
WHO FOOD ADDITIVES SERIES: 53
First draft prepared by
Gladwin Roberts
Science Strategy (Veterinary), Office of Chemical Safety, Department of Health and Ageing, Canberra, Australia
and
Carl E. Cerniglia
Division of Microbiology, National Center for Toxicological Research, Food and Drug Administration, Arkansas, USA
Pirlimycin is a lincosamide antibiotic that is closely related to lincomycin and clindamycin, and that is active against Gram-positive bacteria, including Staphylococcus aureus, Streptococcus agalactiae, Streptococcus dysgalactiae and Streptococcus uberis, which cause mastitis in dairy cows. The general mechanism of action of the lincosamides is through binding to the 50S ribosomal subunit, thereby inhibiting peptidyl transferase, with subsequent inhibition of protein synthesis in susceptible bacteria. Pirlimycin is administered by daily intramammary infusion at a dose of 50 mg of free base equivalents per udder quarter, for 2 days. For extended therapy, daily treatment may be continued for up to 8 consecutive days. Pirlimycin has not been previously evaluated by the Committee.
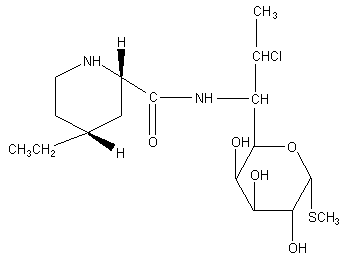
Figure 1. Structure of pirlimycin
The chemical name for pirlimycin is (2S-cis)-methyl 7-chloro-6,7,8-trideoxy-6-
[[(4-ethyl-2-piperidinyl)carbonyl]amino]-l-thio-L-threo-alpha-D-galacto-octopyranoside.
Rats
A group of six male and six female Sprague Dawley rats was given [14C]pirlimycin hydrochloride in aqueous solution at a dose of 29 mg/kg bw per day by gavage. The animals were treated for 5 days and sacrificed 2-4 h after the last dose. Housing was in individual metabolism cages and urine and faeces were collected at intervals of 24 h, from 48 h before treatment until the time of sacrifice. Approximately 88% of the total administered dose was recovered, with about 5-6% in the urine, 60% in the faeces and 21% in the contents of the gastrointestinal tract. The highest tissue concentrations were found in the liver, followed by kidney, muscle and fat. There were no significant differences between the sexes. Analysis of liver residues revealed that 57-76% was unchanged pirlimycin and 21-42% was pirlimycin sulfoxide. The extent of biotransformation appeared to be higher in males. The same two compounds are found in the livers of treated cows. Most of the radiolabel in the urine and faeces was also present as pirlimycin and the sulfoxide metabolite (Nappier et al., 1989).
Cattle
Holstein cows in mid-lactation were given two doses of [14C]pirlimycin hydrochloride by intramammary infusion, with an interval of 24 h between doses. Peak concentrations of radiolabel in blood were reached within 6-12 h, indicating slow transfer from the udder into blood. Approximately half of the administered dose was excreted in the milk, primarily as the parent compound. Limited analysis suggested that in plasma the drug was present in the form of pirlimycin. Liver residues were present as two isomers of pirlimycin sulfoxide (62%), pirlimycin sulfone (10%) and unchanged pirlimycin (25%). The average recovery in excreta was about 10% in the urine and 24% in the faeces. In the urine, 80% of the drug was in the form of pirlimycin and 8% was pirlimycin sulfoxide. In faeces, pirlimycin comprised 45% of the drug substance, 1.5% was pirlimycin sulfoxide, and 50% was a mixture of pirlimycin-adenylate, pirlimycin-uridylate and pirlimycin sulfoxide-adenylate. The nucleotide adducts appear to be formed through the action of microflora in the intestinal tract (Hornish et al., 1992a, 1992b).
Humans
A group of five male volunteers was given oral doses of pirlimycin hydrochloride in capsules. Each man received single doses of 50, 125, 250 and 500 mg, with intervals of 7 days between each treatment. A control group was given a placebo capsule. The drug could not be detected in the serum after the lowest dose and concentration of the drug showed a peak at 2-4 h after the other doses. Mean concentrations were 0.11, 0.23 and 0.62 µg/ml at 125, 250 and 500 mg respectively. The recovery of the drug amounted to 2.8-6.9% in the urine within 24 h, and 29-34% in the faeces within 72 h (Gerard et al., 1982; Brown et al., 1983).
Groups of five men received a single oral dose of pirlimycin hydrochloride of 125 mg in capsules or as a solution in water. Peak plasma concentrations were reached within a few hours, but levels were low, with mean concentrations of 0.11 and 0.18 µg/ml for the groups receiving capsules and solution, respectively. Urinary excretion over 48 h accounted for 4.4% of the dose administered in capsules and 7.3% of the dose administered in solution (Brown & Allen, 1983).
The results of studies of the acute toxicity of pirlimycin are summarized in Table 1. All doses were given as aqueous solutions. Signs of toxicity included dose-related depression in both mice and rats, convulsions in mice and diarrhoea and gastrointestinal irritation in rats.
Table 1. Acute toxicity of pirlimycin hydrochloride
|
Species |
Strain |
Sex |
Route |
LD50 |
Reference |
|
Mouse |
ICR |
Five males and five females |
Intraperitoneal |
537 |
Larsen & Gray (1978) |
|
Mouse |
ICR |
Five males and five females |
Intraperitoneal |
647 |
Larsen & Gray (1978) |
|
Mouse |
B6C3F1 |
Five males and five females |
Intraperitoneal |
616 |
Larsen et al. (1979) |
|
Rat |
Sprague-Dawley |
Five males |
Gavage |
2524 |
Larsen & Gray (1979) |
|
Rat |
Sprague-Dawley |
Five males |
Gavage |
>5000 |
Jackson et al. (1991) |
Rats were given single doses of pirlimycin hydrochloride only or pirlimycin hydrochloride in combination with the 7R-hydroxy degradation product (the hydroxy group (OH) replaces the chloride (Cl)) by gavage. There was no increase in toxicity above that for pirlimycin only when animals were given pirlimycin hydrochloride plus 7R-hydroxy pirlimycin at 1250 mg/kg bw at a total dose of up to 2500 mg/kg bw (Jackson et al., 1991).
Pirlimycin hydrochloride was applied onto the cornea and into the conjunctival sac of both eyes of male New Zealand white rabbits. One rabbit received a single application of 100 mg per eye, the other animal received 20 mg per eye per day for 3 days. One eye in each rabbit was rinsed, 30 s after instillation of the drug. In both cases, severe conjunctival swelling, ocular discharge, iridial capillary injection and corneal opacity and pitting, lasting for 14-21 days, developed in the unrinsed eye. Rinsing reduced the severity of the irritation and the eyes appeared normal within 7 days (Sabaitis et al., 1987a).
Pirlimycin hydrochloride was applied as a paste in water, under an occlusive dressing, onto intact and abraded skin sites of two male New Zealand white rabbits. One rabbit received a single dose of 500 mg applied to each site for 24 h, the other animal received 100 mg per site per day for 5 days. The intact sites appeared normal throughout the study. The abraded sites in both rabbits exhibited moderate erythema and slight oedema for 2-4 days. Both rabbits developed diarrhoea and died on day 4 and 7, respectively, suggesting that there was considerable dermal absorption of pirlimycin. Gross necropsy revealed black/brown pinpoints in the glandular mucosa of the stomach and fluid-filled caecum (Sabaitis et al., 1987b).
Rats
Groups of five male and five female Sprague-Dawley rats were given pirlimycin hydrochloride (in aqueous solution) at a dose of 0 or 500 mg/kg bw per day for 14 days by gavage. Treated females gained more body weight than did controls during the study. Haematology parameters were unaffected. Absolute and relative liver weights were slightly lower in treated males and females. At necropsy, a single streak of congestion in the fundic mucosa of the stomach was present in all treated rats and three out of ten of the controls. Microscopic examination of tissues in two rats of each sex per group revealed evidence of mild inflammatory changes in the non-glandular and fundic gastric mucosa of treated rats, which were suggestive of gastric irritation (Gray et al., 1979).
Groups of 10 male and 10 female Sprague-Dawley rats were given pirlimycin hydrochloride (in aqueous solution) at a dose of 0, 50, 160 or 500 mg/kg bw per day for 30 days by gavage. Measurable levels of antibiotic activity were found in serum, liver and kidney from each treated group. There were no meaningful effects on body-weight gain or haematology. The serum activities of aspartate transaminase and alanine transaminase were slightly higher in all treated groups but there was no dose-response relationship. Adrenal gland weights were elevated in males at 500 mg/kg bw per day. Data on pathology for individual animals were not provided. It was stated that mild inflammatory foci were present in the glandular and non-glandular linings of the stomach in several animals at 500 mg/kg bw per day, and in one rat at 160 mg/kg bw per day. A few myeloid figures and a slight increase in lysosomes in hepatocytes were also found at 500 mg/kg bw per day. Histopathological examination was not performed on tissues of the animals given a dose of 50 mg/kg bw per day and therefore a no-observed-effect level (NOEL) could not be identified (Gray et al., 1981b).
Groups of 20 male and 20 female Sprague-Dawley rats were given pirlimycin hydrochloride (in aqueous solution) at a dose of 0, 10, 30, 100 or 300 mg/kg bw per day for 91 days by gavage. There were no clinical signs of toxicity and no effects on body weight, ophthalmology or urine analysis. Mean corpuscular volume and mean corpuscular haemoglobin were slightly increased in males at >30 mg/kg bw per day. Statistically significant decreases in serum concentrations of total protein, albumin and globulin were recorded in males at >30 mg/kg bw per day, and blood urea nitrogen was decreased in both sexes at 300 mg/kg bw per day. Liver weight was decreased in males at >30 mg/kg bw per day, and kidney weights were decreased in females at 300 mg/kg bw per day. These organ weight changes were not associated with any pathological alterations. The NOEL was 10 mg/kg bw per day (MacKenzie, 1988).
Dogs
Groups of two male and two female beagle dogs were given capsules containing pirlimycin hydrochloride at a dose of 30, 100 or 300 mg/kg bw per day, administered orally as half doses, twice daily, for 30 days. The control group received capsules containing lactose at a dose of 300 mg/kg bw per day. Both females given pirlimycin hydrochloride at a dose of 300 mg/kg bw per day showed frequent vomiting and excessive salivation, and lost 13% and 24% of their body weight. One of these animals was sacrificed after 17 days owing to poor condition. The serum activities of aspartate transaminase and alanine transaminase were elevated in the three survivors of the group at 300 mg/kg bw per day. The results of haematology, urine analysis and measurement of organ weights were similar between groups. Pathological examination revealed changes in the liver consisting of dose-related centrilobular hydropic degeneration and an increase in lysosomes in some dogs at 100 mg/kg bw per day and in all dogs at 300 mg/kg bw per day. Electron microscopy of liver sections identified moderate to large numbers of myeloid bodies in the hepatocytes of these affected animals. The NOEL was 30 mg/kg bw per day (Gray et al., 1981a).
Groups of five male and five female beagle dogs were fed capsules containing pirlimycin hydrochloride at a dose of 0, 4, 16, 40 or 160 mg/kg bw per day for 92 days. Increased salivation and vomiting were observed at 40 and 160 mg/kg bw per day. There were no effects on ophthalmological examination, body weights, haematology, urine analysis, or organ weights. Serum activities of aspartate transaminase and alanine transaminase were slightly elevated at 160 mg/kg bw per day, but liver pathology was unremarkable. Areas of inflammation and lymphoid hyperplasia were more severe in females at 40 and 160 mg/kg bw per day, suggesting gastric irritation. The NOEL was 16 mg/kg bw per day (Jackson et al., 1989).
A battery of tests was conducted to evaluate the genotoxicity of pirlimycin. The results are summarized in Table 2.
Table 2. Results of studies of genotoxicity with pirlimycin
|
End-point |
Test object |
Concentration or dose |
Results |
Reference |
|
In vitro |
||||
|
Reverse mutationa |
S. typhimurium strains |
250, 500, 1000 and 2000 µg/plate ±S9 |
Negative |
Mazurek & Swenson (1983) |
|
Reverse mutationb |
S. typhimurium strains |
625, 1250, 2500 and 5000 µg/plate ±S9 |
Negative |
Aaron & Mazurek (1989) |
|
Forward mutationc |
Mammalian V79 cells |
0.4, 0.8 and 1.6 µg/ml ±S9; 0.25, 0.5 and 1.0 µg/ml -S9 |
Negative |
Harbach et al. (1984) |
|
Forward mutationd |
Chinese hamster ovary cells (As52/Xprt and Hprt loci) |
50, 100, 250, 500, 1000 and 1500 µg/ml +S9; |
Negative |
Stankowski (1990) |
|
Unscheduled DNA synthesise |
Primary hepatocytes from Sprague-Dawley rats |
0.03, 0.1, 0.3 and 1 µ g/ml |
Negative |
Trzos & Swenson (1984) |
|
Unscheduled DNA synthesisf |
Primary hepatocytes from Fischer 344 rats |
1, 3, 10, 30, 100 and 300 µg/ml |
Negative |
Harbach et al. (1989) |
|
Sex-linked recessive lethal mutationsg |
Drosophila melanogaster |
1000 and 7500 mg/kg in feeding solution for 3 days |
Negative |
Foureman (1990) |
|
In vivo |
||||
|
Micronucleus formationh |
Bone-marrow cells from CD-1 mice |
Single intraperitoneal doses of 175, 250 and 375 mg/kg bw |
Negative |
Sorg (1989) |
|
Micronucleus formationi |
Bone-marrow cells from Sprague-Dawley rats |
Two intraperitoneal doses of 50, 100 and 200 mg/kg bw, 24 h apart |
Negative |
Trzos et al. (1984) |
|
S9, 9000 × g supernatant fraction from rodent liver. |
|
|
a |
Positive controls were: 2-acetylaminofluorene for TA98, TA10s0 and TA1538, in both the presence and absence of S9; cyclophosphamide for TA1535, in both the presence and absence of S9; 9-aminoacridine for TA1537, in both the presence and absence of S9. |
|
b |
Positive controls were: 2-aminoanthracene for all strains, in the presence of S9; dexon for TA97; 2-nitrofluorene for TA98 and TA100; cumene hydroperoxide for TA102; sodium azide for TA1535, in the absence of S9. |
|
c |
Positive controls were: 7,12-dimethylbenz(alpha)anthracene in the presence of S9, and 1-methyl-1-nitrosourea in the absence of S9. |
|
d |
Positive controls were: dimethylnitrosamine in the presence of S9; and ethylmethane sulfonate in the absence of S9. |
|
e |
Positive control was ultraviolet light. |
|
f |
Positive control was 2-acetylaminofluorene. |
|
g |
Positive control was dimethylnitrosamine. |
|
h |
Positive control was triethylenemelamine. |
|
1 |
Positive control was cyclophosphamide. |
(a) Multigeneration studies
Rats
Groups of 30 male and 30 female Sprague-Dawley rats were given pirlimycin hydrochloride (in aqueous solution) at a dose of 0, 100, 200 or 400 mg/kg bw per day by gavage. Males were treated from 60 days before mating and during the mating period, while females were treated from 14 days before mating until 21 days after giving birth. One male and one female F1 pup per litter were selected at weaning to produce the F2 generation and were dosed according to the schedule for the F0 generation. All females were allowed to deliver and rear the pups for 21 days after giving birth. In F0 adults, salivation and urogenital/anogenital staining were increased at 200 and 400 mg/kg bw per day and nasal discharge and irritability were increased at 400 mg/kg bw per day. Salivation, nasal discharge and urogenital staining were also observed in F1 adults at 400 mg/kg bw per day. Reduced body-weight gains were evident in both generations of adult males at 400 mg/kg bw per day. The number of F1 dams producing litters was reduced at 200 and 400 mg/kg bw per day, but the frequency was at the lower end of the rane for historical controls for the laboratory. Other fertility and reproductive performance parameters, fetal development, and growth and survival of offspring were unaffected. The NOEL was 100 mg/kg bw per day (Black et al., 1988).
(b) Developmental toxicity
Mice
In two replicate experiments, groups of 43-44 presumed pregnant CD-1 mice were given pirlimycin hydrochloride (in aqueous solution) at a dose of 0, 100, 400 or 1600 mg/kg bw per day by gavage. The animals were treated on days 6-15 of gestation and killed on day 18 of gestation. Diarrhoea and/or soft stools were observed at 1600 mg/kg bw per day. The death of two mice and the early sacrifice due to morbidity in one dam at this dose were associated with these gastrointestinal disturbances. The pregnancy rate was poor and only 28, 23, 23 and 15 litters were available for examination in the control group and in the three groups given increasing drug doses, respectively. There were no effects on body-weight gain in the dams; however fetal body weights were lower at 1600 mg/kg bw per day. Embryonic and fetal development were unaffected. The NOEL for maternal and foetal toxicity was 400 mg/kg bw per day (Marks & Black, 1994).
Rats
Groups of 24 presumed pregnant Sprague-Dawley rats were given pirlimycin hydrochloride (in aqueous solution) at a dose of 0, 200, 400 or 800 mg/kg bw per day by gavage. All dams were treated on days 6-15 of gestation and were killed on day 20 of gestation. Soft stools, post-dosing salivation and urogenital staining were observed at 400 and 800 mg/kg bw per day, with lower body-weight gain at the highest dose. The mean number of implantations was lower at 400 and at 800 mg/kg bw per day, but since implantation would have occurred before dosing, this finding is unlikely to be related to treatment. Resorptions were increased at 800 mg/kg bw per day, primarily due to the resorption of one entire litter. The incidence of incompletely ossified sternebrae was slightly increased in all treated groups, but was within the range for historical controls. Absent or delayed ossification of other skeletal areas was similar in all groups or was higher in controls, in some cases. The NOEL for maternal toxicity was 200 mg/kg bw per day and embryo- and fetal development were not affected at any dose (Terry et al., 1989).
Pirlimycin has been tested for microbiological activity against a range of bacterial species in pure culture. The results obtained in a number of reports have been reviewed and the findings for isolates that are representative of those typically found in the human intestinal microflora are summarized in Table 3. Microbiological activity was measured using bacterial suspensions at cell densities of 105 colony-forming units (CFU)/ml. The most sensitive organisms were B. fragilis, Peptococcus spp., Fusobacterium spp. and Clostridium spp. (Kotarski, 2003).
Table 3. Summary of MIC50s of pirlimycin against major bacterial groups (clinical isolates)
|
Bacterial species |
No. of strains |
MIC50 (mg/ml) |
MIC90 (mg/ml) |
|
Enterococcus spp. |
29 |
>16.000 |
>16 |
|
B. fragilis |
66 |
0.125 |
4 |
|
B. fragilis group |
64 |
0.25 |
1 |
|
Bacteroides spp. |
72 |
0.25 |
1 |
|
Peptococcus spp. |
50 |
0.125 |
1 |
|
Peptostreptococcus spp. |
22 |
0.500 |
1 |
|
Fusobacterium spp. |
19 |
0.125 |
1 |
|
Clostridium spp. |
18 |
0.125 |
4 |
|
Eubacterium spp. |
18 |
1.000 |
4 |
From Kotarski (2003).
MIC, minimum inhibitory concentration at which 50% or 90% of isolates are inhibited.
Minimum inhibitory concentrations (MIC) for pirlimycin were determined against pure cultures of 102 strains of bacteria distributed among 10 genera that are considered to be predominant human intestinal microflora. The activity was measured using bacterial suspensions at cell densities of 108CFU/ml and 1010CFU/ml, densities that are closer to those of cells in faecal flora (1011 CFU/ml). The results are summarized in Table 4. A strong inoculum effect was not noted since most MIC values were similar at both cell densities. The lowest MIC50 at the higher inoculum level was 0.12 µg/ml for Bifidobacterium and Peptococcus/Peptostreptococcus. Enterococci were generally resistant and none of the strains of Escherichia coli were inhibited (Thurn et al., 1995).
Table 4. MIC50s of pirlimycin against predominant human intestinal microflora
|
Bacterial species/genus |
No. of isolates |
Density of innococulum |
|
|
×108 |
×1010 |
||
|
Bacteroides |
15 |
0.25 |
0.25 |
|
Bifidobacterium |
13 |
0.03 |
0.12 |
|
Eubacterium |
10 |
0.25 |
0.50 |
|
Fusobacterium |
6 |
0.06 |
0.50 |
|
Peptococcus and Peptostreptococcus |
16 |
0.06 |
0.12 |
|
Clostridium |
8 |
1.00 |
2.00 |
|
Lactobacillus |
11 |
0.50 |
2.00 |
|
Enterococcus |
10 |
8.00 |
16.00 |
|
Escherichia coli |
13 |
>128.00 |
>128.00 |
MIC50, minimum inhibitory concentration at which 50% of isolates are inhibited.
The microbiological activities of pirlimycin and its sulfoxide metabolite were tested against pure cultures of anaerobic bacteria, Bifidobacterium spp. (15 strains), Eubacterium spp. (13 strains) and Bacteroides fragilis (two strains). At the standard inoculum density (105CFU/ml), pirlimycin exhibited a MIC50 of 0.06 µg/ml for Bifidobacterium spp. and Eubacterium spp. and MIC90s of 0.13 and 2 µg/ml for these genera, respectively. The MICs of pirlimycin against the two Bacteroides fragilis strains were 0.13 and 0.25 µg/ml. Pirlimycin sulfoxide was at least 13-fold less active than pirlimycin against the tested strains (Kennedy et al., 1991). Using an inoculum size (1010-1011 CFU/ml) that more closely resembled the bacterial populations in the human gastrointestinal tract revealed considerably higher MICs. For both compounds, the MIC50 and the MIC90 was >128 µg/ml for each genus tested. On the basis of the individual MIC values, pirlimycin was more active than the sulfoxide (Watts et al., 1992).
The microbiological activities of pirlimycin and a number of its metabolites were compared against pure cultures of a variety of bacterial genera commonly found in the environment. On the basis of MICs, pirlimycin sulfoxide was at least an order of magnitude less active than the parent compound. No microbiological activity was shown by the adenylate metabolite or by three other uncharacterized metabolites (Yancey et al., 1990; Yein, 1989).
Pure broth cultures of anaerobic bacteria (107-109CFU/ml) were incubated with pirlimycin hydrochloride at a concentration of 0, 3 or 6 µg/ml for 6 or 12 h. The genera tested were Bacteroides spp. (six strains), Bifidobacterium spp. (four strains), Clostridium spp. (six strains), Eubacterium spp. (five strains), Fusobacterium prausnitzii (six strains), Lactobacillus spp. (five strains) and Peptococcus/Peptostreptococcus spp. (four strains). A decrease in cell numbers of greater than 1 × log10 was regarded as evidence for a reduction in the viability of the organism. According to this criterion, a total of three of the 36 strains showed decreased viability—one strain of Bacteroides at 6 µg/ml and two strains of Fusobacterium at 3 and 6 µg/ml (Greening et al., 1995).
A single subcutaneous injection of antibiotics at various doses was administered to hamsters in a model of antibiotic-associated pseudomembranous colitis. Treatment with the antibiotic was preceded by oral administration of Clostridium difficile as the colitis-producing agent. The animals were observed for 2 weeks, after which the rate of mortality was used to estimate the dose of antibiotic required to induce death, presumably due to pseudomembranous colitis. The dose calculated to induce colitis in 50% of the hamsters (CID50) was 2.6 mg/kg for pirlimycin, 4 mg/kg for clindamycin and 2.9 mg/kg for lincomycin (Stapert et al., 1983, 1991).
Five male volunteers each received single oral doses of pirlimycin hydrochloride at 50, 125, 250 and 500 mg in capsules. The doses were administered in escalating order, with 7 days between treatments. Five men were given placebo capsules containing starch. Soft stools were noted frequently in one treated man. Clostridium difficile was found in stool samples of two, four, five and three men during the week after treatment with 50, 125, 250 and 500 mg, respectively. C. difficile toxin was consistently detected in the stools of one subject subsequent to the dose at 125 mg. In comparison, there was a single instance of C. difficile found in the stools of a control subject. There were no meaningful effects on haematology, serum chemistry and urine analysis parameters (Brown et al., 1983; Gerard et al., 1982).
The Committee considered the results of studies on pharmacokinetics and metabolism, acute and short-term toxicity, genotoxicity, reproductive and developmental toxicity, microbiological effects, and studies in humans. Most of the studies of toxicity were carried out according to appropriate standards for study protocol and conduct. The studies of acute toxicity reported in the 1970s were conducted before the requirement for compliance with good laboratory practice and have no assurance of quality.
Pirlimycin, when administered orally as pirlimycin hydrochloride, appears to be poorly absorbed in rats. Approximately 5-6% of a radiolabelled dose was excreted in the urine and >80% was recovered in the faeces and gastrointestinal contents. Most of the radioactivity was present as pirlimycin and pirlimycin sulfoxide, the same compounds as found in the livers of treated cows. Nucleotide adducts of pirlimycin and pirlimycin sulfoxide, formed by the activity of intestinal microflora, were detected in cows' faeces. These metabolites were not detected in rats, but because they are not found in edible tissues, they are not relevant for human risk assessment. Similarly, the bioavailability of pirlimycin in humans given an oral dose of pirlimycin hydrochloride appeared to be low.
The acute oral toxicity of pirlimycin hydrochloride was low. Signs of toxicity included depression, diarrhoea and gastrointestinal irritation. In rabbits, local application of pirlimycin hydrochloride resulted in severe ocular irritation and moderate dermal irritation at skin sites that had been abraded.
In a preliminary study, rats received pirlimycin hydrochloride at a dose of 0 or 500 mg/kg bw per day by gavage for 14 days. The treated group had lower liver weights and inflammatory changes in the gastric mucosa that were suggestive of irritation.
Rats were given pirlimycin hydrochloride at a dose of 0, 50, 160 or 500 mg/kg bw per day by gavage for 30 days. Serum activities of aspartate transaminase and alanine transaminase were slightly higher in all treated groups, but the increases were not proportional to the dose. At 500 mg/kg bw per day, a few myeloid bodies and an increase in lysosomes were found in hepatocytes. Inflammatory foci were present in the lining of the stomach in a few animals at 500 mg/kg bw per day and in one rat at 160 mg/kg bw per day. Since histopathology was not performed for animals treated with a dose of 50 mg/kg bw per day, a NOEL could not be identified.
Groups of rats were given pirlimycin hydrochloride at a dose of 0, 10, 30, 100 or 300 mg/kg bw per day by gavage for 91 days. In males receiving a dose of >30 mg/kg bw per day, there were decreases in serum concentrations of total protein, albumin and globulin, and in liver weight. At 300 mg/kg bw per day, blood urea nitrogen was decreased in both sexes and kidney weights were lower in females. The NOEL was 10 mg/kg bw per day.
Groups of two males and two females were given pirlimycin hydrochloride in capsules at a daily dose of 0, 30, 100 or 300 mg/kg bw per day for 30 days. Both females in the group receiving 300 mg/kg bw per day showed frequent vomiting, excessive salivation and lost body weight, leading to the sacrifice of one female after 17 days. Serum activities of aspartate transaminase and alanine transaminase were elevated in the survivors at 300 mg/kg bw per day. Three dogs at 100 mg/kg bw per day and all dogs at 300 mg/kg bw per day exhibited liver changes consisting of centrilobular hydropic degeneration, and increases in lysosomes and myeloid bodies in the hepatocytes. The NOEL was 30 mg/kg bw per day.
Dogs were given pirlimycin hydrochloride in capsules at a dose of 0, 4, 16, 40 or 160 mg/kg bw per day for 92 days. Salivation and vomiting were increased at 40 and 160 mg/kg bw per day and serum activities of aspartate transaminase and alanine transaminase were elevated at 160 mg/kg bw per day. Inflammation and lymphoid hyperplasia of the stomach, which were suggestive of gastric irritation, were more severe in females given a dose of 40 and 160 mg/kg bw per day. The NOEL was 16 mg/kg bw per day.
Assays covering an appropriate range of genotoxic end-points were conducted with pirlimycin hydrochloride in vitro and in vivo. The results of all the assays were negative and the Committee concluded that pirlimycin does not pose a genotoxic hazard.
Long-term studies were not carried out and there are thus no data available on the carcinogenic potential of pirlimycin. However, the drug has no genotoxic potential, is not chemically related to known carcinogens and, in short-term studies, causes no changes that are likely to progress to neoplasia. The Committee therefore concluded that pirlimycin is unlikely to pose a carcinogenic risk, and that studies of carcinogenicity were not necessary.
In a two-generation study of reproductive toxicity in rats, pirlimycin hydrochloride was administered at a dose of 0, 100, 200 or 400 mg/kg bw per day by gavage. Clinical signs of toxicity in adult animals included salivation, nasal discharge and urogenital/anogenital staining at doses of 200 and 400 mg/kg bw per day. The numbers of F1 dams producing a litter were reduced at 200 and 400 mg/kg bw per day, but the frequencies were at the lower end of the range for historical controls. Other fertility and reproduction parameters and the development of offspring were unaffected. The NOEL was 100 mg/kg bw per day.
Developmental toxicity was investigated in mice given pirlimycin hydrochloride at a dose of 0, 100, 400 or 1600 mg/kg bw per day by gavage on days 6-15 of gestation. Diarrhoea and soft stools were noted in dams at 1600 mg/kg bw per day. At this dose, fetal body weight was lower, but development was unaffected. Pirlimycin was not teratogenic in mice. The NOEL for maternal and fetal toxicity was 400 mg/kg bw per day.
In a study of developmental toxicity in rats, pirlimycin hydrochloride was administered at a dose of 0, 200, 400 or 800 mg/kg bw per day by gavage on days 6-15 of gestation. Soft stools, salivation and urogenital staining were observed at 400 and 800 mg/kg bw per day and body-weight gain was lower at 800 mg/kg bw per day. Fetal development was not affected at any dose. Pirlimycin was not teratogenic in rats. The NOEL for maternal toxicity was 200 mg/kg bw per day.
Microbiological activity
Pirlimycin hydrochloride has been tested for its inhibitory activity against microorganisms representative of the human colonic microflora. The most sensitive species were Bifidobacterium spp. and Peptococcus/Peptostreptococcus spp., with a MIC50 of 0.12 µg/ml at an inoculum density of 1010CFU/ml. In another experiment, strains of anaerobic bacteria at cell densities of 107-109CFU were incubated with pirlimycin hydrochloride. Of the 36 strains tested, only three showed evidence of decreased viability at drug concentrations of 3 and 6 µg/ml. Pirlimycin sulfoxide and other unidentified metabolites showed less or no activity against anaerobic bacteria.
In a model of pseudomembranous colitis in hamsters, single subcutaneous injections of pirlimycin hydrochloride were administered after oral treatment with Clostridium difficile as the colitis-producing agent. It was estimated that a dose of 2.6 mg/kg bw induced death in 50% of the treated animals.
Human volunteers received single oral doses of pirlimycin hydrochloride of 0, 50, 125, 250 and 500 mg, at weekly intervals. Clostridium difficile was found in the stool samples of two, four, five and three persons after each dose of the drug, and in one person in the control group. There were no effects on haematology, serum chemistry and urine analysis parameters.
A decision-tree for evaluating the potential effect of veterinary drug residues on human intestinal microflora was developed by the Committee at its fifty-second meeting (Annex 1, reference 140). At its present meeting, the Committee used the decision-tree to answer the following questions in its assessment of pirlimycin:
1. "Does the ingested residue have antimicrobial properties?"
Yes. Pirlimycin is a lincosamide that is active against Gram-positive aerobic and anaerobic cocci (eg. staphylococci, streptococci, peptostreptococci) as well as Gram-negative anaerobes. Pirlimycin is the main residue in the milk of treated cows and pirlimycin sulfoxide is the main residue in the liver. The microbiological activity of pirlimycin sulfoxide is significantly less than that of pirlimycin.
2. "Does the drug residue enter the lower bowel?"
Yes. Pirlimycin appears to be poorly absorbed in humans. Up to 34% of an oral dose was recovered in human faeces. However, since total recovery of the administered dose was only 40%, the fraction of the dose entering the colon is likely to be higher. A conservative estimate of the bioavailability of pirlimycin in the gastrointestinal tract would be 50%.
3. "Is the ingested residue transformed irreversibly to inactive metabolites by chemical transformation, metabolism mediated by the host or intestinal microflora in the bowel and/or by binding to intestinal contents."
No. While there is no specific information on the nature of the substances excreted in human faeces, significant microbiological activity has been detected. Therefore, it is assumed that microbiological activity is likely to be present in the human gastrointestinal tract.
4. "Do data on the effects of the drug on the colonic microflora provide a basis to conclude that the ADI derived from toxicological data is sufficiently low to protect the intestinal flora?"
No. A number of studies have demonstrated the potential for adverse effects of pirlimycin on the intestinal microflora. Studies of toxicity have not identified adverse findings at low oral doses and thus would not be expected to provide adequate protection for the intestinal flora.
5. "Do clinical data from the therapeutic use of the class of drugs in humans or data from in vitro or in vivo model systems indicate that effects could occur in the gastrointestinal tract?"
Yes. Gastrointestinal effects (nausea, vomiting, abdominal cramps, diarrhoea) are the most commonly reported adverse reactions to the therapeutic use of lincosamides (clindamycin and lincomycin) in humans. Pirlimycin has not been used as a therapeutic agent in humans. However, limited experimentation in humans has revealed soft stools and overgrowth of Clostridium difficilein treated persons.
6. "Determine the most sensitive adverse effect(s) of the drug on the human intestinal microflora."
The available data indicate that oral exposure to pirlimycin is associated with disruption of the colonization barrier, rather than emergence of resistance. Other lincosamides are widely used in human medicine and also result in disruption of the colonization barrier. There are no studies available on the likely emergence of resistance to pirlimycin. In a study of the magnitude of and trends in the development of bacterial resistance to lincosamides, the pattern of susceptibility of human isolates of Gram-positive aerobic and anaerobic bacteria changed little over a 12-year period (1971-1983). It was concluded that disruption of the colonization barrier is the most appropriate end-point for the determination of a microbiological acceptable daily intake (ADI).
7. "If disruption of the colonization barrier is the concern, determine the MIC of the drug against 100 strains of predominant intestinal flora and take the geometric mean MIC of the most sensitive genus or genera to derive an ADI using the formula discussed at the forty-seventh meeting of the Committee (Annex 1, reference 125). Other model systems may be used to establish a no-observed-effect concentration to derive an ADI."
Using all relevant data acquired in studies conducted in vitro and in vivo, the Committee considered that the study in humans was the most appropriate to use in determining a microbiological ADI. In this study, a single oral dose of 50 mg of pirlimycin caused a minor change in the intestinal microflora. A safety factor of 10 was used to address variability between human subjects. Because of the limited nature of the human studies (single oral doses, administration in males only, small sample size) the Committee considered that an additional safety factor of 10 was necessary, resulting in an overall safety factor of 100.
|
Upper limit of ADI |
= |
NOEL |
|
SF × bw |
||
|
= |
50mg |
|
|
100 × 60 kg bw |
||
|
= |
8.3 µg/kg bw |
where:
bw = body weight:
SF = safety factor
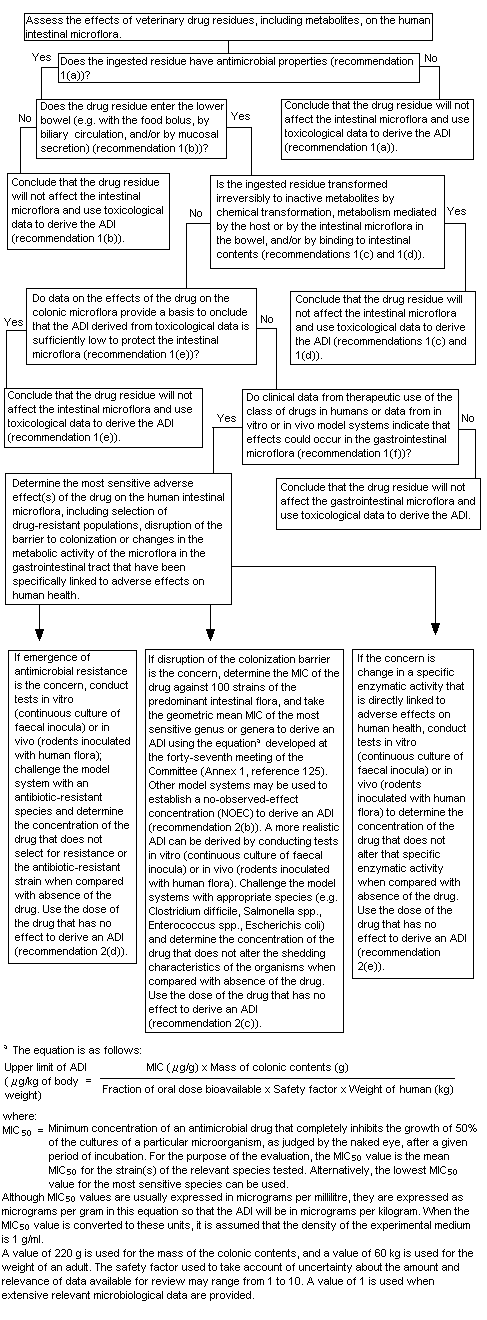
Figure 2. Decision-tree for determining adverse microbiological effects of residues of antimicrobial drugs in food-producing animals
The most appropriate study to use in determining a toxicological NOEL was the 91-day study in rats. Hence, the NOEL for pirlimycin was 10 mg/kg bw per day on the basis of serum biochemical changes. As there were no long-term studies in animals on the toxicological effects of pirlimycin after prolonged exposure in the diet, an extra safety factor in addition to the usual safety factor of 100 was considered necessary. The Committee used a safety factor of 10 to account for the absence of a long-term study, leading to an overall safety factor of 1000. An ADI of 0-10 µg/kg bw was established on the basis of toxicological data.
The Committee noted that pirlimycin belongs to the class of lincosamide antibiotics that are known to be associated with perturbation of the gastrointestinal microflora following administration in humans. The ADI based on MIC data was considered to be extremely conservative considering the findings in a hamster model of pseudomembranous colitis and in humans. Therefore, the Committee established a microbiological ADI of 0-8 µg/kg bw on the basis of the NOEL for gastrointestinal effects in humans. As is its usual practice, the Committee rounded the value of the ADI to one significant figure.
Aaron, C.S. & Mazurek, J.H. (1989) Evaluation of U-57,930E pirlimycin in the Salmonella/microsome test (Ames assay). Unpublished report No. 7227/89/030 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Black, D.L., Marks, T.A., Shaw, C.I., Morris, D.F., Terry, R.D. & Poppe, S.M. (1988) A two-generation reproduction study (oral) in rats given U-57,930E. Unpublished report No. 7227/88/010 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Brown, R.J. & Allen, H.R. (1983) Single-dose oral bioavailability comparison of pirlimycin HCl (U057930E) capsule formulation and oral solution with clindamycin HCl capsules. Unpublished report No. 7254/83/043 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Brown, R.J., Allen, H.R. & Hornbeck, L.S. (1983) Single-dose placebo-controlled tolerance and ADME study of oral pirlimycin HCl (U-57930E) compared to oral clindamycin HCl at four dose levels: Protocol 4601. Unpublished report No. 7254/83/042 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Foureman, P. (1990) Drosophila melanogaster sex-linked recessive lethal assay of U-57,930E. Unpublished report of Project No 126 from the Zoology Department, University of Wisconsin, Madison, WI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Gerard, G.C., Lummis, W.L., Hall, L.T., Patel, R.K. & Spiro, T.E. (1982) The analysis of pirlimycin and clindamycin concentrations in human serum, urine and feces for infectious disease protocol #4601. Unpublished report No. 7262/82/7262/027 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Gray, J.E., Larsen, E.R. & Weaver, R.N. (1979) U-57,930E; 14-day oral tolerance in the white rat. Unpublished report No. 7254/79/7263/009 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Gray, J.E., Larsen, E.R. & Weaver, R.N. (1981a) U-57,930E; 30-day oral toxicity test in the dog. Unpublished report No. 7254/81/7263/004 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Gray, J.E., Weaver, R.N., Larsen, E.R. & Platte, T.F. (1981b) U-57,930E, 30-day oral toxicity test in the rat. Unpublished report No. 7254/81/7263/010 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Greening, R.C., Baker, K.D. & Kotarski, S.F. (1995) Effect of pirlimycin on the viability of anaerobic bacterial species found in the human gastrointestinal tract. Unpublished report No. 782-7922-95-002 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Harbach, P.R., Johnson, M.A. & Swenson, D.H. (1984) The V79 mammalian cell mutation assay with pirlimycin (U-57,930E) with and without an S9 metabolic activation system. Unpublished report No. 7268/84/003 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Harbach, P.R., Wiser, S.K. & Aaron, C.S. (1989) U-57,930E: evaluation in the in vitro unscheduled DNA synthesis (UDS) assay in rat primary hepatocytes. Unpublished report No. 7227/89/024 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Hornish, R.E., Arnold, T.S., Benner, C.P., Cazers, A.R., Callahan, J.K., Chester, S.T., Cox, T.D., Flook, T.F., Hoffman, G.A., Hubbard, V.L., Janose, R.L., Mejeur, R.L. & Pierce, P.A. (1992a) Residue studies of 14C-pirlimycin hydrochloride (U-57,930E) in the lactating dairy cow treated twice in all four quarters at a 24-hour interval with 50 mg/quarter of pirlimycin free base equivalents. Unpublished report No. 782-7926-92-002 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Hornish, R.E., Arnold, T.S., Baczynskyj, L., Chester, S.T., Cox, T.D., Flook, T.F., Janose, R.L., Kloosterman, D.A., Nappier, J.M., Reeves, D.R., Yein, F.S. & Zaya, M.J. (1992b) Pirlimycin in the dairy cow. In: Hutson, D.H., Hawkins, D.R., Paulson, G.D. & Struble, C.B., eds. Xenobiotics and Food Producing Animals, ACS Symposium Series #503. American Chemical Society, Washington DC, pp 132-147.
Jackson, T.A., Cypher, J.J. & Brussee, D.M. (1991) U-57930E: Acute single dose oral toxicity study with a degradation product of pirlimycin (U-62113 [7R-OH-pirlimycin]) in male Sprague-Dawley rats. Unpublished report No. 7220-91-012 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Jackson, T.A., Cypher, J.J., Brussee, D.M. & Mulholland, M.P. (1989) U-57930E; 90-day oral toxicity and drug safety study in the beagle dog. Unpublished report No. 7220-89-006 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Kennedy, M.J., Yancey, R.J., Case, C.A. & Hornish, R.E. (1991) In vitro activity of pirlimycin (U-57,930E) and pirlimycin sulfoxide hydrochloride against Bifidobacterium spp. and Eubacterium spp. from the human gastrointestinal tract. Unpublished report No. 705-7923-91-016 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Kotarski, S.F. (2003) Evaluation of microbiological effects of pirlimycin. Unpublished report from Pfizer Animal Health, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Larsen, E.R. & Gray, J.E. (1978) The LD50 determination of U-57,930E in the mouse. Unpublished report No. 7254/78/7263/009 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Larsen, E.R. & Gray, J.E. (1979) U-57,930E; oral LD50 in the rat. Unpublished report No. 7254/79/7263/010 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Larsen, E.R., Gray, J.E. & Harris, J.H. (1979) The LD50 determination of U-57,930E in B6C3F1 mice. Unpublished report No. 7254/79/7263/001 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
MacKenzie, K.M. (1988) 13-week oral toxicity study in Sprague Dawley rats with U-57,930E. Unpublished report No. HLA 6105-135 from Hazleton Laboratories America, Madison, WI, USA. Upjohn Report No. 7220-88-043. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Marks, T.A. & Black, D.L. (1994) U-57930E: A segment II teratology study (oral) in mice. Unpublished report No. 7224-93-067 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Mazurek, J.H. & Swenson, D.H. (1983) Evaluation of U-57,930E (pirlimicin) in the Salmonella/microsome (Ames assay). Unpublished report No. 7268/83/025 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Nappier, J.M., Hornish, R.E., Kubicek, M.F., Stuart, D.J., Wolthuis, T.L., Reeves, D.R. & Flook, T.F. (1989) Comparative metabolism of pirlimycin hydrochloride (U-57,930E) in rats (oral gavage) and bovine (udder infusion). Unpublished report No. 782-9760-89-001 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Sabaitis, C.P., Leong, B.K.J. & Polderman, E.M. (1987a) Preliminary eye irritation study on pirlimycin hydrochloride (U-57,930E) in albino rabbits. Unpublished report No. 7277/87/043 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Sabaitis, C.P., Leong, B.K.J. & Polderman, E.M. (1987a) Preliminary dermal irritation study on pirlimycin hydrochloride (U-57,930E) in albino rabbits. Unpublished report No. 7277/87/044 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Sorg, R.M. (1989) Micronucleus test (MNT) EPA. Unpublished report of study No. PH 309-UP-006-88 from Pharmakon Research International, Waverly, PA, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Stankowski, L.F. (1990) AS52/XPRT and CHO/HPRT mammalian cell forward gene mutation assays. Unpublished report of study Nos PH 314-UP-004-88 and PH 314-UP-001-89 from Pharmakon Research International, Waverly, PA, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Stapert, D., Hamel, J.C. & Ford, C.W. (1983) Development of a hamster antibiotic-associated colitis model. Unpublished report No. 7254/83/014 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Stapert, D., Hamel, J.C., Lee, J.C., Yancey, R.J. & Ford, C.W. (1991) The hamster model of antibiotic-associated pseudomembranous colitis, an update. Unpublished report No. 7252-91-054 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Terry, R.D., Marks, T.A., Morris, D.F., Black, D.L., Shaw, C.I. & Poppe, S.M. (1989) A segment II teratology study (oral) of U-57,930E in rats. Unpublished report No. 7227/88/129 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Thurn, K.K., Baker, K.D., Greening, R.C. & Kotarski, S.F. (1995) In vitro activity of pirlimycin against bacterial species found in the human gastrointestinal tract. Unpublished report No. 782-7922-95-001 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Trzos, R.J. & Swenson, D.H. (1984) The primary hepatocyte unscheduled DNA synthesis (UDS) assay with U-57,930E, and ultraviolet light. Unpublished report No. 7268/84/020 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Trzos, R.J., Swenson, D.H. & Brown, P.K. (1984) The micronucleus test with U-57,930E (pirlimycin). Unpublished report No. 7268/83/029 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Watts, J.L., Salmon, S.A., Yancey, R.J., Case, C.A. & Kennedy, M.J. (1992) Determination of minimum inhibitory concentrations (MIC) of pirlimycin (U-57,930) and pirlimycin sulfoxide against Eubacterium spp and Bifidobacterium spp. Unpublished report No. 705-7923-92-010 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Yancey, R.J., Kennedy, M.J. & Case, C.A. (1990) Minimal inhibitory concentration (MIC) determinations of pirlimycin (U-57,930E) and its adenylate and sulfoxide derivatives for organisms commonly found in the environment. Unpublished report No. 782-7922-90-001 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
Yein, F.S. (1989) Bioactivity of pirlimycin metabolite. Unpublished memo of 28 March 1989 from The Upjohn Company, Kalamazoo, MI, USA. Submitted to WHO by Pfizer Animal Health, Kalamazoo, MI, USA.
See Also:
Toxicological Abbreviations
PIRLIMYCIN (JECFA Evaluation)