
KRESOXIM-METHYL JMPR 1998
First draft prepared by
K. Fujimori
National Institute of Health Sciences, Tokyo, Japan
Explanation
Evaluation for acceptable daily intake
Biochemical aspects
Absorption, distribution, and excretion
Biotransformation
Toxicological studies
Acute toxicity
Short-term studies of toxicity
Long-term studies of toxicity and carcinogenicity
Genotoxicity
Reproductive toxicity
Multigeneration reproductive toxicity
Developmental toxicity
Special studies
Tumour initiating potential
Tumour promoting potential
Hepatic-cell proliferation
Morphology of hepatic proliferation
Induction of hepatic metabolic enzyme activities
Mechanism of decreased serum enzyme activities
Studies on metabolites
Acute toxicity
Genotoxicity
Comments
Toxicological evaluation
References
Explanation
Kresoxim-methyl, methyl-(E)-2-methoxyimino-2-[2-(2-
methylphenoxymethyl)phenyl] acetate, is a broad-spectrum fungicide and
a member of the strobilurin family, a new class of biologically active
compounds structurally related to strobilurin A, a natural product of
the wood-decaying fungus Strobilurus tenacellus. It is intended for
use as an agricultural spray in the control and treatment of fungal
infections on crops and fruits. Strobilurins are known to bind to the
bcl complex (complex III), one of the oxide reductase proteins of the
electron transport chain in mitochondria. The ester linkage in
kresoxim-methyl is essential for its activity. Kresoxim-methyl was
evaluated for the first time by the present Meeting.
Evaluation for Acceptable Daily Intake
1. Biochemical aspects
(a) Absorption, distribution, and excretion
Kresoxim-methyl labelled with 14C on the phenyl A ring (phenoxy;
radiochemical purity, > 98%) or B ring (phenyl; radiochemical purity,
> 98%) or with 13C on the carbon side-chain was administered to rats
by gavage as a suspension in 0.5% carboxymethyl cellulose (CMC) or
intravenously as a 0.9% saline solution. The design of the study
conformed to good laboratory practice. In groups of five male and five
female rats given [14C-B ring]kresoxim-methyl by gavage at 50 or 500
mg/kg bw, with or without pretreatment with unlabelled
kresoxim-methyl, or [14C-A ring]kresoxim-methyl at a dose of 500
mg/kg bw, the compound was excreted predominantly in faeces. At the
low dose of [14C-B ring]-labelled compound, faecal excretion
represented 65-67% of the administered dose and urinary excretion,
20-28% of the dose within 48 h; less than 1% of the radiolabel was
recovered in urine and faeces at this time. Pretreatment with
unlabelled kresoxim-methyl at the low dose for 14 days did not change
the excretion pattern. At the high dose, faecal excretion represented
80-81% of the dose and urinary excretion, 8-13% within 48 h. The total
radiolabel recovered within 120 h was 97% of the [14C-A ring] and
90-96% of the [14C-B ring], with 62-78% of the A ring and 81% of the
B ring excreted in faeces and 17-33% of the A ring and 9-13% of the B
ring in urine. No radiolabel was detected in exhaled air.
In the groups given the [14C-B ring]-labelled material, peak
concentrations of radiolabel in plasma were reached 0.5-1 h after
dosing at the low dose and 8 h after dosing at the high dose. The
plasma level then declined, with a terminal half-life of 17-19 h at
the low dose and 22-30 h at the high dose. The ratios of the area
under the curve for the high:low dose (10:1) were 2.3 for males and
2.1 for females. Radiolabel concentrations were determined in tissues
0.5, 8, 24, 96, and 120 h after dosing. Except for the
gastrointestinal tract, the highest residual concentration was found
in the liver (0.1 g/g at 120 h and 0.3-1.4 g/g at 24 h after dosing at
50 mg/kg bw). The residual concentrations in other tissues were less
than 0.1 g/g tissue at 120 h after dosing at 50 mg/kg bw. The
concentrations of radiolabel in the tissues were comparable in males
and females, indicating a similar pattern of wide distribution and
elimination.
Groups of five male and five female rats given [14C-B
ring]kresoxim-methyl intravenously as a single dose of 5 mg/kg bw
excreted 49-66% of the radiolabel in urine and 23-48% in faeces within
120 h.
Groups of four male and four female rats with canulated bile
ducts were given the [14C-B ring]-labelled material as a single oral
dose of 50 or 500 mg/kg bw. Biliary excretion accounted for 35-43% of
the radiolabel at the low dose and 14-15% at the high dose within 48
h. Urinary excretion represented 20-28% at the low dose and 8-13% at
the high dose, and faecal excretion represented 65-67% at the low dose
and 80-81% at the high dose within 48 h. Excretion of the [14C-A
ring]-labelled material in bile was not examined (Gans, 1994).
(b) Biotransformation
The samples collected in the experiments described above (Gans,
1994) were analysed for metabolites of kresoxim-methyl, in a study
that conformed to good laboratory practice. After oral administration,
high proportions of parent compound were found in the faeces (Table
1), but none was detected in the bile or in tissues (plasma, liver,
and kidney) sampled about 4 h after administration of the low or high
dose (Table 2). A total of 34 metabolites, including conjugates, was
identified by nuclear magnetic resonance spectroscopy and mass
spectrometry in rat excreta, with 20 in urine, eight in faeces, and 17
in bile. The major metabolites identified in urine and faeces were M1,
a hydrolytic product of the acetyl ester; M2, an oxidative metabolite
of the aryl-methyl moiety of M1; and M9, a hydroxylated metabolite of
the phenoxy ring of M1. M1 and M9 were the major metabolites
identified in tissues. Glucuronated conjugates were detected in
notable quantities in the bile. There was no evidence that the
metabolic pathways were induced by pretreatment with kresoxim-methyl.
A small difference in the metabolite pattern in urine and bile was
observed between males and females, the percentages of M1 and M9 in
urine from females being greater than in urine from males. In summary,
the metabolic pathways of kresoxim-methyl consisted of hydrolytic
cleavages of the ester, the oxime ether, and the benzyl ether bonds;
hydroxylation at the para position of the phenoxy ring; oxidation of
the aryl-methyl group to benzyl alcohol and its subsequent oxidation
to the corresponding carboxylic acid; and conjugation of the resulting
hydroxy groups with glucuronate and sulfate (Kohl, 1994). The proposed
metabolic pathway for kresoxim-methyl in rats is shown in Figure 1.
The major metabolites identified in plants were a hydrolytic
product of the acetyl ester (M1), an oxidative metabolite of the
aryl-methyl moiety (M2), a hydroxylated metabolite of para- or
meta-hydroxylated metabolites of the phenoxy ring of the first
metabolite (M9 or M54), and their conjugates (Grosshans, 1994a,b;
Nelsen et al., 1995).
2. Toxicological studies
(a) Acute toxicity
Studies of the acute toxicity of kresoxim-methyl are summarized
in Table 3. Oral administration of 5000 mg/kg bw kresoxim-methyl as a
suspension of 0.5% CMC produced no deaths or abnormal clinical signs
in mice or rats, and no abnormal changes in organs were seen at
necropsy. Dermal application of 2000 mg/kg bw in a suspension of 0.5%
CMC caused no deaths or signs of clinical toxicity, except for a
slight but definite erythma at the site of application in some
animals. Groups of five male and five female Wistar rats were exposed
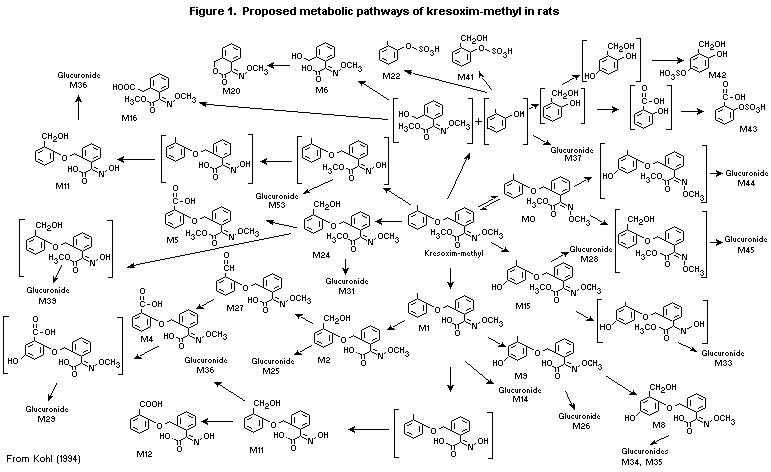 Table 1. Percents of a single oral dose of kresoxim-methyl found as parent compound and
metabolites in rat excreta and tissues
Substance Faeces Urine
50 mg/kg bw 500 mg/kg bw 50 mg/kg bw 500 mg/kg bw
Male Female Male Female Male Female Male Female
Parent 49.5 47.1 74.9 39.5
M1 2.1 0.1 7.1 0.4 2.7 2.8 2.2
M2 2.7 0.5 0.5 5.8 2.0 3.4 1.5 2.0
M4 1.1 0.5 0.3 2.5 mix1 mix1 mix1 mix1
M6 2.8 1.1 1.9 0.5
M8 0.1 0.4 mix3
M9 5.2 6.0 0.9 13.3 5.5 11.0 2.7 4.9
M11 mix2 mix2 mix3
M12 mix2 mix2 mix3
M14 mix1 mix1 mix1 mix1
M15 1.3 2.7 0.1 3.4
M16 0.3 mix3
M20 mix1 mix1 mix1 mix1
M24 0.1 0.1 0.4
M26 mix2 mix2 mix3
mix1 1.4 1.6 0.9 1.1
mix2 0.9 0.8
mix3 1.4
UK1 1.3 0.6 0.4 0.1 0.1 0.2
UK2 0.2 0.1 1.4
UK3 0.1 1.8
UK4 0.3
UK5 0.2
UK5 0.1
Recovery 83.3 86.7 84.1 86.1 99.2 97.4 100.1 99.4
mix1, mixture of M4 + M14 + M20; mix2, M11 + M12 + M26; mix3, M8 + M11 + M12 + M16 + M26; UK, unknown
compound
Table 2. Percents of a single oral dose of kresoxim-methyl found as parent compound and
metabolites in rat bile and tissues
Substance Bile Plasma Liver
50 mg/kg bw 50 mg/kg bw 50 mg/kg bw 500 mg/kg bw
Male Female Male Female Male Female Male Female
Parent 0 0 0 0 0 0 0 0
M1 1.7 1.9 0.386 0.304 0.13 0.07 0.07 0.12
M2 mix5 mix5 0.095 mix8 0.08 0.04 0.04 0.04
M4 0.041 mix8 0.03 0.02 0.04 0.02
M6 0.027 0.006
M9 1.1 1.3 0.173 0.164 0.17 0.07 0.06 0.09
M11 0.002 mix4 mix4 mix4 mix4
mix4 mix4 mix4 mix4 mix4 mix4
M28 0.7 2.9
M31 0.5 1.1
M35 1.7 0.7
M44 0.4 0.3
M45 mix5 mix5
mix4 0.115 0.08 0.02 0.02 0.01
mix5 1.1 1.2
mix6 6.3 3.6
mix7 0.4 0.2
mix8 0.169
UK1 0.02
UK2 0.024 0.02 0.01
Recovery 100.0 100.1 84.8 82.5 87.0 96.8
mix4, M11 + M12 + M16 + M26; mix5, mixture of M2 + M45; mix6, M25+M26+M29+M33+M39; mix7,
M34+M36+M37; mix8, M2 + M4
The tissue samples were collected 3.5-4 h after a single oral adminstration. The values in
plasma are expressed as microgram equivalent per ml.
to a dust aerosol of kresoxim-methyl at concentrations of 2 and 5.6
mg/L through a head-nose inhalation system. The mass median
aerodynamic diameter of the dust aerosol particles was 1.8-2.4 µm. No
deaths occurred; during exposure to either concentration, nonspecific
clinical signs such as accelerated and intermittent respiration,
urine-smeared fur, reddish nose, eye discharge, and reddish eyelid
crust, were observed. These signs disappeared one day after exposure.
In a study conducted according to good laboratory practice, white
Vienna rabbits of each sex received 4-h dermal applications of a
single dose of 0.5 g kresoxim-methyl (purity, 93.7%) as a fine powder
moistened with distilled water. The skin was examined 1, 24, 48, and
72 h after removal of the compound: little or no erythema was observed
(Rossbacher & Kirsch, 1992a).
In another study conducted according to good laboratory practice,
a single dose of 39 mg kresoxim-methyl (purity, 93.7%) in a volume of
0.1 ml was administered to the right eye of white Vienna rabbits of
each sex. The eyes were examined 1, 24, 48, and 72 h after
application, without washing. Some conjunctival redness (score, 0.1-4)
was observed at 1, 24, and 48 h but had disappeared by 72 h after
application (Rossbacher & Kirsch, 1992b).
In a further study conducted according to good laboratory
practice, the skin sensitizing potential of kresoxim-methyl (purity,
93.7%) was tested in female Dunkin Hartley guinea-pigs by the
maximization method. For induction, a 5% suspension of kresoxim-methyl
in 0.5% CMC was applied intradermally, followed by topical application
of a 50% suspension. At challenge, 50% kresoxim-methyl (20 rabbits) or
the vehicle (10 rabbits) was applied dermally. No dermal reaction was
observed in the rabbits challenged with kresoxim-methyl (Rossbacher &
Kirsch, 1993).
(b) Short-term toxicity
Mice
In a range-finding study conducted according to good laboratory
practice, groups of five male and five female B6C3F1(Cr) mice were
given diets containing kresoxim-methyl (purity, 96.6%) at
concentrations of 0, 500, 2000, or 8000 ppm for 28 days, equal to 0,
110, 480, and 2100 mg/kg bw per day for males and 0, 180, 800, and
3800 mg/kg bw per day for females. The animals were observed for
clinical signs, deaths, food consumption, body weight, and clinical
chemical, haematological, and pathological end-points. There were no
deaths or signs of clinical toxicity. At the highest dose,
significantly reduced serum concentrations of triglyceride and
cholesterol were observed in males, and significantly increased
relative liver weights (p < 0.05) were observed in animals of each
sex. No compound-related lesions were observed on histopathological
examination. The NOAEL was 8000 ppm, equal to 2100 mg/kg bw per day,
as the increased relative liver weights were not accompanied by
histopathological changes (Schilling & Hildebrand, 1992b).
Table 3. Acute toxicity of kresoxim-methyl
Species Strain Sex Route LD50 or LC50 Purity Reference
(mg/kg bw or (%)
mg/L air)
Mouse ICR M/F Oral > 5000 94.3 Yamamoto (1994)
Rat Chbb Wistar M/F Oral > 5000 93.7 Kirsch & Hildebrand (1993a)
Rat Chbb Wistar M/F Dermal > 2000 93.7 Kirsch & Hildebrand (1993b)
Rat Chbb Wistar M/F Inhalation > 5.6 96.6 Gamer & Kirsch (1992)
These studies were conducted in accordance with good laboratory practice.
Groups of 10 male and 10 female C57Bl/6N(Cr) mice were given
diets containing kresoxim-methyl (purity, 98.7%) at concentrations of
0, 250, 1000, 4000, or 8000 ppm, equal to 0, 57, 230, 910, and 1900
mg/kg bw per day for males and 0, 80, 330, 1300, and 2600 mg/kg bw per
day for females, for three months. The study was carried out according
to good laboratory practice. The animals were observed for clinical
signs, deaths, food consumption, body weight, clinical chemical
parameters including the activities of serum alanine (ALAT) and
aspartate aminotransferases (ASAT), alkaline phosphatase (AP), and
gamma-glutamyl transferase (GGT), and haematological and pathological
end-points. There were no deaths, signs of clinical toxicity, or
changes in haematological or clinical chemical parameters.
Dose-dependent reductions in terminal body weight, by 4% at 4000 and
7% at 8000 ppm, and body-weight gain, by 11% at 4000 and 24% at 8000
ppm, were seen in males; however, these reductions were not
significant. Significant increases in relative liver weight were
observed in males at 4000 and 8000 ppm, but no compound-related
lesions were observed on histopathological examination. The NOAEL was
8000 ppm, equal to 1900 mg/kg bw per day, on the basis of the absence
of toxicologically significant changes (Mellert & Hildebrand, 1994b)
Rats
In a range-finding study that conformed to good laboratory
practice, groups of five male and five female Wistar (Cr) rats were
given diets containing kresoxim-methyl (purity, 96.55%) at a
concentration of 0, 1000, 4000, or 16 000 ppm for 28 days, equal to 0,
91, 360, and 1400 mg/kg bw per day for males and 0, 95, 380, and 1500
mg/kg bw per day for females. The rats were observed for clinical
signs, deaths, food consumption, body weight, clinical chemical
parameters including the activities of serum ALAT, ASAT, AP, and GGT,
and haematological and pathological end-points. There were no deaths,
signs of clinical toxicity, or changes in haematological parameters.
The terminal body weights were slightly reduced in animals of each sex
at 4000 ppm (by 4% in males and 10% in females) and at 16 000 ppm (by
7% in males and 6% in females), and the absolute liver weights were
slightly increased in males (by 8%) and females (9%) at 16 000 ppm;
however, these changes were not statistically significant. Significant
increases in relative liver weights were observed in females at 16 000
ppm, and significantly increased serum GGT activity and albumin
concentration were observed in males at this dose. No compound-related
lesions were observed on histopathological examination. The NOAEL was
4000 ppm, equal to 360 mg/kg bw per day, on the basis of increased
serum enzyme activity in males and increased relative liver weight in
females (Schilling & Hildebrand, 1992a).
Groups of 10 male and 10 female Wistar (Chbb) rats were given
diets containing kresoxim-methyl (purity, 98.7%) at concentrations of
0, 500, 2000, 8000, or 16 000 ppm, equal to 0, 36, 150, 580, and 1200
mg/kg bw per day in males and 0, 43, 170, 670, and 1400 mg/kg bw per
day in females, for 90 days. The rats were observed for clinical
signs, deaths, food consumption, body weight, clinical chemical
parameters including the activities of serum ALAT, ASAT, AP, and GGT,
and haematological and pathological end-points. Food consumption and
body weights were determined once a week, and enzyme activities were
determined after six weeks and at the end of the study.
There were no deaths, signs of clinical toxicity, changes in food
consumption, or compound-related changes in haematological parameters.
Slight but significant decreases in terminal body weight (7-8% at 8000
and 11-13% at 16 000 ppm) and body-weight gain (7-10% at 8000 and
13-15% at 16 000 ppm) were observed in males. Significant increases in
relative liver weight were observed in males at 16 000 ppm (10%) and
in females at 2000 ppm and higher (10% at 2000, 7% at 8000, and 12% at
16 000 ppm). Significant increases in relative kidney weight were also
observed in males, but the absolute weights were not increased. No
compound-related histopathological lesions were observed in these or
other organs in treated groups. Dose-dependent, statistically
significantly increased activities of GGT were observed in males at
8000 ppm and higher, and significantly decreased activities of AP and
ALAT were observed in males at all doses and in females at 2000 ppm
and higher. These reductions in enzyme activity were considered to be
related to the slight decrease in food consumption on the basis of
mechanistic studies on percent reductions in intestinal and hepatic
isozymes per total serum AP activity (Moss, 1994; Mellert et al.,
1997a). The NOAEL was 2000 ppm, equal to 150 mg/kg bw per day, on the
basis of decreased body weight and body-weight gain and increased GGT
activity in males at higher doses (Mellert & Hildebrand, 1994a).
Groups of five male and five female Wistar (Chbb) rats received
dermal applications of kresoxim-methyl (purity, 94.3%) suspended in
0.5% CMC at a dose of 0 or 1000 mg/kg bw per day under a
semi-occlusive dressing (four layers of absorbent gauze and an elastic
dressing) for 6 h/day for 21 days. The study design corresponded to
good laboratory practice. The rats were observed for clinical signs,
deaths, food consumption, body weight, clinical chemical parameters
including the activities of serum ALAT, ASAT, AP, and GGT,
haematological parameters including clotting times, and pathological
end-points. Blood samples for haematological and clotting analysis and
for clinical chemistry were collected at termination. There were no
compound-related effects on mortality rates, clinical signs,
haematological parameters, clotting times, or clinical chemical
parameters, including serum enzyme activities. There were no
significant changes in body-weight gain or food consumption in the
treated group, and no signs of irritation were observed on treated
skin of test or control animals. No effect on organ weights was
observed, and histopathological examination revealed no
treatment-related alterations in the liver or in any other tissue
examined. The NOAEL was 1000 mg/kg bw per day, the highest dose tested
(Kirsch & Hildebrand, 1994c).
Dogs
Groups of six male and six female beagles, six to nine months
old, were given diets containing kresoxim-methyl (purity, 94-95.9%) at
concentrations of 0, 1000, 5000, or 25 000 ppm, equal to 0, 28, 140,
and 740 mg/kg bw per day for males and 0, 32, 160, and 800 mg/kg bw
per day for females, for three months. The study was conducted
according to the principles of good laboratory practice. The animals
were observed for clinical signs, deaths, food consumption, body
weight, clinical chemical parameters including the activities of serum
ALAT, ASAT, AP, and GGT, and haematological and pathological
end-points. Blood samples for haematological and clinical chemical
analysis were collected during weeks 4 and 13 of treatment.
No deaths or ophthalmological abnormalities were observed. During
the first three weeks, diarrhoea and vomiting were observed frequently
in most animals at 25 000 ppm, and a slight but significant reduction
in body-weight gain was observed in females at this dose throughout
the study. There were no treatment-related changes in haematological
or urinary parameters; slight but significant decreases in the
concentration of total protein were observed in males at 25 000 ppm,
and significant decreases in the concentration of albumin were
observed in females at 5000 ppm and animals at 25 000 ppm. These
changes were observed during week 4 of treatment but had disappeared
by week 13. The changes in albumin and total protein concentration
might not be related to treatment, because they were slight and
transient, and may have been a result of the vomiting and diarrhoea
that occurred during the first weeks of the study. Dose-dependent
increases in the absolute and relative weights of the liver were
observed but were not significant. Histopathological examination
revealed no compound-related lesions in tissues, including the liver.
The NOAEL was 5000 ppm, equal to 140 mg/kg bw per day, on the basis of
vomiting and diarrhoea in animals of each sex and reduced body-weight
gain in females (Mellert & Hildebrand, 1994c).
Groups of six male and six female beagles, six to nine months
old, were given diets containing kresoxim-methyl (purity, 93.7% ) at a
concentration of 0, 1000, 5000, or 25 000 ppm, equal to 0, 27, 140,
and 710 mg/kg bw per day in males and 0, 30, 150, and 760 mg/kg bw per
day in females, for 12 months. The study conformed to good laboratory
practice. The animals were observed for clinical signs, deaths, food
consumption, body weight, ophthalmological end-points, clinical
chemical parameters including the activities of serum ALAT, ASAT, AP,
and GGT, haematological parameters including clotting time, and
pathological end-points. Blood samples were collected for
haematological and clotting analysis and clinical chemistry after 3,
6, and 12 months of treatment.
No deaths or ophthalmological abnormalities were observed.
Diarrhoea and vomiting occurred infrequently in animals of each sex at
25 000 ppm, and the body weights of males at this dose were
significantly reduced at study termination. There was no reduction in
body-weight gain or food consumption at any dose. Significant
increases in the number of platelets were observed in males at all
doses; the values for males at 25 000 ppm were within the range in
historical controls, except for the mean value at the third month.
There were no compound-related changes in clotting time. There were no
compound-related changes in urinary or clinical chemical parameters or
in the activities of serum enzymes. Significant increases in relative
liver weights were observed in males at 5000 ppm, but the absolute
liver weights were not significantly increased. Histopathological
examination revealed no treatment-related alterations in the liver or
in any other tissue examined. The NOAEL was 5000 ppm, equal to 140
mg/kg per day, on the basis of reduced body weight in males (Hellwig &
Hildebrand, 1994b).
(c) Long-term studies of toxicity and carcinogenicity
Mice
Groups of 50 male and 50 female C57Bl/6N (Cr) mice were given
diets containing kresoxim-methyl (mean purity, 96.3% during the first
12 months and 93.2% during the following six months) at concentrations
of 0, 400, 2000, or 8000 ppm for 18 months. Satellite groups of 10
mice of each sex were treated concurrently for 12 months. The doses
were equivalent to 0, 60, 300, and 1300 mg/kg bw per day in males and
0, 81, 400, and 1700 mg/kg bw per day in females in the main groups,
and 0, 61, 320, and 1400 mg/kg bw per day in males and 0, 84, 410, and
1900 mg/kg bw per day in females in the satellite groups. The animals
were observed for clinical signs, deaths, food consumption, body
weight, and haematological and pathological end-points. Blood samples
for haematology were collected during months 12 and 18 of treatment.
The study conformed to good laboratory practice.
No compound-related effects were observed with respect to
mortality rates, clinical signs, food consumption, or haematological
parameters throughout the study. Statistically significant decreases
in terminal body weights and body-weight gains were observed in the
main groups in males at 8000 ppm and in females at 2000 and 8000 ppm
during the final six months. Increased relative liver weights were
observed in females in the satellite group examined at 12 months and
in the main groups examined at 18 months at 8000 ppm. Increased
relative adrenal weights were observed in males at 12 and 18 months
and in females at 18 months. Histopathological examination at
12 months revealed no compound-related lesions in any group treated
for 12 months, but examination at 18 months revealed significantly
increased incidences of centrilobular fatty infiltration (1/50 at 0
and 16/50 at 8000 ppm) in the liver, a significantly increased
incidence and a greater degree of severity of hepatic amyloidosis
(6/50 at 0 and 16/50 at 8000 ppm), and increased incidences of
lymphoid infiltration (16/50 at 0 and 27/50 at 8000 ppm) and papillary
necrosis of the kidney (2/50 at 0 and 13/50 at 8000 ppm) in females.
There was no treatment-related increase in the incidence of neoplastic
lesions. The NOAEL was 400 ppm, equal to 81 mg/kg bw per day, on the
basis of reductions in body weight and body-weight gain in females
(Mellert & Hildebrand, 1994e).
Rats
In a study of toxicity, groups of 20 male and 20 female Wistar
rats were given diets containing kresoxim-methyl (purity, 92.7-96.6%)
at concentrations of 0, 200, 800, 8000, or 16 000 ppm, equal to 0, 9,
36, 370, and 750 mg/kg bw per day in males and 0, 12, 46, 500, and
1000 mg/kg bw per day in females, for 24 months. The animals were
observed for clinical signs, deaths, food consumption, body weight,
ophthalmological end-points, clinical chemical parameters including
the activities of serum ALAT, ASAT, AP, and GGT, and haematological,
urinary, and histopathological end-points. Blood samples for
haematology and clinical chemistry were collected at 3, 6, 12, 18, and
24 months of the treatment. The design of the study conformed to good
laboratory practice.
There were no treatment-related effects on mortality rates,
clinical signs, or ophthalmoscopic parameters. The terminal body
weight and body-weight gain were slightly reduced in males at 16 000
ppm (by 4%) and significantly reduced in females at 8000 ppm (by 9 and
13%, respectively) and 16 000 ppm (by 6 and 10%, respectively). No
significant change in food consumption was observed. Slight but
significant reductions in mean corpuscular volume and mean corpuscular
haemoglobin were observed in males at 16 000 ppm and in females at
> 200 ppm; however, these changes were within the background range
and were not clearly dose-dependent. The activity of serum ALAT was
significantly decreased in animals of each sex at 8000 and 16 000 ppm
and that of serum AP in animals of each sex at > 200 ppm. The
author suggested that these reductions in enzyme activities are not
toxicologically relevant, which is reasonable (Moss, 1994; Mellert et
al., 1997a). The relative liver weights were significantly increased
in males at 8000 and 16 000 ppm, and the absolute liver weights were
significantly increased in males at the highest dose. Significant,
dose-related increases in GGT activity were also observed in males at
> 8000 ppm.
Microscopic examination revealed evidence of neoplasia in the
liver. Increased incidences of hepatocellular carcinoma were observed
in animals of each sex at 8000 and 16 000 ppm (in males, 0/20 at 0,
1/20 at 200 ppm, 1/20 at 800 ppm, 3/20 at 8000 ppm, and 8/20 at 16 000
ppm; in females, 0/20 at 0, 0/20 at 200 ppm, 2/20 at 800 ppm, 6/20 at
8000 ppm, and 6/20 at 16 000 ppm). No hepatocellular adenomas were
observed. The incidence and severity of hepatocellular hypertrophy
were dose-dependent and increased in animals of each sex (males, 0/20
at 0, 3/20 at 800 ppm, 4/20 at 8000 ppm, and 7/20 at 16 000 ppm;
females, 1/20 at 0 and 8/20 at 16 000 ppm); however, statistical
significance was achieved only at 16 000 ppm in animals of each sex.
Significant increases in the incidence and severity of eosinophilic
foci (0/20 at 0, 6/20 at 8000 ppm, and 8/20 at 16 000 ppm) and mixed-
cell foci (0/20 at 0, 4/20 at 8000 ppm, and 5/20 at 16 000 ppm) were
observed in males. Evidence of a proliferative response in bile-duct
cells was associated with increased incidences of biliary cysts in
males at 16 000 ppm (0/20 in controls versus 4/20) and in females at
8000 and 16 000 ppm (3/20 in controls versus 7/20 and 7/20), bile-duct
proliferation in females at 8000 and 16 000 ppm (5/20 in controls
versus 8/20 and 11/20), and pericholangitis of the liver in males at
16 000 ppm (1/20 in controls versus 4/20). Significantly increased
incidences of tubular casts of the kidneys (2/20 in controls versus
10/20) and tubular atrophy of the kidney (4/20 in controls versus
12/20) were seen in females at 16 000 ppm. Increased incidences of
lesions in other tissues were age-related or independent of dose and
were not considered to be toxicologically significant.
The NOAEL for non-neoplastic alterations was 800 ppm, equal to 36
mg/kg bw per day, on the basis of increased activity of serum GGT,
increased relative liver weight, and increased incidence and degree of
severity of eosinophilic foci in males. The NOAEL for neoplasia was
also 800 ppm on the basis of an increased incidence of hepatocellular
carcinoma in animals of each sex (Mellert & Hildebrand, 1994d).
In a study of carcinogenicity, groups of 50 male and 50 female
Wistar rats were fed diets containing kresoxim-methyl (purity,
92.7-96.6%) at concentrations of 0, 200, 800, 8000, or 16 000 ppm,
equal to 0, 9, 36, 380, and 770 mg/kg bw per day for males and 0, 12,
47, 500, and 1000 mg/kg bw per day for females, for 24 months. The
animals were observed for clinical signs, deaths, food consumption,
body weight, and haematological and histopathological end-points.
Blood samples for haematology were collected at the end of the study.
The study was carried according to the principles of good laboratory
practice.
There were no treatment related effects on mortality rates or
clinical signs. The terminal body weights and body-weight gains were
significantly reduced in animals of each sex at 8000 ppm
(9 and 13% in males and 13 and 20% in females, respectively) and 16
000 ppm (9 and 12% in males and 14 and 21% in females, respectively).
No significant change was observed in food consumption. Significantly
increased relative liver weights were observed in males at 16 000 ppm.
Microscopic examination revealed hepatic neoplasia: increased
incidences of hepatocellular carcinoma were observed in animals of
each sex at 8000 and 16 000 ppm (males, 7/50 at 0, 5/50 at 200 ppm,
2/50 at 800 ppm, 18/50 at 8000 ppm, and 11/50 at 16 000 ppm; females,
1/50 at 0, 1/50 at 200 ppm, 2/50 at 800 ppm, 13/50 at 8000 ppm, and
16/50 at 16 000 ppm). The numbers of animals with adenoma plus
carcinoma in the liver were significantly increased among males at
8000 ppm (8/50 at 0, 19/50 at 8000 ppm, and 13/50 at 16 000 ppm) and
among females at 8000 and 16 000 ppm (1/50 in controls versus 15/50
and 17/50). The incidence of hepatocellular hypertrophy was increased
in males at 8000 and 16 000 ppm and in females at 16 000 ppm but
reached statistical significance only in males at 16 000 ppm (males,
3/50 at 0, 5/50 at 8000 ppm, and 10/50 at 16 000 ppm; females, 5/50 at
0, and 7/50 at 16 000 ppm). There were dose-dependent increases in the
incidences of eosinophilic foci (males, 1/50 at 0, 5/50 at 8000 ppm,
and 11/50 at 16 000 ppm; females, 3/50 at 0, 8/50 at 8000 ppm, and
5/50 at 16 000 ppm) and mixed-cell foci in animals of each sex (males,
4/50 at 0, 9/50 at 8000 ppm, and 12/50 at 16 000 ppm; females, 0/50 at
0 and 5/50 at 16 000 ppm); however, significant results were observed
only at 16 000 ppm. There was evidence of alterations in bile-duct
cells, including an increased incidence of bile-duct proliferation in
females at 16 000 ppm (10/50 in controls versus 28/50),
cholangiofibrosis in females at 16 000 ppm (1/50 in controls versus
7/50), and biliary cysts in males at 8000 ppm (males, 1/50 at 0, 7/50
at 8000 ppm, and 6/50 at 16 000 ppm; females, 8/50 at 0, 12/50 at 8000
ppm, and 15/50 at 16 000 ppm). Other non-neoplastic lesions included
tubular mineralization of the kidneys in males at 16 000 ppm (6/50 in
controls versus 18/50), and round-cell infiltration of the adrenal
cortex in males at 8000 ppm (5/50 at 0, 13/50 at 8000 ppm, and 5/50 at
16 000 ppm). The tubular mineralization was dose-related and
considered to be related to treatment. The lesions observed in other
tissues were considered to be independent of dose and age-related.
The NOAEL for non-neoplastic alterations was 800 ppm, equal to 36
mg/kg bw per day, on the basis of reduced body weight and body-weight
gain and hepatic alterations. The NOAEL for neoplasia was also 800 ppm
on the basis of increased incidences of hepatocellular carcinoma
(Mellert & Hildebrand, 1994f).
A histopathological re-evaluation on the heptocellular tumour
incidence in the two studies in rats was conducted by a pathology
working group. The results are shown in Table 4. Concurrent
reassessment revealed similar dose-response relationships in the
occurrence of hepatocellular carcinoma, and the statistically
significant results with the combined data clearly indicate the
hepatic carcinogenic potential of kresoxim-methyl in rats (van
Ravenzwaay, 1996).
(d) Genotoxicity
The results of assays for the genotoxicity of kresoxim-methyl are
summarized in Table 5. No point mutations were observed in vitro in
bacterial or mammalian cells. A significantly increased frequency of
chromosomal damage was observed in Chinese hamster lung cells with an
exogenous metabolic activation system treated with kresoxim-methyl
(purity, 93.7%) at > 100 µg/ml; however, crystals were observed in
medium cultured at 100 µg/ml for 6 h. No chromosomal damage was
observed in human lymphocytes in vitro. Assays for DNA repair and
damage in rat hepatocytes showed marked cytotoxicty, characterized by
altered cell morphology and reduced numbers of live cells at > 10
µg/ml . Kresoxim-methyl at these doses also increased extracellular
lactic dehydrogenase activity. The percent of cells in repair was
slightly increased at > 1 µg/ml (by 1-2% in comparison with 52% in
the positive control), but the authors considered these percentages to
be below their evaluation criteria (net grain, > 5%). Kresoxim-methyl
did not cause DNA damage or repair ex vivo in hepatocytes isolated
from treated rats. It did not induce micronucleus formation in mice or
rats treated in vivo.
Table 4. Incidences of hepatocellular carcinomas and other parameters in
rats in the studies of Mellert & Hildebrand (1994e,f)
Parameter Dose (ppm)
0 200 800 8000 16 000
Study of toxicity
Males
Incidence 0/20 1/20 1/20 3/20 8/20*
% incidence 0 5 5 15 40
Absolute body weight (% of control) 100 106 109 94 96
Body-weight gain (% of control) 100 109 112 91 96
Females
Incidence 1/20 0/20 2/20 6/20 6/20
% incidence 5 0 10 30 30
Absolute body weight (% of control) 100 105 88 91(*) 94(*)
Body-weight gain (% of control) 100 107 81 87(*) 90(*)
Study of carcinogenicity
Males
Incidence 7/50 5/50 2/50 18/50* 13/50*a
% incidence 14 10 4 36 26
Absolute body weight (% of control) 100 101 98 91 (*) 91 (*)
Body-weight gain (% of control) 100 102 98 87 (*) 79 (*)
Females
No of incidence 1/50 1/50 2/50 13/50* 16/50*
% of incidence 2 2 4 26 32
Absolute body weight (% of control) 100 99 98 87(*) 86(*)
Body-weight gain (% of control) 100 98 97 80(*) 79(*)
Terminal absolute body weight and body-weight gain were expressed as percent of
control, but the statistical significance was calculated on the basis of weight.
* Statistically significantly different from control.
a Includes two animals with hepatocholangiocarcinomas
Table 5. Results of assays for the genotoxicity of kresoxim-methyl
End-point Test object Concentration Purity Result Reference
(%)
In vitro
Reverse mutationa,b S. typhimurium TA98, TA100, 20-5000 µg/plate 93.7 Negative Engelhardt &
TA1535, TA1537; E. coli WP2 Hoffmann (1993a)
uvrA
Reverse mutationa,b S. typhimurium TA98, TA100, 20-5000 µg/plate 94.3 Negative Engelhardt &
TA1535, TA1537; E. coli WP2 Hildebrandt (1994)
uvrA
Reverse mutationa,b S. typhimurium TA98, TA100, 20-5000 µg/plate 90.2 Negative Engelhardt (1996)
TA1535, TA1537; E. coli WP2
uvrA
Reverse mutationb,c S. typhimurium TA98, TA100, 51-5000 µg/plate 98.6 Negative Nakajima (1997)
TA1535, TA1537; E. coli WP2
uvrA
DNA repaird B. subtilis rec M45+, H17- 191-6100 µg/plate (-S9) 98.6 Negative Nakajima (1997)
95-3050 µg/plate (+S9) Negative
Gene mutatione Chinese hamster ovary cells, 0.01-100 µg/ml (-S9) 94.3 Negative Polloth & Hoffman
hprt locus 0.1-100 µg/ml (+S9) (1994a)
Chromosomal Human lymphocytes 10-40 µg/ml 98.7 Negative Engelhardt &
aberrationb,f Hoffmann (1993b)
Chromosomal Chinese hamster lung cells 0.45-55 µg/ml (-S9) 93.7 Negative Akanuma et al. (1997)
aberrationg 50-200 µg/ml (+S9) Positive
DNA damage and Wistar rat hepatocytes 0.33-10 µg/ml 94.3 Negative Polloth & Hoffman
repairh (1994b)
Table 5. (continued)
End-point Test object Concentration Purity Result Reference
(%)
In vivo
DNA damage and Wistar rat hepatocytes Single oral gavage, 18 h 94.3 Negative Polloth & Hildebrand
repairi 0, 20, 200, 1000 mg/kg bw (1994c)
DNA damage and Wistar rat hepatocytes 3-week feeding 94.3 Negative Polloth & Hoffman
repairi 0, 200, 16 000 ppm (1994b)
Micronucleus NMRI mouse bone marrow Single i.p, 16 and 48 h, 93.7 Negative Engelhardt &
formationj 0, 500, 1000, 2000 mg/kg bw Hoffmann (1993c)
Micronucleus Wistar rat bone marrow Single i.p, 24 and 48 h, 94.9 Negative Engelhardt &
formationk 0, 500, 1000, 2000 mg/kg bw Hoffmann (1997)
S9, microsomal fraction of rat hepatocytes; i.p., intraperitoneal
All of the tests were carried out according to good laboratory practice, and all of the positive controls produced the expected results.
a In dimethyl sulfoxide; the positive controls were 2-aminoanthracene, N-methyl-N'-nitro-N-nitrosoguanidine,
N-ethyl-N'-nitro-N-nitrosoguanidine, 9-aminoacridine chloride, and 4-nitro-ortho-phenylendiamine.
b In the presence and absence of S9
c In dimethyl sulfoxide; the positive controls were 2-(2-furyl)-3-(5-nitro-2-furyl)acrylamide for TA98, TA100, and WP2uvrA; sodium
azide for TA1535; and 9-aminoacridine chloride for TA1537 -S9 and 2-aminoanthracene + S9.
d Positive controls were mitomycin C -S9 and try-P-1 +S9; negative control was kanamycin -S9.
e Positive controls were ethylmethanesulfonate -S9 and 3-methylcholanthrene +S9.
f Positive controls were mitomycin C -S9 and cyclophosphamide +S9.
g Positive controls were mitomycin C -S9 and benzo[a]pyrene.
h Positive control was 2-acetylaminofluorene.
i Positive control was 2-acetylaminofluorene at a single oral dose of 50 mg/kg bw.
j Positive controls were cyclophosphamide at a single i.p dose of 20 mg/kg bw and vincristine at a single i.p dose of 0.15 mg/kg bw.
k Positive control was cyclophosphamide at a single i.p dose of 20 mg/kg bw.
(e) Reproductive toxicity
(i) Multigeneration reproductive toxicity
Rats
In a two-generation study of reproductive toxicity, which
conformed to good laboratory practice, groups of 25 male and 25 female
Wistar rats were fed diets containing kresoxim-methyl (purity,
> 93.7%) at concentrations of 0, 50, 1000, 4000, or 16 000 ppm. The
F0 generation was exposed directly, the F1a and F1b generations
directly and indirectly, and F2 generation indirectly. The mean daily
intakes of kresoxim-methyl by the F0 generation were 5, 100, 410, and
1600 mg/kg bw per day for males and 6, 120, 480, and 2300 mg/kg bw per
day for females. Female intakes were 6, 110, 440, and 1700 mg/kg bw
per day during premating; and 4, 87, 360, and 1400 mg/kg bw per day
during gestation and 7, 150, 600, and 2400 mg/kg bw per day during
lactation for the F1a and F1b generations. The mean daily intakes by
the F1 generation were 4, 88, 360, and 1500 mg/kg bw per day for
males and 5, 110, 440, and 1800 mg/kg bw per day for females. female
intakes were 5, 100, 420, and 1700 mg/kg bw per day during premating;
4, 85, 350, and 1300 mg/kg bw per day during gestation for F2a; and
7, 140, 560, and 2300 mg/kg bw per day during lactation for F2a.
The parental rats were observed for clinical signs, deaths, food
consumption, body weight, and clinical chemical, histopathological,
and reproductive parameters including mating, fertility, gestation,
and live-birth indices. The litters and pups were observed for
viability, lactation, behaviour, and developmental indices that
included pinna unfolding and opening of the auditory canal and eyes.
The functional tests included grip strength, startle reflex, and
pupillary reflex. Reproductive organs and the pituitary, liver, and
kidney were examined histopathologically. The clinical chemical
end-points included assays for serum ALAT, ASAT, AP, and GGT activity.
No compound-related clinical signs or deaths were observed in the
F0, F1, or F2 generation throughout the study. F0 and F1 parental
animals showed no effects on mating, fertility, gestation, or
live-birth indices, but significant reductions in food consumption
were observed in F0 and F1 males during treatment and in F0 and F1
females during gestation and lactation at 16 000 ppm. Significant
reductions in body weight were seen at doses of 4000 ppm and higher in
F0 and F1 males and in F0 and F1 females during gestation and
lactation of the F1a and F1b generations. Significant reductions in
body-weight gain were also observed in F0 and F1 males at these
doses and in F0 females at 16 000 ppm during the premating period
before the first gestation. Significant reductions in the activities
of ALAT and AP were observed in F0 and F1 parents of each sex,
although these reductions may not be toxicologically relevant (Moss,
1994; Mellert & Hildebrand, 1995). The activity of GGT was
significantly increased in F0 males at 4000 ppm and higher and in F1
animals of each sex at 16 000 ppm. Significantly decreased numbers of
fat storage cells were observed in the livers of F0 and F1 males at
4000 ppm and higher; however, this change may have occurred as a
result of the reduced food consumption at higher doses. Significant
increases in relative kidney weights were observed in F0 males at 16
000 ppm and in F0 females and F1 males at 4000 ppm and higher. No
treatment-related morphological lesions were observed in the liver or
kidney.
No compound-related changes in clinical signs, sex ratio,
viability index, or lactation index were seen in pups of the F1a,
F1b, and F2a generations. Body weights and body-weight gain during
lactation were significantly decreased in F1a, F1b, and F2a pups at
4000 ppm and higher. A significantly lower percentage of F1b pups at
these doses had pinna unfolding; significant retardations in opening
of the auditory canal and eyes were also observed in F1b and F2a pups
at 4000 ppm,but were not dose-dependent. There were no differences in
the results of reflex tests between controls and treated animals in
any generation. Necroscopy of pups revealed no external abnormality.
The NOAEL for parental toxicity was 1000 ppm, equal to 100 mg/kg
bw per day in F0 males and 88 mg/kg bw per day in F1 males, on the
basis of reduced body weight and body-weight gain, increased serum GGT
activity, and increased relative kidney weights. The NOAEL for
reproductive toxicity was 1000 ppm, equal to an overall mean intake of
100 mg/kg bw per day for F1 and F2 pups, on the basis of reduced
body weight and body-weight gain (Hellwig & Hildebrand, 1994a).
(ii) Developmental toxicity
Rats
Groups of 25 female Wistar rats were given kresoxim-methyl
(purity, > 93.7%) suspended in 0.5% CMC by gavage at a dose of 0,
100, 400, or 1000 mg/kg bw per day on days 6-15 of gestation. The
study was conducted in accordance with good laboratory practice. No
treatment-related changes in clinical signs, mortality rates, body
weight, or food consumption were observed in maternal animals. There
were no differences in conception rate, mean number of corpora lutea,
total implantations, resorptions, pre- or post-implantation loss, or
number of live fetuses. No significant differences in fetal sex ratio,
placental weight, or fetal body weight were observed between control
and treated groups. External examination revealed three fetuses with
external malformations: one fetus at 100 mg/kg bw per day had anasarca
and a cleft palate, one fetus at 400 mg/kg bw per day was acaudate,
and one fetus at 1000 mg/kg bw per day had meningocele and unilateral
microphthalmia; however, the incidence of these malformations was
within the range for historical controls. One fetus at 1000 mg/kg bw
per day had hydrocephalus, but this incidence was also within the
historical control range. A significantly increased incidence of
incompletely ossified thoracic vertebral bodies was seen in 23% of all
fetuses and 58% of litters at 1000 mg/kg bw per day; the mean
historical control values were 8% (0-49%) of all fetuses and 23%
(0-100%) of litters. The NOAEL for maternal toxicity was 1000 mg/kg bw
per day, and that for embryo and fetal toxicity was 400 mg/kg bw per
day on the basis of a slight increase in variations in fetuses at 1000
mg/kg bw per day. There was no evidence of teratogenicity at doses
< 1000 mg/kg bw per day (Hellwig, 1994).
Rabbits
Groups of 15 female Himalayan rabbits were given kresoxim-methyl
(purity, 96.6%) suspended in 0.5% CMC by gavage at a dose of 0, 100,
400, or 1000 mg/kg bw on days 7-19 of gestation. The study was
conducted in accordance with good laboratory practice. No
compound-related changes in clinical signs, deaths, body weight, or
food consumption were observed in maternal animals, and there were no
compound-related changes in conception rate, mean numbers of corpora
lutea, total implantations, resorptions, pre- or post-implantation
loss, or live fetuses. No significant differences in fetal sex ratio,
placental weight, or fetal body weight were observed between control
and treated groups. External examination revealed one fetus with
microcephaly and brachygnathia at 100 mg/kg bw per day, but the
incidence was within that of historical controls. Eight fetuses (0 /15
at 0, 2/15 at 100, 2/15 at 400, and 3/15 at 1000 mg/kg bw per day) had
soft-tissue malformations: one at 100 mg/kg bw per day had a septal
defect and one had agnesis of the gall-bladder (2.5% incidence); two
at 400 mg/kg bw per day had a septal defect (1.9%); at 1000 mg/kg bw
per day, one had a septal defect, dilatation of the aortic arch, and a
descending aortic, one had hydrocephaly, and one had agnesis of the
gall-bladder (4.1%). The percent of soft-tissue malformations in
historical controls was 2.2-3.1%. The incidences of ventricular septal
defects in the treated groups were comparable to historical values.
Increased incidences of fused sternebrae were observed in 3/15
controls, 11/15 at 100 mg/kg bw per day, 7/15 at 400 mg/kg bw per day,
and 9/15 at 1000 mg/kg bw per day; the increase at 100 mg/kg bw per
day was significant but was within the historical control range.
Increased total numbers of fetal malformations were also observed in
treated groups but again at incidence rates comparable to those of
historical controls (0% at 0, 4.9% at 100, 3.8% at 400, and 4.1% at
1000 mg/kg bw per day versus 2.9-3.5% for historical controls). The
NOAEL for both maternal and developmental toxicity was thus 1000 mg/kg
bw per day, the highest dose tested (Hellwig & Hildebrand, 1993, GLP)
(f) Special studies
(i) Tumour initiating potential
Groups of 10 Wistar rats of each sex were subjected to a partial
hepatectomy and 14 h later received a single dose of 2388 mg/kg bw
technical-grade kresoxim-methyl (purity, 92.7-94.3%) suspended in 0.5%
CMC by gavage to rats. For promotion, phenobarbital was incorporated
in the diet at a concentration of 500 ppm for eight weeks. Liver
slices were examined histologically on slides stained with
haematoxylin and eosin (H&E) or stained immunochemically for the
placental form of glutathione S-transferase (GST-P). The study
conformed to good laboratory practice. The incidences of
hepatocellular alteration (foci) and of GST-P-positive foci were used
to estimate initiating potential. N-Nitrosomorpholine was used as
the positive control. Hepatocellular hypertrophy was found in almost
all of the phenobarbital-treated animals, and GST-positive foci and
foci of hepatocellular alteration were found in nearly all animals
treated with the positive control. The number of animals with
GST-P-positive foci in groups treated with kresoxim-methyl was
comparable to that of vehicle controls. The numbers of foci per liver
in promoted animals were 0-3 in those given kresoxim-methyl, 0-10 in
vehicle controls, and 3-100 in positive controls. The results suggest
that kresoxim-methyl does not have tumour initiating potential in rats
in this test (Gamer & Hildebrand, 1995).
(ii) Tumour promoting potential
In a medium-term study of promotion, which did not conform to
good laboratory practice, male Fischer rats were initiated with a
single intraperitoneal injection of N-nitrosodiethylamine at a dose
of 299 mg/kg bw. The animals were then maintained on basal diet
ad libitum for 14 days. Five groups of 16 male rats were fed diets
containing 0, 200, 800, 8000, or 16 000 ppm kresoxim-methyl (purity,
95.4%) for six weeks, with average intakes of 0, 11, 42, 430, and 890
mg/kg bw per day (not adjusted for purity). The remaining 16 male rats
were fed a diet containing 500 ppm phenobarbital (28 mg/kg bw per day)
as a positive control for six weeks. The animals were subjected to a
two-thirds partial hepatectomy after the first week of feeding with
kresoxim-methyl or phenobarbital and were observed for clinical signs,
deaths, food consumption, and body weight. The liver was examined
grossly and histopathologically.
There were no compound-related deaths or clinical signs of
toxicity. Body weight and food consumption in groups given
kresoxim-methyl were comparable to those of controls. Significant
increases in the absolute and relative weights of the liver were
observed in groups given kresoxim-methyl at 800 ppm and higher.
Treatment with phenobarbital caused significant increases in body
weight, food consumption, and relative liver weight. Quantification of
hepatic foci with a computer-assisted image analyser revealed
significant, dose-related increases in the number and area of
GST-P-positive hepatocellular foci in groups given kresoxim-methyl at
> 8000 ppm, as well as in the phenobarbital-treated positive
controls. The NOAEL for promotion was 800 ppm (Harada et al., 1997).
(iii) Hepatic-cell proliferation
A series of studies was conducted to investigate the effect of
kresoxim-methyl on hepatic-cell proliferation in rats, by measuring
S-phase DNA synthesis, an indicator of cell proliferation.
Incorporation of bromodeoxyuridine (BrdU) into DNA was measured by
immunohistochemical staining.
In the first study, groups of five young male Wistar rats, 64
days old, were given diets containing kresoxim-methyl (purity, 94.3%)
at concentrations of 0, 200, or 16 000 ppm, equal to 0, 15, and 1100
mg/kg bw per day, for three weeks. Osmotic minipumps filled with
BrdU were implanted subcutaneously one week before necroscopy. the
animals were observed for clinical signs, deaths, food consumption,
and body weight. The livers were examined grossly and
immunohisto-pathologically. Samples of the hepatic lobule and the
jejunum were taken as positive tissues for proliferation and were
stained with H&E and immunochemically with an antibody against BrdU.
Immunopositive and H&E-counterstained hepatocyte nuclei from 11 fields
for each of three lobes were counted. No treatment-related changes in
body weight, food consumption, or clinical signs were seen. A slight
increase in liver weights was observed at 16 000 ppm, but no
treatment-related gross lesions or histopathological changes were
observed in the livers of treated rats. A statistically significant
increase in the number of hepatocytes in which BrdU was incorporated
into the DNA of S-phase cells was observed in the periportal zone
(zone 1) and the intermediate zone (zone 2) of the hepatic lobule in
the group at 16 000 ppm. No significant increase in cell proliferation
was observed in the group at 200 ppm (Polloth & Hildebrand, 1994a).
In a supplementary study with a similar design, groups of five
young male Wistar rats received kresoxim-methyl (purity, 94.9% ) in
the diet at a concentration of 0, 800, or 8000 ppm, equal to 0, 61,
and 600 mg/kg bw per day, for three weeks. Results similar to those
observed at 16 000 ppm in the first study were observed at 8000 ppm.
Statistically significant increases in cell proliferation were
observed in zones 1 and 2 of the hepatic lobule at 8000 ppm, but not
at 800 ppm (Mellert et al., 1997a).
The NOAEL from the combined results of these two studies for
hepatic cell proliferation was 800 ppm, equal to 61 mg/kg bw per day.
In a study of the hepatic proliferating activity of
kresoxim-methyl in the livers of older rats, groups of five male
Wistar (Chbb) rats aged 16 months were given diets containing
kresoxim-methyl (purity, 94.3%) at a concentration of 0, 200, or 16
000 ppm for three weeks. The design of the study was similar to those
described above. No compound-related changes were seen in clinical
signs or body weight, and no compound-related lesions in the liver
were observed by microscopic examination with H&E staining. A
statistically significant increase in cell proliferation was observed
in zone 1 of the hepatic lobule at 16 000 ppm, which was comparable to
that observed in the young rats (Polloth & Hildebrand, 1994d) .
In a study of the hepatic proliferating activity of
kresoxim-methyl in the livers of rats treated for various periods,
groups of five male Wistar (Chbb) rats, 42 days old, were given diets
containing kresoxim-methyl (purity, 92.7%) at a concentration of 0 or
16 000 ppm for 1, 6, or 13 weeks. Groups were were allowed to recover
for two or three weeks. Significant increases in cell proliferation
were observed in the treated groups after one week (zones 1, 2, and 3)
and after six weeks (zone 1). The increase in zone 1 in the group
treated for one week was greater than that in the group treated for
six weeks. This compound-related enhancement of cell proliferation was
significantly reversed in the groups allowed to recover. The zonal
distribution of increased cell proliferation revealed a selective
effect of kresoxim-methyl on hepatocytes in zone 1 (Mellert et al.,
1996a).
In a study of unscheduled DNA synthesis and S-phase response in
rat hepatocytes, groups of three male Wistar (Chbb) rats received a
single oral dose of 0, 20, 200, or 1000 mg/kg bw kresoxim-methyl
(purity, 94.3%) by gavage. 2-Acetylaminofluorene was used as a
positive control, at a dose of 50 mg/kg bw in the assay of unscheduled
DNA synthesis and at 1000 mg/kg bw in the assay of S-phase response.
Hepatocytes were prepared by in-situ hepatic perfusion 18 h after
treatment. The isolated hepatocytes were cultured with 3H-thymidine
for 18 h, and S-phase response and unscheduled DNA synthesis were
evaluated autoradiographically in the labelled cells. Exposure of rats
to kresoxim-methyl in vivo was not cytotoxic to liver cells. Slight
but dose-dependent increases in the number of cells in S-phase were
observed in all treated groups, with 1% at 0, 1.37% at 20, 2.78% at
200, and 2.58% at 1000 mg/kg bw, as well as in the positive control
group (5.87%). The results suggest that kresoxim-methyl induced a
moderate increase in S-phase DNA synthesis at 200 mg/kg bw and has a
weak potential for enhancing hepatic cell proliferation (Polloth &
Hildebrand, 1994c).
(iv) Morphology of hepatic proliferation
Groups of three female Wistar (Chbb) rats, 12 weeks old, received
diets containing kresoxim-methyl (purity, 94.3% ) at concentrations of
0, 200, or 16 000 ppm, equal to 0, 15, and 1200 mg/kg bw per day, for
three weeks. At termination, the livers were fixed in situ by
perfusion, and the peroxisomes in the liver were examined by light and
electron microscopy after staining with diaminobenzidine to detect
catalase activity. There were no compound-related changes in clinical
signs, body weight, or food consumption; reduced body-weight gain was
observed at 16 000 ppm. No compound-related lesions were observed in
the liver, and no difference was seen between treated and control
animals in the numbers of peroxisomes (Mellert et al., 1995a).
Groups of three female Wistar (Chbb) rats, 15 months old,
received diets containing kresoxim-methyl (purity, 94.3%) at
concentrations of 0, 200, or 16 000 ppm for three weeks and were then
fixed in situ by perfusion. Liver samples were examined by light and
electron microscopy. There were no compound-related changes in
clinical signs or body weight, and no compound-related lesions were
observed in the liver on light microscopic examination. Electron
microscopy showed that the amount, shape, and size of hepatocyte
mitochondria in the treated group were comparable to those in
controls. (Mellert et al., 1995b).
(v) Induction of hepatic metabolic enzyme activities
Groups of 10 male and 10 female Wistar rats were fed diets
containing kresoxim-methyl at concentrations of 0, 200, or 16 000 ppm
for three weeks, equal to 0, 13, and 1000 mg/kg bw per day for males
and 0, 15, and 1200 mg/kg bw per day for females. The animals were
observed for clinical signs, deaths, body weight, and food
consumption. Indicators of hepatic enzymes were measured, including
the activities of GGT and drug metabolizing enzymes, the concentration
of glutathione in liver homogenates, and the content of cytochrome
P450 in microsomes. Significant increases in the activities of GGT and
pentoxyresorufin depentylase and in P450 content were observed in
males at 16 000 ppm. The pattern of induction of drug metabolizing
enzyme activities resembled that of phenobarbital. In females, only a
tendency towards induction was observed (Mellert et al., 1996b).
(vi) Mechanism of decreased serum enzyme activities
As marked reductions in the activities of serum AP and ALAT were
reported in short- and long-term studies of toxicity, a series of
experiments was conducted in which groups of five males and five
females were fed diets containing kresoxim-methyl at a concentration
of 8000 ppm for two weeks. These studies did not conform to good
laboratory practice. In the first experiment, AP activity was
determined in serum samples and extracts of liver and small intestine.
The intestinal activity of AP was not changed by treatment, the
estimated ratio of intestinal and hepatic or bone AP isozyme
activities in the serum being 38.5%. The author indicated the
reduction in serum AP activity observed in the kresoxim-methyl treated
groups was mostly due to a reduction in intestinal AP activity. In the
second experiment, serum AP activity was markedly reduced after
fasting and was increased by feeding a diet supplemented with olive
oil. In the third experiment, addition of sera collected from treated
animals to sera collected from untreated animals did not suppress AP
activity, indicating the absence of an inhibitor. The observed
reductions in serum AP and ALAT activities was therefore probably due
to a slight alteration in food absorption in treated rats. (Moss,
1994).
In a second study to investigate the reduced enzyme activities,
groups of 10 male and 10 female Wistar rats were fed diets containing
kresoxim-methyl at a concentration of 0 or 16 000 ppm for two weeks,
equal to 910 mg/kg bw per day for males and 1100 mg/kg bw per day for
females. The animals were observed for clinical signs, deaths, body
weight, and food consumption. ALAT and AP activities in serum and
urine were assayed at the end of the study. There were no
compound-related changes in clinical signs or mortality rates.
Significantly decreased food consumption was observed in treated
animals of each sex. A slight but significant decrease in body weight
was observed in treated males. Significantly reduced activities of
ALAT and AP in serum were observed in animals of each sex, but no
change in the activities of either enzyme was observed in urine. No
change in urinary creatinine or urinary volume was observed in treated
animals, indicating no change in renal function. Thus, the reduced
enzyme activity observed in sera of kresoxim-methyl-treated rats was
not caused by a change in renal excretion of the enzymes (Mellert et
al., 1997b).
(g) Studies on metabolites
(i) Acute toxicity
Metabolites M1, M2, and M9 were given orally to rats in a
suspension of 0.5% CMC. M1 (purity, 98.5%) produced a variety of
abnormal clinical changes including dyspnoea, staggering gait, and
tremor at doses of 2000 mg/kg bw and higher. M2 (purity, 97.7%) caused
no deaths or abnormal symptoms at 5000 mg/kg bw. M9 (purity, 99.6%)
caused dyspnoea and exhaustion in animals of each sex at 5000 mg/kg bw
but resulted in no change in general appearance at 3000 mg/kg bw
(Kirsch & Hildebrand, 1994a,b,c, 1995).
(ii) Genotoxicity
M1, M2, and M9 of the same purities described above did not
induce reverse mutation in bacteria at a concentration of 5000
µg/plate, whereas the positive controls used gave the expected
positive responses (Hoffman & Engelhardt, 1995a,b,c)
Comments
About 60% of an oral dose of 50 mg/kg bw and 25% of a dose of 500
mg/kg bw kresoxim-methyl was absorbed. It was excreted mainly in the
faeces (70% of the low dose and 80% of the high dose), predominantly
via the bile (about 40% of the low dose and 15% of the high dose
within 48 h), with lesser amounts in urine (about 20% of the low dose
and 10% of the high dose). Peak levels of the radiolabel in plasma
were reached 0.5-1 h after the low dose and 8 h after the high dose.
The plasma half-life was 17-19 h at the low dose and 22-31 h at the
high dose. The highest residual concentrations were found in the
liver, but the concentrations in all tissues, including the liver,
were less than 0.1 g equivalent/g tissue after 120 h of treatment at
the low dose.
After oral administration of kresoxim-methyl, a high proportion
of the parent compound was found in the faeces, but none was detected
in tissues or bile examined 4 h after dosing. In rats, 34 metabolites
of kresoxim-methyl were identified. The proposed metabolic pathways
are hydrolytic cleavage of the ester, the oxime ether, and the benzyl
ether bonds, hydroxylation at the para position of the phenoxy ring,
hydroxylation of the aryl-methyl group and its subsequent oxidation to
form the corresponding carboxylic acid, and conjugation of the
resulting hydroxy groups with glucuronate or sulfate. The major
metabolites identified in both rats and plants were the free acid,
code number 490M1 {(E)-methoxyimino[alpha- (ortho-tolyloxy)- ortho-
tolyl]acetic acid}, the hydroxy derivative of this, 490M2
[alpha- (ortho-hydroxymethylphenoxy)- ortho-tolyl
(methoxyimino)acetic acid] formed by hydroxylation of the aryl-methyl
group, the para-hydroxytolyloxy product 490M9
[alpha- (para-hydroxy- ortho-tolyloxy)- ortho-tolyl
(methoxyimino)acetic acid], and their conjugates. 490M1, 490M2, and
490M9 all had low acute toxicity and were not mutagenic.
WHO has not classified kresoxim-methyl for acute toxicity.
In a range-finding study in B6C3F1 mice, kresoxim-methyl was
administered in the diet at concentrations of 0, 500, 2000, or 8000
ppm for 28 days. The NOAEL was 8000 ppm, equal to 2100 mg/kg bw per
day. In a three-month study, C57Bl/6N mice received kresoxim-methyl in
the diet at concentrations of 0, 250, 1000, 4000, or 8000 ppm. The
NOAEL was 8000 ppm, equal to 1900 mg/kg bw per day.
In a range-finding study in rats, kresoxim-methyl was
administered in the diet at concentrations of 0, 1000, 4000, or 16 000
ppm for 28 days. The NOAEL was 4000 ppm in males, equal to
370 mg/kg bw per day, on the basis of increased activities of serum
gamma-glutamyl transferase at 16 000 ppm, equal to 1500 mg/kg bw per
day. In a three-week study of toxicity in rats, kresoxim-methyl was
administered in the diet at concentrations of 0, 10, 50, or 8000 ppm.
The NOAEL was 50 ppm, equal to 3 mg/kg bw per day, on the basis of
increased hepatic gamma-glutamyl transferase activity in males at 8000
ppm. In a 90-day study of toxicity in rats, kresoxim-methyl (purity,
98.7%) was administered in the diet at concentrations of 0, 500, 2000,
8000, or 16 000 ppm. The NOAEL was 500 ppm in females, equal to 43
mg/kg bw per day, based on increased relative liver weight at 2000 ppm
and above, and 2000 ppm in males, equal to 150 mg/kg bw per day, based
on decreased body-weight gain and increased activity of serum
gamma-glutamyl transferase at 8000 ppm and above.
In a three-month study of toxicity in dogs, kresoxim-methyl was
administered at dietary concentrations of 0, 1000, 5000, or 25 000
ppm. The NOAEL was 5000 ppm, equal to 140 mg/kg bw per day, on the
basis of vomiting, diarrhoea, and reduced body-weight gain in animals
of each sex at 25 000 ppm. In a 12-month study in dogs,
kresoxim-methyl was administered at dietary concentrations of 0, 1000,
5000, or 25 000 ppm. The NOAEL was 5000 ppm, equal to 140 mg/kg bw per
day, on the basis of a reduction in body weight in males at 25 000
ppm. No compound-related toxicity was observed in females.
In an assay for carcinogenicity in mice, kresoxim-methyl was
administered at dietary concentrations of 0, 400, 2000, or 8000 ppm
for 18 months. The NOAEL was 400 ppm, equal to 81 mg/kg bw per day, in
females on the basis of reduction in body weight at 2000 ppm. The
NOAEL in males was 2000 ppm, equal to 300 mg/kg bw per day, on the
basis of decreased body weight and increased relative adrenal weight
at 8000 ppm. At this dose, increased incidences of renal papilliary
necrosis and hepatic amyloidosis were observed in females. There was
no evidence of carcinogenicity.
In a two-year study of toxicity in rats, kresoxim-methyl was
administered at dietary concentrations of 0, 200, 800, 8000, or 16 000
ppm. The NOAEL was 800 ppm, equal to 36 mg/kg bw per day, on the basis
of an increased incidence of hepatocellular carcinoma in animals of
each sex, increased serum gamma-glutamyl transferase activity,
increased relative liver weight, an increased incidence and degree of
severity of eosinophilic foci and mixed-cell foci in males, and a
decrease in terminal body weight and body-weight gain in females at
8000 ppm and 16 000 ppm. There was also an increased incidence of
biliary cysts and bile-duct proliferation.
In a study of carcinogenicity in rats, kresoxim-methyl was
administered at dietary concentrations of 0, 200, 800, 8000, or 16 000
ppm for 24 months. Evidence of biliary alterations included increased
incidences of biliary cysts and cholangiofibrosis in females at 16 000
ppm. At this dose, increased relative liver weights and an increased
incidence of hepatocellular hypertrophy were observed in males. The
NOAEL was 800 ppm, equal to 36 mg/kg bw per day, on the basis of
increased incidences of hepatocellular carcinoma, reductions in body
weight and body-weight gain, and an increased incidence of
eosinophilic foci and mixed-cell foci in animals of each sex at 8000
ppm and above. The overall NOAEL for neoplastic and non-neoplastic
effects was 800 ppm, equal to 36 mg/kg bw per day.
It is generally recognized that the process of carcinogenesis is
divided into three stages: initiation, promotion, and progression. A
series of mechanistic studies was conducted with kresoxim-methyl,
including tests for tumour initiating and promoting potential. In a
study on tumour initiating activity, kresoxim-methyl did not increase
the number of liver-cell foci in rats at a single dose of 2400 mg/kg
bw. In a study on the promoting potential of kresoxim-methyl, rats
received an initiating dose of N-nitrosodiethylamine and then a diet
containing 0, 200, 800, 8000, or 16 000 ppm kresoxim-methyl for six
weeks. Quantitative analysis of hepatic foci with a computer-assisted
image analyser revealed significant, dose-dependent increases in the
number and area of placental-type glutathione S-transferase-positive
hepatocellular foci, indicating a promoting effect of kresoxim-methyl
on hepatocarcinogenesis at doses of 8000 ppm and above. The NOAEL for
the promoting effect was 800 ppm, equal to 43 mg/kg bw per day.
Four studies were conducted to investigate the effect of
kresoxim-methyl on hepatic-cell proliferation in rat liver by
measuring bromodeoxyuridine incorporation into hepatocyte DNA during
S-phase DNA synthesis. The results showed a selective cell
proliferation effect of kresoxim-methyl on hepatocytes in the
periportal zone. The NOAEL was 800 ppm, equal to 61 mg/kg bw per day,
while animals treated with 8000 ppm and above showed a statistically
significant increase in cell proliferation. There was no difference in
the sensitivity of young and old rats.
The genotoxic potential of kresoxim-methyl was investigated in a
series of tests, including assays for gene mutation in bacteria and
mammalian cells, unscheduled DNA synthesis, and cytogenicity
in vitro, an assay for micronucleus formation in vivo, and an
assay for unscheduled DNA synthesis ex vivo. Kresoxim-methyl had
moderate potential to induce chromosomal aberrations in vitro with
exogenous metabolic activation, but positive effects were not observed
in any other test, including the assay for micronuclei in rat bone
marrow. The Meeting concluded that kresoxim-methyl is not genotoxic.
The three major metabolites in rats did not induce reverse mutation in
Salmonella typhimurium in vitro.
The increased incidence of liver tumours observed in rats at 8000
ppm and above was considered to be associated with increased cell
proliferation. The mechanistic studies indicated that kresoxim-methyl
has tumour promoting potential at 8000 ppm, which coincides with the
lowest level at which increased liver-cell proliferation was observed.
These results indicate a threshold for the neoplastic mode of action.
The Meeting concluded that a level of 800 ppm kresoxim-methyl has no
carcinogenic potential.
In a two-generation study of reproductive toxicity in rats, the
NOAEL values were 1000 ppm for parental animals of each sex, 100 mg/kg
bw per day for F0 offspring, and 88 mg/kg bw per day for F1
offspring; these were based on reductions in body weight and
body-weight gain and increased serum gamma-glutamyl transferase
activity and relative kidney weight at 4000 ppm and above. The NOAEL
for pups was 1000 ppm, equal to 110 mg/kg bw per day for F1 pups and
97 mg/kg bw per day for F2 pups, on the basis of reductions in body
weight and body-weight gain at 4000 ppm and above.
The NOAEL for embryo- and fetotoxicity in a study of
developmental toxicity in rats was 400 mg/kg bw per day. No maternal
toxicity or teratogenic effects were observed at doses up to and
including the highest one of 1000 mg/kg bw per day. Kresoxim-methyl
did not induce toxicity in a study of developmental toxicity in
rabbits up to and including the highest dose of 1000 mg/kg bw per day.
An ADI of 0-0.4 mg/kg bw was established on the basis of the
NOAEL of 800 ppm, equal to 36 mg/kg bw per day, in the 24-month study
of toxicity and carcinogenicity in rats, and a 100-fold safety factor.
An acute RfD was not allocated because kresoxim-methyl has low
acute toxicity and did not exhibit developmental toxicity. The Meeting
concluded that the acute intake of residues is unlikely to present a
risk to consumers.
Toxicological evaluation
Levels that cause no toxic effect
Mouse: 400 ppm, equal to 81 mg/kg bw per day (18-month study
of toxicity)
Rat: 800 ppm, equal to 36 mg/kg bw per day (two-year study
of toxicity and carcinogenicity )
1000 ppm, equal to 88 mg/kg bw per day (two-generation
study of reproductive toxicity)
400 mg/kg bw per day (study of developmental toxicity)
Rabbit: 1000 mg/kg bw per day (developmental toxicity; highest
dose tested)
Dog: 5000 ppm, equal to 140 mg/kg bw per day (12-month study
of toxicity)
Estimate of acceptable daily intake for humans
0-0.4 mg/kg bw
Estimate of acute reference dose
Not allocated (unnecessary)
Studies that would provide information useful for continued
evaluation of the compound
Observations in humans
List of end-points relevant for setting guidance values for dietary and non-dietary exposure
Absorption, distribution, excretion, and metabolism in mammals
Rate and extent of oral absorption Rapid, 25-60% absorbed
Dermal absorption No data
Distribution Minimum, highest levels in liver
Potential for accumulation Very little
Rate and extent of excretion Rapid/complete, 87-93% within 48 h
Metabolism in animals Extensive. No parent compound in urine, bile, or
tissues; 34 metabolites identified.
Toxicologically significant compounds Parent compound in rat; three major metabolites in
(animals, plants and environment) plants
Acute toxicity
Rat: LD50 oral > 5000 mg/kg bw
Rat: LD50 dermal > 2000 mg/kg bw
Rat: LC50 inhalation > 5.6 mg/L
Skin irritation Not irritating
Eye irritation Not irritating
Skin sensitization Not sensitizing
Short-term toxicity
Target/critical effect Liver: increased relative liver weight (mouse, rat)
Lowest relevant oral NOAEL Rat: 28-day, 43 mg/kg bw per day
Lowest relevant dermal NOAEL No data
Lowest relevant inhalation NOAEL No data
Genotoxicity Not genotoxic
Long-term toxicity and carcinogenicity
Target/critical effect: Hepatocellular carcinoma
Lowest relevant NOAEL Rat: 2-year, 36 mg/kg bw per day, diet
Carcinogenicity Non-genotoxic carcinogen, tumour promoter
Reproductive toxicity
Reproduction target/critical effect Reduction in F0 body weight at parenterally toxic
dose
Lowest relevant reproductive NOAEL Rat: 97 mg/kg bw per day, diet
Developmental target/critical effect None
Lowest relevant developmental NOAEL Rat: 1000 mg/kg bw per day, highest dose tested
Neurotoxicity/Delayed neurotoxicity No data
Other toxicological studies No data
Medical data No data
Summary Value Study Safety factor
ADI 0-0.4 mg/kg bw 2-year study of 100
toxicity and
carcinogenicity, rat
Acute reference dose Not allocated
(unnecessary )
References
Akanuma, M. et al. (1997) Report: In vitro cytogenetic investigations
of Reg. No. 242009 in Chinese hamster cultured cell. Unpublished
report No. IET 97-0021 from Institute for Environmental Toxicology,
Kodaira, Tokyo, Japan. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Engelhardt, G. (1996) Report on the study of Reg. No. 242009 in the
Ames Salmonella/mammalian-microsome mutagenicity test and Escherichia
coli/mammalian-microsome reverse mutation assay (standard plate test
preincubation test). Unpublished report No. 93/10249 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1993a) Report on the study of Reg.
No. 242009 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation assay
(standard plate test preincubation test). Unpublished report No.
93/10249 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1993b) Report: In vitro cytogenetic
investigations of Reg. No. 242009 in human lymphocytes. Unpublished
report No. 93/10493 from BASF Aktiengesellschaft, Ludwigshafen,
Germany. Submitted to WHO by BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1993c) Report: Cytogenetic study in
vivo of Reg. No. 242009 in mice micronucleus test. Single
intraperitoneal sdministration.Unpublished report No. 93/11104 from
BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by
BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1994) Report on the study of Reg. No.
242009 in the Ames test (Salmonella/mammalian-microsome mutagenicity
test--Standard plate test preincubation test). Unpublished report No.
93/10249 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1997) Report: Cytogenetic study in
vivo of Reg. No. 242009 in rat micronucleus test. Single
intraperitoneal administration. Unpublished report No. 06M0180/914384
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Gamer, A.O. & Hildebrand, B. (1995) Report: Study of foci initiating
activity of Reg. No. 242009 in Wistar rats. Unpublished report No.
95/10244 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Gamer, A.O. & Kirsch, P. (1992) Report: Study on the acute oral
toxicity of Reg. No. 242009 in rats. Unpublished report No. 94/10753
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Gans, G. (1994) Study of the biokinetics of [14C]-Reg. No. 242009
([14C]-BAS 490F) in rats. Unpublished report No. 94/10992 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Grosshans, F. (1994a) The metabolism of 14C-242009 in apples. BASF
Reg. No. 94/11760. Unpublished report from BASF Aktiengesellshaft,
Limburgerhof, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Grosshans, F. (1994b) The metabolism of 14C-242009 in wheat. BASF
Reg. No. 94/10685. Unpublished report from BASF Aktiengesellshaft,
Limburgerhof, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Harada, T. et al. (1997) BAS 490F (Reg. No. 242009): Medium term
promotion hepato-carcinogenesis study in rats. Unpublished report No.
97/10918 from Institute for Environmental Toxicology, Kodaira, Tokyo,
Japan. Submitted to WHO by BASF AG, Limbergerhof, Germany.
Hellwig, J. (1994) Report: Study of the prenatal toxicity of of Reg.
No. 242009 in Wistar rats after oral administration (gavage).
Unpublished report No. 93/10833 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Hellwig, J. & Hildebrand, B. (1993) Report: Study of the prenatal
toxicity of Reg. No. 242009 in rabbits after oral administration
(gavage). Unpublished report No. 93/10980 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Hellwig, J. & Hildebrand, B. (1994a) Report: Reproduction toxicity
study with Reg. No. 242009 in Wistar rats. Continuous dietary
administration over 2 generations (2 litters in the first and 1 litter
in the second generation. Unpublished report No. 94/10950 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Hellwig, J. & Hildebrand, B. (1994b) Report on the study of the
toxicity of Reg. No. 242009 in beagle dogs. Administration via the
diet over 12 months. Unpublished report No. 94/10832 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Hoffmann, H.D. & Engelhardt, G. (1995a) Report on the study of Reg.
No. 262451 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation assay
(standard plate test preincubation test). Unpublished report No.
95/10409 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Hoffmann, H.D. & Engelhardt, G.. (1995b) Report on the study of Reg.
No. 292932 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation Assay
(standard plate test preincubation test). Unpublished report No.
95/10026 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Hoffmann, H.D. & Engelhardt, G. (1995c) Report on the study of Reg.
No. 291685 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation assay
(standard plate test preincubation test). Unpublished report No.
95/10027 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1993a) Report: Study on the acute oral
toxicity of Reg. No. 242009 in rats. Unpublished report No. 93/10730
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1993b) Report: Study on the acute dermal
toxicity of Reg. No. 242009 in rats. Unpublished report No. 93/11108
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1994a) Report: Study on the acute oral
toxicity of Reg. No. 291685 in rats. Unpublished report No. 94/11172
prepared by BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted
to WHO by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1994b) Report: Study on the acute oral
toxicity of Reg. No. 292932 in rats. Unpublished report No. 94/11171
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1994c) Report: Study on the acute dermal
toxicity of Reg. No. 242009 in Wistar rats. Application to the intact
skin over 3 weeks (21 applications). Unpublished report No. 94/11070
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1995) Report: Study on the acute oral
toxicity of Reg. No. 262451 in rats. Unpublished report No. 95/10213
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kohl, W. (1994) The metabolism of [14C]-Reg.No.242009 ([14C]-BAS
490F) in rats. Unpublished report No. 94/10981 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994a) Report: Study on the oral
toxicity of Reg. No. 242009 in Wistar rats. Administration in the diet
over 3 months. Unpublished report No. 94/10954 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994b) Report: Study on the oral
toxicity of Reg. No. 242009 in C57BL mice. Administration in the diet
for 3 months. Unpublished report No. 94/10496 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994c) Report: Study on the oral
toxicity of Reg. No. 242009 in beagle dogs. Administration via the
diet over 3 months (Supplementary study to project No.
83M0180/910149). Unpublished report No. 97/10318 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994d) Report: Chronic toxity study with
Reg. No. 242009 in Wistar rats. Administration in the diet for 24
months. Unpublished report No. 94/10951 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. & Hildebrand, B. (1994e) Report: Carcinogenicity study
with Reg. No. 242009 in C57BL mice. Administration in the diet for 24
months. Unpublished report No. 94/10953 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. & Hildebrand, B. (1994f) Report: Carcinogenicity study
with Reg. No. 242009 in Wistar rats. Administration in the diet for 24
months. Unpublished report No. 94/10953 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. & Hildebrand, B. (1995) Test study on enzyme activity
after treatment with Reg. No. 242009 in Wistar rats. Dietary
administration for 3 weeks and recovery of 2 weeks. Unpublished report
No. 95/10853 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Mellert, W., Kaufmann, W. & Hildebrand, B. (1995a) Reg. No. 242009 --
Electron microscopic examinations of liver samples to assess
peroxisomes from Wistar rats treated for 3 weeks in the diet.
Unpublished report No. 95/11106 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. , Kaufmann, W. & Hildebrand, B. (1995b) Reg. No. 242009 --
Electron microscopic examinations of liver samples to assess
mitochondria from old Wistar rats treated for 3 weeks in the diet.
Unpublished report No. 95/11105 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W., Bahnemann, R. & Hildebrand, B. (1996a) Reg. No. 242009 --
S phase response study in male Wistar rats including reversibility.
Administration in the diet up to 13 weeks. Unpublished report No.
96/10053 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Mellert, W. et al. (1996b) Reg. No. 242009 -- Examination of enzyme
activities in the liver of Wistar rats. Administration in the diet for
3 weeks. Unpublished report No. 96/10100 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. et al. (1997a) Report: S-phase response study with BAS
490F (Reg. No. 242009) in Wistar rats after administration in the diet
for 3 weeks. Unpublished report No. 94/10496 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. et al. (1997b) Report: BAS 490F (Reg. No. 242009): Study
of enzyme excretion in urine of Wistar rats after repeated
administration in the diet. Unpublished report No. 97/10317 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Moss, D. (1994) Effects of Reg. No. 242009 on enzyme levels in rat
serum. Unpublished report No. 94/10578 from Royal Postgraduate Medical
School, Hammersmith Hospital, London, United Kingdom. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Nakajima, M. (1997) Reverse mutation assay of Reg. No. 279482.
Unpublished report No.97/11152 from Biosafety Research Center, Foods,
Drugs and Pesticides, Shizuoka, Japan. Submitted to WHO by BASF AG,
Limbergerhof, Germany.
Nelsen, J. et al. (1995) Metabolism of 14C-BAS 490F in grapes. BASF
Reg. No. 92/11760. Unpublished report from BASF Corporation, ARC,
Resesarch Triangle Park, North Carolina, USA. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Polloth, C. & Hildebrand, B. (1994a) Report: Ex vivo unscheduled DNA
synthesis (UDS) assay and S phase response in rat hepatocytes with
Reg. No. 242009. Unpublished report No. 94/10867 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Polloth, C. & Hildebrand, B. (1994b) Report: Ex vivo unscheduled DNA
synthesis (UDS) in rat hepatocytes with Reg. No. 242009. Unpublished
report No. 94/10894 from BASF Aktiengesellschaft, Ludwigshafen,
Germany. Submitted to WHO by BASF AG, Limbergerhof, Germany.
Polloth, C. & Hildebrand, B. (1994c) Report: S phase response with
Reg. No. 242009 in Wistar rat after administration in the diet for 3
weeks. Unpublished report No. 94/10922 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Polloth, C. & Hildebrand, B. (1994d) Report: S phase response with
Reg. No. 242009 in 16 month old Wistar rat after administration in the
diet for 3 weeks. Unpublished report No. 94/10984 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Polloth, C. & Hoffmann, H.D. (1994a) Report: Gene mutation test in
Chinese hamster ovary cells (HPRT locus assay) with Reg. No. 242009.
Unpublished report No. 94/10350 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Polloth, C. & Hoffmann, H.D. (1994b) Report: In vitro unscheduled DNA
synthesis (UDS) assay in rat hepatocytes with Reg. No. 242009.
Unpublished report No. 94/10351 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
van Ravenzwaay, B. (1996) Kresoxim-methyl: Mechanism and assessment of
liver tumor induction. Unpublished report No. 96/10078 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Rossbacher, R. & Kirsch, P. (1992a) Report: Study on the acute dermal
irritation/corrosion of Reg. No. 242009 in the rabbit. Unpublished
report No. 92/11663, BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Rossbacher, R. & Kirsch, P. (1992b) Report: Study on the acute eye
irritation of Reg. No. 242009 in the rabbit. Unpublished report No.
92/11664, BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to
WHO by BASF AG, Limbergerhof, Germany.
Rossbacher, R. & Kirsch, P. (1993) Report on the maximization test for
the sensitizing potential of Reg. No. 242009 in guinea pigs.
Unpublished report No. 93/10014 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Schilling, K. & Hildebrand, B. (1992a) Report: Study on the oral
toxicity of Reg. No. 242009 in Wistar rats. Administration in the diet
over 4 weeks (range finding). Unpublished report No. 92/10551 from
BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by
BASF AG, Limbergerhof, Germany.
Schilling, K. & Hildebrand, B. (1992b) Report: Study on the oral
toxicity of Reg. No. 242009 in B6C3F1 mice. Administration in the diet
over 4 weeks (range finding). Unpublished report No. 92/10539 from
BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by
BASF AG, Limbergerhof, Germany.
Yamamoto, T. (1994) Report: Study on the acute oral toxicity of Reg.
No. 242009 in mice. Unpublished report No.94/ from Biosafety Research
Center, Foods, Drugs and Pesticides, Shizuoka, Japan. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Table 1. Percents of a single oral dose of kresoxim-methyl found as parent compound and
metabolites in rat excreta and tissues
Substance Faeces Urine
50 mg/kg bw 500 mg/kg bw 50 mg/kg bw 500 mg/kg bw
Male Female Male Female Male Female Male Female
Parent 49.5 47.1 74.9 39.5
M1 2.1 0.1 7.1 0.4 2.7 2.8 2.2
M2 2.7 0.5 0.5 5.8 2.0 3.4 1.5 2.0
M4 1.1 0.5 0.3 2.5 mix1 mix1 mix1 mix1
M6 2.8 1.1 1.9 0.5
M8 0.1 0.4 mix3
M9 5.2 6.0 0.9 13.3 5.5 11.0 2.7 4.9
M11 mix2 mix2 mix3
M12 mix2 mix2 mix3
M14 mix1 mix1 mix1 mix1
M15 1.3 2.7 0.1 3.4
M16 0.3 mix3
M20 mix1 mix1 mix1 mix1
M24 0.1 0.1 0.4
M26 mix2 mix2 mix3
mix1 1.4 1.6 0.9 1.1
mix2 0.9 0.8
mix3 1.4
UK1 1.3 0.6 0.4 0.1 0.1 0.2
UK2 0.2 0.1 1.4
UK3 0.1 1.8
UK4 0.3
UK5 0.2
UK5 0.1
Recovery 83.3 86.7 84.1 86.1 99.2 97.4 100.1 99.4
mix1, mixture of M4 + M14 + M20; mix2, M11 + M12 + M26; mix3, M8 + M11 + M12 + M16 + M26; UK, unknown
compound
Table 2. Percents of a single oral dose of kresoxim-methyl found as parent compound and
metabolites in rat bile and tissues
Substance Bile Plasma Liver
50 mg/kg bw 50 mg/kg bw 50 mg/kg bw 500 mg/kg bw
Male Female Male Female Male Female Male Female
Parent 0 0 0 0 0 0 0 0
M1 1.7 1.9 0.386 0.304 0.13 0.07 0.07 0.12
M2 mix5 mix5 0.095 mix8 0.08 0.04 0.04 0.04
M4 0.041 mix8 0.03 0.02 0.04 0.02
M6 0.027 0.006
M9 1.1 1.3 0.173 0.164 0.17 0.07 0.06 0.09
M11 0.002 mix4 mix4 mix4 mix4
mix4 mix4 mix4 mix4 mix4 mix4
M28 0.7 2.9
M31 0.5 1.1
M35 1.7 0.7
M44 0.4 0.3
M45 mix5 mix5
mix4 0.115 0.08 0.02 0.02 0.01
mix5 1.1 1.2
mix6 6.3 3.6
mix7 0.4 0.2
mix8 0.169
UK1 0.02
UK2 0.024 0.02 0.01
Recovery 100.0 100.1 84.8 82.5 87.0 96.8
mix4, M11 + M12 + M16 + M26; mix5, mixture of M2 + M45; mix6, M25+M26+M29+M33+M39; mix7,
M34+M36+M37; mix8, M2 + M4
The tissue samples were collected 3.5-4 h after a single oral adminstration. The values in
plasma are expressed as microgram equivalent per ml.
to a dust aerosol of kresoxim-methyl at concentrations of 2 and 5.6
mg/L through a head-nose inhalation system. The mass median
aerodynamic diameter of the dust aerosol particles was 1.8-2.4 µm. No
deaths occurred; during exposure to either concentration, nonspecific
clinical signs such as accelerated and intermittent respiration,
urine-smeared fur, reddish nose, eye discharge, and reddish eyelid
crust, were observed. These signs disappeared one day after exposure.
In a study conducted according to good laboratory practice, white
Vienna rabbits of each sex received 4-h dermal applications of a
single dose of 0.5 g kresoxim-methyl (purity, 93.7%) as a fine powder
moistened with distilled water. The skin was examined 1, 24, 48, and
72 h after removal of the compound: little or no erythema was observed
(Rossbacher & Kirsch, 1992a).
In another study conducted according to good laboratory practice,
a single dose of 39 mg kresoxim-methyl (purity, 93.7%) in a volume of
0.1 ml was administered to the right eye of white Vienna rabbits of
each sex. The eyes were examined 1, 24, 48, and 72 h after
application, without washing. Some conjunctival redness (score, 0.1-4)
was observed at 1, 24, and 48 h but had disappeared by 72 h after
application (Rossbacher & Kirsch, 1992b).
In a further study conducted according to good laboratory
practice, the skin sensitizing potential of kresoxim-methyl (purity,
93.7%) was tested in female Dunkin Hartley guinea-pigs by the
maximization method. For induction, a 5% suspension of kresoxim-methyl
in 0.5% CMC was applied intradermally, followed by topical application
of a 50% suspension. At challenge, 50% kresoxim-methyl (20 rabbits) or
the vehicle (10 rabbits) was applied dermally. No dermal reaction was
observed in the rabbits challenged with kresoxim-methyl (Rossbacher &
Kirsch, 1993).
(b) Short-term toxicity
Mice
In a range-finding study conducted according to good laboratory
practice, groups of five male and five female B6C3F1(Cr) mice were
given diets containing kresoxim-methyl (purity, 96.6%) at
concentrations of 0, 500, 2000, or 8000 ppm for 28 days, equal to 0,
110, 480, and 2100 mg/kg bw per day for males and 0, 180, 800, and
3800 mg/kg bw per day for females. The animals were observed for
clinical signs, deaths, food consumption, body weight, and clinical
chemical, haematological, and pathological end-points. There were no
deaths or signs of clinical toxicity. At the highest dose,
significantly reduced serum concentrations of triglyceride and
cholesterol were observed in males, and significantly increased
relative liver weights (p < 0.05) were observed in animals of each
sex. No compound-related lesions were observed on histopathological
examination. The NOAEL was 8000 ppm, equal to 2100 mg/kg bw per day,
as the increased relative liver weights were not accompanied by
histopathological changes (Schilling & Hildebrand, 1992b).
Table 3. Acute toxicity of kresoxim-methyl
Species Strain Sex Route LD50 or LC50 Purity Reference
(mg/kg bw or (%)
mg/L air)
Mouse ICR M/F Oral > 5000 94.3 Yamamoto (1994)
Rat Chbb Wistar M/F Oral > 5000 93.7 Kirsch & Hildebrand (1993a)
Rat Chbb Wistar M/F Dermal > 2000 93.7 Kirsch & Hildebrand (1993b)
Rat Chbb Wistar M/F Inhalation > 5.6 96.6 Gamer & Kirsch (1992)
These studies were conducted in accordance with good laboratory practice.
Groups of 10 male and 10 female C57Bl/6N(Cr) mice were given
diets containing kresoxim-methyl (purity, 98.7%) at concentrations of
0, 250, 1000, 4000, or 8000 ppm, equal to 0, 57, 230, 910, and 1900
mg/kg bw per day for males and 0, 80, 330, 1300, and 2600 mg/kg bw per
day for females, for three months. The study was carried out according
to good laboratory practice. The animals were observed for clinical
signs, deaths, food consumption, body weight, clinical chemical
parameters including the activities of serum alanine (ALAT) and
aspartate aminotransferases (ASAT), alkaline phosphatase (AP), and
gamma-glutamyl transferase (GGT), and haematological and pathological
end-points. There were no deaths, signs of clinical toxicity, or
changes in haematological or clinical chemical parameters.
Dose-dependent reductions in terminal body weight, by 4% at 4000 and
7% at 8000 ppm, and body-weight gain, by 11% at 4000 and 24% at 8000
ppm, were seen in males; however, these reductions were not
significant. Significant increases in relative liver weight were
observed in males at 4000 and 8000 ppm, but no compound-related
lesions were observed on histopathological examination. The NOAEL was
8000 ppm, equal to 1900 mg/kg bw per day, on the basis of the absence
of toxicologically significant changes (Mellert & Hildebrand, 1994b)
Rats
In a range-finding study that conformed to good laboratory
practice, groups of five male and five female Wistar (Cr) rats were
given diets containing kresoxim-methyl (purity, 96.55%) at a
concentration of 0, 1000, 4000, or 16 000 ppm for 28 days, equal to 0,
91, 360, and 1400 mg/kg bw per day for males and 0, 95, 380, and 1500
mg/kg bw per day for females. The rats were observed for clinical
signs, deaths, food consumption, body weight, clinical chemical
parameters including the activities of serum ALAT, ASAT, AP, and GGT,
and haematological and pathological end-points. There were no deaths,
signs of clinical toxicity, or changes in haematological parameters.
The terminal body weights were slightly reduced in animals of each sex
at 4000 ppm (by 4% in males and 10% in females) and at 16 000 ppm (by
7% in males and 6% in females), and the absolute liver weights were
slightly increased in males (by 8%) and females (9%) at 16 000 ppm;
however, these changes were not statistically significant. Significant
increases in relative liver weights were observed in females at 16 000
ppm, and significantly increased serum GGT activity and albumin
concentration were observed in males at this dose. No compound-related
lesions were observed on histopathological examination. The NOAEL was
4000 ppm, equal to 360 mg/kg bw per day, on the basis of increased
serum enzyme activity in males and increased relative liver weight in
females (Schilling & Hildebrand, 1992a).
Groups of 10 male and 10 female Wistar (Chbb) rats were given
diets containing kresoxim-methyl (purity, 98.7%) at concentrations of
0, 500, 2000, 8000, or 16 000 ppm, equal to 0, 36, 150, 580, and 1200
mg/kg bw per day in males and 0, 43, 170, 670, and 1400 mg/kg bw per
day in females, for 90 days. The rats were observed for clinical
signs, deaths, food consumption, body weight, clinical chemical
parameters including the activities of serum ALAT, ASAT, AP, and GGT,
and haematological and pathological end-points. Food consumption and
body weights were determined once a week, and enzyme activities were
determined after six weeks and at the end of the study.
There were no deaths, signs of clinical toxicity, changes in food
consumption, or compound-related changes in haematological parameters.
Slight but significant decreases in terminal body weight (7-8% at 8000
and 11-13% at 16 000 ppm) and body-weight gain (7-10% at 8000 and
13-15% at 16 000 ppm) were observed in males. Significant increases in
relative liver weight were observed in males at 16 000 ppm (10%) and
in females at 2000 ppm and higher (10% at 2000, 7% at 8000, and 12% at
16 000 ppm). Significant increases in relative kidney weight were also
observed in males, but the absolute weights were not increased. No
compound-related histopathological lesions were observed in these or
other organs in treated groups. Dose-dependent, statistically
significantly increased activities of GGT were observed in males at
8000 ppm and higher, and significantly decreased activities of AP and
ALAT were observed in males at all doses and in females at 2000 ppm
and higher. These reductions in enzyme activity were considered to be
related to the slight decrease in food consumption on the basis of
mechanistic studies on percent reductions in intestinal and hepatic
isozymes per total serum AP activity (Moss, 1994; Mellert et al.,
1997a). The NOAEL was 2000 ppm, equal to 150 mg/kg bw per day, on the
basis of decreased body weight and body-weight gain and increased GGT
activity in males at higher doses (Mellert & Hildebrand, 1994a).
Groups of five male and five female Wistar (Chbb) rats received
dermal applications of kresoxim-methyl (purity, 94.3%) suspended in
0.5% CMC at a dose of 0 or 1000 mg/kg bw per day under a
semi-occlusive dressing (four layers of absorbent gauze and an elastic
dressing) for 6 h/day for 21 days. The study design corresponded to
good laboratory practice. The rats were observed for clinical signs,
deaths, food consumption, body weight, clinical chemical parameters
including the activities of serum ALAT, ASAT, AP, and GGT,
haematological parameters including clotting times, and pathological
end-points. Blood samples for haematological and clotting analysis and
for clinical chemistry were collected at termination. There were no
compound-related effects on mortality rates, clinical signs,
haematological parameters, clotting times, or clinical chemical
parameters, including serum enzyme activities. There were no
significant changes in body-weight gain or food consumption in the
treated group, and no signs of irritation were observed on treated
skin of test or control animals. No effect on organ weights was
observed, and histopathological examination revealed no
treatment-related alterations in the liver or in any other tissue
examined. The NOAEL was 1000 mg/kg bw per day, the highest dose tested
(Kirsch & Hildebrand, 1994c).
Dogs
Groups of six male and six female beagles, six to nine months
old, were given diets containing kresoxim-methyl (purity, 94-95.9%) at
concentrations of 0, 1000, 5000, or 25 000 ppm, equal to 0, 28, 140,
and 740 mg/kg bw per day for males and 0, 32, 160, and 800 mg/kg bw
per day for females, for three months. The study was conducted
according to the principles of good laboratory practice. The animals
were observed for clinical signs, deaths, food consumption, body
weight, clinical chemical parameters including the activities of serum
ALAT, ASAT, AP, and GGT, and haematological and pathological
end-points. Blood samples for haematological and clinical chemical
analysis were collected during weeks 4 and 13 of treatment.
No deaths or ophthalmological abnormalities were observed. During
the first three weeks, diarrhoea and vomiting were observed frequently
in most animals at 25 000 ppm, and a slight but significant reduction
in body-weight gain was observed in females at this dose throughout
the study. There were no treatment-related changes in haematological
or urinary parameters; slight but significant decreases in the
concentration of total protein were observed in males at 25 000 ppm,
and significant decreases in the concentration of albumin were
observed in females at 5000 ppm and animals at 25 000 ppm. These
changes were observed during week 4 of treatment but had disappeared
by week 13. The changes in albumin and total protein concentration
might not be related to treatment, because they were slight and
transient, and may have been a result of the vomiting and diarrhoea
that occurred during the first weeks of the study. Dose-dependent
increases in the absolute and relative weights of the liver were
observed but were not significant. Histopathological examination
revealed no compound-related lesions in tissues, including the liver.
The NOAEL was 5000 ppm, equal to 140 mg/kg bw per day, on the basis of
vomiting and diarrhoea in animals of each sex and reduced body-weight
gain in females (Mellert & Hildebrand, 1994c).
Groups of six male and six female beagles, six to nine months
old, were given diets containing kresoxim-methyl (purity, 93.7% ) at a
concentration of 0, 1000, 5000, or 25 000 ppm, equal to 0, 27, 140,
and 710 mg/kg bw per day in males and 0, 30, 150, and 760 mg/kg bw per
day in females, for 12 months. The study conformed to good laboratory
practice. The animals were observed for clinical signs, deaths, food
consumption, body weight, ophthalmological end-points, clinical
chemical parameters including the activities of serum ALAT, ASAT, AP,
and GGT, haematological parameters including clotting time, and
pathological end-points. Blood samples were collected for
haematological and clotting analysis and clinical chemistry after 3,
6, and 12 months of treatment.
No deaths or ophthalmological abnormalities were observed.
Diarrhoea and vomiting occurred infrequently in animals of each sex at
25 000 ppm, and the body weights of males at this dose were
significantly reduced at study termination. There was no reduction in
body-weight gain or food consumption at any dose. Significant
increases in the number of platelets were observed in males at all
doses; the values for males at 25 000 ppm were within the range in
historical controls, except for the mean value at the third month.
There were no compound-related changes in clotting time. There were no
compound-related changes in urinary or clinical chemical parameters or
in the activities of serum enzymes. Significant increases in relative
liver weights were observed in males at 5000 ppm, but the absolute
liver weights were not significantly increased. Histopathological
examination revealed no treatment-related alterations in the liver or
in any other tissue examined. The NOAEL was 5000 ppm, equal to 140
mg/kg per day, on the basis of reduced body weight in males (Hellwig &
Hildebrand, 1994b).
(c) Long-term studies of toxicity and carcinogenicity
Mice
Groups of 50 male and 50 female C57Bl/6N (Cr) mice were given
diets containing kresoxim-methyl (mean purity, 96.3% during the first
12 months and 93.2% during the following six months) at concentrations
of 0, 400, 2000, or 8000 ppm for 18 months. Satellite groups of 10
mice of each sex were treated concurrently for 12 months. The doses
were equivalent to 0, 60, 300, and 1300 mg/kg bw per day in males and
0, 81, 400, and 1700 mg/kg bw per day in females in the main groups,
and 0, 61, 320, and 1400 mg/kg bw per day in males and 0, 84, 410, and
1900 mg/kg bw per day in females in the satellite groups. The animals
were observed for clinical signs, deaths, food consumption, body
weight, and haematological and pathological end-points. Blood samples
for haematology were collected during months 12 and 18 of treatment.
The study conformed to good laboratory practice.
No compound-related effects were observed with respect to
mortality rates, clinical signs, food consumption, or haematological
parameters throughout the study. Statistically significant decreases
in terminal body weights and body-weight gains were observed in the
main groups in males at 8000 ppm and in females at 2000 and 8000 ppm
during the final six months. Increased relative liver weights were
observed in females in the satellite group examined at 12 months and
in the main groups examined at 18 months at 8000 ppm. Increased
relative adrenal weights were observed in males at 12 and 18 months
and in females at 18 months. Histopathological examination at
12 months revealed no compound-related lesions in any group treated
for 12 months, but examination at 18 months revealed significantly
increased incidences of centrilobular fatty infiltration (1/50 at 0
and 16/50 at 8000 ppm) in the liver, a significantly increased
incidence and a greater degree of severity of hepatic amyloidosis
(6/50 at 0 and 16/50 at 8000 ppm), and increased incidences of
lymphoid infiltration (16/50 at 0 and 27/50 at 8000 ppm) and papillary
necrosis of the kidney (2/50 at 0 and 13/50 at 8000 ppm) in females.
There was no treatment-related increase in the incidence of neoplastic
lesions. The NOAEL was 400 ppm, equal to 81 mg/kg bw per day, on the
basis of reductions in body weight and body-weight gain in females
(Mellert & Hildebrand, 1994e).
Rats
In a study of toxicity, groups of 20 male and 20 female Wistar
rats were given diets containing kresoxim-methyl (purity, 92.7-96.6%)
at concentrations of 0, 200, 800, 8000, or 16 000 ppm, equal to 0, 9,
36, 370, and 750 mg/kg bw per day in males and 0, 12, 46, 500, and
1000 mg/kg bw per day in females, for 24 months. The animals were
observed for clinical signs, deaths, food consumption, body weight,
ophthalmological end-points, clinical chemical parameters including
the activities of serum ALAT, ASAT, AP, and GGT, and haematological,
urinary, and histopathological end-points. Blood samples for
haematology and clinical chemistry were collected at 3, 6, 12, 18, and
24 months of the treatment. The design of the study conformed to good
laboratory practice.
There were no treatment-related effects on mortality rates,
clinical signs, or ophthalmoscopic parameters. The terminal body
weight and body-weight gain were slightly reduced in males at 16 000
ppm (by 4%) and significantly reduced in females at 8000 ppm (by 9 and
13%, respectively) and 16 000 ppm (by 6 and 10%, respectively). No
significant change in food consumption was observed. Slight but
significant reductions in mean corpuscular volume and mean corpuscular
haemoglobin were observed in males at 16 000 ppm and in females at
> 200 ppm; however, these changes were within the background range
and were not clearly dose-dependent. The activity of serum ALAT was
significantly decreased in animals of each sex at 8000 and 16 000 ppm
and that of serum AP in animals of each sex at > 200 ppm. The
author suggested that these reductions in enzyme activities are not
toxicologically relevant, which is reasonable (Moss, 1994; Mellert et
al., 1997a). The relative liver weights were significantly increased
in males at 8000 and 16 000 ppm, and the absolute liver weights were
significantly increased in males at the highest dose. Significant,
dose-related increases in GGT activity were also observed in males at
> 8000 ppm.
Microscopic examination revealed evidence of neoplasia in the
liver. Increased incidences of hepatocellular carcinoma were observed
in animals of each sex at 8000 and 16 000 ppm (in males, 0/20 at 0,
1/20 at 200 ppm, 1/20 at 800 ppm, 3/20 at 8000 ppm, and 8/20 at 16 000
ppm; in females, 0/20 at 0, 0/20 at 200 ppm, 2/20 at 800 ppm, 6/20 at
8000 ppm, and 6/20 at 16 000 ppm). No hepatocellular adenomas were
observed. The incidence and severity of hepatocellular hypertrophy
were dose-dependent and increased in animals of each sex (males, 0/20
at 0, 3/20 at 800 ppm, 4/20 at 8000 ppm, and 7/20 at 16 000 ppm;
females, 1/20 at 0 and 8/20 at 16 000 ppm); however, statistical
significance was achieved only at 16 000 ppm in animals of each sex.
Significant increases in the incidence and severity of eosinophilic
foci (0/20 at 0, 6/20 at 8000 ppm, and 8/20 at 16 000 ppm) and mixed-
cell foci (0/20 at 0, 4/20 at 8000 ppm, and 5/20 at 16 000 ppm) were
observed in males. Evidence of a proliferative response in bile-duct
cells was associated with increased incidences of biliary cysts in
males at 16 000 ppm (0/20 in controls versus 4/20) and in females at
8000 and 16 000 ppm (3/20 in controls versus 7/20 and 7/20), bile-duct
proliferation in females at 8000 and 16 000 ppm (5/20 in controls
versus 8/20 and 11/20), and pericholangitis of the liver in males at
16 000 ppm (1/20 in controls versus 4/20). Significantly increased
incidences of tubular casts of the kidneys (2/20 in controls versus
10/20) and tubular atrophy of the kidney (4/20 in controls versus
12/20) were seen in females at 16 000 ppm. Increased incidences of
lesions in other tissues were age-related or independent of dose and
were not considered to be toxicologically significant.
The NOAEL for non-neoplastic alterations was 800 ppm, equal to 36
mg/kg bw per day, on the basis of increased activity of serum GGT,
increased relative liver weight, and increased incidence and degree of
severity of eosinophilic foci in males. The NOAEL for neoplasia was
also 800 ppm on the basis of an increased incidence of hepatocellular
carcinoma in animals of each sex (Mellert & Hildebrand, 1994d).
In a study of carcinogenicity, groups of 50 male and 50 female
Wistar rats were fed diets containing kresoxim-methyl (purity,
92.7-96.6%) at concentrations of 0, 200, 800, 8000, or 16 000 ppm,
equal to 0, 9, 36, 380, and 770 mg/kg bw per day for males and 0, 12,
47, 500, and 1000 mg/kg bw per day for females, for 24 months. The
animals were observed for clinical signs, deaths, food consumption,
body weight, and haematological and histopathological end-points.
Blood samples for haematology were collected at the end of the study.
The study was carried according to the principles of good laboratory
practice.
There were no treatment related effects on mortality rates or
clinical signs. The terminal body weights and body-weight gains were
significantly reduced in animals of each sex at 8000 ppm
(9 and 13% in males and 13 and 20% in females, respectively) and 16
000 ppm (9 and 12% in males and 14 and 21% in females, respectively).
No significant change was observed in food consumption. Significantly
increased relative liver weights were observed in males at 16 000 ppm.
Microscopic examination revealed hepatic neoplasia: increased
incidences of hepatocellular carcinoma were observed in animals of
each sex at 8000 and 16 000 ppm (males, 7/50 at 0, 5/50 at 200 ppm,
2/50 at 800 ppm, 18/50 at 8000 ppm, and 11/50 at 16 000 ppm; females,
1/50 at 0, 1/50 at 200 ppm, 2/50 at 800 ppm, 13/50 at 8000 ppm, and
16/50 at 16 000 ppm). The numbers of animals with adenoma plus
carcinoma in the liver were significantly increased among males at
8000 ppm (8/50 at 0, 19/50 at 8000 ppm, and 13/50 at 16 000 ppm) and
among females at 8000 and 16 000 ppm (1/50 in controls versus 15/50
and 17/50). The incidence of hepatocellular hypertrophy was increased
in males at 8000 and 16 000 ppm and in females at 16 000 ppm but
reached statistical significance only in males at 16 000 ppm (males,
3/50 at 0, 5/50 at 8000 ppm, and 10/50 at 16 000 ppm; females, 5/50 at
0, and 7/50 at 16 000 ppm). There were dose-dependent increases in the
incidences of eosinophilic foci (males, 1/50 at 0, 5/50 at 8000 ppm,
and 11/50 at 16 000 ppm; females, 3/50 at 0, 8/50 at 8000 ppm, and
5/50 at 16 000 ppm) and mixed-cell foci in animals of each sex (males,
4/50 at 0, 9/50 at 8000 ppm, and 12/50 at 16 000 ppm; females, 0/50 at
0 and 5/50 at 16 000 ppm); however, significant results were observed
only at 16 000 ppm. There was evidence of alterations in bile-duct
cells, including an increased incidence of bile-duct proliferation in
females at 16 000 ppm (10/50 in controls versus 28/50),
cholangiofibrosis in females at 16 000 ppm (1/50 in controls versus
7/50), and biliary cysts in males at 8000 ppm (males, 1/50 at 0, 7/50
at 8000 ppm, and 6/50 at 16 000 ppm; females, 8/50 at 0, 12/50 at 8000
ppm, and 15/50 at 16 000 ppm). Other non-neoplastic lesions included
tubular mineralization of the kidneys in males at 16 000 ppm (6/50 in
controls versus 18/50), and round-cell infiltration of the adrenal
cortex in males at 8000 ppm (5/50 at 0, 13/50 at 8000 ppm, and 5/50 at
16 000 ppm). The tubular mineralization was dose-related and
considered to be related to treatment. The lesions observed in other
tissues were considered to be independent of dose and age-related.
The NOAEL for non-neoplastic alterations was 800 ppm, equal to 36
mg/kg bw per day, on the basis of reduced body weight and body-weight
gain and hepatic alterations. The NOAEL for neoplasia was also 800 ppm
on the basis of increased incidences of hepatocellular carcinoma
(Mellert & Hildebrand, 1994f).
A histopathological re-evaluation on the heptocellular tumour
incidence in the two studies in rats was conducted by a pathology
working group. The results are shown in Table 4. Concurrent
reassessment revealed similar dose-response relationships in the
occurrence of hepatocellular carcinoma, and the statistically
significant results with the combined data clearly indicate the
hepatic carcinogenic potential of kresoxim-methyl in rats (van
Ravenzwaay, 1996).
(d) Genotoxicity
The results of assays for the genotoxicity of kresoxim-methyl are
summarized in Table 5. No point mutations were observed in vitro in
bacterial or mammalian cells. A significantly increased frequency of
chromosomal damage was observed in Chinese hamster lung cells with an
exogenous metabolic activation system treated with kresoxim-methyl
(purity, 93.7%) at > 100 µg/ml; however, crystals were observed in
medium cultured at 100 µg/ml for 6 h. No chromosomal damage was
observed in human lymphocytes in vitro. Assays for DNA repair and
damage in rat hepatocytes showed marked cytotoxicty, characterized by
altered cell morphology and reduced numbers of live cells at > 10
µg/ml . Kresoxim-methyl at these doses also increased extracellular
lactic dehydrogenase activity. The percent of cells in repair was
slightly increased at > 1 µg/ml (by 1-2% in comparison with 52% in
the positive control), but the authors considered these percentages to
be below their evaluation criteria (net grain, > 5%). Kresoxim-methyl
did not cause DNA damage or repair ex vivo in hepatocytes isolated
from treated rats. It did not induce micronucleus formation in mice or
rats treated in vivo.
Table 4. Incidences of hepatocellular carcinomas and other parameters in
rats in the studies of Mellert & Hildebrand (1994e,f)
Parameter Dose (ppm)
0 200 800 8000 16 000
Study of toxicity
Males
Incidence 0/20 1/20 1/20 3/20 8/20*
% incidence 0 5 5 15 40
Absolute body weight (% of control) 100 106 109 94 96
Body-weight gain (% of control) 100 109 112 91 96
Females
Incidence 1/20 0/20 2/20 6/20 6/20
% incidence 5 0 10 30 30
Absolute body weight (% of control) 100 105 88 91(*) 94(*)
Body-weight gain (% of control) 100 107 81 87(*) 90(*)
Study of carcinogenicity
Males
Incidence 7/50 5/50 2/50 18/50* 13/50*a
% incidence 14 10 4 36 26
Absolute body weight (% of control) 100 101 98 91 (*) 91 (*)
Body-weight gain (% of control) 100 102 98 87 (*) 79 (*)
Females
No of incidence 1/50 1/50 2/50 13/50* 16/50*
% of incidence 2 2 4 26 32
Absolute body weight (% of control) 100 99 98 87(*) 86(*)
Body-weight gain (% of control) 100 98 97 80(*) 79(*)
Terminal absolute body weight and body-weight gain were expressed as percent of
control, but the statistical significance was calculated on the basis of weight.
* Statistically significantly different from control.
a Includes two animals with hepatocholangiocarcinomas
Table 5. Results of assays for the genotoxicity of kresoxim-methyl
End-point Test object Concentration Purity Result Reference
(%)
In vitro
Reverse mutationa,b S. typhimurium TA98, TA100, 20-5000 µg/plate 93.7 Negative Engelhardt &
TA1535, TA1537; E. coli WP2 Hoffmann (1993a)
uvrA
Reverse mutationa,b S. typhimurium TA98, TA100, 20-5000 µg/plate 94.3 Negative Engelhardt &
TA1535, TA1537; E. coli WP2 Hildebrandt (1994)
uvrA
Reverse mutationa,b S. typhimurium TA98, TA100, 20-5000 µg/plate 90.2 Negative Engelhardt (1996)
TA1535, TA1537; E. coli WP2
uvrA
Reverse mutationb,c S. typhimurium TA98, TA100, 51-5000 µg/plate 98.6 Negative Nakajima (1997)
TA1535, TA1537; E. coli WP2
uvrA
DNA repaird B. subtilis rec M45+, H17- 191-6100 µg/plate (-S9) 98.6 Negative Nakajima (1997)
95-3050 µg/plate (+S9) Negative
Gene mutatione Chinese hamster ovary cells, 0.01-100 µg/ml (-S9) 94.3 Negative Polloth & Hoffman
hprt locus 0.1-100 µg/ml (+S9) (1994a)
Chromosomal Human lymphocytes 10-40 µg/ml 98.7 Negative Engelhardt &
aberrationb,f Hoffmann (1993b)
Chromosomal Chinese hamster lung cells 0.45-55 µg/ml (-S9) 93.7 Negative Akanuma et al. (1997)
aberrationg 50-200 µg/ml (+S9) Positive
DNA damage and Wistar rat hepatocytes 0.33-10 µg/ml 94.3 Negative Polloth & Hoffman
repairh (1994b)
Table 5. (continued)
End-point Test object Concentration Purity Result Reference
(%)
In vivo
DNA damage and Wistar rat hepatocytes Single oral gavage, 18 h 94.3 Negative Polloth & Hildebrand
repairi 0, 20, 200, 1000 mg/kg bw (1994c)
DNA damage and Wistar rat hepatocytes 3-week feeding 94.3 Negative Polloth & Hoffman
repairi 0, 200, 16 000 ppm (1994b)
Micronucleus NMRI mouse bone marrow Single i.p, 16 and 48 h, 93.7 Negative Engelhardt &
formationj 0, 500, 1000, 2000 mg/kg bw Hoffmann (1993c)
Micronucleus Wistar rat bone marrow Single i.p, 24 and 48 h, 94.9 Negative Engelhardt &
formationk 0, 500, 1000, 2000 mg/kg bw Hoffmann (1997)
S9, microsomal fraction of rat hepatocytes; i.p., intraperitoneal
All of the tests were carried out according to good laboratory practice, and all of the positive controls produced the expected results.
a In dimethyl sulfoxide; the positive controls were 2-aminoanthracene, N-methyl-N'-nitro-N-nitrosoguanidine,
N-ethyl-N'-nitro-N-nitrosoguanidine, 9-aminoacridine chloride, and 4-nitro-ortho-phenylendiamine.
b In the presence and absence of S9
c In dimethyl sulfoxide; the positive controls were 2-(2-furyl)-3-(5-nitro-2-furyl)acrylamide for TA98, TA100, and WP2uvrA; sodium
azide for TA1535; and 9-aminoacridine chloride for TA1537 -S9 and 2-aminoanthracene + S9.
d Positive controls were mitomycin C -S9 and try-P-1 +S9; negative control was kanamycin -S9.
e Positive controls were ethylmethanesulfonate -S9 and 3-methylcholanthrene +S9.
f Positive controls were mitomycin C -S9 and cyclophosphamide +S9.
g Positive controls were mitomycin C -S9 and benzo[a]pyrene.
h Positive control was 2-acetylaminofluorene.
i Positive control was 2-acetylaminofluorene at a single oral dose of 50 mg/kg bw.
j Positive controls were cyclophosphamide at a single i.p dose of 20 mg/kg bw and vincristine at a single i.p dose of 0.15 mg/kg bw.
k Positive control was cyclophosphamide at a single i.p dose of 20 mg/kg bw.
(e) Reproductive toxicity
(i) Multigeneration reproductive toxicity
Rats
In a two-generation study of reproductive toxicity, which
conformed to good laboratory practice, groups of 25 male and 25 female
Wistar rats were fed diets containing kresoxim-methyl (purity,
> 93.7%) at concentrations of 0, 50, 1000, 4000, or 16 000 ppm. The
F0 generation was exposed directly, the F1a and F1b generations
directly and indirectly, and F2 generation indirectly. The mean daily
intakes of kresoxim-methyl by the F0 generation were 5, 100, 410, and
1600 mg/kg bw per day for males and 6, 120, 480, and 2300 mg/kg bw per
day for females. Female intakes were 6, 110, 440, and 1700 mg/kg bw
per day during premating; and 4, 87, 360, and 1400 mg/kg bw per day
during gestation and 7, 150, 600, and 2400 mg/kg bw per day during
lactation for the F1a and F1b generations. The mean daily intakes by
the F1 generation were 4, 88, 360, and 1500 mg/kg bw per day for
males and 5, 110, 440, and 1800 mg/kg bw per day for females. female
intakes were 5, 100, 420, and 1700 mg/kg bw per day during premating;
4, 85, 350, and 1300 mg/kg bw per day during gestation for F2a; and
7, 140, 560, and 2300 mg/kg bw per day during lactation for F2a.
The parental rats were observed for clinical signs, deaths, food
consumption, body weight, and clinical chemical, histopathological,
and reproductive parameters including mating, fertility, gestation,
and live-birth indices. The litters and pups were observed for
viability, lactation, behaviour, and developmental indices that
included pinna unfolding and opening of the auditory canal and eyes.
The functional tests included grip strength, startle reflex, and
pupillary reflex. Reproductive organs and the pituitary, liver, and
kidney were examined histopathologically. The clinical chemical
end-points included assays for serum ALAT, ASAT, AP, and GGT activity.
No compound-related clinical signs or deaths were observed in the
F0, F1, or F2 generation throughout the study. F0 and F1 parental
animals showed no effects on mating, fertility, gestation, or
live-birth indices, but significant reductions in food consumption
were observed in F0 and F1 males during treatment and in F0 and F1
females during gestation and lactation at 16 000 ppm. Significant
reductions in body weight were seen at doses of 4000 ppm and higher in
F0 and F1 males and in F0 and F1 females during gestation and
lactation of the F1a and F1b generations. Significant reductions in
body-weight gain were also observed in F0 and F1 males at these
doses and in F0 females at 16 000 ppm during the premating period
before the first gestation. Significant reductions in the activities
of ALAT and AP were observed in F0 and F1 parents of each sex,
although these reductions may not be toxicologically relevant (Moss,
1994; Mellert & Hildebrand, 1995). The activity of GGT was
significantly increased in F0 males at 4000 ppm and higher and in F1
animals of each sex at 16 000 ppm. Significantly decreased numbers of
fat storage cells were observed in the livers of F0 and F1 males at
4000 ppm and higher; however, this change may have occurred as a
result of the reduced food consumption at higher doses. Significant
increases in relative kidney weights were observed in F0 males at 16
000 ppm and in F0 females and F1 males at 4000 ppm and higher. No
treatment-related morphological lesions were observed in the liver or
kidney.
No compound-related changes in clinical signs, sex ratio,
viability index, or lactation index were seen in pups of the F1a,
F1b, and F2a generations. Body weights and body-weight gain during
lactation were significantly decreased in F1a, F1b, and F2a pups at
4000 ppm and higher. A significantly lower percentage of F1b pups at
these doses had pinna unfolding; significant retardations in opening
of the auditory canal and eyes were also observed in F1b and F2a pups
at 4000 ppm,but were not dose-dependent. There were no differences in
the results of reflex tests between controls and treated animals in
any generation. Necroscopy of pups revealed no external abnormality.
The NOAEL for parental toxicity was 1000 ppm, equal to 100 mg/kg
bw per day in F0 males and 88 mg/kg bw per day in F1 males, on the
basis of reduced body weight and body-weight gain, increased serum GGT
activity, and increased relative kidney weights. The NOAEL for
reproductive toxicity was 1000 ppm, equal to an overall mean intake of
100 mg/kg bw per day for F1 and F2 pups, on the basis of reduced
body weight and body-weight gain (Hellwig & Hildebrand, 1994a).
(ii) Developmental toxicity
Rats
Groups of 25 female Wistar rats were given kresoxim-methyl
(purity, > 93.7%) suspended in 0.5% CMC by gavage at a dose of 0,
100, 400, or 1000 mg/kg bw per day on days 6-15 of gestation. The
study was conducted in accordance with good laboratory practice. No
treatment-related changes in clinical signs, mortality rates, body
weight, or food consumption were observed in maternal animals. There
were no differences in conception rate, mean number of corpora lutea,
total implantations, resorptions, pre- or post-implantation loss, or
number of live fetuses. No significant differences in fetal sex ratio,
placental weight, or fetal body weight were observed between control
and treated groups. External examination revealed three fetuses with
external malformations: one fetus at 100 mg/kg bw per day had anasarca
and a cleft palate, one fetus at 400 mg/kg bw per day was acaudate,
and one fetus at 1000 mg/kg bw per day had meningocele and unilateral
microphthalmia; however, the incidence of these malformations was
within the range for historical controls. One fetus at 1000 mg/kg bw
per day had hydrocephalus, but this incidence was also within the
historical control range. A significantly increased incidence of
incompletely ossified thoracic vertebral bodies was seen in 23% of all
fetuses and 58% of litters at 1000 mg/kg bw per day; the mean
historical control values were 8% (0-49%) of all fetuses and 23%
(0-100%) of litters. The NOAEL for maternal toxicity was 1000 mg/kg bw
per day, and that for embryo and fetal toxicity was 400 mg/kg bw per
day on the basis of a slight increase in variations in fetuses at 1000
mg/kg bw per day. There was no evidence of teratogenicity at doses
< 1000 mg/kg bw per day (Hellwig, 1994).
Rabbits
Groups of 15 female Himalayan rabbits were given kresoxim-methyl
(purity, 96.6%) suspended in 0.5% CMC by gavage at a dose of 0, 100,
400, or 1000 mg/kg bw on days 7-19 of gestation. The study was
conducted in accordance with good laboratory practice. No
compound-related changes in clinical signs, deaths, body weight, or
food consumption were observed in maternal animals, and there were no
compound-related changes in conception rate, mean numbers of corpora
lutea, total implantations, resorptions, pre- or post-implantation
loss, or live fetuses. No significant differences in fetal sex ratio,
placental weight, or fetal body weight were observed between control
and treated groups. External examination revealed one fetus with
microcephaly and brachygnathia at 100 mg/kg bw per day, but the
incidence was within that of historical controls. Eight fetuses (0 /15
at 0, 2/15 at 100, 2/15 at 400, and 3/15 at 1000 mg/kg bw per day) had
soft-tissue malformations: one at 100 mg/kg bw per day had a septal
defect and one had agnesis of the gall-bladder (2.5% incidence); two
at 400 mg/kg bw per day had a septal defect (1.9%); at 1000 mg/kg bw
per day, one had a septal defect, dilatation of the aortic arch, and a
descending aortic, one had hydrocephaly, and one had agnesis of the
gall-bladder (4.1%). The percent of soft-tissue malformations in
historical controls was 2.2-3.1%. The incidences of ventricular septal
defects in the treated groups were comparable to historical values.
Increased incidences of fused sternebrae were observed in 3/15
controls, 11/15 at 100 mg/kg bw per day, 7/15 at 400 mg/kg bw per day,
and 9/15 at 1000 mg/kg bw per day; the increase at 100 mg/kg bw per
day was significant but was within the historical control range.
Increased total numbers of fetal malformations were also observed in
treated groups but again at incidence rates comparable to those of
historical controls (0% at 0, 4.9% at 100, 3.8% at 400, and 4.1% at
1000 mg/kg bw per day versus 2.9-3.5% for historical controls). The
NOAEL for both maternal and developmental toxicity was thus 1000 mg/kg
bw per day, the highest dose tested (Hellwig & Hildebrand, 1993, GLP)
(f) Special studies
(i) Tumour initiating potential
Groups of 10 Wistar rats of each sex were subjected to a partial
hepatectomy and 14 h later received a single dose of 2388 mg/kg bw
technical-grade kresoxim-methyl (purity, 92.7-94.3%) suspended in 0.5%
CMC by gavage to rats. For promotion, phenobarbital was incorporated
in the diet at a concentration of 500 ppm for eight weeks. Liver
slices were examined histologically on slides stained with
haematoxylin and eosin (H&E) or stained immunochemically for the
placental form of glutathione S-transferase (GST-P). The study
conformed to good laboratory practice. The incidences of
hepatocellular alteration (foci) and of GST-P-positive foci were used
to estimate initiating potential. N-Nitrosomorpholine was used as
the positive control. Hepatocellular hypertrophy was found in almost
all of the phenobarbital-treated animals, and GST-positive foci and
foci of hepatocellular alteration were found in nearly all animals
treated with the positive control. The number of animals with
GST-P-positive foci in groups treated with kresoxim-methyl was
comparable to that of vehicle controls. The numbers of foci per liver
in promoted animals were 0-3 in those given kresoxim-methyl, 0-10 in
vehicle controls, and 3-100 in positive controls. The results suggest
that kresoxim-methyl does not have tumour initiating potential in rats
in this test (Gamer & Hildebrand, 1995).
(ii) Tumour promoting potential
In a medium-term study of promotion, which did not conform to
good laboratory practice, male Fischer rats were initiated with a
single intraperitoneal injection of N-nitrosodiethylamine at a dose
of 299 mg/kg bw. The animals were then maintained on basal diet
ad libitum for 14 days. Five groups of 16 male rats were fed diets
containing 0, 200, 800, 8000, or 16 000 ppm kresoxim-methyl (purity,
95.4%) for six weeks, with average intakes of 0, 11, 42, 430, and 890
mg/kg bw per day (not adjusted for purity). The remaining 16 male rats
were fed a diet containing 500 ppm phenobarbital (28 mg/kg bw per day)
as a positive control for six weeks. The animals were subjected to a
two-thirds partial hepatectomy after the first week of feeding with
kresoxim-methyl or phenobarbital and were observed for clinical signs,
deaths, food consumption, and body weight. The liver was examined
grossly and histopathologically.
There were no compound-related deaths or clinical signs of
toxicity. Body weight and food consumption in groups given
kresoxim-methyl were comparable to those of controls. Significant
increases in the absolute and relative weights of the liver were
observed in groups given kresoxim-methyl at 800 ppm and higher.
Treatment with phenobarbital caused significant increases in body
weight, food consumption, and relative liver weight. Quantification of
hepatic foci with a computer-assisted image analyser revealed
significant, dose-related increases in the number and area of
GST-P-positive hepatocellular foci in groups given kresoxim-methyl at
> 8000 ppm, as well as in the phenobarbital-treated positive
controls. The NOAEL for promotion was 800 ppm (Harada et al., 1997).
(iii) Hepatic-cell proliferation
A series of studies was conducted to investigate the effect of
kresoxim-methyl on hepatic-cell proliferation in rats, by measuring
S-phase DNA synthesis, an indicator of cell proliferation.
Incorporation of bromodeoxyuridine (BrdU) into DNA was measured by
immunohistochemical staining.
In the first study, groups of five young male Wistar rats, 64
days old, were given diets containing kresoxim-methyl (purity, 94.3%)
at concentrations of 0, 200, or 16 000 ppm, equal to 0, 15, and 1100
mg/kg bw per day, for three weeks. Osmotic minipumps filled with
BrdU were implanted subcutaneously one week before necroscopy. the
animals were observed for clinical signs, deaths, food consumption,
and body weight. The livers were examined grossly and
immunohisto-pathologically. Samples of the hepatic lobule and the
jejunum were taken as positive tissues for proliferation and were
stained with H&E and immunochemically with an antibody against BrdU.
Immunopositive and H&E-counterstained hepatocyte nuclei from 11 fields
for each of three lobes were counted. No treatment-related changes in
body weight, food consumption, or clinical signs were seen. A slight
increase in liver weights was observed at 16 000 ppm, but no
treatment-related gross lesions or histopathological changes were
observed in the livers of treated rats. A statistically significant
increase in the number of hepatocytes in which BrdU was incorporated
into the DNA of S-phase cells was observed in the periportal zone
(zone 1) and the intermediate zone (zone 2) of the hepatic lobule in
the group at 16 000 ppm. No significant increase in cell proliferation
was observed in the group at 200 ppm (Polloth & Hildebrand, 1994a).
In a supplementary study with a similar design, groups of five
young male Wistar rats received kresoxim-methyl (purity, 94.9% ) in
the diet at a concentration of 0, 800, or 8000 ppm, equal to 0, 61,
and 600 mg/kg bw per day, for three weeks. Results similar to those
observed at 16 000 ppm in the first study were observed at 8000 ppm.
Statistically significant increases in cell proliferation were
observed in zones 1 and 2 of the hepatic lobule at 8000 ppm, but not
at 800 ppm (Mellert et al., 1997a).
The NOAEL from the combined results of these two studies for
hepatic cell proliferation was 800 ppm, equal to 61 mg/kg bw per day.
In a study of the hepatic proliferating activity of
kresoxim-methyl in the livers of older rats, groups of five male
Wistar (Chbb) rats aged 16 months were given diets containing
kresoxim-methyl (purity, 94.3%) at a concentration of 0, 200, or 16
000 ppm for three weeks. The design of the study was similar to those
described above. No compound-related changes were seen in clinical
signs or body weight, and no compound-related lesions in the liver
were observed by microscopic examination with H&E staining. A
statistically significant increase in cell proliferation was observed
in zone 1 of the hepatic lobule at 16 000 ppm, which was comparable to
that observed in the young rats (Polloth & Hildebrand, 1994d) .
In a study of the hepatic proliferating activity of
kresoxim-methyl in the livers of rats treated for various periods,
groups of five male Wistar (Chbb) rats, 42 days old, were given diets
containing kresoxim-methyl (purity, 92.7%) at a concentration of 0 or
16 000 ppm for 1, 6, or 13 weeks. Groups were were allowed to recover
for two or three weeks. Significant increases in cell proliferation
were observed in the treated groups after one week (zones 1, 2, and 3)
and after six weeks (zone 1). The increase in zone 1 in the group
treated for one week was greater than that in the group treated for
six weeks. This compound-related enhancement of cell proliferation was
significantly reversed in the groups allowed to recover. The zonal
distribution of increased cell proliferation revealed a selective
effect of kresoxim-methyl on hepatocytes in zone 1 (Mellert et al.,
1996a).
In a study of unscheduled DNA synthesis and S-phase response in
rat hepatocytes, groups of three male Wistar (Chbb) rats received a
single oral dose of 0, 20, 200, or 1000 mg/kg bw kresoxim-methyl
(purity, 94.3%) by gavage. 2-Acetylaminofluorene was used as a
positive control, at a dose of 50 mg/kg bw in the assay of unscheduled
DNA synthesis and at 1000 mg/kg bw in the assay of S-phase response.
Hepatocytes were prepared by in-situ hepatic perfusion 18 h after
treatment. The isolated hepatocytes were cultured with 3H-thymidine
for 18 h, and S-phase response and unscheduled DNA synthesis were
evaluated autoradiographically in the labelled cells. Exposure of rats
to kresoxim-methyl in vivo was not cytotoxic to liver cells. Slight
but dose-dependent increases in the number of cells in S-phase were
observed in all treated groups, with 1% at 0, 1.37% at 20, 2.78% at
200, and 2.58% at 1000 mg/kg bw, as well as in the positive control
group (5.87%). The results suggest that kresoxim-methyl induced a
moderate increase in S-phase DNA synthesis at 200 mg/kg bw and has a
weak potential for enhancing hepatic cell proliferation (Polloth &
Hildebrand, 1994c).
(iv) Morphology of hepatic proliferation
Groups of three female Wistar (Chbb) rats, 12 weeks old, received
diets containing kresoxim-methyl (purity, 94.3% ) at concentrations of
0, 200, or 16 000 ppm, equal to 0, 15, and 1200 mg/kg bw per day, for
three weeks. At termination, the livers were fixed in situ by
perfusion, and the peroxisomes in the liver were examined by light and
electron microscopy after staining with diaminobenzidine to detect
catalase activity. There were no compound-related changes in clinical
signs, body weight, or food consumption; reduced body-weight gain was
observed at 16 000 ppm. No compound-related lesions were observed in
the liver, and no difference was seen between treated and control
animals in the numbers of peroxisomes (Mellert et al., 1995a).
Groups of three female Wistar (Chbb) rats, 15 months old,
received diets containing kresoxim-methyl (purity, 94.3%) at
concentrations of 0, 200, or 16 000 ppm for three weeks and were then
fixed in situ by perfusion. Liver samples were examined by light and
electron microscopy. There were no compound-related changes in
clinical signs or body weight, and no compound-related lesions were
observed in the liver on light microscopic examination. Electron
microscopy showed that the amount, shape, and size of hepatocyte
mitochondria in the treated group were comparable to those in
controls. (Mellert et al., 1995b).
(v) Induction of hepatic metabolic enzyme activities
Groups of 10 male and 10 female Wistar rats were fed diets
containing kresoxim-methyl at concentrations of 0, 200, or 16 000 ppm
for three weeks, equal to 0, 13, and 1000 mg/kg bw per day for males
and 0, 15, and 1200 mg/kg bw per day for females. The animals were
observed for clinical signs, deaths, body weight, and food
consumption. Indicators of hepatic enzymes were measured, including
the activities of GGT and drug metabolizing enzymes, the concentration
of glutathione in liver homogenates, and the content of cytochrome
P450 in microsomes. Significant increases in the activities of GGT and
pentoxyresorufin depentylase and in P450 content were observed in
males at 16 000 ppm. The pattern of induction of drug metabolizing
enzyme activities resembled that of phenobarbital. In females, only a
tendency towards induction was observed (Mellert et al., 1996b).
(vi) Mechanism of decreased serum enzyme activities
As marked reductions in the activities of serum AP and ALAT were
reported in short- and long-term studies of toxicity, a series of
experiments was conducted in which groups of five males and five
females were fed diets containing kresoxim-methyl at a concentration
of 8000 ppm for two weeks. These studies did not conform to good
laboratory practice. In the first experiment, AP activity was
determined in serum samples and extracts of liver and small intestine.
The intestinal activity of AP was not changed by treatment, the
estimated ratio of intestinal and hepatic or bone AP isozyme
activities in the serum being 38.5%. The author indicated the
reduction in serum AP activity observed in the kresoxim-methyl treated
groups was mostly due to a reduction in intestinal AP activity. In the
second experiment, serum AP activity was markedly reduced after
fasting and was increased by feeding a diet supplemented with olive
oil. In the third experiment, addition of sera collected from treated
animals to sera collected from untreated animals did not suppress AP
activity, indicating the absence of an inhibitor. The observed
reductions in serum AP and ALAT activities was therefore probably due
to a slight alteration in food absorption in treated rats. (Moss,
1994).
In a second study to investigate the reduced enzyme activities,
groups of 10 male and 10 female Wistar rats were fed diets containing
kresoxim-methyl at a concentration of 0 or 16 000 ppm for two weeks,
equal to 910 mg/kg bw per day for males and 1100 mg/kg bw per day for
females. The animals were observed for clinical signs, deaths, body
weight, and food consumption. ALAT and AP activities in serum and
urine were assayed at the end of the study. There were no
compound-related changes in clinical signs or mortality rates.
Significantly decreased food consumption was observed in treated
animals of each sex. A slight but significant decrease in body weight
was observed in treated males. Significantly reduced activities of
ALAT and AP in serum were observed in animals of each sex, but no
change in the activities of either enzyme was observed in urine. No
change in urinary creatinine or urinary volume was observed in treated
animals, indicating no change in renal function. Thus, the reduced
enzyme activity observed in sera of kresoxim-methyl-treated rats was
not caused by a change in renal excretion of the enzymes (Mellert et
al., 1997b).
(g) Studies on metabolites
(i) Acute toxicity
Metabolites M1, M2, and M9 were given orally to rats in a
suspension of 0.5% CMC. M1 (purity, 98.5%) produced a variety of
abnormal clinical changes including dyspnoea, staggering gait, and
tremor at doses of 2000 mg/kg bw and higher. M2 (purity, 97.7%) caused
no deaths or abnormal symptoms at 5000 mg/kg bw. M9 (purity, 99.6%)
caused dyspnoea and exhaustion in animals of each sex at 5000 mg/kg bw
but resulted in no change in general appearance at 3000 mg/kg bw
(Kirsch & Hildebrand, 1994a,b,c, 1995).
(ii) Genotoxicity
M1, M2, and M9 of the same purities described above did not
induce reverse mutation in bacteria at a concentration of 5000
µg/plate, whereas the positive controls used gave the expected
positive responses (Hoffman & Engelhardt, 1995a,b,c)
Comments
About 60% of an oral dose of 50 mg/kg bw and 25% of a dose of 500
mg/kg bw kresoxim-methyl was absorbed. It was excreted mainly in the
faeces (70% of the low dose and 80% of the high dose), predominantly
via the bile (about 40% of the low dose and 15% of the high dose
within 48 h), with lesser amounts in urine (about 20% of the low dose
and 10% of the high dose). Peak levels of the radiolabel in plasma
were reached 0.5-1 h after the low dose and 8 h after the high dose.
The plasma half-life was 17-19 h at the low dose and 22-31 h at the
high dose. The highest residual concentrations were found in the
liver, but the concentrations in all tissues, including the liver,
were less than 0.1 g equivalent/g tissue after 120 h of treatment at
the low dose.
After oral administration of kresoxim-methyl, a high proportion
of the parent compound was found in the faeces, but none was detected
in tissues or bile examined 4 h after dosing. In rats, 34 metabolites
of kresoxim-methyl were identified. The proposed metabolic pathways
are hydrolytic cleavage of the ester, the oxime ether, and the benzyl
ether bonds, hydroxylation at the para position of the phenoxy ring,
hydroxylation of the aryl-methyl group and its subsequent oxidation to
form the corresponding carboxylic acid, and conjugation of the
resulting hydroxy groups with glucuronate or sulfate. The major
metabolites identified in both rats and plants were the free acid,
code number 490M1 {(E)-methoxyimino[alpha- (ortho-tolyloxy)- ortho-
tolyl]acetic acid}, the hydroxy derivative of this, 490M2
[alpha- (ortho-hydroxymethylphenoxy)- ortho-tolyl
(methoxyimino)acetic acid] formed by hydroxylation of the aryl-methyl
group, the para-hydroxytolyloxy product 490M9
[alpha- (para-hydroxy- ortho-tolyloxy)- ortho-tolyl
(methoxyimino)acetic acid], and their conjugates. 490M1, 490M2, and
490M9 all had low acute toxicity and were not mutagenic.
WHO has not classified kresoxim-methyl for acute toxicity.
In a range-finding study in B6C3F1 mice, kresoxim-methyl was
administered in the diet at concentrations of 0, 500, 2000, or 8000
ppm for 28 days. The NOAEL was 8000 ppm, equal to 2100 mg/kg bw per
day. In a three-month study, C57Bl/6N mice received kresoxim-methyl in
the diet at concentrations of 0, 250, 1000, 4000, or 8000 ppm. The
NOAEL was 8000 ppm, equal to 1900 mg/kg bw per day.
In a range-finding study in rats, kresoxim-methyl was
administered in the diet at concentrations of 0, 1000, 4000, or 16 000
ppm for 28 days. The NOAEL was 4000 ppm in males, equal to
370 mg/kg bw per day, on the basis of increased activities of serum
gamma-glutamyl transferase at 16 000 ppm, equal to 1500 mg/kg bw per
day. In a three-week study of toxicity in rats, kresoxim-methyl was
administered in the diet at concentrations of 0, 10, 50, or 8000 ppm.
The NOAEL was 50 ppm, equal to 3 mg/kg bw per day, on the basis of
increased hepatic gamma-glutamyl transferase activity in males at 8000
ppm. In a 90-day study of toxicity in rats, kresoxim-methyl (purity,
98.7%) was administered in the diet at concentrations of 0, 500, 2000,
8000, or 16 000 ppm. The NOAEL was 500 ppm in females, equal to 43
mg/kg bw per day, based on increased relative liver weight at 2000 ppm
and above, and 2000 ppm in males, equal to 150 mg/kg bw per day, based
on decreased body-weight gain and increased activity of serum
gamma-glutamyl transferase at 8000 ppm and above.
In a three-month study of toxicity in dogs, kresoxim-methyl was
administered at dietary concentrations of 0, 1000, 5000, or 25 000
ppm. The NOAEL was 5000 ppm, equal to 140 mg/kg bw per day, on the
basis of vomiting, diarrhoea, and reduced body-weight gain in animals
of each sex at 25 000 ppm. In a 12-month study in dogs,
kresoxim-methyl was administered at dietary concentrations of 0, 1000,
5000, or 25 000 ppm. The NOAEL was 5000 ppm, equal to 140 mg/kg bw per
day, on the basis of a reduction in body weight in males at 25 000
ppm. No compound-related toxicity was observed in females.
In an assay for carcinogenicity in mice, kresoxim-methyl was
administered at dietary concentrations of 0, 400, 2000, or 8000 ppm
for 18 months. The NOAEL was 400 ppm, equal to 81 mg/kg bw per day, in
females on the basis of reduction in body weight at 2000 ppm. The
NOAEL in males was 2000 ppm, equal to 300 mg/kg bw per day, on the
basis of decreased body weight and increased relative adrenal weight
at 8000 ppm. At this dose, increased incidences of renal papilliary
necrosis and hepatic amyloidosis were observed in females. There was
no evidence of carcinogenicity.
In a two-year study of toxicity in rats, kresoxim-methyl was
administered at dietary concentrations of 0, 200, 800, 8000, or 16 000
ppm. The NOAEL was 800 ppm, equal to 36 mg/kg bw per day, on the basis
of an increased incidence of hepatocellular carcinoma in animals of
each sex, increased serum gamma-glutamyl transferase activity,
increased relative liver weight, an increased incidence and degree of
severity of eosinophilic foci and mixed-cell foci in males, and a
decrease in terminal body weight and body-weight gain in females at
8000 ppm and 16 000 ppm. There was also an increased incidence of
biliary cysts and bile-duct proliferation.
In a study of carcinogenicity in rats, kresoxim-methyl was
administered at dietary concentrations of 0, 200, 800, 8000, or 16 000
ppm for 24 months. Evidence of biliary alterations included increased
incidences of biliary cysts and cholangiofibrosis in females at 16 000
ppm. At this dose, increased relative liver weights and an increased
incidence of hepatocellular hypertrophy were observed in males. The
NOAEL was 800 ppm, equal to 36 mg/kg bw per day, on the basis of
increased incidences of hepatocellular carcinoma, reductions in body
weight and body-weight gain, and an increased incidence of
eosinophilic foci and mixed-cell foci in animals of each sex at 8000
ppm and above. The overall NOAEL for neoplastic and non-neoplastic
effects was 800 ppm, equal to 36 mg/kg bw per day.
It is generally recognized that the process of carcinogenesis is
divided into three stages: initiation, promotion, and progression. A
series of mechanistic studies was conducted with kresoxim-methyl,
including tests for tumour initiating and promoting potential. In a
study on tumour initiating activity, kresoxim-methyl did not increase
the number of liver-cell foci in rats at a single dose of 2400 mg/kg
bw. In a study on the promoting potential of kresoxim-methyl, rats
received an initiating dose of N-nitrosodiethylamine and then a diet
containing 0, 200, 800, 8000, or 16 000 ppm kresoxim-methyl for six
weeks. Quantitative analysis of hepatic foci with a computer-assisted
image analyser revealed significant, dose-dependent increases in the
number and area of placental-type glutathione S-transferase-positive
hepatocellular foci, indicating a promoting effect of kresoxim-methyl
on hepatocarcinogenesis at doses of 8000 ppm and above. The NOAEL for
the promoting effect was 800 ppm, equal to 43 mg/kg bw per day.
Four studies were conducted to investigate the effect of
kresoxim-methyl on hepatic-cell proliferation in rat liver by
measuring bromodeoxyuridine incorporation into hepatocyte DNA during
S-phase DNA synthesis. The results showed a selective cell
proliferation effect of kresoxim-methyl on hepatocytes in the
periportal zone. The NOAEL was 800 ppm, equal to 61 mg/kg bw per day,
while animals treated with 8000 ppm and above showed a statistically
significant increase in cell proliferation. There was no difference in
the sensitivity of young and old rats.
The genotoxic potential of kresoxim-methyl was investigated in a
series of tests, including assays for gene mutation in bacteria and
mammalian cells, unscheduled DNA synthesis, and cytogenicity
in vitro, an assay for micronucleus formation in vivo, and an
assay for unscheduled DNA synthesis ex vivo. Kresoxim-methyl had
moderate potential to induce chromosomal aberrations in vitro with
exogenous metabolic activation, but positive effects were not observed
in any other test, including the assay for micronuclei in rat bone
marrow. The Meeting concluded that kresoxim-methyl is not genotoxic.
The three major metabolites in rats did not induce reverse mutation in
Salmonella typhimurium in vitro.
The increased incidence of liver tumours observed in rats at 8000
ppm and above was considered to be associated with increased cell
proliferation. The mechanistic studies indicated that kresoxim-methyl
has tumour promoting potential at 8000 ppm, which coincides with the
lowest level at which increased liver-cell proliferation was observed.
These results indicate a threshold for the neoplastic mode of action.
The Meeting concluded that a level of 800 ppm kresoxim-methyl has no
carcinogenic potential.
In a two-generation study of reproductive toxicity in rats, the
NOAEL values were 1000 ppm for parental animals of each sex, 100 mg/kg
bw per day for F0 offspring, and 88 mg/kg bw per day for F1
offspring; these were based on reductions in body weight and
body-weight gain and increased serum gamma-glutamyl transferase
activity and relative kidney weight at 4000 ppm and above. The NOAEL
for pups was 1000 ppm, equal to 110 mg/kg bw per day for F1 pups and
97 mg/kg bw per day for F2 pups, on the basis of reductions in body
weight and body-weight gain at 4000 ppm and above.
The NOAEL for embryo- and fetotoxicity in a study of
developmental toxicity in rats was 400 mg/kg bw per day. No maternal
toxicity or teratogenic effects were observed at doses up to and
including the highest one of 1000 mg/kg bw per day. Kresoxim-methyl
did not induce toxicity in a study of developmental toxicity in
rabbits up to and including the highest dose of 1000 mg/kg bw per day.
An ADI of 0-0.4 mg/kg bw was established on the basis of the
NOAEL of 800 ppm, equal to 36 mg/kg bw per day, in the 24-month study
of toxicity and carcinogenicity in rats, and a 100-fold safety factor.
An acute RfD was not allocated because kresoxim-methyl has low
acute toxicity and did not exhibit developmental toxicity. The Meeting
concluded that the acute intake of residues is unlikely to present a
risk to consumers.
Toxicological evaluation
Levels that cause no toxic effect
Mouse: 400 ppm, equal to 81 mg/kg bw per day (18-month study
of toxicity)
Rat: 800 ppm, equal to 36 mg/kg bw per day (two-year study
of toxicity and carcinogenicity )
1000 ppm, equal to 88 mg/kg bw per day (two-generation
study of reproductive toxicity)
400 mg/kg bw per day (study of developmental toxicity)
Rabbit: 1000 mg/kg bw per day (developmental toxicity; highest
dose tested)
Dog: 5000 ppm, equal to 140 mg/kg bw per day (12-month study
of toxicity)
Estimate of acceptable daily intake for humans
0-0.4 mg/kg bw
Estimate of acute reference dose
Not allocated (unnecessary)
Studies that would provide information useful for continued
evaluation of the compound
Observations in humans
List of end-points relevant for setting guidance values for dietary and non-dietary exposure
Absorption, distribution, excretion, and metabolism in mammals
Rate and extent of oral absorption Rapid, 25-60% absorbed
Dermal absorption No data
Distribution Minimum, highest levels in liver
Potential for accumulation Very little
Rate and extent of excretion Rapid/complete, 87-93% within 48 h
Metabolism in animals Extensive. No parent compound in urine, bile, or
tissues; 34 metabolites identified.
Toxicologically significant compounds Parent compound in rat; three major metabolites in
(animals, plants and environment) plants
Acute toxicity
Rat: LD50 oral > 5000 mg/kg bw
Rat: LD50 dermal > 2000 mg/kg bw
Rat: LC50 inhalation > 5.6 mg/L
Skin irritation Not irritating
Eye irritation Not irritating
Skin sensitization Not sensitizing
Short-term toxicity
Target/critical effect Liver: increased relative liver weight (mouse, rat)
Lowest relevant oral NOAEL Rat: 28-day, 43 mg/kg bw per day
Lowest relevant dermal NOAEL No data
Lowest relevant inhalation NOAEL No data
Genotoxicity Not genotoxic
Long-term toxicity and carcinogenicity
Target/critical effect: Hepatocellular carcinoma
Lowest relevant NOAEL Rat: 2-year, 36 mg/kg bw per day, diet
Carcinogenicity Non-genotoxic carcinogen, tumour promoter
Reproductive toxicity
Reproduction target/critical effect Reduction in F0 body weight at parenterally toxic
dose
Lowest relevant reproductive NOAEL Rat: 97 mg/kg bw per day, diet
Developmental target/critical effect None
Lowest relevant developmental NOAEL Rat: 1000 mg/kg bw per day, highest dose tested
Neurotoxicity/Delayed neurotoxicity No data
Other toxicological studies No data
Medical data No data
Summary Value Study Safety factor
ADI 0-0.4 mg/kg bw 2-year study of 100
toxicity and
carcinogenicity, rat
Acute reference dose Not allocated
(unnecessary )
References
Akanuma, M. et al. (1997) Report: In vitro cytogenetic investigations
of Reg. No. 242009 in Chinese hamster cultured cell. Unpublished
report No. IET 97-0021 from Institute for Environmental Toxicology,
Kodaira, Tokyo, Japan. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Engelhardt, G. (1996) Report on the study of Reg. No. 242009 in the
Ames Salmonella/mammalian-microsome mutagenicity test and Escherichia
coli/mammalian-microsome reverse mutation assay (standard plate test
preincubation test). Unpublished report No. 93/10249 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1993a) Report on the study of Reg.
No. 242009 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation assay
(standard plate test preincubation test). Unpublished report No.
93/10249 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1993b) Report: In vitro cytogenetic
investigations of Reg. No. 242009 in human lymphocytes. Unpublished
report No. 93/10493 from BASF Aktiengesellschaft, Ludwigshafen,
Germany. Submitted to WHO by BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1993c) Report: Cytogenetic study in
vivo of Reg. No. 242009 in mice micronucleus test. Single
intraperitoneal sdministration.Unpublished report No. 93/11104 from
BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by
BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1994) Report on the study of Reg. No.
242009 in the Ames test (Salmonella/mammalian-microsome mutagenicity
test--Standard plate test preincubation test). Unpublished report No.
93/10249 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Engelhardt, G. & Hoffmann, H.D. (1997) Report: Cytogenetic study in
vivo of Reg. No. 242009 in rat micronucleus test. Single
intraperitoneal administration. Unpublished report No. 06M0180/914384
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Gamer, A.O. & Hildebrand, B. (1995) Report: Study of foci initiating
activity of Reg. No. 242009 in Wistar rats. Unpublished report No.
95/10244 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Gamer, A.O. & Kirsch, P. (1992) Report: Study on the acute oral
toxicity of Reg. No. 242009 in rats. Unpublished report No. 94/10753
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Gans, G. (1994) Study of the biokinetics of [14C]-Reg. No. 242009
([14C]-BAS 490F) in rats. Unpublished report No. 94/10992 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Grosshans, F. (1994a) The metabolism of 14C-242009 in apples. BASF
Reg. No. 94/11760. Unpublished report from BASF Aktiengesellshaft,
Limburgerhof, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Grosshans, F. (1994b) The metabolism of 14C-242009 in wheat. BASF
Reg. No. 94/10685. Unpublished report from BASF Aktiengesellshaft,
Limburgerhof, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Harada, T. et al. (1997) BAS 490F (Reg. No. 242009): Medium term
promotion hepato-carcinogenesis study in rats. Unpublished report No.
97/10918 from Institute for Environmental Toxicology, Kodaira, Tokyo,
Japan. Submitted to WHO by BASF AG, Limbergerhof, Germany.
Hellwig, J. (1994) Report: Study of the prenatal toxicity of of Reg.
No. 242009 in Wistar rats after oral administration (gavage).
Unpublished report No. 93/10833 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Hellwig, J. & Hildebrand, B. (1993) Report: Study of the prenatal
toxicity of Reg. No. 242009 in rabbits after oral administration
(gavage). Unpublished report No. 93/10980 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Hellwig, J. & Hildebrand, B. (1994a) Report: Reproduction toxicity
study with Reg. No. 242009 in Wistar rats. Continuous dietary
administration over 2 generations (2 litters in the first and 1 litter
in the second generation. Unpublished report No. 94/10950 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Hellwig, J. & Hildebrand, B. (1994b) Report on the study of the
toxicity of Reg. No. 242009 in beagle dogs. Administration via the
diet over 12 months. Unpublished report No. 94/10832 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Hoffmann, H.D. & Engelhardt, G. (1995a) Report on the study of Reg.
No. 262451 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation assay
(standard plate test preincubation test). Unpublished report No.
95/10409 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Hoffmann, H.D. & Engelhardt, G.. (1995b) Report on the study of Reg.
No. 292932 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation Assay
(standard plate test preincubation test). Unpublished report No.
95/10026 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Hoffmann, H.D. & Engelhardt, G. (1995c) Report on the study of Reg.
No. 291685 in the Ames Salmonella/mammalian-microsome mutagenicity
test and Escherichia coli/mammalian-microsome reverse mutation assay
(standard plate test preincubation test). Unpublished report No.
95/10027 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1993a) Report: Study on the acute oral
toxicity of Reg. No. 242009 in rats. Unpublished report No. 93/10730
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1993b) Report: Study on the acute dermal
toxicity of Reg. No. 242009 in rats. Unpublished report No. 93/11108
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1994a) Report: Study on the acute oral
toxicity of Reg. No. 291685 in rats. Unpublished report No. 94/11172
prepared by BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted
to WHO by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1994b) Report: Study on the acute oral
toxicity of Reg. No. 292932 in rats. Unpublished report No. 94/11171
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1994c) Report: Study on the acute dermal
toxicity of Reg. No. 242009 in Wistar rats. Application to the intact
skin over 3 weeks (21 applications). Unpublished report No. 94/11070
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kirsch, P. & Hildebrand, B. (1995) Report: Study on the acute oral
toxicity of Reg. No. 262451 in rats. Unpublished report No. 95/10213
from BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Kohl, W. (1994) The metabolism of [14C]-Reg.No.242009 ([14C]-BAS
490F) in rats. Unpublished report No. 94/10981 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994a) Report: Study on the oral
toxicity of Reg. No. 242009 in Wistar rats. Administration in the diet
over 3 months. Unpublished report No. 94/10954 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994b) Report: Study on the oral
toxicity of Reg. No. 242009 in C57BL mice. Administration in the diet
for 3 months. Unpublished report No. 94/10496 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994c) Report: Study on the oral
toxicity of Reg. No. 242009 in beagle dogs. Administration via the
diet over 3 months (Supplementary study to project No.
83M0180/910149). Unpublished report No. 97/10318 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. & Hildebrand, B. (1994d) Report: Chronic toxity study with
Reg. No. 242009 in Wistar rats. Administration in the diet for 24
months. Unpublished report No. 94/10951 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. & Hildebrand, B. (1994e) Report: Carcinogenicity study
with Reg. No. 242009 in C57BL mice. Administration in the diet for 24
months. Unpublished report No. 94/10953 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. & Hildebrand, B. (1994f) Report: Carcinogenicity study
with Reg. No. 242009 in Wistar rats. Administration in the diet for 24
months. Unpublished report No. 94/10953 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. & Hildebrand, B. (1995) Test study on enzyme activity
after treatment with Reg. No. 242009 in Wistar rats. Dietary
administration for 3 weeks and recovery of 2 weeks. Unpublished report
No. 95/10853 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Mellert, W., Kaufmann, W. & Hildebrand, B. (1995a) Reg. No. 242009 --
Electron microscopic examinations of liver samples to assess
peroxisomes from Wistar rats treated for 3 weeks in the diet.
Unpublished report No. 95/11106 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. , Kaufmann, W. & Hildebrand, B. (1995b) Reg. No. 242009 --
Electron microscopic examinations of liver samples to assess
mitochondria from old Wistar rats treated for 3 weeks in the diet.
Unpublished report No. 95/11105 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W., Bahnemann, R. & Hildebrand, B. (1996a) Reg. No. 242009 --
S phase response study in male Wistar rats including reversibility.
Administration in the diet up to 13 weeks. Unpublished report No.
96/10053 from BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Mellert, W. et al. (1996b) Reg. No. 242009 -- Examination of enzyme
activities in the liver of Wistar rats. Administration in the diet for
3 weeks. Unpublished report No. 96/10100 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Mellert, W. et al. (1997a) Report: S-phase response study with BAS
490F (Reg. No. 242009) in Wistar rats after administration in the diet
for 3 weeks. Unpublished report No. 94/10496 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Mellert, W. et al. (1997b) Report: BAS 490F (Reg. No. 242009): Study
of enzyme excretion in urine of Wistar rats after repeated
administration in the diet. Unpublished report No. 97/10317 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Moss, D. (1994) Effects of Reg. No. 242009 on enzyme levels in rat
serum. Unpublished report No. 94/10578 from Royal Postgraduate Medical
School, Hammersmith Hospital, London, United Kingdom. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
Nakajima, M. (1997) Reverse mutation assay of Reg. No. 279482.
Unpublished report No.97/11152 from Biosafety Research Center, Foods,
Drugs and Pesticides, Shizuoka, Japan. Submitted to WHO by BASF AG,
Limbergerhof, Germany.
Nelsen, J. et al. (1995) Metabolism of 14C-BAS 490F in grapes. BASF
Reg. No. 92/11760. Unpublished report from BASF Corporation, ARC,
Resesarch Triangle Park, North Carolina, USA. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Polloth, C. & Hildebrand, B. (1994a) Report: Ex vivo unscheduled DNA
synthesis (UDS) assay and S phase response in rat hepatocytes with
Reg. No. 242009. Unpublished report No. 94/10867 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Polloth, C. & Hildebrand, B. (1994b) Report: Ex vivo unscheduled DNA
synthesis (UDS) in rat hepatocytes with Reg. No. 242009. Unpublished
report No. 94/10894 from BASF Aktiengesellschaft, Ludwigshafen,
Germany. Submitted to WHO by BASF AG, Limbergerhof, Germany.
Polloth, C. & Hildebrand, B. (1994c) Report: S phase response with
Reg. No. 242009 in Wistar rat after administration in the diet for 3
weeks. Unpublished report No. 94/10922 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Polloth, C. & Hildebrand, B. (1994d) Report: S phase response with
Reg. No. 242009 in 16 month old Wistar rat after administration in the
diet for 3 weeks. Unpublished report No. 94/10984 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Polloth, C. & Hoffmann, H.D. (1994a) Report: Gene mutation test in
Chinese hamster ovary cells (HPRT locus assay) with Reg. No. 242009.
Unpublished report No. 94/10350 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Polloth, C. & Hoffmann, H.D. (1994b) Report: In vitro unscheduled DNA
synthesis (UDS) assay in rat hepatocytes with Reg. No. 242009.
Unpublished report No. 94/10351 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
van Ravenzwaay, B. (1996) Kresoxim-methyl: Mechanism and assessment of
liver tumor induction. Unpublished report No. 96/10078 from BASF
Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by BASF
AG, Limbergerhof, Germany.
Rossbacher, R. & Kirsch, P. (1992a) Report: Study on the acute dermal
irritation/corrosion of Reg. No. 242009 in the rabbit. Unpublished
report No. 92/11663, BASF Aktiengesellschaft, Ludwigshafen, Germany.
Submitted to WHO by BASF AG, Limbergerhof, Germany.
Rossbacher, R. & Kirsch, P. (1992b) Report: Study on the acute eye
irritation of Reg. No. 242009 in the rabbit. Unpublished report No.
92/11664, BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to
WHO by BASF AG, Limbergerhof, Germany.
Rossbacher, R. & Kirsch, P. (1993) Report on the maximization test for
the sensitizing potential of Reg. No. 242009 in guinea pigs.
Unpublished report No. 93/10014 from BASF Aktiengesellschaft,
Ludwigshafen, Germany. Submitted to WHO by BASF AG, Limbergerhof,
Germany.
Schilling, K. & Hildebrand, B. (1992a) Report: Study on the oral
toxicity of Reg. No. 242009 in Wistar rats. Administration in the diet
over 4 weeks (range finding). Unpublished report No. 92/10551 from
BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by
BASF AG, Limbergerhof, Germany.
Schilling, K. & Hildebrand, B. (1992b) Report: Study on the oral
toxicity of Reg. No. 242009 in B6C3F1 mice. Administration in the diet
over 4 weeks (range finding). Unpublished report No. 92/10539 from
BASF Aktiengesellschaft, Ludwigshafen, Germany. Submitted to WHO by
BASF AG, Limbergerhof, Germany.
Yamamoto, T. (1994) Report: Study on the acute oral toxicity of Reg.
No. 242009 in mice. Unpublished report No.94/ from Biosafety Research
Center, Foods, Drugs and Pesticides, Shizuoka, Japan. Submitted to WHO
by BASF AG, Limbergerhof, Germany.
See Also:
Toxicological Abbreviations