
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 210
PRINCIPLES FOR THE ASSESSMENT OF RISKS TO HUMAN HEALTH FROM
EXPOSURE TO CHEMICALS
This report contains the collective views of an international group
of experts and does not necessarily represent the decisions or the
stated policy of the United Nations Environment Programme, the
International Labour Organisation, or the World Health
Organization.
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1999
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO), and the World Health Organization (WHO). The overall
objectives of the IPCS are to establish the scientific basis for
assessment of the risk to human health and the environment from
exposure to chemicals, through international peer review processes, as
a prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization, the United Nations
Institute for Training and Research, and the Organisation for Economic
Co-operation and Development (Participating Organizations), following
recommendations made by the 1992 UN Conference on Environment and
Development to strengthen cooperation and increase coordination in the
field of chemical safety. The purpose of the IOMC is to promote
coordination of the policies and activities pursued by the
Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing-in-Publication Data
Principles for the assessment of risks to human health from exposure
to chemicals.
(Environmental health criteria ; 210)
1.Chemicals - toxicity
2.Chemicals - adverse effects
3.Risk assessment - methods
4.Environmental exposure
5.Toxicity tests
6.Dose-response relationship, Drug
7.No-observed-adverse effect level
I.International Programme on Chemical Safety
II.Series
ISBN 92 4 157210 8 (NLM Classification: QV 602)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1999
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in this
publication do not imply the expression of any opinion whatsoever on
the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
PRINCIPLES FOR THE ASSESSMENT OF RISKS TO HUMAN HEALTH FROM EXPOSURE
TO CHEMICALS
PREAMBLE
ABBREVIATIONS
1. SUMMARY
2. INTRODUCTION
3. HEALTH HAZARD IDENTIFICATION
3.1. Introduction
3.2. Human data
3.2.1. Criteria for establishing causality
3.3. Animal studies
3.4. In vitro studies
3.5. Structure-activity relationships
4. DOSE-RESPONSE
4.1. Introduction
4.2. Considerations in dose-response assessment
4.2.1. Introduction
4.2.2. Inter- and intra-species considerations
4.2.2.1 Introduction
4.2.2.2 Species differences
4.2.2.3 Human variability
4.3. Non-neoplastic (threshold) effects
4.3.1. Characterization of threshold
4.3.1.1 No-observed-adverse-effect level (NOAEL)
4.3.1.2 Benchmark dose/concentration
4.3.1.3 Lowest-observed-adverse-effect level
4.3.2. Uncertainty factors
4.4. Quantitative risk assessment for neoplastic (non-threshold)
effects
4.4.1. Introduction
4.4.2. Linear extrapolation
4.4.3. Estimation of potency in the experimental range
4.4.4. Two-stage clonal expansion model
4.4.5. Proportional analyses - carcinogenic and
non-neoplastic effects
5. EXPOSURE ASSESSMENT
5.1. Definition of exposure and related terms
5.2. Exposure and dose
5.3. Approaches to quantification of exposure
5.3.1. Measurement at point of contact (personal
monitoring)
5.3.2. Scenario evaluation method (time activity and
monitoring/modelling)
5.3.3. Biomarkers of exposure/estimation of internal dose
5.4. Variability and uncertainty
5.4.1. Assessing uncertainty
5.5. Exposure settings
5.5.1. Exposure in the general environment
5.5.2. Occupational settings
5.5.3. Consumer products
6. RISK CHARACTERIZATION AND IMPLICATIONS FOR RISK MANAGEMENT
6.1. General considerations
6.2. Considerations in risk characterization
6.3. Considerations in risk management
6.3.1. Societal factors
6.3.2. Individual and population risks
6.3.3. Comparative risk
6.3.4. Risk perception
6.3.5. Risk and hazard communication
6.3.6. Economic factors
6.3.6.1Cost-benefit analyses
6.3.7. Political factors
6.3.8. Regulatory limits
6.4. Risk management options
6.4.1. Risk reduction
6.4.1.1 Technology-based criteria
REFERENCES
APPENDIX
RÉSUMÉ
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41
22 - 9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number
5 U01 ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial support
from the European Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure
to environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects
of pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and
in vitro studies provide support and are used mainly to supply
evidence missing from human studies. It is mandatory that research on
human subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary -- a review of the salient facts and the risk evaluation
of the chemical
* Identity -- physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
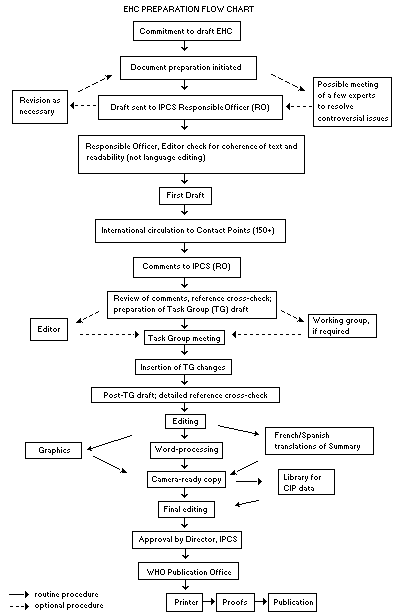 The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
PARTICIPANTS IN THE PLANNING AND TASK GROUP MEETINGS ON PRINCIPLES FOR
THE ASSESSMENT OF RISKS TO HUMAN HEALTH FROM EXPOSURE TO CHEMICALS
Members
Dr A. Aitio, Institute of Occupational Health, Laboratory of
Biochemistry, Helsinki, Finland a,b
Dr N. Aldrige, The Robens Institute of Industrial and Environmental
Health and Safety, University of Guildford, Guildford, Surrey, United
Kingdom (deceased)a,b
Dr D. Anderson, British Industry Biological Research Association
(BIBRA), Carshalton, Surrey, United Kingdoma,b
Professor C.L. Berry, Department of Morbid Anatomy, London Hospital
Medical College, London, United Kingdoma
Dr R. Burnett, Biostatistics and Computer Division, Environmental
Health Directorate, Health and Welfare Canada, Ottawa, Ontario,
Canadaa
Dr J.R.P. Cabral, Unit of Mechanisms of Carcinogenesis, International
Agency for Research on Cancer, Lyon, Francea
Dr E. Cardis, Unit of Biostatistics Research and Informatics,
International Agency for Research on Cancer, Lyon, Francea
Dr M. Cikrt, Institute of Hygiene and Epidemiology, Prague, Czech
Republica
Dr D.B. Clayson, Carp, Ontario, Canada
Mr D.J. Clegg, Pesticide Section, Toxicological Evaluation Division,
Food Directorate, Health Protection Branch, Tunney's Pasture, Ottawa,
Ontario, Canadaa
Professor E. Dybing, Department of Environmental Medicine, National
Institute of Public Health, Oslo, Norwayc
Dr R. Fielder, Department of Health, Elephant and Castle, London
United Kingdomb
Dr L. Fishbein, Fairfax, Virginia, USAc
Dr H. Gibb, US Environmental Protection Agency, Washington, DC,
USAa,b,d
Dr M. Goddard, Biostatistics and Computer Division, Environmental
Health Centre, Health and Welfare Canada, Tunney's Pasture, Ottawa,
Ontario, Canadab
Professor B. Goldstein, Rutgers Medical College, Busch Campus,
Pescataway, New Jersey, USAa
Dr R.F. Hertel, Federal Institute for Consumers, Health Protection and
Veterinary Medicine, FE-821 Bundesgesundheitsamt, BGVV, Berlin,
Germanyc,d
Dr J. Huff, Environmental Carcinogenesis Programme, National Institute
of Environmental Health Sciences, Research Triangle Park, North
Carolina, USAb
Professor M. Ikeda, Department of Environmental Health, Tohoku
University School of Medicine, Sendai, Japana
Dr D. Krewski, Biostatistics and Computer Division, Environmental
Health Directorate, Health and Welfare Canada, Ottawa, Ontario,
Canadaa
Professor R. Kroes, initially National Institute of Public Health
and Environmental Hygiene, Bilthoven, subsequently Research
Institute for Toxicology, University of Utrecht, Utrecht, the
Netherlandsa,c
Professor M. Lotti, University of Padua Medical School, Institute of
Occupational Medicine, Padua, Italya
Dr G.W. Lucier, Division of Biometry and Risk Assessment, National
Institute of Environmental Health Sciences, Research Triangle Park,
North Carolina, USAa
Dr L. Magos, Toxicology Unit, Medical Research Council Laboratories,
Carshalton, Surrey, United Kingdoma
Dr E. McConnell, Raleigh, North Carolina, USAa
Ms M.E. Meek, Environmental Health Directorate, Health Canada, Ottawa,
Ontario, Canadac
Dr R.L. Melnick, National Institute of Environmental Health Sciences,
Division of Biometry and Risk Assessment, Research Triangle Park,
North Carolina, USAa
Professor D.V. Parke, Department of Biochemistry, University of
Surrey, Guildford, Surrey, United Kingdoma
Dr J. Parker, Office of Health and Environmental Assessment, US
Environmental Protection Agency, Washington, DC, USAa
Dr O.E. Paynter, Hazard Evaluation Division, US Environmental
Protection Agency, Washington, DC, USAa
Dr P.K. Ray, Industrial Toxicology Research Centre, Lucknow, Indiaa
Dr A.G. Renwick, Clinical Pharmacology Group, University of
Southampton, Southhampton, Hampshire, United Kingdomc
Dr J. Sekizawa, Division of Information on Chemical Safety, National
Institute of Hygienic Sciences, Tokyo, Japanb
Dr J. Shaum, US Environmental Protection Agency, National Center for
Environmental Assessment, Washington, DC, USAd
Professor J.A. Sokal, Institute of Occupational Medicine and
Environmental Health, Sosnowiec, Polandc
Dr J. Steadman, Department of Health and Social Security, Elephant and
Castle, London, United Kingdoma
Dr L. Strayner, Division of Standards Development and Technology
Transfer, National Institute for Occupational Safety and Health,
Cincinnati, Ohio, USAb
Dr G.M.H. Swaen, Department of Occupational Medicine, University of
Limburg, Maastricht, the Netherlandsa,b
Dr A. Walker, Organisation for Economic Co-operation and Development,
Paris, Francea
Professor R. Walker, Food Safety Group, Division of Toxicology, School
of Biological Sciences, University of Surrey, Guildford, Surrey,
United Kingdomc
Dr J.E. Zejda, Department of Epidemiology, Institute of Occupational
Medicine and Environmental Health, Sosnowiec, Polandc
Observers
Professor G. Di Renzo, International Union of Toxicology, Department
of Neuroscience, Faculty of Medicine and Surgery, University of Naples
"Federico II", Naples, Italyc
Dr M. Jaroszewski, Health and Safety Directorate, Occupational
Medicine and Hygiene Unit, Commission of the European Community,
Luxembourgb
Dr C. Lally, European Council of Chemical Industry Federation (CEFIC),
Procter and Gamble, Strombbek Bever, Belgiumc
Professor A. Mutti, Institute of Clinical Medicine and Nephrology,
Parma, Italyc
Dr J. O'Donoghue (Representing AIHC) Corporate Health and Environment
Laboratories, Eastman Kodak Company, Rochester, New York, USAb
Dr M. Penman, ICI C & P Limited, Occupational Health Division, Wilton,
Middlesborough, Cleveland, United Kingdomc
Mrs M. Richold, European Centre for Ecotoxicology and Toxicology of
Chemicals (ECETOC), Unilever Research Laboratory, Environmental Safety
Laboratory, Sharnbrook, Bedford, United Kingdomc
Mr P. Verschuren, International Life Sciences Institute, Brussels
Belgiumc,b
Secretariat
Dr G.C. Becking, Inter-regional and Research Unit, International
Programme on Chemical Safety, World Health Organization, Research
Triangle Park, North Carolina, USAb
Dr K. Gutschmidt, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerlandd
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerlandc
Dr M. Younes, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerlandd
a Participated in Planning and Working Groups on Scientific
Principles for the Assessment of Risks to Human Health from
Exposure to Chemicals.
b Participated in the WHO Task Group Meeting on the initial draft
of Principles for the Assessment of Risk from Exposure to
Chemicals (British Industry Biological Research Association
(BIBRA), Carshalton, Surrey, United Kingdom, March 1993).
c Participated in the WHO Task Group Meeting on the initial draft
of General Principles and Methods for Chemical Safety (Human
Health Protection (National Institute of Public Health and
Environmental Protection) (RIVM), Bilthoven, the Netherlands, 22-
25 March 1994).
d Participated in the WHO Finalizing Group Meetings on Principles
for the Assessment of Risks to Human Health from Exposure to
Chemicals (World Health Organization, Geneva, Switzerland, 2-5
September 1996 and 18-20 September 1997).
PRINCIPLES FOR THE ASSESSMENT OF RISKS TO HUMAN HEALTH FROM EXPOSURE
TO CHEMICALS
This monograph is an amalgamation of two draft documents
"Principles for the Assessment of Risk from Exposure to Chemicals" and
"General Principles and Methods for Chemical Safety (Human Health
Protection)".
Both documents were planned to cover different aspects of
chemical safety and risk assessment; one dealing with the basic
science for general readers, and the other providing more practical
approaches to risk assessment of chemicals for risk assessors.
However, they turned out to have a substantial amount of overlapping
information and it was therefore decided to use both drafts as a basis
for this new, comprehensive document. The more detailed draft on
"General Principles and Methods for Chemical Safety (Human Health
Protection)" will be published as a separate document for training
purposes.
This Environmental Health Criteria monograph is aimed at
furnishing a practical overview of chemical safety and at providing
the framework of risk assessment for regulatory and research
scientists, as well as risk managers. It is intended to complement
existing Environmental Health Criteria that address methodologies for
the assessment of risks from exposure to chemicals with a view towards
different end-points or to susceptible population groups. It is not
intended as a textbook on toxicology.
This monograph should not be considered as being of a
prescriptive nature. The chapters on exposure assessment and risk
characterization, in particular, provide rather some practical
guidance.
Several planning, working and Task Group meetings took place to
discuss and agree upon the structures and contents of both
Environmental Health Criteria documents.
A WHO Task Group on "Principles for the Assessment of Risk from
Exposure to Chemicals" met at the British Industrial Biological
Research Association (BIBRA), Carshalton, Surrey, United Kingdom, in
March 1993. Dr G.C. Becking, IPCS, welcomed the participants on behalf
of the Director, IPCS, and the three IPCS cooperating organizations
(UNEP/ILO/WHO), and the Task Group reviewed the draft document.
The main contributors to the first draft on Principles for the
Assessment of Risk from Exposure to Chemicals were Dr N. Aldridge,
Robens Institute of Industrial and Environmental Health and Safety,
United Kingdom, Dr H. Gibb, US Environmental Protection Agency, Dr J.
Huff, National Institute of Environmental Health Sciences, USA, Dr L
Stayner, National Institute for Occupational Safety and Health, USA.
A second WHO Task Group met to review the draft monograph on
General Principles and Methods for Chemical Safety (Human Health
Protection). This group met in at the National Institute of Public
Health and Environmental Protection (RIVM), Bilthoven, the
Netherlands, from 22 to 25 November 1995. Dr E. Smith, IPCS, welcomed
the participants on behalf of the Director, IPCS, and the three IPCS
cooperating organizations (UNEP/ILO/WHO), and the Task Group reviewed
the draft document.
The main contributors to the draft on Principles for the
Assessment of Risk from Exposure to Chemicals were Dr D.B. Clayson,
Carp, Canada, Professor E. Dybing, National Institute of Public
Health, Norway, Dr L. Fishbein, Fairfax, Virginia, USA, Dr A.G.
Renwick, University of Southampton, United Kingdom, Professor R.
Walker, University of Surrey, United Kingdom, and Professor J.A Sokal,
Institute of Occupational Health and Environmental Medicine,
Sosnowiec, Poland.
In addition to the Task Group meetings, meetings were held during
1996 and 1997 in Geneva to combine the two documents.
Dr E. Smith and Dr G. Becking, both members of the IPCS, were
responsible for the preparation of the initial draft documents. Dr M.
Younes (IPCS) was responsible for the overall scientific content of
the final monograph and Dr P.G. Jenkins (IPCS) for the technical
editing.
The efforts of all who helped in the preparation and finalization
of the document are gratefully acknowledged.
ABBREVIATIONS
ADD average daily dose
ADI acceptable daily intake
EPI exposure/potency index
GLP good laboratory practice
IARC International Agency for Research on Cancer
LOAEL lowest-observed-adverse-effect level
NOAEL no-observed-adverse-effect level
OECD Organisation for Economic Co-operation and Development
PBPK physiologically based pharmacokinetic
SAR structure-activity relationship
US EPA US Environmental Protection Agency
1. SUMMARY
Control of risks from exposure to chemicals (chemical safety)
requires first of all a scientific, ideally quantitative, assessment
of potential effects at given exposure levels (risk assessment). Based
upon the results of risk assessment, and taking into consideration
other factors, a decision-making process aimed at eliminating or, if
this is not possible, reducing to a minimum the risk to the
chemical(s) under consideration (risk management), can be started.
Risk assessment is a conceptual framework that provides the
mechanism for a structured review of information relevant to
estimating health or environmental outcomes. In conducting risk
assessments, the National Academy of Sciences risk assessment paradigm
has proven to be a useful tool (US NAS, 1983). This paradigm divides
the risk assessment process into four distinct steps: hazard
identification, dose-response assessment, exposure assessment and risk
characterization.
The purpose of hazard identification is to evaluate the weight of
evidence for adverse effects in humans based on assessment of all
available data on toxicity and mode of action. It is designed to
address primarily two questions: (1) whether an agent may pose a
health hazard to human beings, and (2) under what circumstances an
identified hazard may be expressed. Hazard identification is based on
analyses of a variety of data that may range from observations in
humans to analysis of structure-activity relationships. The result of
the hazard identification exercise is a scientific judgement as to
whether the chemical evaluated can, under given exposure conditions,
cause an adverse health effect in humans. Generally, toxicity is
observed in one or more target organ(s). Often, multiple end-points
are observed following exposure to a given chemical. The critical
effect, which is usually the first significant adverse effect that
occurs with increasing dose, is determined.
Dose-response assessment is the process of characterizing the
relationship between the dose of an agent administered or received and
the incidence of an adverse health effect. For most types of toxic
effects (i.e. organ-specific, neurological/behavioural, immunological,
non-genotoxic carcinogenesis, reproductive or developmental), it is
generally considered that there is a dose or concentration below which
adverse effects will not occur (i.e. a threshold). For other types of
toxic effects, it is assumed that there is some probability of harm at
any level of exposure (i.e. that no threshold exists). At the present
time, the latter assumption is generally applied primarily for
mutagenesis and genotoxic carcinogenesis.
If a threshold has been assumed (e.g., for non-neoplastic effects
and non-genotoxic carcinogens), traditionally, a level of exposure
below which it is believed that there are no adverse effects, based on
a no-observed-adverse-effect level (NOAEL) (approximation of the
threshold) and uncertainty factors, has been estimated. Alternatively,
the magnitude by which the no (lowest)-observed-adverse-effect level
(N(L)OAEL) exceeds the estimated exposure (i.e. the "margin of
safety") is considered in light of various sources of uncertainty. In
the past, this approach has often been described as a "safety
evaluation". Therefore, the dose that can be considered as a first
approximation of the threshold, i.e. the NOAEL, is critical.
Increasingly, however, the "benchmark dose", a model-derived estimate
(or its lower confidence limit) of a particular incidence level (e.g.,
5%) for the critical effect, is being proposed for use in quantitative
assessment of the dose-response for such effects.
There is no clear consensus on appropriate methodology for the
risk assessment of chemicals for which the critical effect may not
have a threshold (i.e. genotoxic carcinogens and germ cell mutagens).
Indeed, a number of approaches based largely on characterization of
dose-response have been adopted for assessment in such cases.
Therefore, the critical data points are those that define the slope of
the dose-response relationship (rather than the NOAEL, which is the
first approximation of a threshold).
The third step in the process of risk assessment is the exposure
assessment, which has the aim of determining the nature and extent of
contact with chemical substances experienced or anticipated under
different conditions. Multiple approaches can be used to conduct
exposure assessments. Generally, approaches include indirect and
direct techniques, covering measurement of environmental
concentrations and personal exposures, as well as biomarkers.
Questionnaires and models are also often used. Exposure assessment
requires the determination of the emissions, pathways and rates of
movement of a substance and its transformation or degradation, in
order to estimate the concentrations to which human populations or
environmental spheres (water, soil and air) may be exposed.
Depending on the purpose of an exposure assessment, the numerical
output may be an estimate of either the intensity, rate, duration or
frequency of contact exposure or dose (resulting amount that actually
crosses the boundary). For risk assessments based on dose-response
relationships, the output usually includes an estimate of dose. It is
important to note that the internal dose, not the external exposure
level, determines the toxicological outcome of a given exposure.
Risk characterization is the final step in risk assessment. It is
designed to support risk managers by providing, in plain language, the
essential scientific evidence and rationale about risk that they need
for decision-making. In risk characterization, estimates of the risk
to human health under relevant exposure scenarios are provided. Thus,
a risk characterization is an evaluation and integration of the
available scientific evidence used to estimate the nature, importance,
and often the magnitude of human and/or environmental risk, including
attendant uncertainty, that can reasonably be estimated to result from
exposure to a particular environmental agent under specific
circumstances.
The term "risk management" encompasses all of those activities
required to reach decisions on whether an associated risk requires
elimination or necessary reduction. Risk management strategies/or
options can be broadly classified as regulatory, non-regulatory,
economic, advisory or technological, which are not mutually exclusive.
Thus legislative mandates (statutory guidance), political
considerations, socioeconomic values, cost, technical feasibility,
populations at risk, duration and magnitude of risk, risk comparison,
and possible impact on trade between countries can generally be
considered as a broad panoply of elements that can be factored into
final policy or rule making. Key decision factors such as the size of
the population, the resources, costs of meeting targets and the
scientific quality of risk assessment and subsequent managerial
decisions vary enormously from one decision context to another. It is
also recognized that risk management is a complex multidisciplinary
procedure which is seldom codified or uniform, is frequently
unstructured, but which can respond to evolving input from a wide
variety of sources. Increasingly, risk perception and risk
communication are recognized as important elements, which must also be
considered for the broadest possible public acceptance of risk
management decisions.
Chemicals have become an indispensable part of human life,
sustaining activities and development, preventing and controlling many
diseases, and increasing agricultural productivity. Despite their
benefits, chemicals may, especially when misused, cause adverse
effects on human health and environmental integrity. The widespread
application of chemicals throughout the world increases the potential
of adverse effects. The growth of chemical industries, both in
developing as well as in developed countries, is predicted to continue
to increase. In this context, it is recognized that the assessment and
management of risks from exposure to chemicals are among the highest
priorities in pursuing the principles of sustainable development.
2. INTRODUCTION
Despite the societal benefits that accrue from the use of
chemicals, substantial potential hazards to health may be associated
with exposure during the production, use or disposal of the
approximately 100 000 unique chemicals or 4 million mixtures,
formulations and blends already in commercial use or the several
hundred new synthetic chemicals introduced each year (EC, 1990). This
monograph outlines the nature of the data available and their use in
the assessment of risk in a risk assessment/risk management framework.
It is hoped that scientists, risk assessors and health risk managers
will find this monograph helpful to decision-making in this area.
A number of national and international organizations and agencies
have developed guidance on assessment of exposure and various health
end-points (e.g., carcinogenicity, developmental toxicity, etc.). It
is not the purpose of this monograph to endorse particular approaches
but rather to acquaint the reader with relevant methodology and issues
for consideration.
It is also hoped that the reader will find this monograph useful
in the interpretation of risk assessments on specific chemicals. The
reader is referred to such sources for chemical-specific hazard
identification and, depending on the monograph, dose-response
information. A list of assessments produced by various national and
international agencies is included in ECETOC/UNEP (1996). These
sources do not, of course, provide the exposure information necessary
to characterize risk at the local level. Since exposure will vary
considerably under different circumstances, responsible authorities
are strongly encouraged to characterize risk on the basis of local
measured or predicted exposure scenarios. It is hoped that the general
approaches to exposure assessment described in this monograph will
assist the reader in characterizing risk in specific situations.
In the chapters of this monograph, the following four distinct
and essential components of the risk assessment paradigm are
addressed:
(1) hazard identification - identification of the inherent
capability of a substance to cause adverse effects;
(2) assessment of dose-response relationships involves
characterization of the relationship between the dose of an agent
administered or received and the incidence of an adverse effect;
(3) exposure assessment is the qualitative and/or quantitative
assessment of the chemical nature, form and concentration of a
chemical to which an identified population is exposed from all
sources (air, water, soil and diet);
(4) risk characterization is the synthesis of critically evaluated
information and data from exposure assessment, hazard
identification and dose-response considerations into a summary
that identifies clearly the strengths and weaknesses of the
database, the criteria applied to evaluation and validation of
all aspects of methodology, and the conclusions reached from the
review of scientific information.
The logical consequence of the process of assessment of potential
risk is the application of the information to the development of
practical measures (risk management) for the protection of human
health. Although not the principal focus of this monograph, the
importance of clear understanding and communication of the nature and
limitations of the scientific basis for risk assessment in risk
management is addressed in the final chapter.
In Appendix 1 to this monograph, an example of a hazard
identification scheme for carcinogenicity, developed by the
International Agency for Research on Cancer (IARC), is presented. In
Appendix 2, the currently available and draft guidelines of the
Organisation for Economic Cooperation and Development (OECD) for
testing of chemicals are presented. For sample exposure and risk
characterizations, readers are referred to IPCS (1994).
3. HEALTH HAZARD IDENTIFICATION
3.1 Introduction
The purpose of hazard identification is to evaluate the weight of
evidence for adverse effects in humans based on assessment of all
available data on toxicity and mode of action. It is designed to
address primarily two questions: (a) whether an agent may pose a
health hazard to humans, and (b) under what circumstances an
identified hazard may be expressed. Hazard identification is based on
analyses of a variety of data that may range from observations in
humans to analysis of structure-activity relationships.
In hazard identification, the weight of evidence is assessed on
the basis of combined strength and coherence of inferences
appropriately drawn from all of the available data. This entails
rigorous examination of the quantity, quality and nature of the
results of available toxicological and epidemiological studies and
structure-activity analyses and information on mechanisms of toxicity.
The latter is particularly important with respect to assessment of
relevance to humans.
Several classification schemes provide a framework for assessment
of the weight of evidence for various toxicological end-points (DFG,
1972; IPCS, 1986 (neurotoxicity); US EPA, 1986a, 1996a; IARC, 1987;
EC, 1992; Health Canada, 1994; IPCS, 1996 (immunotoxicity); IPCS, 1997
(delayed hypersensitivity)). An example (the IARC scheme) is presented
in Appendix 1 to illustrate the nature of criteria on which
classification of weight of evidence is based. Such classification
schemes have been helpful in standardizing and communicating the
assessment of hazard identification for particular end-points. In
addition to the classifications themselves, narrative statements to
summarize the nature of and confidence in the evidence based on
limitations and strengths of the database are helpful. Issues that are
often addressed include: the nature, reliability, validity and
consistency of data on response in humans and in laboratory animals,
current knowledge of the mechanistic basis for the response, and, in
the absence of human data, the relevance of responses in experimental
animals to humans.
The result of the hazard identification exercise is a scientific
judgement as to whether the chemical can cause an adverse effect in
humans.
The following is intended to provide the reader with an
appreciation of the complexity of considerations made in assessing
different types of data as a basis for hazard identification in risk
assessment. Fundamentals of epidemiology and toxicity testing are not
addressed here since they are considered in several other sources. An
Environmental Health Criteria monograph on the principles of exposure
assessment is currently in preparation (IPCS, in preparation).
Each source of information (e.g., human data, animal data,
structure-activity relationships) has its advantages and limitations
in contributing to an assessment of weight of evidence, but,
collectively, they permit characterization of potential adverse health
effects.
3.2 Human data
Well-documented observational and clinical epidemiological
studies have the clear advantage over studies in animals in providing
the most relevant information on health effects in the species of
interest, thus avoiding extrapolation from animals to humans. In
addition, epidemiological studies can address hazards to which humans
are exposed in their natural environment, in the presence of
concomitant risk factors such as diet and smoking.
Human populations are heterogeneous in their composition, and
studies of exposed populations are likely to include individuals of
differing susceptibility to the chemical of interest. This may be
viewed as an advantage relative to toxicological studies, which
involve genetically homogeneous populations of test animals.
The database for direct hazard identification in human
populations consists primarily of observational (epidemiological)
studies and case reports. Some information is also available from
ethically conducted human volunteer studies.
In observational studies, the investigator does not control
assignment of study subjects to either exposed or non-exposed groups.
Rather, such studies involve investigation of various individuals or
groups of subjects as they happen to have been exposed, and at no
stage of the study is the exposure of subjects influenced by the
research protocol. Although exposure scenarios are more realistic than
those in the experimental setting, owing to their observational nature
it is often difficult to control for "confounding factors", which may
be contributing to the etiology of the disease being investigated. For
example, variations in smoking between groups may confound
interpretation of observations concerning lung cancer.
Ethical experimental studies in human volunteers offer the
advantage of being better able to control for confounding factors. The
assignment of study subjects to exposure groups is made by the
investigator, who also controls the quality and quantity. Although
such investigations are generally reliable for the establishment of
both causality and exposure-response relationships, they are most
often restricted for ethical reasons to the examination of mild,
temporary effects (e.g., neurobehavioural or biochemical changes) of
short-term exposures in a limited number of subjects. They have
contributed considerably, particularly to our understanding of
kinetics and to the development of air quality guidelines and
standards for traditional pollutants.
Case reports describe a particular effect in an individual or
group of individuals who were exposed to a substance and often
observed by a single physician or group of physicians. These reports
are often anecdotal or highly selected in nature. Owing primarily to
their lack of statistical stability, they are of limited use for
hazard assessment, though helpful in generating hypotheses for further
study. However, reports of cases of the disease or effect of interest
can identify associations, particularly when there are unique features
such as an association with a rare disease or effect of interest
(e.g., vinyl chloride and angiosarcoma or methylmercury and Minamata
disease).
The major types of epidemiological (observational) studies are
analytical and descriptive or correlational studies. Each study type
has well-known strengths and weaknesses that affect interpretation of
study results (Lilienfeld & Lilienfeld, 1979; Mausner & Kramer, 1985;
Kelsey et al., 1986; Rothman, 1986). Analytical epidemiological
studies (that is, cohort and case-control studies), in which exposure
and outcome are examined in individuals rather than in populations,
are generally most reliable in hazard identification as a basis for
risk assessment since it is possible to adjust more rigorously for
confounding factors. The assessment of results of such studies is
based on several features of study design including estimation of
exposure, the role of confounding variables and the measurement of
outcome. Potential limitations, depending upon the nature of the
design, include lack of information on exposure, insufficient sample
size, short length of follow-up and potential bias and confounding.
These factors may limit the usefulness of particular studies for the
purposes of risk assessment.
Epidemiological data demonstrating dose-response, if available,
provide an advantageous basis for analysis, since concerns about
inter-species extrapolation do not arise. Adequacy of human exposure
data for quantification is an important consideration in deciding
whether epidemiological data are the best basis for analysis in a
particular case. If adequate exposure data exist in a well-designed
and well-conducted epidemiological study that detects no effects, it
may be possible to obtain an upper estimate of the potential human
risk to provide a check on plausibility of available estimates based
on animal tumour or other responses (e.g., do confidence limits on one
overlap the point estimate of the other?) (Stayner & Bailer, 1993; US
EPA 1996a).
3.2.1 Criteria for establishing causality
The first step in the evaluation of results of studies in humans
as a basis for hazard identification is the assessment of the
individual results of each separate report. The strengths and
weaknesses of each study must be considered along with potential for
the existence of bias (Gehlbach, 1982), with particular attention to
exposure data, criteria for definition of health outcome under study,
the size of the study population and the statistical power of the
analysis to detect adverse health effects. A set of standardized
criteria for assessing the weight of evidence of causality based on
assessment of the database has been developed (Hill, 1965; Susser,
1977).
Studies in which there is an apparent absence of evidence for a
hypothesized causal relationship between exposure and effect
("negative studies") need to be interpreted carefully (Hernberg,
1980). Such studies should be evaluated for dilution (the inclusion of
unexposed people in an allegedly exposed group of persons),
misclassification (Copeland et al., 1977), omissions, or premature
examination of subjects for diseases that may have long induction
(latency) periods. In addition, the statistical power of the study,
i.e. the probability that the study will be able to demonstrate the
presence of an effect, such as excessive disease or mortality, in a
population if the effect is actually present (Beaumont & Breslow,
1981), must be assessed.
There is no clear-cut criterion to distinguish positive from
negative studies. Although statistical significance has often been
used as the criteria, most epidemiologists believe that it is overly
simplistic to base decisions on arbitrary probability values (Rothman,
1986). For example, when a study fails to detect a statistically
significant effect, this may simply reflect inadequate sample size or
other aspects of study design. Conversely, when the results of a study
are statistically significant, the seemingly positive results may
still be due to confounding or even chance.
A positive association between an agent and an effect may be
interpreted as implying causality, to a greater or lesser extent, if
the following criteria are met: (a) there is not identifiable positive
bias; (b) the possibility of positive confounding has been considered;
(c) the association is unlikely to be due to chance alone; (d) the
association is strong; and (e) there is a dose-response relationship
(IARC, 1990). The following criteria for inferring causality from the
results of epidemiological studies have been developed by Hill (1965):
(a) The strength of the association as measured by the relative risk
In general, epidemiologists have more confidence in their results
when the magnitude of the relative risk is large. However, relative
risks of small magnitude do not necessarily imply lack of causality
and may be important if the disease under study is common (IARC,
1990). In evaluating relative risks, it is important to note the
actual numbers of observed and expected cases.
(b) The consistency of the association
The case for causal inference is strengthened by repetition of
findings "by different investigators, in different places,
circumstances and times" (Hill, 1965). The reproducibility of findings
constitutes one of the strongest arguments for the existence of
causality. If there are discordant results among investigations,
possible reasons such as differences in exposure should be considered
in assessing the results, and data from studies judged to be of high
quality given greater weight than data from studies judged to be
methodologically less sound (IARC, 1990).
(c) The temporal relationship between cause and effect
This principle may be simply restated as exposure must precede
illness. When latency is a factor, exposures must have occurred
sufficiently early to have produced an effect by the time of the
study.
(d) The biological gradient of the association
The evidence for causality is strengthened when the risk of
disease is shown to increase with levels of exposure. Because there
are many possible reasons that an epidemiological study may fail to
detect an exposure-response relationship (e.g., poor exposure data,
lack of adequate exposure gradient), the absence of a dose-response
relationship does not necessarily imply that the relationship is not
causal (IARC, 1990). Strong evidence for causality is provided when a
change in exposure brings about a change in disease frequency
(Hernberg, 1980), e.g., the decrease in risk of lung cancer that
follows cessation of smoking (Doll & Hill, 1956).
(e) the specificity of the association
A highly specific association is one in which the disease under
study is only induced by a particular agent. Specificity of cause is
common in infectious diseases but less common in chronic diseases that
often have a multi-factorial etiology. However, a specific association
may be observed for certain chronic diseases such as between exposure
to crocidolite asbestos and mesothelioma or vinyl chloride and
angiosarcoma. Although the presence of specificity seems to imply
causality, its absence does not exclude it (Fralick, 1983).
(f) biological plausibility of the association
Hill (1965) stated strongly that a proposed causal relationship
should not seriously conflict with knowledge of the biology and
pathophysiology of a disease under study. An epidemiological inference
of causality may be strengthened by data from experimental studies
showing consistency with biological mechanisms. For example, exposure
to ionizing radiation causes cancer in many animal species. However,
the lack of mechanistic or positive animal bioassay data to support an
association observed in an epidemiological study is not, in itself,
sufficient reason to reject causality.
3.3 Animal studies
Owing to the lack of adequate epidemiological data for most
substances, toxicological studies in animal species play an important
role in hazard identification for risk assessment. Toxicity studies
vary widely in purpose, design and conduct, and range from relatively
well-standardized and widely accepted test methods for assaying
various types of toxicity to large numbers of basically
research-oriented investigations employing specialized study designs.
The design, conduct and completeness of reporting of experimental
findings in toxicological studies on mammalian species are of critical
importance in determining the validity and relevance of results.
Toxicological results from adequate animal systems signal anticipated
effects in humans. Thus, negative results cannot be assessed from an
inadequate study, and full evaluation of a positive effect is
confounded by incomplete reporting from poorly designed or poorly
conducted studies. However, positive findings cannot be ignored.
Studies should be of good scientific quality and follow standard
guidelines and recognized good laboratory practices (GLPs) wherever
possible.
Information on the design of specific bioassays, including those
that address acute, short-term, sub-chronic, chronic and developmental
and reproductive toxicity, immunotoxicity and carcinogenicity, are not
presented here but are available in test guidelines, for which
principles of GLP are also specified (IARC, 1986; OECD, 1987, 1998;
Chhabra et al., 1990). A list of currently available OECD Guidelines
is included in Appendix 2. In this section, examples of factors to be
taken into account in assessing these various aspects of study design
for hazard identification are described.
Major end-points in toxicity studies can be grouped into the
following categories (IPCS, 1987a):
* Functional manifestations (weight loss, laxative effects, etc.);
* non-neoplastic lesions with morphological
manifestations/organ-directed toxic effects;
* neoplastic/carcinogenic manifestations.
In addition, a number of specific end-points may require targeted
testing strategies. Such end-points include skin and eye irritation,
reproductive/developmental manifestations, immunotoxicity and
neurotoxicity (including neurodevelopmental effects).
It is important to recognize that there are two types of data
generated in such studies; those in which response is graded, such as
enzyme inhibition (i.e. continuous data), and those in which the
response occurs or does not occur in a single animal, such as a
particular tumour (i.e. quantal data).
In assessing the relevance of various toxicological studies to
hazard identification and risk assessment, several features of study
design are considered, including the purity of the compound
administered, physico-chemical properties (volatility, stability,
solubility), homogeneity of distribution in inhalation experiments,
the size of the study (i.e. the number of exposed and control
animals), whether the study adhered to the principles of GLP, the
relevance of the route of exposure to that of humans, duration of
exposure, the number and suitability of the dose levels administered,
the extent of examination of various toxicological end-points and the
statistical analysis of the data. The types, site, incidence and
severity of effects and the nature of the exposure- or dose-response
relationship are also taken into account. Where data indicate that
there are significant differences in absorption, distribution,
metabolism and elimination of the compound in different animal
species, wherever possible, studies in which the species and strain of
animal are most similar to Homo sapiens in this regard are used
(where relevant human data are available). The consistency of the
results of the principal studies are also considered in the assessment
of the weight of evidence for an effect (e.g., whether similar effects
have been observed in studies in other species or whether such effects
would have been expected based on the structure or properties of the
chemical).
For example, the size of each exposure and concurrent control
group should be large enough for thorough toxicological and
statistical evaluation. The number of animals considered sufficient
depends on the variability, sensitivity and nature (e.g., quantal or
continuous) of the end-point being evaluated. For example, it is
commonly 50 per group in carcinogenicity bioassays where the responses
of interest are quantal in nature and 10 per group in subchronic
studies, where many of the examined end-points are continuous.
Studies in which the route of exposure is similar to that of
humans are most relevant to hazard identification for risk assessment.
For substances of low toxicity, it is important to ensure that when
administered in the diet, the quantities of the substance do not
interfere with normal nutritional needs.
Studies designed and conducted with 3-5 dosed groups plus a
vehicle control group of animals will yield reasonable dose-response
data relevant to hazard identification. The highest concentration of
the chemical should be one that induces a recognizable effect in the
animals such as changes in body or organ weights, enzyme changes or
minor histological changes. Changes such as mortality, gross
pathological changes, and painful or stressful conditions should be
avoided as they may confound the results of the study and may not be
in compliance with national and local animal welfare regulations.
Intermediate dose(s) should be targeted to produce minimally
observable toxic effects. Dose levels should be selected to produce
graded responses; too large intervals may complicate accurate
estimations of the lowest-observed-effect level (LOEL). Ideally, the
lowest dose should not demonstrate any toxicity (e.g., a NOAEL).
To assess fully the toxicological potential of a chemical for
local and systemic effects, all major organ systems should be examined
for dose-related effects and adverse effects in various organs should
be evaluated and described.
3.4 In vitro studies
Isolated cells, tissues and organs can be prepared and maintained
in culture by methods that preserve their in vivo properties and
characteristics. Increasing concern about the ethics of animal
experimentation has served to catalyse efforts leading to the possible
replacement or reduction in the use of animals, and the refinement of
test methods to minimize the stress and suffering to animals (ECETOC,
1989; Gelbke, 1993). In vitro testing contributes particularly to
the assessment of genotoxicity, permitting a decision concerning the
need for further testing.
Over the last decade, in vitro tests have been proposed as a
pre-screen or as an alternative method for other end-points, such as
prenatal toxicity, eye irritation, dermal irritation, tumour promotion
and target organ toxicity (Purchase, 1986; Tennant et al., 1987;
Anderson, 1990; Frazier, 1993; Atterwill, 1995). There has been
particular emphasis on validation programmes for skin and eye
irritation, but most of the tests mentioned above have not yet been
sufficiently validated and the results of validation studies,
especially in the past, have been lacking in consistency. The results
have failed to meet the need for reproducibility and high correlation,
ideally with sound human data but usually, for practical reasons, with
existing animal tests, which they are intended to replace.
Aspects that are important in assessing the adequacy of
in vitro studies include:
* the range of exposure levels, taking into account the toxicity of
the substance in the bacteria/cells, its solubility and, where
appropriate, its effects on the pH and osmolality of the culture
medium;
* whether, in the case of volatile substances, precautions were
taken to ensure the maintenance of effective concentrations of
the substance in the test medium;
* whether (when necessary) an appropriate exogenous metabolism mix
(e.g., S9 from induced rat or hamster liver) was used;
* whether appropriate negative and positive controls were included;
and
* whether there was an adequate number of replicates (within the
tests and of the tests).
Clearly, greater mechanistic understanding would facilitate
moving from purely empirical/correlative approaches to more
mechanistic-based tests. This is likely to facilitate greatly the
chances of adequate validation and acceptance of alternatives for
regulatory purposes.
3.5 Structure-activity relationships
Where epidemiological and toxicological data are not available,
the use of structure-activity relationships (SARs) may be considered.
SARs are based on the assumption that chemical substances that reach
and interact with target sites by the same mechanism do so as a result
of their similar chemical properties.
At present, SAR techniques, particularly those of a quantitative
nature, are not well developed in relation to mammalian toxicity. They
are primarily of value in predicting toxicokinetic properties and in
priority setting for research and evaluation.
4. DOSE-RESPONSE
4.1 Introduction
Approaches to quantification of dose-response vary according to
the scope and purpose of assessments. However, for most types of toxic
effects (i.e. organ-specific, neurological/behavioural, immunological,
non-genotoxic carcinogenesis, reproductive or developmental), it is
generally considered that there is a dose or concentration below which
adverse effects will not occur (i.e. a threshold). For other types of
toxic effects, it is assumed that there is some probability of harm at
any level of exposure (i.e. that no threshold exists); this currently
applies primarily for mutagenesis and carcinogenesis. Some have
restricted the non-threshold assumption to genotoxic carcinogens.
The distinction in approaches for genotoxic carcinogens and other
types of toxic effects is based primarily on the premise that simple
events such as in vitro activation and covalent binding may be
linear over many orders of magnitude. Though it is not possible to
demonstrate experimentally the presence or absence of a threshold,
differences in approach to the dose-response assessment of genotoxic
versus non-genotoxic carcinogens have been adopted in some countries.
However, simple pragmatic distinction on this basis is increasingly
problematic. For example, it is likely that there are thresholds for
aneugenic genotoxic effects.
If a threshold has been assumed (e.g., for non-neoplastic effects
and non-genotoxic carcinogens), traditionally, a level of exposure
below which it is believed that there are no adverse effects, based on
a no-observed-adverse-effect level or NOAEL (approximation of the
threshold) and uncertainty factors, has been estimated (section 4.3).
Alternatively, the magnitude by which the N(L)OAEL exceeds the
estimated exposure (i.e. the "margin of safety"), is considered in
light of various sources of uncertainty (Commission Regulation (EC)
No. 1488/94; Council Regulation (EEC) 793/93) (EC, 1993, 1994). In the
past, this approach has often been described as "safety evaluation".
Therefore, the dose that can be considered as a first approximation of
the threshold, i.e. the NOAEL, is critical. Increasingly, however, the
"benchmark dose", a model-derived estimate (or its lower confidence
limit) of a particular incidence level (e.g., 5%) for the critical
effect, is being proposed for use in quantitative assessment of the
dose-response for such effects.
At present, there is no clear consensus on appropriate
methodology for the risk assessment of chemicals for which the
critical effect may not have a threshold (i.e. genotoxic carcinogens
and germ cell mutagens). Indeed, a number of approaches based largely
on characterization of dose-response have been adopted for assessment
in such cases (section 4.4). Therefore, the critical data points are
those that define the slope of the dose-response relationship (rather
than the NOAEL, which is the first approximation of a threshold).
In North America and some European countries, cancer risks have
traditionally been assessed by mathematical modelling of the
dose-response data in the observable range to estimate the risk at
much lower human intakes or exposures (low dose risk extrapolation).
It should be noted, however, that quantitative estimation of such
risks, particularly those orders of magnitude below the experimental
range (i.e. low dose risk estimation), is uncertain. Owing to this
uncertainty, some countries have chosen not to adopt this approach as
the basis for their regulatory actions for genotoxic carcinogens, and
other countries are increasingly adopting alternative measures of
dose-response. In Canada and the USA, for example, there is,
currently, increasing reliance on specification of the margin between
potency in the experimental range and exposure as the measure of risk
for carcinogens (Health Canada, 1994; US EPA, 1996b). In the United
Kingdom, dose-response for genotoxic carcinogens is not quantified;
instead the goal in risk management is to eliminate exposure or to
reduce levels to as low as is reasonably practical (UK DOH, 1991).
Owing to the increasing reliance on modelling in the experimental
range to characterize dose-response for tumours, which is essentially
similar to the benchmark dose being used increasingly to characterize
dose-response for non-neoplastic effects, approaches to quantitative
risk estimation for carcinogenic and non-neoplastic effects are
converging.
4.2 Considerations in dose-response assessment
4.2.1 Introduction
In considering toxic effects at various dose levels, the dose range of
interest is generally the low-dose range, since it usually reflects
the human exposure situation. Often, however, data on dose-response
are available for higher doses only, and are often derived from animal
experiments only. Therefore, the uncertainty in the dose-response
assessment is larger than the uncertainty in hazard identification, as
it requires extrapolation both from animal to human and from high-dose
to low-dose levels. In certain instances, a distinction is made
between response and effect, with a response being quantal and counted
(e.g., the incidence of a tumour) and an effect being graded and
measured (e.g., relative liver weight).
4.2.2 Inter- and intra-species considerations
4.2.2.1 Introduction
The strains and species of laboratory animals exposed in toxicity
studies have been selected to show minimum inter-individual
variability. In contrast to laboratory animals, humans represent a
very heterogeneous population with both genetic and acquired
diversity.
Therefore, two principal areas are considered when interpreting
data on toxicity acquired in animal species in relation to human risk:
a) Inter-species consideration: comparison of the data for animals
with a representative healthy human. Species differences result
from metabolic, functional and structural variations.
b) Intra-species or inter-individual consideration: comparison of
the representative healthy human with the range of variability
present within the human population in relation to the relevant
parameter(s).
For each of these areas, there are two aspects to be considered
in assessing risk, i.e. toxicokinetics (the delivery of the compound
to the site of action) and toxicodynamics (the inherent sensitivity of
the site of action to the chemical). Any approach that allows for the
incorporation of adequate data on toxicokinetic or toxicodynamic
differences between test animal and humans, or between different
humans, will increase the scientific validity of risk assessment.
Sources of inter-species and inter-individual variations in
toxicokinetics include differences in anatomy (e.g., gastrointestinal
structure and function), physiological function (e.g., cardiac output,
renal and hepatic blood, glomerular filtration rate and gastric pH),
and biochemical differences in, for example, enzymes involved in
xenobiotic metabolism. Sources of inter-species and inter-individual
differences in toxicodynamics (or inherent sensitivity) also include
anatomy. For example, the effect may occur in an organ of questionable
relevance to humans, such as the rodent forestomach. Physiological
differences, such as the hormonal control of the target organ, and
biochemical differences, e.g., species differences in key biochemical
components such as alpha2u-globulin, may also play a role (Flamm &
Lehman-McKeeman, 1991).
In some cases, it may be possible to conclude that effects
detected in animals are unlikely to be relevant to humans. In other
cases, there may be data to indicate that humans are likely to be more
or less sensitive than animal species; this information is important
for consideration in selection of critical effects.
If compound-specific toxicokinetic data are introduced into risk
assessment, then it is essential that these are related to the
species, protocol and active chemical entity (e.g., parent compound or
metabolite) involved in the toxicity that is the basis for the hazard
identification (Monro, 1990, 1993; Renwick, 1993a).
4.2.2.2 Species differences
Metabolism and structural/functional variations are both
important determinants of species differences. Common areas of
metabolic variation between species are digestive tract enzymes,
levels of circulating enzymes, liver enzymes and detoxification
processes.
In extrapolating between species, three aspects need to be
considered: the first relates to differences in body size, which
requires dose normalization or scaling (often done by expressing the
dose per kg body weight). The second relates to differences in
toxicokinetics, particularly bioactivation and/or detoxification
processes. The third aspect concerns the nature and severity of the
target for toxicity. Inter-species normalization (or scaling) is
generally based on physical characteristics (e.g., body weight, body
surface area), although occasionally it is based on caloric demand or,
where there are data in four species, multiple species regression.
When clearance of the parent substance is limited by enzyme
activity rather than blood flow or when metabolites are the toxic
agents, more sophisticated physiologically based pharmacokinetic
models are more appropriate, provided that adequate data are
available. Currently, such data are available for only a small number
of substances.
4.2.2.3 Human variability
Although data from animal studies may provide limited information
on inter-individual variability within the test species, it is the
greater potential variability in the human population that must be
addressed in risk assessment. Sources of inter-individual variability
in human populations include, for example, variations in genetic
composition, nutrition, disease state and lifestyle.
Inter-individual variability may occur in both the toxicokinetics
of the chemical and the sensitivity of the target for toxicity.
4.3 Non-neoplastic (threshold) effects
Although specific aspects vary, comparable schemes have been
developed by various national and international agencies and
organizations to derive levels of exposure considered to present
minimal or no risk for non-neoplastic effects to the general
population. These include: Reference Dose/Concentrations (US
Environmental Protection Agency), Tolerable Daily
Intakes/Concentrations (Health Canada), Minimal Risk Levels (US
ATSDR), Tolerable/Acceptable Daily Intakes (IPCS, 1987a,b, 1990a,b,
1994). In evaluating dose-response for non-neoplastic effects, the
European Union does not derive tolerable intakes; instead effect
levels are compared to estimated exposures ("margin of safety").
In the case of substances for which the critical effect is not
carcinogenicity, it is generally assumed that there is a level of
exposure below which the probability for an adverse effect to occur is
minimal, if not zero (i.e. a threshold). The mechanism underlying this
assumption is that multiple cells (or cell components) must be
irreversibly injured before an adverse effect becomes evident, and
that cellular defence and repair mechanisms are overwhelmed by the
rate at which injury occurs.
4.3.1 Characterization of threshold
For toxic effects, other than heritable mutations and genotoxic
carcinogenicity, considered to have a threshold, i.e. a dose below
which there would be no detectable effect, a number of different
estimates may be used as an approximation of the biological threshold.
4.3.1.1 No-observed-adverse-effect level (NOAEL)
This is a simple estimate of the highest dose in which the
incidence of a toxic effect or change in target organ weight,
histopathology etc., was not significantly different from the
untreated group (from a statistical and biological assessment). It is
based on toxic effects of functional importance or pathological
significance rather than adaptive responses, and is defined as the
highest observed dose or concentration of a substance at which there
is no detectable adverse alteration of morphology, functional
capacity, growth, development or life span of the target (IPCS, 1994).
The NOAEL will depend on the sensitivity of the methods used, the
sizes of the exposed groups and the differences between estimated
exposures or doses. The NOAEL is an observed value which does not take
into account the nature or steepness of the dose-response curve.
In consequence, the NOAEL is not the same as the biological
threshold and may either underestimate or overestimate the true
no-effect level. Though such limitations are recognized and have been
the basis for criticism of the use of the NOAEL (Leisenring & Ryan,
1992; Calabrese & Baldwin, 1994), dose-response relationships are
often so poorly characterized that the NOAEL or LOAEL is the only
quantitative value available as the basis for characterization of
dose-response.
4.3.1.2 Benchmark dose/concentration
This is an alternative method of defining the lower end of the
dose-response curve in the area of the observed threshold
(Crump, 1984). The benchmark dose is the effective dose (or its lower
confidence limit) that produces a certain increase in incidence above
control levels (e.g., 1% or 5% of the maximum toxic response). The
benchmark dose is derived by modelling the data in the observed range
and selecting the point on the curve (or its upper confidence limit)
corresponding to a specified increase in the incidence of an effect.
Any model that fits the empirical data well is likely to provide a
reasonable estimate of the benchmark dose, and choice of the model may
not be critical since estimation is within the observed dose range.
The advantages of the benchmark dose are that it takes into account
the slope of the dose-response curve, the size of the study groups and
the variability in the data. It should be recognized that unless there
are a sufficient number of dose levels at which effects have been
observed, the benchmark dose/concentration offers little advantage
over effect levels as an approximation of the biological threshold.
Statistical modelling of continuous data as a basis for developing
benchmark doses/concentrations is also currently problematic.
4.3.1.3 Lowest-observed-adverse-effect level (LOAEL)
In some studies, there is a significant effect compared to
controls in the lowest dose group. In such cases, there is no NOAEL
and an alternative approach must be adopted. These include estimation
of a benchmark dose or threshold estimate (if the dose-response data
approach zero response) or application of an additional uncertainty
factor.
4.3.2 Uncertainty factors
In deriving tolerable intakes (or RFDs or ADIs), the N(L)OAEL or
benchmark dose/concentrations are divided by uncertainty factors to
account for variabilities and uncertainties. Principal factors applied
relate to extrapolation from animal studies to the human situation and
to inter-individual variability within the response for the human
population. Traditionally, default factors of 10 have been applied to
account for each of these variations. Additional uncertainty factors
have been applied to account for the inadequacy of the database, for
extrapolation from subchronic to chronic exposure and from LOAEL to
NOAEL, and for the severity of a given effect.
Knowledge of actual inter-species differences and
inter-individual variability in the biokinetic behaviour of a given
compound (toxicokinetics) and its target organ (toxicodynamics) would
enable the development of full biologically based dose-response models
or physiologically based pharmacokinetic models. In the absence of
full biological understanding, several approaches have been developed
to incorporate as much scientific information as possible in the
development and application of uncertainty factors. Indeed, a formal
approach to the development of data-derived uncertainty factors has
been developed by Renwick (1993a,b) and proposed by IPCS (IPCS, 1994).
It is presented here as an example of a flexible but structured
approach to the selection of uncertainty factors which reflects the
nature and extent of the database (Lewis, et al., 1990; Renwick,
1993b).
The scheme retains the two 10-fold default uncertainty factors
(for inter-species and inter-individual variation) as the cornerstone
of the structure, in the absence of specific and relevant data on
toxicokinetics or mechanism of action (Renwick, 1993a). However, it
allows for the division of the two default uncertainty factors (for
inter- and intra-species variation) to account for toxicokinetics and
toxicodynamics. The default components of these two factors can then
be replaced by actual quantitative data, when available. This reduces
the extent of uncertainty by allowing the incorporation of appropriate
data on the compound of interest in one or both of these aspects,
where they exist (Fig. 1). There would be very few databases in which
adequate information was available to account quantitatively for both
aspects of either inter-species or of inter-individual differences.
Incorporation of data on one aspect only (e.g., inter-species
toxicokinetics) requires the use of a default factor for the
uncertainty associated with the remaining undefined aspect (e.g.,
inter-species toxicodynamics).
Uncertainty factors often address:
a) Nature of toxicity
Some bodies, e.g., the FAO/WHO Joint Meeting on Pesticide
Residues (JMPR), have used an additional "safety factor" in cases
where the NOAEL is derived for a critical effect that is a severe and
irreversible phenomenon, such as teratogenicity or non-genotoxic
carcinogenicity, especially if the dose-response relationship is
shallow (IPCS, 1987a,b, 1990a,b). This additional factor (of up to 10)
has been applied in such cases to provide a greater margin between the
intake/exposure of any particularly susceptible humans and the
dose-response curve for such toxicity demonstrable in animals.
However, for other types of toxic effect, for example, changes in
organ weight or histopathology, a value of 1 (no further correction)
would be appropriate.
b) Adequacy of the database
A minimum dataset that is considered adequate for risk assessment
is generally established. This will vary according to the purpose of
the assessment (e.g., screening level or full). Additional
deficiencies in a toxicity database that increase the uncertainty of
the extrapolation process have also been recognized by the use of an
additional uncertainty factor. A value of 1 would be applied to an
appropriate and complete database, but a higher factor would be
considered necessary for barely adequate databases.
c) LOAEL to NOAEL extrapolation
In situations where a NOAEL has not been achieved but data are of
sufficient quality to be the basis of the risk assessment, then an
extra uncertainty factor may be applied (Dourson & Stara, 1983). The
magnitude of this factor (e.g., 3 or 10) should be based on the
dose-response data.
d) Inter-species extrapolation
The inter-species uncertainty factor is not necessary if the NOAEL or
risk assessment is based on human data. Where an assessment is based
on data in animals, however, and in situations where there are
appropriate compound-specific toxicokinetic and/or toxicodynamic data,
the relevant default uncertainty factor for inter-species variation
would be replaced by the data-derived factor (Renwick, 1993b). Data on
physiologically based pharmacokinetic (PBPK) modelling should be
included wherever possible; however, such information is available
currently for only a small number of substances. If a data-derived
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
PARTICIPANTS IN THE PLANNING AND TASK GROUP MEETINGS ON PRINCIPLES FOR
THE ASSESSMENT OF RISKS TO HUMAN HEALTH FROM EXPOSURE TO CHEMICALS
Members
Dr A. Aitio, Institute of Occupational Health, Laboratory of
Biochemistry, Helsinki, Finland a,b
Dr N. Aldrige, The Robens Institute of Industrial and Environmental
Health and Safety, University of Guildford, Guildford, Surrey, United
Kingdom (deceased)a,b
Dr D. Anderson, British Industry Biological Research Association
(BIBRA), Carshalton, Surrey, United Kingdoma,b
Professor C.L. Berry, Department of Morbid Anatomy, London Hospital
Medical College, London, United Kingdoma
Dr R. Burnett, Biostatistics and Computer Division, Environmental
Health Directorate, Health and Welfare Canada, Ottawa, Ontario,
Canadaa
Dr J.R.P. Cabral, Unit of Mechanisms of Carcinogenesis, International
Agency for Research on Cancer, Lyon, Francea
Dr E. Cardis, Unit of Biostatistics Research and Informatics,
International Agency for Research on Cancer, Lyon, Francea
Dr M. Cikrt, Institute of Hygiene and Epidemiology, Prague, Czech
Republica
Dr D.B. Clayson, Carp, Ontario, Canada
Mr D.J. Clegg, Pesticide Section, Toxicological Evaluation Division,
Food Directorate, Health Protection Branch, Tunney's Pasture, Ottawa,
Ontario, Canadaa
Professor E. Dybing, Department of Environmental Medicine, National
Institute of Public Health, Oslo, Norwayc
Dr R. Fielder, Department of Health, Elephant and Castle, London
United Kingdomb
Dr L. Fishbein, Fairfax, Virginia, USAc
Dr H. Gibb, US Environmental Protection Agency, Washington, DC,
USAa,b,d
Dr M. Goddard, Biostatistics and Computer Division, Environmental
Health Centre, Health and Welfare Canada, Tunney's Pasture, Ottawa,
Ontario, Canadab
Professor B. Goldstein, Rutgers Medical College, Busch Campus,
Pescataway, New Jersey, USAa
Dr R.F. Hertel, Federal Institute for Consumers, Health Protection and
Veterinary Medicine, FE-821 Bundesgesundheitsamt, BGVV, Berlin,
Germanyc,d
Dr J. Huff, Environmental Carcinogenesis Programme, National Institute
of Environmental Health Sciences, Research Triangle Park, North
Carolina, USAb
Professor M. Ikeda, Department of Environmental Health, Tohoku
University School of Medicine, Sendai, Japana
Dr D. Krewski, Biostatistics and Computer Division, Environmental
Health Directorate, Health and Welfare Canada, Ottawa, Ontario,
Canadaa
Professor R. Kroes, initially National Institute of Public Health
and Environmental Hygiene, Bilthoven, subsequently Research
Institute for Toxicology, University of Utrecht, Utrecht, the
Netherlandsa,c
Professor M. Lotti, University of Padua Medical School, Institute of
Occupational Medicine, Padua, Italya
Dr G.W. Lucier, Division of Biometry and Risk Assessment, National
Institute of Environmental Health Sciences, Research Triangle Park,
North Carolina, USAa
Dr L. Magos, Toxicology Unit, Medical Research Council Laboratories,
Carshalton, Surrey, United Kingdoma
Dr E. McConnell, Raleigh, North Carolina, USAa
Ms M.E. Meek, Environmental Health Directorate, Health Canada, Ottawa,
Ontario, Canadac
Dr R.L. Melnick, National Institute of Environmental Health Sciences,
Division of Biometry and Risk Assessment, Research Triangle Park,
North Carolina, USAa
Professor D.V. Parke, Department of Biochemistry, University of
Surrey, Guildford, Surrey, United Kingdoma
Dr J. Parker, Office of Health and Environmental Assessment, US
Environmental Protection Agency, Washington, DC, USAa
Dr O.E. Paynter, Hazard Evaluation Division, US Environmental
Protection Agency, Washington, DC, USAa
Dr P.K. Ray, Industrial Toxicology Research Centre, Lucknow, Indiaa
Dr A.G. Renwick, Clinical Pharmacology Group, University of
Southampton, Southhampton, Hampshire, United Kingdomc
Dr J. Sekizawa, Division of Information on Chemical Safety, National
Institute of Hygienic Sciences, Tokyo, Japanb
Dr J. Shaum, US Environmental Protection Agency, National Center for
Environmental Assessment, Washington, DC, USAd
Professor J.A. Sokal, Institute of Occupational Medicine and
Environmental Health, Sosnowiec, Polandc
Dr J. Steadman, Department of Health and Social Security, Elephant and
Castle, London, United Kingdoma
Dr L. Strayner, Division of Standards Development and Technology
Transfer, National Institute for Occupational Safety and Health,
Cincinnati, Ohio, USAb
Dr G.M.H. Swaen, Department of Occupational Medicine, University of
Limburg, Maastricht, the Netherlandsa,b
Dr A. Walker, Organisation for Economic Co-operation and Development,
Paris, Francea
Professor R. Walker, Food Safety Group, Division of Toxicology, School
of Biological Sciences, University of Surrey, Guildford, Surrey,
United Kingdomc
Dr J.E. Zejda, Department of Epidemiology, Institute of Occupational
Medicine and Environmental Health, Sosnowiec, Polandc
Observers
Professor G. Di Renzo, International Union of Toxicology, Department
of Neuroscience, Faculty of Medicine and Surgery, University of Naples
"Federico II", Naples, Italyc
Dr M. Jaroszewski, Health and Safety Directorate, Occupational
Medicine and Hygiene Unit, Commission of the European Community,
Luxembourgb
Dr C. Lally, European Council of Chemical Industry Federation (CEFIC),
Procter and Gamble, Strombbek Bever, Belgiumc
Professor A. Mutti, Institute of Clinical Medicine and Nephrology,
Parma, Italyc
Dr J. O'Donoghue (Representing AIHC) Corporate Health and Environment
Laboratories, Eastman Kodak Company, Rochester, New York, USAb
Dr M. Penman, ICI C & P Limited, Occupational Health Division, Wilton,
Middlesborough, Cleveland, United Kingdomc
Mrs M. Richold, European Centre for Ecotoxicology and Toxicology of
Chemicals (ECETOC), Unilever Research Laboratory, Environmental Safety
Laboratory, Sharnbrook, Bedford, United Kingdomc
Mr P. Verschuren, International Life Sciences Institute, Brussels
Belgiumc,b
Secretariat
Dr G.C. Becking, Inter-regional and Research Unit, International
Programme on Chemical Safety, World Health Organization, Research
Triangle Park, North Carolina, USAb
Dr K. Gutschmidt, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerlandd
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerlandc
Dr M. Younes, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerlandd
a Participated in Planning and Working Groups on Scientific
Principles for the Assessment of Risks to Human Health from
Exposure to Chemicals.
b Participated in the WHO Task Group Meeting on the initial draft
of Principles for the Assessment of Risk from Exposure to
Chemicals (British Industry Biological Research Association
(BIBRA), Carshalton, Surrey, United Kingdom, March 1993).
c Participated in the WHO Task Group Meeting on the initial draft
of General Principles and Methods for Chemical Safety (Human
Health Protection (National Institute of Public Health and
Environmental Protection) (RIVM), Bilthoven, the Netherlands, 22-
25 March 1994).
d Participated in the WHO Finalizing Group Meetings on Principles
for the Assessment of Risks to Human Health from Exposure to
Chemicals (World Health Organization, Geneva, Switzerland, 2-5
September 1996 and 18-20 September 1997).
PRINCIPLES FOR THE ASSESSMENT OF RISKS TO HUMAN HEALTH FROM EXPOSURE
TO CHEMICALS
This monograph is an amalgamation of two draft documents
"Principles for the Assessment of Risk from Exposure to Chemicals" and
"General Principles and Methods for Chemical Safety (Human Health
Protection)".
Both documents were planned to cover different aspects of
chemical safety and risk assessment; one dealing with the basic
science for general readers, and the other providing more practical
approaches to risk assessment of chemicals for risk assessors.
However, they turned out to have a substantial amount of overlapping
information and it was therefore decided to use both drafts as a basis
for this new, comprehensive document. The more detailed draft on
"General Principles and Methods for Chemical Safety (Human Health
Protection)" will be published as a separate document for training
purposes.
This Environmental Health Criteria monograph is aimed at
furnishing a practical overview of chemical safety and at providing
the framework of risk assessment for regulatory and research
scientists, as well as risk managers. It is intended to complement
existing Environmental Health Criteria that address methodologies for
the assessment of risks from exposure to chemicals with a view towards
different end-points or to susceptible population groups. It is not
intended as a textbook on toxicology.
This monograph should not be considered as being of a
prescriptive nature. The chapters on exposure assessment and risk
characterization, in particular, provide rather some practical
guidance.
Several planning, working and Task Group meetings took place to
discuss and agree upon the structures and contents of both
Environmental Health Criteria documents.
A WHO Task Group on "Principles for the Assessment of Risk from
Exposure to Chemicals" met at the British Industrial Biological
Research Association (BIBRA), Carshalton, Surrey, United Kingdom, in
March 1993. Dr G.C. Becking, IPCS, welcomed the participants on behalf
of the Director, IPCS, and the three IPCS cooperating organizations
(UNEP/ILO/WHO), and the Task Group reviewed the draft document.
The main contributors to the first draft on Principles for the
Assessment of Risk from Exposure to Chemicals were Dr N. Aldridge,
Robens Institute of Industrial and Environmental Health and Safety,
United Kingdom, Dr H. Gibb, US Environmental Protection Agency, Dr J.
Huff, National Institute of Environmental Health Sciences, USA, Dr L
Stayner, National Institute for Occupational Safety and Health, USA.
A second WHO Task Group met to review the draft monograph on
General Principles and Methods for Chemical Safety (Human Health
Protection). This group met in at the National Institute of Public
Health and Environmental Protection (RIVM), Bilthoven, the
Netherlands, from 22 to 25 November 1995. Dr E. Smith, IPCS, welcomed
the participants on behalf of the Director, IPCS, and the three IPCS
cooperating organizations (UNEP/ILO/WHO), and the Task Group reviewed
the draft document.
The main contributors to the draft on Principles for the
Assessment of Risk from Exposure to Chemicals were Dr D.B. Clayson,
Carp, Canada, Professor E. Dybing, National Institute of Public
Health, Norway, Dr L. Fishbein, Fairfax, Virginia, USA, Dr A.G.
Renwick, University of Southampton, United Kingdom, Professor R.
Walker, University of Surrey, United Kingdom, and Professor J.A Sokal,
Institute of Occupational Health and Environmental Medicine,
Sosnowiec, Poland.
In addition to the Task Group meetings, meetings were held during
1996 and 1997 in Geneva to combine the two documents.
Dr E. Smith and Dr G. Becking, both members of the IPCS, were
responsible for the preparation of the initial draft documents. Dr M.
Younes (IPCS) was responsible for the overall scientific content of
the final monograph and Dr P.G. Jenkins (IPCS) for the technical
editing.
The efforts of all who helped in the preparation and finalization
of the document are gratefully acknowledged.
ABBREVIATIONS
ADD average daily dose
ADI acceptable daily intake
EPI exposure/potency index
GLP good laboratory practice
IARC International Agency for Research on Cancer
LOAEL lowest-observed-adverse-effect level
NOAEL no-observed-adverse-effect level
OECD Organisation for Economic Co-operation and Development
PBPK physiologically based pharmacokinetic
SAR structure-activity relationship
US EPA US Environmental Protection Agency
1. SUMMARY
Control of risks from exposure to chemicals (chemical safety)
requires first of all a scientific, ideally quantitative, assessment
of potential effects at given exposure levels (risk assessment). Based
upon the results of risk assessment, and taking into consideration
other factors, a decision-making process aimed at eliminating or, if
this is not possible, reducing to a minimum the risk to the
chemical(s) under consideration (risk management), can be started.
Risk assessment is a conceptual framework that provides the
mechanism for a structured review of information relevant to
estimating health or environmental outcomes. In conducting risk
assessments, the National Academy of Sciences risk assessment paradigm
has proven to be a useful tool (US NAS, 1983). This paradigm divides
the risk assessment process into four distinct steps: hazard
identification, dose-response assessment, exposure assessment and risk
characterization.
The purpose of hazard identification is to evaluate the weight of
evidence for adverse effects in humans based on assessment of all
available data on toxicity and mode of action. It is designed to
address primarily two questions: (1) whether an agent may pose a
health hazard to human beings, and (2) under what circumstances an
identified hazard may be expressed. Hazard identification is based on
analyses of a variety of data that may range from observations in
humans to analysis of structure-activity relationships. The result of
the hazard identification exercise is a scientific judgement as to
whether the chemical evaluated can, under given exposure conditions,
cause an adverse health effect in humans. Generally, toxicity is
observed in one or more target organ(s). Often, multiple end-points
are observed following exposure to a given chemical. The critical
effect, which is usually the first significant adverse effect that
occurs with increasing dose, is determined.
Dose-response assessment is the process of characterizing the
relationship between the dose of an agent administered or received and
the incidence of an adverse health effect. For most types of toxic
effects (i.e. organ-specific, neurological/behavioural, immunological,
non-genotoxic carcinogenesis, reproductive or developmental), it is
generally considered that there is a dose or concentration below which
adverse effects will not occur (i.e. a threshold). For other types of
toxic effects, it is assumed that there is some probability of harm at
any level of exposure (i.e. that no threshold exists). At the present
time, the latter assumption is generally applied primarily for
mutagenesis and genotoxic carcinogenesis.
If a threshold has been assumed (e.g., for non-neoplastic effects
and non-genotoxic carcinogens), traditionally, a level of exposure
below which it is believed that there are no adverse effects, based on
a no-observed-adverse-effect level (NOAEL) (approximation of the
threshold) and uncertainty factors, has been estimated. Alternatively,
the magnitude by which the no (lowest)-observed-adverse-effect level
(N(L)OAEL) exceeds the estimated exposure (i.e. the "margin of
safety") is considered in light of various sources of uncertainty. In
the past, this approach has often been described as a "safety
evaluation". Therefore, the dose that can be considered as a first
approximation of the threshold, i.e. the NOAEL, is critical.
Increasingly, however, the "benchmark dose", a model-derived estimate
(or its lower confidence limit) of a particular incidence level (e.g.,
5%) for the critical effect, is being proposed for use in quantitative
assessment of the dose-response for such effects.
There is no clear consensus on appropriate methodology for the
risk assessment of chemicals for which the critical effect may not
have a threshold (i.e. genotoxic carcinogens and germ cell mutagens).
Indeed, a number of approaches based largely on characterization of
dose-response have been adopted for assessment in such cases.
Therefore, the critical data points are those that define the slope of
the dose-response relationship (rather than the NOAEL, which is the
first approximation of a threshold).
The third step in the process of risk assessment is the exposure
assessment, which has the aim of determining the nature and extent of
contact with chemical substances experienced or anticipated under
different conditions. Multiple approaches can be used to conduct
exposure assessments. Generally, approaches include indirect and
direct techniques, covering measurement of environmental
concentrations and personal exposures, as well as biomarkers.
Questionnaires and models are also often used. Exposure assessment
requires the determination of the emissions, pathways and rates of
movement of a substance and its transformation or degradation, in
order to estimate the concentrations to which human populations or
environmental spheres (water, soil and air) may be exposed.
Depending on the purpose of an exposure assessment, the numerical
output may be an estimate of either the intensity, rate, duration or
frequency of contact exposure or dose (resulting amount that actually
crosses the boundary). For risk assessments based on dose-response
relationships, the output usually includes an estimate of dose. It is
important to note that the internal dose, not the external exposure
level, determines the toxicological outcome of a given exposure.
Risk characterization is the final step in risk assessment. It is
designed to support risk managers by providing, in plain language, the
essential scientific evidence and rationale about risk that they need
for decision-making. In risk characterization, estimates of the risk
to human health under relevant exposure scenarios are provided. Thus,
a risk characterization is an evaluation and integration of the
available scientific evidence used to estimate the nature, importance,
and often the magnitude of human and/or environmental risk, including
attendant uncertainty, that can reasonably be estimated to result from
exposure to a particular environmental agent under specific
circumstances.
The term "risk management" encompasses all of those activities
required to reach decisions on whether an associated risk requires
elimination or necessary reduction. Risk management strategies/or
options can be broadly classified as regulatory, non-regulatory,
economic, advisory or technological, which are not mutually exclusive.
Thus legislative mandates (statutory guidance), political
considerations, socioeconomic values, cost, technical feasibility,
populations at risk, duration and magnitude of risk, risk comparison,
and possible impact on trade between countries can generally be
considered as a broad panoply of elements that can be factored into
final policy or rule making. Key decision factors such as the size of
the population, the resources, costs of meeting targets and the
scientific quality of risk assessment and subsequent managerial
decisions vary enormously from one decision context to another. It is
also recognized that risk management is a complex multidisciplinary
procedure which is seldom codified or uniform, is frequently
unstructured, but which can respond to evolving input from a wide
variety of sources. Increasingly, risk perception and risk
communication are recognized as important elements, which must also be
considered for the broadest possible public acceptance of risk
management decisions.
Chemicals have become an indispensable part of human life,
sustaining activities and development, preventing and controlling many
diseases, and increasing agricultural productivity. Despite their
benefits, chemicals may, especially when misused, cause adverse
effects on human health and environmental integrity. The widespread
application of chemicals throughout the world increases the potential
of adverse effects. The growth of chemical industries, both in
developing as well as in developed countries, is predicted to continue
to increase. In this context, it is recognized that the assessment and
management of risks from exposure to chemicals are among the highest
priorities in pursuing the principles of sustainable development.
2. INTRODUCTION
Despite the societal benefits that accrue from the use of
chemicals, substantial potential hazards to health may be associated
with exposure during the production, use or disposal of the
approximately 100 000 unique chemicals or 4 million mixtures,
formulations and blends already in commercial use or the several
hundred new synthetic chemicals introduced each year (EC, 1990). This
monograph outlines the nature of the data available and their use in
the assessment of risk in a risk assessment/risk management framework.
It is hoped that scientists, risk assessors and health risk managers
will find this monograph helpful to decision-making in this area.
A number of national and international organizations and agencies
have developed guidance on assessment of exposure and various health
end-points (e.g., carcinogenicity, developmental toxicity, etc.). It
is not the purpose of this monograph to endorse particular approaches
but rather to acquaint the reader with relevant methodology and issues
for consideration.
It is also hoped that the reader will find this monograph useful
in the interpretation of risk assessments on specific chemicals. The
reader is referred to such sources for chemical-specific hazard
identification and, depending on the monograph, dose-response
information. A list of assessments produced by various national and
international agencies is included in ECETOC/UNEP (1996). These
sources do not, of course, provide the exposure information necessary
to characterize risk at the local level. Since exposure will vary
considerably under different circumstances, responsible authorities
are strongly encouraged to characterize risk on the basis of local
measured or predicted exposure scenarios. It is hoped that the general
approaches to exposure assessment described in this monograph will
assist the reader in characterizing risk in specific situations.
In the chapters of this monograph, the following four distinct
and essential components of the risk assessment paradigm are
addressed:
(1) hazard identification - identification of the inherent
capability of a substance to cause adverse effects;
(2) assessment of dose-response relationships involves
characterization of the relationship between the dose of an agent
administered or received and the incidence of an adverse effect;
(3) exposure assessment is the qualitative and/or quantitative
assessment of the chemical nature, form and concentration of a
chemical to which an identified population is exposed from all
sources (air, water, soil and diet);
(4) risk characterization is the synthesis of critically evaluated
information and data from exposure assessment, hazard
identification and dose-response considerations into a summary
that identifies clearly the strengths and weaknesses of the
database, the criteria applied to evaluation and validation of
all aspects of methodology, and the conclusions reached from the
review of scientific information.
The logical consequence of the process of assessment of potential
risk is the application of the information to the development of
practical measures (risk management) for the protection of human
health. Although not the principal focus of this monograph, the
importance of clear understanding and communication of the nature and
limitations of the scientific basis for risk assessment in risk
management is addressed in the final chapter.
In Appendix 1 to this monograph, an example of a hazard
identification scheme for carcinogenicity, developed by the
International Agency for Research on Cancer (IARC), is presented. In
Appendix 2, the currently available and draft guidelines of the
Organisation for Economic Cooperation and Development (OECD) for
testing of chemicals are presented. For sample exposure and risk
characterizations, readers are referred to IPCS (1994).
3. HEALTH HAZARD IDENTIFICATION
3.1 Introduction
The purpose of hazard identification is to evaluate the weight of
evidence for adverse effects in humans based on assessment of all
available data on toxicity and mode of action. It is designed to
address primarily two questions: (a) whether an agent may pose a
health hazard to humans, and (b) under what circumstances an
identified hazard may be expressed. Hazard identification is based on
analyses of a variety of data that may range from observations in
humans to analysis of structure-activity relationships.
In hazard identification, the weight of evidence is assessed on
the basis of combined strength and coherence of inferences
appropriately drawn from all of the available data. This entails
rigorous examination of the quantity, quality and nature of the
results of available toxicological and epidemiological studies and
structure-activity analyses and information on mechanisms of toxicity.
The latter is particularly important with respect to assessment of
relevance to humans.
Several classification schemes provide a framework for assessment
of the weight of evidence for various toxicological end-points (DFG,
1972; IPCS, 1986 (neurotoxicity); US EPA, 1986a, 1996a; IARC, 1987;
EC, 1992; Health Canada, 1994; IPCS, 1996 (immunotoxicity); IPCS, 1997
(delayed hypersensitivity)). An example (the IARC scheme) is presented
in Appendix 1 to illustrate the nature of criteria on which
classification of weight of evidence is based. Such classification
schemes have been helpful in standardizing and communicating the
assessment of hazard identification for particular end-points. In
addition to the classifications themselves, narrative statements to
summarize the nature of and confidence in the evidence based on
limitations and strengths of the database are helpful. Issues that are
often addressed include: the nature, reliability, validity and
consistency of data on response in humans and in laboratory animals,
current knowledge of the mechanistic basis for the response, and, in
the absence of human data, the relevance of responses in experimental
animals to humans.
The result of the hazard identification exercise is a scientific
judgement as to whether the chemical can cause an adverse effect in
humans.
The following is intended to provide the reader with an
appreciation of the complexity of considerations made in assessing
different types of data as a basis for hazard identification in risk
assessment. Fundamentals of epidemiology and toxicity testing are not
addressed here since they are considered in several other sources. An
Environmental Health Criteria monograph on the principles of exposure
assessment is currently in preparation (IPCS, in preparation).
Each source of information (e.g., human data, animal data,
structure-activity relationships) has its advantages and limitations
in contributing to an assessment of weight of evidence, but,
collectively, they permit characterization of potential adverse health
effects.
3.2 Human data
Well-documented observational and clinical epidemiological
studies have the clear advantage over studies in animals in providing
the most relevant information on health effects in the species of
interest, thus avoiding extrapolation from animals to humans. In
addition, epidemiological studies can address hazards to which humans
are exposed in their natural environment, in the presence of
concomitant risk factors such as diet and smoking.
Human populations are heterogeneous in their composition, and
studies of exposed populations are likely to include individuals of
differing susceptibility to the chemical of interest. This may be
viewed as an advantage relative to toxicological studies, which
involve genetically homogeneous populations of test animals.
The database for direct hazard identification in human
populations consists primarily of observational (epidemiological)
studies and case reports. Some information is also available from
ethically conducted human volunteer studies.
In observational studies, the investigator does not control
assignment of study subjects to either exposed or non-exposed groups.
Rather, such studies involve investigation of various individuals or
groups of subjects as they happen to have been exposed, and at no
stage of the study is the exposure of subjects influenced by the
research protocol. Although exposure scenarios are more realistic than
those in the experimental setting, owing to their observational nature
it is often difficult to control for "confounding factors", which may
be contributing to the etiology of the disease being investigated. For
example, variations in smoking between groups may confound
interpretation of observations concerning lung cancer.
Ethical experimental studies in human volunteers offer the
advantage of being better able to control for confounding factors. The
assignment of study subjects to exposure groups is made by the
investigator, who also controls the quality and quantity. Although
such investigations are generally reliable for the establishment of
both causality and exposure-response relationships, they are most
often restricted for ethical reasons to the examination of mild,
temporary effects (e.g., neurobehavioural or biochemical changes) of
short-term exposures in a limited number of subjects. They have
contributed considerably, particularly to our understanding of
kinetics and to the development of air quality guidelines and
standards for traditional pollutants.
Case reports describe a particular effect in an individual or
group of individuals who were exposed to a substance and often
observed by a single physician or group of physicians. These reports
are often anecdotal or highly selected in nature. Owing primarily to
their lack of statistical stability, they are of limited use for
hazard assessment, though helpful in generating hypotheses for further
study. However, reports of cases of the disease or effect of interest
can identify associations, particularly when there are unique features
such as an association with a rare disease or effect of interest
(e.g., vinyl chloride and angiosarcoma or methylmercury and Minamata
disease).
The major types of epidemiological (observational) studies are
analytical and descriptive or correlational studies. Each study type
has well-known strengths and weaknesses that affect interpretation of
study results (Lilienfeld & Lilienfeld, 1979; Mausner & Kramer, 1985;
Kelsey et al., 1986; Rothman, 1986). Analytical epidemiological
studies (that is, cohort and case-control studies), in which exposure
and outcome are examined in individuals rather than in populations,
are generally most reliable in hazard identification as a basis for
risk assessment since it is possible to adjust more rigorously for
confounding factors. The assessment of results of such studies is
based on several features of study design including estimation of
exposure, the role of confounding variables and the measurement of
outcome. Potential limitations, depending upon the nature of the
design, include lack of information on exposure, insufficient sample
size, short length of follow-up and potential bias and confounding.
These factors may limit the usefulness of particular studies for the
purposes of risk assessment.
Epidemiological data demonstrating dose-response, if available,
provide an advantageous basis for analysis, since concerns about
inter-species extrapolation do not arise. Adequacy of human exposure
data for quantification is an important consideration in deciding
whether epidemiological data are the best basis for analysis in a
particular case. If adequate exposure data exist in a well-designed
and well-conducted epidemiological study that detects no effects, it
may be possible to obtain an upper estimate of the potential human
risk to provide a check on plausibility of available estimates based
on animal tumour or other responses (e.g., do confidence limits on one
overlap the point estimate of the other?) (Stayner & Bailer, 1993; US
EPA 1996a).
3.2.1 Criteria for establishing causality
The first step in the evaluation of results of studies in humans
as a basis for hazard identification is the assessment of the
individual results of each separate report. The strengths and
weaknesses of each study must be considered along with potential for
the existence of bias (Gehlbach, 1982), with particular attention to
exposure data, criteria for definition of health outcome under study,
the size of the study population and the statistical power of the
analysis to detect adverse health effects. A set of standardized
criteria for assessing the weight of evidence of causality based on
assessment of the database has been developed (Hill, 1965; Susser,
1977).
Studies in which there is an apparent absence of evidence for a
hypothesized causal relationship between exposure and effect
("negative studies") need to be interpreted carefully (Hernberg,
1980). Such studies should be evaluated for dilution (the inclusion of
unexposed people in an allegedly exposed group of persons),
misclassification (Copeland et al., 1977), omissions, or premature
examination of subjects for diseases that may have long induction
(latency) periods. In addition, the statistical power of the study,
i.e. the probability that the study will be able to demonstrate the
presence of an effect, such as excessive disease or mortality, in a
population if the effect is actually present (Beaumont & Breslow,
1981), must be assessed.
There is no clear-cut criterion to distinguish positive from
negative studies. Although statistical significance has often been
used as the criteria, most epidemiologists believe that it is overly
simplistic to base decisions on arbitrary probability values (Rothman,
1986). For example, when a study fails to detect a statistically
significant effect, this may simply reflect inadequate sample size or
other aspects of study design. Conversely, when the results of a study
are statistically significant, the seemingly positive results may
still be due to confounding or even chance.
A positive association between an agent and an effect may be
interpreted as implying causality, to a greater or lesser extent, if
the following criteria are met: (a) there is not identifiable positive
bias; (b) the possibility of positive confounding has been considered;
(c) the association is unlikely to be due to chance alone; (d) the
association is strong; and (e) there is a dose-response relationship
(IARC, 1990). The following criteria for inferring causality from the
results of epidemiological studies have been developed by Hill (1965):
(a) The strength of the association as measured by the relative risk
In general, epidemiologists have more confidence in their results
when the magnitude of the relative risk is large. However, relative
risks of small magnitude do not necessarily imply lack of causality
and may be important if the disease under study is common (IARC,
1990). In evaluating relative risks, it is important to note the
actual numbers of observed and expected cases.
(b) The consistency of the association
The case for causal inference is strengthened by repetition of
findings "by different investigators, in different places,
circumstances and times" (Hill, 1965). The reproducibility of findings
constitutes one of the strongest arguments for the existence of
causality. If there are discordant results among investigations,
possible reasons such as differences in exposure should be considered
in assessing the results, and data from studies judged to be of high
quality given greater weight than data from studies judged to be
methodologically less sound (IARC, 1990).
(c) The temporal relationship between cause and effect
This principle may be simply restated as exposure must precede
illness. When latency is a factor, exposures must have occurred
sufficiently early to have produced an effect by the time of the
study.
(d) The biological gradient of the association
The evidence for causality is strengthened when the risk of
disease is shown to increase with levels of exposure. Because there
are many possible reasons that an epidemiological study may fail to
detect an exposure-response relationship (e.g., poor exposure data,
lack of adequate exposure gradient), the absence of a dose-response
relationship does not necessarily imply that the relationship is not
causal (IARC, 1990). Strong evidence for causality is provided when a
change in exposure brings about a change in disease frequency
(Hernberg, 1980), e.g., the decrease in risk of lung cancer that
follows cessation of smoking (Doll & Hill, 1956).
(e) the specificity of the association
A highly specific association is one in which the disease under
study is only induced by a particular agent. Specificity of cause is
common in infectious diseases but less common in chronic diseases that
often have a multi-factorial etiology. However, a specific association
may be observed for certain chronic diseases such as between exposure
to crocidolite asbestos and mesothelioma or vinyl chloride and
angiosarcoma. Although the presence of specificity seems to imply
causality, its absence does not exclude it (Fralick, 1983).
(f) biological plausibility of the association
Hill (1965) stated strongly that a proposed causal relationship
should not seriously conflict with knowledge of the biology and
pathophysiology of a disease under study. An epidemiological inference
of causality may be strengthened by data from experimental studies
showing consistency with biological mechanisms. For example, exposure
to ionizing radiation causes cancer in many animal species. However,
the lack of mechanistic or positive animal bioassay data to support an
association observed in an epidemiological study is not, in itself,
sufficient reason to reject causality.
3.3 Animal studies
Owing to the lack of adequate epidemiological data for most
substances, toxicological studies in animal species play an important
role in hazard identification for risk assessment. Toxicity studies
vary widely in purpose, design and conduct, and range from relatively
well-standardized and widely accepted test methods for assaying
various types of toxicity to large numbers of basically
research-oriented investigations employing specialized study designs.
The design, conduct and completeness of reporting of experimental
findings in toxicological studies on mammalian species are of critical
importance in determining the validity and relevance of results.
Toxicological results from adequate animal systems signal anticipated
effects in humans. Thus, negative results cannot be assessed from an
inadequate study, and full evaluation of a positive effect is
confounded by incomplete reporting from poorly designed or poorly
conducted studies. However, positive findings cannot be ignored.
Studies should be of good scientific quality and follow standard
guidelines and recognized good laboratory practices (GLPs) wherever
possible.
Information on the design of specific bioassays, including those
that address acute, short-term, sub-chronic, chronic and developmental
and reproductive toxicity, immunotoxicity and carcinogenicity, are not
presented here but are available in test guidelines, for which
principles of GLP are also specified (IARC, 1986; OECD, 1987, 1998;
Chhabra et al., 1990). A list of currently available OECD Guidelines
is included in Appendix 2. In this section, examples of factors to be
taken into account in assessing these various aspects of study design
for hazard identification are described.
Major end-points in toxicity studies can be grouped into the
following categories (IPCS, 1987a):
* Functional manifestations (weight loss, laxative effects, etc.);
* non-neoplastic lesions with morphological
manifestations/organ-directed toxic effects;
* neoplastic/carcinogenic manifestations.
In addition, a number of specific end-points may require targeted
testing strategies. Such end-points include skin and eye irritation,
reproductive/developmental manifestations, immunotoxicity and
neurotoxicity (including neurodevelopmental effects).
It is important to recognize that there are two types of data
generated in such studies; those in which response is graded, such as
enzyme inhibition (i.e. continuous data), and those in which the
response occurs or does not occur in a single animal, such as a
particular tumour (i.e. quantal data).
In assessing the relevance of various toxicological studies to
hazard identification and risk assessment, several features of study
design are considered, including the purity of the compound
administered, physico-chemical properties (volatility, stability,
solubility), homogeneity of distribution in inhalation experiments,
the size of the study (i.e. the number of exposed and control
animals), whether the study adhered to the principles of GLP, the
relevance of the route of exposure to that of humans, duration of
exposure, the number and suitability of the dose levels administered,
the extent of examination of various toxicological end-points and the
statistical analysis of the data. The types, site, incidence and
severity of effects and the nature of the exposure- or dose-response
relationship are also taken into account. Where data indicate that
there are significant differences in absorption, distribution,
metabolism and elimination of the compound in different animal
species, wherever possible, studies in which the species and strain of
animal are most similar to Homo sapiens in this regard are used
(where relevant human data are available). The consistency of the
results of the principal studies are also considered in the assessment
of the weight of evidence for an effect (e.g., whether similar effects
have been observed in studies in other species or whether such effects
would have been expected based on the structure or properties of the
chemical).
For example, the size of each exposure and concurrent control
group should be large enough for thorough toxicological and
statistical evaluation. The number of animals considered sufficient
depends on the variability, sensitivity and nature (e.g., quantal or
continuous) of the end-point being evaluated. For example, it is
commonly 50 per group in carcinogenicity bioassays where the responses
of interest are quantal in nature and 10 per group in subchronic
studies, where many of the examined end-points are continuous.
Studies in which the route of exposure is similar to that of
humans are most relevant to hazard identification for risk assessment.
For substances of low toxicity, it is important to ensure that when
administered in the diet, the quantities of the substance do not
interfere with normal nutritional needs.
Studies designed and conducted with 3-5 dosed groups plus a
vehicle control group of animals will yield reasonable dose-response
data relevant to hazard identification. The highest concentration of
the chemical should be one that induces a recognizable effect in the
animals such as changes in body or organ weights, enzyme changes or
minor histological changes. Changes such as mortality, gross
pathological changes, and painful or stressful conditions should be
avoided as they may confound the results of the study and may not be
in compliance with national and local animal welfare regulations.
Intermediate dose(s) should be targeted to produce minimally
observable toxic effects. Dose levels should be selected to produce
graded responses; too large intervals may complicate accurate
estimations of the lowest-observed-effect level (LOEL). Ideally, the
lowest dose should not demonstrate any toxicity (e.g., a NOAEL).
To assess fully the toxicological potential of a chemical for
local and systemic effects, all major organ systems should be examined
for dose-related effects and adverse effects in various organs should
be evaluated and described.
3.4 In vitro studies
Isolated cells, tissues and organs can be prepared and maintained
in culture by methods that preserve their in vivo properties and
characteristics. Increasing concern about the ethics of animal
experimentation has served to catalyse efforts leading to the possible
replacement or reduction in the use of animals, and the refinement of
test methods to minimize the stress and suffering to animals (ECETOC,
1989; Gelbke, 1993). In vitro testing contributes particularly to
the assessment of genotoxicity, permitting a decision concerning the
need for further testing.
Over the last decade, in vitro tests have been proposed as a
pre-screen or as an alternative method for other end-points, such as
prenatal toxicity, eye irritation, dermal irritation, tumour promotion
and target organ toxicity (Purchase, 1986; Tennant et al., 1987;
Anderson, 1990; Frazier, 1993; Atterwill, 1995). There has been
particular emphasis on validation programmes for skin and eye
irritation, but most of the tests mentioned above have not yet been
sufficiently validated and the results of validation studies,
especially in the past, have been lacking in consistency. The results
have failed to meet the need for reproducibility and high correlation,
ideally with sound human data but usually, for practical reasons, with
existing animal tests, which they are intended to replace.
Aspects that are important in assessing the adequacy of
in vitro studies include:
* the range of exposure levels, taking into account the toxicity of
the substance in the bacteria/cells, its solubility and, where
appropriate, its effects on the pH and osmolality of the culture
medium;
* whether, in the case of volatile substances, precautions were
taken to ensure the maintenance of effective concentrations of
the substance in the test medium;
* whether (when necessary) an appropriate exogenous metabolism mix
(e.g., S9 from induced rat or hamster liver) was used;
* whether appropriate negative and positive controls were included;
and
* whether there was an adequate number of replicates (within the
tests and of the tests).
Clearly, greater mechanistic understanding would facilitate
moving from purely empirical/correlative approaches to more
mechanistic-based tests. This is likely to facilitate greatly the
chances of adequate validation and acceptance of alternatives for
regulatory purposes.
3.5 Structure-activity relationships
Where epidemiological and toxicological data are not available,
the use of structure-activity relationships (SARs) may be considered.
SARs are based on the assumption that chemical substances that reach
and interact with target sites by the same mechanism do so as a result
of their similar chemical properties.
At present, SAR techniques, particularly those of a quantitative
nature, are not well developed in relation to mammalian toxicity. They
are primarily of value in predicting toxicokinetic properties and in
priority setting for research and evaluation.
4. DOSE-RESPONSE
4.1 Introduction
Approaches to quantification of dose-response vary according to
the scope and purpose of assessments. However, for most types of toxic
effects (i.e. organ-specific, neurological/behavioural, immunological,
non-genotoxic carcinogenesis, reproductive or developmental), it is
generally considered that there is a dose or concentration below which
adverse effects will not occur (i.e. a threshold). For other types of
toxic effects, it is assumed that there is some probability of harm at
any level of exposure (i.e. that no threshold exists); this currently
applies primarily for mutagenesis and carcinogenesis. Some have
restricted the non-threshold assumption to genotoxic carcinogens.
The distinction in approaches for genotoxic carcinogens and other
types of toxic effects is based primarily on the premise that simple
events such as in vitro activation and covalent binding may be
linear over many orders of magnitude. Though it is not possible to
demonstrate experimentally the presence or absence of a threshold,
differences in approach to the dose-response assessment of genotoxic
versus non-genotoxic carcinogens have been adopted in some countries.
However, simple pragmatic distinction on this basis is increasingly
problematic. For example, it is likely that there are thresholds for
aneugenic genotoxic effects.
If a threshold has been assumed (e.g., for non-neoplastic effects
and non-genotoxic carcinogens), traditionally, a level of exposure
below which it is believed that there are no adverse effects, based on
a no-observed-adverse-effect level or NOAEL (approximation of the
threshold) and uncertainty factors, has been estimated (section 4.3).
Alternatively, the magnitude by which the N(L)OAEL exceeds the
estimated exposure (i.e. the "margin of safety"), is considered in
light of various sources of uncertainty (Commission Regulation (EC)
No. 1488/94; Council Regulation (EEC) 793/93) (EC, 1993, 1994). In the
past, this approach has often been described as "safety evaluation".
Therefore, the dose that can be considered as a first approximation of
the threshold, i.e. the NOAEL, is critical. Increasingly, however, the
"benchmark dose", a model-derived estimate (or its lower confidence
limit) of a particular incidence level (e.g., 5%) for the critical
effect, is being proposed for use in quantitative assessment of the
dose-response for such effects.
At present, there is no clear consensus on appropriate
methodology for the risk assessment of chemicals for which the
critical effect may not have a threshold (i.e. genotoxic carcinogens
and germ cell mutagens). Indeed, a number of approaches based largely
on characterization of dose-response have been adopted for assessment
in such cases (section 4.4). Therefore, the critical data points are
those that define the slope of the dose-response relationship (rather
than the NOAEL, which is the first approximation of a threshold).
In North America and some European countries, cancer risks have
traditionally been assessed by mathematical modelling of the
dose-response data in the observable range to estimate the risk at
much lower human intakes or exposures (low dose risk extrapolation).
It should be noted, however, that quantitative estimation of such
risks, particularly those orders of magnitude below the experimental
range (i.e. low dose risk estimation), is uncertain. Owing to this
uncertainty, some countries have chosen not to adopt this approach as
the basis for their regulatory actions for genotoxic carcinogens, and
other countries are increasingly adopting alternative measures of
dose-response. In Canada and the USA, for example, there is,
currently, increasing reliance on specification of the margin between
potency in the experimental range and exposure as the measure of risk
for carcinogens (Health Canada, 1994; US EPA, 1996b). In the United
Kingdom, dose-response for genotoxic carcinogens is not quantified;
instead the goal in risk management is to eliminate exposure or to
reduce levels to as low as is reasonably practical (UK DOH, 1991).
Owing to the increasing reliance on modelling in the experimental
range to characterize dose-response for tumours, which is essentially
similar to the benchmark dose being used increasingly to characterize
dose-response for non-neoplastic effects, approaches to quantitative
risk estimation for carcinogenic and non-neoplastic effects are
converging.
4.2 Considerations in dose-response assessment
4.2.1 Introduction
In considering toxic effects at various dose levels, the dose range of
interest is generally the low-dose range, since it usually reflects
the human exposure situation. Often, however, data on dose-response
are available for higher doses only, and are often derived from animal
experiments only. Therefore, the uncertainty in the dose-response
assessment is larger than the uncertainty in hazard identification, as
it requires extrapolation both from animal to human and from high-dose
to low-dose levels. In certain instances, a distinction is made
between response and effect, with a response being quantal and counted
(e.g., the incidence of a tumour) and an effect being graded and
measured (e.g., relative liver weight).
4.2.2 Inter- and intra-species considerations
4.2.2.1 Introduction
The strains and species of laboratory animals exposed in toxicity
studies have been selected to show minimum inter-individual
variability. In contrast to laboratory animals, humans represent a
very heterogeneous population with both genetic and acquired
diversity.
Therefore, two principal areas are considered when interpreting
data on toxicity acquired in animal species in relation to human risk:
a) Inter-species consideration: comparison of the data for animals
with a representative healthy human. Species differences result
from metabolic, functional and structural variations.
b) Intra-species or inter-individual consideration: comparison of
the representative healthy human with the range of variability
present within the human population in relation to the relevant
parameter(s).
For each of these areas, there are two aspects to be considered
in assessing risk, i.e. toxicokinetics (the delivery of the compound
to the site of action) and toxicodynamics (the inherent sensitivity of
the site of action to the chemical). Any approach that allows for the
incorporation of adequate data on toxicokinetic or toxicodynamic
differences between test animal and humans, or between different
humans, will increase the scientific validity of risk assessment.
Sources of inter-species and inter-individual variations in
toxicokinetics include differences in anatomy (e.g., gastrointestinal
structure and function), physiological function (e.g., cardiac output,
renal and hepatic blood, glomerular filtration rate and gastric pH),
and biochemical differences in, for example, enzymes involved in
xenobiotic metabolism. Sources of inter-species and inter-individual
differences in toxicodynamics (or inherent sensitivity) also include
anatomy. For example, the effect may occur in an organ of questionable
relevance to humans, such as the rodent forestomach. Physiological
differences, such as the hormonal control of the target organ, and
biochemical differences, e.g., species differences in key biochemical
components such as alpha2u-globulin, may also play a role (Flamm &
Lehman-McKeeman, 1991).
In some cases, it may be possible to conclude that effects
detected in animals are unlikely to be relevant to humans. In other
cases, there may be data to indicate that humans are likely to be more
or less sensitive than animal species; this information is important
for consideration in selection of critical effects.
If compound-specific toxicokinetic data are introduced into risk
assessment, then it is essential that these are related to the
species, protocol and active chemical entity (e.g., parent compound or
metabolite) involved in the toxicity that is the basis for the hazard
identification (Monro, 1990, 1993; Renwick, 1993a).
4.2.2.2 Species differences
Metabolism and structural/functional variations are both
important determinants of species differences. Common areas of
metabolic variation between species are digestive tract enzymes,
levels of circulating enzymes, liver enzymes and detoxification
processes.
In extrapolating between species, three aspects need to be
considered: the first relates to differences in body size, which
requires dose normalization or scaling (often done by expressing the
dose per kg body weight). The second relates to differences in
toxicokinetics, particularly bioactivation and/or detoxification
processes. The third aspect concerns the nature and severity of the
target for toxicity. Inter-species normalization (or scaling) is
generally based on physical characteristics (e.g., body weight, body
surface area), although occasionally it is based on caloric demand or,
where there are data in four species, multiple species regression.
When clearance of the parent substance is limited by enzyme
activity rather than blood flow or when metabolites are the toxic
agents, more sophisticated physiologically based pharmacokinetic
models are more appropriate, provided that adequate data are
available. Currently, such data are available for only a small number
of substances.
4.2.2.3 Human variability
Although data from animal studies may provide limited information
on inter-individual variability within the test species, it is the
greater potential variability in the human population that must be
addressed in risk assessment. Sources of inter-individual variability
in human populations include, for example, variations in genetic
composition, nutrition, disease state and lifestyle.
Inter-individual variability may occur in both the toxicokinetics
of the chemical and the sensitivity of the target for toxicity.
4.3 Non-neoplastic (threshold) effects
Although specific aspects vary, comparable schemes have been
developed by various national and international agencies and
organizations to derive levels of exposure considered to present
minimal or no risk for non-neoplastic effects to the general
population. These include: Reference Dose/Concentrations (US
Environmental Protection Agency), Tolerable Daily
Intakes/Concentrations (Health Canada), Minimal Risk Levels (US
ATSDR), Tolerable/Acceptable Daily Intakes (IPCS, 1987a,b, 1990a,b,
1994). In evaluating dose-response for non-neoplastic effects, the
European Union does not derive tolerable intakes; instead effect
levels are compared to estimated exposures ("margin of safety").
In the case of substances for which the critical effect is not
carcinogenicity, it is generally assumed that there is a level of
exposure below which the probability for an adverse effect to occur is
minimal, if not zero (i.e. a threshold). The mechanism underlying this
assumption is that multiple cells (or cell components) must be
irreversibly injured before an adverse effect becomes evident, and
that cellular defence and repair mechanisms are overwhelmed by the
rate at which injury occurs.
4.3.1 Characterization of threshold
For toxic effects, other than heritable mutations and genotoxic
carcinogenicity, considered to have a threshold, i.e. a dose below
which there would be no detectable effect, a number of different
estimates may be used as an approximation of the biological threshold.
4.3.1.1 No-observed-adverse-effect level (NOAEL)
This is a simple estimate of the highest dose in which the
incidence of a toxic effect or change in target organ weight,
histopathology etc., was not significantly different from the
untreated group (from a statistical and biological assessment). It is
based on toxic effects of functional importance or pathological
significance rather than adaptive responses, and is defined as the
highest observed dose or concentration of a substance at which there
is no detectable adverse alteration of morphology, functional
capacity, growth, development or life span of the target (IPCS, 1994).
The NOAEL will depend on the sensitivity of the methods used, the
sizes of the exposed groups and the differences between estimated
exposures or doses. The NOAEL is an observed value which does not take
into account the nature or steepness of the dose-response curve.
In consequence, the NOAEL is not the same as the biological
threshold and may either underestimate or overestimate the true
no-effect level. Though such limitations are recognized and have been
the basis for criticism of the use of the NOAEL (Leisenring & Ryan,
1992; Calabrese & Baldwin, 1994), dose-response relationships are
often so poorly characterized that the NOAEL or LOAEL is the only
quantitative value available as the basis for characterization of
dose-response.
4.3.1.2 Benchmark dose/concentration
This is an alternative method of defining the lower end of the
dose-response curve in the area of the observed threshold
(Crump, 1984). The benchmark dose is the effective dose (or its lower
confidence limit) that produces a certain increase in incidence above
control levels (e.g., 1% or 5% of the maximum toxic response). The
benchmark dose is derived by modelling the data in the observed range
and selecting the point on the curve (or its upper confidence limit)
corresponding to a specified increase in the incidence of an effect.
Any model that fits the empirical data well is likely to provide a
reasonable estimate of the benchmark dose, and choice of the model may
not be critical since estimation is within the observed dose range.
The advantages of the benchmark dose are that it takes into account
the slope of the dose-response curve, the size of the study groups and
the variability in the data. It should be recognized that unless there
are a sufficient number of dose levels at which effects have been
observed, the benchmark dose/concentration offers little advantage
over effect levels as an approximation of the biological threshold.
Statistical modelling of continuous data as a basis for developing
benchmark doses/concentrations is also currently problematic.
4.3.1.3 Lowest-observed-adverse-effect level (LOAEL)
In some studies, there is a significant effect compared to
controls in the lowest dose group. In such cases, there is no NOAEL
and an alternative approach must be adopted. These include estimation
of a benchmark dose or threshold estimate (if the dose-response data
approach zero response) or application of an additional uncertainty
factor.
4.3.2 Uncertainty factors
In deriving tolerable intakes (or RFDs or ADIs), the N(L)OAEL or
benchmark dose/concentrations are divided by uncertainty factors to
account for variabilities and uncertainties. Principal factors applied
relate to extrapolation from animal studies to the human situation and
to inter-individual variability within the response for the human
population. Traditionally, default factors of 10 have been applied to
account for each of these variations. Additional uncertainty factors
have been applied to account for the inadequacy of the database, for
extrapolation from subchronic to chronic exposure and from LOAEL to
NOAEL, and for the severity of a given effect.
Knowledge of actual inter-species differences and
inter-individual variability in the biokinetic behaviour of a given
compound (toxicokinetics) and its target organ (toxicodynamics) would
enable the development of full biologically based dose-response models
or physiologically based pharmacokinetic models. In the absence of
full biological understanding, several approaches have been developed
to incorporate as much scientific information as possible in the
development and application of uncertainty factors. Indeed, a formal
approach to the development of data-derived uncertainty factors has
been developed by Renwick (1993a,b) and proposed by IPCS (IPCS, 1994).
It is presented here as an example of a flexible but structured
approach to the selection of uncertainty factors which reflects the
nature and extent of the database (Lewis, et al., 1990; Renwick,
1993b).
The scheme retains the two 10-fold default uncertainty factors
(for inter-species and inter-individual variation) as the cornerstone
of the structure, in the absence of specific and relevant data on
toxicokinetics or mechanism of action (Renwick, 1993a). However, it
allows for the division of the two default uncertainty factors (for
inter- and intra-species variation) to account for toxicokinetics and
toxicodynamics. The default components of these two factors can then
be replaced by actual quantitative data, when available. This reduces
the extent of uncertainty by allowing the incorporation of appropriate
data on the compound of interest in one or both of these aspects,
where they exist (Fig. 1). There would be very few databases in which
adequate information was available to account quantitatively for both
aspects of either inter-species or of inter-individual differences.
Incorporation of data on one aspect only (e.g., inter-species
toxicokinetics) requires the use of a default factor for the
uncertainty associated with the remaining undefined aspect (e.g.,
inter-species toxicodynamics).
Uncertainty factors often address:
a) Nature of toxicity
Some bodies, e.g., the FAO/WHO Joint Meeting on Pesticide
Residues (JMPR), have used an additional "safety factor" in cases
where the NOAEL is derived for a critical effect that is a severe and
irreversible phenomenon, such as teratogenicity or non-genotoxic
carcinogenicity, especially if the dose-response relationship is
shallow (IPCS, 1987a,b, 1990a,b). This additional factor (of up to 10)
has been applied in such cases to provide a greater margin between the
intake/exposure of any particularly susceptible humans and the
dose-response curve for such toxicity demonstrable in animals.
However, for other types of toxic effect, for example, changes in
organ weight or histopathology, a value of 1 (no further correction)
would be appropriate.
b) Adequacy of the database
A minimum dataset that is considered adequate for risk assessment
is generally established. This will vary according to the purpose of
the assessment (e.g., screening level or full). Additional
deficiencies in a toxicity database that increase the uncertainty of
the extrapolation process have also been recognized by the use of an
additional uncertainty factor. A value of 1 would be applied to an
appropriate and complete database, but a higher factor would be
considered necessary for barely adequate databases.
c) LOAEL to NOAEL extrapolation
In situations where a NOAEL has not been achieved but data are of
sufficient quality to be the basis of the risk assessment, then an
extra uncertainty factor may be applied (Dourson & Stara, 1983). The
magnitude of this factor (e.g., 3 or 10) should be based on the
dose-response data.
d) Inter-species extrapolation
The inter-species uncertainty factor is not necessary if the NOAEL or
risk assessment is based on human data. Where an assessment is based
on data in animals, however, and in situations where there are
appropriate compound-specific toxicokinetic and/or toxicodynamic data,
the relevant default uncertainty factor for inter-species variation
would be replaced by the data-derived factor (Renwick, 1993b). Data on
physiologically based pharmacokinetic (PBPK) modelling should be
included wherever possible; however, such information is available
currently for only a small number of substances. If a data-derived
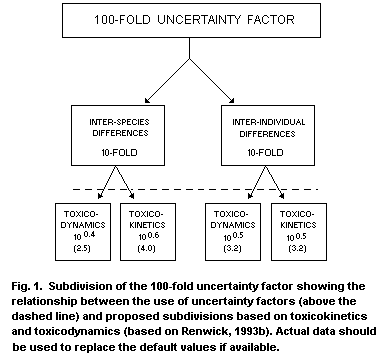 factor is introduced, then the commonly used 10-fold factor would be
replaced by the product of that factor and the remaining default
factor.
The composite default value of 10 has been criticized as
inadequate, for example, to allow for metabolic processes in mice
which can be related to body surface area (Calabrese et al., 1992);
the introduction of data-derived uncertainty factors would allow the
logical future development of more appropriate species specific
defaults.
e) Inter-individual variability in humans
In situations where appropriate toxicokinetic and toxicodynamic
data exist for a particular compound in humans, then the relevant
uncertainty factor should be replaced by the data-derived factor
(Renwick, 1993b). Data on PBPK modelling may also be able to
contribute to this assessment. If a data-derived factor is introduced,
then the commonly used 10-fold factor would be replaced by the product
of the data-derived factor and the remaining default factor.
Although the 10-fold default uncertainty factor is reasonable for
most cases (Dourson & Stara, 1983), it has been criticised as
inadequate for human variability especially when genetically
determined differences in a bioactivation process may be involved
(Calabrese, 1985; Goldstein, 1990). This concern reinforces the
importance of using an approach that allows the incorporation of data
on human variability in either toxicokinetics of the compound or the
sensitivity to its mechanism of action.
In addition to approaches aimed at incorporating as much
biological data as possible in the derivation of uncertainty factors,
probabilistic approaches have been investigated for the
characterization of uncertainty (Baird et al., 1996; Price et al.,
1997). Distributions can be developed on the basis of empirical
relationships observed for, for example, variations between LOAELs and
NOAELs and effect levels in subchronic versus chronic studies. Monte
Carlo techniques can be used to integrate probabilities for the
various areas of uncertainty.
4.4 Quantitative risk assessment for neoplastic (non-threshold)
effects
4.4.1 Introduction
A number of approaches have been adopted for characterization of
dose-response in the assessment of genotoxic neoplastic effects,
including quantitative extrapolation by mathematical modelling of the
dose-response curve to estimate the risk at likely human intakes or
exposures (low-dose risk extrapolation). Traditionally, where
dose-response has been extrapolated into the low-dose range, this has
been accomplished by the use of the linearized Armitage-Doll
multi-stage model. Dose-response may also be estimated in a two-step
process by straight linear extrapolation into the low-dose range from
a modelled point on the dose-response curve. Other measures of
dose-response include estimation of carcinogenic potency in the
experimental range and division of effect levels by a margin of
protection. In more recently developed biological models, different
stages in the process of carcinogenesis have been incorporated and
time to tumour has been taken into account (Moolgavkar et al., 1988),
although currently data are sufficient for application in only a
limited number of cases. In some cases where data permit, the dose
delivered to the target tissue has been incorporated into the
dose-response analysis (PBPK modelling) (IPCS, 1993).
In the same way as approaches adopted for non-neoplastic
(threshold) effects, there are increasingly attempts to incorporate
more of the scientific data in adopted approaches. For example, the
proposed cancer guidelines issued by the US EPA (1996b), updating the
previous guidelines (US EPA, 1986a), put emphasis on the full
integration of mechanistic information and dose-response data.
Depending on the mode of action, linear extrapolation into the
low-dose range or, alternatively, a margin of exposure would be
presented. The adequacy of the latter approach must be judged by
criteria similar to those used in developing tolerable
intakes/exposures for non-cancer effects.
4.4.2 Linear extrapolation
Where data on the mechanism of tumour induction are not
available, as a default, risks are often linearly extrapolated into
the low-dose range. Previously (e.g., US EPA, 1986a) the linearized
multistage model was widely adopted for such extrapolations for data
from studies in animal species, whereas data from epidemiological
studies were generally modelled using a multistage model with a linear
term. More recently, curve fitting within the range of observation
with extrapolation from the lower 95% confidence limits on a dose
associated with a 10% extra risk (the LED10) has been recommended (US
EPA, 1996a). Linear extrapolation is considered to be appropriate if
available evidence supports a mode of action that is anticipated to be
linear or, as a science policy default, there is no evidence of either
linearity or non-linearity.
Other approaches to linear extrapolation have been described in
the literature. Gross et al. (1970) suggested a method based on
discarding data at the upper end of the dose range until a linear
model provides an adequate description of the remaining data. Van
Ryzin (1980) suggested the use of any model that fits the data
reasonably well to estimate the dose producing an excess risk of 1%,
and then using simple linear extrapolation to lower doses. Gaylor &
Kodell (1980) proposed fitting a model to the available data and then
using linear extrapolation below the lowest dose at which observations
were taken. Since the estimates at the lower doses might be unduly
influenced by the choice of the model used in the experimental dose
range, Farmer et al. (1982) suggested linear extrapolation below the
lowest dose or the dose corresponding to an estimated risk of 1%,
whichever was larger.
A model-free procedure based on linear extrapolation below the
lowest dose showing an increased (not necessarily statistically
significant) risk has been proposed by Krewski et al. (1984, 1986)
using linear extrapolation from all doses for which there were no
statistically significant increases in tumour incidence above the
baseline level, and selecting the smallest slope for low-dose risk
estimation. Similarly, Gaylor (1987) considered the smallest slope
obtained from all the possible combinations of data from the doses
where the lowest dose was in the convex portion of the dose-response
curve. In both cases, upper confidence limits on the slopes were used.
A number of arguments have been advanced in support of the
hypothesis of low-dose linearity (Krewski et al., 1986; Murdoch et
al., 1987). For example, the class of additive background models
considered by Crump et al. (1976) predicts low-dose linearity provided
only that the response increases smoothly with dose. However, it is
difficult to prove or disprove low-dose linearity experimentally even
in bioassays involving extremely large numbers of animals (Gaylor et
al., 1985). Indeed, dose-response curves for different types of
tumours in mice following exposure to 2-acetylaminofluorene (2-AAF) in
an ED01 study varied considerably.
Often, linear extrapolation is criticized as being too
conservative. For example, Bailar et al. (1988) demonstrated that a
significant fraction of bioassays conducted for the National
Toxicology Program indicate that, at high experimental doses, observed
response rates are higher than those predicted by a linear model. They
argue that, at low doses, the one-hit model may thus not be
conservative in some cases. However, these observations are not
necessarily inconsistent since, at low doses, the linear term
predominates. Crump et al. (1976), Peto (1978) and Hoel (1980) argue
that low-dose linearity occurs when substances augment existing
carcinogenic processes. The formation of DNA adducts, which may be
predictive of certain tumours induced by genotoxic carcinogens, has
often been observed to be linear at very low doses (Poirier & Beland,
1987). Based on these considerations, it is unclear whether an
estimate based on a linear approximation over- or under-estimates the
true risk.
The outcome of low-dose extrapolation is the resulting lifetime
cancer risk associated with estimated exposure for a particular
population. In view of the considerable uncertainties in extrapolating
results over several orders of magnitude, in the absence of
information on mechanisms of tumour induction, specification of risks
in terms of predicted incidence or numbers of excess deaths per unit
of the population implies a degree of precision that is considered
misleading by some (e.g., Health Canada, 1994).
4.4.3 Estimation of potency in the experimental range
For assessment of Priority Substances under the Canadian
Environmental Protection Act (CEPA), e.g., for genotoxic carcinogens,
a Tumorigenic Dose or Concentration05 (TD5) has been adopted as the
measure of dose-response (Health Canada, 1994; Meek et al., 1994). It
is the intake or concentration associated with a 5% incidence of
tumours in experimental studies on animals or epidemiological studies
on human populations. It serves as the basis for development of an
Exposure/Potency Index (EPI) which is the estimated daily human intake
or exposure divided by the TD5. A calculated EPI of 10-6 represents
a one million fold difference between human exposure and that at the
lower end of the dose-response curve, on which the estimate of potency
is based.
Any model that fits the empirical data well is likely to provide
a reasonable estimate of the TD5. Choice of the model may not be
critical since estimation is within the observed dose range, thereby
avoiding the numerous uncertainties associated with low-dose
extrapolation. Wherever possible, and if considered appropriate,
information on pharmacokinetics, metabolism and mechanisms of
carcinogenicity and mutagenicity is incorporated into the quantitative
estimates of potency derived particularly from studies in animals (to
provide relevant scaling of potency for human populations). The value
of 5% is arbitrary; selection of another value would not affect the
relative potencies for each of a range of compounds. Indeed, in the
literature, others have proposed the TD50 (Peto et al., 1984) and the
TD25 (Allen et al., 1988; Dybing & Huitfeldt, 1992; Dybing et al.,
1997). The Committee on Carcinogenicity of Chemicals in Food, Consumer
Products and the Environment in the United Kingdom has concluded that
the TD50 is the most practical quantitative estimate of carcinogenic
potency for the ranking of genotoxic carcinogens (UK DOH, 1995).
If there is no evidence for linearity, and there is sufficient
evidence to support an assumption of non-linearity for the
carcinogenic response, US EPA (1996a) recommends estimation of a
margin of exposure, which is the LED10 or other point of departure
divided by the environmental exposure of interest. It should be noted,
however, that this contrasts with the approach in Canada and Europe,
where characterization of potency within the experimental range is
considered appropriate for carcinogens, whereas the default in the USA
is linear. Indeed the Committee on Carcinogenicity of Chemicals in
Food, Consumer Products and the Environment in the United Kingdom
concluded that potency indices are not appropriate for the ranking of
non-genotoxic carcinogens. Rather for non-genotoxic compounds, the
emphasis should be on understanding mechanisms and their relevance to
humans.
4.4.4 Two-stage clonal expansion model
This approach is based on the two-stage model of carcinogenesis,
in which it is hypothesized that chemical carcinogenesis occurs in two
steps. Cells are initiated following the occurrence of genetic damage
in one or more cells in the target tissue. Such initiated cells may
then undergo malignant transformation to give rise to a cancerous
lesion. The rate of occurrence of such lesions may be increased by
subsequent exposure to a promoter, which serves to increase the pool
of initiated cells through mechanisms that result in clonal expansion.
Mathematical formulations of this process have been presented by
Moolgavkar et al. (1988) and Chen & Farland (1991). This stochastic
birth-death-mutation model assumes that two mutations, each occurring
at the time of cell division, are necessary for a normal cell to
become malignant. Initiating activity may be quantified in terms of
the rate of occurrence of the first mutation. The overall rate of
occurrence of the second mutation describes progression to a fully
differentiated cancerous lesion. Promotional activity is measured by
the difference in the birth and death rates of initiated cells. In the
absence of promotional effects and variability in the pool of normal
cells, the two-stage birth-death-mutation model reduces to the
classical two-stage model.
It should be noted, however, that there are currently few cases
where data are sufficient to permit application of such a model.
4.4.5 Proportional analyses - carcinogenic and non-neoplastic effects
There have been several investigations of the possibility of
predicting potency for particular types of toxicity from data on other
types of toxicity, including work by Tennant et al. (1987), Portier
(1988), Travis et al. (1990a,b, 1991), Zeiger et al. (1990) and
Haseman & Clark (1990). Such approaches have been necessary due, for
example, to the high cost and degree of difficulty of long-term or
carcinogenic bioassays. However, it is important to note that
correlations between potencies for different types of effects may be
artificially strengthened by dose selection (e.g., the top dose in
carcinogenic bioassays is often the maximum tolerated dose, selected
to elicit small reductions in body weight).
5. EXPOSURE ASSESSMENT
The objective of exposure assessment is to determine the nature
and extent of contact with chemical substances experienced or
anticipated under different conditions. Approaches for assessing
exposure and characterizing uncertainties/variability in resulting
estimates presented here are derived primarily from the Exposure
Assessment Guidelines (US EPA, 1986b, 1992).
5.1 Definition of exposure and related terms
Although there is reasonable agreement that human exposure means
contact with the chemical or agent (Allaby, 1983; Environ, 1988;
Hodgson et al., 1988), there has not yet been widespread agreement as
to whether this means contact with (a) the visible exterior of the
person (skin and openings into the body such as mouth and nostrils),
or (b) the so-called exchange boundaries where absorption takes place
(skin, lung, gastrointestinal tract). These different definitions have
led to some ambiguity in the use of terms and units for quantifying
exposure. In 1992, The US EPA published Guidelines (US EPA, 1992)
defining exposure as taking place at the visible external boundary, as
in (a) above.
Under this definition, it is helpful to think of the human body
as having a hypothetical outer boundary separating inside the body
from outside the body. This outer boundary of the body is the skin and
the openings into the body such as the mouth, the nostrils, and
punctures and lesions in the skin. Exposure to a chemical is the
contact of that chemical with the outer boundary. An exposure
assessment is the quantitative or qualitative evaluation of that
contact, which includes consideration of the intensity, frequency and
duration of contact, the route of exposure (e.g., dermal, oral or
respiratory), rates (chemical intake or uptake rates), the resulting
amount that actually crosses the boundary (a dose), and the amount
absorbed (internal dose). The Commission of the European Communities
(EC, 1996) presented a similar definition for exposure assessment: the
determination of the emissions, pathways and rates of movement of a
substance and its transformation or degradation, in order to estimate
the concentrations/ doses to which human populations or environmental
spheres (water, soil and air) are or may be exposed.
Depending on the purpose of an exposure assessment, the numerical
output may be an estimate of the intensity, rate, duration and
frequency of contact exposure or dose (the resulting amount that
actually crosses the boundary). For risk assessments based on
dose-response relationships, the output usually includes an estimate
of dose.
factor is introduced, then the commonly used 10-fold factor would be
replaced by the product of that factor and the remaining default
factor.
The composite default value of 10 has been criticized as
inadequate, for example, to allow for metabolic processes in mice
which can be related to body surface area (Calabrese et al., 1992);
the introduction of data-derived uncertainty factors would allow the
logical future development of more appropriate species specific
defaults.
e) Inter-individual variability in humans
In situations where appropriate toxicokinetic and toxicodynamic
data exist for a particular compound in humans, then the relevant
uncertainty factor should be replaced by the data-derived factor
(Renwick, 1993b). Data on PBPK modelling may also be able to
contribute to this assessment. If a data-derived factor is introduced,
then the commonly used 10-fold factor would be replaced by the product
of the data-derived factor and the remaining default factor.
Although the 10-fold default uncertainty factor is reasonable for
most cases (Dourson & Stara, 1983), it has been criticised as
inadequate for human variability especially when genetically
determined differences in a bioactivation process may be involved
(Calabrese, 1985; Goldstein, 1990). This concern reinforces the
importance of using an approach that allows the incorporation of data
on human variability in either toxicokinetics of the compound or the
sensitivity to its mechanism of action.
In addition to approaches aimed at incorporating as much
biological data as possible in the derivation of uncertainty factors,
probabilistic approaches have been investigated for the
characterization of uncertainty (Baird et al., 1996; Price et al.,
1997). Distributions can be developed on the basis of empirical
relationships observed for, for example, variations between LOAELs and
NOAELs and effect levels in subchronic versus chronic studies. Monte
Carlo techniques can be used to integrate probabilities for the
various areas of uncertainty.
4.4 Quantitative risk assessment for neoplastic (non-threshold)
effects
4.4.1 Introduction
A number of approaches have been adopted for characterization of
dose-response in the assessment of genotoxic neoplastic effects,
including quantitative extrapolation by mathematical modelling of the
dose-response curve to estimate the risk at likely human intakes or
exposures (low-dose risk extrapolation). Traditionally, where
dose-response has been extrapolated into the low-dose range, this has
been accomplished by the use of the linearized Armitage-Doll
multi-stage model. Dose-response may also be estimated in a two-step
process by straight linear extrapolation into the low-dose range from
a modelled point on the dose-response curve. Other measures of
dose-response include estimation of carcinogenic potency in the
experimental range and division of effect levels by a margin of
protection. In more recently developed biological models, different
stages in the process of carcinogenesis have been incorporated and
time to tumour has been taken into account (Moolgavkar et al., 1988),
although currently data are sufficient for application in only a
limited number of cases. In some cases where data permit, the dose
delivered to the target tissue has been incorporated into the
dose-response analysis (PBPK modelling) (IPCS, 1993).
In the same way as approaches adopted for non-neoplastic
(threshold) effects, there are increasingly attempts to incorporate
more of the scientific data in adopted approaches. For example, the
proposed cancer guidelines issued by the US EPA (1996b), updating the
previous guidelines (US EPA, 1986a), put emphasis on the full
integration of mechanistic information and dose-response data.
Depending on the mode of action, linear extrapolation into the
low-dose range or, alternatively, a margin of exposure would be
presented. The adequacy of the latter approach must be judged by
criteria similar to those used in developing tolerable
intakes/exposures for non-cancer effects.
4.4.2 Linear extrapolation
Where data on the mechanism of tumour induction are not
available, as a default, risks are often linearly extrapolated into
the low-dose range. Previously (e.g., US EPA, 1986a) the linearized
multistage model was widely adopted for such extrapolations for data
from studies in animal species, whereas data from epidemiological
studies were generally modelled using a multistage model with a linear
term. More recently, curve fitting within the range of observation
with extrapolation from the lower 95% confidence limits on a dose
associated with a 10% extra risk (the LED10) has been recommended (US
EPA, 1996a). Linear extrapolation is considered to be appropriate if
available evidence supports a mode of action that is anticipated to be
linear or, as a science policy default, there is no evidence of either
linearity or non-linearity.
Other approaches to linear extrapolation have been described in
the literature. Gross et al. (1970) suggested a method based on
discarding data at the upper end of the dose range until a linear
model provides an adequate description of the remaining data. Van
Ryzin (1980) suggested the use of any model that fits the data
reasonably well to estimate the dose producing an excess risk of 1%,
and then using simple linear extrapolation to lower doses. Gaylor &
Kodell (1980) proposed fitting a model to the available data and then
using linear extrapolation below the lowest dose at which observations
were taken. Since the estimates at the lower doses might be unduly
influenced by the choice of the model used in the experimental dose
range, Farmer et al. (1982) suggested linear extrapolation below the
lowest dose or the dose corresponding to an estimated risk of 1%,
whichever was larger.
A model-free procedure based on linear extrapolation below the
lowest dose showing an increased (not necessarily statistically
significant) risk has been proposed by Krewski et al. (1984, 1986)
using linear extrapolation from all doses for which there were no
statistically significant increases in tumour incidence above the
baseline level, and selecting the smallest slope for low-dose risk
estimation. Similarly, Gaylor (1987) considered the smallest slope
obtained from all the possible combinations of data from the doses
where the lowest dose was in the convex portion of the dose-response
curve. In both cases, upper confidence limits on the slopes were used.
A number of arguments have been advanced in support of the
hypothesis of low-dose linearity (Krewski et al., 1986; Murdoch et
al., 1987). For example, the class of additive background models
considered by Crump et al. (1976) predicts low-dose linearity provided
only that the response increases smoothly with dose. However, it is
difficult to prove or disprove low-dose linearity experimentally even
in bioassays involving extremely large numbers of animals (Gaylor et
al., 1985). Indeed, dose-response curves for different types of
tumours in mice following exposure to 2-acetylaminofluorene (2-AAF) in
an ED01 study varied considerably.
Often, linear extrapolation is criticized as being too
conservative. For example, Bailar et al. (1988) demonstrated that a
significant fraction of bioassays conducted for the National
Toxicology Program indicate that, at high experimental doses, observed
response rates are higher than those predicted by a linear model. They
argue that, at low doses, the one-hit model may thus not be
conservative in some cases. However, these observations are not
necessarily inconsistent since, at low doses, the linear term
predominates. Crump et al. (1976), Peto (1978) and Hoel (1980) argue
that low-dose linearity occurs when substances augment existing
carcinogenic processes. The formation of DNA adducts, which may be
predictive of certain tumours induced by genotoxic carcinogens, has
often been observed to be linear at very low doses (Poirier & Beland,
1987). Based on these considerations, it is unclear whether an
estimate based on a linear approximation over- or under-estimates the
true risk.
The outcome of low-dose extrapolation is the resulting lifetime
cancer risk associated with estimated exposure for a particular
population. In view of the considerable uncertainties in extrapolating
results over several orders of magnitude, in the absence of
information on mechanisms of tumour induction, specification of risks
in terms of predicted incidence or numbers of excess deaths per unit
of the population implies a degree of precision that is considered
misleading by some (e.g., Health Canada, 1994).
4.4.3 Estimation of potency in the experimental range
For assessment of Priority Substances under the Canadian
Environmental Protection Act (CEPA), e.g., for genotoxic carcinogens,
a Tumorigenic Dose or Concentration05 (TD5) has been adopted as the
measure of dose-response (Health Canada, 1994; Meek et al., 1994). It
is the intake or concentration associated with a 5% incidence of
tumours in experimental studies on animals or epidemiological studies
on human populations. It serves as the basis for development of an
Exposure/Potency Index (EPI) which is the estimated daily human intake
or exposure divided by the TD5. A calculated EPI of 10-6 represents
a one million fold difference between human exposure and that at the
lower end of the dose-response curve, on which the estimate of potency
is based.
Any model that fits the empirical data well is likely to provide
a reasonable estimate of the TD5. Choice of the model may not be
critical since estimation is within the observed dose range, thereby
avoiding the numerous uncertainties associated with low-dose
extrapolation. Wherever possible, and if considered appropriate,
information on pharmacokinetics, metabolism and mechanisms of
carcinogenicity and mutagenicity is incorporated into the quantitative
estimates of potency derived particularly from studies in animals (to
provide relevant scaling of potency for human populations). The value
of 5% is arbitrary; selection of another value would not affect the
relative potencies for each of a range of compounds. Indeed, in the
literature, others have proposed the TD50 (Peto et al., 1984) and the
TD25 (Allen et al., 1988; Dybing & Huitfeldt, 1992; Dybing et al.,
1997). The Committee on Carcinogenicity of Chemicals in Food, Consumer
Products and the Environment in the United Kingdom has concluded that
the TD50 is the most practical quantitative estimate of carcinogenic
potency for the ranking of genotoxic carcinogens (UK DOH, 1995).
If there is no evidence for linearity, and there is sufficient
evidence to support an assumption of non-linearity for the
carcinogenic response, US EPA (1996a) recommends estimation of a
margin of exposure, which is the LED10 or other point of departure
divided by the environmental exposure of interest. It should be noted,
however, that this contrasts with the approach in Canada and Europe,
where characterization of potency within the experimental range is
considered appropriate for carcinogens, whereas the default in the USA
is linear. Indeed the Committee on Carcinogenicity of Chemicals in
Food, Consumer Products and the Environment in the United Kingdom
concluded that potency indices are not appropriate for the ranking of
non-genotoxic carcinogens. Rather for non-genotoxic compounds, the
emphasis should be on understanding mechanisms and their relevance to
humans.
4.4.4 Two-stage clonal expansion model
This approach is based on the two-stage model of carcinogenesis,
in which it is hypothesized that chemical carcinogenesis occurs in two
steps. Cells are initiated following the occurrence of genetic damage
in one or more cells in the target tissue. Such initiated cells may
then undergo malignant transformation to give rise to a cancerous
lesion. The rate of occurrence of such lesions may be increased by
subsequent exposure to a promoter, which serves to increase the pool
of initiated cells through mechanisms that result in clonal expansion.
Mathematical formulations of this process have been presented by
Moolgavkar et al. (1988) and Chen & Farland (1991). This stochastic
birth-death-mutation model assumes that two mutations, each occurring
at the time of cell division, are necessary for a normal cell to
become malignant. Initiating activity may be quantified in terms of
the rate of occurrence of the first mutation. The overall rate of
occurrence of the second mutation describes progression to a fully
differentiated cancerous lesion. Promotional activity is measured by
the difference in the birth and death rates of initiated cells. In the
absence of promotional effects and variability in the pool of normal
cells, the two-stage birth-death-mutation model reduces to the
classical two-stage model.
It should be noted, however, that there are currently few cases
where data are sufficient to permit application of such a model.
4.4.5 Proportional analyses - carcinogenic and non-neoplastic effects
There have been several investigations of the possibility of
predicting potency for particular types of toxicity from data on other
types of toxicity, including work by Tennant et al. (1987), Portier
(1988), Travis et al. (1990a,b, 1991), Zeiger et al. (1990) and
Haseman & Clark (1990). Such approaches have been necessary due, for
example, to the high cost and degree of difficulty of long-term or
carcinogenic bioassays. However, it is important to note that
correlations between potencies for different types of effects may be
artificially strengthened by dose selection (e.g., the top dose in
carcinogenic bioassays is often the maximum tolerated dose, selected
to elicit small reductions in body weight).
5. EXPOSURE ASSESSMENT
The objective of exposure assessment is to determine the nature
and extent of contact with chemical substances experienced or
anticipated under different conditions. Approaches for assessing
exposure and characterizing uncertainties/variability in resulting
estimates presented here are derived primarily from the Exposure
Assessment Guidelines (US EPA, 1986b, 1992).
5.1 Definition of exposure and related terms
Although there is reasonable agreement that human exposure means
contact with the chemical or agent (Allaby, 1983; Environ, 1988;
Hodgson et al., 1988), there has not yet been widespread agreement as
to whether this means contact with (a) the visible exterior of the
person (skin and openings into the body such as mouth and nostrils),
or (b) the so-called exchange boundaries where absorption takes place
(skin, lung, gastrointestinal tract). These different definitions have
led to some ambiguity in the use of terms and units for quantifying
exposure. In 1992, The US EPA published Guidelines (US EPA, 1992)
defining exposure as taking place at the visible external boundary, as
in (a) above.
Under this definition, it is helpful to think of the human body
as having a hypothetical outer boundary separating inside the body
from outside the body. This outer boundary of the body is the skin and
the openings into the body such as the mouth, the nostrils, and
punctures and lesions in the skin. Exposure to a chemical is the
contact of that chemical with the outer boundary. An exposure
assessment is the quantitative or qualitative evaluation of that
contact, which includes consideration of the intensity, frequency and
duration of contact, the route of exposure (e.g., dermal, oral or
respiratory), rates (chemical intake or uptake rates), the resulting
amount that actually crosses the boundary (a dose), and the amount
absorbed (internal dose). The Commission of the European Communities
(EC, 1996) presented a similar definition for exposure assessment: the
determination of the emissions, pathways and rates of movement of a
substance and its transformation or degradation, in order to estimate
the concentrations/ doses to which human populations or environmental
spheres (water, soil and air) are or may be exposed.
Depending on the purpose of an exposure assessment, the numerical
output may be an estimate of the intensity, rate, duration and
frequency of contact exposure or dose (the resulting amount that
actually crosses the boundary). For risk assessments based on
dose-response relationships, the output usually includes an estimate
of dose.
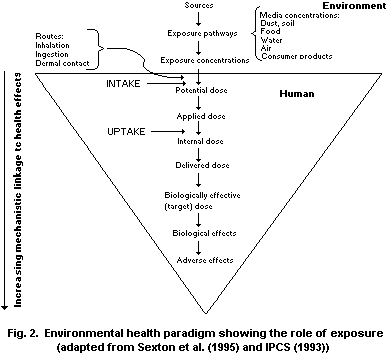 5.2 Exposure and dose
Most of the time, the chemical coming into contact with the outer
boundary of the body is contained in air, water, soil, a product or a
transport or carrier medium; the chemical concentration in these media
at the point of contact is the concentration, on which exposure
estimates are based. Exposure over a period of time can be represented
by a time-dependent profile of the exposure concentration. The area
under the curve of this profile is the magnitude of the exposure, in
concentration-time units (Lioy, 1990; US NRC, 1990):
5.2 Exposure and dose
Most of the time, the chemical coming into contact with the outer
boundary of the body is contained in air, water, soil, a product or a
transport or carrier medium; the chemical concentration in these media
at the point of contact is the concentration, on which exposure
estimates are based. Exposure over a period of time can be represented
by a time-dependent profile of the exposure concentration. The area
under the curve of this profile is the magnitude of the exposure, in
concentration-time units (Lioy, 1990; US NRC, 1990):
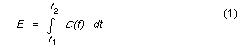 where E is the magnitude of exposure, C(t) is the exposure
concentration as a function of time, and t is time, t2-t1 being the
exposure duration (ED). If ED is a continuous period of time (e.g., a
day, week, year, etc.), then C(t) may be zero during part of this
time. Integrated exposures are done typically for a single individual,
a specific chemical, and a particular pathway or exposure route over a
given time period.
The integrated exposures for a number of different individuals (a
population or population segment, for example), may then be displayed
in a histogram or curve (usually, with integrated exposure increasing
along the abscissa or x-axis, and the number of individuals at that
integrated exposure increasing along the ordinate or y-axis). This
histogram or curve is a presentation of an exposure distribution for
that population or population segment.
Applied dose is the amount of a chemical at the absorption
barrier (skin, lung, gastrointestinal tract) available for absorption.
Usually, it is very difficult to measure the applied dose directly, as
many of the absorption barriers are internal to the human and are not
localized in such a way as to make measurement easy. An approximation
of applied dose can be made, however, using the concept of potential
dose (Lioy, 1990; US NRC, 1990). Potential dose is simply the amount
of the chemical ingested, inhaled or in material applied to the skin.
For the dermal route, potential dose is the amount of chemical
applied or the amount of chemical in the medium applied, e.g., as a
small amount of particulate deposited on the skin. It should be noted
that as not all of the chemical in the particulate is in contact with
the skin, this differs from exposure (the concentration in the
particulate multiplied by the time of contact) and applied dose (the
amount in the layer actually touching the skin).
The applied dose, or the amount that reaches the exchange
boundaries of the skin, lung or gastrointestinal tract, may often be
less than the potential dose if the material is only partly
bioavailable. This will depend, for example, on the form in which the
compound is administered (e.g., neat or in vehicle on skin). Where
data on bioavailability are known, adjustments to the potential dose
to convert it to applied dose and internal dose may be made. For
example, chemicals reaching their target through the gastrointestinal
tract can be metabolized in the anaerobic conditions of the lower
colon prior to absorption. Bioavailability via various routes of
exposure may also vary. For example, intestinal absorption results in
a first pass effect that may lead to metabolic detoxication or
activation by the liver.
The amount of a chemical that has been absorbed and is available
for interaction with biologically significant receptors is called the
internal dose. Once absorbed, the chemical can undergo metabolism,
storage, excretion or transport within the body. The amount
transported to an individual organ, tissue or fluid of interest is
termed the delivered dose. The delivered dose may be only a small part
of the total internal dose. The biologically effective dose, or the
amount that actually reaches cells, sites or membranes where adverse
effects occur (US NRC, 1990), may only be a part of the delivered
dose. Currently, most risk assessments dealing with environmental
chemicals (as opposed to pharmaceutical assessments) use dose-response
relationships based on potential (administered) dose or internal dose,
since the pharmacokinetics necessary to base relationships on the
delivered dose or biologically effective doses are not available. This
may change in the future, as more becomes known about the
pharmacokinetics of environmental chemicals.
Doses are often presented as dose rates, or the amount of a
chemical dose (applied or internal) per unit time (e.g., mg/day), for
instance, as dose rates on a per-unit-body-weight basis (e.g., mg/kg
per day).
The general equation for potential dose for intake processes,
e.g., inhalation and ingestion, is simply the integration of the
chemical intake rate (concentration of the chemical in the medium
multiplied by the intake rate of the medium, C x IR) over time:
where E is the magnitude of exposure, C(t) is the exposure
concentration as a function of time, and t is time, t2-t1 being the
exposure duration (ED). If ED is a continuous period of time (e.g., a
day, week, year, etc.), then C(t) may be zero during part of this
time. Integrated exposures are done typically for a single individual,
a specific chemical, and a particular pathway or exposure route over a
given time period.
The integrated exposures for a number of different individuals (a
population or population segment, for example), may then be displayed
in a histogram or curve (usually, with integrated exposure increasing
along the abscissa or x-axis, and the number of individuals at that
integrated exposure increasing along the ordinate or y-axis). This
histogram or curve is a presentation of an exposure distribution for
that population or population segment.
Applied dose is the amount of a chemical at the absorption
barrier (skin, lung, gastrointestinal tract) available for absorption.
Usually, it is very difficult to measure the applied dose directly, as
many of the absorption barriers are internal to the human and are not
localized in such a way as to make measurement easy. An approximation
of applied dose can be made, however, using the concept of potential
dose (Lioy, 1990; US NRC, 1990). Potential dose is simply the amount
of the chemical ingested, inhaled or in material applied to the skin.
For the dermal route, potential dose is the amount of chemical
applied or the amount of chemical in the medium applied, e.g., as a
small amount of particulate deposited on the skin. It should be noted
that as not all of the chemical in the particulate is in contact with
the skin, this differs from exposure (the concentration in the
particulate multiplied by the time of contact) and applied dose (the
amount in the layer actually touching the skin).
The applied dose, or the amount that reaches the exchange
boundaries of the skin, lung or gastrointestinal tract, may often be
less than the potential dose if the material is only partly
bioavailable. This will depend, for example, on the form in which the
compound is administered (e.g., neat or in vehicle on skin). Where
data on bioavailability are known, adjustments to the potential dose
to convert it to applied dose and internal dose may be made. For
example, chemicals reaching their target through the gastrointestinal
tract can be metabolized in the anaerobic conditions of the lower
colon prior to absorption. Bioavailability via various routes of
exposure may also vary. For example, intestinal absorption results in
a first pass effect that may lead to metabolic detoxication or
activation by the liver.
The amount of a chemical that has been absorbed and is available
for interaction with biologically significant receptors is called the
internal dose. Once absorbed, the chemical can undergo metabolism,
storage, excretion or transport within the body. The amount
transported to an individual organ, tissue or fluid of interest is
termed the delivered dose. The delivered dose may be only a small part
of the total internal dose. The biologically effective dose, or the
amount that actually reaches cells, sites or membranes where adverse
effects occur (US NRC, 1990), may only be a part of the delivered
dose. Currently, most risk assessments dealing with environmental
chemicals (as opposed to pharmaceutical assessments) use dose-response
relationships based on potential (administered) dose or internal dose,
since the pharmacokinetics necessary to base relationships on the
delivered dose or biologically effective doses are not available. This
may change in the future, as more becomes known about the
pharmacokinetics of environmental chemicals.
Doses are often presented as dose rates, or the amount of a
chemical dose (applied or internal) per unit time (e.g., mg/day), for
instance, as dose rates on a per-unit-body-weight basis (e.g., mg/kg
per day).
The general equation for potential dose for intake processes,
e.g., inhalation and ingestion, is simply the integration of the
chemical intake rate (concentration of the chemical in the medium
multiplied by the intake rate of the medium, C x IR) over time:
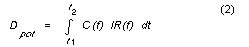 where Dpot is potential dose and IR(t) is the ingestion or inhalation
rate.
The quantity t2-t1, as before, represents the period of time
over which exposure is being examined, or the exposure duration (ED).
The exposure duration may contain times where the chemical is in
contact with the person, and also times when C(t) is zero. Contact
time represents the actual time period where the chemical is in
contact with the person. For cases such as ingestion, where actual
contact with food or water is intermittent, and consequently the
actual contact time may be small, the intake rate is usually expressed
in terms of a frequency of events (e.g., 8 glasses of water consumed
per day) multiplied by the intake per event (e.g., 250 ml of water per
glass of water consumed). Intermittent air exposures (e.g., 8 h
exposed/day multiplied by one cubic metre of air inhaled/hour) can
also be expressed easily using exposure duration rather than contact
time. Hereafter, the term exposure duration will be used in the
examples below to refer to the term t2-t1, since it occurs
frequently in exposure assessments and it is often easier to use.
Equation 2 can also be expressed in discrete form as a summation
of the doses received during various events i:
where Dpot is potential dose and IR(t) is the ingestion or inhalation
rate.
The quantity t2-t1, as before, represents the period of time
over which exposure is being examined, or the exposure duration (ED).
The exposure duration may contain times where the chemical is in
contact with the person, and also times when C(t) is zero. Contact
time represents the actual time period where the chemical is in
contact with the person. For cases such as ingestion, where actual
contact with food or water is intermittent, and consequently the
actual contact time may be small, the intake rate is usually expressed
in terms of a frequency of events (e.g., 8 glasses of water consumed
per day) multiplied by the intake per event (e.g., 250 ml of water per
glass of water consumed). Intermittent air exposures (e.g., 8 h
exposed/day multiplied by one cubic metre of air inhaled/hour) can
also be expressed easily using exposure duration rather than contact
time. Hereafter, the term exposure duration will be used in the
examples below to refer to the term t2-t1, since it occurs
frequently in exposure assessments and it is often easier to use.
Equation 2 can also be expressed in discrete form as a summation
of the doses received during various events i:
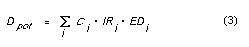 where EDi is the exposure duration for event i. If C and IR are
nearly constant (which is a good approximation if the contact time is
very short), equation 4-3 becomes:
where EDi is the exposure duration for event i. If C and IR are
nearly constant (which is a good approximation if the contact time is
very short), equation 4-3 becomes:
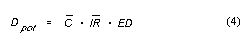 _
where ED is the sum of the exposure durations for all events, and C
__
and IR are the average values for these parameters. Equation 4 will
not necessarily hold in cases where C and IR vary considerably. In
those cases, equation 3 can be used if the exposure can be broken out
into segments where C and IR are approximately constant. If even this
condition cannot be met, equation 2 may be used.
For risk assessments, estimates of dose should be expressed in a
manner that can be compared with available dose-response data.
Frequently, dose-response relationships are based on potential dose
(called administered dose in animal studies), although dose-response
relationships are sometimes based on internal dose.
Doses may be expressed in several different ways. Solving
equations 2, 3 or 4 for example, gives a total dose accumulated over
the time in question. The dose per unit time is the dose rate, which
has units of mass/time (e.g., mg/day). Because intake and uptake can
vary, dose rate is not necessarily constant. An average dose rate over
a period of time is a useful number for many risk assessments.
Exposure assessments take into account the time scale related to
the biological response studied, unless the assessment is intended to
provide data on the range of biological responses (US NRC, 1990). For
developmental toxicity effects, a single short-term exposure can cause
the adverse health effects. For many non-cancer effects, risk
assessments consider the period of time over which the exposure
occurred, and often, if there are no excursions in exposure that would
lead to acute effects, average exposures or doses over the period of
exposure are sufficient for the assessment. These averages are often
in the form of average daily doses (ADDs) expressed, for example, in
mg/kg body weight per day.
An ADD can be calculated from equation 2 by averaging Dpot over
body weight and an averaging time, provided the dosing pattern is
known so that the integral can be solved. It is unusual to have such
data for human exposure and intake over extended periods of time, so
some simplifying assumptions are commonly used. Using equation 4
instead of 2 or 3 involves making steady-state assumptions about C and
IR, but this makes the equation for ADD easier to solve. For intake
processes, then, using equation 4, this becomes:
_
where ED is the sum of the exposure durations for all events, and C
__
and IR are the average values for these parameters. Equation 4 will
not necessarily hold in cases where C and IR vary considerably. In
those cases, equation 3 can be used if the exposure can be broken out
into segments where C and IR are approximately constant. If even this
condition cannot be met, equation 2 may be used.
For risk assessments, estimates of dose should be expressed in a
manner that can be compared with available dose-response data.
Frequently, dose-response relationships are based on potential dose
(called administered dose in animal studies), although dose-response
relationships are sometimes based on internal dose.
Doses may be expressed in several different ways. Solving
equations 2, 3 or 4 for example, gives a total dose accumulated over
the time in question. The dose per unit time is the dose rate, which
has units of mass/time (e.g., mg/day). Because intake and uptake can
vary, dose rate is not necessarily constant. An average dose rate over
a period of time is a useful number for many risk assessments.
Exposure assessments take into account the time scale related to
the biological response studied, unless the assessment is intended to
provide data on the range of biological responses (US NRC, 1990). For
developmental toxicity effects, a single short-term exposure can cause
the adverse health effects. For many non-cancer effects, risk
assessments consider the period of time over which the exposure
occurred, and often, if there are no excursions in exposure that would
lead to acute effects, average exposures or doses over the period of
exposure are sufficient for the assessment. These averages are often
in the form of average daily doses (ADDs) expressed, for example, in
mg/kg body weight per day.
An ADD can be calculated from equation 2 by averaging Dpot over
body weight and an averaging time, provided the dosing pattern is
known so that the integral can be solved. It is unusual to have such
data for human exposure and intake over extended periods of time, so
some simplifying assumptions are commonly used. Using equation 4
instead of 2 or 3 involves making steady-state assumptions about C and
IR, but this makes the equation for ADD easier to solve. For intake
processes, then, using equation 4, this becomes:
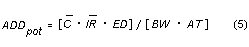 where ADDpot is the average daily potential dose, BW is body weight,
and AT is the time period over which the dose is averaged (converted
_
to days). As with equation 4, the exposure concentration C is best
expressed as an estimate of the arithmetic mean regardless of the
distribution of the data. Again, using average values for C and IR in
equation 5 assumes that C and IR are approximately constant.
For effects such as cancer, where the biological response is
usually described in terms of lifetime probabilities, even though
exposure does not occur over the entire lifetime, doses are often
presented as lifetime average daily doses (LADDs). The LADD takes the
form of equation 6, with lifetime (LT) replacing the averaging time
(AT):
where ADDpot is the average daily potential dose, BW is body weight,
and AT is the time period over which the dose is averaged (converted
_
to days). As with equation 4, the exposure concentration C is best
expressed as an estimate of the arithmetic mean regardless of the
distribution of the data. Again, using average values for C and IR in
equation 5 assumes that C and IR are approximately constant.
For effects such as cancer, where the biological response is
usually described in terms of lifetime probabilities, even though
exposure does not occur over the entire lifetime, doses are often
presented as lifetime average daily doses (LADDs). The LADD takes the
form of equation 6, with lifetime (LT) replacing the averaging time
(AT):
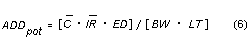 5.3 Approaches to quantification of exposure
Exposure (or dose) is assessed generally by one of the following
approaches:
a) The exposure can be measured at the point of contact (the outer
boundary of the body) while it is taking place, measuring both
exposure concentration and time of contact and integrating them
(point-of-contact or personal measurement);
b) The exposure can be estimated by separately evaluating the
exposure concentration and the time of contact, then combining
this information (scenario evaluation);
c) The exposure can be estimated from dose, which in turn can be
reconstructed through internal indicators (biomarkers, body
burden, excretion levels, etc.) after the exposure has taken
place (reconstruction).
These three approaches to quantification of exposure (or dose)
are independent, as each is based on different data. This offers the
opportunity of checking the accuracy of exposure estimated by one
approach through use of an independent approach, where data permit.
The independence of the three methods is a useful concept in verifying
or validating results. Each of the three has strengths and weaknesses;
using them in combination can considerably strengthen the credibility
of an exposure or risk assessment.
5.3.1 Measurement at point of contact (personal monitoring)
Point-of-contact exposure measurement evaluates the exposure as
it occurs, by measuring the chemical concentrations at the interface
between the person and the environment as a function of time,
resulting in an exposure profile. The best known example of the
point-of-contact measurement is the radiation dosimeter. This small
badge-like device measures exposure to radiation as it occurs and
provides an integrated estimate of exposure for the period of time
over which the measurement has been taken. Another example is the
Total Exposure Assessment Methodology (TEAM) studies (US EPA, 1987a)
conducted by the EPA and similar multimedia exposure studies in Canada
(Otson et al., 1996). In the TEAM studies, a small pump with a
collector and absorbent was attached to a person's clothing to measure
his or her exposure to airborne solvents or other pollutants as it
occurred. A third example is the carbon monoxide (CO) point-of-contact
measurement studies where subjects carried a small CO measuring device
for several days (US EPA, 1984). Dermal patch studies and duplicate
meal studies are also point-of-contact measurement studies. In all of
these examples, the measurements are taken at the interface between
the person and the environment while exposure is occurring. Use of
these data for estimating exposures or doses for periods that differ
from those for which the data are collected (e.g., for estimates of
lifetime exposures) will require some assumptions.
The strength of this method is that it measures exposure
directly, and providing that the measurement devices are accurate, is
likely to give the most accurate exposure value for the period of time
over which the measurement was taken. It is often expensive, however,
and measurement devices and techniques do not currently exist for all
chemicals. This method may also require assumptions to be made
concerning the relationship between short-term sampling and long-term
exposures, if appropriate. This method is also not source-specific, a
limitation when particular sources will need to be addressed by risk
managers.
5.3.2 Scenario evaluation method (time activity and
monitoring/modelling)
In exposure scenario evaluation, the assessor attempts to
determine the concentrations of chemicals in a medium or location and
link this information with the time and ways that individuals or
populations come into contact with the chemical. The set of
assumptions about how this contact takes place is an exposure
scenario.
The first step in a scenario evaluation is usually to
characterize the contaminant concentration in the media of concern at
the point where contact occurs. This is typically accomplished
indirectly by measuring, modelling or using existing data on
concentrations in the bulk media, rather than at the true point of
contact. An example of a scenario evaluation is presented in Table 1.
Since the concentration in the bulk medium is not the same as the
exposure concentration, this is a clear source of potential error in
the exposure estimate. Generally, the closer the medium can be
measured to the point of contact (in both space and time), the less
uncertainty there is in the characterization of exposure
concentration. Where monitoring data are inadequate, fate models are
typically used to estimate chemical concentrations. These models can
span a wide range of complexity in terms of spatial dimensions and
temporal assumptions (i.e. steady-state versus non-steady-state).
Types of fate models include:
* simple dilution models where a measured concentration in an
effluent is divided by a dilution factor or the chemical release
rate is divided by the bulk flow rate of the medium;
* equilibrium models which predict the distribution of a chemical
in the environment based on partitioning ratios or fugacity (the
escaping tendency of a chemical from one environmental phase to
another);
* dispersion models which predict reductions in concentrations from
point sources based on assumed mathematical functions or
dispersion properties of the chemical;
* transport models which predict concentration changes over
distance and can represent dispersion, biochemical degradation
and absorption.
Compilations of existing environmental fate models have been
published (OECD, 1989, 1991a; Braat et al., 1991; ECETOC, 1992, 1993;
RIVM, 1994). The US EPA has produced a software system called the
Integrated Model Evaluation System (IMES) to help assessors select the
fate model best suited to their needs (US EPA, 1992). The software
prompts users to answer a variety of questions about their needs and
then lists the models that have matching features. The system has
information on over 150 models representing all media (air, surface
water and groundwater). Model information includes descriptions of the
model type, computer requirements, validation testing and contact for
obtaining a copy. The Netherlands National Environmental Policy Plan
Uniform System for the Evaluation of Substances (USES) is a
decision-support system for the rapid quantitative assessment of the
hazards and risks of chemicals, including new substances, agricultural
pesticides and biocides (RIVM, 1994). USES has been the basis for the
development of the European Union System for the Evaluation of
Substances (EUSES).
The reliability of modelled estimates of chemical concentration
in the general environment depends on how well the model assumptions
match reality (i.e. how realistic are the assumptions such as
steady-state conditions and homogenous media properties), whether the
model performance has been demonstrated under conditions similar to
those of concern; and the quantity and quality of input data.
Modelling efforts which use input values derived primarily on the
basis of default assumptions are generally most useful for screening
purposes to highlight areas in which specific additional data are
required to estimate exposure more accurately. Further discussion
about model uncertainty can be found below.
The next steps involve identifying who is exposed and developing
estimates of the frequency and duration of exposure. Like chemical
concentration characterization, this is usually done indirectly by use
of demographic data, survey statistics, behaviour observation,
activity diaries, activity models or, in the absence of more
substantive information, assumptions about behaviour. When estimating
potential dose, this step also involves estimating how much contact
occurs. Table 2 shows examples of standardized reference values for
body weights, fluid intake and respiratory volumes. This type of data
is also summarized in the Exposure Factors Handbook (US EPA, 1997).
This Handbook includes information on consumption rates for various
food types, fish ingestion, soil ingestion, dermal contact with soils,
body surface area, lifetime, body weight, inhalation rate, breast milk
ingestion rate, and activity patterns (time spent swimming, bathing
time, time indoors/outdoors, time in vehicles, etc.). For each factor,
descriptions are provided of the average values and the variability in
the general population. Values are recommended for each factor, with a
qualitative indication of the supporting weight of evidence.
Table 1. Estimated daily intake of inorganic fluoride (mg/kg body weight per day), according to age group, by the general
population of Canada (from Liteplo et al., 1994)
Route of exposure 0-6 monthsa 7 months-4 yearsb 5-11 yearsc 12-19 yearsd 20 + yearse
Ambient airf 0.01 0.01 0.01 0.01 0.01
Foodg 14-92 22 16 13 30
Breast milkh 0.5-1.1 - - - -
Soili 0.03-1.6 0.02-1.2 0.01-0.4 0.002-0.1 0.002-0.1
"Fluoridated" drinking-waterj - 45-77 24-42 17-29 16-27
"Non-fluoridated" drinking-waterk - 3.1-12.9 1.7-7.0 1.1-4.8 1.1-4.5
Household productsl - 20-60 8.2-20 2.5 1.1
Total intake of breast-fed infants 0.5-2.6 - - - -
Total intake of formula-fed infants 14-94 - - - -
Total intake ("Fluoridated" water)m - 87-160 49-79 33-45 47-58
Total intake ("Non-fluoridated" water)n - 45-96 26-44 17-21 32-36
a Assumed to weigh 7 kg, breathe 2 m3 air, drink 750 ml of breast milk or infant formula (as food),
and consume 35 mg soil per day.
b Assumed to weigh 13 kg, breathe 5 m3 air, drink 0.8 litres of water, and consume 50 mg soil per day.
c Assumed to weigh 27 kg, breathe 12 m3 air, drink 0.9 litres of water, and consume 35 mg soil per day.
d Assumed to weigh 57 kg, breathe 21 m3 air, drink 1.3 litres of water, and consume 20 mg soil per day.
e Assumed to weigh 70 kg, breathe 23 m3 air, drink 1.5 litres of water, and consume 20 mg soil per day.
f Based on the mean concentration of inorganic (gaseous and particulate) fluoride in ambient air of
0.03 µg/m3, reported for Toronto, Ontario, and assuming the concentration in indoor air is identical
to (outdoor) ambient air.
Table 1 (Continued)
g Formula-fed infants (0-6 months): based on the mean concentrations of inorganic fluoride in infant
formulas purchased in the USA of 0.127 and 0.854 mg/litre reported for ready-to-use, milk-based formula
and soy-based powdered formula (prepared with drinking-water containing 1 ppm fluoride), respectively,
and assuming infants are exclusively formula-fed and consume 750 ml formula per day. General population
(7 months and older): based on levels of inorganic fluoride detected in 109 individual foods from Canada
(and the USA), in the following food groups: 0.01-0.80 µg/g in dairy products, 0.12-1.02 µg/g in cereal
products, 0.01-0.58 µg/g in fruit, 0.01-0.68 µg/g in vegetables, 0.04-4.57 µg/g in meat/fish/eggs;
0.05-0.13 µg/g in fats, 0.11-0.35 µg/g in nuts/legumes, 0.02-0.86 µg/g in foods containing primarily sugar,
0.41-0.84 µg/g in soup, 4.97 µg/g in tea; and the daily intake of each food item by the various age groups
of the general population of Canada.
h Based on the mean concentrations of inorganic fluoride of 4.4 and 9.8 ng/g reported for samples of
breast milk from mothers living in communities served by "non-fluoridated" and "fluoridated" drinking-water,
respectively, assuming the density of breast milk is equal to 1.0 g/ml.
i Based on a range of concentrations of total inorganic fluoride of 6 µg/g reported by Sidhu (1982) for
soil collected in Newfoundland, to 309 µg/g [mean concentration in Canadian surface soil (0-130 cm depth)].
j Based on a range of mean concentrations of inorganic fluoride in "fluoridated" drinking-water of 0.73 mg/litre,
determined from fluoride levels in 3 communities in Newfoundland and Labrador, to 1.25 mg/litre, determined
from 2 communities in the Yukon. "Fluoridated" refers to drinking-water to which inorganic fluoride has been
intentionally added for the prevention of dental caries.
k Based on a range of mean concentrations of inorganic fluoride in "non-fluoridated" drinking-water of (at least)
0.05 mg/litre (reported for 3 communities in British Columbia), to 0.21 mg/litre (reported for an unspecified
number of communities in the Yukon). "Non-fluoridated" refers to drinking-water to which inorganic fluoride
has not been intentionally added for the prevention of dental caries.
l Based on a mean concentration of inorganic fluoride in most dentifrice products of 1000 µg/g and an estimated
intake of dentifrice of 0.26-0.78 g/day for children 7 months to 4 years of age, 0.22-0.54 g/day for children
5 to 11 years of age, 0.14 g/day for adolescents 12 to 19 years of age, and 0.08 g/day for adults 20 + years
of age, assuming an average of 2 brushings per day.
m Estimated total daily intake of inorganic fluoride by individuals consuming "fluoridated" drinking-water in Canada.
n Estimated total daily intake of inorganic fluoride by individuals in Canada consuming drinking-water
that is not "fluoridated".
Table 2. Human contact parameters (from ICRP, 1974)
Body weight, kg
Adult male = 70
Adult female = 58
Average = 64a
Daily fluid intake (milk, tap water, other beverages), ml/day
Normal conditions:
Adults = 1000-2400, representative figure = 1900b
Adult male = 1950
Adult female = 1400
Child (10 years) = 1400
High average temperature (32 °C):
Adults = 2840-3410
moderate activity:
Adults = 3700
Respiratory volumes
8-h respiratory volumes, litres per 8 h resting:
Adult man = 3600
Adult woman = 2900
Child (10 years) = 2300
light/non-occupational activity: Adult man = 9600
Adult woman = 9100
Child (10 years) = 6240
Daily inhalation volume, m3 (8 h resting, 16 h light/non-occupational activity)
Adult male = 23
Adult female = 21
Average adult = 22
Child (10 years) = 15
a WHO uses 60 kg for calculation of acceptable daily intakes
and water quality guidelines (IPCS, 1987b; WHO, 1993).
b WHO uses a daily per capita drinking-water consumption
of 2 litres in calculating water quality guidelines (WHO, 1993).
The chemical concentration and population characterizations are
ultimately combined in an exposure scenario, and there are various
ways to accomplish this. One of the major problems with this approach
is that the limiting assumptions or boundary conditions (e.g.,
steady-state assumptions) do not always hold true. Two ways to address
to this aspect are: (a) to evaluate the exposure or dose equation
under conditions where the limiting assumptions do hold true; or (b)
to deal with the uncertainty caused by the divergence from the
boundary conditions. As an example of the first option, in the
microenvironment method, utilized primarily for evaluating airborne
exposures in the general environment but including contact with the
skin in the occupational environment, segments of time and location
are evaluated where the assumption of constant concentration is
approximately true and then summed over all such time segments for a
total exposure for the respiratory route, effectively removing some of
the boundary conditions. While estimates of exposure concentration and
time-of-contact are still derived indirectly by this method, the
concentration and time-of-contact estimates can be measured for each
microenvironment. This avoids much of the error due to using average
values in cases where concentration varies widely along with
time-of-contact.
As examples of the second approach, there are various tools used
to describe uncertainty caused by parameter variation, such as Monte
Carlo analysis (see below).
One strength of the scenario evaluation approach is that it is
usually the least expensive method of the three. In addition, it is
particularly suited to analysis of the risk consequences of proposed
actions. It is both a strength and a weakness of scenario development
that the evaluation can be performed with little or no data; it is a
technique that is best used when some knowledge exists about the
soundness, validity and uncertainty of the underlying assumptions.
5.3.3 Biomarkers of exposure/estimation of internal dose
Exposure can also be estimated after it has taken place. If a
total dose is known, or can be reconstructed, and information about
intake and uptake rates is available, an average past exposure rate
can be estimated. Reconstruction of dose relies on measuring internal
body indicators after exposure, intake and uptake have already
occurred, and using these measurements to back-calculate dose.
However, the data on body burden levels or biomarkers cannot be used
directly unless a relationship can be established between these levels
or biomarker indications and internal dose, and interfering reactions
(e.g., metabolism of unrelated chemicals) can be accounted for or
ruled out. Biological tissue or fluid measurements that reveal the
presence of a chemical may indicate directly that an exposure has
occurred, provided the chemical is not a metabolite of other
chemicals. These biomarkers of exposure are necessarily limited,
however, to ethical relatively non-invasive techniques.
Biological monitoring can be used to evaluate the amount of a
chemical in the body by measuring one or more of the following items
(not all of these can be measured for every chemical):
* the concentration of the chemical itself in biological tissues or
sera (blood, urine, breath, hair, adipose tissue, etc.);
* the concentration of the chemical's metabolite(s);
* the biological effect that occurs as a result of human exposure
to the chemical (e.g., alkylated haemoglobin or changes in enzyme
induction);
* the amount of a chemical or its metabolites bound to target
molecules.
Biomarkers can be used to estimate chemical uptake during a
specific interval if background levels do not mask the marker and the
relationships between uptake and the marker selected are known. The
time of sampling for biomarkers can be critical. Establishing a
correlation between exposure and the measurement of the marker,
including pharmacokinetics, can help optimize the sampling conditions.
The strengths of this method are that it demonstrates that
exposure to and absorption of the chemical has actually taken place,
and it theoretically can give a good indication of past exposure.
Biomarkers integrate exposure from all sources and take into account
absorption, which may vary considerably due to a variety of factors
including environmental characteristics, genetic predisposition, age,
gender, ethnicity and/or lifestyle factors.
For many environmental pollutants, the flow of events between
exposure and health effects is not well understood. Biomarkers help
address this problem by improving the sensitivity, specificity and
predictive value of detection and quantification of adverse effects at
low dose and early exposure (ECETOC, 1989; Fowle, 1989; Fowle &
Sexton, 1992; US NRC, 1992). Sensitive subpopulations can be better
pinpointed by biomarkers that measure increased absorption rate or a
more severe biological response to a given environmental exposure
(Lauwerys, 1984; ECETOC, 1989; Fowle & Sexton, 1992; Hemminki, 1992;
US NRC, 1992).
Over the last decade, biomarker methods have been developed for
the detection of exposure to carcinogens and other DNA-damaging
agents. These methods involve the detection of the parent compound or
metabolites in body fluids or adducts bound to DNA or protein, such as
haemoglobin and albumin (Shuker, 1989; Wogan, 1989, 1992; Beland &
Poirier, 1993). Methods for detecting exposure to DNA-damaging agents
are classifiable into two categories: a) measurements of levels of
genotoxic chemicals, their metabolites and/or derivatives in cells,
tissues, body fluids or excreta; and b) measurements of biological
responses such as cytogenetic changes in exposed individuals.
Biomarker methods have also been developed to detect exposure
from tobacco use (polycyclic aromatic hydrocarbons (PAHs), aromatic
amines and specific nitrosamines), dietary exposure (aflatoxins,
N-nitrosamines, heterocyclic amines), medicinal exposure (cisplatin,
alkylating agents, 8-methoxypsoralen, ultraviolet photoproducts),
occupational exposure (benzene, ethylene oxide, styrene oxide, vinyl
chloride, aromatic amines, PAHs) and oxidative damage
(8-hydroxyguanine) (Perera, 1987, 1988; Groopman et al., 1988; Wogan,
1989, 1992; Hemminki et al., 1990; Skipper & Tannenbaum, 1990; Beland
& Poirier, 1993).
The drawbacks of the reconstructive method are that it will not
work for every chemical, due to interferences or the reactive nature
of the chemical, it has not been methodologically established for very
many chemicals, data relating internal dose to exposure are needed,
and it may be expensive.
5.4 Variability and uncertainty
Characterization of variability and uncertainty is an integral
component of all steps in risk assessment. However, quantitative
characterization of these aspects is best developed for exposure
estimation. Variability (the receipt of different levels of exposure
by different individuals) is generally distinguished from uncertainty
(the lack of knowledge about the correct value for a specific exposure
measure or estimate). Most of the exposure and risk descriptors deal
with variability directly, but, wherever possible, estimates of the
uncertainty of these descriptors are included. This may be done
qualitatively or quantitatively, and it is beyond the scope of this
report to discuss the mechanics of uncertainty analysis in detail.
Not all approaches historically used to construct measures or
estimates of exposure attempted to distinguish variability and
uncertainty. In particular, in many cases in which estimates were
termed worst case, focusing on the high end of the exposed population
and also selection of high-end values for uncertain physical
quantities resulted in values that were seen to be quite conservative.
By using both the high-end individuals (variability) and upper
confidence bounds on data or physical parameters (uncertainty), these
estimates might be interpreted as "not exceeding an upper bound on
exposures received by certain high-end individuals".
Variability in exposure occurs when some members of the
population are exposed more than others. For example, exposures via
one or more routes to some substances may be elevated for persons
living in the vicinity of point sources (such as industrial
emissions), depending on the form in which these substances are
released and their subsequent environmental transport and
transformation. The intake of some substances by subsistence hunters
or fishermen may also be elevated due to accumulation in the game
species that they consume. Owing to the variation in exposure patterns
at various stages over a lifetime, exposure is often estimated for
various age groups of the general population; for example, Health
Canada (1994) estimates intake for several defined periods of life:
for infants (0-6 months), pre-school children (7 months to 4 years),
elementary school children (5-11 years), teenagers (12-19 years), and
adults (20 years of age and older). Hence, the period up to 6 months
of age is when many infants may be exposed to substances present in
breast milk. In addition, pre-schoolers' exposure to contaminants in
soil may be significantly higher than that for other age groups.
Children of all ages have relatively high intakes of food per unit of
body weight. Adulthood is a period of long-term lower-level exposure
via most environmental media, with relatively high potential exposure
to some substances through activities such as the use of consumer
products. An example of age-stratified estimates of exposure is
presented in Table 1, showing fluoride exposure for five age groups in
the general population.
5.4.1 Assessing uncertainty
Assessing uncertainty may involve simple or very sophisticated
techniques, depending on the requirements of the assessment.
"Uncertainty characterization" generally involves a qualitative
discussion of the thought processes that lead to the selection and
rejection of specific data, estimates, scenarios, etc. For simple
exposure assessments, where not much quantitative information is
available, uncertainty characterization may be all that is necessary.
"Uncertainty assessment" is more quantitative and can include
simpler measures (i.e. ranges) and analytical techniques (i.e.
sensitivity analysis) or, to the extent needed to support the decision
for which the exposure assessment is conducted, more complex measures
and techniques.
Uncertainty in exposure assessment can be classified into three
broad categories:
1. Uncertainty regarding missing or incomplete information needed to
fully define the exposure and dose (scenario uncertainty).
2. Uncertainty regarding some parameter (parameter uncertainty).
3. Uncertainty regarding gaps in scientific theory required to make
predictions on the basis of causal inferences (model
uncertainty).
Identification of the sources of uncertainty in an exposure
assessment is the first step toward eventually determining the type of
action necessary to reduce that uncertainty.
5.5 Exposure settings
Human exposure occurs in the general environment, at occupational
settings or in households/businesses or other areas where consumer
products are used. Each of these settings is discussed below.
5.5.1 Exposure in the general environment
Exposure to environmental substances may occur by inhalation,
ingestion and/or dermal absorption from air, water, food and soil.
Estimation of the total daily intake (often expressed as µg/kg body
weight/day) from all sources is critical in assessing the true
magnitude of risk associated with indirect exposure to substances in
the general environment. This is often referred to as a "multimedia"
approach (Table 1).
The US EPA has sponsored the development of a computer software
programme called Risk Assistant for conducting site-specific risk
assessments for environmental chemicals. The programme prompts the
user to identify the chemicals of concern, the contaminated media and
concentrations in those media. The programme automatically lists the
possible pathways of exposure associated with the contaminated media.
The user can select which of these pathways is of interest. The user
can choose to use default assumptions for exposure parameters or
modify them as desired.
5.5.2 Occupational settings
Workers are exposed in the occupational environment by
inhalation, through dermal contact or by ingestion, although the
latter is not often quantified. Dermal and inhalation monitoring as
well as biological monitoring (biomarkers) are often required to
characterize adequately the exposure of special subgroups of workers
such as mixers, loaders and applicators or pesticides (e.g., farm
families) (WHO, 1986; US EPA, 1987b; Curry & Iyengar, 1992).
Exposure by inhalation in the occupational environment is often
expressed as the concentration of a substance in the breathing zone
averaged over a reference period. This reference period is often 8 h
to represent long-term exposure or 15 min for short-term exposure.
Exposure to the skin is generally expressed as potential dose rate
predominantly to the hands and forearms and is often available only as
output of models.
Measured data on concentrations of chemical substances in the
occupational environment are often available from routine industrial
hygiene or dedicated surveys. The suitability of the use of such
information in estimation of exposure must be carefully assessed based
on consideration of factors such as representation of levels, time
periods and processes.
Cumulative exposure (average intensity multiplied by time) is one
of the most common summary measures for exposure in epidemiological
studies of occupationally exposed populations. However, there may also
be intermittent peak exposures that could be of importance but
difficult to integrate properly in a single concentration-time
exposure model (Ulfvarson, 1992). The elimination rate of a pollutant
is of particular importance in considering the possible impact of peak
versus continuous exposure (Axelson & Westberg, 1992).
Where monitoring data are incomplete or not available,
occupational exposures can also be modelled (EC, 1996), primarily to
highlight areas in which specific additional data are required to
estimate exposure more accurately. To date, these models are
restricted primarily to prediction of mean concentrations over
extended averaging periods (e.g., 8 h). For example, for workplace
exposure modelling in the European Union, criteria to describe broadly
the types of exposure possible address the physical properties of
process chemicals, their use pattern and pattern of control.
Descriptors for the physical properties of process chemicals include,
for example, gas, liquid of high vapour pressure, liquid of medium
vapour pressure, solid respirable dust, solid, granular or aerosol.
Descriptors of use patterns include closed system, within a matrix or
wide dispersive. Descriptors of control patterns include full
containment, local exhaust ventilation, etc. Combinations of various
subsets of these descriptors result in 160 complementary fields to
which numerical ranges of concentrations have been assigned based on
measured data in the United Kingdom National Exposure Database.
Dermal exposure in occupational settings most commonly involves
hands and forearms (approximately 2000 cm2) (EC, 1996). Dermal
exposure to gases and vapours is typically assumed to be very low. The
EU classifies the potential for dermal exposure as none, incidental
(approximately one event per day), intermittent (2 to 10 events per
day) or extensive (>10 events per day). Exposure ranges are estimated
based on several databases and the published literature. Criteria for
both inhalation and dermal exposure are incorporated within a
knowledge-based electronic system (EC, 1996).
5.5.3 Consumer products
A consumer product is one which can be purchased from retail
outlets by members of the general public. People of any age, either
sex, and in any stage of health may be exposed to chemicals in these
products. Much of the discussion below is based on an EU document
providing guidance on assessing exposure to chemicals in consumer
products (EC, 1996).
Exposure to chemicals in consumer products is often considered as
single events, a series of repeated events or as continuous exposure
(e.g., concentrations in indoor air resulting from storage and use of
such products). Routes of exposure are dermal (e.g., cleaning agents,
cosmetics, shampoos), inhalation (e.g., hair spray, powdered
detergents) or by ingestion (e.g., food, drinks or swallowing of tooth
paste; see Table 1 for an example of the latter).
The assessment of the exposure to consumer products can be
conducted following an iterative procedure, which starts with an
initial "screening". This screening would identify if a substance is
used as or in consumer products where further consideration and
possibly quantification of exposure is necessary.
If a substance is used in more than one consumer product, or if
more than one mode of use is employed (e.g., painting and spraying),
or if the product could reasonably be expected to be used in other
ways (e.g., use of a washing machine detergent for washing by hand),
it may be necessary to assess exposure for each case. In addition, if
the substance is used in different consumer products or has different
modes of use, the exposure assessment could examine those uses for
which the highest exposure is expected to occur on a regular basis.
The cumulative exposure expected from the use of the same substance in
different products may also be considered.
To assess the exposure to substances present in consumer
products, information is needed on two sets of parameters: contact
parameters and concentration parameters. The contact parameters denote
where, how long and how often contact with the consumer occurs. The
concentration parameters are needed to estimate the concentration of a
substance in a medium that might come into contact with the body. This
is not necessarily equal to the concentration of the substance in the
product, because a product might be diluted, mixed, undergo
evaporation, etc., before the substance of interest actually reaches
the human body.
By combining the contact parameters with the concentration
estimates, exposure or dose can be estimated. As discussed in section
5.2, exposure and dose can be estimated in a variety of ways. Exposure
to contaminants in air is commonly estimated in concentration-time
units, as shown in equation 1. Exposure to ingested contaminants is
commonly estimated as a potential dose, as shown in equation 2. Dermal
exposures are commonly estimated as an internal dose.
For example, exposure to a component of a hair spray used twice a
day, could be based on assumptions that the weight of product used per
event is 5000 mg, the weight fraction of the chemical substance is 1%,
the inhaled fraction is 70%, the room volume is 2 m3, the volume
inhaled is 0.8m3, and the exposure time is 6 min (EC, 1996). Dermal
exposure to a component of a watch strap could be estimated taking
into consideration the area of contact, the thickness and density of
the material, the weight fraction of the chemical substance, period of
contact per day and fraction likely to migrate from strap to skin, and
fraction or rate that the chemical is absorbed into the body.
For a realistic assessment, the following data would ideally be
available:
a) Contact data
- frequency of product use
- duration of product use per event
- site of product use, including size of room
- air exchange rate
b) Concentration data
- weight fraction of substance in the product
- if available, concentration of substance in the products as
used, e.g., after dilution or evaporation has occurred
c) Product use
- physical form of product (aerosol, dry powder, large
crystals, liquid, gas, etc)
- amount of product used per event
- contact surface (if appropriate)
- intended use of product
The diversity of consumer products does not allow for a single
set of information sources, handbooks or databases to be consulted.
Rather, it is necessary to explore which information sources apply to
the substance of interest. Below, an overview is provided of possible
information sources that may be useful.
i) Product registers are available in some countries and may provide
information on whether the substance under consideration is
present in marketed consumer products.
ii) Specific information on use durations and contact frequencies for
consumer products is often lacking. An estimate of these
parameters can be derived from time budget data where available.
Time budgets comprise information on the behaviour of a
population during a day, week or year. Because time budgets may
vary geographically, it is useful to check if the national
statistical agencies have gathered such data on a regional basis.
iii) Information on actual product use by the consumer is not widely
available. The directions of the manufacturer provide information
on the recommended use, not on the way products may be handled
before or after actual use nor on reasonably foreseeable misuse.
Although information can be gained from Poison Control Centres
and case studies reported in the literature, such data generally
represent the more extreme misuses of the product and might not
be very informative about the normal range of uses.
iv) Information accompanying exposure assessment computer programmes
(see below) may also be useful sources of data.
v) Some countries require manufacturers of certain products (e.g.,
cosmetics, toys, pharmaceuticals, food contact materials,
pesticides) to provide data useful for estimating exposure.
Assessors should use these data, where available and appropriate,
when conducting the exposure assessment.
Measured data useful for exposure assessment may be available for
a number of substances (e.g., concentrations of solvents in room air
as a consequence of the application of consumer products containing a
solvent or of their migration from articles; concentration of polymer
softeners or other additives migrating from food contact materials,
children's toys or other articles).
The reliability and representativeness of the measured exposure
data may be evaluated considering:
* if they represent the whole group of consumers or a certain
subset;
* if they reflect all exposure scenarios of concern;
* if they describe the foreseeable use;
* if they reflect the complete range of reasonable exposure values
or only an isolated value in any part of this range.
The European Union (EC, 1996) has presented a variety of simple
algorithms that can be used to assess consumer exposure for a number
of common exposure scenarios. Many give an exposure value per event
(single use), but are readily adaptable to different situations. In
addition, the European Union (EC, 1996) has summarized a variety of
more complex computer models for assessing consumer exposure
(CONSEXPO, THERdbASE, US EPA household exposure models MCCEM and HOUSE
EXP: SCIES, DERMAL, FLUSH and AMEM).
6. RISK CHARACTERIZATION AND IMPLICATIONS FOR RISK MANAGEMENT
6.1 General considerations
The traditional goal of regulating risks is to protect and
improve public health and well-being. Since 1980, risk assessment has
increasingly formed the methodological basis in many countries,
particularly industrialized nations, for the regulation of chemicals
in the occupational and general environments.
Risk assessment, comprising the elements of hazard
identification, dose-response assessment, exposure assessment and risk
characterization, is now recognized as an essential tool by many
national, regional and international bodies, and it is also recognized
that it is a continuously evolving process which has changed
considerably in the last two decades (US NAS, 1983; Somers, 1987,1993;
UK HSE, 1989; Scala; 1991; Ballantyne et al., 1993; EC, 1996). It
should be recognized as a vital mechanism for the delivery of salient
information to decision-makers.
Risk characterization aims to provide a synthesis of estimates of
exposure levels and health risks; it also summarizes sources of
uncertainty in scientific data and provides the primary basis for
making risk management decisions. The results of a risk assessment (as
summarized in the characterization) are the basis of identification of
chemical exposures that pose no significant health threat and those
that present significant risks. Additionally, to the extent permitted
by available data, risk characterization indicates how risk varies
with exposure, to help risk managers evaluate a range of options. It
assists risk management officials and decision makers in allocating
scarce resources and money to the most important resolvable
uncertainties and reduction of risks. However, the results of risk
assessment, as summarized in the risk characterization, are but one
consideration in health and environmental decision-making.
The term "risk management" encompasses all of those activities
required to reach decisions on whether an associated risk requires
elimination or necessary reduction. Risk management strategies/or
options can be broadly classified as regulatory, non-regulatory,
economic, advisory or technological, which are not mutually exclusive.
Thus legislative mandates (statutory guidance), political
considerations, socioeconomic values, cost, technical feasibility,
populations at risk, duration and magnitude of risk, risk comparison,
and possible impact on trade between countries can generally be
considered as a broad panoply of elements that can be factored into
final policy or rule-making. Key decision factors such as the size of
the population, the resources, costs of meeting targets and the
scientific quality of risk assessment and subsequent managerial
decisions vary enormously from one decision context to another (Stern,
1986; Ricci & Cox, 1987; Somers, 1987, 1993; Environ, 1988;
Munro & Morrison, 1990; Merrill, 1991; Scala, 1991;
Presidential/Congressional Commission on Risk Assessment and Risk
Management, 1997a,b).
It is also recognized that risk management is a complex
multidisciplinary procedure that is seldom codified or uniform,
frequently unstructured, but which can respond to evolving input from
a wide variety of sources (Stern, 1986). Increasingly, risk perception
and risk communication are recognized as important elements that must
also be considered for the broadest possible public acceptance of
risk-management decisions (Krewski et al., 1987; Slovic, 1987, 1993;
Kraus & Slovic, 1988; Konheim, 1988; Cohrssen & Covello, 1989; US NRC,
1989; Pariza, 1992; ILSI/National Safety Council, 1993; Morgan, 1993;
Singer & Endreny, 1993; Sandman et al., 1993; Van Eijndhoven et al.,
1994).
6.2 Considerations in risk characterization
Definitions and guidance for risk characterization have been
published in US EPA (1996b), where it is defined as:
"a summary, integration, and evaluation of the major scientific
evidence, reasoning and conclusions of a risk assessment. It is a
concise description of the estimates of potential risk and the
strengths and weaknesses of those estimates."
Similarly, the European Union defines risk characterization as: "the
estimation of the incidence and severity of the adverse effects likely
to occur in a human population or environmental sphere due to actual
or predicted exposure to a substance, and may include risk estimation,
i.e. the quantification of that likelihood (Hertel, 1996) .
A risk characterization is the final step in risk assessment. It
is designed to support risk managers by providing, in plain language,
the essential scientific evidence and rationale about risk that they
need for decision-making. In risk characterization, estimates of the
risk to human health under relevant exposure scenarios are provided.
Thus, a risk characterization is an evaluation and integration of the
available scientific evidence used to estimate the nature, importance
and, where possible, the magnitude of human and/or environmental risk,
including attendant uncertainty, that can reasonably be estimated to
result from exposure to a particular environmental agent under
specific circumstances. It is important that risk characterizations be
clear, transparent and reasonable.
For the risk manager, a risk characterization answers the
question: What is the impact (in terms of potential occurrence of
adverse effects or increased risk) from exposure to the agent? Along
with the concise description of risk, a characterization addresses the
uncertainty in the underlying data and models. The characterization
provides a sense of the degree of confidence in the risk estimates and
a sense of where the supporting data lie on the continuum between
evidence that is based on humans, or is highly relevant to humans, and
evidence that is based on animals or in vitro experiments.
The following are sample questions of risk managers that are
commonly addressed in risk characterization:
1) What is the bottom line of the risk assessment?
2) Does the risk assessment provide sufficient information to
support a regulatory decision?
3) What is the range of uncertainty around the estimated exposure
level and the projected number of people who may be exposed to
the chemical? Do we know if people are actually being exposed to
the levels identified in the risk assessment? Are these levels of
public health concern?
4) What data gaps are likely to elicit criticism of the risk
estimate and/or selected risk management options? There will
always be data gaps, but which are the ones that may lead to
criticism of the risk assessment or of the risk management
options and decision(s)?
5) Are studies being conducted that will "soon" provide new
information that could fill a critical data gap or gaps?
6) Has the risk assessment been peer reviewed? If so, by whom, and
what was the outcome of the review?
7) Indicate how likely, or if, there is a chance of zero risk. Has
zero risk actually been ruled out?
8) What is the key parameter that drives the analysis? Is there
research on the horizon that will address this key parameter and
reduce its uncertainty? How much interest is there in issues
surrounding this parameter?
9) If studies were excluded, what would be the consequence for the
risk assessment results? What was the rationale for excluding
these studies?
Other questions primarily concern the issue of uncertainty. Data
lie on a continuum from strong evidence in humans (based on extensive
epidemiology and/or other clinical/field observations) to weak
evidence in humans, animals or other test systems (based on incomplete
data in one or a limited number of species, or structure-activity
relationships). Confidence in the conclusions of the risk assessment
and the estimate of risk also lie on a continuum from high to low.
This degree of confidence is based, to a large extent, on the
completeness, quality and consistency of the database (i.e. the weight
of evidence). Where do the results of the risk assessment fit on the
continuum from high to low confidence?
* What are the specific conditions of exposure believed to cause or
contribute to the risk? Have exposures and/or dose been measured
in the population of interest? If so, has it been possible to
relate exposure to actual body burden? If exposures have been
calculated through analogy, modelling, or other estimation
techniques, what evidence is there that the estimates are
realistic?
* What is the degree of confidence in the existence of the risk and
the magnitude of the risk estimate? If the risk is based on
animal models, is there an observable parallel between humans and
the positively responding animal species in terms of the
absorption, metabolism, distribution and excretion of the
chemical of interest? If not, what is the basis for thinking such
a parallel exists? Is there epidemiological evidence indicating
that comparable effects seen in the animal model have been seen
in human populations (e.g., heavily exposed occupational or
environmental settings, accidents)?
* Can population subgroups be identified who are at increased risk
of exposure and/or especially sensitive to such exposures? At a
given exposure or dose level, are there observable differences in
the range of response among different human subgroups (e.g.,
infants, children, healthy adults, the elderly)? If so, have
these differences been evaluated and employed in the models used
to calculate specific risks? If not, what evidence provides the
basis for conclusions drawn about differences in sensitivity
among subpopulations and their (potential) risks?
6.3 Considerations in risk management
Decisions concerning management of risks are made on the basis of
identified and quantified risk(s), and the potential for impact on
individual humans, groups, populations and the environment. This
involves consideration of socioeconomic, political, risk-benefit and
cost-benefit factors.
The analytical tools of risk assessment and management, as
applied to chemicals with a potential for adverse effects on human
health and environmental integrity, have assumed a more critical role
in decision-making in many countries and are having an increasing
impact on the political process. Potentially many jobs, new products
and industrial facilities can be created, threatened or protected by
the outcomes of risk assessment and management.
6.3.1 Societal factors
The actual level of risk considered "acceptable" must be a
societal and political judgement taking into account such factors as
benefit of the chemical or process, and the cost of its replacement or
removal.
There is increasing concern that a disproportionate share of
human health risks, e.g., from environmental pollution, is being
incurred by low-income deprived and minority populations in developed
and developing countries, and that this has not been sufficiently
addressed in requisite risk evaluations and managerial decisions
(Mushak, 1993; Silbergeld, 1993; Zimmerman, 1993). It is important to
recognize, however, that lifestyle factors are often more important in
determining health status in this regard. The term "environmental
equity" has been applied to the perceived unequal burdens borne by
minorities and the poor in terms of where municipal landfills,
incinerators, hazardous waste sites and industries producing toxic
emissions are located. Race and socioeconomic status are also linked
in some studies to chronic exposures to greater than acceptable levels
of environmental pollution such as lead (Mushak, 1993; Silbergeld,
1993). The term "environmental justice" refers to diverse
environmental regulations, environmental law enforcement and
environmental clean-up programmes, including those in the workplace.
Hence a growing body of scientific evidence and political advocacy is
focusing attention on what is increasingly considered in some quarters
as the inequitable distribution of risk in society. The concept of
environmental justice is being built into national and supranational
regulatory policy considerations. Requirements to conduct risk
management are increasingly being incorporated into national and
supranational legislation e.g., European Commission Regulation CEC No.
1488/94, (EC, 1994).
In contrast, it needs to be recognized that regulations that are
too stringent may impact unnecessarily adversely on the socioeconomic
and, hence, health status of populations.
6.3.2 Individual and population risks
Individual risk can be defined as the probability of someone from
a certain group (or sub-group) suffering health effects from exposure
to a toxicant during an established period (e.g., a year or lifetime).
The distinction made between individual risks for persons from a
critical group and that for persons from the whole population is
important because the acceptability of a certain individual risk
varies according to the size of the group running the risk. An
individual risk can be considered when effects are involved for which
no threshold value exists (stochastic effects), e.g., carcinogens, or
when exposures are involved that are higher than existing threshold
values for non-stochastic effects.
Frequently, individual risks are calculated for some or all of
the persons in the population being studied and are then put into the
context of where they fall in the distribution of risks for the entire
population. Key questions often asked when considering strategies for
dealing with individual risk include:
* to what risk levels are the persons at the highest risk
subjected?
* can individuals with a high degree of susceptibility be
identified?
* what is the average individual risk?
* what is the estimate of the probability that an individual will
suffer an adverse effect given a specific set of exposure
circumstances?
It has also been suggested that sub-groups of the population
could be considered in a meaningful risk management scenario. The
different factors predisposing individuals to sensitive responses to
pollutants include: developmental processes, existing disease, prior
exposure to a particular chemical, chemical class or group of
chemicals that can act mechanistically in a similar manner,
nutritional deficiencies, and tobacco smoking and alcohol consumption
(Seidman et al., 1991; US EPA, 1992).
Group or population risk (which generally is calculated) is
defined as the chance that a certain group of individuals in a certain
environment will simultaneously experience the detrimental
consequences of a significant exposure to a toxicant(s) during a
period, e.g., a year or lifetime.
A clear trend has not yet emerged concerning the question as to
whether risks to individuals, risks to groups or populations, or both,
are to be considered in significant risk decisions (Environ, 1988;
Rodricks, 1992; US EPA, 1992). For example, is a large risk to a small
number of individuals more important from a public health perspective
than a small risk to a large number of people (general public
ingesting a food or water contaminant for a considerable time period)?
A suggested first step following any risk evaluation could be a
determination of whether the risk is large enough to threaten the
public health to a significant degree (Environ, 1988). Resources are
limited and there will always be the possibility that some fraction of
the population will respond adversely to a compound or mixture
regardless of the exposure. The ultimate question could be (given the
limited resources in every society) what percentage of individuals is
society unable to protect in this way? Certain sub-groups, for example
idiosyncratic responders, may be given protection by appropriate
product labelling and information programmes.
6.3.3 Comparative risk
Risk implies uncertainty and subsequent risk evaluations and risk
management decisions are concerned with the concept of probability.
There is an apparent lack of consensus concerning the appropriate
background risk with which to make comparisons (Environ, 1988; US NRC,
1989). While many analysts would find it difficult to compare
voluntary assumed risks to involuntarily assumed risks, proponents of
risk comparisons strongly suggest that there should be consolidation
and greater efforts by those engaged in risk evaluation to identify,
assess and compare risks to public health and the environment posed by
the highest risk hazards (Wilson & Crouch, 1987; Wiener, 1993).
Comparisons should be seen as only one of a number of inputs to risk
decisions, not as a primary determinant (US NRC, 1989).
However, it is also suggested that many people do not perceive
the various threats to health and well-being simply as matters of
probability (Slovic, 1987; Kraus & Slovic, 1988; Pariza, 1992; Sandman
et al., 1993). Indeed, estimated risks of death or disease associated
with exposure to chemicals in the general environment are often
similar to those considered rare, such as being struck by lightening
or dying in an airplane crash, although they are not perceived as such
(Wilson, 1990). Moreover, people tend not to be deeply concerned about
risks that are a matter of choice such as smoking or motorcycle
riding. However, they do expect that governments pay attention to
risks that they cannot control, even though these might be
considerably less.
6.3.4 Risk perception
Whereas analysts employ risk assessment, risk evaluation and risk
management to evaluate hazards and formulate strategies and
regulations for their reduction or elimination, the majority of
individuals rely on intuitive judgements typically called "risk
perception". For these people, the experience with hazards tends to
come from the news media, which principally document mishaps and
threats occurring globally (Slovic, 1987, 1993; Kraus & Slovic, 1988;
Cohrssen & Covello, 1989; Sandman et al., 1993; Van Eijndhoven et al.,
1994).
Risk perception is being increasingly recognized as an important
factor influencing both risk evaluation and risk management. A major
factor that influences the complexity of the social debate over
appropriate laws and regulations is the nature and extent of the
perceived threat to health. The message that is frequently conveyed to
the public is that government standards for risk assessment, risk
evaluation and regulatory action are inconsistently applied, subject
to bureaucratic manipulation, and subject to alteration depending on
the degree of economic impact on the affected industry (Munro &
Morrison, 1990).
Different people perceive risks differently, depending on the
likelihood of adverse effects, whom it affects, how familiar,
widespread and dreaded the effects are, how a hazard affects
individuals personally, and whether or not individuals have
voluntarily agreed to bear the risks. Perceptions of risk are also
influenced to a large degree by the supposed benefits derived from
accepting the risk (Slovic, 1987; Krewski, et al., 1987; Kraus &
Slovic, 1988; Cohrssen & Covello, 1989; Pariza, 1992; Morgan, 1993;
Sandman et al., 1993).
Risks perceived as potentially uncontrollable, capable of causing
a catastrophe on a global scale or risking future generations cause
public anxiety. Fig. 3 illustrates a mosaic of public perception of
risks in terms of risk space quadrants; the upper right quadrant of
this space captures uncontrollable risks that are most likely to
provoke calls for government regulation (Morgan, 1993).
Tables 3 and 4 further depict qualitative factors affecting risk
perception (US NRC, 1989; Scala, 1991). While different people weigh
these factors differently in reaching their overall perceptions of the
riskiness of a hazard, the set of factors that are important in
determining relative perceptions of risk go well beyond the
statistical frequency, magnitude and uncertainty of effects. Public
opinion on acceptable risk constantly changes, usually in the
direction of further risk reduction, which provides further impetus
for additional legislation and regulation in many quarters (Munro &
Morrison, 1990).
6.3.5 Risk and hazard communication
Implicit in the process of risk evaluation and management is the
increasingly recognized role of communication (Cohrssen & Covello,
1989; US NRC, 1989; Morgan, 1993; Sandman et al., 1993; Slovic, 1993).
Risk communication is an interactive process of exchange of
information and opinion among individuals, groups and institutions
involving multiple messages about the nature of risk and other
messages, not strictly about risk, that express concerns, opinions or
reactions to risk messages or to legal and institutional arrangements
for risk management (US NRC, 1989).
Until the mid-1980s, there was little research on communicating
risk to the public. There is now a reasonable consensus on the optimum
basic elements of risk communication. These efforts should be more
systematically oriented to the intended audience, addressing the
audience's perspectives and concerns. To the greatest extent possible,
openness, not minimizing the existence of uncertainty, and discussion
of data gaps and areas of significant disagreement among experts is
recommended. The acceptance of any risk is more dependent on public
confidence in risk management than on quantitative estimates of risk.
Although there is as yet no widely agreed structured knowledge on
communication about chemical hazards, analyses of risk communication
efforts and case studies suggest that risk communication problems
arise from message, source, channel and receiver problems (Cohrssen &
Covello, 1989). Message problems relate primarily to deficiencies in
scientific understanding leading to large uncertainties in risk
estimates or highly technical risk analyses that are unintelligible to
lay persons. Source problems include disagreements among scientific
experts, failures to disclose limitations of risk assessments and
resulting uncertainties, and limited understanding of the concerns and
values of public groups and bureaucratic presentation. Channel
problems include selective and biased media reporting that emphasizes
5.3 Approaches to quantification of exposure
Exposure (or dose) is assessed generally by one of the following
approaches:
a) The exposure can be measured at the point of contact (the outer
boundary of the body) while it is taking place, measuring both
exposure concentration and time of contact and integrating them
(point-of-contact or personal measurement);
b) The exposure can be estimated by separately evaluating the
exposure concentration and the time of contact, then combining
this information (scenario evaluation);
c) The exposure can be estimated from dose, which in turn can be
reconstructed through internal indicators (biomarkers, body
burden, excretion levels, etc.) after the exposure has taken
place (reconstruction).
These three approaches to quantification of exposure (or dose)
are independent, as each is based on different data. This offers the
opportunity of checking the accuracy of exposure estimated by one
approach through use of an independent approach, where data permit.
The independence of the three methods is a useful concept in verifying
or validating results. Each of the three has strengths and weaknesses;
using them in combination can considerably strengthen the credibility
of an exposure or risk assessment.
5.3.1 Measurement at point of contact (personal monitoring)
Point-of-contact exposure measurement evaluates the exposure as
it occurs, by measuring the chemical concentrations at the interface
between the person and the environment as a function of time,
resulting in an exposure profile. The best known example of the
point-of-contact measurement is the radiation dosimeter. This small
badge-like device measures exposure to radiation as it occurs and
provides an integrated estimate of exposure for the period of time
over which the measurement has been taken. Another example is the
Total Exposure Assessment Methodology (TEAM) studies (US EPA, 1987a)
conducted by the EPA and similar multimedia exposure studies in Canada
(Otson et al., 1996). In the TEAM studies, a small pump with a
collector and absorbent was attached to a person's clothing to measure
his or her exposure to airborne solvents or other pollutants as it
occurred. A third example is the carbon monoxide (CO) point-of-contact
measurement studies where subjects carried a small CO measuring device
for several days (US EPA, 1984). Dermal patch studies and duplicate
meal studies are also point-of-contact measurement studies. In all of
these examples, the measurements are taken at the interface between
the person and the environment while exposure is occurring. Use of
these data for estimating exposures or doses for periods that differ
from those for which the data are collected (e.g., for estimates of
lifetime exposures) will require some assumptions.
The strength of this method is that it measures exposure
directly, and providing that the measurement devices are accurate, is
likely to give the most accurate exposure value for the period of time
over which the measurement was taken. It is often expensive, however,
and measurement devices and techniques do not currently exist for all
chemicals. This method may also require assumptions to be made
concerning the relationship between short-term sampling and long-term
exposures, if appropriate. This method is also not source-specific, a
limitation when particular sources will need to be addressed by risk
managers.
5.3.2 Scenario evaluation method (time activity and
monitoring/modelling)
In exposure scenario evaluation, the assessor attempts to
determine the concentrations of chemicals in a medium or location and
link this information with the time and ways that individuals or
populations come into contact with the chemical. The set of
assumptions about how this contact takes place is an exposure
scenario.
The first step in a scenario evaluation is usually to
characterize the contaminant concentration in the media of concern at
the point where contact occurs. This is typically accomplished
indirectly by measuring, modelling or using existing data on
concentrations in the bulk media, rather than at the true point of
contact. An example of a scenario evaluation is presented in Table 1.
Since the concentration in the bulk medium is not the same as the
exposure concentration, this is a clear source of potential error in
the exposure estimate. Generally, the closer the medium can be
measured to the point of contact (in both space and time), the less
uncertainty there is in the characterization of exposure
concentration. Where monitoring data are inadequate, fate models are
typically used to estimate chemical concentrations. These models can
span a wide range of complexity in terms of spatial dimensions and
temporal assumptions (i.e. steady-state versus non-steady-state).
Types of fate models include:
* simple dilution models where a measured concentration in an
effluent is divided by a dilution factor or the chemical release
rate is divided by the bulk flow rate of the medium;
* equilibrium models which predict the distribution of a chemical
in the environment based on partitioning ratios or fugacity (the
escaping tendency of a chemical from one environmental phase to
another);
* dispersion models which predict reductions in concentrations from
point sources based on assumed mathematical functions or
dispersion properties of the chemical;
* transport models which predict concentration changes over
distance and can represent dispersion, biochemical degradation
and absorption.
Compilations of existing environmental fate models have been
published (OECD, 1989, 1991a; Braat et al., 1991; ECETOC, 1992, 1993;
RIVM, 1994). The US EPA has produced a software system called the
Integrated Model Evaluation System (IMES) to help assessors select the
fate model best suited to their needs (US EPA, 1992). The software
prompts users to answer a variety of questions about their needs and
then lists the models that have matching features. The system has
information on over 150 models representing all media (air, surface
water and groundwater). Model information includes descriptions of the
model type, computer requirements, validation testing and contact for
obtaining a copy. The Netherlands National Environmental Policy Plan
Uniform System for the Evaluation of Substances (USES) is a
decision-support system for the rapid quantitative assessment of the
hazards and risks of chemicals, including new substances, agricultural
pesticides and biocides (RIVM, 1994). USES has been the basis for the
development of the European Union System for the Evaluation of
Substances (EUSES).
The reliability of modelled estimates of chemical concentration
in the general environment depends on how well the model assumptions
match reality (i.e. how realistic are the assumptions such as
steady-state conditions and homogenous media properties), whether the
model performance has been demonstrated under conditions similar to
those of concern; and the quantity and quality of input data.
Modelling efforts which use input values derived primarily on the
basis of default assumptions are generally most useful for screening
purposes to highlight areas in which specific additional data are
required to estimate exposure more accurately. Further discussion
about model uncertainty can be found below.
The next steps involve identifying who is exposed and developing
estimates of the frequency and duration of exposure. Like chemical
concentration characterization, this is usually done indirectly by use
of demographic data, survey statistics, behaviour observation,
activity diaries, activity models or, in the absence of more
substantive information, assumptions about behaviour. When estimating
potential dose, this step also involves estimating how much contact
occurs. Table 2 shows examples of standardized reference values for
body weights, fluid intake and respiratory volumes. This type of data
is also summarized in the Exposure Factors Handbook (US EPA, 1997).
This Handbook includes information on consumption rates for various
food types, fish ingestion, soil ingestion, dermal contact with soils,
body surface area, lifetime, body weight, inhalation rate, breast milk
ingestion rate, and activity patterns (time spent swimming, bathing
time, time indoors/outdoors, time in vehicles, etc.). For each factor,
descriptions are provided of the average values and the variability in
the general population. Values are recommended for each factor, with a
qualitative indication of the supporting weight of evidence.
Table 1. Estimated daily intake of inorganic fluoride (mg/kg body weight per day), according to age group, by the general
population of Canada (from Liteplo et al., 1994)
Route of exposure 0-6 monthsa 7 months-4 yearsb 5-11 yearsc 12-19 yearsd 20 + yearse
Ambient airf 0.01 0.01 0.01 0.01 0.01
Foodg 14-92 22 16 13 30
Breast milkh 0.5-1.1 - - - -
Soili 0.03-1.6 0.02-1.2 0.01-0.4 0.002-0.1 0.002-0.1
"Fluoridated" drinking-waterj - 45-77 24-42 17-29 16-27
"Non-fluoridated" drinking-waterk - 3.1-12.9 1.7-7.0 1.1-4.8 1.1-4.5
Household productsl - 20-60 8.2-20 2.5 1.1
Total intake of breast-fed infants 0.5-2.6 - - - -
Total intake of formula-fed infants 14-94 - - - -
Total intake ("Fluoridated" water)m - 87-160 49-79 33-45 47-58
Total intake ("Non-fluoridated" water)n - 45-96 26-44 17-21 32-36
a Assumed to weigh 7 kg, breathe 2 m3 air, drink 750 ml of breast milk or infant formula (as food),
and consume 35 mg soil per day.
b Assumed to weigh 13 kg, breathe 5 m3 air, drink 0.8 litres of water, and consume 50 mg soil per day.
c Assumed to weigh 27 kg, breathe 12 m3 air, drink 0.9 litres of water, and consume 35 mg soil per day.
d Assumed to weigh 57 kg, breathe 21 m3 air, drink 1.3 litres of water, and consume 20 mg soil per day.
e Assumed to weigh 70 kg, breathe 23 m3 air, drink 1.5 litres of water, and consume 20 mg soil per day.
f Based on the mean concentration of inorganic (gaseous and particulate) fluoride in ambient air of
0.03 µg/m3, reported for Toronto, Ontario, and assuming the concentration in indoor air is identical
to (outdoor) ambient air.
Table 1 (Continued)
g Formula-fed infants (0-6 months): based on the mean concentrations of inorganic fluoride in infant
formulas purchased in the USA of 0.127 and 0.854 mg/litre reported for ready-to-use, milk-based formula
and soy-based powdered formula (prepared with drinking-water containing 1 ppm fluoride), respectively,
and assuming infants are exclusively formula-fed and consume 750 ml formula per day. General population
(7 months and older): based on levels of inorganic fluoride detected in 109 individual foods from Canada
(and the USA), in the following food groups: 0.01-0.80 µg/g in dairy products, 0.12-1.02 µg/g in cereal
products, 0.01-0.58 µg/g in fruit, 0.01-0.68 µg/g in vegetables, 0.04-4.57 µg/g in meat/fish/eggs;
0.05-0.13 µg/g in fats, 0.11-0.35 µg/g in nuts/legumes, 0.02-0.86 µg/g in foods containing primarily sugar,
0.41-0.84 µg/g in soup, 4.97 µg/g in tea; and the daily intake of each food item by the various age groups
of the general population of Canada.
h Based on the mean concentrations of inorganic fluoride of 4.4 and 9.8 ng/g reported for samples of
breast milk from mothers living in communities served by "non-fluoridated" and "fluoridated" drinking-water,
respectively, assuming the density of breast milk is equal to 1.0 g/ml.
i Based on a range of concentrations of total inorganic fluoride of 6 µg/g reported by Sidhu (1982) for
soil collected in Newfoundland, to 309 µg/g [mean concentration in Canadian surface soil (0-130 cm depth)].
j Based on a range of mean concentrations of inorganic fluoride in "fluoridated" drinking-water of 0.73 mg/litre,
determined from fluoride levels in 3 communities in Newfoundland and Labrador, to 1.25 mg/litre, determined
from 2 communities in the Yukon. "Fluoridated" refers to drinking-water to which inorganic fluoride has been
intentionally added for the prevention of dental caries.
k Based on a range of mean concentrations of inorganic fluoride in "non-fluoridated" drinking-water of (at least)
0.05 mg/litre (reported for 3 communities in British Columbia), to 0.21 mg/litre (reported for an unspecified
number of communities in the Yukon). "Non-fluoridated" refers to drinking-water to which inorganic fluoride
has not been intentionally added for the prevention of dental caries.
l Based on a mean concentration of inorganic fluoride in most dentifrice products of 1000 µg/g and an estimated
intake of dentifrice of 0.26-0.78 g/day for children 7 months to 4 years of age, 0.22-0.54 g/day for children
5 to 11 years of age, 0.14 g/day for adolescents 12 to 19 years of age, and 0.08 g/day for adults 20 + years
of age, assuming an average of 2 brushings per day.
m Estimated total daily intake of inorganic fluoride by individuals consuming "fluoridated" drinking-water in Canada.
n Estimated total daily intake of inorganic fluoride by individuals in Canada consuming drinking-water
that is not "fluoridated".
Table 2. Human contact parameters (from ICRP, 1974)
Body weight, kg
Adult male = 70
Adult female = 58
Average = 64a
Daily fluid intake (milk, tap water, other beverages), ml/day
Normal conditions:
Adults = 1000-2400, representative figure = 1900b
Adult male = 1950
Adult female = 1400
Child (10 years) = 1400
High average temperature (32 °C):
Adults = 2840-3410
moderate activity:
Adults = 3700
Respiratory volumes
8-h respiratory volumes, litres per 8 h resting:
Adult man = 3600
Adult woman = 2900
Child (10 years) = 2300
light/non-occupational activity: Adult man = 9600
Adult woman = 9100
Child (10 years) = 6240
Daily inhalation volume, m3 (8 h resting, 16 h light/non-occupational activity)
Adult male = 23
Adult female = 21
Average adult = 22
Child (10 years) = 15
a WHO uses 60 kg for calculation of acceptable daily intakes
and water quality guidelines (IPCS, 1987b; WHO, 1993).
b WHO uses a daily per capita drinking-water consumption
of 2 litres in calculating water quality guidelines (WHO, 1993).
The chemical concentration and population characterizations are
ultimately combined in an exposure scenario, and there are various
ways to accomplish this. One of the major problems with this approach
is that the limiting assumptions or boundary conditions (e.g.,
steady-state assumptions) do not always hold true. Two ways to address
to this aspect are: (a) to evaluate the exposure or dose equation
under conditions where the limiting assumptions do hold true; or (b)
to deal with the uncertainty caused by the divergence from the
boundary conditions. As an example of the first option, in the
microenvironment method, utilized primarily for evaluating airborne
exposures in the general environment but including contact with the
skin in the occupational environment, segments of time and location
are evaluated where the assumption of constant concentration is
approximately true and then summed over all such time segments for a
total exposure for the respiratory route, effectively removing some of
the boundary conditions. While estimates of exposure concentration and
time-of-contact are still derived indirectly by this method, the
concentration and time-of-contact estimates can be measured for each
microenvironment. This avoids much of the error due to using average
values in cases where concentration varies widely along with
time-of-contact.
As examples of the second approach, there are various tools used
to describe uncertainty caused by parameter variation, such as Monte
Carlo analysis (see below).
One strength of the scenario evaluation approach is that it is
usually the least expensive method of the three. In addition, it is
particularly suited to analysis of the risk consequences of proposed
actions. It is both a strength and a weakness of scenario development
that the evaluation can be performed with little or no data; it is a
technique that is best used when some knowledge exists about the
soundness, validity and uncertainty of the underlying assumptions.
5.3.3 Biomarkers of exposure/estimation of internal dose
Exposure can also be estimated after it has taken place. If a
total dose is known, or can be reconstructed, and information about
intake and uptake rates is available, an average past exposure rate
can be estimated. Reconstruction of dose relies on measuring internal
body indicators after exposure, intake and uptake have already
occurred, and using these measurements to back-calculate dose.
However, the data on body burden levels or biomarkers cannot be used
directly unless a relationship can be established between these levels
or biomarker indications and internal dose, and interfering reactions
(e.g., metabolism of unrelated chemicals) can be accounted for or
ruled out. Biological tissue or fluid measurements that reveal the
presence of a chemical may indicate directly that an exposure has
occurred, provided the chemical is not a metabolite of other
chemicals. These biomarkers of exposure are necessarily limited,
however, to ethical relatively non-invasive techniques.
Biological monitoring can be used to evaluate the amount of a
chemical in the body by measuring one or more of the following items
(not all of these can be measured for every chemical):
* the concentration of the chemical itself in biological tissues or
sera (blood, urine, breath, hair, adipose tissue, etc.);
* the concentration of the chemical's metabolite(s);
* the biological effect that occurs as a result of human exposure
to the chemical (e.g., alkylated haemoglobin or changes in enzyme
induction);
* the amount of a chemical or its metabolites bound to target
molecules.
Biomarkers can be used to estimate chemical uptake during a
specific interval if background levels do not mask the marker and the
relationships between uptake and the marker selected are known. The
time of sampling for biomarkers can be critical. Establishing a
correlation between exposure and the measurement of the marker,
including pharmacokinetics, can help optimize the sampling conditions.
The strengths of this method are that it demonstrates that
exposure to and absorption of the chemical has actually taken place,
and it theoretically can give a good indication of past exposure.
Biomarkers integrate exposure from all sources and take into account
absorption, which may vary considerably due to a variety of factors
including environmental characteristics, genetic predisposition, age,
gender, ethnicity and/or lifestyle factors.
For many environmental pollutants, the flow of events between
exposure and health effects is not well understood. Biomarkers help
address this problem by improving the sensitivity, specificity and
predictive value of detection and quantification of adverse effects at
low dose and early exposure (ECETOC, 1989; Fowle, 1989; Fowle &
Sexton, 1992; US NRC, 1992). Sensitive subpopulations can be better
pinpointed by biomarkers that measure increased absorption rate or a
more severe biological response to a given environmental exposure
(Lauwerys, 1984; ECETOC, 1989; Fowle & Sexton, 1992; Hemminki, 1992;
US NRC, 1992).
Over the last decade, biomarker methods have been developed for
the detection of exposure to carcinogens and other DNA-damaging
agents. These methods involve the detection of the parent compound or
metabolites in body fluids or adducts bound to DNA or protein, such as
haemoglobin and albumin (Shuker, 1989; Wogan, 1989, 1992; Beland &
Poirier, 1993). Methods for detecting exposure to DNA-damaging agents
are classifiable into two categories: a) measurements of levels of
genotoxic chemicals, their metabolites and/or derivatives in cells,
tissues, body fluids or excreta; and b) measurements of biological
responses such as cytogenetic changes in exposed individuals.
Biomarker methods have also been developed to detect exposure
from tobacco use (polycyclic aromatic hydrocarbons (PAHs), aromatic
amines and specific nitrosamines), dietary exposure (aflatoxins,
N-nitrosamines, heterocyclic amines), medicinal exposure (cisplatin,
alkylating agents, 8-methoxypsoralen, ultraviolet photoproducts),
occupational exposure (benzene, ethylene oxide, styrene oxide, vinyl
chloride, aromatic amines, PAHs) and oxidative damage
(8-hydroxyguanine) (Perera, 1987, 1988; Groopman et al., 1988; Wogan,
1989, 1992; Hemminki et al., 1990; Skipper & Tannenbaum, 1990; Beland
& Poirier, 1993).
The drawbacks of the reconstructive method are that it will not
work for every chemical, due to interferences or the reactive nature
of the chemical, it has not been methodologically established for very
many chemicals, data relating internal dose to exposure are needed,
and it may be expensive.
5.4 Variability and uncertainty
Characterization of variability and uncertainty is an integral
component of all steps in risk assessment. However, quantitative
characterization of these aspects is best developed for exposure
estimation. Variability (the receipt of different levels of exposure
by different individuals) is generally distinguished from uncertainty
(the lack of knowledge about the correct value for a specific exposure
measure or estimate). Most of the exposure and risk descriptors deal
with variability directly, but, wherever possible, estimates of the
uncertainty of these descriptors are included. This may be done
qualitatively or quantitatively, and it is beyond the scope of this
report to discuss the mechanics of uncertainty analysis in detail.
Not all approaches historically used to construct measures or
estimates of exposure attempted to distinguish variability and
uncertainty. In particular, in many cases in which estimates were
termed worst case, focusing on the high end of the exposed population
and also selection of high-end values for uncertain physical
quantities resulted in values that were seen to be quite conservative.
By using both the high-end individuals (variability) and upper
confidence bounds on data or physical parameters (uncertainty), these
estimates might be interpreted as "not exceeding an upper bound on
exposures received by certain high-end individuals".
Variability in exposure occurs when some members of the
population are exposed more than others. For example, exposures via
one or more routes to some substances may be elevated for persons
living in the vicinity of point sources (such as industrial
emissions), depending on the form in which these substances are
released and their subsequent environmental transport and
transformation. The intake of some substances by subsistence hunters
or fishermen may also be elevated due to accumulation in the game
species that they consume. Owing to the variation in exposure patterns
at various stages over a lifetime, exposure is often estimated for
various age groups of the general population; for example, Health
Canada (1994) estimates intake for several defined periods of life:
for infants (0-6 months), pre-school children (7 months to 4 years),
elementary school children (5-11 years), teenagers (12-19 years), and
adults (20 years of age and older). Hence, the period up to 6 months
of age is when many infants may be exposed to substances present in
breast milk. In addition, pre-schoolers' exposure to contaminants in
soil may be significantly higher than that for other age groups.
Children of all ages have relatively high intakes of food per unit of
body weight. Adulthood is a period of long-term lower-level exposure
via most environmental media, with relatively high potential exposure
to some substances through activities such as the use of consumer
products. An example of age-stratified estimates of exposure is
presented in Table 1, showing fluoride exposure for five age groups in
the general population.
5.4.1 Assessing uncertainty
Assessing uncertainty may involve simple or very sophisticated
techniques, depending on the requirements of the assessment.
"Uncertainty characterization" generally involves a qualitative
discussion of the thought processes that lead to the selection and
rejection of specific data, estimates, scenarios, etc. For simple
exposure assessments, where not much quantitative information is
available, uncertainty characterization may be all that is necessary.
"Uncertainty assessment" is more quantitative and can include
simpler measures (i.e. ranges) and analytical techniques (i.e.
sensitivity analysis) or, to the extent needed to support the decision
for which the exposure assessment is conducted, more complex measures
and techniques.
Uncertainty in exposure assessment can be classified into three
broad categories:
1. Uncertainty regarding missing or incomplete information needed to
fully define the exposure and dose (scenario uncertainty).
2. Uncertainty regarding some parameter (parameter uncertainty).
3. Uncertainty regarding gaps in scientific theory required to make
predictions on the basis of causal inferences (model
uncertainty).
Identification of the sources of uncertainty in an exposure
assessment is the first step toward eventually determining the type of
action necessary to reduce that uncertainty.
5.5 Exposure settings
Human exposure occurs in the general environment, at occupational
settings or in households/businesses or other areas where consumer
products are used. Each of these settings is discussed below.
5.5.1 Exposure in the general environment
Exposure to environmental substances may occur by inhalation,
ingestion and/or dermal absorption from air, water, food and soil.
Estimation of the total daily intake (often expressed as µg/kg body
weight/day) from all sources is critical in assessing the true
magnitude of risk associated with indirect exposure to substances in
the general environment. This is often referred to as a "multimedia"
approach (Table 1).
The US EPA has sponsored the development of a computer software
programme called Risk Assistant for conducting site-specific risk
assessments for environmental chemicals. The programme prompts the
user to identify the chemicals of concern, the contaminated media and
concentrations in those media. The programme automatically lists the
possible pathways of exposure associated with the contaminated media.
The user can select which of these pathways is of interest. The user
can choose to use default assumptions for exposure parameters or
modify them as desired.
5.5.2 Occupational settings
Workers are exposed in the occupational environment by
inhalation, through dermal contact or by ingestion, although the
latter is not often quantified. Dermal and inhalation monitoring as
well as biological monitoring (biomarkers) are often required to
characterize adequately the exposure of special subgroups of workers
such as mixers, loaders and applicators or pesticides (e.g., farm
families) (WHO, 1986; US EPA, 1987b; Curry & Iyengar, 1992).
Exposure by inhalation in the occupational environment is often
expressed as the concentration of a substance in the breathing zone
averaged over a reference period. This reference period is often 8 h
to represent long-term exposure or 15 min for short-term exposure.
Exposure to the skin is generally expressed as potential dose rate
predominantly to the hands and forearms and is often available only as
output of models.
Measured data on concentrations of chemical substances in the
occupational environment are often available from routine industrial
hygiene or dedicated surveys. The suitability of the use of such
information in estimation of exposure must be carefully assessed based
on consideration of factors such as representation of levels, time
periods and processes.
Cumulative exposure (average intensity multiplied by time) is one
of the most common summary measures for exposure in epidemiological
studies of occupationally exposed populations. However, there may also
be intermittent peak exposures that could be of importance but
difficult to integrate properly in a single concentration-time
exposure model (Ulfvarson, 1992). The elimination rate of a pollutant
is of particular importance in considering the possible impact of peak
versus continuous exposure (Axelson & Westberg, 1992).
Where monitoring data are incomplete or not available,
occupational exposures can also be modelled (EC, 1996), primarily to
highlight areas in which specific additional data are required to
estimate exposure more accurately. To date, these models are
restricted primarily to prediction of mean concentrations over
extended averaging periods (e.g., 8 h). For example, for workplace
exposure modelling in the European Union, criteria to describe broadly
the types of exposure possible address the physical properties of
process chemicals, their use pattern and pattern of control.
Descriptors for the physical properties of process chemicals include,
for example, gas, liquid of high vapour pressure, liquid of medium
vapour pressure, solid respirable dust, solid, granular or aerosol.
Descriptors of use patterns include closed system, within a matrix or
wide dispersive. Descriptors of control patterns include full
containment, local exhaust ventilation, etc. Combinations of various
subsets of these descriptors result in 160 complementary fields to
which numerical ranges of concentrations have been assigned based on
measured data in the United Kingdom National Exposure Database.
Dermal exposure in occupational settings most commonly involves
hands and forearms (approximately 2000 cm2) (EC, 1996). Dermal
exposure to gases and vapours is typically assumed to be very low. The
EU classifies the potential for dermal exposure as none, incidental
(approximately one event per day), intermittent (2 to 10 events per
day) or extensive (>10 events per day). Exposure ranges are estimated
based on several databases and the published literature. Criteria for
both inhalation and dermal exposure are incorporated within a
knowledge-based electronic system (EC, 1996).
5.5.3 Consumer products
A consumer product is one which can be purchased from retail
outlets by members of the general public. People of any age, either
sex, and in any stage of health may be exposed to chemicals in these
products. Much of the discussion below is based on an EU document
providing guidance on assessing exposure to chemicals in consumer
products (EC, 1996).
Exposure to chemicals in consumer products is often considered as
single events, a series of repeated events or as continuous exposure
(e.g., concentrations in indoor air resulting from storage and use of
such products). Routes of exposure are dermal (e.g., cleaning agents,
cosmetics, shampoos), inhalation (e.g., hair spray, powdered
detergents) or by ingestion (e.g., food, drinks or swallowing of tooth
paste; see Table 1 for an example of the latter).
The assessment of the exposure to consumer products can be
conducted following an iterative procedure, which starts with an
initial "screening". This screening would identify if a substance is
used as or in consumer products where further consideration and
possibly quantification of exposure is necessary.
If a substance is used in more than one consumer product, or if
more than one mode of use is employed (e.g., painting and spraying),
or if the product could reasonably be expected to be used in other
ways (e.g., use of a washing machine detergent for washing by hand),
it may be necessary to assess exposure for each case. In addition, if
the substance is used in different consumer products or has different
modes of use, the exposure assessment could examine those uses for
which the highest exposure is expected to occur on a regular basis.
The cumulative exposure expected from the use of the same substance in
different products may also be considered.
To assess the exposure to substances present in consumer
products, information is needed on two sets of parameters: contact
parameters and concentration parameters. The contact parameters denote
where, how long and how often contact with the consumer occurs. The
concentration parameters are needed to estimate the concentration of a
substance in a medium that might come into contact with the body. This
is not necessarily equal to the concentration of the substance in the
product, because a product might be diluted, mixed, undergo
evaporation, etc., before the substance of interest actually reaches
the human body.
By combining the contact parameters with the concentration
estimates, exposure or dose can be estimated. As discussed in section
5.2, exposure and dose can be estimated in a variety of ways. Exposure
to contaminants in air is commonly estimated in concentration-time
units, as shown in equation 1. Exposure to ingested contaminants is
commonly estimated as a potential dose, as shown in equation 2. Dermal
exposures are commonly estimated as an internal dose.
For example, exposure to a component of a hair spray used twice a
day, could be based on assumptions that the weight of product used per
event is 5000 mg, the weight fraction of the chemical substance is 1%,
the inhaled fraction is 70%, the room volume is 2 m3, the volume
inhaled is 0.8m3, and the exposure time is 6 min (EC, 1996). Dermal
exposure to a component of a watch strap could be estimated taking
into consideration the area of contact, the thickness and density of
the material, the weight fraction of the chemical substance, period of
contact per day and fraction likely to migrate from strap to skin, and
fraction or rate that the chemical is absorbed into the body.
For a realistic assessment, the following data would ideally be
available:
a) Contact data
- frequency of product use
- duration of product use per event
- site of product use, including size of room
- air exchange rate
b) Concentration data
- weight fraction of substance in the product
- if available, concentration of substance in the products as
used, e.g., after dilution or evaporation has occurred
c) Product use
- physical form of product (aerosol, dry powder, large
crystals, liquid, gas, etc)
- amount of product used per event
- contact surface (if appropriate)
- intended use of product
The diversity of consumer products does not allow for a single
set of information sources, handbooks or databases to be consulted.
Rather, it is necessary to explore which information sources apply to
the substance of interest. Below, an overview is provided of possible
information sources that may be useful.
i) Product registers are available in some countries and may provide
information on whether the substance under consideration is
present in marketed consumer products.
ii) Specific information on use durations and contact frequencies for
consumer products is often lacking. An estimate of these
parameters can be derived from time budget data where available.
Time budgets comprise information on the behaviour of a
population during a day, week or year. Because time budgets may
vary geographically, it is useful to check if the national
statistical agencies have gathered such data on a regional basis.
iii) Information on actual product use by the consumer is not widely
available. The directions of the manufacturer provide information
on the recommended use, not on the way products may be handled
before or after actual use nor on reasonably foreseeable misuse.
Although information can be gained from Poison Control Centres
and case studies reported in the literature, such data generally
represent the more extreme misuses of the product and might not
be very informative about the normal range of uses.
iv) Information accompanying exposure assessment computer programmes
(see below) may also be useful sources of data.
v) Some countries require manufacturers of certain products (e.g.,
cosmetics, toys, pharmaceuticals, food contact materials,
pesticides) to provide data useful for estimating exposure.
Assessors should use these data, where available and appropriate,
when conducting the exposure assessment.
Measured data useful for exposure assessment may be available for
a number of substances (e.g., concentrations of solvents in room air
as a consequence of the application of consumer products containing a
solvent or of their migration from articles; concentration of polymer
softeners or other additives migrating from food contact materials,
children's toys or other articles).
The reliability and representativeness of the measured exposure
data may be evaluated considering:
* if they represent the whole group of consumers or a certain
subset;
* if they reflect all exposure scenarios of concern;
* if they describe the foreseeable use;
* if they reflect the complete range of reasonable exposure values
or only an isolated value in any part of this range.
The European Union (EC, 1996) has presented a variety of simple
algorithms that can be used to assess consumer exposure for a number
of common exposure scenarios. Many give an exposure value per event
(single use), but are readily adaptable to different situations. In
addition, the European Union (EC, 1996) has summarized a variety of
more complex computer models for assessing consumer exposure
(CONSEXPO, THERdbASE, US EPA household exposure models MCCEM and HOUSE
EXP: SCIES, DERMAL, FLUSH and AMEM).
6. RISK CHARACTERIZATION AND IMPLICATIONS FOR RISK MANAGEMENT
6.1 General considerations
The traditional goal of regulating risks is to protect and
improve public health and well-being. Since 1980, risk assessment has
increasingly formed the methodological basis in many countries,
particularly industrialized nations, for the regulation of chemicals
in the occupational and general environments.
Risk assessment, comprising the elements of hazard
identification, dose-response assessment, exposure assessment and risk
characterization, is now recognized as an essential tool by many
national, regional and international bodies, and it is also recognized
that it is a continuously evolving process which has changed
considerably in the last two decades (US NAS, 1983; Somers, 1987,1993;
UK HSE, 1989; Scala; 1991; Ballantyne et al., 1993; EC, 1996). It
should be recognized as a vital mechanism for the delivery of salient
information to decision-makers.
Risk characterization aims to provide a synthesis of estimates of
exposure levels and health risks; it also summarizes sources of
uncertainty in scientific data and provides the primary basis for
making risk management decisions. The results of a risk assessment (as
summarized in the characterization) are the basis of identification of
chemical exposures that pose no significant health threat and those
that present significant risks. Additionally, to the extent permitted
by available data, risk characterization indicates how risk varies
with exposure, to help risk managers evaluate a range of options. It
assists risk management officials and decision makers in allocating
scarce resources and money to the most important resolvable
uncertainties and reduction of risks. However, the results of risk
assessment, as summarized in the risk characterization, are but one
consideration in health and environmental decision-making.
The term "risk management" encompasses all of those activities
required to reach decisions on whether an associated risk requires
elimination or necessary reduction. Risk management strategies/or
options can be broadly classified as regulatory, non-regulatory,
economic, advisory or technological, which are not mutually exclusive.
Thus legislative mandates (statutory guidance), political
considerations, socioeconomic values, cost, technical feasibility,
populations at risk, duration and magnitude of risk, risk comparison,
and possible impact on trade between countries can generally be
considered as a broad panoply of elements that can be factored into
final policy or rule-making. Key decision factors such as the size of
the population, the resources, costs of meeting targets and the
scientific quality of risk assessment and subsequent managerial
decisions vary enormously from one decision context to another (Stern,
1986; Ricci & Cox, 1987; Somers, 1987, 1993; Environ, 1988;
Munro & Morrison, 1990; Merrill, 1991; Scala, 1991;
Presidential/Congressional Commission on Risk Assessment and Risk
Management, 1997a,b).
It is also recognized that risk management is a complex
multidisciplinary procedure that is seldom codified or uniform,
frequently unstructured, but which can respond to evolving input from
a wide variety of sources (Stern, 1986). Increasingly, risk perception
and risk communication are recognized as important elements that must
also be considered for the broadest possible public acceptance of
risk-management decisions (Krewski et al., 1987; Slovic, 1987, 1993;
Kraus & Slovic, 1988; Konheim, 1988; Cohrssen & Covello, 1989; US NRC,
1989; Pariza, 1992; ILSI/National Safety Council, 1993; Morgan, 1993;
Singer & Endreny, 1993; Sandman et al., 1993; Van Eijndhoven et al.,
1994).
6.2 Considerations in risk characterization
Definitions and guidance for risk characterization have been
published in US EPA (1996b), where it is defined as:
"a summary, integration, and evaluation of the major scientific
evidence, reasoning and conclusions of a risk assessment. It is a
concise description of the estimates of potential risk and the
strengths and weaknesses of those estimates."
Similarly, the European Union defines risk characterization as: "the
estimation of the incidence and severity of the adverse effects likely
to occur in a human population or environmental sphere due to actual
or predicted exposure to a substance, and may include risk estimation,
i.e. the quantification of that likelihood (Hertel, 1996) .
A risk characterization is the final step in risk assessment. It
is designed to support risk managers by providing, in plain language,
the essential scientific evidence and rationale about risk that they
need for decision-making. In risk characterization, estimates of the
risk to human health under relevant exposure scenarios are provided.
Thus, a risk characterization is an evaluation and integration of the
available scientific evidence used to estimate the nature, importance
and, where possible, the magnitude of human and/or environmental risk,
including attendant uncertainty, that can reasonably be estimated to
result from exposure to a particular environmental agent under
specific circumstances. It is important that risk characterizations be
clear, transparent and reasonable.
For the risk manager, a risk characterization answers the
question: What is the impact (in terms of potential occurrence of
adverse effects or increased risk) from exposure to the agent? Along
with the concise description of risk, a characterization addresses the
uncertainty in the underlying data and models. The characterization
provides a sense of the degree of confidence in the risk estimates and
a sense of where the supporting data lie on the continuum between
evidence that is based on humans, or is highly relevant to humans, and
evidence that is based on animals or in vitro experiments.
The following are sample questions of risk managers that are
commonly addressed in risk characterization:
1) What is the bottom line of the risk assessment?
2) Does the risk assessment provide sufficient information to
support a regulatory decision?
3) What is the range of uncertainty around the estimated exposure
level and the projected number of people who may be exposed to
the chemical? Do we know if people are actually being exposed to
the levels identified in the risk assessment? Are these levels of
public health concern?
4) What data gaps are likely to elicit criticism of the risk
estimate and/or selected risk management options? There will
always be data gaps, but which are the ones that may lead to
criticism of the risk assessment or of the risk management
options and decision(s)?
5) Are studies being conducted that will "soon" provide new
information that could fill a critical data gap or gaps?
6) Has the risk assessment been peer reviewed? If so, by whom, and
what was the outcome of the review?
7) Indicate how likely, or if, there is a chance of zero risk. Has
zero risk actually been ruled out?
8) What is the key parameter that drives the analysis? Is there
research on the horizon that will address this key parameter and
reduce its uncertainty? How much interest is there in issues
surrounding this parameter?
9) If studies were excluded, what would be the consequence for the
risk assessment results? What was the rationale for excluding
these studies?
Other questions primarily concern the issue of uncertainty. Data
lie on a continuum from strong evidence in humans (based on extensive
epidemiology and/or other clinical/field observations) to weak
evidence in humans, animals or other test systems (based on incomplete
data in one or a limited number of species, or structure-activity
relationships). Confidence in the conclusions of the risk assessment
and the estimate of risk also lie on a continuum from high to low.
This degree of confidence is based, to a large extent, on the
completeness, quality and consistency of the database (i.e. the weight
of evidence). Where do the results of the risk assessment fit on the
continuum from high to low confidence?
* What are the specific conditions of exposure believed to cause or
contribute to the risk? Have exposures and/or dose been measured
in the population of interest? If so, has it been possible to
relate exposure to actual body burden? If exposures have been
calculated through analogy, modelling, or other estimation
techniques, what evidence is there that the estimates are
realistic?
* What is the degree of confidence in the existence of the risk and
the magnitude of the risk estimate? If the risk is based on
animal models, is there an observable parallel between humans and
the positively responding animal species in terms of the
absorption, metabolism, distribution and excretion of the
chemical of interest? If not, what is the basis for thinking such
a parallel exists? Is there epidemiological evidence indicating
that comparable effects seen in the animal model have been seen
in human populations (e.g., heavily exposed occupational or
environmental settings, accidents)?
* Can population subgroups be identified who are at increased risk
of exposure and/or especially sensitive to such exposures? At a
given exposure or dose level, are there observable differences in
the range of response among different human subgroups (e.g.,
infants, children, healthy adults, the elderly)? If so, have
these differences been evaluated and employed in the models used
to calculate specific risks? If not, what evidence provides the
basis for conclusions drawn about differences in sensitivity
among subpopulations and their (potential) risks?
6.3 Considerations in risk management
Decisions concerning management of risks are made on the basis of
identified and quantified risk(s), and the potential for impact on
individual humans, groups, populations and the environment. This
involves consideration of socioeconomic, political, risk-benefit and
cost-benefit factors.
The analytical tools of risk assessment and management, as
applied to chemicals with a potential for adverse effects on human
health and environmental integrity, have assumed a more critical role
in decision-making in many countries and are having an increasing
impact on the political process. Potentially many jobs, new products
and industrial facilities can be created, threatened or protected by
the outcomes of risk assessment and management.
6.3.1 Societal factors
The actual level of risk considered "acceptable" must be a
societal and political judgement taking into account such factors as
benefit of the chemical or process, and the cost of its replacement or
removal.
There is increasing concern that a disproportionate share of
human health risks, e.g., from environmental pollution, is being
incurred by low-income deprived and minority populations in developed
and developing countries, and that this has not been sufficiently
addressed in requisite risk evaluations and managerial decisions
(Mushak, 1993; Silbergeld, 1993; Zimmerman, 1993). It is important to
recognize, however, that lifestyle factors are often more important in
determining health status in this regard. The term "environmental
equity" has been applied to the perceived unequal burdens borne by
minorities and the poor in terms of where municipal landfills,
incinerators, hazardous waste sites and industries producing toxic
emissions are located. Race and socioeconomic status are also linked
in some studies to chronic exposures to greater than acceptable levels
of environmental pollution such as lead (Mushak, 1993; Silbergeld,
1993). The term "environmental justice" refers to diverse
environmental regulations, environmental law enforcement and
environmental clean-up programmes, including those in the workplace.
Hence a growing body of scientific evidence and political advocacy is
focusing attention on what is increasingly considered in some quarters
as the inequitable distribution of risk in society. The concept of
environmental justice is being built into national and supranational
regulatory policy considerations. Requirements to conduct risk
management are increasingly being incorporated into national and
supranational legislation e.g., European Commission Regulation CEC No.
1488/94, (EC, 1994).
In contrast, it needs to be recognized that regulations that are
too stringent may impact unnecessarily adversely on the socioeconomic
and, hence, health status of populations.
6.3.2 Individual and population risks
Individual risk can be defined as the probability of someone from
a certain group (or sub-group) suffering health effects from exposure
to a toxicant during an established period (e.g., a year or lifetime).
The distinction made between individual risks for persons from a
critical group and that for persons from the whole population is
important because the acceptability of a certain individual risk
varies according to the size of the group running the risk. An
individual risk can be considered when effects are involved for which
no threshold value exists (stochastic effects), e.g., carcinogens, or
when exposures are involved that are higher than existing threshold
values for non-stochastic effects.
Frequently, individual risks are calculated for some or all of
the persons in the population being studied and are then put into the
context of where they fall in the distribution of risks for the entire
population. Key questions often asked when considering strategies for
dealing with individual risk include:
* to what risk levels are the persons at the highest risk
subjected?
* can individuals with a high degree of susceptibility be
identified?
* what is the average individual risk?
* what is the estimate of the probability that an individual will
suffer an adverse effect given a specific set of exposure
circumstances?
It has also been suggested that sub-groups of the population
could be considered in a meaningful risk management scenario. The
different factors predisposing individuals to sensitive responses to
pollutants include: developmental processes, existing disease, prior
exposure to a particular chemical, chemical class or group of
chemicals that can act mechanistically in a similar manner,
nutritional deficiencies, and tobacco smoking and alcohol consumption
(Seidman et al., 1991; US EPA, 1992).
Group or population risk (which generally is calculated) is
defined as the chance that a certain group of individuals in a certain
environment will simultaneously experience the detrimental
consequences of a significant exposure to a toxicant(s) during a
period, e.g., a year or lifetime.
A clear trend has not yet emerged concerning the question as to
whether risks to individuals, risks to groups or populations, or both,
are to be considered in significant risk decisions (Environ, 1988;
Rodricks, 1992; US EPA, 1992). For example, is a large risk to a small
number of individuals more important from a public health perspective
than a small risk to a large number of people (general public
ingesting a food or water contaminant for a considerable time period)?
A suggested first step following any risk evaluation could be a
determination of whether the risk is large enough to threaten the
public health to a significant degree (Environ, 1988). Resources are
limited and there will always be the possibility that some fraction of
the population will respond adversely to a compound or mixture
regardless of the exposure. The ultimate question could be (given the
limited resources in every society) what percentage of individuals is
society unable to protect in this way? Certain sub-groups, for example
idiosyncratic responders, may be given protection by appropriate
product labelling and information programmes.
6.3.3 Comparative risk
Risk implies uncertainty and subsequent risk evaluations and risk
management decisions are concerned with the concept of probability.
There is an apparent lack of consensus concerning the appropriate
background risk with which to make comparisons (Environ, 1988; US NRC,
1989). While many analysts would find it difficult to compare
voluntary assumed risks to involuntarily assumed risks, proponents of
risk comparisons strongly suggest that there should be consolidation
and greater efforts by those engaged in risk evaluation to identify,
assess and compare risks to public health and the environment posed by
the highest risk hazards (Wilson & Crouch, 1987; Wiener, 1993).
Comparisons should be seen as only one of a number of inputs to risk
decisions, not as a primary determinant (US NRC, 1989).
However, it is also suggested that many people do not perceive
the various threats to health and well-being simply as matters of
probability (Slovic, 1987; Kraus & Slovic, 1988; Pariza, 1992; Sandman
et al., 1993). Indeed, estimated risks of death or disease associated
with exposure to chemicals in the general environment are often
similar to those considered rare, such as being struck by lightening
or dying in an airplane crash, although they are not perceived as such
(Wilson, 1990). Moreover, people tend not to be deeply concerned about
risks that are a matter of choice such as smoking or motorcycle
riding. However, they do expect that governments pay attention to
risks that they cannot control, even though these might be
considerably less.
6.3.4 Risk perception
Whereas analysts employ risk assessment, risk evaluation and risk
management to evaluate hazards and formulate strategies and
regulations for their reduction or elimination, the majority of
individuals rely on intuitive judgements typically called "risk
perception". For these people, the experience with hazards tends to
come from the news media, which principally document mishaps and
threats occurring globally (Slovic, 1987, 1993; Kraus & Slovic, 1988;
Cohrssen & Covello, 1989; Sandman et al., 1993; Van Eijndhoven et al.,
1994).
Risk perception is being increasingly recognized as an important
factor influencing both risk evaluation and risk management. A major
factor that influences the complexity of the social debate over
appropriate laws and regulations is the nature and extent of the
perceived threat to health. The message that is frequently conveyed to
the public is that government standards for risk assessment, risk
evaluation and regulatory action are inconsistently applied, subject
to bureaucratic manipulation, and subject to alteration depending on
the degree of economic impact on the affected industry (Munro &
Morrison, 1990).
Different people perceive risks differently, depending on the
likelihood of adverse effects, whom it affects, how familiar,
widespread and dreaded the effects are, how a hazard affects
individuals personally, and whether or not individuals have
voluntarily agreed to bear the risks. Perceptions of risk are also
influenced to a large degree by the supposed benefits derived from
accepting the risk (Slovic, 1987; Krewski, et al., 1987; Kraus &
Slovic, 1988; Cohrssen & Covello, 1989; Pariza, 1992; Morgan, 1993;
Sandman et al., 1993).
Risks perceived as potentially uncontrollable, capable of causing
a catastrophe on a global scale or risking future generations cause
public anxiety. Fig. 3 illustrates a mosaic of public perception of
risks in terms of risk space quadrants; the upper right quadrant of
this space captures uncontrollable risks that are most likely to
provoke calls for government regulation (Morgan, 1993).
Tables 3 and 4 further depict qualitative factors affecting risk
perception (US NRC, 1989; Scala, 1991). While different people weigh
these factors differently in reaching their overall perceptions of the
riskiness of a hazard, the set of factors that are important in
determining relative perceptions of risk go well beyond the
statistical frequency, magnitude and uncertainty of effects. Public
opinion on acceptable risk constantly changes, usually in the
direction of further risk reduction, which provides further impetus
for additional legislation and regulation in many quarters (Munro &
Morrison, 1990).
6.3.5 Risk and hazard communication
Implicit in the process of risk evaluation and management is the
increasingly recognized role of communication (Cohrssen & Covello,
1989; US NRC, 1989; Morgan, 1993; Sandman et al., 1993; Slovic, 1993).
Risk communication is an interactive process of exchange of
information and opinion among individuals, groups and institutions
involving multiple messages about the nature of risk and other
messages, not strictly about risk, that express concerns, opinions or
reactions to risk messages or to legal and institutional arrangements
for risk management (US NRC, 1989).
Until the mid-1980s, there was little research on communicating
risk to the public. There is now a reasonable consensus on the optimum
basic elements of risk communication. These efforts should be more
systematically oriented to the intended audience, addressing the
audience's perspectives and concerns. To the greatest extent possible,
openness, not minimizing the existence of uncertainty, and discussion
of data gaps and areas of significant disagreement among experts is
recommended. The acceptance of any risk is more dependent on public
confidence in risk management than on quantitative estimates of risk.
Although there is as yet no widely agreed structured knowledge on
communication about chemical hazards, analyses of risk communication
efforts and case studies suggest that risk communication problems
arise from message, source, channel and receiver problems (Cohrssen &
Covello, 1989). Message problems relate primarily to deficiencies in
scientific understanding leading to large uncertainties in risk
estimates or highly technical risk analyses that are unintelligible to
lay persons. Source problems include disagreements among scientific
experts, failures to disclose limitations of risk assessments and
resulting uncertainties, and limited understanding of the concerns and
values of public groups and bureaucratic presentation. Channel
problems include selective and biased media reporting that emphasizes
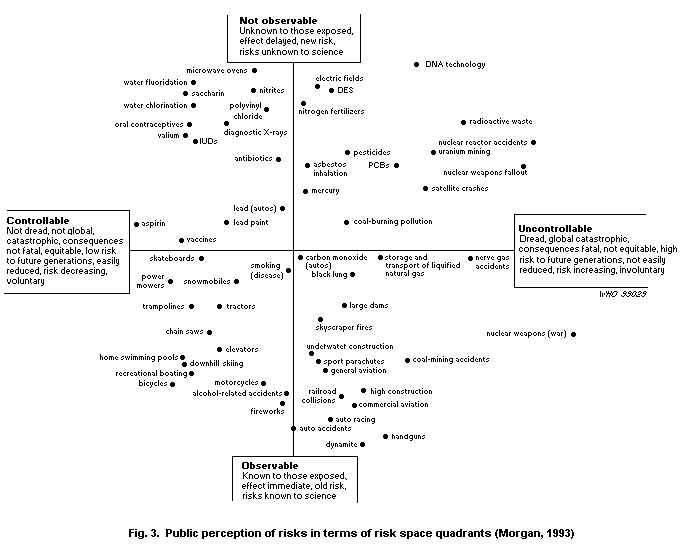 Table 3. Qualitative factors affecting risk perception and evaluation (from: US NRC, 1989)
Factor Conditions associated with increased Conditions associated with
increased public concern decreased public concern
Catastrophic potential Fatalities and injuries grouped Fatalities and injuries
in time and space scattered and random
Familiarity Unfamiliar Familiar
Understanding Mechanisms or process not understood Mechanisms or process understood
Controllability (personal) Uncontrollable Controllable
Voluntariness of exposure Involuntary Voluntary
Effects on children Children specifically at risk Children not specifically at risk
Effects manifestation Delayed effects Immediate effects
Effects on future generations Risk to future generations No risk to future generations
Victim identity Identifiable victims Statistical victims
Dread Effects dreaded Effects not dreaded
Trust in institutions Lack of trust in responsible institutions Trust in responsible institutions
Media attention Much media attention Little media attention
Accident history Major and sometimes minor accidents No major or minor accidents
Equity Inequitable distributions of risks and benefits No major or minor accidents
Benefits Unclear benefits Clear benefits
Reversibility Effects irreversible Effects reversible
Origin Caused by human actions or failures Caused by acts of nature or God
Table 4. Characteristics of risk (from: Scala, 1991)
Characteristic Description Level Examples
Knowledge Society's awareness of risk from activity Little known Food Additivies
Much known Alcoholic drinks
Newness Extent of societal experience Old Guns
New Space travel
Voluntariness Does individual have a choice about Not voluntary Crime
exposure to risk Voluntary Rock climbing
Control Can an individual control exposure, Risk not controlled Natural Disasters
protect himself or control consequences by skill or diligence
Risk controlled Smoking
Dreadedness How much is risk or its consequences feared People do not dread Vaccination
People have much dread Nerve gas
Catastrophic potential Chance of widespread disastrous outcome Not likely Sunbathing
Likely War
Equity Are the benefits and risk shared equally Distributed unequally Hazardous Dump
Distributed equally Skiing
drama, wrongdoing, disagreement, conflict and oversimplification,
distortion, and inaccuracy in interpreting technical risk information.
Receiver problems include inaccurate perception of levels of risk,
strong beliefs and opinions that are resistant to change, and demands
for scientific certainty.
There is a clear need to educate the public, including community
leaders, workers and school children, to enhance awareness so that
they can take voluntarily the action required to reduce or avoid risks
associated with exposure to chemicals in the workplace and general
environments (e.g., indoor air pollutants, pesticides and household
chemicals).
6.3.6 Economic factors
Unlike regulation, which involves strict criteria to be enforced
by regulatory agencies, economic approaches to risk management rely
largely on economic incentives to reduce the levels of pollutants
introduced into the environment (Krewski et al., 1989; Somers, 1993).
The OECD since 1972 has espoused the "Polluter Pays Principle"
(PPP) concept, with the goal of maintaining equitable trading
practices by encouraging polluters to reduce emissions. However, it is
recognized that the consumer ultimately pays the cost required to
accomplish environmental improvements. The main types of economic
instruments in use in OECD countries include charges, subsidies,
deposit-refund schemes, market creation arrangements and financial
enforcement incentives (OECD, 1991b). In 1989, the OECD adopted a
Recommendation on the Application of the PPP to Accidental Pollution,
which links the economic principle and the legal principle to damage
compensation (OECD, 1991b).
6.3.6.1 Cost-benefit analyses
Traditionally, risk reduction has not included a thorough
analysis of costs and benefits (Hammond & Coppock, 1990). Indeed,
there is no widely adopted framework for cost-benefit.
As an example, three major categories of costing relationships
are typically employed in risk reduction by the US EPA, depending on
the situation:
a) benefit/cost analysis weighs the cost of control against monetary
benefits of control;
b) risk/benefit analysis weighs the economic benefits of a polluting
activity against the risks to health and the environment;
c) cost-effectiveness analysis accepts the desirability of
regulation and identifies the least-cost solution to achieve a
given goal, such as a pollution discharge standard (Ris & Preuss,
1988).
The US EPA estimated that the annual compliance cost for USA
federal environmental regulations in 1990 was about 2.1% of the gross
national product (GNP of about 6 trillion dollars). This is expected
to increase to approximately 2.8% of the GNP by the year 2000
(ILSI/National Safety Council, 1993). The benefits of regulation such
as improved quality of life and cleaner environment are often
difficult to quantify in contrast to the enormous costs often cited
for regulatory compliance.
There is broad diversity of opinion as to how costs should be
considered in risk management decisions. Key questions include: How
much can society afford to spend to reduce risks? What is an
acceptable cost per life saved? How should costs be factored into
priority-setting processes? Future success in risk management may to a
large degree depend on ways to weigh benefits and costs and to strike
the appropriate balance in defining how fast to pursue risk
regulations (ILSI/National Safety Council, 1993; Wiener, 1993).
6.3.7 Political factors
Political factors often have an impact on national and local
priorities, drafting of regulatory statutes and introduction of
resulting risk reduction measures. Trade barriers and global
competition also have a considerable impact on risk reduction. For
example, in Canada the decision in 1980 to ban the sale of
urea-formaldehyde foam insulation (UFFI) led to unprecedented public
anger (and anxiety and resentment), great government expense, the
longest civil suit in Canadian history, and appreciable political
consequences. After an 8-year legal trial, it was concluded that there
was not sufficient scientific evidence to substantiate the reported
health problems of UFFI home owners (Somers, 1993).
In 1977, the US Food & Drug Administration (FDA), reacting to
studies that reported the artificial sweetening agent saccharin to be
a bladder carcinogen in rodent feeding studies, proposed to ban the
agent under the Delaney Amendment ("zero-risk") requirement. The
Congress of the USA in November 1977, reacting to the overwhelming
public outcry in support of unrestricted use of saccharin, enacted the
Saccharin Study and Labeling Act (SSLA), which prevented the FDA from
banning saccharin based on the information that was then available.
This made it clear that the public is willing to accept certain risks
from food additives if it perceives that the benefits are high enough
and, possibly, that the risks are low enough (Flamm & Lorentzen,
1988).
6.3.8 Regulatory limits
Traditionally, one avenue of protection of human health has been
through the establishment of exposure limits (variously referred to as
standards, quality criteria, etc.). These are established in a
two-step process, the first involving consideration of the
health-based scientific data and the second involving establishment of
regulatory limits, taking into account the health-based recommendation
along with other factors.
Examples of health-based exposure guidelines include the
Acceptable Daily Intake (ADI), Tolerable Daily Intake (TDI),
Provisional Tolerable Weekly Intake (PTWI), and health-based Maximum
Allowable Concentrations (MAC). Acceptable/Tolerable Intakes are the
amounts of a food additive, contaminant, pesticide or veterinary drug
residue, expressed on a body weight basis, that can be ingested for a
lifetime without appreciable risk to health. The term ADI is commonly
used for additives to food since they impart some beneficial
characteristic (and hence are considered "acceptable") while a TDI
commonly refers to environmental contaminants which are undesirable.
Maximum Allowable Concentrations are either a time-weighted average
concentration of a substance in a medium of exposure that does not
present appreciable hazard for continuing exposure or an upper limit
(ceiling value) which, if exceeded, will have adverse consequences for
health. Often, health-based guidelines are considered, along with
other factors (i.e., technological, socioeconomic, feasibility,
enforcement), to develop operational regulatory limits such as the
Maximum Residue Level (MRL) for pesticides or veterinary drugs, MAC in
exposure media and workplaces, occupational Threshold Limit Values
(TLV), Maximum Workplace Concentrations (MAK), Occupational Exposure
Limits (OEL), Air Quality Standards (AQS), Water Quality Standards
(WQS) or Maximum Use Levels.
Some media of (direct and indirect) exposure and associated
limits are listed below:
Food
* limits for food additives, contaminants, pesticide residues,
veterinary drug residues
* limits for certain chemicals in food packaging materials
* limits for additives and contaminants in animal feed
Cosmetics and other consumer products
* limits for additives and contaminants in cosmetic products (these
include soap and toothpaste)
* limits for other consumer products such as children's toys,
paints and solvents
Water
* drinking-water quality standards
* water quality standards for surface water
* water quality standards for fresh water used for fishing
* water quality standards for estuarine and marine waters
* aqueous effluent standards for industrial effluents and sewage
treatment outfall
* guideline limits for the use of waste water in agriculture and
aquaculture
Air
* air quality (ambient or indoor) limits for gases, vapours,
fibres, particulates
* air quality standards for gaseous or smoke emissions from
industries
Occupational
* occupational exposure limits for gases, vapours, dusts, aerosols
in workplace air and substances absorbed through the skin, mucous
membranes or alimentary tract
* regulatory limits for exposure can be based on appropriate
biomarkers
Soil
* limits for certain chemicals in soil
Agricultural chemicals
* limits for certain contaminants in agrochemicals (fertilizers)
* limits for application rates of pesticides
Chemical waste
* limits for disposal of chemicals as waste products
* waste (including liquid and solid)
* chemical (including mixed industrial), dumps, surface water and
deep well injection
* municipal surface and groundwater contamination, use of sludge in
agriculture
* atmospheric effluents and residual ash from incineration
The two stages and their outputs should not be confused. The
outputs are frequently expressed in different units. For example,
considering pesticide residues in food crops, the ADI is a daily dose
expressed in mg/kg body weight (per day being implicit) whereas the
MRL is a concentration on the crop expressed in mg/kg of the produce.
The MRL may be derived on the basis of Good Agricultural Practice and,
if adhered to, would not result in the ADI being exceeded even if all
the designated crop contained the pesticide at the MRL (an unlikely
postulate). Clearly, to arrive at this conclusion requires information
on daily intakes of the commodities carrying the residue.
6.4 Risk management options
Risk managers can intervene at many points:
a) to prevent the process producing the risk
b) to reduce or eliminate exposures
c) to modify the effects
d) to alter perceptions or valuation, through education and public
relations
e) to compensate for damage after the fact (Morgan, 1993).
6.4.1 Risk reduction
Risk reduction goals can vary considerably and can also be
hampered by the fragmented regulatory structure enforcing
environmental laws in many countries. For example, in the USA, the
regulatory approach to risk reduction depends upon whether a chemical
is a food additive, a food contaminant, a pesticide, a drinking-water
contaminant, an air pollutant, or several of these (Rodricks, 1992).
Increasingly, however, national legislation (such as the Canadian
Environmental Protection Act) that allows for introduction of control
measures for chemicals in a variety of media is being introduced.
Essentially, such legislation enables the development of control
measures in the medium that will contribute most significantly to
reduction of risk. The existing substances regulation of the European
Union also provides the opportunity for concerted action based on
evaluation of risks for different scenarios and routes of exposure
(EEC Council Regulation No.793/93) (EC, 1993).
However, there is no clear consensus on what is considered a risk
of concern. While target risk levels are embodied in some national
legislation, other countries recommend that exposure be reduced as low
as possible for effects for which it is assumed that there is no
threshold.
It is also well recognized that different countries, as well as
different agencies within the same country, often come to different
conclusions in the manner in which they judge and manage a health risk
employing basically the same scientific data (Nilsson et al., 1993;
Somers, 1993). Nilsson et al. (1993) found that 11 countries regulated
the same pesticides to different degrees, which should not be too
surprising recognizing the differing economic interests and statutes
(Somers, 1993).
6.4.1.1 Technology-based criteria
Technology-based criteria for risk reduction are not based on
costs, benefits or rights, but rather the level of technology to
control certain risks. Regulations based on these criteria typically
mandate "the best available technology" (BAT) or emissions that are
"as low as reasonably achievable". Such rules can be difficult to
apply because people seldom agree on the definition of "available" or
"reasonably achievable" (Morgan, 1993). Similar difficulties can arise
with the implementation of "good agricultural practice", "technically
achievable" and "as far as may be reasonably practicable".
REFERENCES
Allaby M (1983) A dictionary of the environment. 2nd ed. New York,
NY, New York University Press, p 195.
Allen BC, Crump KS, & Shipp AM (1988) Correlation between carcinogenic
potency of chemicals in animals and humans. Risk Analysis, 8: 531-544.
Anderson D (1990) In vitro models. Drug Saf; 5 Suppl 1:27-39.
Atterwill CK (1995) Alternative method of assessing toxicity. Methods
Mol Biol 43: 1-9.
Axelson O & Westberg H (1992) Introductory note on the concept of
exposure and dose in occupational epidemiology. Am J Ind Med, 21: 3-5.
Bailar JC III, Crouch EAC, Shaikh R, & Spiegelman D (1988) One-hit
models of carcinogenesis: conservative or not? Risk Analysis,
8: 485-497.
Baird SJS, Cohen JT, Graham JD, Shyakhter A & Evans JS (1996)
Noncancer risk assessment: a probabilistic alternative to current
practice. J Human Ecological Risk Assessment 2 (1): 79-102.
Ballantyne B, Marrs T, & Turner P (1993) General and Applied
Toxicology, Vol. 2, Stockton, Stockton Press, pp 1069-1158.
Beaumont JJ & Breslow NJ (1981) Power consideration in epidemiologic
studies of vinyl chloride workers. Am J Epidemiol, 114: 725-734.
Beland FA & Poirier MC (1993) Significance of DNA adduct studies in
animal models for cancer dosimetry and risk assessment. Environ Health
Perspect, 99: 5-10.
Braat L, Bakema AH, de Boer KF, Meijers RM, & Van Minnen JG (1991)
EXPECT: Outline of an integrated model for environmental scenario
analysis and impact asessment (Report No 259102001). Bilthoven,
National Institute of Public Health and Environmental Protection.
Calabrese EJ (1985) Uncertainty factors and interindividual variation.
Regul Toxicol Pharmacol, 5: 190-196.
Calabrese EJ, Beck BD, & Chappell WR (1992) Does the animal-to-human
uncertainty factor incorporate differences in surface area? Regul
Toxicol Pharmacol, 15: 172-179.
Calabrese EJ & Baldwin LA (1994) Improved method for detection of the
NOAEL. Regul Toxicol Pharmacol, 19: 48-50.
Chhabra RS, Huff JE, Schwetz BS & Selkirk J (1990) An overview of
prechronic and chronic toxicity / carcinogenicity experimental designs
and criteria used by the National Toxicology Program. Environ Hlth
Perspect 86: 313-321.
Chen C & Farland W (1991) Incorporating cell proliferation in
quantative cancer risk assessment: approaches, issues, and
uncertainties. Prog Clin Biol Res, 369: 481-499.
Cohrssen JJ & Covello VT (1989) Risk analysis: A guide to principles
and methods for analyzing health and environmental risks. Washington,
DC, Council on Environmental Quality.
Copeland KT, Checkoway H, McMichael AJ, & Holbrook RH (1977) Bias due
to mis-classification in the estimation of relative risk. Am J
Epidemiol, 105: 488-494.
Crump KS, Hoel DG, Langley CH, & Peto R (1976) Fundamental
carcinogenic processes and their implications for low dose risk
assessment. Cancer Research, 36: 2973-2979.
Crump KS (1984) A new method for determining allowable daily intakes.
Fundam Appl Toxicol, 4: 854-871.
Curry P & Iyengar S (1992) Comparison of exposure assessment
guidelines for pesticides. Revs Environ Contam Toxicol, 129: 80-93.
DFG (German National Research Foundation) (1972) [Hazardous working
compounds: Toxicological-occupational explanation of maximum-allowable
concentrations (MAK-Werte)] in German. Senatskommission zur Prüfung
gesundheitlicher Arbeitsstoffe. D Henschler/H Greim (Hrsg.), Wiley-VCH
Doll R & Hill AB (1956) Lung cancer and other causes of death in
relation to smoking - a second report on the mortality of British
doctors. Br Med J, 2: 1071-1077.
Dourson ML & Stara JF (1983) Regulatory history and experimental
support of uncertainty (safety) factors. Regul Toxicol Pharmacol,
3: 224-238.
Dybing E, Sanner T, Roelfzema H, Kroese D & Tennant RW (1997) T25: a
simplified potency index: description of the system and study of
correlations between carcinogenic potency and species/site specificity
and mutagenicity. Pharmacol-Toxicol, Jun, 80 (6): 272-279.
Dybing E & Huitfeldt HS (1992) Species differences in carcinogen
metabolism and interspecies extrapolation. In: Vainio H, Magee PN
McGregor DB, & McMichael AJ ed. Mechanisms of Carcinogenicity in Risk
Identification. Lyon, France, International Agency for Research on
Cancer, pp 501-522.
EC (1990) EEC Note for Guidance: Good Clinical Practice for trials on
medical products in the European Community. Pharmacology and
Toxicology, 67:361-372.
EC (1992) Council Directive 92/32/EEC of 30th April 1992. Amending
for the 7th time Directive 67/548/EEC on the approximation of the
laws, regulations and administrative provision relating to the
classification, packaging and labelling of dangerous substances.
Official Journal of the European Communities L 154.
EC (1993) Council Regulation (EC) 793/93 of 23 March 1993 on the
evaluation and control of the risks of existing substances. Official
Journal of the European Communities L 84.
EC (1994) Commission Regulation (EC) No 1488/94 of 28 June 1994 laying
down the principles for the assessment of risks to man and the
environment of existing substances in accordance with Council
Regulation (EEC) No 793/93. Official Journal of the European
Communities L 161.
EC (1996) Technical Guidance Document in Support of the Commission
Directive 93/67/EEC on Risk assessment for New Notified Substances and
Commission Regulation 1488/94/EEC on Risk assessment for Existing
Chemicals. Brussels, European Comission.
ECETOC (1989) Alternative approaches for the assessment of
reproductive toxicity. Brussels, Belgium, European Chemical Industry
Ecology and Toxicology Centre (Monograph No 12).
ECETOC (1992) Estimating environmental concentrations of chemicals
using fate and exposure models. Brussels, Belgium, European Chemical
Industry Ecology and Toxicology Centre (Technical Report No 50).
ECETOC (1993) Environmental hazard assessment. Brussels, Belgium,
European Chemical Industry Ecology and Toxicology Centre (Technical
Report No 51).
ECETOC/UNEP (1996) Inventory of Critical Reviews on Chemicals.
Brussels, Belgium, European Chemical Industry Ecology and Toxicology
Centre. United Nations Environment Programme. International Register
of Potentially Toxic Chemicals, Geneva.
Environ (1988) Elements of Toxicology and Chemical Risk Assessment.
Revised Edition. Washington, DC, Environ Corporation, 74 pp.
Farmer JH, Kodell RL, & Gaylor DW (1982) Estimation and extrapolation
of tumor probabilities from a mouse bioassay with survival/sacrifice
components. Risk Analysis, 2: 27-34.
Flamm WG & Lehma-McKeeman LD (1991) The human relevance of the renal
tumor-inducing potential of d-limonene in male rats: implications
for risk assessment. Regul Toxicol Pharmacol, 13: 70-86.
Flamm WG & Lorentzen RJ (1988) Quantitative Risk Assessment (QRA): A
special problem in the approval of new products. In: Cothern CR,
Mehlman MA, & Marcus WL ed. Risk Assessment of Industrial and
Environmental Chemicals. Advances in Modern Toxicology. Princeton, NJ,
Princeton Scientific Publ Co, Vol XV, pp.91-108.
Fowle JR (1989) Summary and perspectives: Panel discussion on
toxicological exposure assessment: State of the art. J Am Coll
Toxicol, 8: 865-870.
Fowle JR & Sexton K (1992) EPA priorities for biological research in
environmental health. Environ Hlth Perspec, 98: 235-241.
Fralick RA (1983) The role of epidemiology in defining an occupational
carcinogen. Occup Health Ontario, 4: 2-16.
Frazier JM (1993) In vitro models for toxicological research and
testing. Toxicol Lett, 68: 73-90.
Gaylor DW & Kodell RL (1980) Linear interpolation algorithm for low
dose risk assessment of toxic substances. J Environ Path Toxicol,
4: 305-312.
Gaylor DW, Greenman DL, & Frith CH (1985) Urinary bladder neoplasms
induced in Balb/C female mice with low doses of 2-acetylaminofluorene.
J Environ Pathol Toxicol Oncol, 6: 127-136.
Gaylor DW (1987) Linear-nonparametric upper limits for low dose
extrapolation. In: Proc Biopharm Sec Amer Statis Asso, pp. 63-66.
Gehlbach SH (1982) Interpreting the medical literature a clinician's
guide. Lexington, Mass. Collamore Press, ISBN: 0669045063.
Gelbke HP (1993) New trends in toxicology and their impact on toxicity
testing-scientific and practical aspects. Human Exp Toxicol,
12: 511-515.
Goldstein BD (1990) The problem with the margin of safety: toward the
concept of protection. Risk Analysis, 10: 7-10.
Groopman JD, Cain LG, & Kensler TW (1988) Aflatoxin exposure in human
populations: measurements and relation to cancer. CRC Crit Revs
Toxicol, 19: 113-145.
Gross MA, Fitzhugh OG, & Mantel N (1970) Evaluation of safety for food
additives: an illustration involving the influence of methyl
salicylate on rat reproduction. Biometrics, 26: 181-194.
Hammond PB & Coppock R (1990) Valuing Health Risks, Costs and Benefits
for Environmental Decision Making. In: Report of a Conference. Hammond
PB & Coppock R ed. Washington, DC, National Research Council, National
Academy Press.
Hasemann JK & Clark (1990) Carcinogenicity results for 114 laboratory
animal studies used to asses the predictivity of four in vitro genetic
toxicity assays for rodent carcinogenicity. Environ Mol Mutagen, 16
Suppl 18: 15-31.
Health Canada (1994) Human Health Risk Assessment for Priority
Substances. Environmental Health Directorate, Health Canada, Canada
Communication Group - Publishing, Cat No En40-215/41E, Ottawa, 1994.
Hemminki K, Grzybowska E, Chorazy M, Twardowska-Saucha K, Sroczynski
JW, Putman KL, Randerath K, Phillips DH, Hewer A, & Santella RM (1990)
DNA adducts in humans environmentally exposed to aromatic compounds in
an industrial area of Poland. Carcinogenesis, 11(7): 1229-1231.
Hemminki K (1992) Significance of DNA and protein adducts. In: Vainio
H, Magee PN, McGregor DB, & McMichael AJ ed. Mechanisms of
Carcinogenesis in Risk Identification. Lyon, France, International
Agency for Research on Cancer, pp 525-534.
Hernberg S (1980) Evaluation of epidemiologic studies in assessing the
long-term effects of occupational noxious agents. Scand J Work Environ
Health, 6: 163-169.
Hertel RF (1996) Outline on risk assessment of existing substances in
the European Union. Env Toxicol Pharmacol 2: 93-96.
Hill AB (1965) The environment and disease. Association or causation?
Proc R Soc Med, 58: 295-300.
Hodgson E, Mailman RB, & Chambers JE (1988) Dictionary of Toxicology.
New York, NY, Van Nostrand Reinhold Co, p 154.
Hoel DG (1980) Incorporation of background in dose-response models.
Fed Proc, 39: 73-75.
IARC (1987) IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, No. 42. Lyon, International Agency for Research on Cancer.
IARC (1990) IARC Scientific Publication on Cancer: Causes, Occurrence
and Control, No. 100. Lyon, International Agency for Research on
Cancer.
ICRP (1974) International Commission on Radiological Protection:
Report of the Task Group on Reference Man. ICRP Publication No. 23.
Oxford, Pergamon Press.
ILSI/National Safety Council (1993) Regulating Risk: The Science and
Politics of Risk: A Conference Summary. Washington DC, ILSI Press,
International Life Sciences Institute.
IPCS (1978) Environmental Health Criteria 6: Principles and methods
for evaluating the toxicity of chemicals. Part I. Geneva, Switzerland,
World Health Organization, International Programme on Chemical Safety.
IPCS (1983) Environmental Health Criteria 27: Guidelines on studies in
environmental epidemiology. Geneva, Switzerland, World Health
Organization,. International Programme on Chemical Safety.
IPCS (1984) Environmental Health Criteria 30: Principles for
evaluating health risks to progency associated with exposure to
chemicals during pregnancy. Geneva, Switzerland, World Health
Organization, International Programme on Chemical Safety.
IPCS (1985a) Environmental Health Criteria 46: Guidelines for the
study of genetic effects in human populations. Geneva, Switzerland,
World Health Organization, International Programme on Chemical Safety.
IPCS (1985b) , Environmental Health Criteria 47: Summary report on the
evaluation of short-term in vitro tests for carcinogens
(collaborative study on in vitro tests). Geneva, Switzerland, World
Health Organization, International Programme on Chemical Safety.
IPCS (1985c) Environmental Health Criteria 51: Guide to short-term
tests for detecting mutagenic and carcinogenic chemicals. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1986a) Environmental Health Criteria 57: Principles of
toxicokinetic studies. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1986b) , Environmental Health Criteria 59: Principles for
evaluating health risks from chemicals during infancy and early
childhood: The need for a special approach. Geneva, Switzerland, World
Health Organization, International Programme on Chemical Safety.
IPCS (1986c) Environmental Health Criteria 60: Principles and methods
for the assessment of neurotoxicity associated with exposure to
chemicals. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1987a) Environmental Health Criteria 70: Principles for the
safety assessment of food additives and contaminants in food. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1987b) Environmental Health Criteria 72: Principles of studies
on diseases of suspected chemical aetiology and their prevention.
Geneva, Switzerland, World Health Organization, International
Programme on Chemical Safety.
IPCS (1990a) Environmental Health Criteria 104: Principles for the
toxicological assessment of pesticide residues in food. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1990b), Environmental Health Criteria 109: Summary report on the
evaluation of short-term in vivo tests for carcinogens
(collaborative study on in vivo tests). Geneva, Switzerland, World
Health Organization, International Programme on Chemical Safety.
IPCS (1991) Environmental Health Criteria 119: Principles and methods
for the assessment of nephrotoxicity associated with exposure to
chemicals. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1992a) Environmental Health Criteria 141: Quality management for
chemical testing. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1992b) Environmental Health Criteria 144: Principles for
evaluating the effects of chemicals on the aged population. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1993) Environmental Health Criteria 155: Biomarkers and risk
assessment: concepts and principles. Geneva, Switzerland, World Health
Organization, International Programme on Chemical Safety.
IPCS (1994) Environmental Health Criteria 170: Assessing human health
risks to chemicals: derivation of guidance values for health-based
exposure limits. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1996) Environmental Health Criteria 180: Principles and methods
for assessing direct immunotoxicity associated with exposure to
chemicals. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1997) Environmental chemicals and respiratory
hypersensitization. Proceedings of an IPCS Workshop, Oslo, Norway, 29
November December. Environ Toxicol Pharmacol, 2/3 (3-special issue),
77-242.
IPCS (1998) Draft Environmental Health Criteria: Principles of human
exposure assessment. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
Kelsey JL, Thompson WD & Evans AS (1986) Methods in observational
epidemiology. Boston, MA, USA: Little, Brown and Company.
Konheim CS (1988) Risk communication in the real world. Risk Anal,
8: 367-373.
Kraus NN & Slovic P (1988) Taxonomic analysis of perceived
risk: Modeling individual and group perceptions within homogenous
hazard domains. Risk Anal, 8: 435-455.
Krewski D, Brown C, & Murdoch D (1984) Determining "safe" levels of
exposure: safety factors or mathematical models? Fund Appl Toxicol,
4: S383-S394.
Krewski D, Murdoch D, & Dewanji A (1986) Statistical modeling and
extrapolation of carcinogenesis data. In: Moolgavkar SH & Prentice RL,
ed. Modern Statistical Methods in Chronic Disease Epidemiology. New
York, NY, John Wiley and Sons, pp 259-282.
Krewski D, Somers E, & Birkwood PL (1987) Risk perceptions in a
decision making context. Environ Carcinogen Revs J Environ Sci Hlth,
C5(2): 175-209.
Krewski D, Thorslund T & Whithey J (1989) Carcinogenic risk assessment
of complex mixtures. Toxicol Ind Health 5: 851-867.
Lauwerys R (1984) Basic concepts of human exposure. In: Berlin A,
Draper M, Hemminki K, & Vainio H ed. Monitoring Exposure to
Carcinogenic and Mutagenic Agents. IARC Scientific Publications, Lyon,
France, International Agency for Research on Cancer, No 59, pp. 31-36.
Leisenring W & Ryan L (1992) Statistical properties of the NOAEL.
Regul Toxicol Pharmacol, 15: 161-171.
Lewis SC, Lynch JR, & Nikiforov AI (1990) New approach to deriving
community exposure guidelines from "No-Observed-Adverse-Effect
Levels". Regul Toxicol Pharmacol, 11: 314-330.
Lilienfeld AM & Lilienfeld D (1979) Foundations of Epidemiology. 2nd
ed. New York, NY: Oxford University Press.
Lioy PJ (1990) Assessing total human exposure to contaminants. Environ
Hlth Perspec, 24: 938-945.
Liteplo RG, Meek ME, Gomes R, & Savard S (1994) Inorganic
fluoride: evaluation of risks to health from environmental exposure
to fluoride in Canada. Environ Carcinog & Ecotox Revs.
c12(2): 327-344.
Mausner JS & Kremer S (1985) Epidemiology, 2nd ed. Philadelphia: WB
Saunders Company.
Meek ME, Newhook R, Liteplo RG & Armstrong VC (1994) Approach to
assessment of risk to human health for Priority Substance under the
Canadian Environmental Protection. J Environ Sci Health
C12(2): 105-134.
Merrill RA (1991) Regulatory toxicology. In: Amdur MO, Doull J, &
Klaasen CD ed. Casarett and Doull's Toxicology. 4th edition. New York,
Oxford, Beijing, Singapore, Toronto, Pergamon Press, pp 970-983.
Monro AM (1990) Interspecies comparisons in toxicology: the utility
and futility of plasma concentrations of the test substance. Regul
Toxicol Pharmacol, 12: 137-160.
Monro A (1993) How useful are chronic (life-span) toxicology studies
in rodents in identifying pharmaceuticals that pose a carcinogenic
risk to humans? Adverse Drug React Toxicol Rev, 12: 5-34.
Montesano R, Bartsch H, Vainio H, Wilbourn & Yamasaki H (eds) (1986)
Long-term and short-term assays for carcinogens. A critical appraisal
(IARC Scientific Publications No. 83), Lyon, France, International
Agency for Research on Cancer (IARC).
Moolgavkar SH, Dewanji A, & Venzon DJ (1988) A stochastic two-stage
model for cancer risk assessment. I: The hazard function and the
probability of tumor. Risk Anal, 8:383-392.
Morgan MG (1993) Risk analysis and management. Sci Amer, 269: 32-41.
Munro IC & Morrison AB (1990) Interaction of science and politics in
toxicological regulation. In: Clayson DB, Munro IC, Shubik P, &
Swenberg JA ed. Progress in Predictive Toxicology. Amsterdam, Elsevier
Science Publ BV, pp 373-387.
Murdoch DJ, Krewski D, & Crump KS (1987) Mathematical models of
carcinogenesis. In: Thompson JR & Brown B ed. Cancer modeling, New
York, NY, Marcel Dekker, pp 61-89.
Mushak B (1993) Environmental equity: A new coalition for justice.
Environ Hlth Persp, 101: 478-483.
Nilsson R, Tasheva M, & Jaeger B (1993) Why different regulatory
decisions when the scientific information base is similar?- Human risk
asessment. Regul Toxicol Pharmacol, 17: 292-332.
OECD (1987) Decision-recommendation of the Council on the systematic
investigation of existing chemicals C (87) 90 (Final). Paris, France,
Organization for Economic Cooperation and Development.
OECD (1989) Compendium of Environmental Exposure Assessment Methods
for Chemicals. Paris, France, Organization for Economic Cooperation
and Development (Environmental Monograph No 27).
OECD (1991a) The OECD Workshop on the Application of Simple Models for
Environmental Assessment [Berlin, 11-13 December]. Paris, France,
Organisation for Economic Co-operation and Development.
OECD (1991b) The State of the Environment. Paris, France, Organisation
for Economic Co-operation and Development, pp.253-265.
OECD (1998) OECD Principles on Good Laboratory Practice (as revised in
1997). OECD Series on Principles of Good Laboratory Practice and
compliance monitoring. Number 1. Environment Directorate, Chemicals
Group and Management Committee. Paris, France, Organisation for
Economic Co-operation and Development, ENV/MC/CHEM (98)17.
Otson R, Fellin P, Chan C (1996) Multi-media exposure study: rational
and design. In: Measurement of Toxic and Related Air pollutants
VIP-64. Air & Waste Management Association, One Gate Center, Third
Floor, Pittsburgh, PA 15222, USA. pp 559-566.
Pariza MW (1992) Risk-benefit perception. In: Food Safety Assessment.
Finley JW, Robinson SF, & Armstrong DJ, eds. American Chemical Society
(ACS) Symposium No 484. Washington, DC, American Chemical Society, pp
36-40.
Perera FP (1987) Molecular cancer epidemiology: A new tool in cancer
prevention. J Natl Cancer Inst, 78: 887-898.
Perera FP (1988) The significance of DNA and protein adducts in human
biomonitoring studies. Mutat Res, 205: 255-269.
Peto R (1978) Carcinogenic effects of chronic exposure to very low
levels of toxic substances, Environ Health Perspect, 22: 155-161.
Peto R, Pike MC, Bernstein L, Gold LS & Ames BN (1984) The TD50: a
proposed general convention for the numerical description of the
carcinogenic potency of chemicals in chronic-exposure animal
experiments. Environ Health Perspective 58, 1-8.
Poirier MC & Beland FA (1987) Determination of carcinogen-induced
macromolecular adducts in animals and humans. Prog Exp Tumor Res,
31: 1-10.
Portier CJ (1988) Species correlation of chemical carcinogens. Risk
Anal, Dec 8 (4): 551-553.
Presidential/Congressional Commission on Risk Assessment and Risk
Management (1997a) Framework for Environmental Health Risk Management.
Final Report Vol. 1, Washington, DC, USA.
Presidential/Congressional Commission on Risk Assessment and Risk
Management (1997b) Risk Assessment and Risk Management in Regulatory
Decision-Making. Final Report Vol. 2, Washington, DC, USA.
Price PS, Keenan RE, Swartout JC, Gillis CA, Carlson-Lynch H, Dourson
ML (1997) An approach for modeling noncancer dose responses with an
emphasis on uncertainty. Risk Anal 17(4): 427-437.
Purchase (1986) Unpredictability: an essay in toxicology. Food Chem
Toxicol, Apr 24 (4): 343-349.
Renwick AG (1993a) Toxicokinetics. In: Ballantyne B, Marrs T, & Turner
P ed. General & Applied Toxicology. New York, Stockton Press, Vol 1,
pp 121-151.
Renwick AG (1993b) Data-derived safety factors for the evaluation of
food additives and environmental contaminants. Fd Addit Contam,
10: 275-305.
Ricci PF & Cox LA (1987) Acceptability of chronic health risks. Toxic
Law Rept, 1: 986-1001.
Ris CH & Preuss PW (1988) Risk assessment and risk management : A
process. In: Cothern CR, Mehlman MA, & Marcus WL ed. Risk Assessment
and Risk Management of Industrial and Environmental Chemicals.
Princeton, NJ, Princeton Scientific Publ Co, Vol XV, pp 1-21.
RIVM (1994) Uniform Systems for the Evaluation of Substances (USES),
version 1.0. The Hague, The Netherlands, Ministry of Housing, Spatial
Planning and Environment (Distribution no 11144/150).
Rodricks JV (1992) Calculated Risks. Cambridge, MA, Cambridge
University Press.
Rothman KT (1986) Modern Epidemiology. Boston, MA, USA: Little, Brown
and Company.
Sandman PM, Muller PM, Johnson BB, & Weinstein NN (1993) Agency
communication, community outrage and perception of risk. Three
simulation experiments. Risk Anal, 13: 585 592.
Scala RA (1991) Risk assessment. In: Amdur MO, Doull J, & Klaasen CD,
ed. Casarett and Doull's Toxicology. 4th edition, New York, Oxford,
Beijing, Frankfurt, Sao Paulo, Sydney, Tokyo, Toronto, Pergamon Press,
pp 985-996.
Seidman BC, Brown SL, De Rosa CT, & Mumtaz MM (1991) Sensitive and
Hyper-susceptible populations: Risk assessment considerations for
exposure to single chemicals and chemical mixtures. In: Zervos C ed.
Risk Analysis. New York, NY, Plenum Press, pp 305-313.
Sexton K, Callahan MA, & Ryan EF (1995) Estimating exposure and dose
to characterize health risks: the role of human tissue monitoring in
exposure assessment. Env Health Health, Suppl 3: 13-30.
Shuker DEG (1989) Nucleic acid-carcinogen adducts in human dosimetry.
Arch Toxicol, (Suppl 13): 55-65.
Silbergeld EK (1993) Risk assessment: The perspective experience of US
environmentalists. Environ Hlth Persp, 101: 100-104.
Singer E & Endreny PM (1993) Reporting on Risk. New York, Russel Sage
Foundation.
Skipper PI & Tannenbaum SR (1990) Protein adducts in the molecular
dosimetry of chemical carcinogens. Carcinogenesis, 11: 507-518.
Slovic P (1987) Perceptions of risk. Science, 236: 280-284.
Slovic P (1993) Perceived risk, trust and democracy. Risk Anal,
13: 675-682.
Somers E (1987) Making decisions with numbers. Regul Toxicol
Pharmacol, 8: 35-42.
Somers E (1993) Perspectives on Risk Management. [Presented at
Managing Risks to Life and Health Meeting, 18 October]. Ottawa, Royal
Society of Canada.
Stayner LT & Bailer AJ (1993) Comparing toxicologic and epidemiologic
studies. Methylene chloride - a case study. Risk Anal, Dec 13
(6): 667-673.
Stern RM (1986) Analysis of the decision making process in chemical
safety. Sci Total Environ, 51: 27-62.
Susser M (1977) Judgement and causal inference: criteria in
epidemiologic studies. Am J Epidemiol, 105: 1- 15.
Tennant RW, Margolin BH, Shelby MD, Zeiger E, Haseman JK, Spalding J,
Caspary W, Resnick M, Stasiewicz S, Anderson B, et al. (1987)
Prediction of chemical carcinogenicity in rodents from in vitro
genetic toxicity assays. Science 236: 933-941.
Travis CC, Pack SA, Saulsbury AW & Yambert MW (1990a) Prediction of
carcinogenic potency from toxicological data. Mutal Res, May 241
(1): 21-36.
Travis CC, Saulsbury AW & Pack SA (1990b) Prediction of cancer potency
using a battery of mutation and toxicity data. Mutagenesis, May 5
(3): 213-219.
Travis CC, Wang LA & Waehner MJ (1991) Quantitative correlation of
carcinogenic potency with four different classes of short-term test
data. Mutagenesis, Sep 6 (5): 353-360.
UK DOH (1991) (Department of Health) Guidelines for the evaluation of
chemicals for carcinogenicity. Committee on Carcinogenicity of
Chemicals in Food, Consumer Products and the Environment. London, Her
Majesty's Stationary Office (HMSO).
UK DOH (1995) (Department of Health) Annual Report of the Committees
on Toxicity, Mutagenicity, Carcinogenicity of Chemicals in Food,
Consumer Products and the Environment. London, Her Majesty's
Stationary Office (HMSO).
UK HSE (1989) Health and Safety Executive. Quantified risk
assessment: Its input to decision making. London, Her Majesty's
Stationery Office (HMSO).
Ulfvarson U (1992) Validation of exposure information in occupational
epidemiology. Am J Ind Med, 21: 125-132.
US EPA (1984) Study of carbon monoxide exposure on residents of
Washington, DC, and Denver, CO. Environmental Monitoring Systems
Laboratory, Office of Research and Development, Research Triangle
Park, NC (EPA-600/S4-84/031, NTIS PB84-183516).
US EPA (1986a) Guidelines for carcinogen risk assessment. Fed Reg,
51: 33992-34003.
US EPA (1986b) Guidelines for Estimating Exposures. Fed Reg, 51
(185): 34042-34054.
US EPA (1987a) The total exposure assessment methodology (TEAM) study.
Vol I: Summary and analysis. Office of Acid Deposition, Environmental
Monitoring and Quality Assurance, Office of Research and Development,
Washington, DC, US Environmental Protection Agency
(EPA-600/6-87/002a).
US EPA (1987b) Pesticide Assessment Guidelines; Subdivision
U-Applicator Exposure. Washington, DC, US Environmental Protection
Agency (EPA PB 87-133286).
US EPA (1992) Guidelines for exposure assessment. Washington, DC, US
Environmental Protection Agency, Federal Register, 57:22888.
US EPA (1996a) Risk Characterization: Practical Guidance for
NCEA - Washington Based Risk Assessors, National Center for
Environmental Assessment, Washington, DC, US Environmental Protection
Agency (NCEAW-0105).
US EPA (1996b) Proposed Guidelines for Carcinogen Risk Assessment.
Washington, DC, US Environmental Protection Agency, Federal Register.
6(79): 17960-18011.
US EPA (1996c) Exposure Factors Handbook. Washington, DC, US
Environmental Protection Agency EPA/600P-95/002Bc, Review Draft.
Update to exposure Factors Handbook EPA/600/8-89/043 - May 1989.
US FDA (1993) Food and Drug Administration: Toxicological Principles
for the Safety Assessment of Direct Food Additives and Color Additives
Used in Food. "Redbook II" Draft. Washington, DC, Center for Food
Safety and Applied Nutrition, US Food and Drug Administration.
US NAS (1983) National Academy of Science: Risk Assessment in the
Federal Government: Managing the Process. National Research Council,
Committee on the Institutional Means for Assessment of Risks to Public
Health. Washington DC, National Academy Press, pp 1-50.
US NRC (1989) National Research Council: Improving Risk Communication.
Washington, DC, National Academy Press.
US NRC (1990) National Research Council: Human exposure assessment for
airborne pollutants: Advances and applications. Committee on Advances
in Assessing Human Exposure to Airborne Pollutants, Committee on
Geosciences, Environment, and Resources. Washington, DC, National
Academy Press.
US NRC (1992) National Research Council: Biological Markers in
Immunotoxicology. Washington, DC, National Academy Press.
Van Eijndhoven JCM, Wetering RAPM, Worrel CW, de Boer J, Van der Pligt
J, & Stallen PJM (1994) Risk communication in the Netherlands: The
monitored introduction of the EC "Post-Seveso" Directive. Risk Anal,
14: 87-96.
Van Ryzin J (1980) Quantitative risk assessment. J Occup Med,
22: 321-326.
WHO (1986) Field Surveys of Exposure to Pesticides. Standard Protocol.
Toxicol Lett, 33: 2234-235.
WHO (1993) Guidelines for drinking-water quality, Recommendations.
Geneva, Switzerland, World Health Organization, Vol 1, 30 pp.
Wiener JO (1993) Risk assessment and cost-benefit analyses: In the
public interst? Environ Hlth Perspect, 101: 408-409.
Wilson R (1990) Analyzing the daily risks of life. In: Readings in
Risk. Glickman TS & Gough M ed. Washington, DC, Resources for the
Future, pp 55-59.
Wilson R & Crouch EAC (1987) Risk assessment and comparisons: An
introduction. Science, 236: 267-270.
Wogan GN (1989) Markers of Exposure to Carcinogens: Methods for Human
Biomonitoring. J Am Coll Toxicol, 8: 871-881.
Wogan GN (1992) Molecular epidemiology in cancer risk and prevention:
Recent progress and avenues for future research. Environ Hlth
Perspect, 98: 167-178.
Zeiger E, Haseman JK, Shelby MD, Margolin BH & Tennant RW (1990)
Evalution of four in vitro genetic toxicity tests for predicting
rodent carcinogenecity: confirmation of earlier results with 41
additional chemicals. Environ Mol Mutagen, 16 Suppl 18: 1-14.
Zimmerman R (1993) Social equity and environmental risk. Risk Anal,
13: 649-666.
APPENDIX 1. PREAMBLE TO THE IARC MONOGRAPHS
The Preamble to the Monographs sets out the objective and scope
of the evaluation programme, the procedures used when making
assessments, and the types of evidence considered and criteria used in
reaching the final evaluations. The list of contents is given here as
is the full text referring to the Background and Evaluation sections.
Full text of the Preamble should always be used when referring to the
list of evaluations provided.
Background
In 1969, the International Agency for Research on Cancer (IARC)
initiated a programme to evaluate the carcinogenic risk of chemicals
to humans and to produce monographs on individual chemicals. The
Monographs programme has since been expanded to include
consideration of exposures to complex mixtures of chemicals (which
occur, for example, in some occupations and as a result of human
habits) and of exposures to other agents, such as radiation and
viruses. With Supplement 6 (IARC, 1987a), the title of the series was
modified from IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans to IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans, in order to reflect the widened
scope of the programme.
The criteria established in 1971 to evaluate carcinogenic risk to
humans were adopted by the working groups whose deliberations resulted
in the first 16 volumes of the IARC Monographs series. Those
criteria were subsequently updated by further ad-hoc working groups
(IARC, 1977, 1978, 1979, 1982, 1983, 1987b, 1988, 1991; Vainio et al.,
1992).
Evaluation
Evaluations of the strength of the evidence for carcinogenicity
arising from human and experimental animal data are made, using
standard terms.
It is recognized that the criteria for these evaluations,
described below, cannot encompass all of the factors that may be
relevant to an evaluation of carcinogenicity. In considering all of
the relevant scientific data, the Working Group may assign the agent,
mixture or exposure circumstance to a higher or lower category than a
strict interpretation of these criteria would indicate.
(a) Degrees of evidence for carcinogenicity in humans and in
experimental animals and supporting evidence
These categories refer only to the strength of the evidence that
an exposure is carcinogenic and not to the extent of its carcinogenic
activity (potency) nor to the mechanisms involved. A classification
may change as new information becomes available.
An evaluation of degree of evidence, whether for a single agent
or a mixture, is limited to the materials tested, as defined
physically, chemically or biologically. When the agents evaluated are
considered by the Working Group to be sufficiently closely related,
they may be grouped together for the purpose of a single evaluation of
degree of evidence.
(i) Carcinogenicity in humans
The applicability of an evaluation of the carcinogenicity of a
mixture, process, occupation or industry on the basis of evidence from
epidemiological studies depends on the variability over time and place
of the mixtures, processes, occupations and industries. The Working
Group seeks to identify the specific exposure, process or activity
which is considered most likely to be responsible for any excess risk.
The evaluation is focused as narrowly as the available data on
exposure and other aspects permit.
The evidence relevant to carcinogenicity from studies in humans
is classified into one of the following categories:
Sufficient evidence of carcinogenicity: The Working Group considers
that a causal relationship has been established between exposure to
the agent, mixture or exposure circumstance and human cancer. That is,
a positive relationship has been observed between the exposure and
cancer in studies in which chance, bias and confounding could be ruled
out with reasonable confidence.
Limited evidence of carcinogenicity: A positive association has been
observed between exposure to the agent, mixture or exposure
circumstance and cancer for which a causal interpretation is
considered by the Working Group to be credible, but chance, bias or
confounding could not be ruled out with reasonable confidence.
Inadequate evidence of carcinogenicity: The available studies are of
insufficient quality, consistency or statistical power to permit a
conclusion regarding the presence or absence of a causal association,
or no data on cancer in humans are available.
Evidence suggesting lack of carcinogenicity: There are several
adequate studies covering the full range of levels of exposure that
human beings are known to encounter, which are mutually consistent in
not showing a positive association between exposure to the agent,
mixture or exposure circumstance and any studied cancer at any
observed level of exposure. A conclusion of evidence suggesting lack
of carcinogenicity is inevitably limited to the cancer sites,
conditions and levels of exposure and length of observation covered by
the available studies. In addition, the possibility of a very small
risk at the levels of exposure studied can never be excluded.
In some instances, the above categories may be used to classify
the degree of evidence related to carcinogenicity in specific organs
or tissues.
(ii) Carcinogenicity in experimental animals
The evidence relevant to carcinogenicity in experimental animals
is classified into one of the following categories:
Sufficient evidence of carcinogenicity: The Working Group considers
that a causal relationship has been established between the agent or
mixture and an increased incidence of malignant neoplasms or of an
appropriate combination of benign and malignant neoplasms in (a) two
or more species of animals or (b) in two or more independent studies
in one species carried out at different times or in different
laboratories or under different protocols.
Exceptionally, a single study in one species might be considered
to provide sufficient evidence of carcinogenicity when malignant
neoplasms occur to an unusual degree with regard to incidence, site,
type of tumour or age at onset.
Limited evidence of carcinogenicity: The data suggest a carcinogenic
effect but are limited for making a definitive evaluation because,
e.g. (a) the evidence of carcinogenicity is restricted to a single
experiment; or (b) there are unresolved questions regarding the
adequacy of the design, conduct or interpretation of the study; or (c)
the agent or mixture increases the incidence only of benign neoplasms
or lesions of uncertain neoplastic potential, or of certain neoplasms
which may occur spontaneously in high incidences in certain strains.
Inadequate evidence of carcinogenicity: The studies cannot be
interpreted as showing either the presence or absence of a
carcinogenic effect because of major qualitative or quantitative
limitations, or no data on cancer in experimental animals are
available.
Evidence suggesting lack of carcinogenicity: Adequate studies
involving at least two species are available which show that, within
the limits of the tests used, the agent or mixture is not
carcinogenic. A conclusion of evidence suggesting lack of
carcinogenicity is inevitably limited to the species, tumour sites and
levels of exposure studied.
(b) Other data relevant to the evaluation of carcinogenicity and
its mechanisms
Other evidence judged to be relevant to an evaluation of
carcinogenicity and of sufficient importance to affect the overall
evaluation is then described. This may include data on preneoplastic
lesions, tumour pathology, genetic and related effects,
structure-activity relationships, metabolism and pharmacokinetics,
physicochemical parameters and analogous biological agents.
Data relevant to mechanisms of the carcinogenic action are also
evaluated. The strength of the evidence that any carcinogenic effect
observed is due to a particular mechanism is assessed, using terms
such as weak, moderate or strong. Then, the Working Group assesses if
that particular mechanism is likely to be operative in humans. The
strongest indications that a particular mechanism operates in humans
come from data on humans or biological specimens obtained from exposed
humans. The data may be considered to be especially relevant if they
show that the agent in question has caused changes in exposed humans
that are on the causal pathway to carcinogenesis. Such data may,
however, never become available, because it is at least conceivable
that certain compounds may be kept from human use solely on the basis
of evidence of their toxicity and/or carcinogenicity in experimental
systems.
For complex exposures, including occupational and industrial
exposures, the chemical composition and the potential contribution of
carcinogens known to be present are considered by the Working Group in
its overall evaluation of human carcinogenicity. The Working Group
also determines the extent to which the materials tested in
experimental systems are related to those to which humans are exposed.
(c) Overall evaluation
Finally, the body of evidence is considered as a whole, in order
to reach an overall evaluation of the carcinogenicity to humans of an
agent, mixture or circumstance of exposure.
An evaluation may be made for a group of chemical compounds that
have been evaluated by the Working Group. In addition, when supporting
data indicate that other, related compounds for which there is no
direct evidence of capacity to induce cancer in humans or in animals
may also be carcinogenic, a statement describing the rationale for
this conclusion is added to the evaluation narrative; an additional
evaluation may be made for this broader group of compounds if the
strength of the evidence warrants it.
The agent, mixture or exposure circumstance is described
according to the wording of one of the following categories, and the
designated group is given. The categorization of an agent, mixture or
exposure circumstance is a matter of scientific judgement, reflecting
the strength of the evidence derived from studies in humans and in
experimental animals and from other relevant data.
* Group 1: The agent (mixture) is carcinogenic to humans. The
exposure circumstance entails exposures that are carcinogenic
to humans.
This category is used when there is sufficient evidence of
carcinogenicity in humans. Exceptionally, an agent (mixture) may be
placed in this category when evidence in humans is less than
sufficient but there is sufficient evidence of carcinogenicity in
experimental animals and strong evidence in exposed humans that the
agent (mixture) acts through a relevant mechanism of carcinogenicity.
* Group 2
This category includes agents, mixtures and exposure
circumstances for which, at one extreme, the degree of evidence of
carcinogenicity in humans is almost sufficient, as well as those for
which, at the other extreme, there are no human data but for which
there is evidence of carcinogenicity in experimental animals. Agents,
mixtures and exposure circumstances are assigned to either group 2A
(probably carcinogenic to humans) or group 2B (possibly carcinogenic
to humans) on the basis of epidemiological and experimental evidence
of carcinogenicity and other relevant data.
* Group 2A: The agent (mixture) is probably carcinogenic to
humans.
The exposure circumstance entails exposures that are probably
carcinogenic to humans.
This category is used when there is limited evidence of
carcinogenicity in humans and sufficient evidence of carcinogenicity
in experimental animals. In some cases, an agent (mixture) may be
classified in this category when there is inadequate evidence of
carcinogenicity in humans and sufficient evidence of carcinogenicity
in experimental animals and strong evidence that the carcinogenesis is
mediated by a mechanism that also operates in humans. Exceptionally,
an agent, mixture or exposure circumstance may be classified in this
category solely on the basis of limited evidence of carcinogenicity in
humans.
* Group 2B: The agent (mixture) is possibly carcinogenic to
humans.
The exposure circumstance entails exposures that are possibly
carcinogenic to humans.
This category is used for agents, mixtures and exposure circumstances
for which there is limited evidence of carcinogenicity in humans and
less than sufficient evidence of carcinogenicity in experimental
animals. It may also be used when there is inadequate evidence of
carcinogenicity in humans but there is sufficient evidence of
carcinogenicity in experimental animals. In some instances, an agent,
mixture or exposure circumstance for which there is
inadequate evidence of carcinogenicity in humans but
limited evidence of carcinogenicity in experimental animals together
with supporting evidence from other relevant data may be placed in
this group.
* Group 3: The agent (mixture or exposure circumstance) is not
classifiable as to its carcinogenicity to humans.
This category is used most commonly for agents, mixtures and
exposure circumstances for which the evidence of carcinogenicity is
inadequate in humans and inadequate or limited in experimental
animals.
Exceptionally, agents (mixtures) for which the evidence of
carcinogenicity is inadequate in humans but sufficient in experimental
animals may be placed in this category when there is strong evidence
that the mechanism of carcinogenicity in experimental animals does not
operate in humans.
Agents, mixtures and exposure circumstances that do not fall into
any other group are also placed in this category.
* Group 4: The agent (mixture) is probably not carcinogenic to
humans.
This category is used for agents or mixtures for which there is
evidence suggesting lack of carcinogenicity in humans and in
experimental animals. In some instances, agents or mixtures for which
there is inadequate evidence of carcinogenicity in humans but
evidence suggesting lack of carcinogenicity in experimental animals,
consistently and strongly supported by a broad range of other relevant
data, may be classified in this group.
References:
IARC (1977) IARC Monographs Programme on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans. Preamble (IARC intern.
tech. Rep. No. 77/002), Lyon
IARC (1978) Chemicals with Sufficient Evidence
of Carcinogenicity in Experimental Animals-IARC Monographs
Volumes 1-17 (IARC intern. tech. Rep. No. 78/003), Lyon
IARC (1979) Criteria to Select Chemicals for IARC Monographs (IARC
intern. tech. Rep. No. 79/003), Lyon
IARC (1982) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Supplement 4,
Chemicals, Industrial Processes and Industries Associated with
Cancer in Humans (IARC Monographs, Volumes 1 to 29), Lyon
IARC (1983) Approaches to Classifying Chemical Carcinogens According
to Mechanism of Action (IARC intern. tech. Rep. No. 83/001), Lyon
IARC (1987a) IARC Monographs on the Evaluation of Carcinogenic Risks
to Humans, Supplement 6, Genetic and Related Effects: An Updating
of Selected IARC Monographs from Volumes 1 to 42, Lyon
IARC (1987b) IARC Monographs on the Evaluation of Carcinogenic Risks
to Humans, Supplement 7, Overall Evaluations of Carcinogenicity:
An Updating of IARC Monographs Volumes 1 to 42, Lyon
IARC (1988) Report of an IARC Working Group to Review the Approaches
and Processes Used to Evaluate the Carcinogenicity of Mixtures and
Groups of Chemicals (IARC intern. tech. Rep.No. 88/002), Lyon
IARC (1991) A Consensus Report of an IARC Monographs Working Group
on the Use of Mechanisms of Carcinogenesis in Risk Identification
(IARC intern. tech. Rep. No. 91/002), Lyon
Vainio, H., Magee, P., McGregor, D. & McMichael, A., eds (1992)
Mechanisms of Carcinogenesis in Risk Identification (IARC Scientific
Publications No. 116), Lyon, IARC
APPENDIX 2. OECD'S GUIDELINES FOR THE TESTING OF CHEMICALS
(from http://www.oecd.org/ehs/test/health.htm)
1. Adopted Test Guidelines
TG 401 Acute Oral Toxicity (Updated Guideline, adopted 24th
February 1987)
TG 402 Acute Dermal Toxicity (Updated Guideline, adopted 24th
February 1987)
TG 403 Acute Inhalation Toxicity (Original Guideline, adopted 12th
May 1981)
TG 404 Acute Dermal Irritation/Corrosion (Updated Guideline,
adopted 17th July 1992)
TG 405 Acute Eye Irritation/Corrosion (Updated Guideline, adopted
24th February 1987)
TG 406 Skin Sensitisation (Updated Guideline, adopted 17th July
1992)
TG 407 Repeated Dose 28-day Oral Toxicity Study in Rodents (Updated
Guideline, adopted 27th July 1995
TG 408 Subchronic Oral Toxicity - Rodent: 90-day Study (Original
Guideline, adopted 12th May 1981)
TG 409 Subchronic Oral Toxicity - Non-Rodent: 90-day Study
(Original Guideline, adopted 12th May 1981)
TG 410 Repeated Dose Dermal Toxicity: 21/28-day Study (Original
Guideline, adopted 12th May 1981)
TG 411 Subchronic Dermal Toxicity: 90-day Study (Original
Guideline, adopted 12th May 1981)
TG 412 Repeated Dose Inhalation Toxicity: 28-day or 14-day Study
(Original Guideline, adopted 12th may 1981)
TG 413 Subchronic Inhalation Toxicity: 90-day Study (Original
Guideline, adopted 12th May 1981
TG 414 Teratogenicity (Original Guideline, adopted 12th May 1981
TG 415 One-Generation Reproduction Toxicity Study (Original
Guideline, adopted 26th May 1983)
TG 416 Two-Generation Reproduction Toxicity Study (Original
Guideline, adopted 26th May 1983
TG 417 Toxicokinetics (Updated Guideline, adopted 4th April 1984)
TG 418 Delayed Neurotoxicity of Organophosphorus Substances
Following Acute Exposure (Updated Guideline, adopted 27th
July 1995)
TG 419 Delayed Neurotoxicity of Organophosphorus Substances: 28-day
Repeated Dose Study (Updated Guideline, adopted 27th July
1995
TG 420 Acute Oral Toxicity - Fixed Dose Method (Original Guideline,
adopted 17th July 1992
TG 421 Reproduction/Developmental Toxicity Screening Test (Original
Guideline, adopted 27th July 1995)
TG 422 Combined Repeated Dose Toxicity Study with the
Reproduction/Developmental Toxicity Screening Test (Original
Guideline, adopted 22nd March 1996)
TG 423 Acute Oral toxicity - Acute Toxic Class Method (Original
Guideline, adopted 22nd March 1996)
TG 424 Neurotoxicity Study in Rodents (Original Guideline, adopted
21st July 1997
TG 451 Carcinogenicity Studies (Original Guideline, adopted 12th
May 1981)
TG 452 Chronic Toxicity Studies (Original Guideline, adopted 12th
May 1981)
TG 453 Combined Chronic Toxicity/Carcinogenicity Studies (Original
Guideline, adopted 12th May 1981
TG 471 Bacterial Reverse Mutation Test (Updated Guideline, adopted
21st July 1997
TG 473 In vitro Mammalian Chromosomal Aberration Test (Updated
Guideline, adopted 21st July 1997
TG 474 Mammalian Erythrocyte Micronucleus Test (Updated Guideline,
adopted 21st July 1997)
TG 475 Mammalian Bone Marrow Chromosomal Aberration Test (Updated
Guideline, adopted 21st July 1997)
TG 476 In vitro Mammalian Cell Gene Mutation Test (Updated
Guideline, adopted 21st July 1997)
TG 477 Genetic Toxicology: Sex-Linked Recessive Lethal Test in
Drosophila melanogaster (Updated Guideline, adopted 4th
April 1984)
TG 478 Genetic Toxicology: Rodent Dominant Lethal Test (Updated
Guideline, adopted 4th April 1984)
TG 479 Genetic Toxicology: In vitro Sister Chromatid Exchange Assay
in Mammalian Cells (Original Guideline, adopted 23rd October
1986)
TG 480 Genetic Toxicology: Saccharomyces cerevisiae, Gene Mutation
Assay (Original Guideline, adopted 23rd October 1986)
TG 481 Genetic Toxicology: Saacharomyces cerevisiae, Miotic
Recombination Assay (Original Guideline, adopted 23rd
October 1986)
TG 482 Genetic Toxicology: DNA Damage and Repair, Unscheduled DNA
Synthesis in Mammalian Cells in vitro (Original Guideline,
adopted 23rd October 1986)
TG 483 Mammalian Spermatogonial Chromosome Aberration Test
(Original Guideline, adopted 21st July 1997) TG 484 Genetic
Toxicology: Mouse Spot Test (Original Guideline, adopted
23rd October 1986)
TG 485 Genetic Toxicology: Mouse Heritable Translocation Assay
(Original Guideline, adopted 23rd October 1986)
TG 486 Unscheduled DNA Synthesis (UDS) Test with Mammalian Liver
Cells in vivo (Original Guideline, adopted 21st July 1997)
2. Draft Test Guidelines
TG 403 Acute Inhalation Toxicity (Draft Updated Guideline, August
1996)a
TG 408 Repeated Dose 90-Day Oral Toxicity Study in Rodents (Draft
Updated Guideline, May 1998, EPOC Document)a
TG 409 Repeated Dose 90-Day Oral Toxicity Study in Non-Rodents
(Draft Updated Guideline, May 1998, EPOC Document)a
TG 414 Prenatal Developmental Toxicity Study (Draft Updated
Guideline, March 1998)a
TG 416 Two-Generation Reproduction Toxicity Study (Draft Updated
Guideline, April 1996)a
TG 425 Acute Oral Toxicity: Up-and-Down Procedure (Draft New
Guideline, May 1998, EPOC Document)a
Somatic Mutation and Recombination Tests (SMART) in Drosophila
melanogaster (Draft New Guideline, May 1994)a
Percutaneous Absorption: in vitro Method (Draft New Guideline, May
1996)a
Percutaneous Absorption: in vivo Method (Draft New Guideline, June
1996)a
Acute Dermal Photoirritation Screening Test (Draft New Guideline,
February 1995)a
Acute Dermal Photoirritation Dose-Response Test (Draft New Guideline,
February 1995)a
In Vitro Syrian Hamster Embryo (SHE) Cell Transformation Assay (Draft
New Guideline, March 1996)a
Acute Dermal Irritation Study in Human Volunteers (Draft New
Guideline, April 1997)a
__________________________
a Available in Portable Document Format or Word 6 Format.
1. RÉSUMÉ
La maîtrise des risques résultant d'une exposition à des produits
chimiques (la sécurité chimique) implique avant tout une évaluation
scientifique - dans le meilleur des cas, quantitative - des effets
potentiels en fonction de l'intensité de l'exposition (l'évaluation du
risque). En s'appuyant sur les résultats de cette évaluation et compte
tenu d'un certain nombre d'autres facteurs, il est possible d'entamer
un processus décisionnel visant à éliminer ou, en cas d'impossibilité,
à réduire au minimum, le ou les risques imputables à la ou aux
substances chimiques en cause (la gestion du risque).
L'évaluation du risque constitue le cadre conceptuel dans lequel
peut s'exercer un processus ordonné d'examen des données permettant
d'apprécier les conséquences sanitaires ou écologiques de l'exposition
à telle ou telle substance. Aux Etats-Unis, l'Académie nationale des
sciences suit, pour ses évaluations du risque, une démarche qui a fait
la preuve de son utilité (US NAS, 1983). Elle distingue quatre phases
distinctes dans le processus d'évaluation: la reconnaissance du
danger, l'évaluation de la relation dose-réponse, l'évaluation de
l'exposition et la caractérisation du risque.
La reconnaissance du danger a pour objet d'apprécier les éléments
qui tendent à prouver l'existence d'effets indésirables pour l'homme,
en s'appuyant sur l'ensemble des données toxicologiques disponibles et
sur tout ce que l'on peut savoir du mode d'action du produit en cause.
Il s'agit essentiellement de répondre à deux questions, à savoir 1) si
l'agent en cause représente un danger pour l'Homme et 2) dans quelles
circonstances ce danger est susceptible de se manifester. La
reconnaissance du danger repose sur l'analyse de diverses données qui
peuvent aller d'observations sur l'Homme à l'étude des relations entre
l'activité de la substance et sa structure. Il doit alors être
possible de se prononcer scientifiquement sur la question de savoir si
la substance à expertiser peut, dans des conditions d'exposition
données, avoir des effets indésirables sur la santé humaine. En
général, les effets toxiques s'observent au niveau d'un ou de
plusieurs organes cibles. Souvent, on s'efforce d'observer les divers
points d'aboutissement de l'action toxique de la substance. On
détermine alors l'effet critique, qui représente habituellement le
premier effet indésirable important à apparaître lorsque la dose
augmente.
L'évaluation de la relation dose-réponse consiste à établir la
relation qui existe entre la dose de produit administrée ou reçue et
la fréquence d'un effet nocif. Pour presque tous les types d'effets
toxiques (c'est-à-dire organospécifiques, neurologiques ou
comportementaux, immunologiques, cancérogènes non génotoxiques,
génésiques ou développementaux), on estime généralement qu'il existe
une dose ou une concentration au-dessous de laquelle aucun effet
indésirable ne se produit (c'est-à-dire qu'il existe un seuil de
toxicité). Pour d'autres types d'effets toxiques, on suppose qu'il
existe une probabilité d'action toxique quelle que soit l'intensité de
l'exposition (autrement dit qu'il n'y a pas de seuil de toxicité). A
l'heure actuelle, cette dernière hypothèse s'applique en général
essentiellement aux effets mutagènes et aux effets cancérogènes
génotoxiques.
Si l'on suppose l'existence d'un seuil (par exemple, dans le cas
d'effets non cancérogènes ou d'effets cancérogènes non génotoxiques),
on a l'habitude de déterminer le niveau d'exposition au-dessous duquel
on estime nulle la probabilité d'effets toxiques et que l'on exprime
par la dose sans effet nocif observable ou NOAEL, compte tenu d'un
certain nombre de facteurs d'incertitude (il s'agit d'une valeur
approchée du seuil de toxicité). On peut aussi déterminer de combien
la dose (la plus faible) sans effet nocif observable dépasse le niveau
d'exposition estimé (c'est-à-dire la "marge de sécurité") en fonction
des diverses sources d'incertitude. C'est une méthode que l'on a pu
souvent qualifier d'"évaluation du degré de sécurité". Par conséquent
la dose que l'on peut considérer en première approximation comme le
seuil de toxicité, c'est-à-dire la NOAEL, constitue la dose critique.
On a toutefois de plus en plus tendance à utiliser la "dose de
référence", une estimation (ou la limite inférieure de l'intervalle de
confiance correspondant), obtenue par modélisation, de la dose
produisant l'effet critique avec une fréquence particulière (par ex.
5%) pour l'évaluation quantitative de la relation dose-réponse dans le
cas de ce genre d'effets.
Il n'y a pas de véritable consensus au sujet de la méthodologie à
adopter pour évaluer le risque dans le cas de substances pour
lesquelles il pourrait ne pas exister de seuil pour l'effet critique
(par exemple les cancérogènes génotoxiques et les mutagènes agissant
au niveau des cellules germinales). De fait, on utilise en pareil cas
un certain nombre de méthodes qui reposent en grande partie sur la
caractérisation de la relation dose-réponse. Dans ces conditions, ce
qui compte, ce sont les points expérimentaux qui définissent la pente
de la courbe dose-réponse (et non pas la NOAEL, qui constitue une
première approximation de la valeur du seuil).
La troisième phase du processus consiste dans l'évaluation de
l'exposition. Elle a pour objet de déterminer la nature et le degré du
contact qui a eu lieu ou qui pourrait avoir lieu avec telle ou telle
substance chimique dans diverses conditions. Différentes méthodes
peuvent être utilisées pour procéder à ce type d'évaluation. En
général il s'agit de méthodes directes ou indirectes comportant la
mesure des concentrations dans l'environnement et celle de
l'exposition individuelle ou de marqueurs biologiques. On fait souvent
appel aussi à des modèles et à des questionnaires. L'évaluation de
l'exposition nécessite la détermination des émissions de produits
chimiques, des voies qu'ils empruntent et de la vitesse de leur
déplacement, de même que leur transformation ou décomposition, afin
d'évaluer la concentration à laquelle les populations humaines ou les
différents compartiments de l'environnement (eau, air, sol) peuvent
être exposés.
Selon le but de l'évaluation, le résultat numérique peut se présenter
sous la forme d'une estimation de l'intensité, de la vitesse,de la
durée ou de la fréquence du contact ou encore d'une estimation de la
dose (quantité de produit qui franchit effectivement la limite). Il
importe de noter que c'est la dose interne, et non le niveau
d'exposition externe, qui détermine l'effet toxique d'une exposition
donnée.
La caractérisation du risque constitue la phase finale du
processus d'évaluation du risque. Elle a pour but de faciliter la
tâche de ceux qui ont la responsabilité de gérer ce risque en leur
fournissant, en langage ordinaire, les données scientifiques
essentielles et les principes de base sur lesquels appuyer leurs
décisions. En particulier, on leur donne une évaluation du risque pour
la santé humaine dans des situations d'exposition appropriées. La
caractérisation du risque revient donc à évaluer et à intégrer les
données scientifiques disponibles pour déterminer la nature,
l'importance - et souvent l'ampleur- du risque biologique ou
écologique qu'une exposition à tel ou tel produit peut faire courir
dans des circonstances précises, compte tenu des incertitudes qui lui
sont attachées.
Par "gestion du risque" on entend l'ensemble des activités à
mettre en oeuvre pour pouvoir décider si le risque associé à une
substance donnée appelle une élimination ou une réduction. Les
stratégies et les options qui s'offrent en la matière peuvent être
classées en gros selon leur nature en réglementaires, non
réglementaires, économiques, conseillées, ou technologiques, les unes
n'excluant pas forcément les autres. Ainsi, les mandats législatifs
(les directives réglementaires), les considérations politiques, les
valeurs socioéconomiques, le coût, la faisabilité technique, les
populations exposées au risque, la durée et l'ampleur du risque et les
conséquences possibles sur les échanges commerciaux internationaux,
constituent toute une panoplie de facteurs dont il pourra être tenu
compte dans la politique ou la réglementation finale. Les déterminants
fondamentaux de la décision tels que la taille de la population, les
ressources, les dépenses à envisager pour atteindre les objectifs de
même que la valeur scientifique de l'évaluation du risque et des
options opérationnelles ultérieures varient considérablement d'un
contexte à l'autre. Il est également admis que la gestion des risques
est une procédure complexe et de nature pluridisciplinaire, qui se
présente rarement sous une forme codifiée ou uniforme, qu'elle est
souvent peu structurée, mais qu'elle est néanmoins susceptible de
prendre en compte des données changeantes émanant des sources les plus
diverses. On estime de plus en plus que la perception du risque et le
problème de la communication sont aussi des éléments importants à
prendre en considération si l'on veut que les décisions soient
acceptées par le public le plus large possible.
Les produits chimiques sont devenus indispensables à l'Homme,
qu'il s'agisse de lui permettre de mener à bien ses activités et son
développement, de prévenir et de combattre de nombreuses maladies et
d'accroître les rendements agricoles. En dépit de tous ces avantages,
les produits chimiques, surtout s'ils sont mal utilisés, peuvent avoir
des effets néfastes sur la santé humaine et sur l'environnement.
L'utilisation généralisée de ces produits dans l'ensemble du monde
augmente le risque d'effets indésirables. On peut s'attendre à ce que
les industries chimiques poursuivent leur croissance dans les pays
développés comme dans les pays en développement. Compte tenu de cela,
l'évaluation et la gestion des risques résultant de l'exposition aux
produits chimiques apparaissent comme des priorités de tout premier
plan dans la recherche d'un développement durable.
1. RESUMEN
El control de los riesgos de exposición a productos químicos
(seguridad química) requiere en primer lugar una evaluación
científica, idealmente cuantitativa, de los efectos potenciales con
determinadas concentraciones de exposición (evaluación del riesgo).
Tomando como base los resultados de la evaluación del riesgo y
teniendo en cuenta otros factores, se puede comenzar un proceso de
adopción de decisiones encaminado a eliminar o, si esto no fuera
posible, reducir al mínimo el riesgo de exposición a los productos
químicos objeto de examen (evaluación del riesgo).
La evaluación del riesgo es un marco conceptual que proporciona
el mecanismo que permite un examen estructurado de la información de
interés para la estimación de los resultados en la salud o en el medio
ambiente. En la realización de las evaluaciones del riesgo, el modelo
de la Academia Nacional de Ciencias ha resultado un instrumento útil
(US NAS, 1983). En este modelo el proceso de evaluación del riesgo se
divide en cuatro etapas distintas: identificación del peligro,
evaluación de la relación dosis-respuesta, evaluación de la exposición
y caracterización del riesgo.
La identificación del peligro tiene por objeto evaluar la
importancia de las pruebas relativas a los efectos adversos en el ser
humano, basándose en la evaluación de todos los datos disponibles
sobre la toxicidad y el mecanismo de acción. Está concebida para
abordar fundamentalmente dos cuestiones:1) si un agente puede
representar un peligro para la salud de los seres humanos y 2) en qué
circunstancias puede manifestarse un peligro identificado. La
identificación del peligro se basa en el análisis de diversos datos,
que pueden ir desde las observaciones en el ser humano hasta el
análisis de las relaciones existentes entre la estructura y la
actividad. El resultado de la práctica de identificación del peligro
es un dictamen científico en cuanto a si el producto químico evaluado
puede, en determinadas condiciones de exposición, causar un efecto
adverso en la salud de los seres humanos. En general, se observa
toxicidad en un órgano destinatario o en más. Con frecuencia se
detectan efectos finales múltiples tras la exposición a un producto
químico concreto. Se determina el efecto crítico, que normalmente es
el primer efecto adverso importante que se produce al aumentar la
dosis.
La evaluación de la relación dosis-respuesta es el proceso de
caracterización de la relación existente entre la dosis de un producto
administrado o recibido y la incidencia de un efecto adverso en la
salud. En la mayor parte de los tipos de efectos tóxicos (es decir,
específicos de órganos, neurológicos/del comportamiento, inmunitarios,
carcinogénesis no genotóxica, en la reproducción o en el desarrollo),
se suele considerar que existe una dosis o concentración por debajo de
la cual no se producen efectos adversos (es decir, un umbral). Para
otros tipos de efectos tóxicos, se supone que existe alguna
probabilidad de peligro en todas las concentraciones de exposición (es
decir, que no existe un umbral). En la actualidad, el último supuesto
se aplica fundamentalmente a la mutagénesis y la carcinogénesis
genotóxica.
Si se supone la existencia de un umbral (por ejemplo, para los
efectos no neoplásicos y para los carcinógenos no genotóxicos),
normalmente se estima que existe un nivel de exposición por debajo del
cual no hay efectos adversos, basado en la concentración sin efectos
adversos observados (NOAEL) (aproximación del umbral) y en factores de
incertidumbre. Otra posibilidad consiste en examinar la magnitud en la
cual la concentración sin efectos adversos observados (o efectos
mínimos) (NOAEL o LOAEL) es superior a la exposición estimada (es
decir, el "margen de seguridad"), teniendo en cuenta distintas fuentes
de incertidumbre. Anteriormente, este método se ha descrito con
frecuencia como una "evaluación de la seguridad". Por consiguiente, es
fundamental la concentración que se puede considerar como una primera
aproximación del umbral, es decir la NOAEL. Sin embargo, en la
evaluación cuantitativa de la relación dosis-respuesta se propone cada
vez más el uso de la "dosis de referencia", estimación derivada de un
modelo (o su límite de confianza más bajo) de un nivel de incidencia
determinado (por ejemplo, del 5%) para el efecto crítico.
No hay un consenso claro sobre la metodología apropiada para la
evaluación del riesgo de los productos químicos sin umbral para el
efecto crítico (es decir, carcinógenos genotóxicos y mutágenos de
células germinales). Es más, en tales casos se han adoptado diversos
métodos basados fundamentalmente en la caracterización de la relación
dosis-respuesta. Por consiguiente, los puntos críticos de los datos
son los que definen la pendiente de la relación dosis-respuesta (más
que la NOAEL, que es la primera aproximación de un umbral).
La tercera etapa en el proceso de evaluación del riesgo es la
evaluación de la exposición, que tiene por objeto determinar la
naturaleza y la amplitud del contacto experimentado o previsto con las
sustancias químicas en distintas condiciones. Se pueden utilizar
numerosos métodos para realizar las evaluaciones de la exposición. En
general, los métodos incluyen técnicas indirectas y directas, que
comprenden la medición de las concentraciones en el medio ambiente y
las exposiciones personales, así como biomarcadores. También se
utilizan con frecuencia cuestionarios y modelos. La evaluación de la
exposición requiere la determinación de las emisiones, las rutas y las
velocidades de desplazamiento de una sustancia y su transformación o
degradación, a fin de estimar las concentraciones a las cuales pueden
estar expuestas poblaciones humanas o las distintas esferas del medio
ambiente (agua, suelo y aire).
En función de la finalidad de una evaluación de la exposición, el
resultado numérico puede ser una estimación de la intensidad, la
velocidad, la duración o la frecuencia de la exposición o la dosis por
contacto (cantidad resultante que realmente cruza la frontera). Para
la evaluación del riesgo basada en la relación dosis-respuesta, el
resultado normalmente incluye una estimación de la dosis. Es
importante señalar que es la dosis interna, no el nivel exposición
externa, la que determina el resultado toxicológico de una exposición
determinada.
La caracterización del riesgo es la última etapa de la evaluación
del riesgo. Está concebida para prestar asistencia a los especialistas
en gestión del riesgo mediante el suministro, en lenguaje sencillo, de
pruebas científicas esenciales y de los fundamentos en relación con el
riesgo que necesitan para adoptar una decisión. En la caracterización
del riesgo se proporcionan estimaciones del riesgo para la salud
humana en los modelos de exposición pertinentes. Así pues, una
caracterización del riesgo es una evaluación e integración de las
pruebas científicas disponibles utilizadas para estimar la naturaleza,
la importancia y con frecuencia la magnitud del riesgo humano y/o para
el medio ambiente, incluidas las incertidumbres pendientes, que
razonablemente se puede estimar que se derivan de la exposición a un
agente concreto del medio ambiente en circunstancias específicas.
El término "gestión del riesgo" comprende todas las actividades
precisas para adoptar una decisión sobre si un riesgo asociado
requiere la eliminación o una reducción necesaria. Las
estrategias/opciones de gestión del riesgo se pueden clasificar a
grandes rasgos como reglamentarias, no reglamentarias, económicas,
consultivas o tecnológicas, que no son excluyentes entre sí. De esta
manera, los mandatos legislativos (orientación reglamentaria), los
aspectos políticos, los valores económicos, el costo, la viabilidad
técnica, las poblaciones con riesgo, la duración y la magnitud del
riesgo, la comparación de los riesgos y las posibles repercusiones en
el comercio entre los países pueden considerarse, en general, como un
amplio abanico de elementos que pueden influir en la formulación final
de políticas o normas. Los factores fundamentales para decisión, como
el tamaño de la población, los recursos, los costos del logro de los
objetivos y la calidad científica de la evaluación del riesgo y las
posteriores decisiones administrativas, varían enormemente del
contexto de una decisión al de otra. Se reconoce asimismo que la
gestión del riesgo es un procedimiento multidisciplinario complejo que
raramente aparece codificado o uniforme y con frecuencia no está
estructurado, pero que puede responder a aportaciones en evolución de
una amplia variedad de fuentes. Cada vez se reconoce con más
frecuencia que la percepción y la comunicación del riesgo son
elementos importantes que también hay que tener en cuenta para lograr
una aceptación pública lo más amplia posible de las decisiones en
materia de gestión del riesgo.
Los productos químicos se han convertido en una parte
indispensable de la vida humana, que sostienen las actividades y el
desarrollo, previenen y combaten numerosas enfermedades y aumentan la
productividad agrícola. A pesar de sus ventajas, los productos
químicos pueden, especialmente cuando se utilizan de manera indebida,
producir efectos adversos en la salud humana y la integridad del medio
ambiente. La aplicación generalizada de productos químicos en todo el
mundo aumenta el potencial de los efectos adversos. Se prevé que
seguirá aumentando el crecimiento de las industrias químicas, tanto en
los países en desarrollo como desarrollados. En esta situación, se
reconoce que la evaluación y la gestión de los riesgos de la
exposición a productos químicos son una de las prioridades más
importantes a la hora de aplicar los principios del desarrollo
sostenible.
Table 3. Qualitative factors affecting risk perception and evaluation (from: US NRC, 1989)
Factor Conditions associated with increased Conditions associated with
increased public concern decreased public concern
Catastrophic potential Fatalities and injuries grouped Fatalities and injuries
in time and space scattered and random
Familiarity Unfamiliar Familiar
Understanding Mechanisms or process not understood Mechanisms or process understood
Controllability (personal) Uncontrollable Controllable
Voluntariness of exposure Involuntary Voluntary
Effects on children Children specifically at risk Children not specifically at risk
Effects manifestation Delayed effects Immediate effects
Effects on future generations Risk to future generations No risk to future generations
Victim identity Identifiable victims Statistical victims
Dread Effects dreaded Effects not dreaded
Trust in institutions Lack of trust in responsible institutions Trust in responsible institutions
Media attention Much media attention Little media attention
Accident history Major and sometimes minor accidents No major or minor accidents
Equity Inequitable distributions of risks and benefits No major or minor accidents
Benefits Unclear benefits Clear benefits
Reversibility Effects irreversible Effects reversible
Origin Caused by human actions or failures Caused by acts of nature or God
Table 4. Characteristics of risk (from: Scala, 1991)
Characteristic Description Level Examples
Knowledge Society's awareness of risk from activity Little known Food Additivies
Much known Alcoholic drinks
Newness Extent of societal experience Old Guns
New Space travel
Voluntariness Does individual have a choice about Not voluntary Crime
exposure to risk Voluntary Rock climbing
Control Can an individual control exposure, Risk not controlled Natural Disasters
protect himself or control consequences by skill or diligence
Risk controlled Smoking
Dreadedness How much is risk or its consequences feared People do not dread Vaccination
People have much dread Nerve gas
Catastrophic potential Chance of widespread disastrous outcome Not likely Sunbathing
Likely War
Equity Are the benefits and risk shared equally Distributed unequally Hazardous Dump
Distributed equally Skiing
drama, wrongdoing, disagreement, conflict and oversimplification,
distortion, and inaccuracy in interpreting technical risk information.
Receiver problems include inaccurate perception of levels of risk,
strong beliefs and opinions that are resistant to change, and demands
for scientific certainty.
There is a clear need to educate the public, including community
leaders, workers and school children, to enhance awareness so that
they can take voluntarily the action required to reduce or avoid risks
associated with exposure to chemicals in the workplace and general
environments (e.g., indoor air pollutants, pesticides and household
chemicals).
6.3.6 Economic factors
Unlike regulation, which involves strict criteria to be enforced
by regulatory agencies, economic approaches to risk management rely
largely on economic incentives to reduce the levels of pollutants
introduced into the environment (Krewski et al., 1989; Somers, 1993).
The OECD since 1972 has espoused the "Polluter Pays Principle"
(PPP) concept, with the goal of maintaining equitable trading
practices by encouraging polluters to reduce emissions. However, it is
recognized that the consumer ultimately pays the cost required to
accomplish environmental improvements. The main types of economic
instruments in use in OECD countries include charges, subsidies,
deposit-refund schemes, market creation arrangements and financial
enforcement incentives (OECD, 1991b). In 1989, the OECD adopted a
Recommendation on the Application of the PPP to Accidental Pollution,
which links the economic principle and the legal principle to damage
compensation (OECD, 1991b).
6.3.6.1 Cost-benefit analyses
Traditionally, risk reduction has not included a thorough
analysis of costs and benefits (Hammond & Coppock, 1990). Indeed,
there is no widely adopted framework for cost-benefit.
As an example, three major categories of costing relationships
are typically employed in risk reduction by the US EPA, depending on
the situation:
a) benefit/cost analysis weighs the cost of control against monetary
benefits of control;
b) risk/benefit analysis weighs the economic benefits of a polluting
activity against the risks to health and the environment;
c) cost-effectiveness analysis accepts the desirability of
regulation and identifies the least-cost solution to achieve a
given goal, such as a pollution discharge standard (Ris & Preuss,
1988).
The US EPA estimated that the annual compliance cost for USA
federal environmental regulations in 1990 was about 2.1% of the gross
national product (GNP of about 6 trillion dollars). This is expected
to increase to approximately 2.8% of the GNP by the year 2000
(ILSI/National Safety Council, 1993). The benefits of regulation such
as improved quality of life and cleaner environment are often
difficult to quantify in contrast to the enormous costs often cited
for regulatory compliance.
There is broad diversity of opinion as to how costs should be
considered in risk management decisions. Key questions include: How
much can society afford to spend to reduce risks? What is an
acceptable cost per life saved? How should costs be factored into
priority-setting processes? Future success in risk management may to a
large degree depend on ways to weigh benefits and costs and to strike
the appropriate balance in defining how fast to pursue risk
regulations (ILSI/National Safety Council, 1993; Wiener, 1993).
6.3.7 Political factors
Political factors often have an impact on national and local
priorities, drafting of regulatory statutes and introduction of
resulting risk reduction measures. Trade barriers and global
competition also have a considerable impact on risk reduction. For
example, in Canada the decision in 1980 to ban the sale of
urea-formaldehyde foam insulation (UFFI) led to unprecedented public
anger (and anxiety and resentment), great government expense, the
longest civil suit in Canadian history, and appreciable political
consequences. After an 8-year legal trial, it was concluded that there
was not sufficient scientific evidence to substantiate the reported
health problems of UFFI home owners (Somers, 1993).
In 1977, the US Food & Drug Administration (FDA), reacting to
studies that reported the artificial sweetening agent saccharin to be
a bladder carcinogen in rodent feeding studies, proposed to ban the
agent under the Delaney Amendment ("zero-risk") requirement. The
Congress of the USA in November 1977, reacting to the overwhelming
public outcry in support of unrestricted use of saccharin, enacted the
Saccharin Study and Labeling Act (SSLA), which prevented the FDA from
banning saccharin based on the information that was then available.
This made it clear that the public is willing to accept certain risks
from food additives if it perceives that the benefits are high enough
and, possibly, that the risks are low enough (Flamm & Lorentzen,
1988).
6.3.8 Regulatory limits
Traditionally, one avenue of protection of human health has been
through the establishment of exposure limits (variously referred to as
standards, quality criteria, etc.). These are established in a
two-step process, the first involving consideration of the
health-based scientific data and the second involving establishment of
regulatory limits, taking into account the health-based recommendation
along with other factors.
Examples of health-based exposure guidelines include the
Acceptable Daily Intake (ADI), Tolerable Daily Intake (TDI),
Provisional Tolerable Weekly Intake (PTWI), and health-based Maximum
Allowable Concentrations (MAC). Acceptable/Tolerable Intakes are the
amounts of a food additive, contaminant, pesticide or veterinary drug
residue, expressed on a body weight basis, that can be ingested for a
lifetime without appreciable risk to health. The term ADI is commonly
used for additives to food since they impart some beneficial
characteristic (and hence are considered "acceptable") while a TDI
commonly refers to environmental contaminants which are undesirable.
Maximum Allowable Concentrations are either a time-weighted average
concentration of a substance in a medium of exposure that does not
present appreciable hazard for continuing exposure or an upper limit
(ceiling value) which, if exceeded, will have adverse consequences for
health. Often, health-based guidelines are considered, along with
other factors (i.e., technological, socioeconomic, feasibility,
enforcement), to develop operational regulatory limits such as the
Maximum Residue Level (MRL) for pesticides or veterinary drugs, MAC in
exposure media and workplaces, occupational Threshold Limit Values
(TLV), Maximum Workplace Concentrations (MAK), Occupational Exposure
Limits (OEL), Air Quality Standards (AQS), Water Quality Standards
(WQS) or Maximum Use Levels.
Some media of (direct and indirect) exposure and associated
limits are listed below:
Food
* limits for food additives, contaminants, pesticide residues,
veterinary drug residues
* limits for certain chemicals in food packaging materials
* limits for additives and contaminants in animal feed
Cosmetics and other consumer products
* limits for additives and contaminants in cosmetic products (these
include soap and toothpaste)
* limits for other consumer products such as children's toys,
paints and solvents
Water
* drinking-water quality standards
* water quality standards for surface water
* water quality standards for fresh water used for fishing
* water quality standards for estuarine and marine waters
* aqueous effluent standards for industrial effluents and sewage
treatment outfall
* guideline limits for the use of waste water in agriculture and
aquaculture
Air
* air quality (ambient or indoor) limits for gases, vapours,
fibres, particulates
* air quality standards for gaseous or smoke emissions from
industries
Occupational
* occupational exposure limits for gases, vapours, dusts, aerosols
in workplace air and substances absorbed through the skin, mucous
membranes or alimentary tract
* regulatory limits for exposure can be based on appropriate
biomarkers
Soil
* limits for certain chemicals in soil
Agricultural chemicals
* limits for certain contaminants in agrochemicals (fertilizers)
* limits for application rates of pesticides
Chemical waste
* limits for disposal of chemicals as waste products
* waste (including liquid and solid)
* chemical (including mixed industrial), dumps, surface water and
deep well injection
* municipal surface and groundwater contamination, use of sludge in
agriculture
* atmospheric effluents and residual ash from incineration
The two stages and their outputs should not be confused. The
outputs are frequently expressed in different units. For example,
considering pesticide residues in food crops, the ADI is a daily dose
expressed in mg/kg body weight (per day being implicit) whereas the
MRL is a concentration on the crop expressed in mg/kg of the produce.
The MRL may be derived on the basis of Good Agricultural Practice and,
if adhered to, would not result in the ADI being exceeded even if all
the designated crop contained the pesticide at the MRL (an unlikely
postulate). Clearly, to arrive at this conclusion requires information
on daily intakes of the commodities carrying the residue.
6.4 Risk management options
Risk managers can intervene at many points:
a) to prevent the process producing the risk
b) to reduce or eliminate exposures
c) to modify the effects
d) to alter perceptions or valuation, through education and public
relations
e) to compensate for damage after the fact (Morgan, 1993).
6.4.1 Risk reduction
Risk reduction goals can vary considerably and can also be
hampered by the fragmented regulatory structure enforcing
environmental laws in many countries. For example, in the USA, the
regulatory approach to risk reduction depends upon whether a chemical
is a food additive, a food contaminant, a pesticide, a drinking-water
contaminant, an air pollutant, or several of these (Rodricks, 1992).
Increasingly, however, national legislation (such as the Canadian
Environmental Protection Act) that allows for introduction of control
measures for chemicals in a variety of media is being introduced.
Essentially, such legislation enables the development of control
measures in the medium that will contribute most significantly to
reduction of risk. The existing substances regulation of the European
Union also provides the opportunity for concerted action based on
evaluation of risks for different scenarios and routes of exposure
(EEC Council Regulation No.793/93) (EC, 1993).
However, there is no clear consensus on what is considered a risk
of concern. While target risk levels are embodied in some national
legislation, other countries recommend that exposure be reduced as low
as possible for effects for which it is assumed that there is no
threshold.
It is also well recognized that different countries, as well as
different agencies within the same country, often come to different
conclusions in the manner in which they judge and manage a health risk
employing basically the same scientific data (Nilsson et al., 1993;
Somers, 1993). Nilsson et al. (1993) found that 11 countries regulated
the same pesticides to different degrees, which should not be too
surprising recognizing the differing economic interests and statutes
(Somers, 1993).
6.4.1.1 Technology-based criteria
Technology-based criteria for risk reduction are not based on
costs, benefits or rights, but rather the level of technology to
control certain risks. Regulations based on these criteria typically
mandate "the best available technology" (BAT) or emissions that are
"as low as reasonably achievable". Such rules can be difficult to
apply because people seldom agree on the definition of "available" or
"reasonably achievable" (Morgan, 1993). Similar difficulties can arise
with the implementation of "good agricultural practice", "technically
achievable" and "as far as may be reasonably practicable".
REFERENCES
Allaby M (1983) A dictionary of the environment. 2nd ed. New York,
NY, New York University Press, p 195.
Allen BC, Crump KS, & Shipp AM (1988) Correlation between carcinogenic
potency of chemicals in animals and humans. Risk Analysis, 8: 531-544.
Anderson D (1990) In vitro models. Drug Saf; 5 Suppl 1:27-39.
Atterwill CK (1995) Alternative method of assessing toxicity. Methods
Mol Biol 43: 1-9.
Axelson O & Westberg H (1992) Introductory note on the concept of
exposure and dose in occupational epidemiology. Am J Ind Med, 21: 3-5.
Bailar JC III, Crouch EAC, Shaikh R, & Spiegelman D (1988) One-hit
models of carcinogenesis: conservative or not? Risk Analysis,
8: 485-497.
Baird SJS, Cohen JT, Graham JD, Shyakhter A & Evans JS (1996)
Noncancer risk assessment: a probabilistic alternative to current
practice. J Human Ecological Risk Assessment 2 (1): 79-102.
Ballantyne B, Marrs T, & Turner P (1993) General and Applied
Toxicology, Vol. 2, Stockton, Stockton Press, pp 1069-1158.
Beaumont JJ & Breslow NJ (1981) Power consideration in epidemiologic
studies of vinyl chloride workers. Am J Epidemiol, 114: 725-734.
Beland FA & Poirier MC (1993) Significance of DNA adduct studies in
animal models for cancer dosimetry and risk assessment. Environ Health
Perspect, 99: 5-10.
Braat L, Bakema AH, de Boer KF, Meijers RM, & Van Minnen JG (1991)
EXPECT: Outline of an integrated model for environmental scenario
analysis and impact asessment (Report No 259102001). Bilthoven,
National Institute of Public Health and Environmental Protection.
Calabrese EJ (1985) Uncertainty factors and interindividual variation.
Regul Toxicol Pharmacol, 5: 190-196.
Calabrese EJ, Beck BD, & Chappell WR (1992) Does the animal-to-human
uncertainty factor incorporate differences in surface area? Regul
Toxicol Pharmacol, 15: 172-179.
Calabrese EJ & Baldwin LA (1994) Improved method for detection of the
NOAEL. Regul Toxicol Pharmacol, 19: 48-50.
Chhabra RS, Huff JE, Schwetz BS & Selkirk J (1990) An overview of
prechronic and chronic toxicity / carcinogenicity experimental designs
and criteria used by the National Toxicology Program. Environ Hlth
Perspect 86: 313-321.
Chen C & Farland W (1991) Incorporating cell proliferation in
quantative cancer risk assessment: approaches, issues, and
uncertainties. Prog Clin Biol Res, 369: 481-499.
Cohrssen JJ & Covello VT (1989) Risk analysis: A guide to principles
and methods for analyzing health and environmental risks. Washington,
DC, Council on Environmental Quality.
Copeland KT, Checkoway H, McMichael AJ, & Holbrook RH (1977) Bias due
to mis-classification in the estimation of relative risk. Am J
Epidemiol, 105: 488-494.
Crump KS, Hoel DG, Langley CH, & Peto R (1976) Fundamental
carcinogenic processes and their implications for low dose risk
assessment. Cancer Research, 36: 2973-2979.
Crump KS (1984) A new method for determining allowable daily intakes.
Fundam Appl Toxicol, 4: 854-871.
Curry P & Iyengar S (1992) Comparison of exposure assessment
guidelines for pesticides. Revs Environ Contam Toxicol, 129: 80-93.
DFG (German National Research Foundation) (1972) [Hazardous working
compounds: Toxicological-occupational explanation of maximum-allowable
concentrations (MAK-Werte)] in German. Senatskommission zur Prüfung
gesundheitlicher Arbeitsstoffe. D Henschler/H Greim (Hrsg.), Wiley-VCH
Doll R & Hill AB (1956) Lung cancer and other causes of death in
relation to smoking - a second report on the mortality of British
doctors. Br Med J, 2: 1071-1077.
Dourson ML & Stara JF (1983) Regulatory history and experimental
support of uncertainty (safety) factors. Regul Toxicol Pharmacol,
3: 224-238.
Dybing E, Sanner T, Roelfzema H, Kroese D & Tennant RW (1997) T25: a
simplified potency index: description of the system and study of
correlations between carcinogenic potency and species/site specificity
and mutagenicity. Pharmacol-Toxicol, Jun, 80 (6): 272-279.
Dybing E & Huitfeldt HS (1992) Species differences in carcinogen
metabolism and interspecies extrapolation. In: Vainio H, Magee PN
McGregor DB, & McMichael AJ ed. Mechanisms of Carcinogenicity in Risk
Identification. Lyon, France, International Agency for Research on
Cancer, pp 501-522.
EC (1990) EEC Note for Guidance: Good Clinical Practice for trials on
medical products in the European Community. Pharmacology and
Toxicology, 67:361-372.
EC (1992) Council Directive 92/32/EEC of 30th April 1992. Amending
for the 7th time Directive 67/548/EEC on the approximation of the
laws, regulations and administrative provision relating to the
classification, packaging and labelling of dangerous substances.
Official Journal of the European Communities L 154.
EC (1993) Council Regulation (EC) 793/93 of 23 March 1993 on the
evaluation and control of the risks of existing substances. Official
Journal of the European Communities L 84.
EC (1994) Commission Regulation (EC) No 1488/94 of 28 June 1994 laying
down the principles for the assessment of risks to man and the
environment of existing substances in accordance with Council
Regulation (EEC) No 793/93. Official Journal of the European
Communities L 161.
EC (1996) Technical Guidance Document in Support of the Commission
Directive 93/67/EEC on Risk assessment for New Notified Substances and
Commission Regulation 1488/94/EEC on Risk assessment for Existing
Chemicals. Brussels, European Comission.
ECETOC (1989) Alternative approaches for the assessment of
reproductive toxicity. Brussels, Belgium, European Chemical Industry
Ecology and Toxicology Centre (Monograph No 12).
ECETOC (1992) Estimating environmental concentrations of chemicals
using fate and exposure models. Brussels, Belgium, European Chemical
Industry Ecology and Toxicology Centre (Technical Report No 50).
ECETOC (1993) Environmental hazard assessment. Brussels, Belgium,
European Chemical Industry Ecology and Toxicology Centre (Technical
Report No 51).
ECETOC/UNEP (1996) Inventory of Critical Reviews on Chemicals.
Brussels, Belgium, European Chemical Industry Ecology and Toxicology
Centre. United Nations Environment Programme. International Register
of Potentially Toxic Chemicals, Geneva.
Environ (1988) Elements of Toxicology and Chemical Risk Assessment.
Revised Edition. Washington, DC, Environ Corporation, 74 pp.
Farmer JH, Kodell RL, & Gaylor DW (1982) Estimation and extrapolation
of tumor probabilities from a mouse bioassay with survival/sacrifice
components. Risk Analysis, 2: 27-34.
Flamm WG & Lehma-McKeeman LD (1991) The human relevance of the renal
tumor-inducing potential of d-limonene in male rats: implications
for risk assessment. Regul Toxicol Pharmacol, 13: 70-86.
Flamm WG & Lorentzen RJ (1988) Quantitative Risk Assessment (QRA): A
special problem in the approval of new products. In: Cothern CR,
Mehlman MA, & Marcus WL ed. Risk Assessment of Industrial and
Environmental Chemicals. Advances in Modern Toxicology. Princeton, NJ,
Princeton Scientific Publ Co, Vol XV, pp.91-108.
Fowle JR (1989) Summary and perspectives: Panel discussion on
toxicological exposure assessment: State of the art. J Am Coll
Toxicol, 8: 865-870.
Fowle JR & Sexton K (1992) EPA priorities for biological research in
environmental health. Environ Hlth Perspec, 98: 235-241.
Fralick RA (1983) The role of epidemiology in defining an occupational
carcinogen. Occup Health Ontario, 4: 2-16.
Frazier JM (1993) In vitro models for toxicological research and
testing. Toxicol Lett, 68: 73-90.
Gaylor DW & Kodell RL (1980) Linear interpolation algorithm for low
dose risk assessment of toxic substances. J Environ Path Toxicol,
4: 305-312.
Gaylor DW, Greenman DL, & Frith CH (1985) Urinary bladder neoplasms
induced in Balb/C female mice with low doses of 2-acetylaminofluorene.
J Environ Pathol Toxicol Oncol, 6: 127-136.
Gaylor DW (1987) Linear-nonparametric upper limits for low dose
extrapolation. In: Proc Biopharm Sec Amer Statis Asso, pp. 63-66.
Gehlbach SH (1982) Interpreting the medical literature a clinician's
guide. Lexington, Mass. Collamore Press, ISBN: 0669045063.
Gelbke HP (1993) New trends in toxicology and their impact on toxicity
testing-scientific and practical aspects. Human Exp Toxicol,
12: 511-515.
Goldstein BD (1990) The problem with the margin of safety: toward the
concept of protection. Risk Analysis, 10: 7-10.
Groopman JD, Cain LG, & Kensler TW (1988) Aflatoxin exposure in human
populations: measurements and relation to cancer. CRC Crit Revs
Toxicol, 19: 113-145.
Gross MA, Fitzhugh OG, & Mantel N (1970) Evaluation of safety for food
additives: an illustration involving the influence of methyl
salicylate on rat reproduction. Biometrics, 26: 181-194.
Hammond PB & Coppock R (1990) Valuing Health Risks, Costs and Benefits
for Environmental Decision Making. In: Report of a Conference. Hammond
PB & Coppock R ed. Washington, DC, National Research Council, National
Academy Press.
Hasemann JK & Clark (1990) Carcinogenicity results for 114 laboratory
animal studies used to asses the predictivity of four in vitro genetic
toxicity assays for rodent carcinogenicity. Environ Mol Mutagen, 16
Suppl 18: 15-31.
Health Canada (1994) Human Health Risk Assessment for Priority
Substances. Environmental Health Directorate, Health Canada, Canada
Communication Group - Publishing, Cat No En40-215/41E, Ottawa, 1994.
Hemminki K, Grzybowska E, Chorazy M, Twardowska-Saucha K, Sroczynski
JW, Putman KL, Randerath K, Phillips DH, Hewer A, & Santella RM (1990)
DNA adducts in humans environmentally exposed to aromatic compounds in
an industrial area of Poland. Carcinogenesis, 11(7): 1229-1231.
Hemminki K (1992) Significance of DNA and protein adducts. In: Vainio
H, Magee PN, McGregor DB, & McMichael AJ ed. Mechanisms of
Carcinogenesis in Risk Identification. Lyon, France, International
Agency for Research on Cancer, pp 525-534.
Hernberg S (1980) Evaluation of epidemiologic studies in assessing the
long-term effects of occupational noxious agents. Scand J Work Environ
Health, 6: 163-169.
Hertel RF (1996) Outline on risk assessment of existing substances in
the European Union. Env Toxicol Pharmacol 2: 93-96.
Hill AB (1965) The environment and disease. Association or causation?
Proc R Soc Med, 58: 295-300.
Hodgson E, Mailman RB, & Chambers JE (1988) Dictionary of Toxicology.
New York, NY, Van Nostrand Reinhold Co, p 154.
Hoel DG (1980) Incorporation of background in dose-response models.
Fed Proc, 39: 73-75.
IARC (1987) IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, No. 42. Lyon, International Agency for Research on Cancer.
IARC (1990) IARC Scientific Publication on Cancer: Causes, Occurrence
and Control, No. 100. Lyon, International Agency for Research on
Cancer.
ICRP (1974) International Commission on Radiological Protection:
Report of the Task Group on Reference Man. ICRP Publication No. 23.
Oxford, Pergamon Press.
ILSI/National Safety Council (1993) Regulating Risk: The Science and
Politics of Risk: A Conference Summary. Washington DC, ILSI Press,
International Life Sciences Institute.
IPCS (1978) Environmental Health Criteria 6: Principles and methods
for evaluating the toxicity of chemicals. Part I. Geneva, Switzerland,
World Health Organization, International Programme on Chemical Safety.
IPCS (1983) Environmental Health Criteria 27: Guidelines on studies in
environmental epidemiology. Geneva, Switzerland, World Health
Organization,. International Programme on Chemical Safety.
IPCS (1984) Environmental Health Criteria 30: Principles for
evaluating health risks to progency associated with exposure to
chemicals during pregnancy. Geneva, Switzerland, World Health
Organization, International Programme on Chemical Safety.
IPCS (1985a) Environmental Health Criteria 46: Guidelines for the
study of genetic effects in human populations. Geneva, Switzerland,
World Health Organization, International Programme on Chemical Safety.
IPCS (1985b) , Environmental Health Criteria 47: Summary report on the
evaluation of short-term in vitro tests for carcinogens
(collaborative study on in vitro tests). Geneva, Switzerland, World
Health Organization, International Programme on Chemical Safety.
IPCS (1985c) Environmental Health Criteria 51: Guide to short-term
tests for detecting mutagenic and carcinogenic chemicals. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1986a) Environmental Health Criteria 57: Principles of
toxicokinetic studies. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1986b) , Environmental Health Criteria 59: Principles for
evaluating health risks from chemicals during infancy and early
childhood: The need for a special approach. Geneva, Switzerland, World
Health Organization, International Programme on Chemical Safety.
IPCS (1986c) Environmental Health Criteria 60: Principles and methods
for the assessment of neurotoxicity associated with exposure to
chemicals. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1987a) Environmental Health Criteria 70: Principles for the
safety assessment of food additives and contaminants in food. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1987b) Environmental Health Criteria 72: Principles of studies
on diseases of suspected chemical aetiology and their prevention.
Geneva, Switzerland, World Health Organization, International
Programme on Chemical Safety.
IPCS (1990a) Environmental Health Criteria 104: Principles for the
toxicological assessment of pesticide residues in food. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1990b), Environmental Health Criteria 109: Summary report on the
evaluation of short-term in vivo tests for carcinogens
(collaborative study on in vivo tests). Geneva, Switzerland, World
Health Organization, International Programme on Chemical Safety.
IPCS (1991) Environmental Health Criteria 119: Principles and methods
for the assessment of nephrotoxicity associated with exposure to
chemicals. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1992a) Environmental Health Criteria 141: Quality management for
chemical testing. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1992b) Environmental Health Criteria 144: Principles for
evaluating the effects of chemicals on the aged population. Geneva,
Switzerland, World Health Organization, International Programme on
Chemical Safety.
IPCS (1993) Environmental Health Criteria 155: Biomarkers and risk
assessment: concepts and principles. Geneva, Switzerland, World Health
Organization, International Programme on Chemical Safety.
IPCS (1994) Environmental Health Criteria 170: Assessing human health
risks to chemicals: derivation of guidance values for health-based
exposure limits. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1996) Environmental Health Criteria 180: Principles and methods
for assessing direct immunotoxicity associated with exposure to
chemicals. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
IPCS (1997) Environmental chemicals and respiratory
hypersensitization. Proceedings of an IPCS Workshop, Oslo, Norway, 29
November December. Environ Toxicol Pharmacol, 2/3 (3-special issue),
77-242.
IPCS (1998) Draft Environmental Health Criteria: Principles of human
exposure assessment. Geneva, Switzerland, World Health Organization,
International Programme on Chemical Safety.
Kelsey JL, Thompson WD & Evans AS (1986) Methods in observational
epidemiology. Boston, MA, USA: Little, Brown and Company.
Konheim CS (1988) Risk communication in the real world. Risk Anal,
8: 367-373.
Kraus NN & Slovic P (1988) Taxonomic analysis of perceived
risk: Modeling individual and group perceptions within homogenous
hazard domains. Risk Anal, 8: 435-455.
Krewski D, Brown C, & Murdoch D (1984) Determining "safe" levels of
exposure: safety factors or mathematical models? Fund Appl Toxicol,
4: S383-S394.
Krewski D, Murdoch D, & Dewanji A (1986) Statistical modeling and
extrapolation of carcinogenesis data. In: Moolgavkar SH & Prentice RL,
ed. Modern Statistical Methods in Chronic Disease Epidemiology. New
York, NY, John Wiley and Sons, pp 259-282.
Krewski D, Somers E, & Birkwood PL (1987) Risk perceptions in a
decision making context. Environ Carcinogen Revs J Environ Sci Hlth,
C5(2): 175-209.
Krewski D, Thorslund T & Whithey J (1989) Carcinogenic risk assessment
of complex mixtures. Toxicol Ind Health 5: 851-867.
Lauwerys R (1984) Basic concepts of human exposure. In: Berlin A,
Draper M, Hemminki K, & Vainio H ed. Monitoring Exposure to
Carcinogenic and Mutagenic Agents. IARC Scientific Publications, Lyon,
France, International Agency for Research on Cancer, No 59, pp. 31-36.
Leisenring W & Ryan L (1992) Statistical properties of the NOAEL.
Regul Toxicol Pharmacol, 15: 161-171.
Lewis SC, Lynch JR, & Nikiforov AI (1990) New approach to deriving
community exposure guidelines from "No-Observed-Adverse-Effect
Levels". Regul Toxicol Pharmacol, 11: 314-330.
Lilienfeld AM & Lilienfeld D (1979) Foundations of Epidemiology. 2nd
ed. New York, NY: Oxford University Press.
Lioy PJ (1990) Assessing total human exposure to contaminants. Environ
Hlth Perspec, 24: 938-945.
Liteplo RG, Meek ME, Gomes R, & Savard S (1994) Inorganic
fluoride: evaluation of risks to health from environmental exposure
to fluoride in Canada. Environ Carcinog & Ecotox Revs.
c12(2): 327-344.
Mausner JS & Kremer S (1985) Epidemiology, 2nd ed. Philadelphia: WB
Saunders Company.
Meek ME, Newhook R, Liteplo RG & Armstrong VC (1994) Approach to
assessment of risk to human health for Priority Substance under the
Canadian Environmental Protection. J Environ Sci Health
C12(2): 105-134.
Merrill RA (1991) Regulatory toxicology. In: Amdur MO, Doull J, &
Klaasen CD ed. Casarett and Doull's Toxicology. 4th edition. New York,
Oxford, Beijing, Singapore, Toronto, Pergamon Press, pp 970-983.
Monro AM (1990) Interspecies comparisons in toxicology: the utility
and futility of plasma concentrations of the test substance. Regul
Toxicol Pharmacol, 12: 137-160.
Monro A (1993) How useful are chronic (life-span) toxicology studies
in rodents in identifying pharmaceuticals that pose a carcinogenic
risk to humans? Adverse Drug React Toxicol Rev, 12: 5-34.
Montesano R, Bartsch H, Vainio H, Wilbourn & Yamasaki H (eds) (1986)
Long-term and short-term assays for carcinogens. A critical appraisal
(IARC Scientific Publications No. 83), Lyon, France, International
Agency for Research on Cancer (IARC).
Moolgavkar SH, Dewanji A, & Venzon DJ (1988) A stochastic two-stage
model for cancer risk assessment. I: The hazard function and the
probability of tumor. Risk Anal, 8:383-392.
Morgan MG (1993) Risk analysis and management. Sci Amer, 269: 32-41.
Munro IC & Morrison AB (1990) Interaction of science and politics in
toxicological regulation. In: Clayson DB, Munro IC, Shubik P, &
Swenberg JA ed. Progress in Predictive Toxicology. Amsterdam, Elsevier
Science Publ BV, pp 373-387.
Murdoch DJ, Krewski D, & Crump KS (1987) Mathematical models of
carcinogenesis. In: Thompson JR & Brown B ed. Cancer modeling, New
York, NY, Marcel Dekker, pp 61-89.
Mushak B (1993) Environmental equity: A new coalition for justice.
Environ Hlth Persp, 101: 478-483.
Nilsson R, Tasheva M, & Jaeger B (1993) Why different regulatory
decisions when the scientific information base is similar?- Human risk
asessment. Regul Toxicol Pharmacol, 17: 292-332.
OECD (1987) Decision-recommendation of the Council on the systematic
investigation of existing chemicals C (87) 90 (Final). Paris, France,
Organization for Economic Cooperation and Development.
OECD (1989) Compendium of Environmental Exposure Assessment Methods
for Chemicals. Paris, France, Organization for Economic Cooperation
and Development (Environmental Monograph No 27).
OECD (1991a) The OECD Workshop on the Application of Simple Models for
Environmental Assessment [Berlin, 11-13 December]. Paris, France,
Organisation for Economic Co-operation and Development.
OECD (1991b) The State of the Environment. Paris, France, Organisation
for Economic Co-operation and Development, pp.253-265.
OECD (1998) OECD Principles on Good Laboratory Practice (as revised in
1997). OECD Series on Principles of Good Laboratory Practice and
compliance monitoring. Number 1. Environment Directorate, Chemicals
Group and Management Committee. Paris, France, Organisation for
Economic Co-operation and Development, ENV/MC/CHEM (98)17.
Otson R, Fellin P, Chan C (1996) Multi-media exposure study: rational
and design. In: Measurement of Toxic and Related Air pollutants
VIP-64. Air & Waste Management Association, One Gate Center, Third
Floor, Pittsburgh, PA 15222, USA. pp 559-566.
Pariza MW (1992) Risk-benefit perception. In: Food Safety Assessment.
Finley JW, Robinson SF, & Armstrong DJ, eds. American Chemical Society
(ACS) Symposium No 484. Washington, DC, American Chemical Society, pp
36-40.
Perera FP (1987) Molecular cancer epidemiology: A new tool in cancer
prevention. J Natl Cancer Inst, 78: 887-898.
Perera FP (1988) The significance of DNA and protein adducts in human
biomonitoring studies. Mutat Res, 205: 255-269.
Peto R (1978) Carcinogenic effects of chronic exposure to very low
levels of toxic substances, Environ Health Perspect, 22: 155-161.
Peto R, Pike MC, Bernstein L, Gold LS & Ames BN (1984) The TD50: a
proposed general convention for the numerical description of the
carcinogenic potency of chemicals in chronic-exposure animal
experiments. Environ Health Perspective 58, 1-8.
Poirier MC & Beland FA (1987) Determination of carcinogen-induced
macromolecular adducts in animals and humans. Prog Exp Tumor Res,
31: 1-10.
Portier CJ (1988) Species correlation of chemical carcinogens. Risk
Anal, Dec 8 (4): 551-553.
Presidential/Congressional Commission on Risk Assessment and Risk
Management (1997a) Framework for Environmental Health Risk Management.
Final Report Vol. 1, Washington, DC, USA.
Presidential/Congressional Commission on Risk Assessment and Risk
Management (1997b) Risk Assessment and Risk Management in Regulatory
Decision-Making. Final Report Vol. 2, Washington, DC, USA.
Price PS, Keenan RE, Swartout JC, Gillis CA, Carlson-Lynch H, Dourson
ML (1997) An approach for modeling noncancer dose responses with an
emphasis on uncertainty. Risk Anal 17(4): 427-437.
Purchase (1986) Unpredictability: an essay in toxicology. Food Chem
Toxicol, Apr 24 (4): 343-349.
Renwick AG (1993a) Toxicokinetics. In: Ballantyne B, Marrs T, & Turner
P ed. General & Applied Toxicology. New York, Stockton Press, Vol 1,
pp 121-151.
Renwick AG (1993b) Data-derived safety factors for the evaluation of
food additives and environmental contaminants. Fd Addit Contam,
10: 275-305.
Ricci PF & Cox LA (1987) Acceptability of chronic health risks. Toxic
Law Rept, 1: 986-1001.
Ris CH & Preuss PW (1988) Risk assessment and risk management : A
process. In: Cothern CR, Mehlman MA, & Marcus WL ed. Risk Assessment
and Risk Management of Industrial and Environmental Chemicals.
Princeton, NJ, Princeton Scientific Publ Co, Vol XV, pp 1-21.
RIVM (1994) Uniform Systems for the Evaluation of Substances (USES),
version 1.0. The Hague, The Netherlands, Ministry of Housing, Spatial
Planning and Environment (Distribution no 11144/150).
Rodricks JV (1992) Calculated Risks. Cambridge, MA, Cambridge
University Press.
Rothman KT (1986) Modern Epidemiology. Boston, MA, USA: Little, Brown
and Company.
Sandman PM, Muller PM, Johnson BB, & Weinstein NN (1993) Agency
communication, community outrage and perception of risk. Three
simulation experiments. Risk Anal, 13: 585 592.
Scala RA (1991) Risk assessment. In: Amdur MO, Doull J, & Klaasen CD,
ed. Casarett and Doull's Toxicology. 4th edition, New York, Oxford,
Beijing, Frankfurt, Sao Paulo, Sydney, Tokyo, Toronto, Pergamon Press,
pp 985-996.
Seidman BC, Brown SL, De Rosa CT, & Mumtaz MM (1991) Sensitive and
Hyper-susceptible populations: Risk assessment considerations for
exposure to single chemicals and chemical mixtures. In: Zervos C ed.
Risk Analysis. New York, NY, Plenum Press, pp 305-313.
Sexton K, Callahan MA, & Ryan EF (1995) Estimating exposure and dose
to characterize health risks: the role of human tissue monitoring in
exposure assessment. Env Health Health, Suppl 3: 13-30.
Shuker DEG (1989) Nucleic acid-carcinogen adducts in human dosimetry.
Arch Toxicol, (Suppl 13): 55-65.
Silbergeld EK (1993) Risk assessment: The perspective experience of US
environmentalists. Environ Hlth Persp, 101: 100-104.
Singer E & Endreny PM (1993) Reporting on Risk. New York, Russel Sage
Foundation.
Skipper PI & Tannenbaum SR (1990) Protein adducts in the molecular
dosimetry of chemical carcinogens. Carcinogenesis, 11: 507-518.
Slovic P (1987) Perceptions of risk. Science, 236: 280-284.
Slovic P (1993) Perceived risk, trust and democracy. Risk Anal,
13: 675-682.
Somers E (1987) Making decisions with numbers. Regul Toxicol
Pharmacol, 8: 35-42.
Somers E (1993) Perspectives on Risk Management. [Presented at
Managing Risks to Life and Health Meeting, 18 October]. Ottawa, Royal
Society of Canada.
Stayner LT & Bailer AJ (1993) Comparing toxicologic and epidemiologic
studies. Methylene chloride - a case study. Risk Anal, Dec 13
(6): 667-673.
Stern RM (1986) Analysis of the decision making process in chemical
safety. Sci Total Environ, 51: 27-62.
Susser M (1977) Judgement and causal inference: criteria in
epidemiologic studies. Am J Epidemiol, 105: 1- 15.
Tennant RW, Margolin BH, Shelby MD, Zeiger E, Haseman JK, Spalding J,
Caspary W, Resnick M, Stasiewicz S, Anderson B, et al. (1987)
Prediction of chemical carcinogenicity in rodents from in vitro
genetic toxicity assays. Science 236: 933-941.
Travis CC, Pack SA, Saulsbury AW & Yambert MW (1990a) Prediction of
carcinogenic potency from toxicological data. Mutal Res, May 241
(1): 21-36.
Travis CC, Saulsbury AW & Pack SA (1990b) Prediction of cancer potency
using a battery of mutation and toxicity data. Mutagenesis, May 5
(3): 213-219.
Travis CC, Wang LA & Waehner MJ (1991) Quantitative correlation of
carcinogenic potency with four different classes of short-term test
data. Mutagenesis, Sep 6 (5): 353-360.
UK DOH (1991) (Department of Health) Guidelines for the evaluation of
chemicals for carcinogenicity. Committee on Carcinogenicity of
Chemicals in Food, Consumer Products and the Environment. London, Her
Majesty's Stationary Office (HMSO).
UK DOH (1995) (Department of Health) Annual Report of the Committees
on Toxicity, Mutagenicity, Carcinogenicity of Chemicals in Food,
Consumer Products and the Environment. London, Her Majesty's
Stationary Office (HMSO).
UK HSE (1989) Health and Safety Executive. Quantified risk
assessment: Its input to decision making. London, Her Majesty's
Stationery Office (HMSO).
Ulfvarson U (1992) Validation of exposure information in occupational
epidemiology. Am J Ind Med, 21: 125-132.
US EPA (1984) Study of carbon monoxide exposure on residents of
Washington, DC, and Denver, CO. Environmental Monitoring Systems
Laboratory, Office of Research and Development, Research Triangle
Park, NC (EPA-600/S4-84/031, NTIS PB84-183516).
US EPA (1986a) Guidelines for carcinogen risk assessment. Fed Reg,
51: 33992-34003.
US EPA (1986b) Guidelines for Estimating Exposures. Fed Reg, 51
(185): 34042-34054.
US EPA (1987a) The total exposure assessment methodology (TEAM) study.
Vol I: Summary and analysis. Office of Acid Deposition, Environmental
Monitoring and Quality Assurance, Office of Research and Development,
Washington, DC, US Environmental Protection Agency
(EPA-600/6-87/002a).
US EPA (1987b) Pesticide Assessment Guidelines; Subdivision
U-Applicator Exposure. Washington, DC, US Environmental Protection
Agency (EPA PB 87-133286).
US EPA (1992) Guidelines for exposure assessment. Washington, DC, US
Environmental Protection Agency, Federal Register, 57:22888.
US EPA (1996a) Risk Characterization: Practical Guidance for
NCEA - Washington Based Risk Assessors, National Center for
Environmental Assessment, Washington, DC, US Environmental Protection
Agency (NCEAW-0105).
US EPA (1996b) Proposed Guidelines for Carcinogen Risk Assessment.
Washington, DC, US Environmental Protection Agency, Federal Register.
6(79): 17960-18011.
US EPA (1996c) Exposure Factors Handbook. Washington, DC, US
Environmental Protection Agency EPA/600P-95/002Bc, Review Draft.
Update to exposure Factors Handbook EPA/600/8-89/043 - May 1989.
US FDA (1993) Food and Drug Administration: Toxicological Principles
for the Safety Assessment of Direct Food Additives and Color Additives
Used in Food. "Redbook II" Draft. Washington, DC, Center for Food
Safety and Applied Nutrition, US Food and Drug Administration.
US NAS (1983) National Academy of Science: Risk Assessment in the
Federal Government: Managing the Process. National Research Council,
Committee on the Institutional Means for Assessment of Risks to Public
Health. Washington DC, National Academy Press, pp 1-50.
US NRC (1989) National Research Council: Improving Risk Communication.
Washington, DC, National Academy Press.
US NRC (1990) National Research Council: Human exposure assessment for
airborne pollutants: Advances and applications. Committee on Advances
in Assessing Human Exposure to Airborne Pollutants, Committee on
Geosciences, Environment, and Resources. Washington, DC, National
Academy Press.
US NRC (1992) National Research Council: Biological Markers in
Immunotoxicology. Washington, DC, National Academy Press.
Van Eijndhoven JCM, Wetering RAPM, Worrel CW, de Boer J, Van der Pligt
J, & Stallen PJM (1994) Risk communication in the Netherlands: The
monitored introduction of the EC "Post-Seveso" Directive. Risk Anal,
14: 87-96.
Van Ryzin J (1980) Quantitative risk assessment. J Occup Med,
22: 321-326.
WHO (1986) Field Surveys of Exposure to Pesticides. Standard Protocol.
Toxicol Lett, 33: 2234-235.
WHO (1993) Guidelines for drinking-water quality, Recommendations.
Geneva, Switzerland, World Health Organization, Vol 1, 30 pp.
Wiener JO (1993) Risk assessment and cost-benefit analyses: In the
public interst? Environ Hlth Perspect, 101: 408-409.
Wilson R (1990) Analyzing the daily risks of life. In: Readings in
Risk. Glickman TS & Gough M ed. Washington, DC, Resources for the
Future, pp 55-59.
Wilson R & Crouch EAC (1987) Risk assessment and comparisons: An
introduction. Science, 236: 267-270.
Wogan GN (1989) Markers of Exposure to Carcinogens: Methods for Human
Biomonitoring. J Am Coll Toxicol, 8: 871-881.
Wogan GN (1992) Molecular epidemiology in cancer risk and prevention:
Recent progress and avenues for future research. Environ Hlth
Perspect, 98: 167-178.
Zeiger E, Haseman JK, Shelby MD, Margolin BH & Tennant RW (1990)
Evalution of four in vitro genetic toxicity tests for predicting
rodent carcinogenecity: confirmation of earlier results with 41
additional chemicals. Environ Mol Mutagen, 16 Suppl 18: 1-14.
Zimmerman R (1993) Social equity and environmental risk. Risk Anal,
13: 649-666.
APPENDIX 1. PREAMBLE TO THE IARC MONOGRAPHS
The Preamble to the Monographs sets out the objective and scope
of the evaluation programme, the procedures used when making
assessments, and the types of evidence considered and criteria used in
reaching the final evaluations. The list of contents is given here as
is the full text referring to the Background and Evaluation sections.
Full text of the Preamble should always be used when referring to the
list of evaluations provided.
Background
In 1969, the International Agency for Research on Cancer (IARC)
initiated a programme to evaluate the carcinogenic risk of chemicals
to humans and to produce monographs on individual chemicals. The
Monographs programme has since been expanded to include
consideration of exposures to complex mixtures of chemicals (which
occur, for example, in some occupations and as a result of human
habits) and of exposures to other agents, such as radiation and
viruses. With Supplement 6 (IARC, 1987a), the title of the series was
modified from IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans to IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans, in order to reflect the widened
scope of the programme.
The criteria established in 1971 to evaluate carcinogenic risk to
humans were adopted by the working groups whose deliberations resulted
in the first 16 volumes of the IARC Monographs series. Those
criteria were subsequently updated by further ad-hoc working groups
(IARC, 1977, 1978, 1979, 1982, 1983, 1987b, 1988, 1991; Vainio et al.,
1992).
Evaluation
Evaluations of the strength of the evidence for carcinogenicity
arising from human and experimental animal data are made, using
standard terms.
It is recognized that the criteria for these evaluations,
described below, cannot encompass all of the factors that may be
relevant to an evaluation of carcinogenicity. In considering all of
the relevant scientific data, the Working Group may assign the agent,
mixture or exposure circumstance to a higher or lower category than a
strict interpretation of these criteria would indicate.
(a) Degrees of evidence for carcinogenicity in humans and in
experimental animals and supporting evidence
These categories refer only to the strength of the evidence that
an exposure is carcinogenic and not to the extent of its carcinogenic
activity (potency) nor to the mechanisms involved. A classification
may change as new information becomes available.
An evaluation of degree of evidence, whether for a single agent
or a mixture, is limited to the materials tested, as defined
physically, chemically or biologically. When the agents evaluated are
considered by the Working Group to be sufficiently closely related,
they may be grouped together for the purpose of a single evaluation of
degree of evidence.
(i) Carcinogenicity in humans
The applicability of an evaluation of the carcinogenicity of a
mixture, process, occupation or industry on the basis of evidence from
epidemiological studies depends on the variability over time and place
of the mixtures, processes, occupations and industries. The Working
Group seeks to identify the specific exposure, process or activity
which is considered most likely to be responsible for any excess risk.
The evaluation is focused as narrowly as the available data on
exposure and other aspects permit.
The evidence relevant to carcinogenicity from studies in humans
is classified into one of the following categories:
Sufficient evidence of carcinogenicity: The Working Group considers
that a causal relationship has been established between exposure to
the agent, mixture or exposure circumstance and human cancer. That is,
a positive relationship has been observed between the exposure and
cancer in studies in which chance, bias and confounding could be ruled
out with reasonable confidence.
Limited evidence of carcinogenicity: A positive association has been
observed between exposure to the agent, mixture or exposure
circumstance and cancer for which a causal interpretation is
considered by the Working Group to be credible, but chance, bias or
confounding could not be ruled out with reasonable confidence.
Inadequate evidence of carcinogenicity: The available studies are of
insufficient quality, consistency or statistical power to permit a
conclusion regarding the presence or absence of a causal association,
or no data on cancer in humans are available.
Evidence suggesting lack of carcinogenicity: There are several
adequate studies covering the full range of levels of exposure that
human beings are known to encounter, which are mutually consistent in
not showing a positive association between exposure to the agent,
mixture or exposure circumstance and any studied cancer at any
observed level of exposure. A conclusion of evidence suggesting lack
of carcinogenicity is inevitably limited to the cancer sites,
conditions and levels of exposure and length of observation covered by
the available studies. In addition, the possibility of a very small
risk at the levels of exposure studied can never be excluded.
In some instances, the above categories may be used to classify
the degree of evidence related to carcinogenicity in specific organs
or tissues.
(ii) Carcinogenicity in experimental animals
The evidence relevant to carcinogenicity in experimental animals
is classified into one of the following categories:
Sufficient evidence of carcinogenicity: The Working Group considers
that a causal relationship has been established between the agent or
mixture and an increased incidence of malignant neoplasms or of an
appropriate combination of benign and malignant neoplasms in (a) two
or more species of animals or (b) in two or more independent studies
in one species carried out at different times or in different
laboratories or under different protocols.
Exceptionally, a single study in one species might be considered
to provide sufficient evidence of carcinogenicity when malignant
neoplasms occur to an unusual degree with regard to incidence, site,
type of tumour or age at onset.
Limited evidence of carcinogenicity: The data suggest a carcinogenic
effect but are limited for making a definitive evaluation because,
e.g. (a) the evidence of carcinogenicity is restricted to a single
experiment; or (b) there are unresolved questions regarding the
adequacy of the design, conduct or interpretation of the study; or (c)
the agent or mixture increases the incidence only of benign neoplasms
or lesions of uncertain neoplastic potential, or of certain neoplasms
which may occur spontaneously in high incidences in certain strains.
Inadequate evidence of carcinogenicity: The studies cannot be
interpreted as showing either the presence or absence of a
carcinogenic effect because of major qualitative or quantitative
limitations, or no data on cancer in experimental animals are
available.
Evidence suggesting lack of carcinogenicity: Adequate studies
involving at least two species are available which show that, within
the limits of the tests used, the agent or mixture is not
carcinogenic. A conclusion of evidence suggesting lack of
carcinogenicity is inevitably limited to the species, tumour sites and
levels of exposure studied.
(b) Other data relevant to the evaluation of carcinogenicity and
its mechanisms
Other evidence judged to be relevant to an evaluation of
carcinogenicity and of sufficient importance to affect the overall
evaluation is then described. This may include data on preneoplastic
lesions, tumour pathology, genetic and related effects,
structure-activity relationships, metabolism and pharmacokinetics,
physicochemical parameters and analogous biological agents.
Data relevant to mechanisms of the carcinogenic action are also
evaluated. The strength of the evidence that any carcinogenic effect
observed is due to a particular mechanism is assessed, using terms
such as weak, moderate or strong. Then, the Working Group assesses if
that particular mechanism is likely to be operative in humans. The
strongest indications that a particular mechanism operates in humans
come from data on humans or biological specimens obtained from exposed
humans. The data may be considered to be especially relevant if they
show that the agent in question has caused changes in exposed humans
that are on the causal pathway to carcinogenesis. Such data may,
however, never become available, because it is at least conceivable
that certain compounds may be kept from human use solely on the basis
of evidence of their toxicity and/or carcinogenicity in experimental
systems.
For complex exposures, including occupational and industrial
exposures, the chemical composition and the potential contribution of
carcinogens known to be present are considered by the Working Group in
its overall evaluation of human carcinogenicity. The Working Group
also determines the extent to which the materials tested in
experimental systems are related to those to which humans are exposed.
(c) Overall evaluation
Finally, the body of evidence is considered as a whole, in order
to reach an overall evaluation of the carcinogenicity to humans of an
agent, mixture or circumstance of exposure.
An evaluation may be made for a group of chemical compounds that
have been evaluated by the Working Group. In addition, when supporting
data indicate that other, related compounds for which there is no
direct evidence of capacity to induce cancer in humans or in animals
may also be carcinogenic, a statement describing the rationale for
this conclusion is added to the evaluation narrative; an additional
evaluation may be made for this broader group of compounds if the
strength of the evidence warrants it.
The agent, mixture or exposure circumstance is described
according to the wording of one of the following categories, and the
designated group is given. The categorization of an agent, mixture or
exposure circumstance is a matter of scientific judgement, reflecting
the strength of the evidence derived from studies in humans and in
experimental animals and from other relevant data.
* Group 1: The agent (mixture) is carcinogenic to humans. The
exposure circumstance entails exposures that are carcinogenic
to humans.
This category is used when there is sufficient evidence of
carcinogenicity in humans. Exceptionally, an agent (mixture) may be
placed in this category when evidence in humans is less than
sufficient but there is sufficient evidence of carcinogenicity in
experimental animals and strong evidence in exposed humans that the
agent (mixture) acts through a relevant mechanism of carcinogenicity.
* Group 2
This category includes agents, mixtures and exposure
circumstances for which, at one extreme, the degree of evidence of
carcinogenicity in humans is almost sufficient, as well as those for
which, at the other extreme, there are no human data but for which
there is evidence of carcinogenicity in experimental animals. Agents,
mixtures and exposure circumstances are assigned to either group 2A
(probably carcinogenic to humans) or group 2B (possibly carcinogenic
to humans) on the basis of epidemiological and experimental evidence
of carcinogenicity and other relevant data.
* Group 2A: The agent (mixture) is probably carcinogenic to
humans.
The exposure circumstance entails exposures that are probably
carcinogenic to humans.
This category is used when there is limited evidence of
carcinogenicity in humans and sufficient evidence of carcinogenicity
in experimental animals. In some cases, an agent (mixture) may be
classified in this category when there is inadequate evidence of
carcinogenicity in humans and sufficient evidence of carcinogenicity
in experimental animals and strong evidence that the carcinogenesis is
mediated by a mechanism that also operates in humans. Exceptionally,
an agent, mixture or exposure circumstance may be classified in this
category solely on the basis of limited evidence of carcinogenicity in
humans.
* Group 2B: The agent (mixture) is possibly carcinogenic to
humans.
The exposure circumstance entails exposures that are possibly
carcinogenic to humans.
This category is used for agents, mixtures and exposure circumstances
for which there is limited evidence of carcinogenicity in humans and
less than sufficient evidence of carcinogenicity in experimental
animals. It may also be used when there is inadequate evidence of
carcinogenicity in humans but there is sufficient evidence of
carcinogenicity in experimental animals. In some instances, an agent,
mixture or exposure circumstance for which there is
inadequate evidence of carcinogenicity in humans but
limited evidence of carcinogenicity in experimental animals together
with supporting evidence from other relevant data may be placed in
this group.
* Group 3: The agent (mixture or exposure circumstance) is not
classifiable as to its carcinogenicity to humans.
This category is used most commonly for agents, mixtures and
exposure circumstances for which the evidence of carcinogenicity is
inadequate in humans and inadequate or limited in experimental
animals.
Exceptionally, agents (mixtures) for which the evidence of
carcinogenicity is inadequate in humans but sufficient in experimental
animals may be placed in this category when there is strong evidence
that the mechanism of carcinogenicity in experimental animals does not
operate in humans.
Agents, mixtures and exposure circumstances that do not fall into
any other group are also placed in this category.
* Group 4: The agent (mixture) is probably not carcinogenic to
humans.
This category is used for agents or mixtures for which there is
evidence suggesting lack of carcinogenicity in humans and in
experimental animals. In some instances, agents or mixtures for which
there is inadequate evidence of carcinogenicity in humans but
evidence suggesting lack of carcinogenicity in experimental animals,
consistently and strongly supported by a broad range of other relevant
data, may be classified in this group.
References:
IARC (1977) IARC Monographs Programme on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans. Preamble (IARC intern.
tech. Rep. No. 77/002), Lyon
IARC (1978) Chemicals with Sufficient Evidence
of Carcinogenicity in Experimental Animals-IARC Monographs
Volumes 1-17 (IARC intern. tech. Rep. No. 78/003), Lyon
IARC (1979) Criteria to Select Chemicals for IARC Monographs (IARC
intern. tech. Rep. No. 79/003), Lyon
IARC (1982) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Supplement 4,
Chemicals, Industrial Processes and Industries Associated with
Cancer in Humans (IARC Monographs, Volumes 1 to 29), Lyon
IARC (1983) Approaches to Classifying Chemical Carcinogens According
to Mechanism of Action (IARC intern. tech. Rep. No. 83/001), Lyon
IARC (1987a) IARC Monographs on the Evaluation of Carcinogenic Risks
to Humans, Supplement 6, Genetic and Related Effects: An Updating
of Selected IARC Monographs from Volumes 1 to 42, Lyon
IARC (1987b) IARC Monographs on the Evaluation of Carcinogenic Risks
to Humans, Supplement 7, Overall Evaluations of Carcinogenicity:
An Updating of IARC Monographs Volumes 1 to 42, Lyon
IARC (1988) Report of an IARC Working Group to Review the Approaches
and Processes Used to Evaluate the Carcinogenicity of Mixtures and
Groups of Chemicals (IARC intern. tech. Rep.No. 88/002), Lyon
IARC (1991) A Consensus Report of an IARC Monographs Working Group
on the Use of Mechanisms of Carcinogenesis in Risk Identification
(IARC intern. tech. Rep. No. 91/002), Lyon
Vainio, H., Magee, P., McGregor, D. & McMichael, A., eds (1992)
Mechanisms of Carcinogenesis in Risk Identification (IARC Scientific
Publications No. 116), Lyon, IARC
APPENDIX 2. OECD'S GUIDELINES FOR THE TESTING OF CHEMICALS
(from http://www.oecd.org/ehs/test/health.htm)
1. Adopted Test Guidelines
TG 401 Acute Oral Toxicity (Updated Guideline, adopted 24th
February 1987)
TG 402 Acute Dermal Toxicity (Updated Guideline, adopted 24th
February 1987)
TG 403 Acute Inhalation Toxicity (Original Guideline, adopted 12th
May 1981)
TG 404 Acute Dermal Irritation/Corrosion (Updated Guideline,
adopted 17th July 1992)
TG 405 Acute Eye Irritation/Corrosion (Updated Guideline, adopted
24th February 1987)
TG 406 Skin Sensitisation (Updated Guideline, adopted 17th July
1992)
TG 407 Repeated Dose 28-day Oral Toxicity Study in Rodents (Updated
Guideline, adopted 27th July 1995
TG 408 Subchronic Oral Toxicity - Rodent: 90-day Study (Original
Guideline, adopted 12th May 1981)
TG 409 Subchronic Oral Toxicity - Non-Rodent: 90-day Study
(Original Guideline, adopted 12th May 1981)
TG 410 Repeated Dose Dermal Toxicity: 21/28-day Study (Original
Guideline, adopted 12th May 1981)
TG 411 Subchronic Dermal Toxicity: 90-day Study (Original
Guideline, adopted 12th May 1981)
TG 412 Repeated Dose Inhalation Toxicity: 28-day or 14-day Study
(Original Guideline, adopted 12th may 1981)
TG 413 Subchronic Inhalation Toxicity: 90-day Study (Original
Guideline, adopted 12th May 1981
TG 414 Teratogenicity (Original Guideline, adopted 12th May 1981
TG 415 One-Generation Reproduction Toxicity Study (Original
Guideline, adopted 26th May 1983)
TG 416 Two-Generation Reproduction Toxicity Study (Original
Guideline, adopted 26th May 1983
TG 417 Toxicokinetics (Updated Guideline, adopted 4th April 1984)
TG 418 Delayed Neurotoxicity of Organophosphorus Substances
Following Acute Exposure (Updated Guideline, adopted 27th
July 1995)
TG 419 Delayed Neurotoxicity of Organophosphorus Substances: 28-day
Repeated Dose Study (Updated Guideline, adopted 27th July
1995
TG 420 Acute Oral Toxicity - Fixed Dose Method (Original Guideline,
adopted 17th July 1992
TG 421 Reproduction/Developmental Toxicity Screening Test (Original
Guideline, adopted 27th July 1995)
TG 422 Combined Repeated Dose Toxicity Study with the
Reproduction/Developmental Toxicity Screening Test (Original
Guideline, adopted 22nd March 1996)
TG 423 Acute Oral toxicity - Acute Toxic Class Method (Original
Guideline, adopted 22nd March 1996)
TG 424 Neurotoxicity Study in Rodents (Original Guideline, adopted
21st July 1997
TG 451 Carcinogenicity Studies (Original Guideline, adopted 12th
May 1981)
TG 452 Chronic Toxicity Studies (Original Guideline, adopted 12th
May 1981)
TG 453 Combined Chronic Toxicity/Carcinogenicity Studies (Original
Guideline, adopted 12th May 1981
TG 471 Bacterial Reverse Mutation Test (Updated Guideline, adopted
21st July 1997
TG 473 In vitro Mammalian Chromosomal Aberration Test (Updated
Guideline, adopted 21st July 1997
TG 474 Mammalian Erythrocyte Micronucleus Test (Updated Guideline,
adopted 21st July 1997)
TG 475 Mammalian Bone Marrow Chromosomal Aberration Test (Updated
Guideline, adopted 21st July 1997)
TG 476 In vitro Mammalian Cell Gene Mutation Test (Updated
Guideline, adopted 21st July 1997)
TG 477 Genetic Toxicology: Sex-Linked Recessive Lethal Test in
Drosophila melanogaster (Updated Guideline, adopted 4th
April 1984)
TG 478 Genetic Toxicology: Rodent Dominant Lethal Test (Updated
Guideline, adopted 4th April 1984)
TG 479 Genetic Toxicology: In vitro Sister Chromatid Exchange Assay
in Mammalian Cells (Original Guideline, adopted 23rd October
1986)
TG 480 Genetic Toxicology: Saccharomyces cerevisiae, Gene Mutation
Assay (Original Guideline, adopted 23rd October 1986)
TG 481 Genetic Toxicology: Saacharomyces cerevisiae, Miotic
Recombination Assay (Original Guideline, adopted 23rd
October 1986)
TG 482 Genetic Toxicology: DNA Damage and Repair, Unscheduled DNA
Synthesis in Mammalian Cells in vitro (Original Guideline,
adopted 23rd October 1986)
TG 483 Mammalian Spermatogonial Chromosome Aberration Test
(Original Guideline, adopted 21st July 1997) TG 484 Genetic
Toxicology: Mouse Spot Test (Original Guideline, adopted
23rd October 1986)
TG 485 Genetic Toxicology: Mouse Heritable Translocation Assay
(Original Guideline, adopted 23rd October 1986)
TG 486 Unscheduled DNA Synthesis (UDS) Test with Mammalian Liver
Cells in vivo (Original Guideline, adopted 21st July 1997)
2. Draft Test Guidelines
TG 403 Acute Inhalation Toxicity (Draft Updated Guideline, August
1996)a
TG 408 Repeated Dose 90-Day Oral Toxicity Study in Rodents (Draft
Updated Guideline, May 1998, EPOC Document)a
TG 409 Repeated Dose 90-Day Oral Toxicity Study in Non-Rodents
(Draft Updated Guideline, May 1998, EPOC Document)a
TG 414 Prenatal Developmental Toxicity Study (Draft Updated
Guideline, March 1998)a
TG 416 Two-Generation Reproduction Toxicity Study (Draft Updated
Guideline, April 1996)a
TG 425 Acute Oral Toxicity: Up-and-Down Procedure (Draft New
Guideline, May 1998, EPOC Document)a
Somatic Mutation and Recombination Tests (SMART) in Drosophila
melanogaster (Draft New Guideline, May 1994)a
Percutaneous Absorption: in vitro Method (Draft New Guideline, May
1996)a
Percutaneous Absorption: in vivo Method (Draft New Guideline, June
1996)a
Acute Dermal Photoirritation Screening Test (Draft New Guideline,
February 1995)a
Acute Dermal Photoirritation Dose-Response Test (Draft New Guideline,
February 1995)a
In Vitro Syrian Hamster Embryo (SHE) Cell Transformation Assay (Draft
New Guideline, March 1996)a
Acute Dermal Irritation Study in Human Volunteers (Draft New
Guideline, April 1997)a
__________________________
a Available in Portable Document Format or Word 6 Format.
1. RÉSUMÉ
La maîtrise des risques résultant d'une exposition à des produits
chimiques (la sécurité chimique) implique avant tout une évaluation
scientifique - dans le meilleur des cas, quantitative - des effets
potentiels en fonction de l'intensité de l'exposition (l'évaluation du
risque). En s'appuyant sur les résultats de cette évaluation et compte
tenu d'un certain nombre d'autres facteurs, il est possible d'entamer
un processus décisionnel visant à éliminer ou, en cas d'impossibilité,
à réduire au minimum, le ou les risques imputables à la ou aux
substances chimiques en cause (la gestion du risque).
L'évaluation du risque constitue le cadre conceptuel dans lequel
peut s'exercer un processus ordonné d'examen des données permettant
d'apprécier les conséquences sanitaires ou écologiques de l'exposition
à telle ou telle substance. Aux Etats-Unis, l'Académie nationale des
sciences suit, pour ses évaluations du risque, une démarche qui a fait
la preuve de son utilité (US NAS, 1983). Elle distingue quatre phases
distinctes dans le processus d'évaluation: la reconnaissance du
danger, l'évaluation de la relation dose-réponse, l'évaluation de
l'exposition et la caractérisation du risque.
La reconnaissance du danger a pour objet d'apprécier les éléments
qui tendent à prouver l'existence d'effets indésirables pour l'homme,
en s'appuyant sur l'ensemble des données toxicologiques disponibles et
sur tout ce que l'on peut savoir du mode d'action du produit en cause.
Il s'agit essentiellement de répondre à deux questions, à savoir 1) si
l'agent en cause représente un danger pour l'Homme et 2) dans quelles
circonstances ce danger est susceptible de se manifester. La
reconnaissance du danger repose sur l'analyse de diverses données qui
peuvent aller d'observations sur l'Homme à l'étude des relations entre
l'activité de la substance et sa structure. Il doit alors être
possible de se prononcer scientifiquement sur la question de savoir si
la substance à expertiser peut, dans des conditions d'exposition
données, avoir des effets indésirables sur la santé humaine. En
général, les effets toxiques s'observent au niveau d'un ou de
plusieurs organes cibles. Souvent, on s'efforce d'observer les divers
points d'aboutissement de l'action toxique de la substance. On
détermine alors l'effet critique, qui représente habituellement le
premier effet indésirable important à apparaître lorsque la dose
augmente.
L'évaluation de la relation dose-réponse consiste à établir la
relation qui existe entre la dose de produit administrée ou reçue et
la fréquence d'un effet nocif. Pour presque tous les types d'effets
toxiques (c'est-à-dire organospécifiques, neurologiques ou
comportementaux, immunologiques, cancérogènes non génotoxiques,
génésiques ou développementaux), on estime généralement qu'il existe
une dose ou une concentration au-dessous de laquelle aucun effet
indésirable ne se produit (c'est-à-dire qu'il existe un seuil de
toxicité). Pour d'autres types d'effets toxiques, on suppose qu'il
existe une probabilité d'action toxique quelle que soit l'intensité de
l'exposition (autrement dit qu'il n'y a pas de seuil de toxicité). A
l'heure actuelle, cette dernière hypothèse s'applique en général
essentiellement aux effets mutagènes et aux effets cancérogènes
génotoxiques.
Si l'on suppose l'existence d'un seuil (par exemple, dans le cas
d'effets non cancérogènes ou d'effets cancérogènes non génotoxiques),
on a l'habitude de déterminer le niveau d'exposition au-dessous duquel
on estime nulle la probabilité d'effets toxiques et que l'on exprime
par la dose sans effet nocif observable ou NOAEL, compte tenu d'un
certain nombre de facteurs d'incertitude (il s'agit d'une valeur
approchée du seuil de toxicité). On peut aussi déterminer de combien
la dose (la plus faible) sans effet nocif observable dépasse le niveau
d'exposition estimé (c'est-à-dire la "marge de sécurité") en fonction
des diverses sources d'incertitude. C'est une méthode que l'on a pu
souvent qualifier d'"évaluation du degré de sécurité". Par conséquent
la dose que l'on peut considérer en première approximation comme le
seuil de toxicité, c'est-à-dire la NOAEL, constitue la dose critique.
On a toutefois de plus en plus tendance à utiliser la "dose de
référence", une estimation (ou la limite inférieure de l'intervalle de
confiance correspondant), obtenue par modélisation, de la dose
produisant l'effet critique avec une fréquence particulière (par ex.
5%) pour l'évaluation quantitative de la relation dose-réponse dans le
cas de ce genre d'effets.
Il n'y a pas de véritable consensus au sujet de la méthodologie à
adopter pour évaluer le risque dans le cas de substances pour
lesquelles il pourrait ne pas exister de seuil pour l'effet critique
(par exemple les cancérogènes génotoxiques et les mutagènes agissant
au niveau des cellules germinales). De fait, on utilise en pareil cas
un certain nombre de méthodes qui reposent en grande partie sur la
caractérisation de la relation dose-réponse. Dans ces conditions, ce
qui compte, ce sont les points expérimentaux qui définissent la pente
de la courbe dose-réponse (et non pas la NOAEL, qui constitue une
première approximation de la valeur du seuil).
La troisième phase du processus consiste dans l'évaluation de
l'exposition. Elle a pour objet de déterminer la nature et le degré du
contact qui a eu lieu ou qui pourrait avoir lieu avec telle ou telle
substance chimique dans diverses conditions. Différentes méthodes
peuvent être utilisées pour procéder à ce type d'évaluation. En
général il s'agit de méthodes directes ou indirectes comportant la
mesure des concentrations dans l'environnement et celle de
l'exposition individuelle ou de marqueurs biologiques. On fait souvent
appel aussi à des modèles et à des questionnaires. L'évaluation de
l'exposition nécessite la détermination des émissions de produits
chimiques, des voies qu'ils empruntent et de la vitesse de leur
déplacement, de même que leur transformation ou décomposition, afin
d'évaluer la concentration à laquelle les populations humaines ou les
différents compartiments de l'environnement (eau, air, sol) peuvent
être exposés.
Selon le but de l'évaluation, le résultat numérique peut se présenter
sous la forme d'une estimation de l'intensité, de la vitesse,de la
durée ou de la fréquence du contact ou encore d'une estimation de la
dose (quantité de produit qui franchit effectivement la limite). Il
importe de noter que c'est la dose interne, et non le niveau
d'exposition externe, qui détermine l'effet toxique d'une exposition
donnée.
La caractérisation du risque constitue la phase finale du
processus d'évaluation du risque. Elle a pour but de faciliter la
tâche de ceux qui ont la responsabilité de gérer ce risque en leur
fournissant, en langage ordinaire, les données scientifiques
essentielles et les principes de base sur lesquels appuyer leurs
décisions. En particulier, on leur donne une évaluation du risque pour
la santé humaine dans des situations d'exposition appropriées. La
caractérisation du risque revient donc à évaluer et à intégrer les
données scientifiques disponibles pour déterminer la nature,
l'importance - et souvent l'ampleur- du risque biologique ou
écologique qu'une exposition à tel ou tel produit peut faire courir
dans des circonstances précises, compte tenu des incertitudes qui lui
sont attachées.
Par "gestion du risque" on entend l'ensemble des activités à
mettre en oeuvre pour pouvoir décider si le risque associé à une
substance donnée appelle une élimination ou une réduction. Les
stratégies et les options qui s'offrent en la matière peuvent être
classées en gros selon leur nature en réglementaires, non
réglementaires, économiques, conseillées, ou technologiques, les unes
n'excluant pas forcément les autres. Ainsi, les mandats législatifs
(les directives réglementaires), les considérations politiques, les
valeurs socioéconomiques, le coût, la faisabilité technique, les
populations exposées au risque, la durée et l'ampleur du risque et les
conséquences possibles sur les échanges commerciaux internationaux,
constituent toute une panoplie de facteurs dont il pourra être tenu
compte dans la politique ou la réglementation finale. Les déterminants
fondamentaux de la décision tels que la taille de la population, les
ressources, les dépenses à envisager pour atteindre les objectifs de
même que la valeur scientifique de l'évaluation du risque et des
options opérationnelles ultérieures varient considérablement d'un
contexte à l'autre. Il est également admis que la gestion des risques
est une procédure complexe et de nature pluridisciplinaire, qui se
présente rarement sous une forme codifiée ou uniforme, qu'elle est
souvent peu structurée, mais qu'elle est néanmoins susceptible de
prendre en compte des données changeantes émanant des sources les plus
diverses. On estime de plus en plus que la perception du risque et le
problème de la communication sont aussi des éléments importants à
prendre en considération si l'on veut que les décisions soient
acceptées par le public le plus large possible.
Les produits chimiques sont devenus indispensables à l'Homme,
qu'il s'agisse de lui permettre de mener à bien ses activités et son
développement, de prévenir et de combattre de nombreuses maladies et
d'accroître les rendements agricoles. En dépit de tous ces avantages,
les produits chimiques, surtout s'ils sont mal utilisés, peuvent avoir
des effets néfastes sur la santé humaine et sur l'environnement.
L'utilisation généralisée de ces produits dans l'ensemble du monde
augmente le risque d'effets indésirables. On peut s'attendre à ce que
les industries chimiques poursuivent leur croissance dans les pays
développés comme dans les pays en développement. Compte tenu de cela,
l'évaluation et la gestion des risques résultant de l'exposition aux
produits chimiques apparaissent comme des priorités de tout premier
plan dans la recherche d'un développement durable.
1. RESUMEN
El control de los riesgos de exposición a productos químicos
(seguridad química) requiere en primer lugar una evaluación
científica, idealmente cuantitativa, de los efectos potenciales con
determinadas concentraciones de exposición (evaluación del riesgo).
Tomando como base los resultados de la evaluación del riesgo y
teniendo en cuenta otros factores, se puede comenzar un proceso de
adopción de decisiones encaminado a eliminar o, si esto no fuera
posible, reducir al mínimo el riesgo de exposición a los productos
químicos objeto de examen (evaluación del riesgo).
La evaluación del riesgo es un marco conceptual que proporciona
el mecanismo que permite un examen estructurado de la información de
interés para la estimación de los resultados en la salud o en el medio
ambiente. En la realización de las evaluaciones del riesgo, el modelo
de la Academia Nacional de Ciencias ha resultado un instrumento útil
(US NAS, 1983). En este modelo el proceso de evaluación del riesgo se
divide en cuatro etapas distintas: identificación del peligro,
evaluación de la relación dosis-respuesta, evaluación de la exposición
y caracterización del riesgo.
La identificación del peligro tiene por objeto evaluar la
importancia de las pruebas relativas a los efectos adversos en el ser
humano, basándose en la evaluación de todos los datos disponibles
sobre la toxicidad y el mecanismo de acción. Está concebida para
abordar fundamentalmente dos cuestiones:1) si un agente puede
representar un peligro para la salud de los seres humanos y 2) en qué
circunstancias puede manifestarse un peligro identificado. La
identificación del peligro se basa en el análisis de diversos datos,
que pueden ir desde las observaciones en el ser humano hasta el
análisis de las relaciones existentes entre la estructura y la
actividad. El resultado de la práctica de identificación del peligro
es un dictamen científico en cuanto a si el producto químico evaluado
puede, en determinadas condiciones de exposición, causar un efecto
adverso en la salud de los seres humanos. En general, se observa
toxicidad en un órgano destinatario o en más. Con frecuencia se
detectan efectos finales múltiples tras la exposición a un producto
químico concreto. Se determina el efecto crítico, que normalmente es
el primer efecto adverso importante que se produce al aumentar la
dosis.
La evaluación de la relación dosis-respuesta es el proceso de
caracterización de la relación existente entre la dosis de un producto
administrado o recibido y la incidencia de un efecto adverso en la
salud. En la mayor parte de los tipos de efectos tóxicos (es decir,
específicos de órganos, neurológicos/del comportamiento, inmunitarios,
carcinogénesis no genotóxica, en la reproducción o en el desarrollo),
se suele considerar que existe una dosis o concentración por debajo de
la cual no se producen efectos adversos (es decir, un umbral). Para
otros tipos de efectos tóxicos, se supone que existe alguna
probabilidad de peligro en todas las concentraciones de exposición (es
decir, que no existe un umbral). En la actualidad, el último supuesto
se aplica fundamentalmente a la mutagénesis y la carcinogénesis
genotóxica.
Si se supone la existencia de un umbral (por ejemplo, para los
efectos no neoplásicos y para los carcinógenos no genotóxicos),
normalmente se estima que existe un nivel de exposición por debajo del
cual no hay efectos adversos, basado en la concentración sin efectos
adversos observados (NOAEL) (aproximación del umbral) y en factores de
incertidumbre. Otra posibilidad consiste en examinar la magnitud en la
cual la concentración sin efectos adversos observados (o efectos
mínimos) (NOAEL o LOAEL) es superior a la exposición estimada (es
decir, el "margen de seguridad"), teniendo en cuenta distintas fuentes
de incertidumbre. Anteriormente, este método se ha descrito con
frecuencia como una "evaluación de la seguridad". Por consiguiente, es
fundamental la concentración que se puede considerar como una primera
aproximación del umbral, es decir la NOAEL. Sin embargo, en la
evaluación cuantitativa de la relación dosis-respuesta se propone cada
vez más el uso de la "dosis de referencia", estimación derivada de un
modelo (o su límite de confianza más bajo) de un nivel de incidencia
determinado (por ejemplo, del 5%) para el efecto crítico.
No hay un consenso claro sobre la metodología apropiada para la
evaluación del riesgo de los productos químicos sin umbral para el
efecto crítico (es decir, carcinógenos genotóxicos y mutágenos de
células germinales). Es más, en tales casos se han adoptado diversos
métodos basados fundamentalmente en la caracterización de la relación
dosis-respuesta. Por consiguiente, los puntos críticos de los datos
son los que definen la pendiente de la relación dosis-respuesta (más
que la NOAEL, que es la primera aproximación de un umbral).
La tercera etapa en el proceso de evaluación del riesgo es la
evaluación de la exposición, que tiene por objeto determinar la
naturaleza y la amplitud del contacto experimentado o previsto con las
sustancias químicas en distintas condiciones. Se pueden utilizar
numerosos métodos para realizar las evaluaciones de la exposición. En
general, los métodos incluyen técnicas indirectas y directas, que
comprenden la medición de las concentraciones en el medio ambiente y
las exposiciones personales, así como biomarcadores. También se
utilizan con frecuencia cuestionarios y modelos. La evaluación de la
exposición requiere la determinación de las emisiones, las rutas y las
velocidades de desplazamiento de una sustancia y su transformación o
degradación, a fin de estimar las concentraciones a las cuales pueden
estar expuestas poblaciones humanas o las distintas esferas del medio
ambiente (agua, suelo y aire).
En función de la finalidad de una evaluación de la exposición, el
resultado numérico puede ser una estimación de la intensidad, la
velocidad, la duración o la frecuencia de la exposición o la dosis por
contacto (cantidad resultante que realmente cruza la frontera). Para
la evaluación del riesgo basada en la relación dosis-respuesta, el
resultado normalmente incluye una estimación de la dosis. Es
importante señalar que es la dosis interna, no el nivel exposición
externa, la que determina el resultado toxicológico de una exposición
determinada.
La caracterización del riesgo es la última etapa de la evaluación
del riesgo. Está concebida para prestar asistencia a los especialistas
en gestión del riesgo mediante el suministro, en lenguaje sencillo, de
pruebas científicas esenciales y de los fundamentos en relación con el
riesgo que necesitan para adoptar una decisión. En la caracterización
del riesgo se proporcionan estimaciones del riesgo para la salud
humana en los modelos de exposición pertinentes. Así pues, una
caracterización del riesgo es una evaluación e integración de las
pruebas científicas disponibles utilizadas para estimar la naturaleza,
la importancia y con frecuencia la magnitud del riesgo humano y/o para
el medio ambiente, incluidas las incertidumbres pendientes, que
razonablemente se puede estimar que se derivan de la exposición a un
agente concreto del medio ambiente en circunstancias específicas.
El término "gestión del riesgo" comprende todas las actividades
precisas para adoptar una decisión sobre si un riesgo asociado
requiere la eliminación o una reducción necesaria. Las
estrategias/opciones de gestión del riesgo se pueden clasificar a
grandes rasgos como reglamentarias, no reglamentarias, económicas,
consultivas o tecnológicas, que no son excluyentes entre sí. De esta
manera, los mandatos legislativos (orientación reglamentaria), los
aspectos políticos, los valores económicos, el costo, la viabilidad
técnica, las poblaciones con riesgo, la duración y la magnitud del
riesgo, la comparación de los riesgos y las posibles repercusiones en
el comercio entre los países pueden considerarse, en general, como un
amplio abanico de elementos que pueden influir en la formulación final
de políticas o normas. Los factores fundamentales para decisión, como
el tamaño de la población, los recursos, los costos del logro de los
objetivos y la calidad científica de la evaluación del riesgo y las
posteriores decisiones administrativas, varían enormemente del
contexto de una decisión al de otra. Se reconoce asimismo que la
gestión del riesgo es un procedimiento multidisciplinario complejo que
raramente aparece codificado o uniforme y con frecuencia no está
estructurado, pero que puede responder a aportaciones en evolución de
una amplia variedad de fuentes. Cada vez se reconoce con más
frecuencia que la percepción y la comunicación del riesgo son
elementos importantes que también hay que tener en cuenta para lograr
una aceptación pública lo más amplia posible de las decisiones en
materia de gestión del riesgo.
Los productos químicos se han convertido en una parte
indispensable de la vida humana, que sostienen las actividades y el
desarrollo, previenen y combaten numerosas enfermedades y aumentan la
productividad agrícola. A pesar de sus ventajas, los productos
químicos pueden, especialmente cuando se utilizan de manera indebida,
producir efectos adversos en la salud humana y la integridad del medio
ambiente. La aplicación generalizada de productos químicos en todo el
mundo aumenta el potencial de los efectos adversos. Se prevé que
seguirá aumentando el crecimiento de las industrias químicas, tanto en
los países en desarrollo como desarrollados. En esta situación, se
reconoce que la evaluación y la gestión de los riesgos de la
exposición a productos químicos son una de las prioridades más
importantes a la hora de aplicar los principios del desarrollo
sostenible.