
CADUSAFOS
First draft prepared by Dr. E. Bosshard
Federal Office of Public Health
Zurich, Switzerland
S,S-di-sec-butyl O-ethyl phosphorodithioate
EXPLANATION
Cadusafos is an organophosphate insecticide, which was evaluated
by the JMPR for the first time at this meeting. It is effective for
controlling attacks by nematodes and soil-borne insects on bananas,
citrus, maize, potatoes and sugar cane. It is formulated for soil
application as granules, an emulsifiable concentrate and a
microemulsion.
EVALUATION FOR ACCEPTABLE DAILY INTAKE
BIOLOGICAL DATA
Biochemical aspects
Absorption, distribution and excretion
Adult rats (Sprague-Dawley, 5/sex) were dosed orally with single
doses of 20 mg/kg bw 14C-labelled cadusafos. Urine and faeces were
collected for 7 days. Animals were then sacrificed and carcass and
tissues were analyzed for residual radioactivity. About 75% of the
applied radioactivity was excreted in urine and about 15% in faeces
over the course of 7 days. Most radioactivity was eliminated within
the first 24 hours after dosing. Highest residues were measured in
liver and in adipose tissue with a mean value of 0.7 ppm. Another
group of rats (5/sex) was monitored for 14CO2. Expiration amounted
to 13% of the applied radioactivity within three days. No sex
difference in the elimination and distribution pattern was observed
(Selim, 1984).
Rats (Crl:CD(SD)BR, 10/sex/group) received one of four dosing
regimens with 14C-labelled cadusafos. Dosing regimens were a single
oral low dose of 1 mg/kg bw, a single iv dose of 0.8 mg/kg bw,
multiple oral low doses of 1 mg/kg bw nonlabelled material over 14
days followed by an additional dose of labelled material and a control
group. Urine and faeces were collected for 7 days and tissues were
then analyzed for remaining radioactivity. Animals for which 14CO2
was monitored were sacrificed 3 days after dosing. For all groups,
more than 90% of the administered radioactivity was eliminated within
48 hours after dosing. Mean total urinary excretion was about 67%,
78% and 71% after oral single, iv and oral multiple dosing,
respectively. Corresponding excretion values in faeces were 10%, 5%
and 7%. 14CO2 expiration varied between 13% and 16% in the three
dosing regimens. Residues in tissues were low. Highest levels were
measured in liver and in fat showing mean concentrations of up to
about 0.07 ppm in liver and 0.03 ppm in fat after oral dosing. In the
iv study mean concentration in lung was highest with mean values of
about 0.05 ppm, followed by a concentration of 0.03 ppm in liver and
fat. No marked sex differences were observed (Puhl, 1987).
Biotransformation
Male and female rats (Sprague-Dawley, 5/sex/dose) were
administered an oral dose of 14C-labelled cadusafos (at the butyl
side chains) at rates of 1 and 21 mg/kg bw. Another group received
multiple oral doses of 1 mg/kg bw (labelled and non-labelled
material). Results from oral dosing were compared to those from iv
dosing consisting of a single dose of about 0.8 mg/kg bw. The
excretion pattern found in previous experiments was confirmed.
Analyses of excretion profiles showed that the majority of 14C
activity was eliminated within the first 24 hours after dosing. In
the fraction of non-conjugated neutral metabolites the majority of the
radioactivity excreted in urine was contributed by methyl-1-methyl-2-
hydroxypropane sulfone. Other metabolites detected were
0-ethyl-S-(2-butyl) phosphorothioic acid, S,S-di-(2-butyl)
phosphorodithioic acid, methyl-2-butyl-sulfone and sulfoxide.
0-ethyl-S-(2-butyl)phosphorothioic acid, methylsulfonic acid, hydroxy
sulfone and sec-butyl sulfonic acid were identified. As major polar
metabolites in the remaining fractions such compounds as
4-hydroxy-2-butyl sulfonic acid, 3-hydroxy-2-butyl sulfonic acid,
sec-butyl sulfonic acids, and S-(2-butyl) phosphorothioic acid were
detected. In faeces the parent compound was found at rates of 6 to 64%
in the different oral dosing regimens with highest values after
administration of a single oral high dose. In the intravenously dosed
group parent compound was not detected in faeces. Major faecal
metabolites were sec-butyl sulfonic acid and monophosphorothioic
acid-related acidic compounds.
Cleavage of the thio-( sec-butyl) group is the initial step
producing sec-butyl mercaptan and 0-ethyl-S-(2-butyl)phosphorothioic
acid as major metabolites. Further cleavage and oxidation reactions
may result in S-(2-butyl)phosphorothioic acid or 0-ethyl
phosphorothioic acid, methyl sec-butyl sulfide, sulfoxide, sulfone
and finally hydroxysulfones. Sec-butyl mercaptan can also be
oxidized to butyl sulfonic acid, ethyl and methyl sulfonic acid.
Formation of CO2 could be derived from either the sec-butyl
mercaptan moiety or the corresponding sulfonic acid. CO2 may then
be incorporated into urea or other endogenous substances (Wu, 1988).
Toxicological studies
Acute toxicity studies
The predominant signs of toxicity were those typical for
cholinesterase inhibition; tremors, loss of muscle control, decreased
locomotion, diarrhoea, lacrimation and salivation. These effects
occurred irrespective of the species and application route. The data
are summarized in Table 1.
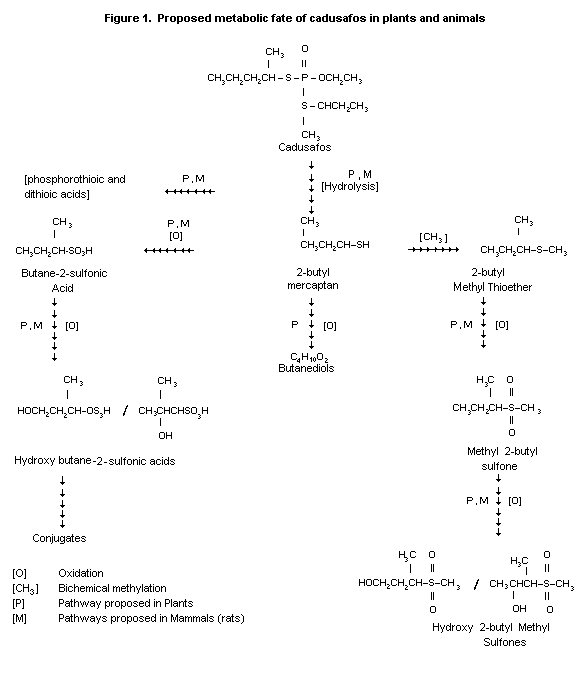 Table 1: Acute toxicity of Cadusafos
Species Sex Route LD50 Reference
(mg/kg bw)
Mouse M&F oral 71 Rand (1983b)
Rat M&F oral 39 (F)a De Prospo (1986)
132 (M)a
42 (F)a Freeman (1987b)
80 (M)a
30 (F)b McCarty (1984c)
48 (M)b
Rabbit M&F dermal 11 Freeman (1987a)
41 (F)b Rand (1983a)
24 (M)b
Rat M&F inh 0.032 mg/lc Dudek (1984)
a 1% (w/v) in corn oil
b 10% (w/v) in corn oil
c 4-hour LC50
Skin and eye irritation testing were performed in rabbits. One
hour after the application of 0.1 ml of Cadusafos (> LD50) into one
eye moderate discharge, miosis and corneal depressions appeared.
Clinical signs including loss of muscle control were observed. All
animals died within two hours of dosing. In a repetition experiment
only 0.01 ml of Cadusafos was applied. Slight irritation was observed
only in the unwashed eyes (McCarty, 1984a).
Primary skin irritation was tested with small doses of 0.015 ml
on each of two test sites for four hours. No skin reaction was
observed, however clinical signs became manifest and most animals died
within 24 hours after treatment (McCarty 1984b).
No sensitizing potential was observed in guinea pigs after
topical application at doses of 0.01 ml.
Short-term studies
Rats
Groups of rats (Sprague-Dawley, 15/sex/group) received Cadusafos
in their diet at concentrations of 0, 0.1, 0.5, 1.0, 5.0 and 800 ppm
over at least 90 days. An additional 10 animals were assigned to the
control and 5 ppm groups to study reversibility of the effects or
delayed toxicity over 28 days after termination of the main study.
Treatment-related effects occurred in animals at 800 ppm and
consisted of decreased locomotion, tremors and emaciation, and splayed
hindlegs. Thirteen of 15 female and 11/15 male animals at 800 ppm died
or were sacrificed moribund prior to termination of the study. No
treatment-related increase in mortality was found in the other dose
groups. Marked reduction with respect to total body weight gain
occurred in the animals dosed at 800 ppm, resulting in values of 50%
of the control animals. No dose-related decrease was observed in the
other dose groups. Animals at 800 ppm also showed reduced food
consumption particularly at the beginning of the study.
Haematological changes observed at 800 ppm included an increase in
platelet count, a decrease of the haemoglobin level and haematocrit
value and, particularly in male animals, a slight reduction in red
blood cell count. Also various clinical chemistry values were changed
at the highest dose level: depression of glucose (in males only),
total protein and, particularly in females, an increase of the blood
urea nitrogen level. Inhibition of the cholinesterase activity in
plasma and erythrocytes was observed at 5 and 800 ppm. At 5 ppm,
maximal inhibition in plasma was about 24% in males, almost 50% in
females. Maximum erythrocyte inhibition was 22% in males and about 25%
in females at 5 ppm. Brain cholinesterase was only marginally
inhibited (6%) at 5 ppm. After the twenty-eight day recovery period
no significant differences between animals treated with 5 ppm and
control animals were found with respect to cholinesterase inhibition.
Severe cholinesterase inhibition occurred at 800 ppm. In plasma,
activity was below the detection limit and in erythrocytes in females
inhibition amounted to 86% whereas males were somewhat less affected.
Brain cholines-terase inhibition was about 85% in both sexes. Changes
in absolute and/or relative organ weights were seen at 800 ppm in
different organs. No compound-related histologic alterations in
tissues were seen.
The no observed adverse effect level (NOAEL) in rats under the
conditions of this study is 1.0 ppm in the diet (70 µg/kg bw)
considering the cholinesterase inhibition in erythrocytes and brain at
higher dose levels as biologically significant (McCarty et al.,
1985).
Dogs
Groups of Beagle dogs (4/sex/dose) received cadusafos by capsule
at dose levels of 0, 10, 30 and 90 µg/kg bw over 91 days. Treatment
had no effect on clinical signs of toxicity, ophthalmoscopic findings,
body weight, food consumption, parameters of clinical chemistry and
haematology, gross or microscopic pathology. Treatment-related
effects consisted of a dose-related inhibition of cholinesterase
activity in plasma at dose levels of 30 and 90 µg/kg bw. Inhibition
resulted in values of about 60% and 40% of the pretest value at 30 and
90 µg/kg bw, respectively. Maximum inhibition at the 10 µg/kg bw
level resulted in an activity corresponding to about 80% of the
pretest value. No dose-related depression of acetylcholinesterase
inhibition was observed in erythrocytes or brain. A sex difference
was not obvious. A decrease in absolute and relative testicular weight
was observed at 30 and 90 µg/kg bw. The difference between these two
dose groups was not dose-related and was not considered to be
treatment-related. No gross pathological or micropathological lesions
attributable to the test compound were found (Seely et al., 1985b).
Groups of Beagle dogs (4/sex/dose) were dosed orally with
cadusafos by capsule at dose levels of 0, 0.2, 1, 5 or 20 µg/kg bw
seven days a week over one year. Dose levels were selected based on
results of a dose-range finding study and the 91-day study performed
previously in this laboratory (Seely et al., 1985a,b). The treatment
had no effect on survival, clinical signs, ophthalmoscopic findings,
body weight food consumption, parameters of clinical chemistry and
haematology, nor organ weights. The only treatment-related effect
observed in males was an inhibition of the plasma cholinesterase
activity at 20 µg/kg bw resulting in a 40% lower value compared to the
pretest activity. In females treated with 5 or 20 µg/kg bw
cholinesterase activity inhibition in plasma was about 25%. No other
compound-related differences in cholinesterase activities were
observed. Gross and microscopic examination revealed no
compound-related lesions in any tissue (Shellenberger, 1986).
A short-term toxicity study comparing cadusafos produced by the
old and new manufacturing processes was performed in dogs. Groups of
Beagle dogs (4/sex/group) were treated with the two products by
capsule at levels of 0, 1, 10 and 100 µg/kg bw over 13 weeks. No
deaths nor clinical signs which were considered to be treatment
related occurred. No differences were observed between the dose groups
with respect to body weight, food consumption, organ weight or gross
necropsy findings. Plasma cholinesterase inhibition was about 20% at
10 µg/kg bw, about 60% at 100 µg/kg bw. No significant difference
occurred between the old and the new material (Dalgard, 1988). The no
observable effect level was 1 µg/kg bw/day.
Long-term/carcinogenicity studies
Mice
Groups of mice (Swiss-Webster, 60/sex/group) received Cadusafos
in the diet at concentrations of 0, 0.1, 0.5, 1.0, and 5.0 ppm over
two years. Ten animals/sex/dose were sacrificed after one year of
treatment for interim investigations. Selection of dietary
concentrations were based on the results of a range finding study
conducted earlier in this laboratory (McCarty et al., 1986).
Feeding of Cadusafos did not influence the survival of the test
animals, body weight, food consumption, parameters of hematology or
organ weights. There were no clinical signs of toxicity other than
decreased locomotor activity in treated animals.
Marked inhibition of cholinesterase activity in plasma and
erythrocytes at 5 ppm was observed (males 76%, females 68%).
Inhibition of cholinesterase activity in erythrocytes was about 26% in
males and about 32% in females. Brain cholinesterase activity was
reduced by 13% only in males. Higher incidences of non-neoplastic
effects included adrenal cortical atrophy in treated animals of both
sexes, focal cortical cell hyperplasia in treated males and duodenal
mucosal hyperplasia in females at 5 ppm. In males necrotizing
arteritis in kidney showed incidences of 6%, 8%, 10%, 22% and 24% in
dose groups 0 to 5 ppm respectively. In male mice a slight increase in
the incidence of lymphoreticular neoplasms was found resulting in
frequencies of 12%, 10%, 10%, 16% and 22% in groups 0, 0.1, 0.5, 1.0,
and 5 ppm, respectively. In another carcinogenicity study with
Swiss-Webster mice conducted in the same laboratory the control
incidence was 14%. In NTP studies, the total mean incidence was 16%
with a standard deviation of 15.1. These tumours were not considered
to have been treatment-related.
A NOAEL of 0.5 ppm, corresponding to about 72 µg/kg bw, was
determined, based on renal necrotizing arteritis occurring in higher
incidences at dose levels of 1 ppm and above in male mice (McCarty
et al., 1987).
Rats
Cadusafos was administered continuously to groups of rats
(Sprague-Dawley, 60/sex/dose) at dietary concentrations of 0, 0.1,
0.5, 1.0 and 5.0 ppm over two years. Dose level selection was based
on the results of a previous range-finding study (Rand, 1986) and a
cholinesterase inhibition titration study (Geiger, 1986). Ten
animals/sex/group were used for interim histopathological examination
after 12 months.
Mortality, body weight, food consumption, parameters of clinical
chemistry and haematology, urinalysis and organ weights were not
affected by the treatment. Ophthalmoscopic examinations did not reveal
dose-related alterations. Cholines-terase activity was inhibited in
plasma and erythrocytes at 5 ppm with maximum inhibition of 37% and
23% respectively in males. Females were somewhat more affected showing
maximum inhibition in plasma and erythrocytes of 52% and 31%,
respectively. Signs of toxicity consisting of decreased locomotion
became apparent in females at 5 ppm. Inhibition of brain
cholinesterase was not found.
Tissues of all animals in the control group and the highest dose
group were examined. However, in the 0.1, 0.5 and 1.0 ppm dose
groups, tissues of only some of the animals (about 60%) were examined
histologically. Based on the incomplete data submitted the incidences
of neoplastic findings including astrocytoma in the brain, pituitary
adenoma, adrenal pheochromocytoma, c-cell adenoma and carcinoma of the
thyroid were higher in the 0.1, 0.5 and 1.0 ppm groups, particularly
in males. However, dose-response relationships were lacking and the
increases were considered to be unrelated to treatment (Weiner
et al., 1986).
The level of 1 ppm (50 µg/kg bw) is identified as the NOAEL based
on significant inhibition of cholinesterase activity in erythrocytes
and clinical signs of toxicity at higher dose levels.
Reproduction study
Cadusafos was administered continuously in the diet to groups of
rats (Sprague-Dawley, 25/sex/group) over two consecutive generations.
Each generation consisted of two litters. Dietary levels were 0, 0.1,
0.5 and 5.0 ppm. Breeding was initiated after 8 weeks of exposure for
the animals of the parental generation (F0) and after 11 weeks of
exposure for the following generation (F1).
All adults and selected weanlings (10/sex/group) were subjected to
a complete necropsy. Microscopic examination of selected tissues was
conducted for parental animals in the 0 and 5 ppm group and for all
weanlings. The treatment did not affect mortality. A slight reduction
in total body weight gain (less than 10%) was observed in F0 males
at 0.5 and 5 ppm without dose-relationship. In the F1 generation body
weight gain was reduced about 10% in both sexes at 5 ppm.
Cholinesterase premating activity in plasma and red blood cells was
inhibited at rates of about 16% and 18%, respectively, in F0 males
fed with 5 ppm. In F0 females at 5 ppm inhibition of plasma
cholinesterase relative to premating activity was 45%, in red blood
cells 18%. Similar values were found at termination. F1 animals
showed a similar inhibition pattern, resulting in cholinesterase
inhibition in plasma (19% and 57%) and in erythrocytes (20% and 25%,
in males and females, respectively) fed with 5 ppm until termination
of the study. There were no treatment-related effects on brain
cholinesterase activities in the F0 or F1 generation. A higher
incidence of stillbirths was observed in one litter of the F1
generation at 0.5 and 5 ppm. The same effect was not found in any
other litter. The apparent dose-related increase probably is a
reflection of the unusually low control incidence in that litter.
There were no gross or microscopic alterations in any generation which
were considered to be treatment-related. The NOAEL was 0.5 ppm
(equivalent to 25 µg/kg bw) based on decreased body weight gain
(DeProspo et al., 1987).
Special studies on embryotoxicity and teratogenicity
Rats
Rats (Sprague-Dawley, 25 female/group) received oral doses of 0,
2.0, 6.0 and 18 mg/kg bw by gavage on days 6 through 15 of gestation.
Caesarean section was performed on day 20 of gestation. At 18 mg/kg bw
signs of toxicity between study days 7 and 20 were observed in all
animals and included tremors, decreased locomotion, alopecia, oral
discharge, lacrimation. The same signs were observed in a few animals
at 6 mg/kg bw but to a lesser extent. Reduced body weight gain and
food consumption were also observed in the 18 mg/kg bw group.
No treatment-related effects were observed regarding the number
of corpora lutea, implantations, resorptions or litter size. A
decrease in fetal body weights was found among fetuses from the 18
mg/kg bw group. Gross external changes were observed as single
findings in pups of the 18 mg/kg bw including an umbilical hernia and
a microphthalmia. Single findings also occurred in fetuses from the
6 mg/kg bw group (one anophthalmia, one atresia of genital papillae,
one acaudate). At 18 mg/kg bw skeletal changes included fused ribs and
vertebrae, absence of metatarsals and sternebrae and partial
ossification. Various other findings were observed sporadically in the
other dose groups. The NOAEL in this study was 2 mg/kg bw for
maternal toxicity and 6 mg/kg bw for fetotoxicity (Freeman, 1985).
Rabbits
Groups of rabbits (New Zealand White, 20 female/group) received
Cadusafos orally by gavage at dosages of 0, 0.1, 0.3, or 0.9 mg/kg bw
on day 7 through 19 of gestation. Caesarean section was performed on
day 29 of gestation. Treatment related clinical signs were observed in
animals at 0.9 mg/kg bw including hypersensitivity, rales, diarrhoea,
dyspnoea, ataxia, loss of muscle control, prostration. In a few
animals at 0.3 mg/kg bw similar clinical signs occurred. One animal
at 0.3 mg/kg bw and two animals at 0.9 mg/kg bw died. One control
animal died from undetermined causes and another animal in this group
was sacrificed due to an early delivery. Two rabbits at 0.9 mg/kg bw
aborted. Early delivery occurred in a control animal and in one
animal at 0.9 mg/kg bw.
Treatment did not influence implantation, litter size or fetal
weights. Malformations observed in pups consisting of fused or
serrated sternebrae, manubrium and xiphoid bone showed incidental
distribution over the dose groups. The incidences of delayed skeletal
ossification did not indicate a treatment relationship.
Cadusafos caused maternal toxicity at levels of 0.3 mg/kg bw and
above, most probably explaining the slight increase in the incidence
of resorptions in the dose groups. The study gave no indication of a
teratogenic effect of cadusafos. The NOAEL in this study was shown to
be 0.1 mg/kg bw (Freeman et al., 1985).
Special studies on genotoxicity
Results of genotoxicity testing are summarized in Table 2.
Cadusafos showed no genotoxic activity in five tests screening for
gene mutations nor one test screening for chromosome aberrations. An
increase in transformation frequency in the presence of exogenous
metabolic activation, however, was found in a transformation test with
BALB/3T3 mouse embryo cells. Positive results were limited to this
single test system, and there were no other supportive indications for
genotoxic activity.
Special study on acute delayed neurotoxicity
Four groups of ten hens were treated with a single oral dose of
8 mg/kg bw, a dose corresponding to the approximate oral LD50.
Atropine was administered to all test birds by intramuscular injection
of 10 mg/kg bw immediately prior to dosing. Retreatment with cadusafos
and atropine was performed after a 21-day observation period. An
additional group was treated with a single oral dose of 500 mg/kg bw.
tri-o-cresylphosphate (TOCP) as positive control, another group only
with corn oil as negative control group. Mortalities (16/40) and
clinical signs of toxicity including unsteadiness, leg stiffness and
weakness were observed in animals treated with Cadusafos. Surviving
birds had recovered by days 4-6 after dosing. Similar observations
were recorded after re-dosing. No clinical signs of neurotoxicity,
assessed by the appearance of ataxia, were recorded in the control or
surviving test birds. All positive control birds dosed with TOCP
developed ataxia within the 21-day observation period (Roberts
et al., 1984).
As recorded in a summary report, treatment did not cause
alterations in nerve tissue in 9/10 birds. The effects observed in
the remaining bird are probably not treatment-related (FMC, 1990a).
Table 2: Results of genotoxicity assays on Cadusafos
Test System Test Object Concentration Results Reference
Ames test Salmonella 3.4-340 µg/plate Negative Haworth et al. (1984)
typhimurium (without activation)
(5 strains) 12-1200 µg/plate
(with activation)
Ames test Salmonella 8-900 µg/plate Negative Lawlor (1985)
typhimurium (with and without
metabolic activation)
CHO/HGPRT Chinese hamster 2.5-75 µg/ml Negative Stankowski et al. (1985)
ovary cells (without activation)
5-125 µg/ml
(with activation)
CHO/HGPRT Chinese hamster 80-110 nl/ml Negative Thilagar et al. (1984c)
ovary cell (without activation)
110-140 nl/ml
(with activation)
Chromosome Chinese hamster 6.25-75 nl/ml Negative Thilagar et al. (1984a)
aberration test ovary cells (with and without
metabolic activation)
Unscheduled Primary rat 10-45 nl/ml Negative Thilagar et al. (1984b)
DNA synthesis hepatocytes
Transformation BALB/3T3 mouse 0.01-0.07 µl/ml Positive Putman et al. (1984)
assay embryo cells (without activation) (activated
0.06.0.09 µl/ml only)
(with activation)
Special studies on antidotal effects
Four groups of rats (Sprague-Dawley, 10/sex/group) were dosed
orally in a 5% (w/v) solution in corn oil at dosage levels of 66 mg/kg
bw (males) and 38 mg/kg bw (females). Following treatment with
cadusafos, one group received no antidotal treatment, one group
received 100 mg/kg bw. atropine, one group 100 mg/kg bw, 2-PAM
(2-pralidoxime) and one group 100 mg/kg bw atropine + 100 mg/kg bw
2-PAM. Observations for toxicity were conducted over 14 days after
treatment. Antidotes were administered immediately after the onset of
clinical signs (Atropine sc injection, 2-PAM im injection). Generally
clinical signs including decreased locomotion, diarrhoea, lacrimation,
mydriasis appeared earlier in females. Atropine treatment alone or in
conjunction with 2-PAM was effective in amelioration of toxicity
induced by oral administration of Cadusafos with respect to mortality,
incidence, duration and severity of clinical signs. 2-PAM therefore
seems not to be effective (Kedderis, 1988a).
A similar study with dose levels of Cadusafos of 66 and 115 mg/kg
bw (males) and 38 or 100 mg/kg bw (females) and the same antidotal
treatment gave generally the same results concerning antidotal effects
(Kedderis, 1988b).
Studies on Cadusafos after the manufacturing change
Short-term and long-term studies presented earlier in this paper
(except for the short-term study in the dog) did not contain the
impurity arising at a rate of 1-3% as a result of manufacturing
change. An additional long-term non-rodent study, an acute oral
study as well as a mutagenicity study with Cadusafos containing the
new impurity have been conducted. Oral LD50 values for rats for the
product after the manufacturing change are reported to be 131 mg/kg bw
for males (vs 47.5 mg/kg bw before the change) and 30 mg/kg bw (vs 39
mg/kg bw) for females. No mutagenic activity was observed when tested
with and without activation in the Ames-test.
The available data do not give evidence of any differences in
toxicity of the new product compared to the old product.
COMMENTS
Following oral administration to rats cadusafos was absorbed and
eliminated mainly via the urine (50-70%), but also via expired air
(10-15%) and faeces (5-10%). Extensive metabolism proceeds by
hydrolysis and oxidation.
Cadusafos has a marked acute oral toxicity with typical signs of
cholinesterase inhibition in the rats and mice tested.
In a 90-day study in rats at dietary concentrations of 0.1, 0.5,
1, 5 or 800 ppm, a NOAEL of 1 ppm (equal to 0.07 mg/kg bw/day) was
determined, with 5 ppm causing inhibition in plasma and erythrocyte
cholinesterase.
In short-term studies in dogs cadusafos was administered by
capsule at dose levels 0, 0.01, 0.03, 0.09 or 0.1 mg/kg bw/day for 91
days or 0.0002, 0.001, 0.005 or 0.02 mg/kg bw/day for 1 year. No
adverse toxicological effects were observed. The NOAEL for dogs,
based on these studies, was 0.1 mg/kg bw/day.
In a 2-year long-term/carcinogenicity study in mice using dietary
concentrations of cadusafos of 0, 0.1, 0.5, 1 or 5 ppm, a NOAEL of 0.5
ppm, equal to 0.089 mg/kg bw/day, was determined, based on the
occurrence of renal necrotizing arteritis at 1 ppm. There was no
evidence of carcinogenicity.
In a 2-year long-term/carcinogenicity study in rats at dietary
concentrations of 0, 0.1, 0.5, 1 or 5 ppm, a NOAEL of 1 ppm, equal to
0.05 and 0.06 mg/kg bw/day for males and females, respectively, was
established. At 5 ppm, erythrocyte acetylcholinesterase was inhibited
without a corresponding inhibition of brain acetylcholinesterase.
However, clinical signs of toxicity were observed in females at 5 ppm.
There was no evidence of carcinogenicity.
In a multi-generation study in rats at dietary concentrations of
0, 0.1, 0.5 or 5 ppm, a NOAEL of 0.5 ppm, equal to 0.03 mg/kg bw/day,
was determined. At 5 ppm, reduction in body-weight gain and
inhibition of cholinesterase in plasma and erythrocytes were noted in
the F1 generation. There were no adverse effects on reproduction.
An oral teratogenicity study in rats at dose levels of 0, 2, 6 or
18 mg/kg bw/day indicated dose-related maternal toxicity including
clinical signs such as tremors, and red oral discharge at 6 and 18
mg/kg bw/day. Embryo/fetotoxicity at 18 mg/kg bw/day was indicated by
a decrease in fetal body-weight gain. There were no teratogenic
effects. The NOAEL was 2 mg/kg bw/day. A teratogenicity study in
rabbits at dose levels of 0, 0.1, 0.3 or 0.9 mg/kg bw/day showed
maternal toxicity and embryotoxic effects at dose levels of 0.3 and
0.9 mg/kg bw/day respectively.
After reviewing the in vitro genotoxicity data it was concluded
that, on the basis of the limited data available, there was no
evidence of genotoxicity. A positive response was limited to a cell
transformation assay with BALB/3T3 mouse embryo cells after
activation.
The ADI was based upon the results of the reproduction study in
rats, using a 100-fold safety factor.
TOXICOLOGICAL EVALUATION
Level causing no toxicological effect
Mouse: 0.5 ppm, equal to 0.089 mg/kg bw/day
Rat: 1 ppm, equivalent to 0.05 mg/kg bw/day
(2-year study)
0.5 ppm, equal to 0.03 mg/kg bw/day
(reproduction study)
Dog: 0.1 mg/kg bw/day
Estimate of acceptable daily intake for humans
0-0.0003 mg/kg bw
Studies which will provide information valuable in the
continued evaluation of the compound
Observations in humans.
REFERENCES
FMC (1990a) FMC 67825 Technical Material Cadusafos. Mammalian
Toxicology Overview prepared by Toxicology Department FMC
Corporation Chemical Research and Development Center, Princeton, New
Jersey. Submitted to WHO by FMC Corporation, NJ, USA.
Dalgard, D.W. (1988) 13-Week Comparative Oral Toxicity Study in Dogs
with FMC 67825 (techn.). Project Number A87-2531. Unpublished report
prepared by Hazleton Laboratories, America, Inc. for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
De Prospo, J.R. et al. (1987) Multi-Generation Reproduction Study
with FMC 67825 Technical in Rats. Unpublished report prepared by
FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
De Prospo, J.R. (1986) Acute Oral Toxicity in Rats with FMC 67825
Technical Material. Study Number A85-1796. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Dudek, B.R. (1984) Four Hour Acute Aerosol Inhalation Toxicity Study
in Rats with FMC 67825 Technical Material. Study Number A84-1231.
Unpublished report prepared by ToxiGenics, Inc., Illinois for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Freeman, Ch. (1985) Teratology Study in Rats with FMC 67825
Technical. Study Number A84-1173. Unpublished report prepared by
FMC Toxicology Laboratory Princeton, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Freeman, Ch. (1985) Teratology Study in Rabbits with FMC 67825
Technical. Study Number A84-1175. Unpublished report prepared by FMC
Toxicology Laboratory, New Jersey.
Freeman, Ch. (1987a) Acute Dermal Toxicity in Rabbits with FMC 67825
Technical Material. Study Number A86-2190. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Freeman, Ch. (1987b) Acute Oral Toxicity in Rats with FMC 67825
Technical. Study Number A86-2191. Unpublished report prepared by
FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
Geiger, L.E. et al. (1986). Cholinesterase Inhibition Titration
Study in Rats with FMC 67825 (techn.). Study Number A84-1169.
Unpublished report prepared by FMC Toxicology Laboratory, New
Jersey. Submitted to WHO by FMC Corporation, NJ, USA.
Haworth, St.R. et al. (1984) Salmonella/Mammalian Microsome
Plate Incorporation Mutagenicity Assay (Ames Test). With FMC 67825
D (techn.). Study Number A83-1155. Unpublished report prepared by
Microbiological Associates, Bethesda, Maryland for FMC Corporation.
Submitted to WHO by FMC Corporation, NJ, USA.
Kedderis, L.B. (1988a) FMC 67825 Technical: Antidotal Study in Rats
with Atropine and/or 2-PAM. Study Number A86-2222. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
Kedderis, L.B. (1988b) FMC 67825 Technical: Antidotal Study in Rats
with Atropine and/or 2-PAM. Study Number A86-2222A. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
Lawlor, T.E. et al. (1985) Salmonella/Mammalian-Microsome Plate
Incorporation Mutagenicity Assay (Ames Test) with FMC 67825
(techn.). Study Number A85-1797. Unpublished report prepared by
Microbiological Associates, Inc., Bethesda, Maryland for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
McCarty, J.D. (1984a) Primary Eye Irritation in Rabbits with FMC
67825 (techn.). Study Number A83-1154. Unpublished report prepared
by FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
McCarty, J.D. (1984b) Primary Skin Irritation in Rabbits with FMC
67825 (techn.). Study Number A84-1238. Unpublished report prepared
by FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
McCarty, J.D. (1984c) Acute Oral Toxicity in Rats with FMC 67825
Technical Material. Study Number A83-1164. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
McCarty, J.D. (1984d) Skin Sensitization Study in Guinea Pigs with
FMC 67825 Technical Material. Study Number A84-1271. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
McCarty, J.D. et al. (1985) Ninety Day Feeding Study in Rats with
FMC 67825 (techn.). Study Number A84-1232. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
McCarty, J.D. et al. (1986) Twenty-Eight Day Range-finding Study
with FMC 67825 Technical in Mice. Study Number A83-1153. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
McCarty, J.D. et al. (1987) Chronic Oncogenicity Study in Mice
with FMC 67825 Technical. Study Number A84-1437. Unpublished report
prepared by FMC Corp Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
Puhl, R.J. (1987) FMC 67825 (techn.) Rat Metabolism Single and
Multiple Low-Dose Test Regimen. Unpublished Report prepared by
Hazelton Laboratories America Inc. Wisconsin. (HLA Study No.
6124-105; FMC Report No. PC-0077) Submitted to FMC Corporation, New
Jersey. Submitted to WHO by FMC Corporation, NJ, USA.
Putman, D.L. et al. (1984) Activity of FMC 67825 (techn.), (RUGBY)
in the Morphological Transformation of BALB/3T3 Mouse Embryo Cells
in the Presence and Absence of Exogenous Metabolic Activation.
Study Number A83-1158. Unpublished report prepared by
Microbiological Associates Inc., Bethesda, Maryland for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Rand, G.M. (1986) Twenty-Eight Day Range-finding Study in Rats with
FMC 67825 (techn.). Study Number A83-1146. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Rand, G.M. (1983a) Acute Dermal Toxicity in Rabbits with FMC 67825
Technical Material. Study Number A83-916. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Rand, G.M. (1983b) Acute Oral Toxicity in Mice with FMC 67825
Technical Material. Study Number A83-915. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Roberts, N.L. et al. (1984) Acute Delayed Neurotoxicity Study in
the Domestic Hen with FMC 67825. Study Number A84-1246. Unpublished
report prepared by Huntingdon Research Centre England for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Seely, J.C. et al. (1985a) 14-Day Range-finding Oral Toxicity
Study in Dogs with FMC 67825 (techn.). Study Number A84-1203.
Unpublished report prepared by Pharmacopathics Research
Laboratories, Inc. Laurel, Maryland for FMC Corporation. Submitted
to WHO by FMC Corporation, NJ, USA.
Seely, J.C. et al. (1985b) 91-Day Subchronic Oral Toxicity Study
in the Dog with FMC 67825 Technical. Study Number A84-1204.
Unpublished report prepared by Pharmacopathics Research
Laboratories, Inc. Laurel, Maryland for FMC Corporation. Submitted
to WHO by FMC Corporation, NJ, USA.
Selim. S. (1984) Rat Balance Study and Tissue Distribution of 14C
Labelled FMC 67825 (techn.). Report (Nr. FM-175T) of the Primate
Research Institute, New Mexico State University, submitted to FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Shellenberger, Th.E. (1986) One Year Oral Toxicity Study in the Dog
with FMC 67825 (techn.). Study Number A84-1538. Unpublished report
prepared by Tegeris Laboratories Inc. (formerly: Pharmacopathics
Research Laboratories, Inc.), Laurel, Maryland for FMC Corporation.
Submitted to WHO by FMC Corporation, NJ, USA.
Stankowski, L.F. et al. (1985) Mammalian Cell Forward Gene
Mutation Assay with FMC 67825 (techn.). Study Number A85-1601.
Unpublished Report prepared by Pharmakon Research International,
Inc., Waverly, Pennsylvania for FMC Corporation. Submitted to WHO
by FMC Corporation, NJ, USA.
Thilagar, A. et al. (1984a) Chromosome Aberrations Assay in
Chinese Hamster Ovary (CHO) Cells. With FMC 67825 (techn.) Study
Number A83-1157. Unpublished report prepared by Microbiological
Associates, Bethesda, Maryland for FMC Corporation. Submitted to
WHO by FMC Corporation, NJ, USA.
Thilagar, A. et al. (1984b) Unscheduled DNA Synthesis in Rat
Primary Hepatocytes with FMC 67825 (techn.). Study Number A83-1159.
Unpublished report prepared by Microbiological Associates Bethesda,
Maryland for FMC Corporation. Submitted to WHO by FMC Corporation,
NJ, USA.
Thilagar, A. et al. (1984c) CHO/HGPRT Mutation Assay in Chinese
Hamster Ovary (CHO) Cells. With FMC 65825 (techn.) (RUGBY). Study
Number T-2199-332. Unpublished report prepared by Microbiological
Associates Inc., Bethesda, Maryland for FMC Corporation. Submitted
to WHO by FMC Corporation, NJ, USA.
Weiner, M. et al. (1986) Chronic Oral toxicity/Oncogenicity Study
in Rats with FMC 67825 Technical. Study Number A84-1287. Unpublished
report prepared by Hazleton Laboratories America Inc., Virginia for
FMC Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Wu, J. (1988) FMC 67825 (techn.) Rat Metabolism: Metabolite
Identification and Distribution.10 Unpublished report prepared by
FMC Corporation, Metabolism Laboratories, New Jersey. Submitted to
WHO by FMC Corporation, NJ, USA.
Table 1: Acute toxicity of Cadusafos
Species Sex Route LD50 Reference
(mg/kg bw)
Mouse M&F oral 71 Rand (1983b)
Rat M&F oral 39 (F)a De Prospo (1986)
132 (M)a
42 (F)a Freeman (1987b)
80 (M)a
30 (F)b McCarty (1984c)
48 (M)b
Rabbit M&F dermal 11 Freeman (1987a)
41 (F)b Rand (1983a)
24 (M)b
Rat M&F inh 0.032 mg/lc Dudek (1984)
a 1% (w/v) in corn oil
b 10% (w/v) in corn oil
c 4-hour LC50
Skin and eye irritation testing were performed in rabbits. One
hour after the application of 0.1 ml of Cadusafos (> LD50) into one
eye moderate discharge, miosis and corneal depressions appeared.
Clinical signs including loss of muscle control were observed. All
animals died within two hours of dosing. In a repetition experiment
only 0.01 ml of Cadusafos was applied. Slight irritation was observed
only in the unwashed eyes (McCarty, 1984a).
Primary skin irritation was tested with small doses of 0.015 ml
on each of two test sites for four hours. No skin reaction was
observed, however clinical signs became manifest and most animals died
within 24 hours after treatment (McCarty 1984b).
No sensitizing potential was observed in guinea pigs after
topical application at doses of 0.01 ml.
Short-term studies
Rats
Groups of rats (Sprague-Dawley, 15/sex/group) received Cadusafos
in their diet at concentrations of 0, 0.1, 0.5, 1.0, 5.0 and 800 ppm
over at least 90 days. An additional 10 animals were assigned to the
control and 5 ppm groups to study reversibility of the effects or
delayed toxicity over 28 days after termination of the main study.
Treatment-related effects occurred in animals at 800 ppm and
consisted of decreased locomotion, tremors and emaciation, and splayed
hindlegs. Thirteen of 15 female and 11/15 male animals at 800 ppm died
or were sacrificed moribund prior to termination of the study. No
treatment-related increase in mortality was found in the other dose
groups. Marked reduction with respect to total body weight gain
occurred in the animals dosed at 800 ppm, resulting in values of 50%
of the control animals. No dose-related decrease was observed in the
other dose groups. Animals at 800 ppm also showed reduced food
consumption particularly at the beginning of the study.
Haematological changes observed at 800 ppm included an increase in
platelet count, a decrease of the haemoglobin level and haematocrit
value and, particularly in male animals, a slight reduction in red
blood cell count. Also various clinical chemistry values were changed
at the highest dose level: depression of glucose (in males only),
total protein and, particularly in females, an increase of the blood
urea nitrogen level. Inhibition of the cholinesterase activity in
plasma and erythrocytes was observed at 5 and 800 ppm. At 5 ppm,
maximal inhibition in plasma was about 24% in males, almost 50% in
females. Maximum erythrocyte inhibition was 22% in males and about 25%
in females at 5 ppm. Brain cholinesterase was only marginally
inhibited (6%) at 5 ppm. After the twenty-eight day recovery period
no significant differences between animals treated with 5 ppm and
control animals were found with respect to cholinesterase inhibition.
Severe cholinesterase inhibition occurred at 800 ppm. In plasma,
activity was below the detection limit and in erythrocytes in females
inhibition amounted to 86% whereas males were somewhat less affected.
Brain cholines-terase inhibition was about 85% in both sexes. Changes
in absolute and/or relative organ weights were seen at 800 ppm in
different organs. No compound-related histologic alterations in
tissues were seen.
The no observed adverse effect level (NOAEL) in rats under the
conditions of this study is 1.0 ppm in the diet (70 µg/kg bw)
considering the cholinesterase inhibition in erythrocytes and brain at
higher dose levels as biologically significant (McCarty et al.,
1985).
Dogs
Groups of Beagle dogs (4/sex/dose) received cadusafos by capsule
at dose levels of 0, 10, 30 and 90 µg/kg bw over 91 days. Treatment
had no effect on clinical signs of toxicity, ophthalmoscopic findings,
body weight, food consumption, parameters of clinical chemistry and
haematology, gross or microscopic pathology. Treatment-related
effects consisted of a dose-related inhibition of cholinesterase
activity in plasma at dose levels of 30 and 90 µg/kg bw. Inhibition
resulted in values of about 60% and 40% of the pretest value at 30 and
90 µg/kg bw, respectively. Maximum inhibition at the 10 µg/kg bw
level resulted in an activity corresponding to about 80% of the
pretest value. No dose-related depression of acetylcholinesterase
inhibition was observed in erythrocytes or brain. A sex difference
was not obvious. A decrease in absolute and relative testicular weight
was observed at 30 and 90 µg/kg bw. The difference between these two
dose groups was not dose-related and was not considered to be
treatment-related. No gross pathological or micropathological lesions
attributable to the test compound were found (Seely et al., 1985b).
Groups of Beagle dogs (4/sex/dose) were dosed orally with
cadusafos by capsule at dose levels of 0, 0.2, 1, 5 or 20 µg/kg bw
seven days a week over one year. Dose levels were selected based on
results of a dose-range finding study and the 91-day study performed
previously in this laboratory (Seely et al., 1985a,b). The treatment
had no effect on survival, clinical signs, ophthalmoscopic findings,
body weight food consumption, parameters of clinical chemistry and
haematology, nor organ weights. The only treatment-related effect
observed in males was an inhibition of the plasma cholinesterase
activity at 20 µg/kg bw resulting in a 40% lower value compared to the
pretest activity. In females treated with 5 or 20 µg/kg bw
cholinesterase activity inhibition in plasma was about 25%. No other
compound-related differences in cholinesterase activities were
observed. Gross and microscopic examination revealed no
compound-related lesions in any tissue (Shellenberger, 1986).
A short-term toxicity study comparing cadusafos produced by the
old and new manufacturing processes was performed in dogs. Groups of
Beagle dogs (4/sex/group) were treated with the two products by
capsule at levels of 0, 1, 10 and 100 µg/kg bw over 13 weeks. No
deaths nor clinical signs which were considered to be treatment
related occurred. No differences were observed between the dose groups
with respect to body weight, food consumption, organ weight or gross
necropsy findings. Plasma cholinesterase inhibition was about 20% at
10 µg/kg bw, about 60% at 100 µg/kg bw. No significant difference
occurred between the old and the new material (Dalgard, 1988). The no
observable effect level was 1 µg/kg bw/day.
Long-term/carcinogenicity studies
Mice
Groups of mice (Swiss-Webster, 60/sex/group) received Cadusafos
in the diet at concentrations of 0, 0.1, 0.5, 1.0, and 5.0 ppm over
two years. Ten animals/sex/dose were sacrificed after one year of
treatment for interim investigations. Selection of dietary
concentrations were based on the results of a range finding study
conducted earlier in this laboratory (McCarty et al., 1986).
Feeding of Cadusafos did not influence the survival of the test
animals, body weight, food consumption, parameters of hematology or
organ weights. There were no clinical signs of toxicity other than
decreased locomotor activity in treated animals.
Marked inhibition of cholinesterase activity in plasma and
erythrocytes at 5 ppm was observed (males 76%, females 68%).
Inhibition of cholinesterase activity in erythrocytes was about 26% in
males and about 32% in females. Brain cholinesterase activity was
reduced by 13% only in males. Higher incidences of non-neoplastic
effects included adrenal cortical atrophy in treated animals of both
sexes, focal cortical cell hyperplasia in treated males and duodenal
mucosal hyperplasia in females at 5 ppm. In males necrotizing
arteritis in kidney showed incidences of 6%, 8%, 10%, 22% and 24% in
dose groups 0 to 5 ppm respectively. In male mice a slight increase in
the incidence of lymphoreticular neoplasms was found resulting in
frequencies of 12%, 10%, 10%, 16% and 22% in groups 0, 0.1, 0.5, 1.0,
and 5 ppm, respectively. In another carcinogenicity study with
Swiss-Webster mice conducted in the same laboratory the control
incidence was 14%. In NTP studies, the total mean incidence was 16%
with a standard deviation of 15.1. These tumours were not considered
to have been treatment-related.
A NOAEL of 0.5 ppm, corresponding to about 72 µg/kg bw, was
determined, based on renal necrotizing arteritis occurring in higher
incidences at dose levels of 1 ppm and above in male mice (McCarty
et al., 1987).
Rats
Cadusafos was administered continuously to groups of rats
(Sprague-Dawley, 60/sex/dose) at dietary concentrations of 0, 0.1,
0.5, 1.0 and 5.0 ppm over two years. Dose level selection was based
on the results of a previous range-finding study (Rand, 1986) and a
cholinesterase inhibition titration study (Geiger, 1986). Ten
animals/sex/group were used for interim histopathological examination
after 12 months.
Mortality, body weight, food consumption, parameters of clinical
chemistry and haematology, urinalysis and organ weights were not
affected by the treatment. Ophthalmoscopic examinations did not reveal
dose-related alterations. Cholines-terase activity was inhibited in
plasma and erythrocytes at 5 ppm with maximum inhibition of 37% and
23% respectively in males. Females were somewhat more affected showing
maximum inhibition in plasma and erythrocytes of 52% and 31%,
respectively. Signs of toxicity consisting of decreased locomotion
became apparent in females at 5 ppm. Inhibition of brain
cholinesterase was not found.
Tissues of all animals in the control group and the highest dose
group were examined. However, in the 0.1, 0.5 and 1.0 ppm dose
groups, tissues of only some of the animals (about 60%) were examined
histologically. Based on the incomplete data submitted the incidences
of neoplastic findings including astrocytoma in the brain, pituitary
adenoma, adrenal pheochromocytoma, c-cell adenoma and carcinoma of the
thyroid were higher in the 0.1, 0.5 and 1.0 ppm groups, particularly
in males. However, dose-response relationships were lacking and the
increases were considered to be unrelated to treatment (Weiner
et al., 1986).
The level of 1 ppm (50 µg/kg bw) is identified as the NOAEL based
on significant inhibition of cholinesterase activity in erythrocytes
and clinical signs of toxicity at higher dose levels.
Reproduction study
Cadusafos was administered continuously in the diet to groups of
rats (Sprague-Dawley, 25/sex/group) over two consecutive generations.
Each generation consisted of two litters. Dietary levels were 0, 0.1,
0.5 and 5.0 ppm. Breeding was initiated after 8 weeks of exposure for
the animals of the parental generation (F0) and after 11 weeks of
exposure for the following generation (F1).
All adults and selected weanlings (10/sex/group) were subjected to
a complete necropsy. Microscopic examination of selected tissues was
conducted for parental animals in the 0 and 5 ppm group and for all
weanlings. The treatment did not affect mortality. A slight reduction
in total body weight gain (less than 10%) was observed in F0 males
at 0.5 and 5 ppm without dose-relationship. In the F1 generation body
weight gain was reduced about 10% in both sexes at 5 ppm.
Cholinesterase premating activity in plasma and red blood cells was
inhibited at rates of about 16% and 18%, respectively, in F0 males
fed with 5 ppm. In F0 females at 5 ppm inhibition of plasma
cholinesterase relative to premating activity was 45%, in red blood
cells 18%. Similar values were found at termination. F1 animals
showed a similar inhibition pattern, resulting in cholinesterase
inhibition in plasma (19% and 57%) and in erythrocytes (20% and 25%,
in males and females, respectively) fed with 5 ppm until termination
of the study. There were no treatment-related effects on brain
cholinesterase activities in the F0 or F1 generation. A higher
incidence of stillbirths was observed in one litter of the F1
generation at 0.5 and 5 ppm. The same effect was not found in any
other litter. The apparent dose-related increase probably is a
reflection of the unusually low control incidence in that litter.
There were no gross or microscopic alterations in any generation which
were considered to be treatment-related. The NOAEL was 0.5 ppm
(equivalent to 25 µg/kg bw) based on decreased body weight gain
(DeProspo et al., 1987).
Special studies on embryotoxicity and teratogenicity
Rats
Rats (Sprague-Dawley, 25 female/group) received oral doses of 0,
2.0, 6.0 and 18 mg/kg bw by gavage on days 6 through 15 of gestation.
Caesarean section was performed on day 20 of gestation. At 18 mg/kg bw
signs of toxicity between study days 7 and 20 were observed in all
animals and included tremors, decreased locomotion, alopecia, oral
discharge, lacrimation. The same signs were observed in a few animals
at 6 mg/kg bw but to a lesser extent. Reduced body weight gain and
food consumption were also observed in the 18 mg/kg bw group.
No treatment-related effects were observed regarding the number
of corpora lutea, implantations, resorptions or litter size. A
decrease in fetal body weights was found among fetuses from the 18
mg/kg bw group. Gross external changes were observed as single
findings in pups of the 18 mg/kg bw including an umbilical hernia and
a microphthalmia. Single findings also occurred in fetuses from the
6 mg/kg bw group (one anophthalmia, one atresia of genital papillae,
one acaudate). At 18 mg/kg bw skeletal changes included fused ribs and
vertebrae, absence of metatarsals and sternebrae and partial
ossification. Various other findings were observed sporadically in the
other dose groups. The NOAEL in this study was 2 mg/kg bw for
maternal toxicity and 6 mg/kg bw for fetotoxicity (Freeman, 1985).
Rabbits
Groups of rabbits (New Zealand White, 20 female/group) received
Cadusafos orally by gavage at dosages of 0, 0.1, 0.3, or 0.9 mg/kg bw
on day 7 through 19 of gestation. Caesarean section was performed on
day 29 of gestation. Treatment related clinical signs were observed in
animals at 0.9 mg/kg bw including hypersensitivity, rales, diarrhoea,
dyspnoea, ataxia, loss of muscle control, prostration. In a few
animals at 0.3 mg/kg bw similar clinical signs occurred. One animal
at 0.3 mg/kg bw and two animals at 0.9 mg/kg bw died. One control
animal died from undetermined causes and another animal in this group
was sacrificed due to an early delivery. Two rabbits at 0.9 mg/kg bw
aborted. Early delivery occurred in a control animal and in one
animal at 0.9 mg/kg bw.
Treatment did not influence implantation, litter size or fetal
weights. Malformations observed in pups consisting of fused or
serrated sternebrae, manubrium and xiphoid bone showed incidental
distribution over the dose groups. The incidences of delayed skeletal
ossification did not indicate a treatment relationship.
Cadusafos caused maternal toxicity at levels of 0.3 mg/kg bw and
above, most probably explaining the slight increase in the incidence
of resorptions in the dose groups. The study gave no indication of a
teratogenic effect of cadusafos. The NOAEL in this study was shown to
be 0.1 mg/kg bw (Freeman et al., 1985).
Special studies on genotoxicity
Results of genotoxicity testing are summarized in Table 2.
Cadusafos showed no genotoxic activity in five tests screening for
gene mutations nor one test screening for chromosome aberrations. An
increase in transformation frequency in the presence of exogenous
metabolic activation, however, was found in a transformation test with
BALB/3T3 mouse embryo cells. Positive results were limited to this
single test system, and there were no other supportive indications for
genotoxic activity.
Special study on acute delayed neurotoxicity
Four groups of ten hens were treated with a single oral dose of
8 mg/kg bw, a dose corresponding to the approximate oral LD50.
Atropine was administered to all test birds by intramuscular injection
of 10 mg/kg bw immediately prior to dosing. Retreatment with cadusafos
and atropine was performed after a 21-day observation period. An
additional group was treated with a single oral dose of 500 mg/kg bw.
tri-o-cresylphosphate (TOCP) as positive control, another group only
with corn oil as negative control group. Mortalities (16/40) and
clinical signs of toxicity including unsteadiness, leg stiffness and
weakness were observed in animals treated with Cadusafos. Surviving
birds had recovered by days 4-6 after dosing. Similar observations
were recorded after re-dosing. No clinical signs of neurotoxicity,
assessed by the appearance of ataxia, were recorded in the control or
surviving test birds. All positive control birds dosed with TOCP
developed ataxia within the 21-day observation period (Roberts
et al., 1984).
As recorded in a summary report, treatment did not cause
alterations in nerve tissue in 9/10 birds. The effects observed in
the remaining bird are probably not treatment-related (FMC, 1990a).
Table 2: Results of genotoxicity assays on Cadusafos
Test System Test Object Concentration Results Reference
Ames test Salmonella 3.4-340 µg/plate Negative Haworth et al. (1984)
typhimurium (without activation)
(5 strains) 12-1200 µg/plate
(with activation)
Ames test Salmonella 8-900 µg/plate Negative Lawlor (1985)
typhimurium (with and without
metabolic activation)
CHO/HGPRT Chinese hamster 2.5-75 µg/ml Negative Stankowski et al. (1985)
ovary cells (without activation)
5-125 µg/ml
(with activation)
CHO/HGPRT Chinese hamster 80-110 nl/ml Negative Thilagar et al. (1984c)
ovary cell (without activation)
110-140 nl/ml
(with activation)
Chromosome Chinese hamster 6.25-75 nl/ml Negative Thilagar et al. (1984a)
aberration test ovary cells (with and without
metabolic activation)
Unscheduled Primary rat 10-45 nl/ml Negative Thilagar et al. (1984b)
DNA synthesis hepatocytes
Transformation BALB/3T3 mouse 0.01-0.07 µl/ml Positive Putman et al. (1984)
assay embryo cells (without activation) (activated
0.06.0.09 µl/ml only)
(with activation)
Special studies on antidotal effects
Four groups of rats (Sprague-Dawley, 10/sex/group) were dosed
orally in a 5% (w/v) solution in corn oil at dosage levels of 66 mg/kg
bw (males) and 38 mg/kg bw (females). Following treatment with
cadusafos, one group received no antidotal treatment, one group
received 100 mg/kg bw. atropine, one group 100 mg/kg bw, 2-PAM
(2-pralidoxime) and one group 100 mg/kg bw atropine + 100 mg/kg bw
2-PAM. Observations for toxicity were conducted over 14 days after
treatment. Antidotes were administered immediately after the onset of
clinical signs (Atropine sc injection, 2-PAM im injection). Generally
clinical signs including decreased locomotion, diarrhoea, lacrimation,
mydriasis appeared earlier in females. Atropine treatment alone or in
conjunction with 2-PAM was effective in amelioration of toxicity
induced by oral administration of Cadusafos with respect to mortality,
incidence, duration and severity of clinical signs. 2-PAM therefore
seems not to be effective (Kedderis, 1988a).
A similar study with dose levels of Cadusafos of 66 and 115 mg/kg
bw (males) and 38 or 100 mg/kg bw (females) and the same antidotal
treatment gave generally the same results concerning antidotal effects
(Kedderis, 1988b).
Studies on Cadusafos after the manufacturing change
Short-term and long-term studies presented earlier in this paper
(except for the short-term study in the dog) did not contain the
impurity arising at a rate of 1-3% as a result of manufacturing
change. An additional long-term non-rodent study, an acute oral
study as well as a mutagenicity study with Cadusafos containing the
new impurity have been conducted. Oral LD50 values for rats for the
product after the manufacturing change are reported to be 131 mg/kg bw
for males (vs 47.5 mg/kg bw before the change) and 30 mg/kg bw (vs 39
mg/kg bw) for females. No mutagenic activity was observed when tested
with and without activation in the Ames-test.
The available data do not give evidence of any differences in
toxicity of the new product compared to the old product.
COMMENTS
Following oral administration to rats cadusafos was absorbed and
eliminated mainly via the urine (50-70%), but also via expired air
(10-15%) and faeces (5-10%). Extensive metabolism proceeds by
hydrolysis and oxidation.
Cadusafos has a marked acute oral toxicity with typical signs of
cholinesterase inhibition in the rats and mice tested.
In a 90-day study in rats at dietary concentrations of 0.1, 0.5,
1, 5 or 800 ppm, a NOAEL of 1 ppm (equal to 0.07 mg/kg bw/day) was
determined, with 5 ppm causing inhibition in plasma and erythrocyte
cholinesterase.
In short-term studies in dogs cadusafos was administered by
capsule at dose levels 0, 0.01, 0.03, 0.09 or 0.1 mg/kg bw/day for 91
days or 0.0002, 0.001, 0.005 or 0.02 mg/kg bw/day for 1 year. No
adverse toxicological effects were observed. The NOAEL for dogs,
based on these studies, was 0.1 mg/kg bw/day.
In a 2-year long-term/carcinogenicity study in mice using dietary
concentrations of cadusafos of 0, 0.1, 0.5, 1 or 5 ppm, a NOAEL of 0.5
ppm, equal to 0.089 mg/kg bw/day, was determined, based on the
occurrence of renal necrotizing arteritis at 1 ppm. There was no
evidence of carcinogenicity.
In a 2-year long-term/carcinogenicity study in rats at dietary
concentrations of 0, 0.1, 0.5, 1 or 5 ppm, a NOAEL of 1 ppm, equal to
0.05 and 0.06 mg/kg bw/day for males and females, respectively, was
established. At 5 ppm, erythrocyte acetylcholinesterase was inhibited
without a corresponding inhibition of brain acetylcholinesterase.
However, clinical signs of toxicity were observed in females at 5 ppm.
There was no evidence of carcinogenicity.
In a multi-generation study in rats at dietary concentrations of
0, 0.1, 0.5 or 5 ppm, a NOAEL of 0.5 ppm, equal to 0.03 mg/kg bw/day,
was determined. At 5 ppm, reduction in body-weight gain and
inhibition of cholinesterase in plasma and erythrocytes were noted in
the F1 generation. There were no adverse effects on reproduction.
An oral teratogenicity study in rats at dose levels of 0, 2, 6 or
18 mg/kg bw/day indicated dose-related maternal toxicity including
clinical signs such as tremors, and red oral discharge at 6 and 18
mg/kg bw/day. Embryo/fetotoxicity at 18 mg/kg bw/day was indicated by
a decrease in fetal body-weight gain. There were no teratogenic
effects. The NOAEL was 2 mg/kg bw/day. A teratogenicity study in
rabbits at dose levels of 0, 0.1, 0.3 or 0.9 mg/kg bw/day showed
maternal toxicity and embryotoxic effects at dose levels of 0.3 and
0.9 mg/kg bw/day respectively.
After reviewing the in vitro genotoxicity data it was concluded
that, on the basis of the limited data available, there was no
evidence of genotoxicity. A positive response was limited to a cell
transformation assay with BALB/3T3 mouse embryo cells after
activation.
The ADI was based upon the results of the reproduction study in
rats, using a 100-fold safety factor.
TOXICOLOGICAL EVALUATION
Level causing no toxicological effect
Mouse: 0.5 ppm, equal to 0.089 mg/kg bw/day
Rat: 1 ppm, equivalent to 0.05 mg/kg bw/day
(2-year study)
0.5 ppm, equal to 0.03 mg/kg bw/day
(reproduction study)
Dog: 0.1 mg/kg bw/day
Estimate of acceptable daily intake for humans
0-0.0003 mg/kg bw
Studies which will provide information valuable in the
continued evaluation of the compound
Observations in humans.
REFERENCES
FMC (1990a) FMC 67825 Technical Material Cadusafos. Mammalian
Toxicology Overview prepared by Toxicology Department FMC
Corporation Chemical Research and Development Center, Princeton, New
Jersey. Submitted to WHO by FMC Corporation, NJ, USA.
Dalgard, D.W. (1988) 13-Week Comparative Oral Toxicity Study in Dogs
with FMC 67825 (techn.). Project Number A87-2531. Unpublished report
prepared by Hazleton Laboratories, America, Inc. for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
De Prospo, J.R. et al. (1987) Multi-Generation Reproduction Study
with FMC 67825 Technical in Rats. Unpublished report prepared by
FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
De Prospo, J.R. (1986) Acute Oral Toxicity in Rats with FMC 67825
Technical Material. Study Number A85-1796. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Dudek, B.R. (1984) Four Hour Acute Aerosol Inhalation Toxicity Study
in Rats with FMC 67825 Technical Material. Study Number A84-1231.
Unpublished report prepared by ToxiGenics, Inc., Illinois for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Freeman, Ch. (1985) Teratology Study in Rats with FMC 67825
Technical. Study Number A84-1173. Unpublished report prepared by
FMC Toxicology Laboratory Princeton, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Freeman, Ch. (1985) Teratology Study in Rabbits with FMC 67825
Technical. Study Number A84-1175. Unpublished report prepared by FMC
Toxicology Laboratory, New Jersey.
Freeman, Ch. (1987a) Acute Dermal Toxicity in Rabbits with FMC 67825
Technical Material. Study Number A86-2190. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Freeman, Ch. (1987b) Acute Oral Toxicity in Rats with FMC 67825
Technical. Study Number A86-2191. Unpublished report prepared by
FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
Geiger, L.E. et al. (1986). Cholinesterase Inhibition Titration
Study in Rats with FMC 67825 (techn.). Study Number A84-1169.
Unpublished report prepared by FMC Toxicology Laboratory, New
Jersey. Submitted to WHO by FMC Corporation, NJ, USA.
Haworth, St.R. et al. (1984) Salmonella/Mammalian Microsome
Plate Incorporation Mutagenicity Assay (Ames Test). With FMC 67825
D (techn.). Study Number A83-1155. Unpublished report prepared by
Microbiological Associates, Bethesda, Maryland for FMC Corporation.
Submitted to WHO by FMC Corporation, NJ, USA.
Kedderis, L.B. (1988a) FMC 67825 Technical: Antidotal Study in Rats
with Atropine and/or 2-PAM. Study Number A86-2222. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
Kedderis, L.B. (1988b) FMC 67825 Technical: Antidotal Study in Rats
with Atropine and/or 2-PAM. Study Number A86-2222A. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
Lawlor, T.E. et al. (1985) Salmonella/Mammalian-Microsome Plate
Incorporation Mutagenicity Assay (Ames Test) with FMC 67825
(techn.). Study Number A85-1797. Unpublished report prepared by
Microbiological Associates, Inc., Bethesda, Maryland for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
McCarty, J.D. (1984a) Primary Eye Irritation in Rabbits with FMC
67825 (techn.). Study Number A83-1154. Unpublished report prepared
by FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
McCarty, J.D. (1984b) Primary Skin Irritation in Rabbits with FMC
67825 (techn.). Study Number A84-1238. Unpublished report prepared
by FMC Toxicology Laboratory, New Jersey. Submitted to WHO by FMC
Corporation, NJ, USA.
McCarty, J.D. (1984c) Acute Oral Toxicity in Rats with FMC 67825
Technical Material. Study Number A83-1164. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
McCarty, J.D. (1984d) Skin Sensitization Study in Guinea Pigs with
FMC 67825 Technical Material. Study Number A84-1271. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
McCarty, J.D. et al. (1985) Ninety Day Feeding Study in Rats with
FMC 67825 (techn.). Study Number A84-1232. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
McCarty, J.D. et al. (1986) Twenty-Eight Day Range-finding Study
with FMC 67825 Technical in Mice. Study Number A83-1153. Unpublished
report prepared by FMC Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
McCarty, J.D. et al. (1987) Chronic Oncogenicity Study in Mice
with FMC 67825 Technical. Study Number A84-1437. Unpublished report
prepared by FMC Corp Toxicology Laboratory, New Jersey. Submitted
to WHO by FMC Corporation, NJ, USA.
Puhl, R.J. (1987) FMC 67825 (techn.) Rat Metabolism Single and
Multiple Low-Dose Test Regimen. Unpublished Report prepared by
Hazelton Laboratories America Inc. Wisconsin. (HLA Study No.
6124-105; FMC Report No. PC-0077) Submitted to FMC Corporation, New
Jersey. Submitted to WHO by FMC Corporation, NJ, USA.
Putman, D.L. et al. (1984) Activity of FMC 67825 (techn.), (RUGBY)
in the Morphological Transformation of BALB/3T3 Mouse Embryo Cells
in the Presence and Absence of Exogenous Metabolic Activation.
Study Number A83-1158. Unpublished report prepared by
Microbiological Associates Inc., Bethesda, Maryland for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Rand, G.M. (1986) Twenty-Eight Day Range-finding Study in Rats with
FMC 67825 (techn.). Study Number A83-1146. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Rand, G.M. (1983a) Acute Dermal Toxicity in Rabbits with FMC 67825
Technical Material. Study Number A83-916. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Rand, G.M. (1983b) Acute Oral Toxicity in Mice with FMC 67825
Technical Material. Study Number A83-915. Unpublished report
prepared by FMC Toxicology Laboratory, New Jersey. Submitted to WHO
by FMC Corporation, NJ, USA.
Roberts, N.L. et al. (1984) Acute Delayed Neurotoxicity Study in
the Domestic Hen with FMC 67825. Study Number A84-1246. Unpublished
report prepared by Huntingdon Research Centre England for FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Seely, J.C. et al. (1985a) 14-Day Range-finding Oral Toxicity
Study in Dogs with FMC 67825 (techn.). Study Number A84-1203.
Unpublished report prepared by Pharmacopathics Research
Laboratories, Inc. Laurel, Maryland for FMC Corporation. Submitted
to WHO by FMC Corporation, NJ, USA.
Seely, J.C. et al. (1985b) 91-Day Subchronic Oral Toxicity Study
in the Dog with FMC 67825 Technical. Study Number A84-1204.
Unpublished report prepared by Pharmacopathics Research
Laboratories, Inc. Laurel, Maryland for FMC Corporation. Submitted
to WHO by FMC Corporation, NJ, USA.
Selim. S. (1984) Rat Balance Study and Tissue Distribution of 14C
Labelled FMC 67825 (techn.). Report (Nr. FM-175T) of the Primate
Research Institute, New Mexico State University, submitted to FMC
Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Shellenberger, Th.E. (1986) One Year Oral Toxicity Study in the Dog
with FMC 67825 (techn.). Study Number A84-1538. Unpublished report
prepared by Tegeris Laboratories Inc. (formerly: Pharmacopathics
Research Laboratories, Inc.), Laurel, Maryland for FMC Corporation.
Submitted to WHO by FMC Corporation, NJ, USA.
Stankowski, L.F. et al. (1985) Mammalian Cell Forward Gene
Mutation Assay with FMC 67825 (techn.). Study Number A85-1601.
Unpublished Report prepared by Pharmakon Research International,
Inc., Waverly, Pennsylvania for FMC Corporation. Submitted to WHO
by FMC Corporation, NJ, USA.
Thilagar, A. et al. (1984a) Chromosome Aberrations Assay in
Chinese Hamster Ovary (CHO) Cells. With FMC 67825 (techn.) Study
Number A83-1157. Unpublished report prepared by Microbiological
Associates, Bethesda, Maryland for FMC Corporation. Submitted to
WHO by FMC Corporation, NJ, USA.
Thilagar, A. et al. (1984b) Unscheduled DNA Synthesis in Rat
Primary Hepatocytes with FMC 67825 (techn.). Study Number A83-1159.
Unpublished report prepared by Microbiological Associates Bethesda,
Maryland for FMC Corporation. Submitted to WHO by FMC Corporation,
NJ, USA.
Thilagar, A. et al. (1984c) CHO/HGPRT Mutation Assay in Chinese
Hamster Ovary (CHO) Cells. With FMC 65825 (techn.) (RUGBY). Study
Number T-2199-332. Unpublished report prepared by Microbiological
Associates Inc., Bethesda, Maryland for FMC Corporation. Submitted
to WHO by FMC Corporation, NJ, USA.
Weiner, M. et al. (1986) Chronic Oral toxicity/Oncogenicity Study
in Rats with FMC 67825 Technical. Study Number A84-1287. Unpublished
report prepared by Hazleton Laboratories America Inc., Virginia for
FMC Corporation. Submitted to WHO by FMC Corporation, NJ, USA.
Wu, J. (1988) FMC 67825 (techn.) Rat Metabolism: Metabolite
Identification and Distribution.10 Unpublished report prepared by
FMC Corporation, Metabolism Laboratories, New Jersey. Submitted to
WHO by FMC Corporation, NJ, USA.
See Also:
Toxicological Abbreviations