
UNITED NATIONS ENVIRONMENT PROGRAMME
INTERNATIONAL LABOUR ORGANISATION
WORLD HEALTH ORGANIZATION
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 202
SELECTED NON-HETEROCYCLIC
POLICYCLIC AROMATIC HYDROCARBONS
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First and second drafts prepared by staff members at the Fraunhofer
Institute of Toxicology and Aerosol Research, Hanover, Germany, under
the coordination of Dr R.F. Hertel, Dr G. Rosner, and Dr J. Kielhorn,
in cooperation with Dr E. Menichini, Italy, Dr P.L. Grover, United
Kingdom, and Dr J. Blok, Netherlands. Dr P. Muller, Canada, and Dr R.
Schoeny and Dr T.L. Mumford, USA, prepared and revised the drafts of
Appendix I.
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals
World Health Organization
Geneva, 1998
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO), and the World Health Organization (WHO). The overall objectives
of the IPCS are to establish the scientific basis for assessment of
the risk to human health and the environment from exposure to
chemicals, through international peer-review processes, as a
prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization, and the Organisation for
Economic Co-operation and Development (Participating Organizations),
following recommendations made by the 1992 United Nations Conference
on Environment and Development, to strengthen cooperation and increase
coordination in the field of chemical safety. The purpose of the IOMC
is to promote coordination of the policies and activities pursued by
the Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing in Publication Data
Selected non-heterocyclic polycyclic aromatic hydrocarbons.
(Environmental health criteria ; 202)
1. Polycyclic hydrocarbons, Aromatic 2.Environmental exposure
3.Occupational exposure 4.Risk assessment - methods
I.INternational Programme on Chemical Safety II.Series
ISBN 92 4 157202 7 (NLM Classification: QD 341.H9)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1998
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city or area or
of its authorities, or concerning the delimitation of its frontiers or
boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
PREAMBLE
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA
FOR SELECTED NON-HETEROCYCLIC POLYCYCLIC AROMATIC HYDROCARBONS
ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED NON-HETEROCYCLIC POLYCYCLIC
AROMATIC HYDROCARBONS
1. SUMMARY
1.1. Selection of compounds for this monograph
1.2. Identity, physical and chemical properties, and analytical
methods
1.3. Sources of human and environmental exposure
1.4. Environmental transport, distribution, and transformation
1.5. Environmental levels and human exposure
1.5.1. Air
1.5.2. Surface water and precipitation
1.5.3. Sediment
1.5.4. Soil
1.5.5. Food
1.5.6. Aquatic organisms
1.5.7. Terrestrial organisms
1.5.8. General population
1.5.9. Occupational exposure
1.6. Kinetics and metabolism
1.7. Effects on laboratory mammals and in vitro
1.8. Effects on humans
1.9. Effects on other organisms in the laboratory and the field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.1.1. Technical products
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling
2.4.1.1 Ambient air
2.4.1.2 Workplace air
2.4.1.3 Combustion effluents
2.4.1.4 Water
2.4.1.5 Solid samples
2.4.2. Preparation
2.4.3. Analysis
2.4.3.1 Gas chromatography
2.4.3.2 High-performance liquid chromatography
2.4.3.3 Thin-layer chromatography
2.4.3.4 Other techniques
2.4.4. Choice of PAH to be quantified
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. PAH in coal and petroleum products
3.2.2. Production levels and processes
3.2.3. Uses of individual PAH
3.2.4. Emissions during production and processing of PAH
3.2.4.1 Emissions to the atmosphere
3.2.4.2 Emissions to the hydrosphere
3.2.5. Emissions during use of individual PAH
3.2.6. Emissions of PAH during processing and use
of coal and petroleum products
3.2.6.1 Emissions to the atmosphere
3.2.6.2 Emissions to the hydrosphere
3.2.6.3 Emissions to the geosphere
3.2.6.4 Emissions to the biosphere
3.2.7. Emissions of PAH caused by incomplete combustion
3.2.7.1 Industrial point sources
3.2.7.2 Other diffuse sources
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Physicochemical parameters that dtermine
environmental transport and distribution
4.1.2. Distribution and transport in the gaseous phase
4.1.3. Volatilization
4.1.4. Adsorption onto soils and sediments
4.1.5. Bioaccumulation
4.1.5.1 Aquatic organisms
4.1.5.2 Terrestrial organisms
4.1.6. Biomagnification
4.2. Transformation
4.2.1. Biotic transformation
4.2.1.1 Biodegradation
4.2.1.2 Biotransformation
4.2.2. Abiotic degradation
4.2.2.1 Photodegradation in the environment
4.2.2.2 Hydrolysis
4.2.3. Ultimate fate after use
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Atmosphere
5.1.1.1 Source identification
5.1.1.2 Background and rural levels
5.1.1.3 Industrial sources
5.1.1.4 Diffuse sources
5.1.2. Hydrosphere
5.1.2.1 Surface and coastal waters
5.1.2.2 Groundwater
5.1.2.3 Drinking-water and water supplies
5.1.2.4 Precipitation
5.1.3. Sediment
5.1.3.1 River sediment
5.1.3.2 Lake sediment
5.1.3.3 Marine sediment
5.1.3.4 Estuarine sediment
5.1.3.5 Harbour sediment
5.1.3.6 Time trends of PAH in sediment
5.1.4. Soil
5.1.4.1 Background values
5.1.4.2 Industrial sources
5.1.4.3 Diffuse sources
5.1.4.4 Time trends of PAH in soil
5.1.5. Food
5.1.5.1 Meat and meat products
5.1.5.2 Fish and marine foods
5.1.5.3 Dairy products: cheese, butter, cream
milk, and related products
5.1.5.4 Vegetables
5.1.5.5 Fruits and confectionery
5.1.5.6 Cereals and dried food products
5.1.5.7 Beverages
5.1.5.8 Vegetable and animal fats and oils
5.1.6. Biota
5.1.7. Animals
5.1.7.1 Aquatic organisms
5.1.7.2 Terrestrial organisms
5.2. Exposure of the general population
5.2.1. Indoor air
5.2.2. Food
5.2.3. Other sources
5.2.4. Intake of PAH by inhalation
5.2.5. Intake of PAH from food and drinking-water
5.3. Occupational exposure
5.3.1. Occupational exposure during processing and use
of coal and petroleum products
5.3.1.1 Coal coking
5.3.1.2 Coal gasification and coal liquefaction
5.3.1.3 Pteroleum refining
5.3.1.4 Road paving
5.3.1.5 Roofing
5.3.1.6 Impregnation of wood with creosotes
5.3.1.7 Other exposures
5.3.2. Occupational exposure resulting from incomplete
combustion of mineral oil, coal, and their products
5.3.2.1 Aluminium production
5.3.2.2 Foundries
5.3.2.3 Other workplaces
6. KINETICS AND METABOLISM IN LABORATORY MAMMALS AND HUMANS
6.1. Absorption
6.1.1. Absorption by inhalation
6.1.2. Absorption in the gastrointestinal tract
6.1.3. Absorption through the skin
6.2. Distribution
6.3. Metabolic transformation
6.3.1. Cytochromes P450 and PAH metabolism
6.3.1.1 Individual cytochrome P450 enzymes
that metabolize PAH
6.3.1.2 Regulation of cytochrome P450 enzymes
that metabolize PAH
6.3.2. Metabolism of benzo [a]pyrene
6.4. Elimination and excretion
6.5. Retention and turnover
6.5.1. Human body burdens of PAH
6.6. Reactions with tissue components
6.6.1. Reactions with proteins
6.6.2. Reactions with nucleic acids
6.7. Analytical methods
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO
7.1. Toxicity after a single exposure
7.1.1. Benzo [a]pyrene
7.1.2. Chrysene
7.1.3. Dibenz [a,h]anthracene
7.1.4. Fluoranthene
7.1.5. Naphthalene
7.1.6. Phenanthrene
7.1.7. Pyrene
7.2. Short-term toxicity
7.2.1. Subacute toxicity
7.2.1.1 Acenaphthene
7.2.1.2 Acenaphthylene
7.2.1.3 Anthracene
7.2.1.4 Benzo [a]pyrene
7.2.1.5 Benz [a]anthracene
7.2.1.6 Dibenz [a,h]anthracene
7.2.1.7 Fluoranthene
7.2.1.8 Naphthalene
7.2.1.9 Phenanthrene
7.2.1.10 Pyrene
7.2.2. Subchronic toxicity
7.2.2.1 Acenaphthene
7.2.2.2 Anthracene
7.2.2.3 Benzo [a]pyrene
7.2.2.4 Fluorene
7.2.2.5 Fluoranthene
7.2.2.6 Naphthalene
7.2.2.7 Pyrene
7.3. Long-term toxicity
7.3.1. Anthracene
7.3.2. Benz [a]anthracene
7.3.3. Dibenz [a,h]anthracene
7.4. Dermal and ocular irritation and dermal sensitization
7.4.1. Anthracene
7.4.2. Benzo [a]pyrene
7.4.3. Naphthalene
7.4.4. Phenanthrene
7.5. Reproductive effects, embryotoxicity, and teratogenicity
7.5.1. Benzo [a]pyrene
7.5.1.1 Teratogenicity in mice of different
genotypes
7.5.1.2 Reproductive toxicity
7.5.1.3 Effects on postnatal development
7.5.1.4 Immunological effects in pregnant
rats and mice
7.5.2. Naphthalene
7.5.2.1 Embryotoxicity
7.5.2.2 Toxicity in cultured embryos
7.6. Mutagenicity and related end-points
7.7. Carcinogenicity
7.7.1. Single substances
7.7.1.1 Benzo [a]pyrene
7.7.1.2 Benzo [e]pyrene
7.7.2. Comparative studies
7.7.2.1 Carcinogenicity
7.7.2.2 Further evidence
7.7.3. PAH in complex mixtures
7.7.4. Transplacental carcinogenicity
7.7.4.1 Benzo [a]pyrene
7.7.4.2 Pyrene
7.8. Special studies
7.8.1. Phototoxicity
7.8.1.1 Anthracene
7.8.1.2 Benzo [a]pyrene
7.8.1.3 Pyrene
7.8.1.4 Comparisons of individual PAH
7.8.2. Immunotoxicity
7.8.2.1 Benzo [a]pyrene
7.8.2.2 Dibenz [a,h]anthracene
7.8.2.3 Fluoranthene
7.8.2.4 Naphthalene
7.8.2.5 Comparisons of individual PAH
7.8.2.6 Exposure in utero
7.8.2.7 Mechanisms of the immunotoxicity of PAH
7.8.3. Hepatotoxicity
7.8.3.1 Benzo [a]pyrene
7.8.3.2 Comparisons of individual PAH
7.8.4. Renal toxicity
7.8.5. Ocular toxicity of naphthalene
7.8.6. Percutaneous absorption
7.8.7. Other studies
7.8.7.1 Benzo [k]fluoranthene
7.8.7.2 Benzo [a]pyrene
7.8.7.3 Phenanthrene
7.8.7.4 Comparisons of individual PAH
7.9. Toxicity of metabolites
7.9.1. Benzo [a]pyrene
7.9.2. 5-Methylchrysene
7.9.3. 1-Methylphenanthrene
7.10. Mechanisms of carcinogenicity
7.10.1. History
7.10.2. Current theories
7.10.3. Theories under discussion
7.10.3.1 Acenaphthene and acenaphthylene
7.10.3.2 Anthracene
7.10.3.3 Benzo [a]pyrene
7.10.3.4 Benz [a]anthracene
7.10.3.5 Benzo [c]phenanthrene
7.10.3.6 Chrysene
7.10.3.7 Cyclopenta [c,d]pyrene
7.10.3.8 Fluorene
7.10.3.9 Indeno[1,2,3- cd]pyrene
7.10.3.10 5-Methylchrysene
7.10.3.11 1-Methylphenanthrene
7.10.3.12 Naphthalene
7.10.3.13 Phenanthrene
7.10.3.14 Investigations of groups of PAH
8. EFFECTS ON HUMANS
8.1. Exposure of the general population
8.1.1. Naphthalene
8.1.1.1 Poisoning incidents
8.1.1.2 Controlled studies
8.1.2. Mixtures of PAH
8.1.2.1 PAH in unvented coal combustion
in homes
8.1.2.2 PAH in cigarette smoke
8.1.2.3 PAH in coal-tar shampoo
8.2. Occupational exposure
8.3. Biomarkers of exposure to PAH
8.3.1. Urinary metabolites in general
8.3.2. 1-Hydroxypyrene
8.3.2.1 Method of determination
8.3.2.2 Concentrations
8.3.2.3 Time course of elimination
8.3.2.4 Suitability as a biomarker
8.3.3. Mutagenicity in urine
8.3.4. Genotoxicity in lymphocytes
8.3.5. DNA adducts
8.3.5.1 Method of determination
8.3.5.2 Concentrations
8.3.5.3 Suitability as a biomarker
8.3.6. Antibodies to DNA adducts
8.3.7. Protein adducts
8.3.8. Activity of cytochrome P450
8.3.9. Cell surface differentiation antigens in lung cancer
8.3.10. Oncogene proteins
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND THE FIELD
9.1. Laboratory experiments
9.1.1. Microorganisms
9.1.1.1 Water
9.1.1.2 Soil
9.1.2. Aquatic organisms
9.1.2.1 Plants
9.1.2.2 Invertebrates
9.1.2.3 Vertebrates
9.1.2.4 Sediment-dwelling organisms
9.1.2.5 Toxicity of combinations of PAH
9.1.3. Terrestrial organisms
9.1.3.1 Plants
9.1.3.2 Invertebrates
9.1.3.3 Vertebrates
9.2. Field observations
9.2.1. Microorganisms
9.2.1.1 Water
9.2.1.2 Soil
9.2.2. Aquatic organisms
9.2.2.1 Plants
9.2.2.2 Invertebrates
9.2.2.3 Vertebrates
9.2.3. Terrestrial organisms
9.2.3.1 Plants
9.2.3.2 Invertebrates
9.2.3.3 Vertebrates
10 EVALUATION OF RISKS TO HUMAN HEALTH AND EFFECTS ON THE
ENVIRONMENT
10.1. Human health
10.1.1. Exposure
10.1.1.1 General population
10.1.1.2 Occupational exposure
10.1..2 Toxic effects
10.1.2.1 Bioavailability
10.1.2.2 Acute toxicity
10.1.2.3 Irritation and allergic sensitization
10.1.2.4 Medium-term toxicity
10.1.2.5 Carcinogenicity
10.1.2.6 Reproductive toxicity
10.1.2.7 Immunotoxicity
10.1.2.8 Genotoxicity
10.2. Environment
10.2.1. Environmental levels and fate
10.2.2. Ecotoxic effects
10.2.2.1 Terrestrial organisms
10.2.2.2 Aquatic organisms
11 RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH AND THE
ENVIRONMENT
11.1. General recommendations
11.2. Protection of human health
11.3. Recommendations for further research
11.3.1. General
11.3.2. Protection of human health
11.3.3. Environmental protection
11.3.4. Risk assessment
12 PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
12.1. International Agency for Research on Cancer
12.2. WHO Water Quality Guidelines
12.3. FAO/WHO Joint Expert Committee on Food Additives
12.4. WHO Regional Office for Europe Air Quality Guidelines
APPENDIX I. SOME APPROACHES TO RISK ASSESSMENT FOR POLYCYCLIC AROMATIC
HYDROCARBONS
I.1 Introduction
I.2 Approaches to risk assessment
I.2.1 Toxicity equivalence factors and related approaches
I.2.1.1 Principle
I.2.1.2 Development and validation
I.2.1.2.1 Derivation of the potency of
benzo [a]pyrene
I2.1.2.2 Derivation of the relative potency of
PAH other than benzo [a]pyrene
I.2.1.3 Application
I.2.2 Comparative potency approach
I.2.2.1 Principle
I.2.2.2 Development and validation
I.2.2.3 Key implicit and explicit assumptions
I.2.2.4 Application
I.2.3 Benzo [a]pyrene as a surrogate for the PAH fraction
of complex mixtures
I.2.3.1 Principle
I.2.3.2 Development and validation
I.2.3.3 PAH profiles of complex mixtures
I.2.3.4 Potency of complex mixtures
I.2.3.5 Key implicit and explicit assumptions
I.2.3.6 Application
I.3 Comparison of the three procedures
I.3.1 Individual PAH approach
I.3.2 Comparative potency approach
I.3.3 Benzo [a]pyrene surrogate approach
APPENDIX II; SOME LIMIT VALUES
II.1 Exposure of the consumer
II.2 Occupational exposure
II.3 Classification
II.3.1 European Union
II.3.2 USA
REFERENCES
RESUME
RESUMEN
Environmental Health Criteria
PREAMBLE
Objectives
The WHO Environmental Health Criteria Programme was initiated in
1973, with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976; numerous assessments of chemicals and
of physical effects have since been produced. Many EHC monographs have
been devoted to toxicological methods, e.g. for genetic, neurotoxic,
teratogenic, and nephrotoxic effects. Other publications have been
concerned with e.g. epidemiological guidelines, evaluation of
short-term tests for carcinogens, biomarkers, and effects on the
elderly.
Since the time of its inauguration, the EHC Programme has widened
its scope, and the importance of environmental effects has been
increasingly emphasized in the total evaluation of chemicals, in
addition to their health effects.
The original impetus for the Programme came from resolutions of
the World Health Assembly and the recommendations of the 1972 United
Nations Conference on the Human Environment. Subsequently, the work
became an integral part of the International Programme on Chemical
Safety (IPCS), a cooperative programme of UNEP, ILO, and WHO. In this
manner, with the strong support of the new partners, the importance of
occupational health and environmental effects was fully recognized.
The EHC monographs have become widely established, used, and
recognized throughout the world.
The recommendations of the 1992 United Nations Conference on
Environment and Development and the subsequent establishment of the
Intergovernmental Forum on Chemical Safety, with priorities for action
in the six programme areas of Chapter 19, Agenda 21, lend further
weight to the need for EHC assessments of the risks of chemicals.
Scope
The Criteria monographs are intended to provide critical reviews
of the effect on human health and the environment of chemicals,
combinations of chemicals, and physical and biological agents. They
include reviews of studies that are of direct relevance for the
evaluation and do not describe every study that has been carried out.
Data obtained worldwide are used, and results are quoted from original
studies, not from abstracts or reviews. Both published and unpublished
reports are considered, and the authors are responsible for assessing
all of the articles cited; however, preference is always given to
published data, and unpublished data are used only when relevant
published data are absent or when the unpublished data are pivotal to
the risk assessment. A detailed policy statement is available that
describes the procedures used for citing unpublished proprietary data,
so that this information can be used in the evaluation without
compromising its confidential nature (WHO, 1990).
In the evaluation of human health risks, sound data on humans,
whenever available, are preferred to data on experimental animals.
Studies of animals and in-vitro systems provide support and are used
mainly to supply evidence missing from human studies. It is mandatory
that research on human subjects be conducted in full accord with
ethical principles, including the provisions of the Helsinki
Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not in any sense recommendations for regulation or
setting standards. The latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary: a review of the salient facts and the risk evaluation of
the chemical
* Identity: physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution, and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in-vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and the field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g. the
International Agency for Research on Cancer, the Joint FAO/WHO
Expert Committee on Food Additives, and the Joint FAO/WHO
Meeting on Pesticide Residues
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of chemicals that are of
priority for subsequent evaluation. Such meetings have been held in
Ispra, Italy (1980); Oxford, United Kingdom (1984); Berlin, Germany
(1987); and North Carolina, United States of America (1995). The
selection of chemicals is based on the following criteria: the
existence of scientific evidence that the substance presents a hazard
to human health and/or the environment; the existence of evidence that
the possible use, persistence, accumulation, or degradation of the
substance involves significant human or environmental exposure; the
existence of evidence that the populations at risk (both human and
other species) and the risks for the environment are of a significant
size and nature; there is international concern, i.e. the substance is
of major interest to several countries; adequate data are available on
the hazards.
If it is proposed to write an EHC monograph on a chemical that is
not on the list of priorities, the IPCS Secretariat first consults
with the cooperating organizations and the participating institutions.
Procedures
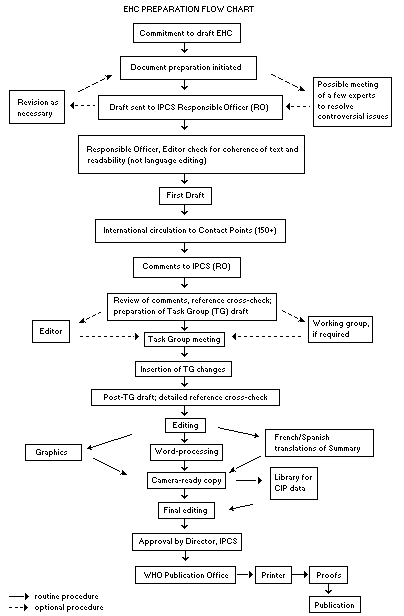
The order of procedures that result in the publication of an EHC
monograph is shown in the following flow chart. A designated staff
member of IPCS, responsible for the scientific quality of the
document, serves as Responsible Officer (RO). The IPCS Editor is
responsible for the layout and language. The first draft, prepared by
consultants or, more usually, staff at an IPCS participating
institution is based initially on data provided from the International
Register of Potentially Toxic Chemicals and reference data bases such
as Medline and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the first draft acceptable,
it is distributed in its unedited form to over 150 EHC contact points
throughout the world for comment on its completeness and accuracy and,
where necessary, to provide additional material. The contact points,
usually designated by governments, may be participating institutions,
IPCS focal points, or individual scientists known for their particular
expertise. Generally, about four months are allowed before the
comments are considered by the RO and author(s). A second draft
incorporating the comments received and approved by the Director,
IPCS, is then distributed to Task Group members, who carry out a peer
review at least six weeks before their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government, or industry. Their
function is to evaluate the accuracy, significance, and relevance of
the information in the document and to assess the risks to health and
the environment from exposure to the chemical. A summary and
recommendations for further research and improved safety are also
drawn up. The composition of the Task Group is dictated by the range
of expertise required for the subject of the meeting and by the need
for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations, so that
representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide valuable contributions to the process, they can speak only
at the invitation of the Chairperson. Observers do not participate in
the final evaluation of the chemical, which is the sole responsibility
of the Task Group members. The Task Group may meet in camera when it
considers that to be appropriate.
All individuals who participate in the preparation of an EHC
monograph as authors, consultants, or advisers must, in addition to
serving in their personal capacity as scientists, inform the RO if at
any time a conflict of interest, whether actual or potential, could be
perceived in their work. They are required to sign a statement to that
effect. This procedure ensures the transparency and probity of the
process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it is edited for language, the references are checked, and
camera-ready copy is prepared. After approval by the Director, IPCS,
the monograph is submitted to the WHO Office of Publications for
printing. At this time, a copy of the final draft is also sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern about
health or environmental effects of the agent because of greater
exposure; an appreciable time has elapsed since the last evaluation.
All participating institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The chairpersons of task groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
 WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED
NON-HETEROCYCLIC POLYCYCLIC AROMATIC HYDROCARBONS
Hanover, Germany, 25-29 September 1995
Members
Dr P.E.T. Douben, Her Majesty's Inspectorate of Pollution, London,
United Kingdom (Chairman)
Dr P.L. Grover, Institute for Cancer Research, Sutton, United Kingdom
Dr R.F. Hertel, Bundesgesinstitut für gesundheitlichen
Verbraucherschutz und Veterinarmedizin, Berlin, Germany
Professor J. Jacob, Biochemisches Institut für Umweltcarcinogene,
Grosshausdorf, Germany
Dr J Kielhorn, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr R.W. Luebke, National Health and Ecology Effects Laboratory, US
Environmental Protection Agency, Research Triangle Park, NC, USA
(Joint Rapporteur)
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood,
Huntingdon, Cambridgeshire, United Kingdom (Joint Rapporteur)
Dr I. Mangelsdorf, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr E. Menichini, Istituto Superiore di Sanita, Rome, Italy
Dr P. Muller, Ministry of Environment and Energy, Toronto, Ontario,
Canada
Dr J.L. Mumford, National Health and Environmental Effects Research
Laboratory, US Environmental Protection Agency, Research Triangle
Park, NC, USA
Dr G. Rosner, Freiburg, Germany
Dr R. Schoeny, National Center for Environmental Assessment, US
Environmental Protection Agency, Cincinnati, OH, USA
Dr T. Sorahan, Institute of Occupational Health, University of
Birmingham, Birmingham, United Kingdom
Dr Kimber L. White, Jr, Medical College of Virginia, Virginia
Commonwealth University, Richmond, VA, USA (Vice-Chairman)
Secretariat
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
Dr. M. Castegnaro, International Agency for Research on Cancer, Lyon,
France
Assisting the Secretariat
Dr S. Artelt, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr A. Boehncke, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr O. Creutzenburg, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
1. SUMMARY
1.1 Selection of compounds for this monograph
Polycyclic aromatic hydrocarbons (PAH) constitute a large class
of compounds, and hundreds of individual substances may be released
during incomplete combustion or pyrolysis of organic matter, an
important source of human exposure. Studies of various environmentally
relevant matrices, such as coal combustion effluents, motor vehicle
exhaust, used motor lubricating oil, and tobacco smoke, have shown
that the PAH in these mixtures are mainly responsible for their
carcinogenic potential.
PAH occur almost always in mixtures. Because the composition of
such mixtures is complex and varies with the generating process, all
mixtures containing PAH could not possible be covered in detail in
this monograph. Thus, 33 individual compounds (31 parent PAH and two
alkyl derivatives) were selected for evaluation on the basis of the
availability of relevant data on toxicological end-points and/or
exposure (Table 1). Since epidemiological studies, which are essential
for risk assessment, were available only for mixtures, however,
Sections 8 and 10 present the results of studies of mixtures of PAH,
in contrast to the rest of the monograph.
Numerous papers and reviews have been published on the
occurrence, distribution, and transformation of PAH in the environment
and on their ecotoxicological and toxicological effects. Only
references from the last 10-15 years are cited in this monograph,
unless no other information was available; reviews are cited for older
studies and for further information.
1.2 Identity, physical and chemical properties, and analytical
methods
The term 'polycyclic aromatic hydrocarbons' commonly refers to a
large class of organic compounds containing two or more fused aromatic
rings made up of carbon and hydrogen atoms. At ambient temperatures,
PAH are solids. The general characteristics common to the class are
high melting- and boiling-points, low vapour pressure, and very low
water solubility which tends to decrease with increasing molecular
mass. PAH are soluble in many organic solvents and are highly
lipophilic. They are chemically rather inert. Reactions that are of
interest with respect to their environmental fate and possible sources
of loss during atmospheric sampling are photodecomposition and
reactions with nitrogen oxides, nitric acid, sulfur oxides, sulfuric
acid, ozone, and hydroxyl radicals.
Ambient air is sampled by collecting suspended particulate matter
on glass-fibre, polytetrafluoroethylene, or quartz-fibre filters by
means of high-volume or passive samplers. Vapour-phase PAH, which
might volatilize from filters during sampling, are commonly trapped by
adsorption on polyurethane foam. The sampling step is by far the most
important source of variability in results.
Table 1. Polycyclic aromatic hydrocarbons evaluated in this monograph
Common name CAS name Synonyma CAS Registry No.
Acenaphthylene Acenaphthylene 91-20-3
Acenaphthene Acenaphthylene, 1,2-dihydro- 208-96-8
Anthanthrene Dibenzo[def,mno]chrysene 191-26-4
Anthracene Anthracene 120-12-7
Benz[a]anthracene Benz[a]anthracene 1,2-Benzanthracene, 56-55-3
tetraphene
Benzo[a]fluorene 11 H-Benzo[a]fluorene 1,2-Benzofluorene 238-84-6
Benzo[b]fluorene 11 H-Benzo[b]fluorene 2,3-Benzofluorene 243-17-4
Benzo[b]fluoranthene Benz[e]acephenanthrylene 3,4-Benzofluoranthene 205-99-2
Benzo[ghi]fluoranthene Benzo[ghi]fluoranthene 2,13-Benzofluoranthene 203-12-3
Benzo[j]fluoranthene Benzo[j]fluoranthene 10,11-Benzofluoranthene 205-82-3
Benzo[k]fluoranthene Benzo[k]fluoranthene 11,12-Benzofluoranthene 207-08-9
Benzo[ghi]perylene Benzo[ghi]perylene 1,12-Benzoperylene 191-24-2
Benzo[c]phenanthrene Benzo[c]phenanthrene 3,4-Benzophenanthrene 195-19-7
Benzo[a]pyrene Benzo[a]pyrene 3,4-Benzopyreneb 50-32-8
Benzo[e]pyrene Benzo[e]pyrene 1,2-Benzopyrene 192-97-2
Chrysene Chrysene 1,2-Benzophenanthrene 218-01-9
Coronene Coronene Hexabenzobenzene 191-07-1
Cyclopenta[cd]pyrene Cyclopenta[cd]pyrene Cyclopenteno[cd]pyrene 27208-37-3
Dibenz[a,h]anthracene Dibenz[a,h]anthracene 1,2:5,6-Dibenzanthracene 53-70-3
Dibenzo[a,e]pyrene Naphtho[1,2,3,4-def]chrysene 1,2:4,5-Dibenzopyrene 192-65-4
Dibenzo[a,h]pyrene Dibenzo[b,def]chrysene 3,4:8,9-Dibenzopyrene 189-64-0
Dibenzo[a,i]pyrene Benzo[rst]pentaphene 3,4:9,10-Dibenzopyrene 189-55-9
Dibenzo[a,l]pyrene Dibenzo[def,p]chrysene 1,2:3,4-Dibenzopyrene 191-30-0
Fluoranthene Fluoranthene 206-44-0
Fluorene 9H-Fluorene 86-73-7
Indeno[1,2,3-cd]pyrene Indeno[1,2,3-cd]-pyrene 2,3-o-Phenylenpyrene 193-39-5
5-Methylchrysene Chrysene, 5-methyl- 3697-24-3
1-Methylphenanthrene Phenanthrene, 1-methyl- 832-69-9
Table 1. (continued)
Common name CAS name Synonyma CAS Registry No.
Naphthalene Naphthalene 91-20-3
Perylene Perylene peri-Dinaphthalene 198-55-0
Phenanthrene Phenanthrene 85-01-8
Pyrene Pyrene Benzo[def]phenanthrene 129-00-0
Triphenylene Triphenylene 9,10-Benzophenanthrene 217-59-4
Extensive lists of synonyms have been imported by the IARC (1983) and Loening & Merritt (1990).
a Common synonym appearing in the literature
b Also reported as benzo[def]chrysene
Air is sampled at the workplace at low flow rates; particles are
collected on glass-fibre or polytetrafluoroethylene filters and
vapours on Amberlite XAD-2 resin. Devices for sampling stack gases are
composed of a glass-fibre or quartz-fibre filter in front of a cooler
to collect condensable matter and an adsorbent (generally XAD-2)
cartridge. Vehicle exhausts are sampled under laboratory conditions,
with standard driving cycles simulating on-road conditions. Emissions
are collected either undiluted or after dilution with filtered cold
air.
Many extraction and purification techniques have been described.
Depending on the matrix, PAH are extracted from samples with a Soxhlet
apparatus, ultrasonically, by liquid-liquid partition, or, after
sample dissolution or alkaline digestion, with a selective solvent.
Supercritical fluid extraction from various environmental solids has
also been used. The efficiency of extraction depends heavily on the
solvent used, and many of the solvents commonly used in the past were
not appropriate. Extracted samples are usually purified by column
chromatography, particularly on alumina, silica gel, or Sephadex LH-20
but also by thin-layer chromatography.
Identification and quantification are routinely performed by gas
chromatography with flame ionization detection or by high-performance
liquid chromatography (HPLC) with ultraviolet and fluorescence
detection, generally in series. In gas chromatography, fused silica
capillary columns are used, with polysiloxanes (SE-54 and SE-52) as
stationary phases; silica-C18 columns are commonly used in HPLC. A
mass spectrometric detector is often coupled to a gas chromatograph in
order to confirm the identity of peaks.
The choice of PAH to be determined depends on the purpose of the
measurement, e.g. for health-orientated or ecotoxicological studies or
to investigate sources. Testing for different sets of compounds may be
required or recommended at national and international levels.
1.3 Sources of human and environmental exposure
Little information is available on the production and processing
of PAH, but it is probable that only small amounts of PAH are released
as a direct result of these activities. The PAH found principally are
used as intermediates in the production of polyvinylchloride and
plasticizers (naphthalene), pigments (acenaphthene, pyrene), dyes
(anthracene, fluoranthene), and pesticides (phenanthrene).
The largest emissions of PAH result from incomplete combustion of
organic materials during industrial processes and other human
activities, including:
- processing of coal, crude oil, and natural gas, including coal
coking, coal conversion, petroleum refining, and production of
carbon blacks, creosote, coal-tar, and bitumen;
- aluminium, iron and steel production in plants and foundries;
- heating in power plants and residences and cooking;
- combustion of refuse;
- motor vehicle traffic; and
- environmental tobacco smoke.
PAH, especially these of higher molecular mass, entering the
environment via the atmosphere are adsorbed onto particulate matter.
The hydrosphere and geosphere are affected secondarily by wet and dry
deposition. Creosote-preserved wood is another source of release of
PAH into the hydrosphere, and deposition of contaminated refuse, like
sewage sludge and fly ash, contributes to emissions of PAH into the
geosphere. Little information is available about the passage of PAH
into the biosphere. PAH occur naturally in peat, lignite, coal, and
crude oil. Most of the PAH in hard coals are tightly bound within the
coal structure and cannot be leached out.
The release of PAH into the environment has been determined by
identification of a characteristic PAH concentration profile, but this
has been possible in only a few cases. Benzo [a]pyrene has frequently
been used as an indicator of PAH, especially in older studies.
Generally, emissions of PAH are only estimates based on more or less
reliable data and give only a rough idea of exposure.
The most important sources of PAH are as follows:
Coal coking: Airborne emissions of PAH from coal coking in
Germany have decreased significantly over the last 10 years as a
result of technical improvements to existing plants, closure of old
plants, and reduced coke production. Similar situations are assumed to
exist in western Europe, Japan, and the USA, but no data were
available.
Production of aluminium (mainly special coal anodes), iron,
and steel and the binding agents used in moulding sand in foundries:
Little information is available.
Domestic and residential heating: Phenanthrene, fluoranthene,
pyrene, and chrysene are emitted as major components. The emissions
from wood stoves are 25-1000 times higher than those from
charcoal-fired stoves, and in areas where wood burning predominates
for domestic heating the major portion of airborne PAH may be derived
from this source, especially in winter. The release of PAH during
residential heating is thus assumed to be an important source in
developing countries where biomass is often burnt in relatively simple
stoves.
Cooking: PAH may be emitted during incomplete combustion of
fuels, from cooking oil, and from food being cooked.
Motor vehicle traffic: The main compounds released from
petrol-fuelled vehicles are fluoranthene and pyrene, while naphthalene
and acenaphthene are abundant in the exhaust of diesel-fuelled
vehicles. Although cyclopenta [cd]-pyrene is emitted at a high rate
from petrol-fuelled engines, its concentration in diesel exhaust is
only just above the limit of detection. The emission rates, which
depend on the substance, the type of vehicle, its engine conditions,
and the test conditions, range from a few nanograms per kilometre to
> 1000 mg/km. PAH emissions from vehicle engines are dramatically
reduced by fitting catalytic converter devices.
Forest fires: In countries with large forest areas, fires can
make an imprtant contribution to PAH emissions.
Coal-fired power plants: PAH released into the atmosphere from
such plants consist mainly of two- and three-ring compounds. In
contaminated areas, the PAH levels in ambient air may be higher than
those in the stack gases.
Incineration of refuse: The PAH emissions in stack gases from
this souce in a number of countries were < 10 mg/m3.
1.4 Environmental transport, distribution, and transformation
Several distribution and transformation processes determine the
fate of both individual PAH and mixtures. Partitioning between water
and air, between water and sediment, and between water and biota are
the most important of the distribution processes.
As PAH are hydrophobic with low solubility in water, their
affinity for the aquatic phase is very low; however, in spite of the
fact that most PAH are released into the environment via the
atmosphere, considerable concentrations are also found in the
hydrosphere because of their low Henry's law constants. As the
affinity of PAH for organic phases is greater than that for water,
their partition coefficients between organic solvents, such as
octanol, and water are high. Their affinity for organic fractions in
sediment, soil, and biota is also high, and PAH thus accumulate in
organisms in water and sediments and in their food. The relative
importance of uptake from food and from water is not clear. In
Daphnia and molluscs, accumulation of PAH from water is positively
correlated with the octanol:water partition coefficient ( Kow). In
fish and algae that can metabolize PAH, however, the internal
concentrations of different PAH are not correlated with the Kow.
Biomagnification - the increase in the concentration of a
substance in animals in successive trophic levels of food chains - of
PAH has not been observed in aquatic systems and would not be expected
to occur, because most organisms have a high biotransformation
potential for PAH. Organisms at higher trophic levels in food chains
show the highest potential biotransformation.
PAH are degraded by photodegradation, biodegradation by
microorganisms, and metabolism in higher biota. Although the last
route of transformation is of minor importance for the overall fate of
PAH in the environment, it is an important pathway for the biota,
since carcinogenic metabolites may be formed. As PAH are chemically
stable, with no reactive groups, hydrolysis plays no role in their
degradation. Few standard tests for the biodegradation of PAH are
available In general, they are biodegraded under aerobic conditions,
the biodegradation rate decreasing drastically with the number of
aromatic rings. Under anaerobic conditions, degradation is much
slower.
PAH are photooxidized in air and water in the presence of
sensitizing radicals like OH, NO3, and O3. Under laboratory
conditions, the half-life of the reaction with airborne OH radicals is
about one day, whereas reactions with NO3 and O3 usually have much
lower velocity constants. The adsorption of high-molecular-mass PAH
onto carbonaceous particles in the environment should stabilize the
reaction with OH radicals. The reaction of two- to four-ring PAH,
which occur mainly in the vapour phase, with NO3 leads to nitro-PAH,
which are known mutagens. The photooxidation of some PAH in water
seems to be more rapid than in air. Calculations based on
physicochemical and degradation parameters indicate that PAH with four
or more aromatic rings persist in the environment.
1.5 Environmental levels and human exposure
PAH are ubiquitous in the environment, and various individual PAH
have been detected in different compartments in numerous studies.
1.5.1 Air
The levels of individual PAH tend to be higher in winter than in
summer by at least one order of magnitude. The predominant source
during winter is residential heating, while that during summer is
urban motor vehicle traffic. Average concentrations of 1-30 ng/m3 of
individual PAH were detected in the ambient air of various urban
areas. In large cities with heavy motor vehicle traffic and extensive
use of biomass fuel, such as Calcutta, levels of up to 200 ng/m3 of
individual PAH were found. Concentrations of 1-50 ng/m3 were detected
in road tunnels. Cyclopenta [cd]pyrene and pyrene were present at
concentrations up to 100 ng/m3. In a subway station, PAH
concentrations of up to 20 ng/m3 were measured. Near industrial
sources, the average concentrations of individual PAH ranged from 1 to
10 ng/m3. Phenanthrene was present at up to a maximum of about 310
ng/m3.
The background values of PAH are at least one or two orders of
magnitude lower than those near sources like motor vehicle traffic.
For example, the levels at 1100 m ranged from 0.004 to 0.03 ng/m3.
1.5.2 Surface water and precipitation
Most of the PAH in water are believed to result from urban
runoff, from atmospheric fallout (smaller particles), and from asphalt
abrasion (larger particles). The major source of PAH varies, however,
in a given body of water. In general, most samples of surface water
contain individual PAH at levels of up to 50 ng/litre, but highly
polluted rivers had concentrations of up to 6000 ng/litre. The PAH
levels in groundwater are within the range 0.02-1.8 ng/litre, and
drinking-water samples contain concentrations of the same order of
magnitude. Major sources of PAH in drinking-water are asphalt-lined
storage tanks and delivery pipes.
The levels of individual PAH in rainwater ranged from 10 to 200
ng/litre, whereas levels of up to 1000 ng/litre have been detected in
snow and fog.
1.5.3 Sediment
The concentrations of individual PAH in sediment were generally
one order of magnitude higher than those in precipitation.
1.5.4 Soil
The main sources of PAH in soil are atmospheric deposition,
carbonization of plant material, and deposition from sewage and
particulate waste. The extent of pollution of soil depends on factors
such as its cultivation, its porosity, and its content of humic
substances.
Near industrial sources, individual PAH levels of up to 1 g/kg
soil have been found. The concentrations in soil from other sources,
such as automobile exhaust, are in the range 2-5 mg/kg. In unpolluted
areas, the PAH levels were 5-100 µg/kg soil.
1.5.5 Food
Raw food does not normally contain high levels of PAH, but they
are formed by processing, roasting, baking, or frying. Vegetables may
be contaminated by the deposition of airborne particles or by growth
in contaminated soil. The levels of individual PAH in meat, fish,
dairy products, vegetables and fruits, cereals and their products,
sweets, beverages, and animal and vegetable fats and oils were within
the range 0.01-10 µg/kg. Concentrations of over 100 µg/kg have been
detected in smoked meat and up to 86 µg/kg in smoked fish; smoked
cereals contained up to 160 µg/kg. Coconut oil contained up to 460
µg/kg. The levels in human breast milk were 0.003-0.03 µg/kg.
1.5.6 Aquatic organisms
Marine organisms are known to adsorb and accumulate PAH from
water. The degree of contamination is related to the extent of
industrial and urban development and shipping movements. PAH
concentrations of up to 7 mg/kg have been detected in aquatic
organisms living near industrial effluents, and the average levels of
PAH in aquatic animals sampled at contaminated sites were 10-500
µg/kg, although levels of up to 5 mg/kg were also detected.
The average levels of PAH in aquatic animals sampled at various
sites with unspecified sources of PAH were 1-100 µg/kg, but
concentrations of up to 1 mg/kg were found, for example, in lobsters
in Canada.
1.5.7 Terrestrial organisms
The concentrations of PAH in insects ranged from 730 to 5500
µg/kg. The PAH content of earthworm faeces depends significantly on
the location: those in a highly industrialized region in eastern
Germany contained benzo [a]pyrene at concentrations up to 2 mg/kg.
1.5.8 General population
The main sources of nonoccupational exposure are: polluted
ambient air, smoke from open fireplaces and cooking, environmental
tobacco smoke, contaminated food and drinking-water, and the use of
PAH-contaminated products. PAH can be found in indoor air as a result
of residential heating and environmental tobacco smoke at average
concentrations of 1-100 ng/m3, with a maximum of 2300 ng/m3.
The intake of individual PAH from food has been estimated to be
0.10-10 µg/day per person. The total daily intake of benzo [a]pyrene
from drinking-water was estimated to be 0.0002 µg/person. Cereals and
cereal products are the main contributors to the intake of PAH from
food because they are a major component of the total diet.
1.5.9 Occupational exposure
Near a coke-oven battery, the levels of benzo [a]pyrene ranged
from < 0.1 to 100-200 µg/m3, with a maximum of about 400 µg/m3. In
modern coal gasification systems, the concentration of PAH is usually
< 1 µg/m3 with a maximum of 30 µg/m3. Personal samples taken from
operators of petroleum refinery equipment showed exposure to 2.6-470
µg/m3. In samples of air taken near bitumen processing plants at
refineries, the total PAH levels were 0.004-50 µg/m3. Near road
paving operations, the total PAH concentrations in personal air
samples were up to 190 µg/m3, and the mean value in area air samples
was 0.13 µg/m3. The PAH levels in personal air samples taken at an
aluminium smelter were 0.05-9.6 µg/m3, but urine samples of workers
at an aluminium plant contained very low levels. Area air samples
contained PAH concentrations of up to 5 µg/m3 in one German foundry,
3-40 µg/m3 at iron mines and 4-530 µg/m3 at copper mines. The
concentrations of PAH in cooking fumes in a food factory ranged from
0.07 to 26 µg/m3.
1.6 Kinetics and metabolism
PAH are absorbed through the pulmonary tract, the
gastrointestinal tract, and the skin. The rate of absorption from the
lungs depends on the type of PAH, the size of the particles on which
they are absorbed, and the composition of the adsorbent. PAH adsorbed
onto particulate matter are cleared from the lungs more slowly than
free hydrocarbons. Absorption from the gastrointestinal tract occurs
rapidly in rodents, but metabolites return to the intestine via
biliary excretion. Studies with 32P-postlabelling of percutaneous
absorption of mixtures of PAH in rodents showed that components of the
mixtures reach the lungs, where they become bound to DNA. The rate of
percutaneous absorption in mice according to the compound.
PAH are widely distributed throughout the organism after
administration by any route and are found in almost all internal
organs, but particularly those rich in lipids. Intravenously injected
PAH are cleared rapidly from the bloodstream of rodents but can cross
the placental barrier and have been detected in fetal tissues.
The metabolism of PAH is complex. In general, parent compounds
are converted via intermediate epoxides to phenols, diols, and
tetrols, which can themselves be conjugated with sulfuric or
glucuronic acids or with glutathione. Most metabolism results in
detoxification, but some PAH are activated to DNA-binding species,
principally diol epoxides, which can initiate tumours.
PAH metabolites and their conjugates are excreted via the urine
and faeces, but conjugates excreted in the bile can be hydrolysed by
enzymes of the gut flora and reabsorbed. It can be inferred from the
available information on the total human body burden that PAH do not
persist in the body and that turnover is rapid. This inference
excludes those PAH moieties that become covalently bound to tissue
constituents, in particular nucleic acids, and are not removed by
repair.
1.7 Effects on laboratory mammals and in vitro
The acute toxicity of PAH appears to be moderate to low. The
well-characterized PAH, naphthalene, showed oral and intravenous LD50
values of 100-500 mg/kg body weight (bw) in mice and a mean oral LD50
of 2700 mg/kg bw in rats. The values for other PAH are similar. Single
high doses of naphthalene induced bronchiolar necrosis in mice, rats,
and hamsters.
Short-term studies showed adverse haematological effects,
expressed as myelotoxicity with benzo [a]pyrene, haemolymphatic
changes with dibenz [a,h]-anthracene, and anaemia with naphthalene;
however, in a seven-day study by oral and intraperitoneal
administration in mice, tolerance to the effect of naphthalene was
observed.
Systemic effects caused by long-term treatment with PAH have been
described only rarely, because the end-point of most studies has been
carcinogenicity. Significant toxic effects are manifested at doses at
which carcinogenic responses are also triggered.
In studies of adverse effects on the skin after dermal
application, non- or weakly carcinogenic PAH such as perylene,
benzo [e]pyrene, phenanthrene, pyrene, anthracene, acenaphthalene,
fluorene, and fluoranthene were inactive, whereas carcinogenic
compounds such as benz [a]anthracene, dibenz [a,h]-anthracene, and
benzo [a]pyrene caused hyperkeratosis. Anthracene and naphthalene
vapours caused mild eye irritation. Benzo [a]pyrene induced contact
hypersensitivity in guinea-pigs and mice.
Benz [a]anthracene, benzo [a]pyrene, dibenz [a,h]anthracene,
and naphthalene were embrotoxic to mice and rats. Benzo [a]pyrene
also had teratogenic and reproductive effects. Intensive efforts have
been made to elucidate the genetic basis of the embryotoxic effect of
benzo [a]pyrene. Fetal death and malformations are observed only if
the cytochrome P450 monooxy-genase system is inducible, either in the
mother (with placental permigration) or in the embryo. Not all of the
effects observed can be explained by genetic predisposition, however:
in mice and rabbits, benzo [a]pyrene had transplacental carcinogenic
activity, resulting in pulmonary adenomas and skin papillomas in the
progeny. Reduced fertility and oocyte destruction were also observed.
PAH have also been studied extensively in assays for genotoxicity
and cell transformation; most of the 33 PAH covered in this monograph
are genotoxic or probably genotoxic. The only compounds for which
negative results were found in all assays were anthracene, fluorene,
and naphthalene. Owing to inconsistent results, phenanthrene and
pyrene could not be reliably classified for genotoxicity.
Comprehensive work on the carcinogenicity of PAH shows that 17 of
the 33 studied are, or are suspected of being, carcinogenic (Table 2).
The best-characterized PAH is benzo [a]pyrene, which has been studied
by all current methods in seven species. PAH that have been the
subject of 12 or more studies are anthanthrene, anthracene,
benz [a]anthracene, chrysene, dibenz [a,h]-anthracene,
dibenzo [a,i]pyrene, 5-methylchrysene, phenanthrene, and pyrene.
Special studies of the phototoxicity, immunotoxicity, and
hepatotoxicity of PAH are supplemented by reports on the ocular
toxicity of naphthalene. Anthracene, benzo [a]pyrene, and some other
PAH were phototoxic to mammalian skin and in cell cultures in vitro
when applied with ultraviolet radiation. PAH have generally been
reported to have immunosuppressive effects. After intraperitoneal
treatment of mice with benzo [a]pyrene, immunological parameters were
strongly suppressed in the progeny for up to 18 months. Increased
liver regeneration and an increase in liver weight have also been
observed. The effect of naphthalene in inducing formation of cataracts
in the rodent eye has been attributed to the inducibility of the
cytochrome P450 system in studies in which genetically different mouse
strains were used.
Theoretical models to predict the carcinogenic potency of PAH
from their structures, based on a large amount of experimental work,
were presented as early as the 1930s. The first model was based on the
high chemical reactivity of certain double bonds (the K-region
theory). A later systematic approach was based on the chemical
synthesis of possible metabolites and their mutagenic activity. This
'bay region' theory proposes that epoxides adjacent to a bay region
yield highly stabilized carbonium ions. Other theoretical approaches
are the 'di-region theory' and the 'radical cation potential theory'.
Many individual PAH are carcinogenic to animals and may be
carcinogenic to humans, and exposure to several PAH-containing
mixtures has been shown to increase the incidence of cancer in human
populations. There is concern that those PAH found to be carcinogenic
in experimental animals are likely to be carcinogenic in humans. PAH
produce tumours both at the site of contact and at distant sites. The
carcinogenic potency of PAH may vary with the route of exposure.
Various approaches to assessing the risk associated with exposure to
PAH, singly and in mixtures, have been proposed. No one approach is
endorsed in this monograph; however, the data requirements,
assumptions, applicability, and other features of three quantitative
risk assessment processes that have been validated to some degree are
described.
1.8 Effects on humans
Because of the complex profile of PAH in the environment and in
workplaces, human exposure to pure, individual PAH has been limited to
scientific experiments with volunteers, except in the case of
naphthalene which is used as a moth-repellant for clothing.
After dermal application, anthracene, fluoranthene, and
phenanthrene induced specific skin reactions, and benzo [a]pyrene
induced reversible, regressive verrucae which were classified as
neoplastic proliferations. The systemic effects of naphthalene are
known from numerous cases of accidental intake, particularly by
children. The lethal oral dose is 5000-15 000 mg for adults and 2000
mg taken over two days for a child. The typical effect after dermal or
oral exposure is acute haemolytic anaemia, which can also affect
fetuses transplacentally.
Table 2. Summary of results of tests for genotoxicity and
carcinogenicity for the 33 polycyclic aromatic hydrocarbons studies
Compound Genotoxicity Carcinogenicity
Acenaphthene (?) (?)
Acenaphthylene (?) No studies
Anthanthrene (+) +
Anthracene - -
Benz[a]anthracene + +
Benzo[b]fluoranthene + +
Benzo[j]fluoranthene + +
Benzo[ghi]fluoranthene (+) (-)
Benzo[k]fluoranthene + +
Benzo[a]fluorene (?) (?)
Benzo[b]fluorene (?) (?)
Benzo[ghi]perylene + -
Benzo[c]phenanthrene (+) +
Benzo[a]pyrene + +
Benzo[e]pyrene + ?
Chrysene + +
Coronene (+) (?)
Cyclopenta[cd]pyrene + +
Dibenz[a,h]anthracene + +
Dibenzo[a,e]pyrene + +
Dibenzo[a,h]pyrene (+) +
Dibenzo[a,i]pyrene + +
Dibenzo[a,l]pyrene (+) +
Fluoranthene + (+)
Fluorene - -
Indeno[1,2,3-cd]pyrene + +
5-Methylchrysene + +
1-Methylphenanthrene + (-)
Naphthalene - (?)
Perylene + (-)
Phenanthrene (?) (?)
Pyrene (?) (?)
Triphenylene + (-)
+, positive; -, negative; ?, questionable
Parentheses, result derived from small database
Tobacco smoking is the most important single factor in the
induction of lung tumours and also for increased incidences of tumours
of the urinary bladder, renal pelvis, mouth, pharynx, larynx, and
oesophagus. The contribution of PAH in the diet to the development of
human cancer is not considered to be high. In highly industrialized
areas, increased body burdens of PAH due to polluted ambient air were
detected. Psoriasis patients treated with coal-tar are also exposed to
PAH.
Occupational exposure to soot as a cause of scrotal cancer was
noted for the first time in 1775. Later, occupational exposure to tars
and paraffins was reported to induce skin cancer. The lung is now the
main site of PAH-induced cancer, whereas skin tumours have become more
rare because of better personal hygiene.
Epidemiological studies have been conducted of workers exposed at
coke ovens during coal coking and coal gasification, at asphalt works,
foundries, and aluminium smelters, and to diesel exhaust. Increased
lung tumour rates due to exposure to PAH have been found in coke-oven
workers, asphalt workers, and workers in Söderberg potrooms of
aluminium reduction plants. The highest risk was found for coke-oven
workers, with a standardized mortality ratio of 195. Dose-response
relationships were found in several studies. In aluminium plants, not
only urinary bladder cancer but also asthma-like symptoms, lung
function abnormalities, and chronic bronchitis have been observed.
Coke-oven workers were found to have decreased serum immunoglobulin
levels and decreased immune function. Occupational exposure to
naphthalene for five years was reported to have caused cataract.
Several methods have been developed to assess internal exposure
to PAH. In most of the studies, PAH metabolites such as urinary
thioethers, 1-naphthol, b-naphthylamine, hydroxyphenanthrenes, and
1-hydroxypyrene were measured in urine. The latter has been used
widely as a biological index of exposure.
The genotoxic effects of PAH have been determined by testing for
mutagenicity in urine and faeces and for the presence of micronuclei,
chromosomal aberrations, and sister chromatid exchange in peripheral
blood lymphocytes. In addition, adducts of benzo [a]pyrene with DNA
in peripheral lymphocytes and other tissues and with proteins like
albumin as well as antibodies to DNA adducts have been measured.
1-Hydroxypyrene in urine and DNA adducts in lymphocytes have been
investigated as markers in several studies. 1-Hydroxpyrene can be
measured more easily than DNA adducts, there is less variation between
individuals, and lower levels of exposure can be detected. Both
markers have been used to assess human exposure in various
environments. Increased 1-hydroxpyrene excretion or DNA adducts were
found at various workplaces in coke plants, aluminum manufacturing,
wood impregnation plants, foundries, and asphalt works. The highest
exposures were those of coke-oven workers and workers impregnating
wood with creosote, who took up 95% of total of PAH through the skin,
in contrast to the general population in whom uptake via food and
tobacco smoking predominate.
Estimates of the risk associated with exposure to PAH and PAH
mixtures are based on estimates of exposure and the results of
epidemiological studies. Data for coke-oven workers resulted in a
relative risk for lung cancer of 15.7. On this basis, the risk of the
general population for developing lung cancer over a lifetime has been
calculated to be 10-4 to 10-5 per ng of benzo [a]pyrene per m3 air.
In other words, about one person in 10 000 or 100 000 would be
expected to develop lung cancer in his or her lifetime as a result of
exposure to benzo [a]pyrene in air.
1.9 Effects on other organisms in the laboratory and the field
PAH are acutely toxic to fish and Daphnia magna in combination
with absorption of ultraviolet radiation and visible light. Metabolism
and degradation alter the toxicity of PAH. At low concentrations, PAH
can stimulate the growth of microorganisms and algae. The most toxic
PAH for algae are benz [a]anthracene (four-ring), the concentration
at which given life parameters are reduced by 50% (EC50) being 1-29
µg/litre, and benzo [a]pyrene (five-ring), with an EC50 of 5-15
µg/litre. The EC50 values for algae for most three-ring PAH are
240-940 µg/litre. Naphthalene (two-ring) is the least toxic, with
EC50 values of 2800-34 000 µg/litre.
No clear difference in sensitivity was found between different
taxonomic groups of invertebrates like crustaceans, insects, molluscs,
polychaetes, and echinoderms. Naphthalene is the least toxic, with
96-h LC50 values of 100-2300 µg/litre. The 96-h LC50 values for
three-ring PAH range between < 1 and 3000 µg/litre. Anthracene may be
more toxic than the other three-ring PAH, with 24-h LC50 values
between < 1 and 260 µg/litre. The 96-h LC50 values for four-, five-,
and six-ring PAH are 0.2-1200 µg/litre. Acute toxicity (LC50) in fish
was seen at concentrations of 110 to > 10 000 µg/litre of
naphthalene, 30-4000 µg/litre of three-ring PAH (anthracene, 2.8-360
µg/litre), and 0.7-26 µg/litre for four- or five-ring PAH.
Contamination of sediments with PAH at concentrations of 250
mg/kg was associated with hepatic tumours in free-living fish. Tumours
have also been induced in fish exposed in the laboratory. Exposure of
fish to certain PAH can also cause physiological changes and affect
their growth, reproduction, swimming performance, and respiration.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Identity
The name 'polycyclic aromatic hydrocarbons' (PAH) commonly refers
to a large class of organic compounds containing two or more fused
aromatic rings, even though in a broad sense non-fused ring systems
should be included. In particular, the term 'PAH' refers to compounds
containing only carbon and hydrogen atoms (i.e. unsubstituted parent
PAH and their alkyl-substituted derivatives), whereas the more general
term 'polycyclic aromatic compounds' also includes the functional
derivatives (e.g. nitro- and hydroxy-PAH) and the heterocyclic
analogues, which contain one or more hetero atoms in the aromatic
structure (aza-, oxa-, and thia-arenes). Some authors refer to
polycyclic aromatic compounds as 'polycyclic organic matter', and the
term 'polynuclear' is frequently used for 'polycyclic', as in
'polynuclear aromatic compounds'.
More than 100 PAH have been identified in atmospheric particulate
matter (Lao et al., 1973; Lee et al., 1976a) and in emissions from
coal-fired residential furnaces (Grimmer et al., 1985), and about 200
have been found in tobacco smoke (Lee et al., 1976b, 1981).
The selection of PAH evaluated in this monograph is discussed in
Section 1. The nomenclature, common names, synonyms, and abbreviations
used are given in Table 1 in that section. The structural formulae are
shown in Figure 1. Molecular formulae, relative molecular masses, and
CAS Registry numbers are given in Table 3.
2.1.1 Technical products
Technical-grade naphthalene, also known as naphthalin and tar
camphor, has a minimum purity of 95%. The impurities reported are
benzo [b]thiophene (thianaphthene) when naphthalene is obtained from
coal-tar and methylindenes when it is derived from petroleum (Society
of German Chemists, 1989).
Commercially available anthracene, also known by the trade name
Tetra Olive N2G (IARC, 1983), has a purity of 90-95% (Hawley, 1987).
The impurities reported are phenanthrene, chrysene, carbazole (Hawley,
1987), tetracene, naphthacene (Budavari et al., 1989), and pyridine at
a maximum of 0.2% (IARC, 1983). The following purities were reported
for other technical-grade products: acenaphthene, 95-99%;
fluoranthene, > 95% (Griesbaum et al., 1989); fluorene, about 95%;
phenanthrene, 90%; and pyrene, about 95% (Franck & Stadelhofer, 1987).
The other compounds are generally produced as chemical
intermediates and for research purposes (see also sections 3.2.2 and
3.2.3). Reference materials certified to be of geater than 99% purity
are available for 22 of the PAH considered (Community Bureau of
Reference, 1992); the remaining compounds are commercially available
as chemical standards, with a purity of 99% or more.
Table 3. Identity of polycyclic aromatic hydrocarbons covered in this
volume ranked according to molecular mass
Compound Molecular Relative CAS
formula molecular Registry
mass No.
Naphthalene C10H8 128.2 91-20-3
Acenaphthylene C12H8 152.2 208-96-8
Acenaphthene C12H10 154.2 83-32-9
Fluorene C13H10 166.2 86-73-7
Anthracene C14H10 178.2 120-12-7
Phenanthrene C14H10 178.2 85-01-8
1-Methylphenanthrene C15H12 192.3 832-69-9
Fluoranthene C16H10 202.3 206-44-0
Pyrene C16H10 202.3 129-00-0
Benzo[a]fluorene C17H12 216.3 238-84-6
Benzo[b]fluorene C17H12 216.3 243-17-4
Benzo[ghi]fluoranthene C18H10 226.3 203-12-3
Cyclopenta[cd]pyrene C18H10 226.3 2720837-3
Benz[a]anthracene C18H12 228.3 56-55-3
Benzo[c]phenanthrene C18H12 228.3 195-19-7
Chrysene C18H12 228.3 218-01-9
Triiphenylene C18H12 228.3 217-59-4
5-Methylchrysene C19H14 242.3 3697-24-3
Benzo[b]fluoranthene C20H12 252.3 205-99-2
Benzo[j]fluoranthene C20H12 252.3 205-82-3
Benzo[k]fluoranthene C20H12 252.3 207-08-9
Benzo[a]pyrene C20H12 252.3 50-32-8
Benzo[e]pyrene C20H12 252.3 192-97-2
Perylene C20H12 252.3 198-55-0
Anthanthrene C22H12 276.3 191-26-4
Benzo[ghi]perylene C22H12 276.3 191-24-2
Indeno[1,2,3-cd]pyrene C22H12 276.3 193-39-5
Dibenz[a,h]anthracene C22H14 278.4 53-70-3
Coronene C24H14 300.4 191-07-1
Dibenzo[a,e]pyrene C24H14 302.4 192-65-4
Dibenzo[a,h]pyrene C24H14 302.4 189-64-0
Dibenzo[a,i]pyrene C24H14 302.4 189-55-9
Dibenzo[a,l]pyrene C24H14 302.4 191-30-0
PAH considered (Community Bureau of Reference, 1992); the remaining
compounds are commercially available as chemical standards, with a purity of
99% or more.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED
NON-HETEROCYCLIC POLYCYCLIC AROMATIC HYDROCARBONS
Hanover, Germany, 25-29 September 1995
Members
Dr P.E.T. Douben, Her Majesty's Inspectorate of Pollution, London,
United Kingdom (Chairman)
Dr P.L. Grover, Institute for Cancer Research, Sutton, United Kingdom
Dr R.F. Hertel, Bundesgesinstitut für gesundheitlichen
Verbraucherschutz und Veterinarmedizin, Berlin, Germany
Professor J. Jacob, Biochemisches Institut für Umweltcarcinogene,
Grosshausdorf, Germany
Dr J Kielhorn, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr R.W. Luebke, National Health and Ecology Effects Laboratory, US
Environmental Protection Agency, Research Triangle Park, NC, USA
(Joint Rapporteur)
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood,
Huntingdon, Cambridgeshire, United Kingdom (Joint Rapporteur)
Dr I. Mangelsdorf, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr E. Menichini, Istituto Superiore di Sanita, Rome, Italy
Dr P. Muller, Ministry of Environment and Energy, Toronto, Ontario,
Canada
Dr J.L. Mumford, National Health and Environmental Effects Research
Laboratory, US Environmental Protection Agency, Research Triangle
Park, NC, USA
Dr G. Rosner, Freiburg, Germany
Dr R. Schoeny, National Center for Environmental Assessment, US
Environmental Protection Agency, Cincinnati, OH, USA
Dr T. Sorahan, Institute of Occupational Health, University of
Birmingham, Birmingham, United Kingdom
Dr Kimber L. White, Jr, Medical College of Virginia, Virginia
Commonwealth University, Richmond, VA, USA (Vice-Chairman)
Secretariat
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
Dr. M. Castegnaro, International Agency for Research on Cancer, Lyon,
France
Assisting the Secretariat
Dr S. Artelt, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr A. Boehncke, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr O. Creutzenburg, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
1. SUMMARY
1.1 Selection of compounds for this monograph
Polycyclic aromatic hydrocarbons (PAH) constitute a large class
of compounds, and hundreds of individual substances may be released
during incomplete combustion or pyrolysis of organic matter, an
important source of human exposure. Studies of various environmentally
relevant matrices, such as coal combustion effluents, motor vehicle
exhaust, used motor lubricating oil, and tobacco smoke, have shown
that the PAH in these mixtures are mainly responsible for their
carcinogenic potential.
PAH occur almost always in mixtures. Because the composition of
such mixtures is complex and varies with the generating process, all
mixtures containing PAH could not possible be covered in detail in
this monograph. Thus, 33 individual compounds (31 parent PAH and two
alkyl derivatives) were selected for evaluation on the basis of the
availability of relevant data on toxicological end-points and/or
exposure (Table 1). Since epidemiological studies, which are essential
for risk assessment, were available only for mixtures, however,
Sections 8 and 10 present the results of studies of mixtures of PAH,
in contrast to the rest of the monograph.
Numerous papers and reviews have been published on the
occurrence, distribution, and transformation of PAH in the environment
and on their ecotoxicological and toxicological effects. Only
references from the last 10-15 years are cited in this monograph,
unless no other information was available; reviews are cited for older
studies and for further information.
1.2 Identity, physical and chemical properties, and analytical
methods
The term 'polycyclic aromatic hydrocarbons' commonly refers to a
large class of organic compounds containing two or more fused aromatic
rings made up of carbon and hydrogen atoms. At ambient temperatures,
PAH are solids. The general characteristics common to the class are
high melting- and boiling-points, low vapour pressure, and very low
water solubility which tends to decrease with increasing molecular
mass. PAH are soluble in many organic solvents and are highly
lipophilic. They are chemically rather inert. Reactions that are of
interest with respect to their environmental fate and possible sources
of loss during atmospheric sampling are photodecomposition and
reactions with nitrogen oxides, nitric acid, sulfur oxides, sulfuric
acid, ozone, and hydroxyl radicals.
Ambient air is sampled by collecting suspended particulate matter
on glass-fibre, polytetrafluoroethylene, or quartz-fibre filters by
means of high-volume or passive samplers. Vapour-phase PAH, which
might volatilize from filters during sampling, are commonly trapped by
adsorption on polyurethane foam. The sampling step is by far the most
important source of variability in results.
Table 1. Polycyclic aromatic hydrocarbons evaluated in this monograph
Common name CAS name Synonyma CAS Registry No.
Acenaphthylene Acenaphthylene 91-20-3
Acenaphthene Acenaphthylene, 1,2-dihydro- 208-96-8
Anthanthrene Dibenzo[def,mno]chrysene 191-26-4
Anthracene Anthracene 120-12-7
Benz[a]anthracene Benz[a]anthracene 1,2-Benzanthracene, 56-55-3
tetraphene
Benzo[a]fluorene 11 H-Benzo[a]fluorene 1,2-Benzofluorene 238-84-6
Benzo[b]fluorene 11 H-Benzo[b]fluorene 2,3-Benzofluorene 243-17-4
Benzo[b]fluoranthene Benz[e]acephenanthrylene 3,4-Benzofluoranthene 205-99-2
Benzo[ghi]fluoranthene Benzo[ghi]fluoranthene 2,13-Benzofluoranthene 203-12-3
Benzo[j]fluoranthene Benzo[j]fluoranthene 10,11-Benzofluoranthene 205-82-3
Benzo[k]fluoranthene Benzo[k]fluoranthene 11,12-Benzofluoranthene 207-08-9
Benzo[ghi]perylene Benzo[ghi]perylene 1,12-Benzoperylene 191-24-2
Benzo[c]phenanthrene Benzo[c]phenanthrene 3,4-Benzophenanthrene 195-19-7
Benzo[a]pyrene Benzo[a]pyrene 3,4-Benzopyreneb 50-32-8
Benzo[e]pyrene Benzo[e]pyrene 1,2-Benzopyrene 192-97-2
Chrysene Chrysene 1,2-Benzophenanthrene 218-01-9
Coronene Coronene Hexabenzobenzene 191-07-1
Cyclopenta[cd]pyrene Cyclopenta[cd]pyrene Cyclopenteno[cd]pyrene 27208-37-3
Dibenz[a,h]anthracene Dibenz[a,h]anthracene 1,2:5,6-Dibenzanthracene 53-70-3
Dibenzo[a,e]pyrene Naphtho[1,2,3,4-def]chrysene 1,2:4,5-Dibenzopyrene 192-65-4
Dibenzo[a,h]pyrene Dibenzo[b,def]chrysene 3,4:8,9-Dibenzopyrene 189-64-0
Dibenzo[a,i]pyrene Benzo[rst]pentaphene 3,4:9,10-Dibenzopyrene 189-55-9
Dibenzo[a,l]pyrene Dibenzo[def,p]chrysene 1,2:3,4-Dibenzopyrene 191-30-0
Fluoranthene Fluoranthene 206-44-0
Fluorene 9H-Fluorene 86-73-7
Indeno[1,2,3-cd]pyrene Indeno[1,2,3-cd]-pyrene 2,3-o-Phenylenpyrene 193-39-5
5-Methylchrysene Chrysene, 5-methyl- 3697-24-3
1-Methylphenanthrene Phenanthrene, 1-methyl- 832-69-9
Table 1. (continued)
Common name CAS name Synonyma CAS Registry No.
Naphthalene Naphthalene 91-20-3
Perylene Perylene peri-Dinaphthalene 198-55-0
Phenanthrene Phenanthrene 85-01-8
Pyrene Pyrene Benzo[def]phenanthrene 129-00-0
Triphenylene Triphenylene 9,10-Benzophenanthrene 217-59-4
Extensive lists of synonyms have been imported by the IARC (1983) and Loening & Merritt (1990).
a Common synonym appearing in the literature
b Also reported as benzo[def]chrysene
Air is sampled at the workplace at low flow rates; particles are
collected on glass-fibre or polytetrafluoroethylene filters and
vapours on Amberlite XAD-2 resin. Devices for sampling stack gases are
composed of a glass-fibre or quartz-fibre filter in front of a cooler
to collect condensable matter and an adsorbent (generally XAD-2)
cartridge. Vehicle exhausts are sampled under laboratory conditions,
with standard driving cycles simulating on-road conditions. Emissions
are collected either undiluted or after dilution with filtered cold
air.
Many extraction and purification techniques have been described.
Depending on the matrix, PAH are extracted from samples with a Soxhlet
apparatus, ultrasonically, by liquid-liquid partition, or, after
sample dissolution or alkaline digestion, with a selective solvent.
Supercritical fluid extraction from various environmental solids has
also been used. The efficiency of extraction depends heavily on the
solvent used, and many of the solvents commonly used in the past were
not appropriate. Extracted samples are usually purified by column
chromatography, particularly on alumina, silica gel, or Sephadex LH-20
but also by thin-layer chromatography.
Identification and quantification are routinely performed by gas
chromatography with flame ionization detection or by high-performance
liquid chromatography (HPLC) with ultraviolet and fluorescence
detection, generally in series. In gas chromatography, fused silica
capillary columns are used, with polysiloxanes (SE-54 and SE-52) as
stationary phases; silica-C18 columns are commonly used in HPLC. A
mass spectrometric detector is often coupled to a gas chromatograph in
order to confirm the identity of peaks.
The choice of PAH to be determined depends on the purpose of the
measurement, e.g. for health-orientated or ecotoxicological studies or
to investigate sources. Testing for different sets of compounds may be
required or recommended at national and international levels.
1.3 Sources of human and environmental exposure
Little information is available on the production and processing
of PAH, but it is probable that only small amounts of PAH are released
as a direct result of these activities. The PAH found principally are
used as intermediates in the production of polyvinylchloride and
plasticizers (naphthalene), pigments (acenaphthene, pyrene), dyes
(anthracene, fluoranthene), and pesticides (phenanthrene).
The largest emissions of PAH result from incomplete combustion of
organic materials during industrial processes and other human
activities, including:
- processing of coal, crude oil, and natural gas, including coal
coking, coal conversion, petroleum refining, and production of
carbon blacks, creosote, coal-tar, and bitumen;
- aluminium, iron and steel production in plants and foundries;
- heating in power plants and residences and cooking;
- combustion of refuse;
- motor vehicle traffic; and
- environmental tobacco smoke.
PAH, especially these of higher molecular mass, entering the
environment via the atmosphere are adsorbed onto particulate matter.
The hydrosphere and geosphere are affected secondarily by wet and dry
deposition. Creosote-preserved wood is another source of release of
PAH into the hydrosphere, and deposition of contaminated refuse, like
sewage sludge and fly ash, contributes to emissions of PAH into the
geosphere. Little information is available about the passage of PAH
into the biosphere. PAH occur naturally in peat, lignite, coal, and
crude oil. Most of the PAH in hard coals are tightly bound within the
coal structure and cannot be leached out.
The release of PAH into the environment has been determined by
identification of a characteristic PAH concentration profile, but this
has been possible in only a few cases. Benzo [a]pyrene has frequently
been used as an indicator of PAH, especially in older studies.
Generally, emissions of PAH are only estimates based on more or less
reliable data and give only a rough idea of exposure.
The most important sources of PAH are as follows:
Coal coking: Airborne emissions of PAH from coal coking in
Germany have decreased significantly over the last 10 years as a
result of technical improvements to existing plants, closure of old
plants, and reduced coke production. Similar situations are assumed to
exist in western Europe, Japan, and the USA, but no data were
available.
Production of aluminium (mainly special coal anodes), iron,
and steel and the binding agents used in moulding sand in foundries:
Little information is available.
Domestic and residential heating: Phenanthrene, fluoranthene,
pyrene, and chrysene are emitted as major components. The emissions
from wood stoves are 25-1000 times higher than those from
charcoal-fired stoves, and in areas where wood burning predominates
for domestic heating the major portion of airborne PAH may be derived
from this source, especially in winter. The release of PAH during
residential heating is thus assumed to be an important source in
developing countries where biomass is often burnt in relatively simple
stoves.
Cooking: PAH may be emitted during incomplete combustion of
fuels, from cooking oil, and from food being cooked.
Motor vehicle traffic: The main compounds released from
petrol-fuelled vehicles are fluoranthene and pyrene, while naphthalene
and acenaphthene are abundant in the exhaust of diesel-fuelled
vehicles. Although cyclopenta [cd]-pyrene is emitted at a high rate
from petrol-fuelled engines, its concentration in diesel exhaust is
only just above the limit of detection. The emission rates, which
depend on the substance, the type of vehicle, its engine conditions,
and the test conditions, range from a few nanograms per kilometre to
> 1000 mg/km. PAH emissions from vehicle engines are dramatically
reduced by fitting catalytic converter devices.
Forest fires: In countries with large forest areas, fires can
make an imprtant contribution to PAH emissions.
Coal-fired power plants: PAH released into the atmosphere from
such plants consist mainly of two- and three-ring compounds. In
contaminated areas, the PAH levels in ambient air may be higher than
those in the stack gases.
Incineration of refuse: The PAH emissions in stack gases from
this souce in a number of countries were < 10 mg/m3.
1.4 Environmental transport, distribution, and transformation
Several distribution and transformation processes determine the
fate of both individual PAH and mixtures. Partitioning between water
and air, between water and sediment, and between water and biota are
the most important of the distribution processes.
As PAH are hydrophobic with low solubility in water, their
affinity for the aquatic phase is very low; however, in spite of the
fact that most PAH are released into the environment via the
atmosphere, considerable concentrations are also found in the
hydrosphere because of their low Henry's law constants. As the
affinity of PAH for organic phases is greater than that for water,
their partition coefficients between organic solvents, such as
octanol, and water are high. Their affinity for organic fractions in
sediment, soil, and biota is also high, and PAH thus accumulate in
organisms in water and sediments and in their food. The relative
importance of uptake from food and from water is not clear. In
Daphnia and molluscs, accumulation of PAH from water is positively
correlated with the octanol:water partition coefficient ( Kow). In
fish and algae that can metabolize PAH, however, the internal
concentrations of different PAH are not correlated with the Kow.
Biomagnification - the increase in the concentration of a
substance in animals in successive trophic levels of food chains - of
PAH has not been observed in aquatic systems and would not be expected
to occur, because most organisms have a high biotransformation
potential for PAH. Organisms at higher trophic levels in food chains
show the highest potential biotransformation.
PAH are degraded by photodegradation, biodegradation by
microorganisms, and metabolism in higher biota. Although the last
route of transformation is of minor importance for the overall fate of
PAH in the environment, it is an important pathway for the biota,
since carcinogenic metabolites may be formed. As PAH are chemically
stable, with no reactive groups, hydrolysis plays no role in their
degradation. Few standard tests for the biodegradation of PAH are
available In general, they are biodegraded under aerobic conditions,
the biodegradation rate decreasing drastically with the number of
aromatic rings. Under anaerobic conditions, degradation is much
slower.
PAH are photooxidized in air and water in the presence of
sensitizing radicals like OH, NO3, and O3. Under laboratory
conditions, the half-life of the reaction with airborne OH radicals is
about one day, whereas reactions with NO3 and O3 usually have much
lower velocity constants. The adsorption of high-molecular-mass PAH
onto carbonaceous particles in the environment should stabilize the
reaction with OH radicals. The reaction of two- to four-ring PAH,
which occur mainly in the vapour phase, with NO3 leads to nitro-PAH,
which are known mutagens. The photooxidation of some PAH in water
seems to be more rapid than in air. Calculations based on
physicochemical and degradation parameters indicate that PAH with four
or more aromatic rings persist in the environment.
1.5 Environmental levels and human exposure
PAH are ubiquitous in the environment, and various individual PAH
have been detected in different compartments in numerous studies.
1.5.1 Air
The levels of individual PAH tend to be higher in winter than in
summer by at least one order of magnitude. The predominant source
during winter is residential heating, while that during summer is
urban motor vehicle traffic. Average concentrations of 1-30 ng/m3 of
individual PAH were detected in the ambient air of various urban
areas. In large cities with heavy motor vehicle traffic and extensive
use of biomass fuel, such as Calcutta, levels of up to 200 ng/m3 of
individual PAH were found. Concentrations of 1-50 ng/m3 were detected
in road tunnels. Cyclopenta [cd]pyrene and pyrene were present at
concentrations up to 100 ng/m3. In a subway station, PAH
concentrations of up to 20 ng/m3 were measured. Near industrial
sources, the average concentrations of individual PAH ranged from 1 to
10 ng/m3. Phenanthrene was present at up to a maximum of about 310
ng/m3.
The background values of PAH are at least one or two orders of
magnitude lower than those near sources like motor vehicle traffic.
For example, the levels at 1100 m ranged from 0.004 to 0.03 ng/m3.
1.5.2 Surface water and precipitation
Most of the PAH in water are believed to result from urban
runoff, from atmospheric fallout (smaller particles), and from asphalt
abrasion (larger particles). The major source of PAH varies, however,
in a given body of water. In general, most samples of surface water
contain individual PAH at levels of up to 50 ng/litre, but highly
polluted rivers had concentrations of up to 6000 ng/litre. The PAH
levels in groundwater are within the range 0.02-1.8 ng/litre, and
drinking-water samples contain concentrations of the same order of
magnitude. Major sources of PAH in drinking-water are asphalt-lined
storage tanks and delivery pipes.
The levels of individual PAH in rainwater ranged from 10 to 200
ng/litre, whereas levels of up to 1000 ng/litre have been detected in
snow and fog.
1.5.3 Sediment
The concentrations of individual PAH in sediment were generally
one order of magnitude higher than those in precipitation.
1.5.4 Soil
The main sources of PAH in soil are atmospheric deposition,
carbonization of plant material, and deposition from sewage and
particulate waste. The extent of pollution of soil depends on factors
such as its cultivation, its porosity, and its content of humic
substances.
Near industrial sources, individual PAH levels of up to 1 g/kg
soil have been found. The concentrations in soil from other sources,
such as automobile exhaust, are in the range 2-5 mg/kg. In unpolluted
areas, the PAH levels were 5-100 µg/kg soil.
1.5.5 Food
Raw food does not normally contain high levels of PAH, but they
are formed by processing, roasting, baking, or frying. Vegetables may
be contaminated by the deposition of airborne particles or by growth
in contaminated soil. The levels of individual PAH in meat, fish,
dairy products, vegetables and fruits, cereals and their products,
sweets, beverages, and animal and vegetable fats and oils were within
the range 0.01-10 µg/kg. Concentrations of over 100 µg/kg have been
detected in smoked meat and up to 86 µg/kg in smoked fish; smoked
cereals contained up to 160 µg/kg. Coconut oil contained up to 460
µg/kg. The levels in human breast milk were 0.003-0.03 µg/kg.
1.5.6 Aquatic organisms
Marine organisms are known to adsorb and accumulate PAH from
water. The degree of contamination is related to the extent of
industrial and urban development and shipping movements. PAH
concentrations of up to 7 mg/kg have been detected in aquatic
organisms living near industrial effluents, and the average levels of
PAH in aquatic animals sampled at contaminated sites were 10-500
µg/kg, although levels of up to 5 mg/kg were also detected.
The average levels of PAH in aquatic animals sampled at various
sites with unspecified sources of PAH were 1-100 µg/kg, but
concentrations of up to 1 mg/kg were found, for example, in lobsters
in Canada.
1.5.7 Terrestrial organisms
The concentrations of PAH in insects ranged from 730 to 5500
µg/kg. The PAH content of earthworm faeces depends significantly on
the location: those in a highly industrialized region in eastern
Germany contained benzo [a]pyrene at concentrations up to 2 mg/kg.
1.5.8 General population
The main sources of nonoccupational exposure are: polluted
ambient air, smoke from open fireplaces and cooking, environmental
tobacco smoke, contaminated food and drinking-water, and the use of
PAH-contaminated products. PAH can be found in indoor air as a result
of residential heating and environmental tobacco smoke at average
concentrations of 1-100 ng/m3, with a maximum of 2300 ng/m3.
The intake of individual PAH from food has been estimated to be
0.10-10 µg/day per person. The total daily intake of benzo [a]pyrene
from drinking-water was estimated to be 0.0002 µg/person. Cereals and
cereal products are the main contributors to the intake of PAH from
food because they are a major component of the total diet.
1.5.9 Occupational exposure
Near a coke-oven battery, the levels of benzo [a]pyrene ranged
from < 0.1 to 100-200 µg/m3, with a maximum of about 400 µg/m3. In
modern coal gasification systems, the concentration of PAH is usually
< 1 µg/m3 with a maximum of 30 µg/m3. Personal samples taken from
operators of petroleum refinery equipment showed exposure to 2.6-470
µg/m3. In samples of air taken near bitumen processing plants at
refineries, the total PAH levels were 0.004-50 µg/m3. Near road
paving operations, the total PAH concentrations in personal air
samples were up to 190 µg/m3, and the mean value in area air samples
was 0.13 µg/m3. The PAH levels in personal air samples taken at an
aluminium smelter were 0.05-9.6 µg/m3, but urine samples of workers
at an aluminium plant contained very low levels. Area air samples
contained PAH concentrations of up to 5 µg/m3 in one German foundry,
3-40 µg/m3 at iron mines and 4-530 µg/m3 at copper mines. The
concentrations of PAH in cooking fumes in a food factory ranged from
0.07 to 26 µg/m3.
1.6 Kinetics and metabolism
PAH are absorbed through the pulmonary tract, the
gastrointestinal tract, and the skin. The rate of absorption from the
lungs depends on the type of PAH, the size of the particles on which
they are absorbed, and the composition of the adsorbent. PAH adsorbed
onto particulate matter are cleared from the lungs more slowly than
free hydrocarbons. Absorption from the gastrointestinal tract occurs
rapidly in rodents, but metabolites return to the intestine via
biliary excretion. Studies with 32P-postlabelling of percutaneous
absorption of mixtures of PAH in rodents showed that components of the
mixtures reach the lungs, where they become bound to DNA. The rate of
percutaneous absorption in mice according to the compound.
PAH are widely distributed throughout the organism after
administration by any route and are found in almost all internal
organs, but particularly those rich in lipids. Intravenously injected
PAH are cleared rapidly from the bloodstream of rodents but can cross
the placental barrier and have been detected in fetal tissues.
The metabolism of PAH is complex. In general, parent compounds
are converted via intermediate epoxides to phenols, diols, and
tetrols, which can themselves be conjugated with sulfuric or
glucuronic acids or with glutathione. Most metabolism results in
detoxification, but some PAH are activated to DNA-binding species,
principally diol epoxides, which can initiate tumours.
PAH metabolites and their conjugates are excreted via the urine
and faeces, but conjugates excreted in the bile can be hydrolysed by
enzymes of the gut flora and reabsorbed. It can be inferred from the
available information on the total human body burden that PAH do not
persist in the body and that turnover is rapid. This inference
excludes those PAH moieties that become covalently bound to tissue
constituents, in particular nucleic acids, and are not removed by
repair.
1.7 Effects on laboratory mammals and in vitro
The acute toxicity of PAH appears to be moderate to low. The
well-characterized PAH, naphthalene, showed oral and intravenous LD50
values of 100-500 mg/kg body weight (bw) in mice and a mean oral LD50
of 2700 mg/kg bw in rats. The values for other PAH are similar. Single
high doses of naphthalene induced bronchiolar necrosis in mice, rats,
and hamsters.
Short-term studies showed adverse haematological effects,
expressed as myelotoxicity with benzo [a]pyrene, haemolymphatic
changes with dibenz [a,h]-anthracene, and anaemia with naphthalene;
however, in a seven-day study by oral and intraperitoneal
administration in mice, tolerance to the effect of naphthalene was
observed.
Systemic effects caused by long-term treatment with PAH have been
described only rarely, because the end-point of most studies has been
carcinogenicity. Significant toxic effects are manifested at doses at
which carcinogenic responses are also triggered.
In studies of adverse effects on the skin after dermal
application, non- or weakly carcinogenic PAH such as perylene,
benzo [e]pyrene, phenanthrene, pyrene, anthracene, acenaphthalene,
fluorene, and fluoranthene were inactive, whereas carcinogenic
compounds such as benz [a]anthracene, dibenz [a,h]-anthracene, and
benzo [a]pyrene caused hyperkeratosis. Anthracene and naphthalene
vapours caused mild eye irritation. Benzo [a]pyrene induced contact
hypersensitivity in guinea-pigs and mice.
Benz [a]anthracene, benzo [a]pyrene, dibenz [a,h]anthracene,
and naphthalene were embrotoxic to mice and rats. Benzo [a]pyrene
also had teratogenic and reproductive effects. Intensive efforts have
been made to elucidate the genetic basis of the embryotoxic effect of
benzo [a]pyrene. Fetal death and malformations are observed only if
the cytochrome P450 monooxy-genase system is inducible, either in the
mother (with placental permigration) or in the embryo. Not all of the
effects observed can be explained by genetic predisposition, however:
in mice and rabbits, benzo [a]pyrene had transplacental carcinogenic
activity, resulting in pulmonary adenomas and skin papillomas in the
progeny. Reduced fertility and oocyte destruction were also observed.
PAH have also been studied extensively in assays for genotoxicity
and cell transformation; most of the 33 PAH covered in this monograph
are genotoxic or probably genotoxic. The only compounds for which
negative results were found in all assays were anthracene, fluorene,
and naphthalene. Owing to inconsistent results, phenanthrene and
pyrene could not be reliably classified for genotoxicity.
Comprehensive work on the carcinogenicity of PAH shows that 17 of
the 33 studied are, or are suspected of being, carcinogenic (Table 2).
The best-characterized PAH is benzo [a]pyrene, which has been studied
by all current methods in seven species. PAH that have been the
subject of 12 or more studies are anthanthrene, anthracene,
benz [a]anthracene, chrysene, dibenz [a,h]-anthracene,
dibenzo [a,i]pyrene, 5-methylchrysene, phenanthrene, and pyrene.
Special studies of the phototoxicity, immunotoxicity, and
hepatotoxicity of PAH are supplemented by reports on the ocular
toxicity of naphthalene. Anthracene, benzo [a]pyrene, and some other
PAH were phototoxic to mammalian skin and in cell cultures in vitro
when applied with ultraviolet radiation. PAH have generally been
reported to have immunosuppressive effects. After intraperitoneal
treatment of mice with benzo [a]pyrene, immunological parameters were
strongly suppressed in the progeny for up to 18 months. Increased
liver regeneration and an increase in liver weight have also been
observed. The effect of naphthalene in inducing formation of cataracts
in the rodent eye has been attributed to the inducibility of the
cytochrome P450 system in studies in which genetically different mouse
strains were used.
Theoretical models to predict the carcinogenic potency of PAH
from their structures, based on a large amount of experimental work,
were presented as early as the 1930s. The first model was based on the
high chemical reactivity of certain double bonds (the K-region
theory). A later systematic approach was based on the chemical
synthesis of possible metabolites and their mutagenic activity. This
'bay region' theory proposes that epoxides adjacent to a bay region
yield highly stabilized carbonium ions. Other theoretical approaches
are the 'di-region theory' and the 'radical cation potential theory'.
Many individual PAH are carcinogenic to animals and may be
carcinogenic to humans, and exposure to several PAH-containing
mixtures has been shown to increase the incidence of cancer in human
populations. There is concern that those PAH found to be carcinogenic
in experimental animals are likely to be carcinogenic in humans. PAH
produce tumours both at the site of contact and at distant sites. The
carcinogenic potency of PAH may vary with the route of exposure.
Various approaches to assessing the risk associated with exposure to
PAH, singly and in mixtures, have been proposed. No one approach is
endorsed in this monograph; however, the data requirements,
assumptions, applicability, and other features of three quantitative
risk assessment processes that have been validated to some degree are
described.
1.8 Effects on humans
Because of the complex profile of PAH in the environment and in
workplaces, human exposure to pure, individual PAH has been limited to
scientific experiments with volunteers, except in the case of
naphthalene which is used as a moth-repellant for clothing.
After dermal application, anthracene, fluoranthene, and
phenanthrene induced specific skin reactions, and benzo [a]pyrene
induced reversible, regressive verrucae which were classified as
neoplastic proliferations. The systemic effects of naphthalene are
known from numerous cases of accidental intake, particularly by
children. The lethal oral dose is 5000-15 000 mg for adults and 2000
mg taken over two days for a child. The typical effect after dermal or
oral exposure is acute haemolytic anaemia, which can also affect
fetuses transplacentally.
Table 2. Summary of results of tests for genotoxicity and
carcinogenicity for the 33 polycyclic aromatic hydrocarbons studies
Compound Genotoxicity Carcinogenicity
Acenaphthene (?) (?)
Acenaphthylene (?) No studies
Anthanthrene (+) +
Anthracene - -
Benz[a]anthracene + +
Benzo[b]fluoranthene + +
Benzo[j]fluoranthene + +
Benzo[ghi]fluoranthene (+) (-)
Benzo[k]fluoranthene + +
Benzo[a]fluorene (?) (?)
Benzo[b]fluorene (?) (?)
Benzo[ghi]perylene + -
Benzo[c]phenanthrene (+) +
Benzo[a]pyrene + +
Benzo[e]pyrene + ?
Chrysene + +
Coronene (+) (?)
Cyclopenta[cd]pyrene + +
Dibenz[a,h]anthracene + +
Dibenzo[a,e]pyrene + +
Dibenzo[a,h]pyrene (+) +
Dibenzo[a,i]pyrene + +
Dibenzo[a,l]pyrene (+) +
Fluoranthene + (+)
Fluorene - -
Indeno[1,2,3-cd]pyrene + +
5-Methylchrysene + +
1-Methylphenanthrene + (-)
Naphthalene - (?)
Perylene + (-)
Phenanthrene (?) (?)
Pyrene (?) (?)
Triphenylene + (-)
+, positive; -, negative; ?, questionable
Parentheses, result derived from small database
Tobacco smoking is the most important single factor in the
induction of lung tumours and also for increased incidences of tumours
of the urinary bladder, renal pelvis, mouth, pharynx, larynx, and
oesophagus. The contribution of PAH in the diet to the development of
human cancer is not considered to be high. In highly industrialized
areas, increased body burdens of PAH due to polluted ambient air were
detected. Psoriasis patients treated with coal-tar are also exposed to
PAH.
Occupational exposure to soot as a cause of scrotal cancer was
noted for the first time in 1775. Later, occupational exposure to tars
and paraffins was reported to induce skin cancer. The lung is now the
main site of PAH-induced cancer, whereas skin tumours have become more
rare because of better personal hygiene.
Epidemiological studies have been conducted of workers exposed at
coke ovens during coal coking and coal gasification, at asphalt works,
foundries, and aluminium smelters, and to diesel exhaust. Increased
lung tumour rates due to exposure to PAH have been found in coke-oven
workers, asphalt workers, and workers in Söderberg potrooms of
aluminium reduction plants. The highest risk was found for coke-oven
workers, with a standardized mortality ratio of 195. Dose-response
relationships were found in several studies. In aluminium plants, not
only urinary bladder cancer but also asthma-like symptoms, lung
function abnormalities, and chronic bronchitis have been observed.
Coke-oven workers were found to have decreased serum immunoglobulin
levels and decreased immune function. Occupational exposure to
naphthalene for five years was reported to have caused cataract.
Several methods have been developed to assess internal exposure
to PAH. In most of the studies, PAH metabolites such as urinary
thioethers, 1-naphthol, b-naphthylamine, hydroxyphenanthrenes, and
1-hydroxypyrene were measured in urine. The latter has been used
widely as a biological index of exposure.
The genotoxic effects of PAH have been determined by testing for
mutagenicity in urine and faeces and for the presence of micronuclei,
chromosomal aberrations, and sister chromatid exchange in peripheral
blood lymphocytes. In addition, adducts of benzo [a]pyrene with DNA
in peripheral lymphocytes and other tissues and with proteins like
albumin as well as antibodies to DNA adducts have been measured.
1-Hydroxypyrene in urine and DNA adducts in lymphocytes have been
investigated as markers in several studies. 1-Hydroxpyrene can be
measured more easily than DNA adducts, there is less variation between
individuals, and lower levels of exposure can be detected. Both
markers have been used to assess human exposure in various
environments. Increased 1-hydroxpyrene excretion or DNA adducts were
found at various workplaces in coke plants, aluminum manufacturing,
wood impregnation plants, foundries, and asphalt works. The highest
exposures were those of coke-oven workers and workers impregnating
wood with creosote, who took up 95% of total of PAH through the skin,
in contrast to the general population in whom uptake via food and
tobacco smoking predominate.
Estimates of the risk associated with exposure to PAH and PAH
mixtures are based on estimates of exposure and the results of
epidemiological studies. Data for coke-oven workers resulted in a
relative risk for lung cancer of 15.7. On this basis, the risk of the
general population for developing lung cancer over a lifetime has been
calculated to be 10-4 to 10-5 per ng of benzo [a]pyrene per m3 air.
In other words, about one person in 10 000 or 100 000 would be
expected to develop lung cancer in his or her lifetime as a result of
exposure to benzo [a]pyrene in air.
1.9 Effects on other organisms in the laboratory and the field
PAH are acutely toxic to fish and Daphnia magna in combination
with absorption of ultraviolet radiation and visible light. Metabolism
and degradation alter the toxicity of PAH. At low concentrations, PAH
can stimulate the growth of microorganisms and algae. The most toxic
PAH for algae are benz [a]anthracene (four-ring), the concentration
at which given life parameters are reduced by 50% (EC50) being 1-29
µg/litre, and benzo [a]pyrene (five-ring), with an EC50 of 5-15
µg/litre. The EC50 values for algae for most three-ring PAH are
240-940 µg/litre. Naphthalene (two-ring) is the least toxic, with
EC50 values of 2800-34 000 µg/litre.
No clear difference in sensitivity was found between different
taxonomic groups of invertebrates like crustaceans, insects, molluscs,
polychaetes, and echinoderms. Naphthalene is the least toxic, with
96-h LC50 values of 100-2300 µg/litre. The 96-h LC50 values for
three-ring PAH range between < 1 and 3000 µg/litre. Anthracene may be
more toxic than the other three-ring PAH, with 24-h LC50 values
between < 1 and 260 µg/litre. The 96-h LC50 values for four-, five-,
and six-ring PAH are 0.2-1200 µg/litre. Acute toxicity (LC50) in fish
was seen at concentrations of 110 to > 10 000 µg/litre of
naphthalene, 30-4000 µg/litre of three-ring PAH (anthracene, 2.8-360
µg/litre), and 0.7-26 µg/litre for four- or five-ring PAH.
Contamination of sediments with PAH at concentrations of 250
mg/kg was associated with hepatic tumours in free-living fish. Tumours
have also been induced in fish exposed in the laboratory. Exposure of
fish to certain PAH can also cause physiological changes and affect
their growth, reproduction, swimming performance, and respiration.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Identity
The name 'polycyclic aromatic hydrocarbons' (PAH) commonly refers
to a large class of organic compounds containing two or more fused
aromatic rings, even though in a broad sense non-fused ring systems
should be included. In particular, the term 'PAH' refers to compounds
containing only carbon and hydrogen atoms (i.e. unsubstituted parent
PAH and their alkyl-substituted derivatives), whereas the more general
term 'polycyclic aromatic compounds' also includes the functional
derivatives (e.g. nitro- and hydroxy-PAH) and the heterocyclic
analogues, which contain one or more hetero atoms in the aromatic
structure (aza-, oxa-, and thia-arenes). Some authors refer to
polycyclic aromatic compounds as 'polycyclic organic matter', and the
term 'polynuclear' is frequently used for 'polycyclic', as in
'polynuclear aromatic compounds'.
More than 100 PAH have been identified in atmospheric particulate
matter (Lao et al., 1973; Lee et al., 1976a) and in emissions from
coal-fired residential furnaces (Grimmer et al., 1985), and about 200
have been found in tobacco smoke (Lee et al., 1976b, 1981).
The selection of PAH evaluated in this monograph is discussed in
Section 1. The nomenclature, common names, synonyms, and abbreviations
used are given in Table 1 in that section. The structural formulae are
shown in Figure 1. Molecular formulae, relative molecular masses, and
CAS Registry numbers are given in Table 3.
2.1.1 Technical products
Technical-grade naphthalene, also known as naphthalin and tar
camphor, has a minimum purity of 95%. The impurities reported are
benzo [b]thiophene (thianaphthene) when naphthalene is obtained from
coal-tar and methylindenes when it is derived from petroleum (Society
of German Chemists, 1989).
Commercially available anthracene, also known by the trade name
Tetra Olive N2G (IARC, 1983), has a purity of 90-95% (Hawley, 1987).
The impurities reported are phenanthrene, chrysene, carbazole (Hawley,
1987), tetracene, naphthacene (Budavari et al., 1989), and pyridine at
a maximum of 0.2% (IARC, 1983). The following purities were reported
for other technical-grade products: acenaphthene, 95-99%;
fluoranthene, > 95% (Griesbaum et al., 1989); fluorene, about 95%;
phenanthrene, 90%; and pyrene, about 95% (Franck & Stadelhofer, 1987).
The other compounds are generally produced as chemical
intermediates and for research purposes (see also sections 3.2.2 and
3.2.3). Reference materials certified to be of geater than 99% purity
are available for 22 of the PAH considered (Community Bureau of
Reference, 1992); the remaining compounds are commercially available
as chemical standards, with a purity of 99% or more.
Table 3. Identity of polycyclic aromatic hydrocarbons covered in this
volume ranked according to molecular mass
Compound Molecular Relative CAS
formula molecular Registry
mass No.
Naphthalene C10H8 128.2 91-20-3
Acenaphthylene C12H8 152.2 208-96-8
Acenaphthene C12H10 154.2 83-32-9
Fluorene C13H10 166.2 86-73-7
Anthracene C14H10 178.2 120-12-7
Phenanthrene C14H10 178.2 85-01-8
1-Methylphenanthrene C15H12 192.3 832-69-9
Fluoranthene C16H10 202.3 206-44-0
Pyrene C16H10 202.3 129-00-0
Benzo[a]fluorene C17H12 216.3 238-84-6
Benzo[b]fluorene C17H12 216.3 243-17-4
Benzo[ghi]fluoranthene C18H10 226.3 203-12-3
Cyclopenta[cd]pyrene C18H10 226.3 2720837-3
Benz[a]anthracene C18H12 228.3 56-55-3
Benzo[c]phenanthrene C18H12 228.3 195-19-7
Chrysene C18H12 228.3 218-01-9
Triiphenylene C18H12 228.3 217-59-4
5-Methylchrysene C19H14 242.3 3697-24-3
Benzo[b]fluoranthene C20H12 252.3 205-99-2
Benzo[j]fluoranthene C20H12 252.3 205-82-3
Benzo[k]fluoranthene C20H12 252.3 207-08-9
Benzo[a]pyrene C20H12 252.3 50-32-8
Benzo[e]pyrene C20H12 252.3 192-97-2
Perylene C20H12 252.3 198-55-0
Anthanthrene C22H12 276.3 191-26-4
Benzo[ghi]perylene C22H12 276.3 191-24-2
Indeno[1,2,3-cd]pyrene C22H12 276.3 193-39-5
Dibenz[a,h]anthracene C22H14 278.4 53-70-3
Coronene C24H14 300.4 191-07-1
Dibenzo[a,e]pyrene C24H14 302.4 192-65-4
Dibenzo[a,h]pyrene C24H14 302.4 189-64-0
Dibenzo[a,i]pyrene C24H14 302.4 189-55-9
Dibenzo[a,l]pyrene C24H14 302.4 191-30-0
PAH considered (Community Bureau of Reference, 1992); the remaining
compounds are commercially available as chemical standards, with a purity of
99% or more.
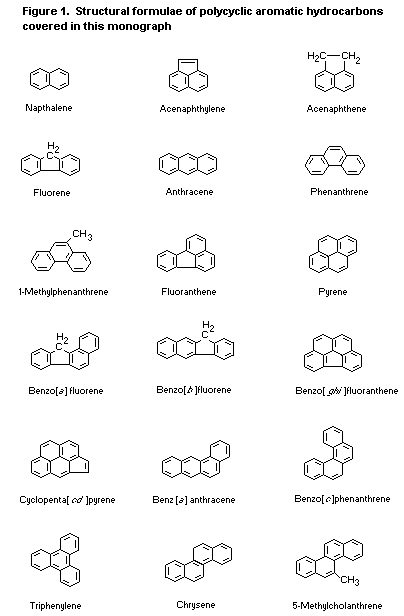
 2.2 Physical and chemical properties
Physical and chemical properties relevant to the toxicological
and ecotoxicological evaluation of the PAH are summarized in Table 4.
It should be kept in mind that the values for any one parameter may be
derived from different sources, with different methods of measurement
or calculation, so that individual values cannot be compared directly
unless the original sources are consulted. In particular, the vapour
pressures reported in the literature for the same PAH vary by up to
several orders of magnitude (Mackay & Shiu, 1981; Lane, 1989).
Variations are also seen in the reported solubility in water of
various PAH, although the values are generally within one order of
magnitude (National Research Council Canada, 1983). Flash-points were
available only for three compounds with high molecular mass (for
naphthalene, 78.9°C by the open-cup method and 87.8°C by the closed-cup
method; anthracene, 121°C by the closed-cup method; and phenanthrene,
171°C by the open-cup method). Explosion limits were available only
for naphthalene (0.9-5.9 vol %) and ananthrene (0.6 vol %) (Lewis,
1992). Vapour density (air = 1) was 4.42 for naphthalene (IARC,
1973), 5.32 for acenaphthene, 6.15 for anthracene (Lewis, 1992), 6.15
for phenanthrene, and 8.7 for benzo[a]pyrene (National Institute for
Occupational Safety and Health and Occupational Safety and Health
Administration, 1981).
The physical and chemical properties are largely determined by
the conjugated alpha-electron systems, which vary fairly regularly
with the number of rings and molecular mass, giving rise to a more or
less wide range of values for each parameter within the whole class.
At room temperature, all PAH are solids. The general characteristics
common to the class are high melting- and boiling-points, low vapour
pressure, and very low solubility in water. PAH are soluble in many
organic solvents (IARC, 1983; Agency for Toxic Substances and Disease
Registry, 1990; Lide, 1991) and are highly lipophilic.
Vapour pressure tends to decrease with increasing molecular mass,
varying by more than 10 orders of magnitude. This characteristic
affects the adsorption of individual PAH onto particulate matter in
the atmosphere and their retention on particulate matter during
sampling on filters (Thrane & Mikalsen, 1981). Vapour pressure
increases markedly with ambient temperature (Murray et al., 1974),
which additionally affects the distribution coefficients between
gaseous and particulate phases (Lane, 1989). Solubility in water tends
to decreases with increasing molecular mass. For additional
information, refer to section 4.1.
PAH are chemically inert compounds (see also section 4.4). When
they react, they undergo two types of reaction: electrophilic
substitution and addition. As the latter destroys the aromatic
character of the benzene ring that is affected, PAH tend to form
derivatives by the former reaction; addition is often followed by
elimination, resulting in net substitution. The chemical and
photochemical reactions of PAH in the atmosphere have been reviewed
Table 4. Physical and chemical properties of polycyclic aromatic compounds covered in this monograph, ranked by molecular mass
Compound Colour Melting- Boiling- Vapour Densityc n-Octanol: Solubility in Henry's law
pointa point pressure water water at 25°C constant at
(°C) (°C) (Pa at 25°C) partition (µg/litre)d 25°C (kPa)
coefficient
(log Kow)
Naphthalene Whiteb 81 217.9c 10.4g 1.15425 h 3.4j 3.17 x 104 4.89 x 10-2 k
Acenaphthylene 92-93 8.9 x 10-1 g 0.89916/2 h 4.07f 114 x 10-3 l
Acenaphthene Whiteb 95 279h 2.9 x 10-1 g 1.02490/4 h 3.92f 3.93 x 103 1.48 x 10-2 k
Fluorene Whitee 115-116 295e 9.0 x 10-2 g 1.2030/4 h 4.18m 1.98 x 103 1.01 x 10-2 n
Anthracene Colourlesso 216.4 342e 8.0 x 10-4 g 1.28325/4 h 4.5j 73 7.3 x 10-2 n
Phenanthrene Colourlessp 100.5 340h 1.6 x 10-2 g 0.9804 h 4.6j 1.29 x 103 3.98 x 10-3 k
1-Methylphenanthrene 123 354-355y 5.07s 255 (24°C)t
Fluoranthene Pale yellowh 108.8 375h 1.2 x 10-3 g 1.2520/4 h 5.22u 260 6.5 x 10-4
(20 °C)w
Pyrene Colourlesse 150.4 393h 6.0 x 10-4 g 1.27123/4 h 5.18j 135 1.1 x 10-3 n
Benzo[a]fluorene Colourlessx 189-190h 399-400y 5.32z 45
Benzo[b]fluorene Colourlessx 213.5 401-402y 1.226aa 5.75z 2.0
Benzo[ghi]fluoranthene Yellowbb 128.4 432cc 1.34523 dd
Cyclopenta[cd]pyrene Orangex 170 439ee
Benz[a]anthracene Colourlessb 160.7 400b 2.8 x 10-5 g 1.226aa 5.61f 14
Benzo[c]phenanthrene Colourlessx 66.1 1.265ff
Chrysene Colourless 253.8 448h 8.4 x 10-5 1.27420/4 e 5.91u 2.0
with blue (20°C)gg
fluoresenceb
Triphenylene Colourlessx 199 425bb 1.3p 5.45hh 43
5-Methylchrysene Colourlessx 117.1 458ii 62 (27°C)jj
Benzo[b]fluoranthene Colourlessi 168.3 481kk 6.7 x 10-5 6.12f 1.2ll 5.1 x 10-5
(20°C)gg (20°C)w
Benzo[j]fluoranthene Yellowb 165.4 480ee 2.0 x 10-6 l 6.12mm 2.5nn
Table 4. (continued)
Compound Colour Melting- Boiling- Vapour Densityc n-Octanol: Solubility in Henry's law
pointa point pressure water water at 25°C constant at
(°C) (°C) (Pa at 25°C) partition (µg/litre)d 25°C (kPa)
coefficient
(log Kow)
Renzo[k]fluoranthene Pale yellowh 215.7 480h 1.3 x 10-8 6.84m 0.76f 4.4 x 10-5
(20°C)oo (20°C)w
Benzo[a]pyrene Yellowishe 178.1 496kk 7.3 x 10-7oo 1.351pp 6.50u 3.8 3.4 x 10-5
(20°C)
Benzo[e]pyrene Pale yellowx 178.7 493kk 7.4 x 10-7qq 6.44rr 5.07 (23°C)tt
Perylene Yellow to 277.5 503ss 1.35v 5.3uu 0.4
colourlessc
Anthanthrene Golden yellowbb 264 547yy 1.39v
Benzo[ghi]perylene Pale yellow- 278.3 545ii 1.4 x 10-8 ww 1.32920 xx 7.10u 0.26 2.7 x 10-5
greenbb (20°C)w
Indeno[1,2,3-cd]pyrene Yellowi 163.6 536yy 1.3 x 10-8 6.58f 62f 2.9 x 10-5
(20°C)gg (20°C)w
Dibenz[a,h]anthracene Colourlessi 266.6 524yy 1.3 x 10-8 1.282i 6.50zz 0.5 (27°C)jj 7 x 10-6 l
(20°C)
Coronene Yellowh 439 525aaa 2.0 x 10-10 qq 1.37b 5.4uu 0.14
Dibenzo[a,e]pyrene Pale yellowh 244.4 592vv
Dibenzo[a,h]pyrene Golden yellowi 317 596vv
Dibenzo[a,i]pyrene Greenish-yellowishi 282 594vv 3.2 x 10-10 mm 7.30hh 0.17l 4.31 x 10-6 l
Dibenzo[a,l]pyrene Pale yellowi 162.4 595vv
a From Karcheret al. (1985); Karcher (1988)
b From Lewis (1992)
c When two temperatures are given as superscripts, they indicate the specific gravity, i.e. the density of the substance at the first
reported temperature relative to the density of water at the second reported temperature. When there is no value, or only one, for
temperature, the datum is in grains per millilitre, at the indicated temperature, if any.
Table 4 (continued)
d From Mackay & Shiu (1977), except where noted
e From Budavari (1989)
f From National Toxicology Program (1993)
g From Sonnefeld et al. (1983)
h From Lide (1991)
i From IARC (1977)
j From Karickhoff et al. (1979)
k From Mackay et al. (1979)
l Calculated by Syracuse Research Center; from National Toxicology Program (1993)
m Calculated as per Leo et al. (1971); from US Environmental Protection Agency (1980)
n From Mackay & Shiu (1981)
o When pure, colourless with violet fluorescence; from Budavari (1989)
p From Hawley (1987)
q From National Institute for Occupational Safety and Health and Occupational Safety and Health Administration (1981)
r From Kruber & Marx (1938)
s Calculated by Karcher et al. (1991)
t From May et al. (1978)
u From Bruggeman et al. (1982)
v At ambient temperature; from Inokuchi & Nakagaki (1959)
w From Ten Hulscher et al. (1992)
x Personal observation by J. Jacob, Germany, on high-purity, certified reference materials
y From Kruber (1937)
z Calculated by Miller et al. (1985)
aa From Schuyer et al. (1953)
bb From IARC (983)
cc From Kruber & Grigoleit (1954)
dd From Ehrlich & Beevers (1956)
ee Reported by Grimmer (1983a)
ff From Beilstein Institute for Organic Chemistry (1993)
gg Reported by Sims & Overcash (1983)
hh Calculated by Yalkowsky & Valvani (1979)
ii Calculated by White (1986)
jj From Davis et al. (1942)
kk From review by Bjorseth (1983); original references cited by White (1986)
ll Temperature not given; reported by Sims & Overcash (1983)
mm Calculated by National Toxicology Program (1993)
nn Temperature not given; unpublished result cited by Wise et al. (1981)
oo From US Environmental Protection Agency (1980)
Table 4 (continued)
pp From Kronberger & Weiss (1944)
qq From review of Santodonato et al. (1981)
rr Calculated by Ruepert et al. (1985)
ss From Verschueren (1983)
tt From Schwarz (1977)
uu From Brooke et al. (1986)
vv From Agency for Toxic Substances and Disease Registry (1990)
xx From White (1948)
yy Estimated from gas chromatographis retention time; from Grimmer (1983a)
zz From Means et al. (1980)
aaa From Von Boente (1955)
(Valerio et al., 1984; Lane, 1989). After photodecomposition in the
presence of air and sunlight, a number of oxidative products are
formed, including quinones and endoperoxides. PAH have been shown
experimentally to react with nitrogen oxides and nitric acid to form
the nitro derivatives of PAH, and to react with sulfur oxides and
sulfuric acid (in solution) to form sulfinic and sulfonic acids. PAH
may also be attacked by ozone and hydroxyl radicals present in the
atmosphere. The formation of nitro-PAH is particularly important owing
to their biological impact and mutagenic activity (IARC, 1984a,
1989a). In general, the above reactions are of interest with regard to
the environmental fate of PAH, but the results of experimental studies
are difficult to interpret because of the complexity of interactions
occurring in environmental mixtures and the difficulty in eliminating
artefacts during analytical determinations. These reactions are also
considered to be responsible for possible losses of PAH during ambient
atmospheric sampling (see section 2.4.1.1).
2.3 Conversion factors
Atmospheric concentrations of PAH are usually expressed as
micrograms or nanograms per cubic meter. At 25°C and 101.3 kPa, the
conversion factors for a compound of given relative molecular mass are
obtained as follows:
ppb = µg/m3 × 24.45/relative molecular mass
µg/m3 = ppb × relative molecular mass/24.45.
For example, for benzo [a]pyrene, 1 ppb = 10.3 µg/m3 and
1 µg/m3 = 0.0969 ppb.
2.4 Analytical methods
Tables 5 and 6 present as examples a limited number of methods
that are applied to 'real' samples of different matrices. The methods
and sources were selected, as far as possible, according to the
following criteria: accessibility of the bibliographic source,
completeness of the description of the procedure, practicability with
common equipment for this type of analysis (even if experienced
personnel are required), recency, and whether it is an official,
validated, or recommended method.
2.4.1 Sampling
2.4.1.1 Ambient air
The physical state of PAH in the atmosphere must be considered
when selecting the sampling apparatus. Compounds with five or more
rings are almost exclusively adsorbed on suspended particulate matter,
whereas lower-molecular-mass PAH are partially or totally present in
the vapour phase (Coutant et al., 1988). When ambient air is
monitored, it is common practice to monitor only particle-bound PAH
Table 5. Analytical methods for polycyclic aromatic hydrocarbons in air
Matrix Sampling, extraction Clean-up Analysis Limit of Reference
detectiona
Ambient air Sampling on GF+PUF, at 45 m3/h; Liquid-liquid partition GC/MS Yamasaki et al.
Soxhlet extraction with cyclohexane with cyclohexane: (1982)
H2O:DMSO, then CC
with SiO2
Sampling on GF+PUF, at 30 m3/h; CC with Al2O3 + HPLC/FL 0.01-0.7 Keller &
Soxhlet extraction with petroleum ether SiO2 ng/m3 Bidleman (1984)
(GF) and DCM (PUF)
Sampling on GF (particle diameter TLC with SiO2 HPLC/UV 0.01-0.3 Greenberg et al.
< 15 µm), at 68 m3/h; Soxhlet extraction + FL ng/m3 (1985)
with cyclohexane, DCM, and acetone
Sampling on GF at 83 m3/h; sonication TLC with SiO2 GC/FID Valerio et al.
(cyclohexane) (1992)
Emissions Sampling by glass wool, condenser, Liquid-liquid partition GC/FID 10 ng/m3 Colmsjo et al.
(municipal and XAD-2; extraction with acetone with DMF (1986a)
incinerator) (glass-wool and XAD-2, by Soxhlet)
Vehicle Sampling by GF and condenser; liquid- CC with SiO2 and GC/FID 2.5-20 ng Grimmer et al.
exhaust liquid partition with acetone:H2O: Selphadex LH-20 per test (1979)
cyclohexane and DMF:H,O:cyclohexane
Sampling in dilution tunnel by Liquid-liquid partition GC/FID or Westerholm et
PTFE-coated GF and condenser; Soxhlet with cyclohexane: GC/MS al. (1988)
extraction of filter (DCM) and H2O:DMF
condensate (acetone); remaining
aqueous phase extracted with DCM
Table 5. (continued)
Matrix Sampling, extraction Clean-up Analysis Limit of Reference
detectiona
Indoor air Sampling on GF (particle diameter TLC with acetyloxylated Spectrofluorescence Lioy at al. (1988)
< 10 µm) at 10 l/min; sonication cellulose (benzo[a]pyrene only)
(cyclohexane)
Sampling on quartz-fibre filtre and GC/MS Chuang at al.
XAD-4 at 226 l/min; Soxhlet extraction (1991)
with DCM
Sampling on PTFE-coated GF at filtration; then CC HPLC/FL 0.02-0.12 Daisey & Gundel
20 l/minfor 24 h; Soxhlet extraction SiO2 cartridge), ng/m3 b (1993)
with DCM optional
Sampling on GF and PUF, at 20 litres/min GC/FID, GC/MS US Environmental
for 24 h; Soxhlet extraction (10% ether: or HPLC/UV + FL Protection Agency
n-hexane) (1990)
Workplace air Sampling on PTFE filter and XAD-2 GC/FID 0.3-0.5 µg NIOSH (1994a,b)
at 2 l/min; sonication or Soxhlet per sample
extraction of filterc, extraction of HPLC/UV 0.05-0.8 µg
XAD-2 with toluene (for GC) or + FL per sample
acetonitrile (for HPLC)
Workplace air Sampling on filter (GF, quartz fibre, CC (XAD-2) GC/FID approx 0.5 German
PTFE or silver membrane) at 2 litres/min; µg/m3 Research
sonication or Soxhlet extraction with Commission
cyclohexane or toluene (1991)
Tobacco Sampling by acetone trap; solvent CC (SiO2 + Sephadex GC/MS + ng/cigarette Lee at al. (1976b)
smoke partition scheme (acids/bases/neutral LH-20); then NMR
compounds/PAH) HPLC/UV
Table 5 (continued)
GC glass fibre; PUF, polyurethane foam; DMSO, dimethyl sulfoxide; CC, column chromatography; GC, gas chromatography;
MS, mass spectrometry; DCM, dichloromethane; HPLC, high-performance liquid chromatography; FL, fluorescence detection;
TLC, thin-layer chromatography; UV, ultraviolet detection; FID, flame-ionization detection; DMF, N-dimethylformamide;
PTFE, polytetrafluoroethylene; NMR, nuclear magnetic resonance
a Various PAH
b The following PAH can be determined: fluoranthene, pyrene, chrysene, benzo[e]pyrene, benzo[b]fluoranthene,
benzo[k]fluoranthene, benzo[a]pyrene, benzo[ghi]perylene, indeno[1,2,3-cd]pyrene.
c Appropriate solvent must be determined by recovery tests on specific samples.
Table 6. Analytical methods for polycyclic aromatic hydrocarbons in matrices other than air
Matrix Extraction Clean-up Analysis Limit of Reference
detectiona
Tap-water Preconcentration on PUF; Liquid-liquid partition GC/FID or TLC 0.1 ng/litre Basu & Saxena
extraction (with acetone and with cyclohexane: (Al2O3: acetyl (1978a)
cyclohexane) H2O:methanol and celluose) with FL
cyclohexane: H2O: detector
DMSO; then CC
OWN)
Groundwater Liquid-liquid partition with CC (SiO2), if needed GC/FID µg/litre level US Environmental
DCM GC/MS 10 µg/litre Protection Agency
HPLC/UV + FL 0-01-2 µg/litre (1986a)
Wastewater Liquid-liquid partition with CC (SiO2), if needed GC/FID or 0.01 -0.2 µg/litre US Environmental
DCM HPLC/UV+FL (by HPLC) Protection Agency
(1984a)
Seawater Liquid-liquid partition with CC (SiO2 + Al2O2) GC/FID or Desideri at al.
n-hexane or CCl4 HPLC/UV (1984)
Soil Sonication with DCM CC (Al2O2); then GC/MS 1 µg/kg Vogt at
liquid-liquid partition al. (1987)
(n-hexane:H2O:DMSO)
Soxhlet extraction with DCM CC (Florisil cartridge) HPLC/UV + FL 1 µg/kg Jones et a[.
(1989a)
Sediment Soxhlet extraction with DCM CC (SiO2 + Sephadex HPLC/DAD/MS Quilliam & Sim
LH20) (1988)
Sonication with acetone: CC (Florisil) HPLC/UV + FL 1-160 µg/kg Marcus et al.
n-hexane (1988)
Table 6. (continued)
Matrix Extraction Clean-up Analysis Limit of Reference
detectiona
Meat and fish (I) digestion (alcoholic KOH), Liquid-liquid partition GC/FID 2.5-20 ng/ Grimmer &
products (I), then liquid-liquid partition with cyclohexane: sample Bohnke (1979b)
vegetable oils (methanol: H2O:cyclohexane) H2O:DMF); then CC
(II), and sewage (II) dissolution in cyclohexane (SiO2 + Sephadex
sludge (III) (III) refluxing with acetone LH20)
Food (total Refluxing with alcoholic KOH, Liquid-liquid partition HPLC/FL 0.002-0.7 µg/kg Dennis et al.
diet) extraction with isooctane (isooctane:H2O:DMF); (1983)
then CC (SiO2 cartridge)
Saponification with alcoholic CC (SiO2) HPLC/FL 0.03-2 µg/kg de Vos et al.
KOH, extraction with (1990)
cyclohexane
Saponikation wit ahoholic CC (Florisil); then TLC/UV+FL 0.02 µg/kg Howard (1979);
KOH, extraction with liquid-liquid partition (benzo[a]pyrene) Fazio (1990)
isooctane isooctane:H2O:DMSO)
Seafood Digestion with alcoholic KOH, CC (Al2O3 + SiO2 + HPLC/FL 0.01-0.6 µg/kg Perfetti et al.
extraction with TCTFE C18 cartridge) (1992)
Smoked food Digestion with alcoholic KOH, CC (Al2O3 + SiO2); HPLC/UV+FL 0.03-0.4 Joe et al. (1984)
extraction with TCTFE liquid-liquid partition µg/kg
(cyclohexane:H2O:DMSO)
Refluxing with cyclohexane or Liquid-liquid partition TLC/FLb (only 0 0.5 ng/kg IUPAC (1987)
TCTFE, extraction with with cyclohexane:H2O: benzo[alpyrene)
methanol:H2O DMF); then CC (SiO2)
Solid waste Soxhlet extraction with DCM CC (SiO2), if needed GC/FID µg/kg level US Environmental
or sonication with GC/MS 1-200 mg/kg Protection Agency
DGM:acetone HPLC/UV + FL µg/kg level (1986b)
Table 6. (continued)
Matrix Extraction Clean-up Analysis Limit of Reference
detectiona
Mineral oil and Liquid-liquid partition with CC (SiO2 + Sephadex GC/FID 100 ng/kg Grimmer &
fuel cyclohexane:H2O:DMF) LH20) Bohnke (1979a)
Medicinal oil Liquid-liquid partition CC (SiO2 + Sephadex HPLC/FL + 0.2-200 ng/kg Geahchan at al.
(cyclohexane: H2O:DMF) LH20) GC/FID (1991)
Plants Sonication (acetonitrile), CC (SiO2) GC/FID Coates et al.
extraction with pentane (1986)
Urine Adjusted to pH3, extraction CC (SiO2 cartridge) HPLC/FLc Becher & Bjorseth
in C18 cartridge, metabolites (1983)
reduced with hydriodic acid
Urine and Addition of HCl, refluxing CC (SiO2) + Sephadex GC/MSd Jacob at al. (1989)
faeces with toluene, addition of LH20
methanol and diazomethanol in
ether (faeces saponified before
acidification)
Tissue Homogenization (benzene: CC (Florisil) GC/MS 5-50 µg/kg Liao et al. (1988)
n-hexane)
Skine Sonication of exposure pads HPLC/FL 6 ng/cm2 Jongeneelen et al.
with DCM, centrifugation (1988a)
Table 6. (continued)
PUF, polyurethane foam; DMSO, dimethyl sulfoxide; CC, column chromatography; GC, gas chromatography; FID, flame ionization detection; FL,
fluorescence detection; DCM, dichloromethane; MS, mass spectrometry; UV, ultraviolet detection; DAD, diode-array detector; DMF,
N-dimethylformamide; TLC, thin-layer chromatography; TCTFE, 1,1,2-trichlorotrifluoroethane
a Various PAH
b Benzo[a]pyrene content estimated to be > 0.6 µg/kg (screening method)
c Determination of unmetabolized and metabolized PAH
d Determination of pyrene and 1-hydroxypyrene
e Measurement of skin contamination with soft polypropylene exposure pads mounted on skin sites
(Menichini, 1992a), probably because of the increased work involved in
trapping volatile compounds, both in assembling the sampling unit and
in analysing samples, and also because lighter compounds are of lesser
toxicological interest. Of the PAH that are classified as 'probably'
and 'possibly' carcinogenic to humans (IARC, 1987), only
benz [a]anthracene is found at significant levels in the vapour phase
(Van Vaeck et al., 1984; Coutant et al., 1988; Baek et al., 1992).
Sampling is generally performed by collecting total suspended
particulate matter for 24 h on glass-fibre filters by means of
high-volume samplers. Other filters that have been used are quartz
fibres (Hawthorne et al., 1992), polytetrafluoroethylene (PTFE)
membranes (Benner et al., 1989; Baek et al., 1992), and, in
comparisons, PTFE-coated glass fibres (Lindskog et al., 1987; De Raat
et al., 1990). The effects of these materials on the decomposition of
PAH during sampling have been compared (see section 2.2). Some studies
indicated that higher recoveries are obtained with PTFE and
PTFE-coated filters (Lee et al., 1980a; Grosjean, 1983); however, more
recent investigations did not confirm this finding (Lindskog et al.,
1987; Ligocki & Pankow, 1989; De Raat et al., 1990). Moreover, when
cellulose acetate membrane filters were compared with glass-fibre
filters, they had similar efficiency for collecting heavier PAH, but
the former had greater efficiency for collecting three- and four-ring
compounds (Spitzer & Dannecker, 1983).
The most widely used method for trapping vapour-phase PAH is
adsorption on plugs of polyurethane foam located behind the filter
(Keller & Bidleman, 1984; Chuang et al., 1987; De Raat et al., 1987a;
Benner et al., 1989; Hawthorne et al., 1992). This method is widely
accepted, probably because of the low pressure drop, the low blanks,
the low cost, and ease of handling. Among the other sorbents tested
(see also reviews by Leinster & Evans, 1986; Davis et al., 1987),
further polymeric materials have received particular attention,
including Amberlite XAD-2 resin, which is a valid alternative to
polyurethane foam (Chuang et al., 1987), Porapak PS, which has been
successfully tested in combination with a silanized glass-fibre filter
at a flow rate of 2 m3/h (Jacob et al., 1990a), and Tenax(R) (Baek
et al., 1992).
The trapped vapours contain both the PAH that were initially
present in the vapour phase and those already collected on the filter
and volatilized during sampling (the 'blowing-off' effect) (Van Vaeck
et al., 1984; Coutant et al., 1988). The amount of PAH found in the
vapour phase increases with ambient temperature (Yamasaki et al.,
1982). Samplers incorporating an annular denuder, as well as a filter
and back-up trap, have been used to investigate phase distribution and
artefact formation (Coutant et al., 1988, 1992).
Sampling times are restricted to 24 h in order to avoid sample
degradation and losses. Grimmer et al. (1982) proposed a useful method
for controlling losses due to chemical degradation and volatilization
from filters which is based on the invariability of PAH profiles (i.e.
the ratio of all PAH to one another) at different collection times.
The adsorption of gas-phase PAH onto a quartz-fibre filter has been
investigated as a possible sampling artefact (Hart & Pankow, 1994);
the results suggested that overestimation of particle-associated PAH
can be avoided by replacing quartz-fibre filters with a PTFE membrane
filters, or can be corrected by using back-up quartz-fibre filters.
Elutriators and cascade impactors have been used to achieve
particle size-selective sampling of PAH (Menichini, 1992a).
Instruments designed as additions to high-volume samplers are
available, including 'PM10' inlets, which allow collection of airborne
particles with a 50% cutoff at the aerodynamic diameter of 10 m (US
Environmental Protection Agency, 1987a; Lioy et al., 1988; Hawthorne
et al., 1992), and cascade impactors (Van Vaeck et al., 1984; Catoggio
et al., 1989).
When PAH are collected in indoor air, samplers operating at 20 or
200 litre/min are commonly used. The filter and sorbent materials are
those used for outdoor air (Wilson et al., 1991; see also Table 5).
The sampling step is by far the most important source of
variability in the results of atmospheric PAH determination. Most
investigations are difficult to compare because of differences in
factors such as season, meteorological conditions, time of day, number
and characteristics of sampling sites, and sampling parameters
(Menichini, 1992a). Passive biological sampling has been investigated
as an approach to long-term sampling of atmospheric PAH (Jacob &
Grimmer, 1992), and preliminary correlation factors have been
determined by comparing the PAH profiles in biological (plants,
particularly) and air samples. Of the matrices tested, spruce sprouts
were found to be the most suitable.
2.4.1.2 Workplace air
The general considerations described for ambient air are also
valid for the working environment. Less volatile PAH may be retained
than in ambient air because of the high temperatures that are often
found at the workplace. In the potroom of an aluminium plant where
Sderberg electrodes were used, 42% of benz [a]anthracene was found
in the vapour phase (Andersson et al., 1983), and in an iron foundry
at a site where the temperature of the PAH source was 600-700°C, four-
to seven-ring PAH represented about 70% of the total in the vapour
phase (Knecht et al., 1986).
Glass-fibre or PTFE filters are usually used to collect
particle-bound PAH. A number of back-up systems can be used to
efficiently trap volatile PAH, including liquid impingers and solid
sorbents such as Tenax(R)-GC, Chromosorb, and XAD-2 (Bjorseth &
Becher, 1986; Davis et al., 1987). The latter seems to be the most
practical. The US National Institute for Occupational Safety and
Health (1994a,b) recommended use of a PTFE-laminated membrane followed
by a tube containing two sections of XAD-2. For sampling in bright
sunlight, opaque or foil-wrapped filter cassettes can be used to
prevent degradation.
The exposure of workers is estimated by taking air samples at
various locations in the workplace or by personal sampling, in which
workplace air is pumped through a filter attached to clothing close to
the breathing zone for a specified time. Both procedures provide an
estimate and not a precise measurement of an individual's exposure.
2.4.1.3 Combustion effluents
The validity of a collected sample, i.e. the degree to which it
reflects the 'true' composition of the emission, is a crucial factor
in the determination of PAH in emissions. The problems associated with
efficient collection of volatile PAH are enhanced when sampling
combustion effluents, such as stack gases and vehicle exhausts,
because of the elevated temperatures at sampling positions.
A sampling device for stack gases is constituted by a glass- or
quartz-fibre filter, followed by a special unit which generally
consists in a cooler for collecting condensable matter and an
adsorbent cartridge (Colmsjö et al., 1986a; Funcke et al., 1988).
Tenax(R) has been used as an adsorbent (Jones et al., 1976), but
XAD-2 seems to be more suitable (Warman, 1985) and is generally
preferred. Two sampling procedures have been described in detail by
the US Environmental Protection Agency (1986c). In the first
('Modified method 5 sampling train'), the unit basically includes a
glass- or quartz-fibre filter kept at around 120°C, a condenser coil
that conditions the gas at a maximum of 20°C, and a bed of XAD-2
jacketed to maintain the internal gas temperature at about 17°C. The
second ('Source assessment sampling system') is often used for
stationary investigations (Warman, 1985). The apparatus consists of a
stainless-steel probe, which enters an oven containing the filter,
preceded by three cyclone separators in series, with cutoff diameters
of 10, 3, and 1 m; the volatile organic compounds are cooled and
trapped on XAD-2. The sorbent is followed by a condensate collection
trap and an impinger train.
Motor vehicle exhausts are sampled under laboratory conditions,
by chassis or engine dynamometer testing. Standard driving cycles are
employed to simulate on-road conditions (Stenberg, 1985; see also
section 3.2.7.2).
Two basic techniques have been used to collect, sample, and
analyse exhaust (Levsen, 1988; IARC, 1989a). In the first-raw gas
sampling-the exhaust pipe is connected directly to the sampling
apparatus; undiluted emissions are cooled in a condenser and then
allowed to pass through a filter for collection of particulates
(Grimmer et al., 1979, 1988a; Society of German Engineers, 1989). A
second technique-dilution tube sampling-is now often used, in which
hot exhaust is diluted with filtered cold air in a tunnel, from which
samples are collected isokinetically. This technique simulates the
process of dilution that occurs under real conditions on the road (US
Environmental Protection Agency, 1992a).
Particles are almost always collected on glass-fibre, glass-fibre
with PTFE binder, quartz-fibre filters, or PTFE membranes; the latter
have been reported to be particularly efficient and chemical inert
(Lee & Schuetzle, 1983). Glass-fibre filters impregnated with liquid
paraffin are also used (Grimmer et al., 1979; Society of German
Engineers, 1989). Vapour-phase PAH (Stenberg, 1985) may be collected
by cryo-condensation (Stenberg et al., 1983) or on an adsorbent trap
with a polymeric material such as XAD-2 (Lee & Schuetzle, 1983).
Artefacts may be introduced during collection on filters as a
result of chemical conversion of PAH, particularly into nitro-PAH and
oxidation products (Lee & Schuetzle, 1983; Schuetzle, 1983; IARC,
1989a). These effects have not been fully evaluated.
2.4.1.4 Water
The concentrations of PAH in uncontaminated groundwater supplies
and in drinking-water are generally very low, at 0.1 and 1 ng/litre
(see sections 5.1.2.1 and 5.1.2.2). This implies that serious errors
arising from adsorption losses and contamination occur during
collection and storage of samples or that a preconcentration step may
be needed to enrich the sample. It is recommended that sampling be
performed on-site, directly in the extraction vessel (Smith et al.,
1981).
Various solid sorbents have been successfully used for
preconcentration (Smith et al., 1981), including Tenax(R)-GC,
prefiltered if necessary (Leoni et al., 1975); XAD resins (Griest &
Caton, 1983); open-pore polyurethane foam (Basu et al., 1987); and
prepacked disposable cartridges of bonded-phase silica gel (Chladek &
Marano, 1984; Van Noort & Wondergem, 1985a). Solid sorbents have
limitations when the sample contains suspended material, since
adsorbed PAH may be lost by filtration (Van Noort & Wondergem, 1985a).
2.4.1.5 Solid samples
Some foodstuffs (Liem et al., 1992), soil, sediment, tissues, and
plants usually require homogenization before a sample is extracted.
2.4.2 Preparation
As most environmental samples contain only small amounts of PAH,
sophisticated techniques are required for their detection and
quantification. Therefore, efficient extraction from the sample matrix
is usually followed by one or more purification steps, so that the
sample to be analysed is as free as possible from impurities and
interference. Many extraction and purification techniques and
combinations ('isolation schemes') have been described, validated, and
recommended, but no single scheme is commonly recognized as 'the best'
for a given matrix. The isolation schemes have been classified
according to groups of matrices (Jacob & Grimmer, 1979; Grimmer,
1983a), as summarized briefly below.
PAH are extracted from a sample (Lee et al., 1981; Santodonato et
al., 1981; Grimmer, 1983a; Griest & Caton, 1983) with:
- a Soxhlet apparatus, from filters loaded with particulate matter,
vehicle exhausts, or sediments;
- directly by liquid-liquid partition, for water samples; or
- after complete dissolution (e.g. fats and vegetable and mineral
oils) or alkaline digestion of samples (e.g. meat products) by a
selective solvent such as N,N-dimethylformamide (Natusch &
Tomkins, 1978) or dimethyl sulfoxide. Complete extraction of PAH
from samples such as soot emitted by diesel engines, carbon
blacks, and other carbonaceous materials is particularly
difficult.
Extraction of PAH from soil, sediment, sewage sludge, and vehicle
exhaust particulates by refluxing with various solvents has been
investigated. In all cases, toluene was found to be the most efficient
solvent, especially for vehicle exhaust (Jacob et al., 1994).
As an alternative to Soxhlet extraction, ultrasonic extraction
(Griest & Caton, 1983) has advantages in terms of reduced time of
extraction (minutes versus hours) and superior recovery efficiency and
reproducibility, particularly for solid samples and filters loaded
with particulate matter. Comparisons of techniques depend, however, on
the matrix, solvent, and experimental conditions.
Supercritical fluid extraction (Langenfeld et al., 1993) has
gained attention as a rapid alternative to conventional liquid
extraction from polyurethane foam sorbents (Hawthorne et al., 1989a),
soil (Wenclawiak et al., 1992), and other environmental solids such as
urban dust, fly ash, and sediment (Hawthorne & Miller, 1987). This
technique can also be directly coupled with on-column gas
chromatography (see section 2.4.3.1); the extract is quantitatively
transferred onto the gas chromatographic column for a rapid (< 1 h)
analysis with maximal sensitivity. This technique has been used for
urban dust samples (Hawthorne et al., 1989b).
Extracted samples are usually purified from interfering
substances by adsorption column chromatography. The classical
sorbents, alumina and silica gel, are widely used. In addition, the
hydrophobic Sephadex LH-20 has been found to be suitable for isolating
PAH from nonaromatic, nonpolar compounds, which is important if the
sample is analysed by gas chromatography (Grimmer & Böhnke, 1979a); It
has also been used in partition chromatography as a carrier of the
stationary phase, to separate PAH from alkyl derivatives (Grimmer &
Böhnke, 1979b). Chromatography on silica gel and Sephadex is often
combined (Jacob & Grimmer, 1979; Grimmer, 1983a).
Clean-up has also been achieved by eluting extracted samples
through XAD-2 (soil samples: Spitzer & Kuwatsuka, 1986), XAD-2 and
Sephadex LH-20 in series (foods: Vaessen et al., 1988), or Florisil
(food, water, and sediment samples: references given in Table 6).
Conventional chromatographic columns may be substituted by
prepacked commercial cartridges, which have advantages in terms of
time and solvents consumed and reproducibility. For example, silica
cartridges have been used to purify foodstuffs (Dennis et al., 1983),
urine (Becher & Bjorseth, 1983), vehicle emissions (Benner et al.,
1989), mineral oil mist (Menichini et al., 1990), and atmospheric
samples (Baek et al., 1992); soil samples have been cleaned up on
Florisil cartridges (Jones et al., 1989a).
Preparative thin-layer chromatography is also used for, e.g. air
particulates (see Table 5) and vegetable oils (Menichini et al.,
1991a).
Handling of samples in the absence of ultraviolet radiation is
recommended at all stages in order to avoid photodecomposition of PAH
(Society of German Engineers, 1989; US Environmental Protection
Agency, 1990; US National Institute for Occupational Safety and
Health, 1994a,b). It is also generally recommended that possible
sources of interference and contamination be controlled, particularly
from solvents (US Environmental Protection Agency, 1984a, 1986b,
1990), and that samples be refrigerated until extraction (US
Environmental Protection Agency, 1984a; US National Institute for
Occupational Safety and Health, 1994a,b).
2.4.3 Analysis
PAH are now routinely identified and quantified by gas
chromatography or high-performance liquid chromatography (HPLC). Each
technique has a number of relative advantages. Both are rather
expensive, particularly HPLC, and require qualified operating
personnel; nevertheless, they are considered necessary in order to
analyse 'real' samples for a large number of PAH with accuracy and
precision.
2.4.3.1 Gas chromatography
Excellent separation (< 3000 plates per meter) is obtained by
the use of commercially available fused silica capillary columns,
making it possible to analyse very complex mixtures containing more
than 100 PAH.
The most widely used stationary phases are the
methylpolylsiloxanes: especially SE-54 (5% phenyl-, 1%
vinyl-substituted) and SE-52 (5% phenyl-substituted), but SE-30 and
OV-101 (unsubstituted), OV-17 (50% phenyl-substituted), Dexsil 300
(carborane-substituted) and their equivalent phases are also used.
Chemically bonded phases are used increasingly because they can be
rinsed to restore column performance and undergo little 'bleeding' at
the high temperatures of analysis (about 300°C) that are required for
determining high-boiling-point compounds.
Nematic liquid crystal phases (Bartle, 1985) have also been used
to separate some isomeric compounds that are poorly resolved by
siloxane phases, such as chrysene and triphenylene on
N,N'-bis (para-methoxy-benzylidene)-a,a'-bi- para-toluidine
(Janini et al., 1975) and
N,N'-bis (para-phenylbenzylidene)-a,a'-bi- para-toluidine (Janini
et al., 1976).
Splitless or on-column injection is necessary to gain sensitivity
in trace analysis, the latter being preferred as it allows better
reproducibility. Flame ionization detectors are almost always used
because of the excellent linearity, sensitivity, and reliability of
their response. Since the signal is related linearly to the carbon
mass of the compound, PAH are recorded in proportion to their
quantities, and the chromatogram shows the quantitative composition of
the sample directly. Because flame ionization detectors are
non-selective, samples for gas chromatography must be highly purified.
Peak identification, which is done routinely from data on retention,
must be confirmed by analysing samples on a different gas
chromatographic column, by an independent technique, such as HPLC, or
by directly coupling a mass spectrometric detector to the gas
chromatograph (Lee et al., 1981; Olufsen & Bjorseth, 1983; Bartle,
1985; Hites, 1989).
Mass spectrometers have gained wide acceptance. They are powerful
tools for identifying compounds, especially when commercially
available libraries of reference spectra are used to match the spectra
obtained and to control the purity of a compound. As isomeric
compounds often have indistinguishable spectra, however, the final
assignment must also be based on retention.
On-line coupling of liquid chromatography, capillary gas
chromatography, and quadrupole mass spectrometry has been used to
determine PAH in vegetable oils (Vreuls et al., 1991).
2.4.3.2 High-performance liquid chromatography
The packing material considered most suitable for separating PAH
consists of silica particles chemically bonded to linear C18
hydrocarbon chains; selection of the appropriate phase has been
discussed in detail by Wise et al. (1993). Typically, 25-cm columns
packed with 5-m particles are used in the gradient elution technique,
and the mobile phase consists of mixtures of acetonitrile and water or
methanol and water ('reversed-phase HPLC'). As the efficiency of
separation that can be achieved with HPLC columns is much lower than
that with capillary gas chromatography, HPLC is generally less
suitable for separating samples containing complex PAH mixtures.
The advantages of HPLC derive from the capabilities of the
detectors with which it is used. Those most widely used for PAH are
ultraviolet and fluorescence detectors, generally arranged in series,
with flow-cell photometers or spectrophotometers. Both, but especially
the latter, are highly specific and sensitive: the detection limits
with fluorescence are at least one order of magnitude lower than those
with ultraviolet detection. The specificity of fluorescence detectors
allows the determination of individual PAH in the presence of other
nonfluorescing substances. In addition, since different PAH have
different absorptivity or different fluorescence spectral
characteristics at given wavelengths, the detectors can be optimized
for maximal response to specific compounds. This may prove
advantageous in the identification of unresolved components. In
particular, wavelength-programmed fluorescence detection, to measure
changes in excitation and emission wavelengths during a
chromatographic run (Hansen et al., 1991a), is being used for the
analysis of environmental samples (Wise et al., 1993). HPLC is
suitable to a limited degree for lower-molecular-mass compounds like
naphthalene, acenaphthene, and acenaphthylene, for which the detection
limits are relatively high (US Environmental Protection Agency,
1984a).
Owing to the selectivity of packing materials, various isomers
that cannot be separated efficiently on the usual capillary gas
chromatographic columns can be resolved at baseline and identified by
HPLC. Such isomers include the pairs chrysene-triphenylene and
benzo [b]fluoranthene-benzo [k]fluorathene (Wise et al., 1980).
Coupling of a mass spectrometer to HPLC has also been used in
detecting PAH (e.g., Quilliam & Sim, 1988).
As much information on isomeric structure can be obtained from
spectra seen during the elution of chromatographic peaks, an
ultraviolet diode-array detector has been used to confirm peaks (Dong
& Greenberg, 1988; Kicinski et al., 1989). For applications of HPLC to
determination of PAH, reference should be made to published reviews
(Lee et al., 1981; Wise, 1983, 1985).
2.4.3.3 Thin-layer chromatography
Thin-layer chromatography is commonly used only for identifying
individual compounds, such as benzo [a]pyrene, during screening
(IUPAC, 1987) or for identifying selected PAH, such as the six PAH
that WHO (1971) recommended be determined in drinking-water (Borneff &
Kunte, 1979). It is an inexpensive, quick analytical technique but has
low separation efficiency. The last parameter is improved by
two-dimensional processes (see, e.g. Borneff & Kunte, 1979).
Quantification may be done by spectrophotometric or
spectrofluorimetric methods in solution after the scrubbed substance
spot has been extracted (Howard, 1979; Fazio, 1990) or in situ by
scanning spectrofluorimetry (Borneff & Kunte, 1979).
Acetylated cellulose is the adsorbent that has been used most
widely for one-step separation of PAH fractions, and mixed aluminium
oxide and acetylated cellulose have been used for two-dimensional
development (Daisey, 1983).
2.4.3.4 Other techniques
A number of unconventional instruments and techniques based on
spectro-scopic principles have been developed as possible alternatives
to the chromatographic methods for PAH. Most of them are, however,
expensive, require skilled personnel, and are not yet considered
useful for the practising analyst (Wehry, 1983; Vo-Dinh, 1989).
Low-temperature luminescence in frozen solutions ('Shpol'skii
effect') has been used for various environmental samples, particularly
to identify methylated PAH isomers (Garrigues & Ewald, 1987; Saber et
al., 1987). This technique was used widely in the countries of former
Soviet Union (Dikun, 1967). Synchronous luminescence and room
temperature phosphorimetry have been reported to be simple,
cost-effective techniques for screening PAH (Vo-Dinh et al., 1984;
Abbott et al., 1986).
Infrared analysis, particularly Fourier transform infrared
spectroscopy coupled to gas chromatography (Stout & Mamantov, 1989),
and capillary supercritical fluid chromatography (Wright & Smith,
1989) have also been used. Various environmental samples have been
analysed by packed column supercritical fluid chromatography, with
rapid separation of PAH (Heaton et al., 1994).
2.4.4 Choice of PAH to be quantified
The choice of PAH depends on the purpose of the measurement. For
example, carcinogenic PAH are of interest in studies of human health,
but other, more abundant PAH may be of interest in ecotoxicological
studies. Quantification of a number of PAH is advantageous when the
profiles are to be correlated with sources and/or effects.
Table 7 lists the PAH that are required or recommended to be
determined at national or international levels. According to an EEC
(1980) Directive, which followed a WHO (1971) recommendation, the
concentrations of six reference compounds (also known as 'Borneff
PAH') must be measured in drinking-water in order to check its
compliance with the cumulative limit value for the PAH class. The
choice of these six PAH by WHO was not based on toxicological
considerations but on the fact that analytical investigations were
then largely confined to these relatively easily detected compounds
(WHO, 1984).
Table 7. Some polycyclic aromatic hydrocarbons required or recommended for determination by various authorities
Compound WHO/EECa US EPAb European Italyd Norwaye
(drinking- (waste Aluminium (air)
water) water) Associationc Health Environment
Acenaphthene X
Acenapthylene X
Anthracene X X X
Anthanthrene X X
Benz[a]anthracene X X X X X
Benzo[a]fluorene X
Benzo[a]pyrene X X X X X
Benzo[b]fluoranthene X X X X X X
Benzo[b]fluorene X
Benzo[c]phenanthrene X X
Benzo[e]pyrene X
Benzo[ghi]perylene X X X X
Benzo[j]fluoranthene X X X X
Benzo[k]fluoranthene X X X X X X
Chrysene X X X X
Cyclopenta[a]pyrene X X
Dibenzo[a,e]pyrene X X X
Dibenz[a,h]anthracene X X X X X
Dibenzo[a,h]pyrene X X X
Dibenzo[a,i]pyrene X X X
Dibenzo[a,l]pyrene X X
Fluoranthene X X X X
Fluorene X
Indeno[1,2,3-cd]pyrene X X X X X X
Naphthalene X X
Phenanthrene X X X
Pyrene X X X
Triphenylene X
Table 7 (continued)
a Recommended by WHO (1971) and required by an EEC (1980) Directive
b Required by the US Environmental Protection Agency (1984a) for the analysis of municipal and industrial
wastewater
c Recommended by the European Aluminium Association, Environmental Health and Safety Secretariat (1990)
d Recommended by the Italian National Advisory Toxicological Committee for health-related studies
(Menichini, 1992b)
e Recommended at the International Workshop on polycyclic aromatic hydrocarbons (State Pollution Control
Authority and Norwegian Food Control Authority, 1992) for studies of health and on the environment
The method required by the US Environmental Protection Agency
(1984a) for the analysis of municipal and industrial wastewater covers
the determination of 16 'priority pollutant PAH' considered to be
representative of the class. Outside the USA, this list of compounds
is often taken as a reference list for the analysis of various
environmental matrices.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
Appraisal
Coal and crude oils contain polycyclic aromatic hydrocarbons
(PAH) in considerable concentrations owing to diagenetic formation in
fossil fuels. The main PAH produced commercially are naphthalene,
acenaphthene, anthracene, phenanthrene, fluoranthene, and pyrene. The
release of PAH during production and processing, predominantly of
plasticizers, dyes, and pigments, is of only minor importance. Most
PAH enter the environment via the atmosphere from incomplete
combustion processes, such as:
- processing of coal and crude oil: e.g. refining, coal
gasification, and coking;
- heating: power plants and residential heating with wood, coal,
and mineral oil;
- fires: e.g. forest, straw, agriculture, and cooking;
- vehicle traffic; and
- tobacco smoking.
Industrial processes such as coal coking, aluminium, iron and
steel production, and foundries make important contributions to the
levels of PAH in the environment. An important indoor source of
exposure to airborne PAH, especially in developing countries, is
cooking fumes (see section 5.2).
The hydrosphere and the geosphere are affected secondarily by wet
and dry deposition. PAH are released directly into the hydrosphere,
for example during wood preservation with creosotes. Deposition of
contaminated refuse like sewage sludge and fly ash may cause further
emissions into the geosphere.
It is very difficult to identify a source on the basis of the
ratio of the measured concentrations of different individual PAH, and
such studies are in most cases inconclusive.
3.1 Natural occurrence
In some geographical areas, forest fires and volcanoes are the
main natural sources of PAH in the environment (Baek et al., 1991). In
Canada, about 2000 tonnes of airborne PAH per year are attributed to
natural forest fires (Environment Canada, 1994). On the basis of
samples from volcanoes, Ilnitsky et al. (1977) estimated that the
worldwide release of benzo [a]pyrene from this source was 1.2-14
t/year; no estimate was given of total PAH emissions from this source.
Coal is generally considered to be an aromatic material. Most of
the PAH in coal are tightly bound in the structure and cannot be
leached out, and the total PAH concentrations tend to be higher in
hard coal than in soft coals, like lignite and brown coal.
Hydroaromatic structures represent 15-25% of the carbon in coal. The
PAH identified include benz [a]anthracene, benzo [a]pyrene,
benzo [e]pyrene, perylene, and phenanthrene (Neff, 1979; Anderson et
al., 1986). Table 8 shows the typical contents of PAH in different
crude oils, such as those derived from coal conversion or from shale.
Table 8. Polycyclic aromatic hydrocarbon content of crude oils
from various sources
Compound PAH content (mg/kg) in crude oil from
Coala Petroleum Shale
Acenaphthene 1700/1800 147-348 147-903
Anthracene 4100 204-321 231-986
Anthanthrene Trace/< 800 NR 0.3
Benz[a]anthracene Trace/< 2200 1-7 1
Benzo[a]fluorene 2100/2500 11-22 53
Benzo[a]pyrene < 500/< 1200 0.1-4 3-192
Benzo[b]fluorene < 1500/3400 < 13 140
Benzo[c]phenanthrene < 600/< 2200 NR NR
Benzo[e]pyrene < 1200/1300 0.5-29 1-19
Benzofluorenesb < 500/< 1300 23 NR
Benzo[ghi]fluoranthene 3200 NR NR
Benzo[ghi]perylene 4300/6600 ND-8 1-5
ND-5
Chrysene < 1500/2500 7-26 3-52
Coronene NR 0.2 NR
Dibenz[a,h]anthracene NR 0.4-0.7 1-5
Fluoranthene < 1900/< 3700 2-326 6-400
Fluorene 5300/9900 106-220 104-381
1-Methylphenanthrene < 1200/< 5100 > 21 NR
Naphthalene 100/2800 402-900 203-1390
Perylene Trace/< 600 6-31 0.3-68
Phenanthrene 12 000/20 400 > 129-322 221-842
Pyrene 14 200/35 000 2-216 18-421
Triphenylene NR 3/13 0.5
From Guerin at al. (1978), Weaver & Gibson (1979), Grimmer at al.
(1983a), Sporstol et al. (1983), IARC (1985, 1989b)
Ranges represent at least three values; NR, not reported; ND, not
detected
a Two crude oils from coal conversion; single measurements
b Isomers not specified
Two rare PAH minerals have been described: the greenish-yellow,
fluorescent curtisite from surface vents of hot springs at Skagg
Springs, California, USA, and the bituminous mercury ore idrialite
from Idria, Yugoslavia, the two main components of which are chrysene
and dibenz [a,h]-anthracene. These minerals are assumed to have been
formed by the pyrolysis of organic material at depths below that at
which petroleum id generated (West et al., 1986).
3.2 Anthropogenic sources
3.2.1 PAH in coal and petroleum products
Commercial processing of coal leads first to coal-tars, which are
further processed to yield pitch, asphalt, impregnating oils
(creosotes for the preservation of wood), and residue oils such as
anthracene oil (IARC, 1985). The concentration of PAH in coal-tars is
generally ¾ 1%; naphthalene and phenanthrene are by far the most
abundant compounds, occurring at concentrations of 5-10%. Comparable
levels were detected in high-temperature coal-tar pitches. The PAH
content of soots is about one order of magnitude lower, and that of
carbon and furnace blacks ranges from about 1 to 500 mg/kg, pyrene
being present at the highest concentration (IARC, 1984a; Nishioka et
al., 1986). The PAH contents of some impregnating oils, bitumens,
asphalts, and roof paints are shown in Table 9. In bitumens, PAH
constitute only a minor part of the total content of polyaromatic
compounds.
Table 9. Polycyclic aromatic hydrocarbon content of impregnating oils, bitumens, asphalts,
and roof paints
Compound Concentration (mg/kg)
Impregnating Bitumens Road tar (asphalt, Roof
oils (oil-derived) coal-derived) paint
Anthracene 1600-22 500 0.01-0.32 4170-14 400 2380
Anthranthrene NR Trace-1.8 NR NR
Benz[a]anthracene 169-11 700 0.14-35 6820-24 100 6640
Benzo[a]pyrene 45-3490 0.1-27 5110-10 400 5950
Benzo[b]fluoranthene 42-3630 5 4490-10 900 5420
Benzo[e]pyrene 65-2020 0.03-52 3300-6750 3820
Benzo[gh]perylene 57-570 Trace-15 2390-2730 3270
Benzo[k]fluoranthene 24-2610 0.024-0.19 3170-7650 4470
Chrysene NR 0.04-34 NR
Chrysene + 779-12 900 NR 6820-26 100 7700
triphenylene
Coronene NR 0.2-2.8 NR NR
Fluoranthene 703-85 900 0.15-5 23 500-61 900 12 100
Fluorene 8040-58 400 NR 6310-15 500 2220
Indeno[1,2,3-cd]pyrene 57-273 Trace 3100-3530 3320
Perylene 66-744 0.08-39 1550-2300 1730
Phenanthrene 7070-159 300 0.32-7.3 20 300-52 500 8180
Pyrene 604-46 400 0.08-38 15 100-42 500 8960
Triphenylene NR 0.3-7.6 NR NR
From IARC (1985), Lehmann et al. (1986), Knecht & Woitowitz (1990);
NR, not reported; ranges represent at least three values
The concentrations of PAH in petrol and diesel fuels for vehicles
and in heating oils are several parts per million. Almost all
compounds are present at < 1 mg/kg; only phenanthrene, anthracene,
and fluoranthene are sometimes found at > 10 mg/kg (Herlan, 1982).
The PAH levels in unused engine lubricating oils are of the same order
of magnitude. During the use of petrol-fuelled engine oils, the PAH
content rises dramatically, by 30-500 times; in comparison, the total
PAH levels in used diesel-fuelled engine oils were only 1.4-6.1 times
greater than that in an unused sample. The major constituents of used
oils are pyrene and fluoranthene, although benzo [b]fluoranthene,
benzo [j]-fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene, and
dibenz [a,h]anthracene were also detected at considerable
concentrations (IARC, 1984a; Carmichael et al., 1990).
PAH have also been found in machine lubricating and cutting oils,
which is of interest for the estimation of exposure in the workplace.
The concentrations were < 7 mg/kg, although phenanthrene may have
been present at a higher level (Grimmer et al., 1981a; Rimatori et
al., 1983; Menichini et al., 1990; Paschke et al., 1992).
PAH were detected in coloured printing oils, the concentrations
of individual compounds varying between < 0.0001 and 63 mg/kg
(Tetzen, 1989). By far the most abundant compounds were fluoranthene
and pyrene (> 1 mg/kg); benzo [ghi]fluoranthene,
cyclopenta [cd]pyrene, benz [a]anthracene, benzo [c]-phenanthrene,
chrysene, triphenylene, benzo [b+j+k]fluoranthenes, benzo [a]pyrene,
benzo [e]pyrene, anthanthrene, benzo [ghi]perylene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene, and coronene were
found at concentrations of < 0.5 mg/kg.
3.2.2 Production levels and processes
Most of the PAH considered in this monograph are formed
unintentionally during combustion and other processes. Only a few are
produced commercially, including naphthalene, acenaphthene, fluorene,
anthracene, phenanthrene, fluoranthene, and pyrene (Franck &
Stadelhofer, 1987). The most important industrial product is
naphthalene (see section 3.2.3). In 1987, about 220 kt of this
compound were produced in western Europe, 190 kt in eastern Europe,
170 kt in Japan, and 110 kt in the USA (Fox et al., 1988); in 1986,
> 1 kt was produced in Canada (Environment Canada, 1994). In 1985,
about 2.5 kt of acenaphthene and 20 kt of anthracene were produced
worldwide (Franck & Stadelhofer, 1987). In 1986, 0.1-1 t anthracene
and 1 t fluorene were produced in Canada (Environment Canada, 1994).
In 1993, a major producer in Germany produced < 5000 t anthracene,
< 1000 t acenaphthene, < 500 t pyrene, < 50 t phenanthrene, and
< 50 t fluoranthene (personal communication, Rütgers-VfT AG, 1994).
The substances are not synthesized chemically for industrial
purposes but are isolated from products of coal processing, mainly
hard coal-tar. The raw material is concentrated and the product
purified by subsequent distillation and crystallization. Only
naphthalene is sometimes isolated from pyrolysis residue oils, olefin
fractions, and petroleum-derived fractions; it is also obtained by
distillation and crystallization (Collin & Höke, 1985; Franck &
Stadelhofer, 1987; Griesbaum et al., 1989; Collin & Höke, 1991). In
the USA in 1970, the distribution of capacity was about 60% coal-tar-
and 40% petroleum-derived naphthalene (Gaydos, 1981); more detailed
data were not available. The purity of the technical-grade products is
90-99% (Collin & Höke, 1985; Franck & Stadelhofer, 1987; Griesbaum et
al., 1989; Collin & Höke, 1991; see also Section 2).
3.2.3 Uses of individual PAH
The uses of commercially produced PAH are as follows (Collin &
Höke, 1985; Franck & Stadelhofer, 1987; Griesbaum et al., 1989; Collin
& Höke, 1991):
- naphthalene: main use: production of phthalic anhydride
(intermediate for polyvinyl chloride plasticizers); also,
production of azo dyes, surfactants and dispersants, tanning
agents, carbaryl (insecticide), alkylnaphthalene solvents (for
carbonless copy paper), and use without processing as a fumigant
(moth repellent) (see Figure 2);
- acenaphthene: main use, production of naphthalic anhydride
(intermediate for pigments); also, for acenaphthylene
(intermediate for resins);
- fluorene: production of fluorenone (mild oxidizing agent);
- anthracene: main use, production of anthraquinone (intermediate
for dyes); also, use without processing as a scintillant (for
detection of high-energy radiation);
- phenanthrene: main use, production of phenanthrenequinone
(intermediate for pesticides); also, for diphenic acid
(intermediate for resins)
- fluoranthene: production of fluorescent and vat dyes;
- pyrene: production of dyes (perinon pigments).
3.2.4 Emissions during production and processing of PAH
The emissions of PAH during industrial production and processing
in developed countries are not thought to be important in comparison
with the release of PAH from incomplete combustion processes, since
closed systems and recycling procedures are usually used. Few data
were available.
3.2.4.1 Emissions to the atmosphere
No data were available.
2.2 Physical and chemical properties
Physical and chemical properties relevant to the toxicological
and ecotoxicological evaluation of the PAH are summarized in Table 4.
It should be kept in mind that the values for any one parameter may be
derived from different sources, with different methods of measurement
or calculation, so that individual values cannot be compared directly
unless the original sources are consulted. In particular, the vapour
pressures reported in the literature for the same PAH vary by up to
several orders of magnitude (Mackay & Shiu, 1981; Lane, 1989).
Variations are also seen in the reported solubility in water of
various PAH, although the values are generally within one order of
magnitude (National Research Council Canada, 1983). Flash-points were
available only for three compounds with high molecular mass (for
naphthalene, 78.9°C by the open-cup method and 87.8°C by the closed-cup
method; anthracene, 121°C by the closed-cup method; and phenanthrene,
171°C by the open-cup method). Explosion limits were available only
for naphthalene (0.9-5.9 vol %) and ananthrene (0.6 vol %) (Lewis,
1992). Vapour density (air = 1) was 4.42 for naphthalene (IARC,
1973), 5.32 for acenaphthene, 6.15 for anthracene (Lewis, 1992), 6.15
for phenanthrene, and 8.7 for benzo[a]pyrene (National Institute for
Occupational Safety and Health and Occupational Safety and Health
Administration, 1981).
The physical and chemical properties are largely determined by
the conjugated alpha-electron systems, which vary fairly regularly
with the number of rings and molecular mass, giving rise to a more or
less wide range of values for each parameter within the whole class.
At room temperature, all PAH are solids. The general characteristics
common to the class are high melting- and boiling-points, low vapour
pressure, and very low solubility in water. PAH are soluble in many
organic solvents (IARC, 1983; Agency for Toxic Substances and Disease
Registry, 1990; Lide, 1991) and are highly lipophilic.
Vapour pressure tends to decrease with increasing molecular mass,
varying by more than 10 orders of magnitude. This characteristic
affects the adsorption of individual PAH onto particulate matter in
the atmosphere and their retention on particulate matter during
sampling on filters (Thrane & Mikalsen, 1981). Vapour pressure
increases markedly with ambient temperature (Murray et al., 1974),
which additionally affects the distribution coefficients between
gaseous and particulate phases (Lane, 1989). Solubility in water tends
to decreases with increasing molecular mass. For additional
information, refer to section 4.1.
PAH are chemically inert compounds (see also section 4.4). When
they react, they undergo two types of reaction: electrophilic
substitution and addition. As the latter destroys the aromatic
character of the benzene ring that is affected, PAH tend to form
derivatives by the former reaction; addition is often followed by
elimination, resulting in net substitution. The chemical and
photochemical reactions of PAH in the atmosphere have been reviewed
Table 4. Physical and chemical properties of polycyclic aromatic compounds covered in this monograph, ranked by molecular mass
Compound Colour Melting- Boiling- Vapour Densityc n-Octanol: Solubility in Henry's law
pointa point pressure water water at 25°C constant at
(°C) (°C) (Pa at 25°C) partition (µg/litre)d 25°C (kPa)
coefficient
(log Kow)
Naphthalene Whiteb 81 217.9c 10.4g 1.15425 h 3.4j 3.17 x 104 4.89 x 10-2 k
Acenaphthylene 92-93 8.9 x 10-1 g 0.89916/2 h 4.07f 114 x 10-3 l
Acenaphthene Whiteb 95 279h 2.9 x 10-1 g 1.02490/4 h 3.92f 3.93 x 103 1.48 x 10-2 k
Fluorene Whitee 115-116 295e 9.0 x 10-2 g 1.2030/4 h 4.18m 1.98 x 103 1.01 x 10-2 n
Anthracene Colourlesso 216.4 342e 8.0 x 10-4 g 1.28325/4 h 4.5j 73 7.3 x 10-2 n
Phenanthrene Colourlessp 100.5 340h 1.6 x 10-2 g 0.9804 h 4.6j 1.29 x 103 3.98 x 10-3 k
1-Methylphenanthrene 123 354-355y 5.07s 255 (24°C)t
Fluoranthene Pale yellowh 108.8 375h 1.2 x 10-3 g 1.2520/4 h 5.22u 260 6.5 x 10-4
(20 °C)w
Pyrene Colourlesse 150.4 393h 6.0 x 10-4 g 1.27123/4 h 5.18j 135 1.1 x 10-3 n
Benzo[a]fluorene Colourlessx 189-190h 399-400y 5.32z 45
Benzo[b]fluorene Colourlessx 213.5 401-402y 1.226aa 5.75z 2.0
Benzo[ghi]fluoranthene Yellowbb 128.4 432cc 1.34523 dd
Cyclopenta[cd]pyrene Orangex 170 439ee
Benz[a]anthracene Colourlessb 160.7 400b 2.8 x 10-5 g 1.226aa 5.61f 14
Benzo[c]phenanthrene Colourlessx 66.1 1.265ff
Chrysene Colourless 253.8 448h 8.4 x 10-5 1.27420/4 e 5.91u 2.0
with blue (20°C)gg
fluoresenceb
Triphenylene Colourlessx 199 425bb 1.3p 5.45hh 43
5-Methylchrysene Colourlessx 117.1 458ii 62 (27°C)jj
Benzo[b]fluoranthene Colourlessi 168.3 481kk 6.7 x 10-5 6.12f 1.2ll 5.1 x 10-5
(20°C)gg (20°C)w
Benzo[j]fluoranthene Yellowb 165.4 480ee 2.0 x 10-6 l 6.12mm 2.5nn
Table 4. (continued)
Compound Colour Melting- Boiling- Vapour Densityc n-Octanol: Solubility in Henry's law
pointa point pressure water water at 25°C constant at
(°C) (°C) (Pa at 25°C) partition (µg/litre)d 25°C (kPa)
coefficient
(log Kow)
Renzo[k]fluoranthene Pale yellowh 215.7 480h 1.3 x 10-8 6.84m 0.76f 4.4 x 10-5
(20°C)oo (20°C)w
Benzo[a]pyrene Yellowishe 178.1 496kk 7.3 x 10-7oo 1.351pp 6.50u 3.8 3.4 x 10-5
(20°C)
Benzo[e]pyrene Pale yellowx 178.7 493kk 7.4 x 10-7qq 6.44rr 5.07 (23°C)tt
Perylene Yellow to 277.5 503ss 1.35v 5.3uu 0.4
colourlessc
Anthanthrene Golden yellowbb 264 547yy 1.39v
Benzo[ghi]perylene Pale yellow- 278.3 545ii 1.4 x 10-8 ww 1.32920 xx 7.10u 0.26 2.7 x 10-5
greenbb (20°C)w
Indeno[1,2,3-cd]pyrene Yellowi 163.6 536yy 1.3 x 10-8 6.58f 62f 2.9 x 10-5
(20°C)gg (20°C)w
Dibenz[a,h]anthracene Colourlessi 266.6 524yy 1.3 x 10-8 1.282i 6.50zz 0.5 (27°C)jj 7 x 10-6 l
(20°C)
Coronene Yellowh 439 525aaa 2.0 x 10-10 qq 1.37b 5.4uu 0.14
Dibenzo[a,e]pyrene Pale yellowh 244.4 592vv
Dibenzo[a,h]pyrene Golden yellowi 317 596vv
Dibenzo[a,i]pyrene Greenish-yellowishi 282 594vv 3.2 x 10-10 mm 7.30hh 0.17l 4.31 x 10-6 l
Dibenzo[a,l]pyrene Pale yellowi 162.4 595vv
a From Karcheret al. (1985); Karcher (1988)
b From Lewis (1992)
c When two temperatures are given as superscripts, they indicate the specific gravity, i.e. the density of the substance at the first
reported temperature relative to the density of water at the second reported temperature. When there is no value, or only one, for
temperature, the datum is in grains per millilitre, at the indicated temperature, if any.
Table 4 (continued)
d From Mackay & Shiu (1977), except where noted
e From Budavari (1989)
f From National Toxicology Program (1993)
g From Sonnefeld et al. (1983)
h From Lide (1991)
i From IARC (1977)
j From Karickhoff et al. (1979)
k From Mackay et al. (1979)
l Calculated by Syracuse Research Center; from National Toxicology Program (1993)
m Calculated as per Leo et al. (1971); from US Environmental Protection Agency (1980)
n From Mackay & Shiu (1981)
o When pure, colourless with violet fluorescence; from Budavari (1989)
p From Hawley (1987)
q From National Institute for Occupational Safety and Health and Occupational Safety and Health Administration (1981)
r From Kruber & Marx (1938)
s Calculated by Karcher et al. (1991)
t From May et al. (1978)
u From Bruggeman et al. (1982)
v At ambient temperature; from Inokuchi & Nakagaki (1959)
w From Ten Hulscher et al. (1992)
x Personal observation by J. Jacob, Germany, on high-purity, certified reference materials
y From Kruber (1937)
z Calculated by Miller et al. (1985)
aa From Schuyer et al. (1953)
bb From IARC (983)
cc From Kruber & Grigoleit (1954)
dd From Ehrlich & Beevers (1956)
ee Reported by Grimmer (1983a)
ff From Beilstein Institute for Organic Chemistry (1993)
gg Reported by Sims & Overcash (1983)
hh Calculated by Yalkowsky & Valvani (1979)
ii Calculated by White (1986)
jj From Davis et al. (1942)
kk From review by Bjorseth (1983); original references cited by White (1986)
ll Temperature not given; reported by Sims & Overcash (1983)
mm Calculated by National Toxicology Program (1993)
nn Temperature not given; unpublished result cited by Wise et al. (1981)
oo From US Environmental Protection Agency (1980)
Table 4 (continued)
pp From Kronberger & Weiss (1944)
qq From review of Santodonato et al. (1981)
rr Calculated by Ruepert et al. (1985)
ss From Verschueren (1983)
tt From Schwarz (1977)
uu From Brooke et al. (1986)
vv From Agency for Toxic Substances and Disease Registry (1990)
xx From White (1948)
yy Estimated from gas chromatographis retention time; from Grimmer (1983a)
zz From Means et al. (1980)
aaa From Von Boente (1955)
(Valerio et al., 1984; Lane, 1989). After photodecomposition in the
presence of air and sunlight, a number of oxidative products are
formed, including quinones and endoperoxides. PAH have been shown
experimentally to react with nitrogen oxides and nitric acid to form
the nitro derivatives of PAH, and to react with sulfur oxides and
sulfuric acid (in solution) to form sulfinic and sulfonic acids. PAH
may also be attacked by ozone and hydroxyl radicals present in the
atmosphere. The formation of nitro-PAH is particularly important owing
to their biological impact and mutagenic activity (IARC, 1984a,
1989a). In general, the above reactions are of interest with regard to
the environmental fate of PAH, but the results of experimental studies
are difficult to interpret because of the complexity of interactions
occurring in environmental mixtures and the difficulty in eliminating
artefacts during analytical determinations. These reactions are also
considered to be responsible for possible losses of PAH during ambient
atmospheric sampling (see section 2.4.1.1).
2.3 Conversion factors
Atmospheric concentrations of PAH are usually expressed as
micrograms or nanograms per cubic meter. At 25°C and 101.3 kPa, the
conversion factors for a compound of given relative molecular mass are
obtained as follows:
ppb = µg/m3 × 24.45/relative molecular mass
µg/m3 = ppb × relative molecular mass/24.45.
For example, for benzo [a]pyrene, 1 ppb = 10.3 µg/m3 and
1 µg/m3 = 0.0969 ppb.
2.4 Analytical methods
Tables 5 and 6 present as examples a limited number of methods
that are applied to 'real' samples of different matrices. The methods
and sources were selected, as far as possible, according to the
following criteria: accessibility of the bibliographic source,
completeness of the description of the procedure, practicability with
common equipment for this type of analysis (even if experienced
personnel are required), recency, and whether it is an official,
validated, or recommended method.
2.4.1 Sampling
2.4.1.1 Ambient air
The physical state of PAH in the atmosphere must be considered
when selecting the sampling apparatus. Compounds with five or more
rings are almost exclusively adsorbed on suspended particulate matter,
whereas lower-molecular-mass PAH are partially or totally present in
the vapour phase (Coutant et al., 1988). When ambient air is
monitored, it is common practice to monitor only particle-bound PAH
Table 5. Analytical methods for polycyclic aromatic hydrocarbons in air
Matrix Sampling, extraction Clean-up Analysis Limit of Reference
detectiona
Ambient air Sampling on GF+PUF, at 45 m3/h; Liquid-liquid partition GC/MS Yamasaki et al.
Soxhlet extraction with cyclohexane with cyclohexane: (1982)
H2O:DMSO, then CC
with SiO2
Sampling on GF+PUF, at 30 m3/h; CC with Al2O3 + HPLC/FL 0.01-0.7 Keller &
Soxhlet extraction with petroleum ether SiO2 ng/m3 Bidleman (1984)
(GF) and DCM (PUF)
Sampling on GF (particle diameter TLC with SiO2 HPLC/UV 0.01-0.3 Greenberg et al.
< 15 µm), at 68 m3/h; Soxhlet extraction + FL ng/m3 (1985)
with cyclohexane, DCM, and acetone
Sampling on GF at 83 m3/h; sonication TLC with SiO2 GC/FID Valerio et al.
(cyclohexane) (1992)
Emissions Sampling by glass wool, condenser, Liquid-liquid partition GC/FID 10 ng/m3 Colmsjo et al.
(municipal and XAD-2; extraction with acetone with DMF (1986a)
incinerator) (glass-wool and XAD-2, by Soxhlet)
Vehicle Sampling by GF and condenser; liquid- CC with SiO2 and GC/FID 2.5-20 ng Grimmer et al.
exhaust liquid partition with acetone:H2O: Selphadex LH-20 per test (1979)
cyclohexane and DMF:H,O:cyclohexane
Sampling in dilution tunnel by Liquid-liquid partition GC/FID or Westerholm et
PTFE-coated GF and condenser; Soxhlet with cyclohexane: GC/MS al. (1988)
extraction of filter (DCM) and H2O:DMF
condensate (acetone); remaining
aqueous phase extracted with DCM
Table 5. (continued)
Matrix Sampling, extraction Clean-up Analysis Limit of Reference
detectiona
Indoor air Sampling on GF (particle diameter TLC with acetyloxylated Spectrofluorescence Lioy at al. (1988)
< 10 µm) at 10 l/min; sonication cellulose (benzo[a]pyrene only)
(cyclohexane)
Sampling on quartz-fibre filtre and GC/MS Chuang at al.
XAD-4 at 226 l/min; Soxhlet extraction (1991)
with DCM
Sampling on PTFE-coated GF at filtration; then CC HPLC/FL 0.02-0.12 Daisey & Gundel
20 l/minfor 24 h; Soxhlet extraction SiO2 cartridge), ng/m3 b (1993)
with DCM optional
Sampling on GF and PUF, at 20 litres/min GC/FID, GC/MS US Environmental
for 24 h; Soxhlet extraction (10% ether: or HPLC/UV + FL Protection Agency
n-hexane) (1990)
Workplace air Sampling on PTFE filter and XAD-2 GC/FID 0.3-0.5 µg NIOSH (1994a,b)
at 2 l/min; sonication or Soxhlet per sample
extraction of filterc, extraction of HPLC/UV 0.05-0.8 µg
XAD-2 with toluene (for GC) or + FL per sample
acetonitrile (for HPLC)
Workplace air Sampling on filter (GF, quartz fibre, CC (XAD-2) GC/FID approx 0.5 German
PTFE or silver membrane) at 2 litres/min; µg/m3 Research
sonication or Soxhlet extraction with Commission
cyclohexane or toluene (1991)
Tobacco Sampling by acetone trap; solvent CC (SiO2 + Sephadex GC/MS + ng/cigarette Lee at al. (1976b)
smoke partition scheme (acids/bases/neutral LH-20); then NMR
compounds/PAH) HPLC/UV
Table 5 (continued)
GC glass fibre; PUF, polyurethane foam; DMSO, dimethyl sulfoxide; CC, column chromatography; GC, gas chromatography;
MS, mass spectrometry; DCM, dichloromethane; HPLC, high-performance liquid chromatography; FL, fluorescence detection;
TLC, thin-layer chromatography; UV, ultraviolet detection; FID, flame-ionization detection; DMF, N-dimethylformamide;
PTFE, polytetrafluoroethylene; NMR, nuclear magnetic resonance
a Various PAH
b The following PAH can be determined: fluoranthene, pyrene, chrysene, benzo[e]pyrene, benzo[b]fluoranthene,
benzo[k]fluoranthene, benzo[a]pyrene, benzo[ghi]perylene, indeno[1,2,3-cd]pyrene.
c Appropriate solvent must be determined by recovery tests on specific samples.
Table 6. Analytical methods for polycyclic aromatic hydrocarbons in matrices other than air
Matrix Extraction Clean-up Analysis Limit of Reference
detectiona
Tap-water Preconcentration on PUF; Liquid-liquid partition GC/FID or TLC 0.1 ng/litre Basu & Saxena
extraction (with acetone and with cyclohexane: (Al2O3: acetyl (1978a)
cyclohexane) H2O:methanol and celluose) with FL
cyclohexane: H2O: detector
DMSO; then CC
OWN)
Groundwater Liquid-liquid partition with CC (SiO2), if needed GC/FID µg/litre level US Environmental
DCM GC/MS 10 µg/litre Protection Agency
HPLC/UV + FL 0-01-2 µg/litre (1986a)
Wastewater Liquid-liquid partition with CC (SiO2), if needed GC/FID or 0.01 -0.2 µg/litre US Environmental
DCM HPLC/UV+FL (by HPLC) Protection Agency
(1984a)
Seawater Liquid-liquid partition with CC (SiO2 + Al2O2) GC/FID or Desideri at al.
n-hexane or CCl4 HPLC/UV (1984)
Soil Sonication with DCM CC (Al2O2); then GC/MS 1 µg/kg Vogt at
liquid-liquid partition al. (1987)
(n-hexane:H2O:DMSO)
Soxhlet extraction with DCM CC (Florisil cartridge) HPLC/UV + FL 1 µg/kg Jones et a[.
(1989a)
Sediment Soxhlet extraction with DCM CC (SiO2 + Sephadex HPLC/DAD/MS Quilliam & Sim
LH20) (1988)
Sonication with acetone: CC (Florisil) HPLC/UV + FL 1-160 µg/kg Marcus et al.
n-hexane (1988)
Table 6. (continued)
Matrix Extraction Clean-up Analysis Limit of Reference
detectiona
Meat and fish (I) digestion (alcoholic KOH), Liquid-liquid partition GC/FID 2.5-20 ng/ Grimmer &
products (I), then liquid-liquid partition with cyclohexane: sample Bohnke (1979b)
vegetable oils (methanol: H2O:cyclohexane) H2O:DMF); then CC
(II), and sewage (II) dissolution in cyclohexane (SiO2 + Sephadex
sludge (III) (III) refluxing with acetone LH20)
Food (total Refluxing with alcoholic KOH, Liquid-liquid partition HPLC/FL 0.002-0.7 µg/kg Dennis et al.
diet) extraction with isooctane (isooctane:H2O:DMF); (1983)
then CC (SiO2 cartridge)
Saponification with alcoholic CC (SiO2) HPLC/FL 0.03-2 µg/kg de Vos et al.
KOH, extraction with (1990)
cyclohexane
Saponikation wit ahoholic CC (Florisil); then TLC/UV+FL 0.02 µg/kg Howard (1979);
KOH, extraction with liquid-liquid partition (benzo[a]pyrene) Fazio (1990)
isooctane isooctane:H2O:DMSO)
Seafood Digestion with alcoholic KOH, CC (Al2O3 + SiO2 + HPLC/FL 0.01-0.6 µg/kg Perfetti et al.
extraction with TCTFE C18 cartridge) (1992)
Smoked food Digestion with alcoholic KOH, CC (Al2O3 + SiO2); HPLC/UV+FL 0.03-0.4 Joe et al. (1984)
extraction with TCTFE liquid-liquid partition µg/kg
(cyclohexane:H2O:DMSO)
Refluxing with cyclohexane or Liquid-liquid partition TLC/FLb (only 0 0.5 ng/kg IUPAC (1987)
TCTFE, extraction with with cyclohexane:H2O: benzo[alpyrene)
methanol:H2O DMF); then CC (SiO2)
Solid waste Soxhlet extraction with DCM CC (SiO2), if needed GC/FID µg/kg level US Environmental
or sonication with GC/MS 1-200 mg/kg Protection Agency
DGM:acetone HPLC/UV + FL µg/kg level (1986b)
Table 6. (continued)
Matrix Extraction Clean-up Analysis Limit of Reference
detectiona
Mineral oil and Liquid-liquid partition with CC (SiO2 + Sephadex GC/FID 100 ng/kg Grimmer &
fuel cyclohexane:H2O:DMF) LH20) Bohnke (1979a)
Medicinal oil Liquid-liquid partition CC (SiO2 + Sephadex HPLC/FL + 0.2-200 ng/kg Geahchan at al.
(cyclohexane: H2O:DMF) LH20) GC/FID (1991)
Plants Sonication (acetonitrile), CC (SiO2) GC/FID Coates et al.
extraction with pentane (1986)
Urine Adjusted to pH3, extraction CC (SiO2 cartridge) HPLC/FLc Becher & Bjorseth
in C18 cartridge, metabolites (1983)
reduced with hydriodic acid
Urine and Addition of HCl, refluxing CC (SiO2) + Sephadex GC/MSd Jacob at al. (1989)
faeces with toluene, addition of LH20
methanol and diazomethanol in
ether (faeces saponified before
acidification)
Tissue Homogenization (benzene: CC (Florisil) GC/MS 5-50 µg/kg Liao et al. (1988)
n-hexane)
Skine Sonication of exposure pads HPLC/FL 6 ng/cm2 Jongeneelen et al.
with DCM, centrifugation (1988a)
Table 6. (continued)
PUF, polyurethane foam; DMSO, dimethyl sulfoxide; CC, column chromatography; GC, gas chromatography; FID, flame ionization detection; FL,
fluorescence detection; DCM, dichloromethane; MS, mass spectrometry; UV, ultraviolet detection; DAD, diode-array detector; DMF,
N-dimethylformamide; TLC, thin-layer chromatography; TCTFE, 1,1,2-trichlorotrifluoroethane
a Various PAH
b Benzo[a]pyrene content estimated to be > 0.6 µg/kg (screening method)
c Determination of unmetabolized and metabolized PAH
d Determination of pyrene and 1-hydroxypyrene
e Measurement of skin contamination with soft polypropylene exposure pads mounted on skin sites
(Menichini, 1992a), probably because of the increased work involved in
trapping volatile compounds, both in assembling the sampling unit and
in analysing samples, and also because lighter compounds are of lesser
toxicological interest. Of the PAH that are classified as 'probably'
and 'possibly' carcinogenic to humans (IARC, 1987), only
benz [a]anthracene is found at significant levels in the vapour phase
(Van Vaeck et al., 1984; Coutant et al., 1988; Baek et al., 1992).
Sampling is generally performed by collecting total suspended
particulate matter for 24 h on glass-fibre filters by means of
high-volume samplers. Other filters that have been used are quartz
fibres (Hawthorne et al., 1992), polytetrafluoroethylene (PTFE)
membranes (Benner et al., 1989; Baek et al., 1992), and, in
comparisons, PTFE-coated glass fibres (Lindskog et al., 1987; De Raat
et al., 1990). The effects of these materials on the decomposition of
PAH during sampling have been compared (see section 2.2). Some studies
indicated that higher recoveries are obtained with PTFE and
PTFE-coated filters (Lee et al., 1980a; Grosjean, 1983); however, more
recent investigations did not confirm this finding (Lindskog et al.,
1987; Ligocki & Pankow, 1989; De Raat et al., 1990). Moreover, when
cellulose acetate membrane filters were compared with glass-fibre
filters, they had similar efficiency for collecting heavier PAH, but
the former had greater efficiency for collecting three- and four-ring
compounds (Spitzer & Dannecker, 1983).
The most widely used method for trapping vapour-phase PAH is
adsorption on plugs of polyurethane foam located behind the filter
(Keller & Bidleman, 1984; Chuang et al., 1987; De Raat et al., 1987a;
Benner et al., 1989; Hawthorne et al., 1992). This method is widely
accepted, probably because of the low pressure drop, the low blanks,
the low cost, and ease of handling. Among the other sorbents tested
(see also reviews by Leinster & Evans, 1986; Davis et al., 1987),
further polymeric materials have received particular attention,
including Amberlite XAD-2 resin, which is a valid alternative to
polyurethane foam (Chuang et al., 1987), Porapak PS, which has been
successfully tested in combination with a silanized glass-fibre filter
at a flow rate of 2 m3/h (Jacob et al., 1990a), and Tenax(R) (Baek
et al., 1992).
The trapped vapours contain both the PAH that were initially
present in the vapour phase and those already collected on the filter
and volatilized during sampling (the 'blowing-off' effect) (Van Vaeck
et al., 1984; Coutant et al., 1988). The amount of PAH found in the
vapour phase increases with ambient temperature (Yamasaki et al.,
1982). Samplers incorporating an annular denuder, as well as a filter
and back-up trap, have been used to investigate phase distribution and
artefact formation (Coutant et al., 1988, 1992).
Sampling times are restricted to 24 h in order to avoid sample
degradation and losses. Grimmer et al. (1982) proposed a useful method
for controlling losses due to chemical degradation and volatilization
from filters which is based on the invariability of PAH profiles (i.e.
the ratio of all PAH to one another) at different collection times.
The adsorption of gas-phase PAH onto a quartz-fibre filter has been
investigated as a possible sampling artefact (Hart & Pankow, 1994);
the results suggested that overestimation of particle-associated PAH
can be avoided by replacing quartz-fibre filters with a PTFE membrane
filters, or can be corrected by using back-up quartz-fibre filters.
Elutriators and cascade impactors have been used to achieve
particle size-selective sampling of PAH (Menichini, 1992a).
Instruments designed as additions to high-volume samplers are
available, including 'PM10' inlets, which allow collection of airborne
particles with a 50% cutoff at the aerodynamic diameter of 10 m (US
Environmental Protection Agency, 1987a; Lioy et al., 1988; Hawthorne
et al., 1992), and cascade impactors (Van Vaeck et al., 1984; Catoggio
et al., 1989).
When PAH are collected in indoor air, samplers operating at 20 or
200 litre/min are commonly used. The filter and sorbent materials are
those used for outdoor air (Wilson et al., 1991; see also Table 5).
The sampling step is by far the most important source of
variability in the results of atmospheric PAH determination. Most
investigations are difficult to compare because of differences in
factors such as season, meteorological conditions, time of day, number
and characteristics of sampling sites, and sampling parameters
(Menichini, 1992a). Passive biological sampling has been investigated
as an approach to long-term sampling of atmospheric PAH (Jacob &
Grimmer, 1992), and preliminary correlation factors have been
determined by comparing the PAH profiles in biological (plants,
particularly) and air samples. Of the matrices tested, spruce sprouts
were found to be the most suitable.
2.4.1.2 Workplace air
The general considerations described for ambient air are also
valid for the working environment. Less volatile PAH may be retained
than in ambient air because of the high temperatures that are often
found at the workplace. In the potroom of an aluminium plant where
Sderberg electrodes were used, 42% of benz [a]anthracene was found
in the vapour phase (Andersson et al., 1983), and in an iron foundry
at a site where the temperature of the PAH source was 600-700°C, four-
to seven-ring PAH represented about 70% of the total in the vapour
phase (Knecht et al., 1986).
Glass-fibre or PTFE filters are usually used to collect
particle-bound PAH. A number of back-up systems can be used to
efficiently trap volatile PAH, including liquid impingers and solid
sorbents such as Tenax(R)-GC, Chromosorb, and XAD-2 (Bjorseth &
Becher, 1986; Davis et al., 1987). The latter seems to be the most
practical. The US National Institute for Occupational Safety and
Health (1994a,b) recommended use of a PTFE-laminated membrane followed
by a tube containing two sections of XAD-2. For sampling in bright
sunlight, opaque or foil-wrapped filter cassettes can be used to
prevent degradation.
The exposure of workers is estimated by taking air samples at
various locations in the workplace or by personal sampling, in which
workplace air is pumped through a filter attached to clothing close to
the breathing zone for a specified time. Both procedures provide an
estimate and not a precise measurement of an individual's exposure.
2.4.1.3 Combustion effluents
The validity of a collected sample, i.e. the degree to which it
reflects the 'true' composition of the emission, is a crucial factor
in the determination of PAH in emissions. The problems associated with
efficient collection of volatile PAH are enhanced when sampling
combustion effluents, such as stack gases and vehicle exhausts,
because of the elevated temperatures at sampling positions.
A sampling device for stack gases is constituted by a glass- or
quartz-fibre filter, followed by a special unit which generally
consists in a cooler for collecting condensable matter and an
adsorbent cartridge (Colmsjö et al., 1986a; Funcke et al., 1988).
Tenax(R) has been used as an adsorbent (Jones et al., 1976), but
XAD-2 seems to be more suitable (Warman, 1985) and is generally
preferred. Two sampling procedures have been described in detail by
the US Environmental Protection Agency (1986c). In the first
('Modified method 5 sampling train'), the unit basically includes a
glass- or quartz-fibre filter kept at around 120°C, a condenser coil
that conditions the gas at a maximum of 20°C, and a bed of XAD-2
jacketed to maintain the internal gas temperature at about 17°C. The
second ('Source assessment sampling system') is often used for
stationary investigations (Warman, 1985). The apparatus consists of a
stainless-steel probe, which enters an oven containing the filter,
preceded by three cyclone separators in series, with cutoff diameters
of 10, 3, and 1 m; the volatile organic compounds are cooled and
trapped on XAD-2. The sorbent is followed by a condensate collection
trap and an impinger train.
Motor vehicle exhausts are sampled under laboratory conditions,
by chassis or engine dynamometer testing. Standard driving cycles are
employed to simulate on-road conditions (Stenberg, 1985; see also
section 3.2.7.2).
Two basic techniques have been used to collect, sample, and
analyse exhaust (Levsen, 1988; IARC, 1989a). In the first-raw gas
sampling-the exhaust pipe is connected directly to the sampling
apparatus; undiluted emissions are cooled in a condenser and then
allowed to pass through a filter for collection of particulates
(Grimmer et al., 1979, 1988a; Society of German Engineers, 1989). A
second technique-dilution tube sampling-is now often used, in which
hot exhaust is diluted with filtered cold air in a tunnel, from which
samples are collected isokinetically. This technique simulates the
process of dilution that occurs under real conditions on the road (US
Environmental Protection Agency, 1992a).
Particles are almost always collected on glass-fibre, glass-fibre
with PTFE binder, quartz-fibre filters, or PTFE membranes; the latter
have been reported to be particularly efficient and chemical inert
(Lee & Schuetzle, 1983). Glass-fibre filters impregnated with liquid
paraffin are also used (Grimmer et al., 1979; Society of German
Engineers, 1989). Vapour-phase PAH (Stenberg, 1985) may be collected
by cryo-condensation (Stenberg et al., 1983) or on an adsorbent trap
with a polymeric material such as XAD-2 (Lee & Schuetzle, 1983).
Artefacts may be introduced during collection on filters as a
result of chemical conversion of PAH, particularly into nitro-PAH and
oxidation products (Lee & Schuetzle, 1983; Schuetzle, 1983; IARC,
1989a). These effects have not been fully evaluated.
2.4.1.4 Water
The concentrations of PAH in uncontaminated groundwater supplies
and in drinking-water are generally very low, at 0.1 and 1 ng/litre
(see sections 5.1.2.1 and 5.1.2.2). This implies that serious errors
arising from adsorption losses and contamination occur during
collection and storage of samples or that a preconcentration step may
be needed to enrich the sample. It is recommended that sampling be
performed on-site, directly in the extraction vessel (Smith et al.,
1981).
Various solid sorbents have been successfully used for
preconcentration (Smith et al., 1981), including Tenax(R)-GC,
prefiltered if necessary (Leoni et al., 1975); XAD resins (Griest &
Caton, 1983); open-pore polyurethane foam (Basu et al., 1987); and
prepacked disposable cartridges of bonded-phase silica gel (Chladek &
Marano, 1984; Van Noort & Wondergem, 1985a). Solid sorbents have
limitations when the sample contains suspended material, since
adsorbed PAH may be lost by filtration (Van Noort & Wondergem, 1985a).
2.4.1.5 Solid samples
Some foodstuffs (Liem et al., 1992), soil, sediment, tissues, and
plants usually require homogenization before a sample is extracted.
2.4.2 Preparation
As most environmental samples contain only small amounts of PAH,
sophisticated techniques are required for their detection and
quantification. Therefore, efficient extraction from the sample matrix
is usually followed by one or more purification steps, so that the
sample to be analysed is as free as possible from impurities and
interference. Many extraction and purification techniques and
combinations ('isolation schemes') have been described, validated, and
recommended, but no single scheme is commonly recognized as 'the best'
for a given matrix. The isolation schemes have been classified
according to groups of matrices (Jacob & Grimmer, 1979; Grimmer,
1983a), as summarized briefly below.
PAH are extracted from a sample (Lee et al., 1981; Santodonato et
al., 1981; Grimmer, 1983a; Griest & Caton, 1983) with:
- a Soxhlet apparatus, from filters loaded with particulate matter,
vehicle exhausts, or sediments;
- directly by liquid-liquid partition, for water samples; or
- after complete dissolution (e.g. fats and vegetable and mineral
oils) or alkaline digestion of samples (e.g. meat products) by a
selective solvent such as N,N-dimethylformamide (Natusch &
Tomkins, 1978) or dimethyl sulfoxide. Complete extraction of PAH
from samples such as soot emitted by diesel engines, carbon
blacks, and other carbonaceous materials is particularly
difficult.
Extraction of PAH from soil, sediment, sewage sludge, and vehicle
exhaust particulates by refluxing with various solvents has been
investigated. In all cases, toluene was found to be the most efficient
solvent, especially for vehicle exhaust (Jacob et al., 1994).
As an alternative to Soxhlet extraction, ultrasonic extraction
(Griest & Caton, 1983) has advantages in terms of reduced time of
extraction (minutes versus hours) and superior recovery efficiency and
reproducibility, particularly for solid samples and filters loaded
with particulate matter. Comparisons of techniques depend, however, on
the matrix, solvent, and experimental conditions.
Supercritical fluid extraction (Langenfeld et al., 1993) has
gained attention as a rapid alternative to conventional liquid
extraction from polyurethane foam sorbents (Hawthorne et al., 1989a),
soil (Wenclawiak et al., 1992), and other environmental solids such as
urban dust, fly ash, and sediment (Hawthorne & Miller, 1987). This
technique can also be directly coupled with on-column gas
chromatography (see section 2.4.3.1); the extract is quantitatively
transferred onto the gas chromatographic column for a rapid (< 1 h)
analysis with maximal sensitivity. This technique has been used for
urban dust samples (Hawthorne et al., 1989b).
Extracted samples are usually purified from interfering
substances by adsorption column chromatography. The classical
sorbents, alumina and silica gel, are widely used. In addition, the
hydrophobic Sephadex LH-20 has been found to be suitable for isolating
PAH from nonaromatic, nonpolar compounds, which is important if the
sample is analysed by gas chromatography (Grimmer & Böhnke, 1979a); It
has also been used in partition chromatography as a carrier of the
stationary phase, to separate PAH from alkyl derivatives (Grimmer &
Böhnke, 1979b). Chromatography on silica gel and Sephadex is often
combined (Jacob & Grimmer, 1979; Grimmer, 1983a).
Clean-up has also been achieved by eluting extracted samples
through XAD-2 (soil samples: Spitzer & Kuwatsuka, 1986), XAD-2 and
Sephadex LH-20 in series (foods: Vaessen et al., 1988), or Florisil
(food, water, and sediment samples: references given in Table 6).
Conventional chromatographic columns may be substituted by
prepacked commercial cartridges, which have advantages in terms of
time and solvents consumed and reproducibility. For example, silica
cartridges have been used to purify foodstuffs (Dennis et al., 1983),
urine (Becher & Bjorseth, 1983), vehicle emissions (Benner et al.,
1989), mineral oil mist (Menichini et al., 1990), and atmospheric
samples (Baek et al., 1992); soil samples have been cleaned up on
Florisil cartridges (Jones et al., 1989a).
Preparative thin-layer chromatography is also used for, e.g. air
particulates (see Table 5) and vegetable oils (Menichini et al.,
1991a).
Handling of samples in the absence of ultraviolet radiation is
recommended at all stages in order to avoid photodecomposition of PAH
(Society of German Engineers, 1989; US Environmental Protection
Agency, 1990; US National Institute for Occupational Safety and
Health, 1994a,b). It is also generally recommended that possible
sources of interference and contamination be controlled, particularly
from solvents (US Environmental Protection Agency, 1984a, 1986b,
1990), and that samples be refrigerated until extraction (US
Environmental Protection Agency, 1984a; US National Institute for
Occupational Safety and Health, 1994a,b).
2.4.3 Analysis
PAH are now routinely identified and quantified by gas
chromatography or high-performance liquid chromatography (HPLC). Each
technique has a number of relative advantages. Both are rather
expensive, particularly HPLC, and require qualified operating
personnel; nevertheless, they are considered necessary in order to
analyse 'real' samples for a large number of PAH with accuracy and
precision.
2.4.3.1 Gas chromatography
Excellent separation (< 3000 plates per meter) is obtained by
the use of commercially available fused silica capillary columns,
making it possible to analyse very complex mixtures containing more
than 100 PAH.
The most widely used stationary phases are the
methylpolylsiloxanes: especially SE-54 (5% phenyl-, 1%
vinyl-substituted) and SE-52 (5% phenyl-substituted), but SE-30 and
OV-101 (unsubstituted), OV-17 (50% phenyl-substituted), Dexsil 300
(carborane-substituted) and their equivalent phases are also used.
Chemically bonded phases are used increasingly because they can be
rinsed to restore column performance and undergo little 'bleeding' at
the high temperatures of analysis (about 300°C) that are required for
determining high-boiling-point compounds.
Nematic liquid crystal phases (Bartle, 1985) have also been used
to separate some isomeric compounds that are poorly resolved by
siloxane phases, such as chrysene and triphenylene on
N,N'-bis (para-methoxy-benzylidene)-a,a'-bi- para-toluidine
(Janini et al., 1975) and
N,N'-bis (para-phenylbenzylidene)-a,a'-bi- para-toluidine (Janini
et al., 1976).
Splitless or on-column injection is necessary to gain sensitivity
in trace analysis, the latter being preferred as it allows better
reproducibility. Flame ionization detectors are almost always used
because of the excellent linearity, sensitivity, and reliability of
their response. Since the signal is related linearly to the carbon
mass of the compound, PAH are recorded in proportion to their
quantities, and the chromatogram shows the quantitative composition of
the sample directly. Because flame ionization detectors are
non-selective, samples for gas chromatography must be highly purified.
Peak identification, which is done routinely from data on retention,
must be confirmed by analysing samples on a different gas
chromatographic column, by an independent technique, such as HPLC, or
by directly coupling a mass spectrometric detector to the gas
chromatograph (Lee et al., 1981; Olufsen & Bjorseth, 1983; Bartle,
1985; Hites, 1989).
Mass spectrometers have gained wide acceptance. They are powerful
tools for identifying compounds, especially when commercially
available libraries of reference spectra are used to match the spectra
obtained and to control the purity of a compound. As isomeric
compounds often have indistinguishable spectra, however, the final
assignment must also be based on retention.
On-line coupling of liquid chromatography, capillary gas
chromatography, and quadrupole mass spectrometry has been used to
determine PAH in vegetable oils (Vreuls et al., 1991).
2.4.3.2 High-performance liquid chromatography
The packing material considered most suitable for separating PAH
consists of silica particles chemically bonded to linear C18
hydrocarbon chains; selection of the appropriate phase has been
discussed in detail by Wise et al. (1993). Typically, 25-cm columns
packed with 5-m particles are used in the gradient elution technique,
and the mobile phase consists of mixtures of acetonitrile and water or
methanol and water ('reversed-phase HPLC'). As the efficiency of
separation that can be achieved with HPLC columns is much lower than
that with capillary gas chromatography, HPLC is generally less
suitable for separating samples containing complex PAH mixtures.
The advantages of HPLC derive from the capabilities of the
detectors with which it is used. Those most widely used for PAH are
ultraviolet and fluorescence detectors, generally arranged in series,
with flow-cell photometers or spectrophotometers. Both, but especially
the latter, are highly specific and sensitive: the detection limits
with fluorescence are at least one order of magnitude lower than those
with ultraviolet detection. The specificity of fluorescence detectors
allows the determination of individual PAH in the presence of other
nonfluorescing substances. In addition, since different PAH have
different absorptivity or different fluorescence spectral
characteristics at given wavelengths, the detectors can be optimized
for maximal response to specific compounds. This may prove
advantageous in the identification of unresolved components. In
particular, wavelength-programmed fluorescence detection, to measure
changes in excitation and emission wavelengths during a
chromatographic run (Hansen et al., 1991a), is being used for the
analysis of environmental samples (Wise et al., 1993). HPLC is
suitable to a limited degree for lower-molecular-mass compounds like
naphthalene, acenaphthene, and acenaphthylene, for which the detection
limits are relatively high (US Environmental Protection Agency,
1984a).
Owing to the selectivity of packing materials, various isomers
that cannot be separated efficiently on the usual capillary gas
chromatographic columns can be resolved at baseline and identified by
HPLC. Such isomers include the pairs chrysene-triphenylene and
benzo [b]fluoranthene-benzo [k]fluorathene (Wise et al., 1980).
Coupling of a mass spectrometer to HPLC has also been used in
detecting PAH (e.g., Quilliam & Sim, 1988).
As much information on isomeric structure can be obtained from
spectra seen during the elution of chromatographic peaks, an
ultraviolet diode-array detector has been used to confirm peaks (Dong
& Greenberg, 1988; Kicinski et al., 1989). For applications of HPLC to
determination of PAH, reference should be made to published reviews
(Lee et al., 1981; Wise, 1983, 1985).
2.4.3.3 Thin-layer chromatography
Thin-layer chromatography is commonly used only for identifying
individual compounds, such as benzo [a]pyrene, during screening
(IUPAC, 1987) or for identifying selected PAH, such as the six PAH
that WHO (1971) recommended be determined in drinking-water (Borneff &
Kunte, 1979). It is an inexpensive, quick analytical technique but has
low separation efficiency. The last parameter is improved by
two-dimensional processes (see, e.g. Borneff & Kunte, 1979).
Quantification may be done by spectrophotometric or
spectrofluorimetric methods in solution after the scrubbed substance
spot has been extracted (Howard, 1979; Fazio, 1990) or in situ by
scanning spectrofluorimetry (Borneff & Kunte, 1979).
Acetylated cellulose is the adsorbent that has been used most
widely for one-step separation of PAH fractions, and mixed aluminium
oxide and acetylated cellulose have been used for two-dimensional
development (Daisey, 1983).
2.4.3.4 Other techniques
A number of unconventional instruments and techniques based on
spectro-scopic principles have been developed as possible alternatives
to the chromatographic methods for PAH. Most of them are, however,
expensive, require skilled personnel, and are not yet considered
useful for the practising analyst (Wehry, 1983; Vo-Dinh, 1989).
Low-temperature luminescence in frozen solutions ('Shpol'skii
effect') has been used for various environmental samples, particularly
to identify methylated PAH isomers (Garrigues & Ewald, 1987; Saber et
al., 1987). This technique was used widely in the countries of former
Soviet Union (Dikun, 1967). Synchronous luminescence and room
temperature phosphorimetry have been reported to be simple,
cost-effective techniques for screening PAH (Vo-Dinh et al., 1984;
Abbott et al., 1986).
Infrared analysis, particularly Fourier transform infrared
spectroscopy coupled to gas chromatography (Stout & Mamantov, 1989),
and capillary supercritical fluid chromatography (Wright & Smith,
1989) have also been used. Various environmental samples have been
analysed by packed column supercritical fluid chromatography, with
rapid separation of PAH (Heaton et al., 1994).
2.4.4 Choice of PAH to be quantified
The choice of PAH depends on the purpose of the measurement. For
example, carcinogenic PAH are of interest in studies of human health,
but other, more abundant PAH may be of interest in ecotoxicological
studies. Quantification of a number of PAH is advantageous when the
profiles are to be correlated with sources and/or effects.
Table 7 lists the PAH that are required or recommended to be
determined at national or international levels. According to an EEC
(1980) Directive, which followed a WHO (1971) recommendation, the
concentrations of six reference compounds (also known as 'Borneff
PAH') must be measured in drinking-water in order to check its
compliance with the cumulative limit value for the PAH class. The
choice of these six PAH by WHO was not based on toxicological
considerations but on the fact that analytical investigations were
then largely confined to these relatively easily detected compounds
(WHO, 1984).
Table 7. Some polycyclic aromatic hydrocarbons required or recommended for determination by various authorities
Compound WHO/EECa US EPAb European Italyd Norwaye
(drinking- (waste Aluminium (air)
water) water) Associationc Health Environment
Acenaphthene X
Acenapthylene X
Anthracene X X X
Anthanthrene X X
Benz[a]anthracene X X X X X
Benzo[a]fluorene X
Benzo[a]pyrene X X X X X
Benzo[b]fluoranthene X X X X X X
Benzo[b]fluorene X
Benzo[c]phenanthrene X X
Benzo[e]pyrene X
Benzo[ghi]perylene X X X X
Benzo[j]fluoranthene X X X X
Benzo[k]fluoranthene X X X X X X
Chrysene X X X X
Cyclopenta[a]pyrene X X
Dibenzo[a,e]pyrene X X X
Dibenz[a,h]anthracene X X X X X
Dibenzo[a,h]pyrene X X X
Dibenzo[a,i]pyrene X X X
Dibenzo[a,l]pyrene X X
Fluoranthene X X X X
Fluorene X
Indeno[1,2,3-cd]pyrene X X X X X X
Naphthalene X X
Phenanthrene X X X
Pyrene X X X
Triphenylene X
Table 7 (continued)
a Recommended by WHO (1971) and required by an EEC (1980) Directive
b Required by the US Environmental Protection Agency (1984a) for the analysis of municipal and industrial
wastewater
c Recommended by the European Aluminium Association, Environmental Health and Safety Secretariat (1990)
d Recommended by the Italian National Advisory Toxicological Committee for health-related studies
(Menichini, 1992b)
e Recommended at the International Workshop on polycyclic aromatic hydrocarbons (State Pollution Control
Authority and Norwegian Food Control Authority, 1992) for studies of health and on the environment
The method required by the US Environmental Protection Agency
(1984a) for the analysis of municipal and industrial wastewater covers
the determination of 16 'priority pollutant PAH' considered to be
representative of the class. Outside the USA, this list of compounds
is often taken as a reference list for the analysis of various
environmental matrices.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
Appraisal
Coal and crude oils contain polycyclic aromatic hydrocarbons
(PAH) in considerable concentrations owing to diagenetic formation in
fossil fuels. The main PAH produced commercially are naphthalene,
acenaphthene, anthracene, phenanthrene, fluoranthene, and pyrene. The
release of PAH during production and processing, predominantly of
plasticizers, dyes, and pigments, is of only minor importance. Most
PAH enter the environment via the atmosphere from incomplete
combustion processes, such as:
- processing of coal and crude oil: e.g. refining, coal
gasification, and coking;
- heating: power plants and residential heating with wood, coal,
and mineral oil;
- fires: e.g. forest, straw, agriculture, and cooking;
- vehicle traffic; and
- tobacco smoking.
Industrial processes such as coal coking, aluminium, iron and
steel production, and foundries make important contributions to the
levels of PAH in the environment. An important indoor source of
exposure to airborne PAH, especially in developing countries, is
cooking fumes (see section 5.2).
The hydrosphere and the geosphere are affected secondarily by wet
and dry deposition. PAH are released directly into the hydrosphere,
for example during wood preservation with creosotes. Deposition of
contaminated refuse like sewage sludge and fly ash may cause further
emissions into the geosphere.
It is very difficult to identify a source on the basis of the
ratio of the measured concentrations of different individual PAH, and
such studies are in most cases inconclusive.
3.1 Natural occurrence
In some geographical areas, forest fires and volcanoes are the
main natural sources of PAH in the environment (Baek et al., 1991). In
Canada, about 2000 tonnes of airborne PAH per year are attributed to
natural forest fires (Environment Canada, 1994). On the basis of
samples from volcanoes, Ilnitsky et al. (1977) estimated that the
worldwide release of benzo [a]pyrene from this source was 1.2-14
t/year; no estimate was given of total PAH emissions from this source.
Coal is generally considered to be an aromatic material. Most of
the PAH in coal are tightly bound in the structure and cannot be
leached out, and the total PAH concentrations tend to be higher in
hard coal than in soft coals, like lignite and brown coal.
Hydroaromatic structures represent 15-25% of the carbon in coal. The
PAH identified include benz [a]anthracene, benzo [a]pyrene,
benzo [e]pyrene, perylene, and phenanthrene (Neff, 1979; Anderson et
al., 1986). Table 8 shows the typical contents of PAH in different
crude oils, such as those derived from coal conversion or from shale.
Table 8. Polycyclic aromatic hydrocarbon content of crude oils
from various sources
Compound PAH content (mg/kg) in crude oil from
Coala Petroleum Shale
Acenaphthene 1700/1800 147-348 147-903
Anthracene 4100 204-321 231-986
Anthanthrene Trace/< 800 NR 0.3
Benz[a]anthracene Trace/< 2200 1-7 1
Benzo[a]fluorene 2100/2500 11-22 53
Benzo[a]pyrene < 500/< 1200 0.1-4 3-192
Benzo[b]fluorene < 1500/3400 < 13 140
Benzo[c]phenanthrene < 600/< 2200 NR NR
Benzo[e]pyrene < 1200/1300 0.5-29 1-19
Benzofluorenesb < 500/< 1300 23 NR
Benzo[ghi]fluoranthene 3200 NR NR
Benzo[ghi]perylene 4300/6600 ND-8 1-5
ND-5
Chrysene < 1500/2500 7-26 3-52
Coronene NR 0.2 NR
Dibenz[a,h]anthracene NR 0.4-0.7 1-5
Fluoranthene < 1900/< 3700 2-326 6-400
Fluorene 5300/9900 106-220 104-381
1-Methylphenanthrene < 1200/< 5100 > 21 NR
Naphthalene 100/2800 402-900 203-1390
Perylene Trace/< 600 6-31 0.3-68
Phenanthrene 12 000/20 400 > 129-322 221-842
Pyrene 14 200/35 000 2-216 18-421
Triphenylene NR 3/13 0.5
From Guerin at al. (1978), Weaver & Gibson (1979), Grimmer at al.
(1983a), Sporstol et al. (1983), IARC (1985, 1989b)
Ranges represent at least three values; NR, not reported; ND, not
detected
a Two crude oils from coal conversion; single measurements
b Isomers not specified
Two rare PAH minerals have been described: the greenish-yellow,
fluorescent curtisite from surface vents of hot springs at Skagg
Springs, California, USA, and the bituminous mercury ore idrialite
from Idria, Yugoslavia, the two main components of which are chrysene
and dibenz [a,h]-anthracene. These minerals are assumed to have been
formed by the pyrolysis of organic material at depths below that at
which petroleum id generated (West et al., 1986).
3.2 Anthropogenic sources
3.2.1 PAH in coal and petroleum products
Commercial processing of coal leads first to coal-tars, which are
further processed to yield pitch, asphalt, impregnating oils
(creosotes for the preservation of wood), and residue oils such as
anthracene oil (IARC, 1985). The concentration of PAH in coal-tars is
generally ¾ 1%; naphthalene and phenanthrene are by far the most
abundant compounds, occurring at concentrations of 5-10%. Comparable
levels were detected in high-temperature coal-tar pitches. The PAH
content of soots is about one order of magnitude lower, and that of
carbon and furnace blacks ranges from about 1 to 500 mg/kg, pyrene
being present at the highest concentration (IARC, 1984a; Nishioka et
al., 1986). The PAH contents of some impregnating oils, bitumens,
asphalts, and roof paints are shown in Table 9. In bitumens, PAH
constitute only a minor part of the total content of polyaromatic
compounds.
Table 9. Polycyclic aromatic hydrocarbon content of impregnating oils, bitumens, asphalts,
and roof paints
Compound Concentration (mg/kg)
Impregnating Bitumens Road tar (asphalt, Roof
oils (oil-derived) coal-derived) paint
Anthracene 1600-22 500 0.01-0.32 4170-14 400 2380
Anthranthrene NR Trace-1.8 NR NR
Benz[a]anthracene 169-11 700 0.14-35 6820-24 100 6640
Benzo[a]pyrene 45-3490 0.1-27 5110-10 400 5950
Benzo[b]fluoranthene 42-3630 5 4490-10 900 5420
Benzo[e]pyrene 65-2020 0.03-52 3300-6750 3820
Benzo[gh]perylene 57-570 Trace-15 2390-2730 3270
Benzo[k]fluoranthene 24-2610 0.024-0.19 3170-7650 4470
Chrysene NR 0.04-34 NR
Chrysene + 779-12 900 NR 6820-26 100 7700
triphenylene
Coronene NR 0.2-2.8 NR NR
Fluoranthene 703-85 900 0.15-5 23 500-61 900 12 100
Fluorene 8040-58 400 NR 6310-15 500 2220
Indeno[1,2,3-cd]pyrene 57-273 Trace 3100-3530 3320
Perylene 66-744 0.08-39 1550-2300 1730
Phenanthrene 7070-159 300 0.32-7.3 20 300-52 500 8180
Pyrene 604-46 400 0.08-38 15 100-42 500 8960
Triphenylene NR 0.3-7.6 NR NR
From IARC (1985), Lehmann et al. (1986), Knecht & Woitowitz (1990);
NR, not reported; ranges represent at least three values
The concentrations of PAH in petrol and diesel fuels for vehicles
and in heating oils are several parts per million. Almost all
compounds are present at < 1 mg/kg; only phenanthrene, anthracene,
and fluoranthene are sometimes found at > 10 mg/kg (Herlan, 1982).
The PAH levels in unused engine lubricating oils are of the same order
of magnitude. During the use of petrol-fuelled engine oils, the PAH
content rises dramatically, by 30-500 times; in comparison, the total
PAH levels in used diesel-fuelled engine oils were only 1.4-6.1 times
greater than that in an unused sample. The major constituents of used
oils are pyrene and fluoranthene, although benzo [b]fluoranthene,
benzo [j]-fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene, and
dibenz [a,h]anthracene were also detected at considerable
concentrations (IARC, 1984a; Carmichael et al., 1990).
PAH have also been found in machine lubricating and cutting oils,
which is of interest for the estimation of exposure in the workplace.
The concentrations were < 7 mg/kg, although phenanthrene may have
been present at a higher level (Grimmer et al., 1981a; Rimatori et
al., 1983; Menichini et al., 1990; Paschke et al., 1992).
PAH were detected in coloured printing oils, the concentrations
of individual compounds varying between < 0.0001 and 63 mg/kg
(Tetzen, 1989). By far the most abundant compounds were fluoranthene
and pyrene (> 1 mg/kg); benzo [ghi]fluoranthene,
cyclopenta [cd]pyrene, benz [a]anthracene, benzo [c]-phenanthrene,
chrysene, triphenylene, benzo [b+j+k]fluoranthenes, benzo [a]pyrene,
benzo [e]pyrene, anthanthrene, benzo [ghi]perylene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene, and coronene were
found at concentrations of < 0.5 mg/kg.
3.2.2 Production levels and processes
Most of the PAH considered in this monograph are formed
unintentionally during combustion and other processes. Only a few are
produced commercially, including naphthalene, acenaphthene, fluorene,
anthracene, phenanthrene, fluoranthene, and pyrene (Franck &
Stadelhofer, 1987). The most important industrial product is
naphthalene (see section 3.2.3). In 1987, about 220 kt of this
compound were produced in western Europe, 190 kt in eastern Europe,
170 kt in Japan, and 110 kt in the USA (Fox et al., 1988); in 1986,
> 1 kt was produced in Canada (Environment Canada, 1994). In 1985,
about 2.5 kt of acenaphthene and 20 kt of anthracene were produced
worldwide (Franck & Stadelhofer, 1987). In 1986, 0.1-1 t anthracene
and 1 t fluorene were produced in Canada (Environment Canada, 1994).
In 1993, a major producer in Germany produced < 5000 t anthracene,
< 1000 t acenaphthene, < 500 t pyrene, < 50 t phenanthrene, and
< 50 t fluoranthene (personal communication, Rütgers-VfT AG, 1994).
The substances are not synthesized chemically for industrial
purposes but are isolated from products of coal processing, mainly
hard coal-tar. The raw material is concentrated and the product
purified by subsequent distillation and crystallization. Only
naphthalene is sometimes isolated from pyrolysis residue oils, olefin
fractions, and petroleum-derived fractions; it is also obtained by
distillation and crystallization (Collin & Höke, 1985; Franck &
Stadelhofer, 1987; Griesbaum et al., 1989; Collin & Höke, 1991). In
the USA in 1970, the distribution of capacity was about 60% coal-tar-
and 40% petroleum-derived naphthalene (Gaydos, 1981); more detailed
data were not available. The purity of the technical-grade products is
90-99% (Collin & Höke, 1985; Franck & Stadelhofer, 1987; Griesbaum et
al., 1989; Collin & Höke, 1991; see also Section 2).
3.2.3 Uses of individual PAH
The uses of commercially produced PAH are as follows (Collin &
Höke, 1985; Franck & Stadelhofer, 1987; Griesbaum et al., 1989; Collin
& Höke, 1991):
- naphthalene: main use: production of phthalic anhydride
(intermediate for polyvinyl chloride plasticizers); also,
production of azo dyes, surfactants and dispersants, tanning
agents, carbaryl (insecticide), alkylnaphthalene solvents (for
carbonless copy paper), and use without processing as a fumigant
(moth repellent) (see Figure 2);
- acenaphthene: main use, production of naphthalic anhydride
(intermediate for pigments); also, for acenaphthylene
(intermediate for resins);
- fluorene: production of fluorenone (mild oxidizing agent);
- anthracene: main use, production of anthraquinone (intermediate
for dyes); also, use without processing as a scintillant (for
detection of high-energy radiation);
- phenanthrene: main use, production of phenanthrenequinone
(intermediate for pesticides); also, for diphenic acid
(intermediate for resins)
- fluoranthene: production of fluorescent and vat dyes;
- pyrene: production of dyes (perinon pigments).
3.2.4 Emissions during production and processing of PAH
The emissions of PAH during industrial production and processing
in developed countries are not thought to be important in comparison
with the release of PAH from incomplete combustion processes, since
closed systems and recycling procedures are usually used. Few data
were available.
3.2.4.1 Emissions to the atmosphere
No data were available.
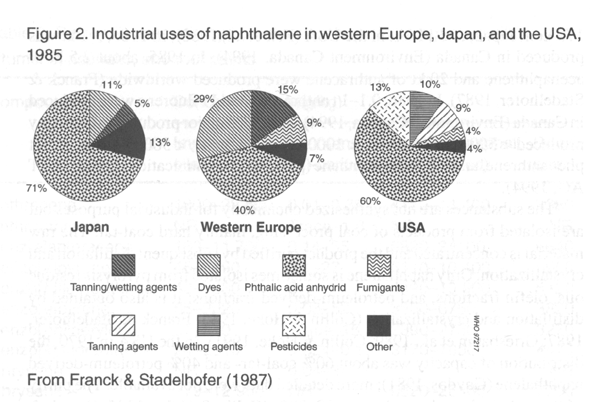 3.2.4.2 Emissions to the hydrosphere
During the refining of aromatic hydrocarbons, and especially hard
coal-tar, 80-190 t/year were estimated to be released to the
hydrosphere in western Germany until 1987. This quantity was reduced
to 8-19 t/year by the installation of new adsorption devices (sand
filtration and adsorbent resin) by the two German hard coal-tar
refineries in 1989 and 1991 (Klassert, 1993).
3.2.5 Emissions during the use of individual PAH
Only naphthalene is used directly (as a moth repellent) without
further processing. On the assumption that all naphthalene-containing
moth repellent is emitted into the atmosphere, the emissions would
have been about 15 000 t/year in western Europe in 1986, about 4400
t/year in Japan in 1987, and about 5500 t/year in the USA in 1987 (Fox
et al., 1988).
3.2.6 Emissions of PAH during processing and use of coal and petroleum
products
Coal coking, coal conversion by gasification and liquefaction,
petroleum refining, and the production and use of carbon blacks,
creosote, coal-tar, and bitumen from fossil fuels may produce
significant quantities of PAH (Anderson et al., 1986). A great deal of
information on emissions of PAH is available in the literature; this
monograph gives an overview of the most reliable values. The emission
profile depends on the source, and specific emission profiles are
detectable only in the direct vicinity of the corresponding source.
Generally, emissions are estimated on the basis of more or less
reliable databases, which are not identified in most publications. The
values reported give only a rough idea of the situation.
3.2.6.1 Emissions to the atmosphere
(a) Coal coking
During coal coking, PAH are released into the ambient air mainly
when an oven is loaded through the charging holes and new coal is
suddenly brought into contact with the hot oven, and from leaks around
oven doors and battery-top lids (Bjorseth & Ramdahl, 1985; Slooff et
al., 1989). The specific emission factor for both benzo [a]pyrene and
benzo [e]pyrene during coal coking was 0.2 mg/kg coal charged (Ahland
et al., 1985). The emission factor for total PAH was estimated to
about 15 mg/kg coal charged (Bjorseth & Ramdahl, 1985).
Stack gases were measured about 8 m away from the aperture
through which coke was discharged at a Belgian coking battery.
Although the effluent may have been slightly diluted with ambient air,
the following PAH concentrations were detected: benz [a]anthracene
plus chrysene, 580 ng/m3; benzo [k]fluoranthene, 500 ng/m3;
benzo [a]pyrene plus benzo [e]pyrene, 470 ng/m3; fluoranthene, 330
ng/m3; pyrene, 180 ng/m3 benzo [ghi]perylene, 140 ng/m3;
anthracene plus phenanthrene, 130 ng/m3; and perylene, 44 ng/m3
(Broddin et al., 1977).
The release of total PAH in 1985 was estimated to about 630
t/year in the USA, 18 t/year in Sweden, and 5.1 t/year in Norway
(Bjorseth & Ramdahl, 1985). The authors emphasized that their data are
subject to uncertainty and should be used only as an indication of the
order of magnitude. In 1990, the total PAH emission in Canada was
estimated to be 13 t/year (Environment Canada, 1994). Further
estimates of total annual emissions of individual PAH compounds during
the coking of coal are shown in Table 10.
Table 10, Estimated annual emissions of polycyclic aromatic hydrocarbons during
coal coking in the Netherlands and western Germany
Compound Annual Year Reference
emission
(t/year)
Netherlands
Anthanthrene 0.5 Before 1989 Slooff at al. (1989)
Benz[a]anthracene 0.3 1988 Slooff at al. (1989)
Benzo[a]pyrene 0.1 Before 1989 Slooff at al. (1989)
Benzo[ghi]perylene 0.2 1988 Slooff et al. (1989)
Benzo[k]fluoranthene 0.1 1988 Slooff at al. (1989)
Chrysene 0.2 1988 Slooff at al. (1989)
Fluoranthene 1.1 1988 Slooff at al. (1989)
lndeno[1,2,3-cd]pyrene 0.1 1988 Slooff et al. (1989)
Naphthalene 1.3 1987 Slooff et al. (1988)
2.0 Before 1989 Slooff et al. (1989)
Phenanthrene 2.1 1988 Slooff et al. (1989)
Western Germany
Benzo[a]pyrene 1.1 1990 Ministers for the
Environment (1992);
1.7 Zimmermeyer et al.
(1991)
Naphthalene 10.0 1987 Society of German
Chemists (1989)
The emission factors for benzo [a]pyrene in the coking industry
in the North-Rhine Westphalia area of Germany have been assumed to
have been reduced to an average of about 60 mg/t coke. The newest
plants have emission factors of 40 mg/t coke (Eisenhut et al., 1990).
The reduction in PAH discharge was brought about by technical
improvements to existing plants, closure of old plants and their
partial replacement by new plants, and a reduction in coke production
(Zimmermeyer et al., 1991). Decreasing trends in the annual emissions
of airborne PAH during coke production are also assumed to have
occurred in other industrialized countries (western Europe, Japan, and
the USA), but no data were available.
(b) Coal conversion
PAH emission factors measured in the USA during gasification of
coal at the end of the 1970s ranged from about 1 µg/g burnt coal for
chrysene and 1500 µg/g burnt coal for naphthalene. Three qualities of
coal were analysed for naphthalene, acenaphthylene, fluorene,
anthracene, phenanthrene, pyrene, benz [a]anthracene, chrysene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]perylene, indeno[1,2,3- cd]pyrene, and
dibenzo [a,h]pyrene (Nichols et al., 1981). In 1981, the stack gas of
one US pilot coal gasification plant with an outdoor filter contained
0.2 and 2.1 µg/m3 naphthalene at two sampling times and 6.8 µg/m3
phenanthrene (Osborn et al., 1984). Acenaphthylene was detected at
concentrations of 0.11-0.12 µg/m3 in the stack gases of two Canadian
pilot coal liquefaction plants (Leach et al., 1987).
(c) Petroleum refining
The average profile of PAH compounds in petroleum refineries
indicates that at least 85% of the total concentration is made up of
two-ring compounds (naphthalene and its derivatives) and 94% of two-
and three-ring compounds. Compounds with five rings or more
contributed less than 0.1% at the catalytic cracking unit. In
turn-round operations on reaction and fractionation towers,
naphthalene and its methyl derivatives accounted for more than 99% of
the total PAH (IARC, 1989b).
Little information is available on the concentrations of PAH in
stack gases. The levels in one French (Masclet et al., 1984) and two
US petroleum refining plants (Karlesky et al., 1987) are available
(Table 11); no information was given about the sampling site in the
French facility, but sampling in the US plants was at the distillation
device and below the cracking tower. The results depended on which
fuel was burnt and the positioning and type of sampling device in the
stack.
Table 11. Polycyclic aromatic hydrocarbon concentrations
in the stack gases of petroleum refinery plants in
France and the USA
Compound Concentration (µg/m3)
France USA
Acenaphthene NR 0.018-0.035
Acenaphthylene NR 0.013/0.019
Anthracene 3.9 0.003-0.041
Benz[a]anthracene 1.6 0.051-0.801
Benzo[a]pyrene 0.4 0.261-3.17
Benzo[b]fluoranthene 1.3 0.323-0.616a
Benzo[e]pyrene 2.8 NR
Benzo[ghi]perylene 0.7 0.23/0.382
Benzo[k]fluoranthene 0.5 NR
Chrysene 1.7 0.021-0.252
Coronene 1.0 NR
Dibenzo[a,h]anthracene NR 0.177
Fluoranthene 2.3 0.030-0.577
Fluorene 2.4 0.041-2.48
Indeno[1,2,3-cd]pyrene 1.2 0.25/0.538
Naphthalene NR 0.052-0.113
Perylene ND ND
Phenanthrene 7.9 0.040-9.13
Pyrene 4.3 0.016-3.56
From Masclet et aL (1984) and Karlesky et al. (1987)
NR, not reported; ND, not detected, limit of detection not
stated; /, single measurements
a Plus benzo[k]fluoranthene
Few data are available on the total release of PAH into the
atmosphere during petroleum refining. In western Germany, the
emissions of naphthalene during petroleum refining, including hard
coal-tar processing, were estimated to be 11 t/year (year not given;
Society of German Chemists, 1989). In the Netherlands, the release of
total PAH in 1988 was estimated to be about 7 t/year; the burning of
pitch contributed 6.6 t/year, regeneration of catalyst, 0.4 t/year,
and refining, < 0.01-0.1 t/year (Slooff et al., 1989). In Canada,
about 0.1 t total PAH were emitted into the atmosphere in 1990
(Environment Canada, 1994).
(d) Other processes
In a US oil-furnace carbon black plant, the following mean
emission factors per kg carbon black produced were found for
individual PAH in three runs in the main vent gas: acenaphthylene,
800 g; pyrene, 500 g; anthracene plus phenanthrene, 70 g;
fluoranthene, 60 g; benzo [ghi]fluoranthene, 40 g;
benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, 30 g; benzo [a]pyrene plus benzo [e]pyrene
plus perylene, 30 g; benzo [ghi]perylene plus anthanthrene, 23 g;
chrysene plus benz [a]anthracene, 9 g; indeno[1,2,3- cd]pyrene,
< 2 g; and benzo [c]phenanthrene, < 2 g. The release of PAH into
ambient air cannot be estimated from these emission factors, however,
as an additional combustion device is fitted in most US carbon-black
plants in which the process vent gases are burnt (Serth & Hughes,
1980).
Compounds with five or more rings (e.g. benzo [a]pyrene)
contributed about 0.3% to the total PAH released from the bitumen
processing unit of a refinery (IARC, 1989b). The emissions of PAH from
batch asphalt mixers are assumed to be low and to occur mainly in
combustion gases (IARC, 1984a), although no experimental data were
available.
Few estimates have been made of the annual emissions of PAH from
processes in which coal and coal products are used. The total release
of PAH to the atmosphere during asphalt production in 1985 was
estimated to be about 4 t in the USA, 0.1 t in Norway, and 0.3 t in
Sweden (Bjorseth & Ramdahl, 1985). In Canada, the amount emitted in
1990 was estimated to be about 2.5 t (Environment Canada, 1994). The
amount released during carbon-black production and processing in 1985
was estimated to be about 3 t in the USA and < 0.1 t in Sweden
(Bjorseth & Ramdahl, 1985). In the Netherlands in 1988, about 3.3 t of
total PAH were emitted during the storage and transport of anthracene
oil, an intermediate in the processing of hard coal-tar (Slooff et
al., 1989).
(e) Use of impregnating oils (creosotes) in wood preservation
Estimates of the total input of PAH into the atmosphere from wood
preservation with creosotes were available only for the Netherlands
for unspecified years, at about 320 t/year (Slooff et al., 1989) and
840 t/year (Berbee, 1992). In 1988, the PAH input during storage of
preserved material was estimated by the same authors to be about 200 t
naphthalene, 110 t phenanthrene, 30 t fluoranthene, 5 t anthracene,
1.1 t benz [a]anthracene, and 0.02 t benzo [k]fluoranthene.
3.2.6.2 Emissions to the hydrosphere
(a) Coal coking
The concentrations of PAH reported in wastewater effluents are
shown in Table 12. The removal of PAH by biological oxidation in two
US coal coking plants was 93 to > 99%. Higher-molecular-mass PAH,
benzo [a]pyrene, dibenz [a,h]anthracene, and benzo [ghi]perylene,
comprised a greater fraction (about 60%) of the total PAH content in
the effluent than in the input stream (Walters & Luthy, 1984). The
total concentration of PAH discharged into the aqueous environment
from a Norwegian coking plant was estimated to be about 23 kg/d
(Berglind, 1982). On the basis of Dutch emission factors, the release
in western Europe in 1985 of fluoranthene was calculated to be about 5
t and that of benzo [a]pyrene about 0.7 t (Berbee, 1992). The total
annual input of PAH into the aqueous environment of the Netherlands
was estimated to be about 1.7 t (year not given; Slooff et al., 1989).
(b) Coal conversion
The PAH content of wastewater from coal and shale conversion was
< 0.5 mg/litre (Guerin, 1977). In raw, untreated wastewaters from a
US pilot coal liquefaction plant, numerous PAH were found to emanate
from the liquefaction section, the untreated hydrogenation section,
and the still bottoms processing device when two kinds of coal were
tested; for example, benzo [a]pyrene was found at a concentration of
0.3-52 µg/litre (Robbins et al., 1981). Numerous PAH were found in raw
wastewater samples from two US pilot coal gasification plants (Walters
& Luthy, 1981; Abbott et al., 1986), the maximum level of
benzo [a]pyrene being 5.0 µg/litre.
No information was available about total PAH emissions into the
aqueous environment from commercial coal conversion plants. In
groundwater near a US in-situ coal gasification site, naphthalene was
found at a concentration of 2.7 µg/litre and acenaphthene and fluorene
at < 0.1 µg/litre (Pellizzari et al., 1979).
Until 1988, the final effluent from the two hard coal-tar
refineries in western Germany contained an average of 50 mg/litre
naphthalene, with a maximum of 120 mg/litre. The annual emission of
this compound was thus calculated to be about 80 t. By 1991, the
estimated release of naphthalene had been reduced to about 8 t/year by
the addition of adsorption devices (Klassert, 1993).
(c) Petroleum refining and offshore oil-well drilling
PAH concentrations in wastewater effluents from these sources are
summarized in Table 13. A refinery-activated sludge unit with a
dual-media filter removed about 95% of the five-ring PAH and 99% of
the four-ring PAH from the effluent of a petroleum refinery (Pancirov
et al., 1980). A similar elimination efficiency was found for
dissolved air flotation treatment of refinery wastewater and
subsequent removal by activated sludge. Air stripping of the compounds
in the sewage plant seemed to be of minor importance (Snider &
Manning, 1982). The concentrations of PAH with more than three rings
were found to be < 0.05 µg/litre even in the input to a sewage device
and < 0.02 µg/litre in the final effluent (German Society for
Mineral-oil and Coal Chemistry, 1984). The authors stated that these
levels were of the same order of magnitude as the background
concentrations in surface waters.
The discharge of total PAH from a Norwegian petroleum refinery
was about 0.26 kg/day (Berglind, 1982). The total concentration of PAH
released into the North Sea from offshore oil-well drilling activities
was about 2.5 t/year in 1987, comprising 2 t/year from drill rinsing
and 0.2 t/year from shipping (Slooff et al., 1989).
(d) Use of impregnating oils (creosotes) in wood preservation
PAH were detected at levels of milligrams per litre in
groundwater under a former wood preserving facility in Florida, USA.
The concentrations of lower-molecular-mass creosote constituents were
smaller in the groundwater than in an unweathered standard, probably
because of greater mobility and biodegradability (Mueller & Lantz,
1993; Middaugh et al., 1994).
Model experiments with fresh and seawater were carried out to
determine the release of PAH from marine pilings made from southern
pine and preserved with creosote (Ingram et al., 1982). The PAH levels
per litre fresh water in the leachate at 20°C after immersion for
three days were: naphthalene, 200-350 g; acenaphthene, 190-230 g;
phenanthrene, 190-230 g; fluorene, 120-150 g; acenaphthylene, 51-88
g; anthracene, 48-76 g; fluoranthene, 27-30 g; pyrene, 12 g; and
benz [a]anthracene, 11-19 g. The concentrations in seawater were
three to four times lower. The amounts of PAH leached increased with
increasing temperature. The concentrations in leachates from pilings
that had been in seawater for 12 years were of the same order of
magnitude. In contrast, rapidly decreasing PAH concentrations were
found three months after the start of the experiment in runoff
rainwater from spruce and pine pilings impregnated with hard coal-tar
(van Dongen, 1987).
The total PAH emissions into water and soil in the Netherlands
from commercial wood preservation were about 28 t/year (year not
given). The release of 10 PAH into water during the storage of
creosote-preserved wood was about 16 t/year; the PAH measured were
naphthalene, anthracene, phenanthrene, fluoranthene,
benz [a]anthracene, benzo [a]pyrene, benzo [ghi]-perylene, and
indeno[1,2,3- cd]pyrene) (Slooff et al., 1989).
In Canada, the maximum release of PAH into water and soil from
creosote-treated wood products was estimated to be 2000 t/year, on the
basis of the PAH content of creosote, the volume of treated wood, the
retention rates of the substances for different wood species, and an
estimated 20% loss of PAH during the time the wood was in service,
i.e. 40 years for pilings and 50 years for railroad ties (Environment
Canada, 1994).
Table 12. Polycyclic aromatic hydrocarbon concentrations (µg/litre) in
wastewater effluents from coal coking plants
Compound [1] [2] [3]a [4] [5]
Acenapthene NR NR NR 0.009-2.5 NR
Acenaphthylene NR NR NR NR NR
Anthracene 0.31 NR NR 0.0-2.0 0.1
Anthanthrene ND NR 0.040/0.600 NR NR
Benzo[j+k]fluoranthene NR NR NR NR NR
Benz[a]anthracene 2.0 11.1 0.504/4.9 NR NR
Benzo[a]fluoranthene 0.8 NR NR NR NR
Benzo[a]pyrene NR 3.8 0.622/4.841 4.7-25 NR
Benzo[b]fluoranthene NR NR NR NR NR
Benzo[a]fluorene 0.81 NR NR NR NR
Benzo[c]phenanthrene ND NR 0.042/0.699 NR NR
Benzo[e]pyrene NR NR 0.323/2.928 NR NR
Benzofluoranthenesb NR 6.9 1.010/8.741 NR NR
Benzo[ghi]fluoranthene ND NR 0.042/0.663 NR NR
Benzo[ghi]perylene 2.0 NR 0.445/2.835 0-9.0 NR
Chrysene NR 7.2 0.732/6.440 1.8-42 NR
Dibenz[a,h]anthracene NR NR NR 0.06-3.0 NR
Fluoranthene 2.8 11.2 NR 1.3-10 NR
Fluorene NR NR NR 0.0-1.0 NR
Indeno[1,2,3-cd]pyrene NR NR 0.371/3.051 NR NR
1-Methylphenanthrene ND NR NR NR NR
Naphthalene NR NR NR 0-4.1 NR
Perylene ND NR 0.117/1.348 NR NR
Phenanthrene 0.4 NR NR 0.45-2.3 0.5
Pyrene 4.0 12.9 NR NR 0.38-60
[1] Effluent channel water from one US coking plant (Griest, 1980);
[2] Effluent channel water from one US coking plant (Griest at al., 1981);
[3] Raw wastewater from two coking plants in western Germany (Grimmer at
al., 1981 b);
[4] Effluents from two US coking plants downstream of company-owned biological
oxidation device (Walters & Luthy, 1984);
[5] Final effluent after biological oxidation; no further information
(Jockers at al., 1988) When the water samples were filtered through solid
sorbents, the results may be underestimates of the actual content of
polycyclic aromatic hydrocarbons (see section 2.4.1.4)
ND, not detected, limit of detection not given; NR not reported
a /, single measurements
b Isomers not specified
Table 13. Polycyclic aromatic hydrocarbons in effluents after wastewater
treatment in petroleum refineries (µg/litre)
Compound [1] [2] [3] [4] [5]
Acenaphthene NR 4.0 < 0.1-6 NR NR
Acenaphthylene NR 1.8 < 0.1-< 1 NR NR
Anthracene NR 11 < 0.01-< 2 0.26 NR
Benz[a]anthracene NR 0.6 < 0.02-< 1 NR NR
Benzo[a]pyrene 0.57 0.1 0.1-2.9 0.11 NR
Benzo[b]fluoranthene < 0.1 0.2 < 0.06 NR NR
Benzo[c]phenanthrene NR 0.2 NR NR NR
Benzo[e]pyrene 0.65 0.3 NR NR NR
Benzo[ghi]fluoranthene < 0.4 NR NR NR NR
Benzo[ghi]perylene 0.36 NR < 0.2-< 1 NR NR
Benzo[j]fluoranthene < 0.2 NR NR NR NR
Benzo[k]fluoranthene < 0.2 0.4a < 0.2 NR NR
Chrysene < 0.03 1.4b < 0.02-1.4 NR NR
Coronene < 0.01 NR NR NR NR
Dibenz[a,h]anthracene NR NR < 0.3-< 1 NR NR
Fluoranthene < 0.2 16.0 < 0.1-< 10 0.26 NR
Fluorene NR 3.4 < 0.1-< 1 1.2 NR
Indeno[1,2,3-cd]pyrene < 0.02 NR < 1 NR NR
1-Methylphenanthrene NR 4.2 NR NR NR
Naphthalene NR 2.4 < 0.1-< 10 15 0.06-9
Perylene 0.14 NR NR NR NR
Phenanthrene NR 111.0 < 0.2-< 0.5 7.1 0.02-1.2
Pyrene 0.07 16.1 < 0.1-7 NR NR
Triphenylene < 0.03 NR NR NR NR
[1] Final effluent from one US petroleum refinery (Pancirov et al., 1980);
[2] Effluent from one Norwegian petroleum refinery after treatment in
oil-separation devices, oil traps, and retention ponds (Berglind, 1982);
[3] Average results for final effluent from 17 US petroleum refineries
(Snider & Manning, 1982);
[4] Final effluent from one Australian petroleum refinery (Symons & Crick,
1983);
[5] Average results for the final effluent from six petroleum refineries
in western Germany (German Society for Mineral-oil and Coal Chemistry,
1984)
When water samples were filtered through solid sorbents, the results may
be underestimates of the actual PAH content (see section 2.4.1.4).
NR, not reported
a With benzo[j]fluoranthene
b With triphenylene
(e) Other sources
PAH may be released into the hydrosphere during leaching of
stocks of coal by rain. In model leaching experiments, naphthalene,
acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene,
pyrene, chrysene, benz [a]anthracene, benzo [k]fluoranthene, and
benzo [a]pyrene were detected at concentrations in the low microgram
per litre range, with a maximum of 100 µg/litre; for example,
benzo [a]pyrene was found at 0.6 µg/litre (Stahl et al., 1984;
Fendinger et al., 1989).
PAH were also found in sludge from US coke processing plants in
the following concentrations (average of five samples): naphthalene,
430 mg/kg; phenanthrene, 260 mg/kg; acenaphthene, 78 mg/kg; pyrene, 30
mg/kg; chrysene, 28 mg/kg; benzo [a]pyrene, 3.8 mg/kg;
benzo [b]fluoranthene, 3.8 mg/kg; and benzo [ghi]perylene, 0.9 mg/kg
(Tucci, 1988).
PAH may also leach into drinking-water from coal-tar or asphalt
coatings on storage tanks and water distribution pipes. Samples from a
five-year-old coal-tar-coated water tank in the USA contained 0.21
µg/litre phenanthrene plus anthracene, 0.081 µg/litre fluoranthene,
0.071 µg/litre pyrene, 0.025 µg/litre naphthalene, and 0.021 µg/litre
fluorene (Alben, 1980). Measurements in numerous US drinking-water
systems showed that PAH accumulate in the water during transport in
these pipes. The total concentration of fluoranthene,
benzo [j]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
indeno[1,2,3- cd]-pyrene, and benzo [ghi]perylene after transport
was in the low nanogram per litre range (Basu et al., 1987). In 1994,
a PAH concentration of 2.7 µg/litre was measured in accordance with
the German Directive on drinking-water (6.9 µg/litre measured in
accordance with US regulations), which was due to transport through a
tar-coated pipe in a central water reservoir; phenanthrene was present
at a concentration of 2.8 µg/litre and pyrene at 1.2 µg/litre (State
Chemical Analysis Institute, Freiburg, 1995). The release of PAH from
this source cannot be estimated from the available data.
During offshore oil and gas production, PAH-containing drilling
muds are discharged directly into the sea. The PAH concentrations at
some oil and gas platforms in the Gulf of Mexico and the North Sea
were found to be 1900 µg/litre for naphthalene and < 0.01 µg/litre
each for chrysene, benzo [b]fluo-ranthene, and
dibenz [a,h]anthracene (van Hattum et al., 1993).
The total PAH passing into the oceans from shipping have not been
estimated. The worldwide discharge of PAH into the oceans from
refineries, marine transportation, and industrial effluents of crude
oil was estimated to be about 6 t/year in 1973 and 4.6 t/year in the
early 1980s (Suess, 1976), but the basis for these estimates is
unknown.
3.2.6.3 Emissions to the geosphere
The average PAH concentrations in soil from more than 20 former
coking sites in Germany were: naphthalene, 1000 mg/kg; phenanthrene,
500 mg/kg; fluoranthene, 200 mg/kg; pyrene, 200 mg/kg; anthracene, 50
mg/kg; and benzo [a]pyrene, 3-5 mg/kg. During vertical leaching, the
compounds are distributed according to their mobility. PAH with
high-boiling points and low water solubility are present at the
highest concentrations at the surface, and more mobile compounds
accumulate in deeper soil layers. Naphthalene is usually leached into
groundwater, in which it is relatively soluble (Hoffmann, 1993).
The sediment of an effluent channel at one US coking plant
contained the following concentrations of PAH (dry weight basis):
fluoranthene, 31 mg/kg; pyrene, 23 mg/kg; benzo [b+j+k]fluoranthenes,
23 mg/kg; benzopyrenes, 19 mg/kg; benz [a]anthracene, 15 mg/kg;
chrysene plus triphenylene, 15 mg/kg; benzo [ghi]perylene, 7.3 mg/kg;
benzo [a]fluorene, 7.2 mg/kg; anthracene, 6.7 mg/kg; perylene, 3.8
mg/kg; phenanthrene, 3.6 mg/kg; benzo [b]fluorene, 3.2 mg/kg;
benzo [ghi]fluoranthene, 2.3 mg/kg; anthanthrene, 2.3 mg/kg;
benzo [c]phenanthrene, 2.1 mg/kg; and 1-methylphenanthrene, 0.71
mg/kg. In the sediment of an effluent from one US petroleum tank farm,
anthracene was detected at 3.4 mg/kg, benz [a]anthracene at 0.13
mg/kg, and benzo [a]pyrene at < 0.049 mg/kg (Griest, 1980).
Oily sludge originating from a dissolved air flotation unit of
the treatment system of a US petrochemical plant effluent was applied
to sandy loam samples seven times during a 920-day active disposal
period followed by a 360-day inactive 'closure' period, and the
decreases in the concentrations of fluorene, phenanthrene, anthracene,
fluoranthene, pyrene, benz [a]anthracene, chrysene, triphenylene,
benzo [ghi]fluoranthene, benzo [b]fluoranthene,
benzo [j]fluoran-thene, benzo [k]fluoranthene, perylene,
benzo [a]pyrene, benzo [e]pyrene, and benzo [ghi]perylene in soil
were determined. The initial PAH levels ranged from 0.9 mg/kg
benzo [j]fluoranthene to 270 mg/kg phenanthrene (dry weight basis).
After 1280 days, the three-ring compounds (fluorene, phenanthrene,
anthracene) had almost completely disappeared, with 0.2-6.9%
remaining, the four-ring substances (fluoranthene,
benz [a]anthracene, chrysene) had been partly degraded, and the
five-ring compounds remained at fairly high concentrations (Bossert et
al., 1984).
PAH may be released into soil from polluted industrial sludges
and during commercial wood preservation; however, no estimates of the
total PAH input into this compartment were available.
3.2.6.4 Emissions into the biosphere
Use of anti-dandruff shampoos containing hard coal-tar may lead
to increased body concentrations of PAH, as measured by urinary
excretion of the PAH metabolite 1-hydroxypyrene. One shampoo had a
total PAH content of 2800 mg/kg, including 290 mg/kg pyrene and 56
mg/kg benzo [a]pyrene (no further specification) (van Schooten et
al., 1994). Application of a 2% crude coal-tar solution in petrolatum
led to significantly increased PAH levels in the blood of five
volunteers (Storer et al., 1984; see also Section 8). Measurements of
hard coal-tar-containing shampoos in Germany showed concentrations of
7-61 mg/kg benzo [a]pyrene. In wood-tar-containing shampoos,
benzo [a]pyrene was detected at concentrations in the low microgram
per kilogram range, but 150 mg benzo [a]pyrene were found in one tar
bath (State Chemical Analysis Institute, Freiburg, 1995).
3.2.7 Emissions of PAH due to incomplete combustion
PAH not only pre-exist in fossil fuels but more are formed during
pyrolysis by a radical mechanism (see Zander, 1980). The domestic
activities that may result in significant emissions of PAH emissions
are vehicle traffic, tobacco smoking, broiling and smoking of foods,
and refuse burning. The industrial activities that result in PAH
release are aluminium production with use of Söderberg electrodes,
iron and steel production, foundries, tyre production, power plants,
incinerators, and stubble burning (Anderson et al., 1986)
3.2.7.1 Industrial point sources
(a) Emissions to the atmosphere
(i) Power plants fired with coal, oil, and gas fossil fuels
PAH emitted into the atmosphere from coal-fired power plants
consist mainly (69-92%) of two- and three-ring compounds, i.e.
naphthalene and phenanthrene and their mono- and dimethyl derivatives.
Naphthalene is by far the major component of PAH fractions (31-35%),
although high concentrations of phenanthrene and fluorene are also
observed (Bonfanti et al., 1988). Specific emission factors of 0.02 g
emitted per kg combusted were measured for benzo [a]pyrene and 0.03
µg/kg for benzo [e]pyrene (Ahland et al., 1985).
The concentrations of PAH in stack gases from comparable coal-
and oil-fired power plants are shown in Table 14. It is difficult to
find a characteristic PAH profile for coal-fired plants. The
concentrations were low during undisturbed combustion (Guggenberger et
al., 1981; Warman, 1985). Low-molecular-mass PAH are found at higher
concentrations than high-molecular-mass compounds in coal combustion
effluents (Warman, 1985); the low-molecular-mass PAH phenanthrene,
fluoranthene, and pyrene were detected at particularly high
concentrations, whereas benzo [a]pyrene was found at a level typical
of that in ambient air (Kanij, 1987). The specific emission factor for
benzo [a]pyrene was 3.5-230 µg/t burnt coal (Ahland & Mertens, 1980).
As the contribution of benzo [a]pyrene to the total release of PAH is
small, it was considered not to be a suitable indicator for this
source (Guggenberger et al., 1981). In contaminated areas, the PAH
concentrations in ambient air may be higher than those in the stack
gases, which result from after-burning (Guggenberger et al., 1981).
Table 14. Concentrations of polycyclic aromatic hydrocarbons (ng/m3) in stack gases of coal- and oil-fired power plants
Compound Fuel [1] [2] [3] [4] [5] [6]a
Acenaphthene Coal NR NR NR NR NR ND-24
Anthracene Coal NR 0.5 < 10-1800 0.4-100 2-65 19-120
Anthanthrene Coal NR NR NR NR < 0.2-< 0.6 NR
Benz[a]anthracene Coal NR 0.6 < 20-1400 NR 1-40 NR
Benzo[a]pyrene Coal < 0.1-0.7b 1.3 0.5-790 0.1-120 0.1-1.9 NR
< 0.5c
Oil < 0.5-7 NR NR NR NR NR
Benzo[b]fluoranthene Coal < 0.1-3b,d 2.0 30/40k NR 0.3-12 NR
< 0.1-0.4c,d (1/880e)
Oil < 0.1-39a NR NR NR NR NR
Benzo[b]fluorene Coal NR NR NR NR < 2-< 6 NR
Benzo[c]phenanthrene Coal NR NR 0.2 NR NR NR
Benzo[e]pyrene Coal NR ND < 10-810 NR 3-< 18 NR
Benzo[ghi]perylene Coal NR NR < 10-1400 NR NR NR
Coal < 0.5-3b 1.2 < 10-< 100 3-22 < 2-< 6 NR
< 0.5c
Oil < 0.5-40 NR NR NR NR NR
Benzo[j]fluoranthene Coal NR NR NR NR < 5-< 13 NR
Benzo[k]fluoranthene Coal < 0.1-2b 0.9 20 NR 1.7-2.5 NR
< 0.1-1.3c
Oil < 0.1-29 NR NR NR NR NR
Chrysene Coal NR 1.8 < 10-< 600 0.1-28 1-41 ND-56
< 10-310e
3.8g
Coronene Coal 1-3b 0.9 < 100 NR NR NR
< 2c
Oil < 2-36 NR NR NR NR NR
Dibenz[a,h]anthracene Coal < 0.5-2b NR < 100 NR NR NR
< 0.5c
Oil < 0.5-26 NR NR NR NR NR
Table 14. (continued)
Compound Fuel [1] [2] [3] [4] [5] [6]a
Fluoranthene Coal NR 4.1 < 10-22 100 0.5-240 20-720 NR
Fluorene Coal NR 1.9 NR NR NR 2-140
Indeno[1,2,3-cd]pyrene Coal NR 1.7 < 10-< 100 NR < 0.1-< 1.4 NR
1-Methylphenanthrene Coal NR NR < 20-90 NR NR NR
Naphthalene Coal NR NR NR 10-1800 NR 420-2100
Perylene Coal < 0.1-0.2b NR NR NR NR NR
< 0.1c
Oil < 0.1-15 ND < 10-< 100 NR < 0.2-0.9 NR
Phenanthrene Coal NR 5.2 < 20-33 200 26-640 32-2930 NR
Pyrene Coal NR 1.3 9-5800 0.2-2850 5-335 ND-311
Triphenyene Coal NR NR NR NR 20-77 NR
[1] Coal- and oil-fired power plants in the former FRG (Guggenberger et al., 1981);
[2] One French coal-fired power plant (Masclet at al., 1984);
[3] 10 Swedish coal-fired power plants (Warman, 1985);
[4] One US coal-fired power plant (Junk at al., 1986);
[5] One Dutch coal-fired power plant (Kanij, 1987);
[6] One German coal-fired power plant with circulating fluid bed combustion (Wienecke at al., 1992)
NR, not reported; ND, not detected, limit of detection not given
a Various coal qualities
b Hard coal
c Brown coal
d With benzo[e]pyrene
e Isomers not specified
f With triphenylene
g With benz[a]anthracene
The inputs of PAH into the atmosphere from power plants were:
about 0.001 t benzo [a]pyrene in western Germany in 1981 (Ahland et
al., 1985) and 0.1 t in 1983 (Grimmer, 1983a); about 1 t/year total
PAH in the USA; 0.1 t in Norway and 6.6 t in Sweden in 1985 (Bjorseth
& Ramdahl, 1985); about 2 t total PAH in the Netherlands in 1988
(Slooff et al., 1989); and about 11 t total PAH in Canada in 1990
(Environment Canada, 1994). These numbers may be subject to
uncertainty and should be used only as an indication of the order of
magnitude of e.g. the concentration in stack gases that is to be
expected from experimental values. Actual information on PAH emissions
from oil- and gas-fired power plants was not available. PAH emissions
from coal-fired power plants have been claimed to be negligible in
Germany due to the installation of appropriate filter systems, despite
the vast amount of stack gases produced (Zimmermeyer et al., 1991;
Ministers for the Environment, 1992).
(ii) Incinerators
Numerous PAH are formed under simulated incinerator conditions
from plastics such as polystyrene, polyethylene, polyvinyl chloride,
and their mixtures (Hawley-Fedder et al., 1984a,b,c, 1987). PAH were
detected at the following concentrations in the stack gases from a
British municipal incinerator: pyrene, 1.6 µg/m3; benz [a]anthracene
plus chrysene, 0.72 µg/m3; fluorene, 0.58 µg/m3;
benzo [ghi]perylene, 0.42 µg/m3; benzo [b]fluoranthene plus
benzo [j]fluoranthene plus benzo [k]fluoranthene, 0.32 µg/m3;
perylene, 0.18 µg/m3; indeno[1,2,3- cd]pyrene, 0.18 µg/m3;
coronene, 0.04 µg/m3; and benzo [a]pyrene plus benzo [e]pyrene,
0.02 µg/m3 (Davies et al., 1976). When PAH were sampled at a height
of about 10 m above the ground in the 110-m chimney of an incineration
plant in Sweden, no measurable amounts of PAH, at a limit of detection
of 10 ng/m3, were found during normal operating conditions or during
start-up in the morning; however, inactivity over a weekend resulted
in detectable concentrations of individual PAH, covering three orders
of magnitude up to around 100 µg/m3 (Colmsjö et al., 1986a).
Comparable results were obtained at a pilot incineration plant in
Canada (Chiu et al., 1991). Only phenanthrene plus anthracene was
found in measurable amounts in the stack gas (limit of detection not
stated). The total release of PAH from this plant was estimated to be
80-100 ng/m3.
The concentrations of PAH emitted in the stack gases from an
Italian municipal solid waste incinerator were: 0.1-1.9 µg/m3
indeno[1,2,3- cd]pyrene, 0.63 µg/m3 acenaphthene, 0.57-2.5 µg/m3
phenanthrene, 0.36-4.4 µg/m3 perylene, 0.35-0.55 µg/m3
benzo [e]pyrene, 0.25-3.6 µg/m3 benz [a]anthracene, 0.23 µg/m3
benzo [k]fluoranthene, 0.22 µg/m3 dibenz [a,h]anthracene, 0.19
µg/m3 benzo [b]fluoranthene, 0.15-0.67 µg/m3 pyrene, 0.15-0.73
µg/m3 acenaphthylene, 0.11-0.23 µg/m3 chrysene, 0.08 µg/m3
anthracene, 0.069 µg/m3 fluorene, 0.068-1.3 µg/m3 fluoranthene,
0.05-1.1 µg/m3 benzo [a]pyrene, and 0.014-0.47 µg/m3
benzo [ghi]perylene, depending on the firing conditions and the
composition of the waste (Morselli & Zappoli, 1988).
The benzo [a]pyrene concentrations in stack gases from
commercial waste incinerators in western Germany were estimated to be
1-6 µg/m3 (Johnke, 1992).
Controlled incineration of automobile tyres for thermal and
electric energy has been estimated to result in considerable release
of PAH into the atmosphere. In laboratory experiments, the following
concentrations were found in flue gas at an incineration temperature
of 677°C (per kg rubber): 930 mg pyrene, 760 mg fluoranthene, 390 mg
phenanthrene, 290 mg anthracene, 220 mg acenaphthylene, 120 mg
chrysene, 84 mg benzo [b]fluoranthene plus benzo [j]fluoranthene
plus benzo [k]fluoranthene, 66 mg benz [a]anthracene, 18 mg
benzo [e]pyrene, 11 mg benzo [a]pyrene, 3.8 mg perylene, 3.3 mg
benzo [ghi]fluoranthene, 2.0 mg dibenz [a,h]anthracene, 1.5 mg
benzo [ghi]perylene, 1.2 mg naphthalene, and 0.5 mg
indeno[1,2,3- cd]pyrene (Jacobs & Billings, 1985). On the basis of
data from Hartung & Koch (1991) on the number of tyres incinerated in
western Germany in 1987, the annual emissions from this source can be
calculated as follows: 160 t pyrene, 130 t fluoranthene, 70 t
phenanthrene, 50 t anthracene, 40 t acenaphthylene, 20 t chrysene, 14
t benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]-fluoranthene, 10 t benz [a]anthracene, 3 t
benzo [e]pyrene, 2 t benzo [a]pyrene, 0.5 t
benzo [ghi]fluoranthene, 0.3 t dibenz [a,h]anthracene, 0.3 t
benzo [ghi]-perylene, 0.2 t naphthalene, and 0.1 t
indeno[1,2,3- cd]pyrene.
The total PAH levels in stack gases from incinerators in
different countries were: Italy, 0.0075-0.21 mg/m3; Japan, 0.002-0.04
mg/m3; Sweden, 0.001 mg/m3; and Canada, 0.00002-0.02 mg/m3 (WHO,
1988). The results for traditional incinerators could not be compared
with those for plants with additional abatement techniques on the
basis of the available data. The total PAH emissions to the atmosphere
resulting from incineration of refuse were about 0.001 t
benzo [a]pyrene in western Germany in 1989 (Ministers for the
Environment, 1992) and about 0.0003 t in 1991 (Johnke, 1992), about 50
t total PAH in the USA, 0.3 t in Norway and 2.2 t in Sweden in 1985
(Bjorseth & Ramdahl, 1985); and about 2.4 t total PAH in Canada in
1990 (Environment Canada, 1994).
In Germany, the contribution of stack gases from commercial
incinerators is estimated to be < 4% of the total stack gas volume
from combustion processes. One of the main confounders of and
contributors to stack gases from combustion is motor vehicle traffic
(Johnke, 1992), indicating that PAH released from incinerators are
probably of minor importance.
(iii) Aluminium production
The production of coal anodes, used in the electrolytic
production of aluminium, from pitch and petroleum coke may still be an
important source of PAH, but confirmatory data are not available.
Estimates of PAH released during the production of aluminium in the
Netherlands in 1988 ranged from about 0.3 t benzo [ghi]perylene to 24
t naphthalene (Slooff et al., 1989). The estimated total airborne PAH
released in 1985 was about 1000 t in the USA, 160 t in Norway, and 35
t in Sweden (Bjorseth & Ramdahl, 1985). In 1990, the input of total
PAH from this source into the atmosphere in Canada was 930 t
(Environment Canada, 1994).
In horizontal and vertical Söderberg aluminium production
processes in Sweden, the emission factors per tonne of aluminium were
0.11 kg benzo [a]pyrene and 4.4 kg total PAH for the horizontal
process and 0.01 kg benzo [a]pyrene and 0.7 kg total PAH for the
vertical process (Alfheim & Wikström, 1984). In a Norwegian vertical
Söderberg aluminium production plant, the emission factors were
0.005-0.015 kg/t aluminium for benzo [a]pyrene and 0.3-0.5 kg/t for
total PAH (European Aluminium Association, 1990).
(iv) Iron and steel production
The total emissions of PAH resulting from iron and steel
production with carbon electrodes containing tar and pitch in Norway
was estimated to be about 34 t in 1985 (Bjorseth & Ramdahl (1985), but
the database for this estimate is limited. The release of total PAH
from metallurgical processes in Canada where similar electrodes were
used, including ferro-alloy smelters but excluding aluminium
production, was estimated to be 19 t in 1990 (Environment Canada,
1994).
(v) Foundries
PAH are formed during casting by thermal decomposition of
carbonaceous ingredients in foundry moulding sand, and they partly
vaporize under the extremely hot reducing conditions at the
mould-metal interface. Thereafter, the compounds are adsorbed onto
soot, fume, or sand particles. Organic binders, coal powder, and other
carbonaceous additives are the predominant sources of PAH in iron and
steel foundries (IARC, 1984b).
In pyrolysis experiments with green-sand additives, the highest
PAH levels were found in coal-tar pitch, with values per kilogram of
additive of 3100 mg benzo [a]pyrene, 3000 mg
benzo [b+j+k]fluoranthenes, 3000 mg pyrene, and 2900 mg fluoranthene;
the lowest levels were found in vegetable product additives, such as
maize starch: 26 mg pyrene, 16 mg fluoranthene, 3 mg
benzo [b+j+k]fluoranthenes, and 2 mg benzo [a]pyrene (Novelli &
Rinaldi, 1979). Less than 0.002 mg/kg benzo [a]pyrene was found in
foundry moulding sand when petrol resin, polystyrol, or polyethylene
was used as the carrier and 7.5 mg/kg when hard coal was used as the
carrier. The PAH content was directly correlated with the amount of
hydrocarbon carrier in the sand (Schimberg et al., 1981).
The following levels of PAH were found in the stack gases of one
French automobile foundry: fluoranthene, 980 ng/m3;
benz [a]anthracene, 830 ng/m3; benzo [a]pyrene, 570 ng/m3;
benzo [b]fluoranthene, 460 ng/m3; indeno[1,2,3- cd]pyrene, 370
ng/m3; anthracene, 250 ng/m3; benzo [k]fluoranthene, 220 ng/m3;
perylene, 160 ng/m3; benzo [ghi]perylene, 130 ng/m3; chrysene, 110
ng/m3; coronene, 28 ng/m3; and pyrene, 15 ng/m3. No further
information was given about the sampling site (Masclet et al., 1984).
The total emission of PAH into the atmosphere from iron foundries in
the Netherlands was estimated to be about 1.3 t in 1988 (Slooff et
al., 1989).
(vi) Other industrial sources
The estimated release of 10 PAH into the atmosphere in the
Netherlands in 1988 was about 1.3 t from sinter processes and 0.2
t/year from phosphorus production (Slooff et al., 1989).
(b) Emissions to the hydrosphere
(i) Aluminium production
PAH levels in wastewater from aluminium production in Norwegian
plants are shown in Table 15. At the beginning of the 1970s, the
release of anthracene and phenanthrene into the aqueous environment
from aluminium production in western Europe was estimated to be 180
t/year (Palmork et al., 1973). About 0.6 t/year are released into
water by the aluminium producing industry in the Netherlands (Slooff
et al., 1989).
(ii) Other industrial sources
No recent data were available on PAH emissions into the aqueous
environment from coal- or oil-fired power plants. PAH were found in
the final effluent from a British municipal incinerator at
concentrations ranging from < 0.01 µg/litre each for coronene and
indeno[1,2,3- cd]pyrene to 0.62 µg/litre fluoranthene. The calculated
daily output of single compounds was in the low milligram range, with
a maximum of 16 mg/d. Actual data were not available (Davies et al.,
1976).
Numerous PAH were detected in the final effluent from a Norwegian
ferro-alloy smelter in which the wastewater from gas scrubbers was
treated by chemical flocculation. The concentrations were 50 µg/litre
phenanthrene, 45 µg/litre pyrene, 40 µg/litre fluoranthene, 39
µg/litre acenaphthylene, 27 µg/litre fluorene, 17 µg/litre
acenaphthene, 13 µg/litre chrysene plus triphenylene, 11 µg/litre
anthracene, 10 µg/litre naphthalene, 10 µg/litre benz [a]anthracene,
9 µg/litre benzo [b]fluoranthene, 6 µg/litre benzo [j]fluoranthene
plus benzo [k]fluoranthene, 6 µg/litre benzo [e]pyrene, 6 µg/litre
benzo [a]pyrene, 3 µg/litre benzo [c]phenanthrene, 3 µg/litre
indeno[1,2,3- cd]pyrene, 3 µg/litre benzo [ghi]perylene, 2 µg/litre
benzo [a]fluorene, 2 µg/litre benzo [b]fluorene, 2 µg/litre
perylene, and 1 µg/litre dibenz [a,h]-anthracene. The PAH contents of
wastewater from gas washers in one Norwegian steel production plant
were of the same order of magnitude (Berglind, 1982).
Table 15. Polycyclic aromatic hydrocarbon concentrations [µg/litre]
in wastewater from aluminium production in Norway
Compound [1] [2] [3]
Acenaphthene NR NR 5
Acenaphthylene NR NR 1
Anthracene 1.1-2.8 0.9 10
Anthenthrene < 1-3.2 NR NR
Benzo[b+k]fluoranthenes 6.8-38.1 NR NR
Benzo[j+k]fluoranthenes NR 10.5 5
Benz[a]anthracene 2.5-5.6 14.6 11
Benzo[a]fluorene 1.5-3.4 8.2 13
Benzo[a]pyrene 1.3-7.4 13.5 4
Benzo[b]fluoranthene NR 21.2 9
Benzo[b]fluorene 1.3-3.0 7.2 2
Benzo[c]phenanthrene NR NR 3
Benzo[e]pyrene 2.6-16.4 17.0 5
Benzo[ghi]perylene NR 8.3 2
Chrysene and triphenylene 5.8-16.0 27.3 17
Coronene < 1-2.0 NR NR
Dibenz[a,h]anthracene NR NR 1
Fluoranthene 12.4-20.8 7.5 124
Fluorene NR NR 3
Indeno[1,2,3-cd]pyrene NR 8.1 2
1-Methyphenanthrene NR 0.4 NR
Naphthalene NR NR 1
Perylene NR 3.2 1
Phenanthrene 14.0-23.1 1.8 34
Pyrene 5.6-15.3 6.4 76
[1] Two samples of wastewater with two runs each from one aluminium
production plant (Kadar at al., 1980);
[2] Wastewater from one aluminiurn production plant; no further
information (Olufsen, 1980);
[3] Effluent from gas washers from one aluminium smelter (Berglind, 1982)
When the water samples were filtered through solid sorbents, the results
may be underestimates of the actual content (see section 2.4.1.4).
NR, not reported
The release of 10 PAH into water from different industries in the
Netherlands was estimated to be 4 t/year (Slooff et al., 1989).
(c) Emissions to the geosphere
The levels of PAH in ash samples from various incinerators are
shown in Table 16. The values given by Eiceman et al. (1979) were
based on the gas chromatographic responses of pyrene and
benzo [a]pyrene. The concentrations of PAH in ashes from coal-fired
power plants were of the same magnitude as the background levels of
these compounds in soil, but fly ash from municipal waste incinerators
may contain significantly higher levels (Guerin, 1977; Kanij, 1987).
The total PAH content of filter residues in incinerators was about
0.20-0.5 µg/g. The compounds are assumed to be tightly bound to
particle surfaces and not mobile in an aqueous environment in the
absence of organic solvents (WHO, 1988). In a comparison of 26
incineration plants, combustion conditions were shown to have a marked
influence on PAH release (Wild et al., 1992).
The material dredged from harbour areas may have a significant
PAH content (see also sections 5.3.3 and 5.3.4). The annual load of
naphthalene, anthracene, phenanthrene, fluoranthene,
benz [a]anthracene, chrysene, benzo [k]-fluoranthene,
benzo [a]pyrene, benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene
in material dredged from Rotterdam harbour was about 12 t (year not
given). The main PAH were fluoranthene and benz [a]anthracene (Slooff
et al., 1989).
3.2.7.2 Other diffuse sources
(a) Atmosphere
(i) Mobile sources
PAH are released into the atmosphere by motor vehicle traffic.
The profile of the PAH released and the quantity of PAH in the exhaust
are fairly similar, independently of the type of engine and the PAH
content of the fuel, indicating that the emitted compounds are formed
predominantly during combustion (Meyer & Grimmer, 1974; Janssen, 1980;
Stenberg, 1985; Williams et al., 1989). PAH accumulate in used engine
oil, but the importance of the PAH content of engine oil on emissions
is still under discussion. Janssen (1980), Pischinger & Lepperhoff
(1980), and Stenberg (1985) assumed that the PAH content of the oil
played only a minor role, but Williams et al. (1989) showed in tests
with diesel fuel that it may contribute considerably to the release of
particulate PAH. There is also doubt about whether PAH emissions are
indepen-dent of the aromaticity of the fuel. Janssen (1980) stated
that release of PAH into the atmosphere is not increased if the
aromaticity does not exceed a concentration of 50% volume (see also
Schuetzle & Frazier, 1986). According to Stenberg (1985), the release
of PAH by automobile traffic is dependent on the:
Table 16. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in ash samples from coal-fired power plants and municipal waste and sewage
sludge incinerators
Compound Coal-fired Municipal waste incinerators Sewage
power plants sludge
incinerators
Netherlands USA Canada Japan Netherlands Canada UK Italy (UK)
[1] [2] [3] [3] [3] [4] [5] (mean) [6] [5]
Acenaphthene + NR NR NR NR NR NR 1-258(7.8) 289-1022i NR
fluoranthene
Acenaphthylene NR NR NR NR NR 3.35 NR 5-1394 NR
Anthracene < 0.14-0.5 NR 10/500 10/10 200 NR 1-62(2.3) 42-651 NR
Anthanthrene < 0.24-< 0.5 NR NR NR NR NR NR NR
Benz[a]anthracene < 0.6-< 1.2 NR NR NR NR NR 1-1646a(12) 280-1278 3a
Benzo[a]fluoranthene NR 36.8 NR NR NR NR NR NR
Benzo[a]pyrene < 0.29-< 1.8 NR ND/400 ND/ND ND NR 1-596(8.2) 1014-3470 3
Benzo[b]fluoranthene < 0.6-< 0.29 NR NR NR NR NR 1-873(5.7) 1818 6
Benzo[b]fluorene < 2.0-< 4 11.8 NR NR NR NR NR NR
Benzo[e]pyrene < 2.9-< 6 NR NR NR NR NR NR 458-1786 NR
Benzo[ghi]perylene < 1.6-1.7 NR NR NR NR NR 10-9507 (62.3) 700-2377 135
Benzo[j]fluoranthene < 4.5-< 9 NR ND/400b ND/NDb NDb NR NR NR
Benzo[k]fluoranthene < 0.15-< 2.8 NR NR NR NR NR 1-276(1.5) 1535 NR
Chrysene < 1.5-< 3 NR NR NR NR NR NR 570-1973 NR
Coronene NR NR NR NR NR NR 3-238 (31.3) 36
Dibenz[a,h]anthracene < 4.2-< 8.2 NR NR NR NR NR 1-167(5.2) 57/69 1
Fluoranthene 1.1-5.2 < 13.4 2/500 3/ND 20 2.14-43.2 1-765 (8.6) 1684-10 890 1
Fluorene NR NR ND/10 ND/ND 60 2.57/4.41 NR 45-522 NR
Indeno[1,2,3-cd]pyrene < 0.82-< 1.6 NR NR NR NR NR NR 478-1343 NR
Naphthalene NR 8.3 NR NR NR NR 4/15(0.2) NR
Perylene < 0.16-< 0.3 NR NR NR NR NR NR 259 NR
Phenanthrene 4.0-43 17.6 NR NR NR 8.76-154c 2-5402 (36.5) 1616-7823 6
Pyrene 0.72-2.9 < 19.0 1/500 1/ND 10 2.47-19.6 1-3407 (45.3) 1863-8799 10
Triphenylene < 2.5-< 5.0 NR NR NR NR 12.7a NR NR
Table 16 (continued)
[1] Pulverized coal ash (Kanij, 1987);
[2] Fly ash (Guerin, 1977);
[3] Fly ash (Eiceman at al., 1979);
[4] Fly ash (Chiu at al., 1991);
[5] Fly ash 26 incinerators with different firing techniques (Wild et al., 1992);
[6] Fly ash from electrostatic precipitator and scrubber (Morselli & Zappoli, 1988)
NR, not reported; ND, not detected; /, single measurements
a With chrysene
b Isomers not specified
c With anthracene
i Only acenaphthene
- aromaticity of the fuel;
- starting temperature: Starting at -10°C results in threefold
higher PAH emissions than a standardized cold start (+ 23°C); the
emission factors measured by Larssen (1985) were significantly
higher in winter than in summer.
- ambient temperature: Low ambient temperatures (5-7°C) increase
PAH emissions from petrol-fuelled vehicles by five to 10 times,
depending on the engine used.
- test conditions: Three standardized test cycles are in general
use: a test developed by the Economic Commission for Europe of
the United Nations (ECE) in Europe; the Federal Test Procedure
(FTP) in the USA; and the Japanese test cycle in Japan. Emissions
at cold start may be lower and those at hot start slightly higher
in the FTP than in the ECE test, but overall agreement between
the tests is good.
- air:fuel ratio (l): Small variations around l = 1, representing
stoichiometric levels of fuel and air, do not affect PAH
emissions significantly; richer mixtures lead to increasing PAH
emissions, and bad ignition at l = 0.8 causes a sharp increase in
PAH emissions.
- type of fuel: Emissions of the sum of phenanthrene,
fluoranthene, pyrene, benzo [ghi]fluoranthene,
cyclopenta [cd]pyrene, benz [a]-anthracene, chrysene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [e]pyrene,
benzo [a]-pyrene, indeno[1,2,3- cd]pyrene,
benzo [ghi]-perylene, and coronene decreased in the FTP cycle as
follows: diesel (total PAH; 960 µg/km) > petrol (170 µg/km) >
petrol containing methanol or ethanol (43-110 µg/km) > methanol
= liquefied petro-leum gas = catalyst-equipped petrol-fuelled
vehicles (6-9 µg/km) (Stenberg, 1985). In comparable
measurements, similar results were obtained but with a much lower
average emission rate for diesel-fuelled vehicles: 186 µg/km for
total PAH, including fluoranthene, pyrene, benz [a]anthracene,
chrysene, benzo [b]-fluoranthene, benzo [e]-pyrene,
benzo [a]pyrene, perylene, indeno[1,2,3- cd]pyrene,
benzo [ghi]-perylene, and coronene. It was not stated whether
the difference in the emission rates was due to the numbers of
PAH chosen for analysis (Lies et al., 1986).
PAH emissions in the exhaust from spark-ignition automobile
engines can be reduced by operation with lean air:fuel ratios, smaller
quenching distances in the combustion chamber, and increased cylinder
wall temperatures in the engine (Pischinger & Lepperhoff, 1980;
Lepperhoff, 1981). Diesel-fuelled engines with low emissions of total
unburnt gaseous hydrocarbons have low rates of PAH emission. Control
can therefore be achieved by using conventional techniques for
reducing unburnt gaseous hydrocarbons (Williams et al., 1989).
Fluoranthene and pyrene constitute 70-80% of total PAH emissions
from vehicles (Lies et al., 1986; Volkswagen AG, 1989; see also Table
17), whereas the emissions from one diesel-fuelled truck consisted
mainly of naphthalene and acenaphthene (Nelson, 1989). Although
cyclopenta [cd]pyrene is emitted at a high rate from petrol-fuelled
engines, its concentration in diesel exhaust is just above the limit
of detection, probably because the oxidizing conditions in
diesel-fuelled engines decompose this relatively reactive compound
(Lies et al., 1986).
The amounts of PAH released from vehicles with three-way
catalytic converters are much lower than those from vehicles without
catalysts (Table 18). The total amount of PAH was increased by a
factor of about 40 between new and used catalytic converters (Hagemann
et al., 1982). PAH emissions from diesel-fuelled vehicles can be
reduced by > 90% by a combination of a catalytic converter and a
particulate trap, as shown by experiments with a heavy-duty
diesel-fuelled truck (Westerholm et al., 1989). Westerholm et al.
(1991) found benz [a]anthracene, benzo [b]fluoranthene,
benzo [k]fluoranthene, benzo [e]-pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, fluoranthene, pyrene,
anthracene, and coronene in much lower amounts than other
investigators, while some other PAH that were not measured by other
investigators, especially phenanthrene and 1-methylphenanthrene, were
detected at quite high concentrations. These differences are possibly
due to the driving cycle used.
Measurements made on particulate matter in the exhausts of light-
and heavy-duty diesel-fuelled vehicles with different fuel qualities
showed concentrations of 1 mg/kg each of benz [a]anthracene,
benzo [b]fluoranthene plus benzo [j]fluoranthene, benzo [a]pyrene
plus benzo [e]pyrene, and benzo [ghi]perylene and 290 mg/kg pyrene.
The results were strongly dependent on the driving cycle and
individual engine conditions (CONCAWE, 1992).
The PAH concentrations measured in the exhaust gases of different
vehicles are shown in Table 19. The differences in PAH emissions from
petrol- and diesel-fuelled vehicles are still under discussion. When
the data of Behn et al. (1985) are compared with those of Klingenberg
et al. (1992), diesel-fuelled vehicles emitted larger amounts of PAH
than petrol-fuelled vehicles. Benzo [a]pyrene was emitted at a rate
of 6 µg/km from a petrol-fuelled vehicle without a catalyst and at 5
µg/km from a diesel-fuelled vehicle (Gibson, 1982). When the PAH
emissions from 10 petrol- and 20 diesel-engined vehicles were measured
under three urban cycles, the mean emission factors (µg/km) for
benzo [a]pyrene were 12 with petrol and 0.56 with diesel in a cold,
low-speed cycle, 0.50 with petrol and 0.37 with diesel in a hot,
low-speed cycle, and 0.37 with petrol and 0.24 with diesel in a hot,
free-flow cycle (Combet et al., 1993). Considerably higher emission
rates were found from four petrol-fuelled passenger cars without
catalysts, 11 with catalysts, and eight diesel-fuelled passenger cars,
two of which had oxidation catalysts, on a chassis dynamometer at the
USA FTP 75 cycle. The diesel-fuelled vehicles emitted about as much
benzo [a]pyrene as the petrol-fuelled vehicles without catalysts
(5-25 µg/km), while the petrol-fuelled vehicles with catalysts had
emission rates significantly below 2 µg/km. The diesel-fuelled
vehicles with oxidation catalysts had emissions of about 5 µg/km
(Klingenberg et al., 1992).
The following emission factors were given for motorcycles and
two-stroke mopeds: 1000 µg/km naphthalene, < 32-650 µg/km
phenanthrene, < 11-170 µg/km anthracene, < 5-110 µg/km fluoranthene,
< 2-11 µg/km chrysene, < 2-11 µg/km indeno[1,2,3- cd]pyrene,
< 1-1200 µg/km benz [a]anthracene, 0-63 µg/km benzo [ghi]perylene,
0-16 µg/km benzo [a]pyrene, and 0-11 µg/km benzo [k]fluoranthene
(Slooff et al., 1989).
Further PAH emissions may result from the abrasion of asphalt by
vehicle traffic, so that PAH in asphalt and bitumens (see section
3.2.1) may contribute considerably to the total PAH emissions due to
automobile traffic. The abrasion caused by spiked tyres in winter was
estimated to be 20-50 mg/km (Lygren et al., 1984).
Another source of PAH from motor vehicle traffic is clutch and
break linings, which are subject to considerable thermal stress,
sometimes resulting in pyrolytic decomposition of abraded particles.
Numerous PAH were found in the abraded dust of brake and clutch
linings in one study, but the values show large standard deviations,
due, presumably, to the fact that the substances are adsorbed onto
asbestos fibres from which they are difficult to separate (Knecht et
al., 1987). Total PAH release from clutch and brake linings cannot be
estimated from the available data.
Rubber vehicle tyres contain highly aromatic oils as softeners.
These oils, which can contain up to 20% PAH, are used at
concentrations of 15-20% in rubber blends (Duus et al., 1994). In
Sweden, it was considered that the input of PAH to the atmosphere from
rubber particles was important (National Chemicals Inspectorate,
1994).
According to estimates for Belgium, western Germany, and the
Netherlands in 1985, the annual PAH input into the atmosphere from
vehicle traffic ranges from < 10 t/year for benzo [ghi]fluoranthene,
benz [a]anthracene, benzo [k]-fluoranthene, benzo [a]pyrene, and
indeno[1,2,3- cd]pyrene, to < 10-20 t/year for anthracene,
fluoranthene, and chrysene, to 10-70 t/year for phenanthrene, to about
100-1000 t/year for naphthalene (Slooff et al., 1989). Values of the
same order of magnitude were reported for emissions of naphthalene in
1987 (Society of German Chemists, 1989) and benzo [a]pyrene in 1989
in western Germany (Ministers for the Environment, 1992) and for total
PAH in 1985 in Norway and Sweden (Bjorseth & Ramdahl, 1985). The total
annual PAH input from vehicle traffic in the USA in 1985 was about
2200 t/year (Bjorseth & Ramdahl, 1985). In Canada, the total PAH input
was estimated to be about 200 t in 1990; 155 t were assumed to be due
to diesel-fuelled and 45 t to petrol-fuelled vehicles (Environment
Canada, 1994).
Table 17. Polycyclic aromatic hydrocarbon emission factors (µg/km) for petrol-fuelled vehicles
Compound [1] [2] [3] [4] [5] [6] [7]
Anthracene NR 0.7/0.7a NR 2/99b NR 21-42 0.6
37/1988c
Anthanthrene NR 0.2/1.3 NR NR NR NR NR
Benzo[b+j+k]fluoranthene NR NR NR NR NR NR 7.6
Benzo[b+k]fluoranthene NR 3.9/7.0 NR NR 0.23-0.54/2.55-9.20 NR NR
Benz[a]anthracene NR 5.7/5.9 3.5-9.0 NR 0.06-0.35/2.5-8.0 5-16 5.1
Benzo[a]pyrene NR 1.9/4.51 1.5-14.5 0.06-2/1-12b 0.06-0.62/1.30-10.4 2-11 3.7
Benzo[e]pyrene NR 2.6/6.2 NR 0.2/2-14b 0.08-0.54/2.54-9.20 NR 5.1
Benzo[ghi]fluoranthene NR 5.6/12 NR NR NR NR 8.8
Benzo[ghi]perylene NR 5.9/13 NR NR 0.19-0.75/1.45-17.5 5-21 18.9
Benzo[j]fluoranthene NR 1.1/0.9 NR NR NR NR NR
Benzo[k]fluoranthene NR NR NR NR NR 0-5 NR
Chrysene NR 6.7/8.7 NR NR 0.12-0.73/2.78-23.1 11-42 7.7
Coronene NR 6.5/12 1.5-20.0 NR NR NR 29.5
Cyclopenta[cd]pyrene NR 2.9/12 NR NR NR NR 16.5
Fluoranthene NR 14/20 NR 3/139-211b 2.7/43.3d 11-158 10.4
ND/186-280c
Indeno[1,2,3-cd]pyrene NR 1.7/3.6 NR NR 0.06-0.43/0.83-6.67 5-21 4.2
Naphthalene 8100-8600a NR NR NR NR 2300f NR
210-2651
Perylene NR 0.3/0.5 NR NR 0.01-0.06/0.25-1.82 NR NR
Phenanthrene NR 2.6/2.9 NR NR NR 84-210 1.8
Pyrene NR 28/31 43-184 4-16/12-268b 2.9/43.0b NR 19.2
ND/124-360c
Table 17 (continued)
NR, not reported; ND not detected (detection limit not stated); /, single measurements
a Two driving distances
b Only particulate phase considered
c Only gaseous phase considered
d Average
e Depending on analytical conditions
f With converter
[1] From measurements in tunnel with converters (Hampton at al., 1983);
[2] One vehicle without converter (Alsberg et al., 1985);
[3] Various tests conducted mainly in the 1970s, some unstandardized, different numbers of vehicles,
without converters (Stenberg, 1985);
[4] FTP cycle only, number of vehicles not given; year of manufacture 1980-85 = petrol-engine vehicles
with converter; 1973-81 = petrol-engine vehicles without converter (Schuetzle & Frazier, 1986);
[5] Various standardized test procedures; four petrol-engine vehicles without, seven with three-way-converter
for each test, all with four or five cylinders (Volkswagen AG, 1988);
[6] No information about test cycle or number of cars tested; city roads, motorways and other roads tested;
no distinction between vehicles with and without converter, unless otherwise stated (Slooff et al.,
1988, 1989);
[7] One petrol-engine vehicle without converter in USFTP test cycle (Strandell at al., 1994)
Table 18. Polycyclic aromatic hydrocarbon emission factors (µg/km) for diesel-fuelled vehicles
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene NR NR NR NR NR NR 41-128 NR
Anthracene 17/63 65-273a 1.2/3.0 NR 21-73b 3.3 2.9-26 4.6
1305-5568c
Benzo[b+j+k]fluoranthene NR NR NR NR NR NR 1.7-12d 5.0
Benzo[b+k]fluoranthene 2.6/47 NR 3.9/6.1 5.57-14.96 NR 0.29 NR NR
Benz[a]anthracene 8/43a NR 4.0/7.0 2.73-3.91 11-21b 0.47 0.7-9.6 2.0
Benzo[a]fluorene NR NR NR NR NR 2.4 NR NR
Benzo[a]pyrene < 1/20 0.6-34a 1.6/2.2 2.09-7.23 1-5 < 0.06 0.5-3.2 1.5
Benzo[e]pyrene 3/38 2-40a 2.5/4.1 2.40-52.8 NR 0.15 1.1-9.9 4.0
Benzo[ghi]fluoranthene NR NR 4.0/12 NR NR 1.5 NR 10.6
Benzo[ghi]perylene < 1/18 NR 1.9/3.1 2.84-26.3 9e < 0.13 0.5-3.7 2.0
Chrysene 14/67 NR 11/25 4.7-21.1 16-42b 2.8f 3.5-28 3.7
Coronene NR NR 0.3/20.7 NR NR < 0.01 NR NR
Cyclopenta[cd]pyrene NR NR 3.6/3.9 NR NR 0.18 NR 4.0
Fluoranthene 58/200 139-580a 13/38 70g 21-105b 17 14-34 43.7
186-771c
Fluorene NR NR NR NR NR NR 38-228 NR
Indeno[1,2,3-cd]pyrene NR NR 1.5/2.3 0.89-7.52 9e < 0.04 NR 1.2
1-Methylphenanthrene NR NR NR NR NR 41 NR NR
Naphthalene NR NR NR NR 2100-6302b NR 1030-1805 NR
Perylene < 1/2 NR NR 0.23-1 NR < 0.01 NR NR
Phenanthrene 295/524 NR 4.6/25 NR NR 2.9 79-308 54.8
Pyrene < 0-9/22 24-734a 20/104 66.9g NR 11 9-30 35.4
702-982c
Table 18 (continued)
NR, not reported; /, single measurements;
[1] ECE test; two passenger cars with < 50 000 and > 100 000 km odometer readings (Scheepers & Bos, 1992);
[2] FTP cycle; number of vehicles not given; year of manufacture, 1980-85 (Schuetzle & Frazier, 1986);
[3] Chassis dynamometer; one heavy-duty vehicle (Westerholm et al., 1986);
[4] Various standardized testing procedures; seven vehicles with four or five cylinders for each test (Volkswagen
AG, 1988);
[5] No information on test cycle or number of cars tested; three traffic situations (Slooff at al., 1989);
[6] Bus cycle simulating public transport (duration 29 min; driving distance, 11.0 km; average speed, 22.9 km/h);
one heavy-duty truck; measurement of particle phase (Westerholm at al., 1991);
[7] Bus cycle (duration, about 10 min after warm-up, each ramp consisting of 10 s acceleration, 10 s constant speed
of 12 km/h, 4.5 s deceleration, 7 s idling); three trucks and two buses without particle trap, two buses with
particle trap (Lowenthal et al., 1994);
[8] US FTP cycle; one passenger car (Strandell at al., 1994)
a Particle phase
b Automobiles and trucks
c Gas phase
d Isomers not specified
e Trucks
f With triphenylene
g Average
Table 19. Polycyclic aromatic hydrocarbon concentrations (µg/m3) in the
exhaust gases of different vehicles
Compound [1] [2] [3]
Acenaphthene NR NR < 0.02-0.81
Acenaphthylene NR NR < 0.02-4.16
Anthracene NR NR < 0.02-6.45
Anthanthrene 0.02-0.07 0.11-0.12 NR
Benz[a]anthracene 1.91-2.24 3.53-4.64 NR
Benzo[a]pyrene 0.46-0.76 2.03-2.33 < 0.02-4.97
Benzo[b]fluoranthene 1.53-2.04a 7.37-8.58a 0.06-6.63
Benzo[b]fluorene NR NR 0.11-12.7
Benzo[e]pyrene 1,07-1.24 2.46-2.90 0.09-6.16
Benzo[ghi]fluoranthene 0.46-0.59 4.81-7.19 NR
Benzo[ghi]perylene 0.76-1.04 3.42-4.41 0.22-1.81
Benzo[k]fluoranthene NR NR < 0.02-2.68
Chrysene 2.37-2.97b 7.37-8.58b 0.07-25.48
Coronene 0.26-0.30 1.82-2.32 < 0.02-1.80
Cyclopenta[cd]pyrene 1.86/2.26 5.80-6.09 NR
Dibenz[a,h]anthracene 0.04-0.07 0.32-0.35 < 0.02-0.44
Fluoranthene 11.83-13.09 20.90-25.30 0.16-35.94
Fluorene NR NR 0.06-2.16
Indeno[1,2,3-cd]pyrene 0.30-0.41 2.89-4.06 < 0.02-0.80
Perylene 0.10-0.26 0.21-0.33 0.13-5.55
Phenanthrene NR NR < 0.02-4.16
Pyrene 6.86-8.96 12.20-15.20 0.06-21.31
NR, not reported; /, single measurements;
[1] One vehicle with spark-ignition engine on chassis dynamometer
at 75% of maximum engine performance (velocity, about 50 km/h)
with varying test periods (Behn et al., 1985);
[2] One turbo-charged diesel-fuelled vehicle on chassis dynamometer
at 75% of maximum engine perfornance (velocity, about 50 km/h)
and a test period of 0.5 h; three tests for each component
(Behn at al., 1985);
[3] Two diesel-fuelled truck engines at different engine speeds
(Moriske at al., 1987)
a With benzo[k]fluoranthene
b With triphenylene
Measurements of PAH concentrations in a Belgian highway tunnel in
1991 were used to calculate emission factors of 2 µg/km for
indeno[1,2,3- cd]pyrene and coronene and 32 µg/km for
benzo [ghi]perylene. The corresponding annual PAH emissions in
Belgium were estimated to be 0.11 t/year for perylene and anthanthrene
and 1.3 t/year for benzo [ghi]perylene; the combined release of
pyrene, benz [a]anthracene, chrysene, benzo [b]fluoranthene,
benzo [j]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [e]pyrene, perylene, anthanthrene, benzo [ghi]perylene,
indeno[1,2,3- cd]pyrene, dibenzo [a,c]anthracene,
dibenzo [a,h]anthracene, and coronene was 8.3 t/year (De Fré et al.,
1994).
The importance of PAH released by aircraft is also under
discussion. While Bjorseth & Ramdahl (1985) classified the maximum
emission in Norway and Sweden in 1985 of 0.1 t/year as small, Slooff
et al. (1989) estimated that the release of naphthalene, anthracene,
phenanthrene, fluoranthene, benz [a]-anthracene, chrysene,
benzo [k]fluoranthene, benzo [a]pyrene, benzo [ghi]-perylene, and
indeno[1,2,3- cd]pyrene was 51 t/year in 1985. The following
concentration ranges were measured in the exhaust gases from two US
by-pass turbine engines at various power settings: naphthalene,
0.77-4.7 µg/m3; phenanthrene, 0.46-1.3 µg/m3; pyrene, 0.15-0.61
µg/m3; fluoranthene, 0.13-0.51 µg/m3; acenaphthene, 0.03-0.21
µg/m3; anthracene, 0.029-0.12 µg/m3; benzofluoranthenes
(unspecified), 0.028-0.096 µg/m3 (isomers not specified); chrysene,
0.026-0.064 µg/m3; benzo [a]pyrene, 0.021-0.073 µg/m3;
benz [a]anthracene, 0.019-0.16 µg/m3; acenaphthylene, 0.017-0.31
µg/m3; benzo [e]pyrene, 0.017-0.057 µg/m3; dibenz [a,h]anthracene,
0.011-0.064 µg/m3; indeno[1,2,3- cd]pyrene, 0.011-0.054 µg/m3; and
benzo [ghi]perylene, 0.011-0.045 µg/m3. Cyclopenta [cd]pyrene was
not detected (limit of detection not stated) (Spicer et al., 1992).
(ii) Domestic residential heating
The main PAH released by domestic slow-combustion furnaces and
hard-coal and brown-coal coal stoves were fluoranthene, pyrene, and
chrysene, which comprised 70-80% of the total PAH in model experiments
(Ahland & Mertens, 1980). The specific emission factors for various
fuels used in residential heating are shown in Table 20 for coal
stoves and Table 21 for wood stoves (Bjorseth & Ramdahl, 1985).
Few data are available on the release of PAH from oil stoves.
Benzo [a]pyrene was detected at a concentration of < 0.05 µg/kg in
one burner-boiler combination (Meyer et al., 1980), and 0.006 and 4
µg/kg benzo [a]pyrene and 0.02 and 15 µg/kg benzo [e]pyrene were
found during testing of atomizer and vaporizer oil heating techniques,
respectively (Ahland et al., 1985). PAH emissions from residential oil
heating seem to be about one order of magnitude lower than those from
coal stoves.
Table 20. Specific polycyclic aromatic hydrocarbon emission factors (mg/kg) for residential
coal stoves
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene NR NR NR NR 65 NR
Acenaphthylene NR NR NR 0.427 NR 7.74
Anthracene 0.0039 NR > 0.595 2.113 26a,b 1.49
Anthanthrene NR NR 0.03-0.08 0.665 NR NR
Benz[a]anthracene NR NR 1.04-3.68 7.181 NR 0.61
Benzo[a]fluorene 0.0009 NR NR 1.366 NR NR
Benzo[a]pyrene 0.0003 0.014-17.4 0.043-1.3 4.303 5c NR
Benzo[b]fluoranthene 0.0002 NR 2.028d 6.102 NR NR
Benzo[b]fluorene 0.0007 NR NR 0.874 NR NR
Benzo[c]phenanthrene NR NR 1.462e 2.215 4 NR
Benzo[e]pyrene 0.0005 0.09-16.2 0.40-1.70 3.994 NR NR
Benzofluoranthenesf NR NR 0.90-3.20 NR 6 NR
Benzo[ghi]fluoranthene NR NR NR 3.323 NR 0.67
Benzo[ghi]perylene 0.0001 NR 0.30-0.50 3.855 NR NR
Benzo[j]fluoranthene NR NR NR 6.782 NR NR
Benzo[k]fluoranthene NR NR 0.569 NA NR NR
Chrysene 0.0016g NR 2.09 9.571 6h 0.68
1.39-5.60g
Coronene NR NR 0.081 1.898 NR NR
Cyclopenta[cd]pyrene NR NR 0.145 3.590 NR NR
Dibenz[a,h]anthracene NR NR 0.113 NR 5 NR
Fluoranthene 0.016 NR 3.30-17.0 28.4 9a 3.47
Fluorene NR NR < 0.065 1.05 44 1.64
Indeno[1,2,3-cd]pyrene 0.0002 0.20-0.60 4.60 NR 4 NR
1-Methylphenanthrene NR NR NR 2.217 NR NR
Naphthalene NR NR NR NR 254 35.7
Perylene NR NR 0.20-0.50 1.134 NR NR
Phenanthrene 0.046 NR > 3.69 3.984 NR 7.42
Pyrene 0.020 NR 2.98-12.0 26.589 8 3.38
Triphenylene NR NR 0.804 NR NR NR
Table 20 (continued)
NR, not reported;
[1] One new residential stove fuelled with charcoal (Ramdahl et al., 1982);
[2] Five coal types: hard-coal and brown-coal briquettes and anthracite (Ahland etal., 1985);
[3] Burning of brown coal in different domestic stoves; single values refer
to one slow-combustion stove; ranges refer to one slow-combustion stove
and one permanent built-in combustion stove at medium load (Grimmer et al.,
1983a);
[4] One slow combustion stove fueled with hard-coal briquettes (Grimmer at al., 1985);
[5] One warm-air furnace and one hot-water boiler fuelled with three different bituminous
coals (Hughes & DeAngelis, 1982);
[6] Samples from chimney of a detached family house with brown-coal heating in Leipzig, Germany
(Engewald et al., 1993)
a In particulate phase
b With phenanthrene
c With benzo[e]pyrene and perylene
d With benzo[j]fluoranthene
e With benzo[ghi]fluoranthene
f Isomers not specified
g With triphenylene
h With benz[a]anthracene
Table 21. Specific polycyclic aromatic hydrocarbon emission factors
(mg/kg) for residential wood stoves
Compound [1] [2] [3]
Anthracene 0.119-1.859 10.4-146.3a 130/3600
Benz[a]anthracene 0.060-0.781 NR 55/740
Benzo[a]fluorene 0.018-0.845 NR NR
Benzo[a]pyrene 0.046-0.617 1.1-11.6b NR
Benzo[b]fluoranthene 0.108-1.016 NR NR
Benzo[b]fluorene 0.011-0.393 NR NR
Benzo[c]phenanthrene NR 0.2-2.3 NR
Benzo[e]pyrene 0.035-0.350 NR NR
Benzofluoranthenesc NR 1.5-15.9 NR
Benzo[ghi]fluoranthene NR 0.4-6.7 NR
Benzo[ghi]perylene 0.034-0.544 1.1-9.9 NR
Chrysene 0.481-0.829d 1.3-37.1e 67/770d
Cyclopenta[cd]pyrene 0.04-0.720 0.5-8.9 15/800
Fluoranthene 0.296-3.245 1.2-31.6 190/2300
Indeno[1,2,3-cd]pyrene 0.033-0.415 NR NR
1-Methylphenanthrene 0.141-2.213 NR NR
Perylene 0.023-0.274 NR NR
Phenanthrene 0.834-8.390 NR 480/7500
Pyrene 0.232-3.822 1.3-24.0 160/2100
NR, not reported; /, single measurements;
[1] One small residential wood stove burning spruce and birch; normal and
slow burning of each kind of wood (Ramdahl at al., 1982);
[2] One zero-clearance fireplace with heat circulation and two airtight
wood stoves (baffled and non-baffled) fuelled with red oak and yellow
pine with different moisture contents (Peters at al., 1981);
[3] One wood-burning stove with and without catalytic combustor (Tan et
al., 1992)
a With phenanthrene
b With benzo[e]pyrene and perylene
c Isomers not specified
d With triphenylene
e With benz[a]anthracene
Numerous PAH, including acenaphthene, acenaphthylene, fluorene,
phenanthrene, anthracene, 1-methylphenanthrene, fluoranthene, pyrene,
benzo [a]fluorene, benzo [ghi]fluoranthene, benzo [c]phenanthrene,
cyclo-penta [cd]pyrene, benz [a]anthracene, chrysene plus
triphenylene, benzo [b]-fluoranthene, benzo [j]fluoranthene,
benzo [j]fluoranthene, benzo [e]pyrene, benzo [a]pyrene, perylene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, and anthanthrene, were
detected in atmospheric emissions from straw-burning residential
stoves, at concentrations mainly in the range of 10 µg/kg to 19 mg/kg
(Ramdahl & Muller, 1983).
The total PAH content of barbecue briquettes was 2.5-13 µg/g
sample. PAH were found in coal and charcoal briquettes but not in lava
stones or pressed sawdust briquettes (Kushwaha et al., 1985).
The PAH content of soot from domestic open fires was 3-240 mg/kg
benzo [a]pyrene, 2-190 mg/kg chrysene, 2-100 mg/kg
benz [a]anthracene, 1-77 mg/kg indeno[1,2,3- cd]pyrene, 2-39 mg/kg
benzo [e]pyrene, 1-29 mg/kg benzo [ghi]perylene, 1-18 mg/kg
coronene, 1-14 mg/kg perylene, and 1-12 mg/kg anthracene (Cretney et
al., 1985).
The amounts of PAH emitted from coal-fired domestic stoves seem
to depend on the quality of the coal used and on the firing technique.
Generally, hard coal has a higher energy content than other fuels;
thus, less total PAH is emitted per unit of gained energy. The lowest
specific emission factors for benzo [a]pyrene and benzo [e]pyrene
were found with anthracite and the highest with gas coal and gas-flame
coal (Ahland et al., 1985). Model experiments with a slow-combustion
stove showed that pitch-bound hard-coal briquettes emitted about 10
times more PAH than bitumen-bound briquettes (Ratajczak et al., 1984).
The use of pitch-bound hard-coal briquettes for domestic heating may
thus be an important source of PAH in the atmosphere. Use of this fuel
was restricted by law to permanent combustion stoves in western
Germany in 1974, and since 1976 only bitumen-bound hard-coal
briquettes have been produced there (Ratajczak et al., 1984). There is
no comparable information for other countries. The levels of airborne
PAH from a permanent combustion stove burning brown coal were two to
four times higher than those from a slow-combustion stove with a
medium load (Grimmer et al., 1983a).
About 25-1000 times more PAH are produced from burning wood than
from the same mass of charcoal. Since the yield of energy per unit
mass is similar, burning wood also produces more PAH per unit of
energy. Burning conditions are apparently the major determinant of
emission and are much more important than the kind of wood (Ramdahl et
al., 1982). In areas where domestic heating is predominantly by wood
burning, most airborne PAH may come from this source, especially in
winter (e.g.Cooper, 1980). Using benz [a]pyrene as an indicator in
extensive measurements in New Jersey, USA, the amounts emitted were
found to be more than 10 times higher during the heating period than
in seasons when heating is not required. An assessment of combustion
source also showed that residential combustion of wood was the
decisive factor (Harkov & Greenberg, 1985). About 43-47% of the total
PAH released in winter in Fairbanks, Alaska, came from residential
wood stoves (Guenther et al., 1988).
The PAH concentrations in gases in the chimney stacks of
residential coal and oil furnaces are given in Table 22. The highest
levels were found during the start of the burning process (Brockhaus &
Tomingas, 1976). Measurements with five qualities of coal showed that
Extrazit(R), a specially treated coal, emitted smaller quantities of
smoke and the lowest PAH levels, and anthracite briquettes emitted the
highest levels. Presumably, the high PAH emissions from anthracite
briquettes are due to the binding agent, hard coal-tar, which has an
especially high PAH content. Furnaces with atomizer oil burners seemed
to emit less PAH than those with vaporizers. Measurements in a
slow-combustion stove and a tiled stove showed that the highest
concentrations of PAH were associated with dust of a particle size of
< 2.1 m. As for residential heating with wood, in areas where the
predominant form of domestic heating is coal burning, a major
proportion of airborne PAH may come from this source, especially in
winter (Moriske et al., 1987).
Table 22. Polycyclic aromatic hydrocarbon concentrations (µg/m3)
in stack gases from residential coal and oil stoves
Compound Coal Oil
Benz[a]anthracene 0.0157-2630 0.2-0.6
Benzo[a]pyrene 0.0016-1270 0.19-0.67
Benzo[b]fluoranthene 0.0188-3270 0.004-0.68
Benzo[e]pyrene 0.0261-3430 0.4-6.9
Benzo[ghi]perylene 0.010-1670 0.41-3.4
Benzo[k]fluoranthene 0.0044-1250 0.18-0.36
Chrysene 0.0142-2590 0.1-0.5
Coronene 0.003-710 0.15-0.47
Dibenz[a,h]anthracene 0.002-410 NR
Fluoranthene 0.0393-6830 0.0134
Perylene 0.0015-2730 0.31-0.8
Pyrene 0.0066-1650 0.1-0.9
From Brockhaus & Tomingas (1976); one permanent combustion stove
burning anthracite and brown-coal briquets and vaporizer and
atomizer oil burners; NR, not reported
Estimates of annual PAH emissions due to residential heating are
available for a few countries:
- In western Germany, the benzo [a]pyrene emissions were about 10
t in 1981 (Ahland et al., 1985), 7 t in 1985, and 2.5 t in 1988,
mainly resulting from coal heating. The reduction in the release
of PAH into the atmosphere due to domestic heating resulting from
increasing use of oil and gas during the last 30-40 years was
estimated to be 90-99% (Zimmermeyer et al., 1991).
- In the Netherlands, the estimated release in 1985 was < 1 t/year
each for benzo [k]fluoranthene and indeno[1,2,3- cd]pyrene,
< 10 t/year each for anthracene, fluoranthene,
benz [a]anthracene, chrysene, benzo [a]-pyrene, and
benzo[ghi]perylene, and 48-70 t/year each for naphthalene and
phenanthrene, mainly resulting from wood heating (Slooff et al.,
1989).
- The total PAH input, mainly from coal and wood heating, was about
63 t in Norway, 130 t in Sweden, and 720 t in the USA in 1985
(Bjorseth & Ramdahl, 1985).
- In Canada in 1990, the total PAH released due to residential
heating, mainly wood burning, was about 500 t (Environment
Canada, 1994).
(iii) Open burning
PAH may be released to the atmosphere during forest and
agricultural fires, burning of accidentally spilled oil, disposal of
road vehicles and especially automobile tyres, open burning of coal
refuse and domestic and municipal waste, and open fires. The release
of PAH into the atmosphere from the burning of wastes, including road
vehicles, in the open is decreasing in industrialized countries due to
comprehensive regulations.
Laboratory experiments with pine needles gave the following
specific PAH emission factors (per kg pine needle): 980-20 000 g
pyrene, 690-15 000 g fluoranthene, 580-12 000 g anthracene plus
phenanthrene, 540-29 000 g chrysene plus benz [a]anthracene,
420-6200 g benzo [ghi]-perylene, 170-4300 g
indeno[1,2,3- cd]pyrene, 140-8800 g benzo [c]-phenanthrene,
130-13 000 g benzofluoranthenes (isomers not specified), 61-800 g
benzo [e]-pyrene, 38-3500 g benzo [a]pyrene, and 24-2100 g
perylene, depending on the amount of needles, area, and type of fire.
Fires moving with the wind and low fuel loading resulted in
significantly smaller amounts of PAH than fires moving against the
wind and high fuel loading (McMahon & Tsoukalas, 1978). The emission
factor for acenaphthene was 230-1000 µg/kg dry straw (Ramdahl &
Mller, 1983) and 660 µg/kg dry wood (Alfheim et al., 1984).
In model experiments with crude oil spilled on water, numerous
PAH were found, including acenaphthene, acenaphthylene, phenanthrene,
anthracene, 1-methylphenanthrene, fluoranthene, pyrene, fluorene,
benzo [a]fluorene, benzo [b]fluorene, benz [a]anthracene, chrysene
plus triphenylene, benzo [b]-fluoranthene, benzo [ghi]fluoranthene,
benzo [e]pyrene, benzo [a]pyrene, perylene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, and coronene, at
concentrations of ¾ 1000 mg/kg individual substance in both the soot
and the burn residue (Benner et al., 1990). Even though the open
burning of oil spilled on water results in a lower PAH content than in
crude oil (see Table 8), this source may be of local importance, e.g.
near tanker accidents.
Between the early and the mid-1970s, the total release of PAH
(including nitrogen-containing analogues and quinone degradation
products) into the atmosphere in the USA due to open burning was
estimated to be about 4000 t/year (Agency for Toxic Substances and
Disease Registry, 1990). The total PAH input from forest and
agricultural fires in 1985 was estimated to be 13 t in Norway, 1.3 t
in Sweden, and 1000 t in the USA, and that from open fires to be 0.4 t
in Norway and 100 t in the USA (Bjorseth & Ramdahl, 1985). The release
of all PAH into the atmosphere from the burning of scrap electrical
cable in 1988 was about 17 t (Slooff et al., 1989). In Canada in 1990,
the total PAH emissions from agricultural burning and open-air fires
were estimated to be about 360 t and those from forest fires to be
about 2000 t (Environment Canada, 1994).
(iv) Other diffuse sources
The total PAH released into the atmosphere in the Netherlands
from roofing tar and asphalt in 1988 was estimated at 0.5 t/year
(Slooff et al., 1989).
(c) Emissions to the hydrosphere
(i) Motor vehicle traffic
The main source of PAH in the aqueous environment as a result of
motor vehicle traffic is highway run-off, which contains asphalt and
soot particles and is washed by rainfall and storm water or snow into
surface waters and soil (see also 3.2.7.2 (a) (i)). The available data
are summarized in Table 23. Higher PAH concentrations were found in
highway run-off in winter than in summer; this was attributed to the
increased abrasion of the road surface due to use of steel-studded
tyres in winter (Berglind, 1982).
It was estimated that an average of < 10 µg/km per vehicle per
day of total PAH are transported via pavement runoff water. Most is
transported to nearby surroundings as small particles of dust (see
also section 3.2.7.2; Lygren et al., 1984). In contrast, storm water
runoff near a US highway was of considerable importance for adjacent
water bodies. In the test area, over 50% of the total PAH input into a
nearby river came from highway runoff. The runoff loading factor was
given as 24 mg/km per vehicle (Hoffman et al., 1985).
Table 23. Polycyclic aromatic hydrocarbon concentrations (µg/litre) in highway runoff
Compound [1] [2] [3] [4] [5]
Acenaphthene 0.016/0.087 0.195/5.126 NR NR NR
Acenaphthylene 0.045 0.557/16.804 NR NR NR
Anthracene 0.042-0.214 0.486/8.917 0.379 0.165 0.246
Benzo[j+k]fluoranthene 0.089/0.277 NR NR NR 0.207
Benz[a]anthracene 0.031-0.139 0.341/0.863 0.677 0.228 NR
Benzo[a]fluorene 0.018-0.170 0.587 NR 0.179 0.396
Benzo[a]pyrene 0.061-0.120 0.537/1.255 0.602 0.250 NR
Benzo[b]fluoranthene 0.129/0.157 NR NR 0.799 1.501
Benzo[b]fluorene 0.033/0.097 0.356/0.366 NR NR 0.192
Benzo[c]phenanthrene NR 0.250 NR NR NR
Benzo[e]pyrene 0.108/0.202 0.238/1.665 0.609 0.360 0.630
Benzofluoranthenesa 0.401/0.695 1.087/2.712 1.171 NR NR
Benzo[e]perylene 0.100-0.299 NR 0.551 0.391 0.319
Chyrsene + triphenylene 0.194-0.433 1.472/2.752 1.147 0.665 1.070
Fluoranthene 0.321-1.573 4.065/15.322 2.665 1.820 3.143
Fluorene 0.0088-0.564 0.432/11.093 0.096 0.485 1.237
Indeno[1,2,3-cd]pyrene 0.061-0.154 0.344/0.666 NR NR NR
1-Methylphenanthrene 0.030-1.073 0.637/2.308 1.366 2.117
Naphthalene NR 2.59 NR 0.123 0.195
Perylene 0.048 NR NR NR NR
Phenanthrene 0.068-2.668 3.297/38.10 1.385 4.055 6.787
Pyrene 0.363-1.449 3.026/12.094 2.002 1.886 3.066
NR, not reported; /, single measurements;
[1] Run-off samples from a Norwegian highway north of Oslo in summer and winter
1980-82 (Berglind, 1982);
[2] Snow 20 and 50 m from the same highway in February 1981 (Berglind, 1982);
[3] Snow from a frozen Norwegian lake 50 m from a highway with high traffic density in
winter 1981-82 (Gjessing at al., 1984);
[4] Snow from a Norwegian highway south of Oslo with concrete pavement, February 1972
(Lygren at al., 1984);
[5] Snow from a Norwegian highway south of Oslo with asphalt pavement, February 1972
(Lygren et al., 1984)
When the water samples were filtered through solid sorbents, the results may be
underestimates of the actual content (see section 2.4.1.4).
a Isomers not specified
(ii) Sewage treatment
The concentrations of PAH in final effluents from municipal
sewage treatment facilities are generally in the low microgram per
litre range and are almost always < 0.1 µg/litre (Nicholls et al.,
1979; Young et al., 1983; van Luin & van Starkenburg, 1984; Kröber &
Häckl, 1989). Maximum values of 29 µg/litre naphthalene and 7 µg/litre
acenaphthene were detected in one US sewage treatment plant, and 8
µg/litre benzo [a]pyrene were found in one German plant (Young et
al., 1983; Kröber & Häckl, 1989), but no explanation was given for
these unusually high concentrations. It was concluded that final
effluents contain PAH at a background level (van Luin & van
Starkenburg, 1984).
Naphthalene was found at a concentration of 9.3 kg/year in the
final effluent from one US municipal sewage plant (Hoffman et al.,
1984). The annual emissions of naphthalene, anthracene, phenanthrene,
fluoranthene, benz [a]anthracene, chrysene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene
from Dutch sewage treatment plants into surface waters were estimated
to be about 0.6 t. The amount of these PAH transported into the
Netherlands from other European countries via the Rhine, Meuse, and
Scheldt rivers was estimated to be 65 t/year (year and database not
given). The main compounds were fluoranthene (18 t/year) and
naphthalene (15 t/year) (Slooff et al., 1989).
(iii) Other sources
PAH have been found in wastewaters from power stations, from
garages with car-wash devices, and from a German car-wash storage tank
at the following concentrations: fluoranthene, 1.3-7.7 µg/litre;
pyrene, 3.5-28 µg/litre; benz [a]anthracene, 0.49-1.9 µg/litre;
chrysene, 1.2-6.0 µg/litre; benzo [e]pyrene, 4.7-16 µg/litre;
benzo [a]pyrene, 0.40-8.8 µg/litre; benzo [b]fluoranthene, 1.2-3.6
µg/litre; and benzo [k]-fluoranthene, 0.51-0.72 µg/litre (Baumung et
al., 1985). Wastewaters from power stations could be an important
local source of PAH.
Numerous PAH were detected in leachate plumes from refuse
landfills in western Germany and the USA (Grimmer et al., 1981b; Götz,
1984; Reinhard et al., 1984). Concentrations < 0.1 µg/litre were
detected of benzo [ghi]-fluoranthene, benz [a]anthracene,
benzo [c]phenanthrene, chrysene, benzofluoranthenes (isomers not
specified), benzo [a]pyrene, benzo [e]pyrene, perylene,
anthanthrene, benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene
(Grimmer et al., 1981b). Naphthalene was found at a concentration >
100 µg/litre, and acenaphthene, fluorene, anthracene, phenanthrene,
and pyrene were found at concentrations of 1-30 µg/litre (Götz, 1984;
Reinhard et al., 1984). The importance of this source for groundwater
pollution cannot be estimated from the available data.
(c) Emissions to the geosphere
(i) Motor vehicle traffic
PAH were deposited within 100 m of a highway at a concentration
of 100-200 µg/km per vehicle per day in winter as small particles of
dust resulting from the abrasion of asphalt by steel-studded tyres
(Lygren et al., 1984). Studies of adsorption on various soil types
showed that most PAH in highway runoff is retained on the soil surface
(Gjessing et al., 1984).
(ii) Open burning
Phenanthrene, fluoranthene, triphenylene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [ghi]perylene, indeno[1,2,3- cd]pyrene, and
coronene were determined in the soil of burning sites in western
Oregon, USA. Before burning, the PAH concentrations in the top 2 cm of
the soil layer ranged from 0.8 ng/g dry weight for benzo [a]pyrene to
4.4 ng/g for fluoranthene and triphenylene. One week after burning,
the concentrations ranged from 0.9 ng/g for benzo [k]fluoranthene to
19 ng/g for triphenylene. The finding that the PAH levels did not
increase appreciably after burning indicates that the bulk of the PAH
were retained within the litter rather than passing into the soil
(Sullivan & Mix, 1983).
(iii) Disposal of sewage sludge and fly ash from incineration
When sewage sludge is applied to soils, adsorbed PAH are added to
the geosphere. The PAH concentrations in municipal aerobic and
anaerobic sewage sludge are given in Table 24.
In a detailed survey of the PAH concentrations in soil to which
anaerobic sludges had been applied between 1942 and 1961 in the United
Kingdom, the total PAH content increased to over 125 mg/kg up to 1948
but had decreased to about 29 mg/kg by 1961. The authors attributed
the declining levels to a decrease in atmospheric PAH contamination
from smoke emissions (Wild et al., 1990). No seasonal variation in the
content or profile of PAH was detected in western Germany by Grimmer
et al. (1980), but Süss (1980) found the highest PAH load in sewage
sludge in January-April and the lowest in July and October. Human
faeces seemed to contribute little to the PAH content of sewage sludge
(Grimmer et al., 1980). The most important emission sources could not
be identified, but McIntyre et al. (1981) concluded that the PAH
content of sewage sludge originating from British treatment works with
significant flows of industrial effluent was higher than that in works
dealing with predominantly domestic effluents.
After application of compost over three years to an agricultural
soil in Spain, no accumulation of PAH was observed (Gonzalez-Vila et
al., 1988). It was shown, however, that the extent of accumulation is
dependent on the duration, frequency, and concentration of
application. After 10 years of sludge spreading, considerable
quantities of PAH were detected in both a sandy loam and a clay soil
Table 24. Polycyclic aromatic hydrocarbons concentrations (mg/kg dry weight) in municipal sewage sludge
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene NR NR NR NR NR NR ND NR
Anthracene NR NR NR 0.89-44 NR NR ND-10.0 NR
Anthanthrene 0.00-2.10 0.03-1.8 NR NR NR NR NR NR
Benz[a]anthracene 0.62-19.0 0.91-17.3 NR NR NR NR ND-2.1 NR
Benzo[a]fluorene 0.28-9.00 0.56-7.9 NR NR NR NR NR NR
Benzo[a]pyrene 0.54-13.3 0.41-14.3 0.12-9.14 NR NR 0.29-2.00 ND-0.64 NR
Benzo[b]fluoranthene NR NR 0.06-9.14 NR < 1-1.3 0.29-1.80 ND-1.100 NR
Benzo[e]pyrene 0.53-12.4 0.48-12.3 NR NR NR NR NR NR
Benzofluoranthenesa 1.07-23.7 1.02-24.8 NR NR NR NR NR NR
Benzo[ghi]perylene 0.40-8.70 0.34-10.9 0.06-9.14 NR NR < 0.1-3.41 ND-1.21 NR
Benzo[k]fluoranthene NR NR 0.06-4.57 NR NR 0.15-1.00 ND-0.500 NR
Chrysene 0.78-23.7 1.24-22.2 NR 0.25-13 NR NR NR NR
Dibenz[a,h]anthracene NR NR NR 13 NR NR ND-0.25 NR
Fluoranthene 0.61-51.6 4.10-28.2 0.34-11.45 0.35-7.1 < 1-10.4 0.54-7.67 0.216-5.14 5.2/5.6b
Fluorene NR NR NR NR NR NR ND-2.9c 3.5/5.8
Indeno[1,2,3-cd]pyrene 0.30-7.40 0.28-9.4 0.06-6.68 NR NR 0.24-2.08 ND-0.640 NR
Naphthalene NR NR NR 0.9-70 NR NR NR 4.5/8.6
Perylene 0.14-6.40 0.09-3.1 NR NR NR NR NR NR
Phenanthrene NR NR NR 0.89-44 NR NR 0.30-40 15.2/18.6d
Pyrene 0.90-47.2 3.20-25.3 NR 0.33-18N R NR ND-7.6 NR
NR, not reported; /, single measurements; ND, not detected (limits of detection, 0.2-1 mg/kg);
[1] Samples from 25 sewage treatment plants in western Germany 1976-78 (Grimmer et al., 1980);
[2] Samples from three sewage treatment facilities in western Germany before 1979 (Suss, 1980);
[3] Samples from 12 British sewage treatment works (McIntyre at al., 1981);
[4] Samples from 20 US sewage treatment works (Naylor & Loehr, 1982);
[5] Samples from six Dutch municipal sewage treatment plants (van Luin & van Starkenburg, 1984);
[6] 31 sludge samples from different sewage treatment works in western Germany (Witte et al., 1988);
[7] Anaerobic sludge samples from 13 sewage treatment plants in western Germany 1985-88 (Krober & Hackl, 1989);
[8] Anaerobic sludge samples from one Spanish sewage treatment facility in spring 1985 and autumn 1986
(Gonzalez-Villa at al., 1988).
a Isomers not specified
b With pyrene
c With acenaphthylene
d With anthracene
(Diercxsens & Tarradellas, 1987). The annual addition of PAH to soil
from sewage sludge in the Netherlands was estimated as follows: 0.1 t
naphthalene, 0.1 t anthracene, 1.5 t phenanthrene, 2.3 t fluoranthene,
0.6 t benzo [a]anthracene, 0.6 t chrysene, 0.4 t
benzo [k]fluoranthene, 0.6 t benz [a]pyrene, 0.6 t
benzo [ghi]-perylene, and 0.6 t indeno[1,2,3- cd]pyrene (year and
database not given; Slooff et al., 1989).
The annual contribution of PAH to landfill in the United Kingdom
from fly ash from coal combustion (see also Table 16) exceeded that
from municipal solid-waste incineration by a factor of about 10, with
the exception of naphthalene, the level of which was about 20 000-fold
higher in fly ash from coal combustion than in that from solid-waste
incineration. The annual PAH loads from solid-waste incineration were
about 0.01 kg naphthalene and 3.5 kg benzo [ghi]perylene, whereas
those from coal combustion were about 15 kg each of anthracene,
benzo [k]fluoranthene, and dibenz [a,h]anthracene and 1200 kg pyrene
(Wild et al., 1992).
(iv) Waste dumping
Soil cores taken from a hazardous waste disposal site in Spain
containing petroleum tar residues and lubricating oils as the major
organic wastes contained 62 mg/kg 1-methylphenanthrene, 53 mg/kg
naphthalene, 52 mg/kg benzo [a]fluorene, 30 mg/kg
benzo [ghi]fluoranthene, 25 mg/kg benzo [c]-phenanthrene, 0.5-0.71
mg/kg acenaphthene, 0.2-48 mg/kg fluorene, 0.2-390 mg/kg phenanthrene,
0.110 mg/kg anthanthrene, 0.1-210 mg/kg pyrene, 0.1-200 mg/kg
acenaphthylene, 0.1-140 mg/kg anthracene, 0.1-140 mg/kg
benzo [e]pyrene, 0.1-145 mg/kg benzo [a]pyrene, 0.1-50 mg/kg
benzo [b]fluorene, 0.08-130 mg/kg chrysene plus triphenylene, 0.08-90
mg/kg indeno[1,2,3- cd]pyrene, 0.06-130 mg/kg benz [a]anthracene,
0.05-290 mg/kg fluoranthene, 0.03-75 mg/kg benzo [ghi]perylene,
0.03-0.2 mg/kg perylene, and 0.01-0.4 mg/kg dibenz [a,h]anthracene
(Navarro et al., 1991).
There can be appreciable movement of PAH into soil from waste
dumping, especially of hazardous refuse. The dumping conditions are
decisive for the amount of PAH released. Annual emissions of PAH in
the Netherlands in 1987 due to the spreading of contaminated composts
onto soils were estimated to be 1 t benz [a]anthracene, 1 t chrysene,
1 t benzo [k]fluoranthene, 0.5 t benzo [ghi]-perylene, 0.5 t
indeno[1,2,3-cd]pyrene, and 0.4 t benzo [a]pyrene (Slooff et al.,
1989).
(d) Biosphere
Perch (Perca fluviatilis) were not significantly contaminated
after an oil spill in Finland due to a tanker accident. The
concentrations of acenaphthene, acenaphthylene, fluorene,
phenanthrene, anthracene, 1-methylphenanthrene, fluoranthene, pyrene,
benzo [a]fluorene, benzo [b]fluorene, chrysene, triphenyl-ene, and
benzofluoranthenes in both contaminated and control groups were
between < 0.1 and 0.2 µg/kg each in muscle and < 0.1 and 16
µg/kg in bile. The investigators concluded that the fish with the
highest load would probably not have survived and others had moved to
less contaminated areas. Additionally, the cold climate caused
clumping of the spilled oil, which then drifted to the coast
(Lindström-Seppä et al., 1989; see also sections 4.1.5.1 and 5.1.7.1).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
Appraisal
The transport and distribution of polycyclic aromatic
hydrocarbons (PAH) in the environment depend on their physicochemical
properties of very low solubility in water and low vapour pressure,
and high partition coefficients for n-octanol:water (log Kow) and
organic carbon:water (log Koc). PAH are stable towards hydrolysis
as they have no reactive groups. In the gaseous phase, PAH and
particularly those of higher molecular mass, are mainly adsorbed to
particulate matter and reach the hydrosphere and geosphere by dry and
wet deposition. Little is volatilized from water phases owing to their
low Henry's law constants. The log Koc values indicate strong
adsorption to the organic matter of soils, so that migration does not
usually occur. The log Kow values indicate high bioaccumulation.
Few experimental data are available on the biodegradation of PAH.
In general, they are biodegradable under aerobic conditions, and the
biodegradation rates decrease drastically with the number of aromatic
rings. Under anaerobic conditions, biodegradation appears to be very
slow.
The bioconcentration factors measured in the water phase vary
widely according to the technique used. High values are seen for some
algae, crustaceans, and molluscs, but those for fish are much lower
owing to rapid biotransformation. The bioaccumulation factors for
aquatic and terrestrial organisms in sediment and soil are generally
very low, probably because of the strong adsorption of PAH onto the
organic matter of soils and sediments, resulting in low
bioavailability.
The photodegradation of PAH in air and water has been studied
intensively. The most important degradation process in both media is
indirect photolysis under the influence of radicals like OH, O3, and
NO3. The measured degradation rate constants vary widely according to
the technique used. Under laboratory conditions, the half-life of the
reaction of PAH with airborne OH radicals is about one day. Adsorption
of high-molecular-mass PAH onto carbonaceous particles in the
environment has a stabilizing effect. Formation of nitro-PAH has been
reported from two- to four-ring PAH in the vapour phase during
photooxidation with NO3. For some PAH, photodegradation in water
seems to be more rapid than in air.
According to model calculations based on physicochemical and
degradation parameters, PAH with four or more aromatic rings persist
in the environment.
4.1 Transport and distribution between media
4.1.1 Physicochemical parameters that determine environmental transport
and distribution
The transport and distribution of PAH in the environment are the
result of the following physicochemical parameters:
- Aqueous solubility: PAH are hydrophobic compounds with very low
solubility in water under environmental conditions: the maximum
at room temperature is 32 mg/litre for naphthalene, and the
minimum is 0.14 µg/litre for coronene (see Table 4).
- Vapour pressure: The vapour pressure of PAH under environmental
conditions is very low: the maximum at room temperature is 10.4
Pa for naphthalene, and the calculated minimum is 3 × 10-12 Pa
for dibenzo [a,i]pyrene (see Table 4).
- n-Octanol:water partition coefficient (log Kow): The
affinity of PAH to organic phases is much higher than that for
water. The log Kow values range from 3.4 for naphthalene to 7.3
for dibenzo [a,i]pyrene (see Table 4), indicating that the
potential for bioaccumulation is high.
- Organic carbon:water partition coefficient (log Koc): The
sorption coefficients of PAH to the organic fraction of sediments
and soils are summarized in Table 25. The high values indicate
that PAH sorb strongly to these fractions. The wide variation in
the results for individual compounds are due to the very long
exposure necessary to reach steady-state or equilibrium
conditions, which can lead to underestimation of sorption
coefficients; furthermore, degradation in the overlying aqueous
phase can lead to overestimates of the actual values.
4.1.2 Distribution and transport in the gaseous phase
PAH are emitted mainly to the atmosphere (see Section 3), where
they can be both transported in the vapour phase and adsorbed onto
particulate matter. The distribution between air and particulate
matter under normal atmospheric conditions depends on the
lipophilicity, vapour pressure, and aqueous solubility of the
substance. Generally, PAH with few (two to four) aromatic rings occur
in the vapour phase and are adsorbed, whereas PAH consisting of more
aromatic rings exist mainly in the adsorbed state (Hoff & Chan, 1987;
McVeety & Hites, 1988; Baker & Eisenreich, 1990). PAH are usually
adsorbed onto particles like fly ash and soot that are emitted during
combustion.
Table 25. Organic carbon normalized sorption coefficients (Koc) of polycyclic aromatic hydrocarbons
Compound log Koc Comments Reference
Acenaphthene 5.38 Average on sediments Kayal & Connell (1990)
3.79 RP-HPLC on CIHAC Szabo et al. (1990)
3.59 RP-HPLC on PIHAC Szabo at al. (1990)
Acenaphthylene 3.83 RP-HPLC on CIHAC Szabo et al. (1990)
3.75 RP-HPLC on PIHAC Szabo et al. (1990)
Anthracene 4.42 Average sorption isotherms on Karickhoff et al. (1979)
sediment
3.74 Suspended particulates Herbes et al. (1980)
4.20 Soil, shake flask, UV Karickhoff (1981)
3.95/4.73 Lake Erie with 9.6 mg C/litre Landrum et al. (1984a)
4.87/5.70 Huron river with 7.8 mg C/litre Landrum et al. (1984a)
4.20 Soil, shake flask, LSC Nkedl-Kizza et al. (1985)
4.93 Fluorescence, quenching interaction Gauthier et al. (1986)
with humic acid
4.38 HPLC Hodson & Williams (1988)
5.76 Average on sediments Kayal & Connell (1990)
4.41 RP-HPLC Pussemier et al. (1990)
4.53 RP-HPLC on CIHAC Szabo at al. (1990)
4.42 RP-HPLC on PIHAC Szabo at al. (1990)
Benz[a]anthracene 4.52 Suspended particles Herbes et al. (1980)
6.30 Average on sediments Kayal & Connell (1990)
7.30 Specified particulate Bromen et al. (1990)
Benzo[a]pyrene 6.66 LSC Eadie et al. (1990)
6.26 Average on sediments Kayal & Connell (1990)
8.3 Specified particulate Broman et al. (1990)
4.0 Predicted to be dissolved Broman et al. (1990)
Benzo[e]pyrene 7.20 Specified particulate Broman at al. (1990)
4.00 Predicted to be dissolved Broman at al. (1990)
Benzo[k]fluoranthene 5.99 Average on sediments Kayal & Connell (1990)
7.00 Specified particulate Broman et al. (1990)
4.00 Predicted to be dissolved Broman at al. (1990)
Table 25. (continued)
Compound log Koc Comments Reference
Chrysene 6.27 Average on sediments Kayal & Connell (1990)
6.90 Specified particulate Broman et al. (1990)
4.0 Predicted to be dissolved Broman at al. (1990)
Coronene 7.80 Specified particulate Broman et al. (1990)
5.0 Predicted to be dissolved Broman et al. (1990)
Dibenz[a,h]anthracene 6.31 Average of 14 soil or sediment Means et al. (1980)
samples, shake flask, LSC
Fluoranthene 6.38 Average on sediments Kayal & Connell (1990)
4.74 RP-HPLC on CIHAC Szabo et al. (1990)
4.62 RP-HPLC on PIHAC Szabo et al. (1990)
6.30 Specified particulate Broman et al. (1990)
4.0 Predicted to be dissolved Broman et al. (1990)
Fluorene 5.47 Average on sediments Kayal & Connell (1990)
3.76 RP-HPLC Pussemier et al. (1990)
4.15 RP-HPLC on CIHAC Szabo et al. (1990)
4.21 RP-HPLC on PIHAC Szabo et al. (1990)
Naphthalene 3.11 Average sorption isotherms on Karickhoff at al. (1979)
sediments
2.38 Suspended particulates Herbes et al. (1980)
2.94 Karickhoff (1981)
3.0 McCarthy & Jimenez (1985);
McCarthy et al. (1985)
2.73-3.91 Aquifer materials Stauffer et al. (1989)
3.15/2.76 Podoll et al. (1989)
5.00 Average on sediments Kayal & Connell (1990)
2.66 Average on sediments Kishi et al. (1990)
3.11 Soil, RP-HPLC Szabo et al. (1990)
3.29 Sandy surface soil Wood et al. (1990)
Table 25. (continued)
Compound log Koc Comments Reference
Phenanthrene 4.36 Average sorption isotherms on Karickhoff et al. (1979)
sediments
4.28 Hodson & Williams (1988)
6.12 Average on sediments Kayal & Connell (1990)
4.22 RP-HPLC on CIHAC Szabo et al. (1990)
4.28 RP-HPLC on PIHAC Szabo et al. (1990)
4.42 Sandy surface soil Wood et al. (1990)
Pyrene 4.92 Average isotherms on sediments Karickhoff et al. (1979)
4.90 Sediment, shake flask, sorption Karickhoff et al. (1979)
isotherm
4.81 Average of soil and sediment Means et al. (1979)
Shake flask, LSC, sorption
isotherms
4.80 Average of 12 soils and sediments Means et al. (1980)
Shake flask, LSC, sorption isotherms
4.78 Soil and sediment; calculated Kow Means at al. (1980)
4.83 Sorption isotherms Karickhoff (1981)
3.11/3.46 Sediment suspensions Karickhoff & Morris (1985)
4.80/5.13 Hodson & Williams (1988)
5.65 LSC Eadie et al. (1990)
5.29 Soil Jury at al. (1990)
6.51 Average on sediments Kayal & Connell (1990)
4.83 RP-HPLC Pussemier et al. (1990)
4.82 RP-HPLC on CIHAC Szabo et al. (1990)
4.77 RP-HPLC on PIHAC Szabo et al. (1990)
6.50 Specified particulate Broman et al. (1990)
4.0 Predicted particulate Broman et al. (1990)
Triphenylene 6.90 Specified particulate Broman et al. (1990)
4.00 Predicted to be dissolved Broman et al. (1990)
RP-HPLC, reversed-phase high-performance liquid chromatography; CIHAC, chemical-induced humic-acid column;
PIHAC, physical-induced humic-acid column; UV, ultraviolet; C, carbon; LSC, liquid scintillation
chromatography
PAH are ubiquitous in the environment, probably because they are
distributed for long distances without significant degradation (Lunde,
1976; De Wiest, 1978; Bjorseth & Sortland Olufsen, 1983; McVeety &
Hites, 1988), e.g. from the United Kingdom and the European continent
to Norway and Sweden during winter (Bjorseth & Lunde, 1979). Washout
ratios calculated from measurements in rain and snow in the area of
northern Lake Superior, during one year showed that airborne PAH
adsorbed onto particulate matter result in effective wet deposition,
while gaseous PAH are removed to only a minor degree (McVeety & Hites,
1988).
4.1.3 Volatilization
Henry's law constant gives a rough estimate of the equilibrium
distribution ratio of concentrations in air and water but cannot
predict the rate at which chemicals are transported between water and
air. The constants for PAH are very low, ranging from 49 Pa .m3/mol
for naphthalene to 0.000449 Pa .m3/mol for dibenzo [a,i]pyrene (see
Table 4). The rates of removal and volatilization of PAH (Table 26)
are strongly dependent on environmental conditions such as the depth
and flow rate of water and wind velocity. Although PAH are released
into the environment mainly in air, considerably higher concentrations
are found in aqueous samples because of the low vapour pressure and
Henry's law constants of PAH.
The volatilization half-life for naphthalene from a 22.5-m water
body was found experimentally to be 6.3 h, whereas the calculated
value was 2.1 h (Klöpffer et al., 1982). Calculations based on a
measured air:water partition coefficient for river water 1 m deep with
a water velocity of 0.5 m/s and a wind velocity of 1 m/s gave a
volatilization half-life of 16 h for naphthalene (Southworth, 1979).
The value calculated for evaporative loss of naphthalene from a 1-m
water layer at 25°C was of the same order of magnitude (Mackay &
Leinonen, 1975). Naphthalene was volatilized from soil at a rate of
30% after 48 h, with neglible loss of PAH with three or more rings
(Park et al., 1990).
4.1.4 Adsorption onto soils and sediments
PAH are adsorbed strongly to the organic fraction of soils and
sediments (see section 4.1.1 and Table 25). Some PAH may be degraded
biologically in the aerobic soil layer, but this process is slow,
because sorption to the organic carbon fraction of the soil reduces
the bioavailability. For the same reason, leaching of PAH from the
soil surface layer to groundwater is assumed to be negligible,
although detectable concentrations have been reported in groundwater
(see section 5.1.2.2).
Table 26. Rates of volatilization of polycyclic aromatic hydrocarbons
Compound Rate constant Half-life (h)a Comments Reference
Anthracene Removal rate constants (estimated) from Southworth (1977)
water column
At 25°C in midsummer sunlight:
0.002 h-1 347 - in deep, slow, somewhat turbid water
0.001 h-1 693 - in deep, slow, muddy water
0.002 h-1 347 - in deep, slow, clear water
0.042 h-1 17 - in shallow, fast, clear water
0.179 h-1 3.9 - in very shallow, fast, clear water
62 Calculated half-life for a river 1 m deep Southworth (1979)
with water velocity of 0.5 m/s and wind
velocity of 1 m/s
Benz[a]anthracene 500 Calculated half-life for a river 1 m deep Southworth (1979)
with water velocity of 0.5 m/s and wind
velocity of 1 m/s
Benzo[a]pyrene 1550 Calculated half-life for a river 1 m deep Southworth (1979)
with water velocity of 0.5 m/s and wind
velocity of 1 m/s
<1 × 10-5 S-1 > 19 Sublimation rate constant from glass Cope & Kalkwarf
surface at 24 °C at an airflow of 3 litre/min (1987)
Naphthalene 1.675 × 10-9 Rate of evaporation estimated at 20 00 Guckel at al. (1973)
mol.cm-2h-1 and air flow of 50 litre/h
7.15 Calculated half-life from 1 m depth of water Mackay & Leinonen
(1975)
16 Half-life for surface waters Southworth
(1979)
200 In a lake, considering current velocity and
wind speed in combination with typical
re-aeration rates
Perylene <1 × 10-5 S-1 > 19 Sublimation rate constant from glass Cope & Kalkwarf
surface at 24°C at an air flow of 3 litre/min (1987)
Pyrene 1.1 × 10-4 S-1 1.8 Sublimation rate constant as loss from Cope & Kalkwarf
glass surface at 24°C at an air flow of 3 litre/min (1987)
Table 26 (continued)
For comparison of results for which only rate constants are reported, half-lives have been estimated from the equation:
t1/2 = In2
k
where t1/2 is the half-life and k is the rate constant. The calculated values are reported in italics.
4.1.5 Bioaccumulation
The ability of a substance to bioconcentrate in organisms in the
aqueous phase is expressed as the bioconcentration factor. For
substances like PAH, with high n-octanol:water partition
coefficients, long exposures are necessary to achieve equilibrium
conditions, so that results obtained under non-equilibrium conditions
can result in underestimates of the bioconcentration factor.
Bioaccumulation may also vary with the metabolic capacity of the
organism (see section 4.2.1.2).
Bioconcentration can also be calculated as the ratio between the
rates of uptake (k1) and depuration (k2). This method has the
advantage that relatively short exposures can be used. It is therefore
preferred for PAH, as constant concentrations of compounds like
benzo [a]pyrene are very difficult to maintain over a long period.
4.1.5.1 Aquatic organisms
Aquatic organisms may accumulate PAH from water, sediments, and
their food. In general, PAH dissolved in pore water are accumulated
from sediment (McElroy & Sisson, 1989), and digestion of sediment may
play an important role in the uptake of PAH by some species. Although
organisms can accumulate PAH from food, the relative importance of
uptake from food and water is not clear (Farrington, 1991).
The bioconcentration factors of PAH in different species are
shown in Table 27; this is not a comprehensive presentation of all of
the available data but provides examples of the accumulation of some
PAH in different groups of organisms. Species that metabolize PAH to
little or no extent, like algae, oligochaetes, molluscs, and the more
primitive invertebrates (protozoans, porifers, and cnidaria),
accumulate high concentrations of PAH, as would be expected from their
log Kowvalues, whereas organisms that metabolize PAH to a great
extent, like fish and higher invertebrates such as arthropods,
echinoderms, and annelids, accumulate little or no PAH (James, 1989).
Remarkably high bioconcentration factors have been measured for
phenanthrene, anthracene, pyrene, benzo [a]anthracene, and
benzo [a]pyrene in the amphipod Pontoporeia hoyi, which has a
20-50% lipid content by wet weight and no capacity to biotransform PAH
(Landrum, 1988).
The ratio of the concentration of an individual PAH in a
bottom-dwelling organism and in the sediment, the bioaccumulation
factor, is usually < 1 when expressed as wet weight. In a coastal
area, the bioaccumulation factors for 16 PAH in polychaete species
varied from 4.9 to 21.8 on a dry-weight basis (Bayona et al., 1991).
Measurements of the concentrations of PAH in P. hoyi and in the
sediment at three sites with different organic carbon contents gave
bioaccumulation factors close to 1 on a wet-weight basis, corrected
for the 64-mm sieved fraction (Eadie et al., 1982). The lipid- and
organic carbon-based bioaccumulation factors in clams (Macoma
baltica) for naphthalene and chrysene added to sediment were 0.78
and 0.16, respectively (Foster et al., 1987). In a study in which
clams were exposed for 28 days to six sediments contaminated with
different concentrations of PAH (and other organic pollutants) and
with an organic carbon content of 0.86-7.4%, the bioaccumulation
factors (normalized with respect to lipid content and organic carbon
content) ranged from 0.15 to 0.85 (Ferraro et al., 1990).
Species that can biotransform PAH have internal concentrations
well below the concentration in the sediment. The average
bioaccumulation factors (normalized with respect to lipid content and
organic carbon content) for eel, pike, and roach at two locations were
0.1 and 0.015. The lowest bioaccumulation factor was found at the site
with the highest PAH concentration (128 mg/kg, organic carbon-based),
probably due to the inductive capability of the fish to biotransform
PAH. This was confirmed by the finding of increased hepatic metabolic
activity for PAH in the fish (Van der Oost et al., 1991).
4.1.5.2 Terrestrial organisms
Little information is available on the accumulation of PAH in
terrestrial organisms. The bioaccumulation factors of 22 PAH in the
earthworm Eisenia foetida at six sites varied from 0.23 to 0.6 on an
ash-free dry-weight basis (Rhett et al., 1988).
The half-life of labelled benzo [a]pyrene in crickets
(Acheta domesticus) was 13 h; after 48 h, 36% of the injected dose
was unchanged benzo [a]pyrene. After topical application of piperonyl
butoxide, a known inhibitor of the mixed-function oxidase system, the
level of polar metabolites in the excreta had decreased by
approximately 75% within 8 h of injection of benzo [a]pyrene. After
articular application of benzo [a]pyrene at 0.29 ng/µl in hexane,
some of the dose accumulated internally; the highest level of polar
metabolites was found after 24 h (Kumi et al., 1991).
The concentration of PAH in vegetation is generally considerably
lower than that in soil, the bioaccumulation factors ranging from
0.0001-0.33 for benzo [a]pyrene and from 0.001-0.18 for 17 other PAH
tested. It was concluded that some terrestrial plants take up PAH
through their roots and/or leaves and translocate them to various
other parts (Edwards, 1983).
When bush beans (Phaseolus vulgaris Pr.) were exposed to
radiolabelled anthracene in a nutrient solution for 30 days during
flowering and seed production, more than 90% of the compound was
metabolized. Of the total 14C radiolabel, 60% was found in the roots,
3% in the stems, 3% in the leaves, 0.1% in the pods, and 17% in the
nutrient solution; 16% was unaccounted for (Edwards, 1986).
Table 27. Measured bioconcentration factors of polycyclic aromatic hydrocarbons in aquatic organisms
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Acenaphthene
Fish
Lepomis 14C S 8.94 28 d 387 Equi Barrows et al.
macrochirus (1980)
Anthracene
Algae
Chlorella fusca HPLC S 50 1 d 7 770a NS Geyer et al.
(1984)
Crustaceans
Daphnia magna 14C, TLC S 35 1 d 511 k1/k2 McCarthy et al.
(1985)
Daphnia magna HPLC S 15 1 d 970 NS Newsted & Giesy
(1987)
Daphnia magna HPLC S 5.58 24 h 2699 NS Oris et al. (1990)
Daphnia pulex Spect S 6 24 h 917 Southworth et al.
(1978)
Hyalella azteca 14C IF 0.0082 8 h/7 h 2089 k1/k2 Landrum &
14C,TLC 1 800 k1/k2 Scavia (1983)
14C IF 0.0066 8 h/7 h 10985 k1/k2
14C, TLC 9096 k1/k2
Pontoporeia hoyi 14C TLC F 4-17 8 W d 16857 k1/k2 Landrum (1982)
Pontoporeia hoyi 14C TLC F 4.6-16.9 6 h/14 d 39727 k1/k2 Landrum (1988)
Oligochaetes
Stylodrilus -C, TLC F < 6 6 hS d 5051 k1/k2 Frank et al.
heringianus (1986)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Fish
Lepomis 14C S 0.7 4 h/60 h 900 k1/k2 Spacie et al.
macrochirus 14C, TLC 675 (1983)
Leuciscus idus UC S 50 3 d 910 NS Freitag A al.
melanotus (1985)
Oncorhynchus 14C HPLC R 12 18h 190 NS Linder &
mykiss 14C HPLC R 12 18 h 270 NS Bergman (1984)
Oncorhynchus 14C HIPLC R 50 72 h/144 h 9000 k1/k2 Linder et al.
mykiss 9200 (1985)
Pimephales HPLC S 6.61 24 In 1016 NS Oris et al. (1990)
promelas
Benz[a]anthracene
Algae
Chlorella fusca 14C S 50 1 d 3180 NS Freitag et al.
(1985)
Crustaceans
Daphnis magna 14C TLC S 0.8 1 d 2920 k1/k2 McCarthy et al.
(1985)
Daphnis pulex Spect S 6 1 d 10109 Southworth et al.
(1978)
Daphnia pulex HPLC S 1.8 1 d 10226 NS Newsted & Giesy
(1987)
Pontoporaia hoyi 14C, TLC F 0.62-1.11 6 h/14 d 63000 k1/k2 Landrum (1988)
Fish
Leuciscus idus 14C S 50 3 d 350 NS Freitag et al.
melanotus (1985)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Benzo[a]fluorene
Crustaceans
Daphnis magna HPLC S 4.8 1d 3668 NS Newsted & Glesy
(1987)
Benzo[b]fluorene
Crustaceans
Daphnia magna HPLC S W 1 d 7725 NS Newsted & Giesy
(1987)
Benzo[a]pyrene
Algae
Periphyton 14C F 1 1 d 9000 NS Leversee et al.
(1981)
Crustaceans
Daphnis magna 14C S/F 1 6 h 2440 k1/k2 Leversee et al.
(1981)
Daphnia magna 14C 3050 NS Leversee et al,
14C HPLC 2837 k1/k2 (1981)
Daphnia magna 14C TLC S 0.63 1 d 5770 k1/k2 McCarthy et al.
(1985)
Daphnia magna HPLC S 1.5 1 d 12761 NS Newsted & Giesy
(1987)
Daphnia pulex 14C S 1.20 24 h 458 NS Trucco et al.
14C S 0.47 24 h 745 NS (1983)
14C S 5.42 24 h 803 NS
14C S 3.21 24 h 1 106 NS
14C S 2.20 24 h 1 259 NS
14C S 1.50 24 h 2 720 NS
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Pontoporeia hoyi 14C, TLC S 0.002-2.6 6 h/14 d 73 000 k1/k2 Landrum (1988)
Oligochaetes
Stylodrilus 14C, TLC F < 0.03 6 h/8 d 7 048 k1/k2 Frank et al.
heringianus (1986)
Molluscs
Mysis relicta 14C F - 6 h/10-26d 8 297 k1/k2 Evans &
Landrum (1989)
Ostrea edulis 14C, GLC S 65.7 3 d 58 NS Riley et al.
Ostrea edulis 14C, GLC S 65.7 3 d 59 NS (1981)
Ostrea edulis 14C, GLC S 65.7 3 d 62 NS
Physa sp. 14C, GLC S 2.5 3 d 2 177 NS Lu et al. (1977)
Rangia cuneata 14C S 30.5 24 h 236 NS Neff & Anderson
14C S 30.5 24 h 187 NS (1975)
Insects
Chironomus 14C S 1 8 h/48 h 970 k1/k2 Leversee et al.
riparius 14C 600 NS (1981)
14C, HPLC 166 NS
Culex pipiens 14C, GLC S 2.5 3 d 37 NS Lu et al. (1977)
quinquefasciatus
Hexagenia limbata 14C, TLC F - 6 h/14 d 5 870 k1/k2 Landrum & Poore
(1988)
Fish
Lepomis 14C-extraction F 1 2 d/4 d 3 208 k1/k2 Jimenez et al.
macrochirus (1987)
Lepomis 14C S/F 1 4 h/4 h 4 700 k1/k2 Leversee et al.
macrochirus 14C 4 h 120 NS (1981)
14C, HPLC 4 h 12.5 NS
Lepomis 14C S 1 4 h/20 h 4 900 k1/k2 Spacie et al.
macrochirus 14C, TLC 490 k1/k2 (1983)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Lepomis 14C, TLC S 0.5 5 h/100 h 2 657 k1/k2 McCarthy &
macrochirus Jimenez (1985)
Leuresthes tenuis Spect S 2 15 d 241 Equi Winkler et al.
(1983)
Oncorhynchus GC-HPLC F 0.4 10 d 920 NS Gerhart &
mykiss Carlson (1978)
Salmo salar 14C S 1 48 h/96 h 2 310 k1/k2 Johnsen et al.
(1989)
Benzo[e]pyrene
Crustaceans
Daphnis magna HPLC S 0.7 1 d 25 200 NS Newsted & Giesy
(1987)
Benzo[ghi]perylene
Crustaceans
Daphnia magna HPLC S 0.2 1 d 28 288 NS Newsted & Giesy
(1987)
Benzo[k]fluoranthene
Crustaceans
Daphnia magna HPLC S 1.4 1 d 13 225 NS Newsted & Giesy
(1987)
Chrysene
Crustaceans
Daphnia magna 14C S 48 48 h/40 h 5 500 NS Eastmond et al.
(1984)
Daphnia magna HPLC S 0.7 1 d 6 088 NS Newsted & Giesy
(1987)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Dibenz[a,h]anthracene
Algae
Chlorella fusca 14C S 50 1 d 2 398 NS Freitag et al.
(1985)
Crustaceans
Daphnia magnia HPLC S 0.4 1 d 50 119 NS Newsted & Giesy
(1987)
Fish
Leuciscus idus 14C S 50 3 d 10 NS Freitag et al.
melanotus (1985)
Fluoranthene
Crustaceans
Crangon HPLC F 2.4 4 d/14 d 180 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magna HPLC S 9 1 d 1 742 NS Newsted & Giesy
(1987)
Molluscs
Mya arenaria HPLC F 2.4 4 d/14 d 4 120 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 2.4 4 d/14 d 5 920 k1/k2 McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 2.4 4 d/14 d 720 k1/k2 McLeese &
Burridge (1987)
Fish
Oncorhynchus GC-HPLC F 3.31 21 d 378 Equi Gerhart &
mykiss Carlson (1978)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Fluorene
Crustaceans
Daphnia magna HPLC S 17 1 d 506 NS Newsted & Giesy
(1987)
Fish
Lepomis - IF 20, 37 30 d 1 800 Equi Finger et al.
macrochirus - IF 86 30 d 700 Equi (1985)
- IF 175, 353 30 d 200 Equi
Naphthalene
Algae
Selenastrum GC S 2,000 1 d 18 000b NS Casserly et al.
capricornutum (1983)
Chlorella fusca 14C S 50 1 d 130a NS Geyer et al.
(1984)
Insects
Somatochlora Spect S 10 48 h 1 548 NS Correa & Coler
cingulata Spect S 100 48 h 178 NS (1983)
Crustaceans
Daphnia magna 14C, HPLC S 1 000 1 d 19.3 k1/k2 McCarthy et al.
(1985)
Daphnia magna 14C S 1 800 48 h/40 h 50 NS Eastmond et al.
(1984)
Daphnia pulex Spect S 1 000 1 d 131 k1/k2 Southworth et al.
(1978)
Daphnia pulex 14C S 2 292 4 h 677 NS Trucco et al.
14C S 0.45 24 h 10 844 NS (1983)
14C S 2.742 4 h 2 337 NS
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Fish
Fundulus 14C S 20 4 h 2.2 NS DiMichele &
heteroclitus Taylor (1978)
Lepomis 14C, HPLC S 1 000 24 h/36 h 310 k1/k2 McCarthy &
macrochirus 14C, HPLC S 100 24 h/36 h 320 k1/k2 Jimenez (1985)
Oncorhynchus 14C S 23 8 h/24 h 253 k1/k2 Melancon & Lech
mykiss (1978)
Perylene
Algae
Chlorella fusca 14C S 50 1 d 2 010 NS Freitag et al.
(1985)
Crustaceans
Crangon HPLC F 0.4 4 d/14 d 175 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magnia HPLC S 0.6 1 d 7 190 NS Newsted & Giesy
(1987)
Molluscs
Mya arenaria HPLC F 0.4 4 d/14 d 100 000 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 0.4 4 d/14 d 105 000 k/q McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 0.4 4 d/14 d 180 k1/k2 McLeese &
Burridge (1987)
Fish
Leuciscus idus 14C S 50 3 d < 10 NS Freitag et al.
melanotus (1985)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Phenanthrene
Bacteria
Mixed Spect S 30-300 2 h 6 300c NS Steen &
Karickhoff (1981)
Algae
Selenastrum GC S 1000 1 d 36 970b NS Casserly et al.
capricornutum (1983)
Chlorella fusca 14C S 50 1 d 1 760a NS Geyer et al.
(1984)
Insects
Hexagenia limbata 14C F - 6 h/14 d 1640 k1/k2 Landrum & Poore
(1988)
Crustaceans
Crangon HPLC F 4.3 4 d/14 d 210 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magna HPLC S 40.1 1 d 323 NS Newsted & Giesy
(1987)
Daphnia magna 14C S 60 48 h/40 h 600 NS Eastmond et al.
(1984)
Daphnia pulex 14C S 6.01 24 h 1 165 NS Trucco et al.
14C S 3.10 24 h 1 032 NS (1983)
14C S 3.45 24 h 1 424 NS
Daphnia pulex Spect S 30 1 d 325 k1/k2 Southworth et al.
(1978)
Pontoporeia hoyi 14C-TLC F 0.7-7.1 6 h/14 d 28 145 k1/k2 Landrum (1988)
Oligochaetes
Stylodrilus 14C-TLC F < 200 6 h/8 d 5 055 k1/k2 Frank et al.
heringianus (1986)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Molluscs
Mya arenaria HPLC F 4.3 4 d/14 d 1 280 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 4.3 4 d/14 d 1 240 k1/k2 McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 4.3 4 d/14 d 500 k1/k2 McLeese &
Burridge (1987)
Pyrene
Bacteria
Mixed Spect S 1-20 2 h 24 600c NS Steen &
Karickhoff (1981)
Algae
Selenastrum GC S 500 1 d 55 800b NS Casserly et al.
capricornutum (1983)
Crustaceans
Crangon HPLC F 1.7 4 d/14 d 225 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnis magna HPLC S 5.7 24 h 2 702 NS Newsted & Giesy
(1987)
Daphnis pulex Sped S 50 24 h 2 702 k1/k2 Southworth et al.
(1978)
Pontoporeia hoyi 14C-TLC F 0.002-0.011 6 h/14 d 16 600 k1/k2 Landrum (1988)
Molluscs
Mya arenaria HPLC F 1.7 4 d/14 d 6 430 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 1.7 4 d/14 d 4 430 k1/k2 McLeese &
Burridge (1987)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Oligochaetes
Stylodrilus 14C-TLC F < 26.4 6 h/8 d 6 588 k1/k2 Frank et al.
heringianus (1986)
Polychaetes
Neiris virens HPLC F 1.7 4 d/14 d 700 k1/k2 McLeese &
Burridge (1987)
Fish
Oncorhynchus GC-HPLC F 3.89 21 d 72.2 Equi Gerhart &
mykiss Carlson (1978)
Triphenylene
Crustaceans
Crangon HPLC F 0.5 4 d/14 d 270 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magna HPLC S 1.7 1 d 9 066 NS Newsted & Giesy
(1987)
Molluscs
Mya arenaria HPLC F 0.5 4 d/14 d 5 540 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 0.5 4 d/14 d 11 390 k1/k2 McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 0.5 4 d/14 d 2 560 k1/k2 McLeese &
Burridge (1987)
Table 27 (continued)
14C, measurement of radioactivity in a liquid scintillation counter: as parent compounds cannot be differentiated from metabolites with
this method, additional extraction is usually performed.
S, static exposure system; Equi, at equilibrium Corg/Cw; HPLC, high-performance liquid chromatography; NS, not steady-state
Corg/Cw;
TLC, thin-layer chromatography; k1/k2, kinetics: uptake rate/depuration rate; Spect, spectroscopy; F, flow-through system;
R, static renewal system; GLC, gas-liquid chromatography; GC, gas chromatography; IF, intermittent flow system
a Based on dry weight (5 × wet weight)
b Based on total suspended solids
c Based on dry weight
4.1.6 Biomagnification
Biomagnification, the increase in the concentration of a
substance in animals in successive trophic levels of food chains, has
been determined in a number of studies. When Daphnia pulex were
exposed to water or algae contaminated with naphthalene, phenanthrene,
benz [a]anthracene, or benzo [a]pyrene, naphthalene accumulated to
the greatest extent from algal food, (bioconcentration factor, 11
000), whereas benz [a]anthracene and benzo [a]pyrene accumulated
more from water (bioconcentration factors, 1100 and 2700,
respectively). It must be emphasized that because of the short
exposure (24 h), the last two compounds would not have reached
equilibrium (Trucco et al., 1983).
In a study of bioaccumulation and biomagnification in closed
laboratory model ecosystems, green algae (Oedogonium cardiacum), D.
magna, mosquito larvae (Culex pipiens quinquefasciatus), snails
(Physa sp.), and mosquito fish (Gambusia affinis) were exposed for
three days to 2 µg/litre of 14C-benzo [a]pyrene. Of the radiolabel
accumulated, 88% was attached to parent compound in snails, 22% in
mosquito larvae, and none in fish. The parent compound represented 46%
of the total extractable radiolabel in mosquito larvae and 90% in
Daphnia. The bioconcentration factors were 5300 for algae, 12 000
for mosquito larvae, 82 000 for snails, 140 000 for Daphnia, and 930
for fish. Despite the apparent absence of bioconcentration in fish,
accumulation is assumed to be due to food-chain transfer, as no
accumulation of benzo [a]pyrene was found in a study of uptake from
water. Biomagnification was also studied in a terrestrial-aquatic
system, by adding 14C-benzo [a]pyrene to Sorghum vulgare seedlings
and allowing them to be eaten by fourth-instar salt-marsh caterpillar
larvae (Estigmene acrea); the labelled products entered the
terrestrial and aquatic phases as products such as faeces. The
food-chain organisms were the same as in the model aquatic ecosystem.
After a 33-day interaction period, the concentrations of
benzo [a]pyrene were 0.01 µg/litre water and 36.1 µg/kg algae, with
bioconcentration factors of 3600, 490, 2100, and 30, respectively.
Most of the radiolabel was found on polar products or as unextractable
radioactivity, which comprised 25% of the total in snails, 63% in
fish, 67% in mosquito larvae, and 79% in algae (Lu et al., 1977).
Trophic transfer of benzo [a]pyrene metabolites between benthic
organisms was studied by feeding Nereis virens 14C-benzo [a]pyrene
and harvesting them five days later. The worm homogenate contained 14%
parent compound, 7.2% organic-soluble metabolites, 58% water-soluble
metabolites, and 21% bound material. Flounder (Pseudiopleuronectes
americanus) were then given doses of 4.8-19 g of either pure
benzo [a]pyrene homogenized in unexposed Nereis or the
worm-metabolite mixture by gavage and analysed after 24 h of
incubation. On the basis of the radiolabel recovered from the fish
tissues, assuming comparable accumulation efficiency, flounder appear
to have at least a limited ability to accumulate polar, conjugated,
and bound metabolic products of benzo [a]pyrene from the diet. The
parent compound represented 5-15% of the radiolabel in liver and 6-7%
in intestine; conjugated metabolites represented 40-60% of the label
in liver and 60-70% in intestine; and bound metabolic products
represented 30% in liver and 10-20% in intestine (McElroy & Sisson,
1989).
4.2 Transformation
On the basis of model calculations, Mackay et al. (1992)
classified some PAH according to their persistence in air, water,
soil, and sediment (Table 28).
Table 28. Suggested half-life classes of polycyclic aromatic
hydrocarbons in various environmental compartments
Class Half-life (h)
Mean Range
1 17 10-30
2 55 30-100
3 170 100-300
4 550 300-1000
5 1 700 1000-3000
6 5 500 3000-10 000
7 17 000 10 000-30 000
8 55 000 > 30 000
Compound Air Water Soil Sediment
Acenalphthylene 2 4 6 7
Anthracene 2 4 6 7
Benz[a]anthracene 3 5 7 8
Benzo[a]pyrene 3 5 7 8
Benzo[k]fluoranthene 3 5 7 8
Chrysene 3 5 7 8
Dibenz[a,h]anthracene 3 5 7 8
Fluoranthene 3 5 7 8
Fluorene 2 4 6 7
Naphthalene 1 3 5 6
Perylene 3 5 7 8
Phenanthrene 2 4 6 7
Pyrene 3 5 7 8
From Mackay et al. (1992)
4.2.1 Biotic transformation
4.2.1.1 Biodegradation
Information on the biodegradation of PAH in water and soil under
aerobic and anaerobic conditions is summarized in Table 29. The few
results available from standard tests for biodegradation in water show
that PAH with up to four aromatic rings are biodegradable under
aerobic conditions but that the biodegradation rate of PAH with more
aromatic rings is very low. Biodegradation under anaerobic conditions
is slow for all components (Neff, 1979). The reactions normally
proceed by the introduction of two hydroxyl groups into the aromatic
nucleus, to form dihydrodiol intermediates. Bacterial degradation
produces cis-dihydrodiols (from a dioxetane intermediate), whereas
metabolism in fungal or mammalian systems produces trans-dihydrodiol
intermediates (from an arene oxide intermediate). The differences in
the metabolic pathways are due to the presence of the cytochrome P450
enzyme system in fungi and mammals. Algae have been reported to
degrade benzo [a]pyrene to oxides, peroxides, and dihydroxydiols (see
below). Owing to the high biotransformation rate (see also section
4.2.1.2), the concentrations of PAH in organisms and water are usually
not in a steady state. Freely dissolved PAH may be rapidly degraded
under natural conditions if sufficient biomass is available and the
turnover rates are fairly high (see Table 29).
Biodegradation is the major mechanism for removal of PAH from
soil. PAH with fewer than four aromatic rings may also be removed by
volatilization and photolysis (see also sections 4.1.4 and 4.2.2.1).
The rate of biodegradation in soil depends on several factors,
including the characteristics of the soil and its microbial population
and the properties of the PAH present. Temperature, pH, oxygen
content, soil type, nutrients, and the presence of other substances
that can act as co-metabolites are also important (Sims & Overcash,
1983). Biodegradation is further affected by the bioavailability of
the PAH. Sorption of PAH by soil organic matter may limit the
biodegradation of compounds that would normally undergo rapid
degradation (Manilal & Alexander, 1991); however, no significant
difference was found in the biodegradation rate of anthracene in water
with 10 and 1000 mg/litre suspended material (Leslie et al., 1987). In
Kidman sandy loam, the biodegradation rates varied between 0.23 h-1
(or 5.5 d-1) for naphthalene and 0.0018 d-1 for fluoranthene (see
Table 29). In a study with sandy loams, forest soil, and roadside soil
partially loaded with sewage sludge from a municipal treatment plant,
the following half-lives (in days) were found: 14-48 for naphthalene,
44-74 for acenaphthene plus fluorene, 83-193 for phenanthrene, 48-210
for anthracene, 110-184 for fluoranthene, 127-320 for pyrene, 106-313
for benz [a]anthracene plus chrysene, 113-282 for
benzo [b]fluoranthene, 143-359 for benzo [k]fluoranthene, 120-258
for benzo [a]pyrene, 365-535 for benzo [ghi]perylene, and 603-2030
for coronene (Wild & Jones, 1993).
Table 29. Biodegradation of polycyclic aromatic hydrocarbons (PAH)
Compound Rate constant Half-life Comments Reference
Acenaphthene 100% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d static flask screening; settled domestic waste (1981)
as inoculum; experiments with 5 and 10 mg/litre
PAH at 25°C; detection by GC
295-2448 h Aerobic half-life; aerobic soil column Kincannon & Lin
(1985)
1180-9792 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation half-life (1991)
0% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 7 d with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
< 3.2 year Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Acenaphthylene 98% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d statis flask screening; settled domestic waste (1981)
as inoculum; 5 or 10 mg/litre PAH at 25°C;
detection by GC
1020-1440 h Aerobic half-life; soil column Kincannon & Lin
(1985)
4080-5760 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
0% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Anthracene 0.061 h-1 10 h Microbial degradation in Third Creek water Southworth
incubated 18 h at 25°C: (1977)
Removal rate constants from water column at
25°C in midsummer sunlight:
0.060 h-1 12 h - in deep, slow, somewhat turbid water
0.030 h-1 23 h - in deep, slow, muddy water
0.061 h-1 11 h - in deep, slow, clear water
0.061 h-1 11 h - in shallow, fast, clear water
0.061 h-1 11 h - in very shallow, fast, clear water
0.035 h-1 20 h Microbial degradation rate constant Herbes et al.
(1980)
51-92% degradation Significant degradation with gradual Tabak et al.
after 7 d adaptation; static flask screening; settled (1981)
domestic waste as inoculum; experiments
with 5 and 10 mg/litre PAH at 25°C; detection
by GC
1200-11 040 h Aerobic half-life; aerobic soil die-away Coover & Sims
(1987)
20O g dry weight of soil at -0.33 bar Park et al.
[33 kPa] soil moisture at 25°C: (1990)
0.0052 d-1 3200 h - Kidman sandy foam; initial test
concentration, 210 mg/kg
0.0138 d-1 1200 h - McLaurin sandy loam; initial test
concentration, 199 mg/kg
4800-44 160 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation half-life (1991)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
1.9% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 2 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
Anthracene 33% after 16 months Degradation in soil in co-metabolic closed Bossert &
bottle with 1-phenyldecane as primary Bartha (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 60%
5% after 56 d Batch test with river water; initial concentration, Fedorak et al.
20 mg/litre related to dissolved organic carbon; (1982)
no mineralization during first 19 days; 20°C
Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons: (1995)
10-60% after 64 d - initial concentration, 31.3 ng/g
- Inoculated with enriched culture of
Mycobacteriarn sp. and initial test concentration
of 37.7 ng/g; biodegradation rate without
enriched culture, 18% after 64 d
Static test in bioreactor in enriched mixed Walter et al.
culture; anthracene oil (38 g/litre) which also (1990)
contained 62 mg/g fluorene; 30°C:
100% after 3 d - under aerobic conditions
90% after 20 d - under anaerobic conditions
17-45 d Aerobic degradation in surface Donneybrook Bulman et al.
sandy loam from Canadian pasture; initial test (1987)
concentrations, 5 and 50 mg/kg; up to 400 days'
exposure at 20 00 and water-holding capacity of
60% of the soil
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
7.9 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benz[a]anthracene 2448-16 320 h Aerobic soil die-away at 10-30°C Groenewegen &
Stolp (1976); Coover
& Sims (1987)
0% degradation No significant degradation under conditions of Tabak et al. (1981)
after 7 d method; static flask sceening; settled domestic
waste as inoculum; experiment with 5 and
10 mg/littre PAH at 25°C; detection by GC
0.0026 d-1 6400 h Kidman sandy loam Park et al. (1990)
9792-65 280 h Anaerobic half-life; estimated unacclimatized Howard et al. (1991)
aqueous aerobic biodegradation
16% after Degradation in soil in co-metabolic closed Bossert & Bartha
16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 18%
0-40% after 64 d Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons; initial (1995)
concentration, 31.3 ng/g
130-240 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
8.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benzo[a]pyrene 0.2-0.9 Aquatic fate rate for bacterial protein Barnsley (1975)
µmol.h-1mg-1
3.5 × 10-5 h-1 19 800 h Estimated rate constant in soil and water Ryan & Cohen
(1986)
1368-12 702 h Aerobic half-life at 10-30°C; soil die-away Coover & Sims
(1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; 33 mg/kg at 25°C:
0.0022 d-1 7416 h - Kidman sandy loam
0.0030 d-1 5496 h - McLaurin sandy loam
5472-50 808 h Anaerobic half-life; estimated unacclimatized Coover & Sims
aqueous aerobic biodegradation (1987)
< 8% after 160 d Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons; initial (1995)
concentration, 105 ng/g
218-347 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
8.2 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Benzo[b]fluoranihene 8640-14 640 h Aerobic half-life; estimated unacclimatized Coover & Sims
aqueous aerobic biodegradation (1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 38 mg/kg at 25°C:
0.0024 d-1 7056 h - Kidman sandy loam
0.0033 d-1 5064 h - McLaurin sandy loam
34 560-58 560 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
9 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benzo[ghi]perylene 14 160-15 600 h Aerobic half-life; aerobic soil dieaway at Coover & Sims
10-30°C (1987)
56 640-62 400 h Anaerobic half-life; aerobic soil dieaway at Coover & Sims
10-30°C (1987)
9.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benzo[k]fluoranthene 21 840-51 360 h Aerobic half-life; aerobic soil dieaway Coover & Sims
(1987)
87 360-205 440 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
8.7 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Chrysene 59% degradation Significant degradation with gradual Tabak et al. (1981)
after 7 d adaptation; static flask screening; settled
domestic waste as inoculum; experiment
with 5 mg/litre PAH at 25°C; detection by GC
38% degradation No significant degradation under conditions of Tabak et al. (1981)
after 7 d method; static flask sceening; settled domestic
waste as inoculum; experiment with 10 mg/litre
PAH at 25°C; detection by GC
8904-24 000 h Aerobic half-life; aerobic soil dieaway Coover & Sims
(1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 100 mg/kg at 25°C:
0.0019 d-1 8904 h - Kidman sandy loam
0.0018 d-1 9288 h - McLaurin sandy loam
35 616-96 000 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
11 % after 16 Degradation in soil in co-metabolic closed Bossert & Bartha
months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 5%
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
224-328 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
8.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Coronene 16.5 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Dibenz[a,h]anthracene 8664-22 560 In Aerobic half-life; aerobic soil die-away Coover & Sims
(1987); Park et al.
(1990)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 13 mg/kg at 25°C:
0.0019 d-1 8664 h - Kidman sandy loam
0.0017 d-1 10 080 h - McLaurin sandy loam
No degradation Degradation in soil in co-metabolic closed Bossert & Bartha
after 16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Fluoranthene 2.2 × 10-3 Aquatic fate rate with bacterial protein Barnsley (1975)
µmol h-1mg-1
100% degradation Significant degradation with gradual adaptation; Tabak et al. (1981)
after 7 d static flask screening; settled domestic waste
as inoculum; experiment with 5 mg/litre PAH
at 25°C; detection by GC
0% degradation No significant degradation under conditions of Tabak et al. (1981)
after 7 d method; static flask screening; settled domestic
waste as inoculum; experiment with 10 mg/litre
PAH at 25°C; detection by GC
3360-10 560 h Aerobic half-life; aerobic soil dieaway Coover & Sims
(1987)
0.19 h-1 3.6 h In atmosphere Dragoescu &
Friedlander (1989)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, 900 mg/kg at 25°C:
0.0018 d-1 9048 h - Kidman sandy loam
0.0026 d-1 6432 h - McLaurin sandy loam
13 440-42 240 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
34-39 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
7.8 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Fluorene 45-77% degradation Significant degnadation with gradual adaptation; Tabak et al. (1981)
after 7 d static flask screening; settled domestic waste
as inoculum; experiment with 5 and 10 mg/litre
PAH at 25°C; detection by GC
Degradation of 30 µg/litre in natural river water Lee & Ryan (1976)
(Skidway River; salinity, 20%):
100% after 1000 d - Turnover time in June at incubation time of
48 h
0% after 72 h - February or May
30% after 1 week Degradation of non-autoclaved groundwater Lee et al. (1984)
samples of ± 0.06 mg/litre by microbes
768-1440 h Aerobic half-life; aerobic soil diaway Coover & Sims
(1987)
3072-5760 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
0% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
100% after 36 h Batch test with enriched culture of Arthrobacter Grifoll et al.
sp.; initial test concentration, 483 µmol/litre; (1992)
22°C
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
< 3.2 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Indeno[1,2,3-cd]pyrene 200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 8 mg/kg at 25°C:
0.0024 d-1 6912 h - Kidman sandy loam
0.0024 d-1 6936 h - McLaurin sandy loam
Naphthalene Degradation in natural river water (Skidway Lee & Ryan
River; salinity, 20%): (1976)
500 d - Turnover time in February at incubation
time of 48 h; test concentration, 40 µg/litre
46 d - Turnover time in May at incubation
time of 24 h; test concentration, 40 µg/litre
79 d - Turnover time in May at incubation time
of 8 h; test concentration, 40 µg/litre
30 d - Turnover time in May at incubation
time of 24 h; test concentration, 130 µg/litre
Degradation of 130 µg/litre in natural water Lee & Ryan
330 d offshore with salinity of 35%: turnover time (1976)
in May at incubation time of 24 h
0.0403.3 × 10-6 At depth of 5-10 m in laboratory water basin Lee & Anderson
g/litre per d (1977)
100% after 8 d In gas-oll-contaminated groundwater Kappeler &
circulated through sand inoculated with Wuhrmann
groundwater under aerobic conditions (1978)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
168 h In oil-polluted estuarine stream Lee (1977)
576 h In clean estuarine stream
1500 h In coastal waters
40 800 h In the Gulf Stream
12h Aerobic half-life; die-away in oil-polluted Walker & Colwell
creek (1976)
Anaerobic half-life: Hambrick et al.
600 h at pH 8 (1980)
6200 h at pH 5
24-216 h In deep, slowly moving, contaminated water Herbes (1981);
Wakeham et al.
(1983)
0.23 h-1 3.O h Microbial degradation rate constant Herbes et al. (1980)
100% degradation Significant degradation with rapid adaptation; Tabak et al. (1981)
after 7 d static flask screening; settled domestic waste
as inoculum; experiments with 6 and 10 mg/litre
PAH at 25°C; detection by GC
100% degradation Degradation of non-autoclaved groundwater Lee et al. (1984)
after 7 d samples of ± 0.04 mg/litre by microbes
0.024 d-1 693 h Groundwater with nutrients and acclimatized Vaishnav & Babeu
microbes (1987)
0.013 d-1 1279 h River water with acclimatized microbes
0.018-1 924 h River water with nutrients and acclimatized
microbes
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[-0.0032 kPa] soil moisture; initial test
concentration, 101 mg/kg at 25°C:
0.377 d-1 50 h - Kidman sandy loam
0.308 d-1 53 h - McLaurin sandy loam
2% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 30 mg/litre PAH and 100 mg/litre sAdge International Trade
and Industry (1992)
< 2.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Perylene No degradation Degradation in soil in co-metabolic closed Bossert & Bartha
after 16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g
Phenanthrene 100% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d static flask screening; settled domestic waste (1981)
as inoculum; experiments with 5 and 10 mg/litre
PAH at 25°C; detection by GC
383-4800 h Aerobic half-life; aerobic soil die-away Coover & Sims
(1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[-0.0032 kPa] soil moisture; initial test
concentration, 900 mg/kg at 25°C:
0.0447 d-1 384 h - Kidman sandy loam
0.0196 d-1 840 h - McLaurin sandy loam
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
1536-19 200 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
96 h Inorganic solution Manilal & Alexander
264 h Kendaia soil (1991)
54% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
> 62 % after Degradation in soil in co-metabolic closed Bossert & Bartha
16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss significant
Serum bottle radiorespirometry in five soils Grosser et al. (1995)
contaminated with hydrocarbons:
38-55% after 64 d - initial concentration, 31.3 ng/g
80% after 32 d - inoculated with enriched culture of
Mycobacterium sp. and an initial test
concentration of 17.9 ng/g
9.7-14 d Aerobic degradation in surface samples of Bulman et al. (1987)
Donneybrook sandy loam from Canadian
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
5.7 years Field tests of rural British soils amended with Wild et al.
metal-enriched sewage sludges with (1991)
0.1-15.1 mg/kg PAH
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Pyrene 100% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d static flask screening; settled domestic waste (1981)
as inoculum; experiment with 5 mg/litre
PAH at 25°C; detection by GC
0% degradation No significant degradation under conditions of Tabak et al.
after 7 d method; static flask screening; settled domestic (1981)
waste as inoculum; experiments with 5 and
10 mg/litre PAH at 25°C; detection by GC
5040-46 600 h Aerobic half-life at 10-30°C; aerobic soil Coover & Sims
die-away (1987)
0.29 h-1 2.4 h In atmosphere Dragoescu &
Friedlander
(1989)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 690 mg/kg at 25°C:
0.0027 d-1 6240 h - Kidman sandy loam
0.0035 d-1 4776 h - McLaurin sandy loam
20 160-182 400 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
70% after 16 months Degradation in soil in co-metabolic closed Bossert & Bartha
bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 27%
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons: (1995)
25-70% after 64 d - initial concentration, 8.5 ng/g
54% after 32 d - inoculated with enriched culture of
Mycobacterium sp. and an initial test
concentration of 7.7 ng/g
52.4% after 96 h Mineralization test with Mycobacterium sp.; Heitkamp et al.
24°C; initial test concentration, 0.5 mg/litre (1988)
48-58 d Aerobic degradation in surface Donneybrook Bulman et al. (1987)
sandy loam from Canadian pasture; initial test
concentrations, 5 and 50 mg/kg; up to 400 days'
exposure at 20°C and water-holding capacity of
60% of the soil
8.5 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
GC, gas chromatography In order to compare numbers when only rate constants are reported, the half-lives were estimated from the formula:
t1/2 = In2
k
where t1/2 is the half-life and k is the rate constant. The calculated values are reported in italics.
After biodegradation of pyrene by a Mycobacterium sp., cis-
and trans-4,5-pyrene dihydrodiol and pyrenol were the initial ring
oxidation products. The main metabolite was 4-phenathroic acid. The
ring fission products were 4-hydroxyperinaphthenone and cinnamic and
phthalic acids (Heitkamp et al., 1988).
The pyrene-metabolizing Mycobacterium sp. can also use
phenanthrene and fluoranthene as the sole source of carbon.
Phenanthrene was degraded and 1-hydroxy-2-naphthoic acid,
ortho-phthalate, and protocatechuate were detected as metabolites.
1-Hydroxy-2-naphthoic acid did not accumulate, indicating that it is
further metabolized (Boldrin et al., 1993).
A strain of Arthobacter sp. was isolated that was capable of
metabolizing fluorene as a sole energy source: 483 nmol/ml were
degraded completely within 36 h, and four major metabolites were
detected: 9-fluorenol, 9 H-fluoren-9-one, 3,4-dihydrocoumarin, and an
unidentified polar-substituted aromatic compound. Fluorenol was not
degraded further, suggesting that it and fluorenone are products of a
separate metabolic pathway from that which produces dihydrocoumarin,
the polar compound, and the energy for cell growth. The bacteria could
also degrade phenanthrene (Grifoll et al., 1992).
The degradation of PAH was studied in a culture made from
activated sludge, polychlorinated biphenyl-degrading bacteria, and
chlorophenol-degrading mixed cultures, adapted to naphthalene. The
metabolites of naphthalene were 2-hydroxybenzoic acid and
1-naphthalenol, those of phenanthrene were 1-phrenanthrenol and
1-hydroxy-2-naphthalenecarboxylic acid, and that of anthracene was
3-hydroxy-2-naphthalenecarboxylic acid. The authors concluded that the
biotransformation pathway proceeds via initial hydroxylation to ring
cleavage, to yield the ortho or meta cleavage intermediates, which
are further metabolized via conventional metabolic pathways (Liu et
al., 1992).
The metabolism of PAH by fungi is similar to that by mammalian
cells. For example, Cunninghamella elegans in culture metabolizes
benzo [a]pyrene to the trans-7,8-diol, the trans-9,10-diol,
3,6-quinone, 9-hydroxybenzo [a]pyrene, 3-hydroxybenzo [a]pyrene, and
7,8-dihydro-7,8-dihydroxybenzo [a]pyrene (Cerniglia, 1984). In a
further experiment, C. elegans metabolized about 69% of added
fluorene after 24 h. The major ethyl acetate-soluble metabolites were
9-fluorenone (62%), 9-fluorenol, and 2-hydroxy-9-fluorenone (together,
7%). The degradation pathway was similar to that in bacteria, with
oxidation at the C9 position of the five-member ring to form an
alcohol and the corresponding ketone. 2-Hydroxy-9-fluorenone had not
been found as a metabolite previously (Pothuluri et al., 1993).
4.2.1.2 Biotransformation
Biotransformation is often advanced as an explanation for the
differences in PAH profiles seen in aquatic organisms and in the
medium to which they were exposed. Furthermore, all of the metabolites
of PAH may not have been identified or quantified. This section
addresses biotransformation in organisms other than bacteria and
fungi, which is discussed in section 4.2.1.1, above.
The uptake of naphthalene and benzo [a]pyrene was studied in
three species of marine fish: the mudsucker or sand goby
(Gillichthys mirabilis), the sculpin (Oligocottus maculosus), and
the sand dab (Citharichthys stigmaeus). In all three species,
biotransformation took place rapidly in the liver. The uptake of
naphthalene was greater than that of benzo [a]pyrene. The major
metabolite of benzo [a]pyrene appeared to be
7,8-dihydroxy-7,8-dihydroxy benzo [a]pyrene, while the major
metabolite of naphthalene was 1,2-dihydro-1,2-dihydroxy-naphthalene.
The gall-bladder was the major storage site for the PAH and their
metabolites. Naphthalene and its metabolites were removed at a higher
rate than benzo [a]pyrene and its metabolites (Lee et al., 1972).
Transformation of naphthalene and benzo [a]pyrene in the
bluegill sunfish Lepomis macrochirus took place very rapidly,
benzo [a]pyrene having the highest rate (McCarthy & Jimenez, 1985).
L. macrochirus were exposed in a flow-through system to 4 nmol/litre
benzo [a]pyrene for 48 h, followed by a 96-h depuration period, at 13
or 23°C in the presence or absence of food. Both polar and nonpolar
metabolites were found. After 48 h, the polar metabolites comprised
10% of the benzo [a]pyrene metabolites in fed fish at 13°C, 20% in
unfed fish at 23°C, and 30% in fed fish at 23°C (Jimenez et al.,
1987). In rainbow trout (Oncorhynchus mykiss) exposed to naphthalene
at 0.5 mg/litre for 24 h, the bile contained 65-70% metabolites, the
liver contained 5-10%, and muscle < 1% (Melancon & Lech, 1978).
In L. macrochirus exposed to 8.9 ± 2.1 µg/litre acenaphthene
for 28 days, the half-life for metabolism was less than one day. No
information was given on metabolites (Barrows et al., 1980).
The depuration of anthracene was investigated in O. mykiss
during simulated day and night cycles of 16 and 8 h, respectively.
After a 96-h clearance period, the metabolites contributed 2-3% of the
depurated substance, half of which came from the bile. No specific
metabolites were reported (Linder & Bergman, 1984). After L.
macrochirus had been exposed to anthracene at 8.9 µg/litre or
benzo [a]pyrene at 0.98 µg/litre for 4 h, the rates of
biotransformation were 0.26 and 0.082 nmol/g per h, respectively, and
8% of the anthracene and 88% of the benzo [a]pyrene were metabolized
(Spacie et al., 1983).
Benzo [a]pyrene is transformed in the Japanese medaka
(Oryzias latipes) and the guppy (Poecilia reticulata), the main
metabolite being the 7,8-diol-9,10-epoxide (Hawkins et al., 1988).
Two benthic organisms, the European fingernail clam (Sphaerium
corneum) and larvae of the midge Chironomus riparius, both
metabolized benzo [a]pyrene. In the larvae, the main metabolite
appeared to be 3-hydroxybenzo [a]pyrene; a quinone isomer was also
found. Only a very small amount of 3-hydroxy-benzo [a]pyrene was
found in the clam. No diol metabolites were found in either species
(Borchert & Westendorf, 1994). After exposure of the benthic
oligochaete Stylodrilus heringianus to either anthracene and pyrene
or phenanthrene and benzo [a]pyrene, 2% degradation of each PAH was
reported within 24 h (Frank et al., 1986).
The half-lives for metabolism in D. magna were 0.5 h for 1.8
mg/litre naphthalene, 9 h for 0.06 mg/litre phenanthrene, and 18 h for
0.023 mg/litre chrysene (Eastmond et al., 1984).
In amphipod Hyalella azteca was exposed to 0.043 nmol/ml
anthracene for 8 h, the rates of biotransformation were 2.2 ± 0.5
nmol/g dry weight per h with no substratum, 3.0 ± 0.8 in the presence
of washed sand from a local lake, and 1.0 ± 0.15 in the presence of
sediment from the lake (Landrum & Scavia, 1983).
The amphipod Rhepoxynius abronius metabolizes benzo [a]pyrene
(Plesha et al., 1988). When two marine amphipods were exposed to a
sediment containing 5.1 ng/mg of this compound, R. abronius
metabolized 49% and Eohaustorius washingtonianus metabolized 27% of
the benzo [a]pyrene after one day. The main metabolites appeared to
be 7,8-dihydro-7,8-dihydroxy-benzo [a]pyrene,
9,10-dihydro-9,10-dihydroxybenzo [a]pyrene,
3-hydroxy-benzo [a]pyrene, and 9-hydroxybenzo [a]pyrene. The ratio
of 7,8-dihydro-7,8-dihydroxybenzo [a]pyrene to
9,10-dihydro-9,10-dihydroxybenzo [a]pyrene in normal-phase
high-performance liquid chromatography was 1.2 for R. abronius and
0.7 for E. washingtonianus (Reichert et al., 1985).
No biotransformation of benzo [a]pyrene or phenanthrene was
found in mayflies (Hexagenia limbata) or in the amphipod
Pontoreia hoyi (Landrum & Poore, 1988).
In a study of the route of metabolism of benzo [a]pyrene in
green algae (Selenastrum capricornutum) exposed to 1.2 µg/litre for
four days, with simulated day and night periods, the major dihydrodiol
metabolites identified were the cis-4,5-diol (< 1%), the
cis-7,8-diol (13%), the 9,10-diol (36%), and the cis-11,12-diol
(50%), demonstrating the presence of a dioxygenase enzyme for this
type of algae (Lindquist & Warshawsky, 1985), as suggested by Cody et
al. (1984). Payne (1977) reported, however, that aryl hydrocarbon
hydroxylase was not present in Fucus and Ascophyllum sp. of marine
algae.
Benzo [a]pyrene was not biotransformed in periphyton after 0.25
or 4 h. In cladocerans (D. magna) exposed to 1.0 µg/litre
benzo [a]pyrene, the biotransformation rate after exposure for 6 h
was 1.07 ± 0.20 nmol/g dry weight per h. In midge larvae (C.
riparius) exposed to 0.6-1.5 µg/litre, the biotrans-formation rate
was 3.6 ± 0.7 nmol/g dry weight per h after exposure for 1 h and 2.7 ±
0.3 after 4 h. In L. macrochirus exposed to 1.0 µg/litre, the
biotransformation rate was 0.20 ± 0.03 nmol/g dry weight per h after 1
h and 0.37 ± 0.04 after 4 h. In chironomids, 3-hydroxybenzo [a]pyrene
was the major metabolite after 8 h, representing 4.4% of the total
water activity; smaller amounts of 7-hydroxy-benzo [a]pyrene and the
9,10- and 7,8-dihydroxydiols of benzo [a]pyrene were also found
(Leversee et al., 1981).
After exposure of benthic species to benzo [a]pyrene for one to
four weeks, the following percentages of metabolites were found: E.
washingtonianus, 22% in the whole body; R. abronius, 74% in the
whole body; clams (Macoma nasuta), < 5% in the body and < 5 in the
hepatopancreas; shrimp (Pandalus platyceros), 94% in the
hepatopancreas; and the English sole (Parophrys vetulus), 94% in the
body, 99% in the liver and > 99% in the bile (Varanasi et al., 1985).
Mosquito larvae (C. pipens quinquefasciatus) were exposed for
three days to 0.002 mg/litre benzo [a]pyrene in the presence or
absence of the mixed-function oxidase inhibitor piperonyl butoxide at
0.0025 mg/litre. Parent benzo [a]pyrene represented 22% of the
excreted PAH in the absence of piperonyl butoxide and 86% in its
presence. After three days' exposure of snails (Physa sp.) to the
same concentration of benzo [a]pyrene with or without piperonyl
butoxide at 0.0025 mg/litre, parent benzo [a]pyrene represented 88%
in the absence of the inhibitor and 85% in its presence. The authors
suggested that snails are deficient in microsomal oxidases. In
mosquito fish (G. affinis) exposed similarly, no parent
benzo [a]pyrene was found in the absence of piperonyl butoxide but
21% in its presence (Lu et al., 1977).
In an aquatic ecosystem, plankton, green algae (Oedogonium
cardiacum), D. magna, mosquito larvae (C. pipiens
quinquefasciatus), snails (Physa sp.), and mosquito fish
(G. affinis) were exposed to 0.002 mg/litre benzo [a]pyrene for
three days. Parent benzo [a]pyrene represented 83, 90, 46, 70, and
55% in the four organisms, respectively. The substance was metabolized
to unidentified hydroxylated polar compounds. The finding of 55%
parent benzo [a]pyrene in the fish was attributed to food-chain
transfer, as none was found after direct exposure. A
terrestrial-aquatic ecosystem was also exposed to benzo [a]pyrene by
applying 0.2 mg of radiolabelled compound to Sorghum vulgare
seedlings to simulate atmospheric fall-out and allowing them to be
consumed by fourth-instar salt-marsh caterpillar larvae (E. acrea).
Faecal products then entered the terrestrial and aquatic ecosystem
described above, which was left for 33 days. The maximum radiolabel
(0.005 ppm) was detected in the aquatic phase after 14 days.
Unmetabolized benzo [a]pyrene accounted for 7.1% of the total
extractable radiolabel in fish, 19% in snails, 32% in algae, and 34%
in mosquitoes. Addition of the mixed-function oxidase inhibitor,
piperonyl butoxide, resulted in 12% parent benzo [a]pyrene in fish,
34% in snails, 48% in the algae, and no change in mosquitoes (Lu et
al., 1977).
The biotransformation of 19 PAH was studied in the food chain
seston (plankton) -> blue mussel (Mytilus edulis L.) -> common
eider duck (Somateria mollissima L.) in the open, northern Baltic
Sea. The concentrations of the PAH in the eider duck showed the
distribution gallbladder > adipose tissue > liver. There was a
high flux of the PAH in the food chain, but the concentration did not
increase with increasing trophic level, indicating that the PAH were
biotransformed rapidly. There was little biotransformation in the
plankton. The distribution of the PAH in blue mussels was different
from that in plankton, perhaps due to metabolic activity in the
mussel. Biotransformation of PAH with a relative molecular mass of 252
was rapid in the ducks (Broman et al., 1990).
In beans (Phaseolus vulgaris L.) exposed to 15 g anthracene
per plant, uptake via the roots was rapid, 90% being metabolized
within 30 days (Edwards, 1986).
These investigations are summarized in Table 30. As the rate of
metabolism depends not only on the species but also on factors such as
temperature, pH, and other experimental conditions, the results are
difficult to compare. Some general conclusions can, however, be drawn:
- The biotransformation potential of aquatic organisms depends on
the activity of cytochrome P450-dependent mixed-function
oxidases, which are important for oxidation, the first step in
the metabolism of xenobiotics such as PAH (James, 1989).
- The tissues in which biotransformation mainly takes place are
liver, lung, kidney, placenta, intestinal tract, and skin
(Cerniglia, 1984).
- The initial transformation step in invertebrates usually occurs
more slowly than in vertebrates (James, 1989). Monoxygenation of
PAH is faster in higher invertebrates like arthropods,
echinoderms, and annelids and slowest in more primitive
invertebrates like protozoa, profina, cnidaria, and molluscs
(Neff, 1979).
- In general, invertebrates excrete PAH metabolites inefficiently
(James, 1989).
- In higher organisms and algae, metabolites are usually produced
by monooxygenase activity, resulting in the formation of
epoxides, phenols, diols, tetrols, quinones, and conjugates.
- It is not clear whether molluscs have cytochrome P450 activity
(Moore et al., 1989).
Table 30. Biotransformation of polycyclic aromatic hydrocarbons by various organisms
Species Compound Biotransformation rate Reference
Fungi
Cunninghamella elegans Benzo[a]pyrene No information Cerniglia (1984)
Algae
Selenastrum capticornutum Benzo[a]pyrene Relatively fast Lindquist & Warshawsky (1985)
Oedogenium cardiacum Benzo[a]pyrene 15% after 3 d in Lu et al. (1977)
aquatic ecosystem
Fucus sp. Various None Payne(1977)
Ascophyllum sp. Various None
Molluscs
Sphaerium corneum Benzo[a]pyrene Very fast (no carcinogenic Borchert & Westendorf (1994)
metabolites)
Physa sp. Benzo[a]pyrene 12% after 3 d Lu et al. (1977)
Mytilus edulis L. Different No information Broman et al. (1990)
Crustaceae
Hyalella azteca Anthracene 2.2 nmol/g dw/h in water Landrum & Scavia (1983)
Hyalella azteca Anthracene 3.0 nmol/g dw/h 5 water/ Landrum & Scavia (1983)
sediment
Daphnia magna Benzo[a]pyrene 1.07 nmol/g dw/h after 6 h Leversee et al. (1981)
Daphnia magna Benzo[a]pyrene 10% after 3 d in aquatic Lu et al. (1977)
ecosystem
Pontoporeia hoyi Benzo[a]pyrene None Landrum & Poore (1988)
Pontoporeia hoyi Benzo[a]pyrene None after 48 h Evans & Landrum (1989)
Mysis relicta Benzo[a]pyrene No information Evans & Landrum (1989)
Rhepoxynius abronius Benzo[a]pyrene No information Plesha et al. (1988)
Rhepoxynius abronius Benzo[a]pyrene 74% after 1-4 weeks Varanasi et al. (1985)
Rhepoxynius abronius Benzo[a]pyrene 49% after 1 d Reichert et al. (1985)
Eohaustorius washingtonianus Benzo[a]pyrene 27% after 1 d Reichert et al. (1985)
Eohaustorius washingtonianus Benzo[a]pyrene 22% after 1-4 weeks Varanasi et al. (1985)
Table 30. (continued)
Species Compound Biotransformation rate Reference
Pandalus platyceros Benzo[a]pyrene < 5% after 1-4 weeks Varanasi et al. (1985)
Parophrys vetulus Benzo[a]pyrene 94% after 1-4 weeks Varanasi et al. (1985)
Daphnia magna Chrysene 50% after 18 h Eastmond et al. (1984)
Daphnia magna Naphthalene 50% after 0.5 h Eastmond et al. (1984)
Daphnia magna Phenanthrene 50% after 9 h Eastmond et al. (1984)
Fish
Lepomis macrochirus Acenaphthene Half-life, < 1 d Barrows et al. (1980)
Lepomis macrochirus Anthracene 8% after 4 h Spacie et al. (1983)
Oncorhynchus mykiss Anthracene 2-3% after 24 h Linder & Bergman (1984)
Gillichthys mirabilis Benzo[a]pyrene Rapid in liver Lee et al. (1972)
Oligocottus maculosus Benzo[a]pyrene Rapid in liver Lee et al. (1972)
Citharichthys stigmaeus Benzo[a]pyrene Rapid in liver Lee et al. (1972)
Lepomis macrochirus Benzo[a]pyrene Very fast McCarthy & Jimenez (1981)
Lapomis macrochirus Benzo[a]pyrene 88% after 4h Spacie et al. (1983)
Lepomis macrochirus Benzo[a]pyrene 0.20-0.37 nmol/g dry Leversee et al. (1981)
weight per h
Oryzias latipes Benzo[a]pyrene No information Hawkins (1988)
Poecilia reticulata Benzo[a]pyrene No information Hawkins (1988)
Rhepoxynius abronius Benzo[a]pyrene None Plesha et al. (1988)
Gambusia affinis Benzo[a]pyrene 100% after 3 d in water Lu et al. (1977)
40% after 3 d in aquatic
ecosystem
Gillichthys mirabilis Naphthalene Rapid in liver Lee et al. (1972)
Oligocottus maculosus Naphthalene Rapid in liver Lee et al. (1972)
Citharichthys stigmaeus Naphthalene Rapid in liver Lee et al. (1972)
Lepomis macrochirus Naphthalene Very fast McCarthy & Jimenez (1981)
Worm
Stylodrilus heringianus Various None Franck et al. (1986)
Table 30. (continued)
Species Compound Biotransformation rate Reference
Insects
Chironomus riparius Benzo[a]pyrene Very fast (no carcinogenic Bochert & Westendorf (1994)
metabolites)
Chironomus riparius Benzo[a]pyrene 2.7-3.6 nmol/g dry weight Leversee et al. (1981)
per h
Hexagenia limbata Benzo[a]pyrene None Landrum & Poore (1983)
Culex pipiens Benzo[a]pyrene 78% after 3 d Lu et al. (1977)
quinquefasciatus
Somatochlora cingulata Naphthalene No information Correa & Coler (1990)
Bird
Somateria mollissima L. Various Fast for PAH with Broman et al. (1990)
molecular mass > 252
Plant
Phaseolus vulgaris L. Anthracene 90% after 30 d Edwards (1986)
- In crustaceans, biotransformation differs greatly between species
and for different PAH. Biotransformation of naphthalene,
anthracene, phenanthrene, and chrysene appears to occur rapidly,
while that of benzo [a]pyrene is generally slower. Only Reichert
et al. (1985) reported significant degradation in R. abronius
(49%) and E. washingtonianus (27%) within one day.
- It is not clear how rapidly biotransformation occurs in insects.
- Too little information was available on algae, plants, and fungi
for conclusions to be drawn.
4.2.2 Abiotic degradation
Abiotic processes may account for the removal of 2-20% of two-
and three-ring PAH from soil (Park et al., 1990). In soils partly
amended with PAH-containing sewage sludge, 24-100% was removed, and
naphthalene was eliminated almost completely by volatilization and
photodegradation (Wild & Jones, 1993).
4.2.2.1 Photodegradation in the environment
PAH can be expected to be photodegraded in air and water but to a
very low extent in soils and sediments, owing to low light intensity.
In natural waters, photodegradation takes place only in the upper few
centimetres of the aqueous phase. Information on the photodegradation
of PAH in air and water is summarized in Table 31; however, as the
testing conditions varied widely, general conclusions cannot be drawn.
PAH are photodegraded in air and water by two processes: direct
photolysis by light with a wavelength < 290 nm and indirect
photolysis by least one oxidizing agent such as OH, O3, and NO3 in
air and ROO radicals in water. In general, indirect photolysis -
photooxidation - is the more important process. The reaction rates of
PAH with airborne OH radicals measured under standard conditions are
given in Table 32, which shows that most of the calculated half-lives
are one day or less. Under environmental conditions, PAH of higher
molecular mass, i.e. those with more aromatic rings, are almost
completely adsorbed onto fine particles (see section 4.1.2); this
reduces the degradation rate markedly.
Degradation half-lives of 3.7-30 days were reported for the
reaction with NOx of various PAH adsorbed onto soot. The degradation
was much slower in the absence of sunlight. PAH did not react
significantly with SO2 (Butler & Crossley, 1981). PAH in wood smoke
and gasoline exhaust did not degrade significantly during winter in
extreme northern and southern latitudes owing to low temperatures and
the low angle of the sun (Kamens et al., 1986a). In summer, however,
at a temperature of 20°C, the half-lives of individual PAH were in the
range of 30-60 min (Kamens et al., 1986b). The degradation rate
increased further with increasing humidity (Kamens et al., 1991).
Table 31. Photodegradation of polycyclic aromatic hydrocarbons
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Acenaphthene Air, particles Determined in rotary photoreactor Behymer &
with 25 µg/g on: Hites (1985)
2.0 - silica gel
2.2 - alumina
44 - fly ash
Water 0.23 h-1 3.0 Rate constant in distilled water Fukuda et al.
(1988)
Acenaphthylene Air, particles Determined in rotary photoreactor Behymer &
with 25 µg/g on: Hites (1985)
0.7 - silica gel
2.2 - alumina
44 - fly ash
Anthracene Air, water 0.58 Measured in atmosphere and water Southworth
from aqueous photolysis rate (1979)
constant for midday summer sunlight
at 35°N
Air, particles Determined with 25 µg/g on: Behymer &
2.9 - silica gel Hites (1985)
0.5 - alumina
48 - fly ash
Water Removal rate constants from water Southworth
at 25°C in midsummer sunlight: (1979)
0.004 h-1 173 - in deep, slow, somewhat turbid
water
<0.001 h-1 > 700 - in deep, slow, muddy water
0.018 h-1 38 - in deep, slow, clear water
0.086 h-1 8 - in shallow, fast, clear water
0.238 h-1 3 - in very shallow, fast, clear water
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water Half-lives calclulated from average Southworth
light intensity over 24 h: (1977)
1.6 - in summer
4.8 - in winter
Water Half-lives calculated for direct Zepp &
sunlight at 40°N at midday in Schlotzhauer
midsummer: (1979)
0.75 - near surface water
108 - inland water
125 - inland water with sediment
partitioning
0.75 - direct photochemical
transformation near water surface
Water 0.66 h-1 1.0 In distilled water Fukuda et al.
(1988)
Benz[a]anthracene Air, particles First-order daytime decay rate Kamens et al.
constants with soot particle loading of: (1988)
0.0125 min-1 0.9 - 1000-2000 ng/mg
0.0250 min-1 0.5 - 30-350 ng/mg
Air, particles Determined with ± 25 µg/g on: Behymer &
4.0 - silica gel Hites (1985)
2.0 - alumina
38 - fly ash
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water Calculated rate constant in pure Mill et al.
water: (1981)
13.4 × 10-5s-1 1.4 - at 366 nm and in sunlight at
23-28°C, early March
2.28 × 1O-5s-1 8.4 - at 313 nm with 1% acetonitrile
in filter-sterilized natural water
5 Early March
Benzo[a]pyrene Air, particles Determined with 25 µg/g on: Behymer &
4.7 silica gel Hites (1985)
1.4 - alumina
31 - fly ash
Air particles First-order daytime decay rate Kamens et al.
constants with soot particle loading of: (1988)
0.0090 min-1 1.3 - 1000-2000 ng/mg
0.0211 min-1 0.54 - 30-350 ng/mg
Air, particles < 6.1 × 10-4 m/s Ozonization rate constant measured Cope &
at 24°C with O3 = 0.16 ppm and Kalkwarf
light intensity of 1.3 kW/m3 (1987)
Air 0.37-1.1 Estimated Lyman et al.
(1982)
Air 1 Sunlight in mid-December Mill & Mabey
(1985)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Air, water Calculated rate constants for Mill et al.
direct photolysis: (1981)
3.86 × 10-4s-1 0.69 - in pure water at 366 nm and in
sunlight at 23-28°C, late January
1.05 × 10-5s-1 1.1 - at 313 nm with 1-20% acetonitrile
in filter-sterilized natural
water, mid-December
Water Computed near-surface half-life for Zepp &
direct photochemical transformation Schlotzhauer
of a natural water body: (1979)
0.54 - latitude 40°N, midday, midsummer
77 - no sedimentmater partitioning
312 - sediment; water partitioning in a
5-m deep inland water body
Air > 1 Summer Valerio et al.
Days Winter (1991)
Methanol 2 Irradiated at 254 nm Lu et al. (1977)
Benzo[b]fluoranthene Air, particles First-order daytime decay rate Kamens et al.
constants with soot particle loading of: (1988)
0.0065 min-1 1.8 - 1000-2000 ng/mg
0.0090 min-1 1.3 - 30-350 ng/mg
Air, water 8.7-720 Based on measured rate of Lane & Katz
photolysis in heptane irradiated with (1977); Muel
light at > 290 nm & Saguem
(1985)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Benzo[ghi]perylene Air, particles Determined with 25 µg/g on: Behymer &
7.0 - silica gel Hites (1985)
2.2 - alumina
29 - fly ash
Air, particles First-order daytime photodegradation Kamens et al.
rate constants for adsorption (1988)
on wood soot particles in an outdoor
Teflon chamber for soot loading of:
0.0077 min-1 1.5 - 1000-2000 ng/mg
0.0116 min-1 1.0 - 30-350 ng/mg
Benzo[k]fluoranthene Air, particles First-order daytime decay constants Kamens et al.
for soot loading of: (1988)
0.0047 min-1 2.5 - 1000-2000 ng/mg
0.0013 min-1 8.9 - 30-350 ng/mg
Air, water 3.8-499 Based on measured rate of photolysis Muel &
in heptane under November Saguem
sunlight, adjusted by ratio of (1985)
sunlight photolysis half-lives in
water: heptane
Chrysene Air, particles Determined with 25 µg/g on: Behymer &
100 - silica gel Hites (1985)
78 - alumina
38 - fly ash
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Air, particles First-order daytime decay constants Kamens et al.
for soot loading of: (1988)
0.0056 min-1 2.1 - 1000-2000 ng/mg
0.0090 min-1 1.3 - 30-350 ng/mg
Air, water 4.4 Calculated for direct photochemical Zepp &
transformation near surface of Schlotzhauer
a water body at 40°N at midday in (1979)
midsummer
Water 13 Estimated on basis of photolysis Lyman et al.
in water in winter (1982)
Dibenzo[a,h]anthracene Air, water 782 Based on measured rate of photolysis Muel &
in heptane in November sun Saguem
6 After adjusting ratio of sunlight (1985)
photolysis in water: heptane
Fluoranthene Air, particles Determined with 25 µg/g on: Behymer &
74 - silica gel Hites (1985)
23 - alumina
44 - fly ash
Air, water 63 Computed, adjusted for approximate Lyman et al.
winter sunlight intensity (1982)
Air, water Calculated photochemical transformation Zepp &
near surface of water body: Schlotzhauer
21 - at 40°N, midday, midsummer (1979)
3800 - 5-m deep inland water body with
no sediment:water partitioning
4800 - with sediment:water partitioning
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water 3800 Summer sunlight in surface water Mill & Mabey
(1985)
Fluorene Air, particles Determined in rotary photoreactor Behymer &
with 25 µg/g on: Hites (1985)
110 - silica gel
62 - alumina
37 - fly ash
Naphthalene Water 13 200 Calculated, 5-m deep inland water Zepp &
Schlotzhauer
(1979)
Water 0.028 h-1 25 Half-life in distilled water Fukuda et al.
(1988)
Perylene Air, particles Determined with 25 µg/g on: Behymer &
3.9 - silica gel Hites (1985)
1.2 - alumina
35 - fly ash
Air, glass < 4.7 × 10-5 m/s Ozonization rate constant measured Cope &
from glass surface at 24°C with 03 Kalkwarf
- 0.16 ppm and light intensity of (1987)
1.3 kW/m2
Phenanthrene Air, particles Determined with 25 µg/g on: Behymer &
150 - silica gel Hites (1985)
45 - alumina
49 - fly ash
Water 3 Based on measured aqueous photolysis Zepp &
quantum yields, midday, mid-summer, Schlotzhauer
40°N (1979)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Air, water 25 Adjusted for approximate winter Lyman et al.
sunlight intensity (1982)
Air, water Calculated, direct sunlight photolysis, Zepp &
midday, midsummer, 40°N: Schlotzhauer
8.4 - near surface water (1970)
1400 - 5-m deep inland water body with
no sediment:water partitioning
1650 - with sedimentmater partitioning
Water 0.11 h-1 6.3 Half-life in distilled water Fukuda et al.
(1988)
Pyrene Air, particles Determined with 25 µg/ml on: Behymer & Hites
21 - on silica gel (1985)
31 - on alumina
46 - on fly ash
Air, particles Adsorption on airborne particles Valerio et al.
by sunlight: (1991)
1 - in summer
Days - in winter
Air, water 1.014 h-1 0.68 Based on measured aqueous photolysis Zepp &
quantum yields, midday, Schlotzhauer
summer, 40°N (1979)
Air, water 2.04 Based on measured aqueous photolysis Lyman et al.
quantum yields, adjusted for (1982)
approximate winter sunlight intensity
Air, glass < 1.05 × 10-4 m/s Ozonization rate on glass surface Cope &
at 24°C with O3 = 0.16 ppm and Kalkwarf
light intensity of 1.3 kW/m2 (1987)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water Calculated, direct sunlight photolysis, Zepp &
midday, midsummer, 40°N: Schlotzhauer
0.58 - near surface water (1979)
100 - 5-m deep inland water body with
no sediment:water partitioning
142 - with sediment:water partitioning
Water 100 Summer sunlight photolysis in Mill & Mabey
surface water (1985)
In order to compare numbers reported only as rate constants, half-lives were estimated from the formula:
t1/2 = In2
k
where t1/2 is the half-life and k is the rate constant. The calculated values are reported in italics.
Table 32. Reactions of polycyclic aromatic hydrocarbons with hydroxy radicals
Compound Oxidation rate Photooxidation Comments Reference
constant half-life (h)
Acenaphthene 1 × 10-10 0.879-8.79 Based on estimated reaction rate Atkinson (1987)
constant with hydroxy radical in air
Acenaphthylene 1.1 × 10-10 0.191-1.27 Based on estimated rate constant for Atkinson (1987)
reaction in air
Anthracene 1.1 × 10-12cm3 58-580 Rate constant for gas-phase reaction Biermann at al.
molec-1s-1 with hydroxy radicals at 298 ± 1 K, based (1985)
the relative rate technique for propane
0.501-5.01 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benz[a]anthracene 0.801-8.01 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[a]pyrene 0.428-4.28 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[b]fluoranthene 1.43-14.3 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[ghi]perylene 0.321-3.21 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[k]fluoranthene 1.1-11 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Chrysene 0.802-8.02 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Dibenz[a,h]anthracene 0.428-4.28 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Fluoranthene 2.02-20.2 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Fluorene 1.3 × 10-11 6.81-68.1 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Table 32. (continued)
Compound Oxidation rate Photooxidation Comments Reference
constant half-life (h)
Naphthalene 2.16 × 10-11 cm3 2.7-27 Rate constant for reaction with hydroxy Atkinson (1989)
molec-1s-1 radicals using relative rate technique
at 294 K
2 × 10-19 cm3 19-321 Upper limit was obtained for reaction
molec-1s-1 with O3
2.35 × 10-11 cm3 2.7-27 Rate constant for gas-phase reaction Biermann et al.
molec-1s-1 with hydroxy radicals at 298 K, based (1985)
on relative rate technique from propene
Phenanthrene 3.4 × 10-11 cm3 2-20 Rate constant for gas-phase reaction Biermann et al.
molec-1s-1 with hydroxy radicals at 298 K, based (1985)
on relative rate technique for propene
3.1 × 10-11 2.01-20.1 Half-life based on measured rate Atkinson (1987)
constants for reaction with hydroxy
radical in air
Pyrene 0.802-8.02 h Based on estimated rate constant for Atkinson (1987);
reactions with hydroxy radical in air and Atkinson & Carter
with hydroxy radical and ozone (1984)
To allow comparison when only rate constants are reported, half-lives were estimated from the following formula:
t1/2 = In 2
[x] × k
where t1/2 is the half-life, [x] is the concentration of the radical with which the compounds react (i.e. hydroxyl or ozone),
and k is the rate constant. The calculated values are reported in italics.
For the concentrations of the radicals, the following ranges of values were used; the lower values are estimates for rural
areas and the higher ones for urban areas (Howard et al., 1991):
[OH]air = 3-30 × 105 radicals/cm3
[O3]air = 3-50 × 1012 molecules/cm3
[OH]water = 5-200 × 10-17 mol/litre
[RO2]water = 1-50 × 10-11 mol/litre
[1O2]water = 1-100 × 10-15 mol/litre
In a study of the fate of 18 PAH on 15 types of fly ash, carbon
black, silica gel, and alumina, the PAH were stabilized, depending on
the colour, which is related to the carbon content: the higher the
carbon content, the more stable the PAH. The authors suggested that
radiation energy is adsorbed by the organic matter of particulates,
and PAH therefore do not achieve the excited state in which they can
be degraded (Behymer & Hites, 1988). The half-lives for direct
photolysis of various PAH adsorbed onto silica gel are in the range of
hours (Vu-Duc & Huynh, 1991).
A two-layer model has been proposed for the behaviour of
naturally occurring PAH on airborne particulate matter, in which
photooxidation takes place in the outer layer, and much slower, 'dark'
oxidation takes place in the inner layer (Valerio et al., 1987). This
model is in line with the results of Kamens et al. (1991), who
reported that PAH on highly loaded particles degrade more slowly than
those on particles with low loads. As PAH occur mainly on particulate
matter with a high carbon content, their degradation in the atmosphere
is slower than that of PAH in the vapour phase under laboratory
conditions or adsorbed on synthetic materials like alumina and silica
gel that have no or a low carbon content.
Formation of nitro-PAH was found from the low-molecular-mass two-
to four-ring PAH that occur in the atmosphere, predominantly in the
vapour phase. The rate constants range from 5.5 × 10-12 cm3/molecule
× s for acenaphthylene to 3.6 × 10-28 cm3/molecule × s for
naphthalene, with corresponding half-lives ranging from 6 min to 1.5
years. The yields were 1% or less (Atkinson et al., 1991; Atkinson &
Arey, 1994).
The rate of degradation of absorbed individual PAH seems to be
independent of their physicochemical characteristics but dependent on
their molecular structure. Thus, activated carbon from graphite
particles effectively stabilized pyrene, phenanthene, fluoranthene,
anthracene, and benzo [a]pyrene adsorbed onto coal fly ash against
photochemical decomposition, but no stabilization was seen for
fluorene, benzo [a]fluorene, benzo [b]fluorene,
9,10-dimethyl-anthracene, or 4-azafluorene. The authors suggested that
PAH that contain benzylic carbon atoms are less reactive than others
(Hughes et al., 1980).
PAH with vinylic bridges appear to degrade by direct photolysis
more rapidly than those with only aromatic rings, both in air and in
the aquatic environment (Hites, 1981).
In measurements of the photodegradation of benz [a]anthracene
and benzo [a]pyrene, addition of humic acids and purging of the
solution with nitrogen reduced the reaction rates significantly (Mill
et al., 1981). The authors concluded that light screening and
quenching occurred with humic acids. The reduction in rate with
exclusion of oxygen was probably due to a decrease in photooxidative
processes. The first metabolites were mainly quinones.
4.2.2.2 Hydrolysis
PAH are chemically stable, with no functional groups that result
in hydrolysis. Under environmental conditions, therefore, hydrolysis
does not contribute to the degradation of PAH (Howard et al., 1991).
4.3 Ultimate fate after use
The main sinks for PAH are sediment and soil. The available
information indicates that high-molecular-mass PAH are especially
persistent in groundwater, soil, and sediment under environmental
conditions.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
Appraisal
Polycyclic aromatic hydrocarbons (PAH) occur in all environmental
compartments. Ambient air, residential heating, and vehicle traffic
are the main sources. The levels of individual substances vary over
several orders of magnitude but are generally in the range < 0.1-100
ng/m3.
Surface waters are contaminated by PAH mainly through atmospheric
deposition, urban runoff, and industrial activities such as coal
coking and aluminium production. Apart from highly industrial polluted
rivers, the concentrations of individual substances are generally
< 50 ng/litre. High concentrations of PAH have been measured in
rainwater and especially in snow and fog. The concentrations of PAH in
sediments are in the low microgram per kilogram range.
PAH levels in soils near industrial sources (e.g. coal coking) are
especially high, sometimes up to grams per kilogram. In contrast,
soils contaminated by atmospheric deposition or runoff have
concentrations of 2-5 mg/kg of individual PAH, and the concentrations
in unpolluted areas are in the low microgram per kilogram range.
PAH have been detected in vegetables but are mainly formed during food
processing, roasting, frying, or baking. The highest levels were
detected in smoked meat and fish, at up to 200 µg/kg food for
individual PAH.
Five-fold increases in the concentrations of PAH in soil have been
observed over a 150-year period, although there are indications that
the concentrations of some PAH are decreasing. Similar findings have
been reported for sediments, perhaps because of measures to reduce
emissions.
Aquatic animals are known to adsorb and accumulate PAH. Especially
high concentrations were found in aquatic organisms from highly
polluted rivers, at levels up to milligrams per kilogram. Of the
terrestrial animals, earthworms are a good indicator of soil pollution
with PAH. The benzo[a]pyrene concentrations in the faeces of
earthworms living in a highly industrialized region were in the low
milligrams per kilogram range.
The main sources of exposure for the general population appear to be
food and air. The estimated intake of individual PAH in the diet is
0.1-8 µg/d. The main contribution appears to be that of cereals and
cereal products, due to the large amounts consumed. In ambient air,
the main sources are residential heating and environmental tobacco
smoke; exposure to PAH from environmental tobacco smoke in indoor air
is estimated to be 6.4 µg/day.
Occupational exposure to PAH occurs via the lung and skin. High
exposure occurs during the processing and use of coal and mineral oil
products, such as in coal coking, petroleum refining, road paving,
asphalt roofing, and impregnation of wood with creosotes; high
concentrations are also found in the air of aluminium production
plants and steel and iron foundries. No measurements were available
for the primary production and processing of PAH.
5.1 Environmental levels
5.1.1 Atmosphere
Relevant data on the occurrence of PAH in ambient air are compiled in
Tables 33-36. The concentrations were determined mainly by gas
chromatography and high-performance liquid chromatography, usually
with enrichment by filtration through a solid sorbent. The amount of
particle-bound PAH is therefore given. In studies in which
vapour-phase PAH were also sampled, the results for the vapour and
particulate phases were combined (for reviews, see Grimmer, 1979;
Ministry of Environment, 1979; Grimmer, 1983b; Lee & Schuetzle, 1983;
Daisey et al., 1986; Baek et al., 1991; Menichini, 1992a).
5.1.1.1 Source identification
Qualitative indications of different sources can be obtained by
comparing the PAH profiles, i.e. the ratio between the total PAH
concentration and that of a selected PAH, in air with those of samples
representative of the emitting sources or by determining PAH that are
emitted mainly from a specific source (Menichini, 1992a). Quantitative
assignments are difficult to make, however, owing to the complexity of
factors that affect the variability of PAH concentrations and
profiles.
Measurements were made at selected sources of PAH in the area of
Chicago, USA, in 1990-92, in order to identify them: Five samples were
taken 100 m directly downwind of a coke plant in an area that was not
affected by steel-making facilities, four samples from diesel buses at
a parking garage, three samples from petrol vehicles under warm-engine
operating conditions at a public parking garage, five samples in
heavily travelled tunnels during evening rush hours, and two samples
from the roof directly downwind of the chimney of fireplaces burning
seasoned oak. The authors give a source distribution pattern in
percent related to the total mass of 20 PAH. Naphthalene made by far
the largest contribution to petrol engine and coke oven emissions (55
and 89%, respectively). The three-ring compounds acenaphthylene,
acenaphthene, fluorene, phenanthrene, anthracene, and retene were
detected in large amounts in diesel motor emissions (56%) and in wood
combustion exhausts (69%). The four-ring fluoranthene, pyrene,
benz [a]anthracene, chrysene, and triphenylene and the five-ring
cyclopenta [cd]pyrene, benzo [b]fluoranthene,
benzo [k]fluoranthene, benzo [a]pyrene, benzo [e]pyrene, and
dibenzo [ghi]perylene together contributed 28% to diesel engine
emissions, 25% to petrol engine emissions, and 20% to wood combustion
emissions (Khalili et al., 1995).
The winter levels of PAH are higher than the summer levels (Gordon,
1976; Lahmann et al., 1984; Greenberg et al., 1985; Chakraborti et
al., 1988; Catoggio et al., 1989), due to more intensive domestic
heating and to meteorological (lower inversions during the winter) and
physicochemical factors (temperature-dependent partition between
gaseous and particulate phases). The ratios of benzo [a]pyrene:CO, in
which CO was used as an 'inert' tracer of automotive emissions, in Los
Angeles, USA, were higher at night (0.18-0.34) than in the day
(0.12-0.14), and substantially more so during winter (0.14-0.34) than
in summer (0.12-0.18), consistent with daytime loss of PAH by chemical
degradation (Grosjean, 1983).
In studies of sources of PAH at commercial, industrial, and urban
sampling sites in Athens, Greece, the effects of wind velocity and
thermal inversion were studied. There seemed to be no direct
correlation between benzo [a]pyrene and lead levels, which would be
expected if exhaust from cars run on leaded petrol were the
preponderant source of PAH (linear regression coefficient, 0.32-0.38)
(Viras et al., 1987).
Differences in the composition of profiles of PAH from different
sources can also be standardized by giving the concentrations relative
to that of a specific PAH. For particle-bound PAH, benzo [e]pyrene
has often been used as a reference compound, since it is
photochemically stable and found mainly in the particulate phase (Baek
et al., 1991).
Cyclopenta [cd]pyrene is emitted particularly from petrol-fuelled
automobiles (Grimmer et al., 1981c). Fluoranthene, pyrene,
benzo [ghi]perylene, and coronene are also found in higher
concentrations in condensates of vehicle exhausts (Baek et al., 1991).
The contribution of vehicles and domestic heating has also been
estimated as the ratio of indeno[1,2,3- cd]pyrene to
benzo [ghi]-perylene concentrations. The ratio should be 0.37 for the
PAH profile in traffic exhaust and 0.90 for domestic heating (Lahmann
et al., 1984; Jaklin & Krenmayr, 1985). In a comparison of the PAH
ratios determined in New Jersey, USA, with those reported in the
literature for samples collected under similar conditions in street
tunnels, the ratios coronene:benzo [a]pyrene and
benzo [ghi]perylene:benzo [a]pyrene indicated that vehicle traffic
was the major source of PAH during the summer (Harkov et al., 1984).
Measurements in ambient air in North Rhine Westphalia, Germany, in
1990 indicated that coronene is the most characteristic PAH for
automobile traffic. At a ratio of benzo [a]pyrene:coronene of < 3.5,
vehicle traffic is the dominant PAH source, whereas emissions with
ratios > 3.5 are influenced by other sources. The benzo [a]pyrene
levels were 0.66-5.0 ng/m3, and those of coronene 0.57-2.5 ng/m3
(Pfeffer, 1994).
In a study of the PAH concentrations during weekdays and weekends in
South Kensington, London, United Kingdom, no distinct differences were
observed in winter, but the average concentrations were 1.5-2.5 times
higher during the week than during the weekends in summer. Likewise,
the diurnal variations appeared to be less distinct during winter than
summer (Baek et al., 1992).
Measurements in streets with high traffic density in Stockholm,
Sweden, showed that the concentration of PAH decreased by 25-50%
during holidays in comparison with weekdays. Benzo [a]pyrene in
street air was all particle-bound, while chrysene and lighter PAH
occurred both on particles and in the vapour phase (Östman et al.,
1991, 1992a,b).
In a study of 15 PAH in the air of various areas in an industrial city
in Germany with 700 000 inhabitants, the highest levels were detected
in air affected by a coke plant, where benzo [a]pyrene was found at
1.4-400 ng/m3 and cyclopenta [cd]pyrene at none detected to 120
ng/m3. The concentrations measured in air affected by vehicle traffic
were 11-110 ng/m3 benzo [a]pyrene and 0.1-440 ng/m3
cyclopenta [cd]pyrene. Within 4 km, the average concentration of 88
ng/m3 cyclopenta [cd]pyrene had dropped to 1.6 ng/m3. The levels
were lower in areas where hand-stoked residential coal heating
predominated (0.37 µg/m3 benzo [a]pyrene and none detected to 39
µg/m3 cyclopenta [cd]pyrene) and where oil heating predominated
(0.2-66 ng/m3 and none detected to 15 ng/m3, respectively). The
concentration of PAH was three to four times higher between 7:43 and
10:00 than between 10:00 and 15:46. Benzo [c]phenanthrene,
cyclopenta [cd]pyrene, benzo [ghi]perylene, and coronene dominated
the PAH in areas with heavy traffic, whereas chrysene,
benzo [b]fluoranthene, and benzo [a]pyrene occurred at the highest
concentrations in an area surrounding a coke plant (Grimmer et al.,
1981c).
The use of receptor-source apportionment modelling was examined,
despite its limited applicability to reactive species, for the PAH
profiles of emissions from a variety of sources (Daisey et al., 1986;
Pistikopoulos et al., 1990). In one study, benzo [b]fluoranthene,
benzo [k]fluoranthene, benzo [a]pyrene, benzo [ghi]perylene,
indeno[1,2,3- cd]pyrene, and coronene were measured in the ambient
air of the centre of Paris, France. The concentrations of PAH varied
from 42% in winter to 72% in summer for petrol-fuelled vehicles, from
25 to 40% for diesel-fuelled vehicles, and from about 30 to 2% for
domestic heating. The winter-summer differences were due mainly to
different emission patterns and not to changes in the rate of decay of
PAH (Pistikopoulos et al., 1990). In another study, the contributions
of PAH from five sources to ambient air were distinguished by use of
fuzzy clustering analysis (Thrane & Wikström, 1984).
The information on PAH levels in ambient air is discussed below
according to possible source: background and rural, industrial
emissions, and diffuse sources like automobile traffic and residential
heating. Attribution of different studies to these sections was
difficult because the sources of PAH emissions are often mixed. For
example, Seifert et al. (1986) determined PAH in Dortmund 200 m from a
coke plant; this study was deemed to relate to PAH levels resulting
from industrial emissions. The concentrations of PAH attributable to
mobile sources can be estimated by monitoring near areas with heavy
traffic in the summer, but it is difficult to estimate the
contribution of home heating, because in winter PAH in ambient air
derive from both mobile sources and home heating. Furthermore,
emissions from mobile sources may differ in winter from those in the
summer because of meteorological and physicochemical factors
(Greenberg et al., 1985; see also section 5.1.1.3).
5.1.1.2 Background and rural levels
The levels in ambient air of rural areas are summarized in Table 33.
Background levels were measured about 25 km from La Paz, Bolivia, at
an altitude of 5200 m (Cautreels & van Cauwenberghe, 1977) and on the
island of Mallorca, Spain, at an altitude of 1100 m (Simó et al.,
1990). The concentrations were generally 0.01-0.1 ng/m3. The average
values in rural areas are usually 0.1-1 ng/m3. Average concentrations
of 0.34 and 0.27 ng/m3 benzo [a]pyrene were measured in two rural
areas in Japan in 1989, with a maximum concentration of 1.1 ng/m3
(Okita et al., 1994).
5.1.1.3 Industrial sources
PAH levels in ambient air resulting mainly from industrial emissions
are summarized in Table 34. The average concentrations of individual
PAH at ground level were 1-10 ng/m3. In general, aluminium smelters
and industrial processes for the pyrolysis of coal, such as coking
operations and steel mills, result in higher levels of PAH than most
other point industrial sources. Furthermore, the levels of PAH are
much higher downwind from major sources than upwind.
The highest levels of individual PAH were measured near an aluminium
smelter in Hoyanger, Norway, with maximum concentrations of 10-100
ng/m3. Phenanthrene was present at very high levels in ambient air
contaminated by industrial emissions (Thrane, 1987). In Sundsvall,
Sweden, near an aluminium production facility, 310 ng/m3
phenanthrene, 190 ng/m3 naphthalene, 120 ng/m3 pyrene, and 84 ng/m3
fluorene were detected (Thrane & Wikström, 1984).
The concentration of benzo [a]pyrene in ambient air near an oil
processing plant in Moscow was up to 13 ng/m3 (Khesina, 1994).
Benzo [a]pyrene was detected at 15-120 ng/m3 and perylene at 3-37
ng/m3 at 39 measuring stations in the heavily polluted area of Upper
Silesia, Poland. The maximum values were 950 ng/m3 for
benzo [a]pyrene and 270 ng/m3 for perylene (Chorazy et al., 1994).
Table 33. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air of background and rural areas
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene 0.32 6.3-23
Anthracene 0.004 0.05 0.03 < 0.05 1.2-3.9 ND-0.05
Anthanathrene 0.004-0.16 0.08 0.07 ND-0.2 ND-0.04
Benz[a]anthracene 0.005 0.12 0.4 0.40 0.07 1.8-3.2 0.16-0.39
Benzo[a]fluorene 0.8-3.3
Benzo[a]pyrene 0.006 0.005 0.002-0.12 0.33/0.47 0.6 ND-0.52 0.45 0.08 0.8-2.5 0.41-0.45
Benzo[b]fluoranthene 0.02 1.2 0.45-0.58
Benzo[b]fluorene 0.24 0.5-2.4
Benzo[c]phenanthrene 0.15-0.20
Benzo[e]pyrene 0.022 0.006 0.007-0.26 0.6 0.59 1.8-5.8 0.44-0.65
Benzo[ghi]fluoranthene ND-0.2
Benzo[ghi]perylene 0.009 0.002 0.005-0.40 0.6 ND-0.58 1.4-3.0 0.89-1.4
Benzo[k]fluoranthene 0.02 0.002-0.088 0.48 0.17-0.25
Chrysene 0.07a 1.0 0.13-0.19
Coronene 0.005-0.23 0.24 ND-0.22 0.4-0.9 0.16-0.26
Cyclopenta[cd]pyrene 0.2 0.16-0.39
Dibenzo[a,h]pyrene 0.14 0.02-0.07
Dibenzo[a,l]pyrene 0.53
Fluoranthene 0.041 0.030 0.18 0.20/0.26 1.2 ND 0.93 1.3 11-47 0.19-0.23
Fluorene 0.45 0.66 14-32
Indeno[1,2,3-cd]pyrene 0.006 0.02 0.7 0.72 0.43-0.65
1-Methylphenanthrene 0.09 0.7-2.8
Naphthalene ND 3.0-98
Perylene 0.001-0.026 0.09 0.08 ND-0.4
Phenanthrene 0.026 2.66 0.4 ND-0.43 4.2 26-70 ND-0.03
Pyrene 0.034 0.024 0.34 0.010-0.15 0.15/0.15 1.3 ND 0.60 0.73 8.8-26 0.16-0.26
Table 33 (continued)
ND, not detected; /, single measurements;
[1] About 25 km from La Paz, Bolivia, at 5200 m (Cautreels & van Cauwenberghe, 1977);
[2] Mallorca, Spain, 1989 (Simo et al.,1991);
[3] Lake Superior, USA, 1986; sum of vapour and particulate phases (Baker & Eisenreich,1990);
[4] Latrobe Valley, Australia, (Lyall at al.,1988);
[5] Belgium, (Van Vaeck et al.,1980);
[6] Denmark (Nielsen, 1984);
[7] Western Germany, 1981 (Pflock et al.,1983);
[8] Oostvoorne, Netherlands, (De Raat et al.,1987b);
[9] Canada, 1989-91 (Environment Canada, 1994);
[10] Sidsjon, Sweden, 1980-81, sum of vapour and particulate phases (Thrane & Wikstrom, 1984);
[11] Folkestone, Ashford, United Kingdom, 1986 (Baek et al., 1992)
a With triphenylene
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled, unless otherwise stated
Table 34. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air near industrial emissions
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene 23 9.8-372 15-122 3.7
Acenaphthylene 747 0.01
Anthracene 2.9/3.4 158 4.5-6.1 4.1-43 0.12/0.15 0.01-3.4 0.08-0.19
Anthanthrene 0.001/3.0 0.2/1.1 ND-3.0 0.15/0.15 0.13-0.22
Benz[a]anthracene 0.28/1.2 7.6 2.0-158 2.5-58 0.8/3.1 0.02-1.2 1.3-4.7
Benzo[a]fluorene 1.1-179
Benzo[a]pyrene 0.002/1.5 0.5/3.5 25/37 6.3-6.7 5.3 1.1-61 2.1-36 0.14/0.11 0.20-0.11 1.8-3.1 1.1-2.6
Benzo[b]fluoranthene 0.9/1.8 4.8 2.7-6.4
Benzo[b]fluorene 0.7-122 0.61-1.4
Benzo[e]pyrene 0.004/1.4 1.8/3.2 11.6 2.5-86 1.3-3.1
Benzo[ghi]fluoranthene ND-0.5 0.26/0.35
Benzo[ghi]perylene 0.003/1.5 4.2/7.1 O.7 2.2-45 0.35/0.33 0.25
Benzo[j]fluoranthene 0.3/0.8
Benzo[k]fluoranthene 0.001/0.67 0.3/1.3 8.0 1.0-2.2
Chrysene 1.6/3.8 14.7 0.22/0.29 0.01-1.6 2.5-7.5
Coronene 0.003/1.5 3.2/2.8 1.3-1.5 ND 0.6-9.0 0.25/0.26
Cyclopenta[cd]pyrene 2.2
Dibenzo[a,h]pyrene ND 277
Dibenzo[a,l]pyrene 1.0-1.5
Fluoranthene 0.8/3.4 88.3 20-812 22-272 0.12/0.20 0.02-10 2.3-3.3
Fluorene 502 27-419 16-46 0.02-0.86
Indeno[1,2,3-cd]pyrene 0.4/0.3 1.1 3.8-38 0.28/0.27 0.10-7.7 1.4-2.4
1-Methylphenanthrene 2.5-58
Naphthalene 22 400 9.0-193 3.1-26 0.03-0.06
Perylene 0.001/0.2 0.3/1.2 0.1-8.3 0.05/0.05 22 0.23-0.61
Phenanthrene 500 54-1760 58-390 0.11/0.16 0.02-152
Pyrene 1.4/3.8 56.3 16-491 14-207 0.17/0.35 0.006-28 1.6-2.1
Table 34 (continued)
ND, not detected; /, single measurements;
[1] Three sampling sites near various industries in Latrobe Valley, Australia (Lyall et al., 1988);
[2] Near various industries, USA, 1971-72 (Gordon & Bryan, 1973);
[3] Near a coke plant, Dortmund, Germany, 1982-83 (Seifert et al., 1986);
[4] Near a coke plant, Dortmund, Germany, 1989 (Buck, 1991);
[5] 100 m directly downwind of a coke plant, Chicago, USA, 1990-92 (Khalili et al., 1995);
[6] Near aluminium smelters, Norway and Sweden, 1980-82 (analytical method not given) (Thrane, 1987); vapour and particulate phase
(Thrane & Wikstrom, 1984);
[7] Near aluminium smelter, Canada, 1989-91 (Environment Canada, 1994);
[8] Near incineration plant, Sweden (Colmsjo et al., 1986a,b);
[9] Near refinery, USA, 1981-83 (Karlesky et al., 1987);
[10] Brown coal industry area, western Germany, 1983 (Seifert et al., 1986);
[11] Near harbours, Netherlands (De Raat et al., 1987b)
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled, unless otherwise stated
In Ontario, Canada, up to 140 ng/m3 benzo [k]fluoranthene, 110
ng/m3 perylene, 110 ng/m3 benzo [a]pyrene, 90 ng/m3
benzo [ghi]perylene, and 43 ng/m3 fluoranthene were found near a
steel mill (Potvin et al., 1980). The benzo [a]pyrene concentrations
near coke ovens in urban areas of the USA were more than double those
in urban areas without coke ovens (Faoro & Manning, 1981). These
results are consistent with those of Grimmer et al. (1981c), who
detected maximum levels of benzo [a]pyrene, chrysene,
benzo [b]fluoranthene, benzo [j]fluoranthene, and
benzo [k]fluoranthene in the area surrounding a coke plant.
The PAH concentrations in ambient air 900 and 2500 m from a municipal
incineration plant were of the same order of magnitude, and no
significant contribution from the plant to the ambient PAH
concentrations was observed (Colmsjö et al., 1986a).
The PAH levels in an industrial area of Ahmedabad City, India, were
significantly higher than those in a residential area. The highest
levels were found during winter, and the rate of degradation of
airborne PAH was predicted to be lowest in the monsoon season. The
most striking finding was the high concentration of
dibenz [a,h]anthracene in urban air (5.3-23 ng/m3) (Raiyani et al.,
1993a). The limited resolution of PAH may have resulted in
overestimation: for instance, the concentrations of
benzo [ghi]perylene and indeno[1,2,3- cd]pyrene reported are one
order of magnitude higher than that of dibenz [a,h]anthracene.
5.1.1.4 Diffuse sources
A special situation of local importance was the pollution of ambient
air in Kuwait after the war in the Persian Gulf, due to burning of oil
fields. The mean concentrations of benzo [a]pyrene at three sampling
sites were 0.27-9.2 ng/m3, and the maximum was 26 ng/m3 (Okita et
al., 1994). These values are within the range of those detected in
urban areas (see below).
(a) Motor vehicle traffic
The concentrations of PAH in the ambient air of various urban areas
are listed in Table 35. The average levels of individual PAH were 1-30
ng/m3. Relatively high concentrations of benzo [a]pyrene,
benzo [ghi]perylene, phenanthrene, fluoranthene, and pyrene were
measured.
Total PAH concentrations of 43-640 ng/m3 were measured in London,
United Kingdom, in 1991, nearly 80% of which consisted of
phenanthrene, fluorene, and fluoranthene; benzo [a]pyrene and
benz [a]anthracene were present at 1% or less (Clayton et al., 1992).
Table 35. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air of urban areas
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthene 0.4-101 2.7-6
Acenaphthlene 0.9-39 4.4-130
Anthracene 34 0.6-36 0.3-2.1 3.5-25
Anthanthrene 2.5 0.1-4.7 30 < 0.1-0.6 0.003-0.76
Benz[a]anthracene 10 0.3-27 1.2-13 0.2-1.4 0.10-25 0.3-7.6
Benzo[a]fluorene 0.1-0.9 0.8-6.9
Benzo[a]pyrene 9.3 0.3-20 29 1.2-11 < 0.1-1.9 0.074-15 0.2-5.7
Benzo[b]fluoranthene 43 1.0-36
Benzo[b]fluorene 0.1-0.8 0.6-7.3
Benzo[c]phenathrene 4.0 0.2-5.0
Benzo[e]pyrene 8.4 0.4-17 16 1.7-15 < 0.1-1.2 0.40-27 0.4-6.5
Benzo[ghi]fluoranthene 12 0.3-5.0 0.1-1.5 0.5-7
Benzo[ghi]perylene 14 0.5-12 1.6/13 27 2.1-11 0.2-3.5 0.45-31 0.9-2.4 0.6-18
Benzo[j]fluoranthene 0.17-13
Benzo[k]fluoranthene 23 0.29-25
Chrysene 0.3-2.5 0.56-29 3.6-5.6 3.3
Coronene 10 0.3-5.5 12 0.88-2.0 0.1-2.4 0.22-3.3 0.4-19
Cyclopenta[cd]pyrene 11 0.1-4.8 71 < 0.1-1.1 0.1-6
Dibenzo[a,h]pyrene 0.22-3.4 0.29-2.8
Fluoranthene 72 6.2-108 0.40/14 1.4-10 0.80-14 1.3-2.0 6.9-38 15
Fluorene 1.3-61 16-86
Indeno[1,2,3-cd]pyrene 8.6 0.4-12 31 < 0.1-2.9 0.39-30 0.4-7.6
1-Methylphenanthrene 0.3-2.5 5-16
Naphthalene 14-63
Perylene 2.3 0.1-4.3 4.8 < 0.1-0.4 0.011-4.4 0.1-1.3
Phenanthrene 153 18-223 3.6-41 32-105 111
Pyrene 74 2.9-67 0.34/12 1.2-5.5 0.34-10 5.5-45 20
Triphenylene 0.15-6.9
Table 35 (continued)
ND, not detected; /, single measurements;
[1] Vienna, Austria, 1983-84; vapour and particulate phase (Jaklin & Krenmayr, 1985);
[2] Linz, Austria, 1985; vapour and particulate phase (Jaklin et al., 1988);
[3] Antwerp, Belgium (Van Vaeck et al., 1980);
[4] Berlin, western Gemany, 1984-85 (Seifert et al., 1986);
[5] Rhein/Ruhr area, western Germany, 1985-88; analytical method not stated (Buck et al., 1989);
[6] Kokkola, Finland (Pyysalo et al., 1987);
[7] St Denis, France, 1979-80 (Muel & Saguem, 1985);
[8] Various cities, Greece, 1984-85 (Viras et al., 1987);
[9] Oslo, Norway, 1981-83, vapour and particulate phase (Larssen, 1985);
[10] Barcelona, Spain, 1988-89, vapour and particulate phase (Albaiges et al., 1991)
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled,
unless otherwise voted
Table 35 (continued)
Compound [11] [12] [13] [14] [15] [16] [17] [18] [19] [20]
Acenaphthene 0.07-3.58 0.05-31.1
Acenaphthylene 9.1 0.8 0.9
Anthracene 21 1.4 2.8 0.01-8.28 0.20-39.8 0.1-0.9 ND-4.8 6.1/11
Anthanthrene 0.63
Benz[a]anthracene 4.1 0.4 1.4 0.24-10.6 0.12-18.5 0.2-5.8 5-21 0.07-2.1
Benzo[a]fluorene 5.0 0.7
Benzo[a]pyrene 2.9 0.2 0.99/1.4 1.6 0.01-7.02 0.18-13.7 0.3-3.4 1-17 0.04-3.2 0.6/1.6
Benzo[b]fluoranthene 1.8 0.01-3.04 0.13-14.8 0.2-3.7 5-30 0.10-3.7
Benzo[c]phenanthrene 2.8
Benzo[e]pyrene 3.5 0.4 1.1/2.0 2.3 2.1/2.1
Benzo[ghi]fluoranthene 7.3 0.8
Benzo[ghi]perylene 6.6 0.5 2.9/3.3 3.3 0.02-6.90 0.15-85.3
Benzo[k]fluoranthene 0.75 0.23-16.5 0.3-0.8 3-22 0.07-0.85
Chrysene 5.1 0.8 1.6 0.04-4.97 0.13-24.3 0.2-5.5 ND-2.3
Coronene 4.1 0.3 2.4/1.7 1.7 0.02-3.72 0.17-6.92 ND-16
Cyclopenta[cd]pyrene 3.9 0.11 4.1
Dibenz[a,h]pyrene 0.12
Fluoranthene 24 3.9 3.5 2.03-62.4 22-23 14-54 0.24-2.0 8.0/9.7
Fluorene 0.07-27.6 0.07-161
Indeno[1,2,3-cd]pyrene 3.8 0.5 1.6 0.3-4.4 4.24
Naphthalene 15/75
Perylene 1.0 0.1 0.2/0.5
Phenanthrene 76 11 5.1 0.06-111 2.25-492 0.1-2.4 78/81
Pyrene 28 32 18 0.39-17.4 0.33-64.4 0.1-7.5 0.48-3.6 8.0/12
Triphenylene
Table 35 (continued)
ND, not detected;/, single measurements;
[11] Stockholm, Sweden, April 1991; vapour and particulate phases (Ostman et al.,1992a,b);
[12] Stockholm, Sweden; 1992 vapour and particulate phases (Ostman et al.,1992a,b);
[13] London, United Kingdom, 1985-87(Baek et al.,1992);
[14] London, United Kingdom, 1987; vapour and particulate phases (Baek et al.,1992);
[15] Manchester, United Kingdom, 1990-91; vapour and particulate phases (Clayton et al.,1992);
[16] Various cities, United Kingdom, 1991-92; vapour and particulate phases (Halsall et al.,1994);
[17] Lake Baikal shore, Russian Federation, 1993-94 (Grachev et al.,1994);
[18] Zagreb, Croatia, 1977-82; determined by thin-layer chromatography and fluorescence detector (Bozicevic et al.,1987);
[19] Los Angeles, USA, 1981-82 (Grosjean, 1983);
[20] Los Angeles basin, USA, 1986; vapour and particulate phases (Arey et al.,1987)
Table 35 (contd)
Compound [21] [22] [23] [24] [25] [26] [27] [28] [29] [30]
Acenaphthene 3.3-9.0 0.06-5.2 0.6
Acenaphthylene < 11-47 1.9
Anthracene 1.9-4.5 0.45-3.8 0.17-0.57 0.12-0.52 0.2 2.5-5.5
Anthanthrene 0.006-3.3 1-11
Benz[a]anthracene 0.07-1.4 0.19-0.40 0.19-4.4 0.99-7.0 0.37-1.7 1.9 20-66
Benzo[a]fluorene 1.8-6.3
Benzo[a]pyrene 0.11-1.6 ND-0.03 0.09-1.7 0.006-1.8 8-38 1.6-8.4 ND-2.3 3.4 30-120
Benzo[b]fluoranthene 0.17-1.7 3.1-12 3.0 109-200
Benzo[b]fluorene 0.19-0.94
Benzo[e]pyrene 0.03-11 ND-0.04 0.016-2.3 4-19 2.7-9.0 2.3 49-182
Benzo[ghi]fluoranthene 0.12-1.3
Benzo[ghi]perylene 0.24-2.7 0.027-4.7 11-33 3.2-12 3.4 34-141
Benzo[j]fluoranthene 0.08-1.1 22-66
Benzo[k]fluoranthene 0.09-0.97 0.005-0.85 1.8-7.7 2.7
Chrysene 0.22-5.3 0.38-0.57 3-15 0.29-1.4 2.4
Coronene 0.14-1.6 0.020-2.3 5.16
Dibenzo[a,a]pyrene 0.06-2.7
Dibenzo[a,h]pyrene 0.46-1.2 5.3-23
Dibenzo[a,l]pyrene 0.05-0.35
Fluoranthene 5.7-10 1.6-11 14-79 1.5-8.3 1.0 11-26
Fluorene 7.4-14 0.94-5.5 0.08-0.15 0.31-1.2 2.8
Indeno[1,2,3-cd]pyrene 0.20-2.9 6-24 2.6-12 3.1
Naphthalene 280-940 ND 4.5-13
Perylene 0.01-0.15 0.001-0.24 2-9 0.51-1.2
Phenanthrene 21-35 2.2-35 0.79-2.6 0.52-2.4 0.7 12-21
Pyrene 0.12-2.8 4.8-10 1.4-6.9 0.008-0.66 16-69 1.5-9.0 0.46-4.0 3.8 20-44
Triphenylene 22-60
Table 35 (continued)
ND, not detected; /, single nwasureme4s;
[21] New Jersey, USA, 1981-82 (Greenberg et al, 1985);
[22] Portland, Oregon, USA, 1984 (Ligocki et al.,1985);
[23] Urban area (not specified), Canada, 1989-91 (Environment Canada,1994);
[24] Latrobe Valley, Australia (Lyall et al., 1988);
[25] Christchurch, New Zealand, 1979 (Cretney et al., 1985);
[26] Osaka, Japan, 1977-78; vapour and particulate phases (Yamasaki et al., 1982);
[27] Osaka, Japan, 1981-82 (Matsumoto & Kashimoto, 1985);
[28] La Plata, Argentina, 1985 (Catoggio et al., 1989);
[29] Ahmedabad City, India, 1984-85 (Raiyani at al.,1993a);
[30] Calcutta, India, 1984 (Chakraborti et al.,1988)
Table 35 (continued)
Compound [31] [32] [33] [34] [35] [36] [37] [38] [39] [40]
Acenaphthene 4.5
Anthraceene 14-16 2.5 1.8 ND-34 8.7-23
Anthanthrene 0.15-0.63 0.001-0.21 2-24
Benz[a]anthracene 2.9-4.8 99-139 23 6.5 0.028-4.8 3.1-9.8
Benzo[alpyrene 3.8-5.5 0.005-1.3 67-73 15 5.6 0.023-4.6 Trace-9.3 ND-44 1.9-7.7 19-72
Benzo[blfluoranthene 1.0-3.1 130-133 0.46-16
Benzo[b]fluorene 0.07-0.18
Benzo[c]phenanthrene 33-37
Benzo[e]pyrene 5.5-7.4 0.016-3.3 96 19 9.1 0.18-8.8 0.17-4.2 ND-370 9-41
Benzo[ghi]fluoranthene 3.0-4.9 0.024-0.98 30-33
Benzo[ghi]perylene 7.0-13 0.004-3.2 49-61 12 7.9 0.21-12 ND-74 11-49
Benzo[j]fluoranthene 2.6-5.5
Benzo[k]fluoranthene 3.4-5.0 0.12-7.4
Chrysene 4.3-6.5 0.34-0.49 237-261 43 16 0.22-8.9 0.22-6.4 ND-170 7-71
Coronene 0.002-1.4 14-16 3.1 2.8 0.14-2.1 Trace-2.1 8-96 4-18
Cyclopenta[cd]pyrene ND 3.1 1.6
Dibenzo[a,h]pyrene 0.012-0.98
Fluoranthene 3.4-4.9 0.14-1.2 0.32-8.6 8-520 15-51
Fluorene 15-26
Indeno[1,2,3-cd]pyrene 5.1-9.1 0.022-2.0 57 11 5.5 0.16-9.6 9-43
Naphthalene 44
Perylene 0.01-0.20 7.6-10 0.004-0.88 ND-28 3-21
Phenanthrene 0.002-1.1 4-170 50-271
Pyrene 3.6-6.6 0.002-0.58 0.13-6.7 0.21-8.6 ND-540 12-49
Triphenylene 1.4-1.9 0.07-0.24 0.11-2.9 ND-50
ND, not detected; /, single measurements;
[31] Various cities, China (Chen et al.,1981);
[32] Various cities, China, 1986-88; determined by thin-layer chromatography and gas chomatography-mass spectroscopy (Chang et
al., 1988; Simoneit et al., 1991);
[33] Various locations with predominantly coal heating; Germany (analytical method not given) (Grimmer, 1980);
[34] Essen, Germany, predominantly coal heating, 1978-79 (Buck, 1983);
[35] Essen, Germany, predominantly oil heating, 1978-79 (Buck, 1983);
[36] Antony, France, 1979-80 (Muel & Saguem, 1985);
[37] Sutton Coldfield, United Kingdom, 1976-78 (Butler & Crossley, 1982);
[38] Barrow, USA, fossil fuel combustion area, 1979 (Daisey et al., 1981);
[39] Wood-heating area, Canada, 1989-91 (Environment Canada, 1994);
[40] Christchurch, New Zealand, 1979 (Cretney et al., 1985)
In Delft, the Netherlands, benzo [a]pyrene levels of up to 140 ng/m3
were measured on a foggy day with low wind velocity near a major road.
High concentrations of pyrene (220 ng/m3), benzo [ghi]perylene (130
ng/m3), and coronene (21 ng/m3) were also found. At border crossings
between the Netherlands and Germany on days with heavy traffic, the
maximum levels of individual PAH were 1-54 ng/m3 (Brasser, 1980).
PAH concentrations were determined in the centre of Paris, France, at
the top of a 55-m tower and thus less likely than ground-level samples
to be affected by traffic emissions and street dust; they can
therefore be considered to be homogeneous and representative. The
maximum levels found were 98 ng/m3 benzo [ghi]perylene, 60 ng/m3
indeno[1,2,3- cd]pyrene, 34 ng/m3 coronene, 28 ng/m3
benzo [b]fluoranthene, 13 ng/m3 benzo [a]pyrene, and 13 ng/m3
benzo [k]fluoranthene (Pistikopoulos et al., 1990).
The average concentration of individual PAH in particulate and vapour
phases during a nine-day photochemical pollution episode in
California, USA, in 1986 was 1 ng/m3. The maximum levels of
acenaphthene, acenaphthylene, fluorene, and phenanthrene ranged from
30 to 64 ng/m3 (Arey et al., 1991).
In 1989, the average benzo [a]pyrene concentrations in five Japanese
cities (Sapporo, Tokyo, Kawasaki, Nagoya, and Osaka) were 1.2-3.1
ng/m3. A maximum level of 15 ng/m3 was detected in Tokyo (Okita et
al., 1994). A detailed examination was undertaken of the molecular
composition of PAH in street-dust samples collected from the Tokyo
metropolitan area. Unsubstituted ring systems (i.e. parent PAH)
ranging from phenanthrene with three rings to benzo [ghi]perylene
with six rings were the primary components, three- and four-ring PAH
(i.e. phenanthrene, fluoranthene, and pyrene) predominating. The
concentrations of total PAH were of the order of a few micrograms per
gram of dust. On the basis of the PAH profile, it was suggested that
PAH in the dust of busy streets arose mainly from automobile exhausts,
while residential areas received a greater contribution from
stationary sources. In both types of dust, asphalt was thought to
contribute to only a minor extent (Takada et al., 1990). Giger &
Schaffner (1978) had come to the same conclusion some 20 years
earlier.
Benzo [a]pyrene was detected in ambient air in Moscow, Russian
Federation, at concentrations of 5.4 ng/m3 at a regular traffic site
and 20 ng/m3 at a crossroads with heavy traffic (Khesina, 1994).
(b) Road tunnels
In road tunnels, the concentrations of individual PAH were usually
1-50 ng/m3 (Table 36). Higher levels were reported in tunnels in
western Germany, with concentrations of 84 and 96 ng/m3
cyclopenta [cd]pyrene (Buck (1983) and 76 ng/m3 (Brasser, 1980) and
110 ng/m3 pyrene (Benner et al., 1989).
Table 36. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air polluted predominantly by vehicle exhaust
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene 168
Acenaphthylene 32 445
Anthracene 8.6/9.8 2.3 55 0.6-12 177
Anthanthrene 7.2 1 500 0.1-4.5 2-82
Benz[a]anthracene 37/44 0.6-1.9 16 20 12 000 102 1.9-2.9 90.2
Benzo[a]fluorene 18 2 800
Benzo[a]pyrene 30 2-14 0.2-0.8 16 12 9 600 66 1.3-26 62.6 0.1-14 1-57
Benzo[b]fluoranthene 2.3 8.8 12 000 43.6
Benzo[e]pyrene 28/32 11 9 600 69 1.5-19 55.5 01-12 3-43
Benzo[ghi]fluoranthene 29 18 3.2-26
Benzo[ghi]perylene 40/47 4-16 0.4-2.6 44 30 19 000 85 1.8-18 17.0 0.6-27 20-213
Benzo[k]fluoranthene 8.1 9.7 9 000 41.2
Chrysene 54/58 25 15 9 500 77.9
Coronene 26/27 2-17 0.3-1.1 29 20 7 500 1.0-10 ND 0.3-14 9-156
Cyclopenta[cd]pyrene 84/96 40 31 7.6-65 100
Dibenzo[a,h]pyrene 14.7
Fluoranthene 35 83 93 6.4-69 117
Fluorene 406
Indeno[1,2,3-cd]pyrene 18/22 0.3-1.3 16 13 9 400 0.3-15 20.0 6-70
1-Methylphenanthrene 2.6-43
Naphthalene 8030
Perylene 3.4 3.1 1 500 1-18
Phenanthrene 8.1 243 4.4-56 300
Pyrene 33-114 47 122 16 000 120 9.7-76 193 0.2-29
Table 36 (continued)
ND, not detected; /, single measurements;
[1] Street tunnel (location not specified), western Germany, 1978-79 (Buck, 1983);
[2] Coen Tunnel, Netherlands (Brasser, 1980);
[3] Street tunnel in Lincoln, Netherlands, 1981 (Kebbekus et al., 1983),
[4] Klara Tunnel, Sweden, 1983 (Colmsjo et al., 1986b);
[5] Soderleds Tunnel, Sweden, 1991; vapour and particulate phases (Ostman et al., 1991);
[6] Craeybeckx Highway Tunnel, Belgium, 1991 (De Fré et al., 1994);
[7] Baltimore Harbor Tunnel, USA, 1975 (Fox & Staley, 1976);
[8] Baltimore Harbor Tunnel, USA, 1985-86 (Benner et al., 1989);
[9] Heavily travelled tunnel, Chicago area, USA, 1990-92 (Khalili et al., 1995);
[10] Diesel bus garage, United Kingdom, 1979 (Waller et al., 1985);
[11] Inside car park, New Zealand (Cretney et al., 1985)
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled, unless otherwise stated
PAH were found at levels of up to 4 ng/m3 in an underground bus
terminal in Stockholm, Sweden; and 21 ng/m3 fluoranthene, 11 ng/m3
pyrene, and 8.1 ng/m3 phenanthrene were found in a subway station
(Colmsjö et al., 1986b).
Very high concentrations of PAH were found in the air of the
Craeybeckx Highway Tunnel in Belgium, which was used daily by an
average of 45 000 vehicles, of which 60% were petrol-fuelled passenger
cars, 20% diesel-fuelled cars, and 20% trucks. Of the cars, only 3%
had three-way catalysts (De Fré et al., 1994).
(c) Residential heating
The PAH levels in ambient air resulting mainly from residential
heating are included in Table 35, as the source cannot be identified
properly (see section 5.1.1.1).
The use of wood and coal for heating was the source of high levels of
benzo [a]pyrene in Calcutta, India (up to 120 ng/m3; Chakraborti et
al., 1988). The concentrations of individual PAH in Calcutta ranged
from 1.3 to 200 ng/m3, the highest levels being those of
benzo [e]pyrene, benzo [ghi]perylene, and benzo [b]fluoranthene.
The average levels of individual PAH resulting from domestic heating
in Christchurch, New Zealand were 1-210 ng/m3, benzo [ghi]perylene
and coronene showing the highest levels (Cretney et al., 1985), and up
to 43 ng/m3 were measured in Essen-Vogelheim, Germany (Buck, 1983).
High concentrations of individual PAH were determined in a residential
area heated primarily by coal, with levels of up to 260 ng/m3
chrysene, benz [a]anthracene, and benzo [b]fluoranthene (Grimmer,
1980).
The following PAH levels were measured on a roof directly downwind of
the chimney of a fireplace burning seasoned oak in the Chicago area,
USA: 1.8 µg/m3 acenaphthylene, 0.40 µg/m3 naphthalene, 0.35 µg/m3
anthracene, 0.22 µg/m3 phenanthrene, 0.20 µg/m3 benzo [a]pyrene,
0.20 µg/m3 benzo [e]pyrene, 0.13 µg/m3 fluorene, 0.10 µg/m3
pyrene, 0.096 µg/m3 fluoranthene, 0.052 µg/m3 acenaphthene, 0.045
µg/m3 benzo [k]fluoranthene, 0.033 µg/m3 chrysene, 0.030 µg/m3
cyclopenta [cd]pyrene, 0.023 µg/m3 benzo [b]fluoranthene, and 0.019
µg/m3 benz [a]anthracene. The levels of indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, and coronene were below
the limit of detection (Khalili et al., 1995).
In a comparison of the PAH concentrations in ambient air in eastern
and western Germany, the concentrations in rural areas were 3-12 times
higher in eastern than in comparable western parts of the country. The
PAH profiles were slightly different: the concentrations of the
lower-boiling-point PAH fluoranthene and pyrene were 110 and 68 ng/m3
in eastern and 36 and 28 ng/m3 in western Germany. The differences
may be due to the different types of brown and hard coal burnt (Jacob
et al., 1993a).
In 1991, PAH were determined in the air of Berchtesgaden, a national
park in Germany, and of the Oberharz (Ministry of Environment, 1993).
The concentration of phenanthrene, fluoranthene, and pyrene (about 14
ng/m3) in the Oberharz was two to three times higher than in
Berchtesgaden, due to the use of brown coal for heating. The levels of
the other PAH were of the same order of magnitude: benz [a]anthracene
and benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, about 5 ng/m3; and benzo [ghi]fluoranthene,
benzo [c]phenanthrene, benzo [e]pyrene, benzo [a]-pyrene,
indeno(1,2,3- cd)pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, anthanthrene, and coronene, < 1 ng/m3.
A model calculation for Germany showed that 5000 oil-heated houses
contributed to the pollution of ambient air by benzo [a]pyrene to the
same extent as one coal-heated house. It was assumed that one German
household consumes annually about 5000 litre of heating oil, producing
a maximum of 5 mg of benzo [a]pyrene (about 1 µg/litre combusted
oil). On the basis of a consumption of a similar amount of hard coal,
the same household would have an output of 25 g benzo [a]pyrene
(about 5000 µg/kg combusted hard coal) annually (J. Jacob, 1994,
personal communication).
5.1.2 Hydrosphere
PAH are found in the hydrosphere (Borneff & Kunte, 1983; Müller,
1987), mostly as a result of urban runoff, with smaller particles from
atmospheric fallout and larger ones from asphalt abrasion (Hoffman et
al., 1984). Long-range atmospheric transport of PAH has been well
documented in different countries (Lunde & Bjrseth, 1977; see also
section 4.1.2). After PAH are emitted into the atmosphere, for example
in motor vehicle exhaust, they are transferred into water by direct
surface contact or as a result of rainfall (Grob & Grob, 1974; Van
Noort & Wondergem, 1985a,b; Kawamura & Kaplan, 1986). The higher
levels of PAH that are found during winter months reflect increased
emissions resulting from domestic heating (Quaghebeur et al., 1983;
Thomas, 1986; see also section 5.1.1.1); however, the major source of
PAH varies for each body of water.
Anthropogenic combustion and pyrolysis and urban runoff containing
atmospheric fallout, asphalt particles, tyre particles, automobile
exhaust condensate and particulates, and lubricating oils and greases
were the major sources of PAH in lakes in Switzerland (Wakeham et al.,
1980a,b).
Comparisons between the levels of individual PAH in precipitation and
those in surface water showed that all of the precipitation samples
were more highly polluted with PAH, because they had been 'washed out'
of the atmosphere. Nearly all of the samples contained > 100 ng/litre
of fluoranthene, benzo [b]fluoranthene, pyrene,
indeno[1,2,3- cd]pyrene, phenanthrene, and naphthalene. The highest
levels of PAH in rainwater were found in Leidschendam, the
Netherlands, where pyrene concentrations < 2000 ng/litre,
fluoranthene concentrations < 1700 ng/litre, and benzo [a]pyrene
and benzo [b]fluoranthene concentrations < 390 ng/litre were
detected (van Noort & Wondergem, 1985b).
Most surface water samples contained concentrations of < 50
ng/litre of individual PAH. The levels in rainwater were 10-200
ng/litre, whereas those in snow were < 1000 µg/kg, with a maximum
of 6800 µg/kg for an individual PAH (Lygren et al., 1984). In one fog
sample, benzo [a]pyrene was found at 880 ng/litre and fluoranthene at
3800 ng/litre (Schrimpff, 1983: see section 5.1.2.4).
In sediment the levels of individual PAH were usually 1000-10 000
µg/kg dry weight, which are one order of magnitude higher than those
in precipitation. Triphenylene was detected in samples of sediment
from the Mediterranean Sea (France) at 2-600 µg/kg (Milano et al.,
1985) and in samples from Lake Geneva (Switzerland) at 25 µg/kg
(Dreier et al., 1985; see section 5.1.3).
5.1.2.1 Surface and coastal waters
The levels of individual PAH found in surface and coastal waters at
various locations are summarized in Table 37. Rivers in Germany
contained some PAH at concentrations of 1-50 ng/litre (Grimmer et al.,
1981b; Ernst et al., 1986; Regional Office for Water and Waste
Disposal, 1986; Kröber & Häckl, 1989) and fluoranthene, pyrene,
chrysene, benzo [a]pyrene, and benzo [e]pyrene at concentrations
< 100 ng/litre. The PAH levels in seawater from the German coast
varied over one order of magnitude depending on the sampling site. In
open seawater, the concentrations of two- to four-ring PAH -
naphthalene, fluorene, phenanthrene, fluoranthene, and pyrene - were
0.1-5 ng/litre, and those of five- to six-ring PAH ranged from < 0.01
to 0.2 ng/litre. Near the coast, the concentration of five- to
six-ring PAH increased with the content of particles, to which they
have greater affinity than two- to four-ring PAH (German Federal
Office for Sea Navigation and Hydrography, 1993).
The maximum levels of PAH in the Rivers Thames and Trent in the United
Kingdom were > 130 ng/litre. The highest levels of individual PAH in
the River Thames were 360 ng/litre fluoranthene, 350 ng/litre
benzo [a]pyrene, 210 ng/litre indeno[1,2,3- cd]pyrene, 160 ng/litre
benzo [ghi]perylene, 140 ng/litre benzo [k]fluoranthene, and 130
ng/litre perylene (Acheson et al., 1976). More recent data were not
available.
In Norway, the levels of most individual PAH were > 100 ng/litre. For
example, surface water from Bislet Creek near Oslo contained
fluoranthene, pyrene, phenanthrene, methylphenanthrene, naphthalene,
acenaphthene, acenaphthylene, and fluorene at concentrations > 1000
ng/litre (Berglind, 1982).
Table 37. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in surface and coastal waters
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthene 14-1232
Acenaphthylene 0.4-0.9 12-1024
Anthracene 1 10 18-932
Anthanthrene 0.2-0.5 15/1.8
Benz[a]anthracene ND 0.16 2.2-6.8 24/66 40/10 71-582
Benzo[a]fluorene 43/330
Benzo[a]pyrene 1-23 0.8 0.39 1.2-7.3 87/25 18 10/60 ND-40 19-311 0.9
Benzo[b]fluoranthene 0.1-0.5 0.07 80/20 ND-42 70-678 0.5-0.9
Benzo[b]fluorene 38 17
Benzo[c]phenanthrene 2.3-4.2 13/34 23-172
Benzo[e]pyrene 2-40 0.06 7.1-11 108/36 40-551
Benzo[ghi]fluoranthene
Benzo[ghi]perylene ND ND < 0.05 3.7-7.0 61/16 50/10 ND-61 33-636 ND
Benzo[k]fluoranthene 0.7-0.8 0.02 3.6-6.1 59/22 40/10 ND-24 0.2-0.5
Chrysene 11-15 36/87 14 10/10
Coronene ND-2.4 15/4.3
Cyclopenta[cd]pyrene ND ND
Dibenzo[a,h]pyrene <0.03 30/10
Fluoranthene 4-616 1.0-3.5 0.35 5.2/9.1 28/102 2.3-13 50/130 2-110 285-3269 3.4-5.1
Fluorene 2 0.63 0.6-1.2 25-1995
Indeno[1,2,3-cd]pyrene Trace < 0.03 2.8-6.1 63/13 50/20 ND-39 17-299 ND
1-Methylphenanthrene 30-1281
5-Methylcholanthrene
Naphthalene 4 50-2090
Perylene 0.8-1.4 27 20 9/28
Phenanthrene 3-136 3.5 1.5-9.1 101-5656
Pyrene 5-402 0.28 4.8/8.5 25/90 2.2-13 100/30 485-3099
Triphenylene
Table 37 (continued)
ND, not detected; /, single measurements;
[1] Lake water, Norway, 1981-82 (Gjessing et al., 1984);
[2] Lake water, Switzerland (Vu Duc & Huynh, 1981);
[3] Lake Superior, USA, 1986 (Baker & Eisenreich, 1990);
[4] Elbe River, Germany, 1980 (Grimmer et al., 1981b);
[5] Elbe River, main drainage channel, Germany, 1980 (Grimmer et al., 1981b);
[6] Water in various rivers, Germany, 1981-83 (Ernst et al., 1986);
[7] Water in various rivers, Germany, 1985; analytical method not given (Regional Office for Water and Waste Disposal,
1986);
[8] Water in various rivers, Germany, 1985-86; analytical method not given (Krober & Hackl (1989);
[9] River water, Norway, 1979 (Berglind, 1982);
[10] River water, Switzerland (Vu Duc & Huynh, 1981)
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated. The results of studies
in which water samples were filtered through solid sorbents may be underestimates of the actual PAH content (see section
2.4.1.4).
Table 37 (continued)
Compound [11] [12] [13] [14] [15] [16] [17] [18] [19] [20]
Acenaphthene ND-3 10 0.08-1.1 50-100
Acenaphthylene ND-5 0.02-1.7 80-1300
Anthracene ND-4 0.2 0.8-9.5 0.01-1.5 < 1-25 ND
Anthanthrene NR
Benz[a]anthracene ND-5 0.3 ND-9.6 0.04-6.8 ND
Benzo[a]fluorene NR
Benzo[a]pyrene 0.1-1.8 130-150 0.1/0.2 ND-10 0.2-1.0 0.03-8.8 ND
Benzo[b]fluoranthene ND-8 0.04-12
Benzo[b]fluorene 4.0-19 NR
Benzo[c]phenanthrene NR
Benzo[e]pyrene 0.02-8.8 ND
Benzo[ghi]fluoranthene NR
Benzo[ghi]perylene 0.2-11 30-160 0.7/0.8 ND-10 0.02-3.8 < 0.3-16 50
Benzo[k]fluoranthene 0.1-1.7 80-140 0.2/0.3 ND-13 0.02-7.7
Chrysene ND-12 NR
Coronene 0.01-1.4 NR
Dibenzo[a,h]pyrene ND-1 100
Fluoranthene 0.7-508 20-360 1.1/3.7 3-12 0.8 10-25 1.4-2.6 0.40-14 NR
Fluorene ND-2 0.7-15 1.9-5.2 0.33-3.2 70-2500
Indeno[1,2,3-cd]pyrene 0.1-8.0 50-210 ND/0.2 ND-8 0.01-3.5 NR
1-Methylphenanthrene NR
5-Methylcholanthrene NR
Naphthalene 4-34 3.6 0.4-9.2 NR
Perylene 40-130 0.01-5.7 NR
Phenanthrene 6-34 21-18 8.0-93 2.4-2.7 0.24-5.8 < 1-3 ND
Pyrene 50-260 1-15 0.3-15 8.8-25 0.82-1.7 0.12-15 < 1-53 10-65
Triphenylene NR
Table 37 (continued)
ND, not detected; /, single measurements;
[11] River water, United Kingdom, 1974 (Lewis, 1975);
[12] Water in various rivers, United Kingdom, analytical method not given (Acheson et al.,1976);
[13] Water in various rivers, United Kingdom; analytical method not given (Sorrell et al., 1980);
[14] River water, USA, 1984 (De Leon et al., 1986);
[15] Surface water, Canada (Environment Canada, 1994);
[16] River water, China, 1981 (Wu et al., 1985);
[17] Coastal water, Germany, 1982 (Ernst et al., 1986);
[18] Seawater, Germany, 1990 (German Federal Office for Sea Navigation and Hydrography, 1993);
[19] Coastal water, Australia, 1983 (Smith et al., 1987);
[20] Water (no further specification), Japan, 1974-91 (Environment Agency, Japan, 1993)
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated. The results of studies
in which water samples were filtered through solid sorbents may be underestimates of the actual PAH content (see section
2.4.1.4).
The highest concentrations of PAH in water in Canada were reported for
water samples from ditches next to utility and railway lines near
Vancouver. The highest mean concentrations were measured near utility
poles treated with creosote, with values of 2000 µg/litre for
fluoranthene, 1600 µg/litre for phenanthrene, and 490 µg/litre for
naphthalene (Environment Canada, 1994).
Four individual PAH were detected in seawater from Green Island,
Australia. The highest levels of PAH found were 53 ng/litre pyrene, 25
ng/litre anthracene, 16 ng/litre benzo [ghi]perylene, and 3 ng/litre
phenanthrene, (Smith et al., 1987).
The total content of phenanthrene, anthracene, fluoranthene, pyrene,
benzo [b]fluorene, and benz [a]anthracene in the Yellow River,
China, was 170 ng/litre (Wu et al., 1985; for individual PAH
concentrations, see Table 37).
The PAH levels found in the River Rhine in Germany and the Netherlands
and in some of its tributaries are summarized in Table 38. Many
investigators have detected PAH in the Rhine. The lowest
concentrations of benzo [a]pyrene, < 10-20 ng/litre, were found in
the Rhine at Lobith and Hagestein in Germany and at Lek in the
Netherlands in 1987-90 (Association of Rhine and Meuse Water Supply
Companies, 1987-90), when the levels of fluoranthene were 70-140
ng/litre. In 1976-79, the Rhine at Lek and Waal contained < 10-580
ng/litre of benzo [a]pyrene (Association of Rhine and Meuse Water
Supply Companies, 1976-79), so that the levels had decreased by one
order of magnitude within 14 years. The sum of fluoranthene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene) was 9-40 ng/litre
at km 30 and 130-5700 ng/litre at km 853, indicating that the level of
pollution increased markedly between the source and the estuary
(Borneff & Kunte (1983). The average concentrations of individual PAH
were 1-50 ng/litre, although individual PAH were found at
concentrations in the range 100-200 ng/litre near Mainz, an
industrialized town (Borneff & Kunte, 1964, 1965). In general, the PAH
levels in the Rhine decreased by a factor of 3 between 1979 and 1989.
The Emscher and Ruhr waterways in Germany have been heavily polluted
(see Table 38). In 1985, the Emscher River contained 6400 ng/litre
fluoranthene, 6000 ng/litre pyrene, 2000 ng/litre benz [a]anthracene,
1100 ng/litre dibenz [a,h]anthracene, 910 ng/litre benzo [a]pyrene,
880 ng/litre chrysene, 630 ng/litre indeno[1,2,3- cd]pyrene, 510
ng/litre benzo [ghi]perylene, 270 ng/litre anthracene, 220 ng/litre
perylene (Regional Office for Water and Waste Disposal, 1986), but by
1989 the levels had decreased by about one order of magnitude
(Regional Office for Water and Waste Disposal, 1990 ). The PAH
concentrations in the Emscher were three times higher than those in
the Rhine near Mainz. Between 1985 and 1989, the PAH levels in the
Emscher decreased further by a factor of 15; however, the levels in
the Ruhr remained about the same or increased slightly between 1979
and 1985 (Regional Office for Water and Waste Disposal, 1986, 1988,
1990).
Table 38. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in the River Rhine and some highly polluted tributaries
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Anthracene 10 270 25-260 10
Anthanthrene 0.9-11 1.3
Benz[a]anthracene 6.1-31 11-50 1970 100-780 13 20
Benzo[a]pyrene 0.8-36 ND-7 6-30 12-40 < 10-20 910 59-280 15 30
Benzo[b]fluoranthene ND-8 7-30 12-40 < 10-30 880 62-310 40
Benzo[c]phenanthrene 1.5-9.1 1.9
Benzo[a]pyrene 18-31 33
Benzo[ghi]fluoranthene 1.0-11 2.2
Benzo[ghi]perylene 15-29 ND-8 6-30 9-30 < 10-20 510 30-210 17 30
Benzo[k]fluoranthene ND-4 2-14 6-20 < 10-40 440 36-150 20
Chrysene 21-62 1080 27 30
Dibenzo[a,h]pyrene 10-40 1100 32-310 30
Fluoranthene 4-18 15-61 25-77 20-140 6420 207-1700 60
Indeno[1,2,3-cd]pyrene 9.5-27 ND-6 2-26 10-40 < 10-20 630 28-220 17 30
Perylene ND-8.1 10 220 13/80 2.1 10
Pyrene 20-50 6010 155-1100 50
ND, not detected; /, single measurements;
[1] Rhine, Germany, 1979 (Grimmer et al.,1981b);
[2] Rhine, Germany, 1985-88, analytical method not given (Krober & Hackl, 1989);
[3] Rhine, Netherlands, 1985-88 (Netherlands' Delegation, 1991);
[4] Rhine, Germany, 1987-89, analytical method not given (Regional Office for Water and Waste Disposal, 1988, 1989, 1990);
[5] Rhine, Netherlands; 1987-90, analytical method not given (Association of Rhine and Meuse Water Supply Companies, 1987-90);
[6] Emscher, Germany, 1985, analytical method not given (Regional Office for Water and Waste Disposal, 1986);
[7] Emscher, Germany, 1987-89, analytical method not given (Regional Office for Water and Waste Disposal, 1988, 1989, 1990);
[8] Ruhr, Germany, 1979 (Grimmer et al., 1981b);
[9] Ruhr, Germany, 1985, analytical method not given (Regional Office for Water and Waste Disposal, 1986)
The PAH levels in the main drainage channels of the River Elbe,
Germany, were one order of magnitude higher than in the river water
(Grimmer et al., 1981b), owing to the high input of rainwater to the
channels.
5.1.2.2 Groundwater
The PAH concentrations in uncontaminated groundwater in the
Netherlands generally did not exceed 0.1 µg/litre, but levels of about
30 µg/litre naphthalene, 10 µg/litre fluoranthene, and 1 µg/litre
benzo [a]pyrene were reported in contaminated groundwater (Luitjen &
Piet, 1983).
Benzo [a]pyrene levels in groundwater in western Germany ranged from
0.1 to 0.6 ng/litre and those of total PAH from 34 to 140 ng/litre
(Andelman & Suess, 1970). Benzo [a]pyrene was also detected at levels
of 0.1-5.0 ng/litre in groundwater (Woidich et al., 1976). More recent
data were not available. Groundwater in the USA contained maximum
concentrations of 0.38-1.8 ng/litre naphthalene, 0.02-0.04 ng/litre
acenaphthene, and 0.008-0.02 ng/litre fluorene (Stuermer et al.,
1982). Near a refinery at Pincher Creek, Alberta, Canada, the pyrene
concentrations in groundwater showed a maximum of 300 µg/litre
(median, 30 µg/litre); the maximum concentration of fluorene was 230
µg/litre (median, 40 µg/litre). At Newcastle, New Brunswick, Canada,
naphthalene was detected at concentrations up to 2.8 µg/litre and
benzo [a]pyrene up to 0.32 µg/litre in groundwater near a
wood-preserving plant (Environment Canada, 1994).
5.1.2.3 Drinking-water and water supplies
PAH levels were determined in drinking-water in samples from Canada,
Scandinavia, and the USA up to 1982. The concentration of naphthalene
was 1.2-8.8 ng/litre, that of benzo [a]pyrene was 0.2-1.6 ng/litre,
and that of the sum of the six 'standard WHO' PAH (fluoranthene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene) was 0.6-24
ng/litre. The highest levels of naphthalene (1300 ng/litre),
benzo [a]pyrene (77 ng/litre), and the six WHO standard PAH (660
ng/litre) were detected in raw water sources in the USA and in the
Great Lakes area of Canada (Müller, 1987). More recent measurements
are given in Table 39. Most samples contained 0.38-16 ng/litre
naphthalene and < 0.04-2.0 ng/litre benzo [a]pyrene. In one set of
water samples from the Netherlands, no PAH were detected, with a limit
of detection for individual PAH of 4 ng/litre (de Vos et al., 1990).
In a study of the changes in PAH concentrations after passage of water
through tar-coated major distribution pipes, the level increased from
an initial concentration of none detected-13 ng/litre to none
detected-62 ng/litre. The finding that water in a few distribution
lines had lower concentrations of PAH may be due to sorption of PAH on
the surfaces of distribution pipes, chemical interaction with oxidants
in water, or a dilution effect (Basu et al., 1987).
Of 101 German drinking-water samples analysed in 1994, four exceeded
the German drinking-water standard of 0.2 µg/litre for the sum of
fluoranthene, benzo [b]fluoranthene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [ghi]-perylene, and indeno[1,2,3- cd]pyrene.
Heavy contamination had occurred after repairs to a pipeline coated
with tar, and one drinking-water sample taken in a household contained
2.7 µg/litre of these PAH, in addition to phenanthrene at 2.8 µg/litre
and pyrene at 1.2 µg/litre (State Chemical Analysis Institute,
Freiburg, 1995). The report stated that abrasion of particles from
tar-coated drinking-water pipelines poses a hazard that is often
difficult to judge since it is often not known what material was used
decades previously.
In Canada, the PAH concentrations in drinking-water were usually below
or near the detection limits of 1-5 ng/litre, although concentrations
of 5.0-21 ng/litre benzo [ghi]perylene, 1.0-12 ng/litre fluoranthene,
1.0-5.0 ng/litre benzo [b]fluoranthene, 1.0-3.0 ng/litre
benzo [k]fluoranthene, and 1.0-3.0 ng/litre benzo [a]pyrene were
detected in some areas (Environment Canada, 1994).
5.1.2.4 Precipitation
(a) Rain
The concentrations of PAH found in precipitation in 1979-91 are
summarized in Table 40. The levels of benzo [a]pyrene were < 1-390
ng/litre. In an analysis of PAH in rainfall in Hanover, Germany,
between July 1989 and March 1990, fluoranthene was the dominant
component, followed by pyrene. The average concentration of all PAH
increased from 351 ng/litre in summer to 765 ng/litre in the autumn of
1989, while a slight decrease was observed in the winter of 1989-90.
These results indicate that the increase in the level of PAH in
precipitation in cold weather is due to an increase in residential
heating and a slower rate of photochemical degradation (Levsen et al.,
1991).
Table 39. Polycyclic aromatic hydrocarbon concentrations (ng/litre) in drinking-water
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 0.6-4.0 7.4-14
Acenaphthylene 0.4-4.4 0.40-1.6
Anthracene 0.5-7 < 1.3-9.7
Anthanthrene 0.2
Benz[a]anthracene ND-1.9 0.4-5.5 0.12-1.5
Benzo[a]fluoranthene 0.1-3.3 0.05-4.2
Benzo[a]pyrene 0.1-0.7 < 0.1-2.0 < 0.04-0.29 Trace-1.9 0.2-0.3 0.2-1.6 < 5.0
Benzo[b]fluoranthene 0.5-1.3 2.4-4.0 0.05-0.34 0.1-14 < 5-40
Benzo[b]fluorene 0.9 0.04-<1.4
Benzo[e]phenanthrene 0.9-1.5 0.28
Benzo[e]pyrene 0.2-4 < 0.1-0.41
Benzo[ghi]fluoranthene 0.36
Benzo[ghi]perylene 0.3-0.9 0.4-1.1 ND 0.4-0.7 0.4-4.0 < 5.0
Benzo[j]fluoranthene 0.03-0.14 0.2-1.2
Benzo[k]fluoranthene 0.2-0.8 0.02-0.10 0.2-4.9 0.1-0.3 0.1-0.7 < 5-40
Chrysene 21-62 1080 27 30
Dibenz[a,h]anthracene 1.2
Fluoranthene 3.5-6.5 1.7-18 < 0.58-24 0.7-3400 3.4-4.2 5-24 2.4-9.0 < 5-623
Fluorene 0.9-4 < 1.1-21 4-16
Indeno[1,2,3-cd]pyrene Trace-0.7 0.4-1.2 ND-1.1 < 0.5 0.7-2.2 < 5.0
1-Methylphenanthrene 0.5-1.0 0.14-13
Naphthalene 1.8-5 < 6.3-8.8 8 6-16
Perylene Trace-0.2 0.2
Phenanthrene 2.5-46 < 2.2-64 24-90
Pyrene 1.6-3.7 1.1-15 < 0.30-12 40/40
Table 39 (continued)
ND, not detected; /, single measurements;
[1] Austria; analytical method, in-situ fluorescence determination (Woidich et al., 1976);
[2] Norway, 1978-80 (Berglind, 1982);
[3] Norway, 1980-81 (Kveseth et al., 1982);
[4] Switzerland, 1973 (Grob & Grob, 1974);
[5] Switzerland (Vu Duc & Huynh, 1981);
[6] United Kingdom; water reservoirs after treatment, 1974 (Lewis, 1975);
[7] USA, 1976; analytical method, high-performance liquid chromtography and gas chromatography
(Thruston, 1978);
[8] USA, 1976-77; analytical method, thin-layer chromatography and gas-liquid chromatography with
flame ionization detection (Basu & Saxena, 1978a,b);
[9] Canada, treated drinking-water, 1987-90 (Environment Canada, 1994)
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated.
The results of studies in which water samples were filtered through sold sorbernts may be
underestimates of the actual PAH content (see section 2.4.1.4).
Table 40. Polycyclic aromatic hydrocarbon concentrations (ng/litre) in rainwater
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 3.2 1.2/16 2.5-8.5
Acenaphthylene 130-200 14 4.7/55 23-59
Anthracene 8-19 0.88/23 2.0-7.9
Benz[a]anthracene 1.2-86 140 6-100 9-33 7-17 20-65 1.6-4.5
Benzo[a]fluoranthene 14-52
Benzo[a]pyrene 5-17 1.1-187 ND-390 10-37 7-26 5-36 ND-0.18
Benzo[b]fluoranthene 2.9-166 15-390 45-70 17-65
Benzo[b]fluorene 15
Benzo[c]phenanthrene 802
Benzo[e]pyrene < 0.5a-149 217-290 7-62 ND-0.51
Benzo[ghi]perylene 7-29 1.7-109 40-70 15-56 22
Benzo[k]fluoranthene 1.0-142 6-190 17-30 9-28
Chrysene 2.9-141 30-120 ND-67 21-29 3.3-12
Dibenz[a,h]anthracene < 0.5a-12 7-20 3-12
Fluoranthene 23-66 23-392 240-270 14-1650 66-180 87-189 115-162 1.7/110 28-70
Fluorene 10-200 6-50 3.2/43 9.1-22
Indeno[1,2,3-cd]pyrene < 0.5a-137 ND-80 50-110 24-72 12
1-Methylphenanthrene 8-26
Naphthalene 8-77 20/72 46-140
Perylene 2
Phenanthrene 130-600 30-133 79-113 158-238 24/140 61-130
Pyrene 9.5-304 25-60 ND-2000 ND-37 36-108 77-175 24-56
Table 40 (continued)
ND, not detected; /, single measurements;
[1] Bavaria, Germany, 1979-80; analytical method, high-performance thin-layer chromatography (Thomas, 1986);
[2] Hanover, Germany, 1989-90 (Levsen et al., 1991);
[3] Italy (Morselli & Zappoli, 1988);
[4] Leidschendam, Netherlands, 1982 (Van Noort & Wondergem, 1985b);
[5] Rotterdam, Netherlands, 1983 (Van Noort & Wondergem, 1985b);
[6] Netherlands, 1983 (Den Hollander et al., 1986);
[7] Oslo, Norway, 1978 (Berglind, 1982);
[8] Oregon, USA, 1982 (Pankow et al., 1984);
[9] Portland, USA, 1984 (Ligocki et al., 1985)
a Detection limit for benzo[a]pyrene
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated. The results
of studies in which water samples were filtered through solid sorbents may be underestimates of the actual PAH
content (see section 2.4.1.4).
The concentrations of phenanthrene and fluoranthene in rainwater were
noticeably higher than those at 200 m when sampled simultaneously, but
no significant differences in the concentrations of
benzo [k]fluoranthene, benzo [b]fluoranthene, benzo [a]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, or
indeno[1,2,3- cd]pyrene were found. The authors suggested that
scavenging in and below clouds was responsible for the presence of PAH
in rainwater (Van Noort & Wondergem, 1985b).
The deposition rates of individual PAH in Cardiff, London, Manchester,
and Stevenage, United Kingdom, were 0.3-20 µg/m2 per day. Anthracene
accounted for about 25% of the deposition in London, followed by
pyrene (16%), benzo [b]fluoranthene (16%), and benz [a]anthracene
(13%) (Clayton et al., 1992).
The rate of precipitation containing PAH after gravitational
deposition by rain, snow, and particles was not affected by the type
or structure of the receiving surface. Precipitation in a beech and
spruce stand contained concentrations of 23-52 ng/litre fluoranthene,
8.9-30 ng/litre benzo [ghi]-perylene, 6.4-27 ng/litre
indeno[1,2,3- cd]pyrene, and 2.0-8.4 ng/litre benzo [a]pyrene. The
deposition of PAH is in general higher under spruce stands because the
rates of interception are higher than those in beech stands.
Substantial amounts of PAH are transferred to the soil by litterfall,
indicating adsorption of PAH on the surfaces of leaves and needles
(Matzner, 1984).
(b) Snow
The concentrations of PAH in snow samples are summarized in Table 41.
A sample collected in Hanover, Germany, contained fluoranthene at 55
ng/litre, pyrene at 31 ng/litre, and other PAH at concentrations up to
9 ng/litre (Levsen et al., 1991). A sample of snow from Bavaria
contained 200 ng/litre fluoranthene, 50 ng/litre benzo [ghi]perylene,
and 29 ng/litre benzo [a]pyrene (Schrimpff et al., 1979).
In Norwegian snow samples, the average concentrations of individual
PAH were 10-100 ng/litre, but levels up to 6800 ng/litre were found of
phenanthrene, 1-methylphenanthrene, fluoranthene,
benzo [b]fluoranthene, and fluorene (Berglind, 1982; Gjessing et al.,
1984; Lygren et al., 1984). Snow taken near a steel plant in Canada
contained average levels of 50-500 ng/litre of individual PAH but
higher amounts of phenanthrene, fluoranthene, and pyrene (Boom &
Marsalek, 1988).
Table 41. Polycyclic aromatic hydrocarbon concentrations (ng/litre) in snow
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene 10-13 <50-98
Acenaphthlene 19-47 <50-153
Anthracene 13-28 9-379 165-246
Benz[a]anthracene 2.6 21-47 15-677 228
Benzo[a]fluoranthene 13 179-396
Benzo[a]pyrene 29 3.0 23-77 54-602 250 <100-558
Benzo[b]fluoranthene 9.2 799-1501 <100-647
Benzo[b]fluorene 11 192
Berzo[e]pyrene 5.5 30-64 609 360-630
Benzo[ghi]perylene 50 4.8 29-85 98-551 319-391 <100-466
Benzo[k]fluoranthene 2.8 <100-990
Chrysene 6.2
Dibenz[a,h]anthracene <0.5a
Fluoranthene 200 55 108-211 86-2665 1820-3143 <50-7020
Fluorene 13-85 96 485-1237 <50-237
Indeno[1,2,3-cd]pyrene <0.5a 20-82 <100-496
I-Methylphenanthrene 1366-2117
Naphthalene 50-94 36-67 123-195
Perylene 12
Phenanthrene 119-276 45-1385 4055-6787 <50-3560
Pyrene 31 68-143 55-2002 <50-3750
Analysed by high-performance liquid chromatography or gas chromatography, unless
otherwise stated. The results of studies in which water samples were filtered through
solid sorbents may be underestimates of the actual PAH content (see section 2.4.1.4).
a Detection limit for benzo[a]pyrene
[1] Bavaria, Germany, 1978; analytical method, high-performence thin-layer chromatography
and gas chromatography-mass spectroscopy (Schrimpff et al., 1979);
[2] Hanover, Germany, 1990 (Levsen et al., 1991);
[3] Norway, 1979-81 (Berglind, 1982);
[4] Norway, 1981-82 (Gjessing et al., 1984);
[5] Norway (Lygren et al., 1984);
[6] Near steel plant, Canada, 1986 (Boom & Marsalek; 1988)
(c) Hail
The PAH levels in a hail sample collected in Hanover, Germany, were of
the same order of magnitude as those in rain samples: fluoranthene,
170 ng/litre; pyrene, 98 ng/litre; benzo [b]fluoranthene, 58
ng/litre; chrysene, 47 ng/litre; benzo [e]pyrene, 40 ng/litre;
indeno[1,2,3- cd]pyrene, 29 ng/litre; benzo [ghi]perylene, 27
ng/litre; benzo [k]fluoranthene, 19 ng/litre; benz [a]an-thracene,
16 ng/litre; benzo [a]pyrene, 12 ng/litre; and
dibenz [a,h]anthracene, 3.3 ng/litre (Levsen et al., 1991).
(d) Fog
The concentrations of PAH in fog are higher than those in rain. A fog
sample collected in western Germany contained 360-3800 ng/litre
fluoranthene and 130-880 ng/litre benzo [a]pyrene (Schrimpff, 1983).
In fog samples collected during the autumn of 1986 in Zürich,
Switzerland, the average concentrations of PAH found were 4400
ng/litre fluoranthene, 2700 ng/litre benzo [b]fluoranthene, 2500
ng/litre pyrene, 2200 ng/litre phenanthrene, 2100 ng/litre
benzo [e]pyrene, 1400 ng/litre benz [a]anthracene, 1400 ng/litre
indeno[1,2,3- cd]pyrene, 1200 ng/litre benzo [a]pyrene, 920 ng/litre
anthracene, 860 ng/litre 1-methylphenanthrene, 750 ng/litre
benzo [b]fluorene, 750 ng/litre perylene, 590 ng/litre
benzo [k]fluoranthene, 540 ng/litre benzo [ghi]perylene, 340
ng/litre anthanthrene, 260 ng/litre fluorene, and 160 ng/litre
benzo [a]fluorene (Capel et al., 1991).
5.1.3 Sediment
PAH levels in sediments from rivers, lakes, seas, estuaries, and
harbours are summarized in Tables 42-46.
5.1.3.1 River sediment
The concentrations of individual PAH in river sediments in 1987-91
(Table 42) varied over a wide range; the maximum values were in the
high nanogram per gram range.
The levels of individual PAH in sediments from German rivers were
about 4000 µg/kg for benzo [a]pyrene, fluoranthene, and
benzo [b]fluoranthene and about 1500 µg/kg for pyrene,
indeno[1,2,3- cd]pyrene, and benz [a]anthracene. The levels of other
PAH generally did not exceed 500 µg/kg (Kröber & Häckl, 1989; Regional
Office for Water and Waste Disposal, 1989). PAH were determined in
many German river sediments. Table 42 gives data for three rivers: the
Rhine and Neckar rivers are highly polluted, whereas the Gersprenz is
relatively uncontaminated.
The concentrations of PAH in the sediments of rivers around Aachen,
Germany, were determined in different size fractions, which allowed
the authors to locate where the sediment became contaminated (Lampe et
al., 1991).
The PAH concentrations in sediment from the River Elbe in Germany in
1991 were of the same order of magnitude as those in Lake Plöner and
Lake Constance, but the river sediment contained more PAH with a low
boiling-point than the lake sediments. The ratio of fluoranthene to
benzo [e]pyrene, taken as a marker of the emission of PAH from the
combustion of brown coal, was 2.8-5.1, similar to those found in the
Elbe sediment. It was concluded that the PAH in the sediment were due
mainly to brown-coal combustion (German Ministry of Environment,
1993).
Table 42. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in river sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12]
Acenaphthene ND-140 14.5 1 100 ND 0.04-130
Acenaphthylene ND 9.7 1 540 ND 0.7-671
Anthracene ND-1010 80-640 670/NR ND/NR 82.1 8-200 152 4 700 10-1200
Benz[a]anthracene 620-1700 10000/NR 50-90/NR 450 ND-100 541 6 600 3.2-2100
Benzo[a]pyrene Q 400-1250 ND-8000/ 20-90/10-80 1-760 70-11 960 454 ND-80 570 4 400 5-3700
ND-5300
Benzo[b]fluoranthene 460-1290 ND-8700/ 50-190/ 620 ND-50
ND-5600 26-150
Benzo[e]pyrene 596 4 900 0.9-1800
Benzo[ghi]fluoranthene 253 NR
Benzo[ghi]perylene Q-578 340-750 ND-2900/ ND 10-70 60-7480 358 ND 353 7 400 3-1310
ND-1900
Benzo[j]fluoranthene 749
Benzo[k]fluoranthene 230-650 ND-4000/ 20-90/10-80 408 ND-60 608
ND-2700
Chrysene ND-1549 6700/NR ND-30/NR 597 904 NR
Coronene 20-260 150-2460 284 NR
Cyclopenta[cd]pyrene 15 1 100 NR
Dibenz[a,h]anthracene 500-1070 2600/NR ND-20/NR 21 ND-200 2 800 8.1-340
Fluoranthene ND-4455 900-2470 ND-19000/ 2-2360 190-29300 904 100-400 1013 13 000 ND-60 NR
100-380/
ND-12 600 52-310
Fluorene ND-260 25.4 ND-2 26 3 000 ND/50 3-130
Indeno[1,2,3-cd]pyrene 360-910 ND-6300/ ND/ND 332 486 16 000 NR
ND-4200
1-Methylphenanthrene 145 NR
Naphthalene ND-2630 7.0 3 800 ND
Perylene 120-320 ND-100 2 400 NR
Phenanthrene ND-220 3300/NR ND-40/NR 361 10-400 563 10 000 ND/220 9-2800
Pyrene ND-2526 680-3450 17000/NR ND-130/NR 736 80-300 940 9200 ND-160 20-3900
Triphenylene 10-80 NR
Table 42 (continued)
NQ not detacted; /, single measurements; NR, not reported; Q, qualitative;
[1] Czechoslovakia, 1988; reference weight not given (Holoubek et al., 1990);
[2] Rhine, Germany, 1982-83 and 1987-88; analytical method and reference weight not given (Regional Office for Water and Waste Disposal, 1989);
[3] Neckar, Germany, 1985-88; fine, unsieved sediment; analytical method not given (Krober & Hackl, 1989);
[4] Gersprenz, Germany, 1985-88; fine, unsieved sediment; analytical method not given (Krober & Hackl, 1989);
[5] Wildbach, Germany, 1989 (Lampe et al., 1991);
[6] Haarbach, Germany, 1989 (Lampe et al., 1991);
[7] River, Bremen, Germany, 1994 (Riess & Wefers, 1990;
[8] Rhone, France, 1985 (Milano & Vernet, 1988);
[9] Sweden, 1985 (Broman et al., 1987);
[10] Black River, USA, 1984 (Fabacher et al., 1991);
[11] Rainy River, Canada, 1986; reference weight not given (Merriman, 1988);
[12] Japan, 1974-91 (Environment Agency, Japan, 1993)
Analysed by high-performance liquid chromatography or gas chromatography and concentration in micrograms per kilogram dry weight
The maximum levels of individual PAH in sediments in Czechoslovakia
were 4500 µg/kg fluoranthene, 2600 µg/kg naphthalene, 2500 µg/kg
pyrene, 1500 µg/kg chrysene, 1000 µg/kg anthracene, 580 µg/kg
benzo [ghi]perylene, 260 µg/kg fluorene, 220 µg/kg phenanthrene, and
140 µg/kg acenaphthene (Holoubek et al., 1990).
The levels of individual PAH in sediments from some of the most
polluted areas in continental USA were summarized by Bieri et al.
(1986). The levels usually ranged from 1000 to 10 000 µg/kg, but that
in sediment from the Elizabeth River, Virginia, contained
concentrations up to 42 000 µg/kg. Up to 39 000 µg/kg wet weight were
found in the Detroit River (Fallon & Horvath, 1985).
The concentrations of individual PAH in sediments from the Trenton
Channel of the Detroit River, a waterway in a highly industrialized
area, connecting Lake St Clair with Lake Erie. varied from not
detected (< 4 µg/kg) to 22 000 µg/kg in different locations.
Sediments from the southwest shore of Grosse Ile had low levels of
contamination, while those in the vicinity of Monguagon Creek had high
levels (Furlong et al., 1988).
5.1.3.2 Lake sediment
The concentrations of individual PAH found in lake sediments in
1984-91 (Table 43) ranged from 1 to about 30 000 µg/kg dry weight. The
total PAH concentrations in surface sediments from Lake Michigan, USA,
were 200-6200 µg/kg dry weight (Helfrich & Armstrong, 1986).
5.1.3.3 Marine sediment
The concentrations of individual PAH in marine sediments in 1985-91
(Table 44) varied widely, with maximum values up to about 4000 µg/kg.
Sediments near power-boat moorings at the coral reef around Green
Island, Australia, were found to contain measurable amounts of several
PAH, strongly suggesting that they originated from fuel spillage or
exhaust emissions (Smith et al., 1987).
The benzo [a]pyrene level was 104-106 times higher in bottom
sediments from the Baltic Sea than in water at the same location. The
bottom sediments also contained more individual PAH than the
corresponding water samples (Veldre & Itra, 1991).
Maximum levels of 460 µg/kg benzo [a]pyrene and 400 µg/kg
benzo [e]pyrene were determined in northern North Sea sediments in
the vicinity of oil fields. The hydrocarbon concentrations were above
the background levels only in water and sediments within a 2-km radius
of platforms, where diesel-coated drill cuttings were dumped. The
contribution of five- and six-ring compounds to the total PAH in
sediments was unexpectedly high in samples unlikely to be contaminated
by oil. Their source was probably windborne combustion products
(Massie et al., 1985).
Table 43. Polycyclic aromatic hydrocarbon concentrations (µg/kg)
in lake sediments
Compound [1] [2] [3] [4]
Anthracene 160 41-620
Benz[a]anthracene ND 150-1700 41
Benzo[a]pyrene 180-2000 45
Benzo[b]fluoranthene 200
Benzo[e]pyrene 80 140-1500 75
Benzo[ghi]fluoranthene 75 18-270
Benzo[ghi]perylene 21-1600 107
Benzo[k]fluoranthene 126
Chrysene 250 124
Coronene 1
Dibenz[a,h]anthracene 70
Fluoranthene 66-248 390 330-3900 103
Fluorene 5.9
Indeno[1,2,3-cd]pyrene 100 25-1500 279
Naphthalene ND
Perylene 50 47-540
Phenanthrene 70-180 100 300-6600 81
Pyrene 110-122 340 210-3500 60
Triphenylene 25
ND, not detected;
[1] Lake Padderudvann, Norway; 1981-82; reference weight not given
(Giessing et al., 1984);
[2] Lake Geneva, Switzerland (Dreier et al., 1985);
[3] Cayuga Lake, USA, 1978; concentrations are given as ng/g
deepwater (Heit, 1985);
[4] Lake Superior, USA (Hamburg Environment Office, 1993)
Analysed by high-performance liquid chromatography or gas
chromatography; concentration in micrograms per kilogram dry weight
Table 44. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in sea sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene ND-6 NR
Acenaphthylene ND-2000 0.6-4.3 NR
Anthracene 3-800 0.3-2.1 6-42 5-313 < 0.06-1.0
Anthanthrene 29-74 NR
Benz[a]anthracene 5-39 1-900 0.8-19 9-150 15-250 < 0.01-6.0
Benzo[a]fluoranthene 2-41 NR
Benzo[a]pyrene 16-25 6-2200 0.4-13 7-160 1100 14-265 0.2-460 < 0.004-4.3
Benzo[b]fluoranthene 13-26 ND-3800 1300 51-490
Benzo[b]fluorene 2-38 NR
Benzo[e]pyrene 5.8-18 0.6-15 9-125 21-345 0.4-396 < 0.1-0.6
Benzo[ghi]perylene ND-400 12-225 700 < 10-623 < 0.01-2.6
Benzo[k]fluoranthene 4.0-9.8 ND-3400 600 10-180 < 0.001-2.5
Chrysene 49 1.0-12 8-165 21-398 < 0.04-0.8
Coronene 11-36 NR
Dibenzo[a,e]pyrene 7-79 NR
Dibenz[a,h]anthracene 2-7 ND-400 0.5-4.2 4-74 NR
Fluoranthene ND/159 4-2000 0.4-31 12-230 2300 36-1913 < 0.1-7.2
Fluorene ND-100 0.5-3.1 1-12 NR
Indeno[1,2,3-cd]pyrene 8-200 17-510
Naphthalene ND-100 0.7-8.6 1-2b 18-1074
Perylene 1-2200 5-105 24-178
Phenanthrene 1-1500 0.8-29 23-93 11-971 < 0.06-4.2
Pyrene 8-160 5-1600 1.6-40 10-145 30-1697 < 0.1-15
Triphenylene 2-600 NR
ND, not detected /, single measurements; NR, not reported;
[1] Baltic Sea, Estonia, reference weight not given (Veldre & Itra, 1991);
[2] Mediterranean Sea, France (Milano et al., 1985);
[3] Adriatic Sea, Italy, 1983 (Marcomini et al., 1986);
[4] Ligurian Sea, Italy (Desideri et al., 1988);
[5] Ketelmeer, Netherlands, 1987 (Netherlands' Delegation, 1991);
[6] North Sea, Netherlands, within 70 km from the coast; 1987-88 (Compaan & Laane, 1992);
[7] North Sea, United Kingdom, 1980 (Massie et al., 1985);
[8] Great Barrier Reef, Australia, 1983 (Smith et al., 1987)
Analysed by high-performance liquid chromatography or gas chromatography
The following background concentrations have been reported in North
Sea sediments: < 0.01-20 µg/kg dry weight benzo [a]pyrene, < 30
µg/kg fluoranthene, < 6 µg/kg benzo (b)fluoranthene plus
benzo (k)fluoranthene, < 5 µg/kg benzo [ghi]-perylene, and < 3
µg/kg indeno[1,2,3- cd]pyrene (Compaan & Laane, 1992).
5.1.3.4 Estuarine sediments
The concentrations of individual PAH in estuarine sediments in 1981-92
(Table 45) varied widely, with maximum values in the high microgram
per gram range. Measurements in sediments from the Continental Shelf
of the Atlantic Ocean and the Gironde Estuary, France, showed
relatively little contamination with PAH when compared with sediments
from more polluted European estuaries (Garrigues et al., 1987). The
levels of PAH in estuarine sediments in the United Kingdom were 10-500
µg/kg. Higher amounts of fluoranthene (1000-1900 µg/kg) and pyrene
(790 µg/kg) were reported in estuaries of the River Mersey and the
River Tamar (Readman et al., 1986).The total PAH concentrations in
sediments from the Penobscot Bay region of the Gulf of Maine, USA,
ranged from 290 to 8800 µg/kg, with a distinct gradient that decreased
seawards. The PAH composition was uniform throughout Penobscot Bay.
Particulates of combustion products transported in the atmosphere were
suggested to be a major source of PAH contamination. The levels in
Penobscot Bay sediments were significantly higher than expected for an
area previously considered to be uncontaminated and fell within the
range found in industrialized regions throughout the world (Johnson et
al., 1985).
The Saguenay Fjord is the major tributary that empties into the St
Lawrence River estuary, and the area is highly industrialized. The PAH
concentrations were maximal near the aluminium smelting plants that
dominate the industrial sector and which were considered to be the
major source of PAH, and the levels decreased with distance from this
industrial zone. The concentrations of benzo [a]pyrene,
benzo [e]pyrene, fluoranthene, benzo [b]fluoranthene,
benzo [j]-fluoranthene, benzo [k]fluoranthene, chrysene and
triphenylene, pyrene, indeno[1,2,3- cd]pyrene, benz [a]anthracene,
dibenz [a,h]anthracene, perylene, benzo [ghi]perylene, and
dibenzo [a,e]pyrene in sediments from the Saguenay Fjord ranged from
2000 to 3800 µg/kg (dry or wet weight basis not given) (Martel et al.,
1986).
5.1.3.5 Harbour sediment
The levels of individual PAH found in harbour sediments (Table 46)
were higher than those in rivers, lakes, or oceans, concentrations
< 650 µg/g being reported.
Table 45. Polycyclic aromatic hydrocarbon concentrations (µg/kg) found in estuarine sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene NR NR 210-670 310
Acenaphthylene NR NR <10-160
Anthracene 0.1-18 10-50 30-210 11-93 ND-49 60-860 610
Benz[a]anthracene 10-790 0.2-68 30-160 30-650 23-189 14-540 70-3200 5-140 2000
Benzo[a]fluoranthene NR NR 2-150
Benzo[a]pyrene 10-560 <0.1-52 30-210 30-760 33-313 10-540 160/7200 4-150 2300 60-6800 20-60
Benzo[b]fluoranthene 0.2-79 100-500 53-346 17-1000
Benzo[e]pyrene 10-620 103 40-180 30-550 56-323 120-8200 1-150 2500
Benzo[ghi]perylene 1-72 120-490 70-410 66-403 23-641 <70-4200 3-96 1300
Benzo[k]fluoranthene <0.1-24 20-100 33-189 14-696
Chrysene 20-1210 0.2-46 30-180 37-263 9-578 2900
Cyclopenta[cd]pyrene NR NR 300/830
Dibenz[a,h]anthracene 0.5-12 NR 8-50 2-120 550-4900 470
Fluoranthene 30-1920 1-100 50-180 80-1880 85-506 156-3700 60-7200 14-410 3900
Fluorene 15 40-120 NR 15-1500 390
Indeno[1,2,3-cd]pyrene 20-630 61 60-240 30-420 50-343 9-228 <130-9000 1800
1-Methylphenanthrene NR NR 240
Naphthalene 43 NR NR 80-2200 400
Perylene 2-52 NR NR 270/880 650 60-4200 50-60
Phenanthrene 30-1470 0.5-74 40-130 60-790 119-413 17-252 60-8700 5-300 2400
Pyrene 20-1980 0.5-102 50-220 60-1510 93-425 16-539 50-5400 4-380 4800
ND, not detected; /, single measurements; NR, not reported;
[1] Estuarine sediment of the River Elbe, Germany (Japenga at al., 1987);
[2] Continental Shelf and Gironde estuary, France (Garrigues et al., 1987);
[3] Wadden Sea, Netherlands, 1988 (Compaan & Laane, 1992);
[4] Mersey, Dee and Tamar estuaries, United Kingdom, 1984 (Readman at al., 1986);
Table 45 (continued)
[5] Humber Estuary/the Wash, United Kingdom, 1990 (Compaan & Laane, 1992);
[6] Gulf of Maine, Penobscot Bay, USA, 1982 (Johnson et al., 1985);
[7] Great Lake tributaries, USA, 1984 (Fabacher at al., 1991);
[8] Chesapeake Bay; USA, 1984-86 (Huggett et al., 1988);
[9] Puget Sound, USA (Varanasi at al., 1992);
[10] Yarra River estuary, Australia, 1976; analytical method: thin-layer chromatography with fluorescence detector (Bagg at al., 1981);
[11] Mallacoota Inlet, Australia, 1976; analytical method: thin-layer chromatography with fluorescence detector (Bagg at al., 1981)
Analysed by high-performance liquid chromatography or gas chromatography and concentration in micrograms per kilogram dry weight, unless
otherwise stated
Table 46. Polycyclic aromatic hydrocarbon concentrations (µg/kg) found in harbour sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene <260-2509 50 3800
Acenaphthlene <240-2700
Anthracene <30-27 200 1800/1700 ND-507 110-17 000 120 10 900
Benz[a]anthracene <50-1991 3400/3400 310-20 000 240 8800/414 000
Benzo[a]pyrene 600-1500 400 <30-16 486 1800/2100 <70-94 984 300-19 000 340 8900/109 000
Benzo[b]fluoranthene 450 <35-17 182 ND-4103 410-15 000
Benzo[e]pyrene 930/930 120-11 000
Benzo[ghi]perylene 300 <35-1079 210-12 000
Benzo[k]fluoranthene 200 <35-1430 150-22 000
Chrysene <30-13 900 3900/3800 580-21 000
Coronene 130
Fluoranthene 2000-3600 850 <70-21 566 900/5800 <5-84 514 640 34 200/60 700
Fluorene <60-24 530 370 810-65 000 100 7000
Indeno[1,2,3-cd]pyrene 300 <50-372 180-14 000 160 157 000/715 000
1-Methylphenanthrene 2100/2300
Naphthalene <310-1564 1300/2000 <10-43 628 400 198 000
Perylene 1100/1200
Phenanthrene <50-5001 4200/4000 45-63 683 510 26 000/655 000
Pyrene <70-5179 6300/6400 196-66 831 610-40 000 740 22 800/413 000
ND, not detected; /, single measurements;
[1] Rotterdam, Netherlands (Japenga et al., 1987);
[2] Rotterdam, Netherlands, 1990 (Netherlands' Delegation, 1991);
[3] Hampton Roads, USA, 1982 (Alden & Butt, 1987);
[4] New York Bight, USA, 1979; reference weight not given (Boehm & Fiest, 1983);
[5] Boston, USA (Shiaris & Jambard-Sweet, 1986);
[6] Black Rock, USA (Rogerson, 1988);
[7] Various harbours of the Rhine, Germany, 1990 (Hamburg Environment Office, 1993);
[8] Vancouver Harbour, Canada (Environment Canada, 1994);
[9] Various harbours near steel mills, Canada (Environment Canada, 1994)
Analysed by high-performance liquid chromatography or gas chromatography and concentration in micrograms per kilogram dry weight, unless
otherwise stated
5.1.3.6 Time trends of PAH in sediment
The PAH levels in sediments taken at various depths indicate changes
and trends in the sources of PAH, e.g. from coal combustion to oil and
gas heating.
Measurements in sediments from Plöner Lake, Germany, showed that the
concentration of PAH in samples from the northern part of the lake,
which is in a populated region situated near a railway, had increased
fivefold since 1920, whereas those in the southern part had remained
constant. The increase in the northern part was attributed to an
increase in the number of PAH emitters. As most of the PAH in the
sediment originated from coal combustion, the concentrations decreased
when coal-fired railway engines were replaced in this area. The
benzo [a]pyrene levels ranged from 240 to 2400 µg/kg dry weight
(Grimmer & Böhnke, 1975). These findings are consistent with the
results of time-dependent analyses of sediments from Lake Constance
(Müller et al., 1977).
A general trend in decreasing PAH concentrations from north to south
was found in bottom sediments from the main stem of Chesapeake Bay,
USA, thought to be due to the higher human population density in the
northern region. Most of the compounds appeared to be derived from the
combustion or high-temperature pyrolysis of carbonaceous fuels rather
than from crude or refined oils. The levels of PAH remained relatively
constant over the period 1979-86 at the locations examined. Naturally
occurring PAH usually comprised less than 20% of the total; the
finding of higher proportions may reflect riverine transport of older
sediments to the area or scouring and removal of recently deposited
sediments. The benzo [a]pyrene concentrations were 12-150 µg/kg dry
weight (Huggett et al., 1988). Similar results were reported for
sediments from Buzzard's Bay, USA (Hites et al., 1977).
In a study of PAH in sediment samples from the lagoon of Venice,
Italy, a historical reconstruction of the PAH depositions in a dated
drilling core made it possible to distinguish between natural and
anthropogenic combustion and between different PAH sources, including
direct petroleum spills and sedimentary diagenesis. The predominance
of unsubstituted homologues and the relative abundance of some
individual components suggested combustion as the predominant source.
The lowest values determined in the deepest strata were assumed to be
background concentrations resulting from pre-industrial pyrolytic
sources, such as forest fires and wood burning. The benzo [a]pyrene
levels were 2.2-17 µg/kg dry weight (Pavoni et al., 1987).
5.1.4 Soil
A rough distinction can be made between local sources of pollution
(point sources) and diffuse sources. Point sources can obviously give
rise to significant local contamination of soil, whereas diffuse
sources usually affect more widespread areas, though to a lesser
extent. The main sources of PAH in soil are:
- atmospheric deposition after local emission, long-range
transport, and pollution from combustion gases emitted by
industry, power plants, domestic heating, and automotive exhausts
(Hembrock-Heger & König, 1990; König et al., 1991) and from
natural combustion like forest fires (Hites et al., 1980);
- deposition from sewage (sewage sludge and irrigation water) and
particulate waste products (compost) (Hembrock-Heger & König,
1990; König et al., 1991); and
- carbonization of plant material (Grimmer et al., 1972).
The extent of soil pollution by PAH also depends on factors such as
the cultivation and use of the soil, its porosity, its lipophilic
surface cover, and its content of humic substances (Windsor & Hites,
1978). There is a correlation between the organic content of a soil
and the PAH concentration: humus contains more PAH than a soil with
little humic content, such as sand (Grimmer et al., 1972; Matzner et
al., 1981; Grimmer, 1993).
This section addresses PAH in soil resulting mainly from industrial
sources, automobile exhaust, and other diffuse sources and gives
background values. Attribution of a study to a particular section was
difficult, as the sources of PAH emissions are often mixed.
5.1.4.1 Background values
Table 47 gives background levels of PAH in soil in rural areas. In
non-polluted areas, PAH concentrations were usually in the range 5-50
µg/kg.
5.1.4.2 Industrial sources
The PAH levels in soil resulting mainly from industrial sources are
given in Table 48.
The PAH levels were determined in soil near one American plant where
animal by-products and brewer's yeast had been processed since 1957.
The operation had subsequently expanded to include the handling of
solvents, flue dust, chips, acids, cyanides, and a wide variety of
industrial waste. Extremely high PAH concentrations were found in the
soil (Aldis et al., 1983).
PAH were detected in the soil at the sites of former coking plants in
Canada (Environment Canada, 1994). For example in Lasalle, Quebec, the
benzo [a]-pyrene levels in 1985 ranged from none detected to 1300
µg/g dry weight. The facility closed in 1976, and by 1991 the
benzo [a]pyrene concentration was below 10 000 µg/kg. In Pincher
Creek, Alberta, high levels of alkylated PAH were measured after a
refinery was dismantled. Maximum concentrations of 260 µg/g dry weight
each of fluoranthene and pyrene were measured; benzo [a]pyrene was
not detected.
Table 47. Polycyclic aromatic hydrocarbon concentrations
(µg/kg dry weight) in soil of background and rural areas
Compound [1] [2] [3] [4]
Acenaphthene 1.7 < 1-21
Acenaphthylene ND/3.0
Anthracene 1.2/4.2
Benzo[a]pyrene 15 6-12 13/22 ND-4.0
Benzo[b]fluoranthene 14/25
Benzo[ghi]perylene 49/28 ND-3.3
Benzo[k]fluoranthene 0.2-3.3
Fluoranthene 22 8-28 35/73 ND-28
Fluorene ND < 1-10
Indeno[1,2,3-cd]pyrene 0.5-4.0
Naphthalene 46 13-60 3.8/11
Phenanthrene 30 17-21 18/39 ND-76
Pyrene 20 9-25 29/42
ND, not detected; /, single measurements;
[1] Norway (depth, 0-10 cm), reference weight not given (Vogt at
al., 1987);
[2] Norway (Aamot et al., 1987);
[3] Wales, United Kingdom (depth, 5 cm) (Jones et al., 1987);
[4] Green Mountain (depth, 0-5 cm), USA (Sullivan & Mix, 1985)
Analysed by high-performance liquid chromatography or gas
chromatography
PAH profiles were found to depend on the depth of soil from which the
samples were taken. A comparison of soil samples from an area of clean
air and from an industrialized area showed that the concentrations of
PAH with lower boiling-points (fluoranthene, chrysene, and pyrene)
decreased with depth, whereas those of PAH with higher boiling-points
(indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene) were relatively greater. The
opposite would have been expected on the basis of the solubility of
these PAH (Jacob et al., 1993b).
Table 48. Polycyclic aromatic hydrocarbon concentrations
(µg/kg dry weight) in soil near industrial emissions
Compound [1] [2] [3] [4]
Acenaphthene 54 5 090 000
Anthracene 144 000 1 600 70
Benz[a]anthracene 79 000 200 000
Benzo[a]pyrene 38 000 321 100
Benzo[b]fluoranthene 200
Benzo[e]pyrene 35 000
Benzo[ghi]perylene 100
Benzo[k]fluoranthene 130 000 100
Chrysene 1 210 000
Fluoranthene 340 000 573 234 000 200
Fluorene 80 8 600 000
Indeno[1,2,3-cd]pyrene 100
Naphthalene 48 5 200 2.4
Perylene 12 000
Phenanthrene 506 000 353 20 000 000 40
Pyrene 208 000 459 16 000 000 100
[1] Near coal gasification plant, Netherlands, concentrations in
µg/kg wet weight (de Leeuw et al., 1986);
[2] Norway, reference weight not given (Vogt et al., 1987);
[3] Near processing plant, USA, 1982; maximum (Aldis et al.,
1983); values, analytical method, and reference weight not
given;
[4] Area of an abandoned coal gasification plant, USA; reference
weight not given (Dong & Greenberg, 1988)
Analysed by high-performance liquid chromatography or gas
chromatography
5.1.4.3 Diffuse sources
(a) Motor vehicle and aircraft exhaust
The concentrations of individual PAH in soil resulting mainly from
motor vehicle exhaust (Table 49) usually range between 1 and 2000
µg/kg. The PAH content of soil often decreased with increasing depth
(Matzner et al., 1981; Wang & Meresz, 1982; Butler et al., 1984). Near
a motorway in the Midlands, United Kingdom, PAH were determined at
depths of 0-4 cm and 4-8 cm. Extremely high concentrations were found
in the surface layer, but soil at a depth of 4-8 cm was two times less
contaminated (Butler et al., 1984). The pollution may have been a
result of airborne transport or of microbial or photochemical
degradation (Hembrock-Heger & König, 1990). Comparably high levels of
PAH were found at Reykjavik Airport, Iceland (Grimmer et al., 1972;
see Table 49).
Table 49. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry
weight) in soil of areas predominantly polluted by vehicle exhaust
Compound [1] [2] [3] [4] [5]
Acenaphthylene 71
Anthracene 0.2 13 11
Anthanthrene 0.4 149
Benz[a]anthracene 2.3 430 169-3297 13
Benzo[a]pyrene 3.2 785 165-3196 38 24
Benzo[b]fluoranthene 41
Benzo[e]pyrene 4.5 870 159-2293 29
Benzo[ghi]perylene 7.1 1450 168 46
Benzo[k]fluoranthene 78
Chrysene 4.1 436 251-2703 39
Coronene 1.8 410 40-322 37
Dibenz[a,h]anthracene 1.1 351 2
Fluoranthene 6.5 1290 200-3703 91 37
Fluorene 5
Indeno[1,2,3-cd]pyrene 36
Naphthalene 3
Perylene 0.6 157 6
Phenanthrene 17 1735 92 45
Pyrene 3.5 1610 145-4515 72 61
[1] Iceland (depth, 20 cm; reference weight not given) (Grimmer et
al., 1972);
[2] Reykjavik Airport, Iceland (surface soil; reference weight not
given) (Grimmer et al., 1972);
[3] United Kingdom, surface soil near motorway; analytical method,
adsorbance measurement, reference weight not given) (Butler et al.,
1984);
[4] United Kingdom (urban soil; depth, 5 cm) (Jones et al., 1987);
[5] Brisbane, Australia (Pathirana et al., 1994)
Analysed by high-performance liquid chromatography or gas chromatography
(b) Other diffuse sources
Table 50 gives the levels of PAH from unpecified sources in soil.
Benzo [a]pyrene levels of 800 µg/kg were found in humus, 100-800
µg/kg in garden soil, 35 µg/kg in forest soil, and 0.8-10 µg/kg in
sand (Fritz, 1971).
Table 50. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry weight) in soil from areas polluted by various diffuse
sources
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthylene NR NR 3.8
Anthracene NR NR ND-1.4 22-70
Anthanthrene 27 0.50 ND 10-38
Benz[a]anthracene 80 0.60 ND 47-101
Benzo[a]pyrene 273 10/6.2 24 0.8/357 116 1.50 ND-1.4 157 54-108
Benzo[b]fluoranthene 49-97
Benzo[e]pyrene 23 20/22 50 143 3.10 ND-5.0 47-116
Benzo[ghi]perylene 106 15/33 32 0.9-339 98 3.0 ND 64-147
Benzo[k]fluoranthene 31-62
Chrysene NR NR ND-2.1 50-128
Coronene 49 0.70 ND-1.7 32-66
Dibenz[a,h]anthracene 266 8.4/22 44 44 0.60 ND-1.4 11-29
Fluoranthene 2.5-444 254 2.1 ND-2.1 83 73-170 0.3-75
Fluorene NR NR 14
Indeno[1,2,3-cd]pyrene 30 6.4/7.9 21.4 1.2-545 127 3.3 32-80
Naphthalene NR NR 58
Perylene 3537 4.0/8.5 5.0 NR NR ND 19-71
Phenanthrene NR NIR ND-18 78 31-106
Pyrene 150 0.80 ND-0.5 90 80-183 0.1-64
ND, not detected; /, single measurements; NR, not reported;
[1] Germany, birch tree peat (Ellwardt, 1976);
[2] Germany, black and white peat (Ellwardt, 1976);
[3] Germany, sandy loam (Ellwardt, 1976);
[4] Soiling mountain, Germany; depth, 0-15 cm; analytical method, high-performance thin-layer chromatography; reference
weight not given (Matzner et al., 1981);
[5] Germany, forest, brown soil, surface layer (Bachmann et al., 1994);
Table 50 (continued)
[6) Germany, forest, brown soil; depth, 0-2 cm (Bachmann et al., 1994);
[7] Iceland; depth, 3-30 cm; reference weight not given (Grimmer et al., 1972);
[8] Norway, bog soil; depth, 0-10 cm; reference weight not given (Vogt et al., 1987);
[9] Toronto, Canada, virgin and cultivated soil; reference weight not given (Wang & Meresz 1982);
[10] Nova Scotia, Canada (Windsor & Hites, 1978)
Analysed by high-performance liquid chromatography or gas chromatography
The PAH concentrations of cultivated soil were slightly higher than
those in virgin soil. For example, the benzo [a]pyrene concentrations
were 65-87 µg/kg in cultivated soil and 54 µg/kg in virgin soil (Wang
& Meresz, 1982). The PAH levels in cultivated soils from German
gardens at a maximum depth of 25 cm decreased from industrial areas
(fluoranthene, 590-2500 µg/kg; benzo [a]pyrene, 220-1400 µg/kg) to
rural areas (fluoranthene, 100-390 µg/kg; benzo [a]pyrene, 30-150
µg/kg) and with soil depth (benzo [a]pyrene concentration, 280-3000
µg/kg at 0-30 cm, 60-4600 µg/kg at 30-60 cm, and 10-7900 µg/kg at
60-100 cm). High PAH concentrations were found at a depth of 100 cm in
soil from an old industrial area and from an area filled with
contaminated soil. In compost soil, benzo [a]pyrene was present at a
concentration of 0.10-2.5 mg/kg in 1986 and 0.02-1.3 mg/kg in 1987
(Crössmann & Wüstemann, 1992).
Fluoranthene and pyrene were measured in soil samples, from a wooded
area in Maine, a marshy area of South Carolina, a grassy, uncultivated
meadow in Nebraska, a mossy area with pine needles in Wyoming, and a
sandy desert area in Nevada, USA, and in dark brown, red clay, and
light brown loam from Samoa. The highest levels of individual PAH (up
to 80 µg/kg) were found in the soil from the wooded area in Maine. The
levels in the marshy and grassy soils of South Carolina and Nebraska
were 8.4-26 µg/kg. The other soils sampled contained fluoranthene and
pyrene at levels < 1 µg/kg (Hites et al., 1980).
In Iceland, the concentrations of individual PAH in lava soil at
depths of 3 and 25 cm were near the limit of detection. Similar levels
were found in vegetable soil at depths of 10 and 30 cm, but the
concentrations at 10 cm were twice as high as those at 30 cm (Grimmer
et al., 1972).
Higher levels of PAH were found in the humus layer of spruce and beech
forest ecosystems than in the mineral soil, but the spruce stand
contained and stored more PAH than the beech stand (Matzner et al.,
1981). Forest soils in Germany contain many PAH in large amounts;
Table 48 shows the PAH concentrations in one forest brown soil. The
first humic layer of the soil had the highest PAH concentration, and
the level decreased with depth to below the limit of detection in
layers below 10 cm (Bachmann et al., 1994).
The concentrations of PAH were no higher in soil that had been treated
with sewage sludge than in untreated soil, indicating that sewage
sludge is not a major source of PAH (Hembrock-Heger & König, 1990;
König et al., 1991).
5.1.4.4 Time trends of PAH in soil
Soil samples collected from Rothamsted Experimental Station in
southeast England over a period of about 140 years (1846-1980) were
analysed for PAH (Jones et al., 1987). All of the soils were collected
from the plough layer (0-3 cm) of an experimental plot for which
atmospheric deposition was the only source of PAH. The total PAH
burden of the plough layer had increased by approximately fivefold
since 1846. The concentrations of most of the individual PAH
(anthracene, anthanthrene, fluorene, benzo [a]pyrene,
benzo [e]pyrene, fluoranthene, benzo [b]fluoranthene,
benzo [k]fluoranthene, chrysene, pyrene, indeno[1,2,3- cd]pyrene,
phenanthrene, and benz [a]-anthracene) had increased by about one
order of magnitude. For example, the benzo [a]pyrene level was 18
µg/kg in 1846 and 130 µg/kg in 1980, and the anthracene level was 3.6
µg/kg in 1846 and 13 µg/kg in 1980. The levels of coronene,
acenaphthylene, acenaphthene, perylene, and benzo [ghi]perylene
remained approximately the same, whereas the naphthalene content
decreased from 39 µg/kg in 1846 to 23 µg/kg in 1980.
5.1.5 Food
In the past, benzo [a]pyrene was the most common PAH determined in
foods and was used as an indicator of the presence of PAH (Tilgner,
1968). The earliest measurements of PAH, in particular of
benzo [a]pyrene, date to 1954; these were reviewed by Lo & Sandi
(1978) and by Howard & Fazio (1980). The levels of individual PAH in
foods in more recent studies are summarized in Tables 51-56.
5.1.5.1 Meat and meat products
The concentrations of individual PAH found in meat are shown in Table
51.
In a comparison of home and commercially smoked meats in Iceland, very
little benzo [a]pyrene was detected in smoked sausage and mutton, but
considerable amounts of benzo [a]pyrene and other PAH were found in
home-smoked mutton and lamb, independently of whether they were
covered with cellophane or muslin. About 60-75% of the total
benzo [a]pyrene was detected in the superficial (outer) layers of the
meat (Thorsteinsson, 1969). These findings are in agreement with those
of Rhee & Bratzler (1970) for smoked bologna and bacon and with those
of Tilgner (1958) and Gorelova et al. (1960).
The amount of PAH formed during roasting, baking, and frying depends
markedly on the conditions (Lijinsky & Shubik, 1964). In an
investigation of the effect of the method of cooking meat, including
broiling (grilling) on electric or gas heat, charcoal broiling, and
broiling over charcoal in a no-drip pan, it was shown that the
formation of PAH can be minimized by avoiding contact of the food with
flames, cooking meat at lower temperatures for a longer time, and
using meat with minimal fat (Lijinsky & Ross, 1967). The most likely
source of PAH is melted fat that drips onto the heat and is pyrolysed
(Lijinsky & Shubik, 1965). The exact chemical mechanism for the
formation of PAH is unknown.
Table 51. Polycyclic aromatic hydrocarbon concentrations (µg/kg fresh weight) in meat and meat products
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13]
Anthracene 0.9 20-31a ND-2 0.5-133
Anthanthrene 5-8 ND ND-66.5
Benz[a]anthracene 0.5 0.5 0.02-0.64 0.03 Trace-0.33a 0.02-0.03 O.04-0.38 0.04-0.13 0.05 16-37 ND-1 0.2-144
Benzo[a]fluorene 17-28 1-2 ND-174
Benzo[a]pyrene 0.1 0.6 0-02-0.45 0.02 0.05 O.01-0.14 0.01-0.04 0.04-0.26 0.03-0.26 0.05 26-42 ND-1 ND-212
Benzo[b]fluoranthene 0.3 1.0 0.30 0.04 16-24 ND-92.3
Benzo[b]fluorene 10-12 2-7 ND-71.9
Benzo[c]phenathrene 1.4 0.03-0.36 0.06 Trace-0.18 0.03-0.04 0.05-0.21 0.05-0.10
Benzo[e]pyrene 0.03 6-9 ND-2 ND-80.9
Benzo[ghi]perylene 0.2 0.6 0.03-0.31 0.03 3.75 Trace-0.12 0.03-0.04 0.06-0.27 0.05-0.19 0.05 10-17 ND-2 ND-153
Benzo[j]fluoranthene 5-7
Benzo[k]fluoranthene 0.2 0.2 0.05 0.01 8-14 ND-172b
Chrysene 0.6 0.15 0.3-140a
Dibenz[a,h]anthracene 0.01 ND-8.8
Fluoranthene 0.9 1.1 7.8 0.48 57-103 6-9 1.1-376
Indeno[1,2,3-cd]pyrene 0.2 0.7 0.04-0.38 0.03 2.5 Trace-0.11 0.01-0.03 0.04-1.40 0.05/0.1 15-22 ND-5 ND-171
1-Methylphenanthrene 4-5 ND-3 0.5-57.6
Perylene ND-3 ND ND-27.9
Phenanthrene 3.0 22-64 10-16 3.5-618
Pyrene 0.55 38-63 5-7 1.2-452
ND, not detected; /, single measurements;
[1] Poultry and eggs, Netherlands, reference weight not given (de Vos et al., 1990);
[2] Meat and meat products, Netherlands, reference weight not given (de Vos et al., 1990);
[3] Smoked beef, Netherlands, reference weight not given (de Vos et al., 1990);
[4] Unsmoked beef, Netherlands (de Vos et al., 1990);
[5] Bacon, United Kingdom (Crosby et al., 1981);
[6] Smoked meat, United Kingdom (McGill et al., 1982);
[7] Unsmoked meat, United Kingdom (McGill et al., 1982);
Table 51 (continued)
[8] Smoked sausages, United Kingdom (McGill et al., 1982);
[9] Unsmoked sausages, United Kingdom (McGill et al., 1982);
[10] Meat, United Kingdom, reference weight not given (Dennis et al., 1983);
[11] Mesquite wood cooked pattie (70-90 % lean), USA, reference weight not given (Maga, 1986);
[12] Hardwood charcoal cooked pattie (70-90% lean), USA, reference weight not given (Maga, 1986);
[13] Grilled sausages, Sweden, reference weight not given (Larsson et al., 1983)
High-performance liquid chromatography or gas chromatography
a In sum with triphenylene
b In sum with benzo[j]fluoranthene
In one study, the highest concentration of benzo [a]pyrene (130
µg/kg) in cooked meat was found in fatty beef, and the concentration
appeared to be proportional to the fat content (Doremire et al.,
1979). Levels of about 50 µg/kg were detected in a charcoal-grilled
T-bone steak (Lijinsky & Ross, 1967), in heavily smoked ham (Toth &
Blaas, 1972), and in various other cooked meats (Potthast, 1980).
Usually, benzo [a]pyrene levels up to 0.5 µg/kg have been found
(Prinsen & Kennedy, 1977).
In meat, poultry, and fish in Canada, benzo [k]fluoranthene was
detected at concentrations up to 0.30 µg/kg and benzo [a]pyrene up to
1.1 µg/kg (Environment Canada, 1994).
Benzo [a]pyrene was found in some German meat products in 1994 at
concentrations generally < 1 µg/kg . The highest concentration, 9.2
µg/kg, was found in a ham from the Black Forest (State Chemical
Analysis Institute, Freiburg, 1995).
5.1.5.2 Fish and other marine foods
Benzo [a]pyrene was found at levels ranging from none detected to 18
µg/kg in smoked fish. The differences were probably due to factors
such as the type of smoke generator, the temperature of combustion,
and the degree of smoking (Draudt, 1963). The highest concentration of
benzo [a]pyrene (130 µg/kg) in seafood was found in mussels from the
Bay of Naples (Bourcart & Mallet, 1965), and a level of about 60 µg/kg
was detected in smoked eel skin. Most of the fish analysed contained
0.1-1.5 µg/kg (Steinig, 1976). Benzo [a]pyrene was also detected at
levels up to 3.3 µg/kg in 21 samples of smoked fish, oysters, and
mussels of various origins (Prinsen & Kennedy, 1977). The levels of
individual PAH are summarized in Table 52.
5.1.5.3 Dairy products: cheese, butter, cream, milk, and related
products
PAH were detected in considerable amounts in smoked cheese (Prinsen &
Kennedy, 1977; Lintas et al., 1979; McGill et al., 1982; Osborne &
Crosby, 1987a). The benzo [a]pyrene content of a smoked Italian
Provola cheese was 1.3 µg/kg (Lintas et al., 1979). Concentrations of
0.01-5.6 µg/kg fresh weight fluoranthene, benz [a]anthracene,
benzo [c]phenanthrene, benzo [a]pyrene, benzo [ghi]perylene, and
indeno[1,2,3- cd]pyrene were found in a smoked cheese sample and
0.01-0.06 µg/kg in unsmoked cheese from the United Kingdom (McGill et
al., 1982). In other unsmoked cheese samples from the United Kingdom,
the individual PAH levels were between < 0.01 µg/kg for
dibenz [a,h]anthracene and 1.5 µg/kg for pyrene. Similar
concentrations of PAH were found in British butter and cream samples
(Dennis et al., 1991).
Table 52. Polycyclic aromatic hydrocarbon concentrations (µg/kg) found in fish and marine foods
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene
Anthracene 0.9 1.3-64.3 1.4-49.6
Benz[a]anthracene 1.3 ND-11.2 ND-6.3 ND-86 Trace-0.09
Benzo[a]pyrene 1.4 ND-5.5 ND-5.4 0.10 ND-18 Trace-0.35
Benzo[b]fluoranthene 2.0 ND-3.9 ND-3.6 0.35
Benzo[c]phenanthrene ND-15 0.01-0.09
Benzo[e]pyrene ND-2.8 ND-3.0
Benzo[ghi]perylene 0.9 ND-2.8 ND-1.8 4.3 ND-25 Trace-0.39
Benzo[k]fluoranthene 0.7 ND-6.7a ND-5.1a 0.10
Chrysene 2.9 ND-13.0b ND-9.4b
Dibenz[a,h]anthracene
Fluoranthene 1.8 1.4-79.9 1.7-48.4 2.4
Fluorene
Naphthalene
Indeno[1,2,3-cd]pyrene 1.6 ND-7.1 ND-2.4 2.7 ND-37 ND-0.33
Perylene ND-1.2 ND-1.0
Phenanthrene 3.5 5-330 10.4-277
Pyrene 1.3-67.8 2.1-38.4
ND, not detected; NR, not reported;
[1] Fish, Netherlands (de Vos et al., 1990);
[2] Herring, whitefish, mackerel, eel, salmon, salmon trout, various fillets; all smoked;
Sweden (Larsson, 1982);
[3] Fish and fish products: sprats, herring, rainbow trout, caviar, herring paste, salmon
paste; all smoked or canned; Sweden (Larsson, 1982);
[4] Kippers, United Kingdom (Crosby et al., 1981);
[5] Fish (smoked), United Kingdom, concentration in µg/kg wet weight (McGill et al., 1982);
[6] Fish, unsmoked, United Kingdom, concentration in µg/kg wet weight (McGill et al., 1982)
Table 52 (continued)
Compound [8] [9] [10] [11] [12] [13] [14]
Acenaphthene < 2-5.13 2.22-22.3
Anthracene < 2-78.4 ND-5.88 ND-0.6 ND-1.9 < 0.05
Benz[a]anthracene 0.14 1.6-7.5 < 2 0.14-5.31 0.8-3.0 0.8-20.9
Benzo[a]pyrene 0.13 t-4.5 < 2-7.63 ND-5.33 0.4-1.0 0.2-12.2 < 0.004
Benzo[b]fluoranthene 0.13 0.13-5.77 4.5-12.2c 1.2-24.3c
Benzo[e]pyrene 0.12 2.4-6.3 0.7-7.6
Benzo[ghi]perylene 0.12 0.17-30.9 0.4-0.8 0.3-5.7
Benzo[k]fluoranthene 0.04 NR NR < 0.002
Chrysene 0.65 < 2 ND-15.9 3.2-8.8b 3.9-30.8b < 0.03
Dibenz[a,h]anthracene 0.03 0.21-39.3 0.1-0.2d <0.1-0.5d
Fluoranthene 0.1 < 2-123.5 ND-32.7 5.1-17.5 4.5-18.7
Fluorene < 2-18.5 ND-65.7
Napthalene < 2-67.4 2.06-156.1
Indeno[1,2,3-cd]pyrene 0.28-28.6 0.3-0.6 0.2-6.4
Perylene 0.2-2.7 0.1-3.1 < 0.05
Phenanthrene < 2-100.8 5.84-87.2 2.1-4.2 1.9-19.6
Pyrene 0.79 < 2-144.9 ND-68.0 3.1-12.4 2.6-11.2 < 0.03
ND, not detected; NR, not reported;
[8] Fish, United Kingdom (Dennis et al., 1983);
[9] Fish, Nigeria (Emerole et al., 1982);
[10] Fresh fish from the Arabian Gulf: andag, sheim, gato, sheiry, faskar, chaniedah; after an oil spill
(Al-Yakoob et al., 1993);
[11] Fresh fish and shrimps, Kuwait, after Gulf war (Saeed et al., 1995);
[12] Fresh oysters, various origins, concentration in µg/kg wet weight (Speer et al., 1990);
[13] Canned or smoked oysters and mussels, various origins, concentration in µg/kg wet weight (Speer at
al., 1990);
[14] Clam, Australia; analytical method: fluorescence spectrophotometry: concentration in µg/kg wet weight
(Smith et al., 1987)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given,
unless otherwise stated
a In sum with benzo[j]fluoranthene
b In sum with triphenylene
c Benzo[b+k]fluoranthenes
d Dibenz[a,h+a,c]anthracenes
In Finnish butter samples, most of the measured PAH (phenanthrene,
1-methylphenanthrene, fluoranthene, pyrene, benzo [a]fluorene,
benzo [ghi]-fluoranthene, cyclopenta [cd]pyrene, perylene,
anthanthrene, benzo [ghi]pyrene, and indeno[1,2,3- cd]pyrene)
occurred at levels < 0.1 µg/kg. The maximum level was 1.4 µg/kg
fluoranthene (Hopia et al., 1986).
The concentrations of fluoranthene, pyrene, benz [a]anthracene,
chrysene, benzo [b]fluoranthene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [e]pyrene, perylene, benzo [ghi]pyrene,
indeno[1,2,3- cd]pyrene, and dibenz [a,h]anthracene were measured in
milk, milk powder, and other dairy products in Canada (Lawrence &
Weber, 1984), the Netherlands (de Vos et al., 1990), and the United
Kingdom (Dennis et al., 1983, 1991). The concentrations ranged from
< 0.01 µg/kg for benzo [k]fluoranthene and dibenz [a,h]anthracene
to 2.7 µg/kg for pyrene.
Canadian infant formula was found to contain 8.0 µg/kg fluoranthene,
4.8 µg/kg pyrene, 1.7 µg/kg benz [a]anthracene, 0.7 µg/kg
benzo [b]fluoranthene, 1.2 µg/kg benzo [a]pyrene, 0.6 µg/kg
perylene, 0.3 µg/kg anthanthrene, and 1.2 µg/kg
indeno[1,2,3- cd]pyrene (Lawrence & Weber, 1984). Slightly lower
levels were detected in British samples in 1982-83 (Dennis et al.,
1991).
PAH were detected at levels of 0.003-0.03 µg/kg in human milk
(Heeschen, 1985).
5.1.5.4 Vegetables
The levels of PAH found in vegetables in recent studies are listed in
Table 53.
Fluoranthene, but no other PAH, was reported to have been found in
unspecified fruits and vegetables in Canada at levels of none detected
to 1.8 µg/kg (Environment Canada, 1994). Kale was found to contain
high concentrations of fluoranthene (120 µg/kg), pyrene (70 µg/kg),
chrysene (62 µg/kg), and benz [a]anthracene (15 µg/kg), and PAH
concentrations up to 7 µg/kg were determined in other vegetables
(Vaessen et al., 1984). The differences in PAH content have been
attributed to variations in the ratio of surface area:weight, in
location (rural or industrialized), and in growing season. Washing (at
20°C) vegetables contaminated by vehicle exhausts did not reduce the
PAH contamination (Grimmer & Hildebrandt, 1965).
In a comparison of the PAH contents of terrestrial plants grown in
chambers containing 'clean air' and in the open field, the
contamination was shown to be due almost exclusively to airborne PAH,
which were not synthesized by the plants (Grimmer & Düvel, 1970) .
Table 53. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in vegetables
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Anthracene 0.09-0.19 <0.1-0.3
Benz[a]anthracene 15 0.7-4.6 0.05-3.17 0.05-3.2 0.4 0.3
Benzo[a]fluoranthene 0.08-2.6
Benzo[a]pyrene 4.2 0.05-1.4 5.6 0.3-6.2 ND-1.42 0.05-3.0 0.2
Benzo[b]fluoranthene 6.1 0.5-7.3 0.9-3.2 0.2
Benzo[b]fluorene 0.11-2.8
Benzo[c]phenanthrene 9.2 0.05-1.5
Benzo[e]pyrene 7.9 0.07-2.2 0.5-6.7 0.2
Benzo[ghi]perylene 7.7 0.13-2.1 10 0.5-10.8 ND-1.39 3.7-10 0.1
Benzo[k]fluoranthene 3.7 ND-17 0.1
Chrysene 62 2.4-4.0 0.8 0.5
Dibenz[a,h]anthracene 1.0 0.04
Dibenzo[a,h]pyrene 0.7
Dibenzo[a,i]pyrene 0.3
Fluoranthene 117 1.1-28 28 2.8-9.1 9.2-17
Indeno[1,2,3-cd]pyrene 7.9 0.14-0.72 2.4 0.3-8.3 ND-1.92 1.8-4.2
1-Methylphenanthrene 0.10-2.1 0.7-1.6
Perylene 0,05-0.75 <0.1-1.7
Phenanthrene 0.47-12 1.8-7.5
Pyrene 70 0.9-18 3.4-10.4
ND, not detected;
[1] Kale, Netherlands (Vaessen et al., 1984);
[2] Lettuce, Finland, concentration in µg/kg fresh weight (Wickstrom et al., 1986);
[3] Lettuce, Germany, from an industrial area (Ministry of Environment, 1994);
[4] Lettuce, Sweden, concentration in µg/kg fresh weight (Larsson & Sahlberg, 1982);
[5] Lettuce and cabbage, United Kingdom, concentration in µg/kg fresh weight (McGill et al., 1982);
[6] Lettuce, India (Lenin, 1994);
[7] Potatoes, Netherlands (de Vos et al., 1990);
[8] Tomatoes, Netherlands (Vaessen et al., 1984)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight
not given, unless otherwise stated
The benzo [a]pyrene levels in potatoes in eastern Germany were
0.2-400 µg/kg. The highest concentrations were detected in the peel of
potatoes grown in soil containing 400 µg/kg benzo [a]pyrene, 750
µg/kg benzo [e]pyrene, 1000 µg/kg benz [a]anthracene, 600 µg/kg
chrysene, 160 µg/kg dibenz [a,h]anthracene, 1000 µg/kg
benzo [b]fluoranthene, 2300 µg/kg phenanthrene, 1800 µg/kg pyrene,
220 µg/kg benzo [k]fluoranthene, 500 µg/kg indeno[1,2,3- cd]pyrene,
2500 µg/kg fluoranthene, and 120 µg/kg anthracene (Fritz, 1971, 1972,
1983).
High concentrations of PAH were detected in lettuce grown close to a
highway; the levels of individual PAH decreased with distance from the
road. Washing the vegetables reduced their content of
high-molecular-mass PAH but not of phenanthrene (Larsson & Sahlberg,
1982). In another study, the profiles of PAH in lettuce were similar
to those in ambient air, indicating that deposition of airborne
particles was the main source of contamination (Wickström et al.,
1986).
PAH concentrations were determined in fenugreek, spinach beet,
spinach, amaranthus, cabbage, onion, lettuce, radish, tomato, and
wheat grown on soil that had been treated with sewage sludge. The
levels of individual PAH in lettuce leaves (Table 53) were one to two
orders of magnitude lower than those in the sewage sludge and the soil
on which the lettuce was grown (Lenin, 1994).
The PAH levels in carrots and beans grown near a German coking plant
were below 0.5 µg/kg wet weight. The levels of fluoranthene were 1.6-
1.7 µg/kg and those of pyrene 1.0-1.1 µg/kg. Vegetables with large,
rough leaf surfaces, such as spinach and lettuce, had PAH levels that
were 10 times higher, perhaps due to deposition from ambient air
(Crössmann & Wüstemann, 1992).
5.1.5.5 Fruits and confectionery (Table 54)
Higher concentrations of PAH were found in fresh fruit than in canned
fruit or juice, and especially high concentrations of phenanthrene (17
µg/kg) and chrysene (69 µg/kg) were found in nuts (de Vos et al.,
1990). In 1982-83 in the United Kingdom, high PAH levels were found in
samples of puddings, biscuits, and cakes, which were probably derived
from vegetable oil. Similar concentrations of individual PAH were
detected in samples of British chocolate (Dennis et al., 1991).
5.1.5.6 Cereals and dried foods
Wheat, corn, oats, and barley grown in areas near industries contained
higher levels of PAH than crops from more remote areas. Drying with
combustion gases increased the contamination by three- to 10-fold; use
of coke as fuel resulted in much less contamination than oil (Bolling,
1964). Cereals contained benzo [a]pyrene at levels of 0.2-4.1 µg/kg
(Table 55). The highest concentrations, up to 160 µg/kg, were found in
smoked cereals (Tuominen et al., 1988).
Table 54. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in fruits and confectionery
Compound [1] [2] [3] [4] [5] [6] [7]
Anthracene 0.4 0.3
Benz[a]anthracene 0.5 0.11 4.2 0.2 4.2 0.08-2.73
Benzo[a]pyrene 0.1 0.07 0.2 0.3 0.4 0.04-2.20
Benzo[b]fluoranthene 0.1 0.1 0.06 0.4 0.4 3.5 0.03-1.27
Benzo[c]phenanthrene 0.5 12 2.2
Benzo[e]pyrene 0.03 0.08-2.92
Benzo[ghi]fluoranthene 0.9 0.9
Benzo[ghi]perylene 0.1 0.06 0.4 1.1 0.2 0.11-2.55
Benzo[k]fluoranthene 0.1 0.1 0.02 0.1 0.1 0.5 0.04-1.36
Chrysene 0.5 0.23 69 0.5 36 0.09-2.84
Dibenzo[a,h]pyrene 0.01 < 0.01-0.23
Fluoranthene 3.6 1.0 0.93 3.0 1.9 2.3 0.52-3.57
Indeno[1,2,3-cd]pyrene 0.4 0.4 0.2 0.10-3.18
Phenanthrene 7.8 17 2.9 3.2
Pyrene 0.83 0.59-2.37
[1] Fresh fruit, Netherlands (de Vos et al., 19900:
[2] Canned fruit and juices, Netherlands (de Vos et al., 1990);
[3] Fruit and sugar, United Kingdom (Dennis et al., 1983);
[4] Nuts, Netherlands (de Vos et al., 1990);
[5] Biscuits, Netherlands (de Vos et al., 1990);
[6] Sugar and sweets, Netherlands (de Vos et al., 1990);
[7] Puddings, biscuits and cakes, United Kingdom (Dennis et al., 1991)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not
given
Table 55. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in cereals and dried foods
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 1.6 NR NR 0.7 2.3
Anthracene 9.4 NR NR 1.3 19/150
Anthanthrene NR NR
Benz[a]anthracene 0.1-42 11 0.69 0.11-0.21 2.5/3.7 0.6 0.3 <0.1/0.2 6.3/110
Benzo[a]pyrene ND-0.3 5.4 0.40 0.10-0.12 0.5/0.8 0.2 0.3/0.4 0.6/160
Benza[b]fluoranthene 0.1-0.5 0.28 0.07-0.09 0.9 0.2 0.1
Benzo[e]pyrene 0.42 0.06-0.17 0.1/0.7
Benzo[ghi]perylene 0.54 0.13-120
Benzo[k]fluoranthene 0.50 0.1-0.14
Dibenz[a,h]anthracene ND-1.2 0.06 0.01-0.02 3.6
Fluoranthene 0.8-26 130 0.71 0.58-0.69 18/28 1.9 1.4 1.5/13 70/790
Fluorene 5.9 NR NR 2.3/2.7 6.4/87
Indeno[1,2,3-cd]pyrene ND-0.4 1.08 0.24-0.33 1.4 0.2
Perylene 0.1-0.4 0.7 NR NR 94 NR NR 14/2983/1
Pyrene 1.1-48 47 0.10 0.38-0.62 20/21 2.2 3.4 1.6/5.4 60/630
ND, not detected; /, single measurements; NR, not reported;
[1] Barley malt, Canada (Lawrence & Weber, 1984);
[2] Bran, Finland (Tuominen et al., 1988);
[3] Bran, United Kingdom (Dennis et al., 1991);
[4] High bran and granary bread, United Kingdom (Dennis et al., 1991);
[5] Bran, Canada (Lawrence & Weber, 1984);
[6] Corn bran, Canada (Lawrence & Weber, 1984);
[7] Flaked milled corn, Canada (Lawrence & Weber, 1984);
[8] Oats, Finland (Tuominen et al., 1988);
[9] Smoked oats, barley and beans, Finland (Tuominen et al., 1988)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given
Table 55 (contd)
Compound [10] [11] [12] [13] [14] [15] [16] [17]
Acenaphthene 0.6/0.7 NR NR 0.6
Anthracene 0.5 NR NR
Anthanthrene 0.05-0.08 NR NR
Benz[a]anthracene 0.4 ND-0.2 0.14-0.25 <0.1/<O.1 0.3-0.8 0.06-0.15 0.33-1.26 0.1
Benzo[a]pyrene < 0.1 0.17-0.30 0.2/0.4 0.1 0.03-0.05 0.15-0.34
Benzo[b]fluoranthene 0.1/0.2 0.02-0.05 0.1-0.27
Benzo[c]phenanthrene NR NR
Benzo[e]pyrene ND-0.1 0.16-0.29 0.2/0.4 0.06-0.16 0.28-0.81
Benzo[ghi]fluoranthene 0.05 NR NR
Benzo[ghi]perylene 0.20-0.35 0.06-0.08 0.15-0.28
Benzo[k]fluoranthene ND-0.2a 0.02-0.07 0.15-0.31
Chrysene 0.3-0.7b NR NR
Coronene 0.03-0.06 NR NR
Cyclopenta[cd]pyrene 0.07-0.13 NR NR
Dibenz[a,h]anthracene 0.03-0.05 3.0 < 0.01 0.01-0.02
Fluoranthene 2.9 0.9-1.3 0.32-0.57 1.8/3.0 1.5-7.4 0.22-0.60 0.82-6.17 3.8
Fluorene 1.3/1.7 NR NR 2.0
Indeno[1,2,3-cd]pyrene 0.16-0.29 3.0 0.08-0.15 0.30-0.65
1-Methylphenanthrene 0.3
Perylene 0.1 < 0.1/0.1 0.1-0.3 NR NR
Phenanthrene 1.3-1.5 9.9/10 NR NR 14
Pyrene 2.8 1.6-2.3 0.22-0.39 1.6/5.5 2.6-8.5 0.26-1.18 1.41-10.86 2.6
ND, not detected; /, single measurements; NR, not reported;
[10] Whole grain oats, Canada (Lawrence & Weber, 1984);
[11] Whole-grain rye, Sweden, concentration in µg/kg fresh weight (Larsson, 1982);
[12] Wheat grain, United Kingdom (Jones et al., 1989b);
[13] Wheat, Finland (Tuominen et al., 1988);
[14] Wheat, Canada (Lawrence & Weber, 1984);
[15] Breakfast cereal, United Kingdom (Dennis et al., 1991);
[16] Bran-enriched cereals, United Kingdom (Dennis et al., 1991);
[17] Bolted rye flour, Finland (Tuominen et al., 1988)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given, unless
otherwise specified
a Benzofluoranthenes
b In sum with triphenylene
Table 55 (contd)
Compound [18] [19] [20] [21] [22] [23] [24] [25]
Acenaphthene NR NR NR
Anthracene NR NR NR
Anthanthrene NR NR NR
Benz[a]anthracene 0.04-0.19 0.64 0.8 0.10-0.14 0.5 0.1 0.4
Benzo[a]pyrene 0.02-0.09 0.43 0.8 0.05-0.15 0.2 0.3 0.8
Benzo[b]fluoranthene 0.02-0.06 0.25 1.2 0.04-0.06 0.5 0.6 1.0 0.05
Benzo[c]phenanthrene NR NR NR 0.7
Benzo[e]pyrene 0.10-0.23 0.35 0.06-0.12
Benzo[ghi]fluoranthene NR NR NR
Benzo[ghi]perylene 0.06-0.19 0.39 0.5 0.04-0.21 0.5 0.9 0.6
Benzo[k]fluoranthene 0.03-0.08 0.35 0.6 0.04-0.1 0.1 0.3 0.5 0.08
Chrysene NR NR 1.0 NR 2.0 1.3 0.4
Coronene NR NR NR
Cyclopenta[cd]pyrene NR NR NR
Dibenz[a,h]anthracene <0.01-011 0.05 <0.01-0.01
Fluoranthene 0.07-0.40 0.66 2.8 0.23-2.03 3.7 0.6 2.5
Fluorene NR NR NR
Indeno[1,2,3-cd]pyrene 0.06-0.24 0.84 0.6 0.11-0.25 0.3 0.6 0.5
1-Methylphenanthrene
Perylene NR NR NR
Phenanthrene NR NR 3 NR 4.2 3.0 2.1
Pyrene 0.04-0.88 0.67 0.23-0.87
NR, not reported;
[18] White four, United Kingdom (Dennis et al., 1991);
[19] Granary flour, United Kingdom (Dennis et al., 1991);
[20] Bread, Netherlands (de Vos et al., 1990);
[21] White bread, 1982-83, United Kingdom (Dennis et al., 1991);
[22] Noodles, pizza, Netherlands (de Vos et al., 1990);
[23] Potato products, Netherlands (de Vos et al., 1990);
[24] Rice, macaroni, Netherlands (de Vos et al., 1990);
[25] Soups, Netherlands (de Vos et al., 1990)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given
The PAH concentration in rye grown near a highway with high traffic
density decreased slightly 7-25 m away from the road (Larsson, 1982).
5.1.5.7 Beverages
Benzo [a]pyrene was found at 0.8 µg/kg in coffee powder, 0.01
µg/litre in brewed coffee, 9.51 µg/kg in tea leaves, and 0.02 µg/litre
in brewed tea (Lintas et al., 1979). In 40 samples of tea leaves from
India, China, and Morocco, the concentration of benzo [a]pyrene was
generally 2.2-60 µg/kg, although concentrations up to 110 µg/kg were
found in smoked teas (Prinsen & Kennedy, 1978).
In samples of whisky and beer, the concentrations of six of 11 PAH
(benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]-perylene, dibenz [a,h]anthracene, and
indeno[1,2,3- cd]pyrene) were below or slightly above 0.01 µg/kg. The
highest level determined (0.24 µg/kg) was that of pyrene (Dennis et
al., 1991). The PAH content of the water used in the preparation of
whisky and beer was not described.
5.1.5.8 Vegetable and animal fats and oils
The levels of PAH in oil products, butter, and margarine are listed in
Table 56. Vegetable oils are reported to be naturally free of PAH, and
contamination is due to technological processes like smoke drying of
oil seeds or environmental sources such as exhaust gases from traffic.
The PAH content of native olive oils was particularly high (Speer et
al., 1990). The PAH content of coconut, soya bean, maize, and rapeseed
oil was radically reduced during refining, particularly by treatment
with activated charcoal (Larsson et al., 1987). This method is now
widely used (Dennis et al., 1991).
Benzo [a]pyrene was detected in 30 vegetable oils from Italy and
France in 1994, including 17 grape-seed oils and one pumpkin-seed oil.
The average concentration was 59 µg/kg, and the maximum value was 140
µg/kg. Benzo [b]fluoranthene, benzo [k]fluoranthene,
dibenz [a,h]anthracene, and indeno[1,2,3- cd]pyrene were also found
in measurable amounts. The source of these high levels was the smoke
in drying ovens (State Chemical Analysis Institute, Freiburg, 1995).
Lard and dripping were found to contain levels of individual PAH
ranging from < 0.01 µg/kg dibenz [a,h]anthracene) to 6.9 µg/kg
fluoranthene (Dennis et al., 1991). High PAH levels were found in
margarine samples in studies in Finland (Hopia et al., 1986), the
Netherlands (Vaessen et al., 1988), New Zealand (Thomson et al.,
1996), and the United Kingdom (Dennis et al., 1991) (see Table 56).
5.1.6 Plants
PAH with low molecular masses are more readily taken up by vegetation
than those with higher molecular masses (Wang & Meresz, 1982).
Table 56. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in vegetable oils and related products
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthene NR < 0.02-45 NR NR NR NR 0.29 NR NR < 0.1 -11
Anthracene NR < 0.02-460 <0.1-0.1 ND-4.8 ND-8 NR 0.04-0.92 NR NR < 0.2-5.6
Anthanthrene Trace-0.1 NR NR NR NR NR 0.03-0.53 NR NR < 0.1-2.7
Benz[a]anthracene NR NR 0.7-6.1 ND-6.1 ND 0.30-7.46 NR 0.22-3.98 0.28-0.96 < 0.1-5.2
Benzo[a]fluorene NR < 0.02-130 NR NR ND-2 NR 0.07-3.8 NR NR NR
Benzo[a]pyrene Trace-0.3 < 0.02-24 0.5-2.3 ND-4.1 ND 0.29-4.92 0.05-2.2 0.19-6.0 0.17-0.83 < 0.2-5.2
Benzo[b]fluoranthene Trace-0.1 < 0.02-91a NR ND-8.9a ND 0.20-2.39 NR 0.16-3.0 0.09-0.37 < 0.2-9.2
Benzo[b]fluorene NR < 0.02-45 NR NR ND NR 0.03-2.1 NR NR NR
Benzo[e]pyrene NR < 0.02-23 0.7-2.4 ND-3.8 ND 0.26-6.06 0.09-2.1 0.42-6.11 0.36-0.87 NR
Benzo[ghi]fluoranthene NR < 0.02-1.3 NR NR ND NR 0.14-4.9 NR NR NR
Benzo[ghi]perylene NR < 0.02-10 0.5-1.7 ND-4.2 NR 0.06-5.23 0.02-1.4 0.38-5.21 0.17-1.16 < 0.2-10.6
Benzo[k]fluoranthene NR NR NR NR ND 0.24-3.17 NR 0.20-3.40 0.16-0.55 < 0.1-11.4
Chrysene NR NR NR 0.1-8.6b ND 0.39-10.3 NR 0.26-7.36 0.31-0.97 < 0.2-7.5
Coronene NR < 0.02 NR NR NR NR NR NR NR NR
Cyclopenta[cd]pyrene NR < 0.02-1.4 NR NR ND NR 0.10-1.1 NR NR NR
Dibenz[a,h]anthracene 0.7-1.1 < 0.02-1.1c NR ND-0.2c NR <0.01-0.82 NR 0.05-1.02 0.04-0.11 < 0.1-9.2
Fluoranthene 0.2-7.5 < 0.02-460 1.2-4.8 0.2-18.2 3-15 0.21-12.4 0.52-9.0 0.09-4.50 0.44-1.56 < 0.1-1.6
Fluorene NR < 0.02-200 NR NR ND-7 NR 0.08-1.6 NR NR < 0.2-2.1
Indeno[1,2,3-cd]pyrene Trace-0.5 < 0.02-0.85 0.3-1.7 ND-4.3 NR 0.59-6.78 0.03-1.1 0.49-9.14 0.43-1.17 < 0.2-9.7
Naphthalene NR NR NR NR NR NR NR NR NR < 0.2-52
1-Methylphenanthrene NR < 0.02-190 NR NR NR NR 0.08-1.8 NR NR NR
Perylene Trace-0.2 < 0.02-5.9 0.1-0.4 ND-0.9 NR NR 0.02-0.57 NR NR NR
Phenanthrene NR 0.09-1400 0.9-1.6 ND-69.4 4-38 NR 0.29-6.0 NR NR < 0.2-4.6
Pyrene 0.2-1.4 < 0.02-330 1.1-4.2 0.1-13.6 2-14 0.58-17.2 0.59-15 0.29-6.03 0.44-1.88 < 0.1-1.7
Table 56 (continued)
ND, not detected; /, single measurements; NR, not reported;
[1] Corn oil, canola, soya bean oil (Lawrence & Weber, 1984);
[2] Corn oil, coconut oil (crude and deodorized), olive oil, soya bean oil, sunflower oil, sesame oil, flaw oil,
wheatseed oil (Hopia et al., 1986);
[3] Coconut oil (pure) (Sagredos et al., 1988);
[4] Various olive oils, safflower oils, sunflower oils, maize germ oils, sesame oil, linseed oil, wheat germ oil
(all native) (Speer et al., 1990);
[5] Various olive oils (Menichini et al., 1991b);
[6] Various unspecified oils (Dennis et al., 1991);
[7] Four cooking margarines, seven table margarines (Hopia et al., 1986);
[8] Margarine (Dennis et al., 1991);
[9] Low-fat spread (Dennis et al., 1991);
[10] Margarine (Thomson et al., 1996)
Analysed by high-performance liquid chromatography or gas chromatography
a Benzo[b+j+k]fluoranthenes
b In sum with triphenylene
c Dibenz[a,h+a,c]anthracenes
In a study of PAH levels in soil (see section 5.1.4), leaf litter, and
soil fauna (see section 5.1.7) from a roadside in Brisbane, Australia,
vegetation height, soil depth, and distance from the roadside were
found to be important in the distribution of PAH. The concentration of
PAH in leaf litter declined exponentially with distance from the
roadway, few PAH being detectable 30 m away. A decrease in PAH levels
with height was found in the roadside vegetation canopy. In leaf
litter, fluorene, phenanthrene, fluoranthene, pyrene, chrysene,
benzo [k]fluoranthene, and benzo [ghi]perylene were present at
concentrations of about 100 µg/kg wet weight. Naphthalene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene, and
indeno[1,2,3- cd]pyrene were present at about 50 µg/kg wet weight,
whereas anthracene was present at concentrations below 10 µg/kg wet
weight. Perylene and dibenz [a,h]anthracene were not detectable. The
tree Casuarina littorina contained high levels of pyrene and
chrysene (100 µg/kg wet weight each) and benzo [k]fluoranthene (72
µg/kg wet weight); the concentrations of fluoranthene, phenanthrene,
and benzo [ghi]-perylene were about 40 µg/kg wet weight. Perylene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene were not detectable (Pathirana et
al., 1994).
The benzo [a]pyrene levels in spruce sprouts from a rural area of
Germany (Bornhövede, Schleswig-Holstein) decreased from 2.6 µg/kg in
1991 to 1.3 µg/kg in 1993. The concentrations of PAH with low
boiling-points significantly decreased between 1991 and 1993; for
example, that of fluoranthene decreased from 44 µg/kg in 1991 to 11
µg/kg in 1993, perhaps due to a decrease in coal burning. The levels
of phenanthrene, fluoranthene, pyrene, and benzo [b]fluoranthene plus
benzo [j]fluoranthene plus benzo [k]fluoranthene were about 10
µg/kg; those of benzo [ghi]fluoranthene, benzo [c]phenanthrene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, and coronene were
about 2 µg/kg; and those of anthracene, dibenz [a,h]anthracene, and
anthanthrene were < 1 µg/kg. The PAH levels in spruce sprouts from
the Saarland, an industrial area in Germany, were about 10 times
higher than those in the Bornhöveder area, although these levels also
decreased between 1991 and 1993: from 5.9 to 4.1 µg/kg for
benzo [a]pyrene and 97 to 58 µg/kg for fluoranthene. the
concentrations of pyrene were 40-50 µg/kg, those of
benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene were 20 µg/kg, and those of
benzo [ghi]perylene, benzo [c]phenanthrene, benz [a]anthracene,
benzo [e]pyrene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, anthanthrene, and
coronene were < 10 µg/kg (Jacob & Grimmer, 1994, 1995). In 1994, the
PAH levels had decreased further. Overall, a 25% decrease in the PAH
levels in spruce sprouts was seen over the previous 10 years (Jacob &
Grimmer, 1995).
The PAH profiles in spruce sprouts and poplar leaves were reasonably
similar in areas with clean air (Bavarian forests) and in
industrialized areas (Saarland) of Germany, indicating that one
emission source is predominantly responsible for air pollution by PAH.
Hard-coal combustion resulted in a characteristic PAH profile (Jacob
et al., 1993a).
The concentrations of PAH in pine needles from Dübener Heide near
Leipzig (Saxony, Germany) were similar to those from the Bornhöveder
area (Schleswig-Holstein, Germany), with an average benzo [a]pyrene
level of 2.3 µg/kg (Jacob & Grimmer, 1995).
Beech leaves from the Harz mountains in Germany contained fluoranthene
at a level of 5 µg/kg, whereas the concentrations of phenanthrene,
pyrene, benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, anthracene, benz [a]anthracene,
benzo [e]pyrene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, anthanthrene, and
coronene were all < 2 µg/kg. Beech sprouts in an industrial area in
eastern Germany contained 10-15 times higher levels of PAH, with
fluoranthene at about 60 µg/kg, pyrene at about 30 µg/kg,
benzo [b]fluoranthene plus benzo [j]-fluoranthene plus
benzo [k]fluoranthene at about 10 µg/kg, and anthracene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, coronene,
dibenz [a,h]anthracene, and anthanthrene at < 2 µg/kg (Jacob &
Grimmer, 1995).
Comparable results were obtained in poplar leaves: those from the
Saarland analysed in 1989, 1991, and 1993 had 10 times lower
concentrations of PAH than those in Dübener Heide. The concentrations
of phenanthrene, fluoranthene, and pyrene were about 20 µg/kg, those
of benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene were about 10 µg/kg, and those of anthracene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, anthanthrene, and coronene were < 5 µg/kg
(Jacob & Grimmer, 1995).
5.1.7 Animals
5.1.7.1 Aquatic organisms
Aquatic invertebrates are known to adsorb and accumulate PAH from
water (see section 4.1.5). The concentrations of PAH in aquatic
organisms collected from various sites are listed in Tables 57-64. All
of the sampling sites listed in Tables 57-60 were contaminated with
industrial effluents, the major components of the PAH profile being
benzo [b]fluoranthene, benz [a]anthracene, benzo [a]pyrene,
benzo [e]pyrene, fluoranthene, pyrene, and phenanthrene. The average
levels of PAH in aquatic organisms from these sites ranged from 1 to
100 µg/kg; the differences in levels generally corresponded to the
degree of industrial and urban development and shipping movements.
Table 57. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry weight)
in bivalves and gastropods; main source, industrial emissions
Compound [1] [2] [3] [4] [5] [6] [7]
Acenaphthene ND ND 7 2.1/8.8
Anthracens 9 9.0/25
Benz[alanthracene 172 203 3 5-41 25-229
Benzo[alpyrene 12 21 1 8.1 2-8 Trace-28 2.6/2.8
Benzo[b]fluoranthene 23 25 3-30 48-90
Benzo[ejpyrene 17 10 Trace-30 231-356
Benzo[ghilperylene ND 4 5
BenzoUlfluoranthene 1.3
Benzolk]fluoranthene 2.3
Chrysene 209 205
Coronene 4
Dibenzo(a,elpyrene 2
Dibenzo[e,ilpyrane 4
Dbenzo[a,lpyrene Trace
Fluoranthene 18 62 7 43-407 300-4992 26/61
Fluorene 2 1.3/6.3
1-Methylphenanthrene 2.9
Naphthalene 15/3
Perylene 8
Phenanthrene 733 462 9 4.4 115-258 55-2542 66/194
Pyrene 85 131 4 32-204 141-3128 23/40
Triphenylene ND
ND, not detected; /, single measurement;
[1] Whole cooked clam (Mya arenaria); oil-contaminated area (tanker accident), Canada, 1979;
concentration in µg/kg wet weight (Sirota & Uthe, 1981);
[2] Whole cooked mussel (Mytilus edulls); oil-contaminated area (tanker accident), Canada,
1979; concentration in µg/kg wet weight (Sirota & Uthe, 1981);
[3] Whole mussel (Mytilus galloprovincialis); Thermaikos Gulf, Aegean Sea, Greece (agricultural
and industrial area); concentration in µg/kg wet weight (Iosifidou et al., 1982);
[4] Whole scallop (Amusium pleuronectes); Gulf of Thailand, Thailand; reference weight not
given (Hungspreugs et al., 1984);
[5] Whole periwinkle (Littorina littorea); moderately polluted parts of North Sea coast,
Norway, 1978-79 (Knutzen & Borland, 1982);
[6] Whole limpet (Patella vulgata); moderately polluted parts of North Sea coast, Norway,
1978-79 (Knutzen & Sortland, 1982);
[7] Whole snails (Thais haemostoma), Pensacola Bay, USA; creosote contaminated; concentration
in µg/kg wet weight (Rostad & Pereira, 1987)
High-performance liquid chromatography or gas chromatography
Table 58. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry weight) in algae
and water plants; main source, industrial emissions
Compound [1] [2] [3] [4] [5] [6]
Benz[a]anthracene 5 4 31-325 45-431 3-40
Benzo[alpyrene 4 5 Trace-64 Trace-<2 2-20
Benzo[b]fluoranthene 4 5 7-76 6-12 5-31
Benzo[e]pyrene 7 14 Trace-100 Trace-8 8-50 410
Benzo[ghi]perylene 4 79
Fluoranthene 45 32 40-412 15-900 <4-236
Phenanthrene 87 34 31-325 45-431 109-146
Pyrene 36 20 28-286 15-388 <4-224 260
[1] Laminaria saccharins (whole); moderately polluted parts of North Sea coast,
Norway, 1978-79 (Knutzen & Sortland, 1982);
[2] Ceramium rubrum (whole), moderately polluted parts of North Sea coast, Norway,
1978-79 (Knutzen & Sortland, 1982);
[3] Bladder wrack (Fucus vesiculosus, whole), moderately polluted parts of North
Sea coast, Norway, 1978-79 (Knutzen & Sortland, 1982);
[4] Knotted wrack (Ascophyllum nodosum, whole), moderately polluted parts of North
Sea coast, Norway, 1978-79 (Knutzen & Sortland, 1982);
[5] Toothed wrack (Fucus serratus, whole), moderately polluted parts of North Sea
coast, Norway, 1978-79 (Knutzen & Sortland, 1982);
[6] Wakame seaweed, Japan (Obana et al., 1981a)
High-performance liquid chromatography or gas chromatography
Table 59. Polycyclic aromatic hydrocarbon concentrations (µg/kg wet weight) in lobsters; main
source, industrial emissions
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene ND ND
Benz[a]anthracene 684 ND/23 1620-23 400 34-604 762-32 700 17-900
Benzo[a]pyrene 24 0.2/2.6 35-1000 2.0-40 711-1430 27-43
Benzo[b]fluoranthene 24 1 155-2350 6-78 1020-3820 29-835
Benzo[e]pyrene 57 5/8 415-9330 15-165 1550-3600 35-36
Benzo[ghi]perylene ND ND/2 7-493 1.6-31 232-769 10-20
Benzo[k]fluoranthene 7.6 0.3/0.6 43-588 1.6-25 502-955 15-26
Chrysene 445 ND 360-5050 5-79 252-1240 15-24
Fluoranthene 318 ND/0.2 1910-12400 103-545 4220-15 200 68-442
Indeno[1,2,3-cd]pyrene 5 38-855 3-45 486-931 12-40
Phenanthrene 1588 ND Trace-3470 Trace-650
Pyrene 488 ND 730-6710 32-265 2910-13 100 59-333
Triphenylene ND/244 2520-23100 Trace-330
ND, not detected; /, single measurements;
[1] Homarus americanus (digestive gland), oil-contaminated area (tanker accident), Canada, 1979
(Sirota & Uthe, 1981);
[2] Homarus americanus (tail muscle), oil-contaminated area (tanker accident), Canada, 1979
(Sirota & Uthe, 1981);
[3] Homarus americanus (hapatopancreas), Sydney Harbour, near coking plant, Canada (Sirota
et al., 1983);
[4] Homarus americanus, (tail muscle), Sydney Harbour, near coking plant, Canada (Sirota
et al., 1983);
[5] Homarus americanus, (digestive gland), Sydney Harbour, near coking plant, Canada, 1982-84
(Uthe & Musial, 1986);
[6] Homarus americanus (tail muscle), Sydney Harbour, near coking plant, Canada, 1982-84
(Uthe & Musial, 1986)
High-performance liquid chromatography or gas chromatography
Table 60. Polycyclic aromatic hydrocarbon levels (µg/kg dry weight) in fish and other aquatic species; main
source, industrial emissions
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 39 Trace-0.9 130 < 25
Acenaphthylene 270 0.1-0.2
Anthracene ND 0.1-0.2 460 < 22 1000
Benz[a]anthracene 22 ND-40 ND-< 0.1 0.1-88 1000 < 21 1-2 800 5
Benzo[a]fluorene 02-0.6 500
Benzo[a]pyrene 7 0.07-8.4 ND-< 0.1 0.1-0.5 570 < 20 ND 8
Benzo[b]fluoranthene <O.1a 28
Benzo[b]fluorene O.1-0.2
Benzo[o]phenanthrene Trace
Benzo[e]pyrene 14 ND-< 0.1 0.1-1.6 840 < 25 25
Benzo[ghi]perylene ND-< 0.1 0.2-18 75 < 25 23
Chrysene 61 < 0.1-2.1b 1500 < 22
Dibenz[a,h]anthracene ND-< 0.1c <100 < 25
Fluoranthene 1800 0.1-9.1 1.2-5.6 4800 < 20 13-18 800 48
Fluorene 0.2-2.4 200 < 25 NDc
Indeno[1,2,3-cd]pyrene ND-< 0.1 0.3-3.7 150 < 25
1-Methylphonanthrene 85 < 10
Naphthalene 2.5-11 610 < 25
Perylene 6 ND-< 0. 1 Trace-0.2 75 < 20
Phenanthrene 2700 28-15 313 0.1-2.4 0.7-9.1 1400 < 20 32-50 900 71
Pyrene 1500 ND-10.0 0.7-3.7 2300 < 20 10-8 800 39
Triphenylene 800
Table 60 (continued)
ND, not detected;
[1] Bullhead catfish (Ictalurus nebulosus, whole); Black River, USA, near coking plant; concentration in
µg/kg wet weight (Vassilaros et al., 1982);
[2] Whole fish (unspecified); Hersey River, USA, creosote polluted; concentration in µg/kg wet weight
(Black et al., 1981);
[3] Bream (fillet and liver); River Elbe, Germany, industrial region of city of Hamburg (Speer et al., 1990);
[4] Dabs (Limanda limanda, muscle, North Sea, United Kingdom, near Beatrice oil platform; concentration in
µg/kg wet weight (McGill et al., 1987);
[5] English sole (Parophrys vetulus, stomach contents); Mukilteo, USA, near petroleum storage tanks (Malins
et al., 1985);
[6] English sole (Parophrys vetufus, liver); Mukilteo, USA, near petroleum storage tanks (Malins et al., 1985);
[7] Whole starfish (Asterias rubens), moderately polluted areas of North Sea coast, Norway, 1978-79 (Knutzen
& Sortland, 1982);
[8] Whole holothurians, Toulon, France; urban sewage (Milano et al., 1986);
[9] Whole crumb-of-bread sponge (Hafichondria panicea); moderately polluted areas of North Sea coast, Norway,
1978-79 (Knutzen & Sortland, 1982)
High-performance liquid chromatography or gas chromatography
a Benzo[b+j+k]fluoranthenes
b In sum with triphenylene
c Dibenz[a,h+a,c]anthracenes
Table 61. Polycyclic aromatic hydrocarbon concentratrations (µg/kg dry weight) in bivalves (mussels and
clams); background values
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene NR 24/46
Acenaphthylene NR 34/130
Anthracene 0.7-19 9-15 149-243 36/43
Benz[a]anthracene NR 0.1-0.8 2.9/42 < 1 31/94
Benzo[a]pyrene 4.6-451 3/5 <0.8-2 3.5/8.7 < 1 1.3/26
Benzo[b]fluoranthene 3.0-120 1.5/12 2.5/18
Benzo[c]phenanthrone 5.3-280 3.1/55 < 1 26/94
Benzo[e]pyrene NR 5-25
Benzo[ghi]perylene 3.4-57 5.4/4.2 3 0.4/8.1
Benzo[k]fluoranthene 1.0-43 1-2 2.6/9.6 1.7/17
Chrysene NR 7.6/27 86
Coronene < 10-24 1.3/2.7 0.7/4.6
Dibenz[a,h]anthracene NR 4.7/6.9 2.1/9.6
Fluoranthene 16-288 23/43 8-23 0.7-7.2 11/111 17 47/180 72
Indeno[1,2,3-cd]pyrene ND-9.9 5.9/3.9 0.3/5.7
1-Methylphenanthrene 22-708
Naphthalene NR 5-4 51/120
Perylene 4.2-59 < 5-26 36
Phenanthrene 21-570 7-109 0.1-1.7 12/155 18 108/216
Pyrene 6.6-394 9-77 15-38 0.3-6.6 6.2/62 23 25/109
Triphenylene 7.5-300 7.9/43 27/106
Table 61 (contd)
ND, not detected; /, single measurements; NR, not reported;
[1] Mussel (Mytilus edulis), Danish, German and Dutch Wadden Sea, 1989 (Compaan & Laane, 1992);
[2] Mussel (Mytilus edulis); Finnish archipelago, Finland, 1978-79; concentration in µg/kg wet weight
(Rainio et al., 1986);
[3] Mussel (Mytilus edulis L.); North Sea coast, Netherlands; concentration in µg/kg wet weight
(Boom, 1987);
[4] Hard shell clam (Mercenaria mercenaria), Rhode Island (seafood stores), USA; concentration in µg/kg
wet weight (Pruell et al., 1984);
[5] Softshell clam (Mya arenaria), Coos Bay, Oregon, USA, 1978-79; reference weight not given (Mix &
Schaffer, 1983);
[6] Clam (Mya mercenaria); Chesapeake Bay, USA, 1984 (Bender & Huggett, 1988);
[7] Mussel (Mytilus edulis); Yaquina Bay, USA, 1979-80; concentration in µg/kg wet weight (Mix & Schaffer,
1983);
[8] Rangia cuneata; Lake Pontchartrain, USA, 1980; concentration in µg/kg wet weight(McFall et al., 1985)
Table 61 (contd)
Compound [9] [10] [11] [12] [13] [14] [15]
Acenaphthene 16
Acenaphthylene 18
Anthracene < 0.05-3.2 <0.05
Benz[a]anthracene < 1-6 < 10 1.0-1.8 ND-2.3
Benzo[a]pyrene 30-168 < 10 < 0.003-0.02 < 0.004 0.41-1.8 0.40-2.6 1.0
Benzo[b]fluoranthene 1.0-1.8 0.83-1.9
Benzo[c]phenanthrene < 1-9
Benzo[e]pyrene
Benzo[ghi]perylene < 1-10 < 0.05-0.3 <0.05 0.53-1.9 0.83-2.3
Benzo[k]fluoranthene < 0.002-0.02 < 0.002 0.29-0.80 0.32-1.2
Chrysene < 0.03-1.4 <0.03
Coronene
Dibenz[a,h]anthracene
Fluoranthene < 1/52 < 1-370 < 0.04-0.70
Fluorene
Indeno[1,2,3-cd]pyrene
1-Methylphenanthrene
Naphthalene
Perylene < 1-10 < 10-300 < 0.01-0.08
Phenanthrene < 1-15 < 1-60 14
Pyrene 17/165 < 1-450 < 0.03-1.4 <0.03
Triphenylene
Table 61 (contd)
[9] Rangia cuneaya, Chesapeake Bay, USA, 1984 (Bender & Huggett, 1988);
[10] Lampsilus radiata, Elliptio complanatus, Anodonata grandis; Lake George, Heats Bay USA
(Heit et al., 1980);
[11] Tridacna maxima, Great Barrier Reef, Australia, 1980-82; concentration in µg/kg wet weight
(Smith et al., 1984);
[12] Clam; Green Island, Great Barrier Reef, Australia, concentration in µg/kg wet weight
(Smith et al., 1984);
[13] Shortnecked clam; near Miyagi Prefecture, Japan, concentration in µg/kg wet weight
(Takatsuki et al., 1985);
[14] Mussel; near Miyagi Prefecture; Japan, reference weight not given (Takatsuki et al., 1985);
[15] Perna viridis; Gulf of Thailand (mussel farm), Thailand, reference weight not given
(Hungspreugs et al., 1984)
High-performance liquid chromatography or gas chromatography;
Table 62. Polycyclic aromatic hydrocarbon concentrations (µg/kg wet weight) in bivalves (Oysters); background values
Compound [1] [2] [3] [4] [5] [6] [7]
Acenaphthene 46 < 0.2-2.0 16
Acenaphthylene 36 < 0.4-3.0
Anthracene 44 < 1-40 < 0.08-0.9 < 0.25-4.2
Benz[a]anthracene 9.9 0.3-12 < 1-135 1.1 1.5-10
Benzo[a]pyrene 0.5-1.6 50-285 < 0.01-5 0.6-2.6 0.78 3.5
Benzo[b]fluoranthene 0.3-5.2 < 0.03-6 3.0-20 2.2
Benzo[c]phenanthrene < 1-70
Benzo[e]pyrene < 1-453 2.8-32
Benzo[ghi]perylene O.4-1.2 < 1-73 < 0.05-5 0.87 < 0.20-2.8
Benzo[k]fluoranthene 12 0.1-0.9 < 0.06-5.1 1.2 < 0.01-< 3
Chrysene 58 1.3-14 < 0.1-3
Dibenz[a,h]anthracene < 1-20 < 0.01-< 4 < 0.06
Fluoranthene 80 0.9-94 < 1-450 0.4-22 470
Fluorene 21 0.1-0.8
Indeno[1,2,3-cd]pyrene 1.7 < 0.01-5
1-Methylphenanthrene 3.5
Naphthalene 35 5-48 0.8-7
Perylene < 1-130
Phenanthrene 220 4.9-77 < 1-45 2-38 6.7
Pyrene 200 1.6-50 < 1-645 < 0.4-15 7.0-52
Triphenylene 0.03
Table 62 (continued)
[1] Crassostrea virginica, Lake Pontchartrain, USA, 1980 (McFall et al., 1985);
[2] Crassostrea virginica; Palmetto Bay (Marina), USA (Marcus & Stokes, 1985);
[3] Crassostrea virginica; Chesapeake Bay, USA, 1983-84; concentration in µg/kg dry weight (Bender & Huggett, 1988);
[4] Saccostrea cucculata, Mermaid Sound, Australia, 1982 (Kagi et al., 1985);
[5] Oyster, Japan (local market); 1977-78 (Obana et al., 1981a);
[6] Oyster, near Miyagi Prefecture, Japan; reference weight not given (Takatsuki et al., 1985);
[7] Ostrea plicatula; Gulf of Thailand, Thailand; reference weight not given (Hungspreugs et al., 1984)
High-performance liquid chromatography or gas chromatography
Table 63. Polycyclic: aromatic hydrocarbon concentrations (µg/kg wet weight)
in crustacea (lobsters); background values
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene ND ND
Benz[a]anthracene 655 179 9-38 Trace-133 6-79 6-17
Benzo[a]pyrene 18 3.8 0.4-2.1 Trace-2 1.6-8 ND-1.6
Benzo[b]fluoranthene 17 28 3-6.5 Trace-5.3 7-16 ND-0.8
Benzo[e]pyrene ND 170 12-23 ND-22 15-29 ND-3.6
Benzo[ghi]perylene 11 63 1.4-6.8 Trace-2.0 2.4-10 ND-0.8
Benzo[k]fluoranthene 2 4.4 0.8-1.9 Trace-11.6 1.9-8 ND-0.8
Chrysene 140 113 2.5-12 ND-14 2-43 ND
Fluoranthene ND 147 46-407 5.5-12 90-162 ND-34
Fluorene ND 194
Indeno[1,2,3-cd]pyrene 22 77 2.1-5.0 ND-3.7 Trace-5 ND-0.8
Phenanthrene ND 1197 20-345 ND-15
Pyrene ND 174 ND-197 ND-5 35-46 ND-22
Triphenylene ND 1373 ND-141 ND-Trace
ND, not detected
[1] Homarus americanus (digestive gland); Port Hood, Canada, 1979 (Sirota & Uthe, 1981);
[2] Homarus americanus (digestive gland); Brown Bank (offshore), Canada, 1979 (Sirota & Uthe, 1981);
[3] Homarus americanus (hepatopancreas); Morien Bay and Mira Bay, Canada (Sirota et al.,1983);
[4] Homarus americanus (tail muscle); Moran Bayand, Mira Bay, Canada (Sirota et al., 1983);
[5] Homarus americanus (digestive gland); Port Morien, Canada, 1982-84 (Uthe & Musial, 1986);
[6] Homarus americanus (tail muscle); Port Morien, Canada, 1982-84 (Uthe & Musial, 1986)
Analysed by high-performance liquid chromatography or gas chromatography
Table 64. Polycyclic aromatic hydrocarbon concentrations (µg/kg wet weight) in fish and other aquatic species
(background values)
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Anenaphthene ND-83 11 7 1-500
Acenaphthylene 43 0.8-24
Anthracene 10 2.0-2.2 ND 20
Benz[a]anthracene 4 4.0-26 1.2 1.6-7.5 20
Benzo[a]fluorene ND
Benzo[e]pyrene 0.04-0.84 1 1.9-15 8 Trace-4.5 5
Benzo[b]fluoranthene 3.2-17
Benzo[e]pyrene ND
Benzo[ghi]perylene 2.0-14 16
Benzo[k]fluoranthene 2.1-11
Chrysene 6 3 3.4-26 NR
Dibenz[a,h]anthracene 1.2-4.13
Fluoranthene 4-95 4 85 9 ND-732 20
Fluorene 8.9 ND-15 1-370 ND
Indeno[1,2,3-cd]pyrene ND-15 NR
1-Methylphenanthrene NR
Naphthalene 45-215 ND-117
Perylene NR
Phenanthrene 8-142 2 157 2.3-35 36 23-43 ND 40
Pyrene 2-62 4 30 31 2.4-74 1.3-9.6 ND
Triphenylene 20
ND, not detected; NR, not reported;
[1] Various seafish (muscle, liver, gall), Finnish archipelago, Finland, 1979 (Rainio et al., 1986);
[2] Edible tissues of various seafish, Arabian Gulf, Iraq (DouAbdul et al., 1987);
[3] Whole bullhead catfish (ictalurus nebulosus), Buckeye Lake, USA (Vassilaros et al., 1982);
[4] Whole bullhead catfish (Ictalurus, nebulosus;, whole), Black River, USA (West et al, 1985);
[5] Whole fish, Hersey River, USA (Black et al., 1981);
[6] Whole striped bass (Morone saxatillis); Potomac River, USA (Vassilaros et al., 1982);
[7] White suckers (Catastomus commersoni); stomach contents; Lake Erie, USA (Maccubbin et al., 1985);
[8] Various fish, Japan, 1970-91 (Environment Agency, Japan, 1993);
[9] Fish bought in market, Ibadan, Nigeria; reference weight not given (Emerole et al., 1982);
[10] Whole holothurians, France; concentration in µg/kg dry weight (Milano et al., 1986)
Analysed by high-performance liquid chromatography or gas chromatography
The levels in holothurians from urban sewage were 1-15 mg/kg (Milano
et al., 1986).
Concentrations of 1-5 mg/kg individual PAH were found in limpets
(Patella vulgata) in the North Sea (Knutzen & Sortland, 1982). The
PAH concentrations in two species of bivalves in Saudafjorden (Norway)
near an iron alloy smelter decreased rapidly with distance from the
source, but the compounds could still be detected more than 15 km
away. High levels of individual PAH were reported in mussels
(Modiolus modiolus), with maximum levels of 57 000 µg/kg
benzo [b]fluoranthene, 25 000 µg/kg benz [a]anthracene, 23 000 µg/kg
benzo [e]pyrene, 21 000 µg/kg benzo [a]pyrene, 20 000 µg/kg
fluoranthene, 8200 µg/kg pyrene, 6000 µg/kg benzo[ghi]perylene, 4000
µg/kg perylene, 2900 µg/kg benzo [a]fluorene, 2300 µg/kg
benzo [b]fluorene, 2200 µg/kg dibenz [a,h]anthracene, 2000 µg/kg
benzo [c]phenanthrene, 1100 µg/kg phenanthrene, 524 µg/kg anthracene,
and 360 µg/kg anthanthrene (Bjrseth, 1979). A very high level of
anthracene (243 µg/kg) was found in mussels (Mytilus edulis L.) in
the North Sea near the Dutch coast (Boom, 1987). Mussels in the USA
frequently contained up to 500 µg/kg of individual PAH (Heit et al.,
1980; Mix & Schaffer, 1983).
The levels of PAH in pooled mussel samples in 1986, 1988, and 1990 in
Germany were about 10 µg/kg for fluoranthene, pyrene, chrysene plus
triphenylene, benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, and benzo [e]pyrene and < 4 µg/kg for
benzo [ghi]fluoran-thene plus benzo [c]phenanthrene,
benz [a]anthracene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, anthanthrene, and
coronene. The levels were high in the winter months and low in summer,
with minima in June and April. The authors concluded that this
seasonal variation was due to more intensive metabolic activity (Jacob
& Grimmer, 1994).
During 1978-79, the average total PAH concentrations in two
subpopulations of softshell clams were 555 µg/kg in the industrialized
bayfront area of Coos Bay, Oregon, and 76 µg/kg in a more remote
environment. During 1979-80, low-molecular-mass, readily water-soluble
PAH were one or two orders of magnitude more concentrated then
high-molecular-mass, less water-soluble PAH in mussels (M. edulis)
(Mix & Schaffer, 1983).
Individual PAH levels of 1-20 mg/kg were found in the hepatopancreas
of lobsters (Homarus americanus) in the south arm of Sydney Harbour,
Canada, near a coking plant (Sirota et al., 1983), and levels of the
same order of magnitude were found in the digestive gland (Uthe &
Musial, 1986). The levels in digestive gland, tail muscle, and
hepatopancreas from lobsters from other areas of Canada were 100-1000
µg/kg (Sirota & Uthe, 1981; Sirota et al., 1983; Uthe & Musial, 1986).
High PAH levels were found in oysters (Crassostrea virginica) in
Chesapeake Bay, USA, with maximum levels of 650 µg/kg pyrene, 450
µg/kg benzo [e]pyrene, 450 µg/kg fluoranthene, 290 µg/kg
benzo [a]pyrene, 130 µg/kg benz [a]anthracene, 130 µg/kg perylene,
73 µg/kg benzo [ghi]-perylene, 70 µg/kg benzo [c]phenanthrene, 48
µg/kg naphthalene, 45 µg/kg phenanthrene, 40 µg/kg anthracene, and 20
µg/kg dibenz [a,h]anthracene. The levels of PAH in clams
(Rangia cuneata) from Chesapeake Bay were 170 µg/kg
benzo [a]pyrene, 170 µg/kg pyrene, 52 µg/kg fluoranthene, 15 µg/kg
phenanthrene, 10 µg/kg perylene, 10 µg/kg benzo [ghi]perylene, 9
µg/kg benzo [c]phenanthrene, and 6 µg/kg benz [a]anthracene (Bender
& Huggett, 1988).
Phenanthrene was found at 15 mg/kg in lampreys (Pteromyzon sp.) in
the Hersey River, USA, which was polluted with creosote used for wood
preservation (Black et al., 1981).
The viviparous blenny (Zoarces viviparus) fish contained 0.06 µg/kg
benzo [a]pyrene and 0.2-3.9 µg/kg phenanthrene and fluoranthene; the
concentrations of other PAH were below the detection limit (0.01
µg/kg). In bream (Abramis brama) the levels were < 0.01-0.15 µg/kg
benzo [a]pyrene and 1.3-15 µg/kg phenanthrene. Mussels (Mytilus
sp.) were shown to accumulate PAH and were thus a better marker for
PAH contamination (Jacob & Grimmer, 1994, 1995).
The concentrations of individual PAH in English sole
(Paraphrys vetulus) taken from near petroleum storage tanks were 1-5
mg/kg (Malins et al., 1985).
5.1.7.2 Terrestrial organisms
The liver of wild deer mice (Peromyscus maniculatus) trapped at a
PAH-contaminated site in South Carolina, USA (Whidbey Island Naval Air
Station) had levels of PAH ranging from 0.075 for
benzo [b]fluoranthene to 4.6 mg/kg for benz [a]anthracene.
Acenaphthylene, acenaphthene, fluorene, benz [a]-anthracene,
chrysene, benzo [b]fluoranthene, benzo [k]fluoranthene,
dibenz [a,h]anthracene, and indeno[1,2,3- cd]pyrene were detected.
Liver from mice at an uncontaminated reference site contained
measurable amounts of only benz [a]anthracene (0.55 mg/kg) and
acenaphthylene (2.2 mg/kg) (Dickerson et al., 1994).
In a study of PAH levels in terrestrial organisms from a roadside in
Brisbane, Australia, 16 PAH were determined: naphthalene, fluorene,
phenanthrene, anthracene, fluoranthene, pyrene, benz [a]anthracene,
chrysene, benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene,
perylene, indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene. In the beetle Laxta
granicollis, pyrene and benzo [ghi]perylene were present at the
highest levels, at 20 µg/kg wet weight each; phenanthrene and
fluoranthene were present at about 10 µg/kg; and the concentrations of
other PAH were < 5 µg/kg. Naphthalene, anthracene,
dibenz [a,h]anthracene, and coronene were not detected. Fluorene, at
a concentration of 11 µg/kg wet weight, was the most abundant PAH in
the beetle Platyzosteria nitida; the concentrations of other PAH
were < 5 µg/kg; whereas naphthalene, dibenz [a,h]anthracene, and
coronene were not detected. In millipedes (myriapods),
benzo [k]fluoranthene was the most abundant PAH (19 µg/kg wet
weight); the pyrene concentration was 12 µg/kg; those of other PAH
were < 5 µg/kg wet weight; and dibenz [a,h]anthracene and coronene
were not detected. In centipedes (Myriaod sp.), no PAH were
detected. In slugs (Arion ater), benzo [k]fluoranthene showed the
highest concentration, at 19 µg/kg wet weight; the pyrene and
naphthalene levels were about 10 µg/kg; those of other PAH were < 5
µg/kg wet weight; and anthracene, perylene, dibenz [a,h]anthracene,
and coronene were not detected. In earthworms (Lumbricus
terrestris), benzo [ghi]perylene was the most abundant PAH (28
µg/kg wet weight); phenanthrene, fluoranthene, pyrene, chrysene,
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene were
present at about 10 µg/kg; and naphthalene and dibenz [a,h]anthracene
were not detected (Pathirana et al., 1994).
The PAH concentrations in earthworms did not seem to be affected by
the location in which the worms lived, but those in the faeces showed
a significant dependence on location. In a survey of earthworm faeces
from the Bornhöveder Lake district in 1988, the concentrations of
phenanthrene, fluoranthene, pyrene, and benzo [b]fluoranthene plus
benzo [j]fluoranthene plus benzo [k]fluoranthene were in the range
of 45 µg/kg; those of benz [a]anthracene, chrysene plus triphenylene,
benzo [e]pyrene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene, and
benzo [ghi]perylene were about 20 µg/kg; and those of anthracene,
benzo [ghi]fluoranthene plus benzo [c]phenanthrene,
dibenz [a,h]anthracene, anthanthrene, and coronene were < 5 µg/kg.
Earthworm faeces in the Saarland contained 250-770 µg/kg
benzo [a]pyrene, and Allolobophora longa earthworm faeces from a
highly industrialized region of eastern Germany (Halle, Leipzig)
contained even higher concentrations: 37-2100 µg/kg benzo [a]pyrene
and 36-1700 µg/kg benzo [e]pyrene. The faeces of the earthworm
Lumbricus terrestris contained 4.6-55 µg/kg benzo [a]pyrene and
6.5-50 µg/kg benzo [e]pyrene (Jacob & Grimmer, 1995).
In insects near the Hersey River, USA, the maximum concentrations of
PAH were 5500 µg/kg phenanthrene, 2900 µg/kg benz [a]anthracene, and
730 µg/kg benzo [a]pyrene (Black et al., 1981).
The lipid fraction of liver from herring gulls (Larus argentatus)
from Pigeon Island and Kingston, Ontario, Canada, contained 0.15 µg/kg
anthracene, 0.082 µg/kg fluoranthene, 0.076 µg/kg pyrene, 0.05 µg/kg
naphthalene, 0.044 µg/kg fluorene, 0.038 µg/kg acenaphthene, and 0.038
µg/kg benzo [a]pyrene (Environment Canada, 1994). The concentrations
of PAH in pooled samples taken from the eggs of herring gulls
(Larus argentatus) on the German North Sea islands Mellum and
Trischen during 1992-93 were below the limit of detection, except for
that of phenanthrene, which was 1 µg/kg wet weight (Jacob & Grimmer,
1994).
5.2 Exposure of the general population
Possible sources of nonoccupational exposure to PAH are:
- polluted ambient air (main emission sources: vehicle traffic,
industrial plants, and residential heating with wood, coal,
mineral oil) (see section 5.1.1);
- polluted indoor air (main emission sources: open stoves and
environmental tobacco smoke) (see Table 65);
- tobacco smoking (see Table 66);
- contaminated food and drinking-water (see sections 5.1.5 and
5.1.2.3)
- use of products containing PAH (coal-tar skin preparations and
coal-tar-containing hair shampoos);
- ingestion of house dust; and
- dermal absorption from contaminated soil and water.
5.2.1 Indoor air, tobacco smoke, and environmental tobacco smoke
PAH are found in indoor air (Table 65) mainly as a result of tobacco
smoking and residential heating with wood, coal, or, in some
developing countries, rural biomass. The PAH levels in indoor air
usually range from 1 to 50 ng/m3. The most abundant PAH were
phenanthrene and naphthalene, with levels of up to 2300 ng/m3. Homes
with gas heating systems had higher indoor levels than those with
electric heating systems (Chuang et al., 1991), and even higher levels
were detected in indoor air near open fireplaces (Alfheim & Ramdahl,
1984). Airtight residential wood-burning stoves seemed to have a minor
effect on the indoor air concentration of PAH (Alfheim & Ramdahl,
1984; Traynor et al., 1987), but in homes with non-airtight wood
stoves, 2-46 times higher PAH concentrations were found during heating
periods than during periods without heating (Daisey et al., 1989).
Emissions from unvented kerosene heaters can significantly affect
indoor air quality in mobile homes, with a maximim value for
naphthalene of 2300 ng/m3. Four of eight heaters investigated emitted
PAH-containing particles at levels that exceeded the USA ambient air
standards for airborne particles, with a 50% cutoff at the aerodynamic
diameter of 10 µm. When the kerosene heaters were in operation, the
concentrations of carcinogenic PAH (with four rings or more) in the
mobile homes were increased by 10-fold (Mumford et al., 1991).
Table 65. Polycyclic: aromatic hydrocarbon concentrations (ng/m3) in indoor air; main source, residential heating
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene NR 589-1649
Acenaphthylene NR 60-592
Anthracene 5-30 408 5-15 84 NR 9.9-11
Benz[a]anthracene 3-9 2-6 3-13 145 NR 0.9-5.5
Benzo[a]pyrene 13-370 0.3-12 1-7 3-23 150 < 0.009-1.34 0.34-3.5 2.0-490 8.5-29
Benzo[b]fluoranthene < 0.007-0.68 0.17-3.8 1.4-420 5.6-21
Benzo[e]pyrene < 0.06-1.36
Benzo[ghi]perylene 14-340 0.4-10 1-7 3-30 125 < 0.01-6.20 0.37-3.7 2.8-450 0.4-7.5
Benzo[k]fluoranthene 5-150 0.07-7 0.6-3 2-10 63 0.005-0.48 0.07-1.9 0.67-200 0.7-21
Chrysene 2-12 3-6 4-13 115 NR
Coronene NR
Cyclopenta[cd]pyrene NR
Dibenzo[a,e]pyrene NR
Dibenz[a,h]anthracene NR 3.3-25
Fluoranthene 16-56 16-24 16-50 208 0.07-1.18 87-268
Fluorene NR
Indeno[1,2,3-cd]pyrene 20-560 1-16 1-8 3-22 130 < 0.02-3.54 1.1-6.1 3.9-740 2.3-11
Phenanthrene 120-400 120-200 140-290 555 NR 31-64
Pyrene 0.02-1.53 1.0-20
ND, not determined; NR, not reported; /, single measurements;
[1] Wood-burning open fire-place, Netherlands (Slooff et al., 1989);
[2] Wood in multi-burner, Netherlands (Slooff et al., 1989);
[3] Coal, Netherlands (Slooff et al., 1989);
[4] Briquettes, Netherlands (Slooff et al., 1989);
[5] 'Icopower' heating, Netherlands (Slooff et al., 1989);
[6] Wood heating in seven homes, USA (Daisey et al., 1989);
[7] Wood burning in one home; volume, 236 m3; airtight stove, Truckee, USA, (elevation, 1800 m) (Traynor et al., 1987);
[8] Wood burning in one home; volume, 236 m3; non-airtight stove, Truckee, USA (elevation, 1800 m) (Traynor et al., 1987);
[9] Wood burning in one home with four different heaters, USA (Knight & Humphreys, 1985)
Analysed by high-performance liquid chromatography or gas chromatography
Table 65 (contd)
Compound [10] [11] [12] [13] [14] [15] [16] [17]
Acenaphthene NR 1-258
Acenaphthylene 10-120 21/68 25-36 NR 1-753
Anthracene 1.5-15 4.2-5.9 NR 0.1-80
Benz[a]anthracene 0.24-3.4 0.72/2.8 0.55-1.0 ND-3.81 25 100 1000 4000 5-1021
Benzo[a]pyrene 0.28-3.3 0.24/2.0 0.54-1.0 ND-4.13 14 700 600 3100 8-1645
Benzo[b]fluoranthene NR 2-930
Benzo[b]pyrene 0.33-10 1.4-3.0 NR 5-1106
Benzo[ghi]perylene 0.32-2.5 0.22/3.7 0.72-1.0 ND-5.4 4-802
Benzo[k]fluoranthene ND-7.81a 4-824
Chrysene 0.58-7.2 1.5/3.1 1.4-2.2 0.18-8.61 7-1439
Coronene 0.31-1.4 0.07/2.3 0.55-0.58 ND-4.75 NR
Cyclopenta[cd]pyrene 0.18-2.0 0.49/4.2 0.36-0.59 ND-2.38 10 700 400 5600 NR
Dibenzo[a,e]pyrene NR 11 700 600 200 NR
Dibenz[a,h]anthracene NR 8-958
Fluoranthene 6.2-23 16/11 11 2.4-37.4 5-1095
Fluorene NR 3-275
Indeno[1,2,3-cd]pyrene 0.24-1.8 0.15/1.3 0.48-0.79 ND-3.53 8400 500 2000 4-670
5-Methylcholanthrene NR 7300 200 200 NR
Naphthalene 750-2200 2300/950 1200-1600 NR NR
Phenanthrene 55-210 48/34 93-110 9.2-210 3-667
Pyrene 3.6-17 9.7/13 6.9-7.6 1.4-18.1 7-850
[10] Gas or electridy, USA (Wilson & Chuang, 1991);
[11] Kerosene; unvented heaters in mobile homes, Apex, USA (Mumford et al., 1991);
[12] Various heating in eight homes, Columbus, USA (Chuang et al., 1991);
[13] Various heating in 33 homes, USA (Wilson et al., 1991);
[14] Smoky coal, Xuan Wei, China (Mumford et al., 1987);
[15] Smokeless coal, Xuan Wei, China (Mumford et al., 1987);
[16] Wood, Xuan Wei, China (Mumford et al., 1987);
[17] Various cooking fuels (cattle dung, wood, kerosene, liquid petroleum gas) in 60 homes, India
(Raiyani et al., 1993b)
a Sum of benzofluranthenes
Table 66. Polycyclic aromatic hydrocarbon concentrations (ng/m3 in indoor air;
main source, environmental tobacco smoke
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene 2.5 36
Acenaphthylene 14 177
Anthracene 2.8 25 1.5 < 1
Anthanthrene 0.5 1.5 < 1 2.5 3
Benz[a]anthracene 1.3 12 15 13
Benzo[a]fluorene 5.5 39
Benzo[a]pyrene 1.8 7.3 14 4.5 0.04-0.16 22
Benzo[b]fluoranthene 0.06-0.08
Benzo[b]fluorene 2.5
Benzo[e]pyrene 2.3 7.1 11 4.5 18
Benzo[ghi]fluoranthene 4.3 18 8.5 14
Benzo[ghi]perylene 2.5 5.8 7 2 0.09-0.36 17
Benzo[k]fluoranthene 0.02-0.06
Coronene 2.0 3.1
Fluoranthene 14 41 5 16 99
Indeno[1,2,3-cd]pyrene 2.3 5.8 1 1.5 0.13-0.45
1-Methylphenanthrene 6.6 38 < 1 3.5
Perylene 0.5 0.8 4 2.5 11
Phananthrene 38 168 3 1
Pyrene 13 32 13 21 66
[1] Office room (volume, 88 m3; ventilation, 176 m3/h; background sample after weekend,
Finland; vapour and particulate phase (Salomaa et al., 1988);
[2] Office room (Volume, 88 m3; ventilation, 176 m3/h; 6 h; 96 cigarettes, American
type, 10 different brands, both medium- and low tar, Finland; vapour and particulate
phase (Salomaa et al., 1988);
[3] House in a forest (room volume, 65 m3; air exchange, 2.0-2.3 turnovers/h); background
sample, Norway (Alfheim & Ramdahl, 1984);
[4] House in a forest (room volume, 65 m3; air exchange, 2.0-2.3 turnovers/h); with
tobacco smoking, Norway (Alfheim & Ramdahl, 1984);
[5] House in a residential, wooded area of Truckee, USA (elevation, 1800 m); volume,
236 m3; no stove (Traynor et al., 1987);
[6] Model room (volume, 36 m3); one air exchange/h, smoking of five cigarattes/h (Ministry
of Environment, 1979))
High-performance liquid chromatography or gas chromatography; concentration of particulate
phase, unless otherwise stated
Emissions from coal and wood combustion in open fires for cooking
purposes in unvented rooms in Xuan Wei County, China, contained
extremely high PAH concentrations (see also section 8). The highest
concentration (benzo [a]pyrene at 15 000 ng/m3) was measured in
fumes from smoky coal combustion. Coal combustion in open fires in
Xuan Wei homes emitted 15 µg/m3 of carcinogenic PAH, while wood
combustion emitted 3.1 µg/m3 (Mumford et al., 1987).
Cooking with rural biomass in open fires also led to high PAH levels
in indoor air, as measured in rural Indian households.
Benzo [a]pyrene was measured at a concentration of about 4 µg/m3
during the cooking period, which occupied about 10% of the household
activities over the year. The cooking fuels included baval, neem,
mango, rayan, and crop residues (Smith et al., 1983). The total
release of PAH into indoor air from this source is unknown but may be
of major importance, especially in developing countries. Very low PAH
emissions were found when liquid petroleum gas was used as a fuel for
cooking (Raiyani et al., 1993b). In contrast, the PAH content of
kitchen air in Berlin, in the industrialized part of Germany, was
similar to that encountered in ambient air (Seifert et al., 1983).
House dust may be another important source of indoor pollution with
PAH. In a study of the homes of four smokers and four nonsmokers in
Columbus, Ohio, USA, the sum of the concentrations of naphthalene,
acenaphthylene, acenaphthene, fluorene, phenanthrene, anthracene,
retene, fluoranthene, pyrene, benz [a]anthracene, chrysene,
cyclopenta [cd]pyrene, benzo [b]fluoranthene,
benzo [j]fluoranthene, benzo [k]fluoranthene, benzo [e]pyrene,
benzo [a]pyrene, indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene in house dust and in soil from the
entryway, the pathway, and the foundation of the houses was 16-580
mg/kg. The concentrations in house dust correlated well with those in
the entryway soil samples, and a weaker correlation was found with the
pathway soil samples, but the relationships were not statistically
significant (Chuang et al., 1995).
A special source of exposure to PAH is wood-heated saunas. The highest
concentrations were found in a smoke sauna, the second highest in a
preheated sauna where the flues were closed before use, and the lowest
concentrations in a sauna heated by continuous burning of wood.
Pyrene, fluoranthene, benz [a]anthracene, and phenanthrene were
present at the highest levels (100-330 µg/m3 air); other PAH were
present at < 50 µg/m3. The concentrations decreased from
benzo [e]pyrene > benzo [a]pyrene > benzo [a]fluorene >
anthra-cene > benzo [b]fluorene > fluorene (Häsänen et al., 1983).
The protocol of a study of total human environmental exposure included
direct monitoring of exposure to benzo [a]pyrene by inhalation and
ingestion during three periods of 14 days. The range and magnitude of
dietary exposure (2-500 ng/day) was much greater than that by
inhalation (10-50 ng/day). The levels of benzo [a]pyrene in indoor
air were closely correlated with the ambient levels in most homes
(Waldman et al., 1991).
Indoor air concentrations of individual PAH due mainly to cigarette
smoke are shown in Table 66, and the levels in mainstream and
sidestream smoke of cigarettes are listed in Table 67. The average PAH
levels ranged from 1 to 50 ng per cigarette, and the major components
were phenanthrene, naphthalene, benzo [a]pyrene, benzo [e]pyrene,
fluoranthene, and pyrene. Sidestream smoke was found to contain 10
times more PAH than mainstream smoke. The levels in sidestream smoke
were 42-2400 ng per cigarette (Grimmer et al., 1987). The PAH
concentrations in the mainstream smoke from filter cigarettes
increased with increasing puff volume (Funcke et al., 1986). In a
pilot study in Columbus, Ohio, USA, naphthalene was the most abundant
PAH; environmental tobacco smoke appeared to be the most significant
source of indoor pollution (Chuang et al., 1991).
Table 67. Concentrations of selected polycyclic aromatic hydrocarbons
in cigarette smoke
Compound Mainstream smoke Sidestream smoke
(µg/100 cigarettes) (µg/100 cigarettes)
Anthracene 2.3-23.5
Anthanthrene 0.2-2.2 3.9
Benz[a]anthracene 0.4-7.6
Benzo[b]fluoranthene 0.4-2.2
Benzo[b]fluoranthene 0.6-2.1
Benzo[k]fluoranthene 0.6-1.2
Benzo[ghi]fluoranthene 0.1-0.4
Benzo[a]fluorene 4.1-18.4 75.0
Benzo[b]fluorene 2.0
Benzo[ghi]perylene 0.3-3.9 9.8
Benzo[c]phenanthrene Present
Benzo[a]pyrene 0.5-7.8 2.5-19.9
Benzo[e]pyrene 0.2-2.5 13.5
Chrysene 0.6-9.6
Coronene 0.1
Dibenz[a,h]anthracene 0.4
Dibenzo[a,e]pyrene Present
Dibenzo[a,h]pyrene Present
Dibenzo[a,i]pyrene 0.17-0.32
Dibenzo[a,l]pyrene Present
Fluoranthene 1.0-27.2 126.0
Fluorene Present
Indeno[1,2,3-cd]pyrene 0.4-2.0
5-Methylcholanthrene 0.06
Perylene 0.3-0.5 3.9
Phenanthrene 8.5-62.4
Pyrene 5.0-27 39.0-101.0
Triphenylene Present
1-Methylphenanthrene 3.2
Adapted from International Agency for Research on Cancer (1985)
In studies in eight healthy male smokers, aged 20-40 years, the
benzo [a]pyrene intake from the smoking of 20 cigarettes per day was
calculated to be 150-750 ng/d, assuming a deposition rate for
particulate matter of 75% (Scherer et al., 1990).
The total concentration of 14 PAH (fluoranthene, pyrene,
benzo [a]fluorene, benz [a]anthracene, chrysene,
benzo [b]fluoranthene, benzo [j]fluoranthene,
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene, perylene,
dibenz [a,h]-anthracene, benzo [ghi]perylene, and anthanthrene)
measured in a 36-m3 room into which sidestream smoke from five German
cigarettes was introduced every hour, with one air change per hour,
was 429 ng/m3. Assuming that the daily inhalation volume for adults
is 18 m3 and that 20 h/d are spent indoors, the volume of indoor air
inhaled daily is 18 m3 × 20/24 = 15 m3. Thus, passive smokers are
exposed daily to 15 × 429 = 6435 ng PAH, including 15 × 22 = 330 ng
benzo [a]pyrene (Ministry of Environment, 1979). An intake of 11 ng
benzo [a]pyrene was estimated in another study on the basis of an
assumed breath volume of 0.5 m3/h , a deposition rate for particulate
matter of 11%, and an exposure time of 8 h, after monitoring in an
unventilated, 45-m3, furnished room (Scherer et al., 1990).
5.2.2 Food
Smoked and barbecued food in particular can contain PAH (Grimmer &
Düvel, 1970; McGill et al., 1982; de Vos et al., 1990; Menichini et
al., 1991b; see also section 5.1.5 and Tables 51-56). Preparation of
food with contaminated drinking-water (see section 5.1.2.3) may also
lead to exposure to PAH.
In 1989 and 1990, the levels of naphthalene and alkylated derivatives,
acenaphthene, acenaphthylene, fluorene, phenanthrene, anthracene,
fluoran-thene, 1-methylphenanthrene, pyrene, benz [a]anthracene,
chrysene, benzo [b]fluoranthene, benzo [k]fluoranthene,
benzo [e]pyrene, benzo [a]pyrene, perylene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene, and
benzo [ghi]-perylene were measured in salmon, herring, cod, rockfish,
and halibut in the area of the Gulf of Alaska where oil spilled from
the tanker Exxon Valdez. As only the sums of the concentrations were
considered, there was no apparent difference from those in fish
samples taken from unpolluted control sites in 1989. In 1990, slightly
elevated PAH concentrations were found at the polluted sampling site.
Nevertheless, the fish from the area were considered to be safe for
human consumption by these investigators (Saxton et al., 1993).
In another special exposure situation, the average daily PAH intake of
the inhabitants of Kuwait due to consumption of seafood after the war
in the Persian Gulf was calculated to be 0.23 µg/day on the basis of
the concentrations monitored in local fish and shrimps (Saed et al.,
1995).
5.2.3 Other sources
Benzo [a]pyrene was detected in coal-tar-containing hair shampoos at
levels of 7000-61 000 µg/kg, and a tar bath lotion contained 150 000
µg/kg benzo [a]pyrene. No PAH were detected in hair shampoos made
from wood tar (State Chemical Analysis Institute Freiburg, 1995). PAH
are absorbed from coal-tar shampoos through the skin during hair
washing. Exposure during one washing with this type of shampoo, which
contains benzo [a]pyrene at 56 mg/kg, for anti-dandruff therapy
results in absorption of 0.45 µg/kg body weight, assuming 20 g
coal-tar, 70 kg body weight, and 3% dermal absorption (van Schooten et
al., 1994; see also section 8).
5.2.4 Intake of PAH by inhalation
Estimates of PAH intake from air are summarized in Table 68.
In an assessment of the risk for cancer due to air pollution in
Germany, the average volume of air inhaled during heavy work was
assumed to be 140 m3 per person per week. The maximum intake of
airborne benzo [a]pyrene per week was thus estimated to be
0.21 µg/week in rural areas, 0.84 µg/week in industrial areas, and
7 µg/week near emission sources (State Committee for Air Pollution
Control, 1992).
On the basis of an average inhalation of 15 m3 air per day, exposure
to benzo [a]pyrene was calculated to be 0.05 µg/d. In industrial
areas, the exposure was calculated to be four times higher (0.19 µg/d)
(Raiyani et al., 1993a).
5.2.5 Intake of PAH from food and drinking-water
Estimates of PAH intake from food are shown in Table 69. The values
for benzo [a]pyrene range from 0.14-1.6 µg/d.
The total dietary intake of some PAH in the United Kingdom was
estimated to be (µg/person per day): 1.1 for pyrene, 0.99 for
fluoranthene, 0.50 for chrysene, 0.25 for benzo [a]pyrene, 0.22 for
benz [a]anthracene, 0.21 for benzo [ghi]perylene, 0.18 for
benzo [b]fluoranthene, 0.17 for benzo [e]pyrene, 0.06 for
benzo [k]fluoranthene, and 0.03 for dibenz [a,h]anthracene. The
major contributors of PAH to the total dietary intake appeared to be
oils and fats, with 28% from butter, 20% from cheese, and 17% from
margarine, in respective dietary survey groups; cereals provided 56%
from white bread and 12% from flour. The oils and fats had the highest
individual PAH levels. Although cereals did not contain high levels of
individual PAH, they were the main contributor by weight to the total
in the diet. Fruits and vegetables contributed most of the rest of the
PAH in the diet, while milk and beverages were of minor importance.
Smoked meat and smoked fish made very small contributions to the food
groups to which they belonged, which themselves were not major
components of the diet (Dennis et al., 1983).
Table 68 Estimated intake of polycyclic aromatic hydrocarbons (µg/day per person) from ambient air
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Anthracene 0.005 0.001
Anthanthrene 0.015
Benz[a]anthracene 0.030 0.013
Benzo[a]pyrene 0.01-0.03a 0.0025-0.025 0.025 0.034a 0.0095-0.0435 0.004a 0.017 0.03-0.05 0.0005-0.20
0.02-0.12b
0.06-1.0c
Benzo[b]fluoranthene 0.060 0.029
Benzo[b]fluorene 0.002 0.002
Benzo[e]pyrene 0.035 0.022
Benzo[ghi]perylene 0.030 0.027
Benzo[j]fluoranthene 0.010
Benzo[k]fluoranthene 0.015 0.015
Chrysene 0.035
Coronene 0.025
Dibenz[a,h]anthracene 0.020 0.004
Fluoranthene 0.040 0.016
Fluorene 0.0005
Indeno[1,2,3-cd]pyrene 0.030 0.024
Perylene 0.015 0.003
Phenanethrene 0.200 0.007
Pyrene 0.040 0.017
Triphenylene 0.220
Table 68 (continued)
[1] Germany (maximum concentrations) (State Committee for Air Pollution Control, 1992);
[2] Italy (Menichini, 1992a);
[3] Netherlands (maximum concentrations) (Guicherit & Schulting, 1985);
[4] United Kingdom (maximum concentrations) (Butler & Crossley, 1979);
[5] USA (Santodonato et al., 1980);
[6] USA (WHO, 1987);
[7] Japan (maximum concentrations) (Matsumoto & Kashimoto, 1985);
[8] China (Chen et al., 1980);
[9] India (Chakraborti et al., 1988)
a Rural areas
b Industrial areas
c Near emission source
Table 69. Estimated intake of polycyciic aromatic hydrocarbons (µg/day per person, maximum values) from food
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Anthracene 5.6
Anthanthrene 0.30
Benz[a]anthracene 0.14
Benzo[a]pyrene 0.36 0.14-1a 0.1-0.3b 0.12-0.42 0.5 0.5 0.48 0.16-1.6
0.2c
Benzo[b]fluoranthene 1.0
Benzo[ghi]perylene 7.6 0.3 0.9
Benzo[j]fluoranthene 0.90
Benzo[k]fluoranthene 0.30
Chrysene 0.90 5.0
Coronene 0.09
Dibenz[a,h]anthracene 0.10
Fluoranthene 4.3 3 10
Indeno[1,2,3-cd]pyrene 0.31 0.4 <0.3
Perylene 0.20
Phenanethrene 2.0
Pyrene 4.0 5.1
[1] Austria (Pfannhauser, 1991);
[2] Germany (State Committee for Pollution Control, 1992);
[3] Italy (Menichini, 1992a);
[4] Netherlands (de Vos et al., 1990);
[5] Market basket study, Netherlands (Vaessen et al., 1984);
[6] Duplicate diet study, Netherlands (Vaessen et al., 1984);
[7] United Kingdom (Dennis et al., 1983);
[8] USA (Santodonato et al., 1980)
a Concentration in µg/week
b Adult non-smoker (70 kg)
c Mean concentration
In Sweden, the annual intake per person of the sum of fluoranthene,
pyrene, benz [a]anthracene, chrysene, triphenylene,
benzo [b]fluoranthene, benzo [j]-fluoranthene,
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene, and
indeno[1,2,3- cd]pyrene was about 1 mg. Cereals again seemed to be
the main contributor (about 34%), followed by vegetables (about 18%)
and oils and fats (about 16%). Although smoked fish and meat products
had the highest PAH levels, they made a modest contribution since they
are minor components of the usual Swedish diet (Larsson, 1986).
5.3 Occupational exposure
PAH have been measured in the air at various workplaces. Studies in
which measurements were reported only as the benzene-soluble fraction
or some other summarizing parameter affected mainly by PAH are not
covered because they do not refer to individual substances. The
presence of PAH metabolites in biological samples (urine, blood) from
workers has been used as a biomarker, and 1-hydroxypyrene seems to be
a suitable marker in some workplaces (see section 8.2.3). No data were
available on occupational exposure during production and use.
Occupational exposure to PAH occurs by both inhalation and dermal
absorption. In coke-oven workers, 75% of their exposure to total
pyrene and 51% of that to benzo [a]pyrene occurs by cutaneous
transfer (Van Rooij et al., 1993a; see also section 6). The exposure
of workers due to deposition of airborne pyrene on the skin, detected
in wipe samples, can be summarized as follows: in refineries,
< 0.0045 µg/cm2 (detection limit), 26 samples below detection limit;
in hot-mix asphalt facilities, < 0.0045 µg/cm2, 25 samples below
detection limit; during paving, < 0.13-0.31 µg/cm2 found in two of
nine samples (assuming a body area of 1.8 m2, equivalent to 5600
µg/person per day); in asphalt roofing manufacture, < 0.0045-0.0091
µg/cm2 found in 1 of 29 samples (assuming a body area of 1.8 m2,
equivalent to 170 µg/person per day); in application of asphalt
roofing, < 0.0045 µg/cm2, 10 samples below detection limit; in a
wood preserving plant, 47-1500 µg pyrene per person per day. These
data indicate that skin penetration is an important factor in
estimating total body exposure to PAH.
5.3.1 Occupational exposure during processing and use of of coal and
petroleum products
The following section is based on data obtained up to the early 1980s
which were compiled by the IARC (1984b, 1985, 1989b). More recent
studies are presented in detail.
5.3.1.1 Coal coking
In studies of pollution of the atmosphere near coke-oven batteries,
the concentration of benzo [a]pyrene varied from < 0.1 in
administrative buildings and a pump house to 100-200 µg/m3 on the
machinery and discharge side of a battery roof. At the top of a coke
battery, the following concentrations of particulate and gaseous PAH
were measured by stationary sampling: naphthalene, 0-4.4
(particulate)/ 280-1200 (gaseous) µg/m3; acenaphthene, 0-17/6.0-100
µg/m3; fluorene, 0-58/23-130 µg/m3; phenanthrene, 27-890/6.7-280
µg/m3; anthracene, 9.6-310/6.0-91 µg/m3; 1-methylphenanthrene,
2.7-21/0-7.0 µg/m3; fluoranthene, 45-430/0-24 µg/m3; pyrene,
35-320/0-14 µg/m3; benzo [a]fluorene, 9.7-90/0-6.8 µg/m3;
benzo [b]fluorene, 3.1-61/0-0.3 µg/m3; benzo [c]phenanthrene,
2.6-49 µg/m3 (particulate); benz [a]anthracene, 5.4-160/< 0.4-1.6
µg/m3; benzo [b]fluoranthene, 5.5-67/0-0.7 µg/m3;
benzo [j]fluoranthene plus benzo [k]fluoranthene, 0-35/0-0.7 µg/m3;
benzo [e]pyrene, 8-73/0-0.2 µg/m3; benzo [a]pyrene, 14-130/0-1.5
µg/m3; perylene, 3.3-19/0-0.1 µg/m3; benzo [ghi]perylene, 8.7-45
µg/m3 (particulate); anthanthrene, 2.6-62 µg/m3 (particulate); and
coronene, 1.0-19 µg/m3 (particulate) (IARC, 1984b).
At eight sites in a German coke plant in 1981, including the top of
the oven and the cabin of a lorry driver, the following PAH
concentrations were measured: 2.7 µg/m3 fluoranthene, 1.9-170 µg/m3
pyrene, 0.38-37 µg/m3 benzo [c]phenanthrene, 0.22-21 µg/m3
cyclopenta [cd]pyrene, 1.2-120 µg/m3 benz [a]anthracene, 0.71-79
µg/m3 benzo [c]pyrene, 0.88-89 µg/m3 benzo [a]pyrene, 0.21-14
µg/m3 perylene, 0.37-27 µg/m3 benzo [ghi]perylene, 0.18-17 µg/m3
anthanthrene, and 0.93-6.5 µg/m3 coronene. The authors pointed out
that the concentrations may have been much higher previously (Manz et
al., 1983).
Measurements with personal air samplers in Germany and Sweden showed
benzo [a]pyrene concentrations varying from 0.16-33 µg/m3 for
coke-oven operators to 4.7-17 µg/m3 for lorry drivers. The ranges of
exposure to all PAH at different workplaces in the 1970s were: lorry
driver, 170-1000 µg/m3; coke-car operator, 4.8-73 µg/m3; jamb
cleaner, 62-240 µg/m3; door cleaner, 9.1-17 µg/m3; push-car
operator, 9.4-62 µg/m3; sweeper, 110 µg/m3; quench-car operator, 5.7
µg/m3; and wharf man, 360 µg/m3 (IARC, 1984b).
Personal air samples taken from 56 Dutch coke-oven workers in 1986
showed pyrene levels of < 0.6 µg/m3 (detection limit) to 9.8 µg/m3
(Jongeneelen et al., 1990). The results of more recent measurements in
personal air samples are shown in Table 70.
5.3.1.2 Coal gasification and coal liquefaction
The levels of individual PAH in area air samples in Norwegian and
British coal gasification plants between the late 1940s and the mid
1950s were in the low microgram per cubic millilitre range. In modern
gasification systems, the concentrations of total PAH are usually <
1 µg/m3, but in one of three plants examined the total aerial PAH
load was about 30 µg/m3. Personal samples taken in modern coal
gasification plants showed similar PAH concentrations (IARC, 1984b).
Table 70. Workplace exposures to polycyclic aromatic hydrocarbons in the atmosphere of coke-oven batteries
(µg/m3), determined from personal air samples
Compound [1] [2] [3] [4] [5] [6] [7]
Acenaphthene 3.8
Acenaphthylene 28
Anthracene 65 16
Anthanthrene 2.4
Benz[a]anthracene 0.11-33.19 96 7.5
Benzo[a]fluorene 70 3.7
Benzo[a]pyrene < 0.01-31.15a 0.03-12.63 0.9-46.02 38 0.1-29 7.3 1300
0.01-22.91b
Benzo[b]fluoranthene 42 1500
Benzo[b]fluorene 4.3
Benzo[c]phenanthrene 1.4
Benzo[e]pyrene 4.7
Benzo[ghi]fluoranthene 1.6
Benzo[ghi]perylene 4.4
Benzo[k]fluoranthene 42
Chrysene 0.08-13.17 72
Coronene 3.2
Cyclopenta[cd]pyrene 1.9
Fluoranthene 0.12-17.00a 144 22 4400
Fluorene 109 14
Indeno[1,2,3-cd]pyrene 4.5
1-Methylphenanthrene 3.4
Naphthalene 28-445a 650
Perylene 1.8
Phenanthrene 0.07-8.53a 195 49
Pyrene 2.36-98.63 17 Trace
Table 70 (continued)
[1] Finland; samples from one plant, 1988-90 (Yrjanheikki et al., 1995);
[2] Italy; samples from 69 workers, six workplaces (Assennato et al., 1993a);
[3] Italy; samples from three workplaces at battery top (Cenni et al., 1993);
[4] Sweden; one typical sample (Andersson et al., 1983);
[5] United Kingdom; samples from 12 plants (Davies et al., 1986);
[6] USA; samples from topside coke-oven workers (Haugen et al., 1986,
[7] India; samples from top of coke oven (Rao et al., 1987)
a Area air samples
b Personal air samples
In a pilot coal liquefaction plant in the United Kingdom, monitoring
of five operators for vapour-phase PAH gave following results:
1900-3300 ng/m3 phenanthrene, 340-670 ng/m3 pyrene, 270-380 ng/m3
fluoranthene, 29-130 ng/m3 anthracene, 22-1700 ng/m3 fluorene,
< 1-1800 ng/m3 naphthalene, < 1-1000 ng/m3 acenaphthene, and
< 1-8 ng/m3 acenaphthylene. The higher-molecular-mass PAH were not
detected (limit of detection, 1 ng/m3). Pyrene was detected in the
particulate phase at concentrations of 630-2900 ng/m3 (Quinlan et
al., 1995a).
5.3.1.3 Petroleum refining
Personal samples from operators of catalytic cracker units and
reaction and fractionation towers in a petroleum refinery showed total
PAH levels of 2.6-470 µg/m3. During performance and turn-round
operations on reaction and fractionation towers, naphthalene and its
methyl derivatives accounted for more than 99% of the total PAH
measured; exposure to anthracene, pyrene, chrysene, and
benzo [a]pyrene was < 1 µg/m3. Area monitoring for these PAH
during normal activities and during shut-down, leak-testing, and
start-up operations after turn-rounds gave total PAH concentrations up
to 400 µg/m3, most of the measurements being < 100 µg/m3 (IARC,
1989b).
The results of personal air sampling of workers at six jobs in seven
American refineries in 1990-91 were as follows (mean and range): 5.5
(< 0.25-10) µg/m3 naphthalene, 3.3 (< 0.44-24) µg/m3 acenaphthene,
3.3 (< 0.19-26) µg/m3 acenaphthylene, 0.98 (< 0.085-7.9) µg/m3
fluoranthene, 0.82 (< 0.055-6.7) µg/m3 phenanthrene, 0.78
(< 0.13-5.3) µg/m3 benzo [e]pyrene, 0.65 (< 0.055-5.2) µg/m3
benzo [b]fluoranthene, 0.47 (< 0.14-2.7) µg/m3 fluorene, 0.29
(< 0.11-1.4) µg/m3 indeno[1,2,3- cd]pyrene, 0.18 (< 0.085-0.69)
µg/m3 benz [a]anthracene, 0.16 (< 0.11-< 0.59) µg/m3
benzo [a]pyrene, 0.063 (< 0.028-0.26) µg/m3 anthracene, < 0.11-
< 0.2 µg/m3 pyrene, < 0.085-< 0.15 µg/m3 chrysene, < 0.085-
< 0.15 µg/m3 benzo [k]fluoran-thene, < 0.11-< 0.2 µg/m3
benzo [ghi]perylene, and < 0.11-< 0.2 µg/m3
dibenz [a,h]anthracene. Dermal wipe samples from the back of the hand
or from the forehead of workers showed PAH levels of < 0.0011-0.29
µg/cm2, with the highest level for naphthalene and the lowest for
anthracene (Radian Corp., 1991).
5.3.1.4 Road paving
In early studies on road paving operations, the total PAH
concentrations reported in personal air samples were 4-190 µg/m3, and
the mean in area air samples was 0.13 µg/m3. The benzo [a]pyrene
concentration in stationary samples was < 0.05-0.19 µg/m3 (IARC,
1985).
The concentrations of individual PAH in fume condensates from paving
asphalt were generally < 2 mg/kg condensate, varying by about seven
times depending on the source of crude oil. The levels of
benzo [a]pyrene, for example, were between 0.09 and 2.0 mg/kg
(Machado et al., 1993).
Fourteen stationary air samples from a road paving site in New Zealand
in 1983 contained: 0.14-52 µg/m3 benz [a]anthracene plus chrysene,
0.2-14 µg/m3 benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, 0.15-9.0 µg/m3 benzo [a]pyrene, 0.31-5.4
µg/m3 benzo [e]pyrene, 0.039-2.2 µg/m3 perylene, 0.24-5.4 µg/m3
benzo [ghi]perylene, and 0.03-6.3 µg/m3 indeno[1,2,3- cd]pyrene
plus dibenz [a,h]anthracene (Swallow & van Noort, 1985). The
concentrations in 17 stationary air samples from a road paving
operation in New Zealand in another study (year not given) were:
1.2-18 µg/m3 benz [a]anthracene plus chrysene, 1.1-11 µg/m3
benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, 0.9-9.0 µg/m3 benzo [a]pyrene, 0.7-5.4
µg/m3 benzo [e]pyrene, and 0.7-6.3 µg/m3 indeno[1,2,3- cd]pyrene
(Darby et al., 1986). Concentrations of up to 1.3 µg/m3 were found
for acenaphthene, < 0.13 µg/m3 for anthracene, and < 0.54
µg/m3 pyrene in road-paving operations. The workers, and especially
the machine driver, were exposed to a mixture of bitumen fumes and
diesel exhaust gases for 4-6 h per day (Monarca et al., 1987).
The PAH concentrations in personal air samples obtained from workers
at six jobs in six paving operations in the USA in 1990 were (mean and
range): 6.5 (1.3-15) µg/m3 naphthalene, 2 (< 0.54-6.9) µg/m3
acenaphthene, 2 (< 0.24-8.1) µg/m3 acenaphthylene, 0.58
(< 0.19-0.98) µg/m3 fluorene, 0.55 (< 0.085-1.3) µg/m3
phenanthrene, 0.26 (< 0.11-0.37) µg/m3 fluoranthene, 0.17
(< 0.13-< 0.31) µg/m3 pyrene, 0.16 (< 0.13-0.27) µg/m3
benzo [e]pyrene, 0.13 (< 0.099-< 0.2) µg/m3 chrysene, 0.052
(< 0.034-0.11) µg/m3 anthracene, < 0.099-< 0.12 µg/m3
benz [a]anthracene, < 0.064-< 0.085 µg/m3 benzo [b]fluoranthene,
< 0.099-< 0.12 µg/m3 benzo [k]fluoranthene, < 0.13-< 0.25 µg/m3
benzo [a]pyrene, < 0.13-< 0.16 µg/m3 benzo [ghi]perylene,
< 0.13-< 0.16 µg/m3 indeno[1,2,3- cd]pyrene, and < 0.13-< 0.16
µg/m3 dibenz [a,h]anthracene. Dermal wipe samples from the back of
the hand and from the forehead of workers contained PAH at <
0.00004-0.43 µg/cm2, with the highest level for naphthalene and the
lowest for anthracene and pyrene (Radian Corp., 1991).
Measurements in the air in France during road paving with different
bitumens and tars showed the highest benzo [a]pyrene concentrations
with hard-coal tar (1-6 µg/m3) and the lowest with petroleum-based
bitumen (0.004-0.007 µg/m3). In general, the benzo [a]pyrene levels
in the workplace atmosphere were two to three orders of magnitude
higher during paving operations with tar products than with bitumen
products (Barat, 1991).
5.3.1.5 Roofing
The concentrations of PAH measured during roofing and roofing
manufacture are shown in Table 71.
The concentrations of individual PAH in fume condensates from roofing
asphalt generated at 232 and 316°C the were usually < 10 mg/kg
condensate, with higher levels only for naphthalene. They varied with
the source of crude oil: those for benzo [a]pyrene were between 0.6
and 2.8 mg/kg (Machado et al., 1993).
Acenaphthene was detected at concentrations of 1.4-2.1 µg/m3 in
personal samples from roofing workers at two US roofing sites in 1985
(Zey & Stephenson, 1986); 0.8-22 µg/m3 phenanthrene were measured at
one US roofing site in 1981 (Reed, 1983). Pyrene was measured at
< 190 µg/m3 at three roofing sites in Canada (year not given)
(Malaiyandi et al., 1986). Personal air samples from 12 roofers at one
US roofing site contained benzo [a]pyrene at 0.53-2.0 µg/m3 in 1987
(Herbert et al., 1990a). The workplace concentrations during bitumen
and coal-tar pitch roofing, waterproofing, and flooring operations
were of the same order of magnitude (IARC, 1985).
Significant, 10-fold differences were found in the levels of
anthracene, fluoranthene, pyrene, benz [a]anthracene,
benzo [b]fluoranthene, benzo [k]-fluoranthene, benzo [a]pyrene, and
benzo [ghi]perylene on skin wipes from the forehead taken before and
after a shift in 10 US roofers in 1987 (Wolff et al., 1989a).
Comparable results for benzo [a]pyrene levels were obtained for 12
roofers at another US roofing site (Herbert et al., 1990a,b).
Dermal wipe samples from the back of the hand or the forehead of
workers at six asphalt roofing manufacturing sites in the USA showed
PAH levels of < 0.12-5.5 µg/cm2, with the highest level for
acenaphthylene and the lowest for fluoranthene, benz [a]anthracene,
benzo [k]fluoranthene, and chrysene. Similar samples from workers at
six asphalt roofing sites in the USA in 1990-91 showed PAH levels of
< 0.0011-0.0045 µg/cm2, with the highest levels for pyrene,
chrysene, and benzo [a]pyrene and the lowest for anthracene (Radian
Corp., 1991).
5.3.1.6 Impregnation of wood with creosotes
Concentrations of PAH ranging from 0.05 µg/m3 benzo [a]pyrene to
650 µg/m3 naphthalene were detected during the handling of
creosote-impregnated wood for railroad ties in Sweden. Naphthalene,
fluorene and phenanthrene were by far the most abundant compounds
(> 100 µg/m3) (Andersson et al., 1983). Concentrations of 0.04-0.28
µg/m3 anthracene and 0.11-7.7 µg/m3 pyrene were found at workplaces
in Finland where railroad ties were manufactured (Korhonen & Mulari,
1983), and concentrations of 1-19 µg/m3 anthracene, 6.5-61 µg/m3
phenanthrene, and 0.6-13 µg/m3 pyrene were measured in one plant
where railroad sleepers were impregnated and in another where poles
Table 71. Exposure to polycyclic aromatic hydrocarbons (µg/m3) during roofing and roofing manufacture
Compound [1] [2] [3] [4]
Acenaphthene < 0.52-3.2 (0.87) < 0.6-6.7 (1.5)
Acenaphthylene < 0.23-29 (7.1) < 0.26-12 (2.9)
Anthracene 0.5/1.5 < 0.033-0.069 (0.043) < 0.037-0.042
Anthanthrene < 0.030
Benz[a]anthracene < 0.03-0.130 1.3/2.5 < 0.099-< 0.13 < 0.11-< 0.13
Benzo[a]fluorene 0.03-0.080
Benzo[a]pyrene < 0.03-0.037 0.9/1.5 < 0.13-< 0.18 < 0.11-< 0.13
Benzo[b]fluoranthene < 0.03-0.093a 0.8/1.2 < 0.065-< 0.38 (0.13) < 0.078-< 0.085
Benzo[b]fluorene 0.051-0.093
Benzo[e]pyrene < 0.03-0.110 < 0.13-3 (0.61) < 0.15-< 0.17
Benzo[ghi]fluoranthene < 0.03
Benzo[ghi]perylene < 0.03-0.069 0.6/0.9 < 0.13-< 0.18 < 0.15-< 0.17
Benzo[k]fluoranthene 0.4/0.7 < 0.099-< 0.13 < 0.099-< 0.12
Chrysene 0.038-0.214 < 0.099-< 0.13 < 0.11-< 0.13
Coronene < 0.03
Dibenz[a,h]anthracene < 0.03 < 0.13-< 0.18 < 0.15-< 0.17
Fluoranthene 0.084-0.234 3.1/7 < 0.099-4 (0.64) < 0.11-0.13
Fluorene < 0.16-14 (2.5) < 0.19-1.1 (0.44)
Indeno[1,2,3-cd]pyrene < 0.030 < 0.13-< 0.18 < 0.15-0.94 (0.16)
Naphthalene < 0.22-9.2 (5.2) 1.2-25 (7.5)
Perylene < 0.030
Phenanthrene < 0.065-1.7 (0.53) < 0.078-1.4 (0.38)
Pyrene 0.035-0.183 2.6/5.4 < 0.13-3.4 (0.76) < 0.15-< 0.73 (0.25)
/, single determinations; mean values shown in parentheses;
[1] Germany; personal and area air samples from one bitumen roofing site (Schmidt, 1992);
[2] USA; personal air samples from nine workers; 1987 (Wolff, M.S. et al., 1989);
[3] USA; personal air samples from six asphalt roofing sites; 1990 (Radian Corp., 1991);
[4] USA; personal air samples from six roofing manufacturing sites; 1990 (Radian Corp., 1991)
a Benzo[b+j+k]fluoranthenes
were preserved (year not given) (Heikkilä et al., 1987). In
measurements of personal air samples from 10 workers in a Dutch plant
for impregnation of railroad sleepers in 1991, 0.3-1.3 µg pyrene/m3
was measured in the breathing zone and 47-1500 µg/d in pads placed on
various areas of the skin of the workers. Dermal exposure was shown to
be reduced by up to 90% by the use of protective clothing (Van Rooij
et al., 1993b).
5.3.1.7 Other exposures
In area air samples taken near the bitumen processing devices of
refineries, the total PAH levels varied from 0.004 to 50 µg/m3 (IARC,
1985, 1989b).
The use of lubricating oils may result in exposure to PAH. At two
Italian glass manufacturing plants, phenanthrene, anthracene, pyrene,
and fluoranthene were found in personal air samples at concentrations
< 3 µg/m3 (year not given) (Menichini et al., 1990). The pyrene
levels resulting from use of lubricating oils in Italian earthenware
factories were 0.02-0.09 µg/m3; the benzo [a]pyrene concentration
was below the limit of detection (Cenni et al., 1993). Measurable
concentrations of individual PAH were detected in indoor air above
asphalt floor tiles in e.g. warehouses, factories, and manufacturing
plants. The concentrations at six sampling sites in Germany were
between < 0.01 ng/m3 for benzo [ghi]perylene and 3.3 ng/m3 for
chrysene. The concentrations of phenanthrene, pyrene, fluoranthene,
chrysene, and benzo [b]fluorene in particular were higher than those
in outdoor air (Luther et al., 1990).
In two Swiss plants for the production of silicon carbide, personal
air samples from four and five workers, respectively, contained the
following PAH levels: 4-140 ng/m3 acenaphthylene, 8-86 ng/m3
acenaphthene, 11-500 ng/m3 fluorene, 88-1400 ng/m3 phenanthrene,
3-250 ng/m3 anthracene, 20-1100 ng/m3 fluoranthene, 30-2500 ng/m3
pyrene, 7-6400 ng/m3 benz [a]-anthracene, 37-14 000 ng/m3 chrysene,
11-3700 ng/m3 benzo [b]fluoranthene plus benzo [j]fluoranthene,
3-470 ng/m3 benzo [k]fluoranthene, 18-3800 ng/m3 benzo [e]pyrene,
4-630 ng/m3 benzo [a]pyrene, 2-250 ng/m3 indeno[1,2,3- cd]pyrene,
2-520 ng/m3 dibenz [a,h]anthracene, 4-550 ng/m3
benzo [ghi]-perylene, and 4-34 ng/m3 coronene (Petry et al., 1994).
5.3.2 Occupational exposure resulting from incomplete combustion of
mineral oil, coal, and their products
5.3.2.1 Aluminium production
Early measurements of atmospheric benzo [a]pyrene at workplaces in
the aluminium industry showed concentrations of 0.02-970 µg/m3 in
personal air samples and 0.03-5.3 µg/m3 in area air samples. In the
atmosphere of an aluminium production plant, naphthalene, fluorene,
phenanthrene, anthracene, fluoranthene, pyrene, benzo [a]fluorene,
benzo [b]fluorene, benzo [c]phenan-threne, benz [a]anthracene,
chrysene, triphenylene, benzo [b]fluoranthene plus
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene,
benzo [ghi]perylene, anthanthrene, and coronene were found at
concentrations < 400 µg/m3. The most abundant compounds were
phenanthrene, naphthalene, fluorene, fluoranthene, and pyrene, at
concentrations > 100 µg/m3. The other substances occurred at
concentrations < 10 µg/m3 (IARC, 1984b).
The following concentrations of PAH were found in four stationary air
samples from an aluminium smelter in New Zealand in 1979: 0.37-9.6
µg/m3 benz [a]anthracene plus chrysene, 0.34-7.6 µg/m3
benzo [b+j+k]fluoranthenes, 0.12-2.6 µg/m3 benzo [e]pyrene,
0.19-4.1 µg/m3 benzo [a]pyrene, 0.05-1.5 µg/m3 perylene, 0.13-2.7
µg/m3 indeno[1,2,3- cd]pyrene plus dibenz [a,h]anthracene, and
0.12-3.3 µg/m3 benzo [ghi]perylene (Swallow & van Noort, 1985).
Similar levels were found in a typical personal air sample from a
Söderberg aluminium plant in Sweden (year not given) with, in
addition, 27 µg/m3 phenanthrene, 20 µg/m3 fluoranthene, 2.8 µg/m3
fluorene, 2.8 µg/m3 anthracene, 2.8 µg/m3 benzo [a]fluorene, and
< 1.0 µg/m3 naphthalene (Andersson et al., 1983).
In personal air samples from 38 workers in the Söderberg potroom of an
aluminium smelter in the humid tropics (location not given), mean
concentrations of < 1.0-48 µg/m3 benzo [a]pyrene and 3.5-130 µg/m3
pyrene were detected (Ny et al., 1993).
The arithmetic mean concentrations of PAH in workplace air samples
from the Canadian aluminium industry were 1100 µg/m3 naphthalene, 130
µg/m3 acenaphthene, 45 µg/m3 fluorene, 30 µg/m3 phenanthrene, 4.5
µg/m3 anthracene, 1.1 µg/m3 fluoranthene, and 0.58 µg/m3 pyrene.
The concentrations of benz [a]anthracene, chrysene, benzo [a]pyrene,
and benzo [e]pyrene were < 0.01 µg/m3 (Lesage et al., 1987).
Personal air samples from 18 workers in a US plant producing anodes
for use in aluminium reduction (year not given) showed pyrene
concentrations of 1.2-7.4 µg/m3 (Tolos et al., 1990).
Urine samples from 11 workers in Norwegian Söderberg aluminium plants
contained very low levels of unchanged PAH, although the
concentrations in the workplace air greatly exceeded the
concentrations in urban air. The total concentration of PAH
metabolites in the samples was 1.5-6 greater than that in a control
group (Becher & Bjrseth, 1983).
The PAH concentrations in the air of aluminium plants is reduced
dramatically by the use of tempered anodes instead of Söderberg
anodes. Measurements of benzo [a]pyrene levels in French factories
showed 1-36 µg/m3 in potrooms with Söderberg anodes and 0.004-0.6
µg/m3 in potrooms with tempered anodes (Barat, 1991).
5.3.2.2 Foundries
In personal air samples from workers in 10 Canadian foundries, mean
concentrations of 0.14-1.8 µg/m3 benz [a]anthracene plus chrysene,
0.09-1.2 µg/m3 benzo [a]pyrene, and 0.09-1.9 µg/m3
dibenz [a,h]anthracene were measured. The benzo [a]pyrene levels in
stationary air samples from six Finnish foundries were 0.01-13 µg/m3,
depending on whether coal-tar pitch or coal powder was used as the
moulding sand additive (IARC, 1984b).
In another study, the highest individual PAH levels were found in coke
making, moulding, and furnaces (Gibson et al., 1977). Personal air
samples from 67 Finnish foundry workers in 1990-91 showed
benzo [a]pyrene concentrations of 2-60 ng/m3 with a mean of 8.6
ng/m3 (Perera et al., 1994). Depending on the foundry process and
sand binder, the mean benzo [a]pyrene level in 29 French foundries
varied from 3 to 2300 ng/m3 (Lafontaine et al., 1990).
Concentrations of PAH measured in foundries are shown in Table 72.
5.3.2.3 Other workplaces
Personal air samples from German chimney sweeps (year not given; 115
samples) showed an average benzo [a]pyrene level of 0.09 µg/m3, but
eight of the samples exceeded 2 µg/m3. With an inhaled air volume of
10 m3 per working day, the daily intake of benzo [a]pyrene was
estimated to be 0.24-2.7 µg, with a median value of 1.3 µg (Knecht et
al., 1989).
In an Italian pyrite mine, pyrene levels of 0.03-0.21 µg/m3 were
measured in personal and area air samples. The benzo [a]pyrene
concentrations were below the limit of detection (Cenni et al., 1993).
Area air samples taken in China showed total PAH levels of 3-40 µg/m3
in two iron mines and 4-530 µg/m3 in four copper mines. Individual
compounds were not identified, but the main components were
naphthalene and acenaphthene in the iron mines and naphthalene,
benz [a]anthracene, benzo [b]fluoranthene, benzo [a]pyrene,
benzo [e]pyrene, and dibenz [a,h]anthracene in the copper mines. The
PAH concentrations probably resulted from the drilling of holes with
hydraulic or pneumatic drills and by the transport of broken ore in
diesel-powered scoops (Wu et al., 1992).
Area and personal air samples from workers in a railway tunnel in
Italy showed pyrene levels of 0.04-0.30 µg/m3. The benzo [a]pyrene
concentrations ranged from below the limit of detection to 0.04 µg/m3
(Cenni et al., 1993).
Table 72. Exposure to polycyclic aromatic hydrocarbons (µg/m3)
in the atmosphere of foundries
Compound [1] [2] [3]
Acenaphthene 0.03
Acenaphthylene ND
Anthracene 2.31 0.05
Anthanthrene 0.64
Benz[a]anthracene 0.008-0.221 0.67 0.01
Benzo[a]fluorene 0.48
Benzo[a]pyrene 0.049-0.152 0.47 0.02
Benzo[b]fluoranthene 0.87a 0.003
Benzo[b]fluorene 0.41
Benzo[e]pyrene 0.48
Benzo[ghi]fluoranthene 0.15
Benzo[ghi]perylene 0.72 0.05
Benzo[k]fluoranthene 0.037-0.458 0.02
Chrysene 0.82b 0.02
Coronene 0.21
Dibenz[a,h]anthracene 0.20 ND
Fluoranthene 1.56 0.13
Fluorene 0.08
Indeno[1,2,3-cd]pyrene 0.81 ND
Naphthalene 9.68
Perylene 0.21
Phenanthrene 4.46 0.32
Pyrene 1.74 0.01
ND, not detected; /, single measurements;
[1] Canada, steel foundry: coke making, moulding, furnaces,
finishing, and cranes (Gibson et al., 1977);
[2] Western Germany, one foundry, area air samples (Knecht et al.,
1986);
[3] Denmark, 70 workers, personal air samples; melting, machine
moulding, casting, sand preparation (Omland et al., 1994)
a In sum with benzo(j+k)fluoranthene
b In sum with triphenylene
In the air of fish and meat smokehouses in Denmark (year not given),
the maximum concentration of naphthalene in stationary air samples was
about 2900 µg/m3. The most abundant compounds were naphthalene,
phenanthrene, pyrene, fluorene, anthracene, and fluoranthene
(> 100 µg/m3) (Nordholm et al., 1986). The minimal values were
< 1 µg/m3, benzo [a]pyrene being detected at minimal levels of
0.08 µg/m3 in meat smokehouses and 0.4 µg/m3 in fish smokehouses
(Hansen et al., 1991b), with a maximum concentration of 78 µg/m3
(Nordholm et al., 1986).
In a further study in nine Danish meat smokehouses, naphthalene was
detected at 21 µg/m3, fluorene at 6.9 µg/m3, fluoranthene at 6.6
µg/m3, phenanthrene at 5.6 µg/m3, acenaphthene at 5.2 µg/m3,
chrysene at 1.2 µg/m3, anthracene at 1.1 µg/m3, pyrene at 0.2
µg/m3, and benzo [ghi]perylene at 0.2 µg/m3 (Hansen et al., 1992).
The concentrations of naphthalene, fluorene, anthracene, phenanthrene,
pyrene, benzo [a]fluorene, chrysene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [e]pyrene, benzo [ghi]perylene, and
dibenz [a,h]anthracene in cooking fumes in a Finnish food factory,
three restaurants, and one bakery (year not given) during the frying
of meat and during deep-frying ranged between < 0.02 µg/m3 (the
limit of detection) and 26 µg/m3. Naphthalene occurred at by far the
highest concentration. Stationary air was sampled as close as possible
to the active working area and the workers' breathing zone (Vainiotalo
& Matveinen, 1993).
6. KINETICS AND METABOLISM IN LABORATORY MAMMALS AND HUMANS
Appraisal
Polycyclic aromatic hydrocarbons (PAH) are lipophilic compounds and
can be absorbed through the lungs, the gastrointestinal tract, and the
skin. In studies of the distribution of PAH in rodents, both the
parent compounds and their metabolites were found in almost all
tissues and particularly those rich in lipids. As a result of
mucociliary clearance and hepatobiliary excretion, they were present,
for example, in the gastrointestinal tract even when administered by
other routes.
The metabolism of PAH to more water-soluble derivatives, which is a
prerequisite for their excretion, is complex. Generally, the process
involves epoxidation of double bonds, a reaction catalysed by
cytochrome P450-dependent mono-oxygenases, rearrangement or hydration
of the epoxides to yield phenols or diols, respectively, and
conjugation of the hydroxylated derivatives. The reaction rates vary
widely: interindividual variations of up to 75-fold have been
observed, for example, with human macrophages, mammary epithelial
cells, and bronchial explants from different donors.
All aspects of the absorption, metabolism, activation, and excretion
of benzo[a]pyrene have been covered exhaustively in the published
literature, but there is a dearth of information on many of the other
PAH considered in this publication, particularly in humans. Thus, this
overview sets out general principles and describes pathways relevant
to benzo[a]pyrene in greater detail.
Most biotransformation leads to detoxification products that are
conjugated and excreted in the urine and faeces. The human body burden
of PAH has not been extensively studied, but tissue samples taken at
autopsy were found in one study to contain benzo[a]pyrene at an
average of 0.3 µg/100 g dry tissue; lung contained 0.2 µg/100 g. In
contrast, the pathways by which several PAH are metabolized to
reactive intermediates that bind covalently to nucleic acids have been
examined in great detail. Although the commonest mechanism in animals
and humans appears to involve the formation of diol epoxides, radical
cations and sulfate esters of hydroxymethyl derivatives may also be
important in certain cases.
6.1 Absorption
PAH are lipophilic compounds, soluble in organic solvents, that are
usually devoid of ionizable or polar groups. Like many other
xenobiotic substances, they would be expected to dissolve readily in,
and be transported through, the external and internal lipoprotein
membranes of mammalian cells. This is confirmed by the uptake of PAH
in vitro from media in which cells are maintained in culture and
modified metabolically by enzymes of the endoplasmic reticulum.
Furthermore, PAH are known to be able to cause biological effects
in vivo in cells and tissues that are distant from their site of
uptake by the organism.
In humans, the major routes of uptake of PAH are thought to be through
(i) the lungs and the respiratory tract after inhalation of
PAH-containing aerosols or of particulates to which a PAH, in the
solid state, has become absorbed; (ii) the gastrointestinal tract
after ingestion of contaminated food or water; and (iii) the skin as a
result of contact with PAH-bearing materials.
6.1.1 Absorption by inhalation
Investigations of the pulmonary absorption of PAH have frequently been
clouded by the existence of the mucociliary clearance mechanism, by
which hydrocarbons absorbed onto particulates that have been inhaled
are swept back up the pulmonary tree and are swallowed, thus entering
the organism through the gastrointestinal tract. Use of isolated
perfused rat lungs, however, provided a clear demonstration that
benzo [a]pyrene is absorbed directly through the pulmonary epithelia.
After intratracheal administration, both the hydrocarbon and its
metabolites were detected in effluent perfusion fluid (Vainio et al.,
1976). Other studies have shown that benzo [a]pyrene administered
in vivo as an aerosol is cleared from the lungs of rats by a
biphasic process in which an initial rapid phase (tracheal clearance)
is followed by a much slower second phase (alveolar clearance)
(Mitchell, 1982). PAH absorbed onto particles may take very much
longer to be cleared from rodent lungs, however, than the free
hydrocarbons, and the factors that affect this clearance rate include
the structure of the hydrocarbon and the dimensions and chemical
nature of the particles onto which the PAH are absorbed (Henry &
Kaufman, 1973; Creasia et al., 1976; Nagel et al., 1976). For example,
while 50% of the benzo [a]pyrene coated onto carbon particles of
15-30 µm was cleared from hamster lungs within 60 h, it took only 10 h
to clear 50% of the benzo [a]pyrene that had been coated onto
0.5-1.0-µm carbon particles. In a comparable experiment, however, when
ferric oxide particles of either 0.5-10 or 15-20 µm were used as
carriers for benzo [a]pyrene, 50% of the hydrocarbon was cleared in
just over 2 h, and carrier particle size did not affect the clearance
rates (Henry & Kaufman, 1973).
Benzo [a]pyrene was metabolized by the epithelia lining the nasal
cavities of hamsters, dogs, and monkeys when 14C-labelled hydrocarbon
was instilled as an aqueous suspension (Dahl et al., 1985;
Petridou-Fischer et al., 1988). From their studies with hamsters, the
authors concluded that when frequent small doses of 650 ng at 10-min
intervals were instilled into the nasal cavity, so as to imitate
inhalation, some 50% of the benzo [a]pyrene was metabolized; a large
fraction of the metabolites could be recovered from the mucus on the
epithelial surfaces; and the nasal epithelia were comparable to those
of the trachea and lungs in their ability to metabolize
benzo [a]pyrene. Metabolites produced nasally would be expected to be
swallowed and then absorbed in the gastrointestinal tract.
In humans, the concentrations of benzo [a]pyrene and pyrene present
in association with soot particles in the lungs were much lower than
would have been expected from the soot content. Thus, only a trace of
benzo [a]pyrene was found in one of 11 lung samples examined, in
which the expected benzo [a]pyrene content ranged from 9 to 200 µg;
in the other 10 samples, no benzo [a]pyrene was detected. Pyrene
disappeared more slowly: all 11 lung samples contained the compound,
at levels of 0.9-4.9 µg, whereas 3-190 µg might have been expected
(Falk et al., 1958). The ability of pulmonary epithelial cells to
metabolize PAH such as chrysene and benzo [a]pyrene to a variety of
hydroxylated derivatives (Jacob et al., 1992) may facilitate the
absorption and clearance of PAH from the lungs.
6.1.2 Absorption in the gastrointestinal tract
Indirect evidence for the gastrointestinal absorption of PAH was
provided by Shay et al. (1949), who found that repeated intragastric
instillation of 3-methylcholanthrene led to the development of mammary
cancer. Mammary tumours can also be induced in rats by intracolonic
adminstration of 7,12-dimethylbenz [a]anthracene (Huggins et al.,
1961). (3-Methylcholanthrene and 7,12-dimethylbenz [a]anthracene are
synthetic PAH that are potent carcinogens.) More direct investigations
by Rees et al. (1971) showed rapid absorption of intragastrically
administered benzo [a]pyrene; the highest levels of hydrocarbon were
found in the thoracic lymph some 3-4 h after administration. In a
report of studies of intact rats and intestinal sacs to examine the
mechanisms involved in benzo [a]pyrene absorption, Rees et al. (1971)
proposed that two sequential steps were involved, in which a phase of
absorption by the mucosa is followed by diffusion through the
intestinal lining. In a study with Sprague-Dawley rats, the presence
of bile was found to increase intestinal absorption of PAH such as
benzo [a]pyrene and 7,12-dimethylbenz [a]anthracene to a greater
degree than that of anthracene and pyrene. The effect may be related
to differences in the aqueous solubility of the PAH examined (Rahman
et al., 1986). The composition of the diet also affects intestinal
absorption of co-administered benzo [a]pyrene. Of the dietary
components studied, soya bean oil and triolein gave rise to the
highest levels of absorption of 14C-benzo [a]pyrene given orally at
a dose of 8.7 µg to Wistar rats, while cellulose, lignin, bread, rice
flake, and potato flake suppressed it (Kawamura et al., 1988).
6.1.3 Absorption through skin
PAH and PAH-containing materials have been applied dermally in
solution in solvents such as acetone and tetrahydrofuran. Dermal
transfer without use of a solvent was achieved by use of reconstituted
vapour-particulate phases emitted from coal-tar and bitumen (Genevois
et al., 1995) and by application in oil (Ingram et al., 1995).
Absorption of PAH through the skin was observed indirectly when it was
found that repeated topical application of 3-methylcholanthrene led to
the appearance of mammary tumours in mice (Maisin & Coolen, 1936;
Englebreth-Holm, 1941). The percutaneous mechanism of absorption is
not universal, however, since although almost all of a dose of
14C-benzo [a]pyrene applied to mouse skin appeared in the faeces
within two weeks, very little dibenz [a,h]anthracene was absorbed in
this way and most was lost through epidermal sloughing (Heidelberger &
Weiss, 1951). Benzo [a]pyrene has been shown to be absorbed
percutaneously in vitro, by absorption from soil into human skin
(Wester et al., 1990) and, after application as a solution in acetone,
into discs of human, mouse, marmoset, rat, rabbit, and guinea-pig skin
(Kao et al., 1985). In the latter experiments, marked interspecies
differences were noted: 10% of the applied dose (10 µg/5 cm2) of
14C-benzo [a]pyrene permeated mouse skin, 3% crossed human skin, and
< 0.5% crossed guinea-pig skin within 24 h. It was concluded that
both diffusional and metabolic processes are involved in the
percutaneous absorption of benzo [a]pyrene.
In Wistar rats that received 14C-pyrene as a solution in acetone on
areas of shaved dorsal skin, the rate of uptake was relatively rapid
(half-life, 0.5-0.8 d). The concentrations of pyrene were highest in
the liver, kidneys, and fat, but those of pyrene metabolites were
highest in the lungs. About 50% of an applied dose of 2, 6, or 15
mg/kg bw was excreted in the urine and faeces during the first six
days after treatment (Withey et al., 1993).
In studies with 32P-postlabelling for the detection of DNA adducts,
when complex mixtures of PAH, such as that present in used lubricating
oil from petrol engines, in coal-tar, or in juniper-tar, were applied
directly to mouse skin, appreciable, persistent levels of DNA adducts
(50-750 amol/µg DNA [1 amol/µg DNA equivalent to 3.3 adducts/1010
nucleotides]) were formed in the lungs (Schoket et al., 1989, 1990).
The level of adducts in mouse skin was inversely related to the
viscosity of the oil applied (Ingram et al., 1995).
Evidence for percutaneous absorption of PAH has also been obtained in
humans in vivo. When 2% coal-tar in petroleum jelly was applied
topically, phenanthrene, anthracene, pyrene, and fluoranthene were
detected in peripheral blood samples (Storer et al., 1984). In
addition, volunteers treated topically with creosote (100 µl) or
pyrene (500 µg, applied as a solution in toluene) and a psoriasis
patient who used a coal-tar shampoo excreted 1-hydroxypyrene in their
urine. In each case, maximal excretion occurred 10-15 h after
treatment (Viau & Vyskocil, 1995).
6.2 Distribution
The whole-body distribution of PAH has been studied in rodents. The
levels found in individual tissues depend on a number of factors,
including the PAH, the route of administration, the vehicle, the times
after treatment at which tissues are assayed, and the presence or
absence of inducers or inhibitors of hydrocarbon metabolism within the
organism. The investigations have shown that (i) detectable levels of
PAH occur in almost all internal organs, (ii) organs rich in adipose
tissue can serve as storage depots from which the hydrocarbons are
gradually released, and (iii) the gastrointestinal tract contains high
levels of hydrocarbon and metabolites, even when PAH are administered
by other routes, as a result of mucociliary clearance and swallowing
or hepatobiliary excretion (Heidelberger & Jones, 1948; Heidelberger &
Weiss, 1951; Kotin et al., 1959; Bock & Dao, 1961; Takahashi &
Yasuhira, 1973; Takahashi, 1978; Mitchell, 1982).
14C-Benzo [a]pyrene injected intravenously at 11 µg/rat was cleared
rapidly from the bloodstream, with a half-life of < 1 min (Kotin et
al., 1959), as confirmed by Schlede et al. (1970a,b), who also noted
that the rate of clearance was increased when animals were pretreated
with 20 mg/kg bw non-radioactive benzo [a]pyrene or 37 mg/kg bw
phenobarbital, both of which can induce metabolism.
The distribution of 3-methylcholanthrene in mice and their fetuses was
studied by whole-body autoradiography. When 1 mg of 14C-labelled
hydrocarbon is injected intravenously, it is not only widely
distributed in maternal tissues but also crosses the placenta and can
be detected in the fetuses (Takahashi & Yasuhira, 1973; Takahashi,
1978), in which it induces pulmonary tumours (Tomatis, 1973; see also
Section 7). The distribution of inhaled and intragastrically or
intravenously administered benzo [a]pyrene and
7,12-dimethylbenz [a]anthracene in rats and mice has also been
studied, with similar results (Shendrikova & Aleksandrov, 1974;
Shendrikova et al., 1973, 1974; Neubert & Tapken, 1988; Withey et al.,
1992). Rapid transfer of radioactive benzo [a]pyrene across the
placenta was confirmed in experiments in which the appearance of
radioactivity in the umbilical vein of pregnant guinea-pigs was
measured (Kelman & Springer, 1982).
Samples of placenta, maternal blood, umbilical cord blood, and milk
from 24 women in south India were examined for the presence of
selected PAH. Although umbilical cord blood and milk showed the
highest levels (benzo [a]pyrene, 0.005-0.41 ppm;
dibenz [a,c]anthracene, 0.013-0.60 ppm; chrysene, 0.002-2.8 ppm),
only 50% of the samples examined contained detectable levels. The
authors concluded that developing fetuses and newborn infants were
exposed to these PAH, probably from the maternal diet (Madhavan &
Naidu, 1995).
After intratracheal administration to mice and rats, the distribution
of PAH was essentially similar to that found after intravenous or
subcutaneous injection (Kotin et al., 1959), except for the expected
high pulmonary levels. Detailed time-concentration curves for several
organs have been obtained after inhalation of 3H-benzo [a]pyrene
aerosols at 500 µg/litre of air (Mitchell, 1982). For example, 1 h
after the end of administration, the highest levels were present in
the stomach and small intestine; as these declined, the amounts of
radioactivity in the large intestine and caecum increased. The
elimination half-times in the respiratory tract were 2-3 h for the
initial rapid phase and 25-50 h for the subsequent slow phase.
6.3 Metabolic transformation
The metabolism of PAH follows the general scheme of xenobiotic
metabolism originally outlined by Williams (1959). The hydrocarbons
are first oxidized to form phase-I metabolites, including primary
metabolites, such as epoxides, phenols, and dihydrodiols, and then
secondary metabolites, such as diol epoxides, tetrahydrotetrols, and
phenol epoxides. The phase-I metabolites are then conjugated with
either glutathione, sulfate, or glucuronic acid to form phase-II
metabolites, which are much more polar and water-soluble than the
parent hydrocarbons.
The metabolism of PAH has been studied in vitro, usually in
microsomal fractions prepared from rat liver, although many other
tissue preparations have also been used. Metabolism in such systems
might be expected to be simpler than that in whole animals because the
enzymes and co-factors necessary for sulfate, glutathione, or
glucuronide conjugate formation may be removed, depleted, or diluted
during tissue fractionation. Use of these systems appears to be
justified, however, because the same types of phase-I metabolites are
formed when animals are treated with simple hydrocarbons such as
naphthalene as when the same hydrocarbon is incubated with hepatic
microsomes or tissue homogenates (Boyland et al., 1964). The
metabolism of PAH has thus been studied extensively in cells and
tissues in culture, which metabolize hydrocarbons to both phase-I and
phase-II metabolites and which probably better represent the
metabolism of PAH that occurs in vivo (for reviews see Conney, 1982;
Cooper et al., 1983; Dipple et al., 1984; Hall & Grover, 1990; Shaw &
Connell, 1994).
Particular attention has been paid to the metabolism of PAH in human
tissues that might be exposed to hydrocarbons present in food and in
the environment and which are, therefore, potential targets for the
carcinogenic action of PAH (Autrup & Harris, 1983). The cells and
tissues examined include the bronchus, the colon, mammary cell
aggregates, keratinocytes, monocytes, and lymphocytes. The metabolism
of PAH by human pulmonary macrophages has also received attention
(Autrup et al., 1978a; Harris et al., 1978a; Marshall et al., 1979)
because it is conceivable that metabolism by these cells might be
responsible, at least in part, for the high incidence of bronchial
cancer in smokers (Wynder et al., 1970). Macrophages can engulf
particulate matter that reaches the terminal airways of the lung and
thus would be expected, especially in smokers, to contain PAH
(Hoffmann et al., 1978). The macrophages and engulfed particulate
matter can then be transported to the bronchi where proximate and
ultimate carcinogens, formed by metabolism in the macrophages, could
leave the macrophages and enter the epithelial cells lining the
bronchi (Autrup et al., 1978a; Harris et al., 1978a). This is an
attractive theoretical mechanism which could account for the high
incidence of respiratory tumours at the junctions of the large bronchi
and which is supported by experimental evidence.
Extracts of organic material from isolated perfused lung tissues of
rabbits that had been exposed intratracheally to benzo [a]pyrene with
or without ferric oxide were analysed for benzo [a]pyrene metabolites
and for mutagenicity. Extracts of lung tissue exposed to
benzo [a]pyrene only were mutagenic and contained benzo [a]pyrene
metabolites. When ferric oxide was co-administered, only the
macrophage extracts were mutagenic, owing to relatively large amounts
of unmetabolized benzo [a]pyrene. These experiments demonstrate that
ferric oxide particles enhance the uptake of benzo [a]pyrene by lung
macrophages and slow its metabolism beyond the 3-h period during which
perfused lung systems can be maintained (Schoeny & Warshawsky, 1983).
Administration of particles in vitro enhances both the uptake and
metabolism of benzo [a]pyrene by hamster alveolar macrophages (Griefe
et al., 1988). Metabolites were found in both the cells and the
culture medium. Subsequent studies showed that concurrent
administration of benzo [a]pyrene and ferric oxide particles resulted
in increased benzo [a]pyrene metabolism and release of superoxides
(Greife & Warshawsky, 1993). In particular, the dihydrodiol fraction
was increased. These studies indicate that particulates may act in
lung cancer by changing the time frame for metabolism, shifting the
site of metabolism to macrophages and enhancing the production of
metabolites that are on the pathway to putative ultimate carcinogenic
forms. In this context, it has been demonstrated that particles of
various sorts exert different toxic effects on rat and hamster
pulmonary macrophages in vitro: ferric oxide and aluminium oxide
particulates were toxic, while crystalline silica was not (Warshawsky
et al., 1994).
The conclusion that the macrophage is the principal metabolizing cell
is further supported by the studies of Ladics et al. (1992a,b), who
demonstrated that the macrophage population was the only one in murine
spleen that could metabolize benzo [a]pyrene, while the other splenic
cell types examined, including B cells, T cells, polymorphonuclear
cells, and the splenic capsule, did not produce benzo [a]pyrene
metabolites above the background level.
Although the same types of metabolite are formed from PAH in many of
the cell and tissue preparations examined in culture, the relative
levels and the rates of formation of these metabolites depend on the
type of tissue or cell that is being studied and on the species and
strain of animal from which the metabolizing systems are prepared.
With heterogeneous populations such as humans, the rate of metabolism
depends on the individual from whom the tissues or cells are prepared.
For example, a 75-fold variation in the extent of hydrocarbon
activation was reported in studies of human bronchus (Harris et al.,
1976), and similar variations were observed among human mammary cell
aggregates (Grover et al., 1980; MacNicoll et al., 1980) and
macrophages (Autrup et al., 1978a). The pattern and role of metabolism
can also be varied by adding inhibitors of the enzymes that are
responsible for metabolism or by pretreating either cells in culture
or the animals from which the metabolizing systems are prepared with
enzyme inducers.
6.3.1 Cytochromes P450 and metabolism of PAH
The cytochromes P450 (CYP) are a superfamily of haemoproteins that
catalyse the oxidation of various endogenous molecules as well as
xenobiotics, including PAH. To date, about 250 genes that encode these
enzymes have been identified in various organisms. For classification
purposes, the CYP have been organized into families and subfamilies
according to their structural homology (Nelson et al., 1993).
Certain CYP belonging to families 1, 2, and 3 are expressed in
mammalian cells and are particularly important in xenobiotic
metabolism, and one or more member of each family is capable of
metabolizing one or more PAH (Guengerich & Shimada, 1991; Gonzalez &
Gelboin, 1994). Most studies to compare the catalytic properties of
different CYP have been carried out with model compounds such as
benzo [a]pyrene. They show that the catalytic properties (e.g. the
Vmax) of different CYP in PAH metabolism can differ essentially
(Shou et al., 1994).
In considering the contribution of a CYP enzyme to PAH metabolism
in vivo, two other parameters in addition to the catalytic
properties should be taken into account: the mode of regulation and
tissue specificity in its expression. Combinations of the three
factors should give an idea of the relative importance of an enzyme in
PAH metabolism.
6.3.1.1 Individual cytochrome P450 enzymes that metabolize PAH
CYP1A: CYP1A appears to be the only enzyme with metabolic capability
towards a wide variety of PAH molecules. It is expressed in various
tissues but at a generally low constitutive level (Guengerich &
Shimada, 1991). The induction of CYP1A1 is controlled by the Ah (aryl
hydrocarbon) receptor, a transcription factor that can be activated by
several ligands such as 2,3,7,8-tetradichlorobenzo- para-dioxin
(TCDD) and PAH, with variable potency (Negishi et al., 1981). Thus,
PAH and material containing PAH can regulate their own metabolism by
inducing CYP1A1. After induction, CYP1A1 expression may reach high
levels, e.g. in the placenta, lung, and peripheral blood cells;
however, in the liver, the principal organ of xenobiotic metabolism,
the level of expression is low even after induction, and other CYP
appear to be more important, at least in the metabolism of
benzo [a]pyrene (Guengerich & Shimada, 1991).
CYP1A2: The other member of the CYP1A family, CYP1A2, also
metabolizes PAH; however, its capacity to metabolize benzo [a]pyrene
to the 3-hydroxy metabolite, for example, is about one-fifth that of
CYP1A1 (Shou et al., 1994). Human CYP1A2 is nevertheless very active
in forming benzo [a]pyrene 7,8-dihydrodiol (Bauer et al., 1995) and
in forming diol epoxides from the 7,8-dihydrodiol (Shou et al., 1994).
There is also evidence that CYP1A2 can activate
7,12-dimethylbenz [a]anthracene to mutagenic species, albeit at a low
rate (Aoyama et al., 1989).
The expression of CYP1A2 is also regulated by the Ah receptor, but in
not exactly the same way as CYP1A1 (Negishi et al., 1981). In the
liver, for example, the level of CYP1A2 expression is much higher than
that of CYP1A1 (Guengerich & Shimada, 1991). While the capacity of
CYP1A2 to oxidize various PAH is more limited than that of CYP1A1, its
role in reactions like diol epoxide formation from benzo [a]pyrene in
the liver could be important because of its high level of expression.
CYP1B: The CYP1B subfamily was discovered only recently. Once the
enzyme had been isolated, it was found to be capable of metabolizing
PAH. Interestingly, its expression is also under the control of the Ah
receptor. Only limited information is available on its expression and
catalytic properties in different tissues, but it seems to be
expressed at least in mouse embryo fibroblasts (Savas et al., 1994),
rat adrenal glands (Bhattacharyya et al., 1995), and several human
tissues (Sutter et al., 1994). A number of PAH may act as substrates
for this enzyme (Shen et al., 1994).
CYP2B: When recombinant gene technology was used to express human
CYP2B6 cDNA in a human lymphoblastoid cell line, this enzyme was shown
to be capable of metabolizing benzo [a]pyrene to 3- and 9-phenols and
trans-dihydrodiols (Shou et al., 1994). In addition, CYP2B enzymes
may be involved in the metabolism of 7,12-dimethylbenz [a]anthracene
(Morrison et al., 1991a).
The constitutive levels of CYP2B enzymes are extremely low in human
liver, but they are strongly induced by phenobarbital and
phenobarbital-type inducers of CYP. Accordingly, immunological studies
of inhibition have shown that the CYP2B enzymes may play a significant
role in the metabolism of PAH, only when they are induced (Hall et
al., 1989; Honkakoski & Lang, 1989).
CYP2C: The CYP2C subfamily contains several members, some of which
are expressed at high levels in human liver. More than one member of
this subfamily may be capable of metabolizing PAH; thus, human CYP2C9
and, to a lesser extent, CYP2C8 metabolize benzo [a]pyrene to 3- and
9-phenols and trans-dihydrodiols (Shou et al., 1994). In addition,
CYP2C enzymes may play an essential role in the metabolism of
benzo [a]pyrene and 7,12-dimethyl-benz [a]anthracene, particularly
in phenobarbital-induced liver (Morrison et al., 1991a,b; Todorovic et
al., 1991). In view of the relative abundance of CYP in human liver
and their role in the metabolism of PAH, it has been suggested that
some CYP2C enzymes play an essential role in hepatic PAH metabolism
(Morrison et al., 1991b; Yun et al., 1992).
CYP3A: CYP3A is one of the most abundant CYP enzymes in human liver,
and it can metabolize benzo [a]pyrene and some of its dihydrodiols to
several metabolic products (Shimada et al., 1989; Yun et al., 1992;
Shou et al., 1994; Bauer et al., 1995). In one study, human CYP3A4 was
the most important single enzyme in the hepatic 3-hydroxylation of
benzo [a]pyrene (Yun et al., 1992).
6.3.1.2 Regulation of cytochrome P450 enzymes that metabolize PAH
All of the enzymes discussed above are inducible, and their level of
expression can be enhanced by external stimuli. CYP1A and CYP1B are
under the transcriptional control of the Ah receptor, which can be
activated by numerous PAH and other planar hydrocarbons, including
dioxins (Negishi et al., 1981; Guengerich & Shimada, 1991)
CYP2B enzymes can also be induced by foreign compounds but not through
the Ah receptor. The mechanism of induction of these enzymes is not
well understood, but their prototype inducer is phenobarbital; several
other drugs used clinically have similar effects (Gonzalez & Gelboin,
1994).
The regulation of CYP2C enzymes is complicated, and both endogenous
factors such as steroid hormones and exogenous factors such as
phenobarbital may be involved. Furthermore, different members of this
subfamily are regulated differently. The CYP3A are also regulated by
endogenous and exogenous factors; typical inducers of this subfamily
are rifampicin, dexamethasone, certain macrolide antibiotics, and
steroid hormones (Guengerich & Shimada, 1991).
Genetic polymorphisms of CYP1A1, CYP1A2, and some CYP2C and CYP3A
enzymes have also been described. Some of the genetic defects leading
to the polymorphism have been identified and can be used to predict an
individual's capacity to metabolize drugs, for example by the
polymerase chain reaction. Genetic polymorphism may lead to dramatic
changes in the capacity to metabolize PAH (Raunio & Pelkonen, 1994).
Studies with a few prototype compounds such as benzo [a]pyrene and
its metabolites and 7,12-dimethylbenz [a]anthracene indicate that
several CYP are involved in PAH metabolism. As each has its own
metabolic capacity, mode of regulation, and tissue-specific
expression, the one that plays a key role in PAH metabolism in vivo
at any one time may vary and will depend on the compound being
metabolized, pre-exposure to inducers of the CYP, the tissue and cell
type where the metabolism is taking place, and the genotype of the
individual in cases of genetic polymorphism.
Many PAH that are metabolized by the CYP-dependent mono-oxygenases
also induce the enzyme system. This ability of hydrocarbons to induce
their own metabolism usually results in lower tissue levels and more
rapid excretion of the hydrocarbon (Schlede et al., 1970b; Aitio,
1974). Although CYP1A1 is mainly responsible for activation of PAH in
the lung and CYP1A2 in the liver, most recent investigations have
shown that other CYP isoforms may also contribute to the metabolism of
PAH in mammals (Jacob et al., 1996). Thus, pretreatment of animals
with inducers of mono-oxygenase systems is frequently associated with
a decreased tumour incidence (Wattenberg, 1978). Conversely, studies
with strains of mice that differ genetically in the capacity of their
mono-oxygenase systems to be induced by PAH indicate that inducibility
may also be associated with an increased tumorigenic or toxicological
response (Nebert, 1980). Induction of the mono-oxygenase system by
different types of inducers can result in different profiles of
hydrocarbon metabolites, although the extent of the effect appears to
be variable (Holder et al., 1974; Jacob et al., 1981a,b; Schmoldt et
al., 1981). The metabolism of benzo [a]pyrene has been investigated
in more detail than that of other hydrocarbons and is used here as an
example.
6.3.2 Metabolism of benzo[a]pyrene
In early studies, the PAH metabolites isolated from or excreted by
experimental animals were shown to consist of hydroxylated
derivatives, commonly in the form of conjugates. Thus, the general
scheme of xenobiotic metabolism outlined above applies to PAH. One of
the principal interests in hydrocarbon metabolism arose, however, from
the realization that hydrocarbons, like many other environmental
carcinogens, are chemically unreactive and that their adverse
biological effects are probably mediated by electrophilic metabolites
capable of covalent interaction with critical macromolecules such as
DNA. Identification of the biologically active metabolites of PAH,
coupled with advances in both the synthesis of known and potential
hydrocarbon metabolites and the analysis of metabolites by
high-performance liquid chromatography, has led in the last two
decades to a greatly enhanced appreciation of the complexity of
hydrocarbon metabolism. Most of these metabolic interrelationships are
illustrated for benzo [a]pyrene in Figure 3; the structures of some
types of metabolites are given in Figure 4. The metabolism of
benzo [a]pyrene and other PAH has been reviewed (for example, Sims &
Grover, 1974, 1981; Conney, 1982; Cooper et al., 1983; Dipple et al.,
1984; Hall & Grover, 1990).
Benzo [a]pyrene is metabolized initially by the microsomal
CYP-dependent mono-oxygenase system to several epoxides (Figure 3).
Once formed, these epoxides (Sims & Grover, 1974) may spontaneously
rearrange to phenols, be hydrated to dihydrodiols in a reaction that
is catalysed by epoxide hydrolase (see review by Oesch 1973), or react
covalently with glutathione, either chemically or in a reaction
catalysed by glutathione S-transferase (Chasseaud, 1979).
6-Hydroxybenzo [a]pyrene is further oxidized either spontaneously or
metabolically to the 1,6-, 3,6-, or 6,12-quinone, and this phenol is
also a presumed intermediate in the oxidation of benzo [a]pyrene to
the three quinones that is catalysed by prostaglandin H synthase. Two
additional phenols may undergo further oxidative metabolism:
3-hydroxybenzo [a]pyrene is metabolized to the 3,6-quinone, and
9-hydroxybenzo [a]pyrene is oxidized to the K-region 4,5-oxide, which
is hydrated to the corresponding 9-hydroxy 4,5-dihydrodiol (Jernström
et al., 1978; for a formula showing a K-region, see Figure 11).
Phenols, quinones, and dihydrodiols can all be conjugated to yield
glucuronides and sulfate esters, and the quinones may also form
glutathione conjugates (Figure 5).
3.2.4.2 Emissions to the hydrosphere
During the refining of aromatic hydrocarbons, and especially hard
coal-tar, 80-190 t/year were estimated to be released to the
hydrosphere in western Germany until 1987. This quantity was reduced
to 8-19 t/year by the installation of new adsorption devices (sand
filtration and adsorbent resin) by the two German hard coal-tar
refineries in 1989 and 1991 (Klassert, 1993).
3.2.5 Emissions during the use of individual PAH
Only naphthalene is used directly (as a moth repellent) without
further processing. On the assumption that all naphthalene-containing
moth repellent is emitted into the atmosphere, the emissions would
have been about 15 000 t/year in western Europe in 1986, about 4400
t/year in Japan in 1987, and about 5500 t/year in the USA in 1987 (Fox
et al., 1988).
3.2.6 Emissions of PAH during processing and use of coal and petroleum
products
Coal coking, coal conversion by gasification and liquefaction,
petroleum refining, and the production and use of carbon blacks,
creosote, coal-tar, and bitumen from fossil fuels may produce
significant quantities of PAH (Anderson et al., 1986). A great deal of
information on emissions of PAH is available in the literature; this
monograph gives an overview of the most reliable values. The emission
profile depends on the source, and specific emission profiles are
detectable only in the direct vicinity of the corresponding source.
Generally, emissions are estimated on the basis of more or less
reliable databases, which are not identified in most publications. The
values reported give only a rough idea of the situation.
3.2.6.1 Emissions to the atmosphere
(a) Coal coking
During coal coking, PAH are released into the ambient air mainly
when an oven is loaded through the charging holes and new coal is
suddenly brought into contact with the hot oven, and from leaks around
oven doors and battery-top lids (Bjorseth & Ramdahl, 1985; Slooff et
al., 1989). The specific emission factor for both benzo [a]pyrene and
benzo [e]pyrene during coal coking was 0.2 mg/kg coal charged (Ahland
et al., 1985). The emission factor for total PAH was estimated to
about 15 mg/kg coal charged (Bjorseth & Ramdahl, 1985).
Stack gases were measured about 8 m away from the aperture
through which coke was discharged at a Belgian coking battery.
Although the effluent may have been slightly diluted with ambient air,
the following PAH concentrations were detected: benz [a]anthracene
plus chrysene, 580 ng/m3; benzo [k]fluoranthene, 500 ng/m3;
benzo [a]pyrene plus benzo [e]pyrene, 470 ng/m3; fluoranthene, 330
ng/m3; pyrene, 180 ng/m3 benzo [ghi]perylene, 140 ng/m3;
anthracene plus phenanthrene, 130 ng/m3; and perylene, 44 ng/m3
(Broddin et al., 1977).
The release of total PAH in 1985 was estimated to about 630
t/year in the USA, 18 t/year in Sweden, and 5.1 t/year in Norway
(Bjorseth & Ramdahl, 1985). The authors emphasized that their data are
subject to uncertainty and should be used only as an indication of the
order of magnitude. In 1990, the total PAH emission in Canada was
estimated to be 13 t/year (Environment Canada, 1994). Further
estimates of total annual emissions of individual PAH compounds during
the coking of coal are shown in Table 10.
Table 10, Estimated annual emissions of polycyclic aromatic hydrocarbons during
coal coking in the Netherlands and western Germany
Compound Annual Year Reference
emission
(t/year)
Netherlands
Anthanthrene 0.5 Before 1989 Slooff at al. (1989)
Benz[a]anthracene 0.3 1988 Slooff at al. (1989)
Benzo[a]pyrene 0.1 Before 1989 Slooff at al. (1989)
Benzo[ghi]perylene 0.2 1988 Slooff et al. (1989)
Benzo[k]fluoranthene 0.1 1988 Slooff at al. (1989)
Chrysene 0.2 1988 Slooff at al. (1989)
Fluoranthene 1.1 1988 Slooff at al. (1989)
lndeno[1,2,3-cd]pyrene 0.1 1988 Slooff et al. (1989)
Naphthalene 1.3 1987 Slooff et al. (1988)
2.0 Before 1989 Slooff et al. (1989)
Phenanthrene 2.1 1988 Slooff et al. (1989)
Western Germany
Benzo[a]pyrene 1.1 1990 Ministers for the
Environment (1992);
1.7 Zimmermeyer et al.
(1991)
Naphthalene 10.0 1987 Society of German
Chemists (1989)
The emission factors for benzo [a]pyrene in the coking industry
in the North-Rhine Westphalia area of Germany have been assumed to
have been reduced to an average of about 60 mg/t coke. The newest
plants have emission factors of 40 mg/t coke (Eisenhut et al., 1990).
The reduction in PAH discharge was brought about by technical
improvements to existing plants, closure of old plants and their
partial replacement by new plants, and a reduction in coke production
(Zimmermeyer et al., 1991). Decreasing trends in the annual emissions
of airborne PAH during coke production are also assumed to have
occurred in other industrialized countries (western Europe, Japan, and
the USA), but no data were available.
(b) Coal conversion
PAH emission factors measured in the USA during gasification of
coal at the end of the 1970s ranged from about 1 µg/g burnt coal for
chrysene and 1500 µg/g burnt coal for naphthalene. Three qualities of
coal were analysed for naphthalene, acenaphthylene, fluorene,
anthracene, phenanthrene, pyrene, benz [a]anthracene, chrysene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]perylene, indeno[1,2,3- cd]pyrene, and
dibenzo [a,h]pyrene (Nichols et al., 1981). In 1981, the stack gas of
one US pilot coal gasification plant with an outdoor filter contained
0.2 and 2.1 µg/m3 naphthalene at two sampling times and 6.8 µg/m3
phenanthrene (Osborn et al., 1984). Acenaphthylene was detected at
concentrations of 0.11-0.12 µg/m3 in the stack gases of two Canadian
pilot coal liquefaction plants (Leach et al., 1987).
(c) Petroleum refining
The average profile of PAH compounds in petroleum refineries
indicates that at least 85% of the total concentration is made up of
two-ring compounds (naphthalene and its derivatives) and 94% of two-
and three-ring compounds. Compounds with five rings or more
contributed less than 0.1% at the catalytic cracking unit. In
turn-round operations on reaction and fractionation towers,
naphthalene and its methyl derivatives accounted for more than 99% of
the total PAH (IARC, 1989b).
Little information is available on the concentrations of PAH in
stack gases. The levels in one French (Masclet et al., 1984) and two
US petroleum refining plants (Karlesky et al., 1987) are available
(Table 11); no information was given about the sampling site in the
French facility, but sampling in the US plants was at the distillation
device and below the cracking tower. The results depended on which
fuel was burnt and the positioning and type of sampling device in the
stack.
Table 11. Polycyclic aromatic hydrocarbon concentrations
in the stack gases of petroleum refinery plants in
France and the USA
Compound Concentration (µg/m3)
France USA
Acenaphthene NR 0.018-0.035
Acenaphthylene NR 0.013/0.019
Anthracene 3.9 0.003-0.041
Benz[a]anthracene 1.6 0.051-0.801
Benzo[a]pyrene 0.4 0.261-3.17
Benzo[b]fluoranthene 1.3 0.323-0.616a
Benzo[e]pyrene 2.8 NR
Benzo[ghi]perylene 0.7 0.23/0.382
Benzo[k]fluoranthene 0.5 NR
Chrysene 1.7 0.021-0.252
Coronene 1.0 NR
Dibenzo[a,h]anthracene NR 0.177
Fluoranthene 2.3 0.030-0.577
Fluorene 2.4 0.041-2.48
Indeno[1,2,3-cd]pyrene 1.2 0.25/0.538
Naphthalene NR 0.052-0.113
Perylene ND ND
Phenanthrene 7.9 0.040-9.13
Pyrene 4.3 0.016-3.56
From Masclet et aL (1984) and Karlesky et al. (1987)
NR, not reported; ND, not detected, limit of detection not
stated; /, single measurements
a Plus benzo[k]fluoranthene
Few data are available on the total release of PAH into the
atmosphere during petroleum refining. In western Germany, the
emissions of naphthalene during petroleum refining, including hard
coal-tar processing, were estimated to be 11 t/year (year not given;
Society of German Chemists, 1989). In the Netherlands, the release of
total PAH in 1988 was estimated to be about 7 t/year; the burning of
pitch contributed 6.6 t/year, regeneration of catalyst, 0.4 t/year,
and refining, < 0.01-0.1 t/year (Slooff et al., 1989). In Canada,
about 0.1 t total PAH were emitted into the atmosphere in 1990
(Environment Canada, 1994).
(d) Other processes
In a US oil-furnace carbon black plant, the following mean
emission factors per kg carbon black produced were found for
individual PAH in three runs in the main vent gas: acenaphthylene,
800 g; pyrene, 500 g; anthracene plus phenanthrene, 70 g;
fluoranthene, 60 g; benzo [ghi]fluoranthene, 40 g;
benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, 30 g; benzo [a]pyrene plus benzo [e]pyrene
plus perylene, 30 g; benzo [ghi]perylene plus anthanthrene, 23 g;
chrysene plus benz [a]anthracene, 9 g; indeno[1,2,3- cd]pyrene,
< 2 g; and benzo [c]phenanthrene, < 2 g. The release of PAH into
ambient air cannot be estimated from these emission factors, however,
as an additional combustion device is fitted in most US carbon-black
plants in which the process vent gases are burnt (Serth & Hughes,
1980).
Compounds with five or more rings (e.g. benzo [a]pyrene)
contributed about 0.3% to the total PAH released from the bitumen
processing unit of a refinery (IARC, 1989b). The emissions of PAH from
batch asphalt mixers are assumed to be low and to occur mainly in
combustion gases (IARC, 1984a), although no experimental data were
available.
Few estimates have been made of the annual emissions of PAH from
processes in which coal and coal products are used. The total release
of PAH to the atmosphere during asphalt production in 1985 was
estimated to be about 4 t in the USA, 0.1 t in Norway, and 0.3 t in
Sweden (Bjorseth & Ramdahl, 1985). In Canada, the amount emitted in
1990 was estimated to be about 2.5 t (Environment Canada, 1994). The
amount released during carbon-black production and processing in 1985
was estimated to be about 3 t in the USA and < 0.1 t in Sweden
(Bjorseth & Ramdahl, 1985). In the Netherlands in 1988, about 3.3 t of
total PAH were emitted during the storage and transport of anthracene
oil, an intermediate in the processing of hard coal-tar (Slooff et
al., 1989).
(e) Use of impregnating oils (creosotes) in wood preservation
Estimates of the total input of PAH into the atmosphere from wood
preservation with creosotes were available only for the Netherlands
for unspecified years, at about 320 t/year (Slooff et al., 1989) and
840 t/year (Berbee, 1992). In 1988, the PAH input during storage of
preserved material was estimated by the same authors to be about 200 t
naphthalene, 110 t phenanthrene, 30 t fluoranthene, 5 t anthracene,
1.1 t benz [a]anthracene, and 0.02 t benzo [k]fluoranthene.
3.2.6.2 Emissions to the hydrosphere
(a) Coal coking
The concentrations of PAH reported in wastewater effluents are
shown in Table 12. The removal of PAH by biological oxidation in two
US coal coking plants was 93 to > 99%. Higher-molecular-mass PAH,
benzo [a]pyrene, dibenz [a,h]anthracene, and benzo [ghi]perylene,
comprised a greater fraction (about 60%) of the total PAH content in
the effluent than in the input stream (Walters & Luthy, 1984). The
total concentration of PAH discharged into the aqueous environment
from a Norwegian coking plant was estimated to be about 23 kg/d
(Berglind, 1982). On the basis of Dutch emission factors, the release
in western Europe in 1985 of fluoranthene was calculated to be about 5
t and that of benzo [a]pyrene about 0.7 t (Berbee, 1992). The total
annual input of PAH into the aqueous environment of the Netherlands
was estimated to be about 1.7 t (year not given; Slooff et al., 1989).
(b) Coal conversion
The PAH content of wastewater from coal and shale conversion was
< 0.5 mg/litre (Guerin, 1977). In raw, untreated wastewaters from a
US pilot coal liquefaction plant, numerous PAH were found to emanate
from the liquefaction section, the untreated hydrogenation section,
and the still bottoms processing device when two kinds of coal were
tested; for example, benzo [a]pyrene was found at a concentration of
0.3-52 µg/litre (Robbins et al., 1981). Numerous PAH were found in raw
wastewater samples from two US pilot coal gasification plants (Walters
& Luthy, 1981; Abbott et al., 1986), the maximum level of
benzo [a]pyrene being 5.0 µg/litre.
No information was available about total PAH emissions into the
aqueous environment from commercial coal conversion plants. In
groundwater near a US in-situ coal gasification site, naphthalene was
found at a concentration of 2.7 µg/litre and acenaphthene and fluorene
at < 0.1 µg/litre (Pellizzari et al., 1979).
Until 1988, the final effluent from the two hard coal-tar
refineries in western Germany contained an average of 50 mg/litre
naphthalene, with a maximum of 120 mg/litre. The annual emission of
this compound was thus calculated to be about 80 t. By 1991, the
estimated release of naphthalene had been reduced to about 8 t/year by
the addition of adsorption devices (Klassert, 1993).
(c) Petroleum refining and offshore oil-well drilling
PAH concentrations in wastewater effluents from these sources are
summarized in Table 13. A refinery-activated sludge unit with a
dual-media filter removed about 95% of the five-ring PAH and 99% of
the four-ring PAH from the effluent of a petroleum refinery (Pancirov
et al., 1980). A similar elimination efficiency was found for
dissolved air flotation treatment of refinery wastewater and
subsequent removal by activated sludge. Air stripping of the compounds
in the sewage plant seemed to be of minor importance (Snider &
Manning, 1982). The concentrations of PAH with more than three rings
were found to be < 0.05 µg/litre even in the input to a sewage device
and < 0.02 µg/litre in the final effluent (German Society for
Mineral-oil and Coal Chemistry, 1984). The authors stated that these
levels were of the same order of magnitude as the background
concentrations in surface waters.
The discharge of total PAH from a Norwegian petroleum refinery
was about 0.26 kg/day (Berglind, 1982). The total concentration of PAH
released into the North Sea from offshore oil-well drilling activities
was about 2.5 t/year in 1987, comprising 2 t/year from drill rinsing
and 0.2 t/year from shipping (Slooff et al., 1989).
(d) Use of impregnating oils (creosotes) in wood preservation
PAH were detected at levels of milligrams per litre in
groundwater under a former wood preserving facility in Florida, USA.
The concentrations of lower-molecular-mass creosote constituents were
smaller in the groundwater than in an unweathered standard, probably
because of greater mobility and biodegradability (Mueller & Lantz,
1993; Middaugh et al., 1994).
Model experiments with fresh and seawater were carried out to
determine the release of PAH from marine pilings made from southern
pine and preserved with creosote (Ingram et al., 1982). The PAH levels
per litre fresh water in the leachate at 20°C after immersion for
three days were: naphthalene, 200-350 g; acenaphthene, 190-230 g;
phenanthrene, 190-230 g; fluorene, 120-150 g; acenaphthylene, 51-88
g; anthracene, 48-76 g; fluoranthene, 27-30 g; pyrene, 12 g; and
benz [a]anthracene, 11-19 g. The concentrations in seawater were
three to four times lower. The amounts of PAH leached increased with
increasing temperature. The concentrations in leachates from pilings
that had been in seawater for 12 years were of the same order of
magnitude. In contrast, rapidly decreasing PAH concentrations were
found three months after the start of the experiment in runoff
rainwater from spruce and pine pilings impregnated with hard coal-tar
(van Dongen, 1987).
The total PAH emissions into water and soil in the Netherlands
from commercial wood preservation were about 28 t/year (year not
given). The release of 10 PAH into water during the storage of
creosote-preserved wood was about 16 t/year; the PAH measured were
naphthalene, anthracene, phenanthrene, fluoranthene,
benz [a]anthracene, benzo [a]pyrene, benzo [ghi]-perylene, and
indeno[1,2,3- cd]pyrene) (Slooff et al., 1989).
In Canada, the maximum release of PAH into water and soil from
creosote-treated wood products was estimated to be 2000 t/year, on the
basis of the PAH content of creosote, the volume of treated wood, the
retention rates of the substances for different wood species, and an
estimated 20% loss of PAH during the time the wood was in service,
i.e. 40 years for pilings and 50 years for railroad ties (Environment
Canada, 1994).
Table 12. Polycyclic aromatic hydrocarbon concentrations (µg/litre) in
wastewater effluents from coal coking plants
Compound [1] [2] [3]a [4] [5]
Acenapthene NR NR NR 0.009-2.5 NR
Acenaphthylene NR NR NR NR NR
Anthracene 0.31 NR NR 0.0-2.0 0.1
Anthanthrene ND NR 0.040/0.600 NR NR
Benzo[j+k]fluoranthene NR NR NR NR NR
Benz[a]anthracene 2.0 11.1 0.504/4.9 NR NR
Benzo[a]fluoranthene 0.8 NR NR NR NR
Benzo[a]pyrene NR 3.8 0.622/4.841 4.7-25 NR
Benzo[b]fluoranthene NR NR NR NR NR
Benzo[a]fluorene 0.81 NR NR NR NR
Benzo[c]phenanthrene ND NR 0.042/0.699 NR NR
Benzo[e]pyrene NR NR 0.323/2.928 NR NR
Benzofluoranthenesb NR 6.9 1.010/8.741 NR NR
Benzo[ghi]fluoranthene ND NR 0.042/0.663 NR NR
Benzo[ghi]perylene 2.0 NR 0.445/2.835 0-9.0 NR
Chrysene NR 7.2 0.732/6.440 1.8-42 NR
Dibenz[a,h]anthracene NR NR NR 0.06-3.0 NR
Fluoranthene 2.8 11.2 NR 1.3-10 NR
Fluorene NR NR NR 0.0-1.0 NR
Indeno[1,2,3-cd]pyrene NR NR 0.371/3.051 NR NR
1-Methylphenanthrene ND NR NR NR NR
Naphthalene NR NR NR 0-4.1 NR
Perylene ND NR 0.117/1.348 NR NR
Phenanthrene 0.4 NR NR 0.45-2.3 0.5
Pyrene 4.0 12.9 NR NR 0.38-60
[1] Effluent channel water from one US coking plant (Griest, 1980);
[2] Effluent channel water from one US coking plant (Griest at al., 1981);
[3] Raw wastewater from two coking plants in western Germany (Grimmer at
al., 1981 b);
[4] Effluents from two US coking plants downstream of company-owned biological
oxidation device (Walters & Luthy, 1984);
[5] Final effluent after biological oxidation; no further information
(Jockers at al., 1988) When the water samples were filtered through solid
sorbents, the results may be underestimates of the actual content of
polycyclic aromatic hydrocarbons (see section 2.4.1.4)
ND, not detected, limit of detection not given; NR not reported
a /, single measurements
b Isomers not specified
Table 13. Polycyclic aromatic hydrocarbons in effluents after wastewater
treatment in petroleum refineries (µg/litre)
Compound [1] [2] [3] [4] [5]
Acenaphthene NR 4.0 < 0.1-6 NR NR
Acenaphthylene NR 1.8 < 0.1-< 1 NR NR
Anthracene NR 11 < 0.01-< 2 0.26 NR
Benz[a]anthracene NR 0.6 < 0.02-< 1 NR NR
Benzo[a]pyrene 0.57 0.1 0.1-2.9 0.11 NR
Benzo[b]fluoranthene < 0.1 0.2 < 0.06 NR NR
Benzo[c]phenanthrene NR 0.2 NR NR NR
Benzo[e]pyrene 0.65 0.3 NR NR NR
Benzo[ghi]fluoranthene < 0.4 NR NR NR NR
Benzo[ghi]perylene 0.36 NR < 0.2-< 1 NR NR
Benzo[j]fluoranthene < 0.2 NR NR NR NR
Benzo[k]fluoranthene < 0.2 0.4a < 0.2 NR NR
Chrysene < 0.03 1.4b < 0.02-1.4 NR NR
Coronene < 0.01 NR NR NR NR
Dibenz[a,h]anthracene NR NR < 0.3-< 1 NR NR
Fluoranthene < 0.2 16.0 < 0.1-< 10 0.26 NR
Fluorene NR 3.4 < 0.1-< 1 1.2 NR
Indeno[1,2,3-cd]pyrene < 0.02 NR < 1 NR NR
1-Methylphenanthrene NR 4.2 NR NR NR
Naphthalene NR 2.4 < 0.1-< 10 15 0.06-9
Perylene 0.14 NR NR NR NR
Phenanthrene NR 111.0 < 0.2-< 0.5 7.1 0.02-1.2
Pyrene 0.07 16.1 < 0.1-7 NR NR
Triphenylene < 0.03 NR NR NR NR
[1] Final effluent from one US petroleum refinery (Pancirov et al., 1980);
[2] Effluent from one Norwegian petroleum refinery after treatment in
oil-separation devices, oil traps, and retention ponds (Berglind, 1982);
[3] Average results for final effluent from 17 US petroleum refineries
(Snider & Manning, 1982);
[4] Final effluent from one Australian petroleum refinery (Symons & Crick,
1983);
[5] Average results for the final effluent from six petroleum refineries
in western Germany (German Society for Mineral-oil and Coal Chemistry,
1984)
When water samples were filtered through solid sorbents, the results may
be underestimates of the actual PAH content (see section 2.4.1.4).
NR, not reported
a With benzo[j]fluoranthene
b With triphenylene
(e) Other sources
PAH may be released into the hydrosphere during leaching of
stocks of coal by rain. In model leaching experiments, naphthalene,
acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene,
pyrene, chrysene, benz [a]anthracene, benzo [k]fluoranthene, and
benzo [a]pyrene were detected at concentrations in the low microgram
per litre range, with a maximum of 100 µg/litre; for example,
benzo [a]pyrene was found at 0.6 µg/litre (Stahl et al., 1984;
Fendinger et al., 1989).
PAH were also found in sludge from US coke processing plants in
the following concentrations (average of five samples): naphthalene,
430 mg/kg; phenanthrene, 260 mg/kg; acenaphthene, 78 mg/kg; pyrene, 30
mg/kg; chrysene, 28 mg/kg; benzo [a]pyrene, 3.8 mg/kg;
benzo [b]fluoranthene, 3.8 mg/kg; and benzo [ghi]perylene, 0.9 mg/kg
(Tucci, 1988).
PAH may also leach into drinking-water from coal-tar or asphalt
coatings on storage tanks and water distribution pipes. Samples from a
five-year-old coal-tar-coated water tank in the USA contained 0.21
µg/litre phenanthrene plus anthracene, 0.081 µg/litre fluoranthene,
0.071 µg/litre pyrene, 0.025 µg/litre naphthalene, and 0.021 µg/litre
fluorene (Alben, 1980). Measurements in numerous US drinking-water
systems showed that PAH accumulate in the water during transport in
these pipes. The total concentration of fluoranthene,
benzo [j]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
indeno[1,2,3- cd]-pyrene, and benzo [ghi]perylene after transport
was in the low nanogram per litre range (Basu et al., 1987). In 1994,
a PAH concentration of 2.7 µg/litre was measured in accordance with
the German Directive on drinking-water (6.9 µg/litre measured in
accordance with US regulations), which was due to transport through a
tar-coated pipe in a central water reservoir; phenanthrene was present
at a concentration of 2.8 µg/litre and pyrene at 1.2 µg/litre (State
Chemical Analysis Institute, Freiburg, 1995). The release of PAH from
this source cannot be estimated from the available data.
During offshore oil and gas production, PAH-containing drilling
muds are discharged directly into the sea. The PAH concentrations at
some oil and gas platforms in the Gulf of Mexico and the North Sea
were found to be 1900 µg/litre for naphthalene and < 0.01 µg/litre
each for chrysene, benzo [b]fluo-ranthene, and
dibenz [a,h]anthracene (van Hattum et al., 1993).
The total PAH passing into the oceans from shipping have not been
estimated. The worldwide discharge of PAH into the oceans from
refineries, marine transportation, and industrial effluents of crude
oil was estimated to be about 6 t/year in 1973 and 4.6 t/year in the
early 1980s (Suess, 1976), but the basis for these estimates is
unknown.
3.2.6.3 Emissions to the geosphere
The average PAH concentrations in soil from more than 20 former
coking sites in Germany were: naphthalene, 1000 mg/kg; phenanthrene,
500 mg/kg; fluoranthene, 200 mg/kg; pyrene, 200 mg/kg; anthracene, 50
mg/kg; and benzo [a]pyrene, 3-5 mg/kg. During vertical leaching, the
compounds are distributed according to their mobility. PAH with
high-boiling points and low water solubility are present at the
highest concentrations at the surface, and more mobile compounds
accumulate in deeper soil layers. Naphthalene is usually leached into
groundwater, in which it is relatively soluble (Hoffmann, 1993).
The sediment of an effluent channel at one US coking plant
contained the following concentrations of PAH (dry weight basis):
fluoranthene, 31 mg/kg; pyrene, 23 mg/kg; benzo [b+j+k]fluoranthenes,
23 mg/kg; benzopyrenes, 19 mg/kg; benz [a]anthracene, 15 mg/kg;
chrysene plus triphenylene, 15 mg/kg; benzo [ghi]perylene, 7.3 mg/kg;
benzo [a]fluorene, 7.2 mg/kg; anthracene, 6.7 mg/kg; perylene, 3.8
mg/kg; phenanthrene, 3.6 mg/kg; benzo [b]fluorene, 3.2 mg/kg;
benzo [ghi]fluoranthene, 2.3 mg/kg; anthanthrene, 2.3 mg/kg;
benzo [c]phenanthrene, 2.1 mg/kg; and 1-methylphenanthrene, 0.71
mg/kg. In the sediment of an effluent from one US petroleum tank farm,
anthracene was detected at 3.4 mg/kg, benz [a]anthracene at 0.13
mg/kg, and benzo [a]pyrene at < 0.049 mg/kg (Griest, 1980).
Oily sludge originating from a dissolved air flotation unit of
the treatment system of a US petrochemical plant effluent was applied
to sandy loam samples seven times during a 920-day active disposal
period followed by a 360-day inactive 'closure' period, and the
decreases in the concentrations of fluorene, phenanthrene, anthracene,
fluoranthene, pyrene, benz [a]anthracene, chrysene, triphenylene,
benzo [ghi]fluoranthene, benzo [b]fluoranthene,
benzo [j]fluoran-thene, benzo [k]fluoranthene, perylene,
benzo [a]pyrene, benzo [e]pyrene, and benzo [ghi]perylene in soil
were determined. The initial PAH levels ranged from 0.9 mg/kg
benzo [j]fluoranthene to 270 mg/kg phenanthrene (dry weight basis).
After 1280 days, the three-ring compounds (fluorene, phenanthrene,
anthracene) had almost completely disappeared, with 0.2-6.9%
remaining, the four-ring substances (fluoranthene,
benz [a]anthracene, chrysene) had been partly degraded, and the
five-ring compounds remained at fairly high concentrations (Bossert et
al., 1984).
PAH may be released into soil from polluted industrial sludges
and during commercial wood preservation; however, no estimates of the
total PAH input into this compartment were available.
3.2.6.4 Emissions into the biosphere
Use of anti-dandruff shampoos containing hard coal-tar may lead
to increased body concentrations of PAH, as measured by urinary
excretion of the PAH metabolite 1-hydroxypyrene. One shampoo had a
total PAH content of 2800 mg/kg, including 290 mg/kg pyrene and 56
mg/kg benzo [a]pyrene (no further specification) (van Schooten et
al., 1994). Application of a 2% crude coal-tar solution in petrolatum
led to significantly increased PAH levels in the blood of five
volunteers (Storer et al., 1984; see also Section 8). Measurements of
hard coal-tar-containing shampoos in Germany showed concentrations of
7-61 mg/kg benzo [a]pyrene. In wood-tar-containing shampoos,
benzo [a]pyrene was detected at concentrations in the low microgram
per kilogram range, but 150 mg benzo [a]pyrene were found in one tar
bath (State Chemical Analysis Institute, Freiburg, 1995).
3.2.7 Emissions of PAH due to incomplete combustion
PAH not only pre-exist in fossil fuels but more are formed during
pyrolysis by a radical mechanism (see Zander, 1980). The domestic
activities that may result in significant emissions of PAH emissions
are vehicle traffic, tobacco smoking, broiling and smoking of foods,
and refuse burning. The industrial activities that result in PAH
release are aluminium production with use of Söderberg electrodes,
iron and steel production, foundries, tyre production, power plants,
incinerators, and stubble burning (Anderson et al., 1986)
3.2.7.1 Industrial point sources
(a) Emissions to the atmosphere
(i) Power plants fired with coal, oil, and gas fossil fuels
PAH emitted into the atmosphere from coal-fired power plants
consist mainly (69-92%) of two- and three-ring compounds, i.e.
naphthalene and phenanthrene and their mono- and dimethyl derivatives.
Naphthalene is by far the major component of PAH fractions (31-35%),
although high concentrations of phenanthrene and fluorene are also
observed (Bonfanti et al., 1988). Specific emission factors of 0.02 g
emitted per kg combusted were measured for benzo [a]pyrene and 0.03
µg/kg for benzo [e]pyrene (Ahland et al., 1985).
The concentrations of PAH in stack gases from comparable coal-
and oil-fired power plants are shown in Table 14. It is difficult to
find a characteristic PAH profile for coal-fired plants. The
concentrations were low during undisturbed combustion (Guggenberger et
al., 1981; Warman, 1985). Low-molecular-mass PAH are found at higher
concentrations than high-molecular-mass compounds in coal combustion
effluents (Warman, 1985); the low-molecular-mass PAH phenanthrene,
fluoranthene, and pyrene were detected at particularly high
concentrations, whereas benzo [a]pyrene was found at a level typical
of that in ambient air (Kanij, 1987). The specific emission factor for
benzo [a]pyrene was 3.5-230 µg/t burnt coal (Ahland & Mertens, 1980).
As the contribution of benzo [a]pyrene to the total release of PAH is
small, it was considered not to be a suitable indicator for this
source (Guggenberger et al., 1981). In contaminated areas, the PAH
concentrations in ambient air may be higher than those in the stack
gases, which result from after-burning (Guggenberger et al., 1981).
Table 14. Concentrations of polycyclic aromatic hydrocarbons (ng/m3) in stack gases of coal- and oil-fired power plants
Compound Fuel [1] [2] [3] [4] [5] [6]a
Acenaphthene Coal NR NR NR NR NR ND-24
Anthracene Coal NR 0.5 < 10-1800 0.4-100 2-65 19-120
Anthanthrene Coal NR NR NR NR < 0.2-< 0.6 NR
Benz[a]anthracene Coal NR 0.6 < 20-1400 NR 1-40 NR
Benzo[a]pyrene Coal < 0.1-0.7b 1.3 0.5-790 0.1-120 0.1-1.9 NR
< 0.5c
Oil < 0.5-7 NR NR NR NR NR
Benzo[b]fluoranthene Coal < 0.1-3b,d 2.0 30/40k NR 0.3-12 NR
< 0.1-0.4c,d (1/880e)
Oil < 0.1-39a NR NR NR NR NR
Benzo[b]fluorene Coal NR NR NR NR < 2-< 6 NR
Benzo[c]phenanthrene Coal NR NR 0.2 NR NR NR
Benzo[e]pyrene Coal NR ND < 10-810 NR 3-< 18 NR
Benzo[ghi]perylene Coal NR NR < 10-1400 NR NR NR
Coal < 0.5-3b 1.2 < 10-< 100 3-22 < 2-< 6 NR
< 0.5c
Oil < 0.5-40 NR NR NR NR NR
Benzo[j]fluoranthene Coal NR NR NR NR < 5-< 13 NR
Benzo[k]fluoranthene Coal < 0.1-2b 0.9 20 NR 1.7-2.5 NR
< 0.1-1.3c
Oil < 0.1-29 NR NR NR NR NR
Chrysene Coal NR 1.8 < 10-< 600 0.1-28 1-41 ND-56
< 10-310e
3.8g
Coronene Coal 1-3b 0.9 < 100 NR NR NR
< 2c
Oil < 2-36 NR NR NR NR NR
Dibenz[a,h]anthracene Coal < 0.5-2b NR < 100 NR NR NR
< 0.5c
Oil < 0.5-26 NR NR NR NR NR
Table 14. (continued)
Compound Fuel [1] [2] [3] [4] [5] [6]a
Fluoranthene Coal NR 4.1 < 10-22 100 0.5-240 20-720 NR
Fluorene Coal NR 1.9 NR NR NR 2-140
Indeno[1,2,3-cd]pyrene Coal NR 1.7 < 10-< 100 NR < 0.1-< 1.4 NR
1-Methylphenanthrene Coal NR NR < 20-90 NR NR NR
Naphthalene Coal NR NR NR 10-1800 NR 420-2100
Perylene Coal < 0.1-0.2b NR NR NR NR NR
< 0.1c
Oil < 0.1-15 ND < 10-< 100 NR < 0.2-0.9 NR
Phenanthrene Coal NR 5.2 < 20-33 200 26-640 32-2930 NR
Pyrene Coal NR 1.3 9-5800 0.2-2850 5-335 ND-311
Triphenyene Coal NR NR NR NR 20-77 NR
[1] Coal- and oil-fired power plants in the former FRG (Guggenberger et al., 1981);
[2] One French coal-fired power plant (Masclet at al., 1984);
[3] 10 Swedish coal-fired power plants (Warman, 1985);
[4] One US coal-fired power plant (Junk at al., 1986);
[5] One Dutch coal-fired power plant (Kanij, 1987);
[6] One German coal-fired power plant with circulating fluid bed combustion (Wienecke at al., 1992)
NR, not reported; ND, not detected, limit of detection not given
a Various coal qualities
b Hard coal
c Brown coal
d With benzo[e]pyrene
e Isomers not specified
f With triphenylene
g With benz[a]anthracene
The inputs of PAH into the atmosphere from power plants were:
about 0.001 t benzo [a]pyrene in western Germany in 1981 (Ahland et
al., 1985) and 0.1 t in 1983 (Grimmer, 1983a); about 1 t/year total
PAH in the USA; 0.1 t in Norway and 6.6 t in Sweden in 1985 (Bjorseth
& Ramdahl, 1985); about 2 t total PAH in the Netherlands in 1988
(Slooff et al., 1989); and about 11 t total PAH in Canada in 1990
(Environment Canada, 1994). These numbers may be subject to
uncertainty and should be used only as an indication of the order of
magnitude of e.g. the concentration in stack gases that is to be
expected from experimental values. Actual information on PAH emissions
from oil- and gas-fired power plants was not available. PAH emissions
from coal-fired power plants have been claimed to be negligible in
Germany due to the installation of appropriate filter systems, despite
the vast amount of stack gases produced (Zimmermeyer et al., 1991;
Ministers for the Environment, 1992).
(ii) Incinerators
Numerous PAH are formed under simulated incinerator conditions
from plastics such as polystyrene, polyethylene, polyvinyl chloride,
and their mixtures (Hawley-Fedder et al., 1984a,b,c, 1987). PAH were
detected at the following concentrations in the stack gases from a
British municipal incinerator: pyrene, 1.6 µg/m3; benz [a]anthracene
plus chrysene, 0.72 µg/m3; fluorene, 0.58 µg/m3;
benzo [ghi]perylene, 0.42 µg/m3; benzo [b]fluoranthene plus
benzo [j]fluoranthene plus benzo [k]fluoranthene, 0.32 µg/m3;
perylene, 0.18 µg/m3; indeno[1,2,3- cd]pyrene, 0.18 µg/m3;
coronene, 0.04 µg/m3; and benzo [a]pyrene plus benzo [e]pyrene,
0.02 µg/m3 (Davies et al., 1976). When PAH were sampled at a height
of about 10 m above the ground in the 110-m chimney of an incineration
plant in Sweden, no measurable amounts of PAH, at a limit of detection
of 10 ng/m3, were found during normal operating conditions or during
start-up in the morning; however, inactivity over a weekend resulted
in detectable concentrations of individual PAH, covering three orders
of magnitude up to around 100 µg/m3 (Colmsjö et al., 1986a).
Comparable results were obtained at a pilot incineration plant in
Canada (Chiu et al., 1991). Only phenanthrene plus anthracene was
found in measurable amounts in the stack gas (limit of detection not
stated). The total release of PAH from this plant was estimated to be
80-100 ng/m3.
The concentrations of PAH emitted in the stack gases from an
Italian municipal solid waste incinerator were: 0.1-1.9 µg/m3
indeno[1,2,3- cd]pyrene, 0.63 µg/m3 acenaphthene, 0.57-2.5 µg/m3
phenanthrene, 0.36-4.4 µg/m3 perylene, 0.35-0.55 µg/m3
benzo [e]pyrene, 0.25-3.6 µg/m3 benz [a]anthracene, 0.23 µg/m3
benzo [k]fluoranthene, 0.22 µg/m3 dibenz [a,h]anthracene, 0.19
µg/m3 benzo [b]fluoranthene, 0.15-0.67 µg/m3 pyrene, 0.15-0.73
µg/m3 acenaphthylene, 0.11-0.23 µg/m3 chrysene, 0.08 µg/m3
anthracene, 0.069 µg/m3 fluorene, 0.068-1.3 µg/m3 fluoranthene,
0.05-1.1 µg/m3 benzo [a]pyrene, and 0.014-0.47 µg/m3
benzo [ghi]perylene, depending on the firing conditions and the
composition of the waste (Morselli & Zappoli, 1988).
The benzo [a]pyrene concentrations in stack gases from
commercial waste incinerators in western Germany were estimated to be
1-6 µg/m3 (Johnke, 1992).
Controlled incineration of automobile tyres for thermal and
electric energy has been estimated to result in considerable release
of PAH into the atmosphere. In laboratory experiments, the following
concentrations were found in flue gas at an incineration temperature
of 677°C (per kg rubber): 930 mg pyrene, 760 mg fluoranthene, 390 mg
phenanthrene, 290 mg anthracene, 220 mg acenaphthylene, 120 mg
chrysene, 84 mg benzo [b]fluoranthene plus benzo [j]fluoranthene
plus benzo [k]fluoranthene, 66 mg benz [a]anthracene, 18 mg
benzo [e]pyrene, 11 mg benzo [a]pyrene, 3.8 mg perylene, 3.3 mg
benzo [ghi]fluoranthene, 2.0 mg dibenz [a,h]anthracene, 1.5 mg
benzo [ghi]perylene, 1.2 mg naphthalene, and 0.5 mg
indeno[1,2,3- cd]pyrene (Jacobs & Billings, 1985). On the basis of
data from Hartung & Koch (1991) on the number of tyres incinerated in
western Germany in 1987, the annual emissions from this source can be
calculated as follows: 160 t pyrene, 130 t fluoranthene, 70 t
phenanthrene, 50 t anthracene, 40 t acenaphthylene, 20 t chrysene, 14
t benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]-fluoranthene, 10 t benz [a]anthracene, 3 t
benzo [e]pyrene, 2 t benzo [a]pyrene, 0.5 t
benzo [ghi]fluoranthene, 0.3 t dibenz [a,h]anthracene, 0.3 t
benzo [ghi]-perylene, 0.2 t naphthalene, and 0.1 t
indeno[1,2,3- cd]pyrene.
The total PAH levels in stack gases from incinerators in
different countries were: Italy, 0.0075-0.21 mg/m3; Japan, 0.002-0.04
mg/m3; Sweden, 0.001 mg/m3; and Canada, 0.00002-0.02 mg/m3 (WHO,
1988). The results for traditional incinerators could not be compared
with those for plants with additional abatement techniques on the
basis of the available data. The total PAH emissions to the atmosphere
resulting from incineration of refuse were about 0.001 t
benzo [a]pyrene in western Germany in 1989 (Ministers for the
Environment, 1992) and about 0.0003 t in 1991 (Johnke, 1992), about 50
t total PAH in the USA, 0.3 t in Norway and 2.2 t in Sweden in 1985
(Bjorseth & Ramdahl, 1985); and about 2.4 t total PAH in Canada in
1990 (Environment Canada, 1994).
In Germany, the contribution of stack gases from commercial
incinerators is estimated to be < 4% of the total stack gas volume
from combustion processes. One of the main confounders of and
contributors to stack gases from combustion is motor vehicle traffic
(Johnke, 1992), indicating that PAH released from incinerators are
probably of minor importance.
(iii) Aluminium production
The production of coal anodes, used in the electrolytic
production of aluminium, from pitch and petroleum coke may still be an
important source of PAH, but confirmatory data are not available.
Estimates of PAH released during the production of aluminium in the
Netherlands in 1988 ranged from about 0.3 t benzo [ghi]perylene to 24
t naphthalene (Slooff et al., 1989). The estimated total airborne PAH
released in 1985 was about 1000 t in the USA, 160 t in Norway, and 35
t in Sweden (Bjorseth & Ramdahl, 1985). In 1990, the input of total
PAH from this source into the atmosphere in Canada was 930 t
(Environment Canada, 1994).
In horizontal and vertical Söderberg aluminium production
processes in Sweden, the emission factors per tonne of aluminium were
0.11 kg benzo [a]pyrene and 4.4 kg total PAH for the horizontal
process and 0.01 kg benzo [a]pyrene and 0.7 kg total PAH for the
vertical process (Alfheim & Wikström, 1984). In a Norwegian vertical
Söderberg aluminium production plant, the emission factors were
0.005-0.015 kg/t aluminium for benzo [a]pyrene and 0.3-0.5 kg/t for
total PAH (European Aluminium Association, 1990).
(iv) Iron and steel production
The total emissions of PAH resulting from iron and steel
production with carbon electrodes containing tar and pitch in Norway
was estimated to be about 34 t in 1985 (Bjorseth & Ramdahl (1985), but
the database for this estimate is limited. The release of total PAH
from metallurgical processes in Canada where similar electrodes were
used, including ferro-alloy smelters but excluding aluminium
production, was estimated to be 19 t in 1990 (Environment Canada,
1994).
(v) Foundries
PAH are formed during casting by thermal decomposition of
carbonaceous ingredients in foundry moulding sand, and they partly
vaporize under the extremely hot reducing conditions at the
mould-metal interface. Thereafter, the compounds are adsorbed onto
soot, fume, or sand particles. Organic binders, coal powder, and other
carbonaceous additives are the predominant sources of PAH in iron and
steel foundries (IARC, 1984b).
In pyrolysis experiments with green-sand additives, the highest
PAH levels were found in coal-tar pitch, with values per kilogram of
additive of 3100 mg benzo [a]pyrene, 3000 mg
benzo [b+j+k]fluoranthenes, 3000 mg pyrene, and 2900 mg fluoranthene;
the lowest levels were found in vegetable product additives, such as
maize starch: 26 mg pyrene, 16 mg fluoranthene, 3 mg
benzo [b+j+k]fluoranthenes, and 2 mg benzo [a]pyrene (Novelli &
Rinaldi, 1979). Less than 0.002 mg/kg benzo [a]pyrene was found in
foundry moulding sand when petrol resin, polystyrol, or polyethylene
was used as the carrier and 7.5 mg/kg when hard coal was used as the
carrier. The PAH content was directly correlated with the amount of
hydrocarbon carrier in the sand (Schimberg et al., 1981).
The following levels of PAH were found in the stack gases of one
French automobile foundry: fluoranthene, 980 ng/m3;
benz [a]anthracene, 830 ng/m3; benzo [a]pyrene, 570 ng/m3;
benzo [b]fluoranthene, 460 ng/m3; indeno[1,2,3- cd]pyrene, 370
ng/m3; anthracene, 250 ng/m3; benzo [k]fluoranthene, 220 ng/m3;
perylene, 160 ng/m3; benzo [ghi]perylene, 130 ng/m3; chrysene, 110
ng/m3; coronene, 28 ng/m3; and pyrene, 15 ng/m3. No further
information was given about the sampling site (Masclet et al., 1984).
The total emission of PAH into the atmosphere from iron foundries in
the Netherlands was estimated to be about 1.3 t in 1988 (Slooff et
al., 1989).
(vi) Other industrial sources
The estimated release of 10 PAH into the atmosphere in the
Netherlands in 1988 was about 1.3 t from sinter processes and 0.2
t/year from phosphorus production (Slooff et al., 1989).
(b) Emissions to the hydrosphere
(i) Aluminium production
PAH levels in wastewater from aluminium production in Norwegian
plants are shown in Table 15. At the beginning of the 1970s, the
release of anthracene and phenanthrene into the aqueous environment
from aluminium production in western Europe was estimated to be 180
t/year (Palmork et al., 1973). About 0.6 t/year are released into
water by the aluminium producing industry in the Netherlands (Slooff
et al., 1989).
(ii) Other industrial sources
No recent data were available on PAH emissions into the aqueous
environment from coal- or oil-fired power plants. PAH were found in
the final effluent from a British municipal incinerator at
concentrations ranging from < 0.01 µg/litre each for coronene and
indeno[1,2,3- cd]pyrene to 0.62 µg/litre fluoranthene. The calculated
daily output of single compounds was in the low milligram range, with
a maximum of 16 mg/d. Actual data were not available (Davies et al.,
1976).
Numerous PAH were detected in the final effluent from a Norwegian
ferro-alloy smelter in which the wastewater from gas scrubbers was
treated by chemical flocculation. The concentrations were 50 µg/litre
phenanthrene, 45 µg/litre pyrene, 40 µg/litre fluoranthene, 39
µg/litre acenaphthylene, 27 µg/litre fluorene, 17 µg/litre
acenaphthene, 13 µg/litre chrysene plus triphenylene, 11 µg/litre
anthracene, 10 µg/litre naphthalene, 10 µg/litre benz [a]anthracene,
9 µg/litre benzo [b]fluoranthene, 6 µg/litre benzo [j]fluoranthene
plus benzo [k]fluoranthene, 6 µg/litre benzo [e]pyrene, 6 µg/litre
benzo [a]pyrene, 3 µg/litre benzo [c]phenanthrene, 3 µg/litre
indeno[1,2,3- cd]pyrene, 3 µg/litre benzo [ghi]perylene, 2 µg/litre
benzo [a]fluorene, 2 µg/litre benzo [b]fluorene, 2 µg/litre
perylene, and 1 µg/litre dibenz [a,h]-anthracene. The PAH contents of
wastewater from gas washers in one Norwegian steel production plant
were of the same order of magnitude (Berglind, 1982).
Table 15. Polycyclic aromatic hydrocarbon concentrations [µg/litre]
in wastewater from aluminium production in Norway
Compound [1] [2] [3]
Acenaphthene NR NR 5
Acenaphthylene NR NR 1
Anthracene 1.1-2.8 0.9 10
Anthenthrene < 1-3.2 NR NR
Benzo[b+k]fluoranthenes 6.8-38.1 NR NR
Benzo[j+k]fluoranthenes NR 10.5 5
Benz[a]anthracene 2.5-5.6 14.6 11
Benzo[a]fluorene 1.5-3.4 8.2 13
Benzo[a]pyrene 1.3-7.4 13.5 4
Benzo[b]fluoranthene NR 21.2 9
Benzo[b]fluorene 1.3-3.0 7.2 2
Benzo[c]phenanthrene NR NR 3
Benzo[e]pyrene 2.6-16.4 17.0 5
Benzo[ghi]perylene NR 8.3 2
Chrysene and triphenylene 5.8-16.0 27.3 17
Coronene < 1-2.0 NR NR
Dibenz[a,h]anthracene NR NR 1
Fluoranthene 12.4-20.8 7.5 124
Fluorene NR NR 3
Indeno[1,2,3-cd]pyrene NR 8.1 2
1-Methyphenanthrene NR 0.4 NR
Naphthalene NR NR 1
Perylene NR 3.2 1
Phenanthrene 14.0-23.1 1.8 34
Pyrene 5.6-15.3 6.4 76
[1] Two samples of wastewater with two runs each from one aluminium
production plant (Kadar at al., 1980);
[2] Wastewater from one aluminiurn production plant; no further
information (Olufsen, 1980);
[3] Effluent from gas washers from one aluminium smelter (Berglind, 1982)
When the water samples were filtered through solid sorbents, the results
may be underestimates of the actual content (see section 2.4.1.4).
NR, not reported
The release of 10 PAH into water from different industries in the
Netherlands was estimated to be 4 t/year (Slooff et al., 1989).
(c) Emissions to the geosphere
The levels of PAH in ash samples from various incinerators are
shown in Table 16. The values given by Eiceman et al. (1979) were
based on the gas chromatographic responses of pyrene and
benzo [a]pyrene. The concentrations of PAH in ashes from coal-fired
power plants were of the same magnitude as the background levels of
these compounds in soil, but fly ash from municipal waste incinerators
may contain significantly higher levels (Guerin, 1977; Kanij, 1987).
The total PAH content of filter residues in incinerators was about
0.20-0.5 µg/g. The compounds are assumed to be tightly bound to
particle surfaces and not mobile in an aqueous environment in the
absence of organic solvents (WHO, 1988). In a comparison of 26
incineration plants, combustion conditions were shown to have a marked
influence on PAH release (Wild et al., 1992).
The material dredged from harbour areas may have a significant
PAH content (see also sections 5.3.3 and 5.3.4). The annual load of
naphthalene, anthracene, phenanthrene, fluoranthene,
benz [a]anthracene, chrysene, benzo [k]-fluoranthene,
benzo [a]pyrene, benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene
in material dredged from Rotterdam harbour was about 12 t (year not
given). The main PAH were fluoranthene and benz [a]anthracene (Slooff
et al., 1989).
3.2.7.2 Other diffuse sources
(a) Atmosphere
(i) Mobile sources
PAH are released into the atmosphere by motor vehicle traffic.
The profile of the PAH released and the quantity of PAH in the exhaust
are fairly similar, independently of the type of engine and the PAH
content of the fuel, indicating that the emitted compounds are formed
predominantly during combustion (Meyer & Grimmer, 1974; Janssen, 1980;
Stenberg, 1985; Williams et al., 1989). PAH accumulate in used engine
oil, but the importance of the PAH content of engine oil on emissions
is still under discussion. Janssen (1980), Pischinger & Lepperhoff
(1980), and Stenberg (1985) assumed that the PAH content of the oil
played only a minor role, but Williams et al. (1989) showed in tests
with diesel fuel that it may contribute considerably to the release of
particulate PAH. There is also doubt about whether PAH emissions are
indepen-dent of the aromaticity of the fuel. Janssen (1980) stated
that release of PAH into the atmosphere is not increased if the
aromaticity does not exceed a concentration of 50% volume (see also
Schuetzle & Frazier, 1986). According to Stenberg (1985), the release
of PAH by automobile traffic is dependent on the:
Table 16. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in ash samples from coal-fired power plants and municipal waste and sewage
sludge incinerators
Compound Coal-fired Municipal waste incinerators Sewage
power plants sludge
incinerators
Netherlands USA Canada Japan Netherlands Canada UK Italy (UK)
[1] [2] [3] [3] [3] [4] [5] (mean) [6] [5]
Acenaphthene + NR NR NR NR NR NR 1-258(7.8) 289-1022i NR
fluoranthene
Acenaphthylene NR NR NR NR NR 3.35 NR 5-1394 NR
Anthracene < 0.14-0.5 NR 10/500 10/10 200 NR 1-62(2.3) 42-651 NR
Anthanthrene < 0.24-< 0.5 NR NR NR NR NR NR NR
Benz[a]anthracene < 0.6-< 1.2 NR NR NR NR NR 1-1646a(12) 280-1278 3a
Benzo[a]fluoranthene NR 36.8 NR NR NR NR NR NR
Benzo[a]pyrene < 0.29-< 1.8 NR ND/400 ND/ND ND NR 1-596(8.2) 1014-3470 3
Benzo[b]fluoranthene < 0.6-< 0.29 NR NR NR NR NR 1-873(5.7) 1818 6
Benzo[b]fluorene < 2.0-< 4 11.8 NR NR NR NR NR NR
Benzo[e]pyrene < 2.9-< 6 NR NR NR NR NR NR 458-1786 NR
Benzo[ghi]perylene < 1.6-1.7 NR NR NR NR NR 10-9507 (62.3) 700-2377 135
Benzo[j]fluoranthene < 4.5-< 9 NR ND/400b ND/NDb NDb NR NR NR
Benzo[k]fluoranthene < 0.15-< 2.8 NR NR NR NR NR 1-276(1.5) 1535 NR
Chrysene < 1.5-< 3 NR NR NR NR NR NR 570-1973 NR
Coronene NR NR NR NR NR NR 3-238 (31.3) 36
Dibenz[a,h]anthracene < 4.2-< 8.2 NR NR NR NR NR 1-167(5.2) 57/69 1
Fluoranthene 1.1-5.2 < 13.4 2/500 3/ND 20 2.14-43.2 1-765 (8.6) 1684-10 890 1
Fluorene NR NR ND/10 ND/ND 60 2.57/4.41 NR 45-522 NR
Indeno[1,2,3-cd]pyrene < 0.82-< 1.6 NR NR NR NR NR NR 478-1343 NR
Naphthalene NR 8.3 NR NR NR NR 4/15(0.2) NR
Perylene < 0.16-< 0.3 NR NR NR NR NR NR 259 NR
Phenanthrene 4.0-43 17.6 NR NR NR 8.76-154c 2-5402 (36.5) 1616-7823 6
Pyrene 0.72-2.9 < 19.0 1/500 1/ND 10 2.47-19.6 1-3407 (45.3) 1863-8799 10
Triphenylene < 2.5-< 5.0 NR NR NR NR 12.7a NR NR
Table 16 (continued)
[1] Pulverized coal ash (Kanij, 1987);
[2] Fly ash (Guerin, 1977);
[3] Fly ash (Eiceman at al., 1979);
[4] Fly ash (Chiu at al., 1991);
[5] Fly ash 26 incinerators with different firing techniques (Wild et al., 1992);
[6] Fly ash from electrostatic precipitator and scrubber (Morselli & Zappoli, 1988)
NR, not reported; ND, not detected; /, single measurements
a With chrysene
b Isomers not specified
c With anthracene
i Only acenaphthene
- aromaticity of the fuel;
- starting temperature: Starting at -10°C results in threefold
higher PAH emissions than a standardized cold start (+ 23°C); the
emission factors measured by Larssen (1985) were significantly
higher in winter than in summer.
- ambient temperature: Low ambient temperatures (5-7°C) increase
PAH emissions from petrol-fuelled vehicles by five to 10 times,
depending on the engine used.
- test conditions: Three standardized test cycles are in general
use: a test developed by the Economic Commission for Europe of
the United Nations (ECE) in Europe; the Federal Test Procedure
(FTP) in the USA; and the Japanese test cycle in Japan. Emissions
at cold start may be lower and those at hot start slightly higher
in the FTP than in the ECE test, but overall agreement between
the tests is good.
- air:fuel ratio (l): Small variations around l = 1, representing
stoichiometric levels of fuel and air, do not affect PAH
emissions significantly; richer mixtures lead to increasing PAH
emissions, and bad ignition at l = 0.8 causes a sharp increase in
PAH emissions.
- type of fuel: Emissions of the sum of phenanthrene,
fluoranthene, pyrene, benzo [ghi]fluoranthene,
cyclopenta [cd]pyrene, benz [a]-anthracene, chrysene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [e]pyrene,
benzo [a]-pyrene, indeno[1,2,3- cd]pyrene,
benzo [ghi]-perylene, and coronene decreased in the FTP cycle as
follows: diesel (total PAH; 960 µg/km) > petrol (170 µg/km) >
petrol containing methanol or ethanol (43-110 µg/km) > methanol
= liquefied petro-leum gas = catalyst-equipped petrol-fuelled
vehicles (6-9 µg/km) (Stenberg, 1985). In comparable
measurements, similar results were obtained but with a much lower
average emission rate for diesel-fuelled vehicles: 186 µg/km for
total PAH, including fluoranthene, pyrene, benz [a]anthracene,
chrysene, benzo [b]-fluoranthene, benzo [e]-pyrene,
benzo [a]pyrene, perylene, indeno[1,2,3- cd]pyrene,
benzo [ghi]-perylene, and coronene. It was not stated whether
the difference in the emission rates was due to the numbers of
PAH chosen for analysis (Lies et al., 1986).
PAH emissions in the exhaust from spark-ignition automobile
engines can be reduced by operation with lean air:fuel ratios, smaller
quenching distances in the combustion chamber, and increased cylinder
wall temperatures in the engine (Pischinger & Lepperhoff, 1980;
Lepperhoff, 1981). Diesel-fuelled engines with low emissions of total
unburnt gaseous hydrocarbons have low rates of PAH emission. Control
can therefore be achieved by using conventional techniques for
reducing unburnt gaseous hydrocarbons (Williams et al., 1989).
Fluoranthene and pyrene constitute 70-80% of total PAH emissions
from vehicles (Lies et al., 1986; Volkswagen AG, 1989; see also Table
17), whereas the emissions from one diesel-fuelled truck consisted
mainly of naphthalene and acenaphthene (Nelson, 1989). Although
cyclopenta [cd]pyrene is emitted at a high rate from petrol-fuelled
engines, its concentration in diesel exhaust is just above the limit
of detection, probably because the oxidizing conditions in
diesel-fuelled engines decompose this relatively reactive compound
(Lies et al., 1986).
The amounts of PAH released from vehicles with three-way
catalytic converters are much lower than those from vehicles without
catalysts (Table 18). The total amount of PAH was increased by a
factor of about 40 between new and used catalytic converters (Hagemann
et al., 1982). PAH emissions from diesel-fuelled vehicles can be
reduced by > 90% by a combination of a catalytic converter and a
particulate trap, as shown by experiments with a heavy-duty
diesel-fuelled truck (Westerholm et al., 1989). Westerholm et al.
(1991) found benz [a]anthracene, benzo [b]fluoranthene,
benzo [k]fluoranthene, benzo [e]-pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, fluoranthene, pyrene,
anthracene, and coronene in much lower amounts than other
investigators, while some other PAH that were not measured by other
investigators, especially phenanthrene and 1-methylphenanthrene, were
detected at quite high concentrations. These differences are possibly
due to the driving cycle used.
Measurements made on particulate matter in the exhausts of light-
and heavy-duty diesel-fuelled vehicles with different fuel qualities
showed concentrations of 1 mg/kg each of benz [a]anthracene,
benzo [b]fluoranthene plus benzo [j]fluoranthene, benzo [a]pyrene
plus benzo [e]pyrene, and benzo [ghi]perylene and 290 mg/kg pyrene.
The results were strongly dependent on the driving cycle and
individual engine conditions (CONCAWE, 1992).
The PAH concentrations measured in the exhaust gases of different
vehicles are shown in Table 19. The differences in PAH emissions from
petrol- and diesel-fuelled vehicles are still under discussion. When
the data of Behn et al. (1985) are compared with those of Klingenberg
et al. (1992), diesel-fuelled vehicles emitted larger amounts of PAH
than petrol-fuelled vehicles. Benzo [a]pyrene was emitted at a rate
of 6 µg/km from a petrol-fuelled vehicle without a catalyst and at 5
µg/km from a diesel-fuelled vehicle (Gibson, 1982). When the PAH
emissions from 10 petrol- and 20 diesel-engined vehicles were measured
under three urban cycles, the mean emission factors (µg/km) for
benzo [a]pyrene were 12 with petrol and 0.56 with diesel in a cold,
low-speed cycle, 0.50 with petrol and 0.37 with diesel in a hot,
low-speed cycle, and 0.37 with petrol and 0.24 with diesel in a hot,
free-flow cycle (Combet et al., 1993). Considerably higher emission
rates were found from four petrol-fuelled passenger cars without
catalysts, 11 with catalysts, and eight diesel-fuelled passenger cars,
two of which had oxidation catalysts, on a chassis dynamometer at the
USA FTP 75 cycle. The diesel-fuelled vehicles emitted about as much
benzo [a]pyrene as the petrol-fuelled vehicles without catalysts
(5-25 µg/km), while the petrol-fuelled vehicles with catalysts had
emission rates significantly below 2 µg/km. The diesel-fuelled
vehicles with oxidation catalysts had emissions of about 5 µg/km
(Klingenberg et al., 1992).
The following emission factors were given for motorcycles and
two-stroke mopeds: 1000 µg/km naphthalene, < 32-650 µg/km
phenanthrene, < 11-170 µg/km anthracene, < 5-110 µg/km fluoranthene,
< 2-11 µg/km chrysene, < 2-11 µg/km indeno[1,2,3- cd]pyrene,
< 1-1200 µg/km benz [a]anthracene, 0-63 µg/km benzo [ghi]perylene,
0-16 µg/km benzo [a]pyrene, and 0-11 µg/km benzo [k]fluoranthene
(Slooff et al., 1989).
Further PAH emissions may result from the abrasion of asphalt by
vehicle traffic, so that PAH in asphalt and bitumens (see section
3.2.1) may contribute considerably to the total PAH emissions due to
automobile traffic. The abrasion caused by spiked tyres in winter was
estimated to be 20-50 mg/km (Lygren et al., 1984).
Another source of PAH from motor vehicle traffic is clutch and
break linings, which are subject to considerable thermal stress,
sometimes resulting in pyrolytic decomposition of abraded particles.
Numerous PAH were found in the abraded dust of brake and clutch
linings in one study, but the values show large standard deviations,
due, presumably, to the fact that the substances are adsorbed onto
asbestos fibres from which they are difficult to separate (Knecht et
al., 1987). Total PAH release from clutch and brake linings cannot be
estimated from the available data.
Rubber vehicle tyres contain highly aromatic oils as softeners.
These oils, which can contain up to 20% PAH, are used at
concentrations of 15-20% in rubber blends (Duus et al., 1994). In
Sweden, it was considered that the input of PAH to the atmosphere from
rubber particles was important (National Chemicals Inspectorate,
1994).
According to estimates for Belgium, western Germany, and the
Netherlands in 1985, the annual PAH input into the atmosphere from
vehicle traffic ranges from < 10 t/year for benzo [ghi]fluoranthene,
benz [a]anthracene, benzo [k]-fluoranthene, benzo [a]pyrene, and
indeno[1,2,3- cd]pyrene, to < 10-20 t/year for anthracene,
fluoranthene, and chrysene, to 10-70 t/year for phenanthrene, to about
100-1000 t/year for naphthalene (Slooff et al., 1989). Values of the
same order of magnitude were reported for emissions of naphthalene in
1987 (Society of German Chemists, 1989) and benzo [a]pyrene in 1989
in western Germany (Ministers for the Environment, 1992) and for total
PAH in 1985 in Norway and Sweden (Bjorseth & Ramdahl, 1985). The total
annual PAH input from vehicle traffic in the USA in 1985 was about
2200 t/year (Bjorseth & Ramdahl, 1985). In Canada, the total PAH input
was estimated to be about 200 t in 1990; 155 t were assumed to be due
to diesel-fuelled and 45 t to petrol-fuelled vehicles (Environment
Canada, 1994).
Table 17. Polycyclic aromatic hydrocarbon emission factors (µg/km) for petrol-fuelled vehicles
Compound [1] [2] [3] [4] [5] [6] [7]
Anthracene NR 0.7/0.7a NR 2/99b NR 21-42 0.6
37/1988c
Anthanthrene NR 0.2/1.3 NR NR NR NR NR
Benzo[b+j+k]fluoranthene NR NR NR NR NR NR 7.6
Benzo[b+k]fluoranthene NR 3.9/7.0 NR NR 0.23-0.54/2.55-9.20 NR NR
Benz[a]anthracene NR 5.7/5.9 3.5-9.0 NR 0.06-0.35/2.5-8.0 5-16 5.1
Benzo[a]pyrene NR 1.9/4.51 1.5-14.5 0.06-2/1-12b 0.06-0.62/1.30-10.4 2-11 3.7
Benzo[e]pyrene NR 2.6/6.2 NR 0.2/2-14b 0.08-0.54/2.54-9.20 NR 5.1
Benzo[ghi]fluoranthene NR 5.6/12 NR NR NR NR 8.8
Benzo[ghi]perylene NR 5.9/13 NR NR 0.19-0.75/1.45-17.5 5-21 18.9
Benzo[j]fluoranthene NR 1.1/0.9 NR NR NR NR NR
Benzo[k]fluoranthene NR NR NR NR NR 0-5 NR
Chrysene NR 6.7/8.7 NR NR 0.12-0.73/2.78-23.1 11-42 7.7
Coronene NR 6.5/12 1.5-20.0 NR NR NR 29.5
Cyclopenta[cd]pyrene NR 2.9/12 NR NR NR NR 16.5
Fluoranthene NR 14/20 NR 3/139-211b 2.7/43.3d 11-158 10.4
ND/186-280c
Indeno[1,2,3-cd]pyrene NR 1.7/3.6 NR NR 0.06-0.43/0.83-6.67 5-21 4.2
Naphthalene 8100-8600a NR NR NR NR 2300f NR
210-2651
Perylene NR 0.3/0.5 NR NR 0.01-0.06/0.25-1.82 NR NR
Phenanthrene NR 2.6/2.9 NR NR NR 84-210 1.8
Pyrene NR 28/31 43-184 4-16/12-268b 2.9/43.0b NR 19.2
ND/124-360c
Table 17 (continued)
NR, not reported; ND not detected (detection limit not stated); /, single measurements
a Two driving distances
b Only particulate phase considered
c Only gaseous phase considered
d Average
e Depending on analytical conditions
f With converter
[1] From measurements in tunnel with converters (Hampton at al., 1983);
[2] One vehicle without converter (Alsberg et al., 1985);
[3] Various tests conducted mainly in the 1970s, some unstandardized, different numbers of vehicles,
without converters (Stenberg, 1985);
[4] FTP cycle only, number of vehicles not given; year of manufacture 1980-85 = petrol-engine vehicles
with converter; 1973-81 = petrol-engine vehicles without converter (Schuetzle & Frazier, 1986);
[5] Various standardized test procedures; four petrol-engine vehicles without, seven with three-way-converter
for each test, all with four or five cylinders (Volkswagen AG, 1988);
[6] No information about test cycle or number of cars tested; city roads, motorways and other roads tested;
no distinction between vehicles with and without converter, unless otherwise stated (Slooff et al.,
1988, 1989);
[7] One petrol-engine vehicle without converter in USFTP test cycle (Strandell at al., 1994)
Table 18. Polycyclic aromatic hydrocarbon emission factors (µg/km) for diesel-fuelled vehicles
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene NR NR NR NR NR NR 41-128 NR
Anthracene 17/63 65-273a 1.2/3.0 NR 21-73b 3.3 2.9-26 4.6
1305-5568c
Benzo[b+j+k]fluoranthene NR NR NR NR NR NR 1.7-12d 5.0
Benzo[b+k]fluoranthene 2.6/47 NR 3.9/6.1 5.57-14.96 NR 0.29 NR NR
Benz[a]anthracene 8/43a NR 4.0/7.0 2.73-3.91 11-21b 0.47 0.7-9.6 2.0
Benzo[a]fluorene NR NR NR NR NR 2.4 NR NR
Benzo[a]pyrene < 1/20 0.6-34a 1.6/2.2 2.09-7.23 1-5 < 0.06 0.5-3.2 1.5
Benzo[e]pyrene 3/38 2-40a 2.5/4.1 2.40-52.8 NR 0.15 1.1-9.9 4.0
Benzo[ghi]fluoranthene NR NR 4.0/12 NR NR 1.5 NR 10.6
Benzo[ghi]perylene < 1/18 NR 1.9/3.1 2.84-26.3 9e < 0.13 0.5-3.7 2.0
Chrysene 14/67 NR 11/25 4.7-21.1 16-42b 2.8f 3.5-28 3.7
Coronene NR NR 0.3/20.7 NR NR < 0.01 NR NR
Cyclopenta[cd]pyrene NR NR 3.6/3.9 NR NR 0.18 NR 4.0
Fluoranthene 58/200 139-580a 13/38 70g 21-105b 17 14-34 43.7
186-771c
Fluorene NR NR NR NR NR NR 38-228 NR
Indeno[1,2,3-cd]pyrene NR NR 1.5/2.3 0.89-7.52 9e < 0.04 NR 1.2
1-Methylphenanthrene NR NR NR NR NR 41 NR NR
Naphthalene NR NR NR NR 2100-6302b NR 1030-1805 NR
Perylene < 1/2 NR NR 0.23-1 NR < 0.01 NR NR
Phenanthrene 295/524 NR 4.6/25 NR NR 2.9 79-308 54.8
Pyrene < 0-9/22 24-734a 20/104 66.9g NR 11 9-30 35.4
702-982c
Table 18 (continued)
NR, not reported; /, single measurements;
[1] ECE test; two passenger cars with < 50 000 and > 100 000 km odometer readings (Scheepers & Bos, 1992);
[2] FTP cycle; number of vehicles not given; year of manufacture, 1980-85 (Schuetzle & Frazier, 1986);
[3] Chassis dynamometer; one heavy-duty vehicle (Westerholm et al., 1986);
[4] Various standardized testing procedures; seven vehicles with four or five cylinders for each test (Volkswagen
AG, 1988);
[5] No information on test cycle or number of cars tested; three traffic situations (Slooff at al., 1989);
[6] Bus cycle simulating public transport (duration 29 min; driving distance, 11.0 km; average speed, 22.9 km/h);
one heavy-duty truck; measurement of particle phase (Westerholm at al., 1991);
[7] Bus cycle (duration, about 10 min after warm-up, each ramp consisting of 10 s acceleration, 10 s constant speed
of 12 km/h, 4.5 s deceleration, 7 s idling); three trucks and two buses without particle trap, two buses with
particle trap (Lowenthal et al., 1994);
[8] US FTP cycle; one passenger car (Strandell at al., 1994)
a Particle phase
b Automobiles and trucks
c Gas phase
d Isomers not specified
e Trucks
f With triphenylene
g Average
Table 19. Polycyclic aromatic hydrocarbon concentrations (µg/m3) in the
exhaust gases of different vehicles
Compound [1] [2] [3]
Acenaphthene NR NR < 0.02-0.81
Acenaphthylene NR NR < 0.02-4.16
Anthracene NR NR < 0.02-6.45
Anthanthrene 0.02-0.07 0.11-0.12 NR
Benz[a]anthracene 1.91-2.24 3.53-4.64 NR
Benzo[a]pyrene 0.46-0.76 2.03-2.33 < 0.02-4.97
Benzo[b]fluoranthene 1.53-2.04a 7.37-8.58a 0.06-6.63
Benzo[b]fluorene NR NR 0.11-12.7
Benzo[e]pyrene 1,07-1.24 2.46-2.90 0.09-6.16
Benzo[ghi]fluoranthene 0.46-0.59 4.81-7.19 NR
Benzo[ghi]perylene 0.76-1.04 3.42-4.41 0.22-1.81
Benzo[k]fluoranthene NR NR < 0.02-2.68
Chrysene 2.37-2.97b 7.37-8.58b 0.07-25.48
Coronene 0.26-0.30 1.82-2.32 < 0.02-1.80
Cyclopenta[cd]pyrene 1.86/2.26 5.80-6.09 NR
Dibenz[a,h]anthracene 0.04-0.07 0.32-0.35 < 0.02-0.44
Fluoranthene 11.83-13.09 20.90-25.30 0.16-35.94
Fluorene NR NR 0.06-2.16
Indeno[1,2,3-cd]pyrene 0.30-0.41 2.89-4.06 < 0.02-0.80
Perylene 0.10-0.26 0.21-0.33 0.13-5.55
Phenanthrene NR NR < 0.02-4.16
Pyrene 6.86-8.96 12.20-15.20 0.06-21.31
NR, not reported; /, single measurements;
[1] One vehicle with spark-ignition engine on chassis dynamometer
at 75% of maximum engine performance (velocity, about 50 km/h)
with varying test periods (Behn et al., 1985);
[2] One turbo-charged diesel-fuelled vehicle on chassis dynamometer
at 75% of maximum engine perfornance (velocity, about 50 km/h)
and a test period of 0.5 h; three tests for each component
(Behn at al., 1985);
[3] Two diesel-fuelled truck engines at different engine speeds
(Moriske at al., 1987)
a With benzo[k]fluoranthene
b With triphenylene
Measurements of PAH concentrations in a Belgian highway tunnel in
1991 were used to calculate emission factors of 2 µg/km for
indeno[1,2,3- cd]pyrene and coronene and 32 µg/km for
benzo [ghi]perylene. The corresponding annual PAH emissions in
Belgium were estimated to be 0.11 t/year for perylene and anthanthrene
and 1.3 t/year for benzo [ghi]perylene; the combined release of
pyrene, benz [a]anthracene, chrysene, benzo [b]fluoranthene,
benzo [j]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [e]pyrene, perylene, anthanthrene, benzo [ghi]perylene,
indeno[1,2,3- cd]pyrene, dibenzo [a,c]anthracene,
dibenzo [a,h]anthracene, and coronene was 8.3 t/year (De Fré et al.,
1994).
The importance of PAH released by aircraft is also under
discussion. While Bjorseth & Ramdahl (1985) classified the maximum
emission in Norway and Sweden in 1985 of 0.1 t/year as small, Slooff
et al. (1989) estimated that the release of naphthalene, anthracene,
phenanthrene, fluoranthene, benz [a]-anthracene, chrysene,
benzo [k]fluoranthene, benzo [a]pyrene, benzo [ghi]-perylene, and
indeno[1,2,3- cd]pyrene was 51 t/year in 1985. The following
concentration ranges were measured in the exhaust gases from two US
by-pass turbine engines at various power settings: naphthalene,
0.77-4.7 µg/m3; phenanthrene, 0.46-1.3 µg/m3; pyrene, 0.15-0.61
µg/m3; fluoranthene, 0.13-0.51 µg/m3; acenaphthene, 0.03-0.21
µg/m3; anthracene, 0.029-0.12 µg/m3; benzofluoranthenes
(unspecified), 0.028-0.096 µg/m3 (isomers not specified); chrysene,
0.026-0.064 µg/m3; benzo [a]pyrene, 0.021-0.073 µg/m3;
benz [a]anthracene, 0.019-0.16 µg/m3; acenaphthylene, 0.017-0.31
µg/m3; benzo [e]pyrene, 0.017-0.057 µg/m3; dibenz [a,h]anthracene,
0.011-0.064 µg/m3; indeno[1,2,3- cd]pyrene, 0.011-0.054 µg/m3; and
benzo [ghi]perylene, 0.011-0.045 µg/m3. Cyclopenta [cd]pyrene was
not detected (limit of detection not stated) (Spicer et al., 1992).
(ii) Domestic residential heating
The main PAH released by domestic slow-combustion furnaces and
hard-coal and brown-coal coal stoves were fluoranthene, pyrene, and
chrysene, which comprised 70-80% of the total PAH in model experiments
(Ahland & Mertens, 1980). The specific emission factors for various
fuels used in residential heating are shown in Table 20 for coal
stoves and Table 21 for wood stoves (Bjorseth & Ramdahl, 1985).
Few data are available on the release of PAH from oil stoves.
Benzo [a]pyrene was detected at a concentration of < 0.05 µg/kg in
one burner-boiler combination (Meyer et al., 1980), and 0.006 and 4
µg/kg benzo [a]pyrene and 0.02 and 15 µg/kg benzo [e]pyrene were
found during testing of atomizer and vaporizer oil heating techniques,
respectively (Ahland et al., 1985). PAH emissions from residential oil
heating seem to be about one order of magnitude lower than those from
coal stoves.
Table 20. Specific polycyclic aromatic hydrocarbon emission factors (mg/kg) for residential
coal stoves
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene NR NR NR NR 65 NR
Acenaphthylene NR NR NR 0.427 NR 7.74
Anthracene 0.0039 NR > 0.595 2.113 26a,b 1.49
Anthanthrene NR NR 0.03-0.08 0.665 NR NR
Benz[a]anthracene NR NR 1.04-3.68 7.181 NR 0.61
Benzo[a]fluorene 0.0009 NR NR 1.366 NR NR
Benzo[a]pyrene 0.0003 0.014-17.4 0.043-1.3 4.303 5c NR
Benzo[b]fluoranthene 0.0002 NR 2.028d 6.102 NR NR
Benzo[b]fluorene 0.0007 NR NR 0.874 NR NR
Benzo[c]phenanthrene NR NR 1.462e 2.215 4 NR
Benzo[e]pyrene 0.0005 0.09-16.2 0.40-1.70 3.994 NR NR
Benzofluoranthenesf NR NR 0.90-3.20 NR 6 NR
Benzo[ghi]fluoranthene NR NR NR 3.323 NR 0.67
Benzo[ghi]perylene 0.0001 NR 0.30-0.50 3.855 NR NR
Benzo[j]fluoranthene NR NR NR 6.782 NR NR
Benzo[k]fluoranthene NR NR 0.569 NA NR NR
Chrysene 0.0016g NR 2.09 9.571 6h 0.68
1.39-5.60g
Coronene NR NR 0.081 1.898 NR NR
Cyclopenta[cd]pyrene NR NR 0.145 3.590 NR NR
Dibenz[a,h]anthracene NR NR 0.113 NR 5 NR
Fluoranthene 0.016 NR 3.30-17.0 28.4 9a 3.47
Fluorene NR NR < 0.065 1.05 44 1.64
Indeno[1,2,3-cd]pyrene 0.0002 0.20-0.60 4.60 NR 4 NR
1-Methylphenanthrene NR NR NR 2.217 NR NR
Naphthalene NR NR NR NR 254 35.7
Perylene NR NR 0.20-0.50 1.134 NR NR
Phenanthrene 0.046 NR > 3.69 3.984 NR 7.42
Pyrene 0.020 NR 2.98-12.0 26.589 8 3.38
Triphenylene NR NR 0.804 NR NR NR
Table 20 (continued)
NR, not reported;
[1] One new residential stove fuelled with charcoal (Ramdahl et al., 1982);
[2] Five coal types: hard-coal and brown-coal briquettes and anthracite (Ahland etal., 1985);
[3] Burning of brown coal in different domestic stoves; single values refer
to one slow-combustion stove; ranges refer to one slow-combustion stove
and one permanent built-in combustion stove at medium load (Grimmer et al.,
1983a);
[4] One slow combustion stove fueled with hard-coal briquettes (Grimmer at al., 1985);
[5] One warm-air furnace and one hot-water boiler fuelled with three different bituminous
coals (Hughes & DeAngelis, 1982);
[6] Samples from chimney of a detached family house with brown-coal heating in Leipzig, Germany
(Engewald et al., 1993)
a In particulate phase
b With phenanthrene
c With benzo[e]pyrene and perylene
d With benzo[j]fluoranthene
e With benzo[ghi]fluoranthene
f Isomers not specified
g With triphenylene
h With benz[a]anthracene
Table 21. Specific polycyclic aromatic hydrocarbon emission factors
(mg/kg) for residential wood stoves
Compound [1] [2] [3]
Anthracene 0.119-1.859 10.4-146.3a 130/3600
Benz[a]anthracene 0.060-0.781 NR 55/740
Benzo[a]fluorene 0.018-0.845 NR NR
Benzo[a]pyrene 0.046-0.617 1.1-11.6b NR
Benzo[b]fluoranthene 0.108-1.016 NR NR
Benzo[b]fluorene 0.011-0.393 NR NR
Benzo[c]phenanthrene NR 0.2-2.3 NR
Benzo[e]pyrene 0.035-0.350 NR NR
Benzofluoranthenesc NR 1.5-15.9 NR
Benzo[ghi]fluoranthene NR 0.4-6.7 NR
Benzo[ghi]perylene 0.034-0.544 1.1-9.9 NR
Chrysene 0.481-0.829d 1.3-37.1e 67/770d
Cyclopenta[cd]pyrene 0.04-0.720 0.5-8.9 15/800
Fluoranthene 0.296-3.245 1.2-31.6 190/2300
Indeno[1,2,3-cd]pyrene 0.033-0.415 NR NR
1-Methylphenanthrene 0.141-2.213 NR NR
Perylene 0.023-0.274 NR NR
Phenanthrene 0.834-8.390 NR 480/7500
Pyrene 0.232-3.822 1.3-24.0 160/2100
NR, not reported; /, single measurements;
[1] One small residential wood stove burning spruce and birch; normal and
slow burning of each kind of wood (Ramdahl at al., 1982);
[2] One zero-clearance fireplace with heat circulation and two airtight
wood stoves (baffled and non-baffled) fuelled with red oak and yellow
pine with different moisture contents (Peters at al., 1981);
[3] One wood-burning stove with and without catalytic combustor (Tan et
al., 1992)
a With phenanthrene
b With benzo[e]pyrene and perylene
c Isomers not specified
d With triphenylene
e With benz[a]anthracene
Numerous PAH, including acenaphthene, acenaphthylene, fluorene,
phenanthrene, anthracene, 1-methylphenanthrene, fluoranthene, pyrene,
benzo [a]fluorene, benzo [ghi]fluoranthene, benzo [c]phenanthrene,
cyclo-penta [cd]pyrene, benz [a]anthracene, chrysene plus
triphenylene, benzo [b]-fluoranthene, benzo [j]fluoranthene,
benzo [j]fluoranthene, benzo [e]pyrene, benzo [a]pyrene, perylene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, and anthanthrene, were
detected in atmospheric emissions from straw-burning residential
stoves, at concentrations mainly in the range of 10 µg/kg to 19 mg/kg
(Ramdahl & Muller, 1983).
The total PAH content of barbecue briquettes was 2.5-13 µg/g
sample. PAH were found in coal and charcoal briquettes but not in lava
stones or pressed sawdust briquettes (Kushwaha et al., 1985).
The PAH content of soot from domestic open fires was 3-240 mg/kg
benzo [a]pyrene, 2-190 mg/kg chrysene, 2-100 mg/kg
benz [a]anthracene, 1-77 mg/kg indeno[1,2,3- cd]pyrene, 2-39 mg/kg
benzo [e]pyrene, 1-29 mg/kg benzo [ghi]perylene, 1-18 mg/kg
coronene, 1-14 mg/kg perylene, and 1-12 mg/kg anthracene (Cretney et
al., 1985).
The amounts of PAH emitted from coal-fired domestic stoves seem
to depend on the quality of the coal used and on the firing technique.
Generally, hard coal has a higher energy content than other fuels;
thus, less total PAH is emitted per unit of gained energy. The lowest
specific emission factors for benzo [a]pyrene and benzo [e]pyrene
were found with anthracite and the highest with gas coal and gas-flame
coal (Ahland et al., 1985). Model experiments with a slow-combustion
stove showed that pitch-bound hard-coal briquettes emitted about 10
times more PAH than bitumen-bound briquettes (Ratajczak et al., 1984).
The use of pitch-bound hard-coal briquettes for domestic heating may
thus be an important source of PAH in the atmosphere. Use of this fuel
was restricted by law to permanent combustion stoves in western
Germany in 1974, and since 1976 only bitumen-bound hard-coal
briquettes have been produced there (Ratajczak et al., 1984). There is
no comparable information for other countries. The levels of airborne
PAH from a permanent combustion stove burning brown coal were two to
four times higher than those from a slow-combustion stove with a
medium load (Grimmer et al., 1983a).
About 25-1000 times more PAH are produced from burning wood than
from the same mass of charcoal. Since the yield of energy per unit
mass is similar, burning wood also produces more PAH per unit of
energy. Burning conditions are apparently the major determinant of
emission and are much more important than the kind of wood (Ramdahl et
al., 1982). In areas where domestic heating is predominantly by wood
burning, most airborne PAH may come from this source, especially in
winter (e.g.Cooper, 1980). Using benz [a]pyrene as an indicator in
extensive measurements in New Jersey, USA, the amounts emitted were
found to be more than 10 times higher during the heating period than
in seasons when heating is not required. An assessment of combustion
source also showed that residential combustion of wood was the
decisive factor (Harkov & Greenberg, 1985). About 43-47% of the total
PAH released in winter in Fairbanks, Alaska, came from residential
wood stoves (Guenther et al., 1988).
The PAH concentrations in gases in the chimney stacks of
residential coal and oil furnaces are given in Table 22. The highest
levels were found during the start of the burning process (Brockhaus &
Tomingas, 1976). Measurements with five qualities of coal showed that
Extrazit(R), a specially treated coal, emitted smaller quantities of
smoke and the lowest PAH levels, and anthracite briquettes emitted the
highest levels. Presumably, the high PAH emissions from anthracite
briquettes are due to the binding agent, hard coal-tar, which has an
especially high PAH content. Furnaces with atomizer oil burners seemed
to emit less PAH than those with vaporizers. Measurements in a
slow-combustion stove and a tiled stove showed that the highest
concentrations of PAH were associated with dust of a particle size of
< 2.1 m. As for residential heating with wood, in areas where the
predominant form of domestic heating is coal burning, a major
proportion of airborne PAH may come from this source, especially in
winter (Moriske et al., 1987).
Table 22. Polycyclic aromatic hydrocarbon concentrations (µg/m3)
in stack gases from residential coal and oil stoves
Compound Coal Oil
Benz[a]anthracene 0.0157-2630 0.2-0.6
Benzo[a]pyrene 0.0016-1270 0.19-0.67
Benzo[b]fluoranthene 0.0188-3270 0.004-0.68
Benzo[e]pyrene 0.0261-3430 0.4-6.9
Benzo[ghi]perylene 0.010-1670 0.41-3.4
Benzo[k]fluoranthene 0.0044-1250 0.18-0.36
Chrysene 0.0142-2590 0.1-0.5
Coronene 0.003-710 0.15-0.47
Dibenz[a,h]anthracene 0.002-410 NR
Fluoranthene 0.0393-6830 0.0134
Perylene 0.0015-2730 0.31-0.8
Pyrene 0.0066-1650 0.1-0.9
From Brockhaus & Tomingas (1976); one permanent combustion stove
burning anthracite and brown-coal briquets and vaporizer and
atomizer oil burners; NR, not reported
Estimates of annual PAH emissions due to residential heating are
available for a few countries:
- In western Germany, the benzo [a]pyrene emissions were about 10
t in 1981 (Ahland et al., 1985), 7 t in 1985, and 2.5 t in 1988,
mainly resulting from coal heating. The reduction in the release
of PAH into the atmosphere due to domestic heating resulting from
increasing use of oil and gas during the last 30-40 years was
estimated to be 90-99% (Zimmermeyer et al., 1991).
- In the Netherlands, the estimated release in 1985 was < 1 t/year
each for benzo [k]fluoranthene and indeno[1,2,3- cd]pyrene,
< 10 t/year each for anthracene, fluoranthene,
benz [a]anthracene, chrysene, benzo [a]-pyrene, and
benzo[ghi]perylene, and 48-70 t/year each for naphthalene and
phenanthrene, mainly resulting from wood heating (Slooff et al.,
1989).
- The total PAH input, mainly from coal and wood heating, was about
63 t in Norway, 130 t in Sweden, and 720 t in the USA in 1985
(Bjorseth & Ramdahl, 1985).
- In Canada in 1990, the total PAH released due to residential
heating, mainly wood burning, was about 500 t (Environment
Canada, 1994).
(iii) Open burning
PAH may be released to the atmosphere during forest and
agricultural fires, burning of accidentally spilled oil, disposal of
road vehicles and especially automobile tyres, open burning of coal
refuse and domestic and municipal waste, and open fires. The release
of PAH into the atmosphere from the burning of wastes, including road
vehicles, in the open is decreasing in industrialized countries due to
comprehensive regulations.
Laboratory experiments with pine needles gave the following
specific PAH emission factors (per kg pine needle): 980-20 000 g
pyrene, 690-15 000 g fluoranthene, 580-12 000 g anthracene plus
phenanthrene, 540-29 000 g chrysene plus benz [a]anthracene,
420-6200 g benzo [ghi]-perylene, 170-4300 g
indeno[1,2,3- cd]pyrene, 140-8800 g benzo [c]-phenanthrene,
130-13 000 g benzofluoranthenes (isomers not specified), 61-800 g
benzo [e]-pyrene, 38-3500 g benzo [a]pyrene, and 24-2100 g
perylene, depending on the amount of needles, area, and type of fire.
Fires moving with the wind and low fuel loading resulted in
significantly smaller amounts of PAH than fires moving against the
wind and high fuel loading (McMahon & Tsoukalas, 1978). The emission
factor for acenaphthene was 230-1000 µg/kg dry straw (Ramdahl &
Mller, 1983) and 660 µg/kg dry wood (Alfheim et al., 1984).
In model experiments with crude oil spilled on water, numerous
PAH were found, including acenaphthene, acenaphthylene, phenanthrene,
anthracene, 1-methylphenanthrene, fluoranthene, pyrene, fluorene,
benzo [a]fluorene, benzo [b]fluorene, benz [a]anthracene, chrysene
plus triphenylene, benzo [b]-fluoranthene, benzo [ghi]fluoranthene,
benzo [e]pyrene, benzo [a]pyrene, perylene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, and coronene, at
concentrations of ¾ 1000 mg/kg individual substance in both the soot
and the burn residue (Benner et al., 1990). Even though the open
burning of oil spilled on water results in a lower PAH content than in
crude oil (see Table 8), this source may be of local importance, e.g.
near tanker accidents.
Between the early and the mid-1970s, the total release of PAH
(including nitrogen-containing analogues and quinone degradation
products) into the atmosphere in the USA due to open burning was
estimated to be about 4000 t/year (Agency for Toxic Substances and
Disease Registry, 1990). The total PAH input from forest and
agricultural fires in 1985 was estimated to be 13 t in Norway, 1.3 t
in Sweden, and 1000 t in the USA, and that from open fires to be 0.4 t
in Norway and 100 t in the USA (Bjorseth & Ramdahl, 1985). The release
of all PAH into the atmosphere from the burning of scrap electrical
cable in 1988 was about 17 t (Slooff et al., 1989). In Canada in 1990,
the total PAH emissions from agricultural burning and open-air fires
were estimated to be about 360 t and those from forest fires to be
about 2000 t (Environment Canada, 1994).
(iv) Other diffuse sources
The total PAH released into the atmosphere in the Netherlands
from roofing tar and asphalt in 1988 was estimated at 0.5 t/year
(Slooff et al., 1989).
(c) Emissions to the hydrosphere
(i) Motor vehicle traffic
The main source of PAH in the aqueous environment as a result of
motor vehicle traffic is highway run-off, which contains asphalt and
soot particles and is washed by rainfall and storm water or snow into
surface waters and soil (see also 3.2.7.2 (a) (i)). The available data
are summarized in Table 23. Higher PAH concentrations were found in
highway run-off in winter than in summer; this was attributed to the
increased abrasion of the road surface due to use of steel-studded
tyres in winter (Berglind, 1982).
It was estimated that an average of < 10 µg/km per vehicle per
day of total PAH are transported via pavement runoff water. Most is
transported to nearby surroundings as small particles of dust (see
also section 3.2.7.2; Lygren et al., 1984). In contrast, storm water
runoff near a US highway was of considerable importance for adjacent
water bodies. In the test area, over 50% of the total PAH input into a
nearby river came from highway runoff. The runoff loading factor was
given as 24 mg/km per vehicle (Hoffman et al., 1985).
Table 23. Polycyclic aromatic hydrocarbon concentrations (µg/litre) in highway runoff
Compound [1] [2] [3] [4] [5]
Acenaphthene 0.016/0.087 0.195/5.126 NR NR NR
Acenaphthylene 0.045 0.557/16.804 NR NR NR
Anthracene 0.042-0.214 0.486/8.917 0.379 0.165 0.246
Benzo[j+k]fluoranthene 0.089/0.277 NR NR NR 0.207
Benz[a]anthracene 0.031-0.139 0.341/0.863 0.677 0.228 NR
Benzo[a]fluorene 0.018-0.170 0.587 NR 0.179 0.396
Benzo[a]pyrene 0.061-0.120 0.537/1.255 0.602 0.250 NR
Benzo[b]fluoranthene 0.129/0.157 NR NR 0.799 1.501
Benzo[b]fluorene 0.033/0.097 0.356/0.366 NR NR 0.192
Benzo[c]phenanthrene NR 0.250 NR NR NR
Benzo[e]pyrene 0.108/0.202 0.238/1.665 0.609 0.360 0.630
Benzofluoranthenesa 0.401/0.695 1.087/2.712 1.171 NR NR
Benzo[e]perylene 0.100-0.299 NR 0.551 0.391 0.319
Chyrsene + triphenylene 0.194-0.433 1.472/2.752 1.147 0.665 1.070
Fluoranthene 0.321-1.573 4.065/15.322 2.665 1.820 3.143
Fluorene 0.0088-0.564 0.432/11.093 0.096 0.485 1.237
Indeno[1,2,3-cd]pyrene 0.061-0.154 0.344/0.666 NR NR NR
1-Methylphenanthrene 0.030-1.073 0.637/2.308 1.366 2.117
Naphthalene NR 2.59 NR 0.123 0.195
Perylene 0.048 NR NR NR NR
Phenanthrene 0.068-2.668 3.297/38.10 1.385 4.055 6.787
Pyrene 0.363-1.449 3.026/12.094 2.002 1.886 3.066
NR, not reported; /, single measurements;
[1] Run-off samples from a Norwegian highway north of Oslo in summer and winter
1980-82 (Berglind, 1982);
[2] Snow 20 and 50 m from the same highway in February 1981 (Berglind, 1982);
[3] Snow from a frozen Norwegian lake 50 m from a highway with high traffic density in
winter 1981-82 (Gjessing at al., 1984);
[4] Snow from a Norwegian highway south of Oslo with concrete pavement, February 1972
(Lygren at al., 1984);
[5] Snow from a Norwegian highway south of Oslo with asphalt pavement, February 1972
(Lygren et al., 1984)
When the water samples were filtered through solid sorbents, the results may be
underestimates of the actual content (see section 2.4.1.4).
a Isomers not specified
(ii) Sewage treatment
The concentrations of PAH in final effluents from municipal
sewage treatment facilities are generally in the low microgram per
litre range and are almost always < 0.1 µg/litre (Nicholls et al.,
1979; Young et al., 1983; van Luin & van Starkenburg, 1984; Kröber &
Häckl, 1989). Maximum values of 29 µg/litre naphthalene and 7 µg/litre
acenaphthene were detected in one US sewage treatment plant, and 8
µg/litre benzo [a]pyrene were found in one German plant (Young et
al., 1983; Kröber & Häckl, 1989), but no explanation was given for
these unusually high concentrations. It was concluded that final
effluents contain PAH at a background level (van Luin & van
Starkenburg, 1984).
Naphthalene was found at a concentration of 9.3 kg/year in the
final effluent from one US municipal sewage plant (Hoffman et al.,
1984). The annual emissions of naphthalene, anthracene, phenanthrene,
fluoranthene, benz [a]anthracene, chrysene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene
from Dutch sewage treatment plants into surface waters were estimated
to be about 0.6 t. The amount of these PAH transported into the
Netherlands from other European countries via the Rhine, Meuse, and
Scheldt rivers was estimated to be 65 t/year (year and database not
given). The main compounds were fluoranthene (18 t/year) and
naphthalene (15 t/year) (Slooff et al., 1989).
(iii) Other sources
PAH have been found in wastewaters from power stations, from
garages with car-wash devices, and from a German car-wash storage tank
at the following concentrations: fluoranthene, 1.3-7.7 µg/litre;
pyrene, 3.5-28 µg/litre; benz [a]anthracene, 0.49-1.9 µg/litre;
chrysene, 1.2-6.0 µg/litre; benzo [e]pyrene, 4.7-16 µg/litre;
benzo [a]pyrene, 0.40-8.8 µg/litre; benzo [b]fluoranthene, 1.2-3.6
µg/litre; and benzo [k]-fluoranthene, 0.51-0.72 µg/litre (Baumung et
al., 1985). Wastewaters from power stations could be an important
local source of PAH.
Numerous PAH were detected in leachate plumes from refuse
landfills in western Germany and the USA (Grimmer et al., 1981b; Götz,
1984; Reinhard et al., 1984). Concentrations < 0.1 µg/litre were
detected of benzo [ghi]-fluoranthene, benz [a]anthracene,
benzo [c]phenanthrene, chrysene, benzofluoranthenes (isomers not
specified), benzo [a]pyrene, benzo [e]pyrene, perylene,
anthanthrene, benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene
(Grimmer et al., 1981b). Naphthalene was found at a concentration >
100 µg/litre, and acenaphthene, fluorene, anthracene, phenanthrene,
and pyrene were found at concentrations of 1-30 µg/litre (Götz, 1984;
Reinhard et al., 1984). The importance of this source for groundwater
pollution cannot be estimated from the available data.
(c) Emissions to the geosphere
(i) Motor vehicle traffic
PAH were deposited within 100 m of a highway at a concentration
of 100-200 µg/km per vehicle per day in winter as small particles of
dust resulting from the abrasion of asphalt by steel-studded tyres
(Lygren et al., 1984). Studies of adsorption on various soil types
showed that most PAH in highway runoff is retained on the soil surface
(Gjessing et al., 1984).
(ii) Open burning
Phenanthrene, fluoranthene, triphenylene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [ghi]perylene, indeno[1,2,3- cd]pyrene, and
coronene were determined in the soil of burning sites in western
Oregon, USA. Before burning, the PAH concentrations in the top 2 cm of
the soil layer ranged from 0.8 ng/g dry weight for benzo [a]pyrene to
4.4 ng/g for fluoranthene and triphenylene. One week after burning,
the concentrations ranged from 0.9 ng/g for benzo [k]fluoranthene to
19 ng/g for triphenylene. The finding that the PAH levels did not
increase appreciably after burning indicates that the bulk of the PAH
were retained within the litter rather than passing into the soil
(Sullivan & Mix, 1983).
(iii) Disposal of sewage sludge and fly ash from incineration
When sewage sludge is applied to soils, adsorbed PAH are added to
the geosphere. The PAH concentrations in municipal aerobic and
anaerobic sewage sludge are given in Table 24.
In a detailed survey of the PAH concentrations in soil to which
anaerobic sludges had been applied between 1942 and 1961 in the United
Kingdom, the total PAH content increased to over 125 mg/kg up to 1948
but had decreased to about 29 mg/kg by 1961. The authors attributed
the declining levels to a decrease in atmospheric PAH contamination
from smoke emissions (Wild et al., 1990). No seasonal variation in the
content or profile of PAH was detected in western Germany by Grimmer
et al. (1980), but Süss (1980) found the highest PAH load in sewage
sludge in January-April and the lowest in July and October. Human
faeces seemed to contribute little to the PAH content of sewage sludge
(Grimmer et al., 1980). The most important emission sources could not
be identified, but McIntyre et al. (1981) concluded that the PAH
content of sewage sludge originating from British treatment works with
significant flows of industrial effluent was higher than that in works
dealing with predominantly domestic effluents.
After application of compost over three years to an agricultural
soil in Spain, no accumulation of PAH was observed (Gonzalez-Vila et
al., 1988). It was shown, however, that the extent of accumulation is
dependent on the duration, frequency, and concentration of
application. After 10 years of sludge spreading, considerable
quantities of PAH were detected in both a sandy loam and a clay soil
Table 24. Polycyclic aromatic hydrocarbons concentrations (mg/kg dry weight) in municipal sewage sludge
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene NR NR NR NR NR NR ND NR
Anthracene NR NR NR 0.89-44 NR NR ND-10.0 NR
Anthanthrene 0.00-2.10 0.03-1.8 NR NR NR NR NR NR
Benz[a]anthracene 0.62-19.0 0.91-17.3 NR NR NR NR ND-2.1 NR
Benzo[a]fluorene 0.28-9.00 0.56-7.9 NR NR NR NR NR NR
Benzo[a]pyrene 0.54-13.3 0.41-14.3 0.12-9.14 NR NR 0.29-2.00 ND-0.64 NR
Benzo[b]fluoranthene NR NR 0.06-9.14 NR < 1-1.3 0.29-1.80 ND-1.100 NR
Benzo[e]pyrene 0.53-12.4 0.48-12.3 NR NR NR NR NR NR
Benzofluoranthenesa 1.07-23.7 1.02-24.8 NR NR NR NR NR NR
Benzo[ghi]perylene 0.40-8.70 0.34-10.9 0.06-9.14 NR NR < 0.1-3.41 ND-1.21 NR
Benzo[k]fluoranthene NR NR 0.06-4.57 NR NR 0.15-1.00 ND-0.500 NR
Chrysene 0.78-23.7 1.24-22.2 NR 0.25-13 NR NR NR NR
Dibenz[a,h]anthracene NR NR NR 13 NR NR ND-0.25 NR
Fluoranthene 0.61-51.6 4.10-28.2 0.34-11.45 0.35-7.1 < 1-10.4 0.54-7.67 0.216-5.14 5.2/5.6b
Fluorene NR NR NR NR NR NR ND-2.9c 3.5/5.8
Indeno[1,2,3-cd]pyrene 0.30-7.40 0.28-9.4 0.06-6.68 NR NR 0.24-2.08 ND-0.640 NR
Naphthalene NR NR NR 0.9-70 NR NR NR 4.5/8.6
Perylene 0.14-6.40 0.09-3.1 NR NR NR NR NR NR
Phenanthrene NR NR NR 0.89-44 NR NR 0.30-40 15.2/18.6d
Pyrene 0.90-47.2 3.20-25.3 NR 0.33-18N R NR ND-7.6 NR
NR, not reported; /, single measurements; ND, not detected (limits of detection, 0.2-1 mg/kg);
[1] Samples from 25 sewage treatment plants in western Germany 1976-78 (Grimmer et al., 1980);
[2] Samples from three sewage treatment facilities in western Germany before 1979 (Suss, 1980);
[3] Samples from 12 British sewage treatment works (McIntyre at al., 1981);
[4] Samples from 20 US sewage treatment works (Naylor & Loehr, 1982);
[5] Samples from six Dutch municipal sewage treatment plants (van Luin & van Starkenburg, 1984);
[6] 31 sludge samples from different sewage treatment works in western Germany (Witte et al., 1988);
[7] Anaerobic sludge samples from 13 sewage treatment plants in western Germany 1985-88 (Krober & Hackl, 1989);
[8] Anaerobic sludge samples from one Spanish sewage treatment facility in spring 1985 and autumn 1986
(Gonzalez-Villa at al., 1988).
a Isomers not specified
b With pyrene
c With acenaphthylene
d With anthracene
(Diercxsens & Tarradellas, 1987). The annual addition of PAH to soil
from sewage sludge in the Netherlands was estimated as follows: 0.1 t
naphthalene, 0.1 t anthracene, 1.5 t phenanthrene, 2.3 t fluoranthene,
0.6 t benzo [a]anthracene, 0.6 t chrysene, 0.4 t
benzo [k]fluoranthene, 0.6 t benz [a]pyrene, 0.6 t
benzo [ghi]-perylene, and 0.6 t indeno[1,2,3- cd]pyrene (year and
database not given; Slooff et al., 1989).
The annual contribution of PAH to landfill in the United Kingdom
from fly ash from coal combustion (see also Table 16) exceeded that
from municipal solid-waste incineration by a factor of about 10, with
the exception of naphthalene, the level of which was about 20 000-fold
higher in fly ash from coal combustion than in that from solid-waste
incineration. The annual PAH loads from solid-waste incineration were
about 0.01 kg naphthalene and 3.5 kg benzo [ghi]perylene, whereas
those from coal combustion were about 15 kg each of anthracene,
benzo [k]fluoranthene, and dibenz [a,h]anthracene and 1200 kg pyrene
(Wild et al., 1992).
(iv) Waste dumping
Soil cores taken from a hazardous waste disposal site in Spain
containing petroleum tar residues and lubricating oils as the major
organic wastes contained 62 mg/kg 1-methylphenanthrene, 53 mg/kg
naphthalene, 52 mg/kg benzo [a]fluorene, 30 mg/kg
benzo [ghi]fluoranthene, 25 mg/kg benzo [c]-phenanthrene, 0.5-0.71
mg/kg acenaphthene, 0.2-48 mg/kg fluorene, 0.2-390 mg/kg phenanthrene,
0.110 mg/kg anthanthrene, 0.1-210 mg/kg pyrene, 0.1-200 mg/kg
acenaphthylene, 0.1-140 mg/kg anthracene, 0.1-140 mg/kg
benzo [e]pyrene, 0.1-145 mg/kg benzo [a]pyrene, 0.1-50 mg/kg
benzo [b]fluorene, 0.08-130 mg/kg chrysene plus triphenylene, 0.08-90
mg/kg indeno[1,2,3- cd]pyrene, 0.06-130 mg/kg benz [a]anthracene,
0.05-290 mg/kg fluoranthene, 0.03-75 mg/kg benzo [ghi]perylene,
0.03-0.2 mg/kg perylene, and 0.01-0.4 mg/kg dibenz [a,h]anthracene
(Navarro et al., 1991).
There can be appreciable movement of PAH into soil from waste
dumping, especially of hazardous refuse. The dumping conditions are
decisive for the amount of PAH released. Annual emissions of PAH in
the Netherlands in 1987 due to the spreading of contaminated composts
onto soils were estimated to be 1 t benz [a]anthracene, 1 t chrysene,
1 t benzo [k]fluoranthene, 0.5 t benzo [ghi]-perylene, 0.5 t
indeno[1,2,3-cd]pyrene, and 0.4 t benzo [a]pyrene (Slooff et al.,
1989).
(d) Biosphere
Perch (Perca fluviatilis) were not significantly contaminated
after an oil spill in Finland due to a tanker accident. The
concentrations of acenaphthene, acenaphthylene, fluorene,
phenanthrene, anthracene, 1-methylphenanthrene, fluoranthene, pyrene,
benzo [a]fluorene, benzo [b]fluorene, chrysene, triphenyl-ene, and
benzofluoranthenes in both contaminated and control groups were
between < 0.1 and 0.2 µg/kg each in muscle and < 0.1 and 16
µg/kg in bile. The investigators concluded that the fish with the
highest load would probably not have survived and others had moved to
less contaminated areas. Additionally, the cold climate caused
clumping of the spilled oil, which then drifted to the coast
(Lindström-Seppä et al., 1989; see also sections 4.1.5.1 and 5.1.7.1).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
Appraisal
The transport and distribution of polycyclic aromatic
hydrocarbons (PAH) in the environment depend on their physicochemical
properties of very low solubility in water and low vapour pressure,
and high partition coefficients for n-octanol:water (log Kow) and
organic carbon:water (log Koc). PAH are stable towards hydrolysis
as they have no reactive groups. In the gaseous phase, PAH and
particularly those of higher molecular mass, are mainly adsorbed to
particulate matter and reach the hydrosphere and geosphere by dry and
wet deposition. Little is volatilized from water phases owing to their
low Henry's law constants. The log Koc values indicate strong
adsorption to the organic matter of soils, so that migration does not
usually occur. The log Kow values indicate high bioaccumulation.
Few experimental data are available on the biodegradation of PAH.
In general, they are biodegradable under aerobic conditions, and the
biodegradation rates decrease drastically with the number of aromatic
rings. Under anaerobic conditions, biodegradation appears to be very
slow.
The bioconcentration factors measured in the water phase vary
widely according to the technique used. High values are seen for some
algae, crustaceans, and molluscs, but those for fish are much lower
owing to rapid biotransformation. The bioaccumulation factors for
aquatic and terrestrial organisms in sediment and soil are generally
very low, probably because of the strong adsorption of PAH onto the
organic matter of soils and sediments, resulting in low
bioavailability.
The photodegradation of PAH in air and water has been studied
intensively. The most important degradation process in both media is
indirect photolysis under the influence of radicals like OH, O3, and
NO3. The measured degradation rate constants vary widely according to
the technique used. Under laboratory conditions, the half-life of the
reaction of PAH with airborne OH radicals is about one day. Adsorption
of high-molecular-mass PAH onto carbonaceous particles in the
environment has a stabilizing effect. Formation of nitro-PAH has been
reported from two- to four-ring PAH in the vapour phase during
photooxidation with NO3. For some PAH, photodegradation in water
seems to be more rapid than in air.
According to model calculations based on physicochemical and
degradation parameters, PAH with four or more aromatic rings persist
in the environment.
4.1 Transport and distribution between media
4.1.1 Physicochemical parameters that determine environmental transport
and distribution
The transport and distribution of PAH in the environment are the
result of the following physicochemical parameters:
- Aqueous solubility: PAH are hydrophobic compounds with very low
solubility in water under environmental conditions: the maximum
at room temperature is 32 mg/litre for naphthalene, and the
minimum is 0.14 µg/litre for coronene (see Table 4).
- Vapour pressure: The vapour pressure of PAH under environmental
conditions is very low: the maximum at room temperature is 10.4
Pa for naphthalene, and the calculated minimum is 3 × 10-12 Pa
for dibenzo [a,i]pyrene (see Table 4).
- n-Octanol:water partition coefficient (log Kow): The
affinity of PAH to organic phases is much higher than that for
water. The log Kow values range from 3.4 for naphthalene to 7.3
for dibenzo [a,i]pyrene (see Table 4), indicating that the
potential for bioaccumulation is high.
- Organic carbon:water partition coefficient (log Koc): The
sorption coefficients of PAH to the organic fraction of sediments
and soils are summarized in Table 25. The high values indicate
that PAH sorb strongly to these fractions. The wide variation in
the results for individual compounds are due to the very long
exposure necessary to reach steady-state or equilibrium
conditions, which can lead to underestimation of sorption
coefficients; furthermore, degradation in the overlying aqueous
phase can lead to overestimates of the actual values.
4.1.2 Distribution and transport in the gaseous phase
PAH are emitted mainly to the atmosphere (see Section 3), where
they can be both transported in the vapour phase and adsorbed onto
particulate matter. The distribution between air and particulate
matter under normal atmospheric conditions depends on the
lipophilicity, vapour pressure, and aqueous solubility of the
substance. Generally, PAH with few (two to four) aromatic rings occur
in the vapour phase and are adsorbed, whereas PAH consisting of more
aromatic rings exist mainly in the adsorbed state (Hoff & Chan, 1987;
McVeety & Hites, 1988; Baker & Eisenreich, 1990). PAH are usually
adsorbed onto particles like fly ash and soot that are emitted during
combustion.
Table 25. Organic carbon normalized sorption coefficients (Koc) of polycyclic aromatic hydrocarbons
Compound log Koc Comments Reference
Acenaphthene 5.38 Average on sediments Kayal & Connell (1990)
3.79 RP-HPLC on CIHAC Szabo et al. (1990)
3.59 RP-HPLC on PIHAC Szabo at al. (1990)
Acenaphthylene 3.83 RP-HPLC on CIHAC Szabo et al. (1990)
3.75 RP-HPLC on PIHAC Szabo et al. (1990)
Anthracene 4.42 Average sorption isotherms on Karickhoff et al. (1979)
sediment
3.74 Suspended particulates Herbes et al. (1980)
4.20 Soil, shake flask, UV Karickhoff (1981)
3.95/4.73 Lake Erie with 9.6 mg C/litre Landrum et al. (1984a)
4.87/5.70 Huron river with 7.8 mg C/litre Landrum et al. (1984a)
4.20 Soil, shake flask, LSC Nkedl-Kizza et al. (1985)
4.93 Fluorescence, quenching interaction Gauthier et al. (1986)
with humic acid
4.38 HPLC Hodson & Williams (1988)
5.76 Average on sediments Kayal & Connell (1990)
4.41 RP-HPLC Pussemier et al. (1990)
4.53 RP-HPLC on CIHAC Szabo at al. (1990)
4.42 RP-HPLC on PIHAC Szabo at al. (1990)
Benz[a]anthracene 4.52 Suspended particles Herbes et al. (1980)
6.30 Average on sediments Kayal & Connell (1990)
7.30 Specified particulate Bromen et al. (1990)
Benzo[a]pyrene 6.66 LSC Eadie et al. (1990)
6.26 Average on sediments Kayal & Connell (1990)
8.3 Specified particulate Broman et al. (1990)
4.0 Predicted to be dissolved Broman et al. (1990)
Benzo[e]pyrene 7.20 Specified particulate Broman at al. (1990)
4.00 Predicted to be dissolved Broman at al. (1990)
Benzo[k]fluoranthene 5.99 Average on sediments Kayal & Connell (1990)
7.00 Specified particulate Broman et al. (1990)
4.00 Predicted to be dissolved Broman at al. (1990)
Table 25. (continued)
Compound log Koc Comments Reference
Chrysene 6.27 Average on sediments Kayal & Connell (1990)
6.90 Specified particulate Broman et al. (1990)
4.0 Predicted to be dissolved Broman at al. (1990)
Coronene 7.80 Specified particulate Broman et al. (1990)
5.0 Predicted to be dissolved Broman et al. (1990)
Dibenz[a,h]anthracene 6.31 Average of 14 soil or sediment Means et al. (1980)
samples, shake flask, LSC
Fluoranthene 6.38 Average on sediments Kayal & Connell (1990)
4.74 RP-HPLC on CIHAC Szabo et al. (1990)
4.62 RP-HPLC on PIHAC Szabo et al. (1990)
6.30 Specified particulate Broman et al. (1990)
4.0 Predicted to be dissolved Broman et al. (1990)
Fluorene 5.47 Average on sediments Kayal & Connell (1990)
3.76 RP-HPLC Pussemier et al. (1990)
4.15 RP-HPLC on CIHAC Szabo et al. (1990)
4.21 RP-HPLC on PIHAC Szabo et al. (1990)
Naphthalene 3.11 Average sorption isotherms on Karickhoff at al. (1979)
sediments
2.38 Suspended particulates Herbes et al. (1980)
2.94 Karickhoff (1981)
3.0 McCarthy & Jimenez (1985);
McCarthy et al. (1985)
2.73-3.91 Aquifer materials Stauffer et al. (1989)
3.15/2.76 Podoll et al. (1989)
5.00 Average on sediments Kayal & Connell (1990)
2.66 Average on sediments Kishi et al. (1990)
3.11 Soil, RP-HPLC Szabo et al. (1990)
3.29 Sandy surface soil Wood et al. (1990)
Table 25. (continued)
Compound log Koc Comments Reference
Phenanthrene 4.36 Average sorption isotherms on Karickhoff et al. (1979)
sediments
4.28 Hodson & Williams (1988)
6.12 Average on sediments Kayal & Connell (1990)
4.22 RP-HPLC on CIHAC Szabo et al. (1990)
4.28 RP-HPLC on PIHAC Szabo et al. (1990)
4.42 Sandy surface soil Wood et al. (1990)
Pyrene 4.92 Average isotherms on sediments Karickhoff et al. (1979)
4.90 Sediment, shake flask, sorption Karickhoff et al. (1979)
isotherm
4.81 Average of soil and sediment Means et al. (1979)
Shake flask, LSC, sorption
isotherms
4.80 Average of 12 soils and sediments Means et al. (1980)
Shake flask, LSC, sorption isotherms
4.78 Soil and sediment; calculated Kow Means at al. (1980)
4.83 Sorption isotherms Karickhoff (1981)
3.11/3.46 Sediment suspensions Karickhoff & Morris (1985)
4.80/5.13 Hodson & Williams (1988)
5.65 LSC Eadie et al. (1990)
5.29 Soil Jury at al. (1990)
6.51 Average on sediments Kayal & Connell (1990)
4.83 RP-HPLC Pussemier et al. (1990)
4.82 RP-HPLC on CIHAC Szabo et al. (1990)
4.77 RP-HPLC on PIHAC Szabo et al. (1990)
6.50 Specified particulate Broman et al. (1990)
4.0 Predicted particulate Broman et al. (1990)
Triphenylene 6.90 Specified particulate Broman et al. (1990)
4.00 Predicted to be dissolved Broman et al. (1990)
RP-HPLC, reversed-phase high-performance liquid chromatography; CIHAC, chemical-induced humic-acid column;
PIHAC, physical-induced humic-acid column; UV, ultraviolet; C, carbon; LSC, liquid scintillation
chromatography
PAH are ubiquitous in the environment, probably because they are
distributed for long distances without significant degradation (Lunde,
1976; De Wiest, 1978; Bjorseth & Sortland Olufsen, 1983; McVeety &
Hites, 1988), e.g. from the United Kingdom and the European continent
to Norway and Sweden during winter (Bjorseth & Lunde, 1979). Washout
ratios calculated from measurements in rain and snow in the area of
northern Lake Superior, during one year showed that airborne PAH
adsorbed onto particulate matter result in effective wet deposition,
while gaseous PAH are removed to only a minor degree (McVeety & Hites,
1988).
4.1.3 Volatilization
Henry's law constant gives a rough estimate of the equilibrium
distribution ratio of concentrations in air and water but cannot
predict the rate at which chemicals are transported between water and
air. The constants for PAH are very low, ranging from 49 Pa .m3/mol
for naphthalene to 0.000449 Pa .m3/mol for dibenzo [a,i]pyrene (see
Table 4). The rates of removal and volatilization of PAH (Table 26)
are strongly dependent on environmental conditions such as the depth
and flow rate of water and wind velocity. Although PAH are released
into the environment mainly in air, considerably higher concentrations
are found in aqueous samples because of the low vapour pressure and
Henry's law constants of PAH.
The volatilization half-life for naphthalene from a 22.5-m water
body was found experimentally to be 6.3 h, whereas the calculated
value was 2.1 h (Klöpffer et al., 1982). Calculations based on a
measured air:water partition coefficient for river water 1 m deep with
a water velocity of 0.5 m/s and a wind velocity of 1 m/s gave a
volatilization half-life of 16 h for naphthalene (Southworth, 1979).
The value calculated for evaporative loss of naphthalene from a 1-m
water layer at 25°C was of the same order of magnitude (Mackay &
Leinonen, 1975). Naphthalene was volatilized from soil at a rate of
30% after 48 h, with neglible loss of PAH with three or more rings
(Park et al., 1990).
4.1.4 Adsorption onto soils and sediments
PAH are adsorbed strongly to the organic fraction of soils and
sediments (see section 4.1.1 and Table 25). Some PAH may be degraded
biologically in the aerobic soil layer, but this process is slow,
because sorption to the organic carbon fraction of the soil reduces
the bioavailability. For the same reason, leaching of PAH from the
soil surface layer to groundwater is assumed to be negligible,
although detectable concentrations have been reported in groundwater
(see section 5.1.2.2).
Table 26. Rates of volatilization of polycyclic aromatic hydrocarbons
Compound Rate constant Half-life (h)a Comments Reference
Anthracene Removal rate constants (estimated) from Southworth (1977)
water column
At 25°C in midsummer sunlight:
0.002 h-1 347 - in deep, slow, somewhat turbid water
0.001 h-1 693 - in deep, slow, muddy water
0.002 h-1 347 - in deep, slow, clear water
0.042 h-1 17 - in shallow, fast, clear water
0.179 h-1 3.9 - in very shallow, fast, clear water
62 Calculated half-life for a river 1 m deep Southworth (1979)
with water velocity of 0.5 m/s and wind
velocity of 1 m/s
Benz[a]anthracene 500 Calculated half-life for a river 1 m deep Southworth (1979)
with water velocity of 0.5 m/s and wind
velocity of 1 m/s
Benzo[a]pyrene 1550 Calculated half-life for a river 1 m deep Southworth (1979)
with water velocity of 0.5 m/s and wind
velocity of 1 m/s
<1 × 10-5 S-1 > 19 Sublimation rate constant from glass Cope & Kalkwarf
surface at 24 °C at an airflow of 3 litre/min (1987)
Naphthalene 1.675 × 10-9 Rate of evaporation estimated at 20 00 Guckel at al. (1973)
mol.cm-2h-1 and air flow of 50 litre/h
7.15 Calculated half-life from 1 m depth of water Mackay & Leinonen
(1975)
16 Half-life for surface waters Southworth
(1979)
200 In a lake, considering current velocity and
wind speed in combination with typical
re-aeration rates
Perylene <1 × 10-5 S-1 > 19 Sublimation rate constant from glass Cope & Kalkwarf
surface at 24°C at an air flow of 3 litre/min (1987)
Pyrene 1.1 × 10-4 S-1 1.8 Sublimation rate constant as loss from Cope & Kalkwarf
glass surface at 24°C at an air flow of 3 litre/min (1987)
Table 26 (continued)
For comparison of results for which only rate constants are reported, half-lives have been estimated from the equation:
t1/2 = In2
k
where t1/2 is the half-life and k is the rate constant. The calculated values are reported in italics.
4.1.5 Bioaccumulation
The ability of a substance to bioconcentrate in organisms in the
aqueous phase is expressed as the bioconcentration factor. For
substances like PAH, with high n-octanol:water partition
coefficients, long exposures are necessary to achieve equilibrium
conditions, so that results obtained under non-equilibrium conditions
can result in underestimates of the bioconcentration factor.
Bioaccumulation may also vary with the metabolic capacity of the
organism (see section 4.2.1.2).
Bioconcentration can also be calculated as the ratio between the
rates of uptake (k1) and depuration (k2). This method has the
advantage that relatively short exposures can be used. It is therefore
preferred for PAH, as constant concentrations of compounds like
benzo [a]pyrene are very difficult to maintain over a long period.
4.1.5.1 Aquatic organisms
Aquatic organisms may accumulate PAH from water, sediments, and
their food. In general, PAH dissolved in pore water are accumulated
from sediment (McElroy & Sisson, 1989), and digestion of sediment may
play an important role in the uptake of PAH by some species. Although
organisms can accumulate PAH from food, the relative importance of
uptake from food and water is not clear (Farrington, 1991).
The bioconcentration factors of PAH in different species are
shown in Table 27; this is not a comprehensive presentation of all of
the available data but provides examples of the accumulation of some
PAH in different groups of organisms. Species that metabolize PAH to
little or no extent, like algae, oligochaetes, molluscs, and the more
primitive invertebrates (protozoans, porifers, and cnidaria),
accumulate high concentrations of PAH, as would be expected from their
log Kowvalues, whereas organisms that metabolize PAH to a great
extent, like fish and higher invertebrates such as arthropods,
echinoderms, and annelids, accumulate little or no PAH (James, 1989).
Remarkably high bioconcentration factors have been measured for
phenanthrene, anthracene, pyrene, benzo [a]anthracene, and
benzo [a]pyrene in the amphipod Pontoporeia hoyi, which has a
20-50% lipid content by wet weight and no capacity to biotransform PAH
(Landrum, 1988).
The ratio of the concentration of an individual PAH in a
bottom-dwelling organism and in the sediment, the bioaccumulation
factor, is usually < 1 when expressed as wet weight. In a coastal
area, the bioaccumulation factors for 16 PAH in polychaete species
varied from 4.9 to 21.8 on a dry-weight basis (Bayona et al., 1991).
Measurements of the concentrations of PAH in P. hoyi and in the
sediment at three sites with different organic carbon contents gave
bioaccumulation factors close to 1 on a wet-weight basis, corrected
for the 64-mm sieved fraction (Eadie et al., 1982). The lipid- and
organic carbon-based bioaccumulation factors in clams (Macoma
baltica) for naphthalene and chrysene added to sediment were 0.78
and 0.16, respectively (Foster et al., 1987). In a study in which
clams were exposed for 28 days to six sediments contaminated with
different concentrations of PAH (and other organic pollutants) and
with an organic carbon content of 0.86-7.4%, the bioaccumulation
factors (normalized with respect to lipid content and organic carbon
content) ranged from 0.15 to 0.85 (Ferraro et al., 1990).
Species that can biotransform PAH have internal concentrations
well below the concentration in the sediment. The average
bioaccumulation factors (normalized with respect to lipid content and
organic carbon content) for eel, pike, and roach at two locations were
0.1 and 0.015. The lowest bioaccumulation factor was found at the site
with the highest PAH concentration (128 mg/kg, organic carbon-based),
probably due to the inductive capability of the fish to biotransform
PAH. This was confirmed by the finding of increased hepatic metabolic
activity for PAH in the fish (Van der Oost et al., 1991).
4.1.5.2 Terrestrial organisms
Little information is available on the accumulation of PAH in
terrestrial organisms. The bioaccumulation factors of 22 PAH in the
earthworm Eisenia foetida at six sites varied from 0.23 to 0.6 on an
ash-free dry-weight basis (Rhett et al., 1988).
The half-life of labelled benzo [a]pyrene in crickets
(Acheta domesticus) was 13 h; after 48 h, 36% of the injected dose
was unchanged benzo [a]pyrene. After topical application of piperonyl
butoxide, a known inhibitor of the mixed-function oxidase system, the
level of polar metabolites in the excreta had decreased by
approximately 75% within 8 h of injection of benzo [a]pyrene. After
articular application of benzo [a]pyrene at 0.29 ng/µl in hexane,
some of the dose accumulated internally; the highest level of polar
metabolites was found after 24 h (Kumi et al., 1991).
The concentration of PAH in vegetation is generally considerably
lower than that in soil, the bioaccumulation factors ranging from
0.0001-0.33 for benzo [a]pyrene and from 0.001-0.18 for 17 other PAH
tested. It was concluded that some terrestrial plants take up PAH
through their roots and/or leaves and translocate them to various
other parts (Edwards, 1983).
When bush beans (Phaseolus vulgaris Pr.) were exposed to
radiolabelled anthracene in a nutrient solution for 30 days during
flowering and seed production, more than 90% of the compound was
metabolized. Of the total 14C radiolabel, 60% was found in the roots,
3% in the stems, 3% in the leaves, 0.1% in the pods, and 17% in the
nutrient solution; 16% was unaccounted for (Edwards, 1986).
Table 27. Measured bioconcentration factors of polycyclic aromatic hydrocarbons in aquatic organisms
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Acenaphthene
Fish
Lepomis 14C S 8.94 28 d 387 Equi Barrows et al.
macrochirus (1980)
Anthracene
Algae
Chlorella fusca HPLC S 50 1 d 7 770a NS Geyer et al.
(1984)
Crustaceans
Daphnia magna 14C, TLC S 35 1 d 511 k1/k2 McCarthy et al.
(1985)
Daphnia magna HPLC S 15 1 d 970 NS Newsted & Giesy
(1987)
Daphnia magna HPLC S 5.58 24 h 2699 NS Oris et al. (1990)
Daphnia pulex Spect S 6 24 h 917 Southworth et al.
(1978)
Hyalella azteca 14C IF 0.0082 8 h/7 h 2089 k1/k2 Landrum &
14C,TLC 1 800 k1/k2 Scavia (1983)
14C IF 0.0066 8 h/7 h 10985 k1/k2
14C, TLC 9096 k1/k2
Pontoporeia hoyi 14C TLC F 4-17 8 W d 16857 k1/k2 Landrum (1982)
Pontoporeia hoyi 14C TLC F 4.6-16.9 6 h/14 d 39727 k1/k2 Landrum (1988)
Oligochaetes
Stylodrilus -C, TLC F < 6 6 hS d 5051 k1/k2 Frank et al.
heringianus (1986)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Fish
Lepomis 14C S 0.7 4 h/60 h 900 k1/k2 Spacie et al.
macrochirus 14C, TLC 675 (1983)
Leuciscus idus UC S 50 3 d 910 NS Freitag A al.
melanotus (1985)
Oncorhynchus 14C HPLC R 12 18h 190 NS Linder &
mykiss 14C HPLC R 12 18 h 270 NS Bergman (1984)
Oncorhynchus 14C HIPLC R 50 72 h/144 h 9000 k1/k2 Linder et al.
mykiss 9200 (1985)
Pimephales HPLC S 6.61 24 In 1016 NS Oris et al. (1990)
promelas
Benz[a]anthracene
Algae
Chlorella fusca 14C S 50 1 d 3180 NS Freitag et al.
(1985)
Crustaceans
Daphnis magna 14C TLC S 0.8 1 d 2920 k1/k2 McCarthy et al.
(1985)
Daphnis pulex Spect S 6 1 d 10109 Southworth et al.
(1978)
Daphnia pulex HPLC S 1.8 1 d 10226 NS Newsted & Giesy
(1987)
Pontoporaia hoyi 14C, TLC F 0.62-1.11 6 h/14 d 63000 k1/k2 Landrum (1988)
Fish
Leuciscus idus 14C S 50 3 d 350 NS Freitag et al.
melanotus (1985)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Benzo[a]fluorene
Crustaceans
Daphnis magna HPLC S 4.8 1d 3668 NS Newsted & Glesy
(1987)
Benzo[b]fluorene
Crustaceans
Daphnia magna HPLC S W 1 d 7725 NS Newsted & Giesy
(1987)
Benzo[a]pyrene
Algae
Periphyton 14C F 1 1 d 9000 NS Leversee et al.
(1981)
Crustaceans
Daphnis magna 14C S/F 1 6 h 2440 k1/k2 Leversee et al.
(1981)
Daphnia magna 14C 3050 NS Leversee et al,
14C HPLC 2837 k1/k2 (1981)
Daphnia magna 14C TLC S 0.63 1 d 5770 k1/k2 McCarthy et al.
(1985)
Daphnia magna HPLC S 1.5 1 d 12761 NS Newsted & Giesy
(1987)
Daphnia pulex 14C S 1.20 24 h 458 NS Trucco et al.
14C S 0.47 24 h 745 NS (1983)
14C S 5.42 24 h 803 NS
14C S 3.21 24 h 1 106 NS
14C S 2.20 24 h 1 259 NS
14C S 1.50 24 h 2 720 NS
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Pontoporeia hoyi 14C, TLC S 0.002-2.6 6 h/14 d 73 000 k1/k2 Landrum (1988)
Oligochaetes
Stylodrilus 14C, TLC F < 0.03 6 h/8 d 7 048 k1/k2 Frank et al.
heringianus (1986)
Molluscs
Mysis relicta 14C F - 6 h/10-26d 8 297 k1/k2 Evans &
Landrum (1989)
Ostrea edulis 14C, GLC S 65.7 3 d 58 NS Riley et al.
Ostrea edulis 14C, GLC S 65.7 3 d 59 NS (1981)
Ostrea edulis 14C, GLC S 65.7 3 d 62 NS
Physa sp. 14C, GLC S 2.5 3 d 2 177 NS Lu et al. (1977)
Rangia cuneata 14C S 30.5 24 h 236 NS Neff & Anderson
14C S 30.5 24 h 187 NS (1975)
Insects
Chironomus 14C S 1 8 h/48 h 970 k1/k2 Leversee et al.
riparius 14C 600 NS (1981)
14C, HPLC 166 NS
Culex pipiens 14C, GLC S 2.5 3 d 37 NS Lu et al. (1977)
quinquefasciatus
Hexagenia limbata 14C, TLC F - 6 h/14 d 5 870 k1/k2 Landrum & Poore
(1988)
Fish
Lepomis 14C-extraction F 1 2 d/4 d 3 208 k1/k2 Jimenez et al.
macrochirus (1987)
Lepomis 14C S/F 1 4 h/4 h 4 700 k1/k2 Leversee et al.
macrochirus 14C 4 h 120 NS (1981)
14C, HPLC 4 h 12.5 NS
Lepomis 14C S 1 4 h/20 h 4 900 k1/k2 Spacie et al.
macrochirus 14C, TLC 490 k1/k2 (1983)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Lepomis 14C, TLC S 0.5 5 h/100 h 2 657 k1/k2 McCarthy &
macrochirus Jimenez (1985)
Leuresthes tenuis Spect S 2 15 d 241 Equi Winkler et al.
(1983)
Oncorhynchus GC-HPLC F 0.4 10 d 920 NS Gerhart &
mykiss Carlson (1978)
Salmo salar 14C S 1 48 h/96 h 2 310 k1/k2 Johnsen et al.
(1989)
Benzo[e]pyrene
Crustaceans
Daphnis magna HPLC S 0.7 1 d 25 200 NS Newsted & Giesy
(1987)
Benzo[ghi]perylene
Crustaceans
Daphnia magna HPLC S 0.2 1 d 28 288 NS Newsted & Giesy
(1987)
Benzo[k]fluoranthene
Crustaceans
Daphnia magna HPLC S 1.4 1 d 13 225 NS Newsted & Giesy
(1987)
Chrysene
Crustaceans
Daphnia magna 14C S 48 48 h/40 h 5 500 NS Eastmond et al.
(1984)
Daphnia magna HPLC S 0.7 1 d 6 088 NS Newsted & Giesy
(1987)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Dibenz[a,h]anthracene
Algae
Chlorella fusca 14C S 50 1 d 2 398 NS Freitag et al.
(1985)
Crustaceans
Daphnia magnia HPLC S 0.4 1 d 50 119 NS Newsted & Giesy
(1987)
Fish
Leuciscus idus 14C S 50 3 d 10 NS Freitag et al.
melanotus (1985)
Fluoranthene
Crustaceans
Crangon HPLC F 2.4 4 d/14 d 180 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magna HPLC S 9 1 d 1 742 NS Newsted & Giesy
(1987)
Molluscs
Mya arenaria HPLC F 2.4 4 d/14 d 4 120 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 2.4 4 d/14 d 5 920 k1/k2 McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 2.4 4 d/14 d 720 k1/k2 McLeese &
Burridge (1987)
Fish
Oncorhynchus GC-HPLC F 3.31 21 d 378 Equi Gerhart &
mykiss Carlson (1978)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Fluorene
Crustaceans
Daphnia magna HPLC S 17 1 d 506 NS Newsted & Giesy
(1987)
Fish
Lepomis - IF 20, 37 30 d 1 800 Equi Finger et al.
macrochirus - IF 86 30 d 700 Equi (1985)
- IF 175, 353 30 d 200 Equi
Naphthalene
Algae
Selenastrum GC S 2,000 1 d 18 000b NS Casserly et al.
capricornutum (1983)
Chlorella fusca 14C S 50 1 d 130a NS Geyer et al.
(1984)
Insects
Somatochlora Spect S 10 48 h 1 548 NS Correa & Coler
cingulata Spect S 100 48 h 178 NS (1983)
Crustaceans
Daphnia magna 14C, HPLC S 1 000 1 d 19.3 k1/k2 McCarthy et al.
(1985)
Daphnia magna 14C S 1 800 48 h/40 h 50 NS Eastmond et al.
(1984)
Daphnia pulex Spect S 1 000 1 d 131 k1/k2 Southworth et al.
(1978)
Daphnia pulex 14C S 2 292 4 h 677 NS Trucco et al.
14C S 0.45 24 h 10 844 NS (1983)
14C S 2.742 4 h 2 337 NS
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Fish
Fundulus 14C S 20 4 h 2.2 NS DiMichele &
heteroclitus Taylor (1978)
Lepomis 14C, HPLC S 1 000 24 h/36 h 310 k1/k2 McCarthy &
macrochirus 14C, HPLC S 100 24 h/36 h 320 k1/k2 Jimenez (1985)
Oncorhynchus 14C S 23 8 h/24 h 253 k1/k2 Melancon & Lech
mykiss (1978)
Perylene
Algae
Chlorella fusca 14C S 50 1 d 2 010 NS Freitag et al.
(1985)
Crustaceans
Crangon HPLC F 0.4 4 d/14 d 175 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magnia HPLC S 0.6 1 d 7 190 NS Newsted & Giesy
(1987)
Molluscs
Mya arenaria HPLC F 0.4 4 d/14 d 100 000 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 0.4 4 d/14 d 105 000 k/q McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 0.4 4 d/14 d 180 k1/k2 McLeese &
Burridge (1987)
Fish
Leuciscus idus 14C S 50 3 d < 10 NS Freitag et al.
melanotus (1985)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Phenanthrene
Bacteria
Mixed Spect S 30-300 2 h 6 300c NS Steen &
Karickhoff (1981)
Algae
Selenastrum GC S 1000 1 d 36 970b NS Casserly et al.
capricornutum (1983)
Chlorella fusca 14C S 50 1 d 1 760a NS Geyer et al.
(1984)
Insects
Hexagenia limbata 14C F - 6 h/14 d 1640 k1/k2 Landrum & Poore
(1988)
Crustaceans
Crangon HPLC F 4.3 4 d/14 d 210 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magna HPLC S 40.1 1 d 323 NS Newsted & Giesy
(1987)
Daphnia magna 14C S 60 48 h/40 h 600 NS Eastmond et al.
(1984)
Daphnia pulex 14C S 6.01 24 h 1 165 NS Trucco et al.
14C S 3.10 24 h 1 032 NS (1983)
14C S 3.45 24 h 1 424 NS
Daphnia pulex Spect S 30 1 d 325 k1/k2 Southworth et al.
(1978)
Pontoporeia hoyi 14C-TLC F 0.7-7.1 6 h/14 d 28 145 k1/k2 Landrum (1988)
Oligochaetes
Stylodrilus 14C-TLC F < 200 6 h/8 d 5 055 k1/k2 Frank et al.
heringianus (1986)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Molluscs
Mya arenaria HPLC F 4.3 4 d/14 d 1 280 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 4.3 4 d/14 d 1 240 k1/k2 McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 4.3 4 d/14 d 500 k1/k2 McLeese &
Burridge (1987)
Pyrene
Bacteria
Mixed Spect S 1-20 2 h 24 600c NS Steen &
Karickhoff (1981)
Algae
Selenastrum GC S 500 1 d 55 800b NS Casserly et al.
capricornutum (1983)
Crustaceans
Crangon HPLC F 1.7 4 d/14 d 225 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnis magna HPLC S 5.7 24 h 2 702 NS Newsted & Giesy
(1987)
Daphnis pulex Sped S 50 24 h 2 702 k1/k2 Southworth et al.
(1978)
Pontoporeia hoyi 14C-TLC F 0.002-0.011 6 h/14 d 16 600 k1/k2 Landrum (1988)
Molluscs
Mya arenaria HPLC F 1.7 4 d/14 d 6 430 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 1.7 4 d/14 d 4 430 k1/k2 McLeese &
Burridge (1987)
Table 27. (continued)
Species Analysis Test Concentration Duration of Bioconcentration Type Reference
system in water exposure factor (in
(µg/litre) or uptake/ wet weight)
depuration
period
Oligochaetes
Stylodrilus 14C-TLC F < 26.4 6 h/8 d 6 588 k1/k2 Frank et al.
heringianus (1986)
Polychaetes
Neiris virens HPLC F 1.7 4 d/14 d 700 k1/k2 McLeese &
Burridge (1987)
Fish
Oncorhynchus GC-HPLC F 3.89 21 d 72.2 Equi Gerhart &
mykiss Carlson (1978)
Triphenylene
Crustaceans
Crangon HPLC F 0.5 4 d/14 d 270 k1/k2 McLeese &
septemspinosa Burridge (1987)
Daphnia magna HPLC S 1.7 1 d 9 066 NS Newsted & Giesy
(1987)
Molluscs
Mya arenaria HPLC F 0.5 4 d/14 d 5 540 k1/k2 McLeese &
Burridge (1987)
Mytilus edulis HPLC F 0.5 4 d/14 d 11 390 k1/k2 McLeese &
Burridge (1987)
Polychaetes
Neiris virens HPLC F 0.5 4 d/14 d 2 560 k1/k2 McLeese &
Burridge (1987)
Table 27 (continued)
14C, measurement of radioactivity in a liquid scintillation counter: as parent compounds cannot be differentiated from metabolites with
this method, additional extraction is usually performed.
S, static exposure system; Equi, at equilibrium Corg/Cw; HPLC, high-performance liquid chromatography; NS, not steady-state
Corg/Cw;
TLC, thin-layer chromatography; k1/k2, kinetics: uptake rate/depuration rate; Spect, spectroscopy; F, flow-through system;
R, static renewal system; GLC, gas-liquid chromatography; GC, gas chromatography; IF, intermittent flow system
a Based on dry weight (5 × wet weight)
b Based on total suspended solids
c Based on dry weight
4.1.6 Biomagnification
Biomagnification, the increase in the concentration of a
substance in animals in successive trophic levels of food chains, has
been determined in a number of studies. When Daphnia pulex were
exposed to water or algae contaminated with naphthalene, phenanthrene,
benz [a]anthracene, or benzo [a]pyrene, naphthalene accumulated to
the greatest extent from algal food, (bioconcentration factor, 11
000), whereas benz [a]anthracene and benzo [a]pyrene accumulated
more from water (bioconcentration factors, 1100 and 2700,
respectively). It must be emphasized that because of the short
exposure (24 h), the last two compounds would not have reached
equilibrium (Trucco et al., 1983).
In a study of bioaccumulation and biomagnification in closed
laboratory model ecosystems, green algae (Oedogonium cardiacum), D.
magna, mosquito larvae (Culex pipiens quinquefasciatus), snails
(Physa sp.), and mosquito fish (Gambusia affinis) were exposed for
three days to 2 µg/litre of 14C-benzo [a]pyrene. Of the radiolabel
accumulated, 88% was attached to parent compound in snails, 22% in
mosquito larvae, and none in fish. The parent compound represented 46%
of the total extractable radiolabel in mosquito larvae and 90% in
Daphnia. The bioconcentration factors were 5300 for algae, 12 000
for mosquito larvae, 82 000 for snails, 140 000 for Daphnia, and 930
for fish. Despite the apparent absence of bioconcentration in fish,
accumulation is assumed to be due to food-chain transfer, as no
accumulation of benzo [a]pyrene was found in a study of uptake from
water. Biomagnification was also studied in a terrestrial-aquatic
system, by adding 14C-benzo [a]pyrene to Sorghum vulgare seedlings
and allowing them to be eaten by fourth-instar salt-marsh caterpillar
larvae (Estigmene acrea); the labelled products entered the
terrestrial and aquatic phases as products such as faeces. The
food-chain organisms were the same as in the model aquatic ecosystem.
After a 33-day interaction period, the concentrations of
benzo [a]pyrene were 0.01 µg/litre water and 36.1 µg/kg algae, with
bioconcentration factors of 3600, 490, 2100, and 30, respectively.
Most of the radiolabel was found on polar products or as unextractable
radioactivity, which comprised 25% of the total in snails, 63% in
fish, 67% in mosquito larvae, and 79% in algae (Lu et al., 1977).
Trophic transfer of benzo [a]pyrene metabolites between benthic
organisms was studied by feeding Nereis virens 14C-benzo [a]pyrene
and harvesting them five days later. The worm homogenate contained 14%
parent compound, 7.2% organic-soluble metabolites, 58% water-soluble
metabolites, and 21% bound material. Flounder (Pseudiopleuronectes
americanus) were then given doses of 4.8-19 g of either pure
benzo [a]pyrene homogenized in unexposed Nereis or the
worm-metabolite mixture by gavage and analysed after 24 h of
incubation. On the basis of the radiolabel recovered from the fish
tissues, assuming comparable accumulation efficiency, flounder appear
to have at least a limited ability to accumulate polar, conjugated,
and bound metabolic products of benzo [a]pyrene from the diet. The
parent compound represented 5-15% of the radiolabel in liver and 6-7%
in intestine; conjugated metabolites represented 40-60% of the label
in liver and 60-70% in intestine; and bound metabolic products
represented 30% in liver and 10-20% in intestine (McElroy & Sisson,
1989).
4.2 Transformation
On the basis of model calculations, Mackay et al. (1992)
classified some PAH according to their persistence in air, water,
soil, and sediment (Table 28).
Table 28. Suggested half-life classes of polycyclic aromatic
hydrocarbons in various environmental compartments
Class Half-life (h)
Mean Range
1 17 10-30
2 55 30-100
3 170 100-300
4 550 300-1000
5 1 700 1000-3000
6 5 500 3000-10 000
7 17 000 10 000-30 000
8 55 000 > 30 000
Compound Air Water Soil Sediment
Acenalphthylene 2 4 6 7
Anthracene 2 4 6 7
Benz[a]anthracene 3 5 7 8
Benzo[a]pyrene 3 5 7 8
Benzo[k]fluoranthene 3 5 7 8
Chrysene 3 5 7 8
Dibenz[a,h]anthracene 3 5 7 8
Fluoranthene 3 5 7 8
Fluorene 2 4 6 7
Naphthalene 1 3 5 6
Perylene 3 5 7 8
Phenanthrene 2 4 6 7
Pyrene 3 5 7 8
From Mackay et al. (1992)
4.2.1 Biotic transformation
4.2.1.1 Biodegradation
Information on the biodegradation of PAH in water and soil under
aerobic and anaerobic conditions is summarized in Table 29. The few
results available from standard tests for biodegradation in water show
that PAH with up to four aromatic rings are biodegradable under
aerobic conditions but that the biodegradation rate of PAH with more
aromatic rings is very low. Biodegradation under anaerobic conditions
is slow for all components (Neff, 1979). The reactions normally
proceed by the introduction of two hydroxyl groups into the aromatic
nucleus, to form dihydrodiol intermediates. Bacterial degradation
produces cis-dihydrodiols (from a dioxetane intermediate), whereas
metabolism in fungal or mammalian systems produces trans-dihydrodiol
intermediates (from an arene oxide intermediate). The differences in
the metabolic pathways are due to the presence of the cytochrome P450
enzyme system in fungi and mammals. Algae have been reported to
degrade benzo [a]pyrene to oxides, peroxides, and dihydroxydiols (see
below). Owing to the high biotransformation rate (see also section
4.2.1.2), the concentrations of PAH in organisms and water are usually
not in a steady state. Freely dissolved PAH may be rapidly degraded
under natural conditions if sufficient biomass is available and the
turnover rates are fairly high (see Table 29).
Biodegradation is the major mechanism for removal of PAH from
soil. PAH with fewer than four aromatic rings may also be removed by
volatilization and photolysis (see also sections 4.1.4 and 4.2.2.1).
The rate of biodegradation in soil depends on several factors,
including the characteristics of the soil and its microbial population
and the properties of the PAH present. Temperature, pH, oxygen
content, soil type, nutrients, and the presence of other substances
that can act as co-metabolites are also important (Sims & Overcash,
1983). Biodegradation is further affected by the bioavailability of
the PAH. Sorption of PAH by soil organic matter may limit the
biodegradation of compounds that would normally undergo rapid
degradation (Manilal & Alexander, 1991); however, no significant
difference was found in the biodegradation rate of anthracene in water
with 10 and 1000 mg/litre suspended material (Leslie et al., 1987). In
Kidman sandy loam, the biodegradation rates varied between 0.23 h-1
(or 5.5 d-1) for naphthalene and 0.0018 d-1 for fluoranthene (see
Table 29). In a study with sandy loams, forest soil, and roadside soil
partially loaded with sewage sludge from a municipal treatment plant,
the following half-lives (in days) were found: 14-48 for naphthalene,
44-74 for acenaphthene plus fluorene, 83-193 for phenanthrene, 48-210
for anthracene, 110-184 for fluoranthene, 127-320 for pyrene, 106-313
for benz [a]anthracene plus chrysene, 113-282 for
benzo [b]fluoranthene, 143-359 for benzo [k]fluoranthene, 120-258
for benzo [a]pyrene, 365-535 for benzo [ghi]perylene, and 603-2030
for coronene (Wild & Jones, 1993).
Table 29. Biodegradation of polycyclic aromatic hydrocarbons (PAH)
Compound Rate constant Half-life Comments Reference
Acenaphthene 100% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d static flask screening; settled domestic waste (1981)
as inoculum; experiments with 5 and 10 mg/litre
PAH at 25°C; detection by GC
295-2448 h Aerobic half-life; aerobic soil column Kincannon & Lin
(1985)
1180-9792 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation half-life (1991)
0% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 7 d with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
< 3.2 year Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Acenaphthylene 98% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d statis flask screening; settled domestic waste (1981)
as inoculum; 5 or 10 mg/litre PAH at 25°C;
detection by GC
1020-1440 h Aerobic half-life; soil column Kincannon & Lin
(1985)
4080-5760 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
0% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Anthracene 0.061 h-1 10 h Microbial degradation in Third Creek water Southworth
incubated 18 h at 25°C: (1977)
Removal rate constants from water column at
25°C in midsummer sunlight:
0.060 h-1 12 h - in deep, slow, somewhat turbid water
0.030 h-1 23 h - in deep, slow, muddy water
0.061 h-1 11 h - in deep, slow, clear water
0.061 h-1 11 h - in shallow, fast, clear water
0.061 h-1 11 h - in very shallow, fast, clear water
0.035 h-1 20 h Microbial degradation rate constant Herbes et al.
(1980)
51-92% degradation Significant degradation with gradual Tabak et al.
after 7 d adaptation; static flask screening; settled (1981)
domestic waste as inoculum; experiments
with 5 and 10 mg/litre PAH at 25°C; detection
by GC
1200-11 040 h Aerobic half-life; aerobic soil die-away Coover & Sims
(1987)
20O g dry weight of soil at -0.33 bar Park et al.
[33 kPa] soil moisture at 25°C: (1990)
0.0052 d-1 3200 h - Kidman sandy foam; initial test
concentration, 210 mg/kg
0.0138 d-1 1200 h - McLaurin sandy loam; initial test
concentration, 199 mg/kg
4800-44 160 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation half-life (1991)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
1.9% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 2 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
Anthracene 33% after 16 months Degradation in soil in co-metabolic closed Bossert &
bottle with 1-phenyldecane as primary Bartha (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 60%
5% after 56 d Batch test with river water; initial concentration, Fedorak et al.
20 mg/litre related to dissolved organic carbon; (1982)
no mineralization during first 19 days; 20°C
Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons: (1995)
10-60% after 64 d - initial concentration, 31.3 ng/g
- Inoculated with enriched culture of
Mycobacteriarn sp. and initial test concentration
of 37.7 ng/g; biodegradation rate without
enriched culture, 18% after 64 d
Static test in bioreactor in enriched mixed Walter et al.
culture; anthracene oil (38 g/litre) which also (1990)
contained 62 mg/g fluorene; 30°C:
100% after 3 d - under aerobic conditions
90% after 20 d - under anaerobic conditions
17-45 d Aerobic degradation in surface Donneybrook Bulman et al.
sandy loam from Canadian pasture; initial test (1987)
concentrations, 5 and 50 mg/kg; up to 400 days'
exposure at 20 00 and water-holding capacity of
60% of the soil
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
7.9 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benz[a]anthracene 2448-16 320 h Aerobic soil die-away at 10-30°C Groenewegen &
Stolp (1976); Coover
& Sims (1987)
0% degradation No significant degradation under conditions of Tabak et al. (1981)
after 7 d method; static flask sceening; settled domestic
waste as inoculum; experiment with 5 and
10 mg/littre PAH at 25°C; detection by GC
0.0026 d-1 6400 h Kidman sandy loam Park et al. (1990)
9792-65 280 h Anaerobic half-life; estimated unacclimatized Howard et al. (1991)
aqueous aerobic biodegradation
16% after Degradation in soil in co-metabolic closed Bossert & Bartha
16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 18%
0-40% after 64 d Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons; initial (1995)
concentration, 31.3 ng/g
130-240 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
8.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benzo[a]pyrene 0.2-0.9 Aquatic fate rate for bacterial protein Barnsley (1975)
µmol.h-1mg-1
3.5 × 10-5 h-1 19 800 h Estimated rate constant in soil and water Ryan & Cohen
(1986)
1368-12 702 h Aerobic half-life at 10-30°C; soil die-away Coover & Sims
(1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; 33 mg/kg at 25°C:
0.0022 d-1 7416 h - Kidman sandy loam
0.0030 d-1 5496 h - McLaurin sandy loam
5472-50 808 h Anaerobic half-life; estimated unacclimatized Coover & Sims
aqueous aerobic biodegradation (1987)
< 8% after 160 d Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons; initial (1995)
concentration, 105 ng/g
218-347 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
8.2 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Benzo[b]fluoranihene 8640-14 640 h Aerobic half-life; estimated unacclimatized Coover & Sims
aqueous aerobic biodegradation (1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 38 mg/kg at 25°C:
0.0024 d-1 7056 h - Kidman sandy loam
0.0033 d-1 5064 h - McLaurin sandy loam
34 560-58 560 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
9 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benzo[ghi]perylene 14 160-15 600 h Aerobic half-life; aerobic soil dieaway at Coover & Sims
10-30°C (1987)
56 640-62 400 h Anaerobic half-life; aerobic soil dieaway at Coover & Sims
10-30°C (1987)
9.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Benzo[k]fluoranthene 21 840-51 360 h Aerobic half-life; aerobic soil dieaway Coover & Sims
(1987)
87 360-205 440 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
8.7 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Chrysene 59% degradation Significant degradation with gradual Tabak et al. (1981)
after 7 d adaptation; static flask screening; settled
domestic waste as inoculum; experiment
with 5 mg/litre PAH at 25°C; detection by GC
38% degradation No significant degradation under conditions of Tabak et al. (1981)
after 7 d method; static flask sceening; settled domestic
waste as inoculum; experiment with 10 mg/litre
PAH at 25°C; detection by GC
8904-24 000 h Aerobic half-life; aerobic soil dieaway Coover & Sims
(1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 100 mg/kg at 25°C:
0.0019 d-1 8904 h - Kidman sandy loam
0.0018 d-1 9288 h - McLaurin sandy loam
35 616-96 000 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
11 % after 16 Degradation in soil in co-metabolic closed Bossert & Bartha
months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 5%
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
224-328 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
8.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Coronene 16.5 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Dibenz[a,h]anthracene 8664-22 560 In Aerobic half-life; aerobic soil die-away Coover & Sims
(1987); Park et al.
(1990)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 13 mg/kg at 25°C:
0.0019 d-1 8664 h - Kidman sandy loam
0.0017 d-1 10 080 h - McLaurin sandy loam
No degradation Degradation in soil in co-metabolic closed Bossert & Bartha
after 16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Fluoranthene 2.2 × 10-3 Aquatic fate rate with bacterial protein Barnsley (1975)
µmol h-1mg-1
100% degradation Significant degradation with gradual adaptation; Tabak et al. (1981)
after 7 d static flask screening; settled domestic waste
as inoculum; experiment with 5 mg/litre PAH
at 25°C; detection by GC
0% degradation No significant degradation under conditions of Tabak et al. (1981)
after 7 d method; static flask screening; settled domestic
waste as inoculum; experiment with 10 mg/litre
PAH at 25°C; detection by GC
3360-10 560 h Aerobic half-life; aerobic soil dieaway Coover & Sims
(1987)
0.19 h-1 3.6 h In atmosphere Dragoescu &
Friedlander (1989)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, 900 mg/kg at 25°C:
0.0018 d-1 9048 h - Kidman sandy loam
0.0026 d-1 6432 h - McLaurin sandy loam
13 440-42 240 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
34-39 d Aerobic degradation in surface samples of Bulman et al.
Donneybrook sandy loam from Canadian (1987)
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
7.8 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Fluorene 45-77% degradation Significant degnadation with gradual adaptation; Tabak et al. (1981)
after 7 d static flask screening; settled domestic waste
as inoculum; experiment with 5 and 10 mg/litre
PAH at 25°C; detection by GC
Degradation of 30 µg/litre in natural river water Lee & Ryan (1976)
(Skidway River; salinity, 20%):
100% after 1000 d - Turnover time in June at incubation time of
48 h
0% after 72 h - February or May
30% after 1 week Degradation of non-autoclaved groundwater Lee et al. (1984)
samples of ± 0.06 mg/litre by microbes
768-1440 h Aerobic half-life; aerobic soil diaway Coover & Sims
(1987)
3072-5760 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
0% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
100% after 36 h Batch test with enriched culture of Arthrobacter Grifoll et al.
sp.; initial test concentration, 483 µmol/litre; (1992)
22°C
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
< 3.2 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Indeno[1,2,3-cd]pyrene 200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 8 mg/kg at 25°C:
0.0024 d-1 6912 h - Kidman sandy loam
0.0024 d-1 6936 h - McLaurin sandy loam
Naphthalene Degradation in natural river water (Skidway Lee & Ryan
River; salinity, 20%): (1976)
500 d - Turnover time in February at incubation
time of 48 h; test concentration, 40 µg/litre
46 d - Turnover time in May at incubation
time of 24 h; test concentration, 40 µg/litre
79 d - Turnover time in May at incubation time
of 8 h; test concentration, 40 µg/litre
30 d - Turnover time in May at incubation
time of 24 h; test concentration, 130 µg/litre
Degradation of 130 µg/litre in natural water Lee & Ryan
330 d offshore with salinity of 35%: turnover time (1976)
in May at incubation time of 24 h
0.0403.3 × 10-6 At depth of 5-10 m in laboratory water basin Lee & Anderson
g/litre per d (1977)
100% after 8 d In gas-oll-contaminated groundwater Kappeler &
circulated through sand inoculated with Wuhrmann
groundwater under aerobic conditions (1978)
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
168 h In oil-polluted estuarine stream Lee (1977)
576 h In clean estuarine stream
1500 h In coastal waters
40 800 h In the Gulf Stream
12h Aerobic half-life; die-away in oil-polluted Walker & Colwell
creek (1976)
Anaerobic half-life: Hambrick et al.
600 h at pH 8 (1980)
6200 h at pH 5
24-216 h In deep, slowly moving, contaminated water Herbes (1981);
Wakeham et al.
(1983)
0.23 h-1 3.O h Microbial degradation rate constant Herbes et al. (1980)
100% degradation Significant degradation with rapid adaptation; Tabak et al. (1981)
after 7 d static flask screening; settled domestic waste
as inoculum; experiments with 6 and 10 mg/litre
PAH at 25°C; detection by GC
100% degradation Degradation of non-autoclaved groundwater Lee et al. (1984)
after 7 d samples of ± 0.04 mg/litre by microbes
0.024 d-1 693 h Groundwater with nutrients and acclimatized Vaishnav & Babeu
microbes (1987)
0.013 d-1 1279 h River water with acclimatized microbes
0.018-1 924 h River water with nutrients and acclimatized
microbes
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[-0.0032 kPa] soil moisture; initial test
concentration, 101 mg/kg at 25°C:
0.377 d-1 50 h - Kidman sandy loam
0.308 d-1 53 h - McLaurin sandy loam
2% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 30 mg/litre PAH and 100 mg/litre sAdge International Trade
and Industry (1992)
< 2.1 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
Perylene No degradation Degradation in soil in co-metabolic closed Bossert & Bartha
after 16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g
Phenanthrene 100% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d static flask screening; settled domestic waste (1981)
as inoculum; experiments with 5 and 10 mg/litre
PAH at 25°C; detection by GC
383-4800 h Aerobic half-life; aerobic soil die-away Coover & Sims
(1987)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[-0.0032 kPa] soil moisture; initial test
concentration, 900 mg/kg at 25°C:
0.0447 d-1 384 h - Kidman sandy loam
0.0196 d-1 840 h - McLaurin sandy loam
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
1536-19 200 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
96 h Inorganic solution Manilal & Alexander
264 h Kendaia soil (1991)
54% degradation Japanese Ministry of Trade and Industry test Japanese Ministry of
after 4 weeks with 100 mg/litre PAH and 30 mg/litre sludge International Trade
and Industry (1992)
> 62 % after Degradation in soil in co-metabolic closed Bossert & Bartha
16 months bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss significant
Serum bottle radiorespirometry in five soils Grosser et al. (1995)
contaminated with hydrocarbons:
38-55% after 64 d - initial concentration, 31.3 ng/g
80% after 32 d - inoculated with enriched culture of
Mycobacterium sp. and an initial test
concentration of 17.9 ng/g
9.7-14 d Aerobic degradation in surface samples of Bulman et al. (1987)
Donneybrook sandy loam from Canadian
pasture; initial test concentrations, 5 and
50 mg/kg; up to 400 days' exposure at 20°C
and water-holding capacity of 60% of the soil
5.7 years Field tests of rural British soils amended with Wild et al.
metal-enriched sewage sludges with (1991)
0.1-15.1 mg/kg PAH
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Pyrene 100% degradation Significant degradation with rapid adaptation; Tabak et al.
after 7 d static flask screening; settled domestic waste (1981)
as inoculum; experiment with 5 mg/litre
PAH at 25°C; detection by GC
0% degradation No significant degradation under conditions of Tabak et al.
after 7 d method; static flask screening; settled domestic (1981)
waste as inoculum; experiments with 5 and
10 mg/litre PAH at 25°C; detection by GC
5040-46 600 h Aerobic half-life at 10-30°C; aerobic soil Coover & Sims
die-away (1987)
0.29 h-1 2.4 h In atmosphere Dragoescu &
Friedlander
(1989)
200 g dry weight of soil at -0.33 bar Park et al. (1990)
[33 kPa] soil moisture; initial test
concentration, ± 690 mg/kg at 25°C:
0.0027 d-1 6240 h - Kidman sandy loam
0.0035 d-1 4776 h - McLaurin sandy loam
20 160-182 400 h Anaerobic half-life; estimated unacclimatized Howard et al.
aqueous aerobic biodegradation (1991)
70% after 16 months Degradation in soil in co-metabolic closed Bossert & Bartha
bottle with 1-phenyldecane as primary (1986)
substrate; 20°C; initial test concentration,
1 mg/g; abiotic loss, 27%
Table 29. (continued)
Compound Rate constant Half-life Comments Reference
Serum bottle radiorespirometry in five soils Grosser et al.
contaminated with hydrocarbons: (1995)
25-70% after 64 d - initial concentration, 8.5 ng/g
54% after 32 d - inoculated with enriched culture of
Mycobacterium sp. and an initial test
concentration of 7.7 ng/g
52.4% after 96 h Mineralization test with Mycobacterium sp.; Heitkamp et al.
24°C; initial test concentration, 0.5 mg/litre (1988)
48-58 d Aerobic degradation in surface Donneybrook Bulman et al. (1987)
sandy loam from Canadian pasture; initial test
concentrations, 5 and 50 mg/kg; up to 400 days'
exposure at 20°C and water-holding capacity of
60% of the soil
8.5 years Field tests of rural British soils amended with Wild et al. (1991)
metal-enriched sewage sludges with
0.1-15.1 mg/kg PAH
GC, gas chromatography In order to compare numbers when only rate constants are reported, the half-lives were estimated from the formula:
t1/2 = In2
k
where t1/2 is the half-life and k is the rate constant. The calculated values are reported in italics.
After biodegradation of pyrene by a Mycobacterium sp., cis-
and trans-4,5-pyrene dihydrodiol and pyrenol were the initial ring
oxidation products. The main metabolite was 4-phenathroic acid. The
ring fission products were 4-hydroxyperinaphthenone and cinnamic and
phthalic acids (Heitkamp et al., 1988).
The pyrene-metabolizing Mycobacterium sp. can also use
phenanthrene and fluoranthene as the sole source of carbon.
Phenanthrene was degraded and 1-hydroxy-2-naphthoic acid,
ortho-phthalate, and protocatechuate were detected as metabolites.
1-Hydroxy-2-naphthoic acid did not accumulate, indicating that it is
further metabolized (Boldrin et al., 1993).
A strain of Arthobacter sp. was isolated that was capable of
metabolizing fluorene as a sole energy source: 483 nmol/ml were
degraded completely within 36 h, and four major metabolites were
detected: 9-fluorenol, 9 H-fluoren-9-one, 3,4-dihydrocoumarin, and an
unidentified polar-substituted aromatic compound. Fluorenol was not
degraded further, suggesting that it and fluorenone are products of a
separate metabolic pathway from that which produces dihydrocoumarin,
the polar compound, and the energy for cell growth. The bacteria could
also degrade phenanthrene (Grifoll et al., 1992).
The degradation of PAH was studied in a culture made from
activated sludge, polychlorinated biphenyl-degrading bacteria, and
chlorophenol-degrading mixed cultures, adapted to naphthalene. The
metabolites of naphthalene were 2-hydroxybenzoic acid and
1-naphthalenol, those of phenanthrene were 1-phrenanthrenol and
1-hydroxy-2-naphthalenecarboxylic acid, and that of anthracene was
3-hydroxy-2-naphthalenecarboxylic acid. The authors concluded that the
biotransformation pathway proceeds via initial hydroxylation to ring
cleavage, to yield the ortho or meta cleavage intermediates, which
are further metabolized via conventional metabolic pathways (Liu et
al., 1992).
The metabolism of PAH by fungi is similar to that by mammalian
cells. For example, Cunninghamella elegans in culture metabolizes
benzo [a]pyrene to the trans-7,8-diol, the trans-9,10-diol,
3,6-quinone, 9-hydroxybenzo [a]pyrene, 3-hydroxybenzo [a]pyrene, and
7,8-dihydro-7,8-dihydroxybenzo [a]pyrene (Cerniglia, 1984). In a
further experiment, C. elegans metabolized about 69% of added
fluorene after 24 h. The major ethyl acetate-soluble metabolites were
9-fluorenone (62%), 9-fluorenol, and 2-hydroxy-9-fluorenone (together,
7%). The degradation pathway was similar to that in bacteria, with
oxidation at the C9 position of the five-member ring to form an
alcohol and the corresponding ketone. 2-Hydroxy-9-fluorenone had not
been found as a metabolite previously (Pothuluri et al., 1993).
4.2.1.2 Biotransformation
Biotransformation is often advanced as an explanation for the
differences in PAH profiles seen in aquatic organisms and in the
medium to which they were exposed. Furthermore, all of the metabolites
of PAH may not have been identified or quantified. This section
addresses biotransformation in organisms other than bacteria and
fungi, which is discussed in section 4.2.1.1, above.
The uptake of naphthalene and benzo [a]pyrene was studied in
three species of marine fish: the mudsucker or sand goby
(Gillichthys mirabilis), the sculpin (Oligocottus maculosus), and
the sand dab (Citharichthys stigmaeus). In all three species,
biotransformation took place rapidly in the liver. The uptake of
naphthalene was greater than that of benzo [a]pyrene. The major
metabolite of benzo [a]pyrene appeared to be
7,8-dihydroxy-7,8-dihydroxy benzo [a]pyrene, while the major
metabolite of naphthalene was 1,2-dihydro-1,2-dihydroxy-naphthalene.
The gall-bladder was the major storage site for the PAH and their
metabolites. Naphthalene and its metabolites were removed at a higher
rate than benzo [a]pyrene and its metabolites (Lee et al., 1972).
Transformation of naphthalene and benzo [a]pyrene in the
bluegill sunfish Lepomis macrochirus took place very rapidly,
benzo [a]pyrene having the highest rate (McCarthy & Jimenez, 1985).
L. macrochirus were exposed in a flow-through system to 4 nmol/litre
benzo [a]pyrene for 48 h, followed by a 96-h depuration period, at 13
or 23°C in the presence or absence of food. Both polar and nonpolar
metabolites were found. After 48 h, the polar metabolites comprised
10% of the benzo [a]pyrene metabolites in fed fish at 13°C, 20% in
unfed fish at 23°C, and 30% in fed fish at 23°C (Jimenez et al.,
1987). In rainbow trout (Oncorhynchus mykiss) exposed to naphthalene
at 0.5 mg/litre for 24 h, the bile contained 65-70% metabolites, the
liver contained 5-10%, and muscle < 1% (Melancon & Lech, 1978).
In L. macrochirus exposed to 8.9 ± 2.1 µg/litre acenaphthene
for 28 days, the half-life for metabolism was less than one day. No
information was given on metabolites (Barrows et al., 1980).
The depuration of anthracene was investigated in O. mykiss
during simulated day and night cycles of 16 and 8 h, respectively.
After a 96-h clearance period, the metabolites contributed 2-3% of the
depurated substance, half of which came from the bile. No specific
metabolites were reported (Linder & Bergman, 1984). After L.
macrochirus had been exposed to anthracene at 8.9 µg/litre or
benzo [a]pyrene at 0.98 µg/litre for 4 h, the rates of
biotransformation were 0.26 and 0.082 nmol/g per h, respectively, and
8% of the anthracene and 88% of the benzo [a]pyrene were metabolized
(Spacie et al., 1983).
Benzo [a]pyrene is transformed in the Japanese medaka
(Oryzias latipes) and the guppy (Poecilia reticulata), the main
metabolite being the 7,8-diol-9,10-epoxide (Hawkins et al., 1988).
Two benthic organisms, the European fingernail clam (Sphaerium
corneum) and larvae of the midge Chironomus riparius, both
metabolized benzo [a]pyrene. In the larvae, the main metabolite
appeared to be 3-hydroxybenzo [a]pyrene; a quinone isomer was also
found. Only a very small amount of 3-hydroxy-benzo [a]pyrene was
found in the clam. No diol metabolites were found in either species
(Borchert & Westendorf, 1994). After exposure of the benthic
oligochaete Stylodrilus heringianus to either anthracene and pyrene
or phenanthrene and benzo [a]pyrene, 2% degradation of each PAH was
reported within 24 h (Frank et al., 1986).
The half-lives for metabolism in D. magna were 0.5 h for 1.8
mg/litre naphthalene, 9 h for 0.06 mg/litre phenanthrene, and 18 h for
0.023 mg/litre chrysene (Eastmond et al., 1984).
In amphipod Hyalella azteca was exposed to 0.043 nmol/ml
anthracene for 8 h, the rates of biotransformation were 2.2 ± 0.5
nmol/g dry weight per h with no substratum, 3.0 ± 0.8 in the presence
of washed sand from a local lake, and 1.0 ± 0.15 in the presence of
sediment from the lake (Landrum & Scavia, 1983).
The amphipod Rhepoxynius abronius metabolizes benzo [a]pyrene
(Plesha et al., 1988). When two marine amphipods were exposed to a
sediment containing 5.1 ng/mg of this compound, R. abronius
metabolized 49% and Eohaustorius washingtonianus metabolized 27% of
the benzo [a]pyrene after one day. The main metabolites appeared to
be 7,8-dihydro-7,8-dihydroxy-benzo [a]pyrene,
9,10-dihydro-9,10-dihydroxybenzo [a]pyrene,
3-hydroxy-benzo [a]pyrene, and 9-hydroxybenzo [a]pyrene. The ratio
of 7,8-dihydro-7,8-dihydroxybenzo [a]pyrene to
9,10-dihydro-9,10-dihydroxybenzo [a]pyrene in normal-phase
high-performance liquid chromatography was 1.2 for R. abronius and
0.7 for E. washingtonianus (Reichert et al., 1985).
No biotransformation of benzo [a]pyrene or phenanthrene was
found in mayflies (Hexagenia limbata) or in the amphipod
Pontoreia hoyi (Landrum & Poore, 1988).
In a study of the route of metabolism of benzo [a]pyrene in
green algae (Selenastrum capricornutum) exposed to 1.2 µg/litre for
four days, with simulated day and night periods, the major dihydrodiol
metabolites identified were the cis-4,5-diol (< 1%), the
cis-7,8-diol (13%), the 9,10-diol (36%), and the cis-11,12-diol
(50%), demonstrating the presence of a dioxygenase enzyme for this
type of algae (Lindquist & Warshawsky, 1985), as suggested by Cody et
al. (1984). Payne (1977) reported, however, that aryl hydrocarbon
hydroxylase was not present in Fucus and Ascophyllum sp. of marine
algae.
Benzo [a]pyrene was not biotransformed in periphyton after 0.25
or 4 h. In cladocerans (D. magna) exposed to 1.0 µg/litre
benzo [a]pyrene, the biotransformation rate after exposure for 6 h
was 1.07 ± 0.20 nmol/g dry weight per h. In midge larvae (C.
riparius) exposed to 0.6-1.5 µg/litre, the biotrans-formation rate
was 3.6 ± 0.7 nmol/g dry weight per h after exposure for 1 h and 2.7 ±
0.3 after 4 h. In L. macrochirus exposed to 1.0 µg/litre, the
biotransformation rate was 0.20 ± 0.03 nmol/g dry weight per h after 1
h and 0.37 ± 0.04 after 4 h. In chironomids, 3-hydroxybenzo [a]pyrene
was the major metabolite after 8 h, representing 4.4% of the total
water activity; smaller amounts of 7-hydroxy-benzo [a]pyrene and the
9,10- and 7,8-dihydroxydiols of benzo [a]pyrene were also found
(Leversee et al., 1981).
After exposure of benthic species to benzo [a]pyrene for one to
four weeks, the following percentages of metabolites were found: E.
washingtonianus, 22% in the whole body; R. abronius, 74% in the
whole body; clams (Macoma nasuta), < 5% in the body and < 5 in the
hepatopancreas; shrimp (Pandalus platyceros), 94% in the
hepatopancreas; and the English sole (Parophrys vetulus), 94% in the
body, 99% in the liver and > 99% in the bile (Varanasi et al., 1985).
Mosquito larvae (C. pipens quinquefasciatus) were exposed for
three days to 0.002 mg/litre benzo [a]pyrene in the presence or
absence of the mixed-function oxidase inhibitor piperonyl butoxide at
0.0025 mg/litre. Parent benzo [a]pyrene represented 22% of the
excreted PAH in the absence of piperonyl butoxide and 86% in its
presence. After three days' exposure of snails (Physa sp.) to the
same concentration of benzo [a]pyrene with or without piperonyl
butoxide at 0.0025 mg/litre, parent benzo [a]pyrene represented 88%
in the absence of the inhibitor and 85% in its presence. The authors
suggested that snails are deficient in microsomal oxidases. In
mosquito fish (G. affinis) exposed similarly, no parent
benzo [a]pyrene was found in the absence of piperonyl butoxide but
21% in its presence (Lu et al., 1977).
In an aquatic ecosystem, plankton, green algae (Oedogonium
cardiacum), D. magna, mosquito larvae (C. pipiens
quinquefasciatus), snails (Physa sp.), and mosquito fish
(G. affinis) were exposed to 0.002 mg/litre benzo [a]pyrene for
three days. Parent benzo [a]pyrene represented 83, 90, 46, 70, and
55% in the four organisms, respectively. The substance was metabolized
to unidentified hydroxylated polar compounds. The finding of 55%
parent benzo [a]pyrene in the fish was attributed to food-chain
transfer, as none was found after direct exposure. A
terrestrial-aquatic ecosystem was also exposed to benzo [a]pyrene by
applying 0.2 mg of radiolabelled compound to Sorghum vulgare
seedlings to simulate atmospheric fall-out and allowing them to be
consumed by fourth-instar salt-marsh caterpillar larvae (E. acrea).
Faecal products then entered the terrestrial and aquatic ecosystem
described above, which was left for 33 days. The maximum radiolabel
(0.005 ppm) was detected in the aquatic phase after 14 days.
Unmetabolized benzo [a]pyrene accounted for 7.1% of the total
extractable radiolabel in fish, 19% in snails, 32% in algae, and 34%
in mosquitoes. Addition of the mixed-function oxidase inhibitor,
piperonyl butoxide, resulted in 12% parent benzo [a]pyrene in fish,
34% in snails, 48% in the algae, and no change in mosquitoes (Lu et
al., 1977).
The biotransformation of 19 PAH was studied in the food chain
seston (plankton) -> blue mussel (Mytilus edulis L.) -> common
eider duck (Somateria mollissima L.) in the open, northern Baltic
Sea. The concentrations of the PAH in the eider duck showed the
distribution gallbladder > adipose tissue > liver. There was a
high flux of the PAH in the food chain, but the concentration did not
increase with increasing trophic level, indicating that the PAH were
biotransformed rapidly. There was little biotransformation in the
plankton. The distribution of the PAH in blue mussels was different
from that in plankton, perhaps due to metabolic activity in the
mussel. Biotransformation of PAH with a relative molecular mass of 252
was rapid in the ducks (Broman et al., 1990).
In beans (Phaseolus vulgaris L.) exposed to 15 g anthracene
per plant, uptake via the roots was rapid, 90% being metabolized
within 30 days (Edwards, 1986).
These investigations are summarized in Table 30. As the rate of
metabolism depends not only on the species but also on factors such as
temperature, pH, and other experimental conditions, the results are
difficult to compare. Some general conclusions can, however, be drawn:
- The biotransformation potential of aquatic organisms depends on
the activity of cytochrome P450-dependent mixed-function
oxidases, which are important for oxidation, the first step in
the metabolism of xenobiotics such as PAH (James, 1989).
- The tissues in which biotransformation mainly takes place are
liver, lung, kidney, placenta, intestinal tract, and skin
(Cerniglia, 1984).
- The initial transformation step in invertebrates usually occurs
more slowly than in vertebrates (James, 1989). Monoxygenation of
PAH is faster in higher invertebrates like arthropods,
echinoderms, and annelids and slowest in more primitive
invertebrates like protozoa, profina, cnidaria, and molluscs
(Neff, 1979).
- In general, invertebrates excrete PAH metabolites inefficiently
(James, 1989).
- In higher organisms and algae, metabolites are usually produced
by monooxygenase activity, resulting in the formation of
epoxides, phenols, diols, tetrols, quinones, and conjugates.
- It is not clear whether molluscs have cytochrome P450 activity
(Moore et al., 1989).
Table 30. Biotransformation of polycyclic aromatic hydrocarbons by various organisms
Species Compound Biotransformation rate Reference
Fungi
Cunninghamella elegans Benzo[a]pyrene No information Cerniglia (1984)
Algae
Selenastrum capticornutum Benzo[a]pyrene Relatively fast Lindquist & Warshawsky (1985)
Oedogenium cardiacum Benzo[a]pyrene 15% after 3 d in Lu et al. (1977)
aquatic ecosystem
Fucus sp. Various None Payne(1977)
Ascophyllum sp. Various None
Molluscs
Sphaerium corneum Benzo[a]pyrene Very fast (no carcinogenic Borchert & Westendorf (1994)
metabolites)
Physa sp. Benzo[a]pyrene 12% after 3 d Lu et al. (1977)
Mytilus edulis L. Different No information Broman et al. (1990)
Crustaceae
Hyalella azteca Anthracene 2.2 nmol/g dw/h in water Landrum & Scavia (1983)
Hyalella azteca Anthracene 3.0 nmol/g dw/h 5 water/ Landrum & Scavia (1983)
sediment
Daphnia magna Benzo[a]pyrene 1.07 nmol/g dw/h after 6 h Leversee et al. (1981)
Daphnia magna Benzo[a]pyrene 10% after 3 d in aquatic Lu et al. (1977)
ecosystem
Pontoporeia hoyi Benzo[a]pyrene None Landrum & Poore (1988)
Pontoporeia hoyi Benzo[a]pyrene None after 48 h Evans & Landrum (1989)
Mysis relicta Benzo[a]pyrene No information Evans & Landrum (1989)
Rhepoxynius abronius Benzo[a]pyrene No information Plesha et al. (1988)
Rhepoxynius abronius Benzo[a]pyrene 74% after 1-4 weeks Varanasi et al. (1985)
Rhepoxynius abronius Benzo[a]pyrene 49% after 1 d Reichert et al. (1985)
Eohaustorius washingtonianus Benzo[a]pyrene 27% after 1 d Reichert et al. (1985)
Eohaustorius washingtonianus Benzo[a]pyrene 22% after 1-4 weeks Varanasi et al. (1985)
Table 30. (continued)
Species Compound Biotransformation rate Reference
Pandalus platyceros Benzo[a]pyrene < 5% after 1-4 weeks Varanasi et al. (1985)
Parophrys vetulus Benzo[a]pyrene 94% after 1-4 weeks Varanasi et al. (1985)
Daphnia magna Chrysene 50% after 18 h Eastmond et al. (1984)
Daphnia magna Naphthalene 50% after 0.5 h Eastmond et al. (1984)
Daphnia magna Phenanthrene 50% after 9 h Eastmond et al. (1984)
Fish
Lepomis macrochirus Acenaphthene Half-life, < 1 d Barrows et al. (1980)
Lepomis macrochirus Anthracene 8% after 4 h Spacie et al. (1983)
Oncorhynchus mykiss Anthracene 2-3% after 24 h Linder & Bergman (1984)
Gillichthys mirabilis Benzo[a]pyrene Rapid in liver Lee et al. (1972)
Oligocottus maculosus Benzo[a]pyrene Rapid in liver Lee et al. (1972)
Citharichthys stigmaeus Benzo[a]pyrene Rapid in liver Lee et al. (1972)
Lepomis macrochirus Benzo[a]pyrene Very fast McCarthy & Jimenez (1981)
Lapomis macrochirus Benzo[a]pyrene 88% after 4h Spacie et al. (1983)
Lepomis macrochirus Benzo[a]pyrene 0.20-0.37 nmol/g dry Leversee et al. (1981)
weight per h
Oryzias latipes Benzo[a]pyrene No information Hawkins (1988)
Poecilia reticulata Benzo[a]pyrene No information Hawkins (1988)
Rhepoxynius abronius Benzo[a]pyrene None Plesha et al. (1988)
Gambusia affinis Benzo[a]pyrene 100% after 3 d in water Lu et al. (1977)
40% after 3 d in aquatic
ecosystem
Gillichthys mirabilis Naphthalene Rapid in liver Lee et al. (1972)
Oligocottus maculosus Naphthalene Rapid in liver Lee et al. (1972)
Citharichthys stigmaeus Naphthalene Rapid in liver Lee et al. (1972)
Lepomis macrochirus Naphthalene Very fast McCarthy & Jimenez (1981)
Worm
Stylodrilus heringianus Various None Franck et al. (1986)
Table 30. (continued)
Species Compound Biotransformation rate Reference
Insects
Chironomus riparius Benzo[a]pyrene Very fast (no carcinogenic Bochert & Westendorf (1994)
metabolites)
Chironomus riparius Benzo[a]pyrene 2.7-3.6 nmol/g dry weight Leversee et al. (1981)
per h
Hexagenia limbata Benzo[a]pyrene None Landrum & Poore (1983)
Culex pipiens Benzo[a]pyrene 78% after 3 d Lu et al. (1977)
quinquefasciatus
Somatochlora cingulata Naphthalene No information Correa & Coler (1990)
Bird
Somateria mollissima L. Various Fast for PAH with Broman et al. (1990)
molecular mass > 252
Plant
Phaseolus vulgaris L. Anthracene 90% after 30 d Edwards (1986)
- In crustaceans, biotransformation differs greatly between species
and for different PAH. Biotransformation of naphthalene,
anthracene, phenanthrene, and chrysene appears to occur rapidly,
while that of benzo [a]pyrene is generally slower. Only Reichert
et al. (1985) reported significant degradation in R. abronius
(49%) and E. washingtonianus (27%) within one day.
- It is not clear how rapidly biotransformation occurs in insects.
- Too little information was available on algae, plants, and fungi
for conclusions to be drawn.
4.2.2 Abiotic degradation
Abiotic processes may account for the removal of 2-20% of two-
and three-ring PAH from soil (Park et al., 1990). In soils partly
amended with PAH-containing sewage sludge, 24-100% was removed, and
naphthalene was eliminated almost completely by volatilization and
photodegradation (Wild & Jones, 1993).
4.2.2.1 Photodegradation in the environment
PAH can be expected to be photodegraded in air and water but to a
very low extent in soils and sediments, owing to low light intensity.
In natural waters, photodegradation takes place only in the upper few
centimetres of the aqueous phase. Information on the photodegradation
of PAH in air and water is summarized in Table 31; however, as the
testing conditions varied widely, general conclusions cannot be drawn.
PAH are photodegraded in air and water by two processes: direct
photolysis by light with a wavelength < 290 nm and indirect
photolysis by least one oxidizing agent such as OH, O3, and NO3 in
air and ROO radicals in water. In general, indirect photolysis -
photooxidation - is the more important process. The reaction rates of
PAH with airborne OH radicals measured under standard conditions are
given in Table 32, which shows that most of the calculated half-lives
are one day or less. Under environmental conditions, PAH of higher
molecular mass, i.e. those with more aromatic rings, are almost
completely adsorbed onto fine particles (see section 4.1.2); this
reduces the degradation rate markedly.
Degradation half-lives of 3.7-30 days were reported for the
reaction with NOx of various PAH adsorbed onto soot. The degradation
was much slower in the absence of sunlight. PAH did not react
significantly with SO2 (Butler & Crossley, 1981). PAH in wood smoke
and gasoline exhaust did not degrade significantly during winter in
extreme northern and southern latitudes owing to low temperatures and
the low angle of the sun (Kamens et al., 1986a). In summer, however,
at a temperature of 20°C, the half-lives of individual PAH were in the
range of 30-60 min (Kamens et al., 1986b). The degradation rate
increased further with increasing humidity (Kamens et al., 1991).
Table 31. Photodegradation of polycyclic aromatic hydrocarbons
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Acenaphthene Air, particles Determined in rotary photoreactor Behymer &
with 25 µg/g on: Hites (1985)
2.0 - silica gel
2.2 - alumina
44 - fly ash
Water 0.23 h-1 3.0 Rate constant in distilled water Fukuda et al.
(1988)
Acenaphthylene Air, particles Determined in rotary photoreactor Behymer &
with 25 µg/g on: Hites (1985)
0.7 - silica gel
2.2 - alumina
44 - fly ash
Anthracene Air, water 0.58 Measured in atmosphere and water Southworth
from aqueous photolysis rate (1979)
constant for midday summer sunlight
at 35°N
Air, particles Determined with 25 µg/g on: Behymer &
2.9 - silica gel Hites (1985)
0.5 - alumina
48 - fly ash
Water Removal rate constants from water Southworth
at 25°C in midsummer sunlight: (1979)
0.004 h-1 173 - in deep, slow, somewhat turbid
water
<0.001 h-1 > 700 - in deep, slow, muddy water
0.018 h-1 38 - in deep, slow, clear water
0.086 h-1 8 - in shallow, fast, clear water
0.238 h-1 3 - in very shallow, fast, clear water
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water Half-lives calclulated from average Southworth
light intensity over 24 h: (1977)
1.6 - in summer
4.8 - in winter
Water Half-lives calculated for direct Zepp &
sunlight at 40°N at midday in Schlotzhauer
midsummer: (1979)
0.75 - near surface water
108 - inland water
125 - inland water with sediment
partitioning
0.75 - direct photochemical
transformation near water surface
Water 0.66 h-1 1.0 In distilled water Fukuda et al.
(1988)
Benz[a]anthracene Air, particles First-order daytime decay rate Kamens et al.
constants with soot particle loading of: (1988)
0.0125 min-1 0.9 - 1000-2000 ng/mg
0.0250 min-1 0.5 - 30-350 ng/mg
Air, particles Determined with ± 25 µg/g on: Behymer &
4.0 - silica gel Hites (1985)
2.0 - alumina
38 - fly ash
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water Calculated rate constant in pure Mill et al.
water: (1981)
13.4 × 10-5s-1 1.4 - at 366 nm and in sunlight at
23-28°C, early March
2.28 × 1O-5s-1 8.4 - at 313 nm with 1% acetonitrile
in filter-sterilized natural water
5 Early March
Benzo[a]pyrene Air, particles Determined with 25 µg/g on: Behymer &
4.7 silica gel Hites (1985)
1.4 - alumina
31 - fly ash
Air particles First-order daytime decay rate Kamens et al.
constants with soot particle loading of: (1988)
0.0090 min-1 1.3 - 1000-2000 ng/mg
0.0211 min-1 0.54 - 30-350 ng/mg
Air, particles < 6.1 × 10-4 m/s Ozonization rate constant measured Cope &
at 24°C with O3 = 0.16 ppm and Kalkwarf
light intensity of 1.3 kW/m3 (1987)
Air 0.37-1.1 Estimated Lyman et al.
(1982)
Air 1 Sunlight in mid-December Mill & Mabey
(1985)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Air, water Calculated rate constants for Mill et al.
direct photolysis: (1981)
3.86 × 10-4s-1 0.69 - in pure water at 366 nm and in
sunlight at 23-28°C, late January
1.05 × 10-5s-1 1.1 - at 313 nm with 1-20% acetonitrile
in filter-sterilized natural
water, mid-December
Water Computed near-surface half-life for Zepp &
direct photochemical transformation Schlotzhauer
of a natural water body: (1979)
0.54 - latitude 40°N, midday, midsummer
77 - no sedimentmater partitioning
312 - sediment; water partitioning in a
5-m deep inland water body
Air > 1 Summer Valerio et al.
Days Winter (1991)
Methanol 2 Irradiated at 254 nm Lu et al. (1977)
Benzo[b]fluoranthene Air, particles First-order daytime decay rate Kamens et al.
constants with soot particle loading of: (1988)
0.0065 min-1 1.8 - 1000-2000 ng/mg
0.0090 min-1 1.3 - 30-350 ng/mg
Air, water 8.7-720 Based on measured rate of Lane & Katz
photolysis in heptane irradiated with (1977); Muel
light at > 290 nm & Saguem
(1985)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Benzo[ghi]perylene Air, particles Determined with 25 µg/g on: Behymer &
7.0 - silica gel Hites (1985)
2.2 - alumina
29 - fly ash
Air, particles First-order daytime photodegradation Kamens et al.
rate constants for adsorption (1988)
on wood soot particles in an outdoor
Teflon chamber for soot loading of:
0.0077 min-1 1.5 - 1000-2000 ng/mg
0.0116 min-1 1.0 - 30-350 ng/mg
Benzo[k]fluoranthene Air, particles First-order daytime decay constants Kamens et al.
for soot loading of: (1988)
0.0047 min-1 2.5 - 1000-2000 ng/mg
0.0013 min-1 8.9 - 30-350 ng/mg
Air, water 3.8-499 Based on measured rate of photolysis Muel &
in heptane under November Saguem
sunlight, adjusted by ratio of (1985)
sunlight photolysis half-lives in
water: heptane
Chrysene Air, particles Determined with 25 µg/g on: Behymer &
100 - silica gel Hites (1985)
78 - alumina
38 - fly ash
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Air, particles First-order daytime decay constants Kamens et al.
for soot loading of: (1988)
0.0056 min-1 2.1 - 1000-2000 ng/mg
0.0090 min-1 1.3 - 30-350 ng/mg
Air, water 4.4 Calculated for direct photochemical Zepp &
transformation near surface of Schlotzhauer
a water body at 40°N at midday in (1979)
midsummer
Water 13 Estimated on basis of photolysis Lyman et al.
in water in winter (1982)
Dibenzo[a,h]anthracene Air, water 782 Based on measured rate of photolysis Muel &
in heptane in November sun Saguem
6 After adjusting ratio of sunlight (1985)
photolysis in water: heptane
Fluoranthene Air, particles Determined with 25 µg/g on: Behymer &
74 - silica gel Hites (1985)
23 - alumina
44 - fly ash
Air, water 63 Computed, adjusted for approximate Lyman et al.
winter sunlight intensity (1982)
Air, water Calculated photochemical transformation Zepp &
near surface of water body: Schlotzhauer
21 - at 40°N, midday, midsummer (1979)
3800 - 5-m deep inland water body with
no sediment:water partitioning
4800 - with sediment:water partitioning
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water 3800 Summer sunlight in surface water Mill & Mabey
(1985)
Fluorene Air, particles Determined in rotary photoreactor Behymer &
with 25 µg/g on: Hites (1985)
110 - silica gel
62 - alumina
37 - fly ash
Naphthalene Water 13 200 Calculated, 5-m deep inland water Zepp &
Schlotzhauer
(1979)
Water 0.028 h-1 25 Half-life in distilled water Fukuda et al.
(1988)
Perylene Air, particles Determined with 25 µg/g on: Behymer &
3.9 - silica gel Hites (1985)
1.2 - alumina
35 - fly ash
Air, glass < 4.7 × 10-5 m/s Ozonization rate constant measured Cope &
from glass surface at 24°C with 03 Kalkwarf
- 0.16 ppm and light intensity of (1987)
1.3 kW/m2
Phenanthrene Air, particles Determined with 25 µg/g on: Behymer &
150 - silica gel Hites (1985)
45 - alumina
49 - fly ash
Water 3 Based on measured aqueous photolysis Zepp &
quantum yields, midday, mid-summer, Schlotzhauer
40°N (1979)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Air, water 25 Adjusted for approximate winter Lyman et al.
sunlight intensity (1982)
Air, water Calculated, direct sunlight photolysis, Zepp &
midday, midsummer, 40°N: Schlotzhauer
8.4 - near surface water (1970)
1400 - 5-m deep inland water body with
no sediment:water partitioning
1650 - with sedimentmater partitioning
Water 0.11 h-1 6.3 Half-life in distilled water Fukuda et al.
(1988)
Pyrene Air, particles Determined with 25 µg/ml on: Behymer & Hites
21 - on silica gel (1985)
31 - on alumina
46 - on fly ash
Air, particles Adsorption on airborne particles Valerio et al.
by sunlight: (1991)
1 - in summer
Days - in winter
Air, water 1.014 h-1 0.68 Based on measured aqueous photolysis Zepp &
quantum yields, midday, Schlotzhauer
summer, 40°N (1979)
Air, water 2.04 Based on measured aqueous photolysis Lyman et al.
quantum yields, adjusted for (1982)
approximate winter sunlight intensity
Air, glass < 1.05 × 10-4 m/s Ozonization rate on glass surface Cope &
at 24°C with O3 = 0.16 ppm and Kalkwarf
light intensity of 1.3 kW/m2 (1987)
Table 31. (continued)
Compound Compartment Photolysis Half-life Comments Reference
rate constant (h)
Water Calculated, direct sunlight photolysis, Zepp &
midday, midsummer, 40°N: Schlotzhauer
0.58 - near surface water (1979)
100 - 5-m deep inland water body with
no sediment:water partitioning
142 - with sediment:water partitioning
Water 100 Summer sunlight photolysis in Mill & Mabey
surface water (1985)
In order to compare numbers reported only as rate constants, half-lives were estimated from the formula:
t1/2 = In2
k
where t1/2 is the half-life and k is the rate constant. The calculated values are reported in italics.
Table 32. Reactions of polycyclic aromatic hydrocarbons with hydroxy radicals
Compound Oxidation rate Photooxidation Comments Reference
constant half-life (h)
Acenaphthene 1 × 10-10 0.879-8.79 Based on estimated reaction rate Atkinson (1987)
constant with hydroxy radical in air
Acenaphthylene 1.1 × 10-10 0.191-1.27 Based on estimated rate constant for Atkinson (1987)
reaction in air
Anthracene 1.1 × 10-12cm3 58-580 Rate constant for gas-phase reaction Biermann at al.
molec-1s-1 with hydroxy radicals at 298 ± 1 K, based (1985)
the relative rate technique for propane
0.501-5.01 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benz[a]anthracene 0.801-8.01 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[a]pyrene 0.428-4.28 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[b]fluoranthene 1.43-14.3 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[ghi]perylene 0.321-3.21 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Benzo[k]fluoranthene 1.1-11 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Chrysene 0.802-8.02 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Dibenz[a,h]anthracene 0.428-4.28 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Fluoranthene 2.02-20.2 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Fluorene 1.3 × 10-11 6.81-68.1 Based on estimated rate constant for Atkinson (1987)
reaction with hydroxy radical in air
Table 32. (continued)
Compound Oxidation rate Photooxidation Comments Reference
constant half-life (h)
Naphthalene 2.16 × 10-11 cm3 2.7-27 Rate constant for reaction with hydroxy Atkinson (1989)
molec-1s-1 radicals using relative rate technique
at 294 K
2 × 10-19 cm3 19-321 Upper limit was obtained for reaction
molec-1s-1 with O3
2.35 × 10-11 cm3 2.7-27 Rate constant for gas-phase reaction Biermann et al.
molec-1s-1 with hydroxy radicals at 298 K, based (1985)
on relative rate technique from propene
Phenanthrene 3.4 × 10-11 cm3 2-20 Rate constant for gas-phase reaction Biermann et al.
molec-1s-1 with hydroxy radicals at 298 K, based (1985)
on relative rate technique for propene
3.1 × 10-11 2.01-20.1 Half-life based on measured rate Atkinson (1987)
constants for reaction with hydroxy
radical in air
Pyrene 0.802-8.02 h Based on estimated rate constant for Atkinson (1987);
reactions with hydroxy radical in air and Atkinson & Carter
with hydroxy radical and ozone (1984)
To allow comparison when only rate constants are reported, half-lives were estimated from the following formula:
t1/2 = In 2
[x] × k
where t1/2 is the half-life, [x] is the concentration of the radical with which the compounds react (i.e. hydroxyl or ozone),
and k is the rate constant. The calculated values are reported in italics.
For the concentrations of the radicals, the following ranges of values were used; the lower values are estimates for rural
areas and the higher ones for urban areas (Howard et al., 1991):
[OH]air = 3-30 × 105 radicals/cm3
[O3]air = 3-50 × 1012 molecules/cm3
[OH]water = 5-200 × 10-17 mol/litre
[RO2]water = 1-50 × 10-11 mol/litre
[1O2]water = 1-100 × 10-15 mol/litre
In a study of the fate of 18 PAH on 15 types of fly ash, carbon
black, silica gel, and alumina, the PAH were stabilized, depending on
the colour, which is related to the carbon content: the higher the
carbon content, the more stable the PAH. The authors suggested that
radiation energy is adsorbed by the organic matter of particulates,
and PAH therefore do not achieve the excited state in which they can
be degraded (Behymer & Hites, 1988). The half-lives for direct
photolysis of various PAH adsorbed onto silica gel are in the range of
hours (Vu-Duc & Huynh, 1991).
A two-layer model has been proposed for the behaviour of
naturally occurring PAH on airborne particulate matter, in which
photooxidation takes place in the outer layer, and much slower, 'dark'
oxidation takes place in the inner layer (Valerio et al., 1987). This
model is in line with the results of Kamens et al. (1991), who
reported that PAH on highly loaded particles degrade more slowly than
those on particles with low loads. As PAH occur mainly on particulate
matter with a high carbon content, their degradation in the atmosphere
is slower than that of PAH in the vapour phase under laboratory
conditions or adsorbed on synthetic materials like alumina and silica
gel that have no or a low carbon content.
Formation of nitro-PAH was found from the low-molecular-mass two-
to four-ring PAH that occur in the atmosphere, predominantly in the
vapour phase. The rate constants range from 5.5 × 10-12 cm3/molecule
× s for acenaphthylene to 3.6 × 10-28 cm3/molecule × s for
naphthalene, with corresponding half-lives ranging from 6 min to 1.5
years. The yields were 1% or less (Atkinson et al., 1991; Atkinson &
Arey, 1994).
The rate of degradation of absorbed individual PAH seems to be
independent of their physicochemical characteristics but dependent on
their molecular structure. Thus, activated carbon from graphite
particles effectively stabilized pyrene, phenanthene, fluoranthene,
anthracene, and benzo [a]pyrene adsorbed onto coal fly ash against
photochemical decomposition, but no stabilization was seen for
fluorene, benzo [a]fluorene, benzo [b]fluorene,
9,10-dimethyl-anthracene, or 4-azafluorene. The authors suggested that
PAH that contain benzylic carbon atoms are less reactive than others
(Hughes et al., 1980).
PAH with vinylic bridges appear to degrade by direct photolysis
more rapidly than those with only aromatic rings, both in air and in
the aquatic environment (Hites, 1981).
In measurements of the photodegradation of benz [a]anthracene
and benzo [a]pyrene, addition of humic acids and purging of the
solution with nitrogen reduced the reaction rates significantly (Mill
et al., 1981). The authors concluded that light screening and
quenching occurred with humic acids. The reduction in rate with
exclusion of oxygen was probably due to a decrease in photooxidative
processes. The first metabolites were mainly quinones.
4.2.2.2 Hydrolysis
PAH are chemically stable, with no functional groups that result
in hydrolysis. Under environmental conditions, therefore, hydrolysis
does not contribute to the degradation of PAH (Howard et al., 1991).
4.3 Ultimate fate after use
The main sinks for PAH are sediment and soil. The available
information indicates that high-molecular-mass PAH are especially
persistent in groundwater, soil, and sediment under environmental
conditions.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
Appraisal
Polycyclic aromatic hydrocarbons (PAH) occur in all environmental
compartments. Ambient air, residential heating, and vehicle traffic
are the main sources. The levels of individual substances vary over
several orders of magnitude but are generally in the range < 0.1-100
ng/m3.
Surface waters are contaminated by PAH mainly through atmospheric
deposition, urban runoff, and industrial activities such as coal
coking and aluminium production. Apart from highly industrial polluted
rivers, the concentrations of individual substances are generally
< 50 ng/litre. High concentrations of PAH have been measured in
rainwater and especially in snow and fog. The concentrations of PAH in
sediments are in the low microgram per kilogram range.
PAH levels in soils near industrial sources (e.g. coal coking) are
especially high, sometimes up to grams per kilogram. In contrast,
soils contaminated by atmospheric deposition or runoff have
concentrations of 2-5 mg/kg of individual PAH, and the concentrations
in unpolluted areas are in the low microgram per kilogram range.
PAH have been detected in vegetables but are mainly formed during food
processing, roasting, frying, or baking. The highest levels were
detected in smoked meat and fish, at up to 200 µg/kg food for
individual PAH.
Five-fold increases in the concentrations of PAH in soil have been
observed over a 150-year period, although there are indications that
the concentrations of some PAH are decreasing. Similar findings have
been reported for sediments, perhaps because of measures to reduce
emissions.
Aquatic animals are known to adsorb and accumulate PAH. Especially
high concentrations were found in aquatic organisms from highly
polluted rivers, at levels up to milligrams per kilogram. Of the
terrestrial animals, earthworms are a good indicator of soil pollution
with PAH. The benzo[a]pyrene concentrations in the faeces of
earthworms living in a highly industrialized region were in the low
milligrams per kilogram range.
The main sources of exposure for the general population appear to be
food and air. The estimated intake of individual PAH in the diet is
0.1-8 µg/d. The main contribution appears to be that of cereals and
cereal products, due to the large amounts consumed. In ambient air,
the main sources are residential heating and environmental tobacco
smoke; exposure to PAH from environmental tobacco smoke in indoor air
is estimated to be 6.4 µg/day.
Occupational exposure to PAH occurs via the lung and skin. High
exposure occurs during the processing and use of coal and mineral oil
products, such as in coal coking, petroleum refining, road paving,
asphalt roofing, and impregnation of wood with creosotes; high
concentrations are also found in the air of aluminium production
plants and steel and iron foundries. No measurements were available
for the primary production and processing of PAH.
5.1 Environmental levels
5.1.1 Atmosphere
Relevant data on the occurrence of PAH in ambient air are compiled in
Tables 33-36. The concentrations were determined mainly by gas
chromatography and high-performance liquid chromatography, usually
with enrichment by filtration through a solid sorbent. The amount of
particle-bound PAH is therefore given. In studies in which
vapour-phase PAH were also sampled, the results for the vapour and
particulate phases were combined (for reviews, see Grimmer, 1979;
Ministry of Environment, 1979; Grimmer, 1983b; Lee & Schuetzle, 1983;
Daisey et al., 1986; Baek et al., 1991; Menichini, 1992a).
5.1.1.1 Source identification
Qualitative indications of different sources can be obtained by
comparing the PAH profiles, i.e. the ratio between the total PAH
concentration and that of a selected PAH, in air with those of samples
representative of the emitting sources or by determining PAH that are
emitted mainly from a specific source (Menichini, 1992a). Quantitative
assignments are difficult to make, however, owing to the complexity of
factors that affect the variability of PAH concentrations and
profiles.
Measurements were made at selected sources of PAH in the area of
Chicago, USA, in 1990-92, in order to identify them: Five samples were
taken 100 m directly downwind of a coke plant in an area that was not
affected by steel-making facilities, four samples from diesel buses at
a parking garage, three samples from petrol vehicles under warm-engine
operating conditions at a public parking garage, five samples in
heavily travelled tunnels during evening rush hours, and two samples
from the roof directly downwind of the chimney of fireplaces burning
seasoned oak. The authors give a source distribution pattern in
percent related to the total mass of 20 PAH. Naphthalene made by far
the largest contribution to petrol engine and coke oven emissions (55
and 89%, respectively). The three-ring compounds acenaphthylene,
acenaphthene, fluorene, phenanthrene, anthracene, and retene were
detected in large amounts in diesel motor emissions (56%) and in wood
combustion exhausts (69%). The four-ring fluoranthene, pyrene,
benz [a]anthracene, chrysene, and triphenylene and the five-ring
cyclopenta [cd]pyrene, benzo [b]fluoranthene,
benzo [k]fluoranthene, benzo [a]pyrene, benzo [e]pyrene, and
dibenzo [ghi]perylene together contributed 28% to diesel engine
emissions, 25% to petrol engine emissions, and 20% to wood combustion
emissions (Khalili et al., 1995).
The winter levels of PAH are higher than the summer levels (Gordon,
1976; Lahmann et al., 1984; Greenberg et al., 1985; Chakraborti et
al., 1988; Catoggio et al., 1989), due to more intensive domestic
heating and to meteorological (lower inversions during the winter) and
physicochemical factors (temperature-dependent partition between
gaseous and particulate phases). The ratios of benzo [a]pyrene:CO, in
which CO was used as an 'inert' tracer of automotive emissions, in Los
Angeles, USA, were higher at night (0.18-0.34) than in the day
(0.12-0.14), and substantially more so during winter (0.14-0.34) than
in summer (0.12-0.18), consistent with daytime loss of PAH by chemical
degradation (Grosjean, 1983).
In studies of sources of PAH at commercial, industrial, and urban
sampling sites in Athens, Greece, the effects of wind velocity and
thermal inversion were studied. There seemed to be no direct
correlation between benzo [a]pyrene and lead levels, which would be
expected if exhaust from cars run on leaded petrol were the
preponderant source of PAH (linear regression coefficient, 0.32-0.38)
(Viras et al., 1987).
Differences in the composition of profiles of PAH from different
sources can also be standardized by giving the concentrations relative
to that of a specific PAH. For particle-bound PAH, benzo [e]pyrene
has often been used as a reference compound, since it is
photochemically stable and found mainly in the particulate phase (Baek
et al., 1991).
Cyclopenta [cd]pyrene is emitted particularly from petrol-fuelled
automobiles (Grimmer et al., 1981c). Fluoranthene, pyrene,
benzo [ghi]perylene, and coronene are also found in higher
concentrations in condensates of vehicle exhausts (Baek et al., 1991).
The contribution of vehicles and domestic heating has also been
estimated as the ratio of indeno[1,2,3- cd]pyrene to
benzo [ghi]-perylene concentrations. The ratio should be 0.37 for the
PAH profile in traffic exhaust and 0.90 for domestic heating (Lahmann
et al., 1984; Jaklin & Krenmayr, 1985). In a comparison of the PAH
ratios determined in New Jersey, USA, with those reported in the
literature for samples collected under similar conditions in street
tunnels, the ratios coronene:benzo [a]pyrene and
benzo [ghi]perylene:benzo [a]pyrene indicated that vehicle traffic
was the major source of PAH during the summer (Harkov et al., 1984).
Measurements in ambient air in North Rhine Westphalia, Germany, in
1990 indicated that coronene is the most characteristic PAH for
automobile traffic. At a ratio of benzo [a]pyrene:coronene of < 3.5,
vehicle traffic is the dominant PAH source, whereas emissions with
ratios > 3.5 are influenced by other sources. The benzo [a]pyrene
levels were 0.66-5.0 ng/m3, and those of coronene 0.57-2.5 ng/m3
(Pfeffer, 1994).
In a study of the PAH concentrations during weekdays and weekends in
South Kensington, London, United Kingdom, no distinct differences were
observed in winter, but the average concentrations were 1.5-2.5 times
higher during the week than during the weekends in summer. Likewise,
the diurnal variations appeared to be less distinct during winter than
summer (Baek et al., 1992).
Measurements in streets with high traffic density in Stockholm,
Sweden, showed that the concentration of PAH decreased by 25-50%
during holidays in comparison with weekdays. Benzo [a]pyrene in
street air was all particle-bound, while chrysene and lighter PAH
occurred both on particles and in the vapour phase (Östman et al.,
1991, 1992a,b).
In a study of 15 PAH in the air of various areas in an industrial city
in Germany with 700 000 inhabitants, the highest levels were detected
in air affected by a coke plant, where benzo [a]pyrene was found at
1.4-400 ng/m3 and cyclopenta [cd]pyrene at none detected to 120
ng/m3. The concentrations measured in air affected by vehicle traffic
were 11-110 ng/m3 benzo [a]pyrene and 0.1-440 ng/m3
cyclopenta [cd]pyrene. Within 4 km, the average concentration of 88
ng/m3 cyclopenta [cd]pyrene had dropped to 1.6 ng/m3. The levels
were lower in areas where hand-stoked residential coal heating
predominated (0.37 µg/m3 benzo [a]pyrene and none detected to 39
µg/m3 cyclopenta [cd]pyrene) and where oil heating predominated
(0.2-66 ng/m3 and none detected to 15 ng/m3, respectively). The
concentration of PAH was three to four times higher between 7:43 and
10:00 than between 10:00 and 15:46. Benzo [c]phenanthrene,
cyclopenta [cd]pyrene, benzo [ghi]perylene, and coronene dominated
the PAH in areas with heavy traffic, whereas chrysene,
benzo [b]fluoranthene, and benzo [a]pyrene occurred at the highest
concentrations in an area surrounding a coke plant (Grimmer et al.,
1981c).
The use of receptor-source apportionment modelling was examined,
despite its limited applicability to reactive species, for the PAH
profiles of emissions from a variety of sources (Daisey et al., 1986;
Pistikopoulos et al., 1990). In one study, benzo [b]fluoranthene,
benzo [k]fluoranthene, benzo [a]pyrene, benzo [ghi]perylene,
indeno[1,2,3- cd]pyrene, and coronene were measured in the ambient
air of the centre of Paris, France. The concentrations of PAH varied
from 42% in winter to 72% in summer for petrol-fuelled vehicles, from
25 to 40% for diesel-fuelled vehicles, and from about 30 to 2% for
domestic heating. The winter-summer differences were due mainly to
different emission patterns and not to changes in the rate of decay of
PAH (Pistikopoulos et al., 1990). In another study, the contributions
of PAH from five sources to ambient air were distinguished by use of
fuzzy clustering analysis (Thrane & Wikström, 1984).
The information on PAH levels in ambient air is discussed below
according to possible source: background and rural, industrial
emissions, and diffuse sources like automobile traffic and residential
heating. Attribution of different studies to these sections was
difficult because the sources of PAH emissions are often mixed. For
example, Seifert et al. (1986) determined PAH in Dortmund 200 m from a
coke plant; this study was deemed to relate to PAH levels resulting
from industrial emissions. The concentrations of PAH attributable to
mobile sources can be estimated by monitoring near areas with heavy
traffic in the summer, but it is difficult to estimate the
contribution of home heating, because in winter PAH in ambient air
derive from both mobile sources and home heating. Furthermore,
emissions from mobile sources may differ in winter from those in the
summer because of meteorological and physicochemical factors
(Greenberg et al., 1985; see also section 5.1.1.3).
5.1.1.2 Background and rural levels
The levels in ambient air of rural areas are summarized in Table 33.
Background levels were measured about 25 km from La Paz, Bolivia, at
an altitude of 5200 m (Cautreels & van Cauwenberghe, 1977) and on the
island of Mallorca, Spain, at an altitude of 1100 m (Simó et al.,
1990). The concentrations were generally 0.01-0.1 ng/m3. The average
values in rural areas are usually 0.1-1 ng/m3. Average concentrations
of 0.34 and 0.27 ng/m3 benzo [a]pyrene were measured in two rural
areas in Japan in 1989, with a maximum concentration of 1.1 ng/m3
(Okita et al., 1994).
5.1.1.3 Industrial sources
PAH levels in ambient air resulting mainly from industrial emissions
are summarized in Table 34. The average concentrations of individual
PAH at ground level were 1-10 ng/m3. In general, aluminium smelters
and industrial processes for the pyrolysis of coal, such as coking
operations and steel mills, result in higher levels of PAH than most
other point industrial sources. Furthermore, the levels of PAH are
much higher downwind from major sources than upwind.
The highest levels of individual PAH were measured near an aluminium
smelter in Hoyanger, Norway, with maximum concentrations of 10-100
ng/m3. Phenanthrene was present at very high levels in ambient air
contaminated by industrial emissions (Thrane, 1987). In Sundsvall,
Sweden, near an aluminium production facility, 310 ng/m3
phenanthrene, 190 ng/m3 naphthalene, 120 ng/m3 pyrene, and 84 ng/m3
fluorene were detected (Thrane & Wikström, 1984).
The concentration of benzo [a]pyrene in ambient air near an oil
processing plant in Moscow was up to 13 ng/m3 (Khesina, 1994).
Benzo [a]pyrene was detected at 15-120 ng/m3 and perylene at 3-37
ng/m3 at 39 measuring stations in the heavily polluted area of Upper
Silesia, Poland. The maximum values were 950 ng/m3 for
benzo [a]pyrene and 270 ng/m3 for perylene (Chorazy et al., 1994).
Table 33. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air of background and rural areas
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene 0.32 6.3-23
Anthracene 0.004 0.05 0.03 < 0.05 1.2-3.9 ND-0.05
Anthanathrene 0.004-0.16 0.08 0.07 ND-0.2 ND-0.04
Benz[a]anthracene 0.005 0.12 0.4 0.40 0.07 1.8-3.2 0.16-0.39
Benzo[a]fluorene 0.8-3.3
Benzo[a]pyrene 0.006 0.005 0.002-0.12 0.33/0.47 0.6 ND-0.52 0.45 0.08 0.8-2.5 0.41-0.45
Benzo[b]fluoranthene 0.02 1.2 0.45-0.58
Benzo[b]fluorene 0.24 0.5-2.4
Benzo[c]phenanthrene 0.15-0.20
Benzo[e]pyrene 0.022 0.006 0.007-0.26 0.6 0.59 1.8-5.8 0.44-0.65
Benzo[ghi]fluoranthene ND-0.2
Benzo[ghi]perylene 0.009 0.002 0.005-0.40 0.6 ND-0.58 1.4-3.0 0.89-1.4
Benzo[k]fluoranthene 0.02 0.002-0.088 0.48 0.17-0.25
Chrysene 0.07a 1.0 0.13-0.19
Coronene 0.005-0.23 0.24 ND-0.22 0.4-0.9 0.16-0.26
Cyclopenta[cd]pyrene 0.2 0.16-0.39
Dibenzo[a,h]pyrene 0.14 0.02-0.07
Dibenzo[a,l]pyrene 0.53
Fluoranthene 0.041 0.030 0.18 0.20/0.26 1.2 ND 0.93 1.3 11-47 0.19-0.23
Fluorene 0.45 0.66 14-32
Indeno[1,2,3-cd]pyrene 0.006 0.02 0.7 0.72 0.43-0.65
1-Methylphenanthrene 0.09 0.7-2.8
Naphthalene ND 3.0-98
Perylene 0.001-0.026 0.09 0.08 ND-0.4
Phenanthrene 0.026 2.66 0.4 ND-0.43 4.2 26-70 ND-0.03
Pyrene 0.034 0.024 0.34 0.010-0.15 0.15/0.15 1.3 ND 0.60 0.73 8.8-26 0.16-0.26
Table 33 (continued)
ND, not detected; /, single measurements;
[1] About 25 km from La Paz, Bolivia, at 5200 m (Cautreels & van Cauwenberghe, 1977);
[2] Mallorca, Spain, 1989 (Simo et al.,1991);
[3] Lake Superior, USA, 1986; sum of vapour and particulate phases (Baker & Eisenreich,1990);
[4] Latrobe Valley, Australia, (Lyall at al.,1988);
[5] Belgium, (Van Vaeck et al.,1980);
[6] Denmark (Nielsen, 1984);
[7] Western Germany, 1981 (Pflock et al.,1983);
[8] Oostvoorne, Netherlands, (De Raat et al.,1987b);
[9] Canada, 1989-91 (Environment Canada, 1994);
[10] Sidsjon, Sweden, 1980-81, sum of vapour and particulate phases (Thrane & Wikstrom, 1984);
[11] Folkestone, Ashford, United Kingdom, 1986 (Baek et al., 1992)
a With triphenylene
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled, unless otherwise stated
Table 34. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air near industrial emissions
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene 23 9.8-372 15-122 3.7
Acenaphthylene 747 0.01
Anthracene 2.9/3.4 158 4.5-6.1 4.1-43 0.12/0.15 0.01-3.4 0.08-0.19
Anthanthrene 0.001/3.0 0.2/1.1 ND-3.0 0.15/0.15 0.13-0.22
Benz[a]anthracene 0.28/1.2 7.6 2.0-158 2.5-58 0.8/3.1 0.02-1.2 1.3-4.7
Benzo[a]fluorene 1.1-179
Benzo[a]pyrene 0.002/1.5 0.5/3.5 25/37 6.3-6.7 5.3 1.1-61 2.1-36 0.14/0.11 0.20-0.11 1.8-3.1 1.1-2.6
Benzo[b]fluoranthene 0.9/1.8 4.8 2.7-6.4
Benzo[b]fluorene 0.7-122 0.61-1.4
Benzo[e]pyrene 0.004/1.4 1.8/3.2 11.6 2.5-86 1.3-3.1
Benzo[ghi]fluoranthene ND-0.5 0.26/0.35
Benzo[ghi]perylene 0.003/1.5 4.2/7.1 O.7 2.2-45 0.35/0.33 0.25
Benzo[j]fluoranthene 0.3/0.8
Benzo[k]fluoranthene 0.001/0.67 0.3/1.3 8.0 1.0-2.2
Chrysene 1.6/3.8 14.7 0.22/0.29 0.01-1.6 2.5-7.5
Coronene 0.003/1.5 3.2/2.8 1.3-1.5 ND 0.6-9.0 0.25/0.26
Cyclopenta[cd]pyrene 2.2
Dibenzo[a,h]pyrene ND 277
Dibenzo[a,l]pyrene 1.0-1.5
Fluoranthene 0.8/3.4 88.3 20-812 22-272 0.12/0.20 0.02-10 2.3-3.3
Fluorene 502 27-419 16-46 0.02-0.86
Indeno[1,2,3-cd]pyrene 0.4/0.3 1.1 3.8-38 0.28/0.27 0.10-7.7 1.4-2.4
1-Methylphenanthrene 2.5-58
Naphthalene 22 400 9.0-193 3.1-26 0.03-0.06
Perylene 0.001/0.2 0.3/1.2 0.1-8.3 0.05/0.05 22 0.23-0.61
Phenanthrene 500 54-1760 58-390 0.11/0.16 0.02-152
Pyrene 1.4/3.8 56.3 16-491 14-207 0.17/0.35 0.006-28 1.6-2.1
Table 34 (continued)
ND, not detected; /, single measurements;
[1] Three sampling sites near various industries in Latrobe Valley, Australia (Lyall et al., 1988);
[2] Near various industries, USA, 1971-72 (Gordon & Bryan, 1973);
[3] Near a coke plant, Dortmund, Germany, 1982-83 (Seifert et al., 1986);
[4] Near a coke plant, Dortmund, Germany, 1989 (Buck, 1991);
[5] 100 m directly downwind of a coke plant, Chicago, USA, 1990-92 (Khalili et al., 1995);
[6] Near aluminium smelters, Norway and Sweden, 1980-82 (analytical method not given) (Thrane, 1987); vapour and particulate phase
(Thrane & Wikstrom, 1984);
[7] Near aluminium smelter, Canada, 1989-91 (Environment Canada, 1994);
[8] Near incineration plant, Sweden (Colmsjo et al., 1986a,b);
[9] Near refinery, USA, 1981-83 (Karlesky et al., 1987);
[10] Brown coal industry area, western Germany, 1983 (Seifert et al., 1986);
[11] Near harbours, Netherlands (De Raat et al., 1987b)
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled, unless otherwise stated
In Ontario, Canada, up to 140 ng/m3 benzo [k]fluoranthene, 110
ng/m3 perylene, 110 ng/m3 benzo [a]pyrene, 90 ng/m3
benzo [ghi]perylene, and 43 ng/m3 fluoranthene were found near a
steel mill (Potvin et al., 1980). The benzo [a]pyrene concentrations
near coke ovens in urban areas of the USA were more than double those
in urban areas without coke ovens (Faoro & Manning, 1981). These
results are consistent with those of Grimmer et al. (1981c), who
detected maximum levels of benzo [a]pyrene, chrysene,
benzo [b]fluoranthene, benzo [j]fluoranthene, and
benzo [k]fluoranthene in the area surrounding a coke plant.
The PAH concentrations in ambient air 900 and 2500 m from a municipal
incineration plant were of the same order of magnitude, and no
significant contribution from the plant to the ambient PAH
concentrations was observed (Colmsjö et al., 1986a).
The PAH levels in an industrial area of Ahmedabad City, India, were
significantly higher than those in a residential area. The highest
levels were found during winter, and the rate of degradation of
airborne PAH was predicted to be lowest in the monsoon season. The
most striking finding was the high concentration of
dibenz [a,h]anthracene in urban air (5.3-23 ng/m3) (Raiyani et al.,
1993a). The limited resolution of PAH may have resulted in
overestimation: for instance, the concentrations of
benzo [ghi]perylene and indeno[1,2,3- cd]pyrene reported are one
order of magnitude higher than that of dibenz [a,h]anthracene.
5.1.1.4 Diffuse sources
A special situation of local importance was the pollution of ambient
air in Kuwait after the war in the Persian Gulf, due to burning of oil
fields. The mean concentrations of benzo [a]pyrene at three sampling
sites were 0.27-9.2 ng/m3, and the maximum was 26 ng/m3 (Okita et
al., 1994). These values are within the range of those detected in
urban areas (see below).
(a) Motor vehicle traffic
The concentrations of PAH in the ambient air of various urban areas
are listed in Table 35. The average levels of individual PAH were 1-30
ng/m3. Relatively high concentrations of benzo [a]pyrene,
benzo [ghi]perylene, phenanthrene, fluoranthene, and pyrene were
measured.
Total PAH concentrations of 43-640 ng/m3 were measured in London,
United Kingdom, in 1991, nearly 80% of which consisted of
phenanthrene, fluorene, and fluoranthene; benzo [a]pyrene and
benz [a]anthracene were present at 1% or less (Clayton et al., 1992).
Table 35. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air of urban areas
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthene 0.4-101 2.7-6
Acenaphthlene 0.9-39 4.4-130
Anthracene 34 0.6-36 0.3-2.1 3.5-25
Anthanthrene 2.5 0.1-4.7 30 < 0.1-0.6 0.003-0.76
Benz[a]anthracene 10 0.3-27 1.2-13 0.2-1.4 0.10-25 0.3-7.6
Benzo[a]fluorene 0.1-0.9 0.8-6.9
Benzo[a]pyrene 9.3 0.3-20 29 1.2-11 < 0.1-1.9 0.074-15 0.2-5.7
Benzo[b]fluoranthene 43 1.0-36
Benzo[b]fluorene 0.1-0.8 0.6-7.3
Benzo[c]phenathrene 4.0 0.2-5.0
Benzo[e]pyrene 8.4 0.4-17 16 1.7-15 < 0.1-1.2 0.40-27 0.4-6.5
Benzo[ghi]fluoranthene 12 0.3-5.0 0.1-1.5 0.5-7
Benzo[ghi]perylene 14 0.5-12 1.6/13 27 2.1-11 0.2-3.5 0.45-31 0.9-2.4 0.6-18
Benzo[j]fluoranthene 0.17-13
Benzo[k]fluoranthene 23 0.29-25
Chrysene 0.3-2.5 0.56-29 3.6-5.6 3.3
Coronene 10 0.3-5.5 12 0.88-2.0 0.1-2.4 0.22-3.3 0.4-19
Cyclopenta[cd]pyrene 11 0.1-4.8 71 < 0.1-1.1 0.1-6
Dibenzo[a,h]pyrene 0.22-3.4 0.29-2.8
Fluoranthene 72 6.2-108 0.40/14 1.4-10 0.80-14 1.3-2.0 6.9-38 15
Fluorene 1.3-61 16-86
Indeno[1,2,3-cd]pyrene 8.6 0.4-12 31 < 0.1-2.9 0.39-30 0.4-7.6
1-Methylphenanthrene 0.3-2.5 5-16
Naphthalene 14-63
Perylene 2.3 0.1-4.3 4.8 < 0.1-0.4 0.011-4.4 0.1-1.3
Phenanthrene 153 18-223 3.6-41 32-105 111
Pyrene 74 2.9-67 0.34/12 1.2-5.5 0.34-10 5.5-45 20
Triphenylene 0.15-6.9
Table 35 (continued)
ND, not detected; /, single measurements;
[1] Vienna, Austria, 1983-84; vapour and particulate phase (Jaklin & Krenmayr, 1985);
[2] Linz, Austria, 1985; vapour and particulate phase (Jaklin et al., 1988);
[3] Antwerp, Belgium (Van Vaeck et al., 1980);
[4] Berlin, western Gemany, 1984-85 (Seifert et al., 1986);
[5] Rhein/Ruhr area, western Germany, 1985-88; analytical method not stated (Buck et al., 1989);
[6] Kokkola, Finland (Pyysalo et al., 1987);
[7] St Denis, France, 1979-80 (Muel & Saguem, 1985);
[8] Various cities, Greece, 1984-85 (Viras et al., 1987);
[9] Oslo, Norway, 1981-83, vapour and particulate phase (Larssen, 1985);
[10] Barcelona, Spain, 1988-89, vapour and particulate phase (Albaiges et al., 1991)
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled,
unless otherwise voted
Table 35 (continued)
Compound [11] [12] [13] [14] [15] [16] [17] [18] [19] [20]
Acenaphthene 0.07-3.58 0.05-31.1
Acenaphthylene 9.1 0.8 0.9
Anthracene 21 1.4 2.8 0.01-8.28 0.20-39.8 0.1-0.9 ND-4.8 6.1/11
Anthanthrene 0.63
Benz[a]anthracene 4.1 0.4 1.4 0.24-10.6 0.12-18.5 0.2-5.8 5-21 0.07-2.1
Benzo[a]fluorene 5.0 0.7
Benzo[a]pyrene 2.9 0.2 0.99/1.4 1.6 0.01-7.02 0.18-13.7 0.3-3.4 1-17 0.04-3.2 0.6/1.6
Benzo[b]fluoranthene 1.8 0.01-3.04 0.13-14.8 0.2-3.7 5-30 0.10-3.7
Benzo[c]phenanthrene 2.8
Benzo[e]pyrene 3.5 0.4 1.1/2.0 2.3 2.1/2.1
Benzo[ghi]fluoranthene 7.3 0.8
Benzo[ghi]perylene 6.6 0.5 2.9/3.3 3.3 0.02-6.90 0.15-85.3
Benzo[k]fluoranthene 0.75 0.23-16.5 0.3-0.8 3-22 0.07-0.85
Chrysene 5.1 0.8 1.6 0.04-4.97 0.13-24.3 0.2-5.5 ND-2.3
Coronene 4.1 0.3 2.4/1.7 1.7 0.02-3.72 0.17-6.92 ND-16
Cyclopenta[cd]pyrene 3.9 0.11 4.1
Dibenz[a,h]pyrene 0.12
Fluoranthene 24 3.9 3.5 2.03-62.4 22-23 14-54 0.24-2.0 8.0/9.7
Fluorene 0.07-27.6 0.07-161
Indeno[1,2,3-cd]pyrene 3.8 0.5 1.6 0.3-4.4 4.24
Naphthalene 15/75
Perylene 1.0 0.1 0.2/0.5
Phenanthrene 76 11 5.1 0.06-111 2.25-492 0.1-2.4 78/81
Pyrene 28 32 18 0.39-17.4 0.33-64.4 0.1-7.5 0.48-3.6 8.0/12
Triphenylene
Table 35 (continued)
ND, not detected;/, single measurements;
[11] Stockholm, Sweden, April 1991; vapour and particulate phases (Ostman et al.,1992a,b);
[12] Stockholm, Sweden; 1992 vapour and particulate phases (Ostman et al.,1992a,b);
[13] London, United Kingdom, 1985-87(Baek et al.,1992);
[14] London, United Kingdom, 1987; vapour and particulate phases (Baek et al.,1992);
[15] Manchester, United Kingdom, 1990-91; vapour and particulate phases (Clayton et al.,1992);
[16] Various cities, United Kingdom, 1991-92; vapour and particulate phases (Halsall et al.,1994);
[17] Lake Baikal shore, Russian Federation, 1993-94 (Grachev et al.,1994);
[18] Zagreb, Croatia, 1977-82; determined by thin-layer chromatography and fluorescence detector (Bozicevic et al.,1987);
[19] Los Angeles, USA, 1981-82 (Grosjean, 1983);
[20] Los Angeles basin, USA, 1986; vapour and particulate phases (Arey et al.,1987)
Table 35 (contd)
Compound [21] [22] [23] [24] [25] [26] [27] [28] [29] [30]
Acenaphthene 3.3-9.0 0.06-5.2 0.6
Acenaphthylene < 11-47 1.9
Anthracene 1.9-4.5 0.45-3.8 0.17-0.57 0.12-0.52 0.2 2.5-5.5
Anthanthrene 0.006-3.3 1-11
Benz[a]anthracene 0.07-1.4 0.19-0.40 0.19-4.4 0.99-7.0 0.37-1.7 1.9 20-66
Benzo[a]fluorene 1.8-6.3
Benzo[a]pyrene 0.11-1.6 ND-0.03 0.09-1.7 0.006-1.8 8-38 1.6-8.4 ND-2.3 3.4 30-120
Benzo[b]fluoranthene 0.17-1.7 3.1-12 3.0 109-200
Benzo[b]fluorene 0.19-0.94
Benzo[e]pyrene 0.03-11 ND-0.04 0.016-2.3 4-19 2.7-9.0 2.3 49-182
Benzo[ghi]fluoranthene 0.12-1.3
Benzo[ghi]perylene 0.24-2.7 0.027-4.7 11-33 3.2-12 3.4 34-141
Benzo[j]fluoranthene 0.08-1.1 22-66
Benzo[k]fluoranthene 0.09-0.97 0.005-0.85 1.8-7.7 2.7
Chrysene 0.22-5.3 0.38-0.57 3-15 0.29-1.4 2.4
Coronene 0.14-1.6 0.020-2.3 5.16
Dibenzo[a,a]pyrene 0.06-2.7
Dibenzo[a,h]pyrene 0.46-1.2 5.3-23
Dibenzo[a,l]pyrene 0.05-0.35
Fluoranthene 5.7-10 1.6-11 14-79 1.5-8.3 1.0 11-26
Fluorene 7.4-14 0.94-5.5 0.08-0.15 0.31-1.2 2.8
Indeno[1,2,3-cd]pyrene 0.20-2.9 6-24 2.6-12 3.1
Naphthalene 280-940 ND 4.5-13
Perylene 0.01-0.15 0.001-0.24 2-9 0.51-1.2
Phenanthrene 21-35 2.2-35 0.79-2.6 0.52-2.4 0.7 12-21
Pyrene 0.12-2.8 4.8-10 1.4-6.9 0.008-0.66 16-69 1.5-9.0 0.46-4.0 3.8 20-44
Triphenylene 22-60
Table 35 (continued)
ND, not detected; /, single nwasureme4s;
[21] New Jersey, USA, 1981-82 (Greenberg et al, 1985);
[22] Portland, Oregon, USA, 1984 (Ligocki et al.,1985);
[23] Urban area (not specified), Canada, 1989-91 (Environment Canada,1994);
[24] Latrobe Valley, Australia (Lyall et al., 1988);
[25] Christchurch, New Zealand, 1979 (Cretney et al., 1985);
[26] Osaka, Japan, 1977-78; vapour and particulate phases (Yamasaki et al., 1982);
[27] Osaka, Japan, 1981-82 (Matsumoto & Kashimoto, 1985);
[28] La Plata, Argentina, 1985 (Catoggio et al., 1989);
[29] Ahmedabad City, India, 1984-85 (Raiyani at al.,1993a);
[30] Calcutta, India, 1984 (Chakraborti et al.,1988)
Table 35 (continued)
Compound [31] [32] [33] [34] [35] [36] [37] [38] [39] [40]
Acenaphthene 4.5
Anthraceene 14-16 2.5 1.8 ND-34 8.7-23
Anthanthrene 0.15-0.63 0.001-0.21 2-24
Benz[a]anthracene 2.9-4.8 99-139 23 6.5 0.028-4.8 3.1-9.8
Benzo[alpyrene 3.8-5.5 0.005-1.3 67-73 15 5.6 0.023-4.6 Trace-9.3 ND-44 1.9-7.7 19-72
Benzo[blfluoranthene 1.0-3.1 130-133 0.46-16
Benzo[b]fluorene 0.07-0.18
Benzo[c]phenanthrene 33-37
Benzo[e]pyrene 5.5-7.4 0.016-3.3 96 19 9.1 0.18-8.8 0.17-4.2 ND-370 9-41
Benzo[ghi]fluoranthene 3.0-4.9 0.024-0.98 30-33
Benzo[ghi]perylene 7.0-13 0.004-3.2 49-61 12 7.9 0.21-12 ND-74 11-49
Benzo[j]fluoranthene 2.6-5.5
Benzo[k]fluoranthene 3.4-5.0 0.12-7.4
Chrysene 4.3-6.5 0.34-0.49 237-261 43 16 0.22-8.9 0.22-6.4 ND-170 7-71
Coronene 0.002-1.4 14-16 3.1 2.8 0.14-2.1 Trace-2.1 8-96 4-18
Cyclopenta[cd]pyrene ND 3.1 1.6
Dibenzo[a,h]pyrene 0.012-0.98
Fluoranthene 3.4-4.9 0.14-1.2 0.32-8.6 8-520 15-51
Fluorene 15-26
Indeno[1,2,3-cd]pyrene 5.1-9.1 0.022-2.0 57 11 5.5 0.16-9.6 9-43
Naphthalene 44
Perylene 0.01-0.20 7.6-10 0.004-0.88 ND-28 3-21
Phenanthrene 0.002-1.1 4-170 50-271
Pyrene 3.6-6.6 0.002-0.58 0.13-6.7 0.21-8.6 ND-540 12-49
Triphenylene 1.4-1.9 0.07-0.24 0.11-2.9 ND-50
ND, not detected; /, single measurements;
[31] Various cities, China (Chen et al.,1981);
[32] Various cities, China, 1986-88; determined by thin-layer chromatography and gas chomatography-mass spectroscopy (Chang et
al., 1988; Simoneit et al., 1991);
[33] Various locations with predominantly coal heating; Germany (analytical method not given) (Grimmer, 1980);
[34] Essen, Germany, predominantly coal heating, 1978-79 (Buck, 1983);
[35] Essen, Germany, predominantly oil heating, 1978-79 (Buck, 1983);
[36] Antony, France, 1979-80 (Muel & Saguem, 1985);
[37] Sutton Coldfield, United Kingdom, 1976-78 (Butler & Crossley, 1982);
[38] Barrow, USA, fossil fuel combustion area, 1979 (Daisey et al., 1981);
[39] Wood-heating area, Canada, 1989-91 (Environment Canada, 1994);
[40] Christchurch, New Zealand, 1979 (Cretney et al., 1985)
In Delft, the Netherlands, benzo [a]pyrene levels of up to 140 ng/m3
were measured on a foggy day with low wind velocity near a major road.
High concentrations of pyrene (220 ng/m3), benzo [ghi]perylene (130
ng/m3), and coronene (21 ng/m3) were also found. At border crossings
between the Netherlands and Germany on days with heavy traffic, the
maximum levels of individual PAH were 1-54 ng/m3 (Brasser, 1980).
PAH concentrations were determined in the centre of Paris, France, at
the top of a 55-m tower and thus less likely than ground-level samples
to be affected by traffic emissions and street dust; they can
therefore be considered to be homogeneous and representative. The
maximum levels found were 98 ng/m3 benzo [ghi]perylene, 60 ng/m3
indeno[1,2,3- cd]pyrene, 34 ng/m3 coronene, 28 ng/m3
benzo [b]fluoranthene, 13 ng/m3 benzo [a]pyrene, and 13 ng/m3
benzo [k]fluoranthene (Pistikopoulos et al., 1990).
The average concentration of individual PAH in particulate and vapour
phases during a nine-day photochemical pollution episode in
California, USA, in 1986 was 1 ng/m3. The maximum levels of
acenaphthene, acenaphthylene, fluorene, and phenanthrene ranged from
30 to 64 ng/m3 (Arey et al., 1991).
In 1989, the average benzo [a]pyrene concentrations in five Japanese
cities (Sapporo, Tokyo, Kawasaki, Nagoya, and Osaka) were 1.2-3.1
ng/m3. A maximum level of 15 ng/m3 was detected in Tokyo (Okita et
al., 1994). A detailed examination was undertaken of the molecular
composition of PAH in street-dust samples collected from the Tokyo
metropolitan area. Unsubstituted ring systems (i.e. parent PAH)
ranging from phenanthrene with three rings to benzo [ghi]perylene
with six rings were the primary components, three- and four-ring PAH
(i.e. phenanthrene, fluoranthene, and pyrene) predominating. The
concentrations of total PAH were of the order of a few micrograms per
gram of dust. On the basis of the PAH profile, it was suggested that
PAH in the dust of busy streets arose mainly from automobile exhausts,
while residential areas received a greater contribution from
stationary sources. In both types of dust, asphalt was thought to
contribute to only a minor extent (Takada et al., 1990). Giger &
Schaffner (1978) had come to the same conclusion some 20 years
earlier.
Benzo [a]pyrene was detected in ambient air in Moscow, Russian
Federation, at concentrations of 5.4 ng/m3 at a regular traffic site
and 20 ng/m3 at a crossroads with heavy traffic (Khesina, 1994).
(b) Road tunnels
In road tunnels, the concentrations of individual PAH were usually
1-50 ng/m3 (Table 36). Higher levels were reported in tunnels in
western Germany, with concentrations of 84 and 96 ng/m3
cyclopenta [cd]pyrene (Buck (1983) and 76 ng/m3 (Brasser, 1980) and
110 ng/m3 pyrene (Benner et al., 1989).
Table 36. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in ambient air polluted predominantly by vehicle exhaust
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene 168
Acenaphthylene 32 445
Anthracene 8.6/9.8 2.3 55 0.6-12 177
Anthanthrene 7.2 1 500 0.1-4.5 2-82
Benz[a]anthracene 37/44 0.6-1.9 16 20 12 000 102 1.9-2.9 90.2
Benzo[a]fluorene 18 2 800
Benzo[a]pyrene 30 2-14 0.2-0.8 16 12 9 600 66 1.3-26 62.6 0.1-14 1-57
Benzo[b]fluoranthene 2.3 8.8 12 000 43.6
Benzo[e]pyrene 28/32 11 9 600 69 1.5-19 55.5 01-12 3-43
Benzo[ghi]fluoranthene 29 18 3.2-26
Benzo[ghi]perylene 40/47 4-16 0.4-2.6 44 30 19 000 85 1.8-18 17.0 0.6-27 20-213
Benzo[k]fluoranthene 8.1 9.7 9 000 41.2
Chrysene 54/58 25 15 9 500 77.9
Coronene 26/27 2-17 0.3-1.1 29 20 7 500 1.0-10 ND 0.3-14 9-156
Cyclopenta[cd]pyrene 84/96 40 31 7.6-65 100
Dibenzo[a,h]pyrene 14.7
Fluoranthene 35 83 93 6.4-69 117
Fluorene 406
Indeno[1,2,3-cd]pyrene 18/22 0.3-1.3 16 13 9 400 0.3-15 20.0 6-70
1-Methylphenanthrene 2.6-43
Naphthalene 8030
Perylene 3.4 3.1 1 500 1-18
Phenanthrene 8.1 243 4.4-56 300
Pyrene 33-114 47 122 16 000 120 9.7-76 193 0.2-29
Table 36 (continued)
ND, not detected; /, single measurements;
[1] Street tunnel (location not specified), western Germany, 1978-79 (Buck, 1983);
[2] Coen Tunnel, Netherlands (Brasser, 1980);
[3] Street tunnel in Lincoln, Netherlands, 1981 (Kebbekus et al., 1983),
[4] Klara Tunnel, Sweden, 1983 (Colmsjo et al., 1986b);
[5] Soderleds Tunnel, Sweden, 1991; vapour and particulate phases (Ostman et al., 1991);
[6] Craeybeckx Highway Tunnel, Belgium, 1991 (De Fré et al., 1994);
[7] Baltimore Harbor Tunnel, USA, 1975 (Fox & Staley, 1976);
[8] Baltimore Harbor Tunnel, USA, 1985-86 (Benner et al., 1989);
[9] Heavily travelled tunnel, Chicago area, USA, 1990-92 (Khalili et al., 1995);
[10] Diesel bus garage, United Kingdom, 1979 (Waller et al., 1985);
[11] Inside car park, New Zealand (Cretney et al., 1985)
Analysed by high-performance liquid chromatography or gas chromatography; only particulates sampled, unless otherwise stated
PAH were found at levels of up to 4 ng/m3 in an underground bus
terminal in Stockholm, Sweden; and 21 ng/m3 fluoranthene, 11 ng/m3
pyrene, and 8.1 ng/m3 phenanthrene were found in a subway station
(Colmsjö et al., 1986b).
Very high concentrations of PAH were found in the air of the
Craeybeckx Highway Tunnel in Belgium, which was used daily by an
average of 45 000 vehicles, of which 60% were petrol-fuelled passenger
cars, 20% diesel-fuelled cars, and 20% trucks. Of the cars, only 3%
had three-way catalysts (De Fré et al., 1994).
(c) Residential heating
The PAH levels in ambient air resulting mainly from residential
heating are included in Table 35, as the source cannot be identified
properly (see section 5.1.1.1).
The use of wood and coal for heating was the source of high levels of
benzo [a]pyrene in Calcutta, India (up to 120 ng/m3; Chakraborti et
al., 1988). The concentrations of individual PAH in Calcutta ranged
from 1.3 to 200 ng/m3, the highest levels being those of
benzo [e]pyrene, benzo [ghi]perylene, and benzo [b]fluoranthene.
The average levels of individual PAH resulting from domestic heating
in Christchurch, New Zealand were 1-210 ng/m3, benzo [ghi]perylene
and coronene showing the highest levels (Cretney et al., 1985), and up
to 43 ng/m3 were measured in Essen-Vogelheim, Germany (Buck, 1983).
High concentrations of individual PAH were determined in a residential
area heated primarily by coal, with levels of up to 260 ng/m3
chrysene, benz [a]anthracene, and benzo [b]fluoranthene (Grimmer,
1980).
The following PAH levels were measured on a roof directly downwind of
the chimney of a fireplace burning seasoned oak in the Chicago area,
USA: 1.8 µg/m3 acenaphthylene, 0.40 µg/m3 naphthalene, 0.35 µg/m3
anthracene, 0.22 µg/m3 phenanthrene, 0.20 µg/m3 benzo [a]pyrene,
0.20 µg/m3 benzo [e]pyrene, 0.13 µg/m3 fluorene, 0.10 µg/m3
pyrene, 0.096 µg/m3 fluoranthene, 0.052 µg/m3 acenaphthene, 0.045
µg/m3 benzo [k]fluoranthene, 0.033 µg/m3 chrysene, 0.030 µg/m3
cyclopenta [cd]pyrene, 0.023 µg/m3 benzo [b]fluoranthene, and 0.019
µg/m3 benz [a]anthracene. The levels of indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, and coronene were below
the limit of detection (Khalili et al., 1995).
In a comparison of the PAH concentrations in ambient air in eastern
and western Germany, the concentrations in rural areas were 3-12 times
higher in eastern than in comparable western parts of the country. The
PAH profiles were slightly different: the concentrations of the
lower-boiling-point PAH fluoranthene and pyrene were 110 and 68 ng/m3
in eastern and 36 and 28 ng/m3 in western Germany. The differences
may be due to the different types of brown and hard coal burnt (Jacob
et al., 1993a).
In 1991, PAH were determined in the air of Berchtesgaden, a national
park in Germany, and of the Oberharz (Ministry of Environment, 1993).
The concentration of phenanthrene, fluoranthene, and pyrene (about 14
ng/m3) in the Oberharz was two to three times higher than in
Berchtesgaden, due to the use of brown coal for heating. The levels of
the other PAH were of the same order of magnitude: benz [a]anthracene
and benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, about 5 ng/m3; and benzo [ghi]fluoranthene,
benzo [c]phenanthrene, benzo [e]pyrene, benzo [a]-pyrene,
indeno(1,2,3- cd)pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, anthanthrene, and coronene, < 1 ng/m3.
A model calculation for Germany showed that 5000 oil-heated houses
contributed to the pollution of ambient air by benzo [a]pyrene to the
same extent as one coal-heated house. It was assumed that one German
household consumes annually about 5000 litre of heating oil, producing
a maximum of 5 mg of benzo [a]pyrene (about 1 µg/litre combusted
oil). On the basis of a consumption of a similar amount of hard coal,
the same household would have an output of 25 g benzo [a]pyrene
(about 5000 µg/kg combusted hard coal) annually (J. Jacob, 1994,
personal communication).
5.1.2 Hydrosphere
PAH are found in the hydrosphere (Borneff & Kunte, 1983; Müller,
1987), mostly as a result of urban runoff, with smaller particles from
atmospheric fallout and larger ones from asphalt abrasion (Hoffman et
al., 1984). Long-range atmospheric transport of PAH has been well
documented in different countries (Lunde & Bjrseth, 1977; see also
section 4.1.2). After PAH are emitted into the atmosphere, for example
in motor vehicle exhaust, they are transferred into water by direct
surface contact or as a result of rainfall (Grob & Grob, 1974; Van
Noort & Wondergem, 1985a,b; Kawamura & Kaplan, 1986). The higher
levels of PAH that are found during winter months reflect increased
emissions resulting from domestic heating (Quaghebeur et al., 1983;
Thomas, 1986; see also section 5.1.1.1); however, the major source of
PAH varies for each body of water.
Anthropogenic combustion and pyrolysis and urban runoff containing
atmospheric fallout, asphalt particles, tyre particles, automobile
exhaust condensate and particulates, and lubricating oils and greases
were the major sources of PAH in lakes in Switzerland (Wakeham et al.,
1980a,b).
Comparisons between the levels of individual PAH in precipitation and
those in surface water showed that all of the precipitation samples
were more highly polluted with PAH, because they had been 'washed out'
of the atmosphere. Nearly all of the samples contained > 100 ng/litre
of fluoranthene, benzo [b]fluoranthene, pyrene,
indeno[1,2,3- cd]pyrene, phenanthrene, and naphthalene. The highest
levels of PAH in rainwater were found in Leidschendam, the
Netherlands, where pyrene concentrations < 2000 ng/litre,
fluoranthene concentrations < 1700 ng/litre, and benzo [a]pyrene
and benzo [b]fluoranthene concentrations < 390 ng/litre were
detected (van Noort & Wondergem, 1985b).
Most surface water samples contained concentrations of < 50
ng/litre of individual PAH. The levels in rainwater were 10-200
ng/litre, whereas those in snow were < 1000 µg/kg, with a maximum
of 6800 µg/kg for an individual PAH (Lygren et al., 1984). In one fog
sample, benzo [a]pyrene was found at 880 ng/litre and fluoranthene at
3800 ng/litre (Schrimpff, 1983: see section 5.1.2.4).
In sediment the levels of individual PAH were usually 1000-10 000
µg/kg dry weight, which are one order of magnitude higher than those
in precipitation. Triphenylene was detected in samples of sediment
from the Mediterranean Sea (France) at 2-600 µg/kg (Milano et al.,
1985) and in samples from Lake Geneva (Switzerland) at 25 µg/kg
(Dreier et al., 1985; see section 5.1.3).
5.1.2.1 Surface and coastal waters
The levels of individual PAH found in surface and coastal waters at
various locations are summarized in Table 37. Rivers in Germany
contained some PAH at concentrations of 1-50 ng/litre (Grimmer et al.,
1981b; Ernst et al., 1986; Regional Office for Water and Waste
Disposal, 1986; Kröber & Häckl, 1989) and fluoranthene, pyrene,
chrysene, benzo [a]pyrene, and benzo [e]pyrene at concentrations
< 100 ng/litre. The PAH levels in seawater from the German coast
varied over one order of magnitude depending on the sampling site. In
open seawater, the concentrations of two- to four-ring PAH -
naphthalene, fluorene, phenanthrene, fluoranthene, and pyrene - were
0.1-5 ng/litre, and those of five- to six-ring PAH ranged from < 0.01
to 0.2 ng/litre. Near the coast, the concentration of five- to
six-ring PAH increased with the content of particles, to which they
have greater affinity than two- to four-ring PAH (German Federal
Office for Sea Navigation and Hydrography, 1993).
The maximum levels of PAH in the Rivers Thames and Trent in the United
Kingdom were > 130 ng/litre. The highest levels of individual PAH in
the River Thames were 360 ng/litre fluoranthene, 350 ng/litre
benzo [a]pyrene, 210 ng/litre indeno[1,2,3- cd]pyrene, 160 ng/litre
benzo [ghi]perylene, 140 ng/litre benzo [k]fluoranthene, and 130
ng/litre perylene (Acheson et al., 1976). More recent data were not
available.
In Norway, the levels of most individual PAH were > 100 ng/litre. For
example, surface water from Bislet Creek near Oslo contained
fluoranthene, pyrene, phenanthrene, methylphenanthrene, naphthalene,
acenaphthene, acenaphthylene, and fluorene at concentrations > 1000
ng/litre (Berglind, 1982).
Table 37. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in surface and coastal waters
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthene 14-1232
Acenaphthylene 0.4-0.9 12-1024
Anthracene 1 10 18-932
Anthanthrene 0.2-0.5 15/1.8
Benz[a]anthracene ND 0.16 2.2-6.8 24/66 40/10 71-582
Benzo[a]fluorene 43/330
Benzo[a]pyrene 1-23 0.8 0.39 1.2-7.3 87/25 18 10/60 ND-40 19-311 0.9
Benzo[b]fluoranthene 0.1-0.5 0.07 80/20 ND-42 70-678 0.5-0.9
Benzo[b]fluorene 38 17
Benzo[c]phenanthrene 2.3-4.2 13/34 23-172
Benzo[e]pyrene 2-40 0.06 7.1-11 108/36 40-551
Benzo[ghi]fluoranthene
Benzo[ghi]perylene ND ND < 0.05 3.7-7.0 61/16 50/10 ND-61 33-636 ND
Benzo[k]fluoranthene 0.7-0.8 0.02 3.6-6.1 59/22 40/10 ND-24 0.2-0.5
Chrysene 11-15 36/87 14 10/10
Coronene ND-2.4 15/4.3
Cyclopenta[cd]pyrene ND ND
Dibenzo[a,h]pyrene <0.03 30/10
Fluoranthene 4-616 1.0-3.5 0.35 5.2/9.1 28/102 2.3-13 50/130 2-110 285-3269 3.4-5.1
Fluorene 2 0.63 0.6-1.2 25-1995
Indeno[1,2,3-cd]pyrene Trace < 0.03 2.8-6.1 63/13 50/20 ND-39 17-299 ND
1-Methylphenanthrene 30-1281
5-Methylcholanthrene
Naphthalene 4 50-2090
Perylene 0.8-1.4 27 20 9/28
Phenanthrene 3-136 3.5 1.5-9.1 101-5656
Pyrene 5-402 0.28 4.8/8.5 25/90 2.2-13 100/30 485-3099
Triphenylene
Table 37 (continued)
ND, not detected; /, single measurements;
[1] Lake water, Norway, 1981-82 (Gjessing et al., 1984);
[2] Lake water, Switzerland (Vu Duc & Huynh, 1981);
[3] Lake Superior, USA, 1986 (Baker & Eisenreich, 1990);
[4] Elbe River, Germany, 1980 (Grimmer et al., 1981b);
[5] Elbe River, main drainage channel, Germany, 1980 (Grimmer et al., 1981b);
[6] Water in various rivers, Germany, 1981-83 (Ernst et al., 1986);
[7] Water in various rivers, Germany, 1985; analytical method not given (Regional Office for Water and Waste Disposal,
1986);
[8] Water in various rivers, Germany, 1985-86; analytical method not given (Krober & Hackl (1989);
[9] River water, Norway, 1979 (Berglind, 1982);
[10] River water, Switzerland (Vu Duc & Huynh, 1981)
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated. The results of studies
in which water samples were filtered through solid sorbents may be underestimates of the actual PAH content (see section
2.4.1.4).
Table 37 (continued)
Compound [11] [12] [13] [14] [15] [16] [17] [18] [19] [20]
Acenaphthene ND-3 10 0.08-1.1 50-100
Acenaphthylene ND-5 0.02-1.7 80-1300
Anthracene ND-4 0.2 0.8-9.5 0.01-1.5 < 1-25 ND
Anthanthrene NR
Benz[a]anthracene ND-5 0.3 ND-9.6 0.04-6.8 ND
Benzo[a]fluorene NR
Benzo[a]pyrene 0.1-1.8 130-150 0.1/0.2 ND-10 0.2-1.0 0.03-8.8 ND
Benzo[b]fluoranthene ND-8 0.04-12
Benzo[b]fluorene 4.0-19 NR
Benzo[c]phenanthrene NR
Benzo[e]pyrene 0.02-8.8 ND
Benzo[ghi]fluoranthene NR
Benzo[ghi]perylene 0.2-11 30-160 0.7/0.8 ND-10 0.02-3.8 < 0.3-16 50
Benzo[k]fluoranthene 0.1-1.7 80-140 0.2/0.3 ND-13 0.02-7.7
Chrysene ND-12 NR
Coronene 0.01-1.4 NR
Dibenzo[a,h]pyrene ND-1 100
Fluoranthene 0.7-508 20-360 1.1/3.7 3-12 0.8 10-25 1.4-2.6 0.40-14 NR
Fluorene ND-2 0.7-15 1.9-5.2 0.33-3.2 70-2500
Indeno[1,2,3-cd]pyrene 0.1-8.0 50-210 ND/0.2 ND-8 0.01-3.5 NR
1-Methylphenanthrene NR
5-Methylcholanthrene NR
Naphthalene 4-34 3.6 0.4-9.2 NR
Perylene 40-130 0.01-5.7 NR
Phenanthrene 6-34 21-18 8.0-93 2.4-2.7 0.24-5.8 < 1-3 ND
Pyrene 50-260 1-15 0.3-15 8.8-25 0.82-1.7 0.12-15 < 1-53 10-65
Triphenylene NR
Table 37 (continued)
ND, not detected; /, single measurements;
[11] River water, United Kingdom, 1974 (Lewis, 1975);
[12] Water in various rivers, United Kingdom, analytical method not given (Acheson et al.,1976);
[13] Water in various rivers, United Kingdom; analytical method not given (Sorrell et al., 1980);
[14] River water, USA, 1984 (De Leon et al., 1986);
[15] Surface water, Canada (Environment Canada, 1994);
[16] River water, China, 1981 (Wu et al., 1985);
[17] Coastal water, Germany, 1982 (Ernst et al., 1986);
[18] Seawater, Germany, 1990 (German Federal Office for Sea Navigation and Hydrography, 1993);
[19] Coastal water, Australia, 1983 (Smith et al., 1987);
[20] Water (no further specification), Japan, 1974-91 (Environment Agency, Japan, 1993)
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated. The results of studies
in which water samples were filtered through solid sorbents may be underestimates of the actual PAH content (see section
2.4.1.4).
The highest concentrations of PAH in water in Canada were reported for
water samples from ditches next to utility and railway lines near
Vancouver. The highest mean concentrations were measured near utility
poles treated with creosote, with values of 2000 µg/litre for
fluoranthene, 1600 µg/litre for phenanthrene, and 490 µg/litre for
naphthalene (Environment Canada, 1994).
Four individual PAH were detected in seawater from Green Island,
Australia. The highest levels of PAH found were 53 ng/litre pyrene, 25
ng/litre anthracene, 16 ng/litre benzo [ghi]perylene, and 3 ng/litre
phenanthrene, (Smith et al., 1987).
The total content of phenanthrene, anthracene, fluoranthene, pyrene,
benzo [b]fluorene, and benz [a]anthracene in the Yellow River,
China, was 170 ng/litre (Wu et al., 1985; for individual PAH
concentrations, see Table 37).
The PAH levels found in the River Rhine in Germany and the Netherlands
and in some of its tributaries are summarized in Table 38. Many
investigators have detected PAH in the Rhine. The lowest
concentrations of benzo [a]pyrene, < 10-20 ng/litre, were found in
the Rhine at Lobith and Hagestein in Germany and at Lek in the
Netherlands in 1987-90 (Association of Rhine and Meuse Water Supply
Companies, 1987-90), when the levels of fluoranthene were 70-140
ng/litre. In 1976-79, the Rhine at Lek and Waal contained < 10-580
ng/litre of benzo [a]pyrene (Association of Rhine and Meuse Water
Supply Companies, 1976-79), so that the levels had decreased by one
order of magnitude within 14 years. The sum of fluoranthene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene) was 9-40 ng/litre
at km 30 and 130-5700 ng/litre at km 853, indicating that the level of
pollution increased markedly between the source and the estuary
(Borneff & Kunte (1983). The average concentrations of individual PAH
were 1-50 ng/litre, although individual PAH were found at
concentrations in the range 100-200 ng/litre near Mainz, an
industrialized town (Borneff & Kunte, 1964, 1965). In general, the PAH
levels in the Rhine decreased by a factor of 3 between 1979 and 1989.
The Emscher and Ruhr waterways in Germany have been heavily polluted
(see Table 38). In 1985, the Emscher River contained 6400 ng/litre
fluoranthene, 6000 ng/litre pyrene, 2000 ng/litre benz [a]anthracene,
1100 ng/litre dibenz [a,h]anthracene, 910 ng/litre benzo [a]pyrene,
880 ng/litre chrysene, 630 ng/litre indeno[1,2,3- cd]pyrene, 510
ng/litre benzo [ghi]perylene, 270 ng/litre anthracene, 220 ng/litre
perylene (Regional Office for Water and Waste Disposal, 1986), but by
1989 the levels had decreased by about one order of magnitude
(Regional Office for Water and Waste Disposal, 1990 ). The PAH
concentrations in the Emscher were three times higher than those in
the Rhine near Mainz. Between 1985 and 1989, the PAH levels in the
Emscher decreased further by a factor of 15; however, the levels in
the Ruhr remained about the same or increased slightly between 1979
and 1985 (Regional Office for Water and Waste Disposal, 1986, 1988,
1990).
Table 38. Polycyclic aromatic hydrocarbon concentrations (ng/m3) in the River Rhine and some highly polluted tributaries
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Anthracene 10 270 25-260 10
Anthanthrene 0.9-11 1.3
Benz[a]anthracene 6.1-31 11-50 1970 100-780 13 20
Benzo[a]pyrene 0.8-36 ND-7 6-30 12-40 < 10-20 910 59-280 15 30
Benzo[b]fluoranthene ND-8 7-30 12-40 < 10-30 880 62-310 40
Benzo[c]phenanthrene 1.5-9.1 1.9
Benzo[a]pyrene 18-31 33
Benzo[ghi]fluoranthene 1.0-11 2.2
Benzo[ghi]perylene 15-29 ND-8 6-30 9-30 < 10-20 510 30-210 17 30
Benzo[k]fluoranthene ND-4 2-14 6-20 < 10-40 440 36-150 20
Chrysene 21-62 1080 27 30
Dibenzo[a,h]pyrene 10-40 1100 32-310 30
Fluoranthene 4-18 15-61 25-77 20-140 6420 207-1700 60
Indeno[1,2,3-cd]pyrene 9.5-27 ND-6 2-26 10-40 < 10-20 630 28-220 17 30
Perylene ND-8.1 10 220 13/80 2.1 10
Pyrene 20-50 6010 155-1100 50
ND, not detected; /, single measurements;
[1] Rhine, Germany, 1979 (Grimmer et al.,1981b);
[2] Rhine, Germany, 1985-88, analytical method not given (Krober & Hackl, 1989);
[3] Rhine, Netherlands, 1985-88 (Netherlands' Delegation, 1991);
[4] Rhine, Germany, 1987-89, analytical method not given (Regional Office for Water and Waste Disposal, 1988, 1989, 1990);
[5] Rhine, Netherlands; 1987-90, analytical method not given (Association of Rhine and Meuse Water Supply Companies, 1987-90);
[6] Emscher, Germany, 1985, analytical method not given (Regional Office for Water and Waste Disposal, 1986);
[7] Emscher, Germany, 1987-89, analytical method not given (Regional Office for Water and Waste Disposal, 1988, 1989, 1990);
[8] Ruhr, Germany, 1979 (Grimmer et al., 1981b);
[9] Ruhr, Germany, 1985, analytical method not given (Regional Office for Water and Waste Disposal, 1986)
The PAH levels in the main drainage channels of the River Elbe,
Germany, were one order of magnitude higher than in the river water
(Grimmer et al., 1981b), owing to the high input of rainwater to the
channels.
5.1.2.2 Groundwater
The PAH concentrations in uncontaminated groundwater in the
Netherlands generally did not exceed 0.1 µg/litre, but levels of about
30 µg/litre naphthalene, 10 µg/litre fluoranthene, and 1 µg/litre
benzo [a]pyrene were reported in contaminated groundwater (Luitjen &
Piet, 1983).
Benzo [a]pyrene levels in groundwater in western Germany ranged from
0.1 to 0.6 ng/litre and those of total PAH from 34 to 140 ng/litre
(Andelman & Suess, 1970). Benzo [a]pyrene was also detected at levels
of 0.1-5.0 ng/litre in groundwater (Woidich et al., 1976). More recent
data were not available. Groundwater in the USA contained maximum
concentrations of 0.38-1.8 ng/litre naphthalene, 0.02-0.04 ng/litre
acenaphthene, and 0.008-0.02 ng/litre fluorene (Stuermer et al.,
1982). Near a refinery at Pincher Creek, Alberta, Canada, the pyrene
concentrations in groundwater showed a maximum of 300 µg/litre
(median, 30 µg/litre); the maximum concentration of fluorene was 230
µg/litre (median, 40 µg/litre). At Newcastle, New Brunswick, Canada,
naphthalene was detected at concentrations up to 2.8 µg/litre and
benzo [a]pyrene up to 0.32 µg/litre in groundwater near a
wood-preserving plant (Environment Canada, 1994).
5.1.2.3 Drinking-water and water supplies
PAH levels were determined in drinking-water in samples from Canada,
Scandinavia, and the USA up to 1982. The concentration of naphthalene
was 1.2-8.8 ng/litre, that of benzo [a]pyrene was 0.2-1.6 ng/litre,
and that of the sum of the six 'standard WHO' PAH (fluoranthene,
benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]perylene, and indeno[1,2,3- cd]pyrene) was 0.6-24
ng/litre. The highest levels of naphthalene (1300 ng/litre),
benzo [a]pyrene (77 ng/litre), and the six WHO standard PAH (660
ng/litre) were detected in raw water sources in the USA and in the
Great Lakes area of Canada (Müller, 1987). More recent measurements
are given in Table 39. Most samples contained 0.38-16 ng/litre
naphthalene and < 0.04-2.0 ng/litre benzo [a]pyrene. In one set of
water samples from the Netherlands, no PAH were detected, with a limit
of detection for individual PAH of 4 ng/litre (de Vos et al., 1990).
In a study of the changes in PAH concentrations after passage of water
through tar-coated major distribution pipes, the level increased from
an initial concentration of none detected-13 ng/litre to none
detected-62 ng/litre. The finding that water in a few distribution
lines had lower concentrations of PAH may be due to sorption of PAH on
the surfaces of distribution pipes, chemical interaction with oxidants
in water, or a dilution effect (Basu et al., 1987).
Of 101 German drinking-water samples analysed in 1994, four exceeded
the German drinking-water standard of 0.2 µg/litre for the sum of
fluoranthene, benzo [b]fluoranthene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [ghi]-perylene, and indeno[1,2,3- cd]pyrene.
Heavy contamination had occurred after repairs to a pipeline coated
with tar, and one drinking-water sample taken in a household contained
2.7 µg/litre of these PAH, in addition to phenanthrene at 2.8 µg/litre
and pyrene at 1.2 µg/litre (State Chemical Analysis Institute,
Freiburg, 1995). The report stated that abrasion of particles from
tar-coated drinking-water pipelines poses a hazard that is often
difficult to judge since it is often not known what material was used
decades previously.
In Canada, the PAH concentrations in drinking-water were usually below
or near the detection limits of 1-5 ng/litre, although concentrations
of 5.0-21 ng/litre benzo [ghi]perylene, 1.0-12 ng/litre fluoranthene,
1.0-5.0 ng/litre benzo [b]fluoranthene, 1.0-3.0 ng/litre
benzo [k]fluoranthene, and 1.0-3.0 ng/litre benzo [a]pyrene were
detected in some areas (Environment Canada, 1994).
5.1.2.4 Precipitation
(a) Rain
The concentrations of PAH found in precipitation in 1979-91 are
summarized in Table 40. The levels of benzo [a]pyrene were < 1-390
ng/litre. In an analysis of PAH in rainfall in Hanover, Germany,
between July 1989 and March 1990, fluoranthene was the dominant
component, followed by pyrene. The average concentration of all PAH
increased from 351 ng/litre in summer to 765 ng/litre in the autumn of
1989, while a slight decrease was observed in the winter of 1989-90.
These results indicate that the increase in the level of PAH in
precipitation in cold weather is due to an increase in residential
heating and a slower rate of photochemical degradation (Levsen et al.,
1991).
Table 39. Polycyclic aromatic hydrocarbon concentrations (ng/litre) in drinking-water
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 0.6-4.0 7.4-14
Acenaphthylene 0.4-4.4 0.40-1.6
Anthracene 0.5-7 < 1.3-9.7
Anthanthrene 0.2
Benz[a]anthracene ND-1.9 0.4-5.5 0.12-1.5
Benzo[a]fluoranthene 0.1-3.3 0.05-4.2
Benzo[a]pyrene 0.1-0.7 < 0.1-2.0 < 0.04-0.29 Trace-1.9 0.2-0.3 0.2-1.6 < 5.0
Benzo[b]fluoranthene 0.5-1.3 2.4-4.0 0.05-0.34 0.1-14 < 5-40
Benzo[b]fluorene 0.9 0.04-<1.4
Benzo[e]phenanthrene 0.9-1.5 0.28
Benzo[e]pyrene 0.2-4 < 0.1-0.41
Benzo[ghi]fluoranthene 0.36
Benzo[ghi]perylene 0.3-0.9 0.4-1.1 ND 0.4-0.7 0.4-4.0 < 5.0
Benzo[j]fluoranthene 0.03-0.14 0.2-1.2
Benzo[k]fluoranthene 0.2-0.8 0.02-0.10 0.2-4.9 0.1-0.3 0.1-0.7 < 5-40
Chrysene 21-62 1080 27 30
Dibenz[a,h]anthracene 1.2
Fluoranthene 3.5-6.5 1.7-18 < 0.58-24 0.7-3400 3.4-4.2 5-24 2.4-9.0 < 5-623
Fluorene 0.9-4 < 1.1-21 4-16
Indeno[1,2,3-cd]pyrene Trace-0.7 0.4-1.2 ND-1.1 < 0.5 0.7-2.2 < 5.0
1-Methylphenanthrene 0.5-1.0 0.14-13
Naphthalene 1.8-5 < 6.3-8.8 8 6-16
Perylene Trace-0.2 0.2
Phenanthrene 2.5-46 < 2.2-64 24-90
Pyrene 1.6-3.7 1.1-15 < 0.30-12 40/40
Table 39 (continued)
ND, not detected; /, single measurements;
[1] Austria; analytical method, in-situ fluorescence determination (Woidich et al., 1976);
[2] Norway, 1978-80 (Berglind, 1982);
[3] Norway, 1980-81 (Kveseth et al., 1982);
[4] Switzerland, 1973 (Grob & Grob, 1974);
[5] Switzerland (Vu Duc & Huynh, 1981);
[6] United Kingdom; water reservoirs after treatment, 1974 (Lewis, 1975);
[7] USA, 1976; analytical method, high-performance liquid chromtography and gas chromatography
(Thruston, 1978);
[8] USA, 1976-77; analytical method, thin-layer chromatography and gas-liquid chromatography with
flame ionization detection (Basu & Saxena, 1978a,b);
[9] Canada, treated drinking-water, 1987-90 (Environment Canada, 1994)
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated.
The results of studies in which water samples were filtered through sold sorbernts may be
underestimates of the actual PAH content (see section 2.4.1.4).
Table 40. Polycyclic aromatic hydrocarbon concentrations (ng/litre) in rainwater
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 3.2 1.2/16 2.5-8.5
Acenaphthylene 130-200 14 4.7/55 23-59
Anthracene 8-19 0.88/23 2.0-7.9
Benz[a]anthracene 1.2-86 140 6-100 9-33 7-17 20-65 1.6-4.5
Benzo[a]fluoranthene 14-52
Benzo[a]pyrene 5-17 1.1-187 ND-390 10-37 7-26 5-36 ND-0.18
Benzo[b]fluoranthene 2.9-166 15-390 45-70 17-65
Benzo[b]fluorene 15
Benzo[c]phenanthrene 802
Benzo[e]pyrene < 0.5a-149 217-290 7-62 ND-0.51
Benzo[ghi]perylene 7-29 1.7-109 40-70 15-56 22
Benzo[k]fluoranthene 1.0-142 6-190 17-30 9-28
Chrysene 2.9-141 30-120 ND-67 21-29 3.3-12
Dibenz[a,h]anthracene < 0.5a-12 7-20 3-12
Fluoranthene 23-66 23-392 240-270 14-1650 66-180 87-189 115-162 1.7/110 28-70
Fluorene 10-200 6-50 3.2/43 9.1-22
Indeno[1,2,3-cd]pyrene < 0.5a-137 ND-80 50-110 24-72 12
1-Methylphenanthrene 8-26
Naphthalene 8-77 20/72 46-140
Perylene 2
Phenanthrene 130-600 30-133 79-113 158-238 24/140 61-130
Pyrene 9.5-304 25-60 ND-2000 ND-37 36-108 77-175 24-56
Table 40 (continued)
ND, not detected; /, single measurements;
[1] Bavaria, Germany, 1979-80; analytical method, high-performance thin-layer chromatography (Thomas, 1986);
[2] Hanover, Germany, 1989-90 (Levsen et al., 1991);
[3] Italy (Morselli & Zappoli, 1988);
[4] Leidschendam, Netherlands, 1982 (Van Noort & Wondergem, 1985b);
[5] Rotterdam, Netherlands, 1983 (Van Noort & Wondergem, 1985b);
[6] Netherlands, 1983 (Den Hollander et al., 1986);
[7] Oslo, Norway, 1978 (Berglind, 1982);
[8] Oregon, USA, 1982 (Pankow et al., 1984);
[9] Portland, USA, 1984 (Ligocki et al., 1985)
a Detection limit for benzo[a]pyrene
Analysed by high-performance liquid chromatography or gas chromatography, unless otherwise stated. The results
of studies in which water samples were filtered through solid sorbents may be underestimates of the actual PAH
content (see section 2.4.1.4).
The concentrations of phenanthrene and fluoranthene in rainwater were
noticeably higher than those at 200 m when sampled simultaneously, but
no significant differences in the concentrations of
benzo [k]fluoranthene, benzo [b]fluoranthene, benzo [a]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, or
indeno[1,2,3- cd]pyrene were found. The authors suggested that
scavenging in and below clouds was responsible for the presence of PAH
in rainwater (Van Noort & Wondergem, 1985b).
The deposition rates of individual PAH in Cardiff, London, Manchester,
and Stevenage, United Kingdom, were 0.3-20 µg/m2 per day. Anthracene
accounted for about 25% of the deposition in London, followed by
pyrene (16%), benzo [b]fluoranthene (16%), and benz [a]anthracene
(13%) (Clayton et al., 1992).
The rate of precipitation containing PAH after gravitational
deposition by rain, snow, and particles was not affected by the type
or structure of the receiving surface. Precipitation in a beech and
spruce stand contained concentrations of 23-52 ng/litre fluoranthene,
8.9-30 ng/litre benzo [ghi]-perylene, 6.4-27 ng/litre
indeno[1,2,3- cd]pyrene, and 2.0-8.4 ng/litre benzo [a]pyrene. The
deposition of PAH is in general higher under spruce stands because the
rates of interception are higher than those in beech stands.
Substantial amounts of PAH are transferred to the soil by litterfall,
indicating adsorption of PAH on the surfaces of leaves and needles
(Matzner, 1984).
(b) Snow
The concentrations of PAH in snow samples are summarized in Table 41.
A sample collected in Hanover, Germany, contained fluoranthene at 55
ng/litre, pyrene at 31 ng/litre, and other PAH at concentrations up to
9 ng/litre (Levsen et al., 1991). A sample of snow from Bavaria
contained 200 ng/litre fluoranthene, 50 ng/litre benzo [ghi]perylene,
and 29 ng/litre benzo [a]pyrene (Schrimpff et al., 1979).
In Norwegian snow samples, the average concentrations of individual
PAH were 10-100 ng/litre, but levels up to 6800 ng/litre were found of
phenanthrene, 1-methylphenanthrene, fluoranthene,
benzo [b]fluoranthene, and fluorene (Berglind, 1982; Gjessing et al.,
1984; Lygren et al., 1984). Snow taken near a steel plant in Canada
contained average levels of 50-500 ng/litre of individual PAH but
higher amounts of phenanthrene, fluoranthene, and pyrene (Boom &
Marsalek, 1988).
Table 41. Polycyclic aromatic hydrocarbon concentrations (ng/litre) in snow
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene 10-13 <50-98
Acenaphthlene 19-47 <50-153
Anthracene 13-28 9-379 165-246
Benz[a]anthracene 2.6 21-47 15-677 228
Benzo[a]fluoranthene 13 179-396
Benzo[a]pyrene 29 3.0 23-77 54-602 250 <100-558
Benzo[b]fluoranthene 9.2 799-1501 <100-647
Benzo[b]fluorene 11 192
Berzo[e]pyrene 5.5 30-64 609 360-630
Benzo[ghi]perylene 50 4.8 29-85 98-551 319-391 <100-466
Benzo[k]fluoranthene 2.8 <100-990
Chrysene 6.2
Dibenz[a,h]anthracene <0.5a
Fluoranthene 200 55 108-211 86-2665 1820-3143 <50-7020
Fluorene 13-85 96 485-1237 <50-237
Indeno[1,2,3-cd]pyrene <0.5a 20-82 <100-496
I-Methylphenanthrene 1366-2117
Naphthalene 50-94 36-67 123-195
Perylene 12
Phenanthrene 119-276 45-1385 4055-6787 <50-3560
Pyrene 31 68-143 55-2002 <50-3750
Analysed by high-performance liquid chromatography or gas chromatography, unless
otherwise stated. The results of studies in which water samples were filtered through
solid sorbents may be underestimates of the actual PAH content (see section 2.4.1.4).
a Detection limit for benzo[a]pyrene
[1] Bavaria, Germany, 1978; analytical method, high-performence thin-layer chromatography
and gas chromatography-mass spectroscopy (Schrimpff et al., 1979);
[2] Hanover, Germany, 1990 (Levsen et al., 1991);
[3] Norway, 1979-81 (Berglind, 1982);
[4] Norway, 1981-82 (Gjessing et al., 1984);
[5] Norway (Lygren et al., 1984);
[6] Near steel plant, Canada, 1986 (Boom & Marsalek; 1988)
(c) Hail
The PAH levels in a hail sample collected in Hanover, Germany, were of
the same order of magnitude as those in rain samples: fluoranthene,
170 ng/litre; pyrene, 98 ng/litre; benzo [b]fluoranthene, 58
ng/litre; chrysene, 47 ng/litre; benzo [e]pyrene, 40 ng/litre;
indeno[1,2,3- cd]pyrene, 29 ng/litre; benzo [ghi]perylene, 27
ng/litre; benzo [k]fluoranthene, 19 ng/litre; benz [a]an-thracene,
16 ng/litre; benzo [a]pyrene, 12 ng/litre; and
dibenz [a,h]anthracene, 3.3 ng/litre (Levsen et al., 1991).
(d) Fog
The concentrations of PAH in fog are higher than those in rain. A fog
sample collected in western Germany contained 360-3800 ng/litre
fluoranthene and 130-880 ng/litre benzo [a]pyrene (Schrimpff, 1983).
In fog samples collected during the autumn of 1986 in Zürich,
Switzerland, the average concentrations of PAH found were 4400
ng/litre fluoranthene, 2700 ng/litre benzo [b]fluoranthene, 2500
ng/litre pyrene, 2200 ng/litre phenanthrene, 2100 ng/litre
benzo [e]pyrene, 1400 ng/litre benz [a]anthracene, 1400 ng/litre
indeno[1,2,3- cd]pyrene, 1200 ng/litre benzo [a]pyrene, 920 ng/litre
anthracene, 860 ng/litre 1-methylphenanthrene, 750 ng/litre
benzo [b]fluorene, 750 ng/litre perylene, 590 ng/litre
benzo [k]fluoranthene, 540 ng/litre benzo [ghi]perylene, 340
ng/litre anthanthrene, 260 ng/litre fluorene, and 160 ng/litre
benzo [a]fluorene (Capel et al., 1991).
5.1.3 Sediment
PAH levels in sediments from rivers, lakes, seas, estuaries, and
harbours are summarized in Tables 42-46.
5.1.3.1 River sediment
The concentrations of individual PAH in river sediments in 1987-91
(Table 42) varied over a wide range; the maximum values were in the
high nanogram per gram range.
The levels of individual PAH in sediments from German rivers were
about 4000 µg/kg for benzo [a]pyrene, fluoranthene, and
benzo [b]fluoranthene and about 1500 µg/kg for pyrene,
indeno[1,2,3- cd]pyrene, and benz [a]anthracene. The levels of other
PAH generally did not exceed 500 µg/kg (Kröber & Häckl, 1989; Regional
Office for Water and Waste Disposal, 1989). PAH were determined in
many German river sediments. Table 42 gives data for three rivers: the
Rhine and Neckar rivers are highly polluted, whereas the Gersprenz is
relatively uncontaminated.
The concentrations of PAH in the sediments of rivers around Aachen,
Germany, were determined in different size fractions, which allowed
the authors to locate where the sediment became contaminated (Lampe et
al., 1991).
The PAH concentrations in sediment from the River Elbe in Germany in
1991 were of the same order of magnitude as those in Lake Plöner and
Lake Constance, but the river sediment contained more PAH with a low
boiling-point than the lake sediments. The ratio of fluoranthene to
benzo [e]pyrene, taken as a marker of the emission of PAH from the
combustion of brown coal, was 2.8-5.1, similar to those found in the
Elbe sediment. It was concluded that the PAH in the sediment were due
mainly to brown-coal combustion (German Ministry of Environment,
1993).
Table 42. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in river sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12]
Acenaphthene ND-140 14.5 1 100 ND 0.04-130
Acenaphthylene ND 9.7 1 540 ND 0.7-671
Anthracene ND-1010 80-640 670/NR ND/NR 82.1 8-200 152 4 700 10-1200
Benz[a]anthracene 620-1700 10000/NR 50-90/NR 450 ND-100 541 6 600 3.2-2100
Benzo[a]pyrene Q 400-1250 ND-8000/ 20-90/10-80 1-760 70-11 960 454 ND-80 570 4 400 5-3700
ND-5300
Benzo[b]fluoranthene 460-1290 ND-8700/ 50-190/ 620 ND-50
ND-5600 26-150
Benzo[e]pyrene 596 4 900 0.9-1800
Benzo[ghi]fluoranthene 253 NR
Benzo[ghi]perylene Q-578 340-750 ND-2900/ ND 10-70 60-7480 358 ND 353 7 400 3-1310
ND-1900
Benzo[j]fluoranthene 749
Benzo[k]fluoranthene 230-650 ND-4000/ 20-90/10-80 408 ND-60 608
ND-2700
Chrysene ND-1549 6700/NR ND-30/NR 597 904 NR
Coronene 20-260 150-2460 284 NR
Cyclopenta[cd]pyrene 15 1 100 NR
Dibenz[a,h]anthracene 500-1070 2600/NR ND-20/NR 21 ND-200 2 800 8.1-340
Fluoranthene ND-4455 900-2470 ND-19000/ 2-2360 190-29300 904 100-400 1013 13 000 ND-60 NR
100-380/
ND-12 600 52-310
Fluorene ND-260 25.4 ND-2 26 3 000 ND/50 3-130
Indeno[1,2,3-cd]pyrene 360-910 ND-6300/ ND/ND 332 486 16 000 NR
ND-4200
1-Methylphenanthrene 145 NR
Naphthalene ND-2630 7.0 3 800 ND
Perylene 120-320 ND-100 2 400 NR
Phenanthrene ND-220 3300/NR ND-40/NR 361 10-400 563 10 000 ND/220 9-2800
Pyrene ND-2526 680-3450 17000/NR ND-130/NR 736 80-300 940 9200 ND-160 20-3900
Triphenylene 10-80 NR
Table 42 (continued)
NQ not detacted; /, single measurements; NR, not reported; Q, qualitative;
[1] Czechoslovakia, 1988; reference weight not given (Holoubek et al., 1990);
[2] Rhine, Germany, 1982-83 and 1987-88; analytical method and reference weight not given (Regional Office for Water and Waste Disposal, 1989);
[3] Neckar, Germany, 1985-88; fine, unsieved sediment; analytical method not given (Krober & Hackl, 1989);
[4] Gersprenz, Germany, 1985-88; fine, unsieved sediment; analytical method not given (Krober & Hackl, 1989);
[5] Wildbach, Germany, 1989 (Lampe et al., 1991);
[6] Haarbach, Germany, 1989 (Lampe et al., 1991);
[7] River, Bremen, Germany, 1994 (Riess & Wefers, 1990;
[8] Rhone, France, 1985 (Milano & Vernet, 1988);
[9] Sweden, 1985 (Broman et al., 1987);
[10] Black River, USA, 1984 (Fabacher et al., 1991);
[11] Rainy River, Canada, 1986; reference weight not given (Merriman, 1988);
[12] Japan, 1974-91 (Environment Agency, Japan, 1993)
Analysed by high-performance liquid chromatography or gas chromatography and concentration in micrograms per kilogram dry weight
The maximum levels of individual PAH in sediments in Czechoslovakia
were 4500 µg/kg fluoranthene, 2600 µg/kg naphthalene, 2500 µg/kg
pyrene, 1500 µg/kg chrysene, 1000 µg/kg anthracene, 580 µg/kg
benzo [ghi]perylene, 260 µg/kg fluorene, 220 µg/kg phenanthrene, and
140 µg/kg acenaphthene (Holoubek et al., 1990).
The levels of individual PAH in sediments from some of the most
polluted areas in continental USA were summarized by Bieri et al.
(1986). The levels usually ranged from 1000 to 10 000 µg/kg, but that
in sediment from the Elizabeth River, Virginia, contained
concentrations up to 42 000 µg/kg. Up to 39 000 µg/kg wet weight were
found in the Detroit River (Fallon & Horvath, 1985).
The concentrations of individual PAH in sediments from the Trenton
Channel of the Detroit River, a waterway in a highly industrialized
area, connecting Lake St Clair with Lake Erie. varied from not
detected (< 4 µg/kg) to 22 000 µg/kg in different locations.
Sediments from the southwest shore of Grosse Ile had low levels of
contamination, while those in the vicinity of Monguagon Creek had high
levels (Furlong et al., 1988).
5.1.3.2 Lake sediment
The concentrations of individual PAH found in lake sediments in
1984-91 (Table 43) ranged from 1 to about 30 000 µg/kg dry weight. The
total PAH concentrations in surface sediments from Lake Michigan, USA,
were 200-6200 µg/kg dry weight (Helfrich & Armstrong, 1986).
5.1.3.3 Marine sediment
The concentrations of individual PAH in marine sediments in 1985-91
(Table 44) varied widely, with maximum values up to about 4000 µg/kg.
Sediments near power-boat moorings at the coral reef around Green
Island, Australia, were found to contain measurable amounts of several
PAH, strongly suggesting that they originated from fuel spillage or
exhaust emissions (Smith et al., 1987).
The benzo [a]pyrene level was 104-106 times higher in bottom
sediments from the Baltic Sea than in water at the same location. The
bottom sediments also contained more individual PAH than the
corresponding water samples (Veldre & Itra, 1991).
Maximum levels of 460 µg/kg benzo [a]pyrene and 400 µg/kg
benzo [e]pyrene were determined in northern North Sea sediments in
the vicinity of oil fields. The hydrocarbon concentrations were above
the background levels only in water and sediments within a 2-km radius
of platforms, where diesel-coated drill cuttings were dumped. The
contribution of five- and six-ring compounds to the total PAH in
sediments was unexpectedly high in samples unlikely to be contaminated
by oil. Their source was probably windborne combustion products
(Massie et al., 1985).
Table 43. Polycyclic aromatic hydrocarbon concentrations (µg/kg)
in lake sediments
Compound [1] [2] [3] [4]
Anthracene 160 41-620
Benz[a]anthracene ND 150-1700 41
Benzo[a]pyrene 180-2000 45
Benzo[b]fluoranthene 200
Benzo[e]pyrene 80 140-1500 75
Benzo[ghi]fluoranthene 75 18-270
Benzo[ghi]perylene 21-1600 107
Benzo[k]fluoranthene 126
Chrysene 250 124
Coronene 1
Dibenz[a,h]anthracene 70
Fluoranthene 66-248 390 330-3900 103
Fluorene 5.9
Indeno[1,2,3-cd]pyrene 100 25-1500 279
Naphthalene ND
Perylene 50 47-540
Phenanthrene 70-180 100 300-6600 81
Pyrene 110-122 340 210-3500 60
Triphenylene 25
ND, not detected;
[1] Lake Padderudvann, Norway; 1981-82; reference weight not given
(Giessing et al., 1984);
[2] Lake Geneva, Switzerland (Dreier et al., 1985);
[3] Cayuga Lake, USA, 1978; concentrations are given as ng/g
deepwater (Heit, 1985);
[4] Lake Superior, USA (Hamburg Environment Office, 1993)
Analysed by high-performance liquid chromatography or gas
chromatography; concentration in micrograms per kilogram dry weight
Table 44. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in sea sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene ND-6 NR
Acenaphthylene ND-2000 0.6-4.3 NR
Anthracene 3-800 0.3-2.1 6-42 5-313 < 0.06-1.0
Anthanthrene 29-74 NR
Benz[a]anthracene 5-39 1-900 0.8-19 9-150 15-250 < 0.01-6.0
Benzo[a]fluoranthene 2-41 NR
Benzo[a]pyrene 16-25 6-2200 0.4-13 7-160 1100 14-265 0.2-460 < 0.004-4.3
Benzo[b]fluoranthene 13-26 ND-3800 1300 51-490
Benzo[b]fluorene 2-38 NR
Benzo[e]pyrene 5.8-18 0.6-15 9-125 21-345 0.4-396 < 0.1-0.6
Benzo[ghi]perylene ND-400 12-225 700 < 10-623 < 0.01-2.6
Benzo[k]fluoranthene 4.0-9.8 ND-3400 600 10-180 < 0.001-2.5
Chrysene 49 1.0-12 8-165 21-398 < 0.04-0.8
Coronene 11-36 NR
Dibenzo[a,e]pyrene 7-79 NR
Dibenz[a,h]anthracene 2-7 ND-400 0.5-4.2 4-74 NR
Fluoranthene ND/159 4-2000 0.4-31 12-230 2300 36-1913 < 0.1-7.2
Fluorene ND-100 0.5-3.1 1-12 NR
Indeno[1,2,3-cd]pyrene 8-200 17-510
Naphthalene ND-100 0.7-8.6 1-2b 18-1074
Perylene 1-2200 5-105 24-178
Phenanthrene 1-1500 0.8-29 23-93 11-971 < 0.06-4.2
Pyrene 8-160 5-1600 1.6-40 10-145 30-1697 < 0.1-15
Triphenylene 2-600 NR
ND, not detected /, single measurements; NR, not reported;
[1] Baltic Sea, Estonia, reference weight not given (Veldre & Itra, 1991);
[2] Mediterranean Sea, France (Milano et al., 1985);
[3] Adriatic Sea, Italy, 1983 (Marcomini et al., 1986);
[4] Ligurian Sea, Italy (Desideri et al., 1988);
[5] Ketelmeer, Netherlands, 1987 (Netherlands' Delegation, 1991);
[6] North Sea, Netherlands, within 70 km from the coast; 1987-88 (Compaan & Laane, 1992);
[7] North Sea, United Kingdom, 1980 (Massie et al., 1985);
[8] Great Barrier Reef, Australia, 1983 (Smith et al., 1987)
Analysed by high-performance liquid chromatography or gas chromatography
The following background concentrations have been reported in North
Sea sediments: < 0.01-20 µg/kg dry weight benzo [a]pyrene, < 30
µg/kg fluoranthene, < 6 µg/kg benzo (b)fluoranthene plus
benzo (k)fluoranthene, < 5 µg/kg benzo [ghi]-perylene, and < 3
µg/kg indeno[1,2,3- cd]pyrene (Compaan & Laane, 1992).
5.1.3.4 Estuarine sediments
The concentrations of individual PAH in estuarine sediments in 1981-92
(Table 45) varied widely, with maximum values in the high microgram
per gram range. Measurements in sediments from the Continental Shelf
of the Atlantic Ocean and the Gironde Estuary, France, showed
relatively little contamination with PAH when compared with sediments
from more polluted European estuaries (Garrigues et al., 1987). The
levels of PAH in estuarine sediments in the United Kingdom were 10-500
µg/kg. Higher amounts of fluoranthene (1000-1900 µg/kg) and pyrene
(790 µg/kg) were reported in estuaries of the River Mersey and the
River Tamar (Readman et al., 1986).The total PAH concentrations in
sediments from the Penobscot Bay region of the Gulf of Maine, USA,
ranged from 290 to 8800 µg/kg, with a distinct gradient that decreased
seawards. The PAH composition was uniform throughout Penobscot Bay.
Particulates of combustion products transported in the atmosphere were
suggested to be a major source of PAH contamination. The levels in
Penobscot Bay sediments were significantly higher than expected for an
area previously considered to be uncontaminated and fell within the
range found in industrialized regions throughout the world (Johnson et
al., 1985).
The Saguenay Fjord is the major tributary that empties into the St
Lawrence River estuary, and the area is highly industrialized. The PAH
concentrations were maximal near the aluminium smelting plants that
dominate the industrial sector and which were considered to be the
major source of PAH, and the levels decreased with distance from this
industrial zone. The concentrations of benzo [a]pyrene,
benzo [e]pyrene, fluoranthene, benzo [b]fluoranthene,
benzo [j]-fluoranthene, benzo [k]fluoranthene, chrysene and
triphenylene, pyrene, indeno[1,2,3- cd]pyrene, benz [a]anthracene,
dibenz [a,h]anthracene, perylene, benzo [ghi]perylene, and
dibenzo [a,e]pyrene in sediments from the Saguenay Fjord ranged from
2000 to 3800 µg/kg (dry or wet weight basis not given) (Martel et al.,
1986).
5.1.3.5 Harbour sediment
The levels of individual PAH found in harbour sediments (Table 46)
were higher than those in rivers, lakes, or oceans, concentrations
< 650 µg/g being reported.
Table 45. Polycyclic aromatic hydrocarbon concentrations (µg/kg) found in estuarine sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11]
Acenaphthene NR NR 210-670 310
Acenaphthylene NR NR <10-160
Anthracene 0.1-18 10-50 30-210 11-93 ND-49 60-860 610
Benz[a]anthracene 10-790 0.2-68 30-160 30-650 23-189 14-540 70-3200 5-140 2000
Benzo[a]fluoranthene NR NR 2-150
Benzo[a]pyrene 10-560 <0.1-52 30-210 30-760 33-313 10-540 160/7200 4-150 2300 60-6800 20-60
Benzo[b]fluoranthene 0.2-79 100-500 53-346 17-1000
Benzo[e]pyrene 10-620 103 40-180 30-550 56-323 120-8200 1-150 2500
Benzo[ghi]perylene 1-72 120-490 70-410 66-403 23-641 <70-4200 3-96 1300
Benzo[k]fluoranthene <0.1-24 20-100 33-189 14-696
Chrysene 20-1210 0.2-46 30-180 37-263 9-578 2900
Cyclopenta[cd]pyrene NR NR 300/830
Dibenz[a,h]anthracene 0.5-12 NR 8-50 2-120 550-4900 470
Fluoranthene 30-1920 1-100 50-180 80-1880 85-506 156-3700 60-7200 14-410 3900
Fluorene 15 40-120 NR 15-1500 390
Indeno[1,2,3-cd]pyrene 20-630 61 60-240 30-420 50-343 9-228 <130-9000 1800
1-Methylphenanthrene NR NR 240
Naphthalene 43 NR NR 80-2200 400
Perylene 2-52 NR NR 270/880 650 60-4200 50-60
Phenanthrene 30-1470 0.5-74 40-130 60-790 119-413 17-252 60-8700 5-300 2400
Pyrene 20-1980 0.5-102 50-220 60-1510 93-425 16-539 50-5400 4-380 4800
ND, not detected; /, single measurements; NR, not reported;
[1] Estuarine sediment of the River Elbe, Germany (Japenga at al., 1987);
[2] Continental Shelf and Gironde estuary, France (Garrigues et al., 1987);
[3] Wadden Sea, Netherlands, 1988 (Compaan & Laane, 1992);
[4] Mersey, Dee and Tamar estuaries, United Kingdom, 1984 (Readman at al., 1986);
Table 45 (continued)
[5] Humber Estuary/the Wash, United Kingdom, 1990 (Compaan & Laane, 1992);
[6] Gulf of Maine, Penobscot Bay, USA, 1982 (Johnson et al., 1985);
[7] Great Lake tributaries, USA, 1984 (Fabacher at al., 1991);
[8] Chesapeake Bay; USA, 1984-86 (Huggett et al., 1988);
[9] Puget Sound, USA (Varanasi at al., 1992);
[10] Yarra River estuary, Australia, 1976; analytical method: thin-layer chromatography with fluorescence detector (Bagg at al., 1981);
[11] Mallacoota Inlet, Australia, 1976; analytical method: thin-layer chromatography with fluorescence detector (Bagg at al., 1981)
Analysed by high-performance liquid chromatography or gas chromatography and concentration in micrograms per kilogram dry weight, unless
otherwise stated
Table 46. Polycyclic aromatic hydrocarbon concentrations (µg/kg) found in harbour sediments
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene <260-2509 50 3800
Acenaphthlene <240-2700
Anthracene <30-27 200 1800/1700 ND-507 110-17 000 120 10 900
Benz[a]anthracene <50-1991 3400/3400 310-20 000 240 8800/414 000
Benzo[a]pyrene 600-1500 400 <30-16 486 1800/2100 <70-94 984 300-19 000 340 8900/109 000
Benzo[b]fluoranthene 450 <35-17 182 ND-4103 410-15 000
Benzo[e]pyrene 930/930 120-11 000
Benzo[ghi]perylene 300 <35-1079 210-12 000
Benzo[k]fluoranthene 200 <35-1430 150-22 000
Chrysene <30-13 900 3900/3800 580-21 000
Coronene 130
Fluoranthene 2000-3600 850 <70-21 566 900/5800 <5-84 514 640 34 200/60 700
Fluorene <60-24 530 370 810-65 000 100 7000
Indeno[1,2,3-cd]pyrene 300 <50-372 180-14 000 160 157 000/715 000
1-Methylphenanthrene 2100/2300
Naphthalene <310-1564 1300/2000 <10-43 628 400 198 000
Perylene 1100/1200
Phenanthrene <50-5001 4200/4000 45-63 683 510 26 000/655 000
Pyrene <70-5179 6300/6400 196-66 831 610-40 000 740 22 800/413 000
ND, not detected; /, single measurements;
[1] Rotterdam, Netherlands (Japenga et al., 1987);
[2] Rotterdam, Netherlands, 1990 (Netherlands' Delegation, 1991);
[3] Hampton Roads, USA, 1982 (Alden & Butt, 1987);
[4] New York Bight, USA, 1979; reference weight not given (Boehm & Fiest, 1983);
[5] Boston, USA (Shiaris & Jambard-Sweet, 1986);
[6] Black Rock, USA (Rogerson, 1988);
[7] Various harbours of the Rhine, Germany, 1990 (Hamburg Environment Office, 1993);
[8] Vancouver Harbour, Canada (Environment Canada, 1994);
[9] Various harbours near steel mills, Canada (Environment Canada, 1994)
Analysed by high-performance liquid chromatography or gas chromatography and concentration in micrograms per kilogram dry weight, unless
otherwise stated
5.1.3.6 Time trends of PAH in sediment
The PAH levels in sediments taken at various depths indicate changes
and trends in the sources of PAH, e.g. from coal combustion to oil and
gas heating.
Measurements in sediments from Plöner Lake, Germany, showed that the
concentration of PAH in samples from the northern part of the lake,
which is in a populated region situated near a railway, had increased
fivefold since 1920, whereas those in the southern part had remained
constant. The increase in the northern part was attributed to an
increase in the number of PAH emitters. As most of the PAH in the
sediment originated from coal combustion, the concentrations decreased
when coal-fired railway engines were replaced in this area. The
benzo [a]pyrene levels ranged from 240 to 2400 µg/kg dry weight
(Grimmer & Böhnke, 1975). These findings are consistent with the
results of time-dependent analyses of sediments from Lake Constance
(Müller et al., 1977).
A general trend in decreasing PAH concentrations from north to south
was found in bottom sediments from the main stem of Chesapeake Bay,
USA, thought to be due to the higher human population density in the
northern region. Most of the compounds appeared to be derived from the
combustion or high-temperature pyrolysis of carbonaceous fuels rather
than from crude or refined oils. The levels of PAH remained relatively
constant over the period 1979-86 at the locations examined. Naturally
occurring PAH usually comprised less than 20% of the total; the
finding of higher proportions may reflect riverine transport of older
sediments to the area or scouring and removal of recently deposited
sediments. The benzo [a]pyrene concentrations were 12-150 µg/kg dry
weight (Huggett et al., 1988). Similar results were reported for
sediments from Buzzard's Bay, USA (Hites et al., 1977).
In a study of PAH in sediment samples from the lagoon of Venice,
Italy, a historical reconstruction of the PAH depositions in a dated
drilling core made it possible to distinguish between natural and
anthropogenic combustion and between different PAH sources, including
direct petroleum spills and sedimentary diagenesis. The predominance
of unsubstituted homologues and the relative abundance of some
individual components suggested combustion as the predominant source.
The lowest values determined in the deepest strata were assumed to be
background concentrations resulting from pre-industrial pyrolytic
sources, such as forest fires and wood burning. The benzo [a]pyrene
levels were 2.2-17 µg/kg dry weight (Pavoni et al., 1987).
5.1.4 Soil
A rough distinction can be made between local sources of pollution
(point sources) and diffuse sources. Point sources can obviously give
rise to significant local contamination of soil, whereas diffuse
sources usually affect more widespread areas, though to a lesser
extent. The main sources of PAH in soil are:
- atmospheric deposition after local emission, long-range
transport, and pollution from combustion gases emitted by
industry, power plants, domestic heating, and automotive exhausts
(Hembrock-Heger & König, 1990; König et al., 1991) and from
natural combustion like forest fires (Hites et al., 1980);
- deposition from sewage (sewage sludge and irrigation water) and
particulate waste products (compost) (Hembrock-Heger & König,
1990; König et al., 1991); and
- carbonization of plant material (Grimmer et al., 1972).
The extent of soil pollution by PAH also depends on factors such as
the cultivation and use of the soil, its porosity, its lipophilic
surface cover, and its content of humic substances (Windsor & Hites,
1978). There is a correlation between the organic content of a soil
and the PAH concentration: humus contains more PAH than a soil with
little humic content, such as sand (Grimmer et al., 1972; Matzner et
al., 1981; Grimmer, 1993).
This section addresses PAH in soil resulting mainly from industrial
sources, automobile exhaust, and other diffuse sources and gives
background values. Attribution of a study to a particular section was
difficult, as the sources of PAH emissions are often mixed.
5.1.4.1 Background values
Table 47 gives background levels of PAH in soil in rural areas. In
non-polluted areas, PAH concentrations were usually in the range 5-50
µg/kg.
5.1.4.2 Industrial sources
The PAH levels in soil resulting mainly from industrial sources are
given in Table 48.
The PAH levels were determined in soil near one American plant where
animal by-products and brewer's yeast had been processed since 1957.
The operation had subsequently expanded to include the handling of
solvents, flue dust, chips, acids, cyanides, and a wide variety of
industrial waste. Extremely high PAH concentrations were found in the
soil (Aldis et al., 1983).
PAH were detected in the soil at the sites of former coking plants in
Canada (Environment Canada, 1994). For example in Lasalle, Quebec, the
benzo [a]-pyrene levels in 1985 ranged from none detected to 1300
µg/g dry weight. The facility closed in 1976, and by 1991 the
benzo [a]pyrene concentration was below 10 000 µg/kg. In Pincher
Creek, Alberta, high levels of alkylated PAH were measured after a
refinery was dismantled. Maximum concentrations of 260 µg/g dry weight
each of fluoranthene and pyrene were measured; benzo [a]pyrene was
not detected.
Table 47. Polycyclic aromatic hydrocarbon concentrations
(µg/kg dry weight) in soil of background and rural areas
Compound [1] [2] [3] [4]
Acenaphthene 1.7 < 1-21
Acenaphthylene ND/3.0
Anthracene 1.2/4.2
Benzo[a]pyrene 15 6-12 13/22 ND-4.0
Benzo[b]fluoranthene 14/25
Benzo[ghi]perylene 49/28 ND-3.3
Benzo[k]fluoranthene 0.2-3.3
Fluoranthene 22 8-28 35/73 ND-28
Fluorene ND < 1-10
Indeno[1,2,3-cd]pyrene 0.5-4.0
Naphthalene 46 13-60 3.8/11
Phenanthrene 30 17-21 18/39 ND-76
Pyrene 20 9-25 29/42
ND, not detected; /, single measurements;
[1] Norway (depth, 0-10 cm), reference weight not given (Vogt at
al., 1987);
[2] Norway (Aamot et al., 1987);
[3] Wales, United Kingdom (depth, 5 cm) (Jones et al., 1987);
[4] Green Mountain (depth, 0-5 cm), USA (Sullivan & Mix, 1985)
Analysed by high-performance liquid chromatography or gas
chromatography
PAH profiles were found to depend on the depth of soil from which the
samples were taken. A comparison of soil samples from an area of clean
air and from an industrialized area showed that the concentrations of
PAH with lower boiling-points (fluoranthene, chrysene, and pyrene)
decreased with depth, whereas those of PAH with higher boiling-points
(indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene) were relatively greater. The
opposite would have been expected on the basis of the solubility of
these PAH (Jacob et al., 1993b).
Table 48. Polycyclic aromatic hydrocarbon concentrations
(µg/kg dry weight) in soil near industrial emissions
Compound [1] [2] [3] [4]
Acenaphthene 54 5 090 000
Anthracene 144 000 1 600 70
Benz[a]anthracene 79 000 200 000
Benzo[a]pyrene 38 000 321 100
Benzo[b]fluoranthene 200
Benzo[e]pyrene 35 000
Benzo[ghi]perylene 100
Benzo[k]fluoranthene 130 000 100
Chrysene 1 210 000
Fluoranthene 340 000 573 234 000 200
Fluorene 80 8 600 000
Indeno[1,2,3-cd]pyrene 100
Naphthalene 48 5 200 2.4
Perylene 12 000
Phenanthrene 506 000 353 20 000 000 40
Pyrene 208 000 459 16 000 000 100
[1] Near coal gasification plant, Netherlands, concentrations in
µg/kg wet weight (de Leeuw et al., 1986);
[2] Norway, reference weight not given (Vogt et al., 1987);
[3] Near processing plant, USA, 1982; maximum (Aldis et al.,
1983); values, analytical method, and reference weight not
given;
[4] Area of an abandoned coal gasification plant, USA; reference
weight not given (Dong & Greenberg, 1988)
Analysed by high-performance liquid chromatography or gas
chromatography
5.1.4.3 Diffuse sources
(a) Motor vehicle and aircraft exhaust
The concentrations of individual PAH in soil resulting mainly from
motor vehicle exhaust (Table 49) usually range between 1 and 2000
µg/kg. The PAH content of soil often decreased with increasing depth
(Matzner et al., 1981; Wang & Meresz, 1982; Butler et al., 1984). Near
a motorway in the Midlands, United Kingdom, PAH were determined at
depths of 0-4 cm and 4-8 cm. Extremely high concentrations were found
in the surface layer, but soil at a depth of 4-8 cm was two times less
contaminated (Butler et al., 1984). The pollution may have been a
result of airborne transport or of microbial or photochemical
degradation (Hembrock-Heger & König, 1990). Comparably high levels of
PAH were found at Reykjavik Airport, Iceland (Grimmer et al., 1972;
see Table 49).
Table 49. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry
weight) in soil of areas predominantly polluted by vehicle exhaust
Compound [1] [2] [3] [4] [5]
Acenaphthylene 71
Anthracene 0.2 13 11
Anthanthrene 0.4 149
Benz[a]anthracene 2.3 430 169-3297 13
Benzo[a]pyrene 3.2 785 165-3196 38 24
Benzo[b]fluoranthene 41
Benzo[e]pyrene 4.5 870 159-2293 29
Benzo[ghi]perylene 7.1 1450 168 46
Benzo[k]fluoranthene 78
Chrysene 4.1 436 251-2703 39
Coronene 1.8 410 40-322 37
Dibenz[a,h]anthracene 1.1 351 2
Fluoranthene 6.5 1290 200-3703 91 37
Fluorene 5
Indeno[1,2,3-cd]pyrene 36
Naphthalene 3
Perylene 0.6 157 6
Phenanthrene 17 1735 92 45
Pyrene 3.5 1610 145-4515 72 61
[1] Iceland (depth, 20 cm; reference weight not given) (Grimmer et
al., 1972);
[2] Reykjavik Airport, Iceland (surface soil; reference weight not
given) (Grimmer et al., 1972);
[3] United Kingdom, surface soil near motorway; analytical method,
adsorbance measurement, reference weight not given) (Butler et al.,
1984);
[4] United Kingdom (urban soil; depth, 5 cm) (Jones et al., 1987);
[5] Brisbane, Australia (Pathirana et al., 1994)
Analysed by high-performance liquid chromatography or gas chromatography
(b) Other diffuse sources
Table 50 gives the levels of PAH from unpecified sources in soil.
Benzo [a]pyrene levels of 800 µg/kg were found in humus, 100-800
µg/kg in garden soil, 35 µg/kg in forest soil, and 0.8-10 µg/kg in
sand (Fritz, 1971).
Table 50. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry weight) in soil from areas polluted by various diffuse
sources
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthylene NR NR 3.8
Anthracene NR NR ND-1.4 22-70
Anthanthrene 27 0.50 ND 10-38
Benz[a]anthracene 80 0.60 ND 47-101
Benzo[a]pyrene 273 10/6.2 24 0.8/357 116 1.50 ND-1.4 157 54-108
Benzo[b]fluoranthene 49-97
Benzo[e]pyrene 23 20/22 50 143 3.10 ND-5.0 47-116
Benzo[ghi]perylene 106 15/33 32 0.9-339 98 3.0 ND 64-147
Benzo[k]fluoranthene 31-62
Chrysene NR NR ND-2.1 50-128
Coronene 49 0.70 ND-1.7 32-66
Dibenz[a,h]anthracene 266 8.4/22 44 44 0.60 ND-1.4 11-29
Fluoranthene 2.5-444 254 2.1 ND-2.1 83 73-170 0.3-75
Fluorene NR NR 14
Indeno[1,2,3-cd]pyrene 30 6.4/7.9 21.4 1.2-545 127 3.3 32-80
Naphthalene NR NR 58
Perylene 3537 4.0/8.5 5.0 NR NR ND 19-71
Phenanthrene NR NIR ND-18 78 31-106
Pyrene 150 0.80 ND-0.5 90 80-183 0.1-64
ND, not detected; /, single measurements; NR, not reported;
[1] Germany, birch tree peat (Ellwardt, 1976);
[2] Germany, black and white peat (Ellwardt, 1976);
[3] Germany, sandy loam (Ellwardt, 1976);
[4] Soiling mountain, Germany; depth, 0-15 cm; analytical method, high-performance thin-layer chromatography; reference
weight not given (Matzner et al., 1981);
[5] Germany, forest, brown soil, surface layer (Bachmann et al., 1994);
Table 50 (continued)
[6) Germany, forest, brown soil; depth, 0-2 cm (Bachmann et al., 1994);
[7] Iceland; depth, 3-30 cm; reference weight not given (Grimmer et al., 1972);
[8] Norway, bog soil; depth, 0-10 cm; reference weight not given (Vogt et al., 1987);
[9] Toronto, Canada, virgin and cultivated soil; reference weight not given (Wang & Meresz 1982);
[10] Nova Scotia, Canada (Windsor & Hites, 1978)
Analysed by high-performance liquid chromatography or gas chromatography
The PAH concentrations of cultivated soil were slightly higher than
those in virgin soil. For example, the benzo [a]pyrene concentrations
were 65-87 µg/kg in cultivated soil and 54 µg/kg in virgin soil (Wang
& Meresz, 1982). The PAH levels in cultivated soils from German
gardens at a maximum depth of 25 cm decreased from industrial areas
(fluoranthene, 590-2500 µg/kg; benzo [a]pyrene, 220-1400 µg/kg) to
rural areas (fluoranthene, 100-390 µg/kg; benzo [a]pyrene, 30-150
µg/kg) and with soil depth (benzo [a]pyrene concentration, 280-3000
µg/kg at 0-30 cm, 60-4600 µg/kg at 30-60 cm, and 10-7900 µg/kg at
60-100 cm). High PAH concentrations were found at a depth of 100 cm in
soil from an old industrial area and from an area filled with
contaminated soil. In compost soil, benzo [a]pyrene was present at a
concentration of 0.10-2.5 mg/kg in 1986 and 0.02-1.3 mg/kg in 1987
(Crössmann & Wüstemann, 1992).
Fluoranthene and pyrene were measured in soil samples, from a wooded
area in Maine, a marshy area of South Carolina, a grassy, uncultivated
meadow in Nebraska, a mossy area with pine needles in Wyoming, and a
sandy desert area in Nevada, USA, and in dark brown, red clay, and
light brown loam from Samoa. The highest levels of individual PAH (up
to 80 µg/kg) were found in the soil from the wooded area in Maine. The
levels in the marshy and grassy soils of South Carolina and Nebraska
were 8.4-26 µg/kg. The other soils sampled contained fluoranthene and
pyrene at levels < 1 µg/kg (Hites et al., 1980).
In Iceland, the concentrations of individual PAH in lava soil at
depths of 3 and 25 cm were near the limit of detection. Similar levels
were found in vegetable soil at depths of 10 and 30 cm, but the
concentrations at 10 cm were twice as high as those at 30 cm (Grimmer
et al., 1972).
Higher levels of PAH were found in the humus layer of spruce and beech
forest ecosystems than in the mineral soil, but the spruce stand
contained and stored more PAH than the beech stand (Matzner et al.,
1981). Forest soils in Germany contain many PAH in large amounts;
Table 48 shows the PAH concentrations in one forest brown soil. The
first humic layer of the soil had the highest PAH concentration, and
the level decreased with depth to below the limit of detection in
layers below 10 cm (Bachmann et al., 1994).
The concentrations of PAH were no higher in soil that had been treated
with sewage sludge than in untreated soil, indicating that sewage
sludge is not a major source of PAH (Hembrock-Heger & König, 1990;
König et al., 1991).
5.1.4.4 Time trends of PAH in soil
Soil samples collected from Rothamsted Experimental Station in
southeast England over a period of about 140 years (1846-1980) were
analysed for PAH (Jones et al., 1987). All of the soils were collected
from the plough layer (0-3 cm) of an experimental plot for which
atmospheric deposition was the only source of PAH. The total PAH
burden of the plough layer had increased by approximately fivefold
since 1846. The concentrations of most of the individual PAH
(anthracene, anthanthrene, fluorene, benzo [a]pyrene,
benzo [e]pyrene, fluoranthene, benzo [b]fluoranthene,
benzo [k]fluoranthene, chrysene, pyrene, indeno[1,2,3- cd]pyrene,
phenanthrene, and benz [a]-anthracene) had increased by about one
order of magnitude. For example, the benzo [a]pyrene level was 18
µg/kg in 1846 and 130 µg/kg in 1980, and the anthracene level was 3.6
µg/kg in 1846 and 13 µg/kg in 1980. The levels of coronene,
acenaphthylene, acenaphthene, perylene, and benzo [ghi]perylene
remained approximately the same, whereas the naphthalene content
decreased from 39 µg/kg in 1846 to 23 µg/kg in 1980.
5.1.5 Food
In the past, benzo [a]pyrene was the most common PAH determined in
foods and was used as an indicator of the presence of PAH (Tilgner,
1968). The earliest measurements of PAH, in particular of
benzo [a]pyrene, date to 1954; these were reviewed by Lo & Sandi
(1978) and by Howard & Fazio (1980). The levels of individual PAH in
foods in more recent studies are summarized in Tables 51-56.
5.1.5.1 Meat and meat products
The concentrations of individual PAH found in meat are shown in Table
51.
In a comparison of home and commercially smoked meats in Iceland, very
little benzo [a]pyrene was detected in smoked sausage and mutton, but
considerable amounts of benzo [a]pyrene and other PAH were found in
home-smoked mutton and lamb, independently of whether they were
covered with cellophane or muslin. About 60-75% of the total
benzo [a]pyrene was detected in the superficial (outer) layers of the
meat (Thorsteinsson, 1969). These findings are in agreement with those
of Rhee & Bratzler (1970) for smoked bologna and bacon and with those
of Tilgner (1958) and Gorelova et al. (1960).
The amount of PAH formed during roasting, baking, and frying depends
markedly on the conditions (Lijinsky & Shubik, 1964). In an
investigation of the effect of the method of cooking meat, including
broiling (grilling) on electric or gas heat, charcoal broiling, and
broiling over charcoal in a no-drip pan, it was shown that the
formation of PAH can be minimized by avoiding contact of the food with
flames, cooking meat at lower temperatures for a longer time, and
using meat with minimal fat (Lijinsky & Ross, 1967). The most likely
source of PAH is melted fat that drips onto the heat and is pyrolysed
(Lijinsky & Shubik, 1965). The exact chemical mechanism for the
formation of PAH is unknown.
Table 51. Polycyclic aromatic hydrocarbon concentrations (µg/kg fresh weight) in meat and meat products
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13]
Anthracene 0.9 20-31a ND-2 0.5-133
Anthanthrene 5-8 ND ND-66.5
Benz[a]anthracene 0.5 0.5 0.02-0.64 0.03 Trace-0.33a 0.02-0.03 O.04-0.38 0.04-0.13 0.05 16-37 ND-1 0.2-144
Benzo[a]fluorene 17-28 1-2 ND-174
Benzo[a]pyrene 0.1 0.6 0-02-0.45 0.02 0.05 O.01-0.14 0.01-0.04 0.04-0.26 0.03-0.26 0.05 26-42 ND-1 ND-212
Benzo[b]fluoranthene 0.3 1.0 0.30 0.04 16-24 ND-92.3
Benzo[b]fluorene 10-12 2-7 ND-71.9
Benzo[c]phenathrene 1.4 0.03-0.36 0.06 Trace-0.18 0.03-0.04 0.05-0.21 0.05-0.10
Benzo[e]pyrene 0.03 6-9 ND-2 ND-80.9
Benzo[ghi]perylene 0.2 0.6 0.03-0.31 0.03 3.75 Trace-0.12 0.03-0.04 0.06-0.27 0.05-0.19 0.05 10-17 ND-2 ND-153
Benzo[j]fluoranthene 5-7
Benzo[k]fluoranthene 0.2 0.2 0.05 0.01 8-14 ND-172b
Chrysene 0.6 0.15 0.3-140a
Dibenz[a,h]anthracene 0.01 ND-8.8
Fluoranthene 0.9 1.1 7.8 0.48 57-103 6-9 1.1-376
Indeno[1,2,3-cd]pyrene 0.2 0.7 0.04-0.38 0.03 2.5 Trace-0.11 0.01-0.03 0.04-1.40 0.05/0.1 15-22 ND-5 ND-171
1-Methylphenanthrene 4-5 ND-3 0.5-57.6
Perylene ND-3 ND ND-27.9
Phenanthrene 3.0 22-64 10-16 3.5-618
Pyrene 0.55 38-63 5-7 1.2-452
ND, not detected; /, single measurements;
[1] Poultry and eggs, Netherlands, reference weight not given (de Vos et al., 1990);
[2] Meat and meat products, Netherlands, reference weight not given (de Vos et al., 1990);
[3] Smoked beef, Netherlands, reference weight not given (de Vos et al., 1990);
[4] Unsmoked beef, Netherlands (de Vos et al., 1990);
[5] Bacon, United Kingdom (Crosby et al., 1981);
[6] Smoked meat, United Kingdom (McGill et al., 1982);
[7] Unsmoked meat, United Kingdom (McGill et al., 1982);
Table 51 (continued)
[8] Smoked sausages, United Kingdom (McGill et al., 1982);
[9] Unsmoked sausages, United Kingdom (McGill et al., 1982);
[10] Meat, United Kingdom, reference weight not given (Dennis et al., 1983);
[11] Mesquite wood cooked pattie (70-90 % lean), USA, reference weight not given (Maga, 1986);
[12] Hardwood charcoal cooked pattie (70-90% lean), USA, reference weight not given (Maga, 1986);
[13] Grilled sausages, Sweden, reference weight not given (Larsson et al., 1983)
High-performance liquid chromatography or gas chromatography
a In sum with triphenylene
b In sum with benzo[j]fluoranthene
In one study, the highest concentration of benzo [a]pyrene (130
µg/kg) in cooked meat was found in fatty beef, and the concentration
appeared to be proportional to the fat content (Doremire et al.,
1979). Levels of about 50 µg/kg were detected in a charcoal-grilled
T-bone steak (Lijinsky & Ross, 1967), in heavily smoked ham (Toth &
Blaas, 1972), and in various other cooked meats (Potthast, 1980).
Usually, benzo [a]pyrene levels up to 0.5 µg/kg have been found
(Prinsen & Kennedy, 1977).
In meat, poultry, and fish in Canada, benzo [k]fluoranthene was
detected at concentrations up to 0.30 µg/kg and benzo [a]pyrene up to
1.1 µg/kg (Environment Canada, 1994).
Benzo [a]pyrene was found in some German meat products in 1994 at
concentrations generally < 1 µg/kg . The highest concentration, 9.2
µg/kg, was found in a ham from the Black Forest (State Chemical
Analysis Institute, Freiburg, 1995).
5.1.5.2 Fish and other marine foods
Benzo [a]pyrene was found at levels ranging from none detected to 18
µg/kg in smoked fish. The differences were probably due to factors
such as the type of smoke generator, the temperature of combustion,
and the degree of smoking (Draudt, 1963). The highest concentration of
benzo [a]pyrene (130 µg/kg) in seafood was found in mussels from the
Bay of Naples (Bourcart & Mallet, 1965), and a level of about 60 µg/kg
was detected in smoked eel skin. Most of the fish analysed contained
0.1-1.5 µg/kg (Steinig, 1976). Benzo [a]pyrene was also detected at
levels up to 3.3 µg/kg in 21 samples of smoked fish, oysters, and
mussels of various origins (Prinsen & Kennedy, 1977). The levels of
individual PAH are summarized in Table 52.
5.1.5.3 Dairy products: cheese, butter, cream, milk, and related
products
PAH were detected in considerable amounts in smoked cheese (Prinsen &
Kennedy, 1977; Lintas et al., 1979; McGill et al., 1982; Osborne &
Crosby, 1987a). The benzo [a]pyrene content of a smoked Italian
Provola cheese was 1.3 µg/kg (Lintas et al., 1979). Concentrations of
0.01-5.6 µg/kg fresh weight fluoranthene, benz [a]anthracene,
benzo [c]phenanthrene, benzo [a]pyrene, benzo [ghi]perylene, and
indeno[1,2,3- cd]pyrene were found in a smoked cheese sample and
0.01-0.06 µg/kg in unsmoked cheese from the United Kingdom (McGill et
al., 1982). In other unsmoked cheese samples from the United Kingdom,
the individual PAH levels were between < 0.01 µg/kg for
dibenz [a,h]anthracene and 1.5 µg/kg for pyrene. Similar
concentrations of PAH were found in British butter and cream samples
(Dennis et al., 1991).
Table 52. Polycyclic aromatic hydrocarbon concentrations (µg/kg) found in fish and marine foods
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene
Anthracene 0.9 1.3-64.3 1.4-49.6
Benz[a]anthracene 1.3 ND-11.2 ND-6.3 ND-86 Trace-0.09
Benzo[a]pyrene 1.4 ND-5.5 ND-5.4 0.10 ND-18 Trace-0.35
Benzo[b]fluoranthene 2.0 ND-3.9 ND-3.6 0.35
Benzo[c]phenanthrene ND-15 0.01-0.09
Benzo[e]pyrene ND-2.8 ND-3.0
Benzo[ghi]perylene 0.9 ND-2.8 ND-1.8 4.3 ND-25 Trace-0.39
Benzo[k]fluoranthene 0.7 ND-6.7a ND-5.1a 0.10
Chrysene 2.9 ND-13.0b ND-9.4b
Dibenz[a,h]anthracene
Fluoranthene 1.8 1.4-79.9 1.7-48.4 2.4
Fluorene
Naphthalene
Indeno[1,2,3-cd]pyrene 1.6 ND-7.1 ND-2.4 2.7 ND-37 ND-0.33
Perylene ND-1.2 ND-1.0
Phenanthrene 3.5 5-330 10.4-277
Pyrene 1.3-67.8 2.1-38.4
ND, not detected; NR, not reported;
[1] Fish, Netherlands (de Vos et al., 1990);
[2] Herring, whitefish, mackerel, eel, salmon, salmon trout, various fillets; all smoked;
Sweden (Larsson, 1982);
[3] Fish and fish products: sprats, herring, rainbow trout, caviar, herring paste, salmon
paste; all smoked or canned; Sweden (Larsson, 1982);
[4] Kippers, United Kingdom (Crosby et al., 1981);
[5] Fish (smoked), United Kingdom, concentration in µg/kg wet weight (McGill et al., 1982);
[6] Fish, unsmoked, United Kingdom, concentration in µg/kg wet weight (McGill et al., 1982)
Table 52 (continued)
Compound [8] [9] [10] [11] [12] [13] [14]
Acenaphthene < 2-5.13 2.22-22.3
Anthracene < 2-78.4 ND-5.88 ND-0.6 ND-1.9 < 0.05
Benz[a]anthracene 0.14 1.6-7.5 < 2 0.14-5.31 0.8-3.0 0.8-20.9
Benzo[a]pyrene 0.13 t-4.5 < 2-7.63 ND-5.33 0.4-1.0 0.2-12.2 < 0.004
Benzo[b]fluoranthene 0.13 0.13-5.77 4.5-12.2c 1.2-24.3c
Benzo[e]pyrene 0.12 2.4-6.3 0.7-7.6
Benzo[ghi]perylene 0.12 0.17-30.9 0.4-0.8 0.3-5.7
Benzo[k]fluoranthene 0.04 NR NR < 0.002
Chrysene 0.65 < 2 ND-15.9 3.2-8.8b 3.9-30.8b < 0.03
Dibenz[a,h]anthracene 0.03 0.21-39.3 0.1-0.2d <0.1-0.5d
Fluoranthene 0.1 < 2-123.5 ND-32.7 5.1-17.5 4.5-18.7
Fluorene < 2-18.5 ND-65.7
Napthalene < 2-67.4 2.06-156.1
Indeno[1,2,3-cd]pyrene 0.28-28.6 0.3-0.6 0.2-6.4
Perylene 0.2-2.7 0.1-3.1 < 0.05
Phenanthrene < 2-100.8 5.84-87.2 2.1-4.2 1.9-19.6
Pyrene 0.79 < 2-144.9 ND-68.0 3.1-12.4 2.6-11.2 < 0.03
ND, not detected; NR, not reported;
[8] Fish, United Kingdom (Dennis et al., 1983);
[9] Fish, Nigeria (Emerole et al., 1982);
[10] Fresh fish from the Arabian Gulf: andag, sheim, gato, sheiry, faskar, chaniedah; after an oil spill
(Al-Yakoob et al., 1993);
[11] Fresh fish and shrimps, Kuwait, after Gulf war (Saeed et al., 1995);
[12] Fresh oysters, various origins, concentration in µg/kg wet weight (Speer et al., 1990);
[13] Canned or smoked oysters and mussels, various origins, concentration in µg/kg wet weight (Speer at
al., 1990);
[14] Clam, Australia; analytical method: fluorescence spectrophotometry: concentration in µg/kg wet weight
(Smith et al., 1987)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given,
unless otherwise stated
a In sum with benzo[j]fluoranthene
b In sum with triphenylene
c Benzo[b+k]fluoranthenes
d Dibenz[a,h+a,c]anthracenes
In Finnish butter samples, most of the measured PAH (phenanthrene,
1-methylphenanthrene, fluoranthene, pyrene, benzo [a]fluorene,
benzo [ghi]-fluoranthene, cyclopenta [cd]pyrene, perylene,
anthanthrene, benzo [ghi]pyrene, and indeno[1,2,3- cd]pyrene)
occurred at levels < 0.1 µg/kg. The maximum level was 1.4 µg/kg
fluoranthene (Hopia et al., 1986).
The concentrations of fluoranthene, pyrene, benz [a]anthracene,
chrysene, benzo [b]fluoranthene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [e]pyrene, perylene, benzo [ghi]pyrene,
indeno[1,2,3- cd]pyrene, and dibenz [a,h]anthracene were measured in
milk, milk powder, and other dairy products in Canada (Lawrence &
Weber, 1984), the Netherlands (de Vos et al., 1990), and the United
Kingdom (Dennis et al., 1983, 1991). The concentrations ranged from
< 0.01 µg/kg for benzo [k]fluoranthene and dibenz [a,h]anthracene
to 2.7 µg/kg for pyrene.
Canadian infant formula was found to contain 8.0 µg/kg fluoranthene,
4.8 µg/kg pyrene, 1.7 µg/kg benz [a]anthracene, 0.7 µg/kg
benzo [b]fluoranthene, 1.2 µg/kg benzo [a]pyrene, 0.6 µg/kg
perylene, 0.3 µg/kg anthanthrene, and 1.2 µg/kg
indeno[1,2,3- cd]pyrene (Lawrence & Weber, 1984). Slightly lower
levels were detected in British samples in 1982-83 (Dennis et al.,
1991).
PAH were detected at levels of 0.003-0.03 µg/kg in human milk
(Heeschen, 1985).
5.1.5.4 Vegetables
The levels of PAH found in vegetables in recent studies are listed in
Table 53.
Fluoranthene, but no other PAH, was reported to have been found in
unspecified fruits and vegetables in Canada at levels of none detected
to 1.8 µg/kg (Environment Canada, 1994). Kale was found to contain
high concentrations of fluoranthene (120 µg/kg), pyrene (70 µg/kg),
chrysene (62 µg/kg), and benz [a]anthracene (15 µg/kg), and PAH
concentrations up to 7 µg/kg were determined in other vegetables
(Vaessen et al., 1984). The differences in PAH content have been
attributed to variations in the ratio of surface area:weight, in
location (rural or industrialized), and in growing season. Washing (at
20°C) vegetables contaminated by vehicle exhausts did not reduce the
PAH contamination (Grimmer & Hildebrandt, 1965).
In a comparison of the PAH contents of terrestrial plants grown in
chambers containing 'clean air' and in the open field, the
contamination was shown to be due almost exclusively to airborne PAH,
which were not synthesized by the plants (Grimmer & Düvel, 1970) .
Table 53. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in vegetables
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Anthracene 0.09-0.19 <0.1-0.3
Benz[a]anthracene 15 0.7-4.6 0.05-3.17 0.05-3.2 0.4 0.3
Benzo[a]fluoranthene 0.08-2.6
Benzo[a]pyrene 4.2 0.05-1.4 5.6 0.3-6.2 ND-1.42 0.05-3.0 0.2
Benzo[b]fluoranthene 6.1 0.5-7.3 0.9-3.2 0.2
Benzo[b]fluorene 0.11-2.8
Benzo[c]phenanthrene 9.2 0.05-1.5
Benzo[e]pyrene 7.9 0.07-2.2 0.5-6.7 0.2
Benzo[ghi]perylene 7.7 0.13-2.1 10 0.5-10.8 ND-1.39 3.7-10 0.1
Benzo[k]fluoranthene 3.7 ND-17 0.1
Chrysene 62 2.4-4.0 0.8 0.5
Dibenz[a,h]anthracene 1.0 0.04
Dibenzo[a,h]pyrene 0.7
Dibenzo[a,i]pyrene 0.3
Fluoranthene 117 1.1-28 28 2.8-9.1 9.2-17
Indeno[1,2,3-cd]pyrene 7.9 0.14-0.72 2.4 0.3-8.3 ND-1.92 1.8-4.2
1-Methylphenanthrene 0.10-2.1 0.7-1.6
Perylene 0,05-0.75 <0.1-1.7
Phenanthrene 0.47-12 1.8-7.5
Pyrene 70 0.9-18 3.4-10.4
ND, not detected;
[1] Kale, Netherlands (Vaessen et al., 1984);
[2] Lettuce, Finland, concentration in µg/kg fresh weight (Wickstrom et al., 1986);
[3] Lettuce, Germany, from an industrial area (Ministry of Environment, 1994);
[4] Lettuce, Sweden, concentration in µg/kg fresh weight (Larsson & Sahlberg, 1982);
[5] Lettuce and cabbage, United Kingdom, concentration in µg/kg fresh weight (McGill et al., 1982);
[6] Lettuce, India (Lenin, 1994);
[7] Potatoes, Netherlands (de Vos et al., 1990);
[8] Tomatoes, Netherlands (Vaessen et al., 1984)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight
not given, unless otherwise stated
The benzo [a]pyrene levels in potatoes in eastern Germany were
0.2-400 µg/kg. The highest concentrations were detected in the peel of
potatoes grown in soil containing 400 µg/kg benzo [a]pyrene, 750
µg/kg benzo [e]pyrene, 1000 µg/kg benz [a]anthracene, 600 µg/kg
chrysene, 160 µg/kg dibenz [a,h]anthracene, 1000 µg/kg
benzo [b]fluoranthene, 2300 µg/kg phenanthrene, 1800 µg/kg pyrene,
220 µg/kg benzo [k]fluoranthene, 500 µg/kg indeno[1,2,3- cd]pyrene,
2500 µg/kg fluoranthene, and 120 µg/kg anthracene (Fritz, 1971, 1972,
1983).
High concentrations of PAH were detected in lettuce grown close to a
highway; the levels of individual PAH decreased with distance from the
road. Washing the vegetables reduced their content of
high-molecular-mass PAH but not of phenanthrene (Larsson & Sahlberg,
1982). In another study, the profiles of PAH in lettuce were similar
to those in ambient air, indicating that deposition of airborne
particles was the main source of contamination (Wickström et al.,
1986).
PAH concentrations were determined in fenugreek, spinach beet,
spinach, amaranthus, cabbage, onion, lettuce, radish, tomato, and
wheat grown on soil that had been treated with sewage sludge. The
levels of individual PAH in lettuce leaves (Table 53) were one to two
orders of magnitude lower than those in the sewage sludge and the soil
on which the lettuce was grown (Lenin, 1994).
The PAH levels in carrots and beans grown near a German coking plant
were below 0.5 µg/kg wet weight. The levels of fluoranthene were 1.6-
1.7 µg/kg and those of pyrene 1.0-1.1 µg/kg. Vegetables with large,
rough leaf surfaces, such as spinach and lettuce, had PAH levels that
were 10 times higher, perhaps due to deposition from ambient air
(Crössmann & Wüstemann, 1992).
5.1.5.5 Fruits and confectionery (Table 54)
Higher concentrations of PAH were found in fresh fruit than in canned
fruit or juice, and especially high concentrations of phenanthrene (17
µg/kg) and chrysene (69 µg/kg) were found in nuts (de Vos et al.,
1990). In 1982-83 in the United Kingdom, high PAH levels were found in
samples of puddings, biscuits, and cakes, which were probably derived
from vegetable oil. Similar concentrations of individual PAH were
detected in samples of British chocolate (Dennis et al., 1991).
5.1.5.6 Cereals and dried foods
Wheat, corn, oats, and barley grown in areas near industries contained
higher levels of PAH than crops from more remote areas. Drying with
combustion gases increased the contamination by three- to 10-fold; use
of coke as fuel resulted in much less contamination than oil (Bolling,
1964). Cereals contained benzo [a]pyrene at levels of 0.2-4.1 µg/kg
(Table 55). The highest concentrations, up to 160 µg/kg, were found in
smoked cereals (Tuominen et al., 1988).
Table 54. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in fruits and confectionery
Compound [1] [2] [3] [4] [5] [6] [7]
Anthracene 0.4 0.3
Benz[a]anthracene 0.5 0.11 4.2 0.2 4.2 0.08-2.73
Benzo[a]pyrene 0.1 0.07 0.2 0.3 0.4 0.04-2.20
Benzo[b]fluoranthene 0.1 0.1 0.06 0.4 0.4 3.5 0.03-1.27
Benzo[c]phenanthrene 0.5 12 2.2
Benzo[e]pyrene 0.03 0.08-2.92
Benzo[ghi]fluoranthene 0.9 0.9
Benzo[ghi]perylene 0.1 0.06 0.4 1.1 0.2 0.11-2.55
Benzo[k]fluoranthene 0.1 0.1 0.02 0.1 0.1 0.5 0.04-1.36
Chrysene 0.5 0.23 69 0.5 36 0.09-2.84
Dibenzo[a,h]pyrene 0.01 < 0.01-0.23
Fluoranthene 3.6 1.0 0.93 3.0 1.9 2.3 0.52-3.57
Indeno[1,2,3-cd]pyrene 0.4 0.4 0.2 0.10-3.18
Phenanthrene 7.8 17 2.9 3.2
Pyrene 0.83 0.59-2.37
[1] Fresh fruit, Netherlands (de Vos et al., 19900:
[2] Canned fruit and juices, Netherlands (de Vos et al., 1990);
[3] Fruit and sugar, United Kingdom (Dennis et al., 1983);
[4] Nuts, Netherlands (de Vos et al., 1990);
[5] Biscuits, Netherlands (de Vos et al., 1990);
[6] Sugar and sweets, Netherlands (de Vos et al., 1990);
[7] Puddings, biscuits and cakes, United Kingdom (Dennis et al., 1991)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not
given
Table 55. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in cereals and dried foods
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 1.6 NR NR 0.7 2.3
Anthracene 9.4 NR NR 1.3 19/150
Anthanthrene NR NR
Benz[a]anthracene 0.1-42 11 0.69 0.11-0.21 2.5/3.7 0.6 0.3 <0.1/0.2 6.3/110
Benzo[a]pyrene ND-0.3 5.4 0.40 0.10-0.12 0.5/0.8 0.2 0.3/0.4 0.6/160
Benza[b]fluoranthene 0.1-0.5 0.28 0.07-0.09 0.9 0.2 0.1
Benzo[e]pyrene 0.42 0.06-0.17 0.1/0.7
Benzo[ghi]perylene 0.54 0.13-120
Benzo[k]fluoranthene 0.50 0.1-0.14
Dibenz[a,h]anthracene ND-1.2 0.06 0.01-0.02 3.6
Fluoranthene 0.8-26 130 0.71 0.58-0.69 18/28 1.9 1.4 1.5/13 70/790
Fluorene 5.9 NR NR 2.3/2.7 6.4/87
Indeno[1,2,3-cd]pyrene ND-0.4 1.08 0.24-0.33 1.4 0.2
Perylene 0.1-0.4 0.7 NR NR 94 NR NR 14/2983/1
Pyrene 1.1-48 47 0.10 0.38-0.62 20/21 2.2 3.4 1.6/5.4 60/630
ND, not detected; /, single measurements; NR, not reported;
[1] Barley malt, Canada (Lawrence & Weber, 1984);
[2] Bran, Finland (Tuominen et al., 1988);
[3] Bran, United Kingdom (Dennis et al., 1991);
[4] High bran and granary bread, United Kingdom (Dennis et al., 1991);
[5] Bran, Canada (Lawrence & Weber, 1984);
[6] Corn bran, Canada (Lawrence & Weber, 1984);
[7] Flaked milled corn, Canada (Lawrence & Weber, 1984);
[8] Oats, Finland (Tuominen et al., 1988);
[9] Smoked oats, barley and beans, Finland (Tuominen et al., 1988)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given
Table 55 (contd)
Compound [10] [11] [12] [13] [14] [15] [16] [17]
Acenaphthene 0.6/0.7 NR NR 0.6
Anthracene 0.5 NR NR
Anthanthrene 0.05-0.08 NR NR
Benz[a]anthracene 0.4 ND-0.2 0.14-0.25 <0.1/<O.1 0.3-0.8 0.06-0.15 0.33-1.26 0.1
Benzo[a]pyrene < 0.1 0.17-0.30 0.2/0.4 0.1 0.03-0.05 0.15-0.34
Benzo[b]fluoranthene 0.1/0.2 0.02-0.05 0.1-0.27
Benzo[c]phenanthrene NR NR
Benzo[e]pyrene ND-0.1 0.16-0.29 0.2/0.4 0.06-0.16 0.28-0.81
Benzo[ghi]fluoranthene 0.05 NR NR
Benzo[ghi]perylene 0.20-0.35 0.06-0.08 0.15-0.28
Benzo[k]fluoranthene ND-0.2a 0.02-0.07 0.15-0.31
Chrysene 0.3-0.7b NR NR
Coronene 0.03-0.06 NR NR
Cyclopenta[cd]pyrene 0.07-0.13 NR NR
Dibenz[a,h]anthracene 0.03-0.05 3.0 < 0.01 0.01-0.02
Fluoranthene 2.9 0.9-1.3 0.32-0.57 1.8/3.0 1.5-7.4 0.22-0.60 0.82-6.17 3.8
Fluorene 1.3/1.7 NR NR 2.0
Indeno[1,2,3-cd]pyrene 0.16-0.29 3.0 0.08-0.15 0.30-0.65
1-Methylphenanthrene 0.3
Perylene 0.1 < 0.1/0.1 0.1-0.3 NR NR
Phenanthrene 1.3-1.5 9.9/10 NR NR 14
Pyrene 2.8 1.6-2.3 0.22-0.39 1.6/5.5 2.6-8.5 0.26-1.18 1.41-10.86 2.6
ND, not detected; /, single measurements; NR, not reported;
[10] Whole grain oats, Canada (Lawrence & Weber, 1984);
[11] Whole-grain rye, Sweden, concentration in µg/kg fresh weight (Larsson, 1982);
[12] Wheat grain, United Kingdom (Jones et al., 1989b);
[13] Wheat, Finland (Tuominen et al., 1988);
[14] Wheat, Canada (Lawrence & Weber, 1984);
[15] Breakfast cereal, United Kingdom (Dennis et al., 1991);
[16] Bran-enriched cereals, United Kingdom (Dennis et al., 1991);
[17] Bolted rye flour, Finland (Tuominen et al., 1988)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given, unless
otherwise specified
a Benzofluoranthenes
b In sum with triphenylene
Table 55 (contd)
Compound [18] [19] [20] [21] [22] [23] [24] [25]
Acenaphthene NR NR NR
Anthracene NR NR NR
Anthanthrene NR NR NR
Benz[a]anthracene 0.04-0.19 0.64 0.8 0.10-0.14 0.5 0.1 0.4
Benzo[a]pyrene 0.02-0.09 0.43 0.8 0.05-0.15 0.2 0.3 0.8
Benzo[b]fluoranthene 0.02-0.06 0.25 1.2 0.04-0.06 0.5 0.6 1.0 0.05
Benzo[c]phenanthrene NR NR NR 0.7
Benzo[e]pyrene 0.10-0.23 0.35 0.06-0.12
Benzo[ghi]fluoranthene NR NR NR
Benzo[ghi]perylene 0.06-0.19 0.39 0.5 0.04-0.21 0.5 0.9 0.6
Benzo[k]fluoranthene 0.03-0.08 0.35 0.6 0.04-0.1 0.1 0.3 0.5 0.08
Chrysene NR NR 1.0 NR 2.0 1.3 0.4
Coronene NR NR NR
Cyclopenta[cd]pyrene NR NR NR
Dibenz[a,h]anthracene <0.01-011 0.05 <0.01-0.01
Fluoranthene 0.07-0.40 0.66 2.8 0.23-2.03 3.7 0.6 2.5
Fluorene NR NR NR
Indeno[1,2,3-cd]pyrene 0.06-0.24 0.84 0.6 0.11-0.25 0.3 0.6 0.5
1-Methylphenanthrene
Perylene NR NR NR
Phenanthrene NR NR 3 NR 4.2 3.0 2.1
Pyrene 0.04-0.88 0.67 0.23-0.87
NR, not reported;
[18] White four, United Kingdom (Dennis et al., 1991);
[19] Granary flour, United Kingdom (Dennis et al., 1991);
[20] Bread, Netherlands (de Vos et al., 1990);
[21] White bread, 1982-83, United Kingdom (Dennis et al., 1991);
[22] Noodles, pizza, Netherlands (de Vos et al., 1990);
[23] Potato products, Netherlands (de Vos et al., 1990);
[24] Rice, macaroni, Netherlands (de Vos et al., 1990);
[25] Soups, Netherlands (de Vos et al., 1990)
Analysed by high-performance liquid chromatography or gas chromatography; reference weight not given
The PAH concentration in rye grown near a highway with high traffic
density decreased slightly 7-25 m away from the road (Larsson, 1982).
5.1.5.7 Beverages
Benzo [a]pyrene was found at 0.8 µg/kg in coffee powder, 0.01
µg/litre in brewed coffee, 9.51 µg/kg in tea leaves, and 0.02 µg/litre
in brewed tea (Lintas et al., 1979). In 40 samples of tea leaves from
India, China, and Morocco, the concentration of benzo [a]pyrene was
generally 2.2-60 µg/kg, although concentrations up to 110 µg/kg were
found in smoked teas (Prinsen & Kennedy, 1978).
In samples of whisky and beer, the concentrations of six of 11 PAH
(benzo [b]fluoranthene, benzo [k]fluoranthene, benzo [a]pyrene,
benzo [ghi]-perylene, dibenz [a,h]anthracene, and
indeno[1,2,3- cd]pyrene) were below or slightly above 0.01 µg/kg. The
highest level determined (0.24 µg/kg) was that of pyrene (Dennis et
al., 1991). The PAH content of the water used in the preparation of
whisky and beer was not described.
5.1.5.8 Vegetable and animal fats and oils
The levels of PAH in oil products, butter, and margarine are listed in
Table 56. Vegetable oils are reported to be naturally free of PAH, and
contamination is due to technological processes like smoke drying of
oil seeds or environmental sources such as exhaust gases from traffic.
The PAH content of native olive oils was particularly high (Speer et
al., 1990). The PAH content of coconut, soya bean, maize, and rapeseed
oil was radically reduced during refining, particularly by treatment
with activated charcoal (Larsson et al., 1987). This method is now
widely used (Dennis et al., 1991).
Benzo [a]pyrene was detected in 30 vegetable oils from Italy and
France in 1994, including 17 grape-seed oils and one pumpkin-seed oil.
The average concentration was 59 µg/kg, and the maximum value was 140
µg/kg. Benzo [b]fluoranthene, benzo [k]fluoranthene,
dibenz [a,h]anthracene, and indeno[1,2,3- cd]pyrene were also found
in measurable amounts. The source of these high levels was the smoke
in drying ovens (State Chemical Analysis Institute, Freiburg, 1995).
Lard and dripping were found to contain levels of individual PAH
ranging from < 0.01 µg/kg dibenz [a,h]anthracene) to 6.9 µg/kg
fluoranthene (Dennis et al., 1991). High PAH levels were found in
margarine samples in studies in Finland (Hopia et al., 1986), the
Netherlands (Vaessen et al., 1988), New Zealand (Thomson et al.,
1996), and the United Kingdom (Dennis et al., 1991) (see Table 56).
5.1.6 Plants
PAH with low molecular masses are more readily taken up by vegetation
than those with higher molecular masses (Wang & Meresz, 1982).
Table 56. Polycyclic aromatic hydrocarbon concentrations (µg/kg) in vegetable oils and related products
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Acenaphthene NR < 0.02-45 NR NR NR NR 0.29 NR NR < 0.1 -11
Anthracene NR < 0.02-460 <0.1-0.1 ND-4.8 ND-8 NR 0.04-0.92 NR NR < 0.2-5.6
Anthanthrene Trace-0.1 NR NR NR NR NR 0.03-0.53 NR NR < 0.1-2.7
Benz[a]anthracene NR NR 0.7-6.1 ND-6.1 ND 0.30-7.46 NR 0.22-3.98 0.28-0.96 < 0.1-5.2
Benzo[a]fluorene NR < 0.02-130 NR NR ND-2 NR 0.07-3.8 NR NR NR
Benzo[a]pyrene Trace-0.3 < 0.02-24 0.5-2.3 ND-4.1 ND 0.29-4.92 0.05-2.2 0.19-6.0 0.17-0.83 < 0.2-5.2
Benzo[b]fluoranthene Trace-0.1 < 0.02-91a NR ND-8.9a ND 0.20-2.39 NR 0.16-3.0 0.09-0.37 < 0.2-9.2
Benzo[b]fluorene NR < 0.02-45 NR NR ND NR 0.03-2.1 NR NR NR
Benzo[e]pyrene NR < 0.02-23 0.7-2.4 ND-3.8 ND 0.26-6.06 0.09-2.1 0.42-6.11 0.36-0.87 NR
Benzo[ghi]fluoranthene NR < 0.02-1.3 NR NR ND NR 0.14-4.9 NR NR NR
Benzo[ghi]perylene NR < 0.02-10 0.5-1.7 ND-4.2 NR 0.06-5.23 0.02-1.4 0.38-5.21 0.17-1.16 < 0.2-10.6
Benzo[k]fluoranthene NR NR NR NR ND 0.24-3.17 NR 0.20-3.40 0.16-0.55 < 0.1-11.4
Chrysene NR NR NR 0.1-8.6b ND 0.39-10.3 NR 0.26-7.36 0.31-0.97 < 0.2-7.5
Coronene NR < 0.02 NR NR NR NR NR NR NR NR
Cyclopenta[cd]pyrene NR < 0.02-1.4 NR NR ND NR 0.10-1.1 NR NR NR
Dibenz[a,h]anthracene 0.7-1.1 < 0.02-1.1c NR ND-0.2c NR <0.01-0.82 NR 0.05-1.02 0.04-0.11 < 0.1-9.2
Fluoranthene 0.2-7.5 < 0.02-460 1.2-4.8 0.2-18.2 3-15 0.21-12.4 0.52-9.0 0.09-4.50 0.44-1.56 < 0.1-1.6
Fluorene NR < 0.02-200 NR NR ND-7 NR 0.08-1.6 NR NR < 0.2-2.1
Indeno[1,2,3-cd]pyrene Trace-0.5 < 0.02-0.85 0.3-1.7 ND-4.3 NR 0.59-6.78 0.03-1.1 0.49-9.14 0.43-1.17 < 0.2-9.7
Naphthalene NR NR NR NR NR NR NR NR NR < 0.2-52
1-Methylphenanthrene NR < 0.02-190 NR NR NR NR 0.08-1.8 NR NR NR
Perylene Trace-0.2 < 0.02-5.9 0.1-0.4 ND-0.9 NR NR 0.02-0.57 NR NR NR
Phenanthrene NR 0.09-1400 0.9-1.6 ND-69.4 4-38 NR 0.29-6.0 NR NR < 0.2-4.6
Pyrene 0.2-1.4 < 0.02-330 1.1-4.2 0.1-13.6 2-14 0.58-17.2 0.59-15 0.29-6.03 0.44-1.88 < 0.1-1.7
Table 56 (continued)
ND, not detected; /, single measurements; NR, not reported;
[1] Corn oil, canola, soya bean oil (Lawrence & Weber, 1984);
[2] Corn oil, coconut oil (crude and deodorized), olive oil, soya bean oil, sunflower oil, sesame oil, flaw oil,
wheatseed oil (Hopia et al., 1986);
[3] Coconut oil (pure) (Sagredos et al., 1988);
[4] Various olive oils, safflower oils, sunflower oils, maize germ oils, sesame oil, linseed oil, wheat germ oil
(all native) (Speer et al., 1990);
[5] Various olive oils (Menichini et al., 1991b);
[6] Various unspecified oils (Dennis et al., 1991);
[7] Four cooking margarines, seven table margarines (Hopia et al., 1986);
[8] Margarine (Dennis et al., 1991);
[9] Low-fat spread (Dennis et al., 1991);
[10] Margarine (Thomson et al., 1996)
Analysed by high-performance liquid chromatography or gas chromatography
a Benzo[b+j+k]fluoranthenes
b In sum with triphenylene
c Dibenz[a,h+a,c]anthracenes
In a study of PAH levels in soil (see section 5.1.4), leaf litter, and
soil fauna (see section 5.1.7) from a roadside in Brisbane, Australia,
vegetation height, soil depth, and distance from the roadside were
found to be important in the distribution of PAH. The concentration of
PAH in leaf litter declined exponentially with distance from the
roadway, few PAH being detectable 30 m away. A decrease in PAH levels
with height was found in the roadside vegetation canopy. In leaf
litter, fluorene, phenanthrene, fluoranthene, pyrene, chrysene,
benzo [k]fluoranthene, and benzo [ghi]perylene were present at
concentrations of about 100 µg/kg wet weight. Naphthalene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene, and
indeno[1,2,3- cd]pyrene were present at about 50 µg/kg wet weight,
whereas anthracene was present at concentrations below 10 µg/kg wet
weight. Perylene and dibenz [a,h]anthracene were not detectable. The
tree Casuarina littorina contained high levels of pyrene and
chrysene (100 µg/kg wet weight each) and benzo [k]fluoranthene (72
µg/kg wet weight); the concentrations of fluoranthene, phenanthrene,
and benzo [ghi]-perylene were about 40 µg/kg wet weight. Perylene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene were not detectable (Pathirana et
al., 1994).
The benzo [a]pyrene levels in spruce sprouts from a rural area of
Germany (Bornhövede, Schleswig-Holstein) decreased from 2.6 µg/kg in
1991 to 1.3 µg/kg in 1993. The concentrations of PAH with low
boiling-points significantly decreased between 1991 and 1993; for
example, that of fluoranthene decreased from 44 µg/kg in 1991 to 11
µg/kg in 1993, perhaps due to a decrease in coal burning. The levels
of phenanthrene, fluoranthene, pyrene, and benzo [b]fluoranthene plus
benzo [j]fluoranthene plus benzo [k]fluoranthene were about 10
µg/kg; those of benzo [ghi]fluoranthene, benzo [c]phenanthrene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, and coronene were
about 2 µg/kg; and those of anthracene, dibenz [a,h]anthracene, and
anthanthrene were < 1 µg/kg. The PAH levels in spruce sprouts from
the Saarland, an industrial area in Germany, were about 10 times
higher than those in the Bornhöveder area, although these levels also
decreased between 1991 and 1993: from 5.9 to 4.1 µg/kg for
benzo [a]pyrene and 97 to 58 µg/kg for fluoranthene. the
concentrations of pyrene were 40-50 µg/kg, those of
benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene were 20 µg/kg, and those of
benzo [ghi]perylene, benzo [c]phenanthrene, benz [a]anthracene,
benzo [e]pyrene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, anthanthrene, and
coronene were < 10 µg/kg (Jacob & Grimmer, 1994, 1995). In 1994, the
PAH levels had decreased further. Overall, a 25% decrease in the PAH
levels in spruce sprouts was seen over the previous 10 years (Jacob &
Grimmer, 1995).
The PAH profiles in spruce sprouts and poplar leaves were reasonably
similar in areas with clean air (Bavarian forests) and in
industrialized areas (Saarland) of Germany, indicating that one
emission source is predominantly responsible for air pollution by PAH.
Hard-coal combustion resulted in a characteristic PAH profile (Jacob
et al., 1993a).
The concentrations of PAH in pine needles from Dübener Heide near
Leipzig (Saxony, Germany) were similar to those from the Bornhöveder
area (Schleswig-Holstein, Germany), with an average benzo [a]pyrene
level of 2.3 µg/kg (Jacob & Grimmer, 1995).
Beech leaves from the Harz mountains in Germany contained fluoranthene
at a level of 5 µg/kg, whereas the concentrations of phenanthrene,
pyrene, benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, anthracene, benz [a]anthracene,
benzo [e]pyrene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, anthanthrene, and
coronene were all < 2 µg/kg. Beech sprouts in an industrial area in
eastern Germany contained 10-15 times higher levels of PAH, with
fluoranthene at about 60 µg/kg, pyrene at about 30 µg/kg,
benzo [b]fluoranthene plus benzo [j]-fluoranthene plus
benzo [k]fluoranthene at about 10 µg/kg, and anthracene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, benzo [ghi]perylene, coronene,
dibenz [a,h]anthracene, and anthanthrene at < 2 µg/kg (Jacob &
Grimmer, 1995).
Comparable results were obtained in poplar leaves: those from the
Saarland analysed in 1989, 1991, and 1993 had 10 times lower
concentrations of PAH than those in Dübener Heide. The concentrations
of phenanthrene, fluoranthene, and pyrene were about 20 µg/kg, those
of benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene were about 10 µg/kg, and those of anthracene,
benz [a]anthracene, benzo [e]pyrene, benzo [a]pyrene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, anthanthrene, and coronene were < 5 µg/kg
(Jacob & Grimmer, 1995).
5.1.7 Animals
5.1.7.1 Aquatic organisms
Aquatic invertebrates are known to adsorb and accumulate PAH from
water (see section 4.1.5). The concentrations of PAH in aquatic
organisms collected from various sites are listed in Tables 57-64. All
of the sampling sites listed in Tables 57-60 were contaminated with
industrial effluents, the major components of the PAH profile being
benzo [b]fluoranthene, benz [a]anthracene, benzo [a]pyrene,
benzo [e]pyrene, fluoranthene, pyrene, and phenanthrene. The average
levels of PAH in aquatic organisms from these sites ranged from 1 to
100 µg/kg; the differences in levels generally corresponded to the
degree of industrial and urban development and shipping movements.
Table 57. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry weight)
in bivalves and gastropods; main source, industrial emissions
Compound [1] [2] [3] [4] [5] [6] [7]
Acenaphthene ND ND 7 2.1/8.8
Anthracens 9 9.0/25
Benz[alanthracene 172 203 3 5-41 25-229
Benzo[alpyrene 12 21 1 8.1 2-8 Trace-28 2.6/2.8
Benzo[b]fluoranthene 23 25 3-30 48-90
Benzo[ejpyrene 17 10 Trace-30 231-356
Benzo[ghilperylene ND 4 5
BenzoUlfluoranthene 1.3
Benzolk]fluoranthene 2.3
Chrysene 209 205
Coronene 4
Dibenzo(a,elpyrene 2
Dibenzo[e,ilpyrane 4
Dbenzo[a,lpyrene Trace
Fluoranthene 18 62 7 43-407 300-4992 26/61
Fluorene 2 1.3/6.3
1-Methylphenanthrene 2.9
Naphthalene 15/3
Perylene 8
Phenanthrene 733 462 9 4.4 115-258 55-2542 66/194
Pyrene 85 131 4 32-204 141-3128 23/40
Triphenylene ND
ND, not detected; /, single measurement;
[1] Whole cooked clam (Mya arenaria); oil-contaminated area (tanker accident), Canada, 1979;
concentration in µg/kg wet weight (Sirota & Uthe, 1981);
[2] Whole cooked mussel (Mytilus edulls); oil-contaminated area (tanker accident), Canada,
1979; concentration in µg/kg wet weight (Sirota & Uthe, 1981);
[3] Whole mussel (Mytilus galloprovincialis); Thermaikos Gulf, Aegean Sea, Greece (agricultural
and industrial area); concentration in µg/kg wet weight (Iosifidou et al., 1982);
[4] Whole scallop (Amusium pleuronectes); Gulf of Thailand, Thailand; reference weight not
given (Hungspreugs et al., 1984);
[5] Whole periwinkle (Littorina littorea); moderately polluted parts of North Sea coast,
Norway, 1978-79 (Knutzen & Borland, 1982);
[6] Whole limpet (Patella vulgata); moderately polluted parts of North Sea coast, Norway,
1978-79 (Knutzen & Sortland, 1982);
[7] Whole snails (Thais haemostoma), Pensacola Bay, USA; creosote contaminated; concentration
in µg/kg wet weight (Rostad & Pereira, 1987)
High-performance liquid chromatography or gas chromatography
Table 58. Polycyclic aromatic hydrocarbon concentrations (µg/kg dry weight) in algae
and water plants; main source, industrial emissions
Compound [1] [2] [3] [4] [5] [6]
Benz[a]anthracene 5 4 31-325 45-431 3-40
Benzo[alpyrene 4 5 Trace-64 Trace-<2 2-20
Benzo[b]fluoranthene 4 5 7-76 6-12 5-31
Benzo[e]pyrene 7 14 Trace-100 Trace-8 8-50 410
Benzo[ghi]perylene 4 79
Fluoranthene 45 32 40-412 15-900 <4-236
Phenanthrene 87 34 31-325 45-431 109-146
Pyrene 36 20 28-286 15-388 <4-224 260
[1] Laminaria saccharins (whole); moderately polluted parts of North Sea coast,
Norway, 1978-79 (Knutzen & Sortland, 1982);
[2] Ceramium rubrum (whole), moderately polluted parts of North Sea coast, Norway,
1978-79 (Knutzen & Sortland, 1982);
[3] Bladder wrack (Fucus vesiculosus, whole), moderately polluted parts of North
Sea coast, Norway, 1978-79 (Knutzen & Sortland, 1982);
[4] Knotted wrack (Ascophyllum nodosum, whole), moderately polluted parts of North
Sea coast, Norway, 1978-79 (Knutzen & Sortland, 1982);
[5] Toothed wrack (Fucus serratus, whole), moderately polluted parts of North Sea
coast, Norway, 1978-79 (Knutzen & Sortland, 1982);
[6] Wakame seaweed, Japan (Obana et al., 1981a)
High-performance liquid chromatography or gas chromatography
Table 59. Polycyclic aromatic hydrocarbon concentrations (µg/kg wet weight) in lobsters; main
source, industrial emissions
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene ND ND
Benz[a]anthracene 684 ND/23 1620-23 400 34-604 762-32 700 17-900
Benzo[a]pyrene 24 0.2/2.6 35-1000 2.0-40 711-1430 27-43
Benzo[b]fluoranthene 24 1 155-2350 6-78 1020-3820 29-835
Benzo[e]pyrene 57 5/8 415-9330 15-165 1550-3600 35-36
Benzo[ghi]perylene ND ND/2 7-493 1.6-31 232-769 10-20
Benzo[k]fluoranthene 7.6 0.3/0.6 43-588 1.6-25 502-955 15-26
Chrysene 445 ND 360-5050 5-79 252-1240 15-24
Fluoranthene 318 ND/0.2 1910-12400 103-545 4220-15 200 68-442
Indeno[1,2,3-cd]pyrene 5 38-855 3-45 486-931 12-40
Phenanthrene 1588 ND Trace-3470 Trace-650
Pyrene 488 ND 730-6710 32-265 2910-13 100 59-333
Triphenylene ND/244 2520-23100 Trace-330
ND, not detected; /, single measurements;
[1] Homarus americanus (digestive gland), oil-contaminated area (tanker accident), Canada, 1979
(Sirota & Uthe, 1981);
[2] Homarus americanus (tail muscle), oil-contaminated area (tanker accident), Canada, 1979
(Sirota & Uthe, 1981);
[3] Homarus americanus (hapatopancreas), Sydney Harbour, near coking plant, Canada (Sirota
et al., 1983);
[4] Homarus americanus, (tail muscle), Sydney Harbour, near coking plant, Canada (Sirota
et al., 1983);
[5] Homarus americanus, (digestive gland), Sydney Harbour, near coking plant, Canada, 1982-84
(Uthe & Musial, 1986);
[6] Homarus americanus (tail muscle), Sydney Harbour, near coking plant, Canada, 1982-84
(Uthe & Musial, 1986)
High-performance liquid chromatography or gas chromatography
Table 60. Polycyclic aromatic hydrocarbon levels (µg/kg dry weight) in fish and other aquatic species; main
source, industrial emissions
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene 39 Trace-0.9 130 < 25
Acenaphthylene 270 0.1-0.2
Anthracene ND 0.1-0.2 460 < 22 1000
Benz[a]anthracene 22 ND-40 ND-< 0.1 0.1-88 1000 < 21 1-2 800 5
Benzo[a]fluorene 02-0.6 500
Benzo[a]pyrene 7 0.07-8.4 ND-< 0.1 0.1-0.5 570 < 20 ND 8
Benzo[b]fluoranthene <O.1a 28
Benzo[b]fluorene O.1-0.2
Benzo[o]phenanthrene Trace
Benzo[e]pyrene 14 ND-< 0.1 0.1-1.6 840 < 25 25
Benzo[ghi]perylene ND-< 0.1 0.2-18 75 < 25 23
Chrysene 61 < 0.1-2.1b 1500 < 22
Dibenz[a,h]anthracene ND-< 0.1c <100 < 25
Fluoranthene 1800 0.1-9.1 1.2-5.6 4800 < 20 13-18 800 48
Fluorene 0.2-2.4 200 < 25 NDc
Indeno[1,2,3-cd]pyrene ND-< 0.1 0.3-3.7 150 < 25
1-Methylphonanthrene 85 < 10
Naphthalene 2.5-11 610 < 25
Perylene 6 ND-< 0. 1 Trace-0.2 75 < 20
Phenanthrene 2700 28-15 313 0.1-2.4 0.7-9.1 1400 < 20 32-50 900 71
Pyrene 1500 ND-10.0 0.7-3.7 2300 < 20 10-8 800 39
Triphenylene 800
Table 60 (continued)
ND, not detected;
[1] Bullhead catfish (Ictalurus nebulosus, whole); Black River, USA, near coking plant; concentration in
µg/kg wet weight (Vassilaros et al., 1982);
[2] Whole fish (unspecified); Hersey River, USA, creosote polluted; concentration in µg/kg wet weight
(Black et al., 1981);
[3] Bream (fillet and liver); River Elbe, Germany, industrial region of city of Hamburg (Speer et al., 1990);
[4] Dabs (Limanda limanda, muscle, North Sea, United Kingdom, near Beatrice oil platform; concentration in
µg/kg wet weight (McGill et al., 1987);
[5] English sole (Parophrys vetulus, stomach contents); Mukilteo, USA, near petroleum storage tanks (Malins
et al., 1985);
[6] English sole (Parophrys vetufus, liver); Mukilteo, USA, near petroleum storage tanks (Malins et al., 1985);
[7] Whole starfish (Asterias rubens), moderately polluted areas of North Sea coast, Norway, 1978-79 (Knutzen
& Sortland, 1982);
[8] Whole holothurians, Toulon, France; urban sewage (Milano et al., 1986);
[9] Whole crumb-of-bread sponge (Hafichondria panicea); moderately polluted areas of North Sea coast, Norway,
1978-79 (Knutzen & Sortland, 1982)
High-performance liquid chromatography or gas chromatography
a Benzo[b+j+k]fluoranthenes
b In sum with triphenylene
c Dibenz[a,h+a,c]anthracenes
Table 61. Polycyclic aromatic hydrocarbon concentratrations (µg/kg dry weight) in bivalves (mussels and
clams); background values
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Acenaphthene NR 24/46
Acenaphthylene NR 34/130
Anthracene 0.7-19 9-15 149-243 36/43
Benz[a]anthracene NR 0.1-0.8 2.9/42 < 1 31/94
Benzo[a]pyrene 4.6-451 3/5 <0.8-2 3.5/8.7 < 1 1.3/26
Benzo[b]fluoranthene 3.0-120 1.5/12 2.5/18
Benzo[c]phenanthrone 5.3-280 3.1/55 < 1 26/94
Benzo[e]pyrene NR 5-25
Benzo[ghi]perylene 3.4-57 5.4/4.2 3 0.4/8.1
Benzo[k]fluoranthene 1.0-43 1-2 2.6/9.6 1.7/17
Chrysene NR 7.6/27 86
Coronene < 10-24 1.3/2.7 0.7/4.6
Dibenz[a,h]anthracene NR 4.7/6.9 2.1/9.6
Fluoranthene 16-288 23/43 8-23 0.7-7.2 11/111 17 47/180 72
Indeno[1,2,3-cd]pyrene ND-9.9 5.9/3.9 0.3/5.7
1-Methylphenanthrene 22-708
Naphthalene NR 5-4 51/120
Perylene 4.2-59 < 5-26 36
Phenanthrene 21-570 7-109 0.1-1.7 12/155 18 108/216
Pyrene 6.6-394 9-77 15-38 0.3-6.6 6.2/62 23 25/109
Triphenylene 7.5-300 7.9/43 27/106
Table 61 (contd)
ND, not detected; /, single measurements; NR, not reported;
[1] Mussel (Mytilus edulis), Danish, German and Dutch Wadden Sea, 1989 (Compaan & Laane, 1992);
[2] Mussel (Mytilus edulis); Finnish archipelago, Finland, 1978-79; concentration in µg/kg wet weight
(Rainio et al., 1986);
[3] Mussel (Mytilus edulis L.); North Sea coast, Netherlands; concentration in µg/kg wet weight
(Boom, 1987);
[4] Hard shell clam (Mercenaria mercenaria), Rhode Island (seafood stores), USA; concentration in µg/kg
wet weight (Pruell et al., 1984);
[5] Softshell clam (Mya arenaria), Coos Bay, Oregon, USA, 1978-79; reference weight not given (Mix &
Schaffer, 1983);
[6] Clam (Mya mercenaria); Chesapeake Bay, USA, 1984 (Bender & Huggett, 1988);
[7] Mussel (Mytilus edulis); Yaquina Bay, USA, 1979-80; concentration in µg/kg wet weight (Mix & Schaffer,
1983);
[8] Rangia cuneata; Lake Pontchartrain, USA, 1980; concentration in µg/kg wet weight(McFall et al., 1985)
Table 61 (contd)
Compound [9] [10] [11] [12] [13] [14] [15]
Acenaphthene 16
Acenaphthylene 18
Anthracene < 0.05-3.2 <0.05
Benz[a]anthracene < 1-6 < 10 1.0-1.8 ND-2.3
Benzo[a]pyrene 30-168 < 10 < 0.003-0.02 < 0.004 0.41-1.8 0.40-2.6 1.0
Benzo[b]fluoranthene 1.0-1.8 0.83-1.9
Benzo[c]phenanthrene < 1-9
Benzo[e]pyrene
Benzo[ghi]perylene < 1-10 < 0.05-0.3 <0.05 0.53-1.9 0.83-2.3
Benzo[k]fluoranthene < 0.002-0.02 < 0.002 0.29-0.80 0.32-1.2
Chrysene < 0.03-1.4 <0.03
Coronene
Dibenz[a,h]anthracene
Fluoranthene < 1/52 < 1-370 < 0.04-0.70
Fluorene
Indeno[1,2,3-cd]pyrene
1-Methylphenanthrene
Naphthalene
Perylene < 1-10 < 10-300 < 0.01-0.08
Phenanthrene < 1-15 < 1-60 14
Pyrene 17/165 < 1-450 < 0.03-1.4 <0.03
Triphenylene
Table 61 (contd)
[9] Rangia cuneaya, Chesapeake Bay, USA, 1984 (Bender & Huggett, 1988);
[10] Lampsilus radiata, Elliptio complanatus, Anodonata grandis; Lake George, Heats Bay USA
(Heit et al., 1980);
[11] Tridacna maxima, Great Barrier Reef, Australia, 1980-82; concentration in µg/kg wet weight
(Smith et al., 1984);
[12] Clam; Green Island, Great Barrier Reef, Australia, concentration in µg/kg wet weight
(Smith et al., 1984);
[13] Shortnecked clam; near Miyagi Prefecture, Japan, concentration in µg/kg wet weight
(Takatsuki et al., 1985);
[14] Mussel; near Miyagi Prefecture; Japan, reference weight not given (Takatsuki et al., 1985);
[15] Perna viridis; Gulf of Thailand (mussel farm), Thailand, reference weight not given
(Hungspreugs et al., 1984)
High-performance liquid chromatography or gas chromatography;
Table 62. Polycyclic aromatic hydrocarbon concentrations (µg/kg wet weight) in bivalves (Oysters); background values
Compound [1] [2] [3] [4] [5] [6] [7]
Acenaphthene 46 < 0.2-2.0 16
Acenaphthylene 36 < 0.4-3.0
Anthracene 44 < 1-40 < 0.08-0.9 < 0.25-4.2
Benz[a]anthracene 9.9 0.3-12 < 1-135 1.1 1.5-10
Benzo[a]pyrene 0.5-1.6 50-285 < 0.01-5 0.6-2.6 0.78 3.5
Benzo[b]fluoranthene 0.3-5.2 < 0.03-6 3.0-20 2.2
Benzo[c]phenanthrene < 1-70
Benzo[e]pyrene < 1-453 2.8-32
Benzo[ghi]perylene O.4-1.2 < 1-73 < 0.05-5 0.87 < 0.20-2.8
Benzo[k]fluoranthene 12 0.1-0.9 < 0.06-5.1 1.2 < 0.01-< 3
Chrysene 58 1.3-14 < 0.1-3
Dibenz[a,h]anthracene < 1-20 < 0.01-< 4 < 0.06
Fluoranthene 80 0.9-94 < 1-450 0.4-22 470
Fluorene 21 0.1-0.8
Indeno[1,2,3-cd]pyrene 1.7 < 0.01-5
1-Methylphenanthrene 3.5
Naphthalene 35 5-48 0.8-7
Perylene < 1-130
Phenanthrene 220 4.9-77 < 1-45 2-38 6.7
Pyrene 200 1.6-50 < 1-645 < 0.4-15 7.0-52
Triphenylene 0.03
Table 62 (continued)
[1] Crassostrea virginica, Lake Pontchartrain, USA, 1980 (McFall et al., 1985);
[2] Crassostrea virginica; Palmetto Bay (Marina), USA (Marcus & Stokes, 1985);
[3] Crassostrea virginica; Chesapeake Bay, USA, 1983-84; concentration in µg/kg dry weight (Bender & Huggett, 1988);
[4] Saccostrea cucculata, Mermaid Sound, Australia, 1982 (Kagi et al., 1985);
[5] Oyster, Japan (local market); 1977-78 (Obana et al., 1981a);
[6] Oyster, near Miyagi Prefecture, Japan; reference weight not given (Takatsuki et al., 1985);
[7] Ostrea plicatula; Gulf of Thailand, Thailand; reference weight not given (Hungspreugs et al., 1984)
High-performance liquid chromatography or gas chromatography
Table 63. Polycyclic: aromatic hydrocarbon concentrations (µg/kg wet weight)
in crustacea (lobsters); background values
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene ND ND
Benz[a]anthracene 655 179 9-38 Trace-133 6-79 6-17
Benzo[a]pyrene 18 3.8 0.4-2.1 Trace-2 1.6-8 ND-1.6
Benzo[b]fluoranthene 17 28 3-6.5 Trace-5.3 7-16 ND-0.8
Benzo[e]pyrene ND 170 12-23 ND-22 15-29 ND-3.6
Benzo[ghi]perylene 11 63 1.4-6.8 Trace-2.0 2.4-10 ND-0.8
Benzo[k]fluoranthene 2 4.4 0.8-1.9 Trace-11.6 1.9-8 ND-0.8
Chrysene 140 113 2.5-12 ND-14 2-43 ND
Fluoranthene ND 147 46-407 5.5-12 90-162 ND-34
Fluorene ND 194
Indeno[1,2,3-cd]pyrene 22 77 2.1-5.0 ND-3.7 Trace-5 ND-0.8
Phenanthrene ND 1197 20-345 ND-15
Pyrene ND 174 ND-197 ND-5 35-46 ND-22
Triphenylene ND 1373 ND-141 ND-Trace
ND, not detected
[1] Homarus americanus (digestive gland); Port Hood, Canada, 1979 (Sirota & Uthe, 1981);
[2] Homarus americanus (digestive gland); Brown Bank (offshore), Canada, 1979 (Sirota & Uthe, 1981);
[3] Homarus americanus (hepatopancreas); Morien Bay and Mira Bay, Canada (Sirota et al.,1983);
[4] Homarus americanus (tail muscle); Moran Bayand, Mira Bay, Canada (Sirota et al., 1983);
[5] Homarus americanus (digestive gland); Port Morien, Canada, 1982-84 (Uthe & Musial, 1986);
[6] Homarus americanus (tail muscle); Port Morien, Canada, 1982-84 (Uthe & Musial, 1986)
Analysed by high-performance liquid chromatography or gas chromatography
Table 64. Polycyclic aromatic hydrocarbon concentrations (µg/kg wet weight) in fish and other aquatic species
(background values)
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
Anenaphthene ND-83 11 7 1-500
Acenaphthylene 43 0.8-24
Anthracene 10 2.0-2.2 ND 20
Benz[a]anthracene 4 4.0-26 1.2 1.6-7.5 20
Benzo[a]fluorene ND
Benzo[e]pyrene 0.04-0.84 1 1.9-15 8 Trace-4.5 5
Benzo[b]fluoranthene 3.2-17
Benzo[e]pyrene ND
Benzo[ghi]perylene 2.0-14 16
Benzo[k]fluoranthene 2.1-11
Chrysene 6 3 3.4-26 NR
Dibenz[a,h]anthracene 1.2-4.13
Fluoranthene 4-95 4 85 9 ND-732 20
Fluorene 8.9 ND-15 1-370 ND
Indeno[1,2,3-cd]pyrene ND-15 NR
1-Methylphenanthrene NR
Naphthalene 45-215 ND-117
Perylene NR
Phenanthrene 8-142 2 157 2.3-35 36 23-43 ND 40
Pyrene 2-62 4 30 31 2.4-74 1.3-9.6 ND
Triphenylene 20
ND, not detected; NR, not reported;
[1] Various seafish (muscle, liver, gall), Finnish archipelago, Finland, 1979 (Rainio et al., 1986);
[2] Edible tissues of various seafish, Arabian Gulf, Iraq (DouAbdul et al., 1987);
[3] Whole bullhead catfish (ictalurus nebulosus), Buckeye Lake, USA (Vassilaros et al., 1982);
[4] Whole bullhead catfish (Ictalurus, nebulosus;, whole), Black River, USA (West et al, 1985);
[5] Whole fish, Hersey River, USA (Black et al., 1981);
[6] Whole striped bass (Morone saxatillis); Potomac River, USA (Vassilaros et al., 1982);
[7] White suckers (Catastomus commersoni); stomach contents; Lake Erie, USA (Maccubbin et al., 1985);
[8] Various fish, Japan, 1970-91 (Environment Agency, Japan, 1993);
[9] Fish bought in market, Ibadan, Nigeria; reference weight not given (Emerole et al., 1982);
[10] Whole holothurians, France; concentration in µg/kg dry weight (Milano et al., 1986)
Analysed by high-performance liquid chromatography or gas chromatography
The levels in holothurians from urban sewage were 1-15 mg/kg (Milano
et al., 1986).
Concentrations of 1-5 mg/kg individual PAH were found in limpets
(Patella vulgata) in the North Sea (Knutzen & Sortland, 1982). The
PAH concentrations in two species of bivalves in Saudafjorden (Norway)
near an iron alloy smelter decreased rapidly with distance from the
source, but the compounds could still be detected more than 15 km
away. High levels of individual PAH were reported in mussels
(Modiolus modiolus), with maximum levels of 57 000 µg/kg
benzo [b]fluoranthene, 25 000 µg/kg benz [a]anthracene, 23 000 µg/kg
benzo [e]pyrene, 21 000 µg/kg benzo [a]pyrene, 20 000 µg/kg
fluoranthene, 8200 µg/kg pyrene, 6000 µg/kg benzo[ghi]perylene, 4000
µg/kg perylene, 2900 µg/kg benzo [a]fluorene, 2300 µg/kg
benzo [b]fluorene, 2200 µg/kg dibenz [a,h]anthracene, 2000 µg/kg
benzo [c]phenanthrene, 1100 µg/kg phenanthrene, 524 µg/kg anthracene,
and 360 µg/kg anthanthrene (Bjrseth, 1979). A very high level of
anthracene (243 µg/kg) was found in mussels (Mytilus edulis L.) in
the North Sea near the Dutch coast (Boom, 1987). Mussels in the USA
frequently contained up to 500 µg/kg of individual PAH (Heit et al.,
1980; Mix & Schaffer, 1983).
The levels of PAH in pooled mussel samples in 1986, 1988, and 1990 in
Germany were about 10 µg/kg for fluoranthene, pyrene, chrysene plus
triphenylene, benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, and benzo [e]pyrene and < 4 µg/kg for
benzo [ghi]fluoran-thene plus benzo [c]phenanthrene,
benz [a]anthracene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene,
dibenz [a,h]anthracene, benzo [ghi]perylene, anthanthrene, and
coronene. The levels were high in the winter months and low in summer,
with minima in June and April. The authors concluded that this
seasonal variation was due to more intensive metabolic activity (Jacob
& Grimmer, 1994).
During 1978-79, the average total PAH concentrations in two
subpopulations of softshell clams were 555 µg/kg in the industrialized
bayfront area of Coos Bay, Oregon, and 76 µg/kg in a more remote
environment. During 1979-80, low-molecular-mass, readily water-soluble
PAH were one or two orders of magnitude more concentrated then
high-molecular-mass, less water-soluble PAH in mussels (M. edulis)
(Mix & Schaffer, 1983).
Individual PAH levels of 1-20 mg/kg were found in the hepatopancreas
of lobsters (Homarus americanus) in the south arm of Sydney Harbour,
Canada, near a coking plant (Sirota et al., 1983), and levels of the
same order of magnitude were found in the digestive gland (Uthe &
Musial, 1986). The levels in digestive gland, tail muscle, and
hepatopancreas from lobsters from other areas of Canada were 100-1000
µg/kg (Sirota & Uthe, 1981; Sirota et al., 1983; Uthe & Musial, 1986).
High PAH levels were found in oysters (Crassostrea virginica) in
Chesapeake Bay, USA, with maximum levels of 650 µg/kg pyrene, 450
µg/kg benzo [e]pyrene, 450 µg/kg fluoranthene, 290 µg/kg
benzo [a]pyrene, 130 µg/kg benz [a]anthracene, 130 µg/kg perylene,
73 µg/kg benzo [ghi]-perylene, 70 µg/kg benzo [c]phenanthrene, 48
µg/kg naphthalene, 45 µg/kg phenanthrene, 40 µg/kg anthracene, and 20
µg/kg dibenz [a,h]anthracene. The levels of PAH in clams
(Rangia cuneata) from Chesapeake Bay were 170 µg/kg
benzo [a]pyrene, 170 µg/kg pyrene, 52 µg/kg fluoranthene, 15 µg/kg
phenanthrene, 10 µg/kg perylene, 10 µg/kg benzo [ghi]perylene, 9
µg/kg benzo [c]phenanthrene, and 6 µg/kg benz [a]anthracene (Bender
& Huggett, 1988).
Phenanthrene was found at 15 mg/kg in lampreys (Pteromyzon sp.) in
the Hersey River, USA, which was polluted with creosote used for wood
preservation (Black et al., 1981).
The viviparous blenny (Zoarces viviparus) fish contained 0.06 µg/kg
benzo [a]pyrene and 0.2-3.9 µg/kg phenanthrene and fluoranthene; the
concentrations of other PAH were below the detection limit (0.01
µg/kg). In bream (Abramis brama) the levels were < 0.01-0.15 µg/kg
benzo [a]pyrene and 1.3-15 µg/kg phenanthrene. Mussels (Mytilus
sp.) were shown to accumulate PAH and were thus a better marker for
PAH contamination (Jacob & Grimmer, 1994, 1995).
The concentrations of individual PAH in English sole
(Paraphrys vetulus) taken from near petroleum storage tanks were 1-5
mg/kg (Malins et al., 1985).
5.1.7.2 Terrestrial organisms
The liver of wild deer mice (Peromyscus maniculatus) trapped at a
PAH-contaminated site in South Carolina, USA (Whidbey Island Naval Air
Station) had levels of PAH ranging from 0.075 for
benzo [b]fluoranthene to 4.6 mg/kg for benz [a]anthracene.
Acenaphthylene, acenaphthene, fluorene, benz [a]-anthracene,
chrysene, benzo [b]fluoranthene, benzo [k]fluoranthene,
dibenz [a,h]anthracene, and indeno[1,2,3- cd]pyrene were detected.
Liver from mice at an uncontaminated reference site contained
measurable amounts of only benz [a]anthracene (0.55 mg/kg) and
acenaphthylene (2.2 mg/kg) (Dickerson et al., 1994).
In a study of PAH levels in terrestrial organisms from a roadside in
Brisbane, Australia, 16 PAH were determined: naphthalene, fluorene,
phenanthrene, anthracene, fluoranthene, pyrene, benz [a]anthracene,
chrysene, benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene,
perylene, indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene. In the beetle Laxta
granicollis, pyrene and benzo [ghi]perylene were present at the
highest levels, at 20 µg/kg wet weight each; phenanthrene and
fluoranthene were present at about 10 µg/kg; and the concentrations of
other PAH were < 5 µg/kg. Naphthalene, anthracene,
dibenz [a,h]anthracene, and coronene were not detected. Fluorene, at
a concentration of 11 µg/kg wet weight, was the most abundant PAH in
the beetle Platyzosteria nitida; the concentrations of other PAH
were < 5 µg/kg; whereas naphthalene, dibenz [a,h]anthracene, and
coronene were not detected. In millipedes (myriapods),
benzo [k]fluoranthene was the most abundant PAH (19 µg/kg wet
weight); the pyrene concentration was 12 µg/kg; those of other PAH
were < 5 µg/kg wet weight; and dibenz [a,h]anthracene and coronene
were not detected. In centipedes (Myriaod sp.), no PAH were
detected. In slugs (Arion ater), benzo [k]fluoranthene showed the
highest concentration, at 19 µg/kg wet weight; the pyrene and
naphthalene levels were about 10 µg/kg; those of other PAH were < 5
µg/kg wet weight; and anthracene, perylene, dibenz [a,h]anthracene,
and coronene were not detected. In earthworms (Lumbricus
terrestris), benzo [ghi]perylene was the most abundant PAH (28
µg/kg wet weight); phenanthrene, fluoranthene, pyrene, chrysene,
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene were
present at about 10 µg/kg; and naphthalene and dibenz [a,h]anthracene
were not detected (Pathirana et al., 1994).
The PAH concentrations in earthworms did not seem to be affected by
the location in which the worms lived, but those in the faeces showed
a significant dependence on location. In a survey of earthworm faeces
from the Bornhöveder Lake district in 1988, the concentrations of
phenanthrene, fluoranthene, pyrene, and benzo [b]fluoranthene plus
benzo [j]fluoranthene plus benzo [k]fluoranthene were in the range
of 45 µg/kg; those of benz [a]anthracene, chrysene plus triphenylene,
benzo [e]pyrene, benzo [a]pyrene, indeno[1,2,3- cd]pyrene, and
benzo [ghi]perylene were about 20 µg/kg; and those of anthracene,
benzo [ghi]fluoranthene plus benzo [c]phenanthrene,
dibenz [a,h]anthracene, anthanthrene, and coronene were < 5 µg/kg.
Earthworm faeces in the Saarland contained 250-770 µg/kg
benzo [a]pyrene, and Allolobophora longa earthworm faeces from a
highly industrialized region of eastern Germany (Halle, Leipzig)
contained even higher concentrations: 37-2100 µg/kg benzo [a]pyrene
and 36-1700 µg/kg benzo [e]pyrene. The faeces of the earthworm
Lumbricus terrestris contained 4.6-55 µg/kg benzo [a]pyrene and
6.5-50 µg/kg benzo [e]pyrene (Jacob & Grimmer, 1995).
In insects near the Hersey River, USA, the maximum concentrations of
PAH were 5500 µg/kg phenanthrene, 2900 µg/kg benz [a]anthracene, and
730 µg/kg benzo [a]pyrene (Black et al., 1981).
The lipid fraction of liver from herring gulls (Larus argentatus)
from Pigeon Island and Kingston, Ontario, Canada, contained 0.15 µg/kg
anthracene, 0.082 µg/kg fluoranthene, 0.076 µg/kg pyrene, 0.05 µg/kg
naphthalene, 0.044 µg/kg fluorene, 0.038 µg/kg acenaphthene, and 0.038
µg/kg benzo [a]pyrene (Environment Canada, 1994). The concentrations
of PAH in pooled samples taken from the eggs of herring gulls
(Larus argentatus) on the German North Sea islands Mellum and
Trischen during 1992-93 were below the limit of detection, except for
that of phenanthrene, which was 1 µg/kg wet weight (Jacob & Grimmer,
1994).
5.2 Exposure of the general population
Possible sources of nonoccupational exposure to PAH are:
- polluted ambient air (main emission sources: vehicle traffic,
industrial plants, and residential heating with wood, coal,
mineral oil) (see section 5.1.1);
- polluted indoor air (main emission sources: open stoves and
environmental tobacco smoke) (see Table 65);
- tobacco smoking (see Table 66);
- contaminated food and drinking-water (see sections 5.1.5 and
5.1.2.3)
- use of products containing PAH (coal-tar skin preparations and
coal-tar-containing hair shampoos);
- ingestion of house dust; and
- dermal absorption from contaminated soil and water.
5.2.1 Indoor air, tobacco smoke, and environmental tobacco smoke
PAH are found in indoor air (Table 65) mainly as a result of tobacco
smoking and residential heating with wood, coal, or, in some
developing countries, rural biomass. The PAH levels in indoor air
usually range from 1 to 50 ng/m3. The most abundant PAH were
phenanthrene and naphthalene, with levels of up to 2300 ng/m3. Homes
with gas heating systems had higher indoor levels than those with
electric heating systems (Chuang et al., 1991), and even higher levels
were detected in indoor air near open fireplaces (Alfheim & Ramdahl,
1984). Airtight residential wood-burning stoves seemed to have a minor
effect on the indoor air concentration of PAH (Alfheim & Ramdahl,
1984; Traynor et al., 1987), but in homes with non-airtight wood
stoves, 2-46 times higher PAH concentrations were found during heating
periods than during periods without heating (Daisey et al., 1989).
Emissions from unvented kerosene heaters can significantly affect
indoor air quality in mobile homes, with a maximim value for
naphthalene of 2300 ng/m3. Four of eight heaters investigated emitted
PAH-containing particles at levels that exceeded the USA ambient air
standards for airborne particles, with a 50% cutoff at the aerodynamic
diameter of 10 µm. When the kerosene heaters were in operation, the
concentrations of carcinogenic PAH (with four rings or more) in the
mobile homes were increased by 10-fold (Mumford et al., 1991).
Table 65. Polycyclic: aromatic hydrocarbon concentrations (ng/m3) in indoor air; main source, residential heating
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Acenaphthene NR 589-1649
Acenaphthylene NR 60-592
Anthracene 5-30 408 5-15 84 NR 9.9-11
Benz[a]anthracene 3-9 2-6 3-13 145 NR 0.9-5.5
Benzo[a]pyrene 13-370 0.3-12 1-7 3-23 150 < 0.009-1.34 0.34-3.5 2.0-490 8.5-29
Benzo[b]fluoranthene < 0.007-0.68 0.17-3.8 1.4-420 5.6-21
Benzo[e]pyrene < 0.06-1.36
Benzo[ghi]perylene 14-340 0.4-10 1-7 3-30 125 < 0.01-6.20 0.37-3.7 2.8-450 0.4-7.5
Benzo[k]fluoranthene 5-150 0.07-7 0.6-3 2-10 63 0.005-0.48 0.07-1.9 0.67-200 0.7-21
Chrysene 2-12 3-6 4-13 115 NR
Coronene NR
Cyclopenta[cd]pyrene NR
Dibenzo[a,e]pyrene NR
Dibenz[a,h]anthracene NR 3.3-25
Fluoranthene 16-56 16-24 16-50 208 0.07-1.18 87-268
Fluorene NR
Indeno[1,2,3-cd]pyrene 20-560 1-16 1-8 3-22 130 < 0.02-3.54 1.1-6.1 3.9-740 2.3-11
Phenanthrene 120-400 120-200 140-290 555 NR 31-64
Pyrene 0.02-1.53 1.0-20
ND, not determined; NR, not reported; /, single measurements;
[1] Wood-burning open fire-place, Netherlands (Slooff et al., 1989);
[2] Wood in multi-burner, Netherlands (Slooff et al., 1989);
[3] Coal, Netherlands (Slooff et al., 1989);
[4] Briquettes, Netherlands (Slooff et al., 1989);
[5] 'Icopower' heating, Netherlands (Slooff et al., 1989);
[6] Wood heating in seven homes, USA (Daisey et al., 1989);
[7] Wood burning in one home; volume, 236 m3; airtight stove, Truckee, USA, (elevation, 1800 m) (Traynor et al., 1987);
[8] Wood burning in one home; volume, 236 m3; non-airtight stove, Truckee, USA (elevation, 1800 m) (Traynor et al., 1987);
[9] Wood burning in one home with four different heaters, USA (Knight & Humphreys, 1985)
Analysed by high-performance liquid chromatography or gas chromatography
Table 65 (contd)
Compound [10] [11] [12] [13] [14] [15] [16] [17]
Acenaphthene NR 1-258
Acenaphthylene 10-120 21/68 25-36 NR 1-753
Anthracene 1.5-15 4.2-5.9 NR 0.1-80
Benz[a]anthracene 0.24-3.4 0.72/2.8 0.55-1.0 ND-3.81 25 100 1000 4000 5-1021
Benzo[a]pyrene 0.28-3.3 0.24/2.0 0.54-1.0 ND-4.13 14 700 600 3100 8-1645
Benzo[b]fluoranthene NR 2-930
Benzo[b]pyrene 0.33-10 1.4-3.0 NR 5-1106
Benzo[ghi]perylene 0.32-2.5 0.22/3.7 0.72-1.0 ND-5.4 4-802
Benzo[k]fluoranthene ND-7.81a 4-824
Chrysene 0.58-7.2 1.5/3.1 1.4-2.2 0.18-8.61 7-1439
Coronene 0.31-1.4 0.07/2.3 0.55-0.58 ND-4.75 NR
Cyclopenta[cd]pyrene 0.18-2.0 0.49/4.2 0.36-0.59 ND-2.38 10 700 400 5600 NR
Dibenzo[a,e]pyrene NR 11 700 600 200 NR
Dibenz[a,h]anthracene NR 8-958
Fluoranthene 6.2-23 16/11 11 2.4-37.4 5-1095
Fluorene NR 3-275
Indeno[1,2,3-cd]pyrene 0.24-1.8 0.15/1.3 0.48-0.79 ND-3.53 8400 500 2000 4-670
5-Methylcholanthrene NR 7300 200 200 NR
Naphthalene 750-2200 2300/950 1200-1600 NR NR
Phenanthrene 55-210 48/34 93-110 9.2-210 3-667
Pyrene 3.6-17 9.7/13 6.9-7.6 1.4-18.1 7-850
[10] Gas or electridy, USA (Wilson & Chuang, 1991);
[11] Kerosene; unvented heaters in mobile homes, Apex, USA (Mumford et al., 1991);
[12] Various heating in eight homes, Columbus, USA (Chuang et al., 1991);
[13] Various heating in 33 homes, USA (Wilson et al., 1991);
[14] Smoky coal, Xuan Wei, China (Mumford et al., 1987);
[15] Smokeless coal, Xuan Wei, China (Mumford et al., 1987);
[16] Wood, Xuan Wei, China (Mumford et al., 1987);
[17] Various cooking fuels (cattle dung, wood, kerosene, liquid petroleum gas) in 60 homes, India
(Raiyani et al., 1993b)
a Sum of benzofluranthenes
Table 66. Polycyclic aromatic hydrocarbon concentrations (ng/m3 in indoor air;
main source, environmental tobacco smoke
Compound [1] [2] [3] [4] [5] [6]
Acenaphthene 2.5 36
Acenaphthylene 14 177
Anthracene 2.8 25 1.5 < 1
Anthanthrene 0.5 1.5 < 1 2.5 3
Benz[a]anthracene 1.3 12 15 13
Benzo[a]fluorene 5.5 39
Benzo[a]pyrene 1.8 7.3 14 4.5 0.04-0.16 22
Benzo[b]fluoranthene 0.06-0.08
Benzo[b]fluorene 2.5
Benzo[e]pyrene 2.3 7.1 11 4.5 18
Benzo[ghi]fluoranthene 4.3 18 8.5 14
Benzo[ghi]perylene 2.5 5.8 7 2 0.09-0.36 17
Benzo[k]fluoranthene 0.02-0.06
Coronene 2.0 3.1
Fluoranthene 14 41 5 16 99
Indeno[1,2,3-cd]pyrene 2.3 5.8 1 1.5 0.13-0.45
1-Methylphenanthrene 6.6 38 < 1 3.5
Perylene 0.5 0.8 4 2.5 11
Phananthrene 38 168 3 1
Pyrene 13 32 13 21 66
[1] Office room (volume, 88 m3; ventilation, 176 m3/h; background sample after weekend,
Finland; vapour and particulate phase (Salomaa et al., 1988);
[2] Office room (Volume, 88 m3; ventilation, 176 m3/h; 6 h; 96 cigarettes, American
type, 10 different brands, both medium- and low tar, Finland; vapour and particulate
phase (Salomaa et al., 1988);
[3] House in a forest (room volume, 65 m3; air exchange, 2.0-2.3 turnovers/h); background
sample, Norway (Alfheim & Ramdahl, 1984);
[4] House in a forest (room volume, 65 m3; air exchange, 2.0-2.3 turnovers/h); with
tobacco smoking, Norway (Alfheim & Ramdahl, 1984);
[5] House in a residential, wooded area of Truckee, USA (elevation, 1800 m); volume,
236 m3; no stove (Traynor et al., 1987);
[6] Model room (volume, 36 m3); one air exchange/h, smoking of five cigarattes/h (Ministry
of Environment, 1979))
High-performance liquid chromatography or gas chromatography; concentration of particulate
phase, unless otherwise stated
Emissions from coal and wood combustion in open fires for cooking
purposes in unvented rooms in Xuan Wei County, China, contained
extremely high PAH concentrations (see also section 8). The highest
concentration (benzo [a]pyrene at 15 000 ng/m3) was measured in
fumes from smoky coal combustion. Coal combustion in open fires in
Xuan Wei homes emitted 15 µg/m3 of carcinogenic PAH, while wood
combustion emitted 3.1 µg/m3 (Mumford et al., 1987).
Cooking with rural biomass in open fires also led to high PAH levels
in indoor air, as measured in rural Indian households.
Benzo [a]pyrene was measured at a concentration of about 4 µg/m3
during the cooking period, which occupied about 10% of the household
activities over the year. The cooking fuels included baval, neem,
mango, rayan, and crop residues (Smith et al., 1983). The total
release of PAH into indoor air from this source is unknown but may be
of major importance, especially in developing countries. Very low PAH
emissions were found when liquid petroleum gas was used as a fuel for
cooking (Raiyani et al., 1993b). In contrast, the PAH content of
kitchen air in Berlin, in the industrialized part of Germany, was
similar to that encountered in ambient air (Seifert et al., 1983).
House dust may be another important source of indoor pollution with
PAH. In a study of the homes of four smokers and four nonsmokers in
Columbus, Ohio, USA, the sum of the concentrations of naphthalene,
acenaphthylene, acenaphthene, fluorene, phenanthrene, anthracene,
retene, fluoranthene, pyrene, benz [a]anthracene, chrysene,
cyclopenta [cd]pyrene, benzo [b]fluoranthene,
benzo [j]fluoranthene, benzo [k]fluoranthene, benzo [e]pyrene,
benzo [a]pyrene, indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene,
benzo [ghi]perylene, and coronene in house dust and in soil from the
entryway, the pathway, and the foundation of the houses was 16-580
mg/kg. The concentrations in house dust correlated well with those in
the entryway soil samples, and a weaker correlation was found with the
pathway soil samples, but the relationships were not statistically
significant (Chuang et al., 1995).
A special source of exposure to PAH is wood-heated saunas. The highest
concentrations were found in a smoke sauna, the second highest in a
preheated sauna where the flues were closed before use, and the lowest
concentrations in a sauna heated by continuous burning of wood.
Pyrene, fluoranthene, benz [a]anthracene, and phenanthrene were
present at the highest levels (100-330 µg/m3 air); other PAH were
present at < 50 µg/m3. The concentrations decreased from
benzo [e]pyrene > benzo [a]pyrene > benzo [a]fluorene >
anthra-cene > benzo [b]fluorene > fluorene (Häsänen et al., 1983).
The protocol of a study of total human environmental exposure included
direct monitoring of exposure to benzo [a]pyrene by inhalation and
ingestion during three periods of 14 days. The range and magnitude of
dietary exposure (2-500 ng/day) was much greater than that by
inhalation (10-50 ng/day). The levels of benzo [a]pyrene in indoor
air were closely correlated with the ambient levels in most homes
(Waldman et al., 1991).
Indoor air concentrations of individual PAH due mainly to cigarette
smoke are shown in Table 66, and the levels in mainstream and
sidestream smoke of cigarettes are listed in Table 67. The average PAH
levels ranged from 1 to 50 ng per cigarette, and the major components
were phenanthrene, naphthalene, benzo [a]pyrene, benzo [e]pyrene,
fluoranthene, and pyrene. Sidestream smoke was found to contain 10
times more PAH than mainstream smoke. The levels in sidestream smoke
were 42-2400 ng per cigarette (Grimmer et al., 1987). The PAH
concentrations in the mainstream smoke from filter cigarettes
increased with increasing puff volume (Funcke et al., 1986). In a
pilot study in Columbus, Ohio, USA, naphthalene was the most abundant
PAH; environmental tobacco smoke appeared to be the most significant
source of indoor pollution (Chuang et al., 1991).
Table 67. Concentrations of selected polycyclic aromatic hydrocarbons
in cigarette smoke
Compound Mainstream smoke Sidestream smoke
(µg/100 cigarettes) (µg/100 cigarettes)
Anthracene 2.3-23.5
Anthanthrene 0.2-2.2 3.9
Benz[a]anthracene 0.4-7.6
Benzo[b]fluoranthene 0.4-2.2
Benzo[b]fluoranthene 0.6-2.1
Benzo[k]fluoranthene 0.6-1.2
Benzo[ghi]fluoranthene 0.1-0.4
Benzo[a]fluorene 4.1-18.4 75.0
Benzo[b]fluorene 2.0
Benzo[ghi]perylene 0.3-3.9 9.8
Benzo[c]phenanthrene Present
Benzo[a]pyrene 0.5-7.8 2.5-19.9
Benzo[e]pyrene 0.2-2.5 13.5
Chrysene 0.6-9.6
Coronene 0.1
Dibenz[a,h]anthracene 0.4
Dibenzo[a,e]pyrene Present
Dibenzo[a,h]pyrene Present
Dibenzo[a,i]pyrene 0.17-0.32
Dibenzo[a,l]pyrene Present
Fluoranthene 1.0-27.2 126.0
Fluorene Present
Indeno[1,2,3-cd]pyrene 0.4-2.0
5-Methylcholanthrene 0.06
Perylene 0.3-0.5 3.9
Phenanthrene 8.5-62.4
Pyrene 5.0-27 39.0-101.0
Triphenylene Present
1-Methylphenanthrene 3.2
Adapted from International Agency for Research on Cancer (1985)
In studies in eight healthy male smokers, aged 20-40 years, the
benzo [a]pyrene intake from the smoking of 20 cigarettes per day was
calculated to be 150-750 ng/d, assuming a deposition rate for
particulate matter of 75% (Scherer et al., 1990).
The total concentration of 14 PAH (fluoranthene, pyrene,
benzo [a]fluorene, benz [a]anthracene, chrysene,
benzo [b]fluoranthene, benzo [j]fluoranthene,
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene, perylene,
dibenz [a,h]-anthracene, benzo [ghi]perylene, and anthanthrene)
measured in a 36-m3 room into which sidestream smoke from five German
cigarettes was introduced every hour, with one air change per hour,
was 429 ng/m3. Assuming that the daily inhalation volume for adults
is 18 m3 and that 20 h/d are spent indoors, the volume of indoor air
inhaled daily is 18 m3 × 20/24 = 15 m3. Thus, passive smokers are
exposed daily to 15 × 429 = 6435 ng PAH, including 15 × 22 = 330 ng
benzo [a]pyrene (Ministry of Environment, 1979). An intake of 11 ng
benzo [a]pyrene was estimated in another study on the basis of an
assumed breath volume of 0.5 m3/h , a deposition rate for particulate
matter of 11%, and an exposure time of 8 h, after monitoring in an
unventilated, 45-m3, furnished room (Scherer et al., 1990).
5.2.2 Food
Smoked and barbecued food in particular can contain PAH (Grimmer &
Düvel, 1970; McGill et al., 1982; de Vos et al., 1990; Menichini et
al., 1991b; see also section 5.1.5 and Tables 51-56). Preparation of
food with contaminated drinking-water (see section 5.1.2.3) may also
lead to exposure to PAH.
In 1989 and 1990, the levels of naphthalene and alkylated derivatives,
acenaphthene, acenaphthylene, fluorene, phenanthrene, anthracene,
fluoran-thene, 1-methylphenanthrene, pyrene, benz [a]anthracene,
chrysene, benzo [b]fluoranthene, benzo [k]fluoranthene,
benzo [e]pyrene, benzo [a]pyrene, perylene,
indeno[1,2,3- cd]pyrene, dibenz [a,h]anthracene, and
benzo [ghi]-perylene were measured in salmon, herring, cod, rockfish,
and halibut in the area of the Gulf of Alaska where oil spilled from
the tanker Exxon Valdez. As only the sums of the concentrations were
considered, there was no apparent difference from those in fish
samples taken from unpolluted control sites in 1989. In 1990, slightly
elevated PAH concentrations were found at the polluted sampling site.
Nevertheless, the fish from the area were considered to be safe for
human consumption by these investigators (Saxton et al., 1993).
In another special exposure situation, the average daily PAH intake of
the inhabitants of Kuwait due to consumption of seafood after the war
in the Persian Gulf was calculated to be 0.23 µg/day on the basis of
the concentrations monitored in local fish and shrimps (Saed et al.,
1995).
5.2.3 Other sources
Benzo [a]pyrene was detected in coal-tar-containing hair shampoos at
levels of 7000-61 000 µg/kg, and a tar bath lotion contained 150 000
µg/kg benzo [a]pyrene. No PAH were detected in hair shampoos made
from wood tar (State Chemical Analysis Institute Freiburg, 1995). PAH
are absorbed from coal-tar shampoos through the skin during hair
washing. Exposure during one washing with this type of shampoo, which
contains benzo [a]pyrene at 56 mg/kg, for anti-dandruff therapy
results in absorption of 0.45 µg/kg body weight, assuming 20 g
coal-tar, 70 kg body weight, and 3% dermal absorption (van Schooten et
al., 1994; see also section 8).
5.2.4 Intake of PAH by inhalation
Estimates of PAH intake from air are summarized in Table 68.
In an assessment of the risk for cancer due to air pollution in
Germany, the average volume of air inhaled during heavy work was
assumed to be 140 m3 per person per week. The maximum intake of
airborne benzo [a]pyrene per week was thus estimated to be
0.21 µg/week in rural areas, 0.84 µg/week in industrial areas, and
7 µg/week near emission sources (State Committee for Air Pollution
Control, 1992).
On the basis of an average inhalation of 15 m3 air per day, exposure
to benzo [a]pyrene was calculated to be 0.05 µg/d. In industrial
areas, the exposure was calculated to be four times higher (0.19 µg/d)
(Raiyani et al., 1993a).
5.2.5 Intake of PAH from food and drinking-water
Estimates of PAH intake from food are shown in Table 69. The values
for benzo [a]pyrene range from 0.14-1.6 µg/d.
The total dietary intake of some PAH in the United Kingdom was
estimated to be (µg/person per day): 1.1 for pyrene, 0.99 for
fluoranthene, 0.50 for chrysene, 0.25 for benzo [a]pyrene, 0.22 for
benz [a]anthracene, 0.21 for benzo [ghi]perylene, 0.18 for
benzo [b]fluoranthene, 0.17 for benzo [e]pyrene, 0.06 for
benzo [k]fluoranthene, and 0.03 for dibenz [a,h]anthracene. The
major contributors of PAH to the total dietary intake appeared to be
oils and fats, with 28% from butter, 20% from cheese, and 17% from
margarine, in respective dietary survey groups; cereals provided 56%
from white bread and 12% from flour. The oils and fats had the highest
individual PAH levels. Although cereals did not contain high levels of
individual PAH, they were the main contributor by weight to the total
in the diet. Fruits and vegetables contributed most of the rest of the
PAH in the diet, while milk and beverages were of minor importance.
Smoked meat and smoked fish made very small contributions to the food
groups to which they belonged, which themselves were not major
components of the diet (Dennis et al., 1983).
Table 68 Estimated intake of polycyclic aromatic hydrocarbons (µg/day per person) from ambient air
Compound [1] [2] [3] [4] [5] [6] [7] [8] [9]
Anthracene 0.005 0.001
Anthanthrene 0.015
Benz[a]anthracene 0.030 0.013
Benzo[a]pyrene 0.01-0.03a 0.0025-0.025 0.025 0.034a 0.0095-0.0435 0.004a 0.017 0.03-0.05 0.0005-0.20
0.02-0.12b
0.06-1.0c
Benzo[b]fluoranthene 0.060 0.029
Benzo[b]fluorene 0.002 0.002
Benzo[e]pyrene 0.035 0.022
Benzo[ghi]perylene 0.030 0.027
Benzo[j]fluoranthene 0.010
Benzo[k]fluoranthene 0.015 0.015
Chrysene 0.035
Coronene 0.025
Dibenz[a,h]anthracene 0.020 0.004
Fluoranthene 0.040 0.016
Fluorene 0.0005
Indeno[1,2,3-cd]pyrene 0.030 0.024
Perylene 0.015 0.003
Phenanethrene 0.200 0.007
Pyrene 0.040 0.017
Triphenylene 0.220
Table 68 (continued)
[1] Germany (maximum concentrations) (State Committee for Air Pollution Control, 1992);
[2] Italy (Menichini, 1992a);
[3] Netherlands (maximum concentrations) (Guicherit & Schulting, 1985);
[4] United Kingdom (maximum concentrations) (Butler & Crossley, 1979);
[5] USA (Santodonato et al., 1980);
[6] USA (WHO, 1987);
[7] Japan (maximum concentrations) (Matsumoto & Kashimoto, 1985);
[8] China (Chen et al., 1980);
[9] India (Chakraborti et al., 1988)
a Rural areas
b Industrial areas
c Near emission source
Table 69. Estimated intake of polycyciic aromatic hydrocarbons (µg/day per person, maximum values) from food
Compound [1] [2] [3] [4] [5] [6] [7] [8]
Anthracene 5.6
Anthanthrene 0.30
Benz[a]anthracene 0.14
Benzo[a]pyrene 0.36 0.14-1a 0.1-0.3b 0.12-0.42 0.5 0.5 0.48 0.16-1.6
0.2c
Benzo[b]fluoranthene 1.0
Benzo[ghi]perylene 7.6 0.3 0.9
Benzo[j]fluoranthene 0.90
Benzo[k]fluoranthene 0.30
Chrysene 0.90 5.0
Coronene 0.09
Dibenz[a,h]anthracene 0.10
Fluoranthene 4.3 3 10
Indeno[1,2,3-cd]pyrene 0.31 0.4 <0.3
Perylene 0.20
Phenanethrene 2.0
Pyrene 4.0 5.1
[1] Austria (Pfannhauser, 1991);
[2] Germany (State Committee for Pollution Control, 1992);
[3] Italy (Menichini, 1992a);
[4] Netherlands (de Vos et al., 1990);
[5] Market basket study, Netherlands (Vaessen et al., 1984);
[6] Duplicate diet study, Netherlands (Vaessen et al., 1984);
[7] United Kingdom (Dennis et al., 1983);
[8] USA (Santodonato et al., 1980)
a Concentration in µg/week
b Adult non-smoker (70 kg)
c Mean concentration
In Sweden, the annual intake per person of the sum of fluoranthene,
pyrene, benz [a]anthracene, chrysene, triphenylene,
benzo [b]fluoranthene, benzo [j]-fluoranthene,
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene, and
indeno[1,2,3- cd]pyrene was about 1 mg. Cereals again seemed to be
the main contributor (about 34%), followed by vegetables (about 18%)
and oils and fats (about 16%). Although smoked fish and meat products
had the highest PAH levels, they made a modest contribution since they
are minor components of the usual Swedish diet (Larsson, 1986).
5.3 Occupational exposure
PAH have been measured in the air at various workplaces. Studies in
which measurements were reported only as the benzene-soluble fraction
or some other summarizing parameter affected mainly by PAH are not
covered because they do not refer to individual substances. The
presence of PAH metabolites in biological samples (urine, blood) from
workers has been used as a biomarker, and 1-hydroxypyrene seems to be
a suitable marker in some workplaces (see section 8.2.3). No data were
available on occupational exposure during production and use.
Occupational exposure to PAH occurs by both inhalation and dermal
absorption. In coke-oven workers, 75% of their exposure to total
pyrene and 51% of that to benzo [a]pyrene occurs by cutaneous
transfer (Van Rooij et al., 1993a; see also section 6). The exposure
of workers due to deposition of airborne pyrene on the skin, detected
in wipe samples, can be summarized as follows: in refineries,
< 0.0045 µg/cm2 (detection limit), 26 samples below detection limit;
in hot-mix asphalt facilities, < 0.0045 µg/cm2, 25 samples below
detection limit; during paving, < 0.13-0.31 µg/cm2 found in two of
nine samples (assuming a body area of 1.8 m2, equivalent to 5600
µg/person per day); in asphalt roofing manufacture, < 0.0045-0.0091
µg/cm2 found in 1 of 29 samples (assuming a body area of 1.8 m2,
equivalent to 170 µg/person per day); in application of asphalt
roofing, < 0.0045 µg/cm2, 10 samples below detection limit; in a
wood preserving plant, 47-1500 µg pyrene per person per day. These
data indicate that skin penetration is an important factor in
estimating total body exposure to PAH.
5.3.1 Occupational exposure during processing and use of of coal and
petroleum products
The following section is based on data obtained up to the early 1980s
which were compiled by the IARC (1984b, 1985, 1989b). More recent
studies are presented in detail.
5.3.1.1 Coal coking
In studies of pollution of the atmosphere near coke-oven batteries,
the concentration of benzo [a]pyrene varied from < 0.1 in
administrative buildings and a pump house to 100-200 µg/m3 on the
machinery and discharge side of a battery roof. At the top of a coke
battery, the following concentrations of particulate and gaseous PAH
were measured by stationary sampling: naphthalene, 0-4.4
(particulate)/ 280-1200 (gaseous) µg/m3; acenaphthene, 0-17/6.0-100
µg/m3; fluorene, 0-58/23-130 µg/m3; phenanthrene, 27-890/6.7-280
µg/m3; anthracene, 9.6-310/6.0-91 µg/m3; 1-methylphenanthrene,
2.7-21/0-7.0 µg/m3; fluoranthene, 45-430/0-24 µg/m3; pyrene,
35-320/0-14 µg/m3; benzo [a]fluorene, 9.7-90/0-6.8 µg/m3;
benzo [b]fluorene, 3.1-61/0-0.3 µg/m3; benzo [c]phenanthrene,
2.6-49 µg/m3 (particulate); benz [a]anthracene, 5.4-160/< 0.4-1.6
µg/m3; benzo [b]fluoranthene, 5.5-67/0-0.7 µg/m3;
benzo [j]fluoranthene plus benzo [k]fluoranthene, 0-35/0-0.7 µg/m3;
benzo [e]pyrene, 8-73/0-0.2 µg/m3; benzo [a]pyrene, 14-130/0-1.5
µg/m3; perylene, 3.3-19/0-0.1 µg/m3; benzo [ghi]perylene, 8.7-45
µg/m3 (particulate); anthanthrene, 2.6-62 µg/m3 (particulate); and
coronene, 1.0-19 µg/m3 (particulate) (IARC, 1984b).
At eight sites in a German coke plant in 1981, including the top of
the oven and the cabin of a lorry driver, the following PAH
concentrations were measured: 2.7 µg/m3 fluoranthene, 1.9-170 µg/m3
pyrene, 0.38-37 µg/m3 benzo [c]phenanthrene, 0.22-21 µg/m3
cyclopenta [cd]pyrene, 1.2-120 µg/m3 benz [a]anthracene, 0.71-79
µg/m3 benzo [c]pyrene, 0.88-89 µg/m3 benzo [a]pyrene, 0.21-14
µg/m3 perylene, 0.37-27 µg/m3 benzo [ghi]perylene, 0.18-17 µg/m3
anthanthrene, and 0.93-6.5 µg/m3 coronene. The authors pointed out
that the concentrations may have been much higher previously (Manz et
al., 1983).
Measurements with personal air samplers in Germany and Sweden showed
benzo [a]pyrene concentrations varying from 0.16-33 µg/m3 for
coke-oven operators to 4.7-17 µg/m3 for lorry drivers. The ranges of
exposure to all PAH at different workplaces in the 1970s were: lorry
driver, 170-1000 µg/m3; coke-car operator, 4.8-73 µg/m3; jamb
cleaner, 62-240 µg/m3; door cleaner, 9.1-17 µg/m3; push-car
operator, 9.4-62 µg/m3; sweeper, 110 µg/m3; quench-car operator, 5.7
µg/m3; and wharf man, 360 µg/m3 (IARC, 1984b).
Personal air samples taken from 56 Dutch coke-oven workers in 1986
showed pyrene levels of < 0.6 µg/m3 (detection limit) to 9.8 µg/m3
(Jongeneelen et al., 1990). The results of more recent measurements in
personal air samples are shown in Table 70.
5.3.1.2 Coal gasification and coal liquefaction
The levels of individual PAH in area air samples in Norwegian and
British coal gasification plants between the late 1940s and the mid
1950s were in the low microgram per cubic millilitre range. In modern
gasification systems, the concentrations of total PAH are usually <
1 µg/m3, but in one of three plants examined the total aerial PAH
load was about 30 µg/m3. Personal samples taken in modern coal
gasification plants showed similar PAH concentrations (IARC, 1984b).
Table 70. Workplace exposures to polycyclic aromatic hydrocarbons in the atmosphere of coke-oven batteries
(µg/m3), determined from personal air samples
Compound [1] [2] [3] [4] [5] [6] [7]
Acenaphthene 3.8
Acenaphthylene 28
Anthracene 65 16
Anthanthrene 2.4
Benz[a]anthracene 0.11-33.19 96 7.5
Benzo[a]fluorene 70 3.7
Benzo[a]pyrene < 0.01-31.15a 0.03-12.63 0.9-46.02 38 0.1-29 7.3 1300
0.01-22.91b
Benzo[b]fluoranthene 42 1500
Benzo[b]fluorene 4.3
Benzo[c]phenanthrene 1.4
Benzo[e]pyrene 4.7
Benzo[ghi]fluoranthene 1.6
Benzo[ghi]perylene 4.4
Benzo[k]fluoranthene 42
Chrysene 0.08-13.17 72
Coronene 3.2
Cyclopenta[cd]pyrene 1.9
Fluoranthene 0.12-17.00a 144 22 4400
Fluorene 109 14
Indeno[1,2,3-cd]pyrene 4.5
1-Methylphenanthrene 3.4
Naphthalene 28-445a 650
Perylene 1.8
Phenanthrene 0.07-8.53a 195 49
Pyrene 2.36-98.63 17 Trace
Table 70 (continued)
[1] Finland; samples from one plant, 1988-90 (Yrjanheikki et al., 1995);
[2] Italy; samples from 69 workers, six workplaces (Assennato et al., 1993a);
[3] Italy; samples from three workplaces at battery top (Cenni et al., 1993);
[4] Sweden; one typical sample (Andersson et al., 1983);
[5] United Kingdom; samples from 12 plants (Davies et al., 1986);
[6] USA; samples from topside coke-oven workers (Haugen et al., 1986,
[7] India; samples from top of coke oven (Rao et al., 1987)
a Area air samples
b Personal air samples
In a pilot coal liquefaction plant in the United Kingdom, monitoring
of five operators for vapour-phase PAH gave following results:
1900-3300 ng/m3 phenanthrene, 340-670 ng/m3 pyrene, 270-380 ng/m3
fluoranthene, 29-130 ng/m3 anthracene, 22-1700 ng/m3 fluorene,
< 1-1800 ng/m3 naphthalene, < 1-1000 ng/m3 acenaphthene, and
< 1-8 ng/m3 acenaphthylene. The higher-molecular-mass PAH were not
detected (limit of detection, 1 ng/m3). Pyrene was detected in the
particulate phase at concentrations of 630-2900 ng/m3 (Quinlan et
al., 1995a).
5.3.1.3 Petroleum refining
Personal samples from operators of catalytic cracker units and
reaction and fractionation towers in a petroleum refinery showed total
PAH levels of 2.6-470 µg/m3. During performance and turn-round
operations on reaction and fractionation towers, naphthalene and its
methyl derivatives accounted for more than 99% of the total PAH
measured; exposure to anthracene, pyrene, chrysene, and
benzo [a]pyrene was < 1 µg/m3. Area monitoring for these PAH
during normal activities and during shut-down, leak-testing, and
start-up operations after turn-rounds gave total PAH concentrations up
to 400 µg/m3, most of the measurements being < 100 µg/m3 (IARC,
1989b).
The results of personal air sampling of workers at six jobs in seven
American refineries in 1990-91 were as follows (mean and range): 5.5
(< 0.25-10) µg/m3 naphthalene, 3.3 (< 0.44-24) µg/m3 acenaphthene,
3.3 (< 0.19-26) µg/m3 acenaphthylene, 0.98 (< 0.085-7.9) µg/m3
fluoranthene, 0.82 (< 0.055-6.7) µg/m3 phenanthrene, 0.78
(< 0.13-5.3) µg/m3 benzo [e]pyrene, 0.65 (< 0.055-5.2) µg/m3
benzo [b]fluoranthene, 0.47 (< 0.14-2.7) µg/m3 fluorene, 0.29
(< 0.11-1.4) µg/m3 indeno[1,2,3- cd]pyrene, 0.18 (< 0.085-0.69)
µg/m3 benz [a]anthracene, 0.16 (< 0.11-< 0.59) µg/m3
benzo [a]pyrene, 0.063 (< 0.028-0.26) µg/m3 anthracene, < 0.11-
< 0.2 µg/m3 pyrene, < 0.085-< 0.15 µg/m3 chrysene, < 0.085-
< 0.15 µg/m3 benzo [k]fluoran-thene, < 0.11-< 0.2 µg/m3
benzo [ghi]perylene, and < 0.11-< 0.2 µg/m3
dibenz [a,h]anthracene. Dermal wipe samples from the back of the hand
or from the forehead of workers showed PAH levels of < 0.0011-0.29
µg/cm2, with the highest level for naphthalene and the lowest for
anthracene (Radian Corp., 1991).
5.3.1.4 Road paving
In early studies on road paving operations, the total PAH
concentrations reported in personal air samples were 4-190 µg/m3, and
the mean in area air samples was 0.13 µg/m3. The benzo [a]pyrene
concentration in stationary samples was < 0.05-0.19 µg/m3 (IARC,
1985).
The concentrations of individual PAH in fume condensates from paving
asphalt were generally < 2 mg/kg condensate, varying by about seven
times depending on the source of crude oil. The levels of
benzo [a]pyrene, for example, were between 0.09 and 2.0 mg/kg
(Machado et al., 1993).
Fourteen stationary air samples from a road paving site in New Zealand
in 1983 contained: 0.14-52 µg/m3 benz [a]anthracene plus chrysene,
0.2-14 µg/m3 benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, 0.15-9.0 µg/m3 benzo [a]pyrene, 0.31-5.4
µg/m3 benzo [e]pyrene, 0.039-2.2 µg/m3 perylene, 0.24-5.4 µg/m3
benzo [ghi]perylene, and 0.03-6.3 µg/m3 indeno[1,2,3- cd]pyrene
plus dibenz [a,h]anthracene (Swallow & van Noort, 1985). The
concentrations in 17 stationary air samples from a road paving
operation in New Zealand in another study (year not given) were:
1.2-18 µg/m3 benz [a]anthracene plus chrysene, 1.1-11 µg/m3
benzo [b]fluoranthene plus benzo [j]fluoranthene plus
benzo [k]fluoranthene, 0.9-9.0 µg/m3 benzo [a]pyrene, 0.7-5.4
µg/m3 benzo [e]pyrene, and 0.7-6.3 µg/m3 indeno[1,2,3- cd]pyrene
(Darby et al., 1986). Concentrations of up to 1.3 µg/m3 were found
for acenaphthene, < 0.13 µg/m3 for anthracene, and < 0.54
µg/m3 pyrene in road-paving operations. The workers, and especially
the machine driver, were exposed to a mixture of bitumen fumes and
diesel exhaust gases for 4-6 h per day (Monarca et al., 1987).
The PAH concentrations in personal air samples obtained from workers
at six jobs in six paving operations in the USA in 1990 were (mean and
range): 6.5 (1.3-15) µg/m3 naphthalene, 2 (< 0.54-6.9) µg/m3
acenaphthene, 2 (< 0.24-8.1) µg/m3 acenaphthylene, 0.58
(< 0.19-0.98) µg/m3 fluorene, 0.55 (< 0.085-1.3) µg/m3
phenanthrene, 0.26 (< 0.11-0.37) µg/m3 fluoranthene, 0.17
(< 0.13-< 0.31) µg/m3 pyrene, 0.16 (< 0.13-0.27) µg/m3
benzo [e]pyrene, 0.13 (< 0.099-< 0.2) µg/m3 chrysene, 0.052
(< 0.034-0.11) µg/m3 anthracene, < 0.099-< 0.12 µg/m3
benz [a]anthracene, < 0.064-< 0.085 µg/m3 benzo [b]fluoranthene,
< 0.099-< 0.12 µg/m3 benzo [k]fluoranthene, < 0.13-< 0.25 µg/m3
benzo [a]pyrene, < 0.13-< 0.16 µg/m3 benzo [ghi]perylene,
< 0.13-< 0.16 µg/m3 indeno[1,2,3- cd]pyrene, and < 0.13-< 0.16
µg/m3 dibenz [a,h]anthracene. Dermal wipe samples from the back of
the hand and from the forehead of workers contained PAH at <
0.00004-0.43 µg/cm2, with the highest level for naphthalene and the
lowest for anthracene and pyrene (Radian Corp., 1991).
Measurements in the air in France during road paving with different
bitumens and tars showed the highest benzo [a]pyrene concentrations
with hard-coal tar (1-6 µg/m3) and the lowest with petroleum-based
bitumen (0.004-0.007 µg/m3). In general, the benzo [a]pyrene levels
in the workplace atmosphere were two to three orders of magnitude
higher during paving operations with tar products than with bitumen
products (Barat, 1991).
5.3.1.5 Roofing
The concentrations of PAH measured during roofing and roofing
manufacture are shown in Table 71.
The concentrations of individual PAH in fume condensates from roofing
asphalt generated at 232 and 316°C the were usually < 10 mg/kg
condensate, with higher levels only for naphthalene. They varied with
the source of crude oil: those for benzo [a]pyrene were between 0.6
and 2.8 mg/kg (Machado et al., 1993).
Acenaphthene was detected at concentrations of 1.4-2.1 µg/m3 in
personal samples from roofing workers at two US roofing sites in 1985
(Zey & Stephenson, 1986); 0.8-22 µg/m3 phenanthrene were measured at
one US roofing site in 1981 (Reed, 1983). Pyrene was measured at
< 190 µg/m3 at three roofing sites in Canada (year not given)
(Malaiyandi et al., 1986). Personal air samples from 12 roofers at one
US roofing site contained benzo [a]pyrene at 0.53-2.0 µg/m3 in 1987
(Herbert et al., 1990a). The workplace concentrations during bitumen
and coal-tar pitch roofing, waterproofing, and flooring operations
were of the same order of magnitude (IARC, 1985).
Significant, 10-fold differences were found in the levels of
anthracene, fluoranthene, pyrene, benz [a]anthracene,
benzo [b]fluoranthene, benzo [k]-fluoranthene, benzo [a]pyrene, and
benzo [ghi]perylene on skin wipes from the forehead taken before and
after a shift in 10 US roofers in 1987 (Wolff et al., 1989a).
Comparable results for benzo [a]pyrene levels were obtained for 12
roofers at another US roofing site (Herbert et al., 1990a,b).
Dermal wipe samples from the back of the hand or the forehead of
workers at six asphalt roofing manufacturing sites in the USA showed
PAH levels of < 0.12-5.5 µg/cm2, with the highest level for
acenaphthylene and the lowest for fluoranthene, benz [a]anthracene,
benzo [k]fluoranthene, and chrysene. Similar samples from workers at
six asphalt roofing sites in the USA in 1990-91 showed PAH levels of
< 0.0011-0.0045 µg/cm2, with the highest levels for pyrene,
chrysene, and benzo [a]pyrene and the lowest for anthracene (Radian
Corp., 1991).
5.3.1.6 Impregnation of wood with creosotes
Concentrations of PAH ranging from 0.05 µg/m3 benzo [a]pyrene to
650 µg/m3 naphthalene were detected during the handling of
creosote-impregnated wood for railroad ties in Sweden. Naphthalene,
fluorene and phenanthrene were by far the most abundant compounds
(> 100 µg/m3) (Andersson et al., 1983). Concentrations of 0.04-0.28
µg/m3 anthracene and 0.11-7.7 µg/m3 pyrene were found at workplaces
in Finland where railroad ties were manufactured (Korhonen & Mulari,
1983), and concentrations of 1-19 µg/m3 anthracene, 6.5-61 µg/m3
phenanthrene, and 0.6-13 µg/m3 pyrene were measured in one plant
where railroad sleepers were impregnated and in another where poles
Table 71. Exposure to polycyclic aromatic hydrocarbons (µg/m3) during roofing and roofing manufacture
Compound [1] [2] [3] [4]
Acenaphthene < 0.52-3.2 (0.87) < 0.6-6.7 (1.5)
Acenaphthylene < 0.23-29 (7.1) < 0.26-12 (2.9)
Anthracene 0.5/1.5 < 0.033-0.069 (0.043) < 0.037-0.042
Anthanthrene < 0.030
Benz[a]anthracene < 0.03-0.130 1.3/2.5 < 0.099-< 0.13 < 0.11-< 0.13
Benzo[a]fluorene 0.03-0.080
Benzo[a]pyrene < 0.03-0.037 0.9/1.5 < 0.13-< 0.18 < 0.11-< 0.13
Benzo[b]fluoranthene < 0.03-0.093a 0.8/1.2 < 0.065-< 0.38 (0.13) < 0.078-< 0.085
Benzo[b]fluorene 0.051-0.093
Benzo[e]pyrene < 0.03-0.110 < 0.13-3 (0.61) < 0.15-< 0.17
Benzo[ghi]fluoranthene < 0.03
Benzo[ghi]perylene < 0.03-0.069 0.6/0.9 < 0.13-< 0.18 < 0.15-< 0.17
Benzo[k]fluoranthene 0.4/0.7 < 0.099-< 0.13 < 0.099-< 0.12
Chrysene 0.038-0.214 < 0.099-< 0.13 < 0.11-< 0.13
Coronene < 0.03
Dibenz[a,h]anthracene < 0.03 < 0.13-< 0.18 < 0.15-< 0.17
Fluoranthene 0.084-0.234 3.1/7 < 0.099-4 (0.64) < 0.11-0.13
Fluorene < 0.16-14 (2.5) < 0.19-1.1 (0.44)
Indeno[1,2,3-cd]pyrene < 0.030 < 0.13-< 0.18 < 0.15-0.94 (0.16)
Naphthalene < 0.22-9.2 (5.2) 1.2-25 (7.5)
Perylene < 0.030
Phenanthrene < 0.065-1.7 (0.53) < 0.078-1.4 (0.38)
Pyrene 0.035-0.183 2.6/5.4 < 0.13-3.4 (0.76) < 0.15-< 0.73 (0.25)
/, single determinations; mean values shown in parentheses;
[1] Germany; personal and area air samples from one bitumen roofing site (Schmidt, 1992);
[2] USA; personal air samples from nine workers; 1987 (Wolff, M.S. et al., 1989);
[3] USA; personal air samples from six asphalt roofing sites; 1990 (Radian Corp., 1991);
[4] USA; personal air samples from six roofing manufacturing sites; 1990 (Radian Corp., 1991)
a Benzo[b+j+k]fluoranthenes
were preserved (year not given) (Heikkilä et al., 1987). In
measurements of personal air samples from 10 workers in a Dutch plant
for impregnation of railroad sleepers in 1991, 0.3-1.3 µg pyrene/m3
was measured in the breathing zone and 47-1500 µg/d in pads placed on
various areas of the skin of the workers. Dermal exposure was shown to
be reduced by up to 90% by the use of protective clothing (Van Rooij
et al., 1993b).
5.3.1.7 Other exposures
In area air samples taken near the bitumen processing devices of
refineries, the total PAH levels varied from 0.004 to 50 µg/m3 (IARC,
1985, 1989b).
The use of lubricating oils may result in exposure to PAH. At two
Italian glass manufacturing plants, phenanthrene, anthracene, pyrene,
and fluoranthene were found in personal air samples at concentrations
< 3 µg/m3 (year not given) (Menichini et al., 1990). The pyrene
levels resulting from use of lubricating oils in Italian earthenware
factories were 0.02-0.09 µg/m3; the benzo [a]pyrene concentration
was below the limit of detection (Cenni et al., 1993). Measurable
concentrations of individual PAH were detected in indoor air above
asphalt floor tiles in e.g. warehouses, factories, and manufacturing
plants. The concentrations at six sampling sites in Germany were
between < 0.01 ng/m3 for benzo [ghi]perylene and 3.3 ng/m3 for
chrysene. The concentrations of phenanthrene, pyrene, fluoranthene,
chrysene, and benzo [b]fluorene in particular were higher than those
in outdoor air (Luther et al., 1990).
In two Swiss plants for the production of silicon carbide, personal
air samples from four and five workers, respectively, contained the
following PAH levels: 4-140 ng/m3 acenaphthylene, 8-86 ng/m3
acenaphthene, 11-500 ng/m3 fluorene, 88-1400 ng/m3 phenanthrene,
3-250 ng/m3 anthracene, 20-1100 ng/m3 fluoranthene, 30-2500 ng/m3
pyrene, 7-6400 ng/m3 benz [a]-anthracene, 37-14 000 ng/m3 chrysene,
11-3700 ng/m3 benzo [b]fluoranthene plus benzo [j]fluoranthene,
3-470 ng/m3 benzo [k]fluoranthene, 18-3800 ng/m3 benzo [e]pyrene,
4-630 ng/m3 benzo [a]pyrene, 2-250 ng/m3 indeno[1,2,3- cd]pyrene,
2-520 ng/m3 dibenz [a,h]anthracene, 4-550 ng/m3
benzo [ghi]-perylene, and 4-34 ng/m3 coronene (Petry et al., 1994).
5.3.2 Occupational exposure resulting from incomplete combustion of
mineral oil, coal, and their products
5.3.2.1 Aluminium production
Early measurements of atmospheric benzo [a]pyrene at workplaces in
the aluminium industry showed concentrations of 0.02-970 µg/m3 in
personal air samples and 0.03-5.3 µg/m3 in area air samples. In the
atmosphere of an aluminium production plant, naphthalene, fluorene,
phenanthrene, anthracene, fluoranthene, pyrene, benzo [a]fluorene,
benzo [b]fluorene, benzo [c]phenan-threne, benz [a]anthracene,
chrysene, triphenylene, benzo [b]fluoranthene plus
benzo [k]fluoranthene, benzo [e]pyrene, benzo [a]pyrene,
benzo [ghi]perylene, anthanthrene, and coronene were found at
concentrations < 400 µg/m3. The most abundant compounds were
phenanthrene, naphthalene, fluorene, fluoranthene, and pyrene, at
concentrations > 100 µg/m3. The other substances occurred at
concentrations < 10 µg/m3 (IARC, 1984b).
The following concentrations of PAH were found in four stationary air
samples from an aluminium smelter in New Zealand in 1979: 0.37-9.6
µg/m3 benz [a]anthracene plus chrysene, 0.34-7.6 µg/m3
benzo [b+j+k]fluoranthenes, 0.12-2.6 µg/m3 benzo [e]pyrene,
0.19-4.1 µg/m3 benzo [a]pyrene, 0.05-1.5 µg/m3 perylene, 0.13-2.7
µg/m3 indeno[1,2,3- cd]pyrene plus dibenz [a,h]anthracene, and
0.12-3.3 µg/m3 benzo [ghi]perylene (Swallow & van Noort, 1985).
Similar levels were found in a typical personal air sample from a
Söderberg aluminium plant in Sweden (year not given) with, in
addition, 27 µg/m3 phenanthrene, 20 µg/m3 fluoranthene, 2.8 µg/m3
fluorene, 2.8 µg/m3 anthracene, 2.8 µg/m3 benzo [a]fluorene, and
< 1.0 µg/m3 naphthalene (Andersson et al., 1983).
In personal air samples from 38 workers in the Söderberg potroom of an
aluminium smelter in the humid tropics (location not given), mean
concentrations of < 1.0-48 µg/m3 benzo [a]pyrene and 3.5-130 µg/m3
pyrene were detected (Ny et al., 1993).
The arithmetic mean concentrations of PAH in workplace air samples
from the Canadian aluminium industry were 1100 µg/m3 naphthalene, 130
µg/m3 acenaphthene, 45 µg/m3 fluorene, 30 µg/m3 phenanthrene, 4.5
µg/m3 anthracene, 1.1 µg/m3 fluoranthene, and 0.58 µg/m3 pyrene.
The concentrations of benz [a]anthracene, chrysene, benzo [a]pyrene,
and benzo [e]pyrene were < 0.01 µg/m3 (Lesage et al., 1987).
Personal air samples from 18 workers in a US plant producing anodes
for use in aluminium reduction (year not given) showed pyrene
concentrations of 1.2-7.4 µg/m3 (Tolos et al., 1990).
Urine samples from 11 workers in Norwegian Söderberg aluminium plants
contained very low levels of unchanged PAH, although the
concentrations in the workplace air greatly exceeded the
concentrations in urban air. The total concentration of PAH
metabolites in the samples was 1.5-6 greater than that in a control
group (Becher & Bjrseth, 1983).
The PAH concentrations in the air of aluminium plants is reduced
dramatically by the use of tempered anodes instead of Söderberg
anodes. Measurements of benzo [a]pyrene levels in French factories
showed 1-36 µg/m3 in potrooms with Söderberg anodes and 0.004-0.6
µg/m3 in potrooms with tempered anodes (Barat, 1991).
5.3.2.2 Foundries
In personal air samples from workers in 10 Canadian foundries, mean
concentrations of 0.14-1.8 µg/m3 benz [a]anthracene plus chrysene,
0.09-1.2 µg/m3 benzo [a]pyrene, and 0.09-1.9 µg/m3
dibenz [a,h]anthracene were measured. The benzo [a]pyrene levels in
stationary air samples from six Finnish foundries were 0.01-13 µg/m3,
depending on whether coal-tar pitch or coal powder was used as the
moulding sand additive (IARC, 1984b).
In another study, the highest individual PAH levels were found in coke
making, moulding, and furnaces (Gibson et al., 1977). Personal air
samples from 67 Finnish foundry workers in 1990-91 showed
benzo [a]pyrene concentrations of 2-60 ng/m3 with a mean of 8.6
ng/m3 (Perera et al., 1994). Depending on the foundry process and
sand binder, the mean benzo [a]pyrene level in 29 French foundries
varied from 3 to 2300 ng/m3 (Lafontaine et al., 1990).
Concentrations of PAH measured in foundries are shown in Table 72.
5.3.2.3 Other workplaces
Personal air samples from German chimney sweeps (year not given; 115
samples) showed an average benzo [a]pyrene level of 0.09 µg/m3, but
eight of the samples exceeded 2 µg/m3. With an inhaled air volume of
10 m3 per working day, the daily intake of benzo [a]pyrene was
estimated to be 0.24-2.7 µg, with a median value of 1.3 µg (Knecht et
al., 1989).
In an Italian pyrite mine, pyrene levels of 0.03-0.21 µg/m3 were
measured in personal and area air samples. The benzo [a]pyrene
concentrations were below the limit of detection (Cenni et al., 1993).
Area air samples taken in China showed total PAH levels of 3-40 µg/m3
in two iron mines and 4-530 µg/m3 in four copper mines. Individual
compounds were not identified, but the main components were
naphthalene and acenaphthene in the iron mines and naphthalene,
benz [a]anthracene, benzo [b]fluoranthene, benzo [a]pyrene,
benzo [e]pyrene, and dibenz [a,h]anthracene in the copper mines. The
PAH concentrations probably resulted from the drilling of holes with
hydraulic or pneumatic drills and by the transport of broken ore in
diesel-powered scoops (Wu et al., 1992).
Area and personal air samples from workers in a railway tunnel in
Italy showed pyrene levels of 0.04-0.30 µg/m3. The benzo [a]pyrene
concentrations ranged from below the limit of detection to 0.04 µg/m3
(Cenni et al., 1993).
Table 72. Exposure to polycyclic aromatic hydrocarbons (µg/m3)
in the atmosphere of foundries
Compound [1] [2] [3]
Acenaphthene 0.03
Acenaphthylene ND
Anthracene 2.31 0.05
Anthanthrene 0.64
Benz[a]anthracene 0.008-0.221 0.67 0.01
Benzo[a]fluorene 0.48
Benzo[a]pyrene 0.049-0.152 0.47 0.02
Benzo[b]fluoranthene 0.87a 0.003
Benzo[b]fluorene 0.41
Benzo[e]pyrene 0.48
Benzo[ghi]fluoranthene 0.15
Benzo[ghi]perylene 0.72 0.05
Benzo[k]fluoranthene 0.037-0.458 0.02
Chrysene 0.82b 0.02
Coronene 0.21
Dibenz[a,h]anthracene 0.20 ND
Fluoranthene 1.56 0.13
Fluorene 0.08
Indeno[1,2,3-cd]pyrene 0.81 ND
Naphthalene 9.68
Perylene 0.21
Phenanthrene 4.46 0.32
Pyrene 1.74 0.01
ND, not detected; /, single measurements;
[1] Canada, steel foundry: coke making, moulding, furnaces,
finishing, and cranes (Gibson et al., 1977);
[2] Western Germany, one foundry, area air samples (Knecht et al.,
1986);
[3] Denmark, 70 workers, personal air samples; melting, machine
moulding, casting, sand preparation (Omland et al., 1994)
a In sum with benzo(j+k)fluoranthene
b In sum with triphenylene
In the air of fish and meat smokehouses in Denmark (year not given),
the maximum concentration of naphthalene in stationary air samples was
about 2900 µg/m3. The most abundant compounds were naphthalene,
phenanthrene, pyrene, fluorene, anthracene, and fluoranthene
(> 100 µg/m3) (Nordholm et al., 1986). The minimal values were
< 1 µg/m3, benzo [a]pyrene being detected at minimal levels of
0.08 µg/m3 in meat smokehouses and 0.4 µg/m3 in fish smokehouses
(Hansen et al., 1991b), with a maximum concentration of 78 µg/m3
(Nordholm et al., 1986).
In a further study in nine Danish meat smokehouses, naphthalene was
detected at 21 µg/m3, fluorene at 6.9 µg/m3, fluoranthene at 6.6
µg/m3, phenanthrene at 5.6 µg/m3, acenaphthene at 5.2 µg/m3,
chrysene at 1.2 µg/m3, anthracene at 1.1 µg/m3, pyrene at 0.2
µg/m3, and benzo [ghi]perylene at 0.2 µg/m3 (Hansen et al., 1992).
The concentrations of naphthalene, fluorene, anthracene, phenanthrene,
pyrene, benzo [a]fluorene, chrysene, benzo [k]fluoranthene,
benzo [a]pyrene, benzo [e]pyrene, benzo [ghi]perylene, and
dibenz [a,h]anthracene in cooking fumes in a Finnish food factory,
three restaurants, and one bakery (year not given) during the frying
of meat and during deep-frying ranged between < 0.02 µg/m3 (the
limit of detection) and 26 µg/m3. Naphthalene occurred at by far the
highest concentration. Stationary air was sampled as close as possible
to the active working area and the workers' breathing zone (Vainiotalo
& Matveinen, 1993).
6. KINETICS AND METABOLISM IN LABORATORY MAMMALS AND HUMANS
Appraisal
Polycyclic aromatic hydrocarbons (PAH) are lipophilic compounds and
can be absorbed through the lungs, the gastrointestinal tract, and the
skin. In studies of the distribution of PAH in rodents, both the
parent compounds and their metabolites were found in almost all
tissues and particularly those rich in lipids. As a result of
mucociliary clearance and hepatobiliary excretion, they were present,
for example, in the gastrointestinal tract even when administered by
other routes.
The metabolism of PAH to more water-soluble derivatives, which is a
prerequisite for their excretion, is complex. Generally, the process
involves epoxidation of double bonds, a reaction catalysed by
cytochrome P450-dependent mono-oxygenases, rearrangement or hydration
of the epoxides to yield phenols or diols, respectively, and
conjugation of the hydroxylated derivatives. The reaction rates vary
widely: interindividual variations of up to 75-fold have been
observed, for example, with human macrophages, mammary epithelial
cells, and bronchial explants from different donors.
All aspects of the absorption, metabolism, activation, and excretion
of benzo[a]pyrene have been covered exhaustively in the published
literature, but there is a dearth of information on many of the other
PAH considered in this publication, particularly in humans. Thus, this
overview sets out general principles and describes pathways relevant
to benzo[a]pyrene in greater detail.
Most biotransformation leads to detoxification products that are
conjugated and excreted in the urine and faeces. The human body burden
of PAH has not been extensively studied, but tissue samples taken at
autopsy were found in one study to contain benzo[a]pyrene at an
average of 0.3 µg/100 g dry tissue; lung contained 0.2 µg/100 g. In
contrast, the pathways by which several PAH are metabolized to
reactive intermediates that bind covalently to nucleic acids have been
examined in great detail. Although the commonest mechanism in animals
and humans appears to involve the formation of diol epoxides, radical
cations and sulfate esters of hydroxymethyl derivatives may also be
important in certain cases.
6.1 Absorption
PAH are lipophilic compounds, soluble in organic solvents, that are
usually devoid of ionizable or polar groups. Like many other
xenobiotic substances, they would be expected to dissolve readily in,
and be transported through, the external and internal lipoprotein
membranes of mammalian cells. This is confirmed by the uptake of PAH
in vitro from media in which cells are maintained in culture and
modified metabolically by enzymes of the endoplasmic reticulum.
Furthermore, PAH are known to be able to cause biological effects
in vivo in cells and tissues that are distant from their site of
uptake by the organism.
In humans, the major routes of uptake of PAH are thought to be through
(i) the lungs and the respiratory tract after inhalation of
PAH-containing aerosols or of particulates to which a PAH, in the
solid state, has become absorbed; (ii) the gastrointestinal tract
after ingestion of contaminated food or water; and (iii) the skin as a
result of contact with PAH-bearing materials.
6.1.1 Absorption by inhalation
Investigations of the pulmonary absorption of PAH have frequently been
clouded by the existence of the mucociliary clearance mechanism, by
which hydrocarbons absorbed onto particulates that have been inhaled
are swept back up the pulmonary tree and are swallowed, thus entering
the organism through the gastrointestinal tract. Use of isolated
perfused rat lungs, however, provided a clear demonstration that
benzo [a]pyrene is absorbed directly through the pulmonary epithelia.
After intratracheal administration, both the hydrocarbon and its
metabolites were detected in effluent perfusion fluid (Vainio et al.,
1976). Other studies have shown that benzo [a]pyrene administered
in vivo as an aerosol is cleared from the lungs of rats by a
biphasic process in which an initial rapid phase (tracheal clearance)
is followed by a much slower second phase (alveolar clearance)
(Mitchell, 1982). PAH absorbed onto particles may take very much
longer to be cleared from rodent lungs, however, than the free
hydrocarbons, and the factors that affect this clearance rate include
the structure of the hydrocarbon and the dimensions and chemical
nature of the particles onto which the PAH are absorbed (Henry &
Kaufman, 1973; Creasia et al., 1976; Nagel et al., 1976). For example,
while 50% of the benzo [a]pyrene coated onto carbon particles of
15-30 µm was cleared from hamster lungs within 60 h, it took only 10 h
to clear 50% of the benzo [a]pyrene that had been coated onto
0.5-1.0-µm carbon particles. In a comparable experiment, however, when
ferric oxide particles of either 0.5-10 or 15-20 µm were used as
carriers for benzo [a]pyrene, 50% of the hydrocarbon was cleared in
just over 2 h, and carrier particle size did not affect the clearance
rates (Henry & Kaufman, 1973).
Benzo [a]pyrene was metabolized by the epithelia lining the nasal
cavities of hamsters, dogs, and monkeys when 14C-labelled hydrocarbon
was instilled as an aqueous suspension (Dahl et al., 1985;
Petridou-Fischer et al., 1988). From their studies with hamsters, the
authors concluded that when frequent small doses of 650 ng at 10-min
intervals were instilled into the nasal cavity, so as to imitate
inhalation, some 50% of the benzo [a]pyrene was metabolized; a large
fraction of the metabolites could be recovered from the mucus on the
epithelial surfaces; and the nasal epithelia were comparable to those
of the trachea and lungs in their ability to metabolize
benzo [a]pyrene. Metabolites produced nasally would be expected to be
swallowed and then absorbed in the gastrointestinal tract.
In humans, the concentrations of benzo [a]pyrene and pyrene present
in association with soot particles in the lungs were much lower than
would have been expected from the soot content. Thus, only a trace of
benzo [a]pyrene was found in one of 11 lung samples examined, in
which the expected benzo [a]pyrene content ranged from 9 to 200 µg;
in the other 10 samples, no benzo [a]pyrene was detected. Pyrene
disappeared more slowly: all 11 lung samples contained the compound,
at levels of 0.9-4.9 µg, whereas 3-190 µg might have been expected
(Falk et al., 1958). The ability of pulmonary epithelial cells to
metabolize PAH such as chrysene and benzo [a]pyrene to a variety of
hydroxylated derivatives (Jacob et al., 1992) may facilitate the
absorption and clearance of PAH from the lungs.
6.1.2 Absorption in the gastrointestinal tract
Indirect evidence for the gastrointestinal absorption of PAH was
provided by Shay et al. (1949), who found that repeated intragastric
instillation of 3-methylcholanthrene led to the development of mammary
cancer. Mammary tumours can also be induced in rats by intracolonic
adminstration of 7,12-dimethylbenz [a]anthracene (Huggins et al.,
1961). (3-Methylcholanthrene and 7,12-dimethylbenz [a]anthracene are
synthetic PAH that are potent carcinogens.) More direct investigations
by Rees et al. (1971) showed rapid absorption of intragastrically
administered benzo [a]pyrene; the highest levels of hydrocarbon were
found in the thoracic lymph some 3-4 h after administration. In a
report of studies of intact rats and intestinal sacs to examine the
mechanisms involved in benzo [a]pyrene absorption, Rees et al. (1971)
proposed that two sequential steps were involved, in which a phase of
absorption by the mucosa is followed by diffusion through the
intestinal lining. In a study with Sprague-Dawley rats, the presence
of bile was found to increase intestinal absorption of PAH such as
benzo [a]pyrene and 7,12-dimethylbenz [a]anthracene to a greater
degree than that of anthracene and pyrene. The effect may be related
to differences in the aqueous solubility of the PAH examined (Rahman
et al., 1986). The composition of the diet also affects intestinal
absorption of co-administered benzo [a]pyrene. Of the dietary
components studied, soya bean oil and triolein gave rise to the
highest levels of absorption of 14C-benzo [a]pyrene given orally at
a dose of 8.7 µg to Wistar rats, while cellulose, lignin, bread, rice
flake, and potato flake suppressed it (Kawamura et al., 1988).
6.1.3 Absorption through skin
PAH and PAH-containing materials have been applied dermally in
solution in solvents such as acetone and tetrahydrofuran. Dermal
transfer without use of a solvent was achieved by use of reconstituted
vapour-particulate phases emitted from coal-tar and bitumen (Genevois
et al., 1995) and by application in oil (Ingram et al., 1995).
Absorption of PAH through the skin was observed indirectly when it was
found that repeated topical application of 3-methylcholanthrene led to
the appearance of mammary tumours in mice (Maisin & Coolen, 1936;
Englebreth-Holm, 1941). The percutaneous mechanism of absorption is
not universal, however, since although almost all of a dose of
14C-benzo [a]pyrene applied to mouse skin appeared in the faeces
within two weeks, very little dibenz [a,h]anthracene was absorbed in
this way and most was lost through epidermal sloughing (Heidelberger &
Weiss, 1951). Benzo [a]pyrene has been shown to be absorbed
percutaneously in vitro, by absorption from soil into human skin
(Wester et al., 1990) and, after application as a solution in acetone,
into discs of human, mouse, marmoset, rat, rabbit, and guinea-pig skin
(Kao et al., 1985). In the latter experiments, marked interspecies
differences were noted: 10% of the applied dose (10 µg/5 cm2) of
14C-benzo [a]pyrene permeated mouse skin, 3% crossed human skin, and
< 0.5% crossed guinea-pig skin within 24 h. It was concluded that
both diffusional and metabolic processes are involved in the
percutaneous absorption of benzo [a]pyrene.
In Wistar rats that received 14C-pyrene as a solution in acetone on
areas of shaved dorsal skin, the rate of uptake was relatively rapid
(half-life, 0.5-0.8 d). The concentrations of pyrene were highest in
the liver, kidneys, and fat, but those of pyrene metabolites were
highest in the lungs. About 50% of an applied dose of 2, 6, or 15
mg/kg bw was excreted in the urine and faeces during the first six
days after treatment (Withey et al., 1993).
In studies with 32P-postlabelling for the detection of DNA adducts,
when complex mixtures of PAH, such as that present in used lubricating
oil from petrol engines, in coal-tar, or in juniper-tar, were applied
directly to mouse skin, appreciable, persistent levels of DNA adducts
(50-750 amol/µg DNA [1 amol/µg DNA equivalent to 3.3 adducts/1010
nucleotides]) were formed in the lungs (Schoket et al., 1989, 1990).
The level of adducts in mouse skin was inversely related to the
viscosity of the oil applied (Ingram et al., 1995).
Evidence for percutaneous absorption of PAH has also been obtained in
humans in vivo. When 2% coal-tar in petroleum jelly was applied
topically, phenanthrene, anthracene, pyrene, and fluoranthene were
detected in peripheral blood samples (Storer et al., 1984). In
addition, volunteers treated topically with creosote (100 µl) or
pyrene (500 µg, applied as a solution in toluene) and a psoriasis
patient who used a coal-tar shampoo excreted 1-hydroxypyrene in their
urine. In each case, maximal excretion occurred 10-15 h after
treatment (Viau & Vyskocil, 1995).
6.2 Distribution
The whole-body distribution of PAH has been studied in rodents. The
levels found in individual tissues depend on a number of factors,
including the PAH, the route of administration, the vehicle, the times
after treatment at which tissues are assayed, and the presence or
absence of inducers or inhibitors of hydrocarbon metabolism within the
organism. The investigations have shown that (i) detectable levels of
PAH occur in almost all internal organs, (ii) organs rich in adipose
tissue can serve as storage depots from which the hydrocarbons are
gradually released, and (iii) the gastrointestinal tract contains high
levels of hydrocarbon and metabolites, even when PAH are administered
by other routes, as a result of mucociliary clearance and swallowing
or hepatobiliary excretion (Heidelberger & Jones, 1948; Heidelberger &
Weiss, 1951; Kotin et al., 1959; Bock & Dao, 1961; Takahashi &
Yasuhira, 1973; Takahashi, 1978; Mitchell, 1982).
14C-Benzo [a]pyrene injected intravenously at 11 µg/rat was cleared
rapidly from the bloodstream, with a half-life of < 1 min (Kotin et
al., 1959), as confirmed by Schlede et al. (1970a,b), who also noted
that the rate of clearance was increased when animals were pretreated
with 20 mg/kg bw non-radioactive benzo [a]pyrene or 37 mg/kg bw
phenobarbital, both of which can induce metabolism.
The distribution of 3-methylcholanthrene in mice and their fetuses was
studied by whole-body autoradiography. When 1 mg of 14C-labelled
hydrocarbon is injected intravenously, it is not only widely
distributed in maternal tissues but also crosses the placenta and can
be detected in the fetuses (Takahashi & Yasuhira, 1973; Takahashi,
1978), in which it induces pulmonary tumours (Tomatis, 1973; see also
Section 7). The distribution of inhaled and intragastrically or
intravenously administered benzo [a]pyrene and
7,12-dimethylbenz [a]anthracene in rats and mice has also been
studied, with similar results (Shendrikova & Aleksandrov, 1974;
Shendrikova et al., 1973, 1974; Neubert & Tapken, 1988; Withey et al.,
1992). Rapid transfer of radioactive benzo [a]pyrene across the
placenta was confirmed in experiments in which the appearance of
radioactivity in the umbilical vein of pregnant guinea-pigs was
measured (Kelman & Springer, 1982).
Samples of placenta, maternal blood, umbilical cord blood, and milk
from 24 women in south India were examined for the presence of
selected PAH. Although umbilical cord blood and milk showed the
highest levels (benzo [a]pyrene, 0.005-0.41 ppm;
dibenz [a,c]anthracene, 0.013-0.60 ppm; chrysene, 0.002-2.8 ppm),
only 50% of the samples examined contained detectable levels. The
authors concluded that developing fetuses and newborn infants were
exposed to these PAH, probably from the maternal diet (Madhavan &
Naidu, 1995).
After intratracheal administration to mice and rats, the distribution
of PAH was essentially similar to that found after intravenous or
subcutaneous injection (Kotin et al., 1959), except for the expected
high pulmonary levels. Detailed time-concentration curves for several
organs have been obtained after inhalation of 3H-benzo [a]pyrene
aerosols at 500 µg/litre of air (Mitchell, 1982). For example, 1 h
after the end of administration, the highest levels were present in
the stomach and small intestine; as these declined, the amounts of
radioactivity in the large intestine and caecum increased. The
elimination half-times in the respiratory tract were 2-3 h for the
initial rapid phase and 25-50 h for the subsequent slow phase.
6.3 Metabolic transformation
The metabolism of PAH follows the general scheme of xenobiotic
metabolism originally outlined by Williams (1959). The hydrocarbons
are first oxidized to form phase-I metabolites, including primary
metabolites, such as epoxides, phenols, and dihydrodiols, and then
secondary metabolites, such as diol epoxides, tetrahydrotetrols, and
phenol epoxides. The phase-I metabolites are then conjugated with
either glutathione, sulfate, or glucuronic acid to form phase-II
metabolites, which are much more polar and water-soluble than the
parent hydrocarbons.
The metabolism of PAH has been studied in vitro, usually in
microsomal fractions prepared from rat liver, although many other
tissue preparations have also been used. Metabolism in such systems
might be expected to be simpler than that in whole animals because the
enzymes and co-factors necessary for sulfate, glutathione, or
glucuronide conjugate formation may be removed, depleted, or diluted
during tissue fractionation. Use of these systems appears to be
justified, however, because the same types of phase-I metabolites are
formed when animals are treated with simple hydrocarbons such as
naphthalene as when the same hydrocarbon is incubated with hepatic
microsomes or tissue homogenates (Boyland et al., 1964). The
metabolism of PAH has thus been studied extensively in cells and
tissues in culture, which metabolize hydrocarbons to both phase-I and
phase-II metabolites and which probably better represent the
metabolism of PAH that occurs in vivo (for reviews see Conney, 1982;
Cooper et al., 1983; Dipple et al., 1984; Hall & Grover, 1990; Shaw &
Connell, 1994).
Particular attention has been paid to the metabolism of PAH in human
tissues that might be exposed to hydrocarbons present in food and in
the environment and which are, therefore, potential targets for the
carcinogenic action of PAH (Autrup & Harris, 1983). The cells and
tissues examined include the bronchus, the colon, mammary cell
aggregates, keratinocytes, monocytes, and lymphocytes. The metabolism
of PAH by human pulmonary macrophages has also received attention
(Autrup et al., 1978a; Harris et al., 1978a; Marshall et al., 1979)
because it is conceivable that metabolism by these cells might be
responsible, at least in part, for the high incidence of bronchial
cancer in smokers (Wynder et al., 1970). Macrophages can engulf
particulate matter that reaches the terminal airways of the lung and
thus would be expected, especially in smokers, to contain PAH
(Hoffmann et al., 1978). The macrophages and engulfed particulate
matter can then be transported to the bronchi where proximate and
ultimate carcinogens, formed by metabolism in the macrophages, could
leave the macrophages and enter the epithelial cells lining the
bronchi (Autrup et al., 1978a; Harris et al., 1978a). This is an
attractive theoretical mechanism which could account for the high
incidence of respiratory tumours at the junctions of the large bronchi
and which is supported by experimental evidence.
Extracts of organic material from isolated perfused lung tissues of
rabbits that had been exposed intratracheally to benzo [a]pyrene with
or without ferric oxide were analysed for benzo [a]pyrene metabolites
and for mutagenicity. Extracts of lung tissue exposed to
benzo [a]pyrene only were mutagenic and contained benzo [a]pyrene
metabolites. When ferric oxide was co-administered, only the
macrophage extracts were mutagenic, owing to relatively large amounts
of unmetabolized benzo [a]pyrene. These experiments demonstrate that
ferric oxide particles enhance the uptake of benzo [a]pyrene by lung
macrophages and slow its metabolism beyond the 3-h period during which
perfused lung systems can be maintained (Schoeny & Warshawsky, 1983).
Administration of particles in vitro enhances both the uptake and
metabolism of benzo [a]pyrene by hamster alveolar macrophages (Griefe
et al., 1988). Metabolites were found in both the cells and the
culture medium. Subsequent studies showed that concurrent
administration of benzo [a]pyrene and ferric oxide particles resulted
in increased benzo [a]pyrene metabolism and release of superoxides
(Greife & Warshawsky, 1993). In particular, the dihydrodiol fraction
was increased. These studies indicate that particulates may act in
lung cancer by changing the time frame for metabolism, shifting the
site of metabolism to macrophages and enhancing the production of
metabolites that are on the pathway to putative ultimate carcinogenic
forms. In this context, it has been demonstrated that particles of
various sorts exert different toxic effects on rat and hamster
pulmonary macrophages in vitro: ferric oxide and aluminium oxide
particulates were toxic, while crystalline silica was not (Warshawsky
et al., 1994).
The conclusion that the macrophage is the principal metabolizing cell
is further supported by the studies of Ladics et al. (1992a,b), who
demonstrated that the macrophage population was the only one in murine
spleen that could metabolize benzo [a]pyrene, while the other splenic
cell types examined, including B cells, T cells, polymorphonuclear
cells, and the splenic capsule, did not produce benzo [a]pyrene
metabolites above the background level.
Although the same types of metabolite are formed from PAH in many of
the cell and tissue preparations examined in culture, the relative
levels and the rates of formation of these metabolites depend on the
type of tissue or cell that is being studied and on the species and
strain of animal from which the metabolizing systems are prepared.
With heterogeneous populations such as humans, the rate of metabolism
depends on the individual from whom the tissues or cells are prepared.
For example, a 75-fold variation in the extent of hydrocarbon
activation was reported in studies of human bronchus (Harris et al.,
1976), and similar variations were observed among human mammary cell
aggregates (Grover et al., 1980; MacNicoll et al., 1980) and
macrophages (Autrup et al., 1978a). The pattern and role of metabolism
can also be varied by adding inhibitors of the enzymes that are
responsible for metabolism or by pretreating either cells in culture
or the animals from which the metabolizing systems are prepared with
enzyme inducers.
6.3.1 Cytochromes P450 and metabolism of PAH
The cytochromes P450 (CYP) are a superfamily of haemoproteins that
catalyse the oxidation of various endogenous molecules as well as
xenobiotics, including PAH. To date, about 250 genes that encode these
enzymes have been identified in various organisms. For classification
purposes, the CYP have been organized into families and subfamilies
according to their structural homology (Nelson et al., 1993).
Certain CYP belonging to families 1, 2, and 3 are expressed in
mammalian cells and are particularly important in xenobiotic
metabolism, and one or more member of each family is capable of
metabolizing one or more PAH (Guengerich & Shimada, 1991; Gonzalez &
Gelboin, 1994). Most studies to compare the catalytic properties of
different CYP have been carried out with model compounds such as
benzo [a]pyrene. They show that the catalytic properties (e.g. the
Vmax) of different CYP in PAH metabolism can differ essentially
(Shou et al., 1994).
In considering the contribution of a CYP enzyme to PAH metabolism
in vivo, two other parameters in addition to the catalytic
properties should be taken into account: the mode of regulation and
tissue specificity in its expression. Combinations of the three
factors should give an idea of the relative importance of an enzyme in
PAH metabolism.
6.3.1.1 Individual cytochrome P450 enzymes that metabolize PAH
CYP1A: CYP1A appears to be the only enzyme with metabolic capability
towards a wide variety of PAH molecules. It is expressed in various
tissues but at a generally low constitutive level (Guengerich &
Shimada, 1991). The induction of CYP1A1 is controlled by the Ah (aryl
hydrocarbon) receptor, a transcription factor that can be activated by
several ligands such as 2,3,7,8-tetradichlorobenzo- para-dioxin
(TCDD) and PAH, with variable potency (Negishi et al., 1981). Thus,
PAH and material containing PAH can regulate their own metabolism by
inducing CYP1A1. After induction, CYP1A1 expression may reach high
levels, e.g. in the placenta, lung, and peripheral blood cells;
however, in the liver, the principal organ of xenobiotic metabolism,
the level of expression is low even after induction, and other CYP
appear to be more important, at least in the metabolism of
benzo [a]pyrene (Guengerich & Shimada, 1991).
CYP1A2: The other member of the CYP1A family, CYP1A2, also
metabolizes PAH; however, its capacity to metabolize benzo [a]pyrene
to the 3-hydroxy metabolite, for example, is about one-fifth that of
CYP1A1 (Shou et al., 1994). Human CYP1A2 is nevertheless very active
in forming benzo [a]pyrene 7,8-dihydrodiol (Bauer et al., 1995) and
in forming diol epoxides from the 7,8-dihydrodiol (Shou et al., 1994).
There is also evidence that CYP1A2 can activate
7,12-dimethylbenz [a]anthracene to mutagenic species, albeit at a low
rate (Aoyama et al., 1989).
The expression of CYP1A2 is also regulated by the Ah receptor, but in
not exactly the same way as CYP1A1 (Negishi et al., 1981). In the
liver, for example, the level of CYP1A2 expression is much higher than
that of CYP1A1 (Guengerich & Shimada, 1991). While the capacity of
CYP1A2 to oxidize various PAH is more limited than that of CYP1A1, its
role in reactions like diol epoxide formation from benzo [a]pyrene in
the liver could be important because of its high level of expression.
CYP1B: The CYP1B subfamily was discovered only recently. Once the
enzyme had been isolated, it was found to be capable of metabolizing
PAH. Interestingly, its expression is also under the control of the Ah
receptor. Only limited information is available on its expression and
catalytic properties in different tissues, but it seems to be
expressed at least in mouse embryo fibroblasts (Savas et al., 1994),
rat adrenal glands (Bhattacharyya et al., 1995), and several human
tissues (Sutter et al., 1994). A number of PAH may act as substrates
for this enzyme (Shen et al., 1994).
CYP2B: When recombinant gene technology was used to express human
CYP2B6 cDNA in a human lymphoblastoid cell line, this enzyme was shown
to be capable of metabolizing benzo [a]pyrene to 3- and 9-phenols and
trans-dihydrodiols (Shou et al., 1994). In addition, CYP2B enzymes
may be involved in the metabolism of 7,12-dimethylbenz [a]anthracene
(Morrison et al., 1991a).
The constitutive levels of CYP2B enzymes are extremely low in human
liver, but they are strongly induced by phenobarbital and
phenobarbital-type inducers of CYP. Accordingly, immunological studies
of inhibition have shown that the CYP2B enzymes may play a significant
role in the metabolism of PAH, only when they are induced (Hall et
al., 1989; Honkakoski & Lang, 1989).
CYP2C: The CYP2C subfamily contains several members, some of which
are expressed at high levels in human liver. More than one member of
this subfamily may be capable of metabolizing PAH; thus, human CYP2C9
and, to a lesser extent, CYP2C8 metabolize benzo [a]pyrene to 3- and
9-phenols and trans-dihydrodiols (Shou et al., 1994). In addition,
CYP2C enzymes may play an essential role in the metabolism of
benzo [a]pyrene and 7,12-dimethyl-benz [a]anthracene, particularly
in phenobarbital-induced liver (Morrison et al., 1991a,b; Todorovic et
al., 1991). In view of the relative abundance of CYP in human liver
and their role in the metabolism of PAH, it has been suggested that
some CYP2C enzymes play an essential role in hepatic PAH metabolism
(Morrison et al., 1991b; Yun et al., 1992).
CYP3A: CYP3A is one of the most abundant CYP enzymes in human liver,
and it can metabolize benzo [a]pyrene and some of its dihydrodiols to
several metabolic products (Shimada et al., 1989; Yun et al., 1992;
Shou et al., 1994; Bauer et al., 1995). In one study, human CYP3A4 was
the most important single enzyme in the hepatic 3-hydroxylation of
benzo [a]pyrene (Yun et al., 1992).
6.3.1.2 Regulation of cytochrome P450 enzymes that metabolize PAH
All of the enzymes discussed above are inducible, and their level of
expression can be enhanced by external stimuli. CYP1A and CYP1B are
under the transcriptional control of the Ah receptor, which can be
activated by numerous PAH and other planar hydrocarbons, including
dioxins (Negishi et al., 1981; Guengerich & Shimada, 1991)
CYP2B enzymes can also be induced by foreign compounds but not through
the Ah receptor. The mechanism of induction of these enzymes is not
well understood, but their prototype inducer is phenobarbital; several
other drugs used clinically have similar effects (Gonzalez & Gelboin,
1994).
The regulation of CYP2C enzymes is complicated, and both endogenous
factors such as steroid hormones and exogenous factors such as
phenobarbital may be involved. Furthermore, different members of this
subfamily are regulated differently. The CYP3A are also regulated by
endogenous and exogenous factors; typical inducers of this subfamily
are rifampicin, dexamethasone, certain macrolide antibiotics, and
steroid hormones (Guengerich & Shimada, 1991).
Genetic polymorphisms of CYP1A1, CYP1A2, and some CYP2C and CYP3A
enzymes have also been described. Some of the genetic defects leading
to the polymorphism have been identified and can be used to predict an
individual's capacity to metabolize drugs, for example by the
polymerase chain reaction. Genetic polymorphism may lead to dramatic
changes in the capacity to metabolize PAH (Raunio & Pelkonen, 1994).
Studies with a few prototype compounds such as benzo [a]pyrene and
its metabolites and 7,12-dimethylbenz [a]anthracene indicate that
several CYP are involved in PAH metabolism. As each has its own
metabolic capacity, mode of regulation, and tissue-specific
expression, the one that plays a key role in PAH metabolism in vivo
at any one time may vary and will depend on the compound being
metabolized, pre-exposure to inducers of the CYP, the tissue and cell
type where the metabolism is taking place, and the genotype of the
individual in cases of genetic polymorphism.
Many PAH that are metabolized by the CYP-dependent mono-oxygenases
also induce the enzyme system. This ability of hydrocarbons to induce
their own metabolism usually results in lower tissue levels and more
rapid excretion of the hydrocarbon (Schlede et al., 1970b; Aitio,
1974). Although CYP1A1 is mainly responsible for activation of PAH in
the lung and CYP1A2 in the liver, most recent investigations have
shown that other CYP isoforms may also contribute to the metabolism of
PAH in mammals (Jacob et al., 1996). Thus, pretreatment of animals
with inducers of mono-oxygenase systems is frequently associated with
a decreased tumour incidence (Wattenberg, 1978). Conversely, studies
with strains of mice that differ genetically in the capacity of their
mono-oxygenase systems to be induced by PAH indicate that inducibility
may also be associated with an increased tumorigenic or toxicological
response (Nebert, 1980). Induction of the mono-oxygenase system by
different types of inducers can result in different profiles of
hydrocarbon metabolites, although the extent of the effect appears to
be variable (Holder et al., 1974; Jacob et al., 1981a,b; Schmoldt et
al., 1981). The metabolism of benzo [a]pyrene has been investigated
in more detail than that of other hydrocarbons and is used here as an
example.
6.3.2 Metabolism of benzo[a]pyrene
In early studies, the PAH metabolites isolated from or excreted by
experimental animals were shown to consist of hydroxylated
derivatives, commonly in the form of conjugates. Thus, the general
scheme of xenobiotic metabolism outlined above applies to PAH. One of
the principal interests in hydrocarbon metabolism arose, however, from
the realization that hydrocarbons, like many other environmental
carcinogens, are chemically unreactive and that their adverse
biological effects are probably mediated by electrophilic metabolites
capable of covalent interaction with critical macromolecules such as
DNA. Identification of the biologically active metabolites of PAH,
coupled with advances in both the synthesis of known and potential
hydrocarbon metabolites and the analysis of metabolites by
high-performance liquid chromatography, has led in the last two
decades to a greatly enhanced appreciation of the complexity of
hydrocarbon metabolism. Most of these metabolic interrelationships are
illustrated for benzo [a]pyrene in Figure 3; the structures of some
types of metabolites are given in Figure 4. The metabolism of
benzo [a]pyrene and other PAH has been reviewed (for example, Sims &
Grover, 1974, 1981; Conney, 1982; Cooper et al., 1983; Dipple et al.,
1984; Hall & Grover, 1990).
Benzo [a]pyrene is metabolized initially by the microsomal
CYP-dependent mono-oxygenase system to several epoxides (Figure 3).
Once formed, these epoxides (Sims & Grover, 1974) may spontaneously
rearrange to phenols, be hydrated to dihydrodiols in a reaction that
is catalysed by epoxide hydrolase (see review by Oesch 1973), or react
covalently with glutathione, either chemically or in a reaction
catalysed by glutathione S-transferase (Chasseaud, 1979).
6-Hydroxybenzo [a]pyrene is further oxidized either spontaneously or
metabolically to the 1,6-, 3,6-, or 6,12-quinone, and this phenol is
also a presumed intermediate in the oxidation of benzo [a]pyrene to
the three quinones that is catalysed by prostaglandin H synthase. Two
additional phenols may undergo further oxidative metabolism:
3-hydroxybenzo [a]pyrene is metabolized to the 3,6-quinone, and
9-hydroxybenzo [a]pyrene is oxidized to the K-region 4,5-oxide, which
is hydrated to the corresponding 9-hydroxy 4,5-dihydrodiol (Jernström
et al., 1978; for a formula showing a K-region, see Figure 11).
Phenols, quinones, and dihydrodiols can all be conjugated to yield
glucuronides and sulfate esters, and the quinones may also form
glutathione conjugates (Figure 5).
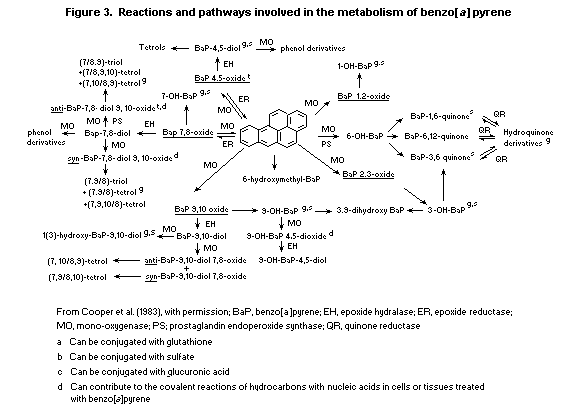
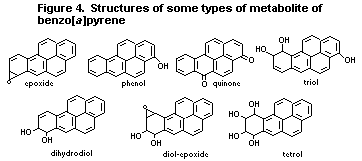
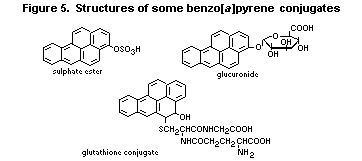 In addition to being conjugated, dihydrodiols can undergo further
oxidative metabolism. The mono-oxygenase system metabolizes
benzo [a]pyrene 4,5-diol to a number of metabolites, while the
9,10-dihydrodiol is metabolized predominantly to its 1- and 3-phenol
derivatives, only minor quantities of a 9,10-diol-7,8-epoxide being
formed. In contrast to 9,10-dihydrodiol metabolism, the principal
route of oxidative metabolism of benzo [a]pyrene 7,8-dihydrodiol is
to a 7,8-diol 9,10-epoxide, and triol formation is a minor pathway.
The diol epoxides can themselves be further metabolized to triol
epoxides and pentols (Dock et al., 1986) and can become conjugated
with glutathione either through chemical reaction or via a glutathione
S-transferase-catalysed reaction (Cooper et al., 1980; Jernström et
al., 1985; Robertson et al., 1986). They may also spontaneously
hydrolyse to tetrols, although epoxide hydrolase does not appear to
catalyse this hydration. Further oxidative metabolism of
benzo [a]pyrene 7,8-diol can also be catalysed by prostaglandin H
synthase (Marnett et al., 1978; Eling et al., 1986; Eling & Curtis,
1992), by a myeloperoxidase system (Mallett et al., 1991), or by
lipoxygenases (Hughes et al., 1989). These reactions may be of
particular importance in situations in which there are relatively low
levels of CYP (i.e. in uninduced cells and tissues) or when chronic
irritation and/or inflammation occurs, as during cigarette smoking
(Kensler et al., 1987; Ji & Marnett, 1992). The products detected have
included diol epoxides (Mallet et al., 1991; Ji & Marnett, 1992) and
tetrols (Sivarajah et al., 1979). Taken together, these reactions
illustrate that benzo [a]pyrene in particular, and PAH in general,
can undergo a multitude of simultaneous or sequential metabolic
transformations; they also illustrate the difficulty in determining
which metabolites are responsible for the various biological effects
resulting from treatment with the parent PAH.
An additional complexity of hydrocarbon metabolism stems from the fact
that the compounds are metabolized to optically active products.
Figure 6 illustrates the stereoselective metabolism of
benzo [a]pyrene to the 7,8-diol-9,10-epoxides. Four isomers may be
generated, since each diastereomer can be resolved into two
enantiomers. In rat liver microsomes, the (+) 7,8-epoxide of
benzo [a]pyrene is formed in excess relative to the (-) isomer, such
that more than 90% of the benzo [a]pyrene 7,8-oxide formed consists
of the (+) enantiomer (Levin et al., 1982). The epoxide is then
metabolized stereospecifically by epoxide hydrolase to the (-)
7,8-dihydrodiol. This metabolically predominant dihydrodiol is
metabolized in turn, primarily to a single diol epoxide isomer, the
(+) anti-benzo [a]pyrene 7,8-diol-9,10-epoxide. The biological
significance of the stereoselective formation of the
7,8-diol-9,10-epoxide isomers is that the metabolically predominant
isomer is also the isomer with the highest tumour-inducing activity
and that found predominantly to be covalently bound to DNA in a
variety of mammalian cells and organs that have been exposed to
benzo [a]pyrene.
In addition to being conjugated, dihydrodiols can undergo further
oxidative metabolism. The mono-oxygenase system metabolizes
benzo [a]pyrene 4,5-diol to a number of metabolites, while the
9,10-dihydrodiol is metabolized predominantly to its 1- and 3-phenol
derivatives, only minor quantities of a 9,10-diol-7,8-epoxide being
formed. In contrast to 9,10-dihydrodiol metabolism, the principal
route of oxidative metabolism of benzo [a]pyrene 7,8-dihydrodiol is
to a 7,8-diol 9,10-epoxide, and triol formation is a minor pathway.
The diol epoxides can themselves be further metabolized to triol
epoxides and pentols (Dock et al., 1986) and can become conjugated
with glutathione either through chemical reaction or via a glutathione
S-transferase-catalysed reaction (Cooper et al., 1980; Jernström et
al., 1985; Robertson et al., 1986). They may also spontaneously
hydrolyse to tetrols, although epoxide hydrolase does not appear to
catalyse this hydration. Further oxidative metabolism of
benzo [a]pyrene 7,8-diol can also be catalysed by prostaglandin H
synthase (Marnett et al., 1978; Eling et al., 1986; Eling & Curtis,
1992), by a myeloperoxidase system (Mallett et al., 1991), or by
lipoxygenases (Hughes et al., 1989). These reactions may be of
particular importance in situations in which there are relatively low
levels of CYP (i.e. in uninduced cells and tissues) or when chronic
irritation and/or inflammation occurs, as during cigarette smoking
(Kensler et al., 1987; Ji & Marnett, 1992). The products detected have
included diol epoxides (Mallet et al., 1991; Ji & Marnett, 1992) and
tetrols (Sivarajah et al., 1979). Taken together, these reactions
illustrate that benzo [a]pyrene in particular, and PAH in general,
can undergo a multitude of simultaneous or sequential metabolic
transformations; they also illustrate the difficulty in determining
which metabolites are responsible for the various biological effects
resulting from treatment with the parent PAH.
An additional complexity of hydrocarbon metabolism stems from the fact
that the compounds are metabolized to optically active products.
Figure 6 illustrates the stereoselective metabolism of
benzo [a]pyrene to the 7,8-diol-9,10-epoxides. Four isomers may be
generated, since each diastereomer can be resolved into two
enantiomers. In rat liver microsomes, the (+) 7,8-epoxide of
benzo [a]pyrene is formed in excess relative to the (-) isomer, such
that more than 90% of the benzo [a]pyrene 7,8-oxide formed consists
of the (+) enantiomer (Levin et al., 1982). The epoxide is then
metabolized stereospecifically by epoxide hydrolase to the (-)
7,8-dihydrodiol. This metabolically predominant dihydrodiol is
metabolized in turn, primarily to a single diol epoxide isomer, the
(+) anti-benzo [a]pyrene 7,8-diol-9,10-epoxide. The biological
significance of the stereoselective formation of the
7,8-diol-9,10-epoxide isomers is that the metabolically predominant
isomer is also the isomer with the highest tumour-inducing activity
and that found predominantly to be covalently bound to DNA in a
variety of mammalian cells and organs that have been exposed to
benzo [a]pyrene.
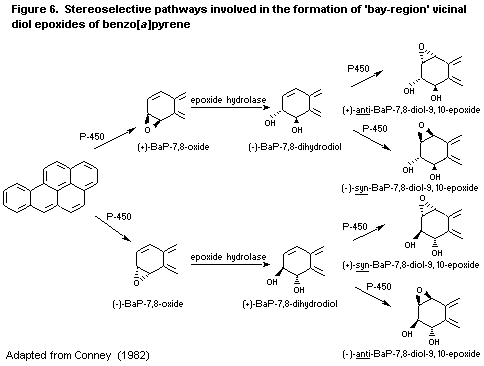 Benzo [a]pyrene metabolism has been examined extensively in human
tissue preparations, including human cells, explant cultures, tissue
homogenates, and microsomal preparations. Table 73 lists some studies
of the metabolism of benzo [a]pyrene in human tissues that included
metabolites soluble in organic solvents and water-soluble conjugates.
The results show that the metabolites produced by different human
tissues are qualitatively similar and that the metabolites detected
are the same as those formed in a variety of animal tissues.
The metabolic profiles reported in human tissues are almost all
identical to those seen for other eukaryotes, indicating the
involvement of similar enzyme systems. The same types of reactive
electrophilic intermediates found in other experimental systems also
appear to be formed in human tissues (Autrup & Harris, 1983). So far,
no differences in the metabolism or activation of benzo [a]pyrene
have been reported that might account for differences in the
susceptibility of different animal and human tissues to its
carcinogenic properties (see Section 7). Studies with cultured cells
and other substrates such as benz [a]anthracene, however, indicate
that bioactivation of PAH is species-dependent (Jacob, 1996).
6.4 Elimination and excretion
Most metabolites of PAH are excreted in faeces and urine. As complete
breakdown of the benzene rings of which unsubstituted PAH are composed
does not occur to any appreciable extent in higher organisms, very
little of an administered dose of an unsubstituted hydrocarbon would
be expected to appear as carbon dioxide in expired air.
The urinary excretion of PAH metabolites has been studied more
extensively than faecal excretion, but the importance of the
enterohepatic circulation of metabolites has led to increased research
on the latter. Detailed studies of the metabolism and excretion of PAH
in whole animals have been restricted mainly to the simpler compounds.
Because of the toxicity of the larger hydrocarbons and the complexity
of their metabolism, most studies on these compounds have been carried
out in hepatic homogenates and microsomal preparations or with
cultured cells (see above).
Metabolism and excretion in whole animals have been examined with
regard to naphthalene (Bourne & Young, 1934; Young, 1947; Booth &
Boyland, 1949; Corner & Young, 1954; Corner et al., 1954; Boyland &
Sims, 1958; Sims, 1959), anthracene (Boyland & Levi, 1935, 1936a,b;
Sims, 1964), phenanthrene (Boyland & Wolf, 1950; Sims, 1962; Boyland &
Sims, 1962a,b; Jacob et al., 1990b; Grimmer et al., 1991a), pyrene
(Harper, 1957, 1958a; Boyland & Sims, 1964a; Jacob et al., 1989,
1990b), benz [a]-anthracene (Harper 1959a,b; Boyland & Sims, 1964b),
and chrysene (Grimmer et al., 1988b, 1990). A limited number of
studies have been published on more complex compounds such as
benzo [a]pyrene (Berenblum & Schoental, 1943; Weigert & Mottram,
1946; Harper, 1958b,c; Falk et al., 1962; Raha, 1972; Jacob et al.,
1990b), dibenz [a,h]anthracene (Dobriner et al., 1939; Boyland et
al., 1941; La Budde & Heidelberger, 1958), and 3-methylcholanthrene
Table 73. Metabolites of benzo[a]pyrene formed by human tissues and cells
Tissue or Type of metabolite detected References
cell type
Dihydrodols Phenols Quinones Tetrols Conjugates
Bronchus + + + + + Pal et al. (1975);
Cohen et al. (1976);
Harris et al. (1977);
Autrup et al. (1978a,
1980)
Colon + + + + + Autrup et al. (1978b);
Autrup (1979)
Endometrium + + + Mass et al. (1981)
Fibroblasts + Baird & Diamond (1978)
Kidney + + + Prough et al. (1979)
Liver + + + + Selkirk et al. (1975);
Prough et al. (1979);
Pelkonen et al. (1977);
Diamond et al. (1980)
Lung + + + + + Cohen et al. (1976);
Stoner et al. (1978);
Mehta et al. (1979);
Prough et al. (1979);
Sipal. et al. (1979)
Lymphocytes + + + Booth et al. (1974);
Selkirk et al. (1975);
Vaught et al. (1978);
Okano et al. (1979);
Gurtoo et al. (1980)
Macrophages + + + + + Autrup et al. (1978a);
Harris et al. (1978a,b);
Autrup et al. (1979);
Marshall et al. (1979)
Table 73 (contd)
Tissue or Type of metabolite detected References
cell type
Dihydrodols Phenols Quinones Tetrols Conjugates
Mammary + Grover et al. (1980);
epithelium MacNicoll et al. (1980)
Monocytes + + + Vaught et al. (1978);
Okano et al. (1979)
Oesophagus + + + + Harris et al. (1979)
Placenta + + + Namkung & Juchau (1980);
Pelkonen & Saarni (1980)
Skin + + + + Fox et al. (1975);
Vermorken et al. (1979);
Parkinson & Newbold (1980);
Kuroki et al. (1980)
(Harper, 1959a; Takahashi & Yasuhira, 1972; Takahashi, 1978). Much of
the earlier qualitative work was reviewed by Boyland & Weigart (1947)
and by Young (1950). The absorption and excretion of different
hydrocarbons in vivo can differ. For example, while almost all of a
topically applied dose of benzo [a]pyrene appeared in mouse faeces
(Heidelberger & Weiss, 1951), little dibenz [a,h]-anthracene was
excreted by this route.
In rats given PAH either singly or as mixtures, the faecal elimination
of chrysene (25% of the dose) was not affected by co-administration of
benz [a]anthracene, but that of benz [a]anthracene was doubled, from
6 to 13% of the dose, when chrysene was given (Bartosek et al., 1984).
Such effects are relevant to human pharmacokinetics, since exposure is
almost always to mixtures of PAH. In workers in a coke plant exposed
to mixtures of PAH, the amounts of phenanthrene, pyrene, and
benzo [a]pyrene inhaled and the amounts of their principal
metabolites excreted in the urine were correlated (Grimmer et al.,
1994).
In rats, the amount of benzo [a]pyrene 7,8-diol excreted in the urine
is related to the susceptibility of individual animals to the
carcinogenic effects of benzo [a]pyrene (Likhachev et al., 1992;
Tyndyk et al., 1994). In studies of the disposition of
benzo [a]pyrene in rats, hamsters, and guinea-pigs after
intratracheal administration, the distribution of the hydrocarbon was
qualitatively similar but quantitatively different. In Sprague-Dawley
and Gunn rats and in guinea-pigs, the rate of excretion was dependent
on the dose administered, but in hamsters the rate of excretion was
independent of dose (0.16 or 350 µg 3H-benzo [a]pyrene) (Weyand &
Bevan 1986, 1987a). Evidence for enterohepatic circulation of
benzo [a]pyrene metabolites was obtained in Sprague-Dawley rats with
bile-duct cannulae treated by intratracheal instillation with 1 µg/kg
bw 3H-benzo [a]pyrene (Weyand & Bevan, 1986). The results of a study
of the pharmacokinetics and bioavailability of pyrene in rats strongly
suggested that enterohepatic recycling took place after oral or
intravenous administration of 14C-labelled compound at 2-15 mg/kg bw
(Withey et al., 1991).
Other studies on the enterohepatic circulation of PAH in rats and
rabbits have also shown that the significant amounts of metabolites
excreted in the bile persist in vivo because of enterohepatic
circulation (Chipman et al., 1981; Chipman, 1982; Boroujerdi et al.,
1981). For example, while some 60% of an intravenous dose of 3 µmol/kg
bw 14C-benzo [a]pyrene was excreted in bile, only 3% appeared in
urine within the first 6 h after injection (Chipman et al., 1981).
Biliary metabolites of xenobiotic compounds are usually polar and
nonreactive, but mutagenic or potentially mutagenic derivatives may be
excreted by this route into the intestine (for a review, see Chipman,
1982). Glucuronic acid conjugates of biliary metabolites can be
hydrolysed by some intestinal flora to potentially reactive species
(Renwick & Drasar, 1976; Chipman et al., 1981; Boroujerdi et al.,
1981; Chipman, 1982). Thio-ether conjugates of hydrocarbons may also
be involved in enterohepatic circulation (Hirom et al., 1983; Bakke et
al., 1983), although there is no evidence that these represent a
mutagenic or carcinogenic hazard to the tissues through which they
pass.
In a controlled study in humans, a 100-250-fold increase in dietary
exposure to PAH, as measured by benzo [a]pyrene intake, resulted in a
4-12-fold increase in urinary excretion of 1-hydroxypyrene. The
authors concluded that dietary exposure to PAH is as substantial as
some occupational exposures (Buckley & Lioy, 1992).
6.5 Retention and turnover
Very little is known about the retention and turnover of PAH in
mammalian species. It can be deduced from the few data available on
hydrocarbon body burdens (see below) that PAH themselves do not
persist for long periods and must therefore turn over reasonably
rapidly. During metabolism, PAH moieties become covalently bound to
tissue constituents such as proteins and nucleic acids. Protein-bound
metabolites are likely to persist, therefore, for periods that do not
exceed the normal lifetime of the protein itself. Nucleic acid adducts
formed from reactions of PAH metabolites can be expected to differ in
their persistence in the body according to whether they are RNA or DNA
adducts. Although most DNA adducts are removed relatively rapidly by
repair, small fractions can persist for long periods. The persistence
of these adducts in tissues such as mouse skin is of considerable
interest since one of the basic features of the two-stage mechanism of
carcinogenesis (Berenblum & Shubik, 1947) is that application of the
tumour promoter can be delayed for many months without markedly
reducing the eventual tumour yield.
The persistence of adducts is also consistent with multistage theories
of carcinogenesis, in which multiple steps in neoplastic
transformation are dependent on the mutagenic and other actions of
carcinogens.
6.5.1 Human body burdens of PAH
Since the effects of chemical carcinogens are likely to be related to
both the dose and the duration of exposure, it is important to
determine the human body load of carcinogens during a lifetime. It has
been estimated that the total intake of PAH over a 70-year lifespan
may amount to the equivalent of 300 mg of benzo [a]pyrene (Lutz &
Schlatter, 1992); however, inhabitants of conurbations are likely to
inhale additional amounts of PAH. Of course, much of the intake of PAH
is metabolized and excreted. Thus, the pulmonary tissues of elderly
town dwellers in Russia contained 1000 times less benzo [a]pyrene
(< 0.1 µg per individual) than might have been expected from the
estimated intake figures alone (Shabad & Dikun, 1959). Some
experiments with cows and domestic fowl fed diets containing added
benzo [a]pyrene tend to confirm this finding, since the meat, milk,
and eggs produced were, after a suitable delay, reported to be much
less heavily contaminated than might have been expected from the
amounts of benzo [a]pyrene administered (Gorelova & Cherepanova,
1970). More recent data are not available.
The average benzo [a]pyrene levels (measured by ultraviolet
spectroscopy) in tissues taken at autopsy from normal people of a wide
age range were 0.32 µg/100 g dry tissue weight in liver, spleen,
kidney, heart, and skeletal muscle and 0.2 µg/100 g in lung (Gräf,
1970; Gräf et al., 1975).
When cancer-free liver and fat from six individuals were assayed for
nine hydrocarbons by co-chromatography with authentic standards,
pyrene, anthracene, benzo [b]fluoranthene, benzo [ghi]perylene,
benzo [k]fluoranthene, and benzo [a]pyrene were detected at average
levels of 380 ppt (0.38 µg/kg wet weight) in liver and 1100 ppt (1.1
µg/kg wet weight) in fat. Pyrene was the most abundant PAH present
(Obana et al., 1981b).
Samples of 24 bronchial carcinomas, taken during surgery or at autopsy
from smokers and nonsmokers with a variety of occupations, were
analysed for the presence of 12 PAH by thin-layer chromatography and
fluorescence spectroscopy. Benzo [a]pyrene, benzo [b]fluoranthene,
fluoranthene, and perylene were detected. Benzo [a]pyrene was
present, but the other three PAH were found in only some of the
samples. The average concentrations of benzo [a]pyrene were 3.5 µg/g
in carcinoma tissue and 0.09 µg/g in tumour-free tissue (Tomingas et
al., 1976).
6.6 Reactions with tissue components
The reactions of metabolites of PAH with tissue constituents
(Weinstein et al., 1978) are relevant because they may indicate the
mechanisms by which the hydrocarbons exert biological effects that
include toxicity and carcinogenesis.
6.6.1 Reactions with proteins
Covalent interactions of PAH with protein in whole animals were first
noted in 1951 (Miller, 1951). It was proposed that reactions with
specific proteins might be involved in the initiation of malignancy in
liver (Miller & Miller, 1953), skin (Abell & Heidelberger, 1962), and
transformable cells in culture (Kuroki & Heidelberger, 1972). These
findings were supported by evidence that hydrocarbon metabolites can
react covalently with protein in microsomal incubates (Grover & Sims,
1968), in preparations of nuclei (Vaught & Bresnick, 1976; Pezzuto et
al., 1976, 1977; Hemminki & Vainio, 1979), and in cells and tissues
maintained in culture, including human tissues (Harris et al., 1978b;
MacNicoll et al., 1980). Although hydrocarbon metabolites often react
at much greater rates with protein than with nucleic acids in the same
biological system, relatively little attention has been paid to the
nature of the hydrocarbon metabolites involved or to the specificity
of these reactions, in terms of which proteins are most extensively
modified and where and the effect that such modification might have on
protein function. The evidence suggests, however, that the reactive
species involved include diol epoxides. Thus, when protein isolated
from the skin of mice that had been treated with benzo [a]pyrene was
hydrolysed, tetrols were liberated, and the patterns of specific
tetrols indicated that both syn and anti isomers of the
benzo [a]pyrene 7,8-diol 9,10-oxides are involved in covalent
reactions with protein (Koreeda et al., 1978). Studies of the covalent
interactions of diol epoxides with nuclear proteins show that a
variety of histones and non-histone proteins are modified (Kootstra &
Slaga, 1979; Kootstra et al., 1979; Whitlock, 1979).
6.6.2 Reactions with nucleic acids
The covalent interactions of electrophilic metabolites of PAH with
nucleic acids have been studied in much greater detail than those with
protein, partly because characterization of the products might, in
theory, be expected to be simpler, partly because the cellular nucleic
acids are, as nucleophiles, more 'homogeneous' than proteins, but
mainly because it has long been suspected that nucleic acid
modifications could lead to a permanent alteration of cell phenotype.
The covalent binding of a PAH (dibenz [a,h]anthracene) to DNA
in vivo was first reported by Heidelberger & Davenport in 1961.
Subsequent studies with naphthalene, dibenz [a,c]anthracene,
dibenz [a,h]anthracene, benzo [a]-pyrene, 3-methylcholanthrene, and
7,12-dimethylbenz [a]anthracene showed that the levels of DNA binding
in mouse skin are correlated with carcinogenic potency, as measured by
Iball's index (Brookes & Lawley, 1964).
6.7 Analytical methods
Of the methods used for the detection of carcinogen-DNA adducts
(Phillips, 1990; Strickland et al., 1993; Weston, 1993), one of the
most widely used is 32P-postlabelling, in which DNA is hydrolysed to
nucleotides, modified nucleotides (i.e. adducts) are labelled with
32P-phosphate, and the post-labelled adducts separated by thin-layer
chromatography and/or high-performance liquid chromatography (for
reviews of the method, see Phillips, 1991, and Phillips et al., 1993).
The main advantages of the 32P-postlabelling assay are its high
sensitivity and the fact that radiolabelled carcinogens and/or their
metabolites need not be synthesized beforehand.
A variety of physical methods have been described for the detection of
adducts, including fluorescence line narrowing spectroscopy,
synchronous fluorescence spectroscopy, and some specialized gas
chromatography-mass spectrometry procedures (Weston, 1993). The
physical methods combine high sensitivity with no requirement for
prior radiolabelling of the carcinogens or their adducts and may be
nondestructive. Sensitive methods involving antisera specific for
carcinogen-DNA adducts have also been developed. These include
radioimmunoassays, enzyme-linked immunosorbent assays, and
immuno-affinity chromatography (Poirier, 1994).
Information on the pathways thought to be involved in the metabolic
activation of several PAH is given in Table 74. For PAH that have been
extensively investigated, reviews are cited. In order to provide an
overall view of activation, the Table also includes data on PAH not
covered elsewhere in this monograph.
Most of the metabolites that have been found to react with nucleic
acids are vicinal diol epoxides, and most of these are diol epoxides
of the 'bay-region' type, although there are certain exceptions (Table
74). For example, activation of benzo [j]fluoranthene in mouse skin
involves a diol epoxide that is not of the bay-region type (Weyand et
al., 1993). Additionally, methyl-substituted PAH may become bound to
hydroxymethyl derivatives which, when conjugated, yield electrophilic
sulfate esters (Surh et al., 1989, 1990a,b).
The sites of attack on nucleic acid bases are usually the extranuclear
amino groups of guanine and adenine. When the reactions of the syn
and anti isomers of benzo [a]pyrene 7,8-diol-9,10-oxide with RNA,
DNA, and homopolymers were examined in experiments in which the
epoxide was incubated with the nucleic acid in a predominantly aqueous
solution, RNA, DNA, poly G, poly A, poly C, poly (dG), poly (dA), and
poly (dC) were modified, but there was little reaction with poly U,
poly I, or poly (dT) (Weinstein et al., 1976; Jennette et al., 1977).
Although many of the hydrocarbon-deoxyribonucleoside adducts formed in
human cells and tissues treated with PAH have not been completely
characterized, the available evidence, which is mostly
chromatographic, suggests that in human bronchial epithelium, colon,
mammary cells in culture, and skin the patterns of adducts formed are
very similar to those formed in corresponding rodent tissues (Autrup
et al., 1978a,b; Harris et al., 1979; Autrup et al., 1980; MacNicoll
et al., 1980; Weston et al., 1983). The rates of reaction of diol
epoxides with nucleic acids was in the general order: poly G > DNA >
poly A > poly C (Jennette et al., 1977).
Diol epoxides are also strongly suspected to react frequently with the
N7 position of guanine. This type of modification has not been
detected more often because N7-alkylated adducts are thought to have a
relatively short half-life at pH 7 and would therefore be lost during
the isolation and hydrolysis of DNA. In experiments in which care was
taken to avoid adduct loss, reactions of benzo [a]pyrene diol epoxide
with both the N2 and N7 positions of guanine residues in DNA were
detected (Osborne et al., 1978). N7 adducts were not, however,
detected in cells treated with anti-benzo [a]pyrene
7,8-diol-9,10-oxide (King et al., 1979).
In studies of the role of radical cations in the activation of PAH
in vitro, adducts were formed in which the 6 position of
benzo [a]pyrene was covalently linked to the C8 and N7 positions of
guanine and the N7 position of adenine, and the 7-methyl position of
7,12-dimethylbenz [a]anthracene was covalently linked to the N7
positions of guanine and adenine (see Figure 7; Cavalieri et al.,
1993; Rogan et al., 1993). All of these adducts are depurination
adducts, which may explain why they were not detected earlier
Table 74. Pathways involved in the metabolic activation of polycyclic aromatic hydrocarbons to form ultimate carcinogens
Compound Derivatives with highest Putative ultimate carcinogen Reference
levels of biological activity
Aceanthrylene 1,2-Oxidea Nesnow et al. (1991)
Benz[j]aceanthrylene ? 1,2-Oxideb Bartczak et al. (1987);
Nesnow et al. (1988)
Benz[l]aceanthrylene ? 1,2-Oxideb,c Nesnow et al. (1984);
Bartzczak et al. (1987);
Nesnow et al. (1988)
Benz[a]anthracene 3,4-Diold,e,f,g 3,4-Diol 1,2-oxldea,b,c,f,g Sims & Grover (1981);
8,9-Diold 8,9-Diol 10,1-oxidea,h Conney (1982);
Wood et al. (1983a)
Benzo[b]fluoranthene 9,10-Dlold,f,i ? 910-Diol-11,12-oxide Geddie et al. (1987);
and 5/6-hydroxy-9,10- Pfau et al. (1992)
diol-11, 12-oxide
Benzo[b]fluoranthene ? 9,10-Diolf,j ? 9,10-Diol 11,12-oxidea Rice et al. (1987);
Weyand et al. (1993)
? 4,5-Diola ? 4,5-Diol 6,6a-oxidea Weyand et al. (1987)
Benzo[c]phenanthrene 3,4-Diold,e,f,g 3,4-Diol 1,2-oxidea,b,c,f,g Conney (1982);
Levin et al. (1986);
Agarwal et al. (1987);
Dipple et al. (1987);
Pruess-Schwartz et al.
(1987)
Benzo[a]pyrene 7,8-Diold,e,f,h 7,8-Diol 9,10-oxidea,b,c,g Cooper et al. (1983);
Osborne & Crosby (1987a)
Table 74. (continued)
Compound Derivatives with highest Putative ultimate carcinogen Reference
levels of biological activity
Benzo[e]pyrene 9,10-Diolf ? 9,10-Diol 11,12-oxideg Osborne & Crosby (1987b)
Chrysene 1,2-Diold,e,f 1,2-Diol 3,4-oxidea,b,c,h Conney (1982);
9-Hydroxy 1,2-diold,e 9-Hydroxy-1,2-diol Hodgson et al. (1983);
3,4-oxideb,c Glatt et al. (1986)
Cyclopenta[cd]pyrene - ? 3,4-oxideb,c,h Gold & Eisenstadt (1980);
Gold et al. (1980)
15,16-Dihydro-11-methylcyclo- 3,4-Diold,f 3,4-Diol 1,2-oxidea Coombs & Bhatt (1987)
penta[a]phenanthren-17-one
15,16-Diydro-1,11-methano- 3,4-Diold 3,4-Diol 1,2-oxide Coombs & Bhatt (1987)
cyclopenta[a]phenanthren-17-one
Dibenz[a,c]anthracene 10,11-Diold ? 10, 11-Diol 12,13-oxide Sims & Grover (1981)
Dibenz[a,h]anthracene 3,4-Diold,f,g,h ? 3,4-Diol 1,2-oxide and Conney (1982);
3,4:10,1 1-bis-diol-epoxides Lecoq et al. (1991, 1992);
Carmichael et al. (1993);
Nesnow et al. (1994)
Dibenzo[a,e]fluoranthene 12,13-Diold,f 12,13-Diol 10-11-oxidea Perin-Roussel et al.
(1983,1984);
3,4-Diold,f 3,4-Diol 1,2-oxidea Saguem et al. (1983a,b);
Zajdela et al. (1987)
Dibenzo[a,h]pyrene 1,2-Diolf,g ? 1,2-Diol 3,4-oxideg Chang et al. (1982)
Dibenzo[a,l]pyrene ? 11,12 Diolf ? 11,12-Diol 13,14-oxide Cavalieri et al. (1991)
Table 74. (continued)
Compound Derivatives with highest Putative ultimate carcinogen Reference
levels of biological activity
Dibenzo[a,i]pyrene 3,4-Diolf,g ? 3,4-Diol 1,2-oxideg Chang et al. (1982)
7,12-Dimethylbenz[a]anthracene 3,4-Diold,e,f,h 3,4-Diol 1,2-oxidea Sims & Grover (1981);
Conney (1982);
Sawicki et al. (1983);
Dipple et al.; 1984)
7-Ethylbenz[a]anthracene 3,4-Diold ? 3,4-Diol 1,2-oxidea,b McKay et al. (1988);
Glatt et al. (1989)
Fluoranthene Z3,Diold 2,3-Diol 1,10b-oxidea La Voie et al. (1982a);
Rastetter et al. (1982);
Babson et al. (1986a);
Hecht et al. (1995)
Indeno[1,2,3-cd]pyrene 1,2-oxideb,f ? Rice et al. (1985)
1,2-Diolf Rice et al. (1986)
8-Hydroxyd
9-Hydroxyd
7-Methylbenz[a]anthracene 3,4-Diold,e,f,h 3,4-Diol 1,2-oxidea,b Sims & Grover (1981);
McKay et al. (1988);
Glatt et al. (1989)
3-Methylcholanthrene 9,10-Diold,f,h ? 9,10-Diol 7,13-oxidea,f Sims & Grover (1981);
? 3-Hydroxymethyl-9,10- Conney (1982);
diol 7,8-oxide DiGiovanni et al. (1985);
Osborne et al. (1986)
5-Methylchrysene 1,2-Diold,f 1,2-Diol 3,4-oxidea,c,h Hecht et al. (1986);
Brookes et al. (1986);
Reardon et al. (1987);
Hecht et al. (1987)
Table 74 (continued)
a DNA adducts characterized
b Directly acting mutagen in S. typhimurium
c Directly acting mutagen in V79 Chinese hamster cells
d Mutagenic to S. typhimurium with metabolic activation
e Mutagenic to V79 Chinese hamster cells with metabolic activation
f Tumour initiator in mouse skin
g Induces tumours in newborn mice
h Transforms cells in culture
i Not detected as a metabolite; activation may therefore occur via a different pathway.
j Although the 45-diol is the most active derivative so far tested, there is some evidence that adducts arise from the 9,1-diol.
in vivo. The formation of apurinic sites in DNA could lead to strand
nicking (Gamper et al., 1977, 1980). When the positions of the nicks
produced as a result of modification by benzo [a]pyrene
7,8-diol-9,10-oxide were investigated with DNA of a defined sequence,
nicking appeared to be the result of the loss of purines and
pyrimidines that had been modified at the N7 position of guanine or at
the N3 position of adenine and cytosine (Haseltine et al., 1980).
In studies of the distribution of covalently bound benzo [a]pyrene
moieties in chromatin, more was bound to the inter-nucleosomal spacer
regions of DNA than to DNA in nucleosomes (Jahn & Litman, 1977, 1979;
Kootstra & Slaga, 1980). One explanation for this finding may be that
nucleosomal DNA is better protected from modification by the presence
of nucleoproteins; results consistent with this suggestion have been
obtained with mitochondrial DNA. Graffi (1940a,b,c) suggested that
lipophilic PAH accumulate in lipid-rich mitochondria. Allen & Coombs
(1980) and Backer & Weinstein (1980) showed much higher levels of
modification of mitochondrial than nuclear DNA in cultured cells
treated with either benzo [a]pyrene or the anti-benzo [a]pyrene
7,8-diol-9,10-oxide.
The molecular properties of adducts of benzo [a]pyrene
7,8-dihydrodiol-9,10-epoxides with DNA have been described (Geacintov
1988; Jernström & Gräslund, 1994). Although the biological
effectiveness of all types of hydrocarbon-nucleic acid adducts has not
been determined, it has been shown that differences in the biological
activities of 7-ethyl- and 7-methylbenz [a]-anthracene are not due to
differences in the mutagenic potential of the adducts formed (Glatt et
al., 1989). Similar conclusions were drawn from work with a series of
bay-region and fjord-region diol epoxides (Phillips et al., 1991; see
section 7.10 for a description of a fjord region). At present,
therefore, all hydrocarbon-deoxyribonucleoside adducts should be
regarded as potentially damaging to the organism.
The relationships between DNA adduct formation and tumour incidence
were examined by Poirier & Beland (1992) on the basis of data from
long-term studies in rodents administered carcinogens. The tumour
incidence was compared with adduct levels measured in target tissues
during the first two months of exposure. In most cases, linear
increases in DNA adduct levels with dose were reflected in linear
increases in tumour incidence, although there were exceptions.
In a comparison of the incidence of lung adenomas in strain A/J mice
240 days after they had received a single intraperitoneal injection of
benzo [a]pyrene, dibenz [a,h]anthracene, benzo [b]fluoranthene,
5-methyl-chrysene, or cyclopenta [cd]pyrene with the levels of DNA
adducts detected in the lungs by 32P-postlabelling between days 1 and
21 after treatment, time-integrated DNA adduct levels were calculated
and plotted against lung adenoma frequency. The slopes obtained were
essentially similar for benzo [a]pyrene, benzo [b]fluoranthene,
5-methylchrysene, and cyclopenta [cd]pyrene but were different for
dibenz [a,h]anthracene. The authors concluded that 'essentially
identical induction of adenomas as a function of [time-integrated DNA
adduct levels] for these PAH suggests that the formation and
persistence of DNA adducts determines their carcinogenic potency'
(Ross et al., 1995).
Benzo [a]pyrene metabolism has been examined extensively in human
tissue preparations, including human cells, explant cultures, tissue
homogenates, and microsomal preparations. Table 73 lists some studies
of the metabolism of benzo [a]pyrene in human tissues that included
metabolites soluble in organic solvents and water-soluble conjugates.
The results show that the metabolites produced by different human
tissues are qualitatively similar and that the metabolites detected
are the same as those formed in a variety of animal tissues.
The metabolic profiles reported in human tissues are almost all
identical to those seen for other eukaryotes, indicating the
involvement of similar enzyme systems. The same types of reactive
electrophilic intermediates found in other experimental systems also
appear to be formed in human tissues (Autrup & Harris, 1983). So far,
no differences in the metabolism or activation of benzo [a]pyrene
have been reported that might account for differences in the
susceptibility of different animal and human tissues to its
carcinogenic properties (see Section 7). Studies with cultured cells
and other substrates such as benz [a]anthracene, however, indicate
that bioactivation of PAH is species-dependent (Jacob, 1996).
6.4 Elimination and excretion
Most metabolites of PAH are excreted in faeces and urine. As complete
breakdown of the benzene rings of which unsubstituted PAH are composed
does not occur to any appreciable extent in higher organisms, very
little of an administered dose of an unsubstituted hydrocarbon would
be expected to appear as carbon dioxide in expired air.
The urinary excretion of PAH metabolites has been studied more
extensively than faecal excretion, but the importance of the
enterohepatic circulation of metabolites has led to increased research
on the latter. Detailed studies of the metabolism and excretion of PAH
in whole animals have been restricted mainly to the simpler compounds.
Because of the toxicity of the larger hydrocarbons and the complexity
of their metabolism, most studies on these compounds have been carried
out in hepatic homogenates and microsomal preparations or with
cultured cells (see above).
Metabolism and excretion in whole animals have been examined with
regard to naphthalene (Bourne & Young, 1934; Young, 1947; Booth &
Boyland, 1949; Corner & Young, 1954; Corner et al., 1954; Boyland &
Sims, 1958; Sims, 1959), anthracene (Boyland & Levi, 1935, 1936a,b;
Sims, 1964), phenanthrene (Boyland & Wolf, 1950; Sims, 1962; Boyland &
Sims, 1962a,b; Jacob et al., 1990b; Grimmer et al., 1991a), pyrene
(Harper, 1957, 1958a; Boyland & Sims, 1964a; Jacob et al., 1989,
1990b), benz [a]-anthracene (Harper 1959a,b; Boyland & Sims, 1964b),
and chrysene (Grimmer et al., 1988b, 1990). A limited number of
studies have been published on more complex compounds such as
benzo [a]pyrene (Berenblum & Schoental, 1943; Weigert & Mottram,
1946; Harper, 1958b,c; Falk et al., 1962; Raha, 1972; Jacob et al.,
1990b), dibenz [a,h]anthracene (Dobriner et al., 1939; Boyland et
al., 1941; La Budde & Heidelberger, 1958), and 3-methylcholanthrene
Table 73. Metabolites of benzo[a]pyrene formed by human tissues and cells
Tissue or Type of metabolite detected References
cell type
Dihydrodols Phenols Quinones Tetrols Conjugates
Bronchus + + + + + Pal et al. (1975);
Cohen et al. (1976);
Harris et al. (1977);
Autrup et al. (1978a,
1980)
Colon + + + + + Autrup et al. (1978b);
Autrup (1979)
Endometrium + + + Mass et al. (1981)
Fibroblasts + Baird & Diamond (1978)
Kidney + + + Prough et al. (1979)
Liver + + + + Selkirk et al. (1975);
Prough et al. (1979);
Pelkonen et al. (1977);
Diamond et al. (1980)
Lung + + + + + Cohen et al. (1976);
Stoner et al. (1978);
Mehta et al. (1979);
Prough et al. (1979);
Sipal. et al. (1979)
Lymphocytes + + + Booth et al. (1974);
Selkirk et al. (1975);
Vaught et al. (1978);
Okano et al. (1979);
Gurtoo et al. (1980)
Macrophages + + + + + Autrup et al. (1978a);
Harris et al. (1978a,b);
Autrup et al. (1979);
Marshall et al. (1979)
Table 73 (contd)
Tissue or Type of metabolite detected References
cell type
Dihydrodols Phenols Quinones Tetrols Conjugates
Mammary + Grover et al. (1980);
epithelium MacNicoll et al. (1980)
Monocytes + + + Vaught et al. (1978);
Okano et al. (1979)
Oesophagus + + + + Harris et al. (1979)
Placenta + + + Namkung & Juchau (1980);
Pelkonen & Saarni (1980)
Skin + + + + Fox et al. (1975);
Vermorken et al. (1979);
Parkinson & Newbold (1980);
Kuroki et al. (1980)
(Harper, 1959a; Takahashi & Yasuhira, 1972; Takahashi, 1978). Much of
the earlier qualitative work was reviewed by Boyland & Weigart (1947)
and by Young (1950). The absorption and excretion of different
hydrocarbons in vivo can differ. For example, while almost all of a
topically applied dose of benzo [a]pyrene appeared in mouse faeces
(Heidelberger & Weiss, 1951), little dibenz [a,h]-anthracene was
excreted by this route.
In rats given PAH either singly or as mixtures, the faecal elimination
of chrysene (25% of the dose) was not affected by co-administration of
benz [a]anthracene, but that of benz [a]anthracene was doubled, from
6 to 13% of the dose, when chrysene was given (Bartosek et al., 1984).
Such effects are relevant to human pharmacokinetics, since exposure is
almost always to mixtures of PAH. In workers in a coke plant exposed
to mixtures of PAH, the amounts of phenanthrene, pyrene, and
benzo [a]pyrene inhaled and the amounts of their principal
metabolites excreted in the urine were correlated (Grimmer et al.,
1994).
In rats, the amount of benzo [a]pyrene 7,8-diol excreted in the urine
is related to the susceptibility of individual animals to the
carcinogenic effects of benzo [a]pyrene (Likhachev et al., 1992;
Tyndyk et al., 1994). In studies of the disposition of
benzo [a]pyrene in rats, hamsters, and guinea-pigs after
intratracheal administration, the distribution of the hydrocarbon was
qualitatively similar but quantitatively different. In Sprague-Dawley
and Gunn rats and in guinea-pigs, the rate of excretion was dependent
on the dose administered, but in hamsters the rate of excretion was
independent of dose (0.16 or 350 µg 3H-benzo [a]pyrene) (Weyand &
Bevan 1986, 1987a). Evidence for enterohepatic circulation of
benzo [a]pyrene metabolites was obtained in Sprague-Dawley rats with
bile-duct cannulae treated by intratracheal instillation with 1 µg/kg
bw 3H-benzo [a]pyrene (Weyand & Bevan, 1986). The results of a study
of the pharmacokinetics and bioavailability of pyrene in rats strongly
suggested that enterohepatic recycling took place after oral or
intravenous administration of 14C-labelled compound at 2-15 mg/kg bw
(Withey et al., 1991).
Other studies on the enterohepatic circulation of PAH in rats and
rabbits have also shown that the significant amounts of metabolites
excreted in the bile persist in vivo because of enterohepatic
circulation (Chipman et al., 1981; Chipman, 1982; Boroujerdi et al.,
1981). For example, while some 60% of an intravenous dose of 3 µmol/kg
bw 14C-benzo [a]pyrene was excreted in bile, only 3% appeared in
urine within the first 6 h after injection (Chipman et al., 1981).
Biliary metabolites of xenobiotic compounds are usually polar and
nonreactive, but mutagenic or potentially mutagenic derivatives may be
excreted by this route into the intestine (for a review, see Chipman,
1982). Glucuronic acid conjugates of biliary metabolites can be
hydrolysed by some intestinal flora to potentially reactive species
(Renwick & Drasar, 1976; Chipman et al., 1981; Boroujerdi et al.,
1981; Chipman, 1982). Thio-ether conjugates of hydrocarbons may also
be involved in enterohepatic circulation (Hirom et al., 1983; Bakke et
al., 1983), although there is no evidence that these represent a
mutagenic or carcinogenic hazard to the tissues through which they
pass.
In a controlled study in humans, a 100-250-fold increase in dietary
exposure to PAH, as measured by benzo [a]pyrene intake, resulted in a
4-12-fold increase in urinary excretion of 1-hydroxypyrene. The
authors concluded that dietary exposure to PAH is as substantial as
some occupational exposures (Buckley & Lioy, 1992).
6.5 Retention and turnover
Very little is known about the retention and turnover of PAH in
mammalian species. It can be deduced from the few data available on
hydrocarbon body burdens (see below) that PAH themselves do not
persist for long periods and must therefore turn over reasonably
rapidly. During metabolism, PAH moieties become covalently bound to
tissue constituents such as proteins and nucleic acids. Protein-bound
metabolites are likely to persist, therefore, for periods that do not
exceed the normal lifetime of the protein itself. Nucleic acid adducts
formed from reactions of PAH metabolites can be expected to differ in
their persistence in the body according to whether they are RNA or DNA
adducts. Although most DNA adducts are removed relatively rapidly by
repair, small fractions can persist for long periods. The persistence
of these adducts in tissues such as mouse skin is of considerable
interest since one of the basic features of the two-stage mechanism of
carcinogenesis (Berenblum & Shubik, 1947) is that application of the
tumour promoter can be delayed for many months without markedly
reducing the eventual tumour yield.
The persistence of adducts is also consistent with multistage theories
of carcinogenesis, in which multiple steps in neoplastic
transformation are dependent on the mutagenic and other actions of
carcinogens.
6.5.1 Human body burdens of PAH
Since the effects of chemical carcinogens are likely to be related to
both the dose and the duration of exposure, it is important to
determine the human body load of carcinogens during a lifetime. It has
been estimated that the total intake of PAH over a 70-year lifespan
may amount to the equivalent of 300 mg of benzo [a]pyrene (Lutz &
Schlatter, 1992); however, inhabitants of conurbations are likely to
inhale additional amounts of PAH. Of course, much of the intake of PAH
is metabolized and excreted. Thus, the pulmonary tissues of elderly
town dwellers in Russia contained 1000 times less benzo [a]pyrene
(< 0.1 µg per individual) than might have been expected from the
estimated intake figures alone (Shabad & Dikun, 1959). Some
experiments with cows and domestic fowl fed diets containing added
benzo [a]pyrene tend to confirm this finding, since the meat, milk,
and eggs produced were, after a suitable delay, reported to be much
less heavily contaminated than might have been expected from the
amounts of benzo [a]pyrene administered (Gorelova & Cherepanova,
1970). More recent data are not available.
The average benzo [a]pyrene levels (measured by ultraviolet
spectroscopy) in tissues taken at autopsy from normal people of a wide
age range were 0.32 µg/100 g dry tissue weight in liver, spleen,
kidney, heart, and skeletal muscle and 0.2 µg/100 g in lung (Gräf,
1970; Gräf et al., 1975).
When cancer-free liver and fat from six individuals were assayed for
nine hydrocarbons by co-chromatography with authentic standards,
pyrene, anthracene, benzo [b]fluoranthene, benzo [ghi]perylene,
benzo [k]fluoranthene, and benzo [a]pyrene were detected at average
levels of 380 ppt (0.38 µg/kg wet weight) in liver and 1100 ppt (1.1
µg/kg wet weight) in fat. Pyrene was the most abundant PAH present
(Obana et al., 1981b).
Samples of 24 bronchial carcinomas, taken during surgery or at autopsy
from smokers and nonsmokers with a variety of occupations, were
analysed for the presence of 12 PAH by thin-layer chromatography and
fluorescence spectroscopy. Benzo [a]pyrene, benzo [b]fluoranthene,
fluoranthene, and perylene were detected. Benzo [a]pyrene was
present, but the other three PAH were found in only some of the
samples. The average concentrations of benzo [a]pyrene were 3.5 µg/g
in carcinoma tissue and 0.09 µg/g in tumour-free tissue (Tomingas et
al., 1976).
6.6 Reactions with tissue components
The reactions of metabolites of PAH with tissue constituents
(Weinstein et al., 1978) are relevant because they may indicate the
mechanisms by which the hydrocarbons exert biological effects that
include toxicity and carcinogenesis.
6.6.1 Reactions with proteins
Covalent interactions of PAH with protein in whole animals were first
noted in 1951 (Miller, 1951). It was proposed that reactions with
specific proteins might be involved in the initiation of malignancy in
liver (Miller & Miller, 1953), skin (Abell & Heidelberger, 1962), and
transformable cells in culture (Kuroki & Heidelberger, 1972). These
findings were supported by evidence that hydrocarbon metabolites can
react covalently with protein in microsomal incubates (Grover & Sims,
1968), in preparations of nuclei (Vaught & Bresnick, 1976; Pezzuto et
al., 1976, 1977; Hemminki & Vainio, 1979), and in cells and tissues
maintained in culture, including human tissues (Harris et al., 1978b;
MacNicoll et al., 1980). Although hydrocarbon metabolites often react
at much greater rates with protein than with nucleic acids in the same
biological system, relatively little attention has been paid to the
nature of the hydrocarbon metabolites involved or to the specificity
of these reactions, in terms of which proteins are most extensively
modified and where and the effect that such modification might have on
protein function. The evidence suggests, however, that the reactive
species involved include diol epoxides. Thus, when protein isolated
from the skin of mice that had been treated with benzo [a]pyrene was
hydrolysed, tetrols were liberated, and the patterns of specific
tetrols indicated that both syn and anti isomers of the
benzo [a]pyrene 7,8-diol 9,10-oxides are involved in covalent
reactions with protein (Koreeda et al., 1978). Studies of the covalent
interactions of diol epoxides with nuclear proteins show that a
variety of histones and non-histone proteins are modified (Kootstra &
Slaga, 1979; Kootstra et al., 1979; Whitlock, 1979).
6.6.2 Reactions with nucleic acids
The covalent interactions of electrophilic metabolites of PAH with
nucleic acids have been studied in much greater detail than those with
protein, partly because characterization of the products might, in
theory, be expected to be simpler, partly because the cellular nucleic
acids are, as nucleophiles, more 'homogeneous' than proteins, but
mainly because it has long been suspected that nucleic acid
modifications could lead to a permanent alteration of cell phenotype.
The covalent binding of a PAH (dibenz [a,h]anthracene) to DNA
in vivo was first reported by Heidelberger & Davenport in 1961.
Subsequent studies with naphthalene, dibenz [a,c]anthracene,
dibenz [a,h]anthracene, benzo [a]-pyrene, 3-methylcholanthrene, and
7,12-dimethylbenz [a]anthracene showed that the levels of DNA binding
in mouse skin are correlated with carcinogenic potency, as measured by
Iball's index (Brookes & Lawley, 1964).
6.7 Analytical methods
Of the methods used for the detection of carcinogen-DNA adducts
(Phillips, 1990; Strickland et al., 1993; Weston, 1993), one of the
most widely used is 32P-postlabelling, in which DNA is hydrolysed to
nucleotides, modified nucleotides (i.e. adducts) are labelled with
32P-phosphate, and the post-labelled adducts separated by thin-layer
chromatography and/or high-performance liquid chromatography (for
reviews of the method, see Phillips, 1991, and Phillips et al., 1993).
The main advantages of the 32P-postlabelling assay are its high
sensitivity and the fact that radiolabelled carcinogens and/or their
metabolites need not be synthesized beforehand.
A variety of physical methods have been described for the detection of
adducts, including fluorescence line narrowing spectroscopy,
synchronous fluorescence spectroscopy, and some specialized gas
chromatography-mass spectrometry procedures (Weston, 1993). The
physical methods combine high sensitivity with no requirement for
prior radiolabelling of the carcinogens or their adducts and may be
nondestructive. Sensitive methods involving antisera specific for
carcinogen-DNA adducts have also been developed. These include
radioimmunoassays, enzyme-linked immunosorbent assays, and
immuno-affinity chromatography (Poirier, 1994).
Information on the pathways thought to be involved in the metabolic
activation of several PAH is given in Table 74. For PAH that have been
extensively investigated, reviews are cited. In order to provide an
overall view of activation, the Table also includes data on PAH not
covered elsewhere in this monograph.
Most of the metabolites that have been found to react with nucleic
acids are vicinal diol epoxides, and most of these are diol epoxides
of the 'bay-region' type, although there are certain exceptions (Table
74). For example, activation of benzo [j]fluoranthene in mouse skin
involves a diol epoxide that is not of the bay-region type (Weyand et
al., 1993). Additionally, methyl-substituted PAH may become bound to
hydroxymethyl derivatives which, when conjugated, yield electrophilic
sulfate esters (Surh et al., 1989, 1990a,b).
The sites of attack on nucleic acid bases are usually the extranuclear
amino groups of guanine and adenine. When the reactions of the syn
and anti isomers of benzo [a]pyrene 7,8-diol-9,10-oxide with RNA,
DNA, and homopolymers were examined in experiments in which the
epoxide was incubated with the nucleic acid in a predominantly aqueous
solution, RNA, DNA, poly G, poly A, poly C, poly (dG), poly (dA), and
poly (dC) were modified, but there was little reaction with poly U,
poly I, or poly (dT) (Weinstein et al., 1976; Jennette et al., 1977).
Although many of the hydrocarbon-deoxyribonucleoside adducts formed in
human cells and tissues treated with PAH have not been completely
characterized, the available evidence, which is mostly
chromatographic, suggests that in human bronchial epithelium, colon,
mammary cells in culture, and skin the patterns of adducts formed are
very similar to those formed in corresponding rodent tissues (Autrup
et al., 1978a,b; Harris et al., 1979; Autrup et al., 1980; MacNicoll
et al., 1980; Weston et al., 1983). The rates of reaction of diol
epoxides with nucleic acids was in the general order: poly G > DNA >
poly A > poly C (Jennette et al., 1977).
Diol epoxides are also strongly suspected to react frequently with the
N7 position of guanine. This type of modification has not been
detected more often because N7-alkylated adducts are thought to have a
relatively short half-life at pH 7 and would therefore be lost during
the isolation and hydrolysis of DNA. In experiments in which care was
taken to avoid adduct loss, reactions of benzo [a]pyrene diol epoxide
with both the N2 and N7 positions of guanine residues in DNA were
detected (Osborne et al., 1978). N7 adducts were not, however,
detected in cells treated with anti-benzo [a]pyrene
7,8-diol-9,10-oxide (King et al., 1979).
In studies of the role of radical cations in the activation of PAH
in vitro, adducts were formed in which the 6 position of
benzo [a]pyrene was covalently linked to the C8 and N7 positions of
guanine and the N7 position of adenine, and the 7-methyl position of
7,12-dimethylbenz [a]anthracene was covalently linked to the N7
positions of guanine and adenine (see Figure 7; Cavalieri et al.,
1993; Rogan et al., 1993). All of these adducts are depurination
adducts, which may explain why they were not detected earlier
Table 74. Pathways involved in the metabolic activation of polycyclic aromatic hydrocarbons to form ultimate carcinogens
Compound Derivatives with highest Putative ultimate carcinogen Reference
levels of biological activity
Aceanthrylene 1,2-Oxidea Nesnow et al. (1991)
Benz[j]aceanthrylene ? 1,2-Oxideb Bartczak et al. (1987);
Nesnow et al. (1988)
Benz[l]aceanthrylene ? 1,2-Oxideb,c Nesnow et al. (1984);
Bartzczak et al. (1987);
Nesnow et al. (1988)
Benz[a]anthracene 3,4-Diold,e,f,g 3,4-Diol 1,2-oxldea,b,c,f,g Sims & Grover (1981);
8,9-Diold 8,9-Diol 10,1-oxidea,h Conney (1982);
Wood et al. (1983a)
Benzo[b]fluoranthene 9,10-Dlold,f,i ? 910-Diol-11,12-oxide Geddie et al. (1987);
and 5/6-hydroxy-9,10- Pfau et al. (1992)
diol-11, 12-oxide
Benzo[b]fluoranthene ? 9,10-Diolf,j ? 9,10-Diol 11,12-oxidea Rice et al. (1987);
Weyand et al. (1993)
? 4,5-Diola ? 4,5-Diol 6,6a-oxidea Weyand et al. (1987)
Benzo[c]phenanthrene 3,4-Diold,e,f,g 3,4-Diol 1,2-oxidea,b,c,f,g Conney (1982);
Levin et al. (1986);
Agarwal et al. (1987);
Dipple et al. (1987);
Pruess-Schwartz et al.
(1987)
Benzo[a]pyrene 7,8-Diold,e,f,h 7,8-Diol 9,10-oxidea,b,c,g Cooper et al. (1983);
Osborne & Crosby (1987a)
Table 74. (continued)
Compound Derivatives with highest Putative ultimate carcinogen Reference
levels of biological activity
Benzo[e]pyrene 9,10-Diolf ? 9,10-Diol 11,12-oxideg Osborne & Crosby (1987b)
Chrysene 1,2-Diold,e,f 1,2-Diol 3,4-oxidea,b,c,h Conney (1982);
9-Hydroxy 1,2-diold,e 9-Hydroxy-1,2-diol Hodgson et al. (1983);
3,4-oxideb,c Glatt et al. (1986)
Cyclopenta[cd]pyrene - ? 3,4-oxideb,c,h Gold & Eisenstadt (1980);
Gold et al. (1980)
15,16-Dihydro-11-methylcyclo- 3,4-Diold,f 3,4-Diol 1,2-oxidea Coombs & Bhatt (1987)
penta[a]phenanthren-17-one
15,16-Diydro-1,11-methano- 3,4-Diold 3,4-Diol 1,2-oxide Coombs & Bhatt (1987)
cyclopenta[a]phenanthren-17-one
Dibenz[a,c]anthracene 10,11-Diold ? 10, 11-Diol 12,13-oxide Sims & Grover (1981)
Dibenz[a,h]anthracene 3,4-Diold,f,g,h ? 3,4-Diol 1,2-oxide and Conney (1982);
3,4:10,1 1-bis-diol-epoxides Lecoq et al. (1991, 1992);
Carmichael et al. (1993);
Nesnow et al. (1994)
Dibenzo[a,e]fluoranthene 12,13-Diold,f 12,13-Diol 10-11-oxidea Perin-Roussel et al.
(1983,1984);
3,4-Diold,f 3,4-Diol 1,2-oxidea Saguem et al. (1983a,b);
Zajdela et al. (1987)
Dibenzo[a,h]pyrene 1,2-Diolf,g ? 1,2-Diol 3,4-oxideg Chang et al. (1982)
Dibenzo[a,l]pyrene ? 11,12 Diolf ? 11,12-Diol 13,14-oxide Cavalieri et al. (1991)
Table 74. (continued)
Compound Derivatives with highest Putative ultimate carcinogen Reference
levels of biological activity
Dibenzo[a,i]pyrene 3,4-Diolf,g ? 3,4-Diol 1,2-oxideg Chang et al. (1982)
7,12-Dimethylbenz[a]anthracene 3,4-Diold,e,f,h 3,4-Diol 1,2-oxidea Sims & Grover (1981);
Conney (1982);
Sawicki et al. (1983);
Dipple et al.; 1984)
7-Ethylbenz[a]anthracene 3,4-Diold ? 3,4-Diol 1,2-oxidea,b McKay et al. (1988);
Glatt et al. (1989)
Fluoranthene Z3,Diold 2,3-Diol 1,10b-oxidea La Voie et al. (1982a);
Rastetter et al. (1982);
Babson et al. (1986a);
Hecht et al. (1995)
Indeno[1,2,3-cd]pyrene 1,2-oxideb,f ? Rice et al. (1985)
1,2-Diolf Rice et al. (1986)
8-Hydroxyd
9-Hydroxyd
7-Methylbenz[a]anthracene 3,4-Diold,e,f,h 3,4-Diol 1,2-oxidea,b Sims & Grover (1981);
McKay et al. (1988);
Glatt et al. (1989)
3-Methylcholanthrene 9,10-Diold,f,h ? 9,10-Diol 7,13-oxidea,f Sims & Grover (1981);
? 3-Hydroxymethyl-9,10- Conney (1982);
diol 7,8-oxide DiGiovanni et al. (1985);
Osborne et al. (1986)
5-Methylchrysene 1,2-Diold,f 1,2-Diol 3,4-oxidea,c,h Hecht et al. (1986);
Brookes et al. (1986);
Reardon et al. (1987);
Hecht et al. (1987)
Table 74 (continued)
a DNA adducts characterized
b Directly acting mutagen in S. typhimurium
c Directly acting mutagen in V79 Chinese hamster cells
d Mutagenic to S. typhimurium with metabolic activation
e Mutagenic to V79 Chinese hamster cells with metabolic activation
f Tumour initiator in mouse skin
g Induces tumours in newborn mice
h Transforms cells in culture
i Not detected as a metabolite; activation may therefore occur via a different pathway.
j Although the 45-diol is the most active derivative so far tested, there is some evidence that adducts arise from the 9,1-diol.
in vivo. The formation of apurinic sites in DNA could lead to strand
nicking (Gamper et al., 1977, 1980). When the positions of the nicks
produced as a result of modification by benzo [a]pyrene
7,8-diol-9,10-oxide were investigated with DNA of a defined sequence,
nicking appeared to be the result of the loss of purines and
pyrimidines that had been modified at the N7 position of guanine or at
the N3 position of adenine and cytosine (Haseltine et al., 1980).
In studies of the distribution of covalently bound benzo [a]pyrene
moieties in chromatin, more was bound to the inter-nucleosomal spacer
regions of DNA than to DNA in nucleosomes (Jahn & Litman, 1977, 1979;
Kootstra & Slaga, 1980). One explanation for this finding may be that
nucleosomal DNA is better protected from modification by the presence
of nucleoproteins; results consistent with this suggestion have been
obtained with mitochondrial DNA. Graffi (1940a,b,c) suggested that
lipophilic PAH accumulate in lipid-rich mitochondria. Allen & Coombs
(1980) and Backer & Weinstein (1980) showed much higher levels of
modification of mitochondrial than nuclear DNA in cultured cells
treated with either benzo [a]pyrene or the anti-benzo [a]pyrene
7,8-diol-9,10-oxide.
The molecular properties of adducts of benzo [a]pyrene
7,8-dihydrodiol-9,10-epoxides with DNA have been described (Geacintov
1988; Jernström & Gräslund, 1994). Although the biological
effectiveness of all types of hydrocarbon-nucleic acid adducts has not
been determined, it has been shown that differences in the biological
activities of 7-ethyl- and 7-methylbenz [a]-anthracene are not due to
differences in the mutagenic potential of the adducts formed (Glatt et
al., 1989). Similar conclusions were drawn from work with a series of
bay-region and fjord-region diol epoxides (Phillips et al., 1991; see
section 7.10 for a description of a fjord region). At present,
therefore, all hydrocarbon-deoxyribonucleoside adducts should be
regarded as potentially damaging to the organism.
The relationships between DNA adduct formation and tumour incidence
were examined by Poirier & Beland (1992) on the basis of data from
long-term studies in rodents administered carcinogens. The tumour
incidence was compared with adduct levels measured in target tissues
during the first two months of exposure. In most cases, linear
increases in DNA adduct levels with dose were reflected in linear
increases in tumour incidence, although there were exceptions.
In a comparison of the incidence of lung adenomas in strain A/J mice
240 days after they had received a single intraperitoneal injection of
benzo [a]pyrene, dibenz [a,h]anthracene, benzo [b]fluoranthene,
5-methyl-chrysene, or cyclopenta [cd]pyrene with the levels of DNA
adducts detected in the lungs by 32P-postlabelling between days 1 and
21 after treatment, time-integrated DNA adduct levels were calculated
and plotted against lung adenoma frequency. The slopes obtained were
essentially similar for benzo [a]pyrene, benzo [b]fluoranthene,
5-methylchrysene, and cyclopenta [cd]pyrene but were different for
dibenz [a,h]anthracene. The authors concluded that 'essentially
identical induction of adenomas as a function of [time-integrated DNA
adduct levels] for these PAH suggests that the formation and
persistence of DNA adducts determines their carcinogenic potency'
(Ross et al., 1995).
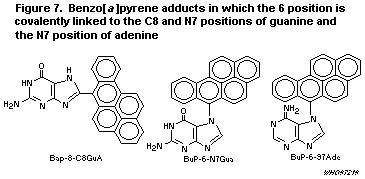 7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO
Appraisal
Single doses of polycyclic aromatic hydrocarbons (PAH) have moderate
to low toxicity, with LD50 values generally > 100 mg/kg bw after
intraperitoneal or intravenous injection and > 500 mg/kg bw after
oral administration. Because most of the experimental studies have
addressed the carcinogenicity of PAH, the database on their short- and
long-term toxicity is quite small. In short-term studies, effects on
the haematopoietic system were observed, e.g. benzo [a]pyrene caused
myelotoxicity and dibenz [a,h]anthracene caused haemolymphatic
alterations in mice. Anaemia is a typical effect of naphthalene.
Values for a no-observed-adverse-effect level (NOAEL) and a
lowest-observed-adverse-effect level (LOAEL) have been obtained in
90-day studies by oral administration. The NOAEL values based on
haematological effects and hepato- and nephrotoxicity were 75-1000
mg/kg bw per day for the noncarcinogenic PAH acenaphthene, anthracene,
fluoranthene, fluorene, and pyrene.
Few studies have been conducted on dermal or ocular irritation. PAH
do, however, have adverse effects after dermal administration, such as
hyperkeratosis, which are correlated with their carcinogenic potency.
Anthracene and naphthalene were reported to cause mild ocular
irritation. The ocular toxicity of naphthalene is characterized by
cataract formation. Benzo [a]pyrene caused skin hypersensitization.
Anthracene and benzo [a]pyrene have been shown to have phototoxic
potential and benzo [a]pyrene, dibenz [a,h]anthracene, and
fluoranthene to have immunotoxic potential.
PAH can cross the placenta and induce adverse effects on the embryo
and fetus. Benz [a]anthracene, benzo [a]pyrene,
dibenz [a,h]anthracene, and naphthalene were found to be embryotoxic.
Benzo [a]pyrene also reduced female fertility and had effects on
oocytes and on postnatal development. Studies on the effects of
benzo [a]pyrene in mice with different genotypes demonstrated the
importance of the genetic predisposition of animals or embryos for the
development of overt toxic effects. A crucial genetic property is the
presence or absence of the arylhydrocarbon (Ah) receptor, which
induces the monooxygenase system; organisms can thus be divided into
Ah responders and Ah non-responders.
Mutagenicity has been investigated intensively in a broad range of
assays. The only compounds that are clearly not mutagenic are
naphthalene, fluorene, and anthracene. The evidence for five PAH is
considered to be questionable because of a limited database, while the
remaining 25 PAH are mutagenic (see Table 87). Mutagenicity is
strictly dependent on metabolic activation of parent compounds. In
bacteria and other cell systems that have no metabolizing system, a
9000 × g microsomal preparation of liver (S9 mix) must be added as a
metabolic activator.
Comprehensive work on the carcinogenicity of these compounds has
yielded negative results for fluorene, anthracene,
1-methylphenanthrene, triphenylene, perylene, benzo[ghi]fluoranthene
and benzo[ghi]perylene, some of which have been shown to be
mutagenic. The evidence for a further nine PAH was classified as
questionable, while the other 17 compounds were carcinogenic.
Generally, the site of tumour development depends on the route of
administration but is not restricted to those sites. Tissues such as
the skin can metabolize PAH to their ultimate metabolites, thus
becoming target organs themselves, and metabolites formed in the liver
can reach various sites of the body via the bloodstream. The
carcinogenic potency of PAH differs by three orders of magnitude, and
toxic equivalence factors have been used to rank individual PAH (see
Appendix I).
The various theories for the mechanism of the carcinogenicity of PAH
take into account chemical structure and ionization potential. The
most prevalent theories are those involving the bay region and radical
cations. The bay-region theory is based on the assumption that diol
epoxides of the parent compounds are the ultimate carcinogens, which
react with electrophilic epoxide groups on N atoms of DNA purines. The
radical cation theory postulates the one-electron oxidation of PAH to
form strong electrophiles which then react with DNA bases. These
theories have been confirmed experimentally by detection of the
corresponding DNA adducts in the PAH that have been investigated.
Nevertheless, there is general agreement that any one theory cannot
cover the mechanisms of action of all PAH.
7.1 Toxicity after a single exposure
Few studies are available on the acute toxicity of PAH, except for
naphthalene. The LD50 values (Table 75) indicate that the acute
toxicity is moderate to low. The results of all of these studies are
summarized, even when a study was old and followed a non-systematic
protocol, in the absence of alternatives.
7.1.1 Benzo [a]pyrene
In young rats, a single intraperitoneal injection of 10 mg
benzo [a]pyrene per animal caused an immediate, sustained reduction
in the growth rate (Haddow et al., 1937). In mice, a single
intraperitoneal injection (dose not specified) resulted in small
spleens, marked cellular depletion, prominent haemosiderosis, and
follicles with large lymphocytes, leading to death (Shubik & Della
Porta, 1957). After a single application of 0.05 ml of a 1% solution
in acetone to the interscapular area of hairless mice (hr/hr strain),
the mitotic rate of epidermal cells was increased (Elgjo, 1968).
7.1.2 Chrysene
In young rats, single intraperitoneal injections of 30 mg chrysene per
animal did not reduce growth (Haddow et al., 1937).
Table 75. Toxicity of single doses of polycyclic: aromatic hydrocarbons
Compound Species Route of LD50 (mg/kg) or Reference
administration LC50 (mg/litre)
Anthracene Mouse Oral 18 000 Montizaan et al. (1989)
Mouse Intraperitoneal > 430 Salamone (1981)
Benzo[a]pyrene Mouse Oral > 1 600 Awogi & Sato (1989)
Mouse Intraperitoneal approx. 250 Salamone (1981)
Mouse Intraperitoneal > 1 600 Awogi & Sato (1989)
Rat Subcutaneous 50 Montizaan et al (1989)
Chrysene Mouse Intraperitoneal > 320 Simmon et al. (1979)
Fluoranthene Rat Oral 2 000 Smyth et al. (1962)
Rabbit Dermal 3 180 Smyth et al. (1962)
Mouse Intravenous 100 Montizaan et al. (1989)
Naphthalene Rat Oral 1 250 Sax & Lewis (1984)
Rat (M) Oral 2 200 Gaines (1969)
Rat (F) Oral 2 400 Gaines (1969)
Rat Oral 9 430 US Environmental Protection
Agency (1978a)
Rat Oral 1 110 Montizaan et al. (1989)
Rat Oral 490 Montizaan et al. (1989)
Rat Oral 1 800 Montizaan et al. (1989)
Rat (M) Dermal > 2 500 Gaines (1969)
Rat (F) Dermal > 2 500 Gaines (1969)
Rat Intraperitoneal approx. 1 000 Bolonova (1967)
Rat (M) Intraperitoneal approx. 1 600 Plopper et al. (1992)
Rat Inhalation > 0.5 mg/litre (8 h) US Environmental Protection
Agency (1978a)
Mouse (F) Oral 354 Plasterer et al. (1985)
Mouse (M) Oral 533 Shopp et al. (1984)
Mouse (F) Oral 710 Shopp et al. (1984)
Mouse Subcutaneous 5 100 Sandmeyer (1981);
Shopp et al. (1984)
Mouse Subcutaneous 969 Sax & Lewis (1984)
Mouse Intraperitoneal 150 Sax & Lewis (1984)
Table 75. (continued)
Compound Species Route of LD50 (mg/kg) or Reference
administration LC50 (mg/litre)
Mouse Intraperitoneal 380 Warren et al. (1982)
Mouse (M) Intraperitoneal approx. 400 Plopper et al. (1992)
Mouse Intravenous 100 Sax & Lewis (1984)
Hamster (M) Intraperitoneal approx. 800 Plopper et al. (1992)
Guinea-pig Oral 1 200 Sax & Lewis (1984)
Phenanthrene Mouse Oral 700 Montizaan et al. (1989)
Mouse Oral 1 000 Montizaan et al. (1989)
Mouse Intraperitoneal 700 Simmon et al. (1979)
Mouse Intravenous 56 Montizaan et al. (1989)
Pyrene Mouse Intraperitoneal 514 (7 d) Salamone (1981)
Mouse Intraperitoneal 678 (4 d) Salamone (1981)
LC50, median lethal concentration; LD50, median lethal dose; M, male; F, female
7.1.3 Dibenz [a,h]anthracene
One or two intraperitoneal injections of 3-90 mg
dibenz [a,h]anthracene per animal within two days led to a reduction
in the growth rate of young rats that persisted for at least 15 weeks
(Haddow et al., 1937).
7.1.4 Fluoranthene
In young rats, a single intraperitoneal injection of 30 mg
fluoranthene per animal did not inhibit growth (Haddow et al., 1937).
7.1.5 Naphthalene
After oral administration of 1-4 g/kg bw naphthalene to dogs or 1-3
g/kg bw to cats, diarrhoea was observed. Rabbits given 1-3 g/kg bw
showed corneal clouding (Flury & Zernik, 1935). After intravenous
injection of 1-6 mg napthalene to white male rabbits weighing 3-4 kg,
no haemolytic effect was seen (Mackell et al., 1951)
In mice, Clara cells of the bronchiolar epithelium are the primary
targets of low doses of naphthalene. Dose-dependent bronchiolar
epithelial cell necrosis was detected after intraperitoneal injection
of a single dose of 50, 100, or 200 mg/kg bw per day to mice (O'Brien
et al., 1989). Severe bronchiolar epithelial cell necrosis was also
seen in mice within 2-4 h after intraperitoneal injection of 200-375
mg/kg bw; hepatic and renal necrosis were not observed (Warren et al.,
1982). Alterations in the morphology of Clara cells were observed as
early as 6 h after intraperitoneal injection of 64 mg/kg bw; ciliated
cells were also affected after 24 and 48 h and at doses up to 256
mg/kg bw. After a 4-h inhalation of 1.0 mg/litre naphthalene,
bronchiolar necrosis was detected in mice but not in rats (Buckpitt &
Franklin, 1989; see also section 7.2.1).
After single injections of 50-400 mg/kg bw to mice, 100-800 mg/kg bw
to hamsters, and 200-1600 mg/kg bw to rats, Clara cells in mice showed
the effects described above; those of rats showed no significant
effects, and minor effects were observed in hamsters. The trachea and
lobar bronchi showed swelling and vacuolation of non-ciliated cells in
mice, no effects in rats, and cytotoxic changes in hamsters. In the
nasal cavity, cytotoxicity to the olfactory epithelium with necrosis
was observed in mice and hamsters at 400 mg/kg bw and in rats at 200
mg/kg bw (Plopper et al., 1992).
Mice injected intraperitoneally with 200-600 mg/kg bw naphthalene
showed dose-dependent abnormalities in the bronchial region (Clara
cells) in studies in which the lungs were examined by scanning
electron micrography. No pulmonary damage was detected at 100 mg/kg
bw. Depletion of pulmonary glutathione, which protects against the
toxicity of xenobiotics, was observed within 6 h of naphthalene
administration (Honda et al., 1990).
The doses and detailed findings of experiments with single doses of
naphthalene are summarized in Table 76.
7.1.6 Phenanthrene
After acute intraperitoneal injection to rats (dose not specified),
liver congestion with a distinct lobular pattern was observed as well
as alterations in some serum parameters (Yoshikawa et al., 1987).
7.1.7 Pyrene
In young rats, single intraperitoneal injections of 10 mg pyrene per
animal did not lead to a reduction in growth rate (Haddow et al.,
1937).
7.2 Short-term toxicity
7.2.1 Subacute toxicity
7.2.1.1 Acenaphthene
Four of five mice given 500 mg/kg bw per day acenaphthene
intraperitoneally for seven days survived (Gerarde, 1960).
7.2.1.2 Acenaphthylene
Nine of 10 mice given 500 mg/kg bw per day acenaphthylene for seven
days survived (Gerarde, 1960).
7.2.1.3 Anthracene
Nine of 10 mice given 500 mg/kg bw per day anthracene for seven days
survived (Gerarde, 1960). Oral administration of 100 mg/kg bw per day
to rats for four days increased carboxylesterase activity in the
intestinal mucosa by 13% (Nousiainen et al., 1984).
7.2.1.4 Benzo [a]pyrene
Death due to myelotoxicity was observed after daily oral
administration of benzo [a]pyrene at 120 mg/kg bw to poor-affinity Ah
receptor DBA/2N mice for one to four weeks, whereas high-affinity C57
Bl/6N mice survived with no myelotoxicity for at least six months
under these conditions (Legraverend et al., 1983).
Rats given 50 or 150 mg/kg bw per day of benzo [a]pyrene orally for
four days showed suppressed carboxylesterase activity in the
intestinal mucosa. The NOAEL with respect to gastric, hepatic, and
renal effects was 150 mg/kg bw per day (Nousiainen et al., 1984)
Table 76. Toxicity of single doses of naphthalene
Species Sex Route of Dose (purity) Effects Reference
(strain) (no./sex administration
per group)
Dog Oral 1000-2000,4000 1000-2000: Light diarrhoea; 4000 mg: Flury & Zernick
or 5000 mg/dog lethal; 5000 my heavy diarrhoea (1935)
Cat Oral 1000-3000 Lethal Flury & Zernick
mg/kg bw (1935)
Rabbit Oral 1000-3000 and 1000-3000 mg: corneal clouding; Flury & Zernick
3000 mg/kg bw 3000 mg death after 24 h (1935)
Dog (1) Oral 400 and 1800 400 mg: weakness, severe anaemia; Zuelzer & Apt
mg/kg bw 1800 mg: weakness, vomiting, diarrhoea, (1949)
slight anaemia; complete recovery within
1-2 weeks
Mouse Inhalation 0.1 mg/litre, 4 h Bronchiolar necrosis Buckpitt & Franklin
(1989)
Mouse M Intraperitoneal 50,100,200, Dose-dependent bronchiolar epithelial-cell O'Brien et al.
(Swiss-Webster 300 mg/kg bw necrosis (1989)
Mouse M (4-35) Intraperitoneal 50,100,200, Dose-dependent bronchiolar necrosis; Plopper et al.
(Swiss-Webster) 300, and 400 300 mg/kg: swollen cells in trachea (1992)
mg/kg bw 400 mg/kg: cytotoxicity in olfactory
(> 99.9%) epithelium
Rat M (4-11) Intraperitoneal 200,400,800, Bronchiolar necrosis not observed; no Plopper et al.
(Sprague-Dawley) and 1600 mg/kg changes in trachea; 200 mg/kg: complete (1992)
bw (> 99.9%) necrosis of olfactory epithelium
Table 76 (continued)
Species Sex Route of Dose (purity) Effects Reference
(strain) (no./sex administration
per group)
Rat M Intraperitoneal 400-1600 mg/kg No damage to lungs, liver, or kidneys O'Brien et al.
(Wistar) bw (1985)
Hamster M (4-6) Intraperitoneal 100,200,400 800 mg/kg: minor alterations in terminal Plopper et al.
(Syrian and 800 mg/kg bronchioles; cytotoxic changes in trachea; (1992)
golden) bw (99.9%) 400 mg/kg: necrosis of olfactory epithelium
Rabbit M Intraperitoneal 0.3-1.7 mg/kg bw No haemolytic effects Mackell et al.
(white) (1951)
M, male
In Fischer 344/Crl rats exposed by inhalation to 7.7 mg/m3 of
benzo [a]pyrene dust for 2 h/day, five days per week for four weeks,
no respiratory tract lesions were observed, as measured by lung
lavage, clearance of tagged particles, and histopathological findings
(Wolff, R.K. et al., 1989).
7.2.1.5 Benz [a]anthracene
When benz [a]anthracene was given orally to rats daily for four days,
the NOAEL with respect to gastric, hepatic, and renal effects was 150
mg/kg bw per day. Carboxylesterase activity in the intestinal mucosa
was suppressed (Nousiainen et al., 1984).
7.2.1.6 Dibenz [a,h]anthracene
Adverse haemolymphatic changes, including the appearance of
extravascular erythrocytes in the lymph spaces and large pigmented
cells, were reported after subcutaneous injection of male rats with
0.28 mg per animal on five days per week for four weeks (Lasnitzki &
Woodhouse, 1944).
7.2.1.7 Fluoranthene
All of 10 mice that received 500 mg/kg bw per day fluoranthene
intraperitoneally for seven days survived (Gerarde, 1960).
7.2.1.8 Naphthalene
Anaemia was induced in three dogs by single oral doses of 3 or 9 g or
a total dose of 10.5 g per animal given over seven days. All three
animals showed neurophysiological symptoms and slight to very severe
changes in haematological parameters. Full recovery was observed
within 7-14 days (Zuelzer & Apt, 1949).
No immunosuppressive effects were observed in a number of test
systems. Tolerance to the effects of naphthalene was reported in mice
after intraperitoneal injection for seven days. A sharp contrast
between single and multiple doses was observed in the effects on the
morphology of the bronchiolar epithelium. When naphthalene was given
intraperitoneally at a dose of 50, 100, or 200 mg/kg bw per day as a
single injection, dose-dependent bronchiolar epithelial cell necrosis
was detected; however, when these doses were given daily for seven
days, no significant effects were observed. Addition of 300 mg/kg bw
on day 8 had no effect, whereas recovered sensitivity was observed
with increasing time between the last dose and the challenge dose. A
single dose of 300 mg/kg bw without pretreatment resulted in
substantial denudation of the bronchiolar epithelium. This pattern was
attributed to a reduction in metabolic activation of naphthalene due
to a decrease in cytochrome P450 mono-oxygenase activity after
multiple dosing. A rough correlation was observed in mouse lung (but
not liver microsomes) between induction of tolerance and decreased
metabolic formation of the 1 R, 2 S-epoxide enantiomer, which is
responsible for tissue-selective toxicity. Such toxicity was
demonstrated in mice both in vivo and in isolated perfused lung
(Buckpitt & Franklin, 1989).
These studies are summarized in Table 77.
7.2.1.9 Phenanthrene
Oral administration of 100 mg/kg bw per day phenanthrene to rats for
four days induced a 30% increase in carboxylesterase activity in the
intestinal mucosa (Nousiainen et al., 1984).
7.2.1.10 Pyrene
Four of five mice injected intraperitoneally with 500 mg/kg bw per day
pyrene for seven days survived (Gerarde, 1960).
7.2.2 Subchronic toxicity
7.2.2.1 Acenaphthene
Administration of 175 mg/kg bw per day acenaphthene to mice by gavage
for 90 days resulted in a NOAEL of 175 mg/kg bw per day and a LOAEL of
350 mg/kg bw per day for hepatotoxicity (US Environmental Protection
Agency, 1989a).
7.2.2.2 Anthracene
Four of five rats given 5 mg per animal anthracene subcutaneously for
four months survived (Gerarde, 1960).
Anthracene was administered to groups of 20 male and female CD-1 (ICR)
BR mice by gavage at a dose of 0, 250, 500, or 1000 mg/kg bw per day
for at least 90 days. No treatment-related effects were noted on
mortality, clinical signs, body weights, food consumption,
ophthalmological findings, the results of haematology and clinical
chemistry, organ weights, organ-to-body weight ratios, and gross
pathological and histopathological findings. The no-observed-effect
level (NOEL) was the highest dose tested, 1000 mg/kg bw per day (US
Environmental Protection Agency, 1989b).
7.2.2.3 Benzo [a]pyrene
Male Syrian golden hamsters were exposed by inhalation to 9.8 or
44.8 mg/m3 benzo [a]pyrene for 4.5 h/day, five days per week for 16
weeks. No neoplastic response was observed in the respiratory tract
(Thyssen et al., 1980).
The growth of rats was inhibited by feeding a diet enriched with
benzo [a]pyrene at 1.1 g/kg for more than 100 days (White & White,
1939).
Table 77. Subacute and subchronic effects of naphthalene
Species Sex Route of Dose (purity) Effects Reference
(strain) (no./sex administration
per group)
Mouse M,F Oral 27, 53, and 267 In all groups, slight alterations in haemato Shopp et al.
(CD-1) (40-112) mg/kg bw, 7 d/ logical parameters; humoral immune response (1984)
week, 14 d not affected. 27 and 53 mg/kg: no significant
effects; 267 mg/kg: 5-10% mortality (m/f);
significantly decreased terminal body weight
(m/f); 30% decrease in thymus weight (m);
significant decrease in weight of spleen (f);
increase in lung weight (f)
Mouse M,F Oral 5.3, 53, and 133 No obvious pulmonary effects or Shopp et al.
(CDO) mg/kg bw, 7 d/ immunotoxicity; significantly decreased (1984)
week, 90 d relative spleen weights (f); tolerance
Mouse M Intraperitoneal 50, 100, and 200 No significant alterations in lung morphology; Buckpitt & Franklin
(Swiss-Webster) mg/kg, 7 d tolerance to 300 mg/kg on day 8 (1989); O'Brien et
al. (1989)
Rat Diet 2 g/kg diet, Inhibition of growth; enlarged, fatty livers White & White
100 d (1939)
Dog (1) Oral 122 g/kg bw per Diarrhoea, weakness, lack of appetite, ataxia, Zuelzer & Apt
day, 7 d very severe anaemia; complete recovery (1949)
within 1-2 weeks
M, male; F, female
7.2.2.4 Fluorene
Groups of 25 male and 25 female CD-1 mice were given 0, 125, 250, or
500 mg/kg bw per day fluorene suspended in corn oil by gavage for 13
weeks. Increased salivation, hypoactivity, and abdomens wetted with
urine were observed in all treated males. The percentage of hypoactive
mice was dose-related. In mice exposed at 500 mg/kg bw per day,
laboured respiration, ptosis (drooping eyelids), and an unkempt
appearance were also observed. A significant decrease in erythrocyte
count and packed cell volume were observed in females treated with 250
mg/kg bw per day fluorene and in males and females treated with 500
mg/kg bw per day. The latter also showed a decreased haemoglobin
concentration and an increased total serum bilirubin level. A
dose-related increase in relative liver weight was observed in treated
mice, and a significant increase in absolute liver weight was observed
in the mice treated with 250 or 500 mg/kg bw per day. Significant
increases in absolute and relative spleen and kidney weights were
observed in males and females exposed to 500 mg/kg bw per day and in
males at 250 mg/kg bw per day. The increases in absolute and relative
liver and spleen weights in animals at the high dose were accompanied
by increases in the amounts of haemosiderin in the spleen and in
Kupffer cells of the liver. No other histopathological lesions were
observed. The LOAEL for haematological effects was 250 mg/kg bw per
day, and the NOAEL was 125 mg/kg bw per day (US Environmental
Protection Agency, 1989c).
In a similar study, fluorene at 35, 50, and 150 mg/kg bw increased the
weight of the liver by about 20% in a dose-dependent fashion and the
mitotic index of hepatocytes by sixfold after 48 h (Danz et al.,
1991).
7.2.2.5 Fluoranthene
Groups of 20 male and 20 female CD-1 mice were given 0, 125, 250, or
500 mg/kg bw per day fluoranthene by gavage for 13 weeks. A fifth
group of 30 male and 30 female mice was used to establish baseline
levels in blood. Body weight, food consumption, and haematological and
serum parameters were recorded regularly throughout the experiment. At
the end of 13 weeks, the animals were killed and autopsied; organs
were weighed and a histological evaluation was made. All treated mice
had dose-dependent nephropathy, increased salivation, and increased
liver enzyme activities, but these effects were either not
significant, not dose-related, or not considered adverse at 125 mg/kg
bw per day. Mice exposed to 500 mg/kg bw per day had increased food
consumption and increased body weight. Mice exposed to the two higher
doses had statistically increased alanine aminotransferase activity
and increased absolute and relative liver weights. Treatment-related
microscopic liver lesions (indicated by pigmentation) were observed in
65% of mice at 250 mg/kg bw per day and 88% of those at the highest
dose. On the basis of the increased alanine aminotransferase activity,
pathological effects in the kidney and liver, and clinical and
haematological changes, the LOAEL was 250 mg/kg bw per day and the
NOAEL 125 mg/kg bw per day (US Environmental Protection Agency, 1988).
7.2.2.6 Naphthalene
In a 90-day study in mice, naphthalene at oral doses up to 133 mg/kg
bw caused neither mortality nor serious changes in organ weights
(Shopp et al., 1984). These authors did not observe haemolytic anaemia
in CD-1 mice after oral uptake, although this effect had been seen in
human patients (Konar et al., 1939; Zuelzer & Apt, 1949; see Section
8). It was suggested that glucose-6-phosphate dehydrogenase deficiency
in erythrocytes, a prerequisite of haemolytic anaemia in adult humans,
was not present in the mice (Shopp et al., 1984).
In rats that ingested 150 mg/kg bw per day naphthalene for the first
three weeks and 200-220 mg/kg bw per day for a further 11 weeks,
reduced weight gain and food intake were observed. Later, the liver
was found to be enlarged, with cell oedema and congestion of the liver
parenchyma, and the kidneys showed signs of inflammation (Kawai,
1979).
The presence of 1 g/kg naphthalene in the feed of rats and rabbits for
46-60 days led to cataracts (US Environmental Protection Agency,
1984b; see also section 7.8).
Administration to rabbits of 0.1-1 mg/kg bw per day naphthalene by
subcutaneous injection for 123 days resulted in severe oedema and a
high degree of vacuolar and collicular degeneration in the brain;
necrosis of nerve cells also occurred. The author suggested that
hypoxaemia resulting from haemolytic anaemia was responsible for this
finding (Suja, 1967; cited by Kawai, 1979).
Subacute and subchronic studies with naphthalene are summarized in
Table 77.
7.2.2.7 Pyrene
The growth of rats was inhibited by feeding a diet enriched with
benzo [a]pyrene at 2 g/kg for more than 100 days. The livers were
enlarged and had a fatty appearance indicating hepatic injury (White &
White, 1939).
Groups of 20 male and 20 female CD-1 mice were given 0, 75, 125, or
250 mg/kg bw per day pyrene in corn oil by gavage for 13 weeks and
then examined for changes in body weight, food consumption, mortality,
clinical pathological manifestations in major organs and tissues, and
changes in haematology and serum chemistry. Nephropathy, characterized
by the presence of multiple foci of renal tubular regeneration, often
accompanied by interstitial lymphocytic infiltrates and/or foci of
interstitial fibrosis, was present in four male control mice, one at
the low dose, one at the medium dose, and nine the high dose. Similar
lesions were seen in two, three, seven, and 10 female mice,
respectively. The renal lesions in all groups were described as
minimal or mild. Relative and absolute kidney weights were reduced in
mice at the two higher doses. On the basis of nephropathy and
decreased kidney weights, the low dose (75 mg/kg bw per day) was
considered to be the NOAEL and 125 mg/kg bw per day the LOAEL (US
Environmental Protection Agency, 1989d).
7.3 Long-term toxicity
Almost all of the long-term studies reported were designed to assess
the carcinogenic potency of PAH and are therefore summarized in
section 7.7. Information about the non-carcinogenic effects, such as
growth inhibition, liver damage, and irritation, which occurred at
concentrations that also caused carcinogenic effects is presented
here. General effects, such as on mortality, body weight, and
pathological findings at sacrifice, were not considered useful.
7.3.1 Anthracene
A group of 28 BD I and BD III rats received anthracene in the diet
from the age of about 100 days, at a daily dose of 5-15 mg per rat.
The experiment was terminated when a total dose of 4.5 g per rat had
been achieved, on day 550. The rats were observed until they died;
some lived for more than 1000 days. No treatment-related effects on
lifespan or on gross or histological appearance of tissues were
observed; haematological parameters were not measured (Schmähl, 1955).
After weekly subcutaneous injections of anthracene at 0.25 mg per
animal for 40 weeks, mice showed deposition of iron in lymph glands
and a reduced number of lymphoid cells (Hoch-Ligeti, 1941).
7.3.2 Benz [a]anthracene
Weekly subcutaneous injection of 0.25 mg per mouse for 40 weeks
resulted in deposition of iron in lymph glands and a reduced number of
lymphoid cells (Hoch-Ligeti, 1941).
7.3.3 Dibenz [a,h]anthracene
Mice given weekly subcutaneous injections of 0.25 mg per animal for
40 weeks had pale, soft, enlarged livers with signs of fatty
degeneration. There was deposition of iron in lymph glands, and the
number of lymphoid cells was reduced (Hoch-Ligeti, 1941).
7.4 Dermal and ocular irritation and dermal sensitization
The adverse dermatological effects observed in animals after acute and
subchronic dermal exposure to PAH included destruction of sebaceous
glands, dermal ulceration, hyperplasia, hyperkeratosis, and
alterations in epidermal cell growth. Perylene, benzo [e]pyrene,
phenanthrene, pyrene, anthracene, naphthalene, acenaphthalene,
fluorene, and fluoranthene did not suppress the sebaceous gland index;
benz [a]anthracene, dibenz [a,h]anthracene, and benzo [a]pyrene
resulted in indices > 1 (Bock & Mund, 1958). In Swiss mice treated
daily for three days with solutions of benzo [a]pyrene in acetone, a
concentration of 0.1% destroyed less than half of the sebaceous
glands, whereas 0.2% destroyed more than 50% (Suntzeff et al., 1955).
7.4.1 Anthracene
Anthracene is a primary irritant, and its fumes can cause mild
irritation of the skin, eyes, mucous membranes, and respiratory tract.
At a concentration of 4.7 mg/m3, mild skin irritation was found in
50% of exposed mice (Montizaan et al., 1989). The median value for
dermal irritant activity (ID50) in the mouse ear was 6.6 × 10-4
mmol or 118 µg/ear; in comparison, the ID50 for benzo [a]pyrene was
5.6 × 10-5 mmol per ear (Brune et al., 1978). Anthracene increases
the sensitivity of skin to solar radiation (Gerarde, 1960). No contact
sensitivity to anthracene was observed (Old et al., 1963).
7.4.2 Benzo [a]pyrene
Four adult female guinea-pigs were injected with a total of 250 µg
benzo [a]pyrene in Freund's adjuvant, and two to three weeks later
were tested for contact sensitivity with solutions of 0.001, 0.01,
0.1, or 1% benzo [a]pyrene in acetone and olive oil. After 24 h, a
slight to severe (0.001-1%) contact hypersensitivity was observed (Old
et al., 1963).
C3H mice were given an epicutaneous administration of 100 µg
benzo [a]pyrene in 0.1% acetone solution into the abdominal skin.
Five days later, contact hypersensitivity was elicited by applying 20
µg benzo [a]pyrene to the dorsal aspect of the ear. The response was
quantified by ear thickness, which reaced a maximum three to five days
after challenge. The LOAEL for allergic contact sensitivity was thus
120 µg (Klemme et al., 1987).
The ID50 value for dermal irritant activity in the mouse ear was 5.6
× 10-5 mmol per ear (Brune et al., 1978).
7.4.3 Naphthalene
A single dose of 100 mg naphthalene to the rabbit eye was slightly
irritating, whereas application of 495 mg to rabbit skin, without
occlusion, caused mild irritation (Sax & Lewis, 1984).
7.4.4 Phenanthrene
No contact sensitization to phenanthrene was observed (Old et al.,
1963).
7.5 Reproductive effects, embryotoxicity, and teratogenicity
The mechanistic aspects of reproductive and embryotoxic effects are
presented in detail and the results summarized in Tables 78-80. The
genotype of mice is decisive for the manifestation of effects.
Studies have been reported on anthracene, benz [a]anthracene,
benzo [a]-pyrene, chrysene, dibenz [a,h]anthracene, and naphthalene.
Embryotoxicity was reported in response to benz [a]anthracene,
benzo [a]pyrene, dibenz [a,h]-anthracene, and naphthalene.
Benzo [a]pyrene also had adverse effects on female fertility,
reproduction, and postnatal development. In a study in young mice, an
NOEL of 150 mg/kg bw per day was obtained for benzo [a]pyrene on the
basis of effects on fertility (sperm in lumen of testes, size of
litters) and embryotoxicity (malformations) (Rigdon & Neal, 1965).
7.5.1 Benzo [a]pyrene
7.5.1.1 Teratogenicity in mice of different genotypes
Benzo [a]pyrene is embryotoxic to mice, and the effect is partly
dependent on the genetically determined induction of the cytochrome
P450 mono-oxygenase receptor, Ah, of the mother and fetus by PAH (see
also section 7.10). In the case of an inducible mother
(Ahb/ Ahb and Ahb/ Ahd, B groups), the genotype of the
fetus is not crucial because the active metabolites formed in the
mother appear to cross the placenta, causing fetal death or
malformation. In contrast, when the mother is non-inducible
(Ahd/ Ahd, D group), the genotype of the fetus is important;
one litter may contain both inducible and non-inducible fetuses.
Another decisive factor is the route by which benzo [a]pyrene is
given to the mother. Three studies of the genetic expression of
effects are summarized below.
Intraperitoneal injection of benzo [a]pyrene at 50 or 300 mg/kg bw on
day 7 or 10 of gestation was more toxic and teratogenic in utero in
genetically inducible C57Bl/6 (Ahb/ Ahb) than in non-inducible
AKR inbred mice (Ahd/ Ahd). In AKR × (C57Bl/6)(AKR)F1 and
(C57Bl/6)(AKR)F1 × AKR back-crosses (father × F1 mother), allelic
differences at the Ah locus in the fetus correlated with
dysmorphogenesis. The inducible fetal Ahb/ Ahd genotype results
in more stillborn and resorbed fetuses,decreased fetal weight,
increased frequency of congenital anomalies, and enhanced
P1-450-mediated covalent binding of benzo [a]pyrene metabolites to
fetal protein and DNA, when compared with fetuses of the non-inducible
Ahd/ Ahd genotype (not-inducible) from the same uterus (see
Table 78). In the case of an inducible mother (Ahb/ Ahd),
however, these parameters do not differ in Ahb/ Ahd and
Ahd/ Ahd individuals in the same uterus, presumably because the
increased benzo [a]pyrene metabolism in maternal tissues and placenta
cancels them out (Shum et al., 1979).
An inducible genotype is not the only factor involved in the
reproductive toxicity of benzo [a]pyrene. In a study in which C57Bl/6
female mice (Ah inducible) were mated with C57Bl/6, DBA/2, or BDF1
male mice (B groups), and DBA/2 females (non-inducible) were mated
with C57Bl/6, DBA/2, or BDF1 males (D groups) and received
intraperitoneal injections of benzo [a]-pyrene, fetal mortality
increased dose-dependently in all groups except the DBA/2 × DBA/2.
Fetal body weight was reduced dose-dependently in all experimental
groups, but the effect was more pronounced in D than B groups, as was
a dose-dependent increase in the frequency of cervical ribs (for
experimental details, see Table 78). These results suggest that
Table 78. Embryotoxicity of polycyclic aromatic hydrocarbons in experimental animals
Species No. per Route of Duration, dose Effects Reference
(strain) group administration
Anthracene
Rat Gavage Day 19 of gestation, F1: no induction of BaP hydroxylase in liver Welch et al.
Sprague- 60 mg/kg compared with control (< 0.2 vs <0.2 units in (1972)
bw controls)
Benz[a]anthracene
Rat 2 Subcutaneous Day 1-11 or 1-15 F0: Day 10 and 12: severe vaginal haemorrhage; Wolfe &
of gestation, 5 mg/ Day 14: intraplacental haemorrhage Bryan (1939)
animal per day F1: fetal death and resorption up to day 18
Rat Gavage Day 19 of gestation, F1: induction of BaP hydroxylase in liver Welch et al.
Sprague-Dawley 60 mg/kg bw (12 vs < 0.2 units in controls) (1972)
Benzo[a]pyrene
Mouse 9 Diet Day 5 or 10 of F1: no malformations Rigdon &
White gestation until Neal (1965)
Swiss delivery, 50 mg/ky bw
Mouse 6-17 Diet Day 2-10 of F1: increased intrauterine toxicity and Legraverend
C57BI/6N, gestation, 120 mg/ malformations in Ahd/Ah7dembryos compared et al. (1984)
AKR/J, and kg per day with Ahb/Ahd embryos in pregnant Ahd/Ahd
back-crosses mice (effect not seen in pregnant Ahb/Ahd mice)
(reciprocal)
Mouse 5-30 Intraperitoneal Day 7, 10, or 12 of 200 mg/kg bw: F1: increase in stillbirths, Shum et al.
C57BI/6, gestation, 50-300 resorptions, malformations (4-fold higher (1979)
AKR and mg/kg bw in pregnant C57BI than in AKR mice)
back-crosses
(reciprocal)
Table 78. (continued)
Species No. per Route of Duration, dose Effects Reference
(strain) group administration
Mouse 20 Intraperitoneal Day 8 of gestation, 150 and 300 mg/kg bw: F0: increased fetal Hoshino et al.
C57BI/6, 150 or 300 mg/kg mortality (except DBA/2 × DBA/2 offspring); (1981)
DBA/2, and reduced fetal body weight; increased number of
back-crosses cervical ribs
(reciprocal) 300 mg/kg: F1: increased malformations
(C57BI/6 × C57BI/6)
Mouse Gavage Day 7-16 of F0: no toxicity MacKenzie &
CIT1 gestation, 10, 40, 160 F1: no toxicity Angevine (1981)
mg/kg bw per day
Rat 17 Subcutaneous Day 1-11 or 16 of F0: Days 10 and 12: profuse vaginal Wolfe & Bryan
gestation, 5 mg/ haemorrhage; day 14: intraplacental (1939)
animal per day haemorrhage; F1: fetal death and resorption
up to day 18
Rat Gavage Day 19 of gestation, F1: induction of BaP-hydroxylase in liver Welch al al.
Sprague-Dawley 60 mg/kg bw (20 vs < 0.2 units in controls) (1972)
Rat 10-15 Subcutaneous Day 6-8 or 6-11 of F1: significant increase in number of resorptions Bui et al.
Sprague-Dawley gestation, 50 mg/kg and fetal wastage (dead fetuses plus resorption); (1986)
bw per day fetal weight reduced
Chrysene
Rat Gavage Day 19 of gestation, F1: induction of BaP hydroxylase in liver Welch et al.
Sprague-Dawley 60 mg/kg bw (6 vs < 0.2 units in controls) (1972)
Dibenzo[a,h]anthracene
Rat Gavage Day 19 of gestation, F1: induction of BaP hydroxylase in liver Welch et al.
Sprague-Dawley 60 mg/kg bw (15 vs < 0.2 units in controls) (1972)
Table 78. (continued)
Species No. per Route of Duration, dose Effects Reference
(strain) group administration
Rat 38 Subcutaneous Day 1-8 or 1-18 of F0: Days 10 and 12: profuse vaginal haemorrhage; Wolfe &
gestation, 5 mg/ day 14: intraplacental haemorrhage Bryan (1939)
animal per day F1: fetal death and resorption up to day 18
Naphthalene
Mouse 50 Gavage Day 7-14 of F0: significant 15% increase in mortality; Plasterer et
CD-1 gestation, 300 mg/ significant reduction in weight gain al. (1985)
kg bw per day F1: significant reduction in number of live
offspring; no malformations
Mouse Gavage Day 6-13 of F0 increased mortality 10/50; control: 0/50); Hardin et al.
CD-1 gestation, 300 mg/ significant reduction in weight gain (1987)
kg bw per day F1: significant reduction in liveborns per litter
Rat 10-15 Intraperitoneal Day 1-15 of F0: no toxicity Hardin et al.
Sprague-Dawley gestation, 395 mg/ F1: no toxicity (1981)
kg per day
For genotypes of the mouse strains used see section 7.5.1.1
Ah-inducible fetuses are more sensitive to lethal events, whereas
those of non-inducible dams are more susceptible to a decrease in body
weight and an increased incidence of cervical ribs. The incidence of
external malformations may, however, differ in mice of different
genotypes after treatment with benzo [a]-pyrene, even if both dams
and fetuses are inducible (Hoshino et al., 1981).
The toxicity of benzo [a]pyrene in utero was investigated in
pregnant Ahd/ Ahd × Ahb/ Ahd F1 and Ahb/ Ahd ×
Ahd/ Ahd F1 back-crossed mice fed benzo [a]pyrene in the
diet at 120 mg/kg daily on days 2-10 of gestation. Embryos of D
females (Ahd/ Ahd genotype; non-inducible) showed more signs
of toxicity and malforma-tions than Ahd/ Ahd embryos. Fetuses
of B females (Ahb/ Ahd genotype) did not show these changes.
The authors suggested that reduced benzo [a]pyrene metabolism in the
intestine had caused high concentrations in the embryos, and more
toxic metabolites (benzo [a]pyrene-1,6- and -3,6-quinones) were
detected in the Ahd/ Ahd embryos than in Ahb/ Ahd
embryos (Legraverend et al., 1984). These results were in contrast to
those reported after intraperitoneal injection by Shum et al. (1979)
and Hoshino et al. (1981). The route of administration can thus affect
the magnitude of the observed effects (see also section 7.8.2.2).
7.5.1.2 Reproductive toxicity
A single intraperitoneal injection of benzo [a]pyrene reduced
fertility and destroyed primordial oocytes of DBA/2N mice in a
dose-dependent manner (Mattison et al., 1980; see also Table 79).
In experiments with B6 (Ah-inducible) and D2 (non-inducible) mice,
primordial oocytes of B6 mice underwent more rapid destruction after
treatment with benzo [a]pyrene than those of D2 mice. This effect
corresponded to a two- to threefold increase in ovarian
arylhydrocarbon hydroxylase (AHH) activity in B6 mice after treatment.
This correlation was not found in analogous experiments with D2B6F1
mice, in which AHH activity was increased by two- to threefold, but
the oocyte destruction was similar to that observed in D2 mice. This
demonstrates an inconsistent consequence of strain differences in
genotype (Mattison & Nightingale, 1980; see also Table 79). The sum of
activation, detoxification, and repair seems to be decisive for the
process of oocyte destruction (Figure 8).
Benzo [a]pyrene and its three metabolites, benzo [a]pyrene
7,8-oxide, benzo [a]pyrene 7,8-diol, and benzo [a]pyrene diol
epoxide, were administered by injection at a single dose of 10 µg into
the right ovary of B6, D2, and D2B6F1 mice. Ovarian volume, weight,
and follicle numbers were measured after two weeks; various reductions
were observed in all strains. There was also compesatory hypertrophy
of the left ovary (Mattison et al., 1989; see also Table 79).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO
Appraisal
Single doses of polycyclic aromatic hydrocarbons (PAH) have moderate
to low toxicity, with LD50 values generally > 100 mg/kg bw after
intraperitoneal or intravenous injection and > 500 mg/kg bw after
oral administration. Because most of the experimental studies have
addressed the carcinogenicity of PAH, the database on their short- and
long-term toxicity is quite small. In short-term studies, effects on
the haematopoietic system were observed, e.g. benzo [a]pyrene caused
myelotoxicity and dibenz [a,h]anthracene caused haemolymphatic
alterations in mice. Anaemia is a typical effect of naphthalene.
Values for a no-observed-adverse-effect level (NOAEL) and a
lowest-observed-adverse-effect level (LOAEL) have been obtained in
90-day studies by oral administration. The NOAEL values based on
haematological effects and hepato- and nephrotoxicity were 75-1000
mg/kg bw per day for the noncarcinogenic PAH acenaphthene, anthracene,
fluoranthene, fluorene, and pyrene.
Few studies have been conducted on dermal or ocular irritation. PAH
do, however, have adverse effects after dermal administration, such as
hyperkeratosis, which are correlated with their carcinogenic potency.
Anthracene and naphthalene were reported to cause mild ocular
irritation. The ocular toxicity of naphthalene is characterized by
cataract formation. Benzo [a]pyrene caused skin hypersensitization.
Anthracene and benzo [a]pyrene have been shown to have phototoxic
potential and benzo [a]pyrene, dibenz [a,h]anthracene, and
fluoranthene to have immunotoxic potential.
PAH can cross the placenta and induce adverse effects on the embryo
and fetus. Benz [a]anthracene, benzo [a]pyrene,
dibenz [a,h]anthracene, and naphthalene were found to be embryotoxic.
Benzo [a]pyrene also reduced female fertility and had effects on
oocytes and on postnatal development. Studies on the effects of
benzo [a]pyrene in mice with different genotypes demonstrated the
importance of the genetic predisposition of animals or embryos for the
development of overt toxic effects. A crucial genetic property is the
presence or absence of the arylhydrocarbon (Ah) receptor, which
induces the monooxygenase system; organisms can thus be divided into
Ah responders and Ah non-responders.
Mutagenicity has been investigated intensively in a broad range of
assays. The only compounds that are clearly not mutagenic are
naphthalene, fluorene, and anthracene. The evidence for five PAH is
considered to be questionable because of a limited database, while the
remaining 25 PAH are mutagenic (see Table 87). Mutagenicity is
strictly dependent on metabolic activation of parent compounds. In
bacteria and other cell systems that have no metabolizing system, a
9000 × g microsomal preparation of liver (S9 mix) must be added as a
metabolic activator.
Comprehensive work on the carcinogenicity of these compounds has
yielded negative results for fluorene, anthracene,
1-methylphenanthrene, triphenylene, perylene, benzo[ghi]fluoranthene
and benzo[ghi]perylene, some of which have been shown to be
mutagenic. The evidence for a further nine PAH was classified as
questionable, while the other 17 compounds were carcinogenic.
Generally, the site of tumour development depends on the route of
administration but is not restricted to those sites. Tissues such as
the skin can metabolize PAH to their ultimate metabolites, thus
becoming target organs themselves, and metabolites formed in the liver
can reach various sites of the body via the bloodstream. The
carcinogenic potency of PAH differs by three orders of magnitude, and
toxic equivalence factors have been used to rank individual PAH (see
Appendix I).
The various theories for the mechanism of the carcinogenicity of PAH
take into account chemical structure and ionization potential. The
most prevalent theories are those involving the bay region and radical
cations. The bay-region theory is based on the assumption that diol
epoxides of the parent compounds are the ultimate carcinogens, which
react with electrophilic epoxide groups on N atoms of DNA purines. The
radical cation theory postulates the one-electron oxidation of PAH to
form strong electrophiles which then react with DNA bases. These
theories have been confirmed experimentally by detection of the
corresponding DNA adducts in the PAH that have been investigated.
Nevertheless, there is general agreement that any one theory cannot
cover the mechanisms of action of all PAH.
7.1 Toxicity after a single exposure
Few studies are available on the acute toxicity of PAH, except for
naphthalene. The LD50 values (Table 75) indicate that the acute
toxicity is moderate to low. The results of all of these studies are
summarized, even when a study was old and followed a non-systematic
protocol, in the absence of alternatives.
7.1.1 Benzo [a]pyrene
In young rats, a single intraperitoneal injection of 10 mg
benzo [a]pyrene per animal caused an immediate, sustained reduction
in the growth rate (Haddow et al., 1937). In mice, a single
intraperitoneal injection (dose not specified) resulted in small
spleens, marked cellular depletion, prominent haemosiderosis, and
follicles with large lymphocytes, leading to death (Shubik & Della
Porta, 1957). After a single application of 0.05 ml of a 1% solution
in acetone to the interscapular area of hairless mice (hr/hr strain),
the mitotic rate of epidermal cells was increased (Elgjo, 1968).
7.1.2 Chrysene
In young rats, single intraperitoneal injections of 30 mg chrysene per
animal did not reduce growth (Haddow et al., 1937).
Table 75. Toxicity of single doses of polycyclic: aromatic hydrocarbons
Compound Species Route of LD50 (mg/kg) or Reference
administration LC50 (mg/litre)
Anthracene Mouse Oral 18 000 Montizaan et al. (1989)
Mouse Intraperitoneal > 430 Salamone (1981)
Benzo[a]pyrene Mouse Oral > 1 600 Awogi & Sato (1989)
Mouse Intraperitoneal approx. 250 Salamone (1981)
Mouse Intraperitoneal > 1 600 Awogi & Sato (1989)
Rat Subcutaneous 50 Montizaan et al (1989)
Chrysene Mouse Intraperitoneal > 320 Simmon et al. (1979)
Fluoranthene Rat Oral 2 000 Smyth et al. (1962)
Rabbit Dermal 3 180 Smyth et al. (1962)
Mouse Intravenous 100 Montizaan et al. (1989)
Naphthalene Rat Oral 1 250 Sax & Lewis (1984)
Rat (M) Oral 2 200 Gaines (1969)
Rat (F) Oral 2 400 Gaines (1969)
Rat Oral 9 430 US Environmental Protection
Agency (1978a)
Rat Oral 1 110 Montizaan et al. (1989)
Rat Oral 490 Montizaan et al. (1989)
Rat Oral 1 800 Montizaan et al. (1989)
Rat (M) Dermal > 2 500 Gaines (1969)
Rat (F) Dermal > 2 500 Gaines (1969)
Rat Intraperitoneal approx. 1 000 Bolonova (1967)
Rat (M) Intraperitoneal approx. 1 600 Plopper et al. (1992)
Rat Inhalation > 0.5 mg/litre (8 h) US Environmental Protection
Agency (1978a)
Mouse (F) Oral 354 Plasterer et al. (1985)
Mouse (M) Oral 533 Shopp et al. (1984)
Mouse (F) Oral 710 Shopp et al. (1984)
Mouse Subcutaneous 5 100 Sandmeyer (1981);
Shopp et al. (1984)
Mouse Subcutaneous 969 Sax & Lewis (1984)
Mouse Intraperitoneal 150 Sax & Lewis (1984)
Table 75. (continued)
Compound Species Route of LD50 (mg/kg) or Reference
administration LC50 (mg/litre)
Mouse Intraperitoneal 380 Warren et al. (1982)
Mouse (M) Intraperitoneal approx. 400 Plopper et al. (1992)
Mouse Intravenous 100 Sax & Lewis (1984)
Hamster (M) Intraperitoneal approx. 800 Plopper et al. (1992)
Guinea-pig Oral 1 200 Sax & Lewis (1984)
Phenanthrene Mouse Oral 700 Montizaan et al. (1989)
Mouse Oral 1 000 Montizaan et al. (1989)
Mouse Intraperitoneal 700 Simmon et al. (1979)
Mouse Intravenous 56 Montizaan et al. (1989)
Pyrene Mouse Intraperitoneal 514 (7 d) Salamone (1981)
Mouse Intraperitoneal 678 (4 d) Salamone (1981)
LC50, median lethal concentration; LD50, median lethal dose; M, male; F, female
7.1.3 Dibenz [a,h]anthracene
One or two intraperitoneal injections of 3-90 mg
dibenz [a,h]anthracene per animal within two days led to a reduction
in the growth rate of young rats that persisted for at least 15 weeks
(Haddow et al., 1937).
7.1.4 Fluoranthene
In young rats, a single intraperitoneal injection of 30 mg
fluoranthene per animal did not inhibit growth (Haddow et al., 1937).
7.1.5 Naphthalene
After oral administration of 1-4 g/kg bw naphthalene to dogs or 1-3
g/kg bw to cats, diarrhoea was observed. Rabbits given 1-3 g/kg bw
showed corneal clouding (Flury & Zernik, 1935). After intravenous
injection of 1-6 mg napthalene to white male rabbits weighing 3-4 kg,
no haemolytic effect was seen (Mackell et al., 1951)
In mice, Clara cells of the bronchiolar epithelium are the primary
targets of low doses of naphthalene. Dose-dependent bronchiolar
epithelial cell necrosis was detected after intraperitoneal injection
of a single dose of 50, 100, or 200 mg/kg bw per day to mice (O'Brien
et al., 1989). Severe bronchiolar epithelial cell necrosis was also
seen in mice within 2-4 h after intraperitoneal injection of 200-375
mg/kg bw; hepatic and renal necrosis were not observed (Warren et al.,
1982). Alterations in the morphology of Clara cells were observed as
early as 6 h after intraperitoneal injection of 64 mg/kg bw; ciliated
cells were also affected after 24 and 48 h and at doses up to 256
mg/kg bw. After a 4-h inhalation of 1.0 mg/litre naphthalene,
bronchiolar necrosis was detected in mice but not in rats (Buckpitt &
Franklin, 1989; see also section 7.2.1).
After single injections of 50-400 mg/kg bw to mice, 100-800 mg/kg bw
to hamsters, and 200-1600 mg/kg bw to rats, Clara cells in mice showed
the effects described above; those of rats showed no significant
effects, and minor effects were observed in hamsters. The trachea and
lobar bronchi showed swelling and vacuolation of non-ciliated cells in
mice, no effects in rats, and cytotoxic changes in hamsters. In the
nasal cavity, cytotoxicity to the olfactory epithelium with necrosis
was observed in mice and hamsters at 400 mg/kg bw and in rats at 200
mg/kg bw (Plopper et al., 1992).
Mice injected intraperitoneally with 200-600 mg/kg bw naphthalene
showed dose-dependent abnormalities in the bronchial region (Clara
cells) in studies in which the lungs were examined by scanning
electron micrography. No pulmonary damage was detected at 100 mg/kg
bw. Depletion of pulmonary glutathione, which protects against the
toxicity of xenobiotics, was observed within 6 h of naphthalene
administration (Honda et al., 1990).
The doses and detailed findings of experiments with single doses of
naphthalene are summarized in Table 76.
7.1.6 Phenanthrene
After acute intraperitoneal injection to rats (dose not specified),
liver congestion with a distinct lobular pattern was observed as well
as alterations in some serum parameters (Yoshikawa et al., 1987).
7.1.7 Pyrene
In young rats, single intraperitoneal injections of 10 mg pyrene per
animal did not lead to a reduction in growth rate (Haddow et al.,
1937).
7.2 Short-term toxicity
7.2.1 Subacute toxicity
7.2.1.1 Acenaphthene
Four of five mice given 500 mg/kg bw per day acenaphthene
intraperitoneally for seven days survived (Gerarde, 1960).
7.2.1.2 Acenaphthylene
Nine of 10 mice given 500 mg/kg bw per day acenaphthylene for seven
days survived (Gerarde, 1960).
7.2.1.3 Anthracene
Nine of 10 mice given 500 mg/kg bw per day anthracene for seven days
survived (Gerarde, 1960). Oral administration of 100 mg/kg bw per day
to rats for four days increased carboxylesterase activity in the
intestinal mucosa by 13% (Nousiainen et al., 1984).
7.2.1.4 Benzo [a]pyrene
Death due to myelotoxicity was observed after daily oral
administration of benzo [a]pyrene at 120 mg/kg bw to poor-affinity Ah
receptor DBA/2N mice for one to four weeks, whereas high-affinity C57
Bl/6N mice survived with no myelotoxicity for at least six months
under these conditions (Legraverend et al., 1983).
Rats given 50 or 150 mg/kg bw per day of benzo [a]pyrene orally for
four days showed suppressed carboxylesterase activity in the
intestinal mucosa. The NOAEL with respect to gastric, hepatic, and
renal effects was 150 mg/kg bw per day (Nousiainen et al., 1984)
Table 76. Toxicity of single doses of naphthalene
Species Sex Route of Dose (purity) Effects Reference
(strain) (no./sex administration
per group)
Dog Oral 1000-2000,4000 1000-2000: Light diarrhoea; 4000 mg: Flury & Zernick
or 5000 mg/dog lethal; 5000 my heavy diarrhoea (1935)
Cat Oral 1000-3000 Lethal Flury & Zernick
mg/kg bw (1935)
Rabbit Oral 1000-3000 and 1000-3000 mg: corneal clouding; Flury & Zernick
3000 mg/kg bw 3000 mg death after 24 h (1935)
Dog (1) Oral 400 and 1800 400 mg: weakness, severe anaemia; Zuelzer & Apt
mg/kg bw 1800 mg: weakness, vomiting, diarrhoea, (1949)
slight anaemia; complete recovery within
1-2 weeks
Mouse Inhalation 0.1 mg/litre, 4 h Bronchiolar necrosis Buckpitt & Franklin
(1989)
Mouse M Intraperitoneal 50,100,200, Dose-dependent bronchiolar epithelial-cell O'Brien et al.
(Swiss-Webster 300 mg/kg bw necrosis (1989)
Mouse M (4-35) Intraperitoneal 50,100,200, Dose-dependent bronchiolar necrosis; Plopper et al.
(Swiss-Webster) 300, and 400 300 mg/kg: swollen cells in trachea (1992)
mg/kg bw 400 mg/kg: cytotoxicity in olfactory
(> 99.9%) epithelium
Rat M (4-11) Intraperitoneal 200,400,800, Bronchiolar necrosis not observed; no Plopper et al.
(Sprague-Dawley) and 1600 mg/kg changes in trachea; 200 mg/kg: complete (1992)
bw (> 99.9%) necrosis of olfactory epithelium
Table 76 (continued)
Species Sex Route of Dose (purity) Effects Reference
(strain) (no./sex administration
per group)
Rat M Intraperitoneal 400-1600 mg/kg No damage to lungs, liver, or kidneys O'Brien et al.
(Wistar) bw (1985)
Hamster M (4-6) Intraperitoneal 100,200,400 800 mg/kg: minor alterations in terminal Plopper et al.
(Syrian and 800 mg/kg bronchioles; cytotoxic changes in trachea; (1992)
golden) bw (99.9%) 400 mg/kg: necrosis of olfactory epithelium
Rabbit M Intraperitoneal 0.3-1.7 mg/kg bw No haemolytic effects Mackell et al.
(white) (1951)
M, male
In Fischer 344/Crl rats exposed by inhalation to 7.7 mg/m3 of
benzo [a]pyrene dust for 2 h/day, five days per week for four weeks,
no respiratory tract lesions were observed, as measured by lung
lavage, clearance of tagged particles, and histopathological findings
(Wolff, R.K. et al., 1989).
7.2.1.5 Benz [a]anthracene
When benz [a]anthracene was given orally to rats daily for four days,
the NOAEL with respect to gastric, hepatic, and renal effects was 150
mg/kg bw per day. Carboxylesterase activity in the intestinal mucosa
was suppressed (Nousiainen et al., 1984).
7.2.1.6 Dibenz [a,h]anthracene
Adverse haemolymphatic changes, including the appearance of
extravascular erythrocytes in the lymph spaces and large pigmented
cells, were reported after subcutaneous injection of male rats with
0.28 mg per animal on five days per week for four weeks (Lasnitzki &
Woodhouse, 1944).
7.2.1.7 Fluoranthene
All of 10 mice that received 500 mg/kg bw per day fluoranthene
intraperitoneally for seven days survived (Gerarde, 1960).
7.2.1.8 Naphthalene
Anaemia was induced in three dogs by single oral doses of 3 or 9 g or
a total dose of 10.5 g per animal given over seven days. All three
animals showed neurophysiological symptoms and slight to very severe
changes in haematological parameters. Full recovery was observed
within 7-14 days (Zuelzer & Apt, 1949).
No immunosuppressive effects were observed in a number of test
systems. Tolerance to the effects of naphthalene was reported in mice
after intraperitoneal injection for seven days. A sharp contrast
between single and multiple doses was observed in the effects on the
morphology of the bronchiolar epithelium. When naphthalene was given
intraperitoneally at a dose of 50, 100, or 200 mg/kg bw per day as a
single injection, dose-dependent bronchiolar epithelial cell necrosis
was detected; however, when these doses were given daily for seven
days, no significant effects were observed. Addition of 300 mg/kg bw
on day 8 had no effect, whereas recovered sensitivity was observed
with increasing time between the last dose and the challenge dose. A
single dose of 300 mg/kg bw without pretreatment resulted in
substantial denudation of the bronchiolar epithelium. This pattern was
attributed to a reduction in metabolic activation of naphthalene due
to a decrease in cytochrome P450 mono-oxygenase activity after
multiple dosing. A rough correlation was observed in mouse lung (but
not liver microsomes) between induction of tolerance and decreased
metabolic formation of the 1 R, 2 S-epoxide enantiomer, which is
responsible for tissue-selective toxicity. Such toxicity was
demonstrated in mice both in vivo and in isolated perfused lung
(Buckpitt & Franklin, 1989).
These studies are summarized in Table 77.
7.2.1.9 Phenanthrene
Oral administration of 100 mg/kg bw per day phenanthrene to rats for
four days induced a 30% increase in carboxylesterase activity in the
intestinal mucosa (Nousiainen et al., 1984).
7.2.1.10 Pyrene
Four of five mice injected intraperitoneally with 500 mg/kg bw per day
pyrene for seven days survived (Gerarde, 1960).
7.2.2 Subchronic toxicity
7.2.2.1 Acenaphthene
Administration of 175 mg/kg bw per day acenaphthene to mice by gavage
for 90 days resulted in a NOAEL of 175 mg/kg bw per day and a LOAEL of
350 mg/kg bw per day for hepatotoxicity (US Environmental Protection
Agency, 1989a).
7.2.2.2 Anthracene
Four of five rats given 5 mg per animal anthracene subcutaneously for
four months survived (Gerarde, 1960).
Anthracene was administered to groups of 20 male and female CD-1 (ICR)
BR mice by gavage at a dose of 0, 250, 500, or 1000 mg/kg bw per day
for at least 90 days. No treatment-related effects were noted on
mortality, clinical signs, body weights, food consumption,
ophthalmological findings, the results of haematology and clinical
chemistry, organ weights, organ-to-body weight ratios, and gross
pathological and histopathological findings. The no-observed-effect
level (NOEL) was the highest dose tested, 1000 mg/kg bw per day (US
Environmental Protection Agency, 1989b).
7.2.2.3 Benzo [a]pyrene
Male Syrian golden hamsters were exposed by inhalation to 9.8 or
44.8 mg/m3 benzo [a]pyrene for 4.5 h/day, five days per week for 16
weeks. No neoplastic response was observed in the respiratory tract
(Thyssen et al., 1980).
The growth of rats was inhibited by feeding a diet enriched with
benzo [a]pyrene at 1.1 g/kg for more than 100 days (White & White,
1939).
Table 77. Subacute and subchronic effects of naphthalene
Species Sex Route of Dose (purity) Effects Reference
(strain) (no./sex administration
per group)
Mouse M,F Oral 27, 53, and 267 In all groups, slight alterations in haemato Shopp et al.
(CD-1) (40-112) mg/kg bw, 7 d/ logical parameters; humoral immune response (1984)
week, 14 d not affected. 27 and 53 mg/kg: no significant
effects; 267 mg/kg: 5-10% mortality (m/f);
significantly decreased terminal body weight
(m/f); 30% decrease in thymus weight (m);
significant decrease in weight of spleen (f);
increase in lung weight (f)
Mouse M,F Oral 5.3, 53, and 133 No obvious pulmonary effects or Shopp et al.
(CDO) mg/kg bw, 7 d/ immunotoxicity; significantly decreased (1984)
week, 90 d relative spleen weights (f); tolerance
Mouse M Intraperitoneal 50, 100, and 200 No significant alterations in lung morphology; Buckpitt & Franklin
(Swiss-Webster) mg/kg, 7 d tolerance to 300 mg/kg on day 8 (1989); O'Brien et
al. (1989)
Rat Diet 2 g/kg diet, Inhibition of growth; enlarged, fatty livers White & White
100 d (1939)
Dog (1) Oral 122 g/kg bw per Diarrhoea, weakness, lack of appetite, ataxia, Zuelzer & Apt
day, 7 d very severe anaemia; complete recovery (1949)
within 1-2 weeks
M, male; F, female
7.2.2.4 Fluorene
Groups of 25 male and 25 female CD-1 mice were given 0, 125, 250, or
500 mg/kg bw per day fluorene suspended in corn oil by gavage for 13
weeks. Increased salivation, hypoactivity, and abdomens wetted with
urine were observed in all treated males. The percentage of hypoactive
mice was dose-related. In mice exposed at 500 mg/kg bw per day,
laboured respiration, ptosis (drooping eyelids), and an unkempt
appearance were also observed. A significant decrease in erythrocyte
count and packed cell volume were observed in females treated with 250
mg/kg bw per day fluorene and in males and females treated with 500
mg/kg bw per day. The latter also showed a decreased haemoglobin
concentration and an increased total serum bilirubin level. A
dose-related increase in relative liver weight was observed in treated
mice, and a significant increase in absolute liver weight was observed
in the mice treated with 250 or 500 mg/kg bw per day. Significant
increases in absolute and relative spleen and kidney weights were
observed in males and females exposed to 500 mg/kg bw per day and in
males at 250 mg/kg bw per day. The increases in absolute and relative
liver and spleen weights in animals at the high dose were accompanied
by increases in the amounts of haemosiderin in the spleen and in
Kupffer cells of the liver. No other histopathological lesions were
observed. The LOAEL for haematological effects was 250 mg/kg bw per
day, and the NOAEL was 125 mg/kg bw per day (US Environmental
Protection Agency, 1989c).
In a similar study, fluorene at 35, 50, and 150 mg/kg bw increased the
weight of the liver by about 20% in a dose-dependent fashion and the
mitotic index of hepatocytes by sixfold after 48 h (Danz et al.,
1991).
7.2.2.5 Fluoranthene
Groups of 20 male and 20 female CD-1 mice were given 0, 125, 250, or
500 mg/kg bw per day fluoranthene by gavage for 13 weeks. A fifth
group of 30 male and 30 female mice was used to establish baseline
levels in blood. Body weight, food consumption, and haematological and
serum parameters were recorded regularly throughout the experiment. At
the end of 13 weeks, the animals were killed and autopsied; organs
were weighed and a histological evaluation was made. All treated mice
had dose-dependent nephropathy, increased salivation, and increased
liver enzyme activities, but these effects were either not
significant, not dose-related, or not considered adverse at 125 mg/kg
bw per day. Mice exposed to 500 mg/kg bw per day had increased food
consumption and increased body weight. Mice exposed to the two higher
doses had statistically increased alanine aminotransferase activity
and increased absolute and relative liver weights. Treatment-related
microscopic liver lesions (indicated by pigmentation) were observed in
65% of mice at 250 mg/kg bw per day and 88% of those at the highest
dose. On the basis of the increased alanine aminotransferase activity,
pathological effects in the kidney and liver, and clinical and
haematological changes, the LOAEL was 250 mg/kg bw per day and the
NOAEL 125 mg/kg bw per day (US Environmental Protection Agency, 1988).
7.2.2.6 Naphthalene
In a 90-day study in mice, naphthalene at oral doses up to 133 mg/kg
bw caused neither mortality nor serious changes in organ weights
(Shopp et al., 1984). These authors did not observe haemolytic anaemia
in CD-1 mice after oral uptake, although this effect had been seen in
human patients (Konar et al., 1939; Zuelzer & Apt, 1949; see Section
8). It was suggested that glucose-6-phosphate dehydrogenase deficiency
in erythrocytes, a prerequisite of haemolytic anaemia in adult humans,
was not present in the mice (Shopp et al., 1984).
In rats that ingested 150 mg/kg bw per day naphthalene for the first
three weeks and 200-220 mg/kg bw per day for a further 11 weeks,
reduced weight gain and food intake were observed. Later, the liver
was found to be enlarged, with cell oedema and congestion of the liver
parenchyma, and the kidneys showed signs of inflammation (Kawai,
1979).
The presence of 1 g/kg naphthalene in the feed of rats and rabbits for
46-60 days led to cataracts (US Environmental Protection Agency,
1984b; see also section 7.8).
Administration to rabbits of 0.1-1 mg/kg bw per day naphthalene by
subcutaneous injection for 123 days resulted in severe oedema and a
high degree of vacuolar and collicular degeneration in the brain;
necrosis of nerve cells also occurred. The author suggested that
hypoxaemia resulting from haemolytic anaemia was responsible for this
finding (Suja, 1967; cited by Kawai, 1979).
Subacute and subchronic studies with naphthalene are summarized in
Table 77.
7.2.2.7 Pyrene
The growth of rats was inhibited by feeding a diet enriched with
benzo [a]pyrene at 2 g/kg for more than 100 days. The livers were
enlarged and had a fatty appearance indicating hepatic injury (White &
White, 1939).
Groups of 20 male and 20 female CD-1 mice were given 0, 75, 125, or
250 mg/kg bw per day pyrene in corn oil by gavage for 13 weeks and
then examined for changes in body weight, food consumption, mortality,
clinical pathological manifestations in major organs and tissues, and
changes in haematology and serum chemistry. Nephropathy, characterized
by the presence of multiple foci of renal tubular regeneration, often
accompanied by interstitial lymphocytic infiltrates and/or foci of
interstitial fibrosis, was present in four male control mice, one at
the low dose, one at the medium dose, and nine the high dose. Similar
lesions were seen in two, three, seven, and 10 female mice,
respectively. The renal lesions in all groups were described as
minimal or mild. Relative and absolute kidney weights were reduced in
mice at the two higher doses. On the basis of nephropathy and
decreased kidney weights, the low dose (75 mg/kg bw per day) was
considered to be the NOAEL and 125 mg/kg bw per day the LOAEL (US
Environmental Protection Agency, 1989d).
7.3 Long-term toxicity
Almost all of the long-term studies reported were designed to assess
the carcinogenic potency of PAH and are therefore summarized in
section 7.7. Information about the non-carcinogenic effects, such as
growth inhibition, liver damage, and irritation, which occurred at
concentrations that also caused carcinogenic effects is presented
here. General effects, such as on mortality, body weight, and
pathological findings at sacrifice, were not considered useful.
7.3.1 Anthracene
A group of 28 BD I and BD III rats received anthracene in the diet
from the age of about 100 days, at a daily dose of 5-15 mg per rat.
The experiment was terminated when a total dose of 4.5 g per rat had
been achieved, on day 550. The rats were observed until they died;
some lived for more than 1000 days. No treatment-related effects on
lifespan or on gross or histological appearance of tissues were
observed; haematological parameters were not measured (Schmähl, 1955).
After weekly subcutaneous injections of anthracene at 0.25 mg per
animal for 40 weeks, mice showed deposition of iron in lymph glands
and a reduced number of lymphoid cells (Hoch-Ligeti, 1941).
7.3.2 Benz [a]anthracene
Weekly subcutaneous injection of 0.25 mg per mouse for 40 weeks
resulted in deposition of iron in lymph glands and a reduced number of
lymphoid cells (Hoch-Ligeti, 1941).
7.3.3 Dibenz [a,h]anthracene
Mice given weekly subcutaneous injections of 0.25 mg per animal for
40 weeks had pale, soft, enlarged livers with signs of fatty
degeneration. There was deposition of iron in lymph glands, and the
number of lymphoid cells was reduced (Hoch-Ligeti, 1941).
7.4 Dermal and ocular irritation and dermal sensitization
The adverse dermatological effects observed in animals after acute and
subchronic dermal exposure to PAH included destruction of sebaceous
glands, dermal ulceration, hyperplasia, hyperkeratosis, and
alterations in epidermal cell growth. Perylene, benzo [e]pyrene,
phenanthrene, pyrene, anthracene, naphthalene, acenaphthalene,
fluorene, and fluoranthene did not suppress the sebaceous gland index;
benz [a]anthracene, dibenz [a,h]anthracene, and benzo [a]pyrene
resulted in indices > 1 (Bock & Mund, 1958). In Swiss mice treated
daily for three days with solutions of benzo [a]pyrene in acetone, a
concentration of 0.1% destroyed less than half of the sebaceous
glands, whereas 0.2% destroyed more than 50% (Suntzeff et al., 1955).
7.4.1 Anthracene
Anthracene is a primary irritant, and its fumes can cause mild
irritation of the skin, eyes, mucous membranes, and respiratory tract.
At a concentration of 4.7 mg/m3, mild skin irritation was found in
50% of exposed mice (Montizaan et al., 1989). The median value for
dermal irritant activity (ID50) in the mouse ear was 6.6 × 10-4
mmol or 118 µg/ear; in comparison, the ID50 for benzo [a]pyrene was
5.6 × 10-5 mmol per ear (Brune et al., 1978). Anthracene increases
the sensitivity of skin to solar radiation (Gerarde, 1960). No contact
sensitivity to anthracene was observed (Old et al., 1963).
7.4.2 Benzo [a]pyrene
Four adult female guinea-pigs were injected with a total of 250 µg
benzo [a]pyrene in Freund's adjuvant, and two to three weeks later
were tested for contact sensitivity with solutions of 0.001, 0.01,
0.1, or 1% benzo [a]pyrene in acetone and olive oil. After 24 h, a
slight to severe (0.001-1%) contact hypersensitivity was observed (Old
et al., 1963).
C3H mice were given an epicutaneous administration of 100 µg
benzo [a]pyrene in 0.1% acetone solution into the abdominal skin.
Five days later, contact hypersensitivity was elicited by applying 20
µg benzo [a]pyrene to the dorsal aspect of the ear. The response was
quantified by ear thickness, which reaced a maximum three to five days
after challenge. The LOAEL for allergic contact sensitivity was thus
120 µg (Klemme et al., 1987).
The ID50 value for dermal irritant activity in the mouse ear was 5.6
× 10-5 mmol per ear (Brune et al., 1978).
7.4.3 Naphthalene
A single dose of 100 mg naphthalene to the rabbit eye was slightly
irritating, whereas application of 495 mg to rabbit skin, without
occlusion, caused mild irritation (Sax & Lewis, 1984).
7.4.4 Phenanthrene
No contact sensitization to phenanthrene was observed (Old et al.,
1963).
7.5 Reproductive effects, embryotoxicity, and teratogenicity
The mechanistic aspects of reproductive and embryotoxic effects are
presented in detail and the results summarized in Tables 78-80. The
genotype of mice is decisive for the manifestation of effects.
Studies have been reported on anthracene, benz [a]anthracene,
benzo [a]-pyrene, chrysene, dibenz [a,h]anthracene, and naphthalene.
Embryotoxicity was reported in response to benz [a]anthracene,
benzo [a]pyrene, dibenz [a,h]-anthracene, and naphthalene.
Benzo [a]pyrene also had adverse effects on female fertility,
reproduction, and postnatal development. In a study in young mice, an
NOEL of 150 mg/kg bw per day was obtained for benzo [a]pyrene on the
basis of effects on fertility (sperm in lumen of testes, size of
litters) and embryotoxicity (malformations) (Rigdon & Neal, 1965).
7.5.1 Benzo [a]pyrene
7.5.1.1 Teratogenicity in mice of different genotypes
Benzo [a]pyrene is embryotoxic to mice, and the effect is partly
dependent on the genetically determined induction of the cytochrome
P450 mono-oxygenase receptor, Ah, of the mother and fetus by PAH (see
also section 7.10). In the case of an inducible mother
(Ahb/ Ahb and Ahb/ Ahd, B groups), the genotype of the
fetus is not crucial because the active metabolites formed in the
mother appear to cross the placenta, causing fetal death or
malformation. In contrast, when the mother is non-inducible
(Ahd/ Ahd, D group), the genotype of the fetus is important;
one litter may contain both inducible and non-inducible fetuses.
Another decisive factor is the route by which benzo [a]pyrene is
given to the mother. Three studies of the genetic expression of
effects are summarized below.
Intraperitoneal injection of benzo [a]pyrene at 50 or 300 mg/kg bw on
day 7 or 10 of gestation was more toxic and teratogenic in utero in
genetically inducible C57Bl/6 (Ahb/ Ahb) than in non-inducible
AKR inbred mice (Ahd/ Ahd). In AKR × (C57Bl/6)(AKR)F1 and
(C57Bl/6)(AKR)F1 × AKR back-crosses (father × F1 mother), allelic
differences at the Ah locus in the fetus correlated with
dysmorphogenesis. The inducible fetal Ahb/ Ahd genotype results
in more stillborn and resorbed fetuses,decreased fetal weight,
increased frequency of congenital anomalies, and enhanced
P1-450-mediated covalent binding of benzo [a]pyrene metabolites to
fetal protein and DNA, when compared with fetuses of the non-inducible
Ahd/ Ahd genotype (not-inducible) from the same uterus (see
Table 78). In the case of an inducible mother (Ahb/ Ahd),
however, these parameters do not differ in Ahb/ Ahd and
Ahd/ Ahd individuals in the same uterus, presumably because the
increased benzo [a]pyrene metabolism in maternal tissues and placenta
cancels them out (Shum et al., 1979).
An inducible genotype is not the only factor involved in the
reproductive toxicity of benzo [a]pyrene. In a study in which C57Bl/6
female mice (Ah inducible) were mated with C57Bl/6, DBA/2, or BDF1
male mice (B groups), and DBA/2 females (non-inducible) were mated
with C57Bl/6, DBA/2, or BDF1 males (D groups) and received
intraperitoneal injections of benzo [a]-pyrene, fetal mortality
increased dose-dependently in all groups except the DBA/2 × DBA/2.
Fetal body weight was reduced dose-dependently in all experimental
groups, but the effect was more pronounced in D than B groups, as was
a dose-dependent increase in the frequency of cervical ribs (for
experimental details, see Table 78). These results suggest that
Table 78. Embryotoxicity of polycyclic aromatic hydrocarbons in experimental animals
Species No. per Route of Duration, dose Effects Reference
(strain) group administration
Anthracene
Rat Gavage Day 19 of gestation, F1: no induction of BaP hydroxylase in liver Welch et al.
Sprague- 60 mg/kg compared with control (< 0.2 vs <0.2 units in (1972)
bw controls)
Benz[a]anthracene
Rat 2 Subcutaneous Day 1-11 or 1-15 F0: Day 10 and 12: severe vaginal haemorrhage; Wolfe &
of gestation, 5 mg/ Day 14: intraplacental haemorrhage Bryan (1939)
animal per day F1: fetal death and resorption up to day 18
Rat Gavage Day 19 of gestation, F1: induction of BaP hydroxylase in liver Welch et al.
Sprague-Dawley 60 mg/kg bw (12 vs < 0.2 units in controls) (1972)
Benzo[a]pyrene
Mouse 9 Diet Day 5 or 10 of F1: no malformations Rigdon &
White gestation until Neal (1965)
Swiss delivery, 50 mg/ky bw
Mouse 6-17 Diet Day 2-10 of F1: increased intrauterine toxicity and Legraverend
C57BI/6N, gestation, 120 mg/ malformations in Ahd/Ah7dembryos compared et al. (1984)
AKR/J, and kg per day with Ahb/Ahd embryos in pregnant Ahd/Ahd
back-crosses mice (effect not seen in pregnant Ahb/Ahd mice)
(reciprocal)
Mouse 5-30 Intraperitoneal Day 7, 10, or 12 of 200 mg/kg bw: F1: increase in stillbirths, Shum et al.
C57BI/6, gestation, 50-300 resorptions, malformations (4-fold higher (1979)
AKR and mg/kg bw in pregnant C57BI than in AKR mice)
back-crosses
(reciprocal)
Table 78. (continued)
Species No. per Route of Duration, dose Effects Reference
(strain) group administration
Mouse 20 Intraperitoneal Day 8 of gestation, 150 and 300 mg/kg bw: F0: increased fetal Hoshino et al.
C57BI/6, 150 or 300 mg/kg mortality (except DBA/2 × DBA/2 offspring); (1981)
DBA/2, and reduced fetal body weight; increased number of
back-crosses cervical ribs
(reciprocal) 300 mg/kg: F1: increased malformations
(C57BI/6 × C57BI/6)
Mouse Gavage Day 7-16 of F0: no toxicity MacKenzie &
CIT1 gestation, 10, 40, 160 F1: no toxicity Angevine (1981)
mg/kg bw per day
Rat 17 Subcutaneous Day 1-11 or 16 of F0: Days 10 and 12: profuse vaginal Wolfe & Bryan
gestation, 5 mg/ haemorrhage; day 14: intraplacental (1939)
animal per day haemorrhage; F1: fetal death and resorption
up to day 18
Rat Gavage Day 19 of gestation, F1: induction of BaP-hydroxylase in liver Welch al al.
Sprague-Dawley 60 mg/kg bw (20 vs < 0.2 units in controls) (1972)
Rat 10-15 Subcutaneous Day 6-8 or 6-11 of F1: significant increase in number of resorptions Bui et al.
Sprague-Dawley gestation, 50 mg/kg and fetal wastage (dead fetuses plus resorption); (1986)
bw per day fetal weight reduced
Chrysene
Rat Gavage Day 19 of gestation, F1: induction of BaP hydroxylase in liver Welch et al.
Sprague-Dawley 60 mg/kg bw (6 vs < 0.2 units in controls) (1972)
Dibenzo[a,h]anthracene
Rat Gavage Day 19 of gestation, F1: induction of BaP hydroxylase in liver Welch et al.
Sprague-Dawley 60 mg/kg bw (15 vs < 0.2 units in controls) (1972)
Table 78. (continued)
Species No. per Route of Duration, dose Effects Reference
(strain) group administration
Rat 38 Subcutaneous Day 1-8 or 1-18 of F0: Days 10 and 12: profuse vaginal haemorrhage; Wolfe &
gestation, 5 mg/ day 14: intraplacental haemorrhage Bryan (1939)
animal per day F1: fetal death and resorption up to day 18
Naphthalene
Mouse 50 Gavage Day 7-14 of F0: significant 15% increase in mortality; Plasterer et
CD-1 gestation, 300 mg/ significant reduction in weight gain al. (1985)
kg bw per day F1: significant reduction in number of live
offspring; no malformations
Mouse Gavage Day 6-13 of F0 increased mortality 10/50; control: 0/50); Hardin et al.
CD-1 gestation, 300 mg/ significant reduction in weight gain (1987)
kg bw per day F1: significant reduction in liveborns per litter
Rat 10-15 Intraperitoneal Day 1-15 of F0: no toxicity Hardin et al.
Sprague-Dawley gestation, 395 mg/ F1: no toxicity (1981)
kg per day
For genotypes of the mouse strains used see section 7.5.1.1
Ah-inducible fetuses are more sensitive to lethal events, whereas
those of non-inducible dams are more susceptible to a decrease in body
weight and an increased incidence of cervical ribs. The incidence of
external malformations may, however, differ in mice of different
genotypes after treatment with benzo [a]-pyrene, even if both dams
and fetuses are inducible (Hoshino et al., 1981).
The toxicity of benzo [a]pyrene in utero was investigated in
pregnant Ahd/ Ahd × Ahb/ Ahd F1 and Ahb/ Ahd ×
Ahd/ Ahd F1 back-crossed mice fed benzo [a]pyrene in the
diet at 120 mg/kg daily on days 2-10 of gestation. Embryos of D
females (Ahd/ Ahd genotype; non-inducible) showed more signs
of toxicity and malforma-tions than Ahd/ Ahd embryos. Fetuses
of B females (Ahb/ Ahd genotype) did not show these changes.
The authors suggested that reduced benzo [a]pyrene metabolism in the
intestine had caused high concentrations in the embryos, and more
toxic metabolites (benzo [a]pyrene-1,6- and -3,6-quinones) were
detected in the Ahd/ Ahd embryos than in Ahb/ Ahd
embryos (Legraverend et al., 1984). These results were in contrast to
those reported after intraperitoneal injection by Shum et al. (1979)
and Hoshino et al. (1981). The route of administration can thus affect
the magnitude of the observed effects (see also section 7.8.2.2).
7.5.1.2 Reproductive toxicity
A single intraperitoneal injection of benzo [a]pyrene reduced
fertility and destroyed primordial oocytes of DBA/2N mice in a
dose-dependent manner (Mattison et al., 1980; see also Table 79).
In experiments with B6 (Ah-inducible) and D2 (non-inducible) mice,
primordial oocytes of B6 mice underwent more rapid destruction after
treatment with benzo [a]pyrene than those of D2 mice. This effect
corresponded to a two- to threefold increase in ovarian
arylhydrocarbon hydroxylase (AHH) activity in B6 mice after treatment.
This correlation was not found in analogous experiments with D2B6F1
mice, in which AHH activity was increased by two- to threefold, but
the oocyte destruction was similar to that observed in D2 mice. This
demonstrates an inconsistent consequence of strain differences in
genotype (Mattison & Nightingale, 1980; see also Table 79). The sum of
activation, detoxification, and repair seems to be decisive for the
process of oocyte destruction (Figure 8).
Benzo [a]pyrene and its three metabolites, benzo [a]pyrene
7,8-oxide, benzo [a]pyrene 7,8-diol, and benzo [a]pyrene diol
epoxide, were administered by injection at a single dose of 10 µg into
the right ovary of B6, D2, and D2B6F1 mice. Ovarian volume, weight,
and follicle numbers were measured after two weeks; various reductions
were observed in all strains. There was also compesatory hypertrophy
of the left ovary (Mattison et al., 1989; see also Table 79).
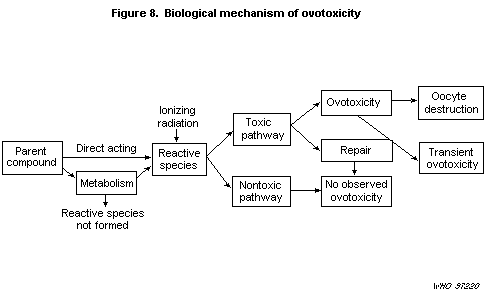 Table 79. Effects of benzo[a]pyrene on fertility in experimental animals
Species Sex/No. Route of Duration, dose Effects Reference
(strain) per administration
group
Mouse M Diet Up to 30 days before mating, NOEL: 150 mg/kg bw per day Rigdon & Neal (1965)
White 5 37.5, 75, or 150 mg/kg bw Parameters: sperm in lumen
per day of testes; number of offspring
Mouse F Diet 20 days before mating NOEL: 150 mg/kg bw per day Rigdon & Neal (1965)
White 5-65 37.5, 75, or 150 mg/kg bw Parameter: number of offspring
Swiss per day
Mouse F Intraperitoneal Day 14 before mating, 10, 100 mg/kg bw: dose-dependent Mattison et al. (1980)
DBA/2N 15 10, 100, 200, or 500 mg/kg decrease in number of pups
bw once 200, 500 mg/kg bw: completely
infertile; threshold: 3.4 mg/kg bw;
50% effect dose: 25.5 mg/kg bw
Mouse F Intraperitoneal Day 21 before sacrifice, Dose-dependent increase in Mattison et al. (1980)
DBX2N 5, 10, 50, 100, or 500 mg/kg primordial oocyte destruction;
bw once 500 mg/kg: 100% destruction;
threshold: 2.7 mg/kg bw; 50%
effect dose: 24.5 mg/kg bw
Mouse F Intraperitoneal Day 13 before sacrifice, 100 mg/kg bw: significant increase Mattison & Nightingale
B6 and D2 5 100 mg/kg bw once in primordial oocyte destruction in (1980)
both genotypes; effects in B6 mice
greater than in D2 mice
Mouse F Intra-ovarian Day 14 before sacrifice, 10 µg: decreased ovarian weight Mattison et al. (1989)
C57BI/6N (136), injection 10 µg/right ovary once (D2); decreased ovarian volume (D2
DBA/2N (D2), and F1); decreased antral follicles
D2B6F1(F1) (F1) decreased number of small
follicles (D2 and F1)
Table 79. (continued)
Species Sex/No. Route of Duration, dose Effects Reference
(strain) per administration
group
Mouse F Intraperitoneal 1, 2, 3, and 4 weeks 500 mg/kg: 35% mortality Swartz & Mattison,
C57BI/6N 5 before sacrifice; 1, 5, 1-500 mg/kg bw: dose- and time- 1985);
10, 50, 100, or 500 dependent decrease in ovarian Miller et al. (1992)
mg/kg bw volume, total volume and number of
corpora lutea/ovary (for last
parameter, after 1 week threshold
was about 1 mg/kq bw and ED50 1.6
mg/kg bw);effect transitory in
low-dose groups, butnot reversible
in two highest by four weeks
For genotypes of the mouse strains used see section 7.5.1.1
7.5.1.3 Effects on postnatal development
Three studies of the postnatal effects of benzo [a]pyrene on mouse
offspring, with administration dermally, intraperitoneally, or orally,
showed adverse effects, including an increased incidence of tumours,
immunological suppression, and reduced fertility (see also Table 80).
7.5.1.4 Immunological effects on pregnant rats and mice
Benzo [a]pyrene given to pregnant rats on day 15 or 19 of gestation
caused alterations at the thymic glucocorticoid receptors in the
offspring, suggesting binding to the pre-encoded hormone receptors and
interference with receptor maturation (Csaba et al., 1991; Csaba &
Inczefi-Gonda, 1992; see also section 7.8.2.6).
Strong suppression of immunological parameters was found in the
progeny of mice that had been treated intraperitoneally with
benzo [a]pyrene at mid-gestation (Urso & Johnson, 1987; see also
section 7.8.2.6).
7.5.2 Naphthalene
7.5.2.1 Embryotoxicity
Naphthalene was administered by gavage at 50, 150, or 450 mg/kg bw per
day to pregnant Sprague-Dawley rats on days 6-15 of gestation, i.e.
during the main period of organogenesis. The dams showed signs of
toxicity including lethargy, slow breathing, prone body posture, and
rooting, and these effects persisted after the end of dosing with the
high dose. The body-weight gain of treated animals was reduced by 31
and 53% in the groups at the two higher doses. Naphthalene did not
induce fetotoxic or teratogenic effects, and the numbers of corpora
lutea per dam, implantation sites per litter, and live fetuses per
litter were within the range in controls. The maternal NOAEL was
< 50 mg/kg bw per day (National Toxicology Program, 1991).
In a second study, doses of 0, 20, 80, or 120 mg/kg bw per day were
given to rabbits by gavage during days 6-19 of gestation. There were
no signs of maternal toxicity, fetotoxicity, or developmental toxicity
(National Toxicology Program, 1992a).
7.5.2.2 Toxicity in cultured embryos
Mice injected intraperitoneally on day 2 of gestation with 14 or 56
mg/kg bw naphthalene were sacrificed 36 h later, and embryos were
cultured in vitro. Maternal doses below the LD50 value inhibited
the viability and implantation capacity of the embryos, and attachment
and embryonic growth in vitro were markedly decreased (Iyer et al.,
1990).
Table 80. Effects of benzo[a]pyrene on postnatal development in experimental animals
Species Sex/No. Route of Duration, dose Effects Reference
(strain) per administration
group
Mouse F Dermal Entire gestation period F1- F4: sensitization of offspring: increased Andrianova
non-inbred 1 drop of 0.5% solution, incidence of papillomas and carcinomas (1971)
twice per week; F1-F4 in all generations compared with animals
treated with BaP, m not treated in utero
1x/week, f 2x/week
Mouse F Intraperitoneal Day 11-13 or 16-18 F1: no difference in birth rate, litter size of Urso &
C3H/Anf 25 of gestation, 100 or progeny compared to controls; severe suppression Gengozian
150 mg/kg of anti-SRBC PFC response up to 78 weeks of life (1980)
(see also section 7.8.2.6); 11-1 fold increase in
tumour incidence (liver, lung, ovaries) after
56-78 weeks
Mouse F Gavage Days 7-16 of F1: 10 mg/kg markedly impaired fertility (by 20%) MacKenzie &
CD-1 gestation, 10, 40, and reduced testis weight (by 40%), 34% sterility Angevine
160 mg/kg bw per day of females; 40 and 160 mg/kg: fertility impaired (1981)
by > 900%/100%; testis weight reduced by > 800%;
100%/100% sterility of females
anti-SRBC PFC, anti-sheep red blood cell antibody (plaque)-forming cells
In a subsequent study, three-day-old whole mouse embryos were
collected at the blastocyst stage, cultured in NCTC 109 medium, and
exposed to naphthalene at 0.16, 0.2, 0.39, or 0.78 mmol/litre for 1 h
with and without S9. They were then transferred to toxicant-free
medium, cultured for 72 h, and evaluated microscopically. Naphthalene
was not directly embryotoxic, but growth and viability were decreased
in the presence of S9, with 100% embryolethality at doses > 0.2
mmol/litre; furthermore, hatching and attachment rates were
significantly decreased. The approximate LC50 in S9-supplemented
media was 0.18 mmol/litre (Iyer et al., 1991).
7.6 Mutagenicity and related end-points
Benzo [a]pyrene has been used extensively as a positive control in a
variety of short-term tests. It is active in assays for the following
end-points: bacterial DNA repair, bacteriophage induction, and
bacterial mutation; mutation in Drosophila melanogaster; DNA
binding, DNA repair, sister chromatid exchange, chromosomal
aberration, point mutation, and transformation in mammalian cells in
culture; and tests in mammals in vivo, including DNA binding, sister
chromatid exchange, chromosomal aberration, sperm abnormalities, and
somatic mutation at specific loci (Hollstein et al., 1979; De Serres &
Ashby, 1981). Positive effects were seen in most assays for the
mutagenicity of benzo [a]pyrene.
A selection of these studies is summarized in Tables 81-88. All of the
data available on the other PAH considered in this monograph were
taken into account. Because of the amount of data, the purities of the
chemicals tested and details of the assay conditions are omitted from
the tables, but they do show the results obtained when S9 was used.
Variations in the S9 metabolic activation component of the assay
system, e.g. the age, sex, and strain of the rats used as a source of
liver and any pretreatment with enzyme inducers such as Aroclor,
3-methylcholanthrene, or phenobarbital, may markedly affect the
results and may account for apparent discrepancies.
DNA binding of benzo [a]pyrene was observed in various species. For
example, adducts were found in cells from hamsters, mice (Arce et al.,
1987), rats (Moore et al., 1982), and chickens (Liotti et al., 1988),
in calf thymus DNA (Cavalieri et al., 1988a), and in human cell
systems (Moore et al., 1982; Harris et al., 1984). Formation of DNA
adducts was inhibited in the presence of scavengers of active oxygen
species like superoxide dismutase, catalase, and citrate-chelated
ferric iron, indicating that reactive oxygen species such as
superoxide, OH radicals, and singlet oxygen may be involved in DNA
binding (Bryla & Weyand, 1991). Benzo [a]pyrene at a total dose of 10
mg/kg bw induced gene mutations in mice, as seen in the coat-colour
spot test (Davidson & Dawson, 1976).
The results of tests for reverse mutation in Salmonella
typhimurimum (Ames test) and for forward mutation in
S. typhimurimum strain TM677 are presented in Table 81. Bacterial
tests for DNA damage in vitro are shown in Table 82. The results of
tests for mutagenicity in yeasts and Drosophila melanogaster,
including host-mediated assays, are shown in Table 83. The results of
various assays carried out on mammalian cells in vitro are
summarized in Tables 82-86. The results of tests in vivo are shown
in Tables 87 and 88.
The activity of PAH in short-term tests is summarized in Table 89,
which gives the evaluations of IARC (1983; see also Section 12) and
the results of studies reported after 1983. Only three of the 33 PAH
considered, i.e. anthracene, fluorene, and naphthalene were inactive
in all short-term tests; 16 had mutagenic effects. Eight PAH showed a
tendency for mutagenic activity, but the data are still too sparse to
permit a final judgement. The available information on acenaphthene,
acenaphthylene, benzo [a]fluorene, and coronene is still inadequate.
As phenanthrene and pyrene showed inconsistent results in various
experiments, they could not be clearly classified as mutagenic.
7.7 Carcinogenicity
Most of the studies that have been conducted on PAH were designed to
assess their carcinogenicity. Studies on various environmentally
relevant matrices such as coal combustion effluents, vehicle exhaust,
used motor lubricating oil, and sidestream tobacco smoke showed that
PAH are the agents predominantly responsible for their carcinogenic
potential (Grimmer et al., 1991b). Because of the abundance of
literature, only studies involving the administration of single PAH
have been taken into account in this monograph.
Benzo [a]pyrene has been tested in a range of species, including
frogs, toads, newts, trout, pigeons, rats, guinea-pigs, rabbits,
ferrets, ground squirrels, tree shrews, marmots, marmosets, and rhesus
monkeys. Tumours have been observed in all experiments with small
animals, and the failure to induce neoplastic responses in large
animals has been attributed to lack of information on the appropriate
route or dose and the inability to observe the animals for a
sufficient time (Osborne & Crosby, 1987a). In studies with other PAH,
benzo [a]pyrene was often used as a positive control and therefore
administered at only one concentration. Benzo [a]pyrene has been
shown to be carcinogenic when given by a variety of routes, including
diet, gavage, inhalation, intratracheal instillation, intraperitoneal,
intravenous, subcutaneous, and intrapulmonary injection, dermal
application, and transplacental administration.
Assessment of the carcinogenic potency of the selected PAH is
restricted for various reasons: Many of the studies performed before
about 1970 were carried out without controls, without clearly defined,
purified test substances, or using experimental designs and facilities
considered today to be inadequate. Despite these shortcomings, all of
the available studies were taken into account, except for those on
dibenz [a,h]anthracene and benzo [a]pyrene. An overview of the
results, as reported by the authors, is given in Table 90. To
facilitate appraisal of the studies, the penultimate column gives a
classification of the substances as positive, negative, or
questionably carcinogenic; indicates whether the tumour incidence was
evaluated statistically; and judges that a study is valid or provides
reasons suggesting that it is unreliable. The criteria used to
classify a study as valid were (i) an appropriate study protocol, i.e.
use of concurrent controls (sham or vehicle), 20 or more animals per
group, and study duration at least six months; and (ii) sufficient
documentation, including detailed description of administration,
results, and the survival of animals. As the use of concurrent
controls is important for making judgements, data for these are given
with the results for treated groups. If control data are not
mentioned, it is because they were not given in the original paper.
In experiments by topical application, the lower, more volatile PAH
partially evaporate, and therefore their doses may have varied. The
substances may also decompose. Both features could lead to
underestimations of carcinogenic potency if they are not taken into
account.
Table 91 shows the classification of the compounds as carcinogenic,
noncarcinogenic, or questionably carcinogenic. In order to make these
classifications, all of the studies were summarized according to
species and route of administration. In cases of doubt, the judgement
was based on valid studies only. For example, despite one positive but
invalid result and two questionable (one valid, one invalid) results
from 17 studies, anthracene was classified as negative; however,
pyrene, for which one positive, valid result and three questionable,
valid results were found in 15 studies, could not be classified as
negative and the compromise 'questionable' was chosen.
The PAH found not to be carcinogenic were anthracene,
benzo [ghi]perylene, fluorene, benzo [ghi]fluoranthene,
1-methylphenanthrene, perylene, and triphenylene. Questionable results
were obtained for acenaphthene, benzo [a]-fluorene,
benzo [b]fluorene, coronene, naphthalene, phenanthrene, and pyrene.
The remaining compounds were found to be carcinogenic.
The dermal route was the commonest mode of administration, followed by
subcutaneous and intramuscular injection. In most studies, the site of
tumour development is related closely to the route of administration,
i.e. dermal application induces skin tumours, inhalation and
intratracheal instillation result in lung tumours, subcutaneous
injection results in sarcomas, and oral administration induces gastric
tumours. Tumour induction is, however, not restricted to the obvious
sites. For example, lung tumours have been observed after oral
administration or subcutaneous injection of benzo [a]pyrene to mice
and liver tumours following intraperitoneal injection. In two studies,
lung tumours were found in mice after intravenous injection of
benzo [a]pyrene and dibenz [a,h]anthracene. Thus, tissues such as
the skin must be able to metabolize PAH to their ultimate metabolites
and itself become a target organ; however, all PAH that reach the
liver via the bloodstream can be metabolized there. The liver in turn
is a depot from which the metabolites are distributed all over the
body (Wall et al., 1991). The carcinogenic potency of the PAH differs
by three orders of magnitude, and several authors have presented
tables of toxic equivalence factors based on experimental results in
order to quantify these differences. Carcinogenic potency cannot be
based only on chemical structure but requires theoretical
considerations and calculations (see section 7.10).
Although this monograph primarily addresses single PAH, it was
considered necessary for risk assessment to present some information
on mixtures of PAH, to which humans are almost always exposed,
predominantly adsorbed onto inhalable particles.
Although the essential results of the studies of carcinogenicity are
summarized in Table 90, special aspects and comparisons of individual
PAH are presented in more detail below.
7.7.1 Single substances
7.7.1.1 Benzo [a]pyrene
Oral administration of benzo [a]pyrene to male and female CFW mice
induced gastric papillomas and squamous-cell carcinomas and increased
the incidence of pulmonary adenomas (Rigdon & Neal, 1966). In other
studies in which mice of the same strain were fed benzo [a]pyrene,
pulmonary adenomas, thymomas, lymphomas, and leukaemia occurred,
indicating that it can cause carcinomas distal to the point of
application (Rigdon & Neal, 1969). The incidence of gastric tumours
was 70% or more in mice fed 50-250 ppm benzo [a]pyrene for four to
six months. No tumours were observed in controls (Rigdon & Neal, 1966;
Neal & Rigdon, 1867; see also Table 90).
In a study of the effects of benzo [a]pyrene given in the diet or by
gavage in conjunction with caffeine, groups of 32 Sprague-Dawley rats
of each sex were fed diets containing 0.15 mg/kg bw benzo [a]pyrene
either five times per week or only on every ninth day. Tumours were
observed in the forestomach, oesophagus, and larynx, at combined
tumour incidences of 3/64, 3/64, and 10/64 in the controls and those
at the low and high doses, respectively. In the study by gavage,
groups of 32 rats of each sex were treated with benzo [a]pyrene at
0.15 mg/kg bw in a 1.5% caffeine solution every ninth day, every third
day, or five times per week. The combined incidences of tumours of the
forestomach, oesophagus, and larynx were 3/64 in controls, 6/64 in
rats at the low dose, 13/64 in those at the medium dose, and 14/64
among those at the high dose (Brune et al., 1981).
In hamsters exposed to 9.5 or 46.5 mg/m3 benzo [a]pyrene by
inhalation for 109 weeks, a dose-response relationship was seen with
tumorigenesis in the nasal cavity, pharynx, larynx, and trachea. The
fact that lung tumours were not detected could not be explained
(Thyssen et al., 1981). Hamster lung tissue can activate
benzo [a]pyrene to carcinogenic derivatives (Dahl et al., 1985).
Table 81. Mutagenicity of polycyclic aromatic hydrocarbons to
Salmonella typhimurium
Compound Result with Reference
Strain metabolic
activation
Acenaphthene
TA98,TA100 - Florin et al. (1980)
TM677 + Kaden et al. (1979)
TA98,TA100 + Epler et al. (1979)
TA100 - Pahlman & Pelkonen
(1987)
Acenaphthylene
TA98,TA100 - Florin et al. (1980)
TM677 + Kaden et al. (1979)
TA98,TA100 - Bos et al. (1988)
Anthanthrene
TA98 + Hermann (1981)
TA100 + LaVoie et al. (1979);
Andrews et al. (1978)
TA98 - Tokiwa et al. (1977)
TM677 + Kaden et al. (1979)
Anthracene
TA98,TA100 - Purchase et al. (1976)
TA98,TA100 - Epler et al. (1979)
TA100 - LaVoie et al. (1979);
Gelboin & Ts'o (1978)
TA98, TA100, - McCann et al. (1975a);
TA1535,TA1537, Salamone et al. (1979);
TA1538 Ho et al. (1981);
Purchase et al.(1976)
TA98,TA100 - Bridges et al, (1981)
TA98,TA100, - Simmon (1979)
TA1535, TA1536,
TA1537,TA1538
TM677 - Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA98,TA100 - Probst et al. (1981)
TA100 + Carver et al. (1986)
TA98,TA100 - LaVoie et al.(1983a(1985)
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TA100 - Pahlman & Pelkonen
(1987)
TA98,TA100 - Bos et al. (1988)
TA98,TA100 - Florin et al. (1980)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benz[a]anthracene
TA100 + Epler et al. (1979);
Bartsch et al. (1980)
TA98,TA100 + McCann et al. (1975a);
Coombs et al. (1976);
Simmon (1979); Salamone
et al. (1979)
TA1535,TA1538 Rosenkranz & Poirier
(1979)
TA100 + Pahlman & Pelkonen
(1987)
TA98,TA100 + Hermann (1981); Carver
et al.(1986)
TA100 + Bartsch et al. (1980)
TM677 + Kaden et al. (1979)
TA100 + Baker et al. (1980)
TA98,TA100 + Bos et al. (1988)
TA98,TA100, + Probst et al. (1981)
TA1535,TA1537
TA98, TA100,
TA1537, TA1538 ± Dunkel et al. (1984)
TA1535 - Dunkel et al. (1984)
TA98,TA100 + Florin et al. (1980)
TA1537,TA1538 - Teranishi et al. (1975)
TA98 + Tokiwa et al. (1977)
Benzo[b]fluoranthene
TA98 + Hermann (1981)
TA100 + LaVoie et al. (1979);
Hecht et al. (1980)
TA100 + Amin et al, (1985a)
TA98,TA100 - Mossanda et al. (1979)
Benzo[j]fluoranthene
TA100 + LaVoie et al. (1980);
Hecht et al. (1980)
TM677 + Kaden et al. (1979)
Benzo[k]fluoranthene
TA100 + LaVoie et al. (1980);
Hecht et al. (1980)
TA98 + Hermann et al. (1980)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benzo[ghi]fluoranthene
TA98 ± Karcher et al. (1984)
TA100 + Karcher et al. (1984)
TA98,TA100 + LaVoie et al. (1979)
Benzo[a]fluorene
TA98, TA100, Salamone et al. (1979)
TA1535, TA1537,
TA1538
TA100 + Epler et al. (1979)
TA100 - LaVoie et al. (1980)
TA98,TA100 - Bos et al. (1988)
TA98 + Tokiwa et al. (1977)
Benzo[b]fluorene
TA98, TA100 - LaVoie et al. (1980)
TA98, TA100, - Salamone et al. (1979)
TA1535, TA1537,
TA1538
TM677 + Kaden et al. (1979)
TA98,TA100 + Bos et al. (1988)
Benzo[ghi]perylene
TA98, TA1538 + Mossanda et al. (1979);
Tokiwa et al. (1977);
Katz et al. (1981)
TA100 + Andrews et al. (1978);
Katz et al. (1981);
LaVoie et al. (1979);
Salamone et al. (1979)
TA1537,TA1538 + Poncelet et al. (1978)
TM677 + Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA100 + Carver et al. (1986)
Benzo[c]phenanthrene
TA98 + Salamone et al. (1979);
Wood et al. (1980)
TA100 + Carver et al. (1986)
TA100 + Wood et al. (1980)
TA98,TA100 + Bos et al. (1988)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benzo[a]pyrene
TA98 + Epler et al. (1979)
TA100 + Andrews et al. (1978)
TA98,TA100 + LaVoie et al. (1979)
TA98,TA100, + McCann et al. 1975a,b)
TA1537,TA1538
TM677 + Kaden et al. (1979)
TM677 + Rastetter et al. (1982)
TM677 + Babson et al. (1 986b)
TA97,TA98, + Sakai et al. (1985)
TA100
TA98,TA100 + Prasanna et al. (1987));
Simmon (1979));
Glatt et al. (1987)
TA1535,TA1538 + Rosenkranz & Poirier
(1979)
TA100 + Norpoth et al. (1984));
Alzieu et al. (1987)); Carver
et al. (1986)); Bos et al.
(1988); Hermann (1981);
Bruce & Heddle (1979);
Marino (1987); Alfheim &
Ramdahl (1984)
TA98 + Lee & Lin (1988)
TA100 + Pahlman & Pelkonen
(1987)
TA97,TA98,TA100 + Marino (1987)
TA97,TA98,TA100 + Sakai et al. (1985)
TA98,TA100 + Devanesan et al. (1990)
TM677 + Skopek & Thilly (1983)
TA98,TA100, + Dunkel et al. (1984)
TA1535, TA1537,
TA1538
TA98, TA100 + Lofroth et al. (1984)
TA98,TA100 + Florin et al. (1980)
TA98 + Tokiwa et al. (1977)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benzo[e]pyrene
TA98 + LaVoie et al. (1979);
Hermann (1981)
TA100 ± Salamone et al. (1979)
TA100 + Andrews et al. (1978);
LaVoie et al., 1979)
TA100 ± McCann et al. (1975a)
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TM677 + Kaden et al. (1979)
TA100 - Epler et al. (1979)
TA98,TA100, + Simmon (1979)
TA1538
TA97, TA100 + Sakai et al. (1985)
TA98, TA100, ± Dunkel et al. (1984)
TA1535,TA1537,
TA1538
TA100 + Carver et al. (1986)
TA100 - Pahlman & Pelkonen
(1987)
TA1537,TA1538 - Teranishi et al. (1975)
TA98 + Tokiwa et al. (1977)
Chrysene
TA100 + McCann et al. (1975a);
LaVoie et al. (1979)
TA98 + McCann et al. (1975a)
TA100 + Wood et al. (1977)
TA100 + Epler et al. (1979);
LaVoie et al. (1979)
TA100 + Salamone et al. (1979)
TA1535,TA1536, - Simmon (1979)
TA1537,TA1538
TA98,TA100 + Bhatia et al. (1987)
TM677 + Kaden et al. (1979)
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TA97,TA100 + Sakai et al (1985)
TA98,TA100 + Bos et al. (1988)
TA98 + Hermann (1981)
TA100 + Carver et al. (1986)
TA100 + Pahlman & Pelkonen
(1987)
TA100 + Florin et al. (1980)
TA98 + Tokiwa et al. (1977)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Coronene
TA98 + Mossanda et al. (1979)
TA98 + Hermann (1981)
TA98 ± Salamone et al. (1979)
TA98 + Florin et al. (1980)
TA98, TA1537, + Poncelet et al. (1978)
TA1538
TA97 ± Sakai et al. (1985)
TM677 - Kaden et al. (1979)
Cyclopenta[cd]pyrene
TA98 + Wood et al. (1980)
TA98,TA100, + Eisenstadt & Gold (1978)
TA1537,TA1538
TM677 + Kaden et al. (1979);
Cavalieri et al. (1981a)
TA98 + Reed et al. (1988)
Dibenz[a,h]anthracene
TA100 + Andrews et al. (1978);
Epler et al. (1979);
McCann et al. (1975a,b)
TA100 + Salamone et al. (1979)
TA98 + Baker et al. (1980)
TA98 + Hermann (1981)
TM677 + Kaden et al. (1979)
TA100 + Wood et al. (1978)
TA100 + Pahlman & Pelkonen
(1987); Carver et al.,
1986)
TA98, TA100, + Probst et al. (1981)
TA1537,TA1538
TA100 + Platt et al. (1990)
TA100 + Lecoq et al. (1989)
TA1537,TA1538 - Teranishi et al. (1975)
Dibenzo[a,e]pyrene
TA100 + LaVoie et al. (1979)
TA1537,TA1538 + Teranishi et al. (1975)
TA98,TA100 +.± Devanesan et al. (1990)
Dibenzo[a,h]pyrene
TA100 ± LaVoie et al. (1979)
TA98,TA100 + Wood et al. (1981)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Dibenzo[a,i]pyrene
TA100 + LaVoie et al. (1979);
McCann et al. (1975a)
TA100 + Baker et al. (1980)
TA98 + Hermann (1981)
TA98 + Wood et al. (1981)
TA1537,TA1538 + Teranishi et al. (1975)
Not specified + Sardella et al. (1981)
Dibenzo[a,l]pyrene
TA98,TA100 + Karcher et al. (1984)
TA98 + Hermann (1981)
TA98,TA100 +,± Devanesan et al. (1990)
Fluoranthene
TA98 + Hermann et al. (1980)
TA98 + Epler et al. (1979)
TA100 - LaVoie et al. (1979)
TA100 + LaVoie et al. (1982a)
TA98, TA100, - Salamone et al. (1979)
TA1535,TA1537,
TA1538
TA98,TA100 + Poncelet et al. (1978)
TA98,TA100 + Mossanda et al. (1979)
TM677 + Kaden et al. (1979)
TM677 + Rastetter et al. (1982)
TM677 + Babson et al. (1986b)
TA97,TA98,TA100 + Sakai et al. (1985)
TA98,TA100 + Bos et al. (1988)
TA100 + Carver et al. (1986);
Hermann (1981);
LaVoie et al., 1979)
TA98,TA100 + Bos et al. (1987)
TA97,TA102, ± Bos et al. (1987)
TA1537
TA1535 - Bos et al. (1987)
TA98,TA100 + Bhatia et al. (1987)
TA98,TA100 - Florin et al. (1980)
TA98 - Tokiwa et al. (1977)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Fluorene
TA98, TA100, - McCann et al. (1975a);
TA1535,TA1537 LaVoie et al. (1979,
1980, 1981a)
TM677 - Kaden et al. (1979)
TA97 - Sakai et al. (1985)
TA98,TA100 - Bos et al. (1988)
TA100 - Pahlman & Pelkonen
(1987)
Indeno[1,2,3-cd]pyrene
TA98 + Hermann et al. (1980)
TA100 + LaVoie et al. (1979)
TA100 + Rice et al. (1985)
5-Methylcholanthrene
TA100 + Coombs et al. (1976);
Gelboin & Ts'o (1978);
LaVoie et al. (1979);
McCann et al. (1975a);
Hecht et al. (1978)
TA100 + Amin et al. (1979)
TA100 + El-Bayoumy et al. (1986)
1-Methylphenanthrene
TA100 + LaVoie et al. (1981b)
TM677 + Kaden et al. (1979)
TA97,TA98,TA100 + Sakai et al. (1985)
TA98,TA100 + LaVoie et al. (1983b)
Naphthalene
TA98,TA100, - Florin et al. (1980)
TA1535,TA1537
TA98, TA100, - McCann et al. (1975a)
TA1535,TA1537,
TA1538
TA98, TA100, - Purchase et al. (1976)
TA1535,TA1538
TA98 - Ho et al. (1981)
TM677 - Kaden et al. (1979)
G46, E. coli K12 - Kramer et al. (1974)
TA98,TA100 - Epler et al. (1979)
TA98,TA100 - Mamber et al. (1984)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
TA97,TA98,TA100 - Sakai et al. (1985)
TA100 - Pahlman & Pelkonen
(1987)
TA98,TA100 - Bos et al. (1988)
Perylene
TA98 + Ho et al. (1981)
TA100 + LaVoie et al. (1979)
TA98,TA100, - Salamone et al. (1979)
TA1535, TA1537,
TA1538
TA98 + Hermann (1981)
TA98 + Florin et al. (1980)
TM677 + Kaden et al. (1979);
Penman et al. (1980)
TA100 + Carver et al. (1986)
TA97,TA100 + Sakai et al. (1985)
TA98,TA100 + Lofroth et al. (1984)
TA100 - Pahlman & Pelkonen
(1987)
Phenanthrene
TA100 + Oesch et al. (1981)
TA100 - Wood et al. (1979)
TA98 + Epler et al. (1979)
TA98 - LaVoie et al. (1979, 1980)
TA100 - LaVoie et al. (1981b)
TA98,TA100 - Probst et al. (1981)
TA100 - LaVoie et al. (1979);
LaVoie et al. (1980);
Gelboin & Ts'o (1978);
McCann et al. (1975a)
TA98, TA100, - McCann et al. (1975a)
TA1535,TA1537
TA100 + Carver et al. (1986)
TM677 - Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA98,TA100 ± Bos et al. (1988)
TA1535,TA1536, - Simmon (1979)
TA1537,TA1538
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TA100 - Pahlman & Pelkonen
(1987)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
TA98, TA100, - Dunkel et al. (1984)
TA1535,TA1537,
TA1538
TA98,TA100 - Florin et al. (1980)
Pyrene
TA98 - Ho et al. (1981);
Rice et al. (1988a)
TA98,TA100, - McCann et al. (1975a);
LaVoie et al. (1979);
TA1535,TA1537 Ho et al. (1981)
TA1537 + Bridges et al. (1981)
TA98,TA100 - Salamone et al. (1979)
TA98,TA100 - Probst et al. (1981)
TA1537 + Epler et al. (1979)
TM677 + Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA98,TA100 ± Bos et al. (1988)
TA100 - Carver et al. (1986);
Hermann (1981)
TA98,TA100 + Bhatia et al. (1987)
TA98, TA100, - Dunkel et al. (1984)
TA1536,TA1537,
TA1538
TA100 - Pahlman & Pelkonen
(1987)
TA98,TA100 - Florin et al. (1980)
Triphenylene
TA98 + Epler et al. (1979)
TA98 + Tokiwa et al. (1977)
TA98,TA100 + Mossanda et al. (1979);
Wood et al. (1980)
TA98 + Hermann (1981)
TA98,TA100 + Poncelet et al. (1978)
TM677 + Kaden et al. (1979)
TA98,TA100 + Bos et al. (1988)
TA100 + Pahlman & Pelkonen
(1987)
TA, used to test reverse mutation to histidine non-auxotrophic mutants);
TM, used to test forward mutation to 8-azaguanine-resistant mutants
+, positive); -, negative); ±, inconclusive
Table 82. DNA damage induced by polycyclic aromatic hydrocarbons in vitro
Test system End-point Metabolica Resultb Reference
activation
Prokaryotes
Anthracene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2, E. coli WP100 R + - Member et al.
(1983)
E. coli WP2, E. coli WP67, R +/- - Tweats (1981)
E. coli CM871
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
E. coli WP2s(lambda) R +/- + Rossman et al.
prophage induction) (1991)
B. subtilis R +/- - Ashby & Kilby
(1981)
B. subtilis R +/- - McCarroll et al.
(1981)
E. coli GY5027 (prophage R + - Mamber et al.
induction) (1984)
Anthranene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benz[a]anthracene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benzo[b]fluoranthene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[ghi]fluoranthene
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Benzo[j]fluoranthene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benzo[a]fluoranthene
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Benzo[b]fluoranthene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Bunzo[ghi]perylene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benzo[a]pyrene
E. coli WP2, E. coli WP100 R + + Mamber et al.
(1983)
E. coli GY5027 R + + Mamber et al.
(1983)
E. coli pol A- R + + Rosenkranz &
Poirier (1979)
E. coli WP2, E. coli WP67, R +/- + Tweats (1981)
E. coli CM871
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli PQ37 R +/- +/+ Mersch-
Sundermann et
al. (1992)
B. subtilis R +/- + McCarroll et al.
(1981)
E. coli WP2s(lambda) R +/- + Rossman et al.
prophago induction) (1991)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[e]pyrene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli WP2s(lambda) R +/- + Rossman et al.
prophage induction) (1991)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Chrysene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli PQ37 R +/- + Mersch-
Sundermann at
al. (1992)
Coronene
E. coli PQ37 R +/- - Mersch-
Sundermann at
al. (1992)
Dibenz[a,h]anthracene
E. coli R +/- + Ichinotsubo et al.
(1977)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
B. subtilis R +/- + McCarroll et al.
(1981)
E. coli WP2s (lambda R +/- + Rossman et al.
prophage induction) (1991)
Dibenzo[a,i]pyrene
E. coli R +/- + Ichinotsubo et al.
(1977)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al.(1992)
B. subtilis R +/- + McCarroll et al.
(1981)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Dibenzo[a,h]pyrene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Dibenzo[a,i]pyrene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Fluoranthene
E. coli WP2s (lambda R +/- - Rossman et al.
prophage induction) (1991)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Fluoranthene
E. coli WP2, E. coli WP100 R + - Mamber et al.
(1983)
E. coli GY5027 R + - Mamber et al.
(1984)
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Indeno[1,2,3-cd]pyrene
E. coli PQ37 R + - Mersch-
Sundermann et
al.(1992)
Naphthalene
E. coli WP2, E. coli WP 100 R + - Mamber et al.
(1983)
E. coli GY5027 R + - Mamber et al.
(1984)
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Parylene
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Phenanthrene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli PQ37 R +/- + Mersch-
Sundermann at
al. (1992)
E. coli WP2s (lambda R +/- + Rossman et al.
prophage induction) (1991)
B. subtilis R +/- - McCarroll et al.
(1981)
Pyrene
E. coli R +/- - Ashby & Kilbey
(1981; De Serres
& Ashby, 1981)
E. coli WP2, E, coli WP100 R + - Mamber et al.
(1983)
E. coli GY5027 R + - Mamber et al.
(1984)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli WP2, E. coli WP67, R +/- - Tweats (1981)
E. coli CM871
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
B. subtilis R +/- - Ashby & Kilbey
(1981)
B. subtilis R +/- - McCarroll et al.
(1981)
E. coli WP2s (lambda R +/- - Rossman et al.
prophage induction) (1991)
Triphenylene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Eukaryotes
Acenaphthene
Rat liver or lung DA - - Beach & Gupta
(1991)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Anthracene
Primary rat hepatocytes UDS - - Williams, 1977;
Probst et al.
(1981)
Primary rat hepatocytes R - - Tong et al.
(1983)
HeLa cells UDS +/- - Martin et al.
(1978; Martin &
McDermid
(1981)
Human skin fibroblasts R - Milo et al. (1978)
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Human peripheral blood DA - - Gupta et al.
lymphocytes (1988)
Benz[a]anthracene
Primary rat hepatocytes UDS - + Probst et al.
(1981)
Primary rat hepatocytes R - + Tong et al.
(1983)
HeLa cells UDS +/- + Martin et al.
(1978)
Rat or human mammary DS - ± Mane et al.
epithelial cells (1990)
Hamster buccal pouch DS - - Nagabhushan et
(epithelial cells (inhibition al. (1990)
of DNA synthesis)
Human peripheral blood DA - + Gupta et al.
lymphocytes (1988)
Benzo[b]fluoranthene
Rat buccal mucosa DA - + Autrup, & Autrup
epithelial cells (1986)
Human leukocytes DA + + Roggeband et al.
(1994a)
Benzo[j]fluoranthene
Rat buccal mucosa DA - + Autrup & Autrup
epithelial cells (1986)
Benzo[k]fluoranthene
Human leukocytes DA + + Roggeband et al.
(1994a)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[a]pyrene
Primary rat hepatocytes UDS - + Probst et al.
(1981)
Primary rat hepatocytes R - + Williams et al.
(1982)
C3H/1OT1/2 mouse clone 8 - ± Lubet et al.
(DNA breaks) (1983a)
Human leukocytes DA + + Roggeband et al.
(1994a)
Hamster or rat trachea DA, - + Roggeband et al.
epithelial cells UDS (1994b)
HeLa cells UDS +/- + Martin et al.
(1978)
Human skin fibroblasts R - + Milo et al. (1978)
Human mammary cells - + Leadon et al.
(oxidative DNA damage) (1988)
Human fibroblasts UDS + + Agrelo & Amos
(1981)
Human fibroblasts WI-38 UDS +/- + Robinson &
Mitchell (1981)
Rat or human mammary R - + Mane et al.
epithelial cells (1990)
Human bronchial cells DA - + Harris et al.
(1984)
Syrian hamster embryo cells R - + Casto (1979)
Hamster buccal pouch - + Nagabhushan et
epithelial cells (inhibition of al. (1990)
DNA synthesis)
Rat buccal mucosa DA - + Autrup & Autrup,
epithelial cells (1986)
Human peripheral DA - + Gupta et al.
lymphocytes (1988)
Primary rat hapatocytes DA - + Monteith &
Gupta (1992)
Primary human hepatocytes DA - + Monteith &
Gupta (1992)
Calf thymus DNA DA - + Bryla & Weyand
(1991)
Primary mouse epidermal DA, - + Gill et al. (1991)
keratinocytes UDS
Primary rat hepatocytes R - + Tong et al.
(SCE) (1983)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[e]pyrene
Primary rat hepatocytes R - - Tong et al.
(SCE) (1983)
HeLa cells (UDS) UDS +/- + Martin et al.
(1978)
Primary rat hepatocytes R - - Williams et al.
(1982)
Rat mammary epithelial - - Mane et al. cells
(DNA synthesis) (1990)
Syrian hamster embryo R - - Casto (1979)
cells
Human skin fibroblasts R - - Milo et al. (1978)
Chrysene
Primary rat hepatocytes R - - Tong et al.
(1983)
Human leukocytes DA + + Roggeband et al.
(1994a)
Cyclopenta[cd]pyrene
Rat liver or lung tissue DA - + Beach & Gupta
(1991)
Calf thymus DNA DA + + Beach & Gupta
(1994)
Dibenz[a,h]anthracene
Primary human foreskin UDS - + Lake et al.
epithelial cells (1978)
HeLa cells UDS +/- + Martin et al.
(1978)
Syrian hamster embryo R - - Casto (1979)
cells
Primary rat hepatocytes UDS - + Probst et al.
(1981)
Mouse liver DNA DA + + Lecoq et al.
(1991)
Human bronchial cells DA - + Harris et al.
(1984)
Hamster embryonic cells + Kuroki &
Heidelberger
(1972)
C3H1OT1/2 mouse clone DA - + Nesnow et al.
8 cells (1994)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Dibenzo[a,i]pyrene
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Fluorene
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Human leukocytes DA + + Roggeband et al.
(1994a)
5-Methylcholanthrene
Primary rat hepatocytes UDS - + Tong et al.
(1981a)
1-Methylphenanthrene
Primary rat hepatocytes UDS - + Tong et al.
(1981a)
Chinese hamster ovary cells DA + + Dunn & Douglas
(1991)
Perylene
Human peripheral blood DA - - Gupta et al.
lymphocytes (1988)
Syrian hamster embryo R - - Casto (1979)
cells
Phenanthrene
Syrian hamster embryo R - - Casto (1979)
cells
Human foreskin epithelial UDS - - Lake et al.
cells (1978)
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Human skin fibroblasts R - Milo et al. (1978)
Pyrene
Syrian hamster embryo R - - Casto (1979)
cells
Human foreskin epithelial UDS - - Lake et al.
cells (1978)
Primary rat hepatocytes UDS - - Probst et al.
(1981)
HeLa cells UDS +/- - Martin et al.
(1978)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Human fibroblast cell line UDS +/- + Robinson &
WI38 Mitchell (1981)
Primary rat hepatocytes R - - Williams et al.
(1982)
Human skin fibroblasts R - Milo et al. (1978)
Human skin fibroblasts UDS + - Agrelo & Amos
(1981)
Primary rat hepatocytes R - - Tong et al.
(1983)
Human peripheral blood DA - - Gupta et al.
lymphocytes (1988)
Triphenylene
Human peripheral blood DA - + Gupta et al.
lymphocytes (1988)
R, DNA repair; DA, DNA adducts; UDS, unscheduled DNA synthesis;
SCE, sister chromatid exchange
a +, tested with metabolic activation; -, tested without metabolic activation;
+/-, tested with and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if
positive only with activation
Table 83. Mutagenicity of polycyclic aromatic hydrocarbons in yeasts and other
eukaryotes, host-mediated mutagenicity, and mutagenicity in Drosophila
Test system End-point Metabolic Resultb Reference
activationa
Yeasts and other eukaryotes
Anthracene
Saccharomyces cerevisiae MGC - - Seibert et al.
D4-RDII (1981)
Saccharomyces cerevisiae MR - - De Serres &
Hoffman (1981)
Benzo[a]pyrene
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Saccharomyces cerevisiae NMR - + DeSerres &
Hoffmann (1981)
Paramecium tetraurelia + + Smith-Sonneborn
(survival) (1983)
Chrysene
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Dibenz[a,h]anthracene
Neurospora crassa - + Barrett & Tatum
(1958)
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Naphthalene
Paramecium tetraurelia + - Smith-Sonneborn
(survival) (1983)
Phenanthrene
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Pyrene
Saccharomyces cerevisiae; NMR; - - De Serres &
S. pombe FM Hoffman (1981)
Host-mediated mutagenicity
Anthracene
Salmonella typhimurium - ± Simmon et al.
TA1530, TA1535,TA1538 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Table 83. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Benz[a]anthracene
Salmonella typhimurium - + Simmon et al.
TA1530,TA1535,TA1538 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Benzo[a]pyrene
Salmonella typhimurium - - Simmon et al.
TA1530,TA1535,TA1538 (1979); Glatt at
al. (1985)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Benzo[e]pyrene
Salmonella typhimurium - - Simmon et al.
TA1538 (1979)
Chrysene
Salmonella typhimurium - - Simmon et al.
TA1530,TA1535,TA1538 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Phenanthrene
Salmonella typhimurium - - Simmon et al.
TA1530,TA1535 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Drosophila malanogaster
Anthracene R - Fujikawa et al.
(1993)
Benz[a]anthracene SLRL + Fahmy & Fahmy
(1973)
Somatic mutation - Fahmy & Fahmy
(1980)
SLRL - Zijistra & Vogel
(1984)
SMART + Frolich & Wurgler
(1990)
R + Fujikawa et al.
(1993)
Table 83. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Benzo[a]pyrene SLRL ± Vogel et al.(1983)
Somatic mutation + Fahmy & Fahmy
(1980)
SLRL - Zijistra & Vogel
(1984)
SLRL - Valencia &
Houtchens (1981)
Somatic mutation + Batiste-Alentorn
et al. (1991)
SMART + Frolich & Wurgler
(1990)
SLRL - Valencia &
Houtchens (1981)
R + Fujikawa et al.
(1993)
Benzo[e]pyrene R - Fujikawa et al.
(1993)
Fluorene R - Fujikawa et al.
(1993)
Pyrene R ± Fujikawa et al.
(1993)
MGC, mitotic gene conversion; NMR, number of mitotic recombinants; MR, mitotic recombination;
SLRL, sex-linked recessive lethal mutation; R, DNA repair; FM, forward mutation;
SMART, somatic mutation and recombination test
a +, tested with metabolic activation; -, tested without metabolic activation; tested with
and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if positive only
with activation
Table 84. Mutagenicity of polycyclic aromatic hydrocarbons in mammalian cells in vitro
Test system End-point Metabolic Resultb Reference
activationa
Anthracene
Chinese hamster V79 HPRT +/- - Knaap et al.
(1981)
Mouse lymphoma L5178Y TK + - Amacher &
Turner (1980);
Amacher et al.
(1980)
Human lymphoblastoid TK6 TK + - Barfknecht et al.
(1981)
Fischer rat embryo OR + - Mishra et al.
(1978)
Human epithelial EUE cells DTR - - Rocchi et al.
(1980)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Benz[a]anthracene
Chinese hamster V79 HPRT + + Krahn &
Heidelberger
(1977); Slaga et
al. (1978)
Chinese hamster V79 HPRT + - Huberman
(1975)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Human epithelial EUE cells DTR - - Rocchi et al.
(1980)
Human keratinocytes HPRT - - Allen-Hoffmann
& Rheinwald
(1984)
Mouse lymphoma L5178Y TK + + Amacher &
Turner (1980);
Amacher et al.
(1980)
Mouse lymphoma L5178Y TK - - Amacher &
(+ hamster hepatocytes) Paillet (1983)
Mouse lymphoma L5178Y TK - + Amacher &
(+ hamster hepatocytes) Paillet (1982)
Rat liver epithelial ARL 18 HPRT - - Tong et al.
(1981a)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Banzo[b]fluoranthene
Chinese hamster V79 HPRT + - Huberman
(1975)
Benzo[a]pyrene
Chinese hamster V79 HPRT + + Arce et al.(1987);
Diamond et al.
1980); Huberman
(1975)
Chinese hamster V79 HPRT + + Krahn &
Heidelberger
(1977)
Mouse lymphoma L5188Y TK - + Amacher &
(+ hamster hepatocytes) Paillet (1982)
Chinese hamster ovary HPRT +/- + Gupta & Singh
(1982)
Fischer rat embryo OR - + Mishra et al.
(1978)
Mouse lymphoma L5178Y TK - + Amacher &
(+ hamster hepatocytes) Paillet (1983)
Mouse lymphoma L5178Y TK +/- + Clive et al.
(1979)
Mouse lymphoma L5178Y TK + + Amacher &
Turner (1980);
Amacher et al.
(1980); Arce et
al.(1987)
Mouse lymphoma L5178Y TK + + Wangenheim &
Bolcsfoldi (1988)
Human lymphoblasts AHH TK - + Crespi & Thilly
(1984)
Human lymphoblasts K6 TK +/- + Crespi et al.
(1985)
Human epithelial EUE cells DTR - + Rocchi et al.
(1980);
Barfknecht et al.
(1982)
Human fibroblasts HS172 DTR +/- + Gupta &
Goldstein (1981)
Human keratinocytes HPRT - + Allen-Hoffmann
& Rheinwald
(1984)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Rat liver epithelial cells - + Tong et al.
ARL 18 (1981a)
Chinese hamster ovary-AS52 + Oberly et al.
(chromosomal mutation) (1992)
Human epithelial teratoma HPRT - + Huberman et al.
P3 (cocultivated with human (1984)
carcinoma BJ cells)
Chinese hamster lung cells HPRT - + Baird et al.
V79 (1984)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary,
(1988)
Mouse lymphoma L5178Y TK +/- + Rees et al.
(1989)
Mouse Balb/c-3T3 OR - + Lubet et al.
(1990)
Mouse lymphoma L5178Y TK +/- + Jotz & Mitchell
(1981)
Benzo[e]pyrene
Chinese hamster V79 HPRT + - Hubermann
(1978)
Rat liver epithelial ARL 18 HPRT - - Tong et al.
(1981a)
Mouse C3H10T1/2 OR - - Gehly et al.
(1982)
Human epithelial teratoma P3 HPRT - - Huberman et al.
(1984)
Chinese hamster lung cells HPRT - - Baird et al.
V79 (1984)
Fischer rat embryo cells OR + - Mishra et al.
(1978)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Mouse lymphoma L517BY TK +/- - Clive et al.
(1979)
Mouse Balb/c-3T3 OR - - Lubet et al.
(1990)
Chrysene
Chinese hamster V79 HPRT + - Huberman &
Sachs (1976)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Human epithelial EUE DTR - - Rocchi et al.
(1980)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Human epithelial teratoma P3 HPRT - + Huberman et al.
(cocultivated with human (1984)
carcinoma BJ cells)
Cyclopenta[cd]pyrene
Human lymphoblastold HH-4 HPRT + + Skopek et al.
(1979)
Mouse lymphoma L5178Y TK +/- + Gold et al. (1980)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Human lymphoblasts AHH1 TK - + Crespi & Thilly
1984)
Chinese hamster V79 HPRT - + Raven et al.
(+ hamster embryo fibroblasts) (1982)
Dibenz[a,h]anthracene
Chinese hamster V79 HPRT + + Huberman &
Sachs (1976);
Huberman
(1978)
Chinese hamster V79 HPRT + + Krahn &
Heidelberger
(1977)
Human epithelial EUE DTR - ± Rocchi et
al., 1980)
Fluoranthene
Human lymphoblastoid HH-4 HPRT + + Thilly et al.
(1980)
Human lymphoblasts AHH1 TK - - Crespi & Thilly
(1984)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Fluorene
Mouse lymphoma L5178Y TK +/- + Wangenheim &
Bolcsfoldi (1988)
1-Methylphenanthrene
Human lymphoblastoid TK6 TK + + Barfknecht et al.
(1981)
Human lymphoblasts AHH1 TK - + Crespi & Thilly
(1984)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Perylene
Human lymphoblastoid TK6 HPRT + - Penman et al.
(1980)
Phenanthrene
Chinese hamster V79 HPRT + - Huberman &
Sachs (1976)
Human lymphoblastoid TK6 TK + + Barfknecht et al.
(1981)
Fischer rat embryo OR + - Mishra et al.
(1978)
Pyrene
Mouse lymphoma L517BY TK +/- + Jotz & Mitchell
(1981)
Fischer rat embryo OR + + Mishra et al.
(1978)
Chinese hamster V79 HPRT + - Huberman
(1975)
Mouse lymphoma L5178Y TK + - Amacher et al.
(1980)
Human lymphoblasts TK6 TK + - Barfknecht et al.
(1982)
Chinese hamster ovary HPRT - Heflich et al.
(1990)
Human epithelial teratoma P3 HPRT - - Huberman et al.
(1984)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Mouse lymphoma L5178Y TK +/- + Wangenheim &
Bolcsfoldi (1988)
Rat liver epithelial ARL18 HPRT - - Tong et al.
(1981a)
Mouse Balb/c-3T3 OR - ± Lubet et al.
(1990)
Triphenylene
Human lymphoblasts TK + + Barfknecht et al.
(1982)
HPRT, hypoxanthine-guanine phosphoribosyl transferase reversion; TK, thymidine kinase
reversion; OR, ouabain resistance; DTR, diphtheria toxin resistance
a +, tested with metabolic activation; - , tested without metabolic activation; +/-,
tested with and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if positive
only with activation
Table 85. Chromosomal effects of polycyclic aromatic hydrocarbons in mammalian cells in vitro
Test system End-point Metabolic Resultb Reference
activationa
Anthracene
Chinese hamster D6 CA, - - Abe & Sasaki
SCE (1977a)
Rat liver epithelial ARL18 SCE - - Tong et al.
(1981b)
Rat liver RL1 CA - - Dean (1981)
Benz[a]anthracene
Chinese hamster ovary SCE - + Pal (1981)
Rat liver epithelial ARL18 SCE - ± Tong et al.
(1981b)
Chinese hamster V79 SCE - ± Mane et al.
(coincubation with rat (1990)
mammary epithelial cells)
Benzo[a]pyrene
Rat liver RL1 CA - + Dean (1981)
Chinese hamster V79-4 CA, - - Popescu et al.
(+ feeder cells) SCE (1977)
Chinese hamster lung CA +/- + Matsuoka et al.
(1979)
Mouse lymphoma L5178Y CA - + Arce et al. (1987)
(+ hamster embryo cells)
Human fibroblasts WI-38 CD +/- + Weinstein et al.
(1977)
Chinese hamster V79 SCE - + Arce et al.(1987);
(+ hamster embryo cells) Wojciechowski et
al. (1981)
Chinese hamster Don-6 SCE - + Abe et al.
(1983a)
Chinese hamster ovary SCE +/- + Husgafvel-
Pursiainen et al.,
1986)
Chinese hamster ovary SCE +/- + Evans & Mitchell
(1981)
Rat pleural mesothelial SCE +/- + Achard et al.
calls (1987)
Rat liver epithelial ARL 18 SCE - + Tong et al.
(1981b)
Rat hepatoma Reuber SCE - + Dean et al.
H4-II-E (1983a)
Rat oesophageal tumour SCE - + Abe et al.
R1 (1983a)
Table 85. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Rat ascites hepatoma SCE - + Abe et al.
AH6&B (1983a)
Human fibroblasts TIG-11 SCE - + Huh et al. (1982)
Human hepatoma cells SCE - + Huh et al. (1982)
Human hepatoma C-HC-4 SCE - + Abe et al.
and C-HC-20 (1983a,b)
Chinese hamster V79 SCE - + Mane et al.
(coincubation rat/human (1990)
mammary epithelial cells)
Primary mouse epidermal UDS - + Gill et al. (1991)
keratinocytes
Human hepatoma (strain SCE, - + Natarajan &
Hap G2) MN Darroudi (1991)
Mouse spleen lymphocytes SCE - + Wielgosz et al.
(1991)
Mouse C3H/10T1/2 clone 8 SCE - + Krolewski et al.
(1986)
Human epithelial teratoma SCE - + Murison (1988)
P3 (coincubation with rat
hepatoma RL-12 cell line)
Chinese hamster epithelial SCE - + DeSalvia et al.
liver (1988)
Human lymphocytes CD +/- + Rees et al.
(1989)
Benzo[e]pyrene
Rat liver epithelial ARL 18 SCE - - Tong et al.
(1981b)
Mouse C3H 10T1/2 CA, - - Gehly et al.
SCE (1982)
Chinese hamster V79 cells SCE - - Mane et al.
(coincubation with rat (1990)
mammary epithelial cells)
Human epithelial teratoma SCE - - Murison (1988)
P3 (coincubation with human
breast carcinoma cells
BJ-015)
Cyclopenta[cd]pyrene
Mouse C3H/10T1/2 clone 8 SCE - + Krolewski et al.
(1986)
Human epithelial teratoma SCE - + Murlson (1988)
P3 (coincubation with human
breast carcinoma cells BJ-015)
Table 85. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Dibenz[a,h]pyrene
Chinese hamster ovary SCE - + Pal (1981)
Fluoranthene
Chinese hamster CHO-1 SCE +/- + Palitti et al.
(1986)
Chinese hamster epithelial SCE - - DeSalvia et al.
liver (1988)
Fluorene
Chinese hamster lung CA +/- + Matsuoka et al.
CHL (1991)
Naphthalene
Mouse embryos (in vitro) CA + + Gollahon (1991);
Gollahon et al.
(1990)
Perylene
Chinese hamster V79 CA - + Popescu et al.
(1977)
Chinese hamster V79 SCE - - Popescu et al.
(1977)
Phenanthrene
Chinese hamster V79-4 SCE - - Popescu et al.
(+ hamster feeder cells) (1977)
Chinese hamster V79-4 CA - + Popescu et al.
(+ hamster feeder cells) (1977)
Chinese hamster Don CA,SCE - Abe & Sasaki
(1977b)
Chinese hamster lung CA - Ishidate &
CHL Odashima
(1977)
Chinese hamster lung CA +/- - Matsuoka et al.
CHL (1979)
Pyrene
Rat liver epithelial ARL 18 SCE - - Tong et al.
(1981b)
Chinese hamster D6 CA, - - Abe & Sasaki
SCE (1977a)
Chinese hamster ovary SCE +/- + Evans & Mitchell
(1981)
Table 85. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Chinese hamster ovary SCE +/- + Perry &
Thomson (1981)
Chinese hamster V79-4 CA - + Popescu et al.
(1977)
Chinese hamster V79-4 SCE - - Popescu et al.
(+ hamster feeder cells) (1977)
Rat liver RL1 CA - - Dean (1981)
Human fibroblasts WI-38 CD +/- - Weinstein et al.
(1977)
Human hepatoma (strain MN, - - Natarajan &
Hep G2) SCE Darroudi (1991)
Chinese hamster epithelial SCE - - DeSalvia et al.
liver (1988)
SCE, sister chromatid exchange; MN, micronucleus formation; CA, chromosomal aberration;
CD, chromosomal damage
a +, tested with metabolic activation; -, tested without metabolic activation;
+/-, tested with and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if positive
only with activation
Table 86. Morphological transformation of mammalian cell in vitro by
polycyclic aromatic hydrocarbons
Test system Resulta Reference
Anthracene
Balb/c3T3 mouse cells - DiPaolo et al. (1972)
Guinea-pig fetal cells - Evans & DiPaolo
(1975)
Neonatal Syrian golden hamster - Purchase et al. (1976)
kidney fibroblasts BHK21 C13
Syrian hamster embryo cells - Pienta et al. (1977)
Hamster BHK21 clone 13 cells - Grab et al. (1980)
Syrian hamster embryo cells - Dunkel et al. (1981)
Balb/3T3 mousecells - Dunkel et al. (1981)
Balb/3T3 mousecells - Peterson et al. (1981);
Lubet et al. (1983a)
Fischer rat embryo cells - Mishra et al. (1978)
Fischer rat embryo cells - Dunkel et al. (1981)
(leukaemia virus-infected)
Fischer rat embryo cells + Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
C3H/10T1/2 mouse clone 8 - Dunkel et al. (1988)
Benz[a]anthracene
Syrian hamster embryo cells + Pienta et al. (1977);
DiPaolo et al. (1969,
1971)
Mouse prostate C3HG23 cells + Marquardt &
Heidelberger (1972)
C3H/10T1/2 mouse cells Nesnow &
Heidelberger (1976)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Hamster embryo cells - Grover et al. (1971)
Syrian hamster embryo cells + Dunkel et al. (1981)
Syrian hamster lung cells FSHL + Emura et al. (1980)
Mouse ventral prostate C3H - Marquardt et al.(1972);
clone G23 cells Grover et al. (1971)
Balb/3T3 mouse clone 1-13 cells + Rundell et al. (1983)
Balb/3T3 mouse cells ± Dunkel et al. (1981)
Fischer rat embryo cells
(Rauscher leukaemia virus infected) ± Freeman et al. (1973)
Fischer rat embryo cells + Dunkel et al. (1981)
(leukaemia virus-infected)
Syrian hamster embryo cells + DiPaolo et al. (1985)
Table 86. (continued)
Test system Resulta Reference
Benzo[b]fluoranthene
Hamster BHP21 clone 13 cells + Greb et al. (1980)
Syrian hamster lung FSHL cells + Emura et al. (1980)
Benzo[k]fluoranthene
Syrian hamster lung FSHL cells - Emura et al. (1980)
Benzo[ghi]perylene
Syrian hamster embryo cells - DiPaolo et al. (1985)
Benzo[a]pyrene
Golden hamster embryo cells + Mager et al. (1977)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Hamster embryo cells (SA7 virus- + Casto et al. (1977)
transformed)
Syrian hamster embryo cells + DiPaolo et al. (1969,
1971)
Syrian hamster embryo cells + Dunkel et al. (1981)
Syrian hamster embryo cells + Casto et al. (1977)
Syrian hamster lung FSHL + Emura et al. (1980,
1987)
Syrian hamster SHE (SA7 virus- + Arce et al. (1987)
transformed)
C3H/10T1/2 mouse + Arce et al. (1987);
Lubet et al. (1983b);
Peterson et al. (1981)
Balb/3T3 mouse + Dunkel et al. (1981)
Balb/3T3 mouse clone A31-1-1 + Little & Vetrovs (1988)
Fischer rat embryo cells + Mishra et al. (1978)
Rat embryo cells (SA7 virus- + DiPaolo & Casto
transformed) (1976)
Fischer rat embryo cells + Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Fischer rat embryo cells + Dunkel et al. (1981)
(leukaemia virus-infected)
Balb/c 3T3 mouse clone A31 cells + Albini et al. (1991)
C3H/10T1/2 mouse clone 8 cells + Dunkel et al. (1988)
C3H/10T1/2 mouse clone 8 cells + Krolewski et al. (1986)
Balb/c3T3 mouse clone 1-13 cells + Rundell et al. (1983)
Benzo[e]pyrene
C3H/10T1/2 mouse cells - Gehly et al. (1982)
Fischer rat embryo cells - Mishra et al. (1978)
Table 86. (continued)
Test system Resulta Reference
Banzo[b]fluranthene
Syrian hamster embryo cells - Pienta et al. (1977)
Syrian hamster embryo cells + DiPaolo et al. (1969)
C3H/10T1/2 mouse clone 8 cells ± Dunkel et al. (1988)
Balb/c-3T3 mouse - Lubet et al. (1990)
Syrian hamster lung FSHL cells ± Emura et al. (1980)
Syrian hamster embryo cells - Dunkel et al. (1981)
Balb/3T3 mouse - Dunkel et al. (1981)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Chrysene
Syrian hamster embryo cells + Pienta et al. (1977)
Mouse prostate C3HG23 cells - Marquardt et al. (1972)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Hamster epithelial lung + Jacob et al. (1993c);
cell line M3E3/C3 Riebelmre et al. (1993)
Cyclopenta[cd]pyrene
C3H10T1/2 mouse clone 8 calls + Gold et al. (1980)
C3H/10T1/2 mouse clone 8 cells + Krolewski et al. (1986)
Dibenz[a,h]anthracene
Syrian hamster embryo cells + DiPaolo et al. (1969);
Pienta et al. (1977)
C3H 10T1/2 mouse cells + Reznikoff et al. (1973)
C3H mouse prostate cells + Chen & Heidelberger
(1969)
C3HG23 mouse prostate cells - Marquardt et al. (1972)
Hamster embryo cells - Grover et al. (1971)
Fischer rat embryo cells + Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Hamster embryo cells (SA7 virus- + Casto (1973); Casto et
transformed) al. (1977)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
C3H/10T1/2 mouse clone 8 cells ± Lubet et al. (1983a,b)
Rat embryo cells (SA7 virus- + DiPaolo & Casto
transformed) (1976)
C3H/10T1/2 mouse clone 8 cells ± Dunkel et al. (1988)
C3H/10T1/2 mouse clone 8 cells + Nesnow et al. (1994)
Fluoranthene
Fischer rat embryo cells - Freeman et al. (1973)
(Rauscher leukaemia virus- infected)
Table 86. (continued)
Test system Resulta Reference
Fluorene
Balb/c3T3 mouse cells ± Tonelli et al. (1979)
Indeno[1,2,3-cd]pyrene
Syrian hamster lung FSHLcells + Emura et al. (1980)
Naphthalene
Fischer rat embryo cells - Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Human lung WI-38 cells - Purchase et al. (1976)
Syrian hamster kidney - Purchase et al. (1976)
BHK-21C13 cells
Balb/c mouse mammary gland - Tonelli et al. (1979)
Balb/c-3T3 mouse cells - Rundell et al. (1983)
Perylene
Syrian hamster embryo cells - DiPaolo et al. (1985)
Syrian hamster embryo cells - Casto (1979)
Phenanthrene
Mouse prostate C3HG23 cells - Marquardt et al. (1972)
Syrian hamster embryo cells - Pienta et al. (1977)
Balb/3T3 mouse cells - Kakunaga (1973)
Fetal guinea-pig cells - Evans & DiPaolo
(1975)
Syrian hamster embryo cells - DiPaolo et al. (1969);
Dunkel et al. (1981)
Hamster BHK21 clone 13 cells - Greb et al. (1980)
Hamster embryo calls (SA7 - Casto et al. (1977)
virus-transformed)
C3H/10T1/2 mouse cells - Peterson et al. (1981)
C3H/10T1/2 mouse cells - Lubet et al. (1983b)
Balb/3T3 mouse cells - Dunkel et al. (1981)
Fischer rat embryo cells - Mishra et al. (1978)
Fischer rat embryo cells Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Fischer rat embryo cells (leukaemia - Dunkel et al. (1981)
virus-infected)
C3H/10T1/2 mouse clone 8 cells - Dunkel et al. (1988)
Table 86. (continued)
Test system Resulta Reference
Pyrene
Syrian hamster embryo cells - DiPaolo et al. (1969);
Pienta et al. (1977);
Casto (1979)
C3H mouse prostate cells - Chen & Heidelberger
(1969)
Balb/C-3T3 mouse cells - DiPaolo et al. (1972);
Kakunaga(1973)
Fetal guinea pig cells - Evans & DiPaolo
(1975)
Fischer rat embryo cells - Mishra et al. (1978)
Hamswr embryo cells (SA7 - Casto et al. (1977)
virus-transformed)
C3H/10T1/2 mouse clone 8 cells ± Dunkel et al. (1988)
Balb/c-3T3 mouse - Lubet et al. (1990)
a Result; +, positive; ±, inconclusive; -, negative
Table 87. Chromosomal effects of polycyclic aromatic hydrocarbons in
mammalian cell systems in vivo, including DNA binding and adducts and
sperm abnormalities
Test system Resulta Reference
Anthracene
Chinese hamster bone marrow: - Roszinsky-Kocher et al.
CA, SCE (1979)
Mouse bone marrow: MN - Salamone et al. (1981)
Mouse: sperm abnormalities - Topham (1980)
Chinese hamster V79 (mouse host- - Sirianni & Huang (1978)
mediated):SCE
Mouse peripheral blood: MN - Oshiro et al. (1992)
Mouse skin : DNA binding - Reddy et al. (1984)
Benz[a]anthracene
Chinese hamster bone marrow: + Roszinsky-Kocher et al.
SCE (1979)
Chinese hamster bone marrow: CA - Roszinsky-Kocher et al.
(1979)
Long-Evans rat bone marrow: CA - Sugiyama (1973)
Chinese hamster bone marrow + Peter et al. (1979)
MN,CA
NMRI mouse(in metaphase II + Peter et al. (1979)
oocytes): CA
Mouse gastrointestinal epithelial - Reddy et al. (1991)
cells: nuclear anomalies
Rat lung: DNA adducts,SCE, MN + Whong et al. (1992)
Mouse skin: DNA binding + Reddy et al. (1984)
Rat bone marrow and spleen + Zhong et al. (1995)
cells: MN
Benzo[b]fluoranthene
Chinese hamster bone marrow: + Roszinsky-Kocher et al.
SCE (1979)
Chinese hamster bone marrow: CA - Roszinsky-Kocher et al.
(1979)
Mouse skin: DNA binding + Weyand et al. (1987)
Lung, liver and peripheral + Ross et al. (1991); Ross
lymphocytes of rats: DNA adducts et al. (1992)
Rat lung, liver; peripheral blood + Ross et al. (1991); Ross
lymphocytes; whole blood cultures: et al. (1992)
SCE
Mouse gastrointestinal epithelial + Reddy et al. (1991)
cells: nuclear anomalies
Rat peripheral blood lymphocytes: + Bryant et al. (1991)
SCE, MN
Table 87. (continued)
Test system Resulta Reference
Mouse gastrointestinal epithelial + Reddy et al. (1991)
cells: nuclear anomalies
Mouse skin: DNA binding + Amin et al. (1991a)
Mouse skin: DNA adducts, MN, UDS + Winker et al. (1995)
Benzo[j]fluoranthene
Mouse lung and liver cells: + Weyand & LaVoie (1988)
DNA adducts
Mouse skin: DNA adducts + Weyand et al. (1993)
Benzo[k]fluoranthene
Mouse skin: DNA binding + Weyand et al. (1987)
Mouse lung and liver cells: + Weyand & LaVoie (1988)
DNA adducts
Benzo[ghi]perylene
Mouse skin: DNA binding + Reddy et al. (1984)
Benzo[a]pyrene
Mouse: dominant lethal mutation + Epstein (1968)
Mouse: dominant lethal mutation + Generoso et al. (1982)
Mouse: spot test + Russell (1977)
Mouse: spot test + Davidson & Dawson
(1976)
Rat hepatocytes: UDS - Miralis et al. (1982)
Mouse germ cells: UDS - Sega (1979)
Mouse skin: DNA binding + Weyand et al. (1987);
Rice et al. (1984)
Chinese hamster bone-marrow +,+ Roszinsky-Kocher et al.
cells: CA, SCE (1979)
Chinese hamster bone-marrow +,+ Bayer (1978)
cells: CA, SCE
Mouse: CA; heritable translocations - Generoso et al. (1982)
Mouse bone marrow: MN + Salamone et al. (1981)
Mouse bone marrow: MN - Bruce & Heddle (1979)
Chinese hamster bone-marrow - Bayer(1978)
cells: MN
Mouse: sperm abnormalities + Topham (1980)
Mouse: sperm abnormalities + Bruce & Heddle (1979)
Chinese hamster V79 + Sirianni & Huang (1978)
(mouse host-mediated): SCE
Mouse epidermal cells: DNA adducts + Albert et al. (1991a,b)
Mouse bone marrow: MN + Shimada et al. (1991)
Mouse keratinocytes: MN + He & Baker (1991)
Mouse lung and liver cells: + Weyand & LaVoie (1988)
Table 87. (continued)
Test system Resulta Reference
DNA adducts
Mouse liver, lung and stomach: + Cummings et al. (1991)
DNA adducts
Rat peripheral lymphocytes: SCE + Li et al. (1991)
Mouse bone marrow: MN + Mavournin et al. (1990)
Mouse bone marrow: MN + Kliesch et al. (1982)
Mouse bone marrow: MN + Harper & Legator (1987)
Mouse peripheral blood cells: MN ± Oshiro et al. (1992)
Mouse gastrointestinal epithelial + Reddy et al. (1991)
cells: nuclear anomalies
Mouse bone marrow: SCE + Wielgosz et al. (1991)
Rat peripheral blood lymphocytes: + Willems et al. (1991)
SCE, DNA adducts
Rat liver cells: DNA adducts + Willems et al. (1991)
Rat peripheral blood lymphocytes: - Willems et al. (1991)
CA
Chinese hamster cells: CA + Matsuoka et al. (1979)
Mouse skin apithelial cells: + Hughes & Phillips (1991)
DNA binding
Human peripheral lymphocytes: + Haugen et al. (1986)
DNA adducts
Rat lung, liver and peripheral + Ross et al. (1991)
lymphocytes: DNA adducts
Mouse bone marrow: MN + Awogi & Sato (1989)
Mouse skin: DNA binding + Reddy et al. (1984)
Mouse skin: DNA adducts + Oueslati et al. (1992)
Mouse and rat bone marrow: MN + Shimada et al. (1992)
Mouse bone marrow: CA + Adler & Ingwersen (1989)
Benzo[e]pyrene
Chinese hamster bone-marrow + Roszinsky-Kocher et al.
cells: SCE (1979)
Chinese hamster bone-marrow - Roszinsky-Kocher et al.
cells: CA (1979)
Mouse gastrointestinal epithelial - Reddy et al. (1991)
cells: nuclear anomalies
Mouse skin: DNA binding Reddy et al. (1984)
Chrysene
Chinese hamster bone-marrow + Roszinsky-Kocher et al.
cells: SCE (1979)
Chinese hamster bone-marrow - Roszinsky-Kocher et al.
cells: CA (1979)
NMRI mice: metaphase II oocytes + Basler et al. (1977)
Mouse keratinocytes: MN + He & Baker (1991)
Mouse skin: DNA binding + Reddy et al. (1984)
Table 87. (continued)
Test system Resulta Reference
Dibenz[a,h]anthracene
Chinese hamster bone-marrow + Roszinsky-Kocher et al.
cells: SCE (1979)
Chinese hamster bone-marrow - Roszinsky-Kocher et al.
cells: CA (1979)
Rat peripheral blood lymphocytes: - Bryant et al. (1990)
SCE, MN
Mouse skin: DNA binding + Lecoq et al. (1991)
Mouse skin: DNA binding + Reddy et al. (1984)
Rat bone-marrow and spleen + Zhong et al. (1995)
cells: MN
Rat lung: DNA adducts, MN, SCE + Whong et al. (1994)
Mouse skin: DNA adducts, MN, + Winker et al. (1995)
UDS
Dibenzo[a,e]pyrene
Mouse skin epithelial cells: + Hughes & Phillips (1991)
DNA binding
Dibenzo[a,i]pyrene
Rat spleen cells: MN + Zhong et al. (1995)
Rat lung: DNA adducts, MN, SCE + Whong et al. (1994)
Fluoranthene
Mouse bone-marrow cells: SCE - Palitti et al. (1986)
Indeno[1,2,3-cd]pyrene
Mouse skin: DNA binding + Weyand et al. (1987)
Mouse skin epithelial cells: + Rice et al. (1990)
DNA binding
5-Methylcholanthrene
Mouse skin: DNA adducts + Amin et al. (1985a)
Naphthalene
Mouse bone-marrow cells: MN - Harper et al. (1984)
Perylene
Mouse skin: DNA binding - Reddy et al. (1984)
Table 87. (continued)
Test system Resulta Reference
Phenanthrene
Chinese hamster bone-marrow - Bayer (1978); Roszinsky
cells: CA Kocher et al. (1979)
Chinese hamster bone-marrow + Bayer (1978); Roszinsky
cells: SCE Kocher et al. (1979)
Chinese hamster bone-marrow - Bayer(1978)
cells: MN
Pyrene
Mouse bone marrow cells: SCE - Paika et al. (1981)
Mouse bone marrow: MIN - Salamone et al. (1981)
Mouse bone marrow cells: MN - Tsuchimoto & Matter
(1981)
Chinese hamster V79 - Sirianni & Huang (1978)
(mouse host-mediated): SCE
Mouse keratinocytes: MN - He & Baker (1991)
Mouse peripheral blood cells: MN - Oshiro et al. (1992)
Mouse gastrointestinal epithelial - Reddy et al. (1991)
cells: nuclear anomalies
Mouse: sperm abnormalities - Topham (1980)
Mouse skin: DNA binding - Reddy et al. (1984)
SCE, sister chromatid exchange; MN, micronucleus assay; CA, chromosomal
aberrations; UDS, unscheduled DNA synthesis
a Result: +, positive; ±, inconclusive; -, negative
Table 88. Effects of polycyclic aromatic hydrocarbons on
morphological transformation of mammalian cells in vivo
Test system Resulta Reference
Anthracene
Mouse bone-marrow cells - Salamone et al. (1981)
Chinese hamster embryo cells - DiPaolo et al. (1973)
Benzo[ghi]perylene
Hamster embryos, transplacental - Quarles et al. (1979)
exposure
Benzo[a]pyrene
Hamster embryos, transplacental + Quarles et al. (1979)
exposure
Phenanthrene
Hamster embryos, transplacental - Quarles et al. (1979)
exposure
a +, positive; ±; inconclusive; -, negative
Table 89. Overview of genotoxicity of polycyclic aromatic
hydrocarbons
Compound Results
Acenaphthene Inconsistent, limited database
Acenaphthylene Inconsistent, limited database
Anthanthrene Positive, limited database
Anthracene Negative, with a few exceptions
Benz[a]anthracene Positive
Benzo[b]fluoranthene Positive
Benzo[k]fluoranthene Positive
Benzo[k]fluoranthene Positive
Benzo[ghi]fluoranthene Positive, limited database
Benzo[a]fluorene Inconsistent, limited database
Benzo[b]fluorene Inconsistent, limited database
Benzo[ghi]perylene Positive
Benzo[c]phenanthrene Positive, limited database
Benzo[a]pyrene Positive
Benzo[e]pyrene Positive
Chrysene Positive
Coronene Positive, limited database
Cyclopenta[cd]pyrene Positive
Dibenz[a,h]anthracene Positive
Dibenzo[a,e]pyrene Positive
Dibenzo[a,h]pyrene Positive, limited database
Dibenzo[a,i]pyrene Positive
Dibenzo[a,l]pyrene Positive, limited database
Fluoranthene Positive
Fluorene Negative, with a few exceptions
Indeno[1,2,3-cd]pyrene Positive
5-Methylchrysene Positive
1-Methylphenanthrene Positive
Naphthalene Negative
Perylene Positive
Phenanthrene Inconsistent
Pyrene Inconsistent
Triphenylene Positive
Table 90. Carcinogenicity of polycyclic aromatic hydrocarbons in experimental animals
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Acenaphthene
Mouse 100 Dermal Dissolved in 90% benzene 9 months No tumours observed n Kennaway
no/lc (1924)
'Pure' Mouse, m 85 Dermal 3 drops, 1x/week of <1 year After 12 months 5/85 survived q Graffi et al.
white approx. 300 solution, 1 with a total of 2 tumours; 0.4 no/ls (1953)
year; initiation experiment tumour/animal; promotor only:
0.08 tumour/animal
Anthanthrene
Mouse 30 Dermal 0.3% in benzene, Life 1/30 lung adenoma n Badger et al.
2 x/week, life no/ld (1940)
Recrystallized Mouse, f 20 Dermal 0.05 or 0.1%, 3 x/week, 15 0/20 with tumours n Hoffmann &
Ha/lcR/Mil 12 months months no/val Wynder (1966)
Recrystallized Mouse, f 30 Dermal 25 Kg/animal, 10 × over 6 months 2/25 papillomas; promotor n Hoffmann &
Swiss 20 days; initiation only: 526 no/val Wynder (1966)
Ha/lcR/Mil experiment
'Rigourously Mouse, f 13 Dermal 0.25 mg/animal, 4 x; 65 weeks 2/13 papillomas; promotor n Van Duuren et
purified' lcR(Ha initiation experiment only: 520 papillomas; control no/val al. (1968)
acetone: 0/20 papillomas
Recrystallized Mouse, f 30 Dermal 43 µg/animal, 2 x/week, < 100 1/30 with skin carcinoma; n Lijinsky &
Swiss 75 weeks control: 2/30 with carcinomas no/val Garcia (1972)
98.65% Mouse, f 40 Dermal 109 µg/animal, 2 x/week, 70 weeks 47% skin-tumour-bearing p Cavalieri et al.
Swiss 30 weeks animals; solvent control: 0% no/val (1977)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
TLC- Mouse, f 30 Dermal 0.69 mg/animal, 1x; 35 weeks 18% with papillomas; p Scribner
purified) CD-1 initiation experiment promoter only: 3% no/val (1973)
> 99% Mouse, f 27 Dermal 221 µg/animal, 1x; 26 weeks 11 % papillomas; solvent n Cavalieri at
Sencar initiation experiment only: 9% yes/val al.(1989)
Mouse, m/f 27 s.c. 0.6 mg/animal, 1x/month, No local sarcomas observed n Lacassagne et
XVII 3 months no/ln, al. (1958)
ld
99.4% Rat f 35 Intrapulm. 0.65 and 3.4 mg/kg, 1x 102/88 1/35 and 19/35 with lung p Deutsch-
Osborne- weeks tumours; control: no tumours yes/val Wenzel et al.
Mendel (1983)
> 99% Rat, f 20 Intra- 1.1 mg/gland, 1x, 8 40 1/20 with mammary tumours; n Cavalieri at
Sprague- mammary glands weeks control: 0/21 or 2120 yes/val al. (1989)
Dawley injection
Anthracene
Mouse 2x100 Dermal 40% suspension/solution 5 months 0/100, 1/100 tumours n Kennaway
no/lc (1924)
Mouse 44 Dermal 5%, 3x/week < 11 No skin tumours n Miescher
months no/lc (1942)
Mouse, 20 Dermal 1.5 mg/animal, 2x/day; 3 21 3/17 with tumours; promotor n Salaman & Roe
'S' days/week; total: 20 x; weeks only: 4/19 yes/val (1956)
initiation experiment
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 5 Dermal 10% solution, 3x/week, < 20 No skin tumours n Wynder &
Swiss life months no/ln, Hoffmann
Millerton ld (1959a)
TLC- Mouse, f 30 Dermal 1.8 mg/animal, 1x; 35 14% with papillomas; q Scribner (1973)
purified CD-1 initiation experiment weeks promoter only: 3% no/val
Mouse, m/f 24 Dermal 4 µg, 1 x/day, 5 days/week, 38 No increased tumour n Forbes et al.
Skh:hair 38 weeks, then 2 h/day weeks frequency compared with yes/val (1976)
less 1, UV controls
outbred
Mouse, f 20 Dermal 100 µg/animal, 10x on 24 weeks 15% with tumours; solvent: n LaVoie et al.,
Swiss alternate days; initiation 10% yes/val (1983a)
albino experiment
(Ha/lcR)
Mouse, m/f 40-50 s.c. 5 mg/animal in tricaprylin; < 22-28 0/26 sarcomas after 5 months n Steiner (1955)
C57BI 1x months yes/ld
Mouse, m 5 i.p. 1000 mg/kg, 1 × < 5 No effects observed n Shubik & Della
Swiss months no/ln Porta (1957)
Rat 31 Oral 6 mg/animal/day, 7x/ 33 22/31 alive after 1 year; no n Schmahl &
(diet) week monthns tumours after 33 months no/lc Reuter, cited by
Gerarde (1960)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Highly Rat, 28 Oral 5-15 mg/animal/day, 700 days 2/28 malignant tumours, n Schmahl (1955)
purified BD I/BD III (diet) 6x/week, 78 weeks no/ld
Rat 10 s.c. 1 mg/animal, 1x/week, < 103 No subcutaneous sarcomas n Boyland &
103 weeks weeks no/ln Burrows (1935)
ld
Rat, 5 s.c. 5 mg/animal, 6-7x 10 No tumours observed n Pollia
Wistar months no/ln (1941)
Highly Rat, 10 s.c. 20 mg/animal, 1x/week, < 29 5/9 tumours (fibromas) at site p Druckrey &
purified BD I/BD III 33 weeks months injection no/ln, Schmahl (1955)
ld
Highly Rat, 10 i.p. 20 mg/animal, 1x/week, > 2 years 1/10 spindle-cell sarcoma q Schmahl (1955)
purified BD I/BD III 33 weeks no/ld
Rat, f 60 Intrapulm. 0.5 mg/animal, 1x/year No lung tumours; control: no n Stanton et al.
Osborne/ tumours no/d (1972)
Mendel
Rabbit 9 Cerebral 4-20 mg/animal, 1 × 20-54 No glioma n Russel (1947)
implant months no/in,
ld
Benz[a]anthracene
Mouse, 8-19 Oral 0.5 mg/animal, 1x, 8x 16 0/13, 1/19 and 1/8 with q Bock & King
C57/BL or 16 × (highest dose), months papillomas; no carcinomas yes/ln (1959)
< 2 months observed; control: 0/12
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m 20 or Oral 1.5 mg/animal, 3x/wk, < 547- 100% hepatomas and 95% p Klein (1963)
B6AF1/J, 40 5 weeks 600 days pulmonary adenomas; solvent no/val
newborn only: 10% hepatomas and
35% pulmonary adenomas
Mouse, m 20 Oral 1.5 mg/animal, 1x/day, < 568 80% hepatomas and 85% p Klein (1963)
B6AF1/J, 2 days days lung adenomas (inadequately no/val
newborn reported)
Purified Mouse 30 Dermal 0.3% in benzene, < 584 1/30 epitheliomas n Barry et al.
2x/week,life days no/ld (1935)
'Pure' Mouse, m 75 Dermal 3 drops, 1x/week of 0.5% < 1 year After 12 months 9/75 survived p Graffi et al.
white solution, 1 year; initiation with a total of 18 tumours; 2 no/val (1953)
experiment tumours/animal; promotor only:
0.08 tumour/animal
Recystallized Mouse, f 30 Dermal 66 µg/animal, 2x/week, 13-15.5 No tumours; solvent only: no n Miller & Miller
albino 20 weeks months tumours no/val (1963)
Mouse, 20 Dermal 0.5% solution, 2x/week, 638 days No tumours; control: no n Stevenson &
C3H 638 days tumours no/val von Haam
(1965)
Recrystallized Mouse, 30-50 Dermal 0.0001-0,5 mg/animal in < 88 Dose-dependent increase in p Bingham & Falk
C3H+e n-dodecane or 0. 1 weeks malignant tumours; solvent yes/val (1969)
mg/animal in toluene, control: no tumours
3x/week, 50 weeks
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized Mouse, f 20 Dermal 1 mg/animal, 1x; initiation 58-60 10/20 with papillomas; p Van Duuren et
Swiss experiment weeks promotor only:1/20; solvent no/val al. (1970)
Millerton control: 0%
ICR/Ha
TLC Mouse, f 30 Dermal 0.5 mg/animal, 1x; 35 weeks 62% with papillomas; p Scribner(1973)
purified CD-1 initiation experiment promotor only: 3% no/val
> 99% Mouse, f 40 Dermal 90 µg/animal, 2x/week, 70 weeks 2.6% skin-tumour bearing n Cavalieri et al.
Swiss 30 weeks animals; solvent control: 0% no/val (1977)
> 99% Mouse, f 30 Dermal 0.46 mg/animal, 1x; 26 weeks 57% with papillomas; p Slaga et al.
CD-1 initiation experiment promotor only: 6% no/val (1978)
Mouse, f 30 Dermal 0.1 and 0.57 mg/animal, 27 weeks 14% and 36% (p < 0.05) with p Levin et al.
CD-1 1x; initiation experiment tumours; solvent control: 7% yes/val (1984)
Mouse, f 30 Dermal 0.23 and 0.57 mg/animal, 27 week 17% and 38% papillomas; p Weyand et al.
CD-11 1x; initiation experiment solvent control: 4% yes/val (1990);Wood et
al. (1980)
Spectrometer Mouse, m/f 50 s.c. 5 mg/animal in tricaprylin; < 22 8/46 sarcomas after 4 months; p Stainer & Falk
C57BI 1x months solvent control: 3/280 no/val (1951)
control
Mouse, m/f 40-50 s.c. 0.05, 0.2, 1, 5, or 10 < 22-28 5/44,11/45,15/44, 20/36 and p Steiner &
C57BI mg/animal in tricaprylin; months 5/16 sarcomas yes/ld Edgcomb
1x (1952); Steiner
(1955)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized Mouse, f 30 s.c. 0.94 mg/animal, 1x < 15 No sarcomas; solvent control: n Miller & Miller
albino months no tumours no/val (1963)
Mouse 20 s.c. 5 mg in tricaprylin, 1x 638 days No tumours; control: no n Stevenson &
C3H tumours no/val von Haam
(1965)
Mouse, m/f 10/10 s.c. 1 mg/animal, 1x/week, 60-80 8/10 m and 6/10 f with p Boyland & Sims
C57BL 10 weeks weeks sarcomas; control: 0/20 m no/val (1967)
and 0/20 f
Mouse, m/f 87 s.c. 0.2 mg/animal in 70-75 70 weeks: 15/15 m and 2/18 f p Grover et al.
Swiss polyethylene glycol weeks with liver tumours, 4/15 m and no/val (1975)
newborn on days 0, 1 and 2 after 10/18 f with lung tumours;
birth corrected control data: 4/22 m
and 1/23 f with liver tumours
and 3/22 m and 1/23 f with lung
tumours
Mouse, m/f 140 i.p. 9.1, 18.2, and 36.4 26 weeks 10/47 m and 4/38 f with n Wislocki et al.
Swiss µg/animal on days 1, 8, pulmonary tumours; solvent no/val (1979)
Webster and 15 after birth control: 7/43 and 2/24
BLU:Ha(ICR),
newborn
Mouse, 11 i.v. 10 mg/kg, 1x 20 weeks 18% lung tumours; n Shimkin &
A control: 21 % no/val Stoner(1975)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 52 Bladder About 2 mg/animal, 1x > 40 17/52 bladder carcinomas p Clayson et al.
C57 × IF F, implant weeks and 1/52 papillomas; control: yes/val (1968)
hybrid 4/89
Rat, f 10 Oral 200 mg/rat, 1x 60 days No tumours in treated animals; n Huggins &
Sprague- control: 8/164 after 310 days no/ln, Yang(1962)
Dawley lc
Rat, m 25 Dermal Saturated solution in < 18 No tumours n Tawfic(1965)
Donryu acetone,dropped at 2x/wk months no/ld
to cover 2 cm2, 5 months
Recrystallized Rat, m 20 s.c. 1.88 mg/animal, 1x > 4 No sarcomas; solvent control: n Miller & Miller
Holtzman months no tumours no/val (1963)
TLC Rat, f 28 i.v. 2 mg/animal (=13 mg/kg), 98 days No mammary tumours n Pataki &
purified Sprague- 3x on day 50, 53, and 56 of no/ld Huggins(1969)
Dawley age
TLC Rat, m 8 i.m. 2.5 mg/animal into hind 270 days No sarcomas; control: no n Pataki &
purified Long- leg, 1 × on day 25 of age spontaneous sarcomas no/ln Huggins(1969)
Evans
>99% Rat, f 20 Intra 0.91 and 3.7 mg into 5th 20 weeks No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1 × control: no tumours no/val (1988a)
Dawley injection
Chromatography Hamster m/f 50 Dermal 8 drops of a 0.5% solution, < 85/61 No tumours n Shubik et al.
control Syrian 2x/week, 10 weeks weeks no/ln, (1960)
golden ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Hamster, m 5 or Dermal 20 mmol/litre solution, < 44 No tumours; control: no n Solt et al.
Syrian 26 (buccal painting 2x/week, 5 or weeks tumours no/val (1987)
golden pouch) 20 weeks
Hamster m 47 or Intra- 0.5 or 3 mg/animal per < 110 No tracheal tumours; control: n Sellakumar
Syrian 33 tracheal week 30 or 15 weeks weeks no tumours yes/val Shubik (1974)
golden
Benzo[b]fluoranthene
Mouse, f 20 Dermal 0.01, 0.1 and 0.5%, < 14,12, 0.01 %: 5% papillomas after p Wynder &
Swiss 3x/week, life and 8 14 months; 0.1%: 65% no/ld Hoffmann
Millerton months papilomas and 85% carcinomas (1959b)
after 12 months;0,5%: 100%
carcinomas after 5 months
Mouse, f 20 Dermal 1 mg, 1x; initiation 63 weeks 18/20 papillomas, 5/20 p Van Duuren et
Swiss experiment carcinomas; promotor only: no/val al. (1966)
ICR/Ha 5/20, 1/20
> 96% Mouse, f 40 Dermal 3.4,5.6,9.2 µg/animal, < 2 years 5/15/540% with local tumours; p Habs et al.
NMRI 2x/week, life control: no tumours yes/val (1980)
Mouse, Dermal 10-100 µg/animal; 20 weeks Dose-related skin tumour p LaVoie et al.
CD-1 initiation experiment incidence yes/val (1982b)
> 99% Mouse, f 20 Dermal 4 and 10 nmol/animal, 34 weeks 45 and 95% tumour incidence; p Amin et al.
Cr1:CD-1 10x every other day; solvent control: 5% yes/val (1985a)
(ICR)BR initiation experiment
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 20 Dermal 0.025 and 0.1 mg/animal, 24 weeks 100 and 100% tumour p Weyand et al.
CD-1 10x every other day; incidence; solvent control: yes/val (1990)
initiation experiment 10%
Mouse, f 20 Dermal 3 and 10 µg/animal, 10x; 34 weeks 65 and 100% with tumours; p Amin et al.
Cr1:CD1 initiation experiment solvent control: 15% yes/val (1991a)
(ICR)BR
Mouse, m/f 16/14 s.c. 0.6mg/animal, 1x/month, approx. 8/16 m and 10/14 f with local p Lacassagne et
XVII nc/Z 3 months 200 days sarcoma no/ld al. (1963a)
> 99% Mouse, m/f 15/17 i.p. 126 µg/animal in DMSO < 52 53% hapatic, 18% lung p LaVoie et al.
CD-1 on days 1,8, and 15 after weeks tumours; control: 6% hepatic yes/val (1987)
newborn birth (total dose) tumours, no lung tumours
99.5% Rat, f 35 Intrapulm. 0.1, 03 and 1 mg/animal, 110/113/ 0/35, 1/35, and 9/35 p Deutsch
Osborne/ 1x 112 pulmonary carcinomas; 1/35, yes/val Wenzel et
Mendel weeks 2/35, and 4/35 pleomorphic al.(1983)
sarcomas; control: no tumours
Hamster, m 47 Intra- 0.5 and 0.5 mg/animal < 110 0/47 and 1/47 tracheal n Sellakumar &
Syrian tracheal per week, 30 weeks weeks tumours; control: no tumours yes/val Shubik (1974)
golden
Berizo[j]fluoranthene
Highly Mouse, f 20 Dermal 0.1 and 0.5%, 3x/week, < 9 and 7 100%/95% with skin p Wynder &
purified Swiss life months carcinomas no/ld Hoffmann
(1959b)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
96% Mouse, f 40 Dermal 3.4, 5.6, 9.2 µg/animal, < 2 years 3, 3, and 5% with local q Habs et al.
NMRI 2x/week, life tumours;controls: 0% yes/val (1980)
> 99% Mouse, f 20 Dermal 3, 10, 100 µg, 10x over 20 24 weeks 30, 55, and 95% with tumours p LaVoie et al.
Cr1:CD1 days; initiation experiment (papillomas/keratinizing yes/val (1982b)
(ICR)BR lesions); 1 malignant lymphoma
Mouse, f 20 Dermal 25, 75 µg, 10x over 20 24 weeks 70 and 90% with papillomas; p Rice et al.
CD-1 days; initiation experiment vehicle control: 10% yes/val (1987)
99% Mouse, m/f 21/18 i.p. 278 µg/animal in DMSO < 52 81% males and 22% females p LaVoie et al.
CD-1 on days 1, 8, and 15 after weeks with liver and lung tumours; yea/val (1987)
newborn birth (total dose) control: 6%:0%
99.9% Rat, f 35 Intrapulm. 0.8, 4, and 20 mg/kg, 1x 110/117/ 1/35, 3/35 and 18/35 p Deutsch-
Osborne/ 89 weeks pulmonary carcinomas; yes/val Wenzel et
Mendel control: no tumours al.(1983)
Benzo[ghi]fluoranthene
Highly Mouse, f 20 Dermal 0.1 and 0.5%, 3x/week, < 13 No skin tumours n Wynder &
purified Swiss life months no/ld Hoffmann
(1959b)
Mouse, f 20 Dermal 1 mg, 1x; initiation 4/20 papillomas, no n Van Duuren et
Swiss experiment carcinomas; promotor only: no/val al. (1966)
ICR/Ha 5/20, 1/20
Benzo[k]fluoranthene
Highly Mouse, f 20 Dermal 0.1 and 0.5%, 3x/week, < 13 0/20 and 2/20 skin papillomas q Wynder &
purified Swiss life months no/ld Hoffmann
(1959b)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 25 Dermal 1 mg/animal (total dose) < 2 years No skin tumours; spontaneous n Mohr (1969)
NMRI in 50 aliquots tumours: 10% na/val
> 96% Mouse, f 40 Dermal 3.4, 5.6, 9.2 µg/animal, < 2 years 3, 0 and 0% with local n Habs et al.
NMRI 2x/week, life tumours; control: no tumours yes/val (1980)
> 99% Mouse, f 20 Dermal 3, 10, 100 µg, 10x over 24 weeks 5, 25, and 75% with tumours p LaVoie et al.
Cr1:CD1 20 days; initiation (papillomas/keratinizing yea/val (1982b)
(ICR)BR experiment lesions)
Mouse m/f 16/14 s.c. 0.6 mg/animal, 1x/month, approx. 8/16 m and 5/14 f with local p Lacassagne et
XVII nc/Z 3 months 200 sarcomas no/ld al.(1963a)
days
> 99% Mouse, m/f 16/18 i.p. 530 µg/animal in DMSO < 52 19% males and 17% females q LaVoie et al.
CD-1 on days 1, 8, and 15 after weeks with tumours; control: 6%:0% yes/val (1987)
newborn birth (total dose) liver and lung tumours
99.5% Rat, f 27-35 Intrapulm. 0.65, 3.4, and 17 mg/kg, 114/95/98 0/35, 3/31 and 12/27 p Deutsch-
Osborne/ 1x weeks pulmonary carcinomas; yes/val Wenzel (1983)
Mendel control: no tumours
Benzo[a]fluorene
Mouse, 20 Dermal 0.3%, 2x/week, life < 20 No skin tumours; 4/20 lung q Badger et al.
stock' months adenoma; 1/20 sebaceous no/ld (1942)
adenoma
> 99.5% Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks 2/20 skin tumours; control: n LaVoie et al.
Swiss initiation experiment 1/20 yes/val (1981c)
Ha/ICR
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 10 s.c. 5 mg/animal, at intervals < 23 1/10 lung adenoma; no n Badger et al.
stock' of a few weeks, life months sarcomas no/ln, (1942)
ld
Benzo[b]fluorene
99.5% Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks 4/20 skin tumours; control: q LaVoie et al.
Swiss initiation experiment 1/20 yes/val (1981c)
Ha/ICR
Benzo[ghi]perylene
Mouse, f 50 Dermal 0.38% solution in benzene, 2/50 with skin tumours; n Lijinsky &
Swiss 3x/week, life control: 1/59 skin carcinomas no/val Saffiotti (1965)
Chromatography Mouse, f 20 Dermal 0.05% and 0.1 %, 3x/week, 15 1/20 and 0/20 skin papillomas; n Hoffmann &
purified Swiss Ha/I 12 months months solvent control: no tumours no/val Wynder (1966)
CR/Mil
Chromatography Mouse, f 30 Dermal 25 µg/animal, 10x over 28 6 months 2/30 papillomas; control: 2/30 n Hoffmann &
purified Swiss Ha/ days; initiation experiment 0/20 no/val Wynder (1966)
ICR/Mil
Mouse, f 50 Dermal 20 µg, 2 mg and 4 mg/ < 22.5 3/50, 6/50, and 4/50 with n Muller (1968)
NMRI animal, 2x/week, 25 weeks months tumours; vehicle control: 7/50 no/val
Mouse, f 50 Dermal 1 and 2 mg/animal, 1x; < 22.5 5/50 and 4/50 with tumours; n Muller (1968)
NMRI initiation experiment months vehicle control: 7/50 no/val
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rigourously Mouse, f 20 Dermal 0.8 mg/animal, 1x; 12-13 3/20 papillomas, 1/20 n Van Duuren et
purified Swiss initiation experiment months squamous-cell carcinoma; no/val al. (1970)
vehicle control: 1/20 with
2 papillomas
Highly Mouse, f 50 Dermal 5.5 and 16.5 µg/animal, 33 weeks No tumours n Goldschmidt et
purified ICR/Ha 3x/week, 33 weeks no/lc al. (1973
Highly Mouse, f 50 Dermal 21 µg/animal, 3x/week, 52 weeks No skin tumours; solvent n Van Duuren &
purified Swiss 52 weeks control: no tumours no/val Goldschmidt
ICR/Ha (1976)
Mouse, f 50 s.c. 0.83 and 16.7 mg/animal, < 22.5 5/50 and 4/40 with tumours; n Muller (1968)
NMRI 1 x/2 weeks, 6 months months control:4/50 no/val
Mouse, f 20 s.c. 0.1, 1 and 10 mg/animal, < 22 No skin or subcutaneous n Muller (1968)
NMRI 1x/2 weeks, 20 weeks months tumours;other tumours same no/val
as control
98.5% Rat, f 34-35 Intrapulm. 0.65, 3.4, and 17 mg/kg, 109/114/ 0/35, 1/35 and 4/34 n Deutsch-
Osborne/ 106 pulmonary carcinomas effect yes/val Wenzel et
Mendel 1x weeks of impurity suggested; al.(1983)
control: no tumours
Benzo[c]phenanthrene
Mouse, 20 Dermal Not specified < 676 7 epitheliomas, 5 papillomas p Barry et al.
days no/ld (1935)
Mouse, 40 Dermal 0.3%, 2x/week, < 19 < 19 1 papilloma, 4 squamous cell q Badger et al.
months months no/ld (1940)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 20 Dermal 0.5% solution, 2x/week, 638 days 3 carcinomas, 2 sarcomas; q Stevenson &
C3H 638 days control: no tumours no/val von Haam
(1965)
Mouse, f 30 Dermal 91 and 457 µg, 1x; 21 weeks 5/30 and 11/30 papillomas; p Levin et al.
CD-1 initiation experiment control:no tumours (1980)
Mouse 10 s.c. 5 mg at intervals of < 15 No injection-site tumours n Badger et al.
several weeks, life months no/ln, (1940)
ld
Mouse 20 s.c. 5 mg in tricaprylin, 1x 638 days 3 sarcomas; controls: no q Stevenson &
C3H tumours no/val von Hearn
(1965)
Rat, 6 s.c. 5 mg/animal; several approx. 1/6 sarcoma at injection site q Badger et al.
repeated doses 18 months no/ln, (1940)
ld
Benzo[a]pyrene
Mouse, f 15 Oral 3 mg/animal in sesame 30 weeks Increased pulmonary tumours p Wattenberg &
A/HeJ oil, 2x (16.6); control: 0.3 yes/val Leong(1970)
Mouse, f 15 Oral 2 mg/animal, 3x, every 26 weeks 15/15 with forestomach p Sparnins et
A/J 2 weeks tumours and 15/15 with yes/val al. (1986)
pulmonary adenomas;no
control
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 25-73 Oral 0.004-1 mg/animal per 140-200 Dose-dependent gastric p Neal & Rigdon
CFW (diet) day, < 110-165 days days tumours (0-90%);control: no no/val (1967)
tumours
Mouse, m/f 9-26 Oral 1-20 mg/animal/day, 150-300 Dose-dependent gastric p Neal & Rigdon,
CFW (diet) < 1-30 days days tumours(0-100%); control: no/val (1967)
no tumours
Mouse, m/f 60-175 Oral 0.25 and 1 mg/g food, < 34 33 and 61 % with stomach p Rigdon & Neal
White (diet) < 34 weeks weeks tumours; 53 and 20% with no/val (1966)
Swiss lung tumours;controls: 1 and
21%
Mouse, f 20-30 Dermal 0.001, 0.005, and 0.01%, < 21, 14, 3 and 43%, 63 and 73%, and p Wynder &
Swiss 3x/week, life and 11 951% and 95% with skin no/ld Hoffmann
carcinomas/papillomas (1959a)
Mouse, f 20 Dermal 0.01, 0.05 and 0.5%, < 12, 6, 85, 95, and 75% with skin p Wynder &
Swiss 3x/week, life and 6 carcinomas no/ld Hoffmann
Millerton months (1959b)
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1%, 3x/week, 15 17/20 and 19/20 skin tumours; p Hoffmann &
Swiss 12 months months solvent control: no tumours no/val Wynder (1966)
Ha/ICR/Mil
Mouse, f 30 Dermal 25 µg/animal, 1/x over 28 6 24/30 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment months only: 2/30 no/val Wynder (1966)
Ha/ICR/Mil
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 50 Dermal 20 and 200 µg/animal, < 22.5 50/50 and 50/50 with skin p Muller (1968)
NMRI 2x/week, 25 weeks months tumours;vehicle control: 7/50 no/val
Mouse, 20-30 Dermal (a) 0.00002% in (a) 21% malignant tumours; p Bingham & Falk
C3H/He n-dodecane/decalin; (b) 50% tumours (three orders yes/val (1969)
(b) 0.02% in decalin, of magnitude difference in dose)
3x/week, 50 weeks
Mouse, f 30 Dermal 5 µg/animal, 10x, 20 days; 24 weeks 19/29 tumour-bearing animals p Hoffmann et al.
Swiss initiation experiment with 67 skin tumours; control: no/val (1972)
Ha/ICR/Mil 1/30
Mouse, f 20 Dermal 0.05 and 0.1 mg/animal; 6 months 13 and 18 with skin tumours; p Masuda &
Swiss ICR 60x no solvent control no/val Kagawa (1972)
Mouse, f 20 Dermal 5 µg/animal, 3x/week, < 72 13/20 with 22 skin tumours; p Hecht et al.
Swiss 72 weeks weeks 4/20 with 4 carcinomas; no/val (1974)
Ha/ICR/Mil solvent control: no tumours
Mouse, f 50 Dermal 5 µg/animal, 3x/week, life 440 days 16 animals with 26 tumours; p Van Duuren &
Swiss control: no tumours no/val Goldschmidt
ICR/Ha (1976)
Mouse, f 20 Dermal 5 and 10 µg/animal, 62 weeks Low dose: 10/20 with 19 skin p Hecht et al.
Swiss 3x/week, 62 weeks tumours, 7/20 with 8 yes/val (1976b)
Ha/ICR carcinomas; high dose:18/20
with 70 skin tumours, 14/20
with 16 carcinomas; solvent
control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
99.9% Mouse, f 40 Dermal 100 µg/animal, 2x/week, 70 weeks 79% skin-tumour bearing P Cavalieri et al.
Swiss 30 weeks animals;solvent control: no no/val (1977)
tumours
Mouse, f 40 Dermal 1.7, 2.8, 4.6 µg/animal, < 2 years 24, 69 and, 61 % with local p Habs et al.
NMRI 2x/week, life tumours(high rate of systemic yes/val (1980)
tumours);control: no tumours
> 99.5% Mouse, f 20 Dermal 30 µg/animal, 10x on 24 weeks 93% with tumours; vehicle p LaVoie et al
Swiss alternate days; initiation control: no tumours no/val (1981b)
Ha/ICR experiment
Mouse, f 20 Dermal 3 µg, 10x over 20 days; 24 weeks 85% with tumours (papillomas/ p LaVoie et al.
Cr1:CD1 initiation experiment keratinizing lesions) yes/val (1982b)
(ICR)BR
> 96% Mouse, f 20 Dermal 2 and 4 µg/animal, 648 and 45% (10% papillomas/35% p Habs et al.
NMRI 2x/week, life 528 days carcinomas) and 85% yes/val (1984)
(mean) (0%/85%) with skin tumours;
control: no tumours
99.5% Mouse, m 50 Dermal 12.5 µg/animal, 2x/week, < 99 94% with malignant skin p Warshawsky &
CH3/HeJ 99 weeks weeks tumours;solvent control: no no/val Barkley (1987)
tumours; untreated control: no
tumours
Mouse, f 24 Dermal 0.8 µmol/mouse, 1x; 24 weeks Enhanced incidence of skin p Cavalieri et al.
Sencar initiation experiment papillomas (80-92%) no/val (1988b)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m 12 Dermal 1.2 mg/animal, 6 days/wk, < 27 Multiple tumours; p Shubik & Della
Swiss 19 weeks weeks squamous-cell carcinomas no/ln Porta (1957)
> 99% Mouse, f 20 Dermal 2.5 µg/animal, 10x over 20 24 weeks 89% tumours, 5.5 skin p Rice et al.
CD-1 days; initiation experiment tumours/animal; control: 5% yes/val (1988b)
> 99% Mouse, f 25 Dermal 2.5 µg/animal, 10x over 20 23 weeks 96% tumours, 3.4 skin p Rice et al.
CD-1 days; initiation experiment tumours/animal; control: no yes/val (1990)
tumours
Chromatography Mouse, f 23-24 Dermal 8.4, 25.2 and 75.7 µg/ 15 weeks 10/23, 17/24 and 21/23 with p Cavalieri et al.
purified Sencar animal, 1x; initiation tumours; control: no tumours yes/val (1991)
experiment
Chromatography Mouse, f 24 Dermal 1, 5 and 25 µg/animal, 1 x; 27 weeks 1/24, 10/24 and 22/24 with p Cavalieri et al.
purified Sencar initiation experiment tumours; control: no tumours yes/val (1991)
Chromatography Mouse, f 24 Dermal 25 µg/animal, 1x; initiation 27 weeks 1/24 with tumours p Cavalieri et al.
purified Sencar experiment without yes/val (1991)
promotion
HPLC Mouse, f 43-50 Dermal 16, 32, or 64 µg/animal, < 35 1, 1.5 and 7.5 tumours/animal p Albert et al.
control ICR/Harlan 1x/week, 29 weeks weeks after 35 weeks no/val (1991a)
Mouse, m 20 Dermal 100 µg/animal, 2x/week, Tumours from 15 weeks p Andrews et al.
Balb/c 3 weeks-5 months onwards no/val (1991)
Mouse, m/f 40-50 s.c. 0.09 mg/animal in < 22-28 16/21 sarcomas after 5 p Steiner (1955)
C57BI tricaprylin; 1x months months yes/ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 14/16 s.c. 0.6 mg/animal, 1x/month, > 129/160 13/14 m and 8/16 f with local p Lacassagne et
XVII 3 months days sarcomas no/ld al. (1958)
(average
latency)
Mouse, m/f 154/ s.c. 0.6 mg/animal, 1x/month, approx. 154/154 m and 112/162 f with p Lacassagne et
XVII nc/Z 162 3 months 110/150 local sarcomas no/ld al. (1963a)
days
Mouse, 20 s.c. 0.1, 1 and 10 mg/animal, 17, 7, 6 All animals with sarcomas at p Muller (1968)
NMRI 1 x/2 weeks, 20 weeks months injection site no/val
Mouse, f 90 s.c. 25, 50, 100, 200 and 400 < 16 25, 50, 55, 75 and 65% with p Pott et al.
NMR1 µg/animal, 1x months tumours; solvent control: no/val (1973)
< 5%
Mouse, m/f 31-38 s.c. 0.01 and 0.1 mg/animal, 30 weeks 16 and 64% with lung p Rippe & Pott
newborn 1x tumours; control: 13% with no/val (1989)
lung tumours
> 99% Mouse, m/f 17/14 i.p. 278 µg/animal in DMSO < 52 76% hepatic and 64% lung p LaVoie et al.
CD-1 on days 1, 8, and 15 after weeks tumours; control: 6% hepatic yes/val (1987)
newborn birth (total dose) tumours, no lung tumours
> 99% Mouse, m/f 28/27 i.p. 59.5 µg/animal on days 26 weeks 46 m, 70, f with lung tumours; p Busby et al.
Swiss- 1, 8, and 15 after birth vehicle control: 14 m, 7 f yes/val (1989)
Webster (total dose)
BLU:Ha(ICR),
newborn
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 10 i.v. 10 mg/kg, 1 × 20 weeks 100% lung tumours; control: p Shimkin &
A 21% no/val Stoner (1975)
Mouse, f 19-22 Intra- 0.05 and 0.15 mg/animal, 27 and 42% with carcinomas p Pott et al.
NMRI tracheal 20x in the respiratory tract; na/val (1978)
control: 9%
99% Mouse, f 45; Intra- 1 mg/animal, 1x/week, < 18 No colonic tumours, 73% lung p Anderson et al.
ICR/Ha 60 colonic 14 weeks months tumours, 94% forestomach yes/val (1983)
contr. instill- tumours, 7% subcutaneous
ation sarcomas, 23% mammary
carcinomas; control: 25% lung
tumours, 20% forestomach
tumours, 9% mammary
carcinomas, no subcutaneous
sarcomas or colonic tumours
99% Mouse, f 38; Intra- 1 mg/animal, 1x/week, < 18 No colonic tumours, 94% fore- p Anderson et al.
C57BI/6 45 colonic 14 weeks months stomach tumours, 16% yes/val (1983)
instill- peritoneal sarcomas, 28%
ation lymphomas; control: 21%
forestomach tumours, no sarcomas
or lymphomas or colonic tumours
Rat, f 9 Oral 100 mg/kg, 1x 60 days 8/9 with mammary tumours; p Huggins &
Sprague- control: 8/164 in 310 days no/ln, Yang(1962)
Dawley lc
Rat f 20 Oral 625 mg/animal, 1x/week, 90 weeks 67-77% with mammary p McCormick et
LEW/Mai 8x; 50 mg/animal, 1 × tumours; control: 30% yes/val al. (1981)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rat, f 50 s.c. 33, 100, 900, and 2700 < 16 10, 15, 70 and 75% with p Pott et al.
Wistar µg/animal, 1x months tumours; solvent only: < 5% no/val (1973)
Rat, f 37 i.p. 5 mg/animal; 1x; in bees' (a) 89% abdominal tumours p Roller et al.
Wistar wax/tricaprylin 25/75 (a) 2 years (mesotheliomas, sarcomas); no/val (1992)
or saline (b) (b) 50%; vehicle controls:
(a) 70%; (b) 3%
Rat, f 13-17 Intra- 0.5, 1, or 2 mg/animal in Life 7, 65, and 92% with lung p Davis et al.
Wistar tracheal infusion; 1x/2 weeks; 18x tumours; control: no tumours yes/val (1975)
Rat, m/f 15/15 Intra- 1 mg/animal, 1x/week, Life 3/13 (m) and 3/14 (f) with p Ishinishi et al.
Wistar tracheal 15x (mean, malignant lung tumours no/val (1976)
491/540 (mean: 22.2%); vehicle control:
days) 0%
Rat, f 36-40 Intra- 1 mg/animal; 20x 19% with lung tumours; p Pott et al.
Wistar tracheal control: no tumours no/val (1987)
Rat, m/f 20/20 Intra- 7 mg/kg, every 14 days, < 781 19/20 m and 18/20 1 with lung p Steinhoff et al.
Sprague- tracheal 22x (total dose: 154 days tumours; vehicle control: 0% no/val (1991)
Dawley mg/kg)
99.1% Rat, f 35 Intrapulm. 0.1, 0.3, or 1 mg/animal, 111/77/54 4/35, 21/35 and 33/35 p Deutsch-
Osborne/ 1x weeks pulmonary carcinomas; 6/35, yes/val Wenzel et
Mendel 2/35 and 0/35 pleomorphic al.(1983)
sarcomas; control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rat, f 35 Intrapulm. 0.05, 0.1, or 0.2 11, 17, and 46% with tumours; p Grimmer et al.
Osborne/ mg/animal, 1X control: no tumours yes/val (1987)
Mendel
99.6% Rat, f 35 Intrapulm. 0.03, 0.1, or 0.3 mg/ < 135 8.6, 31.4, and 77.1% tumour p Wenzel-
Osborne/ animal, 1x weeks incidence; control: no tumours yes/val Hartung et al.
Mendel (1990)
Rat, m 14-15 Intrapulm. 50, 100, or 200 µg/animal < 100 0/10, 3/10 and 4/9 lung p Horikawa et al.
Fischer weeks tumours; control: no tumours no/val (1991)
344/Du Crj
Rat, 94 Intrabr- approx. 3-5 mg/animal, 1x approx. Carcinoma incidence: 17% p Laskin et al.
onchial 5 months no/lc (1970)
pellet
> 99% Rat, f 20 Intra- 1 and 4 mg into 5th 20 weeks 50 and 80% with mammary p Cavalieri et al.
Sprague- mammary mammary gland, 1x tumours; control: no tumours no/val (1988a)
Dawley
Rat f 20 Intra- 63 and 252 µq/gland, 1x, < 24 7/20 and 9/20 with mammary p Cavalieri et al.
Sprague- mammary 8 glands weeks tumours; control: 1/18 yes/val (1991)
Dawley injection
Recrystallized Rat, f 20 Pellet 0.5 and 1 mg; 1x 28 12 and 65% with carcinomas p Topping et al.
Fischer 344 implant- (implanted into tracheal months in tracheal transplants yes/val (1981)
ation transplants)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Hamster, m/f 13 Oral 2.5 mg/animal, 4 < 14 9/13 with forestomach cancer; p Chu &
Syrian (diet) days/week, < 14 months months 2/13 with papillomas no/val Malmgren
golden (1965)
Chromatography Hamster, m/f 15/15 Dermal 4 drops of a 0.8% solution < 99/68 m:1 small nodular melanotic q Shubik et al.
control Syrian in mineral oil, 1x/week, 8 weeks lesion, 2 malignant no/ln, (1960)
golden weeks including a 30-week lymphomas; f: no tumours ld
interval
Chromatography Hamster, m/f 5/5 Dermal 6 drops of 0.01% solution < 70 No skin tumours n Shubik et al.
control Syrian in acetone, 2x/week, 40 weeks no/ln, (1960)
golden weeks ld
Hamster, m 5 or Dermal 20 mmol/litre solution, < 44 10% buccal pouch carcinomas p Solt et al.
Syrian 28 (buccal painting 2x/week, 5 or 20 weeks after 40 week; control: no no/val (1987)
golden pouch) weeks tumours
Hamster, m 10 Inhalation 4.5 h/day, 5 days/week, Life No tumours n Thyssen et al.
Syrian 9.8 mg/m3 for 16 weeks or no/ld (1980)
golden 44.8 mg/m3 for 10 weeks
Hamster, m 24 Inhalation 2.2, 9.5, or 46.5 mg/m3, 109 Dose-dependent tumours in p Thyssen et al.
Syrian 4.5 h/day in the first 10 weeks nasal cavity, pharynx, larynx, no/val (1981)
golden weeks, thereafter 3 h/day, and trachea; also in oesophagus
109 weeks and forestomach (papillomas,
polyps, squamous-cell
carcinomas); no lung tumours;
larynx most affected with 0,
31 and, 52% incidence; control:
no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Pure Hamster, m/f 30/30 Intra- 3 mg/animal, 1x/wk, 15 < 45/60 14/19 and 21/21 with tumours p Saffiotti et al.
Syrian tracheal week (mixed with inert weeks in respiratory tract; control: no/val (1968)
golden dust of haematite no tumours
[ferric oxide]
Hamster, m 30 Intra- 3 mg/animal, 1x/week, < 74 All with bronchioalveolar p Crocker et al.
Syrian tracheal 14 weeks weeks metaplasia; 5/19 no/val (1970)
golden squamous-cell carcinomas,
3/19 adenomas, 1/19 tracheal
tumours
Pure Hamster, m/f 30-50 Intra- 0.25, 0.5, 1, or 2 mg/ Life Dose-related increase in p Saffiotti et al.
Syrian tracheal animal, 1x/week, 30 wks respiratory tract tumours; no/val (1972)
golden (mixed with inert dust of control: no tumours
ferric oxide)
Pure Hamster, m 30 Intra- 0.0625, 0.125, 0.25, 0.5, 78 weeks Dose-related increase in p Feron et al.
Syrian tracheal and 1 mg/animal, 1x/week, respiratory tract tumours no/val (1973)
golden 52 weeks (3-26%); controls: no tumours
Hamster, m/f 25/25 Intra- 0.9 mg/animal per week, < 100 17% (8/46) tumours in p Henry et al.
Syrian tracheal 30 weeks weeks respiratory tract; control: no/val (1975)
golden no tumours
Hamster, Intra- 0.3 or 0.9 mg/animal, < 2 years 17 and 68% with tumours p Pott et al.
Syrian tracheal 1x/week, 20 weeks no/lc (1978)
golden
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Hamster, m 29 Intra- 0.125, 0.25, 0.5, or Life 31, 83, 66, and 31% tumours p Ketkar et al.
Syrian tracheal 1 mg/animal, 1x/week, life in respiratory tract; control; no/val (1979)
golden no tumours
Hamster, m 30 Intra- 5, 20 or 40 µg/animal, Life 4/28, 5/27 and 7/28 with q Kunstler (1983)
Syrian tracheal every 2 weeks, life meta plasia in respiratory no/val
golden tract, malignant neoplasm
and 1 adenoma in high-dose
group; controls: 1/29 or 3/30
Hamster, 97 Intra- 3-5 mg 63/97 with lung cancers p Laskin et al.
bronchial no/val (1970)
pellets
Hamster, Tracheal approx. 0.83 mg/animal, Tracheal papillomas and p Mohr (1971)
Syrian insuff- 3x/week, 1 year carcinomas no/lc
golden lation
Hamster, Bronchial 150 days > 90% with focal cancers p Benfield &
Syrian implants no/lc Hammond
golden (1992)
Dog Paren- > 8 First parenchymal cancer after p Benfield &
chymal months 8 months; 7/12 dogs with no/lc Hammond
implants tumours (1992)
Pig, m/f 1/1 i.m. 4.8 mg/kg, 1x; 6 months 12 No sarcomas n Kallistratos &
German later 2.1 mg/kg, 1x months no/val Pfau (1971)
Edelland-
schwein
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Pig, m/g 1/1 i.m. 6.3 mg/kg, 1x; 6 months 12 No sarcomas n Kallistratos &
mini later 1.9 mg/kg, 1 × months no/val Pfau (1971)
Cattle, m/f 1/1 i.m. 0.95 mg/kg, 1x; 6 months 29 No sarcomas n Kallistratos &
German later 0.75 mg/kg, 1x months no/val Pfau (1971)
black/white
Monkeys m/fm 1/1 s.c. 10 mg/animal, 1x (a) >18 (a) 1/2 with local tumours q Noyes (1969)
(a) /f 1/1 (coadministration with months (b) death within 5 weeks no/ln
Saguinus 10 mg DMBA at other site) (b) < 5
oedipus; weeks
(b) S.
fusciocollis
Monkey, s.c. 1 × (not specified) Fibrosarcomas p Adamson &
Galago no/lc Sieber (1983)
crassus
Monkey, 17 s.c. 30-90 mg/kg, multiple < 18 No tumours observed; n Adamson &
Old world administration (not years survival: 9/17 no/lc Sieber (1983)
specified)
Monkey, m/f 4/2 Intra- 3-15 mg, 1x/week (with 67-69 Bronchioalveolar metaplasia; p Crocker et al.
Galago tracheal ferric oxide), up to 69 weeks 2/3 squamous carcinomas no/val (1970)
crassust weeks arising from bronchus
Benzp[e]pyrene
Mouse, f 20 Dermal 0.1%, 3x/week, life < 13 2/20 papillomas, 3/20 q Wynder &
Swiss months carcinomas no/ld Hoffmann
Millerton (1959a)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 20 Dermal 1 mg, 1x; initiation 64 weeks 2/20 with papillomas; pure q Van Duuren et
Swiss experiment substance: no tumours no/val al. (1968)
ICR/Ha
TLC Mouse, f 20 Dermal 2.5 mg/animal, 1x; 35 weeks 85% with papillomas; p Scribner (1973)
purified CD-1 initiation experiment promotor only: 3% no/val
Highly Mouse, f 50 Dermal 15 µg/animal, 3x/week, 368 days No tumours observed; control: n Van Duuren &
purified ICR/Ha 368 days no tumours no/val Goldschmidt
(1976)
> 99% Mouse, f 30 Dermal 100 µg/animal, 2x/week, 30-40 At 30 wks: 68% papillomas, p Slaga et al.
CD-1 30 weeks weeks at 40 weeks: 24% carcinomas no/val (1979)
> 99% Mouse, f 30 Dermal 100 and 252 µg/animal, 30-40 High dose: at 30 weeks, 19% q Slaga et al.
CD-1 1 x; initiation experiment weeks papillomas; at 40 weeks, no no/val (1979)
carcinomas;vehicle control: et
30 weeks, 14% papillomas
99% Mouse, f 30 Dermal 0.25, 0.63, or 1.5 mg/ 26 weeks 15, 11, or 140% with q Buening et al.
CD-1 animal, 1x;initiation papillomas; vehicle control: no/val (1980)
experiment 7% papillomas
> 95% Mouse, f 30 Dermal 0.5 mg/animal, 1 x; 15 weeks 17% with papillomas; vehicle q Slaga et al.
Sencar initiation experiment control:10% no/val (1980, 1981)
99% Mouse, m/f 30/30 i.p. 0.1, 0.2, 0.4, or 0.2, 0.4, 62-66 21/35 (m), 0/35 (f) or 12/30 q Buening et al.
Swiss-Webster 0.8 mg on days 1, 8, and weeks (m), 0/30 (f) with hepatic no/val (1980)
BLU:Ha(ICR), 15 of life tumours; controls: 11/53 (m),
newborn 0/24 (f)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
99.7% Rat, f 30-35 Intrapulm. 0.8, 4.2, or 20 mg/kg, 1x 117/111/ 1 pulmonary sarcoma at n Deutsch-
Osborne/ 104 4.2 mg/kg; 1 squamous-cell yes/val Wenzel et
Mendel weeks carcinoma at 20 mg/kg; no al.(1983)
tumours in controls
Recrystallized Rat, f 20 Tracheal 1 mg, 1x 28 No tumours n Topping et al.
Fischer pellet months yes/val (1981)
344
Chrysene
Mouse 100 Dermal 1% in 90% benzene < 11 No tumours n Kennaway
months no/ld, (1924)
lc
Purified Mouse Dermal 7.5% in liquid paraffin or 78 or 50 6 or 18 benign, 1 or 9 q Bottomley &
oleic acid, 5x/week, 78 or weeks malignant tumours no/lc Twort (1934
50 weeks
Doubtful Mouse 100 Dermal (a) 0.3% in benzene or < 704 (a) 1/100 papilloma and 1/100 n Barry et al.
Purity 20 (b) 0.3% ln mouse fat, days epithelioma, (b) no tumours no/ld (1935)
2 x/week, life
'Synthesized' Mouse 20 Dermal 0.3% (pure), 2x/week, 440 days No tumours n Barry et al.
440 days no/lc, (1935)
ld
Mouse 50 Dermal (a) 0.3% in benzene, < 797 (a) 2/50 papillomas, n Barry et al.
100 (b) 7.5% in oleic acid, days (b) no tumours no/lc, (1935)
2x/week, life ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
'Pure' Mouse 50 Dermal In benzene, 2x/week, < 276 After 276 days at 11/50 n Schurch &
276 days days survivors, no tumours no/lc, Winterstein
ld (1935)
Mouse, m/f 10/10 Dermal 40 µg/animal, 2x/week, 31 weeks 1/15 carcinomas n Riegel et al.
CF1 31 weeks no/ld (1951)
Mouse, f 20 Dermal 1%, 3x/week, life < 12 9/20 papillomas, 8/20 p Wynder &
Swiss months carcinomas; no solvent no/ld Hoffmann
control (1959a)
Mouse, f 20 Dermal 1 mg, 1x; initiation 63 weeks 16/20 papillomas, 2/20 p Van Duuren et
Swiss experiment carcinomas; promotor only: no/val al. (1966)
ICR/Ha 5/20, 1/20
TLC Mouse, f 30 Dermal 1 mg/animal, 1x; initiation 35 weeks 73% with papillomas; p Scribner(1973)
purified CD-1 experiment promotor only:3% no/val
Mouse, m 20 Dermal 75 µg/animal in decalin, 82 weeks 1/12 papillomas; solvent n Horton &
C3H 2x/week, 82 weeks control:2/13 papillomas no/ld Christian (1974)
Mouse, m 20 Dermal 75 µg/animal in decalin/ 82 weeks 5/19 papillomas; 12/19 p Horton &
C3H dodecane 50/50, 2x/week, carcinomas; solvent control: no/val Christian (1974)
82 weeks; co-carcinogenicity 2/13 papillomas
experiment
> 99.9% Mouse, f 20 Dermal 0.1 mg/animal/day, 110x; 22 weeks 11/18 papillomas/carcinomas; p Hecht et al.
Swiss initiation experiment chrysene only: 4/11 after 72 no/val (1974)
Ha/ICR/Mil weeks; solvent control: no
tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 30 Dermal 0.09, 0.29 and 0.91 mg/ 26 weeks 25, 43 and 52% papillomas; p Levin et al.
CD-1 animal, 1x; initiation promotor only: 7% no/val (1978)
experiment
95% Mouse, f 30 Dermal 0.46 mg/animal, 2x; 26 weeks 21/30 papillomas; promotor p Wood et al.
CD-1 initiation experiment only: 1/30 no/val (1979)
98% Mouse, f 30 Dermal 0.57 mg/animal, 1x; 27 weeks 80% papillomas; promotor p Wood et al.
CD-1 initiation experiment only: 4% yes/val (1980)
> 95% Mouse, f 30 Dermal 0.46 mg/animal, 1x; 15 weeks 21/29 papillomas; promotor p Slaga et al.
Sencar initiation experiment only: 3/30 no/val (1980,1981)
Mouse, f 30 Dermal 0.09 and 0.274 mg/animal, 26 weeks 43, 43% (or 39%) with skin p Chang et al.
CD-1 1 x; initiation experiment papillomas; vehicle control: yes/val (1983)
100%
> 99% Mouse, f 20 Dermal 3.4, 11.4 and 34 µg/ 24 weeks 25, 90 and 95% with tumours; p Rice et al.
CD-1 animal, 10x over 20 days; 0.5, 3, and 4.5 skin tumours/ yes/val (1988b)
initiation experiment animal; control: 20%
Mouse, f 20 Dermal 7.5 µg/animal, 1x; 21 weeks 10% with skin tumours; n Amin et al.
CD-1 initiation experiment solvent control:10% yes/val (1990)
Mouse, m/f 16/16 Dermal 365 µg/animal, 1x; < 100 No skin tumours; solvent n Bhatt &
Sencar initiation experiment weeks control: no tumours no/val Coombs (1990)
Purified Mouse 50 s.c. 2 mg/animal, 1x < 35 No tumours n Bottomley &
weeks no/lc Twort (1934)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Purified Mouse, 30 s.c. 10 mg/animal, 2x 15 No tumours n Shear & Leiter
Jackson A (4-month interval) months no/ld (1941)
Spectrometer Mouse, m/f 50 s.c. 5 mg/animal in tricaprylin; < 22 4/39 sarcomas after 4 months; p Steiner & Falk
control C57BI 1x months solvent control: 3/280 no/val (1951)
Mouse, m/f 40-50 s.c. 5 mg/animal in tricaprylin; < 22-28 5/22 sarcomas after 5 months p Steiner (1955)
C57BI 1x months yes/ld
Mouse, m 20 s.c. 1 mg/animal in arachis oil, 60-80 2/20 injection site tumours; p Boyland & Sims
C57BI 1 x/week, 10 weeks weeks control: no tumours no/val (1967)
Mouse, m/f 104 s.c. 0.1 mg/animal in poly- 70-75 70 weeks: 13/27m liver, 1/27 q Grover et al.,
Swiss ethylene glycol on days weeks m and 1/21 f lung tumours; no/val (1975)
newborn 1,2 and 3 after birth vehicle control: 9/30 m liver,
3/30 in and 1/15 f lung tumours
Mouse, 10 s.c. 1 mg, weekly; later 2 mg 350 days No tumours; control: no n Barry & Cook
at longer intervals tumours no/ln, (1934)
ld
Purified Mouse 50 i.p. 2 mg/animal, 1 × < 45 No tumours n Bottomley &
weeks no/lc Twort (1934)
TLC Mouse, m/f 100 i.p. Total dose 0.32 mg/animal 38-42 5/24 m and 2/11 f pulmonary q Buening et al.
control Swiss- in DMSO on days 1, 8 weeks tumours; 6/24 m liver yes/val (1979)
Webster and 15 after birth tumours; 1/24 m
BLU:Ha(ICR) lymphosarcoma; control:
newborn 2/21 m and 7/38 1 lung tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Repurified, Mouse, m/f 80 i.p. 0.045, 0.09 and 0.18 39-41 Males: 4/27 lung and 6/27 p Chang et al.
256°C Swiss- mg/animal in DMSO on weeks liver tumours; females; 1/11 yes/val (1983)
Webster days 1, 8 and 15 after birth lung and 0/11 liver tumours;
BLU:Ha(ICR) vehicle control: no tumours
newborn
> 98% Mouse, m/f 20-29 i.p. 6.3 and 210 µg/animal 26 7/10% and 15/0% m/f with n Busby et al.
Swiss- (total dose) in 3 aliquots weeks lung tumours; vehicle control: yes/val (1989)
Webster on day 1, 8, and 15 after 14/7% m/f
BLU:Ha(ICR) birth
newborn
Rat 10 s.c. 2 mg/animal, weekly; later < 626 4/10 tumours; control: 2/10 p Barry & Cook
6 mg at longer intervals days sarcomas no/ln, (1934)
ld
Purified Rat 10 s.c. 1 mg/animal, weekly, 103 < 103 No tumours n Boyland &
weeks weeks no/ln Burrows (1935)
ld
Rat, 5 s.c. 5 mg/animal, 7-9x 10 No tumours n Pollia (1941)
Wistar months no/ln
99.6% Rat, f 35 Intrapulm. 1 and 3 mg/animal, 1 × < 135 14.3% and 28.6% tumour p Wenzel-
Osborne/ weeks incidence; control: no tumours no/val Hartung et al.
Mendel (1990)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Coronene
> 96% Mouse, f 40 Dermal 5 or 15 µg/animal, < 104 Low dose:1/39, high dose: n Habs et al.
NMRI 4x/week, 104 weeks weeks 2/40 local tumours at yes/val (1980)
application site; vehicle
control: no tumours
TLC Mouse, f 20 Dermal 0.1 mg, 5x; initiation 65 weeks 6/20 papillomas; promotor q Van Duuren et
control Swiss experiment only: 5/20; coronene only: no/val al. (1968)
ICR/Ha no tumours
Cyclopenta[d]pyrene
> 96% Mouse, f 40 Dermal 1.7, 6.8 and 27.2 µg/ 112 Low dose: no tumours; high q Habs et al.
NMRI animal, 2x/week, 112 weeks dose: 2/38 skin carcinomas, yes/val (1980)
weeks 1/38 sarcomas;control: no
tumours
> 98% Mouse, f 30 Dermal 23, 91, 226, 566 µg/ 27 weeks 10, 21, 30, and 37% p Wood et al.
CD-1 animal, 1x; initiation papillomas; promotor only: 4% yes/val (1980)
experiment
> 99.9% Mouse, f 30 Dermal 45, 136 and 407 µg/ 57 weeks Low dose: 17; med. dose: 11; p Cavalieri et al.
Swiss animal, 2x/week, 30 weeks high dose: 7 skin tumours; no/val (1981b)
control: no tumours
> 99.9% Mouse, f 30 Dermal 4.5, 14 and 41 µg/animal, 44 weeks Low dose: 1/30; med. dose: p Cavalieri et al.
CD-1 every other day, 20 days; 9/29; high dose: 6/29 no/val (1981b)
initiation experiment papillomas; promotor only: 3/29
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 30 Dermal 10, 100 and 200 µg/ 26 weeks Low dose: 11 %; med. dose: p Raveh et al.
Sencar animal, 1x; initiation 39%; high dose: 57% no/val (1982)
experiment papillomas; promotor only:
10%
> 99% Mouse, m/f 8-14 i.p. 0.35, 0.7, 1.05, 1.4, and 26 weeks 62, 60, 56, 70, 86, 93%, p Busby et al.
Swiss- 1.75 mg/animal (total dose) 77, 100, and 89, 100% m/f yes/val (1988)
Webster in 3 aliquots on day 1, 8, with lung tumours; vehicle
BLU:Ha(ICR) and 15 after birth control: 8, 8%
newborn
> 99% Rat, f 20 Intra- 1.8 and 5.4 mg into 4th < 34 No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1x weeks control: no tumours no/val (1988b)
Dawley
Dibenz[a,h]anthracene
Mouse m Oral 1.5 mg/animal in PEG-400, 30 weeks 21 % forestomach papillomas; q Berenblum &
Swiss 1 x; initiation experiment promotor only: 14% no/lc Haran (1955)
Mouse m/f 21/21 Drink- 0.8 mg/day/animal in olive 8-9 14/14 m and 13/13 f with p Snell & Stewart
DBA/2 con- ing- oil, 8-9 months months pulmonary adenomas; 14/14 no/val (1962)
trol: water m and 10/13 f with alveologenic
25/10 carcinomas; control: 1 mouse
with tumour
Mouse, f 20 Dermal 0.001, 0.01, and 0.1%, < 21, 13 0.001%: 30% papillomas, p Wynder &
Swiss 3x/week, life or 9 30% carcinomas; 0.01%: no/ld Hoffmann
Millerton months 95/90% papilloma/carcinoma; (1959a)
0.1%: 90%/75% papilloma/
carcinoma
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m < 50 Dermal 0.02 and 0.16 µg/animal, 32 weeks 33 and 38% with skin p Klein (1960)
Swiss 1x; initiation experiment tumours; acetone control: 13% yes/val
albino
DBA/2Jax
Chromatograph Mouse, f 20 Dermal 38 µg/animal, 2x/wk, < 60 80% with skin tumours; p Lijinsky et al.
recrystallized Swiss 44 weeks, weeks vehicle control: 4% no/val (1965)
Mouse, m/f 30/30 Dermal m: 0.3% solution (= 1.5 < 29/22 m: 26% with papillomas after p Johnson (1968)
IF/Bcr mg/animal), 1x/week, 18 weeks 20 weeks, 100% after 29 no/val
weeks; f: 0.5% (= 1 mg/ weeks; f: 100% with breast
animal), 8x, every 2 weeks tumours after 22 weeks
> 99% Mouse, f 50 Dermal 1 drop, 3x/week, 112 112 6%, 8% and 32% with skin p Platt et al.
NMRI weeks; total doses: 37.8, weeks tumours; controls: 2-4% no/val (1990)
125, and 378 µg/animal
> 99% Mouse, f 16 Dermal 83.5 and 167 µg/animal, 24 weeks 38 and 93% with skin tumours; p Platt et al.
NMRI 1x; initiation experiment vehicle control: no tumours no/val (1990)
Mouse 10 s.c./ 0.2 mg/animal, 2x/week, Life 3/10 with subcutaneous p Boyland &
i.p. 50 weeks alternating sarcomas no/ln Burrow, (1935)
ld
Spectrometer Mouse, m/f 50 s.c. 0.02 mg/animal in < 22 28/48 sarcomas after 4 p Steiner & Falk
control C57BI tricaprylin; 1x months months solvent control: 3/280 no/val (1951)
Mouse, m/f 40-50 s.c. 0.02, 0.04 mg/animal in < 22-28 7/21 and 6/18 sarcomas after p Steiner (1955)
C57BI tricaprylin; 1x months 6 and 5 months yes/ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 20/19 s.c. 1 mg/animal, 1x/week, 60-80 20/20 m and 17/19 f with p Boyland & Sims
C57BL 10 weeks weeks sarcomas; control: no no/val (1967)
sarcomas
Mouse, f 60 s.c. 10, 30, 90, 270 and 810 < 16 40, 35, 65, 75, and 90% with p Pott et al.
NMRI µg/animal, 1x months tumours no/val (1973)
Mouse, m/f 30 s.c. 0.15 and 0.3 mg/animal, 12 months B6 mice: 16/30 and 14/30; D2 p Kouri et al.
B6, D2 (60) 1x mice:1/30 and 0/30 with no/val (1983)
fibrosarcomas
> 99% Mouse, f 47-50 s.c. 10, 30, 86 µg/animal, 1 × 112 52, 46, and 63% with p Platt et al.
NMRI weeks fibrosarcomas; controls: 2-6% no/val (1990)
> 99% Mouse, m/f 40-50 s.c. 11.1 and 111 µg/animal 40 weeks 12/35 with pulmonary tumours; p Platt et al.
NMRI on day 2, 1x controls: 2/33 and 4/41 no/val (1990)
newborn
Mouse, 10 i.v. 10 mg/kg, 1x 20 weeks 100% lung tumours; control: p Shimkin &
A 21% no/val Stoner(1975)
Rat 2x10 s.c. 2 mg/animal, weekly; later < approx. 1/10 and 7/10 with tumours; q Barry & Cook
6 mg at longer intervals 200 control: 2/10 no/ln, (1934)
days ld
Rat 10-18 s.c./ 1 mg/animal, 2x/week, Life 3-6/10 and 9/18 with p Boyland &
(6 i.p. 50 weeks subcutaneous sarcomas no/ln Burrows (1935)
exp.) alterna- ld
ting
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rat, 5 s.c. 5 mg/animal, 4-8x 10 2 with tumours after 8-9 p Pollia (1941)
Wistar months months no/ln
99.3% Rat, f 35 Intrapulm. 0.1 mg/animal, 1x < 123 57.1% tumour incidence; p Wenzel-
Osborne/ weeks control: no tumours no/val Hartung et
Mendel al.(1990)
> 99% Rat, f 20 Intra- 1.1 and 4.5 mg into 5th 20 weeks No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1x control: no tumours no/val (1988a)
Dawley
Chromatography Hamster, m/f 5/5 Dermal 8 drops of a 0.2% solution, < 75 No tumours n Shubik et al.
control Syrian weeks 2x/week, 10 weeks no/ln, (1960)
golden ld
Hamster, m 46 Intra- 0.05 and 0.25 mg/animal, < 110 0/46 and 0/46 respiratory tract q Sellakumar &
Syrian tracheal 1x/week, 30 weeks weeks tumours; control: no tumours yes/val Shubik (11974)
golden
Hamster, Intra- 10.3 and 0.9 mg/animal, < 2 years 55 and 65% with tumours p Pott et al.
Syrian trachea 1x/week, 20 weeks no/val (1978)
golden
Monkey, Not specified No tumours n Adamson &
Old world no/ld, Sieber (1983)
lc
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Dibenzo[a,e]pyrene
Recrystallized Mouse, f 40/20 Dermal 0.05 and 0.1% solution, 15 16/40, 9/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months months and 9/40, 6/20 with no/val Wynder (1966
albino epitheliomas; solvent
Ha/ICR/Mil control: no
Recrystallized Mouse, f 28 Dermal 25 µg/animal, 10x over 20 6 months 10/28 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment only: 2/30 no/val Wynder (1966)
albino
Ha/ICR/Mil
> 99% Mouse, f 21 Dermal 242 µg/animal, 1x; 26 weeks 240% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 9% yes/val (1989)
Mouse, m/f 21/14 s.c. 0.6 mg/animal, 1x/month, < 142 18/21 m and 14/14 f local p Lacassagne et
XVII nc/Z 3 × days m sarcomas; no vehicle control no/val al. (1963b)
or 126
days f
Mouse m/f 12/15 s.c. 0.6 mg/animal, 1x < 196 10/12 m and 10/15 f local p Lacassagne et
days m sarcomas; no vehicle control no/val al. (1963b)
or 220
days f
> 99% Rat f 19 Intra- 10 mg/gland, 1x, 8 glands, < 40 1/19 with mammary tumours; n Cavalieri et al.
Sprague- mammary weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Dibenzo[a,h]pyrene
Mouse 74 Dermal 1 drop of a 0.15% solution 4.5 50% with skin tumours p Kleinenberg
alternate days, 55 or 86 months no/lc (1939)
times
Recystallized Mouse, f 20 Dermal 0.05 and 0.1% solution, 11,15 16/20,15/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months months and 13/20, 15/20 with no/val Wynder (1966)
albino epitheliomas; solvent
Ha/ICR/Mil control: no tumours
Recrystallized Mouse, f 29 Dermal 25 µg/animal, 10x over 20 6 months 21/29 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment only: 2/30 no/val Wynder (1966)
albino
Ha/ICR/Mil
96.6% Mouse, f 40 Dermal 120 µg/animal, 2x/week, 70 weeks 90% tumour incidence; solvent p Cavalieri et al.
Swiss 30 weeks control: no tumours no/val (1977)
Pure Mouse, f 30 Dermal 15.1, 60.5 and 181.4 17 weeks 55, 79, and 72% with skin p Chang et al.
CD-1 µg/animal, 1x; initiation tumours; controls: 0-10% yes/val (1982)
experiment
> 99% Mouse, f 24 Dermal 242 µg/animal, 1x; 26 weeks 75% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 9% yes/val (1989)
Mouse, m/f 35/10 s.c. 0.6 mg/animal, 1x/month, > 111/128 34/35 mand 1/10 f with local p Lacassagne et
XVII 3 months days sarcomas no/ld al. (1958)
(average
latency)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 31 s.c. 0.2 mg/animal, 1x; 27 weeks 26/28 with tumours; solvent p Sardella et al.
CD-1 initiation experiment control: 2/32 no/val (1981)
Mouse, m/f 40 i.p. 3.8, 7.6 and 15.1 µg on 49-54 97% with pulmonary and 44% p Chang et al.
Swiss- days 1, 8 and 15 of life weeks with hepatic tumours; control: yes/val (1982)
Webster pulmonary tumours 27%, no
BLU:Ha(ICR) hepatic tumours
newborn
> 99% Rat f 20 Intra- 12 mg/gland, 1x, 8 glands < 40 19/20 with mammary tumours; p Cavalieri et al.
Sprague- mammary weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Dibenzo[a,i]pyrene
Mouse, m 23 Dermal 1 drop of a saturated > 7 21/23 papillomas and 8/23 p Lacassagne et
XVII solution, 2x/week months epitheliomas;solvent control: no/val al. (1958)
no tumours (14 months)
Mouse, f 20/10 Dermal 0.01 and 0.1 %, 3x/week, < 16 and 0.01%: 10% papillomas, no p Wynder &
Swiss 16 and 13 months 13 months carcinomas; 0.1%: 50% no/ld Hoffmann
Millerton papillomas, 10% carcinomas (1959a)
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1 % solution, 15 months 16/40, 16/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months and 13/20, 15/20 with no/val Wynder (1966)
albino epitheliomas; solvent
Ha/ICR/Mil control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized Mouse, f 30 Dermal 25 µg/animal, 10x over 20 6 months 12/30 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment only: 2/30 no/val Wynder (1966)
albino
Ha/ICR/Mil
Mouse, f 20 Dermal 100 and 500 µg/animal, 22 weeks 40 and 80% with tumours; p Hecht et al.
Swiss 1x; initiation experiment vehicle control: no tumours no/val (1981)
albino
Ha/ICR
Pure Mouse, f 30 Dermal 15.1, 60.5 and 181.4 µg/ 17 weeks 28, 67, and 70% with skin p Chang et al.
CD-1 animal, 1x; initiation tumours; controls: 0-10% yes/val (1982)
experiment
> 99% Mouse, f 24 Dermal 242 µg/animal, 1x; 26 weeks 63% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 95% yes/val (1989)
Mouse, m/f 17/18 s.c. 0.6 mg/animal, 1x/month, 3> 75/82 17/17 m and 16/18(f) with p Lacassagne et
XVII months days local sarcomas no/ld al. (1958)
(average
latency)
Mouse, m/f 8/8 s.c. 2 mg/animal, 1x 2-3 100, 100% with skin tumours; p Waravdekar &
XVII/C57BI months average latency: 74 days no/ln, Ranadive
hybrids ld (1958)
Mouse, m s.c. 0.5 mg/animal, 1x 4-5 100% fibrosarcomas; p Homburger et
C57BL/6 weeks malignant cells identifiable no/ld al. (1962)
after 4-5 weeks
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 50 s.c. 0.1 mg/animal, 1x 75 weeks 40/41 with tumours; solvent p Sardella et al.
CD-1 control: no tumours no/val (1981)
Mouse, m/f 40 i.p. 3.8, 7.6 and 15.1 µg on 49-54 97% with pulmonary and 54% p Chang et al.
Swiss- day 1, 8, and 15 of life weeks with hepatic tumours; control: yes/val (1982)
Webster pulmonary tumours 27%, no
BLU:Ha(ICR) hepatic tumours
newborn
> 99% Rat, f 19 Intra- 12 mg/gland, 1x, 8 < 40 18/19 with mammary tumours; p Cavalieri et al.
Sprague mammary glands weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Hamster m 6-10 s.c. 0.25, 0.5, 1 and 2 mg/ 9-14 55, 90, 100, and 100% with p Wodinsky et al.
Syrian animal, 1x weeks fibrosarcomas; vehicle control: no/val (1964)
(average 0%
latency)
Hamster, m/f 139/ s.c. 1 mg/animal, 1x 11 weeks 99/100% with fibrosarcomas p Wodinsky et al.
Syrian 157 (average no/val (1964)
latency)
Hamster, m 4/34 Intra- 0.5 and 2 mg/animal, < 110 Tumours (i) 6/44 (trachea), p Sellakumar &
Syrian tracheal weekly, 24 and 4 weeks, weeks 37/44 (bronchi), 2/34 yes/val Shubik (1974)
golden respectively (trachea); (ii) 1/34 (larynx),
13/34 (bronchi); control: no
tumours
Hamster, m/f 24/24 Intra- 0.5 and 1 mg/animal, 65 and 75% respiratory p Stenback &
Syrian tracheal 1 x/week, 17 and 12 weeks, tumours (bronchi, trachea); no/ld Sellakumar
golden respectively shortest latency: 8 weeks (1974)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Monkey, Not specified No tumours n Adamson &
Old world no/lc Sieber (1983)
Dibenzo[a,l]pyrene
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1% solution, 11, 14 17/20, 18/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months months and 17/20, 18/20 with no/val Wynder
albino epitheliomas; solvent (1966)
Ha/ICR/Mil control: no tumours
Recrystallized Mouse, 30 Dermal 25 µg/animal, 10x over 20 6 months 18/30 papillomas; 1/30 p Hoffmann &
Swiss days; initiation experiment epitheliomas; promotor no/val Wynder
albino only: 2/30 (1966)
Ha/ICR/Mil
Mouse, f 19-21 Dermal 55, 200, 240, 350 and 700 6 months 20, 19, 21, 19 and 16 with p Masuda &
Swiss ICR µg/animal given in 55, 40, skin tumours; no solvent no/val Kagawa
24, 7 and 7 applications control group (1972)
> 99% Mouse, f 24 Dermal 242 µg/animal, 1x; 26 weeks 92% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 9% yes/val (1989)
Pure, 161- Mouse, f 24 Dermal 10, 30 and 90 µg/animal, 15 weeks 23/24, 22/24 and 24/24 with p Cavalieri et al.
162°C) Sencar 1x; initiation experiment tumours;control; no tumours yes/val (1991)
Pure Mouse, f 24 Dermal 1.2, 6 and 30 µg/animal, 7 weeks 22/24, 20/24 and 20/24 with p Cavalieri et al.
Sencar 1x; initiation experiment tumours; 2 control: no tumours yes/val (1991)
Chomatography Mouse, f 24 Dermal 30 µg/animal, 1x; initiation 27 weeks 7/24 with tumours p Cavalieri et al.
purified Sencar experiment without yes/val (1991)
promotion
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 12/12 s.c. 0.6 mg/animal, 1x/month, < 7 All animals with local p Lacassagne et
XVII 2 months (some animals, months sarcomas (mean latent period: no/val al.(1968a)
nc/ZE 3rd injection after 2 months) 120 days); control: no tumours
> 99% Rat, f 9 Intra- 1.2 mg/gland, 1x, 8 glands < 40 9/9 with mammary tumours; p Cavalieri et al.
Sprague- mammary weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Rat f 20 Intra- 76 and 302 µg/gland, 1x, < 24 20/20 and 19/20 with p Cavalieri et al.
Sprague- mammary 8 glands weeks mammary tumours; control: yes/val (1991)
Dawley 1/18
Fluoranthene
Mouse, 2x10 Dermal 0.3% in benzene, 2x/week, < 501 No tumours n Barry et al.
life days no/ld (1935)
Mouse, f 20 Dermal 0.1 % solution, 3x/week, < 17 No papillomas or carcinomas n Wynder &
Swiss life months no/ld Hoffmann
Millerton (1959a)
Mouse, f 20 Dermal 1%, 3x/week, 12 months 15 At 12 months 0/20 tumours; n Hoffmann et al.
Swiss months no vehicle control no/val (1972)
Ha/ICR/Mil
99.9% Mouse, f 30 Dermal 0.1 mg/animal, 10x over 24 weeks 1/29 skin tumours; solvent n Hoffmann et al.
Swiss 20 days; initiation control: 1/30 no/val (1972)
Ha/ICR/Mil experiment
Recrystallized Mouse, m 15 Dermal 250 µg/animal in decalin, 82 week No papillomas or carcinomas; n Horton &
C3H 2x/week, 82 weeks solvent control: 2/13 no/val Christian (1974)
papillomas
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Purified, Mouse, f 50 Dermal 40 µg/animal, 3x/week, 440 days No tumours observed; n Van Duuren &
107- Swiss life controls: no tumours no/val Goldschmidt
109°C) ICR/Ha (1976)
Mouse, m/f 7/7 s.c. 10 mg/animal, 5x 19 No tumours n Shear (1938)
Jackson A months no/ld,
ln
Mouse, m/f 10/10 s.c. 0.6 mg/animal, 1x/month, No sarcomas n Buu-Hoi (1964)
XVII nc/Z 3x no/ld,
ln
99% Mouse, m/f 20-31 i.p. 0.7 and 3,5 mg/animal 24 weeks 23, 15, and 74, 38% m/f with p Busby et al.
Swiss- (total dose) in 3 aliquots lung tumours; vehicle control: yes/val (1984)
Webster on days 1, 8 and 15 after 4,14%
BLU:Ha(ICR) birth
newborn
> 99.5% Mouse, m/f 22/30 i.p. 0.7 and 3.5 mg/animal 52 weeks 43, 35, and 65, 86% with lung p La Voie et al.
CD-1 (total dose) in 3 aliquots on tumours; 64, 0% and 100, 7% yes/val (1994)
newborn days 1, 8, and 15 after birth with hepatic tumours; vehicle
only: 17, 12% (lung) and 17, 6%
(liver)
Fluorene
Mouse 100 Dermal Dissolved in 90% benzene 9 months No tumours n Kennaway
no/ld, (1924)
lc
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 10/10 Dermal 60 µg/animal, 2x/week, 31 weeks No skin tumours n Riegel et al.
CF1 31 weeks no/ld (1951)
'Pure' Mouse, m 100 Dermal 3 drops, 1x/week of approx. < 1 year After 9 months 10/100 n Graffi et al.
white 3% solution, 1 year; survived, no tumours; promotor no/val (1953)
initiation experiment only: 0.08 tumour/animal
Mouse, m 5 i.p. 1000 mg/kg, 1x < 5 No effects n Shubik & Della
Swiss months no/ld Porta (1957)
ln
Mouse, m 10 s.c. 10 mg/animal, 7x over 19 No tumours n Shear(1938)
Jackson A 16 months months no/ln,
ld
Highly Rat, f 20 Oral 0.05% diet; 4.3 mg/rat per 10.7 2/11 carcinomas (renal pelvis, q Morris et al.
purified Buffalo (diet) day = 796 mg/rat (total months ureter); control: 4/16 with no/ld (1960)
intake) over 6 months carcinomas
Highly Rat, f 18 Oral 0.05% diet; 4.6 mg/rat per < 20.1 7/18 tumours; control: 4/18 or q Morris et al.
purified Buffalo (diet) day = 2553 mg/rat (total months 15/18 tumours no/val (1960)
intake) over 18 months
Recrystallized Mouse, f 30 Dermal 25 µg/animal, 10x over 20 6 months 5/30 papillomas; promotor: q Hoffmann &
Swiss days; initiation experiment 2/30 no/val Wynder (1966)
Ha/ICR/Mil
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1 % solution, 15 Dioxane solvent: no tumours; q Hoffmann &
Swiss 3x/week, 12 months months acetone solvent: dose-related no/val Wynder (1966)
albino tumour increase
Ha/ICR/Mil
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
> 96% Mouse, f 40 Dermal 3.4, 5.6, 9.2 µg/animal, <2 years 3, 0, 0% with local tumours; n Habs at a
NMRI 2x/week, life control: no tumours yes/val (1980)
Mouse, f 25 Dermal 100 µg/animal, 10x over 25 weeks 90% with skin tumours; vehicle p Rice et al.
Crl:CD1 20 days; initiaton control: < 5% yes/val (1986)
(ICR)BR experiment
> 99% Mouse, f 25 Dermal 110 µg/animal, 10x over 23 weeks 72% tumours, 2.1 skin p Rice et al.
CD-1 20 days; initiation tumours/animal; control: yes/val (1990)
experiment no tumours
Mouse, m/f 14/14 s.c. 0.6 mg/animal, 1x/mth, Average, Sarcomas: 10/14 m and 1/14 f p Lacassagne et
XVII 3 months 265 days no/val al. (1963a)
nc/Z m, 145
days f
> 99% Mouse, m/f 11/9 i.p. 580 µg/animal in DMSO < 52 9% hepatic or 0% lung n LaVoie et al.
CD-1 on days 1, 8 and after weeks tumours; controls: 6%/0% yes/val (1987)
newborn birth (total dose)
99.4% Rat, f 35 Intra- 0.16, 0.83 and 4.15 116/109/ 3/35, 8/35 and 21/35 with lung p Deutsch-
Osborne/ pulm. mg/animal, 1x 92 weeks tumours; control: no tumours yes/val Wenzel et al.
Mendel (1983)
5-Methylcholanthrene
> 99.9% Mouse, f 20 Dermal 0.1 mg/animal, 3x/week, 35 weeks 20/20 with 85 skin tumours p Hecht et al.
Swiss 35 weeks (solvent by 25 week; 20/20 with 99 no/val (1974)
Ha/ICR/Mil control: tumours and 12/20 with 37
72 weeks) carcinomas by 35 wks; solvent
control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
> 99.9% Mouse, f 20 Dermal 10, 30 and 100 µg, 10x 24 weeks Low dose: 20/20 mice with p Hecht et al.
Swiss over 20 days; initiation 110 skin tumours; med dose: no/val (1974)
Ha/ICR/Mil experiment 20/20 with 160 skin tumours;
high dose: 17/18 with 96 skin
tumours; solvent control: no
tumours
Mouse, f 20 Dermal 5 and 10 µg/animal, 62 weeks Low dose: 9/20 with 22 skin p Hecht et al.
Swiss 3x/week, 62 weeks tumours, 6/20 with 7 yes/val (1976a)
Ha/ICR carcinomas; high dose: 15/20
with 38 tumours, 10/20 with
12 carcinomas; solvent
control: no tumours
Highly Mouse, f 20 Dermal 1 and 3 µg, 10x over 24 weeks Low dose: 2/20 mice with 2 p Hecht et al.
purified Swiss 20 days; initiation skin tumours; high dose: yes/val (1976a)
Ha/ICR experiment 20/20 with 45 skin tumours
(1 carcinoma); solvent control:
no tumours
> 99.9% Mouse, f 8x20 Dermal 3 and 10 µg, 10 × over 20 24 weeks Low dose: 55-95% of mice p Hecht et al.
Swiss days; initiation experiment with skin tumours; high dose: yes/val (1978)
Ha/ICR/Mil 80-90%; solvent control: no
tumours
> 99.9% Mouse, f 20 Dermal 1 and 3 µg, 10x over 20 24 weeks Low dose: 75% of mice with p Hecht et al.
Swiss Ha/ICR days; initiation experiment skin tumours; high dose: 85% no/val (1979)
outbred
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 20 Dermal 1 or × or 3 µg/animal, 10x 21 weeks 55, 75, and 90% with skin p Amin et al.
Swiss CD-1 over 20 days; initiation tumours; solvent control: 5% yes/val (1981)
experiment
HPLC Mouse, f 20 Dermal 8 and 24 µg/animal, 1x; 26 weeks 80 and 90% tumour-bearing p Hecht et al.
purified CD-1 initiation experiment animals; solvent control: 10% yes/val (1985)
Mouse, f 20 Dermal 8 µg/animal, 1x; initiation 21 weeks 65% with skin tumours; p Amin et al.
CD-1 experiment solvent control: 5% yes/val (1985b)
Mouse, f 20 Dermal 24.2 µg/animal, 1x; 26 weeks 90% with tumours; 5.2 p El-Bayoumy et
CD-1 initiation experiment tumours/animal; solvent no/val al. (1986)
control: 10%/0.1
> 99% Mouse, f 20 Dermal 3.6, 12.1 and 36 µg/ 24 weeks 100, 100 and 100% with p Rice et al.
CD-1 animal, 10x over 20 days; tumours; 9.2, 10.7 and 9.4 yes/val (1988b)
initiation experiment tumours/animal; solvent
control: 20%
Mouse, f 20 Dermal 8 µg/animal, 1x;initiaUon 21 weeks 85% with skin tumours; p Amin et al.
CD-1 experiment solvent control: 10% yes/val (1990)
Mouse, f 20 Dermal 8 µg/animal, 1x; initiation 26 weeks 65% with skin tumours; solvent p Amin et al.
CD-1 experiment control: 10% yes/val (1992)
Mouse, m/m 20/ s.c. 2 mg/animal in tricaprylin, 6 months Swiss mice: no local tumours; q Dunlap &
Swiss/ 2x10 1x 16/20 mice died; C3H mice: no/ld Warren (1943)
C3H 7/10 or 3/10 local sarcomas
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Highly Mouse, m 25 s.c. 50 µg/animal in 32 weeks 22/25 mice with 24 p Hecht et al.
purified 057BL trioctanoin, 1 x/2 weeks, fibrosarcomas; vehicle no/val (1976b)
20 weeks control: no tumours
HPLC Mouse, m/f 35/48 i.p. 1.9 µg/animal on day 1; Weaned 20/21% with pulmonary p Hecht et al.
purified ICR/Ha 3.9 µg on day 8; 7.8 µg after 3 tumours; 23/12% with yes/val (1985)
newborn on day 15 weeks; hepatic tumours; solvent
sacri- control: 4/7% and 2/2%
ficed
after 35
weeks
> 99% Rat f 20 Intra- 0.97 and 3.9 mg into 5th 20 weeks No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1x control: no tumours no/val (1988a)
Dawley
1-Methylphenanthrene
>99.5% Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks No tumours; vehicle control: n LaVoie et al.
Swiss initiaton experiment no tumours no/val (1981b)
Ha/ICR
Naphthalene
Mouse Dermal Several times/wk, < 11 No skin tumours n Kennaway
< 11 months months no/lc (1930)
Highly Mouse, 25; Dermal 0.5% in benzene, 6x/week Life 4/25 with lymphatic q Knake (1956)
purified SW inbred con- for 3 weeks, then 2x/wk for leukaemia; 1/25 lymphosarcoma no/lc,
trol: life of thymus; 4/25 with benign ln
21 tumours; benzene only: 2/21
with sarcomas; 1/21 with lung
adenoma
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 30 Dermal 0.25 mg/animal + 3 µg 78 weeks 5/30 lymphomas; inhibitory q Schmeltz et al.
ICR/Ha benzo[a]pyrene, 3x/wk, effect on skin tumours; no/val (1978)
78 weeks; co-carcinogenicity naphthalene only: no skin
test tumours
98-99% Mouse, f 30 Inha- 0.05 and 0.15 mg/l, 6 months 29 and 30% with pulmonary q Adkins et al.
A/J lation 6 h/day, 5 days/week, tumours; control: 21% yes/val (1986)
6 months (increase in treatment groups
not significant)
> 99% Mouse, m/f 75 Inha- 0.053 and 0.16 mg/litre, 103 Significantly increased q Abdo et al.
B6C3F1 (150) lation 6 h/day, 5 days/week, weeks pulmonary alveolar and yes/val (1992);
103 weeks bronchiolar adenomas in National
females; no cararacts Toxicology
Program
(1992b)
Mouse 23 Bladder 1x (dose unspecified) 7 months 1/23 bladder carcinoma after - Boyland et al.
implant 1 month; "inert" substance: (1964)
higher rate of bladder
carcinoma
Rat, 28 Oral 10-20 mg/animal, Life No tumours n Schmahl
BDI/BDI II (diet) 6x/week, 70 weeks no/ld (1955)
inbred
Rat, 10 s.c. 20 mg/animal, 1x/week, Life No tumours n Schmahl
BDI/BDI II 40 weeks no/ln (1955)
inbred
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Crude, Rat, 38 s.c. 0.5 g/kg, 2x/month, 3.5 Life 5 malignant tumours (4/38 q Knake(1956)
90% 'white' months Imphosarcomas, 1/38 uterine no/val
sarcoma); 1 benign tumour;
vehicle control: 1/38
lymphosarcoma and 1 benign
tumour
Rat, 10 i.p. 20 mg/animal, 1x/week, Life No tumours n Schmahl
BDI/BDI II 40 weeks no/ln (1955)
inbred
Perylene
Recrystalized Mouse, f 20 Dermal 0.8 mg/animal, 1x; 58-60 3/20 papillomas; promotor n Van Duuren et
Swiss initiation experiment weeks only: 1/20 with papillomas; no/val al. (1970)
ICR/Ha pure substance only: no tumours
Recrystallized Mouse, m 20 Dermal 75 µg/animal in decalin, 82 weeks No skin tumours; solvent n Horton &
C3H 2x/week, 82 weeks control: 2/13 papillomas no/val Christian
(1974)
Phenanthrene
Mouse 100 Dermal Dissolved in 90% benzene 9 months No tumours n Kennaway
no/ld, (1924)
lc
'Pure' Mouse, m 100 Dermal 3 drops, 1x/week of approx. < 1 year After 12 months 6/100 n Graft et al.
white 3% solution, 1 year; survived with a total of 1 no/val (1953)
initiation experiment tumour; 0.16 tumour/animal;
promotor only: 0.08
tumour/animal
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 20 Dermal 54 mg/animal, 3x/wk, total: 24 weeks 5/20 survivors with 12 q Salaman & Roe
'S' 10x; initiation experiment papillomas; promotor only: yes/val (1956)
4/19 survivors/4 papillomas
High purity Mouse, m/f 10/10 Dermal 0.3 mg, 4x on days 0, 2, 6 24 weeks 4/19 papillomas; solvent q Roe (1962)
'stock and 8; initiation experiment control: 2/20 yes/val
albino'
TLC Mouse, f 30 Dermal 1.8 mg/animal, 1x; 35 weeks 40% with papillomas; p Scribner (1973)
purified CD-1 initiation experiment promotor only: 3% no/val
> 98% Mouse, f 30 Dermal 1.8 mg/animal, 1x; 36 weeks 5/30 papillomas; solvent q Wood et al.
CD-1 initiation experiment control: 2/30 no/val (1979)
Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks No skin tumours observed; n LaVoie et al.
Swiss initiation experiment vehicle control: no tumours no/val (1981b)
Ha/ICR
Mouse, m/f 40-50 s.c. 5 mg/animal in tricaprylin; < 22-28 No sarcomas after 8 months n Steiner (1955)
C57BI 1x months yes/ld
Mouse, m/f 10/10 s.c. 0.3 mg, 5x on days 0, 2, 24 weeks 3/17 papillomas; solvent n Roe (1962)
'stock 4, 6 and 8; initiation control: 2/20 yes/val
albino' experiment
Mouse, m/f 57 s.c. 40 µg/animal; 1x < 62 3/49 lung adenomas; control: n Grant & Roe
stock administered to neonatal weeks 8/34 and 5/38 yes/val (1963)
albino' mice
newborn
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
> 98% Mouse, 100 i.p. 35, 70 and 140 µg/animal 38-42 6/35 pulmonary adenomas; n Beuning et al.
Swiss- in DMSO on days 1, 8 weeks DMSO only: 9/59 yes/val (1979)
Webster and 15 after birth
BLU:Ha(ICR)
newborn
Rat, f 10 Oral 200 mg/rat, 1x; experiment 60 days No tumours at 60 days; n Huggins &
Sprague- on mammary tumours controls: 8/164 after 310 days no/ln, Yang (1962)
Dawley lc
99.9% Rat, f 35 Intrapulm. 1, 3 and 10 mg/animal, 1x < 135 No tumours; control: no n Wenzel-Hartung
Osborne/ weeks tumours no/val et al.(1990)
Mendel
Pyrene
Mouse 2x20 Dermal 1% in benzene, 2x/week, < 717 1/20 and 1/20 papillomas n Barry et al.
life days no/ld (1935)
Mouse 40 Dermal 0.3% in benzene, 2x/ < 680 No skin lesions n Badger et al.
week, < 680 days days no/ld (1940)
'Pure' Mouse, m 150 Dermal 3 drops, 1x/week of a < 1 year After 6 months 18/150 n Graffi et al.
white 0.3% solution, 1 year; survived with a total of 1 no/val (1953)
initiation experiment tumour; 0.06 tumour/animal;
promotor only: 0.08
tumour/animal
Mouse, 20 Dermal 25 mg/animal, 3x/week; 24 weeks 6/20 mice with 9 papillomas; q Salaman & Roe
'S' total:10x; initiation promotor only: 4/19 mice with yes/val (1956)
experiment 4 papillomas
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 5 Dermal 10%, 3 x/week, life < 18 No skin tumours n Wynder &
Swiss months no/ld, Hoffmann
Millerton ln (1959a)
TLC Mouse, f 30 Dermal 2 mg/animal, 1x; initiation 35 weeks 17% with papillomas; q Scribner (1973)
purified CD-1 experiment promotor only: 3% no/val
High purity Mouse, m 20 Dermal 250 µg/animal in decalin, 82 weeks 3/13 papillomas; solvent q Horton &
C3H 2x/week, 82 weeks control: 2/13 no/val Christian (1974)
Recrystallized Mouse, f 50 Dermal 12 or 40 µg/animal, < 440 No skin tumours observed; n Van Duuren &
Swiss 3 x/week, 368 or 440 days days control: no tumours no/val Goldschmidt
ICR/Ha (1976)
High purity Mouse, f 50 Dermal 4 and 12 µg/animal + 5 µg 33 weeks High dose: 13/50 papillomas, n Goldschmidt et
Swiss benzo[a]pyrene, 3x/week, 5/50 carcinomas; benzo[a]- no/val al. (1973)
ICR/Ha 33 weeks; co-carcinogenicity pyrene only: 6/50 papillomas;
test pyrene only: no tumours
Recrystallized Mouse, f 50 Dermal 4, 12 and 40 µg/animal 368 or 12/26/35 mice with papillomas, p Van Duuren &
Swiss + 5 µg benzo[a]pyrene, 440 days 6/20/26 with squamous cell no/val Goldschmidt
ICR/Ha 3x/week, 368/368/440 carcinomas; positive control: (1976)
days; co-carcinogenicity 15, 11 tumours; solvent control:
test no tumours
> 98% Mouse, f 30 Dermal 20.2 and 80.9 µg/animal, 27 weeks 14 and 10% with tumours; n Wood et al.
CD-1 1x; initiation experiment vehicle control: 10% yes/val (1980)
Crystals Mouse, m/f 30 s.c. 10 mg/animal, 2 × at 4- < 18 No malignant tumours n Shear & Leiter
Jackson A month interval months no/ld (1941)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized, Mouse, m/f 23-28 i.p. 86.1 and 1750 µg/animal 26 weeks 17, 4, and 7, 12% m/f with n Busby et al.
HPLC Swiss- (total dose) in 3 aliquots lung tumours; vehicle control: no/val (1989)
Webster on days 1, 8 and 15 14, 7% m/f
BLU:Ha(lCR) after birth
newborn
> 99% Hamster, m 48 Intra- 3 mg/animal, 1x/week, < 110 1/48 tumours of the trachea, n Sellakumar &
Syrian tracheal 30 weeks weeks 2/48 malignant lymphomas; yes/val Shubik (1974)
golden control: 0/82 and 2/82
Triphenylene
Mouse 10 Dermal 0.3% in benzene, 2x/ < 548 No skin lesions n Barry et al.
week, life days no/ln, (1935)
ld
Recrystallized Mouse, m 20 Dermal 250 µg/animal in decalin, 82 weeks No skin tumours; solvent n Horton &
C3H 2x/week, 82 weeks control: 2/13 papillomas no/val Christian
(1974)
Result: p(ositive), n(egative), q(uestionable); Stat, statistical evaluation: yes or no; Val, validity: val, valid; ld, limited design; lc,
limited documentation; ls, limited survival; ln, limited number of animals
intrapulm., intrapulmonary injection; i.p., intraperitoneal injection; s.c., subcutaneous injection; i.m., intramuscular injection
m, male; f, female
TLC, thin-layer chromatography; DMSO, dimethylsulfoxide; HPLC, high-performance liquid chromatography; DMBA, 7,12-dimethylbenz[a]anthracene
Table 91. Overview of carcinogenicity of polycyclic aromatic hydrocarbons
Compound Carcinogenicity Species Route of administration
(weight of No. of studies with positive, negative, and questionable results
evidence)
Oral Dermal s.c./i.m. i.p./i.v. inh./tr. Other
+ - ± + - ± + - ± + - ± + - ± + - ±
Acenaphthene Questionable Mouse 1 1
Acenaphthylene No studies
Anthanthrene Positive Mouse 2 6 1 1 1
Anthracene Negative Mouse 6 1 1 1
Rat 2 1 2 1 1
Rabbit 1
Benz[a]anthracene Positive Mouse 2 1 7 4 4 2 2 1
Rat 1 1 2 1 1
Hamster 2 1
Benzo[b]fluoranthene Positive Mouse 7 1 1
Rat 1
Hamster 1
Benzo[j]fluoranthene Positive Mouse 3 1 1
Rat 1
Benzo[ghi]fluoranthene (Negative) Mouse 2
Benzo[k]fluoranthene Positive Mouse 1 2 1 1 1
Rat 1
Benzo[a]fluorene (Questionable) Mouse 1 1 1
Benzo[b]fluorene (Questionable) Mouse 1
Benzo[ghi]perylene Negative Mouse 8 2
Rat 1
Benzo[c]phenanthrene (Positive) Mouse 2 2 1 1
Rat 1
Table 91. (continued)
Compound Carcinogenicity Species Route of administration
(weight of No. of studies with positive, negative, and questionable results
evidence)
Oral Dermal s.c./i.m. i.p./i.v. inh./tr. Other
+ - ± + - ± + - ± + - ± + - ± + - ±
Benzo[a]pyrene Positive Mouse 5 26 6 3 1 2
Rat 2 1 1 9 3
Hamster 1 1 1 1 11 1 1
Dog 1
Cattle 1
Pig 2
Monkey 1 1 1 1
Benzo[e]pyrene Questionable Mouse 2 1 5 1
Rat 1 1
Chrysene Positive Mouse 11 9 1 3 3 1 1 2 1
Rat 1 2 1
Coronene (Questionable) Mouse 1 1
Cyclopenta[cd]pyrene Positive Mouse 4 1 1
Rat 1
Dibenz[a,h]anthracene Positive Mouse 1 1 6 8 1
Rat 2 1 1 1
Hamster 1 1 1
Monkey 1
Dibenzo[a,e]pyrene Positive Mouse 3 2
Rat 1
Dibenzo[a,h]pyrene Positive Mouse 6 2 1
Rat 1
Dibenzo[a,i]pyrene Positive Mouse 7 4 1
Rat 1
Hamster 2 2
Monkey 1
Dibenzo[a,l]pyrene Positive Mouse 7 1
Rat 2
Table 91. (continued)
Compound Carcinogenicity Species Route of administration
(weight of No. of studies with positive, negative, and questionable results
evidence)
Oral Dermal s.c./i.m. i.p./i.v. inh./tr. Other
+ - ± + - ± + - ± + - ± + - ± + - ±
Fluoranthene (Positive) Mouse 6 2 3
Fluorene Negative Mouse 3 1 1
Rat 2
+ - ± + - ± + - ± + - ± + - ± + - ±
Indeno[1,2,3-cd]pyrene Positive Mouse 2 1 2 1 1
Rat 1
5-Methylchrysene Positive Mouse 13 1 1 1 1
Rat 1
1-Methylphenanthrene (Negative) Mouse 1
Naphthalene (Questionable) Mouse 1 2 2 1
Rat 1 1 1 1
Perylene (Negative) Mouse 2
Phenanthrene (Questionable) Mouse 1 3 3 3 1
Rat 1 1
Pyrene (Questionable) Mouse 1 7 3 1 1
Hamster 1
Triphenylene (Negative) Mouse 2
+, positive; -, negative; questionable; parentheses, limited number of studies
s.c., subcutaneous; i.m., intramuscular; i.p., intraperitoneal; i.v., intravenous; inh., inhalation; tr., intratracheal
Other: e.g. intramammary injection, bladder implant, bronchial implant
Epidermal cell kinetics, DNA adduct levels, and changes in skin
morphology were measured in ICR/Harlan mice after 29 weekly topical
applications of 16, 32, or 64 µg benzo [a]pyrene for up to 35 weeks.
Initially, there was a linear increase in DNA adducts, which was much
less steep at 64 µg and which did not correlate with the sharp rise in
tumour response at that dose. A dose-dependent increase in the
3H-thymidine labelling index, the mitotic index, and the incidence of
pyknotic and dark cells indicated that benzo [a]pyrene induced
extensive cytotoxicity and cell death, with regenerative
proliferation. Virtually all of the initial tumours were papillomas,
which required an average of eight weeks to progress to carcinomas,
reflecting the tumour-promoting activity of benzo [a]pyrene in this
model (Albert et al., 1991a,b).
In a study with female Sprague-Dawley rats to elucidate whether the
metabolites and DNA adducts of benzo [a]pyrene are formed in the
liver or in target tissues, animals that had received a liver
transplant were compared with normal animals. The liver was found to
serve as a depot for PAH, in this case infused 3H-benzo [a]pyrene,
which was converted into polar metabolites. A few hours later, polar
metabolites and DNA adducts were found in target tissues of both
groups (Wall et al., 1991).
7.7.1.2 Benzo [e]pyrene
The results of studies on benzo [e]pyrene are considered to be
questionable or negative even though positive results were reported in
two studies by dermal application, because no bay-region activation
was found in liver tissue that would result in an 'ultimate'
carcinogen, such as 9,10-dihydroxy-11,12-epoxy-9,10,11,12-
tetrahydrobenzo [e]pyrene (Jacob et al., 1983).
7.7.2 Comparative studies
Comparative studies on the tumorigenic activity of individual PAH that
have been used as the basis for comparative potency factors (see
Appendix I) are summarized below. The detailed results of studies with
individual PAH are given in Table 90. In general, the results of
studies with skin painting and lung implantation were used for
estimating comparative potencies in preference to those from
initiation-promotion experiments and studies by intraperitoneal
injection. No comparative studies have been carried out by oral
administration.
7.7.2.1 Carcinogenicity
(a) Dermal exposure
Skin painting: Solutions of 0.5% benzo [a]pyrene,
benzo [b]fluoranthene, benzo [j]fluoranthene, or
benzo [k]fluoranthene were applied dermally three times weekly to
groups of 20 female Swiss Millerton mice for life, and the number of
skin tumours was determined. The percentages of papillomas/ carcinomas
induced by these compounds after four months were 70/20 with
benzo [a]pyrene, 95/10 with benzo [b]fluoranthene, 40/5 with
benzo [j]-fluoranthene, and none with benzo [k]fluoranthene. Minimal
activity (10% papillomas) was induced by benzo [k]fluoranthene after
11 months (Wynder & Hoffmann, 1959b). The order of potency was thus
benzo [a]pyrene > benzo [b]-fluoranthene > benzo [j]fluoranthene
> benzo [k]fluoranthene.
In a similar regime, 0.01% solutions of benzo [a]pyrene or
dibenz [a,h]anthracene applied dermally to groups of 20 mice induced
10/10% and 15/5% papillomas/carcinomas, respectively after six months.
A 0.1% solution of dibenzo [a,i]pyrene induced 10/0% tumours after
seven months, and a 0.1% solution of benzo [e]pyrene induced 5/0%
tumours after 10 months. A 1% chrysene solution induced 5/5%
papillomas/carcinomas after eight months. A 0.1% solution of
fluoranthene and 10% solutions of anthracene and pyrene had no
activity (Wynder & Hoffmann, 1959a). The order of potency was thus
benzo [a]pyrene = dibenz [a,h]anthracene > dibenzo [a,i]pyrene >
chrysene > benzo [e]pyrene > fluoranthene, anthracene, pyrene.
In a further study, the carcinogenicity of PAH was compared after
dermal application to mice three times weekly, as above. A dose of
0.05% induced the following percentages of papillomas/carcinomas after
eight months: benzo [a]pyrene, 17/17; dibenzo [a,h]pyrene, 14/9;
dibenzo [a,l]pyrene, 10/10; dibenzo [a,i]pyrene, 3/0;
dibenzo [a,e]pyrene, 2/1 after 10 months; indeno[1,2,3- cd]pyrene,
1/1 with 0.5% solution; and benzo [ghi]perylene, 0/0 (Hoffmann &
Wynder, 1966). The order of potency was thus benzo [a]pyrene>
dibenzo [a,h] -pyrene > dibenzo [a,l]pyrene > dibenzo [a,i]pyrene
> dibenzo [a,e]pyrene > indeno[1,2,3- cd]pyrene >
benzo [ghi]perylene.
In a lifetime study by skin painting in female NMRI mice,
benzo [a]pyrene and benzo [b]fluoranthene were carcinogenic,
benzo [j]fluoranthene was weakly carcinogenic, and
benzo [k]fluoranthene, indeno[1,2,3- cd]pyrene, and coronene had no
carcinogenic effect (Habs et al., 1980). The order of potency was thus
benzo [a]pyrene >> benzo [b]fluoranthene > benzo [j]fluoranthene
> benzo [k]-fluoranthene, coronene, indeno[1,2,3- cd]pyrene.
Initiation-promotion: Ten doses of PAH at a total dose of 0.25
mg/mouse were applied every second day to the backs of Swiss Millerton
mice, which were then promoted with 2.5% croton oil in acetone
(Hoffmann & Wynder, 1966). The relative tumour-inducing activity was:
benzo [a]pyrene > dibenzo [a,h]pyrene > dibenzo [a,l]pyrene >
dibenzo [a,i]pyrene > dibenzo [a,e]-pyrene > indeno[1,2,3-
cd]pyrene > benzo [ghi]perylene.
In an assay in CD-1 mice, 30 µg of several PAH were applied in 10
doses over 20 days to the shaven backs of groups of 20 mice. Ten days
after completion of the initiation, promotion was begun by thrice
weekly application of 12- O-tetradecanoylphorbol 13-acetate in 0.1 ml
acetone. The skin tumours induced were predominantly squamous-cell
papillomas. After 20 weeks (10 weeks for benzo [a]pyrene), the
percentages of skin tumour-bearing animals were 85% with
benzo [a]pyrene, 45% with benzo [b]fluoranthene, 30% with
benzo [j]fluoranthene, and 5% with benzo [k]fluoranthene. The
vehicle controls had no tumours (La Voie et al., 1982b).
Sencar mice were treated with the - [a,e]-, - [a,h]-, - [a,i]-, and
- [a,l]- isomers of dibenzopyrene with TPA as a promoter,
anthanthrene as the negative control and with vehicle and sham
controls. Dibenzo [a,e]pyrene was a very weak tumour initiator and
dibenzo [a,h]pyrene and dibenzo [a,i]pyrene were tumorigenic;
dibenzo[a,l]pyrene was highly toxic and an extremely potent carcinogen
(Cavalieri et al., 1989). In a further investigation of
dibenzo[a,l]pyrene, with benzo [a]pyrene as the positive control,
7,12-dimethylbenz [a]anthracene, recognized as the most potent
carcinogenic PAH, was also tested at 4, 20, and 100 nmol of the PAH in
the same regime as above. The tumorigenic activity of
dibenzo[a,l]pyrene in mouse skin was inversely proportional to the
dose, indicating that toxicity interferes with the initiation of
tumours. When the effects of equimolar concentrations were compared,
benzo [a]pyrene was a much weaker tumour initiator than
dibenzo[a,l]pyrene (Cavalieri et al., 1991). The order of potency was
thus dibenzo[a,l]pyrene > dibenzo[a,i]pyrene > dibenzo[a,h]pyrene >
benzo [a]pyrene > dibenzo[a,e]pyrene.
The relationship between the Ah locus and the induction of
subcutaneous fibrosarcomas was studied after administration of
dibenz [a,c]anthracene and dibenz [a,h]anthracene to B6, D2, and
B6D2F1 mice. The doses and results are shown in Table 92.
Dibenz [a,c]anthracene was a weak tumour inducer in all groups
tested. Dibenz [a,h]anthracene was a fairly potent inducer of
subcutaneous tumours in B6 and B6D2F1 mice, but not in D2 mice. These
results, with those of back-crossing experiments, demonstrate a strict
correlation between the tumorigenicity of dibenz [a,h]anthracene and
expression of the Ahb allele (Kouri et al., 1983).
(b) Other routes
Intraperitoneal injection in newborn mice: The tumorigenic activity
of the nonalternant PAH benzo [b]fluoranthene,
benzo [j]fluoranthene, and benzo [k]fluoranthene and of
indeno[1,2,3- cd]pyrene and benzo [a]pyrene were evaluated by
injecting a total of 0.5, 1.1, 2.1, 2.1, or 0.5 µmol of each compound,
respectively, in dimethyl sulfoxide in aliquots of 5, 10, or 20 µl on
days 1, 8, and 15 after birth to CD-1 mice (La Voie et al., 1987).
Direct comparison was not possible owing to the differences in the
total amount injected, but both benzo [b]fluoranthene and
benzo [j]fluoranthene had significant tumorigenic activity, whereas
neither benzo [k]fluoranthene nor indeno[1,2,3- cd]pyrene was
tumorigenic under these conditions. There were problems with the
solubility of indeno[1,2,3- cd]pyrene. The order of potency was thus
benzo [a]pyrene > benzo [b]fluoranthene = benzo [j]fluoranthene >
benzo [k]-fluoranthene, indeno[1,2,3- cd]pyrene.
Intramammary injection: 7,12-Dimethylbenz [a]anthracene and
dibenzopyrene isomers were tested with benzo [a]pyrene by
intramammary injection. Dibenzo[a,e]pyrene was inactive, but
dibenzo [a,l]pyrene was much more carcinogenic than
7,12-dimethylbenz [a]anthracene. At these doses, benzo [a]pyrene had
only marginal activity. Dibenzo [a,l]pyrene thus appears to have the
highest mammary cancer potency of all PAH so far tested (Cavalieri et
al., 1989).
Lung implantation: The relative potencies of PAH to induce
epidermoid carcinomas and pleomorphic sarcomas after intrapulmonary
injection, with benzo [a]pyrene as the reference substance, were
dibenz [a,h] anthracene, 1.91 > benzo [a]pyrene, 1.00 >
anthanthrene, 0.19 > benzo [b]fluoranthene, 0.11 > indeno[1,2,3-
cd]pyrene, 0.08 > chrysene, 0.03 = benzo [k]fluoranthene, 0.03 =
benzo [j]fluoranthene, 0.03 > phenanthrene, 0.001.
Benzo [ghi]perylene and benzo [e]pyrene had no tumour-inducing
effect (Deutsch-Wenzel et al., 1983; Wenzel-Hartung et al., 1990).
Subcutaneous injection: Dose-response curves for benzo [a]pyrene
and dibenz [a,h]anthracene were established after a single
subcutaneous injection of PAH in tricaprylin into the right axilla of
male C3H mice; 99% of the tumours detected were spindle-cell sarcomas.
The responses of vehicle controls were not reported. Under the
conditions in this experiment, dibenz [a,h]anthracene was estimated
to be 4.5 times more potent than benzo [a]pyrene (Bryan & Shimkin,
1943).
Eight of 10 male and six of 10 female C57 black mice had
injection-site tumours 60-80 weeks after 10 weekly subcutaneous
injections of 1 mg benz [a]anthracene, whereas 20/20 males and 17/20
females had tumours after 1 mg dibenz [a,h]anthracene (Boyland &
Sims, 1967).
7.7.2.2 Further evidence
(a) Sebaceous gland assay
Application of carcinogenic PAH to mouse skin leads to the destruction
of sebaceous glands, hyperplasia, hyperkeratosis, and even ulceration
(Bock, 1964). An assay of these glands has been used to screen the
tumorigenic potential of PAH. Acute topical application of
benzo [a]pyrene, benz [a]anthracene, or dibenz [a,h]anthracene was
reported to suppress glandular activity (Bock & Mund, 1958). The order
of potency was benzo [a]pyrene = dibenz [a,h]anthracene >
benz [a]anthracene. In another study, the order of potency was
benzo [a]pyrene > benzo [b]fluoranthene = benzo [j]fluoranthene =
benzo [k]fluoranthene = indeno[1,2,3- cd]pyrene (Habs et al. 1980).
Table 92. Subcutaneous fibrosarcomas induced by a dose of 300 µg per
animal of isomers of dibenzanthracene in different mouse strains
Dibenzanthracene Strain Tumour Carcinogenic
isomer incidence index
a,c B6 1/30 1.1
D2 0/30 0
B6D2F1 1/30 1.2
a,h B6 14/30 24
D2 0/30 0
B6D2F1 33/60 30
B6D2F1 x D2 38/53 29
back-crosses
(Ahb/Ahd phenotype)
B6D2F1 × D2 0/33 0
back-crosses
(Ahd/Ahd phenotype)
From Kouri et al. (1983)
(b) DNA adduct formation
In a 32P-postlabelling test for covalent binding of PAH to DNA in
mouse skin in vivo after a single topical application, the relative
ability to induce DNA adducts was benzo [a]pyrene >
benz [a]anthracene = dibenz [a,h]anthracene = benzo [ghi]perylene
(Reddy et al., 1984). DNA adducts were not induced by pyrene. In a
similar study, the relative ability of PAH to bind covalently to DNA
was benzo [b]fluoranthene > benzo [j]fluoranthene >
benzo [k]fluoranthene > indeno[1,2,3- cd]pyrene (Weyand et al.,
1987). In a study in vitro, the relative ability for covalent
binding of PAH to DNA was reported to be benzo [a]pyrene >
dibenz [a,h]anthracene > benz [a]anthracene > pyrene >
phenanthrene (Grover & Sims, 1968).
7.7.3 PAH in complex mixtures
In a PAH-rich emission mixture prepared by burning tar pitch with
coal, the benzo [a]pyrene content was about 90 µg/m3, two to three
times higher than the concentration measured in old coal plants. The
tumour incidence in rats exposed for 16 h/day on five days per week
for 22 months with a subsequent eight-month exposure to clean air was
18%; the mortality rate was not increased in comparison with controls
exposed to clean air. The lung tumour incidences in mice exposed to
the same atmosphere for 10, 12, or 24 months were 86, 70, and 79%,
respectively, with 3.5, 12.5, and 32% in concurrent controls. An
additive or even potentiating carcinogenic effect with other
respiratory-tract carcinogens was demonstrated. In contrast to a group
exposed concurrently to diesel exhaust, the coal-tar pitch did not
cause particle overload in the lung or impair lung clearance (Heinrich
et al., 1986a,b).
Female Wistar rats were exposed by inhalation to 1.1 (groups 1 and 2)
or 2.6 mg/m3 (groups 3 and 4) of an aerosol of a PAH-rich hard
coal-tar pitch condensate containing 20 or 50 µg/m3 benzo [a]pyrene
(among other PAH), for 17 h per day on five days per week for 10
(groups 1 and 3) and 20 months (groups 2 and 4) and then to clean air
for 20 or 10 months. The aerosol contained benz [a]anthracene and
chrysene at concentrations similar to that of benzo [a]pyrene.
Increased mortality was observed due to the development of large,
multiple tumours in the lungs and not to toxic effects. The lung
tumour rates were 4, 33, 39, and 97% in groups 1,2, 3, and 4,
respectively. Other groups exposed simultaneously to 2 or 6 mg/m3
carbon black, which might serve as a PAH carrier, showed an additional
increase in tumour rates, i.e. 89 and 72% in comparison with 39% in
group 3. A group exposed only to carbon black had a tumour rate of
18%. The authors therefore concluded that there was a more than
additive carcinogenic effect after 10 months of exposure. A 'PAH
depot' effect may be involved, in which the residence time of the PAH
is prolonged due to attachment to the inert carbon black particles,
with an extended period elution of adsorbed PAH. Furthermore, the
irritating, inflammatory, and cell proliferation effects of carbon
black enhance the probability of genotoxic effects in the lungs
(Heinrich, 1989; Heinrich et al., 1994a). Most of the lung tumours
observed after exposure to tar-pitch aerosol with or without carbon
black were classified as squamous-cell carcinomas (Heinrich et al.,
1994b).
The preliminary findings of a study on coal gasification tars have
been reported. Coal-tar is a complex mixture containing over 1000
compounds, of which at least 30 are PAH, including benzo [a]pyrene.
In a two-year bioassay for carcinogenicity, female B6C3F1 mice were
fed up to 100 ppm benzo [a]pyrene or up to 1% coal-tar. Forestomach
tumours were observed in mice fed benzo [a]pyrene, the incidence
increasing sharply at doses between 5 and 25 ppm. Forestomach tumours
were also seen in mice fed coal-tar, with a clear increase at 0.3%;
the incidence was approximately the same at 0.3 and 0.6% but declined
at 1.0%, due to mortality from small intestinal adenocarcinomas, which
were not observed at doses below 0.6%. Steady-state DNA adduct levels
were examined in the forestomachs and small intestines of mice fed
benzo [a]pyrene or coal-tar for four weeks. Those fed
benzo [a]pyrene had one major adduct, which also accounted for 10-25%
of the adducts in the forestomachs of mice fed coal-tar. A linear
dose-response relationship was observed between the dose of
benzo [a]pyrene and the adduct levels in the forestomach. The adduct
levels in the forestomachs of mice fed coal-tar increased in a
relatively linear manner at doses above 0.3%. Total adducts and
benzo [a]pyrene-adduct levels in the small intestine increased up to
the 0.6% dose of coal-tar and then decreased (Culp et al., 1996).
A two-stage study of carcinogenicity in female CD-1 mice was performed
to assess the risk deriving from coal-tar formulations used for human
therapeutic purposes, e.g. against dandruff or psoriasis (see also
section 8.2.3). Mice were treated epicutaneously five times per week
with 50 mg of a 1.5% coal-tar ointment for two weeks ('initiation'),
followed by 'promotion' with 50 mg of a 0.1% dithranol cream, used
against psoriasis, three times per week for 40 weeks. A single dose of
50 µg benzo [a]pyrene was given as an initiator as a positive
control. After 40 weeks of promoter treatment, 4/27 tumours were
observed in the mice treated with coal-tar and 14/28 in those given
benzo [a]pyrene; when coal-tar or dithranol was given alone, no
tumours were observed (Phillips & Alldrick, 1994).
7.7.4 Transplacental carcinogenicity
7.7.4.1 Benzo [a]pyrene
Clastogenic responses to benzo [a]pyrene and AHH inducibility were
measured in 11-day-old embryos of genetically different mice (C57 and
DBA mated inter se and mixed) after transplacental treatment with
150 mg/kg bw by garage 15 h before sacrifice. The rate of chromosomal
aberrations was not correlated quantitatively with AHH activity. It
was concluded that not only activation and detoxification of
benzo [a]pyrene in maternal tissue but also other genetically
controlled processes, such as repair and transformation of primary DNA
lesions into true DNA discontinuities, are involved (Adler et al.,
1989).
Benzo [a]pyrene can cross the placenta in mice and rats (Shendrikova
& Aleksandrov, 1974; Shendrikova et al., 1974; Takahashi, 1974;
Baranova et al., 1976), but the concentration of 14C-benzo [a]pyrene
was one to two orders of magnitude lower in mouse embryonic than
maternal tissues after oral administration (Neubert & Tapken, 1988).
Intraperitoneal administration of benzo [a]pyrene to Ha/ICR mice
during the last half of pregnancy increased the incidences of
pulmonary adenomas and skin papillomas in progeny nursed by foster
mothers, excluding uptake of PAH via the milk (Bulay, 1970; Bulay &
Wattenberg, 1971). A similar result was observed in A and C57B1 mice;
furthermore, lung tumours were induced in Ha/ICR strain mice and liver
tumours in the offspring of all three strains (Nikonova, 1977).
Transplacental carcinogenesis has also been reported in rabbits
(Beniashvili, 1978).
Benzo [a]pyrene was given subcutaneously to strain A and C57B1 mice
at 4 or 6 mg per animal once or twice on days 18 and 19 of gestation.
The progeny of dams given a single dose of 4 mg had a significantly
increased number of adenomas; the 6-mg dose induced 77% lung tumours
in A mice and 12% in controls. The maximum dose, 12 mg, induced 32%
liver tumours in male C57B1 mice and 9% in females, with 1% in
controls (Nikonova, 1977).
7.7.4.2 Pyrene
The tumour incidence in the offspring of strain A mice was not
increased by two subcutaneous injections of 6 mg pyrene on days 18 and
19 of pregnancy (Nikonova, 1977).
7.8 Special studies
Adverse effects of PAH unrelated to cancer have also been seen.
Proliferating tissues such as bone marrow, lymphoid organs, gonads,
and intestinal epithelium are affected, but the major target organs
seemed to be those of the haematopoietic and lymphoid systems.
7.8.1 Phototoxicity
Photodynamic compounds can generate superoxide anion radicals in the
presence of near ultraviolet light. In the absence of oxygen, they act
as photoreducing agents. The main effect is epidermal damage.
7.8.1.1 Anthracene
Increased dermal sensitivity to ultraviolet irradiation was seen in
hairless mice after pretreatment with anthracene, but
photocarcinogenesis was not significantly increased (Forbes et al.,
1976; see also Table 92). In guinea-pigs treated six times on the
dorsal skin with anthracene and then irradiated with ultraviolet
light, a photoirritant reaction was observed that reached a maximum
after a few hours but had faded by 24 h (Lovell & Sanders, 1992).
Petroleum can photosensitize human skin to sunlight, resulting in
erythema and pigmentation. Petroleum also enhanced the
immunomodulatory effects of ultraviolet radiation on mammalian skin,
with depletion of antigen-presenting cells which play a critical role
by presenting antigen to thymus-derived lymphocytes. In tests of PAH
that are present at relatively high concentrations in crude oils,
anthracene but not phenanthrene or benzo [a]pyrene induced
photosensitization of mouse skin in vivo and in vitro. Exposure of
skin sections to anthracene at 5 µg/ml, equivalent to 125 ng/mouse,
reduced the numbers of antigen-presenting cells and of Thy-1-positive
dendritic cells (Burnham & Rahman, 1992).
7.8.1.2 Benzo [a]pyrene
Benzo [a]pyrene in the presence of near-ultraviolet radiation
(290-400 nm) had phototoxic effects, observed as haemolysis of human
erythrocytes and inactivation of Escherichia coli (Kagan et al.,
1989). A significant shortening of tumour-free survival was seen in in
Balb/c mice exposed to ultraviolet irradiation before dermal treatment
with benzo [a]pyrene. Ultraviolet irradiation had a systemic effect,
enhancing subsequent dose-dependent tumour induction by
benzo [a]pyrene (Gensler, 1988).
7.8.1.3 Pyrene
In six guinea-pigs given 5 µmol-5 mmol of pyrene dissolved in ethanol,
a strong phototoxic reaction was observed 20 h after ultraviolet A
irradiation at 1 × 103 J/m2 (320-400 nm) (Kochevar et al., 1982).
When mast cells were isolated from Sprague-Dawley rats, incubated with
25 µmol/litre pyrene, and irradiated with ultraviolet B radiation
(280-320 nm) at 60 kJ/m2, they released 80% of their serotonin
(Gendimenico & Kochevar, 1984).
7.8.1.4 Comparisons of individual PAH
The phototoxic effects of benzo [a]pyrene, benz [a]anthracene,
indeno[1,2,3-cd]pyrene, fluoranthene, and perylene were compared by
treating human fibroblasts with these PAH and then irradiating them
with ultraviolet light (<400 nm). A good correlation was found
between the phototoxic effects and known carcinogenic potency:
benzo [a]pyrene and indeno[1,2,3- cd]pyrene were highly toxic,
benz [a]anthracene was distinctly toxic, fluoranthene slightly toxic,
and perylene not cytotoxic (Bauer et al., 1985).
7.8.2 Immunotoxicity
7.8.2.1 Benzo [a]pyrene
(a) Intraperitoneal and intratracheal injection
Mice injected intraperitoneally with a single dose of 50, 100, or 200
mg/kg bw benzo [a]pyrene showed reduced thymic and splenic weights on
day 5, and the splenocyte antibody-forming response of immunized mice
was reduced by 60-90%. At the higher concentrations, lymphocyte
proliferation was decreased significantly. Benzo [a]pyrene altered
both humoral and cellular immunity; B cells were more susceptible than
T cells (Xue et al., 1991).
In a study of the effect of accumulation of benzo [a]pyrene in
lymphoid organs on humoral immunity, B6C3F1 mice were given
intratracheal instillations of 0.4, 4, or 40 mg/kg bw daily for seven
days and immunized with sheep erythrocytes one day later; then, the
number of antigen-specific, antibody-forming cells was measured. The
spleen showed a decrease, but lung-associated lymph nodes showed
either decreased (up to 60%) or increased (up to 100%) numbers,
depending on whether sheep erythrocytes were given intratracheally or
intraperitoneally (Schnizlein et al., 1987).
(b) Dermal exposure
After dermal exposure of female B6C3F1 mice to 0, 5, 20, or 40 mg/kg
bw per day for 14 days, dose-dependent suppression of the
antibody-forming cell response to sheep erythrocytes was seen both
in vivo and in vitro, the number at 40 mg/kg bw being about 30% of
the control value. The immunosuppression was similar when
benzo [a]pyrene was administered at a high dose subcutaneously in
corn oil. The antibody-forming cell response recovered by about day 60
after dermal exposure, while no recovery was seen after subcutaneous
injection (Parrott et al., 1989).
Female B6C3F1 mice were exposed to benzo [a]pyrene at 0, 0.625, 2.5,
5, 20, or 40 mg/kg bw per day for 28 days and were injected
intravenously with sheep erythrocytes on days 11 and 25. An
antigen-specific enzyme-linked immunosorbent assay to sheep
erythrocyte membranes was used to measure the primary immunoglobulin M
response on day 15 and the secondary immunoglobulin G response on day
30. Significant suppression of the primary immunoglobulin M response
was observed at doses of 5 mg/kg bw per day and more, but the serum
titres of animals treated with 0.625 or 2.5 mg/kg bw per day did not
differ from those of vehicle controls. The secondary immunoglobulin G
response was significantly decreased at all doses (Deal, 1995).
(c) Comparisons of benzo[a]pyrene and benzo[e]pyrene
The immunotoxic effects of benzo [a]pyrene and benzo [e]pyrene have
been compared in several studies. In most, benzo [a]pyrene was
clearly immunosuppressive, whereas benzo [e]pyrene was inactive.
In vivo: After activation of splenic lymphocytes, mice were given
single intraperitoneal injections of benzo [a]pyrene and
benzo [e]pyrene at 2.5, 10, or 50 mg/kg bw for 24 or 48 h before
sacrifice. Mononuclear cell populations were then assayed for AHH
activity, blastogenesis, antigen-specific cell-mediated cytotoxicity,
and the percentage of macrophages. Neither PAH had a significant
effect on blastogenesis. Benzo [a]pyrene suppressed cell-mediated
cytotoxicity of T cells by 40-80%, while benzo [e]pyrene had no
effect. These results suggested that mitogen-activated, AHH-induced
splenic lymphocytes metabolize benzo [a]-pyrene to immunocytotoxic
metabolites. T cells are probably activated by early stimulation of T
suppressor cells accompanied by an increase in T suppressor cell
factors (Wojdani & Alfred, 1984).
B6C3F1 mice were given 10 daily subcutaneous injections of
benzo [a]pyrene and benzo [e]pyrene at doses of 5, 20, or 40 mg/kg
bw over a 14-day period. Three to four days after the last dose,
immune function was evaluated. Benzo [a]pyrene reduced the number of
immunoglobulin M and G antibody plaque-forming cells in response to
sheep erythrocytes and lipopolysaccharide, and the TNP-Ficoll
plaque-forming cell response was depressed by 77%. These changes
indicate altered differentiation and antibody production in mature B
cells. No change in the plaque-forming cell response was observed
after exposure to benzo [e]pyrene (Dean, J.H. et al., 1983).
Mice were given subcutaneous injections of benzo [a]pyrene or
benzo [e]pyrene at 5, 10, or 40 mg/kg bw daily for 11 days, followed
by an injection of sheep erythrocytes. Benzo [a]pyrene suppressed the
antibody response to DNP-Ficoll and sheep erythrocytes but not to
lipopolysaccharide; benzo [e]pyrene was not immunosuppressive (White
& Holsapple, 1984).
In vitro: When benzo [a]pyrene was added to cultured mouse spleen
cells in vitro, a metabolism-activating system was not required to
produce immunosuppression. Furthermore, addition of S9 did not
increase the degree of immunosuppression produced by benzo [a]pyrene
(White & Holsapple, 1984).
In several antibody generating systems in vitro, both
benzo [a]pyrene and benzo [e]pyrene caused a significant,
dose-dependent suppression of the T-dependent and polyclonal antibody
responses. A similar result was found in vitro after a 14-day
exposure of mice to 40 mg/kg bw benzo [a]pyrene in vivo, with 98%
suppression of T-cell-dependent antibody. Benzo [a]pyrene-induced
suppression is multicellular, and the greatest sensitivity is found
early in the immune response. Since benzo [e]pyrene induced
immunosuppression only at high concentrations, the preparation may
have been contaminated with benzo [a]pyrene (Blanton et al., 1986).
Benzo [a]pyrene and seven of its metabolites were evaluated for their
ability to suppress the antibody-forming cell response to sheep
erythrocytes in vitro. Direct addition of benzo [a]pyrene or its
7,8-diol to splenocyte cultures induced similar dose-dependent
suppression of the antibody response to the T-dependent antigen, sheep
erythrocytes. In contrast, exposure to the 4,5-diol, 9,10-diol,
6,12-dione, 3-hydroxy, and 4,5-epoxide metabolites resulted in
decreased antibody responses only with high concentrations; these were
associated with decreased viability of the cultures. In addition,
co-incubation with the cytochrome P450 inhibitor alpha-naphthoflavone
attenuated the suppressive effects of benzo [a]pyrene and
benzo [a]pyrene 7,8-diol (Kawabata & White, 1987).
The anti-sheep erythrocyte plaque-forming cell and mixed lymphocyte
responses were inhibited in murine splenic lymphocytes treated with
benzo [a]pyrene at concentrations ranging from 10-4 to 10-8
mol/litre, with maximal depression at 10-5mol/litre. Benzo [e]pyrene
at the same concentrations did not suppress these responses (Urso et
al., 1986).
7.8.2.2 Dibenz [a,h]anthracene
The immunosuppressive effects of dibenz [a,h]anthracene and
3-methyl-cholanthrene were studied in AHH-inducible (C57B1/6) and
non-inducible (DBA/2N) mice after intraperitoneal injection of 25, 50,
or 100 mg/kg bw five days before challenge with sheep erythrocytes or
after administration by gavage of 10-200 mg/kg bw per day for four
days. Immunosuppression occurred in both strains but was more
pronounced in the C57B1 mice after intraperitoneal injection. The DBA
mice were more susceptible to 3-methyl-cholanthrene given by gavage.
The authors suggest that PAH are rapidly metabolized and excreted
after gavage in AHH-inducible mice, whereas in non-inducible mice they
are absorbed and distributed to the target organs. AHH inducibility
may thus play an important role in the immunosuppressive activity of
PAH (Lubet et al., 1984a,b; see also section 7.5.2.1).
7.8.2.3 Fluoranthene
Murine bone-marrow stromal cells were used as a matrix for the growth
and limited development of precursor B cells in vitro, thus
mimicking B lymphopoiesis in vivo. Fluoranthene acutely suppressed B
lymphopoiesis, and the precursor B cell populations exposed to 50
µg/ml fluoranthene disappeared within two weeks. Lymphotoxicity was
mediated by fluoranthene-induced programmed cell death (apoptosis): 5
µg/ml reduced precursor B cell recoveries by > 95% within one or two
weeks. Lower doses altered the dynamics of B cell lymphopoiesis,
leading to accumulation of precursor B cells (Hinoshita et al., 1992).
7.8.2.4 Naphthalene
Naphthalene did not adversely affect the immune response in CD-1 mice
of each sex given up to 133 mg/kg bw per day orally for three months.
No alteration was seen in the lymphocyte proliferative response to the
T-cell mitogens concanavalin A and phytohaemagglutinin, in the delayed
hypersensitivity response to sheep erythrocytess, or in the popliteal
lymph node response. Furthermore, bone-marrow function was not altered
(Shopp et al., 1984).
Naphthalene is known to induce pulmonary and renal toxicity which is
mediated by its reactive, electrophilic metabolites 1,2-naphthalene
oxide, 1-naphthol, and 1,4-naphthoquinone. The immune response of mice
was, however, unaffected by a 90-day oral treatment with up to 25% of
the LD50 value. Furthermore, the number of antibody-forming cells in
splenocyte cultures was not affected by concentrations of naphthalene
up to 200 µmol/litre; however, 200 µmol/litre of 1-naphthol and 7-20
µmol/litre of 1,4-naphthoqninone suppressed the antibody-forming cell
response and decreased cell viability. Splenic microsomes were unable
to metabolize naphthalene, whereas liver microsomes generated 1,2-
naphthalene diol and 1-naphthol. It was concluded that diffusion of
liver metabolites to the spleen is insufficient to induce
immunotoxicity (Kawabata & White, 1990).
7.8.2.5 Comparisons of individual PAH
The effects of exposure to each of 10 PAH on the immunoglobulin M
antibody response to sheep erythrocytes were examined in B6C3F1 mice,
with a further investigation of the relationship of the
Ah gene complex to the immunosuppression. Mice were given
subcutaneous injections over 14 days, and splenic antibody-forming
cells were evaluated after immunization with sheep erythrocytes.
Significant decreases of 55-91% were observed after treatment with
dibenz [a,c]anthracene, dibenz [a,h]anthracene, benz [a]anthracene,
and benzo [a]pyrene, but no significant effects were observed with
anthracene, benzo [e]pyrene, perylene, or chrysene. Generally, the
structure-activity relationship for immunosuppression was correlated
strongly with that for carcinogenicity. Non-inducible DBA/2 mice had
greater PAH-induced immunosuppression than AHH-inducible B6C3F1 mice
(White et al., 1985).
7.8.2.6 Exposure in utero
Oral administration of benzo [a]pyrene at 2 mg/kg bw to Wistar rats
on day 19 of gestation induced a relative decrease in the number of
thymic glucocorticoid receptors in males and a relative increase in
females at six weeks of age. In animals treated at six weeks of age,
no change in the number or affinity of steroid receptors was seen in
males, but there was a 40% decrease in females. The investigators
concluded that benzo [a]pyrene binds to pre-encoded hormone receptors
and interferes with their maturation. When benzo [a]pyrene was given
on day 15 of gestation, a 30% decrease in the number of receptors was
seen in the offspring (Csaba et al., 1991; Csaba & Inczefi-Gonda,
1992).
Strong suppression of the anti-sheep erythrocytes plaque-forming cell
response, the mixed lymphocyte response, and the graft-versus-host
response in vivo were seen in the progeny of female mice that had
been treated intraperitoneally with benzo [a]pyrene at a dose of 150
mg/kg bw during mid-gestation (11-13 days). Immunodeficiency was seen
within one week after birth and persisted for 18 months.
Benzo [a]pyrene induced marked disorientation of T cells up to four
weeks postnatally. Disruption of T-cell differentiation during
ontogenesis was suggested, implying decreased resistance to the
development of neoplasia (Urso & Gengozian, 1980; Urso & Johnson,
1987). A two- to fourfold reduction in maternal leukocytes was seen
within five days of an intraperitoneal administration of 150 µg
benzo [a]pyrene on day 12 of gestation, which persisted to day 10
post partum and was attributed to lymphocyte depletion. Thus,
benzo [a]pyrene exacerbated the depression of leukocytes seen during
pregnancy. After intraperitoneal injection, it can reach the
lymphocytes, where metabolic activation may take place. Investigation
of the maternal T-cell population, both during pregnancy and post
partum, showed exacerbated decreases in the number of thymocytes
present in the thymus. In the spleen, the number of thymocytes
positive for LytI antigens was also depressed, whereas the number of
Lyt 2+ cells was enhanced, reaching levels > 700 times those of
controls. These results demonstrate disruption of the maternal T-cell
repertoire (Urso et al., 1988; Urso & Johnson, 1988).
7.8.2.7 Mechanisms of the immunotoxicity of PAH
Although the mechanism(s) by which PAH adversely affect the immune
system has not been defined, several have been proposed (Ladics &
White, 1996). The mechanism that is most consistent with that accepted
for the carcinogenesis of PAH is that their immunotoxicity is due not
to the parent compound but to the reactive metabolites formed. These
include the 7,8-diol-9,10-epoxide for benzo [a]pyrene (Kawabata &
White, 1987) and the 3,4-diol-1,2-epoxide for the synthetic compound
7,12-dimethylbenz [a]anthracene (Ladics et al., 1991). Several
investigators have suggested that the mechanism of action of PAH
results from their ability to enter the cell membrane and disrupt
transduction of transmembrane signals and/or alter the conformation of
receptors (Pallardy et al., 1992; Thurmond et al., 1989). It has also
been suggested that PAH alter the immune response as a result of their
ability to induce inappropriate alterations in the levels of various
cytokines, such as interleukins 1 and 2 (Lyte et al., 1987; Meyers et
al., 1988; Pallardy et al., 1989), and this mechanism is under active
investigation. While interaction with the Ah receptor has been
suggested as a possible mechanism of action of PAH, the contradictory
results from immunotoxicological studies with genetically different
strains of mice (high and low AHH responders) must be resolved.
Studies primarily conducted in vitro support the hypothesis that the
immunomodulatory effects of PAH are the result of alterations in
calcium mobilization (Burchiel et al., 1991).
7,12-Dimethylbenz [a]anthracene has been extensively studied as a
prototypic immunotoxicant. Exposure to this compound has been reported
to decrease natural killer cell activity (Kimbar et al., 1986), to
decrease resistance to challenge with tumour cells (Dean et al.,
1986), and to increase susceptibility to chemically induced tumours
(Elmets et al., 1988). In vitro, 7,12-dimethylbenz [a]anthracene
has been shown to suppress cytotoxic T-cell activity, possibly by
inducing defects in antigen recognition (House et al., 1989) and/or in
cytokine production (House et al., 1988). Oral administration of a
cumulative dose of 14 mg/kg bw of 7,12-dimethylbenz [a]anthracene
over 14 days suppressed proliferative responses by about 50% in
splenic lymphocytes and by more than 70% in gut-associated lymphocytes
(Burchiel et al., 1990).
7,12-Dimethylbenz [a]anthracene-mediated immunosuppression was shown
to persist for at least eight weeks (Ward et al., 1986; Burchiel et
al., 1988). This compound also decreased resistance to bacterial (Ward
et al., 1984) and viral (Selgrade et al., 1988) infections. Although
no studies have been carried out on humans in vivo, the IC50 for
inhibition of the response of human tonsillar lymphocytes in culture
to foreign tissue antigens was 10-40 µmol/litre; however, antibody
secretion was affected only at a concentration of 100 µmol/litre (Wood
& Holsapple, 1993).
7.8.3 Hepatotoxicity
7.8.3.1 Benzo [a]pyrene
Non-responsive strains of mice (C57B1/6, C3H/HeN, and Balb/cAnN) had
increased relative liver weights after they were fed for 180 days on a
diet containing benzo [a]pyrene resulting in an intake of 120 mg/kg
bw per day (Robinson et al., 1975).
7.8.3.2 Comparisons of individual PAH
The induction of several enzymes has been correlated with cancer
promotion. Wistar rats treated orally with 100 mg/kg bw per day of
benzo [a]pyrene, benz [a]anthracene, anthracene, chrysene, or
phenanthrene for four days showed induction of cytosolic aldehyde
dehydrogenase activity. Benzo [a]pyrene and benz [a]anthracene were
much more effective than the other substances, increasing liver
weights by 27 and 19%, respectively (Törrönen et al., 1981).
The extent of liver regeneration that determines the ability to induce
a proliferative response was investigated in partially hepatectomized
rats fed diets containing various PAH for 10 days. Doses of 51.4 mg/kg
bw per day acenaphthene or 180 mg/kg bw per day fluorene induced a
significant increase in the rate of liver regeneration, but 15.4 mg/kg
bw per day acenaphthene, 51.4 mg/kg bw per day benzo [a]pyrene, or
514 mg/kg bw per day pyrene, anthracene, or phenanthracene had no
effect (Gershbein, 1975).
7.8.4 Renal toxicity
Rats given 50-150 mg/kg bw benzo [a]pyrene orally over four days
showed moderate induction of renal microsomal carboxylesterase
activity; however, administration of 100 mg/kg bw per day anthracene
or phenanthrene had no effect (Nousiainen et al., 1984).
7.8.5 Ocular toxicity of naphthalene
The development of cataracts in rats, mice, and rabbits after
application of naphthalene is a toxic peculiarity of this compound.
Attempts have been made to clarify the metabolic processes responsible
for the formation of insoluble precipitates in the eye.
Cataracts developed in the eyes of rabbits within a few days of
repeated oral administrations of 0.5-1 g/kg bw per day. Oral
administration was more effective than other modes of application
(Pike, 1944). The oxidation products of naphthalene may reach the eye
via the bloodstream, where 1,2-naphthoquinone is formed which can
react with proteins and other cell components to form insoluble
precipitates with a characteristic brown colour (Van Heyningen &
Pirie, 1967).
C57B1/6 mice, which are susceptible to induction of cytochromes P450,
were given naphthalene by intraperitoneal administration at 500-2000
mg/kg bw. Cataracts were induced in a dose-dependent manner within 8
h. The effect was reduced by pretreatment with P450 inhibitors and
antioxidants and was increased by pretreatment with P450 inducers or
glutathione depletors. Cataracts were also induced in a dose-dependent
manner by intraperitoneal injection of 1,2- or 1,4-naphthoqninone at
5-250 mg/kg bw (molar potency about 10-fold higher). DBA/2 mice, which
are resistant to induction of cytochromes P450, did not develop
cataracts. The authors concluded that P450-dependent bioactivation was
necessary to form reactive intermediates, which are assumed to be
naphthoqninone or a free-radical derivative (Wells et al., 1989).
When a lens cell line from transgenic mice was exposed to the
1,2-dihydrodiol and 1,2-naphthoquinone metabolites of naphthalene, the
dihydrodiol did not appear to be toxic but the naphthoquinone induced
depletion of glutathione levels. Detoxification of naphthoquinone by
the enzyme quinone oxidoreductase prevents formation of a semiquinone
radical by two-electron reduction (Russell et al., 1991).
All rats of various strains given naphthalene orally developed
cataracts. The authors proposed that naphthalene dihydrodiol is
produced in the liver, reaches the aqueous humour, and penetrates the
lens, where it is metabolized to naphthoquinone. Feeding of 1-naphthol
did not induce opacification (Xu et al., 1992).
7.8.6 Percutaneous absorption
As dermal penetration is one of the major routes of entry after
occupational exposure, in-vivo and in-vitro models have been developed
to assess it.
In experiments in vitro on the dermal penetration of
benzo [a]pyrene and pyrene in guinea-pigs, pyrene was absorbed mainly
by passive diffusion, but benzo [a]pyrene was biotransformed during
absorption, and the 7,8,9,10-tetrol metabolite of the putative
ultimate carcinogen was detected in the receptor fluid of diffusion
cells (Ng et al., 1992).
In skin preparations from mice, rats, rabbits, guinea-pigs, marmosets,
and humans treated with benzo [a]pyrene, both the parent compound and
a full spectrum of metabolites were detected, depending on metabolic
viability, i.e. previously frozen skin preparations could not
metabolize benzo [a]pyrene (Kao et al., 1985).
14C-Benzo [a]pyrene was administered to the nuchal area of mice at
doses of 1.25-125 µg/cm2, and the animals were sacrificed seven days
later. Benzo [a]pyrene disappeared rapidly from the application site,
at a rate of 6% within 1 h and 40% within 24 h; after seven days, 7%
remained at the original site. Most of the benzo [a]pyrene was
excreted via the hepatobiliary system and found in the faeces, with
35% after 24 h, 58% after 48 h, and 80% after seven days. Only 10% of
the radiolabel was detected in urine. Uptake was saturated at doses >
15 µg/cm2, implying an enhanced risk for tumour induction in the skin
epithelium (Sanders et al., 1986).
The binding of benzo [a]pyrene to DNA and protein in mouse skin was
15-20 times greater when acetone was used as the vehicle than with a
low-viscosity oil (Ingram & Phillips, 1993). While acetone solutions
of 14C-benzo- [a]pyrene readily penetrated skin from human cadavers,
a significantly smaller amount moved from soil into skin. No
partitioning of benzo [a]pyrene from human skin to plasma was
observed. An experiment with rhesus monkeys in vivo also showed
significantly less absorption from soil (Wester et al., 1990).
7.8.7 Other studies
7.8.7.1 Benzo [k]fluoranthene
Intraperitoneal injections of benzo [k]fluoranthene, a widespread
PAH, to rats for three days induced maximal cytochrome P450/448
activity. Liver microsomes were then prepared, and the metabolic
profile of benzo [k]-fluoranthene was analysed by gas
chromatography-mass spectrometry. trans-5,6-, 8,9-, and mainly
10,11-dihydrodiols were the primary metabolites in noninduced rats.
Pretreatmant with PAH resulted in the generation of 5,6 and 8,9
isomers as the main metabolites, due to induction of monooxygeuases;
secondary metabolism to triols and tetrols was also induced. The
putative ultimate carcinogen, 3,4-diol-1,2-epoxy benz [a]anthracene,
was detected after pretreatment of liver microsomes with
benzo [k]fluoranthene. The authors concluded that
benzo [k]fluoranthene is a relevant component of environmental
pollution and enhanced the carcinogenic risk of benz [a]anthracene
(Schmoldt et al., 1981; Jacob et al., 1981a).
7.8.7.2 Benzo [a]pyrene
Rats were fed a diet containing 400 mg/kg benzo [a]pyrene with or
without 2 g/kg of ß-carotene. Benzo [a]pyrene had no effect on serum
retinol levels, but vitamin A levels were decreased in liver and small
intestine at two weeks, with a 30% decline by four weeks. A similar
effect was not observed in rats fed ß-carotene simultaneously. AHH can
be induced by ß-carotene, but this may not be the mechanism by which
tumours are prevented (Edes et al., 1991).
The role of benzo [a]pyrene in the induction of arteriosclerosis was
investigated by studying its effects on bovine arterial smooth muscle
cells in vitro. The number of cells was unchanged by exposure, but
the secretion of newly synthesized collagen was decreased. Total
cellular DNA was decreased and collagen secretion increased when the
cells were preincubated with platelet factors rather than a serum-free
medium (Stavenow & Pessah-Rasmussen, 1988).
Microsomes from rat aorta transformed benzo [a]pyrene into various
carcinogenic and toxic metabolites after induction with
3-methylcholanthrene. Thus, carcinogenic metabolites of PAH deriving
from cigarette smoke tars could cause endothelial injury, contributing
to the role of cigarette smoking in arteriosclerosis (Thirman et al.,
1994).
7.8.7.3 Phenanthrene
In a study of the oxidation of phenanthrene by liver microsomes from
rats with or without pretreatment with inducers, microsomes from
untreated rats produced only trans-9,10-diol phenanthrene, but
pretreatment with various PAH also led to oxidation at the 1,2 and 3,4
positions. Considerable amounts of the proximal carcinogen 1,2-diol
phenanthrene were detected, but the concentration of the ultimate
carcinogen 1,2-diol-3,4-epoxide was very low. These results are in
accordance with the questionable carcinogenic potency of phenanthrene
(Jacob et al., 1982a; see Table 91).
7.8.7.4 Comparisons of individual PAH
Activation of platelets by calcium ionophore A23187 can mobilize
intracellular stores of calcium ion and stimulate thromboxane
biosynthesis, which can be measured as thromboxane B2 synthesis.
Benz [a]anthracene, chrysene, benzo [a]pyrene, and
benzo [ghi]perylene inhibited thromboxane B2 production in
A-23187-induced, washed platelets from rabbits, while anthracene and
pyrene appeared to stimulate thromboxane B2 synthesis. Fluoranthene,
benzo [b]fluoranthene, benzo [k]fluoranthene, and benzo [e]pyrene
had little or no effect on activation (Yamazaki et al., 1990).
Benzo [a]pyrene, benzo [k]fluoranthene, benzo [b]fluoranthene,
chrysene, benz [a]anthracene, pyrene, phenanthrene, and fluoranthene
were toxic to human hepatoma cell cultures (HepG2), as measured with
neutral red, whereas fluorene, anthracene, acenaphthene, and
acenaphthylene were not (Babich et al., 1988).
7.9 Toxicity of metabolites
Derivatives of parent PAH have been tested for mutagenicity and
carcinogenicity in a number of experiments in order to assign the
effects to definite metabolites or to rank the potency of known
metabolites of a parent compound. Some studies addressed the steric
factors that determine mutagenic or carcinogenic effects, such as the
diastereomers of epoxides and the role of the methyl group in 5- and
6-methylchrysene. These studies are summarized in Tables 93 and 94,
although a few of the studies are reported in detail below.
7.9.1 Benzo [a]pyrene
Benzo [a]pyrene is metabolized to about 20 primary and secondary
oxidized metabolites and to a variety of conjugates. Several
metabolites can induce mutations, transform cells, and bind to
cellular macromolecules, but the 7,8-diol-9,10-epoxides are presently
considered to be the major ultimate carcinogens (DePierre & Ernster,
1978; Pelkonen & Nebert, 1982).
Table 93. Mutagenicity of metabolites of polycyclic aromatic hydrocarbons
Compound Metabolite Test system Results References
Benzo[b]fluoranthene 9,10-Diol Mouse, dermal Positive Amin et al. (1991a)
Benzo[j]fluoranthene 9,10-Diol Mouse, dermal Positive LaVoie et al. (1980; 1982b)
Benzo[k]fluoranthene 9,10-Diol Positive LaVoie et al. (1980)
Benzo[a]pyrene 7,8-Diol-9,10-epoxide Unscheduled DNA binding Positive Gill et al. (1991)
(syn + anti) DNA synthesis Positive
7,8-Diol-9,10-epoxide Hamster embryo cells, Positive Mager et al. (1977)
(2 stereoisomers) transformation
1-Hydroxy Reverse mutation, Positive (±S9) Schoeny et al. (1985)
2-Hydroxy S. typhimurium TA 1538, Negative (S9)
3-Hydroxy TA98,TA100 Positive (+S9)
4-Hydroxy Negative (±S9)
6-Hydroxy Positive (-S9)
7-Hydroxy Positive (+S9)
9-Hydroxy Positive (+S9)
10-Hydroxy Negative (±S9)
12-Hydroxy Positive (±S9)
1,6-Quinone Negative (±S9)
3,6-Quinone Positive (+S9)
4,5-Quinone Negative (±S9)
6,12-Quinone Negative (±S9)
trans-4,5-Diol Negative (±S9)
cis-4,5-Diol Positive (+S9)
trans-7,8-Diol Negative (±S9)
trans-9,10-Diol Negative (±S9)
4,5-Epoxide Positive (±S9)
1-Hydroxy Forward mutation, Positive (+S9) Schoeny et al. (1985)
2-Hydroxy S. typhimurium TM677 Negative (±S9)
3-Hydroxy Negative (±S9)
4-Hydroxy Negative (±S9)
6-Hydroxy Negative (±S9)
7-Hydroxy Positive (±S9)
9-Hydroxy Negative (±S9)
10-Hydroxy Negative (±S9)
Table 93. (continued)
Compound Metabolite Test system Results References
12-Hydroxy Negative (±S9)
1,6-Quinone Negative (±S9)
3,6-Quinone Negative (±S9)
4,5-Quinone Negative (±S9)
6,12-Quinone Negative (±S9)
trans-4,5-Diol Positive (+S9)
cis-4,5-Diol Positive (+S9)
trans-7,8-Diol Positive (+S9)
trans-9,10-Diol Negative (±S9)
cis-7,8-Diol Positive (+S9)
4,5-Epoxide Positive (±S9)
Benzo[e]pyrene 9,10-Diol-11,12-epoxide Bacterial and mammalian cells Weakly positive Wood et al.(1980)
Chrysene 1,2-Diol Bacterial and mammalian cells Positive Wood et al. (1977)
1,2-Diol-3,4-epoxide
5,6-Oxide S. cerevisiae, D4-RDII Positive Siebert et al. (1981)
Cyclopenta[cd]pyrene 3,4-Diol Bacterial and mammalian cells, Positive Gold et al. (1980)
mutagenicity and transformation
trans-3,4-Epoxide Calf thymus DNA, DNA binding Positive Beach et al. (1993)
Fluoranthene 2,3-Diol Bacterial calls Positive LaVoie et al. (1982a)
2,3-Diol-1,10b-epoxide Bacterial calls: Positive Rastetter et al. (1982)
1,10b-Diol-2,3-epoxide S. typhimurium TM677 Weakly positive
or negative
Dibenz[a,h]anthracene 3,4-Diol Bacterial cells Most mutagenic Wood et al. (1978)
compound among
3 dihydrodols
5,6-Epoxide Hamster embryo cells, Positive Huberman et al. (1972);
transformation Marquardt et al. (1972)
Dibenz[a,h]pyrene 1,2-Diol Bacterial cells Positive Wood et al. (1981)
Dibenz[a,i]pyrene 3,4-Diol Bacterial cells Positive Wood et al. (1981)
5-Methylcholanthrene 1,2-Diol and 7,8-diol Bacterial cells Positive Hecht et al. (1978)
5-Hydroxy S. typhimurium TA100 Weakly positive Amin et al. (1979)
1-Methylphenanthrene 1,4-Diol Bacterial cells Positive LaVoie et al. (1981c)
5,6-Diol
Phenanthrene 1,5-Diol,-3,4-epoxide Bacterial and mammalian cells Positive Wood et al. (1979)
9,10-Oxide S. cerevisiae D4-RDII Positive Siebert et al. (1981)
S9, 9000 × g microsomal fraction of liver
Table 94. Carcinogenicity of metabolites of polycydic aromatic hydrocarbons
Compound Metabolite Species, route of Type of Results References
administration investigation,
duration, dose
Benz[a]anthracene 3,4-Diol and Mouse, Carcinogenicity, 3R,4R-Diol: 71% with tumours; Wislocki et al.
3,4-diol-1,2- intraperitoneal 26 weeks other enantiomer not tumorigenic; (1979)
epoxide 3,4-diol-1,2-epoxide: 100% with tumours;
other enantiomer: 42%; control: 13%
Benzo[b]fluoranthene 9,10-Diol Mouse, dermal Initiation Positive Amin et al.
(1991a)
Benzo[j]fluoranthene 9,10-Diol Mouse, dermal Initiation Positive LaVoie et al.
(1980, 1982b)
4,5-Diol Mouse, dermal Initiation 2,3-Diol: 5-11 %; 4,5-diol: 78-100%; Rice et al.
9,10-Diol 9,10-diol: 60%; control: 10% (1987)
2,3-Diol
Benzo[c]phenanthrene 1,2-Diol Mouse, dermal Initiation 1,2-Diol: 3-4% with tumours; Levin et al.
3,4-Diol 3,4-diol: 28-47% with tumours; (1980)
5,6-Diol 5,6-diol: 3-7% with tumours;
1,2-Epoxy- 1,2-epoxy-3,4-diol: 80-90% with tumours;
3,4-diol control: no tumours
3,4-Diol-1,2- Mouse, dermal Initiation 95-100% with tumours; control: 10% Amin et al.
epoxide (1993)
anti-3,4-Diol- Rat, Carcinogenicity 100 % with mammary tumours; Hecht et al.
1,2-epoxide intramammary 1 × 12.2 µmol vehicle control: 3% (1994)
Benzo[a]pyrene 7,8-Diol Mouse,dermal Carcinogenicity, 100% with skin tumours Conney (1982)
43 µg every 2
week; 60 weeks
anti-7,8-Diol- Rat, Carcinogenicity, 47% with mammary tumours; vehicle Hecht et al.
9,10-epoxide intramammary 1 × 12.2 µmol control 3% (1994)
Table 94. (continued)
Compound Metabolite Species, route of Type of Results References
administration investigation,
duration, dose
Benzo[e]pyrene 4,5-Diol Mouse, dermal Initiation No significant effect Buening et al.
9,10-Diol (1980); Slaga
et al. (1980,
1981)
9,10-Diol Mouse, newborn, Carcinogenicity No induction of pulmonary tumours; Buening et al.
9,10-Diol- intrapentoneal significant induction of hepatic (1980); Chang
11,12-epoxide tumours et al. (1981)
Chrysene 1,2-Diol Mouse, dermal Initiation 0.4, 39, 60, and 79% with tumours; Levin et al.
1.25, and 4 control: 7% (1978)
µmol/ animal, 1 x
1,2-Diol Mouse, dermal Initiation 1,2-Diol: positive Slaga et al.
4,5-Diol 3,4-Diol: negative (1980, Chang
1,2-diol, Mouse, newborn Carcinogenicity Induction of pulmonary adenomas Buening et al.
1,2-Diol- (1979); Chang
3,4-epoxide et al. (1983)
Dibenz[a,h]anthracene 3,4-Diol Mouse, dermal Carcinogenicity Induction of skin tumours; induction of Buening et al.
pulmonary tumours in newborn mice (1979); Slaga et
al.(1980), 1981)
5,6-Epoxide Mouse, dermal Initiation Poorly active Van Duuren at
al. (1967)
Dibenzo[a,h]pyrene 1,2-Diol Mouse, dermal, Initiation Positive Wood et al.
intraperitoneal (1981)
Dibenzo[a,h]pyrene 3,4-Diol Mouse, dermal, Initiation Positive Wood et al.
intraperitoneal (1981)
Indeno[1,2,3-cd]pyrene 1,2-Diol, Mouse, dermal Initiation 1,2-Diol: 80%, 1,2-oxide, 80%; Rice et al.
1,2-Oxide 8-hydroxy, 30% tumour-bearing animals (1986)
8-Hydroxy
Table 94. (continued)
Compound Metabolite Species, route of Type of Results References
administration investigation,
duration, dose
5-Methylcholanthrene 1,2-Diol Mouse, dermal Initiation, 1,2-Diol: 19/20 papillomas, 7/20 Hecht et al.
7,8-Diol 3 µg/animal, carcinomas; 7,8-diol: 10/20 (1980)
9,10-Diol 10 × in 20 days papillomas, no carcinomas; 9,10-diol:
no papillomas, no carcinomas
5-Hydroxy Mouse, dermal Initiation 45-950% with skin tumours; solvent Amin et al.
control: 5% (1981)
1,2-Diol- Mouse, newborn Carcinogenicity Induction of pulmonary tumours Amin et al.
3,4-epoxide (1991b)
Phenanthrene 1,2-Diol, Mouse, dermal Initiation Very weakly positive Wood et al.
3,4-Diol (1979)
9,10-Diol
1,2-Diol- Mouse, newborn Carcinogenicity No induction of pulmonary tumours Buening et al.
3,4-epoxide (1979)
Cytochrome P450-dependent metabolizing activity is low in skin. When
14C-benzo [a]pyrene was incubated with arachidonic acid and cytosol
prepared from rat, mouse, or human epidermis, benzo [a]pyrene 1,6-,
3,6-, and 6,12-quinones and other metabolites were formed. These
metabolic reactions were inhibited by selective inhibitors of
lipoxygenase, demonstrating that human and rodent skin can metabolize
benzo [a]pyrene through an arachidonic acid-dependent lipoxygenase
pathway (Agarwal & Mukhtar, 1991). In cultures of normal human
melanocytes, benzo [a]pyrene diols and small amounts of quinones and
phenols were detected in the fraction extractable in organic solvents,
and glucuronide and sulfate conjugates in the water-soluble fraction
(Agarwal et al., 1991).
The (+) anti-7,8-dihydrodiol-9,10-epoxide of benzo [a]pyrene reacts
with DNA to yield almost exclusively the deoxyguanosine adduct, in
which the epoxide function has reacted with the amino group at C2 of
the guanine base. In mouse skin, this one adduct accounts for about
97% of the total binding of benzo [a]pyrene to DNA (Jeffrey et al.,
1976).
Benzo [a]pyrene 7,8-dihydroxy-9,10-epoxide and two other metabolites
(which were detected by trapping with exogenous DNA as described by
Ginsberg & Atherholt, 1989) were present in all serum samples from
mice of three strains and Sprague-Dawley rats after intraperitoneal
injection of 50-200 mg/kg bw benzo [a]pyrene. It was concluded that
transport can occur via the systemic circulation (Garg et al., 1991).
Benzo [a]pyrene or its 4,5-oxide or 7,8-diol-9,10-epoxy metabolite
was administered directly into Swiss mouse embryos at 0.1-4 µg per
embryo on days 10, 12, and 14 of gestation. The 7,8-diol-9,10-epoxy
metabolite was the most potent embryotoxic and teratogenic compound in
fetuses examined on day 18, causing 85% embryolethality and 100%
malformations. Benzo [a]pyrene and the other metabolite did not
increase the incidence of malformations significantly (Barbieri et
al., 1986).
7.9.2 5-Methylchrysene
Investigations of the reaction of 5-methylchrysene diol epoxide
enantiomers with DNA bases and in S. typhimurium showed the
importance of both the absolute configuration and the position of the
methyl group. The 5-methyl-chrysene 1 R,2 S-diol-3 S,4 R-epoxide,
with the methyl group and epoxide ring in the same bay region, were
the most reactive (Melikian et al., 1988).
7.9.3 1-Methylphenanthrene
Incubation of rat liver preparations with 1-methylphenanthrene gave
rise to the 3,4- and 5,6-dihydrodiols of 1-hydroxymethylphenanthrene,
1-methylphenanthrene, 1-hydroxymethyl-phenanthrene, and unidentified
derivatives as metabolites; the dihydrodiols were mutagenic in the
presence of an exogenous metabolizing system (LaVoie et al., 1981b).
7.10 Mechanisms of carcinogenicity
7.10.1 History
In the early 1940s, theoretical chemistry was used to predict the
chemical reactivity of PAH. Pullman (1945,1947) introduced the terms
K- and L-region to describe the reactivity of PAH based on Hückel
molecular orbital calculations (see Figure 9). Later, the term 'bay
region' was introduced for molecular substructures that contribute to
the formation of some ultimate carcinogens. Discrete values of complex
delocalization energies at the K- and L-regions of a PAH were
correlated with its carcinogenic potency. At the time, however, there
was only a limited database on metabolic processes and little
experimental confirmation, and the K-region theory was later found to
be incompatible with the results of experimental work.
7.10.2 Current theories
Miller & Miller (1977) proposed the theory of reactive electrophiles
in chemical carcinogenesis. According to this theory, PAH are
activated by microsomal enzymes to proximate and finally ultimate
carcinogens, which are characterized by an electrophilic centre that
can react with nucleophilic sites on macromolecules such as DNA, RNA,
and protein.
After the discovery that diol epoxides are metabolites of PAH (Sims &
Grover, 1974), a theoretical model was presented by Jerina et al.
(1976), who found that synthesized arene oxides were mutagenic without
metabolic activation. The bay-region theory states two prerequisites
for carcinogenic potency: The epoxide group of an ultimate metabolite
must be part of a bay region (see Figure 10), and the hydroxy groups
of the diol epoxide are preferentially located in the 'pre-bay
region'. The presence of the epoxide group on a saturated benzene ring
in a bay region facilitates ring opening, i.e. the delocalization
energy forming the carbonium ion is higher. This is important for
reactions with DNA via a carbonium ion, which is an alkylating agent.
For example, the metabolic pathway of benzo [a]pyrene is hypothesized
to start with a 7,8 oxidation followed by hydrolysis to
7,8-dihydrodiol, and terminated by 9,10-oxidation, yielding the
ultimate carcinogen 7,8-dihydrodiol-9,10-epoxide. Calculation of the
carbonium ion delocalization energies by the pertubational molecular
orbital method results in a rough correlation with experimentally
determined carcinogenic potency.
The number of PAH tested for carcinogenicity in experimental animals
doubled between the time that Jerina et al. (1976) published their
work and 1980, to 50 compounds. A calculation of the carbonium ion
delocalization energies at that time (Qianhuan, 1980) revealed a
deviation from the energy-carcinogenicity correlation for compounds
that had not been investigated by Jerina et al. To avoid the
shortcomings of the bay-region theory, Qianhuan took into account the
data on all PAH that had been tested for carcinogenicity and
postulated the di-region theory, a bifunctional electrophilic theory
Table 79. Effects of benzo[a]pyrene on fertility in experimental animals
Species Sex/No. Route of Duration, dose Effects Reference
(strain) per administration
group
Mouse M Diet Up to 30 days before mating, NOEL: 150 mg/kg bw per day Rigdon & Neal (1965)
White 5 37.5, 75, or 150 mg/kg bw Parameters: sperm in lumen
per day of testes; number of offspring
Mouse F Diet 20 days before mating NOEL: 150 mg/kg bw per day Rigdon & Neal (1965)
White 5-65 37.5, 75, or 150 mg/kg bw Parameter: number of offspring
Swiss per day
Mouse F Intraperitoneal Day 14 before mating, 10, 100 mg/kg bw: dose-dependent Mattison et al. (1980)
DBA/2N 15 10, 100, 200, or 500 mg/kg decrease in number of pups
bw once 200, 500 mg/kg bw: completely
infertile; threshold: 3.4 mg/kg bw;
50% effect dose: 25.5 mg/kg bw
Mouse F Intraperitoneal Day 21 before sacrifice, Dose-dependent increase in Mattison et al. (1980)
DBX2N 5, 10, 50, 100, or 500 mg/kg primordial oocyte destruction;
bw once 500 mg/kg: 100% destruction;
threshold: 2.7 mg/kg bw; 50%
effect dose: 24.5 mg/kg bw
Mouse F Intraperitoneal Day 13 before sacrifice, 100 mg/kg bw: significant increase Mattison & Nightingale
B6 and D2 5 100 mg/kg bw once in primordial oocyte destruction in (1980)
both genotypes; effects in B6 mice
greater than in D2 mice
Mouse F Intra-ovarian Day 14 before sacrifice, 10 µg: decreased ovarian weight Mattison et al. (1989)
C57BI/6N (136), injection 10 µg/right ovary once (D2); decreased ovarian volume (D2
DBA/2N (D2), and F1); decreased antral follicles
D2B6F1(F1) (F1) decreased number of small
follicles (D2 and F1)
Table 79. (continued)
Species Sex/No. Route of Duration, dose Effects Reference
(strain) per administration
group
Mouse F Intraperitoneal 1, 2, 3, and 4 weeks 500 mg/kg: 35% mortality Swartz & Mattison,
C57BI/6N 5 before sacrifice; 1, 5, 1-500 mg/kg bw: dose- and time- 1985);
10, 50, 100, or 500 dependent decrease in ovarian Miller et al. (1992)
mg/kg bw volume, total volume and number of
corpora lutea/ovary (for last
parameter, after 1 week threshold
was about 1 mg/kq bw and ED50 1.6
mg/kg bw);effect transitory in
low-dose groups, butnot reversible
in two highest by four weeks
For genotypes of the mouse strains used see section 7.5.1.1
7.5.1.3 Effects on postnatal development
Three studies of the postnatal effects of benzo [a]pyrene on mouse
offspring, with administration dermally, intraperitoneally, or orally,
showed adverse effects, including an increased incidence of tumours,
immunological suppression, and reduced fertility (see also Table 80).
7.5.1.4 Immunological effects on pregnant rats and mice
Benzo [a]pyrene given to pregnant rats on day 15 or 19 of gestation
caused alterations at the thymic glucocorticoid receptors in the
offspring, suggesting binding to the pre-encoded hormone receptors and
interference with receptor maturation (Csaba et al., 1991; Csaba &
Inczefi-Gonda, 1992; see also section 7.8.2.6).
Strong suppression of immunological parameters was found in the
progeny of mice that had been treated intraperitoneally with
benzo [a]pyrene at mid-gestation (Urso & Johnson, 1987; see also
section 7.8.2.6).
7.5.2 Naphthalene
7.5.2.1 Embryotoxicity
Naphthalene was administered by gavage at 50, 150, or 450 mg/kg bw per
day to pregnant Sprague-Dawley rats on days 6-15 of gestation, i.e.
during the main period of organogenesis. The dams showed signs of
toxicity including lethargy, slow breathing, prone body posture, and
rooting, and these effects persisted after the end of dosing with the
high dose. The body-weight gain of treated animals was reduced by 31
and 53% in the groups at the two higher doses. Naphthalene did not
induce fetotoxic or teratogenic effects, and the numbers of corpora
lutea per dam, implantation sites per litter, and live fetuses per
litter were within the range in controls. The maternal NOAEL was
< 50 mg/kg bw per day (National Toxicology Program, 1991).
In a second study, doses of 0, 20, 80, or 120 mg/kg bw per day were
given to rabbits by gavage during days 6-19 of gestation. There were
no signs of maternal toxicity, fetotoxicity, or developmental toxicity
(National Toxicology Program, 1992a).
7.5.2.2 Toxicity in cultured embryos
Mice injected intraperitoneally on day 2 of gestation with 14 or 56
mg/kg bw naphthalene were sacrificed 36 h later, and embryos were
cultured in vitro. Maternal doses below the LD50 value inhibited
the viability and implantation capacity of the embryos, and attachment
and embryonic growth in vitro were markedly decreased (Iyer et al.,
1990).
Table 80. Effects of benzo[a]pyrene on postnatal development in experimental animals
Species Sex/No. Route of Duration, dose Effects Reference
(strain) per administration
group
Mouse F Dermal Entire gestation period F1- F4: sensitization of offspring: increased Andrianova
non-inbred 1 drop of 0.5% solution, incidence of papillomas and carcinomas (1971)
twice per week; F1-F4 in all generations compared with animals
treated with BaP, m not treated in utero
1x/week, f 2x/week
Mouse F Intraperitoneal Day 11-13 or 16-18 F1: no difference in birth rate, litter size of Urso &
C3H/Anf 25 of gestation, 100 or progeny compared to controls; severe suppression Gengozian
150 mg/kg of anti-SRBC PFC response up to 78 weeks of life (1980)
(see also section 7.8.2.6); 11-1 fold increase in
tumour incidence (liver, lung, ovaries) after
56-78 weeks
Mouse F Gavage Days 7-16 of F1: 10 mg/kg markedly impaired fertility (by 20%) MacKenzie &
CD-1 gestation, 10, 40, and reduced testis weight (by 40%), 34% sterility Angevine
160 mg/kg bw per day of females; 40 and 160 mg/kg: fertility impaired (1981)
by > 900%/100%; testis weight reduced by > 800%;
100%/100% sterility of females
anti-SRBC PFC, anti-sheep red blood cell antibody (plaque)-forming cells
In a subsequent study, three-day-old whole mouse embryos were
collected at the blastocyst stage, cultured in NCTC 109 medium, and
exposed to naphthalene at 0.16, 0.2, 0.39, or 0.78 mmol/litre for 1 h
with and without S9. They were then transferred to toxicant-free
medium, cultured for 72 h, and evaluated microscopically. Naphthalene
was not directly embryotoxic, but growth and viability were decreased
in the presence of S9, with 100% embryolethality at doses > 0.2
mmol/litre; furthermore, hatching and attachment rates were
significantly decreased. The approximate LC50 in S9-supplemented
media was 0.18 mmol/litre (Iyer et al., 1991).
7.6 Mutagenicity and related end-points
Benzo [a]pyrene has been used extensively as a positive control in a
variety of short-term tests. It is active in assays for the following
end-points: bacterial DNA repair, bacteriophage induction, and
bacterial mutation; mutation in Drosophila melanogaster; DNA
binding, DNA repair, sister chromatid exchange, chromosomal
aberration, point mutation, and transformation in mammalian cells in
culture; and tests in mammals in vivo, including DNA binding, sister
chromatid exchange, chromosomal aberration, sperm abnormalities, and
somatic mutation at specific loci (Hollstein et al., 1979; De Serres &
Ashby, 1981). Positive effects were seen in most assays for the
mutagenicity of benzo [a]pyrene.
A selection of these studies is summarized in Tables 81-88. All of the
data available on the other PAH considered in this monograph were
taken into account. Because of the amount of data, the purities of the
chemicals tested and details of the assay conditions are omitted from
the tables, but they do show the results obtained when S9 was used.
Variations in the S9 metabolic activation component of the assay
system, e.g. the age, sex, and strain of the rats used as a source of
liver and any pretreatment with enzyme inducers such as Aroclor,
3-methylcholanthrene, or phenobarbital, may markedly affect the
results and may account for apparent discrepancies.
DNA binding of benzo [a]pyrene was observed in various species. For
example, adducts were found in cells from hamsters, mice (Arce et al.,
1987), rats (Moore et al., 1982), and chickens (Liotti et al., 1988),
in calf thymus DNA (Cavalieri et al., 1988a), and in human cell
systems (Moore et al., 1982; Harris et al., 1984). Formation of DNA
adducts was inhibited in the presence of scavengers of active oxygen
species like superoxide dismutase, catalase, and citrate-chelated
ferric iron, indicating that reactive oxygen species such as
superoxide, OH radicals, and singlet oxygen may be involved in DNA
binding (Bryla & Weyand, 1991). Benzo [a]pyrene at a total dose of 10
mg/kg bw induced gene mutations in mice, as seen in the coat-colour
spot test (Davidson & Dawson, 1976).
The results of tests for reverse mutation in Salmonella
typhimurimum (Ames test) and for forward mutation in
S. typhimurimum strain TM677 are presented in Table 81. Bacterial
tests for DNA damage in vitro are shown in Table 82. The results of
tests for mutagenicity in yeasts and Drosophila melanogaster,
including host-mediated assays, are shown in Table 83. The results of
various assays carried out on mammalian cells in vitro are
summarized in Tables 82-86. The results of tests in vivo are shown
in Tables 87 and 88.
The activity of PAH in short-term tests is summarized in Table 89,
which gives the evaluations of IARC (1983; see also Section 12) and
the results of studies reported after 1983. Only three of the 33 PAH
considered, i.e. anthracene, fluorene, and naphthalene were inactive
in all short-term tests; 16 had mutagenic effects. Eight PAH showed a
tendency for mutagenic activity, but the data are still too sparse to
permit a final judgement. The available information on acenaphthene,
acenaphthylene, benzo [a]fluorene, and coronene is still inadequate.
As phenanthrene and pyrene showed inconsistent results in various
experiments, they could not be clearly classified as mutagenic.
7.7 Carcinogenicity
Most of the studies that have been conducted on PAH were designed to
assess their carcinogenicity. Studies on various environmentally
relevant matrices such as coal combustion effluents, vehicle exhaust,
used motor lubricating oil, and sidestream tobacco smoke showed that
PAH are the agents predominantly responsible for their carcinogenic
potential (Grimmer et al., 1991b). Because of the abundance of
literature, only studies involving the administration of single PAH
have been taken into account in this monograph.
Benzo [a]pyrene has been tested in a range of species, including
frogs, toads, newts, trout, pigeons, rats, guinea-pigs, rabbits,
ferrets, ground squirrels, tree shrews, marmots, marmosets, and rhesus
monkeys. Tumours have been observed in all experiments with small
animals, and the failure to induce neoplastic responses in large
animals has been attributed to lack of information on the appropriate
route or dose and the inability to observe the animals for a
sufficient time (Osborne & Crosby, 1987a). In studies with other PAH,
benzo [a]pyrene was often used as a positive control and therefore
administered at only one concentration. Benzo [a]pyrene has been
shown to be carcinogenic when given by a variety of routes, including
diet, gavage, inhalation, intratracheal instillation, intraperitoneal,
intravenous, subcutaneous, and intrapulmonary injection, dermal
application, and transplacental administration.
Assessment of the carcinogenic potency of the selected PAH is
restricted for various reasons: Many of the studies performed before
about 1970 were carried out without controls, without clearly defined,
purified test substances, or using experimental designs and facilities
considered today to be inadequate. Despite these shortcomings, all of
the available studies were taken into account, except for those on
dibenz [a,h]anthracene and benzo [a]pyrene. An overview of the
results, as reported by the authors, is given in Table 90. To
facilitate appraisal of the studies, the penultimate column gives a
classification of the substances as positive, negative, or
questionably carcinogenic; indicates whether the tumour incidence was
evaluated statistically; and judges that a study is valid or provides
reasons suggesting that it is unreliable. The criteria used to
classify a study as valid were (i) an appropriate study protocol, i.e.
use of concurrent controls (sham or vehicle), 20 or more animals per
group, and study duration at least six months; and (ii) sufficient
documentation, including detailed description of administration,
results, and the survival of animals. As the use of concurrent
controls is important for making judgements, data for these are given
with the results for treated groups. If control data are not
mentioned, it is because they were not given in the original paper.
In experiments by topical application, the lower, more volatile PAH
partially evaporate, and therefore their doses may have varied. The
substances may also decompose. Both features could lead to
underestimations of carcinogenic potency if they are not taken into
account.
Table 91 shows the classification of the compounds as carcinogenic,
noncarcinogenic, or questionably carcinogenic. In order to make these
classifications, all of the studies were summarized according to
species and route of administration. In cases of doubt, the judgement
was based on valid studies only. For example, despite one positive but
invalid result and two questionable (one valid, one invalid) results
from 17 studies, anthracene was classified as negative; however,
pyrene, for which one positive, valid result and three questionable,
valid results were found in 15 studies, could not be classified as
negative and the compromise 'questionable' was chosen.
The PAH found not to be carcinogenic were anthracene,
benzo [ghi]perylene, fluorene, benzo [ghi]fluoranthene,
1-methylphenanthrene, perylene, and triphenylene. Questionable results
were obtained for acenaphthene, benzo [a]-fluorene,
benzo [b]fluorene, coronene, naphthalene, phenanthrene, and pyrene.
The remaining compounds were found to be carcinogenic.
The dermal route was the commonest mode of administration, followed by
subcutaneous and intramuscular injection. In most studies, the site of
tumour development is related closely to the route of administration,
i.e. dermal application induces skin tumours, inhalation and
intratracheal instillation result in lung tumours, subcutaneous
injection results in sarcomas, and oral administration induces gastric
tumours. Tumour induction is, however, not restricted to the obvious
sites. For example, lung tumours have been observed after oral
administration or subcutaneous injection of benzo [a]pyrene to mice
and liver tumours following intraperitoneal injection. In two studies,
lung tumours were found in mice after intravenous injection of
benzo [a]pyrene and dibenz [a,h]anthracene. Thus, tissues such as
the skin must be able to metabolize PAH to their ultimate metabolites
and itself become a target organ; however, all PAH that reach the
liver via the bloodstream can be metabolized there. The liver in turn
is a depot from which the metabolites are distributed all over the
body (Wall et al., 1991). The carcinogenic potency of the PAH differs
by three orders of magnitude, and several authors have presented
tables of toxic equivalence factors based on experimental results in
order to quantify these differences. Carcinogenic potency cannot be
based only on chemical structure but requires theoretical
considerations and calculations (see section 7.10).
Although this monograph primarily addresses single PAH, it was
considered necessary for risk assessment to present some information
on mixtures of PAH, to which humans are almost always exposed,
predominantly adsorbed onto inhalable particles.
Although the essential results of the studies of carcinogenicity are
summarized in Table 90, special aspects and comparisons of individual
PAH are presented in more detail below.
7.7.1 Single substances
7.7.1.1 Benzo [a]pyrene
Oral administration of benzo [a]pyrene to male and female CFW mice
induced gastric papillomas and squamous-cell carcinomas and increased
the incidence of pulmonary adenomas (Rigdon & Neal, 1966). In other
studies in which mice of the same strain were fed benzo [a]pyrene,
pulmonary adenomas, thymomas, lymphomas, and leukaemia occurred,
indicating that it can cause carcinomas distal to the point of
application (Rigdon & Neal, 1969). The incidence of gastric tumours
was 70% or more in mice fed 50-250 ppm benzo [a]pyrene for four to
six months. No tumours were observed in controls (Rigdon & Neal, 1966;
Neal & Rigdon, 1867; see also Table 90).
In a study of the effects of benzo [a]pyrene given in the diet or by
gavage in conjunction with caffeine, groups of 32 Sprague-Dawley rats
of each sex were fed diets containing 0.15 mg/kg bw benzo [a]pyrene
either five times per week or only on every ninth day. Tumours were
observed in the forestomach, oesophagus, and larynx, at combined
tumour incidences of 3/64, 3/64, and 10/64 in the controls and those
at the low and high doses, respectively. In the study by gavage,
groups of 32 rats of each sex were treated with benzo [a]pyrene at
0.15 mg/kg bw in a 1.5% caffeine solution every ninth day, every third
day, or five times per week. The combined incidences of tumours of the
forestomach, oesophagus, and larynx were 3/64 in controls, 6/64 in
rats at the low dose, 13/64 in those at the medium dose, and 14/64
among those at the high dose (Brune et al., 1981).
In hamsters exposed to 9.5 or 46.5 mg/m3 benzo [a]pyrene by
inhalation for 109 weeks, a dose-response relationship was seen with
tumorigenesis in the nasal cavity, pharynx, larynx, and trachea. The
fact that lung tumours were not detected could not be explained
(Thyssen et al., 1981). Hamster lung tissue can activate
benzo [a]pyrene to carcinogenic derivatives (Dahl et al., 1985).
Table 81. Mutagenicity of polycyclic aromatic hydrocarbons to
Salmonella typhimurium
Compound Result with Reference
Strain metabolic
activation
Acenaphthene
TA98,TA100 - Florin et al. (1980)
TM677 + Kaden et al. (1979)
TA98,TA100 + Epler et al. (1979)
TA100 - Pahlman & Pelkonen
(1987)
Acenaphthylene
TA98,TA100 - Florin et al. (1980)
TM677 + Kaden et al. (1979)
TA98,TA100 - Bos et al. (1988)
Anthanthrene
TA98 + Hermann (1981)
TA100 + LaVoie et al. (1979);
Andrews et al. (1978)
TA98 - Tokiwa et al. (1977)
TM677 + Kaden et al. (1979)
Anthracene
TA98,TA100 - Purchase et al. (1976)
TA98,TA100 - Epler et al. (1979)
TA100 - LaVoie et al. (1979);
Gelboin & Ts'o (1978)
TA98, TA100, - McCann et al. (1975a);
TA1535,TA1537, Salamone et al. (1979);
TA1538 Ho et al. (1981);
Purchase et al.(1976)
TA98,TA100 - Bridges et al, (1981)
TA98,TA100, - Simmon (1979)
TA1535, TA1536,
TA1537,TA1538
TM677 - Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA98,TA100 - Probst et al. (1981)
TA100 + Carver et al. (1986)
TA98,TA100 - LaVoie et al.(1983a(1985)
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TA100 - Pahlman & Pelkonen
(1987)
TA98,TA100 - Bos et al. (1988)
TA98,TA100 - Florin et al. (1980)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benz[a]anthracene
TA100 + Epler et al. (1979);
Bartsch et al. (1980)
TA98,TA100 + McCann et al. (1975a);
Coombs et al. (1976);
Simmon (1979); Salamone
et al. (1979)
TA1535,TA1538 Rosenkranz & Poirier
(1979)
TA100 + Pahlman & Pelkonen
(1987)
TA98,TA100 + Hermann (1981); Carver
et al.(1986)
TA100 + Bartsch et al. (1980)
TM677 + Kaden et al. (1979)
TA100 + Baker et al. (1980)
TA98,TA100 + Bos et al. (1988)
TA98,TA100, + Probst et al. (1981)
TA1535,TA1537
TA98, TA100,
TA1537, TA1538 ± Dunkel et al. (1984)
TA1535 - Dunkel et al. (1984)
TA98,TA100 + Florin et al. (1980)
TA1537,TA1538 - Teranishi et al. (1975)
TA98 + Tokiwa et al. (1977)
Benzo[b]fluoranthene
TA98 + Hermann (1981)
TA100 + LaVoie et al. (1979);
Hecht et al. (1980)
TA100 + Amin et al, (1985a)
TA98,TA100 - Mossanda et al. (1979)
Benzo[j]fluoranthene
TA100 + LaVoie et al. (1980);
Hecht et al. (1980)
TM677 + Kaden et al. (1979)
Benzo[k]fluoranthene
TA100 + LaVoie et al. (1980);
Hecht et al. (1980)
TA98 + Hermann et al. (1980)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benzo[ghi]fluoranthene
TA98 ± Karcher et al. (1984)
TA100 + Karcher et al. (1984)
TA98,TA100 + LaVoie et al. (1979)
Benzo[a]fluorene
TA98, TA100, Salamone et al. (1979)
TA1535, TA1537,
TA1538
TA100 + Epler et al. (1979)
TA100 - LaVoie et al. (1980)
TA98,TA100 - Bos et al. (1988)
TA98 + Tokiwa et al. (1977)
Benzo[b]fluorene
TA98, TA100 - LaVoie et al. (1980)
TA98, TA100, - Salamone et al. (1979)
TA1535, TA1537,
TA1538
TM677 + Kaden et al. (1979)
TA98,TA100 + Bos et al. (1988)
Benzo[ghi]perylene
TA98, TA1538 + Mossanda et al. (1979);
Tokiwa et al. (1977);
Katz et al. (1981)
TA100 + Andrews et al. (1978);
Katz et al. (1981);
LaVoie et al. (1979);
Salamone et al. (1979)
TA1537,TA1538 + Poncelet et al. (1978)
TM677 + Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA100 + Carver et al. (1986)
Benzo[c]phenanthrene
TA98 + Salamone et al. (1979);
Wood et al. (1980)
TA100 + Carver et al. (1986)
TA100 + Wood et al. (1980)
TA98,TA100 + Bos et al. (1988)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benzo[a]pyrene
TA98 + Epler et al. (1979)
TA100 + Andrews et al. (1978)
TA98,TA100 + LaVoie et al. (1979)
TA98,TA100, + McCann et al. 1975a,b)
TA1537,TA1538
TM677 + Kaden et al. (1979)
TM677 + Rastetter et al. (1982)
TM677 + Babson et al. (1 986b)
TA97,TA98, + Sakai et al. (1985)
TA100
TA98,TA100 + Prasanna et al. (1987));
Simmon (1979));
Glatt et al. (1987)
TA1535,TA1538 + Rosenkranz & Poirier
(1979)
TA100 + Norpoth et al. (1984));
Alzieu et al. (1987)); Carver
et al. (1986)); Bos et al.
(1988); Hermann (1981);
Bruce & Heddle (1979);
Marino (1987); Alfheim &
Ramdahl (1984)
TA98 + Lee & Lin (1988)
TA100 + Pahlman & Pelkonen
(1987)
TA97,TA98,TA100 + Marino (1987)
TA97,TA98,TA100 + Sakai et al. (1985)
TA98,TA100 + Devanesan et al. (1990)
TM677 + Skopek & Thilly (1983)
TA98,TA100, + Dunkel et al. (1984)
TA1535, TA1537,
TA1538
TA98, TA100 + Lofroth et al. (1984)
TA98,TA100 + Florin et al. (1980)
TA98 + Tokiwa et al. (1977)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Benzo[e]pyrene
TA98 + LaVoie et al. (1979);
Hermann (1981)
TA100 ± Salamone et al. (1979)
TA100 + Andrews et al. (1978);
LaVoie et al., 1979)
TA100 ± McCann et al. (1975a)
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TM677 + Kaden et al. (1979)
TA100 - Epler et al. (1979)
TA98,TA100, + Simmon (1979)
TA1538
TA97, TA100 + Sakai et al. (1985)
TA98, TA100, ± Dunkel et al. (1984)
TA1535,TA1537,
TA1538
TA100 + Carver et al. (1986)
TA100 - Pahlman & Pelkonen
(1987)
TA1537,TA1538 - Teranishi et al. (1975)
TA98 + Tokiwa et al. (1977)
Chrysene
TA100 + McCann et al. (1975a);
LaVoie et al. (1979)
TA98 + McCann et al. (1975a)
TA100 + Wood et al. (1977)
TA100 + Epler et al. (1979);
LaVoie et al. (1979)
TA100 + Salamone et al. (1979)
TA1535,TA1536, - Simmon (1979)
TA1537,TA1538
TA98,TA100 + Bhatia et al. (1987)
TM677 + Kaden et al. (1979)
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TA97,TA100 + Sakai et al (1985)
TA98,TA100 + Bos et al. (1988)
TA98 + Hermann (1981)
TA100 + Carver et al. (1986)
TA100 + Pahlman & Pelkonen
(1987)
TA100 + Florin et al. (1980)
TA98 + Tokiwa et al. (1977)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Coronene
TA98 + Mossanda et al. (1979)
TA98 + Hermann (1981)
TA98 ± Salamone et al. (1979)
TA98 + Florin et al. (1980)
TA98, TA1537, + Poncelet et al. (1978)
TA1538
TA97 ± Sakai et al. (1985)
TM677 - Kaden et al. (1979)
Cyclopenta[cd]pyrene
TA98 + Wood et al. (1980)
TA98,TA100, + Eisenstadt & Gold (1978)
TA1537,TA1538
TM677 + Kaden et al. (1979);
Cavalieri et al. (1981a)
TA98 + Reed et al. (1988)
Dibenz[a,h]anthracene
TA100 + Andrews et al. (1978);
Epler et al. (1979);
McCann et al. (1975a,b)
TA100 + Salamone et al. (1979)
TA98 + Baker et al. (1980)
TA98 + Hermann (1981)
TM677 + Kaden et al. (1979)
TA100 + Wood et al. (1978)
TA100 + Pahlman & Pelkonen
(1987); Carver et al.,
1986)
TA98, TA100, + Probst et al. (1981)
TA1537,TA1538
TA100 + Platt et al. (1990)
TA100 + Lecoq et al. (1989)
TA1537,TA1538 - Teranishi et al. (1975)
Dibenzo[a,e]pyrene
TA100 + LaVoie et al. (1979)
TA1537,TA1538 + Teranishi et al. (1975)
TA98,TA100 +.± Devanesan et al. (1990)
Dibenzo[a,h]pyrene
TA100 ± LaVoie et al. (1979)
TA98,TA100 + Wood et al. (1981)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Dibenzo[a,i]pyrene
TA100 + LaVoie et al. (1979);
McCann et al. (1975a)
TA100 + Baker et al. (1980)
TA98 + Hermann (1981)
TA98 + Wood et al. (1981)
TA1537,TA1538 + Teranishi et al. (1975)
Not specified + Sardella et al. (1981)
Dibenzo[a,l]pyrene
TA98,TA100 + Karcher et al. (1984)
TA98 + Hermann (1981)
TA98,TA100 +,± Devanesan et al. (1990)
Fluoranthene
TA98 + Hermann et al. (1980)
TA98 + Epler et al. (1979)
TA100 - LaVoie et al. (1979)
TA100 + LaVoie et al. (1982a)
TA98, TA100, - Salamone et al. (1979)
TA1535,TA1537,
TA1538
TA98,TA100 + Poncelet et al. (1978)
TA98,TA100 + Mossanda et al. (1979)
TM677 + Kaden et al. (1979)
TM677 + Rastetter et al. (1982)
TM677 + Babson et al. (1986b)
TA97,TA98,TA100 + Sakai et al. (1985)
TA98,TA100 + Bos et al. (1988)
TA100 + Carver et al. (1986);
Hermann (1981);
LaVoie et al., 1979)
TA98,TA100 + Bos et al. (1987)
TA97,TA102, ± Bos et al. (1987)
TA1537
TA1535 - Bos et al. (1987)
TA98,TA100 + Bhatia et al. (1987)
TA98,TA100 - Florin et al. (1980)
TA98 - Tokiwa et al. (1977)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
Fluorene
TA98, TA100, - McCann et al. (1975a);
TA1535,TA1537 LaVoie et al. (1979,
1980, 1981a)
TM677 - Kaden et al. (1979)
TA97 - Sakai et al. (1985)
TA98,TA100 - Bos et al. (1988)
TA100 - Pahlman & Pelkonen
(1987)
Indeno[1,2,3-cd]pyrene
TA98 + Hermann et al. (1980)
TA100 + LaVoie et al. (1979)
TA100 + Rice et al. (1985)
5-Methylcholanthrene
TA100 + Coombs et al. (1976);
Gelboin & Ts'o (1978);
LaVoie et al. (1979);
McCann et al. (1975a);
Hecht et al. (1978)
TA100 + Amin et al. (1979)
TA100 + El-Bayoumy et al. (1986)
1-Methylphenanthrene
TA100 + LaVoie et al. (1981b)
TM677 + Kaden et al. (1979)
TA97,TA98,TA100 + Sakai et al. (1985)
TA98,TA100 + LaVoie et al. (1983b)
Naphthalene
TA98,TA100, - Florin et al. (1980)
TA1535,TA1537
TA98, TA100, - McCann et al. (1975a)
TA1535,TA1537,
TA1538
TA98, TA100, - Purchase et al. (1976)
TA1535,TA1538
TA98 - Ho et al. (1981)
TM677 - Kaden et al. (1979)
G46, E. coli K12 - Kramer et al. (1974)
TA98,TA100 - Epler et al. (1979)
TA98,TA100 - Mamber et al. (1984)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
TA97,TA98,TA100 - Sakai et al. (1985)
TA100 - Pahlman & Pelkonen
(1987)
TA98,TA100 - Bos et al. (1988)
Perylene
TA98 + Ho et al. (1981)
TA100 + LaVoie et al. (1979)
TA98,TA100, - Salamone et al. (1979)
TA1535, TA1537,
TA1538
TA98 + Hermann (1981)
TA98 + Florin et al. (1980)
TM677 + Kaden et al. (1979);
Penman et al. (1980)
TA100 + Carver et al. (1986)
TA97,TA100 + Sakai et al. (1985)
TA98,TA100 + Lofroth et al. (1984)
TA100 - Pahlman & Pelkonen
(1987)
Phenanthrene
TA100 + Oesch et al. (1981)
TA100 - Wood et al. (1979)
TA98 + Epler et al. (1979)
TA98 - LaVoie et al. (1979, 1980)
TA100 - LaVoie et al. (1981b)
TA98,TA100 - Probst et al. (1981)
TA100 - LaVoie et al. (1979);
LaVoie et al. (1980);
Gelboin & Ts'o (1978);
McCann et al. (1975a)
TA98, TA100, - McCann et al. (1975a)
TA1535,TA1537
TA100 + Carver et al. (1986)
TM677 - Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA98,TA100 ± Bos et al. (1988)
TA1535,TA1536, - Simmon (1979)
TA1537,TA1538
TA1535,TA1538 - Rosenkranz & Poirier
(1979)
TA100 - Pahlman & Pelkonen
(1987)
Table 81. (continued)
Compound Result with Reference
Strain metabolic
activation
TA98, TA100, - Dunkel et al. (1984)
TA1535,TA1537,
TA1538
TA98,TA100 - Florin et al. (1980)
Pyrene
TA98 - Ho et al. (1981);
Rice et al. (1988a)
TA98,TA100, - McCann et al. (1975a);
LaVoie et al. (1979);
TA1535,TA1537 Ho et al. (1981)
TA1537 + Bridges et al. (1981)
TA98,TA100 - Salamone et al. (1979)
TA98,TA100 - Probst et al. (1981)
TA1537 + Epler et al. (1979)
TM677 + Kaden et al. (1979)
TA97 + Sakai et al. (1985)
TA98,TA100 ± Bos et al. (1988)
TA100 - Carver et al. (1986);
Hermann (1981)
TA98,TA100 + Bhatia et al. (1987)
TA98, TA100, - Dunkel et al. (1984)
TA1536,TA1537,
TA1538
TA100 - Pahlman & Pelkonen
(1987)
TA98,TA100 - Florin et al. (1980)
Triphenylene
TA98 + Epler et al. (1979)
TA98 + Tokiwa et al. (1977)
TA98,TA100 + Mossanda et al. (1979);
Wood et al. (1980)
TA98 + Hermann (1981)
TA98,TA100 + Poncelet et al. (1978)
TM677 + Kaden et al. (1979)
TA98,TA100 + Bos et al. (1988)
TA100 + Pahlman & Pelkonen
(1987)
TA, used to test reverse mutation to histidine non-auxotrophic mutants);
TM, used to test forward mutation to 8-azaguanine-resistant mutants
+, positive); -, negative); ±, inconclusive
Table 82. DNA damage induced by polycyclic aromatic hydrocarbons in vitro
Test system End-point Metabolica Resultb Reference
activation
Prokaryotes
Anthracene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2, E. coli WP100 R + - Member et al.
(1983)
E. coli WP2, E. coli WP67, R +/- - Tweats (1981)
E. coli CM871
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
E. coli WP2s(lambda) R +/- + Rossman et al.
prophage induction) (1991)
B. subtilis R +/- - Ashby & Kilby
(1981)
B. subtilis R +/- - McCarroll et al.
(1981)
E. coli GY5027 (prophage R + - Mamber et al.
induction) (1984)
Anthranene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benz[a]anthracene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benzo[b]fluoranthene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[ghi]fluoranthene
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Benzo[j]fluoranthene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benzo[a]fluoranthene
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Benzo[b]fluoranthene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Bunzo[ghi]perylene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Benzo[a]pyrene
E. coli WP2, E. coli WP100 R + + Mamber et al.
(1983)
E. coli GY5027 R + + Mamber et al.
(1983)
E. coli pol A- R + + Rosenkranz &
Poirier (1979)
E. coli WP2, E. coli WP67, R +/- + Tweats (1981)
E. coli CM871
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli PQ37 R +/- +/+ Mersch-
Sundermann et
al. (1992)
B. subtilis R +/- + McCarroll et al.
(1981)
E. coli WP2s(lambda) R +/- + Rossman et al.
prophago induction) (1991)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[e]pyrene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli WP2s(lambda) R +/- + Rossman et al.
prophage induction) (1991)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Chrysene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli PQ37 R +/- + Mersch-
Sundermann at
al. (1992)
Coronene
E. coli PQ37 R +/- - Mersch-
Sundermann at
al. (1992)
Dibenz[a,h]anthracene
E. coli R +/- + Ichinotsubo et al.
(1977)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
B. subtilis R +/- + McCarroll et al.
(1981)
E. coli WP2s (lambda R +/- + Rossman et al.
prophage induction) (1991)
Dibenzo[a,i]pyrene
E. coli R +/- + Ichinotsubo et al.
(1977)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al.(1992)
B. subtilis R +/- + McCarroll et al.
(1981)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Dibenzo[a,h]pyrene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Dibenzo[a,i]pyrene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Fluoranthene
E. coli WP2s (lambda R +/- - Rossman et al.
prophage induction) (1991)
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Fluoranthene
E. coli WP2, E. coli WP100 R + - Mamber et al.
(1983)
E. coli GY5027 R + - Mamber et al.
(1984)
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Indeno[1,2,3-cd]pyrene
E. coli PQ37 R + - Mersch-
Sundermann et
al.(1992)
Naphthalene
E. coli WP2, E. coli WP 100 R + - Mamber et al.
(1983)
E. coli GY5027 R + - Mamber et al.
(1984)
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Parylene
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Phenanthrene
E. coli pol A- R + - Rosenkranz &
Poirier (1979)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli PQ37 R +/- + Mersch-
Sundermann at
al. (1992)
E. coli WP2s (lambda R +/- + Rossman et al.
prophage induction) (1991)
B. subtilis R +/- - McCarroll et al.
(1981)
Pyrene
E. coli R +/- - Ashby & Kilbey
(1981; De Serres
& Ashby, 1981)
E. coli WP2, E, coli WP100 R + - Mamber et al.
(1983)
E. coli GY5027 R + - Mamber et al.
(1984)
E. coli WP2 uvrA R + - Dunkel et al.
(1984)
E. coli WP2, E. coli WP67, R +/- - Tweats (1981)
E. coli CM871
E. coli PQ37 R +/- - Mersch-
Sundermann et
al. (1992)
B. subtilis R +/- - Ashby & Kilbey
(1981)
B. subtilis R +/- - McCarroll et al.
(1981)
E. coli WP2s (lambda R +/- - Rossman et al.
prophage induction) (1991)
Triphenylene
E. coli PQ37 R +/- + Mersch-
Sundermann et
al. (1992)
Eukaryotes
Acenaphthene
Rat liver or lung DA - - Beach & Gupta
(1991)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Anthracene
Primary rat hepatocytes UDS - - Williams, 1977;
Probst et al.
(1981)
Primary rat hepatocytes R - - Tong et al.
(1983)
HeLa cells UDS +/- - Martin et al.
(1978; Martin &
McDermid
(1981)
Human skin fibroblasts R - Milo et al. (1978)
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Human peripheral blood DA - - Gupta et al.
lymphocytes (1988)
Benz[a]anthracene
Primary rat hepatocytes UDS - + Probst et al.
(1981)
Primary rat hepatocytes R - + Tong et al.
(1983)
HeLa cells UDS +/- + Martin et al.
(1978)
Rat or human mammary DS - ± Mane et al.
epithelial cells (1990)
Hamster buccal pouch DS - - Nagabhushan et
(epithelial cells (inhibition al. (1990)
of DNA synthesis)
Human peripheral blood DA - + Gupta et al.
lymphocytes (1988)
Benzo[b]fluoranthene
Rat buccal mucosa DA - + Autrup, & Autrup
epithelial cells (1986)
Human leukocytes DA + + Roggeband et al.
(1994a)
Benzo[j]fluoranthene
Rat buccal mucosa DA - + Autrup & Autrup
epithelial cells (1986)
Benzo[k]fluoranthene
Human leukocytes DA + + Roggeband et al.
(1994a)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[a]pyrene
Primary rat hepatocytes UDS - + Probst et al.
(1981)
Primary rat hepatocytes R - + Williams et al.
(1982)
C3H/1OT1/2 mouse clone 8 - ± Lubet et al.
(DNA breaks) (1983a)
Human leukocytes DA + + Roggeband et al.
(1994a)
Hamster or rat trachea DA, - + Roggeband et al.
epithelial cells UDS (1994b)
HeLa cells UDS +/- + Martin et al.
(1978)
Human skin fibroblasts R - + Milo et al. (1978)
Human mammary cells - + Leadon et al.
(oxidative DNA damage) (1988)
Human fibroblasts UDS + + Agrelo & Amos
(1981)
Human fibroblasts WI-38 UDS +/- + Robinson &
Mitchell (1981)
Rat or human mammary R - + Mane et al.
epithelial cells (1990)
Human bronchial cells DA - + Harris et al.
(1984)
Syrian hamster embryo cells R - + Casto (1979)
Hamster buccal pouch - + Nagabhushan et
epithelial cells (inhibition of al. (1990)
DNA synthesis)
Rat buccal mucosa DA - + Autrup & Autrup,
epithelial cells (1986)
Human peripheral DA - + Gupta et al.
lymphocytes (1988)
Primary rat hapatocytes DA - + Monteith &
Gupta (1992)
Primary human hepatocytes DA - + Monteith &
Gupta (1992)
Calf thymus DNA DA - + Bryla & Weyand
(1991)
Primary mouse epidermal DA, - + Gill et al. (1991)
keratinocytes UDS
Primary rat hepatocytes R - + Tong et al.
(SCE) (1983)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Benzo[e]pyrene
Primary rat hepatocytes R - - Tong et al.
(SCE) (1983)
HeLa cells (UDS) UDS +/- + Martin et al.
(1978)
Primary rat hepatocytes R - - Williams et al.
(1982)
Rat mammary epithelial - - Mane et al. cells
(DNA synthesis) (1990)
Syrian hamster embryo R - - Casto (1979)
cells
Human skin fibroblasts R - - Milo et al. (1978)
Chrysene
Primary rat hepatocytes R - - Tong et al.
(1983)
Human leukocytes DA + + Roggeband et al.
(1994a)
Cyclopenta[cd]pyrene
Rat liver or lung tissue DA - + Beach & Gupta
(1991)
Calf thymus DNA DA + + Beach & Gupta
(1994)
Dibenz[a,h]anthracene
Primary human foreskin UDS - + Lake et al.
epithelial cells (1978)
HeLa cells UDS +/- + Martin et al.
(1978)
Syrian hamster embryo R - - Casto (1979)
cells
Primary rat hepatocytes UDS - + Probst et al.
(1981)
Mouse liver DNA DA + + Lecoq et al.
(1991)
Human bronchial cells DA - + Harris et al.
(1984)
Hamster embryonic cells + Kuroki &
Heidelberger
(1972)
C3H1OT1/2 mouse clone DA - + Nesnow et al.
8 cells (1994)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Dibenzo[a,i]pyrene
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Fluorene
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Human leukocytes DA + + Roggeband et al.
(1994a)
5-Methylcholanthrene
Primary rat hepatocytes UDS - + Tong et al.
(1981a)
1-Methylphenanthrene
Primary rat hepatocytes UDS - + Tong et al.
(1981a)
Chinese hamster ovary cells DA + + Dunn & Douglas
(1991)
Perylene
Human peripheral blood DA - - Gupta et al.
lymphocytes (1988)
Syrian hamster embryo R - - Casto (1979)
cells
Phenanthrene
Syrian hamster embryo R - - Casto (1979)
cells
Human foreskin epithelial UDS - - Lake et al.
cells (1978)
Primary rat hepatocytes UDS - - Probst et al.
(1981)
Human skin fibroblasts R - Milo et al. (1978)
Pyrene
Syrian hamster embryo R - - Casto (1979)
cells
Human foreskin epithelial UDS - - Lake et al.
cells (1978)
Primary rat hepatocytes UDS - - Probst et al.
(1981)
HeLa cells UDS +/- - Martin et al.
(1978)
Table 82. (continued)
Test system End-point Metabolica Resultb Reference
activation
Human fibroblast cell line UDS +/- + Robinson &
WI38 Mitchell (1981)
Primary rat hepatocytes R - - Williams et al.
(1982)
Human skin fibroblasts R - Milo et al. (1978)
Human skin fibroblasts UDS + - Agrelo & Amos
(1981)
Primary rat hepatocytes R - - Tong et al.
(1983)
Human peripheral blood DA - - Gupta et al.
lymphocytes (1988)
Triphenylene
Human peripheral blood DA - + Gupta et al.
lymphocytes (1988)
R, DNA repair; DA, DNA adducts; UDS, unscheduled DNA synthesis;
SCE, sister chromatid exchange
a +, tested with metabolic activation; -, tested without metabolic activation;
+/-, tested with and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if
positive only with activation
Table 83. Mutagenicity of polycyclic aromatic hydrocarbons in yeasts and other
eukaryotes, host-mediated mutagenicity, and mutagenicity in Drosophila
Test system End-point Metabolic Resultb Reference
activationa
Yeasts and other eukaryotes
Anthracene
Saccharomyces cerevisiae MGC - - Seibert et al.
D4-RDII (1981)
Saccharomyces cerevisiae MR - - De Serres &
Hoffman (1981)
Benzo[a]pyrene
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Saccharomyces cerevisiae NMR - + DeSerres &
Hoffmann (1981)
Paramecium tetraurelia + + Smith-Sonneborn
(survival) (1983)
Chrysene
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Dibenz[a,h]anthracene
Neurospora crassa - + Barrett & Tatum
(1958)
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Naphthalene
Paramecium tetraurelia + - Smith-Sonneborn
(survival) (1983)
Phenanthrene
Saccharomyces cerevisiae MGC - - Siebert et al.
D4-RDII (1981)
Pyrene
Saccharomyces cerevisiae; NMR; - - De Serres &
S. pombe FM Hoffman (1981)
Host-mediated mutagenicity
Anthracene
Salmonella typhimurium - ± Simmon et al.
TA1530, TA1535,TA1538 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Table 83. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Benz[a]anthracene
Salmonella typhimurium - + Simmon et al.
TA1530,TA1535,TA1538 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Benzo[a]pyrene
Salmonella typhimurium - - Simmon et al.
TA1530,TA1535,TA1538 (1979); Glatt at
al. (1985)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Benzo[e]pyrene
Salmonella typhimurium - - Simmon et al.
TA1538 (1979)
Chrysene
Salmonella typhimurium - - Simmon et al.
TA1530,TA1535,TA1538 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Phenanthrene
Salmonella typhimurium - - Simmon et al.
TA1530,TA1535 (1979)
Saccharomyces cerevisiae NMR - - Simmon et al.
(1979)
Drosophila malanogaster
Anthracene R - Fujikawa et al.
(1993)
Benz[a]anthracene SLRL + Fahmy & Fahmy
(1973)
Somatic mutation - Fahmy & Fahmy
(1980)
SLRL - Zijistra & Vogel
(1984)
SMART + Frolich & Wurgler
(1990)
R + Fujikawa et al.
(1993)
Table 83. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Benzo[a]pyrene SLRL ± Vogel et al.(1983)
Somatic mutation + Fahmy & Fahmy
(1980)
SLRL - Zijistra & Vogel
(1984)
SLRL - Valencia &
Houtchens (1981)
Somatic mutation + Batiste-Alentorn
et al. (1991)
SMART + Frolich & Wurgler
(1990)
SLRL - Valencia &
Houtchens (1981)
R + Fujikawa et al.
(1993)
Benzo[e]pyrene R - Fujikawa et al.
(1993)
Fluorene R - Fujikawa et al.
(1993)
Pyrene R ± Fujikawa et al.
(1993)
MGC, mitotic gene conversion; NMR, number of mitotic recombinants; MR, mitotic recombination;
SLRL, sex-linked recessive lethal mutation; R, DNA repair; FM, forward mutation;
SMART, somatic mutation and recombination test
a +, tested with metabolic activation; -, tested without metabolic activation; tested with
and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if positive only
with activation
Table 84. Mutagenicity of polycyclic aromatic hydrocarbons in mammalian cells in vitro
Test system End-point Metabolic Resultb Reference
activationa
Anthracene
Chinese hamster V79 HPRT +/- - Knaap et al.
(1981)
Mouse lymphoma L5178Y TK + - Amacher &
Turner (1980);
Amacher et al.
(1980)
Human lymphoblastoid TK6 TK + - Barfknecht et al.
(1981)
Fischer rat embryo OR + - Mishra et al.
(1978)
Human epithelial EUE cells DTR - - Rocchi et al.
(1980)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Benz[a]anthracene
Chinese hamster V79 HPRT + + Krahn &
Heidelberger
(1977); Slaga et
al. (1978)
Chinese hamster V79 HPRT + - Huberman
(1975)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Human epithelial EUE cells DTR - - Rocchi et al.
(1980)
Human keratinocytes HPRT - - Allen-Hoffmann
& Rheinwald
(1984)
Mouse lymphoma L5178Y TK + + Amacher &
Turner (1980);
Amacher et al.
(1980)
Mouse lymphoma L5178Y TK - - Amacher &
(+ hamster hepatocytes) Paillet (1983)
Mouse lymphoma L5178Y TK - + Amacher &
(+ hamster hepatocytes) Paillet (1982)
Rat liver epithelial ARL 18 HPRT - - Tong et al.
(1981a)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Banzo[b]fluoranthene
Chinese hamster V79 HPRT + - Huberman
(1975)
Benzo[a]pyrene
Chinese hamster V79 HPRT + + Arce et al.(1987);
Diamond et al.
1980); Huberman
(1975)
Chinese hamster V79 HPRT + + Krahn &
Heidelberger
(1977)
Mouse lymphoma L5188Y TK - + Amacher &
(+ hamster hepatocytes) Paillet (1982)
Chinese hamster ovary HPRT +/- + Gupta & Singh
(1982)
Fischer rat embryo OR - + Mishra et al.
(1978)
Mouse lymphoma L5178Y TK - + Amacher &
(+ hamster hepatocytes) Paillet (1983)
Mouse lymphoma L5178Y TK +/- + Clive et al.
(1979)
Mouse lymphoma L5178Y TK + + Amacher &
Turner (1980);
Amacher et al.
(1980); Arce et
al.(1987)
Mouse lymphoma L5178Y TK + + Wangenheim &
Bolcsfoldi (1988)
Human lymphoblasts AHH TK - + Crespi & Thilly
(1984)
Human lymphoblasts K6 TK +/- + Crespi et al.
(1985)
Human epithelial EUE cells DTR - + Rocchi et al.
(1980);
Barfknecht et al.
(1982)
Human fibroblasts HS172 DTR +/- + Gupta &
Goldstein (1981)
Human keratinocytes HPRT - + Allen-Hoffmann
& Rheinwald
(1984)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Rat liver epithelial cells - + Tong et al.
ARL 18 (1981a)
Chinese hamster ovary-AS52 + Oberly et al.
(chromosomal mutation) (1992)
Human epithelial teratoma HPRT - + Huberman et al.
P3 (cocultivated with human (1984)
carcinoma BJ cells)
Chinese hamster lung cells HPRT - + Baird et al.
V79 (1984)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary,
(1988)
Mouse lymphoma L5178Y TK +/- + Rees et al.
(1989)
Mouse Balb/c-3T3 OR - + Lubet et al.
(1990)
Mouse lymphoma L5178Y TK +/- + Jotz & Mitchell
(1981)
Benzo[e]pyrene
Chinese hamster V79 HPRT + - Hubermann
(1978)
Rat liver epithelial ARL 18 HPRT - - Tong et al.
(1981a)
Mouse C3H10T1/2 OR - - Gehly et al.
(1982)
Human epithelial teratoma P3 HPRT - - Huberman et al.
(1984)
Chinese hamster lung cells HPRT - - Baird et al.
V79 (1984)
Fischer rat embryo cells OR + - Mishra et al.
(1978)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Mouse lymphoma L517BY TK +/- - Clive et al.
(1979)
Mouse Balb/c-3T3 OR - - Lubet et al.
(1990)
Chrysene
Chinese hamster V79 HPRT + - Huberman &
Sachs (1976)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Human epithelial EUE DTR - - Rocchi et al.
(1980)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Human epithelial teratoma P3 HPRT - + Huberman et al.
(cocultivated with human (1984)
carcinoma BJ cells)
Cyclopenta[cd]pyrene
Human lymphoblastold HH-4 HPRT + + Skopek et al.
(1979)
Mouse lymphoma L5178Y TK +/- + Gold et al. (1980)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Human lymphoblasts AHH1 TK - + Crespi & Thilly
1984)
Chinese hamster V79 HPRT - + Raven et al.
(+ hamster embryo fibroblasts) (1982)
Dibenz[a,h]anthracene
Chinese hamster V79 HPRT + + Huberman &
Sachs (1976);
Huberman
(1978)
Chinese hamster V79 HPRT + + Krahn &
Heidelberger
(1977)
Human epithelial EUE DTR - ± Rocchi et
al., 1980)
Fluoranthene
Human lymphoblastoid HH-4 HPRT + + Thilly et al.
(1980)
Human lymphoblasts AHH1 TK - - Crespi & Thilly
(1984)
Human lymphoblasts TK6 TK + + Barfknecht et al.
(1982)
Fluorene
Mouse lymphoma L5178Y TK +/- + Wangenheim &
Bolcsfoldi (1988)
1-Methylphenanthrene
Human lymphoblastoid TK6 TK + + Barfknecht et al.
(1981)
Human lymphoblasts AHH1 TK - + Crespi & Thilly
(1984)
Table 84. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Perylene
Human lymphoblastoid TK6 HPRT + - Penman et al.
(1980)
Phenanthrene
Chinese hamster V79 HPRT + - Huberman &
Sachs (1976)
Human lymphoblastoid TK6 TK + + Barfknecht et al.
(1981)
Fischer rat embryo OR + - Mishra et al.
(1978)
Pyrene
Mouse lymphoma L517BY TK +/- + Jotz & Mitchell
(1981)
Fischer rat embryo OR + + Mishra et al.
(1978)
Chinese hamster V79 HPRT + - Huberman
(1975)
Mouse lymphoma L5178Y TK + - Amacher et al.
(1980)
Human lymphoblasts TK6 TK + - Barfknecht et al.
(1982)
Chinese hamster ovary HPRT - Heflich et al.
(1990)
Human epithelial teratoma P3 HPRT - - Huberman et al.
(1984)
Mouse lymphoma L5178Y TK +/- + Myhr & Caspary
(1988)
Mouse lymphoma L5178Y TK +/- + Wangenheim &
Bolcsfoldi (1988)
Rat liver epithelial ARL18 HPRT - - Tong et al.
(1981a)
Mouse Balb/c-3T3 OR - ± Lubet et al.
(1990)
Triphenylene
Human lymphoblasts TK + + Barfknecht et al.
(1982)
HPRT, hypoxanthine-guanine phosphoribosyl transferase reversion; TK, thymidine kinase
reversion; OR, ouabain resistance; DTR, diphtheria toxin resistance
a +, tested with metabolic activation; - , tested without metabolic activation; +/-,
tested with and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if positive
only with activation
Table 85. Chromosomal effects of polycyclic aromatic hydrocarbons in mammalian cells in vitro
Test system End-point Metabolic Resultb Reference
activationa
Anthracene
Chinese hamster D6 CA, - - Abe & Sasaki
SCE (1977a)
Rat liver epithelial ARL18 SCE - - Tong et al.
(1981b)
Rat liver RL1 CA - - Dean (1981)
Benz[a]anthracene
Chinese hamster ovary SCE - + Pal (1981)
Rat liver epithelial ARL18 SCE - ± Tong et al.
(1981b)
Chinese hamster V79 SCE - ± Mane et al.
(coincubation with rat (1990)
mammary epithelial cells)
Benzo[a]pyrene
Rat liver RL1 CA - + Dean (1981)
Chinese hamster V79-4 CA, - - Popescu et al.
(+ feeder cells) SCE (1977)
Chinese hamster lung CA +/- + Matsuoka et al.
(1979)
Mouse lymphoma L5178Y CA - + Arce et al. (1987)
(+ hamster embryo cells)
Human fibroblasts WI-38 CD +/- + Weinstein et al.
(1977)
Chinese hamster V79 SCE - + Arce et al.(1987);
(+ hamster embryo cells) Wojciechowski et
al. (1981)
Chinese hamster Don-6 SCE - + Abe et al.
(1983a)
Chinese hamster ovary SCE +/- + Husgafvel-
Pursiainen et al.,
1986)
Chinese hamster ovary SCE +/- + Evans & Mitchell
(1981)
Rat pleural mesothelial SCE +/- + Achard et al.
calls (1987)
Rat liver epithelial ARL 18 SCE - + Tong et al.
(1981b)
Rat hepatoma Reuber SCE - + Dean et al.
H4-II-E (1983a)
Rat oesophageal tumour SCE - + Abe et al.
R1 (1983a)
Table 85. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Rat ascites hepatoma SCE - + Abe et al.
AH6&B (1983a)
Human fibroblasts TIG-11 SCE - + Huh et al. (1982)
Human hepatoma cells SCE - + Huh et al. (1982)
Human hepatoma C-HC-4 SCE - + Abe et al.
and C-HC-20 (1983a,b)
Chinese hamster V79 SCE - + Mane et al.
(coincubation rat/human (1990)
mammary epithelial cells)
Primary mouse epidermal UDS - + Gill et al. (1991)
keratinocytes
Human hepatoma (strain SCE, - + Natarajan &
Hap G2) MN Darroudi (1991)
Mouse spleen lymphocytes SCE - + Wielgosz et al.
(1991)
Mouse C3H/10T1/2 clone 8 SCE - + Krolewski et al.
(1986)
Human epithelial teratoma SCE - + Murison (1988)
P3 (coincubation with rat
hepatoma RL-12 cell line)
Chinese hamster epithelial SCE - + DeSalvia et al.
liver (1988)
Human lymphocytes CD +/- + Rees et al.
(1989)
Benzo[e]pyrene
Rat liver epithelial ARL 18 SCE - - Tong et al.
(1981b)
Mouse C3H 10T1/2 CA, - - Gehly et al.
SCE (1982)
Chinese hamster V79 cells SCE - - Mane et al.
(coincubation with rat (1990)
mammary epithelial cells)
Human epithelial teratoma SCE - - Murison (1988)
P3 (coincubation with human
breast carcinoma cells
BJ-015)
Cyclopenta[cd]pyrene
Mouse C3H/10T1/2 clone 8 SCE - + Krolewski et al.
(1986)
Human epithelial teratoma SCE - + Murlson (1988)
P3 (coincubation with human
breast carcinoma cells BJ-015)
Table 85. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Dibenz[a,h]pyrene
Chinese hamster ovary SCE - + Pal (1981)
Fluoranthene
Chinese hamster CHO-1 SCE +/- + Palitti et al.
(1986)
Chinese hamster epithelial SCE - - DeSalvia et al.
liver (1988)
Fluorene
Chinese hamster lung CA +/- + Matsuoka et al.
CHL (1991)
Naphthalene
Mouse embryos (in vitro) CA + + Gollahon (1991);
Gollahon et al.
(1990)
Perylene
Chinese hamster V79 CA - + Popescu et al.
(1977)
Chinese hamster V79 SCE - - Popescu et al.
(1977)
Phenanthrene
Chinese hamster V79-4 SCE - - Popescu et al.
(+ hamster feeder cells) (1977)
Chinese hamster V79-4 CA - + Popescu et al.
(+ hamster feeder cells) (1977)
Chinese hamster Don CA,SCE - Abe & Sasaki
(1977b)
Chinese hamster lung CA - Ishidate &
CHL Odashima
(1977)
Chinese hamster lung CA +/- - Matsuoka et al.
CHL (1979)
Pyrene
Rat liver epithelial ARL 18 SCE - - Tong et al.
(1981b)
Chinese hamster D6 CA, - - Abe & Sasaki
SCE (1977a)
Chinese hamster ovary SCE +/- + Evans & Mitchell
(1981)
Table 85. (continued)
Test system End-point Metabolic Resultb Reference
activationa
Chinese hamster ovary SCE +/- + Perry &
Thomson (1981)
Chinese hamster V79-4 CA - + Popescu et al.
(1977)
Chinese hamster V79-4 SCE - - Popescu et al.
(+ hamster feeder cells) (1977)
Rat liver RL1 CA - - Dean (1981)
Human fibroblasts WI-38 CD +/- - Weinstein et al.
(1977)
Human hepatoma (strain MN, - - Natarajan &
Hep G2) SCE Darroudi (1991)
Chinese hamster epithelial SCE - - DeSalvia et al.
liver (1988)
SCE, sister chromatid exchange; MN, micronucleus formation; CA, chromosomal aberration;
CD, chromosomal damage
a +, tested with metabolic activation; -, tested without metabolic activation;
+/-, tested with and without metabolic activation
b Result: +, positive; -, negative; ±, inconclusive; positive results shown if positive
only with activation
Table 86. Morphological transformation of mammalian cell in vitro by
polycyclic aromatic hydrocarbons
Test system Resulta Reference
Anthracene
Balb/c3T3 mouse cells - DiPaolo et al. (1972)
Guinea-pig fetal cells - Evans & DiPaolo
(1975)
Neonatal Syrian golden hamster - Purchase et al. (1976)
kidney fibroblasts BHK21 C13
Syrian hamster embryo cells - Pienta et al. (1977)
Hamster BHK21 clone 13 cells - Grab et al. (1980)
Syrian hamster embryo cells - Dunkel et al. (1981)
Balb/3T3 mousecells - Dunkel et al. (1981)
Balb/3T3 mousecells - Peterson et al. (1981);
Lubet et al. (1983a)
Fischer rat embryo cells - Mishra et al. (1978)
Fischer rat embryo cells - Dunkel et al. (1981)
(leukaemia virus-infected)
Fischer rat embryo cells + Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
C3H/10T1/2 mouse clone 8 - Dunkel et al. (1988)
Benz[a]anthracene
Syrian hamster embryo cells + Pienta et al. (1977);
DiPaolo et al. (1969,
1971)
Mouse prostate C3HG23 cells + Marquardt &
Heidelberger (1972)
C3H/10T1/2 mouse cells Nesnow &
Heidelberger (1976)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Hamster embryo cells - Grover et al. (1971)
Syrian hamster embryo cells + Dunkel et al. (1981)
Syrian hamster lung cells FSHL + Emura et al. (1980)
Mouse ventral prostate C3H - Marquardt et al.(1972);
clone G23 cells Grover et al. (1971)
Balb/3T3 mouse clone 1-13 cells + Rundell et al. (1983)
Balb/3T3 mouse cells ± Dunkel et al. (1981)
Fischer rat embryo cells
(Rauscher leukaemia virus infected) ± Freeman et al. (1973)
Fischer rat embryo cells + Dunkel et al. (1981)
(leukaemia virus-infected)
Syrian hamster embryo cells + DiPaolo et al. (1985)
Table 86. (continued)
Test system Resulta Reference
Benzo[b]fluoranthene
Hamster BHP21 clone 13 cells + Greb et al. (1980)
Syrian hamster lung FSHL cells + Emura et al. (1980)
Benzo[k]fluoranthene
Syrian hamster lung FSHL cells - Emura et al. (1980)
Benzo[ghi]perylene
Syrian hamster embryo cells - DiPaolo et al. (1985)
Benzo[a]pyrene
Golden hamster embryo cells + Mager et al. (1977)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Hamster embryo cells (SA7 virus- + Casto et al. (1977)
transformed)
Syrian hamster embryo cells + DiPaolo et al. (1969,
1971)
Syrian hamster embryo cells + Dunkel et al. (1981)
Syrian hamster embryo cells + Casto et al. (1977)
Syrian hamster lung FSHL + Emura et al. (1980,
1987)
Syrian hamster SHE (SA7 virus- + Arce et al. (1987)
transformed)
C3H/10T1/2 mouse + Arce et al. (1987);
Lubet et al. (1983b);
Peterson et al. (1981)
Balb/3T3 mouse + Dunkel et al. (1981)
Balb/3T3 mouse clone A31-1-1 + Little & Vetrovs (1988)
Fischer rat embryo cells + Mishra et al. (1978)
Rat embryo cells (SA7 virus- + DiPaolo & Casto
transformed) (1976)
Fischer rat embryo cells + Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Fischer rat embryo cells + Dunkel et al. (1981)
(leukaemia virus-infected)
Balb/c 3T3 mouse clone A31 cells + Albini et al. (1991)
C3H/10T1/2 mouse clone 8 cells + Dunkel et al. (1988)
C3H/10T1/2 mouse clone 8 cells + Krolewski et al. (1986)
Balb/c3T3 mouse clone 1-13 cells + Rundell et al. (1983)
Benzo[e]pyrene
C3H/10T1/2 mouse cells - Gehly et al. (1982)
Fischer rat embryo cells - Mishra et al. (1978)
Table 86. (continued)
Test system Resulta Reference
Banzo[b]fluranthene
Syrian hamster embryo cells - Pienta et al. (1977)
Syrian hamster embryo cells + DiPaolo et al. (1969)
C3H/10T1/2 mouse clone 8 cells ± Dunkel et al. (1988)
Balb/c-3T3 mouse - Lubet et al. (1990)
Syrian hamster lung FSHL cells ± Emura et al. (1980)
Syrian hamster embryo cells - Dunkel et al. (1981)
Balb/3T3 mouse - Dunkel et al. (1981)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Chrysene
Syrian hamster embryo cells + Pienta et al. (1977)
Mouse prostate C3HG23 cells - Marquardt et al. (1972)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
Hamster epithelial lung + Jacob et al. (1993c);
cell line M3E3/C3 Riebelmre et al. (1993)
Cyclopenta[cd]pyrene
C3H10T1/2 mouse clone 8 calls + Gold et al. (1980)
C3H/10T1/2 mouse clone 8 cells + Krolewski et al. (1986)
Dibenz[a,h]anthracene
Syrian hamster embryo cells + DiPaolo et al. (1969);
Pienta et al. (1977)
C3H 10T1/2 mouse cells + Reznikoff et al. (1973)
C3H mouse prostate cells + Chen & Heidelberger
(1969)
C3HG23 mouse prostate cells - Marquardt et al. (1972)
Hamster embryo cells - Grover et al. (1971)
Fischer rat embryo cells + Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Hamster embryo cells (SA7 virus- + Casto (1973); Casto et
transformed) al. (1977)
Hamster BHK21 clone 13 cells + Greb et al. (1980)
C3H/10T1/2 mouse clone 8 cells ± Lubet et al. (1983a,b)
Rat embryo cells (SA7 virus- + DiPaolo & Casto
transformed) (1976)
C3H/10T1/2 mouse clone 8 cells ± Dunkel et al. (1988)
C3H/10T1/2 mouse clone 8 cells + Nesnow et al. (1994)
Fluoranthene
Fischer rat embryo cells - Freeman et al. (1973)
(Rauscher leukaemia virus- infected)
Table 86. (continued)
Test system Resulta Reference
Fluorene
Balb/c3T3 mouse cells ± Tonelli et al. (1979)
Indeno[1,2,3-cd]pyrene
Syrian hamster lung FSHLcells + Emura et al. (1980)
Naphthalene
Fischer rat embryo cells - Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Human lung WI-38 cells - Purchase et al. (1976)
Syrian hamster kidney - Purchase et al. (1976)
BHK-21C13 cells
Balb/c mouse mammary gland - Tonelli et al. (1979)
Balb/c-3T3 mouse cells - Rundell et al. (1983)
Perylene
Syrian hamster embryo cells - DiPaolo et al. (1985)
Syrian hamster embryo cells - Casto (1979)
Phenanthrene
Mouse prostate C3HG23 cells - Marquardt et al. (1972)
Syrian hamster embryo cells - Pienta et al. (1977)
Balb/3T3 mouse cells - Kakunaga (1973)
Fetal guinea-pig cells - Evans & DiPaolo
(1975)
Syrian hamster embryo cells - DiPaolo et al. (1969);
Dunkel et al. (1981)
Hamster BHK21 clone 13 cells - Greb et al. (1980)
Hamster embryo calls (SA7 - Casto et al. (1977)
virus-transformed)
C3H/10T1/2 mouse cells - Peterson et al. (1981)
C3H/10T1/2 mouse cells - Lubet et al. (1983b)
Balb/3T3 mouse cells - Dunkel et al. (1981)
Fischer rat embryo cells - Mishra et al. (1978)
Fischer rat embryo cells Freeman et al. (1973)
(Rauscher leukaemia virus-infected)
Fischer rat embryo cells (leukaemia - Dunkel et al. (1981)
virus-infected)
C3H/10T1/2 mouse clone 8 cells - Dunkel et al. (1988)
Table 86. (continued)
Test system Resulta Reference
Pyrene
Syrian hamster embryo cells - DiPaolo et al. (1969);
Pienta et al. (1977);
Casto (1979)
C3H mouse prostate cells - Chen & Heidelberger
(1969)
Balb/C-3T3 mouse cells - DiPaolo et al. (1972);
Kakunaga(1973)
Fetal guinea pig cells - Evans & DiPaolo
(1975)
Fischer rat embryo cells - Mishra et al. (1978)
Hamswr embryo cells (SA7 - Casto et al. (1977)
virus-transformed)
C3H/10T1/2 mouse clone 8 cells ± Dunkel et al. (1988)
Balb/c-3T3 mouse - Lubet et al. (1990)
a Result; +, positive; ±, inconclusive; -, negative
Table 87. Chromosomal effects of polycyclic aromatic hydrocarbons in
mammalian cell systems in vivo, including DNA binding and adducts and
sperm abnormalities
Test system Resulta Reference
Anthracene
Chinese hamster bone marrow: - Roszinsky-Kocher et al.
CA, SCE (1979)
Mouse bone marrow: MN - Salamone et al. (1981)
Mouse: sperm abnormalities - Topham (1980)
Chinese hamster V79 (mouse host- - Sirianni & Huang (1978)
mediated):SCE
Mouse peripheral blood: MN - Oshiro et al. (1992)
Mouse skin : DNA binding - Reddy et al. (1984)
Benz[a]anthracene
Chinese hamster bone marrow: + Roszinsky-Kocher et al.
SCE (1979)
Chinese hamster bone marrow: CA - Roszinsky-Kocher et al.
(1979)
Long-Evans rat bone marrow: CA - Sugiyama (1973)
Chinese hamster bone marrow + Peter et al. (1979)
MN,CA
NMRI mouse(in metaphase II + Peter et al. (1979)
oocytes): CA
Mouse gastrointestinal epithelial - Reddy et al. (1991)
cells: nuclear anomalies
Rat lung: DNA adducts,SCE, MN + Whong et al. (1992)
Mouse skin: DNA binding + Reddy et al. (1984)
Rat bone marrow and spleen + Zhong et al. (1995)
cells: MN
Benzo[b]fluoranthene
Chinese hamster bone marrow: + Roszinsky-Kocher et al.
SCE (1979)
Chinese hamster bone marrow: CA - Roszinsky-Kocher et al.
(1979)
Mouse skin: DNA binding + Weyand et al. (1987)
Lung, liver and peripheral + Ross et al. (1991); Ross
lymphocytes of rats: DNA adducts et al. (1992)
Rat lung, liver; peripheral blood + Ross et al. (1991); Ross
lymphocytes; whole blood cultures: et al. (1992)
SCE
Mouse gastrointestinal epithelial + Reddy et al. (1991)
cells: nuclear anomalies
Rat peripheral blood lymphocytes: + Bryant et al. (1991)
SCE, MN
Table 87. (continued)
Test system Resulta Reference
Mouse gastrointestinal epithelial + Reddy et al. (1991)
cells: nuclear anomalies
Mouse skin: DNA binding + Amin et al. (1991a)
Mouse skin: DNA adducts, MN, UDS + Winker et al. (1995)
Benzo[j]fluoranthene
Mouse lung and liver cells: + Weyand & LaVoie (1988)
DNA adducts
Mouse skin: DNA adducts + Weyand et al. (1993)
Benzo[k]fluoranthene
Mouse skin: DNA binding + Weyand et al. (1987)
Mouse lung and liver cells: + Weyand & LaVoie (1988)
DNA adducts
Benzo[ghi]perylene
Mouse skin: DNA binding + Reddy et al. (1984)
Benzo[a]pyrene
Mouse: dominant lethal mutation + Epstein (1968)
Mouse: dominant lethal mutation + Generoso et al. (1982)
Mouse: spot test + Russell (1977)
Mouse: spot test + Davidson & Dawson
(1976)
Rat hepatocytes: UDS - Miralis et al. (1982)
Mouse germ cells: UDS - Sega (1979)
Mouse skin: DNA binding + Weyand et al. (1987);
Rice et al. (1984)
Chinese hamster bone-marrow +,+ Roszinsky-Kocher et al.
cells: CA, SCE (1979)
Chinese hamster bone-marrow +,+ Bayer (1978)
cells: CA, SCE
Mouse: CA; heritable translocations - Generoso et al. (1982)
Mouse bone marrow: MN + Salamone et al. (1981)
Mouse bone marrow: MN - Bruce & Heddle (1979)
Chinese hamster bone-marrow - Bayer(1978)
cells: MN
Mouse: sperm abnormalities + Topham (1980)
Mouse: sperm abnormalities + Bruce & Heddle (1979)
Chinese hamster V79 + Sirianni & Huang (1978)
(mouse host-mediated): SCE
Mouse epidermal cells: DNA adducts + Albert et al. (1991a,b)
Mouse bone marrow: MN + Shimada et al. (1991)
Mouse keratinocytes: MN + He & Baker (1991)
Mouse lung and liver cells: + Weyand & LaVoie (1988)
Table 87. (continued)
Test system Resulta Reference
DNA adducts
Mouse liver, lung and stomach: + Cummings et al. (1991)
DNA adducts
Rat peripheral lymphocytes: SCE + Li et al. (1991)
Mouse bone marrow: MN + Mavournin et al. (1990)
Mouse bone marrow: MN + Kliesch et al. (1982)
Mouse bone marrow: MN + Harper & Legator (1987)
Mouse peripheral blood cells: MN ± Oshiro et al. (1992)
Mouse gastrointestinal epithelial + Reddy et al. (1991)
cells: nuclear anomalies
Mouse bone marrow: SCE + Wielgosz et al. (1991)
Rat peripheral blood lymphocytes: + Willems et al. (1991)
SCE, DNA adducts
Rat liver cells: DNA adducts + Willems et al. (1991)
Rat peripheral blood lymphocytes: - Willems et al. (1991)
CA
Chinese hamster cells: CA + Matsuoka et al. (1979)
Mouse skin apithelial cells: + Hughes & Phillips (1991)
DNA binding
Human peripheral lymphocytes: + Haugen et al. (1986)
DNA adducts
Rat lung, liver and peripheral + Ross et al. (1991)
lymphocytes: DNA adducts
Mouse bone marrow: MN + Awogi & Sato (1989)
Mouse skin: DNA binding + Reddy et al. (1984)
Mouse skin: DNA adducts + Oueslati et al. (1992)
Mouse and rat bone marrow: MN + Shimada et al. (1992)
Mouse bone marrow: CA + Adler & Ingwersen (1989)
Benzo[e]pyrene
Chinese hamster bone-marrow + Roszinsky-Kocher et al.
cells: SCE (1979)
Chinese hamster bone-marrow - Roszinsky-Kocher et al.
cells: CA (1979)
Mouse gastrointestinal epithelial - Reddy et al. (1991)
cells: nuclear anomalies
Mouse skin: DNA binding Reddy et al. (1984)
Chrysene
Chinese hamster bone-marrow + Roszinsky-Kocher et al.
cells: SCE (1979)
Chinese hamster bone-marrow - Roszinsky-Kocher et al.
cells: CA (1979)
NMRI mice: metaphase II oocytes + Basler et al. (1977)
Mouse keratinocytes: MN + He & Baker (1991)
Mouse skin: DNA binding + Reddy et al. (1984)
Table 87. (continued)
Test system Resulta Reference
Dibenz[a,h]anthracene
Chinese hamster bone-marrow + Roszinsky-Kocher et al.
cells: SCE (1979)
Chinese hamster bone-marrow - Roszinsky-Kocher et al.
cells: CA (1979)
Rat peripheral blood lymphocytes: - Bryant et al. (1990)
SCE, MN
Mouse skin: DNA binding + Lecoq et al. (1991)
Mouse skin: DNA binding + Reddy et al. (1984)
Rat bone-marrow and spleen + Zhong et al. (1995)
cells: MN
Rat lung: DNA adducts, MN, SCE + Whong et al. (1994)
Mouse skin: DNA adducts, MN, + Winker et al. (1995)
UDS
Dibenzo[a,e]pyrene
Mouse skin epithelial cells: + Hughes & Phillips (1991)
DNA binding
Dibenzo[a,i]pyrene
Rat spleen cells: MN + Zhong et al. (1995)
Rat lung: DNA adducts, MN, SCE + Whong et al. (1994)
Fluoranthene
Mouse bone-marrow cells: SCE - Palitti et al. (1986)
Indeno[1,2,3-cd]pyrene
Mouse skin: DNA binding + Weyand et al. (1987)
Mouse skin epithelial cells: + Rice et al. (1990)
DNA binding
5-Methylcholanthrene
Mouse skin: DNA adducts + Amin et al. (1985a)
Naphthalene
Mouse bone-marrow cells: MN - Harper et al. (1984)
Perylene
Mouse skin: DNA binding - Reddy et al. (1984)
Table 87. (continued)
Test system Resulta Reference
Phenanthrene
Chinese hamster bone-marrow - Bayer (1978); Roszinsky
cells: CA Kocher et al. (1979)
Chinese hamster bone-marrow + Bayer (1978); Roszinsky
cells: SCE Kocher et al. (1979)
Chinese hamster bone-marrow - Bayer(1978)
cells: MN
Pyrene
Mouse bone marrow cells: SCE - Paika et al. (1981)
Mouse bone marrow: MIN - Salamone et al. (1981)
Mouse bone marrow cells: MN - Tsuchimoto & Matter
(1981)
Chinese hamster V79 - Sirianni & Huang (1978)
(mouse host-mediated): SCE
Mouse keratinocytes: MN - He & Baker (1991)
Mouse peripheral blood cells: MN - Oshiro et al. (1992)
Mouse gastrointestinal epithelial - Reddy et al. (1991)
cells: nuclear anomalies
Mouse: sperm abnormalities - Topham (1980)
Mouse skin: DNA binding - Reddy et al. (1984)
SCE, sister chromatid exchange; MN, micronucleus assay; CA, chromosomal
aberrations; UDS, unscheduled DNA synthesis
a Result: +, positive; ±, inconclusive; -, negative
Table 88. Effects of polycyclic aromatic hydrocarbons on
morphological transformation of mammalian cells in vivo
Test system Resulta Reference
Anthracene
Mouse bone-marrow cells - Salamone et al. (1981)
Chinese hamster embryo cells - DiPaolo et al. (1973)
Benzo[ghi]perylene
Hamster embryos, transplacental - Quarles et al. (1979)
exposure
Benzo[a]pyrene
Hamster embryos, transplacental + Quarles et al. (1979)
exposure
Phenanthrene
Hamster embryos, transplacental - Quarles et al. (1979)
exposure
a +, positive; ±; inconclusive; -, negative
Table 89. Overview of genotoxicity of polycyclic aromatic
hydrocarbons
Compound Results
Acenaphthene Inconsistent, limited database
Acenaphthylene Inconsistent, limited database
Anthanthrene Positive, limited database
Anthracene Negative, with a few exceptions
Benz[a]anthracene Positive
Benzo[b]fluoranthene Positive
Benzo[k]fluoranthene Positive
Benzo[k]fluoranthene Positive
Benzo[ghi]fluoranthene Positive, limited database
Benzo[a]fluorene Inconsistent, limited database
Benzo[b]fluorene Inconsistent, limited database
Benzo[ghi]perylene Positive
Benzo[c]phenanthrene Positive, limited database
Benzo[a]pyrene Positive
Benzo[e]pyrene Positive
Chrysene Positive
Coronene Positive, limited database
Cyclopenta[cd]pyrene Positive
Dibenz[a,h]anthracene Positive
Dibenzo[a,e]pyrene Positive
Dibenzo[a,h]pyrene Positive, limited database
Dibenzo[a,i]pyrene Positive
Dibenzo[a,l]pyrene Positive, limited database
Fluoranthene Positive
Fluorene Negative, with a few exceptions
Indeno[1,2,3-cd]pyrene Positive
5-Methylchrysene Positive
1-Methylphenanthrene Positive
Naphthalene Negative
Perylene Positive
Phenanthrene Inconsistent
Pyrene Inconsistent
Triphenylene Positive
Table 90. Carcinogenicity of polycyclic aromatic hydrocarbons in experimental animals
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Acenaphthene
Mouse 100 Dermal Dissolved in 90% benzene 9 months No tumours observed n Kennaway
no/lc (1924)
'Pure' Mouse, m 85 Dermal 3 drops, 1x/week of <1 year After 12 months 5/85 survived q Graffi et al.
white approx. 300 solution, 1 with a total of 2 tumours; 0.4 no/ls (1953)
year; initiation experiment tumour/animal; promotor only:
0.08 tumour/animal
Anthanthrene
Mouse 30 Dermal 0.3% in benzene, Life 1/30 lung adenoma n Badger et al.
2 x/week, life no/ld (1940)
Recrystallized Mouse, f 20 Dermal 0.05 or 0.1%, 3 x/week, 15 0/20 with tumours n Hoffmann &
Ha/lcR/Mil 12 months months no/val Wynder (1966)
Recrystallized Mouse, f 30 Dermal 25 Kg/animal, 10 × over 6 months 2/25 papillomas; promotor n Hoffmann &
Swiss 20 days; initiation only: 526 no/val Wynder (1966)
Ha/lcR/Mil experiment
'Rigourously Mouse, f 13 Dermal 0.25 mg/animal, 4 x; 65 weeks 2/13 papillomas; promotor n Van Duuren et
purified' lcR(Ha initiation experiment only: 520 papillomas; control no/val al. (1968)
acetone: 0/20 papillomas
Recrystallized Mouse, f 30 Dermal 43 µg/animal, 2 x/week, < 100 1/30 with skin carcinoma; n Lijinsky &
Swiss 75 weeks control: 2/30 with carcinomas no/val Garcia (1972)
98.65% Mouse, f 40 Dermal 109 µg/animal, 2 x/week, 70 weeks 47% skin-tumour-bearing p Cavalieri et al.
Swiss 30 weeks animals; solvent control: 0% no/val (1977)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
TLC- Mouse, f 30 Dermal 0.69 mg/animal, 1x; 35 weeks 18% with papillomas; p Scribner
purified) CD-1 initiation experiment promoter only: 3% no/val (1973)
> 99% Mouse, f 27 Dermal 221 µg/animal, 1x; 26 weeks 11 % papillomas; solvent n Cavalieri at
Sencar initiation experiment only: 9% yes/val al.(1989)
Mouse, m/f 27 s.c. 0.6 mg/animal, 1x/month, No local sarcomas observed n Lacassagne et
XVII 3 months no/ln, al. (1958)
ld
99.4% Rat f 35 Intrapulm. 0.65 and 3.4 mg/kg, 1x 102/88 1/35 and 19/35 with lung p Deutsch-
Osborne- weeks tumours; control: no tumours yes/val Wenzel et al.
Mendel (1983)
> 99% Rat, f 20 Intra- 1.1 mg/gland, 1x, 8 40 1/20 with mammary tumours; n Cavalieri at
Sprague- mammary glands weeks control: 0/21 or 2120 yes/val al. (1989)
Dawley injection
Anthracene
Mouse 2x100 Dermal 40% suspension/solution 5 months 0/100, 1/100 tumours n Kennaway
no/lc (1924)
Mouse 44 Dermal 5%, 3x/week < 11 No skin tumours n Miescher
months no/lc (1942)
Mouse, 20 Dermal 1.5 mg/animal, 2x/day; 3 21 3/17 with tumours; promotor n Salaman & Roe
'S' days/week; total: 20 x; weeks only: 4/19 yes/val (1956)
initiation experiment
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 5 Dermal 10% solution, 3x/week, < 20 No skin tumours n Wynder &
Swiss life months no/ln, Hoffmann
Millerton ld (1959a)
TLC- Mouse, f 30 Dermal 1.8 mg/animal, 1x; 35 14% with papillomas; q Scribner (1973)
purified CD-1 initiation experiment weeks promoter only: 3% no/val
Mouse, m/f 24 Dermal 4 µg, 1 x/day, 5 days/week, 38 No increased tumour n Forbes et al.
Skh:hair 38 weeks, then 2 h/day weeks frequency compared with yes/val (1976)
less 1, UV controls
outbred
Mouse, f 20 Dermal 100 µg/animal, 10x on 24 weeks 15% with tumours; solvent: n LaVoie et al.,
Swiss alternate days; initiation 10% yes/val (1983a)
albino experiment
(Ha/lcR)
Mouse, m/f 40-50 s.c. 5 mg/animal in tricaprylin; < 22-28 0/26 sarcomas after 5 months n Steiner (1955)
C57BI 1x months yes/ld
Mouse, m 5 i.p. 1000 mg/kg, 1 × < 5 No effects observed n Shubik & Della
Swiss months no/ln Porta (1957)
Rat 31 Oral 6 mg/animal/day, 7x/ 33 22/31 alive after 1 year; no n Schmahl &
(diet) week monthns tumours after 33 months no/lc Reuter, cited by
Gerarde (1960)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Highly Rat, 28 Oral 5-15 mg/animal/day, 700 days 2/28 malignant tumours, n Schmahl (1955)
purified BD I/BD III (diet) 6x/week, 78 weeks no/ld
Rat 10 s.c. 1 mg/animal, 1x/week, < 103 No subcutaneous sarcomas n Boyland &
103 weeks weeks no/ln Burrows (1935)
ld
Rat, 5 s.c. 5 mg/animal, 6-7x 10 No tumours observed n Pollia
Wistar months no/ln (1941)
Highly Rat, 10 s.c. 20 mg/animal, 1x/week, < 29 5/9 tumours (fibromas) at site p Druckrey &
purified BD I/BD III 33 weeks months injection no/ln, Schmahl (1955)
ld
Highly Rat, 10 i.p. 20 mg/animal, 1x/week, > 2 years 1/10 spindle-cell sarcoma q Schmahl (1955)
purified BD I/BD III 33 weeks no/ld
Rat, f 60 Intrapulm. 0.5 mg/animal, 1x/year No lung tumours; control: no n Stanton et al.
Osborne/ tumours no/d (1972)
Mendel
Rabbit 9 Cerebral 4-20 mg/animal, 1 × 20-54 No glioma n Russel (1947)
implant months no/in,
ld
Benz[a]anthracene
Mouse, 8-19 Oral 0.5 mg/animal, 1x, 8x 16 0/13, 1/19 and 1/8 with q Bock & King
C57/BL or 16 × (highest dose), months papillomas; no carcinomas yes/ln (1959)
< 2 months observed; control: 0/12
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m 20 or Oral 1.5 mg/animal, 3x/wk, < 547- 100% hepatomas and 95% p Klein (1963)
B6AF1/J, 40 5 weeks 600 days pulmonary adenomas; solvent no/val
newborn only: 10% hepatomas and
35% pulmonary adenomas
Mouse, m 20 Oral 1.5 mg/animal, 1x/day, < 568 80% hepatomas and 85% p Klein (1963)
B6AF1/J, 2 days days lung adenomas (inadequately no/val
newborn reported)
Purified Mouse 30 Dermal 0.3% in benzene, < 584 1/30 epitheliomas n Barry et al.
2x/week,life days no/ld (1935)
'Pure' Mouse, m 75 Dermal 3 drops, 1x/week of 0.5% < 1 year After 12 months 9/75 survived p Graffi et al.
white solution, 1 year; initiation with a total of 18 tumours; 2 no/val (1953)
experiment tumours/animal; promotor only:
0.08 tumour/animal
Recystallized Mouse, f 30 Dermal 66 µg/animal, 2x/week, 13-15.5 No tumours; solvent only: no n Miller & Miller
albino 20 weeks months tumours no/val (1963)
Mouse, 20 Dermal 0.5% solution, 2x/week, 638 days No tumours; control: no n Stevenson &
C3H 638 days tumours no/val von Haam
(1965)
Recrystallized Mouse, 30-50 Dermal 0.0001-0,5 mg/animal in < 88 Dose-dependent increase in p Bingham & Falk
C3H+e n-dodecane or 0. 1 weeks malignant tumours; solvent yes/val (1969)
mg/animal in toluene, control: no tumours
3x/week, 50 weeks
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized Mouse, f 20 Dermal 1 mg/animal, 1x; initiation 58-60 10/20 with papillomas; p Van Duuren et
Swiss experiment weeks promotor only:1/20; solvent no/val al. (1970)
Millerton control: 0%
ICR/Ha
TLC Mouse, f 30 Dermal 0.5 mg/animal, 1x; 35 weeks 62% with papillomas; p Scribner(1973)
purified CD-1 initiation experiment promotor only: 3% no/val
> 99% Mouse, f 40 Dermal 90 µg/animal, 2x/week, 70 weeks 2.6% skin-tumour bearing n Cavalieri et al.
Swiss 30 weeks animals; solvent control: 0% no/val (1977)
> 99% Mouse, f 30 Dermal 0.46 mg/animal, 1x; 26 weeks 57% with papillomas; p Slaga et al.
CD-1 initiation experiment promotor only: 6% no/val (1978)
Mouse, f 30 Dermal 0.1 and 0.57 mg/animal, 27 weeks 14% and 36% (p < 0.05) with p Levin et al.
CD-1 1x; initiation experiment tumours; solvent control: 7% yes/val (1984)
Mouse, f 30 Dermal 0.23 and 0.57 mg/animal, 27 week 17% and 38% papillomas; p Weyand et al.
CD-11 1x; initiation experiment solvent control: 4% yes/val (1990);Wood et
al. (1980)
Spectrometer Mouse, m/f 50 s.c. 5 mg/animal in tricaprylin; < 22 8/46 sarcomas after 4 months; p Stainer & Falk
C57BI 1x months solvent control: 3/280 no/val (1951)
control
Mouse, m/f 40-50 s.c. 0.05, 0.2, 1, 5, or 10 < 22-28 5/44,11/45,15/44, 20/36 and p Steiner &
C57BI mg/animal in tricaprylin; months 5/16 sarcomas yes/ld Edgcomb
1x (1952); Steiner
(1955)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized Mouse, f 30 s.c. 0.94 mg/animal, 1x < 15 No sarcomas; solvent control: n Miller & Miller
albino months no tumours no/val (1963)
Mouse 20 s.c. 5 mg in tricaprylin, 1x 638 days No tumours; control: no n Stevenson &
C3H tumours no/val von Haam
(1965)
Mouse, m/f 10/10 s.c. 1 mg/animal, 1x/week, 60-80 8/10 m and 6/10 f with p Boyland & Sims
C57BL 10 weeks weeks sarcomas; control: 0/20 m no/val (1967)
and 0/20 f
Mouse, m/f 87 s.c. 0.2 mg/animal in 70-75 70 weeks: 15/15 m and 2/18 f p Grover et al.
Swiss polyethylene glycol weeks with liver tumours, 4/15 m and no/val (1975)
newborn on days 0, 1 and 2 after 10/18 f with lung tumours;
birth corrected control data: 4/22 m
and 1/23 f with liver tumours
and 3/22 m and 1/23 f with lung
tumours
Mouse, m/f 140 i.p. 9.1, 18.2, and 36.4 26 weeks 10/47 m and 4/38 f with n Wislocki et al.
Swiss µg/animal on days 1, 8, pulmonary tumours; solvent no/val (1979)
Webster and 15 after birth control: 7/43 and 2/24
BLU:Ha(ICR),
newborn
Mouse, 11 i.v. 10 mg/kg, 1x 20 weeks 18% lung tumours; n Shimkin &
A control: 21 % no/val Stoner(1975)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 52 Bladder About 2 mg/animal, 1x > 40 17/52 bladder carcinomas p Clayson et al.
C57 × IF F, implant weeks and 1/52 papillomas; control: yes/val (1968)
hybrid 4/89
Rat, f 10 Oral 200 mg/rat, 1x 60 days No tumours in treated animals; n Huggins &
Sprague- control: 8/164 after 310 days no/ln, Yang(1962)
Dawley lc
Rat, m 25 Dermal Saturated solution in < 18 No tumours n Tawfic(1965)
Donryu acetone,dropped at 2x/wk months no/ld
to cover 2 cm2, 5 months
Recrystallized Rat, m 20 s.c. 1.88 mg/animal, 1x > 4 No sarcomas; solvent control: n Miller & Miller
Holtzman months no tumours no/val (1963)
TLC Rat, f 28 i.v. 2 mg/animal (=13 mg/kg), 98 days No mammary tumours n Pataki &
purified Sprague- 3x on day 50, 53, and 56 of no/ld Huggins(1969)
Dawley age
TLC Rat, m 8 i.m. 2.5 mg/animal into hind 270 days No sarcomas; control: no n Pataki &
purified Long- leg, 1 × on day 25 of age spontaneous sarcomas no/ln Huggins(1969)
Evans
>99% Rat, f 20 Intra 0.91 and 3.7 mg into 5th 20 weeks No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1 × control: no tumours no/val (1988a)
Dawley injection
Chromatography Hamster m/f 50 Dermal 8 drops of a 0.5% solution, < 85/61 No tumours n Shubik et al.
control Syrian 2x/week, 10 weeks weeks no/ln, (1960)
golden ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Hamster, m 5 or Dermal 20 mmol/litre solution, < 44 No tumours; control: no n Solt et al.
Syrian 26 (buccal painting 2x/week, 5 or weeks tumours no/val (1987)
golden pouch) 20 weeks
Hamster m 47 or Intra- 0.5 or 3 mg/animal per < 110 No tracheal tumours; control: n Sellakumar
Syrian 33 tracheal week 30 or 15 weeks weeks no tumours yes/val Shubik (1974)
golden
Benzo[b]fluoranthene
Mouse, f 20 Dermal 0.01, 0.1 and 0.5%, < 14,12, 0.01 %: 5% papillomas after p Wynder &
Swiss 3x/week, life and 8 14 months; 0.1%: 65% no/ld Hoffmann
Millerton months papilomas and 85% carcinomas (1959b)
after 12 months;0,5%: 100%
carcinomas after 5 months
Mouse, f 20 Dermal 1 mg, 1x; initiation 63 weeks 18/20 papillomas, 5/20 p Van Duuren et
Swiss experiment carcinomas; promotor only: no/val al. (1966)
ICR/Ha 5/20, 1/20
> 96% Mouse, f 40 Dermal 3.4,5.6,9.2 µg/animal, < 2 years 5/15/540% with local tumours; p Habs et al.
NMRI 2x/week, life control: no tumours yes/val (1980)
Mouse, Dermal 10-100 µg/animal; 20 weeks Dose-related skin tumour p LaVoie et al.
CD-1 initiation experiment incidence yes/val (1982b)
> 99% Mouse, f 20 Dermal 4 and 10 nmol/animal, 34 weeks 45 and 95% tumour incidence; p Amin et al.
Cr1:CD-1 10x every other day; solvent control: 5% yes/val (1985a)
(ICR)BR initiation experiment
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 20 Dermal 0.025 and 0.1 mg/animal, 24 weeks 100 and 100% tumour p Weyand et al.
CD-1 10x every other day; incidence; solvent control: yes/val (1990)
initiation experiment 10%
Mouse, f 20 Dermal 3 and 10 µg/animal, 10x; 34 weeks 65 and 100% with tumours; p Amin et al.
Cr1:CD1 initiation experiment solvent control: 15% yes/val (1991a)
(ICR)BR
Mouse, m/f 16/14 s.c. 0.6mg/animal, 1x/month, approx. 8/16 m and 10/14 f with local p Lacassagne et
XVII nc/Z 3 months 200 days sarcoma no/ld al. (1963a)
> 99% Mouse, m/f 15/17 i.p. 126 µg/animal in DMSO < 52 53% hapatic, 18% lung p LaVoie et al.
CD-1 on days 1,8, and 15 after weeks tumours; control: 6% hepatic yes/val (1987)
newborn birth (total dose) tumours, no lung tumours
99.5% Rat, f 35 Intrapulm. 0.1, 03 and 1 mg/animal, 110/113/ 0/35, 1/35, and 9/35 p Deutsch
Osborne/ 1x 112 pulmonary carcinomas; 1/35, yes/val Wenzel et
Mendel weeks 2/35, and 4/35 pleomorphic al.(1983)
sarcomas; control: no tumours
Hamster, m 47 Intra- 0.5 and 0.5 mg/animal < 110 0/47 and 1/47 tracheal n Sellakumar &
Syrian tracheal per week, 30 weeks weeks tumours; control: no tumours yes/val Shubik (1974)
golden
Berizo[j]fluoranthene
Highly Mouse, f 20 Dermal 0.1 and 0.5%, 3x/week, < 9 and 7 100%/95% with skin p Wynder &
purified Swiss life months carcinomas no/ld Hoffmann
(1959b)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
96% Mouse, f 40 Dermal 3.4, 5.6, 9.2 µg/animal, < 2 years 3, 3, and 5% with local q Habs et al.
NMRI 2x/week, life tumours;controls: 0% yes/val (1980)
> 99% Mouse, f 20 Dermal 3, 10, 100 µg, 10x over 20 24 weeks 30, 55, and 95% with tumours p LaVoie et al.
Cr1:CD1 days; initiation experiment (papillomas/keratinizing yes/val (1982b)
(ICR)BR lesions); 1 malignant lymphoma
Mouse, f 20 Dermal 25, 75 µg, 10x over 20 24 weeks 70 and 90% with papillomas; p Rice et al.
CD-1 days; initiation experiment vehicle control: 10% yes/val (1987)
99% Mouse, m/f 21/18 i.p. 278 µg/animal in DMSO < 52 81% males and 22% females p LaVoie et al.
CD-1 on days 1, 8, and 15 after weeks with liver and lung tumours; yea/val (1987)
newborn birth (total dose) control: 6%:0%
99.9% Rat, f 35 Intrapulm. 0.8, 4, and 20 mg/kg, 1x 110/117/ 1/35, 3/35 and 18/35 p Deutsch-
Osborne/ 89 weeks pulmonary carcinomas; yes/val Wenzel et
Mendel control: no tumours al.(1983)
Benzo[ghi]fluoranthene
Highly Mouse, f 20 Dermal 0.1 and 0.5%, 3x/week, < 13 No skin tumours n Wynder &
purified Swiss life months no/ld Hoffmann
(1959b)
Mouse, f 20 Dermal 1 mg, 1x; initiation 4/20 papillomas, no n Van Duuren et
Swiss experiment carcinomas; promotor only: no/val al. (1966)
ICR/Ha 5/20, 1/20
Benzo[k]fluoranthene
Highly Mouse, f 20 Dermal 0.1 and 0.5%, 3x/week, < 13 0/20 and 2/20 skin papillomas q Wynder &
purified Swiss life months no/ld Hoffmann
(1959b)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 25 Dermal 1 mg/animal (total dose) < 2 years No skin tumours; spontaneous n Mohr (1969)
NMRI in 50 aliquots tumours: 10% na/val
> 96% Mouse, f 40 Dermal 3.4, 5.6, 9.2 µg/animal, < 2 years 3, 0 and 0% with local n Habs et al.
NMRI 2x/week, life tumours; control: no tumours yes/val (1980)
> 99% Mouse, f 20 Dermal 3, 10, 100 µg, 10x over 24 weeks 5, 25, and 75% with tumours p LaVoie et al.
Cr1:CD1 20 days; initiation (papillomas/keratinizing yea/val (1982b)
(ICR)BR experiment lesions)
Mouse m/f 16/14 s.c. 0.6 mg/animal, 1x/month, approx. 8/16 m and 5/14 f with local p Lacassagne et
XVII nc/Z 3 months 200 sarcomas no/ld al.(1963a)
days
> 99% Mouse, m/f 16/18 i.p. 530 µg/animal in DMSO < 52 19% males and 17% females q LaVoie et al.
CD-1 on days 1, 8, and 15 after weeks with tumours; control: 6%:0% yes/val (1987)
newborn birth (total dose) liver and lung tumours
99.5% Rat, f 27-35 Intrapulm. 0.65, 3.4, and 17 mg/kg, 114/95/98 0/35, 3/31 and 12/27 p Deutsch-
Osborne/ 1x weeks pulmonary carcinomas; yes/val Wenzel (1983)
Mendel control: no tumours
Benzo[a]fluorene
Mouse, 20 Dermal 0.3%, 2x/week, life < 20 No skin tumours; 4/20 lung q Badger et al.
stock' months adenoma; 1/20 sebaceous no/ld (1942)
adenoma
> 99.5% Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks 2/20 skin tumours; control: n LaVoie et al.
Swiss initiation experiment 1/20 yes/val (1981c)
Ha/ICR
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 10 s.c. 5 mg/animal, at intervals < 23 1/10 lung adenoma; no n Badger et al.
stock' of a few weeks, life months sarcomas no/ln, (1942)
ld
Benzo[b]fluorene
99.5% Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks 4/20 skin tumours; control: q LaVoie et al.
Swiss initiation experiment 1/20 yes/val (1981c)
Ha/ICR
Benzo[ghi]perylene
Mouse, f 50 Dermal 0.38% solution in benzene, 2/50 with skin tumours; n Lijinsky &
Swiss 3x/week, life control: 1/59 skin carcinomas no/val Saffiotti (1965)
Chromatography Mouse, f 20 Dermal 0.05% and 0.1 %, 3x/week, 15 1/20 and 0/20 skin papillomas; n Hoffmann &
purified Swiss Ha/I 12 months months solvent control: no tumours no/val Wynder (1966)
CR/Mil
Chromatography Mouse, f 30 Dermal 25 µg/animal, 10x over 28 6 months 2/30 papillomas; control: 2/30 n Hoffmann &
purified Swiss Ha/ days; initiation experiment 0/20 no/val Wynder (1966)
ICR/Mil
Mouse, f 50 Dermal 20 µg, 2 mg and 4 mg/ < 22.5 3/50, 6/50, and 4/50 with n Muller (1968)
NMRI animal, 2x/week, 25 weeks months tumours; vehicle control: 7/50 no/val
Mouse, f 50 Dermal 1 and 2 mg/animal, 1x; < 22.5 5/50 and 4/50 with tumours; n Muller (1968)
NMRI initiation experiment months vehicle control: 7/50 no/val
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rigourously Mouse, f 20 Dermal 0.8 mg/animal, 1x; 12-13 3/20 papillomas, 1/20 n Van Duuren et
purified Swiss initiation experiment months squamous-cell carcinoma; no/val al. (1970)
vehicle control: 1/20 with
2 papillomas
Highly Mouse, f 50 Dermal 5.5 and 16.5 µg/animal, 33 weeks No tumours n Goldschmidt et
purified ICR/Ha 3x/week, 33 weeks no/lc al. (1973
Highly Mouse, f 50 Dermal 21 µg/animal, 3x/week, 52 weeks No skin tumours; solvent n Van Duuren &
purified Swiss 52 weeks control: no tumours no/val Goldschmidt
ICR/Ha (1976)
Mouse, f 50 s.c. 0.83 and 16.7 mg/animal, < 22.5 5/50 and 4/40 with tumours; n Muller (1968)
NMRI 1 x/2 weeks, 6 months months control:4/50 no/val
Mouse, f 20 s.c. 0.1, 1 and 10 mg/animal, < 22 No skin or subcutaneous n Muller (1968)
NMRI 1x/2 weeks, 20 weeks months tumours;other tumours same no/val
as control
98.5% Rat, f 34-35 Intrapulm. 0.65, 3.4, and 17 mg/kg, 109/114/ 0/35, 1/35 and 4/34 n Deutsch-
Osborne/ 106 pulmonary carcinomas effect yes/val Wenzel et
Mendel 1x weeks of impurity suggested; al.(1983)
control: no tumours
Benzo[c]phenanthrene
Mouse, 20 Dermal Not specified < 676 7 epitheliomas, 5 papillomas p Barry et al.
days no/ld (1935)
Mouse, 40 Dermal 0.3%, 2x/week, < 19 < 19 1 papilloma, 4 squamous cell q Badger et al.
months months no/ld (1940)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 20 Dermal 0.5% solution, 2x/week, 638 days 3 carcinomas, 2 sarcomas; q Stevenson &
C3H 638 days control: no tumours no/val von Haam
(1965)
Mouse, f 30 Dermal 91 and 457 µg, 1x; 21 weeks 5/30 and 11/30 papillomas; p Levin et al.
CD-1 initiation experiment control:no tumours (1980)
Mouse 10 s.c. 5 mg at intervals of < 15 No injection-site tumours n Badger et al.
several weeks, life months no/ln, (1940)
ld
Mouse 20 s.c. 5 mg in tricaprylin, 1x 638 days 3 sarcomas; controls: no q Stevenson &
C3H tumours no/val von Hearn
(1965)
Rat, 6 s.c. 5 mg/animal; several approx. 1/6 sarcoma at injection site q Badger et al.
repeated doses 18 months no/ln, (1940)
ld
Benzo[a]pyrene
Mouse, f 15 Oral 3 mg/animal in sesame 30 weeks Increased pulmonary tumours p Wattenberg &
A/HeJ oil, 2x (16.6); control: 0.3 yes/val Leong(1970)
Mouse, f 15 Oral 2 mg/animal, 3x, every 26 weeks 15/15 with forestomach p Sparnins et
A/J 2 weeks tumours and 15/15 with yes/val al. (1986)
pulmonary adenomas;no
control
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 25-73 Oral 0.004-1 mg/animal per 140-200 Dose-dependent gastric p Neal & Rigdon
CFW (diet) day, < 110-165 days days tumours (0-90%);control: no no/val (1967)
tumours
Mouse, m/f 9-26 Oral 1-20 mg/animal/day, 150-300 Dose-dependent gastric p Neal & Rigdon,
CFW (diet) < 1-30 days days tumours(0-100%); control: no/val (1967)
no tumours
Mouse, m/f 60-175 Oral 0.25 and 1 mg/g food, < 34 33 and 61 % with stomach p Rigdon & Neal
White (diet) < 34 weeks weeks tumours; 53 and 20% with no/val (1966)
Swiss lung tumours;controls: 1 and
21%
Mouse, f 20-30 Dermal 0.001, 0.005, and 0.01%, < 21, 14, 3 and 43%, 63 and 73%, and p Wynder &
Swiss 3x/week, life and 11 951% and 95% with skin no/ld Hoffmann
carcinomas/papillomas (1959a)
Mouse, f 20 Dermal 0.01, 0.05 and 0.5%, < 12, 6, 85, 95, and 75% with skin p Wynder &
Swiss 3x/week, life and 6 carcinomas no/ld Hoffmann
Millerton months (1959b)
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1%, 3x/week, 15 17/20 and 19/20 skin tumours; p Hoffmann &
Swiss 12 months months solvent control: no tumours no/val Wynder (1966)
Ha/ICR/Mil
Mouse, f 30 Dermal 25 µg/animal, 1/x over 28 6 24/30 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment months only: 2/30 no/val Wynder (1966)
Ha/ICR/Mil
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 50 Dermal 20 and 200 µg/animal, < 22.5 50/50 and 50/50 with skin p Muller (1968)
NMRI 2x/week, 25 weeks months tumours;vehicle control: 7/50 no/val
Mouse, 20-30 Dermal (a) 0.00002% in (a) 21% malignant tumours; p Bingham & Falk
C3H/He n-dodecane/decalin; (b) 50% tumours (three orders yes/val (1969)
(b) 0.02% in decalin, of magnitude difference in dose)
3x/week, 50 weeks
Mouse, f 30 Dermal 5 µg/animal, 10x, 20 days; 24 weeks 19/29 tumour-bearing animals p Hoffmann et al.
Swiss initiation experiment with 67 skin tumours; control: no/val (1972)
Ha/ICR/Mil 1/30
Mouse, f 20 Dermal 0.05 and 0.1 mg/animal; 6 months 13 and 18 with skin tumours; p Masuda &
Swiss ICR 60x no solvent control no/val Kagawa (1972)
Mouse, f 20 Dermal 5 µg/animal, 3x/week, < 72 13/20 with 22 skin tumours; p Hecht et al.
Swiss 72 weeks weeks 4/20 with 4 carcinomas; no/val (1974)
Ha/ICR/Mil solvent control: no tumours
Mouse, f 50 Dermal 5 µg/animal, 3x/week, life 440 days 16 animals with 26 tumours; p Van Duuren &
Swiss control: no tumours no/val Goldschmidt
ICR/Ha (1976)
Mouse, f 20 Dermal 5 and 10 µg/animal, 62 weeks Low dose: 10/20 with 19 skin p Hecht et al.
Swiss 3x/week, 62 weeks tumours, 7/20 with 8 yes/val (1976b)
Ha/ICR carcinomas; high dose:18/20
with 70 skin tumours, 14/20
with 16 carcinomas; solvent
control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
99.9% Mouse, f 40 Dermal 100 µg/animal, 2x/week, 70 weeks 79% skin-tumour bearing P Cavalieri et al.
Swiss 30 weeks animals;solvent control: no no/val (1977)
tumours
Mouse, f 40 Dermal 1.7, 2.8, 4.6 µg/animal, < 2 years 24, 69 and, 61 % with local p Habs et al.
NMRI 2x/week, life tumours(high rate of systemic yes/val (1980)
tumours);control: no tumours
> 99.5% Mouse, f 20 Dermal 30 µg/animal, 10x on 24 weeks 93% with tumours; vehicle p LaVoie et al
Swiss alternate days; initiation control: no tumours no/val (1981b)
Ha/ICR experiment
Mouse, f 20 Dermal 3 µg, 10x over 20 days; 24 weeks 85% with tumours (papillomas/ p LaVoie et al.
Cr1:CD1 initiation experiment keratinizing lesions) yes/val (1982b)
(ICR)BR
> 96% Mouse, f 20 Dermal 2 and 4 µg/animal, 648 and 45% (10% papillomas/35% p Habs et al.
NMRI 2x/week, life 528 days carcinomas) and 85% yes/val (1984)
(mean) (0%/85%) with skin tumours;
control: no tumours
99.5% Mouse, m 50 Dermal 12.5 µg/animal, 2x/week, < 99 94% with malignant skin p Warshawsky &
CH3/HeJ 99 weeks weeks tumours;solvent control: no no/val Barkley (1987)
tumours; untreated control: no
tumours
Mouse, f 24 Dermal 0.8 µmol/mouse, 1x; 24 weeks Enhanced incidence of skin p Cavalieri et al.
Sencar initiation experiment papillomas (80-92%) no/val (1988b)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m 12 Dermal 1.2 mg/animal, 6 days/wk, < 27 Multiple tumours; p Shubik & Della
Swiss 19 weeks weeks squamous-cell carcinomas no/ln Porta (1957)
> 99% Mouse, f 20 Dermal 2.5 µg/animal, 10x over 20 24 weeks 89% tumours, 5.5 skin p Rice et al.
CD-1 days; initiation experiment tumours/animal; control: 5% yes/val (1988b)
> 99% Mouse, f 25 Dermal 2.5 µg/animal, 10x over 20 23 weeks 96% tumours, 3.4 skin p Rice et al.
CD-1 days; initiation experiment tumours/animal; control: no yes/val (1990)
tumours
Chromatography Mouse, f 23-24 Dermal 8.4, 25.2 and 75.7 µg/ 15 weeks 10/23, 17/24 and 21/23 with p Cavalieri et al.
purified Sencar animal, 1x; initiation tumours; control: no tumours yes/val (1991)
experiment
Chromatography Mouse, f 24 Dermal 1, 5 and 25 µg/animal, 1 x; 27 weeks 1/24, 10/24 and 22/24 with p Cavalieri et al.
purified Sencar initiation experiment tumours; control: no tumours yes/val (1991)
Chromatography Mouse, f 24 Dermal 25 µg/animal, 1x; initiation 27 weeks 1/24 with tumours p Cavalieri et al.
purified Sencar experiment without yes/val (1991)
promotion
HPLC Mouse, f 43-50 Dermal 16, 32, or 64 µg/animal, < 35 1, 1.5 and 7.5 tumours/animal p Albert et al.
control ICR/Harlan 1x/week, 29 weeks weeks after 35 weeks no/val (1991a)
Mouse, m 20 Dermal 100 µg/animal, 2x/week, Tumours from 15 weeks p Andrews et al.
Balb/c 3 weeks-5 months onwards no/val (1991)
Mouse, m/f 40-50 s.c. 0.09 mg/animal in < 22-28 16/21 sarcomas after 5 p Steiner (1955)
C57BI tricaprylin; 1x months months yes/ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 14/16 s.c. 0.6 mg/animal, 1x/month, > 129/160 13/14 m and 8/16 f with local p Lacassagne et
XVII 3 months days sarcomas no/ld al. (1958)
(average
latency)
Mouse, m/f 154/ s.c. 0.6 mg/animal, 1x/month, approx. 154/154 m and 112/162 f with p Lacassagne et
XVII nc/Z 162 3 months 110/150 local sarcomas no/ld al. (1963a)
days
Mouse, 20 s.c. 0.1, 1 and 10 mg/animal, 17, 7, 6 All animals with sarcomas at p Muller (1968)
NMRI 1 x/2 weeks, 20 weeks months injection site no/val
Mouse, f 90 s.c. 25, 50, 100, 200 and 400 < 16 25, 50, 55, 75 and 65% with p Pott et al.
NMR1 µg/animal, 1x months tumours; solvent control: no/val (1973)
< 5%
Mouse, m/f 31-38 s.c. 0.01 and 0.1 mg/animal, 30 weeks 16 and 64% with lung p Rippe & Pott
newborn 1x tumours; control: 13% with no/val (1989)
lung tumours
> 99% Mouse, m/f 17/14 i.p. 278 µg/animal in DMSO < 52 76% hepatic and 64% lung p LaVoie et al.
CD-1 on days 1, 8, and 15 after weeks tumours; control: 6% hepatic yes/val (1987)
newborn birth (total dose) tumours, no lung tumours
> 99% Mouse, m/f 28/27 i.p. 59.5 µg/animal on days 26 weeks 46 m, 70, f with lung tumours; p Busby et al.
Swiss- 1, 8, and 15 after birth vehicle control: 14 m, 7 f yes/val (1989)
Webster (total dose)
BLU:Ha(ICR),
newborn
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 10 i.v. 10 mg/kg, 1 × 20 weeks 100% lung tumours; control: p Shimkin &
A 21% no/val Stoner (1975)
Mouse, f 19-22 Intra- 0.05 and 0.15 mg/animal, 27 and 42% with carcinomas p Pott et al.
NMRI tracheal 20x in the respiratory tract; na/val (1978)
control: 9%
99% Mouse, f 45; Intra- 1 mg/animal, 1x/week, < 18 No colonic tumours, 73% lung p Anderson et al.
ICR/Ha 60 colonic 14 weeks months tumours, 94% forestomach yes/val (1983)
contr. instill- tumours, 7% subcutaneous
ation sarcomas, 23% mammary
carcinomas; control: 25% lung
tumours, 20% forestomach
tumours, 9% mammary
carcinomas, no subcutaneous
sarcomas or colonic tumours
99% Mouse, f 38; Intra- 1 mg/animal, 1x/week, < 18 No colonic tumours, 94% fore- p Anderson et al.
C57BI/6 45 colonic 14 weeks months stomach tumours, 16% yes/val (1983)
instill- peritoneal sarcomas, 28%
ation lymphomas; control: 21%
forestomach tumours, no sarcomas
or lymphomas or colonic tumours
Rat, f 9 Oral 100 mg/kg, 1x 60 days 8/9 with mammary tumours; p Huggins &
Sprague- control: 8/164 in 310 days no/ln, Yang(1962)
Dawley lc
Rat f 20 Oral 625 mg/animal, 1x/week, 90 weeks 67-77% with mammary p McCormick et
LEW/Mai 8x; 50 mg/animal, 1 × tumours; control: 30% yes/val al. (1981)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rat, f 50 s.c. 33, 100, 900, and 2700 < 16 10, 15, 70 and 75% with p Pott et al.
Wistar µg/animal, 1x months tumours; solvent only: < 5% no/val (1973)
Rat, f 37 i.p. 5 mg/animal; 1x; in bees' (a) 89% abdominal tumours p Roller et al.
Wistar wax/tricaprylin 25/75 (a) 2 years (mesotheliomas, sarcomas); no/val (1992)
or saline (b) (b) 50%; vehicle controls:
(a) 70%; (b) 3%
Rat, f 13-17 Intra- 0.5, 1, or 2 mg/animal in Life 7, 65, and 92% with lung p Davis et al.
Wistar tracheal infusion; 1x/2 weeks; 18x tumours; control: no tumours yes/val (1975)
Rat, m/f 15/15 Intra- 1 mg/animal, 1x/week, Life 3/13 (m) and 3/14 (f) with p Ishinishi et al.
Wistar tracheal 15x (mean, malignant lung tumours no/val (1976)
491/540 (mean: 22.2%); vehicle control:
days) 0%
Rat, f 36-40 Intra- 1 mg/animal; 20x 19% with lung tumours; p Pott et al.
Wistar tracheal control: no tumours no/val (1987)
Rat, m/f 20/20 Intra- 7 mg/kg, every 14 days, < 781 19/20 m and 18/20 1 with lung p Steinhoff et al.
Sprague- tracheal 22x (total dose: 154 days tumours; vehicle control: 0% no/val (1991)
Dawley mg/kg)
99.1% Rat, f 35 Intrapulm. 0.1, 0.3, or 1 mg/animal, 111/77/54 4/35, 21/35 and 33/35 p Deutsch-
Osborne/ 1x weeks pulmonary carcinomas; 6/35, yes/val Wenzel et
Mendel 2/35 and 0/35 pleomorphic al.(1983)
sarcomas; control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rat, f 35 Intrapulm. 0.05, 0.1, or 0.2 11, 17, and 46% with tumours; p Grimmer et al.
Osborne/ mg/animal, 1X control: no tumours yes/val (1987)
Mendel
99.6% Rat, f 35 Intrapulm. 0.03, 0.1, or 0.3 mg/ < 135 8.6, 31.4, and 77.1% tumour p Wenzel-
Osborne/ animal, 1x weeks incidence; control: no tumours yes/val Hartung et al.
Mendel (1990)
Rat, m 14-15 Intrapulm. 50, 100, or 200 µg/animal < 100 0/10, 3/10 and 4/9 lung p Horikawa et al.
Fischer weeks tumours; control: no tumours no/val (1991)
344/Du Crj
Rat, 94 Intrabr- approx. 3-5 mg/animal, 1x approx. Carcinoma incidence: 17% p Laskin et al.
onchial 5 months no/lc (1970)
pellet
> 99% Rat, f 20 Intra- 1 and 4 mg into 5th 20 weeks 50 and 80% with mammary p Cavalieri et al.
Sprague- mammary mammary gland, 1x tumours; control: no tumours no/val (1988a)
Dawley
Rat f 20 Intra- 63 and 252 µq/gland, 1x, < 24 7/20 and 9/20 with mammary p Cavalieri et al.
Sprague- mammary 8 glands weeks tumours; control: 1/18 yes/val (1991)
Dawley injection
Recrystallized Rat, f 20 Pellet 0.5 and 1 mg; 1x 28 12 and 65% with carcinomas p Topping et al.
Fischer 344 implant- (implanted into tracheal months in tracheal transplants yes/val (1981)
ation transplants)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Hamster, m/f 13 Oral 2.5 mg/animal, 4 < 14 9/13 with forestomach cancer; p Chu &
Syrian (diet) days/week, < 14 months months 2/13 with papillomas no/val Malmgren
golden (1965)
Chromatography Hamster, m/f 15/15 Dermal 4 drops of a 0.8% solution < 99/68 m:1 small nodular melanotic q Shubik et al.
control Syrian in mineral oil, 1x/week, 8 weeks lesion, 2 malignant no/ln, (1960)
golden weeks including a 30-week lymphomas; f: no tumours ld
interval
Chromatography Hamster, m/f 5/5 Dermal 6 drops of 0.01% solution < 70 No skin tumours n Shubik et al.
control Syrian in acetone, 2x/week, 40 weeks no/ln, (1960)
golden weeks ld
Hamster, m 5 or Dermal 20 mmol/litre solution, < 44 10% buccal pouch carcinomas p Solt et al.
Syrian 28 (buccal painting 2x/week, 5 or 20 weeks after 40 week; control: no no/val (1987)
golden pouch) weeks tumours
Hamster, m 10 Inhalation 4.5 h/day, 5 days/week, Life No tumours n Thyssen et al.
Syrian 9.8 mg/m3 for 16 weeks or no/ld (1980)
golden 44.8 mg/m3 for 10 weeks
Hamster, m 24 Inhalation 2.2, 9.5, or 46.5 mg/m3, 109 Dose-dependent tumours in p Thyssen et al.
Syrian 4.5 h/day in the first 10 weeks nasal cavity, pharynx, larynx, no/val (1981)
golden weeks, thereafter 3 h/day, and trachea; also in oesophagus
109 weeks and forestomach (papillomas,
polyps, squamous-cell
carcinomas); no lung tumours;
larynx most affected with 0,
31 and, 52% incidence; control:
no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Pure Hamster, m/f 30/30 Intra- 3 mg/animal, 1x/wk, 15 < 45/60 14/19 and 21/21 with tumours p Saffiotti et al.
Syrian tracheal week (mixed with inert weeks in respiratory tract; control: no/val (1968)
golden dust of haematite no tumours
[ferric oxide]
Hamster, m 30 Intra- 3 mg/animal, 1x/week, < 74 All with bronchioalveolar p Crocker et al.
Syrian tracheal 14 weeks weeks metaplasia; 5/19 no/val (1970)
golden squamous-cell carcinomas,
3/19 adenomas, 1/19 tracheal
tumours
Pure Hamster, m/f 30-50 Intra- 0.25, 0.5, 1, or 2 mg/ Life Dose-related increase in p Saffiotti et al.
Syrian tracheal animal, 1x/week, 30 wks respiratory tract tumours; no/val (1972)
golden (mixed with inert dust of control: no tumours
ferric oxide)
Pure Hamster, m 30 Intra- 0.0625, 0.125, 0.25, 0.5, 78 weeks Dose-related increase in p Feron et al.
Syrian tracheal and 1 mg/animal, 1x/week, respiratory tract tumours no/val (1973)
golden 52 weeks (3-26%); controls: no tumours
Hamster, m/f 25/25 Intra- 0.9 mg/animal per week, < 100 17% (8/46) tumours in p Henry et al.
Syrian tracheal 30 weeks weeks respiratory tract; control: no/val (1975)
golden no tumours
Hamster, Intra- 0.3 or 0.9 mg/animal, < 2 years 17 and 68% with tumours p Pott et al.
Syrian tracheal 1x/week, 20 weeks no/lc (1978)
golden
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Hamster, m 29 Intra- 0.125, 0.25, 0.5, or Life 31, 83, 66, and 31% tumours p Ketkar et al.
Syrian tracheal 1 mg/animal, 1x/week, life in respiratory tract; control; no/val (1979)
golden no tumours
Hamster, m 30 Intra- 5, 20 or 40 µg/animal, Life 4/28, 5/27 and 7/28 with q Kunstler (1983)
Syrian tracheal every 2 weeks, life meta plasia in respiratory no/val
golden tract, malignant neoplasm
and 1 adenoma in high-dose
group; controls: 1/29 or 3/30
Hamster, 97 Intra- 3-5 mg 63/97 with lung cancers p Laskin et al.
bronchial no/val (1970)
pellets
Hamster, Tracheal approx. 0.83 mg/animal, Tracheal papillomas and p Mohr (1971)
Syrian insuff- 3x/week, 1 year carcinomas no/lc
golden lation
Hamster, Bronchial 150 days > 90% with focal cancers p Benfield &
Syrian implants no/lc Hammond
golden (1992)
Dog Paren- > 8 First parenchymal cancer after p Benfield &
chymal months 8 months; 7/12 dogs with no/lc Hammond
implants tumours (1992)
Pig, m/f 1/1 i.m. 4.8 mg/kg, 1x; 6 months 12 No sarcomas n Kallistratos &
German later 2.1 mg/kg, 1x months no/val Pfau (1971)
Edelland-
schwein
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Pig, m/g 1/1 i.m. 6.3 mg/kg, 1x; 6 months 12 No sarcomas n Kallistratos &
mini later 1.9 mg/kg, 1 × months no/val Pfau (1971)
Cattle, m/f 1/1 i.m. 0.95 mg/kg, 1x; 6 months 29 No sarcomas n Kallistratos &
German later 0.75 mg/kg, 1x months no/val Pfau (1971)
black/white
Monkeys m/fm 1/1 s.c. 10 mg/animal, 1x (a) >18 (a) 1/2 with local tumours q Noyes (1969)
(a) /f 1/1 (coadministration with months (b) death within 5 weeks no/ln
Saguinus 10 mg DMBA at other site) (b) < 5
oedipus; weeks
(b) S.
fusciocollis
Monkey, s.c. 1 × (not specified) Fibrosarcomas p Adamson &
Galago no/lc Sieber (1983)
crassus
Monkey, 17 s.c. 30-90 mg/kg, multiple < 18 No tumours observed; n Adamson &
Old world administration (not years survival: 9/17 no/lc Sieber (1983)
specified)
Monkey, m/f 4/2 Intra- 3-15 mg, 1x/week (with 67-69 Bronchioalveolar metaplasia; p Crocker et al.
Galago tracheal ferric oxide), up to 69 weeks 2/3 squamous carcinomas no/val (1970)
crassust weeks arising from bronchus
Benzp[e]pyrene
Mouse, f 20 Dermal 0.1%, 3x/week, life < 13 2/20 papillomas, 3/20 q Wynder &
Swiss months carcinomas no/ld Hoffmann
Millerton (1959a)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 20 Dermal 1 mg, 1x; initiation 64 weeks 2/20 with papillomas; pure q Van Duuren et
Swiss experiment substance: no tumours no/val al. (1968)
ICR/Ha
TLC Mouse, f 20 Dermal 2.5 mg/animal, 1x; 35 weeks 85% with papillomas; p Scribner (1973)
purified CD-1 initiation experiment promotor only: 3% no/val
Highly Mouse, f 50 Dermal 15 µg/animal, 3x/week, 368 days No tumours observed; control: n Van Duuren &
purified ICR/Ha 368 days no tumours no/val Goldschmidt
(1976)
> 99% Mouse, f 30 Dermal 100 µg/animal, 2x/week, 30-40 At 30 wks: 68% papillomas, p Slaga et al.
CD-1 30 weeks weeks at 40 weeks: 24% carcinomas no/val (1979)
> 99% Mouse, f 30 Dermal 100 and 252 µg/animal, 30-40 High dose: at 30 weeks, 19% q Slaga et al.
CD-1 1 x; initiation experiment weeks papillomas; at 40 weeks, no no/val (1979)
carcinomas;vehicle control: et
30 weeks, 14% papillomas
99% Mouse, f 30 Dermal 0.25, 0.63, or 1.5 mg/ 26 weeks 15, 11, or 140% with q Buening et al.
CD-1 animal, 1x;initiation papillomas; vehicle control: no/val (1980)
experiment 7% papillomas
> 95% Mouse, f 30 Dermal 0.5 mg/animal, 1 x; 15 weeks 17% with papillomas; vehicle q Slaga et al.
Sencar initiation experiment control:10% no/val (1980, 1981)
99% Mouse, m/f 30/30 i.p. 0.1, 0.2, 0.4, or 0.2, 0.4, 62-66 21/35 (m), 0/35 (f) or 12/30 q Buening et al.
Swiss-Webster 0.8 mg on days 1, 8, and weeks (m), 0/30 (f) with hepatic no/val (1980)
BLU:Ha(ICR), 15 of life tumours; controls: 11/53 (m),
newborn 0/24 (f)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
99.7% Rat, f 30-35 Intrapulm. 0.8, 4.2, or 20 mg/kg, 1x 117/111/ 1 pulmonary sarcoma at n Deutsch-
Osborne/ 104 4.2 mg/kg; 1 squamous-cell yes/val Wenzel et
Mendel weeks carcinoma at 20 mg/kg; no al.(1983)
tumours in controls
Recrystallized Rat, f 20 Tracheal 1 mg, 1x 28 No tumours n Topping et al.
Fischer pellet months yes/val (1981)
344
Chrysene
Mouse 100 Dermal 1% in 90% benzene < 11 No tumours n Kennaway
months no/ld, (1924)
lc
Purified Mouse Dermal 7.5% in liquid paraffin or 78 or 50 6 or 18 benign, 1 or 9 q Bottomley &
oleic acid, 5x/week, 78 or weeks malignant tumours no/lc Twort (1934
50 weeks
Doubtful Mouse 100 Dermal (a) 0.3% in benzene or < 704 (a) 1/100 papilloma and 1/100 n Barry et al.
Purity 20 (b) 0.3% ln mouse fat, days epithelioma, (b) no tumours no/ld (1935)
2 x/week, life
'Synthesized' Mouse 20 Dermal 0.3% (pure), 2x/week, 440 days No tumours n Barry et al.
440 days no/lc, (1935)
ld
Mouse 50 Dermal (a) 0.3% in benzene, < 797 (a) 2/50 papillomas, n Barry et al.
100 (b) 7.5% in oleic acid, days (b) no tumours no/lc, (1935)
2x/week, life ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
'Pure' Mouse 50 Dermal In benzene, 2x/week, < 276 After 276 days at 11/50 n Schurch &
276 days days survivors, no tumours no/lc, Winterstein
ld (1935)
Mouse, m/f 10/10 Dermal 40 µg/animal, 2x/week, 31 weeks 1/15 carcinomas n Riegel et al.
CF1 31 weeks no/ld (1951)
Mouse, f 20 Dermal 1%, 3x/week, life < 12 9/20 papillomas, 8/20 p Wynder &
Swiss months carcinomas; no solvent no/ld Hoffmann
control (1959a)
Mouse, f 20 Dermal 1 mg, 1x; initiation 63 weeks 16/20 papillomas, 2/20 p Van Duuren et
Swiss experiment carcinomas; promotor only: no/val al. (1966)
ICR/Ha 5/20, 1/20
TLC Mouse, f 30 Dermal 1 mg/animal, 1x; initiation 35 weeks 73% with papillomas; p Scribner(1973)
purified CD-1 experiment promotor only:3% no/val
Mouse, m 20 Dermal 75 µg/animal in decalin, 82 weeks 1/12 papillomas; solvent n Horton &
C3H 2x/week, 82 weeks control:2/13 papillomas no/ld Christian (1974)
Mouse, m 20 Dermal 75 µg/animal in decalin/ 82 weeks 5/19 papillomas; 12/19 p Horton &
C3H dodecane 50/50, 2x/week, carcinomas; solvent control: no/val Christian (1974)
82 weeks; co-carcinogenicity 2/13 papillomas
experiment
> 99.9% Mouse, f 20 Dermal 0.1 mg/animal/day, 110x; 22 weeks 11/18 papillomas/carcinomas; p Hecht et al.
Swiss initiation experiment chrysene only: 4/11 after 72 no/val (1974)
Ha/ICR/Mil weeks; solvent control: no
tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 30 Dermal 0.09, 0.29 and 0.91 mg/ 26 weeks 25, 43 and 52% papillomas; p Levin et al.
CD-1 animal, 1x; initiation promotor only: 7% no/val (1978)
experiment
95% Mouse, f 30 Dermal 0.46 mg/animal, 2x; 26 weeks 21/30 papillomas; promotor p Wood et al.
CD-1 initiation experiment only: 1/30 no/val (1979)
98% Mouse, f 30 Dermal 0.57 mg/animal, 1x; 27 weeks 80% papillomas; promotor p Wood et al.
CD-1 initiation experiment only: 4% yes/val (1980)
> 95% Mouse, f 30 Dermal 0.46 mg/animal, 1x; 15 weeks 21/29 papillomas; promotor p Slaga et al.
Sencar initiation experiment only: 3/30 no/val (1980,1981)
Mouse, f 30 Dermal 0.09 and 0.274 mg/animal, 26 weeks 43, 43% (or 39%) with skin p Chang et al.
CD-1 1 x; initiation experiment papillomas; vehicle control: yes/val (1983)
100%
> 99% Mouse, f 20 Dermal 3.4, 11.4 and 34 µg/ 24 weeks 25, 90 and 95% with tumours; p Rice et al.
CD-1 animal, 10x over 20 days; 0.5, 3, and 4.5 skin tumours/ yes/val (1988b)
initiation experiment animal; control: 20%
Mouse, f 20 Dermal 7.5 µg/animal, 1x; 21 weeks 10% with skin tumours; n Amin et al.
CD-1 initiation experiment solvent control:10% yes/val (1990)
Mouse, m/f 16/16 Dermal 365 µg/animal, 1x; < 100 No skin tumours; solvent n Bhatt &
Sencar initiation experiment weeks control: no tumours no/val Coombs (1990)
Purified Mouse 50 s.c. 2 mg/animal, 1x < 35 No tumours n Bottomley &
weeks no/lc Twort (1934)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Purified Mouse, 30 s.c. 10 mg/animal, 2x 15 No tumours n Shear & Leiter
Jackson A (4-month interval) months no/ld (1941)
Spectrometer Mouse, m/f 50 s.c. 5 mg/animal in tricaprylin; < 22 4/39 sarcomas after 4 months; p Steiner & Falk
control C57BI 1x months solvent control: 3/280 no/val (1951)
Mouse, m/f 40-50 s.c. 5 mg/animal in tricaprylin; < 22-28 5/22 sarcomas after 5 months p Steiner (1955)
C57BI 1x months yes/ld
Mouse, m 20 s.c. 1 mg/animal in arachis oil, 60-80 2/20 injection site tumours; p Boyland & Sims
C57BI 1 x/week, 10 weeks weeks control: no tumours no/val (1967)
Mouse, m/f 104 s.c. 0.1 mg/animal in poly- 70-75 70 weeks: 13/27m liver, 1/27 q Grover et al.,
Swiss ethylene glycol on days weeks m and 1/21 f lung tumours; no/val (1975)
newborn 1,2 and 3 after birth vehicle control: 9/30 m liver,
3/30 in and 1/15 f lung tumours
Mouse, 10 s.c. 1 mg, weekly; later 2 mg 350 days No tumours; control: no n Barry & Cook
at longer intervals tumours no/ln, (1934)
ld
Purified Mouse 50 i.p. 2 mg/animal, 1 × < 45 No tumours n Bottomley &
weeks no/lc Twort (1934)
TLC Mouse, m/f 100 i.p. Total dose 0.32 mg/animal 38-42 5/24 m and 2/11 f pulmonary q Buening et al.
control Swiss- in DMSO on days 1, 8 weeks tumours; 6/24 m liver yes/val (1979)
Webster and 15 after birth tumours; 1/24 m
BLU:Ha(ICR) lymphosarcoma; control:
newborn 2/21 m and 7/38 1 lung tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Repurified, Mouse, m/f 80 i.p. 0.045, 0.09 and 0.18 39-41 Males: 4/27 lung and 6/27 p Chang et al.
256°C Swiss- mg/animal in DMSO on weeks liver tumours; females; 1/11 yes/val (1983)
Webster days 1, 8 and 15 after birth lung and 0/11 liver tumours;
BLU:Ha(ICR) vehicle control: no tumours
newborn
> 98% Mouse, m/f 20-29 i.p. 6.3 and 210 µg/animal 26 7/10% and 15/0% m/f with n Busby et al.
Swiss- (total dose) in 3 aliquots weeks lung tumours; vehicle control: yes/val (1989)
Webster on day 1, 8, and 15 after 14/7% m/f
BLU:Ha(ICR) birth
newborn
Rat 10 s.c. 2 mg/animal, weekly; later < 626 4/10 tumours; control: 2/10 p Barry & Cook
6 mg at longer intervals days sarcomas no/ln, (1934)
ld
Purified Rat 10 s.c. 1 mg/animal, weekly, 103 < 103 No tumours n Boyland &
weeks weeks no/ln Burrows (1935)
ld
Rat, 5 s.c. 5 mg/animal, 7-9x 10 No tumours n Pollia (1941)
Wistar months no/ln
99.6% Rat, f 35 Intrapulm. 1 and 3 mg/animal, 1 × < 135 14.3% and 28.6% tumour p Wenzel-
Osborne/ weeks incidence; control: no tumours no/val Hartung et al.
Mendel (1990)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Coronene
> 96% Mouse, f 40 Dermal 5 or 15 µg/animal, < 104 Low dose:1/39, high dose: n Habs et al.
NMRI 4x/week, 104 weeks weeks 2/40 local tumours at yes/val (1980)
application site; vehicle
control: no tumours
TLC Mouse, f 20 Dermal 0.1 mg, 5x; initiation 65 weeks 6/20 papillomas; promotor q Van Duuren et
control Swiss experiment only: 5/20; coronene only: no/val al. (1968)
ICR/Ha no tumours
Cyclopenta[d]pyrene
> 96% Mouse, f 40 Dermal 1.7, 6.8 and 27.2 µg/ 112 Low dose: no tumours; high q Habs et al.
NMRI animal, 2x/week, 112 weeks dose: 2/38 skin carcinomas, yes/val (1980)
weeks 1/38 sarcomas;control: no
tumours
> 98% Mouse, f 30 Dermal 23, 91, 226, 566 µg/ 27 weeks 10, 21, 30, and 37% p Wood et al.
CD-1 animal, 1x; initiation papillomas; promotor only: 4% yes/val (1980)
experiment
> 99.9% Mouse, f 30 Dermal 45, 136 and 407 µg/ 57 weeks Low dose: 17; med. dose: 11; p Cavalieri et al.
Swiss animal, 2x/week, 30 weeks high dose: 7 skin tumours; no/val (1981b)
control: no tumours
> 99.9% Mouse, f 30 Dermal 4.5, 14 and 41 µg/animal, 44 weeks Low dose: 1/30; med. dose: p Cavalieri et al.
CD-1 every other day, 20 days; 9/29; high dose: 6/29 no/val (1981b)
initiation experiment papillomas; promotor only: 3/29
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 30 Dermal 10, 100 and 200 µg/ 26 weeks Low dose: 11 %; med. dose: p Raveh et al.
Sencar animal, 1x; initiation 39%; high dose: 57% no/val (1982)
experiment papillomas; promotor only:
10%
> 99% Mouse, m/f 8-14 i.p. 0.35, 0.7, 1.05, 1.4, and 26 weeks 62, 60, 56, 70, 86, 93%, p Busby et al.
Swiss- 1.75 mg/animal (total dose) 77, 100, and 89, 100% m/f yes/val (1988)
Webster in 3 aliquots on day 1, 8, with lung tumours; vehicle
BLU:Ha(ICR) and 15 after birth control: 8, 8%
newborn
> 99% Rat, f 20 Intra- 1.8 and 5.4 mg into 4th < 34 No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1x weeks control: no tumours no/val (1988b)
Dawley
Dibenz[a,h]anthracene
Mouse m Oral 1.5 mg/animal in PEG-400, 30 weeks 21 % forestomach papillomas; q Berenblum &
Swiss 1 x; initiation experiment promotor only: 14% no/lc Haran (1955)
Mouse m/f 21/21 Drink- 0.8 mg/day/animal in olive 8-9 14/14 m and 13/13 f with p Snell & Stewart
DBA/2 con- ing- oil, 8-9 months months pulmonary adenomas; 14/14 no/val (1962)
trol: water m and 10/13 f with alveologenic
25/10 carcinomas; control: 1 mouse
with tumour
Mouse, f 20 Dermal 0.001, 0.01, and 0.1%, < 21, 13 0.001%: 30% papillomas, p Wynder &
Swiss 3x/week, life or 9 30% carcinomas; 0.01%: no/ld Hoffmann
Millerton months 95/90% papilloma/carcinoma; (1959a)
0.1%: 90%/75% papilloma/
carcinoma
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m < 50 Dermal 0.02 and 0.16 µg/animal, 32 weeks 33 and 38% with skin p Klein (1960)
Swiss 1x; initiation experiment tumours; acetone control: 13% yes/val
albino
DBA/2Jax
Chromatograph Mouse, f 20 Dermal 38 µg/animal, 2x/wk, < 60 80% with skin tumours; p Lijinsky et al.
recrystallized Swiss 44 weeks, weeks vehicle control: 4% no/val (1965)
Mouse, m/f 30/30 Dermal m: 0.3% solution (= 1.5 < 29/22 m: 26% with papillomas after p Johnson (1968)
IF/Bcr mg/animal), 1x/week, 18 weeks 20 weeks, 100% after 29 no/val
weeks; f: 0.5% (= 1 mg/ weeks; f: 100% with breast
animal), 8x, every 2 weeks tumours after 22 weeks
> 99% Mouse, f 50 Dermal 1 drop, 3x/week, 112 112 6%, 8% and 32% with skin p Platt et al.
NMRI weeks; total doses: 37.8, weeks tumours; controls: 2-4% no/val (1990)
125, and 378 µg/animal
> 99% Mouse, f 16 Dermal 83.5 and 167 µg/animal, 24 weeks 38 and 93% with skin tumours; p Platt et al.
NMRI 1x; initiation experiment vehicle control: no tumours no/val (1990)
Mouse 10 s.c./ 0.2 mg/animal, 2x/week, Life 3/10 with subcutaneous p Boyland &
i.p. 50 weeks alternating sarcomas no/ln Burrow, (1935)
ld
Spectrometer Mouse, m/f 50 s.c. 0.02 mg/animal in < 22 28/48 sarcomas after 4 p Steiner & Falk
control C57BI tricaprylin; 1x months months solvent control: 3/280 no/val (1951)
Mouse, m/f 40-50 s.c. 0.02, 0.04 mg/animal in < 22-28 7/21 and 6/18 sarcomas after p Steiner (1955)
C57BI tricaprylin; 1x months 6 and 5 months yes/ld
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 20/19 s.c. 1 mg/animal, 1x/week, 60-80 20/20 m and 17/19 f with p Boyland & Sims
C57BL 10 weeks weeks sarcomas; control: no no/val (1967)
sarcomas
Mouse, f 60 s.c. 10, 30, 90, 270 and 810 < 16 40, 35, 65, 75, and 90% with p Pott et al.
NMRI µg/animal, 1x months tumours no/val (1973)
Mouse, m/f 30 s.c. 0.15 and 0.3 mg/animal, 12 months B6 mice: 16/30 and 14/30; D2 p Kouri et al.
B6, D2 (60) 1x mice:1/30 and 0/30 with no/val (1983)
fibrosarcomas
> 99% Mouse, f 47-50 s.c. 10, 30, 86 µg/animal, 1 × 112 52, 46, and 63% with p Platt et al.
NMRI weeks fibrosarcomas; controls: 2-6% no/val (1990)
> 99% Mouse, m/f 40-50 s.c. 11.1 and 111 µg/animal 40 weeks 12/35 with pulmonary tumours; p Platt et al.
NMRI on day 2, 1x controls: 2/33 and 4/41 no/val (1990)
newborn
Mouse, 10 i.v. 10 mg/kg, 1x 20 weeks 100% lung tumours; control: p Shimkin &
A 21% no/val Stoner(1975)
Rat 2x10 s.c. 2 mg/animal, weekly; later < approx. 1/10 and 7/10 with tumours; q Barry & Cook
6 mg at longer intervals 200 control: 2/10 no/ln, (1934)
days ld
Rat 10-18 s.c./ 1 mg/animal, 2x/week, Life 3-6/10 and 9/18 with p Boyland &
(6 i.p. 50 weeks subcutaneous sarcomas no/ln Burrows (1935)
exp.) alterna- ld
ting
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Rat, 5 s.c. 5 mg/animal, 4-8x 10 2 with tumours after 8-9 p Pollia (1941)
Wistar months months no/ln
99.3% Rat, f 35 Intrapulm. 0.1 mg/animal, 1x < 123 57.1% tumour incidence; p Wenzel-
Osborne/ weeks control: no tumours no/val Hartung et
Mendel al.(1990)
> 99% Rat, f 20 Intra- 1.1 and 4.5 mg into 5th 20 weeks No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1x control: no tumours no/val (1988a)
Dawley
Chromatography Hamster, m/f 5/5 Dermal 8 drops of a 0.2% solution, < 75 No tumours n Shubik et al.
control Syrian weeks 2x/week, 10 weeks no/ln, (1960)
golden ld
Hamster, m 46 Intra- 0.05 and 0.25 mg/animal, < 110 0/46 and 0/46 respiratory tract q Sellakumar &
Syrian tracheal 1x/week, 30 weeks weeks tumours; control: no tumours yes/val Shubik (11974)
golden
Hamster, Intra- 10.3 and 0.9 mg/animal, < 2 years 55 and 65% with tumours p Pott et al.
Syrian trachea 1x/week, 20 weeks no/val (1978)
golden
Monkey, Not specified No tumours n Adamson &
Old world no/ld, Sieber (1983)
lc
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Dibenzo[a,e]pyrene
Recrystallized Mouse, f 40/20 Dermal 0.05 and 0.1% solution, 15 16/40, 9/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months months and 9/40, 6/20 with no/val Wynder (1966
albino epitheliomas; solvent
Ha/ICR/Mil control: no
Recrystallized Mouse, f 28 Dermal 25 µg/animal, 10x over 20 6 months 10/28 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment only: 2/30 no/val Wynder (1966)
albino
Ha/ICR/Mil
> 99% Mouse, f 21 Dermal 242 µg/animal, 1x; 26 weeks 240% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 9% yes/val (1989)
Mouse, m/f 21/14 s.c. 0.6 mg/animal, 1x/month, < 142 18/21 m and 14/14 f local p Lacassagne et
XVII nc/Z 3 × days m sarcomas; no vehicle control no/val al. (1963b)
or 126
days f
Mouse m/f 12/15 s.c. 0.6 mg/animal, 1x < 196 10/12 m and 10/15 f local p Lacassagne et
days m sarcomas; no vehicle control no/val al. (1963b)
or 220
days f
> 99% Rat f 19 Intra- 10 mg/gland, 1x, 8 glands, < 40 1/19 with mammary tumours; n Cavalieri et al.
Sprague- mammary weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Dibenzo[a,h]pyrene
Mouse 74 Dermal 1 drop of a 0.15% solution 4.5 50% with skin tumours p Kleinenberg
alternate days, 55 or 86 months no/lc (1939)
times
Recystallized Mouse, f 20 Dermal 0.05 and 0.1% solution, 11,15 16/20,15/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months months and 13/20, 15/20 with no/val Wynder (1966)
albino epitheliomas; solvent
Ha/ICR/Mil control: no tumours
Recrystallized Mouse, f 29 Dermal 25 µg/animal, 10x over 20 6 months 21/29 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment only: 2/30 no/val Wynder (1966)
albino
Ha/ICR/Mil
96.6% Mouse, f 40 Dermal 120 µg/animal, 2x/week, 70 weeks 90% tumour incidence; solvent p Cavalieri et al.
Swiss 30 weeks control: no tumours no/val (1977)
Pure Mouse, f 30 Dermal 15.1, 60.5 and 181.4 17 weeks 55, 79, and 72% with skin p Chang et al.
CD-1 µg/animal, 1x; initiation tumours; controls: 0-10% yes/val (1982)
experiment
> 99% Mouse, f 24 Dermal 242 µg/animal, 1x; 26 weeks 75% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 9% yes/val (1989)
Mouse, m/f 35/10 s.c. 0.6 mg/animal, 1x/month, > 111/128 34/35 mand 1/10 f with local p Lacassagne et
XVII 3 months days sarcomas no/ld al. (1958)
(average
latency)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 31 s.c. 0.2 mg/animal, 1x; 27 weeks 26/28 with tumours; solvent p Sardella et al.
CD-1 initiation experiment control: 2/32 no/val (1981)
Mouse, m/f 40 i.p. 3.8, 7.6 and 15.1 µg on 49-54 97% with pulmonary and 44% p Chang et al.
Swiss- days 1, 8 and 15 of life weeks with hepatic tumours; control: yes/val (1982)
Webster pulmonary tumours 27%, no
BLU:Ha(ICR) hepatic tumours
newborn
> 99% Rat f 20 Intra- 12 mg/gland, 1x, 8 glands < 40 19/20 with mammary tumours; p Cavalieri et al.
Sprague- mammary weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Dibenzo[a,i]pyrene
Mouse, m 23 Dermal 1 drop of a saturated > 7 21/23 papillomas and 8/23 p Lacassagne et
XVII solution, 2x/week months epitheliomas;solvent control: no/val al. (1958)
no tumours (14 months)
Mouse, f 20/10 Dermal 0.01 and 0.1 %, 3x/week, < 16 and 0.01%: 10% papillomas, no p Wynder &
Swiss 16 and 13 months 13 months carcinomas; 0.1%: 50% no/ld Hoffmann
Millerton papillomas, 10% carcinomas (1959a)
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1 % solution, 15 months 16/40, 16/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months and 13/20, 15/20 with no/val Wynder (1966)
albino epitheliomas; solvent
Ha/ICR/Mil control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized Mouse, f 30 Dermal 25 µg/animal, 10x over 20 6 months 12/30 papillomas; promotor p Hoffmann &
Swiss days; initiation experiment only: 2/30 no/val Wynder (1966)
albino
Ha/ICR/Mil
Mouse, f 20 Dermal 100 and 500 µg/animal, 22 weeks 40 and 80% with tumours; p Hecht et al.
Swiss 1x; initiation experiment vehicle control: no tumours no/val (1981)
albino
Ha/ICR
Pure Mouse, f 30 Dermal 15.1, 60.5 and 181.4 µg/ 17 weeks 28, 67, and 70% with skin p Chang et al.
CD-1 animal, 1x; initiation tumours; controls: 0-10% yes/val (1982)
experiment
> 99% Mouse, f 24 Dermal 242 µg/animal, 1x; 26 weeks 63% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 95% yes/val (1989)
Mouse, m/f 17/18 s.c. 0.6 mg/animal, 1x/month, 3> 75/82 17/17 m and 16/18(f) with p Lacassagne et
XVII months days local sarcomas no/ld al. (1958)
(average
latency)
Mouse, m/f 8/8 s.c. 2 mg/animal, 1x 2-3 100, 100% with skin tumours; p Waravdekar &
XVII/C57BI months average latency: 74 days no/ln, Ranadive
hybrids ld (1958)
Mouse, m s.c. 0.5 mg/animal, 1x 4-5 100% fibrosarcomas; p Homburger et
C57BL/6 weeks malignant cells identifiable no/ld al. (1962)
after 4-5 weeks
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 50 s.c. 0.1 mg/animal, 1x 75 weeks 40/41 with tumours; solvent p Sardella et al.
CD-1 control: no tumours no/val (1981)
Mouse, m/f 40 i.p. 3.8, 7.6 and 15.1 µg on 49-54 97% with pulmonary and 54% p Chang et al.
Swiss- day 1, 8, and 15 of life weeks with hepatic tumours; control: yes/val (1982)
Webster pulmonary tumours 27%, no
BLU:Ha(ICR) hepatic tumours
newborn
> 99% Rat, f 19 Intra- 12 mg/gland, 1x, 8 < 40 18/19 with mammary tumours; p Cavalieri et al.
Sprague mammary glands weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Hamster m 6-10 s.c. 0.25, 0.5, 1 and 2 mg/ 9-14 55, 90, 100, and 100% with p Wodinsky et al.
Syrian animal, 1x weeks fibrosarcomas; vehicle control: no/val (1964)
(average 0%
latency)
Hamster, m/f 139/ s.c. 1 mg/animal, 1x 11 weeks 99/100% with fibrosarcomas p Wodinsky et al.
Syrian 157 (average no/val (1964)
latency)
Hamster, m 4/34 Intra- 0.5 and 2 mg/animal, < 110 Tumours (i) 6/44 (trachea), p Sellakumar &
Syrian tracheal weekly, 24 and 4 weeks, weeks 37/44 (bronchi), 2/34 yes/val Shubik (1974)
golden respectively (trachea); (ii) 1/34 (larynx),
13/34 (bronchi); control: no
tumours
Hamster, m/f 24/24 Intra- 0.5 and 1 mg/animal, 65 and 75% respiratory p Stenback &
Syrian tracheal 1 x/week, 17 and 12 weeks, tumours (bronchi, trachea); no/ld Sellakumar
golden respectively shortest latency: 8 weeks (1974)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Monkey, Not specified No tumours n Adamson &
Old world no/lc Sieber (1983)
Dibenzo[a,l]pyrene
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1% solution, 11, 14 17/20, 18/20 with papillomas p Hoffmann &
Swiss 3x/week, 12 months months and 17/20, 18/20 with no/val Wynder
albino epitheliomas; solvent (1966)
Ha/ICR/Mil control: no tumours
Recrystallized Mouse, 30 Dermal 25 µg/animal, 10x over 20 6 months 18/30 papillomas; 1/30 p Hoffmann &
Swiss days; initiation experiment epitheliomas; promotor no/val Wynder
albino only: 2/30 (1966)
Ha/ICR/Mil
Mouse, f 19-21 Dermal 55, 200, 240, 350 and 700 6 months 20, 19, 21, 19 and 16 with p Masuda &
Swiss ICR µg/animal given in 55, 40, skin tumours; no solvent no/val Kagawa
24, 7 and 7 applications control group (1972)
> 99% Mouse, f 24 Dermal 242 µg/animal, 1x; 26 weeks 92% papillomas; solvent p Cavalieri et al.
Sencar initiation experiment control: 9% yes/val (1989)
Pure, 161- Mouse, f 24 Dermal 10, 30 and 90 µg/animal, 15 weeks 23/24, 22/24 and 24/24 with p Cavalieri et al.
162°C) Sencar 1x; initiation experiment tumours;control; no tumours yes/val (1991)
Pure Mouse, f 24 Dermal 1.2, 6 and 30 µg/animal, 7 weeks 22/24, 20/24 and 20/24 with p Cavalieri et al.
Sencar 1x; initiation experiment tumours; 2 control: no tumours yes/val (1991)
Chomatography Mouse, f 24 Dermal 30 µg/animal, 1x; initiation 27 weeks 7/24 with tumours p Cavalieri et al.
purified Sencar experiment without yes/val (1991)
promotion
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 12/12 s.c. 0.6 mg/animal, 1x/month, < 7 All animals with local p Lacassagne et
XVII 2 months (some animals, months sarcomas (mean latent period: no/val al.(1968a)
nc/ZE 3rd injection after 2 months) 120 days); control: no tumours
> 99% Rat, f 9 Intra- 1.2 mg/gland, 1x, 8 glands < 40 9/9 with mammary tumours; p Cavalieri et al.
Sprague- mammary weeks control: 0/21 or 2/20 yes/val (1989)
Dawley
Rat f 20 Intra- 76 and 302 µg/gland, 1x, < 24 20/20 and 19/20 with p Cavalieri et al.
Sprague- mammary 8 glands weeks mammary tumours; control: yes/val (1991)
Dawley 1/18
Fluoranthene
Mouse, 2x10 Dermal 0.3% in benzene, 2x/week, < 501 No tumours n Barry et al.
life days no/ld (1935)
Mouse, f 20 Dermal 0.1 % solution, 3x/week, < 17 No papillomas or carcinomas n Wynder &
Swiss life months no/ld Hoffmann
Millerton (1959a)
Mouse, f 20 Dermal 1%, 3x/week, 12 months 15 At 12 months 0/20 tumours; n Hoffmann et al.
Swiss months no vehicle control no/val (1972)
Ha/ICR/Mil
99.9% Mouse, f 30 Dermal 0.1 mg/animal, 10x over 24 weeks 1/29 skin tumours; solvent n Hoffmann et al.
Swiss 20 days; initiation control: 1/30 no/val (1972)
Ha/ICR/Mil experiment
Recrystallized Mouse, m 15 Dermal 250 µg/animal in decalin, 82 week No papillomas or carcinomas; n Horton &
C3H 2x/week, 82 weeks solvent control: 2/13 no/val Christian (1974)
papillomas
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Purified, Mouse, f 50 Dermal 40 µg/animal, 3x/week, 440 days No tumours observed; n Van Duuren &
107- Swiss life controls: no tumours no/val Goldschmidt
109°C) ICR/Ha (1976)
Mouse, m/f 7/7 s.c. 10 mg/animal, 5x 19 No tumours n Shear (1938)
Jackson A months no/ld,
ln
Mouse, m/f 10/10 s.c. 0.6 mg/animal, 1x/month, No sarcomas n Buu-Hoi (1964)
XVII nc/Z 3x no/ld,
ln
99% Mouse, m/f 20-31 i.p. 0.7 and 3,5 mg/animal 24 weeks 23, 15, and 74, 38% m/f with p Busby et al.
Swiss- (total dose) in 3 aliquots lung tumours; vehicle control: yes/val (1984)
Webster on days 1, 8 and 15 after 4,14%
BLU:Ha(ICR) birth
newborn
> 99.5% Mouse, m/f 22/30 i.p. 0.7 and 3.5 mg/animal 52 weeks 43, 35, and 65, 86% with lung p La Voie et al.
CD-1 (total dose) in 3 aliquots on tumours; 64, 0% and 100, 7% yes/val (1994)
newborn days 1, 8, and 15 after birth with hepatic tumours; vehicle
only: 17, 12% (lung) and 17, 6%
(liver)
Fluorene
Mouse 100 Dermal Dissolved in 90% benzene 9 months No tumours n Kennaway
no/ld, (1924)
lc
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, m/f 10/10 Dermal 60 µg/animal, 2x/week, 31 weeks No skin tumours n Riegel et al.
CF1 31 weeks no/ld (1951)
'Pure' Mouse, m 100 Dermal 3 drops, 1x/week of approx. < 1 year After 9 months 10/100 n Graffi et al.
white 3% solution, 1 year; survived, no tumours; promotor no/val (1953)
initiation experiment only: 0.08 tumour/animal
Mouse, m 5 i.p. 1000 mg/kg, 1x < 5 No effects n Shubik & Della
Swiss months no/ld Porta (1957)
ln
Mouse, m 10 s.c. 10 mg/animal, 7x over 19 No tumours n Shear(1938)
Jackson A 16 months months no/ln,
ld
Highly Rat, f 20 Oral 0.05% diet; 4.3 mg/rat per 10.7 2/11 carcinomas (renal pelvis, q Morris et al.
purified Buffalo (diet) day = 796 mg/rat (total months ureter); control: 4/16 with no/ld (1960)
intake) over 6 months carcinomas
Highly Rat, f 18 Oral 0.05% diet; 4.6 mg/rat per < 20.1 7/18 tumours; control: 4/18 or q Morris et al.
purified Buffalo (diet) day = 2553 mg/rat (total months 15/18 tumours no/val (1960)
intake) over 18 months
Recrystallized Mouse, f 30 Dermal 25 µg/animal, 10x over 20 6 months 5/30 papillomas; promotor: q Hoffmann &
Swiss days; initiation experiment 2/30 no/val Wynder (1966)
Ha/ICR/Mil
Recrystallized Mouse, f 20 Dermal 0.05 and 0.1 % solution, 15 Dioxane solvent: no tumours; q Hoffmann &
Swiss 3x/week, 12 months months acetone solvent: dose-related no/val Wynder (1966)
albino tumour increase
Ha/ICR/Mil
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
> 96% Mouse, f 40 Dermal 3.4, 5.6, 9.2 µg/animal, <2 years 3, 0, 0% with local tumours; n Habs at a
NMRI 2x/week, life control: no tumours yes/val (1980)
Mouse, f 25 Dermal 100 µg/animal, 10x over 25 weeks 90% with skin tumours; vehicle p Rice et al.
Crl:CD1 20 days; initiaton control: < 5% yes/val (1986)
(ICR)BR experiment
> 99% Mouse, f 25 Dermal 110 µg/animal, 10x over 23 weeks 72% tumours, 2.1 skin p Rice et al.
CD-1 20 days; initiation tumours/animal; control: yes/val (1990)
experiment no tumours
Mouse, m/f 14/14 s.c. 0.6 mg/animal, 1x/mth, Average, Sarcomas: 10/14 m and 1/14 f p Lacassagne et
XVII 3 months 265 days no/val al. (1963a)
nc/Z m, 145
days f
> 99% Mouse, m/f 11/9 i.p. 580 µg/animal in DMSO < 52 9% hepatic or 0% lung n LaVoie et al.
CD-1 on days 1, 8 and after weeks tumours; controls: 6%/0% yes/val (1987)
newborn birth (total dose)
99.4% Rat, f 35 Intra- 0.16, 0.83 and 4.15 116/109/ 3/35, 8/35 and 21/35 with lung p Deutsch-
Osborne/ pulm. mg/animal, 1x 92 weeks tumours; control: no tumours yes/val Wenzel et al.
Mendel (1983)
5-Methylcholanthrene
> 99.9% Mouse, f 20 Dermal 0.1 mg/animal, 3x/week, 35 weeks 20/20 with 85 skin tumours p Hecht et al.
Swiss 35 weeks (solvent by 25 week; 20/20 with 99 no/val (1974)
Ha/ICR/Mil control: tumours and 12/20 with 37
72 weeks) carcinomas by 35 wks; solvent
control: no tumours
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
> 99.9% Mouse, f 20 Dermal 10, 30 and 100 µg, 10x 24 weeks Low dose: 20/20 mice with p Hecht et al.
Swiss over 20 days; initiation 110 skin tumours; med dose: no/val (1974)
Ha/ICR/Mil experiment 20/20 with 160 skin tumours;
high dose: 17/18 with 96 skin
tumours; solvent control: no
tumours
Mouse, f 20 Dermal 5 and 10 µg/animal, 62 weeks Low dose: 9/20 with 22 skin p Hecht et al.
Swiss 3x/week, 62 weeks tumours, 6/20 with 7 yes/val (1976a)
Ha/ICR carcinomas; high dose: 15/20
with 38 tumours, 10/20 with
12 carcinomas; solvent
control: no tumours
Highly Mouse, f 20 Dermal 1 and 3 µg, 10x over 24 weeks Low dose: 2/20 mice with 2 p Hecht et al.
purified Swiss 20 days; initiation skin tumours; high dose: yes/val (1976a)
Ha/ICR experiment 20/20 with 45 skin tumours
(1 carcinoma); solvent control:
no tumours
> 99.9% Mouse, f 8x20 Dermal 3 and 10 µg, 10 × over 20 24 weeks Low dose: 55-95% of mice p Hecht et al.
Swiss days; initiation experiment with skin tumours; high dose: yes/val (1978)
Ha/ICR/Mil 80-90%; solvent control: no
tumours
> 99.9% Mouse, f 20 Dermal 1 and 3 µg, 10x over 20 24 weeks Low dose: 75% of mice with p Hecht et al.
Swiss Ha/ICR days; initiation experiment skin tumours; high dose: 85% no/val (1979)
outbred
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 20 Dermal 1 or × or 3 µg/animal, 10x 21 weeks 55, 75, and 90% with skin p Amin et al.
Swiss CD-1 over 20 days; initiation tumours; solvent control: 5% yes/val (1981)
experiment
HPLC Mouse, f 20 Dermal 8 and 24 µg/animal, 1x; 26 weeks 80 and 90% tumour-bearing p Hecht et al.
purified CD-1 initiation experiment animals; solvent control: 10% yes/val (1985)
Mouse, f 20 Dermal 8 µg/animal, 1x; initiation 21 weeks 65% with skin tumours; p Amin et al.
CD-1 experiment solvent control: 5% yes/val (1985b)
Mouse, f 20 Dermal 24.2 µg/animal, 1x; 26 weeks 90% with tumours; 5.2 p El-Bayoumy et
CD-1 initiation experiment tumours/animal; solvent no/val al. (1986)
control: 10%/0.1
> 99% Mouse, f 20 Dermal 3.6, 12.1 and 36 µg/ 24 weeks 100, 100 and 100% with p Rice et al.
CD-1 animal, 10x over 20 days; tumours; 9.2, 10.7 and 9.4 yes/val (1988b)
initiation experiment tumours/animal; solvent
control: 20%
Mouse, f 20 Dermal 8 µg/animal, 1x;initiaUon 21 weeks 85% with skin tumours; p Amin et al.
CD-1 experiment solvent control: 10% yes/val (1990)
Mouse, f 20 Dermal 8 µg/animal, 1x; initiation 26 weeks 65% with skin tumours; solvent p Amin et al.
CD-1 experiment control: 10% yes/val (1992)
Mouse, m/m 20/ s.c. 2 mg/animal in tricaprylin, 6 months Swiss mice: no local tumours; q Dunlap &
Swiss/ 2x10 1x 16/20 mice died; C3H mice: no/ld Warren (1943)
C3H 7/10 or 3/10 local sarcomas
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Highly Mouse, m 25 s.c. 50 µg/animal in 32 weeks 22/25 mice with 24 p Hecht et al.
purified 057BL trioctanoin, 1 x/2 weeks, fibrosarcomas; vehicle no/val (1976b)
20 weeks control: no tumours
HPLC Mouse, m/f 35/48 i.p. 1.9 µg/animal on day 1; Weaned 20/21% with pulmonary p Hecht et al.
purified ICR/Ha 3.9 µg on day 8; 7.8 µg after 3 tumours; 23/12% with yes/val (1985)
newborn on day 15 weeks; hepatic tumours; solvent
sacri- control: 4/7% and 2/2%
ficed
after 35
weeks
> 99% Rat f 20 Intra- 0.97 and 3.9 mg into 5th 20 weeks No mammary tumours; n Cavalieri et al.
Sprague- mammary mammary gland, 1x control: no tumours no/val (1988a)
Dawley
1-Methylphenanthrene
>99.5% Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks No tumours; vehicle control: n LaVoie et al.
Swiss initiaton experiment no tumours no/val (1981b)
Ha/ICR
Naphthalene
Mouse Dermal Several times/wk, < 11 No skin tumours n Kennaway
< 11 months months no/lc (1930)
Highly Mouse, 25; Dermal 0.5% in benzene, 6x/week Life 4/25 with lymphatic q Knake (1956)
purified SW inbred con- for 3 weeks, then 2x/wk for leukaemia; 1/25 lymphosarcoma no/lc,
trol: life of thymus; 4/25 with benign ln
21 tumours; benzene only: 2/21
with sarcomas; 1/21 with lung
adenoma
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 30 Dermal 0.25 mg/animal + 3 µg 78 weeks 5/30 lymphomas; inhibitory q Schmeltz et al.
ICR/Ha benzo[a]pyrene, 3x/wk, effect on skin tumours; no/val (1978)
78 weeks; co-carcinogenicity naphthalene only: no skin
test tumours
98-99% Mouse, f 30 Inha- 0.05 and 0.15 mg/l, 6 months 29 and 30% with pulmonary q Adkins et al.
A/J lation 6 h/day, 5 days/week, tumours; control: 21% yes/val (1986)
6 months (increase in treatment groups
not significant)
> 99% Mouse, m/f 75 Inha- 0.053 and 0.16 mg/litre, 103 Significantly increased q Abdo et al.
B6C3F1 (150) lation 6 h/day, 5 days/week, weeks pulmonary alveolar and yes/val (1992);
103 weeks bronchiolar adenomas in National
females; no cararacts Toxicology
Program
(1992b)
Mouse 23 Bladder 1x (dose unspecified) 7 months 1/23 bladder carcinoma after - Boyland et al.
implant 1 month; "inert" substance: (1964)
higher rate of bladder
carcinoma
Rat, 28 Oral 10-20 mg/animal, Life No tumours n Schmahl
BDI/BDI II (diet) 6x/week, 70 weeks no/ld (1955)
inbred
Rat, 10 s.c. 20 mg/animal, 1x/week, Life No tumours n Schmahl
BDI/BDI II 40 weeks no/ln (1955)
inbred
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Crude, Rat, 38 s.c. 0.5 g/kg, 2x/month, 3.5 Life 5 malignant tumours (4/38 q Knake(1956)
90% 'white' months Imphosarcomas, 1/38 uterine no/val
sarcoma); 1 benign tumour;
vehicle control: 1/38
lymphosarcoma and 1 benign
tumour
Rat, 10 i.p. 20 mg/animal, 1x/week, Life No tumours n Schmahl
BDI/BDI II 40 weeks no/ln (1955)
inbred
Perylene
Recrystalized Mouse, f 20 Dermal 0.8 mg/animal, 1x; 58-60 3/20 papillomas; promotor n Van Duuren et
Swiss initiation experiment weeks only: 1/20 with papillomas; no/val al. (1970)
ICR/Ha pure substance only: no tumours
Recrystallized Mouse, m 20 Dermal 75 µg/animal in decalin, 82 weeks No skin tumours; solvent n Horton &
C3H 2x/week, 82 weeks control: 2/13 papillomas no/val Christian
(1974)
Phenanthrene
Mouse 100 Dermal Dissolved in 90% benzene 9 months No tumours n Kennaway
no/ld, (1924)
lc
'Pure' Mouse, m 100 Dermal 3 drops, 1x/week of approx. < 1 year After 12 months 6/100 n Graft et al.
white 3% solution, 1 year; survived with a total of 1 no/val (1953)
initiation experiment tumour; 0.16 tumour/animal;
promotor only: 0.08
tumour/animal
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, 20 Dermal 54 mg/animal, 3x/wk, total: 24 weeks 5/20 survivors with 12 q Salaman & Roe
'S' 10x; initiation experiment papillomas; promotor only: yes/val (1956)
4/19 survivors/4 papillomas
High purity Mouse, m/f 10/10 Dermal 0.3 mg, 4x on days 0, 2, 6 24 weeks 4/19 papillomas; solvent q Roe (1962)
'stock and 8; initiation experiment control: 2/20 yes/val
albino'
TLC Mouse, f 30 Dermal 1.8 mg/animal, 1x; 35 weeks 40% with papillomas; p Scribner (1973)
purified CD-1 initiation experiment promotor only: 3% no/val
> 98% Mouse, f 30 Dermal 1.8 mg/animal, 1x; 36 weeks 5/30 papillomas; solvent q Wood et al.
CD-1 initiation experiment control: 2/30 no/val (1979)
Mouse, f 20 Dermal 100 µg, 10x over 20 days; 24 weeks No skin tumours observed; n LaVoie et al.
Swiss initiation experiment vehicle control: no tumours no/val (1981b)
Ha/ICR
Mouse, m/f 40-50 s.c. 5 mg/animal in tricaprylin; < 22-28 No sarcomas after 8 months n Steiner (1955)
C57BI 1x months yes/ld
Mouse, m/f 10/10 s.c. 0.3 mg, 5x on days 0, 2, 24 weeks 3/17 papillomas; solvent n Roe (1962)
'stock 4, 6 and 8; initiation control: 2/20 yes/val
albino' experiment
Mouse, m/f 57 s.c. 40 µg/animal; 1x < 62 3/49 lung adenomas; control: n Grant & Roe
stock administered to neonatal weeks 8/34 and 5/38 yes/val (1963)
albino' mice
newborn
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
> 98% Mouse, 100 i.p. 35, 70 and 140 µg/animal 38-42 6/35 pulmonary adenomas; n Beuning et al.
Swiss- in DMSO on days 1, 8 weeks DMSO only: 9/59 yes/val (1979)
Webster and 15 after birth
BLU:Ha(ICR)
newborn
Rat, f 10 Oral 200 mg/rat, 1x; experiment 60 days No tumours at 60 days; n Huggins &
Sprague- on mammary tumours controls: 8/164 after 310 days no/ln, Yang (1962)
Dawley lc
99.9% Rat, f 35 Intrapulm. 1, 3 and 10 mg/animal, 1x < 135 No tumours; control: no n Wenzel-Hartung
Osborne/ weeks tumours no/val et al.(1990)
Mendel
Pyrene
Mouse 2x20 Dermal 1% in benzene, 2x/week, < 717 1/20 and 1/20 papillomas n Barry et al.
life days no/ld (1935)
Mouse 40 Dermal 0.3% in benzene, 2x/ < 680 No skin lesions n Badger et al.
week, < 680 days days no/ld (1940)
'Pure' Mouse, m 150 Dermal 3 drops, 1x/week of a < 1 year After 6 months 18/150 n Graffi et al.
white 0.3% solution, 1 year; survived with a total of 1 no/val (1953)
initiation experiment tumour; 0.06 tumour/animal;
promotor only: 0.08
tumour/animal
Mouse, 20 Dermal 25 mg/animal, 3x/week; 24 weeks 6/20 mice with 9 papillomas; q Salaman & Roe
'S' total:10x; initiation promotor only: 4/19 mice with yes/val (1956)
experiment 4 papillomas
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Mouse, f 5 Dermal 10%, 3 x/week, life < 18 No skin tumours n Wynder &
Swiss months no/ld, Hoffmann
Millerton ln (1959a)
TLC Mouse, f 30 Dermal 2 mg/animal, 1x; initiation 35 weeks 17% with papillomas; q Scribner (1973)
purified CD-1 experiment promotor only: 3% no/val
High purity Mouse, m 20 Dermal 250 µg/animal in decalin, 82 weeks 3/13 papillomas; solvent q Horton &
C3H 2x/week, 82 weeks control: 2/13 no/val Christian (1974)
Recrystallized Mouse, f 50 Dermal 12 or 40 µg/animal, < 440 No skin tumours observed; n Van Duuren &
Swiss 3 x/week, 368 or 440 days days control: no tumours no/val Goldschmidt
ICR/Ha (1976)
High purity Mouse, f 50 Dermal 4 and 12 µg/animal + 5 µg 33 weeks High dose: 13/50 papillomas, n Goldschmidt et
Swiss benzo[a]pyrene, 3x/week, 5/50 carcinomas; benzo[a]- no/val al. (1973)
ICR/Ha 33 weeks; co-carcinogenicity pyrene only: 6/50 papillomas;
test pyrene only: no tumours
Recrystallized Mouse, f 50 Dermal 4, 12 and 40 µg/animal 368 or 12/26/35 mice with papillomas, p Van Duuren &
Swiss + 5 µg benzo[a]pyrene, 440 days 6/20/26 with squamous cell no/val Goldschmidt
ICR/Ha 3x/week, 368/368/440 carcinomas; positive control: (1976)
days; co-carcinogenicity 15, 11 tumours; solvent control:
test no tumours
> 98% Mouse, f 30 Dermal 20.2 and 80.9 µg/animal, 27 weeks 14 and 10% with tumours; n Wood et al.
CD-1 1x; initiation experiment vehicle control: 10% yes/val (1980)
Crystals Mouse, m/f 30 s.c. 10 mg/animal, 2 × at 4- < 18 No malignant tumours n Shear & Leiter
Jackson A month interval months no/ld (1941)
Table 90. (continued)
Purity Species, Sex No./ Route of Dosage Study Incidence and type of tumour Result Reference
strain sex/ admin. duration Stat./
group at death/ Val.
sacrifice
Recrystallized, Mouse, m/f 23-28 i.p. 86.1 and 1750 µg/animal 26 weeks 17, 4, and 7, 12% m/f with n Busby et al.
HPLC Swiss- (total dose) in 3 aliquots lung tumours; vehicle control: no/val (1989)
Webster on days 1, 8 and 15 14, 7% m/f
BLU:Ha(lCR) after birth
newborn
> 99% Hamster, m 48 Intra- 3 mg/animal, 1x/week, < 110 1/48 tumours of the trachea, n Sellakumar &
Syrian tracheal 30 weeks weeks 2/48 malignant lymphomas; yes/val Shubik (1974)
golden control: 0/82 and 2/82
Triphenylene
Mouse 10 Dermal 0.3% in benzene, 2x/ < 548 No skin lesions n Barry et al.
week, life days no/ln, (1935)
ld
Recrystallized Mouse, m 20 Dermal 250 µg/animal in decalin, 82 weeks No skin tumours; solvent n Horton &
C3H 2x/week, 82 weeks control: 2/13 papillomas no/val Christian
(1974)
Result: p(ositive), n(egative), q(uestionable); Stat, statistical evaluation: yes or no; Val, validity: val, valid; ld, limited design; lc,
limited documentation; ls, limited survival; ln, limited number of animals
intrapulm., intrapulmonary injection; i.p., intraperitoneal injection; s.c., subcutaneous injection; i.m., intramuscular injection
m, male; f, female
TLC, thin-layer chromatography; DMSO, dimethylsulfoxide; HPLC, high-performance liquid chromatography; DMBA, 7,12-dimethylbenz[a]anthracene
Table 91. Overview of carcinogenicity of polycyclic aromatic hydrocarbons
Compound Carcinogenicity Species Route of administration
(weight of No. of studies with positive, negative, and questionable results
evidence)
Oral Dermal s.c./i.m. i.p./i.v. inh./tr. Other
+ - ± + - ± + - ± + - ± + - ± + - ±
Acenaphthene Questionable Mouse 1 1
Acenaphthylene No studies
Anthanthrene Positive Mouse 2 6 1 1 1
Anthracene Negative Mouse 6 1 1 1
Rat 2 1 2 1 1
Rabbit 1
Benz[a]anthracene Positive Mouse 2 1 7 4 4 2 2 1
Rat 1 1 2 1 1
Hamster 2 1
Benzo[b]fluoranthene Positive Mouse 7 1 1
Rat 1
Hamster 1
Benzo[j]fluoranthene Positive Mouse 3 1 1
Rat 1
Benzo[ghi]fluoranthene (Negative) Mouse 2
Benzo[k]fluoranthene Positive Mouse 1 2 1 1 1
Rat 1
Benzo[a]fluorene (Questionable) Mouse 1 1 1
Benzo[b]fluorene (Questionable) Mouse 1
Benzo[ghi]perylene Negative Mouse 8 2
Rat 1
Benzo[c]phenanthrene (Positive) Mouse 2 2 1 1
Rat 1
Table 91. (continued)
Compound Carcinogenicity Species Route of administration
(weight of No. of studies with positive, negative, and questionable results
evidence)
Oral Dermal s.c./i.m. i.p./i.v. inh./tr. Other
+ - ± + - ± + - ± + - ± + - ± + - ±
Benzo[a]pyrene Positive Mouse 5 26 6 3 1 2
Rat 2 1 1 9 3
Hamster 1 1 1 1 11 1 1
Dog 1
Cattle 1
Pig 2
Monkey 1 1 1 1
Benzo[e]pyrene Questionable Mouse 2 1 5 1
Rat 1 1
Chrysene Positive Mouse 11 9 1 3 3 1 1 2 1
Rat 1 2 1
Coronene (Questionable) Mouse 1 1
Cyclopenta[cd]pyrene Positive Mouse 4 1 1
Rat 1
Dibenz[a,h]anthracene Positive Mouse 1 1 6 8 1
Rat 2 1 1 1
Hamster 1 1 1
Monkey 1
Dibenzo[a,e]pyrene Positive Mouse 3 2
Rat 1
Dibenzo[a,h]pyrene Positive Mouse 6 2 1
Rat 1
Dibenzo[a,i]pyrene Positive Mouse 7 4 1
Rat 1
Hamster 2 2
Monkey 1
Dibenzo[a,l]pyrene Positive Mouse 7 1
Rat 2
Table 91. (continued)
Compound Carcinogenicity Species Route of administration
(weight of No. of studies with positive, negative, and questionable results
evidence)
Oral Dermal s.c./i.m. i.p./i.v. inh./tr. Other
+ - ± + - ± + - ± + - ± + - ± + - ±
Fluoranthene (Positive) Mouse 6 2 3
Fluorene Negative Mouse 3 1 1
Rat 2
+ - ± + - ± + - ± + - ± + - ± + - ±
Indeno[1,2,3-cd]pyrene Positive Mouse 2 1 2 1 1
Rat 1
5-Methylchrysene Positive Mouse 13 1 1 1 1
Rat 1
1-Methylphenanthrene (Negative) Mouse 1
Naphthalene (Questionable) Mouse 1 2 2 1
Rat 1 1 1 1
Perylene (Negative) Mouse 2
Phenanthrene (Questionable) Mouse 1 3 3 3 1
Rat 1 1
Pyrene (Questionable) Mouse 1 7 3 1 1
Hamster 1
Triphenylene (Negative) Mouse 2
+, positive; -, negative; questionable; parentheses, limited number of studies
s.c., subcutaneous; i.m., intramuscular; i.p., intraperitoneal; i.v., intravenous; inh., inhalation; tr., intratracheal
Other: e.g. intramammary injection, bladder implant, bronchial implant
Epidermal cell kinetics, DNA adduct levels, and changes in skin
morphology were measured in ICR/Harlan mice after 29 weekly topical
applications of 16, 32, or 64 µg benzo [a]pyrene for up to 35 weeks.
Initially, there was a linear increase in DNA adducts, which was much
less steep at 64 µg and which did not correlate with the sharp rise in
tumour response at that dose. A dose-dependent increase in the
3H-thymidine labelling index, the mitotic index, and the incidence of
pyknotic and dark cells indicated that benzo [a]pyrene induced
extensive cytotoxicity and cell death, with regenerative
proliferation. Virtually all of the initial tumours were papillomas,
which required an average of eight weeks to progress to carcinomas,
reflecting the tumour-promoting activity of benzo [a]pyrene in this
model (Albert et al., 1991a,b).
In a study with female Sprague-Dawley rats to elucidate whether the
metabolites and DNA adducts of benzo [a]pyrene are formed in the
liver or in target tissues, animals that had received a liver
transplant were compared with normal animals. The liver was found to
serve as a depot for PAH, in this case infused 3H-benzo [a]pyrene,
which was converted into polar metabolites. A few hours later, polar
metabolites and DNA adducts were found in target tissues of both
groups (Wall et al., 1991).
7.7.1.2 Benzo [e]pyrene
The results of studies on benzo [e]pyrene are considered to be
questionable or negative even though positive results were reported in
two studies by dermal application, because no bay-region activation
was found in liver tissue that would result in an 'ultimate'
carcinogen, such as 9,10-dihydroxy-11,12-epoxy-9,10,11,12-
tetrahydrobenzo [e]pyrene (Jacob et al., 1983).
7.7.2 Comparative studies
Comparative studies on the tumorigenic activity of individual PAH that
have been used as the basis for comparative potency factors (see
Appendix I) are summarized below. The detailed results of studies with
individual PAH are given in Table 90. In general, the results of
studies with skin painting and lung implantation were used for
estimating comparative potencies in preference to those from
initiation-promotion experiments and studies by intraperitoneal
injection. No comparative studies have been carried out by oral
administration.
7.7.2.1 Carcinogenicity
(a) Dermal exposure
Skin painting: Solutions of 0.5% benzo [a]pyrene,
benzo [b]fluoranthene, benzo [j]fluoranthene, or
benzo [k]fluoranthene were applied dermally three times weekly to
groups of 20 female Swiss Millerton mice for life, and the number of
skin tumours was determined. The percentages of papillomas/ carcinomas
induced by these compounds after four months were 70/20 with
benzo [a]pyrene, 95/10 with benzo [b]fluoranthene, 40/5 with
benzo [j]-fluoranthene, and none with benzo [k]fluoranthene. Minimal
activity (10% papillomas) was induced by benzo [k]fluoranthene after
11 months (Wynder & Hoffmann, 1959b). The order of potency was thus
benzo [a]pyrene > benzo [b]-fluoranthene > benzo [j]fluoranthene
> benzo [k]fluoranthene.
In a similar regime, 0.01% solutions of benzo [a]pyrene or
dibenz [a,h]anthracene applied dermally to groups of 20 mice induced
10/10% and 15/5% papillomas/carcinomas, respectively after six months.
A 0.1% solution of dibenzo [a,i]pyrene induced 10/0% tumours after
seven months, and a 0.1% solution of benzo [e]pyrene induced 5/0%
tumours after 10 months. A 1% chrysene solution induced 5/5%
papillomas/carcinomas after eight months. A 0.1% solution of
fluoranthene and 10% solutions of anthracene and pyrene had no
activity (Wynder & Hoffmann, 1959a). The order of potency was thus
benzo [a]pyrene = dibenz [a,h]anthracene > dibenzo [a,i]pyrene >
chrysene > benzo [e]pyrene > fluoranthene, anthracene, pyrene.
In a further study, the carcinogenicity of PAH was compared after
dermal application to mice three times weekly, as above. A dose of
0.05% induced the following percentages of papillomas/carcinomas after
eight months: benzo [a]pyrene, 17/17; dibenzo [a,h]pyrene, 14/9;
dibenzo [a,l]pyrene, 10/10; dibenzo [a,i]pyrene, 3/0;
dibenzo [a,e]pyrene, 2/1 after 10 months; indeno[1,2,3- cd]pyrene,
1/1 with 0.5% solution; and benzo [ghi]perylene, 0/0 (Hoffmann &
Wynder, 1966). The order of potency was thus benzo [a]pyrene>
dibenzo [a,h] -pyrene > dibenzo [a,l]pyrene > dibenzo [a,i]pyrene
> dibenzo [a,e]pyrene > indeno[1,2,3- cd]pyrene >
benzo [ghi]perylene.
In a lifetime study by skin painting in female NMRI mice,
benzo [a]pyrene and benzo [b]fluoranthene were carcinogenic,
benzo [j]fluoranthene was weakly carcinogenic, and
benzo [k]fluoranthene, indeno[1,2,3- cd]pyrene, and coronene had no
carcinogenic effect (Habs et al., 1980). The order of potency was thus
benzo [a]pyrene >> benzo [b]fluoranthene > benzo [j]fluoranthene
> benzo [k]-fluoranthene, coronene, indeno[1,2,3- cd]pyrene.
Initiation-promotion: Ten doses of PAH at a total dose of 0.25
mg/mouse were applied every second day to the backs of Swiss Millerton
mice, which were then promoted with 2.5% croton oil in acetone
(Hoffmann & Wynder, 1966). The relative tumour-inducing activity was:
benzo [a]pyrene > dibenzo [a,h]pyrene > dibenzo [a,l]pyrene >
dibenzo [a,i]pyrene > dibenzo [a,e]-pyrene > indeno[1,2,3-
cd]pyrene > benzo [ghi]perylene.
In an assay in CD-1 mice, 30 µg of several PAH were applied in 10
doses over 20 days to the shaven backs of groups of 20 mice. Ten days
after completion of the initiation, promotion was begun by thrice
weekly application of 12- O-tetradecanoylphorbol 13-acetate in 0.1 ml
acetone. The skin tumours induced were predominantly squamous-cell
papillomas. After 20 weeks (10 weeks for benzo [a]pyrene), the
percentages of skin tumour-bearing animals were 85% with
benzo [a]pyrene, 45% with benzo [b]fluoranthene, 30% with
benzo [j]fluoranthene, and 5% with benzo [k]fluoranthene. The
vehicle controls had no tumours (La Voie et al., 1982b).
Sencar mice were treated with the - [a,e]-, - [a,h]-, - [a,i]-, and
- [a,l]- isomers of dibenzopyrene with TPA as a promoter,
anthanthrene as the negative control and with vehicle and sham
controls. Dibenzo [a,e]pyrene was a very weak tumour initiator and
dibenzo [a,h]pyrene and dibenzo [a,i]pyrene were tumorigenic;
dibenzo[a,l]pyrene was highly toxic and an extremely potent carcinogen
(Cavalieri et al., 1989). In a further investigation of
dibenzo[a,l]pyrene, with benzo [a]pyrene as the positive control,
7,12-dimethylbenz [a]anthracene, recognized as the most potent
carcinogenic PAH, was also tested at 4, 20, and 100 nmol of the PAH in
the same regime as above. The tumorigenic activity of
dibenzo[a,l]pyrene in mouse skin was inversely proportional to the
dose, indicating that toxicity interferes with the initiation of
tumours. When the effects of equimolar concentrations were compared,
benzo [a]pyrene was a much weaker tumour initiator than
dibenzo[a,l]pyrene (Cavalieri et al., 1991). The order of potency was
thus dibenzo[a,l]pyrene > dibenzo[a,i]pyrene > dibenzo[a,h]pyrene >
benzo [a]pyrene > dibenzo[a,e]pyrene.
The relationship between the Ah locus and the induction of
subcutaneous fibrosarcomas was studied after administration of
dibenz [a,c]anthracene and dibenz [a,h]anthracene to B6, D2, and
B6D2F1 mice. The doses and results are shown in Table 92.
Dibenz [a,c]anthracene was a weak tumour inducer in all groups
tested. Dibenz [a,h]anthracene was a fairly potent inducer of
subcutaneous tumours in B6 and B6D2F1 mice, but not in D2 mice. These
results, with those of back-crossing experiments, demonstrate a strict
correlation between the tumorigenicity of dibenz [a,h]anthracene and
expression of the Ahb allele (Kouri et al., 1983).
(b) Other routes
Intraperitoneal injection in newborn mice: The tumorigenic activity
of the nonalternant PAH benzo [b]fluoranthene,
benzo [j]fluoranthene, and benzo [k]fluoranthene and of
indeno[1,2,3- cd]pyrene and benzo [a]pyrene were evaluated by
injecting a total of 0.5, 1.1, 2.1, 2.1, or 0.5 µmol of each compound,
respectively, in dimethyl sulfoxide in aliquots of 5, 10, or 20 µl on
days 1, 8, and 15 after birth to CD-1 mice (La Voie et al., 1987).
Direct comparison was not possible owing to the differences in the
total amount injected, but both benzo [b]fluoranthene and
benzo [j]fluoranthene had significant tumorigenic activity, whereas
neither benzo [k]fluoranthene nor indeno[1,2,3- cd]pyrene was
tumorigenic under these conditions. There were problems with the
solubility of indeno[1,2,3- cd]pyrene. The order of potency was thus
benzo [a]pyrene > benzo [b]fluoranthene = benzo [j]fluoranthene >
benzo [k]-fluoranthene, indeno[1,2,3- cd]pyrene.
Intramammary injection: 7,12-Dimethylbenz [a]anthracene and
dibenzopyrene isomers were tested with benzo [a]pyrene by
intramammary injection. Dibenzo[a,e]pyrene was inactive, but
dibenzo [a,l]pyrene was much more carcinogenic than
7,12-dimethylbenz [a]anthracene. At these doses, benzo [a]pyrene had
only marginal activity. Dibenzo [a,l]pyrene thus appears to have the
highest mammary cancer potency of all PAH so far tested (Cavalieri et
al., 1989).
Lung implantation: The relative potencies of PAH to induce
epidermoid carcinomas and pleomorphic sarcomas after intrapulmonary
injection, with benzo [a]pyrene as the reference substance, were
dibenz [a,h] anthracene, 1.91 > benzo [a]pyrene, 1.00 >
anthanthrene, 0.19 > benzo [b]fluoranthene, 0.11 > indeno[1,2,3-
cd]pyrene, 0.08 > chrysene, 0.03 = benzo [k]fluoranthene, 0.03 =
benzo [j]fluoranthene, 0.03 > phenanthrene, 0.001.
Benzo [ghi]perylene and benzo [e]pyrene had no tumour-inducing
effect (Deutsch-Wenzel et al., 1983; Wenzel-Hartung et al., 1990).
Subcutaneous injection: Dose-response curves for benzo [a]pyrene
and dibenz [a,h]anthracene were established after a single
subcutaneous injection of PAH in tricaprylin into the right axilla of
male C3H mice; 99% of the tumours detected were spindle-cell sarcomas.
The responses of vehicle controls were not reported. Under the
conditions in this experiment, dibenz [a,h]anthracene was estimated
to be 4.5 times more potent than benzo [a]pyrene (Bryan & Shimkin,
1943).
Eight of 10 male and six of 10 female C57 black mice had
injection-site tumours 60-80 weeks after 10 weekly subcutaneous
injections of 1 mg benz [a]anthracene, whereas 20/20 males and 17/20
females had tumours after 1 mg dibenz [a,h]anthracene (Boyland &
Sims, 1967).
7.7.2.2 Further evidence
(a) Sebaceous gland assay
Application of carcinogenic PAH to mouse skin leads to the destruction
of sebaceous glands, hyperplasia, hyperkeratosis, and even ulceration
(Bock, 1964). An assay of these glands has been used to screen the
tumorigenic potential of PAH. Acute topical application of
benzo [a]pyrene, benz [a]anthracene, or dibenz [a,h]anthracene was
reported to suppress glandular activity (Bock & Mund, 1958). The order
of potency was benzo [a]pyrene = dibenz [a,h]anthracene >
benz [a]anthracene. In another study, the order of potency was
benzo [a]pyrene > benzo [b]fluoranthene = benzo [j]fluoranthene =
benzo [k]fluoranthene = indeno[1,2,3- cd]pyrene (Habs et al. 1980).
Table 92. Subcutaneous fibrosarcomas induced by a dose of 300 µg per
animal of isomers of dibenzanthracene in different mouse strains
Dibenzanthracene Strain Tumour Carcinogenic
isomer incidence index
a,c B6 1/30 1.1
D2 0/30 0
B6D2F1 1/30 1.2
a,h B6 14/30 24
D2 0/30 0
B6D2F1 33/60 30
B6D2F1 x D2 38/53 29
back-crosses
(Ahb/Ahd phenotype)
B6D2F1 × D2 0/33 0
back-crosses
(Ahd/Ahd phenotype)
From Kouri et al. (1983)
(b) DNA adduct formation
In a 32P-postlabelling test for covalent binding of PAH to DNA in
mouse skin in vivo after a single topical application, the relative
ability to induce DNA adducts was benzo [a]pyrene >
benz [a]anthracene = dibenz [a,h]anthracene = benzo [ghi]perylene
(Reddy et al., 1984). DNA adducts were not induced by pyrene. In a
similar study, the relative ability of PAH to bind covalently to DNA
was benzo [b]fluoranthene > benzo [j]fluoranthene >
benzo [k]fluoranthene > indeno[1,2,3- cd]pyrene (Weyand et al.,
1987). In a study in vitro, the relative ability for covalent
binding of PAH to DNA was reported to be benzo [a]pyrene >
dibenz [a,h]anthracene > benz [a]anthracene > pyrene >
phenanthrene (Grover & Sims, 1968).
7.7.3 PAH in complex mixtures
In a PAH-rich emission mixture prepared by burning tar pitch with
coal, the benzo [a]pyrene content was about 90 µg/m3, two to three
times higher than the concentration measured in old coal plants. The
tumour incidence in rats exposed for 16 h/day on five days per week
for 22 months with a subsequent eight-month exposure to clean air was
18%; the mortality rate was not increased in comparison with controls
exposed to clean air. The lung tumour incidences in mice exposed to
the same atmosphere for 10, 12, or 24 months were 86, 70, and 79%,
respectively, with 3.5, 12.5, and 32% in concurrent controls. An
additive or even potentiating carcinogenic effect with other
respiratory-tract carcinogens was demonstrated. In contrast to a group
exposed concurrently to diesel exhaust, the coal-tar pitch did not
cause particle overload in the lung or impair lung clearance (Heinrich
et al., 1986a,b).
Female Wistar rats were exposed by inhalation to 1.1 (groups 1 and 2)
or 2.6 mg/m3 (groups 3 and 4) of an aerosol of a PAH-rich hard
coal-tar pitch condensate containing 20 or 50 µg/m3 benzo [a]pyrene
(among other PAH), for 17 h per day on five days per week for 10
(groups 1 and 3) and 20 months (groups 2 and 4) and then to clean air
for 20 or 10 months. The aerosol contained benz [a]anthracene and
chrysene at concentrations similar to that of benzo [a]pyrene.
Increased mortality was observed due to the development of large,
multiple tumours in the lungs and not to toxic effects. The lung
tumour rates were 4, 33, 39, and 97% in groups 1,2, 3, and 4,
respectively. Other groups exposed simultaneously to 2 or 6 mg/m3
carbon black, which might serve as a PAH carrier, showed an additional
increase in tumour rates, i.e. 89 and 72% in comparison with 39% in
group 3. A group exposed only to carbon black had a tumour rate of
18%. The authors therefore concluded that there was a more than
additive carcinogenic effect after 10 months of exposure. A 'PAH
depot' effect may be involved, in which the residence time of the PAH
is prolonged due to attachment to the inert carbon black particles,
with an extended period elution of adsorbed PAH. Furthermore, the
irritating, inflammatory, and cell proliferation effects of carbon
black enhance the probability of genotoxic effects in the lungs
(Heinrich, 1989; Heinrich et al., 1994a). Most of the lung tumours
observed after exposure to tar-pitch aerosol with or without carbon
black were classified as squamous-cell carcinomas (Heinrich et al.,
1994b).
The preliminary findings of a study on coal gasification tars have
been reported. Coal-tar is a complex mixture containing over 1000
compounds, of which at least 30 are PAH, including benzo [a]pyrene.
In a two-year bioassay for carcinogenicity, female B6C3F1 mice were
fed up to 100 ppm benzo [a]pyrene or up to 1% coal-tar. Forestomach
tumours were observed in mice fed benzo [a]pyrene, the incidence
increasing sharply at doses between 5 and 25 ppm. Forestomach tumours
were also seen in mice fed coal-tar, with a clear increase at 0.3%;
the incidence was approximately the same at 0.3 and 0.6% but declined
at 1.0%, due to mortality from small intestinal adenocarcinomas, which
were not observed at doses below 0.6%. Steady-state DNA adduct levels
were examined in the forestomachs and small intestines of mice fed
benzo [a]pyrene or coal-tar for four weeks. Those fed
benzo [a]pyrene had one major adduct, which also accounted for 10-25%
of the adducts in the forestomachs of mice fed coal-tar. A linear
dose-response relationship was observed between the dose of
benzo [a]pyrene and the adduct levels in the forestomach. The adduct
levels in the forestomachs of mice fed coal-tar increased in a
relatively linear manner at doses above 0.3%. Total adducts and
benzo [a]pyrene-adduct levels in the small intestine increased up to
the 0.6% dose of coal-tar and then decreased (Culp et al., 1996).
A two-stage study of carcinogenicity in female CD-1 mice was performed
to assess the risk deriving from coal-tar formulations used for human
therapeutic purposes, e.g. against dandruff or psoriasis (see also
section 8.2.3). Mice were treated epicutaneously five times per week
with 50 mg of a 1.5% coal-tar ointment for two weeks ('initiation'),
followed by 'promotion' with 50 mg of a 0.1% dithranol cream, used
against psoriasis, three times per week for 40 weeks. A single dose of
50 µg benzo [a]pyrene was given as an initiator as a positive
control. After 40 weeks of promoter treatment, 4/27 tumours were
observed in the mice treated with coal-tar and 14/28 in those given
benzo [a]pyrene; when coal-tar or dithranol was given alone, no
tumours were observed (Phillips & Alldrick, 1994).
7.7.4 Transplacental carcinogenicity
7.7.4.1 Benzo [a]pyrene
Clastogenic responses to benzo [a]pyrene and AHH inducibility were
measured in 11-day-old embryos of genetically different mice (C57 and
DBA mated inter se and mixed) after transplacental treatment with
150 mg/kg bw by garage 15 h before sacrifice. The rate of chromosomal
aberrations was not correlated quantitatively with AHH activity. It
was concluded that not only activation and detoxification of
benzo [a]pyrene in maternal tissue but also other genetically
controlled processes, such as repair and transformation of primary DNA
lesions into true DNA discontinuities, are involved (Adler et al.,
1989).
Benzo [a]pyrene can cross the placenta in mice and rats (Shendrikova
& Aleksandrov, 1974; Shendrikova et al., 1974; Takahashi, 1974;
Baranova et al., 1976), but the concentration of 14C-benzo [a]pyrene
was one to two orders of magnitude lower in mouse embryonic than
maternal tissues after oral administration (Neubert & Tapken, 1988).
Intraperitoneal administration of benzo [a]pyrene to Ha/ICR mice
during the last half of pregnancy increased the incidences of
pulmonary adenomas and skin papillomas in progeny nursed by foster
mothers, excluding uptake of PAH via the milk (Bulay, 1970; Bulay &
Wattenberg, 1971). A similar result was observed in A and C57B1 mice;
furthermore, lung tumours were induced in Ha/ICR strain mice and liver
tumours in the offspring of all three strains (Nikonova, 1977).
Transplacental carcinogenesis has also been reported in rabbits
(Beniashvili, 1978).
Benzo [a]pyrene was given subcutaneously to strain A and C57B1 mice
at 4 or 6 mg per animal once or twice on days 18 and 19 of gestation.
The progeny of dams given a single dose of 4 mg had a significantly
increased number of adenomas; the 6-mg dose induced 77% lung tumours
in A mice and 12% in controls. The maximum dose, 12 mg, induced 32%
liver tumours in male C57B1 mice and 9% in females, with 1% in
controls (Nikonova, 1977).
7.7.4.2 Pyrene
The tumour incidence in the offspring of strain A mice was not
increased by two subcutaneous injections of 6 mg pyrene on days 18 and
19 of pregnancy (Nikonova, 1977).
7.8 Special studies
Adverse effects of PAH unrelated to cancer have also been seen.
Proliferating tissues such as bone marrow, lymphoid organs, gonads,
and intestinal epithelium are affected, but the major target organs
seemed to be those of the haematopoietic and lymphoid systems.
7.8.1 Phototoxicity
Photodynamic compounds can generate superoxide anion radicals in the
presence of near ultraviolet light. In the absence of oxygen, they act
as photoreducing agents. The main effect is epidermal damage.
7.8.1.1 Anthracene
Increased dermal sensitivity to ultraviolet irradiation was seen in
hairless mice after pretreatment with anthracene, but
photocarcinogenesis was not significantly increased (Forbes et al.,
1976; see also Table 92). In guinea-pigs treated six times on the
dorsal skin with anthracene and then irradiated with ultraviolet
light, a photoirritant reaction was observed that reached a maximum
after a few hours but had faded by 24 h (Lovell & Sanders, 1992).
Petroleum can photosensitize human skin to sunlight, resulting in
erythema and pigmentation. Petroleum also enhanced the
immunomodulatory effects of ultraviolet radiation on mammalian skin,
with depletion of antigen-presenting cells which play a critical role
by presenting antigen to thymus-derived lymphocytes. In tests of PAH
that are present at relatively high concentrations in crude oils,
anthracene but not phenanthrene or benzo [a]pyrene induced
photosensitization of mouse skin in vivo and in vitro. Exposure of
skin sections to anthracene at 5 µg/ml, equivalent to 125 ng/mouse,
reduced the numbers of antigen-presenting cells and of Thy-1-positive
dendritic cells (Burnham & Rahman, 1992).
7.8.1.2 Benzo [a]pyrene
Benzo [a]pyrene in the presence of near-ultraviolet radiation
(290-400 nm) had phototoxic effects, observed as haemolysis of human
erythrocytes and inactivation of Escherichia coli (Kagan et al.,
1989). A significant shortening of tumour-free survival was seen in in
Balb/c mice exposed to ultraviolet irradiation before dermal treatment
with benzo [a]pyrene. Ultraviolet irradiation had a systemic effect,
enhancing subsequent dose-dependent tumour induction by
benzo [a]pyrene (Gensler, 1988).
7.8.1.3 Pyrene
In six guinea-pigs given 5 µmol-5 mmol of pyrene dissolved in ethanol,
a strong phototoxic reaction was observed 20 h after ultraviolet A
irradiation at 1 × 103 J/m2 (320-400 nm) (Kochevar et al., 1982).
When mast cells were isolated from Sprague-Dawley rats, incubated with
25 µmol/litre pyrene, and irradiated with ultraviolet B radiation
(280-320 nm) at 60 kJ/m2, they released 80% of their serotonin
(Gendimenico & Kochevar, 1984).
7.8.1.4 Comparisons of individual PAH
The phototoxic effects of benzo [a]pyrene, benz [a]anthracene,
indeno[1,2,3-cd]pyrene, fluoranthene, and perylene were compared by
treating human fibroblasts with these PAH and then irradiating them
with ultraviolet light (<400 nm). A good correlation was found
between the phototoxic effects and known carcinogenic potency:
benzo [a]pyrene and indeno[1,2,3- cd]pyrene were highly toxic,
benz [a]anthracene was distinctly toxic, fluoranthene slightly toxic,
and perylene not cytotoxic (Bauer et al., 1985).
7.8.2 Immunotoxicity
7.8.2.1 Benzo [a]pyrene
(a) Intraperitoneal and intratracheal injection
Mice injected intraperitoneally with a single dose of 50, 100, or 200
mg/kg bw benzo [a]pyrene showed reduced thymic and splenic weights on
day 5, and the splenocyte antibody-forming response of immunized mice
was reduced by 60-90%. At the higher concentrations, lymphocyte
proliferation was decreased significantly. Benzo [a]pyrene altered
both humoral and cellular immunity; B cells were more susceptible than
T cells (Xue et al., 1991).
In a study of the effect of accumulation of benzo [a]pyrene in
lymphoid organs on humoral immunity, B6C3F1 mice were given
intratracheal instillations of 0.4, 4, or 40 mg/kg bw daily for seven
days and immunized with sheep erythrocytes one day later; then, the
number of antigen-specific, antibody-forming cells was measured. The
spleen showed a decrease, but lung-associated lymph nodes showed
either decreased (up to 60%) or increased (up to 100%) numbers,
depending on whether sheep erythrocytes were given intratracheally or
intraperitoneally (Schnizlein et al., 1987).
(b) Dermal exposure
After dermal exposure of female B6C3F1 mice to 0, 5, 20, or 40 mg/kg
bw per day for 14 days, dose-dependent suppression of the
antibody-forming cell response to sheep erythrocytes was seen both
in vivo and in vitro, the number at 40 mg/kg bw being about 30% of
the control value. The immunosuppression was similar when
benzo [a]pyrene was administered at a high dose subcutaneously in
corn oil. The antibody-forming cell response recovered by about day 60
after dermal exposure, while no recovery was seen after subcutaneous
injection (Parrott et al., 1989).
Female B6C3F1 mice were exposed to benzo [a]pyrene at 0, 0.625, 2.5,
5, 20, or 40 mg/kg bw per day for 28 days and were injected
intravenously with sheep erythrocytes on days 11 and 25. An
antigen-specific enzyme-linked immunosorbent assay to sheep
erythrocyte membranes was used to measure the primary immunoglobulin M
response on day 15 and the secondary immunoglobulin G response on day
30. Significant suppression of the primary immunoglobulin M response
was observed at doses of 5 mg/kg bw per day and more, but the serum
titres of animals treated with 0.625 or 2.5 mg/kg bw per day did not
differ from those of vehicle controls. The secondary immunoglobulin G
response was significantly decreased at all doses (Deal, 1995).
(c) Comparisons of benzo[a]pyrene and benzo[e]pyrene
The immunotoxic effects of benzo [a]pyrene and benzo [e]pyrene have
been compared in several studies. In most, benzo [a]pyrene was
clearly immunosuppressive, whereas benzo [e]pyrene was inactive.
In vivo: After activation of splenic lymphocytes, mice were given
single intraperitoneal injections of benzo [a]pyrene and
benzo [e]pyrene at 2.5, 10, or 50 mg/kg bw for 24 or 48 h before
sacrifice. Mononuclear cell populations were then assayed for AHH
activity, blastogenesis, antigen-specific cell-mediated cytotoxicity,
and the percentage of macrophages. Neither PAH had a significant
effect on blastogenesis. Benzo [a]pyrene suppressed cell-mediated
cytotoxicity of T cells by 40-80%, while benzo [e]pyrene had no
effect. These results suggested that mitogen-activated, AHH-induced
splenic lymphocytes metabolize benzo [a]-pyrene to immunocytotoxic
metabolites. T cells are probably activated by early stimulation of T
suppressor cells accompanied by an increase in T suppressor cell
factors (Wojdani & Alfred, 1984).
B6C3F1 mice were given 10 daily subcutaneous injections of
benzo [a]pyrene and benzo [e]pyrene at doses of 5, 20, or 40 mg/kg
bw over a 14-day period. Three to four days after the last dose,
immune function was evaluated. Benzo [a]pyrene reduced the number of
immunoglobulin M and G antibody plaque-forming cells in response to
sheep erythrocytes and lipopolysaccharide, and the TNP-Ficoll
plaque-forming cell response was depressed by 77%. These changes
indicate altered differentiation and antibody production in mature B
cells. No change in the plaque-forming cell response was observed
after exposure to benzo [e]pyrene (Dean, J.H. et al., 1983).
Mice were given subcutaneous injections of benzo [a]pyrene or
benzo [e]pyrene at 5, 10, or 40 mg/kg bw daily for 11 days, followed
by an injection of sheep erythrocytes. Benzo [a]pyrene suppressed the
antibody response to DNP-Ficoll and sheep erythrocytes but not to
lipopolysaccharide; benzo [e]pyrene was not immunosuppressive (White
& Holsapple, 1984).
In vitro: When benzo [a]pyrene was added to cultured mouse spleen
cells in vitro, a metabolism-activating system was not required to
produce immunosuppression. Furthermore, addition of S9 did not
increase the degree of immunosuppression produced by benzo [a]pyrene
(White & Holsapple, 1984).
In several antibody generating systems in vitro, both
benzo [a]pyrene and benzo [e]pyrene caused a significant,
dose-dependent suppression of the T-dependent and polyclonal antibody
responses. A similar result was found in vitro after a 14-day
exposure of mice to 40 mg/kg bw benzo [a]pyrene in vivo, with 98%
suppression of T-cell-dependent antibody. Benzo [a]pyrene-induced
suppression is multicellular, and the greatest sensitivity is found
early in the immune response. Since benzo [e]pyrene induced
immunosuppression only at high concentrations, the preparation may
have been contaminated with benzo [a]pyrene (Blanton et al., 1986).
Benzo [a]pyrene and seven of its metabolites were evaluated for their
ability to suppress the antibody-forming cell response to sheep
erythrocytes in vitro. Direct addition of benzo [a]pyrene or its
7,8-diol to splenocyte cultures induced similar dose-dependent
suppression of the antibody response to the T-dependent antigen, sheep
erythrocytes. In contrast, exposure to the 4,5-diol, 9,10-diol,
6,12-dione, 3-hydroxy, and 4,5-epoxide metabolites resulted in
decreased antibody responses only with high concentrations; these were
associated with decreased viability of the cultures. In addition,
co-incubation with the cytochrome P450 inhibitor alpha-naphthoflavone
attenuated the suppressive effects of benzo [a]pyrene and
benzo [a]pyrene 7,8-diol (Kawabata & White, 1987).
The anti-sheep erythrocyte plaque-forming cell and mixed lymphocyte
responses were inhibited in murine splenic lymphocytes treated with
benzo [a]pyrene at concentrations ranging from 10-4 to 10-8
mol/litre, with maximal depression at 10-5mol/litre. Benzo [e]pyrene
at the same concentrations did not suppress these responses (Urso et
al., 1986).
7.8.2.2 Dibenz [a,h]anthracene
The immunosuppressive effects of dibenz [a,h]anthracene and
3-methyl-cholanthrene were studied in AHH-inducible (C57B1/6) and
non-inducible (DBA/2N) mice after intraperitoneal injection of 25, 50,
or 100 mg/kg bw five days before challenge with sheep erythrocytes or
after administration by gavage of 10-200 mg/kg bw per day for four
days. Immunosuppression occurred in both strains but was more
pronounced in the C57B1 mice after intraperitoneal injection. The DBA
mice were more susceptible to 3-methyl-cholanthrene given by gavage.
The authors suggest that PAH are rapidly metabolized and excreted
after gavage in AHH-inducible mice, whereas in non-inducible mice they
are absorbed and distributed to the target organs. AHH inducibility
may thus play an important role in the immunosuppressive activity of
PAH (Lubet et al., 1984a,b; see also section 7.5.2.1).
7.8.2.3 Fluoranthene
Murine bone-marrow stromal cells were used as a matrix for the growth
and limited development of precursor B cells in vitro, thus
mimicking B lymphopoiesis in vivo. Fluoranthene acutely suppressed B
lymphopoiesis, and the precursor B cell populations exposed to 50
µg/ml fluoranthene disappeared within two weeks. Lymphotoxicity was
mediated by fluoranthene-induced programmed cell death (apoptosis): 5
µg/ml reduced precursor B cell recoveries by > 95% within one or two
weeks. Lower doses altered the dynamics of B cell lymphopoiesis,
leading to accumulation of precursor B cells (Hinoshita et al., 1992).
7.8.2.4 Naphthalene
Naphthalene did not adversely affect the immune response in CD-1 mice
of each sex given up to 133 mg/kg bw per day orally for three months.
No alteration was seen in the lymphocyte proliferative response to the
T-cell mitogens concanavalin A and phytohaemagglutinin, in the delayed
hypersensitivity response to sheep erythrocytess, or in the popliteal
lymph node response. Furthermore, bone-marrow function was not altered
(Shopp et al., 1984).
Naphthalene is known to induce pulmonary and renal toxicity which is
mediated by its reactive, electrophilic metabolites 1,2-naphthalene
oxide, 1-naphthol, and 1,4-naphthoquinone. The immune response of mice
was, however, unaffected by a 90-day oral treatment with up to 25% of
the LD50 value. Furthermore, the number of antibody-forming cells in
splenocyte cultures was not affected by concentrations of naphthalene
up to 200 µmol/litre; however, 200 µmol/litre of 1-naphthol and 7-20
µmol/litre of 1,4-naphthoqninone suppressed the antibody-forming cell
response and decreased cell viability. Splenic microsomes were unable
to metabolize naphthalene, whereas liver microsomes generated 1,2-
naphthalene diol and 1-naphthol. It was concluded that diffusion of
liver metabolites to the spleen is insufficient to induce
immunotoxicity (Kawabata & White, 1990).
7.8.2.5 Comparisons of individual PAH
The effects of exposure to each of 10 PAH on the immunoglobulin M
antibody response to sheep erythrocytes were examined in B6C3F1 mice,
with a further investigation of the relationship of the
Ah gene complex to the immunosuppression. Mice were given
subcutaneous injections over 14 days, and splenic antibody-forming
cells were evaluated after immunization with sheep erythrocytes.
Significant decreases of 55-91% were observed after treatment with
dibenz [a,c]anthracene, dibenz [a,h]anthracene, benz [a]anthracene,
and benzo [a]pyrene, but no significant effects were observed with
anthracene, benzo [e]pyrene, perylene, or chrysene. Generally, the
structure-activity relationship for immunosuppression was correlated
strongly with that for carcinogenicity. Non-inducible DBA/2 mice had
greater PAH-induced immunosuppression than AHH-inducible B6C3F1 mice
(White et al., 1985).
7.8.2.6 Exposure in utero
Oral administration of benzo [a]pyrene at 2 mg/kg bw to Wistar rats
on day 19 of gestation induced a relative decrease in the number of
thymic glucocorticoid receptors in males and a relative increase in
females at six weeks of age. In animals treated at six weeks of age,
no change in the number or affinity of steroid receptors was seen in
males, but there was a 40% decrease in females. The investigators
concluded that benzo [a]pyrene binds to pre-encoded hormone receptors
and interferes with their maturation. When benzo [a]pyrene was given
on day 15 of gestation, a 30% decrease in the number of receptors was
seen in the offspring (Csaba et al., 1991; Csaba & Inczefi-Gonda,
1992).
Strong suppression of the anti-sheep erythrocytes plaque-forming cell
response, the mixed lymphocyte response, and the graft-versus-host
response in vivo were seen in the progeny of female mice that had
been treated intraperitoneally with benzo [a]pyrene at a dose of 150
mg/kg bw during mid-gestation (11-13 days). Immunodeficiency was seen
within one week after birth and persisted for 18 months.
Benzo [a]pyrene induced marked disorientation of T cells up to four
weeks postnatally. Disruption of T-cell differentiation during
ontogenesis was suggested, implying decreased resistance to the
development of neoplasia (Urso & Gengozian, 1980; Urso & Johnson,
1987). A two- to fourfold reduction in maternal leukocytes was seen
within five days of an intraperitoneal administration of 150 µg
benzo [a]pyrene on day 12 of gestation, which persisted to day 10
post partum and was attributed to lymphocyte depletion. Thus,
benzo [a]pyrene exacerbated the depression of leukocytes seen during
pregnancy. After intraperitoneal injection, it can reach the
lymphocytes, where metabolic activation may take place. Investigation
of the maternal T-cell population, both during pregnancy and post
partum, showed exacerbated decreases in the number of thymocytes
present in the thymus. In the spleen, the number of thymocytes
positive for LytI antigens was also depressed, whereas the number of
Lyt 2+ cells was enhanced, reaching levels > 700 times those of
controls. These results demonstrate disruption of the maternal T-cell
repertoire (Urso et al., 1988; Urso & Johnson, 1988).
7.8.2.7 Mechanisms of the immunotoxicity of PAH
Although the mechanism(s) by which PAH adversely affect the immune
system has not been defined, several have been proposed (Ladics &
White, 1996). The mechanism that is most consistent with that accepted
for the carcinogenesis of PAH is that their immunotoxicity is due not
to the parent compound but to the reactive metabolites formed. These
include the 7,8-diol-9,10-epoxide for benzo [a]pyrene (Kawabata &
White, 1987) and the 3,4-diol-1,2-epoxide for the synthetic compound
7,12-dimethylbenz [a]anthracene (Ladics et al., 1991). Several
investigators have suggested that the mechanism of action of PAH
results from their ability to enter the cell membrane and disrupt
transduction of transmembrane signals and/or alter the conformation of
receptors (Pallardy et al., 1992; Thurmond et al., 1989). It has also
been suggested that PAH alter the immune response as a result of their
ability to induce inappropriate alterations in the levels of various
cytokines, such as interleukins 1 and 2 (Lyte et al., 1987; Meyers et
al., 1988; Pallardy et al., 1989), and this mechanism is under active
investigation. While interaction with the Ah receptor has been
suggested as a possible mechanism of action of PAH, the contradictory
results from immunotoxicological studies with genetically different
strains of mice (high and low AHH responders) must be resolved.
Studies primarily conducted in vitro support the hypothesis that the
immunomodulatory effects of PAH are the result of alterations in
calcium mobilization (Burchiel et al., 1991).
7,12-Dimethylbenz [a]anthracene has been extensively studied as a
prototypic immunotoxicant. Exposure to this compound has been reported
to decrease natural killer cell activity (Kimbar et al., 1986), to
decrease resistance to challenge with tumour cells (Dean et al.,
1986), and to increase susceptibility to chemically induced tumours
(Elmets et al., 1988). In vitro, 7,12-dimethylbenz [a]anthracene
has been shown to suppress cytotoxic T-cell activity, possibly by
inducing defects in antigen recognition (House et al., 1989) and/or in
cytokine production (House et al., 1988). Oral administration of a
cumulative dose of 14 mg/kg bw of 7,12-dimethylbenz [a]anthracene
over 14 days suppressed proliferative responses by about 50% in
splenic lymphocytes and by more than 70% in gut-associated lymphocytes
(Burchiel et al., 1990).
7,12-Dimethylbenz [a]anthracene-mediated immunosuppression was shown
to persist for at least eight weeks (Ward et al., 1986; Burchiel et
al., 1988). This compound also decreased resistance to bacterial (Ward
et al., 1984) and viral (Selgrade et al., 1988) infections. Although
no studies have been carried out on humans in vivo, the IC50 for
inhibition of the response of human tonsillar lymphocytes in culture
to foreign tissue antigens was 10-40 µmol/litre; however, antibody
secretion was affected only at a concentration of 100 µmol/litre (Wood
& Holsapple, 1993).
7.8.3 Hepatotoxicity
7.8.3.1 Benzo [a]pyrene
Non-responsive strains of mice (C57B1/6, C3H/HeN, and Balb/cAnN) had
increased relative liver weights after they were fed for 180 days on a
diet containing benzo [a]pyrene resulting in an intake of 120 mg/kg
bw per day (Robinson et al., 1975).
7.8.3.2 Comparisons of individual PAH
The induction of several enzymes has been correlated with cancer
promotion. Wistar rats treated orally with 100 mg/kg bw per day of
benzo [a]pyrene, benz [a]anthracene, anthracene, chrysene, or
phenanthrene for four days showed induction of cytosolic aldehyde
dehydrogenase activity. Benzo [a]pyrene and benz [a]anthracene were
much more effective than the other substances, increasing liver
weights by 27 and 19%, respectively (Törrönen et al., 1981).
The extent of liver regeneration that determines the ability to induce
a proliferative response was investigated in partially hepatectomized
rats fed diets containing various PAH for 10 days. Doses of 51.4 mg/kg
bw per day acenaphthene or 180 mg/kg bw per day fluorene induced a
significant increase in the rate of liver regeneration, but 15.4 mg/kg
bw per day acenaphthene, 51.4 mg/kg bw per day benzo [a]pyrene, or
514 mg/kg bw per day pyrene, anthracene, or phenanthracene had no
effect (Gershbein, 1975).
7.8.4 Renal toxicity
Rats given 50-150 mg/kg bw benzo [a]pyrene orally over four days
showed moderate induction of renal microsomal carboxylesterase
activity; however, administration of 100 mg/kg bw per day anthracene
or phenanthrene had no effect (Nousiainen et al., 1984).
7.8.5 Ocular toxicity of naphthalene
The development of cataracts in rats, mice, and rabbits after
application of naphthalene is a toxic peculiarity of this compound.
Attempts have been made to clarify the metabolic processes responsible
for the formation of insoluble precipitates in the eye.
Cataracts developed in the eyes of rabbits within a few days of
repeated oral administrations of 0.5-1 g/kg bw per day. Oral
administration was more effective than other modes of application
(Pike, 1944). The oxidation products of naphthalene may reach the eye
via the bloodstream, where 1,2-naphthoquinone is formed which can
react with proteins and other cell components to form insoluble
precipitates with a characteristic brown colour (Van Heyningen &
Pirie, 1967).
C57B1/6 mice, which are susceptible to induction of cytochromes P450,
were given naphthalene by intraperitoneal administration at 500-2000
mg/kg bw. Cataracts were induced in a dose-dependent manner within 8
h. The effect was reduced by pretreatment with P450 inhibitors and
antioxidants and was increased by pretreatment with P450 inducers or
glutathione depletors. Cataracts were also induced in a dose-dependent
manner by intraperitoneal injection of 1,2- or 1,4-naphthoqninone at
5-250 mg/kg bw (molar potency about 10-fold higher). DBA/2 mice, which
are resistant to induction of cytochromes P450, did not develop
cataracts. The authors concluded that P450-dependent bioactivation was
necessary to form reactive intermediates, which are assumed to be
naphthoqninone or a free-radical derivative (Wells et al., 1989).
When a lens cell line from transgenic mice was exposed to the
1,2-dihydrodiol and 1,2-naphthoquinone metabolites of naphthalene, the
dihydrodiol did not appear to be toxic but the naphthoquinone induced
depletion of glutathione levels. Detoxification of naphthoquinone by
the enzyme quinone oxidoreductase prevents formation of a semiquinone
radical by two-electron reduction (Russell et al., 1991).
All rats of various strains given naphthalene orally developed
cataracts. The authors proposed that naphthalene dihydrodiol is
produced in the liver, reaches the aqueous humour, and penetrates the
lens, where it is metabolized to naphthoquinone. Feeding of 1-naphthol
did not induce opacification (Xu et al., 1992).
7.8.6 Percutaneous absorption
As dermal penetration is one of the major routes of entry after
occupational exposure, in-vivo and in-vitro models have been developed
to assess it.
In experiments in vitro on the dermal penetration of
benzo [a]pyrene and pyrene in guinea-pigs, pyrene was absorbed mainly
by passive diffusion, but benzo [a]pyrene was biotransformed during
absorption, and the 7,8,9,10-tetrol metabolite of the putative
ultimate carcinogen was detected in the receptor fluid of diffusion
cells (Ng et al., 1992).
In skin preparations from mice, rats, rabbits, guinea-pigs, marmosets,
and humans treated with benzo [a]pyrene, both the parent compound and
a full spectrum of metabolites were detected, depending on metabolic
viability, i.e. previously frozen skin preparations could not
metabolize benzo [a]pyrene (Kao et al., 1985).
14C-Benzo [a]pyrene was administered to the nuchal area of mice at
doses of 1.25-125 µg/cm2, and the animals were sacrificed seven days
later. Benzo [a]pyrene disappeared rapidly from the application site,
at a rate of 6% within 1 h and 40% within 24 h; after seven days, 7%
remained at the original site. Most of the benzo [a]pyrene was
excreted via the hepatobiliary system and found in the faeces, with
35% after 24 h, 58% after 48 h, and 80% after seven days. Only 10% of
the radiolabel was detected in urine. Uptake was saturated at doses >
15 µg/cm2, implying an enhanced risk for tumour induction in the skin
epithelium (Sanders et al., 1986).
The binding of benzo [a]pyrene to DNA and protein in mouse skin was
15-20 times greater when acetone was used as the vehicle than with a
low-viscosity oil (Ingram & Phillips, 1993). While acetone solutions
of 14C-benzo- [a]pyrene readily penetrated skin from human cadavers,
a significantly smaller amount moved from soil into skin. No
partitioning of benzo [a]pyrene from human skin to plasma was
observed. An experiment with rhesus monkeys in vivo also showed
significantly less absorption from soil (Wester et al., 1990).
7.8.7 Other studies
7.8.7.1 Benzo [k]fluoranthene
Intraperitoneal injections of benzo [k]fluoranthene, a widespread
PAH, to rats for three days induced maximal cytochrome P450/448
activity. Liver microsomes were then prepared, and the metabolic
profile of benzo [k]-fluoranthene was analysed by gas
chromatography-mass spectrometry. trans-5,6-, 8,9-, and mainly
10,11-dihydrodiols were the primary metabolites in noninduced rats.
Pretreatmant with PAH resulted in the generation of 5,6 and 8,9
isomers as the main metabolites, due to induction of monooxygeuases;
secondary metabolism to triols and tetrols was also induced. The
putative ultimate carcinogen, 3,4-diol-1,2-epoxy benz [a]anthracene,
was detected after pretreatment of liver microsomes with
benzo [k]fluoranthene. The authors concluded that
benzo [k]fluoranthene is a relevant component of environmental
pollution and enhanced the carcinogenic risk of benz [a]anthracene
(Schmoldt et al., 1981; Jacob et al., 1981a).
7.8.7.2 Benzo [a]pyrene
Rats were fed a diet containing 400 mg/kg benzo [a]pyrene with or
without 2 g/kg of ß-carotene. Benzo [a]pyrene had no effect on serum
retinol levels, but vitamin A levels were decreased in liver and small
intestine at two weeks, with a 30% decline by four weeks. A similar
effect was not observed in rats fed ß-carotene simultaneously. AHH can
be induced by ß-carotene, but this may not be the mechanism by which
tumours are prevented (Edes et al., 1991).
The role of benzo [a]pyrene in the induction of arteriosclerosis was
investigated by studying its effects on bovine arterial smooth muscle
cells in vitro. The number of cells was unchanged by exposure, but
the secretion of newly synthesized collagen was decreased. Total
cellular DNA was decreased and collagen secretion increased when the
cells were preincubated with platelet factors rather than a serum-free
medium (Stavenow & Pessah-Rasmussen, 1988).
Microsomes from rat aorta transformed benzo [a]pyrene into various
carcinogenic and toxic metabolites after induction with
3-methylcholanthrene. Thus, carcinogenic metabolites of PAH deriving
from cigarette smoke tars could cause endothelial injury, contributing
to the role of cigarette smoking in arteriosclerosis (Thirman et al.,
1994).
7.8.7.3 Phenanthrene
In a study of the oxidation of phenanthrene by liver microsomes from
rats with or without pretreatment with inducers, microsomes from
untreated rats produced only trans-9,10-diol phenanthrene, but
pretreatment with various PAH also led to oxidation at the 1,2 and 3,4
positions. Considerable amounts of the proximal carcinogen 1,2-diol
phenanthrene were detected, but the concentration of the ultimate
carcinogen 1,2-diol-3,4-epoxide was very low. These results are in
accordance with the questionable carcinogenic potency of phenanthrene
(Jacob et al., 1982a; see Table 91).
7.8.7.4 Comparisons of individual PAH
Activation of platelets by calcium ionophore A23187 can mobilize
intracellular stores of calcium ion and stimulate thromboxane
biosynthesis, which can be measured as thromboxane B2 synthesis.
Benz [a]anthracene, chrysene, benzo [a]pyrene, and
benzo [ghi]perylene inhibited thromboxane B2 production in
A-23187-induced, washed platelets from rabbits, while anthracene and
pyrene appeared to stimulate thromboxane B2 synthesis. Fluoranthene,
benzo [b]fluoranthene, benzo [k]fluoranthene, and benzo [e]pyrene
had little or no effect on activation (Yamazaki et al., 1990).
Benzo [a]pyrene, benzo [k]fluoranthene, benzo [b]fluoranthene,
chrysene, benz [a]anthracene, pyrene, phenanthrene, and fluoranthene
were toxic to human hepatoma cell cultures (HepG2), as measured with
neutral red, whereas fluorene, anthracene, acenaphthene, and
acenaphthylene were not (Babich et al., 1988).
7.9 Toxicity of metabolites
Derivatives of parent PAH have been tested for mutagenicity and
carcinogenicity in a number of experiments in order to assign the
effects to definite metabolites or to rank the potency of known
metabolites of a parent compound. Some studies addressed the steric
factors that determine mutagenic or carcinogenic effects, such as the
diastereomers of epoxides and the role of the methyl group in 5- and
6-methylchrysene. These studies are summarized in Tables 93 and 94,
although a few of the studies are reported in detail below.
7.9.1 Benzo [a]pyrene
Benzo [a]pyrene is metabolized to about 20 primary and secondary
oxidized metabolites and to a variety of conjugates. Several
metabolites can induce mutations, transform cells, and bind to
cellular macromolecules, but the 7,8-diol-9,10-epoxides are presently
considered to be the major ultimate carcinogens (DePierre & Ernster,
1978; Pelkonen & Nebert, 1982).
Table 93. Mutagenicity of metabolites of polycyclic aromatic hydrocarbons
Compound Metabolite Test system Results References
Benzo[b]fluoranthene 9,10-Diol Mouse, dermal Positive Amin et al. (1991a)
Benzo[j]fluoranthene 9,10-Diol Mouse, dermal Positive LaVoie et al. (1980; 1982b)
Benzo[k]fluoranthene 9,10-Diol Positive LaVoie et al. (1980)
Benzo[a]pyrene 7,8-Diol-9,10-epoxide Unscheduled DNA binding Positive Gill et al. (1991)
(syn + anti) DNA synthesis Positive
7,8-Diol-9,10-epoxide Hamster embryo cells, Positive Mager et al. (1977)
(2 stereoisomers) transformation
1-Hydroxy Reverse mutation, Positive (±S9) Schoeny et al. (1985)
2-Hydroxy S. typhimurium TA 1538, Negative (S9)
3-Hydroxy TA98,TA100 Positive (+S9)
4-Hydroxy Negative (±S9)
6-Hydroxy Positive (-S9)
7-Hydroxy Positive (+S9)
9-Hydroxy Positive (+S9)
10-Hydroxy Negative (±S9)
12-Hydroxy Positive (±S9)
1,6-Quinone Negative (±S9)
3,6-Quinone Positive (+S9)
4,5-Quinone Negative (±S9)
6,12-Quinone Negative (±S9)
trans-4,5-Diol Negative (±S9)
cis-4,5-Diol Positive (+S9)
trans-7,8-Diol Negative (±S9)
trans-9,10-Diol Negative (±S9)
4,5-Epoxide Positive (±S9)
1-Hydroxy Forward mutation, Positive (+S9) Schoeny et al. (1985)
2-Hydroxy S. typhimurium TM677 Negative (±S9)
3-Hydroxy Negative (±S9)
4-Hydroxy Negative (±S9)
6-Hydroxy Negative (±S9)
7-Hydroxy Positive (±S9)
9-Hydroxy Negative (±S9)
10-Hydroxy Negative (±S9)
Table 93. (continued)
Compound Metabolite Test system Results References
12-Hydroxy Negative (±S9)
1,6-Quinone Negative (±S9)
3,6-Quinone Negative (±S9)
4,5-Quinone Negative (±S9)
6,12-Quinone Negative (±S9)
trans-4,5-Diol Positive (+S9)
cis-4,5-Diol Positive (+S9)
trans-7,8-Diol Positive (+S9)
trans-9,10-Diol Negative (±S9)
cis-7,8-Diol Positive (+S9)
4,5-Epoxide Positive (±S9)
Benzo[e]pyrene 9,10-Diol-11,12-epoxide Bacterial and mammalian cells Weakly positive Wood et al.(1980)
Chrysene 1,2-Diol Bacterial and mammalian cells Positive Wood et al. (1977)
1,2-Diol-3,4-epoxide
5,6-Oxide S. cerevisiae, D4-RDII Positive Siebert et al. (1981)
Cyclopenta[cd]pyrene 3,4-Diol Bacterial and mammalian cells, Positive Gold et al. (1980)
mutagenicity and transformation
trans-3,4-Epoxide Calf thymus DNA, DNA binding Positive Beach et al. (1993)
Fluoranthene 2,3-Diol Bacterial calls Positive LaVoie et al. (1982a)
2,3-Diol-1,10b-epoxide Bacterial calls: Positive Rastetter et al. (1982)
1,10b-Diol-2,3-epoxide S. typhimurium TM677 Weakly positive
or negative
Dibenz[a,h]anthracene 3,4-Diol Bacterial cells Most mutagenic Wood et al. (1978)
compound among
3 dihydrodols
5,6-Epoxide Hamster embryo cells, Positive Huberman et al. (1972);
transformation Marquardt et al. (1972)
Dibenz[a,h]pyrene 1,2-Diol Bacterial cells Positive Wood et al. (1981)
Dibenz[a,i]pyrene 3,4-Diol Bacterial cells Positive Wood et al. (1981)
5-Methylcholanthrene 1,2-Diol and 7,8-diol Bacterial cells Positive Hecht et al. (1978)
5-Hydroxy S. typhimurium TA100 Weakly positive Amin et al. (1979)
1-Methylphenanthrene 1,4-Diol Bacterial cells Positive LaVoie et al. (1981c)
5,6-Diol
Phenanthrene 1,5-Diol,-3,4-epoxide Bacterial and mammalian cells Positive Wood et al. (1979)
9,10-Oxide S. cerevisiae D4-RDII Positive Siebert et al. (1981)
S9, 9000 × g microsomal fraction of liver
Table 94. Carcinogenicity of metabolites of polycydic aromatic hydrocarbons
Compound Metabolite Species, route of Type of Results References
administration investigation,
duration, dose
Benz[a]anthracene 3,4-Diol and Mouse, Carcinogenicity, 3R,4R-Diol: 71% with tumours; Wislocki et al.
3,4-diol-1,2- intraperitoneal 26 weeks other enantiomer not tumorigenic; (1979)
epoxide 3,4-diol-1,2-epoxide: 100% with tumours;
other enantiomer: 42%; control: 13%
Benzo[b]fluoranthene 9,10-Diol Mouse, dermal Initiation Positive Amin et al.
(1991a)
Benzo[j]fluoranthene 9,10-Diol Mouse, dermal Initiation Positive LaVoie et al.
(1980, 1982b)
4,5-Diol Mouse, dermal Initiation 2,3-Diol: 5-11 %; 4,5-diol: 78-100%; Rice et al.
9,10-Diol 9,10-diol: 60%; control: 10% (1987)
2,3-Diol
Benzo[c]phenanthrene 1,2-Diol Mouse, dermal Initiation 1,2-Diol: 3-4% with tumours; Levin et al.
3,4-Diol 3,4-diol: 28-47% with tumours; (1980)
5,6-Diol 5,6-diol: 3-7% with tumours;
1,2-Epoxy- 1,2-epoxy-3,4-diol: 80-90% with tumours;
3,4-diol control: no tumours
3,4-Diol-1,2- Mouse, dermal Initiation 95-100% with tumours; control: 10% Amin et al.
epoxide (1993)
anti-3,4-Diol- Rat, Carcinogenicity 100 % with mammary tumours; Hecht et al.
1,2-epoxide intramammary 1 × 12.2 µmol vehicle control: 3% (1994)
Benzo[a]pyrene 7,8-Diol Mouse,dermal Carcinogenicity, 100% with skin tumours Conney (1982)
43 µg every 2
week; 60 weeks
anti-7,8-Diol- Rat, Carcinogenicity, 47% with mammary tumours; vehicle Hecht et al.
9,10-epoxide intramammary 1 × 12.2 µmol control 3% (1994)
Table 94. (continued)
Compound Metabolite Species, route of Type of Results References
administration investigation,
duration, dose
Benzo[e]pyrene 4,5-Diol Mouse, dermal Initiation No significant effect Buening et al.
9,10-Diol (1980); Slaga
et al. (1980,
1981)
9,10-Diol Mouse, newborn, Carcinogenicity No induction of pulmonary tumours; Buening et al.
9,10-Diol- intrapentoneal significant induction of hepatic (1980); Chang
11,12-epoxide tumours et al. (1981)
Chrysene 1,2-Diol Mouse, dermal Initiation 0.4, 39, 60, and 79% with tumours; Levin et al.
1.25, and 4 control: 7% (1978)
µmol/ animal, 1 x
1,2-Diol Mouse, dermal Initiation 1,2-Diol: positive Slaga et al.
4,5-Diol 3,4-Diol: negative (1980, Chang
1,2-diol, Mouse, newborn Carcinogenicity Induction of pulmonary adenomas Buening et al.
1,2-Diol- (1979); Chang
3,4-epoxide et al. (1983)
Dibenz[a,h]anthracene 3,4-Diol Mouse, dermal Carcinogenicity Induction of skin tumours; induction of Buening et al.
pulmonary tumours in newborn mice (1979); Slaga et
al.(1980), 1981)
5,6-Epoxide Mouse, dermal Initiation Poorly active Van Duuren at
al. (1967)
Dibenzo[a,h]pyrene 1,2-Diol Mouse, dermal, Initiation Positive Wood et al.
intraperitoneal (1981)
Dibenzo[a,h]pyrene 3,4-Diol Mouse, dermal, Initiation Positive Wood et al.
intraperitoneal (1981)
Indeno[1,2,3-cd]pyrene 1,2-Diol, Mouse, dermal Initiation 1,2-Diol: 80%, 1,2-oxide, 80%; Rice et al.
1,2-Oxide 8-hydroxy, 30% tumour-bearing animals (1986)
8-Hydroxy
Table 94. (continued)
Compound Metabolite Species, route of Type of Results References
administration investigation,
duration, dose
5-Methylcholanthrene 1,2-Diol Mouse, dermal Initiation, 1,2-Diol: 19/20 papillomas, 7/20 Hecht et al.
7,8-Diol 3 µg/animal, carcinomas; 7,8-diol: 10/20 (1980)
9,10-Diol 10 × in 20 days papillomas, no carcinomas; 9,10-diol:
no papillomas, no carcinomas
5-Hydroxy Mouse, dermal Initiation 45-950% with skin tumours; solvent Amin et al.
control: 5% (1981)
1,2-Diol- Mouse, newborn Carcinogenicity Induction of pulmonary tumours Amin et al.
3,4-epoxide (1991b)
Phenanthrene 1,2-Diol, Mouse, dermal Initiation Very weakly positive Wood et al.
3,4-Diol (1979)
9,10-Diol
1,2-Diol- Mouse, newborn Carcinogenicity No induction of pulmonary tumours Buening et al.
3,4-epoxide (1979)
Cytochrome P450-dependent metabolizing activity is low in skin. When
14C-benzo [a]pyrene was incubated with arachidonic acid and cytosol
prepared from rat, mouse, or human epidermis, benzo [a]pyrene 1,6-,
3,6-, and 6,12-quinones and other metabolites were formed. These
metabolic reactions were inhibited by selective inhibitors of
lipoxygenase, demonstrating that human and rodent skin can metabolize
benzo [a]pyrene through an arachidonic acid-dependent lipoxygenase
pathway (Agarwal & Mukhtar, 1991). In cultures of normal human
melanocytes, benzo [a]pyrene diols and small amounts of quinones and
phenols were detected in the fraction extractable in organic solvents,
and glucuronide and sulfate conjugates in the water-soluble fraction
(Agarwal et al., 1991).
The (+) anti-7,8-dihydrodiol-9,10-epoxide of benzo [a]pyrene reacts
with DNA to yield almost exclusively the deoxyguanosine adduct, in
which the epoxide function has reacted with the amino group at C2 of
the guanine base. In mouse skin, this one adduct accounts for about
97% of the total binding of benzo [a]pyrene to DNA (Jeffrey et al.,
1976).
Benzo [a]pyrene 7,8-dihydroxy-9,10-epoxide and two other metabolites
(which were detected by trapping with exogenous DNA as described by
Ginsberg & Atherholt, 1989) were present in all serum samples from
mice of three strains and Sprague-Dawley rats after intraperitoneal
injection of 50-200 mg/kg bw benzo [a]pyrene. It was concluded that
transport can occur via the systemic circulation (Garg et al., 1991).
Benzo [a]pyrene or its 4,5-oxide or 7,8-diol-9,10-epoxy metabolite
was administered directly into Swiss mouse embryos at 0.1-4 µg per
embryo on days 10, 12, and 14 of gestation. The 7,8-diol-9,10-epoxy
metabolite was the most potent embryotoxic and teratogenic compound in
fetuses examined on day 18, causing 85% embryolethality and 100%
malformations. Benzo [a]pyrene and the other metabolite did not
increase the incidence of malformations significantly (Barbieri et
al., 1986).
7.9.2 5-Methylchrysene
Investigations of the reaction of 5-methylchrysene diol epoxide
enantiomers with DNA bases and in S. typhimurium showed the
importance of both the absolute configuration and the position of the
methyl group. The 5-methyl-chrysene 1 R,2 S-diol-3 S,4 R-epoxide,
with the methyl group and epoxide ring in the same bay region, were
the most reactive (Melikian et al., 1988).
7.9.3 1-Methylphenanthrene
Incubation of rat liver preparations with 1-methylphenanthrene gave
rise to the 3,4- and 5,6-dihydrodiols of 1-hydroxymethylphenanthrene,
1-methylphenanthrene, 1-hydroxymethyl-phenanthrene, and unidentified
derivatives as metabolites; the dihydrodiols were mutagenic in the
presence of an exogenous metabolizing system (LaVoie et al., 1981b).
7.10 Mechanisms of carcinogenicity
7.10.1 History
In the early 1940s, theoretical chemistry was used to predict the
chemical reactivity of PAH. Pullman (1945,1947) introduced the terms
K- and L-region to describe the reactivity of PAH based on Hückel
molecular orbital calculations (see Figure 9). Later, the term 'bay
region' was introduced for molecular substructures that contribute to
the formation of some ultimate carcinogens. Discrete values of complex
delocalization energies at the K- and L-regions of a PAH were
correlated with its carcinogenic potency. At the time, however, there
was only a limited database on metabolic processes and little
experimental confirmation, and the K-region theory was later found to
be incompatible with the results of experimental work.
7.10.2 Current theories
Miller & Miller (1977) proposed the theory of reactive electrophiles
in chemical carcinogenesis. According to this theory, PAH are
activated by microsomal enzymes to proximate and finally ultimate
carcinogens, which are characterized by an electrophilic centre that
can react with nucleophilic sites on macromolecules such as DNA, RNA,
and protein.
After the discovery that diol epoxides are metabolites of PAH (Sims &
Grover, 1974), a theoretical model was presented by Jerina et al.
(1976), who found that synthesized arene oxides were mutagenic without
metabolic activation. The bay-region theory states two prerequisites
for carcinogenic potency: The epoxide group of an ultimate metabolite
must be part of a bay region (see Figure 10), and the hydroxy groups
of the diol epoxide are preferentially located in the 'pre-bay
region'. The presence of the epoxide group on a saturated benzene ring
in a bay region facilitates ring opening, i.e. the delocalization
energy forming the carbonium ion is higher. This is important for
reactions with DNA via a carbonium ion, which is an alkylating agent.
For example, the metabolic pathway of benzo [a]pyrene is hypothesized
to start with a 7,8 oxidation followed by hydrolysis to
7,8-dihydrodiol, and terminated by 9,10-oxidation, yielding the
ultimate carcinogen 7,8-dihydrodiol-9,10-epoxide. Calculation of the
carbonium ion delocalization energies by the pertubational molecular
orbital method results in a rough correlation with experimentally
determined carcinogenic potency.
The number of PAH tested for carcinogenicity in experimental animals
doubled between the time that Jerina et al. (1976) published their
work and 1980, to 50 compounds. A calculation of the carbonium ion
delocalization energies at that time (Qianhuan, 1980) revealed a
deviation from the energy-carcinogenicity correlation for compounds
that had not been investigated by Jerina et al. To avoid the
shortcomings of the bay-region theory, Qianhuan took into account the
data on all PAH that had been tested for carcinogenicity and
postulated the di-region theory, a bifunctional electrophilic theory
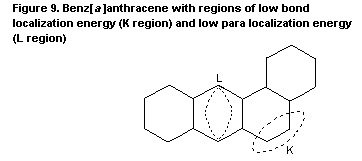
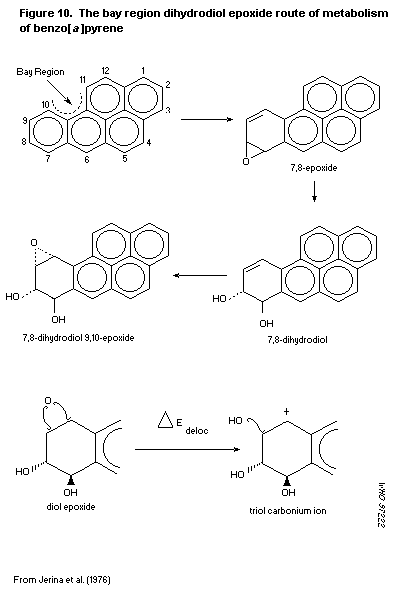 based on the assumption that formation of two carbonium ions on the
same PAH is responsible for carcinogenic activity. A quantitative
equation involving the delocalization energies of the twin active
regions was deduced. Principally, all of the already defined key
regions of PAH (M, E, K, and L; see Figure 11) were used but with
different implications. In this theory, the metabolic activation of
PAH is dependent on two factors, a geometric and an energy factor. The
angular ring, the subangular ring, and an active K region play
decisive roles in carcinogenic potency, and two adequately active,
adjacent regions are required.
PAH are proposed to exert carcinogenicity mainly by DNA complementary
cross-linking. Qualitative and quantitative data have been presented
on the mechanism of formation of PAH-DNA adducts in the radical cation
theory. PAH with relatively low ionization potential, which are the
most potent carcinogens, are activated via cytochrome P450 by
one-electron oxidation (radical cation), whereas PAH with relatively
high ionization potential are activated by mono-oxygenation
(bay-region diol epoxide). In experiments in rat liver microsomes in
which potential DNA adducts were synthesized and used as standards,
four depurination products (one-electron oxidation) and one stable
product (diol epoxide pathway) of benzo [a]pyrene were detected
(Cavalieri & Rogan, 1985; Cavalieri et al., 1993; Rogan et al., 1993).
In a review of the evidence for the four mechanisms of PAH
carcinogenesis, namely, the diol epoxide mechanism, the radical-cation
mechanism, the quinone mechanism, and the benzylic oxidation
mechanism, Harvey (1996) concluded that current research provided
evidence for all four.
7.10.3 Theories under discussion
A molecular geometrical model has been proposed in which the
carcinogenic potency of PAH is predicted from the centre(s) of highest
chemical or biochemical reactivity, with the hypothesized introduction
of a methyl group into the PAH. A good correlation was found between
the predicted carcinogenicity of a series of 50 unsubstituted PAH and
the results found in rats and mice. Bioalkylation is suggested to be
catalysed by cytosolic methyl-transferase with S-adenosyl-L-
methionine. In an experiment to confirm the model, rats were given
subcutaneous injections of benz [a]anthracene, and 24 h later the
tissue at the application site was homogenized and the assumed
metabolites analysed by high-performance liquid chromatography, gas
chromatography, and mass spectrometry. The bioalkylation product
7-methyl-benz [a]anthracene was identified. Six noncarcinogenic
hydrocarbons did not yield alkylated metabolites in this experimental
approach. The authors concluded that bioalkylation, preferably in the
meso-anthranic centres of high reactivity, is a structural
prerequisite of carcinogenicity (Flesher & Myers, 1990, 1991; see also
section 6.6.2).
The binding of 1-hydroxymethylpyrene to DNA after intraperitoneal
injection to rats was similar to the adduct pattern of its active
metabolites 1-hydroxymethylpyrene sulfate and 1-chloromethylpyrene
with isolated DNA, suggesting secondary activation of
hydroxymethyl-PAH sulfates to chloromethyl-PAH (Monnerjahn et al.,
1993).
Transformation of PAH via their proximate carcinogens (e.g.
benzo [a]-pyrene 7,8-diol) to the ultimate carcinogens (e.g.
benzo [a]pyrene 7,8-diol-9,10-epoxide) is reported to be mediated by
cytochrome P450 enzymes; however, two pathways unrelated to P450 have
also been discussed. Peroxidase enzymes can transform benzo [a]pyrene
7,8-diol to benzo [a]pyrene 7,8-diol-9,10-epoxide, but the process
requires the presence of superoxide anions, hydrogen peroxides, and
hydroxyl radicals produced by polymorphonuclear cells. This reaction
was demonstrated in mouse skin after topical treatment with
12- O-tetradecanoylphorbol 13-acetate. The catalytic activity of
myeloperoxi-dase enhances the reactivity of oxygen species. This
alternative mechanism may be important for human exposure to PAH
because simultaneous chronic inflammation (e.g. due to smoking) often
leads to increased numbers of inflammatory cells (Marnett et al.,
1978; Kensler et al., 1987; Ji & Marnett, 1992).
Epoxidation of benzo [a]pyrene 7,8-diol has also been reported to be
mediated by lipoxygenase (Hughes et al., 1989; Agarwal & Muhktar,
1991; see also section 7.9).
7.10.3.1 Acenaphthene and acenaphthylene
B6C3F1 mice were given a single intraperitoneal injection of 300
mg/kg bw acenaphthene or acenaphthylene. Acenaphthylene caused a
> 80-fold induction of hepatic microsomal methoxyresoforin
O-deethylase activity, dependent on the Cyp1a2 gene, which codes for
an enzyme that catalyses the oxidative metabolism of diverse
substrates; acenaphthene increased the activity by > 20-fold. The
tricyclic PAH acenaphthene, acenaphthylene, anthracene, fluorane, and
phenanthrene were not competitive inhibitors at the mouse hepatic
cytosolic Ah receptor when tested together with 3H-labelled
1,2,7,8-tetrachlorodibenzo- para-dioxin or benzo [a]pyrene. The
authors suggested an association between the relatively nontoxic
behaviour of the tricyclic PAH and the observed Ah
receptor-independent induction of hepatic Cyp1a2 expression
(Chaloupka et al., 1994).
7.10.3.2 Anthracene
A single intraperitoneal injection of 300 mg/kg bw anthracene to
B6C3F1 mice caused a > 10-fold induction of hepatic microsomal
methoxyresofurin O-deethylase (Chaloupka et al., 1994).
7.10.3.3 Benzo [a]pyrene
The toxic effects of benzo [a]pyrene in mice vary according to their
genetic constitution (see also section 7.5). The crucial point appears
to be the Ah locus, which determines the inducibility of AHH. For
example, administration of benzo [a]pyrene at 120 mg/kg bw per day in
the diet induced aplastic anaemia and death in nonresponsive AKR/N
mice (Ahd/ Ahdtype) within four weeks, with hypocellular bone
marrow, myeloid precursors, and promegakaryocytes; responsive AKR/N
mice (Ahb/ Ahbtype), however, were still healthy after six
months. In contrast, when benzo [a]pyrene was given intraperitoneally
at 500 mg/kg bw per day, responsive mice survived for a significantly
shorter time than nonresponsive mice (Robinson et al., 1975). In order
to explain these differences, Nebert et al. (1977) proposed that the
gastrointestinal tract and liver of responsive mice have a greater
capacity to detoxify an orally administered dose; however, if
benzo [a]pyrene reaches their bone marrow and other distal tissues,
metabolism there leads to increased formation of toxic metabolites.
Mice with high-affinity Ah receptors showed no myelotoxicity after
administration of 120 mg/kg bw per day benzo [a]pyrene in the diet
for six months, but non-responsive mice at the same dose died within
three weeks due to myelotoxic effects (Legraverand et al., 1983).
After two oral administrations of 10 or 100 mg/kg bw benzo [a]pyrene,
the numbers of sister chromatid exchanges and DNA adducts were
significantly higher in AHH-non-inducible DBA/2 mice than in inducible
C57B1/6 mice (Wielgosz et al., 1991).
Because PAH produce tumours at the site of administration, it was
suggested that they do not require metabolic activation; however, it
was shown later that activation occurs in the target tissue. The
arylkylating agent, for example the 7,8-dihydrodiol-9,10-epoxide of
benzo [a]pyrene, reacts with DNA to yield almost exclusively the
deoxyguanosine adduct. Methyl groups reaching into the bay region can
enhance carcinogenic potency by steric effects (Dipple et al., 1990).
Topical application of the prostaglandin synthetase inhibitor
indomethacin after administration of benzo [a]pyrene to mice delayed
the onset and reduced the size of the skin tumours. It was assumed
that prostaglandin-induced suppression of cellular cutaneous immunity
plays a role in carcinogenesis, as indomethacin can partially restore
cutaneous immunity (Andrews et al., 1991).
7.10.3.4 Benz [a]anthracene
Benz [a]anthracene was not tumorigenic after intravenous or
intramuscular injection in rats. The methyl substituant was shown to
be of great importance in the carcinogenicity of this compound, as
derivatives were highly carcinogenic when they possessed two or three
methyl groups in any combination at position 6, 7, 8, or 12 (Pataki &
Huggins, 1969).
7.10.3.5 Benzo [c]phenanthrene
Benzo [c]phenanthrene is unique among the PAH in that it has no bay
region as such but has a 'fjord' region between positions 1 and 12
(see Figure 12). The synthesized metabolite 3,4-dihydrodiol (but not
the 1,2 or 5,6 derivative) was as mutagenic in the presence of liver
microsomes as the parent compound (Croisy-Delcey et al., 1979).
The four fjord diol epoxides of benzo [c]phenanthrene are very active
tumour initiators. In studies of their reactions with DNA, each diol
epoxide became bound covalently to DNA and showed a unique product
distribution, with either a preference for reaction with
deoxyadenosine residues or a more even distribution between
deoxyguanosine and deoxyadenosine residues. A remarkable feature is
the efficiency of covalent binding to DNA relative to DNA-catalysed
hydrolysis. The authors reported a strong association between
reactivity with adenine in DNA and tumour-initiating activity.
Interaction with DNA can lead directly to activation of the
ras protooncogene and to turnout initiation (Dipple et al., 1987).
In cultures of cells from embryos of Sencar mice, Syrian hamsters, and
Wistar rats, > 74% of all benzo [c]phenanthrene-deoxyribonucleoside
adducts resulted from the 1 R,2 S-epoxy-3 S,4 R; deoxyadenosine
and deoxyguanosine adducts were formed at a ratio of 3:1. The absolute
configuration of the major metabolite and preference for adenosine
residues have been found to be typical for other potent carcinogens
(Pruess-Schwartz et al., 1987).
7.10.3.6 Chrysene
The metabolism of the weak carcinogen chrysene has been investigated
in the presence and absence of other xenobiotics. The putative
ultimate carcinogenic form of chrysene is the 1,2-dihydroxy-3,4-epoxy-
1,2,3,4-tetrahydro metabolite, formed by the inducible cytochrome P450
system. In rats treated with benzo [a]pyrene, benzo [b]fluoranthene,
or benzo [j]fluoranthene, 1,2- and 3,4-oxidation were highly induced,
and the 1,2,3-triol metabolite was produced, which is a derivative of
the 1,2-dihydrodiol-3,4-epoxide. In the absence of induction, chrysene
may not be metabolized to the ultimate carcinogen (Jacob et al.,
1982b).
7.10.3.7 Cyclopenta [cd]pyrene
For most PAH, an initial epoxidation step catalysed by cytochrome
P450-dependent mono-oxygenases is followed by a second epoxidation;
however, cyclopenta [cd]pyrene has no bay region (Figure 13) and has
been suggested to be activated by a single epoxidation at the
cyclopenteno double bond, which may be possible in systems that
generate peroxyl radicals. Reed et al. (1988) found that peroxyl
radicals could activate the mutagenic potential of
cyclopenta [cd]pyrene. This compound is a member of a subclass of PAH
that have a non-aromatic double bond, which may form the centre for
conversion to an ultimate mutagen.
7.10.3.8 Fluorene
A single intraperitoneal injection of 300 mg/kg bw fluorene to B6C3F1
mice caused a greater than fivefold induction of hepatic microsomal
methoxyresofurin O-deethylase activity (Chaloupka et al., 1994; see
also section 7.10.3.1).
7.10.3.9 Indeno[1,2,3- cd]pyrene
The 1,2-diol of indeno[1,2,3- cd]pyrene and its epoxide precursor,
1,2-oxide were found to have similar carcinogenic potency in an
initiation assay. This result is remarkable, because K-region
dihydrodiols such as the 1,2-diol are generally considered to be
detoxification products formed by hydrolysis of K-region oxides.
Further metabolic activation of the 1,2-diol via epoxidation in the
7-10 area was proposed because 8- and 9-hydroxy
indeno[1,2,3- cd]pyrene had been detected as metabolites. If
substantiated, this would be a unique activation mechanism for PAH
(Rice et al., 1990).
7.10.3.10 5-Methylchrysene
The 5-methyl compound was the most tumorigenic of the methylchrysenes,
probably owing to the presence of the methyl group in the same bay
region as the epoxide ring (Hecht et al., 1987). Specific dihydrodiol
epoxides of 5-methylchrysene are formed from their precursor
dihydrodiols after topical application of 5-methylchrysene to mouse
epidermis or injection into newborn mice. (±)- trans-1,2-Dihydroxy-
anti-3,4-epoxy-1,2,3,4-tetrahydro-5-methyl-chrysene was found to be
the ultimate carcinogen (Hecht et al., 1985).
5-Methylchrysene 1,2-diol and the 1,2-diol-3,4-epoxide are major
proximate and ultimate carcinogens; the corresponding
6-methylchrysene-1,2-diol is also a major metabolite but is much less
tumorigenic, perhaps because of the different activity of the
corresponding 3,4-epoxides. The formation of epoxide-type adducts from
6-methylchrysene was only 5% of that observed for 5-methylchrysene
(Amin et al., 1985b). In an investigation of the stereoselectivity of
the metabolic activation of 5- and 6-methylchrysene in mouse skin
in vivo and in rat and mouse liver in vitro, using the resolved
enantiomers as reference compounds, the R,R-enantiomers predominated
(> 90 %), and 5-methyl-chrysene-1 R,2 R-diol was the most
tumorigenic compound in an initiation test (Amin et al., 1987).
5-Methylchrysene is uniquely tumorigenic among the monomethylchrysene
isomers. Its activity is due mainly to the highly tumorigenic diol
epoxide, which has a methyl group and an epoxide ring in the same bay
region. Of the isomers, only 5-methylchrysene can form this type of
'methyl bay region diol epoxide' (Figure 14). Substitutions that
inhibit its formation lead to a decrease in tumorigenicity (Amin et
al., 1990).
based on the assumption that formation of two carbonium ions on the
same PAH is responsible for carcinogenic activity. A quantitative
equation involving the delocalization energies of the twin active
regions was deduced. Principally, all of the already defined key
regions of PAH (M, E, K, and L; see Figure 11) were used but with
different implications. In this theory, the metabolic activation of
PAH is dependent on two factors, a geometric and an energy factor. The
angular ring, the subangular ring, and an active K region play
decisive roles in carcinogenic potency, and two adequately active,
adjacent regions are required.
PAH are proposed to exert carcinogenicity mainly by DNA complementary
cross-linking. Qualitative and quantitative data have been presented
on the mechanism of formation of PAH-DNA adducts in the radical cation
theory. PAH with relatively low ionization potential, which are the
most potent carcinogens, are activated via cytochrome P450 by
one-electron oxidation (radical cation), whereas PAH with relatively
high ionization potential are activated by mono-oxygenation
(bay-region diol epoxide). In experiments in rat liver microsomes in
which potential DNA adducts were synthesized and used as standards,
four depurination products (one-electron oxidation) and one stable
product (diol epoxide pathway) of benzo [a]pyrene were detected
(Cavalieri & Rogan, 1985; Cavalieri et al., 1993; Rogan et al., 1993).
In a review of the evidence for the four mechanisms of PAH
carcinogenesis, namely, the diol epoxide mechanism, the radical-cation
mechanism, the quinone mechanism, and the benzylic oxidation
mechanism, Harvey (1996) concluded that current research provided
evidence for all four.
7.10.3 Theories under discussion
A molecular geometrical model has been proposed in which the
carcinogenic potency of PAH is predicted from the centre(s) of highest
chemical or biochemical reactivity, with the hypothesized introduction
of a methyl group into the PAH. A good correlation was found between
the predicted carcinogenicity of a series of 50 unsubstituted PAH and
the results found in rats and mice. Bioalkylation is suggested to be
catalysed by cytosolic methyl-transferase with S-adenosyl-L-
methionine. In an experiment to confirm the model, rats were given
subcutaneous injections of benz [a]anthracene, and 24 h later the
tissue at the application site was homogenized and the assumed
metabolites analysed by high-performance liquid chromatography, gas
chromatography, and mass spectrometry. The bioalkylation product
7-methyl-benz [a]anthracene was identified. Six noncarcinogenic
hydrocarbons did not yield alkylated metabolites in this experimental
approach. The authors concluded that bioalkylation, preferably in the
meso-anthranic centres of high reactivity, is a structural
prerequisite of carcinogenicity (Flesher & Myers, 1990, 1991; see also
section 6.6.2).
The binding of 1-hydroxymethylpyrene to DNA after intraperitoneal
injection to rats was similar to the adduct pattern of its active
metabolites 1-hydroxymethylpyrene sulfate and 1-chloromethylpyrene
with isolated DNA, suggesting secondary activation of
hydroxymethyl-PAH sulfates to chloromethyl-PAH (Monnerjahn et al.,
1993).
Transformation of PAH via their proximate carcinogens (e.g.
benzo [a]-pyrene 7,8-diol) to the ultimate carcinogens (e.g.
benzo [a]pyrene 7,8-diol-9,10-epoxide) is reported to be mediated by
cytochrome P450 enzymes; however, two pathways unrelated to P450 have
also been discussed. Peroxidase enzymes can transform benzo [a]pyrene
7,8-diol to benzo [a]pyrene 7,8-diol-9,10-epoxide, but the process
requires the presence of superoxide anions, hydrogen peroxides, and
hydroxyl radicals produced by polymorphonuclear cells. This reaction
was demonstrated in mouse skin after topical treatment with
12- O-tetradecanoylphorbol 13-acetate. The catalytic activity of
myeloperoxi-dase enhances the reactivity of oxygen species. This
alternative mechanism may be important for human exposure to PAH
because simultaneous chronic inflammation (e.g. due to smoking) often
leads to increased numbers of inflammatory cells (Marnett et al.,
1978; Kensler et al., 1987; Ji & Marnett, 1992).
Epoxidation of benzo [a]pyrene 7,8-diol has also been reported to be
mediated by lipoxygenase (Hughes et al., 1989; Agarwal & Muhktar,
1991; see also section 7.9).
7.10.3.1 Acenaphthene and acenaphthylene
B6C3F1 mice were given a single intraperitoneal injection of 300
mg/kg bw acenaphthene or acenaphthylene. Acenaphthylene caused a
> 80-fold induction of hepatic microsomal methoxyresoforin
O-deethylase activity, dependent on the Cyp1a2 gene, which codes for
an enzyme that catalyses the oxidative metabolism of diverse
substrates; acenaphthene increased the activity by > 20-fold. The
tricyclic PAH acenaphthene, acenaphthylene, anthracene, fluorane, and
phenanthrene were not competitive inhibitors at the mouse hepatic
cytosolic Ah receptor when tested together with 3H-labelled
1,2,7,8-tetrachlorodibenzo- para-dioxin or benzo [a]pyrene. The
authors suggested an association between the relatively nontoxic
behaviour of the tricyclic PAH and the observed Ah
receptor-independent induction of hepatic Cyp1a2 expression
(Chaloupka et al., 1994).
7.10.3.2 Anthracene
A single intraperitoneal injection of 300 mg/kg bw anthracene to
B6C3F1 mice caused a > 10-fold induction of hepatic microsomal
methoxyresofurin O-deethylase (Chaloupka et al., 1994).
7.10.3.3 Benzo [a]pyrene
The toxic effects of benzo [a]pyrene in mice vary according to their
genetic constitution (see also section 7.5). The crucial point appears
to be the Ah locus, which determines the inducibility of AHH. For
example, administration of benzo [a]pyrene at 120 mg/kg bw per day in
the diet induced aplastic anaemia and death in nonresponsive AKR/N
mice (Ahd/ Ahdtype) within four weeks, with hypocellular bone
marrow, myeloid precursors, and promegakaryocytes; responsive AKR/N
mice (Ahb/ Ahbtype), however, were still healthy after six
months. In contrast, when benzo [a]pyrene was given intraperitoneally
at 500 mg/kg bw per day, responsive mice survived for a significantly
shorter time than nonresponsive mice (Robinson et al., 1975). In order
to explain these differences, Nebert et al. (1977) proposed that the
gastrointestinal tract and liver of responsive mice have a greater
capacity to detoxify an orally administered dose; however, if
benzo [a]pyrene reaches their bone marrow and other distal tissues,
metabolism there leads to increased formation of toxic metabolites.
Mice with high-affinity Ah receptors showed no myelotoxicity after
administration of 120 mg/kg bw per day benzo [a]pyrene in the diet
for six months, but non-responsive mice at the same dose died within
three weeks due to myelotoxic effects (Legraverand et al., 1983).
After two oral administrations of 10 or 100 mg/kg bw benzo [a]pyrene,
the numbers of sister chromatid exchanges and DNA adducts were
significantly higher in AHH-non-inducible DBA/2 mice than in inducible
C57B1/6 mice (Wielgosz et al., 1991).
Because PAH produce tumours at the site of administration, it was
suggested that they do not require metabolic activation; however, it
was shown later that activation occurs in the target tissue. The
arylkylating agent, for example the 7,8-dihydrodiol-9,10-epoxide of
benzo [a]pyrene, reacts with DNA to yield almost exclusively the
deoxyguanosine adduct. Methyl groups reaching into the bay region can
enhance carcinogenic potency by steric effects (Dipple et al., 1990).
Topical application of the prostaglandin synthetase inhibitor
indomethacin after administration of benzo [a]pyrene to mice delayed
the onset and reduced the size of the skin tumours. It was assumed
that prostaglandin-induced suppression of cellular cutaneous immunity
plays a role in carcinogenesis, as indomethacin can partially restore
cutaneous immunity (Andrews et al., 1991).
7.10.3.4 Benz [a]anthracene
Benz [a]anthracene was not tumorigenic after intravenous or
intramuscular injection in rats. The methyl substituant was shown to
be of great importance in the carcinogenicity of this compound, as
derivatives were highly carcinogenic when they possessed two or three
methyl groups in any combination at position 6, 7, 8, or 12 (Pataki &
Huggins, 1969).
7.10.3.5 Benzo [c]phenanthrene
Benzo [c]phenanthrene is unique among the PAH in that it has no bay
region as such but has a 'fjord' region between positions 1 and 12
(see Figure 12). The synthesized metabolite 3,4-dihydrodiol (but not
the 1,2 or 5,6 derivative) was as mutagenic in the presence of liver
microsomes as the parent compound (Croisy-Delcey et al., 1979).
The four fjord diol epoxides of benzo [c]phenanthrene are very active
tumour initiators. In studies of their reactions with DNA, each diol
epoxide became bound covalently to DNA and showed a unique product
distribution, with either a preference for reaction with
deoxyadenosine residues or a more even distribution between
deoxyguanosine and deoxyadenosine residues. A remarkable feature is
the efficiency of covalent binding to DNA relative to DNA-catalysed
hydrolysis. The authors reported a strong association between
reactivity with adenine in DNA and tumour-initiating activity.
Interaction with DNA can lead directly to activation of the
ras protooncogene and to turnout initiation (Dipple et al., 1987).
In cultures of cells from embryos of Sencar mice, Syrian hamsters, and
Wistar rats, > 74% of all benzo [c]phenanthrene-deoxyribonucleoside
adducts resulted from the 1 R,2 S-epoxy-3 S,4 R; deoxyadenosine
and deoxyguanosine adducts were formed at a ratio of 3:1. The absolute
configuration of the major metabolite and preference for adenosine
residues have been found to be typical for other potent carcinogens
(Pruess-Schwartz et al., 1987).
7.10.3.6 Chrysene
The metabolism of the weak carcinogen chrysene has been investigated
in the presence and absence of other xenobiotics. The putative
ultimate carcinogenic form of chrysene is the 1,2-dihydroxy-3,4-epoxy-
1,2,3,4-tetrahydro metabolite, formed by the inducible cytochrome P450
system. In rats treated with benzo [a]pyrene, benzo [b]fluoranthene,
or benzo [j]fluoranthene, 1,2- and 3,4-oxidation were highly induced,
and the 1,2,3-triol metabolite was produced, which is a derivative of
the 1,2-dihydrodiol-3,4-epoxide. In the absence of induction, chrysene
may not be metabolized to the ultimate carcinogen (Jacob et al.,
1982b).
7.10.3.7 Cyclopenta [cd]pyrene
For most PAH, an initial epoxidation step catalysed by cytochrome
P450-dependent mono-oxygenases is followed by a second epoxidation;
however, cyclopenta [cd]pyrene has no bay region (Figure 13) and has
been suggested to be activated by a single epoxidation at the
cyclopenteno double bond, which may be possible in systems that
generate peroxyl radicals. Reed et al. (1988) found that peroxyl
radicals could activate the mutagenic potential of
cyclopenta [cd]pyrene. This compound is a member of a subclass of PAH
that have a non-aromatic double bond, which may form the centre for
conversion to an ultimate mutagen.
7.10.3.8 Fluorene
A single intraperitoneal injection of 300 mg/kg bw fluorene to B6C3F1
mice caused a greater than fivefold induction of hepatic microsomal
methoxyresofurin O-deethylase activity (Chaloupka et al., 1994; see
also section 7.10.3.1).
7.10.3.9 Indeno[1,2,3- cd]pyrene
The 1,2-diol of indeno[1,2,3- cd]pyrene and its epoxide precursor,
1,2-oxide were found to have similar carcinogenic potency in an
initiation assay. This result is remarkable, because K-region
dihydrodiols such as the 1,2-diol are generally considered to be
detoxification products formed by hydrolysis of K-region oxides.
Further metabolic activation of the 1,2-diol via epoxidation in the
7-10 area was proposed because 8- and 9-hydroxy
indeno[1,2,3- cd]pyrene had been detected as metabolites. If
substantiated, this would be a unique activation mechanism for PAH
(Rice et al., 1990).
7.10.3.10 5-Methylchrysene
The 5-methyl compound was the most tumorigenic of the methylchrysenes,
probably owing to the presence of the methyl group in the same bay
region as the epoxide ring (Hecht et al., 1987). Specific dihydrodiol
epoxides of 5-methylchrysene are formed from their precursor
dihydrodiols after topical application of 5-methylchrysene to mouse
epidermis or injection into newborn mice. (±)- trans-1,2-Dihydroxy-
anti-3,4-epoxy-1,2,3,4-tetrahydro-5-methyl-chrysene was found to be
the ultimate carcinogen (Hecht et al., 1985).
5-Methylchrysene 1,2-diol and the 1,2-diol-3,4-epoxide are major
proximate and ultimate carcinogens; the corresponding
6-methylchrysene-1,2-diol is also a major metabolite but is much less
tumorigenic, perhaps because of the different activity of the
corresponding 3,4-epoxides. The formation of epoxide-type adducts from
6-methylchrysene was only 5% of that observed for 5-methylchrysene
(Amin et al., 1985b). In an investigation of the stereoselectivity of
the metabolic activation of 5- and 6-methylchrysene in mouse skin
in vivo and in rat and mouse liver in vitro, using the resolved
enantiomers as reference compounds, the R,R-enantiomers predominated
(> 90 %), and 5-methyl-chrysene-1 R,2 R-diol was the most
tumorigenic compound in an initiation test (Amin et al., 1987).
5-Methylchrysene is uniquely tumorigenic among the monomethylchrysene
isomers. Its activity is due mainly to the highly tumorigenic diol
epoxide, which has a methyl group and an epoxide ring in the same bay
region. Of the isomers, only 5-methylchrysene can form this type of
'methyl bay region diol epoxide' (Figure 14). Substitutions that
inhibit its formation lead to a decrease in tumorigenicity (Amin et
al., 1990).
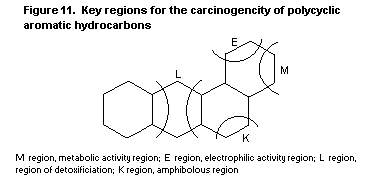
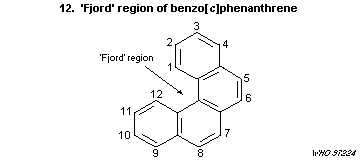
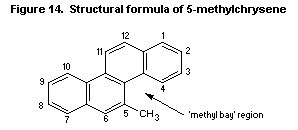 The tumorigenicity of racemic anti-1,2-diol-3,4-epoxides of
chrysene, 5-methyl-, 5-ethyl-, and 5-propylchrysene was determined in
newborn mice. Only the 5-methyl compound was highly tumorigenic,
demonstrating the importance of molecular shape for tumorigenicity. A
methyl group in the same bay region as the epoxide ring leads to
exceptional activity, and this may occur a consequence of DNA adduct
conformation (Amin et al., 1991b).
In a study of the binding of 3H-labelled anti-5- and
anti-6-methylchrysene-1,2-diol-3,4-epoxide to DNA in liver and lung
of newborn mice after intraperitoneal administration on day 1 of life
and sacrifice after 24 h, 1.1 pmol/mg DNA were found with the
benzo [a]pyrene analogue, 0.5 pmol/mg DNA with the 5-methyl compound,
and < 0.01 pmol/mg DNA with the 6-methyl compound, consistent with
their known tumorigenic activities. When the parent compounds were
tested in the same protocol, however, little radiolabel became
associated with DNA adducts. Hence, it can be concluded that the
dihydrodiols are the carcinogens in newborn mice (Melikian et al.,
1991).
The metabolism of 5-methylchrysene has also been investigated in mouse
epidermis in vivo. The diol precursors of 1,2-dihydroxy-3,4-epoxy
5-methylchrysene and 7,8-dihydroxy-9,10-epoxy 5-methylchrysene were
present in equivalent quantities at every time, and the ratio of DNA
adducts with the two precursors was constant over time (Melikian et
al., 1983).
In a study of the correlation of the 5-methylchrysene-DNA adduct
profile in lung tissue with the spectrum of mutations in the K- ras
protooncogene of lung tumours, up to 200 mg/kg bw 5-methylchrysene
were administered to A/J mice and the lungs were analysed for DNA
adducts one to three days later. After a latent period of eight
months, 90% all lung tumours had mutations of K- ras.
N2-Deoxyguanosine was detected as a possible promutagenic adduct
(You et al., 1994).
7.10.3.11 1-Methylphenanthrene
An investigation of an extensive series of alkylated phenanthrenes
suggests that the presence of a methyl substituent at, or adjacent to,
the K region (9,10 position) and an unsubstituted angular ring
adjacent to a free peri position are the prerequisites for mutagenic
activity in S. typhimurium. Substitution at the peri position was
associated with lack of mutagenicity. A non-K-region dihydrodiol
derived from 1-methylphenanthrene was a potent proximate mutugenic
metabolite (LaVoie et al., 1983b).
7.10.3.12 Naphthalene
Severe bronchiolar epithelial-cell necrosis was reported in mice after
intraperitoneal injection of naphthalene. Lung, liver, and kidney
macromolecules were shown by a radiolabelling technique to be the main
targets. Maximal binding of naphthalene was found 2-4 h after
application, and a threshold was found at 200-400 mg/kg bw,
corresponding to glutathione depletion. Covalent binding was highest
in tissues with high cytochrome P450 mono-oxygenase activity, i.e.
lung, liver, and kidney (Warren et al., 1982).
7.10.3.13 Phenanthrene
A single dose of 300 mg/kg of phenanthrene administered
intraperitoneally to B6C3F1 mice caused > 20-fold induction of
hepatic microsomal methoxy-resofurin O-deethylase activity
(Chaloupka et al., 1994; see also section 7.10.3.1).
7.10.3.14 Investigations of groups of PAH
In rats, expression of the Cyp1A1 gene is closely associated with
the inducibility of AHH, an enzyme important for bioactivation of PAH.
Cyp1A1 is regulated by several factors, including the Ah receptor
and the cytosolic 4S PAH-binding protein. The role of the latter
protein was investigated with benzo [a]pyrene and benzo [e]pyrene in
H4-II-E rat hepatoma cells. Both induced gene expression, as measured
by ethoxyresorufin O-deethylase activity, but benzo [a]pyrene was
about 25 times more potent than benzo [e]pyrene. Benzo [a]pyrene
binds to both the Ah receptor and the 4 S protein, and
benzo [e]pyrene only to the protein (Houser et al., 1992).
A series of PAH were investigated in a novel short-term test for the
detection of carcinogens, the initiator tRNA acceptance assay.
Positive responses, i.e. > 15% stimulation, were induced by chrysene,
benzo [c]-phenanthrene, dibenz [a,h]anthracene, benzo [a]pyrene,
and dibenzo [a,i]pyrene; and negative responses were induced by
naphthalene, anthracene, phenanthrene, pyrene, benz [a]anthracene,
benzo [e]pyrene, perylene, and coronene. All of the potent
carcinogens were active in this assay (Hradec et al., 1990).
In a study of the non-carcinogenic PAH anthracene, fluorene, and
naphthalene and several carcinogenic amine derivatives in rats,
naphthalene failed to induce induce the cytochrome P450-dependent
mixed-function oxidases, whereas anthracene was a weak and fluorene an
effective inducer. A relationship was found between carcinogenicity
and the ability to induce hepatic P450 activity. It was assumed that
fluorene is not carcinogenic because it cannot form mutagenic
intermediates (Ayrton et al., 1990).
In a study of the induction of various PAH of the mono-oxygenase
isoenzymes in mouse liver microsomes, the cytochrome P448 and P450
groups were classified by using specific inhibitors in studies of
7-ethoxycoumarin activity. According to the pattern of enzymes
induced, the following groups were distinguished: (i) P448 type,
including dibenz [a,h]anthracene and benzo [k]fluoranthene; (ii)
mixed type, including pyrene, benzo [j]fluoranthene, and
benzo [e]pyrene, in which two inhibitors acted on the enzyme
reaction; and (iii) special P448 type consisting of
indeno[1,2,3- cd]pyrene, in which one inhibitor stimulated the
reaction. The PAH investigated were not a homogeneous group of
selective P448 inducers. No correlation was found with mutagenic
potency (Kemena et al., 1988).
8. EFFECTS ON HUMANS
Appraisal
There is little information on human exposure to single, pure
polycyclic aromatic hydrocarbons (PAH). That which is available
includes reports of accidental exposure to naphthalene and some data
from defined short-term studies of volunteers. All other reports are
of exposure to mixtures of PAH, which also contained other potentially
carcinogenic chemicals, in occupational and environmental situations.
Information on the health effects of these mixtures is confined to
their carcinogenic potential, for which there is evidence from a
number of epidemiological studies, especially for lung cancer and, in
some cases, cancers of the skin and of the urinary bladder. Since
single PAH and PAH mixtures are known to be carcinogenic in
experimental animals, it is plausible to attribute the enhanced cancer
risks seen in humans predominately to the PAH. In addition, the
results of epidemiological studies are important in risk assessment.
Therefore, in contrast to the preceding sections, the results of
studies of PAH mixtures are also presented.
Many workplaces have atmospheres with heavy loads of PAH. The cohorts
affected are gas workers, those exposed at coke ovens, wood
impregnation workers, people working at waste incinerators, workers in
bus garages, workers in nickel and copper refineries and in aluminium
smelters, asphalt workers, and chimney sweeps. Evaluation of the
immunocompetence of coke-oven workers indicated decreased serum
immunoglobulin levels and decreased immune function.
Biomarkers used to assess exposure to PAH include hydroxyphenanthrenes
and 1-hydroxypyrene in urine and DNA adducts in peripheral blood
lymphocytes.
8.1 Exposure of the general population
8.1.1 Naphthalene
Naphthalene is often used in houses as an insect repellant, mainly
against moths, and many incidents of poisoning have been reported.
Acute haemolytic anaemia is a typical systemic effect of oral, dermal,
or inhalation exposure. The lethal oral doses determined in cases of
accidental poisoning are 5-15 g for adults and 2 g within two days for
a six-year old child. Repeated exposure to naphthalene fumes or dust
has led to corneal ulceration, lenticular opacities, and cataracts
(Sandmeyer, 1981). Some case reports are described in more detail
below.
8.1.1.1 Poisoning incidents
(a) Oral exposure
Between 1949 and 1959, 10 cases of the oral poisoning by naphthalene
in children were documented in the United States. The amounts were
usually not specified but were in the order of grams. Some of the
children developed haemolytic anaemia (Anziulewicz et al., 1959).
The symptoms that developed after naphthalene intake included nausea,
vomiting, and convulsions after one to several days, often followed by
diarrhoea. Other symptoms were disturbances of consciousness,
lethargy, ataxia, and, in severe cases, coma and hemiplegia.
Haemolytic anaemia occurred concomitantly, with plasma haemoglobin
contents of up to 40%, often followed by haemoglobinuria. Mild to
severe jaundice can also occur; in one fatal case of poisoning, patchy
liver necrosis was reported. Treatment consists of blood transfusions
and additional alkalization of the urine; following this treatment,
rapid recovery, without persistent damage, was observed (Konar et al.,
1939). In five cases of acute haemolytic anaemia in children of about
two years of age who had eaten moth balls consisting of pure
naphthalene, there was complete recovery within one to four weeks
after transfusion (Zuelzer & Apt, 1949; Mackell et al., 1951).
Tests in vitro revealed that it was not naphthalene itself but its
metabolites a-naphthoquinone and a-naphthol that cause a decrease in
reduced glutathione in erythrocytes. Whole blood from patients
contained erythrocytes with defective glutathione metabolism. This
defect is observed in about 15% of black males and 2% of black females
(Zinkham & Childs, 1958).
(b) Dermal exposure
The effects of skin contact in sensitive individuals range from
irritation to severe dermatitis after exposure to quite small amounts
of naphthalene, such as wearing clothes that had been treated with
moth balls. Workers exposed to naphthalene may develop dermatitis on
their hands, arms, legs, and abdomen (Gerarde, 1960). Cases of
haemolytic anaemia have been reported in babies who absorbed
naphthalene from nappies that had been stored with moth balls
(Anziulewicz et al., 1959).
(c) Inhalation
Haemolytic anaemia was also observed in babies who had inhaled
naphthalene from moth ball-treated wool blankets (Valaes et al.,
1963). The case of a man with exfoliative dermatitis was reported,
which resolved after all contact with naphthalene was eliminated
(Fanburg, 1940).
8.1.1.2 Controlled studies
When the forearm skin of three volunteers was treated with anthracene
in a 2% benzene solution (dose not specified) and irradiated with a
monochromator (340-380 nm), urticarial reactions were seen, with
burning and erythema lasting for several days (Crow et al., 1961).
Twelve healthy white men, 12-26 years old, with fair complexions were
treated dermally with anthracene in an ethanolic acetone solution at
25 µg/cm2 and received ultraviolet irradiation 2 h later. Specific
skin reactions such as transient erythema, delayed erythema, and
whealing were seen. The effect was related both to the anthracene and
the amount of radiation energy. Controls who received no anthracene
treatment but the same irradiation showed no sign of erythema (Kaidbey
& Nonaka, 1984).
Regressive verrucae were reported after up to 120 dermal applications
of 1% benzo [a]pyrene to human skin over four months. The reversible
and benign changes were thought to be neoplastic proliferations, but a
group that did not receive benzene was not evaluated (Cottini &
Mazzone, 1939). Similar epidermal changes and nucleolar enlargement
were reported in volunteers painted daily for four consecutive days on
1-cm2 areas of the upper back (Rhoads et al., 1954).
8.1.2 Mixtures of PAH
8.1.2.1 PAH in unvented coal combustion in homes
Interdisciplinary studies were conducted to investigate exposure to
PAH and the high lung cancer rates in a rural county, Xuan Wei,
located in Yunnan Province, China (Mumford et al., 1987). Mortality
from lung cancer in this county is five times the Chinese national
average, especially among the women, who have the highest rate in
China. Three communes had a mortality rate that was 24 times the
national rate: 126 per 100 000 for women and 118 per 100 000 for men
during 1973-79. An unusual observation in Xuan Wei is the similarity
of the lung cancer rates in men, most of whom are smokers, and women,
most of whom are not (< 0.1% smoke). The mortality rate from lung
cancer was correlated with domestic use of 'smoky' coal
(medium-volatile bituminous coal with low sulfur and high ash) for
cooking and heating, but not with use of wood or smokeless coal.
Monitoring of air during cooking inside the homes showed that women
were exposed to extremely high levels of PAH, with a mean
benzo [a]pyrene concentration of 14.7 µg/m3, comparable to the
levels to which coke-oven workers are exposed. They were also exposed
< 24 mg/m3 of submicron particles containing up to 82% of the
organic matter. The major organic components of smoky coal emissions
are the three- to five-ring alkylated PAH, which contributed 43% of
the organic mass of the particles and 61% of the total mutagenicity in
assays in S. typhimurium. The four-ring PAH were the most
tumorigenic (Chuang et al., 1992). Organic extracts of particles from
coal smoke were more potent in initiating tumours in Sencar mouse skin
than those from wood and smokeless coal combustion and were complete
carcinogens (Mumford et al., 1990). Xuan Wei residents exposed to
smoky coal emissions had significantly more 9-hydroxy benzo [a]pyrene
in their urine, and cells obtained by bronchial alveolar lavage had
more DNA adducts (detected by 32P-postlabelling) than those of
controls (Lewtas et al., 1993). High ratios of the concentrations of
methylated PAH and parent PAH (9.8:1 for women and 5.8:1 for men) in
urine samples from Xuan Wei residents confirmed that they were exposed
to high concentrations of alkylated PAH. Thus, alkylated PAH may play
an important role in the etiology of lung cancer in Xuan Wei (Mumford
et al., 1995).
1-Hydroxypyrene was used as a urinary biomarker to monitor the
exposure of urban populations to PAH originating from coal burning. A
good correlation was found between the concentrations of pyrene and
benzo [a]pyrene in ambient in air and 1-hydroxypyrene in urine (Zhao
et al., 1990; see also section 8.3.2).
8.1.2.2 PAH in cigarette smoke
A large volume of literature exists on the effects of tobacco smoke on
human lungs (see IARC, 1986). On the basis of more than 100
prospective and retrospective studies in more than 15 countries,
cigarette smoke has been shown to be by far the most important single
factor contributing to the development of lung cancer. Other types of
cancer caused by cigarette smoking include cancers of the oral
cavities, larynx, pharynx, oesophagus, bladder, renal pelvis, renal
adenocarcinoma, and pancreas.
Levels of 11 ng per cigarette benzo [a]pyrene were found in
mainstream smoke and 103 ng per cigarette in sidestream smoke; the
corresponding values were 6.8 and 7.6 ng per cigarette for
benzo [e]pyrene, 20 and 497 ng per cigarette for chrysene and
triphenylene, and 13 and 204 ng per cigarette for benz [a]anthracene
(Grimmer et al., 1987). In sidestream smoke, PAH with four or more
rings were responsible for 83% of the total carcinogenic activity
(Grimmer et al., 1988c).
8.1.2.3 PAH in coal-tar shampoo
Of eight commercially available coal-tar shampoos, that with the
highest PAH content (100 times that of the others), containing, e.g.
285 mg/kg pyrene and 56 mg/kg benzo [a]pyrene, was chosen for testing
in 11 healthy people. A dose of 20 g shampoo was used in the evening
(see section 8.2.3), and the internal dose of PAH was assessed as
urinary 1-hydroxypyrene. One day after exposure, the internal dose was
10 times higher than the background level, similar to that measured in
coke-oven workers (van Schooten et al., 1994). The potential
carcinogenicity of coal-tar shampoo formulations has been studied (see
section 7.7). It has been suggested that modest therapeutic doses of
agents containing coal-tar and dithranol are tumorigenic after
combined application and that their use should be reviewed (Phillips &
Alldrick, 1994).
8.2 Occupational exposure
No studies on occupational exposure to single PAH were available, as
in general, industrial workers using or producing coal or coal
products are exposed to mixtures of PAH (see section 5.3). Table 95
lists workplaces in which there is exposure to PAH and the types of
employees exposed. Epidemiological studies have been conducted on
workers exposed at coke ovens in coal coking and coal gasification, at
asphalt works, at foundries, and at aluminium smelters and to diesel
exhaust. Details of the most recent and most important cohort and
case-control studies are given in Tables 96 and 97 (for reviews of
these studies, see also IARC 1983, 1984a,b, 1985).
Levels of exposure to single PAH in these occupations have been
reported (see section 5.3). In the following text, levels of exposure
to 'total PAH' are also given, as reported by the authors; however,
'total PAH' represents only the sum of a limited number of compounds
that have been quantified, i.e. the selection of the investigator, and
such measurements cannot be compared for the purpose of evaluating
levels or risks of pollution.
All benzo [a]pyrene concentrations are reported for comparative
purposes, as it is the only PAH that has been determined in almost all
investigations because of its well-known carcinogenicity. It is
commonly used as an indicator of the level of particle-bound PAH, and
particularly of carcinogenic ones, but PAH profiles may vary according
to source.
The first attribution of a PAH-related cancer to an occupational
exposure was that of Pott in 1775, who described the susceptibility of
English chimney sweeps to scrotal cancer (Pott, 1775); a second was
published by Butlin in 1892. Easily avoidable dermal cancers are
seldom seen today, owing to better personal hygiene and better working
conditions, but the number of respiratory cancers is still
significantly higher in occupational cohorts than in the general
population. In a study of Swedish chimney sweeps who were exposed to
< 9 µg/m3 benzo [a]pyrene, significantly increased rates of lung
tumours were observed, with a standardized mortality rate (SMR) of
2.06 (Table 96; Gustavsson et al., 1988); however, chimney sweeps were
also exposed to arsenic, chromium, cadmium, nickel, sulfur dioxide,
carbon monoxide, organic solvents, and asbestos.
Most important for an evaluation of the possible risk for cancer due
to exposure to PAH are studies of workers exposed at coke ovens in
coke plants or in coal-gasification processes, where the PAH
concentrations are considerable, with levels of 1 mg/m3 total PAH and
300 µg/m3 benzo [a]pyrene (Lindstedt & Sollenberg, 1982; Swaen et
al., 1991; see also section 5.3). The concentrations to which workers
are exposed are not available in most epidemiological studies,
however, and coke-oven workers may be exposed to several other
carcinogenic compounds, such as 2-naphthylamine, arsenic, and benzene.
Table 95. Occupations in which there is exposure to polycyclic
aromatic hydrocarbons
High exposure
- coke ovens
- coal gasification plants
- chimney sweeping
- petroleum refineries (mainly exposed to naphthalene and its methyl
derivatives)
- impregnation of wood with creosotes (mainly exposed to
naphthalene, phenanthrene, and fluorene)
- handling of creosote-impregnated wood (e.g. railroad and utility
workers, carpenters, mainly exposed to naphthalene, phenanthrene,
and fluorene)
Medium exposure
- asphalt and pavement work
- roofing
- aluminium production
- graphite electrode production (e.g. anode production for the
aluminiurn industry)
- founding (processing of e.g. steel and other alloys, from coal
additives in moulding sand)
- smokehouses (processing of meat and fish)
Low exposure
- mechanics, bus garage workers, and machinists (from diesel and
spark-ignition engine exhaust gases)
- mining (from diesel engine exhaust gases)
- use of lubricating and cutting oils (e.g. in steel production)
- cooking
Table 96. Epidemiological studies of lung caner in cohorts exposed to polycyclic aromatic hydrocarbons
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
Coal coking
5321, 10 497 Coal-tar pitch volatiles;b 255 1.95 Risk decreased with period All causes, 1.08 Costantino
coke oven non-oven, 3.2 mg/m3 (topside), (1.59-2.33) follow-up; findings all cancers, 1.34 et al. (1995);
1952-82, steel industry 2.0 (top-side parttime), consistent across racial prostate 1.57 Rockette &
0.88 (side); no categories; strong Redmond
information on smoking correlation with duration of (1985);
habits concentration exposure and exposure Redmond
(1983)
5639 National No information on 62 1.29 Strong correlation with Total 1.19 Swaen et al.
coke plant, population smoking habits (0.99-1.66) exposure concentration; mortality, 1.66 (1991)
1945-84 (Netherlands) risks increased in respiratory 3.08
comparison with workers at disease,
a nitrogen fixation plant liver
536, National information on smoking 25 2.38*** Coke-oven workers: 1.75 All causes, 1.41 Chau et al.
coke plant population habits (2 cases); near oven all cancers, 1.33 (1993)
1963-87 (France) workers: 2.52** (8 cases); cardio- 1.33
unexposed workers: 2.28* vascular
(6 cases) disease
Risk increased (not
significant) for workers in
oldest plant
6767, National Some information on 167 1:17* - - - Hurley et al.
coke plant population smoking habits (1983)
(Scotland,
England, Wales)
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
subcohorts:
1617, coke - - 34 0.94 No clear correlation with - -
oven, duration of exposure; problems
1966-78 in classification of exposure
(risk increase with > 5 years
and > 10 years exposure)
1158, coke - - 32 1.05 Lung cancer risk increased - -
oven, in younger workers; no clear
1967-80 correlation with duration of
exposure and exposure
concentration; problems in
classification of exposure
Coal gasification
2449+1176 National 2-Naphthylamine, 99 3.82b RR given for groups with Bladder 2.35 Doll et al.
(2 cohorts), population 2 µg/m3; some 23 2.72 regular exposure; also cancer (1972)
1953-65 (England and information on increased risk in group with
Wales) smoking habits intermittent exposure;
correlation with exposure
concentration and duration
of exposure
724,1953-80 (a) 3792, same Median of 8 measurements: 68 3.53**a No correlation with All cancers, 1.98 Manz et al.
plant, not at total dust, max. duration of exposure; 88% of urinary 4.35 (1983)
coke-oven 264 mg/m3, BaP, 28 workers with > 10 years' system
(b) 681, µg/m3, max. 89; exposure cancers
office and some information on
administration smoking habits
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
(a) local
population
(Hamburg)
295, Local worker BaP top of ovens: 4 0.82 Owing to incomplete employment All causes 1.27 Gustavsson
1966-86 population 1964: 4.3 µg/m3 (0.22-2.11) registers, only workers & Reuterwall
(0.007-33) with short and recent exposure (1990);
1965: 0.52 µg/m3 selected; no correlation with Gustavsson
(0.021-1.29); no duration of exposure (1989);
differences in smoking Gustavsson
habits between exposed et al. (1987)
and controls
Asphalting
679, paving, National Fume condensate; 25 2.90 SMR for lung cancer given All causes, 1.63 Hansen
1959-86 population flooring: 0.5-260 mg/m3; (1.88-4.29) only for workers aged 40-89 all cancers 2.25 (1989);
(Denmark) median,19.7, manual years; overall mortality Workers Hansen et
road paving: 4.3-3.4 mg/m3; excess primarily in younger aged 40-89 al., 1991c,
total PAH: median, 183 age groups years: liver 4.67 1992;
µg/m3; BaP, 4 µg/m3; cirrhosis Wong et al.
some information on (1992)
smoking habits
2572, paving, National - 7 1.10 Follow-up too short All causes, 0.69 Engholm et
1971-79 to population 8 1.24 stomach 2.01 al. (1991);
1985 (Sweden) (0.53-2.34) cancer Partanen &
Boffetta
(1994)
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
704, roofing, National - 3 2.79 Follow-up too short All causes 0.91 Engholm et
1971-79 to population 4 (0.99-9.31) al. (1991);
1985 (Sweden) Partanen &
Boffetta
(1994)
Paving, - - 332 1.21 Meta-analysis of 11 studies Stomach, 1.33 Partanen &
roofing, (1.08-1.30) bladder, 1.38 Boffetta
others skin, non- 1.74 (1994)
melanoma, 1.41
leukaemia
Paving - - 167 0.87 Meta-analysis of 3 studies Skin, non- 2.18
(0.74-1.01) melanoma
Roofing - - 118 1.96 Meta-analysis of 4 studies Stomach 1.71
(1.46-2.11)
Creosote
impregnation
922 National - 13 0.79 - Skin, non- 2.37 Karlehagen
populations (0.42-1.35) melanoma et al. (1992)
(Sweden and
Norway)
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
Tar distillation
76, 449, PAH: 29 µg/m3, 4 Total rate of tumours 0 - Schunk
1962-72 rubber BaP: 4 µg/m3; some increased (RR 3 and 3.4); (1979)
industry; information on 3 tumours of the digestive
national smoking habits system latency, 10.1-
population 17.5 years
(Germany)
Foundries
2990 General No information on 224 1.44** Respiratory Egan-Baum
(2651 white population smoking habits white white disease: et al. (1981)
males; 339 (USA) males,males; white males 1.10
black males), 39 76** black males 1.24
steel, iron, black black Pneumo-
non-ferrous, males males coniosis:
1961-71 black males 5.76
Respiratory
tuberculosis:
white males 2.32
All cancers:
white males 1.10
black males 1.24
439, Regional Data from 1977; total 21 2.50** Correlation with duration - - Gibson et al.
steel population dust, 1.76-5.11 mg/m3; of exposure; no correlation (1977)
foundry, (Toronto) respirable dust, 0.69-2.65 between latency and exposure
1967-77 mg/m3; no cristobalite concentration
or tridymite; coal-tar pitch
volatiles, 0.19-0.43 mg/m3;
BaP, 0.049-0.152 µg/m3;
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
1718, General Some information on 11 2.04 No clear correlation with - - Moulin et al.
steel or population smoking habits (1.02-3.64) duration of exposure or (1990)
ferro- (France) latency
chromium
production,
31 years
4227, General - 17 0.88 - Liver 1.74 Moulin et al.
steel plant, population (0.51-1.40) cirrhosis (1993)
two cohorts, (France)
1969-84
210, - - 2 0.68 - - -
subcohort: (0.08-2.45)
ferroalloy
workshop
477, - - 11 2.29 No clear correlation with - -
subcohort: (1.14-4.09) duration of exposure or
steel foundry latency; significantly
increased for > 30 years
since first employment
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
10 491, General - 441 1.47*** SMRs increased for all Stomach 1.37 Sorahan
steel population foundry occupations cancer & Cooke
foundry, (England and (fettling shop, pattern, (1989)
20 years, Wales) machine, maintenance,
1946-65 to inspection); some
1985 correlation with latency;
no significant correlation
with duration of exposure
6494, General 2 plants, around 77 0.99 Except for 1 plant, furnace - - Kjuus et al.
production of population furnaces:PAH, 3-49 µg/m3; (0.78-1.24) workers showed no increase (1986)
ferrosilicon (Norway) anode paste plant: 2-20 SIR in rate of lung cancer (8
ferromanganese, µg/m3; total dust: 10-30 cases); increase in anode pass
6 plants, mg/m3; manganese: plant workers (3 cases); no
1953-82 0.5-2 mg/m3 Some correlation with duration
information on smoking of exposure
habits
5579, (a) National - 74 (b) 1.17 Information on Malignant 2.05 Sherson &
iron, steel, population employment available only respiratory Iversen
1967-69 (Denmark), for 1967-69 and 1972-74; tumours, (1986)
non-ferrous (b)Economically misclassification possible; respiratory 1.54
foundries, active some correlation with disease
1972-74 population, duration of exposure excluding
(part to (c) Skilled silicosis,
1980) and unskilled all causes 1.11
manual workers
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
8147, National No information on 72 1.23 white No correlation with duration - - Andjelkovich
iron foundry, population smoldng habits males of exposure; smoking may et al. (1990)
1950-85 (USA) (0.96-1.54) be responsible for lung
Local population 67 cancer
132 non-
white males
(1.02-1.67)
Aluminium production
22 010, General No information on 272 0.964 Highest SMR > 25 years All causes, 85.6 Rockette &
15 plants, population smoking habits of exposure in Soderberg all 88.6 Arena (1983)
1946-77, (USA) proccess: SMR 2.0 based malignant
Soderberg on 5 deaths; some tumours
prebake correlation with duration of
exposure and latency
6455, General Some information on 37 1.14 Electrolysis workers: - - Mur et al.
11 plants, population smoking habits (0.85-1.48) SMR, 1.36 (4 deaths); (1987)
Soderberg (France) no correlation with duration
prebake, of exposure or latency
1950-76
4213, 1 plant General Coal-tar pitch volaties 32 0.93 For > 20 benzene-soluble Brain, 2.17 Spinelli et al.
Soderberg, population low: < 0,2 mg/m3; (0.68-1.25) material of coal-tar pitch bladder(SIR) 1.69 (1991);
1954-85 (British medium: 0.2-1; high: volatiles years: SIR 1.43; Ronneberg &
Columbia) > 1; information on correlated with duration of Langmark
smoking habits exposure; some correlation (1992)
with latency; adjustment for
smoking: no change in lung
cancer risk
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
5406, (a) Regional No information on 101 1.43* SMRs for workers exposed All cancers, 1.23 Gibbs (1985);
3 plants, population smoking habits to tar, SMR significantly respiratory 1.65 Gibbs &
Soderberg, (Quebec) increased compared with disease, Horowitz
prebake, (b) Local local population and pneumonia 1.99 (1979)
1954-85 population never-exposed workers and bronchitis,
Significant correlation oeosophageal 1.53
with duration of exposure; and gastric
correlation with latency; tumours
risk decreased in later
periods (1974-77)
5485, - - 12 1.69 SMR increased in relation - - Gibbs (1985);
Soderberg, to local population Gibbs &
1950-51; Horowitz
1977 (1979)
694 General Some information on 19 1.16 Workers with at least 3 - - Ronneberg &
Soderberg, population smoking habits (0.70-1.81) years of employment; some Andersen
prebake (Norway) (SIR) correlation with duration of (1995)
1962-91 exposure and latency
Other workplaces
2219, General - 29 0.85 - All causes, 0.67 Tate et al.
11 carbon white 0.57-1.21) circulatory 0.60 (1987)
plants, population disease,
1974-83 (USA) respiratory 0.51
disease
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
176, Regional Total dust: 5-100 mg/m3; 1.97 - - - Gustavsson
waste population some information on (1.21-2.75) & Reuterwall
incinerator, smoking habits (1990);
1951-85 Gustavsson
(1989)
Nickel/copper Regional Laboratory fume 50 1.47 Lung tumours correlated - - Verma et al.
smelter and population generation experiments (1.02-1.81) with duration of exposure (1992)
refinery; (Ontario) showed high only it exposed to PAH
special concentrations of PAH
subcohorts (asphalt > tar > mastic);
exposed to no information on smoking
tar habits
OR, odds ratio; RR, relative risk; SMR, standard mortality ratio; SIR, standard incidence ratio; BaP, benzo[a]pyrene
SMR or RR for whole study population is given; results for special subcohorts, subdivided by e.g. level or duration of exposure or
latency, are given under remarks, unless otherwise stated
* Statistically significant at p < 0.01; ** statistically significant at p < 0.01; *** statistically significant at p < 0.001
a Only statistically significant (p < 0.05) increased or decreased rates are shown; unless otherwise stated, refers to mortality
b Benzene-soluble fraction of total particulate matter
Table 97. Case-control studies of cancer types possibly associated with exposure to polycyclic aromatic hydrocarbons PAH)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
Lung tumours
NR Exhaust from Patients with cancer of prostate, 1.41 2 miles from coke oven; some Lyon et al. (1981)
coke plant breast, or brain correlation with distance from
coke oven, highest at 2 and 3
miles distance; effect may also
be due to occupational exposure
Asphalt - - 1.12 Meta-analysis of 5 studies Partanan & Boffetta
workers, (0.93-1.34) (1994)
paving,
roofing,
others,
113 Iron foundry 249 all other deaths in cohort of 2.36* OR for workers aged 42-64; Egan-Baum et al.
cohort of versus steel or foundry workers except non- 1.19 of workers aged > 65, only 65% (1981)
foundry non-ferrous malignant respiratory disease successfully traced
workers foundry and other cancers
12, Occupation in 58 from cohort of steel foundry 4.51 Moulin et al. (1990)
cohort of steel stainless-steel workers
workers versus ferro-
chromium
(a) 51, (b) 47, BaP: 0.1-15 From cohort of iron foundry - More cases exposed to high PAH Tola et al. (1979)
cohort of iron µg/m3; small workers concentrations than controls, not
foundry differences (a) 153, matched by age and statistically significant; some
between intensity of exposure, no cancer information on smoking habits
workplaces cases
Table 97. (continued)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
(b) 47, also same time of entry
into foundry
(c) 27, cases compared with
expected contribution of
exposure of foundry workers
74, cohort of Exposure to 1138, manual workers at same 2.00 Figure given for exposure for Armstrong et al.
aluminium coal-tar pitch plant, not matched, similar (1.33-2.75) 20-41 years at Soderberg pots, (1994)
production volatiles; distribution of birth years no data for exposure to
workers, measurements of benzene-soluble material in
Soderberg benzene-soluble general; strong correlation with
and prebake material; estimated exposure concentration, duration
process concentrations for of exposure and latency;
different job adjustment for smoking: no
categories; 0-3.5 difference in risk
mg/m3
45 butchers Combustion (a) 99 all butchers and 0.84 Smoking habits available Gustavsson (1989)
and slaughter- products, only slaughter-house workers dying (0.40-1.77)
house low, intermittent from malignant disease
workers, 11 exposure to (b) 100 random sample of all
years PAH deceased butchers and
slaughter-house workers
(a) and (b) except tumours related
to chemical exposure
Table 97. (continued)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
Skin tumours
376 patients Occupational 752 general population; 752 1.14 No correlation with duration of Kubasiewicz et al.
with skin exposure to patients from hospitals exposure; no increased risk for (1991)
cancer PAH randomly sampled other exposures related to PAH,
i.e. tar, pitch, soot, coke,
bituminous mass; smoking habits
not availalble
Renal-cell carcinoma
1982-87, Occupational 64 patients with haematuria, 2.54 Exposure to coal, tar, or pitch Sharpe et al. (1989)
hospitals in exposure to 61 controls and cases not treated (0.96-6.99) RR for exposure to coal increased
Montreal, hydrocarbons with haemodialysis 0.29 from 10 to 24 years of age; strong
Canada (predisposes for renal cancer) (0.16-.74) correlation with duration of exposure
and exposure concentration; no
correlation with latency (no case or
control with latency < 20 years);
smoking habits: no significant
difference between cases and
controls; history of smoking > 20
cigarettes per day associated with
tendency to higher stage disease
Table 97. (continued)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
Urinary bladder cancer
138/(66+ 69), Occupational 414, from cohort 2.63 Data consistent with former study; Tremblay et al.
1970-88 exposure to (1.29-5.37), strong correlation with exposure (1995); Theriault et
aluminium benzene-soluble adjusted concentration and duration of al. (1984)
plant, material for smoking exposure highest risk: Soderberg
Soderberg or BaP potroom workers: 5.15; 1930-54:
BaP, max, 51.5 µg/m3;
benzene-soluble material, max
10 mg/m3; 1985-89: BaP, max,
3.1 µg/m3; benzene-soluble
material, max, 0.36 mg/m3
General Contact with From population 2.61 All figures given for 8-28 years Risch et al. (1988)
population, diesel or traffic (0.70-12.5) employed
1979-82 fumes
Aluminium 1.69
smelting (1.24-2.31)
Contact with 3.11
tars, asphalt (1.19-9.68)
OR, odds ratio; RR, relative risk; SMR, standard mortality ratio; SIR, standard incidence ratio; NR, not reported; BaP, benzo[a]pyrene
SMR or RR for whole study population is given; results for special subcohorts, subdivided by e.g. level or duration of exposure,
latency, are given under remarks, unless otherwise stated
a Only statistically significant increased or decreased rates, unless otherwise siated, mortality
* statistically significant at p < 0.05
** statistically significant at p < 0.01
*** statistically significant at p < 0.001
A significantly increased risk for lung cancer (SMR, 1.95) was found
among a cohort of over 5000 workers who were heavily exposed at coke
ovens in coke plants and were followed-up for over 30 years. Although
no data were available on smoking habits, the observed effect is not
likely to be due to smoking since unexposed steel workers in a
comparison group were assumed to have similar smoking habits. In
addition, a high correlation was seen between the risk for respiratory
cancer and the concentration and duration of exposure. The authors
noted however, that the rates of respiratory cancer decreased during
the follow-up period, suggesting that implementation of emission
controls and occupational exposure limits has been beneficial
(Costantino et al., 1995). The increased risk found in this study was
also seen in other studies, including some on coal gasification (Doll
et al., 1972; Manz et al., 1983; Swaen et al., 1991). Other studies,
however, and especially those involving small cohorts, did not show
increased rates for lung cancer among coke-oven workers (Hurley et
al., 1983; Gustavsson & Reuterwall, 1990; Chau et al., 1993).
Coke-oven workers in China who were exposed to PAH were reported to
have decreased titres of immunoglobulins M, G, and A in their serum
and decreased immune function (Lei, 1993).
Several epidemiological studies have been performed on the potential
risks of handling asphalt (for a review, see Partanen & Boffetta,
1994; see also Table 96). The job descriptions included: roofer,
waterproofer, highway maintenance worker, production of hot-lay
asphalt, slater, grader, paver, surfacer, and mastic asphalt worker.
Their exposure to PAH depended on the type of asphalt involved. When
bitumen is used as the binder, the PAH content is relatively low, with
about 200 µg/m3 total PAH and up to about 5 µg/m3 benzo [a]pyrene
(Hansen, 1989; see also section 5.3); bitumen binders contain
primarily asphaltenes, straight and branched aliphatic hydrocarbons,
naphthene aromatics, and resins. Most of the PAH are removed by vacuum
distillation, but they may be formed during cracking operations or be
reintroduced in the flux used in blended or fluxed bitumens. Coal-tars
and coal-tar pitches have been used as binders, especially in the
past, and may contain substantial amounts of PAH (Partanen & Boffetta,
1994). These workers may also be exposed to several other substances,
such as silica, limestone, and asbestos (Chiazze et al., 1991;
Partanen & Boffetta, 1994). In the meta-analysis of Partanen &
Boffetta (1994), increased risks for lung tumours were seen for both
pavers and roofers; tumours of the stomach, bladder, and skin and
leukaemia were also observed. The excess risks were more pronounced
for roofers than for pavers. It is not clear however, if these effects
are due to exposure to PAH, as it could not be determined whether the
carcinogenicity was due to exposure to bitumen or to tar fumes.
Workers are exposed dermally to very high concentrations of PAH when
impregnating wood with creosote, as shown by measuring biomarkers,
especially the excretion of 1-hydroxypyrene in urine (see section
8.3.2). In a cohort study on 922 creosote-exposed workers, a
significant increase in the risk for skin cancer (SIR, 2.37) and
increased risks for lip cancer and malignant lymphoma were observed.
Since the work involves some time outdoors, it cannot be ruled out
that exposure to sunlight contributed to the risks for cancers of the
skin and lip (Karlehagen et al., 1992).
The PAH concentrations in iron, steel, and other ferroalloy foundries
reach levels of 50 µg/m3 and that of benzo [a]pyrene about 10 µg/m3
(Verma et al., 1992; see also section 5.3). Workers in these plants
also have nearly ubiquitous exposure to silica sand. Silicosis and
other chronic respiratory abnormalities have been reported for decades
to be the major health problems of foundry workers. These workers are
also exposed to asbestos, used for heat protection and insulation
around furnaces, to benzene, toluene, formaldehyde, iron, and,
depending on the kind of metal or alloy, to metals such as lead,
chromium, manganese, nickel, copper, cadmium, and zinc. Increased
mortality from lung cancer has been observed consistently in many
studies of foundry workers (Palmer & Scott, 1981; Andjelkovich et al.,
1990). When the results of all the relevant studies are combined, the
SMR is 1.43 (Andjelkovich et al., 1990); however, those authors argue
that the elevated risk is due to smoking, because the SMRs did not
increase with time since first employment in a foundry or with the
length of employment in a foundry. Furthermore, the incidence of
emphysema, which can also be attributed to smoking, is increased in
epidemiological studies of foundry environments.
Information on the possible risks for cancer due to exposure to PAH
can also be obtained from studies of workers in aluminium plants. Two
types of anode are used: the continuous Söderberg anode and the
prebaked anode. Both are manufactured from coal-tar pitch and coke,
and coal-tar pitch volatiles evaporate from them during baking. The
exposure is heavier in potrooms where the Söderberg process is used,
because the anodes are baked continuously. With use of prebaked
anodes, exposure to PAH may occur in the carbon area where the anodes
are prebaked. During Söderberg electrolysis, workers may be exposed to
0.3-3.5 mg/m3 of benzene-soluble organic material and 3-35 µg/m3
benzo [a]pyrene; workers in the carbon area are exposed to
0.4-1.2 mg/m3 benzene-soluble organic material and 0.4-1.2 µg/m3
benzo [a]pyrene. Similar concentrations have been detected in other
parts of such plants (Rnneberg & Langmark, 1992). These workers are
also exposed to fluorides, carbon dioxide, sulfur dioxide, magnetic
fields, hot work environments, and, in some cases, asbestos.
In a large case-control study, an increased risk for lung cancer was
found with exposure in a Söderberg potroom, and a significant
correlation was seen between the increased risk and the duration and
concentration of exposure and latency. Adjustment for smoking did not
alter the correlation (Armstrong et al., 1994). Similar increases were
detected in cohort studies (Rockette & Arena, 1983; Mur et al., 1987).
The studies differed with regard to the magnitude of the risk and the
presence of a dose-response relationship (see also reviews by Abramson
et al., 1989; Rnneberg & Langmark, 1992). In some studies, however,
few cases were available (Rockette & Arena, 1983; Mur et al., 1987;
Rnneberg & Andersen, 1995).
Work in potrooms where Söderberg electrolytic cells were used was also
associated with an increased risk for urinary bladder cancer (Spinelli
et al., 1991; Rnneberg & Langmark, 1992; Tremblay et al., 1995). This
risk may be due to exposure not only to PAH but also to aromatic
amines, which have been detected in the potrooms (Tremblay et al.,
1995).
Asthma-like symptoms, lung function abnormalities, and chronic
bronchitis have also been detected in workers in aluminium plants
(reviewed by Abramson et al., 1989; Kongerud et al., 1994), but the
quality of the studies in which these effects were shown is variable.
These diseases are believed to be due not to PAH but rather to
exposure to alumina, cryolite, carbon, fluorides, and sulfur dioxide;
however, the causative agents have yet to be defined.
Increased risks for lung cancer were found in several studies of
workers exposed to diesel exhausts (WHO, 1996). In comparison with the
occupations described above, the concentrations of PAH to which these
workers are exposed are usually relatively low. The benzo [a]pyrene
concentrations in automobile repair shops and garages reach about 70
ng/m3 (Waller, 1981; Lindstedt & Sollenberg, 1982; Waller et al.,
1985), and truck drivers are exposed to less than 10 ng/m3 (Guillemin
et al., 1992).
The finding of an increased risk for lung cancer must always be
interpreted in relation to the influence of tobacco smoking. The
tobacco smoking habits were seldom known for all of the persons
studied, but there are several reasons for concluding that the
increased risks for lung cancer are not due solely to tobacco smoking:
The general population is often used as a reference group, but their
lung cancer rate is usually lower than that of the working population
because fewer members of the general population are tobacco smokers
(Gibson et al., 1977; Hansen et al., 1989; Andjelkovich et al., 1990;
Moulin et al., 1993). Calculations presented by several authors
indicate that differences in tobacco smoking habits can contribute
only about 20% of the excess risk for lung cancer (Hurley et al.,
1983; Maclaren & Hurley, 1987; Gustavsson et al., 1988; Andjelkovich
et al., 1990; Moulin et al., 1993). Greater contributions are
improbable, since other illnesses usually linked to tobacco smoking
have not been observed to occur more frequently than in controls.
Another reason for excluding tobacco smoking as the major reason for
the increased lung tumour rates is that increased risks also have been
found in studies in which the controls were workers with other
occupations, and not the general population. In this case, it can be
assumed that the tobacco smoking habits are similar, and it is
probable that the increased risks are due to the exposure conditions
and not to tobacco smoking (Costantino et al., 1995). In addition,
several studies show a strong correlation between the risk for
respiratory cancer and the concentration and duration of exposure and
latency. Finally, in studies in which the information on tobacco
smoking habits was good and adjustments could be made for the
influence of tobacco smoking (Spinelli et al., 1991; Armstrong et al.,
1994), tobacco smoking had no effect on the cancer risk. As no
information is available about the comparative cancer risks of
non-smokers and smokers, however, the increased risk for lung cancer
may always represent a combined risk due to tobacco smoking and the
exposure conditions.
Although workers are always exposed to several substances, it seems
plausible to attribute the increased lung cancer risks at least
partially to PAH. The increased risk is seen for workers in several
occupations which have exposure to PAH in common. Although other
carcinogenic chemicals were present, they differed with each
occupation. Airborne high-molecular-mass PAH, which are considered to
be the most carcinogenic, are adsorbed mainly onto particulate matter
(see section 4.1.2), and it was often difficult to distinguish the
toxicological effects caused by particles from those caused by the PAH
themselves.
8.3 Biomarkers of exposure to PAH
Several methods have been developed to assess internal exposure to PAH
after exposure in the environment and in workplaces, which can be used
to evaluate the adequacy of protective regulations. In most studies,
metabolites of PAH were measured in urine, 1-hydroxypyrene being
widely used.
The genotoxic effects of PAH have been determined in tests for
mutagenicity in urine and faeces, micronucleus formation, chromosomal
aberration and sister chromatid exchange in peripheral blood
lymphocytes, adducts of benzo [a]pyrene with DNA in peripheral
lymphocytes, and other tissues and with proteins such as albumin; and
antibodies to DNA adducts. In addition, oncogene expression and
immunostaining of differentiation antigens on lung cell surfaces have
been measured as biological indexes of the risk for lung cancer.
8.3.1 Urinary metabolites in general
The metabolites measured in urine and faeces include urinary
thioethers (Burgaz et al., 1992), 1-naphthol (Bieniek, 1994; Hansen et
al., 1994, 1995), b-naphthylamine (Hansen et al., 1994), hydroxy
phenanthrenes (Martin et al., 1989; Adlkofer et al., 1990; Grimmer et
al., 1994; Mannschreck et al., 1996), and 1-hydroxypyrene.
No difference in thioether excretion in urine was observed between
controls and coke-oven workers or workers in coke and
graphite-electrode-producing plants. It was concluded that the
determination of thioethers in urine is of little value, since smoking
is a strong confounding factor (Ferreira et al., 1994a,b; Reuterwall
et al., 1991).
A good correlation was found with the excretion of hydroxylated
phenanthrenes and 1-hydroxypyrene in urine (Mannschreck et al., 1996).
When phenanthrene, pyrene, and benzo [a]pyrene metabolites are
determined simultaneously, precursors of carcinogens are also
measured, thus providing an estimate of individual risk (Grimmer et
al., 1993).
8.3.2 1-Hydroxypyrene
1-Hydroxypyrene, a metabolite of pyrene, was introduced as a biomarker
of exposure to PAH by Jongeneelen et al. (1986) and has since been
widely used. Its advantages are that pyrene is present in all PAH
mixtures at relatively high concentrations (2-10%), and in certain
environments the pyrene content of the total PAH is fairly constant
(Zhao et al., 1990; Buchet et al., 1992; Jongeneelen, 1994). In
studies at different workplaces, a strong correlation was found
between the pyrene concentrations in air and those of
benzo [a]pyrene, other selected PAH, and total PAH (Jongeneelen et
al., 1990; Tolos et al., 1990; Zhao et al., 1990; Van Rooij et al.,
1992; Ferreira et al., 1994a,b; Jongeneelen, 1994; Elovaara et al.,
1995; Levin et al., 1995; Ovrebo et al., 1995; Quinlan et al., 1995a).
Pyrene is metabolized predominantly to 1-hydroxypyrene (Grimmer et
al., 1993, 1994; Levin et al., 1995), which can be measured easily. In
contrast to other PAH metabolites, which are excreted mainly in
faeces, 1-hydroxypyrene is excreted in urine.
8.3.2.1 Method of determination
The method of determination consists of enzymatic hydrolysis of
conjugated 1-hydroxypyrene in urine samples, followed by solid-phase
extraction and high-performance liquid chromatographic separation with
fluorescence detection (Jongeneelen et al., 1986). An enzyme-linked
immunosorbent assay has also been used (Santella et al., 1994).
In most publications, the concentrations of metabolites in urine are
adjusted to that of creatinine in order to compensate for variations
in urine flow rates; however, it must be borne in mind that creatinine
excretion fluctuates widely because of internal and external factors.
Therefore, correction of the concentrations of chemicals in urine in
this way does not necessarily improve the correlation with exposure
(Levin et al., 1995). For comparisons, a mean urinary creatinine
concentration of 13 mmol/litre can be assumed, giving the
relationship:
1 µmol 1-hydroxypyrene per mol creatinine
= 1.93 µg/g creatinine
= approx. 3 ng/ml urine.
Since most authors give 1-hydroxypyrene concentrations in µmol/mol
creatinine, that unit is used in the following text and tables.
8.3.2.2 Concentrations
(a) General population
The concentrations of 1-hydroxypyrene in urine from the general
population exposed to PAH are compiled in Table 98. The background
concentrations of 1-hydroxypyrene in different countries range from
0.06 to 0.23 µmol/mol creatinine. No difference related to age or sex
was seen (Zhao et al., 1992), and ethanol consumption did not
influence 1-hydroxypyrene concentrations (Van Rooij et al., 1994a).
For nonsmokers, food accounted for 99% of the total daily pyrene
intake (Van Rooij et al., 1994a). Five volunteers who ate low-PAH
meals and high-PAH meals showed 100- to 250-fold increases in
benzo [a]pyrene dose, accompanied by a four- to 12-fold increase in
1-hydroxypyrene excretion in urine (Buckley & Lioy, 1992). A 10- to
80-fold increase was detected in one subject who ate 9 oz (250 g) of
charcoal-grilled beef. In this study of 10 subjects, an eightfold
interindividual variation in urinary excretion of 1-hydroxypyrene was
found after one day. As this variation was not appreciably altered
after adjustment of the 1-hydroxypyrene concentration by urinary
creatinine concentration or individual body weight, it was assumed to
be due to individual differences in the rate of absorption,
metabolism, or excretion of pyrene (Kang et al., 1995).
The intake of pyrene from cigarette smoking (12 nmol/day) is about the
same as the dietary intake from normal food (9.4 nmol/day) (Van Rooij
et al., 1994a). Tobacco smokers who are not otherwise exposed to PAH
have about twice the level of 1-hydroxypyrene in their urine as
nonsmokers (Jongeneelen et al., 1990; Sherson et al., 1992; Van Rooij
et al., 1994a; Levin et al., 1995), although no significant difference
was found in some studies (Jongeneelen et al., 1988b; Zhao et al.,
1992; Ny et al., 1993).
Urine samples from schoolchildren living along arterial roads in Tokyo
had 1.1-1.6 times more urinary 1-hydroxypyrene than those from
children in a less polluted suburban area (Kanoh et al., 1993). Much
higher levels were detected in persons living in highly industrialized
regions in Poland, due to emissions from coke-oven plants. The
concentrations are a maximum of twice as high in winter as in summer
(Jongeneelen, 1994; Ovrebo et al., 1995). 1-Hydroxypyrene levels of up
to 1.5 µmol/mol creatinine have been detected in towns in China (Zhao
et al., 1990, 1992).
Very high exposure to PAH occurs during application of coal-tar
ointments or shampoos by patients with eczema or psoriasis. The mean
1-hydroxypyrene concentrations reached 500 µmol/mol creatinine, and
the maximum value was 5000 µmol/mol creatinine (Santella et al.,
1994).
Table 98. Concentrations of 1-hydroxypyrene in urine (µmol/mol creatinine) in the general
population
Type of exposure or No. of 1-Hydroxypyrene Reference
population investigateda subjects
Median Range
or meanb
Non-smokers
General population, 28 0.06c < 0.02-0.17 Goen et al. (1995)
southern Germany
General population 49 < 0.02c 0.02-0.28 Goen et al. (1995)
Southern Germany
Students and university 24 0.23d - Jongeneelen et
personnel, al. (1986)
Netherlands
University staff, 52 0.26 - Jongeneelen et
Netherlands al. (1988b)
University staff, 39 0.12e 0.04-029 Van Rooij et al.
Netherlands (1994a)
Students and university 15 0.24e - Burgaz et al.
staff, Turkey (1992)
Smokers
General population, 21 0.12c < 0.02-0.68 Goen et al. (1995)
Germany
General population, 20 0.14c < 0.02-0.3 Goen et al. (1995)
Germany
Students and university 22 0.27d - Jongeneelen et
staff, Netherlands al.(1986)
Students and university 38 0.28 - Jongeneelen et
staff, Netherlands al. (1988b)
Students and university 37 0.25e 0.10-0.79 Van Rooij et al.
staff, Netherlands (1994a)
Students and university 14 0.33e - Burgaz et al.
staff, Turkey (1992)
Smoking status not specified
Persons in urban and 27 0.20d - Elovaara et al.
rural areas Estonia (1995)
Persons from rural areas, 81 - 0.19-0.27 Ovrebro et al.
Biala Podlaska, Poland (1995)
Students and university 18 N 0.077d 0.002-0.57 Viau et al. (1993)
staff, Montreal, Canada 3 S
Healthy volunteers, 36 N 0.14e 0.02-0.98 Santella et al.
Columbia, MO, USA 17 S (1994)
Table 98. (continued)
Type of exposure or No. of 1-Hydroxypyrene Reference
population investigateda subjects
Median Range
or meanb
Polluted ambient air
School children, NR - approx.0.4-0.6 Kanoh et al.
Tokyo, Japan, M+F, Nf (1993)
Urban population 74 N 0.68e - Zhao et al. (1992)
Beijing, China 84 S 0.76e
Urban population, 13 N 1.55e - Zhao et al. (1990)
Shenyang, China, F
Urban population, 17 N 1.20e - Zhao et al. (1990)
Taiyuan, China, F
Urban population, 15 N 0.67e - Zhao et al. (1990)
Beijing, China, F
Urban population, 15 N 0.72e - Zhao et al. (1990)
Beijing, China, F (girls)
Urban population, 72 0.66e - Jongeneelen
Bytom, Silesia, (1994)
Poland (boys)
Urban population, 76 0.59e - Jongeneelen
Bytom, Silesia, (1994)
Poland (girls)
Gliwice, Silesia, Polandg 30 - 0.84-1.54 Ovrebro et al.
(1995)
Other exposures
Windsurfers sailing on 6 0.32e 0.16-0.81 Jongeneelen
polluted water, Ketelmeer, (1994)
Netherlands
Therapeutic treatment with coal-tar
Patients with eczema 5 - approx.50-500 Jongeneelen et
al. (1986)
Patients with psoriasis 4 - 13.2-811c Clonfero et al.
(1989)
Patients with psoriasis 53 547e 10-5160 Santella et al.
(1994)
Patient with psoriasis 1 3.45 - Viau et al. (1995)
Table 98. (continued)
Type of exposure or No. of 1-Hydroxypyrene Reference
population investigateda subjects
Median Range
or meanb
Food
Eating 9 oz [250 g] 10 approx.0.5e,h - Kang et al. (1995)
char-broiled beef 0.15-1.2
N, non-smokers; S, smokers; M, males; F, females; NR, not reported
a Unless otherwise stated, male persons were investigated; in some cases, insufficient
characterization of exposure given
b 1-Hydroxypyrene concentration in urine (µmol/mol creatinine), range, and, if available,
median concentration; otherwise, geometric or arithmetic means
c Calculated from 1-hydroxypyrene concentrations given in original reference as µg/g
creatinine.
d Geometric mean
e Arithmetic mean
f Benzo[a]pyrene measured at 0.0006-0.0024 µg/m3 by stationary sampling
g Benzo[a]pyrene measured at 0.009-0.041 µg/m3 by stationary sampling
h Calculated from 1-hydroxypyrene concentrations given in original reference as ng/ml
urine or pmol/ml urine
(b) Workplaces
1-Hydroxypyrene concentrations have been measured in the urine of
persons at various workplaces (Table 99); the urine of concurrent
controls was examined in most investigations. Unexposed workers at the
same plant, such as administrative workers, had slightly higher
1-hydroxypyrene concentrations than the general population (Zhao et
al., 1990; Buchet et al., 1992; Ny et al., 1993; Levin et al., 1995).
The highest 1-hydroxypyrene excretion, up to 90 µmol/mol creatinine,
was found in urine from workers impregnating wood with creosote,
although the PAH levels in the air were quite low. The high exposure
can be attributed to significant dermal uptake, which is several times
higher than that by inhalation (see below).
Other workplaces where there is heavy exposure are coke ovens,
coal-liquefaction plants, aluminium plants, and plants producing
carbon or graphite electrodes. The concentrations of 1-hydroxypyrene
in the urine of workers at these sites were 1-10 µmol/mol creatinine.
Table 99. Concentration of 1-hydroxypyrene in urine (µmol/mol creatinine) at industrial workplaces without and with
exposure to polycyclic aromatic hydrocarbons
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Controls
Unexposed workers in - 120 0.11 < 0.05-1.08 Boogaard and van
petrochemical industry Sittert (1994, 1995)
Office workers at graphite - 9N 0.33d - Buchet et al. (1992)
electrod- producing plant 6S 0.36d
Unexposed workers in coke - 137 0.26e,f 0.01-1.04 Ferreira et al. (1994a,b)
and graphite electrode-
producing plants
Workers in administration of - 13N N: 0.05f < 0.05-0.12 Goen et al. (1995)
municipal waste incineration 8S S: 0.09f < 0.05-0.67
Workers in water supply - 119 0.008 - Hanson et al. (1994)
plants, cotton manufacture,
garbage recycling
Workers in various - 121 0.012e - Hansen et al. (1995)
environments
Unspecified control group - 52N 0.26 - Jongeneelen et al. (1988b)
38S 0.28
Workers in shipping yards - 14N 0.17d - Jongeneelan et al. (1990)
at hot rolling mill 28S 0.51d
Industry, Netherlands - 28 0.51 -
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Coke-oven office workers Levin et al. (1995)
Before renovation - 8N 0.18g -
After renovation - 8N 0.09g -
Construction workers - 34 N 0.4g - Levin et al. (1995)
Water supply workers - 26 N N: 0 0-0.010 Omland et al. (1994)
42S S: 0 0-0.022
Unexposed workers - 10N N: 0.05d,f < 0.03-0.12 Schaller et al. (1993)
10S S: 0.21d,f 0.03-1.2
Workers in water - 20N N: 0.16e 0.1-0.22 Sherson et al. (1992)
purification plants, 26S S: 0.26e 0.18-0.34
Denmark
Guards in aluminium plant - 9 0.31d - Ny et al. (1993)
Maintanance work in - 48 0.61e,f - Van Hummelen et al. (1993)
blast furnace
Office workers in steel - 10 0.51e - Zhao et al. (1990)
plant
Coke ovens
Work at topside oven 0.8-32 3 N N: 5.7d,f - Buchet et al. (1992)
5.9d 3 S S: 6.1d,f
Work at benchside - 4 N N: 1.2d,f -
6 S S: 0.75d,f
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Various occupations 0.39-13 93 N N: 2.7f 0.25-16 Cenni et al. (1993)
near ovens 68 S S: 3.5f 0.29-29
Subgroup topside 2.18 21 6.64f 0.29-29
Subgroup of larry-car - 12 7.76f 3.5-16
operators
Workers with various - 41 N 0.94e - Clonfero et al. (1995)
tasks 65 S 1.53e
Workers at two coke- - 54 0.78e,f 0.01-48 Ferreira et al. (1994a,b)
oven plants
Work at two top side - 19 3.3d 0.8-7.5 Jongeneelen et al. (1990)
ovens 9 2.7d 1.3-6.5
Other work: coke side, - 25 1.9d 0.6-4.1
push side, maintenance
Coke oven 0.9-37 10 4.7g 0.3-30g Levin et al. (1995)
Before renovation, 4
various occupations
After renovation 0.2-6.8 10 1.3g 0.3-5.7g
0.7
Coke workers at 3 plants 0.72-1.5 66 2.45-13.48 - Ovrebro et al. (1995)
Oven workers 33 0.39e,f - van Hummelen et al. (1993)
7 N
26 S
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Subgroup at top side - 7 0.85e,f -
Personnel at coke side, - 13 2.64 0.54-11 Van Rooij et al, (1994b)
push side, top side,
miscellaneous
Workers at top of ovens - 15 S 4.34e - Zhao et al. (1990)
Workers at top or side - 12 S 2.87e -
of ovens
Coal liquefaction
Engineers (control room - 5 8.53 - Quinlan et al. (1995b)
and plant operations)
Technicians (plant - 5 3.74 -
maintenance)
Petrochemical industry
Various operations - - - 0.25-0.68 Boogaard & van Sittert
Workers inspecting furnace, - - - max. 1.56 (1994, 1995)
replacing burners in boilers,
manufacturing rubber grades
Subgroup: production of < 0.01-< 0.22 1
needle coke from ethylene
cracker residue:
Maintenance 3 1.02 0.16-5.51
Operation < 0.17 12 1.13 0.22-13.2
Coal-tar distillation - 4 - 3.7-11.8 Jongeneelen et al. (1986)
operators, cleaner
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Creosote impregnation
Wood impregnation plant - 19 1.6d,e 0.18-10 Viau et al. (1993)
Wood impregnation plant 0.01-0.05 6 97e - Elovaara et al. (1995)
(0.012d)
Wood impregnation plant - 1 20-90 Jongeneelen et al. (1988b)
Aluminum production
Soderberg type anodes:
Potroom workers, 1.9-36 9 0.4-3.6g
respiratory protection 2.8 2.1g - Levin et al. (1995)
50% of time
Potroom workers - - - - Ny et al. (1993)
Electricians, technicians, - 5 0.69d -
engineers, laboratory
workers, industrial
hygiene personnel
Foremen, technicians, 0.88d 4 2.6d -
tappers, crucible cleaner
Crane operators, all- 7.9d 8 14d -
rounders, electrician
Potmen 2.2d 9 31d -
Electrode men 37d 4 40 -
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Prebake type anodes:
Electrode production - 20 0.17-27 Van Rooij et al. (1992)
compartment
Paste plant 1.3 8 3.0d 1.6-7.4
Bake oven 0.3 5 4.4d 0.98-13
Pot relining department 1.2 7 6.0d 1.9-12
Anode bake area - 28 - 0.55-3.6 Tolos et al. (1990)
Anodes from liquid - 17 - 2.1-37f Schaller et al. (1993)
pitch and coke
Anodes not specified - 28 4.2f 0.05-65 Goen et al. (1995)
Production of electrodes
Graphite electrodes - 15 3.2f - van Hummelen et al. (1993)
Graphite electrodes Buchet et al. (1992)
End-product conditioning 0.002-0.4 3N N: 0.55f -
0.03d 7S S: 0.55f
Second thermal treatment 0.002-0.5 8N N: 0.57f -
of electrodes 0.03d 17S S: 0.79f
Progressive heating of raw 0.002-1.9 2N N: 2.53f -
electrodes 0.04d 4S S: 3.13f
Maintenance and repair 0.002-7.5 15N N: 1.21f -
0.21d 2S S: 3.76f
Grinding and mixing of raw 0.57-25 5N N: 2.98f -
components 5.4d 9S S: 2.83f
Electrode impregnation 0.83-73 3N N: 4.14f -
62d 5S S: 4.96f
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Graphite electrodes - 93 1.7e,f 0.03-20 Ferreira et al. (1994a,b)
Carbon electrodes I - 6 5.8f 3.7-43 Goen et al. (1995)
Carbon electrodes II - 3 12.7f 9.4-15
Carbon electrodes III - 14 8.4f 1.1-65
Graphite electrodes Mannschreck et al. (1996)
Crushing 0.09e 2 5.0e,f 0.6-9.4
Baking 1.1-12e 30 12f 0.9-170
Graphitization 0.01-0.11e 24 0.9f 0.1-3.3
Impregnation 0.44-1.1e 9 11f 3.2-42
Conditioning 0.01e 2 1.2e,f 0.9-1.5
Carbon black
Plants manufacturing - 5 - 0.32-0.35 Gardiner et al. (1992)
carbon black
Newspaper printing ink - 1N 0.47 - Jongeneelen et al. (1988b)
1S 0.67
Road paving
Bitumen and coal-tar binders - 43 - 0.9-2.8 Jongeneelen et al. (1988a)
Bitumen and coal-tar binders - 28 - 0.9-3.2 Jongeneelen et al. (1988b)
Bitumen binder - 18 N N: 0.53e - Burgaz et al. (1992)
21S S: 0.67e
Bitumen binder - 3 0.6e - Jongeneelen et al. (1988b)
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Bitumen binder < 0.05 57 0.7g - Levin et al. (1995)
Impregnation of road - 38 4.26f 0.62-22 Goen et al. (1995)
stones with coal-tar
Foundries
Iron foundry, melting, 0.02 25 N N: 0.022 0.006-0.075 Omland et al.(1994)
machine moulding, casting, 45S S: 0.027 0.006-0.16
sand preparations; high
concentration in casting
and mouding
Iron foundry < 0.002 14 2.7e 0.3-6.3 Santella et al. (1993)
0.005-0.012 14 1.8e 0.3-4.2
> 0.012 18 3.6e 0.5-9.7
Iron foundry I 0.00 19 0.013 - Hansen et al. (1994)
0-0.39 14 0.017
Iron foundry II 0.00 13 0.031 -
0-0.039 24 0.022
> 0.039 18 0.027
Iron foundry - 16 N N: 0.11e 0.09-0.13 Sherson et al. (1992)
20S S: 0.42e 0.025-0.59
Steel plant - 12S 1.34e - Zhao et al. (1990)
Diesel exhaust
Railway tunnel under < 0.000-0.04 5N N: 0.08f 0.04-0.31 Cenni et al. (1993)
construction 8S S: 0.18f 0.08-0.38
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Gate-keepers of harbour - 3N N: 0.47e - Jongeneelen et al. (1988b)
terminal for containers 4S S: 0.67e
Waste incinerations
Municipal waste incineration - 53 0.08f < 0.05-0.41 Goen et al. (1995)
Industrial waste incineration - 43 0.06f < 0.05-0.47
Garbage incineration plant - 35N N. 0.12d,f < 0.05-0.41 Schaller et al. (1993)
17S S: 0.22d,f 0.07-0.41
Miscellaneous workplaces
Glass manufacture - 10 0.85 0.2-3.8 Goen et al. (1995)
Lubricating oils in - 7N 0.32f 0.12-0.77 Cenni et al. (1993)
earthenware factories
Clean-up of soil of a dump - 29 0.11d 0.01-0.75 Viau et al. (1993)
contaminated with coal-tars
derived from pyrolysis
of used tyres after major fire
Chimney sweeping - 27 0.05-1.4 - Goen et al. (1995)
0.36f
Meat smoking - 13 < 0.05-0.57 -
0.21f
Fire fighting 0.03-0.7 S - 0.65-1.0
N 0.51-0.6
Table 99 (continued)
N, non-smokers; S, smokers; M, males; F, females
a Unless otherwise stated, male persons were investigated; in some cases, insufficient characterization of
exposure given
b Benzo[a]pyrene, stationary or personal sampling
c 1-Hydroxypyrene concentration in urine (µmol/mol creatinine), range, and, if available, median concentrations;
otherwise, geometric or arithmetic means. Maximum concentrations are given, in post shift samples, in some
studies from the end of the week. Figures are given for the whole study population and on subgroups with high
exposure.
d Geometric mean
e Arithmetic mean
f Calculated from 1-hydroxypyrene concentrations given in original publication as µg/g creatinine or µg/litre.
g Calculated from 1-hydroxypyrene concentrations, given in the original publication as ng/ml urine
The manufacture and handling of bitumens did not result in a
significant increase in urinary excretion of 1-hydroxypyrene
(Jongeneelen et al., 1988a,b; Knecht & Woitowitz, 1990; Burgaz et al.,
1992), suggesting that the main source of exposure to PAH during
paving is the coal-tar used as a binder and not the bitumen itself
(Knecht & Woitowitz, 1990).
In some studies of occupationally exposed individuals, the difference
in urinary 1-hydroxypyrene concentration between smokers and
nonsmokers was greater than expected, suggesting a more than additive
effect of exposure and smoking on the body burden (Jongeneelen et al.,
1990; Sherson et al., 1992; Ovrebo et al., 1994; Clonfero et al.,
1995; Ovrebo et al., 1995; van Schooten et al., 1995). It was
hypothesized that the induced P450 enzymes in smokers result in faster
biotransformation and less efficient ciliary clearance of particles in
the upper airways (Van Rooij et al., 1994a).
The 1-hydroxypyrene concentrations in urine correlated in most cases
with the PAH concentrations in air (Buchet et al., 1992; Levin et al.,
1995; Mannschreck et al., 1996). The weak correlation between the
levels of pyrene in air and 1-hydroxypyrene concentrations in urine
was attributed to extensive dermal uptake of the PAH (Van Rooij et
al., 1992, 1993a,b; Ovrebo et al., 1995). The 1-hydroxypyrene
concentrations in urine correlated quite well with exposure of the
skin, monitored by analysing absorbent pads attached to skin sites
during shifts (Van Rooij et al., 1992, 1993a,b).
Significant dermal uptake, representing up to 95% of the total, was
concluded from the results of several studies of workers exposed at
coke ovens, in coal-liquefaction plants, in the petrochemical
industry, in aluminium reduction plants, in a graphite electrode
plant, in a needle-coke plant, during road paving, and while
impregnating wood with creosote oil (Jongeneelen et al., 1990; Van
Rooij et al., 1992, 1993a,b; Boogaard & van Sittert, 1994; Ferreira et
al., 1994a,b; Van Rooij et al., 1994b; Boogaard & van Sittert, 1995;
Elovaara et al., 1995; Quinlan et al., 1995a,b,c). For example,
workers impregnating wood with creosote had an average, estimated
dermal uptake that was 15 times higher than the estimated respiratory
uptake (Van Rooij et al., 1993b).
Use of dermal protection in the form of impermeable polyvinyl chloride
suits led to a substantial decrease in the urinary concentrations of
1-hydroxy-pyrene (Boogard & van Sittert, 1994, 1995). Frequent changes
of work clothes and underclothes reduced 1-hydroxypyrene excretion by
37-55% (Van Rooij et al., 1994b; Quinlan et al., 1995c).
8.3.2.3 Time course of elimination
The excretion of 1-hydroxypyrene increased significantly between the
beginning and end of a shift and from one day to another during one
week. Decreases were observed between two shifts, but the high values
did not drop to the preshift level of the day before (Jongeneelen et
al., 1988b, 1990; Tolos et al., 1990; Buchet et al., 1992; Van Rooij
et al., 1992; van Hummelen et al., 1993; Van Rooij et al., 1993b;
Omland et al., 1994; Van Rooij et al., 1994b; Elovaara et al., 1995;
Quinlan et al., 1995a,b; van Schooten et al., 1995). After an
exposure-free weekend, the 1-hydroxypyrene concentrations in the urine
of heavily exposed workers did not drop to control levels (Jongeneelen
et al., 1988b, 1990; Viau et al., 1993; Elovaara et al., 1995; Quinlan
et al., 1995b). The baseline values in exposed workers are slightly
higher than those in unexposed controls (Jongeneelen et al., 1988a,
1990; Tolos et al., 1990; Quinlan et al., 1995a,b).
Elimination of 1-hydroxypyrene is biphasic, a moderately rapid phase
being followed by a second, much slower elimination (Jongeneelen et
al., 1988b, 1990; Viau et al., 1995). The half-lives of the first
phase have been determined at various workplaces and for
non-occupationally exposed persons after inhalation, dermal, and oral
exposure. Regardless of the route of exposure, they range from 4.4 to
48 h, most values being about 16 h (Jongeneelen et al., 1988b, 1990;
Buchet et al. 1992; Buckley & Lioy, 1992; Schaller et al., 1993;
Boogard & van Sittert, 1994; Quinlan et al., 1995a,b; Viau & Vyskocil,
1995; Viau et al., 1995). In one study, a half-life of 16 days was
given for the slower phase (Jongeneelen et al. 1988b). This slow
elimination suggests that pyrene accumulates in a secondary
compartment, most probably adipose tissue, from which it is released
only slowly (Jongeneelen et al., 1990).
8.3.2.4 Suitability as a biomarker
When 1-hydroxypyrene was used as a biomarker for exposure to PAH, the
oral, dermal, and inhalation routes were all shown to be important.
Furthermore, low levels of exposure to PAH can be determined. A great
advantage is that the determination of urinary 1-hydroxypyrene is easy
and rapid and thus well suited for use in large-scale epidemiological
studies.
Comparison of different work environments may, however, be difficult,
because the proportion of pyrene in the total PAH or in comparison
with benzo [a]pyrene may vary (Jongeneelen et al., 1990; Buchet et
al., 1992; van Rooij et al., 1993a; Boogard & van Sittert, 1994;
Hansen et al., 1994). For example, the creosote oil used in a wood
impregnation plant contained about 3.4% pyrene and less than 0.0004%
benzo [a]pyrene. Levels of 2-10% pyrene and 0.4-0.6% benzo [a]pyrene
are found in coal-tar, which is the main PAH contaminant in the coke
industry, in the primary aluminium industry, and during road paving
with tar. Polluted ambient air contains about 6.5% benzo [a]pyrene
and 1.8-2.7% pyrene (IARC, 1985; Zhao et al., 1990).
It is not currently possible to assess the risk presented by exposure
to PAH on the basis of urinary 1-hydroxypyrene concentrations, as
epidemiological studies have not demonstrated a relationship with
long-term effects. An indirect dose-response relationship between
urinary 1-hydroxypyrene level and the relative risk for lung cancer
has, however, been estimated for coke-oven workers: 2.3 µmol
1-hydroxypyrene per mol creatinine was estimated to be equal to a
relative risk for lung cancer of approximately 1.3 (Jongeneelen,
1992). Because of the varying composition of PAH mixtures, this risk
estimation cannot be used for other workplaces or ambient air, where a
correction factor may be necessary.
8.3.3 Mutagenicity in urine
The mutagenicity of urine from persons exposed to PAH has been assayed
in a number of studies by Ames' test with Salmonella typhimurium
TA98 or TA100, with and without metabolic activation. In most of these
studies, several urine samples from both control and exposed subjects
could not be assayed because of the toxicity of the urine (Heussner et
al., 1985; Jongeneelen et al., 1986; Clonfero et al., 1989, 1990;
Ferreira et al., 1994b; Santella et al., 1994; Clonfero et al., 1995).
Although tobacco smoke was mutagenic in the presence of metabolic
activation, no increase in mutagenic activity was found in most
studies of workers exposed in occupations such as coking (Reuterwall
et al., 1991; Ferreira et al., 1994a,b), coal-tar distillation
(Jongeneelen et al., 1986), work in Söderberg potrooms of aluminium
plants (Krokje et al., 1988), in anode plants (Clonfero et al., 1984;
Venier et al., 1985; Krokje et al., 1988; Clonfero et al., 1990), and
in a graphite electrode plant (Ferreira et al., 1994a). Only the heavy
exposure of patients with psoriasis to coal-tar applications (Clonfero
et al., 1989, 1990; Santella et al., 1994) and of workers at coke
ovens (Mielzynska & Snit, 1992; Clonfero et al., 1995) and in a carbon
plant (Heussner et al., 1985) resulted in mutagenic urine. Ames' test
therefore appears not to be sensitive enough to detect the presence of
urinary mutagens due to occupational exposure to low levels of PAH
(Becher et al., 1984; Clonfero et al., 1989, 1990).
Expectorate from workers in a coke plant and in Söderberg potrooms in
an aluminium plant showed significantly increased mutagenicity in
Ames' test with S. typhimurium TA98 and TA100 in the presence of
metabolic activation (Krokje et al., 1988; Krokje, 1989).
8.3.4 Genotoxicity in lymphocytes
Genotoxic effects in lymphocytes have been proposed as markers for
exposure to PAH. In studies of iron foundry workers with relatively
low exposure to PAH, elevated frequencies of mutation at the hprt
locus in lymphocytes correlated approximately with the levels of DNA
adducts (Perera et al., 1993, 1994). In one study of coke-oven
workers, significant differences from controls were found in the
number of single-strand DNA breaks; however, there was no difference
between tobacco smokers and nonsmokers (Salagovic et al., 1995).
No increases in the rates of micronuclei, chromosomal aberrations, or
sister chromatid exchange were detected in workers at coke ovens
(Reuterwall et al., 1991), a carbon plant (Heussner et al., 1985), an
aluminium plant (Becher et al., 1984), or a graphite electrode plant
(van Hummelen et al., 1993) or in chimney sweeps (Carstensen et al.,
1993), although in most cases significant effects of smoking could be
detected. In one study in which an increase was found, there was no
difference between tobacco smokers and nonsmokers (Bender et al.,
1988; Salagovic et al., 1995). Environmental pollution in Silesia was
associated with significant increases in the frequencies of sister
chromatid exchange and chromosomal aberration in peripheral blood
cells, independently of smoking (Perera et al., 1992).
8.3.5 DNA adducts
DNA adducts with reactive metabolites (mainly diol epoxides) of
benzo [a]pyrene and other PAH have been identified in numerous
studies (see Section 6). For example, cigarette smokers have higher
levels of adducts with PAH in their lungs than nonsmokers, and there
is a linear relationship between adduct levels and daily or lifetime
cigarette consumption (Phillips et al., 1988).
As binding of electrophilic PAH metabolites to DNA is thought to be a
key step in the initiation of cancer, measurement of DNA adducts could
be an indicator of exposure to PAH and also of cancer risk. As a
surrogate for lung tissue, which is an important target organ for PAH
in humans, the more easily accessible nucleated blood cells and blood
proteins (haemoglobin, albumin) have been investigated.
8.3.5.1 Method of determination
The methods for measuring DNA adducts include immunoassays with
polyclonal and monoclonal antibodies (enzyme-linked immunosorbent
assay [ELISA] and ultrasensitive enzymatic radioimmunoassay),
32P-postlabelling, and synchronous fluorescence spectrophotometry.
Direct comparisons of adduct levels determined by different techniques
may be misleading, however, because different end-points are measured.
For example, polyclonal and monoclonal antisera recognize not only the
benzo [a]pyrene diol epoxide adducts against which they are raised,
but also benz [a]anthracene, chrysene, benzo [k]fluoranthene, and
dibenz [a,c]anthracene, which also form N2 guanine adducts. The
32P-postlabelling assay is even less specific, as it may detect
several aromatic and hydrophobic adducts (Dell'Omo & Lauwerys, 1993).
The detection limits for the three methods are one adduct per 107-108
nucleotides for the immunological methods (Dell'Omo & Lauwerys, 1993)
and synchronous fluorescence spectrometry (Dell'Omo & Lauwerys, 1993;
Rojas et al., 1995) and up to one adduct per 1010 nucleotides in the
32Ppostlabelling assay (Beach & Gupta, 1992; Dell'Omo & Lauwerys,
1993; Ovrebo et al., 1994). DNA adducts may be overlooked with
32P-postlabelling, because of incomplete nuclease P1 digestion,
resistance to 32P-labelling, dephosphorylation of certain adducts, or
co-migration with normal nucleotides (Herbert et al., 1990b; Beach &
Gupta, 1992; Kriek et al., 1993; Segerbäck & Vodicka, 1993; Pavanello
& Levis, 1994). This method is being improved (Segerbäck & Vodicka,
1993; Szyfter et al., 1994).
The results obtained when the different methods were applied in
parallel were usually similar, but the magnitude of the effect
differed (Ovrebo et al., 1990, 1992; Pavanello & Levis, 1994; Perera
et al., 1994). For example, in one study of psoriasis patients treated
with coal-tar, 20-100 times higher levels were found with ELISA than
with the 32P-postlabelling method (Pavanello & Levis, 1994). In
another study, the 32P-postlabelling method was more sensitive than
the ELISA (Kriek et al., 1993). Considerable differences were found in
DNA adduct levels in interlaboratory comparisons (Hemminki et al.,
1990a; Beach & Gupta, 1992; Kriek et al., 1993; Phillips & Castegnaro,
1993).
In the descriptions below, the DNA adduct levels are expressed as
number of adducts per 108 nucleotides or fmol/µg DNA; 33.2 fmol/µg
DNA corresponds to one adduct per 108 nucleotides. Since the levels
in background samples and also in samples from exposed subjects are
sometimes below the limit of detection, the number of positive samples
is often given as well (Herbert et al., 1990a,b; Dell'Omo & Lauwerys,
1993).
8.3.5.2 Concentrations
In general, exposures that lead to the excretion of high
concentrations of 1-hydroxypyrene in urine also lead to elevated DNA
adduct levels. Table 100 gives the DNA adduct levels derived from
studies in which air concentrations and DNA adduct levels were
measured in parallel. Although the concentrations of PAH that occur
under different exposure conditions differ by orders of magnitude (see
section 5.3), the differences in DNA adduct levels are quite small, in
contrast to the results of experiments on excretion of
1-hydroxypyrene. Table 101 summarizes the results of an investigation
of workers in an aluminium reduction plant where the two methods were
applied (van Schooten et al., 1995).
In all populations studied, there is substantial interindividual
variation in PAH-DNA adduct levels, after exposure by inhalation or
orally, which is greater than that described for 1-hydroxypyrene
excretion in urine (Hemminki et al., 1990a,b; Santella et al., 1993;
Szyfter et al., 1994; Rojas et al., 1995). In one study, about 50-fold
interindividual variations were reported among controls and about
100-fold variations among coke-oven workers (Rojas et al., 1995). The
variations are probably due to differences in the induction of AHH
activity in lymphocytes and in the resulting detoxification of
carcinogenic PAH, the ability to repair DNA lesions, and the turnover
of damaged cells (Dell'Omo & Lauwerys, 1993; Szyfter et al., 1994;
Kang et al., 1995; Rojas et al., 1995). These interindividual
variations result in a wide overlap in the ranges of values between
exposed and unexposed subjects in all studies.
Table 100. DNA-polycyclic aromatic hydrocarbon adduct levelsa in various situations of exposure
Population Benzo[a]pyrene Method of Exposed Controls Reference
investigated, (µg/m3) detection
type of emission No. of No. of DNA No. of No. of DNA
subjects adducts/108 subjects adducts/108
nucleotides nucleotides
Polluted ambient air
Industrialized area, 0.015-0.057 32P-Postlabelling 15 13 13 2.3 Hernminki et al.
Silesia, Poland (1990a)
Industrialized area 32P-Postlabelling 19 14 15 4.8 Motykiewicz
Silesia, Poland (1995)
Winter inversion, 0.002-0.008 32P-Postlabelling 29 2.6-6.8 - - Binkova et al.
Teplice, Czech (1995)
Republic
Burning of smoky 19 Immunoassay 18 7.7 18 5.2 Mumford et al.
coal at home, with (1993)
and without
chimneys, China
Coke ovens
Door maintenance 2.3-6.5 Immunoassay 11 5.8b - - Assennato
(1993a)
Work topside 7.3 Fluorescence 13 ND-73b - - Haugen et al.
spectrophotometry (1986)
immunoassay
Battery work 0.54-90 32P-Postlabelling 31 15 13 2.3 Hemminki et al.
(1990a)
Working at high and 0.001-0.009 32P-Postlabelling 31 2.2 22 2.2 Yang et al.
low traffic density (1996)
areas
Table 100. (continued)
Population Benzo[a]pyrene Method of Exposed Controls Reference
investigated, (µg/m3) detection
type of emission No. of No. of DNA No. of No. of DNA
subjects adducts/108 subjects adducts/108
nucleotides nucleotides
Aluminium production
Aluminum plant, prebake 32P-Postlabelling Van Schooten
anode process et al. (1995)
Anode factory 1.5 - 26 - -
Pot relining 1.1 47 - -
Electrode past plant 0.9 32P-Postlabelling 34 11 14 10 Ovrebo et al.
(1994)
Foundries
Foundry 0.02 - - 7.4 - - Perera et al.
(1994)
Iron foundry < 0.005-0.06 Competitive 67 4.4-9.6 - - Santella et al.
ELISA (1993)
Iron foundry < 0.005-0.06 32P-Postlabelling 67 1.9-2.5 - - Perera et al.
(1994)
Iron foundry < 0.05-> 0.2 Fluorescent 35 0.8-21 10 2.2 Perera et al.
ELISA (1988)
Iron foundry < 0.05 32P-Postlabelling 19 7.3 4 4 Szyfter et al.
0.005-0.2 - 63 19 - - (1994)
> 0.2 - 6 29 - -
ND, not detected; ELISA, enzyme-linked immunosorbent assay
a Median or mean values; ranges are from means or medians of several measurements of groups of exposed persons
b In original publication given as fmol/µg DNA. Number of adducts per 108 nucleotides = fmol/µg DNA × 33.2
Table 101. Comparison of methods for measuring exposure to polycyclic aromatic
hydrocarbons in an aluminium plant
Exposure Benzo[a]pyrene Pyrene 1-Hydroxypyrene DNA adducts
(µg/m3)a (µg/m3)a in urine (µmol/mol in leukocytes
creatinine)b (adducts/108
± SD nucleotides)
± SD
Bake oven 0.35 1.5 3.65 ± 2.11 30.1 ± 42.1
Anode factory 1.51 5.6 3.25 ± 1.89 26.2 ± 15.0
Pot relining 1.05 32.3 6.20 ± 8.44 47.3 ± 39.1
Electrolysis 0.03 0.12 0.48 ± 0.27 12.8 ± 10.0
Foundry 0.02 0.04 0.47 ± 0.20 7.4 ± 9.6
From van Schooten et al. (1995)
a Geometric mean
b Arithmetic mean
Significant correlations were found in most studies between the
estimated or measured exposure to PAH and adduct levels (Herbert et
al., 1990a,b; Ovrebo et al., 1990, 1992; Assennato et al., 1993a;
Perera et al., 1994; Szyfter et al., 1994; van Schooten et al., 1995),
but no such correlation was found in others (Herbert et al., 1990a,b;
Kriek et al., 1993; Mumford et al., 1993, Ovrebo et al., 1994; Schoket
et al., 1995).
As shown with 1-hydroxypyrene concentrations in urine, the DNA adduct
concentrations in certain workers may correlate better with dermal
exposure than with PAH concentrations in air (Herbert et al.,
1990a,b).
(a) General population
The levels of DNA adduct in control subjects range from 0.2 to about
10 adducts per 108 nucleotides in leukocytes (Dell'Omo & Lauwerys,
1993). DNA adducts were also found in 43% of placentas and in 27% of
liver samples and 42% of lung specimens from 15 spontaneously aborted
human fetuses. As there was only 60% concordance between placenta and
fetal lung or liver with regard to the presence of detectable adducts,
DNA adducts in the placenta are not a good indicator of adduct
formation in fetal organs. Although several of the mothers were
smokers, none of the fetal samples containing DNA adducts were from
women who smoked during pregnancy, indicating that smoking is unlikely
to have caused adduct formation (Hatch et al., 1990).
Conflicting results have been obtained concerning the effects of
tobacco (cigarette) smoking on DNA adduct levels in peripheral blood
cells. Most investigations of human peripheral lymphocytes have found
no remarkable effect of smoking in control or exposed persons, in
contrast to those of lung and bronchial tissue (Hemminki et al.,
1990a; Herbert et al., 1990a,b; Dell'Omo & Lauwerys, 1993; Binková et
al., 1995; van Schooten et al., 1995; Yang et al., 1996). Some
publications, however, report maximal differences in DNA adduct levels
of about threefold between tobacco smokers and nonsmokers among
controls and exposed individuals (Savela & Hemminki, 1991; Kriek et
al., 1993; Santella et al., 1993; Rojas et al., 1995; van Schooten et
al., 1995). Granulocytes from smokers and nonsmokers showed no
difference in DNA adduct levels, but threefold increases were observed
in T lymphocytes, which have a much longer life than granulocytes
(Savela & Hemminki, 1991).
A synergistic effect of tobacco smoking and occupational exposure to
PAH was reported in two studies (Rojas et al., 1995; van Schooten et
al., 1995). It was hypothesized that tobacco smoke induces AHH in
lymphocytes, resulting in increased formation of benzo [a]pyrene-DNA
adducts in smokers. The large interindividual differences may be due
to the presence of both non-inducible and highly inducible variants in
human lymphocytes (Rojas et al., 1995).
Elevated DNA adduct levels have been detected in the general
populations of industrialized areas in Poland (Silesia) and the Czech
Republic (Teplice) (Hemminki et al., 1990a,b; Perera et al., 1992;
Binková et al., 1995), with levels up to 13 (Hemminki et al., 1990a;
Motykiewicz, 1995) and 5 adducts per 108 nucleotides (Binková et al.,
1995). Levels of 8 adducts per 108 nucleotides were found in
leukocytes from women exposed to high PAH concentrations from burning
smoky coal in China. Although DNA adducts were also detected in
placenta, no dose-response relationships were found between exposure
to benzo [a]pyrene and placental DNA adduct level or the percentage
of samples with detectable DNA adducts (Mumford et al., 1993).
The consumption of charcoal-grilled foods leads to elevated DNA adduct
levels (Rothman et al., 1993; Kang et al., 1995), and such food may
represent a major dietary source of PAH for some populations. Eating
charcoal-grilled beef resulted in a 1.9-3.8-fold increase above the
individual baseline adduct levels in four of 10 subjects (Kang et al.,
1995).
Psoriatic patients undergoing coal-tar treatment had a DNA adduct
level of about 8 per 108 nucleotides (Pavanello & Levis, 1994;
Santella et al., 1995), and levels up to 13 per 108 were found in
skin biopsy samples obtained from patients who had received treatment
with coal-tar ointment. There was no correlation of the adduct levels
after different treatments. No information was given on controls
(Phillips et al., 1990).
(b) Occupational exposure
Workers exposed to PAH had elevated mean levels of adducts and a
higher percentage of positive samples (measured concentrations above
the detection limit) than controls. Elevated DNA adduct levels have
been detected in leukocytes from workers exposed in coke-oven plants
(Assennato et al., 1993a,b; Dell'Omo & Lauwerys, 1993; Harris et al.,
1985; Haugen et al., 1986; Hemminki et al., 1990a,b; Ovrebo et al.,
1992; Kriek et al., 1993a,b; Rojas et al., 1995), aluminium
manufacture (Dell'Omo & Lauwerys, 1993; Kriek et al., 1993a,b; Schoket
et al., 1995; van Schooten et al., 1995), and foundries (Perera et
al., 1988; Dell'Omo & Lauwerys, 1993; Santella, 1993; Santella et al.,
1993; Perera et al., 1994) and among firefighters (Dell'Omo &
Lauwerys, 1993) and roofers (Herbert et al., 1990a,b; Dell'Omo &
Lauwerys, 1993).
In cases of high exposure, for example at coke ovens, 5-70 adducts per
108 nucleotides have been measured. Significant correlations with
exposure concentrations have been found, although the level was no
more than threefold greater than in controls (Hemminki et al.,
1990a,b; Ovrebo et al., 1990; Assennato et al., 1993a).
8.3.5.3 Suitability as a biomarker
DNA adduct levels in the lung may not a reliable indicator of human
cancer risk, although Phillips et al. (1988) found a correlation
between cigarette smoking and DNA adducts in the human lung. Weston et
al. (1993) observed no correlation between lung DNA adduct levels and
a measure of recent tobacco smoking, serum cotinine. Tissue samples
taken from different portions of the same lung showed variations in
DNA adduct levels
The use of lymphocytes as a surrogate for lung cells has also been
questioned, because no correlation has been found between PAH-DNA
adduct levels in human lung and leukocytes (van Schooten et al.,
1992). In studies on rats exposed to coke-oven emissions, the DNA
adduct levels were lower in leukocytes than in lung tissues; in
addition, several types of adducts observed in lung tissue were not
present in leukocytes (Binková et al., 1994). Granulocytes, which form
the majority of peripheral leukocytes, have a relatively short life,
< 24 h, in contrast to lung cells; therefore, adducts are probably
lost within a few days (Savela & Hemminki, 1991; Dell'Omo & Lauwerys,
1993). This can be avoided by using the subfraction of T lymphocytes
which have a half-life of several years. The adduct levels in T
lymphocytes were three times higher than in granulocytes (Savela &
Hemminki, 1991).
DNA adducts are much less sensitive for assessing exposure than
excretion of 1-hydroxypyrene in urine. Additionally, because of the
large interindividual differences in control and exposed groups,
adduct levels can be compared only on a group basis. Thus, PAH-DNA
adducts can be used as a qualitative biomarker of exposure to
combustion emissions but to only a limited extent as a quantitative
marker. This method may, however, allow identification of subjects who
are highly susceptible to the DNA-damaging properties of PAH and are
therefore predisposed to lung cancer. This was seen in one
investigation of lung cancer patients with a family history of lung
cancer. Monocytes from these patients treated in vitro with PAH
showed a slight but significant enhancement of formation of
benzo [a]pyrene-DNA adducts in comparison with controls (Nowak et
al., 1992). It is not yet known whether metabolism in leukocytes is
identical to that in lung cells.
8.3.6 Antibodies to DNA adducts
Antibodies to DNA adducts in leukocytes of exposed workers have also
been found (Harris et al., 1985; Haugen et al., 1986; Vähäkangas et
al., 1992; Santella et al., 1995). In a study of coal-tar-treated
patients, elevated levels were also found in controls (Santella et
al., 1995). Since the initial antigenic stimulus could have occurred
several years previously, antibodies to benzo [a]pyrene-DNA adducts
are considered general indicators of past exposure to PAH.
8.3.7 Protein adducts
Because genotoxic compounds can bind to haemoglobin and serum protein,
the assessment of PAH-blood protein adducts has also been considered
as a possible marker of exposure to PAH or even as a surrogate for the
evaluation of adduct concentrations at the level of the target organs.
This approach has several advantages. Relatively large amounts of
haemoglobin and albumin can be obtained easily from a small volume of
human blood. As the lifetime of haemoglobin in humans is about 120
days and that of albumin 20-24 days, exposure days and weeks
previously can be measured. Albumin is synthesized in the liver, where
PAH are metabolized. Therefore, reactive metabolites may easily gain
access to the proteins. Finally, there may be lower interindividual
variation, because there is no repair, as in the case with DNA
(Dell'Omo & Lauwerys, 1993).
The benzo [a]pyrene-albumin adduct concentrations were similar in
foundry workers and controls, both smokers and nonsmokers (Omland et
al., 1994), and in patients with psoriasis treated with coal-tar
(Santella et al., 1995). Minor effects were detected in foundry
workers and roofers (Lee et al., 1991). No pronounced differences in
haemoglobin adduct levels were detected in workers in steel foundries
and in one graphite electrode producing plant (Ferreira et al.,
1994a,b). In another study, however, significantly increased
benzo [a]pyrene binding was detected in serum proteins from smoking
and non-smoking foundry workers (Sherson et al., 1990).
As protein adducts have been used in relatively few studies, no
conclusion can be drawn about the usefulness of this biomarker.
8.3.8 Activity of cytochrome P450
Increased mRNA levels of the CYP1A1 gene, which belongs to the
P4501A1 family responsible for the metabolism of PAH, including
benzo [a]pyrene, have been proposed as biomarkers for exposure to PAH
(Cosma et al., 1992; Van den Heuvel et al., 1993; see also Section 6).
Although the basal levels were not increased, 3-methylcholanthrene
caused greater induction in lymphocytes from railroad workers exposed
to creosote in cell culture (Cosma et al., 1992).
The activity of the cytochrome P450 CYP 1A2 was also determined by
measuring caffeine metabolites in urine. There were significant
differences between tobacco smokers and nonsmokers, but there was no
difference between nonsmoking and smoking foundry workers and the
respective controls (Sherson et al., 1992).
8.3.9 Differentiation antigens on the surface of lung cells
Lung epithelial cells from sputum have been tested for antigens that
indicate neoplastic transformation (Assennato et al., 1993b). One of
23 coke-oven workers who had differentiation antigens on the cell
surface was a smoker who had severe airways obstruction and moderate
dysplasia of bronchial epithelium cells.
8.3.10 Oncogene proteins
Since oncogene activation may be an early step in the carcinogenic
process, its detection may be a useful marker for identifying
individuals at risk for the development of malignancy. Plasma levels
of ras oncogene-related p21 proteins were elevated in a sample of
male residents of the highly industrialized Silesian region of Poland.
They also had elevated DNA adduct levels in their leukocytes (Perera
et al., 1992). Three of 18 foundry workers screened for the oncogene
proteins sis, fes ß-TGF, int-1, myb, src, myc, mos, and ras in serum
had elevated levels of ras and fes; however, two were smokers. No
unexposed individuals had abnormal serum oncogene protein expression.
The levels of ras and fes proteins were also increased in the serum of
lung cancer patients (Brandt-Rauf et al., 1990).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND THE FIELD
Appraisal
Data on toxicity are available mainly for naphthalene, phenanthrene,
and fluoranthene and are scarce for other polycyclic aromatic
hydrocarbons (PAH). Both metabolism and photooxidation can alter the
toxicity of PAH in the environment, photooxidation tending to increase
toxicity.
At low concentrations, PAH can stimulate the growth of microorganisms
and algae. The highest values for the no-observed-effect concentration
(NOEC) were determined for naphthalene (3100 µg/litre in Anabaena
flos-aquae) and acenaphthene (> 4600 µg/litre in A. flos-aquae).
The NOEC values for fluorene, phenanthrene, fluoranthene, and pyrene
were about one order of magnitude lower. Benz [a]anthracene and
chrysene were the most toxic towards algae (NOEC, < 10 µg/litre). The
effective concentration that caused a 50% change (EC50) was between 5
µg/litre for benzo [a]pyrene and 54 000 µg/litre for fluoranthene.
For invertebrates like crustaceans, insects, molluscs, polychaetes,
and echinoderms, naphthalene is least toxic, with a 48-h median lethal
concentration (LC50) of 700-23 000 µg/litre. For three-ring PAH, the
LC50 values ranged between < 1 and 3000 µg/litre. Anthracene may be
more toxic than the other three-ring PAH, with 24-h LC50 values
between < 1 and 260 µg/litre. The values for most four- and five-ring
PAH are between 0.7 and 1800 µg/litre and 0.4-120 µg/litre,
respectively. For six-ring PAH, only one LC50 is available: 0.2
µg/litre for benzo [ghi]perylene. The values for the NOEC are
slightly lower than those for the LC50. The lowest NOEC reported for
benzo[a]pyrene is 0.14 µg/litre.
Naphthalene, acenaphthene, and fluorene are the least toxic for
vertebrates like fish and amphibians, with 96-h LC50 values of 110 to
> 10 000 µg/litre; for phenanthrene and fluoranthene, the LC50
values are 30-4000 µg/litre. Most of the LC50 values for anthracene
were between 2.8 and 360 µg/litre. For four- and five-ring PAH, the
LC50 values were 0.7-26 µg/litre. The lowest NOEC reported for
benzo [a]pyrene in fish was 2.4 µg/litre. In tests with
sediment-dwelling organisms, LC50 values of 3-15 mg/kg dry weight
were determined for fluoranthene.
In earthworms, an LC50 value for growth was 170-200 mg/kg dry weight
for fluorene, and an LC50 value for reproduction was 150 mg/kg dry
weight for phenanthrene. At 180 mg/kg dry weight, chrysene,
benzo [k]fluoranthene, and benzo [a]pyrene did not affect
reproduction by the earthworm Folsoma candida.
9.1 Laboratory experiments
Data on the toxicity of individual PAH to members of different
taxonomic groups are presented in Tables 102-104; data on the toxicity
of PAH metabolites are not included. The use of solvents in laboratory
tests for toxicity often results in unrealistically high
concentrations of PAH, which exceed their maximum water solubility.
Only data from tests with concentrations that did not exceed 10 times
the estimated solubility were used.
9.1.1 Microorganisms
9.1.1.1 Water
The effects of several three-, four-, and five-ring, unsubstituted PAH
on the growth of the bacterium Escherichia coli were studied at
concentrations of 10-5, 10-6, and 10-7 mol/litre in the growth medium
(Hass & Applegate, 1975). Benz [a]anthracene,
dibenz [a,h]anthracene, and benzo [a]pyrene promoted bacterial
growth; pyrene and phenanthrene had slight promoting effects at low
concentrations (10-7 and 10-6, respectively) and inhibitory effects
at higher concentrations, whereas anthracene and chrysene inhibited
bacterial growth at all concentrations.
The effects of dissolved PAH on the growth rate, lag time before
initiation of growth, and the number of cells at the end of the log
growth phase, measured as maximum light absorbance, were determined in
two species of marine bacteria, Serratia marinorubra and Vibrio
parahaemolyticus. Most of the PAH tested increased the lag time and
decreased the growth rate and cell yield; pyrene, however, increased
the growth rate. The extent of inhibition of growth was a function of
both the concentration of PAH and their inherent toxic properties,
which decreased with solubility. Thus, the toxicity of naphthalene at
a concentration of 13 mg/litre was similar to that of benzo [a]pyrene
at 5 µg/litre (Calder & Lader, 1976).
PAH at concentrations of 5-20 µg/litre stimulated the growth of the
freshwater algae Chlorella vulgaris, Scenedesmus obliquus, and
Ankistrodesmus orauntic (Gräf & Nowak, 1966) and of the marine
dinoflagellate Gyrodinium sp. (Ishio et al., 1977). The growth of
sporelings of the marine algae Antithamnium plumula was
progressively inhibited after exposure to 10-300 µg/litre of
benz [a]anthracene.
The growth of the blue-green alga A. flos-aquae was inhibited by
16-50% in comparison with controls after exposure to 5-29 µg/litre
benz [a]anthracene for 14 days. In the same study, 14 days' exposure
to fluoranthene at concentrations of 38-1100 µg/litre inhibited growth
by 19-65%. The NOEC values ranged from 1 µg/litre for benzo [a]pyrene
to > 4600 for acenaphthene (Bastian & Toetz, 1982). Other data for
cyanobacteria are listed in Table 102.
Table 102. Results of tests for the toxicity of polycyclic aromatic hydrocarbons (PAH) towards algae and plants
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic two-ring PAH
Acenaphthene
Anabaena flos-aquae A S Contnuous light 2 h NOEC > 4600 Nitrogen fixation, Bastian & Toetz (1985)
no solvent acetylene reduction
250 × 103 cells/ml activity
Selenastrum 96 h EC50 520 Cell number US Environmental
capricornutum Protection Agency
(1978a)a
Fluorene
Anabaena flos-aquae A S Continuous light, 2 h NOEC 260 Nitrogen fixation Bastian & Toetz (1985)
no solvent
250 × 103 cells/ml
Dunaliella bioculata N S Continuous light, 72 h EC50 15 500 Growth rate Heldal et al. (1984)
solvent methanol
250 × 103 cells/ml
Naphthalene
Anabaena flos-aquae A S Continuous light, < 14 d NOEC 3100 Growth Bastian & Toetz (1985)
250 × 103 cells/ml
Chlamydomonas 24 h LC61 34 400b US Environmental
angulosa Protection Agency
(1986d)
Chlorella vulgaris 48 h LC61 33 000b Cell number Kauss & Hutchinson
(1975)
Champia parvula N R Solvent,TEG 11-14d MATC < 695 Female growth Thursby et al. (1985)
Number cystocarps
Nitzchia palea A S No solvent 4 h EC50 2820 Assimilation rate Millemann et al. (1984)
Selenastrum A S Solvent, methanol 4 h EC50 2960 Assimilation rate Millemann et al. (1984)
capricornutum
Table 102. (continued)
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic three-ring PAH
Anthracene
Chlamydomonas N S 5 × 104 cells/ml 3 h EC50 239b Photosynthesis Hutchinson et al.
angulosa (log phase) inhibition (1980)
Chlorella vulgaris N S 20 × 104 cells/ml 3 h EC50 535b Photosynthesis Hutchinson et al.
(log phase) inhibition (1980)
Selenastrum N R Solvent, acetonec 28 h EC50 3.9-37 Growth rate Gala & Giesy (1992)
capricornutum (8 h) nitrile UV-A cont.c EC10 1.5-7.8 Growth rate
1 × 105 cell/ml
Selenastrum Cool-white light EC30 40 000d Growth US Environmental
capricornutum Protection Agency
(1987b)
Selenastrum Gold fluorescent NOEC 8000d Growth US Environmental
capricornutum light Protection Agency
(1987b)
Fluoranthene
Anabaena flos-aquae A S Continuous light, 2 h LOEC 230 Nitrogen fixation Bastian & Toetz (1982)
no solvent
Scenedesmus N S Solvent, acetone 96 h EC10 1.6 Growth Kordel et al. (1981)
subspicatus 96 h EC50 12
Selenastrum 96 h EC50 54 400d Cell number US Environmental
capricornutum Protection Agency
(1978b)
Selenastrum 96 h EC50 54 600d Chlorophylla US Environmental
capricornutum Protection Agency
(1978b)
Skeletonema costatum 96 h EC50 45 000d Chlorophyla US Environmental
Protection Agency
(1978b)
Skeletonema costatum 96 h EC50 45 600d Cell number US Environmental
Protection Agency
(1978b)
Table 102. (continued)
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Phenenthrene
Anabaena flos-aquae A S Continuous light, 2 h NOEC 130 Nitrogen fixation Bastian & Toetz (1985)
no solvent,
250 × 103 cells/ml
Nitzschia palea A S No solvent 4 h EC50 870 Assimilation rate Millemann et al. (1984)
Selenastrum A S No solvent 4 h EC50 940 Inhibition of Millemann et al. (1984)
capricornutum photosynthesis
Aromatic four-ring PAH
Benz[a]anthracene
Anabaena flos-aquae A S Continuous light < 14 d NOEC 1 Growth Bastian & Toetz (1982)
Anabaena flos-aquae A S Continuous light, 2 h NOEC 19b Acetylene reduction Bastian & Toetz (1985)
no solvent
250 × 103 cells/ml
Anabaena flos-aquae A S Continuous light < 14 d EC 18-29b Growth Bastian & Toetz (1982)
16-48%
Antithamnium plumula - EC 17% 10 Cell production level Boney & Corner (1962)
in algal medium
Chrysene
Anabaena flos-aquae A S Continuous light, 2 h NOEC 5b Nitrogen fixation Bastian & Toetz (1985)
no solvent,
250 × 103 cells/ml
Anabaena flos-aquae A S Continuous light 14 d EC35 62-96% Growth Bastian & Toetz (1982)
saturation
Pyrene
Anabaena flos-aquae A S Continuous light < 14 d NOEC < 100 Growth Bastian & Toetz (1982)
Chlamydomonas N S 5 × 104 cells/ml 3 h EC50 202b Inhibition of Hutchinson et al.
angulosa (log phase) photosynthesis (1980)
Chlorella vulgaris N S 20 × 104 cell/ml 3 h EC50 332b Inhibition of Hutchinson et al.
(log phase) photosynthesis (1980)
Table 102. (continued)
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic five-ring PAH
Benzo[a]pyrene
Anabaena flos-aquae N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
Antithamnium plumula 96 h EC 49% 10b Cell production Boney & Corner (1962)
96 h EC 54% 100d increase
Chlamydomonas N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
reinhardtii
Euglena gracilis N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
Porphyra tenera 80-320 EC 1000d Cell size decrease Boney (1974)
min
Poteriochromonas N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
malhamensis
Scenedasmus acutus N S 500 × 103 cells/ml 72 h EC50 5b Growth Schoeny et al. (1988)
Scenedesmus N S Solvent, acetone 20 h EC 300d Chlorophyll content Zachleder et al. (1983)
quadracanda
Scenedesmus N S Solvent, acetone 20 h EC 300d Biomass Zachleder et al. (1983)
Perylene
Scenedesmus N S Solvent acetone 96 h EC10 0.0066 Growth Kordel et al. (1981)
subspicatus 96 h EC50 > 0.32
A, analysed concentration; N, nominal concentration; S, static system; F, flow-through system; R (0.5 d), system with renewal
(each half day)
NOEC, no-observed-effect concentration; EC, effect concentration; LC, lethal concentration; MATC, maximum acceptable toxicant
concentration; LOEC, lowest-observed-effect concentration; TEG, triethylene glycol
a From Cairns & Nebeker (1982)
b Concentration higher than solubility but not exceeding it by 10 times
c Explicitly mentioned that organisms were tested for phototoxicity of test substance either by sunlight or artificial UV radiation
d Concentration 10 times higher than the solubility
Naphthalene had no detectable effect on the rate of respiration of
yeast at concentrations up to its maximum solubility (31 mg/litre)
(Haubenstricker et al., 1990).
9.1.1.2 Soil
Few data are available on the toxicity of PAH to microbial communities
in the soil. Application of 500 mg/kg dry weight naphthalene did not
reduce soil microbial respiration or nitrogen mineralization, and 90%
was degraded within 10-20 days (Kirchmann et al., 1991).
9.1.2 Aquatic organisms
9.1.2.1 Plants
For many PAH, the dose that is toxic for algae exceeds the maximum
solubility in water (Table 102). Benz [a]anthracene (four-ring) and
benzo [a]pyrene (five-ring) are considered to be the most toxic PAH,
with EC50 values of 29 µg/litre and 5-15 µg/litre, respectively. The
EC50 values for three-ring PAH are 240-940 µg/litre. Naphthalene and
fluorene (two-ring) are considered to be the least toxic, with EC50
values of 2800-15 000 µg/litre. The ranges of EC50 values are 200-330
for four-ring compounds and 5-> 4000 µg/litre for five-ring PAH.
9.1.2.2 Invertebrates
Data on the toxicity of two- to six-ring PAH are available for
invertebrates such as crustaceans, insects, molluscs, polychaetes, and
echinoderms (Table 103). The data include the results of both
phototoxicity and non-phototoxicity tests, which are poorly comparable
because of the dynamic character of the ultraviolet radiation-induced
oxidation products and the often short exposure time.
Phenanthrene, fluorene, and triphenylene did not cause photoinduced
toxicity to Daphnia and did not absorb light. Benzo [a]fluorene,
benzo [k]-fluorene, and chrysene were considered to be moderately
toxic to Daphnia, whereas anthracene, fluoranthene, pyrene,
benz [a]anthracene, benzo [k]-fluoranthene, benzo [a]pyrene,
benzo [e]pyrene, perylene, dibenz [a]anthracene, and
benzo [ghi]perylene were very toxic and absorbed more energy.
Toxicity was also correlated with the phosphorescence lifetime and
energetic state of the molecule. The authors stated that the
photodynamic response was due to the PAH assimilated into organisms
rather than to external photoproducts. Phototoxicity occurs as a
result of energy transfer from activated (triplet) PAH to ground-state
oxygen. Singlet oxygen is a reactive chemical that can denature
biomolecules within aquatic organisms. The authors estimated that the
internal concentration of PAH that caused the death of 50% of the
Daphnia was 77 nmol/g wet weight in continuous light. No toxic
effects were seen in the absence of light. The phototoxicity of
anthracene was confirmed by experiments with
Daphnia pulex (Newsted & Giesy, 1987).
Table 103. Results of tests for the toxicity of polycyclic aromatic hydrocarbons (PAH) towards invertebrates
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
PROTOZOANS
Aromatic two-ring PAH
Acenaphthylene
Tetrahymena 24 h EC50 6300a Population Yoshioka et al.
puryformis growth (1986)
Aromatic three-ring PAH
Anthracene
Paramecium aurelia 10 000 N S Dark 1.5 h NOEC 1000b Mortality Joshi & Misra
cells/litre 0.75 h sunc 0.75 h EC 100% 100a Mortality (1986)
Paramecium 1 h LC90 1000b US Environmental
caudatum Protection
Agency (1987b)
Aromatic five-ring PAH
Benzo[a]pyrene
Gyrodinium sp. Log phase N S Solvent acetone 12 d EC 20% 5a Cell division Ishio et al. (1977)
period
increase
INSECTS
Aromatic two-ring PAH
Fluoranthene
Chironomus riparius Larvae N S l/d = 16/8 48 h EC50 2350 Reproduction Finger et al.
A IF 30 d NOEC 142 Emergence (1985)
Hexagenia bilineata N S 96 h LC50 5800
Naphthalene
Chironomus 4th instar A F Solvent, ethanol Chronic LOEC 500 Physiology Darville & Wilhm
attenuatus LC50 1300 (1984)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Chironomus tentans Larvae A S Solvent, methanol 48 h LC50 2,810 Millemann et al.
(1984)
Somatochlora Nymph - S 21°C 96 h LC50 1000- Correa & Coler
cingulata 2500 (1983)
Tanytarsus dissimilas Life cycle A F Solvent, ethanol Chronic LOEC 500 Physiology Darville & Wilhm
LC50 1300 (1984)
Anthracene
Aedes aegypti 3rd-4th instar N S Solvent DMSO 1 d LC50 < 1.0 Borovsky et al.
6 h sun, 18 h darkc (1987)
Aedes aegypti < 8 h old N S 1 h UVc 12 h LC50 150a Kagan et al.
(1985)
Aedes 3rd-4th instar N S Solvent, DMSO 1 d LC50 260a Borovsky et al.
taeniorrhynchus 6 h sun, 18 h darkc (1987)
Culex 3rd-4th instar N S Solvent, DMSO 1 d LC50 37 Borovsky et al.
quinquefasciatus 6 h sun, 18 h darkc (1987)
Culex sp. 24 h LC50 26.8 US Environmental
Protection Agency
(1987b)
Fluoranthene
Aedes aegypti 3rd-4th instar N S Solvent, DMSO 24 h LC50 10 Borovsky et al.
6 h sun, 18 h darkc (1987)
Aedes aegypti < 8-h old N S 1 h sunc 12 h LC50 12 Kagan et al.
(1985)
Aedes 3-4 instar N S Solvent, DMSO 24 h LC50 48 Borovsky et al.
taeniorrhynchus 6 h sun, 18 h dark, (1987)
Culex 3rd-4th instar N S Solvent, DMSO 24 h LC50 45 Borovsky et al.
quinquefasciatus 6 h sun, 18 h darkc (1987)
Phenanthrene
Chironomus tentans Larvae A S Solvent, methanol 48 h LC50 490 Millemann et al.
(1984)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic four-ring PAH
Pyrene
Aedes aegypti 1st instar N S 1 h UVc 11 d EC 18% 0.9 Adult Kagan & Kagan
7 d NOEC 0.9 emergence (1986)
N S Dark 11 d NOEC 30
< 8-h old N S 1 h UVc 12 h LC50 20 Kagan et al. (1985)
Aedes aegypti 3rd-4th instar N S Solvent, DMSO 24 h LC50 35 Borovsky et al.
6 h sun, 8 h darkc (1987)
Aedes 3rd-4th instar N S Solvent, DMSO 24 h LC50 60 Borovsky et al.
aeniorrhynchus 6 h sun, 8 h darkc (1987)
Culex 3rd-4th instar N S Solvent, DMSO 24 h LC50 37 Borovsky et al.
quinquefasciatus 6 h sun, 8 h darkc (1987)
Aromatic five-ring PAH
Benzo[a]pyrene
Aades aegypti 1st instar N S 30 min UVc 11 d NOEC 0.14 Adult Kagan & Kagan
1st instar N S Dark 11 d NOEC 0.9 emergence (1986)
4th instar N S Dark 7d NOEC 6700b
4th instar N S 30 min UVc 7 d NOEC 30a
4th instar N S 30 min UVc 7 d LC50 120b
POLYCHAETES
Aromatic two-ring PAH
Fluorene
Neanthes Immature A S Solvent, acetone 96 h LC50 1000 Rossi & Neff
arenacoendata adult artificial seawater (1978)
Naphthalene
Neanthes Immature A S Solvent, acetone 96 h LC50 3800 Rossi & Neff
arenacoendata adult artificial seawater (1978)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic three-ring PAH
Fluoranthene
Neanthes Immature A S Solvent, acetone 96 h LC50 500a Rossi & Neff
arenacoendata adult artificial seawater (1978)
1-Methylphenanthrene
Neanthes Immature A S Solvent, acetone 96 h LC50 300b Rossi & Neff
arenacoendata adult artificial seawater (1978)
Phenanthrene
Neanthes Immature A S Solvent, acetone 96 h LC50 600 Rossi & Neff
arenacoendata adult artificial seawater (1978)
Aromatic four-ring PAH
Benz[a]anthracene
Nereis virens A S Sediment 6 d NOEC 14.4 Oxygen McElroy (1985)
mg/kg consumption
NOEC 14.4 Ammonia
mg/kg extraction
NOEC 14.4 Microsomal
mg/kg AHH activity
Chrysene
Neanthes Immature A S Solvent, acetone 96 h NOEC > 1000b Mortality Rossi & Neff
arenacoendata adult artificial seawater (1978)
Aromatic five-ring PAH
Benzo[a]pyrene
Neanthes Adult A S Solvent, acetone 96 h NOEC > 1000b Mortality Rossi & Neff
arenaceodentata (1978)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Dibenz[a,h]anthracene
Neanthes Immature A S Solvent, acetone 96 h NOEC > 1000b Mortality Rossi & Neff
aranacoendata adult artificial seawater (1978)
MOLLUSCS
Aromatic two-ring PAH
Acenaphthene
Aplexa hypnorum Adult A F Solvent, 6 h LC50 > 2040 Holcombe et al.
isopropanol 9 (1983)
Fluorane
Mudalia potosensis N S 96 h LC50 5600a Finger et al.
(1985)
Naphthalene
Physa gyrina Adult A S No solvent 48 h LC50 5020 Millemann et al.
(1984)
Aromatic three-ring PAH
Fluoranthene
Mytilus edulis 40-50-mm A S Solvent, acetone 9 d EC50 80 Feeding rate Donkin et al.
shell (1989)
Aromatic five-ring PAH
Benzo[a]pyrene
Mercenaria A F Columns 1-15 EC < 0.001 Haemocyte Anderson et al.
mercenaria weeks lysozome (1981)
concentration
increase
8 weeks EC < 0.001 Bacterial
clearance
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Mytilus californianus 24 h EC 20% 10 000b Oxygen Sabourin & Tullis
consumption (1981)
EC 10% 1000b Oxygen
consumption
ECHINODERMS
Aromatic five-ring PAH
Benzo[a]pyrene
Strongylocentrotus Gametes S N Solvent, ethanol 30-45 NOEC 0.5 Cytological Hose (1985)
purpuratus, min abnormality
LOEC 0.5 Mitoses/embryo
NOEC 50b Fertilization
success
Psammechinus Eggs N S Artificial seawater 100 min NOEC 2000b Development Bresch et al.
miliaris (fertililized (1972)
30 min)
CRUSTACEANS
Aromatic two-ring PAH
Acenaphthene
Daphnia magna N S Solvent 48 h LC50 41 000b LeBlanc (1980)
NOEC 600 Mortality LeBlanc (1980)
Fluorene
Daphnia magna A R (0.5 d) No solvent 2 d NOEC 17.0 Mortality Newsted & Giesy
-UV:1 d; +UV:1 dc (1987)
Daphnia magna A IF l/d = 16/8 21 d NOEC 62.5 Reproduction Finger et al.
21 d LOEC 125 Reproduction (1985)
N S 270 mg.l CaCO3 49 h EC50 430 Immobilization
Solvent, acetone
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia pulex N S Solvent, acetone 48 h EC50 212 Immobilization Smith et al. (1988)
Gammurmus N S 96 h LC50 600 Finger et al.
pseudolimnaeus (1985)
Naphthalene
Daphnia magna < 24 h N S Solvent 48 h NOEC 600 Mortality LeBlanc (1980)
Daphnia magna Adult N Solvent, ethanol 4 h LOEC 1000 Behaviour Whitman & Miller
(1982)
Daphnia magna A S No solvent 48 h LC50 2160 Millemann et al.
(1984)
Daphnia magna N S 48 h EC50 4700 Immobilization Smith et al. (1988)
Daphnia magna < 24 h S 48 h EC50 4100 Crider et al. (1982)
Daphnia magna N S Solvent 48 h LC50 8600 LeBlanc (1980)
Daphnia magna 4-6 d N S No solvent 48 h LC50 16 000 Bobra et al. (1983)
Daphnia magna N S Solvent, acetone 48 h LC50 22 600 Eastmond et al.
+ triton-X-100 (1984)
Daphnia pulex 24-h old A R Filtered crystals Chronic NOEC 330 Increased Geiger & Buikema
lifespan & (1982)
reproduction
Chronic LOEC 600 Growth
Daphnia pulax 1.9-2.1 mm N S No solvent 96h LC50 1000 Trucco et al.
(1983)
Daphnia pulex N S 48 h LC50 3400 Geiger & Buikema
(1981)
Daphnia pulex N S Solvent, acetone 48 h EC50 4660 Immobility Smith et al. (1988)
Elasmopus sp. Adult - S 22°C (closed 24 h LC50 5000 Lee & Nicol
bottles) (1978)
Gammarus minus A S 48 h LC50 3930 Millemann et al.
(1984)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Hemigrapsus A F Seawater 8 d LC50 2800 Mortality and Gharrett & Rice
nudus 4 h water/8 h air 18 d EC50 700 locomotion (1987)
A F Seawater 8 d LC50 2200 Mortality and
8 h water/4 h air 18 d EC50 2000 locomotion
A F Seawater 8 d LC50 1100 Mortality and
12 h water/0 h air 18 d EC50 800 locomotion
Neomysis A F Artificial sea- 96 h LC50 850 Smith &
americana water, 25°C Hargreaves
A F Artificial sea- 96 h LC50 1280 (1983)
water, 15°C
Palaemonetes - - - - 24 h LC50 2500 Anderson et al.
penaeus (1974)
Pandalus - - S 12°C 96 h LC50 970 Korn et al. (1979)
goniurus S 8°C 96 h LC50 1020
S 4°C 96 h LC50 2200
Parhyale Adult - S 22°C 24 h LC50 6000 Lee & Nicol
hawaiaensis (1978)
Aromatic three-ring PAH
Anthracene
Artemia salina 1 d N S 1 h UVc 3 h LC50 20 Kagan et al.
(1985)
Artemia salina N S Darkc 24 h LC50 > 50 Abernethy et al.
(1986)
Daphnia magna - - UV-A=0 21 d NOEC 2.2 Population Foran et al. (1991)
UV-A=31c 21 d NOEC 2.2 growth
UV-A=60c 21 d NOEC 2.2
UV-A=117c 21 d NOEC 1.9
Daphnia magna Adult N S 1 h UVc 2 h LC50 20 Kagan et al.
(1985)
Daphnia magna A R (0.5d) No solvent 1.21 d LC50 15 Newsted & Giesy
-UV:1 d; (1987)
+UV:0.21 dc
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia magna 4-6 d N S Dark 48 h LC50 35.6 Abernethy et al.
(1986)
Daphnia magna 4-6 d N S No solvent 48 h LC50 3030b Bobra et al. (1983)
Daphnia magna < 24 h S 48 h LC0 < 500b Eastmond et al.
(1984)
Daphnia pulex A S 0.25 h sun, 24.25 h EC50 1.2 Immobility Allred & Giesy
1 d darkc (1985)
A S 0.17 h sun, 24.17 h EC100 9.6
1 d darkc
A S 0.75 h filtered 24.75 h NOEC 12.7
sun, 1 d darkc EC 75% 26.4
Daphnia pulex N S Solvent, acetone 48h EC50 754b Immobility Smith et al. (1988)
Benzo[a]fluorene
Daphnia magna A R (0.5 d) No solvent 1.96 d LC50 4.8 Newsted & Giesy
-UV:1 d; +UV:0.96 dc (1987)
Benzo[b]fluorine
Daphnia magna A R (0.5 d) No solvent 1.93 d LC50 2.2 Newsted & Giesy
-UV:1 d; +UV:0.93 dc (1987)
Fluoranthene
Artemia salina N S 1 h sunc 3 h LC50 40 Kagan et al.
(1985)
Daphnia magna Adult N S 1 h sunc 2 h LC50 4 Kagan et al.
(1985)
Daphnia magna A R (0.5 d) No solvent 1.45 d LC50 9.0 Newsted & Giesy
-UV:1 d; +UV:0.45 dc (1987)
Daphnia magna N S 48 h LC50 325 000b US Environmental
Protection Agency
(1978b)
Daphnia magna < 24 h N S Solvent 48 h LC50 320 000b LeBlanc (1980)
NOEC < 8800b Mortality
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Mysidopsis bahia 96 h LC50 40 US Environmental
Protection Agency
(1978b)
Phenanthrene
Artemia salina N S Dark 24 h LC50 677 Abernethy et al.
(1986)
Daphnia magna A S 48 h LC50 700 Millemann et al.
(1984)
Daphnia magna N S Solvent, acetone 48 h LC50 840 Eastmond et al.
+ triton-X-100 (1984)
Daphnia magna 4-6 d N S No solvent 48 h LC50 1160 Bobra et al. (1983)
Daphnia magna A R (0.5 d) No solvent 2 d NOEC 40.1 Immobility Newsted & Giesy
-UV:1 d; +UVA dc (1987)
Daphnia magna A F Glass column 21 d LC50 130 Hooftman &
21 d EC50 50 Reproduction Evers-de Ruiter
21 d NOEC 21 Reproduction (1992a)
21 d NOEC 66 Mortality
21 d NOEC 38 Growth,
condition,
behaviour
A R Glass column 21 d EC50 180 Reproduction
21 d NOEC 100 Reproduction
Daphnia magna 4-6 d N S Dark 48 h LC50 207 Abernethy et al.
(1986)
Daphnia pulex 1.9-2.1 mm N S No solvent 96 h LC50 100 Trucco et al.
(1983)
Daphnia pulex N S Solvent, acetone 48 h EC50 350 Immobility Smith et al. (1988)
Daphnia pulex N S 48 h EC50 734 Immobility Passino & Smith
(1987)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia pulex 48 h LC50 960- Geiger & Buikema
1280 (1982)
24 h A R (1 d) No solvent approx. NOEC 110 Reproduction,
50 d growth
Daphnia pulex N R (2-3 d) 97% A.I. 21 d LC33-73% 130 Savino & Tanabe
Solvent, acetone LOEC 60 Reproduction, (1989)
growth
Gammarus minus A S 48 h LC50 460 Millemann et al.
(1984)
Aromatic four-ring PAH
Benz[a]anthracene
Daphnia magna A R (0.5 d) No solvent 1.52 d LC50 1.8 Newsted & Giesy
-UV: 1 d; +UV:0.52 dc (1987)
Daphnia pulex 1.9-2.1 mm N S No solvent 96 h LC50 10 Trucco et al.
photo period: 12 h (1983)
Chrysene
Daphnia magna A R (0.5 d) No solvent 2 d LC50 0.7 Newsted & Giesy
-UV:1 d; +UV:1 dc (1987)
Daphnia magna Juvenile N S Solvent, acetone 2 d NOEC 288b Mortality Eastmond et al.
+ adult l/d = 16 h/8 h (1984)
Pyrene
Artemis salina 1 d N S 1 h UVc 3 h LC50 8 Kagan et al.
(1985)
Artemia salina N S Dark 24 h LC50 > 99 Abernethy et al.
(1986)
Daphnia magna Adult N S 1 h UVc 2 h LC50 4 Kagan et al.
(1985)
Daphnia magna A R (0.5 d) No solvent 1.14 d LC50 5.7 Newsted & Giesy
-UV: 1 d; +UV:O. 14 dc (1987)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia magna 4-6 d old N S Dark 48 h LC50 91 Abernethy et al.
(1986)
Daphnia magna 4-6 d old N S no solvent 48 h LC50 1820b Bobra et al. (1983)
Triphenylene
Daphnia magna A R (15 d) No solvent 2 d NOEC 1.7 Mortality Newsted & Giesy
-UV:1 d; +UV:1 dc (1987)
Aromatic five-ring PAH
Benzo[a]pyrene
Artemia salina Eggs 48 h NOEC 10 000b Viability Kuwabara et al.
(1980)
Calanus Adult - LC 4 Mortality Lee et al. (1972)
heigolandicus
Calanus 7 d EC 50b AHH activity Walters et al.
heigolandicus (1979)
Daphnia magna A R (0.5 d) No solvent 1.19 d LC50 1.5 Stimulation Newsted & Giesy
-UV:1 d; +UV:0.19 dc (1987)
Daphnia pulex 1.9-2.1 mm N S No solvent 96 h LC5O 5a Trucco et al.
photo period: 12 h (1983)
Benzo[e]pyrene
Daphnia magna A R (0.5 d) No solvent 1.64 d LC50 0.7 Newsted & Giesy
-UV:1 d; +UV:0.64 dc (1987)
Benzo[k]fluoranthene
Daphnia magna A R (0.5 d) No solvent 1.54 d LC50 1.4a Newsted & Giesy
-UV: 1 d; +UV:0.54 dc (1987)
Dibenz[a,h]anthracene
Daphnia magna A R (0.5d) No solvent 1.13 d LC50 0.4a Newsted & Giesy
-UV:1 d; +UV:0.13 dc (1987)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Perylene
Daphnia magna A R (0.5 d) No solvent 1.76 d LC50 0.6a Newsted & Giesy
-UV:1 d; +UV:0.76 dc (1987)
Aromatic six-ring PAH
Benzo[ghi]perylene
Daphnia magna A R (0.5 d) No solvent 1.58 d LC50 0.2 Newsted & Giesy
-UV:1 d; +UV:0.58 dc (1987)
A, analysed concentration; N, nominal concentration; S, static system; F, flow-through system; IF, intermittent flow;
R(0.5 d), system with renewal (each half day); S\F, first period in a static system followed by a period in a flow-through system;
l/d, light/dark; +UV, with UV radiation; -UV, without UV radiation; DMSO, dimethyl sulfoxide; EC, exposure concentration;
NOEC, no-observed-effect concentration; LC, lethal concentration; LOEC, lowest-observed-effect concentration
a Concentration higher than solubility but not exceeding it by 10 times
b Concentration 10 times higher than the solubility
c Explicitly mentioned that organisms were tested for phototoxicity of test substance either in sunlight or artificial UV radiation
d From Cairns & Nebeker (1982)
PAH-polluted elutriates derived from polluted sediments were highly
toxic to Daphnia magna when combined with either sunlight or 354-nm
near-ultraviolet radiation, whereas none of the elutriates was toxic
in the absence of light (Davenport & Spacie, 1991).
A 50% decrease in feeding rate was reported in the mussel
Mytilus edulis after nine days' exposure to 80 µg/litre fluoranthene
(Donkin et al., 1989). When Mercenaria mercenaria clams were exposed
in flow-through tanks, in which seawater was pumped through sand
columns adsorbing 50 mg benzo [a]pyrene, the concentrations in the
water were generally below the detection limit (< 0.001 µg/litre),
whereas the tissue concentrations were 2-4 µg/kg (0.15 µg/kg in
control clams). This resulted in an increased intrahaemocytic lysozyme
concentration and significantly impaired ability to clear bacteria.
Thus, resistance to bacterial infection is decreased by PAH (Anderson
et al., 1981).
The 96-h LC50 values in the marine polychaete Neanthes
arenaceodentate were 3800 µg/litre for the two-ring PAH naphthalene
and 1000 µg/litre for fluorene, 600 µg/litre for phenanthrene, and 300
µg/litre for 1-methyl-phenanthrene (three-ring). None of the four- and
five-ring PAH were toxic up to the highest concentration tested, 1000
µg/litre, except fluoranthene, which had a 96-h LC50 of 500 µg/litre
(Rossi & Neff, 1978).
9.1.2.3 Vertebrates
Data on toxicity to vertebrates like fish and amphibians are available
for two- to five-ring PAH (Table 104). Most of the data are derived
from phototoxicity tests.
As discussed in Section 4, fish can metabolize PAH into intermediates
that may have teratogenic, mutagenic, or carcinogenic properties and
are associated with hepatic tumours in free-living fish. In addition,
certain PAH can cause physiological changes that affect the growth,
reproduction, swimming performance, and respiration of fish. The
effect of environmental carcinogens on fish populations depends on the
exposure received at each susceptible life stage, the ability at each
stage to absorb and metabolize the carcinogen and repair the ensuing
damage, and the consequences of tumour induction in vital organs at
each life stage. Additional factors that influence carcinogenicity are
the stage of organism development, the route of exposure, genetic
variation, and cytochrome P450 mixed-function oxygenase activity
(Bailey et al., 1989).
Several PAH can produce cancer-like growths and are teratogenic and
mutagenic to fish. In Oncorhynchus mykiss (former name for
Salmo gairdneri), the liver was the primary target after exposure to
benzo [a]pyrene in the diet and by intraperitoneal injection.
Administration by the latter route, while not directly relevant to
environmental exposure, also produced a fibrosarcoma and a stomach
papilloma in one individual, along with tumours, indicating that the
route of exposure is of some importance in fish (Hendricks et al.,
1985).
Table 104. Results of tests for the toxicity of polycyclic aromatic hydrocarbons (PAH) towards vertebrates
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
FISH
Aromatic two-ring PAH
Acenaphthene
Cyprinodon Juvenile N S 96 h LC50 2200 Heitmuller et al.
variegatus (1981)
Ictalurus punctatus A F solvent 96 h LC50 1720 Holcombe et al.
(1983)
Lepomis macrochirus 0.32-1.2 g N S solvent 96 h LC50 1700 Buccafusco et al.
(1981)
Pimephales promelas Juvenile (32 d) A F solvent 96 h LC50 1600 Holcombe et al.
(1983)
Pimephales promelas Embryo- A F l/d = 16/8 h 32 d NOEC 509 Survival Cairns & Nebeker
juvenile Glass column 96 h LC50 608 (1982)
Solvent, DMF
Oncorhynchus mykiss Juvenile A F Solvent, 96 h LC50 670 Holcombe et al.
isopropanol 48 h LC50 1130 (1983)
Salmo trutta Juvenile A F Solvent, 69 h LC50 580 Holcombe et al.
isopropanol 48 h LC50 650 (1983)
Acenaphthylene
Oryzias latipes 48 h LG50 11 000 Yoshioka et al.
(1986)
Fluorene
Lepomis macrochirus A F 30 d NOEC 19 Predating prey Finger et al.
NOEC 42 Growth (1985)
NOEC 49 Mortality
N S 96 h LC50 910
Oncorhynchus mykiss N S 96 h LC50 820
Pimephales promelas N S 96 h LC50 > 100 000a
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Naphthalene
Abramis bramana 1 year N R 96 h LC50 10 000 Frumin et al. (1992)
Fundulus heteroclitus 30 d NOEC 1600 US Environmental
Protection Agency
(1986d)
Micropterus salmoides Eggs and A F No solvent 7 d NOEC 28 Survival Black et al. (1983)
larvae (to 4 d LC8-35 28-239
posthatch
Embryo-larva A F No solvent 7 d LC50 510
(to 4 d
posthatch)
Micropterus salmoides Eggs-larvae A F No solvent 7 d LC50 680 Millemann et al.
(1984)
Oncorhynchus Fry - S 4-12°C 96 h LC50 1370-1240 Korn et al. (1979)
gorbuscha
Oncorhynchus kisutch 1 g A F 96 h LC50 770 Parasitized Moles (1980)
Oncorhynchus kisutch 1 g A F 40 d EC 670- Growth Moles et al. (1981)
1400
Oncorhynchus kisutch 1 g A F 96 h LC50 2100 Moles (1980)
Oncorhynchus kisutch 1 g A F 96 h LC50 3220 Unparasitized Moles (1980)
Oncorhynchus mykiss Eggs-larvae A F No solvent 23 d NOEC 15 Hatching Black et al. (1983)
(to 4 d post- 27 d NOEC 15 Survival
hatch) LC50 110
Oncorhynchus mykiss Eggs-larvae A F No solvent 27 d NOEC 120 Survival, Millemann et al.
teratogenity (1984)
Oncorhynchus mykiss Adult A F 96 h LC50 1600 DeGraeve et al.
(1982)
Oncorhynchus mykiss 13-21 d N S Solvent, 96 h LC50 4500 Edsall (1991)
acetone
Oncorhynchus mykiss A F 96 h LC50 2300 DeGraeve et al.
(1980)
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Pimephales promelas Embryo-larva A F 30 d NOEC 450 Growth, DeGraeve et al.
EC 850 hatching (1982)
Subchronic LOEC 850 Reproduction
Pimephales promelas Juvenile A S No solvent 96 h LC50 1990 Millemann et al.
(1984)
Pimephales promelas A F 96 h LC50 4900 Reproduction DeGraeve et al.
(1980)
Pimephales promelas A F 96 h LC50 6140 Geiger et al. (1985)
Pimephales promelas A F 96 h LC50 6080 Holcombe et al.
(1984)
Pimephales promelas Adult A F 96 h LC50 7900 DeGraeve et al.
(1982)
Pimephales promelas A F 96 h LC50 8900 DeGraeve et al.
(1980)
Tilapia oreochromis N R 96 h LC50 22 400 Frumin et al.
(1992)
Aromatic three-ring PAH
Anthracene
Lepomis macrochirus 1-1.5 g N F 24 h UV, 200 h NOEC 1.2 Mortality Oris & Giesy
0 h darkb (1986)
Lepomis sp. Juvenile A F Low UV 96 h LC50 2.78 Oris & Giesy
(2-3 cm) intensityb (1985)
Lepomis macrochirus 1-1.5 g N F 24 h UVb 125 h LC50 4.5 Oris & Giesy
96 h LC50 4.5 (1986)
Lepomis macrochirus 96 h LC50 11.9 US Environmental
Protection Agency
(1987b)
Lepomis sp. Juvenile A F High UV 96 h LC50 11.9 Oris & Giesy
(2-3 cm) intensityb (1985)
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Lepomis macrochirus A F Shadowb, 144 h LC0 12.7 Bowling et al.
microcosm + sediment (1983)
2 h sunb 25 h LC50 12.7
12 h sunb 72 h LC100 12.7
Lepomis macrochirus 1-1.5 g N F 12 h UV, 200 h NOEC 15 Mortality Oris & Giesy
12 h darkb (1986)
Lepomis sp. Juvenile A F Medium UV 96 h LC50 18.2 Oris & Giesy
(2-3 cm) intensityb (1985)
Low UV 96 h LC50 26.5
intensityb
Lepomis macrochirus 1-1.5 g N F 6h UV, 96 h LC50 46 Oris & Giesy
18 h darkb (1986)
Pimephales promelas A R (0.5 d) No 1.6d LC50 5.4 Oris & Giesy
solvent; dark: (1987)
1 d; +UV:16 hb
Dark 4 d NOEC 5.4 Mortality
Pimephales promelas Adult A F No sun 9 weeks LOEC 6.6 Egg production Hall & Oris (1991)
F1 A R No sun NOEC 6.6 Deformities
No sun NOEC > 12 Survival and
Sunb LOEC 12 hatching
Pimephales promelas 0.8g N S 0.5 h sunb 24 h LC50 360c Hatching Kagan et al. (1985)
Fluoranthene
Brachydanio rerio Eggs-larvae A F Yellow light 41 d NOEC 4.8 Growth Hooftman & Evers-
Solvent, TBA 41 d NOEC 48 Mortality de Ruiter (1992b)
Cyprinodon variegatus Juvenile N S 96 h LC0 560 000a Heitmuller et al.
(1981)
Lepomis macrochirus N S 96 h LC50 3980a US Environmental
Protection Agency
(1978b)
Lepomnis macrochirus 0.32-1.2 g N S Solvent 96 h LC50 4000a Buccafusco et al.
(1981)
Pimephales promelas 0.8 g N S 0.5 h sunb 24 h LC50 200 Kagan et al. (1985)
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Phenanthrene
Brachydanio redo Eggs-larvae A R Yellow light 28 d NOEC 28 Growth Hooftman & Evers-
(2 d) solvent, TBA 28 d LOEC 49 de Ruiter (1992d)
Gambusia affinis 24 h LC50 150 Neff (1979)
Micropterus salmoides Eggs-larvae A F 7 d LC50 180 Black et al. (1983)
Micropterus salmoides Eggs-larvae A F No solvent 7 d LC50 250 Millemann et al.
(1984)
Oncorhynchus mykiss Eggs-larvae A F No solvent 23 d NOEC 4 Hatching Black et al. (1983)
27 d NOEC 4 Survival
Oncorhynchus mykiss Eggs-larvae A F No solvent 27 d LC50 30 Millemann et al.
(1984)
Oncorhynchus mykiss Eggs-larvae A F 7 d LC50 40 Black et al. (1983)
Oncorhynchus mykiss N S Solvent, 96 h LC50 3200c Edsall (1991)
acetone
Pimephales promelas A R (0.5 d) No 5 d NOEC 10 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 10
Aromatic four-ring PAH
Benz[a]anthracene
Pimephales promelas A R (0.5 d) No 3.7 d LC50 1.8 Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 1.8 Mortality
Poecilia formosa F Injection 9 months EC 23c Thyroid Woodhead et al.
morphology (1982)
Benzo[k]fluoranthene
Brachydanio rerio Eggs-larvae N F Yellow light 42 d LC50 0.68 Hooftman & Evers-
solvent, TBA 42 d NOEC 0.23 Growth de Ruiter (1992c)
42 d NOEC 0.40 Mortality
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Poecilia formosa F Injection 9 months EC 23c Spleen Woodhead et al.
morphology (1982)
Chrysene
Brachydanio rerio Eggs-larvae A R (2 d) Yellow 28 d NOEC > 0.9 Growth, Hooftman & Evers-
light, glass mortality, de Ruiter (1992b)
column hatching
Pyrene
Pimephales promelas A R (0.5 d) No 1.13 d LC50 25.6 Oris & Giesy
solvent; dark: (1987)
1 d; +UV:0.13 db
Dark 4 d NOEC 25.6 Mortality
Pimephales promelas N S Solvent, 24 h NOEC > 10 000a Mortality Kagan et al.
DMSO (1987)
Aromatic five-ring PAH
Benzo[a]pyrene
Brachydanio rerio Eggs-larvae A R (2 d) Yellow 28 d NOEC > 4 Growth, Hooftman & Evers-
light, glass mortality, de Ruiter (1992b)
column hatching
Fundulus grandis F Injection 7 d EC 18% 30 mg/ Liver weight Melius et al. (1980)
kg bw increase
Hippoglossoides Adult Food 5 h EC 21% 8 mg/kg Hatching Hose et al. (1981)
elassodan success
Ictalurus punctatus Juvenile 7-10 EC 1.0 Skeletal Martin (1980)
months structure,
pigmentation
Leuresthes tenuis Embryo A S Solvent, 14 d NOEC 7.0c Hatching Winkler et al.
acetone success (1983)
14 d EC 12% 7.0c Larval
morphology
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Leuresthes tenuis Larvae - EC 100a Growth Puffer et al. (1979)
EC 100a Development
Micropogonias Adult Oral 30 d NOEC 5.7 Behaviour, Thomas (1988)
undulatus 45-60 g mg/kg growth,
(fresh wt) respiration,
locomotion
EC 34% 5.7 Ovarian growth,
mg/kg hormone level
(fresh wt)
Oryzias latipes 6-10 d A R 0.5 mg/litre 2 × 0.25d NOEC 47+11a,c Neoplastic Hawkins et al.
DMF 2 × 0.25e NOEC 47+11a,c lesions (1988,1990)
2 × 0.25f NOEC < 47+11a,c
Oncorhynchus kisutch Embryos N S\F0.1 mmol/litre 24 hg NOEC 11 000a Emergence Ostrander et al.
1 d after DMSO (1988)
fertilization
1 week before NOEC 10 000a Emergence
hatching
1 d after NOEC < 10 000a Orientation
fertilization
1 week before NOEC < 10 000a Orientation
hatching
Oncorhynchus mykiss Eggs Injection EC 4.5 mg/ Carcino- Black et al.
egg genicity (1988)
Oncorhynchus mykiss Embryo-larva A R Column 36 d NOEC 2.4c Morphology Hannah et al.
36 d LOEC 6.7c Morphology (1982)
Parophyrus vetulus Eggs-larvae A S Solvent, 5 d NOEC > 2.1 Development Hose et al.
ethanol (1982)
Pimephales promelas A R (0.5 d) No 2.7 d LC50 5.6c Oris & Giesy
solvent; dark: (1987)
1 d; +UV:40 hb
Darkb 4 d NOEC 5.6 Mortality
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Poecilia reticulata 6-10 d A S 0.5 mg/litre 2 × 0.25e NOEC 32+8 Neoplastic Hawkins et al.
DMF; darkb 2 × 0.25f NOEC 32+8 lesions (1988, 1990)
Poeciliopsis lucida A S Solvent, 1 d/6 LC 70% 3750a Lethal effects Goddard et al.
Poeciliopsis lucida acetone months NOEC 1250a Lethal effects (1987)
Psettichtys Embryo A F Solvent, 5 d EC 50% 0.1 Hatching Hose et al.
melanostAicatus ethanol success (1982)
7 d EC 0.1 Development
Benzo[a]pyrene
Pimephales promelas A R (0.5 d) No 5 d NOEC 2.9 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 2.9 Mortality
Dibenz[a,h]anthracene
Pimephales promelas Larvae A R (0.5 d) No 5 d NOEC 0.15 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 0.15 Mortality
Perylene
Pimephales promelas Larvae A R (0.5 d) No 5 d NOEC 1.7c Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 1.7c Mortality
Aromatic six-ring PAH
Benzo[ghi]perylene
Brachydanio rerio Eggs-larvae A R (2 d) Yellow 28 d NOEC > 0.16 Growth, Hooftman & Evers-
light, glass mortality, de Ruiter (1992d)
column hatching
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Pimephales promelas Larvae A R (0.5 d) No 5 d NOEC 0.15 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5d NOEC 0.15 Mortality
AMPHIBIANS
Aromatic two-ring PAH
Naphthalene
Xenopus laevis Larvae F Fluorescent 96 h LC50 2100 Edmisten &
(3 weeks) light 6 h EC50 1700- Absence of Bantle (1982)
2300 swimming
Aromatic three-ring PAH
Anthracene
Rana pipiens Embryo Sunb 24 h LC50 110c Kagan et al.
(1985)
Rana pipiens 5 h LC50 25 US Environmental
Protection Agency
(1987b)
Fluoranthene
Rana pipiens N S 1 h sunb 24 h LC50 90 Kagan et al;
(1985)
Aromatic four-ring PAH
Pyrene
Rana pipiens Embryo Sunb 24 h LC50 140c Kagan et al.
(1985)
Table 104 (continued)
A, analysed concentration; N, nominal concentration; S, static system; F, flow-through system; IF, intermittent flow; R (0.5 d),
system with renewal (each half day); S\F, first period in a static system, second in a flow-through system; l/d, light/dark;
UV, with UV radiation; -UV, without UV radiation; TBA, tertiary butyl alcohol; DMF, dimethylformamide; DMSO, dimethyl sulfoxide;
LC, lethal concentration; NOEC, no-observed-effect concentration; EC, effect concentration; LOEC, lowest-observed-effect
concentration
a Concentration 10 times higher than solubility
b Explicitly mentioned that organisms were tested for phototoxicity of test substance either by sunlight or artificial UV radiation
c Concentration higher than solubility but not exceeding it by 10 times
d Exposure followed by 168 d depuration
e Exposure was followed by 252 d depuration
f Exposure followed by 365 d depuration
e Exposure followed by 62 d depuration
Painting of benzo [a]pyrene and 3-methylcholanthrene onto the skin of
three freshwater fish twice a week for three to six months caused
epitheliomas in Gasterosteus aculetus (stickleback) and Rhodeus
amarus (bitterling) but not in Cyprinus carpio (carp). In the same
study, 10 injections of 10 mg benzo [a]pyrene in glycerol to G.
aculetus produced injection-site necrosis but no tumours (Ermer,
1970).
The phototoxic potential of PAH in fish is influenced by the amount of
ultraviolet radiation and light absorbed. Adult fathead minnows
(Pimephales promelas) were exposed in the absence of artificial
ultraviolet radiation to 0.6 or 12 µg/litre anthracene for six weeks,
the dose being increased to 20 µg/litre for three weeks in two groups.
Eggs were collected daily, placed in clean water, and exposed or
unexposed to ultraviolet radiation, until 96 h after hatching. All
fish showed impaired egg production. The lethal concentration in eggs
was estimated to be 19 µg/g (Hall & Oris, 1991).
Exposure of the spot Leiostomus xanthurus to PAH-contaminated
sediment derived from a river station resulted in death, with fin
erosion, ulceration of the lateral body surface, and several lesions
of internal organs. The total concentration of the 21 PAH analysed in
the sediment was 21 000 mg/kg dry weight. The 28-day LC50 was
estimated to be 3.2% of contaminated sediment (Roberts et al., 1989).
9.1.2.4 Sediment-dwelling organisms
The 10-day LC50 values for fluoranthene in sediment were 11 mg/kg dry
weight for the marine amphipod Eohaustorius estuarius, 5.1 mg/kg for
the marine amphipod Rhepoxynius abronius, and 15 mg/kg for the
freshwater amphipod Hyalella azteca. The sensitivity of
E. estuarius was not influenced by salinity (DeWitt et al., 1989).
The toxicity of a sediment containing 46% fines and 0.9% total organic
carbon and artificially supplemented with solutions of fluoranthene,
phenanthrene, benz [a]anthracene, benzo [a]pyrene,
2,6-dimethylnaphthalene, 1-methylnaphthalene, and 2-methylnaphthalene
was tested in the amphipod Rhepoxynius abronius. Significant
mortality occurred among amphipods exposed for 10 days to nominal
concentrations of 15, 10, 10, 5, 0.5, 0.5, and 0.5 mg/kg dry weight of
the seven PAH, respectively, whereas at levels five times lower no
toxic effects were observed (Plesha et al., 1988).
The ability of an amphipod species to metabolize xenobiotic compounds
to reactive intermediates appears to influence its sensitivity to
chemical contaminants. Contaminated sediments are generally more toxic
to R. abronius than E. washingtonianus (Plesha et al., 1988). R.
abronius and E. washingtonianus exposed to sediment-associated,
radiolabelled benzo [a]pyrene accumulated similar concentrations of
radiolabel in their tissues after seven days of exposure, but the
proportions of metabolites and the amount of radiolabel bound to
cellular macromolecules were greater in R. abronius than in
E. washingtonianus. One explanation for the greater sensitivity of
R. abronius might therefore be a greater tendency to form reactive
metabolites that are more acutely toxic than the parent compound
(Reichert et al., 1985).
The toxicity of fluoranthene in sediment to the marine amphipods
R. abronius and Corophium spinicorne was determined by equilibrium
partitioning. Toxicity was determined in well-sorted, fine sands
containing organic carbon at 0.18, 0.31, or 0.48% of dry weight. The
epibenthic tube-dwelling C. spinicorne was less sensitive than the
free-burrowing R. abronius, possibly because of different routes of
exposure. The 10-day LC50 values for R. abronius were 3.4, 6.5, and
10.7 mg/kg dry weight in sediments with increasing organic carbon. A
10-day LC50 value for C. spinicorne could be determined only in the
sediment with the lowest organic carbon level, at 5.1 mg/kg dry
weight. An LC50 of 24 µg/litre was calculated on the basis of
equilibrium partitioning. In another experiment, a 10-day LC50 of 4.2
mg/kg dry weight was determined for R. abronius in sediment with
0.26% organic carbon (Swartz et al., 1988).
Diporeia sp. were exposed to a sediment artificially contaminated
with a mixture of labelled phenanthrene and pyrene and nine unlabelled
PAH at total concentrations of 21, 41, 120, and 330 µmol/kg dry
sediment. The amphipods avoided the sediment containing the highest
dose during the first six days; the estimated LC50 at day 26 was
estimated to be 600 mmol/kg dry sediment. Deaths occurred after 26
days' exposure to the two highest concentrations. The concentration of
PAH required to elicit 38% mortality at day 19 was 2.9 mmol/kg
organism. The authors concluded that PAH probably have a non-polar
narcotic mode of action and suggested that their effect is additive,
which is supported by the observation that exposure of Diporeia for
31 days to pyrene resulted in an LD50 of 5.8 mmol/kg organism
(Landrum et al., 1991).
9.1.2.5 Toxicity of combinations of PAH
In the concentration addition model, additive effects were found for
phenanthrene, anthracene, naphthalene, and acenaphthene in Daphnia
magna (Muñoz & Tarazona, 1993). Although the toxic effects of
combined PAH to Brachydanio rerio were also found to be additive,
the concentrations tested were close to the maximum solubility of the
PAH (Hooftman et al., 1993).
9.1.3 Terrestrial organisms
9.1.3.1 Plants
The effects of anthracene on emergence were tested in three native
Australian plant species, heath banksia (Banksia ericifolia),
she-oak (Casuarina distyla), and yellow bloodwood (Eucalyptus
eximia), and in three crop species, oat (Avena sativa), cucumber
(Cucumis sativus), and soya bean (Glycine max). A. sativa and
C. sativus were sensitive, with EC50 values of 30 and 720 mg/kg,
respectively; the the other plants were not sensitive up to the
highest concentration tested (1000 mg/kg dry weight) (Mitchell et al.,
1988).
9.1.3.2 Invertebrates
Porcellio scaber and Oniscus asellus showed little difference in
their sensitivity to benzo [a]pyrene. The growth of both species was
affected after exposure for nine weeks to 100 and 316 mg/kg dry
weight. The only difference in response was that the lipid pool was
reduced in O. asellus and the protein pool was reduced in
P. scaber. No effects were observed at 32 mg/kg dry weight (Van
Brummelen & Stuijfzand, 1993).
The LC50 values for fluorene in four earthworm species,
Allolobophora tuberculata, Eisenia foetida, Eudrilus eugenia, and
Perionyx excavatus, in an artificial soil were 210, 17, 200, and 170
mg/kg dry weight, respectively, after two weeks' exposure in soil. In
the contact test, the LC50 values were 120, 170, 47, and 78 µg/m2,
respectively (Neuhauser et al., 1986). Fluorene at 750 mg/kg dry
weight significantly reduced the reproduction of E. foetida, but no
deaths occurred (Neuhauser & Callahan, 1990).
Benzo [a]pyrene was incorporated in the food of the wood louse
Porcellio scaber, and respiration, growth, and food consumption were
measured for four weeks. Consumption was not affected by
concentrations up to 125 mg/kg, but at the highest dose, male isopods
showed significantly greater assimilation (34%) than controls (26%)
due to an active mechanism, which was not observed in females. Growth
varied considerably between individuals. At the highest
concentrations, however, the growth efficiency of males was
significantly decreased. At 25 mg/kg, no significant effects were
found (Van Straalen & Verweij, 1991).
The toxicity of PAH to the earthworm E. foetida and the springtail
Folsoma candida was studied in a standard soil. As phenanthrene at a
concentration of 1000 mg/kg dry weight appeared to be removed almost
completely from the soil after 14 days' exposure, the soil was renewed
regularly. The 28-day LC50 of phenanthrene in F. candida was 150
mg/kg dry weight, the EC50 was 120 mg/kg, and the NOEC for
reproduction was 75 mg/kg dry weight. The 21-day EC50 for
reproduction of E. foetida was 240 mg/kg dry weight. No effect on
the survival or reproduction of F. candida was seen after 28 days'
exposure to chrysene, benzo [k]fluoranthene, or benzo [a]pyrene at
180 mg/kg dry weight, and no effect on the reproduction or survival of
E. foetida was seen after 14 days' exposure to chrysene at 1000
mg/kg dry weight (Bowmer et al., 1993).
Ingestion of naphthalene, anthracene, benz [a]anthracene, pyrene, or
benzo [a]pyrene at 1000 mg/kg food per day for 18 days caused
significant increases in mortality among Acheta domesticus crickets.
Naphthalene caused 50% mortality within 12 days, and anthracene,
benz [a]anthracene, pyrene, and benzo [a]pyrene caused 39, 26, 23,
and 32% mortality, respectively, after 18 days (Walton, 1980).
9.1.3.3 Vertebrates
Benzo [a]pyrene and chrysene dissolved in oil and covering less than
10% of the surface of duck eggs reduced hatching and had teratogenic
and embryotoxic effects (Hoffmann & Gay, 1981). The 72-h LD50 values
for chick embryos were 14 µg/kg egg for benzo [k]fluoranthen, 39
µg/kg for dibenz [a,h]anthracene, and 79 µg/kg for
benz [a]anthracene (Brunström et al., 1991). The LD50 values for
acenaphthene, anthracene, phenanthrene, and fluorene in red-winged
blackbirds were > 100 mg/kg (Schafer et al., 1983).
9.2 Field observations
9.2.1 Microorganisms
9.2.1.1 Water
The effects on a benthic community were determined in the sediment of
a stream in which a gradient of PAH concentrations was found, with
total PAH contents at four sites of 0, 3100, 39 000, and 49 000 mg/kg
dry weight. Detrital accumulation and the redox potentials increased
with PAH level. Removal of fungi at the polluted sites was probably
the major factor in detrital accumulation, and a reduction in
bacterial biomass was thought to be the primary cause of the increased
redox potentials (Catallo & Gambrell, 1987).
Addition of 1000 mg/kg dry weight naphthalene to anaerobic salt marsh
sediments resulted in significant inhibition of methanogenesis,
sulfate reduction, and evolution of carbon dioxide. Phenanthrene at
the same concentration had no significant effect on these activities,
whereas the same dose of naphthalene inhibited methanogenesis and then
stimulated it relative to controls; however, the sulfate reduction was
sustained. Carbon dioxide evolution was reduced in only one of the
three experiments (Kiene & Capone, 1984).
9.2.1.2 Soil
No data were available
9.2.2 Aquatic organisms
9.2.2.1 Plants
No data were available
9.2.2.2 Invertebrates
In the study of Catallo & Gambrell (1987), described in section
9.2.1.1, the population densities of nematodes, oligochaetes, and
other benthic invertebrates were significantly decreased at the site
with a total PAH content of 3100 mg/kg and were eradicated at the
other two sites.
Ecosystem responses were tested in small, multispecies, aquatic
systems (Leffler microcosm) exposed once to fluorene dissolved in
acetone at a concentration of 0.12, 0.50, 2, 5, or 10 mg/litre. The
estimated half-life was 2.1 days. The LOEL for respiration in the dark
(Rni) and the ratio of net productivity:Rni was 0.12 mg/litre in all
four communities, suggesting that the responses of these microcosms
were not completely independent of their source communities. At 5 and
10 mg/litre, the zooplankton populations were almost eliminated (Stay
et al., 1988).
9.2.2.3 Vertebrates
PAH metabolites were found in English sole (Parophrys vetulus) with
hepatic tumours collected from Puget Sound, Washington, USA (Malins,
1982; Malins et al., 1984). Detectable levels of similar organic free
radicals were found only when microsomes were incubated with the PAH
fraction of extracts of sediment from this area and not after
incubation with the alkane or aromatic polychlorinated biphenyl
fractions (Collier et al., 1992).
A consistent, statistically significant association was found between
the prevalence of hepatic tumours in free-living P. vetulus and the
levels of PAH in bottom sediment from sites where the fish were
captured, in a series of studies conducted over seven years in Puget
Sound. The strongest relationships were found for four categories of
hepatic lesion. Other contaminants such as trace metals,
polychlorinated biphenyls, pesticides, and chlorinated butadienes were
measured in all studies, but the strongest associations with liver
lesions were found with PAH. The concentrations of total PAH in the
sediment ranged from 0.005 mg/kg dry weight at an uncontaminated site
to 540 mg/kg at the most polluted location (Landahl et al., 1990).
9.2.3 Terrestrial organisms
9.2.3.1 Plants
No data were available.
9.2.3.2 Invertebrates
Application of naphthalene at 200 g/m2 to four soils resulted in a
significant reduction in soil arthropods such as Collembola. At 10
g/m2, the densities of most arthropods increased (Best et al., 1978).
9.2.3.3 Vertebrates
No data were available.
10. EVALUATION OF RISKS TO HUMAN HEALTH AND EFFECTS ON THE ENVIRONMENT
10.1 Human health
10.1.1 Exposure
Polycyclic aromatic hydrocarbons (PAH) are released into the
environment as a result of incomplete combustion of organic materials,
especially during industrial processes, incineration of refuse, and
fossil fuel combustion. They are released mainly into the atmosphere,
adsorbed onto particulate matter, which is deposited in the aquatic
and terrestrial environments. Direct contamination may also occur
from, e.g. creosote-preserved wood and deposition of contaminated
refuse such as sewage sludge and fly ash.
Owing to their long-range transport, PAH, and particularly those with
high molecular masses, are ubiquitous in the environment. The levels
in ambient air vary considerably, ranging from several picograms per
cubic metre to < 1 µg/m3, although phenanthrene has been found at
levels of several micrograms per cubic metre.
10.1.1.1 General population
Humans are exposed to various complex mixtures of PAH in the air,
food, water, and soil. The main sources of human exposure are
emissions from the combustion of coal, diesel, petrol, kerosene, wood,
biomass, and synthetic chemicals such as plastics. PAH account for a
significant portion of the carcinogenicity of some mixtures, such as
coal-tar soot, but not of others such as cigarette smoke, diesel
emissions, and urban aerosol. The levels of selected PAH of
toxicological significance in various environmental media are given in
Tables 105 and 106.
Pollution of indoor air by PAH is due mainly to tobacco smoking,
residential heating, and PAH from outdoor ambient air (Table 107).
Extremely high values (e.g. 15 000 ng/m3 benzo [a]pyrene) were found
in unvented rooms with open fireplaces, especially those in which soft
coal was used for cooking and heating. High concentrations have also
been reported in wood-heated saunas.
The predominant sources of PAH pollution in urban areas are motor
vehicle traffic (both petrol- and diesel-fuelled) and residential
heating, especially with wood, coal, and biomass. The concentrations
are up to one order of magnitude higher in winter than in summer. Such
differences may limit the validity of sampling campaigns performed
during only part of the year for the purpose of estimating mean human
exposure in urban areas.
No conclusion can be drawn about the relative PAH emissions from
petrol-fuelled engines (without catalytic converters) and
diesel-fuelled engines, given the limited number of studies in which
emissions were compared under the same conditions of sampling and
analysis.
Table 105. Reported levels of selected polycyclic aromatic hydrocarbons (PAH) in various media
Compound Ambient air Drinking-water Surface water Soil Soil near Sediment
(ng/m3) (ng/litre) (ng/litre)a (µg/kg) industrial (µg/kg)b
sources
(mg/kg)
Acenaphthenec 0.06-370 0.02-14 0.08-1200 1-21 1-5100 0.04-3800
Anthracene 0.004-61 0.5-9.7 0.01-930 0.2-70 0.2-140 0.06-27 000
Benzo[a]pyrene 0.002-780 0.04-2.0 0.03-910 0.8-3200 0.8-38 0.004-110 000
Chrysened 0.01-260 No data 10-1100 2.1-2700 2.1-1200 0.04-21 000
Dibenzo[a,l]pyrene 0.05-1.5 No data No data No data No data No data
Fluoranthene 0.03-810 0.58-3400 0.4-6400 0.3-3700 0.3-340 0.1-610 000
Fluorene 0.02-420 0.008-21 0.33-2500 1-14 1-8600 0.5-6500
Naphthalenec 0.03-940 0.38-8.8 0.4-2100 3-60 3-5.2 0.7-44 000
Phenanthrene 0.002-1800 2.2-90 0.24-5700 17-1700 17-20 000 0.06-65 000
Pyrene 0.002-540 0.3-40 0.12-3100 0.1-4500 0.1-1600 0.1-410 000
Only detected values are given, owing to the variability of limits of detection; data obtained from studies of
road tunnels were excluded even though short-term exposure to such high levels may contribute significantly to
overall daily exposure.
a Concentration may exceed solubility in water owing to presence of particulates in sample
b Highest values usually determined in harbour sediments
c Probably underestimated because of shortcomings in sampling and analytical procedures; data were not provided
from laboratories that found high values for three- to six-ring PAH.
d Most measurements performed with gas chromatography, so that actual levels are overestimates due to analytical
interference by triphenylene.
Table 106. Reported levels (µg/kg) of selected polycyclic aromatic hydrocarbons (PAH) in food
Compound Meat and Fish and Dairy products Oil, fats, and Vegetables Cereals
meat products seafooda margarine and fruit
Acenaphthene No data 0.9-500 No data 0.02-0.45 No data 0.6-0.7
Anthracene 0.9-31 0.05-240 No data 0.02-460 0.09-0.4b 0.5-1.3
Benzo[a]pyrene 0.01-42 (130c) 0.003-290 0.08-1.3 0.02-140 0.05-6.2b 0.1-0.8
Chrysened 0.15-0.6 0.03-210 1.3-1.5 0.1-120 0.5-69b 0.77
Dibenzo[a,l]pyrene No data No data No data No data No data No data
Fluoranthene 0.48-100 0.1-1800 0.01-4.2 (8.0e) 0.02-460 0.93-120b 0.3-28
Fluorene No data 0.2-370 No data 0.02-200 No data 1.3-2.7
Naphthalenec No data 0.8-210 No data No data No data No data
Phenanthrene 3-64 0.1-2700 0.56-0.72 0.09-1400 0.47-17b 9.9-29
Pyrene 0.55-63 0.03-1500 0.04-2.7 (4.8e) 0.02-330 0.83-70b 0.22-21
a Data from industrially polluted areas included as food items from those areas enter the market
b Values detected in vegetables grown on contaminated soil are excluded
c Exceptionally high values found in processed foods, but PAH not determined in these studies
d Most measurements performed with gas chromatography, so that actual levels are overestimates due to analytical
interference by triphenylene.
e Found in infant food
Table 107. Ranges of indoor concentrations of
selected polycyclic aromatic hydrocarbons
Compound Concentration
(ng/m3)
Acenaphthene 2.5-1650
Anthracene 1-410
Fluoranthene 5-270
Naphthalene 300-2300
Pyrene 3.6-32
Benzo[a]pyrene 0.04-370a
Phenanthrene 3-550
Chrysene 0.6-110
a Levels up to 14 700 ng/m3 found in Chinese
houses with open fires
Drinking-water generally contains low levels of individual PAH, up to
some hundreds of nanograms per litre, depending on the compound. The
levels of PAH in beverages, including alcoholic drinks, are usually
< 0.01 µg/kg.
10.1.1.2 Occupational exposure
The concentrations in air to which workers are exposed depend on the
type of industry. The levels in coke ovens, where the highest exposure
may occur, are up to several hundred micrograms per cubic metre. Table
108 shows some levels of occupational exposure.
10.1.2 Toxic effects
10.1.2.1 Bioavailability
Owing to the high lipophilicity of this class of compounds, their
bioavailability after ingestion and inhalation must be considered to
be significant. Dermal adsorption appears to depend on the PAH being
studied and the species evaluated: 3% of an applied dose of
benzo [a]pyrene was absorbed by human skin and 10% by mouse skin
within 24 h.
10.1.2.2 Acute toxicity
Values for the median lethal dose (LD50) indicate that PAH have
moderate to low acute toxicity. For example, the oral LD50 of
benzo [a]pyrene is > 1600 mg/kg in mice, and the oral LD50 of
naphthalene is 350-700 mg/kg bw in mice but 500-9000 mg/kg bw in rats.
Table 108. Ranges of occupational exposure to selected
polycyclic aromatic hydrocarbons
Compound Concentration (µg/m3)
Acenaphthene 0.44 in oil refining to 135 in aluminium
smelting
Anthracene 0.028 in oil refining to 405 in coke ovens
Fluoranthene 0.085 in oil refining to 191 in coke ovens
Naphthalene 0.22 in roofing to 2900 in food smoke-houses
Pyrene 0.11 in oil refining to 333 in coke ovens
Benzo[a]pyrene 0.09 in chimney sweeping to 137 in coke
ovens
Phenanthrene 0.085 in road paving to 1167 in coke ovens
Chrysenea 0.085 in oil refining to 191 in coke ovens
a Most measurements performed by gas chromatography, so that the
actual levels may be overestimates due to analytical
interference by triphenylene
Case reports have shown that exposure to naphthalene results in
haemolytic anaemia, and lethal doses of 2-3 g for children and 5-25 g
for adults have been reported. There appears to be no cause for
concern about any acute toxicity of occupational exposure or exposure
of the general population, with the exception of accidental ingestion.
10.1.2.3 Irritation and allergic sensitization
Anthracene, benzo [a]pyrene, and naphthalene are primary irritants.
Anthracene and benzo [a]pyrene were reported to be sensitizers,
whereas phenanthrene did not induce contact sensitivity.
10.1.2.4 Medium-term toxicity
Oral administration resulted in no-observed-adverse-effect levels of
175 mg/kg bw per day acanaphthene for hepatotoxicity; 125 mg/kg bw per
day fluoranthene for nephropathy, increased relative liver weights,
and haematological and clinical effects; 125 mg/kg bw per day fluorene
for altered haematological parameters; and 75 mg/kg bw per day pyrene
for nephropathy. Anthanthrene at 1000 mg/kg bw per day had no effect.
The daily uptake of PAH by humans is estimated to be 3.7 µg,
corresponding to about 0.05 µg/kg bw per day (70 kg bw). Human
exposure is thus six orders of magnitude lower than the concentrations
administered in studies in mice. There is thus no cause for concern
about any medium-term toxic effects in humans for the PAH tested so
far.
10.1.2.5 Carcinogenicity
The carcinogenicity of individual PAH and PAH-containing mixtures in
experimental animals has been well studied. Virtually no data exist on
the carcinogenicity of individual PAH in humans, although a limited
database on the carcinogenicity of PAH-containing mixtures is
available: these have been shown to increase the incidence of cancer
in exposed human populations. The finding that a number of individual
PAH are carcinogenic to experimental animals indicates that they are
potentially carcinogenic to humans. PAH can produce tumours both at
the site of contact and distantly, and the carcinogenic potency of PAH
may vary with the route of exposure.
(a) Experimental models
Benzo [a]pyrene is the best-studied PAH. It has been tested in
multiple species and by various routes, including orally, by
inhalation, and by skin painting for dermal carcinogenesis. It has
been shown to be carcinogenic by all routes tested in a number of
animal species. Those species in which no tumours were found are
suspected to have been tested at inadequate doses or observed for an
insufficient portion of their life span.
Other PAH have been assayed for dermal carcinogenicity as either
complete carcinogens or as initiators in initiation-promotion models.
Assays used commonly include tests for lung adenomas in newborn mice
treated by intraperitoneal, intrapulmonary, or subcutaneous injection.
There are insufficient data to determine whether other PAH are
carcinogenic.
(b) Epidemiology
Numerous epidemiological studies have been reported of groups of
workers exposed to environments that contain mixtures of PAH, all of
which also contained chemicals other than PAH. Cases of respiratory
diseases such as pneumoconiosis, respiratory tuberculosis, pneumonia,
and bronchitis and diseases of the circulatory system were reported in
these studies, but these effects are not considered to be specific to
PAH because of simultaneous exposure to agents that cause similar
effects. Thus, exposure in iron and steel foundries entails exposure
not only to PAH but also to other potentially carcinogenic materials
such as nickel, chromium, silica, soot, asbestos, and benzene. The
working environment of aluminium smelters is unlikely to include
nickel or chromium but includes alumina, aluminium fluoride, and
aromatic amines. If each of these materials occurred in a separate
location in a factory, classical epidemiological techniques would have
little difficulty in identifying the agent responsible for a
statistically significant cancer excess. This is not the situation,
and, while epidemiological studies have produced convincing evidence
that cancers occur in workers exposed to PAH, the attribution of
exposure to PAH as the cause of these excesses can only be based on
information from animal models.
Studies of workers exposed to mixtures of PAH indicate that the lung
is the target organ after inhalation. Confounding by cigarette smoking
cannot explain the effects observed. Studies of workers at gas and
coke ovens, at aluminium smelters, in iron and steel foundries, and
with bitumen and asphalt consistently show excess risks for lung
cancer. Coke-oven workers are probably exposed to the highest
concentrations, and studies of these populations have provided
evidence of dose-response effects. In a comparison of the mortality of
5321 coke-oven workers with that of 10 497 steel workers at the same
plants, a monotonic positive trend was shown in the relationship
between the risk for lung cancer and the estimated level of cumulative
exposure to coal-tar pitch volatiles, the benzene-soluble fraction of
particulate matter. With a risk of unity for the unexposed group, the
risk increased from 1.2 for those with exposure of 1-49 mg/m3 ×
months to 3.1 for the group with the heaviest exposure of > 650
mg/m3 × months. Analysis of the relative risks and the numbers of
deaths from lung cancer on which they were based resulted in the
conclusion that 124 deaths occurred among these coke-oven workers over
a period of 30 years that can be attributed to exposure to coal-tar
pitch volatiles, i.e. 2.3% of the cohort. Earlier findings from this
study were used by others to estimate a unit risk coefficient of 8.7 ×
10-2 for exposure to benzo [a]pyrene, i.e. the absolute lifetime
risk of lung cancer from a working lifetime exposure to 1 µg/m3 of
benzo [a]pyrene. Given the large number of cancers that occurred in
this cohort, this risk coefficient is probably the best estimate
currently available. It should be recognized, however, that the
reports on which this estimate is based gave relatively little
information on exposure levels, no data on time trends in the level of
exposure, and no data on benzo [a]pyrene levels in the participating
plants.
There is also good evidence that urinary bladder cancer has occurred
in cohorts of aluminium smelters and gas and coke workers, although
the overall findings are not as consistent as those for lung cancer.
In a study of aluminium smelters, a positive trend was observed
between the risk for urinary bladder cancer and the estimated level of
cumulative exposure to benzene-soluble matter. PAH cannot be assumed
to be responsible for this trend, however, because the known bladder
carcinogen 2-naphthylamine and other aromatic amino and nitro
compounds were present in the working environment. Similarly, the
excess of urinary bladder cancer in gas workers is more likely to be a
consequence of exposure to 2- and 1-naphthylamines.
PAH are almost certainly one of the carcinogenic agents responsible
for lung cancers in cigarette smokers, although the role of PAH in the
many other diseases caused by cigarette smoking, including
nonmalignant diseases of the respiratory system, is unknown. Studies
on the rates of mortality from lung cancer in relation to indoor
burning of coal or wood in open fires for cooking and heating add to
the body of evidence that links PAH and the risk for lung cancer.
Workers exposed to diesel or petrol fumes had relatively low exposure
to PAH, and studies of these exposure are not likely to assist in
quantification of the risks for cancer associated with exposure to
PAH.
Quantitative risk assessments were not made for PAH, either
individually or as mixtures; however, Appendix I gives some
comparative features of three approaches to quantitative risk
assessment that have been used and which have been at least partly
validated.
10.1.2.6 Reproductive toxicity
(a) Developmental studies
There are no studies in humans. Embryotoxic effects have been
described in experimental animals exposed to PAH such as
benz [a]anthracene, benzo [a]pyrene, and naphthalene. In mice
treated intraperitoneally with benzo [a]pyrene, increased numbers of
stillborn and resorbed fetuses, decreased fetal weight, and increased
incidences of congenital anomalies were seen at a minimum dose of 50
mg/kg bw per day. In mice given benzo [a]pyrene in the diet,
malformations were found after administration of 120 mg/kg bw per day
on days 2-10 of gestation. In another study in mice fed the compound
during most of the period of gestation, no treatment-related
embryotoxic effects were found after doses up to 133 mg/kg bw per day.
In mice, 1 mg benzo [a]pyrene per gram of food would result in
consumption of 5 g/day. In humans, the total median intake from food,
air, water, and soil for a 70-kg person would be 0.05 µg/kg bw per
day. In view of the inter- and intraspecies differences, there is at
present no reason for concern about effects on development.
(b) Fertility
There are no studies in humans. In mice fed diets containing
benzo [a]pyrene at doses up to 133 mg/kg bw per day, no
treatment-related effects on fertility were seen, again indicating no
concern about effects of PAH on fertility in humans.
10.1.2.7 Immunotoxicity
At least one immunotoxic PAH will probably be present in any mixture
of PAH. Thus, when such mixtures are evaluated for their potential
effects on human health, the immune system should be considered a
primary target organ. Benzo [a]pyrene caused immunosuppression in
mice after dermal application for 28 days at doses as low as 625 µg/kg
bw. In a study in which the immune status of coke-oven workers was
evaluated, suppressed immune status was found, as indicated by
decreased serum antibody levels and functional impairment.
The mechanisms of immunosuppressive action that have been proposed
include formation of active metabolites (including diol epoxides),
alterations in cytokine levels, disruption of membrane fluidity,
interaction with the Ah receptor, and alterations in calcium ion flux.
10.1.2.8 Genotoxicity
PAH have repeatedly been shown to have genotoxic effects both in
in vivo in rodents and in vitro in mammalian (including human)
cell lines and prokaryotes. Some PAH, however, appear not to be
genotoxic. Most of the unsubstituted PAH categorized as genotoxic are
not genotoxic per se but require metabolism to intermediates which
react with DNA to form DNA adducts and induce genotoxic damage.
Genotoxic events are postulated to be a required step in the
carcinogenic process.
10.2 Environment
10.2.1 Environmental levels and fate
Concentrations of up to 100 µg/kg of individual PAH have been detected
in soil, although higher concentrations of pyrene, phenanthrene,
chrysene, and benzo [a]pyrene have been found. The concentrations in
soil near industrial sources of PAH are up to three orders of
magnitude higher.
Concentrations of up to 6 µg/litre have been reported for individual
PAH in surface water, including polluted rivers. High concentrations
have also been reported in sediment, which acts as a sink for PAH. The
concentrations in sediment from rivers, lakes, and seas are generally
< 30 mg/kg dry weight. Concentrations up to 655 mg/kg dry weight have
been reported in sediment from harbours. The individual PAH compounds
detected in environmental samples vary according to their source and
any degradative processes. Phenanthrene is the PAH found in highest
concentrations in aquatic samples. Those that occur at the highest
concentrations in sediment include phenanthrene, fluoranthene, pyrene,
benz [a]anthracene, benzo [b]fluoranthene, benzo [k]fluo-ranthene,
and indeno[1,2,3- cd]pyrene.
PAH are sparingly soluble in water and therefore have affinity for
sediment, soil, and biota. They may be removed from the environment by
biodegradation or photodegradation. The rates of degradation vary and
generally decrease with increasing numbers of aromatic rings. PAH are
inherently biodegradable, and low-molecular-mass compounds can be
completely degraded by acclimated microorganisms. In surface waters
with low numbers of unacclimated microorganisms, PAH can persist for
longer periods of time.
10.2.2 Ecotoxic effects
10.2.2.1 Terrestrial organisms
Few data are available on the effects of PAH on terrestrial organisms,
and none are available on plants, wild mammals, or birds. The values
for the concentration causing 50% lethality (LC50) reported for
earthworm species are 150 mg/kg dry weight for phenanthrene and
170-210 mg/kg for fluorene. The no-observed-effect level for the
survival and reproduction of earthworm species was 180 mg/kg dry soil
for chrysene, benzo [k]fluoranthene, and benzo [a]pyrene. PAH in
soil are unlikely to exert toxic effects on terrestrial invertebrates,
except when the soil is highly contaminated.
10.2.2.2 Aquatic organisms
The toxicity of naphthalene, phenanthrene, and fluoranthene on aquatic
organisms has been well studied in the laboratory, but that of other
PAH has not. The toxicity of PAH to aquatic organisms is affected by
metabolism and photo-oxidation, and they are generally more toxic in
the presence of ultraviolet light. Naphthalene is the least toxic to
invertebrates, with 48-h LC50 values of 700-23 000 µg/litre. The LC50
values for three-ring PAH range from < 1 to 3000 µg/litre, anthracene
being more toxic than the others, with 24-h LC50 values of < 1 to
260 µg/litre. Four-, five-, and six-ring PAH have 48-h LC50 values of
0.2-1800 µg/litre, their toxicity increasing with molecular mass. The
96-h values for acute toxicity in fish were 110 to > 10 000 µg/litre
for naphthalene, 30-4000 µg/litre for three-ring PAH (those for
anthracene being 2.8-360 µg/litre), and 0.7-26 µg/litre for four- and
five-ring PAH.
The no-observed-effect and lowest-observed-effect levels for
invertebrates were 300-1000 µg/litre for naphthalene, 2-600 µg/litre
for three-ring PAH, 5-290 µg/litre for four-ring PAH, and 0.1-50
µg/litre for five-ring PAH. The long-term treatment doses resulting in
toxicity in fish were 15-1600 µg/litre for naphthalene, 1.2-510
µg/litre for three-ring PAH, 0.9-26 µg/litre for four-ring PAH, and
0.15-7 µg/litre for five-ring PAH. Higher no-observed-effect
concentrations have been reported, but in studies in which the PAH
were present at more than than 10 times their maximum aqueous
solubility; these have therefore been ignored. As the concentrations
of PAH reported in surface water are usually in the range of nanograms
per litre, they are unlikely to exert adverse effects on aquatic
organisms, except in cases of heavy exposure, as with creosote.
The LC50 values reported for sediment-dwelling organisms were 3.4-15
mg/kg dry sediment for fluoranthene, 10 mg/kg for phenanthrene, 10
mg/kg for benz [a]anthracene, and 5 mg/kg for benzo [a]pyrene. Since
sediment acts as a sink for PAH, sediment-dwelling organisms may be
adversely affected.
PAH may induce neoplastic effects in aquatic organisms. Tumour
development has been reported in fish exposed to benzo [a]pyrene and
3-methyl-cholanthrene after oral, dermal, or intraperitoneal
administration. Hepatic tumours have been found in wild fish living in
water with sediment containing PAH at a concentration of 250 mg/kg.
Although the levels of PAH in sediment are generally an order of
magnitude lower than this value, the possibility that tumours might be
formed at lower concentrations cannot be excluded. The ecological
significance of the carcinogenic effects of PAH in fish has not been
assessed.
11. RECOMMENDATIONS FOR THE PROTECTION OF HUMAN HEALTH AND THE ENVIRONMENT
11.1 General recommendations
* International agreement on analytical procedures and
interlaboratory quality control studies is strongly recommended.
Sampling strategies and analytical procedures should be optimized
and standardized before surveys of exposure to polycyclic
aromatic hydrocarbons (PAH) are undertaken.
* Emissions and effluents of PAH from both point and diffuse
sources should be monitored and inventories compiled.
* Concentrations of individual PAH should be given rather than
'total PAH'. When PAH are designated as 'not detected', the
relevant limits of detection should be given.
* PAH emissions and effluents should be reduced by:
- filtration and scrubbing of industrial emissions,
- treatment of effluents, and
- use of catalytic converters and particle traps on motor
vehicles.
11.2 Protection of human health
* Owing to their proven immunotoxic effects, coal-tar shampoos
should be used for anti-dandruff therapy only if no other
treatment is available.
* In view of the proven immunotoxic and carcinogenic effects of PAH
in coke-oven workers, exposure to PAH in occupational settings
should be eliminated or minimized by reducing emissions to the
extent possible or, when they cannot be sufficiently reduced, by
providing effective personal protection.
* Public education about the sources and health effects of exposure
to PAH should be improved.
* Use of unvented indoor fires, as in many developing countries,
should be discouraged, and they should be replaced by more
efficient, well-vented combustion devices.
* The risk of exposure to PAH from passive smoking should be
stressed and measures taken to avoid it.
* Urban air pollution should be monitored all year round and not
only seasonally.
11.3 Recommendations for further research
11.3.1 General
- Investigate the suitability of benzo [a]pyrene as an
indicator of the effects of PAH on human health and the
environment and examine the use of other PAH as surrogates.
11.3.2 Protection of human health
- More data should be collected on the human body burden of
PAH and on biomarkers for these compounds.
- The reproductive effects of PAH should be studied further.
- More studies on dermal absorption are required.
- The contribution of the high-molecular-mass PAH to the
overall carcinogenic potential of PAH should be studied.
11.3.3 Environmental protection
- The toxic effects of PAH to plants and earthworms should be
studied.
- The body burdens and possible toxic effects of PAH in wild
mammals and birds should be investigated, as most of these
species can metabolize PAH.
- The extent to which higher-molecular-mass PAH are absorbed
by sediments and serve as a sink and the effects of
disturbing sediment, e.g. by dredging, on aquatic organisms
should be investigated.
- The environmental significance of the tumours that have been
found in fish exposed to PAH must be addressed.
- Reliable data should be collected on environmentally
relevant PAH like dibenzo [a,l]pyrene, particularly with
regard to noncarcinogenic end-points such as effects on the
immune system.
11.3.4 Risk assessment
- Quantitative estimates of the risks presented by PAH should
be compared using various approaches and exposure scenarios,
both for human health and ecological protection.
- The risk estimates obtained by the various approaches to
risk assessment based on data on human exposure should be
compared.
- Risk assessment procedures that allow integrated assessment
of the risks due to inhalation and to oral and dermal
exposure should be developed and validated.
- Comparative risk assessment methods for evaluating
immunotoxicity and other noncancer risks associated with
exposure to PAH should be improved.
- The use of the results of alternative tests, such as those
for genotoxicity and other short-term effects, in assessing
risks due to PAH should be evaluated.
- Human exposure to alkylated PAH in a variety of situations
should be investigated further and data acquired on the
mutagenicity and experimental carcinogenicity of these
compounds.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
12.1 International Agency for Research on Cancer
Polycyclic aromatic hydrocarbons (PAH) have been evaluated by a number
of working groups convened by IARC. The evaluations made between 1973
and 1983 and summarized in Supplement 7 to the IARC Monographs
(IARC, 1987) are shown in Table 109.
12.2 WHO Water Quality Guidelines
PAH have been assessed in the WHO Guidelines for Drinking-water
Quality (WHO, 1984, 1996). A quantitative risk assessment was
conducted using the two-stage birth-death mutation model. The
resulting guidelines for benzo [a]pyrene in drinking-water
corresponding to excess lifetime risks for gastric cancer of 10-4,
10-5, and 10-6 are 7, 0.7, and 0.07 µg/litre.
The data are insufficient to derive guidelines for other PAH, but the
following recommendations are made for the group:
* Because of the close association between PAH and suspended
solids, treatment to achieve the recommended level of turbidity,
when necessary, will ensure that the PAH levels are reduced to a
minimum.
* Water should not be contaminated with PAH during water treatment
or distribution. The use of coal-tar-based and similar materials
for pipe linings and coatings on storage tanks should therefore
be discontinued. It is recognized that it may be impracticable to
remove coal-tar linings from existing pipes, and research is
needed on methods of minimizing the leaching of PAH from such
materials.
* In monitoring levels of PAH, the use of specific compounds as
indicators for the group as a whole is recommended. The choice of
indicator compound will vary in each situation. PAH should be
monitored regularly in order to determine background levels,
against which any changes can be assessed and remedial action
taken, if necessary.
* When drinking-water is known to have been contaminated by PAH,
the specific compounds present and the source of the
contamination should be identified, as the carcinogenic potential
of PAH varies.
12.3 FAO/WHO Joint Expert Committee on Food Additives
Benzo [a]pyrene was assessed at a meeting of this Committee in June
1990 (WHO, 1991). The Committee concluded that, for the purpose of
evaluation, the most significant toxicological effect of
benzo [a]pyrene is its carcinogenicity. It was recognized that
benzo [a]pyrene is only one member of a class of more than 100
compounds and that they should be considered as a class.
The Committee was unable to establish a tolerable intake for
benzo [a]pyrene on the basis of the available data. Nevertheless, the
large difference between the estimated human intake of
benzo [a]pyrene and the doses that induce tumours in animals suggests
that any effects on human health are likely to be small. Despite this,
the considerable uncertainties in risk estimation require that efforts
be made to minimize human exposure to benzo [a]pyrene as far as is
practicable.
The Committee acknowledged the complexity of reducing exposure to
benzo [a]pyrene and other PAH. Furthermore, it noted that exposure to
benzo [a]pyrene constitutes only a fraction of consumers' exposure to
these compounds and that some other members of this class, not
evaluated at the meeting, have toxicological properties similar to
those of benzo [a]pyrene and may thus contribute to the overall
carcinogenic risk. In this regard, strategies to minimize exposure to
benzo [a]pyrene would also be effective in reducing overall exposure
to PAH. These include practices that consumers can effect, such as
washing fruits and vegetables thoroughly to remove any surface
contamination, trimming excess fat prior to barbecuing meats to
minimize 'flare-ups', and cooking in a fashion that prevents contact
of food with flames. Measures that can be taken by the food industry
include use of indirect heating for drying foods, switching to
non-coal-fired roasters (e.g. for roasting coffee beans), using
protective coverings (e.g. cellulose casing) when smoking foods
conventionally, and ensuring compliance with the limits for PAH in
food additives specified by national and international bodies. The
Committee urged application of these measures in order to minimize
contamination of food with PAH, including benzo [a]pyrene.
12.4 WHO Regional Office for Europe Air Quality Guidelines
PAH have been assessed as atmospheric pollutants by the WHO Regional
Office for Europe (WHO, 1987). No guideline was set, but the group
concluded that no safe level of PAH could be recommended, owing to
their carcinogenicity. There is no known threshold for the induction
of cancer by benzo [a]pyrene, the most thoroughly studied PAH, nor is
there an ambient mixture of PAH that does not contain benzo [a]pyrene
and other substances for which there is sufficient evidence of
carcinogenicity in animals.
A number of estimates have been made of the risk presented by PAH,
based primarily on studies in which benzo [a]pyrene was used as the
index compound. The US Environmental Protection Agency (1984d)
proposed an upper-bound lifetime cancer risk of 62 per 100 000 exposed
people per microgram of benzene-soluble coke-oven emission per cubic
metre of ambient air. Assuming a 0.71% content of benzo [a]pyrene in
these emissions, it can be estimated that nine out of 100 000 people
exposed to 1 ng/m3 benzo [a]pyrene over a lifetime would be at risk
of developing cancer.
Table 109. Degree of evidence for carcinogenicity in humans and in experimental animals
and overall evaluations of carcinogenicity to humans for agents evaluated in IARC
Monographs
Compound Degree of evidence Overall IARC Monographs
for carcinogenicity evaluationa volume (year)
Human Animal
Anthanthrene ND L 3 32(1983)
Anthracene ND I 3 32(1983)
Benz[a]anthracene ND S 2A 3 (1973); 32 (1983)
Benzo[b]fluoranthene ND S 2B 3 (1973); 32 (1983)
Benzo[j]fluoranthene ND S 2B 3 (1973); 32 (1983)
Benzo[k]fluoranthene ND S 2B 32(1983)
Benzo[ghi]fluoranthene ND I 3 32(1983)
Benzo[a]fluorene ND I 3 32(1983)
Benzo[b]fluorene ND I 3 32(1983)
Benzo[ghi]perylene ND I 3 32(1983)
Benzo[c]phenanthrene ND I 3 32(1983)
Benzo[a]pyrene ND S 2A 3 (1973); 32 (1983)
Benzo[e]pyrene ND I 3 3 (1973); 32 (1983)
Chrysene ND L 3 3 (1973); 32 (1983)
Coronene ND I 3 32(1983)
Cyclopenta[cd]pyrene ND L 3 32(1983)
Dibenz[a,h]anthracene ND S 2A 3 (1973); 32 (1983)
Dibenzo[a,e]pyrene ND S 2B 3 (1973); 32 (1983)
Dibenzo[a,h]pyrene ND S 2B 3 (1973); 32 (1983)
Dibenzo[a,i]pyrene ND S 2B 3 (1973); 32 (1983)
Dibenzo[a,l]pyrene ND S 2B 3 (1973); 32 (1983)
Fluoranthene ND I 3 32(1983)
Fluorene ND I 3 32(1983)
Indeno[1,2,3-cd]pyrene ND S 2B 3 (1973); 32 (1983)
5-Methylchrysene ND S 2B 32(1983)
1-Methylphenanthrene ND I 3 32(1983)
Perylene ND I 3 32(1983)
Phenanthrene ND I 3 32(1983)
Pyrene ND I 3 32(1983)
Triphenylene ND I 3 32(1983)
Adapted from IARC (1987)
ND, no adequate data; I, inadequate evidence; L, limited evidence; S, sufficient
evidence
a Group 1, the compound is carcinogenic to human; Group 2A, the compound is
probably carcinogenic to humans; Group 2B, the compound is possibly carcinogenic
to humans; Group 3, the compound is not classifiable as to its carcinogenicity
to humans
APPENDIX I
SOME APPROACHES TO RISK ASSESSMENT FOR POLYCYCLIC AROMATIC
HYDROCARBONS
I.1 Introduction
Various environmental and technical problems hinder assessment of the
risk posed by mixtures containing polycyclic aromatic hydrocarbons
(PAH), particularly for carcinogenicity. Ambient environments contain
a wide range of PAH, some of which are highly carcinogenic, while
others are probably not (Table AI.1). Both anthropogenic and natural
sources may contribute to the ambient levels of these compounds, such
as in the air over industrial towns (see Section 3).
As PAH undergo transformation in the environment (see Section 4), it
cannot be taken for granted that the composition of ambient mixtures
is similar to that of the source. No data are available to assess the
potency of individual PAH in humans, but the carcinogenicity of
several mixtures containing PAH has been estimated in epidemiological
studies (Albert et al., 1983). Mixtures of a similar type, such as
from coke ovens, may not always present the same risk, however, even
when the fuel used and the operating conditions are similar.
Furthermore, PAH are often not the only contributors to the
carcinogenic risk presented by a given mixture: other chemicals and
particulate matter (Heinrich, 1995) may also contribute. The basis for
quantification and the way in which the quantity of a mixture is
expressed are also important. Should the levels be expressed in terms
of the total mass of extractable material or as a surrogate? If the
concentration of a chosen surrogate is used as an indicator of the
quantity of the mixture and its toxicity, the surrogate selected must
be predictive of the toxicity of the mixture.
Estimating the risk conferred by exposure to PAH has further problems.
As humans are exposed to mixtures of PAH and other compounds and not
to pure PAH, experimental data must be used to estimate the risk for
exposure to individual PAH and the result extrapolated to the low
doses to which humans are exposed. Such extrapolation is problematic,
because species may differ in the enzymes that activate PAH (Michel et
al., 1995) and in their susceptibility to the tumorigenic effects of
PAH; these differences may otherwise be a simple reflection of
differences in weight, surface area, basal metabolic rate, or
respiratory volume. The degree to which species differences affect
extrapolated human risks is unknown.
Another set of problems stems from the large number of PAH that are
typically found in a complex mixture. Only a small percentage of the
environmentally relevant PAH has been investigated for carcinogenicity
in experimental animals, and the toxicity of most of the PAH in
complex mixtures remains unknown. About a dozen PAH of known toxicity
are monitored in most programmes, and these contribute only a small
proportion of the risk represented by PAH fractions extracted from
complex mixtures (Thorslund & Farrar, 1990a). It is therefore likely
Table AI.1. Evaluations of the carcinogenicity of some polycyclic
aromatic hydrocarbons
Compound IARC US Environmental Task Groupb
(1987)a Protection Agency
(1993)a
Acenaphthene D Questionable
Acenaphthylene Dc
Anthanthrene Positive
Anthracene 3 Dc Negative
Benz[a]anthracene 2A B2c Positive
Benzo[b]fluoranthene 2B B2c Positive
Benzo[j]fluoranthene 2B B2 Positive
Benzo[ghi]fluoranthene (Negative)
Benzo[k]fluoranthene 2B B2c Positive
Benzo[a]fluorene Questionable
Benzo[b]fluorene Questionable
Benzo[ghi]perylene 3 Dc Negative
Benzo[c]phenanthrene (Positive)
Benzo[a]pyrene 2A B2c Positive
Benzo[e]pyrene 3 C Questionable
Chrysene 3 B2c Positive
Coronene Questionable
Cyclopenta[cd]pyrene B2 Positive
Dibenz[a,h]anthracene B2c Positive
Dibenzo[a,e]pyrene B2 Positive
Dibenzo[a,h]pyrene B2 Positive
Dibenzo[a,i]pyrene B2 Positive
Dibenzo[a,l]pyrene B2 Positive
Dibenzo[e,l]pyrene D
Dibenzo[a,e]fluoranthene B2
Dibenzo[a,h]fluoranthene B2
Dibenzo[a,i]fluoranthene B2
Dibenzo[a,l]fluoranthene B2
Fluoranthene 3 Dc (Positive)
Fluorene 3 Dc Negative
Indeno[1,2,3-cd]pyrene B2c Positive
5-Methylchrysene Positive
1-Methophenanthrene (Negative)
Naphthalene Dc/C Questionable
Perylene (Negative)
Phenanthrene 3 Dc Questionable
Pyrene Dc Questionable
Triphenylene (Negative)
a Based on the results of studies in humans and experimental animals
b Based on the results of studies in experimental animals
c Consensus position of the US Environmental Protection Agency;
others have been presented at scientific meetings (Schoeny et al.,
1994; McClure & Schoeny, 1995).
that summing the risks posed by individual PAH of known toxicity does
not accurately reflect the contribution of all the PAH in a mixture.
I.2 Approaches to risk assessment
Three of the most popular approaches for assessing dose-response
relationships for PAH are presented, with their strengths and
weaknesses. These approaches are toxicity equivalence factors,
comparative potency, and use of benzo [a]pyrene as a surrogate.
I.2.1 Toxicity equivalence factors and related approaches
Several approaches to quantification may be considered for assessing
the risks posed by mixtures of agents. When there are sufficient data,
they can be used as a basis for quantitative estimates of risk.
Relatively few mixtures containing PAH have been tested under
conditions that are acceptable for risk assessment. For some
processes, e.g. coal coking, the data on human exposure are
sufficiently complete to allow quantitative estimates of risk;
however, changes in the parameters of the combustion process, such as
temperature and amount of oxygen feedstock, may result in variations
in the types, amounts, and physical status of PAH in the mixture.
These variations may be sufficient to alter the risk posed by the
mixture. One way of resolving the uncertainty inherent in differences
in the composition of mixtures is to base quantitative estimates on
considerations of individual components; this alternative is explored
below.
I.2.1.1 Principle
The approaches are based on an assumption of additive risk, which
leads, in principle, to an estimate of the risk associated with
identified PAH. In practice, the risks attributable to individual PAH
are summed, or the risk posed by individual PAH is expressed relative
to that for benzo [a]pyrene, and then the levels of these equivalents
are summed. The latter process is the toxicity equivalence factor
approach.
The first step is to estimate the potency of a single PAH, which
serves as a standard against which the potency of other compounds is
later derived. In practice, this compound is usually benzo [a]pyrene.
Since no data from studies in humans are available that are suitable
for assessing the potency of individual PAH, the potency of
benzo [a]pyrene in humans is estimated from the results of studies in
animal models. The uncertainty associated with this extrapolation is
discussed above.
The second step is to estimate the potency of the PAH relative to that
of benzo [a]pyrene, in order to obtain a benzo [a]pyrene equivalent.
The estimate is based on the relative potencies of benzo [a]pyrene
and other PAH in experimental animals. The key assumption is that the
relative potency of two PAH in an animal model is the same or similar
to that of the same compounds in humans. This comparative potency
approach has been used in relation to chlorinated
dibenzo- para-dioxins and dibenzofurans (US Environmental Protection
Agency, 1987) and for PAH and PAH-rich mixtures (Albert et al., 1983;
Clement Associates, 1988). Further evidence supports the assumption
made in this approach (Albert et al., 1983; Lewtas, 1985a,b; Nesnow,
1990).
The third step involves summation of risks, which can be done either
by summing the benzo [a]pyrene equivalents and multiplying by the
potency of benzo [a]pyrene or by estimating the potency of each PAH
in humans (risk for cancer) and then adding them. The underlying
assumption is that the individual estimates of risk are additive.
Although there may be interactions between PAH, the risks appear to be
approximately additive, especially at low, environmentally relevant
doses (Krewski et al., 1989; Nesnow et al., 1995). In the absence of
information to the contrary, additivity is assumed.
The toxicity equivalence factor approach is feasible if at least two
pieces of information are available:
- the amount of each PAH in the mixture, or the amount of each
'major bioactive PAH', and
- a quantitative estimate of the risk associated with each
identified PAH.
For the first requirement, good data on the composition of mixtures of
PAH can be generated with existing analytical techniques (see Section
2), although such analyses can be resource-intensive. Quantitative
risk estimates for individual PAH are generally not available, as the
data are insufficient. The deficiencies include the following:
- the data relate to exposures that are not typically used in
deriving quantitative estimates of risk after oral or inhalation
exposure,
- the study populations were inappropriately small,
- the studies involved only one dose,
- dose-response relationships were not reported, and/or
- different PAH were tested in different studies with different
designs.
Environmental mixtures contain many more PAH than can be monitored
feasibly or practically. Furthermore, the toxicity of most
environmentally relevant PAH has not yet been quantified. As the
toxicity equivalence factor approach assumes additivity of the risks
posed by PAH in a mixture, the question remains of the extent to which
the risk of one PAH is representative of the risk of all of those in
the mixture.
I.2.1.2 Development and validation
I.2.1.2.1 Derivation of the potency of benzo [a]pyrene
Quantitative risk estimation is problematic for even the best-studied
PAH, namely benzo [a]pyrene. The risk for carcinogenicity is best
estimated on the basis of data from long-term, usually lifetime,
assays in which several doses are tested in large groups of animals.
The data on benzo [a]pyrene are less than optimal: few lifetime
assays have been conducted with exposure other than dermal and
generally few doses were tested.
Krewski et al. (1989) calculated the probability of occurrence of a
tumour at a specified time after continuous exposure to a mixture of
PAH. The dose of the PAH mixture was described as a benzo [a]pyrene
equivalent dose d as follows:
d = S Ridi + do (1)
where Ri is the relative carcinogenicity of the ith PAH in
comparison with that of benzo [a]pyrene, di is the dose of the
ith PAH, and do is the dose of benzo [a]pyrene.
The tumour probability, P(d), was calculated from a two-stage birth-
death mutation model. For the mixture as a whole, P(d) is given by
the equation:
P (d) = 1-exp (- A( 1 + bd)2) (2)
where the unknown values A and b were estimated from bioassays
with benzo [a]pyrene as A = 0.00616 and b = 3.52 µg-1 for
lifetime exposure.
The US Environmental Protection Agency (1993) made a quantitative risk
estimate for oral exposure to benzo [a]pyrene that consisted of a
range of values, from 4.5 to 11.7 per (mg/kg)/day, with a geometric
mean of 7.3 per (mg/kg)/day. These estimates were obtained by using
three methods of determining an upper bound on a linear low-dose term
from data on the incidence of gastrointestinal tumours in mice exposed
to benzo [a]pyrene in the diet (Neal & Rigdon, 1967). The models used
were a form of the two-stage Moolgavkar, Venzon, and Knudson model
(Moolgavkar & Venzon, 1979; Moolgavkar & Knudson, 1981) and a
Weibull-type model (Rees & Hattis, 1994). The total numbers of tumours
in male and female rats exposed in the diet (Brune et al., 1981) were
used in a linearized multistage procedure to derive an upper bound on
the low-dose term (slope factor), shown in Table AI.2.
The potency of benzo [a]pyrene in humans exposed by inhalation has
been assessed on the basis of extrapolations from the results for
rodents, sometimes exposed other than by respiration. Since the
sensitivity to PAH is likely to differ with the route of exposure,
assessments made on the basis of exposure by inhalation are
preferable; some of these are shown in Table AI.3.
Table AI.2. Slope factors for humans based on the results of
studies in which benzo[a]pyrene was fed to rodents in the diet
Study Slope factor Comments
(per [mg/kg]/day)
Neal & Rigdon 5.9 Two-stage, conditional upper bound
(1967) 9.0 Two-stage, slope from 10% response
4.5 Weilbull-type model
Brune at al. 11.7 Linearized multistage procedure
(1981) applied to oesophageal, laryngeal,
and forestomach tumours in males
and females
From US Environmental Protection Agency (1992b, 1993)
The commonest method of extrapolation is based on the relative surface
areas or body weights of experimental animals and humans, assuming
that these are good predictors of the relative potency of PAH in two
species. While such scaling factors have been validated for other
compounds (Chappell, 1989) and have been used in the case of PAH
(Thorslund & Farrar, 1990b; Collins et al., 1991; Collins & Alexeeff,
1993), this relationship may not hold for PAH. Muller et al. (1995a,b,
1996) compared the actual and extrapolated doses required to produce a
tumorigenic effect of a given magnitude in a particular rodent
species, on the basis of the assumption that if extrapolations based
on surface area and/or body weight hold between rodents to humans, the
same extrapolation should hold even more closely for closely related
rodents. The extrapolated dose was estimated from that required to
induce a similar response in another rodent species under matching
experimental conditions and by extrapolating to the target rodent
species. Examples of the analysis are shown in Tables AI.4 and AI.5.
The extrapolated and actual doses differed by much as two orders of
magnitude, even though mice and rats are closely related, of similar
sizes, and with similar diets, and, furthermore, laboratory rodents
are placed in similar habitats. It would be expected that
extrapolation from one rodent to another is more justified than
extrapolation from rodents to humans, but these analyses indicate that
extrapolation from rodents to humans may lead to even larger errors.
Although some of the discrepancies may be due to methodological
problems, much of the difference is due to the low predictive value of
the two extrapolations. Extrapolations based on surface area or body
weight further imply that species differences in the metabolism of the
parent compound to the primary carcinogen are functions of body weight
or surface area. This assumption is not supported by the available
pharmacokinetic data for PAH (Michel et al., 1995).
Table AI.3. Estimated carcinogenic potency of benzo[a]pyrene in humans exposed by inhalation, on the basis of
extrapolations from the results of studies in experimental animals
Risk Species Route of exposure Assumptions Data source UCL/ Assessor
(ng/m3) MLE
1.7 × 10-6a Hamster Inhalation on salt Risk proportional to Thyssen et UCL US Environmental
particulate (body weight)2/3 and al. (1981) Protection Agency
inhalation rate = 0.037 m3/d (1984a); Collins &
Alexeeff (1993)
1.1 × 10-6 Hamster Inhalation on salt Risk proportional to Thyssen et UCL Collins & Alexeeff
particulate (body welght)2/3 and al. (1991) (1993)
inhalation rate = 0.063 m3/d
4.7 × 10-6 Hamster Intratracheal Risk proportional to Saffiotti at UCL Collins & Alexeeff
instillation with (body weight)2/3 al. (1972) (1993)
ferric oxide
4.4 × 10-6 Hamster Intratracheal Risk proportional to Feron et al. UCL Collins & Alexeeff
instillation with (body weight)2/3 (1973) (1993)
ferric oxide
2.0 × 10-8 Rat Inhalation of Risk equal to that of humans Heinrich at UCL Heinrich et al.
coal-tar/pitch and hamsters at same air al. (1994c) (1994c)
condensation level of benzo[a]pyrene,
aerosolb corrected for rat lifetime
(2 years)
7.0 × 10-9 Hamster Risk proprotional to body Thyssen at MLE Clement Associates
weight al. (1981) (1990)
3.6 × 10-8 Hamster Risk proprotional to body Thyssen et MLE Clement Associates
weight3/4 al. (1981) (1990)
6.2 × 10-8 Hamster Risk proprotional to body Thyssen et MLE Clement Associates
weight2/3 al. (1981) (1990)
Table AI.3. (continued)
Risk Species Route of exposure Assumptions Data source UCL/ Assessor
(ng/m3) MLE
3.9 × 10-8 Hamster Risk equal to that of humans Thyssen et MLE Clement Associates
and hamsters at same air al. (1981) (1990)
level of benzo[a]pyrene
UCL, upper confidence limit; MLE, maximum likelihood estimate
a Recalculated from 6.11 mg/kg bw per day using the assumptions of the US Environmental Protection Agency for humans:
70 kg, 20 m3/d
b Other active polycyclic aromatic hydrocarbons present in the administered aerosol
Table AI.4. Comparison of actual and extrapolated doses of 3-methylcholanthrene and benzo[a]pyrene required to obtain observed
tumour incidence; topical administration
Species Compound Actual dose (mg) Extrapolated dose (mg) Extrapolated/ Reference
actual dose
Surface Body Surface Body
area weight area weight
Rat 3-Methylcholanthrene 58 9.0 20 0.16 0.35 Cavalieri et al.
(1978)a; Zackheim
(1964)
Hamster 3-Methylcholanthrene 5.0b 0.048 0.074 0.0096 0.015 Cavalieri et al.
(1978)a; Bernfeld &
Homburger (1983)
Hamster Benzo[a]pyrene 5.0b 0.019 0.028 0.0038 0.056 Cavalieri et al.
(1978)a; Bernfeld &
Homburger (1983)
a Data for mouse
b No tumours induced at dose tested; assumed 1% response rate
Table AI.5. Comparison of actual and extrapolated doses of 3-methylcholanthrene and benzo[a]pyrene required to obtain observed tumour
incidence; respiratory exposure
Species Compound Actual dose (mg) Extrapolated dose (mg) Extrapolated/ Reference
actual dose
Surface Body Surface Body
area weight area weight
Rat 3-Methylcholanthrene 0.50a 7.0a 15a 14a 30a Nettesheim & Hammons
(1971)b; Hirano et al.
(1974)
Hamster 3-Methylcholanthrene 0.7a 4.7a 7.1 6.7a 10a Nettesheim & Hammons
(1971)b; Hammond et al.
(1987)
Rat Benzo[a]pyrene 15 31 33 2.1 2.2 Furst et al. (1979)b;
Ishinishi et al. (1976)
Hamster Benzo[a]pyrene 30 33 222 1.1 0.73 Furst et al. (1979)b;
Saffiotti et al. (1972)
a Nettesheim & Hammons (1971) used intratracheal instillation, while Hirano et al. (1974) used intrapulmonary pellets to
administer 3-methylcholanthrene to rats. Hammond et al. (1987) applied intrabronchial pellets to hamsters. The fact that
instillation is considered to be less effective in inducing tumours than implantation is considered to be the likely
explanation for the apparent discrepancy between the actual and the extrapolated doses.
b Data for mouse
Some risk assessments are based on extrapolation of the results of
studies in which the compound was deposited directly onto or in close
proximity to the tissues where tumours were later observed. Since the
dose delivered to these tissues is direct and not a reflection of body
weight or surface, it may well be argued that extrapolations based on
body weight or surface area are not appropriate.
It is likely, therefore, that the estimates of the potency of PAH in
humans based on such extrapolations would lead to substantial errors.
It is therefore preferable to use other approaches, like the relative
potency approach, in order to estimate human risk.
I.2.1.2.2 Derivation of relative potencies of PAH other than
benzo [a]pyrene
Some quantitative risk estimates for mixtures of PAH are based on the
assumption that all PAH (or all carcinogenic PAH) have the same
potency as benzo [a]pyrene and that the carcinogenic effect of the
mixture can be estimated by summing the effects of each PAH. Some PAH
are less carcinogenic in animal models than benzo [a]pyrene, and a
few are more active. In order to provide more reasonable estimates of
the carcinogenicity of PAH mixtures, schemes have been devised that
are similar to the toxicity equivalence factor approaches of the US
Environmental Protection Agency and NATO for evaluating chlorinated
dibenzodioxins and dibenzofurans, which are based on a general or
specific hypothesis of relative potency.
In a provisional quantitative risk assessment of PAH, the US
Environmental Protection Agency (1993) used benzo [a]pyrene as a
standard and derived the relative potencies of individual PAH in
increments of order of magnitude by comparison. Only the results of
carcinogenicity bioassays were considered, and these were limited to
those in which benzo [a]pyrene and other PAH were assayed by the same
protocol and within the same time frame. The studies involved various
routes of exposure, including skin painting, intraperitoneal and
subcutaneous injection, and lung implantation (see section 7.7.2).
Maximum likelihood estimates from a two-stage model were used for
comparison, and ranges of estimates were presented. These values for
the results of complete carcinogenesis assays in mouse skin are shown
in Table AI.6. The US Environmental Protection Agency (1993)
considered that the data on PAH did not meet all of the requirements
for application of the toxicity equivalence factor approach and
recommended that the values be applied only to carcinogenicity and not
to other end-points. In order to differentiate between these values
and a toxicity equivalence factor meant for use in evaluating all
types of toxicity, the relative potencies were designated 'estimated
orders of potential potency'. The US Environmental Protection Agency
(1993) further recommended that these values not be used for
evaluating inhaled PAH mixtures, for the following reasons:
- The US Environmental Protection Agency currently has no consensus
value for an inhalation unit risk.
Table AI.6. Estimated orders of potency of selected polycyclic aromatic
hydrocarbons in mouse skin carcinogenesis
Compound Relative potencya Reference
Benzo[a]pyrene 1.0 1.0
Benz[a]anthracene 0.145 0.1 Bingham & Falk (1969)
Benzo[b]fluoranthene 0.167 0.1 Habs et al. (1980)
Benzo[k]fluoranthene 0.020 0.01 Habs et al. (1980)
Chrysene 0.004 0.001 Wynder & Hoffmann
(1959)
Dibenz[a,h]anthracene 1.11 1.0 Wynder & Hoffmann
(1959)
Indeno[1,2,3-cd]pyrene 0.055b 0.1 Habs et al. (1980);
Hoffmann & Wynder (1966)
a Model was P(d) = 1-exp[-A(1 + bd)2] for all except
indeno[1,2,3-cd]-pyrene. Actual figures (left) and rounded to
order of 10 (right)
b Simple mean of relative potencies (0.021 and 0.089), the latter
being derived with the one-hit model
- There is no basis for assuming that the relative order of potency
for PAH is the same after oral and inhalation exposure.
- The co-carcinogenic potential of particulate carriers in the lung
has not been sufficiently elucidated.
Improvements to the estimated orders of potential potency have been
published (McClure & Schoeny, 1995). As the two-stage model requires
estimation of several parameters in the absence of data, other models
were investigated. It was shown that the model used had little effect
on the values when applied consistently; however, the assay type and
data set used could alter the values by orders of magnitude. It was
proposed, therefore, that data from all available appropriate assays
should be modelled and that a central tendency estimate, rounded to
powers of 10, would give a more realistic value. The list of PAH for
which estimated orders of potential potency were estimated was
expanded from that of US Environmental Protection Agency (1993) to
include several more that could be considered probable human
carcinogens (Table AI.7).
In a preliminary validation exercise based on published data on
PAH-containing mixtures, McClure & Schoeny (1995) used the values in
Table AI.7 to estimate the carcinogenicity of two synthetic mixtures
of PAH (Pfeiffer, 1973), both as a sum of their components and as
whole mixtures. A good correlation was found between the two
measurements of potency. These results indicate that the components of
a defined mixture have additive risks.
Table AI.7. Estimated order of carcinogenic potency for 13
polycyclic aromatic hydrocarbons in Group B2 as compared with
benzo[a]pyrene
Compound Estimated order No. of estimates
of potency
Benzo[a]pyrene 1.0
Benz[a]anthracene 0.1 4
Benzo[b]fluoranthene 0.1 8
Benzo[j]fluoranthene 0.1 7
Benzo[k]fluoranthene 0.1 7
Chrysene 0.1 5
Cyclopenta[cd]pyrene 0.1 4
Dibenz[a,h]anthracene 1.0 3
Dibenzo[a,e]fluoranthenea 1.0 3
Dibenzo[a,e]pyrene 1.0 3
Dibenzo[a,h]pyrene 1.0 2
Dibenzo[a,i]pyrene 0.1 3
Dibenzo[a,l]pyrene 100 2
Indeno[1,2,3-cd]pyrene 0.1 4
Adapted from McClure & Schoeny (1995)
a Not evaluated by the Task Group
The additivity of the potency of mixtures of five PAH was investigated
in the mouse lung adenoma model (Nesnow et al., 1995). Different
combinations of PAH with different biological and chemical properties
were tested. Some interaction was found in which the potency of the
compounds was more or less than additive. The differences were no more
than twofold and, therefore, would not be large enough to alter
significantly the outcome of risk assessments, in which uncertainty of
an order of magnitude is not seen as excessive.
Complex environmental mixtures differ from defined synthetic mixtures
in that they contain not only PAH of known carcinogenicity but also
hundreds of PAH and other potentially carcinogenic non-PAH compounds
for which carcinogenicity has not been established. The risk
attributed to a PAH for which data on exposure and carcinogenic
potency exist would be similar to that for the entire mixture only if
the PAH for which the potency is unknown contributed little or nothing
to the potency of the mixture as a whole. McClure & Schoeny (1995)
reported that the carcinogenic activity of a coal liquefaction
material (Mahlum et al., 1984) was similar to that estimated by adding
benzo [a]pyrene equivalents derived from estimated orders of
potential potency for several measured PAH. Similar estimates for coal
flue gas, petrol-engine exhaust, and diesel exhaust, however, resulted
in underestimates of two to three orders of magnitude of the risks
presented by the PAH fractions when the results of studies by either
lung implantation or dermal application in rodents were used for the
calculation (Grimmer et al., 1984; Thorslund & Farrar, 1990a; see
Table AI.8).
Other strategies for risk assessment based on the toxicity equivalence
factor approach for individual PAH have been published. The resulting
estimates are compared in Table AI.9. Krewski et al. (1989) analysed
the published values and derived a new set based largely on estimates
from a two-stage model (Clement Associates, 1988). Applications of
their toxicity equivalence factors to the results of bioassays wth PAH
mixtures (Pfeiffer, 1977; Schmähl et al., 1977) indicated that their
values would be unlikely to underestimate the carcinogenic risk posed
by whole mixtures.
A set of toxicity equivalence factors was derived for the 17 PAH
commonly measured at hazardous waste sites, and a new list (see column
2 of Table AI.9) was calculated on the basis of older work and the
primary literature (Nisbet & LaGoy, 1992). The values tend to
overestimate the carcinogenic risks of mixtures.
The relative potency values in 14 publications were used to classify
PAH into categories of high, medium, low, and very low risk (column 3
of Table AI.9), and 'environmental assessment levels' were calculated
on the basis of the highest potency for a PAH, rounded to an order of
magnitude, relative to that of benzo [a]pyrene (Malcom & Dobson,
1994).
Kalberlah et al. (1995) and others adopted the approach of the US
Environmental Protection Agency Office of Pesticides, Pollution
Prevention and Toxic Substances to determine the relative potencies of
PAH. A panel of five experts made independent reviews of the existing
data on about 150 PAH and scored them as having high, moderate,
marginal, or slight potential carcinogenicity. The panel considered
data from studies of skin painting in mice, the induction of lung and
liver adenomas in newborn mice, mammary tumours in rats, studies by
oral administration, studies of genotoxicity, and structure-activity
relationships. Their evaluation, converted to powers of 10 to
represent levels of concern, is presented in column 4 of Table AI.9.
Columns 5 and 6 of Table AI.9 show the values of the US Environmental
Protection Agency (1993) and McClure & Schoeny (1995), discussed
previously. The results of the six toxicity equivalence factor
approaches show a reasonable degree of agreement for PAH that are
generally considered to be carcinogenic. In all of them,
dibenz [a,h]anthracene appears to be equipotent or somewhat more
potent than benzo [a]pyrene; and, in most, the benzofluoranthenes and
benzanthracene were about 10% as potent as benzo [a]pyrene. The
greatest variation in the estimated relative potency is observed for
chrysene, although all agree that chrysene is not as potent a
carcinogen as benzo [a]pyrene.
Table AI.8. Comparison of the carcinogenic potency of the polycyclic aromatic
hydrocarbon (PAH) fraction of PAH-rich mixtures with the integrated potency
of the eight PAH found in the fraction
Method of estimating risk Potency relative to that of benzo[a]pyrene
Flue gas Diesel-engine Petrol-engine
exhaust exhaust
Fraction of mixture containing 0.38 0.28 0.67
PAH with > three rings
Sum of risk for eight PAH 0.0011 0.0018 0.0007
Fraction/sum 340 150 950
From Thorslund & Farrar (1990a)
Table AI.9. Relative potencies of indicator polycyclic aromatic hydrocarbons
Compound [1] [2] [3] [4] [5] [6] [7]
1-Methylphenanthrene 0.001
Acenaphthene 0.001 0.001 0.001 0
Acenaphthylene 0.001 0.001 0.01
Anthanthrene 0.320 0.28
Anthracene 0.01 0.01 0.01
Benz[a]anthracene 0.145 0.1 0.1 0.1 0.1 0.1 0.014
Benzo[a]pyrene 1.0 1.0 1.0 1.0 1.0 1.0 1.0
Benzo[b]fluoranthene 0.141 0.1 0.1 0.1 0.1 0.1 0.11
Benzo[e]pyrene 0.004 0.01 0
Benzo[ghi]perylene 0.022 0.01 0.01 0.01 0.012
Benzo[j]fluoranthene 0.1 0.1 0.045
Benzo[k]fluoranthene 0.061 0.1 0.1 0.1 0.01 0.1 0.037
Chrysene 0.0044 0.01 0.01 0.01 0.001 0.1 0.026
Coronene 0.001
Cyclopenta[cd]pyrene 0.023 0.1 0.1 0.012
Dibenzo[a,e]pyrene 1.0
Dibenz[a,c]anthracenea 0.1
Dibenz[a,h]anthracene 1.11 5 1.0 1.0 1.0 1.0 0.89
Dibenzo[a,l]pyrene 100 100
Dibenzo[a,e]fluoranthenea 1.0
Dibenzo[a,h]pyrene 1.0 1.2
Dibenzo[a,i]pyrene 0.1
Fluoranthene 0.001 0.001 0.01
Fluorene 0.001 0.001 0
Indeno[1,2,3-cd]pyrene 0.232 0.1 0.1 0.1 0.1 0.1 0.067
Naphthalene 0.001 0.001
Perylene 0.001
Phenanthrene 0.001 0.001 0 0.00064
Pyrene 0.81 0.001 0.001 0.001 0
Table AI.9 (continued)
[1] Krewski et al. (1989);
[2] Nisbet& LaGoy (1992);
[3] Malcolm & Dobson (1994);
[4] Kalberlah et al. (1995);
[5] US Environmental Protection Agency (1993);
[6] McClure & Schoeny (1995);
[7] Muller et al. (1995a,b, 1996)
a Not evaluated by the Task Group
I.2.1.3 Application
Application of the toxicity equivalence factor approach to assessing
the risk posed by dibenzodioxins and dibenzofurans involves the
following steps:
(1) analytical determination of the agents in the environmental
sample;
(2) multiplication of the concentrations of congeners in the sample
by the toxicity equivalence factors to express the concentration
in terms of the standard agent (e.g. benzo [a]pyrene)
equivalents;
(3) summation of the products in step (2) to obtain the equivalents
of the standard agent in the sample;
(4) determination of human exposure to the mixture in question,
expressed in terms of standard chemical equivalents; and
(5) combination of the exposure derived in step 4 with information on
the toxicity (here, carcinogenic potency) of the standard
chemical in order to estimate the risks associated with exposure
to the mixture.
These steps were followed for PAH, using one or more of the toxicity
equivalence factors given in Table AI.9 and benzo [a]pyrene as the
standard.
I.2.2 Comparative potency approach
I.2.2.1 Principle
The comparative potency approach is used to estimate the potency of
the PAH in mixtures without having to identify or quantify the
individual compounds. The carcinogenic potency of an unknown mixture
in humans is estimated from the potency of the mixture in a bioassay
and from the potency of another mixture(s) in the same bioassay and in
humans. It is assumed that the relationship (ratio) between the
potency of a mixture in a bioassay and human cancer risk is constant
for different (PAH-rich) mixtures. The relationship is expressed in
equation (3):
Human risk carcinogen1/ Bioassay potency carcinogen1
= Human risk carcinogen2/ Bioassay potency carcinogen2 (3)
= k
The carcinogenic risk to humans due to exposure to a mixture can
readily be derived by a rearrangement of terms.
The potency in the bioassay and the risk to humans are expressed in
terms of the mass of extractable organic compounds contained in the
mixture. Although this method is intended to predict human risks due
to PAH, in practice it estimates the risk due to all organic compounds
present. This discrepancy may not be significant when estimating the
carcinogenic risk of mixtures rich in polycyclic organic matter, such
as coal-tar, but may be important when estimating the risk of exposure
to cigarette smoke or ambient air, in which PAH do not necessarily
play a major role.
I.2.2.2 Development and validation
The comparative potency approach was initially proposed as part of an
approach to assessing the carcinogenic risk of PAH in diesel emissions
(Albert et al., 1983; Lewtas, 1985a,b; Nesnow, 1990). For source
mixtures such as coal-tar, coke-oven emissions, and diesel and petrol
emissions tested both for skin tumorigenicity in mice (Nesnow et al.,
1982a,b) and in short-term bioassays (Lewtas, 1985a), there was
generally good agreement (Lewtas, 1985a). Furthermore, there appears
to be a good correlation between the potency of mixtures in bioassays
and in humans, although this correlation is based on limited
epidemiological data of good quality (Nesnow, 1990; Lewtas, 1993).
Thus, risk assessments of relatively high quality are currently
available only for cigarette smoke, coke-oven emissions, and coal-tar.
Although the concept of comparative potency has been extensively
validated, some outstanding issues remain, which are discussed below.
I.2.2.3 Key implicit and explicit assumptions
The comparative approach assumes that several distinct sources
contribute PAH to the environment at a given location in an ambient
environment. For example, in an industrial city in winter, the sources
of PAH may include emissions from steel manufacture, from cars,
lorries, and transport from other locations, and from home heating.
Each source is assumed to make a specific contribution to the overall
risk for lung cancer due to PAH. The proportion of the total risk
attributable to each source depends on its potency (risk per unit mass
of organic compounds) and the overall contribution to the mass of
organic compounds in the ambient air. The total risk can be expressed
as follows:
Total risk = (unit risk source1 × mass of organic
compounds source1) + (unit risk source2 × mass of
organic compounds source2) + (unit risk source3 (4)
× mass of organic compounds source3) ...
The unit risks for some sources are listed in Table AI.10.
Table AI.10. Potency of some source mixtures expressed as average
dose causing 50% papilloma incidence in male and female Sencar
mice
Source mixture Organic compounds (mg)
Coke-oven main 0.14
Coke-oven topside 0.16
Roofing tar 2.0
Nissan diesel emission 1.6
Volkswagen Rabbit diesel emission Tumour incidence did not reach
50% with tested doses
From Nesnow et al. (1982a,b)
Implicit in this approach are the assumptions that the composition of
the organic compounds emitted from each source (such as diesel
engines) is constant and that the potency (unit risk) of a given
source mixture is constant. If the potency and composition of the
organic compounds within mixtures from the same source vary widely,
the standard mixture may not accurately represent other mixtures from
a similar source. There is some evidence, however, that mixtures from
similar sources have substantially different compositions. Thus, the
benzo [a]pyrene content of the emissions from four diesel engines
varied over a 600-fold range (Nesnow et al., 1982a,b; see Table
AI.11). Significant differences in the potency of the four mixtures
were also seen. Nevertheless, the potency of the mixtures appears to
correlate reasonably well with their benzo [a]pyrene content (see
Table AI.11), so that the comparative potency approach may be viable
in principle but may not be appropriate for expressing the potency of
the mixtures in terms of the mass of the organic content. A possible
alternative is to express the potency in terms of the level of
benzo [a]pyrene present in the mixture. If this solution is used, the
comparative potency approach becomes essentially the benzo [a]pyrene
surrogate approach discussed in section I.2.3.
The levels of PAH in ambient air may be influenced by multiple
sources, and some PAH may be transformed in the environment. In order
to estimate the carcinogenic risk due to exposure to an ambient
mixture, the contribution of individual sources to the ambient air
levels must be estimated, because contributing sources differ in their
carcinogenicity. Making reliable estimates of the contribution of
individual sources to ambient air levels is still a difficult,
non-routine process.
Table AI.11. Benzo[a]pyrene content of organic fraction extracted from
diesel-engine emissions
Source Benzo[a]pyrene % Mice with tumours/ng
(ng/mg organic fraction) benzo[a]pyrenea
Nissan 1200 0.024
Oldsmobile 2.0 NS
Caterpillar 2.0 NS
Volkswagen Rabbit 26 0.11
From Nesnow et al. (1982a,b); NS, no significant response observed over
the range of doses tested
a Calculated by Muller et al. (1995a,b, 1996)
I.2.2.4 Application
In order to use the comparative potency approach, the carcinogenicity
of the major source mixtures that contribute to a given ambient
environment must be established. The potency of a number of the
mixtures has been estimated (see Table AI.10), and the potency of
source mixtures that affect air has been expressed in terms of risk
per mass of organic compounds per cubic metre. The levels of each
source mixture must then be estimated for a given ambient environment.
The total risk is calculated as shown in equation (4).
I.2.3 Benzo [a]pyrene as a surrogate for the PAH fraction of
complex mixtures
I.2.3.1 Principle
The third approach assumes that the risk due to the PAH component of
complex mixtures and the levels of individual PAH in the mixtures are
proportional to those of benzo [a]pyrene in the mixture and vary
proportionately. Using this approach, the risk due to the PAH
component of mixtures can be estimated as the product of the
environmental levels of benzo [a]pyrene and the estimate of the risk
attributable to mixtures per unit amount of benzo [a]pyrene.
In general, the approach does not predict the potency of an ambient
complex mixture as a whole but merely its PAH component. There is no
reason to believe that benzo [a]pyrene is a good indicator of
chlorinated compounds such as dioxins and dibenzofurans or volatile
organic compounds such as benzene and 1,3-butadiene, which may be
present in some ambient complex mixtures. The contribution of non-PAH
to the overall risk of exposure to complex mixtures must thus be
assessed separately.
I.2.3.2 Development and validation
Benzo [a]pyrene was initially favoured as an indicator of all urban
pollution, and in a number of assessments based primarily (or
entirely) on studies of the general population exposed to ambient air
benzo [a]pyrene was used as an index of exposure to a wider mixture
of materials (Nisbet et al., 1985). There is evidence, however, that
benzo [a]pyrene cannot serve as an indicator of the toxicity of whole
mixtures. Various factors may influence the relative contents of PAH
and other contaminants of ambient air. For instance, the relative
proportions of benzo [a]pyrene and other PAH in ambient urban air has
been declining over the years, while the levels of volatile organic
compounds and others have been rising. In some mixtures, such as
cigarette smoke condensate, PAH probably play only a minor role in
overall toxicity. Pott & Heinrich (1992) showed that mixtures
containing large amounts of carcinogenic compounds other than PAH,
such as cigarette smoke, are much more potent at a given level of
benzo [a]pyrene than mixtures that owe much of their carcinogenicity
to PAH, such as coke-oven emissions. Nisbet et al. (1985) argued
convincingly that benzo [a]pyrene cannot serve as a general indicator
of all pollutants in the ambient air, although it may be a suitable
indicator for the carcinogenic risk posed by four- to seven-ring,
unsubstituted PAH in the mixture.
Muller et al. (1995a,b, 1996) examined the PAH profiles of a wide
range of mixtures from many sources and found that they were generally
similar (see also section I.2.3.3). Furthermore, those mixtures rich
in PAH and in which PAH are likely to contribute a significant
proportion of the risk of the mixture are very similar in potency
expressed per unit amount of benzo [a]pyrene. This observation is
consistent with the notion that the PAH components of these mixtures
are approximately equipotent. (Establishment of the potency of
mixtures is discussed in section I.2.3.4.)
These findings do not imply that all mixtures are similar. Differences
in the PAH profiles of the same mixture have been analysed in order to
establish markers for mixtures from a particular source (Gordon &
Bryan, 1973; Greenberg et al., 1981; Vogt et al., 1987). The levels of
some substituted PAH are clearly not related to the levels of
benzo [a]pyrene in the mixture (Albert et al., 1983). The differences
in the profile of four- to seven-ring, unsubstituted PAH in various
mixtures are probably too small to alter the estimated risks of the
PAH component of the mixtures significantly.
I.2.3.3 PAH profiles of complex mixtures
Information on the levels of PAH in various source mixtures is
provided in Section 5. The levels of PAH in environmental mixtures
used in the following analysis were derived mainly from monitoring in
Canada (Muller et al., 1995a,b, 1996). About 100 mixtures were
classified into different types, such as diesel emissions, coke-oven
emissions, ambient air particulate, soils, and sediments, to
facilitate the analysis, and the profiles of the 15 PAH most commonly
tested in mixtures were compared. The results are expressed as the
ratios of the levels of each PAH relative to benzo [a]pyrene; the
geometric mean, the upper and lower 95% confidence limits, and the
confidence range were calculated for each PAH ratio for a mixture
type. The confidence range was determined by dividing the upper
confidence limit by the lower confidence limit and used as a measure
of the range of the means of the relative levels a given PAH will
assume about 95 times out of 100.
The PAH profiles for petrol exhaust emissions are provided as an
example in Table AI.12. It can be seen that the confidence range for
each PAH is less than 5.0, and many are less than 2.0, indicating that
the levels of these PAH, relative to benzo [a]pyrene, are very stable
and vary little among the sources. They also indicate that any
variation among samples is probably not large enough to alter the
estimated risk. The low variation also means that the level of
benzo [a]pyrene is a good predictor of the levels of the other PAH
that may be present in a given mixture from petrol engines.
The confidence ranges for all types of combustion mixture and all of
the PAH considered are presented in Table AI.13. The confidence ranges
were < 50 for more than 90% of all entries and < 6 for 50% of all
entries. Given the degree of uncertainty usually associated with risk
assessment, the uncertainty presented by the variation in PAH profile
is relatively small. In addition, while some compounds in a given
mixture may be found at higher levels than expected, those of other
compounds may be lower than expected, and there may therefore be
little difference between the estimates of risk based on chemical
analysis and those based on the predicted composition of a mixture.
The ambient mixtures appear to be less variable than the combustion
emission mixtures. For example, Table AI.14 shows that the confidence
ranges for most types of ambient air are similar. The corresponding
levels of PAH relative to benzo [a]pyrene are shown in Table AI.15.
Samples of ambient air were collected in 1982-86 at point sources in
Hamilton, Ontario, Canada, on days when the wind was blowing from
nearby steel-mill operations 50% or more of the time. Mobile sources
and home heating also contributed, but the urban levels of PAH were
much lower on days when the wind was not blowing from the direction of
the steel mills. The average level of benzo [a]pyrene was about 1.8
ng/m3. Samples of ambient air from mobile sources were collected in
Toronto, Ontario, during the summer near the intersection of two busy
multi-lane highways in 1988-92. The average level of benzo [a]pyrene
was about 0.17 ng/m3. The highway intersection is surrounded by
residential areas, and the samples of ambient air associated with home
heating were collected in the same location as the mobile sources by
the same collection and analytical protocol, but in winter. The
average level of benzo [a]pyrene was 0.41 ng/m3. Home heating and
mobile emisions are considered to be the main sources of PAH. In
samples of ambient air collected on Wallpole Island, Ontario, a rural
location with little traffic and no industrial source, the average
levels of benzo [a]pyrene was about 0.093 ng/m3 (T. Dann, personal
communication).
Table AI.12. Confidence ranges for petrol engine exhaust (relative to
benzo[a]pyrene
Compound Mean 95% confidence
Lower limit Upper limit Interval
Anthracene 7.0 9.3 5.3 1.8
Phenanthrene 25 38 17 2.3
Fluoranthene 7.3 14 4.0 3.5
Pyrene 9.5 19 4.7 4.0
Benz[a]anthracene 0.81
Perylene 0.27 0.60 0.12 4.8
Benzo[e]pyrene 1.1 1.4 0.79 1.8
Benzo[ghi]perylene 2.6 3.5 2.0 1.7
Dibenz[a,h]anthracene 0.072
Coronene 2.0 2.7 1.4 1.9
Indeno[1,2,3-cd]pyrene 0.80 1.1 0.60 1.8
Anthanthrene 0.38 0.55 0.27 2.1
Chrysene and triphenylene 3.0 4.6 1.9 2.4
Benzofluoranthenes 1.1 1.5 0.85 1.7
From Muller (1995a,b, 1996), based on data from Hoffmann & Wynder
(1962), Grimmer & Hildebrandt (1975), Grimmer & Bohnke (1978),
Alsberg et al. (1985), and Hagemann et al. (1982)
Table AI.13. Confidence ranges for polycyclic aromatic hydrocarbons in various combustion mixtures (relative to
benzo[a]pyrene
Compound Combustion mixture
Coke Coal-tar Coal-fired Coal Open burning Wood Diesel Petrol Roofing Paving
ovens power plants stoves and fireplaces stoves emissions emissions asphalt asphalt
Anthracene 3.2 440 830 7.6 1.8
Phenanthrene 1.3 32 27 2.7 2.3
Fluoranthene 2.7 4.5 3.2 43 4.9 1.4 6.1 3.5 5.7 5.2
Pyrene 2.6 280 37 18 5.6 1.5 5.3 4.0 19 2.7
Benz[a]anthracene 19 2.8 32 160 2.0 9.0 130 8.5
Perylene 2.3 27 23 8.2 31 4.8 4.6 5.9
Benzo[e]pyrene 1.3 5.5 3.3 35 1.6 1.5 22 1.8
Benzo[ghi]perylene 8.8 12 32 2.7 8.9 1.7 5.1 2.8
Dibenz[a,h]anthracene 310 3.2 2.4
Coronene 7.8 7.1 730 3.2 1.9
Indeno[1,2,3-cd]pyrene 7.4 1.9 7.0 1.8
Anthanthrene 8.7 2.1
Chrysene and 2.1 12 430 42 25 4.3 7.3 2.4 23 6.1
triphenylene
Benzofluoranthenes 5.7 2.0 28 7.1 5.0 3.4 1.7 7.7 120
Incidence of confdence 0 2 2 1 2 0 0 0 1 1
range > 50
Table AI.14. Confidence ranges for particulates extracted from ambient
air (relative to benzo[a]pyrene)
Compound Point Near Home Transport Geometric
source mobile heating mean
source
Anthracene 2.8 5.7 6.7 2.0 20
Phenanthrene 2.3 1.7 2.6 1.4 13
Fluoranthene 2.2 1.5 1.7 1.4 8.1
Pyrene 2.4 1.4 1.7 1.4 6.7
Benz[a]anthracene 2.0 1.4 1.5 1.2 2.3
Perylene 2.7 1.3 1.2 1.9 1.7
Benzo[e]pyrene 2.8 1.3 1.6 1.1 1.3
Benzo[ghi]perylene 2.5 1.5 1.6 1.2 2.4
Indeno[1,2,3-cd]pyrene 1.3 1.4 1.8 1.2 1.2
Anthanthrene 2.0 3.4 1.8 41 1.9
Chrysene and 2.1 1.3 2.0 1.3 1.4
triphenylene
Benzofluoranthenes 2.5 1.3 1.9 1.3 1.7
Table AI.16. Mean profiles of polycyclic aromatic hydrocarbons in
ambient air (relative to benzo[a]pyrene)
Compound Point Near Home Transport Geometric
source mobile heating mean
source
Anthracene 5.5 7.6 1.0 1.8 2.9
Phenanthrene 38 200 39 43 60
Fluoranthene 14 48 12 13 18
Pyrene 9.3 28 11 7.1 12
Benz[a]anthracene 1.4 0.82 1.0 0.78 0.97
Perylene 0.33 0.25 0.22 0.24 0.26
Benzo[e]pyrene 1.5 1.3 1.6 1.4 1.4
Benzo[ghi]perylene 1.4 1.5 2.4 1.3 1.6
Indeno[1,2,3-cd]pyrene 1.5 1.3 1.5 1.4 1.4
Anthanthrene 0.19 0.15 0.13 0.20 0.17
Chrysene and 3.0 2.7 3.5 2.9 3.0
triphenylene
Benzofluoranthenes 3.6 2.9 3.6 4.4 3.6
Table AI.16 presents the average PAH profiles of the combustion
emissions, ambient air particulates, soils, and sediments and the
average profile of the four types of mixture. The confidence ranges
indicate that the four mixtures had fairly similar PAH profiles.
On the basis of this analysis, Muller et al. (1995a,b, 1996) concluded
that a wide variety of mixtures have fairly similar profiles of
commonly assayed PAH. They assumed that the PAH fraction of all
sources of environmental mixtures have profiles similar to the
average, as shown in the sixth column of Table AI.16. This conclusion
does not include substituted PAH, as there is strong evidence that
different mixtures contain different levels of substituted PAH. It
does not imply that there are no real differences due to the source of
the mixture, the type of fuel, and the pyrolysis conditions that
produced it. Furthermore, aerial transport of PAH, degradation in
sunlight or by soil microorganisms, and other factors may alter the
PAH profile. These factors are, however, unlikely to generate large
enough differences in the PAH profiles of mixtures to significantly
alter the estimate of risk for a given mixture.
I.2.3.4 Potency of complex mixtures
If benzo [a]pyrene is a suitable indicator of the carcinogenic
potency of the PAH in a mixture, then the potency of a mixture
expressed as the tumour incidence per nanogram of its benzo [a]pyrene
content should be numerically similar for all mixtures in which PAH
are expected to be the major cause of tumorigenic effects. Nesnow et
al. (1982b) tested a number of PAH-rich mixtures in a
tumour-initiation assay in mouse skin. The different types of mixture
were roughly equipotent when the potency was expressed in terms of
benzo [a]pyrene content (Table AI.17).
I.2.3.5 Key implicit and explicit assumptions
The approach assumes that the levels of individual PAH relative to
benzo [a]pyrene are relatively stable from mixture to mixture. It
also assumes that the risk attributable to PAH in any given mixture is
proportional to the risk due to benzo [a]pyrene. In other words, the
level of benzo [a]pyrene is sufficient to estimate the risk of the
PAH fraction in a mixture.
Differences in the composition of mixtures from different sources have
been used to estimate the contribution of those sources to the ambient
levels of PAH (see Gordon & Bryan, 1973; Greenberg et al., 1981; Vogt
et al., 1987). Other authors have reported transformation of PAH in
the environment. Since some compounds are more photosensitive than
others, the proportion of PAH in ambient air will change over time as
a result of the different transformation rates of different compounds
(Van Cauwenberghe, 1985). Muller et al. (1995a,b, 1996) examined a
wide variety of mixtures from different combustion sources and ambient
mixtures and concluded that the differences in the profiles of four-
to seven-ring unsubstituted PAH relative to the benzo [a]pyrene
content are not large enough to affect the risk posed by the mixtures
Table AI.16. Average profiles for combustion-derived mixtures, ambient air, soil, and sediment
(relative to benzo[a]pyrene)
Compound Source Ambient Soil Sediment Geometric Confidence
mixtures air mean range
Anthracene 3.9 2.9 0.85 0.47 1.6 24
Phenanthrene 18 60 2.8 3.6 4.3 23
Fluoranthene 4.9 18 2.5 3.2 3.8 1.8
Pyrene 4.5 12 3.1 2.4 2.8 2.9
Benz[a]anthracene 1.8 0.97 1.4 1.4 1.2 2.6
Perylene 0.51 0.26 0.34 1.4 0.45 17
Benzo[e]pyrene 1.0 1.4 1.4 1.1 7.4
Benzo[ghi]perylene 0.98 1.6 1.4 0.96 1.0 1.4
Dibenz[a,h]anthracene 0.35 0.45 0.30 0.28 4.2
Coronene 0.35 0.45 0.34 3.9
Indeno[1,2,3-cd]pyrene 0.51 1.4 0.95 1.2 0.86 4.7
Anthanthrene 0.49 0.17 0.19 0.31
Chrysene and 2.3 3.0 1.4 1.2 2.0 3.3
triphenylene
Benzofluoranthenes 1.6 3.6 1.7 2.4 2.5 11
Table A1.17. Potency of various mixtures in an
assay for tumour initiation
Source mixture Incidence per ng
benzo[a]pyrene
Coke-oven main 9.4 × 10-2
Coke-oven topside 7.6 × 10-2
Smoky coal 7.8 × 10-2
Smokeless coal 2.8 × 10-2
Roofing tar 1.1 × 10-2
Wood smoke 5.8 × 10-2
Diesel engine exhaust 2.5 × 10-2
Petrol engine exhaust 5.6 × 10-2
Max/min 8.6
Calculated from data of Nesnow et al. (1982b)
significantly. The PAH profile of a tested mixture may deviate from
the average profile by about an order of magnitude (up or down). Since
the levels of some PAH may be above and those of others below the
expected levels, these differences would tend to cancel each other
out, leading to an error of much less than one order of magnitude.
Such small differences are below the resolution of the risk assessment
process.
I.2.3.6 Application
The first step is to estimate the carcinogenic risk due to exposure to
the PAH present in a typical mixture, and this estimate is used for
all subsequent assessments. WHO (1987) estimated that the risk for
lung cancer due to lifelong exposure to PAH in mixtures by inhalation
was 8.7 × 10-5/ng benzo [a]pyrene per m3. This estimate was based
on the assessment of the risk for lung cancer of coke-oven workers
conducted by the US Environmental Protection Agency (1984d), which
generated an upper bound risk estimate expressed in terms of
benzene-extractable material. WHO (1987) converted the US
Environmental Protection Agency estimate into benzo [a]pyrene levels
by assuming that the benzene extract contained 0.71% benzo [a]pyrene.
Sloof et al. (1989) in the Netherlands estimated the risk for lung
cancer to be 1.0 × 10-4/ng benzo [a]pyrene per m3 on the basis of
the estimate of WHO and the assessments of Pike (1983) and Tuomisto &
Jantunen (1987). That of Pike (1983) was based on the mortality from
lung cancer of gas workers, and that of Tuomisto & Jantunen (1987) was
based on the exposure of Chinese women to smoky coal smoke. Muller et
al. (1995a,b, 1996) proposed 2.3 × 10-5/ng benzo [a]pyrene per m3
as the risk for lung cancer from lifelong exposure to PAH in ambient
mixtures on the basis of the study of the US Environmental Protection
Agency (1984d) on coke-oven workers and assuming that benzo [a]pyrene
represents 1.7 ng/µg of benzene-extractable material from coke-oven
emissions. Rather than the upper bound, Muller et al. used a maximum
likelihood estimate, calculated from the US Environmental Protection
Agency study, of 3.9 × 10-5/µg of benzene-extractable per m3.
The next step is to estimate the environmental levels of
benzo [a]pyrene. In a simplified situation, in which the population
is exposed to a fixed level of environmental benzo [a]pyrene, the
lifetime cancer risk is estimated as the product of the potency of a
typical mixture (expressed as risk per nanogram of benzo [a]pyrene
per cubic metre in the case of air) and the level of benzo [a]pyrene
(expressed as ng/m3) in the environment. For example, using the risk
estimate proposed by Muller et al. (1995a,b, 1996) and assuming that a
population is exposed over a lifetime to benzo [a]pyrene at 0.5
ng/m3, the risk of the population is about 1.2 × 10-5. In other
words, about one person in 100 000 would be expected to develop lung
cancer in his or her lifetime as a result of exposure to PAH in air.
I.3 Comparison of the three procedures
Each approach has its advantages and disadvantages (Table AI.18):
I.3.1 Individual PAH approach
Main advantages:
* Clearly defined chemical species are assessed.
* A good body of scientific literature is available to evaluate it.
* Not affected by variability in the composition of mixtures
* Relatively easy to apply in ambient environments affected by many
sources
* Regulatory experience exists.
Main disadvantages:
* May underestimate risk due to all PAH by considering only a few
compounds
* Depends on extrapolation from animal models to humans
* Resource-intensive, as monitoring and analysis are required
I.3.2 Comparative potency approach
Main advantages:
* The risk of whole mixtures, rather than only a few components, is
estimated.
Table AI.18. Features and properties of three approaches to risk assessment for mixtures containing polycyclic aromatic
hydrocarbons (PAH)
Property or feature Individual PAH approach Comparative potency Benzo[a]pyrene (BaP)
approach surrogate approach
Portion of mixture for which Selected PAH in complex Entire complex mixture Unsubstituted PAH component
cancer risk is estimated mixtures account for only of complex mixtures
portion of risk of PAH fraction
PAH included in assessment Relatively few PAH out of All PAH Most PAH, except PAH for which
hundreds in environment levels do not correlate well with
those of BaP, e,g. substituted
PAH
Other toxicants included None Toxicants present in source Some, e.g. those present in
mixture coke-oven emissions
Assumption of additivity of Yes: good evidence that No: assumption not required No: assumption not required
components of mixture risks of ill health due to
exposure to PAH are
approximately additive;
little known about additivity
of risks due to PAH and
other compounds
Incorporates directly No: requires extrapolation Yes: requires data from Yes: potency derived from data
available data on human from animal models to animal models to estimate on potency of coke-oven
cancer risk estimate potency of all PAH potency of some mixtures emissions in humans
Assumption that mixtures No: not applicable Yes: assumes mixtures from Yes: assumes mixtures from
from similar sources are similar sources are about similar sources are about
about equipotent equipotent when expressed equipotent when expressed
in terms of mass of organic in terms of mass of BaP;
extractable material; basis assumption supported by
for assumption equivocal available data
Table AI.18. (continued)
Property or feature Individual PAH approach Comparative potency Benzo[a]pyrene (BaP)
approach surrogate approach
Assumption that mixtures No: assumption not required No: assumption not required Yes: evidence from studies of
from different sources are animal models supports the
about equipotent assumption; human data are
equivocal and inadequate to
validate the approach
Assumption that environmental No: assumption not required Yes: evidence that PAH Yes: evidence that PAH profile
transformation profile relative to BaP does relative to BaP does not vary
processes do not change the not vary enough to affect the enough to affect estimated risk
PAH profile enough to affect estimated risk significantly significantly
the overall cancer risk
significantly
Assumption that the PAH No: assumption not required No: assumption not required Yes: good evidence to
profile of various mixtures is support this assumption
roughly comparable
Suitable for ambient Yes Requires apportioning of Yes
environments affected by ambient mixture to multiple
multiple sources sources
Monitoring requirements Selected PAH Organic extractable matter BaP
(this information is not
usually reported and methods
not standardized)
Regulatory use Yes No Yes
* A good body of scientific literature is available to evaluate it.
* Takes advantage of existing data on human carcinogenicity
* Simple and requires inexpensive monitoring
Main disadvantages:
* Does not define the contribution of PAH to estimated overall
risk.
* Difficult to use for assessing speciated components of a mixture.
* Risk estimates require estimates of the contributions of
individual sources to the levels of organic compounds in the
ambient environment.
* The assumption that mixtures from the same source are associated
with similar risks may not be supported by the available data.
* The levels of compounds extractable in organic solvents are not
usually reported, and the analytical methods are not
standardized.
I.3.3 Benzo [a]pyrene surrogate approach
Main advantages:
* Can be used to estimate risk of entire PAH component of a mixture
* Simple and based on a few testable assumptions
* Well supported by the available data
* Relatively easy and inexpensive to apply for regulatory purposes
* Regulatory experience exists.
Main disadvantages:
* May result in overestimate of the risk of PAH within a mixture
* Some PAH, such as substituted ones, are not well represented by
benzo [a]pyrene and must be considered separately.
APPENDIX II
SOME LIMIT VALUES
Regulatory decisions about chemicals taken in a country can be fully
understood only within the framework of the legislation of that
country. Furthermore, the regulations and guidelines of all countries
are subject to change and should always be verified with the
appropriate regulatory authorities before application.
II.1 Exposure of the consumer
The concentrations of some components of polycyclic aromatic
hydrocarbons (PAH), especially benzo[a]pyrene, in air, water, and food
and the use of PAH-containing technical products are regulated by law
in many countries. The available limit values are listed in Table
AII.1.
II.2 Occupational exposure
Regulations for limits in the air at different workplaces are compiled
in Table AII.2. Only values for individual substances are given. For
some occupations, e.g. roofers and asphalt workers, limit values are
not given for individual compounds but for the mixture of organic
vapours released, e.g. bitumen fumes, coal-pitch, and coal-tar
volatiles, that are soluble in benzene or hexane. These limit values
were not taken into account.
II.3 Classification
Only classifications based on a toxicological end-point are given
here. Those relevant to exposure in the workplace are shown in Table
AII.3. Especially in industrialized countries, classifications also
exist for industrial emissions into air, water, and soil. As these
are special regulations, which differ from country to country, they
are not included.
Some classifications refer to technical mixtures with a high PAH
content:
II.3.1 European Union
* A maximum of 50 mg/kg benzo[a]pyrene will render coal-tar derived
products carcinogenic (Directive 94/69/EC: European Economic
Community, 1994a).
* For lubricant base oils analyzed by the legally defined method,
the cut-off to define carcinogenicity is 3% of the extract
containing mainly PAH, equivalent to 0.5-1 mg/kg benzo[a]pyrene
(CONCAWE, 1994).
Table AII.1. Limit values for consumer exposure to individual polycyclic aromatic hydrocarbons (PAH) in various countries
Country, year Compound Limit value Reference
Ambient air
Italy EEC (1994)
01.01.1996 to 31.12.1998 Benzo[a]pyrene 2.5 ng/m3
From 01.01.1999 Benzo[a]pyrene 1 ng/m3
Former USSR, 1985 Benzo[a]pyrene 1 ng/m3 Khesina (1994); UNEP
(1994)
Ambient water
USA, 1984 Sum of benzo[a]pyrene, benz[a]anthracene, 0.2 µg/litre Slooff et al. (1989)
benzofluoranthenes, chrysene, fluoranthene,
indeno[1,2,3-cd]pyrene, anthracene, pyrene,
dibenz[a]anthracene
Former USSR, 1990 Benzo[a]pyrene 0.005 µg/litre UNEP (1994)
EEC, 1980 Sum of fluoranthene, benzo[b]fluoranthene, 1.2 µg/litre Slooff et al. (1989)
benzo[k]fluoranthene, benzo[a]pyrene,
benzo[ghi]perylene, indeno[1,2,3-cd]pyrene
Drinking-water
WHO guideline, 1995 Benzo[a]pyrene 0.7 µg/litre WHO (1996)
EEC, 1980 (adopted by most Sum of fluoranthene, benzo[b]fluoranthene, 0.2 µg/litre EEC (1980)
Member States and numerous benzo[k]fluoranthene, benzo[a]pyrene,
other European countries) benzo[ghi]perylene, indeno[1,2,3-cd]pyrene
Former Czechoslovakia, 1991 Sum of PAH expressed as fluoranthene 40 µg/litre UNEP (1994)
Benzo[a]pyrene 0.01 µg/litre
Canada, 1991 Benzo[a]pyrene 0.01 µg/litre UNEP (1994)
Netherlands, 1989 Sum of fluoranthene, benzo[b]fluoranthene, 0.1 µg/litre Slooff et al. (1989)
benzo[k]fluoranthene, benzo[a]pyrene,
benzo[ghi]perylene, indeno[1,2,3-cd]pyrene
Table AII.1. (continued)
Country, year Compound Limit value Reference
Soil
USSR, 1985 Benzo[a]pyrene 0.02 mg/kg UNEP (1994)
Food
EEC, 1991: use of flavourings Benzo[a]pyrene 0.03 µg/kg European Economic
in food Community (1991)
Germany, 1988: meat and meat Benzo[a]pyrene 1 µg/kg EEC (1988)
products
Italy, 1988: food and beverages Benzo[a]pyrene 0.03 µg/kg Anon. (1988)
Other
EEC, 1994: tar-oil products Benzo[a]pyrene 50 mg/kg Appendix I, No. 32 of
for wood preservation guideline 94/60/EG dated
20.12.1994
Germany, 1994: products for Benzo[a]pyrene 5 mg/kg Appendix to s1 of German
wood preservation Chemicals Prohibition
Directive (1994)
Former Czechoslovakia, 1991 Benzo[a]pyrene Prohibited in UNEP (1994)
cosmetics
* Any substance containing benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[j]fluoranthene, benzo[a]pyrene, or
dibenz[a,h]anthracene at a concentration > 0.1% is regarded as carcinogenic (Annex I of Directive 67/548/EEC).
It therefore cannot be supplied to the general public in the European Union (Directive 94/60/EC) but only to
professional users (Von Meyerinck, 1995).
Table AII.2. Limit values for individual polycyclic aromatic hydrocarbons at various workplaces
Country, year Workplace or Compound Limit value Reference
emission source
Finland, 1989 NR skin Benzo[a]pyrene 10 µg/m3 (TWA) UNEP (1994)
absorption
France, 1988 Production of carbon Benzo[a]pyrene 0.15 µg/m3 Lafontaine et al.
electrodes (1990b)
Germany, 1989 Cokeries, oven area Benzo[a]pyrene 5 µg/m3 German Federal
Department for
Worker Safety (1989)
Other workplaces Benzo[a]pyrene 2 µg/m3 German Federal
Department for
Worker Safety (1989)
Sweden,1993 NR Benzo[a]pyrene 2 µg/m3 (LLV) Swedish National
20 µg/m3 (STV); Board of Occupational
Safety & Health (1994)
Former USSR, 1990 NR Benzo[a]pyrene 0.15 µg/m3 (MAC) UNEP (1994)
Argentina, 1991 NR Naphthalene 50 mg/m3 (TWA; MPC) UNEP (1994)
75 mg/m3 (STEL; MPC)
Bulgaria, 1985 NR Naphthalene 20 mg/m3 (MPC) UNEP (1994)
Canada, 1991 NR Naphthalene 50 mg/m3 (TWA; TLV) UNEP (1994)
75 mg/m3 (STEL; TLV)
Germany, 1993 NR Naphthalene 50 mg/m3 (MAK) American Conference of
Governmental Industrial
Hygienists (1995)
Hungary, 1985 NR Naphthalene 20 mg/m3 (TWA; MAC) UNEP (1994)
100 mg/m3 (STEL: MAC)
Italy, 1991 NR Naphthalene 50 µg/m3 EEC (1991)
Mexico, 1991 NR Naphthalene 50 mg/m3 (TWA; MXL) UNEP (1994)
75 mg/m3 (STEL; MXL)
Poland, 1985 NR Naphthahne 20 mg/m3 (TWA; MPC) UNEP (1994)
Romania, 1985 NR Naphthalene 30 mg/m3 (TWA; MPC) UNEP (1994)
40 mg/m3 (MPC)
Table AII.2. (continued)
Country, year Workplace or Compound Limit value Reference
emission source
Sweden, 1991 NR; skin Naphthalene 0.2 mg/m3 (TWA; HLV) UNEP (1994)
absorption 0.6 mg/m3 (STEL; HLV)
Switzerland, 1987 NR Naphthalene 50 mg/m3 (TWA; MAK) UNEP (1994)
United Kingdom, 1992 NR Naphthalene 50 mg/m3 (TWA; OES) UNEP (1994)
75 mg/m3 (STEL; OES)
USA, 1993 NR Naphthalene 52 mg/m3 (TWA) American Conference of
79 mg/m3 (STEL) Governmental Industrial
Hygienists (1995)
Former USSR, 1993 NR Naphthalene 20 mg/m3 American Conference of
Governmental Industrial
Hygienists (1995)
Former Yugoslavia,1985 NR Naphthalene 50 mg/m3 (TWA; MAC) UNEP (1994)
USA, 1993 Cokeries, oven area Phenylene 0.1 mg/m3 (TWA) American Conference of
Governmental Industrial
Hygienists (1995)
Former USSR, 1989 NR Phenylene 0.8 mg/m3 (MAC) UNEP (1994)
USA, 1993 NR Pyrene 0.1 mg/m3 (TWA) American Conference of
Governmental Industrial
Hygienists (1995)
Former USSR, 1989 NR Pyrene 0.03 mg/m3 (MAC) UNEP (1994)
NR, not reported; HLV, hygienic limit value; LLV, level limit value; MAC, maximum allowable concentration; MAK, maximum
workpace concentration; MPC, maximum permissible concentration; MXL, maximum limit; OES, occupational exposure standard;
TWA, time-weighted average; STEL, short-time exposure level; STV, short-term value; TLV, threshold limit value
Table AII.3. Toxicological classifications of polycyclic aromatic hydrocarbons with regard to exposure in
the workplace
Compound ACGIHa (TLV) IARCb EUc German
MAKd
TWA (8 h) STEL (15 min)
Benz[a]anthracene A2 A2 2A Carcinogenic, category 2 A2
Benzo[b]fluoranthene A2 A2 2B Carcinogenic, category 2 A2
Benzo[j]fluoranthene 2B Carcinogenic, category 2 A2
Benzo[k]fluoranthene 2B Carcinogenic, category 2 A2
Benzo[a]pyrene A2 2A Carcinogenic, category 2 A2
Chrysene A2 A2 3 Carcinogenic, category 2 A2
Dibenz[a,h]anthracene 2A Carcinogenic, category 2 A2
Dibenzo[a,e]pyrene 2B A2
Dibenzo[a,h]pyrene 2B A2
Dibenzo[a,i]pyrene 2B A2
Dibenzo[a,l]pyrene 2B A2
Indeno[1,2,3-cd]pyrene 2B A2
5-Methylchrysene 2B
a American Conference of Governmental Industrial Hygienists (1995)
b IARC (1987) see Section 12
c EU-Rili (Appendix I)
d German Dangerous Chemicals Directive (1995)
II.3.2 USA
* Coal-tar and coal-pitch volatiles, which are mixtures of organic
vapours with high PAH levels, are classified as A1, confirmed
human carcinogens (American Conference of Governmental Industrial
Hygienists, 1995).
* Diesel exhaust is considered to be a suspected human carcinogen
(A2), but notice has been given of intended changes (American
Conference of Governmental Industrial Hygienists, 1995).
13. REFERENCES
Aamot E, Krane J, & Steiness E (1987) Determination of trace amounts
of polycyclic aromatic hydrocarbons in soil. Fresenius Z Anal Chem,
328: 569-571.
Abarzua AS, Jonas L, Putzke HP, & Wittenburg E (1983) [Influence of
3,4-benzopyrene on the ultrastructure of the green alga Scenedesmus
quadricauda.] Arch Protistenk, 127: 103-113 (in German).
Abbott DW, Moody RL, Mann RM, & Vo-Dinh T (1986) Synchronous
luminescence screening for polynuclear aromatic compounds in
environmental samples collected at a coal gasification process
development unit. Am Ind Hyg Assoc J, 47: 379-385.
Abdo KM, Eustis SL, McDonald M, Jokinen MP, Adkins B, & Haseman JK
(1992) Naphthalene: A respiratory tract toxicant and carcinogen for
mice. Inhal Toxicol, 4: 393-409.
Abe S & Sasaki M (1977a) Studies on chromosomal aberrations and sister
chromatid exchanges induced by chemicals. Proc Jpn Acad, 53: 46-49.
Abe S & Sasaki M (1977b) Chromosome aberrations and sister chromatid
exchanges in Chinese hamster cells exposed to various chemicals. J
Natl Cancer Inst, 58: 1635-1641.
Abe S, Nemoto N, & Sasaki M (1983a) Sister-chromatid exchange
induction by indirect mutagens/carcinogens, aryl hydrocarbon
hydroxylase activity and benzo [a]pyrene metabolism in cultured human
hepatoma cells. Mutat Res, 109: 83-90.
Abe S, Nemoto N, & Sasaki M (1983b) Comparison of aryl hydrocarbon
hydroxylase activity and inducibility of sister-chromatid exchanges by
polycyclic aromatic hydrocarbons in mammalian cell lines. Mutat Res,
122: 47-51.
Abell CW & Heidelberger C (1962) Interaction of carcinogenic
hydrocarbons with tissues. VIII. Binding of tritium-labelled
hydrocarbons to the soluble proteins of mouse skin. Cancer Res, 22:
931-946.
Abernethy S, Bobra AM, Shiu WY, Wells PG, & Mackay D (1986) Acute
lethal toxicity of hydrocarbons and chlorinated hydrocarbons to two
planktonic crustaceans: The key role of organism-water partitioning.
Aquat Toxicol, 8: 163-174.
Abramson MJ, Wlodarczyl JH, Saunders NA, & Hensley MJ (1989) Does
aluminum smelting cause lung disease? Am Rev Respir Dis, 139:
1042-1057.
Achard S, Perderiset M, & Jaurand MC (1987) Sister chromatid exchanges
in rat pleural mesothelial cells treated with crocidolite,
attapulgite, or benzo 3-4 pyrene. Br J Ind Med, 44: 281-283.
Acheson MA, Harrison RM, Perry R, & Wellings RA (1976) Factors
affecting the extraction and analysis of polynuclear aromatic
hydrocarbons in water. Water Res, 10: 207-212.
Adamson RH & Sieber SM (1983) Chemical carcinogenesis studies in
nonhuman primates. In: Langenbach R, Nesnow S, & Rice JM ed. Organ and
species specificity in chemical carcinogenesis. New York, Plenum
Press, pp 129-156.
Adkins B Jr, van Stee EW, Simmons JE, & Eustis SL (1986) Oncogenic
response of strain A/J mice to inhaled chemicals. J Toxicol Environ
Health, 17: 311-322.
Adler ID & Ingwersen I (1989) Evaluation of chromosomal aberration in
bone marrow of 1C3F1 mice. Mutat Res, 224: 343-345.
Adler ID, Kliesch U, & Kiefer F (1989) Clastogenic effects of
benzo [a]pyrene in postimplantation embryos with different genetic
background. Teratog Carcinog Mutag, 6: 383-392.
Adlkofer F, Scherer G, Von Maltzan C, Von Meyerinck L, Jarczyk L,
Martin F, & Grimmer G (1990) Dietary influences on urinary excretion
of hydroxyphenanthrenes, thioethers and mutagenicity in man. In:
Vainio H, Sorsa M, & McMichael AJ ed. Complex mixtures and cancer
risk. Lyon, International Agency for Research on Cancer, pp 415-420
(IARC Scientific Publications No. 104).
Agarwal R & Mukhtar H (1991) Metabolism of benzo [a]pyrene and
benzo [a]pyrene 7,8-dihydrodiol catalyzed by human and rodent
epidermal lipoxygenase. Proc Am Assoc Cancer Res, 32: 117.
Agarwal SK, Sayer JM, Yeh HJC, Pannell LK, Hilton BD, Pigott MA,
Dipple A, Yagi H, & Jerina DM (1987) Chemical characterization of DNA
adducts derived from the configurationally isomeric
benzo [c]phenanthrene-3,4-diol 1,2-epoxides. J Am Chem Soc, 109:
2497-2504.
Agarwal R, Medrano EE, Khan IU, Nordlund JJ, & Mukhtar H (1991)
Metabolism of benzo(a)pyrene by human melanocytes in culture.
Carcinogenesis, 12: 1963-1966.
Agency for Toxic Substances and Disease Registry (1990) Toxicological
profile for polycyclic aromatic hydrocarbons. Atlanta, Georgia, US
Department of Health and Human Services, 231 pp (Report No. TP-90-20).
Agrelo C & Amos H (1981) DNA repair in human fibroblasts. In: de
Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, pp 528-532 (in Mutation Research, Volume
1).
Ahland E & Mertens H (1980) [PAH emissions from coal fires -
Collection, analysis, results. Air pollution from polycyclic aromatic
hydrocarbons - Registration and evaluation.] Düsseldorf, VDI-Verlag
GmbH, pp 107-111 (VDI Report No. 358) (in German).
Ahland E, Borneff J, Brune H, Grimmer G, Habs M, Heinrich U, Hermann
P, Misfeld J, Mohr U, Pott F, Schmähl D, Thyssen J, & Timm J (1985)
[Air pollution and cancer. Investigation of hazardous constituents
from various emission sources and their carcinogenic impact.] Münch
Med Wochenschr, 127: 218-221 (in German).
Aitio A (1974) Different elimination and effect on mixed function
oxidase of 20-methylcholanthrene after intragastric and
intraperitoneal administration. Res Commun Chem Pathol Pharmacol, 9:
701-710.
Albaiges J, Bayona JM, Fernandez P, Grimalt J, Rosell A, & Simo R
(1991) Vaporparticle partitioning of hydrocarbons in western
Mediterranean urban and marine atmospheres. Mikrochim Acta, 2: 13-27.
Alben K (1980) Coal tar coatings of storage tanks. A source of
contamination of the potable water supply. Environ Sci Technol, 14:
468-470.
Albert RE, Lewtas J, Nesnow S, Thorslund TW, & Anderson E (1983)
Comparative potency method for cancer risk assessment: Application to
diesel particulate emissions. Risk Anal, 3: 101-117.
Albert RE, Miller ML, Cody TE, Andringa A, Shukla R, & Baxter CS
(1991a) Benzo [a]pyrene-induced skin damage and tumor promotion in
the mouse. Carcinogenesis, 12: 1273-1280.
Albert RE, Miller ML, Cody TE, Barkley W, & Shukla R (1991b) Cell
kinetics and benzo [a]pyrene DNA adducts in mouse skin tumorigenesis.
In: Butterworth BE ed. Chemically induced cell proliferation:
Implications for risk assessment. New York, Wiley-Liss, Inc., pp 115-
122 (Progress in Clinical and Biological Research, Volume 369).
Albini A, Colacci A, Melchiori A, Carlone S, Nanni P, Lollini PL, de
Giovanni C, Nicoletti G, Grilli S, & Parodi S (1991) Chemical
carcinogens in tumor progression. Induction of invasive and metastatic
potential in 3T3 cells by benzo [a]pyrene transformation. Proc Am
Assoc Cancer Res, 32: 153.
Alden RW III & Butt AJ (1987) Statistical classification of the
toxicity and polynuclear aromatic hydrocarbon contamination of
sediments from a highly industrialized seaport. Environ Toxicol Chem,
6: 673-684.
Aldis H, Osborn J, & Wolf F (1983) Investigation of soil and water
contamination at Western Processing, King County, Washington. In:
National Conference of Management of Uncontrolled Hazardous Waste
Sites, Silver Spring, 31 October-2 November 1982, Washington DC,
Hazardous Materials Control Research Institute, pp. 43-53.
Alfheim I & Ramdahl T (1984) Contribution of wood combustion to indoor
air pollution as measured by mutagenicity in Salmonella and
polycyclic aromatic hydrocarbon concentration. Environ Mutagen, 6:
121-130.
Alfheim I & Wikström L (1984) Air pollution from aluminium smelting
plants. I. The emission of polycyclic aromatic hydrocarbons and of
mutagens from an aluminium smelting plant using the Söderberg process.
Toxicol Environ Chem, 8: 55-72.
Alfheim I, Becher G, Hongslo JK, & Ramdahl T (1984) Mutagenicity
testing of high performance liquid chromatography fractions from wood
stove emission samples using a modified Salmonella assay requiring
smaller sample volumes. Environ Mutagen, 6: 91-102.
Allen JA & Coombs MM (1980) Covalent binding of polycyclic aromatic
hydrocarbons to mitochondrial and nuclear DNA. Nature, 287: 244-245.
Allen-Hoffmann BL & Rheinwald JG (1984) Polycyclic aromatic
hydrocarbon mutagenesis of human epidermal keratinocytes in culture.
Proc Natl Acad Sci USA, 81: 7802-7806.
Allred PM & Giesy JP (1985) Solar radiation-induced toxicity of
anthracene to Daphnia pulex. Environ Toxicol Chem, 4: 219-226.
Alsberg T, Stenberg U, Westerholm R, Strandell M, Rannug U, Sundvall
A, Romert L, Bernson V, Pettersson B, Toftgard R, Franzen B, Jansson
M, Gustafsson JC, Egebäck KE & Tejle G (1985) Chemical and biological
characterization of organic material from gasoline exhaust particles.
Environ Sci Technol, 19: 43-50.
Al-Yakoob S, Saeed T, & Al-Hashash H (1993) Polycyclic aromatic
hydrocarbons in edible tissue of fish from the Gulf after 1991 oil
spill. Mar Pollut Bull, 27: 297-301.
Alzieu P, Cassand P, Colin C, Grolier P, & Narbonne JF (1987) Effect
of vitamins A, C and glutathione on the mutagenicity of
benzo [a]pyrene mediated by S9 from vitamin A-deficient rats. Mutat
Res, 192: 227-231.
Amacher DE & Paillet SC (1982) Hamster hepatocyte-mediated activation
of procarcinogens to mutagens in the L5178Y/TK mutation assay. Mutat
Res, 106: 305-316.
Amacher DE & Paillet SC (1983) The activation of procarcinogens to
mutagens by cultured rat hepatocytes in the L5178Y/TK mutation assay.
Mutat Res, 113: 77-88.
Amacher D & Turner GN (1980) Promutagen activation by rodent-liver
postmitochondrial fractions in the L5178Y/TK cell mutation assay.
Mutat Res, 74: 485-501.
Amacher DE, Paillett SC, Turner GN, Ray VA, & Salsburg DS (1980) Point
mutations at the thymidine kinase locus in L5178Y mouse lymphoma
cells. II. Test validation and interpretation. Mutat Res, 72: 447-474.
American Conference of Governmental Industrial Hygienists (1995) 1995-
1996 Threshold Limit Values (TLVs) for chemical substances and
physical agents and Biological Exposure Indices (BEIs). Cincinnati,
Ohio, 138 pp.
Amin S, Hecht SS, LaVoie E, & Hoffmann D (1979) Synthesis and
mutagenicity of 5,11-dimethylchrysene and some methyl-oxidized
derivatives of 5-methylchrysene. J Med Chem, 22: 1336-1340.
Amin S, Juchatz A, Furuya K, & Hecht SS (1981) Effects of fluorine
substitution on the tumor initiating activity and metabolism of
5-hydroxymethylchrysene, a tumorigenic metabolite of 5-methylchrysene.
Carcinogenesis, 2: 1027-1032.
Amin S, Huie K, & Hecht SS (1985a) Mutagenicity and tumor-initiating
activity of methylated benzo [b]fluoranthenes. Carcinogenesis, 6:
1023-1025.
Amin S, Huie K, Melikian AA, Leszczynska JM, & Hecht SS (1985b)
Comparative metabolic activation in mouse skin of the weak carcinogen
6-methylchrysene and the strong carcinogen 5-methylchrysene. Cancer
Res, 45: 6406-6412.
Amin S, Huie K, Balanikas G, Hecht SS, Pataki J, & Harvey RG (1987)
High stereoselectivity in mouse skin metabolic activation of
methylchrysenes to tumorigenic dihydrodiols. Cancer Res, 47:
3613-3617.
Amin S, Hecht SS, DiRaddo P, & Harvey RG (1990) Comparative tumor
initiating activities of cyclopentano and methyl derivatives of
5-methylchrysene and chrysene. Cancer Lett, 51: 17-20.
Amin S, Weyand EH, Huie K, Boger E, Neuber E, Hecht SS, & Lavoie EJ
(1991a) Effects of fluorine substitution on benzo(b)fluoranthene
tumorgenicity and DNA adduct formation in mouse skin. In: Cooke M,
Loening K, & Merritt J ed. Polynuclear aromatic hydrocarbons:
Measurements, means and metabolism. Columbus, Ohio, Battelle Press, pp
25-35.
Amin S, Misra B, Braley J, & Hecht SS (1991b) Comparative
tumorigenicity in newborn mice of chrysene and
5-alkylchrysene-1,2-diol-3,4-epoxides. Cancer Lett, 58: 115-118.
Amin S, Desai D, & Hecht SS (1992) Comparative tumorigenicity of
dimethylchrysenes in mouse skin. Chem Res Toxicol, 5: 237-241.
Amin S, Desai D, & Hecht SS (1993) Tumor-initiating activity on mouse
skin of bay region diol-epoxides of 5,6-dimethylchrysene and
benzo(c)phenanthrene. Carcinogenesis, 14: 2033-2037
Andelman JB & Suess MJ (1970) Polynuclear aromatic hydrocarbons in the
water environment. Bull World Health Organ, 43: 479-508.
Anderson JW, Neff JM, Cox BA, Tatem ME, & Hightower GM (1974) The
effects of oil on estuarine animals: Toxicity, uptake and depuration,
respiration. In: Vernberg FJ & Vernberg WB ed. Pollution and
physiology of marine organisms. New York, Academic Press, pp 285-310.
Anderson R, Giam CS, Ray LE, & Tripp MR (1981) Effects of
environmental pollutants on immunological competency of the clam
Mercenaria mercenaria: Impaired bacterial clearance. Aquat Toxicol,
1: 187-195.
Anderson LM, Priest LJ, Deschner EE, & Budinger JM (1983) Carcinogenic
effects of intracolonic benzo [a]pyrene in b-naphthoflavone-induced
mice. Cancer Lett, 20: 117-123.
Anderson JW, Neff JM, & Boehm PD (1986) Sources, fates and effects of
aromatic hydrocarbons in the Alaskan marine environment with
recommendations for monitoring strategies. Sequim, Washington,
Battelle Pacific Northwest Laboratories, 230 pp (Report No.
EPA/600/3-86-018).
Andersson K, Levin J-O, & Nilsson C-A (1983) Sampling and analysis of
particulate and gaseous polycyclic aromatic hydrocarbons from coal tar
sources in the working environment. Chemosphere, 12: 197-207.
Andjelkovich DA, Mathew RM, Richardson RB, & Levine RJ (1990)
Mortality of iron foundry workers: I. Overall findings. J Occup Med,
32: 529-540.
Andrews AW, Thibault LH, & Lijinsky W (1978) The relationship between
carcinogenicity and mutagenicity of some polynuclear hydrocarbons.
Mutat Res, 51: 311-318.
Andrews FJ, Halliday GM, & Muller HK (1991) A role for prostaglandins
in the suppression of cutaneous cellular immmunity and tumor
development in benzo(a)pyrene- but not
dimethylbenz(a)anthracene-treated mice. Clin Exp Immunol, 85: 9-13.
Andrianova MM (1971) Transplacental action of 3-methylcholanthrene and
benz [a]pyrene on four generations of mice. Bull Exp Biol Med, 71:
677-680.
Anziulewicz JA, Dick HJ, & Chiarulli EE (1959) Transplacental
naphthalene poisoning. Am J Obstet Gynecol, 9: 519-521.
Aoyama T, Gonzalez FJ, & Gelboin HV (1989) Human cDNA-expressed
cytochrome P450 1A2: Mutagen activation and substrate specificity. Mol
Carcinog, 2: 192-198.
Arce GT, Allen JW, Doerr CL, Elmore E, Hatch GG, Moore MM, Sharief Y,
Grunberger D, & Nesnow S (1987) Relationships between benzo(a)pyrene-
DNA adduct levels and genotoxic effects in mammalian cells. Cancer
Res, 47: 3388-3395.
Arey J, Zielinska B, Atkinson R, & Winer AM (1987) Polycyclic aromatic
hydrocarbon and nitroarene concentrations in ambient air during a
wintertime high-NOx episode in the Los Angeles basin. Atmos Environ,
21: 1437-1444.
Arey J, Zielinska B, Atkinson R, & Winer AM (1991) Analysis of PAH and
their nitroderivates in ambient air. In: Cooke M, Loening K, & Merritt
J ed. Polynuclear aromatic hydrocarbons: Measurement, means, and
metabolism. Columbus, Ohio, Battelle Press, pp 51-67.
Armstrong B, Tremblay C, Baris D, & Thériault G (1994) Lung cancer
mortality and polynuclear aromatic hydrocarbons: A case-cohort study
of aluminum production workers in Arvida, Québec, Canada. Am J
Epidemiol, 139: 250-262.
Ashby J & Kilbey B (1981) Summary report on the performance of
bacterial repair, phage induction, degranulation, and nuclear
enlargement assays. In: de Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North Holland, pp 33-48
(Progress in Mutation Research, Volume 1).
Assennato G, Ferri GM, Foà V, Strickland P, Poirier M, Pozzoli L, &
Cottica D (1993a) Correlation between PAH airborne concentration and
PAH-DNA adducts levels in coke-oven workers. Int Arch Occup Environ
Health, 65: S143-S145.
Assennato G, Ferri GM, Tockman MS, Poirier MC, Schoket B, Porro A,
Corrado V, & Strickland PT (1993b) Biomarkers of carcinogen exposure
and cancer risk in a coke plant. Environ Health Perspectives, 99: 237-
239.
Association of Rhine and Meuse Water Supply Companies (1976) Annual
Report, Part A, The Rhine, Amsterdam, 82 pp.
Association of Rhine and Meuse Water Supply Companies (1977) Annual
Report, Part A, The Rhine, Amsterdam, 54 pp.
Association of Rhine and Meuse Water Supply Companies (1978) Annual
Report, Part A, The Rhine, Amsterdam, 55 pp.
Association of Rhine and Meuse Water Supply Companies (1979) Annual
Report, Part A, The Rhine, Amsterdam, 60 pp.
Association of Rhine and Meuse Water Supply Companies (1987-88) Annual
Report, Part A, The Rhine, Amsterdam, 164 pp.
Association of Rhine and Meuse Water Supply Companies (1989) Annual
Report, Part A, The Rhine, Amsterdam, 108 pp.
Association of Rhine and Meuse Water Supply Companies (1990) Annual
Report, Part A, The Rhine, Amsterdam, 92 pp.
Atkinson R (1987) Structure-activity relationship for the estimation
of rate constants for the gas-phase reactions of OH radicals with
organic compounds. Int J Chem Kinet, 19: 799-828.
Atkinson, R (1989) Kinetics and mechanisms of the gas-phase reactions
of the hydroxyl radical with organic compounds (26) naphthalene. J
Phys Chem Ref Data Monogr, 1: 237.
Atkinson R & Arey J (1994) Atmospheric chemistry of gas-phase
polycylic aromatic hydrocarbons: Formation of atmospheric mutagens.
Environ Health Perspectives, 102 (suppl 4): 117-126.
Atkinson R & Carter WPL (1984) Kinetics and mechanisms of gas-phase
ozone reaction with organic compounds under atmospheric conditions.
Chem Rev, 84: 437-470.
Atkinson R, Arey J, Zielinska B, & Winer AM (1991) Atmospheric loss
processes for PAH and formation of nitroarenes. In: Cooke M, Loening
K, & Merritt J ed. Polynuclear aromatic hydrocarbons: Measurement,
means, and metabolism. Columbus, Ohio, Batelle Press, pp 69-88.
Autrup H (1979) Separation of water-soluble metabolites of
benzo [a]pyrene formed by cultured human colon. Biochem Pharmacol,
28: 1727-1730.
Autrup JL & Autrup H (1986) Metabolism of tobacco specific carcinogens
in cultured rat buccal mucosa epithelial cells. Acta Pharmacol
Toxicol, 59: 339-344.
Autrup H & Harris CC (1983) Metabolism of chemical carcinogens by
human tissues. In: Harris CC & Autrup H ed. Human carcinogenesis. New
York, Academic Press, pp 169-194.
Autrup H, Harris CC, Stoner GD, Selkirk JK, Schafer PW, & Trump B
(1978a) Metabolism of [3H]benzo [a]pyrene by cultured human bronchus
and cultured human pulmonary alveolar macrophages. Lab Invest, 38:
217-224.
Autrup H, Harris CC, Trump B, & Jeffrey AM (1978b) Metabolism of
benzo [a]pyrene and identification of the major benzo [a]pyrene-DNA
adducts in cultured human colon. Cancer Res, 38: 3689-3696.
Autrup H, Harris CC, Schafer PW, Trump B, Stoner GD, & Hsu IC (1979)
Uptake of benzo [a]pyrene-ferric oxide particulates by human
pulmonary macrophages and release of benzo [a]pyrene and its
metabolites. Proc Soc Exp Biol Med, 161: 280-284.
Autrup H, Wefald FC, Jeffrey AM, Tate H, Schwartz RD, Trump B, &
Harris CC (1980) Metabolism of benzo [a]pyrene by cultured
tracheobronchial tissues from mice, rats, hamsters, bovines and
humans. Int J Cancer, 25: 293-300.
Awogi T & Sato T (1989) Micronucleus test with benzo [a]pyrene using
a single peroral administration and intraperitoneal injection in males
of the MS/Ae and CDl-1 mouse strains. Mutat Res, 223: 353-356.
Ayrton AD, McFarlane M, Walker R, Neville S, & Ioannides C (1990) The
induction of P450 I proteins by aromatic amines may be related to
their carcinogenic potential. Carcinogenesis, 11: 803-810.
Babich H, Sardana MK, & Borenfreund E (1988) Acute cytotoxicities of
polynuclear aromatic hydrocarbons determined in vitro with the human
liver tumor cell line, HepG2. Cell Biol Toxicol, 4: 295-309.
Babson JR, Russo-Rodriguez SE, Rastetter WR, & Wogan GG (1986a)
In vitro DNA-binding of microsomally-activated fluoranthene:
Evidence that the major product is a fluoranthrene N2-deoxyguanosine
adduct. Carcinogenesis, 7: 859-865.
Babson JR, Russo-Rodriguez SE, Wattley RV, Bergstein PL, Rastetter WH,
Liber HL, Andon BM, Thilly WG, & Wogan GN (1986b) Microsomal
activation of fluoranthene to mutagenic metabolites. Toxicol Appl
Pharmacol, 85: 355-366.
Bachmann S, Stolz W, Kantor W, & Kuhnt G (1994) [Texte 6:
Implementation of the soil information system for a survey of forest
soils in Germany.] Berlin, Ministry of the Environment, 445 pp (UBA
Report No. 93-147) (in German)
Backer JM & Weinstein IB (1980) Mitochondrial DNA is a major cellular
target for a dihydrodiol-epoxide derivative of benzo [a]pyrene.
Science, 209: 297-299.
Badger GM, Cook JW, Hewett CL, Kennaway EL, Kennaway NM, Martin RH, &
Robinson AM (1940) The production of cancer by pure hydrocarbons. Part
V. Proc R Soc Med, 129: 439-474.
Badger GM, Cook JW, Hewett CL, Kennaway EL, Kennaway NM, & Martin RH
(1942) The production of cancer by pure hydrocarbons. VI. Proc R Soc
Lond, 131: 170-182.
Baek SO, Field RA, Goldstone ME, Kirk PW, Lester JN, & Perry R (1991)
A review of atmospheric polycyclic aromatic hydrocarbons: Sources,
fate and behavior. Water Air Soil Pollut, 60: 279-300.
Baek SO, Goldstone ME, Kirk PWW, Lester JN, & Perry R (1992)
Concentrations of particulate and gaseous polycyclic aromatic
hydrocarbons in London air following a reduction in the lead content
of petrol in the United Kingdom. Sci Total Environ, 111: 169-199.
Bagg J, Smith D, & Maher WA (1981) Distribution of polycyclic aromatic
hydrocarbons in sediments from estuaries of south-eastern Australia.
Aust J Mar Freshwater Res, 32: 65-73.
Bailey GS, Goeger DE, & Hendricks J (1989) Factors influencing
experimental carcinogenesis in laboratory fish models. In: Varanasi U
ed. Metabolism of polycyclic aromatic hydrocarbons in the aquatic
environment. Boca Raton, Florida, CRC Press, pp 253-268.
Baird WM & Diamond L (1978) Metabolism and DNA binding of polycyclic
aromatic hydrocarbons by human diploid fibroblasts. Int J Cancer, 22:
189-195.
Baird WM, Salmon CP, & Diamond L (1984) Benzo(e)pyrene-induced
alterations in the metabolic activation of benzo(a)pyrene and
7,12-dimethylbenz(a)anthracene by hamster embryo cells. Cancer Res,
44: 1445-1452.
Baker JE & Eisenreich SJ (1990) Concentrations and fluxes of
polycyclic aromatic hydrocarbons and polychlorinated biphenyls across
the air-water interface of Lake Superior. Environ Sci Technol, 24:
342-352.
Baker RSU, Bonin AM, Stupans I, & Holder GM (1980) Comparison of rat
and guinea pig as sources of the S9 fraction in the
Salmonella/mammalian microsome mutagenicity test. Mutat Res, 71: 43-
52.
Bakke J, Struble C, Gustafsson JA, & Gustafsson B (1983) Enterohepatic
circulation and catabolism of mercapturic acid pathway metabolites of
naphthalene. In: Rydstrom J, Montelius J, & Bengtsson M. ed.
Extrahepatic drug metabolism and chemical carcinogenesis. Amsterdam,
Elsevier Science Publishers, pp 257-266.
Baranova LN, Shendrikova IA, Aleksandrov VA, Likhachev AY,
Ivanov-Galitsin MN, Dikun PP, & Napalkov NP (1976) [On the mechanism
of penetration of carcinogenic polycyclic hydrocarbons through the
placenta of rats and mice.] Dokl Akad Nauk SSSR, 228: 733-736 (in
Russian).
Barat F (1991) [Measures for clean air at the workplace.]
Staub-Reinhalt Luft, 51: 243-246 (in German).
Barbieri O, Ognio E, Rossi O, Astigiano S, & Rossi L (1986)
Embryotoxicity of benzo(a)pyrene and some of its synthetic derivatives
in Swiss mice. Cancer Res, 46: 94-98.
Barfknecht TR, Andon BM, Thilly WG, & Hites RA (1981) Soot and
mutation in bacteria and human cells. In: Cooke M & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemical analysis and biological
fate. Columbus, Ohio, Battelle Press, pp 231-242.
Barfknecht TR, Hites RA, Cavaliers EL, & Thilly WG (1982) Human cell
mutagenicity of polycyclic aromatic hydrocarbon components of diesel
emissions. Dev Toxicol Environ Sci, 10: 277-294.
Barnsley EA (1975) The bacterial degradation of fluoranthene and
benzo [a]pyrene. Can J Microbiol, 21: 1004-1008.
Barratt RW & Tatum EL (1958) Carcinogenic mutagens. Ann NY Acad Sci,
71: 1072-1084.
Barrows ME, Petrocelli SR, Macek KJ, & Carroll JJ (1980)
Bioconcentration and elimination of selected water pollutants by
bluegill sunfish Lepomis macrochirus. In: Dynamic, exposure, hazard
assessment of toxic chemicals. Michigan, Ann Arbor Science Publishers,
pp 379-392.
Barry G & Cook CW (1934) A comparison of the action of some polycyclic
aromatic hydrocarbons in producing tumours of connective tissue. Am J
Cancer, 20: 58-69.
Barry G, Cook JW, Haslewood AD, Hewett CL, Hieger I, & Kennaway EL
(1935) The production of cancer by pure hydrocarbons. Part III. Proc R
Soc Med, 117: 318-351.
Bartczak AW, Sangaiah S, Ball LM, Warren SH, & Gold A (1987) Synthesis
and bacterial mutagenicity of the cyclopenta oxides of the four
cyclopenta-fused isomers of benzanthracene. Mutagenesis, 2: 101-105.
Bartle KD (1985) Recent advances in the analysis of polycyclic
aromatic compounds by gas chromatography. In: Bjorseth A & Ramdahl T
ed. Handbook of polycyclic aromatic hydrocarbons. Volume 2. Emission
sources and recent progress in analytical chemistry. New York, Marcel
Dekker, pp 193-236.
Bartosek I, Guaitani A, Modica R, Fiume M, & Urso R (1984) Comparative
kinetics of oral benz(a)anthracene, chrysene und triphenylene in rats:
Study with hydrocarbon mixtures. Toxicol Lett, 23: 333-339.
Bartsch H, Malaveille C, Camus AM, Martel-Planche G, Brun G,
Hautefeuille A, Sabadie N, Barbin A, Kuroki T, Drevon C, Piccoli C, &
Montesano R (1980) Validation and comparative studies on 180 chemicals
with S. typhimurium strains and V79 Chinese hamster cells in the
presence of various metabolizing systems. Mutat Res, 76: 1-50.
Basler A, Herbold B, Peter S, & Röhrborn G (1977) Mutagenicity of
polycyclic hydrocarbons. II. Monitoring genetical hazards of chrysene
in vitro and in vivo. Mutat Res, 48: 249-254.
Bastian MV & Toetz DW (1982) Effect of eight polynuclear hydrocarbons
on growth of Anabaena flos aquae. Bull Environ Contam Toxicol, 29:
531-538.
Bastian MV & Toetz DW (1985) Effect of polynuclear hydrocarbons on
algal nitrogen fixation (acetylene reduction). Bull Environ Contam
Toxicol, 35: 258-265.
Basu DK & Saxena J (1978a) Monitoring of polycyclic aromatic
hydrocarbons in water. II. Extraction and recovery of six
representative compounds with polyurethane foams. Environ Sci Technol,
12: 791-795.
Basu DK & Saxena J (1978b) Polynuclear aromatic hydrocarbons in
selected US drinking water and their raw water sources. Environ Sci
Technol, 12: 795-798.
Basu DK, Saxena J, Stoss FW, Santodonato J, Neal MW, & Kopfler FC
(1987) Comparison of drinking water mutagenicity with leaching of
polycyclic aromatic hydrocarbons from water distribution pipes.
Chemosphere, 16: 2595-2612.
Batiste-Alentorn M, Xamena N, Creus A, & Marcos R (1991) Genotoxicity
studies with the unstable zeste-white (UZ) system of Drosophila
melanogaster: Results with ten carcinogenic compounds. Environ Mol
Mutag, 18: 120-125.
Bauer L, Gräf W, & Mueller LG (1985) The phototoxic effect of
polycyclic aromatic hydrocarbons (PAH) on human fibroblastic cultures.
Zentralbl Bakteriol Hyg I Abt Orig B, 181: 281-294.
Bauer E, Guo Z, Ueng Y-F, Bell LC, Zeldin D, & Guengerich FP (1995)
Oxidation of benzo [a]pyrene by recombinant human cytochrome P450
enzymes. Chem Res Toxicol, 8: 136-142.
Baumung H, Huber L, & Kalbfus W (1985) [Testing of selected components
of wastewater from filling stations and fuel storage depots.] Erdöl
Kohle Erdgas Petrochem, 38: 558-559 (in German).
Bayer U (1978) In vivo induction of sister chromatid exchanges by
three polyaromatic hydrocarbons. Carcinogenesis, 3: 423-428.
Bayona JM, Fernandez P, Porte C, Tolosa I, Valls M, & Albaiges J
(1991) Partitioning of urban wastewater organic microcontaminants
among coastal compartments. Chemosphere, 23: 313-326.
Beach AC & Gupta RC (1991) Analysis of cyclopenta(CP)-fused and
'pseudo-CP' polycyclic aromatic hydrocarbon (PAH)-DNA adducts by
32P-postlabeling. Proc Am Assoc Cancer Res, 32: 98.
Beach AC & Gupta RC (1992) Human biomonitoring and the 32
P-postlabeling assay. Carcinogenesis, 13: 1053-1074.
Beach AC & Gupta RC (1994) DNA adducts of the ubiquitous contaminant
cyclopenta (cd)pyrene. Carcinogenesis, 15: 1065-1072.
Beach AC, Agarwal SC, Lamberg GR, Nesnow S, & Gupta RC (1993) Reaction
of cyclopenta (c,d)pyrene-3,4-epoxide with DNA and desoxynucleotides.
Carcinogenesis, 14: 767-771.
Becher G & Bjorseth A (1983) Determination of exposure to polycyclic
aromatic hydrocarbons by analysis of human urine. Cancer Lett, 17:
301-311.
Becher G, Haugen A, & Bjorseth A (1984) Multimethod determination of
occupational exposure to polycyclic aromatic hydrocarbons in an
aluminium plant. Carcinogenesis, 5: 647-651.
Behn U, Kaschani DT, Lützke K, Rentel S, & Burk HD (1985) [PAH
sampling systems for industrial plants.] Erdöl Kohle Erdgas Petrochem,
38: 131 (in German).
Behymer TD & Hites RA (1985) Photolysis of polycyclic aromatic
hydrocarbons adsorbed on simulated atmospheric particulates. Environ
Sci Technol, 19: 1004-1006.
Behymer TD & Hites RA (1988) Photolysis of polycyclic aromatic
hydrocarbons adsorbed on fly ash. Environ Sci Technol, 22: 1311-1319.
Beilstein Institute for Organic Chemistry (1993) Data base.
Benzo(c)phenanthrene. Frankfurt, 2 pp.
Bender ME & Huggett RJ (1988) Polynuclear aromatic hydrocarbon
residues in shellfish: Species variations and apparent intraspecific
differences. In: Kaiser HE ed. Cancer growth and progression. Volume
5: Comparative aspects of tumor development. Dordrecht, Kluwer
Academic Publishers, pp 226-234.
Bender MA, Leonard RC, White O Jr, Costantino JP, & Redmond CK (1988)
Chromosomal aberrations and sister-chromatid exchanges in lymphocytes
from coke oven workers. Mutat Res, 206: 11-16.
Benfield JR & Hammond WG (1992) Bronchial and pulmonary carcinogenesis
at focal sites in dogs and hamsters. Cancer Res, 52: 2687-2693.
Beniashvili DSH (1978) [A comparative study on the action of some
carcinogens inducing transplacental blastomogenesis in rabbits.] Vopr
Onkol, 24: 77-83 (in Russian with English abstract).
Benner BA Jr, Gordon GE, & Wise SA (1989) Mobile sources of
atmospheric polycyclic aromatic hydrocarbons: A roadway tunnel study.
Environ Sci Technol, 23: 1269-1278.
Benner BA Jr, Bryner NP, Wise SA, Mulholland GW, Lao RC, & Fingas MF
(1990) Polycyclic aromatic hydrocarbon emissions from the combustion
of crude oil on water. Environ Sci Technol, 24: 1418-1427.
Berbee RPM (1992) PAH in the aquatic environment: Sources and
emissions. Summary. Proceedings of the workshop on polycyclic aromatic
hydrocarbons (PAH), Oslo, 11-13 November 1991 (Report TA-816). Oslo,
State Pollution Control Authority, Norwegian Food Control Authority.
Berenblum I & Haran H (1955) The influence of croton oil and of
polyethylene glycol-400 on carcinogenesis in the forestomach of the
mouse. Cancer Res, 15: 510-516.
Berenblum I & Schoental R (1943) The metabolism of 3,4-benzpyrene in
mice and rats. The isolation of a hydroxy and a quinone derivative and
a consideration of their biological significance. Cancer Res, 3: 145-
150.
Berenblum I & Shubik P (1947) The role of croton oil applications,
associated with a single painting of a carcinogen in tumour induction
of the mouse skin. Br J Cancer, 1: 379-382.
Berglind L (1982) Determination of polycyclic aromatic hydrocarbons in
industrial discharges and other aqueous effluents (Nordic PAH
Project). Oslo, Central Institute for Industrial Research, 21 pp
(Report No. 16).
Bernfeld P & Homburger F (1983) Skin painting studies in Syrian
hamsters. Prog Exp Tumor Res, 26: 128-153.
Best GR, Nabholz JV, Ojasti J, & Crossley DA Jr (1978) Response of
micro arthropod populations to naphthalene in three contrasting
habitats. Pedobiologia, 18: 189-201.
Bhatia AL, Tausch, H & Stehlik G (1987) Mutagenicity of chlorinated
polycyclic aromatic compounds. Ecotoxicol Environ Saf, 14: 48-55.
Bhatt TS & Coombs MM (1990) The carcinogenicity of
cyclopenta [a]phenanthrene and chrysene derivatives in the Sencar
mouse. Polycyclic Aromat Compd, 1: 51-58.
Bhattacharyya KK, Brake PB, Eltom SE, Otto SA, & Jefcoate CR (1995)
Identification of a rat adrenal cytochrome P450 active in polycyclic
hydrocarbon metabolism as rat CYP1B1; demonstration of a unique
tissue-specific pattern of hormonal and aryl hydrocarbon
receptor-linked regulation. J Biol Chem, 270: 11595-11602.
Bieniek G (1994) The presence of 1-naphthol in the urine of industrial
workers exposed to naphthalene. Occup Environ Med, 51: 357-359.
Bieri RH, Hein C, Huggett RJ, Shou P, Slone H, Smith C, & Su C (1986)
Polycyclic aromatic hydrocarbons in surface sediments from the
Elizabeth River subestuary. Int J Environ Anal Chem, 26: 97-113.
Biermann HW, MacLeod H, Atkinson R, Winer AM, & Pitts JN Jr (1985)
Kinetics of the gas-phase reactions of the hydroxyl radical with
naphthalene, phenanthrene and anthracene. Environ Sci Technol, 19:
244-248.
Bingham E & Falk HL (1969) Environmental carcinogens. The modifying
effect of cocarcinogens on the threshold response. Arch Environ
Health, 19: 779-783.
Binková B, Dobiás L, Wolff T, & Srám RJ (1994) 32P-Postlabeling
analysis of DNA adducts in tissues of rats exposed to coke-oven
emissions. Mutat Res, 307: 355-363.
Binková B, Lewtas J, Misková I, Lenrcek J, & Srám R (1995) DNA adducts
and personal air monitoring of carcinogenic polycyclic aromatic
hydrocarbons in an environmentally exposed population. Carcinogenesis,
16: 1037-1046.
Bjorseth A (1979) Determination of polycyclic aromatic hydrocarbons in
sediments and mussels from Saudafjord, west Norway, by glass capillary
gas chromatography. Sci Total Environ, 13: 71-86.
Bjorseth A (1983) Appendix: List of dicyclic and polycyclic aromatic
hydrocarbons, their structure, molecular weight, melting and boiling
points. In: Bjorseth A ed. Handbook of polycyclic aromatic
hydrocarbons. New York, Marcel Dekker, pp 709-718.
Bjorseth A & Becher G (1986) PAH in work atmospheres: Occurrence and
determination. Florida, Boca Raton, CRC Press, pp 57-62.
Bjorseth A & Lunde G (1979) Long-range transport of polycyclic
aromatic hydrocarbons. Atmos Environ, 13: 45-53.
Bjorseth A & Olufsen S (1983) Long transport of polycyclic aromatic
hydrocarbons. In: Bjorseth A ed. Polycyclic aromatic hydrocarbons. New
York, Marcel Dekker, pp 507-524.
Bjorseth A & Ramdahl T (1985) Sources and emissions of PAH. In:
Bjorseth A & Ramdahl T ed. Handbook of polycyclic aromatic
hydrocarbons: Volume 2: Emission sources and recent progress in
analytical chemistry. New York, Marcel Dekker, pp 1-20.
Black JJ, Hart TF Jr, & Evans E (1981) HPLC studies of PAH pollution
in a Michigan trout stream. In: Cooke M & Dennis A ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 343-355.
Black JA, Birge WJ, Westerman AG, & Francis PC (1983) Comparative
aquatic toxicology of aromatic hydrocarbons. Fundam Appl Toxicol, 3:
353-358.
Black JJ, Maccubin AE, & Johnston CJ (1988) Carcinogenicity of
benzo [a]pyrene in rainbow trout resulting from embryo micro
injection. Aquat Toxicol, 13: 297-308.
Blanton RH, Lyte M, Myers MJ, & Bick PH (1986) Immunomodulation by
polyaromatic hydrocarbons in mice and murine cells. Cancer Res, 46:
2735-2739.
Bobra A, Shiu WY, & Mackay (1983) A predictive correlation for the
acute toxicity of hydrocarbons and chlorinated hydrocarbons to the
water flea Daphnia magna. Chemosphere, 12: 1121-1129.
Bock FG (1964) Early effects of hydrocarbons on mammalian skin. Progr
Exp Tumor Res, 4: 126-168.
Bock FG & Dao TL (1961) Factors affecting the polynuclear hydrocarbon
level in rat mammary glands. Cancer Res, 21: 1024-1029.
Bock FG & King DW (1959) A study of the sensitivity of the mouse
forestomach toward certain polycyclic hydrocarbons. J Natl Cancer
Inst, 23: 833-838.
Bock FG & Mund R (1958) A survey of compounds for activity in
suppression of mouse sebaceous glands. Cancer Res, 18: 887-892.
Boehm PD & Fiest DL (1983) Ocean dumping of dredged material in the
New York Bight: Organic chemistry studies. In: Kester DR, Ketchum H,
Bostwick I, & Duedal IW ed. Wastes in the ocean. Volume 2:
Dredged-material disposal in the ocean. New York, John Wiley & Sons,
pp 151-169.
Boldrin B, Tiehm A, & Fritzsche C (1993) Degradation of phenanthrene,
fluorene, fluoranthene, and pyrene by a Mycobacterium sp. Appl
Environ Microbiol, 59: 1927-1930.
Bolling H (1964) [Carcinogenic substances in cereals dried by
combustion gas.] Tech Monit Pinerolo, 15: 137-142 (in Italian).
Bolonova LN (1967) [Action of naphthalene and its methyl derivatives
on the ammonia content in rat brain.] Farmakol Toksikol, 30: 484-486
(in Russian).
Boney AD (1974) Aromatic hydrocarbons and the growth of marine algae.
Mar Pollut Bull, 5: 185-186.
Boney AD & Corner EDS (1962) On the effects of some carcinogenic
hydrocarbons on the growth of sporelings of marine red algae. J Mar
Biol Assoc, 42: 579-585.
Bonfanti L, Cioni M, Belli R, & Cappiello A (1988) Determination of
trace organic compounds in effluents from a coal-fired power plant.
Biomed Mass Spectrom, 16: 175-178.
Boogaard PJ & van Sittert NJ (1994) Exposure to polycyclic aromatic
hydrocarbons in petrochemical industries by measurement of urinary
1-hydroxypyrene. Occup Environ Med, 51: 250-258.
Boogaard PJ & van Sittert NJ (1995) Urinary 1-hydroxypyrene as
biomarker of exposure to polycyclic aromatic hydrocarbons in workers
in petrochemical industries: Baseline values and dermal uptake. Sci
Total Environ, 163: 203-209.
Boom A & Marsalek J (1988) Accumulation of polycyclic aromatic
hydrocarbons (PAHs) in an urban snowpack. Sci Total Environ, 74: 133-
148.
Boom MM (1987) The determination of polycyclic aromatic hydrocarbons
in indigenous and transplanted mussels (Mytilus
edulis L) along the Dutch coast. Int J Environ Anal Chem, 31: 251-
261.
Booth J & Boyland E (1949) Metabolism of polycyclic compounds. 5.
Formation of 1,2-dihydroxy-1,2-dihydronaphthalene. Biochem J, 44: 361-
365.
Booth J, Keysell GR, Pal K, & Sims P (1974) The metabolism of
polycyclic hydrocarbons by cultured human lymphocytes. FEBS Lett, 43:
341-344.
Borchert J & Westendorf J (1994) [Kinetics of uptake and metabolism of
sediment-bound benzo(a)pyrene in sediment-inhabiting invertebrates.]
Ber Zent Meeres Klimaforsch, 7: 75-55 (in German).
Borneff J & Kunte H (1964) [Carcinogenic substances in water and soil.
XVI. Determination of polycyclic aromatics in water samples by direct
extraction.] Arch Hyg, 148: 585-597 (in German).
Borneff J & Kunte H (1965) [Carcinogenic substances in water and soil.
XVII. Source and evaluation of polycyclic aromatic hydrocarbons in
water.] Arch Hyg, 149: 226-243 (in German).
Borneff J & Kunte H (1979) Method 1. Analysis of polycyclic aromatic
hydrocarbons in water using thin layer chromatography and
spectrofluorometry. In: Egan H, Castegnaro M, Bogovski P, Kunte H, &
Walker EA ed. Environmental carcinogens: Selected methods of analysis.
Volume 3: Analysis of polycyclic aromatic hydrocarbons in
environmental samples. Lyon, International Agency for Research on
Cancer, pp 129-139 (IARC Scientific Publications No. 29).
Borneff J & Kunte H (1983) Polycyclic aromatic hydrocarbons in river
and lake water, biota, and sediments. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 629-652.
Boroujerdi M, Kung HC, Wilson AGE, & Anderson MW (1981) Metabolism and
DNA binding of benzo [a]pyrene in vivo in the rat. Cancer Res, 41:
951-957.
Borovsky D, Linley JR, & Kagan J (1987) Polycyclic aromatic compounds
as phototoxic mosquito larvicides. J Am Mosq Control Assoc, 3: 246-
250.
Bos RP, Prinsen WJC, van Rooy JGM, Jongeneelen FJ, Theuws JLG, &
Henderson PT (1987) Fluoranthene, a volatile mutagenic compound,
present in creosote and coal tar. Mutat Res, 187: 119-125.
Bos RP, Theuws JLG, Jongeneelen FJ, & Henderson PT (1988) Mutagenicity
of bi-,tri- and tetra-cyclic aromatic hydrocarbons in the 'taped-plate
assay' and in the conventional Salmonella
mutagenicity assay. Mutat Res, 204: 203-206.
Bossert ID & Bartha R (1986) Structure-biodegradability relationships
of polycyclic aromatic hydrocarbons in soil. Bull Environ Contam
Toxicol, 37: 490-495.
Bossert I, Kachel WM, & Bartha R (1984) Fate of hydrocarbons during
oily sludge disposal in soil. Appl Environ Microbiol, 47: 763-767.
Bottomley AC & Twort CC (1934) The carcinogenicity of chrysene and
oleic acid. Am J Cancer, 21: 781-786.
Bourcart J & Mallet L (1965) [Coastal marine pollution in the central
region of the Tyrrhenian Sea (Bay of Naples) by polyaromatic
hydrocarbons of the benzo-3,4-pyrene type.] C R Acad Sci Paris, 260:
3729-3734 (in French).
Bourguet CC, Checkoway H, & Hulka BS (1987) A case-control study of
skin cancer in the tire and rubber manufacturing industry. Am J Ind
Med, 11: 461-473.
Bourne MC & Young L (1934) The metabolism of naphthalene in rabbits.
Biochem J, 28: 803-808.
Bowling J, Leversee G, Landrum PF, & Giesy JP (1983) Acute mortality
of anthracene-contaminated fish exposed to sunlight. Aquat Toxicol, 3
:79-90.
Bowmer CT, Roza P, Henzen L, & Degeling C (1993) The development of
chronic toxicological tests for PAH contaminated soils using the
earthworm Eisenia fetida and the springtail Folsomia candida.
Delft, TNO Institute of Environmental Sciences, 49 pp (TNO Report No.
IMW-R 92/387).
Boyland E & Burrows H (1935) The experimental production of sarcoma in
rats and mice by a colloidal aqueous solution of
1:2:5:6-dibenzanthracene. J Pathol Bacteriol, 41: 231-238.
Boyland E & Levi AA (1935) Metabolism of polycyclic compounds. I.
Production of dihydroxydihydroanthracene from anthracene. Biochem J,
29: 2679-2683.
Boyland E & Levi AA (1936a) Metabolism of polycyclic compounds. II.
Production of dihydroxydihydroanthracene glycuronic acid from
anthracene. Biochem J, 30: 728-731.
Boyland E & Levi AA (1936b) Metabolism of polycyclic compounds. III.
Anthrylmercapturic acid. Biochem J, 30: 1225-1227.
Boyland E & Sims P (1958) Metabolism of polycyclic compounds. 12. An
acid-labile precursor of 1-naphthylmercapturic acid and naphthol: An
N-acetyl-S-(1,2-dihydro-hydroxynaphthyl) L-cysteine. Biochem J, 68:
440-447.
Boyland E & Sims P (1962a) Metabolism of polycyclic compounds. 20. The
metabolism of phenanthrene in rabbits and rats: Mercapturic acids and
related compounds. Biochem J, 84: 564-570.
Boyland E & Sims P (1962b) Metabolism of polycyclic compounds. 21. The
metabolism of phenanthrene in rabbits and rats: Dihydrodihydroxy
compounds and related glucosiduronic acids. Biochem J, 84: 571-582.
Boyland E & Sims P (1964a) Metabolism of polycyclic compounds. 23. The
metabolism of pyrene in rats and rabbits. Biochem J, 90: 391-398.
Boyland E & Sims P (1964b) Metabolism of polycyclic compounds. 24. The
metabolism of benz [a]anthracene. Biochem J, 91: 493-506.
Boyland E & Sims P (1967) The carcinogenic activities in mice of
compounds related to benz [a]anthracene. Int J Cancer, 2: 500-504.
Boyland E & Weigert F (1947) Metabolism of carcinogenic compounds. Br
Med Bull, 4: 354-359.
Boyland E & Wolf G (1950) Metabolism of polycyclic compounds. 6.
Conversion of phenanthrene into dihydroxydihydrophenanthrene. Biochem
J, 47: 64-69.
Boyland E, Levi AA, Mawson EH, & Roe E (1941) Metabolism of polycyclic
compounds. 4. Production of a dihydroxy-1,2,5,6-dibenzanthracene.
Biochem J, 35: 184-191.
Boyland E, Kimura M, & Sims P (1964) Metabolism of polycyclic
compounds. 26. The hydroxylation of some aromatic hydrocarbons by the
ascorbic acid model hydroxylating system and by rat liver microsomes.
Biochem J, 92: 631-638.
Bozicevic Z, Cvitas T, Curic M, Klasinc L, & Pecina P (1987) Airborne
polycyclic aromatic hydrocarbons in the city of Zagreb, Yugoslavia.
Sci Total Environ, 66: 127-136.
Brandt-Rauf PW, Smith S, Perera FP, Niman HL, Yohannan W, Hemminki K,
& Santella RM (1990) Serum oncogene proteins in foundry workers. J Soc
Occup Med, 40: 11-14.
Brasser LJ (1980) [Polycyclic aromatic hydrocarbon concentration in
the Netherlands. Air pollution from polycyclic aromatic hydrocarbons:
Collection and evaluation.] Düsseldorf, VDI-Verlag GmbH, pp 171-180
(VDI Report No. 358) (in German).
Bresch H, Spielhoff R, Mohr U, & Barkemeyer H (1972) Use of the sea
urchin egg for quick screen testing of the biological activities of
substances. I. Influence of fractions of a tobacco smoke condensate on
early development. Proc Soc Exp Biol Med, 141: 747-752.
Bridges BA, Zeiger E, & McGregor DB (1981) Summary report on the
performance of bacterial mutation assays. In: de Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens. Report of the
international collaborative programme. New York, Elsevier North
Holland, pp 49-67 (Progress in Mutation Research, Volume 1).
Brockhaus A & Tomingas R (1976) [Emission of polycyclic hydrocarbons
during burning processes in small heating installations and their
concentration in the atmosphere.] StaubReinhalt Luft, 36: 96-101 (in
German).
Broddin G, Van Vaeck L, & Van Cauwenberghe K (1977) On the size
distribution of polycyclic aromatic hydrocarbon containing particles
from a coke oven emission source. Atmos Environ, 11: 1061-1064.
Broman D, Colmjö A, & Näf C (1987) Characterization of the PAC profile
in settling particulates from the urban waters of Stockholm. Bull
Environ Contam Toxicol, 38: 1020-1028.
Broman D, Naf C, Rolff C, & Zebuhr Y (1990) Occurrence and dynamics of
polychlorinated dibenzo-p-dioxins and dibenzofurans and polycyclic
aromatic hydrocarbons in the mixed surface layer of remote coastal and
offshore water of the Baltic. Environ Sci Technol, 25: 1850-1864.
Brooke DN, Dobbs AJ & Williams N (1986) Octanol:water partition
coefficients (P): Measurement, estimation and interpretation,
particularly for chemicals with P > 105. Ecotoxicol Environ Saf, 11:
251-260.
Brookes P & Lawley PD (1964) Evidence of the binding of polynuclear
aromatic hydrocarbons to the nucleic acids of mouse skin: Relation
between carcinogenic power of hydrocarbons and their binding to
deoxyribonucleic acid. Nature, 202: 781-784.
Brookes P, Ellis MV, Pataki J, & Harvey RG (1986) Mutation in
mammalian cells by isomers of 5-methylchrysene diolepoxide.
Carcinogenesis, 7: 463-466.
Bruce WR & Heddle JA (1979) The mutagenic activity of 61 agents as
determined by the micronucleus, Salmonella, and sperm abnormality
assays. Can J Genet Cytol, 21: 319-334.
Bruggeman WA, Van der Steen J, & Hutzinger O (1982) Reversed-phase
thin-layer chromatography of polynuclear aromatic hydrocarbons and
chlorinated biphenyls. Relationship with hydrophobicity as measured by
aqueous solubility and octanol-water partition coefficient. J
Chromatogr, 238: 335-346
Brune K, Kalin H, Schmidt R, & Hecker E (1978) Inflammatory, tumor
initiating and promoting activities of polycyclic aromatic
hydrocarbons and diterpene esters in mouse skin as compared with their
prostaglandin releasing potency in vitro. Cancer Lett, 4: 333-342.
Brune H, Deutsch-Wenzel RP, Habs M, Ivankovic S, & Schmähl D (1981)
Investigation of the tumorigenic response to benzo [a]pyrene in
aqeous caffeine solution applied orally to Sprague-Dawley rats. J
Cancer Res Clin Oncol, 102: 153-157.
Brunström B, Hakansson H, & Lundberg K (1991) Effects of a technical
PCB preparation and fractions thereof on ethoxyresorufin O-deethylase
activity, vitamin A levels and thymic development in the mink Mustela
vison. Pharmacol Toxicol, 69: 421-426.
Bryan WR & Shimkin MB (1943) Quantitative analysis of dose-response
data obtained with three carcinogenic hydrocarbons in strain C3H male
mice. J Natl Cancer Inst, 3: 503-531.
Bryant MF, Erexson GL, Kwanyuen P, Atwater AL, & Kligerman AD (1990)
Sister chromatid exchange and micronucleus analyses in rat peripheral
blood lymphocytes following in vivo exposure to
dibenz(a,h)anthracene (Abstract 30). Environ Mol Mutag, 15 (suppl 17):
10-11.
Bryant MF, Kwanyuen P, Atwater AL, Erexson GL, & Kligerman AD (1991)
Cytogenetic effects of benzo-b-fluoranthene in Sprague-Dawley rat
peripheral blood lymphocytes after in vivo
exposure (Abstract 33). Environ Mol Mutag, 17 (suppl 19): 13.
Bryla P & Weyand EH (1991) Role of activated oxygen species in
benzo [a]pyrene:DNA adduct formation in vitro. Free Radicals Biol
Med, 11: 17-24.
Buccafusco RJ, Ells SJ, & LeBlanc GA (1981) Acute toxicity of priority
pollutants to bluegill (Lepomis macrochirus). Bull Environ Contam
Toxicol, 26: 446-452.
Buchet JP, Gennart JP, Mercado-Calderon F, Delavignette JP, Cupers L,
& Lauwerys R (1992) Evaluation of exposure to polycyclic aromatic
hydrocarbons in a coke production and a graphite electrode
manufacturing plant: Assessment of urinary excretion of
1-hydroxypyrene as a biological indicator of exposure. Br J Ind Med,
49: 761-768.
Buck M (1983) [Immission measurements of polycyclic aromatic
hydrocarbons (PAH) in the Rhein-Ruhr-Gebiet.] Schriftenr Landesanst
Immissionsschutz Landes Nordrhein-Westfalen. 57: 37-45 (in German).
Buck M (1991) [Methods and results of the measurement of carcinogenic
polycyclic aromatic hydrocarbons.] In: [Carcinogenic compounds in the
environment: Sources, measurement, risk, reduction.] Dusseldorf,
VDI-Verlag, pp 49-70 (VDI Report No. 888) (in German).
Buck M, Ixfeld H, & Ellermann K (1989) [Report on the results of
discontinuous sulfur dioxide and multiple component measurements taken
in the Rhine-Ruhr area in the period from 1 January to 31 December
1988.] In: Summaries of reports, 1988. A series from the State of
North-Rhine Westfalia Pollution Control Office. Düsseldorf, Cornelsen
Verlag Schwann-Girardet, pp 69-130 (in German with English summary)
Buckley TJ & Lioy PJ (1992) An examination of the time course from
human dietary exposure to polycyclic aromatic hydrocarbons to urinary
elimination of 1-hydroxypyrene. Br J Ind Med, 49: 113-124.
Buckpitt AR & Franklin RB (1989) Relationship of naphthalene and
2-methylnaphthalene metabolism to pulmonary bronchiolar epithelial
cell necrosis. Pharmacol Ther, 41: 393-410.
Budavari S, O'Neil MJ, Smith A, & Heckelman PE (1989) The Merck Index:
An encyclopedia of chemicals, drugs, and biologicals, 11th ed. Rahway,
New Jersey, Merck & Co., pp 108, 350, 650, 1008, 1139, 1266.
Buening MK, Levin W, Karle JM, Yagi H, Jerina DM, & Conney AH (1979)
Tumorigenicity of bay-region epoxides and other derivatives of
chrysene and phenanthrene in newborn mice. Cancer Res, 39: 5063-5068.
Buening MK, Levin W, Wood AW, Chang RL, Lehr RE, Taylor CW, Yagi H,
Jerina DM, & Conney AH (1980) Tumorigenicity activity of
benzo(e)pyrene derivatives on mouse skin and in newborn mice. Cancer
Res, 40: 203-206.
Bui QQ, Tran MB, & West WL (1986) A comparative study of the
reproductive effects of methadone and benzo [a]pyrene in the pregnant
and pseudopregnant rat. Toxicology, 42: 195-204.
Bulay OM (1970) The study of development of lung and skin tumors in
mice exposed in utero to polycyclic hydrocarbons. Acta Med Turk, 7: 3-
38.
Bulay OM & Wattenberg LW (1971) Carcinogenic effects of polycyclic
hydrocarbon carcinogen administration to mice during pregnancy on the
progeny. J Natl Cancer Inst, 46: 397-402.
Bulman TL, Lesage S, Fowlie P, & Webber MD (1987) The fate of
polynuclear aromatic hydrocarbons in soil. In: Vandermeulan JH &
Hurley SE ed. Oil in fresh water: chemistry, biology, countermeasure
technology. New York, Pergamon Press, pp 231-251.
Burchiel SW, Hadley WM, Barton SL, Fincher RH, Lauer LD, & Dean JH
(1988) Persistent suppression of humoral immunity produced by
7,12-dimethylbenz [a]anthracene (DMBA) in B6C3F1 mice: Correlation
with changes in spleen cell surface markers detected by flow
cytometry. Int J Immunopharmacol, 10: 369-376.
Burchiel SW, De Davis AP, Gomez MP, Montano RM, Barton SL, & Seamer LC
(1990) Inhibition of lymphocyte activation in splenic and
gut-associated lymphoid tissues followig oral exposure of mice to
7,12-dimethylbenz [a]anthracene. Toxicol Appl Pharmacol, 105:
434-442.
Burchiel SW, Thompson TA, & Davis DP (1991) Alterations in
mitogen-induced calcium mobilization and intracellular free calcium
produced by 7,12-dimethylybenz(a)-anthracene in the Jurkat human T
cell line. Int J Immunopharmacol, 13: 109-115.
Burgaz S, Borm PJA, & Jongeneelen F (1992) Evaluation of urinary
excretion of 1-hydroxypyrene and thioethers in workers exposed to
bitumen fumes. Int Arch Occup Environ Health, 63: 397-401.
Burnham K & Rahman M (1992) Effects of petrochemicals and ultraviolet
radiation on epidermal IA expression in vitro. J Toxicol Environ
Health, 35: 175-185.
Busby WF, Goldman ME, Newberne PM, & Wogan GN (1984) Tumorigenicity of
fluoranthene in a newborn mouse lung adenoma bioassay. Carcinogenesis,
5: 1311-1316.
Busby WF Jr, Stevens EK, Kellenbach ER, Cornelisse J, & Lugtenburg J
(1988) Dose-response relationships of the tumorigenicity of
cyclopenta [c,d]pyrene, benzo [a]pyrene and 6-nitrochrysene in a
newborn mouse lung adenoma bioassay. Carcinogenesis, 9: 741-746.
Busby WF Jr, Stevens EK, Martin CN, Chow FL, & Garner RC (1989)
Comparative lung tumorigenicity of parent and mononitro-polynuclear
aromatic hydrocarbons in the BLU: Ha newborn mouse assay. Toxicol Appl
Pharmacol, 99: 555-563.
Butler JD & Crossley P (1979) An appraisal of relative airborne
suburban concentrations of polycyclic aromatic hydrocarbons monitored
indoors and outdoors. Sci Total Environ, 11: 53-58.
Butler JD & Crossley P (1981) Reactivity of polycyclic aromatic
hydrocarbons adsorbed on soot particles. Atmos Environ, 15: 91-94.
Butler JD & Crossley P (1982) Predicting polycyclic aromatic
hydrocarbon concentrations in urban aerosols by linear multiple
regression analysis. Environ Pollut, B3: 109-123.
Butler JD, Butterworth V, Kellow SC, & Robinson HG (1984) Some
observations on the polycyclic aromatic hydrocarbon (PAH) content of
surface soils in urban areas. Sci Total Environ, 33: 75-85.
Butlin HT (1892) Cancer of the scrotum in chimney-sweeps and others.
Br Med J, ii: 66-71.
Buu-Hoi NP (1964) New developments in chemical carcinogenesis by
polycyclic hydrocarbons and related heterocycles: A review. Cancer
Res, 24: 1511-1523.
Cairns MA & Nebeker AV (1982) Toxicity of acenaphthene and isophorone
to early life stages of fathead minnows. Arch Environ Contam Toxicol,
11: 703-707.
Calder JA & Lader JH (1976) Effect of dissolved aromatic hydrocarbons
on the growth of marine bacteria in batch culture. Appl Environ
Microbiol, 32: 95-101.
Callahan MA, Slimak MW, Gabel NW, May IP, & Fowler CF (1979)
Water-related environmental fate of 129 priority pollutants. Vol II.
Halogenated aliphatic hydrocarbons, halogenated ethers, monocyclic
aromatics, phthalate esters, polycyclic aromatic hydrocarbons,
nitrosamines and miscellaneous compounds Report EPA-440/4-79-029b;
PB80 204381). Washington DC, US Environmental Protection Agency, 673
pp.
Capel PD, Leuenberger C, & Giger W (1991) Hydrophobic organic
chemicals in urban fog. Atmos Environ, 25: 1335-1346.
Carmichael PL, Jacob J, Grimmer G, & Phillips DH (1990) Analysis of
the polycyclic aromatic hydrocarbon content of petrol and diesel
engine lubricating oils and determination of DNA adducts in topically
treated mice by 32P-postlabelling. Carcinogenesis, 11: 2025-2032.
Carmichael PL, Platt KL, Ni She M, Lecoq S, Oesch F, Phillips DH, &
Grover PL (1993) Evidence for the involvement of a bis-diol-epoxide in
the metabolic activation of dibenz [a,h]anthracene to DNA-binding
species in mouse skin. Cancer Res, 53: 944-948.
Carstensen U, Alexandrie AK, Högstedt B, Rannug A, Bratt I, & Hagmar L
(1993) B- and T-lymphocyte micronclei in chimney sweeps with respect
to genetic polymorphism for CIP1A1 and GST1 (class Mu). Mutat Res,
289: 187-195.
Carver JH, Machado ML, & MacGregor JA (1986) Application of modified
Salmonella/microsome prescreen to petroleum-derived complex mixtures
and polynuclear aromatic hydrocarbons (PAH). Mutat Res, 174: 247-253.
Casserly DM, Davis EM, Downs TD, & Guthrie RK (1983) Sorption of
organisms by Selenastrum capricornutum. Water Res, 17: 1591-1594.
Casto BC (1973) Enhancement of adenovirus transformation by treatment
of hamsters with ultraviolet irradiation, DNA base analogs, and
dibenz(a,h)anthracene. Cancer Res, 33: 402-407.
Casto BC (1979) Polycyclic hydrocarbons and Syrian hamster embryo
cells: Cell transformation, enhancement of viral transformation and
analysis of DNA-damage. In: Jones PW & Leber P ed. Polynuclear
aromatic hydrocarbons. Ann Arbor, Michigan, Ann Arbor Science
Publishers, pp 51-66.
Casto BC, Janosko N, & DiPaolo JA (1977) Development of a focus assay
model for transformation of hamster cells in vitro by chemical
carcinogens. Cancer Res, 37: 3508-3515.
Catallo WJ III & Gambrell RP (1987) The effects of high levels of
polycyclic aromatic hydrocarbons on sediment physicochemical
properties and benthic organisms in a polluted stream. Chemosphere,
16: 1053-1064.
Catoggio JA, Succar SD, & Roca AE (1989) Polynuclear aromatic
hydrocarbon content of particulate matter suspended in the atmosphere
of La Plata, Argentina. Sci Total Environ, 79: 43-58.
Cautreels W & van Cauwenberghe K (1977) Comparison between the organic
fraction of suspended matter at a background and an urban station. Sci
Total Environ, 8: 79-88.
Cavalieri E & Rogan E (1985) Role of radical cations in aromatic
hydrocarbon carcinogenesis. Environ Health Perspectives, 64: 69-84.
Cavalieri E, Mailander P, & Pelfrene A (1977) Carcinogenic activity of
anthanthrene on mouse skin. Z Krebsforsch, 89: 113-118.
Cavalieri E, Roth R, Althoff J, Grandjean C, Patil K, Marsh S, &
McLaughlin D (1978) Carcinogenicity and metabolic profiles of
3-methylcholanthrene oxygenated derivatives at the 1 and 2 positions.
Chem-Biol Interactions, 22: 69-81.
Cavalieri E, Rogan E, & Thilly WG (1981a) Carcinogenicity,
mutagenicity and binding studies of the environmental contaminant
cyclopenteno [c,d]pyrene and some of its derivatives. In: Cooke M &
Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemical analysis and
biological fate. Columbus, Ohio, Battelle Press, pp 487-499.
Cavalieri E, Rogan E, Toth B, & Munhall A (1981b) Carcinogenicity of
the environmental pollutants cyclopenteno(c,d)pyrene and
cyclopentano(c,d)pyrene in mouse skin. Carcinogenesis, 2: 277-281.
Cavalieri E, Rogan E, & Sinha D (1988a) Carcinogenicity of aromatic
hydrocarbons directly applied to rat mammary gland. Cancer Res Clin
Oncol, 114: 3-9.
Cavalieri E, Rogan E, Cremonesi P, Higginbotham S, & Salmasi S (1988b)
Tumorigenicity of 6-halogenated derivatives of benzo [a]pyrene in
mouse skin and rat mammary gland. J Cancer Res Clin Oncol, 114: 10-15.
Cavalieri EL, Rogan EG, Higginbotham S, Cremonesi P, & Salmasi S
(1989) Tumor-initiating activity in mouse skin and carcinogenicity in
rat mammary gland of dibenzo[a]pyrenes: The very potent environmental
carcinogen dibenzo(a,l)pyrene. J Cancer Res Clin Oncol, 115: 67-72.
Cavalieri EL, Higginbotham S, Ramakrishna NVS, Devanesan PD, Todorovic
R, Rogan EG, & Salmasi S (1991) Comparative dose-response
tumorigenicity studies of dibenzo [a,l]pyrene versus
7,12-dimethylbenz [a]anthracene, benzo [a]pyrene and two
dibenzo [a,l]pyrene dihydrodiols in mouse skin and rat mammary gland.
Carcinogenesis, 12: 1939-1944.
Cavalieri EL, Rogan EG, Ramakrishna NVS, & Devanesan PD (1993)
Mechanisms of benzo(a)pyrene and 7,12-dimethylbenz(a)anthracene
activation: Qualitative aspects of the stable and depurination DNA
adducts obtained from radical cations and diol epoxides. In: Garrigues
P & Lamotte M ed. Polycyclic aromatic compounds: Synthesis,
properties, analytical measurements, occurrence and biological
effects. Bordeaux, Gordon & Breach Science Publishers, pp 725-732.
Cenni A, Sciarra G, Sartorelli P, & Pappalardo F (1993) Environmental
and biological monitoring of polycyclic aromatic hydrocarbons (PAHs)
in coke plants and other workplaces. Med Lav, 84: 379-386.
Cerniglia CE (1984) Microbial metabolism of polycyclic aromatic
hydrocarbons. In: Cerniglia CE ed. Advances in applied microbiology,
Volume 30. Jefferson, Arkansas, Academic Press, pp 31-71.
Chakraborti D, Van Vaeck L, & Van Espen P (1988) Calcutta pollutants:
Part II. Polynuclear aromatic hydrocarbon and some metal concentration
on air particulates during winter 1984. Int J Environ Anal Chem, 32:
109-120.
Chaloupka K, Santosstefano M, Goldfarb IS, Liu G, Myers MJ, Tsyrolv
IB, Gelboin HV, Krishnan V, & Safe S (1994) Aryl hydrocarbon (ah)
receptor-independent induction of Cyp1a2 gene expression by
acenaphthylene and related compounds in B6C3F1 mice. Carcinogenesis,
15: 2835-2840.
Chang RL, Levin W, Wood AW, Lehr RE, Kumar S, Yagi H, Jerina DM, &
Conney AH (1981) Tumorigenicity of the diastereomeric bay-region
benzo(e)pyrene 9,10-diol-11,12-epoxides in newborn mice. Cancer Res,
41: 915-918.
Chang RL, Levin W, Wood AW, Lehr RE, Kumar S, Yagi H, Jerina DM, &
Conney AH (1982) Tumorigenicity of bay-region diol-epoxides and other
benzo-ring derivatives of dibenzo[a,h]pyrene and dibenzo[a,i]pyrene on
mouse skin and newborn mice. Cancer Res, 42: 25-29.
Chang RL, Levin W, Wood AW, Yagi H, Tada M, Vyas KP, Jerina DM, &
Conney AH (1983) Tumorigenicity of enantiomers of chrysene
1,2-dihydrodiol and of the diastereomeric bay-region chrysene
1,2-diol-3,4-epoxides on mouse skin and in newborn mice. Cancer Res,
43: 192-196.
Chang SC, Chang KT, Keng, YF Lan CF, Hsiao HC, Hsen SH, & Wei YH
(1988) Mutagenicity and polycyclic aromatic hydrocarbons analysis of
airborne particulate matters from Taipei city. Proc Natl Sci Counc
Taiwan, 12: 129-139.
Chappell WR (1989) Interspecific scaling of toxicity data: A question
of interpretation. Risk Anal, 9: 13-14.
Chasseaud LF (1979) The role of glutathione and glutathione
S-transferases in the metabolism of chemical carcinogens and other
electrophilic agents. Adv Cancer Res, 29: 175-274.
Chau N, Bertrand JP, Mur JM, Figueredo A, Patris A, Moulin JJ, & Pham
QT (1993) Mortality in retired coke oven plant workers. Br J Ind Med,
50: 127-135..
Chemical Abstracts Service (1988) Ring systems handbook. Ring Systems
File I RF 1-RF 27595. Columbus, Ohio, American Chemical Society.
Chemical Abstracts Service (1990) Index guide. Part 1, pp. 1-1470;
Part 2, pp 1471-2446. Columbus, Ohio, American Chemical Society.
Chen TT & Heidelberger C (1969) Quantitative studies on the malignant
transformation of mouse prostate cells by carcinogenic hydrocarbons
in vitro. Int J Cancer, 4: 166-178.
Chen PHS, Shieh HH, & Gaw JM (1980) Determination of polycyclic
aromatic hydrocarbons in airborne particulates at various sites in
Taipei city by GC/MS and glass capillary GC. Proc Natl Sci Counc
Taiwan, 4: 280-284.
Chen PHS, Chuang CY Lu YD, & Chung KT (1981) Gas chromatography/mass
spectrometric determination of polycyclic aromatic hydrocarbons in
airborne particulates at various sites in Kaohsiung area. Proc Natl
Sci Counc Taiwan, 5: 262-267.
Chiazze L Jr, Watkins DK, & Amsel J (1991) Asphalt and risk of cancer
in man. Br J Ind Med, 48: 538-542.
Chipman JK (1982) Bile as a source of potential reactive metabolites.
Toxicology, 25: 99-111.
Chipman JK, Hirom PC, Frost GS, & Millburn P (1981) The biliary
excretion and enterohepatic circulation of benzo [a]pyrene and its
metabolites in the rat. Biochem Pharmacol, 30: 937-944.
Chiu CH, Howes PS, Li KC, Poole GK, & Lao RC (1991) The determination
and measurement of PAH in municipal incinerator samples. In: Cooke M,
Loening K, & Merritt J ed. Polynuclear aromatic hydrocarbons:
Measurement, means, and metabolism. Columbus, Ohio, Battelle Press, pp
195-211.
Chladek E & Marano RS (1984) Use of bonded phase silica sorbents for
the sampling of priority pollutants in wastewaters. J Chromatogr Sci,
22: 313-320.
Chorazy M, Szeliga J, Strozyk M, & Cimander B (1994) Ambient air
pollutants in Upper Silesia: Partial chemical composition and
biological activity. Environ Health Perspectives, 102: 61-66.
Chu EW & Malmgren RA (1965) An inhibitory effect of vitamin A on the
induction of tumors of forestomach and cervix in the Syrian hamster by
carcinogenic polynuclear hydrocarbons. Cancer Res, 25: 884-895.
Chuang JC, Hannan SW, & Wilson NK (1987) Field comparison of
polyurethane foam and XAD-2 resin for air sampling for polynuclear
aromatic hydrocarbons. Environ Sci Technol, 21: 798-804.
Chuang JC, Mack GA, Kuhlman MR, & Wilson NK (1991) Polycyclic aromatic
hydrocarbons and their derivatives in indoor and outdoor air in an
eight-home study. Atmos Environ, 25B: 369-380.
Chuang JC, Wise SA, Cao S, & Mumford JL (1992) Chemical
characterization of mutagenic fractions of particles from indoor coal
combustion: A study of lung cancer in Xuan Wie, China. Environ Sci
Technol, 26: 999-1004.
Chuang JC, Callahan PJ, Menton RG, & Gordon SM (1995) Monitoring
methods for polycyclic aromatic hydrocarbons and their distribution in
house dust and track-in soil. Environ Sci Technol, 29: 494-500.
Clayson DB, Pringle JAS, Bonser GM, & Wood M (1968) The technique of
bladder implantation: Further results and an assessment. Br J Cancer,
22: 825-832.
Clayton P, Davis BJ, Jones K, & Jones P (1992) Toxic organic
micropollutants in urban air. Stevenage, Hertfordshire, Warren Spring
Laboratory, 122 pp (Report No LR 904 [PA]).
Clement Associates (1988) Comparative potency approach for estimating
the cancer risk associated with exposure to mixtures of polycyclic
aromatic hydrocarbons. Interim final report. Fairfax, Virginia, 125 pp
(Report No. 68-02-4403).
Clement Associates (1990) Development of a dose-response model for
inhaled B[a]P. Fairfax, Virginia, 28 pp (prepared for US EPA under
contract No. 68-02-4601).
Clive D, Johnson KO, Spector AG, & Brown MMM (1979) Validation and
characterization of the L5178Y/TK+/- mouse lymphoma mutagen assay
system. Mutat Res, 59: 61-108.
Clonfero E, Venier P, Toffolo D, Busi L, & Gava C (1984) Mutagenesis
test on urine of workers exposed to polycyclic aromatic hydrocarbons
in an anode plant. Med Lav, 75: 275-281.
Clonfero E, Zordan M, Venier P, Paleologo M, Levis AG, Cottica D,
Pozzoli L, Jongeneelen FJ, Bos RP, & Anzion RBM (1989) Biological
monitoring of human exposure to coal tar. Urinary excretion of total
polycyclic aromatic hydrocarbons, 1-hydroxypyrene and mutagens in
psoriatic patients. Int Arch Occup Environ Health, 61: 363-368.
Clonfero E, Jongeneelen F, Zordan M, & Levis AG (1990) Biological
monitoring of human exposure to coal tar. Urinary mutagenicity assays
and analytical determination of polycyclic aromatic hydrocarbon
metabolites in urine. In: Vainio H, Sorsa M, & McMichael AJ ed.
Complex mixtures and cancer risk. Lyon, International Agency for
Research on Cancer, pp 215-222 (IARC Scientific Publications No. 104).
Clonfero E, Granella M, Marchioro M, Leopardi Barra E, Nardini B,
Ferri G, & Foà V (1995) Urinary excretion of mutagens in coke oven
workers. Carcinogenesis, 16: 547-554.
Coates JT, Elzerman AW, & Garrison AW (1986) Extraction and
determination of selected polycyclic aromatic hydrocarbons in plant
tissues. J Assoc Off Anal Chem, 69: 110-114.
Cody T, Radike M, & Warshawsky D (1984) The phototoxicity of
benzo [a]pyrene in the green algea Selenastrum capricornutum.
Environ Res, 35: 122-132.
Cohen GM, Haws SM, Moore BP, & Bridges JW (1976) Benzo [a]pyren-3-yl
hydrogen sulphate, a major ethyl acetate-extractable metabolite of
benzo [a]pyrene in human, hamster and rat lung cultures. Biochem
Pharmacol, 25: 2561-2570.
Collier TK, Singh SV, Awasthi YC, & Varanasi U (1992) Short
communication. Hepatic xenobiotic metabolizing enzymes in two species
of benthic fish showing different prevalences of
contaminant-associated liver neoplasms. Toxicol Appl Pharmacol, 113:
319-324.
Collin G & Höke H (1985) Anthracene. In: Elvers B, Hawkins S, & Schulz
G ed. Ullmann's encyclopedia of industrial chemistry, 5th ed.,Volume
A2: Weinheim, Verlagsgesellschaft , pp 343-345.
Collin G & Höke H (1991) Naphthalene and hydronaphthalene. In: Elvers
B, Hawkins S, & Schulz G ed. Ullmann's encyclopedia of industrial
chemistry, 5th ed., Volume A17: Weinheim, Verlagsgesellschaft, pp 1-8.
Collins JF & Alexeeff G (1993) Benzo [a]pyrene as a toxic air
contaminant. Part B: Health effects of benzo [a]pyrene. Berkeley,
California, California Environmental Protection Agency, Office of
Environmental Health Hazard Assessment, 69 pp.
Collins JF, Brown JP, Dawson SV, & Marty MA (1991) Risk assessment for
benzo [a]pyrene. Regul Toxicol Pharmacol, 13: 170-184.
Colmsjö A, Zebühr YU, & Ostman CE (1986a) Polynuclear aromatic
compounds in flue gases and ambient air in the vicinity of a municipal
incineration plant. Atmos Environ, 20: 2279-2282.
Colmsjö AL, Zebühr YU, & Ostman CE (1986b) Polynuclear aromatic
compounds in the ambient air of Stockholm. Chemosphere, 15: 169-182.
Combet E, Jarosz J, Martin-Bouyer M, Paturel L, & Saber A (1993)
[Shpol'skii spectrofluorimetric measurement of unit emissions of PAH
from 30 petrol and diesel light vehicles in eight representative
cycles.] Sci Total Environ, 134: 147-160 (in French).
Community Bureau of Reference (1992) Reference materials. Brussels,
Commission of the European Communities, 2 pp.
Compaan H & Laane RW (1992) Polycyclic aromatic hydrocarbons (PAH) in
the North Sea: An inventory. Delft, TNO Institute of Environmental
Sciences, 130 pp (TNO Report IMW-R 92/392).
CONCAWE (The Oil Companies' European Organization for Environment,
Health and Safety) (1992) The chemical composition of diesel
particulate emissions (Report No. 92/51). The Hague, 24 pp.
CONCAWE (The Oil Companies' European Organization for Environment,
Health and Safety) (1994) Motor vehicle emission regulations and fuel
specifications. 1994 update (Report 4/94). The Hague, 234 pp.
Conney AH (1982) Induction of microsomal enzymes by foreign chemicals
and carcinogenesis by polycyclic aromatic hydrocarbons: G.H.A. Clowes
memorial lecture. Cancer Res, 42: 4875-4917.
Coombs MM & Bhatt TS ed. (1987) Cyclopenta(a)phenanthrenes. Polycyclic
aromatic compounds structurally related to steroids. Cambridge,
Cambridge University Press, 265 pp.
Coombs MM, Dixon C, & Kissonerghis A-M (1976) Evaluation of the
mutagenicity of compounds of known carcinogenicity, belonging to the
benz [a]anthracene, chrysene, and cyclopenta [a]phenanthrene series,
using Ames' test. Cancer Res, 36: 4525-4529.
Cooper JA (1980) Environmental impact of residential wood combustion
emissions and its implications. J Air Pollut Control Assoc, 30: 855-
861.
Cooper CS, Hewer A, Ribeiro O, Grover PL, & Sims P (1980) The
enzyme-catalysed conversion of a non-'bay-region' diol-epoxide of
benz(a)anthracene into a gluthathione conjugate. FEBS Lett, 118: 39-
42.
Cooper CS, Grover PL, & Sims P (1983) The metabolism and activation of
benzo [a]pyrene. Drug Metab, 7: 295-396.
Coover MP & Sims RC (1987) The effect of temperature on polycyclic
aromatic hydrocarbon persistence in an unacclimated agricultural soil.
Hazard Waste Hazard Mater, 4: 69-82.
Cope VW & Kalkwarf DR (1987) Photooxidation of selected polycyclic
aromatic hydrocarbons and pyrenequinones coated on glass surfaces.
Environ Sci Technol, 21: 643-648.
Corner EDS & Young L (1954) Biochemical studies of toxic agents. 7.
The metabolism of naphthalene in animals of different species. Biochem
J, 58: 647-655.
Corner EDS, Billett FS, & Young L (1954) Biochemical studies of toxic
agents. 6. The conversion of naphthalene into
1,2-dihydro-2-hyroxy-l-naphthyl glucosiduronic acid in the rabbit.
Biochem J, 56: 270-274.
Correa M & Coler R (1983) Enhanced oxygen uptake rates in dragonfly
nymphs Somatochlora cingulata as an indication of stress from
naphthalene. Environ Contam Toxicol, 30: 269-276.
Cosma GN, Toniolo P, Currie D, Pasternack BS, & Seymour JG (1992)
Expression of the CYP1A1 gene in peripheral lymphocytes as a marker of
exposure to creosote in railroad workers. Cancer Epidemiol Biomarkers
Prev, 1: 137-142.
Costantino JP, Remond CK, & Bearden A (1995) Occupationally related
cancer risk among coke oven workers: 30 years of follow-up. J Occup
Environ Med, 37: 597-604.
Cottini GB & Mazzone GB (1939) The effects of 3:4-benzpyrene on human
skin. Am J Cancer, 37: 186-195.
Coutant RW, Brown L, Chuang JC, Riggin RM, & Lewis RG (1988) Phase
distribution and artifact formation in ambient air sampling for
polynuclear aromatic hydrocarbons. Atmos Environ, 22: 403-409.
Coutant RW, Callahan PJ, & Chuang JC (1992) Efficiency of
silicone-grease-coated denuders for collection of polynuclear aromatic
hydrocarbons. Atmos Environ, 26A: 2831-2834.
Creasia DA, Poggenburg JK Jr, & Nettesheim P (1976) Elution of
benzo [a]pyrene from carbon particles in the respiratory tract of
mice. J Environ Health, 1: 967-975.
Crespi CL & Thilly WG (1984) Assay for gene mutation in a human
lymphoblast line, AHH-1, competent for xenobiotic metabolism. Mutat
Res, 128: 221-230.
Crespi CL, Liber HL, Behymer TD, Hites RA, & Thilly WG (1985) A human
cell line sensitive to mutation by particle-born chemicals. Mutat Res,
157: 71-75.
Cretney JR, Lee HL, & Wright GJ (1985) Analysis of polycyclic aromatic
hydrocarbons in air particulate matter from a lightly industrialized
urban area. Environ Sci Technol, 19: 397-404.
Crider JY, Wilhm J, & Harmon HJ (1982) Effects of naphthalene on the
hemoglobin concentration and oxygen uptake of Daphnia
magna. Bull Environ Contam Toxicol, 28: 52-57.
Crocker TT, Chase JE, Wells SA, & Nunes LL (1970) Preliminary report
on experimental squamous carcinoma of the lung in hamsters and in a
primate (Galago Crassicauda Tus.). San Diego, California, Office of
the County Veterinarian, pp 317-328 (Symposium Series No. 21).
Croisy-Delcey M, Ittah Y, & Jerina DM (1979) Synthesis of
benzo [c]phenanthrene dihydrodiols. Tetrahedr Lett, 31: 2849-2852.
Crosby NT, Hunt DC, Philp LA, & Patel I (1981) Determination of
polynuclear aromatic hydrocarbons in food, water and smoke using
high-performance liquid chromatography. Analyst, 106: 135-145.
Crössmann G & Wüstemann M (1992) [Loadings in domestic gardens and
allotments by anorganic and organic substances with damaging
potential: Actual documentation. Part I: Soils and garden wastes and
Part II: Vegetables and fruits.] Berlin, Ministry of Environment, pp
40-42, 108-124 (in German).
Crow KD, Alexander E, Buck WHL, Johnson BE, Magnus IA, & Porter AD
(1961) Photosensitivity due to pitch. Br J Dermatol, 73: 220-232.
Csaba G & Inczefi-Gonda A (1992) Benzopyrene exposure at 15 days of
prenatal life reduces the binding capacity of thymic glucocorticoid
receptors in adulthood. Gen Pharmacol, 23: 123-124.
Csaba G, Inczefi-Gonda A, & Szeberenyi S (1991) Lasting impact of a
single benzpyrene treatment in pre-natal and growing age on the thymic
glucocorticoid receptors of rats. Gen Pharmacol, 22: 815-818.
Culp SJ, Gaylor DW, Sheldon WG, Goldstein LS, & Beland FA (1996) DNA
adduct measurements in relation to tumor incidence during the chronic
feeding of coal tar or benzo(a)pyrene to mice. Polycyclic Aromat Compd
11: 161-168.
Cummings DA, Lin ELC, Daniel FB, Klaunig JE, & Schut, HAJ (1991) In
vivo and in vitro binding of benzo [a]pyrene to tissue DNA and
circulating macromolecules in the mouse, monkey and human. Proc Am
Assoc Cancer Res, 32: 159.
Dahl AR, Coslett DC, Bond JA, & Hesseltine GR (1985) Metabolism of
benzo [a]pyrene on the nasal mucosa of Syrian hamsters: Comparison to
other extrahepatic tissues and possible role of nasally produced
metabolites in carcinogenesis. J Natl Cancer Inst, 75: 135-139.
Dai Q (1980) Researches on chemical carcinogens and mechanism of
chemical carcinogenesis. DI-region theory: A quantitative molecular
orbital model of carcinogenic activity for polycyclic aromatic
hydrocarbons. Sci Sin, 23: 453-470.
Daisey JM (1983) Analysis of polycyclic aromatic hydrocarbons by
thin-layer chromatography. In: Bjorseth A ed. Handbook of polycyclic
aromatic hydrocarbons. New York, Marcel Dekker, pp 397-437.
Daisey JM & Gundel LA (1993) Method 20: Determination of extractable
particulate organic matter and selected polycyclic aromatic
hydrocarbons. In: Seifert B, van de Wiel HJ, Dodet B, & O'Neill IK ed.
Environmental carcinogens. Methods of analysis and exposure
measurement: Volume 12. Indoor air. Lyon, International Agency for
Research on Cancer, pp 314-327 (IARC Scientific Publications No 109).
Daisey JM, McCaffrey RJ, & Gallagher RA (1981) Polycyclic aromatic
hydrocarbons and total extractable particulate organic matter in the
arctic aerosol. Atmos Environ, 15: 1353-1361.
Daisey JM, Cheney JL, & Lioy PH (1986) Profiles of organic particulate
emissions from air pollution sources: Status and needs for receptor
source apportionment modeling. J Air Pollut Control Assoc, 36: 17-33.
Daisey JM, Spengler JD, & Kaarakka P (1989) A comparison of the
organic chemical composition of indoor aerosols during woodburning and
non-woodburning periods. Environ Int, 15: 435-442.
Danz M, Hartmann A, Otto M, & Blaszyk H (1991) Hitherto unknown
additive growth effects of fluorene and 2-acetylaminofluorene on bile
duct epithelium and hepatocytes in rats. Arch Toxicol, 14 (suppl): 71-
74.
Darby FW, Willis AF, & Winchester RV (1986) Occupational health
hazards from road construction and sealing work. Ann Occup Hyg, 30:
445-454.
Darville RG & Wilhm JL (1984) The effect of naphthalene on oxygen
consumption and hemoglobin concentration in Chironomus
attenuatus and on oxygen consumption and life cycle of Tanytarsus
dissimilis. Environ Toxicol Chem, 3: 135-142.
Davenport R & Spacie A (1991) Acute phototoxicity of harbor and
tributary sediments from lower Lake Michigan. J Great Lakes Res, 17:
51-56.
Davidson GE & Dawson GWP (1976) Chemically induced presumed somatic
mutations in the mouse. Mutat Res, 38: 151-154.
Davies IW, Harrison RM, Perry R, Ratnayaka D, & Wellings RA (1976)
Municipal incinerator as source of polynuclear aromatic hydrocarbons
in environment. Environ Sci Technol, 10: 451-453.
Davies GM, Hodkinson A, & DiVetta P (1986) Measurement and analysis of
occupational exposures to coke oven emissions. Ann Occup Hyg, 30: 51-
62.
Davis WW, Krahl ME, & Clowes GHA (1942) Solubility of carcinogenic and
related hydrocarbons in water. J Am Chem Soc, 64: 108-110.
Davis BR, Whitehead JK, Gill ME, Lee PN, Butterworth AD, & Roe FJR
(1975) Response of rat lung to 3,4-benzpyrene administered by
intratracheal instillation in infusine with or without carbon black.
Br J Cancer, 31: 443-452.
Davis CS, Fellin P, & Otson R (1987) A review of sampling methods for
polyaromatic hydrocarbons in air. J Am Pollut Control Assoc, 37: 1397-
1408.
Deal CL (1995) The role of metabolic activation in B[a]P-induced
suppression of the humoral immune response. Richmond, Virginia,
Virginia Commonwealth University, Medical College of Richmond, 120 pp
(Doctoral Thesis).
Dean BJ (1981) Activity of 27 coded compounds in the RL, chromosome
assay. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens. Report of the international collaborative programme.
New York, Elsevier North-Holland, pp 570-579 (Progress in Mutation
Research, Volume 1).
Dean RG, Bynum G, Jacobson-Kram D, & Hadley E (1983a) Activation of
polycyclic hydrocarbons in Reuber H4-II-E hepatoma cells. An
in vitro system for the induction of SCEs. Mutat Res, 11: 419-427.
Dean JH, Luster MI, Boorman GA, Lauer LD, & Leubke RW (1983b)
Selective immunosuppression resulting from exposure to the
carcinogenic congener benzopyrene in B6C3F1 mice. Clin Exp Immunol,
52: 199-206.
Dean JH, Ward EC, Murray MJ, Lauer LD, House RV, Stillman W, Hammilton
TA, & Adams DO (1986) Immunosupression following
7,12-dimethylbenz [a]anthracene exposure in B6C3F1 mice. II. Altered
cell-mediated imunity and tumor resistance. Int J Immunopharmacol, 8:
189-198.
De Fré R, Bruynseraede P, & Kretzschmar JG (1994) Air pollution
measurements in traffic tunnels. Environ Health Perspectives, 102: 31-
37.
DeGraeve GM, Geiger DL, Meyer JS, & Bergman HL (1980) Acute and
embryo-larval toxicity of phenolic compounds to aquatic biota. Arch
Environ Contam Toxicol, 9: 557-568.
DeGraeve GM, Elder RG, Woods DC, & Bergman HL (1982) Effects of
naphthalene and benzene on fathead minnows and rainbow trout. Arch
Environ Contam Toxicol, 11: 487-490.
DeLeon IR, Byrne CJ, Peuler EA, Antoine SR, Schaeffer J, & Murphy RC
(1986) Trace organic and heavy metal pollutants in the Mississippi
River. Chemosphere, 15: 795-805.
Dell'Omo M & Lauwerys RR (1993) Adducts of macromolecules in the
biological monitoring of workers exposed to polycyclic aromatic
hydrocarbons. Crit Rev Toxicol, 23: 111-126.
Den Hollander H, Van de Meent D, Van Noort P, & Wondergem E (1986) Wet
deposition of polycyclic aromatic hydrocarbons in the Netherlands. Sci
Total Environ, 52: 211-219.
Dennis MJ, Massey RC, McWeeny DJ, Knowles ME, & Watson D (1983)
Analysis of polycyclic aromatic hydrocarbons in UK total diets. Food
Chem Toxicol, 21: 569-574
Dennis MJ, Massey RC, Cripps G, Venn I, Howarth N, & Lee G (1991)
Factors affecting the polycyclic aromatic hydrocarbons content of
cereals, fats and other food products. Food Addit Contam, 8: 517-530.
DePierre JW & Ernster L (1978) The metabolism of polycyclic
hydrocarbons and its relationship to cancer. Biochim Biophys Acta,
473: 149-186.
De Raat WK, Schulting FL, Burghardt E, & De Meijere FA (1987a)
Application of polyurethane foam for sampling volatile mutagens from
ambient air. Sci Total Environ, 63: 175-189.
De Raat WK, Kooijman SALM, & Gielen JWJ (1987b) Concentrations of
polycyclic aromatic hydrocarbons in airborne particles in the
Netherlands and their correlation with mutagenicity. Sci Total
Environ, 66: 95-114.
De Raat WK, Bakker GL, & De Meijere FA (1990) Comparison of filter
materials used for sampling of mutagens and polycyclic aromatic
hydrocarbons in ambient airborne particles. Atmos Environ, 24A: 2875-
2887.
DeSalvia R, Meschini R, Fiore M, Polani S, Palitti F, Carluccio MA, &
Turchi G (1988) Induction of sister-chromatid exchanges by
procarcinogens in metabolically competent Chinese hamster epithelial
liver cells. Mutat Res, 207: 69-75.
De Serres FJ & Ashby J ed. (1981) Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, 813 pp (Progress in Mutation Research,
Volume 1).
De Serres FJ & Hoffman GR (1981) Summary report on the performance of
yeast assays. In: de Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens. Report of the international collaborative
programme. New York, Elsevier North Holland, pp 68-76 (Progress in
Mutation Research, Volume 1).
Desideri PG, Lepri L, Heimler D, Giannessi S, & Checchini L (1984)
Concentration, separation and determination of hydrocarbons in sea
water. J Chromatogr, 284: 167-178.
Desideri PG, Lepri L, Canovaro M, & Checcini L (1988) Recovery,
identification and determination of organic compounds in marine
sediments. Stud Environ Sci, 34: 317-331.
Deutsch-Wenzel RP (1983) Experimental studies on the carcinogenicity
of five nitrogen containing polynuclear aromatic compounds directly
injected into rat lungs. Cancer Lett, 20: 97-101.
Deutsch-Wenzel RP, Brune H, Grimmer G, Dettbarn G, & Misfeld J (1983)
Experimental studies in rat lungs on the carcinogenicity and dose-
response relationships of eight frequently occuring environmental
polycyclic aromatic hydrocarbons. J Natl Cancer Inst, 71: 539-544.
Devanesan PD, Cremonesi P, Nunnally JE, Rogan EG, & Cavalieri EL
(1990) Metabolism and mutagenicity of dibenzo [a,e]pyrene and the
very potent environmental carcinogen dibenzo [a,l]pyrene. Chem Res
Toxicol, 3: 580-586.
263-268.
De Wiest F (1978) Any factors influencing the dispersion and the
transport of heavy hydrocarbons associated with airborne particles.
Atmos Environ, 12: 1705-1711.
DeWitt TH, Swartz RC, & Lamberson JO (1989) Measuring the acute
toxicity of estuarine sediments. Environ Toxicol Chem, 8: 1035-1048.
Diamond L, Kruszewski F, Aden DP, Knowles BB, & Baird WM (1980)
Metabolic activation of benzo [a]pyrene by a human hepatoma cell
line. Carcinogenesis, 1: 871-875.
Dickerson RL, Hooper MJ, Gard NW, Cobb GP, & Kendall RJ (1994)
Toxicological foundations of ecological risk assessment: Biomarker
development and interpretation based on laboratory and wildlife
species. Environ Health Perspectives, 102 (suppl 12): 65-69.
Diercxsens P & Tarradellas J (1987) Soil contamination by some organic
micropollutants related to sewage sludge spreading. Int J Environ Anal
Chem, 28: 143-159.
DiGiovanni J, Diamond L, Pritchett WP, Fisher EP, & Harvey RG (1985)
Tumor-initiating activity of the 9,10-dihydrodiol- and
9,10-dihydrodiol-7,8-epoxide of 3-methylcholanthrene in Sencar mice.
Cancer Lett, 28: 223-228.
Dikun PP (1967) [Detection of polycyclic aromatic hydrocarbons in
atmospheric contamination and other materials with quasi-linear
fluorescence spectra.] Zh Prikl Spektrosk, 6: 202-209 (in Russian).
DiMichele L & Taylor M (1978) Histopathological and physiological
responses of Fundulus heteroclitus to naphthalene exposure. J Fish
Res Board Can, 35: 1060-1066.
DiPaolo JA & Casto BC (1976) In vitro transformation: Interaction of
chemical carcinogens with viruses and physical agents. In: Montesano
R, Bartsch, H, & Tomatis L ed. Screening tests in chemical
carcinogenesis. Lyon, International Agency for Research on Cancer, pp
415-432 (IARC Scientific Publications No. 12).
DiPaolo JA, Donovan JP, & Nelson RL (1969) Quantitative studies of in
vitro transformation by chemical carcinogens, J Natl Cancer Inst 42:
867-874.
DiPaolo JA, Donovan PJ, & Nelson RL (1971) Transformation of hamster
cells in vitro by polycyclic hydrocarbons without cytotoxicity. Proc
Natl Acad Sci USA, 68: 2958-2961.
DiPaolo JA, Takano K, & Popescu NC (1972) Quantitation of chemically
induced neoplastic transformation of BALB/3T3 cloned cell lines.
Cancer Res, 35: 2686-2695.
DiPaolo JA, Nelson RL, Donovan PJ, & Evans CH (1973) Host-mediated in
vivo-in vitro assay for chemical carcinogenesis. Arch Pathol, 95:
380-385.
DiPaolo JA, Doniger J, Evans CH, & Popescu NC (1985) Enhancement and
inhibition of transformation of Syrian hamster embryo cells.
Carcinogenesis, 8: 319-328.
Dipple A, Moschel RC, & Bigger CAH (1984) Polynuclear aromatic
carcinogens. In: Searle CE ed. Chemical carcinogenesis, 2nd ed.
Washington DC, American Chemical Society, Vol 1ume, pp 41-163.
Dipple A, Pigott MA, Agarwal SK, Yagi H, Sayer JM, & Jerina DM (1987)
Optically active benzo [c]phenanthrene diol epoxides bind extensively
to adenine in DNA. Nature, 327: 535-536.
Dipple A, Cheng SC, & Bigger CA (1990) Polycyclic aromatic hydrocarbon
carcinogens. Prog Clin Biol Res, 347:109-127.
Dobriner K, Rhoads CP, & Lavin GI (1939) Conversion of
1,2,5,6-dibenzanthracene by rabbits, rats and mice. Significance in
carcinogenesis of this conversion. Proc Soc Exp Biol Med, 41: 67-69.
Dock L, Waern F, Martinez M, Grover PL, & Jernstrom B (1986) Studies
on the further activation of benz [a]pyrene diol epoxides by rat
liver microsomes and nuclei. Chem-Biol Interactions, 58: 301-318.
Doll R, Vessey MP, Buckley AR, Fear EC, Gammon EJ, Gunn W, Hughes GO,
Lee K, & Norman-Smith B (1972) Mortality of gasworkers. Final report
of a prospective study. Br J Med, 29: 394-406.
Dong MW & Greenberg A (1988) Liquid chromatographic analysis of
polynuclear aromatic hydrocarbons with diode array detection. J Liq
Chromatogr, 11: 1887-1905.
van Dongen (1987) [Leaching characteristics of pre-impregnated wood
during storage, Parts 1 and 2.]. Zeist, Organisation for Applied
Science (TNO), 170 pp (Report No. HI 87.1178) (in Dutch).
Donkin P, Widdows J, Evans SV, Worrall CM, & Carr M (1989)
Quantitative structure-activity relationships for the effect of
hydrophobic organic chemicals on rate of feeding by mussels Mytilus
edulis. Aquat Toxicol, 14: 277-293.
Doremire ME, Harmon GE, & Pratt DE (1979) 3,4-Benzopyrene in charcoal
grilled meats. J Food Sci, 44: 622-623.
DouAbdul AAZ, Abaychi JK, Al-Edanee TE, Ghani AA, & Al-Saad HT (1987)
Polynuclear aromatic hydrocarbons (PAHs) in fish from the Arabian
Gulf. Bull Environ Contam Toxicol, 38: 546-552.
Dragoescu C & Friedlander S (1989) Dynamics of the aerosol products of
incomplete combustion in urban atmospheres. Aerosol Sci Technol, 10:
249-257.
Draudt HN (1963) The meat smoking process: A review. Food Technol, 17:
85-90.
Dreier F, Siles S, Buchs M, Gülacar FO, & Buchs A (1985) [Polycyclic
aromatic hydrocarbons in sediments of Léman Lake.] Arch Sci Genève,
38: 215-224 (in French).
Druckrey H & Schmähl D (1955) [Carcinogenic effect of anthracene.]
Naturwissen-schaften, 42: 159-160 (in German).
Dunkel VD, Pienta RJ, Sivak A, & Traul KA (1981) Comparative
neoplastic transformation responses of Balb/3T3 cells, Syrian hamster
embryo cells, and Rauscher murine leukemia virus-infected Fischer 344
rat embryo cells to chemical carcinogens. J Natl Cancer Inst, 67:
1303-1315.
Dunkel VC, Zeiger E, Brusick D, McCoy E, McGregor D, Mortelmans K,
Rosenkranz HS, & Simmon VF (1984) Reproducibility of microbial
mutagenicity assays: 1. Tests with Salmonella typhimurium and
Escherichia coli using a standardized protocol. Environ Mutag, 6
(suppl 2): 1-39.
Dunkel VC, Schechtmann LM, Tu AS, Sivak A, Lubet RA, & Cameron TP
(1988) Intralaboratory evaluation of the C3H/10T1/2 cell
transformation assay. Environ Mol Mutag, 12: 21-31.
Dunlap CE & Warren S (1943) The carcinogenic activity of some new
derivatives of aromatic hydrocarbons. I. Compounds related to
chrysene. Cancer Res, 3: 606-607.
Dunn BP & Douglas GR (1991) DNA adducts in cells treated with
1-methylphenanthrene. Proc Am Assoc Cancer Res, 32: 93.
Duus U, Ahlborn J, & Andersson J (1994) Toxic oil in rubber tyres.
Solna, National Chemicals Inspectorate, 4 pp.
Eadie B, Landrum PF, & Faust W (1982) Polycyclic aromatic hydrocarbons
in sediments pore water and the amphipod
Pontoporeia hoyi from Lake Michigan, USA. Chemosphere, 11: 847-858.
Eadie BJ, Morehead NR, & Landrum PF (1990) Three-phase partitioning of
hydrophobic organic compounds in Great Lakes waters. Chemosphere, 20:
161-178.
Eastmond D, Booth G, & Lee M (1984) Toxicity accumulation and
elimination of polycyclic aromatic sulfur heterocycles in
Daphnia magna. Arch Environ Contam Toxicol, 13: 105-112.
Edes TE, Gysbers DG, Buckley CS, & Thornton WH Jr (1991) Exposure to
the carcinogen benzopyrene depletes tissue vitamin A: b-Carotene
prevents depletion. Nutr Cancer, 15: 159-166.
Edmisten G & Bantle J (1982) Use of Xenopus laevis larvae in 96
hour, flow-through toxicity tests with naphthalene. Bull Environ
Contam Toxicol, 29: 392-399.
Edsall CC (1991) Acute toxicities to larval rainbow trout of
representative compounds detected in Great Lakes fish. Bull Environ
Contam Toxicol, 46: 173-178.
Edwards NT (1983) Polycyclic hydrocarbons (PAHs) in the terrestrial
environment. A review. J Environ Qual, 12: 427-441.
Edwards NT (1986) Uptake translocation and metabolism of anthracene in
bush bean Phaseolus vulgaris. Environ Toxicol Chem, 5: 659-666.
Egan-Baum E, Miller BA, & Waxweiler RJ (1981) Lung cancer and other
mortality patterns among foundrymen. Scand J Work Environ Health, 7
(suppl 4): 147-155.
Ehrlich HW & Beevers CA (1956) The crystal structure of
2:13-benzfluoranthene. Acta Crystal, 9: 602-606.
Eiceman GA, Clement RE, & Karasek FW (1979) Analysis of fly ash from
municipal incinerators for trace organic compounds. Anal Chem, 51:
2343-2350.
Eisenhut W, Friedrich F, & Reinke M (1990) Coking plant environment in
West-Germany. Coke Making Int, 1: 74-77.
Eisenstadt E & Gold A (1978) Cyclopenta [c,d]pyrene: A highly
mutagenic polycyclic aromatic hydrocarbon. Proc Natl Acad Sci USA, 75:
1667-1669.
El-Bayoumy K, Amin S, & Hecht SS (1986) Effects of 6-nitro
substitution on 5-methylchrysene tumorigenicity, mutagenicity and
metabolism. Carcinogenesis, 7: 673-676.
Elgjo K (1968) Growth kinetics of the mouse epidermis after a single
application of 3,4-benzopyrene, croton oil, or 1,2-benzopyrene. Acta
Pathol Microbiol Scand, 73: 183-190.
Eling TE & Curtis JF (1992) Xenobiotic metabolism by prostaglandin H
synthase. Pharmacol Ther, 53: 261-273.
Eling T, Curtis J, Battista J, & Marnett LJ (1986) Oxidation of
(+)-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene by mouse keratinocytes:
Evidence for peroxyl radical- and monoxygenase-dependent metabolism.
Carcinogenesis, 12: 1957-1963.
Ellwardt PC (1976) [German Society for Moor and Peat. Determination of
polycyclic aromatic hydrocarbons with and without carcinogenic effect
in peat in comparison with their occurrence in soils and composts].
Ber Dtsch Ges Moor Torfkunde Hanover, 6: 135-144 (in German).
Elmets CA, Khan WA, Klemme JC, & Mukhtar H (1988) Impaired
immunological surveillance by 7,12-dimethylbenz [a]anthracene
augments its skin tumorigenicity in C3H mice. Biochem Biophys Res
Commun, 151: 148-152.
Elovaara E, Heikkilä P, Pyy L, Muranen P, & Riihimäki V (1995)
Significance of dermal and respiratory uptake in creosote workers:
Exposure to polycyclic aromatic hydrocarbons and urinary excretion of
1-hydroxypyrene. Occup Environ Med, 52: 196-203.
Emerole GO, Uwaifo AO, Thabarew MI, & Bababunmi EA (1982) The presence
of aflatoxin and some polycyclic aromatic hydrocarbons in human foods.
Cancer Lett, 15: 123-129.
Emura M, Richter-Reichhelm HB, Schneider P, & Mohr U (1980)
Sensitivity of Syrian golden hamster fetal lung cells to
benzo [a]pyrene and other polycyclic hydrocarbons in vitro.
Toxicology, 17: 149-155.
Emura M, Mohr U, Riebe M, Aufderheide M, & Dungworth DL (1987)
Predispostion of cloned fetal hamster lung epithelial cells to
transformation by a precarcinogen, benzo(a)pyrene, using growth
hormone supplementation and collagen gel substratum. Cancer Res, 47:
1155-1160.
Engelbreth-Holm J (1941) Acceleration of the development of mammary
carcinomas in mice by methylcholanthrene. Cancer Res, 1: 109-112.
Engewald W, Knobloch T, & Efer J (1993) [Volatile organic compounds in
emissions from brown-coal-fired residential stoves.] Z Umweltchem
Okotox, 5: 303-308 (in German).
Engholm G, Englund A, & Linder B (1991) Mortality and cancer incidence
in Swedish road paving asphalt workers and roofers. Eur Asphalt Mag,
1: 62-67.
Environment Agency, Japan (1993) Chemicals in the environment: Summary
of the result of environmental survey for chemical substances 1974 to
1991. Tokyo, 5 pp.
Environment Canada (1994) Canadian Environmental Protection Act.
Priority substances list assessment report: Polycyclic aromatic
hydrocarbons. Ottawa, Ministry of Supply and Services, 61 pp.
Epler JL, Rao TK, & Guerin MR (1979) Evaluation of feasibility of
mutagenic testing of shale oil products and effluents. Environ Health
Perspectives, 30: 179-184.
Epstein S (1968) Chemical mutagens in the human environment. Nature,
219: 385-387.
Ermer M (1970) [Studies with carcinogens in short-lived fish species.]
Zool Anz, 184: 175-193 (in German).
Ernst W, Eder G, Goerke H, Weber K, Weigelt S, & Weigelt V (1986)
[Organic environmental chemicals in German estuaries and coastal
waters: Origin, biotransfer, degradation.] Bremerhaven, Federal
Ministry for Research and Technology, Institute for Oceanography, 78
pp (Report No. M/86-001) (in German).
European Aluminium Association, Environment, Health and Safety
Secretariat (1990) Polycyclic aromatic hydrocarbons. Oslo, 51 pp
(Report of the Working Group on PAH No. ALUM./TEKN.-HYG.UTV No.
80-90).
European Economic Community (1967) Council Directive 67/548/EEC, of 27
June 1967 on the approximation of laws, regulations and administrative
provisions relating to the classification, packaging and labelling of
dangerous substances. Off J Eur Communities, L196: 234-237
European Economic Community (1980) Council Directive 80/778/EEC, of 15
July 1980, relating to the quality of water intended for human
consumption. Off J Eur Communities, L229: 11-29.
European Economic Community (1983) Council Directive of 16 June 1983
amending Council Directive 70/220/EEC on the approximation of the laws
of the Member States relating to measures to be taken against air
pollution by gases from positive ignition engines of motor vehicles
(83/351/EEC). Off J Eur Communities, L 197(I): 1.
European Economic Community (1988) Council Directive, of 22 June 1988,
on the approximation of the laws of member states relating to
flavourings for use in foodstuffs and to source materials for their
production (88/388/EEC). Off J Eur Communities, L184: 61-65.
European Economic Community (1991) Council Directive, of 29 May 1991,
on establishing indicative values by implementing Council Directive
80/1107/EEC on the protection of workers from the risks related to
exposure to chemical, physical and biological agents at work
(91/322/EEC). Off J Eur Communities, L177: 22-24.
European Economic Community (1994a) Council Directive 94/69/EC, of 19
December 1994 adapting to technical progress for the twenty-first time
Council Directive 67/548/EEC on the approximation of laws, regulations
and administrative provisions relating to the classification,
packaging and labelling of dangerous substances. Off J Eur
Communities, L381: 1-241.
European Economic Community (1994b) Council Directive 94/60/EC, of 25
November 1994 updating technical norms for concentration limits, and
attention and alarm levels for atmospheric pollution in urban areas
and provisions for certain pollutants referred to in the ministerial
decree of 15 April 1994. Off J Eur Communities, L290: 1-32.
Evans CH & DiPaolo JA (1975) Neoplastic transformation of guinea pig
fetal cells in culture induced by chemical carcinogens. Cancer Res,
35: 1035-1044.
Evans MS & Landrum PF (1989) Toxicokinetics of DDE, benzo [a]pyrene,
and 2,4,5,2',4',5'-hexachlorobiphenyl in Pontoporeia hoyi and Mysis
relicta. J Great Lakes Res, 15: 589-600.
Evans EL & Mitchell AD (1981) Effects of 20 coded chemicals on sister
chromatid exchange frequencies in cultured Chinese hamster cells. In:
De Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, pp 538-550 (Progress in Mutation
Research, Volume 1).
Fabacher DL, Besser JM, Schmitt CJ, Harshbarger JC, Peterman PH, &
Lebo JA (1991) Contaminated sediments from tributaries of the Great
Lakes: Chemical characterization and carcinogenic effects in medaka
(Oryzias latipes). Arch Environ Contam Toxicol, 21: 17-34.
Fahmy OG & Fahmy MJ (1973) Oxidative activiation of
benz [a]anthrazene and methylated derivatives in mutagenesis and
carcinogenesis. Cancer Res, 33: 2354-2361.
Fahmy MJ & Fahmy OG (1980) Altered control of gene acitivity in the
soma by carcinogens. Mutat Res, 72: 165-172.
Falk HL, Kotin P, & Markul I (1958) The disappearance of carcinogens
from soot in human lungs. Cancer, 11: 482-489.
Falk HL, Kotin P, Lee SS, & Nathan A (1962) Intermediary metabolism of
benzo [a]pyrene in the rat. J Natl Cancer Inst, 28: 699-724.
Fallon ME & Horvath FJ (1985) Preliminary assessment of contaminants
in soft sediments of the Detroit River. J Great Lakes Res, 11: 373-
378.
Fanburg SJ (1940) Exfoliative dermatitis due to naphthalene: Report of
an eruption resembling Mycosis fungoides. Arch Dermatol Syphilol,
42: 53-58.
Faoro RB & Manning JA (1981) Trends in benzo(a)pyrene, 1966-77. J Air
Pollut Control Assoc, 31: 62-64.
Farrington JW (1991) Biochemical processes governing exposure and
uptake of organic pollutant compounds in aquatic organisms. Environ
Health Perspectives, 90: 75-84.
Fazio T (1990) 48. Food additives. Indirect: Polycyclic aromatic
hydrocarbons and benzo [a]pyrene in food. Spectrophotometric method
(No. 973.30). In: Helrich K ed. Official methods of analysis of the
Association of Official Analytical Chemists (AOAC), 15th ed.
Arlington, Virginia, Association of Official Analytical Chemists,
Volume 2, pp 1176-1178.
Fedorak PM, Foght JM, & Westlake DWS (1982) A method for monitoring
mineralization of 14C-labeled compounds in aqueous samples. Water
Res, 16: 1285-1290.
Fendinger NJ, Radway JC, Tuttle JH, & Means JC (1989) Characterization
of organic material leached from coal by simulated rainfall. Environ
Sci Technol, 23: 170-177.
Feron VJ, de Jong D, & Emmelot P (1973) Dose-response correlation for
the induction of respiratory-tract tumors in Syrian golden hamsters by
intratracheal instillations of benzo(a)pyrene. Eur J Cancer, 9: 387-
390.
Ferraro S, Lee I, Ozretich R, & Specht D (1990) Predicting
bioaccumulation potential. A test of a fugacity-based model. Arch
Environ Contam Toxicol, 3: 386-394.
Ferreira MJ Jr, Buchet JP, Burrion JB, Moro J, Cupers L, Delavignette
JP, Jacques J, & Lauwerys R (1994a) Determinants of urinary
thioethers, D-glucaric acid and mutagenicity after exposure to
polycyclic aromatic hydrocarbons assessed by air monitoring and
measurement of 1-hydroxypyrene in urine: A cross-sectional study in
workers of coke and graphite-electrode-producing plants. Int Arch
Occup Environ Health, 65: 329-338.
Ferreira M Jr, Tas S, Dell'Omo M, Goormans G, Buchet JP, & Lauwerys R
(1994b) Determinants of benzo(a)pyrene diol epoxide adducts to
haemoglobin in workers exposed to polycyclic aromatic hydrocarbons.
Occup Environ Med, 51: 451-455.
Feunekes FDJR, Jongeneleen FJ, van der Laan H, & Schoonhof FHG (1997)
Uptake of polycyclic aromatic hydrocarbons among trainers in a
fire-fighting training facility. Am Ind Hyg Assoc J, 58: 23-28.
Finger SE, Little EF, Henry MG, Fairchild JF, & Boyle TP (1985)
Comparison of laboratory and field assessment of fluorene. Part I:
Effects of fluorene on the survival, growth, reproduction, and
behavior of aquatic organisms in laboratory tests. In: Boyle TP ed.
Validation and predictability of laboratory methods for assessing the
fate and effects of contaminants in aquatic ecosystems. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 120-133
(ASTM STP No. 865).
Flesher JW & Myers SR (1990) Bioalkylation of benz(a)anthracene as a
biochemical probe for carcinogenic activity. Drug Metab Disposition,
18: 163-167.
Flesher JW & Myers SR (1991) Rules of molecular geometry for
predicting carcinogenic activity of unsubstituted polynuclear aromatic
hydrocarbons. Teratog Carcinog Mutag, 11: 41-54.
Florin I, Rutberg L, Curvall M, & Enzell CR (1980) Screening of
tobacco smoke constituents for mutagenicity using the Ames' test.
Toxicology, 18: 219-232.
Flury F & Zernik F (1935) [List of the toxic and lethal doses for the
most common poisons in experimental animals.] In: Abderhalden E ed.
[Handbook of biological methods.] Berlin, Urban & Schwarzenberg, pp
12289-14222 (in German).
Foran JA, Holst LL, & Giesy JP (1991) Effects of photoenhanced
toxicity of anthracene on ecological and genetic fitness of Daphnia
magna: A reappraisal. Environ Toxicol Chem, 10: 425-427.
Forbes PD, Davies RE, & Urbach F (1976) Phototoxicity and
photocarcinogenesis: Comparative effects of anthracene and
8-methoxypsoralen in the skin of mice. Food Cosmet Toxicol, 14: 303-
306.
Foster GD, Baksi SM & Means JC (1987) Bioaccumulation of trace organic
contaminants from sediment by Baltic clams (Macoma
balthica) and soft-shell clams (Mya arenaria). Environ Toxicol
Chem, 6: 969-976.
Fox MA & Staley SW (1976) Determination of polycyclic aromatic
hydrocarbons in atmospheric particulate matter by high pressure liquid
chromatography coupled with fluorescence techniques. Anal Chem, 48:
992-998.
Fox CH, Selkirk JK, Price FM, Croy RG, Sandord KK, & Cottler-Fox M
(1975) Metabolism of benzo [a]pyrene by human epithelial cells
in vitro. Cancer Res, 35: 3551-3557.
Fox M, Lutz H-J, Gassman E, & Yoshikawa S (1988) Chemical economics
handbook (CEH): Product review for naphthalene. Menlo Park,
California, SRI International, 31 pp.
Franck HG & Stadelhofer JW (1987) [Industrial aromatic chemistry. Raw
products, processes, products.] Berlin, Springer-Verlag, pp 308-380
(in German).
Frank AP, Landrum PF, & Eadie BJ (1986) Polycyclic aromatic
hydrocarbon: Rates of uptake, depuration, and biotransformation by
Lake Michigan (Stylodrilus heringianus). Chemosphere, 15: 317-330.
Freeman AE, Weisburger EK, Weisburger JH, Wolford RG, Maryak JM &
Huebner RJ (1973) Transformation of cell cultures as an indication of
the carcinogenic potential of chemicals. J Natl Cancer Inst, 51: 799-
807.
Freitag DL, Ballhorn L, Geyer H, & Korte F (1985) Environmental hazard
profile of organic chemicals. An experimental method for the
assessment of the behaviour of organic chemicals in the ecosphere by
means of simple laboratory tests with 14C labelled chemicals.
Chemosphere, 14: 1589-1616.
Fritz W (1971) [The extent of sources of contamination of our food
with carcinogenic hydrocarbons.] Ernährungsforschung, 16: 547-557 (in
German).
Fritz W (1972) [Contamination of food with carcinogenic hydrocarbons
during processing and cooking.] Arch Geschwulstforsch, 40: 81-90 (in
German).
Fritz W (1983) [Analysis and assessment of carcinogenic polycyclic
aromatic hydrocarbons from the food hygiene. Toxicology point of view.
A review]. Nahrung, 27: 965-973 (in German).
Frölich A & Würgler FE (1990) Drosophila wing-spot test: Improved
detectability of genotoxicity of polycyclic aromatic hydrocarbons.
Mutat Res, 234: 71-80.
Frumin G, Chuiko G, Pavlov D, & Menzykova O (1992) New rapid method to
evaluate the median effect concentrations of xenobiotics in
hydrobionts. Bull Environ Contam Toxicol, 49: 361-367.
Fujikawa K, Fort FL, Samejima K, & Sakamoto Y (1993) Gentoxic potency
in Drosophila melanogaster of selected aromatic amines and
polycyclic hydrocarbons as assayed in the DNA repair test. Mutat Res,
290: 175-182.
Fukuda K, Inagaki Y, Maruyama T, Kojima HI, & Yoshida T (1988) On the
photolysis of alkylated naphthalenes in aquatic systems. Chemosphere,
17: 651-659.
Funcke W, König J, Henze W, & Umland F (1986) Determination of
polycyclic aromatic hydrocarbons in mainstream cigarette smoke with
respect to particle size. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemistry, characterization and carcinogenesis.
Columbus, Ohio, Battelle Press, pp 333-341.
Funcke W, König J, & Balfanz E (1988) Determination of polycyclic
aromatic hydrocarbons in flue gases from coal-fired power plants. In:
Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons: A decade of
progress. Columbus, Ohio, Battelle Press, pp 277-284.
Furlong ET, Carter DS, & Hites, RA (1988) Organic contaminants in
sediments from the Trenton Channel of the Detroit River, Michigan. J
Great Lakes Res, 14: 489-501.
Furst A, Kolff B, & Dempsey DA (1979) Pulmonary tumor induction: Three
hydrocarbons compared. Proc West Pharmacol Soc, 22: 269-271.
Gaines TB (1969) Acute toxicity of pesticides. Toxicol Appl Pharmacol,
14: 515-534.
Gala WR & Giesy JP (1992) Photo-induced toxity of anthracene to the
green algae, Selenastrum capricornutum. Arch Environ Contam Toxicol,
23: 316-323.
Gamper HB, Tung ASC, Straub K, Bartholomew JC, & Calvin M (1977) DNA
strand scission by benzo [a]pyrene diol epoxides. Science, 197: 671-
674.
Gamper HB, Bartholomew JC, & Calvin M (1980) Mechanism of
benzo(a)pyrene diol epoxide induced deoxyribonucleic acid strand
scission. Biochemistry, 19: 3948-3956.
Gardiner K, Hale KA, Calvert IA, Rice D, & Harrington JM (1992) The
suitability of the urinary metabolite 1-hydroxypyrene as an index of
polynuclear aromatic hydrocaarbon bioavailability from workers exposed
to carbon black. Ann Occup Hyg, 36: 681-688.
Garg A, Beach A, & Gupta RC (1991) DNA-reactive metabolites (DRM)
detected in the serum of benzo [a]pyrene (BP)-treated rodents by
32P-postlabeling. Proc Am Assoc Cancer Res, 32: 122.
Garrigues P & Ewald M (1987) High resolution emission spectroscopy
(Shpol'skii effect): A new analytical technique for the analysis of
polycyclic aromatic hydrocarbons (PAH) in the environmental samples.
Chemosphere, 16: 485-494.
Garrigues P, Soclo HH, Marniesse MP, & Ewald M (1987) Origin of
polycyclic aromatic hydrocarbons (PAH) in recent sediments from the
continental shelf of the 'Golfe de Gascogne' (Atlantic Ocean) and in
the Gironde estuary. Int J Environ Anal Chem, 28: 121-131.
Gauthier TD, Shane EC, Guerin WF, Seltz WR, & Grant CL (1986)
Fluorescence quenching method for determining equilibrium constants
for polycyclic aromatic hydrocarbons binding to dissolved humic
materials. Environ Sci Technol, 20: 1162-1166.
Gaydos RM (1981) Naphthalene. In: Kirk-Othmer encyclopedia of chemical
technology, 3rd ed. New York, John Wiley & Sons, Volume 15, pp 698-
719.
Geacintov NE (1988) Mechanisms of reaction of polycyclic aromatic
epoxide derivatives with nucleic acids. In: Yang SK & Silverman BD ed.
Polycyclic aromatic hydrocarbon carcinogenesis: Structure-activity
relationships. Boca Raton, Florida, CRC Press, Volume 2, pp 181-206.
Geahchan A, Le Gren I, Chambon P, & Chambon R (1991) Improved method
for determination of polynuclear aromatic hydrocarbons in
pharmacopoeial paraffin and mineral oils. J Assoc Off Anal Chem, 74:
968-973.
Geddie JE, Amin S, Huie K, & Hecht SS (1987) Formation and
tumorigenicity of benzo [b]fluoranthene metabolites in mouse
epidermis. Carcinogenesis, 8: 1579-1584.
Gehly EB, Landolph JR, Heidelberger C, Nagasawa H, & Little JB (1982)
Induction of cytotoxicity, mutation, cytogenetic changes and
neoplastic transformation by benzo(a)pyrene and derivatives in C3H/10T
1/2 clone 8 mouse fibroblasts. Cancer Res, 42: 1866-1875.
Geiger JG Jr & Buikema AL Jr (1981) Oxygen consumption and filtering
rate of Daphnia pulex after exposure to water-soluble fractions of
naphthalene, phenanthrene, No. 2 fuel oil, and coal-tar creosote.
Environ Contam Toxicol, 27: 783-789.
Geiger JG Jr & Buikema AL Jr (1982) Hydrocarbons depress growth and
reproduction of Daphnia pulex (Cladocera). J Fish Aquat Sci, 39:
830-836.
Geiger DL, Northcott CE, Call DJ, & Brooke LT ed. (1985) Acute
toxicities of organic chemicals to fathead minnows Pimephales
promelas. Superior, Wisconsin, University of Wisconsin, Center for
Lake Superior Environmental Studies, Volume 2, pp 211-212.
Gelboin HV & Ts'o POP (1978) Polycyclic hydrocarbons and cancer.
Volume 2: Metabolism and cell biology. New York, Academic Press, 441
pp.
Gendimenico GJ & Kochevar IE (1984) Degranulation of mast cells and
inhibition of the response to secretory agents by phototoxic and
ultraviolet radiation. Toxicol Appl Pharmacol, 76: 374-382
Generoso WM, Cain KT, Hellwig CD, & Cacheiro NLA (1982) Lack of
association between induction of dominant-lethal mutations and
induction of heritable translocations with benzo(a)pyrene in
postmeiotic germ cells of male mice. Mutat Res, 94: 155-163.
Genevois C, Brandt HCA, Bartsch H, Wild CP & Castegnaro M (1995)
Formation of DNA adducts in skin, lung and lymphocytes after skin
painting of rats with undiluted coaltar and bitumen vapor/particulates
condensates. In: Fifteenth international symposium on polycyclic
aromatic compounds: Chemistry, biology and environmental impact,
Belgirate, Italy, 19-22 September 1995. Ispra, Joint Research Centre
European Commission, pp 54-55.
Gensler HL (1988) Enhancement of chemical carcinogenesis in mice by
systemic effects of ultraviolet irradiation. Cancer Res, 48: 620-623.
Gerarde HW (1960) Toxicology and biochemistry of aromatic
hydrocarbons. In: Browning E ed. Elsevier monographs on toxic agents.
Amsterdam, Elsevier Publishing Co., pp 240-321.
Gerhart E & Carlson R (1978) Hepatic mixed-function oxidase activity
in rainbow trout exposed to several polycyclic aromatic compounds.
Environ Res, 17: 284-295.
German Federal Department for Worker Safety (1989) [23.TRK-Wert for
benzo(a)pyrene.] In:[Bundesarbeitsblatt 10.] Dortmund, pp 58-61 (in
German).
German Federal Office for Sea Navigation and Hydrography (1993)
[Monitoring of the sea. Report for the year 1990. Part II: Data.]
Hamburg, pp 79-94 (in German).
German Ministry of Environment (1979) [Air quality criteria for
selected polycyclic aromatic hydrocarbons. PAH as an environmental
carcinogen.] Berlin, Erich Schmidt Verlag, 270 pp (Report No.
UBA-1/79) (in German).
German Ministry of Environment (1993) [Environmental sample bank.
Annual report 1991.] Berlin, 139 pp (Report No. UBA-7/93) (in German).
German Research Commission (1991) Polycyclic aromatic hydrocarbons
(PAH) (particle-bound). In: Kettrup A ed. Commission for the
investigation of health hazards of chemical compounds in the work air,
working group on analytical chemistry: Analyses of hazardous
substances in air. Weinheim, VCH Publishers, Volume 1, pp 41-52.
German Society for Mineral-oil and Coal Chemistry (1984)
[Investigations into the contents of wastewater from oil refineries.]
Hamburg, 95 pp (Report No. 283) (in German).
Gershbein LL (1975) Liver regeneration as influenced by the structure
of aromatic and heterocyclic compounds. Res Commun Chem Pathol
Pharmacol, 11: 445-466.
Gerstle RW, Cuffe ST, Orning AA, & Schwartz CH (1965) Air pollution
emissions from coal-fired power plants. Report No. 2. J Air Pollut
Control Assoc, 15: 59-64.
Geyer H, Politzki G, & Freitag D (1984) Prediction of ecotoxicological
behaviour of chemicals: Relationship between n-octonal/water
partition coefficient and bioaccumulation of organic chemicals by
Alga chlorella. Chemosphere, 13: 269-284.
Gharrett JA & Rice SD (1987) Influence of simulated tidal cycles on
aromatic hudrocarbon uptake and elimination by the shore crab
Hemigrapsus nudus. Mar Biol, 95: 365-370.
Gibbs GW (1985) Mortality of aluminum reduction plant workers, 1950
through 1977. J Occup Med, 27: 761-770.
Gibbs GW & Horowitz I (1979) Lung cancer mortality in aluminum
reduction plant workers. J Occup Med, 21: 347-353.
Gibson TL (1982) Nitro derivates of polynuclear aromatic hydrocarbons
in airborne and source particulate matter. Atmos Environ, 16: 2037-
2040.
Gibson ES, Martin RH, & Lockington JN (1977) Lung cancer mortality in
a steel foundry. J Occup Med, 19: 807-812.
Giger W & Schaffner C (1978) Determination of polycyclic aromatic
hydrocarbons in the environment by glass capillary gas chromatography.
Anal Chem, 50: 243-249.
Gill RD, Butterworth BE, Nettikumara AN, & Digiovanni J (1991)
Relationship between DNA adduct formation and unscheduled DNA
synthesis (UDS) in cultured mouse epidermal keratinocytes. Environ Mol
Mutag, 18: 200-206.
Ginsberg GL & Atherholt TB (1989) Transport of DNA-adducting
metabolites in mouse serum following benzo(a)pyrene administration.
Carcinogenesis, 10: 673-679.
Gjessing E, Lygren E, Berglind L, Gulbrandsen T, & Skaane R (1984)
Effect of highway runoff on lake water quality. Sci Total Environ, 33:
245-257.
Glatt H, Bücker M, Platt KL, & Oesch F (1985) Host-mediated
mutagenicity experiments with benzo [a]pyrene and two of its
metabolites. Mutat Res, 156: 163-169.
Glatt H, Seidel A, Bochnitschek W, Marquardt H, Marquardt H, Hodgson
RM, Grover PL, & Oesch F (1986) Mutagenic and cell-transforming
activities of triol-epoxides as compared to other chrysene
metabolites. Cancer Res, 46: 4556-4565.
Glatt H, Seidel A, Ribeiro O, Kirkhy C, Ilirom P, & Oesch F (1987)
Metabolic activation to a mutagen of
3-hydroxy- trans-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene, a
secondary metabolite of benzo [a]pyrene. Carcinogenesis, 8:
1621-1627.
Glatt H, Harvey RG, Phillips DH, Hewer A, & Grover PL (1989) Influence
of the alkyl substituent on mutagenicity and covalent DNA binding of
bay region diol-epoxides of 7-methyl- and 7-ethylbenz [a]anthracene
in Salmonella and V79 Chinese hamster cells. Cancer Res, 49: 1778-
1782.
Goddard KA, Schultz RJ, Stegeman JJ, Schultz RJ, & Stegemann JJ (1987)
Uptake, toxicity and distribution of benzo [a]pyrene and
monooxygenase induction in the top minnows Poeciliopsis monacha
and Poeciliopsis lucida. Drug Metab Disposition, 15: 449-455.
Göen T, Gündel J, Schaller KH, & Angerer J (1995) The elimination of
1-hydroxypyrene in the urine of the general population and workers
with different occupational exposures to PAH. Sci Total Environ, 163:
195-201.
Gold A & Eisenstadt E (1980) Metabolic activation of
cyclopenta(cd)pyrene to 3,4-epoxycyclopenta(cd)pyrene by rat liver
microsomes. Cancer Res, 40: 3940-3944.
Gold A, Nesnow S, Moore M, Garland H, Curtis G, Howard B, Graham D, &
Eisenstadt E (1980) Mutagenesis and morphological transformation of
mammalian cells by a nonbay-region polycyclic cyclopenta(cd)pyrene and
its 3,4-oxide. Cancer Res, 40: 4482-4484.
Goldschmidt BM, Katz C, & Van Duuren BL (1973) The cocarcinogenic
activity of noncarcinogenic aromatic hydrocarbons. Proc Am Assoc
Cancer Res, 14: 84.
Gollahon L (1991) Chromosomal damage to preimplantation mouse embryos
in vitro by naphthalene and aflatoxin B1. Diss Abstr Int B, 52: 694-
695.
Gollahon LS, Iyer P, Martin JE, & Irvin TR (1990) Chromosomal damage
to preimplantation embryos in vitro by naphthalene. Toxicologist,
10: 274.
Gonzalez FJ & Gelboin HV (1994) Role of human cytochromes P450 in the
metabolic activation of chemical carcinogens and toxins. Drug Metab
Rev, 26: 165-183.
Gonzalez-Vila FJ, Lopez JL, & Martin F (1988) Determination of
polynuclear aromatic compounds in composted municipal refuse and
compost-amended soils by a simple cleanup procedure. Biomed Environ
Mass Spectrom, 16: 423-425.
Gordon RJ (1976) Distribution of airborne polycyclic aromatic
hydrocarbons throughout Los Angeles. Environ Sci Technol, 10: 370-373.
Gordon RJ & Bryan RJ (1973) Patterns in airborne polynuclear
hydrocarbon concentrations at four Los Angeles sites. Environ Sci
Technol, 7: 1050-1053.
Gorelova ND & Cherepanova AI (1970) [Possibility of accumulation of
3,4-benzpyrene in tissues and organs of cows and calves and in milk
when this carcinogen is present in food.] Vopr Onkol, 16: 69-73 (in
Russian).
Gorelova ND, Dikun PP, Solinek VA, & Emshanova AV (1960) [Content of
3,4-benzpyrene in fish smoked by different methods.] Vopr Onkol, 6:
33-37 (in Russian).
Götz R (1984) [Investigations into leakage water from the refuse dump
Georgswerder in Hamburg. Results of analyses up to 1982 inclusive.]
Müll Abfall, 12: 349-356 (in German).
Grachev MA, Baram GI, Hodgher, TV, Gorshow AG, Vodiannikova NI, &
Bartz MP (1994) Determination of the concentration of PAHs in the
atmosphere of the south part of the Lake Baikal coast. Irkutsk,
Limnological Institute, Russian Academy of Sciences (unpublished
report).
Gräf W (1970) Levels of 3,4-benzopyrene in human organs of different
ages. Second communication. Arch Hyg, 154: 331-335.
Gräf W & Nowak W (1966) [Promotion of growth in lower and higher
plants by carcinogenic polycyclic aromatics.] Arch Hyg, 150: 513-528
(in German).
Gräf W, Eff H, & Schormair S (1975) Levels of carcinogenic, polycyclic
aromatic hydrocarbons in human and animal tissues. Third
communication. Zentralbl Bakteriol Hyg Abt Orig B, 161: 85-103.
Graffi A (1940a) [Cellular accumulation of carcinogenic hydrocarbons.]
Z Krebsforschung, 49: 477-495 (in German).
Graffi A (1940b) [Intracellular benzopyrene accumulation in living
normal and tumor cells.] Z Krebsforschung, 50: 196-219 (in German).
Graffi A (1940c) [Some observations on the aetiology of tumors, in
particular on the nature of the active agent in cell-free transferable
chicken tumours.] Z Krebsforschung, 50: 501-551 (in German).
Graffi A, Vlamynck E, Hoffmann F, & Schulz I (1953) [Studies on the
tumor producing effects of different chemicals in combination with
croton oil.] Arch Geschwulstforsch, 5: 110-126 (in German).
Grant G & Roe FJC (1963) The effect of phenanthrene on tumour
induction by 3,4-benzopyrene administered to newly born mice. Br J
Cancer, 17: 261-265.
Greb W, Strobel R & Röhrborn G (1980) Transformation of BHK 21/CL 13
cells by various polycyclic aromatic hydrocarbons using the method of
Styles. Toxicol Lett, 7: 143-148.
Greenberg A, Bozzelli JW, Cannova F, Forstner F, Giorgio P, Stout D, &
Yokoyama R (1981) Correlations between lead and coronene
concentrations at urban, suburban, and industrial sites in New Jersey.
Environ Sci Technol, 15: 566-570.
Greenberg A, Darack F, Harkov R, Lioy P, & Daisey J (1985) Polycyclic
aromatic hydrocarbons in New Jersey: A comparison of winter and summer
concentrations over a two-year period. Atmos Environ, 19: 1325-1339.
Greife AL & Warshawsky D (1993) Influence of the dose levels of
carcinogen ferric oxide on the metabolism of benzo(a)pyrene by
pulmonary alveolar macrophages in suspension culture. J Toxicol
Environ Health, 38: 399-417.
Greife A, Schoeny R, & Warshawsky D (1988) Effect of the cocarcinogen
ferric oxide on benzo(a)pyrene metabolism by hamster alveolar
macrophages. In: Cooke M & Dennis A ed. Polycyclic aromatic
hydrocarbons: Chemical and biological effects. Columbus, Ohio,
Battelle Press, pp 317-327.
Griesbaum K, Behr A, Biedenkapp D, Voges HW, Garbe D, Paetz C, Collin
G, Mayer D, & Höke H (1989) Hydrocarbons. In: Ullmann's encyclopedia
of industrial chemistry, 5th ed. Volume A13. Weinheim, Verlag Chemie,
pp 227-281.
Griest WH (1980) Multicomponent polycyclic aromatic hydrocarbon
analysis of inland water and sediment. Environ Sci Res, 16: 173-183.
Griest WH & Caton JE (1983) Extraction of polycyclic aromatic
hydrocarbons for quantitative analysis. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 95-148.
Griest WH, Maskarinec MP, Herbes SE, & Southworth GR (1981)
Multicomponent methods for the identification and quantification of
polycyclic aromatic hydrocarbons in the aqueous environment. In:
Jackson LP & Wright CC ed. Analysis of waters associated with
alternative fuel production. Philadelphia, Pennsylvania, American
Society for Testing and Materials, pp 167-178 (ASTM STP No. 720).
Grifoll M, Casellas M, Bayona JM, & Solanas AM (1992) Isolation and
characterization of a fluorene-degrading bacterium: Identification of
ring oxidation and ring fission products. Appl Environ Microbiol, 58:
2910-2917.
Grimmer G (1979) Sources and occurrence of polycyclic aromatic
hydrocarbons. In: Egan H, Castegnaro M, Bogovski P, Kunte H, & Walker
EA ed. Environmental carcinogens: Selected methods of analysis. Volume
3. Analysis of polycyclic hydrocarbons in environmental samples. Lyon,
International Agency for Research on Cancer, pp 31-54 (IARC Scientific
Publications No. 29).
Grimmer G (1980) [Analysis and comparison of PAH profile from
environmental samples.] Düsseldorf, VDI-Verlag, pp 39-50 (VDI Report
No. 358) (in German).
Grimmer G (1983a) Chemistry. In: Grimmer G ed. Environmental
carcinogens: Polycyclic aromatic hydrocarbons. Boca Raton, Florida,
CRC Press, pp 27-60.
Grimmer G (1983b) Profile analysis of polycyclic aromatic hydrocarbons
in air. In: Bjorseth A ed. Handbook of polycyclic aromatic
hydrocarbons. New York, Marcel Dekker, pp 149-191.
Grimmer G (1993) [Environmental-toxicological evaluation of PAH in
soil.] Altlasten Spectrum, 2: 85-92 (in German).
Grimmer G & Böhnke H (1975) Profile analysis of polycyclic aromatic
hydrocarbons and metal content in sediment layers of a lake. Cancer
Lett, 1: 75-84.
Grimmer G & Böhnke H (1978) The tumor producing effect of automobile
exhaust condensate and fractions thereof. Part 1: Chemical studies. J
Environ Pathol Toxicol, 1: 661-667.
Grimmer G & Böhnke H (1979a) Method 4: Gas chromatographic profile
analysis of polycyclic aromatic hydrocarbons in (i) high protein
foods, (ii) fats and vegetable oils and (iii) plants, soils and sewage
sludge. In: Egan H, Castegnaro M, Bogovski P, Kunte H, & Walker EA ed.
Environmental carcinogens: Selected methods of analysis. Volume 3.
Analysis of polycyclic aromatic hydrocarbons in environmental samples.
Lyon, International Agency for Research on Cancer, pp 163-173 (IARC
Scientific Publications No. 29).
Grimmer G & Böhnke H (1979b) Method 3: Gas chromatographic profile
analysis of polycyclic aromatic hydrocarbons in lubricating oil,
cutting oil and fuel. In: Egan H, Castegnaro M, Bogovski P, Kunte H, &
Walker EA ed. Environmental carcinogens: Selected methods of analysis.
Volume 3. Analysis of polycyclic aromatic hydrocarbons in
environmental samples. Lyon, International Agency for Research on
Cancer, pp 155-162 (IARC Scientific Publications No. 29).
Grimmer G & Düvel D (1970) [Investigation of biosynthetic formation of
polycyclic hydrocarbons in higher plants.] Z Naturforsch, 256: 1171-
1175 (in German with English summary).
Grimmer G & Hildebrandt A (1965) [The content of polyaromatic
hydrocarbons in various vegetables.] Dtsch Lebensm Rundsch, 61: 237-
239 (in German).
Grimmer G & Hildebrandt A (1975) Investigations on the carcinogenic
burden by air pollution in man. XIII. Assessment of the contribution
of passenger cars to air pollution by carcinogenic polycyclic aromatic
hydrocarbons. Zentralbl Bakteriol Hyg I Orig B, 161: 104-124.
Grimmer G, Jacob J, & Hildebrandt A (1972) [Hydrocarbons in the human
environment. 9. Content of polycyclic hydrocarbons in Icelandic soil.]
Z Krebsforsch, 78: 65-72 (in German).
Grimmer G, Hildebrandt A, & Böhnke H (1979) Method 2: Gas
chromatographic profile analysis of polycyclic aromatic hydrocarbons
in automobile exhaust gas condensate. In: Egan H, Castegnaro M,
Bogovski P, Kunte H, & Walker EA ed. Environmental carcinogens:
Selected methods of analysis. Volume 3. Analysis of polycyclic
aromatic hydrocarbons in environmental samples. Lyon, International
Agency for Research on Cancer, pp 141-154 (IARC Scientific
Publications No. 29).
Grimmer G, Hilge G, & Niemitz W (1980) [Comparison of the profile of
polycyclic aromatic hydrocarbons of sewage sludge samples from 25
filter plants.] Vom Wasser, 54: 255-272 ( in German).
Grimmer G, Jacob J, & Naujack KW (1981a) Profile of the polycyclic
aromatic hydrocarbons from lubricating oils. Inventory by GCGC/MS. PAH
in environmental materials, Part 1. Fresenius Z Anal Chem, 306: 347-
355.
Grimmer G, Schneider D, & Dettbarn G (1981b) [The load of different
rivers in the Federal Republic of Germany by PAH (PAH-profiles of
water surface)]. Vom Wasser, 56: 131-144 (in German).
Grimmer G, Naujack K-W, & Schneider D (1981c) Comparison of the
profiles of polycyclic aromatic hydrocarbons in different areas of a
city by glass-capillary-gas chromatography in the nanogram range. Int
J Environ Chem, 10: 265-276.
Grimmer G, Naujack K-W, & Schneider D (1982) Profile analysis of
polycyclic aromatic hydrocarbons by glass capillary gas chromatography
in atmospheric suspended particulate matter in the nanogram range
collecting 10 m3 of air. Fresenius Z Anal Chem, 311: 475-484.
Grimmer G, Jacob J, & Naujack KW (1983) Profile of the polycyclic
aromatic compounds from crude oils. Inventory by GCGC/MS. PAH in
environmental materials, part 3. Fresenius Z Anal Chem, 314: 29-36.
Grimmer G, Brune H, Deutsch-Wenzel R, Dettbarn G, & Misfeld J (1984)
Contribution of polycyclic aromatic hydrocarbons to the carcinogenic
impact of gasoline engine exhaust condensate evaluated by implantation
into the lungs of rats. J Natl Cancer Inst, 72: 733-739.
Grimmer G, Jacob J, Dettbarn G, & Naujack KW (1985) Determination of
polycyclic aromatic hydrocarbons, azaarenes, and thiaarenes emitted
from coal-fired residential furnaces by gas chromatography/mass
spectrometry. Fresenius Z Anal Chem, 322: 595-602.
Grimmer G, Naujack KW, & Dettbarn G (1987) Gas chromatographic
determination of polycyclic aromatic hydrocarbons, aza-arenes,
aromatic amines in the particle and vapor phase of mainstream and
sidestream smoke of cigarettes. Toxicol Lett, 35: 117-124.
Grimmer G, Jacob J, Dettbarn G, & Naujack K-W (1988a) Effect of the
pH-value of diesel exhaust on the amount of filter-collected
nitro-PAH. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: A decade of progress. Columbus, Ohio, Battelle Press,
pp 341-351.
Grimmer G, Brune H, Dettbarn G, Heinrich U, Jacob J, Mohtashamipur E,
Norpoth K, Pott F, & Wenzel-Hartung R (1988b) Urinary and faecal
excretion of chrysene and chrysene metabolites by rats after oral,
intraperitoneal, intratracheal or intrapulmonary application. Arch
Toxicol, 62: 401-405.
Grimmer G, Brune H, Dettbarn G, Naujack K-W, Mohr U, & Wenzel-Hartung
R (1988c) Contribution of polycyclic aromatic compounds to the
carcinogenicity of sidestream smoke of cigarettes evaluated by
implantation into lungs of rats. Cancer Lett, 43: 173-177.
Grimmer G, Brune H, Dettbarn G, Jacob J, Mohtashamipur E, Norpoth K, &
Wenze-Hartung R (1991a) Urinary and fecal excretion of phenanthrene
and phenanthrols by rats following oral, intraperitoneal, or
intrapulmonary application. Polycyclic Aromat Compd, 2: 39-47.
Grimmer G, Brune H, Dettbarn G, Jacob J, Misfeld J, Mohr U, Naujack
K-W, Timm J, & Wenzel-Hartung R (1991b) Relevance of polycyclic
aromatic hydrocarbons as environmental carcinogens. Fresenius J Anal
Chem, 339: 792-795.
Grimmer G, Dettbarn G, & Naujack KW (1991c) A method for the
determination of phenanthrene and five isomeric hydroxyphenanthrenes
in urine of man and rat. In: Cooke M, Loening K, & Merritt J ed.
Polynuclear aromatic hydrocarbons: Measurements, means, and
metabolism. Columbus, Ohio, Battelle Press, pp 389-403.
Grimmer G, Dettbarn G, & Jacob J (1993) Biomonitoring of polycyclic
aromatic hydrocarbons in highly exposed coke plant workers by
measurement of urinary phenanthrene and pyrene metabolites (phenols
and dihydrodiols). Int Arch Occup Environ Health, 65: 189-199.
Grimmer G, Dettbarn G, Naujack KW, & Jacob J (1994) Relationship
between inhaled PAH and urinary excretion of phenanthrene, pyrene and
benzo [a]pyrene metabolites in coke plant workers. Polycyclic Aromat
Compd, 5: 269-277.
Grob K & Grob G (1974) Organic substances in potable water and in its
precursor. Part II: Applications in the area of Zürich. J Chromatogr,
90: 303-313.
Groenewegen D & Stolp H (1976) [Microbial breakdown of polycyclic
aromatic hydrocarbons.] Zentralbl Bakteriol Parasitenkd Infektionskr
Hyg Abt 1 Orig B, 161: 225-232 (in German).
Grosjean D (1983) Polycyclic aromatic hydrocarbons in Los Angeles air
from samples collected on teflon, glass and quartz filters. Atmos
Environ, 17: 2565-2573.
Grosser RJ, Warshawsky D, & Vestal JR (1995) Mineralization of
polycyclic and N-heterocyclic aromatic compounds in
hydrocarbon-contaminated soils. Environ Toxicol Chem, 14: 375-382.
Grover PL & Sims P (1968) Enzyme-catalysed reactions of polycyclic
hydrocarbons with deoxyribonucleic acid and protein in vitro.
Biochem J, 110: 159-160.
Grover PL, Sims P, Huberman E, Marquardt H, Kuroki T, & Heidelberger C
(1971) In vitro transformation of rodent cells by K-region
derivatives of polycyclic hydrocarbons. Proc Natl Acad Sci USA, 68:
1098-1101.
Grover PL, Sims P, Mitchel BCU, & Roe FJ (1975) The carcinogenicity of
polycyclic hydrocarbon epoxides in newborn mice. Br J Cancer, 31: 182-
188.
Grover PL, MacNicoll AD, Sims P, Easty GC, & Neville AM (1980)
Polycyclic hydrocarbon activation and metabolism in epithelial cell
aggregates prepared from human mammary tissue. Int J Cancer, 26: 467-
475.
Gückel W, Synnatschke R, & Rittig R (1973) A method for determining
the volatility of active ingredients used in plant protection. Pestic
Sci, 4: 137-147.
Guengerich FP & Shimada T (1991) Oxidation of toxic and carcinogenic
chemicals by human cytochrome P-450 enzymes. Chem Res Toxicol, 4: 391-
407.
Guenther FR, Chesler SN, Gordon GE, & Zoller WH (1988) Residential
wood combustion: A source of atmospheric polycyclic aromatic
hydrocarbons. J High Resol Chromatogr Chromatogr Commun, 11: 761-766.
Guerin MR (1977) Energy sources of polycyclic aromatic hydrocarbons.
Oak Ridge, Tennessee, Oak Ridge National Laboratory, Analytical
Chemistry Division, pp 1-78 (EPA Conf/Doc. 770-130-2).
Guerin MR, Epler JL, Griest WH, Clark BR, & Rao TK (1978) Polycyclic
aromatic hydrocarbons from fossil fuel conversion processes. In: Jones
PW & Freudenthal RI ed. Carcinogenesis. Volume 3: Polynuclear aromatic
hydrocarbons. New York, Raven Press, pp 21-33.
Guggenberger J, Krammer G, & Lindenmüller W (1981) [Contribution to
the determination of the emission of polycyclic aromatic hydrocarbons
from large capacity furnaces.] Staub-Reinhalt Luft, 41: 339-344 (in
German).
Guicherit R & Schulting FL (1985) The occurrence of organic chemicals
in the atmosphere of the Netherlands. Sci Total Environ, 43: 193-219.
Guillemin MP, Herrera H, Huynh CK, Droz PO, & Vu Duc T (1992)
Occupational exposure of truck drivers to dust and polynuclear
aromatic hydrocarbons: A pilot study in Geneva, Switzerland. Int Arch
Occup Environ Health, 63: 439-447.
Gupta RS & Goldstein S (1981) Mutagen testing in the human fibroblasts
diphtheria toxin resistance (HF Dipr) system. In: De Serres FJ & Ashby
J ed. Evaluation of short-term tests for carcinogens. Report of the
international collaborative programme. New York, Elsevier North
Holland, pp 614-625 (Progress in Mutation Research, Volume 1).
Gupta RS & Singh B (1982) Mutagenic responses of five independent
genetic loci in CHO cells to a variety of mutagens. Development and
characteristics of a mutagen screening system based on selection for
multiple drug-resistant markers. Mutat Res, 94: 449-466.
Gupta RC, Earley K, & Sharma S (1988) Use of human peripheral blood
lymphocytes to measure DNA binding capacity of chemical carcinogens.
Proc Natl Acad Sci USA, 85: 3513-3517.
Gurtoo HL, Vaught JB, Marinello AJ, Paigen B, Gessner T, & Bolanowska
W (1980) High-pressure liquid chromatographic analysis of
benzo [a]pyrene metabolism by human lymphocytes from donors of
different aryl hydrocarbon hydroxylase inducibility and antipyrine
half-lives. Cancer Res, 40: 1305-1310.
Gustavsson P (1989) Cancer and ischemic heart disease in occupatioal
groups exposed to combustion products. Solna, National Institute of
Occupational Health, pp 1-28 (World and Health series No. 21).
Gustavsson P & Reuterwall C (1990) Mortality and incidence of cancer
among Swedish gas workers. Br J Ind Med, 47: 169-174.
Gustavsson P, Fellenius E, & Hogstedt C (1987) Possible causes of
increased lung cancer incidence among butchers and slaughterhouse
workers. Scand J Work Environ Health, 13: 518-523.
Gustavsson P, Gustavsson A, & Hogstedt C (1988) Excess of cancer in
Swedish chimney sweeps. Br J Ind Hyg, 45: 777-781.
Habs M, Schmähl S, & Misfeld J (1980) Local carcinogenicity of some
environmentally relevant polycyclic aromatic hydrocarbons after
lifelong topical application to mouse skin. Geschwulstforschung, 50:
266-274.
Habs M, Jahn SAA, & Schmähl D (1984) Carcinogenic activity of
condensate from coloquint seeds (Citrullus colocynthis) after
chronic epicutaneous administration to mice. J Cancer Res Clin Oncol,
108: 154-156.
Haddow A, Scott CM, & Scott JD (1937) The influence of certain
carcinogenic and other hydrocarbons on body growth in the rat. Proc R
Soc Ser B, 122: 477-507.
Hagemann R, Virelizier H, Gaudin D, & Pesneau A (1982) Polycyclic
aromatic hydrocarbons in exhaust particles emitted from gasoline and
diesel automobile engines. Toxicol Environ Chem, 5: 227-236.
Hall M & Grover PL (1990) Polycyclic aromatic hydrocarbons:
Metabolism, activation and tumour initiation. In: Cooper CS & Grover
PL ed. Handbook of experimental pharmacology. Berlin, Springer-Verlag,
Volume 94/I, pp 327-372.
Hall AT & Oris JT (1991) Anthracene reduces reproductive potential and
is maternally transferred during long-term exposure in fathead
minnows. Aquat Toxicol, 19: 249-264.
Hall M, Forrester LM, Parker DK, Grover PL, & Wolf CR (1989) Relative
contribution of various forms of cytochrome P450 to the metabolism of
benzo [a]pyrene by human liver microsomes. Carcinogenesis, 10: 1815-
1821.
Halsall CJ, Coleman PJ, Davies BJ, Burnett V, Waterhouse KS,
Harding-Jones P, & Jones KC (1994) Polycyclic aromatic hydrocarbons in
UK urban air. Environ Sci Technol, 28: 2380-2386.
Hambrick GA, Delaume RD, & Patrick WH Jr (1980) Effects of estuarine
sediment, pH and oxidation-reduction potential on microbial
hydrocarbon degradation. Appl Environ Microbiol, 40: 365-369.
Hamburg Environment Office (1993) [Report on an investigation into
polycyclic aromatic hydrocarbons (PAHs) in Hamburg's waters.] Hamburg,
63 (Report 42/93 13-16) (in German).
Hammond WG, Gabriel A, Paladugu RR, Azumi N, Hill RL, & Benfield JR
(1987) Differential susceptibility to bronchial carcinogenesis in
syngeneic hamsters. Cancer Res, 47: 5202-5206.
Hampton CV, Pierson WR, Schuetzle D, & Harvey TM (1983) Hydrocarbon
gases emitted from vehicles on the road. 2. Determination of emission
rates from diesel and spark ignition vehicles. Environ Sci Technol,
17: 699-708.
Hannah JB, Hose JE, Landolt ML, Miller BS, Felton SP, & Iwaoka WT
(1982) Benzo [a]pyrene-induced morphologic and developmental
abnormalities in rainbow trout Salmo gairdneri Richardson. Arch
Environ Contam Toxicol, 11: 727-734.
Hansen ES (1989) Cancer incidence in an occupational cohort exposed to
bitumen fumes. Scand J Work Environ Health, 15: 101-105.
Hansen ÅM, Olsen IB, Holst E, & Poulsen OM (1991a) Validation of a
high-performance liquid chromatography/fluorescence detection method
for the simultaneous quantification of fifteen polycyclic aromatic
hydrocarbons. Ann Occup Hyg, 35: 603-611.
Hansen ÅM, Poulsen OM, & Christensen JM (1991b) Correlation of levels
of volatile versus carcinogenic particulate polycyclic aromatic
hydrocarbons in air samples from smokehouses. Int Arch Occup Environ
Health, 63: 247-252.
Hansen ÅM, Olsen IB, & Poulsen OM (1992) Polycyclic aromatic
hydrocarbons in air samples of meat smokehouses. Sci Total Environ,
126: 17-26.
Hansen ÅM, Omland , Poulsen OM, Sherson D, Sigsgaard T, Christensen
JM, & Overgaard E (1994) Correlation between work process-related
exposure to polycyclic aromatic hydrocarbons and urinary levels of
a-naphthol, b-naphthylamine and 1-hydroxypyrene in iron foundry
workers. Int Arch Occup Environ Health, 65: 385-394.
Hansen AM, Christensen JM, & Sherson D (1995) Estimation of reference
values for urinary 1-hydroxypyrene and a-naphthol in Danish workers.
Sci Total Environ, 163: 211-219.
Hardin BD, Bond GP, Sikov MR, Andrew FD, Beliles RP, & Niemeier RW
(1981) Testing of selected workplace chemicals for teratogenic
potential. Scand J Work Environ Health, 7: 66-75.
Hardin BD, Schuler RL, Burg JR, Booth GM, Hazelden KP, MacKenzie KM,
Piccirillo VJ, & Smith KN (1987) Evaluation of 60 chemicals in a
preliminary developmental toxicity test. Teratog Carcinog Mutag, 7:
29-48.
Harkov R & Greenberg A (1985) Benzo(a)pyrene in New Jersey. Results
from a twenty-seven-site study. J Air Pollut Control Assoc, 35: 238-
243.
Harkov R, Greenberg A, Darack F, Daisey JM, & Lioy PJ (1984)
Summertime variations in polycyclic aromatic hydrocarbons at four
sites in New Jersey. Environ Sci Technol, 18: 187-291.
Harper KH (1957) The metabolism of pyrene. Br J Cancer, 11: 499-507.
Harper KH (1958a) The intermediary metabolism of pyrene. Br J Cancer,
12: 116-120.
Harper KH (1958b) The intermediary metabolism of 3,4-benzpyrene. Br J
Cancer, 12: 121-128.
Harper KH (1958c) The intermediary metabolism of 3,4-benzyprene: The
biosynthesis and identification of the X1 and X2 metabolites. Br J
Cancer, 12: 645-660.
Harper KH (1959a) The intermediary metabolism of polycyclic
hydrocarbons. Br J Cancer, 13: 718-731.
Harper KH (1959b) Metabolism of 1,2-benzanthracene in the rabbit. Br J
Cancer, 13: 746-750.
Harper BL & Legator MS (1987) Pyridine prevents the clastogenicity of
benzene but not of benzo [a]pyrene or cyclophosphamide. Mutat Res,
179: 23-31.
Harper BL, Sadagopa Ramanujam VM, Gad-El-Karim MM & Legator MS (1984)
The influence of simple aromatics on benzene clastogenicity. Mutat
Res, 128: 105-114.
Harris CC, Autrup H, Connor R, Barrett LA, McDowell EM, & Trump BF
(1976) Interindividual variation in binding of benzo [a]pyrene to DNA
in cultured human bronchi. Science, 194: 1067-1069.
Harris CC, Autrup H, Stoner G, Yang SK, Leutz JC, Gelboin HV, Selkirk
JK, Conner RJ, Barrett LA, Jones RT, McDowell E, & Trump BF (1977)
Metabolism of benzo [a]pyrene and 7,12-dimethylbenz [a]anthracene in
cultured human bronchus and pancreatic duct. Cancer Res, 37: 3349-
3355.
Harris CC, Hsu IC, Stoner GD, Trump BF, & Selkirk JK (1978a) Human
pulmonary alveolar macrophages metabolise benzo [a]pyrene to
proximate and ultimate mutagens. Nature, 272: 633-634.
Harris CC, Autrup H, & Stoner G (1978b) Metabolism of benzo [a]pyrene
in cultured human tissues and cells. In: Gelboin HV & Ts'o POP ed.
Polycyclic hydrocarbons and cancer. New York, Academic Press, Volume
2, pp 331-342.
Harris CC, Autrup H, Stoner GD, Trump BF, Hillman E, Schafer PW, &
Jeffrey AM (1979) Metabolism of benzo [a]pyrene,
N-nitrosodimethylamine, and N-nitrosopyrrolidine and
identification of the major carcinogen-DNA adducts formed in cultured
human esophagus. Cancer Res, 39: 4401-4406.
Harris CC, Grafstom RC, Shamsuddin AM, Sinopoli NT, Trump BF, & Autrup
H (1984) Carcinogen metabolism and carcinogen-DNA adducts in human
tissues and cells. In: Greim H, Jung R, Kramer M, Marquardt H, & Oesch
F ed. Biochemical basis of chemical carcinogenesis. New York, Raven
Press, pp 123-135.
Harris CC, Vahakangas K, Newman MJ, Trivers GE, Shamsuddin A, Sinopoli
N, Mann DL, & Wright WE (1985) Detection of benzo [a]pyrene diol
epoxide-DNA adducts in peripheral blood lymphocytes and antibodies to
the adducts in serum from coke oven workers. Proc Natl Acad Sci USA,
82: 6672-6676.
Hart KM & Pankow JF (1994) High-volume air sampler for particle and
gas sampling. 2. Use of backup filters to correct for the adsorption
of gas-phase polycyclic aromatic hydrocarbons to the front filter.
Environ Sci Technol, 28: 655-661.
Hartung H & Koch D (1991) [Environmental research plan from the
Federal Ministry for Environment and Reactor Safety (Research Project
103 03 220/01): Waste disposal: Quantity, recycling and disposal of
used motor tyres in the Federal Republic of Germany.] Berlin, Ministry
of the Environment, pp 73-74 (Report No. UBA-FB 89-149, Text 52/91)
(in German).
Harvey RG (1996) Mechanisms of carcinogenesis of polycyclic aromatic
compounds. Polycyclic Aromat Compd, 9: 1-23.
Häsänen E, Pohjola V, Pyysalo H, & Wickström K (1983) Polycyclic
aromatic hydrocarbons in the Finnish wood-heated sauna. Kemia Kemi,
10: 27-29.
Haseltine WA, Lo KM, & D'Andrea AD (1980) Preferred sites of strand
scission in DNA modified by anti-diol epoxide of benzo [a]pyrene.
Science, 209: 929-931.
Hass BS & Applegate HG (1975) The effects of unsubstituted polycyclic
aromatic hydrocarbons on the growth of Escherichia
coli. Chem-Biol Interactions, 10: 265-268.
Hatch MC, Warburton D, & Santella RM (1990) Polycyclic aromatic
hydrocarbon-DNA adducts in spontaneously aborted fetal tissue.
Carcinogenesis, 11: 1673-1675.
van Hattum B, Cofino WP, & Feenstra JF (1993) Environmental aspects of
produced water discharges from oil and gas production on the Dutch
continental shelf: Part II. A literature review of characteristics of
produced water from offshore platforms. Amsterdam, Oil and Gas
Exploitation and Production Association, 64 pp.
Haubenstricker ME, Meier PG, Mancy KH, & Brabec MJ (1990) Rapid
toxicity testing based on yeast respiratory activity. Bull Environ
Contam Toxicol, 44: 669-674.
Haudenschild R, Poffer J, Schott T, & Deuber A (1990) [Application of
corrosion inhibitor in a bus garage.] Illus Z Arb Sicherheitstech, 37:
6-8 (in German).
Haugen A, Becher G, Benestad C, Vahakangas K, Trivers GE, Newman MJ, &
Harris CC (1986) Determination of polycyclic aromatic hydrocarbons in
the urine, benzo(a)pyrene diol epoxide-DNA adducts in lymphocyte DNA,
and antibodies to the adducts in sera from coke oven workers exposed
to measured amounts of polycyclic aromatic hydrocarbons in the work
atmosphere. Cancer Res, 46: 4178-4183.
Hawkins WE, Walker WW, Overstreet RM, Lytle TF, & Lytle JS (1988)
Dose-related carcinogenic effects of water-borne benzo [a]pyrene on
livers of two small fish species. Ecotoxicol Environ Saf, 16: 219-231.
Hawkins WE, Walker WW, Overstreet RM, Lytle JS, & Lytle TF (1990)
Carcinogenic effects of some polycyclic aromatic hydrocarbons on the
Japanese medaka and guppy in waterborne exposures. Sci Total Environ,
94: 155-167.
Hawley G (1987) Hawley's condensed chemical dictionary, 11th rev ed.
New York, Van Nostrand Reinhold Co., p 6.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1984a) Products obtained
during combustions of polymers under simulated incinerator conditions:
I. Polyethylene. J Chromatogr, 314: 263-273.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1984b) Products obtained
during combustions of polymers under simulated incinerator conditions:
II. Polystyrene. J Chromatogr, 315: 201-210.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1984c) Products obtained
during combustions of polymers under simulated incinerator conditions:
III. Polyvinyl chloride. J Chromatogr, 315: 211-221.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1987) Identification of
organic compounds produced during combustion of a polymer mixture. J
Chromatogr, 387: 207-221.
Hawthorne SB & Miller DJ (1987) Extraction and recovery of polycyclic
aromatic hydrocarbons from environmental solids using supercritical
fluids. Anal Chem, 59: 1705-1708.
Hawthorne SB, Krieger MS, & Miller DJ (1989a) Supercritical carbon
dioxide extraction of polychlorinated biphenyls, polycyclic aromatic
hydrocarbons, heteroatom-containing polycyclic aromatic hydrocarbons,
and n-alkanes from polyurethane foam sorbents. Anal Chem, 61: 736-
740.
Hawthorne SB, Miller DJ, & Krieger MS (1989b) Coupled SFE-GC: A rapid
and simple technique for extracting, identifying, and quantitating
organic analytes from solid and sorbent resins. J Chromatogr Sci, 27:
347-354.
Hawthorne SB, Miller DJ, Langenfeld JJ, & Krieger MS (1992) PM-10
high-volume collection and quantitation of semi- and nonvolatile
phenols, methoxylated phenols, alkanes, and polycyclic aromatic
hydrocarbons from winter urban air and their relationship to wood
smoke emissions. Environ Sci Technol, 26: 2251-2262.
He S & Baker R (1991) Micronuclei in mouse skin cells following
in vivo exposure to benzo [a]pyrene,
7,12-dimethylbenz [a]anthracene, chrysene, pyrene and urethane.
Environ Mol Mutag, 17: 163-168.
Heaton DM, Bartle, KD, Clifford AA, Meyers P, & King BW (1994) Rapid
separation of polycyclic aromatic hydrocarbons by packed column
supercitical fluid chromatography. Chromatographia, 39: 607-611.
Hecht SS, Bondinell WE, & Hoffmann D (1974) Chrysene and
methylchrysenes: Presence in tobacco smoke and carcinogenicity. J Natl
Cancer Inst, 53: 1121-1133.
Hecht SS, Loy M, Maronpot RR, & Hoffmann D (1976a) A study of chemical
carcinogenesis: Comparative carcinogenicity of 5-methylchrysene,
benzo(a)pyrene, and modified chrysenes. Cancer Lett, 1: 147-153.
Hecht SS, Loy M, & Hoffmann D (1976b) On the structure and
carcinogenicity of the methylchrysenes. In: Freudenthal R & Jones PW
ed. Carcinogenesis: A comprehensive survey. Volume 1: Polynuclear
aromatic hydrocarbons chemistry, metabolism, and carcinogenesis. New
York, Raven Press, pp 325-340.
Hecht SS, Loy M, Mazzarese R, & Hoffmann D (1978) On the
carcinogenicity of 5-methylchrysene: Structure-activity studies and
metabolism. Polycyclic Hydrocarbons Cancer, 1: 119-130.
Hecht SS, Grabowski W, & Groth K (1979) Analysis of faeces for
benzo [a]pyrene after consumption of charcoal-broiled beef by rats
and humans. Food Cosmet Toxicol, 17: 223-227.
Hecht SS, LaVoie E, Amin S, Bedenko V, & Hoffmann D (1980) On the
metabolic activation of the benzofluoranthenes. In: Bjorseth A &
Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry and
biological effects. Columbus, Ohio, Battelle Press, pp 417-433.
Hecht SS, LaVoie EJ, Bedenko V, Hoffmann D, Sardella DJ, Boger E, &
Lehr RE (1981) On the metabolic activation of dibenzo [a,i]pyrene and
dibenzo [a,h]pyrene. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: Chemical analysis and biological fate. Columbus, Ohio,
Battelle Press, pp 43-45.
Hecht SS, Radok L, Amin S, Huie K, Melikian AA, Hoffmann D, Pataki J,
& Harvey RG (1985) Tumorigenicity of 5-methylchrysene dihydrodiols and
dihydrodiol epoxides in newborn mice and on mouse skin. Cancer Res,
45: 1449-1452.
Hecht SS, Melikian AA, & Amin S (1986) Methylchrysenes as probes for
the mechanism of metabolic activation of carcinogenic methylated
polynuclear aromatic hydrocarbons. Acc Chem Res, 19: 174-180.
Hecht SS, Amin S, Huie K, Melikian AA, & Harvey RG (1987) Enhancing
effect of a bay region methyl group on tumorigenicity in newborn mice
and mouse skin of enantiomeric bay region diol epoxides formed
stereoselectively from methylchrysenes in mouse epidermis. Cancer Res,
47: 5310-5315.
Hecht SS, El-Bayoumy K, Rivenson A, & Amin S (1994) Potent mammary
carcinogenicity in female CD rats of a fjord region diol-epoxide of
benzo(c)phenanthrene compared to a bay region diol-epoxide of
benzo(a)pyrene. Cancer Res, 54: 21-24.
Hecht SS, Amin S, Lin J-M, Rivenson A, Kurtzke C, & El-Bayoumy K
(1995) Short communication. Mammary carcinogenicity in female CD rats
of a diol epoxide metabolite of fluoranthene, a commonly occurring
environmental pollutant. Carcinogenesis, 16: 1433-1435.
Heeschen WH (1985) [Scientific working group on chemicals in human
milk.] Arch Gynekol, 238: 199-216 (in German).
Heflich RH, Thornton-Manning JR, Kinouchi T, & Beland FA (1990)
Mutagenicity of oxidized microsomal metabolites of 1-nitropyrene in
Chinese hamster ovary cells. Mutagenesis, 5: 151-157.
Heidelberger C & Davenport GR (1961) Local functional components of
carcinogenesis. Acta Unio Int Contra Cancrum, 17: 55-63.
Heidelberger C & Jones HB (1948) The distribution of radioactivity in
the mouse following administration of dibenzanthracene labeled in the
9 and 10 positions with carbon 14*. Cancer, 1: 252-260.
Heidelberger C & Weiss SM (1951) The distribution of radioactivity in
mice following administration of 3,4-benzpyrene-5-C14 and
1,2,5,6-dibenzanthracene-9,10-C14. Cancer Res, 11: 885-891.
Heikkilä PR, Hämeilä M, Pyy L, & Rauni P (1987) Exposure to creosote
in the impregnation and handling of impregnated wood. Scand J Work
Environ Health, 13: 431-437.
Heinrich U (1989) Exhaust-specific carcinogenic effects of polycyclic
aromatic hydrocarbons and their significance for the estimation of the
exhaust exposure-related lung cancer risk. In: Mohr U, Bates DV,
Dungworth DL, Lee PN, McClellan RO, & Roe FJC ed. Assessment of
inhalation hazards: Integration and extrapolation using diverse data.
Berlin, Springer-Verlag, pp 301-313.
Heinrich U (1995) Do PAH contribute to the lung tumor response in
diesel engine exhaust-exposed rats? In: Fifteenth international
symposium on polycyclic aromatic compounds: Chemistry, biology and
environmental impact, Belgirate, Italy, 19-22 September 1995. Ispra,
Joint Research Centre European Commission, p 150.
Heinrich U, Pott F, Mohr U, Fuhst R, & König J (1986a) Lung tumours in
rats and mice after inhalation of PAH-rich emissions. Exp Pathol, 29:
29-34.
Heinrich U, Pott F, & Rittinghausen S (1986b) Comparison of chronic
inhalation effects in rodents after long-term exposure to either coal
oven flue gas mixed with pyrolysed pitch or diesel engine exhaust. In:
Ishinishi N, Koizumi A, McClellan RO, & Stöber W ed. Carcinogenic and
mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier
Science Publishers, pp 441-457.
Heinrich U, Peters L, Creutzenberg O, Dasenbrock C, & Hoymann HG
(1994a) Inhalation exposure of rats to tar/pitch condensation aerosol
or carbon black alone or in combination with irritant gases. In: Mohr
U, Dungworth DL, Mauderly JL, & Oberdörster G ed. Toxic and
carcinogenic effects of solid particles in the respiratory tract.
Washington DC, International Life Sciences Institute Press, pp 433-
441.
Heinrich U, Dungworth DL, Pott F, Peters L, Dasenbrock C, Levsen K,
Koch W, Creutzenberg O, & Schulte A (1994b) The carcinogenic effects
of carbon black particles and tar-pitch condensation aerosol after
inhalation exposure of rats. Ann Occup Hyg, 38 (suppl): 351-356.
Heinrich U, Roller M, & Pott F (1994c) Estimation of a lifetime unit
lung cancer risk for benzo [a]pyrene based on tumor rates in rats
exposed to coaltar/pitch condensation aerosol. Toxicol Lett, 72: 155-
161.
Heit M (1985) The relationship of a coal fired power plant to the
levels of polycyclic aromatic hydrocarbons (PAH) in the sediment of
Cayuga Lake. Water Air Soil Pollut, 24: 41-61.
Heit M, Klusek CS, & Miller KM (1980) Trace element, radionuclide, and
polynuclear aromatic hydrocarbon concentrations in Unionidae mussels
from northern Lake George. Environ Sci Technol, 14: 465-468.
Heitkamp MA, Freeman JP, Miller DW, & Cerniglia CE (1988) Pyrene
degradation by a Mycobacterium sp.: Identification of ring oxidation
and ring fission products. Appl Environ Microbiol, 54: 2556-2565.
Heitmuller PT, Hollister TA, & Parrish PR (1981) Acute toxicity of 54
industrial chemicals to sheepshead minnows Cyprinodon
variegatus. Bull Environ Contam Toxicol, 27: 596-604.
Heldal M, Norland S, Lien T, Knutsen G, Tjessem K, & Aarberg A (1984)
Toxic responses of the green alga Dunaliella bioculata
(Chlorophycea, Volvocales) to selected oxidised hydrocarbons. Environ
Pollut, A34: 119-132.
Helfrich J & Armstrong DE (1986) Polycyclic aromatic hydrocarbons in
sediments of the southern basin of Lake Michigan. J Great Lakes Res,
12: 192-199.
Hembrock-Heger A & König W (1990) Occurrence and transfer of
polycyclic aromatic hydrocarbons in soil and plants.] Düsseldorf,
VDI-Verlag, pp 815-830 (VDI Report No. 837) (in German).
Hemminki K & Vainio H (1979) Preferential binding of benzo [a]pyrene
into nuclear matrix fraction. Cancer Lett, 6: 167-173.
Hemminki K, Grzybowska E, Chorazy M, Twardowska-Saucha K, Srczynski
JW, Putman KL, Randerath K, Phillips DH, Hewer A, Santella RM, &
Perera FP (1990a) DNA adducts in humans related to occupational and
environmental exposure to aromatic compounds. In: Vainio H, Sorsa M, &
McMichael AJ ed. Complex mixtures and cancer risk. Lyon, International
Agency for Research on Cancer, pp 181-192 (IARC Scientific
Publications No. 104).
Hemminki K, Grzybowska E, Chorazy M, Twardowska-Saucha K, Sroczynski
JW, Putman KL, Randerath, Phillips DH, & Hewer A (1990b) Aromatic DNA
adducts in white blood cells of coke workers. Int Arch Occup Environ
Health, 62: 467-470.
Hendricks JP, Meyers TR, Shelton DW, Casteel JL, & Bailey GS (1985)
Hepatocarcinogenicity of benzo [a]pyrene to rainbow trout by dietary
exposure and intraperitoneal injection. J Natl Cancer Inst, 74: 839-
851.
Henry MC & Kaufman DG (1973) Clearance of benzo [a]pyrene from
hamster lungs after administration on coated particles. J Natl Cancer
Inst, 51: 1961-1964.
Henry MC, Port CD & Kauhman DG (1975) Importance of physical
properties of benzo (a)pyrene-ferric oxide mixtures in lung tumor
induction. Cancer Res, 35: 207-217.
Herbert R, Marcus M, Wolff MS, Perera FP, Andrews L, Godbold JH,
Rivera M, Stefanidis M, Lu XQ, Landrigan PJ, & Santella RM (1990a)
Detection of adducts of deoxyribonucleic acid in white blood cells of
roofers by 32P-postlabeling. Scand J Work Environ Health, 16: 135-
143.
Herbert R, Marcus M, Wolff MS, Perera FP, Andrews L, Godbold JH,
Rivera M, Stefanidis M, Lu XQ, Landrigan PJ, & Santella RM (1990b) A
pilot study of detection of DNA adducts in white blood cells of
roofers by 32P-postlabelling. In: Vainio H, SorsaM, & McMichael AJ
ed. Complex mixtures and cancer risk. Lyon, International Agenca for
Research on Cancer, pp 205-214 (IARC Scientific Publications No. 104).
Herbes SE (1981) Rates of microbial transformation of polycyclic
aromatic hydrocarbons in water and sediments in the vicinity of
coal-coking waste water discharge. Appl Environ Microbiol, 41: 20-28.
Herbes SE, Southworth GR, Shaeffer DL, Griest WH, & Maskarinec MP
(1980) Critical pathways of polycyclic aromatic hydrocarbons in
aquatic environments. In: Witschi H ed. The scientific basis of
toxicity assessment.Amsterdam, Elsevier Biomedical Press, pp 113-128.
Herlan A (1982) Content of polycyclic aromatics in middle distillates.
Zentralbl Bakteriol Hyg I Abt Orig B, 176: 249-268.
Hermann M (1981) Synergistic effects of individual polycyclic aromatic
hydrocarbons on the mutagenicity of their mixtures. Mutat Res, 90:
399-409.
Hermann M, Durand JP, Charpentier JM, Chaude O, Hofnung M, Petroff N,
Vandecasteele JP, & Weill N (1980) Correlations of mutagenic activity
with polynuclear aromatic hydrocarbon content of various mineral oils.
In: Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons:
Chemical analysis and biological fate. Columbus, Ohio, Battelle Press,
pp 899-916.
Heussner JC, Ward JB, & Legator MS (1985) Genetic monitoring of
aluminium workers exposed to coal tar pitch volatiles. Mutat Res, 155:
143-155.
Hinoshita F, Hardin JA, & Sherr DH (1992) Fluoranthene induces
programmed cell death and alters growth of immature B cell populations
in bone marrow cultures. Toxicology, 73: 203-218.
Hirano T, Stanton M, & Layard M (1974) Measurement of epidermoid
carcinoma development induced in the lungs of rats by
3-methylcholanthrene-containing beeswax pellets. J Natl Cancer Inst,
53: 1209-1215.
Hirom PC, Chipman JK, Millburn P, & Pue MA (1983) Enterohepatic
circulation of the aromatic hydrocarbons benzo(a)pyrene and
naphthalene. In: Rydstrom J, Montelius J, & Bengtsson M ed.
Extrahepatic drug metabolism and chemical carcinogenesis. Amsterdam,
Elsevier Science Publishers, pp 275-281.
Hites RA (1981) Sources and fates of atmospheric polycyclic aromatic
hydrocarbons. In: Macias ES & Hopke PK ed. Atmospheric aerosol,
source/air quality relationships (ACS Symposium Series 167).
Washington DC, American Chemical Society, pp 187-196.
Hites RA (1989) Mass spectrometry of polycyclic aromatic compounds.
In: Vo-Dinh T ed. Chemical analysis of polycyclic aromatic compounds.
New York, John Wiley & Sons, pp 219-261.
Hites RA, LaFlamme JW, & Farrington JW (1977) Sedimentary polycyclic
aromatic hydrocarbons: The historical record. Science, 198: 829-831.
Hites RA, LaFlamme RE, & Windsor JG Jr (1980) Polycyclic aromatic
hydrocarbons in marine/aquatic sediments: Their ubiquity. In: Petrakis
L & Weiss FT ed. Petroleum in the marine environment (Advances in
Chemistry Series No. 185). Washington DC, American Chemical Society,
pp 289-311.
Ho CH, Clark BR, Guerin MR, Barkenbus BD, Rao TK, & Epler JL (1981)
Analytical and biological analyses of test materials from the
synthetic fuel technologies: IV. Studies of chemical
structure-mutagenic activity relationships of aromatic nitrogen
compounds relevant to synfuels. Mutat Res, 85: 335-345.
Hoch-Ligeti C (1941) Studies on the changes in the lymphoid tissue of
mice treated with carcinogenic and noncarcinogenic hydrocarbons.
Cancer Res, 1: 484-488.
Hodgson RM, Weston A, & Grover PL (1983) Metabolic activation of
chrysene in mouse skin: Evidence for the involvement of a
triol-epoxide. Carcinogenesis, 4: 1639-1643.
Hodson J & Williams NA (1988) The estimation of the adsorption (Koc)
for soils by high performance liquid chromatography. Chemosphere, 17:
67-77.
Hoff RM & Chan KW (1987) Measurement of polycyclic aromatic
hydrocarbons in the air along the Niagara River. Environ Sci Technol,
21: 556-561.
Hoffmann K (1993) [Risk assessment and restoration concept for
PAH-contaminated soil.] Altlasten-Spektrum, 2: 93-99 (in German).
Hoffmann DJ & Gay ML (1981) Embryotoxic effects of benzo [a]pyrene,
chrysene, and 7,12-dimethylbenz [a]anthracene in petroleum
hydrocarbon mixtures in mallard ducks. J Toxicol Environ Health, 7:
775-787.
Hoffmann D & Wynder EL (1962) Analytical and biological studies on
gasoline engine exhaust. Natl Cancer Inst Monogr, 9: 91-110.
Hoffmann D & Wynder EL (1966) [Carcinogenic effect of dibenzopyrenes.]
Z Krebsforsch, 68: 137-149 (in German).
Hoffmann D, Rathkamp G, Nesnow S, & Wynder EL (1972) Fluoranthenes:
Quantitative determination in cigarette smoke, formation by pyrolysis,
and tumor-initiating activity. J Natl Cancer Inst, 49: 1165-1175.
Hoffmann D, Schmeltz I, Hecht SS, & Wynder EL (1978) Tobacco
carcinogenesis. In: Gelboin HV & Ts'o POP ed. Polycyclic hydrocarbons
and cancer, Volume 1. New York, Academic Press, pp 85-117.
Hoffman EJ, Mills GL, Latimer JS, & Quinn JG (1984) Urban runoff as a
source of polycyclic aromatic hydrocarbons to coastal waters. Environ
Sci Technol, 18: 580-587.
Hoffman EJ, Latimer JS, Hunt CD, Mills GL, & Quinn JG (1985)
Stormwater runoff from highways. Water Air Soil Pollut, 25: 349-364
Holcombe GW, Phipps GL, & Fiandt JT (1983) Toxicity of selected
priority pollutants to various aquatic organisms. Ecotoxicol Environ
Saf, 7: 400-409.
Holcombe GW, Phipps GL, Knuth ML, & Felhaber T (1984) The acute
toxicity of selected substituted phenols, benzenes and benzoic acid
esters to fathead minnows Pimephales promelas. Environ Pollut, A35:
367-381.
Holder G, Yagi H, Dansette P, Jerina DM, Levin W, Lu AYH, & Conney AH
(1974) Effects of inducers and epoxide hydrase on the metabolism of
benzo [a]pyrene by liver microsomes and a reconstituted system:
Analysis by high pressure liquid chromatography. Proc Natl Acad Sci
USA, 71: 4356-4360.
Hollstein M, McCann J, Angelosanto FA, & Nichols WW (1979) Short-term
tests for carcinogens and mutagens. Mutat Res, 65: 133-226.
Holman A, Karlsson A, Bratt I, Raihle G, & Hogstedt B (1994) Increased
frequency of micronuclei in lymphocytes of Swedish chimney sweeps. Int
Arch Occup Environ Health, 66: 185-187.
Holoubek I, Housková L, Seda Z, Holoubkova I, Korínek P, Bohácek Z, &
Cáslavsk J (1990) Project TOCOEN. The fate of selected organic
pollutants in the environment. III. Water and sediments 1988. Toxicol
Environ Chem, 29: 29-35.
Homburger F, Russfield AB, Baker JR, & Tregier A (1962) Experimental
chemotherapy in chemically induced mouse tumors and their transplants.
Cancer Res, 22: 368-375.
Honda T, Kiyozumi M, & Kojima S (1990) Alkylnaphthalene: XI. Pulmonary
toxicity of naphthalene, 2-methylnaphthalene, and
isopropylnaphthalenes in mice. Chem Pharm Bull, 38: 3130-3135.
Honkakoski P & Lang MA (1989) Mouse liver phenobarbital-inducible P450
system: Purification, characterization, and differential inducibility
of four cytochrome P450 isozymes from the D2 mouse. Arch Biochem
Biophys, 273: 42-57.
Hooftman RJ & Evers-de Ruiter A (1992a) Investigations into aquatic
toxicity of phenanthrene (cover-report for reproduction tests with the
waterflea Daphnia magna and an early life stage test with the zebra
fish Brachydanio rerio. Delft, The Netherlands, TNO Institute of
Environmental Science, 23 pp (TNO Report No. IMW-R 92/290).
Hooftman RJ & Evers-de Ruiter A (1992b) The toxicity and uptake of
fluoranthene in Brachidanio rerio in an early life stage test.
Delft, TNO Institute of Environmental Science, 36 pp (TNO Report No.
IMW-R 92/207).
Hooftman RJ & Evers-de Ruiter A (1992c) The toxicity and uptake of
benzo[k]fluoranthene using Brachidanio rerio in an early life stage
test. Delft, The Netherlands, TNO Institute of Environmental Science,
33 pp (TNO Report No. IMW-R 92/218).
Hooftman RJ & Evers-de Ruiter A (1992d) Early life stage tests with
Brachidanio rerio and several polycyclic aromatic hydrocarbons using
an intermittent flow-through system. Delft, TNO Institute of
Environmental Science, 35 pp (TNO Report No. IMW-R 92/210).
Hooftman RN, Henzen L, & Roza P (1993) The toxicity of a polycyclic
aromatic mixture in an early stage toxicity test carried out in an
intermittent flow-through system. Delft, TNO Institute of
Environmental Science, 41 pp (TNO Report No. IMW-R 93/253).
Hopia A, Pyysalo H, & Wickström K (1986) Margarines, butter and
vegetable oils as sources of polycyclic aromatic hydrocarbons. J Am
Oil Chem Soc, 63: 889-893.
Horikawa K, Sera N, Otofuji T, Murakami K, Tokiwa H, Hwagawa M, Izumi
K, & Otsuka H (1991) Pulmonary carcinogenicity of 3,9- and
3,7-dinitrofluoranthene, 3-nitrofluoranthene and benzo [a]pyrene in
F344 rats. Carcinogenesis, 12: 1003-1008.
Horton AW & Christian GM (1974) Cocarcinogenic versus incomplete
carcinogenic activity among aromatic hydrocarbons: Contrast between
chrysene and benzo [b]-triphenylene. J Natl Cancer Inst, 53:
1017-1020.
Hose JE (1985) Potential uses of sea-urchin embryos for identifying
toxic chemicals: Description of a bioassay incorporating cytologic,
cytogenetic and embryologic endpoints. J Appl Toxicol, 5: 245-254.
Hose JE, Hannah JB, Landolt ML, Miller BS, Felton SP, & Iwaoka WT
(1981) Uptake of benzo [a]pyrene by gonadal tissue of flatfish family
Plueronectidae and its effects on subsequent egg development. J
Toxicol Environ Health, 11: 991-1000.
Hose JE, Hannah JB, DiJulio D, Landolt ML, Miller BS, Iwaoka WT, &
Felton SP (1982) Effects of benzo [a]pyrene on early development of
flatfish. Arch Environ Contam Toxicol, 11: 167-171.
Hoshino K, Hayashi Y, Takehira Y, & Kameyama Y (1981) Influences of
genetic factors on the teratogenicity of environmental pollutants:
Teratogenic susceptibility to benzo [a]pyrene and Ah locus in mice.
Congenital Anomalies, 21: 97-103.
House RV, Pallardy MJ, Burleson GR, & Dean JH (1988)
7,12-Dimethylbenz [a]-anthracene-induced modulation of cytokines
involved in cytotoxic T-lymphocyte induction. In Vitro Toxicol, 2:
267-278.
House RV, Pallardy MJ, & Dean JH (1989) Suppression of murine
cytotoxic T-lymphocyte induction following exposure to
7,12-dimethylbenz [a]anthracene: Dysfunction of antigen recognition.
Int J Immunopharmacol, 2: 207-215.
Houser WH, Raha A, & Vickers M (1992) Induction of CYP1A1 gene
expression in H4II-E rat hepatoma cells by benzo [e]pyrene. Mol
Carcinog, 5: 232-237.
Howard J (1979) Method 5: Analysis of benzo [a]pyrene and other
polycyclic aromatic hydrocarbons in foods. In: Egan H, Castegnaro M,
Bogovski P, Kunte H, & Walker EA ed. Environmental carcinogens:
Selected methods of analysis. Volume 3. Analysis of polycyclic
aromatic hydrocarbons in environmental samples. Lyon, International
Agency for Research on Cancer, pp 175-191 (IARC Scientific
Publications No. 29).
Howard JW & Fazio T (1980) Review of polycyclic aromatic hydrocarbons
in foods. Analytical methodology and reported findings of polycyclic
aromatic hydrocarbons in foods. J Assoc Off Anal Chem, 63: 1077-1104.
Howard PH, Boethling RS, Jarvis WF, Meylan WM, & Michalenko EM (1991)
In: Printup HT ed. Handbook of environmental degradation rates.
Chelsea, Michigan, Lewis Publishers, 725 pp.
Hradec J, Seidel A, Platt KL, Glatt HR, Oesch F, & Koblyakov V (1990)
The initiator tRNA acceptance assay as a short-term test for
carcinogens. 6. Results with 78 polycyclic aromatic compounds.
Carcinogenesis, 11: 1921-1926.
Huberman E (1975) Mammalian cell transformation and cell-mediated
mutagenesis by carcinogenic polycyclic hydrocarbons. Mutat Res, 29:
285-291.
Huberman E (1978) Cell transformation and mutability of different
genetic loci in mammalian cells by metabolically activated
carcinogenic polycyclic hydrocarbons. In: Gelboin HV & Ts'O POP ed.
Polycyclic hydrocarbons and cancer: Molecular and cell biology, Volume
2. New York, Academic Press, pp 161-174.
Huberman E & Sachs L (1976) Mutability of different genetic loci in
mammalian cells by metabolically activated carcinogenic polycyclic
hydrocarbons. Proc Natl Acad Sci USA, 73: 188-192.
Huberman E, Kuroki T, Marquardt H, Selkirk JK, Heidelberger C, Grover
PL & Sims P (1972) Transformation of hamster embryo cells by epoxides
and other derivatives of polycyclic hydrocarbons. Cancer Res, 32:
1391-1396.
Huberman E, McKeown CK, Jones CA, Hoffman DR, & Murao S (1984)
Induction of mutations by chemical agents at the hypoxanthine-guanine
phosphoribosyl transferase locus in human epithelial teratoma cells.
Mutat Res, 130: 127-137.
Huggett RJ, deFur PO, & Bieri RH (1988) Organic compounds in
Chesapeake Bay sediments. Mar Pollut Bull, 19: 454-458.
Huggins C & Yang NC (1962) Induction and extinction of mammary cancer.
Science, 137: 257-262.
Huggins C, Morii S, & Grand LC (1961) Mammary cancer induced by a
single dose of polynuclear hydrocarbons: Routes of administration. Ann
Surg, 154: 315-318.
Hughes TW & DeAngelis DG (1982) Emissions from coal-fired residential
combustion equipment. In: Proceedings of the International Conference
on Residues of Solid Fuels: Environmental Impacts/ Solutions. Dayton,
Ohio, Monsanto Research Corporation, pp 333-348.
Hughes NC & Phillips DH (1991) Dependence on dose of initial and
persistant levels of benzo(a)pyrene and dibenzo(a,e)pyrene DNA adducts
in mouse tissues. Proc Am Assoc Cancer Res, 32: 98.
Hughes MM, Natusch DFS, Taylor DR, & Zeller MV (1980) Chemical
transformations of particulate polycyclic organic matter. In: Bjorseth
A & Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry and
biological effects. Columbus, Ohio, Battelle Press, pp 1-8.
Hughes MF, Chamulitrat W, Mason RP, & Eling TE (1989) Epoxidation of
7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene via a
hydroperoxide-dependent mechanism catalyzed by lipoxygenases.
Carcinogenesis, 10: 2075-2080.
Huh N, Nemoto N, & Utakoji T (1982) Metabolic activation of
benzo [a]pyrene, aflatoxin B1, and dimethylnitrosamine by a human
hepatoma cell line. Mutat Res, 94: 339-348.
van Hummelen P, Gennart JP, Buchete JP, Lauwerys R, & Kirsch-Volders M
(1993) Biological markers in PAH exposed workers and controls. Mutat
Res, 300: 231-239.
Hungspreugs M, Silpipat S, Tonapong C, Lee RF, Windom HL, & Tenore KR
(1984) Heavy metals and polycyclic hydrocarbon compounds in benthic
organisms of the upper Gulf of Thailand. Mar Pollut Bull, 15: 213-218.
Hurley JF, Archibald RMcL, Collins PL, Fanning DM, Jacobsen M, &
Steele RC (1983) The mortality of coke workers in Britain. Am J Ind
Med, 4: 691-704.
Husgafvel-Pursiainen K, Sorsa M, Mller M, & Benestad C (1986)
Genotoxicity and polynuclear aromatic hydrocarbon analysis of
environmental tobacco smoke samples from restaurants. Mutagenesis, 1:
287-292.
Hutchinson TC, Hellebust JA, Tam D, Mackay D, Mascarenhas RA, & Shiu
WY (1980) The correlation of the toxicity to algae of hydrocarbons and
halogenated hydrocarbons with their physical-chemical properties. In:
Afghan BK & Mackay D ed. Hydrocarbons and halogenated hydrocarbons in
the aquatic environment. New York, Plenum Press, pp 577-586.
IARC (1973) Certain polycyclic aromatic hydrocarbons and heterocyclic
compounds. Lyon, International Agency for Research on Cancer, p 178
(IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Man, Volume 13).
IARC (1983) Polynuclear aromatic compounds. Part 1: Chemical,
environmental and experimental data. Lyon, International Agency for
Research on Cancer, 477 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 32).
IARC (1984a) Polynuclear aromatic hydrocarbons. Part 2: Carbon blacks,
mineral oils (lubricant base oils and derived products) and some
nitroarenes. Lyon, International Agency for Research on Cancer, 365 pp
(IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Volume 33).
IARC (1984b) Polynuclear aromatic compounds. Part 3: Industrial
exposures in aluminium production, coal gasification, coke production,
and iron and steel founding. Lyon, International Agency for Research
on Cancer, 219 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 34).
IARC (1985) Polynuclear aromatic compounds. Part 4: Bitumens,
coal-tars and derived products, shale-oils and soots. Lyon,
International Agency for Research on Cancer, 271 pp (IARC Monographs
on the Evaluation of the Carcinogenic Risk of Chemicals to Humans,
Volume 35).
IARC (1986) Tobacco smoking. Lyon, International Agency for Research
on Cancer, 139 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 38).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC Monographs Volumes 1 to 42. Lyon, International Agency for
Research on Cancer, 403 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Supplement 7).
IARC (1989a) Diesel and gasoline engine exhausts and some nitroarenes.
Lyon, International Agency for Research on Cancer, 322 pp (IARC
Monographs on the Evaluation of Carcinogenic Risk of Chemicals to
Humans, Volume 46).
IARC (1989b) Occupational exposures in petroleum refining: Crude oil
and major petroleum fuels. Lyon, International Agency for Research on
Cancer, 283 pp (IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Man, Volume 45).
Ichinotsubo K, Mower HF, Settliff J, & Mandel M (1977) The use of rec-
bacteria for testing of carcinogenic substances. Mutat Res, 46: 53-
61.
Ilnitsky AP, Mischenko VS, & Shabad LM (1977) New data on volcanoes as
natural sources of carcinogenic substances. Cancer Lett, 3: 227-230.
Ingram AJ & Phillips JC (1993) The dermal bioavailability of
radiolabelled benzo(a)pyrene from acetone or from oils of differing
viscosity, assessed by DNA and protein binding. J Appl Toxicol, 13:
25-32.
Ingram LL Jr, McGinnis GD, Gjovik LR, & Roberson G (1982) Migration of
creosote and its components from treated piling sections in a marine
environment. In: Proceedings of the Seventy-eighth Annual Meeting of
the American Wood-preservers' Association, New Orleans, Louisiana, 2-5
May 1982. Bethesda, Maryland, American Wood-preservers' Association,
Volume 78, pp 120-128.
Ingram AJ, Lee R, & Phillips JC (1995) Factors affecting the
bioavailability of benzo(a)pyrene from oils in mouse skin: Oil
viscosity, grooming, activity and its prevention. J Appl Toxicol, 15:
175-182.
Inokuchi H & Nakagaki M (1959) The density of the polycyclic aromatic
compounds. Bull Chem Soc Jpn, 32: 65-67.
International Programme on Chemical Safety (1993) Environmental health
criteria 155: Biomarkers and risk assessment: Concepts and principles.
Geneva, World Health Organization, 82 pp.
International Union of Pure and Applied Chemistry (1979) Nomenclature
of organic chemistry. Sections A, B, C, D, E, F and H. Oxford,
Pergamon Press, pp 20-31.
International Union of Pure and Applied Chemistry (1987) Recommended
method for a thin-layer-chromatographic screening method for the
determination of benzo(a)pyrene in smoked food. Pure Appl Chem, 59:
1735-1738.
Iosifidou HG, Kilikidis SD, & Kamarianos AP (1982) Analysis for
polycyclic aromatic hydrocarbons in mussels (Mytilus
galloprovincialis) from the Thermaikos Gulf, Greece. Bull Environ
Contam Toxicol, 28: 535-541.
Ishidate M & Odashima S 81977) Chromosome tests with 134 compounds on
Chinese hamster cells in vitro: A screening for chemical
carcinogens. Mutat Res, 48: 337-354.
Ishinishi N, Kodema Y, Kunitake E, Nobutomo K & Fukushima Y (1976) The
carcinogenicity of dusts collected from an open-hearth furnace for the
smelting of iron: A preliminary experimental study. In: Nordberg GF,
ed. Effects and dose-response relationships of toxic metals.
Amsterdam, Elsevier Scientific Publishers, pp 480-488.
Ishio S, Chen JC, & Kawasaki Y (1977) Cell division of
Gyrodinium sp. and mitotic delay induced by causal substances of
algal tumor and carcinogens. Jpn Soc Sci Fish, 43: 507-516.
Iyer P, Gollahon LS, Martin JE, & Irvin TR (1990) Evaluation of the
in vitro growth of rodent preimplantation embryos exposed to
naphthalene in vivo. Toxicologist, 10: 274.
Iyer P, Martin JE, & Irvin TR (1991) Role of biotransformation in in
vitro preimplantation embryotoxicity of naphthalene. Toxicology, 66:
257-270.
Jacob J (1996) Mammalian organism - Cell cultures - Enzymes. A
comparison of in vivo and in vitro systems. In: Mohr M, Adler K,
Dungworth D, Harris C, Plopper C, & Saracci R ed. Correlations between
in vitro and in vivo investigations in inhalation toxicology.
Washington DC, International Life Sciences Institute Press, pp 279-
297.
Jacob J & Grimmer G (1979) Extraction and enrichment of polycyclic
aromatic hydrocarbons (PAH) from environmental matter. In: Egan H,
Castegnaro M, Bogovski P, Kunte H, & Walker EA ed. Environmental
carcinogens: Selected methods of analysis. Volume 3: Analysis of
polycyclic aromatic hydrocarbons in environmental samples. Lyon,
International Agency for Research on Cancer, pp 79-89 (IARC Scientific
Publications No. 29).
Jacob J & Grimmer G (1992) Organic analytical approaches. In: Rossbach
M, Schladot JD, & Ostapczuk P ed. Specimen banking. Berlin,
SpringerVerlag, pp 93-113.
Jacob J & Grimmer G (1994) [Environmental sample bank. PAH analysis in
different matrices. Annual Report 1993.] Grosshansdorf, Biochemical
Institute for Environmental Carcinogens, 50 pp (in German).
Jacob J & Grimmer G (1995) [Environmental sample bank. PAH analysis in
environmental samples. Annual Report 1994.] Grosshansdorf, Biochemical
Institute for Environmental Carcinogens, 50 pp (in German).
Jacob J, Grimmer G, & Schmoldt A (1981a) The influence of polycyclic
aromatic hydrocarbons as inducers of monooxygenases on the metabolic
profile of benz [a]anthracene in the rat liver microsomes. Cancer
Lett, 14: 175-185.
Jacob J, Schmoldt A, & Grimmer G (1981b) Glass-capillary-gas
chromatography/mass spectrometry data of mono- and polyhydroxylated
benz(a)anthracene. Comparison with benz(a)anthracene metabolites from
rat liver microsomes. Hoppe-Seyler's Z Physiol Chem, 362: 1021-1030.
Jacob J, Schmoldt A, & Grimmer G (1982a) Influence of monooxygenase
inducers on the metabolic profile of phenanthrene in rat liver
microsomes. Toxicology, 25: 333-343.
Jacob J, Schmoldt A, & Grimmer G (1982b) Formation of carcinogenic and
inactive chrysene metabilites by rat liver microsomes of various
monooxygenase activities. Arch Toxicol, 51: 255-265.
Jacob J, Schmoldt A & Grimmer G (1983) Benzo(e)pyrene metabolism in
rat liver microsomes: Dependence of the metabolite profile on the
pretreatment of rats with various monooxygenase inducers.
Carcinogenesis, 4: 905-919.
Jacob J, Brune H, Dettbarn G, Grimmer D, Heinrich U, Mohtashamipur E,
Norpoth K, Pott F, & Wenzel-Hartung R (1989) Urinary and faecal
excretion of pyrene and hydroxypyrene by rats after oral,
intraperitoneal, intratracheal or intrapulmonary application. Cancer
Lett, 46: 15-20.
Jacob J, Grimmer G, & Schneider D (1990a) Sampling and analysis of
polycyclic aromatic hydrocarbons present in particulate matter and in
semivolatiles (b.p. > 130°C) of ambient air. Fresenius' J Anal Chem,
37: 73.
Jacob J, Brune H, Grimmer G, Heinrich U, Mohtashamipur E, Norpoth K,
Pott F, & Wenzel-Hartung R (1990b) Urinary and faecal excretion of
metabolites after various modes of administration of polycyclic
aromatic hydrocarbons (PAH) to rats. In: Seemayer NH & Hadnagy W ed.
Environmental hygiene II. Berlin, Springer-Verlag, pp 87-90.
Jacob J, Grimmer G, Mohr U, Emura M, Riebe-Imre M, & Raab G (1992) The
metabolism of chrysene and benzo(a)pyrene in hamster, rat and human
lung cells in culture. In: Seemeyer NH & Hadnagy W ed. Environmental
hygiene III. Berlin, Springer-Verlag, pp 87-90.
Jacob J, Grimmer G, & Hildebrandt A (1993a) Correlation between PAH
concentrations measured in air, biological passive samplers and in
corresponding soil samples in Germany. In: Garrigues P & Lamotte M ed.
Polycyclic aromatic compounds: Synthesis, properties, analytical
measurements, occurrence and biological effects. Bordeaux, Gordon &
Breach Science Publishers, pp 427-433.
Jacob J, Grimmer G, & Hildebrandt A (1993b) The use of passive
samplers for monitoring polycyclic aromatic hydrocarbons in ambient
air. Sci Total Environ, 139/140: 307-321.
Jacob J, Grimmer G, Mohr U, Emura M, Riebe-Imre M, Raab G, & Knebel J
(1993c) Metabolic activation of chrysene and benzo [a]pyrene in
hamster, rat and human in vitro lung cell cultures. In: Garrigues P
& Lamotte M ed. Polycyclic aromatic compounds: Synthesis, properties,
analytical measurements, occurrence and biological effects. Bordeaux,
Gordon & Breach Science Publishers, pp 1175-1182.
Jacob J, Grimmer G, Hanssen HP, & Hildebrand A (1994) Extractability
and bioavailability of PAH from soil and air particulate matter.
Polycyclic Aromat Compd, 5: 209-217.
Jacob J, Doehmer J, Grimmer G, Soballa V, Raab G, Seidel A, & Greim H
(1996) Metabolism of phenanthrene, benz(a)anthracene, benzo(a)pyrene
and benzo(c)-phenanthrene by eight cDNA-expressed human and rat
cytochromes P450. Polycyclic Aromat Compd 10: 1-9.
Jacobs BW & Billings CE (1985) Characterization and temperature
dependence of PAH emissions from a simulated rubber combustion
operation. Am Ind Hyg Assoc J, 46: 547-554.
Jahn CL & Litman GW (1977) Distribution of covalently bound
benzo [a]pyrene in chromatin. Biochem Biophys Res Commun, 76:
534-540.
Jahn CL & Litman GW (1979) Accessibility of deoxyribonucleic acid in
chromatin to the covalent binding of the chemical carcinogen
benzo [a]pyrene. Biochemistry, 18: 1442-1449.
Jaklin J & Krenmayr P (1985) A routine method for the quantitative
determination of polycyclic aromatic hydrocarbons (PAHs) in urban air.
Int J Environ Anal Chem, 21: 33-42.
Jaklin J, Krenmayr P, & Varmuza K (1988) [Polycyclic aromatic
compounds in the atmosphere of Linz (Austria).] Fresenius' Z Anal
Chem, 331: 479-485 (in German).
James MO (1989) Biotransformation and disposition of PAH in aquatic
invertebrates. In: Varanasi U ed. Metabolism of polycyclic aromatic
hydrocarbons in the aquatic environment. Boca Raton, Florida, CRC
Press, pp 69-91.
Janini GM, Johnston K, & Zielinski WL Jr (1975) Use of nematic liquid
crystal for gas-liquid chromatographic separation of polyaromatic
hydrocarbons. Anal Chem, 47: 670-674.
Janini GM, Muschik GM, Schroer JA, & Zielinski WL Jr (1976) Gas-liquid
chromatographic evaluation and gas-chromatography/mass spectrometric
application of new high-temperature liquid crystal stationary phases
for polycyclic aromatic hydrocarbon separations. Anal Chem, 48: 1879-
1883.
Janssen O (1980) [Brief review of investigations on PAH in vehicle
exhaust, round robin tests on profile analysis, role of fuels and
lubricants, field studies. Air pollution from polycyclic aromatic
hydrocarbons. Registration and evaluation.] Düsseldorf, VDI-Verlag, pp
69-79 (VDI Report No. 358) (in German).
Japanese Ministry of International Trade and Industry (1992)
Biodegradation and accumulation, data of existing chemicals based on
the CSCL Japan. Tokyo: Japan Chemical Industry Ecology-toxicology and
Information Center, 467 pp.
Japenga J, Wagenaar WJ, Smedes F, & Salomons W (1987) A new, rapid
clean-up procedure for the simultaneous determination of different
groups of organic micropollutants in sediments. Application in two
European estuarine sediment studies. Environ Technol Lett, 8: 9-20.
Jeffrey AM, Jennette KW, Blobstein SH, Weinstein IB, Beland FA, Harvey
RG, Kasai H, Miura, & Nakananishi K (1976) Benzo [a]pyrene-nucleic
acid derivative found in vivo: Structure of a benzo[-]pyrene
tetrahydrodiol epoxide-guanosine adduct. J Am Chem Soc, 98: 5714-5715.
Jennette KW, Jeffrey AM, Blobstein SH, Beland FA, Harvey RG, &
Weinstein IB (1977) Nucleoside adducts from the in vitro reaction of
benzo [a]pyrene-7,8-dihydrodiol 9,10-oxide or benzo [a]pyrene
4,5-oxide with nucleic acids. Biochemistry, 16: 932-938.
Jerina DM, Lehr RE, Yagi H, Hernandez O, Dansette PM, Wislocki PG,
Wood AW, Chang RI, Levin W, & Conney AH (1976) Mutagenicity of
benzo [a]pyrene derivatives and the description of a quantum
mechanical model which predicts the ease of carbonium ion formation
from diol epoxides. In: De Serres FJ, Fouts JR, & Bend JR ed.
In vitro metabolic activation in mutagenesis testing. New York,
Elsevier North Holland, pp 159-178.
Jernström B & Gräslund A (1994) Covalent binding of benzo(a)pyrene
7,8-dihydrodiol 9,10-epoxides to DNA: Molecular structures, induced
mutations and biological consequences. Biophys Chem, 49: 185-199.
Jernström B, Vadi H, & Orrennius S (1978) Formation of DNA-binding
products from isolated benzo(a)pyrene metabolites in rat liver nuclei.
Chem-Biol Interactions, 20: 311-321.
Jernström B, Martinez M, Meyer DJ, & Ketterer B (1985) Glutathione
conjugation of the carcinogenic and mutagenic electrophile
(±)-7b,8a-dihydroxy-9a,10a-oxy-7,8,9,10-tetra hydrobenzo [a]pyrene
catalyzed by purified rat liver glutathione transferases.
Carcinogenesis, 6: 85-89.
Ji C & Marnett LJ (1992) Oxygen radical-dependent epoxidation of
(7 S,8 S)-dihyroxy-7,8-dihydrobenzo [a]pyrene in mouse skin
in vivo. J Biol Chem, 267: 17842-17848.
Jimenez BD, Cirmo CP, & McCarthy JF (1987) Effects of feeding and
temperature on uptake, elimination and metabolism of benzo(a)pyrene in
the bluegill sunfish (Lepomis macrochirus). Aquat Toxicol, 10: 41-
57.
Jockers R, Patalas N, Schacht S, Rehm H-J, & Freier D (1988)
[Development of biotechnological processes for the treatment of
wastewater from coal refinement: Final report.] Essen,
Bergbau-Forschung, 158 pp (Report No. BMFT FKZ 03 E-6284-A) (in
German).
Joe FL Jr, Salemme J, & Fazio T (1984) Liquid chromatographic
determination of trace residues of polynuclear aromatic hydrocarbons
in smoked foods. J Assoc Off Anal Chem, 67: 1076-1082.
Johnke B (1992) [Waste incineration as dioxin source or sink.]
ENTSORGA-Mag Entsorg.wirtsch, 9: 59-66 (in German).
Johnsen S, Kukkonen J, & Grande M (1989) Influence of natural aquatic
humic substances on the bioavailability of benzo(a)pyrene to Atlantic
salmon. Sci Total Environ, 81/82: 691-702.
Johnson S (1968) Effect of thymectomy on the induction of skin tumours
by dibenzanthracene, and of breast tumours by dimethylbenzanthracene
in mice of the IF strain. Br J Cancer, 22: 755-761.
Johnson AC, Larsen PF, Gadbois DF, & Humason AW (1985) The
distribution of polycyclic aromatic hydrocarbons in the superficial
sediments of Penobscot Bay (Maine, USA) in relation to possible
sources and to other sites worldwide. Mar Environ Res, 15: 1-16.
Jones PW, Giammar RD, Strup PE, & Stanford TB (1976) Efficient
collection of polycyclic organic compounds from combustion effluents.
Environ Sci Technol, 10: 806-810.
Jones KC, Stratford JA, Waterhouse KS, & Johnston AE (1987)
Polynuclear aromatic hydrocarbons in UK soils: Long-term temporal
trends and current levels. In: Hemphill DD ed. Trace substances in
environmental health. XXI. Proceedings of the University of Missouri's
21st Annual Conference on Trace Substances in Environmental Health, St
Louis, Missouri, 25-28 May 1987. Columbia, Missouri, University of
Missouri, pp 140-148.
Jones KC, Stratford JA, Waterhouse, & Vogt NB (1989a) Organic
contaminants in Welsh soils: Polynuclear aromatic hydrocarbons.
Environ Sci Technol, 23: 540-550.
Jones KC, Grimmer G, Jacob J, & Johnston AE (1989b) Changes in the
polynuclear aromatic hydrocarbon content of wheat grain and pasture
grassland over the last century from one site in the UK. Sci Total
Environ, 78: 117-130.
Jones KC, Stratford JA, Waterhouse KS, Furlong ET, Giger W, Hites RA,
Schaffner C, & Johnston AE (1989c) Increases in the polynuclear
aromatic hydrocarbon content of an agricultural soil over the last
century. Environ Sci Technol, 23: 95-101.
Jongeneelen FJ (1992) Biological exposure limit for occupational
exposure to coal tar pitch volatiles at coke ovens. Int Arch Occup
Environ Health, 63: 511-516.
Jongeneelen FJ (1994) Biological monitoring of environmental exposure
to polycyclic aromatic hydrocarbons: 1-Hydroxypyrene in urine of
people. Toxicol Lett, 72: 205-211.
Jongeneelen FJ, Bos RP, Anzion RBM, Theuws JLG, & Henderson PT (1986)
Biological monitoring of polycyclic aromatic hydrocarbons: Metabolites
in urine. Scand J Work Environ Health, 12: 137-143.
Jongeneelen FJ, Scheepers PTJ, Groenendijk A, Van Aerts LAGJM, Anzion
RBM, Bos RP, & Veenstra SJ (1988a) Airborne concentrations, skin
contamination, and urinary metabolite excretion of polycyclic aromatic
hydrocarbons among paving workers exposed to coal tar derived road
tars. Am Ind Hyg Assoc J, 49: 600-607.
Jongeneelen FJ, Anzion RBM, Scheepers PTJ, Bos RP, Henderson PT,
Nijenhuis EH, Veenstra SJ, Brouns RME, & Winkes A (1988b)
1-Hydroxypyrene in urine as a biological indicator of exposure to
polycyclic aromatic hydrocarbons in several work environments. Ann
Occup Hyg, 32: 35-43.
Jongeneelen FJ, van Leeuwen FE, Oosterink S, Anzion RBM, van der Loop
F, Bos RP, & van Veen HG (1990) Ambient and biological monitoring of
coke oven workers: Determinants of the internal dose of polycyclic
aromatic hydrocarbons. Br J Ind Med, 47: 454-461.
Joshi PL & Misra RB (1986) Evaluation of chemically-induced
phototoxicity to aquatic organism using Paramecium as a model.
Biochem Biophys Res Commun, 139: 79-84.
Jotz MM & Mitchell AD (1981) Effects of 20 coded chemicals on the
forward mutation frequency at the thymidine kinase locus in L5178Y
mouse lymphoma cells. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North Holland, pp 580-593
(Progress in Mutation Research, Volume 1).
Junk GA, Richard JJ, Avery MJ, Vick RD, & Norton GA (1986) Organic
compounds from coal combustion. Am Chem Soc Symp Ser, 319: 109-123.
Jury WA, Russo D, Streile G, & El Abd H (1990) Evaluation of
volatilization by organic chemicals residing below the soil surface.
Water Res, 26: 13-26.
Kadar R, Nagy K, & Frimstad D (1980) Determination of polycyclic
aromatic hydrocarbons in industrial waste water at the ng/ml level.
Talanta, 27: 227-230.
Kaden DA, Hites RA, & Thilly WG (1979) Mutagenicity of soot and
associated polycyclic aromatic hydrocarbons to Salmonella
typhimurium. Cancer Res, 39: 4152-4159.
Kagan J & Kagan ED (1986) The toxicity of benzo(a)pyrene and pyrene in
the mosquito Aedes aegypti, in the dark and in the presence of
ultraviolet light. Chemosphere, 15: 243-251.
Kagan J, Kagan ED, Kagan IA, Kagan PA, & Quigley S (1985) The
phototoxicity of non-carcinogenic polycyclic aromatic hydrocarbons in
aquatic organisms. Chemosphere, 14: 1829-1834.
Kagan J, Sinnott D, & Kagan E (1987) The toxicity of pyrene in the
fish Pimephales promelas. Synergism by piperonyl butoxide and by
ultraviolet light. Chemosphere, 16: 2291-2298.
Kagan J, Tuveson RW, & Gong HH (1989) The light-dependent cytotoxicity
of benzo [a]pyrene: Effect on human erythrocytes, Escherichia coli
cells, and Haemophilus influenzae transforming DNA. Mutat Res, 216:
231-242.
Kagi R, Alexander R, & Cumbers M (1985) Polycyclic aromatic
hydrocarbons in rock oysters: A baseline study. Int J Environ Anal
Chem, 22: 135-153.
Kaidbey KH & Nonaka S (1984) Action spectrum for anthracene-induced
photosensitization of human skin. Photochem Photobiol, 39: 375-378.
Kakunaga T (1973) A quantitative system for assay of malignant
transformation by chemical carcinogens using a clone derived from
BALB/3T3. Int J Cancer, 12: 463-473.
Kalberlah F, Frijus-Plessen N, & Hassauer M (1995) [Toxicological
criteria for the risk assessment of polyaromatic hydrocarbons (PAH) in
existing chemicals. Part 1: The use of equivalency factors.]
Altlasten-Spektrum, 5: 231-237 (in German).
Kallistratos G & Pfau A (1971) [Effect of 3,4-benzopyrene on large
mammals.] Naturwissenschaften, 58: 222 (in German).
Kamens RM, Zhishi G, Fulcher JN, & Bell D (1986a) The influence of
temperature on the daytime PAH decay on atmospheric soot particles.
Proceedings of the 79th Air Pollution Control Association Annual
Meeting, 23-27 June (No. 86-77-2), pp 1-19.
Kamens RM, Fulcher JN, & Zhishi G (1986b) Effects of temperature on
wood soot PAH decay in atmospheres with sunlight and low Nox
nitrogen oxides. Atmos Environ, 20: 1579-1587.
Kamens RM, Guo Z, Fulcher JN, & Bell DA (1988) Influence of humidity,
sunlight and temperature on the daytime decay of polyaromatic
hydrocarbons on atmospheric soot particles. Environ Sci Technol, 22:
103-108.
Kamens RM, Zhishi G, Fulcher JN, & Bell DA (1991) The influence of
humidity on the daytime decay of PAH on atmospheric soot particles.
In: Cooke M, Loening K, & Merrit J ed. Polynuclear aromatic
hydrocarbons: Measurement, means and metabolism. Columbus, Ohio,
Battelle Press, pp 435-451.
Kang DH, Rothman N, Poirier, MC, Greenberg A, Hsu CH, Schwartz BS,
Baser ME, Groopman JD, Weston A, & Strickland PT (1995)
Interindividual differences in the concentration of
1-hydroxypyrene-glucuronide in urine and polycyclic aromatic
hydrocarbon-DNA adducts in peripheral white blood cells after
charbroiled beef consumption. Carcinogenesis, 16: 1079-1085.
Kanij JBW (1987) The emission of polycyclic aromatic hydrocarbons by
coal-fired power stations in the Netherlands. Kema Sci Tech Rep, 5:
83-91.
Kanoh T, Fukuda M, Onozuka H, Kinouchi T, & Ohnishi Y (1993) Urinary
1-hydroxypyrene as a marker of exposure to polycyclic aromatic
hydrocarbons in environment. Environ Res, 62: 230-241.
Kao J, Patterson FK, & Hall J (1985) Skin penetration and metabolism
of topically applied chemicals in six mammalian species, including
man: An in vitro study with benzo [a]pyrene and testosterone.
Toxicol Appl Pharmacol, 81: 502-516.
Kappeler T & Wuhrmann K (1978) Microbial degradation of water soluble
fraction of gas oil. Water Res, 12: 327-333.
Karcher W (1988) Spectral atlas of polycyclic aromatic compounds,
Volume 2. Dordrecht, Kluwer Academic Publishers, pp 16-18, 55.
Karcher W, Dubois J, Fordham RJ, Glaude P, Barale R, & Zucconi D
(1984) Molecular spectra and mutagenic activity of some nitrogen
containing heterocyclic aromatic compounds. In: Cooke M & Dennis AJ
ed. Mechanisms, methods and metabolism: Polynuclear aromatic
hydrocarbons. Columbus, Ohio, Battelle Press, pp 685-696
Karcher W, Fordham R, Dubois JJ, Glaude PGJM, & Ligthart JAM (1985)
Spectral atlas of polycyclic aromatic compounds including data on
occurrence and biological activity. Dordrecht, D Reichel Publishing
Co., 818 pp.
Karcher W, Devillers J, Garrigues P, & Jacob J (1991) Spectral atlas
of polycyclic aromatic compounds.Dordrecht, Kluwer Academic
Publishers, p 21.
Karickhoff SW (1981) Semi-empirical estimation of sorption of
hydrophobic pollutants on natural sediments and soils. Chemosphere,
10: 833-846.
Karickhoff SW & Morris KR (1985) Sorption dynamics of hydrophobic
pollutants in sediment suspensions. Environ Toxicol Chem, 4: 469-479.
Karickhoff SW, Brown DS, & Scott TA (1979) Sorption of hydrophobic
pollutants on natural sediments. Water Res, 13: 241-248.
Karlehagen S, Andersen A, & Ohlson CG (1992) Cancer incidence among
creosote-exposed workers. Scand J Work Environ Health, 18: 26-29.
Karlesky DL, Ramelow G, Ueno Y, Warner IM, & Ho C-N (1987) Survey of
polynuclear aromatic compounds in oil refining areas. Environ Pollut,
43: 195-207.
Katz M, Heddle JA, & Salamone MF (1981) Mutagenic activity of
polycyclic aromatic hydrocarbons and other environmental pollutants.
Columbus, Ohio, Battelle Press, pp 519-528.
Kauss PB & Hutchinson TC (1975) The effects of water-soluble petroleum
components on the growth of Chlorella vulgaris
Beijerinck. Environ Pollut, 9: 157-174.
Kawabata TT & White KL (1987) Suppression of the in vitro humoral
immune response of mouse splenocytes by benzo(a)pyrene metabolites and
inhibition of benzo(a)pyrene-induced immunosuppression by
alpha-naphthoflavone. Cancer Res, 47: 2317-2322.
Kawabata TT & White KL (1990) Effects of naphthalene and naphthalene
metabolites on the in vitro humoral immune response. J Toxicol
Environ Health, 30: 53-67.
Kawai M (1979) [Review of toxicological and occupational aspects of
naphthalene.] Aromatics, 31: 168-181 (in Japanese).
Kawamura K & Kaplan IR (1986) Compositional change of organic matter
in rainwater during precipitation events. Atmos Environ, 20: 527-535.
Kawamura Y, Kamata E, Ogawa Y, Kaneko T, Uchiyama S, & Saito Y (1988)
The effect of various foods on the intestinal absorption of
benzo(a)pyrene in rats. J Food Hyg Soc Jpn, 29: 21-25.
Kayal SI & Connell DW. (1990) Partitioning of unsubstituted polycyclic
aromatic hydrocarbons between surface sediments and the water column
in the Brisbane River estuary. Aust J Mar Freshwater Res, 41: 443-456.
Kebbekus B, Greenberg A, Horgan L, Bozzelli J, Darack F, Eveleens C, &
Stangeland L (1983) Concentration of selected vapor and
particulate-phase substances in the Lincoln and Holland tunnels. J Air
Pollut Control Assoc, 33: 328-330.
Keller CD & Bidleman TF (1984) Collection of airborne polycyclic
aromatic hydrocarbons and other organics with a glass fiber
filter-polyurethane foam system. Atmos Environ, 18: 837-845.
Kelman BJ & Springer DL (1982) Movements of benzo [a]pyrene across
the hemochorial placenta of the guinea pig (41307). Proc Soc Exp Biol
Med, 169: 58-62.
Kemena A, Norpoth KH, & Jacob J (1988) Differential induction of the
monooxygenase isoenzymes in mouse liver microsomes by polycyclic
aromatic hydrocarbons. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: A decade of progress. Columbus, Ohio, Battelle
Press, pp 449-460.
Kennaway FL (1924) On the cancer-producing factor in tar. Br Med J, i:
564-567.
Kennaway EL (1930) Further experiments on cancer-producing substances.
Biochem J, 24: 497-504.
Kensler TW, Egner PA, Moore KG, Taffe BG, Twerdok LE, & Trush MA
(1987) Role of inflammatory cells in the metabolic activation of
polycyclic aromatic hydrocarbons in mouse skin. Toxicol Appl
Pharmacol, 90: 337-346.
Ketkar M, Green V, Schneider P, & Mohr U (1979) Investigations on the
carcinogenic burden by air pollution in man. Intratracheal
instillation studies with benzo(a)pyrene in a mixture of Tris buffer
and saline in Syrian golden hamsters. Cancer Lett, 6: 279-284.
Khalili NR, Scheff PA, & Holsen TM (1995) PAH source fingerprints for
coke ovens, diesel and gasoline engines, highway tunnels, and wood
combustion emissions. Atmos Environ, 29: 533-542.
Khesina AY (1994) Urban air pollution by carcinogenic and genotoxic
polyaromatic hydrocarbons in the former USSR. Environ Health Perspect,
102 (suppl 4): 49-53.
Kicinski HG, Adamek S, & Kettrup A (1989) Trace enrichment and HPLC
analysis of polycyclic aromatic hydrocarbons in environmental samples,
using solid phase extraction in connection with UV/VIS diode-array and
fluorescence detection. Chromatographia, 28: 203-208.
Kiene R & Capone D (1984) Effects of organic pollutants on
methanogenesis, sulfate reduction and carbon dioxide evolution in salt
marsh sediments. Mar Environ Res, 13: 141-160.
Kimber I, Jones K, & Vignali DAA (1986) The influence of
7,12-dimethybenzanthracene on natural killer (NK) cell function in
rats. J Clin Lab Immunol, 20: 193-198.
Kincannon DF & Lin YS (1985) Microbial degradation of hazardous wastes
by land treatment. In: Proceedings of the 40th Industrial Waste
Conference, Purdue University, West Lafayette, Indiana, 14-16 May,
1985. Boston, Butterworth Press, pp 607-619.
King HWS, Osborne MR, & Brookes P (1979) The in vitro and
in vivo reaction at the N7 position of guanine of the ultimate
carcinogen derived from benzo [a]pyrene. Chem-Biol Interactions, 24:
345-353.
Kirchmann H, Aström H, & Jönsäll G (1991) Organic pollutants in sewage
sludge. 1. Effect of toluene, naphthalene, 2-methylnaphthalene,
4-n-nonylphenol and di-2-ethylhexyl phthalate on soil biological
processes and their decomposition in soil. Swed J Agric Res, 21: 107-
113.
Kishi H, Kogure N, & Hashimoto Y (1990) Contribution of soil
constituents in adsorption coefficient of aromatic compounds,
halogenated alicyclic and aromatic compounds to soil. Chemosphere, 21:
867-876.
Kjuus H, Andersen A, Langard S, & Knudsen KE (1986) Cancer incidence
among workers in the Norwegian ferroalloy industry. Br J Ind Med, 43:
227-236.
Klassert A (1993) [Demonstration plant for adsorptive purification of
waste water from basic chemical industry.] Berlin, Ministry of
Environment, 93 pp (Report No. UBA 20441-5/2) (in German).
Klein M (1960) A comparison of the initiating and promoting actions of
9,10-dimethyl-1,2-benzanthracene and 1,2,5,6-dibenzanthracene in skin
tumorigenesis. Cancer Res, 20: 1179-1183.
Klein M (1963) Susceptibility of strain B6AF1/J hybrid infant mice to
tumorigenesis with 1,2-benzanthracene, deoxycholic acid, and
3-methylcholanthrene. II. Tumours called forth by painting the skin
with dibenzpyrene. Cancer Res, 23: 1701-1707.
Kleinenberg HE (1939) [Investigations on the blastomogenic effect of
3:4:8:9- dibenzpyrene and some of its derivatives.] Arch Biol Nauk,
56: 39-47 (in Russian).
Klemme JC, Mukhtar H, & Elmets CA (1987) Induction of contact
hypersensitivity to dimethylbenz(a)anthracene and benzo(a)pyrene in
C3H/HeN mice. Cancer Res, 47: 6074-6078.
Kliesch U, Roupova I, & Adler ID (1982) Induction of chromosome damage
in mouse bone marrow by benzo [a]pyrene. Mutat Res, 102: 265-273.
Klingenberg H, Schürmann D, & Lies K-H (1992) Specific air pollutants
from passenger cars: Emission and ambient air concentrations. In:
Toxic air pollutants from mobile sources: Emissions and health
effects. Proceedings of an International Specialty Conference.
Pittsburgh, Pennsylvania, Air & Waste Management Association, pp 82-
93.
Klöpffer W, Rippen G, & Frische R (1982) Physicochemical properties as
useful tools for predicting the environmental fate of organic
chemicals. Ecotoxicol Environ Saf, 6: 294-301.
Klug A (1950) The crystal and molecular structure of triphenylene,
C18H12. Acta Crystallogr, 3: 165-175.
Knaap AGAC, Goze C, & Simons JWIM (1981) Mutagenic activity of seven
coded samples in V79 Chinese hamster cells. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens. Report of the
international collaborative programme. New York, Elsevier/North
Holland, pp 608-613 (Progress in Mutation Research, Volume 1).
Knake E (1956) [Weak tumor-inducing effect of naphthalene and
benzene.] Virchows Arch, 329: 141-116 (in German).
Knecht U & Woitowitz H-J (1990) [Cancer risk from use of tar bitumen
in road building.] Bremerhaven, Wirtschaftsverlag NW Verlag für neue
Wissenschaft GmbH, 47 pp (in German).
Knecht U, Elliehausen H-J, & Woitowitz H-J (1986) Gaseous and adsorbed
PAH in an iron foundry. Br J Ind Med, 43: 834-838.
Knecht U, Elliellausen HJ, Judas W, & Woitowitz HJ (1987) Polycyclic
aromatic hydrocarbons (PAH) in abraded particles of brake and clutch
linings. Int J Environ Anal Chem, 28: 227-236.
Knecht U, Bolm-Audorff U, & Woitowitz HJ (1989) Atmospheric
concentrations of polycyclic aromatic hydrocarbons during chimney
sweeping. Br J Ind Med, 46: 479-481.
Knight CV & Humphreys MP (1985) Polynuclear aromatic hydrocarbons in
indoor residential air resulting from use of conventional and
catalytic wood heaters in a weatherized home. In: Cooke M & Dennis AJ
ed. Mechanisms, methods and metabolism: Polynuclear aromatic
hydrocarbons. Columbus, Ohio, Battelle Press, pp 725-737.
Knutzen J & Sortland B (1982) Polycyclic aromatic hydrocarbons (PAH)
in some algae invertebrates from moderately polluted parts of the
coast of Norway. Water Res, 16: 421-428.
Kochevar IE, Armstrong RB, Einbinder J, Walther RR, & Harber LC (1982)
Coal tar phototoxicity: Active compounds and action spectra. Photochem
Photobiol, 36: 65-69.
Konar NR, Roy HK, & De MN (1939) Naphthalene poisoning. Indian Med
Gaz, 74: 723-725.
Kongerud J, Boe J, Soyseth V, Naalsund A, & Magnus P (1994) Aluminium
potroom asthma: The Norwegian experience. Eur Respir J, 7: 165-172.
König W, Hembrock-Heger A, & Wilkens (1991) [Persistent organic
chemicals in soil - routes of entry and occurrence]. Umweltwissensch
Schadstoff-Forsch, 3: 33-36 (in German).
Kootstra A & Slaga TJ (1979) Differential accessibility of (±)
trans-7b,8a-dihydroxy-9a,10a-epoxy-7,8,9,10-tetrahydrobenzo [a]
pyrene to histone proteins. FEBS Lett, 108: 321-325.
Kootstra A & Slaga TJ (1980) Binding of isomers of benzo [a]pyrene
diol-epoxide to chromatin. Biochem Biophys Res Commun, 93: 954-959.
Kootstra A, Slaga TJ, & Olins DE (1979) Interaction of
benzo [a]pyrene diol-epoxide with nuclei and isolated chromatin.
Chem-Biol Interactions, 28: 225-236.
Kördel W, Schoene K, Bruckert J, Pfeiffer U, Schreiber G, Rittmann D,
Hochrainer D, Otto F, Spiegelberg T, Fingerhut R, Kuhnen-Clausen D, &
König J (1981) [Test for chemicals: Study on the feasibility of the EC
regulatory test methods.] Hanover, Fraunhofer Institute for Toxicology
and Aerosol Research, Volume 1, p 553 (in German).
Koreeda M, Moore PD, Wislocki PG, Levin W, Conney AH, Yagi H, & Jerina
DM (1978) Binding of benzo[a]pyrene 7,8-diol 9,10-epoxides to DNA,
RNA, and protein of mouse skin occurs with high stereoselectivity.
Science, 199: 778-780.
Korhonen K & Mulari K (1983) Airborne pollutants in ties workers.
Lappeenrannan, Finnish Institute of Occupational Health, 23 pp.
Korn S, Moles D, & Rice SD (1979) Effects of temperature on the median
tolerance limit of pink salmon and shrimp exposed to toluene,
naphthalene, and Cook Inlet crude oil. Bull Environ Contam Toxicol,
21: 521-525.
Kotin P, Falk HL, & Busser R (1959) Distribution, retention, and
elimination of C14-3,4-benzpyrene after administration to mice and
rats. J Natl Cancer Inst, 23: 541-555.
Kouri RE, Connolly GM, Nebert DW, & Lubet RA (1983) Association
between susceptibility to dibenzanthracene induced fibrosarcoma
formation and the Ah locus. Int J Cancer, 32: 765-768.
Krahn DF & Heidelberger C (1977) Liver homogenate-mediated mutagenesis
in Chinese hamster V79 cells by polycyclic aromatic hydrocarbons and
aflatoxins. Mutat Res, 46: 27-44.
Krämer M, Bimboes D, & Greim H (1974) Salmonella typhimurium and E.
coli to detect chemical mutagens. Arch Pharmacol, 284: R46.
Krewski D, Thorslund T, & Withey J (1989) Carcinogenic risk assessment
of complex mixtures. Toxicol Ind Health, 5: 851-867.
Kriek E, Van Schooten FJ, Hillebrand MJX, Van Leeuwen FE, Den Engelse
L, De Looff AJA, & Dijkmans APG (1993) DNA adducts as a measure of
lung cancer risk in humans exposed to polycyclic aromatic
hydrocarbons. Environ Health Perspectives, 99: 71-75.
Kröber B & Häckl M (1989) [Report on orientation measurements on
dangerous, organic compounds in wastewater pipes, sewage works and
waters in the Federal State of Hessen (1985-1988)] .Wiesbaden, Hessen
State Office for the Environment, pp 225-288 (Report 96) (in German).
Krokje Å (1989) Mutagenicity of expectorate from workers in a coke
plant. Mutat Res, 223: 213-219.
Krokje Å, Tiltnes A, Mylius E, & Gullvå B (1988) Testing for mutagens
in an aluminium plant. The results of Salmonella
typhimurium tests on urine from exposed workers. Mutat Res, 204:
163-172.
Krolewski B, Nagasawa H, & Little JB (1986) Effect of aliphatic amides
on oncogenic transformation, sister chromatid exchanges, and mutations
induced by cyclopenta [cd]-pyrene. Carcinogenesis, 7: 1647-1650.
Kronberger H & Weiss J (1944) Formation and structure of some organic
molecular compounds. Part III. The dielectric polarisation of some
solid crystalline molecular compounds. J Chem Soc, 146: 464-469.
Kruber O (1937) [Some new constituents of coal tar pitch.] Chem Ber,
70: 1556-1564 (in German).
Kruber O & Grigoleit G (1954) [New compounds isolated from coal tar
pitch.] Chem Ber, 87: 1895-1905 (in German).
Kruber O & Marx A (1938) [The anthracene oil of coal tar.] Chem Ber,
71: 2478-2484 (in German).
Kubasiewicz M, Starzynski Z, & Szymczak W (1991) Case-referent study
on skin cancer and its relation to occupational exposure to polycyclic
aromatic hydrocarbons. II. Study results. Pol J Occup Med Environ
Health, 4: 141-147.
Kunstler K (1983) Failure to induce tumors by intratracheal
instillation of automobile exhaust condensate and fractions thereof in
Syrian golden hamsters. Cancer Lett, 18: 105-108.
Kuroki T & Heidelberger C (1972) Determination of the h-protein in
transformable and transformed cells in culture. Biochemistry, 11:
2116-2124.
Kuroki T, Nemoto N, & Kitano Y (1980) Metabolism of benzo [a]pyrene
in human epidermal keratinocytes in culture. Carcinogenesis, 1: 559-
565.
Kushwaha SC, Clarkson SG & Mehkeri KA (1985) Polycyclic aromatic
hydrocarbons in barbecue briquets. J Food Saf, 7: 177-201.
Kuwabara K, Nakamura A, & Kashimoto T (1980) Effect of petroleum oil,
pesticides, PCBs and other environmental contaminants on the
hatchability of Artemia salina dry eggs. Environ Contam Toxicol, 25:
69-74.
Kveseth K, Sortland B, & Bokn T (1982) Polycyclic aromatic
hydrocarbons in sewage, mussels and tap water. Chemosphere, 11: 623-
639.
La Budde JA & Heidelberger C (1958) The synthesis of the mono- and
dihydroxy derivatives of 1,2,5,6-dibenzanthracene excreted by the
rabbit and of other hydroxylated dibenzanthracene derivatives. J Am
Chem Soc, 80: 1225-1236.
Lacassagne A, Buu-Hoi NP, & Zajdela F (1958) [Relationship between
molecular structure and carcinogenic activity in three series of
hexacyclic aromatic hydrocarbons.] C R Acad Sci Paris, 245: 1477-1481
(in French).
Lacassagne A, Buu-Hoi NP, Zajdela F, Lavit-Lamy D, & Chalvet O (1963a)
[Carcinogenic activity of fluoranthene-based polycyclic aromatic
hydrocarbons.] Acta Unio Int Contra Cancrum, 19: 490-496 (in French).
Lacassagne A, Buu-Hoi NP, Zajdela F, & Lavit-Lamy D (1963b) [High
carcinogenic activity of 1,2:3,4-dibenzopyrene and
1,2:4,5-dibenzopyrene.] C R Acad Sci Paris, 256: 2728-2731 (in
French).
Lacassagne A, Buu-Hoi NP, Zajdela F, & Vingiello FA (1968a) The true
dibenzo [a,l]pyrene, a new, potent carcinogen. Naturwissenschaften,
55: 43.
Lacassagne A, Buu-Hoi NP, & Zajdela F (1968b) [Absence of sarcomagenic
property in dibenz [a,c]anthracene; clear activity of its 10-methyl
derivative.] Eur J Cancer, 4: 123-127 (in French).
Ladics GS & White KL (1996) Immunotoxicity of polyaromatic
hydrocarbons. In: Smialowicz RJ & Holsapple MP ed. Experimental
immunotoxicology. Boca Raton, Florida, CRC Press, pp 331-349.
Ladics GS, Kawabata TT, & White KL Jr (1991) Suppression of the
in vitro humoral immune response of mouse splenocytes by
7,12-dimethylbenz(a)anthracene metabolites and inhibition of
immunosuppression by a-naphthoflavone. Toxicol Appl Pharmacol, 110:
31-44.
Ladics GS, Kawabata TT, Munson AE, & White KL Jr (1992a) Generation of
7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo [a]pyrene by murine
splenic macrophages. Toxicol Appl Pharmacol, 115: 72-79.
Ladics GS, Kawabata TT, Munson AE, & White KL Jr (1992b) Metabolism of
benzo [a]pyrene by murine splenic cell types. Toxicol Appl Pharmacol,
116: 248-257.
Lafontaine M, Attenont H, Hubert G, Taiclet A, & Truy S (1990a)
[Emission of polycyclic aromatic hydrocarbons in foundries.] Cah Notes
Doc, 141: 799-807 (in French).
Lafontaine M, Attenont H, & Truy S (1990b) [Exposure to polycyclic
aromatic hydrocarbons in the electrode-producing industry]. Cah Notes
Doc, 132: 453-455.
Lahmann E, Seifert B, Zhao L, & Bake D (1984) [Immission of polycyclic
aromatic hydrocarbons in Berlin (West).] Staub-Reinhalt Luft, 44: 149-
157 (in German).
Lake RS, Kropko ML, Pezzutti MR, Shoemaker RH, & Igel HJ (1978)
Chemical induction of unscheduled DNA synthesis in human skin
epithelial cell cultures. Water Res, 38: 2091-2098.
Lampe V, Puettmann W, & Kasig W (1991) Analysis of the PAH load of
river sediments from the Aachen area. Contribution to the
determination of emission/immission pathways. Wiss Umwelt, 1: 51-62.
Landahl JT, McCain BB, Myers MS, Rhodes LD, & Brown DW (1990)
Consistent associations between hepatic lesions in English sole
(Parophrys vetulus) and polycyclic aromatic hydrocarbons in bottom
sediment. Environ Health Perspectives, 89: 195-203.
Landrum PF (1982) Uptake, depuration and biotransformation of
anthracene by the scud Pontoporeia hoyi. Chemosphere, 11: 1049-1057.
Landrum PF (1988) Toxicokinetics of organic xenobiotics in the
amphipod Pontoporeia hoyi: Role of physiological and environmental
variables. Aquat Toxicol, 12: 245-271.
Landrum PF & Poore R (1988) Toxicokinetics of selected xenobiotics in
Heagenia limbata. J Great Lakes Res, 14: 427-437.
Landrum PF & Scavia D (1983) Influence of sediment on anthracene
uptake, depuration, and biotransformation by the amphipod
Hyalella azteca. Can J Fish Aquat Sci, 40: 298-305.
Landrum PF, Bartell SM, Giesy JP, Leversee GJ, Bowling JW, Haddock J,
LaGory K, Gerould S, & Bruno M (1984a) Fate of anthracene in an
artificial stream: A case study. Ecotoxicol Environ Saf, 8: 183-201.
Landrum PF, Nihart SR, Edie BJ, & Gardner WS (1984b) Reverse-phase
separation method for determining pollutant binding to Aldrich humic
acid and dissolved organic carbon of natural waters. Environ Sci
Technol, 18: 187-192.
Landrum PF, Eadie BJ, & Faust WR (1991) Toxicokinetics and toxicity of
a mixture of sediment-associated polycyclic aromatic hydrocarbons to
the amphipod Diporeia sp. Environ Toxicol Chem, 10: 35-46.
Lane DA (1989) The fate of polycyclic aromatic compounds in the
atmosphere and during sampling. In: Vo-Dinh T ed. Chemical analysis of
polycyclic aromatic compounds. New York, John Wiley & Sons, pp 31-58.
Lane DA & Katz M (1977) The photomodification of benzo [a]pyrene,
benzo [b]-fluoranthene, and benzo [k]fluoranthene under simulated
atmospheric conditions. Adv Environ Sci Technol, 8: 137-154.
Langenfeld JL, Hawthorne SB, Miller DJ, & Pawliszyn J (1993) Effects
of temperature and pressure on supercritical fluid extraction
efficiencies of polycyclic aromatic hydrocarbons and polychlorinated
biphenyls. Anal Chem, 65: 338-344.
Lao RC, Thomas RS, Oja H, & Dubois L (1973) Application of a gas
chromatograph-mass spectrometer-data processor combination to the
analysis of the polycyclic aromatic hydrocarbon content of airborne
pollutants. Anal Chem, 45: 908-915.
Larssen S (1985) Automotive emission factors: An indirect measurement
method applied to polycyclic aromatic hydrocarbon and lead emissions.
In: Proceedings of the Air Pollution Control Association, 77th Annual
Meeting, San Francisco, 24-29 June 1984. Houston, Texas, Air Pollution
Control Association, pp 1-18.
Larsson BK (1982) Polycyclic aromatic hydrocarbons in smoked fish. Z
Lebensm Untersuch Forsch, 174: 101-107.
Larsson B (1986) Polycyclic aromatic hydrocarbons in Swedish foods:
Aspects on analysis, occurrence and intake. Uppsala, Swedish
University of Agricultural Sciences, 60 pp (Doctoral Thesis).
Larsson B & Sahlberg G (1982) Polycyclic aromatic hydrocarbons in
lettuce. Influence of a highway and an aluminium smelter. In: Cooke M
& Fisher AJ ed. Polynuclear aromatic hydrocarbons: Physical and
biological chemistry. Columbus, Ohio, Battelle Press, pp 417-426.
Larsson BK, Sahlberg GP, Eriksson AT, & Busk LA (1983) Polycyclic
aromatic hydrocarbons in grilled food. J Agric Food Chem, 31: 867-873.
Larsson BK, Eriksson AT, & Cervenka M (1987) Polycyclic aromatic
hydrocarbons in crude and deodorized vegetable oils. J Am Oil Chem
Soc, 64: 365-370.
Laskin S, Kuschner M, & Drew RT (1970) Studies in pulmonary
carcinogenesis. In: Hanna MG, Nettesheim P, & Gilbert J ed. Inhalation
carcinogenesis. Oak Ridge, Tennessee, US Atomic Energy Commission,
Division of Technical Information, pp 321-351 (AEC Symposium Series
No. 18).
Lasnitzki A & Woodhouse DL (1944) The effect of
1:2:5:6-dibenzanthracene on the lymph nodes of the rat. J Anat, 78:
121-128.
LaVoie EJ, Bedenko V, Hirota N, Hecht SS, & Hoffmann D (1979) A
comparison of the mutagenicity, tumor-initiating activity and complete
carcinogenicity of polynuclear aromatic hydrocarbons. In: Jones PW &
Leber P ed. Polynuclear aromatic hydrocarbons. Ann Arbor, Michigan,
Ann Arbor Science Publishers, pp 705-721.
LaVoie EJ, Tulley L, Bedenko V, & Hoffmann D (1980) Mutagenicity,
tumor initiating activity, and metabolism of tricyclic polynuclear
aromatic hydrocarbons. In: Bjorseth A & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemistry and biological effects. Columbus,
Ohio, Battelle Press, pp 1041-1057.
LaVoie EJ, Tulley L, Bedenko V, & Hoffmann D (1981a) Mutagenicity of
methylated fluorenes and benzofluorenes. Mutat Res, 91: 167-176.
LaVoie EJ, Tulley-Freiler L, Bedenko V, & Hoffmann D (1981c)
Mutagenicity, tumor-initiating activity and metabolism of
methylphenanthrenes. Cancer Res, 41: 3441-3447.
LaVoie EJ, Tulley-Freiler L, Bedenko V, Girach Z, & Hoffmann D (1981c)
Comparative studies on the tumor initiating activity and metabolism of
methylfluorenes and methylbenzofluorenes. In: Cooke M & Dennis AJ ed.
Chemical analysis and biologicalfate: Polynuclear aromatic
hydrocarbons. Columbus, Ohio, Battelle Press, pp 417-427.
La Voie EJ, Hecht SS, Bedenko V, & Hoffmann D (1982a) Identification
of the mutagenic metabolites of fluoranthene, 2-methylfluoranthene,
and 3-methylfluoranthene. Carcinogenesis, 3: 841-846.
La Voie EJ, Amin S, Hecht SS, Furuya K, & Hoffmann D (1982b) Tumour
initiating activity of hydrodiols of benzo [b]fluoranthene,
benzo [j]fluoranthene, and benzo [k]-fluoranthene. Carcinogenesis,
3: 49-53.
LaVoie EJ, Coleman DT, Tonne RL, & Hoffmann D (1983a) Mutagenicity,
tumor initiating activity and metabolism of methylated anthracenes.
In: Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons.
Columbus, Ohio, Battelle Press, pp 785-798.
LaVoie EJ, Tulley-Freiler L, Bedenko V, & Hoffmann D (1983b)
Mutagenicity of substituted phenanthrenes in Salmonella
typhimurium. Mutat Res, 116: 91-102.
LaVoie EJ, Coleman DT, Rice JE, Geddie NG & Hoffmann D (1985)
Tumor-initiating activity, mutagenicity, and metabolism of methylated
anthracenes. Carcinogenesis, 6: 1483-1488.
LaVoie EJ, Braley J, Rice JE, & Rivenson A (1987) Tumorigenic activity
of non-alternant polynuclear aromatic hydrocarbons in newborn mice.
Cancer Lett, 34: 15-20.
LaVoie EJ, Cai ZW, Meschter CL, & Weyand EH (1994) Tumorigenic
activity of fluoranthene, 2-methylfluoranthene in newborn CD-1 mice.
Carcinogenesis, 15: 2131-2135.
Lawrence JF & Weber DF (1984) Determination of polycyclic aromatic
hydrocarbons in Canadian samples of processed vegetable and dairy
products by liquid chromatography with fluorescence detection. J Agric
Food Chem, 32: 794-797.
Leach JM, Otson R, & Armstrong V (1987) Airborne contaminants in two
small Canadian coal liquefaction pilot plants. Am Ind Hyg Assoc J, 48:
693-697.
Leadon SA, Stampfer MR, & Bartley J (1988) Production of oxidative DNA
damage during the metabolic activation of benzo [a]pyrene in human
mammary epithelial cells correlates with cell killing. Proc Natl Acad
Sci USA, 85: 4365-4368.
LeBlanc GA (1980) Acute toxicity of priority pollutants to water flea
(Daphnia magna). Environ Contam Toxicol, 24: 684-691.
Lecoq S, Perin F, Plessis MJ, Strapelias H, & Duquesne M (1989)
Comparison of the in vitro metabolisms and mutagenicities of
dibenz [a,c]anthracene, dibenz [a,h]anthrancene, and
dibenz [a,j]anthrancene: Influence of norharman. Carcinogenesis, 10:
461-469.
Lecoq S, Ni She M, Hewer A, Grover PL, Platt KL, Oesch F, & Phillips
DH (1991) The metabolic activation of dibenz [a,h]anthracene in mouse
skin examined by 32P-postlabelling: Minor contribution of the
3,4-diol 1,2-oxides to DNA binding. Carcinogenesis, 12: 1079-1083.
Lecoq S, Pfau W, Grover PL, & Phillips DH (1992) HPLC separation of
32P-postlabelled DNA adducts formed from dibenz [a,h]anthracene in
skin. Chem-Biol Interactions, 85: 173-185.
Lee RF (1977) Fate of petroleum components in estuarine water of
southeastern United States. In: Proceedings of the oil spill
conference: Prevention, behavior, control, cleanup, New Orleans, 8-10
March 1997. Washington DC, American Petroleum Institute, pp 611-616
(API Publication No. 4284).
Lee RF & Andersen JW (1977) Fate and effect of naphthalenes:
Controlled ecosystem pollution experiment. Bull Mar Sci, 27: 127-134.
Lee H & Lin JY (1988) Antimutagenic activity of extracts from
anticancer drugs in Chinese medicine. Mutat Res, 204: 229-234.
Lee WY & Nicol JAC (1978) The effect of naphthalene on survival and
activity of the amphipod Parhyale hawaiensis. Bull Environ Contam
Toxicol, 20: 233-240.
Lee RF & Ryan C (1976) Biodegradation of petroleum hydrocarbons by
marine microbes. In: Sharpley JM & Kaplan AM ed. Biodeterioration of
materials. Milton Keynes, Essex, Applied Science Publishers, pp 119-
125.
Lee FSC & Schuetzle D (1983) Sampling, extraction, and analysis of
polycyclic aromatic hydrocarbons from internal combustion engines. In:
Bjorseth A ed. Handbook of polycyclic aromatic hydrocarbons. New York,
Marcel Dekker, pp 27-94.
Lee RF, Sauerheber R, & Dobbs G (1972) Uptake, metabolism and
discharge of polycyclic aomatic hydrocarbons by marine fish. Mar Biol,
17: 201-208.
Lee ML, Novotny M, & Bartle KD (1976a) Gas chromatography/mass
spectrometric and nuclear magnetic resonance determination of
polynuclear aromatic hydrocarbons in airborne particulates. Anal Chem,
48: 1566-1572.
Lee ML, Novotny M, & Bartle KD (1976b) Gas chromatography/mass
spectrometric and nuclear magnetic resonance spectrometric studies of
carcinogenic polynuclear aromatic hydrocarbons in tobacco and
marijuana smokes condensates. Anal Chem, 48: 405-416.
Lee FS-C, Harvey TM, Prater TJ, Paputa MC, & Schuetzle D (1980)
Chemical analysis of diesel particulate matter and an evaluation of
artifact formation. In: Sampling and analysis of toxic organics in the
atmosphere. Philadelphia, Pennsylvania, American Society for Testing
and Materials, pp 92-110 (ASTM STP 721).
Lee ML, Novotny MV, & Bartle KD (1981) Analytical chemistry of
polycyclic aromatic compounds. New York, Academic Press, pp 35-40, 78-
122, 156-289.
Lee MD, Wilson JT, & Ward CH (1984) Microbial degradation of selected
aromatics in a hazardous waste site. Dev Ind Microbiol, 25: 557-565.
Lee PM, Baoyun Y, Herbert R, Hamminki K, Perera FP, & Santella RM
(1991) Immunologic measurement of polycyclic aromatic
hydrocarbon-albumin adducts in foundry workers and roofers. Scand J
Work Environ Health, 17: 190-194.
de Leeuw JW, de Leer EWB, Sinninghe Damsté JS, & Schuyl PJW (1986)
Screening of anthropogenic compounds in polluted sediments and soils
by flash evaporation/pyrolysis gas chromatography-mass spectrometry.
Anal Chem, 58: 1852-1857.
Legraverend C, Harrison DE, Ruscetti FW, & Nebert DW (1983) Bone
marrow toxicity induced by oral benzo [a]pyrene: Protection resides
at the level of the intestine and liver. Toxicol Appl Pharmacol, 70:
390-401.
Legraverend C, Guenthner TM, & Nebert DW (1984) Importance of the
route of administration for genetic differences in
benzo [a]pyrene-induced in utero toxicity and teratogenicity.
Teratology, 29: 35-47.
Lehmann EJ, Auffarth J, Häger J, Rentel KH, & Altenburg H (1986)
[Concentration profiles of selected PAH in coal-tar derived products.]
Staub-Reinhalt Luft, 46: 128-131 (in German).
Lei ZM (1993) The relationship between concentration of B(a)P in
blood, urine, and immune function of coking workers. Chung Hua Yu Fang
I. Hsueh Tsa Chih, 27: 212-214 (in Chinese).
Leinster P & Evans MJ (1986) Factors affecting the sampling of
airborne polycyclic aromatic hydrocarbons. A review. Ann Occup Hyg,
30: 481-495.
Lenin K (1994) Contamination with polycyclic aromatic hydrocarbons
through sewage irrigation. Doctoral thesis submitted to the Jawaharlal
Nehru University, Delhi, India, 22 pp.
Leo A, Hansch C, & Elkins D (1971) Partition coefficients and their
uses. Chem Rev, 71: 525-616.
Leoni V, Puccetti G, & Grella A (1975) Preliminary results on the use
of Tenax(R) for the extraction of pesticides and polynuclear
aromatic hydrocarbons from surface and drinking waters for analytical
purposes. J Chromatogr, 106: 119-124.
Lepperhoff G (1981) [The PAH emissions of spark ignition engines. Part
I.] Wiss Umwelt, 2: 72-87 (in German).
Lesage J, Perrault G, & Durand P (1987) Evaluation of worker exposure
to polycyclic aromatic hydrocarbons. Am Ind Hyg Assoc J, 48: 753-759.
Leslie TJ, Dickson KL, Jordan JA, & Hopkins DW (1987) Effects of
suspended solids on the water column biotransformation of anthracene.
Arch Environ Contam Toxicol, 16: 637-642.
Leversee GJ, Landrum PJ, Bartell S, Gerould S, Bruno M, Spacie A,
Bowling J, Haddock J, & Fannin T (1981) Disposition of
benzo [a]pyrene in aquatic system components: Periphyton,
Chironomids, Daphnia, fish. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 357-367.
Levin W, Wood AW, Chang RL, Yagi H, Mah HD, Jerina DM, & Conney AH
(1978) Evidence for bay region activation of chrysene 1,2-dihydrodiol
to an ultimate carcinogen. Cancer Res, 38: 1831-1834.
Levin W, Wood AW, Chang RL, Ittah Y, Croisy-Delcey M, Yagi H, Jerina
DM, & Conney AH (1980) Exceptionally high tumor-initiating activity of
benzo(c)phenanthrene bay-region diol-epoxides on mouse skin. Cancer
Res, 40: 3910-3914.
Levin W, Wood A, Chang R, Ryan D, Thomas P, Yagi H, Thakker D, Vyas K,
Boyd C, Chu S-Y, Conney A, & Jerina D (1982) Oxidative metabolism of
polycyclic aromatic hydrocarbons to ultimate carcinogens. Drug Metab
Rev, 13: 555-580.
Levin W, Chang RL, Wood AW, Yagi H, Thakker DR, Jerina DM, & Conney AH
(1984) High stereoselectivity among the optical isomers of the
diastereomeric bay-region diolepoxides of benz(a)anthracene in the
expression of tumorigenic activity in murine tumor models. Cancer Res,
44: 929-933.
Levin W, Chang RL, Wood AW, Thakker DR, Yagi H, Jerina DM, & Conney AH
(1986) Tumorigenicity of optical isomers of the diastereomeric
bay-region 3,4-diol-1,2-epoxides of benzo [c]phenanthrene in murine
tumor models. Cancer Res, 46: 2257-2261.
Levin JO, Rhén M, & Sikström E (1995) Occupational PAH exposure:
Urinary 1-hydroxypyrene levels of coke oven workers, aluminium smelter
pot-room workers, roadpavers, and occupationally non-exposed persons
in Sweden. Sci Total Environ, 193: 169-177.
Levsen K (1988) The analysis of diesel particulate. Fresenius' Z Anal
Chem, 331: 467-478.
Levsen K, Behnert S, Priess B, & Winkeler HD (1991) The contamination
of precipitation in Hannover by hydrocarbons. Vom Wasser, 76: 109-126.
Lewis WM (1975) Polynuclear aromatic hydrocarbons in water. Water
Treat Exam, 23: 243-277.
Lewis RJ Sr (1992) Sax's dangerous properties of industrial materials,
8th ed. New York, Van Nostrand Reinhold Co., 1245 pp.
Lewtas J (1985a) Development of a comparative potency method for
cancer risk assessment of complex mixtures using short-term
in vivo and in vitro bioassays. Toxicol Ind Health, 4: 193-203.
Lewtas J (1985b) Combustion emissions: Characterization and comparison
of their mutagenic and carcinogenic activity. In: Stich HF ed.
Carcinogens and mutagens in the environment, Volume V: The workplace:
Source of carcinogens. Boca Raton, Florida, CRC Press, pp 59-74.
Lewtas J (1993) Complex mixtures of air pollutants: Characterizing the
cancer risk of polycyclic organic matter. Environ Health Perspectives,
100: 211-218.
Lewtas J, Mumford J, Everson RB, Hulka B, Wilcosky T, Kozumbo W,
Thompson C, George M, Dobiàs L, Sràm R, Li X, & Gallagher J (1993)
Comparison of DNA adducts from exposure to complex mixtures in various
human tissues and experimental systems. Environ Health Perspectives,
99: 89-97.
Li Y, Xu H, Song Y, Yang X, & Li Z (1991) [A study on the relationship
between urinary BaP and SCE frequency in rats.] Zhongguo Huanjing
Kexue, 11: 361-364 (in Chinese with English abstract).
Liao W, Smith WD, Chiang TC, & Williams LR (1988) Rapid, low-cost
cleanup procedure for determination of semivolatile organic compounds
in human and bovine adipose tissues. J Assoc Off Anal Chem, 71: 742-
747.
Lide DR ed. (1991) CRC handbook of chemistry and physics, 72nd ed.
Boca Raton, Florida, CRC Press, pp 22-35.
Liem AKD, Baumann RA, de Jong APJM, van der Velde EG, & van Zoonen P
(1992) Analysis of organic micropollutants in the lipid fraction of
foodstuffs. J Chromatogr, 624: 317-339.
Lies KH, Hartung A, Postulka A, Gring H, & Schulze J (1986)
Composition of diesel exhaust with particular reference to particle
bound organics including formation of artifacts. Dev Toxicol Environ
Sci, 13: 65-82.
Ligocki MP & Pankow JF (1989) Measurements of the gas/particle
distributions of atmospheric organic compounds. Environ Sci Technol,
23: 75-83.
Ligocki MP, Leuenberger C, & Pankow JF (1985) Trace organic compounds
in rain. II. Gas scavening of neutral organic compounds. Atmos
Environ, 19: 1609-1617.
Lijinsky W & Garcia H (1972) Skin carcinogenesis tests of hydrogenated
derivatives of anthanthrene and other polynuclear hydrocarbons. Z
Krebsforsch, 77: 226-230.
Lijinsky W & Ross AE (1967) Production of carcinogenic polynuclear
hydrocarbons in the cooking of food. Food Cosmet Toxicol, 5: 343-347.
Lijinsky W & Saffiotti U (1965) Relationships between structure and
skin tumorigenic activity among hydrogenated derivates of several
polycyclic aromatic hydrocarbons. Ann It Dermatol Clin Sper, 19: 34-
44.
Lijinsky W & Shubik P (1964) Benzo(a)pyrene and other polynuclear
hydrocarbons in charcoal-broiled meat. Science, 145: 53-55.
Lijinsky W & Shubik P (1965) Polynuclear hydrocarbon carcinogens in
cooked meat and smoked food. Ind Med Surg, 34: 152-154.
Lijinsky WH, Garcia B, & Terrracini B (1965) Tumorigenic activity of
hydrogenated derivatives of dibenz [a,h]anthracene. J Natl Cancer
Inst, 34:1-6.
Likhachev AJ, Beniashvili DS, Bykov VJ, Dikun PP, Tyndyk ML,
Savochkina IV, Yermilov VB, & Zabezhinski MA (1992) Biomarkers for
individual susceptibility to carcinogenic agents: Excretion and
carcinogenic risk of benzo [a]pyrene metabolites. Environ Health
Perspectives, 98: 211-214.
Linder G & Bergman H (1984) Periodic depuration of anthracene
metabolites by rainbow trout Salmo gairdneri. Trans Am Fish Soc,
113: 513-520.
Linder G, Bergman H, & Meyer J (1985) Anthracene bioconcentration in
rainbow-trout during single-compound and complex-mixture exposures.
Environ Toxicol Chem, 4: 549-558.
Lindquist B & Warshawsky D (1985) Identification of the
11,12-dihydro-11,12-dihydroxybenzo [a]pyrene as a major metabolite
produced by the green alga, Selenastrum capriconutum. Biochem
Biophys Res Commun, 130: 7.
Lindskog A, Brorström-Lundén E, Alfheim I, & Hagen I (1987) Chemical
transformation of PAH on airborne particles by exposure to NO2 during
sampling: A comparison between two filter media. Sci Total Environ,
61: 51-57.
Lindstedt G & Sollenberg J (1982) Polycyclic aromatic hydrocarbons in
the occupational environment - with special reference to
benzo [a]pyrene measurements in Swedish industry. Scand J Work
Environ Health, 3: 1-19.
Lindström-Seppä P, Hänninen O, Tuominen J, & Pyysalo H (1989)
Polycyclic aromatic hydrocarbons in perch (Perca fluviatilis)
following an oil-spill in Vaasa Archipelago, Finland. Toxicol Environ
Chem, 19: 83-86.
Lintas C, De Matthaeis MC, & Merli F (1979) Determination of
benzo(a)pyrene in smoked, cooked and toasted food products. Food
Cosmet Toxicol, 17: 325-328.
Liotti FS, Pelliccia C, & Pezzetti F (1988) Different response of
chicken embryo fibroblasts and hepatocytes to the interference of
certain antioxidants on the binding of [G3H]benzo [a]pyrene to DNA.
Cancer Lett, 41: 235-242.
Lioy PL, Waldman JM, Greenberg A, Harkov R, & Pietarinen C (1988) The
total human environmental exposure study (THEES) to benzo(a)pyrene:
Comparison of the inhalation and food pathways. Arch Environ Health,
43: 304-312.
Little JB & Vetrovs H (1988) Studies of ionizing radiation as a
promoter of neoplastic transformation in vitro. Int J Radiat Biol,
53: 661-666.
Liu D, Maguire RJ, Pacepavicius GJ, & Nagy E (1992) Microbial
degradation of polycyclic aromatic hydrocarbons and polycyclic
aromatic nitrogen heterocyclics. Environ Toxicol Water Qual, 7: 355-
372.
Lo MT & Sandi E (1978) Polycyclic aromatic hydrocarbons (polynuclear)
in foods. Res Rev, 69: 35-85.
Loening KL & Merritt JE (1990) Some aids for naming polycyclic
aromatic hydrocarbons and their heterocyclic analogs. In: Loening K,
Merritt J, Later D, & Wright W ed. Polynuclear aromatic hydrocarbons:
Nomenclature guide. Columbus, Ohio, Battelle Press, pp 1-25.
Löfroth G, Toftgard R, Nilsson L, Agurell E, & Gustafsson JA (1984)
Short-term bioassays of nitro derivatives of benzo [a]pyrene and
perylene. Carcinogenesis, 5: 925-930.
Lovell WW & Sanders DJ (1992) Phototoxicity testing in guinea-pigs.
Food Chem Toxicol, 30: 155-160.
Lowenthal DH, Zielinska B, Chow JC, Watson JG, Gautman M, Ferguson DH,
Neuroth GR, & Stevens KD (1994) Characterizitation of heavy-duty
diesel vehicle emissions. Atmos Environ, 28: 731-743.
Lu PY, Metcalf RL, Plummer N, & Mandel D (1977) The environmental fate
of three carcinogens: Benzo [a]pyrene, benzidine, and vinyl chloride
evaluated in laboratory model ecosystems. Arch Environ Contam Toxicol,
6: 129-142.
Lubet RA, Kiss E, Gallagher MM, Dively C, Konri R, & Schechtman LM
(1983a) Induction of neoplastic transformation and DNA single-strand
breaks in C3H/10T1/2 clone 8 cells by polycyclic hydrocarbons and
alkylating agents. J Natl Cancer Inst, 71: 991-997.
Lubet RA, Connelly GM, & Nebert DW (1983b)
Dibenz [a,h]anthracene-induced subcutaneous tumors in mice. Strain
sensitivity and the role of carcinogen metabolism. Carcinogenesis, 4:
513-517.
Lubet RA, Brunda MJ, & Lemaira B (1984a) Polycyclic hydrocarbon
induced immunotoxicity in mice: Role of the Ah locus. In: Cooke M &
Dennis AJ ed. Polycyclic aromatic hydrocarbons: Mechanisms, methods
and metabolism. Columbus, Ohio, Battelle Press, pp 843-858.
Lubet RA, Brunda MJ, Taramelli D, Dansie D, Nebert DW, & Kouri RE
(1984b) Induction of immunotoxicity by polycyclic hydrocarbons: Role
of the Ah locus. Arch Toxicol, 56: 18-24.
Lubet RA, Kouri RE, Curren RA, Putman DL, & Schechtman LM (1990)
Induction of mutagenesis and transformation in BALB/c-3T3 clone A31-1
cells by diverse chemical carcinogens. Environ Mol Mutag, 16: 13-20.
Luijten JA & Piet GJ (1983) Identity and sources of organic chemicals
in groundwater. In: Proceedings of the 1st Atlantic Workshop,
Nashville, Tennessee. Denver, Colorado, American Water Works
Association, pp 7-18.
van Luin AB & van Starkenburg W (1984) Hazardous substances in waste
water. Water Sci Technol, 17: 843-853.
Lunde G (1976) Long-range aerial transmission of organic
micropollutants. Ambio, 5: 207-208.
Lunde G & Bjorseth A (1977) Polycyclic aromatic hydrocarbons in
long-range transported aerosols. Nature, 268: 518-519.
Luther M, Moriske HJ, & Rüden H (1990) [Indoor air levels of
polycyclic aromatic hydrocarbons from the use of asphalt floor tiles.]
Forum Städte-Hyg, 41: 95-103 (in German).
Lutz WK & Schlatter J (1992) Chemical carcinogens and overnutrition in
diet-related cancer. Carcinogenesis, 13: 2211-2216.
Lyall RJ, Hooper MA, & Mainwaring SJ (1988) Polycyclic aromatic
hydrocarbons in the Latrobe Valley. Atmos Environ, 22: 2549-2555.
Lygren E, Gjessing E, & Berglind L (1984) Pollution transport from a
highway. Sci Total Environ, 33: 147-159.
Lyman WJ, Reehl WF, & Rosenblatt DH ed. (1982) Handbook on chemical
property estimation methods, environmental behavior of organic
compounds. New York, McGrawHill, 960 pp.
Lyon JL, Klauber MR, Graff W, & Chiu G (1981) Cancer clustering around
point sources of pollution. Assessment by a case control methodology.
Environ Res, 25: 29-34.
Lyte Ml, Blanton RH, Myers MJ, & Bick PH (1987) Effect of
in vivo administration of carcinogen benzo(a)pyrene on interleukin-2
and interleukin-3 production. Int J Immunopharmacol, 9: 307-312.
Maccubbin AE, Black P, Trzeciak L, & Black JJ (1985) Evidence for
polynuclear aromatic hydrocarbons in the diet of bottom-feeding fish.
Bull Environ Contam Toxicol, 34: 876-882.
Machado ML, Beatty PW, Fetzer JC, Glickman AH, & McGinnis EL (1993)
Evaluation of the relationship between PAH content and mutagenic
activity of fumes from roofing and paving asphalts and coal tar pitch.
Fundam Appl Toxicol, 21: 492-499.
Mackay D & Leinonen PJ (1975) Rate of evaporation of low-solubility
contaminants from water bodies to atmosphere. Environ Sci Technol, 9:
1178-1180.
Mackay D & Shiu WY (1977) Aqueous solubility of polynuclear aromatic
hydrocarbons. J Chem Eng Data, 22: 399-402.
Mackay D & Shiu WY (1981) A critical review of Henry's law constants
for chemicals of environmental interest. J Phys Chem Ref Data, 10:
1175-1199.
Mackay D, Shiu WY, & Sutherland RP (1979) Determination of air-water
Henry's law constants for hydrophobic pollutants. Environ Sci Technol,
13: 333-337.
Mackay D, Shiu WY, & Ma KC (1992) Illustrated handbook of physical-
chemical properties and environmental fate for organic chemicals.
Volume II: Polynuclear aromatic hydrocarbons, polychlorinated dioxins
and dibenzofurans. Boca Raton, Florida, Lewis Publishers, pp 1-367.
Mackell JV, Rieders F, Brieger H, & Bauer EL (1951) Acute hemolytic
anemia due to ingestion of naphthalene moth balls. Pediatrics, 7: 722-
728.
MacKenzie KM & Angevine DM (1981) Infertility in mice exposed in utero
to benzo(a)pyrene. Biol Reprod, 24: 183-191.
Maclaren WM & Hurley JF (1987) Mortality of tar distillation workers.
Scand J Work Environ Health, 13: 404-411.
MacNicoll AD, Easty GC, Neville AM, Grover PL, & Sims P (1980)
Metabolism and activation of carcinogenic polycyclic hydrocarbons by
human mammary cells. Biochem Biophys Res Commun, 95: 1599-1606.
Madhavan ND& Naidu KA ( 1995) Polycyclic aromatic hydrocarbons in
placenta, maternal blood, umbilical cord blood and milk of Indian
women. Hum Exp Toxicol, 14: 503-506.
Maga JA (1986) Polycyclic aromatic hydrocarbon (PAH) composition of
mesquite (Prosopis fuliflora) smoke and grilled beef. J Agric Food
Chem, 34: 249-251.
Mager R, Huberman E, Yang SK, Gelboin HV, & Sachs L (1977)
Transformation of normal hamster cells by benzo(a)pyrene diol-epoxide.
Int J Cancer, 19: 814-817.
Mahlum D, Wright CW, Chess EK, & Wilson BW (1984) Fractionation of
skin tumor initiating activity in coal liquids. Cancer Res, 44: 5176-
5181.
Maisin J & Coolen M-L (1936) [Carcinogenic potency of
methylcholanthrene.] Comp Soc Biol, 123: 159-160 (in French).
Malaiyandi M, Benedek A, Holko AP, & Bancsi JJ (1986) Measurement of
potentially hazardous polynuclear aromatic hydrocarbons from
occupational exposure during roofing and paving operations. In: Cooke
M, Dennis AJ, & Fisher GL ed. Polynuclear aromatic hydrocarbons:
Physical and biological chemistry. Columbus, Ohio, Battelle Press, pp
471-489.
Malcolm HM & Dobson S (1994) The calculation of an environmental
assessment level (EAL) for atmospheric PAHs using relative
potencies.London, Department of the Environment, 34 pp (Report No.
DoE/HMIP/RR/94/041).
Malins DC (1982) Alterations in the cellular and subcellular structure
of marine teleosts and invertebrates exposed to petroleum in the
laboratory and field: A critical review. Can J Fish Aquat Sci, 39:
877-889.
Malins DC, McCain BB, Brown DW, Chan S-L, Myers MS, Landahl JT,
Prohaska PG, Friedman AJ, Rhodes LD, Burrows DG, Gronlund WD, &
Hodgins HO (1984) Chemical pollutants in sediments and diseases of
bottom-dwelling fish in Puget Sound, Washington. Environ Sci Technol,
18: 705-713.
Malins DC, Krahn MM, Brown DW, Rhodes LD, Myers MS, McCain BB, & Chan
SL (1985) Toxic chemicals in marine sediment and biota from Mukilteo,
Washington: Relationships with hepatic neoplasms and other hepatic
lesions in English sole (Parophrys vetulus). J Natl Cancer Inst, 74:
487-494.
Mallet WG, Mosebrook DR, & Trush MA (1991) Activation of
(±)- trans-7,8-dihydroxy-7,8-dihydrobenzo(a)pyrene to diolepoxides by
human polymorphonuclear leukocytes or myeloperoxidase. Carcinogenesis,
12: 521-524.
Mamber SW, Bryson V, & Katz SE (1983) The Escherichia coli
WP2/WP100 rec assay for detection of potential chemical carcinogens.
Mutat Res, 119: 135-144.
Mamber SW, Bryson V, & Katz SE (1984) Evaluation of the Escherichia
coli K12 inductest for detection of potential chemical carcinogens.
Mutat Res, 130: 141-151.
Mane SS, Purnell DM, & Hsu IC (1990) Genotoxic effects of five
polycyclic aromatic hydrocarbons in human and rat mammary epithelial
cells. Environ Mol Mutagen, 15: 78-82.
Manilal VB & Alexander M (1991) Factors affecting the microbial
degradation of phenanthrene in soil. Appl Microbiol Biotechnol, 35:
410-405.
Mannschreck C, Gündel J, & Angerer K (1996) Occupational exposure to
PAH. Biological monitoring of hydroxylated metabolites. Polycyclic
Aromat Compd 11: 11-18.
Manz A, Berger J, & Waltsgott H (1983) [Occupational cancer in gas
industry workers.] Bremerhaven, German Federal Department for Worker
Safety and Accident Research pp 1-131 (Wirtschaftsverlag NW, Report
No. 352) (in German).
Marcomini A, Pavoni B, Donazzolo R, & Orio AA (1986) Combined
preparative and analytical use of normal-phase and reversed-phase
high-performance liquid chromato-graphy for determination of aliphatic
and polycyclic aromatic hydrocarbons in sediments of the Adriatic Sea.
Mar Chem, 18: 71-84.
Marcus JM & Stokes TP (1985) Polynuclear aromatic hydrocarbons in
oyster tissue around three coastal marinas. Bull Environ Contam
Toxicol, 35: 835-844.
Marcus JM, Swearingen GR, Williams AD, & Heizer DD (1988) Polynuclear
aromatic hydrocarbon and heavy metal concentrations in sediments at
coastal South Carolina marinas. Arch Environ Contam Toxicol, 17: 103-
113.
Marino DJ (1987) Evaluation of Pluronic(R) Polyol F17 as a vehicle
for petroleum hydrocarbons in the Salmonella/microsomal assay. Environ
Mutagen, 9: 307-316.
Marnett LJ, Reed GA, & Dennison DJ (1978) Prostaglandin synthetase
dependent activation of 7,8-dihydro-7,8-dihydroxy-benzo(a)pyrene to
mutagenic derivatives. Biochem Biophys Res Commun, 82: 210-216.
Marquardt H & Heidelberger C (1972) Influence of 'feeder cells' and
inducers and inhibitors of microsomal mixed-function oxidases on
hydrocarbon-induced malignant transfromation of cells derived from C3H
mouse prostate. Cancer Res, 32: 721-725.
Marquardt H, Kuroki T, Huberman E, Selkirk JK, Heidelberger C, Grover
PL, & Sims P (1972) Malignant transformation of cells derived from
mouse prostate by epoxides and other derivatives of polycyclic
hydrocarbons. Cancer Res, 32: 716-720.
Marshall MV, McLemore TL, Martin RR, Jenkins WT, Snodgrass DR, Corson
MA, Arnott MS, Wray NP, & Griffin AC (1979) Patterns of
benzo [a]pyrene metabolism in normal human pulmonary alveolar
macrophages. Cancer Lett, 8: 103-109.
Martel L, Gagnon MJ, Masse R, Leclerc A, & Tremblay L (1986)
Polycyclic aromatic hydrocarbons in sediments from the Saguenay Fjord,
Canada. Bull Environ Contam Toxicol, 37: 133-140.
Martin BJ (1980) Effects of petroleum compounds on estuarine fishes.
Gulf Breeze, Florida, US Environmental Protection Agency, 43 pp
(EPA-600/3-80-019; PB 80-141310).
Martin CN & McDermid AC (1981) Testing of 42 coded compounds for their
ability to induce unscheduled DNA repair synthesis in HeLa cells. In:
DeSerres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, pp 533-537 (Progress in Mutation
Research, Volume 1).
Martin CN, McDermid AC, & Garner RC (1978) Testing of known
carcinogens and noncarcinogens for their ability to induce unscheduled
DNA synthesis in HeLa cells. Cancer Res, 38: 2621-2627.
Martin F, Hoepfner I, Scherer G, Adlkofer F, Dettbarn G, & Grimmer G
(1989) Urinary excretion of hydroxy-phenanthrenes after intake of
polycyclic aromatic hydrocarbons. Environ Int, 15: 41-47.
Masclet P, Nikolaou K, & Mouvier G (1984) [Identification of sources
of particular polycyclic aromatic hydrocarbons in the urban
atmosphere.] In: Versino B & Angeletti ed. Physico-chemical behavior
of atmospheric pollutants. Dordrecht: Reidel Publishing Co., pp 616-
625 (in French).
Mass MJ, Rodgers NT, & Kaufman DG (1981) Benzo [a]pyrene metabolism
in organ culture of human endometrium. Chem-Biol Interactions, 33:
195-205.
Massie LC, Ward AP, & Davies JM (1985) The effects of oil exploration
and production in the northern North Sea: Part 1. The levels of
hydrocarbons in water and sediments in selected areas, 1978-1981. Mar
Environ Res, 15: 165-213.
Masuda Y & Kagawa R (1972) A novel synthesis and carcinogenicity of
dibenzo [a,l]pyrene. Chem Pharm Bull, 20: 2736-2737.
Matsumoto H & Kashimoto T (1985) Average daily respiratory intake of
polycyclic aromatic hydrocarbons in ambient air determined by
capillary gas chromatography. Bull Environ Contam Toxicol, 34: 17-23.
Matsuoka A, Hayashi M, & Ishidate MJ (1979) Chromosomal aberration
tests on 29 chemicals combined with S9 mix in vitro. Mutat Res, 66:
277-290.
Matsuoka A, Sofuni I, Miyata N, & Ishidate M Jr (1991) Clastogenicity
of 1-nitropyrene, dinitropyrenes, fluorene and mononitrofluorenes in
cultered Chinese hamster cells. Mutat Res, 259: 103-110.
Mattison DR & Nightingale MR (1980) The biochemical and genetic
characteristics of murine ovarian aryl hydrocarbon (benzo [a]pyrene)
hydroxylase activity and its relationship to primordial oocyte
destruction by polycyclic aromatic hydrocarbons. Toxicol Appl
Pharmacol, 56: 399-408.
Mattison DR, White NB, & Nightingale MS (1980) The effect of
benzo(a)pyrene on fertility, primordial oocyte number, and ovarian
response to pregnant mare's serum gonadotropin. Pediatr Pharmacol, 1:
143-151.
Mattison DR, Singh H, Takizawa K, & Thomford PJ (1989) Ovarian
toxicity of benzo(a)pyrene and metabolites in mice. Reprod Toxicol, 3:
115-125.
Matzner E (1984) Annual rates of deposition of polycyclic aromatic
hydrocarbons in different forest ecosystems. Water Air Soil Pollut,
21: 425-434.
Matzner B, Hübner D, & Thomas W (1981) Content and storage of
polycyclic aromatic hydrocarbons in two forested ecosystems in
northern Germany. Z Pflanzenernähr Bodenkd, 144: 283-288.
Mavournin KH, Blakey DH, Cimino MC, Salamone MF, & Heddle JA (1990)
The in vivo micronucleus assay in mammalian bone marrow and
peripheral blood. A report of the US Environmental Protection Agency
Gene-Tox Program. Mutat Res, 239: 29-80.
May WE, Wasik SP, & Freeman DH (1978) Determination of the aqueous
solubility of polynuclear aromatic hydrocarbons by a coupled column
liquid chromatographic technique. Anal Chem, 50: 175-179.
McCann J, Choi E, Yamasaki E, & Ames BN (1975a) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc Natl Acad Sci USA, 72: 5135-5139.
McCann J, Spingarn NE, Kobori J, & Ames BN (1975b) Detection of
carcinogens as mutagens: Bacterial tester strains with R factor
plasmids. Proc Natl Acad Sci USA, 72: 979-983.
McCarroll NE, Keech BH, & Piper CE (1981) A microsuspension adaptation
of the Bacillus subtilis 'rec' assay. Environ Mutag, 3: 607-616.
McCarthy JF & Jimenez BD (1985) Reduction in bioavailability to
bluegills of polycyclic aromatic hydrocarbons bound to dissolved humic
material. Environ Toxicol Chem, 4: 511-521.
McCarthy JF, Jimenez BD, & Barbee T (1985) Effect of dissolved humic
material on accumulation of polycyclic aromatic hydrocarbons:
Structure-activity relationships. Aquat Toxicol, 7: 15-24.
McClure P & Schoeny R (1995) Evaluation of a component-based relative
potency approach to cancer risk assessment for exposure to PAH. In:
Fifteenth international symposium on polycyclic aromatic compounds:
Chemistry, biology and environmental impact, Belgirate, Italy, 19-22
September 1995. Ispra, Joint Research Centre European Commission, p
161.
McCormick DL, Burns FJ, & Albert RE (1981) Inhibition of
benzo(a)pyrene-induced mammary carcinogenesis by retinyl acetate. J
Natl Cancer Inst, 66: 559-564.
McElroy AE (1985) In vivo metabolism of benz(a)anthracene by the
polychaete Nereis virens. Mar Environ Res, 17: 133-136.
McElroy AE & Sisson JP (1989) Trophic transfer of benzo [a]pyrene
metabolites between benthic marine organisms. Mar Environ Res, 28:
265-269.
McFall JA, Antoine SR, & DeLeon IR (1985) Base-neutral extractable
organic pollutants in biota and sediments from Lake Pontchartrain.
Chemosphere, 14: 1561-1569.
McGill AS, Mackie PR, Parsons E, Bruce C, & Hardy R (1982) The
polynuclear aromatic hydrocarbon content of smoked foods in the United
Kingdom. In: Cooke M, Dennis AJ, & Fisher GL ed. Polynuclear aromatic
hydrocarbons: Physical and biological chemistry. Columbus, Ohio,
Battelle Press, pp 491-499.
McGill AS, Mackie PR, Howgate P, & McHenery JG (1987) The flavour and
chemical assessment of dabs (Limanda limanda) caught in the vicinity
of the Beatrice oil platform. Mar Pollut Bull, 18: 186-189.
McIntyre AE, Perry R & Lester JN (1981) Analysis of polynuclear
aromatic hydrocarbons in sewage sludges. Anal Lett, 14: 291-309.
McKay S, Phillips DH, Hewer AJ, & Grover PL (1988) Metabolic
activation of 7-ethyl- and 7-methylbenz [a]anthracene in mouse skin.
Carcinogenesis, 9: 141-145.
McLeese DW & Burridge LE (1987) Comparative accumulation of PAHs in
four marine invertebrates. In: Capuzzo IM & Kester DR ed. Oceanic
processes in marine pollution. Malabar, Florida, Kirger RE, pp
109-118.
McMahon CK & Tsoukalas SN (1978) Polynuclear aromatic hydrocarbons in
forest fire smoke. In: Jones PW & Freudenthal RI ed. Carcinogenesis.
Volume 3: Polynuclear aromatic hydrocarbons. New York, Raven Press, pp
61-73.
McVeety BD & Hites RA (1988) Atmospheric deposition of polycyclic
aromatic hydrocarbons to water surfaces: A mass balance approach.
Atmos Environ, 22: 511-536.
Means JC, Hassett JJ, Wood SG, & Banwart WL (1979) Sorption properties
of energy-related pollutants and sediments. In: Jones PW & Leber P ed.
Polynuclear aromatic hydrocarbons. Ann Arbor, Michigan, Ann Arbor
Science Publishers, pp 327-340.
Means JC, Wood SG, Hassett JJ, & Banwart WL (1980) Sorption of
polynuclear aromatic hydrocarbons by sediments and soils. Environ Sci
Technol, 14: 1524-1528.
Mehta R, Meredith-Brown M, & Cohen GM (1979) Metabolism and covalent
binding of benzo [a]pyrene in human peripheral lung. Chem-Biol
Interactions, 28: 345-358.
Melancon M & Lech J (1978) Distribution and elimination of naphthalene
and 2-methylnaphthalene in rainbow trout during short- and long-term
exposures. Arch Environ Contam Toxicol, 7: 207-220.
Melikian AA, LaVoie EJ, Hecht SS, & Hoffmann D (1983) 5-Methylchrysene
metabolism in mouse epidermis in vivo, diol epoxide-DNA adduct
persistence, and diol epoxide reactivity with DNA as potential factors
influencing the predominance of 5-methylchrysene-1,2-diol-3,4-epoxide-
DNA adducts in mouse epidermis. Carcinogenesis, 4: 843-849.
Melikian AA, Amin S, Huie K, Hecht SS, & Harvey G (1988) Reactivity
with DNA bases and mutagenicity toward Salmonella typhimurium of
methylchrysene diol epoxide enantiomers. Cancer Res, 48: 1781-1787.
Melikian AA, Prahalad AK, Amin SG, & Hecht SS (1991) Comparative DNA
binding of benzo(a)pyrene, 5-methylchrysene, 6-methylchrysene and
their corresponding bay-region dihydrodiol epoxides in newborn mouse
lung and liver in vivo. Proc Am Assoc Cancer Res, 32: 96.
Melius P, Elan D, Kilgore M, Tan B, & Schoor WP (1980) Mixed function
oxidase inducibility and polyaromatic hydrocarbon metabolism in the
mullet, sea catfish and Gulf killifish. In: Bjorseth A & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemistry and biological effects.
Columbus, Ohio, Battelle Press, pp 1059-1075.
Menichini E (1992a) Urban air pollution by polycyclic aromatic
hydrocarbons: Levels and sources of variability. Sci Total Environ,
116: 109-135.
Menichini E (1992b) Opinion adopted by the Italian National Advisory
Toxicological Committee on polycyclic aromatic hydrocarbons. Rome,
Istituto Superiore di Sanità, 69 pp (Serie Relazioni 92/4).
Menichini E, Bonanni L, & Merli F (1990) Determination of polycyclic
aromatic hydrocarbons in mineral oils and oil aerosols in glass
manufacturing. Toxicol Environ Chem, 28: 37-51.
Menichini E, di Domenico A, Bonanni L, Corradetti E, Mazzanti L, &
Zucchetti G (1991a) Reliability assessment of a gas chromatographic
method for polycyclic aromatic hydrocarbons in olive oil. J
Chromatogr, 555: 211-220.
Menichini E, Bocca A, Merli F, Ianni D, & Monfredini F (1991b)
Polycyclic aromatic hydrocarbons in olive oils on the Italian market.
Food Addit Contam, 8: 363-369.
Merriman JC (1988) Distribution of organic contaminants in water and
suspended solids of the Rainy River. Water Pollut Res J Can, 23: 590-
600.
Mersch-Sundermann V, Mochayedi S, & Kevekordes S (1992) Genotoxicity
of polycyclic aromatic hydrocarbons in
Escherichia coli PQ37. Mutat Res, 278: 1-9.
Meyer JP & Grimmer G (1974) [The influence of PCA-containing and
PCA-free fuel on the emission of polycyclic aromatic hydrocarbons from
a motor vehicle with spark-ignition engine in the Europe test cycle.]
Hamburg, German Society for Mineral-oil and Coal Chemistry, 22 pp
(BMI-DGMK joint project 4547, Part 1) (in German).
Meyer H-P, Grimmer G, & Behn U (1980) [PAH from oil heating:
Collection, analysis, results. Air pollution from polycylic aromatic
hydrocarbons. Registration and evaluation.] Düsseldorf, VDI-Verlag, pp
101-105 (VDI Report No. 358) (in German).
Meyers MJ, Blanton RH, & Bick PH(1988) Inhibition of IL-2
responsiveness following exposure to benzo(a)pyrene is due to
alterations in accessory cell function. Int J Immunopharmacol, 10:
177-186.
Michel X, Bani MH, & Narbonne JF (1995) Regio-selective metabolism of
benzo [a]pyrene by microsomes from 5 vertebrate species. In:
Fifteenth international symposium on polycyclic aromatic compounds:
Chemistry, biology and environmental impact, Belgirate, Italy, 19-22
September 1995. Ispra, Joint Research Centre European Commission, p
78.
Middaugh DP, Thomas RL, Lantz SE, Heard CS, & Mueller JG (1994) Field
scale testing of a hyperfiltration unit for removal of creosote and
pentachlorophenol from groundwater: Chemical and biological
assessment. Arch Environ Contam Toxicol, 26: 309-319.
Mielzynska D & Snit M (1992) Urine mutagenicity in workers directly
employed in coke production. Pol J Occup Med, 5: 363-371.
Miescher G (1942) [Experimental studies on the formation of tumours by
photosensitization.] Schweiz Med Wochenschr, 72: 1082-1984 (in
German).
Milano JC & Vernet JL (1988) [Size of the contribution of carcinogenic
polyaromatic hydrocarbons by the Rhône.] Oceanis, 14: 133-140 (in
French).
Milano JC, Fache B, & Vernet JL (1985) [Contamination of the Cortiou
site by polyaromatic hydrocarbons in wastewater from the city of
Marseille (western Mediterranean, France).] J Rech Oceanogr, 10: 36-38
(in French).
Milano JC, Fache B, & Vernet JL (1986) [Pollution of the sediment,
fauna and flora by polyaromatic hydrocarbons at Cap Sicié at the
emission site of the city of Toulon (French Mediterranean coast).] J
Rech Oceanogr, 11: 93-96 (in French).
Mill T & Mabey W (1985) Photochemical transformations. In: Neely WB &
Blau GE ed. Environmental exposure from chemicals, Volume 1. Boca
Raton, Florida, CRC Press, pp 175-216.
Mill T, Mabey WR, Lan BY, & Baraze A (1981) Photolysis of polycyclic
aromatic hydrocarbons in water. Chemosphere, 10: 1281-1290.
Millemann RE, Birge WJ, Black JA, Cushman RM, Daniels KL, Franco PJ,
Giddings JM, McCarthy JF, & Stewart AJ (1984) Comparative acute
toxicity of aquatic organisms of components of coal derived synthetic
fuels. Trans Am Fish Soc, 113: 74-85.
Miller EC (1951) Studies on the formation of protein-bound derivatives
of 3,4-benzpyrene in the epidermal fraction of mouse skin. Cancer Res,
11: 100-108.
Miller JA & Miller EC (1953) The carcinogenic aminoazo dyes. Adv
Cancer Res, 1: 339-396.
Miller JA & Miller EC (1963) The carcinogenicities of fluoro
derivatives of 10-methyl- 1,2-benzanthracene. II. Substitution of the
K region and the 3'- 6-, and 7-positions. Cancer Res, 23: 229-239.
Miller JA & Miller (1977) Ultimate chemical carcinogens as reactive
mutagenic electrophiles In: Hiatt HH, Watson JD, & Winston JA ed.
Origins of human cancer, Book B, Mechanisms of carcinogenesis. Cold
Spring Harbor, New York, Cold Spring Harbor Laboratory, pp 605-627.
Miller MM, Wasik SP, Huang G-L, Shiu W-Y, & Mackay D (1985)
Relationships between octanol-water partition coefficient and aqueous
solubility. Environ Sci Technol, 19: 522-529.
Miller MM, Plowchalk DR, Weitzman GA,, London SN & Mattison DR (1992)
The effect of benzo(a)pyrene on murine ovarian and corpora lutea
volumes. Am J Obstet Gynecol, 166: 1535-1541.
Milo GE, Blakeslee J, Yohn DS, & DiPaolo JA (1978) Biochemical
activiation of aryl hydrocarbon hydroxylase activity, cellular
distribution of polynuclear hydrocarbon metabolites, and DNA damage by
polynuclear hydrocarbon products in human cells in vitro.
Cancer Res, 38: 1638-1644.
Miralis JC, Tyson CK, & Butterworth BE (1982) Detection of genotoxic
carcinogens in the in vivo-in vitro hepatocyte DNA repair assay.
Environ Mutag, 4: 553-562.
Mishra NK, Wilson CM, Pant KJ, & Thomas FO (1978) Simultaneous
determination of cellular mutagenesis and transformation by chemical
carcinogens in Fischer rat embryo cells. J Toxicol Environ Health, 4:
79-91.
Mitchell CE (1982) Distribution and retention of benzo [a]pyrene in
rats after inhalation. Toxicol Lett, 11: 35-42
Mitchell RL, Burchett MD, Pulkownik A, & McCluskey L (1988) Effects of
environmentally hazardous chemicals on the emergence and early growth
of selected Australian plants. Plant Soil, 112: 195-199.
Mix MC & Schaffer RL (1983) Concentrations of unsubstituted polycyclic
aromatic hydrocarbons in softshell clams from Coos Bay, Oregon, USA.
Mar Pollut Bull, 14: 94-97.
Mohr U (1969) [Studies on the carcinogenic properties of
3.4,-10.11-,12.13- tribenzofluoranthene by mice.] Arch Hyg, 153: 495-
510 (in German).
Mohr U (1971) [Carcinogenicity of diethylnitrosamine.] Fortschr Med,
89: 251-253 (in German).
Moles A (1980) Sensitivity of parasitized Coho salmon fry to crude
oil, toluene, and naphthalene. Am Fish Soc, 109: 293-297.
Moles A, Bates S, Rice SD, & Korn S (1981) Reduced growth of Coho
salmon fry exposed to two petroleum components, toluene and
naphthalene, in fresh water. Am Fish Soc, 110: 430-436.
Monarca S, Pasquini R, Scassellati Sforzolini G, Savino A, Bauleo FA,
& Angeli G (1987) Environmental monitoring of mutagenic/carcinogenic
hazards during road paving operations with bitumens. Int Arch Occup
Environ Health, 59: 393-402.
Monnerjahn S, Seidel A, Steinberg P, Oesch F, Hinz M, Stezowsky JJ,
Hewer A, Phillips DH & Glatt HR (1993) Formation of DNA adducts from
1-hydroxymethylpyrene in liver cells in vivo and in
vitro. In: Phillips DH, Castegnaro M & Bartsch H ed. Postlabelling
methods for detection of DNA adducts, Lyon, International Agency for
Research on Cancer, pp. 189-193 (IARC Scientific Publications No. 124)
Monteith DK & Gupta RC (1992) Carcinogen-DNA adducts in cultures of
rat and human hepatocytes. Cancer Lett, 62: 87-93.
Montizaan GK, Kramers PGH, Janus JA & Posthumus R (1989) Integrated
criteria document PAH: Effects of 10 selected compounds. Appendix to
Report No. 758474011. Bilthoven, National Institute of Public Health
and Environmental Protection, 180 pp (Re-publication in March 1989 of
addendum to Report No. 758447007).
Moolgavkar SH & Knudson A (1981) Mutation and cancer: A model for
human carcinogenesis. J Natl Cancer Inst, 66: 1037-1052.
Moolgavkar SH & Venzon DJ (1979) Two-event models for carcinogenesis.
Math Biosci, 47: 55-77.
Moore BP, Hicks RM, Knowles MA, & Redgrave S (1982) Metabolism and
binding of benzo(a)pyrene and 2-acetylaminofluorene by short-term
organ cultures of human and rat bladder. Cancer Res, 42: 642-648.
Moore MN, Livingstone DR, & Widdows J (1989) Hydrocarbons in marine
mollusks: Biological effects and ecological consequences. In: Varanasi
U ed. Metabolism of polycyclic aromatic hydrocarbons in the aquatic
environment. Boca Raton, Florida, CRC Press, pp 291-329.
Moriske H-J, Freise R, Schneider C, & Rüden H (1987) [Polar neutral
organic compounds (POCN) in town aerosols. 2. Communication:
Measurement of the emissions from homeheating and cars and from
immission particles in Berlin (West).] Zentralbl Bakteriol Hyg I Abt
Orig B, 185: 72-104 (in German).
Morris HP, Velat CA, Wagner BP, Dahlgard M, & Ray FE (1960) Studies of
carcinogenicity in the rat of derivatives of aromatic amines related
to N-2-fluorenylacetamide. J Natl Cancer Inst, 24: 149-180.
Morrison VM, Burnett AK, & Craft JA (1991a) Metabolism of
7,12-dimethyl-benz [a]anthracene in hepatic microsomal membranes from
rats treated with isoenzyme-selective inducers of cytochromes P450.
Biochem Pharmacol, 41: 1505-1512.
Morrison VM, Burnett AK, Forrester LM, Wolf CR, & Craft JA (1991b) The
contribution of specific cytochromes P450 in the metabolism of
7,12-dimethylbenz [a]anthracene in rat and human liver microsomal
membranes. Chem-Biol Interactions, 79: 179-196.
Morselli L & Zappoli S (1988) PAH determination in samples of
environmental interest. Sci Total Environ, 73: 257-266.
Mossanda K, Poncelet F, Fouassin A, & Mercier A (1979) Short papers.
Detection of mutagenic polycyclic aromatic hydrocarbons in African
smoked fish. Food Cosmet Toxicol, 17: 141-143.
Motykiewicz G (1995) Application of biomarkers in heavily polluted
industrialized areas of countries of central and eastern Europe.
Toxicology, 101: 117-123.
Moulin JJ, Portefaix P, Wild P, Mur JM, Smagghe G, & Mantout B (1990)
Mortality study among workers producing ferroalloys and stainless
steel in France. Br J Ind Med, 47: 537-543.
Moulin JJ, Wild P, Mantout B, Fournier-Betz M, Mur JM, & Smagghe G
(1993) Mortality from lung cancer and cardiovascular diseases among
stainless-steel producing workers. Cancer Causes Control, 4: 75-81.
Muel B & Saguem S (1985) Determination of 23 polycyclic aromatic
hydrocarbons in atmospheric particulate matter of the Paris area and
photolysis by sunlight. Int J Environ Anal Chem, 19: 111-131.
Mueller JG & Lantz SE (1993) Strategy using bioreactors and specially
selected microorganisms for bioremediation of groundwater contaminated
with creosote and pentachlorophenol. Environ Sci Technol, 27: 691-698.
Müller E (1968) [Carcinogenic compounds in water and soil. XX.
Investigation into the carcinogenic properties of benzo(ghi)perylene.]
Arch Hyg, 152: 23-36 (in German with English abstract).
Müller H (1987) Hydrocarbons in the freshwater environment. A
literature review. Arch Hydrobiol, 24: 1-69.
Müller G, Grimmer G & Böhnke H (1977) [Sedimentary record of heavy
metals and polycyclic aromatic hydrocarbons in Lake Constance.].
Naturwissenschaften, 64: 427-431 (in German).
Muller P, Leece B & Raha D (1995a) Estimated risk of cancer from
exposure to PAH fractions of complex mixtures. In: Fifteenth
international symposium on polycyclic aromatic compounds: Chemistry,
biology and environmental impact, Belgirate, Italy, 19-22 September
1995. Ispra, Joint Research Centre European Commission, pp 159-160.
Muller P, Leece B & Raha D (1995b) Dose-response assessment PAH.
Ottawa, Ontario Ministry of the Environment and Energy, 197 pp.
Muller P, Leece B & Raha D (1996) Scientific criteria document for
multimedia environmental standards development: Polycyclic aromatic
hydrocarbons (PAH). Part 1. Dose response assessment. Ottawa, Ontario
Ministry of the Environment and Energy, 203 pp.
Mumford JL, He XZ, Chapman RS, Cao SR, Harris DB, Li XM, Xian YL,
Jiang WZ, Xu CW, Chuang JC, Wilson WE, & Cooke M (1987a) Lung cancer
and indoor air pollution in Xuan Wei, China. Science, 235: 217-235.
Mumford JL, Chapman RS, Harris DB, He XZ, Cao SR, Xian YL, & Li XM
(1987b) Lung cancer and indoor air exposure to unvented coal and wood
combustion emission in Xuan Wie, China. In: Seifert B, Esdorn H,
Fischer M, Rüden H, & Wegner J ed. Indoor Air '87, Volume 3:
Developing countries, guaranteeing adequate indoor air quality,
control measures, ventilation effectiveness, thermal climate and
comfort, policy and strategies. Berlin, Institute for Water, Soil and
Air Hygiene, pp 8-14.
Mumford JL, Helmes CT, Lee X, Seidenberg J, & Nesnow S (1990) Mouse
skin tumorigenicity studies of indoor coal and wood combustion
emissions from homes of residents in Xuan Wie, China, with high lung
cancer mortality. Carcinogenesis, 11: 397-403.
Mumford JL, Williams RW, Walsh DB, Burton RM, Svensgaard DJ, Chuang
JC, Houk VS, & Lewtas J (1991) Indoor air pollutants from unvented
kerosene heater emissions in mobile homes: Studies on particles,
semivolatile organics, carbon monoxide, and mutagenicity. Environ Sci
Technol, 25: 1732-1738.
Mumford JL, Lee X, Lewtas J, Young TL, & Santella RM (1993) DNA
adducts as biomarkers for assessing exposure to polycyclic aromatic
hydrocarbons in tissues from Xuan Wei women with high exposure to coal
combustion emissions and high lung cancer mortality. Environ Health
Perspectives, 99: 83-87.
Mumford JL, Li X, Hu F, Lu XB & Chuang JC (1995) Human exposure and
dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei,
China, with high lung cancer mortality associated with exposure to
unvented coal smoke. Carcinogenesis, 16: 3031-3036.
Munz MJ & Tarazona JV (1993) Synergistic effect of two- and
four-component combinations of the polycyclic aromatic hydrocarbons:
Phenanthrene, anthracene, and acenaphthene on Daphnia magna. Bull
Environ Contam Toxicol, 50: 363-368.
Mur JM, Moulin JJ, Meyer-Bisch C, Massin N, Couplon JP & Loulergue J
(1987) Mortality of aluminium reduction plant workers in France. Int J
Epidemiol, 16: 257-264.
Murison GL (1988) Induction of sister-chromatid exchanges by direct
and indirect agents in a human teratoma cell line. Mutat Res, 203:
347-354.
Murray JJ, Pottie RF, & Pupp C (1974) The vapor pressures and
enthalpies of sublimation of five polycyclic aromatic hydrocarbons.
Can J Chem, 52: 557-563.
Myhr BC & Caspary WJ (1988) Evaluation of the L5178Y mouse lymphoma
cell mutagenesis assay: Intralaboratory results for sixty-three coded
chemicals tested at Litton Bionetics, Inc. Environ Mol Mutag, 12
(suppl 13): 103-194.
Nagabhushan M, Hussong J, Polverini PJ, & Solt DB (1990) Inhibition of
hamster buccal pouch epithelial cell replication during in
vitro exposure to polycyclic aromatic hydrocarbons. Proc Am Assoc
Cancer Res, 31: 86.
Nagel DL, Stenbäck F, Clayson DB, & Wallcave L (1976) Intratracheal
installation studies with 7 H-dibenzo [c,g]carbazole in Syrian
hamster. J Natl Cancer Inst, 57: 119-123.
Namkung MJ & Juchau MR (1980) On the capacity of human placental
enzymes to catalyse the formation of diols from benzo [a]pyrene.
Toxicol Appl Pharmacol, 55: 253-259.
Natarajan AT & Darroudi F (1991) Use of human hepatoma cells for in
vitro metabolic activation of chemical mutagens/carcinogens.
Mutagenesis, 6: 399-403.
National Chemicals Inspectorate (1994) [Report on the use of highly
aromatic oils in tyres in Sweden.] Solna, pp 15-21 (Report No. 6/94)
(in Swedish with English summary).
National Institute for Occupational Health and Safety and Occupational
Safety and Health Administration (1981) Occupational health guideline
for coal tar pitch volatiles. In: Mackison FW, Stricoff RS, &
Partridge LJ Jr ed. Occupational health guidelines for chemical
hazards. Cambridge, Massachusetts, 30 pp [DHHS (NIOSH) Publication No.
81-123; US NTIS PB83-154609, Part 1 of 3].
National Institute for Occupational Health and Safety (1994a)
Polynuclear aromatic hydrocarbons by HPLC: Method No. 5506. In: Eller
PM & Cassinelli ME ed. NIOSH manual of analytical methods, 4th ed.
Cincinnati, Ohio, 8 pp (Publication No. 94-113).
National Institute for Occupational Health and Safety (1994b)
Polynuclear aromatic hydrocarbons by GC: Method No. 5515. In: Eller PM
& Cassinelli ME ed. NIOSH manual of analytical methods, 4th ed.
Cincinnati, Ohio, 7 pp. (Publication No. 94-113).
National Research Council Canada (1983) Polycyclic aromatic
hydrocarbons in the aquatic environment: formation, sources, fate and
effects on aquatic biota. Ottawa, Associate Committee on Scientific
Criteria for Environmental Quality, pp 32-33 (Report NRCC No. 18981).
National Toxicology Program (1991) Final study report and appendix:
Developmental toxicity evaluation of naphthalene (CAS No. 91-20-3)
administered by gavage to Sprague-Dawley (CD) rats on gestational days
6 through 15. Research Triangle Park, North Carolina, US Department of
Health and Human Services, 107 pp (Report TER-91006).
National Toxicology Program (1992a) Final study report and appendix:
Development toxicity evaluation of naphthalene (CAS No. 91-20-3)
administered by gavage to New-Zealand White (NZW) rabbits on
gestational days 6 through 19. Research Triangle Park, North Carolina,
US Department of Health and Human Services, 107 pp (Report TER-91021).
National Toxicology Program (1992b) Toxicology and carcinogenesis
studies of naphthalene (CAS No. 91-20-3) in B6C3F1 mice (inhalation
studies). Research Triangle Park, North Carolina, US Department of
Health and Human Services, 129 pp (NTP TR 410; NIH Publication No.
92-3141).
National Toxicology Program (1993) Hazardous substances data bank.
Bethesda, Maryland, National Library of Medicine, 20 pp.
Natusch DFS & Tomkins BA (1978) Isolation of polycyclic organic
compounds by solvent extraction with dimethyl sulfoxide. Anal Chem,
50: 1429-1434.
Navarro A, Rosell A, Villanueva J, & Grimalt JO (1991) Monitoring of
hazardous waste dumps by the study of metals and solvent-soluble
organic chemicals. Chemosphere, 22: 913-928.
Naylor LM & Loehr RC (1982) Priority pollutants in municipal sewage
sludge. Bio-Cycle, 23: 18-22.
Neal J & Rigdon RH (1967) Gastric tumors in mice fed benzo(a)pyrene: A
quantitative study. Tex Rep Biol Med, 25: 553-557.
Nebert DW (1980) Pharmacogenetics: An approach to understanding
chemical and biological aspects of cancer. J Natl Cancer Inst, 6:
1279-1290.
Nebert DW, Levitt RC, Jensen NM, Lambert GS, & Felton JS (1977) Birth
defects and aplastic anemia: Differences in polycyclic hydrocarbon
toxicity associated with the Ah locus. Arch Toxicol, 39: 109-132.
Neff JM (1979) Polycyclic aromatic hydrocarbons in the aquatic
environment: Sources, fates and biological effects. London, Applied
Science Publishers, 262 pp.
Neff JM & Anderson JW (1975) Accumulation, release, and distribution
of benzo(a)pyrene-C14 in the clam Rangia cuneata.
Proc Conf Prev Control Oil Pollut, 75: 469-471.
Negishi M, Swan DC, Enquist LW, & Nebert DW (1981) Isolation and
characterization of a cloned DNA sequence associated with the murine
Ah locus and a 3-methylcholan-threne-induced form of cytochrome
P450. Proc Natl Acad Sci USA, 78: 800-801.
Nelson PF (1989) Combustion-generated polycyclic aromatic hydrocarbons
in diesel exhaust emissions. Fuel, 68: 283-286.
Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW,
Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O, Okuda K, &
Nebert W (1993) The P450 superfamily: Update on new sequences, gene
mapping, accession numbers, early trivial names of enzymes, and
nomenclature. DNA Cell Biol, 12: 1-51.
Nesnow S (1990) Mouse skin tumors and human lung cancer: Relationships
with complex environmental emissions. In: Vainio H, Sorsa M, &
McMichael A ed. Complex mixtures and cancer risk. Lyon, International
Agency for Research on Cancer, pp 44-54 (IARC Scientific Publications
No. 104).
Nesnow S & Heidelberger C (1976) The effect of modifiers of microsomal
enzymes on chemical oncogenesis in cultures of C3H mouse cell lines.
Cancer Res, 36: 1801-1808.
Nesnow S, Evans C, Stead A, Creason J, Slaga TJ, & Triplett LL (1982a)
Skin carcinogenesis studies of emission extracts. In Lewtas J ed.
Toxicological effects of emissions from diesel engines. Amsterdam,
Elsevier, 295-320.
Nesnow S, Triplett L, & Slaga TJ (1982b) Comparative tumor-initiating
activity of complex mixtures from environmental particulate emissions
on Sencar mouse skin. J Natl Cancer Inst, 68: 829-834.
Nesnow S, Leavitt S, Easterling R, Watts R, Toney SH, Claxton L,
Sangaiah R, Toney GE, Wiley J, Fraher P, & Gold A (1984) Mutagenicity
of cyclopenta-fused isomers of benz [a]anthracene in bacterial and
rodent cells and identification of the major rat liver microsomal
metabolites. Cancer Res, 44: 4993-5003.
Nesnow S, Easterling RE, Ellis S, Watts R, & Ross J (1988) Metabolism
of benz [j]aceanthrylene (cholanthrylene) and benz [1]aceanthrylene
by induced rat liver S9. Cancer Lett, 39: 19-27.
Nesnow S, Ross J, Mohapatra N, Gupta R, Sangaiah R, & Gold A (1991)
Genotoxicity and identification of the major DNA-adducts of
aceanthrylene. In: Cooke M, Loening K, & Merritt J ed. Polynuclear
aromatic hydrocarbons: Measurements, means, and metabolism. Columbus,
Ohio, Battelle Press, pp 629-639.
Nesnow S, Ross J, Beck S, Lasley J, Nelson G, Lamberg G, Platt KL, &
Agarwal SC (1994) Morphological transformation and DNA adduct
formation by dibenz [a,h]-anthracene and its metabolites in
C3H10T1/2Cl8 cells. Carcinogenesis, 15: 2225-2231.
Nesnow S, Mass MJ, Ross JA, Gennings C, Carter WH Jr, & Stoner GD
(1995) Lung tumorigenic interactions of five environmental PAH.
Additivity, synergism and antagonism. In: Fifteenth international
symposium on polycyclic aromatic compounds: Chemistry, biology and
environmental impact, Belgirate, Italy, 19-22 September 1995. Ispra,
Joint Research Centre European Commission, p 151.
Netherlands' Delegation (1991) The problem of polycyclic aromatic
hydrocarbons (PAH) in the Rhine. In: Working Group B of International
Rhine Commission, pp 1-7 (B 5/91 Revision).
Nettesheim P & Hammons AS (1971) Induction of squamous cell carcinoma
in the respiratory tract of mice. J Natl Cancer Inst, 47: 697-701.
Neubert D & Tapken S (1988) Transfer of benzo(a)pyrene into mouse
embryos and fetuses. Arch Toxicol, 62: 236-239.
Neuhauser EF & Callahan CA (1990) Growth and reproduction of the
earthworm Eisenia fetida exposed to sublethal concentrations of
organic chemicals. Soil Biol Biochem, 22: 175-179.
Neuhauser EF, Durkin PR, Malecki MR, & Anatra M (1986) Comparative
toxicity of ten organic chemicals to four earthworm species. Comp
Biochem Phys, C83: 197-200.
Newsted JL & Giesy JP Jr (1987) Predictive models for photoinduced
acute toxicity of polycyclic aromatic hydrocarbons to Daphnia
magna, Strauss Cladocera, Crustacea. Toxicol Chem, 6: 445-461.
Ng KM, Chu I, Bronaugh RL, Franklin CA, & Somers DA (1992)
Percutaneous absorption and metabolism of pyrene, benzo (a)pyrene,
and di(2-ethylhexyl)phthalate: Comparison of in vitro and in
vivo results in the hairless guinea pig. Toxicol Appl Pharmacol,
115: 216-223.
Nicholls TP, Perry R, & Lester JN (1979) The influence of heat
treatment on the metallic and polycyclic aromatic hydrocarbon content
of sewage sludge. Sci Total Environ, 12: 137-150.
Nichols DG, Gangwal SK, & Sparacino CM (1981) Analysis and assessment
of PAH from coal composition and gasification. In: Cooke M & Dennis AJ
ed. Polynuclear aromatic hydrocarbons: Chemical analysis and
biological fate. Columbus, Ohio, Battelle Press, 397-406.
Nielsen T (1984) Reactivity of polycyclic aromatic hydrocarbons toward
nitrating species. Environ Sci Technol, 18: 157-163.
Nikonova TV (1977) Transplacental action of benzo(a)pyrene and pyrene.
Bull Exp Biol Med, 84: 1025-1027.
Nisbet ICT & LaGoy PK (1992) Toxic equivalency factors (TEFs) for
polycyclic aromatic hydrocarbons (PAHs). Regul Toxicol Pharmacol, 16:
290-300.
Nisbet ICT, Schneiderman MA, Karch NJ, & Siegel DM (1985) Review and
evaluation of the evidence for cancer associated with air pollution.
Research Triangle Park, North Carolina, US Environmental Protection
Agency, 295 pp (NTIS/PB85-142016).
Nishioka M, Chang H-C, & Lee ML (1986) Structural characteristics of
polycyclic aromatic hydrocarbon isomers in coal tars and combustion
products. Environ Sci Technol, 20: 1023-1027.
Nkedi-Kizza P, Rao PSC, & Hornsby AG (1985) Influence of organic
cosolvent on sorption of hydrophobic organic chemicals by soils.
Environ Sci Technol, 19: 975-979.
Nordholm L, Espensen I-M, Jensen HS, & Holst E (1986) Polycyclic
aromatic hydrocarbons in smokehouses. Scand J Work Environ Health, 12:
614-618.
Norpoth K, Kemena A, Jacob J, & Schümann C (1984) The influence of 18
environmentally relevant polycyclic aromatic hydrocarbons and Clophen
A50, as liver monooxygenase inducers, on the mutagenic activity of
benz [a]anthracene in the Ames test. Carcinogenesis, 5: 747-752.
Nousiainen U, Törrönen R, & Hänninen O (1984) Differential induction
of various carboxylesterases by certain polycyclic aromatic
hydrocarbons in the rat. Toxicology, 32: 243-251.
Novelli G & Rinaldi A (1979) Further contributions to the study of
coal dust for foundries. Giesserei, 66: 480-482.
Nowak D, Meyer A, Schmidt-Preuss U, Gatzemeyer U, Magnussen H, &
Rüdiger HW (1992) Formation of benzo(a)pyrene-DNA adducts in blood
monocytes from lung cancer patients with a familial history of lung
cancer. J Cancer Res Clin Oncol, 118: 67-71.
Noyes WF (1969) Carcinogen-induced neoplasia with metastasis in a
South American primate, Saguinus oedipus (33845). Proc Soc Exp Biol
Med, 131: 223-225.
Ny ET, Heederik D, Kromhout H, & Jongeneelen F (1993) The relationship
between polycyclic aromatic hydrocarbons in air and in urine of
workers in a Söderberg potroom. Am Ind Hyg Assoc J, 54: 277-284.
Obana H, Hori S, & Kashimoto T (1981a) Determination of polycyclic
aromatic hydrocarbons in marine samples by high-performance liquid
chromatography. Bull Environ Contam Toxicol, 26: 613-620.
Obana H, Hori S, Kashimoto T, & Kunita N (1981b) Polycyclic aromatic
hydrocarbons in human fat and liver. Bull Environ Contam Toxicol, 27:
23-27.
Oberly TJ, Huffman DM, & Garriott ML (1992) An evaluation of
chromosomal mutagens in the CHO-AS52 cell line. Environ Mol Mutag, 19
(suppl 20): 46 (Abstract).
O'Brien KAF, Smith LL, & Cohen GM (1985) Differences in
naphthalene-induced toxicity in the mouse and rat. Chem-Biol
Interactions, 55: 109-122.
O'Brien KAF, Suverkropp C, Kaqnekal S, Plopper CG, & Buckpitt AR
(1989) Tolerance to multiple doses of the pulmonary toxicant,
naphthalene. Toxicol Appl Pharmacol, 99: 487-500.
Oesch F (1973) Mammalian epoxide hydrases: inducible enzymes
catalysing the inactivation of carcinogenic and cytotoxic metabolites
derived from aromatic and olefinic compounds. Xenobiotica, 3: 305-340.
Oesch F, Bücker M, & Glatt HR (1981) Activation of phenanthrene to
mutagenic metabolites and evidence for at least two different
activation pathways. Mutat Res, 81: 110.
Okano P, Miller HN, Robinson RC, & Gelboin HV (1979) Comparison of
benzo [a]pyrene and
(-)-trans-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene metabolism in
human blood monocytes and lymphocytes. Cancer Res, 39: 3184-3193.
Okita T, Yanagihara M, Yoshida K, Iwata M, Tanabe K, & Hara H (1994)
Measurements of air pollution associated with oil fires in Kuwait by a
Japanese research team. Atmos Environ, 28: 2255-2259.
Old LJ, Benacerraf B, & Carswell E (1963) Contact reactivity to
carcinogenic polycyclic hydrocarbons. Nature, 198: 1215-1216.
Olufsen SB (1980) Polynuclear aromatic hydrocarbons in Norwegian
drinking water resources. In: Bjorseth A & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemistry and biological effects. Columbus,
Ohio, Battelle Press, pp 333-343.
Olufsen SB & Bjorseth A (1983) Analysis of polycyclic aromatic
hydrocarbons by gas chromatography. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 257-300.
Omland O, Sherson D, Hansen AM, Sigsgaard T, Autrup H, & Overgaard E (
1994) Exposure of iron foundry workers to polycyclic aromatic
hydrocarbons: benzo(a)pyrene-albumin adducts and 1-hydroxypyrene as
biomarkers for exposure. Occup Environ Med, 51: 513-518.
Oris JT & Giesy JP Jr (1985) The photoenhanced toxicity of anthracene
to juvenile sunfish (Lepomis spp.). Aquat Toxicol, 6: 133-146.
Oris JT & Giesy JP (1986) Photoinduced toxicity of anthracene to
juvenile bluegill sunfish (Lepomis macrochirus Rafinesque):
Photoperiod effects and predictive hazard evaluation. Environ Toxicol
Chem, 5: 761-768.
Oris JT & Giesy JP Jr (1987) The photo-induced toxicity of polycyclic
aromatic hydrocarbons to larvae of the fathead minnow (Pimephales
promelas). Chemosphere, 16: 1395-1404.
Oris JT, Hall AT, & Tylka JD (1990) Humic acids reduce the
photo-induced toxicity of anthracene to fish and Daphnia. Environ
Toxicol Chem, 9: 575-584.
Osborn JF, Santhanam S, Davidson CI, Flotard RD, & Stetter JR (1984)
Characterization of airborne trace metal and trace organic species
from coal gasification. Environ Monit Assess, 4: 317-333.
Osborne MR & Crosby NT (1987a) Binding to proteins and nucleic acids.
In: Benzopyrenes. Cambridge, Cambridge University Press, pp 137-176
(Cambridge Monographs on Cancer Research).
Osborne MR & Crosby NT (1987b) Benzo [e]pyrene. In: Benzopyrenes.
Cambridge, Cambridge University Press, pp 229-250 (Cambridge
Monographs on Cancer Research).
Osborne MR, Harvey RG, & Brookes P (1978) The reaction of
trans-7,8-dihydroxy- anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo
[a]pyrene with DNA involves attack at the N7-position of guanine
moieties. Chem-Biol Interactions, 20: 123-130.
Osborne MR, Brookes P, Lee H, & Harvey RG (1986) The reaction of a
3-methylcholan-threne diol epoxide with DNA in relation to the binding
of 3-methylcholanthrene to the DNA of mammalian cells. Carcinogenesis,
7: 1345-1350.
Oshiro Y, Balwierz PS, Soelter SG, Guzzie PJ, & Rohrbacher E (1992)
Evaluation of mouse peripheral blood micronucleus assay. Environ Mol
Mutag, 19 (suppl 20): 47 (Abstract).
Ostman C, Nilsson U, Carlsson H, Andersson I, & Fahlgren L (1991)
[Polycyclic aromatic compounds (PAC) in work environment. I. PAC in
Stockholm street air: An introductory study.] Solna, Swedish National
Institute of Occupational Health, 16 pp (Research Report No. 21) (in
Swedish).
Ostman C, Nilsson U, Carlsson H, Andersson I, & Fahlgren L (1992a)
[Polycyclic aromatic compounds (PAC) in work environment. II. PAC in
Stockholm street air: Content of PAH in two busy streets.] Solna,
Swedish National Institute of Occupational Health, 41 pp (Research
Report No. 34) (in Swedish with English summary).
Ostman C, Nilsson U, Carlsson H, Andersson I, & Fahlgren L (1992b)
[Polycyclic aromatic compounds (PAC) in work environment. III. PAC in
Stockholm street air: Content of PAH before and after a traffic
diversion.] Solna, Swedish National Institute of Occupational Health,
Division of Analytical Chemistry, 31 pp (Research Report No. 35) (in
Swedish with English summary).
Ostrander GK, Landolt ML, & Kocan RM (1988) The ontogeny of Coho
salmon (Oncorhynchus kisutch) behavior following embryonic exposure
to benzo [a]pyrene. Aquat Toxicol, 13: 325-346.
Oueslati R, Alexandrov K, Chouikha M, & Chouroulinkov I (1992)
Formation and persistence of DNA adducts in epidermal and dermal mouse
skin exposed to benzo(a)pyrene in vivo. In Vivo, 6: 231-236.
Ovrebo S, Hewer A, Phillips DH, & Haugen A (1990) Polycyclic aromatic
hydrocarbon-DNA adducts in coke-oven workers. In: Vainio H, Sorsa M, &
McMichael AJ ed. Complex mixtures and cancer risk. Lyon, International
Agency for Research on Cancer, pp 193-198 (IARC Scientific
Publications No. 104).
Ovrebo S, Haugen A, Phillips DH, & Hewer A (1992) Detection of
polycyclic aromatic hydrocarbon-DNA adducts in white blood cells from
coke oven workers: Correlation with job catagories. Cancer Res, 52:
1510-1514.
Ovrebo S, Haugen A, Fjeldstad PE, Hemminki K, & Szyfter K (1994)
Biological monitoring of exposure to polycyclic aromatic hydrocarbon
in an electrode paste plant. J Occup Med, 36: 303-310.
Ovrebo S, Fjeldstad PE, Grzybowska E, Kure EH, Chorazy M, & Haugen A
(1995) Biological monitoring of polycyclic aromatic hydrocarbon
exposure in a highly polluted area of Poland. Environ Health
Perspectives, 103: 838-843.
Pahlman R & Pelkonen O (1987) Mutagenicity studies of different
polycyclic aromatic hydrocarbons: The significance of enzymatic
factors and molecular structure. Carcinogenesis, 8: 773-778.
Paika IJ, Beauchesne MT, Randall M, Schreck RR, & Latt SA (1981) In
vivo SCE analysis of 20 coded compounds. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests of carcinogens. Report of the
international collaborative programme. New York, Elsevier
North-Holland, pp 672-681 (Progress in Mutation Research, Volume 1).
Pal K (1981) The induction of sister-chromatid exchanges in Chinese
hamster ovary cells by K-region epoxides and some dihydrodiols derived
from benz [a]anthracene, dibenz [a,c]anthracene and
dibenz [a,h]anthracene. Mutat Res, 84: 389-398.
Pal K, Grover PL, & Sims P (1975) The metabolism of carcinogenic
polycyclic hydrocarbons by tissues of the respiratory tract. Biochem
Soc Trans, 3: 174-175.
Palitti F, Cozzi R, Fiore M, Palombo F, Polcaro C, Perez G, & Possagno
E (1986) An in vitro and in vivo study on mutagenic activity of
fluoranthene: Comparison between cytogenetic studies and HPLC
analysis. Mutat Res, 174: 125-130.
Pallardy MJ, House RV, & Dean JH (1989) Molecular mechanism of
7,12-dimethylbenz(a)anthracene induced immunosuppression: Evidence for
action via the interleukin-2 pathway. Mol Pharmacol, 36: 128-133.
Pallardy M, Mishal Z, Lebrec H, & Bohuon C (1992) Immune modification
due to chemical interference with transmembrane signalling:
Application to polycyclic aromatic hydrocarbons. Int J
Immunopharmacol, 14: 377-381.
Palmer WG & Scott WD (1981) Lung cancer in ferrous foundry workers: A
review. Am Ind Hyg Assoc J, 42: 329-340.
Palmork KH, Wilhelmsen S, & Neppelberg T (1973) International Council
for Exploration of the Sea. The contribution of polycyclic aromatic
hydrocarbons (PAH) to the marine environment from different
industries. Bergen, Institute of Marine Research, pp 1-21 (Report No.
C M 1973/E:33).
Pancirov RJ, Searl TD, & Brown RA (1980) Methods of analysis for
polynuclear aromatic hydrocarbons in environmental samples. Adv Chem
Ser, 185: 123-142.
Pankow JF, Isabelle LM, & Asher WE (1984) Trace organic compounds in
rain. 1. Sampler design and analysis by adsorption/thermal desorption
(ATD). Environ Sci Technol, 18: 310-318.
Park KS, Sims RC, Dupont RR, Doucette WJ, & Matthews JE (1990) Fate of
PAH compounds in two soil types: Influence of volatilization, abiotic
loss and biological activity. Environ Toxicol Chem, 9: 187-195.
Parkinson EK & Newbold RF (1980) Benzo [a]pyrene metabolism and DNA
adduct formation in serially cultivated strains of human epidermal
keratinocytes. Int J Cancer, 26: 289-299.
Parrott MC, Kawabata TT, & White KL Jr (1989) Suppression of humoral
immunity following dermal exposure to benzo(a)pyrene in B6C3F1 mice.
Toxicologist, 9: 201 (Abstract 801).
Partanen T & Boffetta P (1994) Cancer risk in asphalt workers and
roofers: Review and meta-analysis of epidemiologic studies. Am J Ind
Med, 26: 721-740.
Paschke A, Herbel W, Steinhard H, Franke S, & Francke W (1992)
Determination of mono- to tetracyclic aromatic hydrocarbons in
lubricating oil. J High Res Chromatogr, 15: 827-833.
Passino DRM & Smith SB (1987) Acute bioassays and hazard evaluation of
representative contaminants detected in Great Lakes fish. Environ
Toxicol Chem, 6: 901-907.
Pataki J & Huggins C (1969) Molecular site of substituents of
benz(a)anthracene related to carcinogenicity. Cancer Res, 29: 506-509.
Pathirana S, Connell W, & Vowles PD (1994) Distribution of polycyclic
aromatic hydrocarbons (PAHs) in an urban roadway system. Ecotoxicol
Environ Saf, 28: 256-269.
Pavanello S & Levis AG (1994) Human peripheral blood lymphocytes as a
cell model to evaluate the genotoxic effect of coal tar treatment.
Environ Health Perspectives, 102: 95-99.
Pavoni B, Sfriso A, & Marcomini A (1987) Concentration and flux
profiles of PCBs, DDTs and PAHs in a dated sediment core from the
lagoon of Venice. Mar Chem, 21: 25-35.
Payne JF (1977) Mixed function oxidases in marine organisms in
relation to petroleum hydrocarbon metabolism and detection. Mar Pollut
Bull, 8: 112-116.
Pelkonen O & Nebert DW (1982) Metabolism of polycyclic aromatic
hydrocarbons: Etiologic role in carcinogenesis. Pharmacol Rev, 34:
189-222.
Pelkonen O & Saarni H (1980) Unusual patterns of benzo [a]pyrene
metabolites and DNA-benzo [a]pyrene adducts produced by human
placental microsomes in vitro. Chem-Biol Interactions, 30: 287-296.
Pelkonen O, Sotaniemi E, & Mokka R (1977) The in vitro oxidative
metabolism of benzo [a]pyrene in human liver measured by different
assays. Chem-Biol Interactions, 16: 13-22.
Pellizzari ED, Castillo NP, Willis S, Smith D, & Bursey JT (1979)
Identification of organic components in aqueous effluents from
energy-related processes. In: Van Hall CE ed. Measurement of organic
pollutants in water and wastewater. Philadelphia, Pennsylvania,
American Society for Testing and Materials, pp 256-274 (ASTM STP 686).
PPenman BW, Kaden DA, Liber HL, Skopek TR, & Thilly WG (1980) Perylene
is a more potent mutagen than benzo [a]pyrene for Salmonella
typhimurium. Mutat Res, 77: 271-277.
Perera FP, Hemminki K, Young TL, Brenner D, Kelly G, & Santella RM
(1988) Detection of polycyclic aromatic hydrocarbon-DNA adducts in
white blood cells of foundry workers. Cancer Res, 48: 2288-2291.
Perera FP, Hemminki K, Gryzbowska E, Motykiewicz G, Michalska J,
Santella RM, Young T, Dickey C, Brandt-Rauf P, DeVivo I, BlanerW, Tsai
W, & Chorazy M (1992) Molecular and genetic damage in humans from
environmental pollution in Poland. Nature, 360: 256-258.
Perera FP, Tang DL, O'Neill JPO, Bigbee WL, Albertini RJ, Santella R,
Ottman R, Tsai WY, Dickey C, Mooney LA, Savela K, & Hemminki K (1993)
HPRT and glycophorin A mutations in foundry workers: Relationship to
PAH exposure and to PAH-DNA adducts. Carcinogenesis, 14: 969-973.
Perera FP, Dickey C, Santella R, O'Neill JP, Albertini RJ, Ottman R,
Tsai WY, Mooney LA, Savela K, & Hemminki K (1994) Carcinogen-DNA
adducts and gene mutation in foundry workers with low-level exposure
to polycyclic aromatic hydrocarbons. Carcinogenesis, 15: 2905-2910.
Perfetti GA, Nyman PJ, Fisher S, Joe FL Jr, & Diachenko GW (1992)
Determination of polynuclear aromatic hydrocarbons in seafood by
liquid chromatography with fluorescence detection. J Assoc Off Anal
Chem, 75: 872-877.
Perin-Roussel O, Saguem S, Ekert B, & Zajdela F (1983) Binding to DNA
of bay-region and pseudo bay region diol-epoxides of
dibenzo [a,e]fluoranthene and comparison with adducts obtained with
dibenzo [a,e]fluoranthene or its dihydrodiols in the presence of
microsomes. Carcinogenesis, 4: 27-32.
Perin-Roussel O, Croisy A, Ekert B, & Zajdela F (1984) The metabolic
activation of dibenzo(a,e)fluoranthene in vitro.
Evidence that its bay-region and pseudo-bay-region diolepoxides react
preferentially with guanosine. Cancer Lett, 22: 289-298.
Perry PE & Thomson EJ (1981) Evaluation of the sister chromatid
exchange method in mammalian cells as a screening system for
carcinogens. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens. Report of the international collaborative
programme. New York, Elsevier North-Holland, pp 560-569 (Progress in
Mutation Research, Volume 1).
Peter S, Palme GE, & Röhrborn G (1979) Mutagenicity of polycyclic
hydrocarbons. III. Monitoring genetic hazards of benz(a)anthracene.
Acta Morphol Acad Sci Hung, 27: 199-204.
Peters JA, DeAngelis DG, & Hughes TW (1981) An environmental
assessment of POM emissions from residential wood-fired stoves and
fireplaces. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: Chemical analysis and biological fate. Columbus, Ohio,
Battelle Press, pp 571-581.
Peterson AR, Landolph JR, Peterson H, Spears CP, & Heidelberger C
(1981) Oncogenic transformation and mutation of C3H/10T1/2 clone 8
mouse embryo fibroblasts by alkylating agents. Cancer Res, 41: 3095-
3099.
Petridou-Fischer J, Whaley SL, & Dahl AR (1988) In vivo
metabolism of nasally instilled benzo [a]pyrene in dogs and monkeys.
Toxicology, 48: 31-40.
Petry TH, Schmid P, & Schlatter CH (1994) Exposure to polycyclic
aromatic hydrocarbons (PAHs) in two different silicon carbide plants.
Ann Occup Hyg, 38: 741-752.
Pezzuto JM, Lea MA, & Yang CS (1976) Binding of metabolically
activated benzo(a)pyrene to nuclear macromolecules. Cancer Res, 36:
3647-3653.
Pezzuto JM, Lea MA, & Yang CS (1977) The role of microsomes and
nuclear envelope in the metabolic activation of benzo [a]pyrene
leading to binding with nuclear macro-molecules. Cancer Res, 37: 3427-
3433.
Pfannhauser W (1991) [Polycyclic aromatic hydrocarbons (PAH) in food
and selected vegetables in Austria.] Mitt Geb Lebensmittelunters Hyg,
82: 66-79 (in German).
Pfau W, Hughes NC, Grover PL, & Phillips DH (1992) HPLC separation of
32P-postlabelled benzo [b]fluoranthene-DNA adducts. Cancer Lett, 65:
159-167.
Pfeffer HU (1994) Ambient air concentrations of pollutants at
traffic-related sites in urban areas of North Rhine-Westphalia,
Germany. Sci Total Environ, 146/147: 263-273.
Pfeiffer EH (1973) Investigation on the carcinogenic burden by air
pollution in man. VII. Studies on the oncogenic interaction of
polycyclic aromatic hydrocarbons. Zentralbl Bakteriol Hyg Abt. Orig B,
158: 69-83.
Pfeiffer EH (1977) Oncogenic interaction of carcinogenic and
non-carcinogenic polycyclic aromatic hydrocarbons. In: Mohr V, Schmähl
D, & Tomatis L eds. Air pollution and cancer in man. Lyon,
International A gency for Research on Cancer, pp 69-77 (IARC
Scientific Publications No. 16).
Pflock H, Georgii HW, & Müller J (1983) [Particulate polycyclic
aromatic hydrocarbons (PAH) in polluted and in clean areas.]
Staub-Reinhalt Luft, 43: 230-234 (in German).
Phillips DH (1990) Modern methods of DNA adduct determination. In:
Cooper CS & Grover PL ed. Handbook of experimental pharmacology,
Volume 94/1. Berlin, Springer-Verlag, pp 503-546.
Phillips DH (1991) DNA-adduct analysis by 32P-postlabeling in the
study of human exposure to carcinogens. In: Groopman JD & Skipper PL
ed. Molecular dosimetry and human cancer. Chapter 9: Analytical,
epidemiological, and social considerations. Boca Raton, Florida, CRC
Press, pp 151-170.
Phillips DH & Alldrick AJ (1994) Tumorigenicity of a combination of
psoriasis therapies. Br J Cancer, 69: 1043-1045.
Phillips DH & Castegnaro M (1993) Results of an interlaboratory trial
of 32P-postlabelling. In: Phillips DH, Castegnaro M & Bartsch H ed.
Postlabelling methods for detection of DNA adducts. Lyon,
International Agency for Research on Cancer, pp 35-49 (IARC Scientific
Publications No. 124)
Phillips DH, Hewer A, Martin CN, Garner RC, & King MM (1988)
Correlation of DNA adduct levels in human lung with cigerette smoking.
Nature, 336: 790-792.
Phillips DH, Schoket B, Hewer A, & Grover PL (1990) DNA adduct
formation in human and mouse skin by mixtures of polycyclic aromatic
hydrocarbons. In: Vainio H, Sorsa M, & McMichael AJ ed. Complex
mixtures and cancer risk. Lyon, International Agency for Research on
Cancer, pp 223-229 (IARC Scientific Publications No. 104).
Phillips DH, Hewer A, Seidel A, Steinbrecher T, Schrode R, Oesch F, &
Glatt H (1991) Relationship between mutagenicity and DNA adduct
formation in mammalian cells for fjord- and bay-region diol-epoxides
of polycyclic aromatic hydrocarbons. Chem-Biol Interactions, 80: 177-
186.
Phillips DH, Castegnaro M & Bartsch H ed. (1993) Postlabelling methods
for detection of DNA adducts. Lyon, International Agency for Research
on Cancer, 388 pp (IARC Scientific Publications No. 124).
Pienta RJ, Poiley JA, & Lebherz WB III (1977) Morphological
transformation of early passage golden Syrian hamster embryo cells
derived from cryopreserved primary cultures as a reliable in
vitro bioassay for identifying diverse carcinogens. Int J Cancer,
19: 642-655.
Pike MH (1944) Ocular pathology due to organic compounds. J Mich State
Soc, 43: 581-584.
Pike MC (1983) Human cancer risk assessment. Polynuclear aromatic
hydrocarbons: Evaluation of sources and effects. Report by the
Committee on Pyrene and Selected Analogues, the Board on Toxicology
and Environmental Health Hazards, the Commission on Life Sciences and
the National Research Council. Washington DC, National Academic Press,
pp C1-C28.
Pischinger F & Lepperhoff G (1980) [Influence of air-fuel ratio on the
PAH emissions from Otto motor engines. Air pollution from polycylic
aromatic hydrocarbons. Registration and evaluation.] Düsseldorf,
VDI-Verlag, pp 59-63 (VDI Report No. 358) (in German).
Pistikopoulos P, Masclet P, & Mouvier G (1990) A receptor model
adapted to reactive species: Polycyclic aromatic hydrocarbons:
Evaluation of source contributions in an open urban site. I. Particle
compounds. Atmos Environ, 24: 1189-1197.
Plasterer MR, Bradshaw WS, Booth GM, Carter MW, Schuler RL, & Hardin
BD (1985) Developmental toxicity of nine selected compounds following
prenatal exposure in the mouse: Naphthalene, p-nitrophenol, sodium
selenite, dimethyl phthalate, ethylenethiourea, and four glycol ether
derivatives. J Toxicol Environ Health, 15: 25-38.
Platt KL, Pfeiffer E, Petrovic P, Friesel H, Beermann D, Hecker E, &
Oesch F (1990) Comparative tumorigenicity of picene and
dibenz [a,h]anthracene in the mouse. Carcinogenesis, 11: 1721-1726.
Plesha P, Stein J, Schiewe M, McCin B, & Varanasi U (1988) Toxicity of
marine sediments supplemented with mixtures of selected chlorinated
and aromatic hydrocarbons to the infaunal amphipod
Rhepoxynius-abronius. Mar Environ Res, 25: 85-98.
Plopper CG, Suverkropp C, Morin D, Nishio S, & Buckpitt A (1992)
Relationship of cytochrome P-450 activity to Clara cell cytotoxicity.
I. Histopathologic comparison of the respiratory tract of mice, rats
and hamsters after parenteral administration of naphthalene. J
Pharmacol Exp Ther, 261: 353-363.
Podoll RT, Irwin KC, & Parish HJ (1989) Dynamic studies of naphthalene
sorption on soil from aqueous solution. Chemosphere, 18: 2399-2412.
Poirier MC (1994) Human exposure monitoring, dosimetry, and cancer
risk assessment: The use of antisera specific for carcinogen-DNA
adducts and carcinogen-modified DNA. Drug Metab Rev, 26: 87-109.
Poirier MC & Beland FA (1992) DNA adduct measurement and tumor
incidence during chronic carcinogen exposure in animal models:
Implications for DNA adduct-based human cancer risk assessment. Chem
Res Toxicol, 5: 749-755.
Pollia JA (1941) Investigation on the possible carcinogenic effect of
anthracene and chrysene and some of their compounds. II. The effect of
subcutaneous injection in rats. J Ind Hyg Toxicol, 223: 449-451.
Poncelet F, Massanda K, Fouassin A, & Mercier M (1978) Mutagenic study
of some polycyclic aromatic hydrocarbons present in smoked fishes from
Africa. Arch Int Phys Biochem, 86: 954-955.
Popescu NC, Turnbull D, & DiPaolo JA (1977) Sister chromatid exchange
and chromosome aberration analysis with the use of several carcinogens
and noncarcinogens: Brief communication. J Natl Cancer Inst, 59: 289-
293.
Pothuluri JV, Freeman JP, Evang FE, & Cerniglia CE (1993)
Biotransformation of fluorene by the fungus Cunninghamella
elegans. Appl Environ Microbiol, 59: 1977-1980.
Pott P (1775) Chirurgical observations relative to the cataract, the
polypus of the nose, the cancer of the scrotum, the different kinds of
ruptures, and the mortification of the toes and feet. London, Hawes,
Clark & Collins, pp 63-68.
Pott F & Heinrich U (1992) [Dust and dust components / polycyclic
aromatic hydrocarbons (PAH).] In: Wichmann HE, Schlipköter HW, &
Fülgraff G ed. [Handbook of environmental medicine, 2nd ed.]
Landsberg, Lech Ecomed mbH, pp 1-23 (in German).
Pott F, Brockhaus A, & Huth F (1973) [Tests on the production of
tumours in animal experiments with polycyclic aromatic hydrocarbons.]
Zentralbl Bakteriol Hyg I Abt Orig B, 157: 34-43 (in German).
Pott F, Mohr U, & Brockhaus A (1978) [Studies on the combined effects
of benzopyrene and dibenz [a]anthracene with SO2 and NO2 inhalation
on the golden hamster.] In: Rothe H ed. [Air hygiene and silicosis
research.] Essen, Girardet, pp 224-226 (in German).
Pott F, Ziem U, Reiffer FJ, Huth F, Ernst H, & Mohr U (1987)
Carcinogenicity studies on fibres, metal compounds, and some other
dusts in rats. Exp Pathol (Jena), 32: 129-152.
Potthast K (1980) [Recent results on the benzo [a]pyrene content of
meat products.] Fleischwirtschaft, 60: 1941-1949 (in German).
Potvin RR, Adamek EG, & Balsillie D (1980) Ambient PAH levels near a
steel mill in northern Ontario. In: Cooke M & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemical analysis and biological
fate. Columbus, Ohio, Battelle Press, pp 741-753.
Prasanna P, Jacobs MM, & Yang Sk (1987) Selenium inhibition of
benzo [a]pyrene, 3-methylcholanthrene, and 3-methylcholanthrylene
mutagenicity in Salmonella typhimurium strains TA98 and TA100. Mutat
Res, 190: 101-105.
Prinsen AJ & Kennedy WBH (1977) [Quantity of alpha-benzopyrene in
smoked foods.] In: [Aroma and flavouring substances.] Bilthoven,
National Institute of Public Health and Environmental Protection, pp
1-11 (Report No. 11) (in Dutch).
Prinsen AJ & Kennedy WBH (1978) [Quantity of alpha benzopyrene in
various tea samples.] In: [Aroma and flavouring substances.]
Bilthoven, National Institute of Public Health and Environmental
Protection, pp 1-5 (Report No. 12) (in Dutch).
Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK, & Neal SB (1981)
Chemically-induced unscheduled DNA synthesis in primary rat hepatocyte
cultures: A comparison with bacterial mutagenicity using 218
compounds. Environ Mutagen, 3: 11-32.
Prough RA, Patrizi VW, Okita RT, Masters BSS, & Jakobsson SW (1979)
Characteristics of benzo [a]pyrene metabolism by kidney, liver, and
lung microsomal fractions from rodents and humans. Cancer Res, 39:
1199-1206.
Pruell RJ, Hoffmann EJ, & Quinn JG (1984) Total hydrocarbons,
polycyclic aromatic hydrocarbons and synthetic organic compounds in
the hard shell clam, Mercenaria mercenaria, purchased at commercial
seafood stores. Mar Environ Res, 11: 163-181.
Pruess-Schwartz D, Baird WM, Yagi H, Jerina DM, Pigott MA, & Dipple A
(1987) Stereochemical specificity in the metabolic activation of
benzo [c]phenanthrene to metabolites that covalently bind to DNA in
rodent embryo cell cultures. Cancer Res, 47: 4032-4037.
Puffer H, Duncan K, Von Hofe E, Brewer G, & Mondal S (1979)
Benz(a)pyrene: Studies of the effects of this ubiquitous pollutant on
fishes. Ocean, 79: 398-400.
Pullman A (1945) [On an electronic theory of the carcinogenic action
of condensed aromatic hydrocarbons.] C R Séance Soc Biol Fil, 139:
1056-1058 (in French).
Pullman A (1947) [Contribution to the study of the electronic
structure of organic molecules. Special study on carcinogenic
hydrocarbons.]. Ann Chim, 2: 7-71 (in French).
Purchase IFH, Longstaff E, Ashby J, Styles JA, Anderson D, Lefevre PA,
& Westwood FR (1976) Evaluation of six short term tests for detecting
organic chemical carcinogens and recommendations for their use.
Nature, 264: 624-627.
Pussemier L, Szabo G, & Bulman RA (1990) Prediction of the soil
adsorption coefficient Koc for aromatic pollutants. Chemosphere, 21:
1199-1212.
Pyysalo H, Tuominen J, Wickström K, Skyttä E, Tikkanen L, Salomaa S,
Sorsa M, Nurmela T, Mattila T, & Pohjola V (1987) Polycyclic organic
material (POM) in urban air. Fractionation, chemical analysis and
genotoxicity of particulate and vapour phases in an industrial town in
Finland. Atmos Environ, 21: 1167-1180.
Quaghebeur D, De Wulf E, Ravelingien MC, & Janssens G (1983)
Polycyclic aromatic hydrocarbons in rainwater. Sci Total Environ, 32:
35-54.
Quarles JM, Sega MW, Schenley CK, & Lijinsky W (1979) Transformation
of hamster fetal cells by nitrosated pesticides in a transplacental
assay. Cancer Res, 39: 4525-4533.
Quilliam MA & Sim PG (1988) Determination of polycyclic aromatic
compounds by high-performance liquid chromatography with simultaneous
mass spectrometry and ultraviolet diode array detection. J Chromatogr
Sci, 26: 160-167.
Quinlan R, Kowalczyz G, Gardiner K, Calvert I, Hale K, & Walton S
(1995a) Polycyclic aromatic hydrocarbon exposure in coal liquefaction
workers: The value of urinary 1-hydroxypyrene excretion in the
development of occupational hygiene control strategies. Ann Occup Hyg,
39: 329-346.
Quinlan R, Kowalczyk G, Gardiner K, Hale K, Walton S, & Calvert I
(1995b) Urinary 1-hydroxypyrene: A biomarker for polycyclic aromatic
hydrocarbon exposure in coal liquefaction workers. Occup Med, 45: 63-
68.
Quinlan R, Kowalczyk G, Gardiner K, & Calvert I (1995c) Exposure to
polycyclic aromatic hydrocarbons in coal liquefaction workers: Impact
of a workwear policy on excretion of urinary 1-hydroxypyrene. Occup
Environ Med, 52: 600-605.
Radian Corp (1991) Asphalt industry cross sectional exposure
assessment study. Final Report. Sacramento, California, 200 pp.
Raha CR (1972) Metabolism of benzo [a]pyrene at the 4,5-position. Ind
J Biochem Biophys, 9: 105-110.
Rahman A, Barrowman JA, & Rahimtula A (1986) The influence of bile on
the bioavailability of polynuclear aromatic hydrocarbons from the rat
intestine. Can J Physiol Pharmacol, 64: 1214-1218.
Rainio K, Linko RR, & Ruotsila L (1986) Polycyclic aromatic
hydrocarbons in mussel and fish from the Finnish archipelago sea. Bull
Environ Contam Toxicol, 37: 337-343.
Raiyani CV, Jani JP, Desai NM, Shah JA, & Kashyap SK (1993a) Levels of
polycyclic aromatic hydrocarbons in ambient environment of Ahmedabad
City. Indian J Environ Prot, 13: 206-215.
Raiyani CV, Jani JP, Desai NM, Shah JA, Shah PG, & Kashyap SK (1993b)
Assessment of indoor exposure to polycyclic aromatic hydrocarbons for
urban poor using variuos types of cooking fuels. Bull Environ Contam
Toxicol, 50: 757-763.
Ramdahl T & Mller M (1983) Chemical and biological characterization
of emissions from a cereal straw burning furnace. Chemosphere, 12: 23-
34.
Ramdahl T, Alfheim I, Rustad S, & Olsen T (1982) Chemical and
biological characterization of emissions from small residential stoves
burning wood and charcoal. Chemosphere, 11: 601-611.
Rao KSM, Phadke KM, & Muthal PI (1987) Estimation of carcinogenic
polycyclic aromatic hyrocarbon concentrations on the top of coke
ovens. Res Ind, 43: 276-281.
Rastetter WH, Nachbar RB, Russo-Rodriguez S, Wattley RV, Thilly WG,
Andon BM, Jorgensen WL, & Ibrahim M (1982) Fluoranthene: Synthesis and
mutagenicity of fluor diol epoxides. J Org Chem, 47: 4873-4878.
Ratajczak EA, Ahland E, Grimmer G, & Dettbarn G (1984) [Reduction of
PAH emissions from hard coal briquettes containing bitumen instead of
pitch binder.] Staub-Reinhalt Luft, 44: 505-509 (in German).
Raunio H & Pelkonen O (1994) Cancer genetics: Genetic factors in the
activation and inactivation of chemical carcinogens. In: Ioannides C
ed. Drugs, diet and disease. Volume 1: Mechanistic approaches to
cancer. New York, Simon & Schuster International, pp 229-258.
Raveh D, Slaga TJ, & Huberman E (1982) Cell-mediated mutagenesis and
tumor-initiating activity of the ubiquitous polycyclic hydrocarbon,
cyclopenta [c,d]pyrene. Carcinogenesis, 3: 763-766.
Readman JW, Preston MR, & Mantoura RFC (1986) An integrated technique
to quantify sewage, oil and PAH pollution in estuarine und coastal
environments. Mar Pollut Bull, 17: 298-308.
Reardon DB, Prakash AS, Hilton BD, Roman JM, Pataki J, Harvey RG, &
Dipple A (1987) Characterization of
5-methylchrysene-1,2-dihydroidiol-3,4-epoxide-DNA adducts.
Carcinogenesis, 8: 1317-1322.
Reddy MV, Gupta RC, Randerath E, & Randerath K (1984)
32P-Postlabeling test for covalent DNA binding of chemicals
in vivo: Application to a variety of aromatic carcinogens and
methylating agents. Carcinogenesis, 5: 231-243.
Reddy MV, Olson JA, Stober GR, & Daniel FB (1991) Induction of nuclear
anomalies in the gastrointestinal tract by polycyclic aromatic
hydrocarbons. Cancer Lett, 56: 215-224.
Redmond CK (1983) Cancer mortality among coke oven workers. Environ
Health Perspectives, 52: 67-73.
Reed L (1983) Health hazard evaluation: Anchor Hocking Glass Company
Roofing Site, Lancaster, Ohio, January 1983. Cincinnati, Ohio,
National Institute for Occupational Safety and Health, pp 1-9 (Report
HETA 82-067-1253; PB 84-173046).
Reed GA, Layton ME, & Ryan MJ (1988) Metabolic activation of
cyclopenteno [c,d]pyrene by peroxyl radicals. Carcinogenesis, 9:
2291-2295.
Rees DC & Hattis D (1994) Developing quantitative strategies for
animal to human extrapolation. In: Hayes AW ed. Principles and methods
of toxicology, 3rd Ed, New York, Raven Press, pp 275-315.
Rees ED, Mandelstam P, Lowry JQ, & Lipscomb H (1971) A study of the
mechanism of intestinal absorption of benzo [a]pyrene. Biochem
Biophys Acta, 225: 96-107.
Rees RW, Brice AJ, Carlton JB, Gilbert PJ & Mitchell I de G (1989)
Optimization of metabolic activation for four mutagens in a bacterial,
fungal and two mammalian cell mutagenesis assays. Carcinogenesis, 4:
335-342.
Regional Office for Water and Waste Disposal (1986) [Water report
1985.] Düsseldorf, Northrhine-Westphalia, pp 24-27 (in German).
Regional Office for Water and Waste Disposal (1988) [Water report
1987] Düsseldorf, Northrhine-Westphalia, Table 10, p 29 (in German).
Regional Office for Water and Waste Disposal (1989) [Water report
1988.] Düsseldorf, Northrhine-Westphalia, pp 28, i-viii (in German).
Regional Office for Water and Waste Disposal (1990) [Water report
1989.] Düsseldorf, Northrhine-Westphalia, pp 25-29 (in German).
Reichert WL, Eberhart BTL, & Varanasi U (1985) Exposure of two species
of deposit-feeding amphipods to sediments-associated
3H-benzo [a]pyrene: Uptake, metabolism and covalent binding to
tissue macromolecules. Aquat Toxicol, 6: 45.
Reinhard M, Goodman NL, & Barker JF (1984) Occurrence and distribution
of organic chemicals in two landfill leachate plumes. Environ Sci
Technol, 18: 953-961.
Renwick AG & Drasar BS (1976) Environmental carcinogens and large
bowel cancer. Nature, 263: 234-235.
Reuterwall C, Aringer L, Elinder CG, Rannung A, Levin JO, Juringe L, &
Onfelt A (1991) Assessment of genotoxic exposure in Swedish coke-oven
work by different methods of biological monitoring. Scand J Work
Environ Health, 17: 123-132.
Reznikoff CA, Bertram JS, Brankow DW, & Heidelberger C (1973)
Quantitative and qualitative studies of chemical transformation of
cloned C3H mouse embryo cells sensitive to postconfluence inhibition
of cell division. Cancer Res, 33: 3239-3249.
Rhee K & Bratzler LJ (1970) Benzo(a)pyrene in smoked meat products. J
Food Sci, 35: 146-149.
Rhett RG, Simmers JW, & Lee CR (1988) Eisenia foetida used as a
biomonitoring tool to predict the potential bioaccumulation of
contaminants from contaminated dredged material. In: Edwards CA &
Neuhauser EF ed. Earthworms in waste and environmental management. The
Hague, SPB Academic Publishers, pp 321-328.
Rhoads CP, Smith WE, Cooper NS & Sullivan RD (1954) Early changes in
the skin or several species including man, after painting with
carcinogenic materials. Proc Am Assoc Cancer Res, 1: 40.
Rice JE, Hosted TJ Jr, & LaVoie EJ (1984) Fluoranthene and pyrene
enhance benzo [a]pyrene-DNA adduct formation in vivo in mouse skin.
Cancer Lett, 24: 327-333.
Rice JE, Coleman DT, Hosted TJ, LaVoie EJ, McCaustland DJ, & Wiley JC
(1985) Identification of mutagenic metabolites of
indeno[1,2,3- cd]pyrene formed in vitro with rat liver enzymes.
Cancer Res, 45: 5421-5425.
Rice JE, Hosted TJ Jr, DeFloria MC, LaVoie EJ, Fischer DL, & Wiley JC
Jr (1986) Tumor-initiating activity of major in vivo
metabolites of indeno[1,2,3- cd]pyrene on mouse skin. Carcinogenesis,
7: 1761-1764.
Rice JE, Weyand EH, Geddie NG, DeFloria MC, & LaVoie EJ (1987)
Identification of tumorigenic metabolites of benzo [j]fluoranthene
formed in vivo in mouse skin. Cancer Res, 47: 6166-6170.
Rice JE, Geddie NG, Defloria MC, & LaVoie EJ (1988a) Structural
requirements favoring mutagenic activity among methylated pyrenes in
S. typhimurium. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: A decade of progress. Columbus, Ohio, Battelle Press, pp
773-785.
Rice JE, Jordan K, Little P, & Hussain N (1988b) Comparative
tumor-initiating activity of methylene-bridged and bay-region
methylated derivatives of benz [a]anthracene and chrysene.
Carcinogenesis, 9: 2275-2278.
Rice JE, Weyand EH, Burrill C, & Lavoie EJ (1990) Fluorine probes for
investigating the mechanism of activation of indeno [1,2,3- cd]pyrene
to a tumorigenic agent. Carcinogenesis, 11: 1971-1974.
Riebe-Imre, M, Peiser, D, Emura M, Aufderheide M, Jacob J, Grimmer G,
& Raab G (1993) Passage-dependent characteristics of PAH-metabolism
and PAH-induced transformation in airway epithelial cells in vitro.
In: Garrigues P & Lamotte M ed. Polycyclic aromatic compounds:
Synthesis, properties, analytical measurements, occurrence and
biological effects. Bordeaux, Gordon & Breach Science Publishers, pp
849-856.
Riegel B, Wartman WB, Hill WT, Reeb BB, Shubik P, & Stanger DW (1951)
Delay of methylcholanthrene skin carcinogenesis in mice by
1,2,5,6-dibenzofluorene. Cancer Res, 11: 301-306.
Riess MH & Wefers H (1994) [Ecotoxicological evaluation of chlorine
and phosphorous organics as well as PAH in sediments of Bremen
waters.] Bremen, Institute for Environmental Chemistry, pp 9-10 (in
German).
Rigdon RH & Neal J (1965) Effects of feeding benzo [a]pyrene on
fertility, embryos, and young mice. J Natl Cancer Inst, 34: 297-305.
Rigdon RH & Neal J (1966) Gastric carcinomas and pulmonary adenomas in
mice fed benzo(a)pyrene. Tex Rep Biol Med, 24: 195-207.
Rigdon RH & Neal J (1969) Relationship of leukemia to lung and stomach
tumors in mice fed benzo(a)pyrene. Proc Soc Exp Biol Med, 130: 146-
148.
Riley RT, Shirazi MA, & Swartz RC (1981) Transport of naphthalene in
the oyster O streaedulis. Mar Biol, 63: 325-330
Rimatori V, Sperduto B, & Iannaccone A (1983) Environmental oil
aerosol. J Aerosol Sci, 14: 253-256.
Rippe RM & Pott F (1989) [Carcinogenicity studies into nitro-PAH
(nitroarenes) in view of their importance in the cancer-inducing
effects of diesel motor emissions.] In: Environmental Hygiene, Volume
21. Düsseldorf, Stefan W Albers Verlag, pp 65-89 (Society for Research
on Air Hygiene and Silicosis, Medical Institute for Environmental
Hygiene, Annual Report 1988/1989) (in German).
Risch HA, Burch JD, Miller AB, Hill GB, Steele R, & Howe GR (1988)
Occupational factors and the incidence of cancer of the bladder in
Canada. Br J Ind Med, 45: 361-367.
Robbins WK, Searl TD, Wasserstrom DH, & Boyer GT (1981) Determination
of polynuclear aromatic hydrocarbons in wastewater from coal
liquefaction processes by the gas chromatography-ultraviolet
spectrometry technique. In: Jackson LP & Wright CC ed. Analysis of
waters associated with alternative fuel production. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 149-166
(ASTM STP 720).
Roberts MH Jr, Hargis WJ Jr, Strobel CJ, & De Lisle PF (1989) Acute
toxicity of PAH contaminated sediments to the estuarine fish,
Leiostomus xanthurus. Bull Environ Contam Toxicol, 42: 142-149.
Robertson IGC, Guthenberg C, Mannervik B, & Jernström B (1986)
Differences in stereoselectivity and catalytic efficiency of three
human glutathione transferases in the conjugation of glutathione with
7b, 8a-dihydroxy-9a,10a-oxy-7,8,9,10-tetrahydrobenzo [a]pyrene.
Cancer Res, 46: 2220-2224.
Robinson DE & Mitchell AD (1981) Unscheduled DNA synthesis response of
human fibroblasts, WI-38 cells, to 20 coded chemicals. In: DeSerres FJ
& Ashby J ed. Evaluation of short-term tests for carcinogens. Report
of the international collaborative programme. New York, Elsevier North
Holland, pp 517-527 (Progress in Mutation Research, Volume 1).
Robinson JR, Felton JS, Levitt RC, Thorgeirsson SS, & Nebert DW (1975)
Relationship between 'aromatic hydrocarbon responsiveness' and the
survival times in mice treated with various drugs and environmental
compounds. Mol Pharmacol, 11: 850-865.
Rocchi P, Ferreri AM, Borgia R, & Prodi G (1980) Polycyclic
hydrocarbons induction of diphtheria toxin-resistant mutants in human
cells. Carcinogenesis, 1: 765-767.
Rockette HE & Arena VC (1983) Mortality studies of aluminum reduction
plant workers: Potroom and carbon department. J Occup Med, 25: 549-
557.
Rockette HE & Redmond CK (1985) Selection, follow-up, and analysis in
the coke oven study. Natl Cancer Inst Monogr, 67: 89-94.
Roe FJC (1962) Effect of phenanthrene on tumour-initiation by
3,4-benzpyrene. Br J Cancer, 16: 503-506.
Roe JC & Grant CA (1964) Test of pyrene and phenanthrene for
incomplete carcinogenic and anticarcinogenic activity. Br Emp Cancer
Campaign, 41: 59.
Rogan EG, Cavalieri EL, Ramakrishna NVS, & Devanesan PD (1993)
Mechanisms of benzo(a)pyrene and 7,12-dimethylbenz(a)anthracene
activation: Quantitative aspects of the stable and depurination DNA
adducts obtained from radical cations and diol epoxides. In: Garrigues
P & Lamotte M ed. Polycyclic aromatic compounds: Synthesis,
properties, analytical measurements, occurrence and biological
effects. Bordeaux, Gordon & Breach Science Publishers, pp 733-740.
Rogerson PF (1988) Organic chemical waste characterization for marine
disposal of Black Rock Harbor dredged materials. In: Lichtenberg JJ,
Winter JA, Weber CI, & Fradkin L ed. Chemical and biological
characterization of municipal sludges, sediments, dredge spoils, and
drilling muds. Philadelphia, Pennsylvania, American Society for
Testing and Materials, pp 213-222.
Roggeband R, Van den Berg PTM, Steenwinkel MJST, Van Delft JHM, Van
der Wulp CJM, & Baan RA (1994a) In situ detection of different PAH-
DNA adducts by means of immunofluorescence microscopy. In: Annual
report on toxicology 1993/1994. Zeist, TNO Nutrition and Food Research
Institute, pp 79-80.
Roggeband R, Wolterbeek APM, Van den Berg PTM, & Baan RA (1994b) DNA
adducts in hamster and rat tracheas exposed to benzo(a)pyrene
in vitro. Toxicol Lett, 72: 105-111.
Rojas M, Alexandrov K, Auburtin G, Wastiaux-Denamur A, Mayer L, Mahieu
B, Sebastien P, & Bartsch H (1995) Anti-benzo(a)pyrene diolepoxide-DNA
adduct levels in pheripheral mononuclear cells from coke oven workers
and the enhancing effect of smoking. Carcinogenesis, 16: 1373-1376.
Roller M, Kamino K, & Rosenbruch M (1992) Carcinogenicity testing of
bladder carcinogens and other organic compounds by the intraperitoneal
and intravesical route. In: Seemayer NH & Hadnagy W ed. Environmental
hygiene. III. Berlin, Springer-Verlag, pp 95-98.
Ronneberg A & Andersen A (1995) Mortality and cancer morbidity in
workers from an aluminum smelter with prebaked carbon anodes. Part II:
Cancer morbidity. Occup Environ Med, 52: 250-254.
Ronneberg A & Langmark F (1992) Epidemiologic evidence of cancer in
aluminum reduction plant workers. Am J Ind Med, 22: 573-590.
Rosenkranz HS & Poirier LA (1979) Evaluation of the mutagenicity and
DNA-modifying activity of carcinogens and noncarcinogens in microbial
systems. J Natl Cancer Inst, 62: 873-891.
Ross J, Nelson G, Erexson G, Kligerman A, Earley K, Gupta RC, & Nesnow
S (1991) DNA adducts in rat lung, liver, and peripheral blood
lymphocytes produced by ip administration of benzo [a]pyrene
metabolites and derivatives. Carcinogenesis, 12: 1953-1955.
Ross JA, Nelson GB, Holden KL, Kligerman AD, Erexson GL, Bryant MF,
Earley K, Beach AC, Gupta RC, & Nesnow S (1992) DNA adducts and
induction of sister chromatid exchanges in the rat following
benzo (b)fluoranthene administration. Carcinogenesis, 13: 1731-1734.
Ross JA, Nelson GB, Wilson KH, Rabinowitz JR, Galati A, Stoner GD,
Nesnow S, & Mass MJ (1995) Adenomas induced by polycyclic aromatic
hydrocarbons in strain A/J mouse lung correlate with time-integrated
DNA adduct levels. Cancer Res, 55: 1039-1044.
Rossi SS & Neff JM (1978) Toxicity of polynuclear aromatic
hydrocarbons to the polychaete Neanthes arenaceodentata. Mar Pollut
Bull, 9: 220-223.
Rossman TG, Molina M, Meyer L, Boone P, Klein CB, Wang Z, Li F, Lin
WC, & Kinney PL (1991) Performance of 133 compounds in the lambda
prophage induction endpoint of the microscreen assay and a comparison
with Salmonella typhimurium mutagenicity and rodent carcinogenicity
assays. Mutat Res, 260: 349-367.
Rostad CE & Pereira WE (1987) Creosote compounds in snails obtained
from Pensacola Bay, Florida, near an onshore hazardous-waste site.
Chemosphere, 16: 2397-2404.
Roszinsky-Köcher G, Basler A, & Röhrborn G (1979) Mutagenicity of
polycyclic hydrocarbons. V. Induction of sister-chromatid exchanges
in vivo. Mutat Res, 66: 65-67.
Rothman N, Poirier MC, Haas RA, Correa-Villasenor A, Ford P, Hansen
JA, O'Toole T, & Strickland PT (1993) Association of PAH-DNA adducts
in peripheral white blood cells with dietary exposure to polyaromatic
hydrocarbons. Environ Health Perspect, 99: 265-267.
Ruepert C, Grinwis A, & Govers H (1985) Prediction of partition
coefficients of unsubstituted polycyclic aromatic hydrocarbons from
C18 chromatographic and structural properties. Chemosphere, 14: 279-
291
Rundell JO, Guntakatta M, & Matthews EJ (1983) Criterion development
for the application of Balb/c-3T3 cells to routine testing for
chemical carcinogenic potential. In: Waters MD, Sandhu SS, Lewtas J,
Claxton L, Chernoff N, & Nesnow S ed. Short-term bioassays in the
analysis of complex environmental mixtures. III. New York, Plenum
Press, pp 309-324.
Russell H (1947) An unsuccessful attempt to induce gliomata in rabbits
with cholanthrene. J Pathol Bacteriol, 59: 481-483.
Russell LB (1977) Validation of the in vivo somatic mutation method
in the mouse as a prescreen for germinal point mutations. Arch
Toxicol, 38: 75-85.
Russell P, Yamada T, Xu GT, Garland D, & Zigler JS Jr (1991) Effects
of naphthalene metabolites on cultured cells from eye lens. Free
Radicals Biol Med, 10: 255-261.
Ryan PA & Cohen Y (1986) Multimedia transport of particle-bound
organics: Benzo [a]pyrene test case. Chemosphere, 15: 21-47.
Saber A, Jarosz J, Martin-Bouyer M, Paturel L, & Vial M (1987)
Analysis of polycyclic aromatic hydrocarbons in lacustral sediments by
high resolution Shpol'skii spectrofluori-metry at 10 K. Int J Environ
Anal Chem, 28: 171-184.
Sabourin TD & Tullis RE (1981) Effect of three aromatic hydrocarbons
on respiration and heart rates of the mussel,
Mytilus californianus. Bull Environ Contam Toxicol, 26: 729-736.
Saed T, Al-Yakoob S, Al-Hashash, & Al-Bahlou M (1995) Preliminary
exposure assessment for Kuwaiti consumers to polycyclic aromatic
hydrocarbons in seafood. Environ Int, 21: 255-263.
Saffiotti U, Cefis F, & Kolb LH (1968) A method for experimental
induction of bronchogenic carcinoma. Cancer Res, 28: 104-113.
Saffiotti U, Montesano R, Sellkumar AR, & Kaufman DG (1972)
Respiratory tract carcinogenesis induced in hamsters by different dose
levels of benzo [a]pyrene and ferric oxide. J Natl Cancer Inst, 49:
1199-1204.
Sagredos AN, Sinha-Roy D, & Thomas A (1988) [Determination, sources
and composition of polycyclic aromatic hydrocarbons in oils and fats.]
Fat Sci Technol, 90: 76-81 (in German).
Saguem S, Mispelter J, Perin-Roussel O, Lhoste JM, & Zajdela F (1983a)
Multi-step metabolism of the carcinogen dibenzo [a,e]fluoranthene. I.
Identification of the metabolites from rat microsomes. Carcinogenesis,
4: 827-835.
Saguem S, Perin-Roussel O, Mispelter J, Lhoste JM, & Zajdela F (1983b)
Multi-step metabolism of the carcinogen dibenzo [a,e]fluoranthene.
II. Metabolism pathways. Carcinogenesis, 4: 837-842.
Sakai M, Yoshida D, & Mizusaki S (1985) Mutagenicity of polycyclic
hydrocarbons and quinones on Salmonella thyphimurium TA97. Mutat
Res, 156: 61-67.
Salagovic J, Kalina I, & Dubayová K (1995) Induction of single strand
DNA breaks in workers professionally exposed to polycyclic aromatic
hydrocarbons. Neoplasma, 42: 115-118.
Salaman MH & Roe FJC (1956) Further tests for tumour-initiating
activity: N,N-Di-(2-chloroethyl)-p-aminophenylbutyric acid (CB1348) as
an initiator of skin tumor formation in the mouse. Br J Cancer, 10:
363-378.
Salamone MF (1981) Toxicity of 41 carcinogens and noncarcinogenic
analogs. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens. Report of the international collaborative programme.
Amsterdam, Elsevier North-Holland, pp 682-685 (Progress in Mutation
Research, Volume 1).
Salamone MF, Heddle JA, & Katz M (1979) The mutagenic activity of
thirty polycyclic aromatic hydrocarbons (PAH) an oxides in urban
airborne particulates. Environ Int, 2: 37-43.
Salamone MF, Heddle JA, & Katz M (1981) Mutagenic activity of 41
compounds in the in vivo micronucleus assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens. Report of
the international collaborative programme. Amsterdam, Elsevier
North-Holland, pp 686-697 (Progress in Mutation Research,Volume 1).
Salomaa S, Tuominen J, & Skyttä E (1988) Genotoxicity and PAC analysis
of particulate and vapour phases of environmental tobacco smoke. Mutat
Res, 204: 173-183.
Sanders CL, Skinner D, & Gelman RA (1986) Percutaneous absorption of
7,10 14C-benzo(a)pyrene and 7,12 14C-dimethylbenz(a)anthracene in
mice. J Environ Pathol Toxicol Oncol, 7: 25-34.
Sandmeyer EE (1981) Aromatic hydrocarbons. In: Clayton GD & Clayton FE
ed. Patty's industrial hygiene and toxicology, 3rd rev Ed. New York,
John Wiley & Sons, pp 3333-3339.
Santella RM (1993) Human DNA and protein adduct dosimetry. Proc Am
Assoc Cancer Res, 34: 604-605.
Santella RM, Hemminki K, Tang DL, Paik M, Ottman R, Young TL, Savela
K, Vodickova L, Dickey C, Whyatt R, & Perera FP (1993) Polycyclic
aromatic hydrocarbon-DNA adducts in white blood cells and urinary
1-hydroxypyrene in foundry workers. Cancer Epidemiol Biomarkers Prev,
2: 59-62.
Santella RM, Nunes MG, Blaskovic R, Perera FP, Tang D, Beachman A, Lin
JH, & DeLeo VA (1994) Quantification of polycyclic aromatic
hydrocarbons, 1-hydroxypyrene, and mutagenicity in urine of coal
tar-treated psoriasis patients and untreated volunteers. Cancer
Epidemiol Biomarkers Prev, 3: 137-140.
Santella RM, Perera FP, Young TL, Zhang YJ, Chiamprasert S, Tang D,
Wang LW, Beachman A, Lin JH, & DeLeo VA (1995) Polycyclic aromatic
hydrocarbon-DNA and protein adducts in coar tar treated patients and
controls and their relationship to glutathione S-transferase genotype.
Mutat Res, 334: 117-124.
Santodonato J, Basu D, & Howard PH (1980) Multimedia human exposure
and carcinogenic risk assessment for environmental PAH. In: Bjorseth A
& Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry and
biological effects. Columbus, Ohio, Battelle Press, 435-454.
Santodonato J, Howard P, & Basu D (1981) In: Lee SD & Grant L ed.
Health and ecological assessment of polynuclear aromatic hydrocarbons.
Park Forest South, Illinois, Pathotox Publishers, 364 pp.
Sardella DJ, Boger E, & Ghoshal PK (1981) Active sites in hexacyclic
carcinogens probed by the fluorine substitution methodology. Columbus,
Ohio, Battelle Press, pp 529-538.
Savas U, Bhattacharyya KK, Christou M, Alexander DL, & Jefcoate CR
(1994) Mouse cytochrome P450EF, representative of a new 1B subfamily
of cytochrome P450s. Cloning, sequence determination, and tissue
expression. J Biol Chem, 269: 14905-14911.
Savela K & Hemminki K (1991) DNA adducts in lymphocytes and
granulocytes of smokers and nonsmokers detected by the
32P-postlabelling assay. Carcinogenesis, 12: 503-508.
Savino JF & Tanabe LL (1989) Sublethal effects of phenanthrene,
nicotine, and pinane on Daphnia pulex. Bull Environ Contam Toxicol,
42: 778-784.
Sawicki JT, Moschel RC, & Dipple A (1983) Involvement of both
syn and antidihydrodiol-epoxides in the binding of
7,12-dimethylbenz [a]anthracene to DNA in mouse embryo cell cultures.
Cancer Res, 43: 3213-3218.
Sax NI & Lewis JR Sr (1984) Dangerous properties of industrial
materials, 7th Ed. New York, Van Nostrand Reinhold Co, pp 2451-2452.
Saxton WL, Newton RT, Rohrberg J, Sutton J, & Johnson LJ (1993)
Polycyclic aromatic hydrocarbons in seafood from the Gulf of Alaska
following a major crude oil spill. Bull Environ Contam Toxicol, 51:
515-522.
Schafer EW Jr, Bowles WA Jr, & Hurlbut J (1983) The acute oral
toxicity, repellency, and hazard potential of 998 chemicals to one or
more species of wild and domestic birds. Arch Environ Contam Toxicol,
12: 355-381.
Schaller KH, Angerer J, & Hausmann N (1993) The determination of
1-hydroxypyrene in urine as a tool for the biological monitoring of
PAH-exposed persons. Polycyclic Aromat Compd, 3 (suppl): 1023-1030.
Scheepers PTJ & Bos RP (1992) Combustion of diesel fuel from a
toxicological perspective 1. Origin of incomplete combustion products.
Int Arch Occup Environ Health, 64: 149-161.
Scherer G, Conze C, Von Meyerinck L, Sorsa M, & Adlkofer F (1990)
Importance of exposure to gaseous and particulate phase components of
tobacco smoke in active and passive smokers. Int Arch Occup Environ
Health, 62: 459-466.
Schimberg RW, Toivonen E, & Tossavainen A (1981) [Polycyclic aromatic
hydrocarbons and other hazardous agents in foundry moulding sand with
various hydrocarbon carriers.] Staub-Reinhalt Luft, 41: 221-224 (in
German).
Schlede E, Kuntzman R, Haber S, & Conney AH (1970a) Effect of enzyme
induction on the metabolism and tissue distribution of
benzo [a]pyrene. Cancer Res, 30: 2893-2897.
Schlede E, Kuntzman R, & Conney AH (1970b) Stimulatory effect of
benzo [a]pyrene and phenobarbital pretreatment on the biliary
excretion of benzo [a]pyrene metabolites in the rat. Cancer Res, 30:
2898-2904.
Schmähl D (1955) [Testing of naphthalene and anthracene for
carcinogenic effects in rats.] Z Krebsforsch, 60: 697-710 (in German).
Schmähl D, Schmidt KG, & Habs M (1977) Syncarcinogenic action of
polycyclic hydrocarbons in automobile exhaust gas condensates. In: Air
pollution and cancer in man. Lyon, International Agency for Research
on Cancer, pp 53-59 (IARC Scientific Publications No. 16).
Schmeltz I, Tosk J, Hilfrich J, Hirota N, Hoffmann D, & Wynder EL
(1978) Bioassays of naphthalene and alkylnaphthalenes for
co-carcinogenic activity. Relation to tobacco carcinogenesis. In:
Jones PW & Freudenthal RI ed. Carcinogenesis, Volume 3: Polynuclear
aromatic hydrocarbons. New York, Raven Press, pp 47-60.
Schmidt H (1992) [Emissions of polycyclic aromatic hydrocarbons in the
processsing of bitumen and polymer bitumen street sections.] Bitumen,
54: 50-53 (in German).
Schmoldt A, Jacob J, & Grimmer G (1981) Dose-dependent induction of
rat liver microsomal aryl hydrocarbon monooxygenase by
benzo [k]fluoranthene. Cancer Lett, 13: 249-257.
Schnizlein CT, Munson AE, & Rhoades RA (1987) Immunomodulation of
local and systemic immunity after subchronic pulmonary exposure of
mice to benzo(a)pyrene. Int J Immunopharmacol, 9: 99-106.
Schoeny R & Warshawsky D (1983) Mutagenicity of benzo(a)pyrene
metabolites generated on the isolated perfused lung following
particulate exposure. Teratog Carcinog Mutag, 151: 151-162.
Schoeny R, Cody T, Radike M, & Warshawsky D (1985) Mutagenicity of
algal metabolites of benzo(a)pyrene for Salmonella typhimurium.
Environ Mutag, 7: 839-855.
Schoeny R, Cody T, Warshawsky D, & Radike M. (1988) Metabolism of
mutagenic polycyclic hydrocarbons by photosynthetic algal species.
Mutat Res, 197: 289-302.
Schoeny R, Lewtas J, McClure P, & Stiteler W (1994) Risk assessment
for polycyclic aromatic hydrocarbons: Options revisited. Dallas,
Texas, Society of Toxicology.
Schoket B, Hewer A, Grover PL, & Phillips DH (1989) 32P-Postlabelling
analysis of DNA adducts in the skin of mice treated with petrol and
diesel engine lubricating oils and exhaust condensates.
Carcinogenesis, 10: 1485-1490.
Schoket B, Horkay I, Kósa A, Páldeák L, Hewer A, Grover PL, & Phillips
DH (1990) Formation of DNA adducts in the skin of psoriasis patients,
in human skin in organ culture, and in mouse skin and lung following
topical application of coal-tar and juniper tar. J Invest Dermatol,
94: 241-246.
Schoket B, Poirier MC, & Vincze I (1995) Biomonitoring of genotoxic
exposure in aluminium plant workers by determination of DNA adducts in
human peripheral blood lymphocytes. Sci Total Environ, 163: 153-163.
van Schooten FJ, Hillebrandt MJX, van Leeuwen FE, van Zandwijk N,
Janssen HM, den Engelse L, & Kriek E (1992) Polycyclic aromatic
hydrocarbon-DNA adducts in white blood cells from lung cancer
patients: No correlation with adduct levels in lung. Carcinogenesis,
13: 987-993.
van Schooten FJ, Moonen EJC, Rhijnsburger E, van Algen B, Thijssen
HHW, & Kleinjans JCS (1994) Dermal uptake of polycyclic aromatic
hydrocarbons after hairwash with coal-tar shampoo. Lancet, 344: 1505-
1506.
van Schooten FJ, Jongeneelen FJ, Hillebrand MJ, van Leeuwen FE, de
Looff AJ, Dijkmans AP, van Rooij JGM, den Engelse L, & Kriek E (1995)
Polycyclic aromatic hydrocarbon-DNA adducts in white blood cell DNA
and 1-hydroxypyrene in the urine from aluminium workers: Relation with
job category and synergistic effect of smoking. Cancer Epidemiol
Biomarkers Prev, 4: 69-77.
Schrimpff E (1983) [Forests dying from high concentrations of
pollutants in fog.] Staub Reinhalt Luft, 43: 240 (in German).
Schrimpff E, Thomas W, & Herrmann R (1979) Regional patterns of
contaminants (PAH, pesticides and trace metals) in snow of northeast
Bavaria and their relationship to human influence and orographic
effects. Water Air Soil Pollut, 11: 481-497.
Schuetzle D (1983) Sampling of vehicle emissions for chemical analysis
and biological testing. Environ Health Perspectives, 47: 65-80.
Schuetzle D & Frazier JA (1986) Factors influencing the emission of
vapor and particulate phase components from diesel engines. In:
Ishinishi N, Koizumi A, McClellan RO, & Stöber W ed. Carcinogenic and
mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier
Science Publishers, pp 41-63.
Schunk W (1979) [Relationship between exposure to polycyclic
hydrocarbons and the increased occurrence of cancer cases in two
chemical companies in Thüringen.] Z Ärztl Fortbild, 73: 84-88 (in
German).
Schürch O & Winterstein A (1935) [The cancer inducing effect of
aromatic hydrocarbons.] Z Physiol Chem, 236: 79-91 (in German).
Schuyer J, Blom L, & Van Krevelen DW (1953) The molar refraction of
condensed aromatic compounds. Trans Faraday Soc, 49: 1391-1401.
Schwarz FP (1977) Determination of temperature dependence of
solubilities of polycyclic aromatic hydrocarbons in aqueous solutions
by a fluorescence method. J Chem Eng Data, 22: 273-277.
Scribner JO (1973) Brief communication: Tumor initiation by apparently
noncarcinogenic polycyclic aromatic hydrocarbons J Natl Cancer Inst,
50: 1717-1719.
Sega GA (1979) Unscheduled DNA synthesis (DNA repair) in the germ
cells of male mice. Its role in the study of mammalian mutagenesis.
Genetics, 92: 49-58.
Segerbäck D & Vodicka P (1993) Recoveries of DNA adducts of polycyclic
aromatic hydrocarbons in the 32P-postlabelling assay. Carcinogenesis,
14: 2463-2469.
Seifert B, Ullrich D, & Schmahl HJ (1983) Occurrence of carcinogenic
organic substances in kitchen air. In: Proceedings of VIth World
Congress on Air Quality, Paris, 16-20 May 1983. Paris, SEFIC, pp 177-
179 (Poster presentation).
Seifert B, Ullrich D, & Liu ZF (1986) [Inorganic and organic air
pollutants at a street intersection in West Berlin. Reports of Society
for Water, Soil, and Air) Schriftenreihe Ver WaBoLu, 67: 211-222 (in
German).
Selgrade MK, Daniels MJ, Burleson GR, Lauer LD, & Dean JH (1988)
Effects of 7,12-dimethylbenz [a]anthracene, benzo [a]pyrene and
cyclosporin A on murine cytomegalo-virus infections: Studies of
resistance mechanisms. Int J Immunopharmacol,10: 811-818.
Selkirk JK, Croy RG, Whitlock JP Jr, & Gelboin HV (1975) In
vitro metabolism of benzo [a]pyrene by human liver microsomes and
lymphocytes. Cancer Res, 35: 3651-3655.
Sellakumar A & Shubik P (1974) Carcinogenicity of different polycyclic
hydrocarbons in the respiratory tract of hamsters. J Natl Cancer Inst,
53: 1713-1719.
Serth RW & Hughes TW (1980) Polycyclic organic matter (POM) and trace
element contents of carbon black vent gas. Environ Sci Technol, 14:
298-301.
Shabad LM & Dikun PP (1959) Atmospheric pollution by the carcinogenic
hydrocarbon 3,4-benzpyrene. Leningrad, Medgiz, 228 pp.
Sharpe CR, Rochon JE, Adam JM, & Siussa S (1989) Case-control study of
hydrocarbon exposures in patients with renal cell carcinoma. Can Med
Assoc J, 140: 1309-1318.
Shaw GR & Connell DW (1994) Prediction and monitoring of the
carcinogenicity of polycyclic aromatic compounds (PACs). In: Ware GW
ed. Reviews of environmental contamination and toxicology. Berlin,
Springer-Verlag, pp 1-62.
Shay H, Aegerter EA, Gruenstein M, & Komarov SA (1949) Development of
adenocarcinoma of the breast in the Wistar rat following the gastric
instillation of methylcholanthrene. J Natl Cancer Inst, 10: 255-266.
Shear MJ (1938) Studies in carcinogenesis. V. Methyl derivatives of
1:2-benzanthracene. Am J Cancer, 33: 499-537.
Shear MJ & Leiter J (1941) Studies in carcinogenesis. XVI. Production
of subcutaneous tumors in mice by miscellaneous polycyclic compounds.
J Natl Cancer Inst, 2: 241-259.
Shen Z, Wells RL, & Elkind MM (1994) Enhanced cytochrome P450
(Cyp1b1) expression, aryl hydrocarbon hydroxylase activity,
cytotoxicity, and transformation of C3H10T1/2 cells by
dimethylbenz (a)anthracene in conditioned medium. Cancer Res, 54:
4052-4056.
Shendrikova IA & Aleksandrov VA (1974) Comparative characteristics of
penetration of polycyclic hydrocarbons through the placenta in the
fetus in rats. Bull Exp Biol Med, 77: 77-79.
Shendrikova IA, Ivanov-Golitsyn MM, Anisimov YN, & Likhachev AYa
(1973) [Dynamics of transplacental penetration of
7,12-dimethylbenz [a]anthracene in mice.] Vopr Onkol, 19: 75-79 (in
Russian with English translation).
Shendrikova IA, Ivanov-Golitsyn MN, & Likjachev AYa (1974) [The
transplacental penetration of benzo [a]pyrene in mice.] Vopr Onkol,
20: 53-56 (in Russian with English abstract).
Sherson D & Iversen E (1986) Mortality among foundry workers in
Denmark due to cancer and respiratory and cardiovascular diseases.
Cancer Res Monogr, 2: 403-414.
Sherson D, Sabro P, Sigsgaard T, Johansen F, & Autrup H (1990)
Biological monitoring of foundry workers exposed to polycylic aromatic
hydrocarbons. Br J Ind Med, 47: 448-453.
Sherson D, Sigsgaard T, Overgaard E, Luft S, Poulsen H, & Jongenellen
FJ (1992) Interaction of smoking, PAH uptake and cytochrome P450IA2
activity among foundry workers. Br J Ind Med, 49:197-202.
Shiaris MP & Jambard-Sweet D (1986) Polycyclic aromatic hydrocarbons
in surficial sediments of Boston Harbour, Massachusetts, USA. Mar
Pollut Bull, 17: 469-472.
Shimada T, Martin MV, Pruess-Schwartz D, Marnett LJ, & Guengerich FP
(1989) Roles of individual human cytochrome P450 enzymes in the
bioactivation of benzo (a)pyrene,
7,8-dihydroxy-7,8-dihydrobenzo (a)pyrene, and other dihydrodiol
derivatives of polycyclic aromatic hydrocarbons. Cancer Res, 49: 6304-
6312.
Shimada H, Satake S, Itoh S, Hattori C, Hayashi M, & Ishidate M Jr
(1991) Multiple dosing effects of benzo [a]pyrene in the mouse bone
marrow micronucleus test. Mutat Res, 252: 107.
Shimada H, Suzuki H, Itoh S, Hattori C, Matsuura Y, Tada S, & Watanabe
C (1992) The micronucleus test of benzo(a)pyrene with mouse and rat
peripheral blood reticulocytes. Mutat Res, 278:165-168.
Shimkin MB & Stoner GD (1975) Lung tumours in mice: Application to
carcinogenesis bioassay. In: Klein G & Weinhouse S ed. Advances in
cancer research, Volume 1. New York, Raven Press, pp 1-58.
Shopp GM, White KL Jr, Holsapple MP, Barnes DW, Duke SS, Anderson AC,
Condie LW Jr, Hayes JR, & Borzelleca JF (1984) Naphthalene toxicity in
CD-1 mice: General toxicology and immunotoxicology. Fundam Appl
Toxicol, 4: 406-419.
Shou M, Korzekwa KR, Crespi CL, Gonzalez FJ, & Gelboin HV (1994) The
role of 12 cDNA-expressed human, rodent, and rabbit cytochromes P450
in the metabolism of benzo [a]pyrene trans-7,8-dihydrodiol. Mol
Carcinog, 1: 159-168.
Shubik P & Della Porta G (1957) Carcinogenesis and acute intoxication
with large doses of polycyclic hydrocarbons. Am Med Assoc Arch Pathol,
64: 691-703.
Shubik P, Pietra G, & Della Porta G (1960) Studies of skin
carcinogenesis in the Syrian golden hamster. Cancer Res, 20: 100-105.
Shum S, Jensen NM, & Nebert DW (1979) The murine Ah locus:
In utero toxicity and teratogenesis with genetic differences in
benzo [a]pyrene metabolism. Teratology, 20: 365-376.
Siebert D, Marquardt H, Friesel H, & Hecker E (1981) Polycyclic
aromatic hydrocarbons and possible metabolites: Convertogenic activity
in yeast and tumor initiating activity in mouse skin. J Cancer Res
Clin Oncol, 102: 127-139.
Simmon VF (1979) In vitro mutagenicity assays of chemical
carcinogens and related compounds with Salmonella typhimurium.
J Natl Cancer Inst, 62: 893-899.
Simmon VF, Rosenkranz HS, Zeiger E, & Poirier LA (1979) Mutagenic
activity of chemical carcinogens and related compounds in the
intraperitoneal host-mediated assay. J Natl Cancer Inst, 62: 911-918.
Simó R, Colom-Altés M, Grimalt JO, & Albaigés J (1991) Background
levels of atmospheric hydrocarbons, sulfate and nitrate over the
western Mediterranean. Atmos Environ, 25A: 1463-1471.
Simoneit BRT, Sheng G, Chen X, Fu J, Zhang J, & Xu Y (1991) Molecular
marker study of extractable organic matter in aerosols from urban
areas of China. Atmos Environ, 25: 2111-2130.
Sims P (1959) Metabolism of polycyclic compounds. 14. The conversion
of naphthalene into compounds related to
trans-1:2-dihydro-1:2-dihydroxynaphthalene by rabbits. Biochem J,
73: 389-395.
Sims P (1962) Metabolism of polycyclic compounds. 19. The metabolism
of phenanthrene in rabbits and rats: Phenols and sulphuric esters.
Biochem J, 84: 558-563.
Sims P (1964) Metabolism of polycyclic compounds. 25. The metabolism
of anthracene and some related compounds in rats. Biochem J, 92: 621-
631.
Sims P & Grover PL (1974) Epoxides in polycyclic aromatic hydrocarbon
metabolism and carcinogenesis. Adv Cancer Res, 20: 165-274.
Sims P & Grover PL (1981) Involvement of dihydrodiols and diol
epoxides in the metabolic activation of polycyclic hydrocarbons other
than benzo [a]pyrene. In: Gelboin HV & Ts'o POP ed. Polycyclic
hydrocarbons and cancer, Volume 3. New York, Academic Press, pp 117-
181.
Sims RC & Overcash MR (1983) Fate of polynuclear aromatic compounds
(PNAs) in soil plant systems. Res Rev, 88: 1-67.
Sipal Z, Ahlenius T, Bergstrand A, Rodriquez L, & Jakobsson SW (1979)
Oxidative biotransformation of benzo [a]pyrene by human lung
microsomal fractions prepared from surgical specimens. Xenobiotica, 9:
633-645.
Sirianni SR & Huang CC (1978) Sister chromatid exchange induced by
promutagens/carcinogens in Chinese hamster cells cultured in diffusion
chambers in mice. Proc Soc Exp Biol Med, 158: 269-274.
Sirota GR & Uthe JF (1981) Polynuclear aromatic hydrocarbon
contamination in marine shellfish. In: Cooke M & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemical analysis and biological
fate. Columbus, Ohio, Battelle Press, pp 329-341.
Sirota R, Uthe JF, Sreedharan A, Matheson R, Musial J, & Hamilton K
(1983) Polynuclear aromatic hydrocarbons in lobster (Homarus
americanus) and sediments in the vicinity of a coking facility. In:
Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons: Formation,
metabolism and measurement. Columbus, Ohio, Battelle Press, pp 1123-
1136.
Sivarajah K, Mukhtar H, & Eling T (1979) Arachidonic acid-dependent
metabolism of (+) trans-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene
(BP-7,8 diol) to 7,10/8,9 tetrols. FEBS Lett, 106: 17-20.
Skopek TR & Thilly WG (1983) Rate of induced forward mutation at 3
genetic loci in Salmonella typhimurium. Mutat Res, 108: 45-56.
Skopek TR, Liber HL, Kaden DA, Hites RA, & Thilly WG (1979) Mutation
of human cells by kerosene soot. J Natl Cancer Inst, 63: 309-312.
Slaga TJ, Hubermann E, Selkirk JK, Harvey RG, & Bracken WM (1978)
Carcinogenicity and mutagenicity of benz(a)anthracene diols and
diol-epoxides. Cancer Res, 38: 1699-1704.
Slaga TJ, Jecker L, Bracken WM, & Weeks CE (1979) The effects of weak
or noncarcinogenic polycyclic hydrocarbons on
7,12-dimethylbenz [a]anthracene and benzo [a]pyrene skin
tumor-initiation. Cancer Lett, 7: 51-59.
Slaga TJ, Gleason GL, Mills C, Ewald L, Fu PP, Lee HM, & Harvey RG
(1980) Comparison of the tumour-initiating activities of dihydrodiols
and diol-epoxides of various polycyclic aromatic hydrocarbons. Cancer
Res, 40: 1981-1984.
Slaga TJ, Iyer RP, Lyga W, Secrist A III, Daub GH, & Harvey RG (1981)
Comparison of the skin tumor-initiating activities of dihydrodiols,
diol-epoxides, and methylated derivatives of various polycyclic
aromatic hydrocarbons. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 753-769.
Slooff W, Janus JA, Matthijsen AJCM, Montizaan GK, & Ros JPM (1989)
Integrated criteria document on PAHs (Report No. 758474011).
Bilthoven, National Institute of Public Health and Environmental
Protection, pp 15-36.
Smith RL & Hargreaves BR (1983) A simple toxicity apparatus for
continuous flow with small volumes: Demonstration with mysids and
naphthalene. Bull Environ Contam Toxicol, 30: 406-412.
Smith SR, Tanaka J, Futoma DJ, & Smith TE (1981) Sampling and
preconcentration methods for the analysis of polycyclic aromatic
hydrocarbons in water systems. CRC Crit Rev Anal Chem, 10: 375-425.
Smith KR, Aggarwal AL, & Dave RM (1983) Air pollution and rural
biomass fuels in developing countries: A pilot village study in India
and implications for research and policy. Atmos Environ, 17: 2343-
2362.
Smith JD, Bagg J, & Bycroft BM (1984) Polycyclic aromatic hydrocarbons
in the clam Tridacna maxima from the Great Barrier Reef, Australia.
Environ Sci Technol, 18: 353-358.
Smith JD, Bagg J, & Sin YO (1987) Aromatic hydrocarbons in seawater,
sediments and clams from Green Island, Great Barrier Reef, Australia.
Aust J Mar Freshwater Res, 38: 501-510.
Smith S, Savino J, & Blouin M (1988) Acute toxicity to Daphnia
pulex of six classes of chemical compounds potentially hazardous to
Great Lakes aquatic biota. J Great Lakes Res, 14: 394-404.
Smith-Sonneborn J (1983) Use of a ciliated protozoan as a model system
to detect toxic and carcinogenic agents. In: Kolber AR, Wong TK, Grant
LD, DeWoskin RS, & Hughes TJ ed. In vitro toxicity testing of
environmental agents: Current and future possibilities. Part A: Survey
of test systems. New York, Plenum Press, pp 113-137.
Smyth HF, Carpenter CP, Weil CS, Pozzani UC, & Striegel JA (1962)
Range-finding toxicity data: List VI. Ind Hyg J, March-April: 95-107.
Snell KC & Stewart HL (1962) Pulmonary adenomatosis induced in DBA/2
mice by oral administration of dibenz [a,h]anthracene. J Natl Cancer
Inst, 28: 1043-1049.
Snider EH & Manning FS (1982) A survey of pollutant emission levels in
wastewaters and residuals from the petroleum refining industry.
Environ Int, 7: 237-258.
Society of German Chemists (Advisory Committee on Existing Chemicals
of Environ-mental Relevance) (1989) [Naphthalene.] Weinheim, VCH
Verlagsgesellschaft, 155 pp (Report No. 39) (in German).
Society of German Engineers (1989) Emission measurement. Measurement
of polycyclic aromatic hydrocarbons (PAH): Measurement of PAH in the
exhaust gas from gasoline and diesel engines of passenger cars. Gas
chromatographic determination. Düsseldorf, Commission for Air Quality
part 1, 27 pp (VDI Report No. 3872).
Solt DB, Polverini PJ, & Calderon L (1987) Carcinogenic response of
hamster buccal pouch epithelium to 4 polycyclic aromatic hydrocarbons.
J Oral Pathol, 16: 294-302.
Sonnefeld WJ, Zoller WH, & May WE (1983) Dynamic coupled-column liquid
chromatographic determination of ambient temperature vapor pressures
of polynuclear aromatic hydrocarbons. Anal Chem, 55: 275-280.
Sorahan T & Cooke MA (1989) Cancer mortality in a cohort of United
Kingdom steel foundry workers: 1946-1985. Br J Ind Med, 46: 74-81.
Sorrell K, Brass HJ, & Reding R (1980) A review of occurrences and
treatment of polynuclear aromatic hydrocarbons in water. Environ Int,
4: 245-254.
Southworth GR (1977) Transport and tranformations of anthracene in
natural waters. In: Marking LL & Kimerle RA ed. Aquatic toxicology.
Philadelphia, Pennsylvania, American Society for Testing and
Materials, pp 359-380 (ASTM ATP 667).
Southworth GR (1979) The role of volatilization in removing polycyclic
aromatic hydrocarbons from aquatic environments. Bull Environ Contam
Toxicol, 21: 507-514.
Southworth GR, Beauchamp JJ, & Schmieder PK (1978) Bioaccumulation
potential of polycyclic aromatic hydrocarbons in Daphnia pulex.
Water Res, 12: 973-977.
Spacie A, Landrum PF, & Leversee GJ (1983) Uptake, depuration, and
biotransformation of anthracene and benzo [a]pyrene in bluegill
sunfish. Ecotoxicol Environ Saf, 7: 330-341.
Sparnins VL, Mott AW, Baraney G, & Wattenberg LW (1986) Effects of
allyl methyl trisulfide on glutathione-S-transferase activity and
BP-induced neoplasia in the mouse. Nutr Cancer, 8: 211-215.
Speer K, Steeg E, Horstmann P, Kühn TH, & Montag A (1990)
Determination and distribution of PAH in native vegetable oils, smoked
fish products, mussels and oysters, and bream from the River Elbe. J
High Resol Chromatogr, 13: 104-111.
Spicer CW, Holdren MW, Smith DL, Hughes DP, & Smith MD (1992) Chemical
composition of exhaust from aircraft turbine engines. J Eng Gas
Turbines Power, 114: 111-117.
Spinelli JJ, Band PR, Svirchev LM, & Gallagher RP (1991) Mortality and
cancer incidence in aluminium reduction plant workers. J Occup Med,
33: 1150-1155.
Spitzer T & Dannecker W (1983) Membrane filters as adsorbents for
polycyclic aromatic hydrocarbons during high-volume sampling of air
particulate matter. Anal Chem, 55: 2226-2228.
Spitzer T & Kuwatsuka S (1986) Simple method for determination of
polynuclear aromatic hydrocarbons in soil by clean-up on XAD-2. J
Chromatogr, 358: 434-437.
Sporstl S, Gjs N, Lichtenthaler RG, Gustavsen KO, Urdal K, Oreld F,
& Skei J (1983) Source identification of aromatic hydrocarbons in
sediments using GC/MS. Environ Sci Technol, 17: 282-286.
Stahl RG, Liehr JG, & Davis EM (1984) Characterization of organic
compounds in simulated rainfall runoffs from model coal piles. Arch
Environ Contam Toxicol, 13: 179-190.
Stanton MF, Miller E, Wrench C, & Blackwell R (1972) Experimental
induction of epidermoid carcinoma in the lungs of rats by cigarette
smoke condensate. J Natl Cancer Inst, 49: 867-877.
State Chemical Analysis Institute, Freiburg (1995) [Food control and
the environment: Annual report 1994.] Freiburg, pp 156-160 (in
German).
State Committee for Air Pollution Control (1992) [Cancer risk from air
pollution.] Development of evaluation criteria for carcinogenic
pollutants. Düsseldorf, Minister for Environment and Agriculture for
Northrhine-Westfalia, 71 pp (in German).
State Pollution Control Authority and Norwegian Food Control Authority
(1992) Recommendations from the workshop on PAH. In: Proceedings of
the workshop on polyaromatic hydrocarbons (PAH), Oslo, 11-13 November
1991. Oslo, p 423 (Report No. TA-816/1992).
Stauffer TB, MacIntyre WG, & Wickman DC (1989) Sorption of nonpolar
organic chemicals on low-carbon-content aquifer materials. Environ
Toxicol Chem, 8: 845-852.
Stavenow L & Pessah-Rasmussen H (1988) Effects of polycyclic aromatic
hydrocarbons on proliferation, collagen secretion and viability of
arterial smooth muscle cells in culture. Artery, 15: 94-108.
Stay FS, Katko A, Rohm CM, Fix MA, & Larsen DP (1988) Effects of
fluorene on microcosms developed from four natural communities.
Environ Toxicol Chem, 7: 635-644.
Steen WC & Karickhoff SW (1981) Biosorption of hydrophobic organic
pollutants by mixed microbial populations. Chemosphere, 10: 27-32.
Steiner PF (1955) Carcinogenicity of multiple chemicals simultaneously
administered. Cancer Res, 15: 632-635.
Steiner PE & Edgcomb JH (1952) Carcinogenicity of 1,2-benzanthracene.
Cancer Res, 12: 657-659.
Steiner PF & Falk HL (1951) Summation and inhibition effects of weak
and strong carcinogenic hydrocarbons: 1:2-Benzanthracene, chrysene,
1:2:5:6-dibenzanthracene, and 20-methylcholanthrene. Cancer Res, 11:
56-63.
Steinhoff D, Mohr U & Hahnemann S (1991) Carcinogenesis studies with
iron oxides. Exp Pathol, 43: 189-194.
Steinig J (1976) 3,4-Benzopyrene contents in smoked fish depending on
smoking procedure. Z Lebensmittelunters Forsch, 162: 235-242.
Stenbäck F & Sellakumar A (1974) Lung tumor induction by
dibenz(a,i)pyrene in the Syrian golden hamster. Z Krebsforsch, 82:
175-182.
Stenberg UR (1985) PAH emissions from automobiles. In: Bjorseth A &
Ramdahl T ed. Handbook of polycyclic aromatic hydrocarbons. Volume 2:
Emission sources and recent progress in analytical chemistry. New
York, Marcel Dekker, pp 87-111.
Stenberg U, Alsberg T, & Westerholm R (1983) Applicability of a
cryogradient technique for the enrichment of PAH from automobile
exhausts: Demonstration of methodology and evaluation experiments.
Environ Health Perspectives, 47: 43-51.
Stevenson JL & Von Haam E (1965) Carcinogenicity of benz(a)anthracene
and benzo(c)phenanthrene derivatives. Am Ind Hyg Assoc J, 26: 475-478.
Stoner GD, Harris CC, Autrup H, Trump BJ, Kingsbury EW, & Myers GA
(1978) Explant cultures of human peripheral lung. I. Metabolism of
benzo [a]pyrene. Lab Invest, 38: 685-692.
Storer JS, DeLeon I, Millikan LE, Laseter JL, & Griffing C (1984)
Human absorption of crude coal tar products. Arch Dermatol, 120: 874-
877.
Stout P & Mamantov G (1989) Recent advances in infrared analysis of
polycyclic aromatic compounds. In: Vo-Dinh T ed. Chemical analysis of
polycyclic aromatic compounds. New York, John Wiley & Sons, pp 411-
432.
Strandell M, Zakrisson S, Alsberg T, Westerholm R, Winquist L, &
Rannug U (1994) Chemical analysis and biological testing of a polar
fraction of ambient air, diesel engine, and gasoline engine
particulate extracts. Environ Health Perspectives, 102 (suppl 4): 85-
92.
Strickland PT, Routledge MN, & Dipple A (1993) Methodologies for
measuring carcinogen adducts in humans. Cancer Epidemiol Biomarkers
Prev, 2: 607-619.
Stuermer DH, Ng DJ, & Morris CJ (1982) Organic contaminants in
groundwater near underground coal gasification site in northeastern
Wyoming. Environ Sci Technol, 16: 582-587.
Suess MJ (1976) The environmental load and cycle of polycyclic
aromatic hydrocarbons. Sci Total Environ, 6: 239-250.
Sugiyama T (1973) Chromosomal aberrations and carcinogenesis by
various benz(a)-anthracene derivatives. Gann, 64: 637-639.
Sullivan TJ & Mix MC (1983) Pyrolytic deposition of polynuclear
aromatic hydrocarbons due to slash burning on clear-cut sites. Bull
Environ Contam Toxicol, 31: 208-215.
Sullivan TJ & Mix MC (1985) Persistence and fate of polynuclear
aromatic hydrocarbons deposited on slash burn sites in the Cascade
Mountains and coast range of Oregon. Arch Environ Contam Toxicol, 14:
187-192.
Suntzeff VA, Cowdry EV, & Croninger A (1955) Microscopic visualization
of the degeneration of sebaceous glands caused by carcinogens. Cancer
Res, 15: 637-640.
Surh YS, Liem A, Miller EC, & Miller JA ( 1989) Metabolic activation
of the carcinogen 6-hydroxymethylbenzo(a)pyrene: Formation of an
electrophilic sulfuric acid ester and benylic DNA adducts in rat liver
in vivo and in reactions in vivo.
Carcinogenesis, 10: 1519-1528.
Surh YJ, Blomquist JC, Liem A, & Miller JA (1990a) Metabolic
activation of 9-hydroxymethyl-10-methylanthracene and
1-hydroxymethylpyrene to electrophilic, mutagenic and tumorigenic
sulfuric acid esters by rat hepatic sulfotransferase activity.
Carcinogenesis, 11: 1451-1460.
Surh, YJ, Liem A, Miller EC, & Miller JA (1990b) The strong
hepatocarcinogenicity of the electrophilic and mutagenic metabolite
6-sulfooxymethylbenzo(a)pyrene and its formation of benzylic DNA
adducts in the livers of infant male B6C3F1 mice. Biochem Biophys Res
Commun, 172: 85-91.
Süss A (1980) [Basic questions for the solution of environmental
problems.] FreisingMunich, Bavarian State Department for Soil and
Horticulture, pp 376-378 (in German).
Sutter TR, Tang YM, Hayes CL, Wo Y-YP, Jabs EW, Li X, Yin H, Cody CW,
& Greenlee WF (1994) Complete cDNA sequence of a human
dioxin-inducible mRNA identifies a new gene subfamily of cytochrome
P450 that maps to chromosome 2. J Biol Chem, 269: 13092-13099.
Swaen GMH, Slangen JJM, Volovics A, Hayes RB, Scheffers T, & Sturmans
F (1991) Mortality of coke plant workers in the Netherlands. Br J Ind
Med, 48: 130-135.
Swallow WH & van Noort RW (1985) Occupational exposure to polycyclic
aromatic hydrocarbons in some New Zealand industries. Wellington,
Department of Scientific and Industrial Research, 17 pp.
Swartz RC, Kemp PF, Schults DW, & Lamberson JO (1988) Effects of
mixtures of sediment contaminants on the marine infaunal amphipod
Rhepoxynius abronius. Environ Toxicol Chem, 7: 1013-1020.
Swartz WJ & Mattison DR (1985) Benzo(a)pyrene inhibits ovulation in
C57Bl/6N mice. Anat Rec, 221: 268-276.
Swedish National Board of Occupational Safety and Health (1994)
Occupational exposure limit values, Solna, National Institute of Work
and Health, p 5.
Symons RK & Crick I (1983) Determination of polynuclear aromatic
hydrocarbons in refinery effluent by high-performance liquid
chromatography. Anal Chim Acta, 151: 237-243.
Szabo G, Prosser SL, & Bulman RA (1990) Determination of the
adsorption coefficient (Koc) of some aromatics for soil by RP-HPLC on
two immobilized humic acid phases. Chemosphere, 21: 777-788.
Szyfter K, Krüger J, Ericson P, Vaca C, Försti A, & Hemminki K (1994)
32P-Postlabelling analysis of DNA adducts in humans: Adduct
distribution and method improvement. Mutat Res, 313: 269-276.
Tabak HH, Quave SA, Mashni CI, & Barth EF (1981) Biodegradability
studies with organic priority pollutant compounds. J Water Pollut
Control Fed, 53: 1503-1518.
Takada H, Onda T, & Ogura N (1990) Determination of polycyclic
aromatic hydrocarbons in urban street dusts and their source materials
by capillary gas chromatography. Environ Sci Technol, 24: 1179-1186.
Takahashi G (1974) Distribution and metabolism of benzo(a)pyrene in
fetal mouse. Bull Chest Dis Res Inst Kyoto Univ, 7: 155-160.
Takahashi G (1978) Distribution and excretion of the hydrocarbon
3-methylcholanthrene in the animal body. In: Gelboin HV and Ts'o POP,
ed. Polycyclic hydrocarbons and cancer, Volume 1. New York, Academic
Press, pp 233-246.
Takahashi G & Yasuhira K (1972) Excretion and conversion of
3-methylcholanthrene metabolites in the intestinal tract of the mouse.
Cancer Res, 32: 710-715.
Takahashi G & Yasuhira K (1973) Macroautoradiographic and radiometric
studies on the distribution of 3-methylcholanthrene in mice and their
fetuses. Cancer Res, 33: 23-28.
Takatsuki K, Suzuki S, Sato N, & Ushizawa I (1985) Liquid
chromatographic determination of polycyclic aromatic hydrocarbons in
fish and shellfish. J Assoc Off Anal Chem, 68: 945-949.
Tan YL, Quanci JF, Borys RD, & Quanci MJ (1992) Polycyclic aromatic
hydrocarbons in smoke particles from wood and duff burning. Atmos
Environ, 26A: 1177-1181.
Tawfic HN (1965) Studies on ear duct tumors in rats. Part II:
Inhibitory effect of methylcholanthrene and 1,2-benzanthracene on
tumor formation by 4-dimethylamino-stilbene. Acta Pathol Jpn, 15: 255-
260.
Ten Hulscher ThEM, Van Der Velde LE, & Bruggeman WA (1992) Temperature
dependence of Henry's law constants for selected chlorobenzenes,
polychlorinated biphenyls and polycyclic aromatic hydrocarbons.
Environ Toxicol Chem, 11: 1595-1603.
Teranishi K, Hamada K, & Watanabe H (1975) Quantitative relationship
between carcinogencity and mutagenicity of polyaromatic hydrocarbons
in Salmonella typhimurium mutants. Mutat Res, 31: 97-102.
Teta MJ, Ott MG, & Schnatter AR ( 1987) Population based mortality
surveillance in carbon products manufacturing plants. Br J Ind Med,
44: 344-350.
Tetzen D (1989) [Environmentally relevant classification of PKWF and
Printosol. Classification of printing ink oils.] Verfkroniek, 62: 469-
472 (in German).
Theriault G, Tremblay C, Cordier S, & Gingras S (1984) Bladder cancer
in the aluminium industry. Lancet, i: 947-950.
Thilly WG, DeLuca JG, Furth EE, Hoppe H IV, Kaden DA, Krolewski JJ,
Liber HL, Skopek TR, Slapikoff SA, Tizard RJ, & Penman BW (1980)
Gene-locus mutation assays in diploid human lymphoblast lines. In: De
Serres FJ & Hollaender A ed. Chemical mutagens: Principles and methods
for their detection, Volume 6. New York, Plenum Press, pp 331-364.
Thirman M, Albrecht JH, Krüger MA, Erickson RR, Cherwitz DL, Park SS,
Gelboin HV, & Holtzmann JL (1994) Induction of cytochrome CYPIA1 and
formation of toxic metabolites of benzo (a)pyrene by rat aorta: A
possible role in atherogenesis. Proc Natl Acad Sci USA, 91: 5397-5401.
Thomas W (1986) Principal component analysis of trace substance
concentrations in rainwater samples. Atmos Environ, 20: 995-1000.
Thomas P (1988) Reproductive endocrine function in female Atlantic
croaker exposed to pollutants. Mar Environ Res, 24: 179-183.
Thomson BM, Lake RJ, & Lilli RE (1996) The contribution of margarine
to cancer risk from polycyclic aromatic hydrocarbons in the New
Zealand diet. Polycyclic Aromat Compd 11: 177-184.
Thorslund TW & Farrar D (1990a) Development of relative potency
estimates for PAHs and hydrocarbon combustion product fractions
compared to benzo(a)pyrene and their use in carcinogenic risk
assessments. Fairfax, Virginia, Clement Associates, 79 pp.
Thorslund TW & Farrar D (1990b) Ingestion dose-response model for
benzo [a]pyrene. Fairfax, Virginia, Clement International Corp, 22 pp
(Contract 68-02-4601).
Thorsteinsson T (1969) Polycyclic hydrocarbons in commercially and
home-smoked food in Iceland. Cancer Lett, 23: 455-457.
Thrane KE (1987) Deposition of polycyclic aromatic hydrocarbons (PAH)
in the surroundings of primary aluminium industry. Water Air Soil
Pollut, 33: 385-393.
Thrane KE & Mikalsen A (1981) High-volume sampling of airborne
polycyclic aromatic hydrocarbons using glass fibre filters and
polyurethane foam. Atmos Environ, 15: 909-918.
Thrane KE & Wikström L (1984) Monitoring of polycyclic aromatic
hydrocarbons in ambient air. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Mechanisms, methods and metabolism. Columbus,
Ohio, Battelle Press, pp 1299-1314.
Thruston AD Jr (1978) High pressure liquid chromatography techniques
for the isolation and identification of organics in drinking water
extracts. J Chromatogr Sci, 16: 254-259.
Thurmond LM, Tucker AN, Rickert DE, Lauer LD, & Dean JH (1989)
Functional and biochemical disposition of
7,12-dimethylbenz(a)anthracene in murine lymphoid cells. Chem-Biol
Interactions, 72: 93-104.
Thursby GB, Steele RL, & Kane ME (1985) Effect of organic chemicals on
growth on reproduction in the marine red alga Champia
parvula. Environ Toxicol Chem, 4: 797-806.
Thyssen J, Althoff J, Kimmerle G, & Mohr U (1980) Investigations on
the carcinogenic burden of air pollution in man. XIX. Effect of
inhaled benzo(a)pyrene in Syrian golden hamsters: A pilot study.
Zentralbl Bakteriol Hyg I Abt Orig B, 171: 441-444.
Thyssen J, Althoff J, Kimmerle G, & Mohr U (1981) Inhalation studies
with benz [a]pyrene in Syrian golden hamsters. J Natl Cancer Inst,
66: 575-577.
Tilgner DJ (1958) [New information about smoking procedures].
Fleischwirtschaft, 10: 649-653 (in German).
Tilgner DJ (1968) Carcinogens in food. Food Manuf, 43: 37-39, 42.
Todorovic R, Devanesan PD, Cavalieri EL, Rogan EG, Park SS, & Gelboin
HV (1991) A monoclonal antibody to rat liver cytochrome P450 IIC11
strongly and regiospecifically inhibits constitutive benzo [a]pyrene
metabolism and DNA binding. Mol Carcinog, 4: 308-314.
Tokiwa H, Morita K, Takeyoshi H, Takahashi K, & Ohnishi Y (1977)
Detection of mutagenic activity in particulate air pollutants. Mutat
Res, 48: 237-248.
Tola S, Koskela R-S, Hernberg S, & Järvinen E (1979) Lung cancer
mortality among iron foundry workers. J Occup Med, 21: 753-760.
Tolos WP, Shaw PB, Lowry LK, MacKenzie BA, Deng J-F, & Markel HL
(1990) 1-Pyrenol: A biomarker for occupational exposure to polycyclic
aromatic hydrocarbons. Appl Occup Environ Hyg, 5: 303-309.
Tomatis L (1973) Transplacental carcinogenesis. In: Raven RW ed.
Modern trends in oncology. Part 1: Research progress. London,
Butterworths, pp 99-126.
Tomingas R, Pott F, & Dehnen W (1976) Polycyclic aromatic hydrocarbons
in human bronchial carcinoma. Cancer Lett, 1: 189-195.
Tonelli QJ, Custer RP, & Sorof S (1979) Transformation of cultured
mouse mammary glands by aromatic amines and amides and their
derivatives. Cancer Res, 39: 1784-1792.
Tong C, Laspia MF, Telang S, & Williams GM (1981a) The use of adult
rat liver cultures in the detection of the genotoxicity of various
polycyclic aromatic hydrocarbons. Environ Mutag, 3: 477-487.
Tong C, Brat SV, & Williams GM (1981b) Sister-chromatid exchange
induction by polycyclic aromatic hydrocarbons in an intact cell system
of adult rat-liver epithelial cells. Mutat Res, 91: 467-473.
Tong C, Brat VS, Telang S, Laspia, MF, Fazio M, Reiss B, & Williams GM
(1983) Effects of genotoxic polycyclic aromatic hydrocarbons in rat
liver culture systems. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Formation; metabolism,and measurement.
Columbus, Ohio, Battelle Press, pp 1189-1203.
Topham JC (1980) Do induced sperm-head abnormalities in mice
specifically identify mammalian mutagens rather than carcinogens?
Mutat Res, 74: 379-387.
Topping DC, Martin DH, & Nettesheim P (1981) Determination of
cocarcinogenic activity of benzo [e]pyrene for respiratory tract
mucosa. Cancer Lett, 11: 315-321.
Törrönen R, Nousianen U, & Hänninen O (1981) Induction of aldehyde
dehydrogenase by polycyclic aromatic hydrocarbons in rats. Chem-Biol
Interactions, 36: 33-44.
Toth L & Blaas W (1972) [The effect of smoking technology on the
content of carcinogenic hydrocarbons in smoked meat products.]
Fleischwirtschaft, 9: 1121-1124 (in German).
Traynor GW, Apte MG, Carruthers AR, Dillworth JF, Grimsrud DT, &
Gundel LA (1987) Indoor air pollution due to emissions from
wood-burning stoves. Environ Sci Technol, 21: 691-697.
Tremblay C, Armstrong B, Thériault G, & Brodeur J (1995) Estimation of
risk of developing bladder cancer among workers exposed to coal tar
pitch volatiles in the primary aluminum industry. Am J Ind Med, 27:
335-348.
Trucco RG, Engelhardt FR, & Stacey B (1983) Toxicity accumulation and
clearance of aromatic hydrocarbons in Daphnia pulex. Environ Pollut,
A31: 191-202.
Tsuchimoto T & Matter BE (1981) Activity of coded compounds in the
micronucleus test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North-Holland, pp 705-711
(Progress in Mutation Research, Volume 1).
Tucci AF (1988) PAH characterization in hazardous waste from coke
processing plants. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: A decade of progress. Columbus, Ohio, Battelle Press, pp
865-870.
Tuominen JP, Pyysalo HS, & Sauri M (1988) Cereal products as a source
of polycyclic aromatic hydrocarbons. J Agric Food Chem, 36: 118-120.
Tuomisto J & Jantunen M (1987) A simple way of comparing carcinogenic
effects of chemical and radioactive emissions. Kuopio, National Public
Health Institute, 18 pp (Report No. NPHI-A2/1987).
Tweats DJ (1981) Activity of 42 coded compounds in a differential
killing test using Escherichia coli strains WP2, WP67 (uvrA polA),
and CM871 (uvrA lexA recA). In: De Serres FJ & Ashby J ed. Evaluation
of short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North-Holland, pp 199-209
(Progress in Mutation Research, Volume 1).
Tyndyk ML, Zabezhinski MA, Bykoy VJ, Dikun PP, Dymochka LA,
Nepomnyaschaya OB, Yatsuk OS, Yermilov VB, & Likhachev AJ (1994)
Individual values of excretion of benzo [a]pyrene metabolites and
susceptibility to its carcinogenic effect in rats. Cancer Lett, 78:
163-170.
UNEP (1994) International register for Potentially Toxic Chemicals, PC
database
Urso P & Gengozian N (1980) Depressed humoral immunity and increased
tumor incidence in mice following in utero exposure to
benzo [a]pyrene. J Toxicol Environ Health, 6: 569-576.
Urso P & Johnson RA (1987) Early changes in T lymphocytes and subsets
of mouse progeny defective as adults in controlling growth of a
syngeneic tumor after in utero insult with benzo(a)pyrene.
Immunopharmacology, 14: 1-10.
Urso P & Johnson RA (1988) Quantitative and functional change in T
cells of primiparous mice following injection of benzo(a)pyrene at the
second trimester of pregnancy. Immunopharmacol Immunotoxicol, 10: 195-
217.
Urso P, Gengozian N, Rossi RM, & Johnson RA (1986) Suppression of
humoral and cellmediated immune responses in vitro by
benzo(a)pyrene. J Immunopharmacol, 8: 223-241.
Urso P, Ryan MC, & Bennett JS (1988) Changes in peripheral blood cells
in mice after injection with benzo(a)pyrene during pregnancy.
Immunopharmacol Immunotoxicol, 10: 179-193.
US Environmental Protection Agency (1978a) Ambient water quality
criteria: Naphthalene. Washington DC, p B/6 (PB-296 786).
US Environmental Protection Agency (1978b) Ambient water quality
criteria: Acenaphthene. Washington DC, p B/7 (PB-296 782).
US Environmental Protection Agency (1978c) Ambient water quality
criteria: Fluoranthene. Washington DC, pp B/5-B/7 (PB-292 433).
US Environmental Protection Agency (1980) Ambient water quality
criteria for polynuclear aromatic hydrocarbons. Washington DC, 7 pp
(EPA 440/5-80-069).
US Environmental Protection Agency (1984a) Guidelines establishing
test procedures for the analysis of pollutants under the clean water
act: Method 610. Polynuclear aromatic hydrocarbons. Fed Reg, 49(209):
43344-43352 (40 CFR Part 136).
US Environmental Protection Agency (1984b) Health effects assessment
for naphthalene. Research Triangle Park, North Carolina, Section 7
(EPA/540/1-86/014).
US Environmental Protection Agency (1984c) Health effects assessment
for polycyclic aromatic hydrocarbons (PAH). Washington DC, Office of
Emergency and Remedial Response, 49 pp (EPA 540/1-86/013; NTIS
PB86-134244/AS).
US Environmental Protection Agency (1984d) Carcinogenic assessment of
coke oven emissions. Final report. Washington DC, Office of Health and
Environmental Assessment, 209 pp (EPA 600/6-82-003F; NTIS
PB84-170182).
US Environmental Protection Agency (1984e) Health effects assessment
for benzo(a)pyrene. Cincinnati, Ohio, 43 pp (EPA/540/1-86/022; PB
86-134335).
US Environmental Protection Agency (1986a) (I) Method 3510 'Separatory
funnel liquid-liquid extraction'; (II) Method 3520 'Continuous liquid-
liquid extraction'; (III) Method 3630 'Silica gel cleanup'; (IV)
Method 8100 'Polynuclear aromatic hydrocarbons'; (V) Method 8270 'Gas
chromatography/mass spectrometry for semivolatile organics: Capillary
column technique'; (VI) Method 8310 'Polynuclear aromatic
hydrocarbons'. In: Test methods for evaluating solid waste, 3rd Ed,
Volume IB. Washington DC, 50 pp (EPA-SW-846).
US Environmental Protection Agency (1986b) (I) Method 3540 'Soxhlet
extraction'; (II) Method 3550 'Sonication extraction'; (III) Method
3630 'Silica gel cleanup'; (IV) Method 8100 'Polynuclear aromatic
hydrocarbons'; (V) Method 8270 'Gas chromatography/mass spectrometry
for semivolatile organics: Capillary column technique'; (VI) Method
8310 'Polynuclear aromatic hydrocarbons'. In: Test methods for
evaluating solid waste, 3rd Ed, Volume IB. Washington DC, 45 pp
(EPA-SW-846).
US Environmental Protection Agency (1986c) (I) Method 0010 'Modified
method 5 sampling train'; (II) Method 0020 'Source assessment sampling
system (SASS)'. In: Test methods for evaluating solid waste, 3rd Ed,
Volume II. Washington DC, 86 pp (EPA-SW846).
US Environmental Protection Agency (1986d) Health and environmental
effects profile for naphthalene. Cincinnati, Ohio, pp 41-54 (PB
88-242383).
US Environmental Protection Agency (1987a) Reference method for the
determination of particulate matter as PM10 in the atmosphere.
Appendix J. Fed Reg, 52(126): 24664-24666.
US Environmental Protection Agency (1987b) Health and environmental
effects profile for anthracene. Cincinnati, Ohio, pp 43-47, 57, 63, 66
(PB 89-118319).
US Environmental Protection Agency (1987c) The risk assessment
guidelines of 1986. Washington DC, Office of Health and Environmental
Assessment (EPA/600/8-87/045).
US Environmental Protection Agency (1988) 13-Week mouse oral
subchronic toxicity study (fluoranthene). Washington DC, 104 pp (TRL
Study No. 042-008).
US Environmental Protection Agency (1989a) Mouse oral subchronic
toxicity study with acenaphthene. Washington DC, 89 pp.
US Environmental Protection Agency (1989b) Subchronic toxicity in mice
with anthracene: Final report. Washington DC, 465 pp (HLA Study No.
2399-131).
US Environmental Protection Agency (1989c) Mouse oral subchronic
toxicity study (fluorene). Washington DC, 38 pp (TRL Study No.
042010).
US Environmental Protection Agency (1989d) Mouse oral subchronic
toxicity of pyrene. Washington DC, 102 pp (TRL Study No. 042-012).
US Environmental Protection Agency (1990) Method IP-7 'Determination
of benzo(a)pyrene and other polynuclear aromatic hydrocarbons in
indoor air'. In: Winberry WT Jr, Forehand L, Murphy NT, Ceroli A,
Phinney B, & Evans A ed. Compendium of methods for the determination
of air pollutants in indoor air. Research Triangle Park, North
Carolina, pp 550-639 (EPA 600/4-90/010; US NTIS PB90-200288).
US Environmental Protection Agency (1992a) Protection of the
environment: Chapter 1, part 86, sections 86-109, 86-110. US Code Fed
Regul, Title 40, Parts 86-99: 377-410.
US Environmental Protection Agency (1992b) Drinking water criteria
document for polycyclic aromatic hydrocarbons (PAHs). Washington DC,
Office of Water, 442 pp.
US Environmental Protection Agency (1993) Provisional guidance for
quantitative risk assessment of polycyclic aromatic hydrocarbons.
Cincinnati, Ohio, Office of Health and Environmental Assessment,
Environmental Criteria and Assessment Office (EPA/600/R-93/089).
Uthe JF & Musial CJ (1986) Polycyclic aromatic hydrocarbon
contamination of American lobster, Homarus americanus, in the
proximity of a coal-coking plant. Bull Environ Contam Toxicol, 37:
730-738.
Vaessen HAMG, Schuller PL, Jekel AA, & Wilbers AAMM (1984) Polycyclic
aromatic hydrocarbons in selected foods: Analysis and occurence.
Toxicol Environ Chem, 7: 297-324.
Vaessen HAMG, Jekel AA, & Wilbers AAMM (1988) Dietary intake of
polycyclic aromatic hydrocarbons. Toxicol Environ Chem, 16: 281-294.
Vähäkangas K, Pyy L, & Yrjänheikki E (1992) Assessment of PAH-exposure
among coke oven workers. Pharmacogenetics, 2: 304-308
Vainio H, Uotila P, Hartiala J, & Pelkonen O (1976) The fate of
intratracheally installed benzo [a]pyrene in the isolated perfused
rat lung of both control and 20-methylcholanthrene pretreated rats.
Res Commun Chem Pathol Pharmacol, 13: 259-271.
Vainiotalo S & Matveinen K (1993) Cooking fumes as a hygienic problem
in the food and catering industries. Am Ind Hyg Assoc J, 54: 376-382.
Vaishnav DD & Babeu L (1987) Comparison of occurrence and rates of
chemical biodegradation in natural waters. Bull Environ Contam
Toxicol, 39: 237-244.
Valaes T, Doxiadis SA, & Fessas P (1963) Acute hemolysis due to
naphthalene inhalation. J. Pediatr, 63: 904-915.
Valencia R & Houtchens K (1981) Mutagenic acitivity of 10 coded
compounds in the Drosophila sex-linked recessive lethal test. In: De
Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme.
Amsterdam, Elsevier North-Holland, pp 652-659 (Progress in Mutation
Research, Volume 1).
Valerio F, Bottino P, Ugolini D, Cimberle MR, Tozzi GA, & Frigerio A
(1984) Chemical and photochemical degradation of polycyclic aromatic
hydrocarbons in the atmosphere. Sci Total Environ, 40: 169-188.
Valerio F, Antolini E, & Lazzarotto A (1987) A model to evaluate
half-lifes of PAHs naturally occurring on airborne particulate. Int J
Environ Anal Chem, 28: 185-196.
Valerio F, Pala M, Brescianini C, Lazarrotto A, & Balducci D (1991)
Effect of sunlight and temperature on concentration of pyrene and
benzo [a]pyrene adsorbed on airborne particulate. Toxicol Environ
Chem, 31/32: 113-118.
Valerio F, Brescianini C, Pala M, Lazzarotto A, Balducci D, & Vincenzo
F (1992) Sources and atmospheric concentrations of polycyclic aromatic
hydrocarbons and heavy metals in two Italian towns (Genoa and La
Spezia). Sci Total Environ, 114: 47-57.
Van Brummelen TC & Stuijfzand SC (1993) Effects of benzo [a]pyrene on
survival, growth and energy reserves in terrestrial isopods Oniscus
asellus and Porcellio scaber. Sci Total Environ, Suppl (Part II):
921-930.
Van Cauwenberghe KA (1985) Atmospheric reactions of PAH. In: Bjorseth
A & Ramdahl T ed. Handbook of polycyclic aromatic hydrocarbons, Volume
2. Emission sources and recent progress in analytical chemistry. New
York, Marcel Dekker, pp 351-384.
Van den Heuvel JPP, Clark GC, Thompson CL, McCoy Z, Miller CR, Lucier
GW, & Bell DA (1993) CYP1A1 mRNA levels as a human exposure biomarker:
Use of quantitative polymerase chain reaction to measure CYP1A1
expression in human peripheral blood lymphocytes. Carcinogenesis, 14:
2003-2006.
Van der Oost R, Heida H, Opperhuizen A, & Vermeulen (1991)
Interrelationships between bioaccumulation of organic trace pollutants
PCBs organochlorine pesticides and PAHs and MFO-induction in fish.
Comp Biochem Phys, C100: 43-48.
Van Duuren BL & Goldschmidt BM (1976) Cocarcinogenic and
tumor-promoting agents in tobacco carcinogenesis. J Natl Cancer Inst,
56: 1237-1242.
Van Duuren BL, Sivak A, Segal A, Orris L, & Langseth L (1966) The
tumor-promoting agents of tobacco leaf and tobacco smoke condensate. J
Natl Cancer Inst, 37: 519-526.
Van Duuren BL, Langseth L, Goldschmidt BM & Orris L (1967)
Carcinogenicity of epoxides, lactones, and peroxy compounds. VI.
Structure and carcinogenic activity. J Natl Cancer Inst, 39:
1217-1228.
Van Duuren BL, Sivak A, Langseth L, Goldschmidt BM, & Segal A (1968)
Initiators and promoters in tobacco carcinogenesis. Natl Cancer Inst
Monogr, 28: 173-180.
Van Duuren BL, Sivak A, Goldschmidt BM, Katz C, & Melchionne S (1970)
Initiating activity of aromatic hydrocarbons in two-stage
carcinogenesis. J Natl Cancer Inst, 44: 1167-1173.
Van Heyningen R & Pirie A (1967) The metabolism of naphthalene and its
toxic effect on the eye. Biochem J, 102: 842-852.
Van Noort PCM & Wondergem E (1985a) The isolation of some polynuclear
aromatic hydrocarbons from aqueous samples by means of reversed-phase
concentrator columns. Anal Chim Acta, 172: 335-340.
Van Noort PCM & Wondergem E (1985b) Scavenging of airborne polycyclic
aromatic hydrocarbons by rain. Environ Sci Technol, 19: 1044-1048.
Van Rooij JGM, Bodelier-Bade MM, De Looff AJA, Dijkmans AGP, &
Jongeneelen FJ (1992) Dermal exposure to polycyclic aromatic
hydrocarbons among primary aluminium workers. Med Lav, 83: 519-529.
Van Rooij JGM, Bodelier-Bade MM, & Jongeneelen FJ (1993a) Estimation
of individual dermal and respiratory uptake of polycyclic aromatic
hydrocarbons in 12 coke oven workers. Br J Ind Med, 50: 623-632.
Van Rooij JGM, Van Lieshout EMA, Bodelier-Bade MM, & Jongeneelen FJ
(1993b) Effect of the reduction of skin contamination on the internal
dose of creosote workers exposed to polycyclic aromatic hydrocarbons.
Scand J Work Environ Health, 19: 200-207.
Van Rooij JGM, Veeger MMS, Bodelier-Bade MM, Scheepers PTJ, &
Jongeneelen FJ (1994a) Smoking and dietary intake of polycyclic
aromatic hydrocarbons as sources of interindividual variability in the
baseline excretion of 1-hydroxypyrene in urine. Int Arch Occup Environ
Health, 66: 55-65.
Van Rooij JGM, Bodelier-Bade MM, Hopmans PMJ, & Jongeneelen FJ (1994b)
Reduction of urinary 1-hydroxypyrene excretion in coke-oven workers
exposed to polycyclic aromatic hydrocarbons due to improved hygienic
skin protective measures. Ann Occup Hyg, 38: 247-256.
Van Straalen NM & Verweij RA (1991) Effects of benzo [a]pyrene on
food assimilation and growth efficiency in Porcellio scaber
(Isopoda). Bull Environ Contam Toxicol, 46: 134-140.
Van Vaeck L, Broddin G, & Van Cauwenberghe K (1980) On the relevance
of air pollution measurements of aliphatic and polyaromatic
hydrocarbons in ambient particulate matter. Biomed Mass Spectrom, 7:
473-483.
Van Vaeck L, Van Cauwenberghe K, & Janssens J (1984) The gas-particle
distribution of organic aerosol constituents: Measurement of the
volatilisation artefact in hi-vol cascade impactor sampling. Atmos
Environ, 18: 417-430.
Varanasi U, Reichert WL, Stein JE, Brown DW, & Sanborn HR (1985)
Bioavailability and biotransformation of aromatic hydrocarbons in
benthic organisms exposed to sediment from an urban estuary. Environ
Sci Technol, 19: 836-841.
Varanasi U, Stein JE, Reichert WL, Tilbury KL, Krahn MM, & Chan SL
(1992) Chlorinated and aromatic hydrocarbons in bottom sediments, fish
and marine mammals in US coastal waters: Laboratory and field studies
of metabolism and accumulation. In: Walker CH & Livingstone DR ed.
Persistent pollutants in marine ecosystems. Oxford, Pergamon Press, pp
82-115.
Vassilaros DL, Stoker PW, Booth GM, & Lee ML (1982) Capillary gas
chromatographic determination of polycyclic aromatic compounds in
vertebrate fish tissue. Anal Chem, 54: 106-112.
Vaught J & Bresnick E (1976) Binding of polycyclic hydrocarbons to
nuclear components in vitro. Biochem Biophys Res Commun, 69:
587-591.
Vaught JB, Gurtoo HL, Paigen B, Minowada J, & Sartori P (1978)
Comparison of benzo [a]pyrene metabolism by human peripheral blood
lymphocytes and monocytes. Cancer Lett, 5: 261-268.
Veldre IA & Itra AR (1991) PAH in the water medium of the Estonian SSR
and their monitoring. In: Cooke M, Loening K, & Merritt J ed.
Polynuclear aromatic hydrocarbons: Measurement, means, and metabolism.
Columbus, Ohio, Battelle Press, pp 939-948.
Venier P, Clonfero E, Cottica D, Gava C, Zordan M, Pozzoli L, & Levis
AG (1985) Mutagenic activity and polycyclic aromatic hydrocarbon level
in urine of workers exposed to coal tar pitch volatiles in an anode
plant. Carcinogenesis, 6: 749-752.
Verma DK, Julian JA, Roberts RS, Muir DC, Jadon N, & Shaw DS (1992)
Polycyclic aromatic hydrocarbons (PAHs): A possible cause of lung
cancer mortality among nickel/copper smelter and refinery workers. Am
Ind Hyg Assoc J, 53: 317-324.
Vermorken AJM, Goos CMAA, Roelofs HMJ, Henderson PT, & Bloemendal H
(1979) Metabolism of benzo [a]pyrene in isolated human scalp hair
follicles. Toxicology, 14: 109-116.
Verschueren K (1983) Handbook of environmental data on organic
chemicals, 2nd Ed. New York, Van Nostrand Reinhold Co, pp 460, 760,
968.
Viau C & Vyskocil A (1995) Patterns of 1-hydroxypyrene excretion in
volunteers exposed to pyrene by the dermal route. Sci Total Environ,
163: 187-190.
Viau C, Vyskoçil A, Tremblay C, & Morissette L (1993) Urinary
excretion of 1-hydroxypyrene in workers exposed to polycyclic aromatic
hydrocarbon mixtures. J Occup Med Toxicol, 2: 267-276.
Viau C, Carrier G, Vyskoçil A, & Dodd C (1995) Urinary excretion
kinetics of 1-hydroxypyrene in volunteers exposed to pyrene by the
oral and dermal route. Sci Total Environ, 163: 179-186.
Viras LG, Siskos PA, & Stephanou E (1987) Determination of polycyclic
aromatic hydrocarbons in Athens atmosphere. Int J Environ Anal Chem,
28: 71-85.
Vo-Dinh T (1989) Chemical analysis of polycyclic aromatic compounds.
New York, John Wiley & Sons, 487 pp.
Vo-Dinh T, Bruewer TJ, Colovos GC, Wagner TJ, & Jungers RH (1984)
Field evaluation of a cost-effective screening procedure for
polynuclear aromatic pollutants in ambient air samples. Environ Sci
Technol, 18: 477-482.
Vogel EW, Zijlstra JA, & Blijleven WGH (1983) Mutagenic activity of
selected aromatic amines and polycyclic hydrobarbons in
Drosophila melanogaster. Mutat Res, 107: 53-77.
Vogt NB, Brakstad F, Thrane K, Nordenson S, Krane J, Aamot E, Kolset
K, Esbensen K, & Steinnes E (1987) Polycyclic aromatic hydrocarbons in
soil and air: Statistical analysis and classification by the SIMCA
method. Environ Sci Technol, 21: 35-44.
Volkswagen AG (1989) Unregulated motor vehicle exhaust gas components.
Wolfsburg, Research and Development, 128 pp.
Von Boente L (1955) [Role of coronene in the products of high-pressure
hydrogenation.] Brennstoff-Chemie, 36: 210-214 (in German).
Von Meyerinck (1995)
de Vos (1980)
de Vos RH, Van Dokkum W, Schouten A, & De Jong-Berkhout P (1990)
Polycyclic aromatic hydrocarbons in Dutch total diet samples
(1984-1986). Food Chem Toxicol, 28:
Vreuls JJ, De Jong GJ, & Brinkman UAT (1991) On-line coupling of
liquid chromatography, capillary gas chromatography and mass
spectrometry for the determination and identification of polycyclic
aromatic hydrocarbons in vegetable oils. Chromatographia, 31: 113-118.
Vu Duc T & Huynh CK (1981) [Organic micropolluants in water.
Preliminary results on polycyclic aromatic haloforms and
hydrocarbons.] Méd Soc Prév, 26: 315-316 (in French).
Vu Duc T & Huynh CK (1991) Photodecomposition rates of adsorbed PAH
and mutagenic activity of irradiated synthetic mixtures. In: Cooke M,
Loening K, & Merritt J ed. Polynuclear aromatic hydrocarabons:
Measurement, means and metabolism. Columbus, Ohio, Battelle Press, pp
979-994.
Wakeham SG, Schaffner C, & Giger W (1980a) Polycyclic aromatic
hydrocarbons in recent lake sediments. I. Compounds having
anthropogenic origins. Geochim Cosmochim Acta, 44: 403-413.
Wakeham SG, Schaffner C, & Giger W (1980b) Diagenetic polycyclic
aromatic hydrocarbons in recent sediments: Structural information
obtained by high performance liquid chromatography. Phys Chem Earth,
12: 353-363.
Wakeham SG, Davis AC, & Karas J (1983) Mesocosm experiments to
determine the fate and persistence of volatile organic compounds in
coastal seawater. Environ Sci Technol, 17: 611-617.
Waldman JM, Lioy PJ, Greenberg A, & Butler JP (1991) Analysis of human
exposure to benzo(a)pyrene via inhalation and food ingestion in the
Total Human Environment Exposure Study (THEES). J Exposure Anal
Environ Epidemiol, 1: 193-225.
Walker JD & Colwell RR (1976) Measuring the potential activity of
hydrocarbon-degrading bacteria. Appl Environ Microbiol, 31: 189-197.
Wall KL, Gao W, Te Koppele JM, Kwei GY, Kauffman FC, & Thurman RG
(1991) The liver plays a central role in the mechanism of chemical
carcinogenesis due to polycyclic aromatic hydrocarbons.
Carcinogenesis, 12: 783-786.
Waller RE (1981) Trends in lung cancer in London in relation to
exposure to diesel fumes. Environ Int, 5: 479-483.
Waller RE, Hampton L, & Lawther PJ (1985) A further study of air
pollution in diesel bus garages. Br J Ind Med, 42: 824-830.
Walter U, Beyer M, Klein J, & Rehm HJ (1990) Biodegradation of
polycyclic aromatic hydrocarbons by a bacterial mixed culture.
Biotechnol Conf, 4: 489-492.
Walters RW & Luthy RG (1981) Physicochemical behavior of PAH in coal
conversion liquid effluents. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 539-550.
Walters RW & Luthy RG (1984) Liquid/suspended solid phase partitioning
of polycyclic aromatic hydrocarbons in coal coking wastewaters. Water
Res, 18: 795-809.
Walton B (1980) Influence of route of entry on toxicity of polycyclic
aromatic hydrocarbons to the cricket (Acheta
domesticus). Bull Environ Contam Toxicol, 25: 289-293.
Wang DT & Meresz O (1982) Occurrence and potential uptake of
polynuclear aromatic hydrocarbons of highway traffic origin by
proximally grown food crops. In: Cooke M, Dennis AJ & Fisher GL ed.
Polynuclear aromatic hydrocarbons: Physical and biological chemistry.
Columbus, Ohio, Battelle Press, pp 885-896.
Wangenheim J & Bolcsfoldi G (1988) Mouse lymphoma L5178Y thymidine
kinase locus assay of 50 compounds. Mutagenesis, 3: 193-205.
Waravdekar SS & Ranadive KJ (1958) Biologic testing of
3,4,9,10-dibenzpyrene. J Natl Cancer Inst, 21: 1151-1159.
Ward EC, Murray MJ, Lauer LD, House RV, Irons R, & Dean JH (1984)
Immunosuppres-sion following 7,12-dimethylbenz [a]anthracene exposure
in B6C3F1 mice. I. Effects on humoral immunity and host resistance.
Toxicol Appl Pharmacol, 75: 299-308.
Ward EC, Murray MJ, Lauer LD, House RV, & Dean JH (1986) Persistent
suppression of humoral and cell-mediated immunity in mice following
exposure to the polycyclic aromatic hydrocarbon
7,12-dimethylbenz [a]anthracene. Int J Immunopharmacol, 8: 13-22.
Warman K (1985) PAH emissions from coal-fired plants. In: Bjorseth A &
Ramdahl T ed. Handbook of polycyclic aromatic hydrocarbons, Volume 2:
Emission sources and recent progress in analytical chemistry. New
York, Marcel Dekker, pp 21-59.
Warren DL, Brown DR Jr, & Buckpitt AR (1982) Evidence for cytochrome
P450 mediated metabolism in the bronchiolar damage by naphthalene.
Chem-Biol Interactions, 40: 287-303.
Warshawsky D & Barkley W (1987) Comparative carcinogenic potencies of
7H-dibenzo [c,g]carbazole, dibenz [a,j]acridine and benzo [a]pyrene
in mouse skin. Cancer Lett, 37: 337-344.
Warshawsky D, Barkley W, Miller ML, LaDow K, & Andringa A (1994)
Carcinogenicity of 7H-dibenzo(c,g)carbazole, dibenz(a,j)acridine and
benzo(a)pyene in mouse skin and liver following topical application.
Toxicology, 93: 135-149.
Warshawsky D, Barkley W, Miller ML, LaDow K, & Andringa A (1995)
Erratum to 'Carcinogenicity of 7H-dibenz(a)pyrene in mouse skin and
liver following topical application' (Toxicology 93 (1994) 135-149).
Toxicology, 96: 239-240.
Wattenberg LW (1978) Inhibitors of chemical carcinogenesis. Adv Cancer
Res, 26: 197-226.
Wattenberg LW & Leong JL (1970) Inhibition of the carcinogenic action
of benzo(a)pyrene by flavones. Cancer Res, 30: 1922-1925.
Weaver NK & Gibson RL (1979) The US oil shale industry: A health
perspective. Am Ind Hyg Assoc J, 40: 460-467.
Wehry EL (1983) Optical spectrometric techniques for determination of
polycyclic aromatic hydrocarbons. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 323-396.
Weigert F & Mottram JC (1946) The biochemistry of benzpyrene. II. The
course of its metabolism and the chemical nature of the metabolites.
Cancer Res, 6: 109-120.
Weinstein IB, Jeffrey AM, Jennette KW, Blobstein SH, Harvey RG, Harris
C, Autrup H, Kasai H, & Nakanishi K (1976) Benzo [a]pyrene diol
epoxides as intermediates in nucleic acid binding in vitro
and in vivo. Science, 193: 592-595.
Weinstein D, Katz ML, & Kazmer S (1977) Chromosomal effects of
carcinogens and noncarcinogens on WI-38 after short term exposures
with and without metabolic acitivation. Mutat Res, 46: 297-304.
Weinstein IB, Jeffrey AM, Leffler S, Pulkrabek P, Yamasaki H, &
Grunberger D (1978) Interactions between polycyclic aromatic
hydrocarbons and cellular macromolecules. In: Gelboin HV & Ts'o POP
ed. Polycyclic hydrocarbons and cancer, Volume 2. New York, Academic
Press, pp 3-36.
Welch RM, Gommi B, Alvares AP, & Conney AH (1972) Effect of
enzyme-induction on the metabolsim of benzo(a)pyrene and
3-methyl-4-monomethylaminoazobenzene in the pregnant and fetal rat.
Cancer Res, 32: 973-978.
Wells PG, Wilson B, & Lubek BM (1989) In vivo murine studies on the
biochemical mechanism of naphthalene cataractogenesis. Toxicol Appl
Pharmacol, 99: 466-473.
Wenclawiak B, Rathmann C, & Teuber A (1992) Supercritical-fluid
extraction of soil samples and determination of polycyclic aromatic
hydrocarbons (PAHs) by HPLC. Fresenius' J Anal Chem, 344: 497-500.
Wenzel-Hartung R, Brune H, Grimmer G, Germann P, Timm J, & Wosniok W
(1990) Evaluation of the carcinogenic potency of four environmental
polycyclic aromatic compounds following intrapulmonary application in
rats. Exp Pathol, 40: 221-227.
West WR, Smith PA, Stoker PW, Booth GM, Smith-Oliver T, Butterworth
BE, & Lee ML (1985) Analysis and genotoxicity of a PAC-polluted river
sediment. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: Mechanisms, methods, and metabolism. Columbus, Ohio,
Battelle Press, pp 1395-1411.
West WR, Wise SA, Campbell RM, Bartle KD & Lee ML (1986) The analysis
of polycyclic hydrocarbon minerals curtisite and idrialite by high
resolution gas and liquid chromatographic techniques. In: Cooke M &
Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry,
characterization and carcinogenesis. Columbus, Battelle Press, pp 995-
1009.
Wester RC, Maibach HI, Bucks DAW, Sedik L, Melendres J, Liao C, &
DiZio S (1990) Percutaneous absorption of [14C] DDT and [14C]
benzo [a]pyrene from soil. Fundam Appl Toxicol, 15: 510-516.
Westerholm R, Alsberg T, Strandell M, Frommelin Å, Grigoriadis V,
Hantzaridou A & Maitra G (1986) Chemical analysis and biological
testing of emissions from a heavy duty diesel truck with and without
two different particulate traps. Warrendale, Pennsylvania, Society of
Automotive Engineers, pp 74-82 (SAE Technical Paper Series No.
860014).
Westerholm R, Stenberg U, & Alsberg T (1988) Some aspects of the
distribution of polycyclic aromatic hydroarbons (PAH) between
particles and gas phase from diluted gasoline exhausts generated with
the use of a dilution tunnel, and its validity for measurement in
ambient air. Atmos Environ, 22: 1005-1010.
Westerholm R, Hang L, Egebäck K-E, & Grägg K (1989) Exhaust emission
reduction from a heavy duty diesel truck, using a catalyst and a
particulate trap. Fuel, 68: 856-860.
Westerholm RN, Almen J, Li H, Rannug JU, Egebäck K-E, & Grägg K (1991)
Chemical and biological characterization of particulate-,
semivolatile-, and gas-phase-associated compounds: A comparison of
three different semivolatile-phase samplers. Environ Sci Technol, 25:
332-338.
Weston A (1993) Physical methods for the detection of carcinogen-DNA
adducts in humans. Mutat Res, 288: 19-29.
Weston A, Grover PL, & Sims P (1983) Metabolic activation of
benzo [a]pyrene in human skin maintained in short-term organ culture.
Chem-Biol Interactions, 45: 359-371.
Weston A, Bowman ED, Shields PG, Trivers GE, Poirier MC, Santella RM,
& Manchester DK (1993) Detection of polycyclic aromatic hycrocarbon-
DNA adducts in human lung. Environ Health Perspectives, 99: 257-259.
Weyand EH & Bevan DR (1986) Benzo(a)pyrene disposition, and metabolism
in rats following intratracheal instillation. Cancer Res, 46: 5655-
5661.
Weyand EH & Bevan DR (1987) Species differences in disposition of
benzo [a]pyrene. Drug Metab Disposition, 15: 442-448.
Weyand EH & LaVoie EJ (1988) Comparison of PAH DNA adduct formation
and tumor initiating activity in newborn mice. Proc Am Assoc Cancer
Res, 29: 98.
Weyand EH, Rice JE, Hussain N, & LaVoie EJ (1987) Detection of DNA
adducts of tumorigenic nonalternant polycyclic aromatic hydrocarbons
by 32P-postlabelling. Proc Am Assoc Cancer Res, 28: 102.
Weyand EH, Patel S, LaVoie EJ, Cho B, & Harvey RG (1990) Relative
tumor initiating activity of benzo [a]fluoranthene,
benzo [b]fluoranthene, naphtho[1,2- b]fluoranthene and
naphtho[2,1- a]fluoranthene on mouse skin. Cancer Lett, 52: 229-233.
Weyand EH, Bryla P, Wu Y, He Z-M, & LaVoie EJ (1993) Detection of the
major DNA adducts of benzo(j)fluoranthene in mouse skin: Nonclassical
dihydrodiol epoxides. Chem Res Toxicol, 6: 117-124.
White JG (1948) The crystal structure of 1:12-benzperylene: A
quantitatve X-ray investigation. J Chem Soc, 1: 1398-1408.
White CM (1986) Prediction of the boiling point, heat of vaporization,
and vapor pressure at various temperatures for polycyclic aromatic
hydrocarbons. J Chem Eng Data, 31: 198-203.
White KL & Holsapple MP (1984) Direct suppression of in vitro
antibody production by mouse spleen cells by the carcinogen
benzo(a)pyrene but not by the noncarcinogenic congener benzo(e)pyrene.
Cancer Res, 44: 3388-3393.
White J & White A (1939) Inhibition of growth of the rat by oral
administration of methylcholanthrene, benzpyrene, or pyrene and the
effects of various dietary supplements. J Biol Chem, 131: 149-161.
White KL, Lysy HH, & Holsapple MP (1985) Immunosuppression by
polycyclic aromatic hydrocarbons: A structure-activity relationship in
B6C3F1 and DBA/2 mice. Immunopharmacology, 9: 155-164.
Whitlock JP (1979) The conformation of the chromatin core particle is
ionic strength dependent. J Biol Chem, 254: 5684-5689.
Whitman LJ & Miller RJ (1982) The phototactic behavior of
Daphnia magna as an indicator of chronic toxicity. Proc Oklahoma
Acad Sci, 62: 22-33.
WHO (1971) International drinking-water standards, 3rd Ed, p 55.
WHO (1984) Guidelines for drinking-water quality, Volume 1:
Recommendations, pp 66-68; Volume 2: Health criteria and other
supporting information. Geneva, pp 185-186.
WHO (1987) Polynuclear aromatic hydrocarbons (PAH). In: Air quality
guidelines for Europe. Copenhagen, Regional Office for Europe, pp 105-
117 (WHO Regional Publications, European Series, No. 23).
WHO (1988) Emissions of heavy metals and PAH from municipal solid
waste incinerators: Control technology and health impact. Copenhagen,
Regional Office for Europe, 67 pp (Environmental Health Series, No
32WHO/FADL).
WHO (1991) Evaluation of certain food additives and contaminants.
Thirty-seventh report of the Joint FAO/WHO Expert Committee on Food
Additives. Geneva, pp 27-29 (WHO Technical Report Series 806).
WHO (1996) Guidelines for drinking-water quality, Volume 2: Health
criteria and other supporting information. Geneva, pp 495-505.
Whong WZ, Stewart JD, Cutler D, & Ong T (1992) Comparative study of
DNA adduct formation and cytogenic effects of two constituents in coke
oven emissions with an in vivo rat lung cell system. Environ Mol
Mutag, 19 (Suppl 20): 70.
Whong WZ, Stewart DJ, Cutler D, & Ong T (1994) Induction of in
vivo DNA adducts by 4 industrial by-products in the rat lung-cell
system. Mutat Res, 312:165-172.
Wickström K, Pyysalo H, Plaami-Heikkilä S, & Tuominen J (1986)
Polycyclic aromatic compounds (PAC) in leaf lettuce. Z
Lebensmittelunters Forsch, 183: 182-185.
Wielgosz SM, Brauze D, & Pawlak AL (1991) Ah locus-associated
differences in induction of sister-chromatid exchanges and in DNA
adducts by benzo [a]pyrene in mice. Mutat Res, 246: 129-137.
Wienecke J, Kruse H, & Wassermann O (1992) Organic compounds in the
flue gas from a power station with circulating fluid bed combustion of
coal. Chemosphere, 25: 1889-1895.
Wild SR & Jones KC (1993) Biological and abiotic losses of polynuclear
aromatic hydrocarbons (PAHs) from soils freshly amended with sewage
sludge. Environ Toxicol Chem, 12: 5-12.
Wild SR, McGrath SP, & Jones KC (1990) The polynuclear aromatic
hydrocarbon (PAH) content of archived sewage sludges. Chemosphere, 20:
703-716.
Wild SR, Berrow ML, & Jones KC (1991) The persistence of polynuclear
aromatic hydrocarbons (PAH's) in sewage sludge amended agricultural
soils. Environ Pollut, 72: 141-157.
Wild SR, Mitchell DJ, Yelland CM, & Jones KC (1992) Arrested municipal
solid waste incinerator fly ash as a source of polynuclear aromatic
hydrocarbons (PAHs) to the environment. Waste Manage Res, 10: 99-111.
Willems MI, Roggeband R, Baan RA, Wilmer JWGM, De Raat WK, & Lohman
PHM (1991) Monitoring the exposure of rats to benzo [a]pyrene by the
determination of mutagenic acitivity in excreta, chromosome
aberrations and sister chromatid exchanges in peripheral blood cells,
and DNA adducts in peripheral blood lymphocytes and liver.
Mutagenesis, 6: 151-158.
Williams RT (1959) Detoxication mechanisms. The metabolism and
detoxication of drugs, toxic substances and other organic compounds,
2nd Ed. London, Chapman & Hall, 768 pp.
Williams GM (1977) Detection of chemical carcinogens by unscheduled
DNA synthesis in rat liver primary cell cultures. Cancer Res, 37:
1845-1851.
Williams GM, Laspia MF & Dunkel VC (1982) Reliability of the
hepatocyte primary culture / DNA repair test in testing of coded
carcinogens and noncarcinogens. Mutat Res, 97: 359-370.
Williams PT, Abbass MK, Andrews GE, & Bartle KD (1989) Diesel
particulate emissions: The role of unburned fuel. Combust Flame, 75:
1-24.
Wilson NK & Chuang JC (1991) Indoor air levels of polynuclear aromatic
hydrocarbons and related compounds in an eight-home pilot study. In:
Cooke M, Loening K, & Merritt J ed. Polynuclear aromatic hydrocarbons:
Measurements, means and metabolism. Columbus, Ohio, Battelle Press, pp
1053-1064.
Wilson NK, Chuang JC, & Kuhlman MR (1991) Sampling polycyclic aromatic
hydrocarbons and related semivolatile organic compounds in indoor air.
Indoor Air, 4: 513-521.
Windsor JG Jr & Hites RA (1978) Polycyclic aromatic hydrocarbons in
Gulf of Maine sediments and Nova Scotia soils. Geochim Cosmochim Acta,
41: 27-33.
Winker N, Weniger P, Klein W, Ott E, Kocsis F, Schoket B, & Korpert K
(1995) Detection of polycyclic aromatic hydrocarbon exposure damage
using different methods in laboratory animals. J Appl Toxicol, 15: 59-
62.
Winkler DL, Duncan KL, Hose JE, & Puffer HW (1983) Effects of
benzo(a)pyrene on the early development of California grunion,
Leuresthes tenuis (Pisces, Atherinidae). Fish Bull, 81: 473-481.
Wise SA (1983) High-performance liquid chromatography for the
determination of polycyclic aromatic hydrocarbons. In: Bjorseth A ed.
Handbook of polycyclic aromatic hydrocarbons, Volume 2: Emission,
sources and recent progress in analytical chemistry. New York, Marcel
Dekker, pp 183-256.
Wise SA (1985) Recent progress in the determination of PAH by high
performance liquid chromatography. In: Bjorseth A & Ramdahl T ed.
Handbook of polycyclic aromatic hydrocarbons, Volume 2. New York,
Marcel Dekker, pp 113-191.
Wise SA, Bonnett WJ, & May WE (1980) Normal- and reverse-phase liquid
chromatographic separations of polycyclic aromatic hydrocarbons. In:
Bjorseth A & Dennis AJ ed. Polynuclear aromatic hydrocarbons:
Chemistry and biological effects. Columbus, Ohio, Battelle Press, pp
791-806.
Wise SA, Bonnett WJ, Guenther FR, & May WE (1981) A relationship
between reversed-phase C18 liquid cromatographic retention and the
shape of polycyclic aromatic hydrocarbons. J Chromatogr Sci, 19: 457-
465.
Wise SA, Sander LC, & May WE (1993) Determination of polycyclic
aromatic hydrocarbons by liquid chromatography. J Chromatogr, 642:
329-349.
Wislocki PG, Buening MK, Levin W, Lehr RE, Thakkes DR, Jerina DM, &
Conney AH (1979) Tumorigenicity of the diastereomeric
benz [a]anthracene 3,4-diol-1,2-epoxides and the (+)-and
(-)-enantiomers of benz [a]anthracene 3,4-dihydrodiol in newborn
mice. J Natl Cancer Inst, 63: 201-204.
Withey JR, Law FCP, & Endrenyi L (1991) Pharmacokinetics and
bioavailability of pyrene in the rat. J. Toxicol Environ Health, 32:
429-447.
Withey JR, Shedden J, Law FPC, & Abedini S (1992) Distribution to the
fetus and major organs of the rat following inhalation exposure to
pyrene. J Appl Toxicol, 12: 223-231.
Withey JR, Law FCP, & Endrenyi L (1993) Percutaneous uptake,
distribution, and excretion of pyrene in rats. J Toxicol Environ
Health, 40: 601-612.
Witte H, Langenohl T, & Offenbächer G (1988) [Investigation of the
entry of organic pollutants into soils and plants through the use of
sewage sludge in agriculture. Part A: Organic pollutant load in sewage
sludge.] Korresp Abwasser, 5: 440-448 (in German).
Wodinsky I, Helinski A, & Kensler CJ (1964) Susceptibility of Syrian
hamsters to induction of fibrosarcomas with a single injection of
3,4,9,10-dibenzpyrene. Nature, 203: 308-309.
Woidich W, Pfannhauser W, Blaicher G, & Tiefenbacher K (1976)
[Analysis of polycyclic aromatic hydrocarbons in drinking and utility
water.] Lebensmittelchem Gerichtl Chem, 30: 141-146 (in German).
Wojciechowski JP, Kaur P & Sabharwal PS (1981) Comparison of metabolic
systems required to activate pro-mutagens/carcinogens
in vitro for sister-chromatid exchange studies. Mutat Res, 88: 89-
97.
Wojdani A & Alfred LJ (1984) Alterations in cell-mediated immune
functions induced in mouse splenic lymphocytes by polycyclic aromatic
hydrocarbons. Cancer Res, 44: 942-945.
Wolfe JM & Bryan WR (1939) Effects induced in pregnant rats by
injection of chemically pure carcinogenic agents. Am J Cancer, 36:
359-368.
Wolff MS, Herbert R, Marcus M, Rivera M, Landrigan PJ, & Andrews LR
(1989) Polycyclic aromatic hydrocarbons (PAH) residues on skin in
relation to air levels among roofers. Arch Environ Health, 44:
157-163.
Wolff RK, Griffith WC, Henderson RF, Hahn FF, Harkema JR, Rebar AH,
Eidson AF, & McClellan RO (1989) Effects of repeated inhalation
exposures to 1-nitropyrene, benzo(a)pyrene, Ga203 particles, and
S02 alone and in combinations of particle clearance, bronchoalveolar
lavage fluid composition, and histopathology. J Toxicol Environ
Health, 27: 123-138.
Wong O, Bailey WJ, & Amsel J (1992) Cancer mortality and incidence in
mastic asphalt workers. Scand J Work Environ Health, 18: 133-135.
Wood SC & Holsapple MP (1993) Effects of
7,12-dimethylbenz [a]anthracene on the superantigen toxic shock
syndrome toxin (TSST-1)-induced proliferation and antibody secretion
by human lymphocytes. Fundam Appl Toxicol, 20: 280-287.
Wood AW, Levin W, Ryan D, Thomas PE, Yagi H, Mah HD, Thakker DR,
Jerina DM, & Conney AH (1977) High mutagenicity of metabolically
activated chrysene 1,2-dihydrodiol: Evidence for bay region activation
of chrysene. Biochem Biophys Res Commun, 78: 847-854.
Wood AW, Levin W, Thomas PE, Ryan D, Karle JM, Yagi H, Jerina DM, &
Conney AH (1978) Metabolic activation of dibenzo(a,h)anthracene and
its dihydrodiols to bacterial mutagens. Cancer Res, 38: 1967-1973.
Wood AW, Chang RL, Levin W, Ryan DE, Thomas PE, Mah HD, Karle JM, Yagi
H, Jerina DM, & Conney AH (1979) Mutagenicity and tumorigenicity of
phenanthrene and chrysene epoxides and diol epoxides. Cancer Res, 39:
4069-4077.
Wood AW, Levin W, Chang RL, Huang M-T, Ryan DE, Thomas PE, Lehr RE,
Kumar S, Koreeda M, & Akagi, H (1980) Mutagenicity and
tumor-initiating activity of cyclopenta(c,d)pyrene and structurally
related compounds. Cancer Res, 40: 642-649.
Wood AW, Chang RL, Levin W, Ryan DE, Thomas PE, Lehr RE, Kumar S,
Sardella DJ, Boger E, Yagi H, Sayer JM, Jerina DM, & Conney AH (1981)
Mutagenicity of the bayregion diol-epoxides and other benzo-ring
derivatives of dibenzo(a,h)pyrene and dibenzo(a,i)pyrene. Cancer Res,
41: 2589-2597.
Wood AW, Chang RL, Levin W, Yagi H, Thakker DR, van Bladeren PJ,
Jerina DM, & Conney AH (1983) Mutagenicity of the enantiomers of the
diastereomeric bay-region benz [a]anthracene 3,4-diol-1,2-epoxides in
bacterial and mammalian cells. Cancer Res, 43: 5821-5825.
Wood AL, Bouchard DC, Brusseau ML, & Rao PSC (1990) Cosolvent effects
on sorption and mobility of organic contaminants in soil. Chemosphere,
21: 575-587.
Woodhead AD, Setlow RB, & Pond V (1982) Effects of polycyclic aromatic
hydrocarbons on the proliferation of ectopic thyroid tissue in
Poecilia formosa, the Amazon molly. J Fish Biol, 20: 455-463.
Wright BW & Smith RD (1989) Capillary supercritical fluid
chromatography methods. In: Vo-Dinh T ed. Chemical analysis of
polycyclic aromatic compounds. New York, John Wiley & Sons, pp
111-149.
Wu R, Jiang Y-M, Ge N-C, Bai S-M, & Qiao S-J (1985) Determination of
trace amounts of organic pollutants in the Yellow River by capillary
column gas chromatography-mass spectrometry. Int J Environ Anal Chem,
22: 115-126.
Wu Z, Hearl FJ, Peng K, McCawley MA, Chen A, Palassis J, Dosemeci M,
Chen J, McLaughlin JK, Rexing SH, & Blot WJ (1992) Occupational
hygiene around the world: Current occupational exposures in Chinese
iron and copper mines. Appl Occup Environ Health, 7: 735-743.
Wynder EL & Hoffmann D (1959a) A study of tobacco carcinogenesis. VII.
The role of higher polycyclic hydrocarbons. Cancer, 12: 1079-1086.
Wynder EL & Hoffmann D (1959b) The carcinogenicity of
benzofluoranthenes. Cancer, 12: 1194-1199.
Wynder EL, Mabuchi K, & Beattie EJ (1970) The epidemiology of lung
cancer. Recent trends. J Am Med Assoc, 213: 2221-2228.
Xu GT, Zigler JS Jr, & Lou MF (1992) The possible mechanism of
naphthalene cataract in rat and its prevention by an aldose reductase
inhibitor (AL01576). Exp Eye Res, 54: 63-72.
Xue B, Lei ZM, Zhao XL, & Yang XH (1991) [Acute immunotoxicity induced
by benzo(a)pyrene in mice.] Zhonggou Yaolixue Dulixue Zazhi, 5: 221-
222 (in Chinese with English abstract).
Yalkowsky SH & Valvani SC (1979) Solubilities and partitioning. 2.
Relationships between aqueous solubilities, partition coefficients,
and molecular surface areas of rigid aromatic hydrocarbons. J Chem Eng
Data, 24: 127-129.
Yamasaki H, Kuwata K, & Miyamoto H (1982) Effects of ambient
temperature on aspects of airborne polycyclic aromatic hydrocarbons.
Environ Sci Technol, 16: 189-194.
Yamazaki H, Imamura E, Kamei S, Yamauchi A, Kakiuchi Y, & Tai HH
(1990) Polycyclic aromatic hydrocarbons affect the calcium ionophore
induced activation of rabbit platelet. Chemosphere, 21: 21-28.
Yang K, Airoldi L, Pastorelli R, Restano J, Guanci M, & Hemminki K
(1996) Aromatic DNA adducts in lymphocytes of humans working at high
and low traffic density areas. Chem-Biol Interactions, 101: 127-136.
Yoshikawa T, Ruhr LP, Flory W, Banton MJ, Giamalva D, Church DF, &
Pryor WA (1987) Toxicity of polycyclic aromatic hydrocarbons. III.
Effects of beta-naphthoflavone pretreatment on hepatotoxicity of
compounds produced in the ozonation or NO2-nitration of phenanthrene
and pyrene in rats. Vet Hum Toxicol, 29: 113-117.
Yoshioka Y, Nagase H, Ose Y, & Sato T (1986) Evaluation of the test
method 'activated sludge, respiration inhibition test' proposed by the
OECD. Ecotoxicol Environ Saf, 12: 209-212.
You L, Wang D, Galati A, Ross JA, Mass MJ, Nelson GB, Wilson KH, Amin
S, Stoner JC, Nesnow S, & Stoner GD (1994) Tumor multiplicity, DNA
adducts and K- ras mutation pattern of 5-methylchrysene in strain A/J
mouse lung. Carcinogenesis, 15: 2613-2618.
Young L (1947) The metabolic conversion of naphthalene to
1:2-dihydronaphthalene-1:2-diol. Biochem J, 41: 417-422.
Young L (1950) The oxidation of polycyclic hydrocarbons in the animal
body. Biochem Soc Symp, 5: 27-39.
Young DR, Gossett RW, Baird RB, Brown DA, Taylor PA, & Miille MJ
(1983) Wastewater inputs and marine bioaccumulation of priority
pollutant organics off southern California. Water Chlorination, 4:
871-884.
Yrjänheikki E, Pyy L, Hakala E, Lapinlampi T, Lisko A, & Vähäkangas K
(1995) Exposure to polycyclic aromatic hydrocarbons in a new coking
plant. Am Ind Hyg Assoc J, 56: 782-787.
Yun C-H, Shimada T, & Guengerich FP (1992) Roles of human liver
cytochrome P4502C and 3A enzymes in the 3-hydroxylation of
benzo (a)pyrene. Cancer Res, 52: 1868-1874.
Zachleder V, Abarzua S, & Wittenburg E (1983) Effects of
3,4-benzopyrene on the course of cell cycle events in the chlorococcal
alga Scenedesmus quadricauda. Planta, 157: 432-440.
Zackheim HS (1964) Comparative cutaneous carcinogenesis in the rat.
Oncologia, 17: 236-246
Zajdela F, Perin-Roussel O, & Saguem S (1987) Marked differences
between mutagenicity in Salmonella and tumour-initiating activities
of dibenzo [a,e]fluoranthene proximate metabolites; initiation
inhibiting activity of norharman. Carcinogenesis, 8: 461-464.
Zander M (1980) [Origins, chemical and physical properties of PAH.]
In: [Air pollution through polycyclic aromatic hydrocarbons:
Registration and evaluation.] Düsseldorf, VDI-Verlag, pp 11-21 (VDI
Report No. 358) (in German).
Zepp RG & Schlotzhauer PF (1979) Photoreactivity of selected aromatic
hydrocarbons in water. In: Jones PW & Leber P ed. Polynuclear aromatic
hydrocarbons. Carcinogenesis and mutagenesis. Ann Arbor, Michigan, Ann
Arbor Science Publishers, pp 141-158.
Zey JN & Stephenson R (1986) NIOSH health hazard evaluation: Roofing
and waterproofing sites, Chicago, Illinois. Cincinnati, Ohio, National
Institute for Occupational Safety and Health, 29 pp (Report No.
HETA-85-416-1742; PB 87-174207).
Zhao Z-H, Quan W-Y, & Tian D (1990) Urinary 1-hydroxypyrene as an
indicator of human exposure to ambient polycylic aromatic hydrocarbons
in a coal-burning environment. Sci Total Environ, 92: 145-154.
Zhao Z-H, Quan WY, & Tian DH (1992) Experiments on the effects of
several factors on the 1-hydroxypyrene level in human urine as an
indicator of exposure to polycyclic aromatic hydrocarbons. Sci Total
Environ, 133: 197-207.
Zhong BZ, Gu ZW, Stewart J, & Ong T (1995) Micronucleus formation
induced by three polycyclic aromatic hydrocarbons in rat bone marrow
and spleen erythrocytes following intratracheal instillation. Mutat
Res, 326: 147-153.
Zijlstra JA & Vogel EW (1984) Mutagenicity of
7,12-dimethylbenz [a]anthracene and some other aromatic mutagens in
Drosophila melanogaster. Mutat Res, 125: 243-261.
Zimmermeyer G, Roge G, & Schabronath J (1991) [Reduction of PAH
emissions: For example, some stationary plants.] In: [Carcinogenic
compounds in the environment: Sources, measurement, risk, reduction.]
Düsseldorf, VDI-Verlag, pp 93-115 (VDI Report No. 888) (in German).
Zinkham WH & Childs B (1958) A defect of glutathione metabolism in
erythrocytes from patients with a naphthalene-induced hemolytic
anemia. Pediatrics, 14: 461-471.
Zuelzer WW & Apt L (1949) Acute hemolytic anemia due to naphthalene
poisoning. J Am Med Assoc, 141: 185-190.
1. RÉSUMÉ
1.1 Choix des composés pour la monographie
Les hydrocarbures aromatiques polycycliques forment un vaste groupe de
composés et c'est par centaines qu'ils peuvent être libérés dans
l'environnement lors de la combustion incomplète ou de la pyrolyse des
matières organiques, constituant ainsi une source importante
d'exposition humaine. L'étude des matrices susceptibles d'être
importantes sur le plan écologique tels que les résidus de combustion
du charbon, les gaz d'échappement des véhicules à moteur, les huiles
lubrifiantes usées et la fumée de tabac, montre que leur activité
cancérogène est essentiellement liée à leur teneur en HAP.
Les HAP se présentent presque toujours sous la forme de mélanges.
Etant donné que leur composition est complexe et qu'elle dépend du
processus qui leur a donné naissance, il n'a pas été possible de
passer en revue tous les mélanges susceptibles de contenir des
hydrocarbures aromatiques polycycliques. C'est pourquoi on a choisi 33
composés (31 composés originaux et 2 dérivés alkylés) en vue d'une
évaluation sur la base des données pertinentes relatives aux points
d'aboutissement toxicologiques retenus ou à l'exposition (Tableau 1).
Toutefois, étant donné que l'on ne disposait d'études épidémiologiques
que pour les mélanges et que ces études sont indispensables pour
l'évaluation du risque, les sections 8 et 10 exposent les résultats
relatifs à des mélanges d'HAP, contrairement au reste de la
monographie.
Nombre d'articles et de mises au point ont été publiées au sujet de la
présence, de la distribution et de la transformation des HAP dans
l'environnement ainsi que de leurs effets toxicologiques et
écotoxicologiques. Sauf indication contraire, seules les références
des 10 à 15 dernières années sont prises en compte dans la
monographie. En revanche, les mises au point consacrées à des études
plus anciennes sont citées à titre de complément d'information.
1.2 Identité, propriétés physiques et chimiques et méthodes d'analyse
On désigne généralement par hydrocarbures aromatiques polycycliques un
vaste ensemble de composés contenant un ou plusieurs noyaux
aromatiques condensés et constitués de carbone et d'hydrogène. Ces
hydrocarbures sont solides à la température ambiante. Ils ont pour
caractéristiques communes d'avoir un point de fusion et un point
d'ébullition élevés, une faible tension de vapeur et d'être très peu
solubles dans l'eau-d'autant moins que leur masse moléculaire est plus
élevée. Ils sont solubles dans de nombreux solvants organiques et très
lipophiles. Chimiquement, ils sont relativement inertes. Les réactions
intéressantes du point de vue de leur devenir dans l'environnement et
des possibilités de pertes au cours des prélèvements d'air sont celles
qui comportent une photodécomposition ou dans lesquelles interviennent
les oxydes d'azote, l'acide nitrique, les oxydes de soufre, l'acide
sulfurique, l'ozone et les radicaux hydroxyles.
Tableau 1. Les hydrocarbures aromatiques polycycliques évalués dans cette monographie
Nom commun Nom CAS Synonymea No d'enregistrement CAS
Acenaphthylene Acenaphthylene 91-20-3
Acenaphthene Acenaphthylene, 1,2-dihydro- 208-96-8
Anthanthrene Dibenzo[def,mno]chrysene 191-26-4
Anthracene Anthracene 120-12-7
Benz[a]anthracene Benz[a]anthracene 1,2-Benzanthracene, 56-55-3
tetraphene
Benzo[a]fluorene 11H-Benzo[a]fluorene 1,2-Benzofluorene 238-84-6
Benzo[b]fluorene 11H-Benzo[b]fluorene 2,3-Benzofluorene 243-17-4
Benzo[b]fluoranthene Benz[e]acephenanthrylene 3,4-Benzofluoranthene 205-99-2
Benzo[ghi]fluoranthene Benzo[ghi]fluoranthene 2,13-Benzofluoranthene 203-12-3
Benzo[j]fluoranthene Benzo[j]fluoranthene 10,11-Benzofluoranthene 205-82-3
Benzo[k]fluoranthene Benzo[k]fluoranthene 11,12-Benzofluoranthene 207-08-9
Benzo[ghi]perylene Benzo[ghi]perylene 1,12-Benzoperylene 191-24-2
Benzo[c]phenanthrene Benzo[c]phenanthrene 3,4-Benzophenanthrene 195-19-7
Benzo[a]pyrene Benzo[a]pyrene 3,4-Benzopyreneb 50-32-8
Benzo[e]pyrene Benzo[e]pyrene 1,2-Benzopyrene 192-97-2
Chrysene Chrysene 1,2-Benzophenanthrene 218-01-9
Coronene Coronene Hexabenzobenzene 191-07-1
Cyclopenta[cd]pyrene Cyclopenta[cd]pyrene Cyclopenteno[cd]pyrene 27208-37-3
Dibenz[a,h]anthracene Dibenz[a,h]anthracene 1,2:5,6-Dibenzanthracene 53-70-3
Dibenzo[a,e]pyrene Naphtho[1,2,3,4-def]chrysene 1,2:4,5-Dibenzopyrene 192-65-4
Dibenzo[a,h]pyrene Dibenzo[b,def]chrysene 3,4:8,9-Dibenzopyrene 189-64-0
Dibenzo[a,i]pyrene Benzo[rst]pentaphene 3,4:9,10-Dibenzopyrene 189-55-9
Dibenzo[a,l]pyrene Dibenzo[def,p]chrysene 1,2:3,4-Dibenzopyrene 191-30-0
Fluoranthene Fluoranthene 206-44-0
Fluorene 9H-Fluorene 86-73-7
Indeno[1,2,3-cd]pyrene Indeno[1,2,3-cd]-pyrene 2,3-o-Phenylenpyrene 193-39-5
5-Methylchrysene Chrysene, 5-methyl- 3697-24-3
1-Methylphenanthrene Phenanthrene, 1-methyl- 832-69-9
Tableau 1. (cont.)
Nom commun Nom CAS Synonymea No d'enregistrement CAS
Naphthalene Naphthalene 91-20-3
Perylene Perylene peri-Dinaphthalene 198-55-0
Phenanthrene Phenanthrene 85-01-8
Pyrene Pyrene Benzo[def]phenanthrene 129-00-0
Triphenylene Triphenylene 9,10-Benzophenanthrene 217-59-4
Des listes assez complètes de synonymes ont également été publiées par le CIRC (1989) et par Loening & Merritt (1990).
a Synonyme commun utilisé dans la littérature
b On le trouve également sous le nom de benzo(def)chrysène.
L'échantillonnage dans l'air ambiant s'effectue par recueil de
matières particulaires sur filtres eu fibre de verre,
polytétrafluoréthylène, ou fibre de quartz, au moyen
d'échantillonneras de grand volume ou d'échantillonneurs passifs.
Comme il y a risque de volatilisation des hydrocarbures de la phase
gazeuse, qui pourraient s'évaporer des filtres lors de
l'échantillonnage, on a l'habitude de les piéger par adsorption sur
mousse de polyuréthane. La variabilité des résultats provient
essentiellement de ce processus d'échantillonnage.
Sur les lieux de travail, les prélèvements d'air se font à faible
débit; les particules sont recueillies sur des filtres de fibre de
verre ou de polytétrafluoréthylène et la phase gazeuse sur résine
Amberlite XAD-2. Les collecteurs de gaz de cheminées sont constitués
de filtres en fibre de verre ou en quartz placés devant un réfrigérant
destiné à retenir le condensant et une cartouche d'adsorbant (en
général, de l'XAD-2). Les gaz d'échappement des véhicules à moteur
sont recueillis au laboratoire, pendant un cycle de fonctionnement
normalisé simulant les conditions réelles. Les émissions sont
recueillies telles quelles ou après dilution dans de l'air froid
filtré.
De nombreuses techniques d'extraction et de purification ont été
décrites. En fonction de la matrice, on peut extraire les HAP au
Soxhlet, par action des ultrasons, par partage liquide-liquide ou,
après dissolution et digestion alcaline, au moyen d'un solvant
sélectif. On utilise aussi l'extraction par fluide supercritique pour
diverses substances solides présentes dans l'environnement.
L'efficacité de l'extraction dépend largement du solvant et nombre de
solvants utilisés par le passé se sont révélés inappropriés. Après
l'extraction, on procède généralement à une purification par
chromatographie sur colonne - d'alumine, de gel de silice ou de
sephadex LH-20- mais aussi par chromatographie sur couche mince.
La recherche et le dosage s'effectuent classiquement par
chromatographie en phase gazeuse avec détection par ionisation de
flamme ou encore par chromatographie en phase liquide à haute
performance avec détection en UV ou fluorescence, généralement en
série. Pour la chromatographie en phase gazeuse, on utilise des
colonnes capillaires en silice fondue, avec des polysiloxanes comme
phase stationnaire (SE-54 et SE-52); pour la chromatographie en phase
liquide, on utilise couramment des colonnes de gel de silice C-18.
Pour confirmer l'identité des pics chromatographiques, on couple
généralement un spectromètre de masse au chromatographie.
Le choix des HAP à doser dépend de l'objectif de la mesure: étude à
visée sanitaire, investigation écotoxicologique ou recherche d'une
source de pollution. La recherche et le dosage de divers groupes de
composés peuvent être demandés au niveau national ou international.
1.3 Sources d'exposition humaine et environnementale
On sait peu de choses sur la production des hydrocarbures aromatiques
polycycliques et sur les processus auxquels ils peuvent être soumis,
mais il est probable que ces activités n'entraînent la libération que
de petites quantités d'hydrocarbures. Ceux que l'on retrouve dans
l'environnement sont principalement utilisés comme intermédiaires dans
la préparation du chlorure de polyvinyle et de divers plastifiants
(naphtalène), de pigments (acénaphtène, pyrène), de colorants
(anthracène, fluoranthène) et de pesticides (phénanthrène).
Les émissions les plus importantes d'hydrocarbures aromatiques
polycycliques sont dues à la combustion incomplète de matières
organiques lors de divers processus industriels ou d'autres activités
humaines, notamment:
- les diverses opérations effectuées sur la houille, le pétrole
brut et le gaz naturel, y compris la cokéfaction, la conversion
de la houille, le raffinage du pétrole et la production de noirs
de carbone, de créosote, de goudron de houille et de bitume;
- la production d'aluminium, de fer et d'acier dans les divers
ateliers et fonderies;
- la génération d'énergie calorifique par les centrales thermiques,
le chauffage des habitations et la cuisine;
- le brûlage des déchets;
- la circulation automobile; et
- la fumée de tabac dispersée dans l'environnement.
Les HAP, notamment ceux qui ont une masse moléculaire élevée,
s'adsorbent sur les particules de matière une fois qu'ils sont libérés
dans l'environnement par la voie atmosphérique. La pénétration dans
l'hydrosphère et la géosphère s'effectue ensuite selon un processus de
dépôt par voie humide ou par voie sèche. La conservation du bois par
traitement au créosote constitue une autre source de pénétration d'HAP
dans l'hydrosphère et les dépôts de déchets contaminés, comme par
exemple les boues d'égout et les cendres volantes, contribuent à
l'introduction de ces composés dans la géosphère. On possède peu de
renseignements sur le passage des HAP dans la biosphère. Des
hydrocarbures aromatiques polycycliques sont présents à l'état naturel
dans la tourbe, le lignite, la houille et le pétrole brut. La plupart
de ceux que l'on trouve dans l'anthracite sont fermement liés à la
structure carbonée et ne peuvent pas en être extraits par lessivage.
On a pu déterminer le passage d'HAP dans l'environnement en mettant en
évidence un profil de concentration caractéristique, mais cela n'a été
possible que dans quelques cas. Le benzo(a)pyrène est fréquemment
utilisé comme indicateur de la présence d'HAP, notamment dans les
études un peu anciennes. En général, les chiffres concernant les
émissions d'HAP ne sont que des estimations basées sur des données
plus ou moins fiables et elles ne donnent qu'une idée approximative de
l'exposition.
Les sources d'HAP les plus importantes sont les suivantes:
La cokéfaction de la houille: les émissions atmosphériques d'HAP
résultant de la cokéfaction de la houille ont sensiblement diminué en
Allemagne au cours des 10 dernières années par suite des améliorations
techniques apportées aux installations industrielles, à la fermeture
des anciennes usines et au recul de la production de coke. On pense
que la situation est à peu près la même dans le reste de l'Europe
occidentale, au Japon, et aux Etats-Unis, sans toutefois disposer de
données à ce sujet.
La production d'aluminium (notamment en raison de l'utilisation
d'anodes spéciales en graphite), de fer et d'acier ainsi que les
liants utilisés en fonderie dans les moules à sable. On ne possède
guère d'informations à ce sujet.
Le chauffage collectif ou individuel: les émissions sont
principalement constituées de phénanthrène, de pyrène et de chrysène.
Les poëles à bois en émettent 25 à 1000 fois plus que les poëles à
charbon et dans les régions où l'on se chauffe surtout au bois, la
majeure partie des émissions atmosphériques d'HAP ont ce mode de
chauffage pour origine, principalement l'hiver. On considère donc que
le chauffage des habitations est une source importante d'hydrocarbures
aromatiques polycycliques dans les pays en développement où l'on brûle
de la biomasse dans des poëles assez rudimentaires.
La cuisine: la combustion incomplète des matières combustibles peut
produire des HAP, de même que le chauffage de l'huile de cuisine et la
cuisson des denrées alimentaires elles-mêmes.
La circulation des véhicules à moteur: les principaux hydrocarbures
rejetés par les véhicules dotés de moteurs à essence sont le
fluoranthène et le pyrène, tandis que le naphtalène et l'acénaphtène
prédominent dans les gaz d'échappement des moteurs diesel. Le
cyclopenta(cd)-pyrène est émis en grande quantité par les moteurs à
essence mais il est juste au-dessus de la limite de détection dans les
gaz d'échappement des moteurs diesel. Le taux d'émission, qui dépend
du composé, du type de véhicule et de l'état de son moteur, de même
que des conditions dans lesquelles l'essai se déroule, varie de
quelques nanogrammes par kilomètre à > 1000 mg/km. La pose d'un
catalyseur réduit de façon spectaculaire les émissions d'HAP par les
moteurs d'automobiles.
Les feux de forêt: Dans les pays où la forêt couvre de vases
étendues de territoire, les feux de forêt peuvent contribuer de façon
importante à l'émission d'HAP.
Centrales thermiques à charbon: Les HAP libérés dans l'atmosphère
par ces centrales sont principalement des composés bi-et tricycliques.
Dans les zones contaminées, la teneur en HAP de l'air ambiant peut
être supérieure à celle qui résulte des émission de cheminées.
Incinération des déchets: les émissions d'HAP dans les gaz de
cheminée des usines d'incinération sont inférieures à 10 mg/m3 dans
un certain nombre de pays.
1.4 Transport, distribution et transformation dans l'environnement
La destinée des HAP, qu'ils soient seuls ou en mélange, dépend d'un
certain nombre de processus de distribution et de transformation. Les
processus de distribution les plus importants sont le partage entre
l'eau et l'air, entre l'eau et les sédiments et entre l'eau et les
organismes vivants.
Comme ces hydrocarbures sont hydrophobes et très peu solubles dans
l'eau, il n'ont qu'une très faible affinité pour la phase aqueuse;
toutefois, bien qu'ils soient libérés dans l'environnement par la voie
atmosphérique, on les retrouve également en concentration importante
dans l'hydrosphère, du fait que leur constante de Henry est faible.
Etant donné que leur affinité est plus grande pour la phase organique
que pour la phase aqueuse, leur coefficient de partage entre les
solvants organiques- comme l'octanol- et l'eau est élevé. Ils ont
également une forte affinité pour les fractions organiques des
sédiments, du sol et des organismes vivants, de sorte qu'ils
s'accumulent dans les organismes aquatiques et sédimentaires ainsi que
dans leur nourriture. On ne connaît pas avec certitude l'importance
relative de leur fixation à partir de la nourriture et de l'eau. Chez
la daphnie et les mollusques, il y a corrélation positive entre
l'accumulation d'HAP provenant de l'eau et le coefficient de partage
entre l'octanol et l'eau (Kow). Par contre, chez les poissons et les
algues qui sont capables de métaboliser ces hydrocarbures, il n'y a
pas de corrélation entre la concentration des différents HAP et le
Kow.
Le phénomène de bioamplification, c'est-à-dire la concentration d'une
substance dans l'organisme d'animaux à chaque niveau trophique
successif de la chaîne alimentaire, n'a pas été observé en milieu
aquatique et il ne semble pas qu'il puisse se produire, car la plupart
des organismes sont tout à fait à même de métaboliser les HAP. Ce sont
les organismes qui occupent les niveaux trophiques les plus élevés qui
possèdent la plus grande capacité de biotransformation.
La dégradation des HAP s'opère par photodécomposition, par
biodégradation microbienne et, chez les organismes supérieurs, par
métabolisation. Cette dernière voie de transformation n'a guère
d'influence sur la destinée globale des HAP dans l'environnement, mais
elle joue néanmoins un rôle biologique important du fait que des
métabolites cancérogènes sont susceptibles de se former. Comme les HAP
sont chimiquement stables et dépourvus de groupements réactifs,
l'hydrolyse n'intervient pas dans leur décomposition. Il n'existe
guère d'épreuves classiques pour l'étude de la biodégradation des HAP.
Celle-ci s'opère généralement en aérobiose, la vitesse du processus
diminuant fortement avec le nombre de cycles aromatiques. En
anaérobiose, la dégradation est beaucoup plus lente.
Dans l'air et dans l'eau, les HAP subissent une photo-oxydation en
présence de radicaux ou molécules sensibilisateurs comme OH, NO3 ou
O3. Au laboratoire, le temps de demi-réaction avec les radicaux OH
présents dans l'air est d'environ 1 jour; en revanche la constante de
vitesse est généralement beaucoup plus faible dans le cas des
réactions avec NO3 et O3. En principe, l'adsorption des HAP de
masse moléculaire élevée sur les particules carbonées devrait
stabiliser leur réaction avec les radicaux OH. La réaction des HAP
possédant 2 à 4 cycles avec NO3, réaction qui a lieu principalement
en phase gazeuse, conduit à la nitration de ces hydrocarbures, c'est-
à-dire à la formation de produits notoirement cancérogènes. Pour
certains d'entre eux, la photo-oxydation semble être plus rapide dans
l'eau que dans l'air. Les calculs basés sur la physico-chimie et la
biodégradabilité de ces hydrocarbures montrent que ceux qui possèdent
quatre cycles aromatiques ou davantage persistent dans
l'environnement.
1.5 Concentrations dans l'environnement et exposition humaine
Les HAP sont présent dans tout l'environnement et de nombreuses études
ont permis d'en mettre un certain nombre en évidence dans divers
compartiments.
1.5.1 Air
La concentration des HAP a tendance à être environ 10 fois plus élevée
l'hiver que l'été. En hiver, la source principale d'hydrocarbures
aromatiques polycyclique est le chauffage des habitations, alors qu'en
été, ce sont les gaz d'échappement des véhicules à moteur qui sont les
principaux responsables. Dans l'air de diverses agglomérations, on a
mis en évidence, pour un certain nombre d'hydrocarbures aromatiques
polycycliques, des concentrations moyennes de 1 à 30 ng/m3. Dans
certains grands centres urbains, où la circulation automobile est très
intense et où l'on utilise beaucoup la biomasse pour le chauffage des
habitations, comme Calcutta par exemple, on a trouvé des teneurs
allant jusqu'à 200 ng/m3 pour divers HAP. Sous les tunnels routiers,
on a mesuré des concentrations de 1 à 50 ng/m3. Le
cyclopenta(cd)pyrène et le pyrène étaient présents à des
concentrations atteignant 100 ng/m3. Dans une station de métro, on a
mesuré des concentrations allant jusqu'à 20 ng/m3. A proximité de
sources de pollution d'origine industrielle, la concentration moyenne
des divers HAP s'étalait de 1 à 10 ng/m3. La concentration du
phénanthrène pouvait atteindre environ 310 ng/m3.
La concentration de fond des HAP est d'au moins deux ordres de
grandeur plus faible qu'à proximité de sources telles que les
véhicules à moteur. A titre d'exemple, à 1100 m d'altitude, on a
obtenu des valeurs comprises entre 0,004 et 0,03 ng/m3.
1.5.2 Eaux superficielles et précipitations
On pense que la majeure partie des HAP présents dans l'eau y ont été
entraînés par ruissellement ou résultent de retombées atmosphériques
(petites particules) ou encore de l'abrasion de l'asphalte (grosses
particules). Il reste que, pour une étendue d'eau donnée, la majeure
partie des HAP n'a pas toujours la même origine. Eu général, la
plupart des échantillons d'eaux superficielles contiennent divers HAP
à des concentrations pouvant atteindre 50 ng/litre, mais dans des
cours d'eau extrêmement pollués, on a relevé des concentrations allant
jusqu'à 6 000 ng/litre. La teneur des eaux souterraine en HAP se situe
entre 0,02 et 1,8 ng/litre et dans des échantillons d'eau destinée à
la boisson, on a trouvé des valeurs du même ordre de grandeur. Les HAP
présents dans l'eau de boisson ont principalement pour origine le
revêtement d'asphalte des réservoirs et des canalisations.
Dans l'eau de pluie, on relève des concentrations comprises entre 10
et 200 ng/litre, et des teneurs allant jusqu'à 1000 ng/litre ont été
mesurées dans la neige et le brouillard.
1.5.3 Sédiments
La concentration des HAP dans les sédiments est généralement 10 fois
plus forte que dans les précipitations.
1.5.4 Sol
Les HAP présents dans le sol proviennent principalement des dépôts
atmosphériques, de la carbonisation des végétaux et du dépôt de
particules en suspension dans les effluents ou divers types de
déchets. L'ampleur de la pollution des sols dépend de facteurs tels
que le type de cultures auxquels ils sont soumis, la porosité et la
teneur en substances humiques.
A proximité des sources industrielles, on a trouvé, pour les divers
HAP, des concentrations dans le sol pouvant atteindre 1 g/kg. Pour les
HAP ayant une autre origine, par exemple les gaz d'échappement des
véhicules à moteur, la concentration dans le sol va de 2 à 5 mg/kg.
Dans les zones non polluées, la teneur du sol en HAP se situe entre 5
et 100 µg/kg.
1.5.5 Denrées alimentaires
Les denrées alimentaires crues ou plus généralement, non transformées,
ne contiennent normalement pas de grandes quantités d'HAP, mais il
peut s'en former lorsqu'on fait rôtir, griller, frire ou que l'on
prépare d'une manière ou d'une autre, ces produits. Les légumes
peuvent être contaminés par le dépôt de particules aéroportées ou par
le sol sur lequel ils ont poussé. D'après diverses mesures, la
concentration d'un certain nombre d'HAP dans la viande, le poisson,
les produits laitiers, les légumes, les fruits, les céréales et les
produits céréaliers, les pâtisseries et confiseries, les boissons
ainsi que les huiles et graisses animales et végétales, se situe entre
0,01 et 10 µg/kg. On a trouvé des concentrations supérieures à 100
µg/kg dans de la viande fumée et pouvant aller jusqu'à 86 µg/kg dans
du poisson fumé; dans des céréales fumées, les valeurs relevées
atteignaient 160 µg/kg. Dans de l'huile de coco on en a trouvé jusqu'à
460 µg/kg. Dans le lait maternel, les mesures ont donné des valeurs
comprises entre 0,003 et 0,03 µg/kg.
1.5.6 Organismes aquatiques
On sait que les organismes marins absorbent et accumulent les HAP
présents dans l'eau. Le degré de contamination dépend du développement
industriel et urbain ainsi que du trafic maritime dans la zone. Des
concentrations allant jusqu'à 7 mg/kg out été mises en évidence dans
des organismes aquatiques vivant à proximité de points de décharge
d'effluents industriels et la concentration moyenne d'HAP dans
l'organisme d'animaux aquatiques prélevés dans des zones contaminées
s'est révélée comprise entre 10 et 500 µg/kg, avec des pointes à 5
mg/kg.
Chez des animaux aquatiques prélevés dans des zones où la pollution
par des HAP n'avait pas d'origine précise, on a trouvé des valeurs
moyennes de 1 à 100 µg/kg, mais des valeurs supérieures sont
possibles, par exemple 1 mg/kg chez des homards du Canada.
1.5.7 Organismes terrestres
Chez des insectes, on a relevé des concentrations comprises entre 730
et 5 500 µg/kg. La teneur en HAP des déjections de lombrics varie
sensiblement selon le lieu: dans une région fortement industrialisée
de l'est de l'Allemagne, la teneur en benzo(a)pyrène des déjections de
lombrics atteignait 2 mg/kg.
1.5.8 Population générale
Les principales sources d'exposition non professionnelle sont les
suivantes: air pollué, fumée de feux et foyers non couverts, fumée de
tabac dispersée dans l'environnement, denrées alimentaires et eau de
boisson contaminées, utilisation de produits contaminés par des HAP.
On peut trouver dans l'air à l'intérieur des habitations des HAP qui
proviennent du chauffage ou de la présence de fumée de tabac, à des
concentrations moyennes de 1 à 100 ng/m3, avec des pointes à 2300
ng/m3.
On estime que l'apport d'HAP d'origine alimentaire est de 0,10 à 10 µg
par jour et par personne. La quantité totale de benzo(a)pyrène
absorbée quotidiennement avec l'eau de boisson est estimée à 0,0002 µg
par personne. Ce sont les céréales et les produits céréaliers qui
contribuent le plus à cet apport car ils sont un constituant majeur de
la ration alimentaire totale.
1.5.9 Exposition professionnelle
A proximité d'une batterie de fours à coke, on a relevé des teneurs en
benzo(a)pyrène allant de <0,1 à 100-200 µg/m3, avec des pointes à
environ 400 µg/m3. Dans les installations modernes de gazéification
de la houille, la concentration des HAP est en général de < 1 µg/m3
et de 30 µg/m3 au maximum. Des échantillons prélevés individuellement
dans la zone de travail de personnes affectées à divers postes dans
des raffineries de pétrole, ont révélé une exposition comprise entre
2,6 et 470 µg/m3. Dans des échantillons d'air prélevés dans des
ateliers de préparation de bitume, on a obtenu une concentration en
HAP totaux de 0,004 à 50 µg/m3. Lors de travaux d'enrobage de
chaussées, on a relevé sur le site des concentrations atmosphériques
en HAP totaux pouvant atteindre 190 µg/m3, avec une concentration
moyenne de 0,13 µg/m3. Dans une fonderie d'aluminium, la dosimétrie
individuelle indiquait des valeurs de 0,05-9,6 µg/m3 pour les HAP
totaux, mais dans les urines d'ouvriers travaillant dans une unité de
production d'aluminium, on n'en a relevé que de très petites
quantités. Dans une fonderie allemande, les échantillons d'air ambiant
contenaient des HAP à des concentrations pouvant atteindre 5 µg/m3.
Elles étaient respectivement égales à 3-40 µg/m3 dans des mines de
fer et à 4-530 µg/m3 dans des mines de cuivre. Dans les vapeurs de
cuisson d'une usine de produits alimentaires, la concentration en HAP
était comprise entre 0,07 et 26 µg/m3.
1.6 Cinétique et métabolisme
Les HAP sont absorbés au niveau des poumons, des voies digestives et
par la voie percutanée. La vitesse de résorption au niveau pulmonaire
dépend de la nature de l'hydrocarbure, de la granulométrie des
particules sur lesquelles il est adsorbé et de la composition du
substrat adsorbant. Les hydrocarbures adsorbés sur des particules sont
éliminés plus lentement des poumons que les hydrocarbures libres. Chez
les rongeurs, la résorption est rapide dans les voies digestives, mais
les métabolites, excrétés par la voie biliaire, finissent par
retourner dans l'intestin. Des études effectuées sur des rongeurs avec
des mélanges d'HAP ayant subi un postmarquage au 32p ont montré
qu'après absorption par la voie percutanée, les hydrocarbures
parvenaient jusqu'aux poumons où ils se liaient à l'ADN. Chez la
souris, la vitesse d'absorption percutanée dépend de la nature du
composé.
Quelle que soit la voie d'administration, les HAP se répartissent dans
tout l'organisme et on en retrouve dans pratiquement tous les organes
internes, mais plus particulièrement dans ceux qui sont riches en
lipides. Après injection intraveineuse à des rongeurs, les HAP sont
rapidement éliminés du courant sanguin, mais ils sont capables de
traverser la barrière foeto-placentaire et on en a décelé la présence
dans les tissus foetaux.
Les HAP ont un métabolisme complexe. En général, le composé initial
est époxydé puis transformé en phénol, diol ou tétrol qui peut
lui-même se conjuguer à l'acide sulfurique ou à l'acide glucuronique
(sulfuro- ou glucuro-conjugaison) ou encore au glutathion. La plupart
du temps, la métabolisation d'un hydrocarbure aromatique polycyclique
entraîne sa détoxication, mais certains d'entre eux subissent une
activation en composés susceptibles de se lier à l'ADN, principalement
des époxydes-diols, qui sont capables d'amorcer un processus de
cancérisation. Les métabolites et leurs conjugués sont excrétés dans
les urines et les matières fécales, mais les conjugués excrétés dans
la bile peuvent être hydrolysés par les enzymes de la flore
intestinale et être ensuite réabsorbés. On peut déduire des données
disponibles au sujet de la charge totale de l'organisme humain en HAP,
que ces hydrocarbures ne s'accumulent pas dans l'organisme et qu'ils
se renouvellent rapidement. Il faut exclure de cette conclusion les
HAP qui forment des liaisons covalentes avec certain constituants
tissulaires, notamment les acides nucléiques et que les processus de
réparation ne permettent pas d'éliminer.
1.7 Effets sur les mammifères de laboratoire et effets in vitro
La toxicité aiguë des HAP est faible à modérée. Un HAP bien
caractérisé, le naphtalène, a donné des valeurs de la DL50 par voie
orale ou intraveineuse égales à 100-500 mg/kg pc chez la souris et une
DL50 moyenne par voie orale de 2700 mg/kg pc chez le rat. Pour les
autres HAP, on a obtenu des valeurs similaires. Du naphtalène
administré en doses uniques à des souris, des rats et des hamsters, a
provoqué l'apparition d'une nécrose des bronchioles.
Des études à court terme ont révélé des anomalies hématologiques, dues
à une myélotoxicité dans le cas du benzo(a)pyrène et qui, en ce qui
concerne le dibenz(a,h)anthracène, consistaient en modifications
hémolymphatiques. Dans le cas du naphtalène, on a constaté une anémie.
Toutefois, une étude de 7 jours au cours de laquelle on a administré
du naphtalène à des souris par voie orale et intrapéritonéale, a
révélé l'existence d'une tolérance aux effets de cet hydrocarbure. On
n'a que rarement décrit des effets généraux dus à une longue
exposition à des HAP, car l'effet toxicologique retenu dans la plupart
des études correspondantes était la cancérisation. Aux doses où se
déclenche un processus de cancérisation, on observe également des
effets toxiques importants.
En étudiant les effets cutanés indésirables des HAP après application
sur l'épiderme, on a constaté que des hydrocarbures faiblement ou non
cancérogènes comme le pérylène, le benzo(e)pyrène, le phénanthrène, le
pyrène, l'anthracène, l'acénaphtène, le fluorène et le fluoranthène
étaient inactifs, alors que les hydrocarbures cancérogènes comme le
benz(a) anthracène, le dibenz(a,h)-anthracène et le benzo(a)pyrène
provoquaient une hyperkératose. Les vapeurs d'anthracène et de
naphtalène peuvent causer une légère irritation oculaire. Le
benzo(a)pyrène a provoqué une hypersensibilité de contact chez des
cobayes et des souris. Le benz(a)anthracène, le benzo(a)pyrène, le
dibenz(a,h)anthracène et le naphtalène se sont révélés embryotoxiques
chez la souris et le benzo(a)pyrène a également eu des effets
tératogènes et des effets sur la reproduction. On a fait de gros
efforts pour tenter de d'élucider les bases génétiques des effets
embryotoxiques du benzo(a)pyrène. On n'observe de morts foetales et de
malformations que si le système des monooxygénases du cytochrome p450
est inductible, chez la mère (avec migration transplacentaire) ou chez
l'embryon. On ne peut expliquer tous les effets observés par une
prédisposition génétique, mais chez la souris et le lapin, cet
hydrocarbure a une activité transplacentaire qui se traduit par des
adénomes pulmonaires et des papillomes cutanés dans la descendance des
animaux traités. On a également observé une diminution de la fécondité
et la destruction des ovocytes.
On a également procédé à de nombreuses études sur la génotoxicité des
HAP et sur leur aptitude à provoquer une transformation cellulaire. La
plupart des 33 HAP qui font l'objet de cette monographie sont
génotoxiques ou ont des chances de l'être. Les seuls composés pour
lesquels on ait obtenu des résultats négatifs dans toutes les épreuves
sont l'anthracène, le fluorène et le naphtalène. Dans le cas du
phénanthrène et du pyrène, l'irrégularité des résultats ne permet pas
de se prononcer avec certitude sur leur génotoxicité.
Les travaux très complets qui ont été consacrés à la cancérogénicité
des hydrocarbures aromatiques polycycliques montrent que 26 des 33
composés qui font l'objet de la présente monographie sont
effectivement cancérogènes ou soupçonnés de l'être (Tableau 2). Le
mieux connu de tous est le benzo(a)pyrène, qui a été étudié par toutes
les méthodes existantes sur sept espèces. Plus d'une douzaine d'études
ont été consacrées à l'anthracène, à l'anthanthrène, au
benz(a)anthracène, au chrysène, au dibenz(a,h)anthracène, au
dibenzo(a,i)pyrène, au 5-méthylehrysène, au phénanthrène et au pyrène.
Aux études spéciales sur l'immunotoxicité, la phototoxicité et
l'hépatotoxicité des HAP s'ajoutent un certain nombre d'articles sur
la toxicité oculaire du naphtalène. L'anthracène, le benzo(a)pyrène et
un certain nombre d'autres HAP sont phototoxiques pour la peau des
mammifères ou les cultures cellulaires in vitro, lorsqu'on les
applique sous rayonnement ultraviolet. D'une façon générale, on
considère que les HAP ont un effet immunodépresseur. Après
administration de benzo(a)pyrène à des souris, on a observé une forte
immunodépression dans la descendance de ces animaux pendant une
période pouvant atteindre 18 mois. On a également constaté un
accroissement de la régénération du tissu hépatique et une
augmentation du poids du foie. La formation de cataractes sous l'effet
du naphtalène chez des souris appartenant à des souches génétiquement
différentes, a été attribuée à l'inductibilité du cytochrome P 450.
Dès les années 30, on a proposé des modèles théoriques prenant en
compte un grand nombre de résultats expérimentaux, pour tenter de
prévoir le pouvoir cancérogène des hydrocarbures aromatiques
polycycliques à partir de leur structure moléculaire. Le premier de
ces modèles se fondait sur la forte réactivité chimique de certaines
doubles liaisons (théorie de la région K). Ultérieurement, on a tenté
une approche systématique du problème basée sur la synthèse chimique
des divers métabolites possibles et l'étude de leur activité mutagène.
Tableau 2. Résultats des épreuves de génotoxicité et de cancérogénicité
effectuées sur les 33 hydrocarbures aromatiques polycycliques étudiés.
Compound Genotoxicity Carcinogenicity
Acenaphthene (?) (?)
Acenaphthylene (?) No studies
Anthracene - -
Benz[a]anthracene + +
Benzo[a]fluorene (?) (?)
Benzo[a]pyrene + +
Benzo[b]fluoranthene + +
Benzo[b]fluorene (?) (?)
Benzo[c]phenanthrene (+) +
Benzo[e]pyrene + ?
Benzo[ghi]fluoranthene (+) (-)
Benzo[ghi]perylene + -
Benzo[j]fluoranthene + +
Benzo[l]fluoranthene + +
Chrysene + +
Coronene (+) (?)
Cyclopenta[cd]pyrene + +
Dibenzo[a,e]pyrene + +
Dibenz[a,h]anthracene + +
Dibenzo[a,h]pyrene (+) +
Dibenzo[a,i]pyrene + +
Dibenzo[a,l]pyrene (+) +
Fluoranthene + (+)
Fluorene - -
Indeno[1,2,3-cd]pyrene + +
1-Methylphenanthrene + -
5-Methylchrysene + +
Naphthalene - ?
Perylene + -
Phenanthrene (?) (?)
Pyrene (?) -
Triphenylene + -
+, résultat positif; -, résultat négatif; ?, résultat douteux
Parenthèses, résultat tiré d'une petite base de données.
Selon cette théorie, dite de la "région en baie", les époxydes
adjacents à une région en baie conduisent à la formation d'ions
carbonium très stables qui peuvent alkyler les bases nucléiques. Parmi
les autres théories, on peut citer la théorie de la "di-région" et
celle du "cation radical potentiel".
De nombreux HAP sont cancérogènes pour l'animal et pourraient
également l'être pour l'Homme. D'ailleurs, on a montré que
l'exposition à divers mélanges contenant des HAP augmentait
l'incidence des cancers dans les populations humaines en cause. Ce qui
est préoccupant, c'est que les HAP dont l'étude expérimentale a révélé
l'activité cancérogène chez l'animal, sont probablement aussi
cancérogènes pour l'Homme. Ces composés font apparaître des tumeurs
non seulement au point de contact, mais aussi à distance de ce point.
Le pouvoir cancérogène peut varier avec la voie d'administration. On a
proposé plusieurs méthodes pour évaluer le risque associé à une
exposition à ces composés, seuls ou en mélange. Sans se prononcer en
faveur de telle ou telle méthode, la monographie indique les données
nécessaires, les hypothèses et les conditions de validité etc. pour
trois d'entre elles qui ont été plus ou moins validées en vue d'une
évaluation quantitative du risque.
1.8 Effets sur l'Homme
Les HAP sont présents dans l'environnement et sur les lieux de travail
dans des conditions d'une telle complexité que pour étudier
l'exposition humaine à chacun de ces composés à l'état pur, on s'est
limité à des expériences sur des voluntaires, sauf dans le cas du
naphtalène que l'on utilise comme anti-mite sur les vêtements.
L'application cutanée d'anthracène, de fluoranthène et de phénanthrène
provoque des réaction cutanées spécifiques et le benzo(a)pyrène donne
naissance à des verrues régressives et réversibles que l'on a classées
comme étant de nature néoplasique. On connaît les effets généraux du
naphtalène en raison des nombreux cas d'ingestion accidentelle
auxquels il a donné lieu, notamment chez des enfants. La dose létale
est de 5 000 à 15 000 mg pour un adulte et de 2 000 mg sur deux jours
pour un enfant. Après contact cutané ou ingestion, l'intoxication se
caractérise par une anémie hémolytique aiguë, qui peut également
toucher le foetus par la voie transplacentaire.
On sait que le tabagisme est la cause la plus importante de cancers du
poumon et qu'il accroît également l'incidence des tumeurs de la
vessie, du bassinet du rein, de la cavité buccale, du pharynx, du
larynx et de l'oesophage. On estime en revanche que les HAP présents
dans les denrées alimentaires ne jouent pas un rôle important dans
l'apparition des cancers chez l'Homme. Dans les zones très
industrialisées, on a observé un accroissement de la charge de
l'organisme en HAP par suite de la pollution de l'air ambiant. Les
malades dont on traite le psoriasis par des applications de goudron,
sont également exposés à des HAP.
C'est en 1775 que l'on a, pour la première fois, avancé que
l'exposition professionnelle à la suie était une cause de cancer du
scrotum. Par la suite, on a remarqué que l'exposition aux goudrons et
aux paraffines provoquait des cancers de la peau. Le poumon est
désormais la principale localisation des cancers dus aux HAP, les
cancers cutanés étant devenus plus rares par suite des progrès de
l'hygiène individuelle.
On a effectué des études épidémiologiques sur l'exposition aux HAP
chez des ouvriers de fours à coke pendant la cokéfaction et la
gazéification de la houille, chez des ouvriers d'ateliers de
préparation d'asphalte, de fonderies, de fours à aluminium ou encore
chez des travailleurs exposés aux gaz d'échappement de moteurs diesel.
On a constaté une augmentation des tumeurs pulmonaires dues aux HAP
chez les ouvriers des fours à coke et des ateliers de préparation de
l'asphalte, de même que chez ceux qui travaillaient près des cuves de
réduction électrolytique de l'alumine par le procédé Söderberg. C'est
chez les ouvriers des fours à coke que l'on a mis en évidence le
risque le plus élevé, avec un rapport comparatif de mortalité (SMR) de
195. Plusieurs études ont permis d'établir des relations dose-réponse.
Dans les usine d'aluminium, on a observé non seulement des cancers de
la vessie, mais aussi des symptômes asthmatiformes, des anomalies de
la fonction pulmonaire et des bronchites chroniques. Chez les ouvriers
des fours à coke, il y avait diminution des taux d'immunoglobulines
sériques et dépression des fonctions immunitaires. Par ailleurs, ou a
signalé l'apparition d'une cataracte après cinq ans d'exposition au
naphtalène.
On a mis au point un certain nombre de méthodes pour évaluer
l'exposition interne aux HAP. Dans la plupart de ces études, on a
procédé au dosage des métabolites urinaires de ces composés:
thioéthers, 1-naphtol, ß-naphtylamine, hydroxyphénantrènes et
1-hydroxypyrène. Ce dernier composé est largement utilisé comme
indicateur biologique d'une exposition à des HAP.
Les effets génotoxiques des HAP ont été étudiés en recherchant la
présence de substances mutagènes dans les urines et les matières
fécales et celle de micronoyaux, d'aberrations chromosomiques et
d'échanges entre chromatides-soeurs dans les lymphocytes du sang
périphérique. En outre, on a dosé les adduits du benzo(a)pyrène et de
l'ADN dans les lymphocytes du sang périphérique et dans d'autres
tissus, ainsi que ceux que cet HAP forme avec des protéines comme
l'albumine. On a également procédé au dosage des anticorps dirigés
contre les adduits de l'ADN.
Plusieurs études se sont donné pour but de rechercher s'il était
possible d'utiliser la présence de 1-hydroxypyrène dans l'urine et
d'adduits de l'ADN dans les lymphocytes comme marqueurs d'une
exposition à des HAP. Il est plus facile de doser le 1-hydroxypyrène
urinaire que les adduits de l'ADN. Ce marqueur a en outre l'avantage
d'être moins sujet aux variations interindividuelles et de permettre
de repérer des expositions plus faibles. Les deux types de marqueurs
ont été utilisés pour évaluer l'exposition humaine dans divers
environnements. Ainsi, on a relevé une augmentation de la
concentration urinaire de 1-hydroxypyrène sur les divers lieux de
travail de cokeries, d'usines de production d'aluminium, d'ateliers
d'imprégnation du bois, de fonderies et d'ateliers de préparation
d'asphalte. L'exposition la plus forte a été observée chez les
ouvriers des fours à coke et chez ceux qui travaillent à
l'imprégnation du bois avec du créosote. Chez ces travailleurs,
l'exposition est à 95% percutanée, alors que dans le reste de la
population, les HAP sont absorbés principalement par la voie
alimentaire et par la voie respiratoire lors de la consommation de
tabac.
L'estimation du risque associé à une exposition à des HAP repose sur
l'évaluation de cette exposition et sur les résultats des études
épidémiologiques. En ce qui concerne les ouvriers des fours à coke, on
aboutit à un risque relatif de cancer du poumon égal à 15,7. En
procédant de la même manière pour la population générale, on trouve
que le risque, pour un individu donné, de faire un cancer du poumon au
cours de son existence est de 10-4 à 10-5 par ng de benzo(a)pyrène
par m3 d'air. En d'autres termes, il y a environ une personne sur 10
000 ou 100 000 qui va faire un cancer au cours de sa vie par suite de
la présence de benzo(a)pyrène dans l'air ambiant.
1.9 Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
S'il y a absorption simultanée de lumière UV ou de lumière visible,
les HAP peuvent provoquer des effets toxiques aigus sur les poissons
et sur des invertébrés aquatiques comme les daphnies. La toxicité des
HAP peut être modifiée par dégradation et métabolisation. A faible
concentration, les HAP peuvent stimuler la croissance des
microorganismes et des algues. Le composé le plus toxique pour les
algues est le benz(a)anthracène, un hydrocarbure tétracyclique. La
valeur de la CE50 pour ce composé (réduction de 50% des paramètres
vitaux) est égale à 1-29 µg/litre. Dans le cas du benzo(a)pyrène, un
composé pentacyclique, elle est égale à 5-15 µg/litre. Toujours en ce
qui concerne les algues, la CE50 pour la plupart des HAP
tricycliques est égale à 240-290 µg/litre. Le naphtalène, qui est
bicyclique, est le moins toxique, avec une CE50 de 2 800-34 000
µg/litre.
Il n'y a pas de différence de sensibilité bien nette entre les divers
groupes taxonomiques d'invertébrés tels que les crustacés, les
insectes, les mollusques, les polychètes et les échinodermes. Le
naphtalène est le moins toxique avec une CL50 à 96 h de 100 à 2
300/µg/litre. Pour trois HAP tricycliques, la valeur de ce même
paramètre varie de <1 à 3 000 µg/litre. L'anthracène pourrait être
plus toxique que les autres HAP tricycliques avec une CL50 à 24 h
comprise entre <1 et 260 µg/litre. Pour les composés à quatre, cinq et
six noyaux aromatiques, la CL50 à 96 h est comprise entre 0,2 et 1
200 µg/litre. Des effets toxiques aigus (CL50) ont été observés chez
des poissons à des concentrations de 110 à > 10 000 µg/litre de
naphtalène, de 30 à 40 000 µg/litre d'HAP tricycliques (anthracène,
2,8-360 µg/litre) et de 0,7 à 26 µg/litre d'HAP tétra- et
pentacycliques.
Chez des poissons vivant à l'état sauvage, on a attribué la présence
de tumeurs hépatiques à la contamination des sédiments par des HAP à
la concentration de 250 mg/kg. On a également provoqué l'apparition de
tumeurs chez des poissons par exposition en laboratoire. L'exposition
des poissons à certains HAP peut aussi provoquer chez eux des
anomalies physiologiques et perturber leur croissance, leur
reproduction, leur locomotion et leur respiration.
1. RESUMEN
1.1 Selección de compuestos para esta monografía
Los hidrocarburos aromáticos policiclicos (HAP) constituyen una clase
muy amplia de compuestos, y durante la combustión incompleta o la
pirolisis de materia orgánica pueden liberarse cientos de sustancias
distintas, que son una fuente importante de exposición humana. Los
estudios de diversas matrices aplicables al medio ambiente, como los
efluentes de la combustión de carbón, los gases de escape de los
vehículos de motor, el aceite lubricante de motores usados y el humo
del tabaco, han demostrado que los HAP de esas mezclas son los
principales responsables de su potencial carcinogénico.
En las mezclas casi siempre hay presentes HAP. Debido a que la
composición de tales muestras es compleja y varía con el proceso de
formación, no es posible examinar con detalle en la presente
monografía todas las mezclas que contienen HAP. Así pues, se
seleccionaron 33 compuestos distintos (31 HAP originales y dos
derivados alquilo) para evaluarlos tomando como base la disponibilidad
de datos pertinentes sobre los efectos finales toxicológicos y/o la
exposición (Cuadro 1). Sin embargo, dado que sólo se disponía para las
mezclas de estudios epidemiológicos, que son imprescindibles para la
evaluación del riesgo, en las Secciones 8 y 10 se presentan los
resultados de estudios de mezclas de HAP, en contraposición con el
resto de la monografía.
Se han publicado numerosos artículos y reseñas sobre la presencia,
distribución y transformación de HAP en el medio ambiente y sobre sus
efectos ecotoxicológicos y toxicológicos. Solamente se citan en la
presente monografía referencias de los 10-15 últimos años, a menos que
no se dispusiera de otra información; en relación con los estudios más
antiguos y con otra información se citan reseñas.
1.2 Identidad, propiedades físicas y químicas y métodos analíticos
El término "hidrocarburos aromáticos policíclicos" se refiere en
general a una clase muy amplia de compuestos orgánicos que contienen
dos o más anillos aromáticos condensados formados por átomos de
carbono y de hidrógeno. A temperatura ambiente, los HAP son sólidos.
Las características comunes de la clase son puntos de fusión y de
ebullición elevados, presión de vapor baja y solubilidad en agua muy
baja, que tiende a disminuir con el aumento del peso molecular. Los
HAP son solubles en muchos disolventes orgánicos y muy lipófilos.
Desde el punto de vista químico son bastante inertes. Las reacciones
que tienen interés con respecto a su destino en el medio ambiente y a
las posibles fuentes de pérdidas durante el muestren atmosférico son
la fotodescomposición y las reacciones con óxidos de nitrógeno, ácido
nítrico, óxidos de azufre, ácido sulfúrico, ozono y radicales
hidroxilo.
Cuadro 1. Hidrocarburos aromáticos policíclicos evaluados en esta monografía
Nombre común Nombre CAS Sinónimoa No de registro CAS
Acenaftileno Acenaftileno 91-20-3
Acenafteno Acenaftileno, 1.2-dihidro- 208-96-8
Antantreno Dibenzo[def,mno]criseno 191-264
Antraceno Antraceno 120-12-7
Benz[a]antraceno Benz[a]antraceno 1,2-Benzantraceno, tetrafeno 56-55-3
Benzo[a]fluoreno 11H-Benzo[a]fluoreno 1,2-Benzofluoreno 238-84-6
Benzo[b]fluoreno 11H-Benzo[b]fluoreno 2,3-Benzofluoreno 243-17-4
Benzo[b]fluoranteno Benz[e]acefenantrileno 3,4-Benzofluoranteno 205-99-2
Benzo[ghl]fluoranteno Benzo[ghl]fluoranteno 2,13-Benzofluoranteno 203-12-3
Benzo[j]fluoranteno Benzo[j]fluoranteno 10,11-Benzofluoranteno 205-82-3
Benzo[k]fluoranteno Benzo[k]fluoranteno 11,12-Benzofluoranteno 207-08-9
Benzo[ghi]perileno Benzo[ghi]perileno 1,12-Benzoperileno 191-24-2
Benzo[c]fenantreno Benzo[c]fenantreno 3,4-Benzofenantreno 195-19-7
Benzo[a]pireno Benzo[a]pireno 3,4-Benzopirenob 50-32-8
Benzo[e]pireno Benzo[e]pireno 1.2-Benzopireno 192-97-2
Criseno Criseno 1,2-Benzofenantreno 218-01-9
Coroneno Coroneno Hexabenzobencano 191-07-1
Ciclopenta[cd]pireno Ciclopenta[cd]pireno Ciclopenteno[cd]pireno 27208-37-3
Dibenz[a,h]antraceno Dibenz[a,h]antraceno 1,2:5.6-Dibenzantraceno 53-70-3
Dibenzo[a,e]pireno Naftol[1,2:3,4-def]criseno 1,2:4,5-Dibenzopireno 192-65-4
Dibenzo[a,h]pireno Dibenzo[b,def]criseno 3,4:8,9-Dibenzopireno 189-64-0
Dibenzo[a,i]pireno Benzo[rsf]pentafeno 3,4:9,10-Dibenzopireno 189-55-9
Dibenzo[a,l]pireno Dibenzo[def,p]criseno 1,2:3,4-Dibenzopireno 191-30-0
Fluoranteno Fluoranteno 206-44-0
Fluoreno 9H-Fluoreno 86-73-7
Indeno[1,2,3-cd]pireno Indeno[1,2,3-cd]pireno 2,3-o-Fenilenpireno 193-39-5
5-Metilcriseno Criseno, 5-metil- 3697-24-3
1-Metilfenantreno Fenantreno, 1-metil- 832-69-9
Naftaleno Naftaleno 91-20-3
Perileno Perileno peri-Dinaftaleno 198-55-0
Penantreno Fenantreno 85-01-8
Pireno Pireno Benzo[def]fenantreno 129-00-0
Trifenileno Trifenileno 9,10-Benzofenantreno 217-59-4
Cuadro 1 (sigue).
Han notificado listas muy amplias de sinónimos el CIIC (1983) y Loening y Merritt (1990).
a Sinónimo común que aparece en la bibliografia.
c También denominado benzo[def]criseno.
El aire del ambiente se muestrea recogiendo partículas suspendidas en
filtros de fibra de vidrio, de politetrafluoroetileno o de fibra de
cuarzo mediante muestreadores de alto volumen o pasivos. Los HAP en
fase de vapor, que podrían volatilizarse de los filtros durante el
muestreo, se suelen retener por adsorción en espuma de poliuretano. La
fase de muestren es con diferencia la fuente más importante de
variabilidad de los resultados.
En el lugar de trabajo se toman muestras con tasas de flujo bajas; se
recogen las partículas en filtros de fibra de vidrio o de
politetrafluoroetileno y los vapores en resina XAD-2 de amberlita. Los
dispositivos para el muestren de gases de chimenea constan de un
filtro de fibra de vidrio o de fibra de cuarzo en la parte frontal de
un refrigerador para recoger la materia condensable y un cartucho
adsorbente (por lo general XAD-2). Las muestras de gases de escape de
los vehículos se toman en condiciones de laboratorio, con ciclos de
conducción normalizados que simulan las condiciones en carretera. Las
emisiones se recogen sin diluir o bien una vez diluidas con aire frío
filtrado.
Se han descrito numerosas técnicas de extracción y purificación. En
función de la matriz, los HAP se extraen de las muestras con un
aparato de Soxhlet, por ultrasonidos, mediante reparto liquido-liquido
o, tras la disolución o la digestión alcalina de la muestra, con un
disolvente selectivo. También se ha utilizado la extracción de fluidos
supercríticos a partir de diversos sólidos del medio ambiente. La
eficacia de la extracción depende en gran medida del disolvente
utilizado, y muchos de los que se utilizaban en el pasado no eran
apropiados. Las muestras extraídas se suelen purificar por
cromatografía en columna, en particular sobre alúmina, silicagel o
Sephadex LH-20, pero también por cromatografía en capa fina.
La identificación y la cuantificación se realizan habitualmente
mediante cromatografía de gases con detección por ionización de llama
o mediante cromatografía liquida de alta resolución (HPLC) con
detección por ultravioleta o fluorescencia, generalmente en serie. En
la cromatografía de gases se utilizan columnas capilares de sílice
fundido con polisiloxanos (SE-54 y SE-52) como fases estacionarias; en
la HPLC se suelen utilizar columnas de sílice-C18. Con frecuencia se
acopla un detector de espectrometría de masas a una columna de fase
gaseosa a fin de confirmar la identidad de los picos.
La elección de los HAP que se han de determinar depende de la
finalidad de la medición, por ejemplo para estudios orientados a la
salud o ecotoxicológicos, o bien para investigar las fuentes. Es
posible que se exija o se recomiende la realización de pruebas para
distintos conjuntos de compuestos a nivel nacional e internacional.
1.3 Fuentes de exposición humana y ambiental
Es poca la información disponible acerca de la producción y
elaboración de HAP, pero es probable que sólo se desprendan pequeñas
cantidades como resultado directo de esas actividades. Los HAP que se
encuentran se utilizan sobre todo como productos intermedios en la
producción de cloruro de polivinilo y de agentes plastificantes
(naftaleno), pigmentos (acenafteno, pireno), tintes (antraceno,
fluoranteno) y plaguicidas (fenantreno).
Las mayores emisiones de HAP se derivan de la combustión incompleta de
materia orgánica durante procesos industriales y en otras actividades
humanas, en particular:
- elaboración de carbón, de petróleo crudo y de gas natural,
incluida la coquificación de carbón, la conversión de carbón, el
refinado de petróleo y la producción de negro de humo, de
creosota, de alquitrán de hulla y de betún;
- producción de aluminio, de hierro y de acero en fábricas y
fundiciones;
- calefacción en centrales de energía y en residencias y ocinado;
- combustión de basuras;
- tráfico de vehículos de motor; y
- humo de tabaco en el medio ambiente.
Los HAP, especialmente los de mayor peso molecular, cuando se
incorporen al medio ambiente a través de la atmósfera se adsorben en
las partículas en suspensión. La hidrosfera y la geosfera se ven
afectadas de manera secundaria por la deposición húmeda y seca. La
madera conservada con creosota es otro fuente de liberación de HAP en
la hidrosfera, y la deposición de desechos contaminados, como fangos
de alcantarillado y cenizas en suspensión, contribuye a las emisiones
de HAP en la geosfera. Hay poca información acerca del paso de HAP a
la biosfera. Hay HAP presentes naturalmente en la turba, el lignito,
el carbón y el petróleo crudo. La mayoría de los HAP de las antracitas
están fuertemente unidos a la estructura del carbón y no se pueden
lixiviar.
La liberación de HAP en el medio ambiente se ha determinado mediante
la identificación de un perfil característico de su concentración,
pero esto sólo ha sido posible en un pequeño número de casos. Con
frecuencia se ha utilizado el benzo[a]pireno como indicador de HAP,
especialmente en estudios más antiguos. En general, las emisiones de
HAP son solamente estimaciones basadas en datos más o menos fidedignos
y apenas den una idea general de la exposición.
Las fuentes más importantes de HAP son las siguientes:
Coquificación de carbón: Las emisiones de HAP en suspensión en el
aire procedentes de la coquificación de carbón en Alemania han
disminuido considerablemente en los 10 últimos años gracias a las
mejoras técnicas de las instalaciones existentes, al cierre de otras
antiguas y a la menor producción de coque. Se supone que la situación
es análoga en el resto de Europa occidental, el Japón y los Estados
Unidos, pero no se dispone de datos.
Producción de aluminio (principalmente en ánodos especiales de
carbón), de hierro y de acero y los aglutinantes utilizados en la
arena de moldeo de las fundiciones: La información disponible es
escasa.
Cocinas y calefacción de viviendas: Los principales componentes que
se emiten son fenantreno, fluoranteno, pireno y criseno. Las emisiones
de los hornillos de leña son 25-1000 veces superiores a las que se
producen en los de carbón, y en las zonas donde predomina el uso de
leña en las viviendas la mayor proporción de HAP en suspensión puede
derivarse de esta fuente, especialmente en invierno. Por consiguiente,
se supone que la liberación de HAP en la calefacción de las viviendas
es una fuente importante en los países en desarrollo, donde con
frecuencia se quema biomasa en hornillos relativamente simples.
Cocinado: Pueden emitirse HAP durante la combustión incompleta de
los combustibles, del aceite de cocinar y de los alimentos que se
cocinen.
Tráfico de vehículos de motor: Los principales compuestos que se
liberar de los vehículos de gasolina son el fluoranteno y el pireno,
mientras que en los gases de escape de los vehículos de motor diesel
abundan el naftaleno y el acenafteno. Aunque los motores de gasolina
emiten una proporción elevada de ciclopenta[cd]pireno, su
concentración en los gases de escape de los motores diesel está apenas
por encima del límite de detección. Las tasas de emisión, que dependen
de la sustancia, el tipo de vehículo, el estado de su motor y las
condiciones de la prueba, oscilan entre unos pocos nanogramos por
kilómetro y > 1000 mg/km. Las emisiones de HAP de los vehículos de
motor se reducen enormemente con la instalación de catalizadores.
Incendios forestales: En los países con grandes superficies
forestales, los incendios pueden contribuir de manera importante a las
emisiones de HAP.
Centrales eléctricas de carbón: Los HAP que se liberar en la
atmósfera a partir de dichas centrales son sobre todo compuestos de
dos y tres anillos. En las zonas contaminadas, los niveles de HAP en
el aire pueden ser más elevados que los de los gases de chimenea.
Incineración de basuras: Las emisiones de HAP en los gases
procedentes de este tipo de incineración fueron en varios países < 10
mg/m3.
1.4 Transporte, distribución y transformación en el medio ambiente
Son varios los factores de distribución y de transformación de los que
depende el destino tanto de los HAP por separado como de las mezclas.
Los procesos de distribución más importantes son el reparto entre el
agua y el aire, entre el agua y los sedimentos y entre el agua y la
biota.
Puesto que los HAP son hidrófobos, con escasa solubilidad en el agua,
su afinidad por la fase acuática es muy pequeña; sin embargo, a pesar
del hecho de que la mayoría de los HAP se liberan en el medio ambiente
a través de la atmósfera, también se encuentran concentraciones
considerables en la hidrosfera, debido a sus bajas constantes de la
ley de Henry. Como la afinidad de los HAP por las fases orgánicas es
mayor que la que tienen por el agua, sus coeficientes de reparto entre
disolventes orgánicos como el octanol y el agua son elevados. Su
afinidad por las fracciones orgánicas de los sedimentos, del suelo y
de la biota es también alta, por lo que se acumulan en los organismos
del agua y los sedimentos y en sus alimentos. No se conoce con
claridad la importancia relativa de su ingesta con los alimentos y el
agua. En Daphnia y en los moluscos, hay una correlación positiva
entre la acumulación de HAP procedentes del agua y el coeficiente de
reparto octanol:agua (Kow). Sin embargo, en los peces y las algas
capaces de metabolizar los HAP no hay correlación entre las
concentraciones internas de distintos HAP y el Kow.
No se ha observado bioamplificación -aumento de la concentración de
una sustancia en animales de niveles tróficos sucesivos de las cadenas
alimentarias- en sistemas acuáticos y no cabe prever que se produzca,
puesto que la mayoría de los organismos tienen un potencial de
biotransformación elevado para los HAP. El mayor potencial de
biotransformación se observa en los organismos de los niveles tróficos
más altos de las cadenas alimentarias.
Los HAP se descomponen por fotodegradación, biodegradación por
microorganismos y metabolismo en la biota de niveles más altos. Aunque
la última rata de transformación tiene escasa importancia para el
destino global de los HAP en el medio ambiente, es una vía importante
para la biota, debido a que pueden formarse metabolitos
carcinogénicos. Dado que los HAP son químicamente estables, sin grupos
reactivos, la hidrólisis no interviene en su degradación. Hay pocas
pruebas normalizadas para la biodegradación de los HAP. En general, se
biodegradan en condiciones aerobias, registrando un fuerte aumento la
tasa de biodegradación con el número de anillos aromáticos. En
condiciones anaerobias la degradación es mucho más lenta.
Los HAP se fotooxidan en el aire y en el agua en presencia de
radicales sensibilizantes como OH, NO3 y O3. En condiciones de
laboratorio, la semivida de la reacción con radicales OH presentes en
el aire es de alrededor de un día, mientras que las reacciones con
NO3 y O3 suelen tener unas constantes de velocidad mucho más
bajas. La adsorción de HAP de peso molecular alto en partículas
carbonosas en el medio ambiente debería estabilizar la reacción con
radicales OH. La reacción, que tiene lugar sobre todo en la fase
gaseosa, de HAP de entre dos y cuatro anillos con NO3 da lugar a la
formación de nitro-HAP, que son conocidos como mutágenos. Parece que
la fotooxidación de algunos HAP en el agua es más rápida que en el
aire. Los cálculos basados en parámetros fisicoquímicos y de
degradación indican que los HAP con cuatro o más anillos aromáticos
persisten en el medio ambiente.
1.5 Niveles ambientales y exposición humana
Los HAP están omnipresentes en el medio ambiente, habiéndose detectado
en numerosos estudios diversos HAP por separado en distintos
compartimentos.
1.5.1 Aire
Los niveles de cada uno de los HAP tiendan a ser más elevados en
invierno que en verano por lo menos en un orden de magnitud. La fuente
predominante durante el invierno es la calefacción de las viviendas y
la del verano el tráfico urbano de vehículos de motor. Se detectaron
concentraciones medias de distintos HAP de 1-30 ng/m3 en la
atmósfera de diversas zonas urbanas. En grandes ciudades con un
tráfico intenso de vehículos de motor y utilización abundante de
combustibles a base de biomasa, como Calcuta, se encontraron niveles
de hasta 200 ng/m3 de distintos HAP. En túneles de carreteras se
detectaron concentraciones de hasta 1-50 ng/m3. Había
ciclopenta[cd]-pireno y pireno en concentraciones de hasta 100
ng/m3. En una estación de metro se midieron concentraciones de HAP
de hasta 20 ng/m3. En las cercanías de fuentes industriales, las
concentraciones medias de los distintos HAP oscilaban entre 1 y 10
ng/m3. Había fenantreno presente hasta un máximo aproximado de 310
ng/m3.
Los valores básicos de los HAP son por lo menos uno o dos órdenes de
magnitud menores que los obtenidos cerca de fuentes como el tráfico de
vehículos de motor. Por ejemplo, los niveles a 1100 m oscilaban entre
0,004 y 0,03 ng/m3.
1.5.2 Agua superficial y precipitación
La mayor parte de los HAP presentes en el agua proceden al parecer del
agua de escorrentía urbana, de la precipitación atmosférica
(partículas más pequeñas) y de la abrasión del asfalto (partículas
mayores). La principal fuente de HAP, sin embargo, varia para
distintas masas de agua. En general, la mayoría de las muestras de
agua superficial contienen distintos HAP en concentraciones de hasta
50 ng/litro, pero los ríos muy contaminados tenían concentraciones de
hasta 6000 ng/litro. Los niveles de HAP en el agua freática son del
orden de 0,02-1,8 ng/litro, y las muestras de agua potable contienen
concentraciones del mismo orden de magnitud. Las principales fuentes
de los HAP presentes en el agua potable son los depósitos y las
tuberías con revestimiento de asfalto.
Los niveles de HAP por separado en el agua de lluvia oscilaban entre
10 y 200 ng/litro, mientras que en la nieve y la niebla se detectaron
concentraciones de hasta 1000 ng/litro.
1.5.3 Sedimentos
Las concentraciones de los distintos HAP cm los sedimentos eran por lo
general un orden de magnitud superiores a las presentes cm la
precipitación.
1.5.4 Suelo
Las principales fuentes de los HAP presentes en el suelo son la
deposición atmosférica, la carbonización de material vegetal y la
deposición a partir de aguas residuales y desechos particulados. El
grado de contaminación del suelo depende de factores como su cultivo,
su porosidad y su contenido de sustancias húmicas.
Cerca de fuentes industriales se han encontrado concentraciones de
distintos HAP de hasta 1 g/kg de suelo. La concentración en el suelo a
partir de otras fuentes, como los gases de escape de los automóviles,
son del orden de 2-5 mg/kg. En zonas no contaminadas, los niveles de
HAP eran de 5-100 µg/kg de suelo.
1.5.5 Alimentos
Los alimentos credos no suelen contener niveles elevados de HAP, pero
se forman al elaborarlos, asados o cocerlos en horno o freírlos. Las
hortalizas pueden contaminarse por la deposición de partículas de la
atmósfera o por el crecimiento en suelo contaminado. Las
concentraciones de los distintos HAP en la carne, el pescado, los
productos lácteos, las frutas y hortalizas, los cereales y sus
productos, los dulces, las bebidas y las grasas y los aceites animales
y vegetales eran del orden de 0,01-10 µg/kg. Se han detectado
concentraciones de más de 100 µg/kg en carne ahumada y de hasta 86
µg/kg cm pescado ahumado; los cereales ahumados contenían hasta 160
µg/kg. En el aceite de coco se encontraron concentraciones de hasta
460 µg/kg. Los niveles en la leche materna humana eran de 0,003-0,03
µg/kg.
1.5.6 Organismos acuáticos
Se sabe que los organismos marinos adsorben y acumulan HAP del agua.
El grado de contaminación depende del desarrollo industrial y urbano y
de los movimientos de transporte marítimo. Se han detectado
concentraciones de HAP de hasta 7 mg/kg en organismos acuáticos que
vivían cerca de efluentes industriales, y los niveles medios de HAP en
los animales acuáticos muestreados en lugares contaminados fueron de
10-500 µg/kg, aunque también se detectaron niveles de hasta 5 mg/kg.
Los niveles medios de HAP en animales acuáticos muestreados en
diversos lugares con fuentes sin especificar de dichos compuestos
fueron de 1-100 µg/kg, pero se encontraron concentraciones de hasta 1
mg/kg, por ejemplo en langostas en el Canadá.
1.5.7 Organismos terrestres
Las concentraciones de HAP en insectos oscilaban entre 730 y 5500
µg/kg. El contenido de las heces de las lombrices de tierra depende
considerablemente del lugar: las de una región muy industrializada de
Alemania oriental contenían concentraciones de benzo[a]pireno de hasta
2 mg/kg.
1.5.8 Población general
Las principales fuentes de exposición no profesional son las
siguientes: atmósfera contaminada, humo de fuegos abiertos y del
cocinado, humo de tabaco en el medio ambiente, alimentos y agua
potable contaminados y utilización de productos contaminados. Pueden
encontrarse HAP en el aire de espacios cerrados como consecuencia de
la calefacción de las viviendas y del humo del tabaco del medio
ambiente en concentraciones medias de 1-100 ng/m3, con un máximo de
2300 ng/m3.
Se ha estimado que la ingesta de distintos HAP con los alimentos es de
0,10-10 µg/día por persona. La ingesta diaria total de benzo[a]-pireno
con el agua potable se estimó en 0,0002 µg/persona. Los cereales y
productos derivados son los que más contribuyen a la ingesta de HAP
con los alimentos, por ser el principal componente de la alimentación
total.
1.5.9 Exposición profesional
Cerca de una batería de hornos de coque, los niveles de
benzo[a]-pireno oscilaban entre < 0,1 y 100-200 µg/m3, con un máximo
aproximado de 400 µg/m3. En los sistemas modernos de gasificación
del carbón, la concentración de HAP suele ser < 1 µg/m3, con un
máximo de 30 µg/m3. Las muestras personales tomadas de operadores de
equipo de refinerías de petróleo mostraron una exposición a 2,6470
µg/m3. En muestras de aire tomadas cerca de instalaciones de
elaboración de asfalto en refinerías, los niveles totales de HAP
fueron de 0,004-50 µg/m3. En las proximidades de obras de
pavimentación de carreteras, las concentraciones totales de HAP en
muestras personales de aire eran de hasta 190 µg/m3, con un valor
medio en las muestras de aire de la zona de 0,13 µg/m3. Los niveles
de HAP en las muestras personales de aire tomadas en una fundición de
aluminio eran de 0,059,6 µg/m3, pero las muestras de orina de los
trabajadores de una fábrica de aluminio contenían niveles muy bajos.
Las muestras de aire de la zona contenían concentraciones de HAP de
hasta 5 µg/m3 en una fundición alemana, 3-40 µg/m3 en minas de
hierro µg/m3 y 4530 µg/m3 en minas de cobre. Las concentraciones
de HAP en humos de cocinado en una fábrica de productos alimenticios
oscilaban entre 0,07 y 26 µg/m3.
1.6 Cinética y metabolismo
Los HAP se absorben por las vías respiratorias, el aparato digestivo y
la piel. La tasa de absorción por los pulmones depende del tipo de
HAP, el tamaño de las partículas sobre las que están adsorbidos y la
composición del adsorbente. Los HAP adsorbidos sobre partículas se e
'laminan de los pulmones con mayor lentitud que los hidrocarburos
libres. En el aparato digestivo se produce una absorción rápida en los
roedores, pero los metabolitos vuelven al intestino mediante la
excreción biliar. En estudios de absorción percutánea de mezclas de
HAP marcados con 32P en roedoras se observó que los componentes de las
mezclas negaban a los pulmones, donde se unían al ADN. La tasa de
absorción percutánea en ratones varia en función del compuesto.
Los HAP se distribuyen ampliamente en todo el organismo tras la
administración por cualquier vía y se encuentran en casi todos los
órganos internos, particularmente en los ricos en lípidos. Los HAP
inyectados por vía intravenosa se eliminan con rapidez de la corriente
sanguínea en los roedores, pero pueden atravesar la barrera
placentaria y se han detectado en tejidos fetales.
El metabolismo de los HAP es complejo. En general, los compuestos
originales se convierten mediante epóxidos intermedios en fenoles,
dioles y tetroles, que a su vez pueden conjugarse con los ácidos
sulfúrico y glucurónico o con el glutatión. El metabolismo produce en
su mayor parte una desintoxicación, pero algunos HAP se activan a
especies que se unen al ADN, principalmente diolepóxidos, que pueden
inducir tumores. Los metabolitos de los HAP y sus cunjugados se
excretan en la orina y las heces, pero los conjugados que se excretan
en la bilis pueden hidrolizarse por la acción de enzimas de la flora
intestinal y reabsorberse. De la información disponible acerca de la
carga total en el cuerpo humano cabe deducir que los HAP no persisten
en el organismo y que su ciclo metabólico es rápido. De la deducción
anterior están excluidos los grupos de HAP que se unen por enlaces
covalentes a elementos constitutivos de los tejidos, en particular
ácidos nucleicos, y que no se eliminan por reparación.
1.7 Efectos en mamíferos de laboratorio y en sistemas de prueba in vitro
La toxicidad aguda de los HAP parece ser de moderada a baja. Un
producto bien caracterizado, el naftaleno, mostró valores para la
DL50 por vía oral e intravenosa de 100-500 mg/kg de peso corporal en
ratones y una DL50 media por vía oral de 2700 mg/kg de peso corporal
en ratas. Los valores para otros HAP son semejantes. Con dosis altas
únicas de naftaleno se indujo en ratones, ratas y hámsteres necrosis
bronquiolar.
En estudios de corta duración se pusieron de manifiesto efectos
hematológicos adversos en forma de mielotoxicidad con el
benzo[a]-pireno, cambios hemolinfáticos con el dibenz[a,h]antraceno y
anemia con el naftaleno; sin embargo, en un estudio de siete días en
el que se administró a ratones naftaleno por vía oral e
intraperitoneal se observó tolerancia al efecto de este producto.
Sólo raramente se han descrito efectos sistémicos provocados por un
tratamiento prolongado con HAP, porque el efecto final de la mayor
parte de los estudios ha sido la carcinogenicidad. Se manifiestan
efectos tóxicos importantes a dosis en las cuales se desencadenan
también respuestas carcinogénicas
En estudios de los efectos adversos en la piel tras la aplicación
cutánea, los productos con carcinogenicidad nula o débil, como el
perileno, el benzo[e]pireno, el fenantreno, el pireno, el antraceno,
el acenaftaleno, el fluoreno y el fluoranteno, fueron inactivos,
mientras que los compuestos carcinógenos; como el benz[a]antraceno, el
dibenz[a,h]antraceno y el banzo[a]pireno provocaron hiperqueratosis.
Los vapores de antraceno y de naftaleno provocaron irritación ocular
leve. El benzo[a]pireno indujo hipersensibilidad por contacto en
cobayas y ratones.
El benz[a]antraceno, el benzo[a]pireno, el dibenz[a,h]antraceno y el
naftaleno fueron embriotóxicos para ratones y ratas. El benzo[a]-
pireno mostró asimismo teratogenicidad y efectos en la reproducción.
Se han realizado grandes esfuerzos para aclarar la base genética de
los efectos embriotóxicos del benzo[a]pireno. Sólo se observan
mortalidad fetal y malformaciones si es inducible el sistema citocromo
P-450 monooxigenasa, bien en la madre (con permigración placentaria) o
bien en el embrión. No todos los efectos observados se pueden explicar
por la predisposición genética, pero en ratones y conejos el
benzo[a]pireno mostró actividad carcinogénica transplacentaria,
produciendo adenomas pulmonares y papilomas cutáneos en la
descendencia. Se observó asimismo reducción de la fecundidad y
destrucción de oocitos.
Se han estudiado también ampliamente los HAP en valoraciones de la
genotoxicidad y de la transformación celular; la mayor parte de los 33
HAP comprendidos en la presente monografía son genotóxicos o
probablemente genotóxicos. Los únicos compuestos para los cuales se
obtuvieron resultados negativos en todas las valoraciones fueron el
antraceno, el fluoreno y el naftaleno. Habida cuenta de la falta de
uniformidad de los resultados, el fenantreno y el pireno no podrían
clasificarse de manera fidedigna como genotóxicos.
En un trabajo amplio sobre la carcinogenicidad de los HAP se ha puesto
de manifiesto que 26 de los 33 productos estudiados son, o se sospecha
que son, carcinogénicos (Cuadro 2). El compuesto mejor caracterizado
es el benzo[a]pireno, que se ha estudiado aplicando todos los métodos
actuales en siete especies. Los HAP que han sido objeto de 12 estudios
o más son el antantreno, el antraceno, el benz[a]-antraceno, el
criseno, el dibenz[a,h]antraceno, el dibenzo[a,i]pireno, el 5-
metilcriseno, el fenantreno y el pireno. Los estudios especiales de
fototoxicidad, inmunotoxicidad y hepatotoxicidad de los HAP se
complementan con informes sobre la toxicidad ocular del naftaleno.
Cuadro 2. Resumen de los resultados de las pruebas de genotoxicidad
y carcinogenicidad de los 33 hidrocarburos aromáticos policiclicos
estudiados
Compuesto Genotoxicidad Carcinogenicidad
Acenafteno (?) (?)
Acenaftileno (?) No hay estudios
Antraceno - -
Benz[a]antraceno + +
Benzo[a]fluoreno (?) (?)
Benzo[a]pireno + +
Benzo[b]fluoranteno + +
Benzo[b]fluoreno (?) (?)
Benzo[c]fenantreno (+) +
Benzo[e]pireno + ?
Benzo[ghi]fluoranteno (+) (-)
Benzo[ghi]perileno + -
Benzo[j]fluoranteno + +
Benzo[k]fluoranteno + +
Criseno + +
Coroneno (+) (?)
Ciclopenta[cd]pireno + +
Dibenzo[a,e]pireno + +
Dibenz[a,h]antraceno + +
Dibenzo[a,h]pireno (+) +
Dibenzo[a,i]pireno + +
Dibenzo[a,l]pireno (+) +
Fluoranteno + (+)
Fluoreno - -
Indeno[1,2.3-cd]pireno + +
1-Metilfenantreno + -
5-Metilcriseno + +
Naftaleno ?
Perileno + -
Fenantreno (?) (?)
Pireno (?) -
Trifenileno + -
+, positivo; -, negativo; ?, dudoso
Paréntesis, resultado derivado de un número de datos pequeño
El antraceno, el benzo[a]pireno y algunos otros HAP mostraron
fototoxicidad en la piel de mamíferos y en cultivos de células in
vitro cuando se aplicaron con radiación ultravioleta. Se ha notificado
que en general estos compuestos tienen efectos inmunosupresores. Tras
la administración intraperitoneal de benzo[a]pireno a ratones, se
manifestó una fuerte supresión de los parámetros inmunitarios en la
descendencia durante un período de hasta 18 meses. Se observó asimismo
una mayor regeneración hepática y un aumento del peso del hígado. El
efecto del naftaleno en la aparición de cataratas en un roedor se ha
atribuido a su capacidad de inducción del sistema del citocromo P-450
en estudios realizados con estirpes de ratones genéticamente
diferentes.
Los modelos teóricos para pronosticar la actividad carcinogénica de
los HAP a partir de sus estructuras, basados en una gran cantidad de
trabajos experimentales, se presentaron ya en los años treinta. El
primer modelo se basaba en la elevada reactividad química de
determinados dobles enlaces (teoría de la región K). Más tarde se
aplicó un método más sistemático, basado en la síntesis química de
posibles metabolitos y en su actividad mutagénica. Según esta teoría
de la región "bay", los epóxidos adyacentes a una región "bay" dan
lugar a iones carbonio muy estables. Otros métodos teóricos son la
"teoría de la región di" y la "teoría del potencial del radical
catiónico".
Muchos de los distintos HAP son carcinógenos para los animales y
pueden serio para el ser humano y se ha observado que la exposición a
varias mezclas con HAP aumenta la incidencia de cáncer en poblaciones
humanas. Existe la preocupación de que los HAP cuya carcinogenicidad
se ha demostrado en animales de experimentación puedan serio también
para el ser humano. Los HAP producen rumores, tanto en el lugar del
contacto como en otros lejanos. Su actividad carcinogénica puede
variar en función de la vía de exposición. Se han propuesto diversos
métodos para evaluar el riesgo asociado a la exposición a estos
productos aislados y en mezclas. En la presente monografía no se
respalda ningún método; sin embargo, se describen las necesidades de
datos, las hipótesis, la aplicabilidad y otras características de tres
procesos de evaluación cuantitativa del riesgo que han sido validados
en cierta medida.
1.8 Efectos en el ser humano
Habida cuenta de la complejidad del perfil de los HAP en el medio
ambiente y en los lugares de trabajo, la exposición humana a productos
puros por separado se ha limitado a experimentos científicos con
voluntarios, salvo en el caso del naftaleno, que se utiliza como
antipolilla para la ropa.
Tras la aplicación cutánea, el antraceno, el fluoranteno y el
fenantreno indujeron reacciones específicas en la piel, y el benzo[a]-
pireno produjo la formación de verrugas regresivas reversibles que se
clasificaron como proliferaciones neoplásicas. Los efectos sistémicos
del naftaleno se conocen por los numerosos casos de ingesta
accidental, particularmente de niños. La dosis letal por vía oral es
de 5000-15 000 mg para los adultos y 2000 mg ingeridos durante dos
días en los niños. El efecto normal tras la exposición por vía cutánea
u oral es una anemia hemolítica aguda que, a través de la placenta,
puede afectar también a los fetos.
El humo del tabaco es el factor aislado más importante en la inducción
de tumores de pulmón y también de un aumento de la incidencia de
tumores de la vejiga urinaria, la pelvis renal, la boca, la faringe,
la laringe y el esófago. No se considera que sea elevada la
contribución de los HAP en los alimentos a la aparición de tumores
humanos. En zonas fuertemente industrializadas se detectó también una
mayor carga corporal de HAP debido a la contaminación del aire. Están
expuestos asimismo a estos compuestos los enfermos de psoriasis
tratados con alquitrán de hulla.
La exposición profesional al hollín como causa de cáncer de escroto se
observó por primera vez en 1775. Más tarde se informó que la
exposición a los alquitranes y la parafina inducían cáncer cutáneo.
Ahora el cáncer inducido por HAP afecta principalmente al pulmón,
mientras que el cáncer cutáneo es más raro gracias a una mayor higiene
personal.
Se han realizado estudios epidemiológicos de trabajadores expuestos en
hornos de coque durante la coquificación del carbón y su gasificación,
en obras de asfaltado, fundiciones e instalaciones de aluminio y a los
gases de escape de los motores diesel. Se ha detectado un índice de
cáncer de pulmón más elevado debido a su exposición a los HAP en los
trabajadores de hornos de coque, los que utilizan asfalto y los de las
salas de crisoles de Söderberg de las instalaciones de reducción de
aluminio. El riesgo más elevado se observó en los trabajadores de
hornos de coque, con una razón de mortalidad normalizada de 195. En
varios estudios se hallaron relaciones dosis-respuesta. En las
instalaciones de aluminio no sólo se detectó cáncer de la vejiga
urinaria, sino también síntomas semejantes al asma, anomalías
funcionales de los pulmones y bronquitis crónica. En los trabajadores
de hornos de coque se observó asimismo una disminución de la
concentración de inmunoglobulina en el suero y una reducción de la
función immunitaria. Se notificó que una exposición durante cinco años
al naftaleno había provocado cataratas.
Se han elaborado varios métodos para evaluar la exposición interna a
los HAP. En la mayoría de los estudios, se midieron en orina
metabolitos de los HAP como los tioéteres urinarios, el 1-naftol, la
ß-naftilamina, los hidroxifenantrenos y el 1-hidroxipireno. Este
último se ha utilizado con frecuencia como indice biológico de
exposición.
Se han determinado los efectos genotóxicos de los HAP mediante pruebas
de mutagenicidad en orina y heces y de la presencia de m/cm-núcleos,
aberraciones cromosómicas e intercambio de cromátidas hermanas en
linfocitos de la sangre periférica. Además, se han medido aductos de
benzo[a]pireno con ADN en linfocitos periféricos y en otros tejidos y
con proteínas como la albúmina, así como anticuerpos de aductos de
ADN.
En varios estudios se han investigado el 1-hidroxipireno en orina y
los aductos de ADN en linfocitos como marcadores. El primero se puede
medir más fácilmente que los segundos, presenta menos variación entre
los individuos y es posible detectar niveles de exposición más bajos.
Ambos marcadores se han Utilizado para evaluar la exposición humana en
diversas condiciones. En varios puestos de trabajo de instalaciones de
coque, de fabricación de aluminio, de instalaciones de impregnación de
la madera, de fundiciones y de obras de asfaltado se observó un
aumento de la excreción de 1-hidroxipireno o de aductos de ADN. Las
exposiciones más elevadas se detectaron en los trabajadores de hornos
de coque y en los de impregnación de la madera con creosota, que
absorbían hasta el 95% del total de HAP a través de la piel, a
diferencia de la población general, en la que predomina la absorción
mediante los alimentos y el humo del tabaco.
Las estimaciones del riesgo relacionado con la exposición a HAP y sus
mezclas se basan en estimaciones de la exposición y en los resultados
de estudios epidemiológicos. Los datos obtenidos de trabajadores de
hornos de coque pusieron de manifiesto un riesgo relativo de cáncer de
pulmón de 15,7. Teniendo esto en cuenta, se ha calculado que el riesgo
de aparición de cáncer de pulmón en la población general durante toda
la vida es de 104 a 10-s por ng de banzo[a]pireno por m3 de aire.
Dicho de otra manera, como consecuencia de la exposición al
benzo[a]pireno en el aire cabra esperar la aparición de cáncer de
pulmón a lo largo de toda la vida en una persona de cada 10 000 a 100
000.
1.9 Efectos en otros organismos en el laboratorio y en el medio
ambiente
Los HAP tienen toxicidad aguda para los peces y para Daphnia magna en
combinación con la absorción de radiación ultravioleta y luz visible.
El metabolismo y la degradación alteran la toxicidad de los HAP. A
concentraciones bajas, los HAP pueden estimular el crecimiento de
microorganismos y algas. Los HAY más tóxicos para las algas son el
benz[a]antraceno (cuatro anillos), con una CE50 (concentración a la
cual determinados parámetros vitales se reducen a la mitad) de 1-29
µg/litro, y el benzo[a]pireno (cinco anillos), con una CE50 de 545
µg/litro. Los valores de la CL50 en las algas para la mayor parte de
los HAP de tres anillos son de 240-940 µg/litro. El naftaleno (dos
anillos) es el menos tóxico, con valores de la CE50 de 2800-34 000
µg/litro.
No se han observado diferencias claras de sensibilidad entre distintos
grupos taxonómicos de invertebrados, como los crustáceos, los
insectos, los moluscos, los poliquetos y los equinodermos. El
naftaleno es el menos tóxico, con valores de la CL50 en 96 horas de
100-2300 µg/litro. Los valores de la CL50 en 96 horas para los HAP
de tres anillos oscilan entre <1 y 3000 µg/litro. El antraceno puede
ser más tóxico que los otros HA/' de tres anillos, con CL50 en 96
horas entre <1 y 260 µg/litro: Los valores de la CL50 en 96 horas
para los HAP de cuatro, cinco y seis anillos son de 0,2-1200 µg/litro.
Se observó toxicidad aguda (CL50) en peces a concentraciones de 110
a >10 000 µg/litro de naftaleno, 30-4000 µg/litro de HAP de tres
anillos (antraceno, 2,8-360 µg/litro) y 0,7-26 µg/litro para los de
cuatro o cinco altillos.
La contaminación de los sedimentos con HAP a concentraciones de 250
mg/kg se asoció a tumores hepáticos en peces vivos libres. Se han
inducido asimismo tumores en peces expuestos en el laboratorio. La
exposición de los peces a determinados HAP puede producir también
cambios fisiológicos y afectar a su crecimiento, reproducción,
capacidad natatoria y respiración.
The tumorigenicity of racemic anti-1,2-diol-3,4-epoxides of
chrysene, 5-methyl-, 5-ethyl-, and 5-propylchrysene was determined in
newborn mice. Only the 5-methyl compound was highly tumorigenic,
demonstrating the importance of molecular shape for tumorigenicity. A
methyl group in the same bay region as the epoxide ring leads to
exceptional activity, and this may occur a consequence of DNA adduct
conformation (Amin et al., 1991b).
In a study of the binding of 3H-labelled anti-5- and
anti-6-methylchrysene-1,2-diol-3,4-epoxide to DNA in liver and lung
of newborn mice after intraperitoneal administration on day 1 of life
and sacrifice after 24 h, 1.1 pmol/mg DNA were found with the
benzo [a]pyrene analogue, 0.5 pmol/mg DNA with the 5-methyl compound,
and < 0.01 pmol/mg DNA with the 6-methyl compound, consistent with
their known tumorigenic activities. When the parent compounds were
tested in the same protocol, however, little radiolabel became
associated with DNA adducts. Hence, it can be concluded that the
dihydrodiols are the carcinogens in newborn mice (Melikian et al.,
1991).
The metabolism of 5-methylchrysene has also been investigated in mouse
epidermis in vivo. The diol precursors of 1,2-dihydroxy-3,4-epoxy
5-methylchrysene and 7,8-dihydroxy-9,10-epoxy 5-methylchrysene were
present in equivalent quantities at every time, and the ratio of DNA
adducts with the two precursors was constant over time (Melikian et
al., 1983).
In a study of the correlation of the 5-methylchrysene-DNA adduct
profile in lung tissue with the spectrum of mutations in the K- ras
protooncogene of lung tumours, up to 200 mg/kg bw 5-methylchrysene
were administered to A/J mice and the lungs were analysed for DNA
adducts one to three days later. After a latent period of eight
months, 90% all lung tumours had mutations of K- ras.
N2-Deoxyguanosine was detected as a possible promutagenic adduct
(You et al., 1994).
7.10.3.11 1-Methylphenanthrene
An investigation of an extensive series of alkylated phenanthrenes
suggests that the presence of a methyl substituent at, or adjacent to,
the K region (9,10 position) and an unsubstituted angular ring
adjacent to a free peri position are the prerequisites for mutagenic
activity in S. typhimurium. Substitution at the peri position was
associated with lack of mutagenicity. A non-K-region dihydrodiol
derived from 1-methylphenanthrene was a potent proximate mutugenic
metabolite (LaVoie et al., 1983b).
7.10.3.12 Naphthalene
Severe bronchiolar epithelial-cell necrosis was reported in mice after
intraperitoneal injection of naphthalene. Lung, liver, and kidney
macromolecules were shown by a radiolabelling technique to be the main
targets. Maximal binding of naphthalene was found 2-4 h after
application, and a threshold was found at 200-400 mg/kg bw,
corresponding to glutathione depletion. Covalent binding was highest
in tissues with high cytochrome P450 mono-oxygenase activity, i.e.
lung, liver, and kidney (Warren et al., 1982).
7.10.3.13 Phenanthrene
A single dose of 300 mg/kg of phenanthrene administered
intraperitoneally to B6C3F1 mice caused > 20-fold induction of
hepatic microsomal methoxy-resofurin O-deethylase activity
(Chaloupka et al., 1994; see also section 7.10.3.1).
7.10.3.14 Investigations of groups of PAH
In rats, expression of the Cyp1A1 gene is closely associated with
the inducibility of AHH, an enzyme important for bioactivation of PAH.
Cyp1A1 is regulated by several factors, including the Ah receptor
and the cytosolic 4S PAH-binding protein. The role of the latter
protein was investigated with benzo [a]pyrene and benzo [e]pyrene in
H4-II-E rat hepatoma cells. Both induced gene expression, as measured
by ethoxyresorufin O-deethylase activity, but benzo [a]pyrene was
about 25 times more potent than benzo [e]pyrene. Benzo [a]pyrene
binds to both the Ah receptor and the 4 S protein, and
benzo [e]pyrene only to the protein (Houser et al., 1992).
A series of PAH were investigated in a novel short-term test for the
detection of carcinogens, the initiator tRNA acceptance assay.
Positive responses, i.e. > 15% stimulation, were induced by chrysene,
benzo [c]-phenanthrene, dibenz [a,h]anthracene, benzo [a]pyrene,
and dibenzo [a,i]pyrene; and negative responses were induced by
naphthalene, anthracene, phenanthrene, pyrene, benz [a]anthracene,
benzo [e]pyrene, perylene, and coronene. All of the potent
carcinogens were active in this assay (Hradec et al., 1990).
In a study of the non-carcinogenic PAH anthracene, fluorene, and
naphthalene and several carcinogenic amine derivatives in rats,
naphthalene failed to induce induce the cytochrome P450-dependent
mixed-function oxidases, whereas anthracene was a weak and fluorene an
effective inducer. A relationship was found between carcinogenicity
and the ability to induce hepatic P450 activity. It was assumed that
fluorene is not carcinogenic because it cannot form mutagenic
intermediates (Ayrton et al., 1990).
In a study of the induction of various PAH of the mono-oxygenase
isoenzymes in mouse liver microsomes, the cytochrome P448 and P450
groups were classified by using specific inhibitors in studies of
7-ethoxycoumarin activity. According to the pattern of enzymes
induced, the following groups were distinguished: (i) P448 type,
including dibenz [a,h]anthracene and benzo [k]fluoranthene; (ii)
mixed type, including pyrene, benzo [j]fluoranthene, and
benzo [e]pyrene, in which two inhibitors acted on the enzyme
reaction; and (iii) special P448 type consisting of
indeno[1,2,3- cd]pyrene, in which one inhibitor stimulated the
reaction. The PAH investigated were not a homogeneous group of
selective P448 inducers. No correlation was found with mutagenic
potency (Kemena et al., 1988).
8. EFFECTS ON HUMANS
Appraisal
There is little information on human exposure to single, pure
polycyclic aromatic hydrocarbons (PAH). That which is available
includes reports of accidental exposure to naphthalene and some data
from defined short-term studies of volunteers. All other reports are
of exposure to mixtures of PAH, which also contained other potentially
carcinogenic chemicals, in occupational and environmental situations.
Information on the health effects of these mixtures is confined to
their carcinogenic potential, for which there is evidence from a
number of epidemiological studies, especially for lung cancer and, in
some cases, cancers of the skin and of the urinary bladder. Since
single PAH and PAH mixtures are known to be carcinogenic in
experimental animals, it is plausible to attribute the enhanced cancer
risks seen in humans predominately to the PAH. In addition, the
results of epidemiological studies are important in risk assessment.
Therefore, in contrast to the preceding sections, the results of
studies of PAH mixtures are also presented.
Many workplaces have atmospheres with heavy loads of PAH. The cohorts
affected are gas workers, those exposed at coke ovens, wood
impregnation workers, people working at waste incinerators, workers in
bus garages, workers in nickel and copper refineries and in aluminium
smelters, asphalt workers, and chimney sweeps. Evaluation of the
immunocompetence of coke-oven workers indicated decreased serum
immunoglobulin levels and decreased immune function.
Biomarkers used to assess exposure to PAH include hydroxyphenanthrenes
and 1-hydroxypyrene in urine and DNA adducts in peripheral blood
lymphocytes.
8.1 Exposure of the general population
8.1.1 Naphthalene
Naphthalene is often used in houses as an insect repellant, mainly
against moths, and many incidents of poisoning have been reported.
Acute haemolytic anaemia is a typical systemic effect of oral, dermal,
or inhalation exposure. The lethal oral doses determined in cases of
accidental poisoning are 5-15 g for adults and 2 g within two days for
a six-year old child. Repeated exposure to naphthalene fumes or dust
has led to corneal ulceration, lenticular opacities, and cataracts
(Sandmeyer, 1981). Some case reports are described in more detail
below.
8.1.1.1 Poisoning incidents
(a) Oral exposure
Between 1949 and 1959, 10 cases of the oral poisoning by naphthalene
in children were documented in the United States. The amounts were
usually not specified but were in the order of grams. Some of the
children developed haemolytic anaemia (Anziulewicz et al., 1959).
The symptoms that developed after naphthalene intake included nausea,
vomiting, and convulsions after one to several days, often followed by
diarrhoea. Other symptoms were disturbances of consciousness,
lethargy, ataxia, and, in severe cases, coma and hemiplegia.
Haemolytic anaemia occurred concomitantly, with plasma haemoglobin
contents of up to 40%, often followed by haemoglobinuria. Mild to
severe jaundice can also occur; in one fatal case of poisoning, patchy
liver necrosis was reported. Treatment consists of blood transfusions
and additional alkalization of the urine; following this treatment,
rapid recovery, without persistent damage, was observed (Konar et al.,
1939). In five cases of acute haemolytic anaemia in children of about
two years of age who had eaten moth balls consisting of pure
naphthalene, there was complete recovery within one to four weeks
after transfusion (Zuelzer & Apt, 1949; Mackell et al., 1951).
Tests in vitro revealed that it was not naphthalene itself but its
metabolites a-naphthoquinone and a-naphthol that cause a decrease in
reduced glutathione in erythrocytes. Whole blood from patients
contained erythrocytes with defective glutathione metabolism. This
defect is observed in about 15% of black males and 2% of black females
(Zinkham & Childs, 1958).
(b) Dermal exposure
The effects of skin contact in sensitive individuals range from
irritation to severe dermatitis after exposure to quite small amounts
of naphthalene, such as wearing clothes that had been treated with
moth balls. Workers exposed to naphthalene may develop dermatitis on
their hands, arms, legs, and abdomen (Gerarde, 1960). Cases of
haemolytic anaemia have been reported in babies who absorbed
naphthalene from nappies that had been stored with moth balls
(Anziulewicz et al., 1959).
(c) Inhalation
Haemolytic anaemia was also observed in babies who had inhaled
naphthalene from moth ball-treated wool blankets (Valaes et al.,
1963). The case of a man with exfoliative dermatitis was reported,
which resolved after all contact with naphthalene was eliminated
(Fanburg, 1940).
8.1.1.2 Controlled studies
When the forearm skin of three volunteers was treated with anthracene
in a 2% benzene solution (dose not specified) and irradiated with a
monochromator (340-380 nm), urticarial reactions were seen, with
burning and erythema lasting for several days (Crow et al., 1961).
Twelve healthy white men, 12-26 years old, with fair complexions were
treated dermally with anthracene in an ethanolic acetone solution at
25 µg/cm2 and received ultraviolet irradiation 2 h later. Specific
skin reactions such as transient erythema, delayed erythema, and
whealing were seen. The effect was related both to the anthracene and
the amount of radiation energy. Controls who received no anthracene
treatment but the same irradiation showed no sign of erythema (Kaidbey
& Nonaka, 1984).
Regressive verrucae were reported after up to 120 dermal applications
of 1% benzo [a]pyrene to human skin over four months. The reversible
and benign changes were thought to be neoplastic proliferations, but a
group that did not receive benzene was not evaluated (Cottini &
Mazzone, 1939). Similar epidermal changes and nucleolar enlargement
were reported in volunteers painted daily for four consecutive days on
1-cm2 areas of the upper back (Rhoads et al., 1954).
8.1.2 Mixtures of PAH
8.1.2.1 PAH in unvented coal combustion in homes
Interdisciplinary studies were conducted to investigate exposure to
PAH and the high lung cancer rates in a rural county, Xuan Wei,
located in Yunnan Province, China (Mumford et al., 1987). Mortality
from lung cancer in this county is five times the Chinese national
average, especially among the women, who have the highest rate in
China. Three communes had a mortality rate that was 24 times the
national rate: 126 per 100 000 for women and 118 per 100 000 for men
during 1973-79. An unusual observation in Xuan Wei is the similarity
of the lung cancer rates in men, most of whom are smokers, and women,
most of whom are not (< 0.1% smoke). The mortality rate from lung
cancer was correlated with domestic use of 'smoky' coal
(medium-volatile bituminous coal with low sulfur and high ash) for
cooking and heating, but not with use of wood or smokeless coal.
Monitoring of air during cooking inside the homes showed that women
were exposed to extremely high levels of PAH, with a mean
benzo [a]pyrene concentration of 14.7 µg/m3, comparable to the
levels to which coke-oven workers are exposed. They were also exposed
< 24 mg/m3 of submicron particles containing up to 82% of the
organic matter. The major organic components of smoky coal emissions
are the three- to five-ring alkylated PAH, which contributed 43% of
the organic mass of the particles and 61% of the total mutagenicity in
assays in S. typhimurium. The four-ring PAH were the most
tumorigenic (Chuang et al., 1992). Organic extracts of particles from
coal smoke were more potent in initiating tumours in Sencar mouse skin
than those from wood and smokeless coal combustion and were complete
carcinogens (Mumford et al., 1990). Xuan Wei residents exposed to
smoky coal emissions had significantly more 9-hydroxy benzo [a]pyrene
in their urine, and cells obtained by bronchial alveolar lavage had
more DNA adducts (detected by 32P-postlabelling) than those of
controls (Lewtas et al., 1993). High ratios of the concentrations of
methylated PAH and parent PAH (9.8:1 for women and 5.8:1 for men) in
urine samples from Xuan Wei residents confirmed that they were exposed
to high concentrations of alkylated PAH. Thus, alkylated PAH may play
an important role in the etiology of lung cancer in Xuan Wei (Mumford
et al., 1995).
1-Hydroxypyrene was used as a urinary biomarker to monitor the
exposure of urban populations to PAH originating from coal burning. A
good correlation was found between the concentrations of pyrene and
benzo [a]pyrene in ambient in air and 1-hydroxypyrene in urine (Zhao
et al., 1990; see also section 8.3.2).
8.1.2.2 PAH in cigarette smoke
A large volume of literature exists on the effects of tobacco smoke on
human lungs (see IARC, 1986). On the basis of more than 100
prospective and retrospective studies in more than 15 countries,
cigarette smoke has been shown to be by far the most important single
factor contributing to the development of lung cancer. Other types of
cancer caused by cigarette smoking include cancers of the oral
cavities, larynx, pharynx, oesophagus, bladder, renal pelvis, renal
adenocarcinoma, and pancreas.
Levels of 11 ng per cigarette benzo [a]pyrene were found in
mainstream smoke and 103 ng per cigarette in sidestream smoke; the
corresponding values were 6.8 and 7.6 ng per cigarette for
benzo [e]pyrene, 20 and 497 ng per cigarette for chrysene and
triphenylene, and 13 and 204 ng per cigarette for benz [a]anthracene
(Grimmer et al., 1987). In sidestream smoke, PAH with four or more
rings were responsible for 83% of the total carcinogenic activity
(Grimmer et al., 1988c).
8.1.2.3 PAH in coal-tar shampoo
Of eight commercially available coal-tar shampoos, that with the
highest PAH content (100 times that of the others), containing, e.g.
285 mg/kg pyrene and 56 mg/kg benzo [a]pyrene, was chosen for testing
in 11 healthy people. A dose of 20 g shampoo was used in the evening
(see section 8.2.3), and the internal dose of PAH was assessed as
urinary 1-hydroxypyrene. One day after exposure, the internal dose was
10 times higher than the background level, similar to that measured in
coke-oven workers (van Schooten et al., 1994). The potential
carcinogenicity of coal-tar shampoo formulations has been studied (see
section 7.7). It has been suggested that modest therapeutic doses of
agents containing coal-tar and dithranol are tumorigenic after
combined application and that their use should be reviewed (Phillips &
Alldrick, 1994).
8.2 Occupational exposure
No studies on occupational exposure to single PAH were available, as
in general, industrial workers using or producing coal or coal
products are exposed to mixtures of PAH (see section 5.3). Table 95
lists workplaces in which there is exposure to PAH and the types of
employees exposed. Epidemiological studies have been conducted on
workers exposed at coke ovens in coal coking and coal gasification, at
asphalt works, at foundries, and at aluminium smelters and to diesel
exhaust. Details of the most recent and most important cohort and
case-control studies are given in Tables 96 and 97 (for reviews of
these studies, see also IARC 1983, 1984a,b, 1985).
Levels of exposure to single PAH in these occupations have been
reported (see section 5.3). In the following text, levels of exposure
to 'total PAH' are also given, as reported by the authors; however,
'total PAH' represents only the sum of a limited number of compounds
that have been quantified, i.e. the selection of the investigator, and
such measurements cannot be compared for the purpose of evaluating
levels or risks of pollution.
All benzo [a]pyrene concentrations are reported for comparative
purposes, as it is the only PAH that has been determined in almost all
investigations because of its well-known carcinogenicity. It is
commonly used as an indicator of the level of particle-bound PAH, and
particularly of carcinogenic ones, but PAH profiles may vary according
to source.
The first attribution of a PAH-related cancer to an occupational
exposure was that of Pott in 1775, who described the susceptibility of
English chimney sweeps to scrotal cancer (Pott, 1775); a second was
published by Butlin in 1892. Easily avoidable dermal cancers are
seldom seen today, owing to better personal hygiene and better working
conditions, but the number of respiratory cancers is still
significantly higher in occupational cohorts than in the general
population. In a study of Swedish chimney sweeps who were exposed to
< 9 µg/m3 benzo [a]pyrene, significantly increased rates of lung
tumours were observed, with a standardized mortality rate (SMR) of
2.06 (Table 96; Gustavsson et al., 1988); however, chimney sweeps were
also exposed to arsenic, chromium, cadmium, nickel, sulfur dioxide,
carbon monoxide, organic solvents, and asbestos.
Most important for an evaluation of the possible risk for cancer due
to exposure to PAH are studies of workers exposed at coke ovens in
coke plants or in coal-gasification processes, where the PAH
concentrations are considerable, with levels of 1 mg/m3 total PAH and
300 µg/m3 benzo [a]pyrene (Lindstedt & Sollenberg, 1982; Swaen et
al., 1991; see also section 5.3). The concentrations to which workers
are exposed are not available in most epidemiological studies,
however, and coke-oven workers may be exposed to several other
carcinogenic compounds, such as 2-naphthylamine, arsenic, and benzene.
Table 95. Occupations in which there is exposure to polycyclic
aromatic hydrocarbons
High exposure
- coke ovens
- coal gasification plants
- chimney sweeping
- petroleum refineries (mainly exposed to naphthalene and its methyl
derivatives)
- impregnation of wood with creosotes (mainly exposed to
naphthalene, phenanthrene, and fluorene)
- handling of creosote-impregnated wood (e.g. railroad and utility
workers, carpenters, mainly exposed to naphthalene, phenanthrene,
and fluorene)
Medium exposure
- asphalt and pavement work
- roofing
- aluminium production
- graphite electrode production (e.g. anode production for the
aluminiurn industry)
- founding (processing of e.g. steel and other alloys, from coal
additives in moulding sand)
- smokehouses (processing of meat and fish)
Low exposure
- mechanics, bus garage workers, and machinists (from diesel and
spark-ignition engine exhaust gases)
- mining (from diesel engine exhaust gases)
- use of lubricating and cutting oils (e.g. in steel production)
- cooking
Table 96. Epidemiological studies of lung caner in cohorts exposed to polycyclic aromatic hydrocarbons
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
Coal coking
5321, 10 497 Coal-tar pitch volatiles;b 255 1.95 Risk decreased with period All causes, 1.08 Costantino
coke oven non-oven, 3.2 mg/m3 (topside), (1.59-2.33) follow-up; findings all cancers, 1.34 et al. (1995);
1952-82, steel industry 2.0 (top-side parttime), consistent across racial prostate 1.57 Rockette &
0.88 (side); no categories; strong Redmond
information on smoking correlation with duration of (1985);
habits concentration exposure and exposure Redmond
(1983)
5639 National No information on 62 1.29 Strong correlation with Total 1.19 Swaen et al.
coke plant, population smoking habits (0.99-1.66) exposure concentration; mortality, 1.66 (1991)
1945-84 (Netherlands) risks increased in respiratory 3.08
comparison with workers at disease,
a nitrogen fixation plant liver
536, National information on smoking 25 2.38*** Coke-oven workers: 1.75 All causes, 1.41 Chau et al.
coke plant population habits (2 cases); near oven all cancers, 1.33 (1993)
1963-87 (France) workers: 2.52** (8 cases); cardio- 1.33
unexposed workers: 2.28* vascular
(6 cases) disease
Risk increased (not
significant) for workers in
oldest plant
6767, National Some information on 167 1:17* - - - Hurley et al.
coke plant population smoking habits (1983)
(Scotland,
England, Wales)
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
subcohorts:
1617, coke - - 34 0.94 No clear correlation with - -
oven, duration of exposure; problems
1966-78 in classification of exposure
(risk increase with > 5 years
and > 10 years exposure)
1158, coke - - 32 1.05 Lung cancer risk increased - -
oven, in younger workers; no clear
1967-80 correlation with duration of
exposure and exposure
concentration; problems in
classification of exposure
Coal gasification
2449+1176 National 2-Naphthylamine, 99 3.82b RR given for groups with Bladder 2.35 Doll et al.
(2 cohorts), population 2 µg/m3; some 23 2.72 regular exposure; also cancer (1972)
1953-65 (England and information on increased risk in group with
Wales) smoking habits intermittent exposure;
correlation with exposure
concentration and duration
of exposure
724,1953-80 (a) 3792, same Median of 8 measurements: 68 3.53**a No correlation with All cancers, 1.98 Manz et al.
plant, not at total dust, max. duration of exposure; 88% of urinary 4.35 (1983)
coke-oven 264 mg/m3, BaP, 28 workers with > 10 years' system
(b) 681, µg/m3, max. 89; exposure cancers
office and some information on
administration smoking habits
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
(a) local
population
(Hamburg)
295, Local worker BaP top of ovens: 4 0.82 Owing to incomplete employment All causes 1.27 Gustavsson
1966-86 population 1964: 4.3 µg/m3 (0.22-2.11) registers, only workers & Reuterwall
(0.007-33) with short and recent exposure (1990);
1965: 0.52 µg/m3 selected; no correlation with Gustavsson
(0.021-1.29); no duration of exposure (1989);
differences in smoking Gustavsson
habits between exposed et al. (1987)
and controls
Asphalting
679, paving, National Fume condensate; 25 2.90 SMR for lung cancer given All causes, 1.63 Hansen
1959-86 population flooring: 0.5-260 mg/m3; (1.88-4.29) only for workers aged 40-89 all cancers 2.25 (1989);
(Denmark) median,19.7, manual years; overall mortality Workers Hansen et
road paving: 4.3-3.4 mg/m3; excess primarily in younger aged 40-89 al., 1991c,
total PAH: median, 183 age groups years: liver 4.67 1992;
µg/m3; BaP, 4 µg/m3; cirrhosis Wong et al.
some information on (1992)
smoking habits
2572, paving, National - 7 1.10 Follow-up too short All causes, 0.69 Engholm et
1971-79 to population 8 1.24 stomach 2.01 al. (1991);
1985 (Sweden) (0.53-2.34) cancer Partanen &
Boffetta
(1994)
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
704, roofing, National - 3 2.79 Follow-up too short All causes 0.91 Engholm et
1971-79 to population 4 (0.99-9.31) al. (1991);
1985 (Sweden) Partanen &
Boffetta
(1994)
Paving, - - 332 1.21 Meta-analysis of 11 studies Stomach, 1.33 Partanen &
roofing, (1.08-1.30) bladder, 1.38 Boffetta
others skin, non- 1.74 (1994)
melanoma, 1.41
leukaemia
Paving - - 167 0.87 Meta-analysis of 3 studies Skin, non- 2.18
(0.74-1.01) melanoma
Roofing - - 118 1.96 Meta-analysis of 4 studies Stomach 1.71
(1.46-2.11)
Creosote
impregnation
922 National - 13 0.79 - Skin, non- 2.37 Karlehagen
populations (0.42-1.35) melanoma et al. (1992)
(Sweden and
Norway)
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
Tar distillation
76, 449, PAH: 29 µg/m3, 4 Total rate of tumours 0 - Schunk
1962-72 rubber BaP: 4 µg/m3; some increased (RR 3 and 3.4); (1979)
industry; information on 3 tumours of the digestive
national smoking habits system latency, 10.1-
population 17.5 years
(Germany)
Foundries
2990 General No information on 224 1.44** Respiratory Egan-Baum
(2651 white population smoking habits white white disease: et al. (1981)
males; 339 (USA) males,males; white males 1.10
black males), 39 76** black males 1.24
steel, iron, black black Pneumo-
non-ferrous, males males coniosis:
1961-71 black males 5.76
Respiratory
tuberculosis:
white males 2.32
All cancers:
white males 1.10
black males 1.24
439, Regional Data from 1977; total 21 2.50** Correlation with duration - - Gibson et al.
steel population dust, 1.76-5.11 mg/m3; of exposure; no correlation (1977)
foundry, (Toronto) respirable dust, 0.69-2.65 between latency and exposure
1967-77 mg/m3; no cristobalite concentration
or tridymite; coal-tar pitch
volatiles, 0.19-0.43 mg/m3;
BaP, 0.049-0.152 µg/m3;
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
1718, General Some information on 11 2.04 No clear correlation with - - Moulin et al.
steel or population smoking habits (1.02-3.64) duration of exposure or (1990)
ferro- (France) latency
chromium
production,
31 years
4227, General - 17 0.88 - Liver 1.74 Moulin et al.
steel plant, population (0.51-1.40) cirrhosis (1993)
two cohorts, (France)
1969-84
210, - - 2 0.68 - - -
subcohort: (0.08-2.45)
ferroalloy
workshop
477, - - 11 2.29 No clear correlation with - -
subcohort: (1.14-4.09) duration of exposure or
steel foundry latency; significantly
increased for > 30 years
since first employment
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
10 491, General - 441 1.47*** SMRs increased for all Stomach 1.37 Sorahan
steel population foundry occupations cancer & Cooke
foundry, (England and (fettling shop, pattern, (1989)
20 years, Wales) machine, maintenance,
1946-65 to inspection); some
1985 correlation with latency;
no significant correlation
with duration of exposure
6494, General 2 plants, around 77 0.99 Except for 1 plant, furnace - - Kjuus et al.
production of population furnaces:PAH, 3-49 µg/m3; (0.78-1.24) workers showed no increase (1986)
ferrosilicon (Norway) anode paste plant: 2-20 SIR in rate of lung cancer (8
ferromanganese, µg/m3; total dust: 10-30 cases); increase in anode pass
6 plants, mg/m3; manganese: plant workers (3 cases); no
1953-82 0.5-2 mg/m3 Some correlation with duration
information on smoking of exposure
habits
5579, (a) National - 74 (b) 1.17 Information on Malignant 2.05 Sherson &
iron, steel, population employment available only respiratory Iversen
1967-69 (Denmark), for 1967-69 and 1972-74; tumours, (1986)
non-ferrous (b)Economically misclassification possible; respiratory 1.54
foundries, active some correlation with disease
1972-74 population, duration of exposure excluding
(part to (c) Skilled silicosis,
1980) and unskilled all causes 1.11
manual workers
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
8147, National No information on 72 1.23 white No correlation with duration - - Andjelkovich
iron foundry, population smoldng habits males of exposure; smoking may et al. (1990)
1950-85 (USA) (0.96-1.54) be responsible for lung
Local population 67 cancer
132 non-
white males
(1.02-1.67)
Aluminium production
22 010, General No information on 272 0.964 Highest SMR > 25 years All causes, 85.6 Rockette &
15 plants, population smoking habits of exposure in Soderberg all 88.6 Arena (1983)
1946-77, (USA) proccess: SMR 2.0 based malignant
Soderberg on 5 deaths; some tumours
prebake correlation with duration of
exposure and latency
6455, General Some information on 37 1.14 Electrolysis workers: - - Mur et al.
11 plants, population smoking habits (0.85-1.48) SMR, 1.36 (4 deaths); (1987)
Soderberg (France) no correlation with duration
prebake, of exposure or latency
1950-76
4213, 1 plant General Coal-tar pitch volaties 32 0.93 For > 20 benzene-soluble Brain, 2.17 Spinelli et al.
Soderberg, population low: < 0,2 mg/m3; (0.68-1.25) material of coal-tar pitch bladder(SIR) 1.69 (1991);
1954-85 (British medium: 0.2-1; high: volatiles years: SIR 1.43; Ronneberg &
Columbia) > 1; information on correlated with duration of Langmark
smoking habits exposure; some correlation (1992)
with latency; adjustment for
smoking: no change in lung
cancer risk
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
5406, (a) Regional No information on 101 1.43* SMRs for workers exposed All cancers, 1.23 Gibbs (1985);
3 plants, population smoking habits to tar, SMR significantly respiratory 1.65 Gibbs &
Soderberg, (Quebec) increased compared with disease, Horowitz
prebake, (b) Local local population and pneumonia 1.99 (1979)
1954-85 population never-exposed workers and bronchitis,
Significant correlation oeosophageal 1.53
with duration of exposure; and gastric
correlation with latency; tumours
risk decreased in later
periods (1974-77)
5485, - - 12 1.69 SMR increased in relation - - Gibbs (1985);
Soderberg, to local population Gibbs &
1950-51; Horowitz
1977 (1979)
694 General Some information on 19 1.16 Workers with at least 3 - - Ronneberg &
Soderberg, population smoking habits (0.70-1.81) years of employment; some Andersen
prebake (Norway) (SIR) correlation with duration of (1995)
1962-91 exposure and latency
Other workplaces
2219, General - 29 0.85 - All causes, 0.67 Tate et al.
11 carbon white 0.57-1.21) circulatory 0.60 (1987)
plants, population disease,
1974-83 (USA) respiratory 0.51
disease
Table 96. (continued)
Group, no., Comparison Exposure concentration, Deaths Dose-response, remarks Other tumour sites Reference
workplace, group, no. exposure to other and diseasesa
study period workplace chemicals, smoking habits No. SMR, RR
(95% Cl) Type SMR,
RR
176, Regional Total dust: 5-100 mg/m3; 1.97 - - - Gustavsson
waste population some information on (1.21-2.75) & Reuterwall
incinerator, smoking habits (1990);
1951-85 Gustavsson
(1989)
Nickel/copper Regional Laboratory fume 50 1.47 Lung tumours correlated - - Verma et al.
smelter and population generation experiments (1.02-1.81) with duration of exposure (1992)
refinery; (Ontario) showed high only it exposed to PAH
special concentrations of PAH
subcohorts (asphalt > tar > mastic);
exposed to no information on smoking
tar habits
OR, odds ratio; RR, relative risk; SMR, standard mortality ratio; SIR, standard incidence ratio; BaP, benzo[a]pyrene
SMR or RR for whole study population is given; results for special subcohorts, subdivided by e.g. level or duration of exposure or
latency, are given under remarks, unless otherwise stated
* Statistically significant at p < 0.01; ** statistically significant at p < 0.01; *** statistically significant at p < 0.001
a Only statistically significant (p < 0.05) increased or decreased rates are shown; unless otherwise stated, refers to mortality
b Benzene-soluble fraction of total particulate matter
Table 97. Case-control studies of cancer types possibly associated with exposure to polycyclic aromatic hydrocarbons PAH)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
Lung tumours
NR Exhaust from Patients with cancer of prostate, 1.41 2 miles from coke oven; some Lyon et al. (1981)
coke plant breast, or brain correlation with distance from
coke oven, highest at 2 and 3
miles distance; effect may also
be due to occupational exposure
Asphalt - - 1.12 Meta-analysis of 5 studies Partanan & Boffetta
workers, (0.93-1.34) (1994)
paving,
roofing,
others,
113 Iron foundry 249 all other deaths in cohort of 2.36* OR for workers aged 42-64; Egan-Baum et al.
cohort of versus steel or foundry workers except non- 1.19 of workers aged > 65, only 65% (1981)
foundry non-ferrous malignant respiratory disease successfully traced
workers foundry and other cancers
12, Occupation in 58 from cohort of steel foundry 4.51 Moulin et al. (1990)
cohort of steel stainless-steel workers
workers versus ferro-
chromium
(a) 51, (b) 47, BaP: 0.1-15 From cohort of iron foundry - More cases exposed to high PAH Tola et al. (1979)
cohort of iron µg/m3; small workers concentrations than controls, not
foundry differences (a) 153, matched by age and statistically significant; some
between intensity of exposure, no cancer information on smoking habits
workplaces cases
Table 97. (continued)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
(b) 47, also same time of entry
into foundry
(c) 27, cases compared with
expected contribution of
exposure of foundry workers
74, cohort of Exposure to 1138, manual workers at same 2.00 Figure given for exposure for Armstrong et al.
aluminium coal-tar pitch plant, not matched, similar (1.33-2.75) 20-41 years at Soderberg pots, (1994)
production volatiles; distribution of birth years no data for exposure to
workers, measurements of benzene-soluble material in
Soderberg benzene-soluble general; strong correlation with
and prebake material; estimated exposure concentration, duration
process concentrations for of exposure and latency;
different job adjustment for smoking: no
categories; 0-3.5 difference in risk
mg/m3
45 butchers Combustion (a) 99 all butchers and 0.84 Smoking habits available Gustavsson (1989)
and slaughter- products, only slaughter-house workers dying (0.40-1.77)
house low, intermittent from malignant disease
workers, 11 exposure to (b) 100 random sample of all
years PAH deceased butchers and
slaughter-house workers
(a) and (b) except tumours related
to chemical exposure
Table 97. (continued)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
Skin tumours
376 patients Occupational 752 general population; 752 1.14 No correlation with duration of Kubasiewicz et al.
with skin exposure to patients from hospitals exposure; no increased risk for (1991)
cancer PAH randomly sampled other exposures related to PAH,
i.e. tar, pitch, soot, coke,
bituminous mass; smoking habits
not availalble
Renal-cell carcinoma
1982-87, Occupational 64 patients with haematuria, 2.54 Exposure to coal, tar, or pitch Sharpe et al. (1989)
hospitals in exposure to 61 controls and cases not treated (0.96-6.99) RR for exposure to coal increased
Montreal, hydrocarbons with haemodialysis 0.29 from 10 to 24 years of age; strong
Canada (predisposes for renal cancer) (0.16-.74) correlation with duration of exposure
and exposure concentration; no
correlation with latency (no case or
control with latency < 20 years);
smoking habits: no significant
difference between cases and
controls; history of smoking > 20
cigarettes per day associated with
tendency to higher stage disease
Table 97. (continued)
Cases, no., Exposure: Controls: no., matching Odds ratio Remarks Reference
tumour type, PAH analysed, (95% Cl)
study group exposure
concentration
Urinary bladder cancer
138/(66+ 69), Occupational 414, from cohort 2.63 Data consistent with former study; Tremblay et al.
1970-88 exposure to (1.29-5.37), strong correlation with exposure (1995); Theriault et
aluminium benzene-soluble adjusted concentration and duration of al. (1984)
plant, material for smoking exposure highest risk: Soderberg
Soderberg or BaP potroom workers: 5.15; 1930-54:
BaP, max, 51.5 µg/m3;
benzene-soluble material, max
10 mg/m3; 1985-89: BaP, max,
3.1 µg/m3; benzene-soluble
material, max, 0.36 mg/m3
General Contact with From population 2.61 All figures given for 8-28 years Risch et al. (1988)
population, diesel or traffic (0.70-12.5) employed
1979-82 fumes
Aluminium 1.69
smelting (1.24-2.31)
Contact with 3.11
tars, asphalt (1.19-9.68)
OR, odds ratio; RR, relative risk; SMR, standard mortality ratio; SIR, standard incidence ratio; NR, not reported; BaP, benzo[a]pyrene
SMR or RR for whole study population is given; results for special subcohorts, subdivided by e.g. level or duration of exposure,
latency, are given under remarks, unless otherwise stated
a Only statistically significant increased or decreased rates, unless otherwise siated, mortality
* statistically significant at p < 0.05
** statistically significant at p < 0.01
*** statistically significant at p < 0.001
A significantly increased risk for lung cancer (SMR, 1.95) was found
among a cohort of over 5000 workers who were heavily exposed at coke
ovens in coke plants and were followed-up for over 30 years. Although
no data were available on smoking habits, the observed effect is not
likely to be due to smoking since unexposed steel workers in a
comparison group were assumed to have similar smoking habits. In
addition, a high correlation was seen between the risk for respiratory
cancer and the concentration and duration of exposure. The authors
noted however, that the rates of respiratory cancer decreased during
the follow-up period, suggesting that implementation of emission
controls and occupational exposure limits has been beneficial
(Costantino et al., 1995). The increased risk found in this study was
also seen in other studies, including some on coal gasification (Doll
et al., 1972; Manz et al., 1983; Swaen et al., 1991). Other studies,
however, and especially those involving small cohorts, did not show
increased rates for lung cancer among coke-oven workers (Hurley et
al., 1983; Gustavsson & Reuterwall, 1990; Chau et al., 1993).
Coke-oven workers in China who were exposed to PAH were reported to
have decreased titres of immunoglobulins M, G, and A in their serum
and decreased immune function (Lei, 1993).
Several epidemiological studies have been performed on the potential
risks of handling asphalt (for a review, see Partanen & Boffetta,
1994; see also Table 96). The job descriptions included: roofer,
waterproofer, highway maintenance worker, production of hot-lay
asphalt, slater, grader, paver, surfacer, and mastic asphalt worker.
Their exposure to PAH depended on the type of asphalt involved. When
bitumen is used as the binder, the PAH content is relatively low, with
about 200 µg/m3 total PAH and up to about 5 µg/m3 benzo [a]pyrene
(Hansen, 1989; see also section 5.3); bitumen binders contain
primarily asphaltenes, straight and branched aliphatic hydrocarbons,
naphthene aromatics, and resins. Most of the PAH are removed by vacuum
distillation, but they may be formed during cracking operations or be
reintroduced in the flux used in blended or fluxed bitumens. Coal-tars
and coal-tar pitches have been used as binders, especially in the
past, and may contain substantial amounts of PAH (Partanen & Boffetta,
1994). These workers may also be exposed to several other substances,
such as silica, limestone, and asbestos (Chiazze et al., 1991;
Partanen & Boffetta, 1994). In the meta-analysis of Partanen &
Boffetta (1994), increased risks for lung tumours were seen for both
pavers and roofers; tumours of the stomach, bladder, and skin and
leukaemia were also observed. The excess risks were more pronounced
for roofers than for pavers. It is not clear however, if these effects
are due to exposure to PAH, as it could not be determined whether the
carcinogenicity was due to exposure to bitumen or to tar fumes.
Workers are exposed dermally to very high concentrations of PAH when
impregnating wood with creosote, as shown by measuring biomarkers,
especially the excretion of 1-hydroxypyrene in urine (see section
8.3.2). In a cohort study on 922 creosote-exposed workers, a
significant increase in the risk for skin cancer (SIR, 2.37) and
increased risks for lip cancer and malignant lymphoma were observed.
Since the work involves some time outdoors, it cannot be ruled out
that exposure to sunlight contributed to the risks for cancers of the
skin and lip (Karlehagen et al., 1992).
The PAH concentrations in iron, steel, and other ferroalloy foundries
reach levels of 50 µg/m3 and that of benzo [a]pyrene about 10 µg/m3
(Verma et al., 1992; see also section 5.3). Workers in these plants
also have nearly ubiquitous exposure to silica sand. Silicosis and
other chronic respiratory abnormalities have been reported for decades
to be the major health problems of foundry workers. These workers are
also exposed to asbestos, used for heat protection and insulation
around furnaces, to benzene, toluene, formaldehyde, iron, and,
depending on the kind of metal or alloy, to metals such as lead,
chromium, manganese, nickel, copper, cadmium, and zinc. Increased
mortality from lung cancer has been observed consistently in many
studies of foundry workers (Palmer & Scott, 1981; Andjelkovich et al.,
1990). When the results of all the relevant studies are combined, the
SMR is 1.43 (Andjelkovich et al., 1990); however, those authors argue
that the elevated risk is due to smoking, because the SMRs did not
increase with time since first employment in a foundry or with the
length of employment in a foundry. Furthermore, the incidence of
emphysema, which can also be attributed to smoking, is increased in
epidemiological studies of foundry environments.
Information on the possible risks for cancer due to exposure to PAH
can also be obtained from studies of workers in aluminium plants. Two
types of anode are used: the continuous Söderberg anode and the
prebaked anode. Both are manufactured from coal-tar pitch and coke,
and coal-tar pitch volatiles evaporate from them during baking. The
exposure is heavier in potrooms where the Söderberg process is used,
because the anodes are baked continuously. With use of prebaked
anodes, exposure to PAH may occur in the carbon area where the anodes
are prebaked. During Söderberg electrolysis, workers may be exposed to
0.3-3.5 mg/m3 of benzene-soluble organic material and 3-35 µg/m3
benzo [a]pyrene; workers in the carbon area are exposed to
0.4-1.2 mg/m3 benzene-soluble organic material and 0.4-1.2 µg/m3
benzo [a]pyrene. Similar concentrations have been detected in other
parts of such plants (Rnneberg & Langmark, 1992). These workers are
also exposed to fluorides, carbon dioxide, sulfur dioxide, magnetic
fields, hot work environments, and, in some cases, asbestos.
In a large case-control study, an increased risk for lung cancer was
found with exposure in a Söderberg potroom, and a significant
correlation was seen between the increased risk and the duration and
concentration of exposure and latency. Adjustment for smoking did not
alter the correlation (Armstrong et al., 1994). Similar increases were
detected in cohort studies (Rockette & Arena, 1983; Mur et al., 1987).
The studies differed with regard to the magnitude of the risk and the
presence of a dose-response relationship (see also reviews by Abramson
et al., 1989; Rnneberg & Langmark, 1992). In some studies, however,
few cases were available (Rockette & Arena, 1983; Mur et al., 1987;
Rnneberg & Andersen, 1995).
Work in potrooms where Söderberg electrolytic cells were used was also
associated with an increased risk for urinary bladder cancer (Spinelli
et al., 1991; Rnneberg & Langmark, 1992; Tremblay et al., 1995). This
risk may be due to exposure not only to PAH but also to aromatic
amines, which have been detected in the potrooms (Tremblay et al.,
1995).
Asthma-like symptoms, lung function abnormalities, and chronic
bronchitis have also been detected in workers in aluminium plants
(reviewed by Abramson et al., 1989; Kongerud et al., 1994), but the
quality of the studies in which these effects were shown is variable.
These diseases are believed to be due not to PAH but rather to
exposure to alumina, cryolite, carbon, fluorides, and sulfur dioxide;
however, the causative agents have yet to be defined.
Increased risks for lung cancer were found in several studies of
workers exposed to diesel exhausts (WHO, 1996). In comparison with the
occupations described above, the concentrations of PAH to which these
workers are exposed are usually relatively low. The benzo [a]pyrene
concentrations in automobile repair shops and garages reach about 70
ng/m3 (Waller, 1981; Lindstedt & Sollenberg, 1982; Waller et al.,
1985), and truck drivers are exposed to less than 10 ng/m3 (Guillemin
et al., 1992).
The finding of an increased risk for lung cancer must always be
interpreted in relation to the influence of tobacco smoking. The
tobacco smoking habits were seldom known for all of the persons
studied, but there are several reasons for concluding that the
increased risks for lung cancer are not due solely to tobacco smoking:
The general population is often used as a reference group, but their
lung cancer rate is usually lower than that of the working population
because fewer members of the general population are tobacco smokers
(Gibson et al., 1977; Hansen et al., 1989; Andjelkovich et al., 1990;
Moulin et al., 1993). Calculations presented by several authors
indicate that differences in tobacco smoking habits can contribute
only about 20% of the excess risk for lung cancer (Hurley et al.,
1983; Maclaren & Hurley, 1987; Gustavsson et al., 1988; Andjelkovich
et al., 1990; Moulin et al., 1993). Greater contributions are
improbable, since other illnesses usually linked to tobacco smoking
have not been observed to occur more frequently than in controls.
Another reason for excluding tobacco smoking as the major reason for
the increased lung tumour rates is that increased risks also have been
found in studies in which the controls were workers with other
occupations, and not the general population. In this case, it can be
assumed that the tobacco smoking habits are similar, and it is
probable that the increased risks are due to the exposure conditions
and not to tobacco smoking (Costantino et al., 1995). In addition,
several studies show a strong correlation between the risk for
respiratory cancer and the concentration and duration of exposure and
latency. Finally, in studies in which the information on tobacco
smoking habits was good and adjustments could be made for the
influence of tobacco smoking (Spinelli et al., 1991; Armstrong et al.,
1994), tobacco smoking had no effect on the cancer risk. As no
information is available about the comparative cancer risks of
non-smokers and smokers, however, the increased risk for lung cancer
may always represent a combined risk due to tobacco smoking and the
exposure conditions.
Although workers are always exposed to several substances, it seems
plausible to attribute the increased lung cancer risks at least
partially to PAH. The increased risk is seen for workers in several
occupations which have exposure to PAH in common. Although other
carcinogenic chemicals were present, they differed with each
occupation. Airborne high-molecular-mass PAH, which are considered to
be the most carcinogenic, are adsorbed mainly onto particulate matter
(see section 4.1.2), and it was often difficult to distinguish the
toxicological effects caused by particles from those caused by the PAH
themselves.
8.3 Biomarkers of exposure to PAH
Several methods have been developed to assess internal exposure to PAH
after exposure in the environment and in workplaces, which can be used
to evaluate the adequacy of protective regulations. In most studies,
metabolites of PAH were measured in urine, 1-hydroxypyrene being
widely used.
The genotoxic effects of PAH have been determined in tests for
mutagenicity in urine and faeces, micronucleus formation, chromosomal
aberration and sister chromatid exchange in peripheral blood
lymphocytes, adducts of benzo [a]pyrene with DNA in peripheral
lymphocytes, and other tissues and with proteins such as albumin; and
antibodies to DNA adducts. In addition, oncogene expression and
immunostaining of differentiation antigens on lung cell surfaces have
been measured as biological indexes of the risk for lung cancer.
8.3.1 Urinary metabolites in general
The metabolites measured in urine and faeces include urinary
thioethers (Burgaz et al., 1992), 1-naphthol (Bieniek, 1994; Hansen et
al., 1994, 1995), b-naphthylamine (Hansen et al., 1994), hydroxy
phenanthrenes (Martin et al., 1989; Adlkofer et al., 1990; Grimmer et
al., 1994; Mannschreck et al., 1996), and 1-hydroxypyrene.
No difference in thioether excretion in urine was observed between
controls and coke-oven workers or workers in coke and
graphite-electrode-producing plants. It was concluded that the
determination of thioethers in urine is of little value, since smoking
is a strong confounding factor (Ferreira et al., 1994a,b; Reuterwall
et al., 1991).
A good correlation was found with the excretion of hydroxylated
phenanthrenes and 1-hydroxypyrene in urine (Mannschreck et al., 1996).
When phenanthrene, pyrene, and benzo [a]pyrene metabolites are
determined simultaneously, precursors of carcinogens are also
measured, thus providing an estimate of individual risk (Grimmer et
al., 1993).
8.3.2 1-Hydroxypyrene
1-Hydroxypyrene, a metabolite of pyrene, was introduced as a biomarker
of exposure to PAH by Jongeneelen et al. (1986) and has since been
widely used. Its advantages are that pyrene is present in all PAH
mixtures at relatively high concentrations (2-10%), and in certain
environments the pyrene content of the total PAH is fairly constant
(Zhao et al., 1990; Buchet et al., 1992; Jongeneelen, 1994). In
studies at different workplaces, a strong correlation was found
between the pyrene concentrations in air and those of
benzo [a]pyrene, other selected PAH, and total PAH (Jongeneelen et
al., 1990; Tolos et al., 1990; Zhao et al., 1990; Van Rooij et al.,
1992; Ferreira et al., 1994a,b; Jongeneelen, 1994; Elovaara et al.,
1995; Levin et al., 1995; Ovrebo et al., 1995; Quinlan et al., 1995a).
Pyrene is metabolized predominantly to 1-hydroxypyrene (Grimmer et
al., 1993, 1994; Levin et al., 1995), which can be measured easily. In
contrast to other PAH metabolites, which are excreted mainly in
faeces, 1-hydroxypyrene is excreted in urine.
8.3.2.1 Method of determination
The method of determination consists of enzymatic hydrolysis of
conjugated 1-hydroxypyrene in urine samples, followed by solid-phase
extraction and high-performance liquid chromatographic separation with
fluorescence detection (Jongeneelen et al., 1986). An enzyme-linked
immunosorbent assay has also been used (Santella et al., 1994).
In most publications, the concentrations of metabolites in urine are
adjusted to that of creatinine in order to compensate for variations
in urine flow rates; however, it must be borne in mind that creatinine
excretion fluctuates widely because of internal and external factors.
Therefore, correction of the concentrations of chemicals in urine in
this way does not necessarily improve the correlation with exposure
(Levin et al., 1995). For comparisons, a mean urinary creatinine
concentration of 13 mmol/litre can be assumed, giving the
relationship:
1 µmol 1-hydroxypyrene per mol creatinine
= 1.93 µg/g creatinine
= approx. 3 ng/ml urine.
Since most authors give 1-hydroxypyrene concentrations in µmol/mol
creatinine, that unit is used in the following text and tables.
8.3.2.2 Concentrations
(a) General population
The concentrations of 1-hydroxypyrene in urine from the general
population exposed to PAH are compiled in Table 98. The background
concentrations of 1-hydroxypyrene in different countries range from
0.06 to 0.23 µmol/mol creatinine. No difference related to age or sex
was seen (Zhao et al., 1992), and ethanol consumption did not
influence 1-hydroxypyrene concentrations (Van Rooij et al., 1994a).
For nonsmokers, food accounted for 99% of the total daily pyrene
intake (Van Rooij et al., 1994a). Five volunteers who ate low-PAH
meals and high-PAH meals showed 100- to 250-fold increases in
benzo [a]pyrene dose, accompanied by a four- to 12-fold increase in
1-hydroxypyrene excretion in urine (Buckley & Lioy, 1992). A 10- to
80-fold increase was detected in one subject who ate 9 oz (250 g) of
charcoal-grilled beef. In this study of 10 subjects, an eightfold
interindividual variation in urinary excretion of 1-hydroxypyrene was
found after one day. As this variation was not appreciably altered
after adjustment of the 1-hydroxypyrene concentration by urinary
creatinine concentration or individual body weight, it was assumed to
be due to individual differences in the rate of absorption,
metabolism, or excretion of pyrene (Kang et al., 1995).
The intake of pyrene from cigarette smoking (12 nmol/day) is about the
same as the dietary intake from normal food (9.4 nmol/day) (Van Rooij
et al., 1994a). Tobacco smokers who are not otherwise exposed to PAH
have about twice the level of 1-hydroxypyrene in their urine as
nonsmokers (Jongeneelen et al., 1990; Sherson et al., 1992; Van Rooij
et al., 1994a; Levin et al., 1995), although no significant difference
was found in some studies (Jongeneelen et al., 1988b; Zhao et al.,
1992; Ny et al., 1993).
Urine samples from schoolchildren living along arterial roads in Tokyo
had 1.1-1.6 times more urinary 1-hydroxypyrene than those from
children in a less polluted suburban area (Kanoh et al., 1993). Much
higher levels were detected in persons living in highly industrialized
regions in Poland, due to emissions from coke-oven plants. The
concentrations are a maximum of twice as high in winter as in summer
(Jongeneelen, 1994; Ovrebo et al., 1995). 1-Hydroxypyrene levels of up
to 1.5 µmol/mol creatinine have been detected in towns in China (Zhao
et al., 1990, 1992).
Very high exposure to PAH occurs during application of coal-tar
ointments or shampoos by patients with eczema or psoriasis. The mean
1-hydroxypyrene concentrations reached 500 µmol/mol creatinine, and
the maximum value was 5000 µmol/mol creatinine (Santella et al.,
1994).
Table 98. Concentrations of 1-hydroxypyrene in urine (µmol/mol creatinine) in the general
population
Type of exposure or No. of 1-Hydroxypyrene Reference
population investigateda subjects
Median Range
or meanb
Non-smokers
General population, 28 0.06c < 0.02-0.17 Goen et al. (1995)
southern Germany
General population 49 < 0.02c 0.02-0.28 Goen et al. (1995)
Southern Germany
Students and university 24 0.23d - Jongeneelen et
personnel, al. (1986)
Netherlands
University staff, 52 0.26 - Jongeneelen et
Netherlands al. (1988b)
University staff, 39 0.12e 0.04-029 Van Rooij et al.
Netherlands (1994a)
Students and university 15 0.24e - Burgaz et al.
staff, Turkey (1992)
Smokers
General population, 21 0.12c < 0.02-0.68 Goen et al. (1995)
Germany
General population, 20 0.14c < 0.02-0.3 Goen et al. (1995)
Germany
Students and university 22 0.27d - Jongeneelen et
staff, Netherlands al.(1986)
Students and university 38 0.28 - Jongeneelen et
staff, Netherlands al. (1988b)
Students and university 37 0.25e 0.10-0.79 Van Rooij et al.
staff, Netherlands (1994a)
Students and university 14 0.33e - Burgaz et al.
staff, Turkey (1992)
Smoking status not specified
Persons in urban and 27 0.20d - Elovaara et al.
rural areas Estonia (1995)
Persons from rural areas, 81 - 0.19-0.27 Ovrebro et al.
Biala Podlaska, Poland (1995)
Students and university 18 N 0.077d 0.002-0.57 Viau et al. (1993)
staff, Montreal, Canada 3 S
Healthy volunteers, 36 N 0.14e 0.02-0.98 Santella et al.
Columbia, MO, USA 17 S (1994)
Table 98. (continued)
Type of exposure or No. of 1-Hydroxypyrene Reference
population investigateda subjects
Median Range
or meanb
Polluted ambient air
School children, NR - approx.0.4-0.6 Kanoh et al.
Tokyo, Japan, M+F, Nf (1993)
Urban population 74 N 0.68e - Zhao et al. (1992)
Beijing, China 84 S 0.76e
Urban population, 13 N 1.55e - Zhao et al. (1990)
Shenyang, China, F
Urban population, 17 N 1.20e - Zhao et al. (1990)
Taiyuan, China, F
Urban population, 15 N 0.67e - Zhao et al. (1990)
Beijing, China, F
Urban population, 15 N 0.72e - Zhao et al. (1990)
Beijing, China, F (girls)
Urban population, 72 0.66e - Jongeneelen
Bytom, Silesia, (1994)
Poland (boys)
Urban population, 76 0.59e - Jongeneelen
Bytom, Silesia, (1994)
Poland (girls)
Gliwice, Silesia, Polandg 30 - 0.84-1.54 Ovrebro et al.
(1995)
Other exposures
Windsurfers sailing on 6 0.32e 0.16-0.81 Jongeneelen
polluted water, Ketelmeer, (1994)
Netherlands
Therapeutic treatment with coal-tar
Patients with eczema 5 - approx.50-500 Jongeneelen et
al. (1986)
Patients with psoriasis 4 - 13.2-811c Clonfero et al.
(1989)
Patients with psoriasis 53 547e 10-5160 Santella et al.
(1994)
Patient with psoriasis 1 3.45 - Viau et al. (1995)
Table 98. (continued)
Type of exposure or No. of 1-Hydroxypyrene Reference
population investigateda subjects
Median Range
or meanb
Food
Eating 9 oz [250 g] 10 approx.0.5e,h - Kang et al. (1995)
char-broiled beef 0.15-1.2
N, non-smokers; S, smokers; M, males; F, females; NR, not reported
a Unless otherwise stated, male persons were investigated; in some cases, insufficient
characterization of exposure given
b 1-Hydroxypyrene concentration in urine (µmol/mol creatinine), range, and, if available,
median concentration; otherwise, geometric or arithmetic means
c Calculated from 1-hydroxypyrene concentrations given in original reference as µg/g
creatinine.
d Geometric mean
e Arithmetic mean
f Benzo[a]pyrene measured at 0.0006-0.0024 µg/m3 by stationary sampling
g Benzo[a]pyrene measured at 0.009-0.041 µg/m3 by stationary sampling
h Calculated from 1-hydroxypyrene concentrations given in original reference as ng/ml
urine or pmol/ml urine
(b) Workplaces
1-Hydroxypyrene concentrations have been measured in the urine of
persons at various workplaces (Table 99); the urine of concurrent
controls was examined in most investigations. Unexposed workers at the
same plant, such as administrative workers, had slightly higher
1-hydroxypyrene concentrations than the general population (Zhao et
al., 1990; Buchet et al., 1992; Ny et al., 1993; Levin et al., 1995).
The highest 1-hydroxypyrene excretion, up to 90 µmol/mol creatinine,
was found in urine from workers impregnating wood with creosote,
although the PAH levels in the air were quite low. The high exposure
can be attributed to significant dermal uptake, which is several times
higher than that by inhalation (see below).
Other workplaces where there is heavy exposure are coke ovens,
coal-liquefaction plants, aluminium plants, and plants producing
carbon or graphite electrodes. The concentrations of 1-hydroxypyrene
in the urine of workers at these sites were 1-10 µmol/mol creatinine.
Table 99. Concentration of 1-hydroxypyrene in urine (µmol/mol creatinine) at industrial workplaces without and with
exposure to polycyclic aromatic hydrocarbons
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Controls
Unexposed workers in - 120 0.11 < 0.05-1.08 Boogaard and van
petrochemical industry Sittert (1994, 1995)
Office workers at graphite - 9N 0.33d - Buchet et al. (1992)
electrod- producing plant 6S 0.36d
Unexposed workers in coke - 137 0.26e,f 0.01-1.04 Ferreira et al. (1994a,b)
and graphite electrode-
producing plants
Workers in administration of - 13N N: 0.05f < 0.05-0.12 Goen et al. (1995)
municipal waste incineration 8S S: 0.09f < 0.05-0.67
Workers in water supply - 119 0.008 - Hanson et al. (1994)
plants, cotton manufacture,
garbage recycling
Workers in various - 121 0.012e - Hansen et al. (1995)
environments
Unspecified control group - 52N 0.26 - Jongeneelen et al. (1988b)
38S 0.28
Workers in shipping yards - 14N 0.17d - Jongeneelan et al. (1990)
at hot rolling mill 28S 0.51d
Industry, Netherlands - 28 0.51 -
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Coke-oven office workers Levin et al. (1995)
Before renovation - 8N 0.18g -
After renovation - 8N 0.09g -
Construction workers - 34 N 0.4g - Levin et al. (1995)
Water supply workers - 26 N N: 0 0-0.010 Omland et al. (1994)
42S S: 0 0-0.022
Unexposed workers - 10N N: 0.05d,f < 0.03-0.12 Schaller et al. (1993)
10S S: 0.21d,f 0.03-1.2
Workers in water - 20N N: 0.16e 0.1-0.22 Sherson et al. (1992)
purification plants, 26S S: 0.26e 0.18-0.34
Denmark
Guards in aluminium plant - 9 0.31d - Ny et al. (1993)
Maintanance work in - 48 0.61e,f - Van Hummelen et al. (1993)
blast furnace
Office workers in steel - 10 0.51e - Zhao et al. (1990)
plant
Coke ovens
Work at topside oven 0.8-32 3 N N: 5.7d,f - Buchet et al. (1992)
5.9d 3 S S: 6.1d,f
Work at benchside - 4 N N: 1.2d,f -
6 S S: 0.75d,f
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Various occupations 0.39-13 93 N N: 2.7f 0.25-16 Cenni et al. (1993)
near ovens 68 S S: 3.5f 0.29-29
Subgroup topside 2.18 21 6.64f 0.29-29
Subgroup of larry-car - 12 7.76f 3.5-16
operators
Workers with various - 41 N 0.94e - Clonfero et al. (1995)
tasks 65 S 1.53e
Workers at two coke- - 54 0.78e,f 0.01-48 Ferreira et al. (1994a,b)
oven plants
Work at two top side - 19 3.3d 0.8-7.5 Jongeneelen et al. (1990)
ovens 9 2.7d 1.3-6.5
Other work: coke side, - 25 1.9d 0.6-4.1
push side, maintenance
Coke oven 0.9-37 10 4.7g 0.3-30g Levin et al. (1995)
Before renovation, 4
various occupations
After renovation 0.2-6.8 10 1.3g 0.3-5.7g
0.7
Coke workers at 3 plants 0.72-1.5 66 2.45-13.48 - Ovrebro et al. (1995)
Oven workers 33 0.39e,f - van Hummelen et al. (1993)
7 N
26 S
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Subgroup at top side - 7 0.85e,f -
Personnel at coke side, - 13 2.64 0.54-11 Van Rooij et al, (1994b)
push side, top side,
miscellaneous
Workers at top of ovens - 15 S 4.34e - Zhao et al. (1990)
Workers at top or side - 12 S 2.87e -
of ovens
Coal liquefaction
Engineers (control room - 5 8.53 - Quinlan et al. (1995b)
and plant operations)
Technicians (plant - 5 3.74 -
maintenance)
Petrochemical industry
Various operations - - - 0.25-0.68 Boogaard & van Sittert
Workers inspecting furnace, - - - max. 1.56 (1994, 1995)
replacing burners in boilers,
manufacturing rubber grades
Subgroup: production of < 0.01-< 0.22 1
needle coke from ethylene
cracker residue:
Maintenance 3 1.02 0.16-5.51
Operation < 0.17 12 1.13 0.22-13.2
Coal-tar distillation - 4 - 3.7-11.8 Jongeneelen et al. (1986)
operators, cleaner
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Creosote impregnation
Wood impregnation plant - 19 1.6d,e 0.18-10 Viau et al. (1993)
Wood impregnation plant 0.01-0.05 6 97e - Elovaara et al. (1995)
(0.012d)
Wood impregnation plant - 1 20-90 Jongeneelen et al. (1988b)
Aluminum production
Soderberg type anodes:
Potroom workers, 1.9-36 9 0.4-3.6g
respiratory protection 2.8 2.1g - Levin et al. (1995)
50% of time
Potroom workers - - - - Ny et al. (1993)
Electricians, technicians, - 5 0.69d -
engineers, laboratory
workers, industrial
hygiene personnel
Foremen, technicians, 0.88d 4 2.6d -
tappers, crucible cleaner
Crane operators, all- 7.9d 8 14d -
rounders, electrician
Potmen 2.2d 9 31d -
Electrode men 37d 4 40 -
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Prebake type anodes:
Electrode production - 20 0.17-27 Van Rooij et al. (1992)
compartment
Paste plant 1.3 8 3.0d 1.6-7.4
Bake oven 0.3 5 4.4d 0.98-13
Pot relining department 1.2 7 6.0d 1.9-12
Anode bake area - 28 - 0.55-3.6 Tolos et al. (1990)
Anodes from liquid - 17 - 2.1-37f Schaller et al. (1993)
pitch and coke
Anodes not specified - 28 4.2f 0.05-65 Goen et al. (1995)
Production of electrodes
Graphite electrodes - 15 3.2f - van Hummelen et al. (1993)
Graphite electrodes Buchet et al. (1992)
End-product conditioning 0.002-0.4 3N N: 0.55f -
0.03d 7S S: 0.55f
Second thermal treatment 0.002-0.5 8N N: 0.57f -
of electrodes 0.03d 17S S: 0.79f
Progressive heating of raw 0.002-1.9 2N N: 2.53f -
electrodes 0.04d 4S S: 3.13f
Maintenance and repair 0.002-7.5 15N N: 1.21f -
0.21d 2S S: 3.76f
Grinding and mixing of raw 0.57-25 5N N: 2.98f -
components 5.4d 9S S: 2.83f
Electrode impregnation 0.83-73 3N N: 4.14f -
62d 5S S: 4.96f
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Graphite electrodes - 93 1.7e,f 0.03-20 Ferreira et al. (1994a,b)
Carbon electrodes I - 6 5.8f 3.7-43 Goen et al. (1995)
Carbon electrodes II - 3 12.7f 9.4-15
Carbon electrodes III - 14 8.4f 1.1-65
Graphite electrodes Mannschreck et al. (1996)
Crushing 0.09e 2 5.0e,f 0.6-9.4
Baking 1.1-12e 30 12f 0.9-170
Graphitization 0.01-0.11e 24 0.9f 0.1-3.3
Impregnation 0.44-1.1e 9 11f 3.2-42
Conditioning 0.01e 2 1.2e,f 0.9-1.5
Carbon black
Plants manufacturing - 5 - 0.32-0.35 Gardiner et al. (1992)
carbon black
Newspaper printing ink - 1N 0.47 - Jongeneelen et al. (1988b)
1S 0.67
Road paving
Bitumen and coal-tar binders - 43 - 0.9-2.8 Jongeneelen et al. (1988a)
Bitumen and coal-tar binders - 28 - 0.9-3.2 Jongeneelen et al. (1988b)
Bitumen binder - 18 N N: 0.53e - Burgaz et al. (1992)
21S S: 0.67e
Bitumen binder - 3 0.6e - Jongeneelen et al. (1988b)
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Bitumen binder < 0.05 57 0.7g - Levin et al. (1995)
Impregnation of road - 38 4.26f 0.62-22 Goen et al. (1995)
stones with coal-tar
Foundries
Iron foundry, melting, 0.02 25 N N: 0.022 0.006-0.075 Omland et al.(1994)
machine moulding, casting, 45S S: 0.027 0.006-0.16
sand preparations; high
concentration in casting
and mouding
Iron foundry < 0.002 14 2.7e 0.3-6.3 Santella et al. (1993)
0.005-0.012 14 1.8e 0.3-4.2
> 0.012 18 3.6e 0.5-9.7
Iron foundry I 0.00 19 0.013 - Hansen et al. (1994)
0-0.39 14 0.017
Iron foundry II 0.00 13 0.031 -
0-0.039 24 0.022
> 0.039 18 0.027
Iron foundry - 16 N N: 0.11e 0.09-0.13 Sherson et al. (1992)
20S S: 0.42e 0.025-0.59
Steel plant - 12S 1.34e - Zhao et al. (1990)
Diesel exhaust
Railway tunnel under < 0.000-0.04 5N N: 0.08f 0.04-0.31 Cenni et al. (1993)
construction 8S S: 0.18f 0.08-0.38
Table 99. (continued)
Type of exposure, Benzo[a]pyrene No. of 1-Hydroxypyrene Reference
population investigateda (µg/m3)b subjects
Median Range
or meanc
Gate-keepers of harbour - 3N N: 0.47e - Jongeneelen et al. (1988b)
terminal for containers 4S S: 0.67e
Waste incinerations
Municipal waste incineration - 53 0.08f < 0.05-0.41 Goen et al. (1995)
Industrial waste incineration - 43 0.06f < 0.05-0.47
Garbage incineration plant - 35N N. 0.12d,f < 0.05-0.41 Schaller et al. (1993)
17S S: 0.22d,f 0.07-0.41
Miscellaneous workplaces
Glass manufacture - 10 0.85 0.2-3.8 Goen et al. (1995)
Lubricating oils in - 7N 0.32f 0.12-0.77 Cenni et al. (1993)
earthenware factories
Clean-up of soil of a dump - 29 0.11d 0.01-0.75 Viau et al. (1993)
contaminated with coal-tars
derived from pyrolysis
of used tyres after major fire
Chimney sweeping - 27 0.05-1.4 - Goen et al. (1995)
0.36f
Meat smoking - 13 < 0.05-0.57 -
0.21f
Fire fighting 0.03-0.7 S - 0.65-1.0
N 0.51-0.6
Table 99 (continued)
N, non-smokers; S, smokers; M, males; F, females
a Unless otherwise stated, male persons were investigated; in some cases, insufficient characterization of
exposure given
b Benzo[a]pyrene, stationary or personal sampling
c 1-Hydroxypyrene concentration in urine (µmol/mol creatinine), range, and, if available, median concentrations;
otherwise, geometric or arithmetic means. Maximum concentrations are given, in post shift samples, in some
studies from the end of the week. Figures are given for the whole study population and on subgroups with high
exposure.
d Geometric mean
e Arithmetic mean
f Calculated from 1-hydroxypyrene concentrations given in original publication as µg/g creatinine or µg/litre.
g Calculated from 1-hydroxypyrene concentrations, given in the original publication as ng/ml urine
The manufacture and handling of bitumens did not result in a
significant increase in urinary excretion of 1-hydroxypyrene
(Jongeneelen et al., 1988a,b; Knecht & Woitowitz, 1990; Burgaz et al.,
1992), suggesting that the main source of exposure to PAH during
paving is the coal-tar used as a binder and not the bitumen itself
(Knecht & Woitowitz, 1990).
In some studies of occupationally exposed individuals, the difference
in urinary 1-hydroxypyrene concentration between smokers and
nonsmokers was greater than expected, suggesting a more than additive
effect of exposure and smoking on the body burden (Jongeneelen et al.,
1990; Sherson et al., 1992; Ovrebo et al., 1994; Clonfero et al.,
1995; Ovrebo et al., 1995; van Schooten et al., 1995). It was
hypothesized that the induced P450 enzymes in smokers result in faster
biotransformation and less efficient ciliary clearance of particles in
the upper airways (Van Rooij et al., 1994a).
The 1-hydroxypyrene concentrations in urine correlated in most cases
with the PAH concentrations in air (Buchet et al., 1992; Levin et al.,
1995; Mannschreck et al., 1996). The weak correlation between the
levels of pyrene in air and 1-hydroxypyrene concentrations in urine
was attributed to extensive dermal uptake of the PAH (Van Rooij et
al., 1992, 1993a,b; Ovrebo et al., 1995). The 1-hydroxypyrene
concentrations in urine correlated quite well with exposure of the
skin, monitored by analysing absorbent pads attached to skin sites
during shifts (Van Rooij et al., 1992, 1993a,b).
Significant dermal uptake, representing up to 95% of the total, was
concluded from the results of several studies of workers exposed at
coke ovens, in coal-liquefaction plants, in the petrochemical
industry, in aluminium reduction plants, in a graphite electrode
plant, in a needle-coke plant, during road paving, and while
impregnating wood with creosote oil (Jongeneelen et al., 1990; Van
Rooij et al., 1992, 1993a,b; Boogaard & van Sittert, 1994; Ferreira et
al., 1994a,b; Van Rooij et al., 1994b; Boogaard & van Sittert, 1995;
Elovaara et al., 1995; Quinlan et al., 1995a,b,c). For example,
workers impregnating wood with creosote had an average, estimated
dermal uptake that was 15 times higher than the estimated respiratory
uptake (Van Rooij et al., 1993b).
Use of dermal protection in the form of impermeable polyvinyl chloride
suits led to a substantial decrease in the urinary concentrations of
1-hydroxy-pyrene (Boogard & van Sittert, 1994, 1995). Frequent changes
of work clothes and underclothes reduced 1-hydroxypyrene excretion by
37-55% (Van Rooij et al., 1994b; Quinlan et al., 1995c).
8.3.2.3 Time course of elimination
The excretion of 1-hydroxypyrene increased significantly between the
beginning and end of a shift and from one day to another during one
week. Decreases were observed between two shifts, but the high values
did not drop to the preshift level of the day before (Jongeneelen et
al., 1988b, 1990; Tolos et al., 1990; Buchet et al., 1992; Van Rooij
et al., 1992; van Hummelen et al., 1993; Van Rooij et al., 1993b;
Omland et al., 1994; Van Rooij et al., 1994b; Elovaara et al., 1995;
Quinlan et al., 1995a,b; van Schooten et al., 1995). After an
exposure-free weekend, the 1-hydroxypyrene concentrations in the urine
of heavily exposed workers did not drop to control levels (Jongeneelen
et al., 1988b, 1990; Viau et al., 1993; Elovaara et al., 1995; Quinlan
et al., 1995b). The baseline values in exposed workers are slightly
higher than those in unexposed controls (Jongeneelen et al., 1988a,
1990; Tolos et al., 1990; Quinlan et al., 1995a,b).
Elimination of 1-hydroxypyrene is biphasic, a moderately rapid phase
being followed by a second, much slower elimination (Jongeneelen et
al., 1988b, 1990; Viau et al., 1995). The half-lives of the first
phase have been determined at various workplaces and for
non-occupationally exposed persons after inhalation, dermal, and oral
exposure. Regardless of the route of exposure, they range from 4.4 to
48 h, most values being about 16 h (Jongeneelen et al., 1988b, 1990;
Buchet et al. 1992; Buckley & Lioy, 1992; Schaller et al., 1993;
Boogard & van Sittert, 1994; Quinlan et al., 1995a,b; Viau & Vyskocil,
1995; Viau et al., 1995). In one study, a half-life of 16 days was
given for the slower phase (Jongeneelen et al. 1988b). This slow
elimination suggests that pyrene accumulates in a secondary
compartment, most probably adipose tissue, from which it is released
only slowly (Jongeneelen et al., 1990).
8.3.2.4 Suitability as a biomarker
When 1-hydroxypyrene was used as a biomarker for exposure to PAH, the
oral, dermal, and inhalation routes were all shown to be important.
Furthermore, low levels of exposure to PAH can be determined. A great
advantage is that the determination of urinary 1-hydroxypyrene is easy
and rapid and thus well suited for use in large-scale epidemiological
studies.
Comparison of different work environments may, however, be difficult,
because the proportion of pyrene in the total PAH or in comparison
with benzo [a]pyrene may vary (Jongeneelen et al., 1990; Buchet et
al., 1992; van Rooij et al., 1993a; Boogard & van Sittert, 1994;
Hansen et al., 1994). For example, the creosote oil used in a wood
impregnation plant contained about 3.4% pyrene and less than 0.0004%
benzo [a]pyrene. Levels of 2-10% pyrene and 0.4-0.6% benzo [a]pyrene
are found in coal-tar, which is the main PAH contaminant in the coke
industry, in the primary aluminium industry, and during road paving
with tar. Polluted ambient air contains about 6.5% benzo [a]pyrene
and 1.8-2.7% pyrene (IARC, 1985; Zhao et al., 1990).
It is not currently possible to assess the risk presented by exposure
to PAH on the basis of urinary 1-hydroxypyrene concentrations, as
epidemiological studies have not demonstrated a relationship with
long-term effects. An indirect dose-response relationship between
urinary 1-hydroxypyrene level and the relative risk for lung cancer
has, however, been estimated for coke-oven workers: 2.3 µmol
1-hydroxypyrene per mol creatinine was estimated to be equal to a
relative risk for lung cancer of approximately 1.3 (Jongeneelen,
1992). Because of the varying composition of PAH mixtures, this risk
estimation cannot be used for other workplaces or ambient air, where a
correction factor may be necessary.
8.3.3 Mutagenicity in urine
The mutagenicity of urine from persons exposed to PAH has been assayed
in a number of studies by Ames' test with Salmonella typhimurium
TA98 or TA100, with and without metabolic activation. In most of these
studies, several urine samples from both control and exposed subjects
could not be assayed because of the toxicity of the urine (Heussner et
al., 1985; Jongeneelen et al., 1986; Clonfero et al., 1989, 1990;
Ferreira et al., 1994b; Santella et al., 1994; Clonfero et al., 1995).
Although tobacco smoke was mutagenic in the presence of metabolic
activation, no increase in mutagenic activity was found in most
studies of workers exposed in occupations such as coking (Reuterwall
et al., 1991; Ferreira et al., 1994a,b), coal-tar distillation
(Jongeneelen et al., 1986), work in Söderberg potrooms of aluminium
plants (Krokje et al., 1988), in anode plants (Clonfero et al., 1984;
Venier et al., 1985; Krokje et al., 1988; Clonfero et al., 1990), and
in a graphite electrode plant (Ferreira et al., 1994a). Only the heavy
exposure of patients with psoriasis to coal-tar applications (Clonfero
et al., 1989, 1990; Santella et al., 1994) and of workers at coke
ovens (Mielzynska & Snit, 1992; Clonfero et al., 1995) and in a carbon
plant (Heussner et al., 1985) resulted in mutagenic urine. Ames' test
therefore appears not to be sensitive enough to detect the presence of
urinary mutagens due to occupational exposure to low levels of PAH
(Becher et al., 1984; Clonfero et al., 1989, 1990).
Expectorate from workers in a coke plant and in Söderberg potrooms in
an aluminium plant showed significantly increased mutagenicity in
Ames' test with S. typhimurium TA98 and TA100 in the presence of
metabolic activation (Krokje et al., 1988; Krokje, 1989).
8.3.4 Genotoxicity in lymphocytes
Genotoxic effects in lymphocytes have been proposed as markers for
exposure to PAH. In studies of iron foundry workers with relatively
low exposure to PAH, elevated frequencies of mutation at the hprt
locus in lymphocytes correlated approximately with the levels of DNA
adducts (Perera et al., 1993, 1994). In one study of coke-oven
workers, significant differences from controls were found in the
number of single-strand DNA breaks; however, there was no difference
between tobacco smokers and nonsmokers (Salagovic et al., 1995).
No increases in the rates of micronuclei, chromosomal aberrations, or
sister chromatid exchange were detected in workers at coke ovens
(Reuterwall et al., 1991), a carbon plant (Heussner et al., 1985), an
aluminium plant (Becher et al., 1984), or a graphite electrode plant
(van Hummelen et al., 1993) or in chimney sweeps (Carstensen et al.,
1993), although in most cases significant effects of smoking could be
detected. In one study in which an increase was found, there was no
difference between tobacco smokers and nonsmokers (Bender et al.,
1988; Salagovic et al., 1995). Environmental pollution in Silesia was
associated with significant increases in the frequencies of sister
chromatid exchange and chromosomal aberration in peripheral blood
cells, independently of smoking (Perera et al., 1992).
8.3.5 DNA adducts
DNA adducts with reactive metabolites (mainly diol epoxides) of
benzo [a]pyrene and other PAH have been identified in numerous
studies (see Section 6). For example, cigarette smokers have higher
levels of adducts with PAH in their lungs than nonsmokers, and there
is a linear relationship between adduct levels and daily or lifetime
cigarette consumption (Phillips et al., 1988).
As binding of electrophilic PAH metabolites to DNA is thought to be a
key step in the initiation of cancer, measurement of DNA adducts could
be an indicator of exposure to PAH and also of cancer risk. As a
surrogate for lung tissue, which is an important target organ for PAH
in humans, the more easily accessible nucleated blood cells and blood
proteins (haemoglobin, albumin) have been investigated.
8.3.5.1 Method of determination
The methods for measuring DNA adducts include immunoassays with
polyclonal and monoclonal antibodies (enzyme-linked immunosorbent
assay [ELISA] and ultrasensitive enzymatic radioimmunoassay),
32P-postlabelling, and synchronous fluorescence spectrophotometry.
Direct comparisons of adduct levels determined by different techniques
may be misleading, however, because different end-points are measured.
For example, polyclonal and monoclonal antisera recognize not only the
benzo [a]pyrene diol epoxide adducts against which they are raised,
but also benz [a]anthracene, chrysene, benzo [k]fluoranthene, and
dibenz [a,c]anthracene, which also form N2 guanine adducts. The
32P-postlabelling assay is even less specific, as it may detect
several aromatic and hydrophobic adducts (Dell'Omo & Lauwerys, 1993).
The detection limits for the three methods are one adduct per 107-108
nucleotides for the immunological methods (Dell'Omo & Lauwerys, 1993)
and synchronous fluorescence spectrometry (Dell'Omo & Lauwerys, 1993;
Rojas et al., 1995) and up to one adduct per 1010 nucleotides in the
32Ppostlabelling assay (Beach & Gupta, 1992; Dell'Omo & Lauwerys,
1993; Ovrebo et al., 1994). DNA adducts may be overlooked with
32P-postlabelling, because of incomplete nuclease P1 digestion,
resistance to 32P-labelling, dephosphorylation of certain adducts, or
co-migration with normal nucleotides (Herbert et al., 1990b; Beach &
Gupta, 1992; Kriek et al., 1993; Segerbäck & Vodicka, 1993; Pavanello
& Levis, 1994). This method is being improved (Segerbäck & Vodicka,
1993; Szyfter et al., 1994).
The results obtained when the different methods were applied in
parallel were usually similar, but the magnitude of the effect
differed (Ovrebo et al., 1990, 1992; Pavanello & Levis, 1994; Perera
et al., 1994). For example, in one study of psoriasis patients treated
with coal-tar, 20-100 times higher levels were found with ELISA than
with the 32P-postlabelling method (Pavanello & Levis, 1994). In
another study, the 32P-postlabelling method was more sensitive than
the ELISA (Kriek et al., 1993). Considerable differences were found in
DNA adduct levels in interlaboratory comparisons (Hemminki et al.,
1990a; Beach & Gupta, 1992; Kriek et al., 1993; Phillips & Castegnaro,
1993).
In the descriptions below, the DNA adduct levels are expressed as
number of adducts per 108 nucleotides or fmol/µg DNA; 33.2 fmol/µg
DNA corresponds to one adduct per 108 nucleotides. Since the levels
in background samples and also in samples from exposed subjects are
sometimes below the limit of detection, the number of positive samples
is often given as well (Herbert et al., 1990a,b; Dell'Omo & Lauwerys,
1993).
8.3.5.2 Concentrations
In general, exposures that lead to the excretion of high
concentrations of 1-hydroxypyrene in urine also lead to elevated DNA
adduct levels. Table 100 gives the DNA adduct levels derived from
studies in which air concentrations and DNA adduct levels were
measured in parallel. Although the concentrations of PAH that occur
under different exposure conditions differ by orders of magnitude (see
section 5.3), the differences in DNA adduct levels are quite small, in
contrast to the results of experiments on excretion of
1-hydroxypyrene. Table 101 summarizes the results of an investigation
of workers in an aluminium reduction plant where the two methods were
applied (van Schooten et al., 1995).
In all populations studied, there is substantial interindividual
variation in PAH-DNA adduct levels, after exposure by inhalation or
orally, which is greater than that described for 1-hydroxypyrene
excretion in urine (Hemminki et al., 1990a,b; Santella et al., 1993;
Szyfter et al., 1994; Rojas et al., 1995). In one study, about 50-fold
interindividual variations were reported among controls and about
100-fold variations among coke-oven workers (Rojas et al., 1995). The
variations are probably due to differences in the induction of AHH
activity in lymphocytes and in the resulting detoxification of
carcinogenic PAH, the ability to repair DNA lesions, and the turnover
of damaged cells (Dell'Omo & Lauwerys, 1993; Szyfter et al., 1994;
Kang et al., 1995; Rojas et al., 1995). These interindividual
variations result in a wide overlap in the ranges of values between
exposed and unexposed subjects in all studies.
Table 100. DNA-polycyclic aromatic hydrocarbon adduct levelsa in various situations of exposure
Population Benzo[a]pyrene Method of Exposed Controls Reference
investigated, (µg/m3) detection
type of emission No. of No. of DNA No. of No. of DNA
subjects adducts/108 subjects adducts/108
nucleotides nucleotides
Polluted ambient air
Industrialized area, 0.015-0.057 32P-Postlabelling 15 13 13 2.3 Hernminki et al.
Silesia, Poland (1990a)
Industrialized area 32P-Postlabelling 19 14 15 4.8 Motykiewicz
Silesia, Poland (1995)
Winter inversion, 0.002-0.008 32P-Postlabelling 29 2.6-6.8 - - Binkova et al.
Teplice, Czech (1995)
Republic
Burning of smoky 19 Immunoassay 18 7.7 18 5.2 Mumford et al.
coal at home, with (1993)
and without
chimneys, China
Coke ovens
Door maintenance 2.3-6.5 Immunoassay 11 5.8b - - Assennato
(1993a)
Work topside 7.3 Fluorescence 13 ND-73b - - Haugen et al.
spectrophotometry (1986)
immunoassay
Battery work 0.54-90 32P-Postlabelling 31 15 13 2.3 Hemminki et al.
(1990a)
Working at high and 0.001-0.009 32P-Postlabelling 31 2.2 22 2.2 Yang et al.
low traffic density (1996)
areas
Table 100. (continued)
Population Benzo[a]pyrene Method of Exposed Controls Reference
investigated, (µg/m3) detection
type of emission No. of No. of DNA No. of No. of DNA
subjects adducts/108 subjects adducts/108
nucleotides nucleotides
Aluminium production
Aluminum plant, prebake 32P-Postlabelling Van Schooten
anode process et al. (1995)
Anode factory 1.5 - 26 - -
Pot relining 1.1 47 - -
Electrode past plant 0.9 32P-Postlabelling 34 11 14 10 Ovrebo et al.
(1994)
Foundries
Foundry 0.02 - - 7.4 - - Perera et al.
(1994)
Iron foundry < 0.005-0.06 Competitive 67 4.4-9.6 - - Santella et al.
ELISA (1993)
Iron foundry < 0.005-0.06 32P-Postlabelling 67 1.9-2.5 - - Perera et al.
(1994)
Iron foundry < 0.05-> 0.2 Fluorescent 35 0.8-21 10 2.2 Perera et al.
ELISA (1988)
Iron foundry < 0.05 32P-Postlabelling 19 7.3 4 4 Szyfter et al.
0.005-0.2 - 63 19 - - (1994)
> 0.2 - 6 29 - -
ND, not detected; ELISA, enzyme-linked immunosorbent assay
a Median or mean values; ranges are from means or medians of several measurements of groups of exposed persons
b In original publication given as fmol/µg DNA. Number of adducts per 108 nucleotides = fmol/µg DNA × 33.2
Table 101. Comparison of methods for measuring exposure to polycyclic aromatic
hydrocarbons in an aluminium plant
Exposure Benzo[a]pyrene Pyrene 1-Hydroxypyrene DNA adducts
(µg/m3)a (µg/m3)a in urine (µmol/mol in leukocytes
creatinine)b (adducts/108
± SD nucleotides)
± SD
Bake oven 0.35 1.5 3.65 ± 2.11 30.1 ± 42.1
Anode factory 1.51 5.6 3.25 ± 1.89 26.2 ± 15.0
Pot relining 1.05 32.3 6.20 ± 8.44 47.3 ± 39.1
Electrolysis 0.03 0.12 0.48 ± 0.27 12.8 ± 10.0
Foundry 0.02 0.04 0.47 ± 0.20 7.4 ± 9.6
From van Schooten et al. (1995)
a Geometric mean
b Arithmetic mean
Significant correlations were found in most studies between the
estimated or measured exposure to PAH and adduct levels (Herbert et
al., 1990a,b; Ovrebo et al., 1990, 1992; Assennato et al., 1993a;
Perera et al., 1994; Szyfter et al., 1994; van Schooten et al., 1995),
but no such correlation was found in others (Herbert et al., 1990a,b;
Kriek et al., 1993; Mumford et al., 1993, Ovrebo et al., 1994; Schoket
et al., 1995).
As shown with 1-hydroxypyrene concentrations in urine, the DNA adduct
concentrations in certain workers may correlate better with dermal
exposure than with PAH concentrations in air (Herbert et al.,
1990a,b).
(a) General population
The levels of DNA adduct in control subjects range from 0.2 to about
10 adducts per 108 nucleotides in leukocytes (Dell'Omo & Lauwerys,
1993). DNA adducts were also found in 43% of placentas and in 27% of
liver samples and 42% of lung specimens from 15 spontaneously aborted
human fetuses. As there was only 60% concordance between placenta and
fetal lung or liver with regard to the presence of detectable adducts,
DNA adducts in the placenta are not a good indicator of adduct
formation in fetal organs. Although several of the mothers were
smokers, none of the fetal samples containing DNA adducts were from
women who smoked during pregnancy, indicating that smoking is unlikely
to have caused adduct formation (Hatch et al., 1990).
Conflicting results have been obtained concerning the effects of
tobacco (cigarette) smoking on DNA adduct levels in peripheral blood
cells. Most investigations of human peripheral lymphocytes have found
no remarkable effect of smoking in control or exposed persons, in
contrast to those of lung and bronchial tissue (Hemminki et al.,
1990a; Herbert et al., 1990a,b; Dell'Omo & Lauwerys, 1993; Binková et
al., 1995; van Schooten et al., 1995; Yang et al., 1996). Some
publications, however, report maximal differences in DNA adduct levels
of about threefold between tobacco smokers and nonsmokers among
controls and exposed individuals (Savela & Hemminki, 1991; Kriek et
al., 1993; Santella et al., 1993; Rojas et al., 1995; van Schooten et
al., 1995). Granulocytes from smokers and nonsmokers showed no
difference in DNA adduct levels, but threefold increases were observed
in T lymphocytes, which have a much longer life than granulocytes
(Savela & Hemminki, 1991).
A synergistic effect of tobacco smoking and occupational exposure to
PAH was reported in two studies (Rojas et al., 1995; van Schooten et
al., 1995). It was hypothesized that tobacco smoke induces AHH in
lymphocytes, resulting in increased formation of benzo [a]pyrene-DNA
adducts in smokers. The large interindividual differences may be due
to the presence of both non-inducible and highly inducible variants in
human lymphocytes (Rojas et al., 1995).
Elevated DNA adduct levels have been detected in the general
populations of industrialized areas in Poland (Silesia) and the Czech
Republic (Teplice) (Hemminki et al., 1990a,b; Perera et al., 1992;
Binková et al., 1995), with levels up to 13 (Hemminki et al., 1990a;
Motykiewicz, 1995) and 5 adducts per 108 nucleotides (Binková et al.,
1995). Levels of 8 adducts per 108 nucleotides were found in
leukocytes from women exposed to high PAH concentrations from burning
smoky coal in China. Although DNA adducts were also detected in
placenta, no dose-response relationships were found between exposure
to benzo [a]pyrene and placental DNA adduct level or the percentage
of samples with detectable DNA adducts (Mumford et al., 1993).
The consumption of charcoal-grilled foods leads to elevated DNA adduct
levels (Rothman et al., 1993; Kang et al., 1995), and such food may
represent a major dietary source of PAH for some populations. Eating
charcoal-grilled beef resulted in a 1.9-3.8-fold increase above the
individual baseline adduct levels in four of 10 subjects (Kang et al.,
1995).
Psoriatic patients undergoing coal-tar treatment had a DNA adduct
level of about 8 per 108 nucleotides (Pavanello & Levis, 1994;
Santella et al., 1995), and levels up to 13 per 108 were found in
skin biopsy samples obtained from patients who had received treatment
with coal-tar ointment. There was no correlation of the adduct levels
after different treatments. No information was given on controls
(Phillips et al., 1990).
(b) Occupational exposure
Workers exposed to PAH had elevated mean levels of adducts and a
higher percentage of positive samples (measured concentrations above
the detection limit) than controls. Elevated DNA adduct levels have
been detected in leukocytes from workers exposed in coke-oven plants
(Assennato et al., 1993a,b; Dell'Omo & Lauwerys, 1993; Harris et al.,
1985; Haugen et al., 1986; Hemminki et al., 1990a,b; Ovrebo et al.,
1992; Kriek et al., 1993a,b; Rojas et al., 1995), aluminium
manufacture (Dell'Omo & Lauwerys, 1993; Kriek et al., 1993a,b; Schoket
et al., 1995; van Schooten et al., 1995), and foundries (Perera et
al., 1988; Dell'Omo & Lauwerys, 1993; Santella, 1993; Santella et al.,
1993; Perera et al., 1994) and among firefighters (Dell'Omo &
Lauwerys, 1993) and roofers (Herbert et al., 1990a,b; Dell'Omo &
Lauwerys, 1993).
In cases of high exposure, for example at coke ovens, 5-70 adducts per
108 nucleotides have been measured. Significant correlations with
exposure concentrations have been found, although the level was no
more than threefold greater than in controls (Hemminki et al.,
1990a,b; Ovrebo et al., 1990; Assennato et al., 1993a).
8.3.5.3 Suitability as a biomarker
DNA adduct levels in the lung may not a reliable indicator of human
cancer risk, although Phillips et al. (1988) found a correlation
between cigarette smoking and DNA adducts in the human lung. Weston et
al. (1993) observed no correlation between lung DNA adduct levels and
a measure of recent tobacco smoking, serum cotinine. Tissue samples
taken from different portions of the same lung showed variations in
DNA adduct levels
The use of lymphocytes as a surrogate for lung cells has also been
questioned, because no correlation has been found between PAH-DNA
adduct levels in human lung and leukocytes (van Schooten et al.,
1992). In studies on rats exposed to coke-oven emissions, the DNA
adduct levels were lower in leukocytes than in lung tissues; in
addition, several types of adducts observed in lung tissue were not
present in leukocytes (Binková et al., 1994). Granulocytes, which form
the majority of peripheral leukocytes, have a relatively short life,
< 24 h, in contrast to lung cells; therefore, adducts are probably
lost within a few days (Savela & Hemminki, 1991; Dell'Omo & Lauwerys,
1993). This can be avoided by using the subfraction of T lymphocytes
which have a half-life of several years. The adduct levels in T
lymphocytes were three times higher than in granulocytes (Savela &
Hemminki, 1991).
DNA adducts are much less sensitive for assessing exposure than
excretion of 1-hydroxypyrene in urine. Additionally, because of the
large interindividual differences in control and exposed groups,
adduct levels can be compared only on a group basis. Thus, PAH-DNA
adducts can be used as a qualitative biomarker of exposure to
combustion emissions but to only a limited extent as a quantitative
marker. This method may, however, allow identification of subjects who
are highly susceptible to the DNA-damaging properties of PAH and are
therefore predisposed to lung cancer. This was seen in one
investigation of lung cancer patients with a family history of lung
cancer. Monocytes from these patients treated in vitro with PAH
showed a slight but significant enhancement of formation of
benzo [a]pyrene-DNA adducts in comparison with controls (Nowak et
al., 1992). It is not yet known whether metabolism in leukocytes is
identical to that in lung cells.
8.3.6 Antibodies to DNA adducts
Antibodies to DNA adducts in leukocytes of exposed workers have also
been found (Harris et al., 1985; Haugen et al., 1986; Vähäkangas et
al., 1992; Santella et al., 1995). In a study of coal-tar-treated
patients, elevated levels were also found in controls (Santella et
al., 1995). Since the initial antigenic stimulus could have occurred
several years previously, antibodies to benzo [a]pyrene-DNA adducts
are considered general indicators of past exposure to PAH.
8.3.7 Protein adducts
Because genotoxic compounds can bind to haemoglobin and serum protein,
the assessment of PAH-blood protein adducts has also been considered
as a possible marker of exposure to PAH or even as a surrogate for the
evaluation of adduct concentrations at the level of the target organs.
This approach has several advantages. Relatively large amounts of
haemoglobin and albumin can be obtained easily from a small volume of
human blood. As the lifetime of haemoglobin in humans is about 120
days and that of albumin 20-24 days, exposure days and weeks
previously can be measured. Albumin is synthesized in the liver, where
PAH are metabolized. Therefore, reactive metabolites may easily gain
access to the proteins. Finally, there may be lower interindividual
variation, because there is no repair, as in the case with DNA
(Dell'Omo & Lauwerys, 1993).
The benzo [a]pyrene-albumin adduct concentrations were similar in
foundry workers and controls, both smokers and nonsmokers (Omland et
al., 1994), and in patients with psoriasis treated with coal-tar
(Santella et al., 1995). Minor effects were detected in foundry
workers and roofers (Lee et al., 1991). No pronounced differences in
haemoglobin adduct levels were detected in workers in steel foundries
and in one graphite electrode producing plant (Ferreira et al.,
1994a,b). In another study, however, significantly increased
benzo [a]pyrene binding was detected in serum proteins from smoking
and non-smoking foundry workers (Sherson et al., 1990).
As protein adducts have been used in relatively few studies, no
conclusion can be drawn about the usefulness of this biomarker.
8.3.8 Activity of cytochrome P450
Increased mRNA levels of the CYP1A1 gene, which belongs to the
P4501A1 family responsible for the metabolism of PAH, including
benzo [a]pyrene, have been proposed as biomarkers for exposure to PAH
(Cosma et al., 1992; Van den Heuvel et al., 1993; see also Section 6).
Although the basal levels were not increased, 3-methylcholanthrene
caused greater induction in lymphocytes from railroad workers exposed
to creosote in cell culture (Cosma et al., 1992).
The activity of the cytochrome P450 CYP 1A2 was also determined by
measuring caffeine metabolites in urine. There were significant
differences between tobacco smokers and nonsmokers, but there was no
difference between nonsmoking and smoking foundry workers and the
respective controls (Sherson et al., 1992).
8.3.9 Differentiation antigens on the surface of lung cells
Lung epithelial cells from sputum have been tested for antigens that
indicate neoplastic transformation (Assennato et al., 1993b). One of
23 coke-oven workers who had differentiation antigens on the cell
surface was a smoker who had severe airways obstruction and moderate
dysplasia of bronchial epithelium cells.
8.3.10 Oncogene proteins
Since oncogene activation may be an early step in the carcinogenic
process, its detection may be a useful marker for identifying
individuals at risk for the development of malignancy. Plasma levels
of ras oncogene-related p21 proteins were elevated in a sample of
male residents of the highly industrialized Silesian region of Poland.
They also had elevated DNA adduct levels in their leukocytes (Perera
et al., 1992). Three of 18 foundry workers screened for the oncogene
proteins sis, fes ß-TGF, int-1, myb, src, myc, mos, and ras in serum
had elevated levels of ras and fes; however, two were smokers. No
unexposed individuals had abnormal serum oncogene protein expression.
The levels of ras and fes proteins were also increased in the serum of
lung cancer patients (Brandt-Rauf et al., 1990).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND THE FIELD
Appraisal
Data on toxicity are available mainly for naphthalene, phenanthrene,
and fluoranthene and are scarce for other polycyclic aromatic
hydrocarbons (PAH). Both metabolism and photooxidation can alter the
toxicity of PAH in the environment, photooxidation tending to increase
toxicity.
At low concentrations, PAH can stimulate the growth of microorganisms
and algae. The highest values for the no-observed-effect concentration
(NOEC) were determined for naphthalene (3100 µg/litre in Anabaena
flos-aquae) and acenaphthene (> 4600 µg/litre in A. flos-aquae).
The NOEC values for fluorene, phenanthrene, fluoranthene, and pyrene
were about one order of magnitude lower. Benz [a]anthracene and
chrysene were the most toxic towards algae (NOEC, < 10 µg/litre). The
effective concentration that caused a 50% change (EC50) was between 5
µg/litre for benzo [a]pyrene and 54 000 µg/litre for fluoranthene.
For invertebrates like crustaceans, insects, molluscs, polychaetes,
and echinoderms, naphthalene is least toxic, with a 48-h median lethal
concentration (LC50) of 700-23 000 µg/litre. For three-ring PAH, the
LC50 values ranged between < 1 and 3000 µg/litre. Anthracene may be
more toxic than the other three-ring PAH, with 24-h LC50 values
between < 1 and 260 µg/litre. The values for most four- and five-ring
PAH are between 0.7 and 1800 µg/litre and 0.4-120 µg/litre,
respectively. For six-ring PAH, only one LC50 is available: 0.2
µg/litre for benzo [ghi]perylene. The values for the NOEC are
slightly lower than those for the LC50. The lowest NOEC reported for
benzo[a]pyrene is 0.14 µg/litre.
Naphthalene, acenaphthene, and fluorene are the least toxic for
vertebrates like fish and amphibians, with 96-h LC50 values of 110 to
> 10 000 µg/litre; for phenanthrene and fluoranthene, the LC50
values are 30-4000 µg/litre. Most of the LC50 values for anthracene
were between 2.8 and 360 µg/litre. For four- and five-ring PAH, the
LC50 values were 0.7-26 µg/litre. The lowest NOEC reported for
benzo [a]pyrene in fish was 2.4 µg/litre. In tests with
sediment-dwelling organisms, LC50 values of 3-15 mg/kg dry weight
were determined for fluoranthene.
In earthworms, an LC50 value for growth was 170-200 mg/kg dry weight
for fluorene, and an LC50 value for reproduction was 150 mg/kg dry
weight for phenanthrene. At 180 mg/kg dry weight, chrysene,
benzo [k]fluoranthene, and benzo [a]pyrene did not affect
reproduction by the earthworm Folsoma candida.
9.1 Laboratory experiments
Data on the toxicity of individual PAH to members of different
taxonomic groups are presented in Tables 102-104; data on the toxicity
of PAH metabolites are not included. The use of solvents in laboratory
tests for toxicity often results in unrealistically high
concentrations of PAH, which exceed their maximum water solubility.
Only data from tests with concentrations that did not exceed 10 times
the estimated solubility were used.
9.1.1 Microorganisms
9.1.1.1 Water
The effects of several three-, four-, and five-ring, unsubstituted PAH
on the growth of the bacterium Escherichia coli were studied at
concentrations of 10-5, 10-6, and 10-7 mol/litre in the growth medium
(Hass & Applegate, 1975). Benz [a]anthracene,
dibenz [a,h]anthracene, and benzo [a]pyrene promoted bacterial
growth; pyrene and phenanthrene had slight promoting effects at low
concentrations (10-7 and 10-6, respectively) and inhibitory effects
at higher concentrations, whereas anthracene and chrysene inhibited
bacterial growth at all concentrations.
The effects of dissolved PAH on the growth rate, lag time before
initiation of growth, and the number of cells at the end of the log
growth phase, measured as maximum light absorbance, were determined in
two species of marine bacteria, Serratia marinorubra and Vibrio
parahaemolyticus. Most of the PAH tested increased the lag time and
decreased the growth rate and cell yield; pyrene, however, increased
the growth rate. The extent of inhibition of growth was a function of
both the concentration of PAH and their inherent toxic properties,
which decreased with solubility. Thus, the toxicity of naphthalene at
a concentration of 13 mg/litre was similar to that of benzo [a]pyrene
at 5 µg/litre (Calder & Lader, 1976).
PAH at concentrations of 5-20 µg/litre stimulated the growth of the
freshwater algae Chlorella vulgaris, Scenedesmus obliquus, and
Ankistrodesmus orauntic (Gräf & Nowak, 1966) and of the marine
dinoflagellate Gyrodinium sp. (Ishio et al., 1977). The growth of
sporelings of the marine algae Antithamnium plumula was
progressively inhibited after exposure to 10-300 µg/litre of
benz [a]anthracene.
The growth of the blue-green alga A. flos-aquae was inhibited by
16-50% in comparison with controls after exposure to 5-29 µg/litre
benz [a]anthracene for 14 days. In the same study, 14 days' exposure
to fluoranthene at concentrations of 38-1100 µg/litre inhibited growth
by 19-65%. The NOEC values ranged from 1 µg/litre for benzo [a]pyrene
to > 4600 for acenaphthene (Bastian & Toetz, 1982). Other data for
cyanobacteria are listed in Table 102.
Table 102. Results of tests for the toxicity of polycyclic aromatic hydrocarbons (PAH) towards algae and plants
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic two-ring PAH
Acenaphthene
Anabaena flos-aquae A S Contnuous light 2 h NOEC > 4600 Nitrogen fixation, Bastian & Toetz (1985)
no solvent acetylene reduction
250 × 103 cells/ml activity
Selenastrum 96 h EC50 520 Cell number US Environmental
capricornutum Protection Agency
(1978a)a
Fluorene
Anabaena flos-aquae A S Continuous light, 2 h NOEC 260 Nitrogen fixation Bastian & Toetz (1985)
no solvent
250 × 103 cells/ml
Dunaliella bioculata N S Continuous light, 72 h EC50 15 500 Growth rate Heldal et al. (1984)
solvent methanol
250 × 103 cells/ml
Naphthalene
Anabaena flos-aquae A S Continuous light, < 14 d NOEC 3100 Growth Bastian & Toetz (1985)
250 × 103 cells/ml
Chlamydomonas 24 h LC61 34 400b US Environmental
angulosa Protection Agency
(1986d)
Chlorella vulgaris 48 h LC61 33 000b Cell number Kauss & Hutchinson
(1975)
Champia parvula N R Solvent,TEG 11-14d MATC < 695 Female growth Thursby et al. (1985)
Number cystocarps
Nitzchia palea A S No solvent 4 h EC50 2820 Assimilation rate Millemann et al. (1984)
Selenastrum A S Solvent, methanol 4 h EC50 2960 Assimilation rate Millemann et al. (1984)
capricornutum
Table 102. (continued)
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic three-ring PAH
Anthracene
Chlamydomonas N S 5 × 104 cells/ml 3 h EC50 239b Photosynthesis Hutchinson et al.
angulosa (log phase) inhibition (1980)
Chlorella vulgaris N S 20 × 104 cells/ml 3 h EC50 535b Photosynthesis Hutchinson et al.
(log phase) inhibition (1980)
Selenastrum N R Solvent, acetonec 28 h EC50 3.9-37 Growth rate Gala & Giesy (1992)
capricornutum (8 h) nitrile UV-A cont.c EC10 1.5-7.8 Growth rate
1 × 105 cell/ml
Selenastrum Cool-white light EC30 40 000d Growth US Environmental
capricornutum Protection Agency
(1987b)
Selenastrum Gold fluorescent NOEC 8000d Growth US Environmental
capricornutum light Protection Agency
(1987b)
Fluoranthene
Anabaena flos-aquae A S Continuous light, 2 h LOEC 230 Nitrogen fixation Bastian & Toetz (1982)
no solvent
Scenedesmus N S Solvent, acetone 96 h EC10 1.6 Growth Kordel et al. (1981)
subspicatus 96 h EC50 12
Selenastrum 96 h EC50 54 400d Cell number US Environmental
capricornutum Protection Agency
(1978b)
Selenastrum 96 h EC50 54 600d Chlorophylla US Environmental
capricornutum Protection Agency
(1978b)
Skeletonema costatum 96 h EC50 45 000d Chlorophyla US Environmental
Protection Agency
(1978b)
Skeletonema costatum 96 h EC50 45 600d Cell number US Environmental
Protection Agency
(1978b)
Table 102. (continued)
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Phenenthrene
Anabaena flos-aquae A S Continuous light, 2 h NOEC 130 Nitrogen fixation Bastian & Toetz (1985)
no solvent,
250 × 103 cells/ml
Nitzschia palea A S No solvent 4 h EC50 870 Assimilation rate Millemann et al. (1984)
Selenastrum A S No solvent 4 h EC50 940 Inhibition of Millemann et al. (1984)
capricornutum photosynthesis
Aromatic four-ring PAH
Benz[a]anthracene
Anabaena flos-aquae A S Continuous light < 14 d NOEC 1 Growth Bastian & Toetz (1982)
Anabaena flos-aquae A S Continuous light, 2 h NOEC 19b Acetylene reduction Bastian & Toetz (1985)
no solvent
250 × 103 cells/ml
Anabaena flos-aquae A S Continuous light < 14 d EC 18-29b Growth Bastian & Toetz (1982)
16-48%
Antithamnium plumula - EC 17% 10 Cell production level Boney & Corner (1962)
in algal medium
Chrysene
Anabaena flos-aquae A S Continuous light, 2 h NOEC 5b Nitrogen fixation Bastian & Toetz (1985)
no solvent,
250 × 103 cells/ml
Anabaena flos-aquae A S Continuous light 14 d EC35 62-96% Growth Bastian & Toetz (1982)
saturation
Pyrene
Anabaena flos-aquae A S Continuous light < 14 d NOEC < 100 Growth Bastian & Toetz (1982)
Chlamydomonas N S 5 × 104 cells/ml 3 h EC50 202b Inhibition of Hutchinson et al.
angulosa (log phase) photosynthesis (1980)
Chlorella vulgaris N S 20 × 104 cell/ml 3 h EC50 332b Inhibition of Hutchinson et al.
(log phase) photosynthesis (1980)
Table 102. (continued)
Compound, species Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic five-ring PAH
Benzo[a]pyrene
Anabaena flos-aquae N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
Antithamnium plumula 96 h EC 49% 10b Cell production Boney & Corner (1962)
96 h EC 54% 100d increase
Chlamydomonas N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
reinhardtii
Euglena gracilis N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
Porphyra tenera 80-320 EC 1000d Cell size decrease Boney (1974)
min
Poteriochromonas N S 500 × 103 cells/ml 72 h EC50 > 4000d Growth Schoeny et al. (1988)
malhamensis
Scenedasmus acutus N S 500 × 103 cells/ml 72 h EC50 5b Growth Schoeny et al. (1988)
Scenedesmus N S Solvent, acetone 20 h EC 300d Chlorophyll content Zachleder et al. (1983)
quadracanda
Scenedesmus N S Solvent, acetone 20 h EC 300d Biomass Zachleder et al. (1983)
Perylene
Scenedesmus N S Solvent acetone 96 h EC10 0.0066 Growth Kordel et al. (1981)
subspicatus 96 h EC50 > 0.32
A, analysed concentration; N, nominal concentration; S, static system; F, flow-through system; R (0.5 d), system with renewal
(each half day)
NOEC, no-observed-effect concentration; EC, effect concentration; LC, lethal concentration; MATC, maximum acceptable toxicant
concentration; LOEC, lowest-observed-effect concentration; TEG, triethylene glycol
a From Cairns & Nebeker (1982)
b Concentration higher than solubility but not exceeding it by 10 times
c Explicitly mentioned that organisms were tested for phototoxicity of test substance either by sunlight or artificial UV radiation
d Concentration 10 times higher than the solubility
Naphthalene had no detectable effect on the rate of respiration of
yeast at concentrations up to its maximum solubility (31 mg/litre)
(Haubenstricker et al., 1990).
9.1.1.2 Soil
Few data are available on the toxicity of PAH to microbial communities
in the soil. Application of 500 mg/kg dry weight naphthalene did not
reduce soil microbial respiration or nitrogen mineralization, and 90%
was degraded within 10-20 days (Kirchmann et al., 1991).
9.1.2 Aquatic organisms
9.1.2.1 Plants
For many PAH, the dose that is toxic for algae exceeds the maximum
solubility in water (Table 102). Benz [a]anthracene (four-ring) and
benzo [a]pyrene (five-ring) are considered to be the most toxic PAH,
with EC50 values of 29 µg/litre and 5-15 µg/litre, respectively. The
EC50 values for three-ring PAH are 240-940 µg/litre. Naphthalene and
fluorene (two-ring) are considered to be the least toxic, with EC50
values of 2800-15 000 µg/litre. The ranges of EC50 values are 200-330
for four-ring compounds and 5-> 4000 µg/litre for five-ring PAH.
9.1.2.2 Invertebrates
Data on the toxicity of two- to six-ring PAH are available for
invertebrates such as crustaceans, insects, molluscs, polychaetes, and
echinoderms (Table 103). The data include the results of both
phototoxicity and non-phototoxicity tests, which are poorly comparable
because of the dynamic character of the ultraviolet radiation-induced
oxidation products and the often short exposure time.
Phenanthrene, fluorene, and triphenylene did not cause photoinduced
toxicity to Daphnia and did not absorb light. Benzo [a]fluorene,
benzo [k]-fluorene, and chrysene were considered to be moderately
toxic to Daphnia, whereas anthracene, fluoranthene, pyrene,
benz [a]anthracene, benzo [k]-fluoranthene, benzo [a]pyrene,
benzo [e]pyrene, perylene, dibenz [a]anthracene, and
benzo [ghi]perylene were very toxic and absorbed more energy.
Toxicity was also correlated with the phosphorescence lifetime and
energetic state of the molecule. The authors stated that the
photodynamic response was due to the PAH assimilated into organisms
rather than to external photoproducts. Phototoxicity occurs as a
result of energy transfer from activated (triplet) PAH to ground-state
oxygen. Singlet oxygen is a reactive chemical that can denature
biomolecules within aquatic organisms. The authors estimated that the
internal concentration of PAH that caused the death of 50% of the
Daphnia was 77 nmol/g wet weight in continuous light. No toxic
effects were seen in the absence of light. The phototoxicity of
anthracene was confirmed by experiments with
Daphnia pulex (Newsted & Giesy, 1987).
Table 103. Results of tests for the toxicity of polycyclic aromatic hydrocarbons (PAH) towards invertebrates
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
PROTOZOANS
Aromatic two-ring PAH
Acenaphthylene
Tetrahymena 24 h EC50 6300a Population Yoshioka et al.
puryformis growth (1986)
Aromatic three-ring PAH
Anthracene
Paramecium aurelia 10 000 N S Dark 1.5 h NOEC 1000b Mortality Joshi & Misra
cells/litre 0.75 h sunc 0.75 h EC 100% 100a Mortality (1986)
Paramecium 1 h LC90 1000b US Environmental
caudatum Protection
Agency (1987b)
Aromatic five-ring PAH
Benzo[a]pyrene
Gyrodinium sp. Log phase N S Solvent acetone 12 d EC 20% 5a Cell division Ishio et al. (1977)
period
increase
INSECTS
Aromatic two-ring PAH
Fluoranthene
Chironomus riparius Larvae N S l/d = 16/8 48 h EC50 2350 Reproduction Finger et al.
A IF 30 d NOEC 142 Emergence (1985)
Hexagenia bilineata N S 96 h LC50 5800
Naphthalene
Chironomus 4th instar A F Solvent, ethanol Chronic LOEC 500 Physiology Darville & Wilhm
attenuatus LC50 1300 (1984)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Chironomus tentans Larvae A S Solvent, methanol 48 h LC50 2,810 Millemann et al.
(1984)
Somatochlora Nymph - S 21°C 96 h LC50 1000- Correa & Coler
cingulata 2500 (1983)
Tanytarsus dissimilas Life cycle A F Solvent, ethanol Chronic LOEC 500 Physiology Darville & Wilhm
LC50 1300 (1984)
Anthracene
Aedes aegypti 3rd-4th instar N S Solvent DMSO 1 d LC50 < 1.0 Borovsky et al.
6 h sun, 18 h darkc (1987)
Aedes aegypti < 8 h old N S 1 h UVc 12 h LC50 150a Kagan et al.
(1985)
Aedes 3rd-4th instar N S Solvent, DMSO 1 d LC50 260a Borovsky et al.
taeniorrhynchus 6 h sun, 18 h darkc (1987)
Culex 3rd-4th instar N S Solvent, DMSO 1 d LC50 37 Borovsky et al.
quinquefasciatus 6 h sun, 18 h darkc (1987)
Culex sp. 24 h LC50 26.8 US Environmental
Protection Agency
(1987b)
Fluoranthene
Aedes aegypti 3rd-4th instar N S Solvent, DMSO 24 h LC50 10 Borovsky et al.
6 h sun, 18 h darkc (1987)
Aedes aegypti < 8-h old N S 1 h sunc 12 h LC50 12 Kagan et al.
(1985)
Aedes 3-4 instar N S Solvent, DMSO 24 h LC50 48 Borovsky et al.
taeniorrhynchus 6 h sun, 18 h dark, (1987)
Culex 3rd-4th instar N S Solvent, DMSO 24 h LC50 45 Borovsky et al.
quinquefasciatus 6 h sun, 18 h darkc (1987)
Phenanthrene
Chironomus tentans Larvae A S Solvent, methanol 48 h LC50 490 Millemann et al.
(1984)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic four-ring PAH
Pyrene
Aedes aegypti 1st instar N S 1 h UVc 11 d EC 18% 0.9 Adult Kagan & Kagan
7 d NOEC 0.9 emergence (1986)
N S Dark 11 d NOEC 30
< 8-h old N S 1 h UVc 12 h LC50 20 Kagan et al. (1985)
Aedes aegypti 3rd-4th instar N S Solvent, DMSO 24 h LC50 35 Borovsky et al.
6 h sun, 8 h darkc (1987)
Aedes 3rd-4th instar N S Solvent, DMSO 24 h LC50 60 Borovsky et al.
aeniorrhynchus 6 h sun, 8 h darkc (1987)
Culex 3rd-4th instar N S Solvent, DMSO 24 h LC50 37 Borovsky et al.
quinquefasciatus 6 h sun, 8 h darkc (1987)
Aromatic five-ring PAH
Benzo[a]pyrene
Aades aegypti 1st instar N S 30 min UVc 11 d NOEC 0.14 Adult Kagan & Kagan
1st instar N S Dark 11 d NOEC 0.9 emergence (1986)
4th instar N S Dark 7d NOEC 6700b
4th instar N S 30 min UVc 7 d NOEC 30a
4th instar N S 30 min UVc 7 d LC50 120b
POLYCHAETES
Aromatic two-ring PAH
Fluorene
Neanthes Immature A S Solvent, acetone 96 h LC50 1000 Rossi & Neff
arenacoendata adult artificial seawater (1978)
Naphthalene
Neanthes Immature A S Solvent, acetone 96 h LC50 3800 Rossi & Neff
arenacoendata adult artificial seawater (1978)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Aromatic three-ring PAH
Fluoranthene
Neanthes Immature A S Solvent, acetone 96 h LC50 500a Rossi & Neff
arenacoendata adult artificial seawater (1978)
1-Methylphenanthrene
Neanthes Immature A S Solvent, acetone 96 h LC50 300b Rossi & Neff
arenacoendata adult artificial seawater (1978)
Phenanthrene
Neanthes Immature A S Solvent, acetone 96 h LC50 600 Rossi & Neff
arenacoendata adult artificial seawater (1978)
Aromatic four-ring PAH
Benz[a]anthracene
Nereis virens A S Sediment 6 d NOEC 14.4 Oxygen McElroy (1985)
mg/kg consumption
NOEC 14.4 Ammonia
mg/kg extraction
NOEC 14.4 Microsomal
mg/kg AHH activity
Chrysene
Neanthes Immature A S Solvent, acetone 96 h NOEC > 1000b Mortality Rossi & Neff
arenacoendata adult artificial seawater (1978)
Aromatic five-ring PAH
Benzo[a]pyrene
Neanthes Adult A S Solvent, acetone 96 h NOEC > 1000b Mortality Rossi & Neff
arenaceodentata (1978)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Dibenz[a,h]anthracene
Neanthes Immature A S Solvent, acetone 96 h NOEC > 1000b Mortality Rossi & Neff
aranacoendata adult artificial seawater (1978)
MOLLUSCS
Aromatic two-ring PAH
Acenaphthene
Aplexa hypnorum Adult A F Solvent, 6 h LC50 > 2040 Holcombe et al.
isopropanol 9 (1983)
Fluorane
Mudalia potosensis N S 96 h LC50 5600a Finger et al.
(1985)
Naphthalene
Physa gyrina Adult A S No solvent 48 h LC50 5020 Millemann et al.
(1984)
Aromatic three-ring PAH
Fluoranthene
Mytilus edulis 40-50-mm A S Solvent, acetone 9 d EC50 80 Feeding rate Donkin et al.
shell (1989)
Aromatic five-ring PAH
Benzo[a]pyrene
Mercenaria A F Columns 1-15 EC < 0.001 Haemocyte Anderson et al.
mercenaria weeks lysozome (1981)
concentration
increase
8 weeks EC < 0.001 Bacterial
clearance
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Mytilus californianus 24 h EC 20% 10 000b Oxygen Sabourin & Tullis
consumption (1981)
EC 10% 1000b Oxygen
consumption
ECHINODERMS
Aromatic five-ring PAH
Benzo[a]pyrene
Strongylocentrotus Gametes S N Solvent, ethanol 30-45 NOEC 0.5 Cytological Hose (1985)
purpuratus, min abnormality
LOEC 0.5 Mitoses/embryo
NOEC 50b Fertilization
success
Psammechinus Eggs N S Artificial seawater 100 min NOEC 2000b Development Bresch et al.
miliaris (fertililized (1972)
30 min)
CRUSTACEANS
Aromatic two-ring PAH
Acenaphthene
Daphnia magna N S Solvent 48 h LC50 41 000b LeBlanc (1980)
NOEC 600 Mortality LeBlanc (1980)
Fluorene
Daphnia magna A R (0.5 d) No solvent 2 d NOEC 17.0 Mortality Newsted & Giesy
-UV:1 d; +UV:1 dc (1987)
Daphnia magna A IF l/d = 16/8 21 d NOEC 62.5 Reproduction Finger et al.
21 d LOEC 125 Reproduction (1985)
N S 270 mg.l CaCO3 49 h EC50 430 Immobilization
Solvent, acetone
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia pulex N S Solvent, acetone 48 h EC50 212 Immobilization Smith et al. (1988)
Gammurmus N S 96 h LC50 600 Finger et al.
pseudolimnaeus (1985)
Naphthalene
Daphnia magna < 24 h N S Solvent 48 h NOEC 600 Mortality LeBlanc (1980)
Daphnia magna Adult N Solvent, ethanol 4 h LOEC 1000 Behaviour Whitman & Miller
(1982)
Daphnia magna A S No solvent 48 h LC50 2160 Millemann et al.
(1984)
Daphnia magna N S 48 h EC50 4700 Immobilization Smith et al. (1988)
Daphnia magna < 24 h S 48 h EC50 4100 Crider et al. (1982)
Daphnia magna N S Solvent 48 h LC50 8600 LeBlanc (1980)
Daphnia magna 4-6 d N S No solvent 48 h LC50 16 000 Bobra et al. (1983)
Daphnia magna N S Solvent, acetone 48 h LC50 22 600 Eastmond et al.
+ triton-X-100 (1984)
Daphnia pulex 24-h old A R Filtered crystals Chronic NOEC 330 Increased Geiger & Buikema
lifespan & (1982)
reproduction
Chronic LOEC 600 Growth
Daphnia pulax 1.9-2.1 mm N S No solvent 96h LC50 1000 Trucco et al.
(1983)
Daphnia pulex N S 48 h LC50 3400 Geiger & Buikema
(1981)
Daphnia pulex N S Solvent, acetone 48 h EC50 4660 Immobility Smith et al. (1988)
Elasmopus sp. Adult - S 22°C (closed 24 h LC50 5000 Lee & Nicol
bottles) (1978)
Gammarus minus A S 48 h LC50 3930 Millemann et al.
(1984)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Hemigrapsus A F Seawater 8 d LC50 2800 Mortality and Gharrett & Rice
nudus 4 h water/8 h air 18 d EC50 700 locomotion (1987)
A F Seawater 8 d LC50 2200 Mortality and
8 h water/4 h air 18 d EC50 2000 locomotion
A F Seawater 8 d LC50 1100 Mortality and
12 h water/0 h air 18 d EC50 800 locomotion
Neomysis A F Artificial sea- 96 h LC50 850 Smith &
americana water, 25°C Hargreaves
A F Artificial sea- 96 h LC50 1280 (1983)
water, 15°C
Palaemonetes - - - - 24 h LC50 2500 Anderson et al.
penaeus (1974)
Pandalus - - S 12°C 96 h LC50 970 Korn et al. (1979)
goniurus S 8°C 96 h LC50 1020
S 4°C 96 h LC50 2200
Parhyale Adult - S 22°C 24 h LC50 6000 Lee & Nicol
hawaiaensis (1978)
Aromatic three-ring PAH
Anthracene
Artemia salina 1 d N S 1 h UVc 3 h LC50 20 Kagan et al.
(1985)
Artemia salina N S Darkc 24 h LC50 > 50 Abernethy et al.
(1986)
Daphnia magna - - UV-A=0 21 d NOEC 2.2 Population Foran et al. (1991)
UV-A=31c 21 d NOEC 2.2 growth
UV-A=60c 21 d NOEC 2.2
UV-A=117c 21 d NOEC 1.9
Daphnia magna Adult N S 1 h UVc 2 h LC50 20 Kagan et al.
(1985)
Daphnia magna A R (0.5d) No solvent 1.21 d LC50 15 Newsted & Giesy
-UV:1 d; (1987)
+UV:0.21 dc
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia magna 4-6 d N S Dark 48 h LC50 35.6 Abernethy et al.
(1986)
Daphnia magna 4-6 d N S No solvent 48 h LC50 3030b Bobra et al. (1983)
Daphnia magna < 24 h S 48 h LC0 < 500b Eastmond et al.
(1984)
Daphnia pulex A S 0.25 h sun, 24.25 h EC50 1.2 Immobility Allred & Giesy
1 d darkc (1985)
A S 0.17 h sun, 24.17 h EC100 9.6
1 d darkc
A S 0.75 h filtered 24.75 h NOEC 12.7
sun, 1 d darkc EC 75% 26.4
Daphnia pulex N S Solvent, acetone 48h EC50 754b Immobility Smith et al. (1988)
Benzo[a]fluorene
Daphnia magna A R (0.5 d) No solvent 1.96 d LC50 4.8 Newsted & Giesy
-UV:1 d; +UV:0.96 dc (1987)
Benzo[b]fluorine
Daphnia magna A R (0.5 d) No solvent 1.93 d LC50 2.2 Newsted & Giesy
-UV:1 d; +UV:0.93 dc (1987)
Fluoranthene
Artemia salina N S 1 h sunc 3 h LC50 40 Kagan et al.
(1985)
Daphnia magna Adult N S 1 h sunc 2 h LC50 4 Kagan et al.
(1985)
Daphnia magna A R (0.5 d) No solvent 1.45 d LC50 9.0 Newsted & Giesy
-UV:1 d; +UV:0.45 dc (1987)
Daphnia magna N S 48 h LC50 325 000b US Environmental
Protection Agency
(1978b)
Daphnia magna < 24 h N S Solvent 48 h LC50 320 000b LeBlanc (1980)
NOEC < 8800b Mortality
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Mysidopsis bahia 96 h LC50 40 US Environmental
Protection Agency
(1978b)
Phenanthrene
Artemia salina N S Dark 24 h LC50 677 Abernethy et al.
(1986)
Daphnia magna A S 48 h LC50 700 Millemann et al.
(1984)
Daphnia magna N S Solvent, acetone 48 h LC50 840 Eastmond et al.
+ triton-X-100 (1984)
Daphnia magna 4-6 d N S No solvent 48 h LC50 1160 Bobra et al. (1983)
Daphnia magna A R (0.5 d) No solvent 2 d NOEC 40.1 Immobility Newsted & Giesy
-UV:1 d; +UVA dc (1987)
Daphnia magna A F Glass column 21 d LC50 130 Hooftman &
21 d EC50 50 Reproduction Evers-de Ruiter
21 d NOEC 21 Reproduction (1992a)
21 d NOEC 66 Mortality
21 d NOEC 38 Growth,
condition,
behaviour
A R Glass column 21 d EC50 180 Reproduction
21 d NOEC 100 Reproduction
Daphnia magna 4-6 d N S Dark 48 h LC50 207 Abernethy et al.
(1986)
Daphnia pulex 1.9-2.1 mm N S No solvent 96 h LC50 100 Trucco et al.
(1983)
Daphnia pulex N S Solvent, acetone 48 h EC50 350 Immobility Smith et al. (1988)
Daphnia pulex N S 48 h EC50 734 Immobility Passino & Smith
(1987)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia pulex 48 h LC50 960- Geiger & Buikema
1280 (1982)
24 h A R (1 d) No solvent approx. NOEC 110 Reproduction,
50 d growth
Daphnia pulex N R (2-3 d) 97% A.I. 21 d LC33-73% 130 Savino & Tanabe
Solvent, acetone LOEC 60 Reproduction, (1989)
growth
Gammarus minus A S 48 h LC50 460 Millemann et al.
(1984)
Aromatic four-ring PAH
Benz[a]anthracene
Daphnia magna A R (0.5 d) No solvent 1.52 d LC50 1.8 Newsted & Giesy
-UV: 1 d; +UV:0.52 dc (1987)
Daphnia pulex 1.9-2.1 mm N S No solvent 96 h LC50 10 Trucco et al.
photo period: 12 h (1983)
Chrysene
Daphnia magna A R (0.5 d) No solvent 2 d LC50 0.7 Newsted & Giesy
-UV:1 d; +UV:1 dc (1987)
Daphnia magna Juvenile N S Solvent, acetone 2 d NOEC 288b Mortality Eastmond et al.
+ adult l/d = 16 h/8 h (1984)
Pyrene
Artemis salina 1 d N S 1 h UVc 3 h LC50 8 Kagan et al.
(1985)
Artemia salina N S Dark 24 h LC50 > 99 Abernethy et al.
(1986)
Daphnia magna Adult N S 1 h UVc 2 h LC50 4 Kagan et al.
(1985)
Daphnia magna A R (0.5 d) No solvent 1.14 d LC50 5.7 Newsted & Giesy
-UV: 1 d; +UV:O. 14 dc (1987)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Daphnia magna 4-6 d old N S Dark 48 h LC50 91 Abernethy et al.
(1986)
Daphnia magna 4-6 d old N S no solvent 48 h LC50 1820b Bobra et al. (1983)
Triphenylene
Daphnia magna A R (15 d) No solvent 2 d NOEC 1.7 Mortality Newsted & Giesy
-UV:1 d; +UV:1 dc (1987)
Aromatic five-ring PAH
Benzo[a]pyrene
Artemia salina Eggs 48 h NOEC 10 000b Viability Kuwabara et al.
(1980)
Calanus Adult - LC 4 Mortality Lee et al. (1972)
heigolandicus
Calanus 7 d EC 50b AHH activity Walters et al.
heigolandicus (1979)
Daphnia magna A R (0.5 d) No solvent 1.19 d LC50 1.5 Stimulation Newsted & Giesy
-UV:1 d; +UV:0.19 dc (1987)
Daphnia pulex 1.9-2.1 mm N S No solvent 96 h LC5O 5a Trucco et al.
photo period: 12 h (1983)
Benzo[e]pyrene
Daphnia magna A R (0.5 d) No solvent 1.64 d LC50 0.7 Newsted & Giesy
-UV:1 d; +UV:0.64 dc (1987)
Benzo[k]fluoranthene
Daphnia magna A R (0.5 d) No solvent 1.54 d LC50 1.4a Newsted & Giesy
-UV: 1 d; +UV:0.54 dc (1987)
Dibenz[a,h]anthracene
Daphnia magna A R (0.5d) No solvent 1.13 d LC50 0.4a Newsted & Giesy
-UV:1 d; +UV:0.13 dc (1987)
Table 103. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Perylene
Daphnia magna A R (0.5 d) No solvent 1.76 d LC50 0.6a Newsted & Giesy
-UV:1 d; +UV:0.76 dc (1987)
Aromatic six-ring PAH
Benzo[ghi]perylene
Daphnia magna A R (0.5 d) No solvent 1.58 d LC50 0.2 Newsted & Giesy
-UV:1 d; +UV:0.58 dc (1987)
A, analysed concentration; N, nominal concentration; S, static system; F, flow-through system; IF, intermittent flow;
R(0.5 d), system with renewal (each half day); S\F, first period in a static system followed by a period in a flow-through system;
l/d, light/dark; +UV, with UV radiation; -UV, without UV radiation; DMSO, dimethyl sulfoxide; EC, exposure concentration;
NOEC, no-observed-effect concentration; LC, lethal concentration; LOEC, lowest-observed-effect concentration
a Concentration higher than solubility but not exceeding it by 10 times
b Concentration 10 times higher than the solubility
c Explicitly mentioned that organisms were tested for phototoxicity of test substance either in sunlight or artificial UV radiation
d From Cairns & Nebeker (1982)
PAH-polluted elutriates derived from polluted sediments were highly
toxic to Daphnia magna when combined with either sunlight or 354-nm
near-ultraviolet radiation, whereas none of the elutriates was toxic
in the absence of light (Davenport & Spacie, 1991).
A 50% decrease in feeding rate was reported in the mussel
Mytilus edulis after nine days' exposure to 80 µg/litre fluoranthene
(Donkin et al., 1989). When Mercenaria mercenaria clams were exposed
in flow-through tanks, in which seawater was pumped through sand
columns adsorbing 50 mg benzo [a]pyrene, the concentrations in the
water were generally below the detection limit (< 0.001 µg/litre),
whereas the tissue concentrations were 2-4 µg/kg (0.15 µg/kg in
control clams). This resulted in an increased intrahaemocytic lysozyme
concentration and significantly impaired ability to clear bacteria.
Thus, resistance to bacterial infection is decreased by PAH (Anderson
et al., 1981).
The 96-h LC50 values in the marine polychaete Neanthes
arenaceodentate were 3800 µg/litre for the two-ring PAH naphthalene
and 1000 µg/litre for fluorene, 600 µg/litre for phenanthrene, and 300
µg/litre for 1-methyl-phenanthrene (three-ring). None of the four- and
five-ring PAH were toxic up to the highest concentration tested, 1000
µg/litre, except fluoranthene, which had a 96-h LC50 of 500 µg/litre
(Rossi & Neff, 1978).
9.1.2.3 Vertebrates
Data on toxicity to vertebrates like fish and amphibians are available
for two- to five-ring PAH (Table 104). Most of the data are derived
from phototoxicity tests.
As discussed in Section 4, fish can metabolize PAH into intermediates
that may have teratogenic, mutagenic, or carcinogenic properties and
are associated with hepatic tumours in free-living fish. In addition,
certain PAH can cause physiological changes that affect the growth,
reproduction, swimming performance, and respiration of fish. The
effect of environmental carcinogens on fish populations depends on the
exposure received at each susceptible life stage, the ability at each
stage to absorb and metabolize the carcinogen and repair the ensuing
damage, and the consequences of tumour induction in vital organs at
each life stage. Additional factors that influence carcinogenicity are
the stage of organism development, the route of exposure, genetic
variation, and cytochrome P450 mixed-function oxygenase activity
(Bailey et al., 1989).
Several PAH can produce cancer-like growths and are teratogenic and
mutagenic to fish. In Oncorhynchus mykiss (former name for
Salmo gairdneri), the liver was the primary target after exposure to
benzo [a]pyrene in the diet and by intraperitoneal injection.
Administration by the latter route, while not directly relevant to
environmental exposure, also produced a fibrosarcoma and a stomach
papilloma in one individual, along with tumours, indicating that the
route of exposure is of some importance in fish (Hendricks et al.,
1985).
Table 104. Results of tests for the toxicity of polycyclic aromatic hydrocarbons (PAH) towards vertebrates
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
FISH
Aromatic two-ring PAH
Acenaphthene
Cyprinodon Juvenile N S 96 h LC50 2200 Heitmuller et al.
variegatus (1981)
Ictalurus punctatus A F solvent 96 h LC50 1720 Holcombe et al.
(1983)
Lepomis macrochirus 0.32-1.2 g N S solvent 96 h LC50 1700 Buccafusco et al.
(1981)
Pimephales promelas Juvenile (32 d) A F solvent 96 h LC50 1600 Holcombe et al.
(1983)
Pimephales promelas Embryo- A F l/d = 16/8 h 32 d NOEC 509 Survival Cairns & Nebeker
juvenile Glass column 96 h LC50 608 (1982)
Solvent, DMF
Oncorhynchus mykiss Juvenile A F Solvent, 96 h LC50 670 Holcombe et al.
isopropanol 48 h LC50 1130 (1983)
Salmo trutta Juvenile A F Solvent, 69 h LC50 580 Holcombe et al.
isopropanol 48 h LC50 650 (1983)
Acenaphthylene
Oryzias latipes 48 h LG50 11 000 Yoshioka et al.
(1986)
Fluorene
Lepomis macrochirus A F 30 d NOEC 19 Predating prey Finger et al.
NOEC 42 Growth (1985)
NOEC 49 Mortality
N S 96 h LC50 910
Oncorhynchus mykiss N S 96 h LC50 820
Pimephales promelas N S 96 h LC50 > 100 000a
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Naphthalene
Abramis bramana 1 year N R 96 h LC50 10 000 Frumin et al. (1992)
Fundulus heteroclitus 30 d NOEC 1600 US Environmental
Protection Agency
(1986d)
Micropterus salmoides Eggs and A F No solvent 7 d NOEC 28 Survival Black et al. (1983)
larvae (to 4 d LC8-35 28-239
posthatch
Embryo-larva A F No solvent 7 d LC50 510
(to 4 d
posthatch)
Micropterus salmoides Eggs-larvae A F No solvent 7 d LC50 680 Millemann et al.
(1984)
Oncorhynchus Fry - S 4-12°C 96 h LC50 1370-1240 Korn et al. (1979)
gorbuscha
Oncorhynchus kisutch 1 g A F 96 h LC50 770 Parasitized Moles (1980)
Oncorhynchus kisutch 1 g A F 40 d EC 670- Growth Moles et al. (1981)
1400
Oncorhynchus kisutch 1 g A F 96 h LC50 2100 Moles (1980)
Oncorhynchus kisutch 1 g A F 96 h LC50 3220 Unparasitized Moles (1980)
Oncorhynchus mykiss Eggs-larvae A F No solvent 23 d NOEC 15 Hatching Black et al. (1983)
(to 4 d post- 27 d NOEC 15 Survival
hatch) LC50 110
Oncorhynchus mykiss Eggs-larvae A F No solvent 27 d NOEC 120 Survival, Millemann et al.
teratogenity (1984)
Oncorhynchus mykiss Adult A F 96 h LC50 1600 DeGraeve et al.
(1982)
Oncorhynchus mykiss 13-21 d N S Solvent, 96 h LC50 4500 Edsall (1991)
acetone
Oncorhynchus mykiss A F 96 h LC50 2300 DeGraeve et al.
(1980)
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Pimephales promelas Embryo-larva A F 30 d NOEC 450 Growth, DeGraeve et al.
EC 850 hatching (1982)
Subchronic LOEC 850 Reproduction
Pimephales promelas Juvenile A S No solvent 96 h LC50 1990 Millemann et al.
(1984)
Pimephales promelas A F 96 h LC50 4900 Reproduction DeGraeve et al.
(1980)
Pimephales promelas A F 96 h LC50 6140 Geiger et al. (1985)
Pimephales promelas A F 96 h LC50 6080 Holcombe et al.
(1984)
Pimephales promelas Adult A F 96 h LC50 7900 DeGraeve et al.
(1982)
Pimephales promelas A F 96 h LC50 8900 DeGraeve et al.
(1980)
Tilapia oreochromis N R 96 h LC50 22 400 Frumin et al.
(1992)
Aromatic three-ring PAH
Anthracene
Lepomis macrochirus 1-1.5 g N F 24 h UV, 200 h NOEC 1.2 Mortality Oris & Giesy
0 h darkb (1986)
Lepomis sp. Juvenile A F Low UV 96 h LC50 2.78 Oris & Giesy
(2-3 cm) intensityb (1985)
Lepomis macrochirus 1-1.5 g N F 24 h UVb 125 h LC50 4.5 Oris & Giesy
96 h LC50 4.5 (1986)
Lepomis macrochirus 96 h LC50 11.9 US Environmental
Protection Agency
(1987b)
Lepomis sp. Juvenile A F High UV 96 h LC50 11.9 Oris & Giesy
(2-3 cm) intensityb (1985)
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Lepomis macrochirus A F Shadowb, 144 h LC0 12.7 Bowling et al.
microcosm + sediment (1983)
2 h sunb 25 h LC50 12.7
12 h sunb 72 h LC100 12.7
Lepomis macrochirus 1-1.5 g N F 12 h UV, 200 h NOEC 15 Mortality Oris & Giesy
12 h darkb (1986)
Lepomis sp. Juvenile A F Medium UV 96 h LC50 18.2 Oris & Giesy
(2-3 cm) intensityb (1985)
Low UV 96 h LC50 26.5
intensityb
Lepomis macrochirus 1-1.5 g N F 6h UV, 96 h LC50 46 Oris & Giesy
18 h darkb (1986)
Pimephales promelas A R (0.5 d) No 1.6d LC50 5.4 Oris & Giesy
solvent; dark: (1987)
1 d; +UV:16 hb
Dark 4 d NOEC 5.4 Mortality
Pimephales promelas Adult A F No sun 9 weeks LOEC 6.6 Egg production Hall & Oris (1991)
F1 A R No sun NOEC 6.6 Deformities
No sun NOEC > 12 Survival and
Sunb LOEC 12 hatching
Pimephales promelas 0.8g N S 0.5 h sunb 24 h LC50 360c Hatching Kagan et al. (1985)
Fluoranthene
Brachydanio rerio Eggs-larvae A F Yellow light 41 d NOEC 4.8 Growth Hooftman & Evers-
Solvent, TBA 41 d NOEC 48 Mortality de Ruiter (1992b)
Cyprinodon variegatus Juvenile N S 96 h LC0 560 000a Heitmuller et al.
(1981)
Lepomis macrochirus N S 96 h LC50 3980a US Environmental
Protection Agency
(1978b)
Lepomnis macrochirus 0.32-1.2 g N S Solvent 96 h LC50 4000a Buccafusco et al.
(1981)
Pimephales promelas 0.8 g N S 0.5 h sunb 24 h LC50 200 Kagan et al. (1985)
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Phenanthrene
Brachydanio redo Eggs-larvae A R Yellow light 28 d NOEC 28 Growth Hooftman & Evers-
(2 d) solvent, TBA 28 d LOEC 49 de Ruiter (1992d)
Gambusia affinis 24 h LC50 150 Neff (1979)
Micropterus salmoides Eggs-larvae A F 7 d LC50 180 Black et al. (1983)
Micropterus salmoides Eggs-larvae A F No solvent 7 d LC50 250 Millemann et al.
(1984)
Oncorhynchus mykiss Eggs-larvae A F No solvent 23 d NOEC 4 Hatching Black et al. (1983)
27 d NOEC 4 Survival
Oncorhynchus mykiss Eggs-larvae A F No solvent 27 d LC50 30 Millemann et al.
(1984)
Oncorhynchus mykiss Eggs-larvae A F 7 d LC50 40 Black et al. (1983)
Oncorhynchus mykiss N S Solvent, 96 h LC50 3200c Edsall (1991)
acetone
Pimephales promelas A R (0.5 d) No 5 d NOEC 10 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 10
Aromatic four-ring PAH
Benz[a]anthracene
Pimephales promelas A R (0.5 d) No 3.7 d LC50 1.8 Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 1.8 Mortality
Poecilia formosa F Injection 9 months EC 23c Thyroid Woodhead et al.
morphology (1982)
Benzo[k]fluoranthene
Brachydanio rerio Eggs-larvae N F Yellow light 42 d LC50 0.68 Hooftman & Evers-
solvent, TBA 42 d NOEC 0.23 Growth de Ruiter (1992c)
42 d NOEC 0.40 Mortality
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Poecilia formosa F Injection 9 months EC 23c Spleen Woodhead et al.
morphology (1982)
Chrysene
Brachydanio rerio Eggs-larvae A R (2 d) Yellow 28 d NOEC > 0.9 Growth, Hooftman & Evers-
light, glass mortality, de Ruiter (1992b)
column hatching
Pyrene
Pimephales promelas A R (0.5 d) No 1.13 d LC50 25.6 Oris & Giesy
solvent; dark: (1987)
1 d; +UV:0.13 db
Dark 4 d NOEC 25.6 Mortality
Pimephales promelas N S Solvent, 24 h NOEC > 10 000a Mortality Kagan et al.
DMSO (1987)
Aromatic five-ring PAH
Benzo[a]pyrene
Brachydanio rerio Eggs-larvae A R (2 d) Yellow 28 d NOEC > 4 Growth, Hooftman & Evers-
light, glass mortality, de Ruiter (1992b)
column hatching
Fundulus grandis F Injection 7 d EC 18% 30 mg/ Liver weight Melius et al. (1980)
kg bw increase
Hippoglossoides Adult Food 5 h EC 21% 8 mg/kg Hatching Hose et al. (1981)
elassodan success
Ictalurus punctatus Juvenile 7-10 EC 1.0 Skeletal Martin (1980)
months structure,
pigmentation
Leuresthes tenuis Embryo A S Solvent, 14 d NOEC 7.0c Hatching Winkler et al.
acetone success (1983)
14 d EC 12% 7.0c Larval
morphology
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Leuresthes tenuis Larvae - EC 100a Growth Puffer et al. (1979)
EC 100a Development
Micropogonias Adult Oral 30 d NOEC 5.7 Behaviour, Thomas (1988)
undulatus 45-60 g mg/kg growth,
(fresh wt) respiration,
locomotion
EC 34% 5.7 Ovarian growth,
mg/kg hormone level
(fresh wt)
Oryzias latipes 6-10 d A R 0.5 mg/litre 2 × 0.25d NOEC 47+11a,c Neoplastic Hawkins et al.
DMF 2 × 0.25e NOEC 47+11a,c lesions (1988,1990)
2 × 0.25f NOEC < 47+11a,c
Oncorhynchus kisutch Embryos N S\F0.1 mmol/litre 24 hg NOEC 11 000a Emergence Ostrander et al.
1 d after DMSO (1988)
fertilization
1 week before NOEC 10 000a Emergence
hatching
1 d after NOEC < 10 000a Orientation
fertilization
1 week before NOEC < 10 000a Orientation
hatching
Oncorhynchus mykiss Eggs Injection EC 4.5 mg/ Carcino- Black et al.
egg genicity (1988)
Oncorhynchus mykiss Embryo-larva A R Column 36 d NOEC 2.4c Morphology Hannah et al.
36 d LOEC 6.7c Morphology (1982)
Parophyrus vetulus Eggs-larvae A S Solvent, 5 d NOEC > 2.1 Development Hose et al.
ethanol (1982)
Pimephales promelas A R (0.5 d) No 2.7 d LC50 5.6c Oris & Giesy
solvent; dark: (1987)
1 d; +UV:40 hb
Darkb 4 d NOEC 5.6 Mortality
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Poecilia reticulata 6-10 d A S 0.5 mg/litre 2 × 0.25e NOEC 32+8 Neoplastic Hawkins et al.
DMF; darkb 2 × 0.25f NOEC 32+8 lesions (1988, 1990)
Poeciliopsis lucida A S Solvent, 1 d/6 LC 70% 3750a Lethal effects Goddard et al.
Poeciliopsis lucida acetone months NOEC 1250a Lethal effects (1987)
Psettichtys Embryo A F Solvent, 5 d EC 50% 0.1 Hatching Hose et al.
melanostAicatus ethanol success (1982)
7 d EC 0.1 Development
Benzo[a]pyrene
Pimephales promelas A R (0.5 d) No 5 d NOEC 2.9 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 2.9 Mortality
Dibenz[a,h]anthracene
Pimephales promelas Larvae A R (0.5 d) No 5 d NOEC 0.15 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 0.15 Mortality
Perylene
Pimephales promelas Larvae A R (0.5 d) No 5 d NOEC 1.7c Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5 d NOEC 1.7c Mortality
Aromatic six-ring PAH
Benzo[ghi]perylene
Brachydanio rerio Eggs-larvae A R (2 d) Yellow 28 d NOEC > 0.16 Growth, Hooftman & Evers-
light, glass mortality, de Ruiter (1992d)
column hatching
Table 104. (continued)
PAH, species Life stage Test conditions Duration Effect Concentration End-point Reference
(µg/litre)
Pimephales promelas Larvae A R (0.5 d) No 5 d NOEC 0.15 Mortality Oris & Giesy
solvent; dark: (1987)
1 d; +UV:4 db
Dark 5d NOEC 0.15 Mortality
AMPHIBIANS
Aromatic two-ring PAH
Naphthalene
Xenopus laevis Larvae F Fluorescent 96 h LC50 2100 Edmisten &
(3 weeks) light 6 h EC50 1700- Absence of Bantle (1982)
2300 swimming
Aromatic three-ring PAH
Anthracene
Rana pipiens Embryo Sunb 24 h LC50 110c Kagan et al.
(1985)
Rana pipiens 5 h LC50 25 US Environmental
Protection Agency
(1987b)
Fluoranthene
Rana pipiens N S 1 h sunb 24 h LC50 90 Kagan et al;
(1985)
Aromatic four-ring PAH
Pyrene
Rana pipiens Embryo Sunb 24 h LC50 140c Kagan et al.
(1985)
Table 104 (continued)
A, analysed concentration; N, nominal concentration; S, static system; F, flow-through system; IF, intermittent flow; R (0.5 d),
system with renewal (each half day); S\F, first period in a static system, second in a flow-through system; l/d, light/dark;
UV, with UV radiation; -UV, without UV radiation; TBA, tertiary butyl alcohol; DMF, dimethylformamide; DMSO, dimethyl sulfoxide;
LC, lethal concentration; NOEC, no-observed-effect concentration; EC, effect concentration; LOEC, lowest-observed-effect
concentration
a Concentration 10 times higher than solubility
b Explicitly mentioned that organisms were tested for phototoxicity of test substance either by sunlight or artificial UV radiation
c Concentration higher than solubility but not exceeding it by 10 times
d Exposure followed by 168 d depuration
e Exposure was followed by 252 d depuration
f Exposure followed by 365 d depuration
e Exposure followed by 62 d depuration
Painting of benzo [a]pyrene and 3-methylcholanthrene onto the skin of
three freshwater fish twice a week for three to six months caused
epitheliomas in Gasterosteus aculetus (stickleback) and Rhodeus
amarus (bitterling) but not in Cyprinus carpio (carp). In the same
study, 10 injections of 10 mg benzo [a]pyrene in glycerol to G.
aculetus produced injection-site necrosis but no tumours (Ermer,
1970).
The phototoxic potential of PAH in fish is influenced by the amount of
ultraviolet radiation and light absorbed. Adult fathead minnows
(Pimephales promelas) were exposed in the absence of artificial
ultraviolet radiation to 0.6 or 12 µg/litre anthracene for six weeks,
the dose being increased to 20 µg/litre for three weeks in two groups.
Eggs were collected daily, placed in clean water, and exposed or
unexposed to ultraviolet radiation, until 96 h after hatching. All
fish showed impaired egg production. The lethal concentration in eggs
was estimated to be 19 µg/g (Hall & Oris, 1991).
Exposure of the spot Leiostomus xanthurus to PAH-contaminated
sediment derived from a river station resulted in death, with fin
erosion, ulceration of the lateral body surface, and several lesions
of internal organs. The total concentration of the 21 PAH analysed in
the sediment was 21 000 mg/kg dry weight. The 28-day LC50 was
estimated to be 3.2% of contaminated sediment (Roberts et al., 1989).
9.1.2.4 Sediment-dwelling organisms
The 10-day LC50 values for fluoranthene in sediment were 11 mg/kg dry
weight for the marine amphipod Eohaustorius estuarius, 5.1 mg/kg for
the marine amphipod Rhepoxynius abronius, and 15 mg/kg for the
freshwater amphipod Hyalella azteca. The sensitivity of
E. estuarius was not influenced by salinity (DeWitt et al., 1989).
The toxicity of a sediment containing 46% fines and 0.9% total organic
carbon and artificially supplemented with solutions of fluoranthene,
phenanthrene, benz [a]anthracene, benzo [a]pyrene,
2,6-dimethylnaphthalene, 1-methylnaphthalene, and 2-methylnaphthalene
was tested in the amphipod Rhepoxynius abronius. Significant
mortality occurred among amphipods exposed for 10 days to nominal
concentrations of 15, 10, 10, 5, 0.5, 0.5, and 0.5 mg/kg dry weight of
the seven PAH, respectively, whereas at levels five times lower no
toxic effects were observed (Plesha et al., 1988).
The ability of an amphipod species to metabolize xenobiotic compounds
to reactive intermediates appears to influence its sensitivity to
chemical contaminants. Contaminated sediments are generally more toxic
to R. abronius than E. washingtonianus (Plesha et al., 1988). R.
abronius and E. washingtonianus exposed to sediment-associated,
radiolabelled benzo [a]pyrene accumulated similar concentrations of
radiolabel in their tissues after seven days of exposure, but the
proportions of metabolites and the amount of radiolabel bound to
cellular macromolecules were greater in R. abronius than in
E. washingtonianus. One explanation for the greater sensitivity of
R. abronius might therefore be a greater tendency to form reactive
metabolites that are more acutely toxic than the parent compound
(Reichert et al., 1985).
The toxicity of fluoranthene in sediment to the marine amphipods
R. abronius and Corophium spinicorne was determined by equilibrium
partitioning. Toxicity was determined in well-sorted, fine sands
containing organic carbon at 0.18, 0.31, or 0.48% of dry weight. The
epibenthic tube-dwelling C. spinicorne was less sensitive than the
free-burrowing R. abronius, possibly because of different routes of
exposure. The 10-day LC50 values for R. abronius were 3.4, 6.5, and
10.7 mg/kg dry weight in sediments with increasing organic carbon. A
10-day LC50 value for C. spinicorne could be determined only in the
sediment with the lowest organic carbon level, at 5.1 mg/kg dry
weight. An LC50 of 24 µg/litre was calculated on the basis of
equilibrium partitioning. In another experiment, a 10-day LC50 of 4.2
mg/kg dry weight was determined for R. abronius in sediment with
0.26% organic carbon (Swartz et al., 1988).
Diporeia sp. were exposed to a sediment artificially contaminated
with a mixture of labelled phenanthrene and pyrene and nine unlabelled
PAH at total concentrations of 21, 41, 120, and 330 µmol/kg dry
sediment. The amphipods avoided the sediment containing the highest
dose during the first six days; the estimated LC50 at day 26 was
estimated to be 600 mmol/kg dry sediment. Deaths occurred after 26
days' exposure to the two highest concentrations. The concentration of
PAH required to elicit 38% mortality at day 19 was 2.9 mmol/kg
organism. The authors concluded that PAH probably have a non-polar
narcotic mode of action and suggested that their effect is additive,
which is supported by the observation that exposure of Diporeia for
31 days to pyrene resulted in an LD50 of 5.8 mmol/kg organism
(Landrum et al., 1991).
9.1.2.5 Toxicity of combinations of PAH
In the concentration addition model, additive effects were found for
phenanthrene, anthracene, naphthalene, and acenaphthene in Daphnia
magna (Muñoz & Tarazona, 1993). Although the toxic effects of
combined PAH to Brachydanio rerio were also found to be additive,
the concentrations tested were close to the maximum solubility of the
PAH (Hooftman et al., 1993).
9.1.3 Terrestrial organisms
9.1.3.1 Plants
The effects of anthracene on emergence were tested in three native
Australian plant species, heath banksia (Banksia ericifolia),
she-oak (Casuarina distyla), and yellow bloodwood (Eucalyptus
eximia), and in three crop species, oat (Avena sativa), cucumber
(Cucumis sativus), and soya bean (Glycine max). A. sativa and
C. sativus were sensitive, with EC50 values of 30 and 720 mg/kg,
respectively; the the other plants were not sensitive up to the
highest concentration tested (1000 mg/kg dry weight) (Mitchell et al.,
1988).
9.1.3.2 Invertebrates
Porcellio scaber and Oniscus asellus showed little difference in
their sensitivity to benzo [a]pyrene. The growth of both species was
affected after exposure for nine weeks to 100 and 316 mg/kg dry
weight. The only difference in response was that the lipid pool was
reduced in O. asellus and the protein pool was reduced in
P. scaber. No effects were observed at 32 mg/kg dry weight (Van
Brummelen & Stuijfzand, 1993).
The LC50 values for fluorene in four earthworm species,
Allolobophora tuberculata, Eisenia foetida, Eudrilus eugenia, and
Perionyx excavatus, in an artificial soil were 210, 17, 200, and 170
mg/kg dry weight, respectively, after two weeks' exposure in soil. In
the contact test, the LC50 values were 120, 170, 47, and 78 µg/m2,
respectively (Neuhauser et al., 1986). Fluorene at 750 mg/kg dry
weight significantly reduced the reproduction of E. foetida, but no
deaths occurred (Neuhauser & Callahan, 1990).
Benzo [a]pyrene was incorporated in the food of the wood louse
Porcellio scaber, and respiration, growth, and food consumption were
measured for four weeks. Consumption was not affected by
concentrations up to 125 mg/kg, but at the highest dose, male isopods
showed significantly greater assimilation (34%) than controls (26%)
due to an active mechanism, which was not observed in females. Growth
varied considerably between individuals. At the highest
concentrations, however, the growth efficiency of males was
significantly decreased. At 25 mg/kg, no significant effects were
found (Van Straalen & Verweij, 1991).
The toxicity of PAH to the earthworm E. foetida and the springtail
Folsoma candida was studied in a standard soil. As phenanthrene at a
concentration of 1000 mg/kg dry weight appeared to be removed almost
completely from the soil after 14 days' exposure, the soil was renewed
regularly. The 28-day LC50 of phenanthrene in F. candida was 150
mg/kg dry weight, the EC50 was 120 mg/kg, and the NOEC for
reproduction was 75 mg/kg dry weight. The 21-day EC50 for
reproduction of E. foetida was 240 mg/kg dry weight. No effect on
the survival or reproduction of F. candida was seen after 28 days'
exposure to chrysene, benzo [k]fluoranthene, or benzo [a]pyrene at
180 mg/kg dry weight, and no effect on the reproduction or survival of
E. foetida was seen after 14 days' exposure to chrysene at 1000
mg/kg dry weight (Bowmer et al., 1993).
Ingestion of naphthalene, anthracene, benz [a]anthracene, pyrene, or
benzo [a]pyrene at 1000 mg/kg food per day for 18 days caused
significant increases in mortality among Acheta domesticus crickets.
Naphthalene caused 50% mortality within 12 days, and anthracene,
benz [a]anthracene, pyrene, and benzo [a]pyrene caused 39, 26, 23,
and 32% mortality, respectively, after 18 days (Walton, 1980).
9.1.3.3 Vertebrates
Benzo [a]pyrene and chrysene dissolved in oil and covering less than
10% of the surface of duck eggs reduced hatching and had teratogenic
and embryotoxic effects (Hoffmann & Gay, 1981). The 72-h LD50 values
for chick embryos were 14 µg/kg egg for benzo [k]fluoranthen, 39
µg/kg for dibenz [a,h]anthracene, and 79 µg/kg for
benz [a]anthracene (Brunström et al., 1991). The LD50 values for
acenaphthene, anthracene, phenanthrene, and fluorene in red-winged
blackbirds were > 100 mg/kg (Schafer et al., 1983).
9.2 Field observations
9.2.1 Microorganisms
9.2.1.1 Water
The effects on a benthic community were determined in the sediment of
a stream in which a gradient of PAH concentrations was found, with
total PAH contents at four sites of 0, 3100, 39 000, and 49 000 mg/kg
dry weight. Detrital accumulation and the redox potentials increased
with PAH level. Removal of fungi at the polluted sites was probably
the major factor in detrital accumulation, and a reduction in
bacterial biomass was thought to be the primary cause of the increased
redox potentials (Catallo & Gambrell, 1987).
Addition of 1000 mg/kg dry weight naphthalene to anaerobic salt marsh
sediments resulted in significant inhibition of methanogenesis,
sulfate reduction, and evolution of carbon dioxide. Phenanthrene at
the same concentration had no significant effect on these activities,
whereas the same dose of naphthalene inhibited methanogenesis and then
stimulated it relative to controls; however, the sulfate reduction was
sustained. Carbon dioxide evolution was reduced in only one of the
three experiments (Kiene & Capone, 1984).
9.2.1.2 Soil
No data were available
9.2.2 Aquatic organisms
9.2.2.1 Plants
No data were available
9.2.2.2 Invertebrates
In the study of Catallo & Gambrell (1987), described in section
9.2.1.1, the population densities of nematodes, oligochaetes, and
other benthic invertebrates were significantly decreased at the site
with a total PAH content of 3100 mg/kg and were eradicated at the
other two sites.
Ecosystem responses were tested in small, multispecies, aquatic
systems (Leffler microcosm) exposed once to fluorene dissolved in
acetone at a concentration of 0.12, 0.50, 2, 5, or 10 mg/litre. The
estimated half-life was 2.1 days. The LOEL for respiration in the dark
(Rni) and the ratio of net productivity:Rni was 0.12 mg/litre in all
four communities, suggesting that the responses of these microcosms
were not completely independent of their source communities. At 5 and
10 mg/litre, the zooplankton populations were almost eliminated (Stay
et al., 1988).
9.2.2.3 Vertebrates
PAH metabolites were found in English sole (Parophrys vetulus) with
hepatic tumours collected from Puget Sound, Washington, USA (Malins,
1982; Malins et al., 1984). Detectable levels of similar organic free
radicals were found only when microsomes were incubated with the PAH
fraction of extracts of sediment from this area and not after
incubation with the alkane or aromatic polychlorinated biphenyl
fractions (Collier et al., 1992).
A consistent, statistically significant association was found between
the prevalence of hepatic tumours in free-living P. vetulus and the
levels of PAH in bottom sediment from sites where the fish were
captured, in a series of studies conducted over seven years in Puget
Sound. The strongest relationships were found for four categories of
hepatic lesion. Other contaminants such as trace metals,
polychlorinated biphenyls, pesticides, and chlorinated butadienes were
measured in all studies, but the strongest associations with liver
lesions were found with PAH. The concentrations of total PAH in the
sediment ranged from 0.005 mg/kg dry weight at an uncontaminated site
to 540 mg/kg at the most polluted location (Landahl et al., 1990).
9.2.3 Terrestrial organisms
9.2.3.1 Plants
No data were available.
9.2.3.2 Invertebrates
Application of naphthalene at 200 g/m2 to four soils resulted in a
significant reduction in soil arthropods such as Collembola. At 10
g/m2, the densities of most arthropods increased (Best et al., 1978).
9.2.3.3 Vertebrates
No data were available.
10. EVALUATION OF RISKS TO HUMAN HEALTH AND EFFECTS ON THE ENVIRONMENT
10.1 Human health
10.1.1 Exposure
Polycyclic aromatic hydrocarbons (PAH) are released into the
environment as a result of incomplete combustion of organic materials,
especially during industrial processes, incineration of refuse, and
fossil fuel combustion. They are released mainly into the atmosphere,
adsorbed onto particulate matter, which is deposited in the aquatic
and terrestrial environments. Direct contamination may also occur
from, e.g. creosote-preserved wood and deposition of contaminated
refuse such as sewage sludge and fly ash.
Owing to their long-range transport, PAH, and particularly those with
high molecular masses, are ubiquitous in the environment. The levels
in ambient air vary considerably, ranging from several picograms per
cubic metre to < 1 µg/m3, although phenanthrene has been found at
levels of several micrograms per cubic metre.
10.1.1.1 General population
Humans are exposed to various complex mixtures of PAH in the air,
food, water, and soil. The main sources of human exposure are
emissions from the combustion of coal, diesel, petrol, kerosene, wood,
biomass, and synthetic chemicals such as plastics. PAH account for a
significant portion of the carcinogenicity of some mixtures, such as
coal-tar soot, but not of others such as cigarette smoke, diesel
emissions, and urban aerosol. The levels of selected PAH of
toxicological significance in various environmental media are given in
Tables 105 and 106.
Pollution of indoor air by PAH is due mainly to tobacco smoking,
residential heating, and PAH from outdoor ambient air (Table 107).
Extremely high values (e.g. 15 000 ng/m3 benzo [a]pyrene) were found
in unvented rooms with open fireplaces, especially those in which soft
coal was used for cooking and heating. High concentrations have also
been reported in wood-heated saunas.
The predominant sources of PAH pollution in urban areas are motor
vehicle traffic (both petrol- and diesel-fuelled) and residential
heating, especially with wood, coal, and biomass. The concentrations
are up to one order of magnitude higher in winter than in summer. Such
differences may limit the validity of sampling campaigns performed
during only part of the year for the purpose of estimating mean human
exposure in urban areas.
No conclusion can be drawn about the relative PAH emissions from
petrol-fuelled engines (without catalytic converters) and
diesel-fuelled engines, given the limited number of studies in which
emissions were compared under the same conditions of sampling and
analysis.
Table 105. Reported levels of selected polycyclic aromatic hydrocarbons (PAH) in various media
Compound Ambient air Drinking-water Surface water Soil Soil near Sediment
(ng/m3) (ng/litre) (ng/litre)a (µg/kg) industrial (µg/kg)b
sources
(mg/kg)
Acenaphthenec 0.06-370 0.02-14 0.08-1200 1-21 1-5100 0.04-3800
Anthracene 0.004-61 0.5-9.7 0.01-930 0.2-70 0.2-140 0.06-27 000
Benzo[a]pyrene 0.002-780 0.04-2.0 0.03-910 0.8-3200 0.8-38 0.004-110 000
Chrysened 0.01-260 No data 10-1100 2.1-2700 2.1-1200 0.04-21 000
Dibenzo[a,l]pyrene 0.05-1.5 No data No data No data No data No data
Fluoranthene 0.03-810 0.58-3400 0.4-6400 0.3-3700 0.3-340 0.1-610 000
Fluorene 0.02-420 0.008-21 0.33-2500 1-14 1-8600 0.5-6500
Naphthalenec 0.03-940 0.38-8.8 0.4-2100 3-60 3-5.2 0.7-44 000
Phenanthrene 0.002-1800 2.2-90 0.24-5700 17-1700 17-20 000 0.06-65 000
Pyrene 0.002-540 0.3-40 0.12-3100 0.1-4500 0.1-1600 0.1-410 000
Only detected values are given, owing to the variability of limits of detection; data obtained from studies of
road tunnels were excluded even though short-term exposure to such high levels may contribute significantly to
overall daily exposure.
a Concentration may exceed solubility in water owing to presence of particulates in sample
b Highest values usually determined in harbour sediments
c Probably underestimated because of shortcomings in sampling and analytical procedures; data were not provided
from laboratories that found high values for three- to six-ring PAH.
d Most measurements performed with gas chromatography, so that actual levels are overestimates due to analytical
interference by triphenylene.
Table 106. Reported levels (µg/kg) of selected polycyclic aromatic hydrocarbons (PAH) in food
Compound Meat and Fish and Dairy products Oil, fats, and Vegetables Cereals
meat products seafooda margarine and fruit
Acenaphthene No data 0.9-500 No data 0.02-0.45 No data 0.6-0.7
Anthracene 0.9-31 0.05-240 No data 0.02-460 0.09-0.4b 0.5-1.3
Benzo[a]pyrene 0.01-42 (130c) 0.003-290 0.08-1.3 0.02-140 0.05-6.2b 0.1-0.8
Chrysened 0.15-0.6 0.03-210 1.3-1.5 0.1-120 0.5-69b 0.77
Dibenzo[a,l]pyrene No data No data No data No data No data No data
Fluoranthene 0.48-100 0.1-1800 0.01-4.2 (8.0e) 0.02-460 0.93-120b 0.3-28
Fluorene No data 0.2-370 No data 0.02-200 No data 1.3-2.7
Naphthalenec No data 0.8-210 No data No data No data No data
Phenanthrene 3-64 0.1-2700 0.56-0.72 0.09-1400 0.47-17b 9.9-29
Pyrene 0.55-63 0.03-1500 0.04-2.7 (4.8e) 0.02-330 0.83-70b 0.22-21
a Data from industrially polluted areas included as food items from those areas enter the market
b Values detected in vegetables grown on contaminated soil are excluded
c Exceptionally high values found in processed foods, but PAH not determined in these studies
d Most measurements performed with gas chromatography, so that actual levels are overestimates due to analytical
interference by triphenylene.
e Found in infant food
Table 107. Ranges of indoor concentrations of
selected polycyclic aromatic hydrocarbons
Compound Concentration
(ng/m3)
Acenaphthene 2.5-1650
Anthracene 1-410
Fluoranthene 5-270
Naphthalene 300-2300
Pyrene 3.6-32
Benzo[a]pyrene 0.04-370a
Phenanthrene 3-550
Chrysene 0.6-110
a Levels up to 14 700 ng/m3 found in Chinese
houses with open fires
Drinking-water generally contains low levels of individual PAH, up to
some hundreds of nanograms per litre, depending on the compound. The
levels of PAH in beverages, including alcoholic drinks, are usually
< 0.01 µg/kg.
10.1.1.2 Occupational exposure
The concentrations in air to which workers are exposed depend on the
type of industry. The levels in coke ovens, where the highest exposure
may occur, are up to several hundred micrograms per cubic metre. Table
108 shows some levels of occupational exposure.
10.1.2 Toxic effects
10.1.2.1 Bioavailability
Owing to the high lipophilicity of this class of compounds, their
bioavailability after ingestion and inhalation must be considered to
be significant. Dermal adsorption appears to depend on the PAH being
studied and the species evaluated: 3% of an applied dose of
benzo [a]pyrene was absorbed by human skin and 10% by mouse skin
within 24 h.
10.1.2.2 Acute toxicity
Values for the median lethal dose (LD50) indicate that PAH have
moderate to low acute toxicity. For example, the oral LD50 of
benzo [a]pyrene is > 1600 mg/kg in mice, and the oral LD50 of
naphthalene is 350-700 mg/kg bw in mice but 500-9000 mg/kg bw in rats.
Table 108. Ranges of occupational exposure to selected
polycyclic aromatic hydrocarbons
Compound Concentration (µg/m3)
Acenaphthene 0.44 in oil refining to 135 in aluminium
smelting
Anthracene 0.028 in oil refining to 405 in coke ovens
Fluoranthene 0.085 in oil refining to 191 in coke ovens
Naphthalene 0.22 in roofing to 2900 in food smoke-houses
Pyrene 0.11 in oil refining to 333 in coke ovens
Benzo[a]pyrene 0.09 in chimney sweeping to 137 in coke
ovens
Phenanthrene 0.085 in road paving to 1167 in coke ovens
Chrysenea 0.085 in oil refining to 191 in coke ovens
a Most measurements performed by gas chromatography, so that the
actual levels may be overestimates due to analytical
interference by triphenylene
Case reports have shown that exposure to naphthalene results in
haemolytic anaemia, and lethal doses of 2-3 g for children and 5-25 g
for adults have been reported. There appears to be no cause for
concern about any acute toxicity of occupational exposure or exposure
of the general population, with the exception of accidental ingestion.
10.1.2.3 Irritation and allergic sensitization
Anthracene, benzo [a]pyrene, and naphthalene are primary irritants.
Anthracene and benzo [a]pyrene were reported to be sensitizers,
whereas phenanthrene did not induce contact sensitivity.
10.1.2.4 Medium-term toxicity
Oral administration resulted in no-observed-adverse-effect levels of
175 mg/kg bw per day acanaphthene for hepatotoxicity; 125 mg/kg bw per
day fluoranthene for nephropathy, increased relative liver weights,
and haematological and clinical effects; 125 mg/kg bw per day fluorene
for altered haematological parameters; and 75 mg/kg bw per day pyrene
for nephropathy. Anthanthrene at 1000 mg/kg bw per day had no effect.
The daily uptake of PAH by humans is estimated to be 3.7 µg,
corresponding to about 0.05 µg/kg bw per day (70 kg bw). Human
exposure is thus six orders of magnitude lower than the concentrations
administered in studies in mice. There is thus no cause for concern
about any medium-term toxic effects in humans for the PAH tested so
far.
10.1.2.5 Carcinogenicity
The carcinogenicity of individual PAH and PAH-containing mixtures in
experimental animals has been well studied. Virtually no data exist on
the carcinogenicity of individual PAH in humans, although a limited
database on the carcinogenicity of PAH-containing mixtures is
available: these have been shown to increase the incidence of cancer
in exposed human populations. The finding that a number of individual
PAH are carcinogenic to experimental animals indicates that they are
potentially carcinogenic to humans. PAH can produce tumours both at
the site of contact and distantly, and the carcinogenic potency of PAH
may vary with the route of exposure.
(a) Experimental models
Benzo [a]pyrene is the best-studied PAH. It has been tested in
multiple species and by various routes, including orally, by
inhalation, and by skin painting for dermal carcinogenesis. It has
been shown to be carcinogenic by all routes tested in a number of
animal species. Those species in which no tumours were found are
suspected to have been tested at inadequate doses or observed for an
insufficient portion of their life span.
Other PAH have been assayed for dermal carcinogenicity as either
complete carcinogens or as initiators in initiation-promotion models.
Assays used commonly include tests for lung adenomas in newborn mice
treated by intraperitoneal, intrapulmonary, or subcutaneous injection.
There are insufficient data to determine whether other PAH are
carcinogenic.
(b) Epidemiology
Numerous epidemiological studies have been reported of groups of
workers exposed to environments that contain mixtures of PAH, all of
which also contained chemicals other than PAH. Cases of respiratory
diseases such as pneumoconiosis, respiratory tuberculosis, pneumonia,
and bronchitis and diseases of the circulatory system were reported in
these studies, but these effects are not considered to be specific to
PAH because of simultaneous exposure to agents that cause similar
effects. Thus, exposure in iron and steel foundries entails exposure
not only to PAH but also to other potentially carcinogenic materials
such as nickel, chromium, silica, soot, asbestos, and benzene. The
working environment of aluminium smelters is unlikely to include
nickel or chromium but includes alumina, aluminium fluoride, and
aromatic amines. If each of these materials occurred in a separate
location in a factory, classical epidemiological techniques would have
little difficulty in identifying the agent responsible for a
statistically significant cancer excess. This is not the situation,
and, while epidemiological studies have produced convincing evidence
that cancers occur in workers exposed to PAH, the attribution of
exposure to PAH as the cause of these excesses can only be based on
information from animal models.
Studies of workers exposed to mixtures of PAH indicate that the lung
is the target organ after inhalation. Confounding by cigarette smoking
cannot explain the effects observed. Studies of workers at gas and
coke ovens, at aluminium smelters, in iron and steel foundries, and
with bitumen and asphalt consistently show excess risks for lung
cancer. Coke-oven workers are probably exposed to the highest
concentrations, and studies of these populations have provided
evidence of dose-response effects. In a comparison of the mortality of
5321 coke-oven workers with that of 10 497 steel workers at the same
plants, a monotonic positive trend was shown in the relationship
between the risk for lung cancer and the estimated level of cumulative
exposure to coal-tar pitch volatiles, the benzene-soluble fraction of
particulate matter. With a risk of unity for the unexposed group, the
risk increased from 1.2 for those with exposure of 1-49 mg/m3 ×
months to 3.1 for the group with the heaviest exposure of > 650
mg/m3 × months. Analysis of the relative risks and the numbers of
deaths from lung cancer on which they were based resulted in the
conclusion that 124 deaths occurred among these coke-oven workers over
a period of 30 years that can be attributed to exposure to coal-tar
pitch volatiles, i.e. 2.3% of the cohort. Earlier findings from this
study were used by others to estimate a unit risk coefficient of 8.7 ×
10-2 for exposure to benzo [a]pyrene, i.e. the absolute lifetime
risk of lung cancer from a working lifetime exposure to 1 µg/m3 of
benzo [a]pyrene. Given the large number of cancers that occurred in
this cohort, this risk coefficient is probably the best estimate
currently available. It should be recognized, however, that the
reports on which this estimate is based gave relatively little
information on exposure levels, no data on time trends in the level of
exposure, and no data on benzo [a]pyrene levels in the participating
plants.
There is also good evidence that urinary bladder cancer has occurred
in cohorts of aluminium smelters and gas and coke workers, although
the overall findings are not as consistent as those for lung cancer.
In a study of aluminium smelters, a positive trend was observed
between the risk for urinary bladder cancer and the estimated level of
cumulative exposure to benzene-soluble matter. PAH cannot be assumed
to be responsible for this trend, however, because the known bladder
carcinogen 2-naphthylamine and other aromatic amino and nitro
compounds were present in the working environment. Similarly, the
excess of urinary bladder cancer in gas workers is more likely to be a
consequence of exposure to 2- and 1-naphthylamines.
PAH are almost certainly one of the carcinogenic agents responsible
for lung cancers in cigarette smokers, although the role of PAH in the
many other diseases caused by cigarette smoking, including
nonmalignant diseases of the respiratory system, is unknown. Studies
on the rates of mortality from lung cancer in relation to indoor
burning of coal or wood in open fires for cooking and heating add to
the body of evidence that links PAH and the risk for lung cancer.
Workers exposed to diesel or petrol fumes had relatively low exposure
to PAH, and studies of these exposure are not likely to assist in
quantification of the risks for cancer associated with exposure to
PAH.
Quantitative risk assessments were not made for PAH, either
individually or as mixtures; however, Appendix I gives some
comparative features of three approaches to quantitative risk
assessment that have been used and which have been at least partly
validated.
10.1.2.6 Reproductive toxicity
(a) Developmental studies
There are no studies in humans. Embryotoxic effects have been
described in experimental animals exposed to PAH such as
benz [a]anthracene, benzo [a]pyrene, and naphthalene. In mice
treated intraperitoneally with benzo [a]pyrene, increased numbers of
stillborn and resorbed fetuses, decreased fetal weight, and increased
incidences of congenital anomalies were seen at a minimum dose of 50
mg/kg bw per day. In mice given benzo [a]pyrene in the diet,
malformations were found after administration of 120 mg/kg bw per day
on days 2-10 of gestation. In another study in mice fed the compound
during most of the period of gestation, no treatment-related
embryotoxic effects were found after doses up to 133 mg/kg bw per day.
In mice, 1 mg benzo [a]pyrene per gram of food would result in
consumption of 5 g/day. In humans, the total median intake from food,
air, water, and soil for a 70-kg person would be 0.05 µg/kg bw per
day. In view of the inter- and intraspecies differences, there is at
present no reason for concern about effects on development.
(b) Fertility
There are no studies in humans. In mice fed diets containing
benzo [a]pyrene at doses up to 133 mg/kg bw per day, no
treatment-related effects on fertility were seen, again indicating no
concern about effects of PAH on fertility in humans.
10.1.2.7 Immunotoxicity
At least one immunotoxic PAH will probably be present in any mixture
of PAH. Thus, when such mixtures are evaluated for their potential
effects on human health, the immune system should be considered a
primary target organ. Benzo [a]pyrene caused immunosuppression in
mice after dermal application for 28 days at doses as low as 625 µg/kg
bw. In a study in which the immune status of coke-oven workers was
evaluated, suppressed immune status was found, as indicated by
decreased serum antibody levels and functional impairment.
The mechanisms of immunosuppressive action that have been proposed
include formation of active metabolites (including diol epoxides),
alterations in cytokine levels, disruption of membrane fluidity,
interaction with the Ah receptor, and alterations in calcium ion flux.
10.1.2.8 Genotoxicity
PAH have repeatedly been shown to have genotoxic effects both in
in vivo in rodents and in vitro in mammalian (including human)
cell lines and prokaryotes. Some PAH, however, appear not to be
genotoxic. Most of the unsubstituted PAH categorized as genotoxic are
not genotoxic per se but require metabolism to intermediates which
react with DNA to form DNA adducts and induce genotoxic damage.
Genotoxic events are postulated to be a required step in the
carcinogenic process.
10.2 Environment
10.2.1 Environmental levels and fate
Concentrations of up to 100 µg/kg of individual PAH have been detected
in soil, although higher concentrations of pyrene, phenanthrene,
chrysene, and benzo [a]pyrene have been found. The concentrations in
soil near industrial sources of PAH are up to three orders of
magnitude higher.
Concentrations of up to 6 µg/litre have been reported for individual
PAH in surface water, including polluted rivers. High concentrations
have also been reported in sediment, which acts as a sink for PAH. The
concentrations in sediment from rivers, lakes, and seas are generally
< 30 mg/kg dry weight. Concentrations up to 655 mg/kg dry weight have
been reported in sediment from harbours. The individual PAH compounds
detected in environmental samples vary according to their source and
any degradative processes. Phenanthrene is the PAH found in highest
concentrations in aquatic samples. Those that occur at the highest
concentrations in sediment include phenanthrene, fluoranthene, pyrene,
benz [a]anthracene, benzo [b]fluoranthene, benzo [k]fluo-ranthene,
and indeno[1,2,3- cd]pyrene.
PAH are sparingly soluble in water and therefore have affinity for
sediment, soil, and biota. They may be removed from the environment by
biodegradation or photodegradation. The rates of degradation vary and
generally decrease with increasing numbers of aromatic rings. PAH are
inherently biodegradable, and low-molecular-mass compounds can be
completely degraded by acclimated microorganisms. In surface waters
with low numbers of unacclimated microorganisms, PAH can persist for
longer periods of time.
10.2.2 Ecotoxic effects
10.2.2.1 Terrestrial organisms
Few data are available on the effects of PAH on terrestrial organisms,
and none are available on plants, wild mammals, or birds. The values
for the concentration causing 50% lethality (LC50) reported for
earthworm species are 150 mg/kg dry weight for phenanthrene and
170-210 mg/kg for fluorene. The no-observed-effect level for the
survival and reproduction of earthworm species was 180 mg/kg dry soil
for chrysene, benzo [k]fluoranthene, and benzo [a]pyrene. PAH in
soil are unlikely to exert toxic effects on terrestrial invertebrates,
except when the soil is highly contaminated.
10.2.2.2 Aquatic organisms
The toxicity of naphthalene, phenanthrene, and fluoranthene on aquatic
organisms has been well studied in the laboratory, but that of other
PAH has not. The toxicity of PAH to aquatic organisms is affected by
metabolism and photo-oxidation, and they are generally more toxic in
the presence of ultraviolet light. Naphthalene is the least toxic to
invertebrates, with 48-h LC50 values of 700-23 000 µg/litre. The LC50
values for three-ring PAH range from < 1 to 3000 µg/litre, anthracene
being more toxic than the others, with 24-h LC50 values of < 1 to
260 µg/litre. Four-, five-, and six-ring PAH have 48-h LC50 values of
0.2-1800 µg/litre, their toxicity increasing with molecular mass. The
96-h values for acute toxicity in fish were 110 to > 10 000 µg/litre
for naphthalene, 30-4000 µg/litre for three-ring PAH (those for
anthracene being 2.8-360 µg/litre), and 0.7-26 µg/litre for four- and
five-ring PAH.
The no-observed-effect and lowest-observed-effect levels for
invertebrates were 300-1000 µg/litre for naphthalene, 2-600 µg/litre
for three-ring PAH, 5-290 µg/litre for four-ring PAH, and 0.1-50
µg/litre for five-ring PAH. The long-term treatment doses resulting in
toxicity in fish were 15-1600 µg/litre for naphthalene, 1.2-510
µg/litre for three-ring PAH, 0.9-26 µg/litre for four-ring PAH, and
0.15-7 µg/litre for five-ring PAH. Higher no-observed-effect
concentrations have been reported, but in studies in which the PAH
were present at more than than 10 times their maximum aqueous
solubility; these have therefore been ignored. As the concentrations
of PAH reported in surface water are usually in the range of nanograms
per litre, they are unlikely to exert adverse effects on aquatic
organisms, except in cases of heavy exposure, as with creosote.
The LC50 values reported for sediment-dwelling organisms were 3.4-15
mg/kg dry sediment for fluoranthene, 10 mg/kg for phenanthrene, 10
mg/kg for benz [a]anthracene, and 5 mg/kg for benzo [a]pyrene. Since
sediment acts as a sink for PAH, sediment-dwelling organisms may be
adversely affected.
PAH may induce neoplastic effects in aquatic organisms. Tumour
development has been reported in fish exposed to benzo [a]pyrene and
3-methyl-cholanthrene after oral, dermal, or intraperitoneal
administration. Hepatic tumours have been found in wild fish living in
water with sediment containing PAH at a concentration of 250 mg/kg.
Although the levels of PAH in sediment are generally an order of
magnitude lower than this value, the possibility that tumours might be
formed at lower concentrations cannot be excluded. The ecological
significance of the carcinogenic effects of PAH in fish has not been
assessed.
11. RECOMMENDATIONS FOR THE PROTECTION OF HUMAN HEALTH AND THE ENVIRONMENT
11.1 General recommendations
* International agreement on analytical procedures and
interlaboratory quality control studies is strongly recommended.
Sampling strategies and analytical procedures should be optimized
and standardized before surveys of exposure to polycyclic
aromatic hydrocarbons (PAH) are undertaken.
* Emissions and effluents of PAH from both point and diffuse
sources should be monitored and inventories compiled.
* Concentrations of individual PAH should be given rather than
'total PAH'. When PAH are designated as 'not detected', the
relevant limits of detection should be given.
* PAH emissions and effluents should be reduced by:
- filtration and scrubbing of industrial emissions,
- treatment of effluents, and
- use of catalytic converters and particle traps on motor
vehicles.
11.2 Protection of human health
* Owing to their proven immunotoxic effects, coal-tar shampoos
should be used for anti-dandruff therapy only if no other
treatment is available.
* In view of the proven immunotoxic and carcinogenic effects of PAH
in coke-oven workers, exposure to PAH in occupational settings
should be eliminated or minimized by reducing emissions to the
extent possible or, when they cannot be sufficiently reduced, by
providing effective personal protection.
* Public education about the sources and health effects of exposure
to PAH should be improved.
* Use of unvented indoor fires, as in many developing countries,
should be discouraged, and they should be replaced by more
efficient, well-vented combustion devices.
* The risk of exposure to PAH from passive smoking should be
stressed and measures taken to avoid it.
* Urban air pollution should be monitored all year round and not
only seasonally.
11.3 Recommendations for further research
11.3.1 General
- Investigate the suitability of benzo [a]pyrene as an
indicator of the effects of PAH on human health and the
environment and examine the use of other PAH as surrogates.
11.3.2 Protection of human health
- More data should be collected on the human body burden of
PAH and on biomarkers for these compounds.
- The reproductive effects of PAH should be studied further.
- More studies on dermal absorption are required.
- The contribution of the high-molecular-mass PAH to the
overall carcinogenic potential of PAH should be studied.
11.3.3 Environmental protection
- The toxic effects of PAH to plants and earthworms should be
studied.
- The body burdens and possible toxic effects of PAH in wild
mammals and birds should be investigated, as most of these
species can metabolize PAH.
- The extent to which higher-molecular-mass PAH are absorbed
by sediments and serve as a sink and the effects of
disturbing sediment, e.g. by dredging, on aquatic organisms
should be investigated.
- The environmental significance of the tumours that have been
found in fish exposed to PAH must be addressed.
- Reliable data should be collected on environmentally
relevant PAH like dibenzo [a,l]pyrene, particularly with
regard to noncarcinogenic end-points such as effects on the
immune system.
11.3.4 Risk assessment
- Quantitative estimates of the risks presented by PAH should
be compared using various approaches and exposure scenarios,
both for human health and ecological protection.
- The risk estimates obtained by the various approaches to
risk assessment based on data on human exposure should be
compared.
- Risk assessment procedures that allow integrated assessment
of the risks due to inhalation and to oral and dermal
exposure should be developed and validated.
- Comparative risk assessment methods for evaluating
immunotoxicity and other noncancer risks associated with
exposure to PAH should be improved.
- The use of the results of alternative tests, such as those
for genotoxicity and other short-term effects, in assessing
risks due to PAH should be evaluated.
- Human exposure to alkylated PAH in a variety of situations
should be investigated further and data acquired on the
mutagenicity and experimental carcinogenicity of these
compounds.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
12.1 International Agency for Research on Cancer
Polycyclic aromatic hydrocarbons (PAH) have been evaluated by a number
of working groups convened by IARC. The evaluations made between 1973
and 1983 and summarized in Supplement 7 to the IARC Monographs
(IARC, 1987) are shown in Table 109.
12.2 WHO Water Quality Guidelines
PAH have been assessed in the WHO Guidelines for Drinking-water
Quality (WHO, 1984, 1996). A quantitative risk assessment was
conducted using the two-stage birth-death mutation model. The
resulting guidelines for benzo [a]pyrene in drinking-water
corresponding to excess lifetime risks for gastric cancer of 10-4,
10-5, and 10-6 are 7, 0.7, and 0.07 µg/litre.
The data are insufficient to derive guidelines for other PAH, but the
following recommendations are made for the group:
* Because of the close association between PAH and suspended
solids, treatment to achieve the recommended level of turbidity,
when necessary, will ensure that the PAH levels are reduced to a
minimum.
* Water should not be contaminated with PAH during water treatment
or distribution. The use of coal-tar-based and similar materials
for pipe linings and coatings on storage tanks should therefore
be discontinued. It is recognized that it may be impracticable to
remove coal-tar linings from existing pipes, and research is
needed on methods of minimizing the leaching of PAH from such
materials.
* In monitoring levels of PAH, the use of specific compounds as
indicators for the group as a whole is recommended. The choice of
indicator compound will vary in each situation. PAH should be
monitored regularly in order to determine background levels,
against which any changes can be assessed and remedial action
taken, if necessary.
* When drinking-water is known to have been contaminated by PAH,
the specific compounds present and the source of the
contamination should be identified, as the carcinogenic potential
of PAH varies.
12.3 FAO/WHO Joint Expert Committee on Food Additives
Benzo [a]pyrene was assessed at a meeting of this Committee in June
1990 (WHO, 1991). The Committee concluded that, for the purpose of
evaluation, the most significant toxicological effect of
benzo [a]pyrene is its carcinogenicity. It was recognized that
benzo [a]pyrene is only one member of a class of more than 100
compounds and that they should be considered as a class.
The Committee was unable to establish a tolerable intake for
benzo [a]pyrene on the basis of the available data. Nevertheless, the
large difference between the estimated human intake of
benzo [a]pyrene and the doses that induce tumours in animals suggests
that any effects on human health are likely to be small. Despite this,
the considerable uncertainties in risk estimation require that efforts
be made to minimize human exposure to benzo [a]pyrene as far as is
practicable.
The Committee acknowledged the complexity of reducing exposure to
benzo [a]pyrene and other PAH. Furthermore, it noted that exposure to
benzo [a]pyrene constitutes only a fraction of consumers' exposure to
these compounds and that some other members of this class, not
evaluated at the meeting, have toxicological properties similar to
those of benzo [a]pyrene and may thus contribute to the overall
carcinogenic risk. In this regard, strategies to minimize exposure to
benzo [a]pyrene would also be effective in reducing overall exposure
to PAH. These include practices that consumers can effect, such as
washing fruits and vegetables thoroughly to remove any surface
contamination, trimming excess fat prior to barbecuing meats to
minimize 'flare-ups', and cooking in a fashion that prevents contact
of food with flames. Measures that can be taken by the food industry
include use of indirect heating for drying foods, switching to
non-coal-fired roasters (e.g. for roasting coffee beans), using
protective coverings (e.g. cellulose casing) when smoking foods
conventionally, and ensuring compliance with the limits for PAH in
food additives specified by national and international bodies. The
Committee urged application of these measures in order to minimize
contamination of food with PAH, including benzo [a]pyrene.
12.4 WHO Regional Office for Europe Air Quality Guidelines
PAH have been assessed as atmospheric pollutants by the WHO Regional
Office for Europe (WHO, 1987). No guideline was set, but the group
concluded that no safe level of PAH could be recommended, owing to
their carcinogenicity. There is no known threshold for the induction
of cancer by benzo [a]pyrene, the most thoroughly studied PAH, nor is
there an ambient mixture of PAH that does not contain benzo [a]pyrene
and other substances for which there is sufficient evidence of
carcinogenicity in animals.
A number of estimates have been made of the risk presented by PAH,
based primarily on studies in which benzo [a]pyrene was used as the
index compound. The US Environmental Protection Agency (1984d)
proposed an upper-bound lifetime cancer risk of 62 per 100 000 exposed
people per microgram of benzene-soluble coke-oven emission per cubic
metre of ambient air. Assuming a 0.71% content of benzo [a]pyrene in
these emissions, it can be estimated that nine out of 100 000 people
exposed to 1 ng/m3 benzo [a]pyrene over a lifetime would be at risk
of developing cancer.
Table 109. Degree of evidence for carcinogenicity in humans and in experimental animals
and overall evaluations of carcinogenicity to humans for agents evaluated in IARC
Monographs
Compound Degree of evidence Overall IARC Monographs
for carcinogenicity evaluationa volume (year)
Human Animal
Anthanthrene ND L 3 32(1983)
Anthracene ND I 3 32(1983)
Benz[a]anthracene ND S 2A 3 (1973); 32 (1983)
Benzo[b]fluoranthene ND S 2B 3 (1973); 32 (1983)
Benzo[j]fluoranthene ND S 2B 3 (1973); 32 (1983)
Benzo[k]fluoranthene ND S 2B 32(1983)
Benzo[ghi]fluoranthene ND I 3 32(1983)
Benzo[a]fluorene ND I 3 32(1983)
Benzo[b]fluorene ND I 3 32(1983)
Benzo[ghi]perylene ND I 3 32(1983)
Benzo[c]phenanthrene ND I 3 32(1983)
Benzo[a]pyrene ND S 2A 3 (1973); 32 (1983)
Benzo[e]pyrene ND I 3 3 (1973); 32 (1983)
Chrysene ND L 3 3 (1973); 32 (1983)
Coronene ND I 3 32(1983)
Cyclopenta[cd]pyrene ND L 3 32(1983)
Dibenz[a,h]anthracene ND S 2A 3 (1973); 32 (1983)
Dibenzo[a,e]pyrene ND S 2B 3 (1973); 32 (1983)
Dibenzo[a,h]pyrene ND S 2B 3 (1973); 32 (1983)
Dibenzo[a,i]pyrene ND S 2B 3 (1973); 32 (1983)
Dibenzo[a,l]pyrene ND S 2B 3 (1973); 32 (1983)
Fluoranthene ND I 3 32(1983)
Fluorene ND I 3 32(1983)
Indeno[1,2,3-cd]pyrene ND S 2B 3 (1973); 32 (1983)
5-Methylchrysene ND S 2B 32(1983)
1-Methylphenanthrene ND I 3 32(1983)
Perylene ND I 3 32(1983)
Phenanthrene ND I 3 32(1983)
Pyrene ND I 3 32(1983)
Triphenylene ND I 3 32(1983)
Adapted from IARC (1987)
ND, no adequate data; I, inadequate evidence; L, limited evidence; S, sufficient
evidence
a Group 1, the compound is carcinogenic to human; Group 2A, the compound is
probably carcinogenic to humans; Group 2B, the compound is possibly carcinogenic
to humans; Group 3, the compound is not classifiable as to its carcinogenicity
to humans
APPENDIX I
SOME APPROACHES TO RISK ASSESSMENT FOR POLYCYCLIC AROMATIC
HYDROCARBONS
I.1 Introduction
Various environmental and technical problems hinder assessment of the
risk posed by mixtures containing polycyclic aromatic hydrocarbons
(PAH), particularly for carcinogenicity. Ambient environments contain
a wide range of PAH, some of which are highly carcinogenic, while
others are probably not (Table AI.1). Both anthropogenic and natural
sources may contribute to the ambient levels of these compounds, such
as in the air over industrial towns (see Section 3).
As PAH undergo transformation in the environment (see Section 4), it
cannot be taken for granted that the composition of ambient mixtures
is similar to that of the source. No data are available to assess the
potency of individual PAH in humans, but the carcinogenicity of
several mixtures containing PAH has been estimated in epidemiological
studies (Albert et al., 1983). Mixtures of a similar type, such as
from coke ovens, may not always present the same risk, however, even
when the fuel used and the operating conditions are similar.
Furthermore, PAH are often not the only contributors to the
carcinogenic risk presented by a given mixture: other chemicals and
particulate matter (Heinrich, 1995) may also contribute. The basis for
quantification and the way in which the quantity of a mixture is
expressed are also important. Should the levels be expressed in terms
of the total mass of extractable material or as a surrogate? If the
concentration of a chosen surrogate is used as an indicator of the
quantity of the mixture and its toxicity, the surrogate selected must
be predictive of the toxicity of the mixture.
Estimating the risk conferred by exposure to PAH has further problems.
As humans are exposed to mixtures of PAH and other compounds and not
to pure PAH, experimental data must be used to estimate the risk for
exposure to individual PAH and the result extrapolated to the low
doses to which humans are exposed. Such extrapolation is problematic,
because species may differ in the enzymes that activate PAH (Michel et
al., 1995) and in their susceptibility to the tumorigenic effects of
PAH; these differences may otherwise be a simple reflection of
differences in weight, surface area, basal metabolic rate, or
respiratory volume. The degree to which species differences affect
extrapolated human risks is unknown.
Another set of problems stems from the large number of PAH that are
typically found in a complex mixture. Only a small percentage of the
environmentally relevant PAH has been investigated for carcinogenicity
in experimental animals, and the toxicity of most of the PAH in
complex mixtures remains unknown. About a dozen PAH of known toxicity
are monitored in most programmes, and these contribute only a small
proportion of the risk represented by PAH fractions extracted from
complex mixtures (Thorslund & Farrar, 1990a). It is therefore likely
Table AI.1. Evaluations of the carcinogenicity of some polycyclic
aromatic hydrocarbons
Compound IARC US Environmental Task Groupb
(1987)a Protection Agency
(1993)a
Acenaphthene D Questionable
Acenaphthylene Dc
Anthanthrene Positive
Anthracene 3 Dc Negative
Benz[a]anthracene 2A B2c Positive
Benzo[b]fluoranthene 2B B2c Positive
Benzo[j]fluoranthene 2B B2 Positive
Benzo[ghi]fluoranthene (Negative)
Benzo[k]fluoranthene 2B B2c Positive
Benzo[a]fluorene Questionable
Benzo[b]fluorene Questionable
Benzo[ghi]perylene 3 Dc Negative
Benzo[c]phenanthrene (Positive)
Benzo[a]pyrene 2A B2c Positive
Benzo[e]pyrene 3 C Questionable
Chrysene 3 B2c Positive
Coronene Questionable
Cyclopenta[cd]pyrene B2 Positive
Dibenz[a,h]anthracene B2c Positive
Dibenzo[a,e]pyrene B2 Positive
Dibenzo[a,h]pyrene B2 Positive
Dibenzo[a,i]pyrene B2 Positive
Dibenzo[a,l]pyrene B2 Positive
Dibenzo[e,l]pyrene D
Dibenzo[a,e]fluoranthene B2
Dibenzo[a,h]fluoranthene B2
Dibenzo[a,i]fluoranthene B2
Dibenzo[a,l]fluoranthene B2
Fluoranthene 3 Dc (Positive)
Fluorene 3 Dc Negative
Indeno[1,2,3-cd]pyrene B2c Positive
5-Methylchrysene Positive
1-Methophenanthrene (Negative)
Naphthalene Dc/C Questionable
Perylene (Negative)
Phenanthrene 3 Dc Questionable
Pyrene Dc Questionable
Triphenylene (Negative)
a Based on the results of studies in humans and experimental animals
b Based on the results of studies in experimental animals
c Consensus position of the US Environmental Protection Agency;
others have been presented at scientific meetings (Schoeny et al.,
1994; McClure & Schoeny, 1995).
that summing the risks posed by individual PAH of known toxicity does
not accurately reflect the contribution of all the PAH in a mixture.
I.2 Approaches to risk assessment
Three of the most popular approaches for assessing dose-response
relationships for PAH are presented, with their strengths and
weaknesses. These approaches are toxicity equivalence factors,
comparative potency, and use of benzo [a]pyrene as a surrogate.
I.2.1 Toxicity equivalence factors and related approaches
Several approaches to quantification may be considered for assessing
the risks posed by mixtures of agents. When there are sufficient data,
they can be used as a basis for quantitative estimates of risk.
Relatively few mixtures containing PAH have been tested under
conditions that are acceptable for risk assessment. For some
processes, e.g. coal coking, the data on human exposure are
sufficiently complete to allow quantitative estimates of risk;
however, changes in the parameters of the combustion process, such as
temperature and amount of oxygen feedstock, may result in variations
in the types, amounts, and physical status of PAH in the mixture.
These variations may be sufficient to alter the risk posed by the
mixture. One way of resolving the uncertainty inherent in differences
in the composition of mixtures is to base quantitative estimates on
considerations of individual components; this alternative is explored
below.
I.2.1.1 Principle
The approaches are based on an assumption of additive risk, which
leads, in principle, to an estimate of the risk associated with
identified PAH. In practice, the risks attributable to individual PAH
are summed, or the risk posed by individual PAH is expressed relative
to that for benzo [a]pyrene, and then the levels of these equivalents
are summed. The latter process is the toxicity equivalence factor
approach.
The first step is to estimate the potency of a single PAH, which
serves as a standard against which the potency of other compounds is
later derived. In practice, this compound is usually benzo [a]pyrene.
Since no data from studies in humans are available that are suitable
for assessing the potency of individual PAH, the potency of
benzo [a]pyrene in humans is estimated from the results of studies in
animal models. The uncertainty associated with this extrapolation is
discussed above.
The second step is to estimate the potency of the PAH relative to that
of benzo [a]pyrene, in order to obtain a benzo [a]pyrene equivalent.
The estimate is based on the relative potencies of benzo [a]pyrene
and other PAH in experimental animals. The key assumption is that the
relative potency of two PAH in an animal model is the same or similar
to that of the same compounds in humans. This comparative potency
approach has been used in relation to chlorinated
dibenzo- para-dioxins and dibenzofurans (US Environmental Protection
Agency, 1987) and for PAH and PAH-rich mixtures (Albert et al., 1983;
Clement Associates, 1988). Further evidence supports the assumption
made in this approach (Albert et al., 1983; Lewtas, 1985a,b; Nesnow,
1990).
The third step involves summation of risks, which can be done either
by summing the benzo [a]pyrene equivalents and multiplying by the
potency of benzo [a]pyrene or by estimating the potency of each PAH
in humans (risk for cancer) and then adding them. The underlying
assumption is that the individual estimates of risk are additive.
Although there may be interactions between PAH, the risks appear to be
approximately additive, especially at low, environmentally relevant
doses (Krewski et al., 1989; Nesnow et al., 1995). In the absence of
information to the contrary, additivity is assumed.
The toxicity equivalence factor approach is feasible if at least two
pieces of information are available:
- the amount of each PAH in the mixture, or the amount of each
'major bioactive PAH', and
- a quantitative estimate of the risk associated with each
identified PAH.
For the first requirement, good data on the composition of mixtures of
PAH can be generated with existing analytical techniques (see Section
2), although such analyses can be resource-intensive. Quantitative
risk estimates for individual PAH are generally not available, as the
data are insufficient. The deficiencies include the following:
- the data relate to exposures that are not typically used in
deriving quantitative estimates of risk after oral or inhalation
exposure,
- the study populations were inappropriately small,
- the studies involved only one dose,
- dose-response relationships were not reported, and/or
- different PAH were tested in different studies with different
designs.
Environmental mixtures contain many more PAH than can be monitored
feasibly or practically. Furthermore, the toxicity of most
environmentally relevant PAH has not yet been quantified. As the
toxicity equivalence factor approach assumes additivity of the risks
posed by PAH in a mixture, the question remains of the extent to which
the risk of one PAH is representative of the risk of all of those in
the mixture.
I.2.1.2 Development and validation
I.2.1.2.1 Derivation of the potency of benzo [a]pyrene
Quantitative risk estimation is problematic for even the best-studied
PAH, namely benzo [a]pyrene. The risk for carcinogenicity is best
estimated on the basis of data from long-term, usually lifetime,
assays in which several doses are tested in large groups of animals.
The data on benzo [a]pyrene are less than optimal: few lifetime
assays have been conducted with exposure other than dermal and
generally few doses were tested.
Krewski et al. (1989) calculated the probability of occurrence of a
tumour at a specified time after continuous exposure to a mixture of
PAH. The dose of the PAH mixture was described as a benzo [a]pyrene
equivalent dose d as follows:
d = S Ridi + do (1)
where Ri is the relative carcinogenicity of the ith PAH in
comparison with that of benzo [a]pyrene, di is the dose of the
ith PAH, and do is the dose of benzo [a]pyrene.
The tumour probability, P(d), was calculated from a two-stage birth-
death mutation model. For the mixture as a whole, P(d) is given by
the equation:
P (d) = 1-exp (- A( 1 + bd)2) (2)
where the unknown values A and b were estimated from bioassays
with benzo [a]pyrene as A = 0.00616 and b = 3.52 µg-1 for
lifetime exposure.
The US Environmental Protection Agency (1993) made a quantitative risk
estimate for oral exposure to benzo [a]pyrene that consisted of a
range of values, from 4.5 to 11.7 per (mg/kg)/day, with a geometric
mean of 7.3 per (mg/kg)/day. These estimates were obtained by using
three methods of determining an upper bound on a linear low-dose term
from data on the incidence of gastrointestinal tumours in mice exposed
to benzo [a]pyrene in the diet (Neal & Rigdon, 1967). The models used
were a form of the two-stage Moolgavkar, Venzon, and Knudson model
(Moolgavkar & Venzon, 1979; Moolgavkar & Knudson, 1981) and a
Weibull-type model (Rees & Hattis, 1994). The total numbers of tumours
in male and female rats exposed in the diet (Brune et al., 1981) were
used in a linearized multistage procedure to derive an upper bound on
the low-dose term (slope factor), shown in Table AI.2.
The potency of benzo [a]pyrene in humans exposed by inhalation has
been assessed on the basis of extrapolations from the results for
rodents, sometimes exposed other than by respiration. Since the
sensitivity to PAH is likely to differ with the route of exposure,
assessments made on the basis of exposure by inhalation are
preferable; some of these are shown in Table AI.3.
Table AI.2. Slope factors for humans based on the results of
studies in which benzo[a]pyrene was fed to rodents in the diet
Study Slope factor Comments
(per [mg/kg]/day)
Neal & Rigdon 5.9 Two-stage, conditional upper bound
(1967) 9.0 Two-stage, slope from 10% response
4.5 Weilbull-type model
Brune at al. 11.7 Linearized multistage procedure
(1981) applied to oesophageal, laryngeal,
and forestomach tumours in males
and females
From US Environmental Protection Agency (1992b, 1993)
The commonest method of extrapolation is based on the relative surface
areas or body weights of experimental animals and humans, assuming
that these are good predictors of the relative potency of PAH in two
species. While such scaling factors have been validated for other
compounds (Chappell, 1989) and have been used in the case of PAH
(Thorslund & Farrar, 1990b; Collins et al., 1991; Collins & Alexeeff,
1993), this relationship may not hold for PAH. Muller et al. (1995a,b,
1996) compared the actual and extrapolated doses required to produce a
tumorigenic effect of a given magnitude in a particular rodent
species, on the basis of the assumption that if extrapolations based
on surface area and/or body weight hold between rodents to humans, the
same extrapolation should hold even more closely for closely related
rodents. The extrapolated dose was estimated from that required to
induce a similar response in another rodent species under matching
experimental conditions and by extrapolating to the target rodent
species. Examples of the analysis are shown in Tables AI.4 and AI.5.
The extrapolated and actual doses differed by much as two orders of
magnitude, even though mice and rats are closely related, of similar
sizes, and with similar diets, and, furthermore, laboratory rodents
are placed in similar habitats. It would be expected that
extrapolation from one rodent to another is more justified than
extrapolation from rodents to humans, but these analyses indicate that
extrapolation from rodents to humans may lead to even larger errors.
Although some of the discrepancies may be due to methodological
problems, much of the difference is due to the low predictive value of
the two extrapolations. Extrapolations based on surface area or body
weight further imply that species differences in the metabolism of the
parent compound to the primary carcinogen are functions of body weight
or surface area. This assumption is not supported by the available
pharmacokinetic data for PAH (Michel et al., 1995).
Table AI.3. Estimated carcinogenic potency of benzo[a]pyrene in humans exposed by inhalation, on the basis of
extrapolations from the results of studies in experimental animals
Risk Species Route of exposure Assumptions Data source UCL/ Assessor
(ng/m3) MLE
1.7 × 10-6a Hamster Inhalation on salt Risk proportional to Thyssen et UCL US Environmental
particulate (body weight)2/3 and al. (1981) Protection Agency
inhalation rate = 0.037 m3/d (1984a); Collins &
Alexeeff (1993)
1.1 × 10-6 Hamster Inhalation on salt Risk proportional to Thyssen et UCL Collins & Alexeeff
particulate (body welght)2/3 and al. (1991) (1993)
inhalation rate = 0.063 m3/d
4.7 × 10-6 Hamster Intratracheal Risk proportional to Saffiotti at UCL Collins & Alexeeff
instillation with (body weight)2/3 al. (1972) (1993)
ferric oxide
4.4 × 10-6 Hamster Intratracheal Risk proportional to Feron et al. UCL Collins & Alexeeff
instillation with (body weight)2/3 (1973) (1993)
ferric oxide
2.0 × 10-8 Rat Inhalation of Risk equal to that of humans Heinrich at UCL Heinrich et al.
coal-tar/pitch and hamsters at same air al. (1994c) (1994c)
condensation level of benzo[a]pyrene,
aerosolb corrected for rat lifetime
(2 years)
7.0 × 10-9 Hamster Risk proprotional to body Thyssen at MLE Clement Associates
weight al. (1981) (1990)
3.6 × 10-8 Hamster Risk proprotional to body Thyssen et MLE Clement Associates
weight3/4 al. (1981) (1990)
6.2 × 10-8 Hamster Risk proprotional to body Thyssen et MLE Clement Associates
weight2/3 al. (1981) (1990)
Table AI.3. (continued)
Risk Species Route of exposure Assumptions Data source UCL/ Assessor
(ng/m3) MLE
3.9 × 10-8 Hamster Risk equal to that of humans Thyssen et MLE Clement Associates
and hamsters at same air al. (1981) (1990)
level of benzo[a]pyrene
UCL, upper confidence limit; MLE, maximum likelihood estimate
a Recalculated from 6.11 mg/kg bw per day using the assumptions of the US Environmental Protection Agency for humans:
70 kg, 20 m3/d
b Other active polycyclic aromatic hydrocarbons present in the administered aerosol
Table AI.4. Comparison of actual and extrapolated doses of 3-methylcholanthrene and benzo[a]pyrene required to obtain observed
tumour incidence; topical administration
Species Compound Actual dose (mg) Extrapolated dose (mg) Extrapolated/ Reference
actual dose
Surface Body Surface Body
area weight area weight
Rat 3-Methylcholanthrene 58 9.0 20 0.16 0.35 Cavalieri et al.
(1978)a; Zackheim
(1964)
Hamster 3-Methylcholanthrene 5.0b 0.048 0.074 0.0096 0.015 Cavalieri et al.
(1978)a; Bernfeld &
Homburger (1983)
Hamster Benzo[a]pyrene 5.0b 0.019 0.028 0.0038 0.056 Cavalieri et al.
(1978)a; Bernfeld &
Homburger (1983)
a Data for mouse
b No tumours induced at dose tested; assumed 1% response rate
Table AI.5. Comparison of actual and extrapolated doses of 3-methylcholanthrene and benzo[a]pyrene required to obtain observed tumour
incidence; respiratory exposure
Species Compound Actual dose (mg) Extrapolated dose (mg) Extrapolated/ Reference
actual dose
Surface Body Surface Body
area weight area weight
Rat 3-Methylcholanthrene 0.50a 7.0a 15a 14a 30a Nettesheim & Hammons
(1971)b; Hirano et al.
(1974)
Hamster 3-Methylcholanthrene 0.7a 4.7a 7.1 6.7a 10a Nettesheim & Hammons
(1971)b; Hammond et al.
(1987)
Rat Benzo[a]pyrene 15 31 33 2.1 2.2 Furst et al. (1979)b;
Ishinishi et al. (1976)
Hamster Benzo[a]pyrene 30 33 222 1.1 0.73 Furst et al. (1979)b;
Saffiotti et al. (1972)
a Nettesheim & Hammons (1971) used intratracheal instillation, while Hirano et al. (1974) used intrapulmonary pellets to
administer 3-methylcholanthrene to rats. Hammond et al. (1987) applied intrabronchial pellets to hamsters. The fact that
instillation is considered to be less effective in inducing tumours than implantation is considered to be the likely
explanation for the apparent discrepancy between the actual and the extrapolated doses.
b Data for mouse
Some risk assessments are based on extrapolation of the results of
studies in which the compound was deposited directly onto or in close
proximity to the tissues where tumours were later observed. Since the
dose delivered to these tissues is direct and not a reflection of body
weight or surface, it may well be argued that extrapolations based on
body weight or surface area are not appropriate.
It is likely, therefore, that the estimates of the potency of PAH in
humans based on such extrapolations would lead to substantial errors.
It is therefore preferable to use other approaches, like the relative
potency approach, in order to estimate human risk.
I.2.1.2.2 Derivation of relative potencies of PAH other than
benzo [a]pyrene
Some quantitative risk estimates for mixtures of PAH are based on the
assumption that all PAH (or all carcinogenic PAH) have the same
potency as benzo [a]pyrene and that the carcinogenic effect of the
mixture can be estimated by summing the effects of each PAH. Some PAH
are less carcinogenic in animal models than benzo [a]pyrene, and a
few are more active. In order to provide more reasonable estimates of
the carcinogenicity of PAH mixtures, schemes have been devised that
are similar to the toxicity equivalence factor approaches of the US
Environmental Protection Agency and NATO for evaluating chlorinated
dibenzodioxins and dibenzofurans, which are based on a general or
specific hypothesis of relative potency.
In a provisional quantitative risk assessment of PAH, the US
Environmental Protection Agency (1993) used benzo [a]pyrene as a
standard and derived the relative potencies of individual PAH in
increments of order of magnitude by comparison. Only the results of
carcinogenicity bioassays were considered, and these were limited to
those in which benzo [a]pyrene and other PAH were assayed by the same
protocol and within the same time frame. The studies involved various
routes of exposure, including skin painting, intraperitoneal and
subcutaneous injection, and lung implantation (see section 7.7.2).
Maximum likelihood estimates from a two-stage model were used for
comparison, and ranges of estimates were presented. These values for
the results of complete carcinogenesis assays in mouse skin are shown
in Table AI.6. The US Environmental Protection Agency (1993)
considered that the data on PAH did not meet all of the requirements
for application of the toxicity equivalence factor approach and
recommended that the values be applied only to carcinogenicity and not
to other end-points. In order to differentiate between these values
and a toxicity equivalence factor meant for use in evaluating all
types of toxicity, the relative potencies were designated 'estimated
orders of potential potency'. The US Environmental Protection Agency
(1993) further recommended that these values not be used for
evaluating inhaled PAH mixtures, for the following reasons:
- The US Environmental Protection Agency currently has no consensus
value for an inhalation unit risk.
Table AI.6. Estimated orders of potency of selected polycyclic aromatic
hydrocarbons in mouse skin carcinogenesis
Compound Relative potencya Reference
Benzo[a]pyrene 1.0 1.0
Benz[a]anthracene 0.145 0.1 Bingham & Falk (1969)
Benzo[b]fluoranthene 0.167 0.1 Habs et al. (1980)
Benzo[k]fluoranthene 0.020 0.01 Habs et al. (1980)
Chrysene 0.004 0.001 Wynder & Hoffmann
(1959)
Dibenz[a,h]anthracene 1.11 1.0 Wynder & Hoffmann
(1959)
Indeno[1,2,3-cd]pyrene 0.055b 0.1 Habs et al. (1980);
Hoffmann & Wynder (1966)
a Model was P(d) = 1-exp[-A(1 + bd)2] for all except
indeno[1,2,3-cd]-pyrene. Actual figures (left) and rounded to
order of 10 (right)
b Simple mean of relative potencies (0.021 and 0.089), the latter
being derived with the one-hit model
- There is no basis for assuming that the relative order of potency
for PAH is the same after oral and inhalation exposure.
- The co-carcinogenic potential of particulate carriers in the lung
has not been sufficiently elucidated.
Improvements to the estimated orders of potential potency have been
published (McClure & Schoeny, 1995). As the two-stage model requires
estimation of several parameters in the absence of data, other models
were investigated. It was shown that the model used had little effect
on the values when applied consistently; however, the assay type and
data set used could alter the values by orders of magnitude. It was
proposed, therefore, that data from all available appropriate assays
should be modelled and that a central tendency estimate, rounded to
powers of 10, would give a more realistic value. The list of PAH for
which estimated orders of potential potency were estimated was
expanded from that of US Environmental Protection Agency (1993) to
include several more that could be considered probable human
carcinogens (Table AI.7).
In a preliminary validation exercise based on published data on
PAH-containing mixtures, McClure & Schoeny (1995) used the values in
Table AI.7 to estimate the carcinogenicity of two synthetic mixtures
of PAH (Pfeiffer, 1973), both as a sum of their components and as
whole mixtures. A good correlation was found between the two
measurements of potency. These results indicate that the components of
a defined mixture have additive risks.
Table AI.7. Estimated order of carcinogenic potency for 13
polycyclic aromatic hydrocarbons in Group B2 as compared with
benzo[a]pyrene
Compound Estimated order No. of estimates
of potency
Benzo[a]pyrene 1.0
Benz[a]anthracene 0.1 4
Benzo[b]fluoranthene 0.1 8
Benzo[j]fluoranthene 0.1 7
Benzo[k]fluoranthene 0.1 7
Chrysene 0.1 5
Cyclopenta[cd]pyrene 0.1 4
Dibenz[a,h]anthracene 1.0 3
Dibenzo[a,e]fluoranthenea 1.0 3
Dibenzo[a,e]pyrene 1.0 3
Dibenzo[a,h]pyrene 1.0 2
Dibenzo[a,i]pyrene 0.1 3
Dibenzo[a,l]pyrene 100 2
Indeno[1,2,3-cd]pyrene 0.1 4
Adapted from McClure & Schoeny (1995)
a Not evaluated by the Task Group
The additivity of the potency of mixtures of five PAH was investigated
in the mouse lung adenoma model (Nesnow et al., 1995). Different
combinations of PAH with different biological and chemical properties
were tested. Some interaction was found in which the potency of the
compounds was more or less than additive. The differences were no more
than twofold and, therefore, would not be large enough to alter
significantly the outcome of risk assessments, in which uncertainty of
an order of magnitude is not seen as excessive.
Complex environmental mixtures differ from defined synthetic mixtures
in that they contain not only PAH of known carcinogenicity but also
hundreds of PAH and other potentially carcinogenic non-PAH compounds
for which carcinogenicity has not been established. The risk
attributed to a PAH for which data on exposure and carcinogenic
potency exist would be similar to that for the entire mixture only if
the PAH for which the potency is unknown contributed little or nothing
to the potency of the mixture as a whole. McClure & Schoeny (1995)
reported that the carcinogenic activity of a coal liquefaction
material (Mahlum et al., 1984) was similar to that estimated by adding
benzo [a]pyrene equivalents derived from estimated orders of
potential potency for several measured PAH. Similar estimates for coal
flue gas, petrol-engine exhaust, and diesel exhaust, however, resulted
in underestimates of two to three orders of magnitude of the risks
presented by the PAH fractions when the results of studies by either
lung implantation or dermal application in rodents were used for the
calculation (Grimmer et al., 1984; Thorslund & Farrar, 1990a; see
Table AI.8).
Other strategies for risk assessment based on the toxicity equivalence
factor approach for individual PAH have been published. The resulting
estimates are compared in Table AI.9. Krewski et al. (1989) analysed
the published values and derived a new set based largely on estimates
from a two-stage model (Clement Associates, 1988). Applications of
their toxicity equivalence factors to the results of bioassays wth PAH
mixtures (Pfeiffer, 1977; Schmähl et al., 1977) indicated that their
values would be unlikely to underestimate the carcinogenic risk posed
by whole mixtures.
A set of toxicity equivalence factors was derived for the 17 PAH
commonly measured at hazardous waste sites, and a new list (see column
2 of Table AI.9) was calculated on the basis of older work and the
primary literature (Nisbet & LaGoy, 1992). The values tend to
overestimate the carcinogenic risks of mixtures.
The relative potency values in 14 publications were used to classify
PAH into categories of high, medium, low, and very low risk (column 3
of Table AI.9), and 'environmental assessment levels' were calculated
on the basis of the highest potency for a PAH, rounded to an order of
magnitude, relative to that of benzo [a]pyrene (Malcom & Dobson,
1994).
Kalberlah et al. (1995) and others adopted the approach of the US
Environmental Protection Agency Office of Pesticides, Pollution
Prevention and Toxic Substances to determine the relative potencies of
PAH. A panel of five experts made independent reviews of the existing
data on about 150 PAH and scored them as having high, moderate,
marginal, or slight potential carcinogenicity. The panel considered
data from studies of skin painting in mice, the induction of lung and
liver adenomas in newborn mice, mammary tumours in rats, studies by
oral administration, studies of genotoxicity, and structure-activity
relationships. Their evaluation, converted to powers of 10 to
represent levels of concern, is presented in column 4 of Table AI.9.
Columns 5 and 6 of Table AI.9 show the values of the US Environmental
Protection Agency (1993) and McClure & Schoeny (1995), discussed
previously. The results of the six toxicity equivalence factor
approaches show a reasonable degree of agreement for PAH that are
generally considered to be carcinogenic. In all of them,
dibenz [a,h]anthracene appears to be equipotent or somewhat more
potent than benzo [a]pyrene; and, in most, the benzofluoranthenes and
benzanthracene were about 10% as potent as benzo [a]pyrene. The
greatest variation in the estimated relative potency is observed for
chrysene, although all agree that chrysene is not as potent a
carcinogen as benzo [a]pyrene.
Table AI.8. Comparison of the carcinogenic potency of the polycyclic aromatic
hydrocarbon (PAH) fraction of PAH-rich mixtures with the integrated potency
of the eight PAH found in the fraction
Method of estimating risk Potency relative to that of benzo[a]pyrene
Flue gas Diesel-engine Petrol-engine
exhaust exhaust
Fraction of mixture containing 0.38 0.28 0.67
PAH with > three rings
Sum of risk for eight PAH 0.0011 0.0018 0.0007
Fraction/sum 340 150 950
From Thorslund & Farrar (1990a)
Table AI.9. Relative potencies of indicator polycyclic aromatic hydrocarbons
Compound [1] [2] [3] [4] [5] [6] [7]
1-Methylphenanthrene 0.001
Acenaphthene 0.001 0.001 0.001 0
Acenaphthylene 0.001 0.001 0.01
Anthanthrene 0.320 0.28
Anthracene 0.01 0.01 0.01
Benz[a]anthracene 0.145 0.1 0.1 0.1 0.1 0.1 0.014
Benzo[a]pyrene 1.0 1.0 1.0 1.0 1.0 1.0 1.0
Benzo[b]fluoranthene 0.141 0.1 0.1 0.1 0.1 0.1 0.11
Benzo[e]pyrene 0.004 0.01 0
Benzo[ghi]perylene 0.022 0.01 0.01 0.01 0.012
Benzo[j]fluoranthene 0.1 0.1 0.045
Benzo[k]fluoranthene 0.061 0.1 0.1 0.1 0.01 0.1 0.037
Chrysene 0.0044 0.01 0.01 0.01 0.001 0.1 0.026
Coronene 0.001
Cyclopenta[cd]pyrene 0.023 0.1 0.1 0.012
Dibenzo[a,e]pyrene 1.0
Dibenz[a,c]anthracenea 0.1
Dibenz[a,h]anthracene 1.11 5 1.0 1.0 1.0 1.0 0.89
Dibenzo[a,l]pyrene 100 100
Dibenzo[a,e]fluoranthenea 1.0
Dibenzo[a,h]pyrene 1.0 1.2
Dibenzo[a,i]pyrene 0.1
Fluoranthene 0.001 0.001 0.01
Fluorene 0.001 0.001 0
Indeno[1,2,3-cd]pyrene 0.232 0.1 0.1 0.1 0.1 0.1 0.067
Naphthalene 0.001 0.001
Perylene 0.001
Phenanthrene 0.001 0.001 0 0.00064
Pyrene 0.81 0.001 0.001 0.001 0
Table AI.9 (continued)
[1] Krewski et al. (1989);
[2] Nisbet& LaGoy (1992);
[3] Malcolm & Dobson (1994);
[4] Kalberlah et al. (1995);
[5] US Environmental Protection Agency (1993);
[6] McClure & Schoeny (1995);
[7] Muller et al. (1995a,b, 1996)
a Not evaluated by the Task Group
I.2.1.3 Application
Application of the toxicity equivalence factor approach to assessing
the risk posed by dibenzodioxins and dibenzofurans involves the
following steps:
(1) analytical determination of the agents in the environmental
sample;
(2) multiplication of the concentrations of congeners in the sample
by the toxicity equivalence factors to express the concentration
in terms of the standard agent (e.g. benzo [a]pyrene)
equivalents;
(3) summation of the products in step (2) to obtain the equivalents
of the standard agent in the sample;
(4) determination of human exposure to the mixture in question,
expressed in terms of standard chemical equivalents; and
(5) combination of the exposure derived in step 4 with information on
the toxicity (here, carcinogenic potency) of the standard
chemical in order to estimate the risks associated with exposure
to the mixture.
These steps were followed for PAH, using one or more of the toxicity
equivalence factors given in Table AI.9 and benzo [a]pyrene as the
standard.
I.2.2 Comparative potency approach
I.2.2.1 Principle
The comparative potency approach is used to estimate the potency of
the PAH in mixtures without having to identify or quantify the
individual compounds. The carcinogenic potency of an unknown mixture
in humans is estimated from the potency of the mixture in a bioassay
and from the potency of another mixture(s) in the same bioassay and in
humans. It is assumed that the relationship (ratio) between the
potency of a mixture in a bioassay and human cancer risk is constant
for different (PAH-rich) mixtures. The relationship is expressed in
equation (3):
Human risk carcinogen1/ Bioassay potency carcinogen1
= Human risk carcinogen2/ Bioassay potency carcinogen2 (3)
= k
The carcinogenic risk to humans due to exposure to a mixture can
readily be derived by a rearrangement of terms.
The potency in the bioassay and the risk to humans are expressed in
terms of the mass of extractable organic compounds contained in the
mixture. Although this method is intended to predict human risks due
to PAH, in practice it estimates the risk due to all organic compounds
present. This discrepancy may not be significant when estimating the
carcinogenic risk of mixtures rich in polycyclic organic matter, such
as coal-tar, but may be important when estimating the risk of exposure
to cigarette smoke or ambient air, in which PAH do not necessarily
play a major role.
I.2.2.2 Development and validation
The comparative potency approach was initially proposed as part of an
approach to assessing the carcinogenic risk of PAH in diesel emissions
(Albert et al., 1983; Lewtas, 1985a,b; Nesnow, 1990). For source
mixtures such as coal-tar, coke-oven emissions, and diesel and petrol
emissions tested both for skin tumorigenicity in mice (Nesnow et al.,
1982a,b) and in short-term bioassays (Lewtas, 1985a), there was
generally good agreement (Lewtas, 1985a). Furthermore, there appears
to be a good correlation between the potency of mixtures in bioassays
and in humans, although this correlation is based on limited
epidemiological data of good quality (Nesnow, 1990; Lewtas, 1993).
Thus, risk assessments of relatively high quality are currently
available only for cigarette smoke, coke-oven emissions, and coal-tar.
Although the concept of comparative potency has been extensively
validated, some outstanding issues remain, which are discussed below.
I.2.2.3 Key implicit and explicit assumptions
The comparative approach assumes that several distinct sources
contribute PAH to the environment at a given location in an ambient
environment. For example, in an industrial city in winter, the sources
of PAH may include emissions from steel manufacture, from cars,
lorries, and transport from other locations, and from home heating.
Each source is assumed to make a specific contribution to the overall
risk for lung cancer due to PAH. The proportion of the total risk
attributable to each source depends on its potency (risk per unit mass
of organic compounds) and the overall contribution to the mass of
organic compounds in the ambient air. The total risk can be expressed
as follows:
Total risk = (unit risk source1 × mass of organic
compounds source1) + (unit risk source2 × mass of
organic compounds source2) + (unit risk source3 (4)
× mass of organic compounds source3) ...
The unit risks for some sources are listed in Table AI.10.
Table AI.10. Potency of some source mixtures expressed as average
dose causing 50% papilloma incidence in male and female Sencar
mice
Source mixture Organic compounds (mg)
Coke-oven main 0.14
Coke-oven topside 0.16
Roofing tar 2.0
Nissan diesel emission 1.6
Volkswagen Rabbit diesel emission Tumour incidence did not reach
50% with tested doses
From Nesnow et al. (1982a,b)
Implicit in this approach are the assumptions that the composition of
the organic compounds emitted from each source (such as diesel
engines) is constant and that the potency (unit risk) of a given
source mixture is constant. If the potency and composition of the
organic compounds within mixtures from the same source vary widely,
the standard mixture may not accurately represent other mixtures from
a similar source. There is some evidence, however, that mixtures from
similar sources have substantially different compositions. Thus, the
benzo [a]pyrene content of the emissions from four diesel engines
varied over a 600-fold range (Nesnow et al., 1982a,b; see Table
AI.11). Significant differences in the potency of the four mixtures
were also seen. Nevertheless, the potency of the mixtures appears to
correlate reasonably well with their benzo [a]pyrene content (see
Table AI.11), so that the comparative potency approach may be viable
in principle but may not be appropriate for expressing the potency of
the mixtures in terms of the mass of the organic content. A possible
alternative is to express the potency in terms of the level of
benzo [a]pyrene present in the mixture. If this solution is used, the
comparative potency approach becomes essentially the benzo [a]pyrene
surrogate approach discussed in section I.2.3.
The levels of PAH in ambient air may be influenced by multiple
sources, and some PAH may be transformed in the environment. In order
to estimate the carcinogenic risk due to exposure to an ambient
mixture, the contribution of individual sources to the ambient air
levels must be estimated, because contributing sources differ in their
carcinogenicity. Making reliable estimates of the contribution of
individual sources to ambient air levels is still a difficult,
non-routine process.
Table AI.11. Benzo[a]pyrene content of organic fraction extracted from
diesel-engine emissions
Source Benzo[a]pyrene % Mice with tumours/ng
(ng/mg organic fraction) benzo[a]pyrenea
Nissan 1200 0.024
Oldsmobile 2.0 NS
Caterpillar 2.0 NS
Volkswagen Rabbit 26 0.11
From Nesnow et al. (1982a,b); NS, no significant response observed over
the range of doses tested
a Calculated by Muller et al. (1995a,b, 1996)
I.2.2.4 Application
In order to use the comparative potency approach, the carcinogenicity
of the major source mixtures that contribute to a given ambient
environment must be established. The potency of a number of the
mixtures has been estimated (see Table AI.10), and the potency of
source mixtures that affect air has been expressed in terms of risk
per mass of organic compounds per cubic metre. The levels of each
source mixture must then be estimated for a given ambient environment.
The total risk is calculated as shown in equation (4).
I.2.3 Benzo [a]pyrene as a surrogate for the PAH fraction of
complex mixtures
I.2.3.1 Principle
The third approach assumes that the risk due to the PAH component of
complex mixtures and the levels of individual PAH in the mixtures are
proportional to those of benzo [a]pyrene in the mixture and vary
proportionately. Using this approach, the risk due to the PAH
component of mixtures can be estimated as the product of the
environmental levels of benzo [a]pyrene and the estimate of the risk
attributable to mixtures per unit amount of benzo [a]pyrene.
In general, the approach does not predict the potency of an ambient
complex mixture as a whole but merely its PAH component. There is no
reason to believe that benzo [a]pyrene is a good indicator of
chlorinated compounds such as dioxins and dibenzofurans or volatile
organic compounds such as benzene and 1,3-butadiene, which may be
present in some ambient complex mixtures. The contribution of non-PAH
to the overall risk of exposure to complex mixtures must thus be
assessed separately.
I.2.3.2 Development and validation
Benzo [a]pyrene was initially favoured as an indicator of all urban
pollution, and in a number of assessments based primarily (or
entirely) on studies of the general population exposed to ambient air
benzo [a]pyrene was used as an index of exposure to a wider mixture
of materials (Nisbet et al., 1985). There is evidence, however, that
benzo [a]pyrene cannot serve as an indicator of the toxicity of whole
mixtures. Various factors may influence the relative contents of PAH
and other contaminants of ambient air. For instance, the relative
proportions of benzo [a]pyrene and other PAH in ambient urban air has
been declining over the years, while the levels of volatile organic
compounds and others have been rising. In some mixtures, such as
cigarette smoke condensate, PAH probably play only a minor role in
overall toxicity. Pott & Heinrich (1992) showed that mixtures
containing large amounts of carcinogenic compounds other than PAH,
such as cigarette smoke, are much more potent at a given level of
benzo [a]pyrene than mixtures that owe much of their carcinogenicity
to PAH, such as coke-oven emissions. Nisbet et al. (1985) argued
convincingly that benzo [a]pyrene cannot serve as a general indicator
of all pollutants in the ambient air, although it may be a suitable
indicator for the carcinogenic risk posed by four- to seven-ring,
unsubstituted PAH in the mixture.
Muller et al. (1995a,b, 1996) examined the PAH profiles of a wide
range of mixtures from many sources and found that they were generally
similar (see also section I.2.3.3). Furthermore, those mixtures rich
in PAH and in which PAH are likely to contribute a significant
proportion of the risk of the mixture are very similar in potency
expressed per unit amount of benzo [a]pyrene. This observation is
consistent with the notion that the PAH components of these mixtures
are approximately equipotent. (Establishment of the potency of
mixtures is discussed in section I.2.3.4.)
These findings do not imply that all mixtures are similar. Differences
in the PAH profiles of the same mixture have been analysed in order to
establish markers for mixtures from a particular source (Gordon &
Bryan, 1973; Greenberg et al., 1981; Vogt et al., 1987). The levels of
some substituted PAH are clearly not related to the levels of
benzo [a]pyrene in the mixture (Albert et al., 1983). The differences
in the profile of four- to seven-ring, unsubstituted PAH in various
mixtures are probably too small to alter the estimated risks of the
PAH component of the mixtures significantly.
I.2.3.3 PAH profiles of complex mixtures
Information on the levels of PAH in various source mixtures is
provided in Section 5. The levels of PAH in environmental mixtures
used in the following analysis were derived mainly from monitoring in
Canada (Muller et al., 1995a,b, 1996). About 100 mixtures were
classified into different types, such as diesel emissions, coke-oven
emissions, ambient air particulate, soils, and sediments, to
facilitate the analysis, and the profiles of the 15 PAH most commonly
tested in mixtures were compared. The results are expressed as the
ratios of the levels of each PAH relative to benzo [a]pyrene; the
geometric mean, the upper and lower 95% confidence limits, and the
confidence range were calculated for each PAH ratio for a mixture
type. The confidence range was determined by dividing the upper
confidence limit by the lower confidence limit and used as a measure
of the range of the means of the relative levels a given PAH will
assume about 95 times out of 100.
The PAH profiles for petrol exhaust emissions are provided as an
example in Table AI.12. It can be seen that the confidence range for
each PAH is less than 5.0, and many are less than 2.0, indicating that
the levels of these PAH, relative to benzo [a]pyrene, are very stable
and vary little among the sources. They also indicate that any
variation among samples is probably not large enough to alter the
estimated risk. The low variation also means that the level of
benzo [a]pyrene is a good predictor of the levels of the other PAH
that may be present in a given mixture from petrol engines.
The confidence ranges for all types of combustion mixture and all of
the PAH considered are presented in Table AI.13. The confidence ranges
were < 50 for more than 90% of all entries and < 6 for 50% of all
entries. Given the degree of uncertainty usually associated with risk
assessment, the uncertainty presented by the variation in PAH profile
is relatively small. In addition, while some compounds in a given
mixture may be found at higher levels than expected, those of other
compounds may be lower than expected, and there may therefore be
little difference between the estimates of risk based on chemical
analysis and those based on the predicted composition of a mixture.
The ambient mixtures appear to be less variable than the combustion
emission mixtures. For example, Table AI.14 shows that the confidence
ranges for most types of ambient air are similar. The corresponding
levels of PAH relative to benzo [a]pyrene are shown in Table AI.15.
Samples of ambient air were collected in 1982-86 at point sources in
Hamilton, Ontario, Canada, on days when the wind was blowing from
nearby steel-mill operations 50% or more of the time. Mobile sources
and home heating also contributed, but the urban levels of PAH were
much lower on days when the wind was not blowing from the direction of
the steel mills. The average level of benzo [a]pyrene was about 1.8
ng/m3. Samples of ambient air from mobile sources were collected in
Toronto, Ontario, during the summer near the intersection of two busy
multi-lane highways in 1988-92. The average level of benzo [a]pyrene
was about 0.17 ng/m3. The highway intersection is surrounded by
residential areas, and the samples of ambient air associated with home
heating were collected in the same location as the mobile sources by
the same collection and analytical protocol, but in winter. The
average level of benzo [a]pyrene was 0.41 ng/m3. Home heating and
mobile emisions are considered to be the main sources of PAH. In
samples of ambient air collected on Wallpole Island, Ontario, a rural
location with little traffic and no industrial source, the average
levels of benzo [a]pyrene was about 0.093 ng/m3 (T. Dann, personal
communication).
Table AI.12. Confidence ranges for petrol engine exhaust (relative to
benzo[a]pyrene
Compound Mean 95% confidence
Lower limit Upper limit Interval
Anthracene 7.0 9.3 5.3 1.8
Phenanthrene 25 38 17 2.3
Fluoranthene 7.3 14 4.0 3.5
Pyrene 9.5 19 4.7 4.0
Benz[a]anthracene 0.81
Perylene 0.27 0.60 0.12 4.8
Benzo[e]pyrene 1.1 1.4 0.79 1.8
Benzo[ghi]perylene 2.6 3.5 2.0 1.7
Dibenz[a,h]anthracene 0.072
Coronene 2.0 2.7 1.4 1.9
Indeno[1,2,3-cd]pyrene 0.80 1.1 0.60 1.8
Anthanthrene 0.38 0.55 0.27 2.1
Chrysene and triphenylene 3.0 4.6 1.9 2.4
Benzofluoranthenes 1.1 1.5 0.85 1.7
From Muller (1995a,b, 1996), based on data from Hoffmann & Wynder
(1962), Grimmer & Hildebrandt (1975), Grimmer & Bohnke (1978),
Alsberg et al. (1985), and Hagemann et al. (1982)
Table AI.13. Confidence ranges for polycyclic aromatic hydrocarbons in various combustion mixtures (relative to
benzo[a]pyrene
Compound Combustion mixture
Coke Coal-tar Coal-fired Coal Open burning Wood Diesel Petrol Roofing Paving
ovens power plants stoves and fireplaces stoves emissions emissions asphalt asphalt
Anthracene 3.2 440 830 7.6 1.8
Phenanthrene 1.3 32 27 2.7 2.3
Fluoranthene 2.7 4.5 3.2 43 4.9 1.4 6.1 3.5 5.7 5.2
Pyrene 2.6 280 37 18 5.6 1.5 5.3 4.0 19 2.7
Benz[a]anthracene 19 2.8 32 160 2.0 9.0 130 8.5
Perylene 2.3 27 23 8.2 31 4.8 4.6 5.9
Benzo[e]pyrene 1.3 5.5 3.3 35 1.6 1.5 22 1.8
Benzo[ghi]perylene 8.8 12 32 2.7 8.9 1.7 5.1 2.8
Dibenz[a,h]anthracene 310 3.2 2.4
Coronene 7.8 7.1 730 3.2 1.9
Indeno[1,2,3-cd]pyrene 7.4 1.9 7.0 1.8
Anthanthrene 8.7 2.1
Chrysene and 2.1 12 430 42 25 4.3 7.3 2.4 23 6.1
triphenylene
Benzofluoranthenes 5.7 2.0 28 7.1 5.0 3.4 1.7 7.7 120
Incidence of confdence 0 2 2 1 2 0 0 0 1 1
range > 50
Table AI.14. Confidence ranges for particulates extracted from ambient
air (relative to benzo[a]pyrene)
Compound Point Near Home Transport Geometric
source mobile heating mean
source
Anthracene 2.8 5.7 6.7 2.0 20
Phenanthrene 2.3 1.7 2.6 1.4 13
Fluoranthene 2.2 1.5 1.7 1.4 8.1
Pyrene 2.4 1.4 1.7 1.4 6.7
Benz[a]anthracene 2.0 1.4 1.5 1.2 2.3
Perylene 2.7 1.3 1.2 1.9 1.7
Benzo[e]pyrene 2.8 1.3 1.6 1.1 1.3
Benzo[ghi]perylene 2.5 1.5 1.6 1.2 2.4
Indeno[1,2,3-cd]pyrene 1.3 1.4 1.8 1.2 1.2
Anthanthrene 2.0 3.4 1.8 41 1.9
Chrysene and 2.1 1.3 2.0 1.3 1.4
triphenylene
Benzofluoranthenes 2.5 1.3 1.9 1.3 1.7
Table AI.16. Mean profiles of polycyclic aromatic hydrocarbons in
ambient air (relative to benzo[a]pyrene)
Compound Point Near Home Transport Geometric
source mobile heating mean
source
Anthracene 5.5 7.6 1.0 1.8 2.9
Phenanthrene 38 200 39 43 60
Fluoranthene 14 48 12 13 18
Pyrene 9.3 28 11 7.1 12
Benz[a]anthracene 1.4 0.82 1.0 0.78 0.97
Perylene 0.33 0.25 0.22 0.24 0.26
Benzo[e]pyrene 1.5 1.3 1.6 1.4 1.4
Benzo[ghi]perylene 1.4 1.5 2.4 1.3 1.6
Indeno[1,2,3-cd]pyrene 1.5 1.3 1.5 1.4 1.4
Anthanthrene 0.19 0.15 0.13 0.20 0.17
Chrysene and 3.0 2.7 3.5 2.9 3.0
triphenylene
Benzofluoranthenes 3.6 2.9 3.6 4.4 3.6
Table AI.16 presents the average PAH profiles of the combustion
emissions, ambient air particulates, soils, and sediments and the
average profile of the four types of mixture. The confidence ranges
indicate that the four mixtures had fairly similar PAH profiles.
On the basis of this analysis, Muller et al. (1995a,b, 1996) concluded
that a wide variety of mixtures have fairly similar profiles of
commonly assayed PAH. They assumed that the PAH fraction of all
sources of environmental mixtures have profiles similar to the
average, as shown in the sixth column of Table AI.16. This conclusion
does not include substituted PAH, as there is strong evidence that
different mixtures contain different levels of substituted PAH. It
does not imply that there are no real differences due to the source of
the mixture, the type of fuel, and the pyrolysis conditions that
produced it. Furthermore, aerial transport of PAH, degradation in
sunlight or by soil microorganisms, and other factors may alter the
PAH profile. These factors are, however, unlikely to generate large
enough differences in the PAH profiles of mixtures to significantly
alter the estimate of risk for a given mixture.
I.2.3.4 Potency of complex mixtures
If benzo [a]pyrene is a suitable indicator of the carcinogenic
potency of the PAH in a mixture, then the potency of a mixture
expressed as the tumour incidence per nanogram of its benzo [a]pyrene
content should be numerically similar for all mixtures in which PAH
are expected to be the major cause of tumorigenic effects. Nesnow et
al. (1982b) tested a number of PAH-rich mixtures in a
tumour-initiation assay in mouse skin. The different types of mixture
were roughly equipotent when the potency was expressed in terms of
benzo [a]pyrene content (Table AI.17).
I.2.3.5 Key implicit and explicit assumptions
The approach assumes that the levels of individual PAH relative to
benzo [a]pyrene are relatively stable from mixture to mixture. It
also assumes that the risk attributable to PAH in any given mixture is
proportional to the risk due to benzo [a]pyrene. In other words, the
level of benzo [a]pyrene is sufficient to estimate the risk of the
PAH fraction in a mixture.
Differences in the composition of mixtures from different sources have
been used to estimate the contribution of those sources to the ambient
levels of PAH (see Gordon & Bryan, 1973; Greenberg et al., 1981; Vogt
et al., 1987). Other authors have reported transformation of PAH in
the environment. Since some compounds are more photosensitive than
others, the proportion of PAH in ambient air will change over time as
a result of the different transformation rates of different compounds
(Van Cauwenberghe, 1985). Muller et al. (1995a,b, 1996) examined a
wide variety of mixtures from different combustion sources and ambient
mixtures and concluded that the differences in the profiles of four-
to seven-ring unsubstituted PAH relative to the benzo [a]pyrene
content are not large enough to affect the risk posed by the mixtures
Table AI.16. Average profiles for combustion-derived mixtures, ambient air, soil, and sediment
(relative to benzo[a]pyrene)
Compound Source Ambient Soil Sediment Geometric Confidence
mixtures air mean range
Anthracene 3.9 2.9 0.85 0.47 1.6 24
Phenanthrene 18 60 2.8 3.6 4.3 23
Fluoranthene 4.9 18 2.5 3.2 3.8 1.8
Pyrene 4.5 12 3.1 2.4 2.8 2.9
Benz[a]anthracene 1.8 0.97 1.4 1.4 1.2 2.6
Perylene 0.51 0.26 0.34 1.4 0.45 17
Benzo[e]pyrene 1.0 1.4 1.4 1.1 7.4
Benzo[ghi]perylene 0.98 1.6 1.4 0.96 1.0 1.4
Dibenz[a,h]anthracene 0.35 0.45 0.30 0.28 4.2
Coronene 0.35 0.45 0.34 3.9
Indeno[1,2,3-cd]pyrene 0.51 1.4 0.95 1.2 0.86 4.7
Anthanthrene 0.49 0.17 0.19 0.31
Chrysene and 2.3 3.0 1.4 1.2 2.0 3.3
triphenylene
Benzofluoranthenes 1.6 3.6 1.7 2.4 2.5 11
Table A1.17. Potency of various mixtures in an
assay for tumour initiation
Source mixture Incidence per ng
benzo[a]pyrene
Coke-oven main 9.4 × 10-2
Coke-oven topside 7.6 × 10-2
Smoky coal 7.8 × 10-2
Smokeless coal 2.8 × 10-2
Roofing tar 1.1 × 10-2
Wood smoke 5.8 × 10-2
Diesel engine exhaust 2.5 × 10-2
Petrol engine exhaust 5.6 × 10-2
Max/min 8.6
Calculated from data of Nesnow et al. (1982b)
significantly. The PAH profile of a tested mixture may deviate from
the average profile by about an order of magnitude (up or down). Since
the levels of some PAH may be above and those of others below the
expected levels, these differences would tend to cancel each other
out, leading to an error of much less than one order of magnitude.
Such small differences are below the resolution of the risk assessment
process.
I.2.3.6 Application
The first step is to estimate the carcinogenic risk due to exposure to
the PAH present in a typical mixture, and this estimate is used for
all subsequent assessments. WHO (1987) estimated that the risk for
lung cancer due to lifelong exposure to PAH in mixtures by inhalation
was 8.7 × 10-5/ng benzo [a]pyrene per m3. This estimate was based
on the assessment of the risk for lung cancer of coke-oven workers
conducted by the US Environmental Protection Agency (1984d), which
generated an upper bound risk estimate expressed in terms of
benzene-extractable material. WHO (1987) converted the US
Environmental Protection Agency estimate into benzo [a]pyrene levels
by assuming that the benzene extract contained 0.71% benzo [a]pyrene.
Sloof et al. (1989) in the Netherlands estimated the risk for lung
cancer to be 1.0 × 10-4/ng benzo [a]pyrene per m3 on the basis of
the estimate of WHO and the assessments of Pike (1983) and Tuomisto &
Jantunen (1987). That of Pike (1983) was based on the mortality from
lung cancer of gas workers, and that of Tuomisto & Jantunen (1987) was
based on the exposure of Chinese women to smoky coal smoke. Muller et
al. (1995a,b, 1996) proposed 2.3 × 10-5/ng benzo [a]pyrene per m3
as the risk for lung cancer from lifelong exposure to PAH in ambient
mixtures on the basis of the study of the US Environmental Protection
Agency (1984d) on coke-oven workers and assuming that benzo [a]pyrene
represents 1.7 ng/µg of benzene-extractable material from coke-oven
emissions. Rather than the upper bound, Muller et al. used a maximum
likelihood estimate, calculated from the US Environmental Protection
Agency study, of 3.9 × 10-5/µg of benzene-extractable per m3.
The next step is to estimate the environmental levels of
benzo [a]pyrene. In a simplified situation, in which the population
is exposed to a fixed level of environmental benzo [a]pyrene, the
lifetime cancer risk is estimated as the product of the potency of a
typical mixture (expressed as risk per nanogram of benzo [a]pyrene
per cubic metre in the case of air) and the level of benzo [a]pyrene
(expressed as ng/m3) in the environment. For example, using the risk
estimate proposed by Muller et al. (1995a,b, 1996) and assuming that a
population is exposed over a lifetime to benzo [a]pyrene at 0.5
ng/m3, the risk of the population is about 1.2 × 10-5. In other
words, about one person in 100 000 would be expected to develop lung
cancer in his or her lifetime as a result of exposure to PAH in air.
I.3 Comparison of the three procedures
Each approach has its advantages and disadvantages (Table AI.18):
I.3.1 Individual PAH approach
Main advantages:
* Clearly defined chemical species are assessed.
* A good body of scientific literature is available to evaluate it.
* Not affected by variability in the composition of mixtures
* Relatively easy to apply in ambient environments affected by many
sources
* Regulatory experience exists.
Main disadvantages:
* May underestimate risk due to all PAH by considering only a few
compounds
* Depends on extrapolation from animal models to humans
* Resource-intensive, as monitoring and analysis are required
I.3.2 Comparative potency approach
Main advantages:
* The risk of whole mixtures, rather than only a few components, is
estimated.
Table AI.18. Features and properties of three approaches to risk assessment for mixtures containing polycyclic aromatic
hydrocarbons (PAH)
Property or feature Individual PAH approach Comparative potency Benzo[a]pyrene (BaP)
approach surrogate approach
Portion of mixture for which Selected PAH in complex Entire complex mixture Unsubstituted PAH component
cancer risk is estimated mixtures account for only of complex mixtures
portion of risk of PAH fraction
PAH included in assessment Relatively few PAH out of All PAH Most PAH, except PAH for which
hundreds in environment levels do not correlate well with
those of BaP, e,g. substituted
PAH
Other toxicants included None Toxicants present in source Some, e.g. those present in
mixture coke-oven emissions
Assumption of additivity of Yes: good evidence that No: assumption not required No: assumption not required
components of mixture risks of ill health due to
exposure to PAH are
approximately additive;
little known about additivity
of risks due to PAH and
other compounds
Incorporates directly No: requires extrapolation Yes: requires data from Yes: potency derived from data
available data on human from animal models to animal models to estimate on potency of coke-oven
cancer risk estimate potency of all PAH potency of some mixtures emissions in humans
Assumption that mixtures No: not applicable Yes: assumes mixtures from Yes: assumes mixtures from
from similar sources are similar sources are about similar sources are about
about equipotent equipotent when expressed equipotent when expressed
in terms of mass of organic in terms of mass of BaP;
extractable material; basis assumption supported by
for assumption equivocal available data
Table AI.18. (continued)
Property or feature Individual PAH approach Comparative potency Benzo[a]pyrene (BaP)
approach surrogate approach
Assumption that mixtures No: assumption not required No: assumption not required Yes: evidence from studies of
from different sources are animal models supports the
about equipotent assumption; human data are
equivocal and inadequate to
validate the approach
Assumption that environmental No: assumption not required Yes: evidence that PAH Yes: evidence that PAH profile
transformation profile relative to BaP does relative to BaP does not vary
processes do not change the not vary enough to affect the enough to affect estimated risk
PAH profile enough to affect estimated risk significantly significantly
the overall cancer risk
significantly
Assumption that the PAH No: assumption not required No: assumption not required Yes: good evidence to
profile of various mixtures is support this assumption
roughly comparable
Suitable for ambient Yes Requires apportioning of Yes
environments affected by ambient mixture to multiple
multiple sources sources
Monitoring requirements Selected PAH Organic extractable matter BaP
(this information is not
usually reported and methods
not standardized)
Regulatory use Yes No Yes
* A good body of scientific literature is available to evaluate it.
* Takes advantage of existing data on human carcinogenicity
* Simple and requires inexpensive monitoring
Main disadvantages:
* Does not define the contribution of PAH to estimated overall
risk.
* Difficult to use for assessing speciated components of a mixture.
* Risk estimates require estimates of the contributions of
individual sources to the levels of organic compounds in the
ambient environment.
* The assumption that mixtures from the same source are associated
with similar risks may not be supported by the available data.
* The levels of compounds extractable in organic solvents are not
usually reported, and the analytical methods are not
standardized.
I.3.3 Benzo [a]pyrene surrogate approach
Main advantages:
* Can be used to estimate risk of entire PAH component of a mixture
* Simple and based on a few testable assumptions
* Well supported by the available data
* Relatively easy and inexpensive to apply for regulatory purposes
* Regulatory experience exists.
Main disadvantages:
* May result in overestimate of the risk of PAH within a mixture
* Some PAH, such as substituted ones, are not well represented by
benzo [a]pyrene and must be considered separately.
APPENDIX II
SOME LIMIT VALUES
Regulatory decisions about chemicals taken in a country can be fully
understood only within the framework of the legislation of that
country. Furthermore, the regulations and guidelines of all countries
are subject to change and should always be verified with the
appropriate regulatory authorities before application.
II.1 Exposure of the consumer
The concentrations of some components of polycyclic aromatic
hydrocarbons (PAH), especially benzo[a]pyrene, in air, water, and food
and the use of PAH-containing technical products are regulated by law
in many countries. The available limit values are listed in Table
AII.1.
II.2 Occupational exposure
Regulations for limits in the air at different workplaces are compiled
in Table AII.2. Only values for individual substances are given. For
some occupations, e.g. roofers and asphalt workers, limit values are
not given for individual compounds but for the mixture of organic
vapours released, e.g. bitumen fumes, coal-pitch, and coal-tar
volatiles, that are soluble in benzene or hexane. These limit values
were not taken into account.
II.3 Classification
Only classifications based on a toxicological end-point are given
here. Those relevant to exposure in the workplace are shown in Table
AII.3. Especially in industrialized countries, classifications also
exist for industrial emissions into air, water, and soil. As these
are special regulations, which differ from country to country, they
are not included.
Some classifications refer to technical mixtures with a high PAH
content:
II.3.1 European Union
* A maximum of 50 mg/kg benzo[a]pyrene will render coal-tar derived
products carcinogenic (Directive 94/69/EC: European Economic
Community, 1994a).
* For lubricant base oils analyzed by the legally defined method,
the cut-off to define carcinogenicity is 3% of the extract
containing mainly PAH, equivalent to 0.5-1 mg/kg benzo[a]pyrene
(CONCAWE, 1994).
Table AII.1. Limit values for consumer exposure to individual polycyclic aromatic hydrocarbons (PAH) in various countries
Country, year Compound Limit value Reference
Ambient air
Italy EEC (1994)
01.01.1996 to 31.12.1998 Benzo[a]pyrene 2.5 ng/m3
From 01.01.1999 Benzo[a]pyrene 1 ng/m3
Former USSR, 1985 Benzo[a]pyrene 1 ng/m3 Khesina (1994); UNEP
(1994)
Ambient water
USA, 1984 Sum of benzo[a]pyrene, benz[a]anthracene, 0.2 µg/litre Slooff et al. (1989)
benzofluoranthenes, chrysene, fluoranthene,
indeno[1,2,3-cd]pyrene, anthracene, pyrene,
dibenz[a]anthracene
Former USSR, 1990 Benzo[a]pyrene 0.005 µg/litre UNEP (1994)
EEC, 1980 Sum of fluoranthene, benzo[b]fluoranthene, 1.2 µg/litre Slooff et al. (1989)
benzo[k]fluoranthene, benzo[a]pyrene,
benzo[ghi]perylene, indeno[1,2,3-cd]pyrene
Drinking-water
WHO guideline, 1995 Benzo[a]pyrene 0.7 µg/litre WHO (1996)
EEC, 1980 (adopted by most Sum of fluoranthene, benzo[b]fluoranthene, 0.2 µg/litre EEC (1980)
Member States and numerous benzo[k]fluoranthene, benzo[a]pyrene,
other European countries) benzo[ghi]perylene, indeno[1,2,3-cd]pyrene
Former Czechoslovakia, 1991 Sum of PAH expressed as fluoranthene 40 µg/litre UNEP (1994)
Benzo[a]pyrene 0.01 µg/litre
Canada, 1991 Benzo[a]pyrene 0.01 µg/litre UNEP (1994)
Netherlands, 1989 Sum of fluoranthene, benzo[b]fluoranthene, 0.1 µg/litre Slooff et al. (1989)
benzo[k]fluoranthene, benzo[a]pyrene,
benzo[ghi]perylene, indeno[1,2,3-cd]pyrene
Table AII.1. (continued)
Country, year Compound Limit value Reference
Soil
USSR, 1985 Benzo[a]pyrene 0.02 mg/kg UNEP (1994)
Food
EEC, 1991: use of flavourings Benzo[a]pyrene 0.03 µg/kg European Economic
in food Community (1991)
Germany, 1988: meat and meat Benzo[a]pyrene 1 µg/kg EEC (1988)
products
Italy, 1988: food and beverages Benzo[a]pyrene 0.03 µg/kg Anon. (1988)
Other
EEC, 1994: tar-oil products Benzo[a]pyrene 50 mg/kg Appendix I, No. 32 of
for wood preservation guideline 94/60/EG dated
20.12.1994
Germany, 1994: products for Benzo[a]pyrene 5 mg/kg Appendix to s1 of German
wood preservation Chemicals Prohibition
Directive (1994)
Former Czechoslovakia, 1991 Benzo[a]pyrene Prohibited in UNEP (1994)
cosmetics
* Any substance containing benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[j]fluoranthene, benzo[a]pyrene, or
dibenz[a,h]anthracene at a concentration > 0.1% is regarded as carcinogenic (Annex I of Directive 67/548/EEC).
It therefore cannot be supplied to the general public in the European Union (Directive 94/60/EC) but only to
professional users (Von Meyerinck, 1995).
Table AII.2. Limit values for individual polycyclic aromatic hydrocarbons at various workplaces
Country, year Workplace or Compound Limit value Reference
emission source
Finland, 1989 NR skin Benzo[a]pyrene 10 µg/m3 (TWA) UNEP (1994)
absorption
France, 1988 Production of carbon Benzo[a]pyrene 0.15 µg/m3 Lafontaine et al.
electrodes (1990b)
Germany, 1989 Cokeries, oven area Benzo[a]pyrene 5 µg/m3 German Federal
Department for
Worker Safety (1989)
Other workplaces Benzo[a]pyrene 2 µg/m3 German Federal
Department for
Worker Safety (1989)
Sweden,1993 NR Benzo[a]pyrene 2 µg/m3 (LLV) Swedish National
20 µg/m3 (STV); Board of Occupational
Safety & Health (1994)
Former USSR, 1990 NR Benzo[a]pyrene 0.15 µg/m3 (MAC) UNEP (1994)
Argentina, 1991 NR Naphthalene 50 mg/m3 (TWA; MPC) UNEP (1994)
75 mg/m3 (STEL; MPC)
Bulgaria, 1985 NR Naphthalene 20 mg/m3 (MPC) UNEP (1994)
Canada, 1991 NR Naphthalene 50 mg/m3 (TWA; TLV) UNEP (1994)
75 mg/m3 (STEL; TLV)
Germany, 1993 NR Naphthalene 50 mg/m3 (MAK) American Conference of
Governmental Industrial
Hygienists (1995)
Hungary, 1985 NR Naphthalene 20 mg/m3 (TWA; MAC) UNEP (1994)
100 mg/m3 (STEL: MAC)
Italy, 1991 NR Naphthalene 50 µg/m3 EEC (1991)
Mexico, 1991 NR Naphthalene 50 mg/m3 (TWA; MXL) UNEP (1994)
75 mg/m3 (STEL; MXL)
Poland, 1985 NR Naphthahne 20 mg/m3 (TWA; MPC) UNEP (1994)
Romania, 1985 NR Naphthalene 30 mg/m3 (TWA; MPC) UNEP (1994)
40 mg/m3 (MPC)
Table AII.2. (continued)
Country, year Workplace or Compound Limit value Reference
emission source
Sweden, 1991 NR; skin Naphthalene 0.2 mg/m3 (TWA; HLV) UNEP (1994)
absorption 0.6 mg/m3 (STEL; HLV)
Switzerland, 1987 NR Naphthalene 50 mg/m3 (TWA; MAK) UNEP (1994)
United Kingdom, 1992 NR Naphthalene 50 mg/m3 (TWA; OES) UNEP (1994)
75 mg/m3 (STEL; OES)
USA, 1993 NR Naphthalene 52 mg/m3 (TWA) American Conference of
79 mg/m3 (STEL) Governmental Industrial
Hygienists (1995)
Former USSR, 1993 NR Naphthalene 20 mg/m3 American Conference of
Governmental Industrial
Hygienists (1995)
Former Yugoslavia,1985 NR Naphthalene 50 mg/m3 (TWA; MAC) UNEP (1994)
USA, 1993 Cokeries, oven area Phenylene 0.1 mg/m3 (TWA) American Conference of
Governmental Industrial
Hygienists (1995)
Former USSR, 1989 NR Phenylene 0.8 mg/m3 (MAC) UNEP (1994)
USA, 1993 NR Pyrene 0.1 mg/m3 (TWA) American Conference of
Governmental Industrial
Hygienists (1995)
Former USSR, 1989 NR Pyrene 0.03 mg/m3 (MAC) UNEP (1994)
NR, not reported; HLV, hygienic limit value; LLV, level limit value; MAC, maximum allowable concentration; MAK, maximum
workpace concentration; MPC, maximum permissible concentration; MXL, maximum limit; OES, occupational exposure standard;
TWA, time-weighted average; STEL, short-time exposure level; STV, short-term value; TLV, threshold limit value
Table AII.3. Toxicological classifications of polycyclic aromatic hydrocarbons with regard to exposure in
the workplace
Compound ACGIHa (TLV) IARCb EUc German
MAKd
TWA (8 h) STEL (15 min)
Benz[a]anthracene A2 A2 2A Carcinogenic, category 2 A2
Benzo[b]fluoranthene A2 A2 2B Carcinogenic, category 2 A2
Benzo[j]fluoranthene 2B Carcinogenic, category 2 A2
Benzo[k]fluoranthene 2B Carcinogenic, category 2 A2
Benzo[a]pyrene A2 2A Carcinogenic, category 2 A2
Chrysene A2 A2 3 Carcinogenic, category 2 A2
Dibenz[a,h]anthracene 2A Carcinogenic, category 2 A2
Dibenzo[a,e]pyrene 2B A2
Dibenzo[a,h]pyrene 2B A2
Dibenzo[a,i]pyrene 2B A2
Dibenzo[a,l]pyrene 2B A2
Indeno[1,2,3-cd]pyrene 2B A2
5-Methylchrysene 2B
a American Conference of Governmental Industrial Hygienists (1995)
b IARC (1987) see Section 12
c EU-Rili (Appendix I)
d German Dangerous Chemicals Directive (1995)
II.3.2 USA
* Coal-tar and coal-pitch volatiles, which are mixtures of organic
vapours with high PAH levels, are classified as A1, confirmed
human carcinogens (American Conference of Governmental Industrial
Hygienists, 1995).
* Diesel exhaust is considered to be a suspected human carcinogen
(A2), but notice has been given of intended changes (American
Conference of Governmental Industrial Hygienists, 1995).
13. REFERENCES
Aamot E, Krane J, & Steiness E (1987) Determination of trace amounts
of polycyclic aromatic hydrocarbons in soil. Fresenius Z Anal Chem,
328: 569-571.
Abarzua AS, Jonas L, Putzke HP, & Wittenburg E (1983) [Influence of
3,4-benzopyrene on the ultrastructure of the green alga Scenedesmus
quadricauda.] Arch Protistenk, 127: 103-113 (in German).
Abbott DW, Moody RL, Mann RM, & Vo-Dinh T (1986) Synchronous
luminescence screening for polynuclear aromatic compounds in
environmental samples collected at a coal gasification process
development unit. Am Ind Hyg Assoc J, 47: 379-385.
Abdo KM, Eustis SL, McDonald M, Jokinen MP, Adkins B, & Haseman JK
(1992) Naphthalene: A respiratory tract toxicant and carcinogen for
mice. Inhal Toxicol, 4: 393-409.
Abe S & Sasaki M (1977a) Studies on chromosomal aberrations and sister
chromatid exchanges induced by chemicals. Proc Jpn Acad, 53: 46-49.
Abe S & Sasaki M (1977b) Chromosome aberrations and sister chromatid
exchanges in Chinese hamster cells exposed to various chemicals. J
Natl Cancer Inst, 58: 1635-1641.
Abe S, Nemoto N, & Sasaki M (1983a) Sister-chromatid exchange
induction by indirect mutagens/carcinogens, aryl hydrocarbon
hydroxylase activity and benzo [a]pyrene metabolism in cultured human
hepatoma cells. Mutat Res, 109: 83-90.
Abe S, Nemoto N, & Sasaki M (1983b) Comparison of aryl hydrocarbon
hydroxylase activity and inducibility of sister-chromatid exchanges by
polycyclic aromatic hydrocarbons in mammalian cell lines. Mutat Res,
122: 47-51.
Abell CW & Heidelberger C (1962) Interaction of carcinogenic
hydrocarbons with tissues. VIII. Binding of tritium-labelled
hydrocarbons to the soluble proteins of mouse skin. Cancer Res, 22:
931-946.
Abernethy S, Bobra AM, Shiu WY, Wells PG, & Mackay D (1986) Acute
lethal toxicity of hydrocarbons and chlorinated hydrocarbons to two
planktonic crustaceans: The key role of organism-water partitioning.
Aquat Toxicol, 8: 163-174.
Abramson MJ, Wlodarczyl JH, Saunders NA, & Hensley MJ (1989) Does
aluminum smelting cause lung disease? Am Rev Respir Dis, 139:
1042-1057.
Achard S, Perderiset M, & Jaurand MC (1987) Sister chromatid exchanges
in rat pleural mesothelial cells treated with crocidolite,
attapulgite, or benzo 3-4 pyrene. Br J Ind Med, 44: 281-283.
Acheson MA, Harrison RM, Perry R, & Wellings RA (1976) Factors
affecting the extraction and analysis of polynuclear aromatic
hydrocarbons in water. Water Res, 10: 207-212.
Adamson RH & Sieber SM (1983) Chemical carcinogenesis studies in
nonhuman primates. In: Langenbach R, Nesnow S, & Rice JM ed. Organ and
species specificity in chemical carcinogenesis. New York, Plenum
Press, pp 129-156.
Adkins B Jr, van Stee EW, Simmons JE, & Eustis SL (1986) Oncogenic
response of strain A/J mice to inhaled chemicals. J Toxicol Environ
Health, 17: 311-322.
Adler ID & Ingwersen I (1989) Evaluation of chromosomal aberration in
bone marrow of 1C3F1 mice. Mutat Res, 224: 343-345.
Adler ID, Kliesch U, & Kiefer F (1989) Clastogenic effects of
benzo [a]pyrene in postimplantation embryos with different genetic
background. Teratog Carcinog Mutag, 6: 383-392.
Adlkofer F, Scherer G, Von Maltzan C, Von Meyerinck L, Jarczyk L,
Martin F, & Grimmer G (1990) Dietary influences on urinary excretion
of hydroxyphenanthrenes, thioethers and mutagenicity in man. In:
Vainio H, Sorsa M, & McMichael AJ ed. Complex mixtures and cancer
risk. Lyon, International Agency for Research on Cancer, pp 415-420
(IARC Scientific Publications No. 104).
Agarwal R & Mukhtar H (1991) Metabolism of benzo [a]pyrene and
benzo [a]pyrene 7,8-dihydrodiol catalyzed by human and rodent
epidermal lipoxygenase. Proc Am Assoc Cancer Res, 32: 117.
Agarwal SK, Sayer JM, Yeh HJC, Pannell LK, Hilton BD, Pigott MA,
Dipple A, Yagi H, & Jerina DM (1987) Chemical characterization of DNA
adducts derived from the configurationally isomeric
benzo [c]phenanthrene-3,4-diol 1,2-epoxides. J Am Chem Soc, 109:
2497-2504.
Agarwal R, Medrano EE, Khan IU, Nordlund JJ, & Mukhtar H (1991)
Metabolism of benzo(a)pyrene by human melanocytes in culture.
Carcinogenesis, 12: 1963-1966.
Agency for Toxic Substances and Disease Registry (1990) Toxicological
profile for polycyclic aromatic hydrocarbons. Atlanta, Georgia, US
Department of Health and Human Services, 231 pp (Report No. TP-90-20).
Agrelo C & Amos H (1981) DNA repair in human fibroblasts. In: de
Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, pp 528-532 (in Mutation Research, Volume
1).
Ahland E & Mertens H (1980) [PAH emissions from coal fires -
Collection, analysis, results. Air pollution from polycyclic aromatic
hydrocarbons - Registration and evaluation.] Düsseldorf, VDI-Verlag
GmbH, pp 107-111 (VDI Report No. 358) (in German).
Ahland E, Borneff J, Brune H, Grimmer G, Habs M, Heinrich U, Hermann
P, Misfeld J, Mohr U, Pott F, Schmähl D, Thyssen J, & Timm J (1985)
[Air pollution and cancer. Investigation of hazardous constituents
from various emission sources and their carcinogenic impact.] Münch
Med Wochenschr, 127: 218-221 (in German).
Aitio A (1974) Different elimination and effect on mixed function
oxidase of 20-methylcholanthrene after intragastric and
intraperitoneal administration. Res Commun Chem Pathol Pharmacol, 9:
701-710.
Albaiges J, Bayona JM, Fernandez P, Grimalt J, Rosell A, & Simo R
(1991) Vaporparticle partitioning of hydrocarbons in western
Mediterranean urban and marine atmospheres. Mikrochim Acta, 2: 13-27.
Alben K (1980) Coal tar coatings of storage tanks. A source of
contamination of the potable water supply. Environ Sci Technol, 14:
468-470.
Albert RE, Lewtas J, Nesnow S, Thorslund TW, & Anderson E (1983)
Comparative potency method for cancer risk assessment: Application to
diesel particulate emissions. Risk Anal, 3: 101-117.
Albert RE, Miller ML, Cody TE, Andringa A, Shukla R, & Baxter CS
(1991a) Benzo [a]pyrene-induced skin damage and tumor promotion in
the mouse. Carcinogenesis, 12: 1273-1280.
Albert RE, Miller ML, Cody TE, Barkley W, & Shukla R (1991b) Cell
kinetics and benzo [a]pyrene DNA adducts in mouse skin tumorigenesis.
In: Butterworth BE ed. Chemically induced cell proliferation:
Implications for risk assessment. New York, Wiley-Liss, Inc., pp 115-
122 (Progress in Clinical and Biological Research, Volume 369).
Albini A, Colacci A, Melchiori A, Carlone S, Nanni P, Lollini PL, de
Giovanni C, Nicoletti G, Grilli S, & Parodi S (1991) Chemical
carcinogens in tumor progression. Induction of invasive and metastatic
potential in 3T3 cells by benzo [a]pyrene transformation. Proc Am
Assoc Cancer Res, 32: 153.
Alden RW III & Butt AJ (1987) Statistical classification of the
toxicity and polynuclear aromatic hydrocarbon contamination of
sediments from a highly industrialized seaport. Environ Toxicol Chem,
6: 673-684.
Aldis H, Osborn J, & Wolf F (1983) Investigation of soil and water
contamination at Western Processing, King County, Washington. In:
National Conference of Management of Uncontrolled Hazardous Waste
Sites, Silver Spring, 31 October-2 November 1982, Washington DC,
Hazardous Materials Control Research Institute, pp. 43-53.
Alfheim I & Ramdahl T (1984) Contribution of wood combustion to indoor
air pollution as measured by mutagenicity in Salmonella and
polycyclic aromatic hydrocarbon concentration. Environ Mutagen, 6:
121-130.
Alfheim I & Wikström L (1984) Air pollution from aluminium smelting
plants. I. The emission of polycyclic aromatic hydrocarbons and of
mutagens from an aluminium smelting plant using the Söderberg process.
Toxicol Environ Chem, 8: 55-72.
Alfheim I, Becher G, Hongslo JK, & Ramdahl T (1984) Mutagenicity
testing of high performance liquid chromatography fractions from wood
stove emission samples using a modified Salmonella assay requiring
smaller sample volumes. Environ Mutagen, 6: 91-102.
Allen JA & Coombs MM (1980) Covalent binding of polycyclic aromatic
hydrocarbons to mitochondrial and nuclear DNA. Nature, 287: 244-245.
Allen-Hoffmann BL & Rheinwald JG (1984) Polycyclic aromatic
hydrocarbon mutagenesis of human epidermal keratinocytes in culture.
Proc Natl Acad Sci USA, 81: 7802-7806.
Allred PM & Giesy JP (1985) Solar radiation-induced toxicity of
anthracene to Daphnia pulex. Environ Toxicol Chem, 4: 219-226.
Alsberg T, Stenberg U, Westerholm R, Strandell M, Rannug U, Sundvall
A, Romert L, Bernson V, Pettersson B, Toftgard R, Franzen B, Jansson
M, Gustafsson JC, Egebäck KE & Tejle G (1985) Chemical and biological
characterization of organic material from gasoline exhaust particles.
Environ Sci Technol, 19: 43-50.
Al-Yakoob S, Saeed T, & Al-Hashash H (1993) Polycyclic aromatic
hydrocarbons in edible tissue of fish from the Gulf after 1991 oil
spill. Mar Pollut Bull, 27: 297-301.
Alzieu P, Cassand P, Colin C, Grolier P, & Narbonne JF (1987) Effect
of vitamins A, C and glutathione on the mutagenicity of
benzo [a]pyrene mediated by S9 from vitamin A-deficient rats. Mutat
Res, 192: 227-231.
Amacher DE & Paillet SC (1982) Hamster hepatocyte-mediated activation
of procarcinogens to mutagens in the L5178Y/TK mutation assay. Mutat
Res, 106: 305-316.
Amacher DE & Paillet SC (1983) The activation of procarcinogens to
mutagens by cultured rat hepatocytes in the L5178Y/TK mutation assay.
Mutat Res, 113: 77-88.
Amacher D & Turner GN (1980) Promutagen activation by rodent-liver
postmitochondrial fractions in the L5178Y/TK cell mutation assay.
Mutat Res, 74: 485-501.
Amacher DE, Paillett SC, Turner GN, Ray VA, & Salsburg DS (1980) Point
mutations at the thymidine kinase locus in L5178Y mouse lymphoma
cells. II. Test validation and interpretation. Mutat Res, 72: 447-474.
American Conference of Governmental Industrial Hygienists (1995) 1995-
1996 Threshold Limit Values (TLVs) for chemical substances and
physical agents and Biological Exposure Indices (BEIs). Cincinnati,
Ohio, 138 pp.
Amin S, Hecht SS, LaVoie E, & Hoffmann D (1979) Synthesis and
mutagenicity of 5,11-dimethylchrysene and some methyl-oxidized
derivatives of 5-methylchrysene. J Med Chem, 22: 1336-1340.
Amin S, Juchatz A, Furuya K, & Hecht SS (1981) Effects of fluorine
substitution on the tumor initiating activity and metabolism of
5-hydroxymethylchrysene, a tumorigenic metabolite of 5-methylchrysene.
Carcinogenesis, 2: 1027-1032.
Amin S, Huie K, & Hecht SS (1985a) Mutagenicity and tumor-initiating
activity of methylated benzo [b]fluoranthenes. Carcinogenesis, 6:
1023-1025.
Amin S, Huie K, Melikian AA, Leszczynska JM, & Hecht SS (1985b)
Comparative metabolic activation in mouse skin of the weak carcinogen
6-methylchrysene and the strong carcinogen 5-methylchrysene. Cancer
Res, 45: 6406-6412.
Amin S, Huie K, Balanikas G, Hecht SS, Pataki J, & Harvey RG (1987)
High stereoselectivity in mouse skin metabolic activation of
methylchrysenes to tumorigenic dihydrodiols. Cancer Res, 47:
3613-3617.
Amin S, Hecht SS, DiRaddo P, & Harvey RG (1990) Comparative tumor
initiating activities of cyclopentano and methyl derivatives of
5-methylchrysene and chrysene. Cancer Lett, 51: 17-20.
Amin S, Weyand EH, Huie K, Boger E, Neuber E, Hecht SS, & Lavoie EJ
(1991a) Effects of fluorine substitution on benzo(b)fluoranthene
tumorgenicity and DNA adduct formation in mouse skin. In: Cooke M,
Loening K, & Merritt J ed. Polynuclear aromatic hydrocarbons:
Measurements, means and metabolism. Columbus, Ohio, Battelle Press, pp
25-35.
Amin S, Misra B, Braley J, & Hecht SS (1991b) Comparative
tumorigenicity in newborn mice of chrysene and
5-alkylchrysene-1,2-diol-3,4-epoxides. Cancer Lett, 58: 115-118.
Amin S, Desai D, & Hecht SS (1992) Comparative tumorigenicity of
dimethylchrysenes in mouse skin. Chem Res Toxicol, 5: 237-241.
Amin S, Desai D, & Hecht SS (1993) Tumor-initiating activity on mouse
skin of bay region diol-epoxides of 5,6-dimethylchrysene and
benzo(c)phenanthrene. Carcinogenesis, 14: 2033-2037
Andelman JB & Suess MJ (1970) Polynuclear aromatic hydrocarbons in the
water environment. Bull World Health Organ, 43: 479-508.
Anderson JW, Neff JM, Cox BA, Tatem ME, & Hightower GM (1974) The
effects of oil on estuarine animals: Toxicity, uptake and depuration,
respiration. In: Vernberg FJ & Vernberg WB ed. Pollution and
physiology of marine organisms. New York, Academic Press, pp 285-310.
Anderson R, Giam CS, Ray LE, & Tripp MR (1981) Effects of
environmental pollutants on immunological competency of the clam
Mercenaria mercenaria: Impaired bacterial clearance. Aquat Toxicol,
1: 187-195.
Anderson LM, Priest LJ, Deschner EE, & Budinger JM (1983) Carcinogenic
effects of intracolonic benzo [a]pyrene in b-naphthoflavone-induced
mice. Cancer Lett, 20: 117-123.
Anderson JW, Neff JM, & Boehm PD (1986) Sources, fates and effects of
aromatic hydrocarbons in the Alaskan marine environment with
recommendations for monitoring strategies. Sequim, Washington,
Battelle Pacific Northwest Laboratories, 230 pp (Report No.
EPA/600/3-86-018).
Andersson K, Levin J-O, & Nilsson C-A (1983) Sampling and analysis of
particulate and gaseous polycyclic aromatic hydrocarbons from coal tar
sources in the working environment. Chemosphere, 12: 197-207.
Andjelkovich DA, Mathew RM, Richardson RB, & Levine RJ (1990)
Mortality of iron foundry workers: I. Overall findings. J Occup Med,
32: 529-540.
Andrews AW, Thibault LH, & Lijinsky W (1978) The relationship between
carcinogenicity and mutagenicity of some polynuclear hydrocarbons.
Mutat Res, 51: 311-318.
Andrews FJ, Halliday GM, & Muller HK (1991) A role for prostaglandins
in the suppression of cutaneous cellular immmunity and tumor
development in benzo(a)pyrene- but not
dimethylbenz(a)anthracene-treated mice. Clin Exp Immunol, 85: 9-13.
Andrianova MM (1971) Transplacental action of 3-methylcholanthrene and
benz [a]pyrene on four generations of mice. Bull Exp Biol Med, 71:
677-680.
Anziulewicz JA, Dick HJ, & Chiarulli EE (1959) Transplacental
naphthalene poisoning. Am J Obstet Gynecol, 9: 519-521.
Aoyama T, Gonzalez FJ, & Gelboin HV (1989) Human cDNA-expressed
cytochrome P450 1A2: Mutagen activation and substrate specificity. Mol
Carcinog, 2: 192-198.
Arce GT, Allen JW, Doerr CL, Elmore E, Hatch GG, Moore MM, Sharief Y,
Grunberger D, & Nesnow S (1987) Relationships between benzo(a)pyrene-
DNA adduct levels and genotoxic effects in mammalian cells. Cancer
Res, 47: 3388-3395.
Arey J, Zielinska B, Atkinson R, & Winer AM (1987) Polycyclic aromatic
hydrocarbon and nitroarene concentrations in ambient air during a
wintertime high-NOx episode in the Los Angeles basin. Atmos Environ,
21: 1437-1444.
Arey J, Zielinska B, Atkinson R, & Winer AM (1991) Analysis of PAH and
their nitroderivates in ambient air. In: Cooke M, Loening K, & Merritt
J ed. Polynuclear aromatic hydrocarbons: Measurement, means, and
metabolism. Columbus, Ohio, Battelle Press, pp 51-67.
Armstrong B, Tremblay C, Baris D, & Thériault G (1994) Lung cancer
mortality and polynuclear aromatic hydrocarbons: A case-cohort study
of aluminum production workers in Arvida, Québec, Canada. Am J
Epidemiol, 139: 250-262.
Ashby J & Kilbey B (1981) Summary report on the performance of
bacterial repair, phage induction, degranulation, and nuclear
enlargement assays. In: de Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North Holland, pp 33-48
(Progress in Mutation Research, Volume 1).
Assennato G, Ferri GM, Foà V, Strickland P, Poirier M, Pozzoli L, &
Cottica D (1993a) Correlation between PAH airborne concentration and
PAH-DNA adducts levels in coke-oven workers. Int Arch Occup Environ
Health, 65: S143-S145.
Assennato G, Ferri GM, Tockman MS, Poirier MC, Schoket B, Porro A,
Corrado V, & Strickland PT (1993b) Biomarkers of carcinogen exposure
and cancer risk in a coke plant. Environ Health Perspectives, 99: 237-
239.
Association of Rhine and Meuse Water Supply Companies (1976) Annual
Report, Part A, The Rhine, Amsterdam, 82 pp.
Association of Rhine and Meuse Water Supply Companies (1977) Annual
Report, Part A, The Rhine, Amsterdam, 54 pp.
Association of Rhine and Meuse Water Supply Companies (1978) Annual
Report, Part A, The Rhine, Amsterdam, 55 pp.
Association of Rhine and Meuse Water Supply Companies (1979) Annual
Report, Part A, The Rhine, Amsterdam, 60 pp.
Association of Rhine and Meuse Water Supply Companies (1987-88) Annual
Report, Part A, The Rhine, Amsterdam, 164 pp.
Association of Rhine and Meuse Water Supply Companies (1989) Annual
Report, Part A, The Rhine, Amsterdam, 108 pp.
Association of Rhine and Meuse Water Supply Companies (1990) Annual
Report, Part A, The Rhine, Amsterdam, 92 pp.
Atkinson R (1987) Structure-activity relationship for the estimation
of rate constants for the gas-phase reactions of OH radicals with
organic compounds. Int J Chem Kinet, 19: 799-828.
Atkinson, R (1989) Kinetics and mechanisms of the gas-phase reactions
of the hydroxyl radical with organic compounds (26) naphthalene. J
Phys Chem Ref Data Monogr, 1: 237.
Atkinson R & Arey J (1994) Atmospheric chemistry of gas-phase
polycylic aromatic hydrocarbons: Formation of atmospheric mutagens.
Environ Health Perspectives, 102 (suppl 4): 117-126.
Atkinson R & Carter WPL (1984) Kinetics and mechanisms of gas-phase
ozone reaction with organic compounds under atmospheric conditions.
Chem Rev, 84: 437-470.
Atkinson R, Arey J, Zielinska B, & Winer AM (1991) Atmospheric loss
processes for PAH and formation of nitroarenes. In: Cooke M, Loening
K, & Merritt J ed. Polynuclear aromatic hydrocarbons: Measurement,
means, and metabolism. Columbus, Ohio, Batelle Press, pp 69-88.
Autrup H (1979) Separation of water-soluble metabolites of
benzo [a]pyrene formed by cultured human colon. Biochem Pharmacol,
28: 1727-1730.
Autrup JL & Autrup H (1986) Metabolism of tobacco specific carcinogens
in cultured rat buccal mucosa epithelial cells. Acta Pharmacol
Toxicol, 59: 339-344.
Autrup H & Harris CC (1983) Metabolism of chemical carcinogens by
human tissues. In: Harris CC & Autrup H ed. Human carcinogenesis. New
York, Academic Press, pp 169-194.
Autrup H, Harris CC, Stoner GD, Selkirk JK, Schafer PW, & Trump B
(1978a) Metabolism of [3H]benzo [a]pyrene by cultured human bronchus
and cultured human pulmonary alveolar macrophages. Lab Invest, 38:
217-224.
Autrup H, Harris CC, Trump B, & Jeffrey AM (1978b) Metabolism of
benzo [a]pyrene and identification of the major benzo [a]pyrene-DNA
adducts in cultured human colon. Cancer Res, 38: 3689-3696.
Autrup H, Harris CC, Schafer PW, Trump B, Stoner GD, & Hsu IC (1979)
Uptake of benzo [a]pyrene-ferric oxide particulates by human
pulmonary macrophages and release of benzo [a]pyrene and its
metabolites. Proc Soc Exp Biol Med, 161: 280-284.
Autrup H, Wefald FC, Jeffrey AM, Tate H, Schwartz RD, Trump B, &
Harris CC (1980) Metabolism of benzo [a]pyrene by cultured
tracheobronchial tissues from mice, rats, hamsters, bovines and
humans. Int J Cancer, 25: 293-300.
Awogi T & Sato T (1989) Micronucleus test with benzo [a]pyrene using
a single peroral administration and intraperitoneal injection in males
of the MS/Ae and CDl-1 mouse strains. Mutat Res, 223: 353-356.
Ayrton AD, McFarlane M, Walker R, Neville S, & Ioannides C (1990) The
induction of P450 I proteins by aromatic amines may be related to
their carcinogenic potential. Carcinogenesis, 11: 803-810.
Babich H, Sardana MK, & Borenfreund E (1988) Acute cytotoxicities of
polynuclear aromatic hydrocarbons determined in vitro with the human
liver tumor cell line, HepG2. Cell Biol Toxicol, 4: 295-309.
Babson JR, Russo-Rodriguez SE, Rastetter WR, & Wogan GG (1986a)
In vitro DNA-binding of microsomally-activated fluoranthene:
Evidence that the major product is a fluoranthrene N2-deoxyguanosine
adduct. Carcinogenesis, 7: 859-865.
Babson JR, Russo-Rodriguez SE, Wattley RV, Bergstein PL, Rastetter WH,
Liber HL, Andon BM, Thilly WG, & Wogan GN (1986b) Microsomal
activation of fluoranthene to mutagenic metabolites. Toxicol Appl
Pharmacol, 85: 355-366.
Bachmann S, Stolz W, Kantor W, & Kuhnt G (1994) [Texte 6:
Implementation of the soil information system for a survey of forest
soils in Germany.] Berlin, Ministry of the Environment, 445 pp (UBA
Report No. 93-147) (in German)
Backer JM & Weinstein IB (1980) Mitochondrial DNA is a major cellular
target for a dihydrodiol-epoxide derivative of benzo [a]pyrene.
Science, 209: 297-299.
Badger GM, Cook JW, Hewett CL, Kennaway EL, Kennaway NM, Martin RH, &
Robinson AM (1940) The production of cancer by pure hydrocarbons. Part
V. Proc R Soc Med, 129: 439-474.
Badger GM, Cook JW, Hewett CL, Kennaway EL, Kennaway NM, & Martin RH
(1942) The production of cancer by pure hydrocarbons. VI. Proc R Soc
Lond, 131: 170-182.
Baek SO, Field RA, Goldstone ME, Kirk PW, Lester JN, & Perry R (1991)
A review of atmospheric polycyclic aromatic hydrocarbons: Sources,
fate and behavior. Water Air Soil Pollut, 60: 279-300.
Baek SO, Goldstone ME, Kirk PWW, Lester JN, & Perry R (1992)
Concentrations of particulate and gaseous polycyclic aromatic
hydrocarbons in London air following a reduction in the lead content
of petrol in the United Kingdom. Sci Total Environ, 111: 169-199.
Bagg J, Smith D, & Maher WA (1981) Distribution of polycyclic aromatic
hydrocarbons in sediments from estuaries of south-eastern Australia.
Aust J Mar Freshwater Res, 32: 65-73.
Bailey GS, Goeger DE, & Hendricks J (1989) Factors influencing
experimental carcinogenesis in laboratory fish models. In: Varanasi U
ed. Metabolism of polycyclic aromatic hydrocarbons in the aquatic
environment. Boca Raton, Florida, CRC Press, pp 253-268.
Baird WM & Diamond L (1978) Metabolism and DNA binding of polycyclic
aromatic hydrocarbons by human diploid fibroblasts. Int J Cancer, 22:
189-195.
Baird WM, Salmon CP, & Diamond L (1984) Benzo(e)pyrene-induced
alterations in the metabolic activation of benzo(a)pyrene and
7,12-dimethylbenz(a)anthracene by hamster embryo cells. Cancer Res,
44: 1445-1452.
Baker JE & Eisenreich SJ (1990) Concentrations and fluxes of
polycyclic aromatic hydrocarbons and polychlorinated biphenyls across
the air-water interface of Lake Superior. Environ Sci Technol, 24:
342-352.
Baker RSU, Bonin AM, Stupans I, & Holder GM (1980) Comparison of rat
and guinea pig as sources of the S9 fraction in the
Salmonella/mammalian microsome mutagenicity test. Mutat Res, 71: 43-
52.
Bakke J, Struble C, Gustafsson JA, & Gustafsson B (1983) Enterohepatic
circulation and catabolism of mercapturic acid pathway metabolites of
naphthalene. In: Rydstrom J, Montelius J, & Bengtsson M. ed.
Extrahepatic drug metabolism and chemical carcinogenesis. Amsterdam,
Elsevier Science Publishers, pp 257-266.
Baranova LN, Shendrikova IA, Aleksandrov VA, Likhachev AY,
Ivanov-Galitsin MN, Dikun PP, & Napalkov NP (1976) [On the mechanism
of penetration of carcinogenic polycyclic hydrocarbons through the
placenta of rats and mice.] Dokl Akad Nauk SSSR, 228: 733-736 (in
Russian).
Barat F (1991) [Measures for clean air at the workplace.]
Staub-Reinhalt Luft, 51: 243-246 (in German).
Barbieri O, Ognio E, Rossi O, Astigiano S, & Rossi L (1986)
Embryotoxicity of benzo(a)pyrene and some of its synthetic derivatives
in Swiss mice. Cancer Res, 46: 94-98.
Barfknecht TR, Andon BM, Thilly WG, & Hites RA (1981) Soot and
mutation in bacteria and human cells. In: Cooke M & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemical analysis and biological
fate. Columbus, Ohio, Battelle Press, pp 231-242.
Barfknecht TR, Hites RA, Cavaliers EL, & Thilly WG (1982) Human cell
mutagenicity of polycyclic aromatic hydrocarbon components of diesel
emissions. Dev Toxicol Environ Sci, 10: 277-294.
Barnsley EA (1975) The bacterial degradation of fluoranthene and
benzo [a]pyrene. Can J Microbiol, 21: 1004-1008.
Barratt RW & Tatum EL (1958) Carcinogenic mutagens. Ann NY Acad Sci,
71: 1072-1084.
Barrows ME, Petrocelli SR, Macek KJ, & Carroll JJ (1980)
Bioconcentration and elimination of selected water pollutants by
bluegill sunfish Lepomis macrochirus. In: Dynamic, exposure, hazard
assessment of toxic chemicals. Michigan, Ann Arbor Science Publishers,
pp 379-392.
Barry G & Cook CW (1934) A comparison of the action of some polycyclic
aromatic hydrocarbons in producing tumours of connective tissue. Am J
Cancer, 20: 58-69.
Barry G, Cook JW, Haslewood AD, Hewett CL, Hieger I, & Kennaway EL
(1935) The production of cancer by pure hydrocarbons. Part III. Proc R
Soc Med, 117: 318-351.
Bartczak AW, Sangaiah S, Ball LM, Warren SH, & Gold A (1987) Synthesis
and bacterial mutagenicity of the cyclopenta oxides of the four
cyclopenta-fused isomers of benzanthracene. Mutagenesis, 2: 101-105.
Bartle KD (1985) Recent advances in the analysis of polycyclic
aromatic compounds by gas chromatography. In: Bjorseth A & Ramdahl T
ed. Handbook of polycyclic aromatic hydrocarbons. Volume 2. Emission
sources and recent progress in analytical chemistry. New York, Marcel
Dekker, pp 193-236.
Bartosek I, Guaitani A, Modica R, Fiume M, & Urso R (1984) Comparative
kinetics of oral benz(a)anthracene, chrysene und triphenylene in rats:
Study with hydrocarbon mixtures. Toxicol Lett, 23: 333-339.
Bartsch H, Malaveille C, Camus AM, Martel-Planche G, Brun G,
Hautefeuille A, Sabadie N, Barbin A, Kuroki T, Drevon C, Piccoli C, &
Montesano R (1980) Validation and comparative studies on 180 chemicals
with S. typhimurium strains and V79 Chinese hamster cells in the
presence of various metabolizing systems. Mutat Res, 76: 1-50.
Basler A, Herbold B, Peter S, & Röhrborn G (1977) Mutagenicity of
polycyclic hydrocarbons. II. Monitoring genetical hazards of chrysene
in vitro and in vivo. Mutat Res, 48: 249-254.
Bastian MV & Toetz DW (1982) Effect of eight polynuclear hydrocarbons
on growth of Anabaena flos aquae. Bull Environ Contam Toxicol, 29:
531-538.
Bastian MV & Toetz DW (1985) Effect of polynuclear hydrocarbons on
algal nitrogen fixation (acetylene reduction). Bull Environ Contam
Toxicol, 35: 258-265.
Basu DK & Saxena J (1978a) Monitoring of polycyclic aromatic
hydrocarbons in water. II. Extraction and recovery of six
representative compounds with polyurethane foams. Environ Sci Technol,
12: 791-795.
Basu DK & Saxena J (1978b) Polynuclear aromatic hydrocarbons in
selected US drinking water and their raw water sources. Environ Sci
Technol, 12: 795-798.
Basu DK, Saxena J, Stoss FW, Santodonato J, Neal MW, & Kopfler FC
(1987) Comparison of drinking water mutagenicity with leaching of
polycyclic aromatic hydrocarbons from water distribution pipes.
Chemosphere, 16: 2595-2612.
Batiste-Alentorn M, Xamena N, Creus A, & Marcos R (1991) Genotoxicity
studies with the unstable zeste-white (UZ) system of Drosophila
melanogaster: Results with ten carcinogenic compounds. Environ Mol
Mutag, 18: 120-125.
Bauer L, Gräf W, & Mueller LG (1985) The phototoxic effect of
polycyclic aromatic hydrocarbons (PAH) on human fibroblastic cultures.
Zentralbl Bakteriol Hyg I Abt Orig B, 181: 281-294.
Bauer E, Guo Z, Ueng Y-F, Bell LC, Zeldin D, & Guengerich FP (1995)
Oxidation of benzo [a]pyrene by recombinant human cytochrome P450
enzymes. Chem Res Toxicol, 8: 136-142.
Baumung H, Huber L, & Kalbfus W (1985) [Testing of selected components
of wastewater from filling stations and fuel storage depots.] Erdöl
Kohle Erdgas Petrochem, 38: 558-559 (in German).
Bayer U (1978) In vivo induction of sister chromatid exchanges by
three polyaromatic hydrocarbons. Carcinogenesis, 3: 423-428.
Bayona JM, Fernandez P, Porte C, Tolosa I, Valls M, & Albaiges J
(1991) Partitioning of urban wastewater organic microcontaminants
among coastal compartments. Chemosphere, 23: 313-326.
Beach AC & Gupta RC (1991) Analysis of cyclopenta(CP)-fused and
'pseudo-CP' polycyclic aromatic hydrocarbon (PAH)-DNA adducts by
32P-postlabeling. Proc Am Assoc Cancer Res, 32: 98.
Beach AC & Gupta RC (1992) Human biomonitoring and the 32
P-postlabeling assay. Carcinogenesis, 13: 1053-1074.
Beach AC & Gupta RC (1994) DNA adducts of the ubiquitous contaminant
cyclopenta (cd)pyrene. Carcinogenesis, 15: 1065-1072.
Beach AC, Agarwal SC, Lamberg GR, Nesnow S, & Gupta RC (1993) Reaction
of cyclopenta (c,d)pyrene-3,4-epoxide with DNA and desoxynucleotides.
Carcinogenesis, 14: 767-771.
Becher G & Bjorseth A (1983) Determination of exposure to polycyclic
aromatic hydrocarbons by analysis of human urine. Cancer Lett, 17:
301-311.
Becher G, Haugen A, & Bjorseth A (1984) Multimethod determination of
occupational exposure to polycyclic aromatic hydrocarbons in an
aluminium plant. Carcinogenesis, 5: 647-651.
Behn U, Kaschani DT, Lützke K, Rentel S, & Burk HD (1985) [PAH
sampling systems for industrial plants.] Erdöl Kohle Erdgas Petrochem,
38: 131 (in German).
Behymer TD & Hites RA (1985) Photolysis of polycyclic aromatic
hydrocarbons adsorbed on simulated atmospheric particulates. Environ
Sci Technol, 19: 1004-1006.
Behymer TD & Hites RA (1988) Photolysis of polycyclic aromatic
hydrocarbons adsorbed on fly ash. Environ Sci Technol, 22: 1311-1319.
Beilstein Institute for Organic Chemistry (1993) Data base.
Benzo(c)phenanthrene. Frankfurt, 2 pp.
Bender ME & Huggett RJ (1988) Polynuclear aromatic hydrocarbon
residues in shellfish: Species variations and apparent intraspecific
differences. In: Kaiser HE ed. Cancer growth and progression. Volume
5: Comparative aspects of tumor development. Dordrecht, Kluwer
Academic Publishers, pp 226-234.
Bender MA, Leonard RC, White O Jr, Costantino JP, & Redmond CK (1988)
Chromosomal aberrations and sister-chromatid exchanges in lymphocytes
from coke oven workers. Mutat Res, 206: 11-16.
Benfield JR & Hammond WG (1992) Bronchial and pulmonary carcinogenesis
at focal sites in dogs and hamsters. Cancer Res, 52: 2687-2693.
Beniashvili DSH (1978) [A comparative study on the action of some
carcinogens inducing transplacental blastomogenesis in rabbits.] Vopr
Onkol, 24: 77-83 (in Russian with English abstract).
Benner BA Jr, Gordon GE, & Wise SA (1989) Mobile sources of
atmospheric polycyclic aromatic hydrocarbons: A roadway tunnel study.
Environ Sci Technol, 23: 1269-1278.
Benner BA Jr, Bryner NP, Wise SA, Mulholland GW, Lao RC, & Fingas MF
(1990) Polycyclic aromatic hydrocarbon emissions from the combustion
of crude oil on water. Environ Sci Technol, 24: 1418-1427.
Berbee RPM (1992) PAH in the aquatic environment: Sources and
emissions. Summary. Proceedings of the workshop on polycyclic aromatic
hydrocarbons (PAH), Oslo, 11-13 November 1991 (Report TA-816). Oslo,
State Pollution Control Authority, Norwegian Food Control Authority.
Berenblum I & Haran H (1955) The influence of croton oil and of
polyethylene glycol-400 on carcinogenesis in the forestomach of the
mouse. Cancer Res, 15: 510-516.
Berenblum I & Schoental R (1943) The metabolism of 3,4-benzpyrene in
mice and rats. The isolation of a hydroxy and a quinone derivative and
a consideration of their biological significance. Cancer Res, 3: 145-
150.
Berenblum I & Shubik P (1947) The role of croton oil applications,
associated with a single painting of a carcinogen in tumour induction
of the mouse skin. Br J Cancer, 1: 379-382.
Berglind L (1982) Determination of polycyclic aromatic hydrocarbons in
industrial discharges and other aqueous effluents (Nordic PAH
Project). Oslo, Central Institute for Industrial Research, 21 pp
(Report No. 16).
Bernfeld P & Homburger F (1983) Skin painting studies in Syrian
hamsters. Prog Exp Tumor Res, 26: 128-153.
Best GR, Nabholz JV, Ojasti J, & Crossley DA Jr (1978) Response of
micro arthropod populations to naphthalene in three contrasting
habitats. Pedobiologia, 18: 189-201.
Bhatia AL, Tausch, H & Stehlik G (1987) Mutagenicity of chlorinated
polycyclic aromatic compounds. Ecotoxicol Environ Saf, 14: 48-55.
Bhatt TS & Coombs MM (1990) The carcinogenicity of
cyclopenta [a]phenanthrene and chrysene derivatives in the Sencar
mouse. Polycyclic Aromat Compd, 1: 51-58.
Bhattacharyya KK, Brake PB, Eltom SE, Otto SA, & Jefcoate CR (1995)
Identification of a rat adrenal cytochrome P450 active in polycyclic
hydrocarbon metabolism as rat CYP1B1; demonstration of a unique
tissue-specific pattern of hormonal and aryl hydrocarbon
receptor-linked regulation. J Biol Chem, 270: 11595-11602.
Bieniek G (1994) The presence of 1-naphthol in the urine of industrial
workers exposed to naphthalene. Occup Environ Med, 51: 357-359.
Bieri RH, Hein C, Huggett RJ, Shou P, Slone H, Smith C, & Su C (1986)
Polycyclic aromatic hydrocarbons in surface sediments from the
Elizabeth River subestuary. Int J Environ Anal Chem, 26: 97-113.
Biermann HW, MacLeod H, Atkinson R, Winer AM, & Pitts JN Jr (1985)
Kinetics of the gas-phase reactions of the hydroxyl radical with
naphthalene, phenanthrene and anthracene. Environ Sci Technol, 19:
244-248.
Bingham E & Falk HL (1969) Environmental carcinogens. The modifying
effect of cocarcinogens on the threshold response. Arch Environ
Health, 19: 779-783.
Binková B, Dobiás L, Wolff T, & Srám RJ (1994) 32P-Postlabeling
analysis of DNA adducts in tissues of rats exposed to coke-oven
emissions. Mutat Res, 307: 355-363.
Binková B, Lewtas J, Misková I, Lenrcek J, & Srám R (1995) DNA adducts
and personal air monitoring of carcinogenic polycyclic aromatic
hydrocarbons in an environmentally exposed population. Carcinogenesis,
16: 1037-1046.
Bjorseth A (1979) Determination of polycyclic aromatic hydrocarbons in
sediments and mussels from Saudafjord, west Norway, by glass capillary
gas chromatography. Sci Total Environ, 13: 71-86.
Bjorseth A (1983) Appendix: List of dicyclic and polycyclic aromatic
hydrocarbons, their structure, molecular weight, melting and boiling
points. In: Bjorseth A ed. Handbook of polycyclic aromatic
hydrocarbons. New York, Marcel Dekker, pp 709-718.
Bjorseth A & Becher G (1986) PAH in work atmospheres: Occurrence and
determination. Florida, Boca Raton, CRC Press, pp 57-62.
Bjorseth A & Lunde G (1979) Long-range transport of polycyclic
aromatic hydrocarbons. Atmos Environ, 13: 45-53.
Bjorseth A & Olufsen S (1983) Long transport of polycyclic aromatic
hydrocarbons. In: Bjorseth A ed. Polycyclic aromatic hydrocarbons. New
York, Marcel Dekker, pp 507-524.
Bjorseth A & Ramdahl T (1985) Sources and emissions of PAH. In:
Bjorseth A & Ramdahl T ed. Handbook of polycyclic aromatic
hydrocarbons: Volume 2: Emission sources and recent progress in
analytical chemistry. New York, Marcel Dekker, pp 1-20.
Black JJ, Hart TF Jr, & Evans E (1981) HPLC studies of PAH pollution
in a Michigan trout stream. In: Cooke M & Dennis A ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 343-355.
Black JA, Birge WJ, Westerman AG, & Francis PC (1983) Comparative
aquatic toxicology of aromatic hydrocarbons. Fundam Appl Toxicol, 3:
353-358.
Black JJ, Maccubin AE, & Johnston CJ (1988) Carcinogenicity of
benzo [a]pyrene in rainbow trout resulting from embryo micro
injection. Aquat Toxicol, 13: 297-308.
Blanton RH, Lyte M, Myers MJ, & Bick PH (1986) Immunomodulation by
polyaromatic hydrocarbons in mice and murine cells. Cancer Res, 46:
2735-2739.
Bobra A, Shiu WY, & Mackay (1983) A predictive correlation for the
acute toxicity of hydrocarbons and chlorinated hydrocarbons to the
water flea Daphnia magna. Chemosphere, 12: 1121-1129.
Bock FG (1964) Early effects of hydrocarbons on mammalian skin. Progr
Exp Tumor Res, 4: 126-168.
Bock FG & Dao TL (1961) Factors affecting the polynuclear hydrocarbon
level in rat mammary glands. Cancer Res, 21: 1024-1029.
Bock FG & King DW (1959) A study of the sensitivity of the mouse
forestomach toward certain polycyclic hydrocarbons. J Natl Cancer
Inst, 23: 833-838.
Bock FG & Mund R (1958) A survey of compounds for activity in
suppression of mouse sebaceous glands. Cancer Res, 18: 887-892.
Boehm PD & Fiest DL (1983) Ocean dumping of dredged material in the
New York Bight: Organic chemistry studies. In: Kester DR, Ketchum H,
Bostwick I, & Duedal IW ed. Wastes in the ocean. Volume 2:
Dredged-material disposal in the ocean. New York, John Wiley & Sons,
pp 151-169.
Boldrin B, Tiehm A, & Fritzsche C (1993) Degradation of phenanthrene,
fluorene, fluoranthene, and pyrene by a Mycobacterium sp. Appl
Environ Microbiol, 59: 1927-1930.
Bolling H (1964) [Carcinogenic substances in cereals dried by
combustion gas.] Tech Monit Pinerolo, 15: 137-142 (in Italian).
Bolonova LN (1967) [Action of naphthalene and its methyl derivatives
on the ammonia content in rat brain.] Farmakol Toksikol, 30: 484-486
(in Russian).
Boney AD (1974) Aromatic hydrocarbons and the growth of marine algae.
Mar Pollut Bull, 5: 185-186.
Boney AD & Corner EDS (1962) On the effects of some carcinogenic
hydrocarbons on the growth of sporelings of marine red algae. J Mar
Biol Assoc, 42: 579-585.
Bonfanti L, Cioni M, Belli R, & Cappiello A (1988) Determination of
trace organic compounds in effluents from a coal-fired power plant.
Biomed Mass Spectrom, 16: 175-178.
Boogaard PJ & van Sittert NJ (1994) Exposure to polycyclic aromatic
hydrocarbons in petrochemical industries by measurement of urinary
1-hydroxypyrene. Occup Environ Med, 51: 250-258.
Boogaard PJ & van Sittert NJ (1995) Urinary 1-hydroxypyrene as
biomarker of exposure to polycyclic aromatic hydrocarbons in workers
in petrochemical industries: Baseline values and dermal uptake. Sci
Total Environ, 163: 203-209.
Boom A & Marsalek J (1988) Accumulation of polycyclic aromatic
hydrocarbons (PAHs) in an urban snowpack. Sci Total Environ, 74: 133-
148.
Boom MM (1987) The determination of polycyclic aromatic hydrocarbons
in indigenous and transplanted mussels (Mytilus
edulis L) along the Dutch coast. Int J Environ Anal Chem, 31: 251-
261.
Booth J & Boyland E (1949) Metabolism of polycyclic compounds. 5.
Formation of 1,2-dihydroxy-1,2-dihydronaphthalene. Biochem J, 44: 361-
365.
Booth J, Keysell GR, Pal K, & Sims P (1974) The metabolism of
polycyclic hydrocarbons by cultured human lymphocytes. FEBS Lett, 43:
341-344.
Borchert J & Westendorf J (1994) [Kinetics of uptake and metabolism of
sediment-bound benzo(a)pyrene in sediment-inhabiting invertebrates.]
Ber Zent Meeres Klimaforsch, 7: 75-55 (in German).
Borneff J & Kunte H (1964) [Carcinogenic substances in water and soil.
XVI. Determination of polycyclic aromatics in water samples by direct
extraction.] Arch Hyg, 148: 585-597 (in German).
Borneff J & Kunte H (1965) [Carcinogenic substances in water and soil.
XVII. Source and evaluation of polycyclic aromatic hydrocarbons in
water.] Arch Hyg, 149: 226-243 (in German).
Borneff J & Kunte H (1979) Method 1. Analysis of polycyclic aromatic
hydrocarbons in water using thin layer chromatography and
spectrofluorometry. In: Egan H, Castegnaro M, Bogovski P, Kunte H, &
Walker EA ed. Environmental carcinogens: Selected methods of analysis.
Volume 3: Analysis of polycyclic aromatic hydrocarbons in
environmental samples. Lyon, International Agency for Research on
Cancer, pp 129-139 (IARC Scientific Publications No. 29).
Borneff J & Kunte H (1983) Polycyclic aromatic hydrocarbons in river
and lake water, biota, and sediments. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 629-652.
Boroujerdi M, Kung HC, Wilson AGE, & Anderson MW (1981) Metabolism and
DNA binding of benzo [a]pyrene in vivo in the rat. Cancer Res, 41:
951-957.
Borovsky D, Linley JR, & Kagan J (1987) Polycyclic aromatic compounds
as phototoxic mosquito larvicides. J Am Mosq Control Assoc, 3: 246-
250.
Bos RP, Prinsen WJC, van Rooy JGM, Jongeneelen FJ, Theuws JLG, &
Henderson PT (1987) Fluoranthene, a volatile mutagenic compound,
present in creosote and coal tar. Mutat Res, 187: 119-125.
Bos RP, Theuws JLG, Jongeneelen FJ, & Henderson PT (1988) Mutagenicity
of bi-,tri- and tetra-cyclic aromatic hydrocarbons in the 'taped-plate
assay' and in the conventional Salmonella
mutagenicity assay. Mutat Res, 204: 203-206.
Bossert ID & Bartha R (1986) Structure-biodegradability relationships
of polycyclic aromatic hydrocarbons in soil. Bull Environ Contam
Toxicol, 37: 490-495.
Bossert I, Kachel WM, & Bartha R (1984) Fate of hydrocarbons during
oily sludge disposal in soil. Appl Environ Microbiol, 47: 763-767.
Bottomley AC & Twort CC (1934) The carcinogenicity of chrysene and
oleic acid. Am J Cancer, 21: 781-786.
Bourcart J & Mallet L (1965) [Coastal marine pollution in the central
region of the Tyrrhenian Sea (Bay of Naples) by polyaromatic
hydrocarbons of the benzo-3,4-pyrene type.] C R Acad Sci Paris, 260:
3729-3734 (in French).
Bourguet CC, Checkoway H, & Hulka BS (1987) A case-control study of
skin cancer in the tire and rubber manufacturing industry. Am J Ind
Med, 11: 461-473.
Bourne MC & Young L (1934) The metabolism of naphthalene in rabbits.
Biochem J, 28: 803-808.
Bowling J, Leversee G, Landrum PF, & Giesy JP (1983) Acute mortality
of anthracene-contaminated fish exposed to sunlight. Aquat Toxicol, 3
:79-90.
Bowmer CT, Roza P, Henzen L, & Degeling C (1993) The development of
chronic toxicological tests for PAH contaminated soils using the
earthworm Eisenia fetida and the springtail Folsomia candida.
Delft, TNO Institute of Environmental Sciences, 49 pp (TNO Report No.
IMW-R 92/387).
Boyland E & Burrows H (1935) The experimental production of sarcoma in
rats and mice by a colloidal aqueous solution of
1:2:5:6-dibenzanthracene. J Pathol Bacteriol, 41: 231-238.
Boyland E & Levi AA (1935) Metabolism of polycyclic compounds. I.
Production of dihydroxydihydroanthracene from anthracene. Biochem J,
29: 2679-2683.
Boyland E & Levi AA (1936a) Metabolism of polycyclic compounds. II.
Production of dihydroxydihydroanthracene glycuronic acid from
anthracene. Biochem J, 30: 728-731.
Boyland E & Levi AA (1936b) Metabolism of polycyclic compounds. III.
Anthrylmercapturic acid. Biochem J, 30: 1225-1227.
Boyland E & Sims P (1958) Metabolism of polycyclic compounds. 12. An
acid-labile precursor of 1-naphthylmercapturic acid and naphthol: An
N-acetyl-S-(1,2-dihydro-hydroxynaphthyl) L-cysteine. Biochem J, 68:
440-447.
Boyland E & Sims P (1962a) Metabolism of polycyclic compounds. 20. The
metabolism of phenanthrene in rabbits and rats: Mercapturic acids and
related compounds. Biochem J, 84: 564-570.
Boyland E & Sims P (1962b) Metabolism of polycyclic compounds. 21. The
metabolism of phenanthrene in rabbits and rats: Dihydrodihydroxy
compounds and related glucosiduronic acids. Biochem J, 84: 571-582.
Boyland E & Sims P (1964a) Metabolism of polycyclic compounds. 23. The
metabolism of pyrene in rats and rabbits. Biochem J, 90: 391-398.
Boyland E & Sims P (1964b) Metabolism of polycyclic compounds. 24. The
metabolism of benz [a]anthracene. Biochem J, 91: 493-506.
Boyland E & Sims P (1967) The carcinogenic activities in mice of
compounds related to benz [a]anthracene. Int J Cancer, 2: 500-504.
Boyland E & Weigert F (1947) Metabolism of carcinogenic compounds. Br
Med Bull, 4: 354-359.
Boyland E & Wolf G (1950) Metabolism of polycyclic compounds. 6.
Conversion of phenanthrene into dihydroxydihydrophenanthrene. Biochem
J, 47: 64-69.
Boyland E, Levi AA, Mawson EH, & Roe E (1941) Metabolism of polycyclic
compounds. 4. Production of a dihydroxy-1,2,5,6-dibenzanthracene.
Biochem J, 35: 184-191.
Boyland E, Kimura M, & Sims P (1964) Metabolism of polycyclic
compounds. 26. The hydroxylation of some aromatic hydrocarbons by the
ascorbic acid model hydroxylating system and by rat liver microsomes.
Biochem J, 92: 631-638.
Bozicevic Z, Cvitas T, Curic M, Klasinc L, & Pecina P (1987) Airborne
polycyclic aromatic hydrocarbons in the city of Zagreb, Yugoslavia.
Sci Total Environ, 66: 127-136.
Brandt-Rauf PW, Smith S, Perera FP, Niman HL, Yohannan W, Hemminki K,
& Santella RM (1990) Serum oncogene proteins in foundry workers. J Soc
Occup Med, 40: 11-14.
Brasser LJ (1980) [Polycyclic aromatic hydrocarbon concentration in
the Netherlands. Air pollution from polycyclic aromatic hydrocarbons:
Collection and evaluation.] Düsseldorf, VDI-Verlag GmbH, pp 171-180
(VDI Report No. 358) (in German).
Bresch H, Spielhoff R, Mohr U, & Barkemeyer H (1972) Use of the sea
urchin egg for quick screen testing of the biological activities of
substances. I. Influence of fractions of a tobacco smoke condensate on
early development. Proc Soc Exp Biol Med, 141: 747-752.
Bridges BA, Zeiger E, & McGregor DB (1981) Summary report on the
performance of bacterial mutation assays. In: de Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens. Report of the
international collaborative programme. New York, Elsevier North
Holland, pp 49-67 (Progress in Mutation Research, Volume 1).
Brockhaus A & Tomingas R (1976) [Emission of polycyclic hydrocarbons
during burning processes in small heating installations and their
concentration in the atmosphere.] StaubReinhalt Luft, 36: 96-101 (in
German).
Broddin G, Van Vaeck L, & Van Cauwenberghe K (1977) On the size
distribution of polycyclic aromatic hydrocarbon containing particles
from a coke oven emission source. Atmos Environ, 11: 1061-1064.
Broman D, Colmjö A, & Näf C (1987) Characterization of the PAC profile
in settling particulates from the urban waters of Stockholm. Bull
Environ Contam Toxicol, 38: 1020-1028.
Broman D, Naf C, Rolff C, & Zebuhr Y (1990) Occurrence and dynamics of
polychlorinated dibenzo-p-dioxins and dibenzofurans and polycyclic
aromatic hydrocarbons in the mixed surface layer of remote coastal and
offshore water of the Baltic. Environ Sci Technol, 25: 1850-1864.
Brooke DN, Dobbs AJ & Williams N (1986) Octanol:water partition
coefficients (P): Measurement, estimation and interpretation,
particularly for chemicals with P > 105. Ecotoxicol Environ Saf, 11:
251-260.
Brookes P & Lawley PD (1964) Evidence of the binding of polynuclear
aromatic hydrocarbons to the nucleic acids of mouse skin: Relation
between carcinogenic power of hydrocarbons and their binding to
deoxyribonucleic acid. Nature, 202: 781-784.
Brookes P, Ellis MV, Pataki J, & Harvey RG (1986) Mutation in
mammalian cells by isomers of 5-methylchrysene diolepoxide.
Carcinogenesis, 7: 463-466.
Bruce WR & Heddle JA (1979) The mutagenic activity of 61 agents as
determined by the micronucleus, Salmonella, and sperm abnormality
assays. Can J Genet Cytol, 21: 319-334.
Bruggeman WA, Van der Steen J, & Hutzinger O (1982) Reversed-phase
thin-layer chromatography of polynuclear aromatic hydrocarbons and
chlorinated biphenyls. Relationship with hydrophobicity as measured by
aqueous solubility and octanol-water partition coefficient. J
Chromatogr, 238: 335-346
Brune K, Kalin H, Schmidt R, & Hecker E (1978) Inflammatory, tumor
initiating and promoting activities of polycyclic aromatic
hydrocarbons and diterpene esters in mouse skin as compared with their
prostaglandin releasing potency in vitro. Cancer Lett, 4: 333-342.
Brune H, Deutsch-Wenzel RP, Habs M, Ivankovic S, & Schmähl D (1981)
Investigation of the tumorigenic response to benzo [a]pyrene in
aqeous caffeine solution applied orally to Sprague-Dawley rats. J
Cancer Res Clin Oncol, 102: 153-157.
Brunström B, Hakansson H, & Lundberg K (1991) Effects of a technical
PCB preparation and fractions thereof on ethoxyresorufin O-deethylase
activity, vitamin A levels and thymic development in the mink Mustela
vison. Pharmacol Toxicol, 69: 421-426.
Bryan WR & Shimkin MB (1943) Quantitative analysis of dose-response
data obtained with three carcinogenic hydrocarbons in strain C3H male
mice. J Natl Cancer Inst, 3: 503-531.
Bryant MF, Erexson GL, Kwanyuen P, Atwater AL, & Kligerman AD (1990)
Sister chromatid exchange and micronucleus analyses in rat peripheral
blood lymphocytes following in vivo exposure to
dibenz(a,h)anthracene (Abstract 30). Environ Mol Mutag, 15 (suppl 17):
10-11.
Bryant MF, Kwanyuen P, Atwater AL, Erexson GL, & Kligerman AD (1991)
Cytogenetic effects of benzo-b-fluoranthene in Sprague-Dawley rat
peripheral blood lymphocytes after in vivo
exposure (Abstract 33). Environ Mol Mutag, 17 (suppl 19): 13.
Bryla P & Weyand EH (1991) Role of activated oxygen species in
benzo [a]pyrene:DNA adduct formation in vitro. Free Radicals Biol
Med, 11: 17-24.
Buccafusco RJ, Ells SJ, & LeBlanc GA (1981) Acute toxicity of priority
pollutants to bluegill (Lepomis macrochirus). Bull Environ Contam
Toxicol, 26: 446-452.
Buchet JP, Gennart JP, Mercado-Calderon F, Delavignette JP, Cupers L,
& Lauwerys R (1992) Evaluation of exposure to polycyclic aromatic
hydrocarbons in a coke production and a graphite electrode
manufacturing plant: Assessment of urinary excretion of
1-hydroxypyrene as a biological indicator of exposure. Br J Ind Med,
49: 761-768.
Buck M (1983) [Immission measurements of polycyclic aromatic
hydrocarbons (PAH) in the Rhein-Ruhr-Gebiet.] Schriftenr Landesanst
Immissionsschutz Landes Nordrhein-Westfalen. 57: 37-45 (in German).
Buck M (1991) [Methods and results of the measurement of carcinogenic
polycyclic aromatic hydrocarbons.] In: [Carcinogenic compounds in the
environment: Sources, measurement, risk, reduction.] Dusseldorf,
VDI-Verlag, pp 49-70 (VDI Report No. 888) (in German).
Buck M, Ixfeld H, & Ellermann K (1989) [Report on the results of
discontinuous sulfur dioxide and multiple component measurements taken
in the Rhine-Ruhr area in the period from 1 January to 31 December
1988.] In: Summaries of reports, 1988. A series from the State of
North-Rhine Westfalia Pollution Control Office. Düsseldorf, Cornelsen
Verlag Schwann-Girardet, pp 69-130 (in German with English summary)
Buckley TJ & Lioy PJ (1992) An examination of the time course from
human dietary exposure to polycyclic aromatic hydrocarbons to urinary
elimination of 1-hydroxypyrene. Br J Ind Med, 49: 113-124.
Buckpitt AR & Franklin RB (1989) Relationship of naphthalene and
2-methylnaphthalene metabolism to pulmonary bronchiolar epithelial
cell necrosis. Pharmacol Ther, 41: 393-410.
Budavari S, O'Neil MJ, Smith A, & Heckelman PE (1989) The Merck Index:
An encyclopedia of chemicals, drugs, and biologicals, 11th ed. Rahway,
New Jersey, Merck & Co., pp 108, 350, 650, 1008, 1139, 1266.
Buening MK, Levin W, Karle JM, Yagi H, Jerina DM, & Conney AH (1979)
Tumorigenicity of bay-region epoxides and other derivatives of
chrysene and phenanthrene in newborn mice. Cancer Res, 39: 5063-5068.
Buening MK, Levin W, Wood AW, Chang RL, Lehr RE, Taylor CW, Yagi H,
Jerina DM, & Conney AH (1980) Tumorigenicity activity of
benzo(e)pyrene derivatives on mouse skin and in newborn mice. Cancer
Res, 40: 203-206.
Bui QQ, Tran MB, & West WL (1986) A comparative study of the
reproductive effects of methadone and benzo [a]pyrene in the pregnant
and pseudopregnant rat. Toxicology, 42: 195-204.
Bulay OM (1970) The study of development of lung and skin tumors in
mice exposed in utero to polycyclic hydrocarbons. Acta Med Turk, 7: 3-
38.
Bulay OM & Wattenberg LW (1971) Carcinogenic effects of polycyclic
hydrocarbon carcinogen administration to mice during pregnancy on the
progeny. J Natl Cancer Inst, 46: 397-402.
Bulman TL, Lesage S, Fowlie P, & Webber MD (1987) The fate of
polynuclear aromatic hydrocarbons in soil. In: Vandermeulan JH &
Hurley SE ed. Oil in fresh water: chemistry, biology, countermeasure
technology. New York, Pergamon Press, pp 231-251.
Burchiel SW, Hadley WM, Barton SL, Fincher RH, Lauer LD, & Dean JH
(1988) Persistent suppression of humoral immunity produced by
7,12-dimethylbenz [a]anthracene (DMBA) in B6C3F1 mice: Correlation
with changes in spleen cell surface markers detected by flow
cytometry. Int J Immunopharmacol, 10: 369-376.
Burchiel SW, De Davis AP, Gomez MP, Montano RM, Barton SL, & Seamer LC
(1990) Inhibition of lymphocyte activation in splenic and
gut-associated lymphoid tissues followig oral exposure of mice to
7,12-dimethylbenz [a]anthracene. Toxicol Appl Pharmacol, 105:
434-442.
Burchiel SW, Thompson TA, & Davis DP (1991) Alterations in
mitogen-induced calcium mobilization and intracellular free calcium
produced by 7,12-dimethylybenz(a)-anthracene in the Jurkat human T
cell line. Int J Immunopharmacol, 13: 109-115.
Burgaz S, Borm PJA, & Jongeneelen F (1992) Evaluation of urinary
excretion of 1-hydroxypyrene and thioethers in workers exposed to
bitumen fumes. Int Arch Occup Environ Health, 63: 397-401.
Burnham K & Rahman M (1992) Effects of petrochemicals and ultraviolet
radiation on epidermal IA expression in vitro. J Toxicol Environ
Health, 35: 175-185.
Busby WF, Goldman ME, Newberne PM, & Wogan GN (1984) Tumorigenicity of
fluoranthene in a newborn mouse lung adenoma bioassay. Carcinogenesis,
5: 1311-1316.
Busby WF Jr, Stevens EK, Kellenbach ER, Cornelisse J, & Lugtenburg J
(1988) Dose-response relationships of the tumorigenicity of
cyclopenta [c,d]pyrene, benzo [a]pyrene and 6-nitrochrysene in a
newborn mouse lung adenoma bioassay. Carcinogenesis, 9: 741-746.
Busby WF Jr, Stevens EK, Martin CN, Chow FL, & Garner RC (1989)
Comparative lung tumorigenicity of parent and mononitro-polynuclear
aromatic hydrocarbons in the BLU: Ha newborn mouse assay. Toxicol Appl
Pharmacol, 99: 555-563.
Butler JD & Crossley P (1979) An appraisal of relative airborne
suburban concentrations of polycyclic aromatic hydrocarbons monitored
indoors and outdoors. Sci Total Environ, 11: 53-58.
Butler JD & Crossley P (1981) Reactivity of polycyclic aromatic
hydrocarbons adsorbed on soot particles. Atmos Environ, 15: 91-94.
Butler JD & Crossley P (1982) Predicting polycyclic aromatic
hydrocarbon concentrations in urban aerosols by linear multiple
regression analysis. Environ Pollut, B3: 109-123.
Butler JD, Butterworth V, Kellow SC, & Robinson HG (1984) Some
observations on the polycyclic aromatic hydrocarbon (PAH) content of
surface soils in urban areas. Sci Total Environ, 33: 75-85.
Butlin HT (1892) Cancer of the scrotum in chimney-sweeps and others.
Br Med J, ii: 66-71.
Buu-Hoi NP (1964) New developments in chemical carcinogenesis by
polycyclic hydrocarbons and related heterocycles: A review. Cancer
Res, 24: 1511-1523.
Cairns MA & Nebeker AV (1982) Toxicity of acenaphthene and isophorone
to early life stages of fathead minnows. Arch Environ Contam Toxicol,
11: 703-707.
Calder JA & Lader JH (1976) Effect of dissolved aromatic hydrocarbons
on the growth of marine bacteria in batch culture. Appl Environ
Microbiol, 32: 95-101.
Callahan MA, Slimak MW, Gabel NW, May IP, & Fowler CF (1979)
Water-related environmental fate of 129 priority pollutants. Vol II.
Halogenated aliphatic hydrocarbons, halogenated ethers, monocyclic
aromatics, phthalate esters, polycyclic aromatic hydrocarbons,
nitrosamines and miscellaneous compounds Report EPA-440/4-79-029b;
PB80 204381). Washington DC, US Environmental Protection Agency, 673
pp.
Capel PD, Leuenberger C, & Giger W (1991) Hydrophobic organic
chemicals in urban fog. Atmos Environ, 25: 1335-1346.
Carmichael PL, Jacob J, Grimmer G, & Phillips DH (1990) Analysis of
the polycyclic aromatic hydrocarbon content of petrol and diesel
engine lubricating oils and determination of DNA adducts in topically
treated mice by 32P-postlabelling. Carcinogenesis, 11: 2025-2032.
Carmichael PL, Platt KL, Ni She M, Lecoq S, Oesch F, Phillips DH, &
Grover PL (1993) Evidence for the involvement of a bis-diol-epoxide in
the metabolic activation of dibenz [a,h]anthracene to DNA-binding
species in mouse skin. Cancer Res, 53: 944-948.
Carstensen U, Alexandrie AK, Högstedt B, Rannug A, Bratt I, & Hagmar L
(1993) B- and T-lymphocyte micronclei in chimney sweeps with respect
to genetic polymorphism for CIP1A1 and GST1 (class Mu). Mutat Res,
289: 187-195.
Carver JH, Machado ML, & MacGregor JA (1986) Application of modified
Salmonella/microsome prescreen to petroleum-derived complex mixtures
and polynuclear aromatic hydrocarbons (PAH). Mutat Res, 174: 247-253.
Casserly DM, Davis EM, Downs TD, & Guthrie RK (1983) Sorption of
organisms by Selenastrum capricornutum. Water Res, 17: 1591-1594.
Casto BC (1973) Enhancement of adenovirus transformation by treatment
of hamsters with ultraviolet irradiation, DNA base analogs, and
dibenz(a,h)anthracene. Cancer Res, 33: 402-407.
Casto BC (1979) Polycyclic hydrocarbons and Syrian hamster embryo
cells: Cell transformation, enhancement of viral transformation and
analysis of DNA-damage. In: Jones PW & Leber P ed. Polynuclear
aromatic hydrocarbons. Ann Arbor, Michigan, Ann Arbor Science
Publishers, pp 51-66.
Casto BC, Janosko N, & DiPaolo JA (1977) Development of a focus assay
model for transformation of hamster cells in vitro by chemical
carcinogens. Cancer Res, 37: 3508-3515.
Catallo WJ III & Gambrell RP (1987) The effects of high levels of
polycyclic aromatic hydrocarbons on sediment physicochemical
properties and benthic organisms in a polluted stream. Chemosphere,
16: 1053-1064.
Catoggio JA, Succar SD, & Roca AE (1989) Polynuclear aromatic
hydrocarbon content of particulate matter suspended in the atmosphere
of La Plata, Argentina. Sci Total Environ, 79: 43-58.
Cautreels W & van Cauwenberghe K (1977) Comparison between the organic
fraction of suspended matter at a background and an urban station. Sci
Total Environ, 8: 79-88.
Cavalieri E & Rogan E (1985) Role of radical cations in aromatic
hydrocarbon carcinogenesis. Environ Health Perspectives, 64: 69-84.
Cavalieri E, Mailander P, & Pelfrene A (1977) Carcinogenic activity of
anthanthrene on mouse skin. Z Krebsforsch, 89: 113-118.
Cavalieri E, Roth R, Althoff J, Grandjean C, Patil K, Marsh S, &
McLaughlin D (1978) Carcinogenicity and metabolic profiles of
3-methylcholanthrene oxygenated derivatives at the 1 and 2 positions.
Chem-Biol Interactions, 22: 69-81.
Cavalieri E, Rogan E, & Thilly WG (1981a) Carcinogenicity,
mutagenicity and binding studies of the environmental contaminant
cyclopenteno [c,d]pyrene and some of its derivatives. In: Cooke M &
Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemical analysis and
biological fate. Columbus, Ohio, Battelle Press, pp 487-499.
Cavalieri E, Rogan E, Toth B, & Munhall A (1981b) Carcinogenicity of
the environmental pollutants cyclopenteno(c,d)pyrene and
cyclopentano(c,d)pyrene in mouse skin. Carcinogenesis, 2: 277-281.
Cavalieri E, Rogan E, & Sinha D (1988a) Carcinogenicity of aromatic
hydrocarbons directly applied to rat mammary gland. Cancer Res Clin
Oncol, 114: 3-9.
Cavalieri E, Rogan E, Cremonesi P, Higginbotham S, & Salmasi S (1988b)
Tumorigenicity of 6-halogenated derivatives of benzo [a]pyrene in
mouse skin and rat mammary gland. J Cancer Res Clin Oncol, 114: 10-15.
Cavalieri EL, Rogan EG, Higginbotham S, Cremonesi P, & Salmasi S
(1989) Tumor-initiating activity in mouse skin and carcinogenicity in
rat mammary gland of dibenzo[a]pyrenes: The very potent environmental
carcinogen dibenzo(a,l)pyrene. J Cancer Res Clin Oncol, 115: 67-72.
Cavalieri EL, Higginbotham S, Ramakrishna NVS, Devanesan PD, Todorovic
R, Rogan EG, & Salmasi S (1991) Comparative dose-response
tumorigenicity studies of dibenzo [a,l]pyrene versus
7,12-dimethylbenz [a]anthracene, benzo [a]pyrene and two
dibenzo [a,l]pyrene dihydrodiols in mouse skin and rat mammary gland.
Carcinogenesis, 12: 1939-1944.
Cavalieri EL, Rogan EG, Ramakrishna NVS, & Devanesan PD (1993)
Mechanisms of benzo(a)pyrene and 7,12-dimethylbenz(a)anthracene
activation: Qualitative aspects of the stable and depurination DNA
adducts obtained from radical cations and diol epoxides. In: Garrigues
P & Lamotte M ed. Polycyclic aromatic compounds: Synthesis,
properties, analytical measurements, occurrence and biological
effects. Bordeaux, Gordon & Breach Science Publishers, pp 725-732.
Cenni A, Sciarra G, Sartorelli P, & Pappalardo F (1993) Environmental
and biological monitoring of polycyclic aromatic hydrocarbons (PAHs)
in coke plants and other workplaces. Med Lav, 84: 379-386.
Cerniglia CE (1984) Microbial metabolism of polycyclic aromatic
hydrocarbons. In: Cerniglia CE ed. Advances in applied microbiology,
Volume 30. Jefferson, Arkansas, Academic Press, pp 31-71.
Chakraborti D, Van Vaeck L, & Van Espen P (1988) Calcutta pollutants:
Part II. Polynuclear aromatic hydrocarbon and some metal concentration
on air particulates during winter 1984. Int J Environ Anal Chem, 32:
109-120.
Chaloupka K, Santosstefano M, Goldfarb IS, Liu G, Myers MJ, Tsyrolv
IB, Gelboin HV, Krishnan V, & Safe S (1994) Aryl hydrocarbon (ah)
receptor-independent induction of Cyp1a2 gene expression by
acenaphthylene and related compounds in B6C3F1 mice. Carcinogenesis,
15: 2835-2840.
Chang RL, Levin W, Wood AW, Lehr RE, Kumar S, Yagi H, Jerina DM, &
Conney AH (1981) Tumorigenicity of the diastereomeric bay-region
benzo(e)pyrene 9,10-diol-11,12-epoxides in newborn mice. Cancer Res,
41: 915-918.
Chang RL, Levin W, Wood AW, Lehr RE, Kumar S, Yagi H, Jerina DM, &
Conney AH (1982) Tumorigenicity of bay-region diol-epoxides and other
benzo-ring derivatives of dibenzo[a,h]pyrene and dibenzo[a,i]pyrene on
mouse skin and newborn mice. Cancer Res, 42: 25-29.
Chang RL, Levin W, Wood AW, Yagi H, Tada M, Vyas KP, Jerina DM, &
Conney AH (1983) Tumorigenicity of enantiomers of chrysene
1,2-dihydrodiol and of the diastereomeric bay-region chrysene
1,2-diol-3,4-epoxides on mouse skin and in newborn mice. Cancer Res,
43: 192-196.
Chang SC, Chang KT, Keng, YF Lan CF, Hsiao HC, Hsen SH, & Wei YH
(1988) Mutagenicity and polycyclic aromatic hydrocarbons analysis of
airborne particulate matters from Taipei city. Proc Natl Sci Counc
Taiwan, 12: 129-139.
Chappell WR (1989) Interspecific scaling of toxicity data: A question
of interpretation. Risk Anal, 9: 13-14.
Chasseaud LF (1979) The role of glutathione and glutathione
S-transferases in the metabolism of chemical carcinogens and other
electrophilic agents. Adv Cancer Res, 29: 175-274.
Chau N, Bertrand JP, Mur JM, Figueredo A, Patris A, Moulin JJ, & Pham
QT (1993) Mortality in retired coke oven plant workers. Br J Ind Med,
50: 127-135..
Chemical Abstracts Service (1988) Ring systems handbook. Ring Systems
File I RF 1-RF 27595. Columbus, Ohio, American Chemical Society.
Chemical Abstracts Service (1990) Index guide. Part 1, pp. 1-1470;
Part 2, pp 1471-2446. Columbus, Ohio, American Chemical Society.
Chen TT & Heidelberger C (1969) Quantitative studies on the malignant
transformation of mouse prostate cells by carcinogenic hydrocarbons
in vitro. Int J Cancer, 4: 166-178.
Chen PHS, Shieh HH, & Gaw JM (1980) Determination of polycyclic
aromatic hydrocarbons in airborne particulates at various sites in
Taipei city by GC/MS and glass capillary GC. Proc Natl Sci Counc
Taiwan, 4: 280-284.
Chen PHS, Chuang CY Lu YD, & Chung KT (1981) Gas chromatography/mass
spectrometric determination of polycyclic aromatic hydrocarbons in
airborne particulates at various sites in Kaohsiung area. Proc Natl
Sci Counc Taiwan, 5: 262-267.
Chiazze L Jr, Watkins DK, & Amsel J (1991) Asphalt and risk of cancer
in man. Br J Ind Med, 48: 538-542.
Chipman JK (1982) Bile as a source of potential reactive metabolites.
Toxicology, 25: 99-111.
Chipman JK, Hirom PC, Frost GS, & Millburn P (1981) The biliary
excretion and enterohepatic circulation of benzo [a]pyrene and its
metabolites in the rat. Biochem Pharmacol, 30: 937-944.
Chiu CH, Howes PS, Li KC, Poole GK, & Lao RC (1991) The determination
and measurement of PAH in municipal incinerator samples. In: Cooke M,
Loening K, & Merritt J ed. Polynuclear aromatic hydrocarbons:
Measurement, means, and metabolism. Columbus, Ohio, Battelle Press, pp
195-211.
Chladek E & Marano RS (1984) Use of bonded phase silica sorbents for
the sampling of priority pollutants in wastewaters. J Chromatogr Sci,
22: 313-320.
Chorazy M, Szeliga J, Strozyk M, & Cimander B (1994) Ambient air
pollutants in Upper Silesia: Partial chemical composition and
biological activity. Environ Health Perspectives, 102: 61-66.
Chu EW & Malmgren RA (1965) An inhibitory effect of vitamin A on the
induction of tumors of forestomach and cervix in the Syrian hamster by
carcinogenic polynuclear hydrocarbons. Cancer Res, 25: 884-895.
Chuang JC, Hannan SW, & Wilson NK (1987) Field comparison of
polyurethane foam and XAD-2 resin for air sampling for polynuclear
aromatic hydrocarbons. Environ Sci Technol, 21: 798-804.
Chuang JC, Mack GA, Kuhlman MR, & Wilson NK (1991) Polycyclic aromatic
hydrocarbons and their derivatives in indoor and outdoor air in an
eight-home study. Atmos Environ, 25B: 369-380.
Chuang JC, Wise SA, Cao S, & Mumford JL (1992) Chemical
characterization of mutagenic fractions of particles from indoor coal
combustion: A study of lung cancer in Xuan Wie, China. Environ Sci
Technol, 26: 999-1004.
Chuang JC, Callahan PJ, Menton RG, & Gordon SM (1995) Monitoring
methods for polycyclic aromatic hydrocarbons and their distribution in
house dust and track-in soil. Environ Sci Technol, 29: 494-500.
Clayson DB, Pringle JAS, Bonser GM, & Wood M (1968) The technique of
bladder implantation: Further results and an assessment. Br J Cancer,
22: 825-832.
Clayton P, Davis BJ, Jones K, & Jones P (1992) Toxic organic
micropollutants in urban air. Stevenage, Hertfordshire, Warren Spring
Laboratory, 122 pp (Report No LR 904 [PA]).
Clement Associates (1988) Comparative potency approach for estimating
the cancer risk associated with exposure to mixtures of polycyclic
aromatic hydrocarbons. Interim final report. Fairfax, Virginia, 125 pp
(Report No. 68-02-4403).
Clement Associates (1990) Development of a dose-response model for
inhaled B[a]P. Fairfax, Virginia, 28 pp (prepared for US EPA under
contract No. 68-02-4601).
Clive D, Johnson KO, Spector AG, & Brown MMM (1979) Validation and
characterization of the L5178Y/TK+/- mouse lymphoma mutagen assay
system. Mutat Res, 59: 61-108.
Clonfero E, Venier P, Toffolo D, Busi L, & Gava C (1984) Mutagenesis
test on urine of workers exposed to polycyclic aromatic hydrocarbons
in an anode plant. Med Lav, 75: 275-281.
Clonfero E, Zordan M, Venier P, Paleologo M, Levis AG, Cottica D,
Pozzoli L, Jongeneelen FJ, Bos RP, & Anzion RBM (1989) Biological
monitoring of human exposure to coal tar. Urinary excretion of total
polycyclic aromatic hydrocarbons, 1-hydroxypyrene and mutagens in
psoriatic patients. Int Arch Occup Environ Health, 61: 363-368.
Clonfero E, Jongeneelen F, Zordan M, & Levis AG (1990) Biological
monitoring of human exposure to coal tar. Urinary mutagenicity assays
and analytical determination of polycyclic aromatic hydrocarbon
metabolites in urine. In: Vainio H, Sorsa M, & McMichael AJ ed.
Complex mixtures and cancer risk. Lyon, International Agency for
Research on Cancer, pp 215-222 (IARC Scientific Publications No. 104).
Clonfero E, Granella M, Marchioro M, Leopardi Barra E, Nardini B,
Ferri G, & Foà V (1995) Urinary excretion of mutagens in coke oven
workers. Carcinogenesis, 16: 547-554.
Coates JT, Elzerman AW, & Garrison AW (1986) Extraction and
determination of selected polycyclic aromatic hydrocarbons in plant
tissues. J Assoc Off Anal Chem, 69: 110-114.
Cody T, Radike M, & Warshawsky D (1984) The phototoxicity of
benzo [a]pyrene in the green algea Selenastrum capricornutum.
Environ Res, 35: 122-132.
Cohen GM, Haws SM, Moore BP, & Bridges JW (1976) Benzo [a]pyren-3-yl
hydrogen sulphate, a major ethyl acetate-extractable metabolite of
benzo [a]pyrene in human, hamster and rat lung cultures. Biochem
Pharmacol, 25: 2561-2570.
Collier TK, Singh SV, Awasthi YC, & Varanasi U (1992) Short
communication. Hepatic xenobiotic metabolizing enzymes in two species
of benthic fish showing different prevalences of
contaminant-associated liver neoplasms. Toxicol Appl Pharmacol, 113:
319-324.
Collin G & Höke H (1985) Anthracene. In: Elvers B, Hawkins S, & Schulz
G ed. Ullmann's encyclopedia of industrial chemistry, 5th ed.,Volume
A2: Weinheim, Verlagsgesellschaft , pp 343-345.
Collin G & Höke H (1991) Naphthalene and hydronaphthalene. In: Elvers
B, Hawkins S, & Schulz G ed. Ullmann's encyclopedia of industrial
chemistry, 5th ed., Volume A17: Weinheim, Verlagsgesellschaft, pp 1-8.
Collins JF & Alexeeff G (1993) Benzo [a]pyrene as a toxic air
contaminant. Part B: Health effects of benzo [a]pyrene. Berkeley,
California, California Environmental Protection Agency, Office of
Environmental Health Hazard Assessment, 69 pp.
Collins JF, Brown JP, Dawson SV, & Marty MA (1991) Risk assessment for
benzo [a]pyrene. Regul Toxicol Pharmacol, 13: 170-184.
Colmsjö A, Zebühr YU, & Ostman CE (1986a) Polynuclear aromatic
compounds in flue gases and ambient air in the vicinity of a municipal
incineration plant. Atmos Environ, 20: 2279-2282.
Colmsjö AL, Zebühr YU, & Ostman CE (1986b) Polynuclear aromatic
compounds in the ambient air of Stockholm. Chemosphere, 15: 169-182.
Combet E, Jarosz J, Martin-Bouyer M, Paturel L, & Saber A (1993)
[Shpol'skii spectrofluorimetric measurement of unit emissions of PAH
from 30 petrol and diesel light vehicles in eight representative
cycles.] Sci Total Environ, 134: 147-160 (in French).
Community Bureau of Reference (1992) Reference materials. Brussels,
Commission of the European Communities, 2 pp.
Compaan H & Laane RW (1992) Polycyclic aromatic hydrocarbons (PAH) in
the North Sea: An inventory. Delft, TNO Institute of Environmental
Sciences, 130 pp (TNO Report IMW-R 92/392).
CONCAWE (The Oil Companies' European Organization for Environment,
Health and Safety) (1992) The chemical composition of diesel
particulate emissions (Report No. 92/51). The Hague, 24 pp.
CONCAWE (The Oil Companies' European Organization for Environment,
Health and Safety) (1994) Motor vehicle emission regulations and fuel
specifications. 1994 update (Report 4/94). The Hague, 234 pp.
Conney AH (1982) Induction of microsomal enzymes by foreign chemicals
and carcinogenesis by polycyclic aromatic hydrocarbons: G.H.A. Clowes
memorial lecture. Cancer Res, 42: 4875-4917.
Coombs MM & Bhatt TS ed. (1987) Cyclopenta(a)phenanthrenes. Polycyclic
aromatic compounds structurally related to steroids. Cambridge,
Cambridge University Press, 265 pp.
Coombs MM, Dixon C, & Kissonerghis A-M (1976) Evaluation of the
mutagenicity of compounds of known carcinogenicity, belonging to the
benz [a]anthracene, chrysene, and cyclopenta [a]phenanthrene series,
using Ames' test. Cancer Res, 36: 4525-4529.
Cooper JA (1980) Environmental impact of residential wood combustion
emissions and its implications. J Air Pollut Control Assoc, 30: 855-
861.
Cooper CS, Hewer A, Ribeiro O, Grover PL, & Sims P (1980) The
enzyme-catalysed conversion of a non-'bay-region' diol-epoxide of
benz(a)anthracene into a gluthathione conjugate. FEBS Lett, 118: 39-
42.
Cooper CS, Grover PL, & Sims P (1983) The metabolism and activation of
benzo [a]pyrene. Drug Metab, 7: 295-396.
Coover MP & Sims RC (1987) The effect of temperature on polycyclic
aromatic hydrocarbon persistence in an unacclimated agricultural soil.
Hazard Waste Hazard Mater, 4: 69-82.
Cope VW & Kalkwarf DR (1987) Photooxidation of selected polycyclic
aromatic hydrocarbons and pyrenequinones coated on glass surfaces.
Environ Sci Technol, 21: 643-648.
Corner EDS & Young L (1954) Biochemical studies of toxic agents. 7.
The metabolism of naphthalene in animals of different species. Biochem
J, 58: 647-655.
Corner EDS, Billett FS, & Young L (1954) Biochemical studies of toxic
agents. 6. The conversion of naphthalene into
1,2-dihydro-2-hyroxy-l-naphthyl glucosiduronic acid in the rabbit.
Biochem J, 56: 270-274.
Correa M & Coler R (1983) Enhanced oxygen uptake rates in dragonfly
nymphs Somatochlora cingulata as an indication of stress from
naphthalene. Environ Contam Toxicol, 30: 269-276.
Cosma GN, Toniolo P, Currie D, Pasternack BS, & Seymour JG (1992)
Expression of the CYP1A1 gene in peripheral lymphocytes as a marker of
exposure to creosote in railroad workers. Cancer Epidemiol Biomarkers
Prev, 1: 137-142.
Costantino JP, Remond CK, & Bearden A (1995) Occupationally related
cancer risk among coke oven workers: 30 years of follow-up. J Occup
Environ Med, 37: 597-604.
Cottini GB & Mazzone GB (1939) The effects of 3:4-benzpyrene on human
skin. Am J Cancer, 37: 186-195.
Coutant RW, Brown L, Chuang JC, Riggin RM, & Lewis RG (1988) Phase
distribution and artifact formation in ambient air sampling for
polynuclear aromatic hydrocarbons. Atmos Environ, 22: 403-409.
Coutant RW, Callahan PJ, & Chuang JC (1992) Efficiency of
silicone-grease-coated denuders for collection of polynuclear aromatic
hydrocarbons. Atmos Environ, 26A: 2831-2834.
Creasia DA, Poggenburg JK Jr, & Nettesheim P (1976) Elution of
benzo [a]pyrene from carbon particles in the respiratory tract of
mice. J Environ Health, 1: 967-975.
Crespi CL & Thilly WG (1984) Assay for gene mutation in a human
lymphoblast line, AHH-1, competent for xenobiotic metabolism. Mutat
Res, 128: 221-230.
Crespi CL, Liber HL, Behymer TD, Hites RA, & Thilly WG (1985) A human
cell line sensitive to mutation by particle-born chemicals. Mutat Res,
157: 71-75.
Cretney JR, Lee HL, & Wright GJ (1985) Analysis of polycyclic aromatic
hydrocarbons in air particulate matter from a lightly industrialized
urban area. Environ Sci Technol, 19: 397-404.
Crider JY, Wilhm J, & Harmon HJ (1982) Effects of naphthalene on the
hemoglobin concentration and oxygen uptake of Daphnia
magna. Bull Environ Contam Toxicol, 28: 52-57.
Crocker TT, Chase JE, Wells SA, & Nunes LL (1970) Preliminary report
on experimental squamous carcinoma of the lung in hamsters and in a
primate (Galago Crassicauda Tus.). San Diego, California, Office of
the County Veterinarian, pp 317-328 (Symposium Series No. 21).
Croisy-Delcey M, Ittah Y, & Jerina DM (1979) Synthesis of
benzo [c]phenanthrene dihydrodiols. Tetrahedr Lett, 31: 2849-2852.
Crosby NT, Hunt DC, Philp LA, & Patel I (1981) Determination of
polynuclear aromatic hydrocarbons in food, water and smoke using
high-performance liquid chromatography. Analyst, 106: 135-145.
Crössmann G & Wüstemann M (1992) [Loadings in domestic gardens and
allotments by anorganic and organic substances with damaging
potential: Actual documentation. Part I: Soils and garden wastes and
Part II: Vegetables and fruits.] Berlin, Ministry of Environment, pp
40-42, 108-124 (in German).
Crow KD, Alexander E, Buck WHL, Johnson BE, Magnus IA, & Porter AD
(1961) Photosensitivity due to pitch. Br J Dermatol, 73: 220-232.
Csaba G & Inczefi-Gonda A (1992) Benzopyrene exposure at 15 days of
prenatal life reduces the binding capacity of thymic glucocorticoid
receptors in adulthood. Gen Pharmacol, 23: 123-124.
Csaba G, Inczefi-Gonda A, & Szeberenyi S (1991) Lasting impact of a
single benzpyrene treatment in pre-natal and growing age on the thymic
glucocorticoid receptors of rats. Gen Pharmacol, 22: 815-818.
Culp SJ, Gaylor DW, Sheldon WG, Goldstein LS, & Beland FA (1996) DNA
adduct measurements in relation to tumor incidence during the chronic
feeding of coal tar or benzo(a)pyrene to mice. Polycyclic Aromat Compd
11: 161-168.
Cummings DA, Lin ELC, Daniel FB, Klaunig JE, & Schut, HAJ (1991) In
vivo and in vitro binding of benzo [a]pyrene to tissue DNA and
circulating macromolecules in the mouse, monkey and human. Proc Am
Assoc Cancer Res, 32: 159.
Dahl AR, Coslett DC, Bond JA, & Hesseltine GR (1985) Metabolism of
benzo [a]pyrene on the nasal mucosa of Syrian hamsters: Comparison to
other extrahepatic tissues and possible role of nasally produced
metabolites in carcinogenesis. J Natl Cancer Inst, 75: 135-139.
Dai Q (1980) Researches on chemical carcinogens and mechanism of
chemical carcinogenesis. DI-region theory: A quantitative molecular
orbital model of carcinogenic activity for polycyclic aromatic
hydrocarbons. Sci Sin, 23: 453-470.
Daisey JM (1983) Analysis of polycyclic aromatic hydrocarbons by
thin-layer chromatography. In: Bjorseth A ed. Handbook of polycyclic
aromatic hydrocarbons. New York, Marcel Dekker, pp 397-437.
Daisey JM & Gundel LA (1993) Method 20: Determination of extractable
particulate organic matter and selected polycyclic aromatic
hydrocarbons. In: Seifert B, van de Wiel HJ, Dodet B, & O'Neill IK ed.
Environmental carcinogens. Methods of analysis and exposure
measurement: Volume 12. Indoor air. Lyon, International Agency for
Research on Cancer, pp 314-327 (IARC Scientific Publications No 109).
Daisey JM, McCaffrey RJ, & Gallagher RA (1981) Polycyclic aromatic
hydrocarbons and total extractable particulate organic matter in the
arctic aerosol. Atmos Environ, 15: 1353-1361.
Daisey JM, Cheney JL, & Lioy PH (1986) Profiles of organic particulate
emissions from air pollution sources: Status and needs for receptor
source apportionment modeling. J Air Pollut Control Assoc, 36: 17-33.
Daisey JM, Spengler JD, & Kaarakka P (1989) A comparison of the
organic chemical composition of indoor aerosols during woodburning and
non-woodburning periods. Environ Int, 15: 435-442.
Danz M, Hartmann A, Otto M, & Blaszyk H (1991) Hitherto unknown
additive growth effects of fluorene and 2-acetylaminofluorene on bile
duct epithelium and hepatocytes in rats. Arch Toxicol, 14 (suppl): 71-
74.
Darby FW, Willis AF, & Winchester RV (1986) Occupational health
hazards from road construction and sealing work. Ann Occup Hyg, 30:
445-454.
Darville RG & Wilhm JL (1984) The effect of naphthalene on oxygen
consumption and hemoglobin concentration in Chironomus
attenuatus and on oxygen consumption and life cycle of Tanytarsus
dissimilis. Environ Toxicol Chem, 3: 135-142.
Davenport R & Spacie A (1991) Acute phototoxicity of harbor and
tributary sediments from lower Lake Michigan. J Great Lakes Res, 17:
51-56.
Davidson GE & Dawson GWP (1976) Chemically induced presumed somatic
mutations in the mouse. Mutat Res, 38: 151-154.
Davies IW, Harrison RM, Perry R, Ratnayaka D, & Wellings RA (1976)
Municipal incinerator as source of polynuclear aromatic hydrocarbons
in environment. Environ Sci Technol, 10: 451-453.
Davies GM, Hodkinson A, & DiVetta P (1986) Measurement and analysis of
occupational exposures to coke oven emissions. Ann Occup Hyg, 30: 51-
62.
Davis WW, Krahl ME, & Clowes GHA (1942) Solubility of carcinogenic and
related hydrocarbons in water. J Am Chem Soc, 64: 108-110.
Davis BR, Whitehead JK, Gill ME, Lee PN, Butterworth AD, & Roe FJR
(1975) Response of rat lung to 3,4-benzpyrene administered by
intratracheal instillation in infusine with or without carbon black.
Br J Cancer, 31: 443-452.
Davis CS, Fellin P, & Otson R (1987) A review of sampling methods for
polyaromatic hydrocarbons in air. J Am Pollut Control Assoc, 37: 1397-
1408.
Deal CL (1995) The role of metabolic activation in B[a]P-induced
suppression of the humoral immune response. Richmond, Virginia,
Virginia Commonwealth University, Medical College of Richmond, 120 pp
(Doctoral Thesis).
Dean BJ (1981) Activity of 27 coded compounds in the RL, chromosome
assay. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens. Report of the international collaborative programme.
New York, Elsevier North-Holland, pp 570-579 (Progress in Mutation
Research, Volume 1).
Dean RG, Bynum G, Jacobson-Kram D, & Hadley E (1983a) Activation of
polycyclic hydrocarbons in Reuber H4-II-E hepatoma cells. An
in vitro system for the induction of SCEs. Mutat Res, 11: 419-427.
Dean JH, Luster MI, Boorman GA, Lauer LD, & Leubke RW (1983b)
Selective immunosuppression resulting from exposure to the
carcinogenic congener benzopyrene in B6C3F1 mice. Clin Exp Immunol,
52: 199-206.
Dean JH, Ward EC, Murray MJ, Lauer LD, House RV, Stillman W, Hammilton
TA, & Adams DO (1986) Immunosupression following
7,12-dimethylbenz [a]anthracene exposure in B6C3F1 mice. II. Altered
cell-mediated imunity and tumor resistance. Int J Immunopharmacol, 8:
189-198.
De Fré R, Bruynseraede P, & Kretzschmar JG (1994) Air pollution
measurements in traffic tunnels. Environ Health Perspectives, 102: 31-
37.
DeGraeve GM, Geiger DL, Meyer JS, & Bergman HL (1980) Acute and
embryo-larval toxicity of phenolic compounds to aquatic biota. Arch
Environ Contam Toxicol, 9: 557-568.
DeGraeve GM, Elder RG, Woods DC, & Bergman HL (1982) Effects of
naphthalene and benzene on fathead minnows and rainbow trout. Arch
Environ Contam Toxicol, 11: 487-490.
DeLeon IR, Byrne CJ, Peuler EA, Antoine SR, Schaeffer J, & Murphy RC
(1986) Trace organic and heavy metal pollutants in the Mississippi
River. Chemosphere, 15: 795-805.
Dell'Omo M & Lauwerys RR (1993) Adducts of macromolecules in the
biological monitoring of workers exposed to polycyclic aromatic
hydrocarbons. Crit Rev Toxicol, 23: 111-126.
Den Hollander H, Van de Meent D, Van Noort P, & Wondergem E (1986) Wet
deposition of polycyclic aromatic hydrocarbons in the Netherlands. Sci
Total Environ, 52: 211-219.
Dennis MJ, Massey RC, McWeeny DJ, Knowles ME, & Watson D (1983)
Analysis of polycyclic aromatic hydrocarbons in UK total diets. Food
Chem Toxicol, 21: 569-574
Dennis MJ, Massey RC, Cripps G, Venn I, Howarth N, & Lee G (1991)
Factors affecting the polycyclic aromatic hydrocarbons content of
cereals, fats and other food products. Food Addit Contam, 8: 517-530.
DePierre JW & Ernster L (1978) The metabolism of polycyclic
hydrocarbons and its relationship to cancer. Biochim Biophys Acta,
473: 149-186.
De Raat WK, Schulting FL, Burghardt E, & De Meijere FA (1987a)
Application of polyurethane foam for sampling volatile mutagens from
ambient air. Sci Total Environ, 63: 175-189.
De Raat WK, Kooijman SALM, & Gielen JWJ (1987b) Concentrations of
polycyclic aromatic hydrocarbons in airborne particles in the
Netherlands and their correlation with mutagenicity. Sci Total
Environ, 66: 95-114.
De Raat WK, Bakker GL, & De Meijere FA (1990) Comparison of filter
materials used for sampling of mutagens and polycyclic aromatic
hydrocarbons in ambient airborne particles. Atmos Environ, 24A: 2875-
2887.
DeSalvia R, Meschini R, Fiore M, Polani S, Palitti F, Carluccio MA, &
Turchi G (1988) Induction of sister-chromatid exchanges by
procarcinogens in metabolically competent Chinese hamster epithelial
liver cells. Mutat Res, 207: 69-75.
De Serres FJ & Ashby J ed. (1981) Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, 813 pp (Progress in Mutation Research,
Volume 1).
De Serres FJ & Hoffman GR (1981) Summary report on the performance of
yeast assays. In: de Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens. Report of the international collaborative
programme. New York, Elsevier North Holland, pp 68-76 (Progress in
Mutation Research, Volume 1).
Desideri PG, Lepri L, Heimler D, Giannessi S, & Checchini L (1984)
Concentration, separation and determination of hydrocarbons in sea
water. J Chromatogr, 284: 167-178.
Desideri PG, Lepri L, Canovaro M, & Checcini L (1988) Recovery,
identification and determination of organic compounds in marine
sediments. Stud Environ Sci, 34: 317-331.
Deutsch-Wenzel RP (1983) Experimental studies on the carcinogenicity
of five nitrogen containing polynuclear aromatic compounds directly
injected into rat lungs. Cancer Lett, 20: 97-101.
Deutsch-Wenzel RP, Brune H, Grimmer G, Dettbarn G, & Misfeld J (1983)
Experimental studies in rat lungs on the carcinogenicity and dose-
response relationships of eight frequently occuring environmental
polycyclic aromatic hydrocarbons. J Natl Cancer Inst, 71: 539-544.
Devanesan PD, Cremonesi P, Nunnally JE, Rogan EG, & Cavalieri EL
(1990) Metabolism and mutagenicity of dibenzo [a,e]pyrene and the
very potent environmental carcinogen dibenzo [a,l]pyrene. Chem Res
Toxicol, 3: 580-586.
263-268.
De Wiest F (1978) Any factors influencing the dispersion and the
transport of heavy hydrocarbons associated with airborne particles.
Atmos Environ, 12: 1705-1711.
DeWitt TH, Swartz RC, & Lamberson JO (1989) Measuring the acute
toxicity of estuarine sediments. Environ Toxicol Chem, 8: 1035-1048.
Diamond L, Kruszewski F, Aden DP, Knowles BB, & Baird WM (1980)
Metabolic activation of benzo [a]pyrene by a human hepatoma cell
line. Carcinogenesis, 1: 871-875.
Dickerson RL, Hooper MJ, Gard NW, Cobb GP, & Kendall RJ (1994)
Toxicological foundations of ecological risk assessment: Biomarker
development and interpretation based on laboratory and wildlife
species. Environ Health Perspectives, 102 (suppl 12): 65-69.
Diercxsens P & Tarradellas J (1987) Soil contamination by some organic
micropollutants related to sewage sludge spreading. Int J Environ Anal
Chem, 28: 143-159.
DiGiovanni J, Diamond L, Pritchett WP, Fisher EP, & Harvey RG (1985)
Tumor-initiating activity of the 9,10-dihydrodiol- and
9,10-dihydrodiol-7,8-epoxide of 3-methylcholanthrene in Sencar mice.
Cancer Lett, 28: 223-228.
Dikun PP (1967) [Detection of polycyclic aromatic hydrocarbons in
atmospheric contamination and other materials with quasi-linear
fluorescence spectra.] Zh Prikl Spektrosk, 6: 202-209 (in Russian).
DiMichele L & Taylor M (1978) Histopathological and physiological
responses of Fundulus heteroclitus to naphthalene exposure. J Fish
Res Board Can, 35: 1060-1066.
DiPaolo JA & Casto BC (1976) In vitro transformation: Interaction of
chemical carcinogens with viruses and physical agents. In: Montesano
R, Bartsch, H, & Tomatis L ed. Screening tests in chemical
carcinogenesis. Lyon, International Agency for Research on Cancer, pp
415-432 (IARC Scientific Publications No. 12).
DiPaolo JA, Donovan JP, & Nelson RL (1969) Quantitative studies of in
vitro transformation by chemical carcinogens, J Natl Cancer Inst 42:
867-874.
DiPaolo JA, Donovan PJ, & Nelson RL (1971) Transformation of hamster
cells in vitro by polycyclic hydrocarbons without cytotoxicity. Proc
Natl Acad Sci USA, 68: 2958-2961.
DiPaolo JA, Takano K, & Popescu NC (1972) Quantitation of chemically
induced neoplastic transformation of BALB/3T3 cloned cell lines.
Cancer Res, 35: 2686-2695.
DiPaolo JA, Nelson RL, Donovan PJ, & Evans CH (1973) Host-mediated in
vivo-in vitro assay for chemical carcinogenesis. Arch Pathol, 95:
380-385.
DiPaolo JA, Doniger J, Evans CH, & Popescu NC (1985) Enhancement and
inhibition of transformation of Syrian hamster embryo cells.
Carcinogenesis, 8: 319-328.
Dipple A, Moschel RC, & Bigger CAH (1984) Polynuclear aromatic
carcinogens. In: Searle CE ed. Chemical carcinogenesis, 2nd ed.
Washington DC, American Chemical Society, Vol 1ume, pp 41-163.
Dipple A, Pigott MA, Agarwal SK, Yagi H, Sayer JM, & Jerina DM (1987)
Optically active benzo [c]phenanthrene diol epoxides bind extensively
to adenine in DNA. Nature, 327: 535-536.
Dipple A, Cheng SC, & Bigger CA (1990) Polycyclic aromatic hydrocarbon
carcinogens. Prog Clin Biol Res, 347:109-127.
Dobriner K, Rhoads CP, & Lavin GI (1939) Conversion of
1,2,5,6-dibenzanthracene by rabbits, rats and mice. Significance in
carcinogenesis of this conversion. Proc Soc Exp Biol Med, 41: 67-69.
Dock L, Waern F, Martinez M, Grover PL, & Jernstrom B (1986) Studies
on the further activation of benz [a]pyrene diol epoxides by rat
liver microsomes and nuclei. Chem-Biol Interactions, 58: 301-318.
Doll R, Vessey MP, Buckley AR, Fear EC, Gammon EJ, Gunn W, Hughes GO,
Lee K, & Norman-Smith B (1972) Mortality of gasworkers. Final report
of a prospective study. Br J Med, 29: 394-406.
Dong MW & Greenberg A (1988) Liquid chromatographic analysis of
polynuclear aromatic hydrocarbons with diode array detection. J Liq
Chromatogr, 11: 1887-1905.
van Dongen (1987) [Leaching characteristics of pre-impregnated wood
during storage, Parts 1 and 2.]. Zeist, Organisation for Applied
Science (TNO), 170 pp (Report No. HI 87.1178) (in Dutch).
Donkin P, Widdows J, Evans SV, Worrall CM, & Carr M (1989)
Quantitative structure-activity relationships for the effect of
hydrophobic organic chemicals on rate of feeding by mussels Mytilus
edulis. Aquat Toxicol, 14: 277-293.
Doremire ME, Harmon GE, & Pratt DE (1979) 3,4-Benzopyrene in charcoal
grilled meats. J Food Sci, 44: 622-623.
DouAbdul AAZ, Abaychi JK, Al-Edanee TE, Ghani AA, & Al-Saad HT (1987)
Polynuclear aromatic hydrocarbons (PAHs) in fish from the Arabian
Gulf. Bull Environ Contam Toxicol, 38: 546-552.
Dragoescu C & Friedlander S (1989) Dynamics of the aerosol products of
incomplete combustion in urban atmospheres. Aerosol Sci Technol, 10:
249-257.
Draudt HN (1963) The meat smoking process: A review. Food Technol, 17:
85-90.
Dreier F, Siles S, Buchs M, Gülacar FO, & Buchs A (1985) [Polycyclic
aromatic hydrocarbons in sediments of Léman Lake.] Arch Sci Genève,
38: 215-224 (in French).
Druckrey H & Schmähl D (1955) [Carcinogenic effect of anthracene.]
Naturwissen-schaften, 42: 159-160 (in German).
Dunkel VD, Pienta RJ, Sivak A, & Traul KA (1981) Comparative
neoplastic transformation responses of Balb/3T3 cells, Syrian hamster
embryo cells, and Rauscher murine leukemia virus-infected Fischer 344
rat embryo cells to chemical carcinogens. J Natl Cancer Inst, 67:
1303-1315.
Dunkel VC, Zeiger E, Brusick D, McCoy E, McGregor D, Mortelmans K,
Rosenkranz HS, & Simmon VF (1984) Reproducibility of microbial
mutagenicity assays: 1. Tests with Salmonella typhimurium and
Escherichia coli using a standardized protocol. Environ Mutag, 6
(suppl 2): 1-39.
Dunkel VC, Schechtmann LM, Tu AS, Sivak A, Lubet RA, & Cameron TP
(1988) Intralaboratory evaluation of the C3H/10T1/2 cell
transformation assay. Environ Mol Mutag, 12: 21-31.
Dunlap CE & Warren S (1943) The carcinogenic activity of some new
derivatives of aromatic hydrocarbons. I. Compounds related to
chrysene. Cancer Res, 3: 606-607.
Dunn BP & Douglas GR (1991) DNA adducts in cells treated with
1-methylphenanthrene. Proc Am Assoc Cancer Res, 32: 93.
Duus U, Ahlborn J, & Andersson J (1994) Toxic oil in rubber tyres.
Solna, National Chemicals Inspectorate, 4 pp.
Eadie B, Landrum PF, & Faust W (1982) Polycyclic aromatic hydrocarbons
in sediments pore water and the amphipod
Pontoporeia hoyi from Lake Michigan, USA. Chemosphere, 11: 847-858.
Eadie BJ, Morehead NR, & Landrum PF (1990) Three-phase partitioning of
hydrophobic organic compounds in Great Lakes waters. Chemosphere, 20:
161-178.
Eastmond D, Booth G, & Lee M (1984) Toxicity accumulation and
elimination of polycyclic aromatic sulfur heterocycles in
Daphnia magna. Arch Environ Contam Toxicol, 13: 105-112.
Edes TE, Gysbers DG, Buckley CS, & Thornton WH Jr (1991) Exposure to
the carcinogen benzopyrene depletes tissue vitamin A: b-Carotene
prevents depletion. Nutr Cancer, 15: 159-166.
Edmisten G & Bantle J (1982) Use of Xenopus laevis larvae in 96
hour, flow-through toxicity tests with naphthalene. Bull Environ
Contam Toxicol, 29: 392-399.
Edsall CC (1991) Acute toxicities to larval rainbow trout of
representative compounds detected in Great Lakes fish. Bull Environ
Contam Toxicol, 46: 173-178.
Edwards NT (1983) Polycyclic hydrocarbons (PAHs) in the terrestrial
environment. A review. J Environ Qual, 12: 427-441.
Edwards NT (1986) Uptake translocation and metabolism of anthracene in
bush bean Phaseolus vulgaris. Environ Toxicol Chem, 5: 659-666.
Egan-Baum E, Miller BA, & Waxweiler RJ (1981) Lung cancer and other
mortality patterns among foundrymen. Scand J Work Environ Health, 7
(suppl 4): 147-155.
Ehrlich HW & Beevers CA (1956) The crystal structure of
2:13-benzfluoranthene. Acta Crystal, 9: 602-606.
Eiceman GA, Clement RE, & Karasek FW (1979) Analysis of fly ash from
municipal incinerators for trace organic compounds. Anal Chem, 51:
2343-2350.
Eisenhut W, Friedrich F, & Reinke M (1990) Coking plant environment in
West-Germany. Coke Making Int, 1: 74-77.
Eisenstadt E & Gold A (1978) Cyclopenta [c,d]pyrene: A highly
mutagenic polycyclic aromatic hydrocarbon. Proc Natl Acad Sci USA, 75:
1667-1669.
El-Bayoumy K, Amin S, & Hecht SS (1986) Effects of 6-nitro
substitution on 5-methylchrysene tumorigenicity, mutagenicity and
metabolism. Carcinogenesis, 7: 673-676.
Elgjo K (1968) Growth kinetics of the mouse epidermis after a single
application of 3,4-benzopyrene, croton oil, or 1,2-benzopyrene. Acta
Pathol Microbiol Scand, 73: 183-190.
Eling TE & Curtis JF (1992) Xenobiotic metabolism by prostaglandin H
synthase. Pharmacol Ther, 53: 261-273.
Eling T, Curtis J, Battista J, & Marnett LJ (1986) Oxidation of
(+)-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene by mouse keratinocytes:
Evidence for peroxyl radical- and monoxygenase-dependent metabolism.
Carcinogenesis, 12: 1957-1963.
Ellwardt PC (1976) [German Society for Moor and Peat. Determination of
polycyclic aromatic hydrocarbons with and without carcinogenic effect
in peat in comparison with their occurrence in soils and composts].
Ber Dtsch Ges Moor Torfkunde Hanover, 6: 135-144 (in German).
Elmets CA, Khan WA, Klemme JC, & Mukhtar H (1988) Impaired
immunological surveillance by 7,12-dimethylbenz [a]anthracene
augments its skin tumorigenicity in C3H mice. Biochem Biophys Res
Commun, 151: 148-152.
Elovaara E, Heikkilä P, Pyy L, Muranen P, & Riihimäki V (1995)
Significance of dermal and respiratory uptake in creosote workers:
Exposure to polycyclic aromatic hydrocarbons and urinary excretion of
1-hydroxypyrene. Occup Environ Med, 52: 196-203.
Emerole GO, Uwaifo AO, Thabarew MI, & Bababunmi EA (1982) The presence
of aflatoxin and some polycyclic aromatic hydrocarbons in human foods.
Cancer Lett, 15: 123-129.
Emura M, Richter-Reichhelm HB, Schneider P, & Mohr U (1980)
Sensitivity of Syrian golden hamster fetal lung cells to
benzo [a]pyrene and other polycyclic hydrocarbons in vitro.
Toxicology, 17: 149-155.
Emura M, Mohr U, Riebe M, Aufderheide M, & Dungworth DL (1987)
Predispostion of cloned fetal hamster lung epithelial cells to
transformation by a precarcinogen, benzo(a)pyrene, using growth
hormone supplementation and collagen gel substratum. Cancer Res, 47:
1155-1160.
Engelbreth-Holm J (1941) Acceleration of the development of mammary
carcinomas in mice by methylcholanthrene. Cancer Res, 1: 109-112.
Engewald W, Knobloch T, & Efer J (1993) [Volatile organic compounds in
emissions from brown-coal-fired residential stoves.] Z Umweltchem
Okotox, 5: 303-308 (in German).
Engholm G, Englund A, & Linder B (1991) Mortality and cancer incidence
in Swedish road paving asphalt workers and roofers. Eur Asphalt Mag,
1: 62-67.
Environment Agency, Japan (1993) Chemicals in the environment: Summary
of the result of environmental survey for chemical substances 1974 to
1991. Tokyo, 5 pp.
Environment Canada (1994) Canadian Environmental Protection Act.
Priority substances list assessment report: Polycyclic aromatic
hydrocarbons. Ottawa, Ministry of Supply and Services, 61 pp.
Epler JL, Rao TK, & Guerin MR (1979) Evaluation of feasibility of
mutagenic testing of shale oil products and effluents. Environ Health
Perspectives, 30: 179-184.
Epstein S (1968) Chemical mutagens in the human environment. Nature,
219: 385-387.
Ermer M (1970) [Studies with carcinogens in short-lived fish species.]
Zool Anz, 184: 175-193 (in German).
Ernst W, Eder G, Goerke H, Weber K, Weigelt S, & Weigelt V (1986)
[Organic environmental chemicals in German estuaries and coastal
waters: Origin, biotransfer, degradation.] Bremerhaven, Federal
Ministry for Research and Technology, Institute for Oceanography, 78
pp (Report No. M/86-001) (in German).
European Aluminium Association, Environment, Health and Safety
Secretariat (1990) Polycyclic aromatic hydrocarbons. Oslo, 51 pp
(Report of the Working Group on PAH No. ALUM./TEKN.-HYG.UTV No.
80-90).
European Economic Community (1967) Council Directive 67/548/EEC, of 27
June 1967 on the approximation of laws, regulations and administrative
provisions relating to the classification, packaging and labelling of
dangerous substances. Off J Eur Communities, L196: 234-237
European Economic Community (1980) Council Directive 80/778/EEC, of 15
July 1980, relating to the quality of water intended for human
consumption. Off J Eur Communities, L229: 11-29.
European Economic Community (1983) Council Directive of 16 June 1983
amending Council Directive 70/220/EEC on the approximation of the laws
of the Member States relating to measures to be taken against air
pollution by gases from positive ignition engines of motor vehicles
(83/351/EEC). Off J Eur Communities, L 197(I): 1.
European Economic Community (1988) Council Directive, of 22 June 1988,
on the approximation of the laws of member states relating to
flavourings for use in foodstuffs and to source materials for their
production (88/388/EEC). Off J Eur Communities, L184: 61-65.
European Economic Community (1991) Council Directive, of 29 May 1991,
on establishing indicative values by implementing Council Directive
80/1107/EEC on the protection of workers from the risks related to
exposure to chemical, physical and biological agents at work
(91/322/EEC). Off J Eur Communities, L177: 22-24.
European Economic Community (1994a) Council Directive 94/69/EC, of 19
December 1994 adapting to technical progress for the twenty-first time
Council Directive 67/548/EEC on the approximation of laws, regulations
and administrative provisions relating to the classification,
packaging and labelling of dangerous substances. Off J Eur
Communities, L381: 1-241.
European Economic Community (1994b) Council Directive 94/60/EC, of 25
November 1994 updating technical norms for concentration limits, and
attention and alarm levels for atmospheric pollution in urban areas
and provisions for certain pollutants referred to in the ministerial
decree of 15 April 1994. Off J Eur Communities, L290: 1-32.
Evans CH & DiPaolo JA (1975) Neoplastic transformation of guinea pig
fetal cells in culture induced by chemical carcinogens. Cancer Res,
35: 1035-1044.
Evans MS & Landrum PF (1989) Toxicokinetics of DDE, benzo [a]pyrene,
and 2,4,5,2',4',5'-hexachlorobiphenyl in Pontoporeia hoyi and Mysis
relicta. J Great Lakes Res, 15: 589-600.
Evans EL & Mitchell AD (1981) Effects of 20 coded chemicals on sister
chromatid exchange frequencies in cultured Chinese hamster cells. In:
De Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, pp 538-550 (Progress in Mutation
Research, Volume 1).
Fabacher DL, Besser JM, Schmitt CJ, Harshbarger JC, Peterman PH, &
Lebo JA (1991) Contaminated sediments from tributaries of the Great
Lakes: Chemical characterization and carcinogenic effects in medaka
(Oryzias latipes). Arch Environ Contam Toxicol, 21: 17-34.
Fahmy OG & Fahmy MJ (1973) Oxidative activiation of
benz [a]anthrazene and methylated derivatives in mutagenesis and
carcinogenesis. Cancer Res, 33: 2354-2361.
Fahmy MJ & Fahmy OG (1980) Altered control of gene acitivity in the
soma by carcinogens. Mutat Res, 72: 165-172.
Falk HL, Kotin P, & Markul I (1958) The disappearance of carcinogens
from soot in human lungs. Cancer, 11: 482-489.
Falk HL, Kotin P, Lee SS, & Nathan A (1962) Intermediary metabolism of
benzo [a]pyrene in the rat. J Natl Cancer Inst, 28: 699-724.
Fallon ME & Horvath FJ (1985) Preliminary assessment of contaminants
in soft sediments of the Detroit River. J Great Lakes Res, 11: 373-
378.
Fanburg SJ (1940) Exfoliative dermatitis due to naphthalene: Report of
an eruption resembling Mycosis fungoides. Arch Dermatol Syphilol,
42: 53-58.
Faoro RB & Manning JA (1981) Trends in benzo(a)pyrene, 1966-77. J Air
Pollut Control Assoc, 31: 62-64.
Farrington JW (1991) Biochemical processes governing exposure and
uptake of organic pollutant compounds in aquatic organisms. Environ
Health Perspectives, 90: 75-84.
Fazio T (1990) 48. Food additives. Indirect: Polycyclic aromatic
hydrocarbons and benzo [a]pyrene in food. Spectrophotometric method
(No. 973.30). In: Helrich K ed. Official methods of analysis of the
Association of Official Analytical Chemists (AOAC), 15th ed.
Arlington, Virginia, Association of Official Analytical Chemists,
Volume 2, pp 1176-1178.
Fedorak PM, Foght JM, & Westlake DWS (1982) A method for monitoring
mineralization of 14C-labeled compounds in aqueous samples. Water
Res, 16: 1285-1290.
Fendinger NJ, Radway JC, Tuttle JH, & Means JC (1989) Characterization
of organic material leached from coal by simulated rainfall. Environ
Sci Technol, 23: 170-177.
Feron VJ, de Jong D, & Emmelot P (1973) Dose-response correlation for
the induction of respiratory-tract tumors in Syrian golden hamsters by
intratracheal instillations of benzo(a)pyrene. Eur J Cancer, 9: 387-
390.
Ferraro S, Lee I, Ozretich R, & Specht D (1990) Predicting
bioaccumulation potential. A test of a fugacity-based model. Arch
Environ Contam Toxicol, 3: 386-394.
Ferreira MJ Jr, Buchet JP, Burrion JB, Moro J, Cupers L, Delavignette
JP, Jacques J, & Lauwerys R (1994a) Determinants of urinary
thioethers, D-glucaric acid and mutagenicity after exposure to
polycyclic aromatic hydrocarbons assessed by air monitoring and
measurement of 1-hydroxypyrene in urine: A cross-sectional study in
workers of coke and graphite-electrode-producing plants. Int Arch
Occup Environ Health, 65: 329-338.
Ferreira M Jr, Tas S, Dell'Omo M, Goormans G, Buchet JP, & Lauwerys R
(1994b) Determinants of benzo(a)pyrene diol epoxide adducts to
haemoglobin in workers exposed to polycyclic aromatic hydrocarbons.
Occup Environ Med, 51: 451-455.
Feunekes FDJR, Jongeneleen FJ, van der Laan H, & Schoonhof FHG (1997)
Uptake of polycyclic aromatic hydrocarbons among trainers in a
fire-fighting training facility. Am Ind Hyg Assoc J, 58: 23-28.
Finger SE, Little EF, Henry MG, Fairchild JF, & Boyle TP (1985)
Comparison of laboratory and field assessment of fluorene. Part I:
Effects of fluorene on the survival, growth, reproduction, and
behavior of aquatic organisms in laboratory tests. In: Boyle TP ed.
Validation and predictability of laboratory methods for assessing the
fate and effects of contaminants in aquatic ecosystems. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 120-133
(ASTM STP No. 865).
Flesher JW & Myers SR (1990) Bioalkylation of benz(a)anthracene as a
biochemical probe for carcinogenic activity. Drug Metab Disposition,
18: 163-167.
Flesher JW & Myers SR (1991) Rules of molecular geometry for
predicting carcinogenic activity of unsubstituted polynuclear aromatic
hydrocarbons. Teratog Carcinog Mutag, 11: 41-54.
Florin I, Rutberg L, Curvall M, & Enzell CR (1980) Screening of
tobacco smoke constituents for mutagenicity using the Ames' test.
Toxicology, 18: 219-232.
Flury F & Zernik F (1935) [List of the toxic and lethal doses for the
most common poisons in experimental animals.] In: Abderhalden E ed.
[Handbook of biological methods.] Berlin, Urban & Schwarzenberg, pp
12289-14222 (in German).
Foran JA, Holst LL, & Giesy JP (1991) Effects of photoenhanced
toxicity of anthracene on ecological and genetic fitness of Daphnia
magna: A reappraisal. Environ Toxicol Chem, 10: 425-427.
Forbes PD, Davies RE, & Urbach F (1976) Phototoxicity and
photocarcinogenesis: Comparative effects of anthracene and
8-methoxypsoralen in the skin of mice. Food Cosmet Toxicol, 14: 303-
306.
Foster GD, Baksi SM & Means JC (1987) Bioaccumulation of trace organic
contaminants from sediment by Baltic clams (Macoma
balthica) and soft-shell clams (Mya arenaria). Environ Toxicol
Chem, 6: 969-976.
Fox MA & Staley SW (1976) Determination of polycyclic aromatic
hydrocarbons in atmospheric particulate matter by high pressure liquid
chromatography coupled with fluorescence techniques. Anal Chem, 48:
992-998.
Fox CH, Selkirk JK, Price FM, Croy RG, Sandord KK, & Cottler-Fox M
(1975) Metabolism of benzo [a]pyrene by human epithelial cells
in vitro. Cancer Res, 35: 3551-3557.
Fox M, Lutz H-J, Gassman E, & Yoshikawa S (1988) Chemical economics
handbook (CEH): Product review for naphthalene. Menlo Park,
California, SRI International, 31 pp.
Franck HG & Stadelhofer JW (1987) [Industrial aromatic chemistry. Raw
products, processes, products.] Berlin, Springer-Verlag, pp 308-380
(in German).
Frank AP, Landrum PF, & Eadie BJ (1986) Polycyclic aromatic
hydrocarbon: Rates of uptake, depuration, and biotransformation by
Lake Michigan (Stylodrilus heringianus). Chemosphere, 15: 317-330.
Freeman AE, Weisburger EK, Weisburger JH, Wolford RG, Maryak JM &
Huebner RJ (1973) Transformation of cell cultures as an indication of
the carcinogenic potential of chemicals. J Natl Cancer Inst, 51: 799-
807.
Freitag DL, Ballhorn L, Geyer H, & Korte F (1985) Environmental hazard
profile of organic chemicals. An experimental method for the
assessment of the behaviour of organic chemicals in the ecosphere by
means of simple laboratory tests with 14C labelled chemicals.
Chemosphere, 14: 1589-1616.
Fritz W (1971) [The extent of sources of contamination of our food
with carcinogenic hydrocarbons.] Ernährungsforschung, 16: 547-557 (in
German).
Fritz W (1972) [Contamination of food with carcinogenic hydrocarbons
during processing and cooking.] Arch Geschwulstforsch, 40: 81-90 (in
German).
Fritz W (1983) [Analysis and assessment of carcinogenic polycyclic
aromatic hydrocarbons from the food hygiene. Toxicology point of view.
A review]. Nahrung, 27: 965-973 (in German).
Frölich A & Würgler FE (1990) Drosophila wing-spot test: Improved
detectability of genotoxicity of polycyclic aromatic hydrocarbons.
Mutat Res, 234: 71-80.
Frumin G, Chuiko G, Pavlov D, & Menzykova O (1992) New rapid method to
evaluate the median effect concentrations of xenobiotics in
hydrobionts. Bull Environ Contam Toxicol, 49: 361-367.
Fujikawa K, Fort FL, Samejima K, & Sakamoto Y (1993) Gentoxic potency
in Drosophila melanogaster of selected aromatic amines and
polycyclic hydrocarbons as assayed in the DNA repair test. Mutat Res,
290: 175-182.
Fukuda K, Inagaki Y, Maruyama T, Kojima HI, & Yoshida T (1988) On the
photolysis of alkylated naphthalenes in aquatic systems. Chemosphere,
17: 651-659.
Funcke W, König J, Henze W, & Umland F (1986) Determination of
polycyclic aromatic hydrocarbons in mainstream cigarette smoke with
respect to particle size. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemistry, characterization and carcinogenesis.
Columbus, Ohio, Battelle Press, pp 333-341.
Funcke W, König J, & Balfanz E (1988) Determination of polycyclic
aromatic hydrocarbons in flue gases from coal-fired power plants. In:
Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons: A decade of
progress. Columbus, Ohio, Battelle Press, pp 277-284.
Furlong ET, Carter DS, & Hites, RA (1988) Organic contaminants in
sediments from the Trenton Channel of the Detroit River, Michigan. J
Great Lakes Res, 14: 489-501.
Furst A, Kolff B, & Dempsey DA (1979) Pulmonary tumor induction: Three
hydrocarbons compared. Proc West Pharmacol Soc, 22: 269-271.
Gaines TB (1969) Acute toxicity of pesticides. Toxicol Appl Pharmacol,
14: 515-534.
Gala WR & Giesy JP (1992) Photo-induced toxity of anthracene to the
green algae, Selenastrum capricornutum. Arch Environ Contam Toxicol,
23: 316-323.
Gamper HB, Tung ASC, Straub K, Bartholomew JC, & Calvin M (1977) DNA
strand scission by benzo [a]pyrene diol epoxides. Science, 197: 671-
674.
Gamper HB, Bartholomew JC, & Calvin M (1980) Mechanism of
benzo(a)pyrene diol epoxide induced deoxyribonucleic acid strand
scission. Biochemistry, 19: 3948-3956.
Gardiner K, Hale KA, Calvert IA, Rice D, & Harrington JM (1992) The
suitability of the urinary metabolite 1-hydroxypyrene as an index of
polynuclear aromatic hydrocaarbon bioavailability from workers exposed
to carbon black. Ann Occup Hyg, 36: 681-688.
Garg A, Beach A, & Gupta RC (1991) DNA-reactive metabolites (DRM)
detected in the serum of benzo [a]pyrene (BP)-treated rodents by
32P-postlabeling. Proc Am Assoc Cancer Res, 32: 122.
Garrigues P & Ewald M (1987) High resolution emission spectroscopy
(Shpol'skii effect): A new analytical technique for the analysis of
polycyclic aromatic hydrocarbons (PAH) in the environmental samples.
Chemosphere, 16: 485-494.
Garrigues P, Soclo HH, Marniesse MP, & Ewald M (1987) Origin of
polycyclic aromatic hydrocarbons (PAH) in recent sediments from the
continental shelf of the 'Golfe de Gascogne' (Atlantic Ocean) and in
the Gironde estuary. Int J Environ Anal Chem, 28: 121-131.
Gauthier TD, Shane EC, Guerin WF, Seltz WR, & Grant CL (1986)
Fluorescence quenching method for determining equilibrium constants
for polycyclic aromatic hydrocarbons binding to dissolved humic
materials. Environ Sci Technol, 20: 1162-1166.
Gaydos RM (1981) Naphthalene. In: Kirk-Othmer encyclopedia of chemical
technology, 3rd ed. New York, John Wiley & Sons, Volume 15, pp 698-
719.
Geacintov NE (1988) Mechanisms of reaction of polycyclic aromatic
epoxide derivatives with nucleic acids. In: Yang SK & Silverman BD ed.
Polycyclic aromatic hydrocarbon carcinogenesis: Structure-activity
relationships. Boca Raton, Florida, CRC Press, Volume 2, pp 181-206.
Geahchan A, Le Gren I, Chambon P, & Chambon R (1991) Improved method
for determination of polynuclear aromatic hydrocarbons in
pharmacopoeial paraffin and mineral oils. J Assoc Off Anal Chem, 74:
968-973.
Geddie JE, Amin S, Huie K, & Hecht SS (1987) Formation and
tumorigenicity of benzo [b]fluoranthene metabolites in mouse
epidermis. Carcinogenesis, 8: 1579-1584.
Gehly EB, Landolph JR, Heidelberger C, Nagasawa H, & Little JB (1982)
Induction of cytotoxicity, mutation, cytogenetic changes and
neoplastic transformation by benzo(a)pyrene and derivatives in C3H/10T
1/2 clone 8 mouse fibroblasts. Cancer Res, 42: 1866-1875.
Geiger JG Jr & Buikema AL Jr (1981) Oxygen consumption and filtering
rate of Daphnia pulex after exposure to water-soluble fractions of
naphthalene, phenanthrene, No. 2 fuel oil, and coal-tar creosote.
Environ Contam Toxicol, 27: 783-789.
Geiger JG Jr & Buikema AL Jr (1982) Hydrocarbons depress growth and
reproduction of Daphnia pulex (Cladocera). J Fish Aquat Sci, 39:
830-836.
Geiger DL, Northcott CE, Call DJ, & Brooke LT ed. (1985) Acute
toxicities of organic chemicals to fathead minnows Pimephales
promelas. Superior, Wisconsin, University of Wisconsin, Center for
Lake Superior Environmental Studies, Volume 2, pp 211-212.
Gelboin HV & Ts'o POP (1978) Polycyclic hydrocarbons and cancer.
Volume 2: Metabolism and cell biology. New York, Academic Press, 441
pp.
Gendimenico GJ & Kochevar IE (1984) Degranulation of mast cells and
inhibition of the response to secretory agents by phototoxic and
ultraviolet radiation. Toxicol Appl Pharmacol, 76: 374-382
Generoso WM, Cain KT, Hellwig CD, & Cacheiro NLA (1982) Lack of
association between induction of dominant-lethal mutations and
induction of heritable translocations with benzo(a)pyrene in
postmeiotic germ cells of male mice. Mutat Res, 94: 155-163.
Genevois C, Brandt HCA, Bartsch H, Wild CP & Castegnaro M (1995)
Formation of DNA adducts in skin, lung and lymphocytes after skin
painting of rats with undiluted coaltar and bitumen vapor/particulates
condensates. In: Fifteenth international symposium on polycyclic
aromatic compounds: Chemistry, biology and environmental impact,
Belgirate, Italy, 19-22 September 1995. Ispra, Joint Research Centre
European Commission, pp 54-55.
Gensler HL (1988) Enhancement of chemical carcinogenesis in mice by
systemic effects of ultraviolet irradiation. Cancer Res, 48: 620-623.
Gerarde HW (1960) Toxicology and biochemistry of aromatic
hydrocarbons. In: Browning E ed. Elsevier monographs on toxic agents.
Amsterdam, Elsevier Publishing Co., pp 240-321.
Gerhart E & Carlson R (1978) Hepatic mixed-function oxidase activity
in rainbow trout exposed to several polycyclic aromatic compounds.
Environ Res, 17: 284-295.
German Federal Department for Worker Safety (1989) [23.TRK-Wert for
benzo(a)pyrene.] In:[Bundesarbeitsblatt 10.] Dortmund, pp 58-61 (in
German).
German Federal Office for Sea Navigation and Hydrography (1993)
[Monitoring of the sea. Report for the year 1990. Part II: Data.]
Hamburg, pp 79-94 (in German).
German Ministry of Environment (1979) [Air quality criteria for
selected polycyclic aromatic hydrocarbons. PAH as an environmental
carcinogen.] Berlin, Erich Schmidt Verlag, 270 pp (Report No.
UBA-1/79) (in German).
German Ministry of Environment (1993) [Environmental sample bank.
Annual report 1991.] Berlin, 139 pp (Report No. UBA-7/93) (in German).
German Research Commission (1991) Polycyclic aromatic hydrocarbons
(PAH) (particle-bound). In: Kettrup A ed. Commission for the
investigation of health hazards of chemical compounds in the work air,
working group on analytical chemistry: Analyses of hazardous
substances in air. Weinheim, VCH Publishers, Volume 1, pp 41-52.
German Society for Mineral-oil and Coal Chemistry (1984)
[Investigations into the contents of wastewater from oil refineries.]
Hamburg, 95 pp (Report No. 283) (in German).
Gershbein LL (1975) Liver regeneration as influenced by the structure
of aromatic and heterocyclic compounds. Res Commun Chem Pathol
Pharmacol, 11: 445-466.
Gerstle RW, Cuffe ST, Orning AA, & Schwartz CH (1965) Air pollution
emissions from coal-fired power plants. Report No. 2. J Air Pollut
Control Assoc, 15: 59-64.
Geyer H, Politzki G, & Freitag D (1984) Prediction of ecotoxicological
behaviour of chemicals: Relationship between n-octonal/water
partition coefficient and bioaccumulation of organic chemicals by
Alga chlorella. Chemosphere, 13: 269-284.
Gharrett JA & Rice SD (1987) Influence of simulated tidal cycles on
aromatic hudrocarbon uptake and elimination by the shore crab
Hemigrapsus nudus. Mar Biol, 95: 365-370.
Gibbs GW (1985) Mortality of aluminum reduction plant workers, 1950
through 1977. J Occup Med, 27: 761-770.
Gibbs GW & Horowitz I (1979) Lung cancer mortality in aluminum
reduction plant workers. J Occup Med, 21: 347-353.
Gibson TL (1982) Nitro derivates of polynuclear aromatic hydrocarbons
in airborne and source particulate matter. Atmos Environ, 16: 2037-
2040.
Gibson ES, Martin RH, & Lockington JN (1977) Lung cancer mortality in
a steel foundry. J Occup Med, 19: 807-812.
Giger W & Schaffner C (1978) Determination of polycyclic aromatic
hydrocarbons in the environment by glass capillary gas chromatography.
Anal Chem, 50: 243-249.
Gill RD, Butterworth BE, Nettikumara AN, & Digiovanni J (1991)
Relationship between DNA adduct formation and unscheduled DNA
synthesis (UDS) in cultured mouse epidermal keratinocytes. Environ Mol
Mutag, 18: 200-206.
Ginsberg GL & Atherholt TB (1989) Transport of DNA-adducting
metabolites in mouse serum following benzo(a)pyrene administration.
Carcinogenesis, 10: 673-679.
Gjessing E, Lygren E, Berglind L, Gulbrandsen T, & Skaane R (1984)
Effect of highway runoff on lake water quality. Sci Total Environ, 33:
245-257.
Glatt H, Bücker M, Platt KL, & Oesch F (1985) Host-mediated
mutagenicity experiments with benzo [a]pyrene and two of its
metabolites. Mutat Res, 156: 163-169.
Glatt H, Seidel A, Bochnitschek W, Marquardt H, Marquardt H, Hodgson
RM, Grover PL, & Oesch F (1986) Mutagenic and cell-transforming
activities of triol-epoxides as compared to other chrysene
metabolites. Cancer Res, 46: 4556-4565.
Glatt H, Seidel A, Ribeiro O, Kirkhy C, Ilirom P, & Oesch F (1987)
Metabolic activation to a mutagen of
3-hydroxy- trans-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene, a
secondary metabolite of benzo [a]pyrene. Carcinogenesis, 8:
1621-1627.
Glatt H, Harvey RG, Phillips DH, Hewer A, & Grover PL (1989) Influence
of the alkyl substituent on mutagenicity and covalent DNA binding of
bay region diol-epoxides of 7-methyl- and 7-ethylbenz [a]anthracene
in Salmonella and V79 Chinese hamster cells. Cancer Res, 49: 1778-
1782.
Goddard KA, Schultz RJ, Stegeman JJ, Schultz RJ, & Stegemann JJ (1987)
Uptake, toxicity and distribution of benzo [a]pyrene and
monooxygenase induction in the top minnows Poeciliopsis monacha
and Poeciliopsis lucida. Drug Metab Disposition, 15: 449-455.
Göen T, Gündel J, Schaller KH, & Angerer J (1995) The elimination of
1-hydroxypyrene in the urine of the general population and workers
with different occupational exposures to PAH. Sci Total Environ, 163:
195-201.
Gold A & Eisenstadt E (1980) Metabolic activation of
cyclopenta(cd)pyrene to 3,4-epoxycyclopenta(cd)pyrene by rat liver
microsomes. Cancer Res, 40: 3940-3944.
Gold A, Nesnow S, Moore M, Garland H, Curtis G, Howard B, Graham D, &
Eisenstadt E (1980) Mutagenesis and morphological transformation of
mammalian cells by a nonbay-region polycyclic cyclopenta(cd)pyrene and
its 3,4-oxide. Cancer Res, 40: 4482-4484.
Goldschmidt BM, Katz C, & Van Duuren BL (1973) The cocarcinogenic
activity of noncarcinogenic aromatic hydrocarbons. Proc Am Assoc
Cancer Res, 14: 84.
Gollahon L (1991) Chromosomal damage to preimplantation mouse embryos
in vitro by naphthalene and aflatoxin B1. Diss Abstr Int B, 52: 694-
695.
Gollahon LS, Iyer P, Martin JE, & Irvin TR (1990) Chromosomal damage
to preimplantation embryos in vitro by naphthalene. Toxicologist,
10: 274.
Gonzalez FJ & Gelboin HV (1994) Role of human cytochromes P450 in the
metabolic activation of chemical carcinogens and toxins. Drug Metab
Rev, 26: 165-183.
Gonzalez-Vila FJ, Lopez JL, & Martin F (1988) Determination of
polynuclear aromatic compounds in composted municipal refuse and
compost-amended soils by a simple cleanup procedure. Biomed Environ
Mass Spectrom, 16: 423-425.
Gordon RJ (1976) Distribution of airborne polycyclic aromatic
hydrocarbons throughout Los Angeles. Environ Sci Technol, 10: 370-373.
Gordon RJ & Bryan RJ (1973) Patterns in airborne polynuclear
hydrocarbon concentrations at four Los Angeles sites. Environ Sci
Technol, 7: 1050-1053.
Gorelova ND & Cherepanova AI (1970) [Possibility of accumulation of
3,4-benzpyrene in tissues and organs of cows and calves and in milk
when this carcinogen is present in food.] Vopr Onkol, 16: 69-73 (in
Russian).
Gorelova ND, Dikun PP, Solinek VA, & Emshanova AV (1960) [Content of
3,4-benzpyrene in fish smoked by different methods.] Vopr Onkol, 6:
33-37 (in Russian).
Götz R (1984) [Investigations into leakage water from the refuse dump
Georgswerder in Hamburg. Results of analyses up to 1982 inclusive.]
Müll Abfall, 12: 349-356 (in German).
Grachev MA, Baram GI, Hodgher, TV, Gorshow AG, Vodiannikova NI, &
Bartz MP (1994) Determination of the concentration of PAHs in the
atmosphere of the south part of the Lake Baikal coast. Irkutsk,
Limnological Institute, Russian Academy of Sciences (unpublished
report).
Gräf W (1970) Levels of 3,4-benzopyrene in human organs of different
ages. Second communication. Arch Hyg, 154: 331-335.
Gräf W & Nowak W (1966) [Promotion of growth in lower and higher
plants by carcinogenic polycyclic aromatics.] Arch Hyg, 150: 513-528
(in German).
Gräf W, Eff H, & Schormair S (1975) Levels of carcinogenic, polycyclic
aromatic hydrocarbons in human and animal tissues. Third
communication. Zentralbl Bakteriol Hyg Abt Orig B, 161: 85-103.
Graffi A (1940a) [Cellular accumulation of carcinogenic hydrocarbons.]
Z Krebsforschung, 49: 477-495 (in German).
Graffi A (1940b) [Intracellular benzopyrene accumulation in living
normal and tumor cells.] Z Krebsforschung, 50: 196-219 (in German).
Graffi A (1940c) [Some observations on the aetiology of tumors, in
particular on the nature of the active agent in cell-free transferable
chicken tumours.] Z Krebsforschung, 50: 501-551 (in German).
Graffi A, Vlamynck E, Hoffmann F, & Schulz I (1953) [Studies on the
tumor producing effects of different chemicals in combination with
croton oil.] Arch Geschwulstforsch, 5: 110-126 (in German).
Grant G & Roe FJC (1963) The effect of phenanthrene on tumour
induction by 3,4-benzopyrene administered to newly born mice. Br J
Cancer, 17: 261-265.
Greb W, Strobel R & Röhrborn G (1980) Transformation of BHK 21/CL 13
cells by various polycyclic aromatic hydrocarbons using the method of
Styles. Toxicol Lett, 7: 143-148.
Greenberg A, Bozzelli JW, Cannova F, Forstner F, Giorgio P, Stout D, &
Yokoyama R (1981) Correlations between lead and coronene
concentrations at urban, suburban, and industrial sites in New Jersey.
Environ Sci Technol, 15: 566-570.
Greenberg A, Darack F, Harkov R, Lioy P, & Daisey J (1985) Polycyclic
aromatic hydrocarbons in New Jersey: A comparison of winter and summer
concentrations over a two-year period. Atmos Environ, 19: 1325-1339.
Greife AL & Warshawsky D (1993) Influence of the dose levels of
carcinogen ferric oxide on the metabolism of benzo(a)pyrene by
pulmonary alveolar macrophages in suspension culture. J Toxicol
Environ Health, 38: 399-417.
Greife A, Schoeny R, & Warshawsky D (1988) Effect of the cocarcinogen
ferric oxide on benzo(a)pyrene metabolism by hamster alveolar
macrophages. In: Cooke M & Dennis A ed. Polycyclic aromatic
hydrocarbons: Chemical and biological effects. Columbus, Ohio,
Battelle Press, pp 317-327.
Griesbaum K, Behr A, Biedenkapp D, Voges HW, Garbe D, Paetz C, Collin
G, Mayer D, & Höke H (1989) Hydrocarbons. In: Ullmann's encyclopedia
of industrial chemistry, 5th ed. Volume A13. Weinheim, Verlag Chemie,
pp 227-281.
Griest WH (1980) Multicomponent polycyclic aromatic hydrocarbon
analysis of inland water and sediment. Environ Sci Res, 16: 173-183.
Griest WH & Caton JE (1983) Extraction of polycyclic aromatic
hydrocarbons for quantitative analysis. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 95-148.
Griest WH, Maskarinec MP, Herbes SE, & Southworth GR (1981)
Multicomponent methods for the identification and quantification of
polycyclic aromatic hydrocarbons in the aqueous environment. In:
Jackson LP & Wright CC ed. Analysis of waters associated with
alternative fuel production. Philadelphia, Pennsylvania, American
Society for Testing and Materials, pp 167-178 (ASTM STP No. 720).
Grifoll M, Casellas M, Bayona JM, & Solanas AM (1992) Isolation and
characterization of a fluorene-degrading bacterium: Identification of
ring oxidation and ring fission products. Appl Environ Microbiol, 58:
2910-2917.
Grimmer G (1979) Sources and occurrence of polycyclic aromatic
hydrocarbons. In: Egan H, Castegnaro M, Bogovski P, Kunte H, & Walker
EA ed. Environmental carcinogens: Selected methods of analysis. Volume
3. Analysis of polycyclic hydrocarbons in environmental samples. Lyon,
International Agency for Research on Cancer, pp 31-54 (IARC Scientific
Publications No. 29).
Grimmer G (1980) [Analysis and comparison of PAH profile from
environmental samples.] Düsseldorf, VDI-Verlag, pp 39-50 (VDI Report
No. 358) (in German).
Grimmer G (1983a) Chemistry. In: Grimmer G ed. Environmental
carcinogens: Polycyclic aromatic hydrocarbons. Boca Raton, Florida,
CRC Press, pp 27-60.
Grimmer G (1983b) Profile analysis of polycyclic aromatic hydrocarbons
in air. In: Bjorseth A ed. Handbook of polycyclic aromatic
hydrocarbons. New York, Marcel Dekker, pp 149-191.
Grimmer G (1993) [Environmental-toxicological evaluation of PAH in
soil.] Altlasten Spectrum, 2: 85-92 (in German).
Grimmer G & Böhnke H (1975) Profile analysis of polycyclic aromatic
hydrocarbons and metal content in sediment layers of a lake. Cancer
Lett, 1: 75-84.
Grimmer G & Böhnke H (1978) The tumor producing effect of automobile
exhaust condensate and fractions thereof. Part 1: Chemical studies. J
Environ Pathol Toxicol, 1: 661-667.
Grimmer G & Böhnke H (1979a) Method 4: Gas chromatographic profile
analysis of polycyclic aromatic hydrocarbons in (i) high protein
foods, (ii) fats and vegetable oils and (iii) plants, soils and sewage
sludge. In: Egan H, Castegnaro M, Bogovski P, Kunte H, & Walker EA ed.
Environmental carcinogens: Selected methods of analysis. Volume 3.
Analysis of polycyclic aromatic hydrocarbons in environmental samples.
Lyon, International Agency for Research on Cancer, pp 163-173 (IARC
Scientific Publications No. 29).
Grimmer G & Böhnke H (1979b) Method 3: Gas chromatographic profile
analysis of polycyclic aromatic hydrocarbons in lubricating oil,
cutting oil and fuel. In: Egan H, Castegnaro M, Bogovski P, Kunte H, &
Walker EA ed. Environmental carcinogens: Selected methods of analysis.
Volume 3. Analysis of polycyclic aromatic hydrocarbons in
environmental samples. Lyon, International Agency for Research on
Cancer, pp 155-162 (IARC Scientific Publications No. 29).
Grimmer G & Düvel D (1970) [Investigation of biosynthetic formation of
polycyclic hydrocarbons in higher plants.] Z Naturforsch, 256: 1171-
1175 (in German with English summary).
Grimmer G & Hildebrandt A (1965) [The content of polyaromatic
hydrocarbons in various vegetables.] Dtsch Lebensm Rundsch, 61: 237-
239 (in German).
Grimmer G & Hildebrandt A (1975) Investigations on the carcinogenic
burden by air pollution in man. XIII. Assessment of the contribution
of passenger cars to air pollution by carcinogenic polycyclic aromatic
hydrocarbons. Zentralbl Bakteriol Hyg I Orig B, 161: 104-124.
Grimmer G, Jacob J, & Hildebrandt A (1972) [Hydrocarbons in the human
environment. 9. Content of polycyclic hydrocarbons in Icelandic soil.]
Z Krebsforsch, 78: 65-72 (in German).
Grimmer G, Hildebrandt A, & Böhnke H (1979) Method 2: Gas
chromatographic profile analysis of polycyclic aromatic hydrocarbons
in automobile exhaust gas condensate. In: Egan H, Castegnaro M,
Bogovski P, Kunte H, & Walker EA ed. Environmental carcinogens:
Selected methods of analysis. Volume 3. Analysis of polycyclic
aromatic hydrocarbons in environmental samples. Lyon, International
Agency for Research on Cancer, pp 141-154 (IARC Scientific
Publications No. 29).
Grimmer G, Hilge G, & Niemitz W (1980) [Comparison of the profile of
polycyclic aromatic hydrocarbons of sewage sludge samples from 25
filter plants.] Vom Wasser, 54: 255-272 ( in German).
Grimmer G, Jacob J, & Naujack KW (1981a) Profile of the polycyclic
aromatic hydrocarbons from lubricating oils. Inventory by GCGC/MS. PAH
in environmental materials, Part 1. Fresenius Z Anal Chem, 306: 347-
355.
Grimmer G, Schneider D, & Dettbarn G (1981b) [The load of different
rivers in the Federal Republic of Germany by PAH (PAH-profiles of
water surface)]. Vom Wasser, 56: 131-144 (in German).
Grimmer G, Naujack K-W, & Schneider D (1981c) Comparison of the
profiles of polycyclic aromatic hydrocarbons in different areas of a
city by glass-capillary-gas chromatography in the nanogram range. Int
J Environ Chem, 10: 265-276.
Grimmer G, Naujack K-W, & Schneider D (1982) Profile analysis of
polycyclic aromatic hydrocarbons by glass capillary gas chromatography
in atmospheric suspended particulate matter in the nanogram range
collecting 10 m3 of air. Fresenius Z Anal Chem, 311: 475-484.
Grimmer G, Jacob J, & Naujack KW (1983) Profile of the polycyclic
aromatic compounds from crude oils. Inventory by GCGC/MS. PAH in
environmental materials, part 3. Fresenius Z Anal Chem, 314: 29-36.
Grimmer G, Brune H, Deutsch-Wenzel R, Dettbarn G, & Misfeld J (1984)
Contribution of polycyclic aromatic hydrocarbons to the carcinogenic
impact of gasoline engine exhaust condensate evaluated by implantation
into the lungs of rats. J Natl Cancer Inst, 72: 733-739.
Grimmer G, Jacob J, Dettbarn G, & Naujack KW (1985) Determination of
polycyclic aromatic hydrocarbons, azaarenes, and thiaarenes emitted
from coal-fired residential furnaces by gas chromatography/mass
spectrometry. Fresenius Z Anal Chem, 322: 595-602.
Grimmer G, Naujack KW, & Dettbarn G (1987) Gas chromatographic
determination of polycyclic aromatic hydrocarbons, aza-arenes,
aromatic amines in the particle and vapor phase of mainstream and
sidestream smoke of cigarettes. Toxicol Lett, 35: 117-124.
Grimmer G, Jacob J, Dettbarn G, & Naujack K-W (1988a) Effect of the
pH-value of diesel exhaust on the amount of filter-collected
nitro-PAH. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: A decade of progress. Columbus, Ohio, Battelle Press,
pp 341-351.
Grimmer G, Brune H, Dettbarn G, Heinrich U, Jacob J, Mohtashamipur E,
Norpoth K, Pott F, & Wenzel-Hartung R (1988b) Urinary and faecal
excretion of chrysene and chrysene metabolites by rats after oral,
intraperitoneal, intratracheal or intrapulmonary application. Arch
Toxicol, 62: 401-405.
Grimmer G, Brune H, Dettbarn G, Naujack K-W, Mohr U, & Wenzel-Hartung
R (1988c) Contribution of polycyclic aromatic compounds to the
carcinogenicity of sidestream smoke of cigarettes evaluated by
implantation into lungs of rats. Cancer Lett, 43: 173-177.
Grimmer G, Brune H, Dettbarn G, Jacob J, Mohtashamipur E, Norpoth K, &
Wenze-Hartung R (1991a) Urinary and fecal excretion of phenanthrene
and phenanthrols by rats following oral, intraperitoneal, or
intrapulmonary application. Polycyclic Aromat Compd, 2: 39-47.
Grimmer G, Brune H, Dettbarn G, Jacob J, Misfeld J, Mohr U, Naujack
K-W, Timm J, & Wenzel-Hartung R (1991b) Relevance of polycyclic
aromatic hydrocarbons as environmental carcinogens. Fresenius J Anal
Chem, 339: 792-795.
Grimmer G, Dettbarn G, & Naujack KW (1991c) A method for the
determination of phenanthrene and five isomeric hydroxyphenanthrenes
in urine of man and rat. In: Cooke M, Loening K, & Merritt J ed.
Polynuclear aromatic hydrocarbons: Measurements, means, and
metabolism. Columbus, Ohio, Battelle Press, pp 389-403.
Grimmer G, Dettbarn G, & Jacob J (1993) Biomonitoring of polycyclic
aromatic hydrocarbons in highly exposed coke plant workers by
measurement of urinary phenanthrene and pyrene metabolites (phenols
and dihydrodiols). Int Arch Occup Environ Health, 65: 189-199.
Grimmer G, Dettbarn G, Naujack KW, & Jacob J (1994) Relationship
between inhaled PAH and urinary excretion of phenanthrene, pyrene and
benzo [a]pyrene metabolites in coke plant workers. Polycyclic Aromat
Compd, 5: 269-277.
Grob K & Grob G (1974) Organic substances in potable water and in its
precursor. Part II: Applications in the area of Zürich. J Chromatogr,
90: 303-313.
Groenewegen D & Stolp H (1976) [Microbial breakdown of polycyclic
aromatic hydrocarbons.] Zentralbl Bakteriol Parasitenkd Infektionskr
Hyg Abt 1 Orig B, 161: 225-232 (in German).
Grosjean D (1983) Polycyclic aromatic hydrocarbons in Los Angeles air
from samples collected on teflon, glass and quartz filters. Atmos
Environ, 17: 2565-2573.
Grosser RJ, Warshawsky D, & Vestal JR (1995) Mineralization of
polycyclic and N-heterocyclic aromatic compounds in
hydrocarbon-contaminated soils. Environ Toxicol Chem, 14: 375-382.
Grover PL & Sims P (1968) Enzyme-catalysed reactions of polycyclic
hydrocarbons with deoxyribonucleic acid and protein in vitro.
Biochem J, 110: 159-160.
Grover PL, Sims P, Huberman E, Marquardt H, Kuroki T, & Heidelberger C
(1971) In vitro transformation of rodent cells by K-region
derivatives of polycyclic hydrocarbons. Proc Natl Acad Sci USA, 68:
1098-1101.
Grover PL, Sims P, Mitchel BCU, & Roe FJ (1975) The carcinogenicity of
polycyclic hydrocarbon epoxides in newborn mice. Br J Cancer, 31: 182-
188.
Grover PL, MacNicoll AD, Sims P, Easty GC, & Neville AM (1980)
Polycyclic hydrocarbon activation and metabolism in epithelial cell
aggregates prepared from human mammary tissue. Int J Cancer, 26: 467-
475.
Gückel W, Synnatschke R, & Rittig R (1973) A method for determining
the volatility of active ingredients used in plant protection. Pestic
Sci, 4: 137-147.
Guengerich FP & Shimada T (1991) Oxidation of toxic and carcinogenic
chemicals by human cytochrome P-450 enzymes. Chem Res Toxicol, 4: 391-
407.
Guenther FR, Chesler SN, Gordon GE, & Zoller WH (1988) Residential
wood combustion: A source of atmospheric polycyclic aromatic
hydrocarbons. J High Resol Chromatogr Chromatogr Commun, 11: 761-766.
Guerin MR (1977) Energy sources of polycyclic aromatic hydrocarbons.
Oak Ridge, Tennessee, Oak Ridge National Laboratory, Analytical
Chemistry Division, pp 1-78 (EPA Conf/Doc. 770-130-2).
Guerin MR, Epler JL, Griest WH, Clark BR, & Rao TK (1978) Polycyclic
aromatic hydrocarbons from fossil fuel conversion processes. In: Jones
PW & Freudenthal RI ed. Carcinogenesis. Volume 3: Polynuclear aromatic
hydrocarbons. New York, Raven Press, pp 21-33.
Guggenberger J, Krammer G, & Lindenmüller W (1981) [Contribution to
the determination of the emission of polycyclic aromatic hydrocarbons
from large capacity furnaces.] Staub-Reinhalt Luft, 41: 339-344 (in
German).
Guicherit R & Schulting FL (1985) The occurrence of organic chemicals
in the atmosphere of the Netherlands. Sci Total Environ, 43: 193-219.
Guillemin MP, Herrera H, Huynh CK, Droz PO, & Vu Duc T (1992)
Occupational exposure of truck drivers to dust and polynuclear
aromatic hydrocarbons: A pilot study in Geneva, Switzerland. Int Arch
Occup Environ Health, 63: 439-447.
Gupta RS & Goldstein S (1981) Mutagen testing in the human fibroblasts
diphtheria toxin resistance (HF Dipr) system. In: De Serres FJ & Ashby
J ed. Evaluation of short-term tests for carcinogens. Report of the
international collaborative programme. New York, Elsevier North
Holland, pp 614-625 (Progress in Mutation Research, Volume 1).
Gupta RS & Singh B (1982) Mutagenic responses of five independent
genetic loci in CHO cells to a variety of mutagens. Development and
characteristics of a mutagen screening system based on selection for
multiple drug-resistant markers. Mutat Res, 94: 449-466.
Gupta RC, Earley K, & Sharma S (1988) Use of human peripheral blood
lymphocytes to measure DNA binding capacity of chemical carcinogens.
Proc Natl Acad Sci USA, 85: 3513-3517.
Gurtoo HL, Vaught JB, Marinello AJ, Paigen B, Gessner T, & Bolanowska
W (1980) High-pressure liquid chromatographic analysis of
benzo [a]pyrene metabolism by human lymphocytes from donors of
different aryl hydrocarbon hydroxylase inducibility and antipyrine
half-lives. Cancer Res, 40: 1305-1310.
Gustavsson P (1989) Cancer and ischemic heart disease in occupatioal
groups exposed to combustion products. Solna, National Institute of
Occupational Health, pp 1-28 (World and Health series No. 21).
Gustavsson P & Reuterwall C (1990) Mortality and incidence of cancer
among Swedish gas workers. Br J Ind Med, 47: 169-174.
Gustavsson P, Fellenius E, & Hogstedt C (1987) Possible causes of
increased lung cancer incidence among butchers and slaughterhouse
workers. Scand J Work Environ Health, 13: 518-523.
Gustavsson P, Gustavsson A, & Hogstedt C (1988) Excess of cancer in
Swedish chimney sweeps. Br J Ind Hyg, 45: 777-781.
Habs M, Schmähl S, & Misfeld J (1980) Local carcinogenicity of some
environmentally relevant polycyclic aromatic hydrocarbons after
lifelong topical application to mouse skin. Geschwulstforschung, 50:
266-274.
Habs M, Jahn SAA, & Schmähl D (1984) Carcinogenic activity of
condensate from coloquint seeds (Citrullus colocynthis) after
chronic epicutaneous administration to mice. J Cancer Res Clin Oncol,
108: 154-156.
Haddow A, Scott CM, & Scott JD (1937) The influence of certain
carcinogenic and other hydrocarbons on body growth in the rat. Proc R
Soc Ser B, 122: 477-507.
Hagemann R, Virelizier H, Gaudin D, & Pesneau A (1982) Polycyclic
aromatic hydrocarbons in exhaust particles emitted from gasoline and
diesel automobile engines. Toxicol Environ Chem, 5: 227-236.
Hall M & Grover PL (1990) Polycyclic aromatic hydrocarbons:
Metabolism, activation and tumour initiation. In: Cooper CS & Grover
PL ed. Handbook of experimental pharmacology. Berlin, Springer-Verlag,
Volume 94/I, pp 327-372.
Hall AT & Oris JT (1991) Anthracene reduces reproductive potential and
is maternally transferred during long-term exposure in fathead
minnows. Aquat Toxicol, 19: 249-264.
Hall M, Forrester LM, Parker DK, Grover PL, & Wolf CR (1989) Relative
contribution of various forms of cytochrome P450 to the metabolism of
benzo [a]pyrene by human liver microsomes. Carcinogenesis, 10: 1815-
1821.
Halsall CJ, Coleman PJ, Davies BJ, Burnett V, Waterhouse KS,
Harding-Jones P, & Jones KC (1994) Polycyclic aromatic hydrocarbons in
UK urban air. Environ Sci Technol, 28: 2380-2386.
Hambrick GA, Delaume RD, & Patrick WH Jr (1980) Effects of estuarine
sediment, pH and oxidation-reduction potential on microbial
hydrocarbon degradation. Appl Environ Microbiol, 40: 365-369.
Hamburg Environment Office (1993) [Report on an investigation into
polycyclic aromatic hydrocarbons (PAHs) in Hamburg's waters.] Hamburg,
63 (Report 42/93 13-16) (in German).
Hammond WG, Gabriel A, Paladugu RR, Azumi N, Hill RL, & Benfield JR
(1987) Differential susceptibility to bronchial carcinogenesis in
syngeneic hamsters. Cancer Res, 47: 5202-5206.
Hampton CV, Pierson WR, Schuetzle D, & Harvey TM (1983) Hydrocarbon
gases emitted from vehicles on the road. 2. Determination of emission
rates from diesel and spark ignition vehicles. Environ Sci Technol,
17: 699-708.
Hannah JB, Hose JE, Landolt ML, Miller BS, Felton SP, & Iwaoka WT
(1982) Benzo [a]pyrene-induced morphologic and developmental
abnormalities in rainbow trout Salmo gairdneri Richardson. Arch
Environ Contam Toxicol, 11: 727-734.
Hansen ES (1989) Cancer incidence in an occupational cohort exposed to
bitumen fumes. Scand J Work Environ Health, 15: 101-105.
Hansen ÅM, Olsen IB, Holst E, & Poulsen OM (1991a) Validation of a
high-performance liquid chromatography/fluorescence detection method
for the simultaneous quantification of fifteen polycyclic aromatic
hydrocarbons. Ann Occup Hyg, 35: 603-611.
Hansen ÅM, Poulsen OM, & Christensen JM (1991b) Correlation of levels
of volatile versus carcinogenic particulate polycyclic aromatic
hydrocarbons in air samples from smokehouses. Int Arch Occup Environ
Health, 63: 247-252.
Hansen ÅM, Olsen IB, & Poulsen OM (1992) Polycyclic aromatic
hydrocarbons in air samples of meat smokehouses. Sci Total Environ,
126: 17-26.
Hansen ÅM, Omland , Poulsen OM, Sherson D, Sigsgaard T, Christensen
JM, & Overgaard E (1994) Correlation between work process-related
exposure to polycyclic aromatic hydrocarbons and urinary levels of
a-naphthol, b-naphthylamine and 1-hydroxypyrene in iron foundry
workers. Int Arch Occup Environ Health, 65: 385-394.
Hansen AM, Christensen JM, & Sherson D (1995) Estimation of reference
values for urinary 1-hydroxypyrene and a-naphthol in Danish workers.
Sci Total Environ, 163: 211-219.
Hardin BD, Bond GP, Sikov MR, Andrew FD, Beliles RP, & Niemeier RW
(1981) Testing of selected workplace chemicals for teratogenic
potential. Scand J Work Environ Health, 7: 66-75.
Hardin BD, Schuler RL, Burg JR, Booth GM, Hazelden KP, MacKenzie KM,
Piccirillo VJ, & Smith KN (1987) Evaluation of 60 chemicals in a
preliminary developmental toxicity test. Teratog Carcinog Mutag, 7:
29-48.
Harkov R & Greenberg A (1985) Benzo(a)pyrene in New Jersey. Results
from a twenty-seven-site study. J Air Pollut Control Assoc, 35: 238-
243.
Harkov R, Greenberg A, Darack F, Daisey JM, & Lioy PJ (1984)
Summertime variations in polycyclic aromatic hydrocarbons at four
sites in New Jersey. Environ Sci Technol, 18: 187-291.
Harper KH (1957) The metabolism of pyrene. Br J Cancer, 11: 499-507.
Harper KH (1958a) The intermediary metabolism of pyrene. Br J Cancer,
12: 116-120.
Harper KH (1958b) The intermediary metabolism of 3,4-benzpyrene. Br J
Cancer, 12: 121-128.
Harper KH (1958c) The intermediary metabolism of 3,4-benzyprene: The
biosynthesis and identification of the X1 and X2 metabolites. Br J
Cancer, 12: 645-660.
Harper KH (1959a) The intermediary metabolism of polycyclic
hydrocarbons. Br J Cancer, 13: 718-731.
Harper KH (1959b) Metabolism of 1,2-benzanthracene in the rabbit. Br J
Cancer, 13: 746-750.
Harper BL & Legator MS (1987) Pyridine prevents the clastogenicity of
benzene but not of benzo [a]pyrene or cyclophosphamide. Mutat Res,
179: 23-31.
Harper BL, Sadagopa Ramanujam VM, Gad-El-Karim MM & Legator MS (1984)
The influence of simple aromatics on benzene clastogenicity. Mutat
Res, 128: 105-114.
Harris CC, Autrup H, Connor R, Barrett LA, McDowell EM, & Trump BF
(1976) Interindividual variation in binding of benzo [a]pyrene to DNA
in cultured human bronchi. Science, 194: 1067-1069.
Harris CC, Autrup H, Stoner G, Yang SK, Leutz JC, Gelboin HV, Selkirk
JK, Conner RJ, Barrett LA, Jones RT, McDowell E, & Trump BF (1977)
Metabolism of benzo [a]pyrene and 7,12-dimethylbenz [a]anthracene in
cultured human bronchus and pancreatic duct. Cancer Res, 37: 3349-
3355.
Harris CC, Hsu IC, Stoner GD, Trump BF, & Selkirk JK (1978a) Human
pulmonary alveolar macrophages metabolise benzo [a]pyrene to
proximate and ultimate mutagens. Nature, 272: 633-634.
Harris CC, Autrup H, & Stoner G (1978b) Metabolism of benzo [a]pyrene
in cultured human tissues and cells. In: Gelboin HV & Ts'o POP ed.
Polycyclic hydrocarbons and cancer. New York, Academic Press, Volume
2, pp 331-342.
Harris CC, Autrup H, Stoner GD, Trump BF, Hillman E, Schafer PW, &
Jeffrey AM (1979) Metabolism of benzo [a]pyrene,
N-nitrosodimethylamine, and N-nitrosopyrrolidine and
identification of the major carcinogen-DNA adducts formed in cultured
human esophagus. Cancer Res, 39: 4401-4406.
Harris CC, Grafstom RC, Shamsuddin AM, Sinopoli NT, Trump BF, & Autrup
H (1984) Carcinogen metabolism and carcinogen-DNA adducts in human
tissues and cells. In: Greim H, Jung R, Kramer M, Marquardt H, & Oesch
F ed. Biochemical basis of chemical carcinogenesis. New York, Raven
Press, pp 123-135.
Harris CC, Vahakangas K, Newman MJ, Trivers GE, Shamsuddin A, Sinopoli
N, Mann DL, & Wright WE (1985) Detection of benzo [a]pyrene diol
epoxide-DNA adducts in peripheral blood lymphocytes and antibodies to
the adducts in serum from coke oven workers. Proc Natl Acad Sci USA,
82: 6672-6676.
Hart KM & Pankow JF (1994) High-volume air sampler for particle and
gas sampling. 2. Use of backup filters to correct for the adsorption
of gas-phase polycyclic aromatic hydrocarbons to the front filter.
Environ Sci Technol, 28: 655-661.
Hartung H & Koch D (1991) [Environmental research plan from the
Federal Ministry for Environment and Reactor Safety (Research Project
103 03 220/01): Waste disposal: Quantity, recycling and disposal of
used motor tyres in the Federal Republic of Germany.] Berlin, Ministry
of the Environment, pp 73-74 (Report No. UBA-FB 89-149, Text 52/91)
(in German).
Harvey RG (1996) Mechanisms of carcinogenesis of polycyclic aromatic
compounds. Polycyclic Aromat Compd, 9: 1-23.
Häsänen E, Pohjola V, Pyysalo H, & Wickström K (1983) Polycyclic
aromatic hydrocarbons in the Finnish wood-heated sauna. Kemia Kemi,
10: 27-29.
Haseltine WA, Lo KM, & D'Andrea AD (1980) Preferred sites of strand
scission in DNA modified by anti-diol epoxide of benzo [a]pyrene.
Science, 209: 929-931.
Hass BS & Applegate HG (1975) The effects of unsubstituted polycyclic
aromatic hydrocarbons on the growth of Escherichia
coli. Chem-Biol Interactions, 10: 265-268.
Hatch MC, Warburton D, & Santella RM (1990) Polycyclic aromatic
hydrocarbon-DNA adducts in spontaneously aborted fetal tissue.
Carcinogenesis, 11: 1673-1675.
van Hattum B, Cofino WP, & Feenstra JF (1993) Environmental aspects of
produced water discharges from oil and gas production on the Dutch
continental shelf: Part II. A literature review of characteristics of
produced water from offshore platforms. Amsterdam, Oil and Gas
Exploitation and Production Association, 64 pp.
Haubenstricker ME, Meier PG, Mancy KH, & Brabec MJ (1990) Rapid
toxicity testing based on yeast respiratory activity. Bull Environ
Contam Toxicol, 44: 669-674.
Haudenschild R, Poffer J, Schott T, & Deuber A (1990) [Application of
corrosion inhibitor in a bus garage.] Illus Z Arb Sicherheitstech, 37:
6-8 (in German).
Haugen A, Becher G, Benestad C, Vahakangas K, Trivers GE, Newman MJ, &
Harris CC (1986) Determination of polycyclic aromatic hydrocarbons in
the urine, benzo(a)pyrene diol epoxide-DNA adducts in lymphocyte DNA,
and antibodies to the adducts in sera from coke oven workers exposed
to measured amounts of polycyclic aromatic hydrocarbons in the work
atmosphere. Cancer Res, 46: 4178-4183.
Hawkins WE, Walker WW, Overstreet RM, Lytle TF, & Lytle JS (1988)
Dose-related carcinogenic effects of water-borne benzo [a]pyrene on
livers of two small fish species. Ecotoxicol Environ Saf, 16: 219-231.
Hawkins WE, Walker WW, Overstreet RM, Lytle JS, & Lytle TF (1990)
Carcinogenic effects of some polycyclic aromatic hydrocarbons on the
Japanese medaka and guppy in waterborne exposures. Sci Total Environ,
94: 155-167.
Hawley G (1987) Hawley's condensed chemical dictionary, 11th rev ed.
New York, Van Nostrand Reinhold Co., p 6.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1984a) Products obtained
during combustions of polymers under simulated incinerator conditions:
I. Polyethylene. J Chromatogr, 314: 263-273.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1984b) Products obtained
during combustions of polymers under simulated incinerator conditions:
II. Polystyrene. J Chromatogr, 315: 201-210.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1984c) Products obtained
during combustions of polymers under simulated incinerator conditions:
III. Polyvinyl chloride. J Chromatogr, 315: 211-221.
Hawley-Fedder RA, Parsons ML, & Karasek FW (1987) Identification of
organic compounds produced during combustion of a polymer mixture. J
Chromatogr, 387: 207-221.
Hawthorne SB & Miller DJ (1987) Extraction and recovery of polycyclic
aromatic hydrocarbons from environmental solids using supercritical
fluids. Anal Chem, 59: 1705-1708.
Hawthorne SB, Krieger MS, & Miller DJ (1989a) Supercritical carbon
dioxide extraction of polychlorinated biphenyls, polycyclic aromatic
hydrocarbons, heteroatom-containing polycyclic aromatic hydrocarbons,
and n-alkanes from polyurethane foam sorbents. Anal Chem, 61: 736-
740.
Hawthorne SB, Miller DJ, & Krieger MS (1989b) Coupled SFE-GC: A rapid
and simple technique for extracting, identifying, and quantitating
organic analytes from solid and sorbent resins. J Chromatogr Sci, 27:
347-354.
Hawthorne SB, Miller DJ, Langenfeld JJ, & Krieger MS (1992) PM-10
high-volume collection and quantitation of semi- and nonvolatile
phenols, methoxylated phenols, alkanes, and polycyclic aromatic
hydrocarbons from winter urban air and their relationship to wood
smoke emissions. Environ Sci Technol, 26: 2251-2262.
He S & Baker R (1991) Micronuclei in mouse skin cells following
in vivo exposure to benzo [a]pyrene,
7,12-dimethylbenz [a]anthracene, chrysene, pyrene and urethane.
Environ Mol Mutag, 17: 163-168.
Heaton DM, Bartle, KD, Clifford AA, Meyers P, & King BW (1994) Rapid
separation of polycyclic aromatic hydrocarbons by packed column
supercitical fluid chromatography. Chromatographia, 39: 607-611.
Hecht SS, Bondinell WE, & Hoffmann D (1974) Chrysene and
methylchrysenes: Presence in tobacco smoke and carcinogenicity. J Natl
Cancer Inst, 53: 1121-1133.
Hecht SS, Loy M, Maronpot RR, & Hoffmann D (1976a) A study of chemical
carcinogenesis: Comparative carcinogenicity of 5-methylchrysene,
benzo(a)pyrene, and modified chrysenes. Cancer Lett, 1: 147-153.
Hecht SS, Loy M, & Hoffmann D (1976b) On the structure and
carcinogenicity of the methylchrysenes. In: Freudenthal R & Jones PW
ed. Carcinogenesis: A comprehensive survey. Volume 1: Polynuclear
aromatic hydrocarbons chemistry, metabolism, and carcinogenesis. New
York, Raven Press, pp 325-340.
Hecht SS, Loy M, Mazzarese R, & Hoffmann D (1978) On the
carcinogenicity of 5-methylchrysene: Structure-activity studies and
metabolism. Polycyclic Hydrocarbons Cancer, 1: 119-130.
Hecht SS, Grabowski W, & Groth K (1979) Analysis of faeces for
benzo [a]pyrene after consumption of charcoal-broiled beef by rats
and humans. Food Cosmet Toxicol, 17: 223-227.
Hecht SS, LaVoie E, Amin S, Bedenko V, & Hoffmann D (1980) On the
metabolic activation of the benzofluoranthenes. In: Bjorseth A &
Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry and
biological effects. Columbus, Ohio, Battelle Press, pp 417-433.
Hecht SS, LaVoie EJ, Bedenko V, Hoffmann D, Sardella DJ, Boger E, &
Lehr RE (1981) On the metabolic activation of dibenzo [a,i]pyrene and
dibenzo [a,h]pyrene. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: Chemical analysis and biological fate. Columbus, Ohio,
Battelle Press, pp 43-45.
Hecht SS, Radok L, Amin S, Huie K, Melikian AA, Hoffmann D, Pataki J,
& Harvey RG (1985) Tumorigenicity of 5-methylchrysene dihydrodiols and
dihydrodiol epoxides in newborn mice and on mouse skin. Cancer Res,
45: 1449-1452.
Hecht SS, Melikian AA, & Amin S (1986) Methylchrysenes as probes for
the mechanism of metabolic activation of carcinogenic methylated
polynuclear aromatic hydrocarbons. Acc Chem Res, 19: 174-180.
Hecht SS, Amin S, Huie K, Melikian AA, & Harvey RG (1987) Enhancing
effect of a bay region methyl group on tumorigenicity in newborn mice
and mouse skin of enantiomeric bay region diol epoxides formed
stereoselectively from methylchrysenes in mouse epidermis. Cancer Res,
47: 5310-5315.
Hecht SS, El-Bayoumy K, Rivenson A, & Amin S (1994) Potent mammary
carcinogenicity in female CD rats of a fjord region diol-epoxide of
benzo(c)phenanthrene compared to a bay region diol-epoxide of
benzo(a)pyrene. Cancer Res, 54: 21-24.
Hecht SS, Amin S, Lin J-M, Rivenson A, Kurtzke C, & El-Bayoumy K
(1995) Short communication. Mammary carcinogenicity in female CD rats
of a diol epoxide metabolite of fluoranthene, a commonly occurring
environmental pollutant. Carcinogenesis, 16: 1433-1435.
Heeschen WH (1985) [Scientific working group on chemicals in human
milk.] Arch Gynekol, 238: 199-216 (in German).
Heflich RH, Thornton-Manning JR, Kinouchi T, & Beland FA (1990)
Mutagenicity of oxidized microsomal metabolites of 1-nitropyrene in
Chinese hamster ovary cells. Mutagenesis, 5: 151-157.
Heidelberger C & Davenport GR (1961) Local functional components of
carcinogenesis. Acta Unio Int Contra Cancrum, 17: 55-63.
Heidelberger C & Jones HB (1948) The distribution of radioactivity in
the mouse following administration of dibenzanthracene labeled in the
9 and 10 positions with carbon 14*. Cancer, 1: 252-260.
Heidelberger C & Weiss SM (1951) The distribution of radioactivity in
mice following administration of 3,4-benzpyrene-5-C14 and
1,2,5,6-dibenzanthracene-9,10-C14. Cancer Res, 11: 885-891.
Heikkilä PR, Hämeilä M, Pyy L, & Rauni P (1987) Exposure to creosote
in the impregnation and handling of impregnated wood. Scand J Work
Environ Health, 13: 431-437.
Heinrich U (1989) Exhaust-specific carcinogenic effects of polycyclic
aromatic hydrocarbons and their significance for the estimation of the
exhaust exposure-related lung cancer risk. In: Mohr U, Bates DV,
Dungworth DL, Lee PN, McClellan RO, & Roe FJC ed. Assessment of
inhalation hazards: Integration and extrapolation using diverse data.
Berlin, Springer-Verlag, pp 301-313.
Heinrich U (1995) Do PAH contribute to the lung tumor response in
diesel engine exhaust-exposed rats? In: Fifteenth international
symposium on polycyclic aromatic compounds: Chemistry, biology and
environmental impact, Belgirate, Italy, 19-22 September 1995. Ispra,
Joint Research Centre European Commission, p 150.
Heinrich U, Pott F, Mohr U, Fuhst R, & König J (1986a) Lung tumours in
rats and mice after inhalation of PAH-rich emissions. Exp Pathol, 29:
29-34.
Heinrich U, Pott F, & Rittinghausen S (1986b) Comparison of chronic
inhalation effects in rodents after long-term exposure to either coal
oven flue gas mixed with pyrolysed pitch or diesel engine exhaust. In:
Ishinishi N, Koizumi A, McClellan RO, & Stöber W ed. Carcinogenic and
mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier
Science Publishers, pp 441-457.
Heinrich U, Peters L, Creutzenberg O, Dasenbrock C, & Hoymann HG
(1994a) Inhalation exposure of rats to tar/pitch condensation aerosol
or carbon black alone or in combination with irritant gases. In: Mohr
U, Dungworth DL, Mauderly JL, & Oberdörster G ed. Toxic and
carcinogenic effects of solid particles in the respiratory tract.
Washington DC, International Life Sciences Institute Press, pp 433-
441.
Heinrich U, Dungworth DL, Pott F, Peters L, Dasenbrock C, Levsen K,
Koch W, Creutzenberg O, & Schulte A (1994b) The carcinogenic effects
of carbon black particles and tar-pitch condensation aerosol after
inhalation exposure of rats. Ann Occup Hyg, 38 (suppl): 351-356.
Heinrich U, Roller M, & Pott F (1994c) Estimation of a lifetime unit
lung cancer risk for benzo [a]pyrene based on tumor rates in rats
exposed to coaltar/pitch condensation aerosol. Toxicol Lett, 72: 155-
161.
Heit M (1985) The relationship of a coal fired power plant to the
levels of polycyclic aromatic hydrocarbons (PAH) in the sediment of
Cayuga Lake. Water Air Soil Pollut, 24: 41-61.
Heit M, Klusek CS, & Miller KM (1980) Trace element, radionuclide, and
polynuclear aromatic hydrocarbon concentrations in Unionidae mussels
from northern Lake George. Environ Sci Technol, 14: 465-468.
Heitkamp MA, Freeman JP, Miller DW, & Cerniglia CE (1988) Pyrene
degradation by a Mycobacterium sp.: Identification of ring oxidation
and ring fission products. Appl Environ Microbiol, 54: 2556-2565.
Heitmuller PT, Hollister TA, & Parrish PR (1981) Acute toxicity of 54
industrial chemicals to sheepshead minnows Cyprinodon
variegatus. Bull Environ Contam Toxicol, 27: 596-604.
Heldal M, Norland S, Lien T, Knutsen G, Tjessem K, & Aarberg A (1984)
Toxic responses of the green alga Dunaliella bioculata
(Chlorophycea, Volvocales) to selected oxidised hydrocarbons. Environ
Pollut, A34: 119-132.
Helfrich J & Armstrong DE (1986) Polycyclic aromatic hydrocarbons in
sediments of the southern basin of Lake Michigan. J Great Lakes Res,
12: 192-199.
Hembrock-Heger A & König W (1990) Occurrence and transfer of
polycyclic aromatic hydrocarbons in soil and plants.] Düsseldorf,
VDI-Verlag, pp 815-830 (VDI Report No. 837) (in German).
Hemminki K & Vainio H (1979) Preferential binding of benzo [a]pyrene
into nuclear matrix fraction. Cancer Lett, 6: 167-173.
Hemminki K, Grzybowska E, Chorazy M, Twardowska-Saucha K, Srczynski
JW, Putman KL, Randerath K, Phillips DH, Hewer A, Santella RM, &
Perera FP (1990a) DNA adducts in humans related to occupational and
environmental exposure to aromatic compounds. In: Vainio H, Sorsa M, &
McMichael AJ ed. Complex mixtures and cancer risk. Lyon, International
Agency for Research on Cancer, pp 181-192 (IARC Scientific
Publications No. 104).
Hemminki K, Grzybowska E, Chorazy M, Twardowska-Saucha K, Sroczynski
JW, Putman KL, Randerath, Phillips DH, & Hewer A (1990b) Aromatic DNA
adducts in white blood cells of coke workers. Int Arch Occup Environ
Health, 62: 467-470.
Hendricks JP, Meyers TR, Shelton DW, Casteel JL, & Bailey GS (1985)
Hepatocarcinogenicity of benzo [a]pyrene to rainbow trout by dietary
exposure and intraperitoneal injection. J Natl Cancer Inst, 74: 839-
851.
Henry MC & Kaufman DG (1973) Clearance of benzo [a]pyrene from
hamster lungs after administration on coated particles. J Natl Cancer
Inst, 51: 1961-1964.
Henry MC, Port CD & Kauhman DG (1975) Importance of physical
properties of benzo (a)pyrene-ferric oxide mixtures in lung tumor
induction. Cancer Res, 35: 207-217.
Herbert R, Marcus M, Wolff MS, Perera FP, Andrews L, Godbold JH,
Rivera M, Stefanidis M, Lu XQ, Landrigan PJ, & Santella RM (1990a)
Detection of adducts of deoxyribonucleic acid in white blood cells of
roofers by 32P-postlabeling. Scand J Work Environ Health, 16: 135-
143.
Herbert R, Marcus M, Wolff MS, Perera FP, Andrews L, Godbold JH,
Rivera M, Stefanidis M, Lu XQ, Landrigan PJ, & Santella RM (1990b) A
pilot study of detection of DNA adducts in white blood cells of
roofers by 32P-postlabelling. In: Vainio H, SorsaM, & McMichael AJ
ed. Complex mixtures and cancer risk. Lyon, International Agenca for
Research on Cancer, pp 205-214 (IARC Scientific Publications No. 104).
Herbes SE (1981) Rates of microbial transformation of polycyclic
aromatic hydrocarbons in water and sediments in the vicinity of
coal-coking waste water discharge. Appl Environ Microbiol, 41: 20-28.
Herbes SE, Southworth GR, Shaeffer DL, Griest WH, & Maskarinec MP
(1980) Critical pathways of polycyclic aromatic hydrocarbons in
aquatic environments. In: Witschi H ed. The scientific basis of
toxicity assessment.Amsterdam, Elsevier Biomedical Press, pp 113-128.
Herlan A (1982) Content of polycyclic aromatics in middle distillates.
Zentralbl Bakteriol Hyg I Abt Orig B, 176: 249-268.
Hermann M (1981) Synergistic effects of individual polycyclic aromatic
hydrocarbons on the mutagenicity of their mixtures. Mutat Res, 90:
399-409.
Hermann M, Durand JP, Charpentier JM, Chaude O, Hofnung M, Petroff N,
Vandecasteele JP, & Weill N (1980) Correlations of mutagenic activity
with polynuclear aromatic hydrocarbon content of various mineral oils.
In: Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons:
Chemical analysis and biological fate. Columbus, Ohio, Battelle Press,
pp 899-916.
Heussner JC, Ward JB, & Legator MS (1985) Genetic monitoring of
aluminium workers exposed to coal tar pitch volatiles. Mutat Res, 155:
143-155.
Hinoshita F, Hardin JA, & Sherr DH (1992) Fluoranthene induces
programmed cell death and alters growth of immature B cell populations
in bone marrow cultures. Toxicology, 73: 203-218.
Hirano T, Stanton M, & Layard M (1974) Measurement of epidermoid
carcinoma development induced in the lungs of rats by
3-methylcholanthrene-containing beeswax pellets. J Natl Cancer Inst,
53: 1209-1215.
Hirom PC, Chipman JK, Millburn P, & Pue MA (1983) Enterohepatic
circulation of the aromatic hydrocarbons benzo(a)pyrene and
naphthalene. In: Rydstrom J, Montelius J, & Bengtsson M ed.
Extrahepatic drug metabolism and chemical carcinogenesis. Amsterdam,
Elsevier Science Publishers, pp 275-281.
Hites RA (1981) Sources and fates of atmospheric polycyclic aromatic
hydrocarbons. In: Macias ES & Hopke PK ed. Atmospheric aerosol,
source/air quality relationships (ACS Symposium Series 167).
Washington DC, American Chemical Society, pp 187-196.
Hites RA (1989) Mass spectrometry of polycyclic aromatic compounds.
In: Vo-Dinh T ed. Chemical analysis of polycyclic aromatic compounds.
New York, John Wiley & Sons, pp 219-261.
Hites RA, LaFlamme JW, & Farrington JW (1977) Sedimentary polycyclic
aromatic hydrocarbons: The historical record. Science, 198: 829-831.
Hites RA, LaFlamme RE, & Windsor JG Jr (1980) Polycyclic aromatic
hydrocarbons in marine/aquatic sediments: Their ubiquity. In: Petrakis
L & Weiss FT ed. Petroleum in the marine environment (Advances in
Chemistry Series No. 185). Washington DC, American Chemical Society,
pp 289-311.
Ho CH, Clark BR, Guerin MR, Barkenbus BD, Rao TK, & Epler JL (1981)
Analytical and biological analyses of test materials from the
synthetic fuel technologies: IV. Studies of chemical
structure-mutagenic activity relationships of aromatic nitrogen
compounds relevant to synfuels. Mutat Res, 85: 335-345.
Hoch-Ligeti C (1941) Studies on the changes in the lymphoid tissue of
mice treated with carcinogenic and noncarcinogenic hydrocarbons.
Cancer Res, 1: 484-488.
Hodgson RM, Weston A, & Grover PL (1983) Metabolic activation of
chrysene in mouse skin: Evidence for the involvement of a
triol-epoxide. Carcinogenesis, 4: 1639-1643.
Hodson J & Williams NA (1988) The estimation of the adsorption (Koc)
for soils by high performance liquid chromatography. Chemosphere, 17:
67-77.
Hoff RM & Chan KW (1987) Measurement of polycyclic aromatic
hydrocarbons in the air along the Niagara River. Environ Sci Technol,
21: 556-561.
Hoffmann K (1993) [Risk assessment and restoration concept for
PAH-contaminated soil.] Altlasten-Spektrum, 2: 93-99 (in German).
Hoffmann DJ & Gay ML (1981) Embryotoxic effects of benzo [a]pyrene,
chrysene, and 7,12-dimethylbenz [a]anthracene in petroleum
hydrocarbon mixtures in mallard ducks. J Toxicol Environ Health, 7:
775-787.
Hoffmann D & Wynder EL (1962) Analytical and biological studies on
gasoline engine exhaust. Natl Cancer Inst Monogr, 9: 91-110.
Hoffmann D & Wynder EL (1966) [Carcinogenic effect of dibenzopyrenes.]
Z Krebsforsch, 68: 137-149 (in German).
Hoffmann D, Rathkamp G, Nesnow S, & Wynder EL (1972) Fluoranthenes:
Quantitative determination in cigarette smoke, formation by pyrolysis,
and tumor-initiating activity. J Natl Cancer Inst, 49: 1165-1175.
Hoffmann D, Schmeltz I, Hecht SS, & Wynder EL (1978) Tobacco
carcinogenesis. In: Gelboin HV & Ts'o POP ed. Polycyclic hydrocarbons
and cancer, Volume 1. New York, Academic Press, pp 85-117.
Hoffman EJ, Mills GL, Latimer JS, & Quinn JG (1984) Urban runoff as a
source of polycyclic aromatic hydrocarbons to coastal waters. Environ
Sci Technol, 18: 580-587.
Hoffman EJ, Latimer JS, Hunt CD, Mills GL, & Quinn JG (1985)
Stormwater runoff from highways. Water Air Soil Pollut, 25: 349-364
Holcombe GW, Phipps GL, & Fiandt JT (1983) Toxicity of selected
priority pollutants to various aquatic organisms. Ecotoxicol Environ
Saf, 7: 400-409.
Holcombe GW, Phipps GL, Knuth ML, & Felhaber T (1984) The acute
toxicity of selected substituted phenols, benzenes and benzoic acid
esters to fathead minnows Pimephales promelas. Environ Pollut, A35:
367-381.
Holder G, Yagi H, Dansette P, Jerina DM, Levin W, Lu AYH, & Conney AH
(1974) Effects of inducers and epoxide hydrase on the metabolism of
benzo [a]pyrene by liver microsomes and a reconstituted system:
Analysis by high pressure liquid chromatography. Proc Natl Acad Sci
USA, 71: 4356-4360.
Hollstein M, McCann J, Angelosanto FA, & Nichols WW (1979) Short-term
tests for carcinogens and mutagens. Mutat Res, 65: 133-226.
Holman A, Karlsson A, Bratt I, Raihle G, & Hogstedt B (1994) Increased
frequency of micronuclei in lymphocytes of Swedish chimney sweeps. Int
Arch Occup Environ Health, 66: 185-187.
Holoubek I, Housková L, Seda Z, Holoubkova I, Korínek P, Bohácek Z, &
Cáslavsk J (1990) Project TOCOEN. The fate of selected organic
pollutants in the environment. III. Water and sediments 1988. Toxicol
Environ Chem, 29: 29-35.
Homburger F, Russfield AB, Baker JR, & Tregier A (1962) Experimental
chemotherapy in chemically induced mouse tumors and their transplants.
Cancer Res, 22: 368-375.
Honda T, Kiyozumi M, & Kojima S (1990) Alkylnaphthalene: XI. Pulmonary
toxicity of naphthalene, 2-methylnaphthalene, and
isopropylnaphthalenes in mice. Chem Pharm Bull, 38: 3130-3135.
Honkakoski P & Lang MA (1989) Mouse liver phenobarbital-inducible P450
system: Purification, characterization, and differential inducibility
of four cytochrome P450 isozymes from the D2 mouse. Arch Biochem
Biophys, 273: 42-57.
Hooftman RJ & Evers-de Ruiter A (1992a) Investigations into aquatic
toxicity of phenanthrene (cover-report for reproduction tests with the
waterflea Daphnia magna and an early life stage test with the zebra
fish Brachydanio rerio. Delft, The Netherlands, TNO Institute of
Environmental Science, 23 pp (TNO Report No. IMW-R 92/290).
Hooftman RJ & Evers-de Ruiter A (1992b) The toxicity and uptake of
fluoranthene in Brachidanio rerio in an early life stage test.
Delft, TNO Institute of Environmental Science, 36 pp (TNO Report No.
IMW-R 92/207).
Hooftman RJ & Evers-de Ruiter A (1992c) The toxicity and uptake of
benzo[k]fluoranthene using Brachidanio rerio in an early life stage
test. Delft, The Netherlands, TNO Institute of Environmental Science,
33 pp (TNO Report No. IMW-R 92/218).
Hooftman RJ & Evers-de Ruiter A (1992d) Early life stage tests with
Brachidanio rerio and several polycyclic aromatic hydrocarbons using
an intermittent flow-through system. Delft, TNO Institute of
Environmental Science, 35 pp (TNO Report No. IMW-R 92/210).
Hooftman RN, Henzen L, & Roza P (1993) The toxicity of a polycyclic
aromatic mixture in an early stage toxicity test carried out in an
intermittent flow-through system. Delft, TNO Institute of
Environmental Science, 41 pp (TNO Report No. IMW-R 93/253).
Hopia A, Pyysalo H, & Wickström K (1986) Margarines, butter and
vegetable oils as sources of polycyclic aromatic hydrocarbons. J Am
Oil Chem Soc, 63: 889-893.
Horikawa K, Sera N, Otofuji T, Murakami K, Tokiwa H, Hwagawa M, Izumi
K, & Otsuka H (1991) Pulmonary carcinogenicity of 3,9- and
3,7-dinitrofluoranthene, 3-nitrofluoranthene and benzo [a]pyrene in
F344 rats. Carcinogenesis, 12: 1003-1008.
Horton AW & Christian GM (1974) Cocarcinogenic versus incomplete
carcinogenic activity among aromatic hydrocarbons: Contrast between
chrysene and benzo [b]-triphenylene. J Natl Cancer Inst, 53:
1017-1020.
Hose JE (1985) Potential uses of sea-urchin embryos for identifying
toxic chemicals: Description of a bioassay incorporating cytologic,
cytogenetic and embryologic endpoints. J Appl Toxicol, 5: 245-254.
Hose JE, Hannah JB, Landolt ML, Miller BS, Felton SP, & Iwaoka WT
(1981) Uptake of benzo [a]pyrene by gonadal tissue of flatfish family
Plueronectidae and its effects on subsequent egg development. J
Toxicol Environ Health, 11: 991-1000.
Hose JE, Hannah JB, DiJulio D, Landolt ML, Miller BS, Iwaoka WT, &
Felton SP (1982) Effects of benzo [a]pyrene on early development of
flatfish. Arch Environ Contam Toxicol, 11: 167-171.
Hoshino K, Hayashi Y, Takehira Y, & Kameyama Y (1981) Influences of
genetic factors on the teratogenicity of environmental pollutants:
Teratogenic susceptibility to benzo [a]pyrene and Ah locus in mice.
Congenital Anomalies, 21: 97-103.
House RV, Pallardy MJ, Burleson GR, & Dean JH (1988)
7,12-Dimethylbenz [a]-anthracene-induced modulation of cytokines
involved in cytotoxic T-lymphocyte induction. In Vitro Toxicol, 2:
267-278.
House RV, Pallardy MJ, & Dean JH (1989) Suppression of murine
cytotoxic T-lymphocyte induction following exposure to
7,12-dimethylbenz [a]anthracene: Dysfunction of antigen recognition.
Int J Immunopharmacol, 2: 207-215.
Houser WH, Raha A, & Vickers M (1992) Induction of CYP1A1 gene
expression in H4II-E rat hepatoma cells by benzo [e]pyrene. Mol
Carcinog, 5: 232-237.
Howard J (1979) Method 5: Analysis of benzo [a]pyrene and other
polycyclic aromatic hydrocarbons in foods. In: Egan H, Castegnaro M,
Bogovski P, Kunte H, & Walker EA ed. Environmental carcinogens:
Selected methods of analysis. Volume 3. Analysis of polycyclic
aromatic hydrocarbons in environmental samples. Lyon, International
Agency for Research on Cancer, pp 175-191 (IARC Scientific
Publications No. 29).
Howard JW & Fazio T (1980) Review of polycyclic aromatic hydrocarbons
in foods. Analytical methodology and reported findings of polycyclic
aromatic hydrocarbons in foods. J Assoc Off Anal Chem, 63: 1077-1104.
Howard PH, Boethling RS, Jarvis WF, Meylan WM, & Michalenko EM (1991)
In: Printup HT ed. Handbook of environmental degradation rates.
Chelsea, Michigan, Lewis Publishers, 725 pp.
Hradec J, Seidel A, Platt KL, Glatt HR, Oesch F, & Koblyakov V (1990)
The initiator tRNA acceptance assay as a short-term test for
carcinogens. 6. Results with 78 polycyclic aromatic compounds.
Carcinogenesis, 11: 1921-1926.
Huberman E (1975) Mammalian cell transformation and cell-mediated
mutagenesis by carcinogenic polycyclic hydrocarbons. Mutat Res, 29:
285-291.
Huberman E (1978) Cell transformation and mutability of different
genetic loci in mammalian cells by metabolically activated
carcinogenic polycyclic hydrocarbons. In: Gelboin HV & Ts'O POP ed.
Polycyclic hydrocarbons and cancer: Molecular and cell biology, Volume
2. New York, Academic Press, pp 161-174.
Huberman E & Sachs L (1976) Mutability of different genetic loci in
mammalian cells by metabolically activated carcinogenic polycyclic
hydrocarbons. Proc Natl Acad Sci USA, 73: 188-192.
Huberman E, Kuroki T, Marquardt H, Selkirk JK, Heidelberger C, Grover
PL & Sims P (1972) Transformation of hamster embryo cells by epoxides
and other derivatives of polycyclic hydrocarbons. Cancer Res, 32:
1391-1396.
Huberman E, McKeown CK, Jones CA, Hoffman DR, & Murao S (1984)
Induction of mutations by chemical agents at the hypoxanthine-guanine
phosphoribosyl transferase locus in human epithelial teratoma cells.
Mutat Res, 130: 127-137.
Huggett RJ, deFur PO, & Bieri RH (1988) Organic compounds in
Chesapeake Bay sediments. Mar Pollut Bull, 19: 454-458.
Huggins C & Yang NC (1962) Induction and extinction of mammary cancer.
Science, 137: 257-262.
Huggins C, Morii S, & Grand LC (1961) Mammary cancer induced by a
single dose of polynuclear hydrocarbons: Routes of administration. Ann
Surg, 154: 315-318.
Hughes TW & DeAngelis DG (1982) Emissions from coal-fired residential
combustion equipment. In: Proceedings of the International Conference
on Residues of Solid Fuels: Environmental Impacts/ Solutions. Dayton,
Ohio, Monsanto Research Corporation, pp 333-348.
Hughes NC & Phillips DH (1991) Dependence on dose of initial and
persistant levels of benzo(a)pyrene and dibenzo(a,e)pyrene DNA adducts
in mouse tissues. Proc Am Assoc Cancer Res, 32: 98.
Hughes MM, Natusch DFS, Taylor DR, & Zeller MV (1980) Chemical
transformations of particulate polycyclic organic matter. In: Bjorseth
A & Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry and
biological effects. Columbus, Ohio, Battelle Press, pp 1-8.
Hughes MF, Chamulitrat W, Mason RP, & Eling TE (1989) Epoxidation of
7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene via a
hydroperoxide-dependent mechanism catalyzed by lipoxygenases.
Carcinogenesis, 10: 2075-2080.
Huh N, Nemoto N, & Utakoji T (1982) Metabolic activation of
benzo [a]pyrene, aflatoxin B1, and dimethylnitrosamine by a human
hepatoma cell line. Mutat Res, 94: 339-348.
van Hummelen P, Gennart JP, Buchete JP, Lauwerys R, & Kirsch-Volders M
(1993) Biological markers in PAH exposed workers and controls. Mutat
Res, 300: 231-239.
Hungspreugs M, Silpipat S, Tonapong C, Lee RF, Windom HL, & Tenore KR
(1984) Heavy metals and polycyclic hydrocarbon compounds in benthic
organisms of the upper Gulf of Thailand. Mar Pollut Bull, 15: 213-218.
Hurley JF, Archibald RMcL, Collins PL, Fanning DM, Jacobsen M, &
Steele RC (1983) The mortality of coke workers in Britain. Am J Ind
Med, 4: 691-704.
Husgafvel-Pursiainen K, Sorsa M, Mller M, & Benestad C (1986)
Genotoxicity and polynuclear aromatic hydrocarbon analysis of
environmental tobacco smoke samples from restaurants. Mutagenesis, 1:
287-292.
Hutchinson TC, Hellebust JA, Tam D, Mackay D, Mascarenhas RA, & Shiu
WY (1980) The correlation of the toxicity to algae of hydrocarbons and
halogenated hydrocarbons with their physical-chemical properties. In:
Afghan BK & Mackay D ed. Hydrocarbons and halogenated hydrocarbons in
the aquatic environment. New York, Plenum Press, pp 577-586.
IARC (1973) Certain polycyclic aromatic hydrocarbons and heterocyclic
compounds. Lyon, International Agency for Research on Cancer, p 178
(IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Man, Volume 13).
IARC (1983) Polynuclear aromatic compounds. Part 1: Chemical,
environmental and experimental data. Lyon, International Agency for
Research on Cancer, 477 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 32).
IARC (1984a) Polynuclear aromatic hydrocarbons. Part 2: Carbon blacks,
mineral oils (lubricant base oils and derived products) and some
nitroarenes. Lyon, International Agency for Research on Cancer, 365 pp
(IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Volume 33).
IARC (1984b) Polynuclear aromatic compounds. Part 3: Industrial
exposures in aluminium production, coal gasification, coke production,
and iron and steel founding. Lyon, International Agency for Research
on Cancer, 219 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 34).
IARC (1985) Polynuclear aromatic compounds. Part 4: Bitumens,
coal-tars and derived products, shale-oils and soots. Lyon,
International Agency for Research on Cancer, 271 pp (IARC Monographs
on the Evaluation of the Carcinogenic Risk of Chemicals to Humans,
Volume 35).
IARC (1986) Tobacco smoking. Lyon, International Agency for Research
on Cancer, 139 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Volume 38).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC Monographs Volumes 1 to 42. Lyon, International Agency for
Research on Cancer, 403 pp (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Supplement 7).
IARC (1989a) Diesel and gasoline engine exhausts and some nitroarenes.
Lyon, International Agency for Research on Cancer, 322 pp (IARC
Monographs on the Evaluation of Carcinogenic Risk of Chemicals to
Humans, Volume 46).
IARC (1989b) Occupational exposures in petroleum refining: Crude oil
and major petroleum fuels. Lyon, International Agency for Research on
Cancer, 283 pp (IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Man, Volume 45).
Ichinotsubo K, Mower HF, Settliff J, & Mandel M (1977) The use of rec-
bacteria for testing of carcinogenic substances. Mutat Res, 46: 53-
61.
Ilnitsky AP, Mischenko VS, & Shabad LM (1977) New data on volcanoes as
natural sources of carcinogenic substances. Cancer Lett, 3: 227-230.
Ingram AJ & Phillips JC (1993) The dermal bioavailability of
radiolabelled benzo(a)pyrene from acetone or from oils of differing
viscosity, assessed by DNA and protein binding. J Appl Toxicol, 13:
25-32.
Ingram LL Jr, McGinnis GD, Gjovik LR, & Roberson G (1982) Migration of
creosote and its components from treated piling sections in a marine
environment. In: Proceedings of the Seventy-eighth Annual Meeting of
the American Wood-preservers' Association, New Orleans, Louisiana, 2-5
May 1982. Bethesda, Maryland, American Wood-preservers' Association,
Volume 78, pp 120-128.
Ingram AJ, Lee R, & Phillips JC (1995) Factors affecting the
bioavailability of benzo(a)pyrene from oils in mouse skin: Oil
viscosity, grooming, activity and its prevention. J Appl Toxicol, 15:
175-182.
Inokuchi H & Nakagaki M (1959) The density of the polycyclic aromatic
compounds. Bull Chem Soc Jpn, 32: 65-67.
International Programme on Chemical Safety (1993) Environmental health
criteria 155: Biomarkers and risk assessment: Concepts and principles.
Geneva, World Health Organization, 82 pp.
International Union of Pure and Applied Chemistry (1979) Nomenclature
of organic chemistry. Sections A, B, C, D, E, F and H. Oxford,
Pergamon Press, pp 20-31.
International Union of Pure and Applied Chemistry (1987) Recommended
method for a thin-layer-chromatographic screening method for the
determination of benzo(a)pyrene in smoked food. Pure Appl Chem, 59:
1735-1738.
Iosifidou HG, Kilikidis SD, & Kamarianos AP (1982) Analysis for
polycyclic aromatic hydrocarbons in mussels (Mytilus
galloprovincialis) from the Thermaikos Gulf, Greece. Bull Environ
Contam Toxicol, 28: 535-541.
Ishidate M & Odashima S 81977) Chromosome tests with 134 compounds on
Chinese hamster cells in vitro: A screening for chemical
carcinogens. Mutat Res, 48: 337-354.
Ishinishi N, Kodema Y, Kunitake E, Nobutomo K & Fukushima Y (1976) The
carcinogenicity of dusts collected from an open-hearth furnace for the
smelting of iron: A preliminary experimental study. In: Nordberg GF,
ed. Effects and dose-response relationships of toxic metals.
Amsterdam, Elsevier Scientific Publishers, pp 480-488.
Ishio S, Chen JC, & Kawasaki Y (1977) Cell division of
Gyrodinium sp. and mitotic delay induced by causal substances of
algal tumor and carcinogens. Jpn Soc Sci Fish, 43: 507-516.
Iyer P, Gollahon LS, Martin JE, & Irvin TR (1990) Evaluation of the
in vitro growth of rodent preimplantation embryos exposed to
naphthalene in vivo. Toxicologist, 10: 274.
Iyer P, Martin JE, & Irvin TR (1991) Role of biotransformation in in
vitro preimplantation embryotoxicity of naphthalene. Toxicology, 66:
257-270.
Jacob J (1996) Mammalian organism - Cell cultures - Enzymes. A
comparison of in vivo and in vitro systems. In: Mohr M, Adler K,
Dungworth D, Harris C, Plopper C, & Saracci R ed. Correlations between
in vitro and in vivo investigations in inhalation toxicology.
Washington DC, International Life Sciences Institute Press, pp 279-
297.
Jacob J & Grimmer G (1979) Extraction and enrichment of polycyclic
aromatic hydrocarbons (PAH) from environmental matter. In: Egan H,
Castegnaro M, Bogovski P, Kunte H, & Walker EA ed. Environmental
carcinogens: Selected methods of analysis. Volume 3: Analysis of
polycyclic aromatic hydrocarbons in environmental samples. Lyon,
International Agency for Research on Cancer, pp 79-89 (IARC Scientific
Publications No. 29).
Jacob J & Grimmer G (1992) Organic analytical approaches. In: Rossbach
M, Schladot JD, & Ostapczuk P ed. Specimen banking. Berlin,
SpringerVerlag, pp 93-113.
Jacob J & Grimmer G (1994) [Environmental sample bank. PAH analysis in
different matrices. Annual Report 1993.] Grosshansdorf, Biochemical
Institute for Environmental Carcinogens, 50 pp (in German).
Jacob J & Grimmer G (1995) [Environmental sample bank. PAH analysis in
environmental samples. Annual Report 1994.] Grosshansdorf, Biochemical
Institute for Environmental Carcinogens, 50 pp (in German).
Jacob J, Grimmer G, & Schmoldt A (1981a) The influence of polycyclic
aromatic hydrocarbons as inducers of monooxygenases on the metabolic
profile of benz [a]anthracene in the rat liver microsomes. Cancer
Lett, 14: 175-185.
Jacob J, Schmoldt A, & Grimmer G (1981b) Glass-capillary-gas
chromatography/mass spectrometry data of mono- and polyhydroxylated
benz(a)anthracene. Comparison with benz(a)anthracene metabolites from
rat liver microsomes. Hoppe-Seyler's Z Physiol Chem, 362: 1021-1030.
Jacob J, Schmoldt A, & Grimmer G (1982a) Influence of monooxygenase
inducers on the metabolic profile of phenanthrene in rat liver
microsomes. Toxicology, 25: 333-343.
Jacob J, Schmoldt A, & Grimmer G (1982b) Formation of carcinogenic and
inactive chrysene metabilites by rat liver microsomes of various
monooxygenase activities. Arch Toxicol, 51: 255-265.
Jacob J, Schmoldt A & Grimmer G (1983) Benzo(e)pyrene metabolism in
rat liver microsomes: Dependence of the metabolite profile on the
pretreatment of rats with various monooxygenase inducers.
Carcinogenesis, 4: 905-919.
Jacob J, Brune H, Dettbarn G, Grimmer D, Heinrich U, Mohtashamipur E,
Norpoth K, Pott F, & Wenzel-Hartung R (1989) Urinary and faecal
excretion of pyrene and hydroxypyrene by rats after oral,
intraperitoneal, intratracheal or intrapulmonary application. Cancer
Lett, 46: 15-20.
Jacob J, Grimmer G, & Schneider D (1990a) Sampling and analysis of
polycyclic aromatic hydrocarbons present in particulate matter and in
semivolatiles (b.p. > 130°C) of ambient air. Fresenius' J Anal Chem,
37: 73.
Jacob J, Brune H, Grimmer G, Heinrich U, Mohtashamipur E, Norpoth K,
Pott F, & Wenzel-Hartung R (1990b) Urinary and faecal excretion of
metabolites after various modes of administration of polycyclic
aromatic hydrocarbons (PAH) to rats. In: Seemayer NH & Hadnagy W ed.
Environmental hygiene II. Berlin, Springer-Verlag, pp 87-90.
Jacob J, Grimmer G, Mohr U, Emura M, Riebe-Imre M, & Raab G (1992) The
metabolism of chrysene and benzo(a)pyrene in hamster, rat and human
lung cells in culture. In: Seemeyer NH & Hadnagy W ed. Environmental
hygiene III. Berlin, Springer-Verlag, pp 87-90.
Jacob J, Grimmer G, & Hildebrandt A (1993a) Correlation between PAH
concentrations measured in air, biological passive samplers and in
corresponding soil samples in Germany. In: Garrigues P & Lamotte M ed.
Polycyclic aromatic compounds: Synthesis, properties, analytical
measurements, occurrence and biological effects. Bordeaux, Gordon &
Breach Science Publishers, pp 427-433.
Jacob J, Grimmer G, & Hildebrandt A (1993b) The use of passive
samplers for monitoring polycyclic aromatic hydrocarbons in ambient
air. Sci Total Environ, 139/140: 307-321.
Jacob J, Grimmer G, Mohr U, Emura M, Riebe-Imre M, Raab G, & Knebel J
(1993c) Metabolic activation of chrysene and benzo [a]pyrene in
hamster, rat and human in vitro lung cell cultures. In: Garrigues P
& Lamotte M ed. Polycyclic aromatic compounds: Synthesis, properties,
analytical measurements, occurrence and biological effects. Bordeaux,
Gordon & Breach Science Publishers, pp 1175-1182.
Jacob J, Grimmer G, Hanssen HP, & Hildebrand A (1994) Extractability
and bioavailability of PAH from soil and air particulate matter.
Polycyclic Aromat Compd, 5: 209-217.
Jacob J, Doehmer J, Grimmer G, Soballa V, Raab G, Seidel A, & Greim H
(1996) Metabolism of phenanthrene, benz(a)anthracene, benzo(a)pyrene
and benzo(c)-phenanthrene by eight cDNA-expressed human and rat
cytochromes P450. Polycyclic Aromat Compd 10: 1-9.
Jacobs BW & Billings CE (1985) Characterization and temperature
dependence of PAH emissions from a simulated rubber combustion
operation. Am Ind Hyg Assoc J, 46: 547-554.
Jahn CL & Litman GW (1977) Distribution of covalently bound
benzo [a]pyrene in chromatin. Biochem Biophys Res Commun, 76:
534-540.
Jahn CL & Litman GW (1979) Accessibility of deoxyribonucleic acid in
chromatin to the covalent binding of the chemical carcinogen
benzo [a]pyrene. Biochemistry, 18: 1442-1449.
Jaklin J & Krenmayr P (1985) A routine method for the quantitative
determination of polycyclic aromatic hydrocarbons (PAHs) in urban air.
Int J Environ Anal Chem, 21: 33-42.
Jaklin J, Krenmayr P, & Varmuza K (1988) [Polycyclic aromatic
compounds in the atmosphere of Linz (Austria).] Fresenius' Z Anal
Chem, 331: 479-485 (in German).
James MO (1989) Biotransformation and disposition of PAH in aquatic
invertebrates. In: Varanasi U ed. Metabolism of polycyclic aromatic
hydrocarbons in the aquatic environment. Boca Raton, Florida, CRC
Press, pp 69-91.
Janini GM, Johnston K, & Zielinski WL Jr (1975) Use of nematic liquid
crystal for gas-liquid chromatographic separation of polyaromatic
hydrocarbons. Anal Chem, 47: 670-674.
Janini GM, Muschik GM, Schroer JA, & Zielinski WL Jr (1976) Gas-liquid
chromatographic evaluation and gas-chromatography/mass spectrometric
application of new high-temperature liquid crystal stationary phases
for polycyclic aromatic hydrocarbon separations. Anal Chem, 48: 1879-
1883.
Janssen O (1980) [Brief review of investigations on PAH in vehicle
exhaust, round robin tests on profile analysis, role of fuels and
lubricants, field studies. Air pollution from polycyclic aromatic
hydrocarbons. Registration and evaluation.] Düsseldorf, VDI-Verlag, pp
69-79 (VDI Report No. 358) (in German).
Japanese Ministry of International Trade and Industry (1992)
Biodegradation and accumulation, data of existing chemicals based on
the CSCL Japan. Tokyo: Japan Chemical Industry Ecology-toxicology and
Information Center, 467 pp.
Japenga J, Wagenaar WJ, Smedes F, & Salomons W (1987) A new, rapid
clean-up procedure for the simultaneous determination of different
groups of organic micropollutants in sediments. Application in two
European estuarine sediment studies. Environ Technol Lett, 8: 9-20.
Jeffrey AM, Jennette KW, Blobstein SH, Weinstein IB, Beland FA, Harvey
RG, Kasai H, Miura, & Nakananishi K (1976) Benzo [a]pyrene-nucleic
acid derivative found in vivo: Structure of a benzo[-]pyrene
tetrahydrodiol epoxide-guanosine adduct. J Am Chem Soc, 98: 5714-5715.
Jennette KW, Jeffrey AM, Blobstein SH, Beland FA, Harvey RG, &
Weinstein IB (1977) Nucleoside adducts from the in vitro reaction of
benzo [a]pyrene-7,8-dihydrodiol 9,10-oxide or benzo [a]pyrene
4,5-oxide with nucleic acids. Biochemistry, 16: 932-938.
Jerina DM, Lehr RE, Yagi H, Hernandez O, Dansette PM, Wislocki PG,
Wood AW, Chang RI, Levin W, & Conney AH (1976) Mutagenicity of
benzo [a]pyrene derivatives and the description of a quantum
mechanical model which predicts the ease of carbonium ion formation
from diol epoxides. In: De Serres FJ, Fouts JR, & Bend JR ed.
In vitro metabolic activation in mutagenesis testing. New York,
Elsevier North Holland, pp 159-178.
Jernström B & Gräslund A (1994) Covalent binding of benzo(a)pyrene
7,8-dihydrodiol 9,10-epoxides to DNA: Molecular structures, induced
mutations and biological consequences. Biophys Chem, 49: 185-199.
Jernström B, Vadi H, & Orrennius S (1978) Formation of DNA-binding
products from isolated benzo(a)pyrene metabolites in rat liver nuclei.
Chem-Biol Interactions, 20: 311-321.
Jernström B, Martinez M, Meyer DJ, & Ketterer B (1985) Glutathione
conjugation of the carcinogenic and mutagenic electrophile
(±)-7b,8a-dihydroxy-9a,10a-oxy-7,8,9,10-tetra hydrobenzo [a]pyrene
catalyzed by purified rat liver glutathione transferases.
Carcinogenesis, 6: 85-89.
Ji C & Marnett LJ (1992) Oxygen radical-dependent epoxidation of
(7 S,8 S)-dihyroxy-7,8-dihydrobenzo [a]pyrene in mouse skin
in vivo. J Biol Chem, 267: 17842-17848.
Jimenez BD, Cirmo CP, & McCarthy JF (1987) Effects of feeding and
temperature on uptake, elimination and metabolism of benzo(a)pyrene in
the bluegill sunfish (Lepomis macrochirus). Aquat Toxicol, 10: 41-
57.
Jockers R, Patalas N, Schacht S, Rehm H-J, & Freier D (1988)
[Development of biotechnological processes for the treatment of
wastewater from coal refinement: Final report.] Essen,
Bergbau-Forschung, 158 pp (Report No. BMFT FKZ 03 E-6284-A) (in
German).
Joe FL Jr, Salemme J, & Fazio T (1984) Liquid chromatographic
determination of trace residues of polynuclear aromatic hydrocarbons
in smoked foods. J Assoc Off Anal Chem, 67: 1076-1082.
Johnke B (1992) [Waste incineration as dioxin source or sink.]
ENTSORGA-Mag Entsorg.wirtsch, 9: 59-66 (in German).
Johnsen S, Kukkonen J, & Grande M (1989) Influence of natural aquatic
humic substances on the bioavailability of benzo(a)pyrene to Atlantic
salmon. Sci Total Environ, 81/82: 691-702.
Johnson S (1968) Effect of thymectomy on the induction of skin tumours
by dibenzanthracene, and of breast tumours by dimethylbenzanthracene
in mice of the IF strain. Br J Cancer, 22: 755-761.
Johnson AC, Larsen PF, Gadbois DF, & Humason AW (1985) The
distribution of polycyclic aromatic hydrocarbons in the superficial
sediments of Penobscot Bay (Maine, USA) in relation to possible
sources and to other sites worldwide. Mar Environ Res, 15: 1-16.
Jones PW, Giammar RD, Strup PE, & Stanford TB (1976) Efficient
collection of polycyclic organic compounds from combustion effluents.
Environ Sci Technol, 10: 806-810.
Jones KC, Stratford JA, Waterhouse KS, & Johnston AE (1987)
Polynuclear aromatic hydrocarbons in UK soils: Long-term temporal
trends and current levels. In: Hemphill DD ed. Trace substances in
environmental health. XXI. Proceedings of the University of Missouri's
21st Annual Conference on Trace Substances in Environmental Health, St
Louis, Missouri, 25-28 May 1987. Columbia, Missouri, University of
Missouri, pp 140-148.
Jones KC, Stratford JA, Waterhouse, & Vogt NB (1989a) Organic
contaminants in Welsh soils: Polynuclear aromatic hydrocarbons.
Environ Sci Technol, 23: 540-550.
Jones KC, Grimmer G, Jacob J, & Johnston AE (1989b) Changes in the
polynuclear aromatic hydrocarbon content of wheat grain and pasture
grassland over the last century from one site in the UK. Sci Total
Environ, 78: 117-130.
Jones KC, Stratford JA, Waterhouse KS, Furlong ET, Giger W, Hites RA,
Schaffner C, & Johnston AE (1989c) Increases in the polynuclear
aromatic hydrocarbon content of an agricultural soil over the last
century. Environ Sci Technol, 23: 95-101.
Jongeneelen FJ (1992) Biological exposure limit for occupational
exposure to coal tar pitch volatiles at coke ovens. Int Arch Occup
Environ Health, 63: 511-516.
Jongeneelen FJ (1994) Biological monitoring of environmental exposure
to polycyclic aromatic hydrocarbons: 1-Hydroxypyrene in urine of
people. Toxicol Lett, 72: 205-211.
Jongeneelen FJ, Bos RP, Anzion RBM, Theuws JLG, & Henderson PT (1986)
Biological monitoring of polycyclic aromatic hydrocarbons: Metabolites
in urine. Scand J Work Environ Health, 12: 137-143.
Jongeneelen FJ, Scheepers PTJ, Groenendijk A, Van Aerts LAGJM, Anzion
RBM, Bos RP, & Veenstra SJ (1988a) Airborne concentrations, skin
contamination, and urinary metabolite excretion of polycyclic aromatic
hydrocarbons among paving workers exposed to coal tar derived road
tars. Am Ind Hyg Assoc J, 49: 600-607.
Jongeneelen FJ, Anzion RBM, Scheepers PTJ, Bos RP, Henderson PT,
Nijenhuis EH, Veenstra SJ, Brouns RME, & Winkes A (1988b)
1-Hydroxypyrene in urine as a biological indicator of exposure to
polycyclic aromatic hydrocarbons in several work environments. Ann
Occup Hyg, 32: 35-43.
Jongeneelen FJ, van Leeuwen FE, Oosterink S, Anzion RBM, van der Loop
F, Bos RP, & van Veen HG (1990) Ambient and biological monitoring of
coke oven workers: Determinants of the internal dose of polycyclic
aromatic hydrocarbons. Br J Ind Med, 47: 454-461.
Joshi PL & Misra RB (1986) Evaluation of chemically-induced
phototoxicity to aquatic organism using Paramecium as a model.
Biochem Biophys Res Commun, 139: 79-84.
Jotz MM & Mitchell AD (1981) Effects of 20 coded chemicals on the
forward mutation frequency at the thymidine kinase locus in L5178Y
mouse lymphoma cells. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North Holland, pp 580-593
(Progress in Mutation Research, Volume 1).
Junk GA, Richard JJ, Avery MJ, Vick RD, & Norton GA (1986) Organic
compounds from coal combustion. Am Chem Soc Symp Ser, 319: 109-123.
Jury WA, Russo D, Streile G, & El Abd H (1990) Evaluation of
volatilization by organic chemicals residing below the soil surface.
Water Res, 26: 13-26.
Kadar R, Nagy K, & Frimstad D (1980) Determination of polycyclic
aromatic hydrocarbons in industrial waste water at the ng/ml level.
Talanta, 27: 227-230.
Kaden DA, Hites RA, & Thilly WG (1979) Mutagenicity of soot and
associated polycyclic aromatic hydrocarbons to Salmonella
typhimurium. Cancer Res, 39: 4152-4159.
Kagan J & Kagan ED (1986) The toxicity of benzo(a)pyrene and pyrene in
the mosquito Aedes aegypti, in the dark and in the presence of
ultraviolet light. Chemosphere, 15: 243-251.
Kagan J, Kagan ED, Kagan IA, Kagan PA, & Quigley S (1985) The
phototoxicity of non-carcinogenic polycyclic aromatic hydrocarbons in
aquatic organisms. Chemosphere, 14: 1829-1834.
Kagan J, Sinnott D, & Kagan E (1987) The toxicity of pyrene in the
fish Pimephales promelas. Synergism by piperonyl butoxide and by
ultraviolet light. Chemosphere, 16: 2291-2298.
Kagan J, Tuveson RW, & Gong HH (1989) The light-dependent cytotoxicity
of benzo [a]pyrene: Effect on human erythrocytes, Escherichia coli
cells, and Haemophilus influenzae transforming DNA. Mutat Res, 216:
231-242.
Kagi R, Alexander R, & Cumbers M (1985) Polycyclic aromatic
hydrocarbons in rock oysters: A baseline study. Int J Environ Anal
Chem, 22: 135-153.
Kaidbey KH & Nonaka S (1984) Action spectrum for anthracene-induced
photosensitization of human skin. Photochem Photobiol, 39: 375-378.
Kakunaga T (1973) A quantitative system for assay of malignant
transformation by chemical carcinogens using a clone derived from
BALB/3T3. Int J Cancer, 12: 463-473.
Kalberlah F, Frijus-Plessen N, & Hassauer M (1995) [Toxicological
criteria for the risk assessment of polyaromatic hydrocarbons (PAH) in
existing chemicals. Part 1: The use of equivalency factors.]
Altlasten-Spektrum, 5: 231-237 (in German).
Kallistratos G & Pfau A (1971) [Effect of 3,4-benzopyrene on large
mammals.] Naturwissenschaften, 58: 222 (in German).
Kamens RM, Zhishi G, Fulcher JN, & Bell D (1986a) The influence of
temperature on the daytime PAH decay on atmospheric soot particles.
Proceedings of the 79th Air Pollution Control Association Annual
Meeting, 23-27 June (No. 86-77-2), pp 1-19.
Kamens RM, Fulcher JN, & Zhishi G (1986b) Effects of temperature on
wood soot PAH decay in atmospheres with sunlight and low Nox
nitrogen oxides. Atmos Environ, 20: 1579-1587.
Kamens RM, Guo Z, Fulcher JN, & Bell DA (1988) Influence of humidity,
sunlight and temperature on the daytime decay of polyaromatic
hydrocarbons on atmospheric soot particles. Environ Sci Technol, 22:
103-108.
Kamens RM, Zhishi G, Fulcher JN, & Bell DA (1991) The influence of
humidity on the daytime decay of PAH on atmospheric soot particles.
In: Cooke M, Loening K, & Merrit J ed. Polynuclear aromatic
hydrocarbons: Measurement, means and metabolism. Columbus, Ohio,
Battelle Press, pp 435-451.
Kang DH, Rothman N, Poirier, MC, Greenberg A, Hsu CH, Schwartz BS,
Baser ME, Groopman JD, Weston A, & Strickland PT (1995)
Interindividual differences in the concentration of
1-hydroxypyrene-glucuronide in urine and polycyclic aromatic
hydrocarbon-DNA adducts in peripheral white blood cells after
charbroiled beef consumption. Carcinogenesis, 16: 1079-1085.
Kanij JBW (1987) The emission of polycyclic aromatic hydrocarbons by
coal-fired power stations in the Netherlands. Kema Sci Tech Rep, 5:
83-91.
Kanoh T, Fukuda M, Onozuka H, Kinouchi T, & Ohnishi Y (1993) Urinary
1-hydroxypyrene as a marker of exposure to polycyclic aromatic
hydrocarbons in environment. Environ Res, 62: 230-241.
Kao J, Patterson FK, & Hall J (1985) Skin penetration and metabolism
of topically applied chemicals in six mammalian species, including
man: An in vitro study with benzo [a]pyrene and testosterone.
Toxicol Appl Pharmacol, 81: 502-516.
Kappeler T & Wuhrmann K (1978) Microbial degradation of water soluble
fraction of gas oil. Water Res, 12: 327-333.
Karcher W (1988) Spectral atlas of polycyclic aromatic compounds,
Volume 2. Dordrecht, Kluwer Academic Publishers, pp 16-18, 55.
Karcher W, Dubois J, Fordham RJ, Glaude P, Barale R, & Zucconi D
(1984) Molecular spectra and mutagenic activity of some nitrogen
containing heterocyclic aromatic compounds. In: Cooke M & Dennis AJ
ed. Mechanisms, methods and metabolism: Polynuclear aromatic
hydrocarbons. Columbus, Ohio, Battelle Press, pp 685-696
Karcher W, Fordham R, Dubois JJ, Glaude PGJM, & Ligthart JAM (1985)
Spectral atlas of polycyclic aromatic compounds including data on
occurrence and biological activity. Dordrecht, D Reichel Publishing
Co., 818 pp.
Karcher W, Devillers J, Garrigues P, & Jacob J (1991) Spectral atlas
of polycyclic aromatic compounds.Dordrecht, Kluwer Academic
Publishers, p 21.
Karickhoff SW (1981) Semi-empirical estimation of sorption of
hydrophobic pollutants on natural sediments and soils. Chemosphere,
10: 833-846.
Karickhoff SW & Morris KR (1985) Sorption dynamics of hydrophobic
pollutants in sediment suspensions. Environ Toxicol Chem, 4: 469-479.
Karickhoff SW, Brown DS, & Scott TA (1979) Sorption of hydrophobic
pollutants on natural sediments. Water Res, 13: 241-248.
Karlehagen S, Andersen A, & Ohlson CG (1992) Cancer incidence among
creosote-exposed workers. Scand J Work Environ Health, 18: 26-29.
Karlesky DL, Ramelow G, Ueno Y, Warner IM, & Ho C-N (1987) Survey of
polynuclear aromatic compounds in oil refining areas. Environ Pollut,
43: 195-207.
Katz M, Heddle JA, & Salamone MF (1981) Mutagenic activity of
polycyclic aromatic hydrocarbons and other environmental pollutants.
Columbus, Ohio, Battelle Press, pp 519-528.
Kauss PB & Hutchinson TC (1975) The effects of water-soluble petroleum
components on the growth of Chlorella vulgaris
Beijerinck. Environ Pollut, 9: 157-174.
Kawabata TT & White KL (1987) Suppression of the in vitro humoral
immune response of mouse splenocytes by benzo(a)pyrene metabolites and
inhibition of benzo(a)pyrene-induced immunosuppression by
alpha-naphthoflavone. Cancer Res, 47: 2317-2322.
Kawabata TT & White KL (1990) Effects of naphthalene and naphthalene
metabolites on the in vitro humoral immune response. J Toxicol
Environ Health, 30: 53-67.
Kawai M (1979) [Review of toxicological and occupational aspects of
naphthalene.] Aromatics, 31: 168-181 (in Japanese).
Kawamura K & Kaplan IR (1986) Compositional change of organic matter
in rainwater during precipitation events. Atmos Environ, 20: 527-535.
Kawamura Y, Kamata E, Ogawa Y, Kaneko T, Uchiyama S, & Saito Y (1988)
The effect of various foods on the intestinal absorption of
benzo(a)pyrene in rats. J Food Hyg Soc Jpn, 29: 21-25.
Kayal SI & Connell DW. (1990) Partitioning of unsubstituted polycyclic
aromatic hydrocarbons between surface sediments and the water column
in the Brisbane River estuary. Aust J Mar Freshwater Res, 41: 443-456.
Kebbekus B, Greenberg A, Horgan L, Bozzelli J, Darack F, Eveleens C, &
Stangeland L (1983) Concentration of selected vapor and
particulate-phase substances in the Lincoln and Holland tunnels. J Air
Pollut Control Assoc, 33: 328-330.
Keller CD & Bidleman TF (1984) Collection of airborne polycyclic
aromatic hydrocarbons and other organics with a glass fiber
filter-polyurethane foam system. Atmos Environ, 18: 837-845.
Kelman BJ & Springer DL (1982) Movements of benzo [a]pyrene across
the hemochorial placenta of the guinea pig (41307). Proc Soc Exp Biol
Med, 169: 58-62.
Kemena A, Norpoth KH, & Jacob J (1988) Differential induction of the
monooxygenase isoenzymes in mouse liver microsomes by polycyclic
aromatic hydrocarbons. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: A decade of progress. Columbus, Ohio, Battelle
Press, pp 449-460.
Kennaway FL (1924) On the cancer-producing factor in tar. Br Med J, i:
564-567.
Kennaway EL (1930) Further experiments on cancer-producing substances.
Biochem J, 24: 497-504.
Kensler TW, Egner PA, Moore KG, Taffe BG, Twerdok LE, & Trush MA
(1987) Role of inflammatory cells in the metabolic activation of
polycyclic aromatic hydrocarbons in mouse skin. Toxicol Appl
Pharmacol, 90: 337-346.
Ketkar M, Green V, Schneider P, & Mohr U (1979) Investigations on the
carcinogenic burden by air pollution in man. Intratracheal
instillation studies with benzo(a)pyrene in a mixture of Tris buffer
and saline in Syrian golden hamsters. Cancer Lett, 6: 279-284.
Khalili NR, Scheff PA, & Holsen TM (1995) PAH source fingerprints for
coke ovens, diesel and gasoline engines, highway tunnels, and wood
combustion emissions. Atmos Environ, 29: 533-542.
Khesina AY (1994) Urban air pollution by carcinogenic and genotoxic
polyaromatic hydrocarbons in the former USSR. Environ Health Perspect,
102 (suppl 4): 49-53.
Kicinski HG, Adamek S, & Kettrup A (1989) Trace enrichment and HPLC
analysis of polycyclic aromatic hydrocarbons in environmental samples,
using solid phase extraction in connection with UV/VIS diode-array and
fluorescence detection. Chromatographia, 28: 203-208.
Kiene R & Capone D (1984) Effects of organic pollutants on
methanogenesis, sulfate reduction and carbon dioxide evolution in salt
marsh sediments. Mar Environ Res, 13: 141-160.
Kimber I, Jones K, & Vignali DAA (1986) The influence of
7,12-dimethybenzanthracene on natural killer (NK) cell function in
rats. J Clin Lab Immunol, 20: 193-198.
Kincannon DF & Lin YS (1985) Microbial degradation of hazardous wastes
by land treatment. In: Proceedings of the 40th Industrial Waste
Conference, Purdue University, West Lafayette, Indiana, 14-16 May,
1985. Boston, Butterworth Press, pp 607-619.
King HWS, Osborne MR, & Brookes P (1979) The in vitro and
in vivo reaction at the N7 position of guanine of the ultimate
carcinogen derived from benzo [a]pyrene. Chem-Biol Interactions, 24:
345-353.
Kirchmann H, Aström H, & Jönsäll G (1991) Organic pollutants in sewage
sludge. 1. Effect of toluene, naphthalene, 2-methylnaphthalene,
4-n-nonylphenol and di-2-ethylhexyl phthalate on soil biological
processes and their decomposition in soil. Swed J Agric Res, 21: 107-
113.
Kishi H, Kogure N, & Hashimoto Y (1990) Contribution of soil
constituents in adsorption coefficient of aromatic compounds,
halogenated alicyclic and aromatic compounds to soil. Chemosphere, 21:
867-876.
Kjuus H, Andersen A, Langard S, & Knudsen KE (1986) Cancer incidence
among workers in the Norwegian ferroalloy industry. Br J Ind Med, 43:
227-236.
Klassert A (1993) [Demonstration plant for adsorptive purification of
waste water from basic chemical industry.] Berlin, Ministry of
Environment, 93 pp (Report No. UBA 20441-5/2) (in German).
Klein M (1960) A comparison of the initiating and promoting actions of
9,10-dimethyl-1,2-benzanthracene and 1,2,5,6-dibenzanthracene in skin
tumorigenesis. Cancer Res, 20: 1179-1183.
Klein M (1963) Susceptibility of strain B6AF1/J hybrid infant mice to
tumorigenesis with 1,2-benzanthracene, deoxycholic acid, and
3-methylcholanthrene. II. Tumours called forth by painting the skin
with dibenzpyrene. Cancer Res, 23: 1701-1707.
Kleinenberg HE (1939) [Investigations on the blastomogenic effect of
3:4:8:9- dibenzpyrene and some of its derivatives.] Arch Biol Nauk,
56: 39-47 (in Russian).
Klemme JC, Mukhtar H, & Elmets CA (1987) Induction of contact
hypersensitivity to dimethylbenz(a)anthracene and benzo(a)pyrene in
C3H/HeN mice. Cancer Res, 47: 6074-6078.
Kliesch U, Roupova I, & Adler ID (1982) Induction of chromosome damage
in mouse bone marrow by benzo [a]pyrene. Mutat Res, 102: 265-273.
Klingenberg H, Schürmann D, & Lies K-H (1992) Specific air pollutants
from passenger cars: Emission and ambient air concentrations. In:
Toxic air pollutants from mobile sources: Emissions and health
effects. Proceedings of an International Specialty Conference.
Pittsburgh, Pennsylvania, Air & Waste Management Association, pp 82-
93.
Klöpffer W, Rippen G, & Frische R (1982) Physicochemical properties as
useful tools for predicting the environmental fate of organic
chemicals. Ecotoxicol Environ Saf, 6: 294-301.
Klug A (1950) The crystal and molecular structure of triphenylene,
C18H12. Acta Crystallogr, 3: 165-175.
Knaap AGAC, Goze C, & Simons JWIM (1981) Mutagenic activity of seven
coded samples in V79 Chinese hamster cells. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens. Report of the
international collaborative programme. New York, Elsevier/North
Holland, pp 608-613 (Progress in Mutation Research, Volume 1).
Knake E (1956) [Weak tumor-inducing effect of naphthalene and
benzene.] Virchows Arch, 329: 141-116 (in German).
Knecht U & Woitowitz H-J (1990) [Cancer risk from use of tar bitumen
in road building.] Bremerhaven, Wirtschaftsverlag NW Verlag für neue
Wissenschaft GmbH, 47 pp (in German).
Knecht U, Elliehausen H-J, & Woitowitz H-J (1986) Gaseous and adsorbed
PAH in an iron foundry. Br J Ind Med, 43: 834-838.
Knecht U, Elliellausen HJ, Judas W, & Woitowitz HJ (1987) Polycyclic
aromatic hydrocarbons (PAH) in abraded particles of brake and clutch
linings. Int J Environ Anal Chem, 28: 227-236.
Knecht U, Bolm-Audorff U, & Woitowitz HJ (1989) Atmospheric
concentrations of polycyclic aromatic hydrocarbons during chimney
sweeping. Br J Ind Med, 46: 479-481.
Knight CV & Humphreys MP (1985) Polynuclear aromatic hydrocarbons in
indoor residential air resulting from use of conventional and
catalytic wood heaters in a weatherized home. In: Cooke M & Dennis AJ
ed. Mechanisms, methods and metabolism: Polynuclear aromatic
hydrocarbons. Columbus, Ohio, Battelle Press, pp 725-737.
Knutzen J & Sortland B (1982) Polycyclic aromatic hydrocarbons (PAH)
in some algae invertebrates from moderately polluted parts of the
coast of Norway. Water Res, 16: 421-428.
Kochevar IE, Armstrong RB, Einbinder J, Walther RR, & Harber LC (1982)
Coal tar phototoxicity: Active compounds and action spectra. Photochem
Photobiol, 36: 65-69.
Konar NR, Roy HK, & De MN (1939) Naphthalene poisoning. Indian Med
Gaz, 74: 723-725.
Kongerud J, Boe J, Soyseth V, Naalsund A, & Magnus P (1994) Aluminium
potroom asthma: The Norwegian experience. Eur Respir J, 7: 165-172.
König W, Hembrock-Heger A, & Wilkens (1991) [Persistent organic
chemicals in soil - routes of entry and occurrence]. Umweltwissensch
Schadstoff-Forsch, 3: 33-36 (in German).
Kootstra A & Slaga TJ (1979) Differential accessibility of (±)
trans-7b,8a-dihydroxy-9a,10a-epoxy-7,8,9,10-tetrahydrobenzo [a]
pyrene to histone proteins. FEBS Lett, 108: 321-325.
Kootstra A & Slaga TJ (1980) Binding of isomers of benzo [a]pyrene
diol-epoxide to chromatin. Biochem Biophys Res Commun, 93: 954-959.
Kootstra A, Slaga TJ, & Olins DE (1979) Interaction of
benzo [a]pyrene diol-epoxide with nuclei and isolated chromatin.
Chem-Biol Interactions, 28: 225-236.
Kördel W, Schoene K, Bruckert J, Pfeiffer U, Schreiber G, Rittmann D,
Hochrainer D, Otto F, Spiegelberg T, Fingerhut R, Kuhnen-Clausen D, &
König J (1981) [Test for chemicals: Study on the feasibility of the EC
regulatory test methods.] Hanover, Fraunhofer Institute for Toxicology
and Aerosol Research, Volume 1, p 553 (in German).
Koreeda M, Moore PD, Wislocki PG, Levin W, Conney AH, Yagi H, & Jerina
DM (1978) Binding of benzo[a]pyrene 7,8-diol 9,10-epoxides to DNA,
RNA, and protein of mouse skin occurs with high stereoselectivity.
Science, 199: 778-780.
Korhonen K & Mulari K (1983) Airborne pollutants in ties workers.
Lappeenrannan, Finnish Institute of Occupational Health, 23 pp.
Korn S, Moles D, & Rice SD (1979) Effects of temperature on the median
tolerance limit of pink salmon and shrimp exposed to toluene,
naphthalene, and Cook Inlet crude oil. Bull Environ Contam Toxicol,
21: 521-525.
Kotin P, Falk HL, & Busser R (1959) Distribution, retention, and
elimination of C14-3,4-benzpyrene after administration to mice and
rats. J Natl Cancer Inst, 23: 541-555.
Kouri RE, Connolly GM, Nebert DW, & Lubet RA (1983) Association
between susceptibility to dibenzanthracene induced fibrosarcoma
formation and the Ah locus. Int J Cancer, 32: 765-768.
Krahn DF & Heidelberger C (1977) Liver homogenate-mediated mutagenesis
in Chinese hamster V79 cells by polycyclic aromatic hydrocarbons and
aflatoxins. Mutat Res, 46: 27-44.
Krämer M, Bimboes D, & Greim H (1974) Salmonella typhimurium and E.
coli to detect chemical mutagens. Arch Pharmacol, 284: R46.
Krewski D, Thorslund T, & Withey J (1989) Carcinogenic risk assessment
of complex mixtures. Toxicol Ind Health, 5: 851-867.
Kriek E, Van Schooten FJ, Hillebrand MJX, Van Leeuwen FE, Den Engelse
L, De Looff AJA, & Dijkmans APG (1993) DNA adducts as a measure of
lung cancer risk in humans exposed to polycyclic aromatic
hydrocarbons. Environ Health Perspectives, 99: 71-75.
Kröber B & Häckl M (1989) [Report on orientation measurements on
dangerous, organic compounds in wastewater pipes, sewage works and
waters in the Federal State of Hessen (1985-1988)] .Wiesbaden, Hessen
State Office for the Environment, pp 225-288 (Report 96) (in German).
Krokje Å (1989) Mutagenicity of expectorate from workers in a coke
plant. Mutat Res, 223: 213-219.
Krokje Å, Tiltnes A, Mylius E, & Gullvå B (1988) Testing for mutagens
in an aluminium plant. The results of Salmonella
typhimurium tests on urine from exposed workers. Mutat Res, 204:
163-172.
Krolewski B, Nagasawa H, & Little JB (1986) Effect of aliphatic amides
on oncogenic transformation, sister chromatid exchanges, and mutations
induced by cyclopenta [cd]-pyrene. Carcinogenesis, 7: 1647-1650.
Kronberger H & Weiss J (1944) Formation and structure of some organic
molecular compounds. Part III. The dielectric polarisation of some
solid crystalline molecular compounds. J Chem Soc, 146: 464-469.
Kruber O (1937) [Some new constituents of coal tar pitch.] Chem Ber,
70: 1556-1564 (in German).
Kruber O & Grigoleit G (1954) [New compounds isolated from coal tar
pitch.] Chem Ber, 87: 1895-1905 (in German).
Kruber O & Marx A (1938) [The anthracene oil of coal tar.] Chem Ber,
71: 2478-2484 (in German).
Kubasiewicz M, Starzynski Z, & Szymczak W (1991) Case-referent study
on skin cancer and its relation to occupational exposure to polycyclic
aromatic hydrocarbons. II. Study results. Pol J Occup Med Environ
Health, 4: 141-147.
Kunstler K (1983) Failure to induce tumors by intratracheal
instillation of automobile exhaust condensate and fractions thereof in
Syrian golden hamsters. Cancer Lett, 18: 105-108.
Kuroki T & Heidelberger C (1972) Determination of the h-protein in
transformable and transformed cells in culture. Biochemistry, 11:
2116-2124.
Kuroki T, Nemoto N, & Kitano Y (1980) Metabolism of benzo [a]pyrene
in human epidermal keratinocytes in culture. Carcinogenesis, 1: 559-
565.
Kushwaha SC, Clarkson SG & Mehkeri KA (1985) Polycyclic aromatic
hydrocarbons in barbecue briquets. J Food Saf, 7: 177-201.
Kuwabara K, Nakamura A, & Kashimoto T (1980) Effect of petroleum oil,
pesticides, PCBs and other environmental contaminants on the
hatchability of Artemia salina dry eggs. Environ Contam Toxicol, 25:
69-74.
Kveseth K, Sortland B, & Bokn T (1982) Polycyclic aromatic
hydrocarbons in sewage, mussels and tap water. Chemosphere, 11: 623-
639.
La Budde JA & Heidelberger C (1958) The synthesis of the mono- and
dihydroxy derivatives of 1,2,5,6-dibenzanthracene excreted by the
rabbit and of other hydroxylated dibenzanthracene derivatives. J Am
Chem Soc, 80: 1225-1236.
Lacassagne A, Buu-Hoi NP, & Zajdela F (1958) [Relationship between
molecular structure and carcinogenic activity in three series of
hexacyclic aromatic hydrocarbons.] C R Acad Sci Paris, 245: 1477-1481
(in French).
Lacassagne A, Buu-Hoi NP, Zajdela F, Lavit-Lamy D, & Chalvet O (1963a)
[Carcinogenic activity of fluoranthene-based polycyclic aromatic
hydrocarbons.] Acta Unio Int Contra Cancrum, 19: 490-496 (in French).
Lacassagne A, Buu-Hoi NP, Zajdela F, & Lavit-Lamy D (1963b) [High
carcinogenic activity of 1,2:3,4-dibenzopyrene and
1,2:4,5-dibenzopyrene.] C R Acad Sci Paris, 256: 2728-2731 (in
French).
Lacassagne A, Buu-Hoi NP, Zajdela F, & Vingiello FA (1968a) The true
dibenzo [a,l]pyrene, a new, potent carcinogen. Naturwissenschaften,
55: 43.
Lacassagne A, Buu-Hoi NP, & Zajdela F (1968b) [Absence of sarcomagenic
property in dibenz [a,c]anthracene; clear activity of its 10-methyl
derivative.] Eur J Cancer, 4: 123-127 (in French).
Ladics GS & White KL (1996) Immunotoxicity of polyaromatic
hydrocarbons. In: Smialowicz RJ & Holsapple MP ed. Experimental
immunotoxicology. Boca Raton, Florida, CRC Press, pp 331-349.
Ladics GS, Kawabata TT, & White KL Jr (1991) Suppression of the
in vitro humoral immune response of mouse splenocytes by
7,12-dimethylbenz(a)anthracene metabolites and inhibition of
immunosuppression by a-naphthoflavone. Toxicol Appl Pharmacol, 110:
31-44.
Ladics GS, Kawabata TT, Munson AE, & White KL Jr (1992a) Generation of
7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo [a]pyrene by murine
splenic macrophages. Toxicol Appl Pharmacol, 115: 72-79.
Ladics GS, Kawabata TT, Munson AE, & White KL Jr (1992b) Metabolism of
benzo [a]pyrene by murine splenic cell types. Toxicol Appl Pharmacol,
116: 248-257.
Lafontaine M, Attenont H, Hubert G, Taiclet A, & Truy S (1990a)
[Emission of polycyclic aromatic hydrocarbons in foundries.] Cah Notes
Doc, 141: 799-807 (in French).
Lafontaine M, Attenont H, & Truy S (1990b) [Exposure to polycyclic
aromatic hydrocarbons in the electrode-producing industry]. Cah Notes
Doc, 132: 453-455.
Lahmann E, Seifert B, Zhao L, & Bake D (1984) [Immission of polycyclic
aromatic hydrocarbons in Berlin (West).] Staub-Reinhalt Luft, 44: 149-
157 (in German).
Lake RS, Kropko ML, Pezzutti MR, Shoemaker RH, & Igel HJ (1978)
Chemical induction of unscheduled DNA synthesis in human skin
epithelial cell cultures. Water Res, 38: 2091-2098.
Lampe V, Puettmann W, & Kasig W (1991) Analysis of the PAH load of
river sediments from the Aachen area. Contribution to the
determination of emission/immission pathways. Wiss Umwelt, 1: 51-62.
Landahl JT, McCain BB, Myers MS, Rhodes LD, & Brown DW (1990)
Consistent associations between hepatic lesions in English sole
(Parophrys vetulus) and polycyclic aromatic hydrocarbons in bottom
sediment. Environ Health Perspectives, 89: 195-203.
Landrum PF (1982) Uptake, depuration and biotransformation of
anthracene by the scud Pontoporeia hoyi. Chemosphere, 11: 1049-1057.
Landrum PF (1988) Toxicokinetics of organic xenobiotics in the
amphipod Pontoporeia hoyi: Role of physiological and environmental
variables. Aquat Toxicol, 12: 245-271.
Landrum PF & Poore R (1988) Toxicokinetics of selected xenobiotics in
Heagenia limbata. J Great Lakes Res, 14: 427-437.
Landrum PF & Scavia D (1983) Influence of sediment on anthracene
uptake, depuration, and biotransformation by the amphipod
Hyalella azteca. Can J Fish Aquat Sci, 40: 298-305.
Landrum PF, Bartell SM, Giesy JP, Leversee GJ, Bowling JW, Haddock J,
LaGory K, Gerould S, & Bruno M (1984a) Fate of anthracene in an
artificial stream: A case study. Ecotoxicol Environ Saf, 8: 183-201.
Landrum PF, Nihart SR, Edie BJ, & Gardner WS (1984b) Reverse-phase
separation method for determining pollutant binding to Aldrich humic
acid and dissolved organic carbon of natural waters. Environ Sci
Technol, 18: 187-192.
Landrum PF, Eadie BJ, & Faust WR (1991) Toxicokinetics and toxicity of
a mixture of sediment-associated polycyclic aromatic hydrocarbons to
the amphipod Diporeia sp. Environ Toxicol Chem, 10: 35-46.
Lane DA (1989) The fate of polycyclic aromatic compounds in the
atmosphere and during sampling. In: Vo-Dinh T ed. Chemical analysis of
polycyclic aromatic compounds. New York, John Wiley & Sons, pp 31-58.
Lane DA & Katz M (1977) The photomodification of benzo [a]pyrene,
benzo [b]-fluoranthene, and benzo [k]fluoranthene under simulated
atmospheric conditions. Adv Environ Sci Technol, 8: 137-154.
Langenfeld JL, Hawthorne SB, Miller DJ, & Pawliszyn J (1993) Effects
of temperature and pressure on supercritical fluid extraction
efficiencies of polycyclic aromatic hydrocarbons and polychlorinated
biphenyls. Anal Chem, 65: 338-344.
Lao RC, Thomas RS, Oja H, & Dubois L (1973) Application of a gas
chromatograph-mass spectrometer-data processor combination to the
analysis of the polycyclic aromatic hydrocarbon content of airborne
pollutants. Anal Chem, 45: 908-915.
Larssen S (1985) Automotive emission factors: An indirect measurement
method applied to polycyclic aromatic hydrocarbon and lead emissions.
In: Proceedings of the Air Pollution Control Association, 77th Annual
Meeting, San Francisco, 24-29 June 1984. Houston, Texas, Air Pollution
Control Association, pp 1-18.
Larsson BK (1982) Polycyclic aromatic hydrocarbons in smoked fish. Z
Lebensm Untersuch Forsch, 174: 101-107.
Larsson B (1986) Polycyclic aromatic hydrocarbons in Swedish foods:
Aspects on analysis, occurrence and intake. Uppsala, Swedish
University of Agricultural Sciences, 60 pp (Doctoral Thesis).
Larsson B & Sahlberg G (1982) Polycyclic aromatic hydrocarbons in
lettuce. Influence of a highway and an aluminium smelter. In: Cooke M
& Fisher AJ ed. Polynuclear aromatic hydrocarbons: Physical and
biological chemistry. Columbus, Ohio, Battelle Press, pp 417-426.
Larsson BK, Sahlberg GP, Eriksson AT, & Busk LA (1983) Polycyclic
aromatic hydrocarbons in grilled food. J Agric Food Chem, 31: 867-873.
Larsson BK, Eriksson AT, & Cervenka M (1987) Polycyclic aromatic
hydrocarbons in crude and deodorized vegetable oils. J Am Oil Chem
Soc, 64: 365-370.
Laskin S, Kuschner M, & Drew RT (1970) Studies in pulmonary
carcinogenesis. In: Hanna MG, Nettesheim P, & Gilbert J ed. Inhalation
carcinogenesis. Oak Ridge, Tennessee, US Atomic Energy Commission,
Division of Technical Information, pp 321-351 (AEC Symposium Series
No. 18).
Lasnitzki A & Woodhouse DL (1944) The effect of
1:2:5:6-dibenzanthracene on the lymph nodes of the rat. J Anat, 78:
121-128.
LaVoie EJ, Bedenko V, Hirota N, Hecht SS, & Hoffmann D (1979) A
comparison of the mutagenicity, tumor-initiating activity and complete
carcinogenicity of polynuclear aromatic hydrocarbons. In: Jones PW &
Leber P ed. Polynuclear aromatic hydrocarbons. Ann Arbor, Michigan,
Ann Arbor Science Publishers, pp 705-721.
LaVoie EJ, Tulley L, Bedenko V, & Hoffmann D (1980) Mutagenicity,
tumor initiating activity, and metabolism of tricyclic polynuclear
aromatic hydrocarbons. In: Bjorseth A & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemistry and biological effects. Columbus,
Ohio, Battelle Press, pp 1041-1057.
LaVoie EJ, Tulley L, Bedenko V, & Hoffmann D (1981a) Mutagenicity of
methylated fluorenes and benzofluorenes. Mutat Res, 91: 167-176.
LaVoie EJ, Tulley-Freiler L, Bedenko V, & Hoffmann D (1981c)
Mutagenicity, tumor-initiating activity and metabolism of
methylphenanthrenes. Cancer Res, 41: 3441-3447.
LaVoie EJ, Tulley-Freiler L, Bedenko V, Girach Z, & Hoffmann D (1981c)
Comparative studies on the tumor initiating activity and metabolism of
methylfluorenes and methylbenzofluorenes. In: Cooke M & Dennis AJ ed.
Chemical analysis and biologicalfate: Polynuclear aromatic
hydrocarbons. Columbus, Ohio, Battelle Press, pp 417-427.
La Voie EJ, Hecht SS, Bedenko V, & Hoffmann D (1982a) Identification
of the mutagenic metabolites of fluoranthene, 2-methylfluoranthene,
and 3-methylfluoranthene. Carcinogenesis, 3: 841-846.
La Voie EJ, Amin S, Hecht SS, Furuya K, & Hoffmann D (1982b) Tumour
initiating activity of hydrodiols of benzo [b]fluoranthene,
benzo [j]fluoranthene, and benzo [k]-fluoranthene. Carcinogenesis,
3: 49-53.
LaVoie EJ, Coleman DT, Tonne RL, & Hoffmann D (1983a) Mutagenicity,
tumor initiating activity and metabolism of methylated anthracenes.
In: Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons.
Columbus, Ohio, Battelle Press, pp 785-798.
LaVoie EJ, Tulley-Freiler L, Bedenko V, & Hoffmann D (1983b)
Mutagenicity of substituted phenanthrenes in Salmonella
typhimurium. Mutat Res, 116: 91-102.
LaVoie EJ, Coleman DT, Rice JE, Geddie NG & Hoffmann D (1985)
Tumor-initiating activity, mutagenicity, and metabolism of methylated
anthracenes. Carcinogenesis, 6: 1483-1488.
LaVoie EJ, Braley J, Rice JE, & Rivenson A (1987) Tumorigenic activity
of non-alternant polynuclear aromatic hydrocarbons in newborn mice.
Cancer Lett, 34: 15-20.
LaVoie EJ, Cai ZW, Meschter CL, & Weyand EH (1994) Tumorigenic
activity of fluoranthene, 2-methylfluoranthene in newborn CD-1 mice.
Carcinogenesis, 15: 2131-2135.
Lawrence JF & Weber DF (1984) Determination of polycyclic aromatic
hydrocarbons in Canadian samples of processed vegetable and dairy
products by liquid chromatography with fluorescence detection. J Agric
Food Chem, 32: 794-797.
Leach JM, Otson R, & Armstrong V (1987) Airborne contaminants in two
small Canadian coal liquefaction pilot plants. Am Ind Hyg Assoc J, 48:
693-697.
Leadon SA, Stampfer MR, & Bartley J (1988) Production of oxidative DNA
damage during the metabolic activation of benzo [a]pyrene in human
mammary epithelial cells correlates with cell killing. Proc Natl Acad
Sci USA, 85: 4365-4368.
LeBlanc GA (1980) Acute toxicity of priority pollutants to water flea
(Daphnia magna). Environ Contam Toxicol, 24: 684-691.
Lecoq S, Perin F, Plessis MJ, Strapelias H, & Duquesne M (1989)
Comparison of the in vitro metabolisms and mutagenicities of
dibenz [a,c]anthracene, dibenz [a,h]anthrancene, and
dibenz [a,j]anthrancene: Influence of norharman. Carcinogenesis, 10:
461-469.
Lecoq S, Ni She M, Hewer A, Grover PL, Platt KL, Oesch F, & Phillips
DH (1991) The metabolic activation of dibenz [a,h]anthracene in mouse
skin examined by 32P-postlabelling: Minor contribution of the
3,4-diol 1,2-oxides to DNA binding. Carcinogenesis, 12: 1079-1083.
Lecoq S, Pfau W, Grover PL, & Phillips DH (1992) HPLC separation of
32P-postlabelled DNA adducts formed from dibenz [a,h]anthracene in
skin. Chem-Biol Interactions, 85: 173-185.
Lee RF (1977) Fate of petroleum components in estuarine water of
southeastern United States. In: Proceedings of the oil spill
conference: Prevention, behavior, control, cleanup, New Orleans, 8-10
March 1997. Washington DC, American Petroleum Institute, pp 611-616
(API Publication No. 4284).
Lee RF & Andersen JW (1977) Fate and effect of naphthalenes:
Controlled ecosystem pollution experiment. Bull Mar Sci, 27: 127-134.
Lee H & Lin JY (1988) Antimutagenic activity of extracts from
anticancer drugs in Chinese medicine. Mutat Res, 204: 229-234.
Lee WY & Nicol JAC (1978) The effect of naphthalene on survival and
activity of the amphipod Parhyale hawaiensis. Bull Environ Contam
Toxicol, 20: 233-240.
Lee RF & Ryan C (1976) Biodegradation of petroleum hydrocarbons by
marine microbes. In: Sharpley JM & Kaplan AM ed. Biodeterioration of
materials. Milton Keynes, Essex, Applied Science Publishers, pp 119-
125.
Lee FSC & Schuetzle D (1983) Sampling, extraction, and analysis of
polycyclic aromatic hydrocarbons from internal combustion engines. In:
Bjorseth A ed. Handbook of polycyclic aromatic hydrocarbons. New York,
Marcel Dekker, pp 27-94.
Lee RF, Sauerheber R, & Dobbs G (1972) Uptake, metabolism and
discharge of polycyclic aomatic hydrocarbons by marine fish. Mar Biol,
17: 201-208.
Lee ML, Novotny M, & Bartle KD (1976a) Gas chromatography/mass
spectrometric and nuclear magnetic resonance determination of
polynuclear aromatic hydrocarbons in airborne particulates. Anal Chem,
48: 1566-1572.
Lee ML, Novotny M, & Bartle KD (1976b) Gas chromatography/mass
spectrometric and nuclear magnetic resonance spectrometric studies of
carcinogenic polynuclear aromatic hydrocarbons in tobacco and
marijuana smokes condensates. Anal Chem, 48: 405-416.
Lee FS-C, Harvey TM, Prater TJ, Paputa MC, & Schuetzle D (1980)
Chemical analysis of diesel particulate matter and an evaluation of
artifact formation. In: Sampling and analysis of toxic organics in the
atmosphere. Philadelphia, Pennsylvania, American Society for Testing
and Materials, pp 92-110 (ASTM STP 721).
Lee ML, Novotny MV, & Bartle KD (1981) Analytical chemistry of
polycyclic aromatic compounds. New York, Academic Press, pp 35-40, 78-
122, 156-289.
Lee MD, Wilson JT, & Ward CH (1984) Microbial degradation of selected
aromatics in a hazardous waste site. Dev Ind Microbiol, 25: 557-565.
Lee PM, Baoyun Y, Herbert R, Hamminki K, Perera FP, & Santella RM
(1991) Immunologic measurement of polycyclic aromatic
hydrocarbon-albumin adducts in foundry workers and roofers. Scand J
Work Environ Health, 17: 190-194.
de Leeuw JW, de Leer EWB, Sinninghe Damsté JS, & Schuyl PJW (1986)
Screening of anthropogenic compounds in polluted sediments and soils
by flash evaporation/pyrolysis gas chromatography-mass spectrometry.
Anal Chem, 58: 1852-1857.
Legraverend C, Harrison DE, Ruscetti FW, & Nebert DW (1983) Bone
marrow toxicity induced by oral benzo [a]pyrene: Protection resides
at the level of the intestine and liver. Toxicol Appl Pharmacol, 70:
390-401.
Legraverend C, Guenthner TM, & Nebert DW (1984) Importance of the
route of administration for genetic differences in
benzo [a]pyrene-induced in utero toxicity and teratogenicity.
Teratology, 29: 35-47.
Lehmann EJ, Auffarth J, Häger J, Rentel KH, & Altenburg H (1986)
[Concentration profiles of selected PAH in coal-tar derived products.]
Staub-Reinhalt Luft, 46: 128-131 (in German).
Lei ZM (1993) The relationship between concentration of B(a)P in
blood, urine, and immune function of coking workers. Chung Hua Yu Fang
I. Hsueh Tsa Chih, 27: 212-214 (in Chinese).
Leinster P & Evans MJ (1986) Factors affecting the sampling of
airborne polycyclic aromatic hydrocarbons. A review. Ann Occup Hyg,
30: 481-495.
Lenin K (1994) Contamination with polycyclic aromatic hydrocarbons
through sewage irrigation. Doctoral thesis submitted to the Jawaharlal
Nehru University, Delhi, India, 22 pp.
Leo A, Hansch C, & Elkins D (1971) Partition coefficients and their
uses. Chem Rev, 71: 525-616.
Leoni V, Puccetti G, & Grella A (1975) Preliminary results on the use
of Tenax(R) for the extraction of pesticides and polynuclear
aromatic hydrocarbons from surface and drinking waters for analytical
purposes. J Chromatogr, 106: 119-124.
Lepperhoff G (1981) [The PAH emissions of spark ignition engines. Part
I.] Wiss Umwelt, 2: 72-87 (in German).
Lesage J, Perrault G, & Durand P (1987) Evaluation of worker exposure
to polycyclic aromatic hydrocarbons. Am Ind Hyg Assoc J, 48: 753-759.
Leslie TJ, Dickson KL, Jordan JA, & Hopkins DW (1987) Effects of
suspended solids on the water column biotransformation of anthracene.
Arch Environ Contam Toxicol, 16: 637-642.
Leversee GJ, Landrum PJ, Bartell S, Gerould S, Bruno M, Spacie A,
Bowling J, Haddock J, & Fannin T (1981) Disposition of
benzo [a]pyrene in aquatic system components: Periphyton,
Chironomids, Daphnia, fish. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 357-367.
Levin W, Wood AW, Chang RL, Yagi H, Mah HD, Jerina DM, & Conney AH
(1978) Evidence for bay region activation of chrysene 1,2-dihydrodiol
to an ultimate carcinogen. Cancer Res, 38: 1831-1834.
Levin W, Wood AW, Chang RL, Ittah Y, Croisy-Delcey M, Yagi H, Jerina
DM, & Conney AH (1980) Exceptionally high tumor-initiating activity of
benzo(c)phenanthrene bay-region diol-epoxides on mouse skin. Cancer
Res, 40: 3910-3914.
Levin W, Wood A, Chang R, Ryan D, Thomas P, Yagi H, Thakker D, Vyas K,
Boyd C, Chu S-Y, Conney A, & Jerina D (1982) Oxidative metabolism of
polycyclic aromatic hydrocarbons to ultimate carcinogens. Drug Metab
Rev, 13: 555-580.
Levin W, Chang RL, Wood AW, Yagi H, Thakker DR, Jerina DM, & Conney AH
(1984) High stereoselectivity among the optical isomers of the
diastereomeric bay-region diolepoxides of benz(a)anthracene in the
expression of tumorigenic activity in murine tumor models. Cancer Res,
44: 929-933.
Levin W, Chang RL, Wood AW, Thakker DR, Yagi H, Jerina DM, & Conney AH
(1986) Tumorigenicity of optical isomers of the diastereomeric
bay-region 3,4-diol-1,2-epoxides of benzo [c]phenanthrene in murine
tumor models. Cancer Res, 46: 2257-2261.
Levin JO, Rhén M, & Sikström E (1995) Occupational PAH exposure:
Urinary 1-hydroxypyrene levels of coke oven workers, aluminium smelter
pot-room workers, roadpavers, and occupationally non-exposed persons
in Sweden. Sci Total Environ, 193: 169-177.
Levsen K (1988) The analysis of diesel particulate. Fresenius' Z Anal
Chem, 331: 467-478.
Levsen K, Behnert S, Priess B, & Winkeler HD (1991) The contamination
of precipitation in Hannover by hydrocarbons. Vom Wasser, 76: 109-126.
Lewis WM (1975) Polynuclear aromatic hydrocarbons in water. Water
Treat Exam, 23: 243-277.
Lewis RJ Sr (1992) Sax's dangerous properties of industrial materials,
8th ed. New York, Van Nostrand Reinhold Co., 1245 pp.
Lewtas J (1985a) Development of a comparative potency method for
cancer risk assessment of complex mixtures using short-term
in vivo and in vitro bioassays. Toxicol Ind Health, 4: 193-203.
Lewtas J (1985b) Combustion emissions: Characterization and comparison
of their mutagenic and carcinogenic activity. In: Stich HF ed.
Carcinogens and mutagens in the environment, Volume V: The workplace:
Source of carcinogens. Boca Raton, Florida, CRC Press, pp 59-74.
Lewtas J (1993) Complex mixtures of air pollutants: Characterizing the
cancer risk of polycyclic organic matter. Environ Health Perspectives,
100: 211-218.
Lewtas J, Mumford J, Everson RB, Hulka B, Wilcosky T, Kozumbo W,
Thompson C, George M, Dobiàs L, Sràm R, Li X, & Gallagher J (1993)
Comparison of DNA adducts from exposure to complex mixtures in various
human tissues and experimental systems. Environ Health Perspectives,
99: 89-97.
Li Y, Xu H, Song Y, Yang X, & Li Z (1991) [A study on the relationship
between urinary BaP and SCE frequency in rats.] Zhongguo Huanjing
Kexue, 11: 361-364 (in Chinese with English abstract).
Liao W, Smith WD, Chiang TC, & Williams LR (1988) Rapid, low-cost
cleanup procedure for determination of semivolatile organic compounds
in human and bovine adipose tissues. J Assoc Off Anal Chem, 71: 742-
747.
Lide DR ed. (1991) CRC handbook of chemistry and physics, 72nd ed.
Boca Raton, Florida, CRC Press, pp 22-35.
Liem AKD, Baumann RA, de Jong APJM, van der Velde EG, & van Zoonen P
(1992) Analysis of organic micropollutants in the lipid fraction of
foodstuffs. J Chromatogr, 624: 317-339.
Lies KH, Hartung A, Postulka A, Gring H, & Schulze J (1986)
Composition of diesel exhaust with particular reference to particle
bound organics including formation of artifacts. Dev Toxicol Environ
Sci, 13: 65-82.
Ligocki MP & Pankow JF (1989) Measurements of the gas/particle
distributions of atmospheric organic compounds. Environ Sci Technol,
23: 75-83.
Ligocki MP, Leuenberger C, & Pankow JF (1985) Trace organic compounds
in rain. II. Gas scavening of neutral organic compounds. Atmos
Environ, 19: 1609-1617.
Lijinsky W & Garcia H (1972) Skin carcinogenesis tests of hydrogenated
derivatives of anthanthrene and other polynuclear hydrocarbons. Z
Krebsforsch, 77: 226-230.
Lijinsky W & Ross AE (1967) Production of carcinogenic polynuclear
hydrocarbons in the cooking of food. Food Cosmet Toxicol, 5: 343-347.
Lijinsky W & Saffiotti U (1965) Relationships between structure and
skin tumorigenic activity among hydrogenated derivates of several
polycyclic aromatic hydrocarbons. Ann It Dermatol Clin Sper, 19: 34-
44.
Lijinsky W & Shubik P (1964) Benzo(a)pyrene and other polynuclear
hydrocarbons in charcoal-broiled meat. Science, 145: 53-55.
Lijinsky W & Shubik P (1965) Polynuclear hydrocarbon carcinogens in
cooked meat and smoked food. Ind Med Surg, 34: 152-154.
Lijinsky WH, Garcia B, & Terrracini B (1965) Tumorigenic activity of
hydrogenated derivatives of dibenz [a,h]anthracene. J Natl Cancer
Inst, 34:1-6.
Likhachev AJ, Beniashvili DS, Bykov VJ, Dikun PP, Tyndyk ML,
Savochkina IV, Yermilov VB, & Zabezhinski MA (1992) Biomarkers for
individual susceptibility to carcinogenic agents: Excretion and
carcinogenic risk of benzo [a]pyrene metabolites. Environ Health
Perspectives, 98: 211-214.
Linder G & Bergman H (1984) Periodic depuration of anthracene
metabolites by rainbow trout Salmo gairdneri. Trans Am Fish Soc,
113: 513-520.
Linder G, Bergman H, & Meyer J (1985) Anthracene bioconcentration in
rainbow-trout during single-compound and complex-mixture exposures.
Environ Toxicol Chem, 4: 549-558.
Lindquist B & Warshawsky D (1985) Identification of the
11,12-dihydro-11,12-dihydroxybenzo [a]pyrene as a major metabolite
produced by the green alga, Selenastrum capriconutum. Biochem
Biophys Res Commun, 130: 7.
Lindskog A, Brorström-Lundén E, Alfheim I, & Hagen I (1987) Chemical
transformation of PAH on airborne particles by exposure to NO2 during
sampling: A comparison between two filter media. Sci Total Environ,
61: 51-57.
Lindstedt G & Sollenberg J (1982) Polycyclic aromatic hydrocarbons in
the occupational environment - with special reference to
benzo [a]pyrene measurements in Swedish industry. Scand J Work
Environ Health, 3: 1-19.
Lindström-Seppä P, Hänninen O, Tuominen J, & Pyysalo H (1989)
Polycyclic aromatic hydrocarbons in perch (Perca fluviatilis)
following an oil-spill in Vaasa Archipelago, Finland. Toxicol Environ
Chem, 19: 83-86.
Lintas C, De Matthaeis MC, & Merli F (1979) Determination of
benzo(a)pyrene in smoked, cooked and toasted food products. Food
Cosmet Toxicol, 17: 325-328.
Liotti FS, Pelliccia C, & Pezzetti F (1988) Different response of
chicken embryo fibroblasts and hepatocytes to the interference of
certain antioxidants on the binding of [G3H]benzo [a]pyrene to DNA.
Cancer Lett, 41: 235-242.
Lioy PL, Waldman JM, Greenberg A, Harkov R, & Pietarinen C (1988) The
total human environmental exposure study (THEES) to benzo(a)pyrene:
Comparison of the inhalation and food pathways. Arch Environ Health,
43: 304-312.
Little JB & Vetrovs H (1988) Studies of ionizing radiation as a
promoter of neoplastic transformation in vitro. Int J Radiat Biol,
53: 661-666.
Liu D, Maguire RJ, Pacepavicius GJ, & Nagy E (1992) Microbial
degradation of polycyclic aromatic hydrocarbons and polycyclic
aromatic nitrogen heterocyclics. Environ Toxicol Water Qual, 7: 355-
372.
Lo MT & Sandi E (1978) Polycyclic aromatic hydrocarbons (polynuclear)
in foods. Res Rev, 69: 35-85.
Loening KL & Merritt JE (1990) Some aids for naming polycyclic
aromatic hydrocarbons and their heterocyclic analogs. In: Loening K,
Merritt J, Later D, & Wright W ed. Polynuclear aromatic hydrocarbons:
Nomenclature guide. Columbus, Ohio, Battelle Press, pp 1-25.
Löfroth G, Toftgard R, Nilsson L, Agurell E, & Gustafsson JA (1984)
Short-term bioassays of nitro derivatives of benzo [a]pyrene and
perylene. Carcinogenesis, 5: 925-930.
Lovell WW & Sanders DJ (1992) Phototoxicity testing in guinea-pigs.
Food Chem Toxicol, 30: 155-160.
Lowenthal DH, Zielinska B, Chow JC, Watson JG, Gautman M, Ferguson DH,
Neuroth GR, & Stevens KD (1994) Characterizitation of heavy-duty
diesel vehicle emissions. Atmos Environ, 28: 731-743.
Lu PY, Metcalf RL, Plummer N, & Mandel D (1977) The environmental fate
of three carcinogens: Benzo [a]pyrene, benzidine, and vinyl chloride
evaluated in laboratory model ecosystems. Arch Environ Contam Toxicol,
6: 129-142.
Lubet RA, Kiss E, Gallagher MM, Dively C, Konri R, & Schechtman LM
(1983a) Induction of neoplastic transformation and DNA single-strand
breaks in C3H/10T1/2 clone 8 cells by polycyclic hydrocarbons and
alkylating agents. J Natl Cancer Inst, 71: 991-997.
Lubet RA, Connelly GM, & Nebert DW (1983b)
Dibenz [a,h]anthracene-induced subcutaneous tumors in mice. Strain
sensitivity and the role of carcinogen metabolism. Carcinogenesis, 4:
513-517.
Lubet RA, Brunda MJ, & Lemaira B (1984a) Polycyclic hydrocarbon
induced immunotoxicity in mice: Role of the Ah locus. In: Cooke M &
Dennis AJ ed. Polycyclic aromatic hydrocarbons: Mechanisms, methods
and metabolism. Columbus, Ohio, Battelle Press, pp 843-858.
Lubet RA, Brunda MJ, Taramelli D, Dansie D, Nebert DW, & Kouri RE
(1984b) Induction of immunotoxicity by polycyclic hydrocarbons: Role
of the Ah locus. Arch Toxicol, 56: 18-24.
Lubet RA, Kouri RE, Curren RA, Putman DL, & Schechtman LM (1990)
Induction of mutagenesis and transformation in BALB/c-3T3 clone A31-1
cells by diverse chemical carcinogens. Environ Mol Mutag, 16: 13-20.
Luijten JA & Piet GJ (1983) Identity and sources of organic chemicals
in groundwater. In: Proceedings of the 1st Atlantic Workshop,
Nashville, Tennessee. Denver, Colorado, American Water Works
Association, pp 7-18.
van Luin AB & van Starkenburg W (1984) Hazardous substances in waste
water. Water Sci Technol, 17: 843-853.
Lunde G (1976) Long-range aerial transmission of organic
micropollutants. Ambio, 5: 207-208.
Lunde G & Bjorseth A (1977) Polycyclic aromatic hydrocarbons in
long-range transported aerosols. Nature, 268: 518-519.
Luther M, Moriske HJ, & Rüden H (1990) [Indoor air levels of
polycyclic aromatic hydrocarbons from the use of asphalt floor tiles.]
Forum Städte-Hyg, 41: 95-103 (in German).
Lutz WK & Schlatter J (1992) Chemical carcinogens and overnutrition in
diet-related cancer. Carcinogenesis, 13: 2211-2216.
Lyall RJ, Hooper MA, & Mainwaring SJ (1988) Polycyclic aromatic
hydrocarbons in the Latrobe Valley. Atmos Environ, 22: 2549-2555.
Lygren E, Gjessing E, & Berglind L (1984) Pollution transport from a
highway. Sci Total Environ, 33: 147-159.
Lyman WJ, Reehl WF, & Rosenblatt DH ed. (1982) Handbook on chemical
property estimation methods, environmental behavior of organic
compounds. New York, McGrawHill, 960 pp.
Lyon JL, Klauber MR, Graff W, & Chiu G (1981) Cancer clustering around
point sources of pollution. Assessment by a case control methodology.
Environ Res, 25: 29-34.
Lyte Ml, Blanton RH, Myers MJ, & Bick PH (1987) Effect of
in vivo administration of carcinogen benzo(a)pyrene on interleukin-2
and interleukin-3 production. Int J Immunopharmacol, 9: 307-312.
Maccubbin AE, Black P, Trzeciak L, & Black JJ (1985) Evidence for
polynuclear aromatic hydrocarbons in the diet of bottom-feeding fish.
Bull Environ Contam Toxicol, 34: 876-882.
Machado ML, Beatty PW, Fetzer JC, Glickman AH, & McGinnis EL (1993)
Evaluation of the relationship between PAH content and mutagenic
activity of fumes from roofing and paving asphalts and coal tar pitch.
Fundam Appl Toxicol, 21: 492-499.
Mackay D & Leinonen PJ (1975) Rate of evaporation of low-solubility
contaminants from water bodies to atmosphere. Environ Sci Technol, 9:
1178-1180.
Mackay D & Shiu WY (1977) Aqueous solubility of polynuclear aromatic
hydrocarbons. J Chem Eng Data, 22: 399-402.
Mackay D & Shiu WY (1981) A critical review of Henry's law constants
for chemicals of environmental interest. J Phys Chem Ref Data, 10:
1175-1199.
Mackay D, Shiu WY, & Sutherland RP (1979) Determination of air-water
Henry's law constants for hydrophobic pollutants. Environ Sci Technol,
13: 333-337.
Mackay D, Shiu WY, & Ma KC (1992) Illustrated handbook of physical-
chemical properties and environmental fate for organic chemicals.
Volume II: Polynuclear aromatic hydrocarbons, polychlorinated dioxins
and dibenzofurans. Boca Raton, Florida, Lewis Publishers, pp 1-367.
Mackell JV, Rieders F, Brieger H, & Bauer EL (1951) Acute hemolytic
anemia due to ingestion of naphthalene moth balls. Pediatrics, 7: 722-
728.
MacKenzie KM & Angevine DM (1981) Infertility in mice exposed in utero
to benzo(a)pyrene. Biol Reprod, 24: 183-191.
Maclaren WM & Hurley JF (1987) Mortality of tar distillation workers.
Scand J Work Environ Health, 13: 404-411.
MacNicoll AD, Easty GC, Neville AM, Grover PL, & Sims P (1980)
Metabolism and activation of carcinogenic polycyclic hydrocarbons by
human mammary cells. Biochem Biophys Res Commun, 95: 1599-1606.
Madhavan ND& Naidu KA ( 1995) Polycyclic aromatic hydrocarbons in
placenta, maternal blood, umbilical cord blood and milk of Indian
women. Hum Exp Toxicol, 14: 503-506.
Maga JA (1986) Polycyclic aromatic hydrocarbon (PAH) composition of
mesquite (Prosopis fuliflora) smoke and grilled beef. J Agric Food
Chem, 34: 249-251.
Mager R, Huberman E, Yang SK, Gelboin HV, & Sachs L (1977)
Transformation of normal hamster cells by benzo(a)pyrene diol-epoxide.
Int J Cancer, 19: 814-817.
Mahlum D, Wright CW, Chess EK, & Wilson BW (1984) Fractionation of
skin tumor initiating activity in coal liquids. Cancer Res, 44: 5176-
5181.
Maisin J & Coolen M-L (1936) [Carcinogenic potency of
methylcholanthrene.] Comp Soc Biol, 123: 159-160 (in French).
Malaiyandi M, Benedek A, Holko AP, & Bancsi JJ (1986) Measurement of
potentially hazardous polynuclear aromatic hydrocarbons from
occupational exposure during roofing and paving operations. In: Cooke
M, Dennis AJ, & Fisher GL ed. Polynuclear aromatic hydrocarbons:
Physical and biological chemistry. Columbus, Ohio, Battelle Press, pp
471-489.
Malcolm HM & Dobson S (1994) The calculation of an environmental
assessment level (EAL) for atmospheric PAHs using relative
potencies.London, Department of the Environment, 34 pp (Report No.
DoE/HMIP/RR/94/041).
Malins DC (1982) Alterations in the cellular and subcellular structure
of marine teleosts and invertebrates exposed to petroleum in the
laboratory and field: A critical review. Can J Fish Aquat Sci, 39:
877-889.
Malins DC, McCain BB, Brown DW, Chan S-L, Myers MS, Landahl JT,
Prohaska PG, Friedman AJ, Rhodes LD, Burrows DG, Gronlund WD, &
Hodgins HO (1984) Chemical pollutants in sediments and diseases of
bottom-dwelling fish in Puget Sound, Washington. Environ Sci Technol,
18: 705-713.
Malins DC, Krahn MM, Brown DW, Rhodes LD, Myers MS, McCain BB, & Chan
SL (1985) Toxic chemicals in marine sediment and biota from Mukilteo,
Washington: Relationships with hepatic neoplasms and other hepatic
lesions in English sole (Parophrys vetulus). J Natl Cancer Inst, 74:
487-494.
Mallet WG, Mosebrook DR, & Trush MA (1991) Activation of
(±)- trans-7,8-dihydroxy-7,8-dihydrobenzo(a)pyrene to diolepoxides by
human polymorphonuclear leukocytes or myeloperoxidase. Carcinogenesis,
12: 521-524.
Mamber SW, Bryson V, & Katz SE (1983) The Escherichia coli
WP2/WP100 rec assay for detection of potential chemical carcinogens.
Mutat Res, 119: 135-144.
Mamber SW, Bryson V, & Katz SE (1984) Evaluation of the Escherichia
coli K12 inductest for detection of potential chemical carcinogens.
Mutat Res, 130: 141-151.
Mane SS, Purnell DM, & Hsu IC (1990) Genotoxic effects of five
polycyclic aromatic hydrocarbons in human and rat mammary epithelial
cells. Environ Mol Mutagen, 15: 78-82.
Manilal VB & Alexander M (1991) Factors affecting the microbial
degradation of phenanthrene in soil. Appl Microbiol Biotechnol, 35:
410-405.
Mannschreck C, Gündel J, & Angerer K (1996) Occupational exposure to
PAH. Biological monitoring of hydroxylated metabolites. Polycyclic
Aromat Compd 11: 11-18.
Manz A, Berger J, & Waltsgott H (1983) [Occupational cancer in gas
industry workers.] Bremerhaven, German Federal Department for Worker
Safety and Accident Research pp 1-131 (Wirtschaftsverlag NW, Report
No. 352) (in German).
Marcomini A, Pavoni B, Donazzolo R, & Orio AA (1986) Combined
preparative and analytical use of normal-phase and reversed-phase
high-performance liquid chromato-graphy for determination of aliphatic
and polycyclic aromatic hydrocarbons in sediments of the Adriatic Sea.
Mar Chem, 18: 71-84.
Marcus JM & Stokes TP (1985) Polynuclear aromatic hydrocarbons in
oyster tissue around three coastal marinas. Bull Environ Contam
Toxicol, 35: 835-844.
Marcus JM, Swearingen GR, Williams AD, & Heizer DD (1988) Polynuclear
aromatic hydrocarbon and heavy metal concentrations in sediments at
coastal South Carolina marinas. Arch Environ Contam Toxicol, 17: 103-
113.
Marino DJ (1987) Evaluation of Pluronic(R) Polyol F17 as a vehicle
for petroleum hydrocarbons in the Salmonella/microsomal assay. Environ
Mutagen, 9: 307-316.
Marnett LJ, Reed GA, & Dennison DJ (1978) Prostaglandin synthetase
dependent activation of 7,8-dihydro-7,8-dihydroxy-benzo(a)pyrene to
mutagenic derivatives. Biochem Biophys Res Commun, 82: 210-216.
Marquardt H & Heidelberger C (1972) Influence of 'feeder cells' and
inducers and inhibitors of microsomal mixed-function oxidases on
hydrocarbon-induced malignant transfromation of cells derived from C3H
mouse prostate. Cancer Res, 32: 721-725.
Marquardt H, Kuroki T, Huberman E, Selkirk JK, Heidelberger C, Grover
PL, & Sims P (1972) Malignant transformation of cells derived from
mouse prostate by epoxides and other derivatives of polycyclic
hydrocarbons. Cancer Res, 32: 716-720.
Marshall MV, McLemore TL, Martin RR, Jenkins WT, Snodgrass DR, Corson
MA, Arnott MS, Wray NP, & Griffin AC (1979) Patterns of
benzo [a]pyrene metabolism in normal human pulmonary alveolar
macrophages. Cancer Lett, 8: 103-109.
Martel L, Gagnon MJ, Masse R, Leclerc A, & Tremblay L (1986)
Polycyclic aromatic hydrocarbons in sediments from the Saguenay Fjord,
Canada. Bull Environ Contam Toxicol, 37: 133-140.
Martin BJ (1980) Effects of petroleum compounds on estuarine fishes.
Gulf Breeze, Florida, US Environmental Protection Agency, 43 pp
(EPA-600/3-80-019; PB 80-141310).
Martin CN & McDermid AC (1981) Testing of 42 coded compounds for their
ability to induce unscheduled DNA repair synthesis in HeLa cells. In:
DeSerres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme. New
York, Elsevier North Holland, pp 533-537 (Progress in Mutation
Research, Volume 1).
Martin CN, McDermid AC, & Garner RC (1978) Testing of known
carcinogens and noncarcinogens for their ability to induce unscheduled
DNA synthesis in HeLa cells. Cancer Res, 38: 2621-2627.
Martin F, Hoepfner I, Scherer G, Adlkofer F, Dettbarn G, & Grimmer G
(1989) Urinary excretion of hydroxy-phenanthrenes after intake of
polycyclic aromatic hydrocarbons. Environ Int, 15: 41-47.
Masclet P, Nikolaou K, & Mouvier G (1984) [Identification of sources
of particular polycyclic aromatic hydrocarbons in the urban
atmosphere.] In: Versino B & Angeletti ed. Physico-chemical behavior
of atmospheric pollutants. Dordrecht: Reidel Publishing Co., pp 616-
625 (in French).
Mass MJ, Rodgers NT, & Kaufman DG (1981) Benzo [a]pyrene metabolism
in organ culture of human endometrium. Chem-Biol Interactions, 33:
195-205.
Massie LC, Ward AP, & Davies JM (1985) The effects of oil exploration
and production in the northern North Sea: Part 1. The levels of
hydrocarbons in water and sediments in selected areas, 1978-1981. Mar
Environ Res, 15: 165-213.
Masuda Y & Kagawa R (1972) A novel synthesis and carcinogenicity of
dibenzo [a,l]pyrene. Chem Pharm Bull, 20: 2736-2737.
Matsumoto H & Kashimoto T (1985) Average daily respiratory intake of
polycyclic aromatic hydrocarbons in ambient air determined by
capillary gas chromatography. Bull Environ Contam Toxicol, 34: 17-23.
Matsuoka A, Hayashi M, & Ishidate MJ (1979) Chromosomal aberration
tests on 29 chemicals combined with S9 mix in vitro. Mutat Res, 66:
277-290.
Matsuoka A, Sofuni I, Miyata N, & Ishidate M Jr (1991) Clastogenicity
of 1-nitropyrene, dinitropyrenes, fluorene and mononitrofluorenes in
cultered Chinese hamster cells. Mutat Res, 259: 103-110.
Mattison DR & Nightingale MR (1980) The biochemical and genetic
characteristics of murine ovarian aryl hydrocarbon (benzo [a]pyrene)
hydroxylase activity and its relationship to primordial oocyte
destruction by polycyclic aromatic hydrocarbons. Toxicol Appl
Pharmacol, 56: 399-408.
Mattison DR, White NB, & Nightingale MS (1980) The effect of
benzo(a)pyrene on fertility, primordial oocyte number, and ovarian
response to pregnant mare's serum gonadotropin. Pediatr Pharmacol, 1:
143-151.
Mattison DR, Singh H, Takizawa K, & Thomford PJ (1989) Ovarian
toxicity of benzo(a)pyrene and metabolites in mice. Reprod Toxicol, 3:
115-125.
Matzner E (1984) Annual rates of deposition of polycyclic aromatic
hydrocarbons in different forest ecosystems. Water Air Soil Pollut,
21: 425-434.
Matzner B, Hübner D, & Thomas W (1981) Content and storage of
polycyclic aromatic hydrocarbons in two forested ecosystems in
northern Germany. Z Pflanzenernähr Bodenkd, 144: 283-288.
Mavournin KH, Blakey DH, Cimino MC, Salamone MF, & Heddle JA (1990)
The in vivo micronucleus assay in mammalian bone marrow and
peripheral blood. A report of the US Environmental Protection Agency
Gene-Tox Program. Mutat Res, 239: 29-80.
May WE, Wasik SP, & Freeman DH (1978) Determination of the aqueous
solubility of polynuclear aromatic hydrocarbons by a coupled column
liquid chromatographic technique. Anal Chem, 50: 175-179.
McCann J, Choi E, Yamasaki E, & Ames BN (1975a) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc Natl Acad Sci USA, 72: 5135-5139.
McCann J, Spingarn NE, Kobori J, & Ames BN (1975b) Detection of
carcinogens as mutagens: Bacterial tester strains with R factor
plasmids. Proc Natl Acad Sci USA, 72: 979-983.
McCarroll NE, Keech BH, & Piper CE (1981) A microsuspension adaptation
of the Bacillus subtilis 'rec' assay. Environ Mutag, 3: 607-616.
McCarthy JF & Jimenez BD (1985) Reduction in bioavailability to
bluegills of polycyclic aromatic hydrocarbons bound to dissolved humic
material. Environ Toxicol Chem, 4: 511-521.
McCarthy JF, Jimenez BD, & Barbee T (1985) Effect of dissolved humic
material on accumulation of polycyclic aromatic hydrocarbons:
Structure-activity relationships. Aquat Toxicol, 7: 15-24.
McClure P & Schoeny R (1995) Evaluation of a component-based relative
potency approach to cancer risk assessment for exposure to PAH. In:
Fifteenth international symposium on polycyclic aromatic compounds:
Chemistry, biology and environmental impact, Belgirate, Italy, 19-22
September 1995. Ispra, Joint Research Centre European Commission, p
161.
McCormick DL, Burns FJ, & Albert RE (1981) Inhibition of
benzo(a)pyrene-induced mammary carcinogenesis by retinyl acetate. J
Natl Cancer Inst, 66: 559-564.
McElroy AE (1985) In vivo metabolism of benz(a)anthracene by the
polychaete Nereis virens. Mar Environ Res, 17: 133-136.
McElroy AE & Sisson JP (1989) Trophic transfer of benzo [a]pyrene
metabolites between benthic marine organisms. Mar Environ Res, 28:
265-269.
McFall JA, Antoine SR, & DeLeon IR (1985) Base-neutral extractable
organic pollutants in biota and sediments from Lake Pontchartrain.
Chemosphere, 14: 1561-1569.
McGill AS, Mackie PR, Parsons E, Bruce C, & Hardy R (1982) The
polynuclear aromatic hydrocarbon content of smoked foods in the United
Kingdom. In: Cooke M, Dennis AJ, & Fisher GL ed. Polynuclear aromatic
hydrocarbons: Physical and biological chemistry. Columbus, Ohio,
Battelle Press, pp 491-499.
McGill AS, Mackie PR, Howgate P, & McHenery JG (1987) The flavour and
chemical assessment of dabs (Limanda limanda) caught in the vicinity
of the Beatrice oil platform. Mar Pollut Bull, 18: 186-189.
McIntyre AE, Perry R & Lester JN (1981) Analysis of polynuclear
aromatic hydrocarbons in sewage sludges. Anal Lett, 14: 291-309.
McKay S, Phillips DH, Hewer AJ, & Grover PL (1988) Metabolic
activation of 7-ethyl- and 7-methylbenz [a]anthracene in mouse skin.
Carcinogenesis, 9: 141-145.
McLeese DW & Burridge LE (1987) Comparative accumulation of PAHs in
four marine invertebrates. In: Capuzzo IM & Kester DR ed. Oceanic
processes in marine pollution. Malabar, Florida, Kirger RE, pp
109-118.
McMahon CK & Tsoukalas SN (1978) Polynuclear aromatic hydrocarbons in
forest fire smoke. In: Jones PW & Freudenthal RI ed. Carcinogenesis.
Volume 3: Polynuclear aromatic hydrocarbons. New York, Raven Press, pp
61-73.
McVeety BD & Hites RA (1988) Atmospheric deposition of polycyclic
aromatic hydrocarbons to water surfaces: A mass balance approach.
Atmos Environ, 22: 511-536.
Means JC, Hassett JJ, Wood SG, & Banwart WL (1979) Sorption properties
of energy-related pollutants and sediments. In: Jones PW & Leber P ed.
Polynuclear aromatic hydrocarbons. Ann Arbor, Michigan, Ann Arbor
Science Publishers, pp 327-340.
Means JC, Wood SG, Hassett JJ, & Banwart WL (1980) Sorption of
polynuclear aromatic hydrocarbons by sediments and soils. Environ Sci
Technol, 14: 1524-1528.
Mehta R, Meredith-Brown M, & Cohen GM (1979) Metabolism and covalent
binding of benzo [a]pyrene in human peripheral lung. Chem-Biol
Interactions, 28: 345-358.
Melancon M & Lech J (1978) Distribution and elimination of naphthalene
and 2-methylnaphthalene in rainbow trout during short- and long-term
exposures. Arch Environ Contam Toxicol, 7: 207-220.
Melikian AA, LaVoie EJ, Hecht SS, & Hoffmann D (1983) 5-Methylchrysene
metabolism in mouse epidermis in vivo, diol epoxide-DNA adduct
persistence, and diol epoxide reactivity with DNA as potential factors
influencing the predominance of 5-methylchrysene-1,2-diol-3,4-epoxide-
DNA adducts in mouse epidermis. Carcinogenesis, 4: 843-849.
Melikian AA, Amin S, Huie K, Hecht SS, & Harvey G (1988) Reactivity
with DNA bases and mutagenicity toward Salmonella typhimurium of
methylchrysene diol epoxide enantiomers. Cancer Res, 48: 1781-1787.
Melikian AA, Prahalad AK, Amin SG, & Hecht SS (1991) Comparative DNA
binding of benzo(a)pyrene, 5-methylchrysene, 6-methylchrysene and
their corresponding bay-region dihydrodiol epoxides in newborn mouse
lung and liver in vivo. Proc Am Assoc Cancer Res, 32: 96.
Melius P, Elan D, Kilgore M, Tan B, & Schoor WP (1980) Mixed function
oxidase inducibility and polyaromatic hydrocarbon metabolism in the
mullet, sea catfish and Gulf killifish. In: Bjorseth A & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemistry and biological effects.
Columbus, Ohio, Battelle Press, pp 1059-1075.
Menichini E (1992a) Urban air pollution by polycyclic aromatic
hydrocarbons: Levels and sources of variability. Sci Total Environ,
116: 109-135.
Menichini E (1992b) Opinion adopted by the Italian National Advisory
Toxicological Committee on polycyclic aromatic hydrocarbons. Rome,
Istituto Superiore di Sanità, 69 pp (Serie Relazioni 92/4).
Menichini E, Bonanni L, & Merli F (1990) Determination of polycyclic
aromatic hydrocarbons in mineral oils and oil aerosols in glass
manufacturing. Toxicol Environ Chem, 28: 37-51.
Menichini E, di Domenico A, Bonanni L, Corradetti E, Mazzanti L, &
Zucchetti G (1991a) Reliability assessment of a gas chromatographic
method for polycyclic aromatic hydrocarbons in olive oil. J
Chromatogr, 555: 211-220.
Menichini E, Bocca A, Merli F, Ianni D, & Monfredini F (1991b)
Polycyclic aromatic hydrocarbons in olive oils on the Italian market.
Food Addit Contam, 8: 363-369.
Merriman JC (1988) Distribution of organic contaminants in water and
suspended solids of the Rainy River. Water Pollut Res J Can, 23: 590-
600.
Mersch-Sundermann V, Mochayedi S, & Kevekordes S (1992) Genotoxicity
of polycyclic aromatic hydrocarbons in
Escherichia coli PQ37. Mutat Res, 278: 1-9.
Meyer JP & Grimmer G (1974) [The influence of PCA-containing and
PCA-free fuel on the emission of polycyclic aromatic hydrocarbons from
a motor vehicle with spark-ignition engine in the Europe test cycle.]
Hamburg, German Society for Mineral-oil and Coal Chemistry, 22 pp
(BMI-DGMK joint project 4547, Part 1) (in German).
Meyer H-P, Grimmer G, & Behn U (1980) [PAH from oil heating:
Collection, analysis, results. Air pollution from polycylic aromatic
hydrocarbons. Registration and evaluation.] Düsseldorf, VDI-Verlag, pp
101-105 (VDI Report No. 358) (in German).
Meyers MJ, Blanton RH, & Bick PH(1988) Inhibition of IL-2
responsiveness following exposure to benzo(a)pyrene is due to
alterations in accessory cell function. Int J Immunopharmacol, 10:
177-186.
Michel X, Bani MH, & Narbonne JF (1995) Regio-selective metabolism of
benzo [a]pyrene by microsomes from 5 vertebrate species. In:
Fifteenth international symposium on polycyclic aromatic compounds:
Chemistry, biology and environmental impact, Belgirate, Italy, 19-22
September 1995. Ispra, Joint Research Centre European Commission, p
78.
Middaugh DP, Thomas RL, Lantz SE, Heard CS, & Mueller JG (1994) Field
scale testing of a hyperfiltration unit for removal of creosote and
pentachlorophenol from groundwater: Chemical and biological
assessment. Arch Environ Contam Toxicol, 26: 309-319.
Mielzynska D & Snit M (1992) Urine mutagenicity in workers directly
employed in coke production. Pol J Occup Med, 5: 363-371.
Miescher G (1942) [Experimental studies on the formation of tumours by
photosensitization.] Schweiz Med Wochenschr, 72: 1082-1984 (in
German).
Milano JC & Vernet JL (1988) [Size of the contribution of carcinogenic
polyaromatic hydrocarbons by the Rhône.] Oceanis, 14: 133-140 (in
French).
Milano JC, Fache B, & Vernet JL (1985) [Contamination of the Cortiou
site by polyaromatic hydrocarbons in wastewater from the city of
Marseille (western Mediterranean, France).] J Rech Oceanogr, 10: 36-38
(in French).
Milano JC, Fache B, & Vernet JL (1986) [Pollution of the sediment,
fauna and flora by polyaromatic hydrocarbons at Cap Sicié at the
emission site of the city of Toulon (French Mediterranean coast).] J
Rech Oceanogr, 11: 93-96 (in French).
Mill T & Mabey W (1985) Photochemical transformations. In: Neely WB &
Blau GE ed. Environmental exposure from chemicals, Volume 1. Boca
Raton, Florida, CRC Press, pp 175-216.
Mill T, Mabey WR, Lan BY, & Baraze A (1981) Photolysis of polycyclic
aromatic hydrocarbons in water. Chemosphere, 10: 1281-1290.
Millemann RE, Birge WJ, Black JA, Cushman RM, Daniels KL, Franco PJ,
Giddings JM, McCarthy JF, & Stewart AJ (1984) Comparative acute
toxicity of aquatic organisms of components of coal derived synthetic
fuels. Trans Am Fish Soc, 113: 74-85.
Miller EC (1951) Studies on the formation of protein-bound derivatives
of 3,4-benzpyrene in the epidermal fraction of mouse skin. Cancer Res,
11: 100-108.
Miller JA & Miller EC (1953) The carcinogenic aminoazo dyes. Adv
Cancer Res, 1: 339-396.
Miller JA & Miller EC (1963) The carcinogenicities of fluoro
derivatives of 10-methyl- 1,2-benzanthracene. II. Substitution of the
K region and the 3'- 6-, and 7-positions. Cancer Res, 23: 229-239.
Miller JA & Miller (1977) Ultimate chemical carcinogens as reactive
mutagenic electrophiles In: Hiatt HH, Watson JD, & Winston JA ed.
Origins of human cancer, Book B, Mechanisms of carcinogenesis. Cold
Spring Harbor, New York, Cold Spring Harbor Laboratory, pp 605-627.
Miller MM, Wasik SP, Huang G-L, Shiu W-Y, & Mackay D (1985)
Relationships between octanol-water partition coefficient and aqueous
solubility. Environ Sci Technol, 19: 522-529.
Miller MM, Plowchalk DR, Weitzman GA,, London SN & Mattison DR (1992)
The effect of benzo(a)pyrene on murine ovarian and corpora lutea
volumes. Am J Obstet Gynecol, 166: 1535-1541.
Milo GE, Blakeslee J, Yohn DS, & DiPaolo JA (1978) Biochemical
activiation of aryl hydrocarbon hydroxylase activity, cellular
distribution of polynuclear hydrocarbon metabolites, and DNA damage by
polynuclear hydrocarbon products in human cells in vitro.
Cancer Res, 38: 1638-1644.
Miralis JC, Tyson CK, & Butterworth BE (1982) Detection of genotoxic
carcinogens in the in vivo-in vitro hepatocyte DNA repair assay.
Environ Mutag, 4: 553-562.
Mishra NK, Wilson CM, Pant KJ, & Thomas FO (1978) Simultaneous
determination of cellular mutagenesis and transformation by chemical
carcinogens in Fischer rat embryo cells. J Toxicol Environ Health, 4:
79-91.
Mitchell CE (1982) Distribution and retention of benzo [a]pyrene in
rats after inhalation. Toxicol Lett, 11: 35-42
Mitchell RL, Burchett MD, Pulkownik A, & McCluskey L (1988) Effects of
environmentally hazardous chemicals on the emergence and early growth
of selected Australian plants. Plant Soil, 112: 195-199.
Mix MC & Schaffer RL (1983) Concentrations of unsubstituted polycyclic
aromatic hydrocarbons in softshell clams from Coos Bay, Oregon, USA.
Mar Pollut Bull, 14: 94-97.
Mohr U (1969) [Studies on the carcinogenic properties of
3.4,-10.11-,12.13- tribenzofluoranthene by mice.] Arch Hyg, 153: 495-
510 (in German).
Mohr U (1971) [Carcinogenicity of diethylnitrosamine.] Fortschr Med,
89: 251-253 (in German).
Moles A (1980) Sensitivity of parasitized Coho salmon fry to crude
oil, toluene, and naphthalene. Am Fish Soc, 109: 293-297.
Moles A, Bates S, Rice SD, & Korn S (1981) Reduced growth of Coho
salmon fry exposed to two petroleum components, toluene and
naphthalene, in fresh water. Am Fish Soc, 110: 430-436.
Monarca S, Pasquini R, Scassellati Sforzolini G, Savino A, Bauleo FA,
& Angeli G (1987) Environmental monitoring of mutagenic/carcinogenic
hazards during road paving operations with bitumens. Int Arch Occup
Environ Health, 59: 393-402.
Monnerjahn S, Seidel A, Steinberg P, Oesch F, Hinz M, Stezowsky JJ,
Hewer A, Phillips DH & Glatt HR (1993) Formation of DNA adducts from
1-hydroxymethylpyrene in liver cells in vivo and in
vitro. In: Phillips DH, Castegnaro M & Bartsch H ed. Postlabelling
methods for detection of DNA adducts, Lyon, International Agency for
Research on Cancer, pp. 189-193 (IARC Scientific Publications No. 124)
Monteith DK & Gupta RC (1992) Carcinogen-DNA adducts in cultures of
rat and human hepatocytes. Cancer Lett, 62: 87-93.
Montizaan GK, Kramers PGH, Janus JA & Posthumus R (1989) Integrated
criteria document PAH: Effects of 10 selected compounds. Appendix to
Report No. 758474011. Bilthoven, National Institute of Public Health
and Environmental Protection, 180 pp (Re-publication in March 1989 of
addendum to Report No. 758447007).
Moolgavkar SH & Knudson A (1981) Mutation and cancer: A model for
human carcinogenesis. J Natl Cancer Inst, 66: 1037-1052.
Moolgavkar SH & Venzon DJ (1979) Two-event models for carcinogenesis.
Math Biosci, 47: 55-77.
Moore BP, Hicks RM, Knowles MA, & Redgrave S (1982) Metabolism and
binding of benzo(a)pyrene and 2-acetylaminofluorene by short-term
organ cultures of human and rat bladder. Cancer Res, 42: 642-648.
Moore MN, Livingstone DR, & Widdows J (1989) Hydrocarbons in marine
mollusks: Biological effects and ecological consequences. In: Varanasi
U ed. Metabolism of polycyclic aromatic hydrocarbons in the aquatic
environment. Boca Raton, Florida, CRC Press, pp 291-329.
Moriske H-J, Freise R, Schneider C, & Rüden H (1987) [Polar neutral
organic compounds (POCN) in town aerosols. 2. Communication:
Measurement of the emissions from homeheating and cars and from
immission particles in Berlin (West).] Zentralbl Bakteriol Hyg I Abt
Orig B, 185: 72-104 (in German).
Morris HP, Velat CA, Wagner BP, Dahlgard M, & Ray FE (1960) Studies of
carcinogenicity in the rat of derivatives of aromatic amines related
to N-2-fluorenylacetamide. J Natl Cancer Inst, 24: 149-180.
Morrison VM, Burnett AK, & Craft JA (1991a) Metabolism of
7,12-dimethyl-benz [a]anthracene in hepatic microsomal membranes from
rats treated with isoenzyme-selective inducers of cytochromes P450.
Biochem Pharmacol, 41: 1505-1512.
Morrison VM, Burnett AK, Forrester LM, Wolf CR, & Craft JA (1991b) The
contribution of specific cytochromes P450 in the metabolism of
7,12-dimethylbenz [a]anthracene in rat and human liver microsomal
membranes. Chem-Biol Interactions, 79: 179-196.
Morselli L & Zappoli S (1988) PAH determination in samples of
environmental interest. Sci Total Environ, 73: 257-266.
Mossanda K, Poncelet F, Fouassin A, & Mercier A (1979) Short papers.
Detection of mutagenic polycyclic aromatic hydrocarbons in African
smoked fish. Food Cosmet Toxicol, 17: 141-143.
Motykiewicz G (1995) Application of biomarkers in heavily polluted
industrialized areas of countries of central and eastern Europe.
Toxicology, 101: 117-123.
Moulin JJ, Portefaix P, Wild P, Mur JM, Smagghe G, & Mantout B (1990)
Mortality study among workers producing ferroalloys and stainless
steel in France. Br J Ind Med, 47: 537-543.
Moulin JJ, Wild P, Mantout B, Fournier-Betz M, Mur JM, & Smagghe G
(1993) Mortality from lung cancer and cardiovascular diseases among
stainless-steel producing workers. Cancer Causes Control, 4: 75-81.
Muel B & Saguem S (1985) Determination of 23 polycyclic aromatic
hydrocarbons in atmospheric particulate matter of the Paris area and
photolysis by sunlight. Int J Environ Anal Chem, 19: 111-131.
Mueller JG & Lantz SE (1993) Strategy using bioreactors and specially
selected microorganisms for bioremediation of groundwater contaminated
with creosote and pentachlorophenol. Environ Sci Technol, 27: 691-698.
Müller E (1968) [Carcinogenic compounds in water and soil. XX.
Investigation into the carcinogenic properties of benzo(ghi)perylene.]
Arch Hyg, 152: 23-36 (in German with English abstract).
Müller H (1987) Hydrocarbons in the freshwater environment. A
literature review. Arch Hydrobiol, 24: 1-69.
Müller G, Grimmer G & Böhnke H (1977) [Sedimentary record of heavy
metals and polycyclic aromatic hydrocarbons in Lake Constance.].
Naturwissenschaften, 64: 427-431 (in German).
Muller P, Leece B & Raha D (1995a) Estimated risk of cancer from
exposure to PAH fractions of complex mixtures. In: Fifteenth
international symposium on polycyclic aromatic compounds: Chemistry,
biology and environmental impact, Belgirate, Italy, 19-22 September
1995. Ispra, Joint Research Centre European Commission, pp 159-160.
Muller P, Leece B & Raha D (1995b) Dose-response assessment PAH.
Ottawa, Ontario Ministry of the Environment and Energy, 197 pp.
Muller P, Leece B & Raha D (1996) Scientific criteria document for
multimedia environmental standards development: Polycyclic aromatic
hydrocarbons (PAH). Part 1. Dose response assessment. Ottawa, Ontario
Ministry of the Environment and Energy, 203 pp.
Mumford JL, He XZ, Chapman RS, Cao SR, Harris DB, Li XM, Xian YL,
Jiang WZ, Xu CW, Chuang JC, Wilson WE, & Cooke M (1987a) Lung cancer
and indoor air pollution in Xuan Wei, China. Science, 235: 217-235.
Mumford JL, Chapman RS, Harris DB, He XZ, Cao SR, Xian YL, & Li XM
(1987b) Lung cancer and indoor air exposure to unvented coal and wood
combustion emission in Xuan Wie, China. In: Seifert B, Esdorn H,
Fischer M, Rüden H, & Wegner J ed. Indoor Air '87, Volume 3:
Developing countries, guaranteeing adequate indoor air quality,
control measures, ventilation effectiveness, thermal climate and
comfort, policy and strategies. Berlin, Institute for Water, Soil and
Air Hygiene, pp 8-14.
Mumford JL, Helmes CT, Lee X, Seidenberg J, & Nesnow S (1990) Mouse
skin tumorigenicity studies of indoor coal and wood combustion
emissions from homes of residents in Xuan Wie, China, with high lung
cancer mortality. Carcinogenesis, 11: 397-403.
Mumford JL, Williams RW, Walsh DB, Burton RM, Svensgaard DJ, Chuang
JC, Houk VS, & Lewtas J (1991) Indoor air pollutants from unvented
kerosene heater emissions in mobile homes: Studies on particles,
semivolatile organics, carbon monoxide, and mutagenicity. Environ Sci
Technol, 25: 1732-1738.
Mumford JL, Lee X, Lewtas J, Young TL, & Santella RM (1993) DNA
adducts as biomarkers for assessing exposure to polycyclic aromatic
hydrocarbons in tissues from Xuan Wei women with high exposure to coal
combustion emissions and high lung cancer mortality. Environ Health
Perspectives, 99: 83-87.
Mumford JL, Li X, Hu F, Lu XB & Chuang JC (1995) Human exposure and
dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei,
China, with high lung cancer mortality associated with exposure to
unvented coal smoke. Carcinogenesis, 16: 3031-3036.
Munz MJ & Tarazona JV (1993) Synergistic effect of two- and
four-component combinations of the polycyclic aromatic hydrocarbons:
Phenanthrene, anthracene, and acenaphthene on Daphnia magna. Bull
Environ Contam Toxicol, 50: 363-368.
Mur JM, Moulin JJ, Meyer-Bisch C, Massin N, Couplon JP & Loulergue J
(1987) Mortality of aluminium reduction plant workers in France. Int J
Epidemiol, 16: 257-264.
Murison GL (1988) Induction of sister-chromatid exchanges by direct
and indirect agents in a human teratoma cell line. Mutat Res, 203:
347-354.
Murray JJ, Pottie RF, & Pupp C (1974) The vapor pressures and
enthalpies of sublimation of five polycyclic aromatic hydrocarbons.
Can J Chem, 52: 557-563.
Myhr BC & Caspary WJ (1988) Evaluation of the L5178Y mouse lymphoma
cell mutagenesis assay: Intralaboratory results for sixty-three coded
chemicals tested at Litton Bionetics, Inc. Environ Mol Mutag, 12
(suppl 13): 103-194.
Nagabhushan M, Hussong J, Polverini PJ, & Solt DB (1990) Inhibition of
hamster buccal pouch epithelial cell replication during in
vitro exposure to polycyclic aromatic hydrocarbons. Proc Am Assoc
Cancer Res, 31: 86.
Nagel DL, Stenbäck F, Clayson DB, & Wallcave L (1976) Intratracheal
installation studies with 7 H-dibenzo [c,g]carbazole in Syrian
hamster. J Natl Cancer Inst, 57: 119-123.
Namkung MJ & Juchau MR (1980) On the capacity of human placental
enzymes to catalyse the formation of diols from benzo [a]pyrene.
Toxicol Appl Pharmacol, 55: 253-259.
Natarajan AT & Darroudi F (1991) Use of human hepatoma cells for in
vitro metabolic activation of chemical mutagens/carcinogens.
Mutagenesis, 6: 399-403.
National Chemicals Inspectorate (1994) [Report on the use of highly
aromatic oils in tyres in Sweden.] Solna, pp 15-21 (Report No. 6/94)
(in Swedish with English summary).
National Institute for Occupational Health and Safety and Occupational
Safety and Health Administration (1981) Occupational health guideline
for coal tar pitch volatiles. In: Mackison FW, Stricoff RS, &
Partridge LJ Jr ed. Occupational health guidelines for chemical
hazards. Cambridge, Massachusetts, 30 pp [DHHS (NIOSH) Publication No.
81-123; US NTIS PB83-154609, Part 1 of 3].
National Institute for Occupational Health and Safety (1994a)
Polynuclear aromatic hydrocarbons by HPLC: Method No. 5506. In: Eller
PM & Cassinelli ME ed. NIOSH manual of analytical methods, 4th ed.
Cincinnati, Ohio, 8 pp (Publication No. 94-113).
National Institute for Occupational Health and Safety (1994b)
Polynuclear aromatic hydrocarbons by GC: Method No. 5515. In: Eller PM
& Cassinelli ME ed. NIOSH manual of analytical methods, 4th ed.
Cincinnati, Ohio, 7 pp. (Publication No. 94-113).
National Research Council Canada (1983) Polycyclic aromatic
hydrocarbons in the aquatic environment: formation, sources, fate and
effects on aquatic biota. Ottawa, Associate Committee on Scientific
Criteria for Environmental Quality, pp 32-33 (Report NRCC No. 18981).
National Toxicology Program (1991) Final study report and appendix:
Developmental toxicity evaluation of naphthalene (CAS No. 91-20-3)
administered by gavage to Sprague-Dawley (CD) rats on gestational days
6 through 15. Research Triangle Park, North Carolina, US Department of
Health and Human Services, 107 pp (Report TER-91006).
National Toxicology Program (1992a) Final study report and appendix:
Development toxicity evaluation of naphthalene (CAS No. 91-20-3)
administered by gavage to New-Zealand White (NZW) rabbits on
gestational days 6 through 19. Research Triangle Park, North Carolina,
US Department of Health and Human Services, 107 pp (Report TER-91021).
National Toxicology Program (1992b) Toxicology and carcinogenesis
studies of naphthalene (CAS No. 91-20-3) in B6C3F1 mice (inhalation
studies). Research Triangle Park, North Carolina, US Department of
Health and Human Services, 129 pp (NTP TR 410; NIH Publication No.
92-3141).
National Toxicology Program (1993) Hazardous substances data bank.
Bethesda, Maryland, National Library of Medicine, 20 pp.
Natusch DFS & Tomkins BA (1978) Isolation of polycyclic organic
compounds by solvent extraction with dimethyl sulfoxide. Anal Chem,
50: 1429-1434.
Navarro A, Rosell A, Villanueva J, & Grimalt JO (1991) Monitoring of
hazardous waste dumps by the study of metals and solvent-soluble
organic chemicals. Chemosphere, 22: 913-928.
Naylor LM & Loehr RC (1982) Priority pollutants in municipal sewage
sludge. Bio-Cycle, 23: 18-22.
Neal J & Rigdon RH (1967) Gastric tumors in mice fed benzo(a)pyrene: A
quantitative study. Tex Rep Biol Med, 25: 553-557.
Nebert DW (1980) Pharmacogenetics: An approach to understanding
chemical and biological aspects of cancer. J Natl Cancer Inst, 6:
1279-1290.
Nebert DW, Levitt RC, Jensen NM, Lambert GS, & Felton JS (1977) Birth
defects and aplastic anemia: Differences in polycyclic hydrocarbon
toxicity associated with the Ah locus. Arch Toxicol, 39: 109-132.
Neff JM (1979) Polycyclic aromatic hydrocarbons in the aquatic
environment: Sources, fates and biological effects. London, Applied
Science Publishers, 262 pp.
Neff JM & Anderson JW (1975) Accumulation, release, and distribution
of benzo(a)pyrene-C14 in the clam Rangia cuneata.
Proc Conf Prev Control Oil Pollut, 75: 469-471.
Negishi M, Swan DC, Enquist LW, & Nebert DW (1981) Isolation and
characterization of a cloned DNA sequence associated with the murine
Ah locus and a 3-methylcholan-threne-induced form of cytochrome
P450. Proc Natl Acad Sci USA, 78: 800-801.
Nelson PF (1989) Combustion-generated polycyclic aromatic hydrocarbons
in diesel exhaust emissions. Fuel, 68: 283-286.
Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW,
Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O, Okuda K, &
Nebert W (1993) The P450 superfamily: Update on new sequences, gene
mapping, accession numbers, early trivial names of enzymes, and
nomenclature. DNA Cell Biol, 12: 1-51.
Nesnow S (1990) Mouse skin tumors and human lung cancer: Relationships
with complex environmental emissions. In: Vainio H, Sorsa M, &
McMichael A ed. Complex mixtures and cancer risk. Lyon, International
Agency for Research on Cancer, pp 44-54 (IARC Scientific Publications
No. 104).
Nesnow S & Heidelberger C (1976) The effect of modifiers of microsomal
enzymes on chemical oncogenesis in cultures of C3H mouse cell lines.
Cancer Res, 36: 1801-1808.
Nesnow S, Evans C, Stead A, Creason J, Slaga TJ, & Triplett LL (1982a)
Skin carcinogenesis studies of emission extracts. In Lewtas J ed.
Toxicological effects of emissions from diesel engines. Amsterdam,
Elsevier, 295-320.
Nesnow S, Triplett L, & Slaga TJ (1982b) Comparative tumor-initiating
activity of complex mixtures from environmental particulate emissions
on Sencar mouse skin. J Natl Cancer Inst, 68: 829-834.
Nesnow S, Leavitt S, Easterling R, Watts R, Toney SH, Claxton L,
Sangaiah R, Toney GE, Wiley J, Fraher P, & Gold A (1984) Mutagenicity
of cyclopenta-fused isomers of benz [a]anthracene in bacterial and
rodent cells and identification of the major rat liver microsomal
metabolites. Cancer Res, 44: 4993-5003.
Nesnow S, Easterling RE, Ellis S, Watts R, & Ross J (1988) Metabolism
of benz [j]aceanthrylene (cholanthrylene) and benz [1]aceanthrylene
by induced rat liver S9. Cancer Lett, 39: 19-27.
Nesnow S, Ross J, Mohapatra N, Gupta R, Sangaiah R, & Gold A (1991)
Genotoxicity and identification of the major DNA-adducts of
aceanthrylene. In: Cooke M, Loening K, & Merritt J ed. Polynuclear
aromatic hydrocarbons: Measurements, means, and metabolism. Columbus,
Ohio, Battelle Press, pp 629-639.
Nesnow S, Ross J, Beck S, Lasley J, Nelson G, Lamberg G, Platt KL, &
Agarwal SC (1994) Morphological transformation and DNA adduct
formation by dibenz [a,h]-anthracene and its metabolites in
C3H10T1/2Cl8 cells. Carcinogenesis, 15: 2225-2231.
Nesnow S, Mass MJ, Ross JA, Gennings C, Carter WH Jr, & Stoner GD
(1995) Lung tumorigenic interactions of five environmental PAH.
Additivity, synergism and antagonism. In: Fifteenth international
symposium on polycyclic aromatic compounds: Chemistry, biology and
environmental impact, Belgirate, Italy, 19-22 September 1995. Ispra,
Joint Research Centre European Commission, p 151.
Netherlands' Delegation (1991) The problem of polycyclic aromatic
hydrocarbons (PAH) in the Rhine. In: Working Group B of International
Rhine Commission, pp 1-7 (B 5/91 Revision).
Nettesheim P & Hammons AS (1971) Induction of squamous cell carcinoma
in the respiratory tract of mice. J Natl Cancer Inst, 47: 697-701.
Neubert D & Tapken S (1988) Transfer of benzo(a)pyrene into mouse
embryos and fetuses. Arch Toxicol, 62: 236-239.
Neuhauser EF & Callahan CA (1990) Growth and reproduction of the
earthworm Eisenia fetida exposed to sublethal concentrations of
organic chemicals. Soil Biol Biochem, 22: 175-179.
Neuhauser EF, Durkin PR, Malecki MR, & Anatra M (1986) Comparative
toxicity of ten organic chemicals to four earthworm species. Comp
Biochem Phys, C83: 197-200.
Newsted JL & Giesy JP Jr (1987) Predictive models for photoinduced
acute toxicity of polycyclic aromatic hydrocarbons to Daphnia
magna, Strauss Cladocera, Crustacea. Toxicol Chem, 6: 445-461.
Ng KM, Chu I, Bronaugh RL, Franklin CA, & Somers DA (1992)
Percutaneous absorption and metabolism of pyrene, benzo (a)pyrene,
and di(2-ethylhexyl)phthalate: Comparison of in vitro and in
vivo results in the hairless guinea pig. Toxicol Appl Pharmacol,
115: 216-223.
Nicholls TP, Perry R, & Lester JN (1979) The influence of heat
treatment on the metallic and polycyclic aromatic hydrocarbon content
of sewage sludge. Sci Total Environ, 12: 137-150.
Nichols DG, Gangwal SK, & Sparacino CM (1981) Analysis and assessment
of PAH from coal composition and gasification. In: Cooke M & Dennis AJ
ed. Polynuclear aromatic hydrocarbons: Chemical analysis and
biological fate. Columbus, Ohio, Battelle Press, 397-406.
Nielsen T (1984) Reactivity of polycyclic aromatic hydrocarbons toward
nitrating species. Environ Sci Technol, 18: 157-163.
Nikonova TV (1977) Transplacental action of benzo(a)pyrene and pyrene.
Bull Exp Biol Med, 84: 1025-1027.
Nisbet ICT & LaGoy PK (1992) Toxic equivalency factors (TEFs) for
polycyclic aromatic hydrocarbons (PAHs). Regul Toxicol Pharmacol, 16:
290-300.
Nisbet ICT, Schneiderman MA, Karch NJ, & Siegel DM (1985) Review and
evaluation of the evidence for cancer associated with air pollution.
Research Triangle Park, North Carolina, US Environmental Protection
Agency, 295 pp (NTIS/PB85-142016).
Nishioka M, Chang H-C, & Lee ML (1986) Structural characteristics of
polycyclic aromatic hydrocarbon isomers in coal tars and combustion
products. Environ Sci Technol, 20: 1023-1027.
Nkedi-Kizza P, Rao PSC, & Hornsby AG (1985) Influence of organic
cosolvent on sorption of hydrophobic organic chemicals by soils.
Environ Sci Technol, 19: 975-979.
Nordholm L, Espensen I-M, Jensen HS, & Holst E (1986) Polycyclic
aromatic hydrocarbons in smokehouses. Scand J Work Environ Health, 12:
614-618.
Norpoth K, Kemena A, Jacob J, & Schümann C (1984) The influence of 18
environmentally relevant polycyclic aromatic hydrocarbons and Clophen
A50, as liver monooxygenase inducers, on the mutagenic activity of
benz [a]anthracene in the Ames test. Carcinogenesis, 5: 747-752.
Nousiainen U, Törrönen R, & Hänninen O (1984) Differential induction
of various carboxylesterases by certain polycyclic aromatic
hydrocarbons in the rat. Toxicology, 32: 243-251.
Novelli G & Rinaldi A (1979) Further contributions to the study of
coal dust for foundries. Giesserei, 66: 480-482.
Nowak D, Meyer A, Schmidt-Preuss U, Gatzemeyer U, Magnussen H, &
Rüdiger HW (1992) Formation of benzo(a)pyrene-DNA adducts in blood
monocytes from lung cancer patients with a familial history of lung
cancer. J Cancer Res Clin Oncol, 118: 67-71.
Noyes WF (1969) Carcinogen-induced neoplasia with metastasis in a
South American primate, Saguinus oedipus (33845). Proc Soc Exp Biol
Med, 131: 223-225.
Ny ET, Heederik D, Kromhout H, & Jongeneelen F (1993) The relationship
between polycyclic aromatic hydrocarbons in air and in urine of
workers in a Söderberg potroom. Am Ind Hyg Assoc J, 54: 277-284.
Obana H, Hori S, & Kashimoto T (1981a) Determination of polycyclic
aromatic hydrocarbons in marine samples by high-performance liquid
chromatography. Bull Environ Contam Toxicol, 26: 613-620.
Obana H, Hori S, Kashimoto T, & Kunita N (1981b) Polycyclic aromatic
hydrocarbons in human fat and liver. Bull Environ Contam Toxicol, 27:
23-27.
Oberly TJ, Huffman DM, & Garriott ML (1992) An evaluation of
chromosomal mutagens in the CHO-AS52 cell line. Environ Mol Mutag, 19
(suppl 20): 46 (Abstract).
O'Brien KAF, Smith LL, & Cohen GM (1985) Differences in
naphthalene-induced toxicity in the mouse and rat. Chem-Biol
Interactions, 55: 109-122.
O'Brien KAF, Suverkropp C, Kaqnekal S, Plopper CG, & Buckpitt AR
(1989) Tolerance to multiple doses of the pulmonary toxicant,
naphthalene. Toxicol Appl Pharmacol, 99: 487-500.
Oesch F (1973) Mammalian epoxide hydrases: inducible enzymes
catalysing the inactivation of carcinogenic and cytotoxic metabolites
derived from aromatic and olefinic compounds. Xenobiotica, 3: 305-340.
Oesch F, Bücker M, & Glatt HR (1981) Activation of phenanthrene to
mutagenic metabolites and evidence for at least two different
activation pathways. Mutat Res, 81: 110.
Okano P, Miller HN, Robinson RC, & Gelboin HV (1979) Comparison of
benzo [a]pyrene and
(-)-trans-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene metabolism in
human blood monocytes and lymphocytes. Cancer Res, 39: 3184-3193.
Okita T, Yanagihara M, Yoshida K, Iwata M, Tanabe K, & Hara H (1994)
Measurements of air pollution associated with oil fires in Kuwait by a
Japanese research team. Atmos Environ, 28: 2255-2259.
Old LJ, Benacerraf B, & Carswell E (1963) Contact reactivity to
carcinogenic polycyclic hydrocarbons. Nature, 198: 1215-1216.
Olufsen SB (1980) Polynuclear aromatic hydrocarbons in Norwegian
drinking water resources. In: Bjorseth A & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemistry and biological effects. Columbus,
Ohio, Battelle Press, pp 333-343.
Olufsen SB & Bjorseth A (1983) Analysis of polycyclic aromatic
hydrocarbons by gas chromatography. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 257-300.
Omland O, Sherson D, Hansen AM, Sigsgaard T, Autrup H, & Overgaard E (
1994) Exposure of iron foundry workers to polycyclic aromatic
hydrocarbons: benzo(a)pyrene-albumin adducts and 1-hydroxypyrene as
biomarkers for exposure. Occup Environ Med, 51: 513-518.
Oris JT & Giesy JP Jr (1985) The photoenhanced toxicity of anthracene
to juvenile sunfish (Lepomis spp.). Aquat Toxicol, 6: 133-146.
Oris JT & Giesy JP (1986) Photoinduced toxicity of anthracene to
juvenile bluegill sunfish (Lepomis macrochirus Rafinesque):
Photoperiod effects and predictive hazard evaluation. Environ Toxicol
Chem, 5: 761-768.
Oris JT & Giesy JP Jr (1987) The photo-induced toxicity of polycyclic
aromatic hydrocarbons to larvae of the fathead minnow (Pimephales
promelas). Chemosphere, 16: 1395-1404.
Oris JT, Hall AT, & Tylka JD (1990) Humic acids reduce the
photo-induced toxicity of anthracene to fish and Daphnia. Environ
Toxicol Chem, 9: 575-584.
Osborn JF, Santhanam S, Davidson CI, Flotard RD, & Stetter JR (1984)
Characterization of airborne trace metal and trace organic species
from coal gasification. Environ Monit Assess, 4: 317-333.
Osborne MR & Crosby NT (1987a) Binding to proteins and nucleic acids.
In: Benzopyrenes. Cambridge, Cambridge University Press, pp 137-176
(Cambridge Monographs on Cancer Research).
Osborne MR & Crosby NT (1987b) Benzo [e]pyrene. In: Benzopyrenes.
Cambridge, Cambridge University Press, pp 229-250 (Cambridge
Monographs on Cancer Research).
Osborne MR, Harvey RG, & Brookes P (1978) The reaction of
trans-7,8-dihydroxy- anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo
[a]pyrene with DNA involves attack at the N7-position of guanine
moieties. Chem-Biol Interactions, 20: 123-130.
Osborne MR, Brookes P, Lee H, & Harvey RG (1986) The reaction of a
3-methylcholan-threne diol epoxide with DNA in relation to the binding
of 3-methylcholanthrene to the DNA of mammalian cells. Carcinogenesis,
7: 1345-1350.
Oshiro Y, Balwierz PS, Soelter SG, Guzzie PJ, & Rohrbacher E (1992)
Evaluation of mouse peripheral blood micronucleus assay. Environ Mol
Mutag, 19 (suppl 20): 47 (Abstract).
Ostman C, Nilsson U, Carlsson H, Andersson I, & Fahlgren L (1991)
[Polycyclic aromatic compounds (PAC) in work environment. I. PAC in
Stockholm street air: An introductory study.] Solna, Swedish National
Institute of Occupational Health, 16 pp (Research Report No. 21) (in
Swedish).
Ostman C, Nilsson U, Carlsson H, Andersson I, & Fahlgren L (1992a)
[Polycyclic aromatic compounds (PAC) in work environment. II. PAC in
Stockholm street air: Content of PAH in two busy streets.] Solna,
Swedish National Institute of Occupational Health, 41 pp (Research
Report No. 34) (in Swedish with English summary).
Ostman C, Nilsson U, Carlsson H, Andersson I, & Fahlgren L (1992b)
[Polycyclic aromatic compounds (PAC) in work environment. III. PAC in
Stockholm street air: Content of PAH before and after a traffic
diversion.] Solna, Swedish National Institute of Occupational Health,
Division of Analytical Chemistry, 31 pp (Research Report No. 35) (in
Swedish with English summary).
Ostrander GK, Landolt ML, & Kocan RM (1988) The ontogeny of Coho
salmon (Oncorhynchus kisutch) behavior following embryonic exposure
to benzo [a]pyrene. Aquat Toxicol, 13: 325-346.
Oueslati R, Alexandrov K, Chouikha M, & Chouroulinkov I (1992)
Formation and persistence of DNA adducts in epidermal and dermal mouse
skin exposed to benzo(a)pyrene in vivo. In Vivo, 6: 231-236.
Ovrebo S, Hewer A, Phillips DH, & Haugen A (1990) Polycyclic aromatic
hydrocarbon-DNA adducts in coke-oven workers. In: Vainio H, Sorsa M, &
McMichael AJ ed. Complex mixtures and cancer risk. Lyon, International
Agency for Research on Cancer, pp 193-198 (IARC Scientific
Publications No. 104).
Ovrebo S, Haugen A, Phillips DH, & Hewer A (1992) Detection of
polycyclic aromatic hydrocarbon-DNA adducts in white blood cells from
coke oven workers: Correlation with job catagories. Cancer Res, 52:
1510-1514.
Ovrebo S, Haugen A, Fjeldstad PE, Hemminki K, & Szyfter K (1994)
Biological monitoring of exposure to polycyclic aromatic hydrocarbon
in an electrode paste plant. J Occup Med, 36: 303-310.
Ovrebo S, Fjeldstad PE, Grzybowska E, Kure EH, Chorazy M, & Haugen A
(1995) Biological monitoring of polycyclic aromatic hydrocarbon
exposure in a highly polluted area of Poland. Environ Health
Perspectives, 103: 838-843.
Pahlman R & Pelkonen O (1987) Mutagenicity studies of different
polycyclic aromatic hydrocarbons: The significance of enzymatic
factors and molecular structure. Carcinogenesis, 8: 773-778.
Paika IJ, Beauchesne MT, Randall M, Schreck RR, & Latt SA (1981) In
vivo SCE analysis of 20 coded compounds. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests of carcinogens. Report of the
international collaborative programme. New York, Elsevier
North-Holland, pp 672-681 (Progress in Mutation Research, Volume 1).
Pal K (1981) The induction of sister-chromatid exchanges in Chinese
hamster ovary cells by K-region epoxides and some dihydrodiols derived
from benz [a]anthracene, dibenz [a,c]anthracene and
dibenz [a,h]anthracene. Mutat Res, 84: 389-398.
Pal K, Grover PL, & Sims P (1975) The metabolism of carcinogenic
polycyclic hydrocarbons by tissues of the respiratory tract. Biochem
Soc Trans, 3: 174-175.
Palitti F, Cozzi R, Fiore M, Palombo F, Polcaro C, Perez G, & Possagno
E (1986) An in vitro and in vivo study on mutagenic activity of
fluoranthene: Comparison between cytogenetic studies and HPLC
analysis. Mutat Res, 174: 125-130.
Pallardy MJ, House RV, & Dean JH (1989) Molecular mechanism of
7,12-dimethylbenz(a)anthracene induced immunosuppression: Evidence for
action via the interleukin-2 pathway. Mol Pharmacol, 36: 128-133.
Pallardy M, Mishal Z, Lebrec H, & Bohuon C (1992) Immune modification
due to chemical interference with transmembrane signalling:
Application to polycyclic aromatic hydrocarbons. Int J
Immunopharmacol, 14: 377-381.
Palmer WG & Scott WD (1981) Lung cancer in ferrous foundry workers: A
review. Am Ind Hyg Assoc J, 42: 329-340.
Palmork KH, Wilhelmsen S, & Neppelberg T (1973) International Council
for Exploration of the Sea. The contribution of polycyclic aromatic
hydrocarbons (PAH) to the marine environment from different
industries. Bergen, Institute of Marine Research, pp 1-21 (Report No.
C M 1973/E:33).
Pancirov RJ, Searl TD, & Brown RA (1980) Methods of analysis for
polynuclear aromatic hydrocarbons in environmental samples. Adv Chem
Ser, 185: 123-142.
Pankow JF, Isabelle LM, & Asher WE (1984) Trace organic compounds in
rain. 1. Sampler design and analysis by adsorption/thermal desorption
(ATD). Environ Sci Technol, 18: 310-318.
Park KS, Sims RC, Dupont RR, Doucette WJ, & Matthews JE (1990) Fate of
PAH compounds in two soil types: Influence of volatilization, abiotic
loss and biological activity. Environ Toxicol Chem, 9: 187-195.
Parkinson EK & Newbold RF (1980) Benzo [a]pyrene metabolism and DNA
adduct formation in serially cultivated strains of human epidermal
keratinocytes. Int J Cancer, 26: 289-299.
Parrott MC, Kawabata TT, & White KL Jr (1989) Suppression of humoral
immunity following dermal exposure to benzo(a)pyrene in B6C3F1 mice.
Toxicologist, 9: 201 (Abstract 801).
Partanen T & Boffetta P (1994) Cancer risk in asphalt workers and
roofers: Review and meta-analysis of epidemiologic studies. Am J Ind
Med, 26: 721-740.
Paschke A, Herbel W, Steinhard H, Franke S, & Francke W (1992)
Determination of mono- to tetracyclic aromatic hydrocarbons in
lubricating oil. J High Res Chromatogr, 15: 827-833.
Passino DRM & Smith SB (1987) Acute bioassays and hazard evaluation of
representative contaminants detected in Great Lakes fish. Environ
Toxicol Chem, 6: 901-907.
Pataki J & Huggins C (1969) Molecular site of substituents of
benz(a)anthracene related to carcinogenicity. Cancer Res, 29: 506-509.
Pathirana S, Connell W, & Vowles PD (1994) Distribution of polycyclic
aromatic hydrocarbons (PAHs) in an urban roadway system. Ecotoxicol
Environ Saf, 28: 256-269.
Pavanello S & Levis AG (1994) Human peripheral blood lymphocytes as a
cell model to evaluate the genotoxic effect of coal tar treatment.
Environ Health Perspectives, 102: 95-99.
Pavoni B, Sfriso A, & Marcomini A (1987) Concentration and flux
profiles of PCBs, DDTs and PAHs in a dated sediment core from the
lagoon of Venice. Mar Chem, 21: 25-35.
Payne JF (1977) Mixed function oxidases in marine organisms in
relation to petroleum hydrocarbon metabolism and detection. Mar Pollut
Bull, 8: 112-116.
Pelkonen O & Nebert DW (1982) Metabolism of polycyclic aromatic
hydrocarbons: Etiologic role in carcinogenesis. Pharmacol Rev, 34:
189-222.
Pelkonen O & Saarni H (1980) Unusual patterns of benzo [a]pyrene
metabolites and DNA-benzo [a]pyrene adducts produced by human
placental microsomes in vitro. Chem-Biol Interactions, 30: 287-296.
Pelkonen O, Sotaniemi E, & Mokka R (1977) The in vitro oxidative
metabolism of benzo [a]pyrene in human liver measured by different
assays. Chem-Biol Interactions, 16: 13-22.
Pellizzari ED, Castillo NP, Willis S, Smith D, & Bursey JT (1979)
Identification of organic components in aqueous effluents from
energy-related processes. In: Van Hall CE ed. Measurement of organic
pollutants in water and wastewater. Philadelphia, Pennsylvania,
American Society for Testing and Materials, pp 256-274 (ASTM STP 686).
PPenman BW, Kaden DA, Liber HL, Skopek TR, & Thilly WG (1980) Perylene
is a more potent mutagen than benzo [a]pyrene for Salmonella
typhimurium. Mutat Res, 77: 271-277.
Perera FP, Hemminki K, Young TL, Brenner D, Kelly G, & Santella RM
(1988) Detection of polycyclic aromatic hydrocarbon-DNA adducts in
white blood cells of foundry workers. Cancer Res, 48: 2288-2291.
Perera FP, Hemminki K, Gryzbowska E, Motykiewicz G, Michalska J,
Santella RM, Young T, Dickey C, Brandt-Rauf P, DeVivo I, BlanerW, Tsai
W, & Chorazy M (1992) Molecular and genetic damage in humans from
environmental pollution in Poland. Nature, 360: 256-258.
Perera FP, Tang DL, O'Neill JPO, Bigbee WL, Albertini RJ, Santella R,
Ottman R, Tsai WY, Dickey C, Mooney LA, Savela K, & Hemminki K (1993)
HPRT and glycophorin A mutations in foundry workers: Relationship to
PAH exposure and to PAH-DNA adducts. Carcinogenesis, 14: 969-973.
Perera FP, Dickey C, Santella R, O'Neill JP, Albertini RJ, Ottman R,
Tsai WY, Mooney LA, Savela K, & Hemminki K (1994) Carcinogen-DNA
adducts and gene mutation in foundry workers with low-level exposure
to polycyclic aromatic hydrocarbons. Carcinogenesis, 15: 2905-2910.
Perfetti GA, Nyman PJ, Fisher S, Joe FL Jr, & Diachenko GW (1992)
Determination of polynuclear aromatic hydrocarbons in seafood by
liquid chromatography with fluorescence detection. J Assoc Off Anal
Chem, 75: 872-877.
Perin-Roussel O, Saguem S, Ekert B, & Zajdela F (1983) Binding to DNA
of bay-region and pseudo bay region diol-epoxides of
dibenzo [a,e]fluoranthene and comparison with adducts obtained with
dibenzo [a,e]fluoranthene or its dihydrodiols in the presence of
microsomes. Carcinogenesis, 4: 27-32.
Perin-Roussel O, Croisy A, Ekert B, & Zajdela F (1984) The metabolic
activation of dibenzo(a,e)fluoranthene in vitro.
Evidence that its bay-region and pseudo-bay-region diolepoxides react
preferentially with guanosine. Cancer Lett, 22: 289-298.
Perry PE & Thomson EJ (1981) Evaluation of the sister chromatid
exchange method in mammalian cells as a screening system for
carcinogens. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens. Report of the international collaborative
programme. New York, Elsevier North-Holland, pp 560-569 (Progress in
Mutation Research, Volume 1).
Peter S, Palme GE, & Röhrborn G (1979) Mutagenicity of polycyclic
hydrocarbons. III. Monitoring genetic hazards of benz(a)anthracene.
Acta Morphol Acad Sci Hung, 27: 199-204.
Peters JA, DeAngelis DG, & Hughes TW (1981) An environmental
assessment of POM emissions from residential wood-fired stoves and
fireplaces. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: Chemical analysis and biological fate. Columbus, Ohio,
Battelle Press, pp 571-581.
Peterson AR, Landolph JR, Peterson H, Spears CP, & Heidelberger C
(1981) Oncogenic transformation and mutation of C3H/10T1/2 clone 8
mouse embryo fibroblasts by alkylating agents. Cancer Res, 41: 3095-
3099.
Petridou-Fischer J, Whaley SL, & Dahl AR (1988) In vivo
metabolism of nasally instilled benzo [a]pyrene in dogs and monkeys.
Toxicology, 48: 31-40.
Petry TH, Schmid P, & Schlatter CH (1994) Exposure to polycyclic
aromatic hydrocarbons (PAHs) in two different silicon carbide plants.
Ann Occup Hyg, 38: 741-752.
Pezzuto JM, Lea MA, & Yang CS (1976) Binding of metabolically
activated benzo(a)pyrene to nuclear macromolecules. Cancer Res, 36:
3647-3653.
Pezzuto JM, Lea MA, & Yang CS (1977) The role of microsomes and
nuclear envelope in the metabolic activation of benzo [a]pyrene
leading to binding with nuclear macro-molecules. Cancer Res, 37: 3427-
3433.
Pfannhauser W (1991) [Polycyclic aromatic hydrocarbons (PAH) in food
and selected vegetables in Austria.] Mitt Geb Lebensmittelunters Hyg,
82: 66-79 (in German).
Pfau W, Hughes NC, Grover PL, & Phillips DH (1992) HPLC separation of
32P-postlabelled benzo [b]fluoranthene-DNA adducts. Cancer Lett, 65:
159-167.
Pfeffer HU (1994) Ambient air concentrations of pollutants at
traffic-related sites in urban areas of North Rhine-Westphalia,
Germany. Sci Total Environ, 146/147: 263-273.
Pfeiffer EH (1973) Investigation on the carcinogenic burden by air
pollution in man. VII. Studies on the oncogenic interaction of
polycyclic aromatic hydrocarbons. Zentralbl Bakteriol Hyg Abt. Orig B,
158: 69-83.
Pfeiffer EH (1977) Oncogenic interaction of carcinogenic and
non-carcinogenic polycyclic aromatic hydrocarbons. In: Mohr V, Schmähl
D, & Tomatis L eds. Air pollution and cancer in man. Lyon,
International A gency for Research on Cancer, pp 69-77 (IARC
Scientific Publications No. 16).
Pflock H, Georgii HW, & Müller J (1983) [Particulate polycyclic
aromatic hydrocarbons (PAH) in polluted and in clean areas.]
Staub-Reinhalt Luft, 43: 230-234 (in German).
Phillips DH (1990) Modern methods of DNA adduct determination. In:
Cooper CS & Grover PL ed. Handbook of experimental pharmacology,
Volume 94/1. Berlin, Springer-Verlag, pp 503-546.
Phillips DH (1991) DNA-adduct analysis by 32P-postlabeling in the
study of human exposure to carcinogens. In: Groopman JD & Skipper PL
ed. Molecular dosimetry and human cancer. Chapter 9: Analytical,
epidemiological, and social considerations. Boca Raton, Florida, CRC
Press, pp 151-170.
Phillips DH & Alldrick AJ (1994) Tumorigenicity of a combination of
psoriasis therapies. Br J Cancer, 69: 1043-1045.
Phillips DH & Castegnaro M (1993) Results of an interlaboratory trial
of 32P-postlabelling. In: Phillips DH, Castegnaro M & Bartsch H ed.
Postlabelling methods for detection of DNA adducts. Lyon,
International Agency for Research on Cancer, pp 35-49 (IARC Scientific
Publications No. 124)
Phillips DH, Hewer A, Martin CN, Garner RC, & King MM (1988)
Correlation of DNA adduct levels in human lung with cigerette smoking.
Nature, 336: 790-792.
Phillips DH, Schoket B, Hewer A, & Grover PL (1990) DNA adduct
formation in human and mouse skin by mixtures of polycyclic aromatic
hydrocarbons. In: Vainio H, Sorsa M, & McMichael AJ ed. Complex
mixtures and cancer risk. Lyon, International Agency for Research on
Cancer, pp 223-229 (IARC Scientific Publications No. 104).
Phillips DH, Hewer A, Seidel A, Steinbrecher T, Schrode R, Oesch F, &
Glatt H (1991) Relationship between mutagenicity and DNA adduct
formation in mammalian cells for fjord- and bay-region diol-epoxides
of polycyclic aromatic hydrocarbons. Chem-Biol Interactions, 80: 177-
186.
Phillips DH, Castegnaro M & Bartsch H ed. (1993) Postlabelling methods
for detection of DNA adducts. Lyon, International Agency for Research
on Cancer, 388 pp (IARC Scientific Publications No. 124).
Pienta RJ, Poiley JA, & Lebherz WB III (1977) Morphological
transformation of early passage golden Syrian hamster embryo cells
derived from cryopreserved primary cultures as a reliable in
vitro bioassay for identifying diverse carcinogens. Int J Cancer,
19: 642-655.
Pike MH (1944) Ocular pathology due to organic compounds. J Mich State
Soc, 43: 581-584.
Pike MC (1983) Human cancer risk assessment. Polynuclear aromatic
hydrocarbons: Evaluation of sources and effects. Report by the
Committee on Pyrene and Selected Analogues, the Board on Toxicology
and Environmental Health Hazards, the Commission on Life Sciences and
the National Research Council. Washington DC, National Academic Press,
pp C1-C28.
Pischinger F & Lepperhoff G (1980) [Influence of air-fuel ratio on the
PAH emissions from Otto motor engines. Air pollution from polycylic
aromatic hydrocarbons. Registration and evaluation.] Düsseldorf,
VDI-Verlag, pp 59-63 (VDI Report No. 358) (in German).
Pistikopoulos P, Masclet P, & Mouvier G (1990) A receptor model
adapted to reactive species: Polycyclic aromatic hydrocarbons:
Evaluation of source contributions in an open urban site. I. Particle
compounds. Atmos Environ, 24: 1189-1197.
Plasterer MR, Bradshaw WS, Booth GM, Carter MW, Schuler RL, & Hardin
BD (1985) Developmental toxicity of nine selected compounds following
prenatal exposure in the mouse: Naphthalene, p-nitrophenol, sodium
selenite, dimethyl phthalate, ethylenethiourea, and four glycol ether
derivatives. J Toxicol Environ Health, 15: 25-38.
Platt KL, Pfeiffer E, Petrovic P, Friesel H, Beermann D, Hecker E, &
Oesch F (1990) Comparative tumorigenicity of picene and
dibenz [a,h]anthracene in the mouse. Carcinogenesis, 11: 1721-1726.
Plesha P, Stein J, Schiewe M, McCin B, & Varanasi U (1988) Toxicity of
marine sediments supplemented with mixtures of selected chlorinated
and aromatic hydrocarbons to the infaunal amphipod
Rhepoxynius-abronius. Mar Environ Res, 25: 85-98.
Plopper CG, Suverkropp C, Morin D, Nishio S, & Buckpitt A (1992)
Relationship of cytochrome P-450 activity to Clara cell cytotoxicity.
I. Histopathologic comparison of the respiratory tract of mice, rats
and hamsters after parenteral administration of naphthalene. J
Pharmacol Exp Ther, 261: 353-363.
Podoll RT, Irwin KC, & Parish HJ (1989) Dynamic studies of naphthalene
sorption on soil from aqueous solution. Chemosphere, 18: 2399-2412.
Poirier MC (1994) Human exposure monitoring, dosimetry, and cancer
risk assessment: The use of antisera specific for carcinogen-DNA
adducts and carcinogen-modified DNA. Drug Metab Rev, 26: 87-109.
Poirier MC & Beland FA (1992) DNA adduct measurement and tumor
incidence during chronic carcinogen exposure in animal models:
Implications for DNA adduct-based human cancer risk assessment. Chem
Res Toxicol, 5: 749-755.
Pollia JA (1941) Investigation on the possible carcinogenic effect of
anthracene and chrysene and some of their compounds. II. The effect of
subcutaneous injection in rats. J Ind Hyg Toxicol, 223: 449-451.
Poncelet F, Massanda K, Fouassin A, & Mercier M (1978) Mutagenic study
of some polycyclic aromatic hydrocarbons present in smoked fishes from
Africa. Arch Int Phys Biochem, 86: 954-955.
Popescu NC, Turnbull D, & DiPaolo JA (1977) Sister chromatid exchange
and chromosome aberration analysis with the use of several carcinogens
and noncarcinogens: Brief communication. J Natl Cancer Inst, 59: 289-
293.
Pothuluri JV, Freeman JP, Evang FE, & Cerniglia CE (1993)
Biotransformation of fluorene by the fungus Cunninghamella
elegans. Appl Environ Microbiol, 59: 1977-1980.
Pott P (1775) Chirurgical observations relative to the cataract, the
polypus of the nose, the cancer of the scrotum, the different kinds of
ruptures, and the mortification of the toes and feet. London, Hawes,
Clark & Collins, pp 63-68.
Pott F & Heinrich U (1992) [Dust and dust components / polycyclic
aromatic hydrocarbons (PAH).] In: Wichmann HE, Schlipköter HW, &
Fülgraff G ed. [Handbook of environmental medicine, 2nd ed.]
Landsberg, Lech Ecomed mbH, pp 1-23 (in German).
Pott F, Brockhaus A, & Huth F (1973) [Tests on the production of
tumours in animal experiments with polycyclic aromatic hydrocarbons.]
Zentralbl Bakteriol Hyg I Abt Orig B, 157: 34-43 (in German).
Pott F, Mohr U, & Brockhaus A (1978) [Studies on the combined effects
of benzopyrene and dibenz [a]anthracene with SO2 and NO2 inhalation
on the golden hamster.] In: Rothe H ed. [Air hygiene and silicosis
research.] Essen, Girardet, pp 224-226 (in German).
Pott F, Ziem U, Reiffer FJ, Huth F, Ernst H, & Mohr U (1987)
Carcinogenicity studies on fibres, metal compounds, and some other
dusts in rats. Exp Pathol (Jena), 32: 129-152.
Potthast K (1980) [Recent results on the benzo [a]pyrene content of
meat products.] Fleischwirtschaft, 60: 1941-1949 (in German).
Potvin RR, Adamek EG, & Balsillie D (1980) Ambient PAH levels near a
steel mill in northern Ontario. In: Cooke M & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemical analysis and biological
fate. Columbus, Ohio, Battelle Press, pp 741-753.
Prasanna P, Jacobs MM, & Yang Sk (1987) Selenium inhibition of
benzo [a]pyrene, 3-methylcholanthrene, and 3-methylcholanthrylene
mutagenicity in Salmonella typhimurium strains TA98 and TA100. Mutat
Res, 190: 101-105.
Prinsen AJ & Kennedy WBH (1977) [Quantity of alpha-benzopyrene in
smoked foods.] In: [Aroma and flavouring substances.] Bilthoven,
National Institute of Public Health and Environmental Protection, pp
1-11 (Report No. 11) (in Dutch).
Prinsen AJ & Kennedy WBH (1978) [Quantity of alpha benzopyrene in
various tea samples.] In: [Aroma and flavouring substances.]
Bilthoven, National Institute of Public Health and Environmental
Protection, pp 1-5 (Report No. 12) (in Dutch).
Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK, & Neal SB (1981)
Chemically-induced unscheduled DNA synthesis in primary rat hepatocyte
cultures: A comparison with bacterial mutagenicity using 218
compounds. Environ Mutagen, 3: 11-32.
Prough RA, Patrizi VW, Okita RT, Masters BSS, & Jakobsson SW (1979)
Characteristics of benzo [a]pyrene metabolism by kidney, liver, and
lung microsomal fractions from rodents and humans. Cancer Res, 39:
1199-1206.
Pruell RJ, Hoffmann EJ, & Quinn JG (1984) Total hydrocarbons,
polycyclic aromatic hydrocarbons and synthetic organic compounds in
the hard shell clam, Mercenaria mercenaria, purchased at commercial
seafood stores. Mar Environ Res, 11: 163-181.
Pruess-Schwartz D, Baird WM, Yagi H, Jerina DM, Pigott MA, & Dipple A
(1987) Stereochemical specificity in the metabolic activation of
benzo [c]phenanthrene to metabolites that covalently bind to DNA in
rodent embryo cell cultures. Cancer Res, 47: 4032-4037.
Puffer H, Duncan K, Von Hofe E, Brewer G, & Mondal S (1979)
Benz(a)pyrene: Studies of the effects of this ubiquitous pollutant on
fishes. Ocean, 79: 398-400.
Pullman A (1945) [On an electronic theory of the carcinogenic action
of condensed aromatic hydrocarbons.] C R Séance Soc Biol Fil, 139:
1056-1058 (in French).
Pullman A (1947) [Contribution to the study of the electronic
structure of organic molecules. Special study on carcinogenic
hydrocarbons.]. Ann Chim, 2: 7-71 (in French).
Purchase IFH, Longstaff E, Ashby J, Styles JA, Anderson D, Lefevre PA,
& Westwood FR (1976) Evaluation of six short term tests for detecting
organic chemical carcinogens and recommendations for their use.
Nature, 264: 624-627.
Pussemier L, Szabo G, & Bulman RA (1990) Prediction of the soil
adsorption coefficient Koc for aromatic pollutants. Chemosphere, 21:
1199-1212.
Pyysalo H, Tuominen J, Wickström K, Skyttä E, Tikkanen L, Salomaa S,
Sorsa M, Nurmela T, Mattila T, & Pohjola V (1987) Polycyclic organic
material (POM) in urban air. Fractionation, chemical analysis and
genotoxicity of particulate and vapour phases in an industrial town in
Finland. Atmos Environ, 21: 1167-1180.
Quaghebeur D, De Wulf E, Ravelingien MC, & Janssens G (1983)
Polycyclic aromatic hydrocarbons in rainwater. Sci Total Environ, 32:
35-54.
Quarles JM, Sega MW, Schenley CK, & Lijinsky W (1979) Transformation
of hamster fetal cells by nitrosated pesticides in a transplacental
assay. Cancer Res, 39: 4525-4533.
Quilliam MA & Sim PG (1988) Determination of polycyclic aromatic
compounds by high-performance liquid chromatography with simultaneous
mass spectrometry and ultraviolet diode array detection. J Chromatogr
Sci, 26: 160-167.
Quinlan R, Kowalczyz G, Gardiner K, Calvert I, Hale K, & Walton S
(1995a) Polycyclic aromatic hydrocarbon exposure in coal liquefaction
workers: The value of urinary 1-hydroxypyrene excretion in the
development of occupational hygiene control strategies. Ann Occup Hyg,
39: 329-346.
Quinlan R, Kowalczyk G, Gardiner K, Hale K, Walton S, & Calvert I
(1995b) Urinary 1-hydroxypyrene: A biomarker for polycyclic aromatic
hydrocarbon exposure in coal liquefaction workers. Occup Med, 45: 63-
68.
Quinlan R, Kowalczyk G, Gardiner K, & Calvert I (1995c) Exposure to
polycyclic aromatic hydrocarbons in coal liquefaction workers: Impact
of a workwear policy on excretion of urinary 1-hydroxypyrene. Occup
Environ Med, 52: 600-605.
Radian Corp (1991) Asphalt industry cross sectional exposure
assessment study. Final Report. Sacramento, California, 200 pp.
Raha CR (1972) Metabolism of benzo [a]pyrene at the 4,5-position. Ind
J Biochem Biophys, 9: 105-110.
Rahman A, Barrowman JA, & Rahimtula A (1986) The influence of bile on
the bioavailability of polynuclear aromatic hydrocarbons from the rat
intestine. Can J Physiol Pharmacol, 64: 1214-1218.
Rainio K, Linko RR, & Ruotsila L (1986) Polycyclic aromatic
hydrocarbons in mussel and fish from the Finnish archipelago sea. Bull
Environ Contam Toxicol, 37: 337-343.
Raiyani CV, Jani JP, Desai NM, Shah JA, & Kashyap SK (1993a) Levels of
polycyclic aromatic hydrocarbons in ambient environment of Ahmedabad
City. Indian J Environ Prot, 13: 206-215.
Raiyani CV, Jani JP, Desai NM, Shah JA, Shah PG, & Kashyap SK (1993b)
Assessment of indoor exposure to polycyclic aromatic hydrocarbons for
urban poor using variuos types of cooking fuels. Bull Environ Contam
Toxicol, 50: 757-763.
Ramdahl T & Mller M (1983) Chemical and biological characterization
of emissions from a cereal straw burning furnace. Chemosphere, 12: 23-
34.
Ramdahl T, Alfheim I, Rustad S, & Olsen T (1982) Chemical and
biological characterization of emissions from small residential stoves
burning wood and charcoal. Chemosphere, 11: 601-611.
Rao KSM, Phadke KM, & Muthal PI (1987) Estimation of carcinogenic
polycyclic aromatic hyrocarbon concentrations on the top of coke
ovens. Res Ind, 43: 276-281.
Rastetter WH, Nachbar RB, Russo-Rodriguez S, Wattley RV, Thilly WG,
Andon BM, Jorgensen WL, & Ibrahim M (1982) Fluoranthene: Synthesis and
mutagenicity of fluor diol epoxides. J Org Chem, 47: 4873-4878.
Ratajczak EA, Ahland E, Grimmer G, & Dettbarn G (1984) [Reduction of
PAH emissions from hard coal briquettes containing bitumen instead of
pitch binder.] Staub-Reinhalt Luft, 44: 505-509 (in German).
Raunio H & Pelkonen O (1994) Cancer genetics: Genetic factors in the
activation and inactivation of chemical carcinogens. In: Ioannides C
ed. Drugs, diet and disease. Volume 1: Mechanistic approaches to
cancer. New York, Simon & Schuster International, pp 229-258.
Raveh D, Slaga TJ, & Huberman E (1982) Cell-mediated mutagenesis and
tumor-initiating activity of the ubiquitous polycyclic hydrocarbon,
cyclopenta [c,d]pyrene. Carcinogenesis, 3: 763-766.
Readman JW, Preston MR, & Mantoura RFC (1986) An integrated technique
to quantify sewage, oil and PAH pollution in estuarine und coastal
environments. Mar Pollut Bull, 17: 298-308.
Reardon DB, Prakash AS, Hilton BD, Roman JM, Pataki J, Harvey RG, &
Dipple A (1987) Characterization of
5-methylchrysene-1,2-dihydroidiol-3,4-epoxide-DNA adducts.
Carcinogenesis, 8: 1317-1322.
Reddy MV, Gupta RC, Randerath E, & Randerath K (1984)
32P-Postlabeling test for covalent DNA binding of chemicals
in vivo: Application to a variety of aromatic carcinogens and
methylating agents. Carcinogenesis, 5: 231-243.
Reddy MV, Olson JA, Stober GR, & Daniel FB (1991) Induction of nuclear
anomalies in the gastrointestinal tract by polycyclic aromatic
hydrocarbons. Cancer Lett, 56: 215-224.
Redmond CK (1983) Cancer mortality among coke oven workers. Environ
Health Perspectives, 52: 67-73.
Reed L (1983) Health hazard evaluation: Anchor Hocking Glass Company
Roofing Site, Lancaster, Ohio, January 1983. Cincinnati, Ohio,
National Institute for Occupational Safety and Health, pp 1-9 (Report
HETA 82-067-1253; PB 84-173046).
Reed GA, Layton ME, & Ryan MJ (1988) Metabolic activation of
cyclopenteno [c,d]pyrene by peroxyl radicals. Carcinogenesis, 9:
2291-2295.
Rees DC & Hattis D (1994) Developing quantitative strategies for
animal to human extrapolation. In: Hayes AW ed. Principles and methods
of toxicology, 3rd Ed, New York, Raven Press, pp 275-315.
Rees ED, Mandelstam P, Lowry JQ, & Lipscomb H (1971) A study of the
mechanism of intestinal absorption of benzo [a]pyrene. Biochem
Biophys Acta, 225: 96-107.
Rees RW, Brice AJ, Carlton JB, Gilbert PJ & Mitchell I de G (1989)
Optimization of metabolic activation for four mutagens in a bacterial,
fungal and two mammalian cell mutagenesis assays. Carcinogenesis, 4:
335-342.
Regional Office for Water and Waste Disposal (1986) [Water report
1985.] Düsseldorf, Northrhine-Westphalia, pp 24-27 (in German).
Regional Office for Water and Waste Disposal (1988) [Water report
1987] Düsseldorf, Northrhine-Westphalia, Table 10, p 29 (in German).
Regional Office for Water and Waste Disposal (1989) [Water report
1988.] Düsseldorf, Northrhine-Westphalia, pp 28, i-viii (in German).
Regional Office for Water and Waste Disposal (1990) [Water report
1989.] Düsseldorf, Northrhine-Westphalia, pp 25-29 (in German).
Reichert WL, Eberhart BTL, & Varanasi U (1985) Exposure of two species
of deposit-feeding amphipods to sediments-associated
3H-benzo [a]pyrene: Uptake, metabolism and covalent binding to
tissue macromolecules. Aquat Toxicol, 6: 45.
Reinhard M, Goodman NL, & Barker JF (1984) Occurrence and distribution
of organic chemicals in two landfill leachate plumes. Environ Sci
Technol, 18: 953-961.
Renwick AG & Drasar BS (1976) Environmental carcinogens and large
bowel cancer. Nature, 263: 234-235.
Reuterwall C, Aringer L, Elinder CG, Rannung A, Levin JO, Juringe L, &
Onfelt A (1991) Assessment of genotoxic exposure in Swedish coke-oven
work by different methods of biological monitoring. Scand J Work
Environ Health, 17: 123-132.
Reznikoff CA, Bertram JS, Brankow DW, & Heidelberger C (1973)
Quantitative and qualitative studies of chemical transformation of
cloned C3H mouse embryo cells sensitive to postconfluence inhibition
of cell division. Cancer Res, 33: 3239-3249.
Rhee K & Bratzler LJ (1970) Benzo(a)pyrene in smoked meat products. J
Food Sci, 35: 146-149.
Rhett RG, Simmers JW, & Lee CR (1988) Eisenia foetida used as a
biomonitoring tool to predict the potential bioaccumulation of
contaminants from contaminated dredged material. In: Edwards CA &
Neuhauser EF ed. Earthworms in waste and environmental management. The
Hague, SPB Academic Publishers, pp 321-328.
Rhoads CP, Smith WE, Cooper NS & Sullivan RD (1954) Early changes in
the skin or several species including man, after painting with
carcinogenic materials. Proc Am Assoc Cancer Res, 1: 40.
Rice JE, Hosted TJ Jr, & LaVoie EJ (1984) Fluoranthene and pyrene
enhance benzo [a]pyrene-DNA adduct formation in vivo in mouse skin.
Cancer Lett, 24: 327-333.
Rice JE, Coleman DT, Hosted TJ, LaVoie EJ, McCaustland DJ, & Wiley JC
(1985) Identification of mutagenic metabolites of
indeno[1,2,3- cd]pyrene formed in vitro with rat liver enzymes.
Cancer Res, 45: 5421-5425.
Rice JE, Hosted TJ Jr, DeFloria MC, LaVoie EJ, Fischer DL, & Wiley JC
Jr (1986) Tumor-initiating activity of major in vivo
metabolites of indeno[1,2,3- cd]pyrene on mouse skin. Carcinogenesis,
7: 1761-1764.
Rice JE, Weyand EH, Geddie NG, DeFloria MC, & LaVoie EJ (1987)
Identification of tumorigenic metabolites of benzo [j]fluoranthene
formed in vivo in mouse skin. Cancer Res, 47: 6166-6170.
Rice JE, Geddie NG, Defloria MC, & LaVoie EJ (1988a) Structural
requirements favoring mutagenic activity among methylated pyrenes in
S. typhimurium. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: A decade of progress. Columbus, Ohio, Battelle Press, pp
773-785.
Rice JE, Jordan K, Little P, & Hussain N (1988b) Comparative
tumor-initiating activity of methylene-bridged and bay-region
methylated derivatives of benz [a]anthracene and chrysene.
Carcinogenesis, 9: 2275-2278.
Rice JE, Weyand EH, Burrill C, & Lavoie EJ (1990) Fluorine probes for
investigating the mechanism of activation of indeno [1,2,3- cd]pyrene
to a tumorigenic agent. Carcinogenesis, 11: 1971-1974.
Riebe-Imre, M, Peiser, D, Emura M, Aufderheide M, Jacob J, Grimmer G,
& Raab G (1993) Passage-dependent characteristics of PAH-metabolism
and PAH-induced transformation in airway epithelial cells in vitro.
In: Garrigues P & Lamotte M ed. Polycyclic aromatic compounds:
Synthesis, properties, analytical measurements, occurrence and
biological effects. Bordeaux, Gordon & Breach Science Publishers, pp
849-856.
Riegel B, Wartman WB, Hill WT, Reeb BB, Shubik P, & Stanger DW (1951)
Delay of methylcholanthrene skin carcinogenesis in mice by
1,2,5,6-dibenzofluorene. Cancer Res, 11: 301-306.
Riess MH & Wefers H (1994) [Ecotoxicological evaluation of chlorine
and phosphorous organics as well as PAH in sediments of Bremen
waters.] Bremen, Institute for Environmental Chemistry, pp 9-10 (in
German).
Rigdon RH & Neal J (1965) Effects of feeding benzo [a]pyrene on
fertility, embryos, and young mice. J Natl Cancer Inst, 34: 297-305.
Rigdon RH & Neal J (1966) Gastric carcinomas and pulmonary adenomas in
mice fed benzo(a)pyrene. Tex Rep Biol Med, 24: 195-207.
Rigdon RH & Neal J (1969) Relationship of leukemia to lung and stomach
tumors in mice fed benzo(a)pyrene. Proc Soc Exp Biol Med, 130: 146-
148.
Riley RT, Shirazi MA, & Swartz RC (1981) Transport of naphthalene in
the oyster O streaedulis. Mar Biol, 63: 325-330
Rimatori V, Sperduto B, & Iannaccone A (1983) Environmental oil
aerosol. J Aerosol Sci, 14: 253-256.
Rippe RM & Pott F (1989) [Carcinogenicity studies into nitro-PAH
(nitroarenes) in view of their importance in the cancer-inducing
effects of diesel motor emissions.] In: Environmental Hygiene, Volume
21. Düsseldorf, Stefan W Albers Verlag, pp 65-89 (Society for Research
on Air Hygiene and Silicosis, Medical Institute for Environmental
Hygiene, Annual Report 1988/1989) (in German).
Risch HA, Burch JD, Miller AB, Hill GB, Steele R, & Howe GR (1988)
Occupational factors and the incidence of cancer of the bladder in
Canada. Br J Ind Med, 45: 361-367.
Robbins WK, Searl TD, Wasserstrom DH, & Boyer GT (1981) Determination
of polynuclear aromatic hydrocarbons in wastewater from coal
liquefaction processes by the gas chromatography-ultraviolet
spectrometry technique. In: Jackson LP & Wright CC ed. Analysis of
waters associated with alternative fuel production. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 149-166
(ASTM STP 720).
Roberts MH Jr, Hargis WJ Jr, Strobel CJ, & De Lisle PF (1989) Acute
toxicity of PAH contaminated sediments to the estuarine fish,
Leiostomus xanthurus. Bull Environ Contam Toxicol, 42: 142-149.
Robertson IGC, Guthenberg C, Mannervik B, & Jernström B (1986)
Differences in stereoselectivity and catalytic efficiency of three
human glutathione transferases in the conjugation of glutathione with
7b, 8a-dihydroxy-9a,10a-oxy-7,8,9,10-tetrahydrobenzo [a]pyrene.
Cancer Res, 46: 2220-2224.
Robinson DE & Mitchell AD (1981) Unscheduled DNA synthesis response of
human fibroblasts, WI-38 cells, to 20 coded chemicals. In: DeSerres FJ
& Ashby J ed. Evaluation of short-term tests for carcinogens. Report
of the international collaborative programme. New York, Elsevier North
Holland, pp 517-527 (Progress in Mutation Research, Volume 1).
Robinson JR, Felton JS, Levitt RC, Thorgeirsson SS, & Nebert DW (1975)
Relationship between 'aromatic hydrocarbon responsiveness' and the
survival times in mice treated with various drugs and environmental
compounds. Mol Pharmacol, 11: 850-865.
Rocchi P, Ferreri AM, Borgia R, & Prodi G (1980) Polycyclic
hydrocarbons induction of diphtheria toxin-resistant mutants in human
cells. Carcinogenesis, 1: 765-767.
Rockette HE & Arena VC (1983) Mortality studies of aluminum reduction
plant workers: Potroom and carbon department. J Occup Med, 25: 549-
557.
Rockette HE & Redmond CK (1985) Selection, follow-up, and analysis in
the coke oven study. Natl Cancer Inst Monogr, 67: 89-94.
Roe FJC (1962) Effect of phenanthrene on tumour-initiation by
3,4-benzpyrene. Br J Cancer, 16: 503-506.
Roe JC & Grant CA (1964) Test of pyrene and phenanthrene for
incomplete carcinogenic and anticarcinogenic activity. Br Emp Cancer
Campaign, 41: 59.
Rogan EG, Cavalieri EL, Ramakrishna NVS, & Devanesan PD (1993)
Mechanisms of benzo(a)pyrene and 7,12-dimethylbenz(a)anthracene
activation: Quantitative aspects of the stable and depurination DNA
adducts obtained from radical cations and diol epoxides. In: Garrigues
P & Lamotte M ed. Polycyclic aromatic compounds: Synthesis,
properties, analytical measurements, occurrence and biological
effects. Bordeaux, Gordon & Breach Science Publishers, pp 733-740.
Rogerson PF (1988) Organic chemical waste characterization for marine
disposal of Black Rock Harbor dredged materials. In: Lichtenberg JJ,
Winter JA, Weber CI, & Fradkin L ed. Chemical and biological
characterization of municipal sludges, sediments, dredge spoils, and
drilling muds. Philadelphia, Pennsylvania, American Society for
Testing and Materials, pp 213-222.
Roggeband R, Van den Berg PTM, Steenwinkel MJST, Van Delft JHM, Van
der Wulp CJM, & Baan RA (1994a) In situ detection of different PAH-
DNA adducts by means of immunofluorescence microscopy. In: Annual
report on toxicology 1993/1994. Zeist, TNO Nutrition and Food Research
Institute, pp 79-80.
Roggeband R, Wolterbeek APM, Van den Berg PTM, & Baan RA (1994b) DNA
adducts in hamster and rat tracheas exposed to benzo(a)pyrene
in vitro. Toxicol Lett, 72: 105-111.
Rojas M, Alexandrov K, Auburtin G, Wastiaux-Denamur A, Mayer L, Mahieu
B, Sebastien P, & Bartsch H (1995) Anti-benzo(a)pyrene diolepoxide-DNA
adduct levels in pheripheral mononuclear cells from coke oven workers
and the enhancing effect of smoking. Carcinogenesis, 16: 1373-1376.
Roller M, Kamino K, & Rosenbruch M (1992) Carcinogenicity testing of
bladder carcinogens and other organic compounds by the intraperitoneal
and intravesical route. In: Seemayer NH & Hadnagy W ed. Environmental
hygiene. III. Berlin, Springer-Verlag, pp 95-98.
Ronneberg A & Andersen A (1995) Mortality and cancer morbidity in
workers from an aluminum smelter with prebaked carbon anodes. Part II:
Cancer morbidity. Occup Environ Med, 52: 250-254.
Ronneberg A & Langmark F (1992) Epidemiologic evidence of cancer in
aluminum reduction plant workers. Am J Ind Med, 22: 573-590.
Rosenkranz HS & Poirier LA (1979) Evaluation of the mutagenicity and
DNA-modifying activity of carcinogens and noncarcinogens in microbial
systems. J Natl Cancer Inst, 62: 873-891.
Ross J, Nelson G, Erexson G, Kligerman A, Earley K, Gupta RC, & Nesnow
S (1991) DNA adducts in rat lung, liver, and peripheral blood
lymphocytes produced by ip administration of benzo [a]pyrene
metabolites and derivatives. Carcinogenesis, 12: 1953-1955.
Ross JA, Nelson GB, Holden KL, Kligerman AD, Erexson GL, Bryant MF,
Earley K, Beach AC, Gupta RC, & Nesnow S (1992) DNA adducts and
induction of sister chromatid exchanges in the rat following
benzo (b)fluoranthene administration. Carcinogenesis, 13: 1731-1734.
Ross JA, Nelson GB, Wilson KH, Rabinowitz JR, Galati A, Stoner GD,
Nesnow S, & Mass MJ (1995) Adenomas induced by polycyclic aromatic
hydrocarbons in strain A/J mouse lung correlate with time-integrated
DNA adduct levels. Cancer Res, 55: 1039-1044.
Rossi SS & Neff JM (1978) Toxicity of polynuclear aromatic
hydrocarbons to the polychaete Neanthes arenaceodentata. Mar Pollut
Bull, 9: 220-223.
Rossman TG, Molina M, Meyer L, Boone P, Klein CB, Wang Z, Li F, Lin
WC, & Kinney PL (1991) Performance of 133 compounds in the lambda
prophage induction endpoint of the microscreen assay and a comparison
with Salmonella typhimurium mutagenicity and rodent carcinogenicity
assays. Mutat Res, 260: 349-367.
Rostad CE & Pereira WE (1987) Creosote compounds in snails obtained
from Pensacola Bay, Florida, near an onshore hazardous-waste site.
Chemosphere, 16: 2397-2404.
Roszinsky-Köcher G, Basler A, & Röhrborn G (1979) Mutagenicity of
polycyclic hydrocarbons. V. Induction of sister-chromatid exchanges
in vivo. Mutat Res, 66: 65-67.
Rothman N, Poirier MC, Haas RA, Correa-Villasenor A, Ford P, Hansen
JA, O'Toole T, & Strickland PT (1993) Association of PAH-DNA adducts
in peripheral white blood cells with dietary exposure to polyaromatic
hydrocarbons. Environ Health Perspect, 99: 265-267.
Ruepert C, Grinwis A, & Govers H (1985) Prediction of partition
coefficients of unsubstituted polycyclic aromatic hydrocarbons from
C18 chromatographic and structural properties. Chemosphere, 14: 279-
291
Rundell JO, Guntakatta M, & Matthews EJ (1983) Criterion development
for the application of Balb/c-3T3 cells to routine testing for
chemical carcinogenic potential. In: Waters MD, Sandhu SS, Lewtas J,
Claxton L, Chernoff N, & Nesnow S ed. Short-term bioassays in the
analysis of complex environmental mixtures. III. New York, Plenum
Press, pp 309-324.
Russell H (1947) An unsuccessful attempt to induce gliomata in rabbits
with cholanthrene. J Pathol Bacteriol, 59: 481-483.
Russell LB (1977) Validation of the in vivo somatic mutation method
in the mouse as a prescreen for germinal point mutations. Arch
Toxicol, 38: 75-85.
Russell P, Yamada T, Xu GT, Garland D, & Zigler JS Jr (1991) Effects
of naphthalene metabolites on cultured cells from eye lens. Free
Radicals Biol Med, 10: 255-261.
Ryan PA & Cohen Y (1986) Multimedia transport of particle-bound
organics: Benzo [a]pyrene test case. Chemosphere, 15: 21-47.
Saber A, Jarosz J, Martin-Bouyer M, Paturel L, & Vial M (1987)
Analysis of polycyclic aromatic hydrocarbons in lacustral sediments by
high resolution Shpol'skii spectrofluori-metry at 10 K. Int J Environ
Anal Chem, 28: 171-184.
Sabourin TD & Tullis RE (1981) Effect of three aromatic hydrocarbons
on respiration and heart rates of the mussel,
Mytilus californianus. Bull Environ Contam Toxicol, 26: 729-736.
Saed T, Al-Yakoob S, Al-Hashash, & Al-Bahlou M (1995) Preliminary
exposure assessment for Kuwaiti consumers to polycyclic aromatic
hydrocarbons in seafood. Environ Int, 21: 255-263.
Saffiotti U, Cefis F, & Kolb LH (1968) A method for experimental
induction of bronchogenic carcinoma. Cancer Res, 28: 104-113.
Saffiotti U, Montesano R, Sellkumar AR, & Kaufman DG (1972)
Respiratory tract carcinogenesis induced in hamsters by different dose
levels of benzo [a]pyrene and ferric oxide. J Natl Cancer Inst, 49:
1199-1204.
Sagredos AN, Sinha-Roy D, & Thomas A (1988) [Determination, sources
and composition of polycyclic aromatic hydrocarbons in oils and fats.]
Fat Sci Technol, 90: 76-81 (in German).
Saguem S, Mispelter J, Perin-Roussel O, Lhoste JM, & Zajdela F (1983a)
Multi-step metabolism of the carcinogen dibenzo [a,e]fluoranthene. I.
Identification of the metabolites from rat microsomes. Carcinogenesis,
4: 827-835.
Saguem S, Perin-Roussel O, Mispelter J, Lhoste JM, & Zajdela F (1983b)
Multi-step metabolism of the carcinogen dibenzo [a,e]fluoranthene.
II. Metabolism pathways. Carcinogenesis, 4: 837-842.
Sakai M, Yoshida D, & Mizusaki S (1985) Mutagenicity of polycyclic
hydrocarbons and quinones on Salmonella thyphimurium TA97. Mutat
Res, 156: 61-67.
Salagovic J, Kalina I, & Dubayová K (1995) Induction of single strand
DNA breaks in workers professionally exposed to polycyclic aromatic
hydrocarbons. Neoplasma, 42: 115-118.
Salaman MH & Roe FJC (1956) Further tests for tumour-initiating
activity: N,N-Di-(2-chloroethyl)-p-aminophenylbutyric acid (CB1348) as
an initiator of skin tumor formation in the mouse. Br J Cancer, 10:
363-378.
Salamone MF (1981) Toxicity of 41 carcinogens and noncarcinogenic
analogs. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens. Report of the international collaborative programme.
Amsterdam, Elsevier North-Holland, pp 682-685 (Progress in Mutation
Research, Volume 1).
Salamone MF, Heddle JA, & Katz M (1979) The mutagenic activity of
thirty polycyclic aromatic hydrocarbons (PAH) an oxides in urban
airborne particulates. Environ Int, 2: 37-43.
Salamone MF, Heddle JA, & Katz M (1981) Mutagenic activity of 41
compounds in the in vivo micronucleus assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens. Report of
the international collaborative programme. Amsterdam, Elsevier
North-Holland, pp 686-697 (Progress in Mutation Research,Volume 1).
Salomaa S, Tuominen J, & Skyttä E (1988) Genotoxicity and PAC analysis
of particulate and vapour phases of environmental tobacco smoke. Mutat
Res, 204: 173-183.
Sanders CL, Skinner D, & Gelman RA (1986) Percutaneous absorption of
7,10 14C-benzo(a)pyrene and 7,12 14C-dimethylbenz(a)anthracene in
mice. J Environ Pathol Toxicol Oncol, 7: 25-34.
Sandmeyer EE (1981) Aromatic hydrocarbons. In: Clayton GD & Clayton FE
ed. Patty's industrial hygiene and toxicology, 3rd rev Ed. New York,
John Wiley & Sons, pp 3333-3339.
Santella RM (1993) Human DNA and protein adduct dosimetry. Proc Am
Assoc Cancer Res, 34: 604-605.
Santella RM, Hemminki K, Tang DL, Paik M, Ottman R, Young TL, Savela
K, Vodickova L, Dickey C, Whyatt R, & Perera FP (1993) Polycyclic
aromatic hydrocarbon-DNA adducts in white blood cells and urinary
1-hydroxypyrene in foundry workers. Cancer Epidemiol Biomarkers Prev,
2: 59-62.
Santella RM, Nunes MG, Blaskovic R, Perera FP, Tang D, Beachman A, Lin
JH, & DeLeo VA (1994) Quantification of polycyclic aromatic
hydrocarbons, 1-hydroxypyrene, and mutagenicity in urine of coal
tar-treated psoriasis patients and untreated volunteers. Cancer
Epidemiol Biomarkers Prev, 3: 137-140.
Santella RM, Perera FP, Young TL, Zhang YJ, Chiamprasert S, Tang D,
Wang LW, Beachman A, Lin JH, & DeLeo VA (1995) Polycyclic aromatic
hydrocarbon-DNA and protein adducts in coar tar treated patients and
controls and their relationship to glutathione S-transferase genotype.
Mutat Res, 334: 117-124.
Santodonato J, Basu D, & Howard PH (1980) Multimedia human exposure
and carcinogenic risk assessment for environmental PAH. In: Bjorseth A
& Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry and
biological effects. Columbus, Ohio, Battelle Press, 435-454.
Santodonato J, Howard P, & Basu D (1981) In: Lee SD & Grant L ed.
Health and ecological assessment of polynuclear aromatic hydrocarbons.
Park Forest South, Illinois, Pathotox Publishers, 364 pp.
Sardella DJ, Boger E, & Ghoshal PK (1981) Active sites in hexacyclic
carcinogens probed by the fluorine substitution methodology. Columbus,
Ohio, Battelle Press, pp 529-538.
Savas U, Bhattacharyya KK, Christou M, Alexander DL, & Jefcoate CR
(1994) Mouse cytochrome P450EF, representative of a new 1B subfamily
of cytochrome P450s. Cloning, sequence determination, and tissue
expression. J Biol Chem, 269: 14905-14911.
Savela K & Hemminki K (1991) DNA adducts in lymphocytes and
granulocytes of smokers and nonsmokers detected by the
32P-postlabelling assay. Carcinogenesis, 12: 503-508.
Savino JF & Tanabe LL (1989) Sublethal effects of phenanthrene,
nicotine, and pinane on Daphnia pulex. Bull Environ Contam Toxicol,
42: 778-784.
Sawicki JT, Moschel RC, & Dipple A (1983) Involvement of both
syn and antidihydrodiol-epoxides in the binding of
7,12-dimethylbenz [a]anthracene to DNA in mouse embryo cell cultures.
Cancer Res, 43: 3213-3218.
Sax NI & Lewis JR Sr (1984) Dangerous properties of industrial
materials, 7th Ed. New York, Van Nostrand Reinhold Co, pp 2451-2452.
Saxton WL, Newton RT, Rohrberg J, Sutton J, & Johnson LJ (1993)
Polycyclic aromatic hydrocarbons in seafood from the Gulf of Alaska
following a major crude oil spill. Bull Environ Contam Toxicol, 51:
515-522.
Schafer EW Jr, Bowles WA Jr, & Hurlbut J (1983) The acute oral
toxicity, repellency, and hazard potential of 998 chemicals to one or
more species of wild and domestic birds. Arch Environ Contam Toxicol,
12: 355-381.
Schaller KH, Angerer J, & Hausmann N (1993) The determination of
1-hydroxypyrene in urine as a tool for the biological monitoring of
PAH-exposed persons. Polycyclic Aromat Compd, 3 (suppl): 1023-1030.
Scheepers PTJ & Bos RP (1992) Combustion of diesel fuel from a
toxicological perspective 1. Origin of incomplete combustion products.
Int Arch Occup Environ Health, 64: 149-161.
Scherer G, Conze C, Von Meyerinck L, Sorsa M, & Adlkofer F (1990)
Importance of exposure to gaseous and particulate phase components of
tobacco smoke in active and passive smokers. Int Arch Occup Environ
Health, 62: 459-466.
Schimberg RW, Toivonen E, & Tossavainen A (1981) [Polycyclic aromatic
hydrocarbons and other hazardous agents in foundry moulding sand with
various hydrocarbon carriers.] Staub-Reinhalt Luft, 41: 221-224 (in
German).
Schlede E, Kuntzman R, Haber S, & Conney AH (1970a) Effect of enzyme
induction on the metabolism and tissue distribution of
benzo [a]pyrene. Cancer Res, 30: 2893-2897.
Schlede E, Kuntzman R, & Conney AH (1970b) Stimulatory effect of
benzo [a]pyrene and phenobarbital pretreatment on the biliary
excretion of benzo [a]pyrene metabolites in the rat. Cancer Res, 30:
2898-2904.
Schmähl D (1955) [Testing of naphthalene and anthracene for
carcinogenic effects in rats.] Z Krebsforsch, 60: 697-710 (in German).
Schmähl D, Schmidt KG, & Habs M (1977) Syncarcinogenic action of
polycyclic hydrocarbons in automobile exhaust gas condensates. In: Air
pollution and cancer in man. Lyon, International Agency for Research
on Cancer, pp 53-59 (IARC Scientific Publications No. 16).
Schmeltz I, Tosk J, Hilfrich J, Hirota N, Hoffmann D, & Wynder EL
(1978) Bioassays of naphthalene and alkylnaphthalenes for
co-carcinogenic activity. Relation to tobacco carcinogenesis. In:
Jones PW & Freudenthal RI ed. Carcinogenesis, Volume 3: Polynuclear
aromatic hydrocarbons. New York, Raven Press, pp 47-60.
Schmidt H (1992) [Emissions of polycyclic aromatic hydrocarbons in the
processsing of bitumen and polymer bitumen street sections.] Bitumen,
54: 50-53 (in German).
Schmoldt A, Jacob J, & Grimmer G (1981) Dose-dependent induction of
rat liver microsomal aryl hydrocarbon monooxygenase by
benzo [k]fluoranthene. Cancer Lett, 13: 249-257.
Schnizlein CT, Munson AE, & Rhoades RA (1987) Immunomodulation of
local and systemic immunity after subchronic pulmonary exposure of
mice to benzo(a)pyrene. Int J Immunopharmacol, 9: 99-106.
Schoeny R & Warshawsky D (1983) Mutagenicity of benzo(a)pyrene
metabolites generated on the isolated perfused lung following
particulate exposure. Teratog Carcinog Mutag, 151: 151-162.
Schoeny R, Cody T, Radike M, & Warshawsky D (1985) Mutagenicity of
algal metabolites of benzo(a)pyrene for Salmonella typhimurium.
Environ Mutag, 7: 839-855.
Schoeny R, Cody T, Warshawsky D, & Radike M. (1988) Metabolism of
mutagenic polycyclic hydrocarbons by photosynthetic algal species.
Mutat Res, 197: 289-302.
Schoeny R, Lewtas J, McClure P, & Stiteler W (1994) Risk assessment
for polycyclic aromatic hydrocarbons: Options revisited. Dallas,
Texas, Society of Toxicology.
Schoket B, Hewer A, Grover PL, & Phillips DH (1989) 32P-Postlabelling
analysis of DNA adducts in the skin of mice treated with petrol and
diesel engine lubricating oils and exhaust condensates.
Carcinogenesis, 10: 1485-1490.
Schoket B, Horkay I, Kósa A, Páldeák L, Hewer A, Grover PL, & Phillips
DH (1990) Formation of DNA adducts in the skin of psoriasis patients,
in human skin in organ culture, and in mouse skin and lung following
topical application of coal-tar and juniper tar. J Invest Dermatol,
94: 241-246.
Schoket B, Poirier MC, & Vincze I (1995) Biomonitoring of genotoxic
exposure in aluminium plant workers by determination of DNA adducts in
human peripheral blood lymphocytes. Sci Total Environ, 163: 153-163.
van Schooten FJ, Hillebrandt MJX, van Leeuwen FE, van Zandwijk N,
Janssen HM, den Engelse L, & Kriek E (1992) Polycyclic aromatic
hydrocarbon-DNA adducts in white blood cells from lung cancer
patients: No correlation with adduct levels in lung. Carcinogenesis,
13: 987-993.
van Schooten FJ, Moonen EJC, Rhijnsburger E, van Algen B, Thijssen
HHW, & Kleinjans JCS (1994) Dermal uptake of polycyclic aromatic
hydrocarbons after hairwash with coal-tar shampoo. Lancet, 344: 1505-
1506.
van Schooten FJ, Jongeneelen FJ, Hillebrand MJ, van Leeuwen FE, de
Looff AJ, Dijkmans AP, van Rooij JGM, den Engelse L, & Kriek E (1995)
Polycyclic aromatic hydrocarbon-DNA adducts in white blood cell DNA
and 1-hydroxypyrene in the urine from aluminium workers: Relation with
job category and synergistic effect of smoking. Cancer Epidemiol
Biomarkers Prev, 4: 69-77.
Schrimpff E (1983) [Forests dying from high concentrations of
pollutants in fog.] Staub Reinhalt Luft, 43: 240 (in German).
Schrimpff E, Thomas W, & Herrmann R (1979) Regional patterns of
contaminants (PAH, pesticides and trace metals) in snow of northeast
Bavaria and their relationship to human influence and orographic
effects. Water Air Soil Pollut, 11: 481-497.
Schuetzle D (1983) Sampling of vehicle emissions for chemical analysis
and biological testing. Environ Health Perspectives, 47: 65-80.
Schuetzle D & Frazier JA (1986) Factors influencing the emission of
vapor and particulate phase components from diesel engines. In:
Ishinishi N, Koizumi A, McClellan RO, & Stöber W ed. Carcinogenic and
mutagenic effects of diesel engine exhaust. Amsterdam, Elsevier
Science Publishers, pp 41-63.
Schunk W (1979) [Relationship between exposure to polycyclic
hydrocarbons and the increased occurrence of cancer cases in two
chemical companies in Thüringen.] Z Ärztl Fortbild, 73: 84-88 (in
German).
Schürch O & Winterstein A (1935) [The cancer inducing effect of
aromatic hydrocarbons.] Z Physiol Chem, 236: 79-91 (in German).
Schuyer J, Blom L, & Van Krevelen DW (1953) The molar refraction of
condensed aromatic compounds. Trans Faraday Soc, 49: 1391-1401.
Schwarz FP (1977) Determination of temperature dependence of
solubilities of polycyclic aromatic hydrocarbons in aqueous solutions
by a fluorescence method. J Chem Eng Data, 22: 273-277.
Scribner JO (1973) Brief communication: Tumor initiation by apparently
noncarcinogenic polycyclic aromatic hydrocarbons J Natl Cancer Inst,
50: 1717-1719.
Sega GA (1979) Unscheduled DNA synthesis (DNA repair) in the germ
cells of male mice. Its role in the study of mammalian mutagenesis.
Genetics, 92: 49-58.
Segerbäck D & Vodicka P (1993) Recoveries of DNA adducts of polycyclic
aromatic hydrocarbons in the 32P-postlabelling assay. Carcinogenesis,
14: 2463-2469.
Seifert B, Ullrich D, & Schmahl HJ (1983) Occurrence of carcinogenic
organic substances in kitchen air. In: Proceedings of VIth World
Congress on Air Quality, Paris, 16-20 May 1983. Paris, SEFIC, pp 177-
179 (Poster presentation).
Seifert B, Ullrich D, & Liu ZF (1986) [Inorganic and organic air
pollutants at a street intersection in West Berlin. Reports of Society
for Water, Soil, and Air) Schriftenreihe Ver WaBoLu, 67: 211-222 (in
German).
Selgrade MK, Daniels MJ, Burleson GR, Lauer LD, & Dean JH (1988)
Effects of 7,12-dimethylbenz [a]anthracene, benzo [a]pyrene and
cyclosporin A on murine cytomegalo-virus infections: Studies of
resistance mechanisms. Int J Immunopharmacol,10: 811-818.
Selkirk JK, Croy RG, Whitlock JP Jr, & Gelboin HV (1975) In
vitro metabolism of benzo [a]pyrene by human liver microsomes and
lymphocytes. Cancer Res, 35: 3651-3655.
Sellakumar A & Shubik P (1974) Carcinogenicity of different polycyclic
hydrocarbons in the respiratory tract of hamsters. J Natl Cancer Inst,
53: 1713-1719.
Serth RW & Hughes TW (1980) Polycyclic organic matter (POM) and trace
element contents of carbon black vent gas. Environ Sci Technol, 14:
298-301.
Shabad LM & Dikun PP (1959) Atmospheric pollution by the carcinogenic
hydrocarbon 3,4-benzpyrene. Leningrad, Medgiz, 228 pp.
Sharpe CR, Rochon JE, Adam JM, & Siussa S (1989) Case-control study of
hydrocarbon exposures in patients with renal cell carcinoma. Can Med
Assoc J, 140: 1309-1318.
Shaw GR & Connell DW (1994) Prediction and monitoring of the
carcinogenicity of polycyclic aromatic compounds (PACs). In: Ware GW
ed. Reviews of environmental contamination and toxicology. Berlin,
Springer-Verlag, pp 1-62.
Shay H, Aegerter EA, Gruenstein M, & Komarov SA (1949) Development of
adenocarcinoma of the breast in the Wistar rat following the gastric
instillation of methylcholanthrene. J Natl Cancer Inst, 10: 255-266.
Shear MJ (1938) Studies in carcinogenesis. V. Methyl derivatives of
1:2-benzanthracene. Am J Cancer, 33: 499-537.
Shear MJ & Leiter J (1941) Studies in carcinogenesis. XVI. Production
of subcutaneous tumors in mice by miscellaneous polycyclic compounds.
J Natl Cancer Inst, 2: 241-259.
Shen Z, Wells RL, & Elkind MM (1994) Enhanced cytochrome P450
(Cyp1b1) expression, aryl hydrocarbon hydroxylase activity,
cytotoxicity, and transformation of C3H10T1/2 cells by
dimethylbenz (a)anthracene in conditioned medium. Cancer Res, 54:
4052-4056.
Shendrikova IA & Aleksandrov VA (1974) Comparative characteristics of
penetration of polycyclic hydrocarbons through the placenta in the
fetus in rats. Bull Exp Biol Med, 77: 77-79.
Shendrikova IA, Ivanov-Golitsyn MM, Anisimov YN, & Likhachev AYa
(1973) [Dynamics of transplacental penetration of
7,12-dimethylbenz [a]anthracene in mice.] Vopr Onkol, 19: 75-79 (in
Russian with English translation).
Shendrikova IA, Ivanov-Golitsyn MN, & Likjachev AYa (1974) [The
transplacental penetration of benzo [a]pyrene in mice.] Vopr Onkol,
20: 53-56 (in Russian with English abstract).
Sherson D & Iversen E (1986) Mortality among foundry workers in
Denmark due to cancer and respiratory and cardiovascular diseases.
Cancer Res Monogr, 2: 403-414.
Sherson D, Sabro P, Sigsgaard T, Johansen F, & Autrup H (1990)
Biological monitoring of foundry workers exposed to polycylic aromatic
hydrocarbons. Br J Ind Med, 47: 448-453.
Sherson D, Sigsgaard T, Overgaard E, Luft S, Poulsen H, & Jongenellen
FJ (1992) Interaction of smoking, PAH uptake and cytochrome P450IA2
activity among foundry workers. Br J Ind Med, 49:197-202.
Shiaris MP & Jambard-Sweet D (1986) Polycyclic aromatic hydrocarbons
in surficial sediments of Boston Harbour, Massachusetts, USA. Mar
Pollut Bull, 17: 469-472.
Shimada T, Martin MV, Pruess-Schwartz D, Marnett LJ, & Guengerich FP
(1989) Roles of individual human cytochrome P450 enzymes in the
bioactivation of benzo (a)pyrene,
7,8-dihydroxy-7,8-dihydrobenzo (a)pyrene, and other dihydrodiol
derivatives of polycyclic aromatic hydrocarbons. Cancer Res, 49: 6304-
6312.
Shimada H, Satake S, Itoh S, Hattori C, Hayashi M, & Ishidate M Jr
(1991) Multiple dosing effects of benzo [a]pyrene in the mouse bone
marrow micronucleus test. Mutat Res, 252: 107.
Shimada H, Suzuki H, Itoh S, Hattori C, Matsuura Y, Tada S, & Watanabe
C (1992) The micronucleus test of benzo(a)pyrene with mouse and rat
peripheral blood reticulocytes. Mutat Res, 278:165-168.
Shimkin MB & Stoner GD (1975) Lung tumours in mice: Application to
carcinogenesis bioassay. In: Klein G & Weinhouse S ed. Advances in
cancer research, Volume 1. New York, Raven Press, pp 1-58.
Shopp GM, White KL Jr, Holsapple MP, Barnes DW, Duke SS, Anderson AC,
Condie LW Jr, Hayes JR, & Borzelleca JF (1984) Naphthalene toxicity in
CD-1 mice: General toxicology and immunotoxicology. Fundam Appl
Toxicol, 4: 406-419.
Shou M, Korzekwa KR, Crespi CL, Gonzalez FJ, & Gelboin HV (1994) The
role of 12 cDNA-expressed human, rodent, and rabbit cytochromes P450
in the metabolism of benzo [a]pyrene trans-7,8-dihydrodiol. Mol
Carcinog, 1: 159-168.
Shubik P & Della Porta G (1957) Carcinogenesis and acute intoxication
with large doses of polycyclic hydrocarbons. Am Med Assoc Arch Pathol,
64: 691-703.
Shubik P, Pietra G, & Della Porta G (1960) Studies of skin
carcinogenesis in the Syrian golden hamster. Cancer Res, 20: 100-105.
Shum S, Jensen NM, & Nebert DW (1979) The murine Ah locus:
In utero toxicity and teratogenesis with genetic differences in
benzo [a]pyrene metabolism. Teratology, 20: 365-376.
Siebert D, Marquardt H, Friesel H, & Hecker E (1981) Polycyclic
aromatic hydrocarbons and possible metabolites: Convertogenic activity
in yeast and tumor initiating activity in mouse skin. J Cancer Res
Clin Oncol, 102: 127-139.
Simmon VF (1979) In vitro mutagenicity assays of chemical
carcinogens and related compounds with Salmonella typhimurium.
J Natl Cancer Inst, 62: 893-899.
Simmon VF, Rosenkranz HS, Zeiger E, & Poirier LA (1979) Mutagenic
activity of chemical carcinogens and related compounds in the
intraperitoneal host-mediated assay. J Natl Cancer Inst, 62: 911-918.
Simó R, Colom-Altés M, Grimalt JO, & Albaigés J (1991) Background
levels of atmospheric hydrocarbons, sulfate and nitrate over the
western Mediterranean. Atmos Environ, 25A: 1463-1471.
Simoneit BRT, Sheng G, Chen X, Fu J, Zhang J, & Xu Y (1991) Molecular
marker study of extractable organic matter in aerosols from urban
areas of China. Atmos Environ, 25: 2111-2130.
Sims P (1959) Metabolism of polycyclic compounds. 14. The conversion
of naphthalene into compounds related to
trans-1:2-dihydro-1:2-dihydroxynaphthalene by rabbits. Biochem J,
73: 389-395.
Sims P (1962) Metabolism of polycyclic compounds. 19. The metabolism
of phenanthrene in rabbits and rats: Phenols and sulphuric esters.
Biochem J, 84: 558-563.
Sims P (1964) Metabolism of polycyclic compounds. 25. The metabolism
of anthracene and some related compounds in rats. Biochem J, 92: 621-
631.
Sims P & Grover PL (1974) Epoxides in polycyclic aromatic hydrocarbon
metabolism and carcinogenesis. Adv Cancer Res, 20: 165-274.
Sims P & Grover PL (1981) Involvement of dihydrodiols and diol
epoxides in the metabolic activation of polycyclic hydrocarbons other
than benzo [a]pyrene. In: Gelboin HV & Ts'o POP ed. Polycyclic
hydrocarbons and cancer, Volume 3. New York, Academic Press, pp 117-
181.
Sims RC & Overcash MR (1983) Fate of polynuclear aromatic compounds
(PNAs) in soil plant systems. Res Rev, 88: 1-67.
Sipal Z, Ahlenius T, Bergstrand A, Rodriquez L, & Jakobsson SW (1979)
Oxidative biotransformation of benzo [a]pyrene by human lung
microsomal fractions prepared from surgical specimens. Xenobiotica, 9:
633-645.
Sirianni SR & Huang CC (1978) Sister chromatid exchange induced by
promutagens/carcinogens in Chinese hamster cells cultured in diffusion
chambers in mice. Proc Soc Exp Biol Med, 158: 269-274.
Sirota GR & Uthe JF (1981) Polynuclear aromatic hydrocarbon
contamination in marine shellfish. In: Cooke M & Dennis AJ ed.
Polynuclear aromatic hydrocarbons: Chemical analysis and biological
fate. Columbus, Ohio, Battelle Press, pp 329-341.
Sirota R, Uthe JF, Sreedharan A, Matheson R, Musial J, & Hamilton K
(1983) Polynuclear aromatic hydrocarbons in lobster (Homarus
americanus) and sediments in the vicinity of a coking facility. In:
Cooke M & Dennis AJ ed. Polynuclear aromatic hydrocarbons: Formation,
metabolism and measurement. Columbus, Ohio, Battelle Press, pp 1123-
1136.
Sivarajah K, Mukhtar H, & Eling T (1979) Arachidonic acid-dependent
metabolism of (+) trans-7,8-dihydroxy-7,8-dihydrobenzo [a]pyrene
(BP-7,8 diol) to 7,10/8,9 tetrols. FEBS Lett, 106: 17-20.
Skopek TR & Thilly WG (1983) Rate of induced forward mutation at 3
genetic loci in Salmonella typhimurium. Mutat Res, 108: 45-56.
Skopek TR, Liber HL, Kaden DA, Hites RA, & Thilly WG (1979) Mutation
of human cells by kerosene soot. J Natl Cancer Inst, 63: 309-312.
Slaga TJ, Hubermann E, Selkirk JK, Harvey RG, & Bracken WM (1978)
Carcinogenicity and mutagenicity of benz(a)anthracene diols and
diol-epoxides. Cancer Res, 38: 1699-1704.
Slaga TJ, Jecker L, Bracken WM, & Weeks CE (1979) The effects of weak
or noncarcinogenic polycyclic hydrocarbons on
7,12-dimethylbenz [a]anthracene and benzo [a]pyrene skin
tumor-initiation. Cancer Lett, 7: 51-59.
Slaga TJ, Gleason GL, Mills C, Ewald L, Fu PP, Lee HM, & Harvey RG
(1980) Comparison of the tumour-initiating activities of dihydrodiols
and diol-epoxides of various polycyclic aromatic hydrocarbons. Cancer
Res, 40: 1981-1984.
Slaga TJ, Iyer RP, Lyga W, Secrist A III, Daub GH, & Harvey RG (1981)
Comparison of the skin tumor-initiating activities of dihydrodiols,
diol-epoxides, and methylated derivatives of various polycyclic
aromatic hydrocarbons. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 753-769.
Slooff W, Janus JA, Matthijsen AJCM, Montizaan GK, & Ros JPM (1989)
Integrated criteria document on PAHs (Report No. 758474011).
Bilthoven, National Institute of Public Health and Environmental
Protection, pp 15-36.
Smith RL & Hargreaves BR (1983) A simple toxicity apparatus for
continuous flow with small volumes: Demonstration with mysids and
naphthalene. Bull Environ Contam Toxicol, 30: 406-412.
Smith SR, Tanaka J, Futoma DJ, & Smith TE (1981) Sampling and
preconcentration methods for the analysis of polycyclic aromatic
hydrocarbons in water systems. CRC Crit Rev Anal Chem, 10: 375-425.
Smith KR, Aggarwal AL, & Dave RM (1983) Air pollution and rural
biomass fuels in developing countries: A pilot village study in India
and implications for research and policy. Atmos Environ, 17: 2343-
2362.
Smith JD, Bagg J, & Bycroft BM (1984) Polycyclic aromatic hydrocarbons
in the clam Tridacna maxima from the Great Barrier Reef, Australia.
Environ Sci Technol, 18: 353-358.
Smith JD, Bagg J, & Sin YO (1987) Aromatic hydrocarbons in seawater,
sediments and clams from Green Island, Great Barrier Reef, Australia.
Aust J Mar Freshwater Res, 38: 501-510.
Smith S, Savino J, & Blouin M (1988) Acute toxicity to Daphnia
pulex of six classes of chemical compounds potentially hazardous to
Great Lakes aquatic biota. J Great Lakes Res, 14: 394-404.
Smith-Sonneborn J (1983) Use of a ciliated protozoan as a model system
to detect toxic and carcinogenic agents. In: Kolber AR, Wong TK, Grant
LD, DeWoskin RS, & Hughes TJ ed. In vitro toxicity testing of
environmental agents: Current and future possibilities. Part A: Survey
of test systems. New York, Plenum Press, pp 113-137.
Smyth HF, Carpenter CP, Weil CS, Pozzani UC, & Striegel JA (1962)
Range-finding toxicity data: List VI. Ind Hyg J, March-April: 95-107.
Snell KC & Stewart HL (1962) Pulmonary adenomatosis induced in DBA/2
mice by oral administration of dibenz [a,h]anthracene. J Natl Cancer
Inst, 28: 1043-1049.
Snider EH & Manning FS (1982) A survey of pollutant emission levels in
wastewaters and residuals from the petroleum refining industry.
Environ Int, 7: 237-258.
Society of German Chemists (Advisory Committee on Existing Chemicals
of Environ-mental Relevance) (1989) [Naphthalene.] Weinheim, VCH
Verlagsgesellschaft, 155 pp (Report No. 39) (in German).
Society of German Engineers (1989) Emission measurement. Measurement
of polycyclic aromatic hydrocarbons (PAH): Measurement of PAH in the
exhaust gas from gasoline and diesel engines of passenger cars. Gas
chromatographic determination. Düsseldorf, Commission for Air Quality
part 1, 27 pp (VDI Report No. 3872).
Solt DB, Polverini PJ, & Calderon L (1987) Carcinogenic response of
hamster buccal pouch epithelium to 4 polycyclic aromatic hydrocarbons.
J Oral Pathol, 16: 294-302.
Sonnefeld WJ, Zoller WH, & May WE (1983) Dynamic coupled-column liquid
chromatographic determination of ambient temperature vapor pressures
of polynuclear aromatic hydrocarbons. Anal Chem, 55: 275-280.
Sorahan T & Cooke MA (1989) Cancer mortality in a cohort of United
Kingdom steel foundry workers: 1946-1985. Br J Ind Med, 46: 74-81.
Sorrell K, Brass HJ, & Reding R (1980) A review of occurrences and
treatment of polynuclear aromatic hydrocarbons in water. Environ Int,
4: 245-254.
Southworth GR (1977) Transport and tranformations of anthracene in
natural waters. In: Marking LL & Kimerle RA ed. Aquatic toxicology.
Philadelphia, Pennsylvania, American Society for Testing and
Materials, pp 359-380 (ASTM ATP 667).
Southworth GR (1979) The role of volatilization in removing polycyclic
aromatic hydrocarbons from aquatic environments. Bull Environ Contam
Toxicol, 21: 507-514.
Southworth GR, Beauchamp JJ, & Schmieder PK (1978) Bioaccumulation
potential of polycyclic aromatic hydrocarbons in Daphnia pulex.
Water Res, 12: 973-977.
Spacie A, Landrum PF, & Leversee GJ (1983) Uptake, depuration, and
biotransformation of anthracene and benzo [a]pyrene in bluegill
sunfish. Ecotoxicol Environ Saf, 7: 330-341.
Sparnins VL, Mott AW, Baraney G, & Wattenberg LW (1986) Effects of
allyl methyl trisulfide on glutathione-S-transferase activity and
BP-induced neoplasia in the mouse. Nutr Cancer, 8: 211-215.
Speer K, Steeg E, Horstmann P, Kühn TH, & Montag A (1990)
Determination and distribution of PAH in native vegetable oils, smoked
fish products, mussels and oysters, and bream from the River Elbe. J
High Resol Chromatogr, 13: 104-111.
Spicer CW, Holdren MW, Smith DL, Hughes DP, & Smith MD (1992) Chemical
composition of exhaust from aircraft turbine engines. J Eng Gas
Turbines Power, 114: 111-117.
Spinelli JJ, Band PR, Svirchev LM, & Gallagher RP (1991) Mortality and
cancer incidence in aluminium reduction plant workers. J Occup Med,
33: 1150-1155.
Spitzer T & Dannecker W (1983) Membrane filters as adsorbents for
polycyclic aromatic hydrocarbons during high-volume sampling of air
particulate matter. Anal Chem, 55: 2226-2228.
Spitzer T & Kuwatsuka S (1986) Simple method for determination of
polynuclear aromatic hydrocarbons in soil by clean-up on XAD-2. J
Chromatogr, 358: 434-437.
Sporstl S, Gjs N, Lichtenthaler RG, Gustavsen KO, Urdal K, Oreld F,
& Skei J (1983) Source identification of aromatic hydrocarbons in
sediments using GC/MS. Environ Sci Technol, 17: 282-286.
Stahl RG, Liehr JG, & Davis EM (1984) Characterization of organic
compounds in simulated rainfall runoffs from model coal piles. Arch
Environ Contam Toxicol, 13: 179-190.
Stanton MF, Miller E, Wrench C, & Blackwell R (1972) Experimental
induction of epidermoid carcinoma in the lungs of rats by cigarette
smoke condensate. J Natl Cancer Inst, 49: 867-877.
State Chemical Analysis Institute, Freiburg (1995) [Food control and
the environment: Annual report 1994.] Freiburg, pp 156-160 (in
German).
State Committee for Air Pollution Control (1992) [Cancer risk from air
pollution.] Development of evaluation criteria for carcinogenic
pollutants. Düsseldorf, Minister for Environment and Agriculture for
Northrhine-Westfalia, 71 pp (in German).
State Pollution Control Authority and Norwegian Food Control Authority
(1992) Recommendations from the workshop on PAH. In: Proceedings of
the workshop on polyaromatic hydrocarbons (PAH), Oslo, 11-13 November
1991. Oslo, p 423 (Report No. TA-816/1992).
Stauffer TB, MacIntyre WG, & Wickman DC (1989) Sorption of nonpolar
organic chemicals on low-carbon-content aquifer materials. Environ
Toxicol Chem, 8: 845-852.
Stavenow L & Pessah-Rasmussen H (1988) Effects of polycyclic aromatic
hydrocarbons on proliferation, collagen secretion and viability of
arterial smooth muscle cells in culture. Artery, 15: 94-108.
Stay FS, Katko A, Rohm CM, Fix MA, & Larsen DP (1988) Effects of
fluorene on microcosms developed from four natural communities.
Environ Toxicol Chem, 7: 635-644.
Steen WC & Karickhoff SW (1981) Biosorption of hydrophobic organic
pollutants by mixed microbial populations. Chemosphere, 10: 27-32.
Steiner PF (1955) Carcinogenicity of multiple chemicals simultaneously
administered. Cancer Res, 15: 632-635.
Steiner PE & Edgcomb JH (1952) Carcinogenicity of 1,2-benzanthracene.
Cancer Res, 12: 657-659.
Steiner PF & Falk HL (1951) Summation and inhibition effects of weak
and strong carcinogenic hydrocarbons: 1:2-Benzanthracene, chrysene,
1:2:5:6-dibenzanthracene, and 20-methylcholanthrene. Cancer Res, 11:
56-63.
Steinhoff D, Mohr U & Hahnemann S (1991) Carcinogenesis studies with
iron oxides. Exp Pathol, 43: 189-194.
Steinig J (1976) 3,4-Benzopyrene contents in smoked fish depending on
smoking procedure. Z Lebensmittelunters Forsch, 162: 235-242.
Stenbäck F & Sellakumar A (1974) Lung tumor induction by
dibenz(a,i)pyrene in the Syrian golden hamster. Z Krebsforsch, 82:
175-182.
Stenberg UR (1985) PAH emissions from automobiles. In: Bjorseth A &
Ramdahl T ed. Handbook of polycyclic aromatic hydrocarbons. Volume 2:
Emission sources and recent progress in analytical chemistry. New
York, Marcel Dekker, pp 87-111.
Stenberg U, Alsberg T, & Westerholm R (1983) Applicability of a
cryogradient technique for the enrichment of PAH from automobile
exhausts: Demonstration of methodology and evaluation experiments.
Environ Health Perspectives, 47: 43-51.
Stevenson JL & Von Haam E (1965) Carcinogenicity of benz(a)anthracene
and benzo(c)phenanthrene derivatives. Am Ind Hyg Assoc J, 26: 475-478.
Stoner GD, Harris CC, Autrup H, Trump BJ, Kingsbury EW, & Myers GA
(1978) Explant cultures of human peripheral lung. I. Metabolism of
benzo [a]pyrene. Lab Invest, 38: 685-692.
Storer JS, DeLeon I, Millikan LE, Laseter JL, & Griffing C (1984)
Human absorption of crude coal tar products. Arch Dermatol, 120: 874-
877.
Stout P & Mamantov G (1989) Recent advances in infrared analysis of
polycyclic aromatic compounds. In: Vo-Dinh T ed. Chemical analysis of
polycyclic aromatic compounds. New York, John Wiley & Sons, pp 411-
432.
Strandell M, Zakrisson S, Alsberg T, Westerholm R, Winquist L, &
Rannug U (1994) Chemical analysis and biological testing of a polar
fraction of ambient air, diesel engine, and gasoline engine
particulate extracts. Environ Health Perspectives, 102 (suppl 4): 85-
92.
Strickland PT, Routledge MN, & Dipple A (1993) Methodologies for
measuring carcinogen adducts in humans. Cancer Epidemiol Biomarkers
Prev, 2: 607-619.
Stuermer DH, Ng DJ, & Morris CJ (1982) Organic contaminants in
groundwater near underground coal gasification site in northeastern
Wyoming. Environ Sci Technol, 16: 582-587.
Suess MJ (1976) The environmental load and cycle of polycyclic
aromatic hydrocarbons. Sci Total Environ, 6: 239-250.
Sugiyama T (1973) Chromosomal aberrations and carcinogenesis by
various benz(a)-anthracene derivatives. Gann, 64: 637-639.
Sullivan TJ & Mix MC (1983) Pyrolytic deposition of polynuclear
aromatic hydrocarbons due to slash burning on clear-cut sites. Bull
Environ Contam Toxicol, 31: 208-215.
Sullivan TJ & Mix MC (1985) Persistence and fate of polynuclear
aromatic hydrocarbons deposited on slash burn sites in the Cascade
Mountains and coast range of Oregon. Arch Environ Contam Toxicol, 14:
187-192.
Suntzeff VA, Cowdry EV, & Croninger A (1955) Microscopic visualization
of the degeneration of sebaceous glands caused by carcinogens. Cancer
Res, 15: 637-640.
Surh YS, Liem A, Miller EC, & Miller JA ( 1989) Metabolic activation
of the carcinogen 6-hydroxymethylbenzo(a)pyrene: Formation of an
electrophilic sulfuric acid ester and benylic DNA adducts in rat liver
in vivo and in reactions in vivo.
Carcinogenesis, 10: 1519-1528.
Surh YJ, Blomquist JC, Liem A, & Miller JA (1990a) Metabolic
activation of 9-hydroxymethyl-10-methylanthracene and
1-hydroxymethylpyrene to electrophilic, mutagenic and tumorigenic
sulfuric acid esters by rat hepatic sulfotransferase activity.
Carcinogenesis, 11: 1451-1460.
Surh, YJ, Liem A, Miller EC, & Miller JA (1990b) The strong
hepatocarcinogenicity of the electrophilic and mutagenic metabolite
6-sulfooxymethylbenzo(a)pyrene and its formation of benzylic DNA
adducts in the livers of infant male B6C3F1 mice. Biochem Biophys Res
Commun, 172: 85-91.
Süss A (1980) [Basic questions for the solution of environmental
problems.] FreisingMunich, Bavarian State Department for Soil and
Horticulture, pp 376-378 (in German).
Sutter TR, Tang YM, Hayes CL, Wo Y-YP, Jabs EW, Li X, Yin H, Cody CW,
& Greenlee WF (1994) Complete cDNA sequence of a human
dioxin-inducible mRNA identifies a new gene subfamily of cytochrome
P450 that maps to chromosome 2. J Biol Chem, 269: 13092-13099.
Swaen GMH, Slangen JJM, Volovics A, Hayes RB, Scheffers T, & Sturmans
F (1991) Mortality of coke plant workers in the Netherlands. Br J Ind
Med, 48: 130-135.
Swallow WH & van Noort RW (1985) Occupational exposure to polycyclic
aromatic hydrocarbons in some New Zealand industries. Wellington,
Department of Scientific and Industrial Research, 17 pp.
Swartz RC, Kemp PF, Schults DW, & Lamberson JO (1988) Effects of
mixtures of sediment contaminants on the marine infaunal amphipod
Rhepoxynius abronius. Environ Toxicol Chem, 7: 1013-1020.
Swartz WJ & Mattison DR (1985) Benzo(a)pyrene inhibits ovulation in
C57Bl/6N mice. Anat Rec, 221: 268-276.
Swedish National Board of Occupational Safety and Health (1994)
Occupational exposure limit values, Solna, National Institute of Work
and Health, p 5.
Symons RK & Crick I (1983) Determination of polynuclear aromatic
hydrocarbons in refinery effluent by high-performance liquid
chromatography. Anal Chim Acta, 151: 237-243.
Szabo G, Prosser SL, & Bulman RA (1990) Determination of the
adsorption coefficient (Koc) of some aromatics for soil by RP-HPLC on
two immobilized humic acid phases. Chemosphere, 21: 777-788.
Szyfter K, Krüger J, Ericson P, Vaca C, Försti A, & Hemminki K (1994)
32P-Postlabelling analysis of DNA adducts in humans: Adduct
distribution and method improvement. Mutat Res, 313: 269-276.
Tabak HH, Quave SA, Mashni CI, & Barth EF (1981) Biodegradability
studies with organic priority pollutant compounds. J Water Pollut
Control Fed, 53: 1503-1518.
Takada H, Onda T, & Ogura N (1990) Determination of polycyclic
aromatic hydrocarbons in urban street dusts and their source materials
by capillary gas chromatography. Environ Sci Technol, 24: 1179-1186.
Takahashi G (1974) Distribution and metabolism of benzo(a)pyrene in
fetal mouse. Bull Chest Dis Res Inst Kyoto Univ, 7: 155-160.
Takahashi G (1978) Distribution and excretion of the hydrocarbon
3-methylcholanthrene in the animal body. In: Gelboin HV and Ts'o POP,
ed. Polycyclic hydrocarbons and cancer, Volume 1. New York, Academic
Press, pp 233-246.
Takahashi G & Yasuhira K (1972) Excretion and conversion of
3-methylcholanthrene metabolites in the intestinal tract of the mouse.
Cancer Res, 32: 710-715.
Takahashi G & Yasuhira K (1973) Macroautoradiographic and radiometric
studies on the distribution of 3-methylcholanthrene in mice and their
fetuses. Cancer Res, 33: 23-28.
Takatsuki K, Suzuki S, Sato N, & Ushizawa I (1985) Liquid
chromatographic determination of polycyclic aromatic hydrocarbons in
fish and shellfish. J Assoc Off Anal Chem, 68: 945-949.
Tan YL, Quanci JF, Borys RD, & Quanci MJ (1992) Polycyclic aromatic
hydrocarbons in smoke particles from wood and duff burning. Atmos
Environ, 26A: 1177-1181.
Tawfic HN (1965) Studies on ear duct tumors in rats. Part II:
Inhibitory effect of methylcholanthrene and 1,2-benzanthracene on
tumor formation by 4-dimethylamino-stilbene. Acta Pathol Jpn, 15: 255-
260.
Ten Hulscher ThEM, Van Der Velde LE, & Bruggeman WA (1992) Temperature
dependence of Henry's law constants for selected chlorobenzenes,
polychlorinated biphenyls and polycyclic aromatic hydrocarbons.
Environ Toxicol Chem, 11: 1595-1603.
Teranishi K, Hamada K, & Watanabe H (1975) Quantitative relationship
between carcinogencity and mutagenicity of polyaromatic hydrocarbons
in Salmonella typhimurium mutants. Mutat Res, 31: 97-102.
Teta MJ, Ott MG, & Schnatter AR ( 1987) Population based mortality
surveillance in carbon products manufacturing plants. Br J Ind Med,
44: 344-350.
Tetzen D (1989) [Environmentally relevant classification of PKWF and
Printosol. Classification of printing ink oils.] Verfkroniek, 62: 469-
472 (in German).
Theriault G, Tremblay C, Cordier S, & Gingras S (1984) Bladder cancer
in the aluminium industry. Lancet, i: 947-950.
Thilly WG, DeLuca JG, Furth EE, Hoppe H IV, Kaden DA, Krolewski JJ,
Liber HL, Skopek TR, Slapikoff SA, Tizard RJ, & Penman BW (1980)
Gene-locus mutation assays in diploid human lymphoblast lines. In: De
Serres FJ & Hollaender A ed. Chemical mutagens: Principles and methods
for their detection, Volume 6. New York, Plenum Press, pp 331-364.
Thirman M, Albrecht JH, Krüger MA, Erickson RR, Cherwitz DL, Park SS,
Gelboin HV, & Holtzmann JL (1994) Induction of cytochrome CYPIA1 and
formation of toxic metabolites of benzo (a)pyrene by rat aorta: A
possible role in atherogenesis. Proc Natl Acad Sci USA, 91: 5397-5401.
Thomas W (1986) Principal component analysis of trace substance
concentrations in rainwater samples. Atmos Environ, 20: 995-1000.
Thomas P (1988) Reproductive endocrine function in female Atlantic
croaker exposed to pollutants. Mar Environ Res, 24: 179-183.
Thomson BM, Lake RJ, & Lilli RE (1996) The contribution of margarine
to cancer risk from polycyclic aromatic hydrocarbons in the New
Zealand diet. Polycyclic Aromat Compd 11: 177-184.
Thorslund TW & Farrar D (1990a) Development of relative potency
estimates for PAHs and hydrocarbon combustion product fractions
compared to benzo(a)pyrene and their use in carcinogenic risk
assessments. Fairfax, Virginia, Clement Associates, 79 pp.
Thorslund TW & Farrar D (1990b) Ingestion dose-response model for
benzo [a]pyrene. Fairfax, Virginia, Clement International Corp, 22 pp
(Contract 68-02-4601).
Thorsteinsson T (1969) Polycyclic hydrocarbons in commercially and
home-smoked food in Iceland. Cancer Lett, 23: 455-457.
Thrane KE (1987) Deposition of polycyclic aromatic hydrocarbons (PAH)
in the surroundings of primary aluminium industry. Water Air Soil
Pollut, 33: 385-393.
Thrane KE & Mikalsen A (1981) High-volume sampling of airborne
polycyclic aromatic hydrocarbons using glass fibre filters and
polyurethane foam. Atmos Environ, 15: 909-918.
Thrane KE & Wikström L (1984) Monitoring of polycyclic aromatic
hydrocarbons in ambient air. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Mechanisms, methods and metabolism. Columbus,
Ohio, Battelle Press, pp 1299-1314.
Thruston AD Jr (1978) High pressure liquid chromatography techniques
for the isolation and identification of organics in drinking water
extracts. J Chromatogr Sci, 16: 254-259.
Thurmond LM, Tucker AN, Rickert DE, Lauer LD, & Dean JH (1989)
Functional and biochemical disposition of
7,12-dimethylbenz(a)anthracene in murine lymphoid cells. Chem-Biol
Interactions, 72: 93-104.
Thursby GB, Steele RL, & Kane ME (1985) Effect of organic chemicals on
growth on reproduction in the marine red alga Champia
parvula. Environ Toxicol Chem, 4: 797-806.
Thyssen J, Althoff J, Kimmerle G, & Mohr U (1980) Investigations on
the carcinogenic burden of air pollution in man. XIX. Effect of
inhaled benzo(a)pyrene in Syrian golden hamsters: A pilot study.
Zentralbl Bakteriol Hyg I Abt Orig B, 171: 441-444.
Thyssen J, Althoff J, Kimmerle G, & Mohr U (1981) Inhalation studies
with benz [a]pyrene in Syrian golden hamsters. J Natl Cancer Inst,
66: 575-577.
Tilgner DJ (1958) [New information about smoking procedures].
Fleischwirtschaft, 10: 649-653 (in German).
Tilgner DJ (1968) Carcinogens in food. Food Manuf, 43: 37-39, 42.
Todorovic R, Devanesan PD, Cavalieri EL, Rogan EG, Park SS, & Gelboin
HV (1991) A monoclonal antibody to rat liver cytochrome P450 IIC11
strongly and regiospecifically inhibits constitutive benzo [a]pyrene
metabolism and DNA binding. Mol Carcinog, 4: 308-314.
Tokiwa H, Morita K, Takeyoshi H, Takahashi K, & Ohnishi Y (1977)
Detection of mutagenic activity in particulate air pollutants. Mutat
Res, 48: 237-248.
Tola S, Koskela R-S, Hernberg S, & Järvinen E (1979) Lung cancer
mortality among iron foundry workers. J Occup Med, 21: 753-760.
Tolos WP, Shaw PB, Lowry LK, MacKenzie BA, Deng J-F, & Markel HL
(1990) 1-Pyrenol: A biomarker for occupational exposure to polycyclic
aromatic hydrocarbons. Appl Occup Environ Hyg, 5: 303-309.
Tomatis L (1973) Transplacental carcinogenesis. In: Raven RW ed.
Modern trends in oncology. Part 1: Research progress. London,
Butterworths, pp 99-126.
Tomingas R, Pott F, & Dehnen W (1976) Polycyclic aromatic hydrocarbons
in human bronchial carcinoma. Cancer Lett, 1: 189-195.
Tonelli QJ, Custer RP, & Sorof S (1979) Transformation of cultured
mouse mammary glands by aromatic amines and amides and their
derivatives. Cancer Res, 39: 1784-1792.
Tong C, Laspia MF, Telang S, & Williams GM (1981a) The use of adult
rat liver cultures in the detection of the genotoxicity of various
polycyclic aromatic hydrocarbons. Environ Mutag, 3: 477-487.
Tong C, Brat SV, & Williams GM (1981b) Sister-chromatid exchange
induction by polycyclic aromatic hydrocarbons in an intact cell system
of adult rat-liver epithelial cells. Mutat Res, 91: 467-473.
Tong C, Brat VS, Telang S, Laspia, MF, Fazio M, Reiss B, & Williams GM
(1983) Effects of genotoxic polycyclic aromatic hydrocarbons in rat
liver culture systems. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Formation; metabolism,and measurement.
Columbus, Ohio, Battelle Press, pp 1189-1203.
Topham JC (1980) Do induced sperm-head abnormalities in mice
specifically identify mammalian mutagens rather than carcinogens?
Mutat Res, 74: 379-387.
Topping DC, Martin DH, & Nettesheim P (1981) Determination of
cocarcinogenic activity of benzo [e]pyrene for respiratory tract
mucosa. Cancer Lett, 11: 315-321.
Törrönen R, Nousianen U, & Hänninen O (1981) Induction of aldehyde
dehydrogenase by polycyclic aromatic hydrocarbons in rats. Chem-Biol
Interactions, 36: 33-44.
Toth L & Blaas W (1972) [The effect of smoking technology on the
content of carcinogenic hydrocarbons in smoked meat products.]
Fleischwirtschaft, 9: 1121-1124 (in German).
Traynor GW, Apte MG, Carruthers AR, Dillworth JF, Grimsrud DT, &
Gundel LA (1987) Indoor air pollution due to emissions from
wood-burning stoves. Environ Sci Technol, 21: 691-697.
Tremblay C, Armstrong B, Thériault G, & Brodeur J (1995) Estimation of
risk of developing bladder cancer among workers exposed to coal tar
pitch volatiles in the primary aluminum industry. Am J Ind Med, 27:
335-348.
Trucco RG, Engelhardt FR, & Stacey B (1983) Toxicity accumulation and
clearance of aromatic hydrocarbons in Daphnia pulex. Environ Pollut,
A31: 191-202.
Tsuchimoto T & Matter BE (1981) Activity of coded compounds in the
micronucleus test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North-Holland, pp 705-711
(Progress in Mutation Research, Volume 1).
Tucci AF (1988) PAH characterization in hazardous waste from coke
processing plants. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: A decade of progress. Columbus, Ohio, Battelle Press, pp
865-870.
Tuominen JP, Pyysalo HS, & Sauri M (1988) Cereal products as a source
of polycyclic aromatic hydrocarbons. J Agric Food Chem, 36: 118-120.
Tuomisto J & Jantunen M (1987) A simple way of comparing carcinogenic
effects of chemical and radioactive emissions. Kuopio, National Public
Health Institute, 18 pp (Report No. NPHI-A2/1987).
Tweats DJ (1981) Activity of 42 coded compounds in a differential
killing test using Escherichia coli strains WP2, WP67 (uvrA polA),
and CM871 (uvrA lexA recA). In: De Serres FJ & Ashby J ed. Evaluation
of short-term tests for carcinogens. Report of the international
collaborative programme. New York, Elsevier North-Holland, pp 199-209
(Progress in Mutation Research, Volume 1).
Tyndyk ML, Zabezhinski MA, Bykoy VJ, Dikun PP, Dymochka LA,
Nepomnyaschaya OB, Yatsuk OS, Yermilov VB, & Likhachev AJ (1994)
Individual values of excretion of benzo [a]pyrene metabolites and
susceptibility to its carcinogenic effect in rats. Cancer Lett, 78:
163-170.
UNEP (1994) International register for Potentially Toxic Chemicals, PC
database
Urso P & Gengozian N (1980) Depressed humoral immunity and increased
tumor incidence in mice following in utero exposure to
benzo [a]pyrene. J Toxicol Environ Health, 6: 569-576.
Urso P & Johnson RA (1987) Early changes in T lymphocytes and subsets
of mouse progeny defective as adults in controlling growth of a
syngeneic tumor after in utero insult with benzo(a)pyrene.
Immunopharmacology, 14: 1-10.
Urso P & Johnson RA (1988) Quantitative and functional change in T
cells of primiparous mice following injection of benzo(a)pyrene at the
second trimester of pregnancy. Immunopharmacol Immunotoxicol, 10: 195-
217.
Urso P, Gengozian N, Rossi RM, & Johnson RA (1986) Suppression of
humoral and cellmediated immune responses in vitro by
benzo(a)pyrene. J Immunopharmacol, 8: 223-241.
Urso P, Ryan MC, & Bennett JS (1988) Changes in peripheral blood cells
in mice after injection with benzo(a)pyrene during pregnancy.
Immunopharmacol Immunotoxicol, 10: 179-193.
US Environmental Protection Agency (1978a) Ambient water quality
criteria: Naphthalene. Washington DC, p B/6 (PB-296 786).
US Environmental Protection Agency (1978b) Ambient water quality
criteria: Acenaphthene. Washington DC, p B/7 (PB-296 782).
US Environmental Protection Agency (1978c) Ambient water quality
criteria: Fluoranthene. Washington DC, pp B/5-B/7 (PB-292 433).
US Environmental Protection Agency (1980) Ambient water quality
criteria for polynuclear aromatic hydrocarbons. Washington DC, 7 pp
(EPA 440/5-80-069).
US Environmental Protection Agency (1984a) Guidelines establishing
test procedures for the analysis of pollutants under the clean water
act: Method 610. Polynuclear aromatic hydrocarbons. Fed Reg, 49(209):
43344-43352 (40 CFR Part 136).
US Environmental Protection Agency (1984b) Health effects assessment
for naphthalene. Research Triangle Park, North Carolina, Section 7
(EPA/540/1-86/014).
US Environmental Protection Agency (1984c) Health effects assessment
for polycyclic aromatic hydrocarbons (PAH). Washington DC, Office of
Emergency and Remedial Response, 49 pp (EPA 540/1-86/013; NTIS
PB86-134244/AS).
US Environmental Protection Agency (1984d) Carcinogenic assessment of
coke oven emissions. Final report. Washington DC, Office of Health and
Environmental Assessment, 209 pp (EPA 600/6-82-003F; NTIS
PB84-170182).
US Environmental Protection Agency (1984e) Health effects assessment
for benzo(a)pyrene. Cincinnati, Ohio, 43 pp (EPA/540/1-86/022; PB
86-134335).
US Environmental Protection Agency (1986a) (I) Method 3510 'Separatory
funnel liquid-liquid extraction'; (II) Method 3520 'Continuous liquid-
liquid extraction'; (III) Method 3630 'Silica gel cleanup'; (IV)
Method 8100 'Polynuclear aromatic hydrocarbons'; (V) Method 8270 'Gas
chromatography/mass spectrometry for semivolatile organics: Capillary
column technique'; (VI) Method 8310 'Polynuclear aromatic
hydrocarbons'. In: Test methods for evaluating solid waste, 3rd Ed,
Volume IB. Washington DC, 50 pp (EPA-SW-846).
US Environmental Protection Agency (1986b) (I) Method 3540 'Soxhlet
extraction'; (II) Method 3550 'Sonication extraction'; (III) Method
3630 'Silica gel cleanup'; (IV) Method 8100 'Polynuclear aromatic
hydrocarbons'; (V) Method 8270 'Gas chromatography/mass spectrometry
for semivolatile organics: Capillary column technique'; (VI) Method
8310 'Polynuclear aromatic hydrocarbons'. In: Test methods for
evaluating solid waste, 3rd Ed, Volume IB. Washington DC, 45 pp
(EPA-SW-846).
US Environmental Protection Agency (1986c) (I) Method 0010 'Modified
method 5 sampling train'; (II) Method 0020 'Source assessment sampling
system (SASS)'. In: Test methods for evaluating solid waste, 3rd Ed,
Volume II. Washington DC, 86 pp (EPA-SW846).
US Environmental Protection Agency (1986d) Health and environmental
effects profile for naphthalene. Cincinnati, Ohio, pp 41-54 (PB
88-242383).
US Environmental Protection Agency (1987a) Reference method for the
determination of particulate matter as PM10 in the atmosphere.
Appendix J. Fed Reg, 52(126): 24664-24666.
US Environmental Protection Agency (1987b) Health and environmental
effects profile for anthracene. Cincinnati, Ohio, pp 43-47, 57, 63, 66
(PB 89-118319).
US Environmental Protection Agency (1987c) The risk assessment
guidelines of 1986. Washington DC, Office of Health and Environmental
Assessment (EPA/600/8-87/045).
US Environmental Protection Agency (1988) 13-Week mouse oral
subchronic toxicity study (fluoranthene). Washington DC, 104 pp (TRL
Study No. 042-008).
US Environmental Protection Agency (1989a) Mouse oral subchronic
toxicity study with acenaphthene. Washington DC, 89 pp.
US Environmental Protection Agency (1989b) Subchronic toxicity in mice
with anthracene: Final report. Washington DC, 465 pp (HLA Study No.
2399-131).
US Environmental Protection Agency (1989c) Mouse oral subchronic
toxicity study (fluorene). Washington DC, 38 pp (TRL Study No.
042010).
US Environmental Protection Agency (1989d) Mouse oral subchronic
toxicity of pyrene. Washington DC, 102 pp (TRL Study No. 042-012).
US Environmental Protection Agency (1990) Method IP-7 'Determination
of benzo(a)pyrene and other polynuclear aromatic hydrocarbons in
indoor air'. In: Winberry WT Jr, Forehand L, Murphy NT, Ceroli A,
Phinney B, & Evans A ed. Compendium of methods for the determination
of air pollutants in indoor air. Research Triangle Park, North
Carolina, pp 550-639 (EPA 600/4-90/010; US NTIS PB90-200288).
US Environmental Protection Agency (1992a) Protection of the
environment: Chapter 1, part 86, sections 86-109, 86-110. US Code Fed
Regul, Title 40, Parts 86-99: 377-410.
US Environmental Protection Agency (1992b) Drinking water criteria
document for polycyclic aromatic hydrocarbons (PAHs). Washington DC,
Office of Water, 442 pp.
US Environmental Protection Agency (1993) Provisional guidance for
quantitative risk assessment of polycyclic aromatic hydrocarbons.
Cincinnati, Ohio, Office of Health and Environmental Assessment,
Environmental Criteria and Assessment Office (EPA/600/R-93/089).
Uthe JF & Musial CJ (1986) Polycyclic aromatic hydrocarbon
contamination of American lobster, Homarus americanus, in the
proximity of a coal-coking plant. Bull Environ Contam Toxicol, 37:
730-738.
Vaessen HAMG, Schuller PL, Jekel AA, & Wilbers AAMM (1984) Polycyclic
aromatic hydrocarbons in selected foods: Analysis and occurence.
Toxicol Environ Chem, 7: 297-324.
Vaessen HAMG, Jekel AA, & Wilbers AAMM (1988) Dietary intake of
polycyclic aromatic hydrocarbons. Toxicol Environ Chem, 16: 281-294.
Vähäkangas K, Pyy L, & Yrjänheikki E (1992) Assessment of PAH-exposure
among coke oven workers. Pharmacogenetics, 2: 304-308
Vainio H, Uotila P, Hartiala J, & Pelkonen O (1976) The fate of
intratracheally installed benzo [a]pyrene in the isolated perfused
rat lung of both control and 20-methylcholanthrene pretreated rats.
Res Commun Chem Pathol Pharmacol, 13: 259-271.
Vainiotalo S & Matveinen K (1993) Cooking fumes as a hygienic problem
in the food and catering industries. Am Ind Hyg Assoc J, 54: 376-382.
Vaishnav DD & Babeu L (1987) Comparison of occurrence and rates of
chemical biodegradation in natural waters. Bull Environ Contam
Toxicol, 39: 237-244.
Valaes T, Doxiadis SA, & Fessas P (1963) Acute hemolysis due to
naphthalene inhalation. J. Pediatr, 63: 904-915.
Valencia R & Houtchens K (1981) Mutagenic acitivity of 10 coded
compounds in the Drosophila sex-linked recessive lethal test. In: De
Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens. Report of the international collaborative programme.
Amsterdam, Elsevier North-Holland, pp 652-659 (Progress in Mutation
Research, Volume 1).
Valerio F, Bottino P, Ugolini D, Cimberle MR, Tozzi GA, & Frigerio A
(1984) Chemical and photochemical degradation of polycyclic aromatic
hydrocarbons in the atmosphere. Sci Total Environ, 40: 169-188.
Valerio F, Antolini E, & Lazzarotto A (1987) A model to evaluate
half-lifes of PAHs naturally occurring on airborne particulate. Int J
Environ Anal Chem, 28: 185-196.
Valerio F, Pala M, Brescianini C, Lazarrotto A, & Balducci D (1991)
Effect of sunlight and temperature on concentration of pyrene and
benzo [a]pyrene adsorbed on airborne particulate. Toxicol Environ
Chem, 31/32: 113-118.
Valerio F, Brescianini C, Pala M, Lazzarotto A, Balducci D, & Vincenzo
F (1992) Sources and atmospheric concentrations of polycyclic aromatic
hydrocarbons and heavy metals in two Italian towns (Genoa and La
Spezia). Sci Total Environ, 114: 47-57.
Van Brummelen TC & Stuijfzand SC (1993) Effects of benzo [a]pyrene on
survival, growth and energy reserves in terrestrial isopods Oniscus
asellus and Porcellio scaber. Sci Total Environ, Suppl (Part II):
921-930.
Van Cauwenberghe KA (1985) Atmospheric reactions of PAH. In: Bjorseth
A & Ramdahl T ed. Handbook of polycyclic aromatic hydrocarbons, Volume
2. Emission sources and recent progress in analytical chemistry. New
York, Marcel Dekker, pp 351-384.
Van den Heuvel JPP, Clark GC, Thompson CL, McCoy Z, Miller CR, Lucier
GW, & Bell DA (1993) CYP1A1 mRNA levels as a human exposure biomarker:
Use of quantitative polymerase chain reaction to measure CYP1A1
expression in human peripheral blood lymphocytes. Carcinogenesis, 14:
2003-2006.
Van der Oost R, Heida H, Opperhuizen A, & Vermeulen (1991)
Interrelationships between bioaccumulation of organic trace pollutants
PCBs organochlorine pesticides and PAHs and MFO-induction in fish.
Comp Biochem Phys, C100: 43-48.
Van Duuren BL & Goldschmidt BM (1976) Cocarcinogenic and
tumor-promoting agents in tobacco carcinogenesis. J Natl Cancer Inst,
56: 1237-1242.
Van Duuren BL, Sivak A, Segal A, Orris L, & Langseth L (1966) The
tumor-promoting agents of tobacco leaf and tobacco smoke condensate. J
Natl Cancer Inst, 37: 519-526.
Van Duuren BL, Langseth L, Goldschmidt BM & Orris L (1967)
Carcinogenicity of epoxides, lactones, and peroxy compounds. VI.
Structure and carcinogenic activity. J Natl Cancer Inst, 39:
1217-1228.
Van Duuren BL, Sivak A, Langseth L, Goldschmidt BM, & Segal A (1968)
Initiators and promoters in tobacco carcinogenesis. Natl Cancer Inst
Monogr, 28: 173-180.
Van Duuren BL, Sivak A, Goldschmidt BM, Katz C, & Melchionne S (1970)
Initiating activity of aromatic hydrocarbons in two-stage
carcinogenesis. J Natl Cancer Inst, 44: 1167-1173.
Van Heyningen R & Pirie A (1967) The metabolism of naphthalene and its
toxic effect on the eye. Biochem J, 102: 842-852.
Van Noort PCM & Wondergem E (1985a) The isolation of some polynuclear
aromatic hydrocarbons from aqueous samples by means of reversed-phase
concentrator columns. Anal Chim Acta, 172: 335-340.
Van Noort PCM & Wondergem E (1985b) Scavenging of airborne polycyclic
aromatic hydrocarbons by rain. Environ Sci Technol, 19: 1044-1048.
Van Rooij JGM, Bodelier-Bade MM, De Looff AJA, Dijkmans AGP, &
Jongeneelen FJ (1992) Dermal exposure to polycyclic aromatic
hydrocarbons among primary aluminium workers. Med Lav, 83: 519-529.
Van Rooij JGM, Bodelier-Bade MM, & Jongeneelen FJ (1993a) Estimation
of individual dermal and respiratory uptake of polycyclic aromatic
hydrocarbons in 12 coke oven workers. Br J Ind Med, 50: 623-632.
Van Rooij JGM, Van Lieshout EMA, Bodelier-Bade MM, & Jongeneelen FJ
(1993b) Effect of the reduction of skin contamination on the internal
dose of creosote workers exposed to polycyclic aromatic hydrocarbons.
Scand J Work Environ Health, 19: 200-207.
Van Rooij JGM, Veeger MMS, Bodelier-Bade MM, Scheepers PTJ, &
Jongeneelen FJ (1994a) Smoking and dietary intake of polycyclic
aromatic hydrocarbons as sources of interindividual variability in the
baseline excretion of 1-hydroxypyrene in urine. Int Arch Occup Environ
Health, 66: 55-65.
Van Rooij JGM, Bodelier-Bade MM, Hopmans PMJ, & Jongeneelen FJ (1994b)
Reduction of urinary 1-hydroxypyrene excretion in coke-oven workers
exposed to polycyclic aromatic hydrocarbons due to improved hygienic
skin protective measures. Ann Occup Hyg, 38: 247-256.
Van Straalen NM & Verweij RA (1991) Effects of benzo [a]pyrene on
food assimilation and growth efficiency in Porcellio scaber
(Isopoda). Bull Environ Contam Toxicol, 46: 134-140.
Van Vaeck L, Broddin G, & Van Cauwenberghe K (1980) On the relevance
of air pollution measurements of aliphatic and polyaromatic
hydrocarbons in ambient particulate matter. Biomed Mass Spectrom, 7:
473-483.
Van Vaeck L, Van Cauwenberghe K, & Janssens J (1984) The gas-particle
distribution of organic aerosol constituents: Measurement of the
volatilisation artefact in hi-vol cascade impactor sampling. Atmos
Environ, 18: 417-430.
Varanasi U, Reichert WL, Stein JE, Brown DW, & Sanborn HR (1985)
Bioavailability and biotransformation of aromatic hydrocarbons in
benthic organisms exposed to sediment from an urban estuary. Environ
Sci Technol, 19: 836-841.
Varanasi U, Stein JE, Reichert WL, Tilbury KL, Krahn MM, & Chan SL
(1992) Chlorinated and aromatic hydrocarbons in bottom sediments, fish
and marine mammals in US coastal waters: Laboratory and field studies
of metabolism and accumulation. In: Walker CH & Livingstone DR ed.
Persistent pollutants in marine ecosystems. Oxford, Pergamon Press, pp
82-115.
Vassilaros DL, Stoker PW, Booth GM, & Lee ML (1982) Capillary gas
chromatographic determination of polycyclic aromatic compounds in
vertebrate fish tissue. Anal Chem, 54: 106-112.
Vaught J & Bresnick E (1976) Binding of polycyclic hydrocarbons to
nuclear components in vitro. Biochem Biophys Res Commun, 69:
587-591.
Vaught JB, Gurtoo HL, Paigen B, Minowada J, & Sartori P (1978)
Comparison of benzo [a]pyrene metabolism by human peripheral blood
lymphocytes and monocytes. Cancer Lett, 5: 261-268.
Veldre IA & Itra AR (1991) PAH in the water medium of the Estonian SSR
and their monitoring. In: Cooke M, Loening K, & Merritt J ed.
Polynuclear aromatic hydrocarbons: Measurement, means, and metabolism.
Columbus, Ohio, Battelle Press, pp 939-948.
Venier P, Clonfero E, Cottica D, Gava C, Zordan M, Pozzoli L, & Levis
AG (1985) Mutagenic activity and polycyclic aromatic hydrocarbon level
in urine of workers exposed to coal tar pitch volatiles in an anode
plant. Carcinogenesis, 6: 749-752.
Verma DK, Julian JA, Roberts RS, Muir DC, Jadon N, & Shaw DS (1992)
Polycyclic aromatic hydrocarbons (PAHs): A possible cause of lung
cancer mortality among nickel/copper smelter and refinery workers. Am
Ind Hyg Assoc J, 53: 317-324.
Vermorken AJM, Goos CMAA, Roelofs HMJ, Henderson PT, & Bloemendal H
(1979) Metabolism of benzo [a]pyrene in isolated human scalp hair
follicles. Toxicology, 14: 109-116.
Verschueren K (1983) Handbook of environmental data on organic
chemicals, 2nd Ed. New York, Van Nostrand Reinhold Co, pp 460, 760,
968.
Viau C & Vyskocil A (1995) Patterns of 1-hydroxypyrene excretion in
volunteers exposed to pyrene by the dermal route. Sci Total Environ,
163: 187-190.
Viau C, Vyskoçil A, Tremblay C, & Morissette L (1993) Urinary
excretion of 1-hydroxypyrene in workers exposed to polycyclic aromatic
hydrocarbon mixtures. J Occup Med Toxicol, 2: 267-276.
Viau C, Carrier G, Vyskoçil A, & Dodd C (1995) Urinary excretion
kinetics of 1-hydroxypyrene in volunteers exposed to pyrene by the
oral and dermal route. Sci Total Environ, 163: 179-186.
Viras LG, Siskos PA, & Stephanou E (1987) Determination of polycyclic
aromatic hydrocarbons in Athens atmosphere. Int J Environ Anal Chem,
28: 71-85.
Vo-Dinh T (1989) Chemical analysis of polycyclic aromatic compounds.
New York, John Wiley & Sons, 487 pp.
Vo-Dinh T, Bruewer TJ, Colovos GC, Wagner TJ, & Jungers RH (1984)
Field evaluation of a cost-effective screening procedure for
polynuclear aromatic pollutants in ambient air samples. Environ Sci
Technol, 18: 477-482.
Vogel EW, Zijlstra JA, & Blijleven WGH (1983) Mutagenic activity of
selected aromatic amines and polycyclic hydrobarbons in
Drosophila melanogaster. Mutat Res, 107: 53-77.
Vogt NB, Brakstad F, Thrane K, Nordenson S, Krane J, Aamot E, Kolset
K, Esbensen K, & Steinnes E (1987) Polycyclic aromatic hydrocarbons in
soil and air: Statistical analysis and classification by the SIMCA
method. Environ Sci Technol, 21: 35-44.
Volkswagen AG (1989) Unregulated motor vehicle exhaust gas components.
Wolfsburg, Research and Development, 128 pp.
Von Boente L (1955) [Role of coronene in the products of high-pressure
hydrogenation.] Brennstoff-Chemie, 36: 210-214 (in German).
Von Meyerinck (1995)
de Vos (1980)
de Vos RH, Van Dokkum W, Schouten A, & De Jong-Berkhout P (1990)
Polycyclic aromatic hydrocarbons in Dutch total diet samples
(1984-1986). Food Chem Toxicol, 28:
Vreuls JJ, De Jong GJ, & Brinkman UAT (1991) On-line coupling of
liquid chromatography, capillary gas chromatography and mass
spectrometry for the determination and identification of polycyclic
aromatic hydrocarbons in vegetable oils. Chromatographia, 31: 113-118.
Vu Duc T & Huynh CK (1981) [Organic micropolluants in water.
Preliminary results on polycyclic aromatic haloforms and
hydrocarbons.] Méd Soc Prév, 26: 315-316 (in French).
Vu Duc T & Huynh CK (1991) Photodecomposition rates of adsorbed PAH
and mutagenic activity of irradiated synthetic mixtures. In: Cooke M,
Loening K, & Merritt J ed. Polynuclear aromatic hydrocarabons:
Measurement, means and metabolism. Columbus, Ohio, Battelle Press, pp
979-994.
Wakeham SG, Schaffner C, & Giger W (1980a) Polycyclic aromatic
hydrocarbons in recent lake sediments. I. Compounds having
anthropogenic origins. Geochim Cosmochim Acta, 44: 403-413.
Wakeham SG, Schaffner C, & Giger W (1980b) Diagenetic polycyclic
aromatic hydrocarbons in recent sediments: Structural information
obtained by high performance liquid chromatography. Phys Chem Earth,
12: 353-363.
Wakeham SG, Davis AC, & Karas J (1983) Mesocosm experiments to
determine the fate and persistence of volatile organic compounds in
coastal seawater. Environ Sci Technol, 17: 611-617.
Waldman JM, Lioy PJ, Greenberg A, & Butler JP (1991) Analysis of human
exposure to benzo(a)pyrene via inhalation and food ingestion in the
Total Human Environment Exposure Study (THEES). J Exposure Anal
Environ Epidemiol, 1: 193-225.
Walker JD & Colwell RR (1976) Measuring the potential activity of
hydrocarbon-degrading bacteria. Appl Environ Microbiol, 31: 189-197.
Wall KL, Gao W, Te Koppele JM, Kwei GY, Kauffman FC, & Thurman RG
(1991) The liver plays a central role in the mechanism of chemical
carcinogenesis due to polycyclic aromatic hydrocarbons.
Carcinogenesis, 12: 783-786.
Waller RE (1981) Trends in lung cancer in London in relation to
exposure to diesel fumes. Environ Int, 5: 479-483.
Waller RE, Hampton L, & Lawther PJ (1985) A further study of air
pollution in diesel bus garages. Br J Ind Med, 42: 824-830.
Walter U, Beyer M, Klein J, & Rehm HJ (1990) Biodegradation of
polycyclic aromatic hydrocarbons by a bacterial mixed culture.
Biotechnol Conf, 4: 489-492.
Walters RW & Luthy RG (1981) Physicochemical behavior of PAH in coal
conversion liquid effluents. In: Cooke M & Dennis AJ ed. Polynuclear
aromatic hydrocarbons: Chemical analysis and biological fate.
Columbus, Ohio, Battelle Press, pp 539-550.
Walters RW & Luthy RG (1984) Liquid/suspended solid phase partitioning
of polycyclic aromatic hydrocarbons in coal coking wastewaters. Water
Res, 18: 795-809.
Walton B (1980) Influence of route of entry on toxicity of polycyclic
aromatic hydrocarbons to the cricket (Acheta
domesticus). Bull Environ Contam Toxicol, 25: 289-293.
Wang DT & Meresz O (1982) Occurrence and potential uptake of
polynuclear aromatic hydrocarbons of highway traffic origin by
proximally grown food crops. In: Cooke M, Dennis AJ & Fisher GL ed.
Polynuclear aromatic hydrocarbons: Physical and biological chemistry.
Columbus, Ohio, Battelle Press, pp 885-896.
Wangenheim J & Bolcsfoldi G (1988) Mouse lymphoma L5178Y thymidine
kinase locus assay of 50 compounds. Mutagenesis, 3: 193-205.
Waravdekar SS & Ranadive KJ (1958) Biologic testing of
3,4,9,10-dibenzpyrene. J Natl Cancer Inst, 21: 1151-1159.
Ward EC, Murray MJ, Lauer LD, House RV, Irons R, & Dean JH (1984)
Immunosuppres-sion following 7,12-dimethylbenz [a]anthracene exposure
in B6C3F1 mice. I. Effects on humoral immunity and host resistance.
Toxicol Appl Pharmacol, 75: 299-308.
Ward EC, Murray MJ, Lauer LD, House RV, & Dean JH (1986) Persistent
suppression of humoral and cell-mediated immunity in mice following
exposure to the polycyclic aromatic hydrocarbon
7,12-dimethylbenz [a]anthracene. Int J Immunopharmacol, 8: 13-22.
Warman K (1985) PAH emissions from coal-fired plants. In: Bjorseth A &
Ramdahl T ed. Handbook of polycyclic aromatic hydrocarbons, Volume 2:
Emission sources and recent progress in analytical chemistry. New
York, Marcel Dekker, pp 21-59.
Warren DL, Brown DR Jr, & Buckpitt AR (1982) Evidence for cytochrome
P450 mediated metabolism in the bronchiolar damage by naphthalene.
Chem-Biol Interactions, 40: 287-303.
Warshawsky D & Barkley W (1987) Comparative carcinogenic potencies of
7H-dibenzo [c,g]carbazole, dibenz [a,j]acridine and benzo [a]pyrene
in mouse skin. Cancer Lett, 37: 337-344.
Warshawsky D, Barkley W, Miller ML, LaDow K, & Andringa A (1994)
Carcinogenicity of 7H-dibenzo(c,g)carbazole, dibenz(a,j)acridine and
benzo(a)pyene in mouse skin and liver following topical application.
Toxicology, 93: 135-149.
Warshawsky D, Barkley W, Miller ML, LaDow K, & Andringa A (1995)
Erratum to 'Carcinogenicity of 7H-dibenz(a)pyrene in mouse skin and
liver following topical application' (Toxicology 93 (1994) 135-149).
Toxicology, 96: 239-240.
Wattenberg LW (1978) Inhibitors of chemical carcinogenesis. Adv Cancer
Res, 26: 197-226.
Wattenberg LW & Leong JL (1970) Inhibition of the carcinogenic action
of benzo(a)pyrene by flavones. Cancer Res, 30: 1922-1925.
Weaver NK & Gibson RL (1979) The US oil shale industry: A health
perspective. Am Ind Hyg Assoc J, 40: 460-467.
Wehry EL (1983) Optical spectrometric techniques for determination of
polycyclic aromatic hydrocarbons. In: Bjorseth A ed. Handbook of
polycyclic aromatic hydrocarbons. New York, Marcel Dekker, pp 323-396.
Weigert F & Mottram JC (1946) The biochemistry of benzpyrene. II. The
course of its metabolism and the chemical nature of the metabolites.
Cancer Res, 6: 109-120.
Weinstein IB, Jeffrey AM, Jennette KW, Blobstein SH, Harvey RG, Harris
C, Autrup H, Kasai H, & Nakanishi K (1976) Benzo [a]pyrene diol
epoxides as intermediates in nucleic acid binding in vitro
and in vivo. Science, 193: 592-595.
Weinstein D, Katz ML, & Kazmer S (1977) Chromosomal effects of
carcinogens and noncarcinogens on WI-38 after short term exposures
with and without metabolic acitivation. Mutat Res, 46: 297-304.
Weinstein IB, Jeffrey AM, Leffler S, Pulkrabek P, Yamasaki H, &
Grunberger D (1978) Interactions between polycyclic aromatic
hydrocarbons and cellular macromolecules. In: Gelboin HV & Ts'o POP
ed. Polycyclic hydrocarbons and cancer, Volume 2. New York, Academic
Press, pp 3-36.
Welch RM, Gommi B, Alvares AP, & Conney AH (1972) Effect of
enzyme-induction on the metabolsim of benzo(a)pyrene and
3-methyl-4-monomethylaminoazobenzene in the pregnant and fetal rat.
Cancer Res, 32: 973-978.
Wells PG, Wilson B, & Lubek BM (1989) In vivo murine studies on the
biochemical mechanism of naphthalene cataractogenesis. Toxicol Appl
Pharmacol, 99: 466-473.
Wenclawiak B, Rathmann C, & Teuber A (1992) Supercritical-fluid
extraction of soil samples and determination of polycyclic aromatic
hydrocarbons (PAHs) by HPLC. Fresenius' J Anal Chem, 344: 497-500.
Wenzel-Hartung R, Brune H, Grimmer G, Germann P, Timm J, & Wosniok W
(1990) Evaluation of the carcinogenic potency of four environmental
polycyclic aromatic compounds following intrapulmonary application in
rats. Exp Pathol, 40: 221-227.
West WR, Smith PA, Stoker PW, Booth GM, Smith-Oliver T, Butterworth
BE, & Lee ML (1985) Analysis and genotoxicity of a PAC-polluted river
sediment. In: Cooke M & Dennis AJ ed. Polynuclear aromatic
hydrocarbons: Mechanisms, methods, and metabolism. Columbus, Ohio,
Battelle Press, pp 1395-1411.
West WR, Wise SA, Campbell RM, Bartle KD & Lee ML (1986) The analysis
of polycyclic hydrocarbon minerals curtisite and idrialite by high
resolution gas and liquid chromatographic techniques. In: Cooke M &
Dennis AJ ed. Polynuclear aromatic hydrocarbons: Chemistry,
characterization and carcinogenesis. Columbus, Battelle Press, pp 995-
1009.
Wester RC, Maibach HI, Bucks DAW, Sedik L, Melendres J, Liao C, &
DiZio S (1990) Percutaneous absorption of [14C] DDT and [14C]
benzo [a]pyrene from soil. Fundam Appl Toxicol, 15: 510-516.
Westerholm R, Alsberg T, Strandell M, Frommelin Å, Grigoriadis V,
Hantzaridou A & Maitra G (1986) Chemical analysis and biological
testing of emissions from a heavy duty diesel truck with and without
two different particulate traps. Warrendale, Pennsylvania, Society of
Automotive Engineers, pp 74-82 (SAE Technical Paper Series No.
860014).
Westerholm R, Stenberg U, & Alsberg T (1988) Some aspects of the
distribution of polycyclic aromatic hydroarbons (PAH) between
particles and gas phase from diluted gasoline exhausts generated with
the use of a dilution tunnel, and its validity for measurement in
ambient air. Atmos Environ, 22: 1005-1010.
Westerholm R, Hang L, Egebäck K-E, & Grägg K (1989) Exhaust emission
reduction from a heavy duty diesel truck, using a catalyst and a
particulate trap. Fuel, 68: 856-860.
Westerholm RN, Almen J, Li H, Rannug JU, Egebäck K-E, & Grägg K (1991)
Chemical and biological characterization of particulate-,
semivolatile-, and gas-phase-associated compounds: A comparison of
three different semivolatile-phase samplers. Environ Sci Technol, 25:
332-338.
Weston A (1993) Physical methods for the detection of carcinogen-DNA
adducts in humans. Mutat Res, 288: 19-29.
Weston A, Grover PL, & Sims P (1983) Metabolic activation of
benzo [a]pyrene in human skin maintained in short-term organ culture.
Chem-Biol Interactions, 45: 359-371.
Weston A, Bowman ED, Shields PG, Trivers GE, Poirier MC, Santella RM,
& Manchester DK (1993) Detection of polycyclic aromatic hycrocarbon-
DNA adducts in human lung. Environ Health Perspectives, 99: 257-259.
Weyand EH & Bevan DR (1986) Benzo(a)pyrene disposition, and metabolism
in rats following intratracheal instillation. Cancer Res, 46: 5655-
5661.
Weyand EH & Bevan DR (1987) Species differences in disposition of
benzo [a]pyrene. Drug Metab Disposition, 15: 442-448.
Weyand EH & LaVoie EJ (1988) Comparison of PAH DNA adduct formation
and tumor initiating activity in newborn mice. Proc Am Assoc Cancer
Res, 29: 98.
Weyand EH, Rice JE, Hussain N, & LaVoie EJ (1987) Detection of DNA
adducts of tumorigenic nonalternant polycyclic aromatic hydrocarbons
by 32P-postlabelling. Proc Am Assoc Cancer Res, 28: 102.
Weyand EH, Patel S, LaVoie EJ, Cho B, & Harvey RG (1990) Relative
tumor initiating activity of benzo [a]fluoranthene,
benzo [b]fluoranthene, naphtho[1,2- b]fluoranthene and
naphtho[2,1- a]fluoranthene on mouse skin. Cancer Lett, 52: 229-233.
Weyand EH, Bryla P, Wu Y, He Z-M, & LaVoie EJ (1993) Detection of the
major DNA adducts of benzo(j)fluoranthene in mouse skin: Nonclassical
dihydrodiol epoxides. Chem Res Toxicol, 6: 117-124.
White JG (1948) The crystal structure of 1:12-benzperylene: A
quantitatve X-ray investigation. J Chem Soc, 1: 1398-1408.
White CM (1986) Prediction of the boiling point, heat of vaporization,
and vapor pressure at various temperatures for polycyclic aromatic
hydrocarbons. J Chem Eng Data, 31: 198-203.
White KL & Holsapple MP (1984) Direct suppression of in vitro
antibody production by mouse spleen cells by the carcinogen
benzo(a)pyrene but not by the noncarcinogenic congener benzo(e)pyrene.
Cancer Res, 44: 3388-3393.
White J & White A (1939) Inhibition of growth of the rat by oral
administration of methylcholanthrene, benzpyrene, or pyrene and the
effects of various dietary supplements. J Biol Chem, 131: 149-161.
White KL, Lysy HH, & Holsapple MP (1985) Immunosuppression by
polycyclic aromatic hydrocarbons: A structure-activity relationship in
B6C3F1 and DBA/2 mice. Immunopharmacology, 9: 155-164.
Whitlock JP (1979) The conformation of the chromatin core particle is
ionic strength dependent. J Biol Chem, 254: 5684-5689.
Whitman LJ & Miller RJ (1982) The phototactic behavior of
Daphnia magna as an indicator of chronic toxicity. Proc Oklahoma
Acad Sci, 62: 22-33.
WHO (1971) International drinking-water standards, 3rd Ed, p 55.
WHO (1984) Guidelines for drinking-water quality, Volume 1:
Recommendations, pp 66-68; Volume 2: Health criteria and other
supporting information. Geneva, pp 185-186.
WHO (1987) Polynuclear aromatic hydrocarbons (PAH). In: Air quality
guidelines for Europe. Copenhagen, Regional Office for Europe, pp 105-
117 (WHO Regional Publications, European Series, No. 23).
WHO (1988) Emissions of heavy metals and PAH from municipal solid
waste incinerators: Control technology and health impact. Copenhagen,
Regional Office for Europe, 67 pp (Environmental Health Series, No
32WHO/FADL).
WHO (1991) Evaluation of certain food additives and contaminants.
Thirty-seventh report of the Joint FAO/WHO Expert Committee on Food
Additives. Geneva, pp 27-29 (WHO Technical Report Series 806).
WHO (1996) Guidelines for drinking-water quality, Volume 2: Health
criteria and other supporting information. Geneva, pp 495-505.
Whong WZ, Stewart JD, Cutler D, & Ong T (1992) Comparative study of
DNA adduct formation and cytogenic effects of two constituents in coke
oven emissions with an in vivo rat lung cell system. Environ Mol
Mutag, 19 (Suppl 20): 70.
Whong WZ, Stewart DJ, Cutler D, & Ong T (1994) Induction of in
vivo DNA adducts by 4 industrial by-products in the rat lung-cell
system. Mutat Res, 312:165-172.
Wickström K, Pyysalo H, Plaami-Heikkilä S, & Tuominen J (1986)
Polycyclic aromatic compounds (PAC) in leaf lettuce. Z
Lebensmittelunters Forsch, 183: 182-185.
Wielgosz SM, Brauze D, & Pawlak AL (1991) Ah locus-associated
differences in induction of sister-chromatid exchanges and in DNA
adducts by benzo [a]pyrene in mice. Mutat Res, 246: 129-137.
Wienecke J, Kruse H, & Wassermann O (1992) Organic compounds in the
flue gas from a power station with circulating fluid bed combustion of
coal. Chemosphere, 25: 1889-1895.
Wild SR & Jones KC (1993) Biological and abiotic losses of polynuclear
aromatic hydrocarbons (PAHs) from soils freshly amended with sewage
sludge. Environ Toxicol Chem, 12: 5-12.
Wild SR, McGrath SP, & Jones KC (1990) The polynuclear aromatic
hydrocarbon (PAH) content of archived sewage sludges. Chemosphere, 20:
703-716.
Wild SR, Berrow ML, & Jones KC (1991) The persistence of polynuclear
aromatic hydrocarbons (PAH's) in sewage sludge amended agricultural
soils. Environ Pollut, 72: 141-157.
Wild SR, Mitchell DJ, Yelland CM, & Jones KC (1992) Arrested municipal
solid waste incinerator fly ash as a source of polynuclear aromatic
hydrocarbons (PAHs) to the environment. Waste Manage Res, 10: 99-111.
Willems MI, Roggeband R, Baan RA, Wilmer JWGM, De Raat WK, & Lohman
PHM (1991) Monitoring the exposure of rats to benzo [a]pyrene by the
determination of mutagenic acitivity in excreta, chromosome
aberrations and sister chromatid exchanges in peripheral blood cells,
and DNA adducts in peripheral blood lymphocytes and liver.
Mutagenesis, 6: 151-158.
Williams RT (1959) Detoxication mechanisms. The metabolism and
detoxication of drugs, toxic substances and other organic compounds,
2nd Ed. London, Chapman & Hall, 768 pp.
Williams GM (1977) Detection of chemical carcinogens by unscheduled
DNA synthesis in rat liver primary cell cultures. Cancer Res, 37:
1845-1851.
Williams GM, Laspia MF & Dunkel VC (1982) Reliability of the
hepatocyte primary culture / DNA repair test in testing of coded
carcinogens and noncarcinogens. Mutat Res, 97: 359-370.
Williams PT, Abbass MK, Andrews GE, & Bartle KD (1989) Diesel
particulate emissions: The role of unburned fuel. Combust Flame, 75:
1-24.
Wilson NK & Chuang JC (1991) Indoor air levels of polynuclear aromatic
hydrocarbons and related compounds in an eight-home pilot study. In:
Cooke M, Loening K, & Merritt J ed. Polynuclear aromatic hydrocarbons:
Measurements, means and metabolism. Columbus, Ohio, Battelle Press, pp
1053-1064.
Wilson NK, Chuang JC, & Kuhlman MR (1991) Sampling polycyclic aromatic
hydrocarbons and related semivolatile organic compounds in indoor air.
Indoor Air, 4: 513-521.
Windsor JG Jr & Hites RA (1978) Polycyclic aromatic hydrocarbons in
Gulf of Maine sediments and Nova Scotia soils. Geochim Cosmochim Acta,
41: 27-33.
Winker N, Weniger P, Klein W, Ott E, Kocsis F, Schoket B, & Korpert K
(1995) Detection of polycyclic aromatic hydrocarbon exposure damage
using different methods in laboratory animals. J Appl Toxicol, 15: 59-
62.
Winkler DL, Duncan KL, Hose JE, & Puffer HW (1983) Effects of
benzo(a)pyrene on the early development of California grunion,
Leuresthes tenuis (Pisces, Atherinidae). Fish Bull, 81: 473-481.
Wise SA (1983) High-performance liquid chromatography for the
determination of polycyclic aromatic hydrocarbons. In: Bjorseth A ed.
Handbook of polycyclic aromatic hydrocarbons, Volume 2: Emission,
sources and recent progress in analytical chemistry. New York, Marcel
Dekker, pp 183-256.
Wise SA (1985) Recent progress in the determination of PAH by high
performance liquid chromatography. In: Bjorseth A & Ramdahl T ed.
Handbook of polycyclic aromatic hydrocarbons, Volume 2. New York,
Marcel Dekker, pp 113-191.
Wise SA, Bonnett WJ, & May WE (1980) Normal- and reverse-phase liquid
chromatographic separations of polycyclic aromatic hydrocarbons. In:
Bjorseth A & Dennis AJ ed. Polynuclear aromatic hydrocarbons:
Chemistry and biological effects. Columbus, Ohio, Battelle Press, pp
791-806.
Wise SA, Bonnett WJ, Guenther FR, & May WE (1981) A relationship
between reversed-phase C18 liquid cromatographic retention and the
shape of polycyclic aromatic hydrocarbons. J Chromatogr Sci, 19: 457-
465.
Wise SA, Sander LC, & May WE (1993) Determination of polycyclic
aromatic hydrocarbons by liquid chromatography. J Chromatogr, 642:
329-349.
Wislocki PG, Buening MK, Levin W, Lehr RE, Thakkes DR, Jerina DM, &
Conney AH (1979) Tumorigenicity of the diastereomeric
benz [a]anthracene 3,4-diol-1,2-epoxides and the (+)-and
(-)-enantiomers of benz [a]anthracene 3,4-dihydrodiol in newborn
mice. J Natl Cancer Inst, 63: 201-204.
Withey JR, Law FCP, & Endrenyi L (1991) Pharmacokinetics and
bioavailability of pyrene in the rat. J. Toxicol Environ Health, 32:
429-447.
Withey JR, Shedden J, Law FPC, & Abedini S (1992) Distribution to the
fetus and major organs of the rat following inhalation exposure to
pyrene. J Appl Toxicol, 12: 223-231.
Withey JR, Law FCP, & Endrenyi L (1993) Percutaneous uptake,
distribution, and excretion of pyrene in rats. J Toxicol Environ
Health, 40: 601-612.
Witte H, Langenohl T, & Offenbächer G (1988) [Investigation of the
entry of organic pollutants into soils and plants through the use of
sewage sludge in agriculture. Part A: Organic pollutant load in sewage
sludge.] Korresp Abwasser, 5: 440-448 (in German).
Wodinsky I, Helinski A, & Kensler CJ (1964) Susceptibility of Syrian
hamsters to induction of fibrosarcomas with a single injection of
3,4,9,10-dibenzpyrene. Nature, 203: 308-309.
Woidich W, Pfannhauser W, Blaicher G, & Tiefenbacher K (1976)
[Analysis of polycyclic aromatic hydrocarbons in drinking and utility
water.] Lebensmittelchem Gerichtl Chem, 30: 141-146 (in German).
Wojciechowski JP, Kaur P & Sabharwal PS (1981) Comparison of metabolic
systems required to activate pro-mutagens/carcinogens
in vitro for sister-chromatid exchange studies. Mutat Res, 88: 89-
97.
Wojdani A & Alfred LJ (1984) Alterations in cell-mediated immune
functions induced in mouse splenic lymphocytes by polycyclic aromatic
hydrocarbons. Cancer Res, 44: 942-945.
Wolfe JM & Bryan WR (1939) Effects induced in pregnant rats by
injection of chemically pure carcinogenic agents. Am J Cancer, 36:
359-368.
Wolff MS, Herbert R, Marcus M, Rivera M, Landrigan PJ, & Andrews LR
(1989) Polycyclic aromatic hydrocarbons (PAH) residues on skin in
relation to air levels among roofers. Arch Environ Health, 44:
157-163.
Wolff RK, Griffith WC, Henderson RF, Hahn FF, Harkema JR, Rebar AH,
Eidson AF, & McClellan RO (1989) Effects of repeated inhalation
exposures to 1-nitropyrene, benzo(a)pyrene, Ga203 particles, and
S02 alone and in combinations of particle clearance, bronchoalveolar
lavage fluid composition, and histopathology. J Toxicol Environ
Health, 27: 123-138.
Wong O, Bailey WJ, & Amsel J (1992) Cancer mortality and incidence in
mastic asphalt workers. Scand J Work Environ Health, 18: 133-135.
Wood SC & Holsapple MP (1993) Effects of
7,12-dimethylbenz [a]anthracene on the superantigen toxic shock
syndrome toxin (TSST-1)-induced proliferation and antibody secretion
by human lymphocytes. Fundam Appl Toxicol, 20: 280-287.
Wood AW, Levin W, Ryan D, Thomas PE, Yagi H, Mah HD, Thakker DR,
Jerina DM, & Conney AH (1977) High mutagenicity of metabolically
activated chrysene 1,2-dihydrodiol: Evidence for bay region activation
of chrysene. Biochem Biophys Res Commun, 78: 847-854.
Wood AW, Levin W, Thomas PE, Ryan D, Karle JM, Yagi H, Jerina DM, &
Conney AH (1978) Metabolic activation of dibenzo(a,h)anthracene and
its dihydrodiols to bacterial mutagens. Cancer Res, 38: 1967-1973.
Wood AW, Chang RL, Levin W, Ryan DE, Thomas PE, Mah HD, Karle JM, Yagi
H, Jerina DM, & Conney AH (1979) Mutagenicity and tumorigenicity of
phenanthrene and chrysene epoxides and diol epoxides. Cancer Res, 39:
4069-4077.
Wood AW, Levin W, Chang RL, Huang M-T, Ryan DE, Thomas PE, Lehr RE,
Kumar S, Koreeda M, & Akagi, H (1980) Mutagenicity and
tumor-initiating activity of cyclopenta(c,d)pyrene and structurally
related compounds. Cancer Res, 40: 642-649.
Wood AW, Chang RL, Levin W, Ryan DE, Thomas PE, Lehr RE, Kumar S,
Sardella DJ, Boger E, Yagi H, Sayer JM, Jerina DM, & Conney AH (1981)
Mutagenicity of the bayregion diol-epoxides and other benzo-ring
derivatives of dibenzo(a,h)pyrene and dibenzo(a,i)pyrene. Cancer Res,
41: 2589-2597.
Wood AW, Chang RL, Levin W, Yagi H, Thakker DR, van Bladeren PJ,
Jerina DM, & Conney AH (1983) Mutagenicity of the enantiomers of the
diastereomeric bay-region benz [a]anthracene 3,4-diol-1,2-epoxides in
bacterial and mammalian cells. Cancer Res, 43: 5821-5825.
Wood AL, Bouchard DC, Brusseau ML, & Rao PSC (1990) Cosolvent effects
on sorption and mobility of organic contaminants in soil. Chemosphere,
21: 575-587.
Woodhead AD, Setlow RB, & Pond V (1982) Effects of polycyclic aromatic
hydrocarbons on the proliferation of ectopic thyroid tissue in
Poecilia formosa, the Amazon molly. J Fish Biol, 20: 455-463.
Wright BW & Smith RD (1989) Capillary supercritical fluid
chromatography methods. In: Vo-Dinh T ed. Chemical analysis of
polycyclic aromatic compounds. New York, John Wiley & Sons, pp
111-149.
Wu R, Jiang Y-M, Ge N-C, Bai S-M, & Qiao S-J (1985) Determination of
trace amounts of organic pollutants in the Yellow River by capillary
column gas chromatography-mass spectrometry. Int J Environ Anal Chem,
22: 115-126.
Wu Z, Hearl FJ, Peng K, McCawley MA, Chen A, Palassis J, Dosemeci M,
Chen J, McLaughlin JK, Rexing SH, & Blot WJ (1992) Occupational
hygiene around the world: Current occupational exposures in Chinese
iron and copper mines. Appl Occup Environ Health, 7: 735-743.
Wynder EL & Hoffmann D (1959a) A study of tobacco carcinogenesis. VII.
The role of higher polycyclic hydrocarbons. Cancer, 12: 1079-1086.
Wynder EL & Hoffmann D (1959b) The carcinogenicity of
benzofluoranthenes. Cancer, 12: 1194-1199.
Wynder EL, Mabuchi K, & Beattie EJ (1970) The epidemiology of lung
cancer. Recent trends. J Am Med Assoc, 213: 2221-2228.
Xu GT, Zigler JS Jr, & Lou MF (1992) The possible mechanism of
naphthalene cataract in rat and its prevention by an aldose reductase
inhibitor (AL01576). Exp Eye Res, 54: 63-72.
Xue B, Lei ZM, Zhao XL, & Yang XH (1991) [Acute immunotoxicity induced
by benzo(a)pyrene in mice.] Zhonggou Yaolixue Dulixue Zazhi, 5: 221-
222 (in Chinese with English abstract).
Yalkowsky SH & Valvani SC (1979) Solubilities and partitioning. 2.
Relationships between aqueous solubilities, partition coefficients,
and molecular surface areas of rigid aromatic hydrocarbons. J Chem Eng
Data, 24: 127-129.
Yamasaki H, Kuwata K, & Miyamoto H (1982) Effects of ambient
temperature on aspects of airborne polycyclic aromatic hydrocarbons.
Environ Sci Technol, 16: 189-194.
Yamazaki H, Imamura E, Kamei S, Yamauchi A, Kakiuchi Y, & Tai HH
(1990) Polycyclic aromatic hydrocarbons affect the calcium ionophore
induced activation of rabbit platelet. Chemosphere, 21: 21-28.
Yang K, Airoldi L, Pastorelli R, Restano J, Guanci M, & Hemminki K
(1996) Aromatic DNA adducts in lymphocytes of humans working at high
and low traffic density areas. Chem-Biol Interactions, 101: 127-136.
Yoshikawa T, Ruhr LP, Flory W, Banton MJ, Giamalva D, Church DF, &
Pryor WA (1987) Toxicity of polycyclic aromatic hydrocarbons. III.
Effects of beta-naphthoflavone pretreatment on hepatotoxicity of
compounds produced in the ozonation or NO2-nitration of phenanthrene
and pyrene in rats. Vet Hum Toxicol, 29: 113-117.
Yoshioka Y, Nagase H, Ose Y, & Sato T (1986) Evaluation of the test
method 'activated sludge, respiration inhibition test' proposed by the
OECD. Ecotoxicol Environ Saf, 12: 209-212.
You L, Wang D, Galati A, Ross JA, Mass MJ, Nelson GB, Wilson KH, Amin
S, Stoner JC, Nesnow S, & Stoner GD (1994) Tumor multiplicity, DNA
adducts and K- ras mutation pattern of 5-methylchrysene in strain A/J
mouse lung. Carcinogenesis, 15: 2613-2618.
Young L (1947) The metabolic conversion of naphthalene to
1:2-dihydronaphthalene-1:2-diol. Biochem J, 41: 417-422.
Young L (1950) The oxidation of polycyclic hydrocarbons in the animal
body. Biochem Soc Symp, 5: 27-39.
Young DR, Gossett RW, Baird RB, Brown DA, Taylor PA, & Miille MJ
(1983) Wastewater inputs and marine bioaccumulation of priority
pollutant organics off southern California. Water Chlorination, 4:
871-884.
Yrjänheikki E, Pyy L, Hakala E, Lapinlampi T, Lisko A, & Vähäkangas K
(1995) Exposure to polycyclic aromatic hydrocarbons in a new coking
plant. Am Ind Hyg Assoc J, 56: 782-787.
Yun C-H, Shimada T, & Guengerich FP (1992) Roles of human liver
cytochrome P4502C and 3A enzymes in the 3-hydroxylation of
benzo (a)pyrene. Cancer Res, 52: 1868-1874.
Zachleder V, Abarzua S, & Wittenburg E (1983) Effects of
3,4-benzopyrene on the course of cell cycle events in the chlorococcal
alga Scenedesmus quadricauda. Planta, 157: 432-440.
Zackheim HS (1964) Comparative cutaneous carcinogenesis in the rat.
Oncologia, 17: 236-246
Zajdela F, Perin-Roussel O, & Saguem S (1987) Marked differences
between mutagenicity in Salmonella and tumour-initiating activities
of dibenzo [a,e]fluoranthene proximate metabolites; initiation
inhibiting activity of norharman. Carcinogenesis, 8: 461-464.
Zander M (1980) [Origins, chemical and physical properties of PAH.]
In: [Air pollution through polycyclic aromatic hydrocarbons:
Registration and evaluation.] Düsseldorf, VDI-Verlag, pp 11-21 (VDI
Report No. 358) (in German).
Zepp RG & Schlotzhauer PF (1979) Photoreactivity of selected aromatic
hydrocarbons in water. In: Jones PW & Leber P ed. Polynuclear aromatic
hydrocarbons. Carcinogenesis and mutagenesis. Ann Arbor, Michigan, Ann
Arbor Science Publishers, pp 141-158.
Zey JN & Stephenson R (1986) NIOSH health hazard evaluation: Roofing
and waterproofing sites, Chicago, Illinois. Cincinnati, Ohio, National
Institute for Occupational Safety and Health, 29 pp (Report No.
HETA-85-416-1742; PB 87-174207).
Zhao Z-H, Quan W-Y, & Tian D (1990) Urinary 1-hydroxypyrene as an
indicator of human exposure to ambient polycylic aromatic hydrocarbons
in a coal-burning environment. Sci Total Environ, 92: 145-154.
Zhao Z-H, Quan WY, & Tian DH (1992) Experiments on the effects of
several factors on the 1-hydroxypyrene level in human urine as an
indicator of exposure to polycyclic aromatic hydrocarbons. Sci Total
Environ, 133: 197-207.
Zhong BZ, Gu ZW, Stewart J, & Ong T (1995) Micronucleus formation
induced by three polycyclic aromatic hydrocarbons in rat bone marrow
and spleen erythrocytes following intratracheal instillation. Mutat
Res, 326: 147-153.
Zijlstra JA & Vogel EW (1984) Mutagenicity of
7,12-dimethylbenz [a]anthracene and some other aromatic mutagens in
Drosophila melanogaster. Mutat Res, 125: 243-261.
Zimmermeyer G, Roge G, & Schabronath J (1991) [Reduction of PAH
emissions: For example, some stationary plants.] In: [Carcinogenic
compounds in the environment: Sources, measurement, risk, reduction.]
Düsseldorf, VDI-Verlag, pp 93-115 (VDI Report No. 888) (in German).
Zinkham WH & Childs B (1958) A defect of glutathione metabolism in
erythrocytes from patients with a naphthalene-induced hemolytic
anemia. Pediatrics, 14: 461-471.
Zuelzer WW & Apt L (1949) Acute hemolytic anemia due to naphthalene
poisoning. J Am Med Assoc, 141: 185-190.
1. RÉSUMÉ
1.1 Choix des composés pour la monographie
Les hydrocarbures aromatiques polycycliques forment un vaste groupe de
composés et c'est par centaines qu'ils peuvent être libérés dans
l'environnement lors de la combustion incomplète ou de la pyrolyse des
matières organiques, constituant ainsi une source importante
d'exposition humaine. L'étude des matrices susceptibles d'être
importantes sur le plan écologique tels que les résidus de combustion
du charbon, les gaz d'échappement des véhicules à moteur, les huiles
lubrifiantes usées et la fumée de tabac, montre que leur activité
cancérogène est essentiellement liée à leur teneur en HAP.
Les HAP se présentent presque toujours sous la forme de mélanges.
Etant donné que leur composition est complexe et qu'elle dépend du
processus qui leur a donné naissance, il n'a pas été possible de
passer en revue tous les mélanges susceptibles de contenir des
hydrocarbures aromatiques polycycliques. C'est pourquoi on a choisi 33
composés (31 composés originaux et 2 dérivés alkylés) en vue d'une
évaluation sur la base des données pertinentes relatives aux points
d'aboutissement toxicologiques retenus ou à l'exposition (Tableau 1).
Toutefois, étant donné que l'on ne disposait d'études épidémiologiques
que pour les mélanges et que ces études sont indispensables pour
l'évaluation du risque, les sections 8 et 10 exposent les résultats
relatifs à des mélanges d'HAP, contrairement au reste de la
monographie.
Nombre d'articles et de mises au point ont été publiées au sujet de la
présence, de la distribution et de la transformation des HAP dans
l'environnement ainsi que de leurs effets toxicologiques et
écotoxicologiques. Sauf indication contraire, seules les références
des 10 à 15 dernières années sont prises en compte dans la
monographie. En revanche, les mises au point consacrées à des études
plus anciennes sont citées à titre de complément d'information.
1.2 Identité, propriétés physiques et chimiques et méthodes d'analyse
On désigne généralement par hydrocarbures aromatiques polycycliques un
vaste ensemble de composés contenant un ou plusieurs noyaux
aromatiques condensés et constitués de carbone et d'hydrogène. Ces
hydrocarbures sont solides à la température ambiante. Ils ont pour
caractéristiques communes d'avoir un point de fusion et un point
d'ébullition élevés, une faible tension de vapeur et d'être très peu
solubles dans l'eau-d'autant moins que leur masse moléculaire est plus
élevée. Ils sont solubles dans de nombreux solvants organiques et très
lipophiles. Chimiquement, ils sont relativement inertes. Les réactions
intéressantes du point de vue de leur devenir dans l'environnement et
des possibilités de pertes au cours des prélèvements d'air sont celles
qui comportent une photodécomposition ou dans lesquelles interviennent
les oxydes d'azote, l'acide nitrique, les oxydes de soufre, l'acide
sulfurique, l'ozone et les radicaux hydroxyles.
Tableau 1. Les hydrocarbures aromatiques polycycliques évalués dans cette monographie
Nom commun Nom CAS Synonymea No d'enregistrement CAS
Acenaphthylene Acenaphthylene 91-20-3
Acenaphthene Acenaphthylene, 1,2-dihydro- 208-96-8
Anthanthrene Dibenzo[def,mno]chrysene 191-26-4
Anthracene Anthracene 120-12-7
Benz[a]anthracene Benz[a]anthracene 1,2-Benzanthracene, 56-55-3
tetraphene
Benzo[a]fluorene 11H-Benzo[a]fluorene 1,2-Benzofluorene 238-84-6
Benzo[b]fluorene 11H-Benzo[b]fluorene 2,3-Benzofluorene 243-17-4
Benzo[b]fluoranthene Benz[e]acephenanthrylene 3,4-Benzofluoranthene 205-99-2
Benzo[ghi]fluoranthene Benzo[ghi]fluoranthene 2,13-Benzofluoranthene 203-12-3
Benzo[j]fluoranthene Benzo[j]fluoranthene 10,11-Benzofluoranthene 205-82-3
Benzo[k]fluoranthene Benzo[k]fluoranthene 11,12-Benzofluoranthene 207-08-9
Benzo[ghi]perylene Benzo[ghi]perylene 1,12-Benzoperylene 191-24-2
Benzo[c]phenanthrene Benzo[c]phenanthrene 3,4-Benzophenanthrene 195-19-7
Benzo[a]pyrene Benzo[a]pyrene 3,4-Benzopyreneb 50-32-8
Benzo[e]pyrene Benzo[e]pyrene 1,2-Benzopyrene 192-97-2
Chrysene Chrysene 1,2-Benzophenanthrene 218-01-9
Coronene Coronene Hexabenzobenzene 191-07-1
Cyclopenta[cd]pyrene Cyclopenta[cd]pyrene Cyclopenteno[cd]pyrene 27208-37-3
Dibenz[a,h]anthracene Dibenz[a,h]anthracene 1,2:5,6-Dibenzanthracene 53-70-3
Dibenzo[a,e]pyrene Naphtho[1,2,3,4-def]chrysene 1,2:4,5-Dibenzopyrene 192-65-4
Dibenzo[a,h]pyrene Dibenzo[b,def]chrysene 3,4:8,9-Dibenzopyrene 189-64-0
Dibenzo[a,i]pyrene Benzo[rst]pentaphene 3,4:9,10-Dibenzopyrene 189-55-9
Dibenzo[a,l]pyrene Dibenzo[def,p]chrysene 1,2:3,4-Dibenzopyrene 191-30-0
Fluoranthene Fluoranthene 206-44-0
Fluorene 9H-Fluorene 86-73-7
Indeno[1,2,3-cd]pyrene Indeno[1,2,3-cd]-pyrene 2,3-o-Phenylenpyrene 193-39-5
5-Methylchrysene Chrysene, 5-methyl- 3697-24-3
1-Methylphenanthrene Phenanthrene, 1-methyl- 832-69-9
Tableau 1. (cont.)
Nom commun Nom CAS Synonymea No d'enregistrement CAS
Naphthalene Naphthalene 91-20-3
Perylene Perylene peri-Dinaphthalene 198-55-0
Phenanthrene Phenanthrene 85-01-8
Pyrene Pyrene Benzo[def]phenanthrene 129-00-0
Triphenylene Triphenylene 9,10-Benzophenanthrene 217-59-4
Des listes assez complètes de synonymes ont également été publiées par le CIRC (1989) et par Loening & Merritt (1990).
a Synonyme commun utilisé dans la littérature
b On le trouve également sous le nom de benzo(def)chrysène.
L'échantillonnage dans l'air ambiant s'effectue par recueil de
matières particulaires sur filtres eu fibre de verre,
polytétrafluoréthylène, ou fibre de quartz, au moyen
d'échantillonneras de grand volume ou d'échantillonneurs passifs.
Comme il y a risque de volatilisation des hydrocarbures de la phase
gazeuse, qui pourraient s'évaporer des filtres lors de
l'échantillonnage, on a l'habitude de les piéger par adsorption sur
mousse de polyuréthane. La variabilité des résultats provient
essentiellement de ce processus d'échantillonnage.
Sur les lieux de travail, les prélèvements d'air se font à faible
débit; les particules sont recueillies sur des filtres de fibre de
verre ou de polytétrafluoréthylène et la phase gazeuse sur résine
Amberlite XAD-2. Les collecteurs de gaz de cheminées sont constitués
de filtres en fibre de verre ou en quartz placés devant un réfrigérant
destiné à retenir le condensant et une cartouche d'adsorbant (en
général, de l'XAD-2). Les gaz d'échappement des véhicules à moteur
sont recueillis au laboratoire, pendant un cycle de fonctionnement
normalisé simulant les conditions réelles. Les émissions sont
recueillies telles quelles ou après dilution dans de l'air froid
filtré.
De nombreuses techniques d'extraction et de purification ont été
décrites. En fonction de la matrice, on peut extraire les HAP au
Soxhlet, par action des ultrasons, par partage liquide-liquide ou,
après dissolution et digestion alcaline, au moyen d'un solvant
sélectif. On utilise aussi l'extraction par fluide supercritique pour
diverses substances solides présentes dans l'environnement.
L'efficacité de l'extraction dépend largement du solvant et nombre de
solvants utilisés par le passé se sont révélés inappropriés. Après
l'extraction, on procède généralement à une purification par
chromatographie sur colonne - d'alumine, de gel de silice ou de
sephadex LH-20- mais aussi par chromatographie sur couche mince.
La recherche et le dosage s'effectuent classiquement par
chromatographie en phase gazeuse avec détection par ionisation de
flamme ou encore par chromatographie en phase liquide à haute
performance avec détection en UV ou fluorescence, généralement en
série. Pour la chromatographie en phase gazeuse, on utilise des
colonnes capillaires en silice fondue, avec des polysiloxanes comme
phase stationnaire (SE-54 et SE-52); pour la chromatographie en phase
liquide, on utilise couramment des colonnes de gel de silice C-18.
Pour confirmer l'identité des pics chromatographiques, on couple
généralement un spectromètre de masse au chromatographie.
Le choix des HAP à doser dépend de l'objectif de la mesure: étude à
visée sanitaire, investigation écotoxicologique ou recherche d'une
source de pollution. La recherche et le dosage de divers groupes de
composés peuvent être demandés au niveau national ou international.
1.3 Sources d'exposition humaine et environnementale
On sait peu de choses sur la production des hydrocarbures aromatiques
polycycliques et sur les processus auxquels ils peuvent être soumis,
mais il est probable que ces activités n'entraînent la libération que
de petites quantités d'hydrocarbures. Ceux que l'on retrouve dans
l'environnement sont principalement utilisés comme intermédiaires dans
la préparation du chlorure de polyvinyle et de divers plastifiants
(naphtalène), de pigments (acénaphtène, pyrène), de colorants
(anthracène, fluoranthène) et de pesticides (phénanthrène).
Les émissions les plus importantes d'hydrocarbures aromatiques
polycycliques sont dues à la combustion incomplète de matières
organiques lors de divers processus industriels ou d'autres activités
humaines, notamment:
- les diverses opérations effectuées sur la houille, le pétrole
brut et le gaz naturel, y compris la cokéfaction, la conversion
de la houille, le raffinage du pétrole et la production de noirs
de carbone, de créosote, de goudron de houille et de bitume;
- la production d'aluminium, de fer et d'acier dans les divers
ateliers et fonderies;
- la génération d'énergie calorifique par les centrales thermiques,
le chauffage des habitations et la cuisine;
- le brûlage des déchets;
- la circulation automobile; et
- la fumée de tabac dispersée dans l'environnement.
Les HAP, notamment ceux qui ont une masse moléculaire élevée,
s'adsorbent sur les particules de matière une fois qu'ils sont libérés
dans l'environnement par la voie atmosphérique. La pénétration dans
l'hydrosphère et la géosphère s'effectue ensuite selon un processus de
dépôt par voie humide ou par voie sèche. La conservation du bois par
traitement au créosote constitue une autre source de pénétration d'HAP
dans l'hydrosphère et les dépôts de déchets contaminés, comme par
exemple les boues d'égout et les cendres volantes, contribuent à
l'introduction de ces composés dans la géosphère. On possède peu de
renseignements sur le passage des HAP dans la biosphère. Des
hydrocarbures aromatiques polycycliques sont présents à l'état naturel
dans la tourbe, le lignite, la houille et le pétrole brut. La plupart
de ceux que l'on trouve dans l'anthracite sont fermement liés à la
structure carbonée et ne peuvent pas en être extraits par lessivage.
On a pu déterminer le passage d'HAP dans l'environnement en mettant en
évidence un profil de concentration caractéristique, mais cela n'a été
possible que dans quelques cas. Le benzo(a)pyrène est fréquemment
utilisé comme indicateur de la présence d'HAP, notamment dans les
études un peu anciennes. En général, les chiffres concernant les
émissions d'HAP ne sont que des estimations basées sur des données
plus ou moins fiables et elles ne donnent qu'une idée approximative de
l'exposition.
Les sources d'HAP les plus importantes sont les suivantes:
La cokéfaction de la houille: les émissions atmosphériques d'HAP
résultant de la cokéfaction de la houille ont sensiblement diminué en
Allemagne au cours des 10 dernières années par suite des améliorations
techniques apportées aux installations industrielles, à la fermeture
des anciennes usines et au recul de la production de coke. On pense
que la situation est à peu près la même dans le reste de l'Europe
occidentale, au Japon, et aux Etats-Unis, sans toutefois disposer de
données à ce sujet.
La production d'aluminium (notamment en raison de l'utilisation
d'anodes spéciales en graphite), de fer et d'acier ainsi que les
liants utilisés en fonderie dans les moules à sable. On ne possède
guère d'informations à ce sujet.
Le chauffage collectif ou individuel: les émissions sont
principalement constituées de phénanthrène, de pyrène et de chrysène.
Les poëles à bois en émettent 25 à 1000 fois plus que les poëles à
charbon et dans les régions où l'on se chauffe surtout au bois, la
majeure partie des émissions atmosphériques d'HAP ont ce mode de
chauffage pour origine, principalement l'hiver. On considère donc que
le chauffage des habitations est une source importante d'hydrocarbures
aromatiques polycycliques dans les pays en développement où l'on brûle
de la biomasse dans des poëles assez rudimentaires.
La cuisine: la combustion incomplète des matières combustibles peut
produire des HAP, de même que le chauffage de l'huile de cuisine et la
cuisson des denrées alimentaires elles-mêmes.
La circulation des véhicules à moteur: les principaux hydrocarbures
rejetés par les véhicules dotés de moteurs à essence sont le
fluoranthène et le pyrène, tandis que le naphtalène et l'acénaphtène
prédominent dans les gaz d'échappement des moteurs diesel. Le
cyclopenta(cd)-pyrène est émis en grande quantité par les moteurs à
essence mais il est juste au-dessus de la limite de détection dans les
gaz d'échappement des moteurs diesel. Le taux d'émission, qui dépend
du composé, du type de véhicule et de l'état de son moteur, de même
que des conditions dans lesquelles l'essai se déroule, varie de
quelques nanogrammes par kilomètre à > 1000 mg/km. La pose d'un
catalyseur réduit de façon spectaculaire les émissions d'HAP par les
moteurs d'automobiles.
Les feux de forêt: Dans les pays où la forêt couvre de vases
étendues de territoire, les feux de forêt peuvent contribuer de façon
importante à l'émission d'HAP.
Centrales thermiques à charbon: Les HAP libérés dans l'atmosphère
par ces centrales sont principalement des composés bi-et tricycliques.
Dans les zones contaminées, la teneur en HAP de l'air ambiant peut
être supérieure à celle qui résulte des émission de cheminées.
Incinération des déchets: les émissions d'HAP dans les gaz de
cheminée des usines d'incinération sont inférieures à 10 mg/m3 dans
un certain nombre de pays.
1.4 Transport, distribution et transformation dans l'environnement
La destinée des HAP, qu'ils soient seuls ou en mélange, dépend d'un
certain nombre de processus de distribution et de transformation. Les
processus de distribution les plus importants sont le partage entre
l'eau et l'air, entre l'eau et les sédiments et entre l'eau et les
organismes vivants.
Comme ces hydrocarbures sont hydrophobes et très peu solubles dans
l'eau, il n'ont qu'une très faible affinité pour la phase aqueuse;
toutefois, bien qu'ils soient libérés dans l'environnement par la voie
atmosphérique, on les retrouve également en concentration importante
dans l'hydrosphère, du fait que leur constante de Henry est faible.
Etant donné que leur affinité est plus grande pour la phase organique
que pour la phase aqueuse, leur coefficient de partage entre les
solvants organiques- comme l'octanol- et l'eau est élevé. Ils ont
également une forte affinité pour les fractions organiques des
sédiments, du sol et des organismes vivants, de sorte qu'ils
s'accumulent dans les organismes aquatiques et sédimentaires ainsi que
dans leur nourriture. On ne connaît pas avec certitude l'importance
relative de leur fixation à partir de la nourriture et de l'eau. Chez
la daphnie et les mollusques, il y a corrélation positive entre
l'accumulation d'HAP provenant de l'eau et le coefficient de partage
entre l'octanol et l'eau (Kow). Par contre, chez les poissons et les
algues qui sont capables de métaboliser ces hydrocarbures, il n'y a
pas de corrélation entre la concentration des différents HAP et le
Kow.
Le phénomène de bioamplification, c'est-à-dire la concentration d'une
substance dans l'organisme d'animaux à chaque niveau trophique
successif de la chaîne alimentaire, n'a pas été observé en milieu
aquatique et il ne semble pas qu'il puisse se produire, car la plupart
des organismes sont tout à fait à même de métaboliser les HAP. Ce sont
les organismes qui occupent les niveaux trophiques les plus élevés qui
possèdent la plus grande capacité de biotransformation.
La dégradation des HAP s'opère par photodécomposition, par
biodégradation microbienne et, chez les organismes supérieurs, par
métabolisation. Cette dernière voie de transformation n'a guère
d'influence sur la destinée globale des HAP dans l'environnement, mais
elle joue néanmoins un rôle biologique important du fait que des
métabolites cancérogènes sont susceptibles de se former. Comme les HAP
sont chimiquement stables et dépourvus de groupements réactifs,
l'hydrolyse n'intervient pas dans leur décomposition. Il n'existe
guère d'épreuves classiques pour l'étude de la biodégradation des HAP.
Celle-ci s'opère généralement en aérobiose, la vitesse du processus
diminuant fortement avec le nombre de cycles aromatiques. En
anaérobiose, la dégradation est beaucoup plus lente.
Dans l'air et dans l'eau, les HAP subissent une photo-oxydation en
présence de radicaux ou molécules sensibilisateurs comme OH, NO3 ou
O3. Au laboratoire, le temps de demi-réaction avec les radicaux OH
présents dans l'air est d'environ 1 jour; en revanche la constante de
vitesse est généralement beaucoup plus faible dans le cas des
réactions avec NO3 et O3. En principe, l'adsorption des HAP de
masse moléculaire élevée sur les particules carbonées devrait
stabiliser leur réaction avec les radicaux OH. La réaction des HAP
possédant 2 à 4 cycles avec NO3, réaction qui a lieu principalement
en phase gazeuse, conduit à la nitration de ces hydrocarbures, c'est-
à-dire à la formation de produits notoirement cancérogènes. Pour
certains d'entre eux, la photo-oxydation semble être plus rapide dans
l'eau que dans l'air. Les calculs basés sur la physico-chimie et la
biodégradabilité de ces hydrocarbures montrent que ceux qui possèdent
quatre cycles aromatiques ou davantage persistent dans
l'environnement.
1.5 Concentrations dans l'environnement et exposition humaine
Les HAP sont présent dans tout l'environnement et de nombreuses études
ont permis d'en mettre un certain nombre en évidence dans divers
compartiments.
1.5.1 Air
La concentration des HAP a tendance à être environ 10 fois plus élevée
l'hiver que l'été. En hiver, la source principale d'hydrocarbures
aromatiques polycyclique est le chauffage des habitations, alors qu'en
été, ce sont les gaz d'échappement des véhicules à moteur qui sont les
principaux responsables. Dans l'air de diverses agglomérations, on a
mis en évidence, pour un certain nombre d'hydrocarbures aromatiques
polycycliques, des concentrations moyennes de 1 à 30 ng/m3. Dans
certains grands centres urbains, où la circulation automobile est très
intense et où l'on utilise beaucoup la biomasse pour le chauffage des
habitations, comme Calcutta par exemple, on a trouvé des teneurs
allant jusqu'à 200 ng/m3 pour divers HAP. Sous les tunnels routiers,
on a mesuré des concentrations de 1 à 50 ng/m3. Le
cyclopenta(cd)pyrène et le pyrène étaient présents à des
concentrations atteignant 100 ng/m3. Dans une station de métro, on a
mesuré des concentrations allant jusqu'à 20 ng/m3. A proximité de
sources de pollution d'origine industrielle, la concentration moyenne
des divers HAP s'étalait de 1 à 10 ng/m3. La concentration du
phénanthrène pouvait atteindre environ 310 ng/m3.
La concentration de fond des HAP est d'au moins deux ordres de
grandeur plus faible qu'à proximité de sources telles que les
véhicules à moteur. A titre d'exemple, à 1100 m d'altitude, on a
obtenu des valeurs comprises entre 0,004 et 0,03 ng/m3.
1.5.2 Eaux superficielles et précipitations
On pense que la majeure partie des HAP présents dans l'eau y ont été
entraînés par ruissellement ou résultent de retombées atmosphériques
(petites particules) ou encore de l'abrasion de l'asphalte (grosses
particules). Il reste que, pour une étendue d'eau donnée, la majeure
partie des HAP n'a pas toujours la même origine. Eu général, la
plupart des échantillons d'eaux superficielles contiennent divers HAP
à des concentrations pouvant atteindre 50 ng/litre, mais dans des
cours d'eau extrêmement pollués, on a relevé des concentrations allant
jusqu'à 6 000 ng/litre. La teneur des eaux souterraine en HAP se situe
entre 0,02 et 1,8 ng/litre et dans des échantillons d'eau destinée à
la boisson, on a trouvé des valeurs du même ordre de grandeur. Les HAP
présents dans l'eau de boisson ont principalement pour origine le
revêtement d'asphalte des réservoirs et des canalisations.
Dans l'eau de pluie, on relève des concentrations comprises entre 10
et 200 ng/litre, et des teneurs allant jusqu'à 1000 ng/litre ont été
mesurées dans la neige et le brouillard.
1.5.3 Sédiments
La concentration des HAP dans les sédiments est généralement 10 fois
plus forte que dans les précipitations.
1.5.4 Sol
Les HAP présents dans le sol proviennent principalement des dépôts
atmosphériques, de la carbonisation des végétaux et du dépôt de
particules en suspension dans les effluents ou divers types de
déchets. L'ampleur de la pollution des sols dépend de facteurs tels
que le type de cultures auxquels ils sont soumis, la porosité et la
teneur en substances humiques.
A proximité des sources industrielles, on a trouvé, pour les divers
HAP, des concentrations dans le sol pouvant atteindre 1 g/kg. Pour les
HAP ayant une autre origine, par exemple les gaz d'échappement des
véhicules à moteur, la concentration dans le sol va de 2 à 5 mg/kg.
Dans les zones non polluées, la teneur du sol en HAP se situe entre 5
et 100 µg/kg.
1.5.5 Denrées alimentaires
Les denrées alimentaires crues ou plus généralement, non transformées,
ne contiennent normalement pas de grandes quantités d'HAP, mais il
peut s'en former lorsqu'on fait rôtir, griller, frire ou que l'on
prépare d'une manière ou d'une autre, ces produits. Les légumes
peuvent être contaminés par le dépôt de particules aéroportées ou par
le sol sur lequel ils ont poussé. D'après diverses mesures, la
concentration d'un certain nombre d'HAP dans la viande, le poisson,
les produits laitiers, les légumes, les fruits, les céréales et les
produits céréaliers, les pâtisseries et confiseries, les boissons
ainsi que les huiles et graisses animales et végétales, se situe entre
0,01 et 10 µg/kg. On a trouvé des concentrations supérieures à 100
µg/kg dans de la viande fumée et pouvant aller jusqu'à 86 µg/kg dans
du poisson fumé; dans des céréales fumées, les valeurs relevées
atteignaient 160 µg/kg. Dans de l'huile de coco on en a trouvé jusqu'à
460 µg/kg. Dans le lait maternel, les mesures ont donné des valeurs
comprises entre 0,003 et 0,03 µg/kg.
1.5.6 Organismes aquatiques
On sait que les organismes marins absorbent et accumulent les HAP
présents dans l'eau. Le degré de contamination dépend du développement
industriel et urbain ainsi que du trafic maritime dans la zone. Des
concentrations allant jusqu'à 7 mg/kg out été mises en évidence dans
des organismes aquatiques vivant à proximité de points de décharge
d'effluents industriels et la concentration moyenne d'HAP dans
l'organisme d'animaux aquatiques prélevés dans des zones contaminées
s'est révélée comprise entre 10 et 500 µg/kg, avec des pointes à 5
mg/kg.
Chez des animaux aquatiques prélevés dans des zones où la pollution
par des HAP n'avait pas d'origine précise, on a trouvé des valeurs
moyennes de 1 à 100 µg/kg, mais des valeurs supérieures sont
possibles, par exemple 1 mg/kg chez des homards du Canada.
1.5.7 Organismes terrestres
Chez des insectes, on a relevé des concentrations comprises entre 730
et 5 500 µg/kg. La teneur en HAP des déjections de lombrics varie
sensiblement selon le lieu: dans une région fortement industrialisée
de l'est de l'Allemagne, la teneur en benzo(a)pyrène des déjections de
lombrics atteignait 2 mg/kg.
1.5.8 Population générale
Les principales sources d'exposition non professionnelle sont les
suivantes: air pollué, fumée de feux et foyers non couverts, fumée de
tabac dispersée dans l'environnement, denrées alimentaires et eau de
boisson contaminées, utilisation de produits contaminés par des HAP.
On peut trouver dans l'air à l'intérieur des habitations des HAP qui
proviennent du chauffage ou de la présence de fumée de tabac, à des
concentrations moyennes de 1 à 100 ng/m3, avec des pointes à 2300
ng/m3.
On estime que l'apport d'HAP d'origine alimentaire est de 0,10 à 10 µg
par jour et par personne. La quantité totale de benzo(a)pyrène
absorbée quotidiennement avec l'eau de boisson est estimée à 0,0002 µg
par personne. Ce sont les céréales et les produits céréaliers qui
contribuent le plus à cet apport car ils sont un constituant majeur de
la ration alimentaire totale.
1.5.9 Exposition professionnelle
A proximité d'une batterie de fours à coke, on a relevé des teneurs en
benzo(a)pyrène allant de <0,1 à 100-200 µg/m3, avec des pointes à
environ 400 µg/m3. Dans les installations modernes de gazéification
de la houille, la concentration des HAP est en général de < 1 µg/m3
et de 30 µg/m3 au maximum. Des échantillons prélevés individuellement
dans la zone de travail de personnes affectées à divers postes dans
des raffineries de pétrole, ont révélé une exposition comprise entre
2,6 et 470 µg/m3. Dans des échantillons d'air prélevés dans des
ateliers de préparation de bitume, on a obtenu une concentration en
HAP totaux de 0,004 à 50 µg/m3. Lors de travaux d'enrobage de
chaussées, on a relevé sur le site des concentrations atmosphériques
en HAP totaux pouvant atteindre 190 µg/m3, avec une concentration
moyenne de 0,13 µg/m3. Dans une fonderie d'aluminium, la dosimétrie
individuelle indiquait des valeurs de 0,05-9,6 µg/m3 pour les HAP
totaux, mais dans les urines d'ouvriers travaillant dans une unité de
production d'aluminium, on n'en a relevé que de très petites
quantités. Dans une fonderie allemande, les échantillons d'air ambiant
contenaient des HAP à des concentrations pouvant atteindre 5 µg/m3.
Elles étaient respectivement égales à 3-40 µg/m3 dans des mines de
fer et à 4-530 µg/m3 dans des mines de cuivre. Dans les vapeurs de
cuisson d'une usine de produits alimentaires, la concentration en HAP
était comprise entre 0,07 et 26 µg/m3.
1.6 Cinétique et métabolisme
Les HAP sont absorbés au niveau des poumons, des voies digestives et
par la voie percutanée. La vitesse de résorption au niveau pulmonaire
dépend de la nature de l'hydrocarbure, de la granulométrie des
particules sur lesquelles il est adsorbé et de la composition du
substrat adsorbant. Les hydrocarbures adsorbés sur des particules sont
éliminés plus lentement des poumons que les hydrocarbures libres. Chez
les rongeurs, la résorption est rapide dans les voies digestives, mais
les métabolites, excrétés par la voie biliaire, finissent par
retourner dans l'intestin. Des études effectuées sur des rongeurs avec
des mélanges d'HAP ayant subi un postmarquage au 32p ont montré
qu'après absorption par la voie percutanée, les hydrocarbures
parvenaient jusqu'aux poumons où ils se liaient à l'ADN. Chez la
souris, la vitesse d'absorption percutanée dépend de la nature du
composé.
Quelle que soit la voie d'administration, les HAP se répartissent dans
tout l'organisme et on en retrouve dans pratiquement tous les organes
internes, mais plus particulièrement dans ceux qui sont riches en
lipides. Après injection intraveineuse à des rongeurs, les HAP sont
rapidement éliminés du courant sanguin, mais ils sont capables de
traverser la barrière foeto-placentaire et on en a décelé la présence
dans les tissus foetaux.
Les HAP ont un métabolisme complexe. En général, le composé initial
est époxydé puis transformé en phénol, diol ou tétrol qui peut
lui-même se conjuguer à l'acide sulfurique ou à l'acide glucuronique
(sulfuro- ou glucuro-conjugaison) ou encore au glutathion. La plupart
du temps, la métabolisation d'un hydrocarbure aromatique polycyclique
entraîne sa détoxication, mais certains d'entre eux subissent une
activation en composés susceptibles de se lier à l'ADN, principalement
des époxydes-diols, qui sont capables d'amorcer un processus de
cancérisation. Les métabolites et leurs conjugués sont excrétés dans
les urines et les matières fécales, mais les conjugués excrétés dans
la bile peuvent être hydrolysés par les enzymes de la flore
intestinale et être ensuite réabsorbés. On peut déduire des données
disponibles au sujet de la charge totale de l'organisme humain en HAP,
que ces hydrocarbures ne s'accumulent pas dans l'organisme et qu'ils
se renouvellent rapidement. Il faut exclure de cette conclusion les
HAP qui forment des liaisons covalentes avec certain constituants
tissulaires, notamment les acides nucléiques et que les processus de
réparation ne permettent pas d'éliminer.
1.7 Effets sur les mammifères de laboratoire et effets in vitro
La toxicité aiguë des HAP est faible à modérée. Un HAP bien
caractérisé, le naphtalène, a donné des valeurs de la DL50 par voie
orale ou intraveineuse égales à 100-500 mg/kg pc chez la souris et une
DL50 moyenne par voie orale de 2700 mg/kg pc chez le rat. Pour les
autres HAP, on a obtenu des valeurs similaires. Du naphtalène
administré en doses uniques à des souris, des rats et des hamsters, a
provoqué l'apparition d'une nécrose des bronchioles.
Des études à court terme ont révélé des anomalies hématologiques, dues
à une myélotoxicité dans le cas du benzo(a)pyrène et qui, en ce qui
concerne le dibenz(a,h)anthracène, consistaient en modifications
hémolymphatiques. Dans le cas du naphtalène, on a constaté une anémie.
Toutefois, une étude de 7 jours au cours de laquelle on a administré
du naphtalène à des souris par voie orale et intrapéritonéale, a
révélé l'existence d'une tolérance aux effets de cet hydrocarbure. On
n'a que rarement décrit des effets généraux dus à une longue
exposition à des HAP, car l'effet toxicologique retenu dans la plupart
des études correspondantes était la cancérisation. Aux doses où se
déclenche un processus de cancérisation, on observe également des
effets toxiques importants.
En étudiant les effets cutanés indésirables des HAP après application
sur l'épiderme, on a constaté que des hydrocarbures faiblement ou non
cancérogènes comme le pérylène, le benzo(e)pyrène, le phénanthrène, le
pyrène, l'anthracène, l'acénaphtène, le fluorène et le fluoranthène
étaient inactifs, alors que les hydrocarbures cancérogènes comme le
benz(a) anthracène, le dibenz(a,h)-anthracène et le benzo(a)pyrène
provoquaient une hyperkératose. Les vapeurs d'anthracène et de
naphtalène peuvent causer une légère irritation oculaire. Le
benzo(a)pyrène a provoqué une hypersensibilité de contact chez des
cobayes et des souris. Le benz(a)anthracène, le benzo(a)pyrène, le
dibenz(a,h)anthracène et le naphtalène se sont révélés embryotoxiques
chez la souris et le benzo(a)pyrène a également eu des effets
tératogènes et des effets sur la reproduction. On a fait de gros
efforts pour tenter de d'élucider les bases génétiques des effets
embryotoxiques du benzo(a)pyrène. On n'observe de morts foetales et de
malformations que si le système des monooxygénases du cytochrome p450
est inductible, chez la mère (avec migration transplacentaire) ou chez
l'embryon. On ne peut expliquer tous les effets observés par une
prédisposition génétique, mais chez la souris et le lapin, cet
hydrocarbure a une activité transplacentaire qui se traduit par des
adénomes pulmonaires et des papillomes cutanés dans la descendance des
animaux traités. On a également observé une diminution de la fécondité
et la destruction des ovocytes.
On a également procédé à de nombreuses études sur la génotoxicité des
HAP et sur leur aptitude à provoquer une transformation cellulaire. La
plupart des 33 HAP qui font l'objet de cette monographie sont
génotoxiques ou ont des chances de l'être. Les seuls composés pour
lesquels on ait obtenu des résultats négatifs dans toutes les épreuves
sont l'anthracène, le fluorène et le naphtalène. Dans le cas du
phénanthrène et du pyrène, l'irrégularité des résultats ne permet pas
de se prononcer avec certitude sur leur génotoxicité.
Les travaux très complets qui ont été consacrés à la cancérogénicité
des hydrocarbures aromatiques polycycliques montrent que 26 des 33
composés qui font l'objet de la présente monographie sont
effectivement cancérogènes ou soupçonnés de l'être (Tableau 2). Le
mieux connu de tous est le benzo(a)pyrène, qui a été étudié par toutes
les méthodes existantes sur sept espèces. Plus d'une douzaine d'études
ont été consacrées à l'anthracène, à l'anthanthrène, au
benz(a)anthracène, au chrysène, au dibenz(a,h)anthracène, au
dibenzo(a,i)pyrène, au 5-méthylehrysène, au phénanthrène et au pyrène.
Aux études spéciales sur l'immunotoxicité, la phototoxicité et
l'hépatotoxicité des HAP s'ajoutent un certain nombre d'articles sur
la toxicité oculaire du naphtalène. L'anthracène, le benzo(a)pyrène et
un certain nombre d'autres HAP sont phototoxiques pour la peau des
mammifères ou les cultures cellulaires in vitro, lorsqu'on les
applique sous rayonnement ultraviolet. D'une façon générale, on
considère que les HAP ont un effet immunodépresseur. Après
administration de benzo(a)pyrène à des souris, on a observé une forte
immunodépression dans la descendance de ces animaux pendant une
période pouvant atteindre 18 mois. On a également constaté un
accroissement de la régénération du tissu hépatique et une
augmentation du poids du foie. La formation de cataractes sous l'effet
du naphtalène chez des souris appartenant à des souches génétiquement
différentes, a été attribuée à l'inductibilité du cytochrome P 450.
Dès les années 30, on a proposé des modèles théoriques prenant en
compte un grand nombre de résultats expérimentaux, pour tenter de
prévoir le pouvoir cancérogène des hydrocarbures aromatiques
polycycliques à partir de leur structure moléculaire. Le premier de
ces modèles se fondait sur la forte réactivité chimique de certaines
doubles liaisons (théorie de la région K). Ultérieurement, on a tenté
une approche systématique du problème basée sur la synthèse chimique
des divers métabolites possibles et l'étude de leur activité mutagène.
Tableau 2. Résultats des épreuves de génotoxicité et de cancérogénicité
effectuées sur les 33 hydrocarbures aromatiques polycycliques étudiés.
Compound Genotoxicity Carcinogenicity
Acenaphthene (?) (?)
Acenaphthylene (?) No studies
Anthracene - -
Benz[a]anthracene + +
Benzo[a]fluorene (?) (?)
Benzo[a]pyrene + +
Benzo[b]fluoranthene + +
Benzo[b]fluorene (?) (?)
Benzo[c]phenanthrene (+) +
Benzo[e]pyrene + ?
Benzo[ghi]fluoranthene (+) (-)
Benzo[ghi]perylene + -
Benzo[j]fluoranthene + +
Benzo[l]fluoranthene + +
Chrysene + +
Coronene (+) (?)
Cyclopenta[cd]pyrene + +
Dibenzo[a,e]pyrene + +
Dibenz[a,h]anthracene + +
Dibenzo[a,h]pyrene (+) +
Dibenzo[a,i]pyrene + +
Dibenzo[a,l]pyrene (+) +
Fluoranthene + (+)
Fluorene - -
Indeno[1,2,3-cd]pyrene + +
1-Methylphenanthrene + -
5-Methylchrysene + +
Naphthalene - ?
Perylene + -
Phenanthrene (?) (?)
Pyrene (?) -
Triphenylene + -
+, résultat positif; -, résultat négatif; ?, résultat douteux
Parenthèses, résultat tiré d'une petite base de données.
Selon cette théorie, dite de la "région en baie", les époxydes
adjacents à une région en baie conduisent à la formation d'ions
carbonium très stables qui peuvent alkyler les bases nucléiques. Parmi
les autres théories, on peut citer la théorie de la "di-région" et
celle du "cation radical potentiel".
De nombreux HAP sont cancérogènes pour l'animal et pourraient
également l'être pour l'Homme. D'ailleurs, on a montré que
l'exposition à divers mélanges contenant des HAP augmentait
l'incidence des cancers dans les populations humaines en cause. Ce qui
est préoccupant, c'est que les HAP dont l'étude expérimentale a révélé
l'activité cancérogène chez l'animal, sont probablement aussi
cancérogènes pour l'Homme. Ces composés font apparaître des tumeurs
non seulement au point de contact, mais aussi à distance de ce point.
Le pouvoir cancérogène peut varier avec la voie d'administration. On a
proposé plusieurs méthodes pour évaluer le risque associé à une
exposition à ces composés, seuls ou en mélange. Sans se prononcer en
faveur de telle ou telle méthode, la monographie indique les données
nécessaires, les hypothèses et les conditions de validité etc. pour
trois d'entre elles qui ont été plus ou moins validées en vue d'une
évaluation quantitative du risque.
1.8 Effets sur l'Homme
Les HAP sont présents dans l'environnement et sur les lieux de travail
dans des conditions d'une telle complexité que pour étudier
l'exposition humaine à chacun de ces composés à l'état pur, on s'est
limité à des expériences sur des voluntaires, sauf dans le cas du
naphtalène que l'on utilise comme anti-mite sur les vêtements.
L'application cutanée d'anthracène, de fluoranthène et de phénanthrène
provoque des réaction cutanées spécifiques et le benzo(a)pyrène donne
naissance à des verrues régressives et réversibles que l'on a classées
comme étant de nature néoplasique. On connaît les effets généraux du
naphtalène en raison des nombreux cas d'ingestion accidentelle
auxquels il a donné lieu, notamment chez des enfants. La dose létale
est de 5 000 à 15 000 mg pour un adulte et de 2 000 mg sur deux jours
pour un enfant. Après contact cutané ou ingestion, l'intoxication se
caractérise par une anémie hémolytique aiguë, qui peut également
toucher le foetus par la voie transplacentaire.
On sait que le tabagisme est la cause la plus importante de cancers du
poumon et qu'il accroît également l'incidence des tumeurs de la
vessie, du bassinet du rein, de la cavité buccale, du pharynx, du
larynx et de l'oesophage. On estime en revanche que les HAP présents
dans les denrées alimentaires ne jouent pas un rôle important dans
l'apparition des cancers chez l'Homme. Dans les zones très
industrialisées, on a observé un accroissement de la charge de
l'organisme en HAP par suite de la pollution de l'air ambiant. Les
malades dont on traite le psoriasis par des applications de goudron,
sont également exposés à des HAP.
C'est en 1775 que l'on a, pour la première fois, avancé que
l'exposition professionnelle à la suie était une cause de cancer du
scrotum. Par la suite, on a remarqué que l'exposition aux goudrons et
aux paraffines provoquait des cancers de la peau. Le poumon est
désormais la principale localisation des cancers dus aux HAP, les
cancers cutanés étant devenus plus rares par suite des progrès de
l'hygiène individuelle.
On a effectué des études épidémiologiques sur l'exposition aux HAP
chez des ouvriers de fours à coke pendant la cokéfaction et la
gazéification de la houille, chez des ouvriers d'ateliers de
préparation d'asphalte, de fonderies, de fours à aluminium ou encore
chez des travailleurs exposés aux gaz d'échappement de moteurs diesel.
On a constaté une augmentation des tumeurs pulmonaires dues aux HAP
chez les ouvriers des fours à coke et des ateliers de préparation de
l'asphalte, de même que chez ceux qui travaillaient près des cuves de
réduction électrolytique de l'alumine par le procédé Söderberg. C'est
chez les ouvriers des fours à coke que l'on a mis en évidence le
risque le plus élevé, avec un rapport comparatif de mortalité (SMR) de
195. Plusieurs études ont permis d'établir des relations dose-réponse.
Dans les usine d'aluminium, on a observé non seulement des cancers de
la vessie, mais aussi des symptômes asthmatiformes, des anomalies de
la fonction pulmonaire et des bronchites chroniques. Chez les ouvriers
des fours à coke, il y avait diminution des taux d'immunoglobulines
sériques et dépression des fonctions immunitaires. Par ailleurs, ou a
signalé l'apparition d'une cataracte après cinq ans d'exposition au
naphtalène.
On a mis au point un certain nombre de méthodes pour évaluer
l'exposition interne aux HAP. Dans la plupart de ces études, on a
procédé au dosage des métabolites urinaires de ces composés:
thioéthers, 1-naphtol, ß-naphtylamine, hydroxyphénantrènes et
1-hydroxypyrène. Ce dernier composé est largement utilisé comme
indicateur biologique d'une exposition à des HAP.
Les effets génotoxiques des HAP ont été étudiés en recherchant la
présence de substances mutagènes dans les urines et les matières
fécales et celle de micronoyaux, d'aberrations chromosomiques et
d'échanges entre chromatides-soeurs dans les lymphocytes du sang
périphérique. En outre, on a dosé les adduits du benzo(a)pyrène et de
l'ADN dans les lymphocytes du sang périphérique et dans d'autres
tissus, ainsi que ceux que cet HAP forme avec des protéines comme
l'albumine. On a également procédé au dosage des anticorps dirigés
contre les adduits de l'ADN.
Plusieurs études se sont donné pour but de rechercher s'il était
possible d'utiliser la présence de 1-hydroxypyrène dans l'urine et
d'adduits de l'ADN dans les lymphocytes comme marqueurs d'une
exposition à des HAP. Il est plus facile de doser le 1-hydroxypyrène
urinaire que les adduits de l'ADN. Ce marqueur a en outre l'avantage
d'être moins sujet aux variations interindividuelles et de permettre
de repérer des expositions plus faibles. Les deux types de marqueurs
ont été utilisés pour évaluer l'exposition humaine dans divers
environnements. Ainsi, on a relevé une augmentation de la
concentration urinaire de 1-hydroxypyrène sur les divers lieux de
travail de cokeries, d'usines de production d'aluminium, d'ateliers
d'imprégnation du bois, de fonderies et d'ateliers de préparation
d'asphalte. L'exposition la plus forte a été observée chez les
ouvriers des fours à coke et chez ceux qui travaillent à
l'imprégnation du bois avec du créosote. Chez ces travailleurs,
l'exposition est à 95% percutanée, alors que dans le reste de la
population, les HAP sont absorbés principalement par la voie
alimentaire et par la voie respiratoire lors de la consommation de
tabac.
L'estimation du risque associé à une exposition à des HAP repose sur
l'évaluation de cette exposition et sur les résultats des études
épidémiologiques. En ce qui concerne les ouvriers des fours à coke, on
aboutit à un risque relatif de cancer du poumon égal à 15,7. En
procédant de la même manière pour la population générale, on trouve
que le risque, pour un individu donné, de faire un cancer du poumon au
cours de son existence est de 10-4 à 10-5 par ng de benzo(a)pyrène
par m3 d'air. En d'autres termes, il y a environ une personne sur 10
000 ou 100 000 qui va faire un cancer au cours de sa vie par suite de
la présence de benzo(a)pyrène dans l'air ambiant.
1.9 Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
S'il y a absorption simultanée de lumière UV ou de lumière visible,
les HAP peuvent provoquer des effets toxiques aigus sur les poissons
et sur des invertébrés aquatiques comme les daphnies. La toxicité des
HAP peut être modifiée par dégradation et métabolisation. A faible
concentration, les HAP peuvent stimuler la croissance des
microorganismes et des algues. Le composé le plus toxique pour les
algues est le benz(a)anthracène, un hydrocarbure tétracyclique. La
valeur de la CE50 pour ce composé (réduction de 50% des paramètres
vitaux) est égale à 1-29 µg/litre. Dans le cas du benzo(a)pyrène, un
composé pentacyclique, elle est égale à 5-15 µg/litre. Toujours en ce
qui concerne les algues, la CE50 pour la plupart des HAP
tricycliques est égale à 240-290 µg/litre. Le naphtalène, qui est
bicyclique, est le moins toxique, avec une CE50 de 2 800-34 000
µg/litre.
Il n'y a pas de différence de sensibilité bien nette entre les divers
groupes taxonomiques d'invertébrés tels que les crustacés, les
insectes, les mollusques, les polychètes et les échinodermes. Le
naphtalène est le moins toxique avec une CL50 à 96 h de 100 à 2
300/µg/litre. Pour trois HAP tricycliques, la valeur de ce même
paramètre varie de <1 à 3 000 µg/litre. L'anthracène pourrait être
plus toxique que les autres HAP tricycliques avec une CL50 à 24 h
comprise entre <1 et 260 µg/litre. Pour les composés à quatre, cinq et
six noyaux aromatiques, la CL50 à 96 h est comprise entre 0,2 et 1
200 µg/litre. Des effets toxiques aigus (CL50) ont été observés chez
des poissons à des concentrations de 110 à > 10 000 µg/litre de
naphtalène, de 30 à 40 000 µg/litre d'HAP tricycliques (anthracène,
2,8-360 µg/litre) et de 0,7 à 26 µg/litre d'HAP tétra- et
pentacycliques.
Chez des poissons vivant à l'état sauvage, on a attribué la présence
de tumeurs hépatiques à la contamination des sédiments par des HAP à
la concentration de 250 mg/kg. On a également provoqué l'apparition de
tumeurs chez des poissons par exposition en laboratoire. L'exposition
des poissons à certains HAP peut aussi provoquer chez eux des
anomalies physiologiques et perturber leur croissance, leur
reproduction, leur locomotion et leur respiration.
1. RESUMEN
1.1 Selección de compuestos para esta monografía
Los hidrocarburos aromáticos policiclicos (HAP) constituyen una clase
muy amplia de compuestos, y durante la combustión incompleta o la
pirolisis de materia orgánica pueden liberarse cientos de sustancias
distintas, que son una fuente importante de exposición humana. Los
estudios de diversas matrices aplicables al medio ambiente, como los
efluentes de la combustión de carbón, los gases de escape de los
vehículos de motor, el aceite lubricante de motores usados y el humo
del tabaco, han demostrado que los HAP de esas mezclas son los
principales responsables de su potencial carcinogénico.
En las mezclas casi siempre hay presentes HAP. Debido a que la
composición de tales muestras es compleja y varía con el proceso de
formación, no es posible examinar con detalle en la presente
monografía todas las mezclas que contienen HAP. Así pues, se
seleccionaron 33 compuestos distintos (31 HAP originales y dos
derivados alquilo) para evaluarlos tomando como base la disponibilidad
de datos pertinentes sobre los efectos finales toxicológicos y/o la
exposición (Cuadro 1). Sin embargo, dado que sólo se disponía para las
mezclas de estudios epidemiológicos, que son imprescindibles para la
evaluación del riesgo, en las Secciones 8 y 10 se presentan los
resultados de estudios de mezclas de HAP, en contraposición con el
resto de la monografía.
Se han publicado numerosos artículos y reseñas sobre la presencia,
distribución y transformación de HAP en el medio ambiente y sobre sus
efectos ecotoxicológicos y toxicológicos. Solamente se citan en la
presente monografía referencias de los 10-15 últimos años, a menos que
no se dispusiera de otra información; en relación con los estudios más
antiguos y con otra información se citan reseñas.
1.2 Identidad, propiedades físicas y químicas y métodos analíticos
El término "hidrocarburos aromáticos policíclicos" se refiere en
general a una clase muy amplia de compuestos orgánicos que contienen
dos o más anillos aromáticos condensados formados por átomos de
carbono y de hidrógeno. A temperatura ambiente, los HAP son sólidos.
Las características comunes de la clase son puntos de fusión y de
ebullición elevados, presión de vapor baja y solubilidad en agua muy
baja, que tiende a disminuir con el aumento del peso molecular. Los
HAP son solubles en muchos disolventes orgánicos y muy lipófilos.
Desde el punto de vista químico son bastante inertes. Las reacciones
que tienen interés con respecto a su destino en el medio ambiente y a
las posibles fuentes de pérdidas durante el muestren atmosférico son
la fotodescomposición y las reacciones con óxidos de nitrógeno, ácido
nítrico, óxidos de azufre, ácido sulfúrico, ozono y radicales
hidroxilo.
Cuadro 1. Hidrocarburos aromáticos policíclicos evaluados en esta monografía
Nombre común Nombre CAS Sinónimoa No de registro CAS
Acenaftileno Acenaftileno 91-20-3
Acenafteno Acenaftileno, 1.2-dihidro- 208-96-8
Antantreno Dibenzo[def,mno]criseno 191-264
Antraceno Antraceno 120-12-7
Benz[a]antraceno Benz[a]antraceno 1,2-Benzantraceno, tetrafeno 56-55-3
Benzo[a]fluoreno 11H-Benzo[a]fluoreno 1,2-Benzofluoreno 238-84-6
Benzo[b]fluoreno 11H-Benzo[b]fluoreno 2,3-Benzofluoreno 243-17-4
Benzo[b]fluoranteno Benz[e]acefenantrileno 3,4-Benzofluoranteno 205-99-2
Benzo[ghl]fluoranteno Benzo[ghl]fluoranteno 2,13-Benzofluoranteno 203-12-3
Benzo[j]fluoranteno Benzo[j]fluoranteno 10,11-Benzofluoranteno 205-82-3
Benzo[k]fluoranteno Benzo[k]fluoranteno 11,12-Benzofluoranteno 207-08-9
Benzo[ghi]perileno Benzo[ghi]perileno 1,12-Benzoperileno 191-24-2
Benzo[c]fenantreno Benzo[c]fenantreno 3,4-Benzofenantreno 195-19-7
Benzo[a]pireno Benzo[a]pireno 3,4-Benzopirenob 50-32-8
Benzo[e]pireno Benzo[e]pireno 1.2-Benzopireno 192-97-2
Criseno Criseno 1,2-Benzofenantreno 218-01-9
Coroneno Coroneno Hexabenzobencano 191-07-1
Ciclopenta[cd]pireno Ciclopenta[cd]pireno Ciclopenteno[cd]pireno 27208-37-3
Dibenz[a,h]antraceno Dibenz[a,h]antraceno 1,2:5.6-Dibenzantraceno 53-70-3
Dibenzo[a,e]pireno Naftol[1,2:3,4-def]criseno 1,2:4,5-Dibenzopireno 192-65-4
Dibenzo[a,h]pireno Dibenzo[b,def]criseno 3,4:8,9-Dibenzopireno 189-64-0
Dibenzo[a,i]pireno Benzo[rsf]pentafeno 3,4:9,10-Dibenzopireno 189-55-9
Dibenzo[a,l]pireno Dibenzo[def,p]criseno 1,2:3,4-Dibenzopireno 191-30-0
Fluoranteno Fluoranteno 206-44-0
Fluoreno 9H-Fluoreno 86-73-7
Indeno[1,2,3-cd]pireno Indeno[1,2,3-cd]pireno 2,3-o-Fenilenpireno 193-39-5
5-Metilcriseno Criseno, 5-metil- 3697-24-3
1-Metilfenantreno Fenantreno, 1-metil- 832-69-9
Naftaleno Naftaleno 91-20-3
Perileno Perileno peri-Dinaftaleno 198-55-0
Penantreno Fenantreno 85-01-8
Pireno Pireno Benzo[def]fenantreno 129-00-0
Trifenileno Trifenileno 9,10-Benzofenantreno 217-59-4
Cuadro 1 (sigue).
Han notificado listas muy amplias de sinónimos el CIIC (1983) y Loening y Merritt (1990).
a Sinónimo común que aparece en la bibliografia.
c También denominado benzo[def]criseno.
El aire del ambiente se muestrea recogiendo partículas suspendidas en
filtros de fibra de vidrio, de politetrafluoroetileno o de fibra de
cuarzo mediante muestreadores de alto volumen o pasivos. Los HAP en
fase de vapor, que podrían volatilizarse de los filtros durante el
muestreo, se suelen retener por adsorción en espuma de poliuretano. La
fase de muestren es con diferencia la fuente más importante de
variabilidad de los resultados.
En el lugar de trabajo se toman muestras con tasas de flujo bajas; se
recogen las partículas en filtros de fibra de vidrio o de
politetrafluoroetileno y los vapores en resina XAD-2 de amberlita. Los
dispositivos para el muestren de gases de chimenea constan de un
filtro de fibra de vidrio o de fibra de cuarzo en la parte frontal de
un refrigerador para recoger la materia condensable y un cartucho
adsorbente (por lo general XAD-2). Las muestras de gases de escape de
los vehículos se toman en condiciones de laboratorio, con ciclos de
conducción normalizados que simulan las condiciones en carretera. Las
emisiones se recogen sin diluir o bien una vez diluidas con aire frío
filtrado.
Se han descrito numerosas técnicas de extracción y purificación. En
función de la matriz, los HAP se extraen de las muestras con un
aparato de Soxhlet, por ultrasonidos, mediante reparto liquido-liquido
o, tras la disolución o la digestión alcalina de la muestra, con un
disolvente selectivo. También se ha utilizado la extracción de fluidos
supercríticos a partir de diversos sólidos del medio ambiente. La
eficacia de la extracción depende en gran medida del disolvente
utilizado, y muchos de los que se utilizaban en el pasado no eran
apropiados. Las muestras extraídas se suelen purificar por
cromatografía en columna, en particular sobre alúmina, silicagel o
Sephadex LH-20, pero también por cromatografía en capa fina.
La identificación y la cuantificación se realizan habitualmente
mediante cromatografía de gases con detección por ionización de llama
o mediante cromatografía liquida de alta resolución (HPLC) con
detección por ultravioleta o fluorescencia, generalmente en serie. En
la cromatografía de gases se utilizan columnas capilares de sílice
fundido con polisiloxanos (SE-54 y SE-52) como fases estacionarias; en
la HPLC se suelen utilizar columnas de sílice-C18. Con frecuencia se
acopla un detector de espectrometría de masas a una columna de fase
gaseosa a fin de confirmar la identidad de los picos.
La elección de los HAP que se han de determinar depende de la
finalidad de la medición, por ejemplo para estudios orientados a la
salud o ecotoxicológicos, o bien para investigar las fuentes. Es
posible que se exija o se recomiende la realización de pruebas para
distintos conjuntos de compuestos a nivel nacional e internacional.
1.3 Fuentes de exposición humana y ambiental
Es poca la información disponible acerca de la producción y
elaboración de HAP, pero es probable que sólo se desprendan pequeñas
cantidades como resultado directo de esas actividades. Los HAP que se
encuentran se utilizan sobre todo como productos intermedios en la
producción de cloruro de polivinilo y de agentes plastificantes
(naftaleno), pigmentos (acenafteno, pireno), tintes (antraceno,
fluoranteno) y plaguicidas (fenantreno).
Las mayores emisiones de HAP se derivan de la combustión incompleta de
materia orgánica durante procesos industriales y en otras actividades
humanas, en particular:
- elaboración de carbón, de petróleo crudo y de gas natural,
incluida la coquificación de carbón, la conversión de carbón, el
refinado de petróleo y la producción de negro de humo, de
creosota, de alquitrán de hulla y de betún;
- producción de aluminio, de hierro y de acero en fábricas y
fundiciones;
- calefacción en centrales de energía y en residencias y ocinado;
- combustión de basuras;
- tráfico de vehículos de motor; y
- humo de tabaco en el medio ambiente.
Los HAP, especialmente los de mayor peso molecular, cuando se
incorporen al medio ambiente a través de la atmósfera se adsorben en
las partículas en suspensión. La hidrosfera y la geosfera se ven
afectadas de manera secundaria por la deposición húmeda y seca. La
madera conservada con creosota es otro fuente de liberación de HAP en
la hidrosfera, y la deposición de desechos contaminados, como fangos
de alcantarillado y cenizas en suspensión, contribuye a las emisiones
de HAP en la geosfera. Hay poca información acerca del paso de HAP a
la biosfera. Hay HAP presentes naturalmente en la turba, el lignito,
el carbón y el petróleo crudo. La mayoría de los HAP de las antracitas
están fuertemente unidos a la estructura del carbón y no se pueden
lixiviar.
La liberación de HAP en el medio ambiente se ha determinado mediante
la identificación de un perfil característico de su concentración,
pero esto sólo ha sido posible en un pequeño número de casos. Con
frecuencia se ha utilizado el benzo[a]pireno como indicador de HAP,
especialmente en estudios más antiguos. En general, las emisiones de
HAP son solamente estimaciones basadas en datos más o menos fidedignos
y apenas den una idea general de la exposición.
Las fuentes más importantes de HAP son las siguientes:
Coquificación de carbón: Las emisiones de HAP en suspensión en el
aire procedentes de la coquificación de carbón en Alemania han
disminuido considerablemente en los 10 últimos años gracias a las
mejoras técnicas de las instalaciones existentes, al cierre de otras
antiguas y a la menor producción de coque. Se supone que la situación
es análoga en el resto de Europa occidental, el Japón y los Estados
Unidos, pero no se dispone de datos.
Producción de aluminio (principalmente en ánodos especiales de
carbón), de hierro y de acero y los aglutinantes utilizados en la
arena de moldeo de las fundiciones: La información disponible es
escasa.
Cocinas y calefacción de viviendas: Los principales componentes que
se emiten son fenantreno, fluoranteno, pireno y criseno. Las emisiones
de los hornillos de leña son 25-1000 veces superiores a las que se
producen en los de carbón, y en las zonas donde predomina el uso de
leña en las viviendas la mayor proporción de HAP en suspensión puede
derivarse de esta fuente, especialmente en invierno. Por consiguiente,
se supone que la liberación de HAP en la calefacción de las viviendas
es una fuente importante en los países en desarrollo, donde con
frecuencia se quema biomasa en hornillos relativamente simples.
Cocinado: Pueden emitirse HAP durante la combustión incompleta de
los combustibles, del aceite de cocinar y de los alimentos que se
cocinen.
Tráfico de vehículos de motor: Los principales compuestos que se
liberar de los vehículos de gasolina son el fluoranteno y el pireno,
mientras que en los gases de escape de los vehículos de motor diesel
abundan el naftaleno y el acenafteno. Aunque los motores de gasolina
emiten una proporción elevada de ciclopenta[cd]pireno, su
concentración en los gases de escape de los motores diesel está apenas
por encima del límite de detección. Las tasas de emisión, que dependen
de la sustancia, el tipo de vehículo, el estado de su motor y las
condiciones de la prueba, oscilan entre unos pocos nanogramos por
kilómetro y > 1000 mg/km. Las emisiones de HAP de los vehículos de
motor se reducen enormemente con la instalación de catalizadores.
Incendios forestales: En los países con grandes superficies
forestales, los incendios pueden contribuir de manera importante a las
emisiones de HAP.
Centrales eléctricas de carbón: Los HAP que se liberar en la
atmósfera a partir de dichas centrales son sobre todo compuestos de
dos y tres anillos. En las zonas contaminadas, los niveles de HAP en
el aire pueden ser más elevados que los de los gases de chimenea.
Incineración de basuras: Las emisiones de HAP en los gases
procedentes de este tipo de incineración fueron en varios países < 10
mg/m3.
1.4 Transporte, distribución y transformación en el medio ambiente
Son varios los factores de distribución y de transformación de los que
depende el destino tanto de los HAP por separado como de las mezclas.
Los procesos de distribución más importantes son el reparto entre el
agua y el aire, entre el agua y los sedimentos y entre el agua y la
biota.
Puesto que los HAP son hidrófobos, con escasa solubilidad en el agua,
su afinidad por la fase acuática es muy pequeña; sin embargo, a pesar
del hecho de que la mayoría de los HAP se liberan en el medio ambiente
a través de la atmósfera, también se encuentran concentraciones
considerables en la hidrosfera, debido a sus bajas constantes de la
ley de Henry. Como la afinidad de los HAP por las fases orgánicas es
mayor que la que tienen por el agua, sus coeficientes de reparto entre
disolventes orgánicos como el octanol y el agua son elevados. Su
afinidad por las fracciones orgánicas de los sedimentos, del suelo y
de la biota es también alta, por lo que se acumulan en los organismos
del agua y los sedimentos y en sus alimentos. No se conoce con
claridad la importancia relativa de su ingesta con los alimentos y el
agua. En Daphnia y en los moluscos, hay una correlación positiva
entre la acumulación de HAP procedentes del agua y el coeficiente de
reparto octanol:agua (Kow). Sin embargo, en los peces y las algas
capaces de metabolizar los HAP no hay correlación entre las
concentraciones internas de distintos HAP y el Kow.
No se ha observado bioamplificación -aumento de la concentración de
una sustancia en animales de niveles tróficos sucesivos de las cadenas
alimentarias- en sistemas acuáticos y no cabe prever que se produzca,
puesto que la mayoría de los organismos tienen un potencial de
biotransformación elevado para los HAP. El mayor potencial de
biotransformación se observa en los organismos de los niveles tróficos
más altos de las cadenas alimentarias.
Los HAP se descomponen por fotodegradación, biodegradación por
microorganismos y metabolismo en la biota de niveles más altos. Aunque
la última rata de transformación tiene escasa importancia para el
destino global de los HAP en el medio ambiente, es una vía importante
para la biota, debido a que pueden formarse metabolitos
carcinogénicos. Dado que los HAP son químicamente estables, sin grupos
reactivos, la hidrólisis no interviene en su degradación. Hay pocas
pruebas normalizadas para la biodegradación de los HAP. En general, se
biodegradan en condiciones aerobias, registrando un fuerte aumento la
tasa de biodegradación con el número de anillos aromáticos. En
condiciones anaerobias la degradación es mucho más lenta.
Los HAP se fotooxidan en el aire y en el agua en presencia de
radicales sensibilizantes como OH, NO3 y O3. En condiciones de
laboratorio, la semivida de la reacción con radicales OH presentes en
el aire es de alrededor de un día, mientras que las reacciones con
NO3 y O3 suelen tener unas constantes de velocidad mucho más
bajas. La adsorción de HAP de peso molecular alto en partículas
carbonosas en el medio ambiente debería estabilizar la reacción con
radicales OH. La reacción, que tiene lugar sobre todo en la fase
gaseosa, de HAP de entre dos y cuatro anillos con NO3 da lugar a la
formación de nitro-HAP, que son conocidos como mutágenos. Parece que
la fotooxidación de algunos HAP en el agua es más rápida que en el
aire. Los cálculos basados en parámetros fisicoquímicos y de
degradación indican que los HAP con cuatro o más anillos aromáticos
persisten en el medio ambiente.
1.5 Niveles ambientales y exposición humana
Los HAP están omnipresentes en el medio ambiente, habiéndose detectado
en numerosos estudios diversos HAP por separado en distintos
compartimentos.
1.5.1 Aire
Los niveles de cada uno de los HAP tiendan a ser más elevados en
invierno que en verano por lo menos en un orden de magnitud. La fuente
predominante durante el invierno es la calefacción de las viviendas y
la del verano el tráfico urbano de vehículos de motor. Se detectaron
concentraciones medias de distintos HAP de 1-30 ng/m3 en la
atmósfera de diversas zonas urbanas. En grandes ciudades con un
tráfico intenso de vehículos de motor y utilización abundante de
combustibles a base de biomasa, como Calcuta, se encontraron niveles
de hasta 200 ng/m3 de distintos HAP. En túneles de carreteras se
detectaron concentraciones de hasta 1-50 ng/m3. Había
ciclopenta[cd]-pireno y pireno en concentraciones de hasta 100
ng/m3. En una estación de metro se midieron concentraciones de HAP
de hasta 20 ng/m3. En las cercanías de fuentes industriales, las
concentraciones medias de los distintos HAP oscilaban entre 1 y 10
ng/m3. Había fenantreno presente hasta un máximo aproximado de 310
ng/m3.
Los valores básicos de los HAP son por lo menos uno o dos órdenes de
magnitud menores que los obtenidos cerca de fuentes como el tráfico de
vehículos de motor. Por ejemplo, los niveles a 1100 m oscilaban entre
0,004 y 0,03 ng/m3.
1.5.2 Agua superficial y precipitación
La mayor parte de los HAP presentes en el agua proceden al parecer del
agua de escorrentía urbana, de la precipitación atmosférica
(partículas más pequeñas) y de la abrasión del asfalto (partículas
mayores). La principal fuente de HAP, sin embargo, varia para
distintas masas de agua. En general, la mayoría de las muestras de
agua superficial contienen distintos HAP en concentraciones de hasta
50 ng/litro, pero los ríos muy contaminados tenían concentraciones de
hasta 6000 ng/litro. Los niveles de HAP en el agua freática son del
orden de 0,02-1,8 ng/litro, y las muestras de agua potable contienen
concentraciones del mismo orden de magnitud. Las principales fuentes
de los HAP presentes en el agua potable son los depósitos y las
tuberías con revestimiento de asfalto.
Los niveles de HAP por separado en el agua de lluvia oscilaban entre
10 y 200 ng/litro, mientras que en la nieve y la niebla se detectaron
concentraciones de hasta 1000 ng/litro.
1.5.3 Sedimentos
Las concentraciones de los distintos HAP cm los sedimentos eran por lo
general un orden de magnitud superiores a las presentes cm la
precipitación.
1.5.4 Suelo
Las principales fuentes de los HAP presentes en el suelo son la
deposición atmosférica, la carbonización de material vegetal y la
deposición a partir de aguas residuales y desechos particulados. El
grado de contaminación del suelo depende de factores como su cultivo,
su porosidad y su contenido de sustancias húmicas.
Cerca de fuentes industriales se han encontrado concentraciones de
distintos HAP de hasta 1 g/kg de suelo. La concentración en el suelo a
partir de otras fuentes, como los gases de escape de los automóviles,
son del orden de 2-5 mg/kg. En zonas no contaminadas, los niveles de
HAP eran de 5-100 µg/kg de suelo.
1.5.5 Alimentos
Los alimentos credos no suelen contener niveles elevados de HAP, pero
se forman al elaborarlos, asados o cocerlos en horno o freírlos. Las
hortalizas pueden contaminarse por la deposición de partículas de la
atmósfera o por el crecimiento en suelo contaminado. Las
concentraciones de los distintos HAP en la carne, el pescado, los
productos lácteos, las frutas y hortalizas, los cereales y sus
productos, los dulces, las bebidas y las grasas y los aceites animales
y vegetales eran del orden de 0,01-10 µg/kg. Se han detectado
concentraciones de más de 100 µg/kg en carne ahumada y de hasta 86
µg/kg cm pescado ahumado; los cereales ahumados contenían hasta 160
µg/kg. En el aceite de coco se encontraron concentraciones de hasta
460 µg/kg. Los niveles en la leche materna humana eran de 0,003-0,03
µg/kg.
1.5.6 Organismos acuáticos
Se sabe que los organismos marinos adsorben y acumulan HAP del agua.
El grado de contaminación depende del desarrollo industrial y urbano y
de los movimientos de transporte marítimo. Se han detectado
concentraciones de HAP de hasta 7 mg/kg en organismos acuáticos que
vivían cerca de efluentes industriales, y los niveles medios de HAP en
los animales acuáticos muestreados en lugares contaminados fueron de
10-500 µg/kg, aunque también se detectaron niveles de hasta 5 mg/kg.
Los niveles medios de HAP en animales acuáticos muestreados en
diversos lugares con fuentes sin especificar de dichos compuestos
fueron de 1-100 µg/kg, pero se encontraron concentraciones de hasta 1
mg/kg, por ejemplo en langostas en el Canadá.
1.5.7 Organismos terrestres
Las concentraciones de HAP en insectos oscilaban entre 730 y 5500
µg/kg. El contenido de las heces de las lombrices de tierra depende
considerablemente del lugar: las de una región muy industrializada de
Alemania oriental contenían concentraciones de benzo[a]pireno de hasta
2 mg/kg.
1.5.8 Población general
Las principales fuentes de exposición no profesional son las
siguientes: atmósfera contaminada, humo de fuegos abiertos y del
cocinado, humo de tabaco en el medio ambiente, alimentos y agua
potable contaminados y utilización de productos contaminados. Pueden
encontrarse HAP en el aire de espacios cerrados como consecuencia de
la calefacción de las viviendas y del humo del tabaco del medio
ambiente en concentraciones medias de 1-100 ng/m3, con un máximo de
2300 ng/m3.
Se ha estimado que la ingesta de distintos HAP con los alimentos es de
0,10-10 µg/día por persona. La ingesta diaria total de benzo[a]-pireno
con el agua potable se estimó en 0,0002 µg/persona. Los cereales y
productos derivados son los que más contribuyen a la ingesta de HAP
con los alimentos, por ser el principal componente de la alimentación
total.
1.5.9 Exposición profesional
Cerca de una batería de hornos de coque, los niveles de
benzo[a]-pireno oscilaban entre < 0,1 y 100-200 µg/m3, con un máximo
aproximado de 400 µg/m3. En los sistemas modernos de gasificación
del carbón, la concentración de HAP suele ser < 1 µg/m3, con un
máximo de 30 µg/m3. Las muestras personales tomadas de operadores de
equipo de refinerías de petróleo mostraron una exposición a 2,6470
µg/m3. En muestras de aire tomadas cerca de instalaciones de
elaboración de asfalto en refinerías, los niveles totales de HAP
fueron de 0,004-50 µg/m3. En las proximidades de obras de
pavimentación de carreteras, las concentraciones totales de HAP en
muestras personales de aire eran de hasta 190 µg/m3, con un valor
medio en las muestras de aire de la zona de 0,13 µg/m3. Los niveles
de HAP en las muestras personales de aire tomadas en una fundición de
aluminio eran de 0,059,6 µg/m3, pero las muestras de orina de los
trabajadores de una fábrica de aluminio contenían niveles muy bajos.
Las muestras de aire de la zona contenían concentraciones de HAP de
hasta 5 µg/m3 en una fundición alemana, 3-40 µg/m3 en minas de
hierro µg/m3 y 4530 µg/m3 en minas de cobre. Las concentraciones
de HAP en humos de cocinado en una fábrica de productos alimenticios
oscilaban entre 0,07 y 26 µg/m3.
1.6 Cinética y metabolismo
Los HAP se absorben por las vías respiratorias, el aparato digestivo y
la piel. La tasa de absorción por los pulmones depende del tipo de
HAP, el tamaño de las partículas sobre las que están adsorbidos y la
composición del adsorbente. Los HAP adsorbidos sobre partículas se e
'laminan de los pulmones con mayor lentitud que los hidrocarburos
libres. En el aparato digestivo se produce una absorción rápida en los
roedores, pero los metabolitos vuelven al intestino mediante la
excreción biliar. En estudios de absorción percutánea de mezclas de
HAP marcados con 32P en roedoras se observó que los componentes de las
mezclas negaban a los pulmones, donde se unían al ADN. La tasa de
absorción percutánea en ratones varia en función del compuesto.
Los HAP se distribuyen ampliamente en todo el organismo tras la
administración por cualquier vía y se encuentran en casi todos los
órganos internos, particularmente en los ricos en lípidos. Los HAP
inyectados por vía intravenosa se eliminan con rapidez de la corriente
sanguínea en los roedores, pero pueden atravesar la barrera
placentaria y se han detectado en tejidos fetales.
El metabolismo de los HAP es complejo. En general, los compuestos
originales se convierten mediante epóxidos intermedios en fenoles,
dioles y tetroles, que a su vez pueden conjugarse con los ácidos
sulfúrico y glucurónico o con el glutatión. El metabolismo produce en
su mayor parte una desintoxicación, pero algunos HAP se activan a
especies que se unen al ADN, principalmente diolepóxidos, que pueden
inducir tumores. Los metabolitos de los HAP y sus cunjugados se
excretan en la orina y las heces, pero los conjugados que se excretan
en la bilis pueden hidrolizarse por la acción de enzimas de la flora
intestinal y reabsorberse. De la información disponible acerca de la
carga total en el cuerpo humano cabe deducir que los HAP no persisten
en el organismo y que su ciclo metabólico es rápido. De la deducción
anterior están excluidos los grupos de HAP que se unen por enlaces
covalentes a elementos constitutivos de los tejidos, en particular
ácidos nucleicos, y que no se eliminan por reparación.
1.7 Efectos en mamíferos de laboratorio y en sistemas de prueba in vitro
La toxicidad aguda de los HAP parece ser de moderada a baja. Un
producto bien caracterizado, el naftaleno, mostró valores para la
DL50 por vía oral e intravenosa de 100-500 mg/kg de peso corporal en
ratones y una DL50 media por vía oral de 2700 mg/kg de peso corporal
en ratas. Los valores para otros HAP son semejantes. Con dosis altas
únicas de naftaleno se indujo en ratones, ratas y hámsteres necrosis
bronquiolar.
En estudios de corta duración se pusieron de manifiesto efectos
hematológicos adversos en forma de mielotoxicidad con el
benzo[a]-pireno, cambios hemolinfáticos con el dibenz[a,h]antraceno y
anemia con el naftaleno; sin embargo, en un estudio de siete días en
el que se administró a ratones naftaleno por vía oral e
intraperitoneal se observó tolerancia al efecto de este producto.
Sólo raramente se han descrito efectos sistémicos provocados por un
tratamiento prolongado con HAP, porque el efecto final de la mayor
parte de los estudios ha sido la carcinogenicidad. Se manifiestan
efectos tóxicos importantes a dosis en las cuales se desencadenan
también respuestas carcinogénicas
En estudios de los efectos adversos en la piel tras la aplicación
cutánea, los productos con carcinogenicidad nula o débil, como el
perileno, el benzo[e]pireno, el fenantreno, el pireno, el antraceno,
el acenaftaleno, el fluoreno y el fluoranteno, fueron inactivos,
mientras que los compuestos carcinógenos; como el benz[a]antraceno, el
dibenz[a,h]antraceno y el banzo[a]pireno provocaron hiperqueratosis.
Los vapores de antraceno y de naftaleno provocaron irritación ocular
leve. El benzo[a]pireno indujo hipersensibilidad por contacto en
cobayas y ratones.
El benz[a]antraceno, el benzo[a]pireno, el dibenz[a,h]antraceno y el
naftaleno fueron embriotóxicos para ratones y ratas. El benzo[a]-
pireno mostró asimismo teratogenicidad y efectos en la reproducción.
Se han realizado grandes esfuerzos para aclarar la base genética de
los efectos embriotóxicos del benzo[a]pireno. Sólo se observan
mortalidad fetal y malformaciones si es inducible el sistema citocromo
P-450 monooxigenasa, bien en la madre (con permigración placentaria) o
bien en el embrión. No todos los efectos observados se pueden explicar
por la predisposición genética, pero en ratones y conejos el
benzo[a]pireno mostró actividad carcinogénica transplacentaria,
produciendo adenomas pulmonares y papilomas cutáneos en la
descendencia. Se observó asimismo reducción de la fecundidad y
destrucción de oocitos.
Se han estudiado también ampliamente los HAP en valoraciones de la
genotoxicidad y de la transformación celular; la mayor parte de los 33
HAP comprendidos en la presente monografía son genotóxicos o
probablemente genotóxicos. Los únicos compuestos para los cuales se
obtuvieron resultados negativos en todas las valoraciones fueron el
antraceno, el fluoreno y el naftaleno. Habida cuenta de la falta de
uniformidad de los resultados, el fenantreno y el pireno no podrían
clasificarse de manera fidedigna como genotóxicos.
En un trabajo amplio sobre la carcinogenicidad de los HAP se ha puesto
de manifiesto que 26 de los 33 productos estudiados son, o se sospecha
que son, carcinogénicos (Cuadro 2). El compuesto mejor caracterizado
es el benzo[a]pireno, que se ha estudiado aplicando todos los métodos
actuales en siete especies. Los HAP que han sido objeto de 12 estudios
o más son el antantreno, el antraceno, el benz[a]-antraceno, el
criseno, el dibenz[a,h]antraceno, el dibenzo[a,i]pireno, el 5-
metilcriseno, el fenantreno y el pireno. Los estudios especiales de
fototoxicidad, inmunotoxicidad y hepatotoxicidad de los HAP se
complementan con informes sobre la toxicidad ocular del naftaleno.
Cuadro 2. Resumen de los resultados de las pruebas de genotoxicidad
y carcinogenicidad de los 33 hidrocarburos aromáticos policiclicos
estudiados
Compuesto Genotoxicidad Carcinogenicidad
Acenafteno (?) (?)
Acenaftileno (?) No hay estudios
Antraceno - -
Benz[a]antraceno + +
Benzo[a]fluoreno (?) (?)
Benzo[a]pireno + +
Benzo[b]fluoranteno + +
Benzo[b]fluoreno (?) (?)
Benzo[c]fenantreno (+) +
Benzo[e]pireno + ?
Benzo[ghi]fluoranteno (+) (-)
Benzo[ghi]perileno + -
Benzo[j]fluoranteno + +
Benzo[k]fluoranteno + +
Criseno + +
Coroneno (+) (?)
Ciclopenta[cd]pireno + +
Dibenzo[a,e]pireno + +
Dibenz[a,h]antraceno + +
Dibenzo[a,h]pireno (+) +
Dibenzo[a,i]pireno + +
Dibenzo[a,l]pireno (+) +
Fluoranteno + (+)
Fluoreno - -
Indeno[1,2.3-cd]pireno + +
1-Metilfenantreno + -
5-Metilcriseno + +
Naftaleno ?
Perileno + -
Fenantreno (?) (?)
Pireno (?) -
Trifenileno + -
+, positivo; -, negativo; ?, dudoso
Paréntesis, resultado derivado de un número de datos pequeño
El antraceno, el benzo[a]pireno y algunos otros HAP mostraron
fototoxicidad en la piel de mamíferos y en cultivos de células in
vitro cuando se aplicaron con radiación ultravioleta. Se ha notificado
que en general estos compuestos tienen efectos inmunosupresores. Tras
la administración intraperitoneal de benzo[a]pireno a ratones, se
manifestó una fuerte supresión de los parámetros inmunitarios en la
descendencia durante un período de hasta 18 meses. Se observó asimismo
una mayor regeneración hepática y un aumento del peso del hígado. El
efecto del naftaleno en la aparición de cataratas en un roedor se ha
atribuido a su capacidad de inducción del sistema del citocromo P-450
en estudios realizados con estirpes de ratones genéticamente
diferentes.
Los modelos teóricos para pronosticar la actividad carcinogénica de
los HAP a partir de sus estructuras, basados en una gran cantidad de
trabajos experimentales, se presentaron ya en los años treinta. El
primer modelo se basaba en la elevada reactividad química de
determinados dobles enlaces (teoría de la región K). Más tarde se
aplicó un método más sistemático, basado en la síntesis química de
posibles metabolitos y en su actividad mutagénica. Según esta teoría
de la región "bay", los epóxidos adyacentes a una región "bay" dan
lugar a iones carbonio muy estables. Otros métodos teóricos son la
"teoría de la región di" y la "teoría del potencial del radical
catiónico".
Muchos de los distintos HAP son carcinógenos para los animales y
pueden serio para el ser humano y se ha observado que la exposición a
varias mezclas con HAP aumenta la incidencia de cáncer en poblaciones
humanas. Existe la preocupación de que los HAP cuya carcinogenicidad
se ha demostrado en animales de experimentación puedan serio también
para el ser humano. Los HAP producen rumores, tanto en el lugar del
contacto como en otros lejanos. Su actividad carcinogénica puede
variar en función de la vía de exposición. Se han propuesto diversos
métodos para evaluar el riesgo asociado a la exposición a estos
productos aislados y en mezclas. En la presente monografía no se
respalda ningún método; sin embargo, se describen las necesidades de
datos, las hipótesis, la aplicabilidad y otras características de tres
procesos de evaluación cuantitativa del riesgo que han sido validados
en cierta medida.
1.8 Efectos en el ser humano
Habida cuenta de la complejidad del perfil de los HAP en el medio
ambiente y en los lugares de trabajo, la exposición humana a productos
puros por separado se ha limitado a experimentos científicos con
voluntarios, salvo en el caso del naftaleno, que se utiliza como
antipolilla para la ropa.
Tras la aplicación cutánea, el antraceno, el fluoranteno y el
fenantreno indujeron reacciones específicas en la piel, y el benzo[a]-
pireno produjo la formación de verrugas regresivas reversibles que se
clasificaron como proliferaciones neoplásicas. Los efectos sistémicos
del naftaleno se conocen por los numerosos casos de ingesta
accidental, particularmente de niños. La dosis letal por vía oral es
de 5000-15 000 mg para los adultos y 2000 mg ingeridos durante dos
días en los niños. El efecto normal tras la exposición por vía cutánea
u oral es una anemia hemolítica aguda que, a través de la placenta,
puede afectar también a los fetos.
El humo del tabaco es el factor aislado más importante en la inducción
de tumores de pulmón y también de un aumento de la incidencia de
tumores de la vejiga urinaria, la pelvis renal, la boca, la faringe,
la laringe y el esófago. No se considera que sea elevada la
contribución de los HAP en los alimentos a la aparición de tumores
humanos. En zonas fuertemente industrializadas se detectó también una
mayor carga corporal de HAP debido a la contaminación del aire. Están
expuestos asimismo a estos compuestos los enfermos de psoriasis
tratados con alquitrán de hulla.
La exposición profesional al hollín como causa de cáncer de escroto se
observó por primera vez en 1775. Más tarde se informó que la
exposición a los alquitranes y la parafina inducían cáncer cutáneo.
Ahora el cáncer inducido por HAP afecta principalmente al pulmón,
mientras que el cáncer cutáneo es más raro gracias a una mayor higiene
personal.
Se han realizado estudios epidemiológicos de trabajadores expuestos en
hornos de coque durante la coquificación del carbón y su gasificación,
en obras de asfaltado, fundiciones e instalaciones de aluminio y a los
gases de escape de los motores diesel. Se ha detectado un índice de
cáncer de pulmón más elevado debido a su exposición a los HAP en los
trabajadores de hornos de coque, los que utilizan asfalto y los de las
salas de crisoles de Söderberg de las instalaciones de reducción de
aluminio. El riesgo más elevado se observó en los trabajadores de
hornos de coque, con una razón de mortalidad normalizada de 195. En
varios estudios se hallaron relaciones dosis-respuesta. En las
instalaciones de aluminio no sólo se detectó cáncer de la vejiga
urinaria, sino también síntomas semejantes al asma, anomalías
funcionales de los pulmones y bronquitis crónica. En los trabajadores
de hornos de coque se observó asimismo una disminución de la
concentración de inmunoglobulina en el suero y una reducción de la
función immunitaria. Se notificó que una exposición durante cinco años
al naftaleno había provocado cataratas.
Se han elaborado varios métodos para evaluar la exposición interna a
los HAP. En la mayoría de los estudios, se midieron en orina
metabolitos de los HAP como los tioéteres urinarios, el 1-naftol, la
ß-naftilamina, los hidroxifenantrenos y el 1-hidroxipireno. Este
último se ha utilizado con frecuencia como indice biológico de
exposición.
Se han determinado los efectos genotóxicos de los HAP mediante pruebas
de mutagenicidad en orina y heces y de la presencia de m/cm-núcleos,
aberraciones cromosómicas e intercambio de cromátidas hermanas en
linfocitos de la sangre periférica. Además, se han medido aductos de
benzo[a]pireno con ADN en linfocitos periféricos y en otros tejidos y
con proteínas como la albúmina, así como anticuerpos de aductos de
ADN.
En varios estudios se han investigado el 1-hidroxipireno en orina y
los aductos de ADN en linfocitos como marcadores. El primero se puede
medir más fácilmente que los segundos, presenta menos variación entre
los individuos y es posible detectar niveles de exposición más bajos.
Ambos marcadores se han Utilizado para evaluar la exposición humana en
diversas condiciones. En varios puestos de trabajo de instalaciones de
coque, de fabricación de aluminio, de instalaciones de impregnación de
la madera, de fundiciones y de obras de asfaltado se observó un
aumento de la excreción de 1-hidroxipireno o de aductos de ADN. Las
exposiciones más elevadas se detectaron en los trabajadores de hornos
de coque y en los de impregnación de la madera con creosota, que
absorbían hasta el 95% del total de HAP a través de la piel, a
diferencia de la población general, en la que predomina la absorción
mediante los alimentos y el humo del tabaco.
Las estimaciones del riesgo relacionado con la exposición a HAP y sus
mezclas se basan en estimaciones de la exposición y en los resultados
de estudios epidemiológicos. Los datos obtenidos de trabajadores de
hornos de coque pusieron de manifiesto un riesgo relativo de cáncer de
pulmón de 15,7. Teniendo esto en cuenta, se ha calculado que el riesgo
de aparición de cáncer de pulmón en la población general durante toda
la vida es de 104 a 10-s por ng de banzo[a]pireno por m3 de aire.
Dicho de otra manera, como consecuencia de la exposición al
benzo[a]pireno en el aire cabra esperar la aparición de cáncer de
pulmón a lo largo de toda la vida en una persona de cada 10 000 a 100
000.
1.9 Efectos en otros organismos en el laboratorio y en el medio
ambiente
Los HAP tienen toxicidad aguda para los peces y para Daphnia magna en
combinación con la absorción de radiación ultravioleta y luz visible.
El metabolismo y la degradación alteran la toxicidad de los HAP. A
concentraciones bajas, los HAP pueden estimular el crecimiento de
microorganismos y algas. Los HAY más tóxicos para las algas son el
benz[a]antraceno (cuatro anillos), con una CE50 (concentración a la
cual determinados parámetros vitales se reducen a la mitad) de 1-29
µg/litro, y el benzo[a]pireno (cinco anillos), con una CE50 de 545
µg/litro. Los valores de la CL50 en las algas para la mayor parte de
los HAP de tres anillos son de 240-940 µg/litro. El naftaleno (dos
anillos) es el menos tóxico, con valores de la CE50 de 2800-34 000
µg/litro.
No se han observado diferencias claras de sensibilidad entre distintos
grupos taxonómicos de invertebrados, como los crustáceos, los
insectos, los moluscos, los poliquetos y los equinodermos. El
naftaleno es el menos tóxico, con valores de la CL50 en 96 horas de
100-2300 µg/litro. Los valores de la CL50 en 96 horas para los HAP
de tres anillos oscilan entre <1 y 3000 µg/litro. El antraceno puede
ser más tóxico que los otros HA/' de tres anillos, con CL50 en 96
horas entre <1 y 260 µg/litro: Los valores de la CL50 en 96 horas
para los HAP de cuatro, cinco y seis anillos son de 0,2-1200 µg/litro.
Se observó toxicidad aguda (CL50) en peces a concentraciones de 110
a >10 000 µg/litro de naftaleno, 30-4000 µg/litro de HAP de tres
anillos (antraceno, 2,8-360 µg/litro) y 0,7-26 µg/litro para los de
cuatro o cinco altillos.
La contaminación de los sedimentos con HAP a concentraciones de 250
mg/kg se asoció a tumores hepáticos en peces vivos libres. Se han
inducido asimismo tumores en peces expuestos en el laboratorio. La
exposición de los peces a determinados HAP puede producir también
cambios fisiológicos y afectar a su crecimiento, reproducción,
capacidad natatoria y respiración.