
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization.
Concise International Chemical Assessment Document 61
First draft prepared by Prof. Fina Petrova Simeonova, Consultant, National Center of Hygiene, Medical Ecology and Nutrition, Sofia, Bulgaria; and Dr Lawrence Fishbein, Fairfax, Virginia, USA
Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 2004
The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
Hydrogen cyanide and cyanides : human health aspects.
(Concise international chemical assessment document ; 61)
1.Cyanides - adverse effects 2.Hydrogen cyanide - adverse effects 3.Risk assessment
4.Environmental exposure I.International Programme on Chemical Safety II.Series
ISBN 92 4 153061 8 (LC/NLM Classification: QV 632)
ISSN 1020-6167
©World Health Organization 2004
All rights reserved. Publications of the World Health Organization can be obtained from Marketing and Dissemination, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 2476; fax: +41 22 791 4857; email: bookorders@who.int). Requests for permission to reproduce or translate WHO publications — whether for sale or for noncommercial distribution — should be addressed to Publications, at the above address (fax: +41 22 791 4806; email: permissions@who.int).
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
The World Health Organization does not warrant that the information contained in this publication is complete and correct and shall not be liable for any damages incurred as a result of its use.
Risk assessment activities of the International Programme on Chemical Safety, including the production of Concise International Chemical Assessment Documents, are supported financially by the Department of Health and Department for Environment, Food & Rural Affairs, UK, Environmental Protection Agency, Food and Drug Administration, and National Institute of Environmental Health Sciences, USA, European Commission, German Federal Ministry of Environment, Nature Conservation and Nuclear Safety, Health Canada, Japanese Ministry of Health, Labour and Welfare, and the Swiss Agency for Environment, Forests and Landscape.
Technically and linguistically edited by Marla Sheffer, Ottawa, Canada, and printed by Wissenchaftliche Verlagsgesellschaft mbH, Stuttgart, Germany
|
6. COMPARATIVE KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS |
|
9.2 Criteria for setting tolerable intakes/concentrations for cyanides |
Concise International Chemical Assessment Documents (CICADs) are the latest in a family of publications from the International Programme on Chemical Safety (IPCS) — a cooperative programme of the World Health Organization (WHO), the International Labour Organization (ILO), and the United Nations Environment Programme (UNEP). CICADs join the Environmental Health Criteria documents (EHCs) as authoritative documents on the risk assessment of chemicals.
International Chemical Safety Cards on the relevant chemical(s) are attached at the end of the CICAD, to provide the reader with concise information on the protection of human health and on emergency action. They are produced in a separate peer-reviewed procedure at IPCS. They may be complemented by information from IPCS Poison Information Monographs (PIM), similarly produced separately from the CICAD process.
CICADs are concise documents that provide summaries of the relevant scientific information concerning the potential effects of chemicals upon human health and/or the environment. They are usually based on selected national or regional evaluation documents or on existing EHCs. Before acceptance for publication as CICADs by IPCS, these documents undergo extensive peer review by internationally selected experts to ensure their completeness, accuracy in the way in which the original data are represented, and the validity of the conclusions drawn.
The primary objective of CICADs is characterization of hazard and dose–response from exposure to a chemical. CICADs are not a summary of all available data on a particular chemical; rather, they include only that information considered critical for characterization of the risk posed by the chemical. The critical studies are, however, presented in sufficient detail to support the conclusions drawn. For additional information, the reader should consult the identified source documents upon which the CICAD has been based.
Risks to human health and the environment will vary considerably depending upon the type and extent of exposure. Responsible authorities are strongly encouraged to characterize risk on the basis of locally measured or predicted exposure scenarios. To assist the reader, examples of exposure estimation and risk characterization are provided in CICADs, whenever possible. These examples cannot be considered as representing all possible exposure situations, but are provided as guidance only. The reader is referred to EHC 170.1
While every effort is made to ensure that CICADs represent the current status of knowledge, new information is being developed constantly. Unless otherwise stated, CICADs are based on a search of the scientific literature to the date shown in the executive summary. In the event that a reader becomes aware of new information that would change the conclusions drawn in a CICAD, the reader is requested to contact IPCS to inform it of the new information.
Procedures
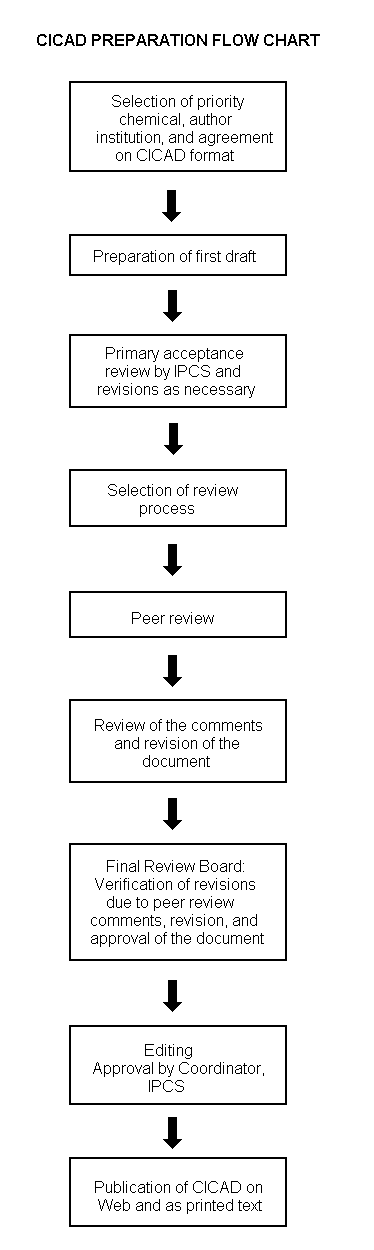
The flow chart on page 2 shows the procedures followed to produce a CICAD. These procedures are designed to take advantage of the expertise that exists around the world — expertise that is required to produce the high-quality evaluations of toxicological, exposure, and other data that are necessary for assessing risks to human health and/or the environment. The IPCS Risk Assessment Steering Group advises the Coordinator, IPCS, on the selection of chemicals for an IPCS risk assessment based on the following criteria:
Thus, it is typical of a priority chemical that
|
Advice from Risk Assessment Steering Group Criteria of priority:
Thus, it is typical of a priority chemical that
Special emphasis is placed on avoiding duplication of effort by WHO and other international organizations. A prerequisite of the production of a CICAD is the availability of a recent high-quality national/regional risk assessment document = source document. The source document and the CICAD may be produced in parallel. If the source document does not contain an environmental section, this may be produced de novo, provided it is not controversial. If no source document is available, IPCS may produce a de novo risk assessment document if the cost is justified. Depending on the complexity and extent of controversy of the issues involved, the steering group may advise on different levels of peer review:
|
The Steering Group will also advise IPCS on the appropriate form of the document (i.e., a standard CICAD or a de novo CICAD) and which institution bears the responsibility of the document production, as well as on the type and extent of the international peer review.
The first draft is usually based on an existing national, regional, or international review. When no appropriate source document is available, a CICAD may be produced de novo. Authors of the first draft are usually, but not necessarily, from the institution that developed the original review. A standard outline has been developed to encourage consistency in form. The first draft undergoes primary review by IPCS to ensure that it meets the specified criteria for CICADs.
The second stage involves international peer review by scientists known for their particular expertise and by scientists selected from an international roster compiled by IPCS through recommendations from IPCS national Contact Points and from IPCS Participating Institutions. Adequate time is allowed for the selected experts to undertake a thorough review. Authors are required to take reviewers’ comments into account and revise their draft, if necessary. The resulting second draft is submitted to a Final Review Board together with the reviewers’ comments. At any stage in the international review process, a consultative group may be necessary to address specific areas of the science. When a CICAD is prepared de novo, a consultative group is normally convened.
The CICAD Final Review Board has several important functions:
Board members serve in their personal capacity, not as representatives of any organization, government, or industry. They are selected because of their expertise in human and environmental toxicology or because of their experience in the regulation of chemicals. Boards are chosen according to the range of expertise required for a meeting and the need for balanced geographic representation.
Board members, authors, reviewers, consultants, and advisers who participate in the preparation of a CICAD are required to declare any real or potential conflict of interest in relation to the subjects under discussion at any stage of the process. Representatives of nongovernmental organizations may be invited to observe the proceedings of the Final Review Board. Observers may participate in Board discussions only at the invitation of the Chairperson, and they may not participate in the final decision-making process.
This CICAD on hydrogen cyanide and cyanides (human health aspects) was prepared by Prof. Fina Petrova Simeonova and Dr Lawrence Fishbein, based principally on the Agency for Toxic Substances and Disease Registry toxicological profile for cyanide (ATSDR, 1997) and the Joint FAO/WHO Expert Committee on Food Additives document on cyanogenic glycosides (JECFA, 1993). The source documents and a description of their review processes are presented in Appendix 1. A comprehensive literature search of several online databases was performed in October 2002 to identify any relevant references published subsequent to those cited in the source documents. This CICAD was first discussed at the 10th Final Review Board meeting, held in Monks Wood, United Kingdom, on 16–19 September 2002. Following revision, it was discussed again and approved as an international assessment at the 11th Final Review Board meeting, held in Varna, Bulgaria, on 8–11 September 2003. Participants at the 10th and 11th Final Review Board meetings are listed in Appendices 2 and 3. The drafts discussed at these meetings were peer reviewed before the meetings; information on the peer review process is presented in Appendix 4. The International Chemical Safety Cards on hydrogen cyanide, sodium cyanide, potassium cyanide, calcium cyanide, cyanogen, cyanogen chloride, acetone cyanohydrin, and potassium ferricyanide, produced by the International Programme on Chemical Safety (IPCS, 1999a,b, 2000b, 2001, 2002a,b,c,d), have also been reproduced in this document.
Cyanides comprise a wide range of compounds of varying degrees of chemical complexity, all of which contain a CN moiety, to which humans are exposed in gas, liquid, and solid form from a broad range of natural and anthropogenic sources. While many chemical forms of cyanide are used in industrial application or are present in the environment, the cyanide anion CN– is the primary toxic agent, regardless of origin.
Hydrogen cyanide is a colourless or pale blue liquid or gas with a faint bitter almond-like odour. Hydrogen cyanide is used primarily in the production of substances such as adiponitrile, methyl methacrylate, chelating agents, cyanuric chloride, methionine and its hydroxylated analogues, and sodium and potassium cyanide. Hydrogen cyanide is also used as a fumigant in ships, railroad cars, large buildings, grain silos, and flour mills, as well as in the fumigation of peas and seeds in vacuum chambers.
Other cyanides, such as sodium and potassium cyanide, are solid or crystalline hygroscopic salts widely used in ore extracting processes for the recovery of gold and silver, electroplating, case-hardening of steel, base metal flotation, metal degreasing, dyeing, printing, and photography. They are also widely used in the synthesis of organic and inorganic chemicals (e.g., nitriles, carboxylic acids, amides, esters, and amines; heavy metal cyanides) and in the production of chelating agents.
Anthropogenic sources of cyanide release to the environment are diverse. Releases to air include chemical manufacturing and processing industries, such as metallurgical industries and metal plating, and extraction of gold and silver from low-grade ores. Other sources include volatilization from cyanide wastes disposed of in landfills and waste ponds, emissions from municipal solid waste incinerators, biomass burning, fossil fuel combustion, including vehicle emissions, fumigation operations, and the production of coke or other coal carbonization procedures.
Hydrogen cyanide is formed during the incomplete combustion of nitrogen-containing polymers, such as certain plastics, polyurethanes, and wool. Hydrogen cyanide is present in cigarette smoke.
Non-point sources of cyanide released to water can result from runoff from cyanide-containing anti-caking salts used on roads, migration from landfills, and agricultural and atmospheric fallout and washout. Point sources of releases to water include discharges from gold mining plants, wastewater treatment works, iron and steel production, and organic chemical industries.
Principal natural sources of cyanides are over 2000 plant species, including fruits and vegetables, that contain cyanogenic glycosides, which can release cyanide on hydrolysis when ingested. Among them, cassava (tapioca, manioc) and sorghum are staple foods for hundreds of millions of people in many tropical countries. Known cyanogenic glycosides in plants include amygdalin, linamarin, prunasin, dhurrin, lotaustralin, and taxiphyllin. Hydrogen cyanide is released into the atmosphere from natural biogenic processes from higher plants, bacteria, and fungi.
In air, cyanide is present as gaseous hydrogen cyanide, with a small amount present in fine dust particles. Cyanides have the potential to be transported over long distances from their respective emission sources.
The majority of the population is exposed to very low levels of cyanide in the general environment. There are, however, specific subgroups with higher potential for exposure. These include individuals involved in large-scale processing of cassava and those consuming significant quantities of improperly prepared foods containing cyanogenic glycosides, such as cassava, speciality foods such as apricot pits, and bitter almonds. Other subgroups with greatest potential for exposure include those in the vicinity of accidental or intended releases from point sources, active and passive smokers, and fire-related smoke inhalation victims.
Workers may be exposed to cyanides during fumigation operations and the production and use of cyanides in many industrial processes — for example, electroplating, case-hardening of steel, and extraction of gold and silver from ores.
Cyanides are well absorbed via the gastrointestinal tract or skin and rapidly absorbed via the respiratory tract. Once absorbed, cyanide is rapidly and ubiquitously distributed throughout the body, although the highest levels are typically found in the liver, lungs, blood, and brain. There is no accumulation of cyanide in the blood or tissues following chronic or repeated exposure.
Approximately 80% of absorbed cyanide is metabolized to thiocyanate in the liver by the mitochondrial sulfur transferase enzyme rhodanese and other sulfur transferases. Thiocyanate is excreted in the urine. Minor pathways for cyanide detoxification involve reaction with cystine to produce aminothiazoline- and iminothiazolidinecarboxylic acids and combination with hydroxycobalamin (vitamin B12a) to form cyanocobalamin (vitamin B12); these end-products are also excreted in the urine.
The principal features of the toxicity profile for cyanide are its high acute toxicity by all routes of administration, with a very steep and rate-dependent dose–effect curve, and chronic toxicity, probably mediated through the main metabolite and detoxification product, thiocyanate. The toxic effects of cyanide ion in humans and animals are generally similar and are believed to result from inactivation of cytochrome oxidase and inhibition of cellular respiration and consequent histotoxic anoxia. The primary targets of cyanide toxicity in humans and animals are the cardiovascular, respiratory, and central nervous systems. The endocrine system is also a potential target for long-term toxicity, as a function of continued exposure to thiocyanate, which prevents the uptake of iodine in the thyroid and acts as a goitrogenic agent.
In humans, whereas slight effects occur at exposure levels of 20–40 mg/m3, 50–60 mg/m3 can be tolerated without immediate or late effects for 20 min to 1 h, 120–150 mg/m3 may lead to death after 0.5–1 h, 150 mg/m3 is likely to be fatal within 30 min, 200 mg/m3 is likely fatal after 10 min, and 300 mg/m3 is immediately fatal. The lowest reported oral lethal dose for humans is 0.54 mg/kg body weight, and the average absorbed dose at the time of death has been estimated at 1.4 mg/kg body weight (calculated as hydrogen cyanide). Sequelae after severe acute intoxications may include neuropsychiatric manifestations and Parkinson-type disease. Cyanide from tobacco smoke has been implicated as a contributing factor in tobacco–alcohol amblyopia. Long-term exposure to lower concentrations of cyanide in occupational settings can result in a variety of symptoms related to central nervous system effects.
Long-term consumption of cassava containing high levels of cyanogenic glycosides has been associated with tropical ataxic neuropathy, spastic paraparesis, and, in areas with low iodine intake, development of hypothyroidism, goitre, and cretinism. While exposure to cyanide has been crudely estimated to be 15–50 mg/day in endemic areas in some such cases, owing to the limitations of data on exposure and potential impact of confounders such as malnutrition, low protein content of the diet, vitamin deficiencies, and iodine status, the available data do not provide meaningful information on dose–response for cyanide.
Data on end-points other than acute toxicity are somewhat limited. This is attributable in large part to difficulties in conducting, for example, investigations of repeated-dose or chronic toxicity due to the high acute toxicity of the compound. Cyanides are weakly irritating to the skin and eye; data on sensitizing properties or carcinogenicity of hydrogen cyanide or its alkali salts have not been identified. Although somewhat limited, the weight of evidence of available data indicates that cyanide is not genotoxic and that it induces developmental effects only at doses or concentrations that are overtly toxic to the mothers.
Available data in human populations are considered inadequate as a basis for characterization of dose–response for chronic ingestion of cyanide. In a 13-week repeated-dose toxicity study in which cyanide was administered in drinking-water, there were no clinical signs associated with central nervous system effects or histopathological effects in the brain or thyroid of rats or mice exposed to doses up to 12.5 mg and 26 mg cyanide/kg body weight per day, respectively. At 12.5 mg cyanide/kg body weight per day, there were slight changes in the reproductive tract in male rats, which, although they apparently would not affect fertility in rats, are possibly significant to humans. The no-observed-adverse-effect level (NOAEL) for these effects was 4.5 mg/kg body weight per day. The examination of neurotoxicity in this study was limited to clinical observation and optical microscopy in autopsy. The few available studies specifically intended to investigate neurotoxicity, while reporting adverse effects at exposure levels of 1.2 mg cyanide/kg body weight per day in rats and 0.48 mg cyanide/kg body weight per day in goats, suffer from weaknesses that preclude their quantitative assessment.
In relation to characterization of concentration–response for repeated-dose toxicity for inhalation (relevant principally to the occupational environment), in three separate studies in rats, there were no adverse systemic effects in rats exposed to acetone cyanohydrin, which is rapidly hydrolysed to hydrogen cyanide at physiological pH, at concentrations up to 211 mg/m3 (corresponding to a concentration of 67 mg hydrogen cyanide/m3). The steepness of the dose–effect curve is illustrated by the observation of 30% mortality among rats exposed part of the day to 225 mg acetone cyanohydrin/m3 (71 mg hydrogen cyanide/m3).
Adverse effects of exposure to the low concentrations of cyanide that are generally present in the general environment (<1 µg/m3 in ambient air; <10 µg/litre in water) are unlikely. Acute cyanide intoxications may arise from eating apricot kernels, choke cherries, and other stone fruit kernels with high concentrations of cyanogenic glycosides. Inadequately prepared cassava, when constituting the major part of the diet, may be hazardous.
Hydrogen cyanide (HCN) is a colourless or pale blue liquid or gas with a faint bitter almond-like odour. Common synonyms are hydrocyanic acid and prussic acid. Hydrogen cyanide is a very weak acid, with a pKa value of 9.22 at 25 °C. It is soluble in water and alcohol. Hydrogen cyanide is commercially available as a gas or as a technical-grade liquid in concentrations of 5, 10, and 96–99.5%. Phosphoric acid is added to liquid hydrogen cyanide as a stabilizer to prevent decomposition and explosion (ATSDR, 1997). Some important physical and chemical properties of hydrogen cyanide are summarized in Table 1.
The conversion factors2 for hydrogen cyanide in air (at 20 °C and 101.3 kPa) are as follows:
1 ppm = 1.12 mg/m3
1 mg/m3 = 0.890 ppm
Table 1: Physical and chemical properties of hydrogen cyanide (CAS No.
|
Property |
Value |
|
Relative molecular mass |
27.03 |
|
Boiling point (°C) |
25.70 |
|
Solubility (30 °C) |
Miscible with water; soluble in ethanol |
|
Specific density: vapours (31 °C) |
0.937 |
|
Odour threshold |
0.7 mg/m3 in air |
|
Henry’s law constant (dimensionless) |
180–300b |
|
Octanol/water partition coefficient (log Kow) |
0.66 |
|
Vapour pressure (kPa) |
35.2 at 0 °C |
a From ACGIH (2001); DECOS (2002).
b Hine & Weimar (1965); Edwards et al. (1978); Gaffney et al. (1987).
Sodium cyanide (NaCN) is a white hygroscopic crystalline powder with a faint bitter almond-like odour. Common synonyms are cyanide of sodium and hydrocyanic acid, sodium. Commercially available sodium cyanide generally achieves a purity of 95–98%. The aqueous solution of sodium cyanide is strongly alkaline and rapidly decomposes. Sodium cyanide produces hydrogen cyanide on contact with acids or acid salts.
Potassium cyanide (KCN) is a white deliquescent solid with an odour of hydrogen cyanide. Common synonyms are hydrocyanic acid, potassium salt and cyanide of potassium. Potassium cyanide is commercially available at a 95% purity. An aqueous solution of potassium cyanide in water is strongly alkaline. Potassium cyanide also produces hydrogen cyanide on contact with acids or acid salts.
Calcium cyanide (Ca(CN)2), also commonly called cyanide of calcium, calcid, or calsyan, is a white crystalline solid. Its aqueous solution gradually liberates hydrogen cyanide. Cyanides such as sodium cyanide, potassium cyanide, and calcium cyanide form strong complexes with many metals (Table 2).
Cyanogen is a colourless toxic gas with an almond-like odour. Common synonyms are carbon nitrile, dicyanogen, ethane dinitrile, and oxalic acid dinitrile. Cyanogen is slowly hydrolysed in aqueous solution, yielding oxalic acid and ammonia. The conversion factors for cyanogen in air at 20 °C and 101.3 kPa are as follows:
1 ppm = 2.16 mg/m3
1 mg/m3 = 0.462 ppm
Table 2: Physical and chemical properties of selected cyanide compounds.a
|
Species |
CAS |
Molecular formula |
Relative molecular mass |
Common |
Boiling point |
Solubility |
|
Sodium cyanide |
|
NaCN |
49.02 |
Cyanide of sodium |
Soluble in water, slightly soluble in alcohol |
|
|
Potassium cyanide |
|
KCN |
65.11 |
Cyanide of potassium |
Soluble in water, slightly soluble in alcohol |
|
|
Calcium cyanide |
|
Ca(CN)2 |
92.12 |
Calcid; calsyan |
Soluble in water, slightly soluble in alcohol |
|
|
Copper cyanide |
54-92-3 |
CuCN |
89.56 |
Cupricin |
Insoluble in water |
|
|
Potassium silver cyanide |
501-61-6 |
KAg(CN)2 |
198.01 |
Potassium dicyanoargentate |
Soluble in water, slightly soluble in ether |
|
|
Sodium ferrocyanide |
|
Na4Fe(CN)6 |
303.91 |
Sodium hexacyanoferrate (II) |
Soluble in water |
|
|
Potassium ferrocyanide |
13943-57-3 |
K4Fe(CN)6 |
368.35 |
Yellow prussiate of potash |
Soluble in water |
|
|
Potassium ferricyanide |
|
K3Fe(CN)6 |
329.95 |
Red prussiate of potash |
Slowly soluble in 2.5 parts of cold water; decomposes slowly in water |
|
|
Cyanogen |
|
NCCN |
52.04 |
Carbon nitrile; dicyanogen |
–20.7 |
Soluble in water, alcohol, and ether |
|
Cyanogen chloride |
|
CNCl |
61.47 |
Chlorine cyanide |
13.8 |
Soluble in water and alcohol |
|
Acetone cyanohydrin |
|
(CH3)2C(OH)CN |
85.10 |
ACH; methyllactonitrile |
82 |
Soluble in water |
|
Sodium nitroprusside |
|
Na2[Fe(CN)5NO] |
261.97 |
Sodium nitroferrocyanide; sodium nitrosyl pentacyanoferrate (III) |
Soluble in 2.3 parts of water, slightly soluble in alcohol |
a From Windholz (1983); ACGIH (2001); ECETOC (2004).
Cyanogen chloride is a colourless gas. Its common synonym is chlorine cyanide, and its common trade name is Caswell No. 267. Cyanogen chloride releases hydrogen cyanide by hydrolysis. Its conversion factors in air are:
1 ppm = 2.56 mg/m3
1 mg/m3 = 0.391 ppm
Common synonyms of acetone cyanohydrin are ACH, 2-cyano-2-propanol, 2-methyllactonitrile, and 2-hydroxy-2-methyl propanenitrile. It dissociates on standing to liberate hydrogen cyanide. Its boiling point is 120 °C (with decomposition to hydrogen cyanide and acetone). Its conversion factors in air are:
1 ppm = 3.54 mg/m3
1 mg/m3 = 0.283 ppm
The half-time of ACH in water was reported to be 9 min (Ellington et al., 1986); further studies reported that this hydrolysis to acetone and hydrogen cyanide was pH dependent, and half-times of 58, 27, and 8 min were observed at pH 4.8, 6.3, and 6.8 (ICI, 1993). In a more recent study, similar findings were reported (half-times of 54.7, 31.2, 5.4, and 4.0 min at pH 6.00, 6.40, 6.86, and 7.00, respectively) (Frank et al., 2002).
Some chemical properties of other cyanides are given in Table 2. Copper cyanide is a white to cream-coloured solid. Its common name is cuprous cyanide, and its synonym is cupricin. Potassium silver cyanide occurs as white crystals; its common synonym is potassium dicyanoargentate. It is sensitive to light. Sodium ferrocyanide decomposes at 435 °C, forming sodium cyanide.
Cyanogenic glycosides are produced naturally by many plants; when hydrolysed, they produce hydrogen cyanide. Chemical structures of some commonly occurring cyanogenic glycosides are depicted in Figure 1.
Further chemical and physical properties of hydrogen cyanide and some cyanides are summarized in the International Chemical Safety Cards included in this document.
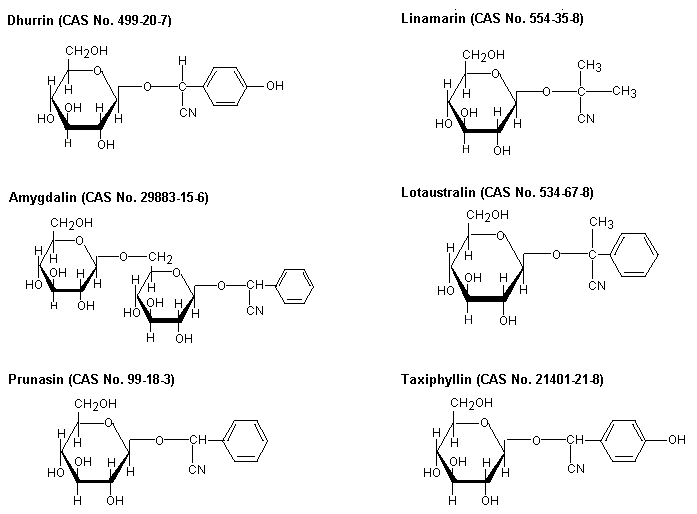
Fig. 1: Cyanogenic glycosides in major edible plants (JECFA, 1993)
Amygdalin occurs in (among others) almonds, dhurrin in sorghum, linamarin in cassava, lotaustralin in cassava and lima beans, prunasin in stone fruits, and taxiphyllin in bamboo shoots.
Cyanides in environmental media are usually collected in sodium or potassium hydroxide solution and measured by spectrophotometry (Agrawal et al., 1991), colorimetry, or ion-specific electrode or by headspace gas chromatography with a nitrogen-specific detector or electron capture detector (Maseda et al., 1989; Seto et al, 1993). Cyanide in aqueous matrices is usually measured by colorimetric, titrimetric (US EPA, 1983), or electrochemical methods after pretreatment to produce hydrogen cyanide and absorption in sodium hydroxide solution. Total cyanide (irrespective of origin) includes all of the available cyanide in a sample; in drinking-water, it is measured by semi-automated colorimetry (EPA Method 335.4) as well as by selective electrode, ultraviolet/distillation/spectrophotometry, and ion chromatography (EPA Method 300.0) (US EPA, 1993a). Free cyanide can also be determined by one method (SM-4500-CN-F) approved for drinking-water compliance monitoring analysis that does not require distillation, the specific ion electrode method (US EPA, 2003a). Weak acid dissociable cyanide analysis (used principally by the precious metals mining industry) includes those cyanide species liberated at moderate pH 4.5, such as aqueous hydrogen cyanide and cyanide anion, the majority of copper, cadmium, nickel, zinc, silver, and tin complexes, and others with similar low dissociation constants. Weak acid dissociable cyanide can be determined in wastewaters by a ligand exchange/flow injection/amperometric technique (EPA Method 1677) (Milosavlievic et al., 1995; US EPA, 1997).
A chromatographic technique with fluorescence detection is used to detect trace amounts of cyanide in blood cells (Chinaka et al., 1998). Cyanide in biological tissue and fluids can be measured spectrophotometrically after reaction with methaemoglobin.
Since many cyanides are unstable and emit volatile hydrogen cyanide gas, sampling, storage, and analysis must be done with caution, preferably immediately upon collection.
The three commonly used techniques (colorimetric, titrimetric, and electrochemical) may all suffer from interference problems, unless proper precautions are taken (ATSDR, 1989).
Metals suppress the transformation of cyanide to formic acid, thus lowering the measured hydrogen cyanide concentration (Dolzine et al., 1982). Carbonyl compounds also decrease the hydrogen cyanide recovery (Honig et al., 1983), as in the case with soybean samples, in which carbonyl compounds occur naturally.
Sodium thiosulfate can interfere with potentiometric (Sylvester et al., 1982) or colorimetric analysis (Ganjeloo et al., 1980). Care should be taken, since it is often used as an antidote to treat chemical poisoning.
Continuous monitoring of cyanide is also available using equipment based on diffusion and amperometric detection of hydrogen cyanide (NIOSH, 1976).
Detection limits for the different methods for hydrogen cyanide range from 0.8 to 400 mg/m3 for air samples, from 0.04 to 200 µg/litre for aqueous samples, and from 0.8 to 300 µg/litre for biological samples. NIOSH Method 7904 for workplace air has a limit of detection of 2.5 µg cyanide (NIOSH, 1994).
Hydrogen cyanide is ubiquitous in nature. It is found in the stratosphere and non-urban troposphere (US EPA, 1990). It is released into the atmosphere from biomass burning, volcanoes, and natural biogenic processes from higher plants, bacteria, algae, and fungi (Fiksel et al., 1981; Cicerone & Zellner, 1983; Way, 1984; ATSDR, 1997; Li et al., 2000). An estimate of the amount of cyanide released to the environment from natural biogenic processes is not available (ATSDR, 1997).
Cyanide occurs naturally as cyanogenic glycosides in at least 2000 plants (Figure 1). Amygdalin (d-mandelonitrile-beta-d-glucoside-6-beta-d-glucoside) has been found in about 1000 species of plants, including cassava (tapioca, manioc), sweet potato, corn, cabbage, linseed, millet, and bamboo, in pits of stone fruits, such as cherries, peaches, and apricots, and in apple seeds (JECFA, 1993; Sharma, 1993; Padmaja, 1995). It is also present in bitter almonds and American white lima beans (Ermans et al., 1972). After ingestion, linamarin can be hydrolysed by either cassava linamarase or an endogenous beta-glucosidase to yield d-glucose and ACH (Frakes et al., 1986a).
Hydrogen cyanide is principally produced by two synthetic catalytic processes involving the reaction of ammonia and natural gas (or methane) with or without air. It is also obtained as a by-product in the production of acrylonitrile by the ammoxidation of propylene, which accounts for approximately 30% of the worldwide production of hydrogen cyanide.
Sodium and potassium cyanides are principally prepared by the direct reaction of hydrogen cyanide with the respective alkali in closed systems (European Chemicals Bureau, 2000a,b). Sodium cyanide is also prepared to a lesser extent by melting sodium chloride with calcium cyanamide or by heating sodium amide salt with carbon.
Calcium cyanide is produced by the reaction of coke, coal, and limestone.
Cyanogen chloride is a reaction product of organic precursors with hypochlorous acid in the presence of ammonia and may be formed as a by-product of the chloramination of water (WHO, 1996; IPCS, 2000a).
ACH was first produced in the 1930s as an intermediate in the production of methyl methacrylate from hydrogen cyanide. It is currently produced from the liquid-phase reaction of hydrogen cyanide and acetone in the presence of an alkali catalyst at atmospheric pressure (ECETOC, 2004).
Hydrogen cyanide capacity is generally treated as the sum of purposeful direct synthesis and that derived as a by-product of acrylonitrile production. Annual US hydrogen cyanide capacity by 11 companies in 1991 was 666 000 tonnes. US production of hydrogen cyanide from 1983 to 1989 rose from 300 000 to 445 000 tonnes (Pesce, 1993). Output of hydrogen cyanide in the USA was 545 000 tonnes in 1992 (Cohrssen, 2001). Worldwide annual production and capacity of hydrogen cyanide in 1992 were estimated to be 950 000 and 1 320 000 tonnes, respectively (Pesce, 1993; Cohrssen, 2001). It has been estimated that the present total annual production of hydrogen cyanide worldwide is 1.4 million tonnes (Mudder & Botz, 2000).
In 1983, the major end uses of hydrogen cyanide in the USA were in the production of adiponitrile (200 000 tonnes), ACH (128 000 tonnes), cyanuric chloride (28 500 tonnes), sodium cyanide (69 000 tonnes), chelating agents (15 800 tonnes), and nitrilotriacetic acid (10 100 tonnes) and for miscellaneous uses (20 000 tonnes) (US EPA, 1990). Hydrogen cyanide is also used in the production of methyl methacrylate, methionine and its hydroxylated analogues, and potassium cyanide (ATSDR, 1997; ECETOC, 2004).
Sodium cyanide is extensively employed in a large number of industrial processes, including electroplating and case-hardening of metals; the extraction (cyanidation) of gold and silver from ores; base metal flotation; coal gasification; and the fumigation of ships, railroad cars, buildings, grain silos, flour mills, seeds in vacuum chambers, and soil. Large quantities of sodium cyanide are used to introduce cyano groups into organic compounds, in particular through a reaction with organic halogen compounds to yield nitriles. The nitriles can then be converted to a variety of carboxylic acids, amides, esters, and amines. Potassium cyanide is used for electrolytic refining of platinum, for metal colouring, and as an electrolyte for the separation of gold, silver, and copper from platinum (Eisler et al., 1999; Patnaik, 1999; ACGIH, 2001; ECETOC, 2004). Cyanide salts are used as chelating agents, and the complex cyanides of copper, zinc, and cadmium are used in electroplating processes, principally the plating of iron, steel, and zinc (ECETOC, 2004).
Calcium cyanide is used chiefly as a fumigant, because it readily releases hydrogen cyanide when exposed to air; as a fertilizer, defoliant, herbicide, and rodenticide; as a stabilizer for cement; and in stainless steel manufacture (ACGIH, 2001).
Cyanogen is used as a fumigant, as a fuel gas for welding and cutting heat-resistant metals, and as a rocket and missile propellant (ATSDR, 1997).
Cyanogen chloride is used as a fumigant gas and as a reagent in chemical synthesis.
Cuprous cyanide is used in plating baths for silver, brass, and copper–tin alloy plating (ATSDR, 1997), as an antifouling agent in marine paint, and as an insecticide and fungicide (Windholz, 1983).
Potassium silver cyanide is used in silver plating and as a bactericide.
Potassium ferricyanide is used chiefly for blueprints, in photography, for staining wood, in calico printing, and in electroplating.
Sodium ferrocyanide is used in ore flotation, as an anti-caking agent in rock salt, and in photography for bleaching, toning, and fixing.
Sodium nitroprusside has been used as an antihypertensive agent and in congestive heart failure and is used for deliberate induction of hypotension during certain neurosurgical procedures.
ACH is used in preparative transcyanohydrination reactions.
More than 30 large-scale accidental releases of cyanide to water systems have been reported since 1975; these include transportation accidents, pipe failures, and tailings dam-related releases (Korte et al., 2000; Mudder & Botz, 2000).
Non-point sources of cyanide released to water can result from runoff from cyanide-containing anti-caking salts (i.e., sodium ferrocyanide) used on roads, migration from landfills, and agricultural and atmospheric fallout and washout (ATSDR, 1997).
The extraction of gold from low-grade ores by cyanidation processes was estimated to result in a worldwide emission of 20 000 tonnes of hydrogen cyanide into the atmosphere (Korte & Coulston, 1998). Another estimate suggested that currently 45 300 tonnes of cyanide are used in the USA in the cyanidation process. The wastes from these processes result in large cyanide-containing ponds near the mining operations (Clark & Hothem, 1991; Henny et al., 1994; Ma & Pritsos, 1997; Eisler et al., 1999).
The major point sources of cyanide release to water are discharges from gold mining plants, publicly owned wastewater treatment plants, iron and steel production, and the organic chemical industries. An estimated 3 billion litres (i.e., 3 × 109 litres) of wastes containing cyanides were generated in the USA in 1983, principally from spent cyanide plating bath solutions from electroplating operations (except for precious metals) and from spent stripping and cleaning bath solutions from electroplating operations (Grosse, 1986).
During cassava starch production, large amounts of cyanoglycosides are released and hydrolysed by plant-borne enzymes, leading to cyanide concentrations in wastewater as high as 200 mg/litre (Siller & Winter, 1998).
The major sources of cyanide released to air, in addition to exhaust from vehicle emissions, are diverse, including chemical manufacturing (hydrogen cyanide, methyl methacrylate, acrylonitrile); processing industries, such as metallurgical industries and metal plating (i.e., electroplating metals and finishing [metal polishes]); extraction of gold and silver from low-grade ores; volatilization from cyanide wastes disposed of in landfills and waste ponds; the production of coke or other coal carbonization procedures; emissions from municipal solid waste incinerators; and direct release of cyanides to the atmosphere resulting from fumigation operations, combustion of polyurethanes, acrylonitrile, and polyamide plastics, and combustion of wool, silk, and fibres (Carotti & Kaiser, 1972; Fiksel et al., 1981; ATSDR, 1997; Eisler et al., 1999).
An estimated total of 1 million tonnes of hydrogen cyanide, amounting to 73.1% of the total environmental releases in the USA, was discharged to the air from manufacturing and processing facilities (ATSDR, 1997).
The estimated amounts of hydrogen cyanide released to air in 1976 from the most common non-industrial sources were as follows: agricultural pest control, 62 tonnes; incineration, 8.2–82 tonnes; and tobacco smoke, 5.9–340 tonnes (Fiksel et al., 1981; ATSDR, 1997).
In 2001, from various locations in the USA, about 1300 tonnes of hydrogen cyanide were released on- and off-site; 540 tonnes were emitted to the atmosphere, 0.1 tonne was released to surface waters, 770 tonnes were injected into Class I wells,3 and 0.42 tonne was released to land (US EPA, 2003c). In 2001, from various locations in the USA, approximately 3400 tonnes of cyanides (not otherwise specified) were released on- and off-site; 220 tonnes were emitted to the atmosphere, 47 tonnes were released to surface waters, 1800 tonnes were injected into Class I wells, and 1300 tonnes were released to land (US EPA, 2003c).
Hydrogen cyanide has been found following the combustion of a number of synthetic polymers. The maximum yield of hydrogen cyanide per gram of polyurethane foam ranged from 0.37 to 0.93 mg under non-flaming conditions and from 0.5 to 1.02 mg under flaming combustion (Sklarew & Hayes, 1984). Hydrogen cyanide concentrations in the off-gas from the shale oil retorting process ranged from 7 to 44 mg/m3 (Sklarew & Hayes, 1984).
One cigarette without a filter liberates 500 µg hydrogen cyanide, while filter cigarettes liberate only 100 µg in mainstream smoke. Hydrogen cyanide concentrations in mainstream and sidestream smoke ranging from 280 to 550 µg/cigarette and from 53 to 111 µg/cigarette, respectively, have been reported; sidestream:mainstream ratios of hydrogen cyanide concentrations ranged from 0.06 to 0.50 (ATSDR, 1997). The level of hydrogen cyanide found in Canadian cigarette smoke under International Organization for Standardization standard smoking conditions were as follows: mainstream smoke, 32–156 µg/cigarette; and sidestream smoke, 77–136 µg/cigarette (Health Canada, 2002).
The average rate of emission of hydrogen cyanide by automobile exhaust was reported to be 7–9 mg/km for cars not equipped with catalytic converters and on the order of 0.6 mg/km for cars with catalytic converters operating under optimum conditions in the mid- to late 1970s (ATSDR, 1997).
Cyanogen chloride is formed as a reaction product of organic precursors with hypochlorous acid in the presence of ammonia and may be formed as a by-product of the chloramination of water (e.g., via the reaction of humic substances with chlorine and chloramine used for water disinfection) (Ohya & Kanno 1987; WHO, 1996; IPCS, 2000a). In the USA, 35% of the surface water plants and 23% of the groundwater plants using chloramine as a primary or secondary disinfectant report cyanogen chloride formation (US EPA, 2002).
Cyanogen is generated in the combustion of nitrogen–carbon compounds and appears in automobile exhaust gases and gases from blast furnaces (CHEMINFO, 1998).
Cyanide is present in the air mostly as a gas, and cyanides have the potential to be transported over long distances from their respective emission sources.
Cyanide is found in ambient air as hydrogen cyanide and to a smaller extent in particulate matter. The concentration of hydrogen cyanide measured since 1981 in the northern hemisphere’s non-urban troposphere ranged from 180 to 190 ng/m3 (Cicerone & Zellner, 1983; Jaramillo et al., 1989).
Ambient air monitoring data for cyanides in Bulgaria in areas near petrochemical plants showed concentrations ranging from 0.2 to 0.8 µg/m3 (annual average value) (Kaloyanova et al., 1985).
Cyanide has been detected at levels of 20–46 mg/m3 in the air near large-scale cassava processing facilities in Nigeria (Okafor & Maduagwu, 2000).
Cyanides, reported as cyanide, hydrogen cyanide, sodium cyanide, potassium cyanide, calcium cyanide, or copper(I) cyanide, have been detected in surface water samples at 70 of the 154 hazardous waste sites where they were studied in the USA; they have also been detected in groundwater samples at 191 of the 419 waste sites studied and in leachate samples of 16 of the 52 sites studied. The median concentrations in the positive samples were 160 µg/litre for groundwater, 70 µg/litre for surface water, and 479 µg/litre for the leachates (HazDat, 2003).
Data from the US National Urban Runoff Program in 1982 revealed that 16% of urban runoff samples collected from four cities across the USA contained cyanides at levels of 2–33 µg/litre (ATSDR, 1997).
According to the US Environmental Protection Agency’s (EPA) STORET database, the mean cyanide concentration in most surface waters in the USA is less than 3.5 µg/litre. Data from the late 1970s to early 1980s indicated that the levels are higher only in limited areas and may exceed 200 µg/litre (ATSDR, 1997).
In 1978, a US EPA survey of drinking-water supplies showed that about 7% of the supplies had cyanide concentrations greater than 10 µg/litre (US EPA, 1993a). Cyanogen chloride is one of the 18 compounds that occur most frequently (8 of 10 city surveys) in potable water within the framework of the US National Organic Reconnaissance Survey (Bedding et al., 1982). In a survey in 1987 of over 35 drinking-water supplies, the quarterly median cyanogen chloride concentrations in drinking-water ranged from 0.45 to 0.80 µg/litre (from 0.19 to 0.34 µg cyanide/litre) (Krasner et al., 1989; ATSDR, 1997). More current data regarding the cyanide and cyanogen chloride levels in drinking-water are lacking.
Levels of 1.58–7.89 mg cyanide/litre have been found in natural water sources near large-scale cassava processing facilities in Nigeria (Okafor et al., 2001).
Cyanide has been identified in the soil of hazardous waste sites in the USA; the median concentrations for the positive sites were 0.8 mg/kg in the subsurface soil (found at 77 sites of the 124 studied) and 0.4 mg/kg in the topsoil (51 positive sites out of 91 sites) (HazDat, 2003).
Cyanide-containing wastes are commonly found in soils at former manufactured gas plant sites in the USA. Most concentrations of cyanide compounds at the manufactured gas plant sites are below 2000 mg/kg. The most prevalent types of cyanide compounds are iron-complexed forms, e.g., ferric ferrocyanide (Prussian blue), rather than the highly toxic free cyanide forms. Iron-complexed cyanides, dominated by the ferrocyanide ion, comprise over 97% of total cyanides in either weathered or unweathered soils (Shifrin et al., 1996).
Many edible plants contain cyanogenic glycosides, whose concentrations can vary widely as a result of genetic and environmental factors, location, season, and soil types (Ermans et al., 1980; JECFA, 1993). Some of the foodstuffs and their cyanide contents are shown in Table 3. Cassava tubers vary widely in their cyanogenic glycoside content, although most varieties contain 15–400 mg cyanide/kg fresh weight. Occasionally varieties of cassava tubers contain 1300–2000 mg cyanide/kg fresh weight, and cassava leaves contain 1000–2000 mg cyanogenic glucosides/kg on a dry matter basis (Padmaja, 1995). Fermentation of cassava pulp for 96 h during gari production reduced the hydrogen cyanide content by 50%; soaking of sliced cassava for 24 h, 40%; and sun-drying, some 15% (Kendirim et al., 1995). It should be noted that the ranges of cyanide concentrations shown in Table 3 are very broad in several cases (i.e., cereals and their products, soy protein products, and apricot pits), which may be due to their different sources and differences in analytical procedures; as well, the values may reflect the older literature.
Hydrogen cyanide can be produced by hydrolytic reaction catalysed by one or more enzymes from the plants containing cyanogenic glycosides. In kernels, for example, this reaction is catalysed by the enzyme emulsin (Lasch & El Shawa, 1981) when the seeds are crushed and moistened. Amygdalin (which is also present in cassava, bitter almonds, and peach stones) is converted to glucose, benzaldehyde, and hydrogen cyanide (Figure 2) (IPCS, 1992). Hydrogen cyanide release can occur during maceration, which activates intracellular beta-glucosidases. This reaction can also result from chewing, which causes the enzyme and the cyanogenic glycosides stored in different compartments to combine (Ermans et al., 1980; Nahrstedt, 1993). The reaction occurs rapidly in an alkaline environment, and the hydrolysis is complete in 10 min. Hydrolysis is possible in an acid solution and takes place slowly.
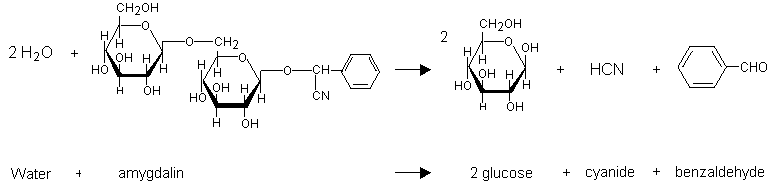
Fig. 2: Hydrolysis of amygdalin
Liberation of hydrogen cyanide from cyanogenic glycosides occurs usually after ingestion and hydrolysis by the glycosidases of the intestinal microflora and, to a lesser degree, by glucosidases of the liver and other tissues (Padmaja, 1995). However, hydrolysis may also occur during the preparation of the food, which may account for the short interval between ingestion and the appearance of signs of poisoning in some accidents (Lasch & El Shawa, 1981).
Table 3: Cyanide concentrations in food products.a
|
Type of product |
Cyanide concentration |
|
Cereal grains and their products |
0.001–0.45 |
|
Soy protein products |
0.07–0.3 |
|
Soybean hulls |
1.24 |
|
Apricot pits, wet weight |
89–2170 |
|
Home-made cherry juice from pitted fruits |
5.1 |
|
Home-made cherry juice containing 100% crushed pits |
23 |
|
Commercial fruit juices |
|
|
Cherry |
4.6 |
|
Apricot |
2.2 |
|
Prune |
1.9 |
|
Tropical foodstuffs |
|
|
Cassava (bitter) / dried root cortex |
2360 |
|
Cassava (bitter) / leaves |
300 |
|
Cassava (bitter) / whole tubers |
380 |
|
Cassava (sweet) / leaves |
451 |
|
Cassava (sweet) / whole tubers |
445 |
|
Gari flour (Nigeria) |
10.6–22.1 |
|
Sorghum / whole immature plant |
2400 |
|
Bamboo / immature shoot tip |
7700 |
|
Lima beans from Java (coloured) |
3000 |
|
Lima beans fom Puerto Rico (black) |
2900 |
|
Lima beans from Burma (white) |
2000 |
a From Nartey (1980); Honig et al. (1983); JECFA (1993); ATSDR (1997).
Laetrile (another name for amygdalin derived from apricot kernels), which was formerly used as an anticancer agent, releases cyanide upon metabolism. Bitter almonds and apricot pits containing cyanogenic glycosides are still sold in health food stores and over the Internet (Suchard et al., 1998). Other drugs, such as sodium nitroprusside, which is used as an antihypertensive and in congestive heart failure (Guiha et al., 1974; Tinker, 1976; Aitken et al., 1977; Schultz, 1984; Rindone & Sloane 1992), also liberate hydrogen cyanide in the body. In sodium nitroprusside, the CN– moiety represents 44% by weight of the molecule. Some aliphatic nitriles that are widely used in the chemical industry — i.e., acetonitrile (IPCS, 1993), acrylonitrile (IARC, 1999), succinonitrile, and adiponitrile — also release cyanide upon metabolism (Willhite & Smith, 1981).
The general population may be exposed to cyanide from ambient air, drinking-water, and food.
Based on an atmospheric hydrogen cyanide concentration of 190 ng/m3 and an average daily inhalation of 20 m3 air, the inhalation exposure of the general US non-urban, non-smoking population to hydrogen cyanide is estimated to be 3.8 µg/day (ATSDR, 1997).
Based on a daily drinking-water consumption of 2 litres for an adult, the daily intake of cyanogen chloride is estimated to be 0.9–1.6 µg (equivalent to 0.4–0.7 µg of cyanide) (ATSDR, 1997) for cyanogen chloride concentrations in water of 0.45–0.80 µg/litre (0.19–0.34 µg cyanide/litre).
Among the general population, subgroups with the highest potential for exposure to cyanide include active and passive smokers, individuals involved in large-scale processing of foods high in cyanogenic glycosides, individuals consuming foods high in cyanogenic glycosides, and, to a lesser degree, fire-related smoke inhalation victims.
Human exposure to cyanide by dietary intake is estimated to be potentially of major significance for cassava-consuming populations; cassava has been estimated to be the staple food for 500 million people. However, data on the concentrations of cyanides in the total diet are lacking; hence, the daily cyanide intake from food cannot be calculated. For human consumption, cassava can be eaten raw, cooked, or grated and roasted into flour and eaten as "gari," which is the common form in Nigeria (Kendirim et al., 1995). In Mozambique, it was estimated that in families affected by the "mantakassa" disease (spastic paraparesis; see section 8), the daily intake of cyanogens was 14–30 mg (as cyanide) at the time of a mantakassa epidemic in 1981 (Ministry of Health, Mozambique, 1984b). In Nigeria, it was estimated that the intake of hydrogen cyanide in the tropical ataxia-endemic areas may be as high as 50 mg/day (Osuntokun, 1981).
Urinary excretion of thiocyanate has been applied in the biological monitoring of exposure to cyanogenic glycosides, especially among cassava-consuming populations. The average urinary thiocyanate concentration among children in the Bandundu region of the Democratic Republic of the Congo (formerly Zaire) was 757 µmol/litre in the south and 50 µmol/litre in the north (both populations consumed cassava as their staple diet, but the cassava was well processed in the north and inadequately processed in the south). These concentrations can be compared with an average of 31 µmol/litre in a non-smoking Swedish reference population (Banea-Mayambu et al., 2000). In the same Bandundu region, it was shown that there was a marked seasonal variation in urinary thiocyanate concentrations in the villages with a high "konzo" (spastic paraparesis) incidence (563–627 µmol/litre in the dry season and 344–381 in the wet season), while the average in non-konzo areas was 241 µmol/litre (Banea-Mayambu et al., 1997). In Mozambique, the average urinary thiocyanate levels among healthy children from areas with epidemic spastic paraparesis varied between 33 and 1175 µmol/litre, whereas levels in areas with no paraparesis were between 18 and 400 µmol/litre (Casadei et al., 1990). In Nampula province in Mozambique, where spastic paraparesis epidemics had been observed in 1981–1982 and during the civil war in 1992–1993, average urinary thiocyanate concentrations among schoolchildren in five areas were between 225 and 384 µmol/litre in October 1999 (Ernesto et al., 2002). In Malawi, in an area where cassava was typically soaked for 3–6 days for processing to flour, urinary thiocyanate concentrations were between 2 and 410 µmol/litre, with a median of 32 µmol/litre (Chiwona-Karltun et al., 2000).
The principal routes of occupational exposure to cyanides are via inhalation and, to a lesser degree, skin absorption. Skin absorption may be significant under some circumstances — for example, when airborne concentrations are very high, such as in fumigation operations. It may occur as well when personal protection is inadequate and operators are splashed.
Workers involved in electroplating, metallurgy, pesticide application, firefighting, gas works operations, tanning, blacksmithing, metal cleaning, photoengraving, photography, and the manufacture of steel, cyanides, adiponitrile and other nitriles, methyl methacrylate, cyanuric acid, dyes, pharmaceuticals, or chelating agents have the potential to be occupationally exposed to higher concentrations of cyanide (Prohorenkov & Kolpakov, 1978; Philips, 1989; IPCS, 1992; Banerjee et al., 1997).
A number of illustrative levels of cyanide in the breathing zone of workers in working environments monitored at different production facilities in the USA during the period 1976–1982 have been reported. The concentration of cyanide in air at a plating facility of a national airline was 0.001–0.004 mg/m3 (NIOSH, 1982). Concentrations of hydrogen cyanide in air at a plating facility of an electrical and electronic company in Virginia, USA, ranged from 0.07 mg/m3 in a salt pot room to 4.3 mg/m3 in a stripping tank (NIOSH, 1976). The concentration of cyanide in air at a plating facility in Ohio, USA, was 1.7 mg/m3 (NIOSH, 1978).
Hydrogen cyanide is readily absorbed following inhalation, oral, and dermal exposure. Following exposure to cyanide in the atmosphere, toxic amounts of cyanide are absorbed with great rapidity through the bronchial mucosa and alveoli (ATSDR, 1997). Humans retained 58% of the hydrogen cyanide in the lungs after inhaling the gas through normal breathing (Landahl & Herrmann, 1950; ATSDR, 1997). Alkali metal cyanides are rapidly absorbed from the gastrointestinal tract. Absorption is affected by the presence of food in the gut, the pH of the gut, and the lipid solubility of the cyanide compound.
Gastrointestinal absorption of inorganic cyanide salts is slower than pulmonary absorption, and the onset of symptoms is delayed and the severity of symptoms diminished compared with inhalation. When simple cyanide salts such as potassium and sodium cyanide are ingested, free cyanide ion can rapidly bind hydrogen ion to form hydrogen cyanide in the highly acidic medium of the stomach. Essentially all cyanide ingested as cyanide salts will form hydrogen cyanide and will be quickly absorbed. However, after oral intake, only part of the dose reaches the blood due to first-pass metabolism by the liver (ECETOC, 2004).
Liquid cyanide compounds are easily absorbed through intact skin upon direct contact due to their lipid solubility and rapid epidermal penetration. Skin absorption of vapours of hydrogen cyanide is also possible when the air concentrations are high. The amount and rate of absorption of cyanides from aqueous solutions or atmospheric hydrogen cyanide depend upon the presence of moisture in the skin, concentration and pH of the solution, the surface area of contact, and the duration of contact (Dugard, 1987). In vitro studies with human skin have shown that penetration of sodium cyanide in aqueous solution through skin decreases with increasing pH (increasing dissociation), reflecting the more rapid absorption of the undissociated hydrogen cyanide. The permeability constant measured for the cyanide ion in aqueous solution was 3.5 × 10–4 cm/h, and that calculated for hydrogen cyanide was 1 × 10–4 cm/h (Dugard, 1987).
Hydrogen cyanide has a pKa of 9.22; thus, at physiological pH (about pH 7), hydrocyanic acid is distributed in the body as hydrogen cyanide and is not present as the free cyanide ion. Hence, the form of cyanide to which exposure occurs, the salt or the free acid, does not influence distribution, metabolism, or excretion from the body (ECETOC, 2004). Inhaled or percutaneously absorbed hydrogen cyanide passes immediately into the systemic circulation. The distribution of cyanide to the various tissues is rapid and fairly uniform. Somewhat higher levels are generally found in the liver, lungs, blood, and brain. The tissue levels of hydrogen cyanide were 0.75, 0.42, 0.41, 0.33, and 0.32 mg/100 g of tissue in lung, heart, blood, kidney, and brain, respectively, in a man who died following inhalation exposure to hydrogen cyanide gas (Gettler & Baine, 1938; Ballantyne, 1983a; ATSDR, 1997; ECETOC, 2004). In contrast, high proportions of ingested sodium and potassium cyanide will pass through the liver and are detoxified by the first-pass effect.
The major portion of cyanide in blood is sequestered in the erythrocytes, and a relatively small proportion is transported via the plasma to target organs. Cyanide is concentrated in red blood cells at a red blood cell to plasma ratio of 199:1; levels in plasma reflect tissue levels better than levels in whole blood or erythrocytes. Small but significant levels of cyanide are found in normal blood plasma (<140 µg/litre) and other tissues (<0.5 mg cyanide/kg) of humans without known occupational cyanide exposure (Feldstein & Klendshoj, 1954). These levels are related mostly to exposure to cyanogenic food, vitamin B12, and tobacco smoke. A detailed survey of normal plasma cyanide levels in 10 cases showed a maximum level of 106 µg/litre, with a mean of 48 µg/litre (Feldstein & Klendshoj, 1954). After cessation of exposure, plasma cyanide levels tend to return to normal within 4–8 h (Feldstein & Klendshoj, 1954; Ansell & Lewis, 1970).
In rats dosed by gavage, highest concentrations of cyanide were found in the liver, followed by the lungs and blood (Yamamoto et al., 1982). After inhalation exposure, the highest concentrations of cyanide in rats were found in the lungs, followed by the blood and liver.
Cyanide has not been shown to accumulate in the blood and tissues following oral exposue to inorganic cyanide (ATSDR, 1997), and no cumulative effect on the organism during repeated exposure has been demonstrated. There is a cumulative effect of exposure to thiocyanate (from the breakdown of cyanogenic glycosides in food plants), resulting in thyroid toxicity, including goitre and cretinism (Nahrstedt, 1993).
A number of illustrative levels of cyanide in organs and blood after oral intake in humans (Ansell & Lewis, 1970; ATSDR, 1997) and rabbits (Ballantyne, 1983a) have been reported. For a given exposure route, whole blood and serum cyanide levels are quite similar for different species (Ballantyne, 1983a).
Although cyanide can interact with substances such as methaemoglobin in the bloodstream, the majority of cyanide metabolism occurs within the tissues. Cyanide is metabolized in mammalian systems by one major route and several minor routes. The major route of metabolism for hydrogen cyanide and cyanides is detoxification in the liver by the mitochondrial enzyme rhodanese, which catalyses the transfer of the sulfane sulfur of thiosulfate to the cyanide ion to form thiocyanate (Figure 3) (Williams, 1959; Ansell & Lewis, 1970). About 80% of cyanide is detoxified by this route. The rate-limiting step is the amount of thiosulfate. While rhodanese is present in the mitochondria of all tissues, the species and tissue distributions of rhodanese are highly variable. In general, the highest concentrations of rhodanese are found in the liver, kidney, brain, and muscle, but the supply of thiosulfate is limited (Aminlari et al., 1994). Rhodanese is present in rat nasal mucosal tissues, particularly in the olfactory region, at a 7-fold higher concentration (on a per milligram of mitochondrial protein basis) than in the liver (Dahl, 1989). Dogs have a lower overall activity of rhodanese than monkeys, rats, and rabbits (ATSDR, 1997).
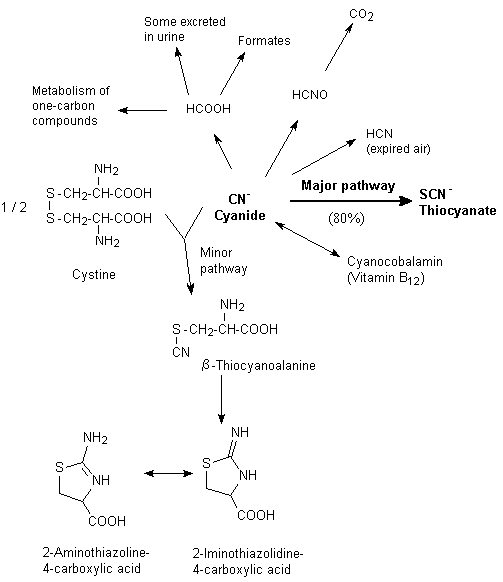
Fig. 3: Basic processes involved in the metabolism of cyanide (ATSDR, 1997)
A number of other sulfur transferases can also metabolize cyanide, and albumin, which carries elemental sulfur in the body in the sulfane form, can assist in the catalysis of cyanide to thiocyanate as well (Sylvester et al., 1982; Westley et al., 1983). Cyanide and thiocyanate can also be metabolized by several minor routes, including the combination of cyanide with hydroxycobalamin (vitamin B12a) to yield cyanocobalamin (vitamin B12) (Boxer & Rickards, 1952) and the non-enzymatic combination of cyanide with cystine, forming 2-iminothiazoline-4-carboxylic acid, which appears to be excreted without further change (Rieders, 1971) (Figure 3).
In studies with rats orally administered potassium cyanide and maintained for up to 4 weeks on either a balanced diet or a diet lacking the sulfur amino acids L-cystine and L-methionine, a strongly positive linear relationship was found between blood cyanide and plasma cyanate (OCN–) concentration (Tor-Agbidye et al., 1999). It was suggested that in Africa, where there are protein-deficient populations whose levels of sulfur-containing amino acids are low, cyanide (from prolonged use of cassava) may conceivably be converted to cyanate, which is known to cause neurodegenerative disease in humans and animals.
While absorbed cyanide is principally excreted as thiocyanate in the urine, traces of free hydrogen cyanide may also be excreted unchanged in the lungs, saliva, sweat, or urine (Hartung, 1982), as carbon dioxide in expired air, or as beta-thiocyanoalanine in saliva and sweat (Friedberg & Schwartzkopf, 1969; Hartung, 1982; JECFA, 1993).
Thiocyanate was found in the urine of non-exposed people at average concentrations of 2.16 mg/litre urine for non-smokers and 3.2 mg/litre urine for smokers (Chandra et al., 1980). Urinary excretion of thiocyanate was monitored in a man after ingestion of about 3–5 g potassium cyanide (15–25 mg cyanide/kg body weight) (Liebowitz & Schwartz, 1948; ATSDR, 1997). The results indicated that the patient excreted 237 mg of thiocyanate over a 72-h period. This quantity was substantially more than the normal average amount of thiocyanate in urine, which varies from 0.85 to 14 mg/24 h (ATSDR, 1997).
The limiting factor in cyanide metabolism is the low concentration of the sulfur-containing substrates in the body — primarily thiosulfate, but also cystine and cysteine. The rate of spontaneous detoxification of cyanide in humans is about 1 µg/kg body weight per minute (Schultz et al., 1982), which is considerably slower than in small rodents (Schubert & Brill, 1968) or dogs (Lawrence, 1947).
After administration of an intravenous dose of 3–4 mg potassium cyanide to beagle dogs, blood levels decreased in a manner consistent with first-order elimination kinetics for the first 80 min (Bright & Marrs, 1988). The half-time for this phase was about 24 min, corresponding to an elimination rate constant of 0.03/min. After 80 min, the blood cyanide concentrations fell at a slower rate, with a half-time of 5.5 h. In rats, after a single oral dose, the blood elimination half-time of cyanide was 14.1 min, corresponding to a rate constant of 0.05/min (Leuschner et al., 1991).
Rats treated orally with 2 mg cyanide/kg body weight excreted 47% of the dose in the urine within 24 h (Farooqui & Ahmed, 1982). A [14C]cyanide intake study with rats (exposed to a regular intake of cyanide in the diet for 3 weeks) indicated the existence of a gastrointestinal circulation of thiocyanate, in which a substantial amount of thiocyanate, which was excreted into the stomach contents of the rat, was reabsorbed by the intestine into the body fluid, to be partly excreted in the urine and partly resecreted into the gastric contents (Okoh & Pitt, 1982). The relative proportion of cyanide to thiocyanate in body fluids is about 1:1000 (Pettigrew & Fell, 1973). The half-time for hydrogen cyanide elimination is about 1 h (Ansell & Lewis, 1970; IPCS, 1992).
Half-time values of the principal metabolite thiocyanate in humans have been reported as 4 h (Blaschle & Melmon, 1980), 2 days (Bödigheimer et al., 1979), and 2.7 days (Schultz et al., 1979). In patients with renal insufficiency, a mean half-time of 9 days was reported (Bödigheimer et al., 1979).
Levels of the cyanide metabolite thiocyanate in blood serum and plasma and urine have been employed as indicators of high cyanide exposure in humans (Lauwerys & Hoet, 2001). However, at low levels of occupational exposure, the relationship between exposure and urinary thiocyanate concentrations shows a wide inter- and intraindividual variation due to a variety of factors (e.g., diet); therefore, measuring cyanide and/or thiocyanate levels in blood and urine is not a reliable biomarker for exposure to low concentrations of cyanide.
Symptoms of cyanide toxicity can occur within seconds of inhalation of hydrogen cyanide or within minutes of ingestion of cyanide salts. Onset may be delayed up to 12 h after ingestion of cyanogenic glycosides, nitriles, or thiocyanates.
Inhalation LC50 values of hydrogen cyanide in rats ranged from 158 mg/m3 for 60 min to 3778 mg/m3 for 10 s (Ballantyne, 1983a). Exposure of mice to cyanide resulted in similar LC50 values (Higgins et al., 1972; Matijak-Schaper & Alarie, 1982), and LC50 values of hydrogen cyanide in rabbits ranged from 2432 mg/m3 for 45 s to 208 mg/m3 for 35 min (Ballantyne, 1983a). The concentration of hydrogen cyanide in inhaled air markedly affects the acute toxicity: the total amount of hydrogen cyanide inhaled leading to death is disproportionately larger at low exposure levels than at high exposure levels (and thus the time leading to death is disproportionately longer) (Table 4). A similar dose rate dependence of acute toxicity was observed in hamsters: (pregnant) hamsters did not show signs of toxicity until they had received a total dose of 30–40 times the single subcutaneous LD50 when given sodium cyanide subcutaneously by an osmotic minipump (Doherty et al., 1982).
Table 4: Acute inhalation toxicity of hydrogen cyanide vapour in rats.a
|
Exposure time |
Median lethal toxicity |
|
|
as LC50 |
as total doseb |
|
|
10 s |
3778 |
631 |
|
1 min |
1471 |
1129 |
|
5 min |
493 |
2463 |
|
30 min |
173 |
5070 |
|
60 min |
158 |
9441 |
a From Ballantyne (1983a).
b Details of calculation not provided.
A similar dose rate dependence has also been noted for oral exposure. While the single-dose gavage LD50 of potassium cyanide was 10 mg/kg body weight in Sherman rats, no mortality was observed when a dose of 250 mg/kg body weight was given in the diet for 90 days. The author ascribed this remarkable difference to the difference in dose rate (bolus vs. dietary exposure): at the low dose rate, the liver is capable of detoxifying cyanide before it reaches the general circulation (Hayes, 1967).
The information on the relative sensitivity of various animals to hydrogen cyanide vapours is mainly based on early studies by Barcroft (1931). In an extensive series of inhalation experiments, in which different species of animals were exposed to usually 5–8 different concentrations of hydrogen cyanide and followed until death, he showed that the lethal time (time for 50% of animals to die at 1000 mg/m3) was 0.8, 1.0, 1.0, 1.0, 2.0, 2.0, 3.0, and 3.5 min for dogs, mice, cats, rabbits, rats, guinea-pigs, goats, and monkeys. Extrapolation to zero mortality gave the following maximal non-lethal concentrations to dogs, rats, mice, rabbits, monkeys, cats, goats, and guinea-pigs: 100, 100, 140, 180, 180, 180, 240, and 400 mg/m3. There was thus a rough inverse relationship between sensitivity to hydrogen cyanide and body size, dogs being a notable exception.
Severe dyspnoea has been observed in dogs exposed to 170–740 mg hydrogen cyanide/m3 for 2–12 min. Pulmonary oedema was found in some dogs at necropsy (Haymaker et al., 1952). Exposure of cynomolgus monkeys to 110–180 mg hydrogen cyanide/m3 led quickly to incapacitation; the time to incapacitation was inversely related to the level of exposure, being 8 min for 180 mg/m3 and 19 min for 110 mg/m3 (Purser et al., 1984). At 70 mg/m3 for 30 min, slight depression of the central nervous system was reported (Purser, 1984).
Following oral administration to rats, LD50s of hydrogen cyanide, sodium cyanide, and potassium cyanide are very similar: 0.156, 0.117, and 0.115 mmol/kg body weight, respectively, i.e., 3–4 mg cyanide/kg body weight (Ballantyne, 1983a4). In mice, an LD50 of 15.8 mg potassium cyanide (corresponding to 6 mg cyanide)/kg body weight has been reported (Ferguson, 1962). In rabbits, hydrogen cyanide, potassium cyanide, and sodium cyanide appear equitoxic on a molar basis (LD50s of 0.092, 0.104, and 0.090 mmol/kg for hydrogen cyanide, sodium cyanide, and potassium cyanide, respectively); rabbits appeared to be somewhat more susceptible to cyanides than mice or rats (Ballantyne, 1983a).
Following application of cyanides in aqueous solution to the intact skin of New Zealand rabbits, the dermal LD50s of hydrogen cyanide, sodium cyanide, and potassium cyanide were 0.260, 0.298, and 0.343 mmol/kg body weight, respectively (corresponding to 6.8, 7.7, and 8.9 mg cyanide/kg body weight) (Ballantyne, 1983a). The dermal toxicity of cyanide, especially of hydrogen cyanide, is markedly greater following application on abraded skin, which enhances the penetration of cyanide (LD50s of 0.087, 0.220, and 0.30 mmol/kg for hydrogen cyanide, sodium cyanide, and potassium cyanide, respectively) (Ballantyne, 1987).5 Local contact may produce mild burns (IPCS, 1992).
In a range-finding toxicity study on ACH, the oral LD50 in rats and dermal LD50 in rabbits were both 17 mg ACH (5.2 mg cyanide)/kg body weight; 4-h inhalation mortality in rats was 2/6 at 220 mg/m3 and 6/6 at 440 mg/m3 (Smyth et al., 1962). In a 4-week inhalation study, in which the average measured concentrations of ACH were 33, 106, and 211 mg/m3, 3/10 male rats died after the first 6-h exposure at the highest concentration (Monsanto Co., 1985c). On this first day of exposure, the four measured concentrations of ACH in the chamber were 196, 214, 225, and 225 mg/m3, 225 mg/m3 being the highest individual concentration measured during the 1-month exposure (when no further mortality was observed). No similar acute mortality was observed in a 14-week study at exposures up to 204 mg/m3, in the male fertility study (202 mg/m3), or in the female fertility study (207 mg/m3) (see sections 7.3 and 7.6) (Monsanto Co., 1984a, 1985a,b).
There are no qualitative differences in acute poisoning between cyanide compounds, since the cyanide ion is the common agent that primarily inhibits tissue cytochrome oxidase activity in rats, mice, and rabbits, with resulting anoxia (Way, 1984; US EPA, 1988). Although acute oral doses of cyanide cause cardiovascular, respiratory, and neuroelectric alterations, many studies have shown that the brain is the organ most sensitive to cyanide toxicity. Death from cyanide poisoning is believed to result from central nervous system depression, subsequent to inhibition of brain cytochrome oxidase activity (Way, 1984). Typical signs of toxicity after inhalation of hydrogen cyanide in test species include rapid breathing, weak and ataxic movements, convulsions, loss of voluntary movement, coma, and decrease and irregularities in respiratory rate and depth preceding death (Ballantyne, 1983b; European Chemicals Bureau, 2000a,b).
Forty-six male adult inbred Wistar rats were used in four experimental groups and one control group and treated with 0, 0.3, 0.9, 3.0, or 9.0 mg potassium cyanide/kg body weight per day in the drinking-water for 15 days. This was equivalent to 0, 0.12, 0.36, 1.2, and 3.6 mg cyanide/kg body weight per day. The high-dose group exhibited a 70% lower body weight gain than the control animals. In qualitative histological analysis, without statistical treatment or morphometric analysis, changes were observed in the kidney, liver, and thyroid. Cytoplasmic vacuolation, considered to reflect hydropic degeneration of proximal tubular epithelial cells, was noted in animals treated at doses of 3.0–9.0 mg potassium cyanide/kg body weight per day and in hepatocytes of those animals treated at a dose of 9.0 mg potassium cyanide/kg body weight per day. A dose-dependent increase in the number of reabsorption vacuoles on follicular colloid in the thyroid gland was noted in all animals of the experimental groups. No changes were observed in serum triiiodothyronine (T3), thyroxine (T4), creatinine, or urea levels; a decrease was observed in serum alanine aminotransferase (ALAT) activity at the two lowest exposure levels. Serum aspartate aminotransferase (ASAT) was elevated by 30% at the two lowest dose levels and by 21% at the 3.0 mg potassium cyanide/kg body weight per day dose; it was decreased by 29% at the highest dose level (Sousa et al., 2002).
No statistically significant increase in the incidence of histopathological changes in the lungs or cardiovascular tissues (e.g., myocardial ultrastructure) compared with the controls was noted in rabbits continuously exposed to 0.6 mg hydrogen cyanide/m3 for 1 or 4 weeks (Hugod, 1981).
Dogs exposed to 50 mg hydrogen cyanide/m3 for 30 min every third day for 28 days exhibited extensive vascular and cellular central nervous system lesions, including vasodilation and haemorrhages (Valade, 1952).
Sprague-Dawley rats (10 per sex and dose level) were exposed to ACH in the atmosphere at average concentrations of 33, 106, or 211 mg/m3 for 6 h/day, 5 days/week, for 4 weeks. This was equivalent to hydrogen cyanide concentrations of 10, 34, and 67 mg/m3 (Monsanto Co., 1985c). On the first exposure day, the highest concentration of ACH in the chamber was 225 mg/m3, corresponding to 71 mg hydrogen cyanide/m3, and 3 out of the 10 males at the highest dose level died after the first exposure (see section 7.1). Irritation of the nose and eyes was observed at the two highest exposure levels. High-dose females had decreased blood haemoglobin and elevated blood urea nitrogen concentrations, and total serum protein levels were decreased in high- and mid-dose males; these laboratory changes were within the reference range. No gross or microscopic changes were observed in a wide range of organs at autopsy. No changes were observed in thyroid function, with the exception of an elevated T3 level in the mid-dose males. The NOAEL (the observed effect being irritation) reported from the study was 33 mg ACH/m3 (10 mg hydrogen cyanide/m3); this can be estimated to correspond to a daily dose of 2.7 mg cyanide/kg body weight per day.6
In 13-week studies, groups of F344/N rats and B6C3F1 mice (10 of each sex) were administered 0, 3, 10, 30, 100, or 300 mg sodium cyanide/litre in drinking-water (NTP, 1993). The equivalent cyanide ion doses were 0, 0.2, 0.5, 1.4, 4.5, and 12.5 (in males) and 0, 0.2, 0.5, 1.7, 4.9, and 12.5 (in females) mg cyanide/kg body weight per day for rats and 0, 0.3, 1, 3, 9, and 26 mg cyanide/kg body weight per day for mice. No deaths, clinically significant effects on body or organ weights, or histopathological or clinical pathology changes were noted in either rats or mice. In particular, no lesions were found in the brain or thyroid gland. Effects on the reproductive organs were analysed in animals in the three highest dose groups. A slight (7–13%) but statistically significant reduction was observed in sperm motility and in the weight of cauda epididymidis in all studied groups of male rats. In the males of the 300 mg/litre group (12.5 mg cyanide/kg body weight per day), a statistically significant decrease was observed in the weight of the left epididymis, left cauda epididymidis, left testis, and the number of spermatid heads per testis. Sodium cyanide at concentrations of 100 and 300 mg/litre (4.9 and 12.5 mg cyanide/kg body weight per day, respectively) caused a statistically significant increase in the time spent by female rats in proestrus and diestrus relative to estrus and metestrus. In male mice, a statistically significant decrease in the left cauda epididymidis weights was noted at 26 mg cyanide/kg body weight per day, but no changes in sperm motility or spermatid head density were observed. No changes in the estrous cycle length in female mice were noted. The authors noted that the changes in male rats are consistent with a small but measurable adverse effect on reproduction. While these changes are insufficient to decrease fertility in rats, the relative sensitivity of humans to such changes is considered to be greater than that of rats; therefore, a potential for adverse reproductive effects exists in humans (NTP, 1993). One of the source documents (ATSDR, 1997) identified 12.5 mg cyanide/kg body weight per day as the lowest-observed-adverse-effect level (LOAEL), based on all the effects on reproductive organs observed in the male rats, and 4.5 mg cyanide/kg body weight per day as the NOAEL; the findings in female rats were not considered adverse (ATSDR, 1997).7
In a 13-week study, male Sprague-Dawley rats were administered potassium cyanide in drinking-water at a dose level of 40, 80, or 160/140 mg/kg body weight per day. These doses correspond to 16, 32, and 64/56 mg cyanide/kg body weight per day (Leuschner et al., 1989). Histopathological investigation of the brain, heart, liver, testes, thyroid, and kidneys did not reveal adverse effects. Urinary protein excretion was increased in dosed animals, and dose-dependent increases were observed in organ weights; these were interpreted to have arisen from decreased food and water consumption caused by decreased palatability.8
All rats survived, but there was a dose-dependent loss of body weight, an increase in thyroid weight, and a decrease of blood haemoglobin and serum T4 levels in rats after 14 weeks on a diet containing 5 or 10 g potassium cyanide/100 g diet (corresponding to approximately 800 and 1600 mg cyanide/kg body weight per day) (Olusi et al., 1979).
In a 3-month study, weanling male Wistar rats were given potassium cyanide at 0, 0.15, 0.3, or 0.6 mg/kg body weight per day daily (0, 0.06, 0.12, and 0.24 mg cyanide/kg body weight per day) by gavage (Soto-Blanco et al., 2002b). In plasma samples collected on the last day of the administration, no changes were observed in the concentrations of T3, T4, or glucose, while a decrease was observed in the concentration of cholesterol, significant at the highest dose (45%, P < 0.05). The authors made a qualitative statement that dose-dependent neuropathological findings were observed, including spheroids on the ventral horn of the spinal cord, neuron loss in the hippocampus, damaged Purkinje cells, and loss of cerebellar matter. No further details or statistical analysis was presented.
No effects were noted in Sprague-Dawley rats fed potassium cyanide at concentrations up to 187.5 mg/100 g diet (750 mg cyanide/kg diet) for 56 days. On protein-deficient diets, the lowest body weight gain was obtained at the highest dietary cyanide concentration (Tewe & Maner, 1985).
In a 40-week study in rabbits, the animals were fed potassium cyanide at a level of 1.76 g/kg diet (corresponding to 24–17 mg cyanide/kg body weight per day) (Okolie & Osagie, 1999). The weight gain of the treated animals was decreased by 33%; at the end of the experimental period, serum urea and creatinine levels were elevated, as were the activities of serum lactate dehydrogenase, sorbitol dehydrogenase, ALAT, and alkaline phosphatase.
In a neuropathological study (Soto-Blanco et al., 2002a), goats, 30–45 days old at the beginning of the study, were given potassium cyanide in milk (until weaning) and in drinking-water thereafter at a dose level of 0.3, 0.6, 1.2, or 3.0 mg (0.12, 0.24, 0.48, or 1.2 mg cyanide)/kg body weight per day for 5 months. In a qualitative morphological and immunohistochemical study, presence of gliosis and spongiosis in the medulla oblongata and spinal cord and gliosis in the pons and damage to Purkinje cells in the cerebellum were observed at the highest dose, but no increase in apoptotic cells was reported. Congestion and haemorrhage in the cerebellum were observed at the 0.48 mg cyanide/kg body weight per day group. No quantification or statistical analysis of the findings was presented.
Hyperactivity tremors, convulsions, and laboured breathing were noted in Sprague-Dawley rats exposed to 7.8 mg cyanide/kg body weight per day as copper cyanide for 90 days by gavage. No similar effects were reported at 1.45 mg cyanide/kg body weight per day. Laboured respiration occurred in rats exposed at a lower dose of 0.8 mg cyanide/kg body weight per day when administered potassium silver cyanide for 90 days (Gerhart, 1987).
The only sign of toxicity observed in male rats exposed by gavage to copper cyanide (14.5 mg cyanide/kg body weight per day) (Gerhart, 1986) or potassium silver cyanide (2.6 mg cyanide/kg body weight per day) for 90 days was an increase in testis weight (Gerhart, 1987). No effects were observed in female rats in either study. The NOAELs were 4.35 mg cyanide/kg body weight per day (Gerhart, 1986) and 0.8 mg cyanide/kg body weight per day (Gerhart, 1987).
Kamalu and co-workers (Kamalu & Agharanya, 1991; Kamalu, 1993) compared the effects of cassava containing linamarin with those of a diet containing an equivalent amount of sodium cyanide in three groups of growing dogs, each comprising six animals, for 14 weeks. One group was fed cassava (gari) as the carbohydrate source, which was expected to release 10.8 mg hydrogen cyanide/kg cooked food; another group was fed on the control diet with rice as the carbohydrate source, to which enough sodium cyanide was added at feeding time to release 10.8 mg hydrogen cyanide/kg cooked food (for both, the daily dose was 1.08 mg hydrogen cyanide/kg body weight). The control group was fed this rice diet without added sodium cyanide. Nephrosis and changed plasma free amino acid profile were observed in the sodium cyanide-treated group, while no effect was observed in the plasma glutamyltransferase, ALAT, or isocitrate dehydrogenase activities or in the histology of the liver, kidney, or myocardium. Adrenal hyperplasia and hypertrophy and pancreatic necrosis and fibrosis were observed. (In contrast, the gari diet caused generalized congestion and haemorrhage, periportal vacuolation of the liver, swelling, vacuolation, and rupture of the epithelial cells of the proximal convoluted tubule of the kidney, myocardial degeneration, adrenal gland degeneration, and pancreatic haemorrhage, necrosis, and fibrosis.) A 36% decrease in the serum T3 concentration was noted, together with histological changes of the thyroid consistent with parenchymatous goitre. A significantly reduced frequency of testicular tubules in stage 8 of the spermatogenic cycle as well as marked testicular germ cell sloughing and degeneration were also observed.
Groups of 30 male albino rats (Charles River) were each exposed for 6 months (6 h/day, 5 days/week) by inhalation to 0, 24, or 54 mg cyanogen/m3 (corresponding to 0, 25, and 56 mg hydrogen cyanide/m3). There were no effects on haematological or clinical chemistry parameters, gross pathology, or histopathology (liver, kidney, cardiovascular system) attributable to the cyanogen exposure. Body weights were significantly lower in rats exposed to 54 mg cyanogen/m3 than in the controls (Lewis et al., 1984).
In an inhalation study, groups of five rhesus monkeys (Macacca mulatta) were exposed to 24 or 54 mg cyanogen/m3 for 6 h/day, 5 days/week, for 6 months. This corresponded to 25 and 56 mg hydrogen cyanide/m3. There were no effects on haematological or clinical chemistry parameters attributable to the inhalation exposure. Total lung moisture content was significantly lower in both treatment groups than in control animals (Lewis et al., 1984).
Sprague-Dawley rats (15 per sex and dose level) were exposed to ACH at concentrations of 0, 36, 101, or 204 mg ACH/m3, 6 h/day, 5 days/week, for 14 weeks. The exposures were equivalent to 0, 11, 32, and 65 mg hydrogen cyanide/m3 (Monsanto Co., 1984a). There were no treatment-related deaths or significant changes in body weight gain or haematology. Irritation of the nose and eyes was observed, but no more in exposed than in non-exposed animals. A decrease in blood glucose was recorded in high- and mid-exposure females, and a decrease in total serum protein and globulin concentrations was noted in the mid- and low-dose females. A comprehensive microscopic evaluation of tissues revealed no abnormalities, and no changes in serum T3 or T4 levels were observed. The NOAEL reported from the study was 204 mg ACH/m3, corresponding to 65 mg hydrogen cyanide/m3. This can be estimated to correspond to a daily dose of 15 mg cyanide/kg body weight per day.9
In the male fertility study described in section 7.6.1, no mortality, clinical signs of toxicity, changes in body weight, or changes in gross necropsy were observed after 48 exposures to up to 202 mg ACH (64 mg hydrogen cyanide)/m3, 6 h/day, 5 days/week (Monsanto Co., 1985a).
In the female fertility study described in section 7.6.1, there was a dose-dependent increase in red nasal discharge of encrustation, but no other clinical signs of toxicity, mortality, changes in body weight, or changes in gross necropsy during or after 34–36 exposures to up to 207 mg ACH (66 mg hydrogen cyanide)/m3, 6 h/day, 7 days/week (Monsanto Co., 1985b).
Few data exist on the effects of chronic cyanide exposure. In a 2-year dietary study, weanling albino rats (10 per sex per group) were administered food fumigated with hydrogen cyanide (special jars were used in order to limit volatilization of hydrogen cyanide from the feed) (Howard & Hanzal, 1955). The average concentrations of cyanide in the feed were 0, 73, and 183 mg/kg diet, as estimated (US EPA, 1993b) based on the authors’ data for concentrations at the beginning and end of each food preparation and by assuming a first-order rate of loss for the intervening period and on the corresponding daily doses of 4.3 and 10.8 mg cyanide/kg body weight per day. No treatment-related effects on survival or growth rate, signs of toxicity, or haematological or histopathological changes in the organs examined (heart, lung, liver, spleen, gastrointestinal tract, kidneys, adrenals, thyroid, testes, uterus, ovaries, cerebrum, cerebellum, and brain) were observed in the treated male or female animals. A NOAEL of 10.8 mg cyanide/kg body weight per day was established.
The effects of cyanide on thyroid function were investigated in groups of 10 male weanling rats fed a semipurified casein-based diet for 11.5 months either supplemented by added methionine, vitamin B12, and potassium iodide or without added vitamin B12 and potassium iodide and with the methionine addition restricted to a third. Both dietary groups were divided into three: one served as the control, the second received 1500 mg potassium cyanide/kg, and the third received 2240 mg potassium thiocyanate/kg. The rats given the potassium cyanide would have received doses of 30 mg cyanide/kg body weight per day. Cyanide, but not thiocyanate, caused a consistent reduction in weight gain in the complete and restricted diet fed animals. Both cyanide and thiocyanate caused decreased thyroid gland activity in young rats, particularly in those fed the restricted diet. Depression of both plasma T4 and the T4 secretion rate, suggestive of depressed thyroid function, was found at 4 months, but to a lesser degree after 1 year. At autopsy, the animals were found to have enlarged thyroids, which suggested a mechanism of adaptation (Philbrick et al., 1979).
In order to study the possible contribution of cyanide exposure to malnutrition-related diabetes mellitus (see section 8), Okolie & Osagie (1999, 2000) fed New Zealand White rabbits potassium cyanide for 10 months (702 mg cyanide/kg diet, corresponding to approximately 20 mg/kg body weight per day). No effects were observed on the serum amylase activity, blood glucose concentration, or the morphology of the pancreas, while degenerative changes were reported in the liver and kidney. Similarly, 1-year feeding of cassava to rats induced no changes in blood glucose homeostasis or pancreatic histology (Mathangi et al., 2000).
Because of the small group size and limited exposure time in most of them, these studies are not informative with regard to the possible carcinogenicity of cyanides.
Sodium cyanide was not mutagenic in Salmonella typhimurium strains TA100, TA1535, TA97, or TA98 with or without exogenous metabolic activation (NTP, 1993). Potassium cyanide was not mutagenic in five strains (TA1535, TA1537, TA1538, TA98, and TA100) of S. typhimurium with or without metabolic activation (De Flora, 1981). Hydrogen cyanide induced mutations in S. typhimurium TA100 in the absence of S9 activation, but was not mutagenic to strain TA98 with or without S9 activation (Kushi et al., 1983). ACH did not induce mutations in S. typhimurium strains TA1535, TA1537, TA1538, TA100, or TA98 in a plate incorporation assay or at concentratons of 100–950 µg/ml in a CHO/HGPRT assay (Monsanto Co., 1983b,c).
DNA repair tests in Escherichia coli WP67, CM871, and WP2 with potassium cyanide were negative (De Flora et al., 1984). Potassium cyanide induced both time- and dose-dependent DNA fragmentation accompanied by cytotoxicity in rat thymocytes in vitro. Cyanide also induced DNA damage in baby hamster kidney cells (BHK-21) in vitro, where, unlike thymocytes, internucleosome DNA fragmentation was not observed (Bhattacharya & Rao, 1997). The cytotoxic mode of double strand breaks in the pathogenesis of DNA fragmentation was studied by Vock et al. (1998), employing an A549 human epithelial-like lung carcinoma cell line treated with potassium cyanide. Induction of double strand breaks by potassium cyanide was observed only after cell viability was reduced to less than 60%, indicating that double strand breaks were the consequence of extragenomic damage, as a secondary effect of high cytotoxicity in combination with cell lethality.
Sodium cyanide was a highly effective inducer of germline aneuploidy in Drosophila (Osgood & Sterling, 1991).
No statistically significant increases in the frequency of chromosomal aberrations or changes in mitotic index compared with control values were found in bone marrow cells from four groups of 24 male and 24 female Sprague-Dawley rats administered a single dose of ACH by oral gavage at levels of 0, 1.5, 5, or 15 mg/kg body weight with preparation intervals of 6, 12, and 24 h post-administration (Monsanto Co., 1984b).
No testicular DNA synthesis inhibition was detected in mice after a single oral potassium cyanide dose of 1 mg/kg body weight (Friedman & Staub, 1976).
Relatively few data are available on the reproductive and developmental toxicity of cyanides. No reproductive and developmental toxicity studies are available for hydrogen cyanide.
After 2 weeks on a diet containing 5 or 10 g potassium cyanide/100 g diet, female rats (10 per group) were mated with untreated males. No pregnancies resulted. The dose corresponds roughly to 1000 and 2000 mg cyanide/kg body weight per day (Olusi et al., 1979). There was a dose-dependent decrease in body weight gain, blood haemoglobin (18% and 23%), and serum T4 concentration (54% and 75%).
In a male fertility study (Monsanto Co., 1985a), Sprague-Dawley rats (n = 15) were exposed by inhalation to ACH (0, 35, 101, or 202 mg/m3; 0, 11, 32, or 64 mg hydrogen cyanide/m3), 6 h/day, 5 days/week, during a period of 69 days (i.e., 48 exposure days). After the treatment period, the males were mated with three non-exposed females each. There were no effects on the mean body weight, clinical signs of toxicity, or anatomical changes in gross necropsy. The mating efficiency, number of live implants, and pre- and post-implantation losses were not different between treated and control groups.
Female Sprague-Dawley rats were exposed by inhalation (6 h/day, 7 days/week) for 21 days to ACH at at 38, 108, or 207 mg/m3 and then mated with untreated males. Exposure of the females, which was equivalent to 12, 34, and 66 mg hydrogen cyanide/m3, was continued until the day of mating, and the females were sacrificed at mid-gestation (gestation days 13–15). No treatment-related effects on female fertility were observed in any of the exposure groups. The only frequently observed clinical sign post-exposure was red nasal discharge or encrustation. The highest exposure in the study could be considered as the no-observed-effect level (NOEL) for female reproductive effects (Monsanto Co., 1985b).
Preliminary experiments with pregnant Golden Syrian hamsters showed that at a dose rate of 0.125 mmol/kg body weight per hour, no effects on the fetus were observed, while at a dose rate of 0.133 mmol/kg body weight per hour or more, there was a 100% resorption rate and maternal deaths. Toxicity to dams increased with increasing dose levels and included shortness of breath, incoordination, reduced body temperature, and loss of body weight. Co-administration of thiosulfate eliminated the teratogenic effect, protecting the dams and fetuses from the toxic effects of cyanide (Doherty et al., 1982). In a follow-up experiment, pregnant Golden Syrian hamsters (5–7 animals per group) were continuously exposed to sodium cyanide from day 6 to day 9 of gestation at 0, 0.126, 0.1275, or 0.1295 mmol/kg body weight per hour by using osmotic minipumps implanted subcutaneously. These doses are equivalent to 0, 3.28, 3.32, and 3.37 mg cyanide/kg body weight per hour or 0, 78.7, 79.6, and 80.9 mg/kg body weight per day. The treatment induced a remarkable increase in resorptions as well as fetal malformations. These included non-closure of the neural tube, exencephaly, encephalocoele, and malformations of the heart and limbs or tail (Doherty et al., 1982).
Pregnant Wistar rats (10 animals per group) were fed a cassava diet liberating 21 mg hydrogen cyanide/kg diet, fortified with 500 mg potassium cyanide/kg diet, throughout gestation and lactation. This is equivalent to an estimated daily dose of 16 mg cyanide/kg body weight. No effects were observed on the number, mortality at birth, or body weight of offspring or weight gain of pups during lactation (Tewe & Maner, 1981a).
In equivalent studies with pregnant Yorkshire pigs, three groups of six animals were given potassium cyanide in the diet (30.3, 277, or 521 mg cyanide/kg diet)10 throughout gestation. No effects were seen on the number or weight of offspring or subsequent lactational performance. Pregnant sows treated at the highest dose level had proliferative changes in the kidney glomeruli and increased thyroid weights. Fetal spleen to body weight and head to body weight ratios in the high-cyanide group were significantly reduced (P > 0.05) compared with the low-cyanide exposed group (Tewe & Maner, 1981b).
In a preliminary study with limited experimental details, pregnant albino rats were fed milled cassava powder as 50% and 80% of their diet during the 15 days of gestation. Growth retardation and an increased frequency of resorptions were observed at both dose levels; in addition, limb defects were observed at the high dose. The weight gain of the dams was lowered; no further information on maternal toxicity was given. No indication of the cyanide content of the cassava diet was given. The effects observed may have resulted from nutritional deficiencies, such as the low protein content of cassava (Singh, 1981).
Groups of pregnant hamsters were fed diets consisting of two types of cassava meal, either a "low-cyanide" (sweet cassava meal) or a "high-cyanide" (bitter cassava meal) variety. These were mixed (80:20) with laboratory chow and administered on days 3–14 of gestation. The cyanide concentration of the sweet cassava meals was 0.6–0.7 mmol/kg; that of the bitter cassava meal was 5–11 (mean 7.9) mmol/kg.11 Cassava-fed dams gained significantly less weight than did control animals (fed diet similar in nutritional value as cassava, but without cyanogenic glycosides), and their offspring showed evidence of fetotoxicity (reduced fetal body weight and reduced ossification of sacrocaudal vertebrae, metatarsals, and sternebrae). The bitter cassava also produced a significant increase in the number of runts compared with litters from dams fed either low-protein12 or laboratory-stock diets. The only teratogenic effects noted were hydrocephalus in three animals in the low-cyanide (sweet cassava) test group and one encephalocoele found in one animal in the high-cyanide (bitter cassava) test group (Frakes et al., 1986b).
In a teratogenicity study, hamsters were given a single oral dose of linamarin (0, 70, 100, 120, or 140 mg/kg body weight, corresponding to 0, 7.4, 11, 13, or 15 mg cyanide/kg body weight per day) on day 8 of the pregnancy. The animals were killed on day 15 of the pregnancy, and the numbers of resorption sites, dead fetuses, and living fetuses were recorded. Living fetuses were examined for gross external malformations and for internal malformations using histopathological methods. Linamarin had no effect on fetal body weight, ossification, embryonic mortality, or litter size. At the two highest doses, which caused overt maternal toxicity (dyspnoea, hyperpnoea, ataxia, tremor, and hypothermia), vertebral and rib anomalies and encephalocoeles were observed (Frakes et al., 1985).
A single oral dose of d,l-amygdalin at gestational day 8 resulted in exencephaly, encephalocoele, and skeletal malformations at doses of >250 mg/kg body weight (>14 mg cyanide/kg body weight) in hamsters (these doses were also clearly toxic to the mothers). At the lowest dose tested, 200 mg/kg body weight (11 mg cyanide/kg body weight), fused ribs were observed in two offspring of one mother (maternal toxicity not reported). Encephalocoele and rib anomalies were also observed after a dose of d-prunasin (177 mg/kg body weight [16 mg cyanide/kg body weight]) in the absence of maternal toxicity in hamsters. No teratogenic effects were noted when hamsters received d,l-amygdalin (275 mg/kg body weight [16 mg cyanide/kg body weight]) intravenously (Willhite, 1982). The teratogenic effects found were considered to be due to cyanide released by bacterial beta-glucosidase in the gastrointestinal tract (Willhite, 1982).
Groups of 25 pregnant Sprague-Dawley rats were dosed by gavage on days 6–15 of gestation with 0, 1, 3, or 10 mg ACH/kg body weight (equivalent to 0, 0.3, 0.9, or 3 mg cyanide/kg body weight). Maternal toxicity was evidenced by slight reductions in body weight gain in the mid- and high-exposure groups, and statistically significant differences between the high-dose group and controls were found for the number of corpora lutea implantations per dam. There were no comparable differences in the number of viable fetuses per dam, post-implantation losses per dam, mean fetal body weight, or fetal sex distribution for all dose groups and the controls. The incidences of fetal malformations and developmental variations for all fetuses of treated animals and controls were also comparable. It was concluded that 10 mg ACH/kg body weight (3 mg cyanide/kg body weight) was not teratogenic in the rat in the presence of maternal toxicity (Monsanto Co., 1982, 1983a).
The effects of cyanide on behaviour were studied in fasted 25-week-old miniature pigs (12 litter mates: 5 females and 7 castrated males) randomized in four groups. The animals were dosed daily for 24 weeks with a single bolus of cyanide as aqueous potassium cyanide just prior to the daily feeding. The doses were 0, 0.4, 0.7, or 1.2 mg cyanide/kg body weight, chosen to be equivalent to those consumed by West Africans in their diet (Jackson et al., 1985). Every 6 weeks, thyroid function (T3 and T4) and fasting blood glucose were measured, but not thyroid-stimulating hormone (TSH). Daily observations were made of clinical signs and various behavioural measurements, including social, antagonistic, exploratory, learning, feeding, and excretory behaviour. In all treatment groups, dose-related decreases were evident from week 6 in blood levels of T3 and T4, and an increase in fasting blood glucose was noted, particularly in top-dose animals. Statistical analysis was not provided for each dose group versus control, but changes in top-dose animals appeared significant by week 18; by week 24, decreases of 35% for T3 and 15% for T4 and an increase of 60% in fasting blood glucose were observed. Behavioural observations revealed a picture of decreased high energy-demanding behaviour, such as exploration and aggression, slower eating, more frequent drinking, and shivering consistent with the decreased thyroid activity. A LOAEL of 1.2 mg/kg body weight per day could be suggested from this study (Jackson, 1988).
Dogs administered sodium cyanide in capsules at levels of 0, 0.5, 2, or 1–4 mg/kg body weight (one dog at each dose level) daily for 13–15 months showed severe signs of acute cyanide poisoning right after the daily dosing (the dog at the lowest dose died). In the autopsy, the only significant findings were degenerative changes in ganglia cells of the central nervous system, interpreted to be caused by multiple episodes of acute cerebral hypoxia (Hertting et al., 1960).
Transient behavioural changes in monkeys but not in rats exposed for 6 months to 54 mg cyanogen/m3 have been reported (Lewis et al., 1984).
Potentiation to noise-induced hearing loss by exposure of rats to low concentrations of hydrogen cyanide was reported by Fechter et al. (2002). It was suggested that hydrogen cyanide (34 mg/m3 for 3.5 h) plus noise produced impaired auditory function by producing significant oxidative stress in the cochlea. Hydrogen cyanide alone did not cause significant hearing loss or hair cell loss.
A single intraperitoneal dose and 25 repeated intraperitoneal doses of sodium cyanide (2 mg/kg body weight [1 mg cyanide/kg body weight]), stated to represent 25% of the LD50, administered to Wistar strain albino rats resulted in similar reductions of memory (T-maze test), along with reductions in the levels of dopamine and 5-hydroxytryptamine and increases in norepinephrine and epinephrine levels in the hippocampus, measured after a month of treatment (Mathangi & Namasivayam, 2000).
Local effects of cyanides on the eye are demonstrated by conjunctival hyperaemia with mild chemosis, lacrimation, photophobia, and tingling sensation (IPCS, 1992). No information was located on the sensitizing capacity of cyanides.
As a respiratory poison, free cyanide (hydrogen cyanide or cyanide ion) has high acute toxicity due to its primary toxic effect of inhibiting cytochrome oxidase (by binding haem iron), the terminal enzyme of the mitochondrial electron transport chain (Isom & Way, 1974). Tissue utilization of oxygen is impaired, and, with time, a state of histotoxic anoxia occurs (oxidative metabolism is brought to complete cessation). Cyanide can also inhibit approximately 40 enzymes, including a number of other important metalloenzymes containing, for the most part, iron, copper, or molybdenum (e.g., alkaline phosphatase, carbonic anhydrase, catalase, peroxidase, ascorbic acid oxidase, xanthine oxidase, and succinic dehydrogenase); these reactions may also contribute to cyanide toxicity (Rieders, 1971; Ardelt et al., 1989; US EPA, 1990; ATSDR, 1997).
Due to its high dependency on oxidative metabolism and limited anaerobic capacity, the central nervous system is particularly vulnerable to cyanide intoxication (Way, 1984). The central nervous system symptoms observed in cyanide toxicity parallel those observed following accumulation of calcium in the brain. Potassium cyanide also alters calcium homeostasis in a neuronal model, the PC-12 cell, and cytosolic calcium ion overload has been implicated as an intracellular mediator of cellular injury during and after anoxic hypoxia (Maduh et al., 1988; Pettersen & Cohen, 1993).
Hydroperoxide generation with subsequent peroxidation of lipids and subsequent changes in structure and function of certain membranes have been suggested as a possible further mechanism of cyanide toxicity (Ardelt et al., 1989).
A number of animal and human cyanide and cyanogenic glycoside toxicological studies have cited evidence of poor diet, low protein, and vitamin B12 and folic acid deficiencies and suggested that these factors may have confounded or exacerbated the observed toxicities. Although the role of vitamin B12 deficiency, for example, has not been fully elaborated, it has been suggested that vitamin B12 may be a protective factor in cyanide neurotoxic effects. Experimentally, vitamin B12 restriction is hypothesized to sensitize the animal to cyanide toxic reactions (Philbrick et al., 1979). One study has demonstrated depletion of stores of protein-bound cobalamin in cyanide-treated animals, and it is theoretically possible that cyanide exposure might "chelate" vitamin B12, leading to its eventual depletion and to secondary folate effects (Blanc et al., 1985).
The dose–effect curve of the acute effects in humans is steep. Whereas slight effects occur at exposure to hydrogen cyanide levels of 20–40 mg/m3, 50–60 mg/m3 can be tolerated without immediate or late effects for 20 min to 1 h, 120–150 mg/m3 is dangerous to life and may lead to death after 0.5–1 h, 150 mg/m3 is likely to be fatal within 30 min, 200 mg/m3 is likely to be fatal after 10 min, and 300 mg/m3 is immediately fatal. It should be emphasized that this represents crude average exposure estimates, based on various studies (DECOS, 2002).
A fire fatalities study in Maryland, USA, covering mostly residential fires over a 6-year period during which 523 fire fatalities occurred as a result of 392 fires, was reported by Birky & Clarke (1981). Although the predominant cause of death was attributed to carbon monoxide, toxic levels of hydrogen cyanide were found in the blood of a substantial percentage of the victims. A study of blood cyanide and carboxyhaemoglobin concentrations in 18 victims found dead in buildings after fires indicated that 50% of the victims had been exposed to toxic levels of hydrogen cyanide and 90% to toxic levels of carbon monoxide (Lundquist et al., 1989). Eighty-eight per cent of the fatalities in fire deaths in Glasgow, Scotland, during the period 1976–1979 had elevated blood cyanide levels; 31% had toxic levels of cyanide, and 12% would have shown severe cyanide poisoning (Anderson & Harland, 1982). Alarie (2002) reviewed the carboxyhaemoglobin and cyanide in blood of fire and non-fire victims resulting from 15 major episodes during the years 1971–1990 in France, the USA, and the United Kingdom. Analysis revealed that hydrogen cyanide is likely to be present in appreciable amounts in the blood of fire victims in modern fires. A review of the resultant mechanism of action of acute carbon monoxide and cyanide exposure and how they may interact concluded that it remains difficult to attribute death in fires to inhalation of hydrogen cyanide per se, given the complexity of interactions of smoke components (principally carbon monoxide).
Acute exposure to cyanide has occurred most frequently by the oral route from attempted suicides and homicides by ingestion of sodium or potassium cyanide or by accidental poisonings due to ingestion of apricot kernels or almond seeds (Rieders, 1971; NIOSH, 1976; US EPA, 1990; ATSDR, 1991; Alarie, 2002). Based on analyses of cyanide contents in tissues and in gastrointestinal tract contents among fatal (oral) poisoning cases (and comparative kinetics with dogs), Gettler & Baine (1938) estimated that death occurred after absorption of an average of 1.4 mg hydrogen cyanide/kg body weight; the lowest fatal absorbed dose was 0.54 mg hydrogen cyanide/kg body weight. In most poisoning cases, a large part of the ingested cyanide remained in the gastrointestinal tract (thus, using the dose ingested as an indicator of the lethality of cyanide is misleading). Some individuals ingesting 1–3 g of cyanide salts have survived (ATSDR, 1991).
The effects of acute cyanide exposure are dominated by central nervous system and cardiovascular disturbances (ATSDR, 1991). Typical signs of acute cyanide poisoning include tachypnoea, headache, vertigo, lack of motor coordination, weak pulse, cardiac arrhythmias, vomiting, stupor, convulsions, and coma (Ballantyne, 1983b; Way, 1984; Johnson & Mellors, 1988). Pathological findings may include tracheal congestion with haemorrhage, cerebral and pulmonary oedema, gastric erosions, and petechiae of the brain meninges and pericardium (Way, 1984). Sequelae of severe acute cyanide exposure may also include Parkinson-like syndromes and cardiovascular signs of delayed post-hypoxic myocardial lesions, as well as neuropsychiatric manifestations similar to those seen with post-hypoxic post-carbon monoxide encephalopathy (Uitti et al., 1985; Carella et al., 1988; Kadushin et al., 1988; ATSDR, 1991).
Dermal absorption of hydrogen cyanide is much slower than pulmonary absorption, and the amount and speed of absorption through human skin are dependent on the amount of skin moisture and duration of skin contact (see also section 6.1). An average LD50 value for dermal exposure of 100 mg/kg body weight was estimated for humans (Rieders, 1971). Concentrations of 7000–12 000 mg/m3 were estimated to be fatal after a 5-min exposure of workers with self-contained respirators without effective skin protection (Minkina, 1988).
Even healthy individuals have a small amount of cyanide in their bodies; concentrations up to 50 µg/100 g tissue have been found in different organs. A survey of plasma cyanide levels in 10 cases showed a maximum level of 106 µg/litre, with a mean of 48 µg/litre. The presence of cyanides in the plasma among "non-exposed" people has been variously attributed to factors including the breakdown of cyanogenic foods, vitamin B12, and heavy smoking (Ansell & Lewis, 1970).
No additive or synergistic effect was observed in the fatalities between cyanide and other factors, such as carbon monoxide, alcohol, age of victims, and presence of heart disease (US EPA, 1990).
Among patients with a serious hypertensive disease, intravenous administration of sodium nitroprusside was reported to lead to lowered serum T4 levels in approximately two-thirds of the patients; this effect became apparent at serum thiocyanate levels in excess of approximately 18 mg/litre, which was reported to correspond approximately to a daily dose of 200 mg sodium nitroprusside (20 mg cyanide) (Bödigheimer et al., 1979). Elevation of cyanide levels in blood was observed in patients receiving more than 2 µg nitroprusside/kg body weight per hour intravenously (Schultz et al., 1982).
Cyanide in tobacco smoke together with large amounts of alcohol and malnutrition have been proposed as culprits in tobacco–alcohol amblyopia, a syndrome at present seldom seen in Western countries, but still occasionally reported elsewhere (Solberg et al., 1998).
Although acute cassava poisoning — sometimes leading to the death of whole families — has been occasionally reported after the consumption of inadequately processed cassava (Osuntokun, 1981; Cliff & Countinho, 1995), a much larger literature is available on the effects of long-term exposure to food containing cyanogenic glycosides. Clinical signs are often confounded by dietary deficiencies, including lack of protein, iodine, and vitamin B12.
Accidental poisonings have been reported in children (and, exceptionally, in adults) who had ingested apricot kernels or seeds or candy made from apricot kernels containing d,l-amygdalin, which, after hydrolysis, yields cyanide (Sayre & Kaymakcalan, 1964; Lasch & El Shawa, 1981; Suchard et al., 1998). Presumably because of lower body weight, children are especially vulnerable, with several fatal poisonings occurring after they had consumed apricot seeds. It has been estimated that, depending on the total cyanogenic potential of apricot seeds, 10 or more seeds could be fatal to a child (Nahrstedt, 1993).
Accidental choke cherry poisonings (attributed to d,l-amygdalin) have also been reported (Pijoan, 1942; Pentore et al., 1996). Pentore et al. (1996) described a case of a 56-year-old woman in Italy who was accidentally poisoned when she ingested choke cherries whose pulp contained cyanide (amygdalin). After recovery from coma, the patient showed signs resembling Parkinson disease, retrobulbar neuritis, and sensorimotor neuropathy. The choke cherries showed cyanide levels ranging from 4.7 to 15 mg/kg in the cherries and from 43 to 45 mg/kg in the spirit. The quantity of cyanide was reported to depend on the ripeness of the cherries and the year in which they were harvested.
Consumption of food containing cyanogenic glycosides has been linked to several different diseases affecting mainly the nervous system, such as tropical ataxic neuropathy in Nigeria, spastic paraparesis (called mantakassa in Mozambique and konzo in the Democratic Republic of the Congo) in Cameroon, Central African Republic, Mozambique, Tanzania, and the Democratic Republic of the Congo (formerly Zaire), as well as retrobulbar neuritis and optic atrophy associated with pernicious anaemia. Cyanides have also been implicated in tobacco–alcohol amblyopia and thyroid effects such as goitre and even cretinism (Osuntokun et al., 1969; Ermans et al., 1972; Makene & Osuntokun, 1972, 1980; Wilson, 1972, 1983; Howlett et al., 1990; US EPA, 1990; ATSDR, 1991; Tylleskär et al., 1994; Boivin, 1997; Lantum, 1998; Ernesto et al., 2002).
Tropical ataxic neuropathy, an upper motor neuron disease characterized by irreversible paraparesis (Ernesto et al., 2002), was described in Nigeria in the 1930s, and dietary cassava was proposed to be the causative factor in 1934. The essential neurological components of the disease are myelopathy, bilateral optic atrophy, bilateral perceptive deafness, and polyneuropathy. The peak incidence is in the 5th and 6th decades of life, and the disease occurs rarely in children under 10 years of age. Patients usually give a history of almost total dependence on a monotonous diet of cassava derivatives. The plasma thiocyanate level in patients within 48 h of admission to hospital was 113 ± 0.2 µmol/litre, while in the referents it was 2.4 ± 0.15 µmol/litre (Osuntokun, 1981). However, the role of cyanide exposure as the only causative agent in tropical neuropathy is made questionable by the finding that when comparing two villages in Nigeria, one with a high prevalence (490/10 000) of tropical ataxic neuropathy and another with a low prevalence (17/10 000) (giving an age-adjusted prevalence ratio of 4), the estimated intake of cassava foods was higher in the latter, and no difference was observed in the urinary thiocyanate excretion between the two villages (Oluwole et al., 2002).
An epidemic of spastic paraparesis occurred in a drought-stricken cassava staple area of Mozambique in 1981–1982. Altogether, 1102 cases were identified. The highest recorded village prevalence rate found by active case detection was 29 per 1000 inhabitants; 65% of the cases were under 15 years of age. In contrast to tropical neuropathy, the onset of mantakassa was acute. General symptoms around the time of onset included fever, pain (especially in the legs), paraesthesiae, headache, dizziness, and vomiting; many patients also complained of weakness in the arms and difficulty in speaking and in seeing. Some mothers said that their children had difficulty in hearing. Neurological investigation revealed symmetrical spastic paraparesis of the lower extremities, symmetrically increased upper limb reflexes, diminished visual acuity, and dysarthria. Some of the patients had sensory changes as well. The mean hydrogen cyanide contents (mg/kg) of cassava from the affected area were as follows: fresh bitter cassava leaves, 377; fresh sweet cassava leaves, 347; fresh bitter cassava roots, 327; fresh sweet cassava roots, 138; dried bitter cassava roots, 95; dried sweet cassava roots, 46; cassava flour, 40; and cooked cassava, 10. The estimated intake of cyanide was 14–30 mg/day. The mean thiocyanate level in 246 specimens of blood and serum from patients from the whole area was 330 µmol/litre. In the village with the most patients, the mean thiocyanate level among the patients was 324 ± 18 µmol/litre, while 22 controls from this village showed serum thiocyanate levels of 288 ± 23 µmol/litre. There was no correlation between the disease severity and serum thiocyanate level. Also, because of the drought, there was a general lack of food, specifically of protein-rich food, with many cases of kwashiorkor13 appearing in February 1982 (Ministry of Health, Mozambique, 1984a,b).
Outbreaks of konzo have been reported in the Democratic Republic of the Congo (formerly Zaire) since 1938. The outbreaks have occurred during droughts and dry seasons. Again, the affected populations have relied almost exclusively on bitter cassava roots as the staple food (Tylleskär et al., 1991, 1992). In konzo-affected villages, the urinary thiocyanate levels were 563–629 µmol/litre during the dry season and 344–381 µmol/litre during the wet season; in reference villages without konzo, the levels were on average 241 µmol/litre. However, the urinary concentrations of linamarin showed a closer association with the disease than those of thiocyanate, and the authors interpreted it to indicate that more important than cyanide in the causation of konzo might be the neurotoxic action of linamarin itself (Banea-Mayambu et al., 1997).
Iodine deficiency and goitre, hypothyroidism, and cretinism are endemic in many areas of Africa. Several surveys in the endemic areas have demonstrated that there is also a strong correlation between cassava consumption and the thyroid effects (Delange & Ermans, 1971; Delange et al., 1971; Ermans et al., 1972; JECFA, 1993; Abuye et al., 1998). A cassava meal also diminished the uptake of 131I in the thyroid (Delange et al., 1971). A study in rural Mozambique found that in a population suffering from endemic spastic paraparesis, adequate iodine intake mitigated against the development of hypothyroidism or goitre, and high levels of dietary cyanogenic glycosides fom cassava could be tolerated (Cliff et al., 1986).
Originally based on a geographical link between the prevalence of diabetes and cassava consumption (McGlashan, 1967), dietary exposure to cyanides has been linked to the malnutrition-related diabetes mellitus (WHO, 1985), also known as the "type-J" or "type-Z" diabetes (Hugh-Jones, 1955; Zuidema, 1959). The very existence of this third type of diabetes (in addition to the juvenile-onset and maturity-onset types) has been controversial (Gill, 1996), and not all studies have detected a relationship between cassava consumption and diabetes prevalence (Cooles, 1988; Swai et al., 1992). The results of the standard glucose tolerance test were no more often abnormal among 88 Nigerian patients with tropical neuropathy than among 88 referents (Famuyiwa et al., 1995).
Chronic occupational exposure to hydrogen cyanide per se resulting in serious injury is rather rare. Symptoms of such poisonings include headache, dizziness, confusion, muscular weakness, poor vision, slurred speech, gastrointestinal tract disturbances, trauma, and enlarged thyroid.
A cross-sectional study was performed on the health effects of long-term cyanide exposure from a plating bath that contained 3% copper cyanide, 3% sodium cyanide, and 1% sodium chloride in the electroplating sections of three factories in Egypt that employed 36 male workers (non-smokers) with 5–15 years of experience; cyanide concentrations in the breathing zones of workers (15-min averaging time) ranged from 5 to 14 mg/m3, the averages in the three factories being 12, 7, and 9 mg/m3 at the time of the study. There was also exposure to petrol fumes, solutions of strong soap and alkalis, and hydrochloric acid. The exposed group reported symptoms such as headache, weakness, changes in taste and smell, giddiness, irritation of the throat, vomiting, effort dyspnoea, lacrimation, salivation, and precordial pain more frequently than controls. Twenty of the exposed workers (56%) exhibited thyroid enlargement to a mild or moderate degree. None of the workers had clinical manifestations of hypo- or hyperthyroidism, but the exposed group showed a lower uptake of radiolabelled iodine in the thyroid; there was no difference in the protein-bound 131I. The exposed workers had significantly higher haemoglobin and cyanomethaemoglobin values and lymphocyte counts compared with 20 male unexposed controls. Punctate basophilia of erythrocytes was present in 28 of 36 subjects (El Ghawabi et al., 1975). The contribution of the other exposures to the findings is difficult to discern.
A retrospective examination employing a questionnaire was performed with 36 former male workers (employees who could be reached and who volunteered, out of an unknown number of people actually employed) of a silver-reclaiming facility in the USA in 1983, which had been closed after the death of a worker because of cyanide poisoning. The only quantitative information on the concentrations of cyanide in the air came from a 24-h measurement 1 day after the factory had been closed; it was 17 mg/m3. The study revealed a high prevalence of symptoms, including eye irritation, fatigue, dizziness, headache, disturbed sleep, ringing in ears, paraesthesia of extremities, nausea, vomiting, dyspnoea, chest pain, palpitation, and weight loss (about 14% of workers reported palpitations, and 31% reported chest pain). Mild subclinical abnormalities in vitamin B12, folate, TSH levels, and thyroid function were found in silver-reclaiming workers 7 months after cyanide exposure had ceased. It was noted that inhalation of hydrogen cyanide was not the only possible route of exposure of these workers in this occupational setting, as the questionnaire disclosed that more than half reported frequent direct contact with liquids containing cyanide and 22% of exposed workers were at risk of inadvertent cyanide ingestion from food and drink in the production area (Blanc et al., 1985).
Effects of occupational exposure (5–19 years) of 111 workers and 30 non-exposed referents to hydrogen cyanide were studied in two large case-hardening and electroplating facilities in India (Chandra et al., 1988). From a daily work profile and air cyanide measurements, the workers were categorized in exposure groups between 1.11 and 4.66 "cyanide-hours" (mg/m3 × h). An abnormal psychological test result overall score (composite score of "delayed memory, visual ability, visual learning, and psychomotor ability") was observed in 31.5% of the exposed subjects, and an increase in the overall number of symptoms (headaches/heaviness in head, giddiness, lacrimation, itching of eyes, congestion of eyes, coated tongue) was found in 12.5% of the exposed workers. "Moderate" impairment in health-related scores showed an increase (no statistical analysis) at exposure levels in excess of 2.5 mg/m3 × h in one factory and 4.35 mg/m3 × h in the other, while findings classified as "diseased" were observed at levels in excess of 2.9 mg/m3 × h. The authors did not provide the incidences of these findings among referents or actual measurements of cyanide concentrations in the air, and few details on the carrying out of the investigations were given.14
Thiocyanate, the major detoxification product of cyanide, prevents the uptake of iodine and acts as a goitrogenic agent. This effect is more pronounced in individuals with decreased capacity to excrete thiocyanate in urine due to kidney dysfunction, etc. (VanderLaan & Bissell, 1946; Schultz, 1984; US EPA, 1990; ATSDR, 1993).
Banerjee et al. (1997) reported a study of 35 male cyanide workers (of 201 male workers) who had worked in an Indian electroplating process of a cable industry for more than 5 consecutive years. Thirty-five non-exposed workers who had worked outside the manufacturing building were matched with the exposed workers for age and dietary habits. The mean serum thiocyanate concentration of the 35 non-smoking exposed employees was 316 ± 15 µmol/litre, which was significantly higher (P < 0.01) than that of the control subjects (90 ± 9.02 µmol/litre). Cyanide exposure resulted in a decrease of serum T4 and T3 concentrations (P < 0.05) and an increase in TSH concentration (P < 0.05) compared with the control subjects.
A case of cerebrovascular accident and hyperthyroidism with struma and another with struma and hypothyroidism were reported among metal case-hardeners. No information on the level of exposure to cyanide was given (Hardy et al., 1950).
Leeser et al. (1990) reported a cross-sectional study, carried out between April and September 1986, of the health of sodium, copper, and potassium cyanide salt production workers from plants in a facility in the United Kingdom. Sixty-three employees from these cyanide salt plants were compared in a controlled study with 100 employees from a diphenyl oxide plant in the same facility. The breathing-zone cyanide concentrations ranged between 0.01 and 3.6 mg/m3. Cyanide workers were examined before and after a block of six shifts in the spring and autumn, while diphenyl oxide workers were seen during their shifts. Haemoglobin and lymphocyte levels tended to be higher in the cyanide workers, although neither was pathologically raised, and no relationship between exposure and haematological findings was found. The absence of a dose–response relationship would suggest that cyanide work was not causal. Thyroid function was normal in both groups, and no goitres were found. Vitamin B12 and T4 levels revealed no differences between cyanide and diphenyl oxide exposure groups.
Biochemical effects of occupational and dietary exposure to cyanide were investigated in a preliminary study of cyanide poisoning from large-scale cassava processing and ingestion of cassava foods in Nigeria (Okafor et al., 2002). The study population included 20 volunteers (female non-smokers, 24–50 years old and without overt signs of sickness or disease; 10 were cassava processors, 5 were "frequent" consumers of cassava, and 5 were "infrequent" consumers of cassava). The mean urinary thiocyanate level of the cassava processors (mean ± SD: 153.50 ± 25.2 µmol/litre) was 2.2 and 2.6 times higher than that of frequent (mean ± SD: 70.1 ± 21.8 µmol/litre) and infrequent (mean ± SD: 59.3 ± 17.0 µmol/litre) cassava consumers, respectively. An increase in plasma activity by 10% above normal ASAT (not statistically significant) was observed in 40% of the cassava processors, whereas it was within normal range in all consumers. No change was observed in the ALAT, alkaline phosphatase, or serum creatinine values.
There are no reported data on the carcinogenicity of cyanides in exposed human populations.
The principal features of the toxicity profile for cyanide are its high acute toxicity with a very steep dose–effect curve and chronic toxicity, probably mediated through the main metabolite and detoxification product, thiocyanate.
Consistent with the results of studies in experimental animals, available data from case reports and the limited data from occupationally exposed populations and populations principally exposed to dietary cyanogenic glycosides from consumption of cassava indicate that the cardiovascular, respiratory, central nervous, and endocrine systems are the major targets of acute and chronic cyanide exposure by all routes.
The generally similar toxic effects of cyanide, whether acute or chronic, in animals and humans are believed to result primarily from inhibition of cellular respiration and consequent histotoxic anoxia. Effects on the thyroid resulting from long-term exposure are likely to be caused by thiocyanate.
Human data on the concentration–effect relationship for acute cyanide toxicity are derived principally from case-studies of poisonings (accidental or intentional); as a result, characterization of concentration–response is uncertain. Whereas slight effects occur at inhalation exposure levels of 20–40 mg hydrogen cyanide/m3, 50–60 mg/m3 can be tolerated without immediate or late effects for 20 min to 1 h, 120–150 mg/m3 may lead to death after 0.5–1 h, 150 mg/m3 is likely to be fatal within 30 min, 200 mg/m3 is likely to be fatal after 10 min, and 300 mg/m3 is immediately fatal (DECOS, 2002). Based on analyses of cyanide contents in tissues and in gastrointestinal tract contents from fatal poisoning cases (and comparative kinetics with dogs), Gettler & Baine (1938) estimated that death occurred after absorption of an average of 1.4 mg hydrogen cyanide/kg body weight; the lowest fatal absorbed dose was 0.54 mg hydrogen cyanide/kg body weight. Fatal intoxications have been reported after ingestion of apricot pits containing cyanogenic glycosides, especially in children.
Intravenous administration of nitroprusside, which liberates cyanide, has been reported to affect thyroid function in patients (Bödigheimer et al., 1979). In some (but not all) studies, thyroid dysfunction and goitre have been reported among cyanide-exposed workers (El Ghawabi et al., 1975; Blanc et al., 1985; Leeser et al., 1990; Banerjee et al., 1997). Thyroid dysfunction in some of these individuals was reported to be accompanied by neurological symptoms, including headache, dizziness, and confusion, as well as mild subclinical abnormalities in vitamin B12 and folate (El Ghawabi et al., 1975; Blanc et al., 1985). Although this neurological and thyroid gland damage could not be unequivocally ascribed to cyanide, since other exposures may have occurred, the similarity of these lesions in humans to those produced in experimental animals treated with cyanide makes a strong case for the role of cyanide in these conditions. Limited information on exposure and confounding factors precludes the use of studies on people exposed at work in hazard characterization.
Long-term consumption of cassava containing high levels of cyanogenic glycosides, usually when constituting the principal source of calories, and associated with malnutrition and protein and vitamin deficiencies, has been associated with neurological diseases involving tropical ataxic neuropathy and endemic spastic paraparesis. In areas with low iodine intake, development of hypothyroidism and goitre, sometimes accompanied by the neurological diseases, has also been linked to cassava. While daily cyanide exposure has been crudely estimated to be 15–50 mg/day in endemic areas, owing to the limitations of data on exposure, which is likely to be quite variable, and the potential impact of confounders, such as general malnutrition, low protein content of the diet, and iodine status, the available data do not provide meaningful information on dose–response for cyanide.
By extrapolating from time to death at different concentrations in single inhalation exposures, maximal non-lethal concentrations for dogs, rats, mice, rabbits, monkeys, cats, goats, and guinea-pigs were estimated to be 100–180 mg hydrogen cyanide/m3 (Barcroft, 1931).
Cyanide is slightly irritating to the skin and eye (IPCS, 1992); data on the sensitizing properties of hydrogen cyanide and its alkali salts have not been identified.
Few repeated-dose toxicity studies have been performed (mainly because of the high acute toxicity), and the information on medium- to long-term effects is limited both for expected target organs and in the range of end-points studied. The possibility of mechanisms of chronic toxicity different from those of acute toxicity cannot, therefore, be totally excluded.
In a 13-week repeated-dose toxicity study (NTP, 1993) in which cyanide was administered in drinking-water, there were no clinical signs (associated with central nervous system effects) or histopathological effects in the brain or thyroid of rats and mice exposed to doses up to 12.5 and 26 mg/kg body weight per day, respectively. In contrast, effects on testicular and epididymal weight and testicular sperm counts and epididymal sperm motility were observed in male rats at the highest dose level studied, 12.5 mg/kg body weight.
In a 14-week repeated-dose toxicity study and in fertility toxicity studies in male and female rats, inhalation exposure to ACH (which is rapidly hydrolysed to hydrogen cyanide at physiological pH) resulted in no systemic toxicity at average ACH concentrations of 202, 204, or 207 mg/m3 (corresponding to 64, 65, and 66 mg hydrogen cyanide/m3). In a similar 4-week inhalation study at an average ACH concentration of 211 mg/m3 (67 mg hydrogen cyanide/m3), no systemic effects were observed, except on the first day of exposure, when the concentrations in the chamber fluctuated and reached values of 225 mg ACH/m3 (71 mg hydrogen cyanide/m3): 3 out of 10 animals died (Monsanto Co., 1984a, 1985c).
Although somewhat limited, the weight of evidence of available data indicates that cyanide is not genotoxic and that it induces developmental effects only at doses or concentrations that are overtly toxic to the mothers. No data on cyanide carcinogenicity have been identified.
The high acute toxicity of cyanide complicates the interpretation of available data as a basis for development of tolerable daily intakes or concentrations for potential effects associated with long-term low-level exposure to cyanide.
Available data in human populations are considered inadequate as a basis for characterization of dose–response for long-term ingestion of cyanide because of very uncertain information on level and duration of exposure, likely confounding by simultaneous other exposures and dietary deficiencies, limited end-points studied, limited statistical power, and limited reporting.
Rather, effect levels in the animal studies considered most robust in the characterization of longer-term effects associated with cyanide are presented here as a basis for comparison with specific exposure levels (i.e., to facilitate development of margins of exposure for assessment of risk in specific, local, or regional circumstances).
In a 13-week repeated-dose toxicity study (NTP, 1993) in which sodium cyanide was administered in drinking-water, there were no mortalities or clinical signs (associated with central nervous system effects) or histopathological effects in the brain, thyroid, or other organs of rats or mice exposed to doses up to 12.5 and 26 mg/kg body weight per day, respectively. The reproductive tract in the males was the most sensitive organ to cyanide exposure in this study: lowered weight of testis and epididymis, together with a decrease in the number of spermatid heads in the testis and decreased motility of epididymal sperm, were observed at the highest dose level. The NOAEL in this study, 4.5 mg/kg body weight per day,15 is consistent with the only available long-term study, in that no adverse effects were observed in rats after 2 years of exposure to cyanide in the diet at 10.8 mg cyanide/kg body weight per day (Howard & Hanzal, 1955). It is also roughly consistent with the NOAEL in the 14-week inhalation study (Monsanto Co., 1984a) in which the airborne systemic no-effect level of 204 mg ACH/m3 can be estimated to correspond to a daily dose of approximately 15 mg cyanide/kg body weight per day.
The examination of neurotoxicity in the NTP (1993) study was limited to clinical observation and optical microscopy at autopsy. The few available studies specifically intended to investigate neurotoxicity suffer from major weaknesses: the study in miniature pigs (Jackson, 1988), which reported slight functional thyroid changes and behavioural aberrations at 1.2 mg cyanide/kg body weight per day, used a bolus dose with consequent high peaks of cyanide concentration in the organism and involved a very small number of animals (three animals in four groups) and limited statistical analysis. Similarly, the neuropathological study in goats (Soto-Blanco et al., 2002a), which reported ultrastructural changes in animals given >0.48 mg cyanide/kg body weight per day in drinking-water for 5 months, did not present quantitative data or statistical analysis.
Inhalation exposure is relevant principally to the occupational environment and in the vicinity of cassava processing facilities. For the reasons stated above, data on people exposed at work cannot be used to characterize the hazard for inhalation exposure. Information on repeated-exposure studies in animals exposed to hydrogen cyanide itself is scanty. In contrast, several studies are available on ACH, which is rapidly hydrolysed at physiological pH (Monsanto Co., 1984a, 1985c). Three separate studies give a systemic no-effect level of 202–207 mg ACH/m3, corresponding to 64–66 mg hydrogen cyanide/m3. However, at an only slightly higher concentration of 225 mg ACH/m3 (71 mg hydrogen cyanide/m3), there was 30% mortality in one of the studies. The dose–effect relationship for cyanide is thus very steep for inhalation exposure. Because in these studies the effects observed were due to acute toxicity exclusively, they do not provide an adequate basis for the derivation of a tolerable concentration for any exposure of longer duration.
Information on levels of cyanide in air and water is limited and tends to be old. Adverse effects are not expected at the low levels reported in ambient air (usually below 1 µg/m3). In the USA, concentrations in surface water and drinking-water are generally below 10 µg/litre; at even the highest concentrations reported (200 µg/litre), adverse effects are not anticipated.
In the vicinity of cassava processing facilities, concentrations considerably in excess of those associated with adverse effects have been reported in both surface water and ambient air. Inadequately prepared cassava, when constituting the major part of the diet, may be hazardous. Concentrations of cyanogens in some other staple food items have been reported to be even higher than those in inadequately prepared cassava and are, therefore, potentially dangerous, although adverse health effects have not been reported.
Apricot and choke cherry kernels contain enough cyanogens to cause acute intoxication, and fatalities have been reported, especially in children.
Studies on humans deal mainly with acute effects of hydrogen cyanide/cyanides or, where longer term, lack adequate information on exposure and thus cannot be used in quantitative hazard characterization. Similarities in the likely mode of action and outcome from humans and experimental studies on animals, however, tend to confirm the most important end-points of hydrogen cyanide toxicity.
The available database on repeated-dose or chronic toxicity of cyanide is limited, primarily due to the high acute toxicity of the compound. While data are lacking on several relevant end-points such as carcinogenicity, significant longer-term adverse effects have not been induced at doses that have not been acutely toxic.
The contribution of factors other than cyanide in cassava-induced health impairment cannot be fully assessed.
The WHO Guidelines for Drinking-water Quality (WHO, 2003) derived a tolerable daily intake of 12 µg/kg body weight by considering 1.2 mg/kg body weight in the Jackson (1988) study as a LOAEL and applying an uncertainty factor of 100. By allocation of 20% of the tolerable daily intake to drinking-water, a guideline value of 0.07 mg/litre was reached.
JECFA (1993) concluded that because of lack of quantitative toxicological and epidemiological information, a safe level of intake of cyanogenic glycosides could not be estimated. However, the committee concluded that a level of up to 10 mg hydrogen cyanide/kg in the Codex Standard for cassava flour is not associated with acute toxicity (JECFA, 1993).
Abuye C, Kelbessa U, Wolde-Gebriel S (1998) Health effects of cassava consumption in south Ethiopia. East African Medical Journal, 75:166–170.
ACGIH (2001) Hydrogen cyanide and cyanide salts. In: Documentation of the threshold values and biological exposure indices, 8th ed. Cincinnati, OH, American Conference of Governmental Industrial Hygienists, pp. 1–6.
Agrawal V, Cherian L, Gupta VK (1991) Extraction spectrophotometric method for the determination of hydrogen cyanide in environmental samples using 4-aminosalicylic acid. International Journal of Environmental Analytical Chemistry, 45:235–244.
Aitken D, West D, Smith F, Poznanski W, Cowan J, Hurtig J, Peterson E, Benoit B (1977) Cyanide toxicity following nitroprusside induced hypotension. Canadian Anaesthetists Society Journal, 24:651–660.
Alarie Y (2002) Toxicity of fire smoke. Critical Reviews in Toxicology, 32:259–289.
Aminlari M, Vaseghi T, Kargar MA (1994) The cyanide-metabolizing enzyme rhodanese in different parts of the respiratory systems in sheep and dog. Toxicology and Applied Pharmacology, 124:64–71.
Anderson RA, Harland WA (1982) Fire deaths in the Glasgow area. III. The role of hydrogen cyanide. Medicine, Science and the Law, 22:35–40.
Ansell M, Lewis FAS (1970) A review of cyanide concentrations found in human organs: A survey of literature concerning cyanide metabolism, "normal," non-fatal, and fatal body cyanide levels. Journal of Forensic Medicine, 17:148–155.
Ardelt BK, Borowitz JL, Isom GE (1989) Brain lipid peroxidation and antioxidant protectant mechanisms following acute cyanide intoxication. Toxicology, 59:147–154.
ATSDR (1989) Toxicological profile for cyanide. Atlanta, GA, US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry.
ATSDR (1991) Case studies in environmental medicine. Atlanta, GA, US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry.
ATSDR (1993) Toxicological profile for cyanide. Prepared by Syracuse Research Corporation under subcontract to Clement International Corporation (Contract No. 205-88-0608).
ATSDR (1997) Toxicological profile for cyanide. Atlanta, GA, US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry.
Ballantyne B (1983a) The influence of exposure route and species on the acute lethal toxicity and tissue concentrations of cyanides. In: Hayes AW, Schnell RC, Miya TS, eds. Developments in the science and practice of toxicology. New York, NY, Elsevier Science Publishers, pp. 583–586.
Ballantyne B (1983b) Acute systemic toxicity of cyanides by topical application to the eye. Journal of Toxicology — Cutaneous and Ocular Toxicology, 2:119–129.
Ballantyne B (1987) Toxicology of cyanides. In: Ballantyne B, Marrs TC, eds. Clinical and experimental toxicology of cyanides. Wright, Bristol, IOP Publishing, pp. 41–126.
Banea-Mayambu JP, Tylleskar T, Gitebo N, Matadi N, Gebre-Medhin M, Rosling H (1997) Geographical and seasonal association between linamarin and cyanide exposure from cassava and the upper motor neurone disease konzo in former Zaire. Tropical Medicine and International Health, 2:1143–1151.
Banea-Mayambu JP, Tylleskar T, Tylleskar K, Gebre-Medhin M, Rosling H (2000) Dietary cyanide from insufficiently processed cassava and growth retardation in children in the Democratic Republic of Congo (formerly Zaire). Annals of Tropical Paediatrics, 20:34–40.
Banerjee KK, Bishayee B, Marimuthu P (1997) Evaluation of cyanide exposure and its effect on thyroid function of workers in a cable industry. Journal of Occupational Medicine, 39:255–260.
Barcroft L (1931) The toxicity of atmospheres containing hydrocyanic gas. Journal of Hygiene, 31:1–34.
Bedding ND, McIntyre AC, Perry RS, Lester JN (1982) Organic contaminants in the aquatic environment. I. Sources and occurrence. The Science of the Total Environment, 25:143–167.
Bhattacharya R, Rao PVL (1997) Cyanide induced DNA fragmentation in mammalian cell cultures. Toxicology, 123:207–215.
Birky MM, Clarke FB (1981) Inhalation of toxic products from fires. Bulletin of the New York Academy of Medicine, 57:997–1013.
Blanc P, Hogan M, Mallin K, Hryhorczuk D, Hessl S, Bernard B (1985) Cyanide intoxication among silver-reclaiming workers. Journal of the American Medical Association, 253:367–371.
Blaschle TF, Melmon KL (1980) Antihypertensive agents and the drug therapy of hypertension. In: Goodman LS, ed. Goodman and Gilman’s the pharmacological basis of therapeutics, 6th ed. New York, NY, Macmillan Publishing Co., pp. 805–806.
Bödigheimer K, Nowak F, Schoenborn W (1979) Pharmakokinetik und thyreotoxizität des nitroprussid-Natrium-Metaboliten Thiocyanat. Deutsche Medizinische Wochenschrift, 104:939–943.
Boivin MJ (1997) An ecological paradigm for a health behavior analysis of "Konzo," a paralytic disease of Zaire from toxic cassava. Social Science and Medicine, 45:1853–1862.
Boxer GE, Rickards JC (1952) Studies on the metabolism of the carbon of cyanide and thiocyanate. Archives of Biochemistry and Biophysics, 36:7–26.
Bright JE, Marrs TC (1988) Pharmacokinetics of intravenous potassium cyanide. Human Toxicology, 7:183–186.
Carella F, Grassi MP, Savoiardo M, Contri P, Rapuzzi B, Mangoni A (1988) Dystonic-parkinsonian syndrome after cyanide poisoning: clinical and MRI findings. Journal of Neurosurgery and Psychiatry, 51:1345–1348.
Carotti AA, Kaiser ER (1972) Concentrations of twenty gaseous chemical species in the flue gas of a municipal incinerator. Journal of the Air Pollution Control Association, 22:224–253.
Casadei E, Cliff J, Neves J (1990) Surveillance of urinary thiocyanate concentration after epidemic spastic paraparesis in Mozambique. Journal of Tropical Medicine and Hygiene, 93:257–261.
Chandra H, Gupta BN, Bhargava SK, Clerk SH, Mahendre PN (1980) Chronic cyanide exposure: a biochemical and industrial hygiene study. Journal of Analytical Toxicology, 4:161–165.
Chandra H, Gupta BN, Mathur N (1988) Threshold limit value of cyanide: a reappraisal in Indian context. Indian Journal of Environmental Protection, 8:170–174.
CHEMINFO (1998) Chemical profiles created by CCOHS: Cyanogen. Hamilton, Ontario, Canadian Centre for Occupational Health and Safety. Available at http:www.intox.org/databank/documents/chemical/cyanogen/cie430.htm.
Chinaka S, Takayama N, Michigama Y, Uneda K (1998) Simultaneous determination of cyanide and thiocyanate in blood by ion chromatography with fluorescence and ultraviolet detection. Journal of Chromatography B, 713:353–359.
Chiwona-Karltun L, Tylleskar T, Mkumbira J, Gebre-Medhin M, Rosling H (2000) Low dietary cyanogen exposure from frequent consumption of potentially toxic cassava in Malawi. International Journal of Food Sciences and Nutrition, 51:33–43.
Cicerone RJ, Zellner R (1983) The atmospheric chemistry of hydrogen cyanide (HCN). Journal of Geophysical Research, 88:689–696.
Clark DR Jr, Hothem RL (1991) Mammal mortality at Arizona, California, and Nevada gold mines using cyanide extraction. California Fish and Game, 77:61–69.
Cliff J, Coutinho J (1995) Acute intoxication from newly-introduced cassava during drought in Mozambique. Tropical Doctor, 25:193.
Cliff J, Lundquist P, Rosling H, Sobro B, Wide L (1986) Thyroid function in a cassava-eating population affected by epidemic spastic paraparesis. Acta Endocrinologica, 113:523–528.
Cohrssen B (2001) Cyanides and nitriles. In: Bingham E, Cohrssen B, Powell CH, eds. Patty’s toxicology, 5th ed. Vol. 4. New York, NY, John Wiley & Sons, pp. 1406–1410.
Cooles P (1988) Diabetes and cassava in Dominica. Tropical and Geographical Medicine, 40:272–273.
Dahl AR (1989) The cyanide-metabolizing enzyme rhodanese in rat nasal respiratory and olfactory mucosa. Toxicology Letters, 45:199–205.
DECOS (2002) Hydrogen cyanide, sodium cyanide and potassium cyanide. The Hague, Minister and State Secretary of Social Affairs and Employment, Dutch Expert Committee on Occupational Standards.
De Flora S (1981) Study of 106 organic and inorganic compounds in Salmonella/microsome test. Carcinogenesis, 2:283–298.
De Flora S, Camoirano A, Zanacchi P, Bennicelli C (1984) Mutagenicity testing with TA97 and TA102 of 30 DNA-damaging compounds, negative with other Salmonella strains. Mutation Research, 134:159–165.
Delange F, Ermans AM (1971) Role of a dietary goitrogen in the etiology of endemic goiter on Idjwi Island. American Journal of Clinical Nutrition, 24:1354–1360.
Delange F, Hershman JM, Ermans AM (1971) Relationship between the serum thyrotropin level, the prevalence of goiter and the pattern of iodine metabolism in Idjwi Island. Journal of Clinical Endocrinology and Metabolism, 33:261–268.
Doherty PA, Ferm V, Smith RP (1982) Congenital malformations induced by infusion of sodium cyanide in the golden hamster. Toxicology and Applied Pharmacology, 64:456–464.
Dolzine TW, Esposito GG, Rinehart DS (1982) Determination of hydrogen cyanide in air by ion chromatography. Analytical Chemistry, 54:470–473.
Dugard PH (1987) The absorption of cyanide through human skin in vitro from solutions of sodium cyanide and gaseous HCN. In: Ballantyne B, Marrs TC, eds. Clinical and experimental toxicology of cyanides. Wright, Bristol, IOP Publishing, pp. 127–137.
ECETOC (2004) Hydrogen cyanide, sodium and potassium cyanides and acetone cyanohydrin. Brussels, European Centre for Ecotoxicology and Toxicology of Chemicals, in press (ECETOC Joint Assessment of Commodity Chemicals).
Edwards TJ, Maurer G, Newman J, Prausnitz JM (1978) Vapor–liquid equilibria in multicomponent aqueous solutions of volatile weak electrolytes. AICHE Journal, 24:966–976.
Eisler R, Clarke DR Jr, Wiemeyer SN, Henry CJ (1999) Sodium cyanide hazards to fish and other wildlife from gold mining operations. In: Azcue JM, ed. Environmental impacts of mining activities. Berlin, Springer, pp. 55–67.
El Ghawabi SH, Gaafar MA, El-Sharati AA, Ahmed SH, Malash KK, Fares R (1975) Chronic cyanide exposure: a clinical, radioisotope, and laboratory study. British Journal of Industrial Medicine, 32:215–219.
Ellington JJ, Stancil FE, Payne WD (1986) Measurement of hydrolysis rate constants for evaluation of hazardous waste land disposal. Vol. 1. Washington, DC, US Environmental Protection Agency (EPA 600/3-86/043; NTIS PB87-140349) [cited in OECD (1997) Acetone cyanohydrin. Screening Information Data Set (SIDS) for high production volume chemicals. Organisation for Economic Co-operation and Development Initial Assessment, Vol. 4, Part 1, pp. 2–14.]
Ermans AM, Delange F, Van Der Velden M, Kinthaert S (1972) Possible role of cyanide and thiocyanate in the etiology of endemic cretinism. Advances in Experimental Medicine and Biology, 30:455–486.
Ermans AM, Mbulamoko NM, Delange F, Ahluwalia R, eds. (1980) Role of cassava in the etiology of endemic goitre and cretinism. Ottawa, Ontario, International Development Research Centre, 182 pp.
Ernesto M, Cardoso AP, Nicala D, Mirione E, Massaza F, Cliff J, Haque MR, Bradbury JH (2002) Persistent konzo and cyanogen toxicity from cassava in northern Mozambique. Acta Tropica, 82:357–362.
European Chemicals Bureau (2000a) Sodium cyanide. Ispra, IUCLID (International Uniform Chemical Information Database).
European Chemicals Bureau (2000b) Potassium cyanide. Ispra, IUCLID (International Uniform Chemical Information Database).
Famuyiwa OO, Akanji AO, Osuntokun BO (1995) Carbohydrate tolerance in patients with tropical ataxic neuropathy — A human model of chronic cyanide intoxication. African Journal of Medicine and Medical Sciences, 24:151–157.
Farooqui MYH, Ahmed AE (1982) Molecular interaction of acrylonitrile and potassium cyanide with rat blood. Chemico-Biological Interactions, 38:145–159.
Fechter LD, Chen GD, Johnson DL (2002) Potentiation of noise-induced hearing loss by low concentrations of hydrogen cyanide in rats. Toxicological Sciences, 66:131–138.
Feldstein M, Klendshoj NC (1954) The determination of cyanide in biologic fluids by microdiffusion analysis. Journal of Laboratory and Clinical Medicine, 44:166–170.
Ferguson HC (1962) Dilution of dose and acute oral toxicity. Toxicology and Applied Pharmacology, 4:759–762.
Fiksel J, Cooper C, Eschenroeder A, Goyer M, Perwak J (1981) Exposure and risk assessment for cyanide. Washington, DC, US Environmental Protection Agency (EPA/440/4-85/008; NTIS PB85-220572).
Frakes RA, Sharma RP, Willhite CC (1985) Developmental toxicity of the cyanogenic glycoside linamarin in the golden hamster. Teratology, 31:241–246.
Frakes RA, Sharma RP, Willhite CC (1986a) Comparative metabolism of linamarin and amygdalin in hamsters. Food and Chemical Toxicology, 24:417–420.
Frakes RA, Sharma RP, Willhite CC, Gomez G (1986b) Effect of cyanogenic glycosides and protein content in cassava diets on hamster prenatal development. Fundamental and Applied Toxicology, 7:191–198.
Frank SJ, Tindale N, Everall N (2002) Decomposition of low levels of acetone cyanohydrin in water. Cleveland, United Kingdom, Lucite International (Unpublished Laboratory Report No. ACRY/Wilton/R+T/2002/04).
Friedberg KD, Schwarzkopf HA (1969) [The exhalation of hydrocyanic acid in cyanide poisoning.] Archives of Toxicology, 24:235–242 (in German).
Friedman MA, Staub J (1976) Inhibition of mouse testicular DNA synthesis by mutagens and carcinogens as a potential mammalian assay for mutagenesis. Mutation Research, 37:67–76.
Gaffney JS, Streit GE, Spall WD, Hall JH (1987) Beyond acid rain. Environmental Science and Technology, 21:519–524.
Ganjeloo A, Isom GE, Morgan RL, Way JL (1980) Fluorometric determination of cyanide in biological fluids with p-benzoquinone. Toxicology and Applied Pharmacology, 55:103–107.
Gerhart JM (1986) Ninety-day oral toxicity study of copper cyanide (CuCN) in Sprague-Dawley rats. Prepared for the Dynamac Corporation, Rockville, MD, by IIT Research Institute, Chicago, IL (IITRI Project No. L06183, Study No. 3) [cited in ATSDR, 1997].
Gerhart JM (1987) Ninety-day oral toxicity study of potassium silver cyanide [KAg(CN)2] in Sprague-Dawley rats. Prepared for the Dynamac Corporation, Rockville, MD, by IIT Research Institute, Chicago, IL (IITRI Project No. L06183, Study No. 4) [cited in ATSDR, 1997].
Gettler AO, Baine JO (1938) The toxicity of cyanide. American Journal of Medical Science, 195:182–198.
Gill G (1996) Tropical diabetes? Tropical Doctor, 26:1–3.
Greim H, ed. (2003) Hydrogen cyanide, potassium cyanide and sodium cyanide. In: Occupational toxicants. Critical data evaluation for MAK values and classification of carcinogens. Vol. 19. Weinheim, Wiley WCH, pp. 189–210 (German version published in 2001).
Grosse DW (1986) Treatment technologies for hazardous wastes. Part IV. A review of alternative treatment processes for metal-bearing hazardous waste streams. Journal of the Air Pollution Control Association, 36:603–614.
Guiha NH, Cohn JN, Mikulic E, Franciosh JA, Limas CT (1974) Treatment of refactory heart failure with infusion of nitroprusside. New England Journal of Medicine, 291:587–592.
Hardy HL, Jeffries WM, Wasserman MM, Waddell WR (1950) Thiocyanate effect following industrial cyanide exposure. New England Journal of Medicine, 242:968–972.
Hartung R (1982) Cyanide and nitriles. In: Clayton GD, Clayton FE, eds. Patty’s industrial hygiene and toxicology, 3rd ed. Vol. II C. New York, NY, John Wiley & Sons, pp. 4845–4906.
Hayes WJ (1967) The 90 day LD50 and chronicity factors as a measure of toxicity. Toxicology and Applied Pharmacology, 11:327–335.
Haymaker W, Ginzler AM, Ferguson RL (1952) Residual neuropathological effects of cyanide poisoning: A study of the central nervous system of 23 dogs exposed to cyanide compounds. The Military Surgeon, 3:231–246.
HazDat (2003) HazDat database. Atlanta, GA, US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry. Available at http://www.atsdr.cdc.gov/hazdat.html#A3.1.2.
Health Canada (2002) Where there’s smoke there’s hydrogen cyanide. Ottawa, Ontario, Health Canada, Healthy Environments and Consumer Safety Branch. Available at http://www.hc-sc.gc.ca/hecs-sesc/tobacco/legislation/warnings/e_o.html#3.
Henny CJ, Hallock K, Hill EF (1994) Cyanide and migratory birds at gold mines in Nevada, USA. Ecotoxicology, 3:45–58.
Hertting GO, Kraupp E, Schnetz E, Wuketich ST (1960) Investigation about the consequences of a chronic administration of acutely toxic doses of sodium cyanide to dogs. Acta Pharmacologica et Toxicologica, 17:27–43 (in German).
Higgins EA, Fiorca V, Thomas AA, Davis HV (1972) Acute toxicity of brief exposures to HF, HCl, NO2, and HCN with and without CO. Fire Technology, 8:120–130.
Hine J, Weimar RD (1965) Carbon basicity. Journal of the American Chemical Society, 87:3387–3396.
Honig DH, Hockridge ME, Gould RM, Rackis JJ (1983) Determination of cyanide in soybeans and soybean products. Journal of Agricultural and Food Chemistry, 31:272–275.
Howard JW, Hanzal RF (1955) Chronic toxicity for rats of food treated with hydrogen cyanide. Agricultural and Food Chemistry, 3:325–329.
Howlett WP, Brubaker GR, Mlingi N, Rosling H (1990) Konzo, an epidemic upper motor neuron disease studied in Tanzania. Brain, 113:223–235.
Hugh-Jones P (1955) Diabetes in Jamaica. Lancet, 2:891–897.
Hugod C (1981) Myocardial morphology in rabbits exposed to various gas-phase constituents of tobacco smoke: an ultrastructural study. Atherosclerosis, 40:181–190.
IARC (1999) Re-evaluation of some organic chemicals, hydrazine and hydrogen peroxide (part one). Lyon, International Agency for Research on Cancer, pp. 67–71 (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71).
ICI (1993) Kinetics for the dissociation of acetone cyanohydrin in water (unpublished report) [cited in OECD (1997) Acetone cyanohydrin. Screening Information Data Set (SIDS) for high production volume chemicals. Organisation for Economic Co-operation and Development Initial Assessment, Vol. 4. Part 1, pp. 2–14].
IPCS (1992) Poison Information Monograph — Cyanides. Geneva, World Health Organization, International Programme on Chemical Safety, September (IPCS/INTOX/PIM 159).
IPCS (1993) Acetonitrile. Geneva, World Health Organization, International Programme on Chemical Safety (Environmental Health Criteria 154).
IPCS (1999a) Calcium cyanide. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 0407).
IPCS (1999b) Acetone cyanohydrin. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 0611).
IPCS (2000a) Disinfectants and disinfectant by-products. Geneva, World Health Organization, International Programme on Chemical Safety (Environmental Health Criteria 216).
IPCS (2000b) Cyanogen chloride. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 1053).
IPCS (2001) Cyanogen. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 1390).
IPCS (2002a) Hydrogen cyanide, liquefied. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 0492).
IPCS (2002b) Potassium cyanide. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 0671).
IPCS (2002c) Sodium cyanide. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 1118).
IPCS (2002d) Potassium ferricyanide. Geneva, World Health Organization, International Programme on Chemical Safety (International Chemical Safety Card 1132).
Isom GE, Way JL (1974) Effect of oxygen on cyanide intoxication. VI. Reactivation of cyanide-inhibited glucose metabolism. Journal of Pharmacology and Experimental Therapeutics, 189:235–243.
Jackson LC (1988) Behavioural effects of chronic sublethal dietary cyanide in an animal model: Implications for humans consuming cassava (Manihot esculenta). Human Biology, 60:597–614.
Jackson LC, Bloch EF, Jackson RT, Chandler JP, Kim YL, Malveaux F (1985) Influence of dietary cyanide on immunoglobulin and thiocyanate levels in the serum of Liberian adults. Journal of the National Medical Association, 77:777–782.
Jaramillo M, Dezafra RL, Barrett J, Emmons LK, Solomon PM, Parrish A (1989) Measurements of stratospheric hydrogen cyanide and McMurdo Station, Antarctica: further evidence of winter stratospheric subsidence? Journal of Geophysical Research, 94:16 773–16 777.
JECFA (1993) Cyanogenic glycosides. In: Toxicological evaluation of certain food additives and naturally occurring toxicants. Geneva, World Health Organization, 39th Meeting of the Joint FAO/WHO Expert Committee on Food Additives (WHO Food Additives Series 30). Available at http://www.inchem.org/documents/jecfa/jecmono/v30je18.htm.
Johnson RP, Mellors JW (1988) Arteriolization of venous blood gases: A clue to the diagnosis of cyanide poisoning. Journal of Emergency Medicine, 6:401–404.
Kadushin FS, Bronstein AC, Riddle MW, Gilmore DA (1988) Cyanide induced parkinsonism. Neuropsychological and radiological findings. Veterinary and Human Toxicology, 30(4):359–360.
Kaloyanova F, Basmadjieva K, Popov M, eds. (1985) [Ecology and health.] Sofia, Meditzina i Fizkultura (in Bulgarian).
Kamalu BP (1991) The effect of a nutritionally-balanced cassava (Manihot esculenta Crantz) diet on endocrine function using the dog as a model. 1. Pancreas. British Journal of Nutrition, 65:365–372.
Kamalu BP (1993) Pathological changes in growing dogs fed on a balanced cassava (Manihot esculenta Crantz) diet. British Journal of Nutrition, 69:921–934.
Kamalu BP, Agharanya JC (1991) The effect of a nutritionally-balanced cassava (Manihot esculenta Crantz) diet on endocrine function using the dog as a model. 2. Thyroid. British Journal of Nutrition, 65:373–379.
Kendirim OC, Chukwu OA, Achinewhu SC (1995) Effect of traditional processing of cassava on the cyanide content of gari and cassava flour. Plant Food for Human Nutrition, 48:335–339.
Korte F, Coulston F (1998) Some considerations on the impact on ecological chemical principles in practice with emphasis on gold mining and cyanide. Ecotoxicology and Environmental Safety, 41:119–129.
Korte F, Spiteller M, Coulston F (2000) The cyanide leaching gold recovery process is a nonsustainable technology with unacceptable impacts on ecosystems and humans: the disaster in Romania. Ecotoxicology and Environmental Safety, 46:241–245.
Krasner SW, McGuire MJ, Jacangelo JG, Patania NL, Reagan KM, Aieta ME (1989) The occurrence of disinfection by-products in U.S. drinking water. Journal of the American Water Works Association, 81:41–53.
Kushi A, Matsumoto T, Yoshida D (1983) Mutagen from gaseous phase of protein pyrolyzate. Agricultural and Biological Chemistry, 47:1979–1982.
Landahl HD, Herrmann RG (1950) Retention of vapours and gases in the human nose and lung. Archives of Industrial Hygiene and Occupational Medicine, 1:36–45.
Lantum H (1998) Spastic paraparesis–konzo in the Garoua Boulai Health District, East Province–Cameroon: A hidden endemic disease. Republic of Cameroon, Yaoundé, p. 85 [cited in Ernesto et al., 2002].
Lasch EE, El Shawa R (1981) Multiple cases of cyanide poisoning by apricot kernels in children from Gaza. Pediatrics, 68:5–7.
Lauwerys RR, Hoet P (2001) Industrial chemical exposure. Guidelines for biological monitoring, 3rd ed. Boca Raton, FL, Lewis Publishers.
Lawrence WS (1947) The toxicity of sodium cyanide at slow rates of infusion. Federation Proceedings, 6(1):349.
Leeser JE, Tomenson JA, Bryson DD (1990) A cross-sectional study of the health of cyanide salt production workers. Macclesfield, ICI Central Toxicology Laboratory.
Leuschner F, Neumann BW, Otto H, Möller E (1989) 13-week toxicity study of potassium cyanide administered to Sprague-Dawley rats in the drinking water. Unpublished study, Laboratory of Pharmacology and Toxicology, July [cited in JECFA, 1993].
Leuschner J, Winkler A, Leuschner F (1991) Toxicokinetic aspects of chronic cyanide exposure in the rat. Toxicology Letters, 57:195–201.
Lewis TR, Anger WK, Te Vault RK (1984) Toxicity evaluation of sub-chronic exposures to cyanogen in monkeys and rats. Journal of Environmental Pathology, Toxicology and Oncology, 5:151–163.
Li Q, Jacob DJ, Bey I, Yantosch RM, Zhao Y, Kondo Y, Notholt J (2000) Atmospheric hydrogen cyanide (HCN): Biomass burning source, ocean sink? Geophysical Research Letters, 27:357–360.
Liebowitz D, Schwartz H (1948) Cyanide poisoning: Report of a case with recovery. American Journal of Clinical Pathology, 18:965–970.
Lundquist P, Rammer L, Sorbo B (1989) The role of hydrogen cyanide and carbon monoxide in fire casualties: a prospective study. Forensic Science International, 43:9–14.
Ma J, Pritsos CA (1997) Tissue specific bioenergetic effects and increased enzymatic activities following acute sublethal peroral exposure to cyanide in the mallard duck. Toxicology and Applied Pharmacology, 142:297–302.
Maduh EU, Johnson JD, Ardelt BK, Borowitz JL, Isom GE (1988) Cyanide-induced neurotoxicity: Mechanisms of attenuation by chlorpromazine. Toxicology and Applied Pharmacology, 96:60–67.
Makene WJ, Wilson J (1972) Biochemical studies in Tanzanian patients with ataxic tropical neuropathy. Journal of Neurology, Neurosurgery and Psychiatry, 35:31–33.
Maseda C, Matsubara K, Shiono H (1989) Improved gas chromatography with electron-capture detection using a reaction pre-column from the determination of blood cyanide: a higher content in the left ventricle of fire victims. Journal of Chromatography, 490:319–327.
Mathangi DC, Namasivayam A (2000) Effect of chronic cyanide intoxication on memory in albino rats. Food and Chemical Toxicology, 38:51–55.
Mathangi DC, Deepa R, Mohan V, Govindarajan M, Namasivayam A (2000) Long-term ingestion of cassava (tapioca) does not produce diabetes or pancreatitis in the rat model. International Journal of Pancreatology, 27:203–208.
Matijak-Schaper M, Alarie Y (1982) Toxicity of carbon monoxide, hydrogen cyanide and low oxygen. Combustion Toxicology, 9:21–61.
McGlashan ND (1967) Geographical evidence on medical hypotheses. Tropical and Geographical Medicine, 19:333–344.
Milosavlievic EB, Solujic L, Hendrix JL (1995) Rapid distillationless "free cyanide" determination by a flow injection ligand exchange method. Environmental Science and Technology, 29:426–430.
Ministry of Health, Mozambique (1984a) Mantakassa: An epidemic of spastic paraparesis associated with chronic cyanide intoxication in a cassava staple area of Mozambique. 1. Epidemiology and clinical and laboratory findings in patients. Bulletin of the World Health Organization, 62:477–484.
Ministry of Health, Mozambique (1984b) Mantakassa: An epidemic of spastic paraparesis associated with chronic cyanide intoxication in a cassava staple area of Mozambique. 2. Nutritional factors and hydrocyanic acid content of cassava products. Bulletin of the World Health Organization, 62:485–492.
Minkina NA (1988) [Hydrogen cyanide and cyanides.] In: Filov VA, ed. [Harmful chemical substances — inorganic compounds of elements of I–IV groups.] St Petersburg, Himiya, pp. 331–352 (in Russian).
Monsanto Co. (1982) Range finding teratology study in the rat. St. Louis, MO, Monsanto Co. (Report IL-83-094; US EPA/OPTS Public Files No. 878216399).
Monsanto Co. (1983a) Teratology study in rats. St. Louis, MO, Monsanto Co. (Report IL-83-105; US EPA/OPTS Public Files No. 878216401).
Monsanto Co. (1983b) In-vivo bone marrow chromosome study in rats. St. Louis, MO, Monsanto Co. (Report HL-83-195; US EPA/OPTS Public Files No. 878216400).
Monsanto Co. (1983c) Salmonella typhimurium/mammalian microsome plate incorporation assay with compound (ACH). St. Louis, MO, Monsanto Co. (Report PK-83-204; US EPA/OPTS Public Files No. 878216403).
Monsanto Co. (1984a) Three-month inhalation toxicity of acetone cyanohydrin in male and female Sprague-Dawley rats. St. Louis, MO, Monsanto Co. (Report ML-82-143; US EPA/OPTS Public Files No. 878216397).
Monsanto Co. (1984b) CHO/HGPRT mammalian cell forward mutation assay, acetone cyanohydrin. St. Louis, MO, Monsanto Co. (Report PR-82-204).
Monsanto Co. (1985a) Male fertility study of Sprague-Dawley rats exposed by the inhalation route to acetone cyanohydrin. St. Louis, MO, Monsanto Co. (Report ML-82-144; US EPA/OPTS Public Files No. 878216404).
Monsanto Co. (1985b) Female fertility study of Sprague-Dawley rats exposed by the inhalation route to acetone cyanohydrin. St. Louis, MO, Monsanto Co. (Report ML-82-145; US EPA/OPTS Public Files No. 878216396).
Monsanto Co. (1985c) One-month inhalation toxicity of acetone cyanohydrin in male and female Sprague-Dawley rats. St. Louis, MO, Monsanto Co. (Report ML-81-178/810068; US EPA/OPTS Public Files No. 878216393).
Mudder TI, Botz M (2000) A global perspective of cyanide. A background paper of the UNEP/ICME Industry Codes of Practice Workshop: Cyanide Management Paris, 26–27 May 2000. Available at http://www.mineralresourcesforum.org/.
Nahrstedt AF (1993) Cyanogenesis and food plants. In: van Beek TA, Breteler H, eds. Proceedings of the International Symposium on Phytochemistry and Agriculture, 22–24 April 1992, Wageningen. Oxford, Oxford University Press, pp. 107–129.
Nartey F (1980) Toxicological aspects of cyanogenesis in tropical foods. In: Smith RL, Bababumni ER, eds. Toxicology in the tropics. London, Taylor & Francis, pp. 53–73 [cited in JECFA, 1993].
NIOSH (1976) Health Hazard Evaluation Report No. 74-129-268. Cincinnati, OH, US Department of Health, Education, and Welfare, Center for Disease Control, National Institute for Occupational Safety and Health.
NIOSH (1978) Health Hazard Evaluation Report No. 77-88-457. Cincinnati, OH, US Department of Health, Education, and Welfare, Center for Disease Control, National Institute for Occupational Safety and Health.
NIOSH (1982) In-depth survey report of American Airlines plating facility. Cincinnati, OH, US Department of Health and Human Services, Centers for Disease Control, National Institute for Occupational Safety and Health (PB83-187799).
NIOSH (1994) Cyanides, aerosol and gas. NIOSH Method 7904. In: Cassinelli ME, O’Connor PF, eds. NIOSH manual of analytical methods, 4th ed. Washington, DC, US Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, August. Available at http://www.cdc.gov/niosh/nmam/pdfs/7904.pdf (DHHS (NIOSH) Publication 94-113).
NTP (1993) Sodium cyanide administered in drinking water to F344/N rats and B6C3F1 mice. Research Triangle Park, NC, National Institutes of Health, National Toxicology Program (Toxicology Report Series No. 37; NIH Publication 94-3386).
Ohya T, Kanno S (1987) Formation of cyanogen chloride during the chlorination of water containing aromatic compounds and ammonium ion. Journal of Pharmaceutical Sciences, 76:S128.
Okafor PN, Maduagwu EN (2000) Cyanide contamination of the atmospheric air during large scale "gari" processing and the toxicity effects of such cyanide equivalent on rat. African Journal of Biomedical Research, 3:19–23.
Okafor PN, Okorowko CO, Alawem FO, Maduagwu EN (2001) Cyanide contamination of natural water sources during large scale cassava processing. African Journal of Biomedical Research, 4:25–27.
Okafor PN, Okorowko CO, Maduagwu EN (2002) Occupational and dietary exposures of humans to cyanide poisoning from large-scale cassava processing and ingestion of cassava foods. Food and Chemical Toxicology, 49:1001–1005.
Okoh PN, Pitt GAJ (1982) The metabolism of cyanide and the gastrointestinal circulation of the resulting thiocyanate under conditions of chronic cyanide intake in the rat. Canadian Journal of Physiology and Pharmacology, 60:381–386.
Okolie NP, Osagie AU (1999) Liver and kidney lesions and associated enzme changes induced in rabbits by chronic cyanide exposure. Food and Chemical Toxicology, 37:745–750.
Okolie NP, Osagie AU (2000) Differential effects of chronic cyanide intoxication on heart, lung and pancreatic tissues. Food and Chemical Toxicology, 38:543–548.
Olusi SO, Oke OL, Odusote A (1979) Effects of cyanogenic agents on reproduction and neonatal development in rats. Biology of the Neonate, 36:233–234.
Oluwole OSA, Onabolu AO, Cotgreave IA, Rosling H, Persson A, Link H (2002) Low prevalence of ataxic polyneuropathy in a community with high exposure to cyanide from cassava foods. Journal of Neurology, 249:1034–1040.
Osgood C, Sterling D (1991) Dichloroacetonitrile, a by-product of water chlorination, induces aneuploidy in Drosophila. Mutation Research, 261(2):85–91.
Osuntokun BO (1972) Chronic cyanide neurotoxicity and neuropathy in Nigerians. Plant Foods for Human Nutrition, 2:215–266.
Osuntokun BO (1980) A degenerative neuropathy with blindness and chronic cyanide intoxication of dietary origin: the evidence in the Nigerians. In: Smith RL, Bababunmi EA, eds. Toxicology in the tropics. London, Taylor and Francis, pp. 16–52.
Osuntokun BO (1981) Cassava diet, chronic cyanide intoxication and neuropathy in the Nigerian Africans. World Review of Nutrition and Dietetics, 36:141–173.
Osuntokun BO, Monekosso GL, Wilson J (1969) Relationship of a degenerative neuropathy to diet: report of a field survey. British Medical Journal, 1:547–550.
Padmaja G (1995) Cyanide detoxification in cassava for food and feed use. Critical Reviews in Food Sciences and Nutrition, 35:259–339.
Patnaik P (1999) A comprehensive guide to the hazardous properties of chemical substances, 2nd ed. New York, NY, Wiley & Sons, pp. 288–295.
Pentore R, Venneri A, Nichelli P (1996) Accidental choke cherry poisoning: early symptoms and neurological sequelae of an unusual case of cyanide intoxication. Italian Journal of Neurological Science, 17:233–235.
Pesce LD (1993) Cyanides. In: Kirk-Othmer encyclopedia of chemical technology, 4th ed. Vol. 7. New York, NY, John Wiley & Sons, pp. 753–782.
Pettersen JC, Cohen SD (1993) The effects of cyanide on brain mitochondrial cytochrome oxidase and respiratory activities. Journal of Applied Toxicology, 13:9–14.
Pettigrew AR, Fell GS (1973) Microdiffusion method for estimation of cyanide in whole blood and its application to the study of conversion of cyanide to thiocyanate. Clinical Chemistry, 19:466–471.
Philbrick DJ, Hopkins JB, Hill DC, Alexander JC, Thomson RG (1979) Effects of prolonged cyanide and thiocyanate feeding in rats. Journal of Toxicology and Environmental Health, 5:579–592.
Philips A (1989) Chemical hazards at electroplating processes. Scheffield, Health and Safety Executive, Library and Information Services, pp. 3–13.
Pijoan M (1942) Cyanide poisoning from choke cherry seed. American Journal of Medical Science, 204:550.
Prohorenkov VI, Kolpakov FI (1978) [Pathogenesis of dermatosis in gold-reclaiming plants.] Gigiena Truda i Profzabolevania, 12:44–45 (in Russian).
Purser DA (1984) A bioassay model for testing the incapacitating effects of exposure to combustion product atmospheres using cynomolgus monkeys. Journal of Fire Sciences, 2:20–36.
Purser DA, Grimshaw P, Berrill KR (1984) Intoxication by cyanide in fires: A study in monkeys using polyacrylonitrile. Archives of Environmental Health, 39:394–400.
Rieders F (1971) Noxious gases and vapors. I: Carbon monoxide, cyanides, methemoglobin, and sulfhemoglobin. In: De Palma JR, ed. Drill’s pharmacology in medicine, 4th ed. New York, NY, McGraw-Hill Book Company, pp. 1180–1205.
Rindone JP, Sloane EP (1992) Cyanide toxicity from sodium nitroprusside: Risk and management. Annals of Pharmacotherapy, 26:515–519.
Sayre JW, Kaymakcalan S (1964) Cyanide poisoning from apricot seeds among children in Central Turkey. New England Journal of Medicine, 270:1964.
Schubert J, Brill WA (1968) Antagonism of experimental cyanide toxicity in relation to the in vivo activity of cytochrome oxidase. Journal of Pharmacology and Experimental Therapeutics, 162:352–359.
Schultz V (1984) Clinical pharmacokinetics of nitroprusside, cyanide, thiosulfate and thiocyanate. Clinical Pharmacokinetics, 9:239–251.
Schultz V, Bonn R, Kindler J (1979) [Kinetics of elimination of thiocyanate in 7 healthy subjects and 8 subjects with renal failure.] Klinische Wochenschrift, 57:243–247 (in German).
Schultz V, Gross R, Pasch T, Busse J, Loescheke G (1982) Cyanide toxicity of sodium nitroprusside in therapeutic use with and without sodium thiosulfate. Klinische Wochenschrift, 60:1393–1400.
Seto Y, Tsunoda H, Ohta H, Shinohara T (1993) Determination of blood cyanide by headspace gas chromatography with nitrogen phosphorus detection and using a megabore capillary column. Analytica Chimica Acta, 276:247–259.
Sharma RP (1993) Cyanide containing foods and potential for fetal malformations. In: Sharma RP, ed. Dietary factors and birth defects. San Francisco, CA, American Association for the Advancement of Science, Pacific Division, pp. 332–348.
Shifrin NS, Beck BD, Gauthier TD, Chapnick SD, Goodman G (1996) Chemistry, toxicology and human risk of cyanide compounds in soils at former manufactured gas plant sites. Regulatory Toxicology and Pharmacology, 23:106–116.
Siller H, Winter J (1998) Degradation of cyanide in agroindustrial or industrial wastewater in an acidification reactor or in a single-step methane reactor by bacteria enriched from soil and peels of cassava. Applied Microbiology and Biotechnology, 50:384–389.
Singh JD (1981) The teratogenic effects of dietary cassava on the pregnant albino rat: preliminary report. Teratology, 24:289–291.
Sklarew DS, Hayes DJ (1984) Trace nitrogen-containing species in the offgas from 2 oil shale retorting processes. Environmental Science and Technology, 18:600–603.
Smyth HF Jr, Carpenter CP, Weil CS, Pozzani UC, Striegel JA (1962) Range-finding toxicity data: List VI. Industrial Hygiene Journal, 23:95–107.
Solberg Y, Rosner M, Belkin M (1998) The association between cigarette smoking and ocular diseases. Survey of Ophthalmology, 42:535–547.
Soto-Blanco B, Maiorka PC, Gorniak SL (2002a) Neuropathologic study of long term cyanide administration to goats. Food and Chemical Toxicology, 40:1693–1698.
Soto-Blanco B, Marioka PC, Gorniak SL (2002b) Effects of long-term low-dose cyanide administration to rats. Ecotoxicology and Environmental Safety, 53:37–41.
Sousa AB, Soto-Blanco B, Guerra JL, Kimura ET, Gorniak SL (2002) Does prolonged oral exposure to cyanide promote hepatotoxicity and nephrotoxicity? Toxicology, 174:87–95.
Suchard JR, Wallace KL, Gerkina D (1998) Acute cyanide toxicity caused by apricot kernel ingestion. Annals of Emergency Medicine, 32:742–744.
Swai AB, McLarty DG, Mtinangi BL, Tatala S, Kitange HM, Mlingi N, Rosling H, Howlett WP, Brubaker GR, Alberti KG (1992) Diabetes is not caused by cassava toxicity. A study in a Tanzanian community. Diabetes Care, 15:1378–1385.
Sylvester DM, Holmes RK, Sander C, Way JL (1982) Interference of thiosulfate with potentiometric analysis of cyanide in blood and its elimination. Toxicology and Applied Pharmacology, 65:116–121.
Tewe OO, Maner JH (1981a) Long-term and carry-over effect of dietary inorganic cyanide (KCN) in the life cycle performance and metabolism of rats. Toxicology and Applied Pharmacology, 58:1–12.
Tewe OO, Maner JH (1981b) Performance and pathophysiological changes in pregnant pigs fed cassava diets containing different levels of cyanide. Research in Veterinary Science, 30:147–151.
Tewe OO, Maner JH (1985) Cyanide, protein and iodine interactions in the performance and metabolism of rats. Journal of Environmental Pathology and Toxicology, 6:69–77.
Tinker JH (1976) Sodium nitroprusside: Pharmacology, toxicology and therapeutics. Anesthesiology, 45:340–354.
Tor-Agbidye J, Palmer VS, Lasarev MR, Craig AM, Blythe LL, Sabri MI, Spencer PS (1999) Bioactivation of cyanide to cyanate in sulfur amino acid deficiency: relevance to neurological disease in humans subsisting on cassava. Toxicological Sciences, 50:228–235.
Tylleskär T, Banea N, Fresco L, Persson LÅ, Rosling H (1991) Epidemiological evidence from Zaire for a dietary etiology of konzo, an upper motor neuron disease. Bulletin of the World Health Organization, 69:581–589.
Tylleskär T, Banea M, Bikangi N, Cooke RD, Poulter NH, Rosling H (1992) Cassava cyanogens and konzo, an upper motoneuron disease found in Africa. Lancet, 339:208–211.
Tylleskär T, Legue E, Peterson K, Kpinzingui E, Stecker P (1994) Konzo in the Central African Republic. Neurology, 44:959–961.
Uitti RJ, Rajput AH, Ashenhurst EM, Rozdilsky B (1985) Cyanide-induced parkinsonism: A clinicopathologic report. Neurology, 35:921–925 (abstract).
US EPA (1983) Methods for chemical analysis of water and waste. Method 335.2. Cyanide, total. Cincinnati, OH, US Environmental Protection Agency, Environmental Monitoring and Support Laboratory.
US EPA (1988) Calcium cyanide: Tolerances for residues. Code of Federal Regulations 40 CFR 180.125, US Environmental Protection Agency.
US EPA (1990) Summary review of health effects associated with hydrogen cyanide. Health issue assessment. Research Triangle Park, NC, US Environmental Protection Agency, Office of Research and Development (EPA/600/8-90/002F).
US EPA (1993a) Methods for the determination of inorganic substances in environmental samples. Washington, DC, US Environmental Protection Agency, Office of Research and Development, August (EPA/600/R-93/100).
US EPA (1993b) Hydrogen cyanide (CASRN
US EPA (1997) EPA Draft Method 1677. Ligand exchange/flow injection/amperometric technique for cyanide analysis. Cincinnati, OH, US Environmental Protection Agency, National Risk Management Research Laboratory.
US EPA (2002) Drinking water criteria document for cyanogen chloride and potential metabolites. Prepared for Health & Ecological Criteria, Division of Science & Technology, Office of Water, US Environmental Protection Agency, Washington, DC.
US EPA (2003a) Ground water & drinking water. Cyanide clarification. LabCert Bulletin, March. US Environmental Protection Agency. Available at http://www.epa.gov/safewater/certlab/labcert0303.html.
US EPA (2003b) Classes of underground injection wells. US Environmental Protection Agency. Available at http://www.epa.gov/region4/water/uic/uictable.htm.
US EPA (2003c) Toxics Release Inventory chemical report. US Environmental Protection Agency (TRI Explorer Report). Available at http://www.epa.gov/tri/tridata/.
Valade MP (1952) [Central nervous system lesions in chronic experimental poisoning with gaseous hydrocyanic acid.] Bulletin de l’Academie Nationale de Médecine, Paris, 136:280–225 (in French).
VanderLaan WP, Bissell A (1946) Effects of propylthiouracil and of potassium thiocyanate on the uptake of iodine by the thyroid gland of the rat. Endocrinology, 39:157–160.
Vock EH, Lutz WK, Hormes P, Hoffmann HD, Vamvakass A (1998) Discrimination between genotoxicity and cytotoxicity in the induction of DNA double-strand breaks in cells treated with etoposide, melphalan, cisplatin, potassium cyanide, Triton X-100, and gamma-irradiation. Mutation Research, 413:83–94.
Way JL (1984) Cyanide intoxication and its mechanism of antagonism. Annual Review of Pharmacology and Toxicology, 24:451–481.
Westley J, Adler H, Westley L, Nishida C (1983) The sulfurtransferases. Fundamental and Applied Toxicology, 3:377–382.
WHO (1985) Diabetes mellitus. Report of a WHO Study Group. Geneva, World Health Organization, 131 pp. Available at http://whqlibdoc.who.int/trs/WHO_TRS_727.pdf (WHO Technical Report Series 727).
WHO (2003) Guidelines for drinking-water quality, 3rd ed. Vol. 1. Recommendations. Geneva, World Health Organization.
Willhite CC (1982) Congenital malformations induced by laetrile. Science, 215:1513–1515.
Willhite CC, Smith RP (1981) The role of cyanide liberation in the acute toxicity of aliphatic nitriles. Toxicology and Applied Pharmacology, 59:589–602.
Williams RT (1959) Detoxification mechanisms, 2nd ed. London, Chapman and Hall, p. 393.
Wilson J (1983) Cyanide in human disease: A review of clinical and laboratory evidence. Fundamental and Applied Toxicology, 3:397–399.
Windholz M, ed. (1983) The Merck index, 10th ed. Rahway, NJ, Merck & Co.
Yamamoto K, Yamamoto Y, Hattori H, Samori T (1982) Effects of routes of administration on the cyanide concentration distribution in the various organs of cyanide-intoxicated rats. Tohoku Journal of Experimental Medicine, 137:73–78.
Zuidema PJ (1959) Cirrhosis and disseminated calcification of the pancreas in patients with malnutrition. Tropical and Geographical Medicine, 11:70–74.
ATSDR (1997) Toxicological profile for cyanide. Atlanta, GA, US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry
The ATSDR Toxicological profile for cyanide may be ordered from:
National Technical Information Service
5285 Port Royal Road
Springfield, VA 22161
USA
Telephone: (800) 553-6847 or (703) 605-6000
It is also available online at
http://www.atsdr.cdc.gov/toxprofiles/tp8.html.
Chemical managers/authors of the ATSDR Toxicological profile on cyanide were:
Carolyn Harper, Division of Toxicology, ATSDR, Atlanta, GA, USA
Susan Goldhaber, Research Triangle Institute, Research Triangle Park, NC, USA
The profile has undergone the following ATSDR internal reviews:
Green Border Review: Green Border Review assures consistency with ATSDR policy.
Health Effects Review: The Health Effects Review Committee examines the health effects chapter of each profile for consistency and accuracy in interpreting health effects and classifying end-points.
Minimal Risk Level Review: The Minimal Risk Level Workgroup considers issues relevant to substance-specific minimal risk levels (MRLs), reviews the health effects database of each profile, and makes recommendations for derivation of MRLs.
The peer review panel for cyanide consisted of the following members:
Dr Joseph Borowitz, Professor of Pathology, Department of Pharmacology and Toxicology, School of Pharmacy and Pharmaceutical Sciences, Purdue University, West Lafayette, IN, USA
Dr Gary Isom, Professor of Toxicology, Department of Pharmacology and Toxicology, Purdue University, West Lafayette, IN, USA
Dr James Way, Professor of Pharmacology and Toxicology, Department of Medical Pharmacology and Toxicology, Texas A&M University, College Station, TX, USA
JECFA (1993) Cyanogenic glycosides. In: Toxicological evaluation of certain food additives and naturally occurring toxicants. Geneva, World Health Organization, 39th Meeting of the Joint FAO/WHO Expert Committee on Food Additives (WHO Food Additives Series 30)
The JECFA monograph Cyanogenic glycosides is available from:
Marketing and Dissemination
World Health Organization
1211 Geneva 27
Switzerland
or at bookorders@who.int. It is also available online at
http://www.inchem.org/documents/jecfa/jecmono/v30je18.htm.
The first draft of the document was prepared by Dr G. Speijers, Laboratory for Toxicology, National Institute of Public Health and Environmental Protection, Bilthoven, The Netherlands. The document was reviewed at the 39th meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA).
The procedure for the preparation of the JECFA monographs is described at
http://www.who.int/pcs/jecfa/FA%20procedural%20guidelines.pdf.
The two consecutive drafts of the CICAD on hydrogen cyanide and cyanides (human health aspects) were sent for review to IPCS national Contact Points and Participating Institutions, as well as to identified experts. Comments were received from:
M. Baril, Institut de Recherche en Santé et en Sécurité du Travail, Montreal, Canada
R. Benson, Drinking Water Program, US Environmental Protection Agency, Denver, CO, USA
D. Chakrabarti, Jadavpur University, Calcutta, India
R.S. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
N. Chiu, Office of Water, US Environmental Protection Agency, Washington, DC, USA
M. Cikrt, National Institute of Public Health, Prague, Czech Republic
C. Cooke, Health and Safety Executive, Bootle, Merseyside, United Kingdom
I. Desi, University of Szeged, Szeged, Hungary
G. Dura, National Institute of Environmental Health of Jozsef Fodor, Budapest, Hungary
C. Elliot-Minty, Health and Safety Executive, Bootle, Merseyside, United Kingdom
H. Greim, National Research Center for Environment and Health, Technical University of Munich, Munich, Germany
M. Gully, Health Canada, Ottawa, Canada
G.K. Hatfield, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
R.F. Hertel, Federal Institute for Risk Assessment, Berlin, Germany
J. Hopkins, Toxicology Advice & Consulting Ltd, Sutton, Surrey, United Kingdom
P. Howe, Centre for Ecology and Hydrology, Monks Wood, United Kingdom
S. Jacobi, Cyanides Task Force, European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC), and European Chemical Industry Council (CEFIC), Brussels, Belgium
U. Kierdorf, Justus-Liebig-University of Giessen, Giessen, Germany
G. Koennecker, Fraunhofer Institute for Toxicology and Experimental Medicine, Hanover, Germany
T. Kuiper-Goodman, Health Canada, Ottawa, Canada
S. Melching-Kollmuβ, Fraunhofer Institute for Toxicology and Experimental Medicine, Hanover, Germany
L. Murthy, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
M.A. Pemberton, Cyanides Task Force, European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC), and European Chemical Industry Council (CEFIC), Brussels, Belgium
J. Rantanen, Institute of Occupational Health, Helsinki, Finland
T. Santonen, Institute of Occupational Health, Helsinki, Finland
S. Schmidt, Fraunhofer Institute for Toxicology and Experimental Medicine, Hanover, Germany
P. Schulte, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
J.C. Stadler, DuPont Haskell Laboratory, Newark, DE, USA
J.L. Stauber, Commonwealth Scientific and Industrial Research Organisation (CSIRO) Energy Technology, Bangor, NSW, Australia
U. Stenius, Karolinska Institute, Stockholm, Sweden
J.H.M. Temmink, Wageningen Agricultural University, Wageningen, The Netherlands
S. Vainiotalo, Institute of Occupational Health, Helsinki, Finland
A. Vyskocil, University of Montreal, Montreal, Canada
D. Willcocks, Existing Chemicals, National Industrial Chemicals Notification and Assessment Scheme (NICNAS), Sydney, NSW, Australia
K. Ziegler-Skylakakis, Commission of European Communities, Luxembourg
Monks Wood, United Kingdom
16–19 September 2002
Members
Dr R. Benson, US Environmental Protection Agency, Region VIII, Denver, CO, USA
Mr R. Cary, Health and Safety Executive, Bootle, Merseyside, United Kingdom
Dr R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
Dr S. Chou, Agency for Toxic Substances and Disease Registry (ATSDR), Atlanta, GA, USA
Dr S. Czerczak, Nofer Institute of Occupational Medicine, Lodz, Poland
Dr S. Dobson, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr G. Dura, National Institute of Environmental Health, Jozsef Fodor Public Health Centre, Budapest, Hungary
Dr L. Fishbein, Fairfax, VA, USA
Dr H. Gibb, National Center for Environmental Assessment, US Environmental Protection Agency, Washington, DC, USA
Dr Y. Hayashi, Division of Chem-Bio Informatics, National Institute of Health Sciences, Ministry of Health, Labour and Welfare, Tokyo, Japan
Dr R.F. Hertel, Federal Institute for Health Protection of Consumers and Veterinary Medicine, Berlin, Germany
Dr A. Hirose, Division of Risk Assessment, National Institute of Health Sciences, Tokyo, Japan
Mr P. Howe, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Prof. J. Jeyaratnam, Colombo, Sri Lanka
Dr J. Kielhorn, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany
Prof. Y.-X. Liang, School of Public Health, Fudan University, Shanghai Medical College, Shanghai, People’s Republic of China
Dr R. Liteplo, Existing Substances Division, Environmental Contaminants Bureau, Health Canada, Ottawa, Ontario, Canada
Ms M.E. Meek, Existing Substances Division, Safe Environments Programme, Health Canada, Ottawa, Ontario, Canada
Mr F.K. Muchiri, Directorate of Occupational Health and Safety Services, Nairobi, Kenya
Dr O. Sabzevari, Department of Toxicology & Pharmacology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
Dr J. Sekizawa, Division of Chem-Bio Informatics, National Institute of Health Sciences, Tokyo, Japan
Dr F.P. Simeonova, Sofia, Bulgaria
Dr J. Stauber, CSIRO Energy Technology, Centre for Advanced Analytical Chemistry, Bangor, Australia
Dr M.H. Sweeney, Document Development Branch, Education and Information Division, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
Dr K. Ziegler-Skylakakis, European Commission, DG Employment & Social Affairs, Luxembourg
Resource Persons
Dr C. Cowles, Health and Safety Executive, Industrial Chemicals Unit HD, Bootle, Merseyside, United Kingdom
Dr C. Elliott-Minty, Health and Safety Executive, Industrial Chemicals Unit HD, Bootle, Merseyside, United Kingdom
Dr K. Fuller, Health and Safety Executive, Industrial Chemicals Unit HD, Bootle, Merseyside, United Kingdom
Observers
Mr A.G. Berends, Solvay S.A., Brussels, Belgium; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr W. Gulledge, American Chemistry Council, Arlington, VA, USA
Mr C. Newsome, Dow Chemical Company Limited, West Drayton, Middlesex, United Kingdom; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr M.A. Pemberton, Wilmslow, United Kingdom; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr W. Stott, Dow Chemical Company, Midland, MI, USA; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Mr J.M. Waechter, Jr, The Dow Chemical Company, Midland, MI, USA; European Chemical Industry Council / European Centre for Ecotoxicology and Toxicology of Chemicals (CEFIC/ECETOC)
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr T. Ehara, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr H. Malcolm, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Ms C. Vickers, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Varna, Bulgaria
8–11 September 2003
Members
Dr I. Benchev, Sofia, Bulgaria
Dr R. Chhabra, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA
Dr C. De Rosa, Agency for Toxic Substances and Disease Registry, Centers for Disease Control and Prevention, Atlanta, GA, USA
Dr S. Dobson, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr G. Dura, National Institute of Environment, József Fodor Public Health Centre, Budapest, Hungary
Dr L. Fishbein, Fairfax, VA, USA
Dr H. Gibb, National Center for Environmental Assessment, US Environmental Protection Agency, Washington, DC, USA
Dr R.F. Hertel, Federal Institute for Risk Assessment, Berlin, Germany
Mr P. Howe, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom
Dr S. Ishimitsu, Division of Safety Information on Drug, Food and Chemicals, National Institute of Hygienic Sciences, Tokyo, Japan
Dr D. Kanungo, Central Insecticides Board, Directorate of Plant Protection, Quarantine & Storage, Ministry of Agriculture, Haryana, India
Dr J. Kielhorn, Fraunhofer Institute for Toxicology and Experimental Medicine, Hanover, Germany
Ms B. Meek, Environmental Health Directorate, Health Canada, Ottawa, Ontario, Canada
Dr T. Morita, Division of Safety Information on Drug, Food and Chemicals, National Institute of Hygienic Sciences, Tokyo, Japan
Mr F.K. Muchiri, Directorate of Occupational Health and Safety Services, Nairobi, Kenya
Dr L. Olsen, Biological Monitoring & Health Assessment Branch, Division of Applied Research & Technology, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
Dr N. Rizov, National Center of Hygiene, Medical Ecology and Nutrition, Sofia, Bulgaria
Dr P. Schulte, Education and Information Division, National Institute for Occupational Safety and Health, Cincinnati, OH, USA
Dr J. Sekizawa, Faculty of Integrated Arts and Sciences, Tokushima University, Tokushima, Japan
Dr F.P. Simeonova, Sofia, Bulgaria
Dr S. Soliman, Faculty of Agriculture, Alexandria University, El Shatby, Alexandria, Egypt
Dr J. Stauber, CSIRO Energy Technology, Centre for Advanced Analytical Chemistry, Bangor, NSW, Australia
Mr P. Watts, Toxicology Advice & Consulting Ltd, Surrey, United Kingdom
Ms D. Willcocks, National Industrial Chemicals Notification and Assessment Scheme, Sydney, NSW, Australia
Dr K. Ziegler-Skylakakis, European Commission, Luxembourg
Observers
Dr S. Jacobi, Degussa AG, Fine Chemicals, Hanau-Wolfgang, Germany
Mr M. Southern, Shell International Petroleum Company Ltd, London, United Kingdom
Dr W. ten Berge, DSM, Heerlen, The Netherlands
Secretariat
Dr A. Aitio, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
Mr T. Ehara, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
|
ACH |
acetone cyanohydrin |
|
ALAT |
alanine aminotransferase |
|
ASAT |
aspartate aminotransferase |
|
ATSDR |
Agency for Toxic Substances and Disease Registry (USA) |
|
CAS |
Chemical Abstracts Service |
|
CHO |
Chinese hamster ovary |
|
CICAD |
Concise International Chemical Assessment Document |
|
DNA |
deoxyribonucleic acid |
|
EHC |
Environmental Health Criteria |
|
EPA |
Environmental Protection Agency (USA) |
|
FAO |
Food and Agriculture Organization of the United Nations |
|
HGPRT |
hypoxanthine–guanine phosphoribosyltransferase |
|
ILO |
International Labour Organization |
|
IPCS |
International Programme on Chemical Safety |
|
JECFA |
Joint FAO/WHO Expert Committee on Food Additives |
|
Kow |
octanol/water partition coefficient |
|
LC50 |
median lethal concentration |
|
LD50 |
median lethal dose |
|
LOAEL |
lowest-observed-adverse-effect level |
|
MAK |
Maximum Allowable Concentration (Germany) |
|
MRL |
minimal risk level |
|
NIOSH |
National Institute for Occupational Safety and Health (USA) |
|
NOAEL |
no-observed-adverse-effect level |
|
NOEL |
no-observed-effect level |
|
PIM |
Poison Information Monograph |
|
SD |
standard deviation |
|
SI |
International System of Units (Système international d’unités) |
|
T3 |
triiodothyronine |
|
T4 |
thyroxine |
|
TSH |
thyroid-stimulating hormone |
|
UNEP |
United Nations Environment Programme |
|
USA |
United States of America |
|
US EPA |
United States Environmental Protection Agency |
|
WHO |
World Health Organization |
HYDROGEN CYANIDE, LIQUEFIED ICSC:0492
POTASSIUM FERRICYANIDE ICSC:1132
Le présent CICAD relatif au cyanure d’hydrogène (acide cyanhydrique) et aux cyanures (aspects touchant la santé humaine) a été préparé par le Pr Fina Petrova Simeonova et le Dr Lawrence Fishbein, principalement sur la base du profil toxicologique des cyanures établi par l’Agency for Toxic Substances and Disease Registry (ATSDR, 1997) et d’un document du Comité mixte FAO/OMS d’experts des additifs alimentaires portant sur les glucosides cyanogéniques (JECFA, 1993). L’appendice 1 est consacré à ces documents de base ainsi qu’à la manière dont ils ont été passés en revue. En octobre 2002, il a été procédé à un dépouillement exhaustif de la bibliographie contenue dans plusieurs bases de données en ligne à la recherche d’articles publiés postérieurement à ceux qui sont cités dans les documents de base. Ce CICAD a fait l’objet d’un premier examen lors de la dixième réunion du Comité d’évaluation finale qui s’est tenue à Monks Wood
(Royaume-Uni) du 16 au 19 septembre 2002. Après avoir été révisé, il a été réexaminé et approuvé en tant qu’évaluation internationale lors de la onzième réunion du Comité d’évaluation finale qui a eu lieu à Varna (Bulgarie) du 8 au 11 septembre 2003. La liste des participants à la dixième et à la onzième réunion du Comité d’évaluation finale figure aux appendices 2 et 3. Les documents préparatoires discutés lors de ces réunions avaient été préalablement examinés par des pairs; des renseignements sur ces examens sont donnés à l’appendice 4. Les fiches internationale sur la sécurité chimique du cyanure d’hydrogène, du cyanure de sodium, du cyanure de potassium, du cyanure de calcium, du cyanogène, du chlorure de cyanogène, de la cyanhydrine d’acétone - plus couramment appelée acétone cyanhydrine - et du ferricyanure de potassium établies par le Programme international sur la sécurité chimique (IPCS, 1999a,b, 2000b, 2001, 2002a,b,c,d) sont également reproduites dans le présent document.
On désigne sous le nom de cyanures des composés très divers et de complexité chimique variable, mais qui contiennent tous un groupement CN et auxquels l’Homme peut se trouver exposé lorsqu’ils sont produits à l’état gazeux, liquide ou solide par toutes sortes de sources naturelles ou anthropogéniques. Différents types de cyanures sont utilisés dans un certain nombre d’applications industrielles ou sont présents dans l’environnement, mais quelle qu’en soit l’origine, c’est l’anion cyanure CN– qui représente l’agent toxique essentiel.
Le cyanure d’hydrogène se présente sous la forme d’un liquide ou d’un gaz incolore à bleu pâle dégageant une légère odeur d’amande amère. On l’utilise principalement pour la préparation de substances telles que l’adiponitrile, le méthacrylate de méthyle, divers agents chélatants, le chlorure cyanurique (2,4,6-trichlorotriazine), la méthionine et ses analogues hydroxylés, ainsi que le cyanure de potassium et le cyanure de sodium. On s’en sert également comme fumigant à l’intérieur des navires, des voitures de chemin de fer, des grands bâtiments, des silos à grain et des minoteries ainsi que pour traiter les pois et les semences en chambre étanche.
D’autres cyanures, comme le cyanure de potassium et le cyanure de sodium se présentent sous la forme de sels solides ou de cristaux hygroscopiques dont on fait grand usage pour extraire l’or et l’argent de leurs minerais, en galvanoplastie, pour la cémentation de l’acier, la flottation des métaux de base et le dégraissage des métaux, la teinture, l’impression et la photographie. Ils sont également très largement utilisés pour la synthèse de composés organiques ou minéraux (comme les nitriles, les acides carboxyliques, les amides, les esters et les amines ou les cyanures de métaux lourds) ainsi que pour la production d’agents chélatants.
Il existe différentes sources anthropogéniques qui libèrent des cyanures dans l’environnement. Ils peuvent par exemple être rejetés dans l’atmosphère par les industries qui les produisent ou les utilisent à diverses fins, comme les industries métallurgiques et celles qui pratiquent la galvanoplastie ou l’extraction de l’or et de l’argent à partir de minerais à faible teneur. Ils peuvent également s’échapper dans l’environnement par volatilisation à partir de déchets enfouis dans des décharges ou rejetés dans des bassins, émission à partir d’incinérateurs municipaux qui brûlent des déchets solides, combustion de biomasse ou de combustibles fossiles (notamment dans les gaz d’échappement des véhicules à moteur), lors d’opérations de fumigation ou encore pendant la production de coke ou autres opérations de carbonisation de la houille.
Il se forme du cyanure d’hydrogène lors de la combustion incomplète des polymères azotés, comme certains plastiques, les polyuréthanes et la laine. La fumée de cigarette en contient également.
Les cyanures qui passent dans le compartiment aquatique à partir de sources non ponctuelles peuvent être transportés par les eaux de ruissellement contenant des sels antiagglomérants cyanurés épandus sur les routes, provenir de déchets enfouis ou encore du lessivage de divers dépôts d’origine agricole ou atmosphérique. Au nombre des sources ponctuelles de cyanures figurent les installations d’extraction de l’or, les usines de traitement de l’eau, la sidérurgie et l’industrie chimique organique.
On compte plus de 2000 espèces végétales (notamment des fruits et des légumes) contenant des glucosides cyanogéniques qui constituent des sources naturelles de cyanures et qui sont capables de libérer des ions cyanure par hydrolyse lorsqu’ils sont ingérés. Parmi ces denrées, on peut citer la cassave (tapioca, manioc) et le sorgho qui, dans de nombreux pays tropicaux, représentent l’alimentation de base de millions de gens. L’amygdaline, la linamarine, la prunasine, la dhurrine, la lotaustraline et la taxiphylline sont des glucosides cyanogéniques bien connus. Certains processus biologiques qui se déroulent dans les plantes supérieures, les champignons et les bactéries conduisent également à la libération de cyanures dans l’atmosphère.
Dans l’air, les cyanures sont présent sous la forme de cyanure d’hydrogène gazeux, avec une petite fraction sous forme de fines particules de poussière. Les cyanures sont susceptibles d’être transportés sur de longues distances à partir de leur source d’émission.
Dans sa majorité, la population n’est exposée qu’à de très faibles quantités de cyanures présents dans l’environnement général. Toutefois, pour certains groupes, la possibilité d’exposition est plus grande. Il s’agit notamment de personnes qui travaillent à la transformation à grande échelle du manioc ou qui consomment des quantités importantes d’aliments mal préparés contenant des glucosides cyanogéniques, comme le manioc ou encore certaines denrées particulières comme les noyaux d’abricots et les amandes amères. Parmi les autres groupes de population qui risquent le plus d’être exposés aux cyanures, on peut citer les personnes qui résident à proximité de sources ponctuelles à partir desquelles ces substances peuvent être libérées volontairement ou accidentellement dans l’environnement, les fumeurs actifs et passifs ou encore les personnes qui ont inhalé de la fumée à la suite d’un incendie.
Les travailleurs peuvent être exposés aux cyanures au cours d’opérations de fumigation, lors de la production de ces substances ou encore lors de nombreuses activités industrielles au cours desquelles elles sont utilisées - par exemple lors d’opérations de galvanoplastie ou de cémentation de l’acier ou lors de l’extraction de l’or et de l’argent de leurs minerais.
Bien résorbés au niveau des voies digestives ou par passage transcutané, les cyanures sont rapidement absorbés par la voie respiratoire. Une fois absorbés, ils se répandent rapidement dans tout l’organisme, mais les teneurs les plus élevées sont habituellement retrouvées au niveau du foie, des poumons, dans le sang et dans l’encéphale. Une exposition chronique ou répétée ne provoque pas d’accumulation de cyanures dans le sang ou les tissus.
Une fois absorbés, les cyanures sont métabolisés dans la proportion d’environ 80 % en thiocyanates au niveau du foie sous l’action d’une sulfo-transférase mitochondriale, la rhodanase, et d’autres sulfo-transférases. Les thiocyanates sont excrétés dans les urines. Il existe des voies métaboliques mineures de détoxication des cyanures qui comportent une réaction avec la cystine conduisant aux acides aminothiazoline- et iminothiazolidinecarboxyliques ou une combinaison avec l’hydroxycobalamine (vitamine B12a) conduisant à la formation de cyanocobalamine (vitamine B12). Ces produits finals sont également excrétés par la voie urinaire.
Les cyanures sont principalement caractérisés par une toxicité élevée quelle que soit la voie d’administration, avec une courbe dose-effet à très forte pente dépendant de la vitesse d’administration, ainsi que par une toxicité chronique dans laquelle intervient probablement l’ion thiocyanate, qui est leur principal métabolite et produit de détoxication. Les effets toxiques de l’ion cyanure sont généralement similaires chez l’Homme et l’animal et on pense qu’ils sont dus à l’inactivation de la cytochrome-oxydase qui conduit à l’inhibition de la respiration et à une anoxie histotoxique. Les principales cibles de l’action toxique de l’ion cyanure sont les appareils cardiovasculaire et respiratoire ainsi que le système nerveux central. Le système endocrinien est également une cible potentielle de l’activité toxique à long terme de l’ion cyanure, qui résulte d’une exposition permanente à l’ion thiocyanate ayant pour effet d’inhiber la fixation de l’iode par la thyroïde et par suite, de conduire à la formation d’un goitre.
Chez l’Homme, si de légers effets peuvent être notés aux doses de 20 à 40 mg/m3, des doses de 50 à 60 mg/m3 sont supportables pendant 20 minutes à 1 heure sans effets immédiats ou retardés. Une dose de l’ordre de 120 à 150 mg/m3 peut entraîner la mort au bout d’une demi-heure à une heure et à partir de 150 mg/m3, la mort peut survenir en l’espace de 30 minutes. Une exposition à 200 mg/m3 est probablement mortelle au bout de 10 minutes et à partir de 300 mg/m3, la mort est immédiate. Chez l’Homme, la dose la plus faible qui se soit révélée mortelle par voie orale est de 0,54 mg/kg de poids corporel et on estime que dans les cas d’empoisonnement, la dose moyenne absorbée au moment de la mort est de 1,4 mg/kg p.c. (calculée en cyanure d’hydrogène). Après une intoxication grave, les séquelles peuvent être de nature neuropsychiatrique ou de type parkinsonien. On estime que les cyanures présents dans la fumée de tabac contribuent à l’amblyopie alcoolo-tabagique. Une exposition prolongée à de faibles concentrations de cyanures sur le lieu de travail peut déterminer divers symptômes neurologiques centraux.
On attribue à la consommation prolongée de manioc riche en glucosides cyanogéniques diverses pathologies telles que la neuropathie ataxique tropicale ou la paraparésie spastique et, dans les régions où l’apport d’iode est insuffisant, l’hypothyroïdie, le goitre et le crétinisme. Si l’on a pu quelquefois estimer l’exposition journalière aux cyanures dans les régions d’endémie à approximativement 15-50 mg, le caractère limité des informations sur l’exposition et l’influence possible de facteurs de confusion comme la malnutrition, la faible teneur en protéines de l’alimentation, les carences vitaminiques et l’insuffisance du bilan iodé font que les données ne permettent pas d’obtenir des renseignements valables sur la relation dose-réponse dans le cas des cyanures.
Les données concernant des points d’aboutissement de l’action toxique autres que la toxicité aiguë sont assez limitées. On peut attribuer pour une grande part cet état de choses à la difficulté d’effectuer, par exemple, des études avec des doses répétées ou des études de toxicité chronique sur une substance présentant une toxicité aiguë aussi forte. Les cyanures sont légèrement irritants pour la peau et la muqueuse oculaire. On n’a pas trouvé de données concernant d’éventuelles propriétés sensibilisantes ou une quelconque activité cancérogène de l’acide cyanhydrique ou de ses sels alcalins. Les éléments d’appréciation dont on dispose sont quelque peu limités, mais montrent cependant que les cyanures ne sont pas génotoxiques et qu’ils ne produisent d’effets sur le développement qu’à des doses toxiques pour la mère.
Les données relatives aux populations humaines ne sont pas suffisantes pour servir de base à la caractérisation de la relation dose-réponse dans le cas d’une ingestion chronique de cyanures. Lors d’une étude de 13 semaines au cours de laquelle on a administré à plusieurs reprises à des rats et à des souris du cyanure dissous dans leur eau de boisson, on n’a pas observé de signes neurologiques centraux ni d’effets histopathologiques au niveau de l’encéphale ou de la thyroïde lorsque les animaux on reçu des doses journalières de cette substance allant respectivement jusqu’à 12,5 et 26 mg par kg de poids corporel. A la dose journalière de 12,5 mg/kg p.c., on a noté de petites anomalies au niveau des voies génitales chez rats mâles, anomalies qui, si elle n’avaient apparemment pas d’effet sur la fécondité des animaux, ne seraient peut-être pas négligeables chez l’Homme. La dose journalière sans effet nocif observable (NOAEL) pour ce type d’effets était de 4,5 mg/kg de poids corporel. Les examens neurologiques effectués dans le cadre de cette étude se sont limités à des observations cliniques et à un examen au microscope optique lors de l’autopsie. Les rares études existantes qui soient spécialement consacrées à l’investigation de la neurotoxicité, font état d’effets nocifs chez le rat à la dose journalière de 1,2 mg de cyanure/kg de poids corporel et à celle de 0,48 mg/kg chez la chèvre, mais elles souffrent de faiblesses qui empêchent d’en tirer des conclusions quantitatives.
Lors de trois études distinctes effectuées sur des rats dans le but de caractériser la relation dose-réponse dans le cas de l’inhalation répétée de cyanure d’hydrogène (principalement sur le lieu de travail), on n’a pas relevé d’effet toxique général chez les animaux exposés à de l’acétone cyanhydrine, composé qui est rapidement hydrolysé en cyanure d’hydrogène au pH physiologique, pour des concentrations allant jusqu’à 211 mg/m3 (soit l’équivalent de 67 mg de cyanure d’hydrogène par m3). La mortalité de 30 % observée chez les rats exposés une partie de la journée à 225 mg/m3 d’acétone cyanhydrine (soit l’équivalent de 71 mg cyanure d’hydrogène par m3) traduit la forte pente de la courbe dose-effet.
Il est peu probable que l’exposition à la faible concentration de cyanures habituellement présents dans l’environnement général (< 1 µg/m3 dans l’air ambiant; < 10 µg/litre dans l’eau) ait des effets nocifs. Des intoxications aiguës par les cyanures peuvent se produire à la suite de la consommation de noyaux d’abricots, de cerises de Virginie ou d’autres drupes riches en glucosides cyanogéniques. Du manioc mal préparé peut être dangereux lorsqu’il constitue l’essentiel de l’alimentation.
Este CICAD sobre el cianuro de hidrógeno y otros cianuros (aspectos relativos a la salud humana), preparado por la Profesora Petrova Simeonova y el Dr. Lawrence Fishbein, se basó principalmente en el perfil toxicológico de los cianuros de la Agencia para el Registro de Sustancias Tóxicas y Enfermedades (ATSDR, 1997) y el documento sobre glucósidos cianogénicos del Comité Mixto FAO/OMS de Expertos en Aditivos Alimentarios (JECFA, 1993). Los documentos originales y la descripción de sus procesos de examen se presentan en el apéndice 1. En octubre de 2002 se realizó una búsqueda bibliográfica amplia de varias bases de datos en línea para localizar cualquier referencia pertinente publicada después de las citadas en los documentos originales. Este CICAD se debatió en primer lugar en la 10ª reunión de la Junta de Evaluación Final, celebrada en Monks Wood (Reino Unido) del 16 al 19 de septiembre de 2002. Tras la revisión, se debatió de nuevo y se aprobó como evaluación internacional en la 11ª reunión de la Junta de Evaluación Final, celebrada en Varna (Bulgaria) del 8 al 11 de septiembre de 2003. Las listas de participantes en las reuniones 10ª y 11ª de la Junta de Evaluación Final figuran en los apéndices 2 y 3. Los proyectos debatidos fueron objeto de un examen colegiado antes de dichas reuniones; la información sobre el proceso del examen colegiado aparece en el apéndice 4. También se reproducen en este documento las Fichas internacionales de seguridad química para el cianuro de hidrógeno, el cianuro de sodio, el cianuro de potasio, el cianuro de calcio, el cianógeno, el cianuro de cloro, la cianohidrina de acetona y el ferricianuro de potasio, preparadas por el Programa Internacional de Seguridad de las Sustancias Químicas (IPCS, 1999a,b, 2000b, 2001, 2002a,b,c,d).
Los cianuros comprenden una amplia gama de compuestos con distintos grados de complejidad química, que se caracterizan por contener todos ellos un grupo CN, a los cuales están expuestas las personas ya sea en forma gaseosa, liquida o sólida a partir de fuentes naturales y antropogénicas. Si bien hay muchas formas químicas de cianuro que se utilizan en aplicaciones industriales o están presentes en el medio ambiente, el agente tóxico principal, con independencia del origen, es el anión cianuro (CN–).
El cianuro de hidrógeno es un líquido o gas incoloro o de tono azul pálido con un ligero olor a almendras amargas. Se utiliza fundamentalmente en la producción de diversas sustancias, por ejemplo adiponitrilo, metacrilato de metilo, agentes quelantes, cianuro de cloro, metionina y sus compuestos análogos hidroxilados y cianuro de sodio y de potasio. También se utiliza como fumigante en embarcaciones, vagones de ferrocarril, edificios grandes, silos de cereales y molinos de harina, así como para la fumigación de guisantes y semillas en cámaras de vacío.
Otros cianuros, como los de sodio y potasio, son sales higroscópicas sólidas o cristalinas que se utilizan ampliamente en los procesos de extracción de minerales para la recuperación del oro y la plata, la galvanoplastia, el endurecimiento superficial del acero, la flotación de metales no preciosos, el desengrasado, teñido e impresión de metales y la fotografía. También se utilizan con frecuencia en la síntesis de sustancias químicas orgánicas e inorgánicas (por ejemplo, nitrilos, ácidos carboxílicos, amidas, ésteres y aminas; cianuros de metales pesados) y en la producción de agentes quelantes.
El cianuro que se libera en el medio ambiente procede de fuentes antropogénicas diversas. Las emisiones al aire se deben a las industrias de fabricación y elaboración de productos químicos, como las metalúrgicas y de chapado metálico, y la extracción de oro y plata a partir de minerales de baja calidad. Otras fuentes son la volatilización de los desechos de cianuro existentes en los vertederos y estanques utilizados para su eliminación, las emisiones de los incineradores municipales de residuos sólidos, la combustión de biomasa, el consumo de combustibles fósiles, en particular las emisiones de los vehículos, las operaciones de fumigación y la producción de coque u otros procedimientos de carbonización del carbón.
Se forma cianuro de hidrógeno durante la combustión incompleta de polímeros que contienen nitrógeno, como ciertos plásticos, poliuretanos y lana. También está presente en el humo de los cigarrillos.
Puede haber fuentes no puntuales de emisiones de cianuro al agua a partir de la escorrentía de sales antiaglutinantes con cianuro utilizadas en las carreteras, la migración desde los vertederos y el arrastre y lavado de los cultivos agrícolas y de la atmósfera. Entre las fuentes puntuales de emisiones al agua están las descargas de instalaciones de extracción de oro, las operaciones de tratamiento de las aguas residuales, la producción de hierro y acero y otras industrias de fabricación de productos químicos orgánicos.
Las principales fuentes naturales de cianuro son más de 2000 especies de plantas, con inclusión de frutas y hortalizas, que contienen glucósidos cianogénicos, que cuando se ingieren pueden liberar cianuro por hidrólisis. Entre ellas, la yuca (tapioca, mandioca) y el sorgo son alimentos básicos de cientos de millones de personas en numerosos países tropicales. Son glucósidos cianogénicos conocidos de plantas la amigdalina, la linamarina, la prunasina, la durrina, la lotaustralina y la taxifilina. Los procesos biogénicos naturales de las plantas superiores, las bacterias y los hongos desprenden cianuro de hidrógeno en la atmósfera.
El cianuro está presente en el aire como cianuro de hidrógeno gaseoso, con una pequeña cantidad en partículas finas de polvo. Los cianuros pueden recorrer largas distancias desde sus respectivas fuentes de emisión.
La mayoría de la población está expuesta a niveles muy bajos de cianuro en el medio ambiente general. Sin embargo, hay subgrupos específicos con un potencial más elevado de exposición. Entre ellos figuran las personas que intervienen en la elaboración de la yuca en gran escala o quienes consumen cantidades importantes de alimentos que contienen glucósidos cianogénicos preparados de manera inadecuada, como la yuca y alimentos especiales, como los huesos de albaricoque y las almendras amargas. Otros subgrupos con un gran potencial de exposición son los que se encuentran en las cercanías de emisiones accidentales o intencionadas de fuentes puntuales, los fumadores activos y pasivos y las víctimas de inhalación del humo a causa de incendios.
Los trabajadores pueden estar expuestos a los cianuros durante las operaciones de fumigación y durante la producción y utilización de cianuros en numerosos procesos industriales, por ejemplo la galvanoplastia, el endurecimiento superficial del acero y la extracción de oro y plata de los minerales.
Los cianuros se absorben bien a través del tracto gastrointestinal o la piel y con gran rapidez en el tracto respiratorio. Una vez absorbido, se distribuye de manera rápida y ubicua por todo el organismo, aunque las concentraciones más elevadas se suelen encontrar en el hígado, los pulmones, la sangre y el cerebro. No hay acumulación de cianuro en la sangre o los tejidos tras la exposición crónica o repetida.
La rodanasa, enzima sulfotransferasa mitocondrial, y otras sulfotransferasas metabolizan en el hígado a tiocianato alrededor del 80% del cianuro absorbido. El tiocianato se excreta en la orina. Son vías secundarias para la desintoxicación del cianuro la reacción con la cistina para formar los ácidos aminotiazolincarboxílico e iminotiazolidincarboxílico y la combinación con la hidroxicobalamina (vitamina B12a) para forrmar cianocobalamina (vitamina B12); estos productos finales también se excretan en la orina.
Las principales características del perfil toxicológico del cianuro son su elevada toxicidad aguda por todas las vías de administración, con una curva de la relación dosis-efecto de pendiente muy pronunciada y dependiente de la tasa de aplicación, y su toxicidad crónica, probablemente mediada por el tiocianato, metabolito principal y producto de la desintoxicación. Los efectos tóxicos del ión cianuro en las personas y los animales son en general semejantes y parece que se deben a la inactivación de la citocromo oxidasa y la inhibición de la respiración celular y la consiguiente anoxia histotóxica. La toxicidad del cianuro en las personas y los animales afecta fundamentalmente a los sistemas cardiovascular, respiratorio y nervioso central. El sistema endocrino también puede verse afectado por la toxicidad prolongada, debido a la exposición continuada al tiocianato, que impide la absorción de yodo en el tiroides y actúa como agente bociógeno.
En las personas, si bien con niveles de exposición de 20-40 mg/m3 se pueden producir efectos ligeros, son tolerables sin efectos inmediatos o tardíos concentraciones de 50-60 mg/m3 durante un intervalo de 20 minutos a una hora, 120-150 mg/m3 pueden provocar la muerte tras un intervalo de 0,5 a una hora, 150 mg/m3 probablemente son mortales en 30 minutos, 200 mg/m3 probablemente son mortales después de 10 minutos y 300 mg/m3 son mortales de manera inmediata. La dosis letal más baja por vía oral notificada en personas es de 0,54 mg/kg de peso corporal y la dosis media absorbida en el momento de la muerte se ha estimado en 1,4 mg/kg de peso corporal (calculada como cianuro de hidrógeno). Entre las secuelas que aparecen tras una intoxicación aguda grave figuran manifestaciones neuropsiquiátricas y una enfermedad del tipo del Parkinson. Se ha señalado que el cianuro del humo del tabaco es un factor que contribuye a la ambliopía debida al alcohol y el tabaco. La exposición prolongada a concentraciones más bajas de cianuro en los entornos ocupacionales puede provocar diversos síntomas relacionados con efectos en el sistema nervioso central.
El consumo prolongado de yuca, que contiene niveles elevados de glucósidos cianogénicos, se ha relacionado con la presencia de neuropatía atáxica tropical y paraparesia espástica y, en zonas con un consumo bajo de yodo, la aparición de hipotiroidismo, bocio y cretinismo. Si bien en zonas endémicas en algunos de esos casos se ha estimado la exposición al cianuro en alrededor de 15-50 mg/día, debido a las limitaciones de los datos sobre la exposición y el efecto potencial de factores de confusión, como la malnutrición, el bajo contenido en proteínas de la alimentación, las deficiencias de vitaminas y la concentración de yodo, los datos disponibles no proporcionan información fidedigna sobre la relación dosis-respuesta para el cianuro.
Los datos sobre los efectos finales distintos de la toxicidad aguda son algo limitados. Esto se puede atribuir en gran parte a las dificultades para realizar, por ejemplo, investigaciones de dosis repetidas o de toxicidad crónica debido a la elevada toxicidad aguda del compuesto. Los cianuros son ligeramente irritantes de la piel y los ojos; no se han localizado datos sobre las propiedades de sensibilización o carcinogenicidad del cianuro de hidrógeno o de sus sales alcalinas. Aunque los datos disponibles son algo limitados, su valor demostrativo indica que el cianuro no es genotóxico y que induce efectos en el desarrollo sólo a dosis o concentraciones que son claramente tóxicas para las madres.
Los datos disponibles sobre poblaciones humanas se consideran insuficientes como base para una caracterización de la relación dosis-respuesta en el caso de la ingestión crónica de cianuro. En un estudio de toxicidad con dosis repetidas de 13 semanas en el que se administró cianuro en el agua de bebida, no se detectaron signos clínicos asociados con efectos en el sistema nervioso central ni efectos histopatológicos en el cerebro o el tiroides de ratas o ratones expuestos a dosis de hasta 12,5 mg y 26 mg de cianuro/kg de peso corporal al día, respectivamente. Con 12,5 mg de cianuro/kg de peso corporal al día, se observaron ligeros cambios en el tracto reproductivo de las ratas macho, que, aunque aparentemente no afectarían a la fecundidad de las ratas, probablemente son importantes para las personas. La concentración sin efectos adversos observados (NOAEL) para estos efectos fue de 4,5 mg/kg de peso corporal al día. El examen de la neurotoxicidad en este estudio se limitó a la observación clínica y la microscopía óptica en la autopsia. Los escasos estudios disponibles orientados específicamente a investigar la neurotoxicidad, si bien informaban de efectos adversos con niveles de exposición de 1,2 mg de cianuro/kg de peso corporal al día en ratas y de 0,48 mg de cianuro/kg de peso corporal al día en cabras, tienen deficiencias que impiden realizar su evaluación cuantitativa.
En tres estudios separados con ratas relativos a la caracterización de la relación concentración-respuesta para la toxicidad de dosis repetidas por inhalación (importante en particular para el entorno ocupacional), no se observaron efectos sistémicos adversos en las ratas expuestas a cianohidrina de acetona, que se hidroliza con rapidez a pH fisiológico para formar cianuro de hidrógeno, en concentraciones de hasta 211 mg/m3 (equivalentes a una concentración de 67 mg de cianuro de hidrógeno/m3). La pendiente de la curva de la relación dosis-respuesta se explica por la observación de una mortalidad del 30% entre las ratas expuestas durante una parte del día a 225 mg de cianohidrina de acetona/m3 (71 mg de cianuro de hidrógeno/m3).
Es poco probable la aparición de efectos adversos por la exposición a las bajas concentraciones de cianuro que normalmente se dan en el medio ambiente (<1 µg/m3 en el aire exterior; <10 µg/l en el agua). Se pueden producir intoxicaciones agudas por cianuro comiendo almendras de albaricoque, cerezas de Virginia y almendras de otros frutos con concentraciones elevadas de glucósidos cianogénicos. La yuca preparada de manera inadecuada puede ser peligrosa cuando constituye la parte más importante de la alimentación.
ENDNOTES:
See Also:
Toxicological Abbreviations