 |
0.12 |
0.002 |
30 |
34 |
|
3-Methyl-5-propyl-2-cyclohexen-1-one (1113) |
|||||
|
Europe |
0.80 |
0.11 |
0.002 |
||
|
USA |
31 |
4.1 |
0.07 |
– |
NA |
|
3-Methyl-2-(2-pentenyl)-2-cyclopenten-1-one (1114) |
|||||
|
Europe |
110 |
16 |
0.26 |
||
|
USA |
54 |
7.2 |
0.12 |
970 |
18 |
|
Isojasmone (1115) |
|||||
|
Europe |
3.0 |
0.43 |
0.01 |
||
|
USA |
0.05 |
0.01 |
0.0002 |
– |
NA |
|
(E)-2-(2-Octenyl)cyclopentanone (1116)e |
|||||
|
Europe |
50 |
7.1 |
0.12 |
||
|
USA |
50 |
6.6 |
0.11 |
– |
NA |
|
2-(3,7-Dimethyl-2,6-octadienyl) cyclopentanone (1117)e |
|||||
|
Europe |
50 |
7.1 |
0.12 |
||
|
USA |
50 |
6.6 |
0.11 |
– |
NA |
|
Total |
|||||
|
Europe |
480 |
||||
|
USA |
280 |
||||
|
NR, no data reported; NA, not applicable; +, reported to occur naturally in foods (Maarse et al., 1999) but no quantitative data available; –, not reported to occur naturally in foods |
|
|
a |
From International Organization of the Flavor Industry (1995) and Lucas et al. (1999) |
|
b |
Intake (µg/person per day) was calculated as follows: [(annual volume, kg) × (1 × 109 µg/kg)/(population × survey correction factor × 365 days)], where population (10%, ‘eaters only’) = 32 × 106 for Europe and 26 × 106 for the USA. The correction factor = 0.6 for Europe and 0.8 for the USA, representing the assumption that only 60% and 80% of the annual production volume of the flavour, resepctively, was reported in the poundage surveys. Intake (µg/kg bw per day) calculated as follows: [(µg/person per day)/body weight], where body weight = 60 kg. Slight variations may occur from rounding. |
|
c |
Quantitative data from Stofberg & Grundschober (1987) |
|
d |
Calculated as follows: (annual consumption in food, kg)/(most recently reported volume as a flavouring agent, kg) |
|
e |
Anticipated annual volume in the USA as reported by the Flavor and Extract Manufacturers Association of the United States |
The unsubstituted monocyclic esters (e.g., cyclohexyl acetate, No. 1093) hydrolyse rapidly to cyclohexanol and the component aliphatic carboxylic acids by classes of enzymes recognized as carboxylesterases (Ford & Moran, 1978; Heymann, 1980; White et al., 1990), the most important of which are the beta-esterases. These enzymes occur in most mammalian tissues (Heymann, 1980; Anders, 1989) but predominate in hepatocytes (Heymann, 1980).
cis and trans-para-1(7),8-Menthadien-2-yl acetate (No. 1098) was rapidly hydrolysed in vitro in the presence of a rat liver homogenate (Salzer, 1998). Incubation of the ester resulted in 92% hydrolysis after 15 min and 100% after 60 min (Salzer, 1998). The structurally related (–)-menthol carbonate esters, (–)-menthol ethylene glycol carbonate and (–)-menthol propylene glycol carbonate, were almost completely hydrolysed after incubation for 1 h with rat liver homogenate (Emberger, 1994).
Sterically hindered esters were also hydrolysed in rat liver homogenate. The sterically hindered ester 3,5,5-trimethyl-[2,3-3H]-cyclohexanyl-[14C]mandelate (cyclandelate) was completely hydrolysed to 3,5,5-trimethyl-[2,3-3H]-cyclohexanol and [14C]mandelic acid within 5 min of incubation with rat hepatocytes. By 20 min, 80% of the alcohol had disappeared, with a concomitant linear increase in a beta-glucuronidase-reactive substance. The resulting alcohol was presumably conjugated with glucuronic acid (White et al., 1990).
In urine collected 18 h after administration of cyclohex-1-en-1-yl acetate at a dose of 350 mg/kg bw to rabbits, 39% of the dose was hydrolysed and then conjugated with glucuronic acid (Elliott et al., 1959). This information indicates that all the esters in this group will be hydrolysed in vivo to yield the corresponding cyclohexanol derivative.
Once formed, the alicyclic terpenoid secondary alcohols and ketones are rapidly absorbed, metabolized and excreted, mainly in the urine as glucuronic acid and, in the case of alpha,beta-unsaturated ketones, as glutathione conjugates (Williams, 1959; Portoghese et al., 1989).
Male Sprague-Dawley rats were exposed to atmospheres containing cyclohexanone (No. 1100) at either 400 ppm [approximately 240 mg/kg bw (Fassett, 2001)] or 1600 ppm [approximately 980 mg/kg bw (Fassett, 2001)] for 6 h (unpublished data). Blood samples obtained at termination showed average plasma concentrations of cyclohexanone and cyclohexanol of 26 and 20 µg/ml, respectively, at 400 ppm and 120 and 140 µg/ml, respectively, at 1600 ppm. Total urinary excretion of cyclohexanol in samples collected 24, 48 and 72 h after exposure was at least 10 times greater than that of cyclohexanone, with 16 and 15 µg/ml cyclohexanone and 140 and 260 µg/ml cyclohexanol at 400 and 1600 ppm, respectively; 13 and 72 mg/ml of conjugated cyclohexanol were excreted within 72 h after exposure to 400 and 1600 ppm, respectively (Topping et al., 1994).
Menthol (5-methyl-2-isopropylcyclohexanol), which is structurally similar to 4-isopropyl-2-cyclohexanone (No. 1110), is rapidly absorbed and excreted in rabbits and rats. Urine and faeces were collected 24 and 48 h after dosing from rats given 3-[3H]menthol at a dose of 500 mg/kg bw (128 µCi/mg) by gavage. Both intact and bile duct-cannulated rats excreted > 70% of the dose within 48 h. Several metabolites of menthol were detected in urine and faecal extracts, but no unchanged menthol was found. The glucuronic acid conjugate of menthol was the main metabolite in bile, while the glucuronic acid conjugate and metabolites formed by side-chain oxidation (e.g., 3,8-menthanediol) were detected in urine (Yamaguchi et al., 1994). Rabbits given a maximum toxic dose of 3.5 g of menthol over 10 h by gavage eliminated 86% of the dose as a glucuronic acid conjugate in the urine (Annex 1, reference 138).
Isophorone (3,5,5-trimethyl-2-cyclohexenone, No. 1112) was partially excreted as the parent compound in exhaled air and urine of rabbits. Rabbits and rats given isophorone at a dose of 1000 mg/kg bw by gavage in water and olive oil, respectively, had untransformed isophorone in expired air and glucuronic acid conjugates in the urine 24 h later (Dutertre-Catella, 1978).
Polar alicyclic ketones of low relative molecular mass and secondary alcohols [e.g., cyclopentanone (No. 1101] were rapidly absorbed, metabolized, conjugated and excreted, mainly in the urine. Treatment with beta-glucuronidase of urine collected from rabbits given cyclopentanone at a dose of 190 mg/kg bw orally showed that the major urinary component was a glucuronic acid conjugate of cyclopentanol (James & Waring, 1971). Similar results were obtained with cyclohexanone (No. 1100). Urine collected from rabbits 18 h after dosing showed that 66% of the oral dose of 250 mg/kg bw had been excreted as the glucuronic acid conjugate of cyclohexanol. The authors concluded that cyclohexanone is first reduced to cyclohexanol and then conjugated with glucuronic acid prior to excretion in the urine (Elliott et al., 1959).
Four groups of male beagles were given cyclohexanone at a dose of 280 mg/kg bw per day by intravenous injection at various concentrations and injection rates for 18–21 days. When given at the highest concentration, cyclohexanol was detected in plasma within 30 min of injection, with mean distribution and elimination half-lives of 6.6 and 81 min, respectively. The mean steady-state volume of distribution of cyclohexanone was 2.6 l/kg, and the mean total body clearance was 27 ml/kg per min, indicating that cyclohexanone is rapidly cleared from the body. To clarify the pharmacokinetics of cyclohexanol, one dog was given 330 mg/kg bw by intravenous injection in a separate study. The plasma half-life was 99 min, the apparent distribution volume was 1.2 l/kg, and the total body clearance was 8.8 ml/kg per min; therefore, cyclohexanol was rapidly cleared. About 60% of an administered dose of cyclohexanone was recovered in 24-h urine as a glucuronide conjugate of cyclohexanol. Direct renal clearance of unmodified cyclohexanone and cyclohexanol is a minor route of elimination, accounting for < 1% of an administered dose. It has been proposed that 74–100% of cyclohexanone is converted to cyclohexanol and further metabolized before elimination. The authors proposed that some cyclohexanone is exhaled through the lungs (Martis et al., 1980; Koeferl et al., 1981).
Four men and four women volunteers were exposed to cyclohexanone at an atmospheric concentration of 100, 210 or 410 mg/m3 for 8 h. Urine collected at 2-h intervals during exposure and for 72 h afterwards showed the presence of glucuronic acid conjugates of cyclohexanediol, with a peak excretion rate about 16 h after exposure. About 60% of the dose was excreted within 72 h (Mraz et al., 1994).
Premature neonates fed a solution of dextrose parenterally showed the presence of trans-1,2-cyclohexanediol, with lesser amounts of trans-1,4-cyclohexanediol and cis-1,3-cyclohexanediol in their urine. It was determined that cyclohexanone had leached from the containers used to deliver the dextrose solution at an average of 0.89 g/24 h (Mills & Walker, 1990). The absence of glucuronide conjugates in the urine of these neonates was explained by decreased activity of microsomal glucuronosyltransferase in premature neonatal liver tissue (Boreus, 1982).
After consuming 720 ml of saké (ethanol content, 10% w/v), a man ingested 100 ml of liquid adhesive containing 39% cyclohexanone. The cyclohexanone was rapidly absorbed as cyclohexanol, presumably by conversion of cyclohexanone catalysed by alcohol dehydrogenase. The plasma and urine concentrations of cyclohexanone and its metabolites were unaffected by gastric lavage (5.5 l of saline), two plasma exchanges (2.4 l each) and haemoperfusion, as compared with pre-treatment values. Cyclohexanol and cyclohexanone were detected in plasma for up to 25 h after ingestion. The concentration of cyclohexanone was at the lower limit of detection but was highest (220 µg/ml) 5 h after ingestion and had decreased to < 10 µg/ml after 20 h. High levels of cyclohexanol glucuronide were detected in the urine for up to 47 h. Urinary excretion of the parent ketone was described as minimal. The elimination half-life of cyclohexanone in human plasma was determined to be 4.8 h, and the rate of elimination (Ke) was 0.14 µg/ml per h. The mechanism of elimination in humans thus involves conversion of cyclohexanone to cyclohexanol, followed by conjugation with glucuronic acid (Sakata et al., 1989).
In summary, the esters of alicyclic secondary alcohols are readily hydrolysed. The resulting secondary alcohols are interconvertible with their corresponding ketones. In the principal excretion pathway, the secondary alcohols are conjugated with glucuronic acid and excreted primarily in the urine.
As indicated above, the major metabolic pathway involves reduction of the alicyclic ketones to yield the corresponding secondary alcohols, which are subsequently excreted primarily as the glucuronic acid conjugates (Williams, 1959; Lington & Bevan, 1994; Topping, 1994). If a double-bond is present, it may be reduced to the corresponding dihydro derivative (Krasavage et al., 1982), presumably by the gut microflora for metabolites excreted into the bile. Endocyclic double-bonds are more prone to reduction than exocyclic double-bonds. In addition to reductive pathways, alicyclic ketones and secondary alcohols containing an alkyl side-chain also undergo oxidation of the side-chain to form polar metabolites, which are excreted as the glucuronide or sulfate conjugates mainly in the urine. In the case of substances of higher relative molecular mass, more lipophilic ketones or sterically hindered functional groups, oxidation of a ring position by cytochrome P450 may compete with reduction of the ketone function or oxidation of the alcohol function (Hildebrandt, 1902; Nelson et al., 1992).
(i) Alicyclicketones and secondary alcohols (Nos 1100 and 1101)
The metabolic fate of cyclohexanol is similar to that of simple acyclic aliphatic secondary alcohols (see Figure 1) (Lington & Bevan, 1994; Topping, 1994).
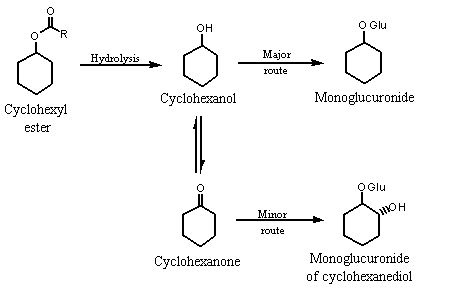
Figure 1. Metabolic fate of cyclohexyl esters in humans
In rabbits, 66% of a dose of cyclohexanone of 190 mg/kg bw and 47% of a dose of cyclopentanone of 190 mg/kg bw administered by gavage were reduced to the corresponding secondary alcohol and excreted in urine as the glucuronic acid conjugate (Elliot et al., 1959; James & Waring, 1971). Trace amounts of mercapturic acid conjugate of the 2-hydroxycyclohexyl derivative were also detected, indicating that the epoxide may be the intermediary metabolite formed from dehydration (James & Waring, 1971). Cyclohexene is formed by dehydration of cyclohexanol, a known contaminant of commercially available cyclohexanone. Hepatic cytochrome P450 monooxygenases catalyse the epoxidation of cycloalkenes. The epoxide may hydrolyse to form diols or undergo nucleophilic attack by mercapturic acid to form the 2-hydroxycyclohexyl mercapturic acid conjugate.
Eighteen-hour urine samples from rabbits given 1500 mg of cyclohexanone by gavage contained 65% cyclohexanol and 6% trans-cyclohexane-1,2-diol as monoglucuronide conjugates (Elliott et al., 1959). Presumably, the diol forms by hydroxylation at the alpha position and subsequent reduction of cyclohexanone. When cyclohexane at 460 mg/kg bw, cyclohexanol at 260 mg/kg bw or cyclohex-1-en-1-yl acetate at 350 mg/kg bw was fed to rabbits, the alcohol and to a lesser extent the diol were common urinary metabolites (Elliott et al., 1959).
The urine of rabbits given a dose of 1200 mg/kg bw of cyclohexanol orally showed the presence of glucuronic acid conjugates. Similar results were obtained in rabbits given cyclohexanone at a dose of 890 mg/kg bw orally (Treon et al., 1943). The glucuronic acid conjugates of cyclohexanol (1.6 mg/l) and cyclohexanone (0.23 mg/l) were found in the urine of workers exposed to a mixture of atmospheric hexanes including 460 mg/m3 of cyclohexane (Perbellini et al., 1980; Governa et al., 1987). The authors concluded that cyclohexane is transformed to cyclohexanol, which subsequently forms glucuronic acid and sulfate conjugates.
(ii) Alkyl substituted alicyclic ketones and secondary alcohols (Nos 1099–1117)
When rabbits were given 2-, 3- or 4-methylcyclohexanone orally at doses of 200–3200 mg/kg bw, the glucuronic acid and sulfate conjugates of the corresponding secondary alcohols were excreted in their urine (Treon et al., 1943; Elliott et al., 1959; Tao & Elliott, 1962).
When a secondary alcohol or ketone function is located on alicyclic-containing alkyl substituents, as in 2-methylcyclohexanone (No. 1102), oxidation of the alkyl substituents competes with oxidation of the alcohol or reduction of the ketone function. When the substance contains allylic or tertiary hydrogen (e.g., 2-hexylidene cyclopentanone, No. 1106), the rate of side-chain oxidation increases, often leading to polyoxygenated metabolites (Dutertre-Catella, 1978, Nelson et al., 1992). The number of possible polyoxygenated metabolites increases with the number of types of alkyl ring substituents (e.g., methyl and isopropyl substituents) and the presence of an alkene (endocyclic and/or exocyclic), in which hydrogenation may precede or succeed oxidation of the substituents (Madyastha & Raj, 1990; Nelson et al., 1992; Yamaguchi et al., 1994). More lipophilic substances may undergo oxidation of the secondary alcohol function to the corresponding ketone, in addition to oxidation of alkyl substituents (Asakawa et al., 1986).
Urine samples collected over up to 4 days from rabbits given isophorone (No. 1112, 3,5,5-trimethyl-2-cyclohexen-1-one) by gavage at a dose of 1000 mg/kg bw contained several metabolites: the major one was 5,5-dimethyl-1-cyclohexene-3-one-1-carboxylic acid, formed by methyl-group oxidation at an exocyclic allylic position; 3,5,5-trimethyl-2-cyclohexen-1-ol (isophorol) was formed by reduction of the ketone group and then conjugation with glucuronic acid; 3,5,5-trimethylcyclohexanone (dihydroisophorone) was formed by hydrogenation of the endocyclic double-bond; and cis- and trans-3,5,5-trimethylcyclohexanol were formed by hydrogenation of the endocyclic double-bond and reduction of the ketone group (see Figure 2) (Truhaut et al., 1970; Dutertre-Catella, 1978).
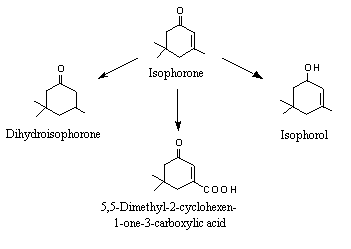
Figure 2. Recognized metabolic fates of isophorone in rabbits
Carvone (2-methyl-5-(1-methylethenyl)-2-cyclohexen-1-one), a structurally related alpha,beta-unsaturated ketone, was partially excreted as the parent compound in both humans and rats (Tamura et al., 1962; Zlatkis et al., 1973). Allylic oxidation products, namely 9-hydroxycarvone, have also been detected in rabbits (see Figure 3) (Williams, 1959; Ishida et al., 1989). Carvone has been shown to induce cytosolic glutathione transferase activity in mice (Zheng et al., 1992), suggesting that it undergoes conjugation with glutathione at the beta position (Portoghese et al., 1989). In rabbits, carvone was mainly reduced to yield carveol, which was then converted to the glucuronic acid conjugate and excreted in urine (Fisher & Bielig, 1940). Unchanged dihydrocarveol l (Fisher & Bielig, 1940) and its glucuronic acid conjugate (Hamalainen, 1912) were detected in the urine of rabbits given carvone.
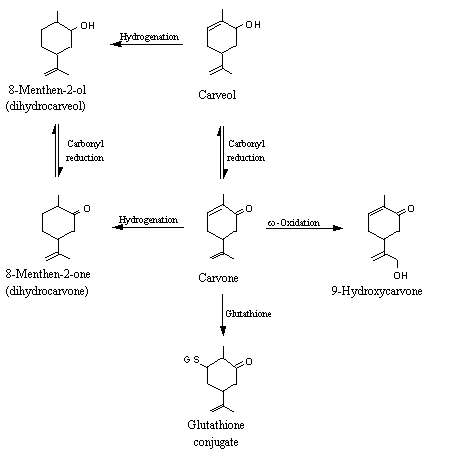
Figure 3. Metabolic fates of carvone in animals
In summary, these 25 alicyclic ketones, secondary alcohols and related esters are metabolized primarily by reduction of the ketone to yield the corresponding secondary alcohol, followed by conjugation with glucuronic acid and excretion in urine. There is also evidence that small amounts are excreted as sulfate conjugates. If side-chains are present, side-chain oxidation may occur. If the ketone is alpha,beta-unsaturated, it may be conjugated with glutathione or undergo allylic oxidation to yield polar oxygenated metabolites that can be excreted in urine.
Oral LD50 values have been reported for 10 of the 25 substances in this group (see Table 3). In rats, the values ranged from 1200 mg/kg bw for cyclopentanone (No. 1101) to > 5000 mg/kg bw for cyclohexyl acetate (No. 1093), 2-hexylidenecyclo-pentanone (No. 1106), isojasmone (No. 1115) and 2-(3,7-dimethyl-2,6-octadienyl)-cyclopentanone (No. 1117), demonstrating that the acute oral toxicity of alicyclic ketones, secondary alcohols and related esters is extremely low.
Table 3. Studies of acute toxicity of alicyclic ketones, secondary alcohols and related esters used as flavouring agents
|
No. |
Agent |
Species |
Sex1 |
Route |
LD50 |
Reference |
|
1093 |
Cyclohexyl acetate |
Rat |
M,F |
Oral |
> 5000 |
Moreno (1977) |
|
1094 |
Cyclohexyl butyrate |
Mouse |
M,F |
Oral |
> 5000 |
Moreno (1982) |
|
1100 |
Cyclohexanone |
Rat |
M,F |
Oral |
1700 |
Kohli et al. (1967) |
|
1100 |
Cyclohexanone |
Rat |
M,F |
Oral |
1800 |
Deichmann & LeBlanc (1943) |
|
1100 |
Cyclohexanone |
Rat |
M,F |
Oral |
1500 |
Smyth et al. (1969) |
|
1100 |
Cyclohexanone |
Rat |
M,F |
Oral |
1800 |
Gupta et al. (1979) |
|
1100 |
Cyclohexanone |
Mouse |
M |
Oral |
2100 |
Gupta et al. (1979) |
|
1100 |
Cyclohexanone |
Mouse |
F |
Oral |
2100 |
Gupta et al. (1979) |
|
1101 |
Cyclopentanone |
Rat |
M,F |
Oral |
1200 |
Levenstein (1976) |
|
1101 |
Cyclopentanone |
Rat |
M |
Oral |
2700 |
Exxon Chemical Americas (1982) |
|
1106 |
2-Hexylidene cyclopentanone |
Rat |
M,F |
Oral |
> 5000 |
Moreno (1980) |
|
1109 |
2-sec-Butylcyclohexanone |
Rat |
NR |
Oral |
2300 |
Leberco Laboratories (1965) |
|
1109 |
2-sec-Butylcyclohexanone |
Rat |
M,F |
Oral |
2400 |
Moreno (1978) |
|
1112 |
Isophorone |
Rat |
M,F |
Oral |
3400 |
Exxon Chemical Americas (1982) |
|
1113 |
3-Methyl-5-propyl-2-cyclohexen-1-one |
Rat |
M,F |
Oral |
2700 |
Moreno (1982) |
|
1115 |
Isojasmone |
Rat |
M,F |
Oral |
> 5000 |
Moreno (1974) |
|
1117 |
2-(3,7-Dimethyl-2,6-octadienyl)cyclopentanone |
Rat |
M,F |
Oral |
> 5000 |
Gabriel (1991) |
M, male; F, female; NR, not reported
In mice, the oral LD50 values were reported to range from 2100 mg/kg bw for cyclohexanone (No. 1100) to > 5000 mg/kg bw for cyclohexyl butyrate (No. 1094), confirming the extremely low acute toxicity of these alicyclic ketones, secondary alcohols and related esters.
Ninety-day or 13-week studies were available for five of the 25 alicyclic ketones and secondary alcohols in this group (Table 4). Two of the five studies were performed at a single target level of intake that was at least 100 times the estimated possible average daily intake from use of the substance as a flavouring agent (Oser & Hall, 1977). The possible average daily intake is determined by multiplying usual use levels in each of 33 food categories (e.g., baked goods and meat products) by the average amount of that food category consumed daily and summing the intake over all 33 food categories (Department of Agriculture, 1965). The possible average daily intake of the vast majority of flavouring agents with low reported annual volumes of use (National Academy of Sciences, 1987; Lucas et al., 1999) is a gross exaggeration of the average daily intake. The calculation is based on the assumption that all foods in a category always contain the substance and that the food category is consumed each day (Oser & Hall, 1977). Therefore, the dietary concentrations in these studies were many orders of magnitude greater than actual levels of intake of alicyclic ketone, secondary alcohol and related esters as flavouring agents.
Table 4. Results of short-term, long-term and carcinogenicity studies of aicyclic ketones, secondary alcohols and related esters used as flavouring substances
|
No. |
Substance |
Species, sex |
No. test groupsa/no. per groupb |
Route |
Duration |
NOEL |
Reference |
|
1100 |
Cyclohexanone |
Mouse, M,F |
7/20 |
Drinking-water |
91 |
1600 (M) |
Lijinsky & Kovatch (1986) |
|
3200 (F) |
|||||||
|
1100 |
Cyclohexanone |
Mouse, M,F |
3c /96–102 |
Drinking-water |
730 |
ND (M) |
Lijinsky & Kovatch (1986) |
|
1600 (F) |
|||||||
|
1100 |
Cyclohexanone |
Rat, M,F |
7/10 |
Drinking-water |
175 |
500 (M) |
Lijinsky & Kovatch (1986) |
|
720 (F) |
|||||||
|
1100 |
Cyclohexanone |
Rat, M,F |
2/104 |
Drinking-water |
730 |
650 |
Lijinsky & Kovatch (1986) |
|
1106 |
2-Hexylidene cyclopentanone |
Rat, M,F |
1/10–32 |
Diet |
90 |
2.9 (M) |
Posternak et al. (1969) |
|
3.4 (F) |
|||||||
|
1109 |
2-sec-Butyl cyclohexanone |
Rat |
3/NR |
Diet |
91 |
370 |
Hummler (1969) |
|
1111 |
Tetramethyl ethyl cyclohexenone |
Rat, M,F |
1/10–32 |
Diet |
90 |
40 (M) |
Posternak et al. (1969) |
|
48 (F) |
|||||||
|
1112 |
Isophorone |
Mouse, M, F |
5/20 |
Gavage |
91 |
1000 (M) |
Bucher et al. (1986); National Toxicology Program (1986) |
|
500 (F) |
|||||||
|
1112 |
Isophorone |
Mouse, M,F |
2/100 |
Gavage |
730 |
250d |
Bucher et al. (1986); National Toxicology Program (1986) |
|
a |
Does not include control animals |
|
b |
Includes both male and female animals |
|
c |
At the highest dose (6.2 mg/kg bw per day), only females (41) were tested |
|
d |
Performed at either a single or multiple doses that produced no adverse effects. Therefore, this dose level is not a true NOEL but the highest dose tested that produced no adverse effects. The actual NOEL would be higher. |
An alicyclic ketone, cyclohexanone (No. 1100), was used in two studies (Lijinsky & Kovatch, 1986); alkyl-substituted ketones and secondary alcohols, 2-sec-butylcyclohexanone (No. 1109), tetramethylethylcyclohexenone (isomeric mixture) (No. 1111) and 2-hexylidenecyclopentanone (No. 1106) in two (Hummler, 1969; Posternak et al., 1969); and an alpha,beta-unsaturated alicyclic ketone, isophorone (No. 1112) in one (Bucher et al., 1986; National Toxicology Program, 1986). Additionally, 13-week studies were performed with structurally related alicyclics, DL-menthol [(1S-1alpha,2beta,5alpha)-5-methyl-2-(1-methylethyl)cyclohexanol] (National Cancer Institute, 1978) and D-carvone (2-methyl-5-(1-methylethenyl)-2-cyclohexenone) (Hagan et al., 1967; National Toxicology Program, 1990). No histopathological changes were found. The only consistent effect found at high doses in these studies was a decrease in body-weight gain in treated groups.
(i) Alicyclic ketones and secondary alcohols
Cyclohexanone (No. 1100) and related secondary alcohols: A 13-week study was performed in which groups of 10 B6C3F1 mice of each sex received drinking-water containing cyclohexanone (No. 1100) at a concentration of 0, 400, 2300, 6500, 13 000, 25 000 or 47 000 ppm, corresponding to 0, 100, 580, 1600, 3200, 6200, 8500 and 12 000 mg/kg bw per day, respectively (Food and Drug Administration, 1993). The drinking-water was acidified to pH 2.5 to suppress bacterial growth. Food and water were provided ad libitum. The mice were monitored daily for survival and weekly for changes in body weight. At necropsy, at least 28 tissues were examined histologically.
No effects were reported at 100, 580 or 1600 mg/kg bw per day. Male mice given 3200 mg/kg bw per day gained slightly less weight, and those at 6200 mg/kg bw per day dose showed a 19% decrease in body-weight gain when compared with controls. Among animals at 8500 mg/kg bw per day, one male died before 13 weeks, and weight gain was depressed by 15% in females and 24% in males. At the highest dose, 6/10 males and 3/10 females died, some mice showed focal coagulative liver necrosis (number and sex not reported), and two females showed hyperplasia of the thymus. The maximal tolerated dose was estimated by the authors to be 6200 mg/kg bw per day for females and 3200 mg/kg bw per day for males (Lijinsky & Kovach, 1986). The concentration of 3200 mg/kg bw per day is more than 10 000 000 times the daily per capita intake ("eaters only") of 0.002 µg/kg bw of cyclohexanone from its use as a flavouring agent in Europe and the USA (Table 2).
A 25-week study was performed in which groups of five Fischer 344 rats of each sex were given drinking-water containing cyclohexanone (No. 1100) at a concentration of 0, 190, 400, 800, 1600, 3300, 4700 or 6500 ppm, corresponding to 0, 30, 60, 120, 240, 500, 720 and 1000 mg/kg bw per day, respectively (Food and Drug Administration, 1993). The drinking-water was acidified to pH 2.5 to suppress bacterial growth. Food and water were provided ad libitum. The rats were monitored daily for survival and weekly for changes in body weight. At necropsy, major organs and tissues were examined histologically, and the observations were used to determine the maximal tolerated dose for this species. All the rats survived to termination of the study at 25 weeks. No observable effects were reported in male or female rats at doses up to 500 mg/kg bw per day. Two males at 720 mg/kg bw per day developed degenerative changes of the thyroid gland that were not seen in other animals in this study. Animals at the highest dose had a 10% depression in weight gain. No other adverse effects were observed. The maximal tolerated dose was calculated to be 1000 mg/kg bw per day (Lijinsky & Kovatch, 1986). This concentration is more than 5 000 000 times the daily per capita intake2 ("eaters only") of 0.002 µg/kg bw of cyclohexanone from its use as a flavouring agent in Europe and the USA (Table 2).
(ii) Alkyl substituted ketones and secondary alcohols
2-sec-Butylcyclohexanone (No. 1109): In a 13-week study, an unspecified number of rats of an unspecified strain were given diets containing 2-sec-butylcyclohexanone (No. 1109) at a concentration calculated to provide an average daily intake of 0, 160, 370 or 900 mg/kg bw per day. Some dose-dependent depression in body-weight gain was reported, which was attributed in part by the authors to the presumed unpalatability of the food. No adverse effects were observed at the two lower doses. An increased mortality rate was reported among rats at 900 mg/kg bw per day, but no details were given (Hummler, 1969). The dose of 370 mg/kg bw per day that resulted in no adverse effects is more than 1 000 000 times the daily per capita intake ("eaters only") of 0.1 µg/kg bw from its use as a flavouring agent in Europe and the USA (Table 2).
Tetramethylethylcyclohexenone (isomeric mixture) (No. 1111) and 2-hexylidene-cyclopentanone (No. 1106): These two alicyclic ketones were studied by the same protocol in two 90-day studies. A control and a test group, each consisting of 10–16 male and 10–16 female Charles River CD rats, were housed in pairs of the same sex and given access to water and food ad libitum. The concentration of the test material in the diet was adjusted during the study to maintain constant levels of intake of tetramethylethylcyclohexenone (isomeric mixture) (No. 1111) of 40 mg/kg bw per day for males and 48 mg/kg bw per day for females, and levels of 2-hexylidenecyclopentanone (No. 1106) of 2.9 mg/kg bw per day for males and 3.4 mg/kg bw per day for females. These dietary levels were calculated to be > 100 times an exaggerated estimate of possible average daily intake from use as flavouring agents. Clinical observations were recorded daily, and food consumption and body weights were determined weekly. Haematological and clinical chemical determinations (blood urea) performed at weeks 7 and 13 revealed elevated blood urea at week 7 in rats given 2-hexylidenecyclopentanone but depressed blood urea at week 14. When the values were compared with those for a composite rather than individual control groups, the differences were not significant. At necropsy, liver and kidney weights were measured; a wide range of tissues and organs from each animal were preserved, and major organs and tissues were examined microscopically. Observations of growth, food intake, haematological and clinical chemical parameters, organ weights and histopathological examination revealed no significant differences between control animals and treated groups given tetramethylethylcyclohexenone (isomeric mixture) or 2-hexylidenecyclopentanone (Posternak et al., 1969). The dose of 48 mg/kg bw per day of tetramethylethyl-cyclohexenone that resulted in no adverse effects is more than 100 000 times the estimated total daily per capita intakes ("eaters only") of up to 0.15 µg/kg bw from use of this substance as a flavouring agent in Europe and the USA (Table 2).
Isophorone (No. 1112): In a 13-week study, groups of 10 B6C3F1 mice and Fischer 344/N rats of each sex received isophorone (No. 1112) in corn oil by gavage at a dose of 0, 62, 120, 250, 500 or 1000 mg/kg bw per day for 5 days per week. Food and water were provided ad libitum. Moribund animals were killed. Body weights were recorded weekly. At termination of the experiment, survivors were killed and necropsied. Three female mice receiving the highest dose died before the end of the study. The body weights of male mice receiving 250, 500 or 1000 mg/kg bw per day were reported to be approximately 10% less than those of male controls; however, no such difference was observed in females, and there was no correlation between final mean body weights and dose. No compound-related gross or microscopic changes were observed. The kidneys of animals at the highest dose were reviewed on two separate occasions, and no evidence of nephropathy was found. One female rat given the highest dose died, but no other adverse effects or dose-related responses were noted. A comprehensive examination of the kidneys of animals at the highest dose revealed no evidence of nephrotoxicity (Bucher et al., 1986; National Toxicology Program, 1986).
(i) Alicyclic ketones and secondary alcohols
Cyclohexanone (No. 1100): Groups of 41–52 B6C3F1 mice of each sex were given drinking-water containing cyclohexanone at a concentration of 0, 6500, 13 000 or 25 000 ppm (females only), corresponding to dietary intakes of 0, 1600, 3200 and 6200 mg/kg bw per day, respectively. The pH of the drinking-water was maintained at 2.5 to suppress bacterial growth. The mice were monitored daily for survival and weekly for body-weight changes. At death or termination, they were subjected to complete necropsy and histopathology. Mice at the lowest dose and females at 3200 mg/kg bw per day showed no significant changes in weight when compared with controls. The body weights of male mice at 3200 mg/kg bw per day and of females at 6200 mg/kg bw per day were 15–20% lower than those of controls during most of the study. No differences in survival rates were reported for animals receiving 1600 mg/kg bw per day; 80% of those at 3200 mg/kg bw per day were alive at week 90. The survival of female mice at the two higher doses was poor: only half the females at the highest dose survived to 50 weeks and < 20% to 75 weeks, and of females at 3200 mg/kg bw per day, only 50% survived to 75 weeks and only 40% to 90 weeks. Histopathological examination of male mice at 1600 mg/kg bw per day revealed an increased incidence of proliferative lesions of the liver and lung, combined with a statistically significant (p = 0.041) increase in the incidence of benign and malignant hepatocellular neoplasms (25/51, 49%) when compared with controls (16/52, 31%). No benign or malignant neoplasms were seen at the two higher concentrations. Female mice at 1600 mg/kg bw per day had an increased incidence of malignant lymphomas (17/50, 34%), which was significant in the life-table (p = 0.036) and incidental tumour tests (p = 0.040), suggesting a weak carcinogenic effect (Lijinsky & Kovatch, 1986). The increased incidence of malignant lymphomas among females at the lowest dose may be attributable to the high, variable background incidence of this lesion in control B6C3F1 mice used for National Toxicology Program studies, which has been increasing with time (Haseman et al., 1986, 1997, 1998). The incidence in female B6C3F1 mice used as controls in studies sponsored by the National Toxicology Program (2001) was 6–30%. The background incidence in female control mice in this study (15%, 8/52) (Lijinsky & Kovatch, 1986) was thus within the range for this tumour type in control B6C3F1 female mice.
In a separate 2-year study with the same protocol, groups of 52 Fischer 344/N rats of each sex were given drinking-water containing cyclohexanone at a concentration of 0, 3300 or 6500 ppm, corresponding to 0, 330 or 650 mg/kg bw per day (Food and Drug Administration, 1993). Rats in both treated groups failed to reach the mean body weight of the control group. More than 85% of the rats at 650 mg/kg bw per day survived to week 90, and 70% survived to the end of the study. The authors noted that the incidences and types of many neoplasms were similar in treated and control groups. A statistically significant increase in the incidence of adrenal cortex adenomas (7/52, 13%; p = 0.030) was reported in males at 330 mg/kg bw per day when compared with controls (1/52, 2%), but no increase was found at 650 mg/kg bw per day (1/51, 2%). The authors noted that the incidence of adrenal cortex adenomas in Fischer 344/N rats in National Toxicology Program laboratories was approximately 1%. They concluded that, in the absence of a dose–response relationship, the increased incidence of benign neoplasms was not indicative of a carcinogenic response (Lijinsky & Kovatch, 1986).
Thus, no dose-dependent effects were observed in either mice or rats, and histopathological examination revealed no lesions that could be associated with administration of the test material. The authors concluded that the doses of 3200 mg/kg bw per day for mice of each sex and 330 mg/kg bw per day for male rats were weakly carcinogenic. These conclusions should be evaluated in relation to the finding of no evidence of carcinogenicity with higher doses in female mice (6200 mg/kg bw per day) and in rats (650 mg/kg bw per day). These data were evaluated independently by a working group convened by the International Agency for Research on Cancer (1989), which determined that cyclohexanone was "not classifiable as to its carcinogenicity to humans".
Isophorone (No. 1112): A 2-year bioassay was conducted in which isophorone (No. 1112) was tested in mice and rats with the standardized National Toxicology Program protocol. The doses were determined from the results of a 13-week study. In the 2-year study, groups of 50 B6C3F1 mice and Fischer 344/N rats of each sex received isophorone in corn oil by gavage at a dose of 0, 250 or 500 mg/kg bw per day on 5 days per week for 103 weeks. Food and water were provided ad libitum. Moribund animals were killed. Body weights were recorded weekly. At the end of the experiment, survivors were killed and necropsied. No clinical signs of toxicity were reported, but significant numbers of animals (36/300) died due to gavage errors. The survival of male rats at the higher dose was reported to be lower than that of controls, and the mean body weights of rats at this dose were reported to be significantly lower than those of controls. No other treatment-related clinical signs were reported. In mice, no treatment-related clinical signs were reported, and isophorone did not adversely affect survival rates. The mean body weights of females at the higher dose were reported to be 5% lower than those of controls during the second year of the study (Bucher et al., 1986; National Toxicology Program, 1986).
Neoplastic and non-neoplastic lesions associated with administration of isophorone to male mice developed principally in the liver (see Table 5). Male mice at 500 mg/kg bw per day showed a significantly increased incidence of coagulative necrosis and hepatocytomegaly, but females showed a decrease. Male mice at the higher dose showed a statistically significant increase in the incidence of combined hepatocellular adenomas and carcinomas (29/50, 58%; p = 0.033) over that in controls (18/48, 38%). No significant difference in the incidence of hepatocellular neoplasms was found in the group at the lower dose. Female B6C3F1 mice showed no evidence of hepatocellular neoplasms at either dose. The authors concluded that there was equivocal evidence of carcinogenic activity in male B6C3F1 mice at 500 mg/kg bw per day, as shown by an increased incidence of hepatocellular adenoma and carcinoma (combined), but no evidence of carcinogenicity in female B6C3F1 mice at 250 or 500 mg/kg per day (National Toxicology Program, 1986).
Table 5. Incidences of hepatocellular neoplasms associated with administration of isophorone to mice by gavage for 103 weeks
|
Lesion |
Dose (mg/kg bw per day) |
||
|
Control |
250 |
500 |
|
|
Males |
|||
|
Hepatocellular adenoma |
6/48 |
7/50 |
13/50 |
|
Hepatocellular carcinoma |
14/48 |
13/50 |
22/50 |
|
Combined* |
18/48 (38%) |
18/50 (36%) |
29/50 (58%) |
|
p = 0.033 |
|||
|
Females |
|||
|
Hepatocellular adenoma |
2/50 |
4/50 |
6/50 |
|
Hepatocellular carcinoma |
2/50 |
2/50 |
2/50 |
|
Combined* |
4/50 (8%) |
6/50 (12%) |
8/50 (16%) |
*Historical incidence rate: 335/1034 (32% ± 9.4%)
As stated previously, the primary neoplastic effects observed in male mice in the National Toxicology Program study were in the liver. The incidences of hepatocellular adenoma in control and high-dose groups of male mice (6/48 and 13/50, respectively) and of carcinomas (14/48 and 22/50, respectively) demonstrate the susceptibility and sensitivity of the B6C3F1 male mouse liver to carcinogenic agents. This conclusion is supported by the fact that the combined incidence of adenomas and carcinomas in control males was greater than in any group of treated females (6/50 and 8/50, respectively). These responses are consistent with the high incidences of hepatocellular neoplasms found in control B6C3F1 male mice in other studies (Maronpot & Boorman, 1982; Maronpot et al., 1987).
The observation of hepatic neoplasms in mice in the National Toxicology Program bioassay are not relevant to the safety evaluation of isophorone in humans at low levels of intake from use as a flavour ingredient. This conclusion is based on the high incidence of spontaneous hepatocellular neoplasms (adenomas and carcinomas) in the strain of mice studied, the absence of a dose–response relationship, the lack of hepatocellular neoplastic effects in rats and the relatively high doses administered, as compared with intake levels from use as a flavour ingredient. The dose of 250 mg/kg bw per day used in the 2-year rat bioassay is > 1 000 000 times the estimated daily per capita intake ("eaters only") of 2 ng/kg of isophorone from its use as a flavour ingredient.
A statistically significant increase in the incidence of mesenchymal neoplasms (14/50, 28%; p = 0.011), primarily subcutaneous fibrosarcomas, was reported in male mice at the higher dose (500 mg/kg bw per day) when compared with the control group (6/48, 13%) (Bucher et al., 1986; National Toxicology Program, 1986). More recent bioassays in mice in which corn oil was used as the vehicle showed a high background incidence of subcutaneous fibrosarcomas. The incidence of combined subcutaneous fibrosarcomas, fibromas and fibrosarcomas in control groups of male mice has been reported to be as high as 20% (National Toxicology Program, 1992b). The observation of mesenchymal neoplasms in mice in this bioassay is also not relevant to the safety evaluation of isophorone in humans at low levels of intake. This conclusion is based on the high background incidence of spontaneous fibrosarcomas in the strain and sex of mice studied, the lack of similar neoplastic effects in rats and the relatively high dose (500 mg/kg bw per day) administered as compared with intake levels from use as a flavour ingredient.
Nephropathy was seen in both treated and control rats of each sex after natural death or at termination. In male rats at 250 and 500 mg/kg bw per day, increased incidences of mineral deposits in renal collecting ducts (31/50, 62%, and 20/50, 40%), tubular-cell hyperplasia (1/50, 2%, and 4/50, 8%), adenomas (0/50 and 2/50, 8%) and adenocarcinomas (3/50, 6%, and 1/50, 2%) were observed, but these were not found not in female rats (see Table 6). Tubule mineralization was characterized by basophilic aggregates in the medullary collecting ducts, often occurring coincidentally with lesions of chronic nephropathy. The authors of the report concluded that there was some evidence for the carcinogenicity of isophorone in male Fischer 344/N rats, as shown by the occurrence of renal tubule cell adenomas and adenocarcinomas in animals given 250 or 500 mg/kg per day; however, there was no evidence of carcinogenicity in female rats at these doses (Bucher et al., 1986; National Toxicology Program, 1986).
Table 6. Incidences of renal neoplasms associated with administration of isophorone to rats by gavage for 103 weeks
|
Lesion |
Dose (mg/kg bw per day) |
||
|
Control |
250 |
500 |
|
|
Males |
|||
|
Nephropathy |
49/50 |
47/50 |
46/50 |
|
Tubule mineralization |
1/50 |
31/50 |
20/50 |
|
Renal tubule hyperplasia |
0/50 |
1/50 |
4/50 |
|
Renal tubule adenoma* |
0/50 |
0/50 |
2/50 |
|
Renal tubule adenocarcinoma* |
0/50 |
3/50 |
1/50 |
|
Females |
|||
|
Nephropathy |
21/50 |
39/50 |
32/50 |
|
Tubule mineralization |
10/50 |
4/50 |
2/50 |
|
Renal tubule hyperplasia |
0/50 |
0/50 |
1/50 |
|
Renal tubule adenoma* |
0/50 |
0/50 |
0/50 |
|
Renal tubule adenocarcinoma* |
0/50 |
0/50 |
0/50 |
* Incidence of tubule cell adenoma or adenocarcinoma in other controls: 4/1091 (0.4%); p < 0.05
It has been shown that renal lesions result from the accumulation of aggregates of alpha-2u-globulin (a protein of low relative molecular mass synthesized in the liver) and isophorone or its metabolites in the P2 segment of the renal proximal tubule. This phenomenon has been observed only in male Fischer 344/N rats and has been referred to as alpha-2u-globulin nephropathy (Strasser et al., 1988; Borghoff et al., 1990). The gene that encodes alpha-2u-globulin has been isolated and the sequence deduced (Untermann et al., 1981). These proteins are expressed in the liver under hormonal control and are the major urinary protein in adult male rats (Roy & Neuhaus, 1967; Wang & Hodgetts, 1998). alpha-2u-Globulin belongs to a superfamily of proteins characterized by a unique hydrophobic binding pocket (Swenberg et al., 1989). The characteristic renal lesions have not been reported in female Fischer 344/N rats or in humans (Bucher et al., 1986). Subsequent investigations have shown that the alpha-2u-globulin nephropathy found in male Fischer 344/N rats does not develop in mammals that do not express the hepatic form of alpha-2u-globulin (Swenberg et al., 1989), such as other strains of rats (Dietrich & Swenberg, 1991), mice (Bucher et al., 1986; National Toxicology Program, 1986; Lehman-McKeeman & Caudill, 1994) and dogs (Webb et al., 1990).
While humans produce serum proteins of low relative molecular mass which are resorbed by the kidney, there is no evidence that alpha-2u-globulin is produced (Swenberg et al., 1989; Olson et al., 1990). Comparison of urine collected from adult male Fischer 344 rats and humans revealed no evidence of alpha-2u-globulin production in humans (Olson et al., 1990). The authors concluded that the very low protein content of human urine, the relatively small proportion of cationic to total proteins and the high relative molecular mass of the most abundant human urinary proteins form a biological basis for suggesting that humans are not at risk for developing the alpha-2u-globulin nephropathy observed in male Fischer 344/N rats.
It is not known whether any human serum proteins have a binding site similar to that of alpha-2u-globulin. Although this is a possibility, it appears remote, since female rats, mice and dogs do not show the renal changes seen in male rats exposed to isophorone. There is a class of human proteins referred to as the alpha-2u-globulin-related proteins, which appear to have no functional relationship to adult male rat urine proteins. The human protein has a higher relative molecular mass, 25 kDa, and is a component of a neutrophil gelatinase complex (Triebel et al., 1992; Kjeldsen et al., 2000). An extensive review of the current scientific literature and genome databases revealed no native protein or biological entity that has the nephropathic effects of alpha-2u-globulin in mature male rats. The accumulated evidence indicates that the unique anatomical, physiological and biochemical properties of the male rat kidney, especially the proximal convoluted tubule, allow isophorone to interfere with renal processing of the strain-specific alpha-2u-globulin. Therefore, this process is not predictive of human carcinogenicity. In a comprehensive review of alpha-2u-globulin nephropathy and associated renal tubule tumours produced in male Fischer 344/N rats exposed to isophorone and other simple chemical substances (e.g., limonene, decalin and methyl isobutyl ketone), it was concluded that the Fischer 344/N rat is not an appropriate model for assessing human renal carcinogenic risk (Environmental Protection Agency, 1991). It has also been concluded that the mechanisms leading to renal carcinogenicity in male Fischer 344/N rats are largely known and strongly indicate that the alpha-2u-globulin nephropathy associated with compounds such as isophorone have no significance for human risk (Burdock et al., 1990).
Preputial gland carcinomas were observed in 5/50 (10%) male rats at the higher dose, and clitoral gland adenomas were reported in 2/50 (4%) female rats at the lower dose, as compared with the incidence of 0/50 in vehicle control groups of each sex. The preputial lesions were believed to be significant, in the apparent absence of lesions in vehicle controls (0/50), males at the lower dose (0/50) and females at the higher dose (0/50) and the low incidence of this lesion in controls given corn oil in other experiments (12/1094). However, the authors emphasized that the actual incidence of lesions of the prepuce and clitoris was unknown since only animals with visible lesions were examined histologically (Bucher et al., 1986).
Macroscopic preputial tumours have been reported sporadically in vehicle controls in previous studies of the National Toxicology Program (1986). In more recent studies, in which histopathological examination of the prepuce was performed on all male rats, the incidence of preputial neoplasms in control rats exceeded the incidence reported in the isophorone-treated male rats (5/50; p < 0.05). For example, the combined incidence of preputial adenomas and carcinomas in control male rats treated only with corn oil by gavage was as high as 23% (11/47; p = 0.218) (National Toxicology Program, 1993a), while the incidence of preputial carcinomas in other control males was reported to be as high as 12% (6/50; p = 0.092) (National Toxicology Program, 1993b). In addition, the high background incidence of clitoral gland adenomas, 6% (3/47; p = 0.346) to 14% (7/49; p = 0.093), in female rats serving as vehicle controls in studies with corn oil by gavage (National Toxicology Program, 1993a,b, 1994) supports the conclusion that the clitoral gland adenomas reported in two females at the lower dose in the study of isophorone (National Toxicology Program, 1986) are of no relevance to humans.
This conclusion was confirmed in a similar study to determine whether there is a link between preputial gland carcinomas and alpha-2u-globulin and/or isophorone (No. 1112). A series of experiments was performed that included measurements of binding of alpha-2u-globulin to DNA, with [14C]isophorone as tracer. [32P]DNA post-labelling was used to detect adduct formation and cell proliferation. No DNA adducts were detected. An increase in cell proliferation was observed after 1 week of exposure in both male and female rats, but only a slight increase was seen in the males after 4 weeks (data not shown). The authors concluded that preputial carcinomas are specific to rats and are not relevant to human risk assessment (Morishita et al., 1997).
The studies of genotoxicity conducted with this group of substances are summarized in Table 7.
Table 7. Studies of genotoxicity with alicyclic ketones, secondary alcohols and related esters used as flavouring agents
|
No. |
Agent |
End-point |
Test object |
Maximum concentration |
Results |
Reference |
|
In vitro |
||||||
|
1093 |
Cyclohexyl acetate |
DNA damage |
B. subtilis H17(rec+), M45 (rec–) |
19 mg/disc |
Negativea |
Yoo (1986) |
|
1094 |
Cyclohexyl butyrate |
DNA damage |
B. subtilis H17(rec+), M45 (rec-) |
19 mg/plate |
Negativea |
Oda et al. (1979 |
|
1100 |
Cyclohexanone |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537 |
33–10 000 mg/plate |
Negativea |
Haworth et al. (1983) |
|
1100 |
Cyclohexanone |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537 |
2.9–2900 mg/plate |
Negativea |
Florin et al. (1980) |
|
1100 |
Cyclohexanone |
Chromosomal |
Chinese hamster ovary cells aberration |
7.5 µl/ml |
Negativea |
Aaron et al. (1985) |
|
1100 |
Cyclohexanone |
Chromosomal |
Human lymphocytes aberration |
9.8–980 mg/ml |
Positivea |
Lederer et al. (1971) |
|
1100 |
Cyclohexanone |
Chromosomal |
Human lymphocytes aberration |
0.005–0.1 mg/ml |
Positivea |
Dyshlovoi et al. (1981) |
|
1100 |
Cyclohexanone |
Sister chromatid exchange |
Chinese hamster ovary cells |
7.5 µl/ml |
Negativeb |
Aaron et al. (1985) |
|
Positivec |
||||||
|
1101 |
Cycopentanone |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537 |
2.5–2500 mg/plate |
Negativea |
Florin et al. (1980) |
|
1106 |
2-Hexylidene cyclo-pentanone |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537, TA1538 |
3600 mg/plate |
Negativea |
Wild et al. (1983) |
|
1108 |
2,2,6-Trimethyl cyclo-hexanone |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535,TA1537 |
4.2–3600 mg/plate |
Negativea |
Florin et al. (1980) |
|
1111 |
Tetramethyethyl cyclohexanone (mixed isomers) |
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537, TA1538 |
3600 mg/plate |
Negativea |
Wild et al. (1983) |
|
1112 |
Isophorone |
Reverse mutation |
S. typhimurium TA97, TA98, TA100, TA1535, TA1537 |
33–10 000 mg/plate |
Negativea |
Mortelmans et al. (1986) |
|
1112 |
Isophorone |
Mutation |
S. typhimurium TA98, TA100, TA1535, TA1537 |
33–10 000 mg/plate |
Negativea |
National Toxicology Program (1986) |
|
1112 |
Isophorone |
Mutation |
L5178YTk+/– mouse lymphoma cells |
67–810 mg/ml |
Negativeb |
McKee et al. (1987) |
|
130–1300 mg/ml |
Negativec |
|||||
|
1112 |
Isophorone |
Mutation |
L5178YTk+/– mouse lymphoma cells |
0.089–0.89 ml/ml |
Negativeb |
O’Donoghue et al. (1988) |
|
0.13–1.3 ml/ml |
Negativec |
|||||
|
1112 |
Isophorone |
Mutation |
L5178YTk+/– mouse lymphoma cells |
800 mg/mld |
Positivec |
McGregor et al. (1988) |
|
1112 |
Isophorone |
Mutation |
L5178YTk+/– mouse lymphoma cells |
1200 mg/ml |
Negativeb |
National Toxicology Program (1986) |
|
Positivec |
||||||
|
1112 |
Isophorone |
Chromosomal aberration |
Chinese hamster ovary cells |
5–1600 mg/ml |
Negativea |
Gulati et al. (1989) |
|
1112 |
Isophorone |
Chromosomal aberration |
Chinese hamster ovary cells |
250–1600 mg/ml |
Negativea |
National Toxicology Program (1986) |
|
1112 |
Isophorone |
Chromosomal aberration |
Chinese hamster lung fibroblasts |
1200c, 1500b mg/ml |
Positivea |
Matsuoka et al. (1996) |
|
250–1000 mg/ml |
Negativea |
|||||
|
1112 |
Isophorone |
Sister chromatid exchange |
Chinese hamster ovary cells |
5–1600 mg/ml |
Positiveb,e |
Gulati et al. (1989) |
|
1112 |
Isophorone |
Sister chromatid exchange |
Chinese hamster ovary cells |
160–1000 mg/ml |
Negativea |
National Toxicology Program (1986) |
|
1112 |
Isophorone |
Unscheduled DNA |
Rat hepatocytes |
0.005–0.4 ml/ml |
Negativea |
O’Donoghue et al. |
|
synthesis |
(1988) |
|||||
|
1112 |
Isophorone |
Unscheduled DNA synthesis |
Rat hepatocytes |
200 ml/ml |
Negativea |
McKee et al. (1987) |
|
In vivo |
||||||
|
1100 |
Cyclohexanone |
Sex-linked recessive lethal mutation |
D. melanogaster |
0.1 ml/100 ml |
Negative |
Goncharova (1970) |
|
1106 |
2-Hexylidene cyclo-pentanone |
Sex-linked recessive lethal mutation |
D. Melanogaster |
5 mmol/l |
Negative |
Wild et al. (1983) |
|
1106 |
2-Hexylidene cyclo-pentanone |
Micronucleus formation |
Mouse bone marrow |
170, 330, 500 mg/kg bw |
Negative |
Wild et al. (1983) |
|
1111 |
Tetramethyethyl cyclo-hexanone (mixed isomers) |
Sex-linked recessive lethal mutation |
D. melanogaster |
10 mM |
Negative |
Wild et al. (1983) |
|
1111 |
Tetramethyethyl cyclo-hexanone (mixed isomers) |
Micronucleus formation |
Mouse bone marrow |
180, 307, 450 mg/kg bw |
Negative |
Wild et al. (1983) |
|
1112 |
Isophorone |
Sex-linked recessive lethal mutation |
D. melanogaster |
2000f and 12 500g ppm |
Negative |
Foureman et al.(1994) |
|
1112 |
Isophorone |
Micronucleus formation |
CD-1 mice |
540 mg/kg bw |
Negative |
McKee et al. (1987) |
|
1112 |
Isophorone |
Micronucleus formation |
CD-1 mice |
0.54 ml/kg bw |
Negative |
O’Donoghue et al. (1988) |
a With and without metabolic activation
b Without metabolic activation
c With metabolic activation
d Cytotoxic at next highest dose tested (1600 mg/ml)
e A positive response was obtained only in the absence of metabolic activation and only after additional culture time (6–13 h)
f Oral administration
g Injection
(i) In vitro
Eight alicyclic ketones, secondary alcohols and related esters have been tested for genotoxicity. Overall, negative results were reported in the standard assay for reverse mutation when various strains of Salmonella typhimurium (TA98, TA100, TA1535, TA1537 and TA1538) were incubated with up to 10 000 µg/plate of cyclohexanone (No. 1100) or isophorone (No. 1112), 2.5–2500 µg/plate of cyclopentanone (No. 1101), up to 4200 µg/plate of 2,2,6-trimethyl cyclohexanone (No. 1108) or up to 3600 µg/plate of 2-hexylidene cyclopentanone (No. 1106) or tetramethylethylcyclohexanone (No. 1111), with or without metabolic activation (Florin et al., 1980; Haworth et al., 1983; Wild et al., 1983; Mortelmans et al., 1986). In another test for reverse mutation with S. typhimurium TA98, TA100, TA1535 and TA1537 (only an abstract), cyclohexanone was reported to produce ‘a large number of revertants’ in TA98, with no further elaboration and no results for the other strains. The concentrations and test conditions used were not specified (Massoud et al., 1980).
Both cyclohexyl acetate (No. 1093) and cyclohexyl butyrate (No. 1094) gave negative results for mutation in Bacillus subtilis M45 (rec–) and H17 (rec+) (Oda et al., 1979; Yoo, 1986). Positive results were reported with cyclohexanone in an assay for forward mutation assay in B. subtilis (Massoud et al., 1980); however, as previously stated, no concentrations or test conditions were reported in the abstract.
The results for forward mutation in mouse lymphoma cells were generally negative with isophorone, with or without metabolic activation (National Toxicology Program, 1986; McKee et al., 1987; O’Donoghue et al., 1988). An increased mutation frequency was reported in L5178Y Tk+/- mouse lymphoma cells without metabolic activation at concentrations of 400 and 800 µg/ml. Isophorone was lethal at 1600 µg/ml (McGregor et al., 1988).
Cyclohexanone (No. 1100) at concentrations up to 980 µg/ml induced chromosomal aberrations in human lymphocytes with or without metabolic activation (Collin et al., 1971; Lederer et al., 1971; Dyshlovoi et al., 1981). It did not induce chromosomal aberrations in Chinese hamster ovary cells at a concentration of 7.5 µl/ml, with or without metabolic activation (Aaron et al., 1985). Isophorone (No. 1112) gave equivocal results in Chinese hamster ovary cells. In one study, no chromosomal aberrations were induced with or without metabolic activation at concentrations up to 1600 µg/ml (Gulati et al., 1989), whereas in another study isophorone at a concentration of 1200 µg/ml without metabolic activation or at a concentration of 1500 µg/ml with metabolic activation induced chromosomal aberrations (Matsuoka et al., 1996); however, lower concentrations of 250–1000 µg/ml tested without metabolic activation did not. In an assay for sister chromatid exchange, cyclohexanone at a concentration of 7.5 µl/ml gave weakly positive results in Chinese hamster ovary cells in the absence of metabolic activation and negative results in the presence of metabolic activation (Aaron et al., 1985). Similarly, isophorone induced sister chromatid exchange in Chinese hamster ovary cells only when tested without metabolic activation at concentrations of 500–1000 µg/ml and then only after delayed harvesting due to the cytostatic effect of isophorone (Gulati et al., 1989). At lower concentrations tested without metabolic activation or at concentrations up to 1600 µg/mL tested with metabolic activation, isophorone did not induce sister chromatid exchange (National Toxicology Program, 1986; Gulati et al., 1989). In an assay for unscheduled DNA synthesis in rat hepatocytes, isophorone showed no sign of genotoxicity at concentrations up to 200 µl/ml (McKee et al., 1987; O’Donoghue et al., 1988).
(ii) In vivo
When cyclohexanone (No. 1100), 2-hexylidene cyclopentanone (No. 1106), tetramethylethyl cyclohexanone (No. 1111) or isophorone (No. 1112) was fed to adult Drosophila melanogaster for 3 days, no mutations were observed (Goncharova, 1970; Wild et al., 1983; Foureman et al., 1994). In addition, negative results were obtained when D. melanogaster were injected with a single dose of 12 500 µg of isophorone (Foureman et al., 1994).
There was no increase in the frequency of micronucleated polychromatic erythrocytes in the bone marrow of male or female CD-1 mice given isophorone (No. 1112) at a dose of 540 µg/kg bw by intraperitoneal injection (McKee et al., 1987; O’Donoghue et al., 1988) or in NMRI mice injected intraperitoneally with 2-hexylidenecyclopentanone at a dose of 170, 330 or 500 mg/kg bw or tetramethylethylcyclopentenone at a dose of 180, 310 or 450 mg/kg bw (Wild et al., 1983).
(iii) Conclusion
Cyclohexyl acetate (1093), cyclohexyl butyrate (No. 1094), cyclopentanone (No. 1101), 2-hexylidene cyclopentanone (No. 1106), 2,2,6-trimethyl cyclohexanone (No. 1108) and tetramethylethylcyclohexanone (mixed isomers) (No. 1111) gave negative results in assays for genotoxicity in vitro. The results reported for the genotoxicity of cyclohexanone (No. 1100) and isophorone (No. 1112) are conflicting. Most of the assays were conducted before 1986, when the pH and ionic strength of test media were often not adequately maintained. Mammalian cells in situ rely on complex regulatory mechanisms to maintain homeostatic conditions, and those in culture are not equipped to respond to environmental changes; therefore, it is important that the culture media used in mammalian cell assays be maintained at a pH of approximately 6.8–7.5. A lower pH or changes in osmolality due to the test agents can give rise to false-positive results, especially when metabolic activation systems are added. Acidity facilitates the breakdown of the components of such systems into mutagenic agents (Brusick, 1986).
The equivocal results of the assays for genotoxicity with cyclohexanone in vitro can be interpreted in terms of physiochemical properties. Compounds that are structurally similar to cyclohexanone have excellent membrane permeability and hydrogen bonding potential (Slater, 1963, 1970; Moreland, 1994). When cyclohexanone and related substances are tested in vitro, they may induce membrane expansion, leading to multiple effects on membrane-related processes. Membrane expansion may increase cell volume and lipid storage vacuoles, block ionic conductance channels, limit the availability of ATP and alter ion fluxes and metabolite distribution between the cytoplasm and organelles. Given these physiochemical properties, it is highly unlikely that any consistent pattern of genotoxicity would result from a battery of assays in bacterial and mammalian cells.
Overall, the tests for genotoxicity yielded mainly negative results. Positive results were reported in mammalian cells at cytotoxic concentrations, usually in the absence of biotransformation enzymes. The results of assays in vivo were negative.
(i) Cyclohexanone (No. 1100)
Postnatal behaviour of rats was examined on the basis of activity in a figure-eight maze. CD-1 mice (number not specified) were given cyclohexanone at a dose of 800 mg/kg bw per day orally on days 8–12 of gestation, and the offspring were tested for motor activity in the maze on days 22, 58 and 200 after parturition. No effect on motor activity was seen (Gray et al., 1986).
A group of 28 ICR/SIM mice were given cyclohexanone orally at a dose of 2200 mg/kg bw per day on days 8–12 of gestation, and all treated animals and their offspring were examined for toxic effects. Six of the 28 mice died, and the remaining mice showed a significant decrease in body-weight gain. The litter size and number and the 2-day survival of the neonates were unaffected by treatment, but there was a significant decrease in live birth weight. The authors reported that the treatment was maternally toxic, as evidenced by > 7% mortality, overt clinical signs of toxicity or significantly reduced body-weight gain (Seidenberg et al., 1986; Seidenberg & Becker, 1987). The dose of 2200 mg/kg bw per day that was maternally toxic is > 1 100 000 000 times the daily per capita ("eaters only") intake of 2 ng/ kg from use of cyclohexanone as a flavouring agent.
In another study, female CF1 mice were given cyclohexanone at a dose of 50 mg/day intraperitoneally for 28 days and were mated on day 10 of this treatment. The numbers of pregnancies and viable fetuses were similar to those of controls. The authors concluded that cyclohexanone had no effect on fertility in mice (Hall et al., 1974).
(ii) Cyclopentanone (No. 1101)
Female CF1 mice were given cyclopentanone at a dose of 50 mg/day intraperitoneally for 28 days and were mated on day 10 of this treatment. The numbers of pregnancies, viable fetuses and resorptions were similar to those of controls. The authors concluded that cyclopentanone had no effect on fertility in mice (Hall et al., 1974).
Groups of 25 COBS CD female rats received cyclopentanone at a dose of 0, 50 or 300 mg/kg bw per day by gavage in corn oil on days 6–15 of gestation. On day 20 of gestation, the fetuses were removed surgically, and intrauterine survival, fetal development and skeletal and visceral morphology were recorded. No maternal, embryonal or fetal effects were seen at either dose. A slight decrease in mean fetal body weight was reported in the group at the higher dose but was not statistically significant. Although an increase in the number of litters with the fetal variant malaligned sternebrae was reported at 50 mg/kg bw per day, no such effect was seen at 300 mg/kg bw per day. The authors concluded that the NOEL was 50 mg/kg bw per day (Rusch et al., 1988).
(iii) Isophorone (No. 1112)
Groups of 12 Fischer 344 rats and 12 CD-1 mice were exposed to atmospheres containing isophorone at a dose of up to 150 ppm (260 mg/kg bw per day) on days 6–15 of gestation. Slight maternal toxicity, in the form of increased fetal resorptions and decreased food consumption resulting in decreased mean body weight, was observed in both species only at the highest concentration of 150 ppm. No developmental effects were seen (Phillips, 1985). In a similar study, rats were exposed to 500 ppm (860 mg/kg bw per day) of isophorone for 6 h/day for 5 days per week. No reproductive or developmental effects were reported (Dutertre-Catella, 1989).
Cyclohexanol (No. 1100)
Seven Sprague-Dawley rats received cyclohexanol (No. 1100) as a 20% solution in peanut oil in two intraperitoneal injections of 200 mg/kg bw each, 5 days per week for up to 6 weeks. A second group of seven rats received the same total dose (400 mg/kg bw per day) on 5 days/ per week for 13 weeks. A control group of five rats received the peanut oil vehicle only. The animals were monitored for changes in body weight and examined by electrophysiology during the study and by neuropathology at the end. The group that received cyclohexanol showed a slight decrease in weight, but no other effects were observed. No evidence of neurotoxicity or neuropathology was reported (Perbellini et al., 1981).
Aaron, C.S., Brewen, J.G., Stetka, D.G., Bleicher, W.T. & Spahn, M.C. (1985) Comparative mutagenesis in mammalian cells (CHO) in culture: Multiple genetic endpoint analysis of cyclohexanone in vitro (Abstract). Environ. Mutag., 7 (Suppl. 3), 60–61.
Anders, M.W. (1989) Biotransformation and bioactivation of xenobiotics by the kidney. In: Hutson, D.H., Caldwell, J. & Paulson, G.D., eds, Intermediary Xenobiotic Metabolism in Animals, New York: Taylor & Francis, pp. 81–97.
Asakawa, Y., Ishida, T., Toyota, M. & Takemoto, T. (1986) Terpenoid transformation in mammals. IV. Biotransformation of (+)-longifolene, (–)-caryophyllene, (–)-caryophyllene oxide, (–)-cyclocolorenone, (+)-nootkatone, (–)-elemol, (–)-abietic acid, and (+)-dehydroabietic acid in rabbits. Xenobiotica, 16, 753–767.
Boreus, L.O. (1982) Biotransformation. In: Boreus, L.O., ed., Principles of Pediatric Pharmacology, New York: Churchill Livingstone, pp. 1–114.
Borghoff, S.J., Short, B.G. & Swenberg, J.A. (1990) Biochemical mechanisms and pathobiology of nephropathy. Annu. Rev. Pharmacol. Toxicol., 30, 349–367.
Brusick, D (1986) Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations. Environ. Mutag., 8, 879–886.
Bucher, J.R., Huff, J. & Kluwe, W.M. (1986) Toxicology and carcinogenesis studies of isophorone in F344 rats and B6C3F1 mice. Toxicology, 39, 207–219.
Burdock, G.A., Wagner, B.M., Smith, R.L., Munro, I.C. & Newberne, P.M. (1990) Recent progress in the consideration of flavoring ingredients under the food additives amendment. 15. GRAS substances. Food Technol., 44, 78–86.
Cramer, G.M., Ford, R.A. & Hall, R.L. (1978) Estimation of toxic hazard—A decision tree approach. Food Cosmet. Toxicol., 16, 255–276.
Deichmann, W.B. & LeBlanc, T.J. (1943) Determination of the approximate lethal dose with about six animals. J. Ind. Hyg. Toxicol., 25, 415–417.
Dietrich, D.R. & Swenberg, J.A. (1991) NCI-Reiter (NBR) male rats fail to develop renal disease following exposure to agents that induce alpha-2u-globulin (alpha2u) nephropathy. Fundam. Appl. Toxicol., 16, 749–762.
Dutertre-Catella, H. (1989) Contributions to the Analytical Toxicological and Biochemical Study of Isophorone, Thesis for Doctorate in Pharmacology, Université Réné Descartes, Paris.
Dutertre-Catella, H., Nguyen, P., Dang, Q. & Truhaut, R. (1978) Metabolic transformations of the trimethyl-3,5,5, cyclohexene-2, one-1 (isophorone). Toxicol. Eur. Res., 1, 209–216.
Dyshlovoi, V.D., Boiko, N.L., Shemetun, A.M. & Kharchenko, T.I. (1981) Cytogenetic action of cyclohexanone. Gig. Sanit., 5, 76–77 (in Russian with English abstract).
Elliott, T.H., Parke, D.V. & Williams, R.T. (1959) Studies in detoxication. The metabolism of cyclo[14C]hexane and its derivatives. Biochem. J., 72, 193–200.
Emberger, D. (1994) In vitro hydrolysis test on menthyl propyleneglycol carbonate. Private communication. Submitted to WHO by Flavor and Extract Manufacturers’ Association of the United States.
Environmental Protection Agency (1991) Risk Assessment Forum. Alpha 2u-globulin: Association with Chemically Induced Renal Toxicity and Neoplasia in the Male Rat, Publication No. EPA/625/3-91019F, Washington DC.
Exxon Chemical Americas (1982) Unpublished report submitted to Environmental Protection Agency.
Felsted, R.L. & Bachur, N.R. (1980) Ketone reductases. In: Jakoby, W.B., ed., Enzymatic Basis of Detoxication, 2nd Ed., New York: Academic Press, pp. 281–293.
Fischer, F.G. & Bielig, H.J. (1940) Biochemical hydrogenations. VII. Hydrogenation of unsaturated compounds in the animal body. Hoppe-Seyler’s Z. Physiol. Chem., 266, 73–98.
Florin, I., Rutberg, L., Curvall, M. & Enzell, C.R. (1980) Screening of tobacco smoke constituents for mutagenicity testing using Ames test. Toxicology, 18, 219–232.
Food and Drug Administration (1993) Conversion table for test chemical treatment doses used in PAFA. In: Priority-based Assessment of Food Additives (PAFA) Database, Washington DC: Center for Food Safety and Applied Nutrition, p. 58.
Ford, D.M. & Moran, E.J. (1978) Preliminary indications of in vitro hydrolysis of two flavor chemical esters. Private communication. Submitted to WHO by Flavor and Extract Manufacturers’ Association of the United States.
Foureman, P., Mason, J.M., Valencia, R. & Zimmering, S. (1994) Chemical mutagenesis testing in Drosophila. X. Results of 70 coded chemicals tested for the National Toxicology Program. Environ. Mol. Mutag., 23, 208–227.
Gabriel, D. (1991) Acute oral toxicity study of 2-(3,7-dimethyl-2,6-octadienyl)cyclopentanone in rats. Unpublished report from Bedoukian Research Inc.
Goncharova, R.I. (1970) Genetic activity of some cyclohexane derivatives. Genet. Tsitol., ?, 137–142.
Governa, M., Calisti, R., Coppa, G., Tagliavento, G., Colombi, A. & Troni, W. (1987) Urinary excretion of 2,5-hexanedione and peripheral polyneuropathies in workers exposed to hexane. J. Toxicol. Environ. Health, 20, 219.
Gray, L.E., Kavlock, R.J., Ostby, J., Ferrell, J., Rogers, J. & Gray, K. (1986) An evaluation of figure-eight maze activity and general behavioral development following prenatal exposure to forty chemicals: Effects of cytosine arabinoside, dinocap, nitrofen, and vitamin A. Neurotoxicology, 7, 449–462.
Gulati, D.K., Witt, K., Anderson, B., Zeiger, E. & Shelby, M.D. (1989) Chromosomal aberration and sister chromatid exchange tests in Chinese hamster ovary cells in vitro. III: Results with 27 chemicals. Environ. Mol. Mutag., 13, 133–193.
Gupta, P.K., Lawrence, W.H., Turner, J.E. & Autian, J. (1979) Toxicological aspects of cyclohexanone. Toxicol. Appl. Pharmacol., 49, 525–533.
Hall, I.H., Carlson, G.L., Abernethy, G.S. & Piantadosi, C. (1974) Cycloalkanones. 4. Antifertility activity. J. Med. Chem., 17, 1253–1257.
Hagan, E.C., Hansen, W.H., Fitzhugh, O.G., Jenner, P.M., Jones, W.I., Taylor, J.M., Long, E.L., Nelson, A.M. & Brouwer, J.B. (1967) Food flavorings and compounds of related structure. II. Subacute and chronic toxicity. Food Cosmet. Toxicol., 5, 141–157.
Hamalainen, J. (1912) Concerning the behavior of acyclic compounds with the glucuronic acid pairing in the organism. Skand. Arch. Physiol., 27, 141–226.
Haseman, J.K., Winbush, J.S. & O’Donnell, M.W., Jr (1986) Use of dual control groups to estimate false positive rates in laboratory animal carcinogenicity studies. Fundam. Appl. Toxicol., 7, 573–584.
Haseman, J.K., Boorman, G.A. & Huff, J. (1997) Value of historical control data and other issues related to the evaluation of long-term rodent carcinogenicity studies. Toxicol. Pathol., 25, 524–527.
Haseman, J.K., Hailey, J.R. & Morris, R.W. (1998) Spontaneous neoplasm incidences in Fischer 344 rats and B6C3F1 mice in two-year carcinogenicity studies: A National Toxicology Program update. Toxicol. Pathol., 26, 428–441.
Haworth, S., Lawlor, T., Mortelmans, K., Speck, W. & Zeigler, E. (1983) Salmonella mutagenicity test results for 250 chemicals. Environ. Mutag., 5 (Suppl. 1), 3–142.
Heymann, E. (1980) Carboxylesterases and amidases. In: Jakoby, W.B., ed., Enzymatic Basis of Detoxication, 2nd Ed., New York: Academic Press, pp. 291–323.
Hildebrandt, H. (1902) On the behavior of carvone and santalol in the animal body. Hoppe Selye’s Z. Physiol. Chem., 36, 452–461.
Hummler, H. (1969) Private communication. Submitted to WHO by Flavor and Extract Manufacturers’ Association of the United States.
International Agency for Research on Cancer (1989) IARC Monographs on the Evaluatiion of the Carcinogenic Risk of Chemicals to Humans, Vol. 47, pp. 157–169.
International Organization of the Flavor Industry (1995) European inquiry on volume use. Private communication. Submitted to WHO by Flavor and Extract Manufacturers’ Association of the United States.
Ishida, T., Toyota, M. & Asakawa, Y. (1989) Terpenoid biotransformation in mammals. V. Metabolism of (+)-citronellal, (±)-7-hydroxycitronellal, citral, (–)-perillaldehyde, (–)-myrtenal, cuminalehyde, thujone, and (±)-carvone in rabbits. Xenobiotica, 19, 843–855.
James, S.P. & Waring, R.H. (1971) The metabolism of alicyclic ketones in the rabbit and rat. Xenobiotica, 1, 573–580.
Kjeldsen, L., Cowland, J.B. & Borregaard, N. (2000) Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta, 482, 272–283.
Koeferl, M.T., Miller, T.R., Fisher, J.D., Martis, L., Garvin, P.J. & Dorner, J.L. (1981) Influence of concentration and rate of intravenous administration on the toxicity of cyclohexanone in beagle dogs. Toxicol. Appl. Pharmacol., 59, 215–229.
Kohli, R.P., Kishor, K., Dua, P.R. & Saxena, R.C. (1967) Anticonvulsant activity of some carbonyl containing compounds. Indian J. Med. Res., 55, 1221–1225.
Krasavage, W.J., O’Donoghue, J.L. & Divincenzo, G.D. (1982) Ketones. In: Clayton, G.D. & Clayton, F.E., eds, Patty’s Industrial Hygiene and Toxicology, Vol. IIC, Toxicology, New York: John Wiley and Sons, pp. 4709–4800.
Leberco Laboratories (1965) Unpublished report. Submitted to WHO by Flavor and Extract Manufacturers’ Association of the United States.
Lederer, J., Collin, J.P., Pottier-Arnould, A.M. & Gondry, E. (1971) Cytogenetic and teratogenetic effect of cyclamate and its metabolites Therapeutique, 47, 357–363 (in French).
Lehman-McKeeman, L.D. & Caudill, D. (1994) d-Limonene induced hyaline droplet nephropathy in alpha-2u-globulin transgenic mice. Fundam. Appl. Toxicol., 23, 562–568.
Levenstein, I. (1976) Acute oral toxicity study in rats and acute dermal toxicity studies in rabbits. Unpublished report to RIFM. Submitted to WHO by Flavor and Extract Manufacturers’ Association of the United States.
Lijinsky, W. & Kovatch, R.M. (1986) Chronic toxicity study of cyclohehanone in rats and mice. J. Natl Cancer Inst., 77, 941–949.
Lington, A.W. & Bevan, C. (1994) Alcohols. In: Clayton, G.D. & Clayton, F.E., eds, Patty’s Industrial Hygiene and Toxicology, 4th Ed., New York: John Wiley & Sons, Vol. IID, pp. 2585–2760.
Lucas, C.D., Putnam, J.M. & Hallagan, J.B. (1999) Flavor and Extract Manufacturers Association of the United States 1995 Poundage and Technical Effects Update Survey, Washington DC: Flavor and Extract Manufacturers’ Association of the United States..
Maarse, H., Visscher, C.A., Willemsens, L.C. & Boelens, M.H. (1999) Volatile Components in Food—Qualitative and Quantitative Data, Zeist: Centraal Instituut Voor Voedingsonderzioek TNO.
Madyastha, K.M. & Raj, C.P. (1990) Biotransformations of R-(+)-pulegone and menthofuran in vitro: Chemical basis for toxicity. Biochem. Biophys. Res. Communications, 3, 1086–1092.
Maronpot, R.R. & Boorman, G.A. (1982) Interpretation of rodent hepatocellular proliferative alterations and hepatocellular tumors in chemical safety assessment. Toxicol. Pathol., 10, 71–80.
Maronpot, R.R., Haseman, J.K., Boorman, G.A., Eustis, S.E., Rao, G.N. & Huff, J.E. (1987) Liver lesions in B6C3F1 mice: The National Toxicology Program, experience, and position. Arch. Toxicol., 10 (Suppl.), 10–26.
Martis, L., Tolhurst, T, Koeferl, M.T., Miller, T.R. & Darby, T.D. (1980) Disposition kinetics of cyclohexanone in beagle dogs. Toxicol. Appl. Pharmacol., 59, 215–229.
Matsuoka, A., Yamakage, K., Kusakabe, H., Wakuri, S., Asakura, M., Noguchi, T., Sugiyama, T., Shimada, H., Nakayama, S., Kasahara, Y., Takahashi, Y., Miura, K.F., Hatanaka, M., Ishidate, M., Jr, Morita, T., Watanabe, K., Hara, M., Odawara, K., Tanaka, N., Hayashi, M. & Sofuni, T. (1996) Re-evaluation of chromosomal aberration induction on nine mouse lymphoma assay ‘unique positive’ NTP carcinogens. Mutat. Res., 369, 243–252.
McGregor, D.B., Brown, A., Cattanach, P., Edwards, I., McBride, D., Riach, C. & Caspary, W.J. (1988) Responses of the L5178Y tk+/tk– mouse lymphoma cell forward mutation assay: III. 72 coded chemicals. Environ. Mol. Mutag., 12, 85–153.
McKee, R.H., Phillips, R.D., Lerman, S.A., Slesinski, R.S., Rogers-Back, A.M., Curren, R.D. & Putman, D.L. (1987) The genotoxic potential of isophorone. Environ. Mutag., 9, 71.
McMahon, R.E. (1982) Alcohols, aldehydes and ketones. In: Jakoby, W.B., Bend, J.R. & Caldwell, J., eds, Metabolic Basis for Detoxification. Metabolism of Functional Groups, New York: Academic Press, p. 91.
Mills, G. & Walker, V. (1990) Urinary excretion of cyclohexanediol, a metabolite of the solvent cyclohexanone, by infants in a special care unit. Clin. Chem., 36, 870–874.
Moreland, D.E. (1994) Effects of toxicants on oxidative phosphorylation and photophosphorylation. In: Hodgson, E. & Levi, P.E., eds, Introduction to Biochemical Toxicology, 2nd Ed., Norwalk, Connecticut: Appleton & Lange, pp. 345–366.
Moreno, O.M. (1974) Acute toxicity studies. Unpublished report to RIFM. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Moreno, O.M. (1977) Acute toxicity studies in rats for cyclohexyl acetate. Unpublished report No. 1692 (07/22) to RIFM. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Moreno, O.M. (1978) Acute toxicity studies in rats, mice, rabbits and guinea pigs. Unpublished report to RIFM. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Moreno, O.M. (1980) Acute toxicity studies. Unpublished report to RIFM. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Moreno, O.M. (1982) Acute toxicity studies. Unpublished report to RIFM. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Morishita, K., Schoonhoven, R. & Swenberg, J.A. (1997) Mechanistic studies on isophorone and preputial gland carcinomas. Toxicologist, 36, 94.
Mortelmans, K., Haworth, S., Lawlor, T., Speck, W., Tainer, W. & Zeiger, E. (1986) Salmonella mutagenicity tests: II. Results from the testing of 270 chemicals. Environ. Mutag., 8, 1–119.
Mraz, J., Galova, E., Nohova, H. & Vitkova, D. (1994) Uptake, metabolism and elimination of cyclohexanone in humans. Int. Arch. Occup. Environ. Health, 66, 203–208.
National Cancer Institute (1978) Bioassay of dl-Menthol for Possible Carcinogenicity, Report No. NCI-CG-TR-98; NIH publication 78-2665, Research Triangle Park, North Carolina.
National Toxicology Program (1986) Toxicology and Carcinogenesis Studies of Isophorone in F344/N Rats and B6C3F1 Mice (Gavage Studies), Report No. NTP-TR-291; NIH Publication No. 86-2547, Research Triangle Park, North Carolina.
National Toxicology Program (1990a) Toxicology and Carcinogenesis Studies of d-Limonene in F344/N Rats and B6C3F1 Mice (Gavage Studies), Report No. NTP-TR-347; NIH Publication No. 88-2902, Research Triangle Park, North Carolina.
National Toxicology Program (1990b) National Toxicology Program Technical Report on the Toxicology and Carcinogenesis Studies of d-Carvone in B6C3F1 Mice (Gavage Studies), Report No. NTP TR 381, Research Triangle Park, NC.
National Toxicology Program (1992b) Toxicology and Carcinogenesis Studies of gamma-Butyrolactone in F344/N Rats and B6C3F1 Mice (Gavage Studies), Report No. NTP-TR-406; NIH Publication No. 92-3137, Research Triangle Park, North Carolina.
National Toxicology Program (1993a) Toxicology and Carcinogenesis Studies of 3,4-Dihydrocoumarin in F344/N Rats and B6C3F1 Mice (Gavage Studies), Report No. NTP-TR-423; NIH Publication No. 93-3154, Research Triangle Park, North Carolina.
National Toxicology Program (1993b) Toxicology and Carcinogenesis Studies of Pentachloroanisole in F344/N Rats and B6C3F1 Mice (Gavage Studies), Report No. NTP-TR-414; NIH Publication No. 93-3145, Research Triangle Park, North Carolina.
National Toxicology Program (1994) Toxicology and Carcinogenesis Studies of o-Benzyl-p-chlorophenol in F344/N Rats and B6C3F1 Mice (Gavage Studies), Report No. NTP-TR-424; NIH Publication No. 94-3155, Research Triangle Park, North Carolina.
National Toxicology Program (2001) Toxicology and Carcinogenesis Studies of Citral (Microencapsulated) in F344/N Rats and B6C3F1 Mice (Feed Studies), Report No. NTP-TR-505; NIH Publication No. 01-4439, Research Triangle Park, North Carolina.
Nelson, S.D., McClanahan, R.H., Thomassen, D., Gordon, W.P. & Knebel, N. (1992) Investigations of mechanisms of reactive metabolite formation from (R)-(+)-pulegone. Xenobiotica, 22, 1157–1164.
Oda, Y., Hamano, Y., Inoue, K., Yamamoto, H. Nihara, T. & Kunita, N. (1979) Mutagenicity of food flavors in bacteria. Shokuhin Eisei Hen, 9, 177–181 (in Japanese).
O’Donoghue, J.L., Haworth, S.R., Curren, R.D., Kirby, P.E., Lawlor, T., Moran, E.J., Phillips, R.D., Putnam, D.L., Rogers-Back, A.M., Slesinski, R.S. & Thilagar, A. (1988) Mutagenicity studies on ketone solvents: Methyl ethyl ketone, methyl isobutyl ketone, and isophorone. Mutat. Res., 206, 149–161.
Olson, M.J., Johnson, J.T. & Reidy, C.A. (1990) A comparison of male rat and human urinary proteins: Implications for human resistance to hyaline droplet nephropathy. Toxicol. Appl. Pharmacol., 102, 524–536.
Oser, B.L. & Hall, R.L. (1977) Criteria employed be the expert panel of Flavor and Extract Manufacturers’ Association of the United States for the GRAS evaluation of flavouring substances. Food Cosmet. Toxicol., 15, 457–466.
Perbellini, L., Brugnone, F. & Pavan, I. (1980) Identification of the metabolites of n-hexane, cyclohexane and their isomers in men’s urine. Toxicol. Appl. Pharmacol., 53, 220–229.
Perbellini, L., De Grandis, D., Semenzato, F. & Bongiovanni, L.G. (1981) [Experimental study on the neurotoxicity of cyclohexanol and cyclohexanone.] Med. Lav., 2, 102–107 (in Italian).
Phillips, R.D. (1985) Ketone program panel. Inhalation teratology study in rats and mice—Isophorone. Report No. USEPA/OTS Doc. 408555049. TSCA negiotated testing agreement submissions. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Portoghese, P.S., Kedziora, G.S., Larson, D.L., Bernard, B.K. & Hall, R.L. (1989) Reactivity of glutathione with alpha,beta-unsaturated ketone flavouring substances. Food Chem. Toxicol., 27, 773–776.
Posternak, J.M., Linder, A. & Vodoz, C.A. (1969) Summaries of toxicological data. Toxicoligical tests on flavoring matters. Food Cosmet. Toxicol., 7, 405–407.
Roy, A.K. & Neuhaus, O.W. (1967) Androgenic control of a sex dependent protein in rat. Nature, 214, 618–620.
Rusch, G.M., Rodwell, D.E., Nemec, M.D. & Tasker, E.J. (1988) A teratology screening study in rats with cyclopentanone. Toxicologist, 8, 213.
Sakata, M., Kikuchi, J. & Haga, M. (1989) Disposition of acetone, methyl ethyl ketone and cyclohexanone in acute poisoning. Clin. Toxicol., 27, 67–77.
Salzer, D. (1998) In vitro hydrolysis test cis and trans-p-1(7),8-menthadien-2-yl acetate. Private communication. Submitted to WHO by Flavor and Manufacturers’ Association of the United States.
Seidenberg, J.M. & Becker, R.A. (1987) A summary of the results of 55 chemicals screened for developmental toxicity in mice. Teratog. Carcinog. Mutag., 7, 17–28.
Seidenberg, J.M., Anderson, D.G. & Becker, R.A. (1986) Validation of an in-vivo developmental toxicity screen in the mouse. Teratog. Carcinog. Mutag., 6, 361–374.
Slater, E.C. (1963) Uncouplers and inhibitors of oxidative phosphorylation. In: Hochester, R.M. & Quaste, J.J., eds, Metabolic Inhibitors, Vol. 2, New York: Academic Press, pp. 503–516.
Slater, E.C. (1970) Application of inhibitors and uncouplers for a study of oxidative phosphorylation. Meth. Enzymol., 10, 48.
Smyth, H.F., Jr, Carpenter, C.P., Weil, C.S., Pozzani, U.C., Striegel, J.A. & Nycum, J.S. (1969) Range-finding toxicity data: List VII. Am. Ind. Hyg.Assoc. J., 30, 470–476.
Stofberg, J. & Grundschober, F. (1987) Consumption ratio and food predominance of flavoring materials. Perfumer Flavorist, 12, 27.
Stofberg, J. & Kirschman, J.C. (1985) The consumption ratio of flavoring materials: A mechanism for setting priorities for safety evaluation. Food Chem. Toxicol., 23, 857–860.
Strasser, J., Jr, Charbonneau, M., Borghoff, S.J., Turner, M.J. & Swenberg, J.A. (1988) Renal protein droplet formation in male Fischer 344 rats after isophorone (IPH) treatment. Toxicologist, 8, 136.
Swenberg, J.A., Short, B., Borghoff, S., Strasser, J. & Charbonneau, M. (1989) The comparative pathobiology of nephropathy. Toxicol. Appl. Pharmacol., 97, 35–46.
Tamura, S., Tsustumi, S. & Kizu, K. (1962) Studies on glucuronic acid metabolism. I. The influence of borneol, ionone and carvone on the urinary excretion of glucuronic acid and ascorbic acid. Fol. Pharmacol. Jpn., 58, 323–336.
Tao, C.C. & Elliot, T.H. (1962) The metabolism of 14C-methylcyclohexane. J. Biochem., 84, 38–39.
Topping, D.C, Morgott, D.A., David, R.M. & O’Donoghue, J.L. (1994) Ketones. In: Clayton, G.D. & Clayton, F.E., eds, Patty’s Industrial Hygiene and Toxicology, 4th Ed., New York, John Wiley & Sons, Vol. IIC, pp. 1739–1878.
Triebel, S., Blaser, J., Reinke, H. & Tschesche, H. (1992) A 25 kDa alpha-2-microglobulin-related protein is a component of the 125 kDa form of gelatinase. FEBS Lett., 314, 386–388.
Treon, J.F., Crutchfield, W.E., Jr & Kitzmiller, K.V.. (1943) J. Ind. Hyg. Toxicol., 25, 199–214.
Truhaut, R., Dutertre-Catella, H. & Nguyen, P. (1970) Metabolic study of an industrial solvent, isophorone, in the rabbit. C.R. Hebd. Seances Acad. Sci. Ser. D: Sci. Nat., 271, 1333–1336.
Unterman, R.D., Lynch, K.R., Nakhasi, H.L., Dolan, K.P., Hamilton, J.W., Cohn, D.V. & Feigelson, P. (1981) Cloning and sequencing of several alpha-2-microglobulin cDNAs. Proc. Natl Acad. Sci. USA, 78, 3478–3482.
Wang, K.S. & Hodgetts, R.B. (1998) The analysis of regulatory sequences required for the high level induction of rat alpha-2u-globulin gene expression by dexamethasone. J. Mol. Endocrinol., 20, 129–139.
Webb, D.R., Kanerva, R.L., Hysell, D.K., Alden, C.L. & Lehman-McKeeman, L.D. (1990) Assessment of the subchronic oral toxicity of d-limonene in dogs. Food Chem. Toxicol., 28, 669–675.
White, D.A., Heffron, A., Miciak, B., Middleton, B., Knights, S. & Knights, D. (1990) Chemical synthesis of dual radiolabelled cyclandelate and its metabolism in rat hepatocytes and mouse J774 cells. Xenobiotica, 20, 71.
Wild, D., King, M.T., Gocke, E. & Eckhardt, K. (1983) Study of artificial flavouring substances for mutagenicity in the Salmonella/microsome, Basc, and micronucleus tests. Food Chem. Toxicol., 21, 707–719.
Williams, R.T. (1959) Detoxification Mechanisms. The Metabolism and Detoxification of Drugs, Toxic Substances and Other Organic Compounds, 2nd Ed., London: Chapman & Hall, pp. 4, 114–126.
Yamaguchi, T., Caldwell, J. & Farmer, P.B. (1994) Metabolic fate of [3H]–l–menthol in the rat. Drug Metab. Disposition, 22, 616–624.
Yoo, Y.S. (1986) Mutagenic and antimutagenic activities of flavoring agents used in foodstuffs. J. Osaka City Med. Center, 34, 267–288 (in Japanese).
Zheng, G.-Q., Kenney, P.M. & Lam, K.L.T. (1992) Effects of carvone compounds on glutathione S-transferase activity in A/J mice. J. Agric. Food Chem., 40, 751–755.
Zlatkis, A., Wolfgang, B., Lichtenstein, H.A., Tishbee, A. & Shunbo, F. (1973) Profile of volatile metabolites in urine by gas chromatography–mass spectrometry. Anal. Chem., 45, 783–787.
See Also:
Toxicological Abbreviations