
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
Environmental Health Criteria 208
CARBON TETRACHLORIDE
This report contains the collective views of an international group
of experts and does not necessarily represent the decisions or the
stated policy of the United Nations Environment Programme, the
International Labour Organisation, or the World Health
Organization.
First draft prepared by Ms J. de Fouw, National Institute of Public
Health and the Environment, Bilthoven, the Netherlands
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1999
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO), and the World Health Organization (WHO). The overall
objectives of the IPCS are to establish the scientific basis for
assessment of the risk to human health and the environment from
exposure to chemicals, through international peer review processes, as
a prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization, the United Nations
Institute for Training and Research, and the Organisation for Economic
Co-operation and Development (Participating Organizations), following
recommendations made by the 1992 UN Conference on Environment and
Development to strengthen cooperation and increase coordination in the
field of chemical safety. The purpose of the IOMC is to promote
coordination of the policies and activities pursued by the
Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing in Publication Data
Carbon tetrachloride.
(Environmental health criteria ; 208)
1.Carbon tetrachloride - toxicity 2.Environmental exposure
I.International Programme on Chemical Safety II.Series
ISBN 92 4 157208 6 (NLM Classification: QD 305.H5)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1999
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR CARBON TETRACHLORIDE
PREAMBLE
ABBREVIATIONS
1. SUMMARY
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling and analysis in air
2.4.2. Sampling and analysis in water
2.4.3. Sampling and analysis in biological samples
2.4.3.1 Blood and tissues
2.4.3.2 Urine
2.4.3.3 Fish
2.4.4. Sampling and analysis in foodstuffs
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production
3.2.1.1 Direct production and procedures
3.2.1.2 Indirect production
3.2.1.3 Emissions
3.2.2. Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Transport
4.1.2. Distribution
4.1.3. Removal from the atmosphere; global
warming potential
4.1.4. Removal from water
4.1.5. Removal from soil
4.2. Abiotic degradation
4.2.1. Degradation in atmosphere
4.2.1.1 Photodegradation
4.2.1.2 Photolysis
4.2.1.3 Ozone-depletion potential
4.2.2. Degradation in water
4.2.3. Other degradation processes
4.3. Biotic degradation
4.3.1. Aerobic
4.3.2. Anaerobic
4.4. Bioaccumulation
5. CONCENTRATIONS IN THE ENVIRONMENT AND EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.3. Soil and sediment
5.1.4. Biota
5.2. General population exposure
5.2.1. Outdoor air
5.2.2. Indoor air
5.2.3. Drinking-water
5.2.4. Foodstuffs
5.2.5. Intake averages
5.3. Occupational exposure
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1. Pharmacokinetics
6.1.1. Absorption
6.1.1.1 Oral
6.1.1.2 Dermal
6.1.1.3 Inhalation
6.1.2. Distribution
6.1.3. Elimination and fate
6.1.4. Physiologically based pharmacokinetic modelling
6.2. Biotransformation and covalent binding of metabolites
6.3. Human studies
6.3.1. Uptake
6.3.1.1 Dermal
6.3.1.2 Inhalation
6.3.2. Elimination
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Lethality
7.1.2. Non-lethal effects
7.1.2.1 Oral exposure
7.1.2.2 Inhalation exposure
7.1.2.3 Subcutaneous and intraperitoneal
exposure
7.1.2.4 Dermal exposure
7.2. Short-term exposure
7.2.1. Oral exposure
7.2.2. Inhalation exposure
7.2.3. Intraperitoneal exposure
7.3. Long-term exposure
7.4. Irritation
7.4.1. Skin irritation
7.4.2. Eye irritation
7.5. Toxicity to the reproductive system, embryotoxicity,
teratogenicity
7.5.1. Reproduction
7.5.2. Embryotoxicity and teratogenicity
7.5.2.1 Oral exposure
7.5.2.2 Inhalation exposure
7.6. Mutagenicity
7.7. Carcinogenicity
7.7.1. Mice
7.7.2. Rats
7.8. Special studies
7.8.1. Immunotoxicity
7.8.2. Influence of oxygen levels
7.9. Factors modifying toxicity
7.9.1. Dosing vehicles
7.9.2. Diet
7.9.3. Alcohol
7.9.4. Enhancement of carbon tetrachloride-induced
hepatotoxicity by various compounds
7.9.5. Reduction of carbon tetrachloride-induced
hepatotoxicity by various compounds
7.10. Mode of action
8. EFFECTS ON HUMANS
8.1. Controlled studies
8.1.1. Inhalation
8.1.2. Dermal
8.2. Case reports
8.3. Epidemiology
8.3.1. Non-cancer epidemiology
8.3.2. Cancer epidemiology
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Toxicity to microorganisms
9.2. Aquatic toxicity
9.2.1. Algae
9.2.2. Invertebrates
9.2.3. Vertebrates
9.3. Terrestrial toxicity
9.3.1. Earthworms
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health risks
10.1.1. Exposure
10.1.2. Health effects
10.1.3. Approaches to health risk assessment
10.1.3.1 Calculation of a TDI based
on oral data
10.1.3.2 Calculation of a TC based on inhalation
data
10.1.3.3 Summary of the results of risk
assessment
10.1.3.4 Conclusions based on exposure and health
risk assessment
10.2. Evaluation of effects on the environment
11. FURTHER RESEARCH
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
REFERENCES
RÉSUMÉ
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41
22 - 9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number 5 U01
ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial support
from the European Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
Cevaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and
in vitro studies provide support and are used mainly to supply
evidence missing from human studies. It is mandatory that research on
human subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary -- a review of the salient facts and the risk evaluation
of the chemical
* Identity -- physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
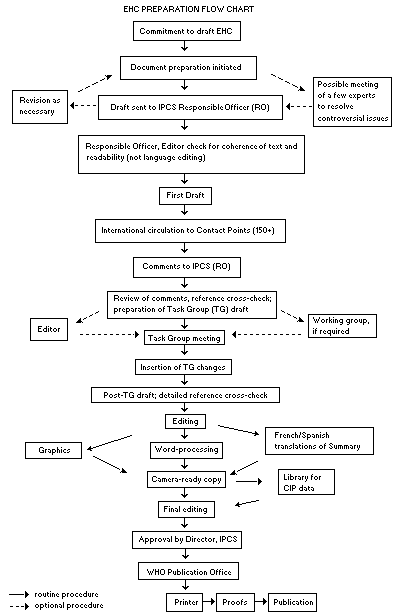
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
 WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR CARBON
TETRACHLORIDE
Members
Dr D. Anderson, British Industry Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United Kingdom
(Chairperson)
Dr E. Elovaara, Finnish Institute for Occupational Health, Helsinki,
Finland
Dr E. Frantik, National Institute of Public Health, Center of
Industrial Hygiene and Occupational Diseases, Prague, Czech Republic
Dr B. Gilbert, Ministry of Health, Far-Manguinhas-FIOCRUZ,
Rio de Janeiro, Brazil (Co-Rapporteur)
Mr M. Greenberg, National Center for Environmental Assessment, Office
of Research and Development, US Environmental Protection Agency,
Research Triangle Park, North Carolina, USA
Mr P. Howe, Institute of Terrestrial Ecology, Monks Wood, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom
Professor H. Kappus, Virchow Klinikum der Humboldt Universitat,
Berlin, Germany
Dr D. McGregor, Unit of Carcinogen Identification and Evaluation,
International Agency for Research on Cancer, Lyon, France
(Co-Rapporteur)
Dr P. Parsons, Health and Safety Executive, Bootle, Merseyside, United
Kingdom
Professor J.A. Sokal, Institute of Occupational Medicine and
Environmental Health, Sosnowiec, Poland
Secretariat
Dr J. de Fouw, Centre for Substances and Risk Assessment, National
Institute of Public Health and the Environment, Bilthoven, The
Netherlands
Professor F. Valic, IPCS Scientific Adviser, Andrija Stampar School
of Public Health, Zagreb University, Zagreb, Croatia (Responsible
Officer and Secretary of Meeting)
ENVIRONMENTAL HEALTH CRITERIA FOR CARBON TETRACHLORIDE
A Task Group on Environmental Health Criteria for Carbon
Tetrachloride met at the British Industrial and Biological Research
Association (BIBRA), Carshalton, United Kingdom, from 2 to 6 March
1998. Dr D. Anderson, welcomed the participants on behalf of the host
institution, and Professor F. Valic opened the Meeting on behalf of
the heads of the three cooperating organizations of the IPCS
(UNEP/ILO/WHO). The Task Group reviewed and revised the draft
monograph and made an evaluation of the risks for human health from
exposure to carbon tetrachloride.
The first draft of this monograph was prepared by Ms J. de Fouw,
Centre for Substances and Risk Assessment, National Institute of
Public Health and the Environment, Bilthoven, the Netherlands.
Professor Valic, Zagreb University, Croatia, was responsible for
the overall scientific content of the monograph and for the
organization of the Meeting, and Dr P.G. Jenkins, IPCS Central Unit,
for the technical editing of the monograph.
The efforts of all who helped in the preparation and finalization
of the monograph are greatfully acknowledged.
ABBREVIATIONS
ALAT alanine aminotransferase
AP alkaline phosphatase
ASAT aspartate aminotransferase
ATPase adenosine triphosphatase
ATSDR Agency for Toxic Substances and Disease Registry
CNS central nervous system
CPK creatine phosphokinase
CYP cytochrome P-450
Hb haemoglobin
Ht haematocrit
ip intraperitoneal
LDH lactate dehydrogenase
LOAEL lowest-observed-adverse-effect level
MPV mean packed volume
NADPH reduced nicotinamide adenine dinucleotide phosphate
NIOSH National Institute for Occupational Safety and Health (USA)
NOAEL no-observed-adverse-effect level
PBB polybrominated biphenyl
PCB polychlorinated biphenyl
RBC red blood cell
SDH sorbitol dehydrogenase
SRBC sheep red blood cells
TC tolerable concentration
TDI tolerable daily intake
1. SUMMARY
Carbon tetrachloride is a clear, colourless, volatile liquid with
a characteristic, sweet odour. It is miscible with most aliphatic
solvents and is itself a solvent. The solubility in water is low.
Carbon tetrachloride is non-flammable and is stable in the presence of
air and light. Decomposition may produce phosgene, carbon dioxide and
hydrochloric acid.
The source of carbon tetrachloride in the environment is likely
to be almost exclusively anthropogenic in origin. Most of the carbon
tetrachloride produced is used in the production of
chlorofluorocarbons (CFCs) and other chlorinated hydrocarbons. The
global production of carbon tetrachloride amounted to 960 000 tonnes
in 1987. However, since the Montreal Protocol on Substances that
Deplete the Ozone Layer (1987) and its amendments (1990 and 1992) have
established a timetable for the phase-out of the production and
consumption of carbon tetrachloride, manufacture has dropped and will
continue to drop.
Several sufficiently sensitive and accurate analytical methods
for determining carbon tetrachloride in air, water and biological
samples have been developed. The majority of these methods are based
on direct injection into a gas chromatograph or adsorption on
activated charcoal, then desorption or evaporation and subsequent gas
chromatographic detection.
Nearly all carbon tetrachloride released to the environment will
ultimately be present in the atmosphere, owing to its volatility.
Since the atmospheric residence time of carbon tetrachloride is long,
it is widely distributed. During the period 1980-1990, atmospheric
levels were around 0.5-1.0 µg/m3. Estimates of atmospheric lifetime
are variable, but 45-50 years is accepted as the most reasonable
value. Carbon tetrachloride contributes both to ozone depletion and to
global warming. It is in general resistant to aerobic biodegradation
but less so to anaerobic. Acclimation increases biodegradation rates.
Although the octanol-water partition coefficient indicates moderate
potential for bioaccumulation, short tissue lifetime reduces this
tendency.
In water, reports have indicated levels of less than 10 ng/litre
in the ocean and generally less than 1 µg/litre in fresh water, but
much higher values close to release sites. Levels of up to 60 µg/kg
have been recorded in foods processed with carbon tetrachloride, but
this practice has now ceased.
The general population is exposed to carbon tetrachloride mainly
via air. On the basis of the reported concentrations in ambient air,
foodstuffs and drinking-water, a daily carbon tetrachloride intake of
around 1 µg/kg body weight has been estimated. This estimate is
probably rather high for the present day, because the use of carbon
tetrachloride as a fumigant of grain has stopped and the carbon
tetrachloride values reported for food and used in the calculation
were especially those found in fatty and grain-based foods. Values of
0.1 to 0.27 µg/kg body weight for daily exposure of the general
population have been reported elsewhere. Exposure to higher levels of
carbon tetrachloride can occur in the workplace as a result of
accidental spillage.
Carbon tetrachloride is well absorbed from the gastrointestinal
and respiratory tract in animals and humans. Dermal absorption of
liquid carbon tetrachloride is possible, but dermal absorption of the
vapour is slow.
Carbon tetrachloride is distributed throughout the whole body,
with highest concentrations in liver, brain, kidney, muscle, fat and
blood. The parent compound is eliminated primarily in exhaled air,
while minimal amounts are excreted in the faeces and urine.
The first step in the biotransformation of carbon tetrachloride
is catalysed by cytochrome P-450 enzymes, leading to the formation of
the reactive trichloromethyl radical. Oxidative biotransformation is
the most important pathway in the elimination of the radical, forming
the even more reactive trichloromethylperoxyl radical, which can react
further to form phosgene. Phosgene may be detoxified by reaction with
water to produce carbon dioxide or with glutathione or cysteine.
Formation of chloroform and dichlorocarbene occurs under anaerobic
conditions.
Covalent binding to macromolecules and lipid peroxidation occur
via metabolic intermediates of carbon tetrachloride.
The liver and kidney are target organs for carbon tetrachloride
toxicity. The severity of the effects on the liver depends on a number
of factors such as species susceptibility, route and mode of exposure,
diet or co-exposure to other compounds, in particular ethanol.
Furthermore, it appears that pretreatment with various compounds, such
as phenobarbital and vitamin A, enhances hepatotoxicity, while other
compounds, such as vitamin E, reduce the hepatotoxic action of carbon
tetrachloride.
Moderate irritation after dermal application was seen on the
skins of rabbits and guinea-pigs, and there was a mild reaction after
application into the rabbit eye.
The lowest LD50 of 2391 mg/kg body weight (14-day period) was
reported in a study on dogs involving intraperitoneal administration.
In rats the LD50 values ranged from 2821 to 10 054 mg/kg body weight.
In a 12-week oral study on rats (5 days/week), the
no-observed-adverse-effect level (NOAEL) was 1 mg/kg body weight. The
lowest-observed-adverse-effect level (LOAEL) reported in this study
was 10 mg/kg body weight, showing a slight, but significant increase
in sorbitol dehydrogenase (SDH) activity and mild hepatic
centrilobular vacuolization. A similar NOAEL of 1.2 mg/kg body weight
(5 days/ week) was found in a 90-day oral study on mice, with a LOAEL
of 12 mg/kg body weight, where hepatotoxicity occurred.
When rats were exposed to carbon tetrachloride by inhalation for
approximately 6 months, 5 days/week, 7 h/day, a NOAEL of 32 mg/m3 was
reported. The LOAEL, based on changes in the liver morphology, was
reported to be 63 mg/m3. In another 90-day study on rats, a NOAEL of
6.1 mg/m3 was found after continuous exposure to carbon
tetrachloride. The lowest exposure level of 32 mg/m3 (the lowest
concentration studied) in a 2-year inhalation study on rats caused
marginal effects.
The only oral long-term toxicity study available was a 2-year
study in rats, which were exposed to 0, 80 or 200 mg carbon
tetrachloride/kg feed. Owing to chronic respiratory disease in all
animals beginning at 14 months, which resulted in increased mortality,
the results reported upon necropsy at 2 years are inadequate for a
health risk evaluation.
It was concluded that carbon tetrachloride can induce embryotoxic
and embryolethal effects, but only at doses that are maternally toxic,
as observed in inhalation studies in rats and mice. Carbon
tetrachloride is not teratogenic.
Many genotoxicity assays have been conducted with carbon
tetrachloride. On the basis of available data, carbon tetrachloride
can be considered as a non-genotoxic compound.
Carbon tetrachloride induces hepatomas and hepatocellular
carcinomas in mice and rats. The doses inducing hepatic tumours are
higher than those inducing cell toxicity.
In humans, acute symptoms after carbon tetrachloride exposure are
independent of the route of intake and are characterized by
gastrointestinal and neurological symptoms, such as nausea, vomiting,
headache, dizziness, dyspnoea and death. Liver damage appears after 24
h or more. Kidney damage is evident often only 2 to 3 weeks following
the poisoning.
Epidemiological studies have not established an association
between carbon tetrachloride exposure and increased risk of mortality,
neoplasia or liver disease. Some studies have suggested an association
with increased risk of non-Hodgkin's lymphoma and, in one study, with
mortality and liver cirrhosis. However, not all of these studies
pinpointed specific exposure to carbon tetrachloride, and the
statistical associations were not strong.
In general carbon tetrachloride appears to be of low toxicity to
bacteria, protozoa and algae; the lowest toxic concentration reported
was for methanogenic bacteria with an IC50 of 6.4 mg/litre. For
aquatic invertebrates acute LC50 values range from 28 to > 770
mg/litre. In freshwater fish the lowest acute LC50 value of 13
mg/litre was found in the golden orfe (Leuciscus idus melanotus),
and for marine species an LC50 value of 50 mg/litre was reported for
the dab (Limanda limanda). Carbon tetrachloride appears to be more
toxic to embryo-larval stages of fish and amphibians than to adults.
The common bullfrog (Rana catesbeiara) is the most susceptible
species, the LC50 being 0.92 mg/litre (fertilization to 4 days after
hatching).
The available data indicate that hepatic tumours are induced by a
non-genotoxic mechanism, and it therefore seems acceptable to develop
a tolerable daily intake (TDI) and a tolerable daily concentration in
air (TC) for carbon tetrachloride.
On the basis of the study of Bruckner et al. (1986), in which a
NOAEL of 1 mg/kg body weight was observed in a 12-week oral study on
rats, and incorporating a conversion factor of 5/7 for daily dosing
and applying an uncertainty factor of 500 (100 for inter- and
intraspecies variation, 10 for duration of the study, and modifying
factor 0.5 because it was a bolus study), a TDI of 1.42 µg/kg body
weight is obtained.
On the basis of a 90-day inhalation study on rats (Prendergast et
al., 1967), in which a NOAEL of 6.1 mg/m3 was reported, a TC of 6.1
µg/m3 was calculated using the factors 7/24 and 5/7 to convert to
continuous exposure and an uncertainty factor of 1000 (100 for
inter- and intraspecies variation and 10 for the duration of the
study). This TC corresponds to a TDI of 0.85 µg/kg body weight.
Comparing the estimated upper limit of prevailing human daily
intake of 0.2 µg/kg body weight with the lowest TDI value (0.85 µg/kg
body weight), the conclusion can be drawn that the currently
prevailing exposure of the general population to carbon tetrachloride
from all sources is unlikely to cause excessive intake of the
chemical.
In general, the risk to aquatic organisms from carbon
tetrachloride is low. However, it may present a risk to embryo-larval
stages at, or near, sites of industrial discharges or spills.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND
ANALYTICAL METHODS
2.1 Identity
Chemical formula: CCl4
Chemical structure:
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR CARBON
TETRACHLORIDE
Members
Dr D. Anderson, British Industry Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United Kingdom
(Chairperson)
Dr E. Elovaara, Finnish Institute for Occupational Health, Helsinki,
Finland
Dr E. Frantik, National Institute of Public Health, Center of
Industrial Hygiene and Occupational Diseases, Prague, Czech Republic
Dr B. Gilbert, Ministry of Health, Far-Manguinhas-FIOCRUZ,
Rio de Janeiro, Brazil (Co-Rapporteur)
Mr M. Greenberg, National Center for Environmental Assessment, Office
of Research and Development, US Environmental Protection Agency,
Research Triangle Park, North Carolina, USA
Mr P. Howe, Institute of Terrestrial Ecology, Monks Wood, Abbots
Ripton, Huntingdon, Cambridgeshire, United Kingdom
Professor H. Kappus, Virchow Klinikum der Humboldt Universitat,
Berlin, Germany
Dr D. McGregor, Unit of Carcinogen Identification and Evaluation,
International Agency for Research on Cancer, Lyon, France
(Co-Rapporteur)
Dr P. Parsons, Health and Safety Executive, Bootle, Merseyside, United
Kingdom
Professor J.A. Sokal, Institute of Occupational Medicine and
Environmental Health, Sosnowiec, Poland
Secretariat
Dr J. de Fouw, Centre for Substances and Risk Assessment, National
Institute of Public Health and the Environment, Bilthoven, The
Netherlands
Professor F. Valic, IPCS Scientific Adviser, Andrija Stampar School
of Public Health, Zagreb University, Zagreb, Croatia (Responsible
Officer and Secretary of Meeting)
ENVIRONMENTAL HEALTH CRITERIA FOR CARBON TETRACHLORIDE
A Task Group on Environmental Health Criteria for Carbon
Tetrachloride met at the British Industrial and Biological Research
Association (BIBRA), Carshalton, United Kingdom, from 2 to 6 March
1998. Dr D. Anderson, welcomed the participants on behalf of the host
institution, and Professor F. Valic opened the Meeting on behalf of
the heads of the three cooperating organizations of the IPCS
(UNEP/ILO/WHO). The Task Group reviewed and revised the draft
monograph and made an evaluation of the risks for human health from
exposure to carbon tetrachloride.
The first draft of this monograph was prepared by Ms J. de Fouw,
Centre for Substances and Risk Assessment, National Institute of
Public Health and the Environment, Bilthoven, the Netherlands.
Professor Valic, Zagreb University, Croatia, was responsible for
the overall scientific content of the monograph and for the
organization of the Meeting, and Dr P.G. Jenkins, IPCS Central Unit,
for the technical editing of the monograph.
The efforts of all who helped in the preparation and finalization
of the monograph are greatfully acknowledged.
ABBREVIATIONS
ALAT alanine aminotransferase
AP alkaline phosphatase
ASAT aspartate aminotransferase
ATPase adenosine triphosphatase
ATSDR Agency for Toxic Substances and Disease Registry
CNS central nervous system
CPK creatine phosphokinase
CYP cytochrome P-450
Hb haemoglobin
Ht haematocrit
ip intraperitoneal
LDH lactate dehydrogenase
LOAEL lowest-observed-adverse-effect level
MPV mean packed volume
NADPH reduced nicotinamide adenine dinucleotide phosphate
NIOSH National Institute for Occupational Safety and Health (USA)
NOAEL no-observed-adverse-effect level
PBB polybrominated biphenyl
PCB polychlorinated biphenyl
RBC red blood cell
SDH sorbitol dehydrogenase
SRBC sheep red blood cells
TC tolerable concentration
TDI tolerable daily intake
1. SUMMARY
Carbon tetrachloride is a clear, colourless, volatile liquid with
a characteristic, sweet odour. It is miscible with most aliphatic
solvents and is itself a solvent. The solubility in water is low.
Carbon tetrachloride is non-flammable and is stable in the presence of
air and light. Decomposition may produce phosgene, carbon dioxide and
hydrochloric acid.
The source of carbon tetrachloride in the environment is likely
to be almost exclusively anthropogenic in origin. Most of the carbon
tetrachloride produced is used in the production of
chlorofluorocarbons (CFCs) and other chlorinated hydrocarbons. The
global production of carbon tetrachloride amounted to 960 000 tonnes
in 1987. However, since the Montreal Protocol on Substances that
Deplete the Ozone Layer (1987) and its amendments (1990 and 1992) have
established a timetable for the phase-out of the production and
consumption of carbon tetrachloride, manufacture has dropped and will
continue to drop.
Several sufficiently sensitive and accurate analytical methods
for determining carbon tetrachloride in air, water and biological
samples have been developed. The majority of these methods are based
on direct injection into a gas chromatograph or adsorption on
activated charcoal, then desorption or evaporation and subsequent gas
chromatographic detection.
Nearly all carbon tetrachloride released to the environment will
ultimately be present in the atmosphere, owing to its volatility.
Since the atmospheric residence time of carbon tetrachloride is long,
it is widely distributed. During the period 1980-1990, atmospheric
levels were around 0.5-1.0 µg/m3. Estimates of atmospheric lifetime
are variable, but 45-50 years is accepted as the most reasonable
value. Carbon tetrachloride contributes both to ozone depletion and to
global warming. It is in general resistant to aerobic biodegradation
but less so to anaerobic. Acclimation increases biodegradation rates.
Although the octanol-water partition coefficient indicates moderate
potential for bioaccumulation, short tissue lifetime reduces this
tendency.
In water, reports have indicated levels of less than 10 ng/litre
in the ocean and generally less than 1 µg/litre in fresh water, but
much higher values close to release sites. Levels of up to 60 µg/kg
have been recorded in foods processed with carbon tetrachloride, but
this practice has now ceased.
The general population is exposed to carbon tetrachloride mainly
via air. On the basis of the reported concentrations in ambient air,
foodstuffs and drinking-water, a daily carbon tetrachloride intake of
around 1 µg/kg body weight has been estimated. This estimate is
probably rather high for the present day, because the use of carbon
tetrachloride as a fumigant of grain has stopped and the carbon
tetrachloride values reported for food and used in the calculation
were especially those found in fatty and grain-based foods. Values of
0.1 to 0.27 µg/kg body weight for daily exposure of the general
population have been reported elsewhere. Exposure to higher levels of
carbon tetrachloride can occur in the workplace as a result of
accidental spillage.
Carbon tetrachloride is well absorbed from the gastrointestinal
and respiratory tract in animals and humans. Dermal absorption of
liquid carbon tetrachloride is possible, but dermal absorption of the
vapour is slow.
Carbon tetrachloride is distributed throughout the whole body,
with highest concentrations in liver, brain, kidney, muscle, fat and
blood. The parent compound is eliminated primarily in exhaled air,
while minimal amounts are excreted in the faeces and urine.
The first step in the biotransformation of carbon tetrachloride
is catalysed by cytochrome P-450 enzymes, leading to the formation of
the reactive trichloromethyl radical. Oxidative biotransformation is
the most important pathway in the elimination of the radical, forming
the even more reactive trichloromethylperoxyl radical, which can react
further to form phosgene. Phosgene may be detoxified by reaction with
water to produce carbon dioxide or with glutathione or cysteine.
Formation of chloroform and dichlorocarbene occurs under anaerobic
conditions.
Covalent binding to macromolecules and lipid peroxidation occur
via metabolic intermediates of carbon tetrachloride.
The liver and kidney are target organs for carbon tetrachloride
toxicity. The severity of the effects on the liver depends on a number
of factors such as species susceptibility, route and mode of exposure,
diet or co-exposure to other compounds, in particular ethanol.
Furthermore, it appears that pretreatment with various compounds, such
as phenobarbital and vitamin A, enhances hepatotoxicity, while other
compounds, such as vitamin E, reduce the hepatotoxic action of carbon
tetrachloride.
Moderate irritation after dermal application was seen on the
skins of rabbits and guinea-pigs, and there was a mild reaction after
application into the rabbit eye.
The lowest LD50 of 2391 mg/kg body weight (14-day period) was
reported in a study on dogs involving intraperitoneal administration.
In rats the LD50 values ranged from 2821 to 10 054 mg/kg body weight.
In a 12-week oral study on rats (5 days/week), the
no-observed-adverse-effect level (NOAEL) was 1 mg/kg body weight. The
lowest-observed-adverse-effect level (LOAEL) reported in this study
was 10 mg/kg body weight, showing a slight, but significant increase
in sorbitol dehydrogenase (SDH) activity and mild hepatic
centrilobular vacuolization. A similar NOAEL of 1.2 mg/kg body weight
(5 days/ week) was found in a 90-day oral study on mice, with a LOAEL
of 12 mg/kg body weight, where hepatotoxicity occurred.
When rats were exposed to carbon tetrachloride by inhalation for
approximately 6 months, 5 days/week, 7 h/day, a NOAEL of 32 mg/m3 was
reported. The LOAEL, based on changes in the liver morphology, was
reported to be 63 mg/m3. In another 90-day study on rats, a NOAEL of
6.1 mg/m3 was found after continuous exposure to carbon
tetrachloride. The lowest exposure level of 32 mg/m3 (the lowest
concentration studied) in a 2-year inhalation study on rats caused
marginal effects.
The only oral long-term toxicity study available was a 2-year
study in rats, which were exposed to 0, 80 or 200 mg carbon
tetrachloride/kg feed. Owing to chronic respiratory disease in all
animals beginning at 14 months, which resulted in increased mortality,
the results reported upon necropsy at 2 years are inadequate for a
health risk evaluation.
It was concluded that carbon tetrachloride can induce embryotoxic
and embryolethal effects, but only at doses that are maternally toxic,
as observed in inhalation studies in rats and mice. Carbon
tetrachloride is not teratogenic.
Many genotoxicity assays have been conducted with carbon
tetrachloride. On the basis of available data, carbon tetrachloride
can be considered as a non-genotoxic compound.
Carbon tetrachloride induces hepatomas and hepatocellular
carcinomas in mice and rats. The doses inducing hepatic tumours are
higher than those inducing cell toxicity.
In humans, acute symptoms after carbon tetrachloride exposure are
independent of the route of intake and are characterized by
gastrointestinal and neurological symptoms, such as nausea, vomiting,
headache, dizziness, dyspnoea and death. Liver damage appears after 24
h or more. Kidney damage is evident often only 2 to 3 weeks following
the poisoning.
Epidemiological studies have not established an association
between carbon tetrachloride exposure and increased risk of mortality,
neoplasia or liver disease. Some studies have suggested an association
with increased risk of non-Hodgkin's lymphoma and, in one study, with
mortality and liver cirrhosis. However, not all of these studies
pinpointed specific exposure to carbon tetrachloride, and the
statistical associations were not strong.
In general carbon tetrachloride appears to be of low toxicity to
bacteria, protozoa and algae; the lowest toxic concentration reported
was for methanogenic bacteria with an IC50 of 6.4 mg/litre. For
aquatic invertebrates acute LC50 values range from 28 to > 770
mg/litre. In freshwater fish the lowest acute LC50 value of 13
mg/litre was found in the golden orfe (Leuciscus idus melanotus),
and for marine species an LC50 value of 50 mg/litre was reported for
the dab (Limanda limanda). Carbon tetrachloride appears to be more
toxic to embryo-larval stages of fish and amphibians than to adults.
The common bullfrog (Rana catesbeiara) is the most susceptible
species, the LC50 being 0.92 mg/litre (fertilization to 4 days after
hatching).
The available data indicate that hepatic tumours are induced by a
non-genotoxic mechanism, and it therefore seems acceptable to develop
a tolerable daily intake (TDI) and a tolerable daily concentration in
air (TC) for carbon tetrachloride.
On the basis of the study of Bruckner et al. (1986), in which a
NOAEL of 1 mg/kg body weight was observed in a 12-week oral study on
rats, and incorporating a conversion factor of 5/7 for daily dosing
and applying an uncertainty factor of 500 (100 for inter- and
intraspecies variation, 10 for duration of the study, and modifying
factor 0.5 because it was a bolus study), a TDI of 1.42 µg/kg body
weight is obtained.
On the basis of a 90-day inhalation study on rats (Prendergast et
al., 1967), in which a NOAEL of 6.1 mg/m3 was reported, a TC of 6.1
µg/m3 was calculated using the factors 7/24 and 5/7 to convert to
continuous exposure and an uncertainty factor of 1000 (100 for
inter- and intraspecies variation and 10 for the duration of the
study). This TC corresponds to a TDI of 0.85 µg/kg body weight.
Comparing the estimated upper limit of prevailing human daily
intake of 0.2 µg/kg body weight with the lowest TDI value (0.85 µg/kg
body weight), the conclusion can be drawn that the currently
prevailing exposure of the general population to carbon tetrachloride
from all sources is unlikely to cause excessive intake of the
chemical.
In general, the risk to aquatic organisms from carbon
tetrachloride is low. However, it may present a risk to embryo-larval
stages at, or near, sites of industrial discharges or spills.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND
ANALYTICAL METHODS
2.1 Identity
Chemical formula: CCl4
Chemical structure:
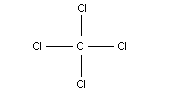 Common name: carbon tetrachloride
Common synonyms: Carbona, carbon chloride, tetrachloromethane,
carbon tet, methane tetrachloride,
perchloromethane, tetrachlorocarbon
Trade names: Benzinoform, Fasciolin, Flukoids, Freon 10,
Halon 104, Necatorina, Necatorine,
Tetrafinol, Tetraform, Tetrasol, Univerm,
Vermoestricid
CAS chemical name: tetrachloromethane
CAS registry number: 56-23-5
RTECS registry number: FG 4900000
2.2 Physical and chemical properties
The most important physical properties of carbon tetrachloride
are given in Table 1.
Table 1. Physical properties of carbon tetrachloridea
Colour colourless
Relative molecular mass 153.8
Boiling point at 101.3 kPa, 20°C 76.72 °C
Melting point at 101.3 kPa, 20°C -22.92 °C
Density (25°C) 1.594 g/ml
Table 1. (Continued)
Density of solid at - 186 °C 1831 kg/m3
- 80 °C 1809 kg/m3
Refractive index at 20 °C 1.4607
Vapour pressure at 20 °C 91.3 mmHg; 12.2 kPa
at 0 °C 32.9 mmHg; 4.4 kPa
Autoignition temperature > 1000 °C
Critical pressure 4.6 MPa
Critical temperature 283.2 °C
Solubility in water at 25 °C 785 mg/litre
Solubility of water in carbon
tetrachloride at 25 °C 0.13 g/kg
Henry's law constant at 24.8 °C 2.3 × 10-2 atm-m3/mol
Heat of evaporation 194.7 kJ/kg
Log Kow 2.64
Log Koc 2.04
a From Kenaga (1980); US EPA (1984b); Huiskamp et al. (1986);
ATSDR (1994).
Carbon tetrachloride is a volatile colourless clear heavy liquid
with a characteristic sweet non-irritant odour. The odour threshold in
water is 0.52 mg/litre and in air is > 10 ppm. Carbon tetrachloride
is miscible with most aliphatic solvents and it is a solvent for
benzyl resins, bitumen, chlorinated rubber, rubber-based gums, oils
and fats. The chemical is non-flammable and fairly stable in the
presence of air and light. Upon heating by a flame or hot metal
surface in air, toxic phosgene is produced. Thermal dissociation in
the absence of air proceeds slowly at about 400°C and is extensive at
temperatures ranging from 900 to 1300°C with the formation of
perchloroethylene, hexachloroethane and some molecular chlorine. A
mixture of carbon tetrachloride and excess of water vapour decomposes
at 250°C to carbon dioxide and hydrochloric acid. When the amount of
water in the mixture is limited, phosgene will be formed too. This
decomposition also occurs when moist or wet carbon tetrachloride is
exposed to UV radiation (253.7 nm). Like other chloromethanes, carbon
tetrachloride reacts (sometimes explosively) with aluminium and its
alloys. Similar violent reaction may occur with metals, such as
barium, magnesium and zinc, boranes and silanes, and, in the presence
of peroxides or light, with unsaturated compounds (such as ethene).
Carbon tetrachloride may be reduced to chloroform when treated with
zinc and acid, and to methane when treated with potassium amalgam and
water (Huiskamp et al., 1986).
2.3 Conversion factors
1 mg carbon tetrachloride/m3 air = 0.156 ppm at 20°C and 101.3
kPa (760 mmHg)
1 ppm = 6.41 mg carbon tetrachloride/m3
2.4 Analytical methods
Procedures used for the sampling and determination of carbon
tetrachloride in different media are summarized in Table 2.
The preferred analytical technique is gas chromatography (GC)
using either electron capture detection (ECD), ion trap detection,
flame or photo ionisation detection or mass spectrometry. Only one
method, reported by Lioy & Lioy (1983), depends on the use of
MIRAN-infrared spectrometry, a method of very poor sensitivity.
2.4.1 Sampling and analysis in air
Methods reported in Table 2 for detecting carbon tetrachloride in
air are of four types.
a) Direct measurement
These methods are simple, because the air is aspirated or
injected directly into the measuring instrument, but they can only be
used when carbon tetrachloride is present in the air at relatively
high levels.
b) Adsorption - liquid desorption
In this type of method, air samples are passed through an
activated adsorbing agent. The adsorbed carbon tetrachloride is
desorbed with an appropriate solvent and then passed through the gas
chromatograph. Activated carbon has been described as superior to
other adsorbents for adsorption. Elution from the carbon is achieved
with carbon disulfide (Morele et al., 1989; ATSDR, 1994).
c) Adsorption - thermal desorption
After adsorption on an activated adsorbing agent, the carbon
tetrachloride is thermally desorbed and driven into the gas
chromatograph.
Table 2. Sampling and analysis of carbon tetrachloridea
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Air aspiration velocity: 28 l/min MIRAN infrared 400 µg/m3 Lioy & Lioy,
optical path: 20 m spectrometry 1983
Air direct injection GC with 2 ECD's 0.4 µg/m3 8 ml injected Lillian &
in series (estimated) Singh, 1974
Air direct injection GC - ECD 0.2 µg/m3 2 ml injected BIT-SC, 1976
Air direct injection GC - ECD 0.06 µg/m3 5 ml injected Lasa et al.,
1979
Air direct injection, methane GC - ECD 0.01 µg/m3 12 ml injected thorough purification of Makide &
added carrier gas and apparatus Yokohata, 1983
required
Air adsorption on Porapak-N GC - ECD 1 µg/m3 20 litres advantage of using Van Tassel et
liquid desorption (methanol) methanol over CS2 is the al., 1981
absence of a background
signal in the ECD
Air adsorption on activated GC - ECD 0.2 µg up to 30 litres activated charcoal shown Morele et al.,
charcoal, liquid desorption can be sampled to be more efficient 1989
(ethanol) trichloroethylene trapping material than
used as IS XADs, Tenax or
Chromosorbs
adsorption on activated GC - FID ca. 0.15 mg
charcoal liquid desorption (detector
(CS2) methylcyclohexane sensitivity)
used as IS
Table 2. (Continued)
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Air adsorption on activated GC - FID 0.01 mg 5-15 litres NIOSH, 1977,
charcoal, liquid desorption 1984
(CS2)
Air adsorption on Chromosorb GC - ECD 0.003 µg/m3 20 ml Makide et al.,
102 or Silicone OV 101 (at 1979
-35 °C), thermal desorption
Air adsorption on Porapak-N, GC - ECD 0.005 µg/m3 0.3-3 litres confirmation of results by Russell &
thermal desorption at 200 °C use of GC - MS Shadoff, 1977
Air adsorption on Chromosorb GC - ECD 0.01 µg/m3 1 litre Elias, 1977
102, thermal desorption at (collection tube (estimated)
200 °C already connected
to GC)
Air adsorption on Carbopak-B at GC - ECD 0.01 µg/m3 1 litre calibration with Crescentini et
78 °C, thermal desorption permeation tubes al., 1981
Air adsorption on Chromosorb-102 GC - ECD - FID ca. 0.06 µg/m3 1-3 litres Heil et al.,
and activated charcoal, (2 detectors in 1979
thermal desorption at 150 °C parallel)
Air adsorption on Tenax-GC, GC - MS 0.2 µg/m3 20 litres compounds were Krost et al.,
thermal desorption at 270 °C cryofocused 1982
Air adsorption on Carbopak-C, GC - MS 0.1 µg/m3 300 ml Crescentini et
thermal desorption at 100 °C al., 1983
Air adsorption on activated GC - ECD followed 0.7 µg/m3 24 h sample Coutant &
charcoal, liquid desorption by a PID Scott, 1982
(5% CS2 in methanol)
Table 2. (Continued)
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Air cold trap (liquid oxygen), GC - ECD 0.006 µg/m3 30 ml aliquot measurement of air Harsch &
heating in trap samples from the Cronn, 1978
stratosphere
Air injection in cold trap, heating GC - MS (SIM) 0.04 µg/m3 100 ml Cronn &
Harsch, 1979
Air cold trap (-173 °C), heating to GC - PID - ECD - 0.006 µg/m3 0.5-1.7 litres column is kept at -103 °C Rudolph &
257 °C FID (3 detectors (cryofocusing) Jebsen, 1983
in series)
Water dibromomethane used as IS GC - ECD 0.001 µg/litre 500 µl injected suitable for routine Herzfeld et al.,
analysis of river waters 1989
Water direct aqueous injection GC - MS (SIM) 2 µg/litre 10 µl injected Fujii, 1977
Water direct aqueous injection GC - ECD 0.015 µg/litre 2 µl injected suitable for halocarbons Grob, 1984
in water in the 0.01 to
10 µg/litre range
Water direct aqueous injection, GC - ECD 0.05 µg/litre 5-20 µl Simmonds &
water removal by injected Kerns, 1979
permeaselective membrane
Water liquid-liquid extraction GC - ECD 0.10 µg/litre 10-20 ml Van Rensburg
(using hexane) et al., 1978
Water liquid-liquid extraction GC - ECD 0.2 µg/litre Inoko et al.,
(using xylene) 1984
Water liquid-liquid extraction GC - ECD 0.05 µg/litre Kroneld, 1985
(using pentane)
Table 2. (Continued)
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Water purge and trap technique, GC - ITD 0.1 µg/litre 5 ml Eichelberger
thermal desorption, et al., 1990
fluorobenzene as IS
Grain codistillation of carbon GC - ECD 1 µg/kg De Vries et al.,
tetrachloride in food sample 1985
and mixture of
1,2-dichloropropane and
1,2-dibromopropane as IS in
hexane
Adipose purge and trap technique GC - MS < 1.3 µg/litre 200-500 mg Peoples et al.,
tissue (Tenax-silica gel), thermal liquefied fat 1979
desorption samples
Blood purge and trap technique GC - MS < 1.3 µg/litre 0.5 ml water- Peoples et al.,
(Tenax-silica gel), thermal serum sample 1979
desorption
Blood warming and passing an inert GC - MS 3 µg/litre 10 ml sample Pellizzari
gas, vapours trapped on et al., 1985
Tenax-GC, thermal desorption
Urine liquid-liquid extraction using GC - ECD (20% < 1 µg/litre 10 ml sample Youssefi et al.,
pentane (adding 2.6 g SP-2100/0.1% 1978
ammonium carbonate) Carbowax 1500
column)
Fish extraction with pentane and GC - ECD 0.1 µg/kg in Baumann
isopropanol, with fresh material Ofstad et al.,
bromotrichloromethane used as IS 1981
a Abbreviations: GC = gas chromatography; MS = mass spectrometry; ECD = electron capture detector; SIM = single (selected) ion monitoring;
FID = flame ionisation detector; ITD = ion trap detector; PID = photo ionisation detector; IS = internal standard.
d) Cold trap - heating
In this type of procedure, air samples are injected into a cold
trap. The trap is then heated and the carbon tetrachloride content
transferred into the column of a gas chromatograph.
2.4.2 Sampling and analysis in water
Several methods for sampling analysing the carbon tetrachloride
content in water are included in Table 2. Most of these methods are
based on direct injection techniques or on liquid-liquid extraction by
means of a non-polar non-halogenated solvent.
2.4.3 Sampling and analysis in biological samples
2.4.3.1 Blood and tissues
Peoples et al. (1979) developed a method to determine carbon
tetrachloride in adipose tissue and blood. In both cases the carbon
tetrachloride is purged and trapped on Tenax-silica gel and determined
by mass spectrometry after thermal desorption.
Pellizzari et al. (1985) similarly passed an inert gas over a
warmed plasma sample with adsorption of the vapour on a Tenax-GC
cartridge, and then recovered the carbon tetrachloride by thermal
desorption.
2.4.3.2 Urine
The only method listed in Table 2 for measuring carbon tetra
chloride concentrations in urine is based on an extraction technique
with pentane and direct gas chromatographic analysis of the pentane
extract (Youssefi et al., 1978).
2.4.3.3 Fish
Baumann Ofstad et al. (1981) developed a method for the analysis
of volatile halogenated hydrocarbons in biological samples and used
this method for the analysis of fish samples. It should be noted that
the identification and quantification of carbon tetrachloride is
especially vulnerable to contamination, so the practical usefulness of
this method is very limited.
2.4.4 Sampling and analysis in foodstuffs
A method for the determination of 22 compounds (including carbon
tetrachloride) in a variety of foods was described by Daft (1988). In
this method the samples are extracted with isooctane, and cleaned up
according to fat content and food type. Most samples (6-10 µl) are
injected for GC with ECD and Hall-electron conductivity detection
immediately following the initial extraction or dilution.
De Vries et al. (1985) provided a method for analysis of carbon
tetrachloride in grain and grain-based products containing 1-2000
µg/kg. A food sample is mixed with water and an internal standard
mixture of 1,2-dichloropropane and 1,2-dibromopropane is added. The
water is then distilled until 1 ml has been collected under hexane.
The hexane is then separated, dried and injected (2 µl) into the GC
column.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
It has been suggested that carbon tetrachloride can be formed in
the troposphere by the solar-induced photochemical reactions of
chlorinated alkenes (Singh et al., 1975). However, so far this
reaction has only been demonstrated in the laboratory, and, even if it
could happen in nature, it is not certain that it would be a major
source of environmental carbon tetrachloride. Carbon tetrachloride has
been detected in volcanic emission gases (Isidorov et al., 1990).
Several studies have shown that global atmospheric levels of carbon
tetrachloride can be explained by anthropogenic sources alone (Singh
et al., 1976).
3.2 Anthropogenic sources
3.2.1 Production
3.2.1.1 Direct production and procedures
Production of carbon tetrachloride began in about 1907 in the
USA. It can be produced by chlorination of methane, methanol, carbon
disulfide, propane, 1,2-dichloroethane and higher hydrocarbons.
The world production of carbon tetrachloride ranged from 850 to
960 kilotonnes over the years 1980-1988. Table 3 provides some data on
past production and production capacities of carbon tetrachloride.
These data are based on information in the ECDIN database (ECDIN,
1992) and BUA-Stoffbericht 45 (BUA, 1990).
Since 1990 the production of carbon tetrachloride has dropped.
The Montreal Protocol of 1990 and its subsequent amendments
established the phase-out by 1996 of production and use of carbon
tetrachloride and of chlorofluorocarbons (CFCs) by major manufacturing
countries. Special conditions were allowed for developing countries,
where consumption of controlled substances under Annex B (including
carbon tetrachloride) was required to be reduced by 85% of its
1998-2000 average level (or a calculated consumption level of 0.2 kg
per capita, whichever is lower) by 2005 and completely stopped by 2010
(UNEP, 1996).
3.2.1.2 Indirect production
Carbon tetrachloride can be produced as a by-product during the
manufacture of other products and compounds (US EPA, 1984a) and during
wood pulp bleaching.
Table 3. Past production and production capacity of carbon
tetrachloride
Country Year Production Capacity
(in kilotonnes) (in kilotonnes)
France 1988 - 90
Italy 1987 95 -
1988 - 130
Germany (former FRG) 1985 150 -
1987 180 -
1988 170 180
EEC 1985 - 520
1987 480 -
1988 478 540
Japan 1985 - 72
1987 52 -
1988 - 70
United Kingdom 1988 - 75
USA 1986 286 -
1987 340 -
1988 - 281
1991 143 -
World 1985 - 1200
1987 960 -
1988 - 1100
3.2.1.3 Emissions
According to US EPA (1991), in 1989 approximately 2000 tonnes of
carbon tetrachloride were released during manufacturing and processing
to the air in the USA. US EPA (1984a) reported emission factors for
carbon tetrachloride arising during the chlorination of hydrocarbons
ranging from 0.9 kg/tonne of carbon tetrachloride (controlled) to 2.8
kg/tonne of carbon tetrachloride (uncontrolled). Furthermore,
emissions may result from industrial water treatment or from old
landfill sites.
3.2.2 Uses
Most of the carbon tetrachloride produced is used in the
production of CFCs, which were primarily used as refrigerants,
propellants, foam-blowing agents and solvents and in the production of
other chlorinated hydrocarbons.
The use of carbon tetrachloride increased in the EEC as well as
in the USA during the years 1980-1987. However, this use has decreased
in recent years due to the Copenhagen Amendment to the Montreal
Protocol (1992) (UNEP, 1996). A survey in Japan could detect no use of
carbon tetrachloride in small to medium scale industries in 1996 (Ukai
et al., 1997).
Carbon tetrachloride has been used as a grain fumigant,
pesticide, solvent for oils and fats, metal degreaser, fire
extinguisher and flame retardant, and in the production of paint, ink,
plastics, semi-conductors and petrol additives. It was previously also
widely used as a cleaning agent. All these uses have tended to be
phased-out as production has dropped (ECDIN, 1992; ATSDR, 1994).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Transport
Carbon tetrachloride introduced into water resources is
transported by movement of surface water and groundwater. Because of
its volatility, evaporation is considered to be the main process for
the removal of carbon tetrachloride from aquatic systems. The amount
of carbon tetrachloride dissolved in the oceans is reported to be less
than 1-3% of that in the atmosphere (Galbally, 1976; Singh et al.,
1976). Practically all the carbon tetrachloride released to the
environment is thus present in the atmosphere (US EPA, 1991). Because
carbon tetrachloride does not degrade readily in the atmosphere,
significant global transport is expected.
Following releases to soil, most carbon tetrachloride is expected
to evaporate rapidly due to its high vapour pressure. A small fraction
of carbon tetrachloride may adsorb to organic matter, based on a
calculated soil adsorption coefficient of 100 (log Koc = 2.04)
(Kenaga, 1980).
Walton et al. (1992) studied the adsorption of carbon
tetrachloride from solution onto two soils, a silt loam (1.49% organic
carbon) and a sandy loam (0.66% organic carbon). The soil was shaken
with several concentrations of carbon tetrachloride (100 to 650 mg/kg
soil) for 18 h. The Koc values determined were 143.6 for the silt
loam and 48.9 for the sandy loam. Duffy et al. (1997) studied the
downward movement of carbon tetrachloride in 3 horizons of a fine
montmorillonitic soil. Koc values of 55, 77.6 and 269 were calculated
for the modern A, buried A and loess C soil horizons. However, the
authors point out that Koc values are unreliable in soils with low
organic carbon and high clay content. Therefore, the highest Koc
value should be treated with some caution.
4.1.2 Distribution
The evidence that the residence time of carbon tetrachloride in
the atmosphere is long (see section 4.1.3) and that nearly all of the
compound is found in this compartment explains the relatively even
distribution over the globe as is recorded in Table 4.
Only a very small proportion of carbon tetrachloride will remain
in water and soil.
Table 4. Levels of carbon tetrachloride in air
Location Year Mean level (µg/m3) Reference
(range in
parentheses)
Northern 1974 0.71 Cox et al. (1976)
hemisphere 1976 0.73 Singh et al. (1977)
1977 0.78 Singh et al. (1979)
1979-1981 0.87 Singh et al. (1983)
Southern 1974 0.44 Cox et al. (1976)
hemisphere 1977 0.76 Singh et al. (1979)
1979-1981 0.82 Singh et al. (1983)
North-West 1975-1980 0.91 Rasmussen et al.
Pacific (0.83-0.99) (1981)
1975-1985 0.77 Rasmussen & Khalil
(0.67-0.83) (1986)
1976 0.78 Cronn et al. (1977)
Antarctica 1975-1980 0.8 Rasmussen et al.
(0.77-0.87) (1981)
1975-1985 0.69 Rasmussen & Khalil
(0.62-0.76) (1986)
Arctic 1982 0.97 Rasmussen & Khalil
(1983)
North Atlantic 1983 0.56 von Düszeln &
Thiemann (1985)
North America 1976 0.86 Pierotti et al.
(0.33-0.99) (1980)
California, USA 1976 0.76-0.86 Singh et al.
(0.66-1.85) (1977)
Bochum, Germany 1978 0.8 (0.1-1.2) Bauer (1981)
Germany (cities) 1980-1981 0.6 von Düszeln &
Thiemann (1985)
Table 4. (Continued)
Location Year Mean level (µg/m3) Reference
(range in
parentheses)
South-West 1986-1988 0.5-0.6 Frank & Frank
Germany (1990)
The Netherlands 1980 0.83-1.0 Guicherit &
(max. 2.2-3.2) Schulting (1985)
Turin, Italy 1988 0.96 (0.17-1.94)a Gilli et al. (1990)
1988 0.47
(0.19-1.17)b Gilli et al. (1990)
Japan 1979 0.69
(0.62-0.72) Makide et al. (1979)
1994-1995 0.53c Sugama et al. (1995)
a cold months
b warm months
c concentrations were higher in winter than during summer
4.1.3 Removal from the atmosphere; global warming potential
The troposphere to stratosphere turnover time has been estimated
at around 30 years (Versar Inc., 1979). This is a shorter period of
time than is estimated for the degradation processes of carbon
tetrachloride in the troposphere (see section 4.2). Therefore
tropospheric carbon tetrachloride will attain significant
concentration in the stratosphere.
Cupitt (1980) calculated that deposition of carbon tetrachloride
from the atmosphere will be very slow.
Estimates of the atmospheric lifetime (the overall persistence of
carbon tetrachloride in the troposphere and the stratosphere combined)
are variable, but most values range from 25 to 100 years (Molina &
Rowland, 1974; Galbally, 1976; Singh et al., 1979; Edwards et al.,
1982a,b; Simmonds et al., 1983, 1988; Rowland, 1985; Huiskamp et al.,
1986; Howard, 1990; IPCC, 1990, 1995; WMO, 1991) with 45-50 years
generally being accepted as the most reasonable value.
The Global Warming Potential (GWP) of carbon tetrachloride,
relative to CO2, is estimated (IPCC, 1995) to be 2000, 1400 and 500
at integration time horizons of 20, 100 and 500 years. Its
contribution to total warming may be 0.3% as integrated effect over a
time horizon of 100 years (IPCC, 1995). Relative to CFC 12, the GWP of
carbon tetrachloride has been estimated to be 0.12 (UNEP, 1989).
4.1.4 Removal from water
The major removal process from water is volatilization to the
atmosphere. This was indicated by laboratory tests performed by
Dilling et al. (1975). These tests showed that a 1 ppm concentration
of low-molecular-weight chlorinated hydrocarbons will not persist in
agitated natural water bodies due to evaporation. In 29 min 50% of the
amount of carbon tetrachloride was evaporated, and in 97 min 90% was
evaporated. Zoeteman et al. (1980) calculated a half-life of carbon
tetrachloride in rivers of 0.3-3 days and in lakes and groundwaters of
30-300 days.
4.1.5 Removal from soil
Anderson et al. (1991) studied the loss of carbon tetrachloride
from two different soil types, a silt loam (1.49% organic carbon) and
a sandy loam (0.66% organic carbon). Carbon tetrachloride was applied
to the soil at a concentration of 100 mg/kg (dry weight) and the soil
was incubated in the dark at 20°C for 7 days. The mean half-life for
disappearance of carbon tetrachloride was 5 days. There was no
significant difference between the loss from sterile and non-sterile
systems indicating that volatilization was the likely removal process.
Jury et al. (1984) predicted that carbon tetrachloride would have
a volatilization half-life of 0.2 days at a depth of 1 cm and 0.8 days
at a depth of 10 cm in soil, based on volatilization tests and
assuming a uniform distribution of the chemical with depth.
4.2 Abiotic degradation
4.2.1 Degradation in atmosphere
4.2.1.1 Photodegradation
Carbon tetrachloride is very stable in the troposphere (Lillian
et al., 1975; Cox et al., 1976; Singh et al., 1980). This is primarily
because carbon tetrachloride, in contrast to most other volatile
halocarbons, has low reactivity towards hydroxyl radicals. This is
evident from rate constants determined by several authors (Howard
Carleton & Evenson, 1976; Cox et al., 1976; Clyne & Holt, 1978). Based
on these rate constants, half-lives of > 3.9 to 137 years can be
calculated for the decomposition of carbon tetrachloride in the
troposphere (Lyman et al., 1982).
Cox et al. (1976) found an even higher tropospheric half-life of
> 330 years.
4.2.1.2 Photolysis
Edwards et al. (1982b) estimated a lifetime in the troposphere
due to photolysis of the order of 500 years.
The principal degradation process for carbon tetrachloride occurs
in the stratosphere, where it is dissociated by short wave length
(190- 220 nm) UV radiation to form the trichloromethyl radical and
chlorine atoms. Simmonds et al. (1983) estimated a half-life of 18-80
years for this photodissociation process.
4.2.1.3 Ozone-depletion potential
The chlorine atoms in carbon tetrachloride interact with oxygen
or ozone to produce ClO* groups (Singh et al., 1975). The chlorine
atoms and ClO* groups attack the surrounding ozone in a reaction in
which they act as catalysts until scavenged by some other chemical
reaction (Isaksen & Stordal, 1981; Rowland, 1985; Ember et al., 1986).
This effect is reflected in an ODP (ozone depletion potential) of 1.08
(WMO, 1991) and 1.1 (UNEP, 1996), compared with the chlorofluorocarbon
CFC-11, and was responsible for the inclusion of carbon tetrachloride
in the amended Montreal Protocol of 1990 (UNEP, 1996).
Catalytic breakdown of ozone by chloride-containing radicals:
CCl4 + h nu -> *CCl3 + *Cl
*CCl3 + O2 -> -> COCl2 + ClO*
*Cl + O3 -> ClO* + O2
ClO* + O -> *Cl + O2
4.2.2 Degradation in water
Carbon tetrachloride dissolved in water does not photodegrade or
oxidize in any measurable amount (Howard et al., 1991). The rate of
hydrolysis was thought to be second order with respect to carbon
tetrachloride with a calculated half-life of 7000 years at a
concentration of 1 ppm (Mabey & Mill, 1978). However, Jeffers et al.
(1996) found that the rate of hydrolysis for dilute solutions of
carbon tetrachloride was first-order and estimated the half-life to be
40 years. The authors reanalysed data previously stated as
second-order kinetics and found it to be consistent with a first-order
rate of hydrolysis.
4.2.3 Other degradation processes
Photodissociation of carbon tetrachloride adsorbed on to
silicates has been observed in the laboratory by Ausloos et al.
(1977).
Gäb et al. (1980) found experimentally that carbon tetrachloride
degraded over sand, silica gel and Al2O3. The degradation rate
depended, among other factors, on the laboratory conditions. Under the
conditions representative for deserts, degradation was about 4.5%
after exposure for 115 days.
4.3 Biotic degradation
4.3.1 Aerobic
Carbon tetrachloride has been shown to be resistant to aerobic
biodegradation by mixed bacterial cultures growing on methane as the
carbon source. No degradation of carbon tetrachloride was observed in
a mixed culture of methane-utilizing bacteria isolated from soil and
incubated in the dark for 6 days (Cochran et al., 1988). Oldenhuis et
al. (1989) reported no degradation of carbon tetrachloride by the
methanotrophic bacterium Methylosinus trichlosporium in the presence
of formate and oxygen.
Vannelli et al. (1990) found that carbon tetrachloride was not
degraded by the ammonia-oxidizing bacterium
Nitrosomonas europea when incubated at 1 mg/litre for 24 h.
In contrast, Tabak et al. (1981) found carbon tetrachloride to be
significantly degradable under aerobic conditions, with rapid
adaptation. Carbon tetrachloride (5 and 10 mg/litre) was incubated at
25°C for 7 days in static culture containing yeast extract inoculated
with settled domestic wastewater. Eighty to eighty-seven per cent of
the initial concentration disappeared within 7 days in the first
culture. An abiotic control showed that 5-23% of this loss could be
due to volatilization. In three subsequent cultures, carbon
tetrachloride was degraded to concentrations below the detection limit
(< 0.1 mg/litre) in the same period.
4.3.2 Anaerobic
The biodegradation of carbon tetrachloride has been studied under
methanogenic conditions. In batch cultures, carbon tetrachloride at a
concentration of 200 µg/litre was incubated in the dark at 35°C with
mixed methanogenic bacteria derived from a laboratory-scale digester
fed with activated sludge. Carbon tetrachloride was found to be
degraded to below the detection limit (< 0.1 µg/litre) within 16
days; carbon dioxide was the only degradation product identified. In a
continuous-flow column study, columns were initially seeded with an
inoculum of methanogenic bacteria from rum distillery wastewater.
Acetate (100 mg/litre) was fed to the column as primary growth
substrate and carbon tetrachloride was fed as a secondary substrate.
The column had a 2 day retention time, and it was found that carbon
tetrachloride was 99% degraded in the column; carbon dioxide being the
major degradation product (Bouwer & McCarty, 1983a).
Bouwer & McCarty (1983b) studied the biodegradation of carbon
tetrachloride under denitrifying conditions. Using batch cultures
seeded with primary sewage effluent and containing nitrate as an
electron acceptor, carbon tetrachloride (75 µg/litre) was found to be
degraded rapidly with no detectable lag period when incubated in the
dark at 25°C for 8 weeks. Chloroform and carbon dioxide were the
degradation products identified.
The biodegradation of carbon tetrachloride using aquifer material
has been studied (Parsons et al., 1985). Microcosms were constructed
containing groundwater and sediment contaminated with trichloroethene.
The concentration of carbon tetrachloride was 4 mg/litre and
incubation was carried out in the dark at 25°C. Reductive
dehalogenation of carbon tetrachloride to chloroform was found to
occur, and 700 µg chloroform/litre was detected after 8 weeks.
Egli et al. (1987) observed that pure cultures of
Desulfobacterium autotrophicum dechlorinated carbon tetrachloride to
trichloromethane and dichloromethane within 6 days.
Klecka & Gonsior (1984) provided evidence that reductive
dehalogenation of carbon tetrachloride in aqueous solution under
anaerobic conditions could be achieved with naturally occurring iron
porphyrins and other reducing agents. Carbon tetrachloride (1
mg/litre) was rapidly degraded to chloroform when incubated at 25°C
with an iron porphyrin (haematin) and sulfide.
Bioremediation studies have shown that anaerobic biodegradation
is enhanced by increasing the concentration of primary substrates
(such as glucose and acetate) and by lowering the redox potential
(providing a relatively higher electron activity which facilitates
dechlorination) (Doong & Wu, 1995, 1996; Doong et al., 1996; Jin &
Englarde, 1996).
4.4 Bioaccumulation
The log octanol-water partition coefficient (Kow) of carbon
tetrachloride is 2.64 indicating a moderate potential for
bioaccumulation under conditions of constant exposure. However,
studies have shown that the compound's short tissue lifetime reduces
this tendency. Barrows et al. (1980) reported a bioconcentration
factor of 30 for bluegill sunfish (Lepomis macrochirus) with a
tissue half-life of less than one day. A similar bioconcentration
factor of 30 (whole body; fresh weight) was reported by Veith (1978)
in bluegill. Neely et al. (1974) found a bioconcentration factor of
17.7 for muscle tissue of rainbow trout (Oncorhynchus mykiss). A
higher bioconcentration factor of 300 (wet weight) has been measured
for carbon tetrachloride in the green alga Chlorella fusca exposed
to 50 µg/litre for at least 24 h (Geyer, 1984). No significant
bioaccumulation in marine food chains was found in an extensive study
by Pearson & McConnell (1975) (see Table 6, section 5.1.4).
Some plants, due to their lipid content, take up carbon
tetrachloride from the air. Thus studies of the equilibrium
partitioning of carbon tetrachloride between the gas phase and conifer
needles (Pinus sylvestris and Picea abies) on the one hand and
hexane-extractable leaf waxes on the other hand showed partition
ratios (g/m3 needle; g/m3 air) of 9-17 and 90-400, respectively
(Frank & Frank, 1986; Brown et al., 1998).
5. CONCENTRATIONS IN THE ENVIRONMENT AND EXPOSURE
5.1 Environmental levels
5.1.1 Air
Reported concentrations of carbon tetrachloride measured in
ambient air are presented in Table 4.
As seen in Table 4, mean global levels of atmospheric carbon
tetrachloride usually lie in the range of 0.5-1.0 µg/m3. Based on an
analysis of 4913 ambient air samples (including remote, rural,
industrial and source-dominated sites in the USA), the average
concentration of carbon tetrachloride was 1.1 µg/m3 (Shah &
Heyerdahl, 1988). Urban atmospheric carbon tetrachloride levels and
levels in industrial areas can be considerably higher as shown by the
measurements by Lillian et al. (1975), Singh et al. (1980, 1982) and
Bozzelli & Kebbekus (1982). These authors reported mean levels of 2-3
µg/m3 (several hundred measurements) with maximum levels up to 6
µg/m3. Near a production facility in the United Kingdom, Pearson &
McConnell (1975) recorded levels an order of magnitude higher.
It has been estimated that concentrations of carbon tetrachloride
were increasing worldwide until recently (Simmonds et al., 1988;
Howard, 1990). The Intergovernmental Panel on Climate Change (IPCC)
has estimated the atmospheric concentration to be about 0.94 µg/m3
and the annual rate of increase to be 1.5% (IPCC, 1990). However, the
accumulation of the substance in the atmosphere seems to have stopped
(Fraser et al., 1994) and even started to decline (Fraser & Derek,
1994).
5.1.2 Water
Some reported aquatic concentrations of carbon tetrachloride are
summarized in Table 5.
As seen in Table 5, remote oceanic levels of carbon tetrachloride
are usually in the range of 0.0005-0.0008 µg/litre. As sites nearer to
effluent sources are examined, higher levels are observed. Thus in
estuaries, levels from 0.01 to 2.7 µg/litre have been observed, and in
remote freshwater sites from 0.0002 to 0.025 µg/litre, while nearer to
industrial facilities mean levels in the range of < 0.1-24.2 µg/litre
have been recorded.
Even higher values, e.g., 160-1500 µg/litre in the River Rhine
and a mean of 75 µg/litre in the River Main, recorded in 1976 in
Germany, were the result of direct waste release (BUA, 1990).
Groundwater levels range from undetectable to a maximum of 80
µg/litre.
Table 5. Levels of carbon tetrachloride in surface water
Area Mean level (µg/litre)
(range in parentheses) Reference
Marine
East Pacific ocean 0.0005 Su & Goldberg
(1976)
East Pacific ocean 0.0007 Singh et al. (1983)
Arctic ocean 0.0008 Fogelqvist (1985)
Estuarine
Scheldt Estuary, 0.01-0.02a van Zoest & van
The Netherlands (max. 0.29) Eck (1991)
Mersey Estuary, UK 2.7 Edwards et al.
(1982a)
Freshwater
Lake Zurich, Switzerland 0.025 (0.02-0.035) Giger et al. (1978)
Lake Ontario, Canada (< 0.0002-0.005) Kaiser et al. (1983)
Niagara River, Canada 0.0029 (max. 0.018) Kaiser et al. (1983)
River Weaver, UK < 0.1 Rogers et al. (1992)
River Gowry, UK 0.9 Rogers et al. (1992)
River Rhine, Lobith, 1.5 (0.4-2.8) Bauer (1981)
Germany
Manchester Ship Canal, UK 3.8 Edwards et al.
(1982a)
Manchester Ship Canal, UK 24.2 Rogers et al. (1992)
Groundwater
Zurich (industrial area) (< 0.05-3.6) Giger et al. (1978)
Birmingham aquifer, UK (0.02-1) Rivett et al.
(1990a,b)
Coventry aquifer, UK 4.9 (max. 80) Burston et al.
(1993)
Washington, New Jersey, (ND-34) Suffet et al. (1985)
USA
Gibbstown, New Jersey, (1.4-1.8) Rosen et al. (1992)
USA
a range of medians
Based on analysis of data from STORET database, carbon
tetrachloride was detectable in 1063 of 8858 ambient water samples,
with a median concentration of 0.1 µg/litre (Staples et al., 1985).
Rain water and snow concentrations of carbon tetrachloride are
generally in the range of 0.3 to 2.8 µg/litre (Su & Goldberg, 1976),
but a level as high as 300 µg/litre was observed in rainwater
collected near a production site in the United Kingdom (Pearson &
McConnell, 1975).
5.1.3 Soil and sediment
Carbon tetrachloride might occur in soil due to spills, runoff
and leaching. However, only 0.8% of 361 measured soil/sediment samples
appeared to contain carbon tetrachloride. The concentration was
reported to be less than 5.0 mg carbon tetrachloride/kg dry weight
soil or sediment (Staples et al., 1985).
5.1.4 Biota
Levels of carbon tetrachloride in biota are summarized in Table
6.
5.2 General population exposure
The general population can be exposed to carbon tetrachloride
through air, foodstuffs and drinking-water.
5.2.1 Outdoor air
Levels in ambient air to which the general population may be
exposed are recorded in Table 4.
5.2.2 Indoor air
Because of its volatility, carbon tetrachloride tends to
volatilize from tap water. Although, human exposure by inhalation of
carbon tetrachloride transferred to the indoor air from showers and
baths, toilets, washing and cooking is conceivable, no experimental
data have been reported (McKone, 1987).
Several reports on carbon tetrachloride levels in dwellings have
been published. Taketomo & Grimsrud (1977) found values ranging
between 0.6 and 1.3 µg/m3 for various types of dwellings, which is in
agreement with the maximum indoor concentration of 1.2 µg/m3 reported
by Clark (1981) and the range of 0.9-1.8 µg/m3 found in a US EPA
study. In addition, several measurements have been made in garages,
shops, supermarkets, swimming pools, restaurants, etc. (Taketomo &
Grimsrud, 1977; Ullrich, 1982). The observed concentrations usually
ranged between 0.6 and 2.0 µg/m3. The highest concentration, 10
µg/m3, was found in a dry-cleaning establishment.
Wallace (1986) reported typical concentrations in homes in
several cities in the USA of about 1 µg/m3; a maximum value of 9
µg/m3 was found. Shah & Heyerdahl (1988) found an average carbon
tetrachloride level of 2.6 µg/m3, based on 2120 indoor samples. It
should be noted, however, that carbon tetrachloride was not detected
in more than half the samples.
Table 6. Levels of carbon tetrachloride in biota
Organism Location Organ Level (µg/kg) Reference
Plankton Liverpool Bay, UK whole body 0.04-0.09 wet weight Pearson & McConnell (1975)
Molluscs Firth of Forth, UK whole body 2 wet weight Pearson & McConnell (1975)
Liverpool Bay, UK whole body 0.4-1 wet weight Pearson & McConnell (1975)
Thames Estuary, UK whole body 0.1-0.9 wet weight Pearson & McConnell (1975)
Irish Sea muscle 5-28 dry weight Dickson & Riley (1976)
digestive tissue 8-20 dry weight Dickson & Riley (1976)
gill 14 dry weight Dickson & Riley (1976)
ovary 16 dry weight Dickson & Riley (1976)
mantle 2-114 dry weight Dickson & Riley (1976)
Crustaceans Firth of Forth, UK whole body 1-3 wet weight Pearson & McConnell (1975)
Liverpool Bay, UK whole body 3-5 wet weight Pearson & McConnell (1975)
Thames Estuary, UK whole body 0.2 wet weight Pearson & McConnell (1975)
Fish Liverpool Bay, UK flesh 2 wet weight Pearson & McConnell (1975)
liver ND-0.3 wet weight Pearson & McConnell (1975)
Thames Estuary, UK flesh 0.3-6 wet weight Pearson & McConnell (1975)
Irish Sea brain 15-191 dry weight Dickson & Riley (1976)
gill 3-209 dry weight Dickson & Riley (1976)
gut 9-44 dry weight Dickson & Riley (1976)
liver 4-51 dry weight Dickson & Riley (1976)
muscle 7-83 dry weight Dickson & Riley (1976)
skeletal tissue 7-22 dry weight Dickson & Riley (1976)
heart 10-40 dry weight Dickson & Riley (1976)
ND = not detected.
5.2.3 Drinking-water
The National Organics Monitoring Survey (NOMS) in the USA
detected carbon tetrachloride (range of 2.4-6.4 ng/litre) in public
drinking-water systems in 10% of the 113 samples surveyed (US EPA,
1984b). In 30 out of 954 drinking-water samples from various cities in
the USA carbon tetrachloride could be detected. Median concentrations
in different groups ranged from 0.3 to 0.7 µg/litre while maximum
concentrations reached 16 µg/litre (Westrick et al., 1984). Bauer
(1981) reported that drinking-water in Germany contained an average of
less than 0.1 µg/litre although a maximum level of 1.4 µg/litre was
found (average of 100 towns in 1977). Lahl et al. (1981) reported a
carbon tetrachloride concentration less than 0.1 µg/litre in the
drinking-water of 50 cities in Germany. In the United Kingdom,
< 0.01-2.3 µg/litre was measured in drinking-water (Reynolds et al.,
1982; Reynolds & Harrison, 1982).
Values as high as a median of 3 µg/litre and a maximum of 39.5
µg/litre were reported in Galicia, Spain (Freiria-Gándara et al.,
1992).
5.2.4 Foodstuffs
According to investigations carried out in Europe and USA between
1973 and 1989, many foodstuffs contained carbon tetrachloride at
concentrations of a few µg/litre or µg/kg.
The following concentrations of carbon tetrachloride in
foodstuffs in the United Kingdom in 1973 were reported: meat, 7-9
µg/kg; edible oils, 16-18 µg/kg; tea, 4 µg/kg; and fruits and
vegetables, 3-8 µg/kg (McConnell et al., 1975). Values in a similar
range were found for dairy products, other edible oils, fats,
beverages, other fruits and bread, but here carbon tetrachloride and
1,1,1-trichloroethane could not be separated (McConnell et al., 1975).
According to a study conducted in Germany, carbon tetrachloride
can be present in decaffeinated coffee (4.9-60 µg/kg), milled cereal,
flour and starch products (levels in 21 samples ranged from less than
0.1 to 26 µg/kg). The origin in the first case is the
caffeine-extraction procedure, and in the second case in all
probability fumigation of the raw cereals. The use of carbon
tetrachloride for fumigation of stored foodstuffs and decaffeination
of coffee appears to have generally ceased and it is unlikely that its
occurrence in food stuff will be of significance. Less than 1 µg/kg
was found in sugar, fruit, vegetables, beverages, bread, toast,
potatoes, olives, oils, milk, butter, eggs, yoghurt, (cream) cheese,
meat and fish. In cough mixtures 0.1 to 1.8 µg/kg was found (Bauer,
1981).
Entz et al. (1982) and Entz & Hollifield (1982), in analyses of
various foods for a series of volatile halogenated hydrocarbons, did
not find carbon tetrachloride at a detection limit of 0.5 to 3 µg/kg,
depending on the type of product. Decaffeinated coffee and flour
products were not included in the studies.
Kroneld (1989) detected carbon tetrachloride in meat (0.9 µg/kg),
fish (0.6 µg/kg) and juice (0.3 µg/kg) in Finland in 1987.
Carbon tetrachloride levels in table-ready foods in the USA were
reported by Heikes (1987). He found up to 2.2 µg/kg in four sorts of
cheese, 0.10-0.34 µg/kg in cereals, 1.7-5.7 µg/kg in fish sticks and
up to 6.0 µg/kg in butter.
In a survey by Daft (1989, 1991) carbon tetrachloride was
detected in 44 out of 549 food items from the USA, most often in fatty
and grain-based foods. The mean level in food items with detectable
levels was 31 µg/kg (with a range of 2 to 210 µg/kg).
5.2.5 Intake averages
The daily average intake of carbon tetrachloride in Japan by
inhalation was calculated to be 7.7 µg/day (based on a daily
inhalation volume of 15 m3/day and assuming a 100% absorption) and by
ingestion less than 0.1 µg/day (Yoshida, 1993). If adjusted to a daily
inhalation volume of 22 m3/day, an absorption of 40% and a body
weight of 64 kg, the daily intake would be 11.4 µg/day or 0.18 µg/kg
per day.
The ATSDR (1994) estimated the daily intake by inhalation to be
0.1 µg/kg body weight based on ambient air level of about 1 µg/m3
(assuming inhalation of 20 m3/day, a body weight of 70 kg and an
absorption of 40% based on measurements in monkeys and humans). The
daily intake via drinking-water was estimated to be about 0.01 µg/kg
body weight based on a typical carbon tetrachloride concentration of
0.5 µg/litre (assuming a water consumption of 2 litres/day and a body
weight of 70 kg).
In an earlier study of about 500 foodstuffs, an average daily
intake via foods and drinks of 8.63 µg/person per day was calculated
for inhabitants of Germany (Lahl, 1983). Because the intake by
inhalation is expected to be at least as much (BUA, 1990), the total
daily average intake would be estimated to be 17.26 µg/person (0.27
µg/kg body weight for a person of 64 kg). This calculation refers to a
period when carbon tetrachloride was still used in food processing or
in fumigation of grain.
5.3 Occupational exposure
The most likely route of exposure in the workplace is by
inhalation. Workers may be exposed to carbon tetrachloride during, for
example, the production of carbon tetrachloride itself, the synthesis
of compounds using carbon tetrachloride as a starting material and the
use of carbon tetrachloride as a solvent. Furthermore, workers have
been exposed to carbon tetrachloride at grain (due to fumigation) and
water treatment facilities. The National Institute for Occupational
Safety and Health estimated that in the USA around 58 000 workers were
potentially exposed to carbon tetrachloride, based on a national
survey conducted from 1981 to 1983 (National Library of Medicine,
1992).
A few studies on concentrations of carbon tetrachloride in
factories, and grain and water treatment facilities have been
reported. For water treatment facilities, Lurker et al. (1983)
reported exposure concentrations of 0.01-0.23 mg/m3; Clark (1981)
reported concen trations ranging from 0 to 1.1 mg/m3.
A peak exposure to an inspector during handling of grain at a
facility in the USA reached 277 mg/m3. Few employees, however, had a
mean exposure above 641 µg/m3 (Deer et al., 1987). Use of carbon
tetrachloride in open beakers resulted in exposure levels of 290-620
mg/m3 at a United Kingdom quartz crystal processing plant. Levels
were reduced to 50-60 mg/m3 by closing the beakers (Kazantzis &
Bomford, 1960).
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1 Pharmacokinetics
6.1.1 Absorption
Carbon tetrachloride is absorbed readily from the
gastrointestinal and respiratory tract. Dermal absorption of carbon
tetrachloride, either in vapour or in liquid phase, is possible, but
the dermal absorption of the vapour appears to be very low.
6.1.1.1 Oral
Carbon tetrachloride is relatively insoluble in water, a source
of exposure relevant to environmental scenarios and human health risk.
As a result, many studies examining the hepatotoxicity of carbon
tetrachloride used corn oil as a dosing vehicle for laboratory animals
(Paul & Rubinstein, 1963; Larson & Plaa, 1965; Marchand et al., 1970).
Corn oil has been found to delay markedly the absorption of carbon
tetrachloride (Kim et al., 1990a) as well as other halocarbons (Withey
et al., 1983) from the gastrointestinal tract.
In part, because carbon tetrachloride in water is directly
relevant to human exposure studies, recent studies in laboratory
animals employed Emulphor(R), a polyethoxylated oil, at
concentrations up to 10%, as an aqueous vehicle for carbon
tetrachloride. Aqueous solutions of carbon tetrachloride in
Emulphor(R) were administered to Sprague-Dawley rats both as a bolus
and during gastric infusion at a constant rate during a 2-h period
(Sanzgiri et al., 1995). Uptake and tissue levels of carbon
tetrachloride after gastric infusion were less than after bolus
dosing. When the concentration of Emulphor(R) was varied up to 10%,
absorption (and distribution) of carbon tetrachloride was not affected
(Sanzgiri & Bruckner, 1997).
A comparison of the uptake of carbon tetrachloride in corn oil
and aqueous emulsions is discussed in section 7.9. Tissue levels of
carbon tetrachloride associated with bolus dosing, gastric infusion,
and inhalation are discussed in section 6.1.2. The relationship of
dosing vehicle, dose rate, and route of exposure to hepatotoxicity is
discussed in section 7.9.
6.1.1.2 Dermal
Liquid carbon tetrachloride on the intact mouse skin was absorbed
at a rate of 8.3 µg/cm2 per minute (Tsuruta, 1975). Jakobson et al.
(1982) examined the percutaneous uptake of liquid carbon tetrachloride
(1 ml) in guinea-pigs (carbon tetrachloride in a glass depot, covering
3.1 cm2 of clipped skin). A peak blood level of about 1 mg carbon
tetrachloride/litre was reached within 1 h. Despite continuation of
the exposure the blood levels declined during the following h,
possibly due to local vasoconstriction, rapid transport from blood to
adipose tissues or biotransformation processes.
Wahlberg & Boman (1979) applied 0.5 or 2 ml of carbon
tetrachloride in a closed glass container on the skin (3.1 cm2) of
guinea-pigs. The deposits were completely absorbed within a few days.
McCollister et al. (1951), who exposed the clipped skin of one
male and one female monkey to [14C]carbon tetrachloride vapour (whole
body exposure), detected radioactivity in the blood and in the expired
air. After an exposure of 3 h at 3056 mg/m3, the blood of the female
contained a carbon tetrachloride level of 12 µg/100 g and the expired
air contained 0.8 µg/litre. After exposure to 7230 mg/m3 for 3.5 h
the blood of the male contained a carbon tetrachloride level of 30
µg/100 g and the expired air contained 3 µg/litre.
6.1.1.3 Inhalation
In rats exposed by inhalation to carbon tetrachloride
concentrations of 100 or 1000 ppm (641 or 6410 mg/m3) for 2 h, the
total amounts systemically absorbed were 17.5 and 179 mg/kg body
weight. The Cmax values (mg/ml) were approximately 1 and 13,
respectively, and the AUC values (mg.min/ml) were approximately 120
and 1900, respectively (Sanzgiri et al., 1995).
Steady-state carbon tetrachloride concentrations in the blood of
approximately 320 mg/litre were reached within about 5 h when dogs
were exposed to a carbon tetrachloride concentration in air of 15 000
ppm (96 150 mg/m3) for several hours (Von Oettingen et al., 1950).
McCollister et al. (1951) exposed three female rhesus monkeys to
an average [14C]carbon tetrachloride concentration of 46 ppm (295
mg/m3) via air for 139, 244 or 300 min, respectively. Within 300 min,
30% of the inhaled quantity was absorbed but in the blood no
steady-state concentration of radioactivity was reached. The
radioactivity level in the blood at that moment corresponded to 3 mg
carbon tetrachloride/litre blood and was distributed over carbon
tetrachloride (56.2%), "acid volatile" carbonates (16.5%) and
non-volatile material (27.3%).
The US EPA Iris Program uses 40% absorption as a mean for the
calculation of human respiratory intake. The determined values ranged
from 30% to 65% (US EPA, 1991).
6.1.2 Distribution
The tissue distribution of carbon tetrachloride has been
investigated in mice after inhalation (Bergman, 1984), in rats after
oral administration (Marchand et al., 1970; Teschke et al., 1983;
Watanabe et al., 1986) and after inhalation (Paustenbach et al.,
1986a), in rabbits after oral administration (Fowler, 1969), in dogs
after inhalation (Von Oettingen et al., 1950) and in monkeys after
inhalation (McCollister et al., 1951).
Bergman (1984) investigated the distribution of [14C]carbon
tetrachloride in the mouse after a single inhalation exposure (10 min;
256 000 mg/m3 air). Immediately after the exposure, high levels of
radioactivity were found in fat, bone marrow, white matter of the
brain, spinal cord and nerves, liver, kidneys, salivary glands and
gastrointestinal mucosa. The radioactivity in bronchi, liver, kidneys,
salivary glands and the gastrointestinal mucosa (particularly in the
mucosa of the glandular part of the stomach and of the colon and
rectum) was to a large extent non-volatile. A similar pattern of
distribution was observed 30 min after the exposure, except in the
liver where a more pronounced accumulation of non-volatile
radioactivity was seen than observed immediately after inhalation. A
large part of the non-volatile radioactivity in the liver and kidneys
appeared to be non-extractable, which may indicate covalent binding to
tissue components (see section 6.2). Non-extractable radioactivity was
also present in the bronchi and nasal mucosa. Non-volatile and
non-extractable radioactivity was present in the vaginal and uterine
mucosa and interstitially in the testis.
The tissue distribution in rats (in order of decreasing
radio-activity), 3 h after oral administration, as reported by
Watanabe et al. (1986) was: liver, kidney, brain, muscle and blood.
Carbon tetrachloride tends to accumulate in fat. Maximal fat tissue
carbon tetrachloride concentrations exceeded the maximal blood levels
by a factor of 60 after oral administration to rats (Marchand et al.,
1970).
Peak levels of carbon tetrachloride were observed 3-6 h following
an acute oral carbon tetrachloride dose (1.5 ml/kg body weight
administered in olive oil) in the blood (26 mg/litre), liver and fat
of female Wistar rats. Subsequently, the carbon tetrachloride levels
declined rapidly (Teschke et al., 1983).
Peak blood levels of carbon tetrachloride after a 12-h inhalation
exposure of rats were 12 mg/litre blood at an airborne concentration
of 2 mg/litre (320 ppm), 20 mg/litre blood at 4 mg/litre (640 ppm) and
36 mg/litre blood at 8 mg/litre (1280 ppm). The blood level attained
50% of this value after 60 min. A 4-h exposure to a concentration of
2.6 mg/litre (406 ppm) led to a blood level of 10.5 mg/litre, which
dropped to 50% of this peak value within 30 min after exposure
(Frantik & Benes, 1984).
Paustenbach et al. (1986a) found the highest concentration of
carbon tetrachloride equivalents in the fat, liver, lungs and adrenals
of male Sprague-Dawley rats repeatedly exposed to 100 ppm (641 mg/m3)
of [14C]carbon tetrachloride vapour for 8 or 11.5 h/day for periods
of 1 to 10 days.
Fowler (1969) administered 1 ml carbon tetrachloride/kg body
weight to rabbits by stomach tube as a 20% (v/v) solution in olive
oil. Five rabbits were killed 6, 24 and 48 h after receiving carbon
tetrachloride and the concentration in fat, liver, kidney and muscle
tissue was determined. Two rabbits receiving olive oil were killed as
control animals. The highest carbon tetrachloride concentration after
6, 24 and 48 h was found in fat tissue, but the amount found in the
fat as well as in the other tissues after 6 h diminished rapidly
during the subsequent 42 h.
Von Oettingen et al. (1950) studied the distribution of carbon
tetrachloride in Beagle dogs after exposure to 15 000 ppm (96 150
mg/m3) and reported a lowest concentration in the blood followed in
increasing order by liver, heart and brain.
The pattern of distribution immediately after a 5-h inhalation of
46 ppm (295 mg) [14C]carbon tetrachloride/m3 in monkeys (McCollister
et al., 1951) was (tissues in order of decreasing concentration of
total radioactivity): fat, liver, bone marrow, blood, brain, kidneys,
heart, spleen, muscle, lungs, bone.
Sanzgiri et al. (1995) demonstrated that the tissue
pharmacokinetic profile was influenced by the route and rate of
administration of carbon tetrachloride. Inhalation exposure of rats to
1000 ppm (6410 mg/m3) carbon tetrachloride for 2 h resulted in a
systemic dose of 179 mg/kg body weight. This dose was subsequently
administered as an oral bolus or a constant gastric infusion over 2 h.
In all cases tissue levels were highest in fat with levels in all
tissues being higher after an oral bolus dose than after inhalation
exposure or gastric infusion. For the liver Cmax was higher after an
oral bolus dose (58 mg/g) than after inhalation (20 mg/g) or gastric
infusion (0.5 µg/g). The authors speculate that the capacity of
first-pass metabolism can be exceeded following a large single bolus
oral dose, although not during gastric infusion of the same dose over
2 h.
6.1.3 Elimination and fate
In a study by Reynolds et al. (1984), all routes of elimination
were investigated simultaneously after a single oral administration of
[14C]carbon tetrachloride to fasted rats at dose levels ranging from
15.4 to 4004 mg/kg body weight. The exhalation of unchanged carbon
tetrachloride increased at higher dose levels (70-90% after
administration of 46.2 mg/kg body weight or more). This result might
be explained by a saturation of the first pass metabolism, or by an
impairment of the overall carbon tetrachloride metabolism due to a
breakdown of cytochrome P-450, which is induced by carbon
tetrachloride-metabolites (as reported by Noguchi et al., 1982a,b).
Both the amount of carbon tetrachloride excreted and the time-course
of excretion depended on the dose, tending to become slower as the
dose increased. For example, the half-life for exhalation of carbon
tetrachloride was 1.3 h at 46.2 mg/kg body weight but was 6.3 h at
4004 mg/kg body weight.
Page & Carlson (1994) examined whether faecal excretion, either
biliary or by direct exsorption, contributed significantly to carbon
tetrachloride elimination from the body of rats. It appeared that
biliary and non-biliary mechanisms contributed to the faecal
elimination of [14C]carbon tetrachloride, but that this route
accounted for less than 1% of the administered dose of 1 mmol/kg body
weight in rats. Thus faecal elimination of carbon tetrachloride (as
parent compound) does not significantly contribute to the overall
elimination of carbon tetrachloride.
The carbon tetrachloride levels in blood declined with a
half-life of 4 to 5 h during the first 24 h after oral administration
of 1.25 ml carbon tetrachloride/kg body weight (Larson & Plaa, 1965)
or 2 ml (0.1 mCi) [14C]carbon tetrachloride/kg body weight (Marchand
et al., 1970). Carbon tetrachloride levels in the liver declined with
a half-life of about 7 h after administration by gastric intubation of
2.5 ml carbon tetrachloride/kg body weight (Dingell & Heimberg, 1968).
Kim et al. (1990a) found a half-life for carbon tetrachloride in
the blood of 98 min and a whole body clearance of 0.13 ml/min per g
when 25 mg carbon tetrachloride/kg body weight was orally administered
in four different vehicles to male Sprague-Dawley rats. The
elimination appeared to be the same in all the different vehicle
groups, whereas the absorption differed (see section 6.1.1.1).
According to Paustenbach et al. (1986a) the rate of carbon
tetrachloride clearance in rats after inhalation exposure is biphasic,
with an initial half-life of 7 to 10 h. Exposure for longer periods of
time led to a decreased rate of clearance (and to higher
concentrations in the fat) (Paustenbach et al., 1986a,b, 1988). In the
study of Sanzgiri et al. (1995) (see section 6.1.1.3) the apparent
clearance values after inhalation doses delivering 17.5 or 179 mg/kg
body weight, respectively, were 150 and 100 ml/min per kg and the
half-life value was about 164 min.
Veng-Pedersen et al. (1987) exposed rats repeatedly by inhalation
to 100 ppm [14C]carbon tetrachloride (641 mg/m3) for either 8 h/day
for 5 days or 11.5 h/day for 4 days. The pulmonary excretion of
[14C]activity was clearly biphasic for both dosing regimens, with
mean half-lives for the first and second phase being 84 and 400 min
for the 8-h exposure and of 91 and 496 min for the 11.5-h exposure,
respectively. This indicates that the second phase of the 11.5-h group
was longer than the second phase of the 8-h group. This observation
suggests that during longer exposure periods a greater fraction of the
inhaled carbon tetrachloride is distributed to poorly perfused tissues
like fat, thus altering the elimination.
McCollister et al. (1951) demonstrated in monkeys that, after an
inhalation exposure to [14C]carbon tetrachloride, radioactive
material was excreted in faeces, urine and expired air. According to
the authors the compounds in the urine consisted of urea, bicarbonate
and an acid hydrolysable, non-amino acid substance.
6.1.4 Physiologically based pharmacokinetic modelling
A biphasic kinetic in the biotransformation of carbon
tetrachloride has been observed in several inhalation studies. The
relationship of arterial blood and inhaled carbon tetrachloride
concentrations, as found in male Sprague-Dawley rats, suggested that
carbon tetrachloride metabolism is limited by blood perfusion of the
liver at inhaled concentrations below 100 ppm (641 mg/m3) and that it
is saturated at inhaled concentrations above 100 ppm. The estimated
rate of reaction (Vmax) measured in the blood was 2.7 mg/kg body
weight per hour. This rate gradually decreased during the exposure
period of 5 h, which could be due to rapid loss of cytochrome P-450
content. The Vmax in rats pretreated with 100 µl carbon tetrachloride
(oral administration, 24 h before inhalation exposure) decreased about
57%, which was in good agreement with the decrease of the cytochrome
P-450 content. (Uemitsu, 1986).
Gargas et al. (1986) calculated for carbon tetrachloride a Vmax
of 0.63 mg/kg body weight per hour in an inhalation study in male
Fischer-344 rats.
Applications of pharmacokinetic models for the inhalation
exposure of rats have provided Vmax and Km estimates in rats of 0.63
mg/h per kg and 0.25 mg/litre (Gargas et al., 1986) and 0.37 mg/h/kg
and 1.3 mg/litre (Evans et al., 1994).
Paustenbach et al. (1988) constructed a physiologically based
pharmacokinetic model (PB-PK) for inhaled carbon tetrachloride and
used this model to predict the pharmacokinetics of inhaled
[14C]carbon tetrachloride in male Sprague-Dawley rats exposed for 8
or 11.5 h/day for 1 or 2 weeks. The simulations were compared with
actual laboratory data (Paustenbach et al., 1986a,b). The model
accurately predicted the behaviour of carbon tetrachloride and its
metabolites. Metabolites were partitioned in three compartments: the
amounts to be excreted in the breath (as [14C]CO2), urine and
faeces. Of total carbon tetrachloride metabolites, 6.5, 9.5 and 84%
were formed via the pathways leading to CO2, urinary and faecal
metabolites, respectively.
The PB-PK model suggests that at concentrations up to 100 ppm
(641 mg/m3), rats, monkeys and humans metabolize and eliminate carbon
tetrachloride in a similar manner. Most species convert 2-5% to CO2,
eliminate 4-8% in the urine, and eliminate 40-50% unchanged in the
breath.
6.2 Biotransformation and covalent binding of metabolites
Metabolism of carbon tetrachloride is initiated by cytochrome
P-450-mediated transfer of an electron to the C-Cl bond, forming an
anion radical that eliminates chloride, thus forming the
trichloromethyl radical. This radical may undergo both oxidative and
reductive biotransformation (see Fig. 1). The isoenzymes implicated in
this process are CYP2E1 and CYP2B1/2B2 (Raucy et al., 1993; Gruebele
et al., 1996). Some isoforms may be preferentially susceptible to
degradation by carbon tetrachloride (Tierney et al., 1992). Evidence
that carbon tetrachloride inactivates CYP2E1 and reduces total CYP2E1
protein in a cell line that constitutively expresses human CYP2E1 has
been obtained by Dai & Cederbaum (1995). When protein synthesis was
blocked, inactivation and degradation of CYP2E1 by carbon
tetrachloride was more pronounced. Free radical scavengers were unable
to prevent CYP2E1 degradation, suggesting that carbon tetrachloride
metabolites react at the active site of CYP2E1. Antioxidants prevented
carbon tetrachloride-induced lipid peroxidation, but not CYP2E1
degradation, suggesting that these processes are disassociated.
The formation of the radical has been demonstrated convincingly
in vitro as well as in vivo in electron spin resonance experiments
and is mediated by a particular cytochrome P-450, of which the haem
moiety is destroyed after carbon tetrachloride exposure (Reiner et
al., 1972; Sipes et al., 1977; Poyer et al., 1978, 1980; Lai et al.,
1979; Tomasi et al., 1980; Fernández et al., 1982; Noguchi et al.,
1982a,b; McCay et al., 1984). The formation of carbon
tetrachloride/cytochrome P-450 complexes has been demonstrated by
Uehleke et al. (1973), Wolf et al. (1977), Ahr et al. (1980) and
Fernández et al. (1982).
The most important pathway in the elimination of trichloromethyl
radicals is the reaction with molecular oxygen, resulting in the
formation of trichloromethylperoxyl radicals (CCl3OO*), as proposed
by Packer et al. (1978), Shah et al. (1979), Mico & Pohl (1983), Pohl
et al. (1984) and McCay et al. (1984). This intermediate, which is
even more reactive than the trichloromethyl radical (Dianzani, 1984),
may interact with lipids, causing lipid peroxidation along with the
production of 4-hydroxyalkenals (Benedetti et al., 1982; Comporti et
al., 1984). Radical-induced lipid peroxidation is also a presumed
source of a variety of metabolites, such as acetone, propanal, butanal
and malondialdehyde, which appear in rat urine 24 h after exposure to
carbon tetrachloride (de Zwart et al., 1997).
It is supposed that the trichloromethylperoxyl radical will react
further to produce phosgene, which again may interact with tissue
macromolecules or with water, finally producing hydrochloric acid and
carbon dioxide (Pohl et al., 1984). Carbon tetrachloride has been
reported to be metabolised to carbon dioxide in liver homogenates by
Rubinstein & Kanics (1964). The biotransformation of carbon
tetrachloride to carbon dioxide in vivo has been reported by
Reynolds et al. (1984).
Common name: carbon tetrachloride
Common synonyms: Carbona, carbon chloride, tetrachloromethane,
carbon tet, methane tetrachloride,
perchloromethane, tetrachlorocarbon
Trade names: Benzinoform, Fasciolin, Flukoids, Freon 10,
Halon 104, Necatorina, Necatorine,
Tetrafinol, Tetraform, Tetrasol, Univerm,
Vermoestricid
CAS chemical name: tetrachloromethane
CAS registry number: 56-23-5
RTECS registry number: FG 4900000
2.2 Physical and chemical properties
The most important physical properties of carbon tetrachloride
are given in Table 1.
Table 1. Physical properties of carbon tetrachloridea
Colour colourless
Relative molecular mass 153.8
Boiling point at 101.3 kPa, 20°C 76.72 °C
Melting point at 101.3 kPa, 20°C -22.92 °C
Density (25°C) 1.594 g/ml
Table 1. (Continued)
Density of solid at - 186 °C 1831 kg/m3
- 80 °C 1809 kg/m3
Refractive index at 20 °C 1.4607
Vapour pressure at 20 °C 91.3 mmHg; 12.2 kPa
at 0 °C 32.9 mmHg; 4.4 kPa
Autoignition temperature > 1000 °C
Critical pressure 4.6 MPa
Critical temperature 283.2 °C
Solubility in water at 25 °C 785 mg/litre
Solubility of water in carbon
tetrachloride at 25 °C 0.13 g/kg
Henry's law constant at 24.8 °C 2.3 × 10-2 atm-m3/mol
Heat of evaporation 194.7 kJ/kg
Log Kow 2.64
Log Koc 2.04
a From Kenaga (1980); US EPA (1984b); Huiskamp et al. (1986);
ATSDR (1994).
Carbon tetrachloride is a volatile colourless clear heavy liquid
with a characteristic sweet non-irritant odour. The odour threshold in
water is 0.52 mg/litre and in air is > 10 ppm. Carbon tetrachloride
is miscible with most aliphatic solvents and it is a solvent for
benzyl resins, bitumen, chlorinated rubber, rubber-based gums, oils
and fats. The chemical is non-flammable and fairly stable in the
presence of air and light. Upon heating by a flame or hot metal
surface in air, toxic phosgene is produced. Thermal dissociation in
the absence of air proceeds slowly at about 400°C and is extensive at
temperatures ranging from 900 to 1300°C with the formation of
perchloroethylene, hexachloroethane and some molecular chlorine. A
mixture of carbon tetrachloride and excess of water vapour decomposes
at 250°C to carbon dioxide and hydrochloric acid. When the amount of
water in the mixture is limited, phosgene will be formed too. This
decomposition also occurs when moist or wet carbon tetrachloride is
exposed to UV radiation (253.7 nm). Like other chloromethanes, carbon
tetrachloride reacts (sometimes explosively) with aluminium and its
alloys. Similar violent reaction may occur with metals, such as
barium, magnesium and zinc, boranes and silanes, and, in the presence
of peroxides or light, with unsaturated compounds (such as ethene).
Carbon tetrachloride may be reduced to chloroform when treated with
zinc and acid, and to methane when treated with potassium amalgam and
water (Huiskamp et al., 1986).
2.3 Conversion factors
1 mg carbon tetrachloride/m3 air = 0.156 ppm at 20°C and 101.3
kPa (760 mmHg)
1 ppm = 6.41 mg carbon tetrachloride/m3
2.4 Analytical methods
Procedures used for the sampling and determination of carbon
tetrachloride in different media are summarized in Table 2.
The preferred analytical technique is gas chromatography (GC)
using either electron capture detection (ECD), ion trap detection,
flame or photo ionisation detection or mass spectrometry. Only one
method, reported by Lioy & Lioy (1983), depends on the use of
MIRAN-infrared spectrometry, a method of very poor sensitivity.
2.4.1 Sampling and analysis in air
Methods reported in Table 2 for detecting carbon tetrachloride in
air are of four types.
a) Direct measurement
These methods are simple, because the air is aspirated or
injected directly into the measuring instrument, but they can only be
used when carbon tetrachloride is present in the air at relatively
high levels.
b) Adsorption - liquid desorption
In this type of method, air samples are passed through an
activated adsorbing agent. The adsorbed carbon tetrachloride is
desorbed with an appropriate solvent and then passed through the gas
chromatograph. Activated carbon has been described as superior to
other adsorbents for adsorption. Elution from the carbon is achieved
with carbon disulfide (Morele et al., 1989; ATSDR, 1994).
c) Adsorption - thermal desorption
After adsorption on an activated adsorbing agent, the carbon
tetrachloride is thermally desorbed and driven into the gas
chromatograph.
Table 2. Sampling and analysis of carbon tetrachloridea
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Air aspiration velocity: 28 l/min MIRAN infrared 400 µg/m3 Lioy & Lioy,
optical path: 20 m spectrometry 1983
Air direct injection GC with 2 ECD's 0.4 µg/m3 8 ml injected Lillian &
in series (estimated) Singh, 1974
Air direct injection GC - ECD 0.2 µg/m3 2 ml injected BIT-SC, 1976
Air direct injection GC - ECD 0.06 µg/m3 5 ml injected Lasa et al.,
1979
Air direct injection, methane GC - ECD 0.01 µg/m3 12 ml injected thorough purification of Makide &
added carrier gas and apparatus Yokohata, 1983
required
Air adsorption on Porapak-N GC - ECD 1 µg/m3 20 litres advantage of using Van Tassel et
liquid desorption (methanol) methanol over CS2 is the al., 1981
absence of a background
signal in the ECD
Air adsorption on activated GC - ECD 0.2 µg up to 30 litres activated charcoal shown Morele et al.,
charcoal, liquid desorption can be sampled to be more efficient 1989
(ethanol) trichloroethylene trapping material than
used as IS XADs, Tenax or
Chromosorbs
adsorption on activated GC - FID ca. 0.15 mg
charcoal liquid desorption (detector
(CS2) methylcyclohexane sensitivity)
used as IS
Table 2. (Continued)
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Air adsorption on activated GC - FID 0.01 mg 5-15 litres NIOSH, 1977,
charcoal, liquid desorption 1984
(CS2)
Air adsorption on Chromosorb GC - ECD 0.003 µg/m3 20 ml Makide et al.,
102 or Silicone OV 101 (at 1979
-35 °C), thermal desorption
Air adsorption on Porapak-N, GC - ECD 0.005 µg/m3 0.3-3 litres confirmation of results by Russell &
thermal desorption at 200 °C use of GC - MS Shadoff, 1977
Air adsorption on Chromosorb GC - ECD 0.01 µg/m3 1 litre Elias, 1977
102, thermal desorption at (collection tube (estimated)
200 °C already connected
to GC)
Air adsorption on Carbopak-B at GC - ECD 0.01 µg/m3 1 litre calibration with Crescentini et
78 °C, thermal desorption permeation tubes al., 1981
Air adsorption on Chromosorb-102 GC - ECD - FID ca. 0.06 µg/m3 1-3 litres Heil et al.,
and activated charcoal, (2 detectors in 1979
thermal desorption at 150 °C parallel)
Air adsorption on Tenax-GC, GC - MS 0.2 µg/m3 20 litres compounds were Krost et al.,
thermal desorption at 270 °C cryofocused 1982
Air adsorption on Carbopak-C, GC - MS 0.1 µg/m3 300 ml Crescentini et
thermal desorption at 100 °C al., 1983
Air adsorption on activated GC - ECD followed 0.7 µg/m3 24 h sample Coutant &
charcoal, liquid desorption by a PID Scott, 1982
(5% CS2 in methanol)
Table 2. (Continued)
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Air cold trap (liquid oxygen), GC - ECD 0.006 µg/m3 30 ml aliquot measurement of air Harsch &
heating in trap samples from the Cronn, 1978
stratosphere
Air injection in cold trap, heating GC - MS (SIM) 0.04 µg/m3 100 ml Cronn &
Harsch, 1979
Air cold trap (-173 °C), heating to GC - PID - ECD - 0.006 µg/m3 0.5-1.7 litres column is kept at -103 °C Rudolph &
257 °C FID (3 detectors (cryofocusing) Jebsen, 1983
in series)
Water dibromomethane used as IS GC - ECD 0.001 µg/litre 500 µl injected suitable for routine Herzfeld et al.,
analysis of river waters 1989
Water direct aqueous injection GC - MS (SIM) 2 µg/litre 10 µl injected Fujii, 1977
Water direct aqueous injection GC - ECD 0.015 µg/litre 2 µl injected suitable for halocarbons Grob, 1984
in water in the 0.01 to
10 µg/litre range
Water direct aqueous injection, GC - ECD 0.05 µg/litre 5-20 µl Simmonds &
water removal by injected Kerns, 1979
permeaselective membrane
Water liquid-liquid extraction GC - ECD 0.10 µg/litre 10-20 ml Van Rensburg
(using hexane) et al., 1978
Water liquid-liquid extraction GC - ECD 0.2 µg/litre Inoko et al.,
(using xylene) 1984
Water liquid-liquid extraction GC - ECD 0.05 µg/litre Kroneld, 1985
(using pentane)
Table 2. (Continued)
Medium Sampling method Analytical method Detection limit Sample size Comments Reference
Water purge and trap technique, GC - ITD 0.1 µg/litre 5 ml Eichelberger
thermal desorption, et al., 1990
fluorobenzene as IS
Grain codistillation of carbon GC - ECD 1 µg/kg De Vries et al.,
tetrachloride in food sample 1985
and mixture of
1,2-dichloropropane and
1,2-dibromopropane as IS in
hexane
Adipose purge and trap technique GC - MS < 1.3 µg/litre 200-500 mg Peoples et al.,
tissue (Tenax-silica gel), thermal liquefied fat 1979
desorption samples
Blood purge and trap technique GC - MS < 1.3 µg/litre 0.5 ml water- Peoples et al.,
(Tenax-silica gel), thermal serum sample 1979
desorption
Blood warming and passing an inert GC - MS 3 µg/litre 10 ml sample Pellizzari
gas, vapours trapped on et al., 1985
Tenax-GC, thermal desorption
Urine liquid-liquid extraction using GC - ECD (20% < 1 µg/litre 10 ml sample Youssefi et al.,
pentane (adding 2.6 g SP-2100/0.1% 1978
ammonium carbonate) Carbowax 1500
column)
Fish extraction with pentane and GC - ECD 0.1 µg/kg in Baumann
isopropanol, with fresh material Ofstad et al.,
bromotrichloromethane used as IS 1981
a Abbreviations: GC = gas chromatography; MS = mass spectrometry; ECD = electron capture detector; SIM = single (selected) ion monitoring;
FID = flame ionisation detector; ITD = ion trap detector; PID = photo ionisation detector; IS = internal standard.
d) Cold trap - heating
In this type of procedure, air samples are injected into a cold
trap. The trap is then heated and the carbon tetrachloride content
transferred into the column of a gas chromatograph.
2.4.2 Sampling and analysis in water
Several methods for sampling analysing the carbon tetrachloride
content in water are included in Table 2. Most of these methods are
based on direct injection techniques or on liquid-liquid extraction by
means of a non-polar non-halogenated solvent.
2.4.3 Sampling and analysis in biological samples
2.4.3.1 Blood and tissues
Peoples et al. (1979) developed a method to determine carbon
tetrachloride in adipose tissue and blood. In both cases the carbon
tetrachloride is purged and trapped on Tenax-silica gel and determined
by mass spectrometry after thermal desorption.
Pellizzari et al. (1985) similarly passed an inert gas over a
warmed plasma sample with adsorption of the vapour on a Tenax-GC
cartridge, and then recovered the carbon tetrachloride by thermal
desorption.
2.4.3.2 Urine
The only method listed in Table 2 for measuring carbon tetra
chloride concentrations in urine is based on an extraction technique
with pentane and direct gas chromatographic analysis of the pentane
extract (Youssefi et al., 1978).
2.4.3.3 Fish
Baumann Ofstad et al. (1981) developed a method for the analysis
of volatile halogenated hydrocarbons in biological samples and used
this method for the analysis of fish samples. It should be noted that
the identification and quantification of carbon tetrachloride is
especially vulnerable to contamination, so the practical usefulness of
this method is very limited.
2.4.4 Sampling and analysis in foodstuffs
A method for the determination of 22 compounds (including carbon
tetrachloride) in a variety of foods was described by Daft (1988). In
this method the samples are extracted with isooctane, and cleaned up
according to fat content and food type. Most samples (6-10 µl) are
injected for GC with ECD and Hall-electron conductivity detection
immediately following the initial extraction or dilution.
De Vries et al. (1985) provided a method for analysis of carbon
tetrachloride in grain and grain-based products containing 1-2000
µg/kg. A food sample is mixed with water and an internal standard
mixture of 1,2-dichloropropane and 1,2-dibromopropane is added. The
water is then distilled until 1 ml has been collected under hexane.
The hexane is then separated, dried and injected (2 µl) into the GC
column.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
It has been suggested that carbon tetrachloride can be formed in
the troposphere by the solar-induced photochemical reactions of
chlorinated alkenes (Singh et al., 1975). However, so far this
reaction has only been demonstrated in the laboratory, and, even if it
could happen in nature, it is not certain that it would be a major
source of environmental carbon tetrachloride. Carbon tetrachloride has
been detected in volcanic emission gases (Isidorov et al., 1990).
Several studies have shown that global atmospheric levels of carbon
tetrachloride can be explained by anthropogenic sources alone (Singh
et al., 1976).
3.2 Anthropogenic sources
3.2.1 Production
3.2.1.1 Direct production and procedures
Production of carbon tetrachloride began in about 1907 in the
USA. It can be produced by chlorination of methane, methanol, carbon
disulfide, propane, 1,2-dichloroethane and higher hydrocarbons.
The world production of carbon tetrachloride ranged from 850 to
960 kilotonnes over the years 1980-1988. Table 3 provides some data on
past production and production capacities of carbon tetrachloride.
These data are based on information in the ECDIN database (ECDIN,
1992) and BUA-Stoffbericht 45 (BUA, 1990).
Since 1990 the production of carbon tetrachloride has dropped.
The Montreal Protocol of 1990 and its subsequent amendments
established the phase-out by 1996 of production and use of carbon
tetrachloride and of chlorofluorocarbons (CFCs) by major manufacturing
countries. Special conditions were allowed for developing countries,
where consumption of controlled substances under Annex B (including
carbon tetrachloride) was required to be reduced by 85% of its
1998-2000 average level (or a calculated consumption level of 0.2 kg
per capita, whichever is lower) by 2005 and completely stopped by 2010
(UNEP, 1996).
3.2.1.2 Indirect production
Carbon tetrachloride can be produced as a by-product during the
manufacture of other products and compounds (US EPA, 1984a) and during
wood pulp bleaching.
Table 3. Past production and production capacity of carbon
tetrachloride
Country Year Production Capacity
(in kilotonnes) (in kilotonnes)
France 1988 - 90
Italy 1987 95 -
1988 - 130
Germany (former FRG) 1985 150 -
1987 180 -
1988 170 180
EEC 1985 - 520
1987 480 -
1988 478 540
Japan 1985 - 72
1987 52 -
1988 - 70
United Kingdom 1988 - 75
USA 1986 286 -
1987 340 -
1988 - 281
1991 143 -
World 1985 - 1200
1987 960 -
1988 - 1100
3.2.1.3 Emissions
According to US EPA (1991), in 1989 approximately 2000 tonnes of
carbon tetrachloride were released during manufacturing and processing
to the air in the USA. US EPA (1984a) reported emission factors for
carbon tetrachloride arising during the chlorination of hydrocarbons
ranging from 0.9 kg/tonne of carbon tetrachloride (controlled) to 2.8
kg/tonne of carbon tetrachloride (uncontrolled). Furthermore,
emissions may result from industrial water treatment or from old
landfill sites.
3.2.2 Uses
Most of the carbon tetrachloride produced is used in the
production of CFCs, which were primarily used as refrigerants,
propellants, foam-blowing agents and solvents and in the production of
other chlorinated hydrocarbons.
The use of carbon tetrachloride increased in the EEC as well as
in the USA during the years 1980-1987. However, this use has decreased
in recent years due to the Copenhagen Amendment to the Montreal
Protocol (1992) (UNEP, 1996). A survey in Japan could detect no use of
carbon tetrachloride in small to medium scale industries in 1996 (Ukai
et al., 1997).
Carbon tetrachloride has been used as a grain fumigant,
pesticide, solvent for oils and fats, metal degreaser, fire
extinguisher and flame retardant, and in the production of paint, ink,
plastics, semi-conductors and petrol additives. It was previously also
widely used as a cleaning agent. All these uses have tended to be
phased-out as production has dropped (ECDIN, 1992; ATSDR, 1994).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Transport
Carbon tetrachloride introduced into water resources is
transported by movement of surface water and groundwater. Because of
its volatility, evaporation is considered to be the main process for
the removal of carbon tetrachloride from aquatic systems. The amount
of carbon tetrachloride dissolved in the oceans is reported to be less
than 1-3% of that in the atmosphere (Galbally, 1976; Singh et al.,
1976). Practically all the carbon tetrachloride released to the
environment is thus present in the atmosphere (US EPA, 1991). Because
carbon tetrachloride does not degrade readily in the atmosphere,
significant global transport is expected.
Following releases to soil, most carbon tetrachloride is expected
to evaporate rapidly due to its high vapour pressure. A small fraction
of carbon tetrachloride may adsorb to organic matter, based on a
calculated soil adsorption coefficient of 100 (log Koc = 2.04)
(Kenaga, 1980).
Walton et al. (1992) studied the adsorption of carbon
tetrachloride from solution onto two soils, a silt loam (1.49% organic
carbon) and a sandy loam (0.66% organic carbon). The soil was shaken
with several concentrations of carbon tetrachloride (100 to 650 mg/kg
soil) for 18 h. The Koc values determined were 143.6 for the silt
loam and 48.9 for the sandy loam. Duffy et al. (1997) studied the
downward movement of carbon tetrachloride in 3 horizons of a fine
montmorillonitic soil. Koc values of 55, 77.6 and 269 were calculated
for the modern A, buried A and loess C soil horizons. However, the
authors point out that Koc values are unreliable in soils with low
organic carbon and high clay content. Therefore, the highest Koc
value should be treated with some caution.
4.1.2 Distribution
The evidence that the residence time of carbon tetrachloride in
the atmosphere is long (see section 4.1.3) and that nearly all of the
compound is found in this compartment explains the relatively even
distribution over the globe as is recorded in Table 4.
Only a very small proportion of carbon tetrachloride will remain
in water and soil.
Table 4. Levels of carbon tetrachloride in air
Location Year Mean level (µg/m3) Reference
(range in
parentheses)
Northern 1974 0.71 Cox et al. (1976)
hemisphere 1976 0.73 Singh et al. (1977)
1977 0.78 Singh et al. (1979)
1979-1981 0.87 Singh et al. (1983)
Southern 1974 0.44 Cox et al. (1976)
hemisphere 1977 0.76 Singh et al. (1979)
1979-1981 0.82 Singh et al. (1983)
North-West 1975-1980 0.91 Rasmussen et al.
Pacific (0.83-0.99) (1981)
1975-1985 0.77 Rasmussen & Khalil
(0.67-0.83) (1986)
1976 0.78 Cronn et al. (1977)
Antarctica 1975-1980 0.8 Rasmussen et al.
(0.77-0.87) (1981)
1975-1985 0.69 Rasmussen & Khalil
(0.62-0.76) (1986)
Arctic 1982 0.97 Rasmussen & Khalil
(1983)
North Atlantic 1983 0.56 von Düszeln &
Thiemann (1985)
North America 1976 0.86 Pierotti et al.
(0.33-0.99) (1980)
California, USA 1976 0.76-0.86 Singh et al.
(0.66-1.85) (1977)
Bochum, Germany 1978 0.8 (0.1-1.2) Bauer (1981)
Germany (cities) 1980-1981 0.6 von Düszeln &
Thiemann (1985)
Table 4. (Continued)
Location Year Mean level (µg/m3) Reference
(range in
parentheses)
South-West 1986-1988 0.5-0.6 Frank & Frank
Germany (1990)
The Netherlands 1980 0.83-1.0 Guicherit &
(max. 2.2-3.2) Schulting (1985)
Turin, Italy 1988 0.96 (0.17-1.94)a Gilli et al. (1990)
1988 0.47
(0.19-1.17)b Gilli et al. (1990)
Japan 1979 0.69
(0.62-0.72) Makide et al. (1979)
1994-1995 0.53c Sugama et al. (1995)
a cold months
b warm months
c concentrations were higher in winter than during summer
4.1.3 Removal from the atmosphere; global warming potential
The troposphere to stratosphere turnover time has been estimated
at around 30 years (Versar Inc., 1979). This is a shorter period of
time than is estimated for the degradation processes of carbon
tetrachloride in the troposphere (see section 4.2). Therefore
tropospheric carbon tetrachloride will attain significant
concentration in the stratosphere.
Cupitt (1980) calculated that deposition of carbon tetrachloride
from the atmosphere will be very slow.
Estimates of the atmospheric lifetime (the overall persistence of
carbon tetrachloride in the troposphere and the stratosphere combined)
are variable, but most values range from 25 to 100 years (Molina &
Rowland, 1974; Galbally, 1976; Singh et al., 1979; Edwards et al.,
1982a,b; Simmonds et al., 1983, 1988; Rowland, 1985; Huiskamp et al.,
1986; Howard, 1990; IPCC, 1990, 1995; WMO, 1991) with 45-50 years
generally being accepted as the most reasonable value.
The Global Warming Potential (GWP) of carbon tetrachloride,
relative to CO2, is estimated (IPCC, 1995) to be 2000, 1400 and 500
at integration time horizons of 20, 100 and 500 years. Its
contribution to total warming may be 0.3% as integrated effect over a
time horizon of 100 years (IPCC, 1995). Relative to CFC 12, the GWP of
carbon tetrachloride has been estimated to be 0.12 (UNEP, 1989).
4.1.4 Removal from water
The major removal process from water is volatilization to the
atmosphere. This was indicated by laboratory tests performed by
Dilling et al. (1975). These tests showed that a 1 ppm concentration
of low-molecular-weight chlorinated hydrocarbons will not persist in
agitated natural water bodies due to evaporation. In 29 min 50% of the
amount of carbon tetrachloride was evaporated, and in 97 min 90% was
evaporated. Zoeteman et al. (1980) calculated a half-life of carbon
tetrachloride in rivers of 0.3-3 days and in lakes and groundwaters of
30-300 days.
4.1.5 Removal from soil
Anderson et al. (1991) studied the loss of carbon tetrachloride
from two different soil types, a silt loam (1.49% organic carbon) and
a sandy loam (0.66% organic carbon). Carbon tetrachloride was applied
to the soil at a concentration of 100 mg/kg (dry weight) and the soil
was incubated in the dark at 20°C for 7 days. The mean half-life for
disappearance of carbon tetrachloride was 5 days. There was no
significant difference between the loss from sterile and non-sterile
systems indicating that volatilization was the likely removal process.
Jury et al. (1984) predicted that carbon tetrachloride would have
a volatilization half-life of 0.2 days at a depth of 1 cm and 0.8 days
at a depth of 10 cm in soil, based on volatilization tests and
assuming a uniform distribution of the chemical with depth.
4.2 Abiotic degradation
4.2.1 Degradation in atmosphere
4.2.1.1 Photodegradation
Carbon tetrachloride is very stable in the troposphere (Lillian
et al., 1975; Cox et al., 1976; Singh et al., 1980). This is primarily
because carbon tetrachloride, in contrast to most other volatile
halocarbons, has low reactivity towards hydroxyl radicals. This is
evident from rate constants determined by several authors (Howard
Carleton & Evenson, 1976; Cox et al., 1976; Clyne & Holt, 1978). Based
on these rate constants, half-lives of > 3.9 to 137 years can be
calculated for the decomposition of carbon tetrachloride in the
troposphere (Lyman et al., 1982).
Cox et al. (1976) found an even higher tropospheric half-life of
> 330 years.
4.2.1.2 Photolysis
Edwards et al. (1982b) estimated a lifetime in the troposphere
due to photolysis of the order of 500 years.
The principal degradation process for carbon tetrachloride occurs
in the stratosphere, where it is dissociated by short wave length
(190- 220 nm) UV radiation to form the trichloromethyl radical and
chlorine atoms. Simmonds et al. (1983) estimated a half-life of 18-80
years for this photodissociation process.
4.2.1.3 Ozone-depletion potential
The chlorine atoms in carbon tetrachloride interact with oxygen
or ozone to produce ClO* groups (Singh et al., 1975). The chlorine
atoms and ClO* groups attack the surrounding ozone in a reaction in
which they act as catalysts until scavenged by some other chemical
reaction (Isaksen & Stordal, 1981; Rowland, 1985; Ember et al., 1986).
This effect is reflected in an ODP (ozone depletion potential) of 1.08
(WMO, 1991) and 1.1 (UNEP, 1996), compared with the chlorofluorocarbon
CFC-11, and was responsible for the inclusion of carbon tetrachloride
in the amended Montreal Protocol of 1990 (UNEP, 1996).
Catalytic breakdown of ozone by chloride-containing radicals:
CCl4 + h nu -> *CCl3 + *Cl
*CCl3 + O2 -> -> COCl2 + ClO*
*Cl + O3 -> ClO* + O2
ClO* + O -> *Cl + O2
4.2.2 Degradation in water
Carbon tetrachloride dissolved in water does not photodegrade or
oxidize in any measurable amount (Howard et al., 1991). The rate of
hydrolysis was thought to be second order with respect to carbon
tetrachloride with a calculated half-life of 7000 years at a
concentration of 1 ppm (Mabey & Mill, 1978). However, Jeffers et al.
(1996) found that the rate of hydrolysis for dilute solutions of
carbon tetrachloride was first-order and estimated the half-life to be
40 years. The authors reanalysed data previously stated as
second-order kinetics and found it to be consistent with a first-order
rate of hydrolysis.
4.2.3 Other degradation processes
Photodissociation of carbon tetrachloride adsorbed on to
silicates has been observed in the laboratory by Ausloos et al.
(1977).
Gäb et al. (1980) found experimentally that carbon tetrachloride
degraded over sand, silica gel and Al2O3. The degradation rate
depended, among other factors, on the laboratory conditions. Under the
conditions representative for deserts, degradation was about 4.5%
after exposure for 115 days.
4.3 Biotic degradation
4.3.1 Aerobic
Carbon tetrachloride has been shown to be resistant to aerobic
biodegradation by mixed bacterial cultures growing on methane as the
carbon source. No degradation of carbon tetrachloride was observed in
a mixed culture of methane-utilizing bacteria isolated from soil and
incubated in the dark for 6 days (Cochran et al., 1988). Oldenhuis et
al. (1989) reported no degradation of carbon tetrachloride by the
methanotrophic bacterium Methylosinus trichlosporium in the presence
of formate and oxygen.
Vannelli et al. (1990) found that carbon tetrachloride was not
degraded by the ammonia-oxidizing bacterium
Nitrosomonas europea when incubated at 1 mg/litre for 24 h.
In contrast, Tabak et al. (1981) found carbon tetrachloride to be
significantly degradable under aerobic conditions, with rapid
adaptation. Carbon tetrachloride (5 and 10 mg/litre) was incubated at
25°C for 7 days in static culture containing yeast extract inoculated
with settled domestic wastewater. Eighty to eighty-seven per cent of
the initial concentration disappeared within 7 days in the first
culture. An abiotic control showed that 5-23% of this loss could be
due to volatilization. In three subsequent cultures, carbon
tetrachloride was degraded to concentrations below the detection limit
(< 0.1 mg/litre) in the same period.
4.3.2 Anaerobic
The biodegradation of carbon tetrachloride has been studied under
methanogenic conditions. In batch cultures, carbon tetrachloride at a
concentration of 200 µg/litre was incubated in the dark at 35°C with
mixed methanogenic bacteria derived from a laboratory-scale digester
fed with activated sludge. Carbon tetrachloride was found to be
degraded to below the detection limit (< 0.1 µg/litre) within 16
days; carbon dioxide was the only degradation product identified. In a
continuous-flow column study, columns were initially seeded with an
inoculum of methanogenic bacteria from rum distillery wastewater.
Acetate (100 mg/litre) was fed to the column as primary growth
substrate and carbon tetrachloride was fed as a secondary substrate.
The column had a 2 day retention time, and it was found that carbon
tetrachloride was 99% degraded in the column; carbon dioxide being the
major degradation product (Bouwer & McCarty, 1983a).
Bouwer & McCarty (1983b) studied the biodegradation of carbon
tetrachloride under denitrifying conditions. Using batch cultures
seeded with primary sewage effluent and containing nitrate as an
electron acceptor, carbon tetrachloride (75 µg/litre) was found to be
degraded rapidly with no detectable lag period when incubated in the
dark at 25°C for 8 weeks. Chloroform and carbon dioxide were the
degradation products identified.
The biodegradation of carbon tetrachloride using aquifer material
has been studied (Parsons et al., 1985). Microcosms were constructed
containing groundwater and sediment contaminated with trichloroethene.
The concentration of carbon tetrachloride was 4 mg/litre and
incubation was carried out in the dark at 25°C. Reductive
dehalogenation of carbon tetrachloride to chloroform was found to
occur, and 700 µg chloroform/litre was detected after 8 weeks.
Egli et al. (1987) observed that pure cultures of
Desulfobacterium autotrophicum dechlorinated carbon tetrachloride to
trichloromethane and dichloromethane within 6 days.
Klecka & Gonsior (1984) provided evidence that reductive
dehalogenation of carbon tetrachloride in aqueous solution under
anaerobic conditions could be achieved with naturally occurring iron
porphyrins and other reducing agents. Carbon tetrachloride (1
mg/litre) was rapidly degraded to chloroform when incubated at 25°C
with an iron porphyrin (haematin) and sulfide.
Bioremediation studies have shown that anaerobic biodegradation
is enhanced by increasing the concentration of primary substrates
(such as glucose and acetate) and by lowering the redox potential
(providing a relatively higher electron activity which facilitates
dechlorination) (Doong & Wu, 1995, 1996; Doong et al., 1996; Jin &
Englarde, 1996).
4.4 Bioaccumulation
The log octanol-water partition coefficient (Kow) of carbon
tetrachloride is 2.64 indicating a moderate potential for
bioaccumulation under conditions of constant exposure. However,
studies have shown that the compound's short tissue lifetime reduces
this tendency. Barrows et al. (1980) reported a bioconcentration
factor of 30 for bluegill sunfish (Lepomis macrochirus) with a
tissue half-life of less than one day. A similar bioconcentration
factor of 30 (whole body; fresh weight) was reported by Veith (1978)
in bluegill. Neely et al. (1974) found a bioconcentration factor of
17.7 for muscle tissue of rainbow trout (Oncorhynchus mykiss). A
higher bioconcentration factor of 300 (wet weight) has been measured
for carbon tetrachloride in the green alga Chlorella fusca exposed
to 50 µg/litre for at least 24 h (Geyer, 1984). No significant
bioaccumulation in marine food chains was found in an extensive study
by Pearson & McConnell (1975) (see Table 6, section 5.1.4).
Some plants, due to their lipid content, take up carbon
tetrachloride from the air. Thus studies of the equilibrium
partitioning of carbon tetrachloride between the gas phase and conifer
needles (Pinus sylvestris and Picea abies) on the one hand and
hexane-extractable leaf waxes on the other hand showed partition
ratios (g/m3 needle; g/m3 air) of 9-17 and 90-400, respectively
(Frank & Frank, 1986; Brown et al., 1998).
5. CONCENTRATIONS IN THE ENVIRONMENT AND EXPOSURE
5.1 Environmental levels
5.1.1 Air
Reported concentrations of carbon tetrachloride measured in
ambient air are presented in Table 4.
As seen in Table 4, mean global levels of atmospheric carbon
tetrachloride usually lie in the range of 0.5-1.0 µg/m3. Based on an
analysis of 4913 ambient air samples (including remote, rural,
industrial and source-dominated sites in the USA), the average
concentration of carbon tetrachloride was 1.1 µg/m3 (Shah &
Heyerdahl, 1988). Urban atmospheric carbon tetrachloride levels and
levels in industrial areas can be considerably higher as shown by the
measurements by Lillian et al. (1975), Singh et al. (1980, 1982) and
Bozzelli & Kebbekus (1982). These authors reported mean levels of 2-3
µg/m3 (several hundred measurements) with maximum levels up to 6
µg/m3. Near a production facility in the United Kingdom, Pearson &
McConnell (1975) recorded levels an order of magnitude higher.
It has been estimated that concentrations of carbon tetrachloride
were increasing worldwide until recently (Simmonds et al., 1988;
Howard, 1990). The Intergovernmental Panel on Climate Change (IPCC)
has estimated the atmospheric concentration to be about 0.94 µg/m3
and the annual rate of increase to be 1.5% (IPCC, 1990). However, the
accumulation of the substance in the atmosphere seems to have stopped
(Fraser et al., 1994) and even started to decline (Fraser & Derek,
1994).
5.1.2 Water
Some reported aquatic concentrations of carbon tetrachloride are
summarized in Table 5.
As seen in Table 5, remote oceanic levels of carbon tetrachloride
are usually in the range of 0.0005-0.0008 µg/litre. As sites nearer to
effluent sources are examined, higher levels are observed. Thus in
estuaries, levels from 0.01 to 2.7 µg/litre have been observed, and in
remote freshwater sites from 0.0002 to 0.025 µg/litre, while nearer to
industrial facilities mean levels in the range of < 0.1-24.2 µg/litre
have been recorded.
Even higher values, e.g., 160-1500 µg/litre in the River Rhine
and a mean of 75 µg/litre in the River Main, recorded in 1976 in
Germany, were the result of direct waste release (BUA, 1990).
Groundwater levels range from undetectable to a maximum of 80
µg/litre.
Table 5. Levels of carbon tetrachloride in surface water
Area Mean level (µg/litre)
(range in parentheses) Reference
Marine
East Pacific ocean 0.0005 Su & Goldberg
(1976)
East Pacific ocean 0.0007 Singh et al. (1983)
Arctic ocean 0.0008 Fogelqvist (1985)
Estuarine
Scheldt Estuary, 0.01-0.02a van Zoest & van
The Netherlands (max. 0.29) Eck (1991)
Mersey Estuary, UK 2.7 Edwards et al.
(1982a)
Freshwater
Lake Zurich, Switzerland 0.025 (0.02-0.035) Giger et al. (1978)
Lake Ontario, Canada (< 0.0002-0.005) Kaiser et al. (1983)
Niagara River, Canada 0.0029 (max. 0.018) Kaiser et al. (1983)
River Weaver, UK < 0.1 Rogers et al. (1992)
River Gowry, UK 0.9 Rogers et al. (1992)
River Rhine, Lobith, 1.5 (0.4-2.8) Bauer (1981)
Germany
Manchester Ship Canal, UK 3.8 Edwards et al.
(1982a)
Manchester Ship Canal, UK 24.2 Rogers et al. (1992)
Groundwater
Zurich (industrial area) (< 0.05-3.6) Giger et al. (1978)
Birmingham aquifer, UK (0.02-1) Rivett et al.
(1990a,b)
Coventry aquifer, UK 4.9 (max. 80) Burston et al.
(1993)
Washington, New Jersey, (ND-34) Suffet et al. (1985)
USA
Gibbstown, New Jersey, (1.4-1.8) Rosen et al. (1992)
USA
a range of medians
Based on analysis of data from STORET database, carbon
tetrachloride was detectable in 1063 of 8858 ambient water samples,
with a median concentration of 0.1 µg/litre (Staples et al., 1985).
Rain water and snow concentrations of carbon tetrachloride are
generally in the range of 0.3 to 2.8 µg/litre (Su & Goldberg, 1976),
but a level as high as 300 µg/litre was observed in rainwater
collected near a production site in the United Kingdom (Pearson &
McConnell, 1975).
5.1.3 Soil and sediment
Carbon tetrachloride might occur in soil due to spills, runoff
and leaching. However, only 0.8% of 361 measured soil/sediment samples
appeared to contain carbon tetrachloride. The concentration was
reported to be less than 5.0 mg carbon tetrachloride/kg dry weight
soil or sediment (Staples et al., 1985).
5.1.4 Biota
Levels of carbon tetrachloride in biota are summarized in Table
6.
5.2 General population exposure
The general population can be exposed to carbon tetrachloride
through air, foodstuffs and drinking-water.
5.2.1 Outdoor air
Levels in ambient air to which the general population may be
exposed are recorded in Table 4.
5.2.2 Indoor air
Because of its volatility, carbon tetrachloride tends to
volatilize from tap water. Although, human exposure by inhalation of
carbon tetrachloride transferred to the indoor air from showers and
baths, toilets, washing and cooking is conceivable, no experimental
data have been reported (McKone, 1987).
Several reports on carbon tetrachloride levels in dwellings have
been published. Taketomo & Grimsrud (1977) found values ranging
between 0.6 and 1.3 µg/m3 for various types of dwellings, which is in
agreement with the maximum indoor concentration of 1.2 µg/m3 reported
by Clark (1981) and the range of 0.9-1.8 µg/m3 found in a US EPA
study. In addition, several measurements have been made in garages,
shops, supermarkets, swimming pools, restaurants, etc. (Taketomo &
Grimsrud, 1977; Ullrich, 1982). The observed concentrations usually
ranged between 0.6 and 2.0 µg/m3. The highest concentration, 10
µg/m3, was found in a dry-cleaning establishment.
Wallace (1986) reported typical concentrations in homes in
several cities in the USA of about 1 µg/m3; a maximum value of 9
µg/m3 was found. Shah & Heyerdahl (1988) found an average carbon
tetrachloride level of 2.6 µg/m3, based on 2120 indoor samples. It
should be noted, however, that carbon tetrachloride was not detected
in more than half the samples.
Table 6. Levels of carbon tetrachloride in biota
Organism Location Organ Level (µg/kg) Reference
Plankton Liverpool Bay, UK whole body 0.04-0.09 wet weight Pearson & McConnell (1975)
Molluscs Firth of Forth, UK whole body 2 wet weight Pearson & McConnell (1975)
Liverpool Bay, UK whole body 0.4-1 wet weight Pearson & McConnell (1975)
Thames Estuary, UK whole body 0.1-0.9 wet weight Pearson & McConnell (1975)
Irish Sea muscle 5-28 dry weight Dickson & Riley (1976)
digestive tissue 8-20 dry weight Dickson & Riley (1976)
gill 14 dry weight Dickson & Riley (1976)
ovary 16 dry weight Dickson & Riley (1976)
mantle 2-114 dry weight Dickson & Riley (1976)
Crustaceans Firth of Forth, UK whole body 1-3 wet weight Pearson & McConnell (1975)
Liverpool Bay, UK whole body 3-5 wet weight Pearson & McConnell (1975)
Thames Estuary, UK whole body 0.2 wet weight Pearson & McConnell (1975)
Fish Liverpool Bay, UK flesh 2 wet weight Pearson & McConnell (1975)
liver ND-0.3 wet weight Pearson & McConnell (1975)
Thames Estuary, UK flesh 0.3-6 wet weight Pearson & McConnell (1975)
Irish Sea brain 15-191 dry weight Dickson & Riley (1976)
gill 3-209 dry weight Dickson & Riley (1976)
gut 9-44 dry weight Dickson & Riley (1976)
liver 4-51 dry weight Dickson & Riley (1976)
muscle 7-83 dry weight Dickson & Riley (1976)
skeletal tissue 7-22 dry weight Dickson & Riley (1976)
heart 10-40 dry weight Dickson & Riley (1976)
ND = not detected.
5.2.3 Drinking-water
The National Organics Monitoring Survey (NOMS) in the USA
detected carbon tetrachloride (range of 2.4-6.4 ng/litre) in public
drinking-water systems in 10% of the 113 samples surveyed (US EPA,
1984b). In 30 out of 954 drinking-water samples from various cities in
the USA carbon tetrachloride could be detected. Median concentrations
in different groups ranged from 0.3 to 0.7 µg/litre while maximum
concentrations reached 16 µg/litre (Westrick et al., 1984). Bauer
(1981) reported that drinking-water in Germany contained an average of
less than 0.1 µg/litre although a maximum level of 1.4 µg/litre was
found (average of 100 towns in 1977). Lahl et al. (1981) reported a
carbon tetrachloride concentration less than 0.1 µg/litre in the
drinking-water of 50 cities in Germany. In the United Kingdom,
< 0.01-2.3 µg/litre was measured in drinking-water (Reynolds et al.,
1982; Reynolds & Harrison, 1982).
Values as high as a median of 3 µg/litre and a maximum of 39.5
µg/litre were reported in Galicia, Spain (Freiria-Gándara et al.,
1992).
5.2.4 Foodstuffs
According to investigations carried out in Europe and USA between
1973 and 1989, many foodstuffs contained carbon tetrachloride at
concentrations of a few µg/litre or µg/kg.
The following concentrations of carbon tetrachloride in
foodstuffs in the United Kingdom in 1973 were reported: meat, 7-9
µg/kg; edible oils, 16-18 µg/kg; tea, 4 µg/kg; and fruits and
vegetables, 3-8 µg/kg (McConnell et al., 1975). Values in a similar
range were found for dairy products, other edible oils, fats,
beverages, other fruits and bread, but here carbon tetrachloride and
1,1,1-trichloroethane could not be separated (McConnell et al., 1975).
According to a study conducted in Germany, carbon tetrachloride
can be present in decaffeinated coffee (4.9-60 µg/kg), milled cereal,
flour and starch products (levels in 21 samples ranged from less than
0.1 to 26 µg/kg). The origin in the first case is the
caffeine-extraction procedure, and in the second case in all
probability fumigation of the raw cereals. The use of carbon
tetrachloride for fumigation of stored foodstuffs and decaffeination
of coffee appears to have generally ceased and it is unlikely that its
occurrence in food stuff will be of significance. Less than 1 µg/kg
was found in sugar, fruit, vegetables, beverages, bread, toast,
potatoes, olives, oils, milk, butter, eggs, yoghurt, (cream) cheese,
meat and fish. In cough mixtures 0.1 to 1.8 µg/kg was found (Bauer,
1981).
Entz et al. (1982) and Entz & Hollifield (1982), in analyses of
various foods for a series of volatile halogenated hydrocarbons, did
not find carbon tetrachloride at a detection limit of 0.5 to 3 µg/kg,
depending on the type of product. Decaffeinated coffee and flour
products were not included in the studies.
Kroneld (1989) detected carbon tetrachloride in meat (0.9 µg/kg),
fish (0.6 µg/kg) and juice (0.3 µg/kg) in Finland in 1987.
Carbon tetrachloride levels in table-ready foods in the USA were
reported by Heikes (1987). He found up to 2.2 µg/kg in four sorts of
cheese, 0.10-0.34 µg/kg in cereals, 1.7-5.7 µg/kg in fish sticks and
up to 6.0 µg/kg in butter.
In a survey by Daft (1989, 1991) carbon tetrachloride was
detected in 44 out of 549 food items from the USA, most often in fatty
and grain-based foods. The mean level in food items with detectable
levels was 31 µg/kg (with a range of 2 to 210 µg/kg).
5.2.5 Intake averages
The daily average intake of carbon tetrachloride in Japan by
inhalation was calculated to be 7.7 µg/day (based on a daily
inhalation volume of 15 m3/day and assuming a 100% absorption) and by
ingestion less than 0.1 µg/day (Yoshida, 1993). If adjusted to a daily
inhalation volume of 22 m3/day, an absorption of 40% and a body
weight of 64 kg, the daily intake would be 11.4 µg/day or 0.18 µg/kg
per day.
The ATSDR (1994) estimated the daily intake by inhalation to be
0.1 µg/kg body weight based on ambient air level of about 1 µg/m3
(assuming inhalation of 20 m3/day, a body weight of 70 kg and an
absorption of 40% based on measurements in monkeys and humans). The
daily intake via drinking-water was estimated to be about 0.01 µg/kg
body weight based on a typical carbon tetrachloride concentration of
0.5 µg/litre (assuming a water consumption of 2 litres/day and a body
weight of 70 kg).
In an earlier study of about 500 foodstuffs, an average daily
intake via foods and drinks of 8.63 µg/person per day was calculated
for inhabitants of Germany (Lahl, 1983). Because the intake by
inhalation is expected to be at least as much (BUA, 1990), the total
daily average intake would be estimated to be 17.26 µg/person (0.27
µg/kg body weight for a person of 64 kg). This calculation refers to a
period when carbon tetrachloride was still used in food processing or
in fumigation of grain.
5.3 Occupational exposure
The most likely route of exposure in the workplace is by
inhalation. Workers may be exposed to carbon tetrachloride during, for
example, the production of carbon tetrachloride itself, the synthesis
of compounds using carbon tetrachloride as a starting material and the
use of carbon tetrachloride as a solvent. Furthermore, workers have
been exposed to carbon tetrachloride at grain (due to fumigation) and
water treatment facilities. The National Institute for Occupational
Safety and Health estimated that in the USA around 58 000 workers were
potentially exposed to carbon tetrachloride, based on a national
survey conducted from 1981 to 1983 (National Library of Medicine,
1992).
A few studies on concentrations of carbon tetrachloride in
factories, and grain and water treatment facilities have been
reported. For water treatment facilities, Lurker et al. (1983)
reported exposure concentrations of 0.01-0.23 mg/m3; Clark (1981)
reported concen trations ranging from 0 to 1.1 mg/m3.
A peak exposure to an inspector during handling of grain at a
facility in the USA reached 277 mg/m3. Few employees, however, had a
mean exposure above 641 µg/m3 (Deer et al., 1987). Use of carbon
tetrachloride in open beakers resulted in exposure levels of 290-620
mg/m3 at a United Kingdom quartz crystal processing plant. Levels
were reduced to 50-60 mg/m3 by closing the beakers (Kazantzis &
Bomford, 1960).
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1 Pharmacokinetics
6.1.1 Absorption
Carbon tetrachloride is absorbed readily from the
gastrointestinal and respiratory tract. Dermal absorption of carbon
tetrachloride, either in vapour or in liquid phase, is possible, but
the dermal absorption of the vapour appears to be very low.
6.1.1.1 Oral
Carbon tetrachloride is relatively insoluble in water, a source
of exposure relevant to environmental scenarios and human health risk.
As a result, many studies examining the hepatotoxicity of carbon
tetrachloride used corn oil as a dosing vehicle for laboratory animals
(Paul & Rubinstein, 1963; Larson & Plaa, 1965; Marchand et al., 1970).
Corn oil has been found to delay markedly the absorption of carbon
tetrachloride (Kim et al., 1990a) as well as other halocarbons (Withey
et al., 1983) from the gastrointestinal tract.
In part, because carbon tetrachloride in water is directly
relevant to human exposure studies, recent studies in laboratory
animals employed Emulphor(R), a polyethoxylated oil, at
concentrations up to 10%, as an aqueous vehicle for carbon
tetrachloride. Aqueous solutions of carbon tetrachloride in
Emulphor(R) were administered to Sprague-Dawley rats both as a bolus
and during gastric infusion at a constant rate during a 2-h period
(Sanzgiri et al., 1995). Uptake and tissue levels of carbon
tetrachloride after gastric infusion were less than after bolus
dosing. When the concentration of Emulphor(R) was varied up to 10%,
absorption (and distribution) of carbon tetrachloride was not affected
(Sanzgiri & Bruckner, 1997).
A comparison of the uptake of carbon tetrachloride in corn oil
and aqueous emulsions is discussed in section 7.9. Tissue levels of
carbon tetrachloride associated with bolus dosing, gastric infusion,
and inhalation are discussed in section 6.1.2. The relationship of
dosing vehicle, dose rate, and route of exposure to hepatotoxicity is
discussed in section 7.9.
6.1.1.2 Dermal
Liquid carbon tetrachloride on the intact mouse skin was absorbed
at a rate of 8.3 µg/cm2 per minute (Tsuruta, 1975). Jakobson et al.
(1982) examined the percutaneous uptake of liquid carbon tetrachloride
(1 ml) in guinea-pigs (carbon tetrachloride in a glass depot, covering
3.1 cm2 of clipped skin). A peak blood level of about 1 mg carbon
tetrachloride/litre was reached within 1 h. Despite continuation of
the exposure the blood levels declined during the following h,
possibly due to local vasoconstriction, rapid transport from blood to
adipose tissues or biotransformation processes.
Wahlberg & Boman (1979) applied 0.5 or 2 ml of carbon
tetrachloride in a closed glass container on the skin (3.1 cm2) of
guinea-pigs. The deposits were completely absorbed within a few days.
McCollister et al. (1951), who exposed the clipped skin of one
male and one female monkey to [14C]carbon tetrachloride vapour (whole
body exposure), detected radioactivity in the blood and in the expired
air. After an exposure of 3 h at 3056 mg/m3, the blood of the female
contained a carbon tetrachloride level of 12 µg/100 g and the expired
air contained 0.8 µg/litre. After exposure to 7230 mg/m3 for 3.5 h
the blood of the male contained a carbon tetrachloride level of 30
µg/100 g and the expired air contained 3 µg/litre.
6.1.1.3 Inhalation
In rats exposed by inhalation to carbon tetrachloride
concentrations of 100 or 1000 ppm (641 or 6410 mg/m3) for 2 h, the
total amounts systemically absorbed were 17.5 and 179 mg/kg body
weight. The Cmax values (mg/ml) were approximately 1 and 13,
respectively, and the AUC values (mg.min/ml) were approximately 120
and 1900, respectively (Sanzgiri et al., 1995).
Steady-state carbon tetrachloride concentrations in the blood of
approximately 320 mg/litre were reached within about 5 h when dogs
were exposed to a carbon tetrachloride concentration in air of 15 000
ppm (96 150 mg/m3) for several hours (Von Oettingen et al., 1950).
McCollister et al. (1951) exposed three female rhesus monkeys to
an average [14C]carbon tetrachloride concentration of 46 ppm (295
mg/m3) via air for 139, 244 or 300 min, respectively. Within 300 min,
30% of the inhaled quantity was absorbed but in the blood no
steady-state concentration of radioactivity was reached. The
radioactivity level in the blood at that moment corresponded to 3 mg
carbon tetrachloride/litre blood and was distributed over carbon
tetrachloride (56.2%), "acid volatile" carbonates (16.5%) and
non-volatile material (27.3%).
The US EPA Iris Program uses 40% absorption as a mean for the
calculation of human respiratory intake. The determined values ranged
from 30% to 65% (US EPA, 1991).
6.1.2 Distribution
The tissue distribution of carbon tetrachloride has been
investigated in mice after inhalation (Bergman, 1984), in rats after
oral administration (Marchand et al., 1970; Teschke et al., 1983;
Watanabe et al., 1986) and after inhalation (Paustenbach et al.,
1986a), in rabbits after oral administration (Fowler, 1969), in dogs
after inhalation (Von Oettingen et al., 1950) and in monkeys after
inhalation (McCollister et al., 1951).
Bergman (1984) investigated the distribution of [14C]carbon
tetrachloride in the mouse after a single inhalation exposure (10 min;
256 000 mg/m3 air). Immediately after the exposure, high levels of
radioactivity were found in fat, bone marrow, white matter of the
brain, spinal cord and nerves, liver, kidneys, salivary glands and
gastrointestinal mucosa. The radioactivity in bronchi, liver, kidneys,
salivary glands and the gastrointestinal mucosa (particularly in the
mucosa of the glandular part of the stomach and of the colon and
rectum) was to a large extent non-volatile. A similar pattern of
distribution was observed 30 min after the exposure, except in the
liver where a more pronounced accumulation of non-volatile
radioactivity was seen than observed immediately after inhalation. A
large part of the non-volatile radioactivity in the liver and kidneys
appeared to be non-extractable, which may indicate covalent binding to
tissue components (see section 6.2). Non-extractable radioactivity was
also present in the bronchi and nasal mucosa. Non-volatile and
non-extractable radioactivity was present in the vaginal and uterine
mucosa and interstitially in the testis.
The tissue distribution in rats (in order of decreasing
radio-activity), 3 h after oral administration, as reported by
Watanabe et al. (1986) was: liver, kidney, brain, muscle and blood.
Carbon tetrachloride tends to accumulate in fat. Maximal fat tissue
carbon tetrachloride concentrations exceeded the maximal blood levels
by a factor of 60 after oral administration to rats (Marchand et al.,
1970).
Peak levels of carbon tetrachloride were observed 3-6 h following
an acute oral carbon tetrachloride dose (1.5 ml/kg body weight
administered in olive oil) in the blood (26 mg/litre), liver and fat
of female Wistar rats. Subsequently, the carbon tetrachloride levels
declined rapidly (Teschke et al., 1983).
Peak blood levels of carbon tetrachloride after a 12-h inhalation
exposure of rats were 12 mg/litre blood at an airborne concentration
of 2 mg/litre (320 ppm), 20 mg/litre blood at 4 mg/litre (640 ppm) and
36 mg/litre blood at 8 mg/litre (1280 ppm). The blood level attained
50% of this value after 60 min. A 4-h exposure to a concentration of
2.6 mg/litre (406 ppm) led to a blood level of 10.5 mg/litre, which
dropped to 50% of this peak value within 30 min after exposure
(Frantik & Benes, 1984).
Paustenbach et al. (1986a) found the highest concentration of
carbon tetrachloride equivalents in the fat, liver, lungs and adrenals
of male Sprague-Dawley rats repeatedly exposed to 100 ppm (641 mg/m3)
of [14C]carbon tetrachloride vapour for 8 or 11.5 h/day for periods
of 1 to 10 days.
Fowler (1969) administered 1 ml carbon tetrachloride/kg body
weight to rabbits by stomach tube as a 20% (v/v) solution in olive
oil. Five rabbits were killed 6, 24 and 48 h after receiving carbon
tetrachloride and the concentration in fat, liver, kidney and muscle
tissue was determined. Two rabbits receiving olive oil were killed as
control animals. The highest carbon tetrachloride concentration after
6, 24 and 48 h was found in fat tissue, but the amount found in the
fat as well as in the other tissues after 6 h diminished rapidly
during the subsequent 42 h.
Von Oettingen et al. (1950) studied the distribution of carbon
tetrachloride in Beagle dogs after exposure to 15 000 ppm (96 150
mg/m3) and reported a lowest concentration in the blood followed in
increasing order by liver, heart and brain.
The pattern of distribution immediately after a 5-h inhalation of
46 ppm (295 mg) [14C]carbon tetrachloride/m3 in monkeys (McCollister
et al., 1951) was (tissues in order of decreasing concentration of
total radioactivity): fat, liver, bone marrow, blood, brain, kidneys,
heart, spleen, muscle, lungs, bone.
Sanzgiri et al. (1995) demonstrated that the tissue
pharmacokinetic profile was influenced by the route and rate of
administration of carbon tetrachloride. Inhalation exposure of rats to
1000 ppm (6410 mg/m3) carbon tetrachloride for 2 h resulted in a
systemic dose of 179 mg/kg body weight. This dose was subsequently
administered as an oral bolus or a constant gastric infusion over 2 h.
In all cases tissue levels were highest in fat with levels in all
tissues being higher after an oral bolus dose than after inhalation
exposure or gastric infusion. For the liver Cmax was higher after an
oral bolus dose (58 mg/g) than after inhalation (20 mg/g) or gastric
infusion (0.5 µg/g). The authors speculate that the capacity of
first-pass metabolism can be exceeded following a large single bolus
oral dose, although not during gastric infusion of the same dose over
2 h.
6.1.3 Elimination and fate
In a study by Reynolds et al. (1984), all routes of elimination
were investigated simultaneously after a single oral administration of
[14C]carbon tetrachloride to fasted rats at dose levels ranging from
15.4 to 4004 mg/kg body weight. The exhalation of unchanged carbon
tetrachloride increased at higher dose levels (70-90% after
administration of 46.2 mg/kg body weight or more). This result might
be explained by a saturation of the first pass metabolism, or by an
impairment of the overall carbon tetrachloride metabolism due to a
breakdown of cytochrome P-450, which is induced by carbon
tetrachloride-metabolites (as reported by Noguchi et al., 1982a,b).
Both the amount of carbon tetrachloride excreted and the time-course
of excretion depended on the dose, tending to become slower as the
dose increased. For example, the half-life for exhalation of carbon
tetrachloride was 1.3 h at 46.2 mg/kg body weight but was 6.3 h at
4004 mg/kg body weight.
Page & Carlson (1994) examined whether faecal excretion, either
biliary or by direct exsorption, contributed significantly to carbon
tetrachloride elimination from the body of rats. It appeared that
biliary and non-biliary mechanisms contributed to the faecal
elimination of [14C]carbon tetrachloride, but that this route
accounted for less than 1% of the administered dose of 1 mmol/kg body
weight in rats. Thus faecal elimination of carbon tetrachloride (as
parent compound) does not significantly contribute to the overall
elimination of carbon tetrachloride.
The carbon tetrachloride levels in blood declined with a
half-life of 4 to 5 h during the first 24 h after oral administration
of 1.25 ml carbon tetrachloride/kg body weight (Larson & Plaa, 1965)
or 2 ml (0.1 mCi) [14C]carbon tetrachloride/kg body weight (Marchand
et al., 1970). Carbon tetrachloride levels in the liver declined with
a half-life of about 7 h after administration by gastric intubation of
2.5 ml carbon tetrachloride/kg body weight (Dingell & Heimberg, 1968).
Kim et al. (1990a) found a half-life for carbon tetrachloride in
the blood of 98 min and a whole body clearance of 0.13 ml/min per g
when 25 mg carbon tetrachloride/kg body weight was orally administered
in four different vehicles to male Sprague-Dawley rats. The
elimination appeared to be the same in all the different vehicle
groups, whereas the absorption differed (see section 6.1.1.1).
According to Paustenbach et al. (1986a) the rate of carbon
tetrachloride clearance in rats after inhalation exposure is biphasic,
with an initial half-life of 7 to 10 h. Exposure for longer periods of
time led to a decreased rate of clearance (and to higher
concentrations in the fat) (Paustenbach et al., 1986a,b, 1988). In the
study of Sanzgiri et al. (1995) (see section 6.1.1.3) the apparent
clearance values after inhalation doses delivering 17.5 or 179 mg/kg
body weight, respectively, were 150 and 100 ml/min per kg and the
half-life value was about 164 min.
Veng-Pedersen et al. (1987) exposed rats repeatedly by inhalation
to 100 ppm [14C]carbon tetrachloride (641 mg/m3) for either 8 h/day
for 5 days or 11.5 h/day for 4 days. The pulmonary excretion of
[14C]activity was clearly biphasic for both dosing regimens, with
mean half-lives for the first and second phase being 84 and 400 min
for the 8-h exposure and of 91 and 496 min for the 11.5-h exposure,
respectively. This indicates that the second phase of the 11.5-h group
was longer than the second phase of the 8-h group. This observation
suggests that during longer exposure periods a greater fraction of the
inhaled carbon tetrachloride is distributed to poorly perfused tissues
like fat, thus altering the elimination.
McCollister et al. (1951) demonstrated in monkeys that, after an
inhalation exposure to [14C]carbon tetrachloride, radioactive
material was excreted in faeces, urine and expired air. According to
the authors the compounds in the urine consisted of urea, bicarbonate
and an acid hydrolysable, non-amino acid substance.
6.1.4 Physiologically based pharmacokinetic modelling
A biphasic kinetic in the biotransformation of carbon
tetrachloride has been observed in several inhalation studies. The
relationship of arterial blood and inhaled carbon tetrachloride
concentrations, as found in male Sprague-Dawley rats, suggested that
carbon tetrachloride metabolism is limited by blood perfusion of the
liver at inhaled concentrations below 100 ppm (641 mg/m3) and that it
is saturated at inhaled concentrations above 100 ppm. The estimated
rate of reaction (Vmax) measured in the blood was 2.7 mg/kg body
weight per hour. This rate gradually decreased during the exposure
period of 5 h, which could be due to rapid loss of cytochrome P-450
content. The Vmax in rats pretreated with 100 µl carbon tetrachloride
(oral administration, 24 h before inhalation exposure) decreased about
57%, which was in good agreement with the decrease of the cytochrome
P-450 content. (Uemitsu, 1986).
Gargas et al. (1986) calculated for carbon tetrachloride a Vmax
of 0.63 mg/kg body weight per hour in an inhalation study in male
Fischer-344 rats.
Applications of pharmacokinetic models for the inhalation
exposure of rats have provided Vmax and Km estimates in rats of 0.63
mg/h per kg and 0.25 mg/litre (Gargas et al., 1986) and 0.37 mg/h/kg
and 1.3 mg/litre (Evans et al., 1994).
Paustenbach et al. (1988) constructed a physiologically based
pharmacokinetic model (PB-PK) for inhaled carbon tetrachloride and
used this model to predict the pharmacokinetics of inhaled
[14C]carbon tetrachloride in male Sprague-Dawley rats exposed for 8
or 11.5 h/day for 1 or 2 weeks. The simulations were compared with
actual laboratory data (Paustenbach et al., 1986a,b). The model
accurately predicted the behaviour of carbon tetrachloride and its
metabolites. Metabolites were partitioned in three compartments: the
amounts to be excreted in the breath (as [14C]CO2), urine and
faeces. Of total carbon tetrachloride metabolites, 6.5, 9.5 and 84%
were formed via the pathways leading to CO2, urinary and faecal
metabolites, respectively.
The PB-PK model suggests that at concentrations up to 100 ppm
(641 mg/m3), rats, monkeys and humans metabolize and eliminate carbon
tetrachloride in a similar manner. Most species convert 2-5% to CO2,
eliminate 4-8% in the urine, and eliminate 40-50% unchanged in the
breath.
6.2 Biotransformation and covalent binding of metabolites
Metabolism of carbon tetrachloride is initiated by cytochrome
P-450-mediated transfer of an electron to the C-Cl bond, forming an
anion radical that eliminates chloride, thus forming the
trichloromethyl radical. This radical may undergo both oxidative and
reductive biotransformation (see Fig. 1). The isoenzymes implicated in
this process are CYP2E1 and CYP2B1/2B2 (Raucy et al., 1993; Gruebele
et al., 1996). Some isoforms may be preferentially susceptible to
degradation by carbon tetrachloride (Tierney et al., 1992). Evidence
that carbon tetrachloride inactivates CYP2E1 and reduces total CYP2E1
protein in a cell line that constitutively expresses human CYP2E1 has
been obtained by Dai & Cederbaum (1995). When protein synthesis was
blocked, inactivation and degradation of CYP2E1 by carbon
tetrachloride was more pronounced. Free radical scavengers were unable
to prevent CYP2E1 degradation, suggesting that carbon tetrachloride
metabolites react at the active site of CYP2E1. Antioxidants prevented
carbon tetrachloride-induced lipid peroxidation, but not CYP2E1
degradation, suggesting that these processes are disassociated.
The formation of the radical has been demonstrated convincingly
in vitro as well as in vivo in electron spin resonance experiments
and is mediated by a particular cytochrome P-450, of which the haem
moiety is destroyed after carbon tetrachloride exposure (Reiner et
al., 1972; Sipes et al., 1977; Poyer et al., 1978, 1980; Lai et al.,
1979; Tomasi et al., 1980; Fernández et al., 1982; Noguchi et al.,
1982a,b; McCay et al., 1984). The formation of carbon
tetrachloride/cytochrome P-450 complexes has been demonstrated by
Uehleke et al. (1973), Wolf et al. (1977), Ahr et al. (1980) and
Fernández et al. (1982).
The most important pathway in the elimination of trichloromethyl
radicals is the reaction with molecular oxygen, resulting in the
formation of trichloromethylperoxyl radicals (CCl3OO*), as proposed
by Packer et al. (1978), Shah et al. (1979), Mico & Pohl (1983), Pohl
et al. (1984) and McCay et al. (1984). This intermediate, which is
even more reactive than the trichloromethyl radical (Dianzani, 1984),
may interact with lipids, causing lipid peroxidation along with the
production of 4-hydroxyalkenals (Benedetti et al., 1982; Comporti et
al., 1984). Radical-induced lipid peroxidation is also a presumed
source of a variety of metabolites, such as acetone, propanal, butanal
and malondialdehyde, which appear in rat urine 24 h after exposure to
carbon tetrachloride (de Zwart et al., 1997).
It is supposed that the trichloromethylperoxyl radical will react
further to produce phosgene, which again may interact with tissue
macromolecules or with water, finally producing hydrochloric acid and
carbon dioxide (Pohl et al., 1984). Carbon tetrachloride has been
reported to be metabolised to carbon dioxide in liver homogenates by
Rubinstein & Kanics (1964). The biotransformation of carbon
tetrachloride to carbon dioxide in vivo has been reported by
Reynolds et al. (1984).
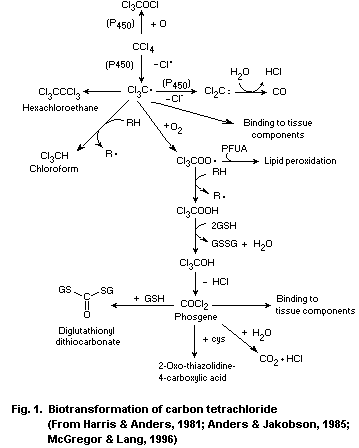 Condensation of phosgene with cysteine leads to the formation of
2-oxothiazolidine-4-carboxylic acid (Shah et al., 1979; Kubic &
Anders, 1980). The condensation of phosgene with glutathione (GSH),
resulting in diglutathionyl dithiocarbonate, has been demonstrated by
Pohl et al. (1981) in in vivo experiments.
Formation of chloroform and CCl2-carbene occur under
O2-deficient circumstances (Reiner et al., 1972; Shah et al., 1979;
Pohl et al., 1984). Under in vivo conditions CCl2-carbene is of
minor importance as an intermediate. Castro et al. (1990) examined the
biotransformation of carbon tetrachloride to chloroform by liver
nuclear preparations of three different species: C3H mice,
Sprague-Dawley rats and Syrian golden hamsters. All species were able
to transform carbon tetrachloride to chloroform. This ability was not
NADPH dependent and proceeded to an equal extent under nitrogen and
air. The relative transforming intensity was mice > hamsters > rats
under anaerobic and hamsters >> mice > rats under aerobic
conditions, respectively. More detailed experiments with preparations
of C3H mice suggested the presence of enzymatic and non-enzymatic
pathways of carbon tetrachloride transformation, as revealed by their
heat susceptibility and the inhibitory effects of EDTA.
Many studies on covalent binding of carbon tetrachloride
metabolites to tissue macromolecules have been carried out, but most
of them have measured only radioactivity and not identified the adduct
formed. Many studies ignored the radioactive impurities present in the
carbon tetrachloride used as well as the possible incorporation of
14C-radioactivity via carbon dioxide riginating from carbon
tetrachloride. For these reasons, the binding of carbon tetrachloride
should be considered as an association of radioactivity, unless
further information is provided.
According to Shertzer et al. (1988), the active radical metabolic
intermediate of carbon tetrachloride may covalently bind to
macromolecules, produce lipid peroxidation, and result in the loss of
intrahepatic calcium homoeostasis.
Cambon-Gros et al. (1986) showed that the fetal rat liver during
the last days of pregnancy, as well as the mother liver, can
metabolize carbon tetrachloride into a free radical: CCl3*. This
radical may bind covalently to the microsomal membranes and cause the
destruction of cytochrome P-450 as well as the inhibition of one of
the main microsomal activities, the ability to store Ca2+. Contrary
to the situation in adults, these radicals do not provoke a membrane
phospholipid peroxidation in the fetus. The animals used in the study
were nulliparous pregnant female Sprague-Dawley rats (twentieth day of
gestation).
Tjälve & Löfberg (1983) showed that covalent binding (probably
due to metabolic activation) occurred in many tissues of exposed rats
(liver, kidney cortex, mucosae of the respiratory tract, the mouth
cavity and the oesophagus).
The non-extractable part of non-volatile radioactivity in liver
and kidney may indicate covalent binding to tissue components
(Bergman, 1984). Association of radioactivity to tissue components
also appeared to occur in the testis, the uterine and vaginal mucosa
and in the nasal mucosa.
A similar pattern of association with tissue components to that
observed by Bergman (1984) has been found by Tjälve & Löfberg (1983)
after intravenous and intraperitoneal administration, indicating that
the tissue binding in the nasal mucosa is not specific to the route of
administration.
Díaz Gómez et al. (1975a) investigated the relations between
liver carbon tetrachloride levels, lipid peroxidation, the covalent
binding to liver lipids and hepatic centrilobular necrosis after
in vivo adminis tration of equimolar doses of carbon tetrachloride
in different animal species. The results support the hypothesis that
carbon tetrachloride-induced lipid peroxidation is not the only
mechanism of its toxic action. In fact, there seems to be a better
correlation between irreversible association with tissue components
and carbon tetrachloride toxicity than between lipid peroxidation and
carbon tetrachloride toxicity.
According to Villarruel et al. (1977) association of carbon
tetrachloride metabolites with lipids occurs mostly in the liver and
kidney cortex and medulla. Ansari et al. (1982) demonstrated the
binding of trichloromethyl radicals originating from carbon
tetrachloride to cholesterol. Binding to membrane lipids, eventually
leading to cross-linking, has been demonstrated by Link et al. (1984).
Association of carbon tetrachloride derivatives with
macromolecules in vitro has been found mainly in microsomal systems,
but binding to lipids and proteins also occurs in purified nuclear
preparations (Díaz Gómez & Castro, 1980b).
The covalent binding of carbon tetrachloride reactive metabolites
to different nuclear and microsomal lipids was studied by Fanelli &
Castro (1995) in male Sprague Dawley and Osborne Mendel rats, strains
with a marked difference in the carcinogenic response to carbon
tetrachloride, the Sprague-Dawley being non-susceptible and the
Osborne-Mendel being responsive. The intensity of covalent binding to
microsomal lipids in vivo and in vitro was higher in the Osborne
Mendel rats. Most of the covalent binding of carbon tetrachloride
reactive metabolites in both rat strains occurs in the phospholipid
and in the cholesterol/cholesterol ester fractions. The covalent
binding to phospholipids is higher in the Sprague Dawley strain, while
binding to cholesterol and cholesterol ester is more intense in the
Osborne Mendel rat.
After administration of [14C]carbon tetrachloride to rats and
mice (9 µmol/kg body weight), the quantities of label associated with
DNA at 6 h post-dosing were 0.52 and 0.72 pmol/mg DNA, respectively.
In this study binding also occurred to nuclear proteins and lipids,
especially to phospholipids (diphosphatidylglycerol) and diglycerides
(Díaz Gómez & Castro, 1980a).
The results of the study of Oraumbo & Van Duuren (1989) indicated
that under aerobic incubation conditions, carbon tetrachloride is
metabolized to one or more electrophilic metabolites, which bind
covalently to chromatin DNA in a dose- and time-dependent manner. In
this study chromatin was isolated from male B6C3F1 hybrid mice and
incubated with [14C]carbon tetrachloride in the presence of hepatic
microsomes from the same animals and a NADPH-regenerating system. The
study was carried out with various carbon tetrachloride concentrations
and incubation times.
6.3 Human studies
6.3.1 Uptake
6.3.1.1 Dermal
Immersion of a thumb in liquid carbon tetrachloride for 30 min
produced a maximum alveolar carbon tetrachloride concentration of
about 3.8 mg/m3 air 30 min after the end of exposure (Stewart & Dodd,
1964). The concentration declined with a half-life of about 2.5 h.
6.3.1.2 Inhalation
Lehmann & Schmidt-Kehl (1936) reported that 60% of the quantity
of carbon tetrachloride inhaled was retained in an experiment
involving a 30-min exposure of a volunteer to 4200 mg/m3.
6.3.2 Elimination
The pulmonary excretion of 33% of the absorbed quantity of
[38Cl] carbon tetrachloride occurred during the first hour after a
single breath by a volunteer (Morgan et al., 1970). Erickson (1981)
found carbon tetrachloride in mother's milk (concentration and
exposure not specified).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Lethality
LD50 values of carbon tetrachloride for various mammalian
species are represented in Table 7.
For female OF1 mice a LC50 of 7176 ppm (45 998 mg/m3) was
reported after a 6-h inhalation exposure to carbon tetrachloride (14
days observation period) (Gradiski et al., 1978). Svirbely et al.
(1947) reported a LC50 of 50 000 mg/m3 after a 7-h inhalation
exposure (8-h observation period) for male and female Swiss mice.
A 100% death rate of Wistar rats due to anaesthesia was observed
by Adams et al. (1952) after inhalation exposure to carbon
tetrachloride at concentrations of 121 600 mg/m3 for 2.2 h or 46 700
mg/m3 for 8 h.
Roudabush et al. (1965) reported that the acute dermal LD50
values of carbon tetrachloride for rabbits and guinea-pigs were in
excess of 15 g/kg body weight. Data on mortality and observation
period were not given.
Wahlberg & Boman (1979) applied carbon tetrachloride in
quantities of 800 and 3200 mg (approximately 2100 and 8500 mg/kg body
weight) to the skin of guinea-pigs. After 21 days 5/20 and 13/20 of
the animals had died in the 800 mg and 3200 mg groups, respectively.
7.1.2 Non-lethal effects
7.1.2.1 Oral exposure
Effects on the liver with changes in several enzyme levels are
reported to be the major effects resulting from an acute oral exposure
to carbon tetrachloride. In addition to the effects on the liver,
effects have also been reported in other organs such as lungs and
kidneys.
a) Mice
Akahori et al. (1983) examined the biochemical alterations in
liver and blood, and the histological findings in the liver of female
C57BL/6J mice after a single oral administration of carbon
tetrachloride in liquid paraffin at doses of 0.02, 0.5 or 1.5 ml/kg
body weight (calculated to be 32, 797 or 2391 mg/kg body weight) at
various times from 15 to 327 h after administration. The biochemical
changes in the liver (decreases in protein, glucose, phospholipids,
DNA and RNA concentrations; increases in triglycerides, glycogen, and
Table 7. LD50 values (mg/kg body weight) for mammals
Species/strain Sex Route Vehicle Observed period LD50a Reference
Mice
Swiss Webster male intraperitoneal corn oil 24 h 4144 Klaassen & Plaa (1967a)
female intraperitoneal corn oil 24 h 4463 Klaassen & Plaa (1967a)
Strain and sex
unknown oral unknown not reported 12 100-14 400 IARC (1979)
Swiss Webster female intraperitoneal corn oil 24 h 4676 Gehring (1968)
OF1 (SPF) female intraperitoneal olive oil 14 days 3350 Gradiski et al. (1974)
Rats
Wistar female oral unknown 14 days 2821 Smyth et al. (1970)
Sprague-Dawley male intraperitoneal corn oil 48 h 4463 Klingensmith et al. (1983)
Sprague-Dawley male intraperitoneal corn oil 24 h 3029 Klaassen & Plaa (1969)
Sprague-Dawley female intraperitoneal peanut oil 24 h 6603 Lundberg et al. (1986)
14 days 2824 Lundberg et al. (1986)
Charles River male oral corn oil 14 days 10 054 Kennedy et al. (1986)
Dogs
Mongrel male/female intraperitoneal corn oil 24 h 2391 Klaassen & Plaa (1967b)
a Most of the values are calculated values because they were reported as ml/kg body weight
free and esterified cholesterol concentrations) and in blood (an
increase in serum aspartate aminotransferase (ASAT) activity and free
and esterified cholesterol concentration; a decrease in glucose
concentration) were generally dose-related but occurred more slowly in
the highest dose groups. The biochemical alterations were reflected in
the histological findings in the liver (centrilobular necrosis in the
low-dose and a mild midzonal necrosis in the mid- and high-dose
groups). These histological findings occurred later in the high-dose
group.
A dose-related increase in the serum angiotensin converting
enzyme level, indicative of pulmonary endothelial cell injury, was
reported by Hollinger (1982), who administered carbon tetrachloride in
vegetable oil at doses of 0.1 to 2.8 ml/kg body weight (159 to 4463
mg/kg body weight) to male Swiss-Webster mice.
Boyd et al. (1980) found morphological effects on the pulmonary
Clara cells of mice, including severe dilations of endoplasmic
reticulum and occasional cellular necrosis, after administration of
2.5 ml/kg body weight (3985 mg/kg body weight) of carbon tetrachloride
in sesame oil. Oral doses of less than 1 ml/kg body weight (1594 mg/kg
body weight) failed to produce visible pulmonary lesions.
b) Rats
Murphy & Malley (1969) reported dose-related increases in liver
and serum alanine aminotransferase (ALAT), liver tyrosine transaminase
and alkaline phosphatase activities after a single oral dose of 0.5 to
2 ml/kg body weight (797 to 3188 mg/kg body weight) of undiluted
carbon tetrachloride in male Holtzman rats.
In a study by Korsrud et al. (1972), fasted male Wistar rats
received carbon tetrachloride at a dose ranging from 0 to 2.5 ml/kg
body weight (0 to 3985 mg/kg body weight) in corn oil. The rats were
killed 18 h later. At a dose of 0.0125 ml/kg body weight (19.9 mg/kg),
there was histopathological evidence of toxic effects on the liver. At
0.025 ml/kg body weight (39.9 mg/kg), liver fat and weight, serum urea
and the activities of sorbitol dehydrogenase, fructose-l-P-aldolase,
isocitrate dehydrogenase, ALAT and aspartate aminotransferase (ASAT)
were increased.
According to Teschke et al. (1984), liver enzymes such as ALAT,
ASAT and glutamate dehydrogenase measured in serum reached maximal
activities 12-48 h following a single intragastric dose of carbon
tetrachloride (1.5 ml/kg body weight) to female Wistar rats.
A maximum increase of ASAT and ALAT after 48 h was reported by
Nakata et al. (1985) after administration of 5 ml/kg body weight (7970
mg/kg body weight) of carbon tetrachloride in corn oil to male Wistar
rats. Regeneration of the liver was characterized by a normalization
of the ASAT and ALAT levels and an increase in hepatic thymidylate
synthetase and thymidine kinase levels, two enzymes that are believed
to be indicative of tissue regeneration.
A single oral bolus of carbon tetrachloride (17.5 or 179 mg/kg)
to male Sprague-Dawley rats induced a dose-dependent increase in serum
sorbitol dehydrogenase and ALAT activities, and a decrease in the
hepatic cytochrome P-450 content and glucose-6-phosphatase activity.
When the same dose was given as a gastric infusion for 2 h, or by
inhalation exposure, the effects were much smaller (Sanzgiri et al.,
1995). No statistically significant difference was observed in the
toxicity of carbon tetrachloride administered orally in either corn
oil, Emulphor, or Tween-85 (Raymond & Plaa, 1997).
Significant increases in alpha-GSH, a cytosolic enzyme of short
half- life found in high concentration throughout the liver, were
detected 2 h after gavage dosing of male Sprague-Dawley rats (Clarke
et al., 1997). It was concluded that alpha-GST is a more sensitive and
accurate measure of carbon tetrachloride hepatotoxicity than ASAT.
Lowrey et al. (1981) reported a dose-dependent decreased capacity
of rat liver microsomes to sequester calcium 5 min after the
administration of carbon tetrachloride to fasted male Sprague-Dawley
rats at doses ranging from 0.025 to 5 ml/kg body weight (40 to 7970
mg/kg body weight). At 10 min after a carbon tetrachloride dose of 2.5
ml/kg body weight (3985 mg/kg body weight), the microsomal calcium
uptake was reduced to 15% of the control levels.
Chen et al. (1977) observed marked decreases in cytochrome P-450
content and P-450-related N-demethylation of dimethylaniline in the
microsomes of the lungs of male Sprague-Dawley rats after an oral
carbon tetrachloride dose in mineral oil of 2.5 ml/kg body weight
(3985 mg/kg body weight).
Shinozuka (1971) found alterations of the rough endoplasmic
reticulum membranes of rat hepatic cells, such as detachment of
ribosomes, narrowing of cisternal spaces, fusion of membranes and
eventual collapse, within 30 min after an administration of 5 ml/kg
body weight (7970 mg/kg body weight) of carbon tetrachloride in
mineral oil to Wistar rats.
Boyd et al. (1980) found lesions in the lungs of male
Sprague-Dawley rats that were similar to the lesions found in mice
(enlarged pulmonary Clara cells with dilations of the endoplasmic
reticulum, occasional cellular necrosis) after carbon tetrachloride
administration at doses of 2.4, 3.2 or 4.5 ml/kg body weight (3825,
5100 or 7173 mg/kg body weight) in sesame oil. These lesions, however,
were less pronounced and less frequent than in mice.
Striker et al. (1968) observed reversible lesions limited to the
proximal tubules in the kidneys of male Sprague-Dawley rats after
administration of 0.25 ml/kg body weight (400 mg/kg body weight) of
carbon tetrachloride in mineral oil. The earliest morphological change
was seen in the mitochondria, followed by cellular swelling, loss of
basilar interdigitations and swollen microvilli. Proliferation of the
smooth endoplasmic reticulum occurred later. Serum parameters, such as
creatinine, blood urea nitrogen and bilirubin, temporarily increased.
Furthermore, a decrease in the ability to preserve sodium ions and
water was observed, accompanied by a reduction of succinate
dehydrogenase activity.
Rats administered carbon tetrachloride in corn oil as a single
intraperitoneal dose had significantly prolonged clotting times that
appeared prior to liver necrosis (Pritchard et al., 1987).
c) Rabbits
Rabbits (strain unspecified), given an intragastric dose of 0.15
ml/kg body weight of a 33% carbon tetrachloride solution in liquid
paraffin (equivalent to 239 mg/kg body weight) showed an abnormal
electrophoretic lipoprotein pattern. This correlated with the degree
of liver injury as measured by ASAT and ALAT activities and plasma
lipid levels (Kanaghinis et al., 1982).
d) Monkeys
Centrilobular hepatocellular necrosis was observed in two out of
four monkeys 24 h after administration of a single oral dose of carbon
tetrachloride (1590 mg/kg).
e) Dogs
Dogs were administered single oral doses of 159, 318 and 477
mg/kg. Increased serum ALAT and ASAT activities were observed at 318
mg/kg or more.
7.1.2.2 Inhalation exposure
a) Mice
Boyd et al. (1980) exposed male Swiss mice to carbon
tetrachloride concentrations of 0.46 or 0.92 mmol/litre air for 1 h,
1.84 mmol/litre air for 12 min, and 3.68 mmol/litre air for 2 min
(70 750, 141 500, 283 000 and 566 000 mg/m3 air). All exposures
produced marked Clara cell lesions, similar to those caused by oral
exposure, and hepatic necrosis.
b) Rats
Brondeau et al. (1983) exposed male Sprague-Dawley rats (IFFA
CREDO; 8 males/group) to carbon tetrachloride at concentrations of
259, 531, 967 and 1459 ppm (1660, 3404, 6198 and 9352 mg/m3 air) for
4 h and examined the ASAT, ALAT, SDH and glutamate dehydrogenase
activities in the serum 24 h post-exposure. At the lowest exposure
level only the glutamate dehydrogenase activity was increased, whereas
at the higher exposure levels increases were observed in all enzyme
activities. Similar increases in serum activities of liver enzymes
have been reported in other rat strains (Magos et al., 1982
(Porton-Wistar and Fischer rats); Jaeger et al., 1975 (Holtzman rats);
Siegers et al., 1985 (Wistar rats)).
When male Sprague-Dawley rats were exposed to carbon
tetrachloride under conditions of various combinations of
concentration (; 1350 to 6900 ppm) and exposure time, it appeared that
the concentration had more influence on the hepatotoxicity than the
exposure time (Uemitsu et al., 1985).
Chen et al. (1977) observed a decrease in the cytochrome P-450
content and P-450-related demethylation of dimethylaniline in the
microsomes of lungs of male Sprague-Dawley rats exposed for 30 min to
air containing 4.38% carbon tetrachloride (280 758 mg/m3).
Morphological analysis of the lungs revealed focal changes in
pulmonary architecture consisting of alveolar collapse, septal
thickening and atypical type II pneumocyte configuration.
c) Cats
Wong & DiStefano (1966) exposed cats to carbon tetrachloride at a
concentration of 10 000 ppm (64 100 mg/m3) for 15, 30, 60 and 240
min. After 15 min the renal lipid content reached maximal levels. An
increase of kidney weight occurred within 60 min and was maintained
throughout the 24-h observation period. The total lipid content of the
liver had increased significantly at 3, 12 and 24 h after the 4-h
exposure period. Increased liver weight was observed 24 h after the
withdrawal of carbon tetrachloride. According to the authors, the
early increases in both the weight and fat content of the kidney
suggests that the renal changes precede the liver damage.
7.1.2.3 Subcutaneous and intraperitoneal exposure
a) Mice
A subcutaneous dose of 28 mg/kg body weight of carbon
tetrachloride in olive oil to male Swiss mice (10/group) appeared to
be the ED50 for causing prolongation of pentobarbital-induced
sleeping time. Histological examination showed changes in the liver
after administration of 77 mg/kg body weight. (Kutob & Plaa, 1962).
Intraperitoneal administration of carbon tetrachloride in corn
oil induced an elevation of the ALAT activity at calculated dose
levels of 11.2 to 15.9 mg/kg body weight in female Swiss mice and at
14.4 to 15.9 mg/kg body weight in male Swiss mice (Klaassen & Plaa,
1967a).
Bhathal et al. (1983) reported striking differences in the degree
of hepatic cell injury among four different strains of mice upon
histological examination after subcutaneous injection of 0.3 ml/mouse
of olive oil containing 5, 10 and 20% carbon tetrachloride. The SJL/J
strain appeared to be the least susceptible and the BALB/c strain the
most susceptible one. The hepatic lesions in the C3H and C57BL/6
strains were intermediate.
b) Rats
The study of Smejkalová et al. (1985) showed the existence of sex
differences in the sensitivity of the liver to carbon tetrachloride,
including a difference in the rate and quality of liver regeneration.
It appeared that in male Wistar rats the biochemical changes occurred
earlier (as early as 6 h after intraperitoneal administration of 1200
mg/kg body weight) and persisted longer (reaching a maximum after 12 h
and persisting for more than 72 h) than in females. In females these
changes reached a maximum after 24 h, and after 72 h the levels were
identical to the control values. Whereas in females the liver
regeneration started sooner than in males and led to complete healing
of the liver tissue, the regeneration in males started more slowly and
healing followed a different course, showing the development of
fibrosis.
Carbon tetrachloride dissolved in olive oil was injected
intraperitoneally to male Fischer rats at doses of 30 to 1000 mg/kg
body weight. Free and esterified cholesterol, triglycerides,
phospholipids and total lipids in plasma were reduced in a
dose-dependent manner. Cholesterol, triglyceride, phospholipid and
total lipid concentrations in the plasma were significantly lower in
rats given 30 mg/kg body weight than in control rats (p < 0.01)
(Honma, 1990).
When male Wistar rats received carbon tetrachloride as an
intraperitoneal injection of 16 or 96 mg/kg body weight in olive oil,
the calcium ion (Ca2+) content of liver microsomes was significantly
increased by 20% in rats treated with 96 mg/kg body weight. The
mitochondrial Ca2+ content was increased in both the 16 and 96 mg/kg
body weight group (600% and 1100%, respectively, 3 h after
administration) (Yamamoto, 1990).
c) Guinea-pigs
Divincenzo & Krasavage (1974) administered intraperitoneally 5,
25, 50, 75 or 150 mg/kg body weight to guinea-pigs. At 25 mg/kg or
more, increased ornithine decarboxylase activity in the serum was
found, an effect that was reflected by histological changes in the
liver.
d) Hamsters
Carbon tetrachloride produced injury to ciliated and non-ciliated
tracheal cells (swollen, loss of staining capacity, diluted nuclei) of
adult male Syrian golden hamsters that received carbon tetrachloride
intraperitoneally at a dose of 2.5 ml/kg body weight (3985 mg/kg body
weight). Groups of three hamsters were killed 1, 4, 12 or 24 h after
treatment. The number of damaged cells increased markedly after 1 h in
the lower trachea, but not until after 4 h in the upper trachea. By 24
h the number of injured cells approached normal values. Effects were
consistent within each group (Ahmadizadeh et al., 1990).
e) Dogs
When five mongrel dogs were given carbon tetrachloride at
different intraperitoneal doses, increases in ALAT were observed. The
calculated ED50 24-h after exposure was 32 mg/kg (Klaassen & Plaa,
1967b).
De Zwart et al. (1997) have identified eight urinary degradation
products of carbon tetrachloride-induced lipid peroxidation as
potentially useful biomarkers of in vivo hepatocellular damage. Male
Wistar rats were injected intraperitoneally with single doses of 38,
77 and 154 mg/kg body weight and the following substances were
identified in urine 12 to 48 h later: formaldehyde, acetaldehyde,
acetone, propanol, butanol, pentanal, hexanal and malondialdehyde. A
dose-dependent increase in histological and clinical chemistry
evidence of hepatocellular damage, along with these degradation
products, was observed. Increases in urinary concentrations of all
eight products were statistically significant at doses of 77 and 154
mg/kg. At 38 mg/kg, acetaldehyde and propanol were the only urinary
markers to exhibit a statistically significant increase.
7.1.2.4 Dermal exposure
The histopathology of the skin, liver, and kidney in the
guinea-pig (weighing 440 to 570 g) was studied by Kronevi et al.
(1979) at 15 min and 1, 4 and 16 h after occlusive epicutaneous
administration of 1 ml of carbon tetrachloride. After 15 min, some
degenerative changes in the epidermis, such a moderate karyopyknosis,
marked spongiosis and perinuclear oedema was observed. These changes
became more obvious with time, and at 16 h a slight karyolysis also
was seen. A junctional separation and cellular infiltration in the
dermis was observed after 4 and 16 h. Carbon tetrachloride exposure
caused hepatic centrilobular hydropic changes and, in addition, a
tendency to necrotic lesions after 16 h. Kidney histology was normal
for all exposed guinea-pigs.
7.2 Short-term exposure
7.2.1 Oral exposure
a) Mice
Hayes et al. (1986) administered carbon tetrachloride in corn oil
to CD-1 ICR mice (20/sex/group) for 14 consecutive days at dose levels
of 0, 625, 1250 or 2500 mg/kg body weight and for 90 consecutive days
at dose levels of 0, 12, 120, 540 or 1200 mg/kg body weight. No
compound-related deaths were seen in the 90-day study whereas, in the
14-day study, 6, 8 and 12 males and 0, 1 and 2 females died within 2-4
days in the 625, 1250 and 2500 mg/kg body weight groups, respectively.
Dose-dependent effects in the 14-day study consisted of decreased
fibrinogen and lymphocyte levels, increased LDH, ALAT and ASAT levels,
increased absolute and relative liver weights in both sexes, and
decreased lung, thymus and kidney weights in males. In the 90-day
study LDH, ASAT, ALAT and AP, cholesterol and bilirubin levels in the
blood were increased in a dose-dependent manner while blood glucose
levels were decreased at all dose levels. In both sexes and in all
90-day dose groups absolute and relative liver, spleen and thymus
weights were increased and liver damage was observed.
The results of a 90-day oral study in CD-1 mice by Condie et al.
(1986), indicated that the no-observed-adverse-effect level (NOAEL)
for hepatotoxic effects after administration of carbon tetrachloride
in corn oil was 1.2 mg/kg body weight (see also section 7.9).
b) Rats
Four groups of weanling rats (six males and six females per
group) were fed diet containing 0, 150, 225 or 520 mg/kg; estimated
daily doses were 0, 7-13, 13-24, 21-27 mg/kg body weight. The fat
content of liver was increased significantly in two higher dose groups
in both males (exposed for 6 weeks) and females (exposed for 5 weeks);
the difference compared to the control group was 50 to 200% (Alumot et
al., 1976).
Carbon tetrachloride treatment for 5 consecutive days at a dose
level of 400 mg/kg body weight in corn oil to male Fischer-344 rats
(CD F/CrlBr) increased the relative liver weight, decreased the CYP
enzyme concentration and activity in the liver, and increased the ALAT
levels (Dent & Graichen, 1982).
Bruckner et al. (1986) administered carbon tetrachloride in corn
oil to male Sprague-Dawley rats (5/group) for 5 consecutive days for
12 weeks, then 2 days without dosing followed by another 4 consecutive
days of dosing. Groups of rats weighing 300 to 350 g received 0, 20,
40 or 80 mg/kg body weight, whereas groups of rats weighing 200 to 250
g received 0, 20, 80 or 160 mg/kg body weight. In rats weighing
300-350 g, 20 mg/kg body weight caused vacuolization of hepatocytes
adjacent to the central vein of most liver lobules. The other dose
levels in this group produced comparable increases in SDH and ALAT
activities, and vacuolization of 25 to 35% of the hepatocytes in each
liver lobule. Carbon tetrachloride appeared to be more toxic to the
rats weighing 200-250 g with regard to necrotic cells, which were
rarely seen in livers of the 300-350 g rats, while in each 200-250 g
rat given 80 mg/kg body weight necrosis was observed.
Groups of 15-16 male Sprague-Dawley rats weighing 200 to 250 g
were given carbon tetrachloride in corn oil at doses of 0, 1, 10 and
33 mg/kg body weight for 5 days a week. The rats were dosed during the
dark part of their light cycle. After 12 weeks, 7 to 9 rats/group were
killed. The remaining animals were killed 13 days after exposure. At
10 mg/kg body weight a slightly but significantly increased SDH
activity and mild hepatic centrilobular vacuolization was seen.
Administration of 33 mg/kg body weight caused elevated serum levels of
SDH, ornithine-carbamyl transferase (OCT) and ALAT, which returned to
normal during the recovery period except for the OCT. Histopathology
of the livers of the 33 mg/kg body weight group revealed cirrhosis,
characterized by bile duct proliferation, fibrosis, lobular
distortion, parenchymal regeneration, hyperplastic nodules and
single-cell necrosis. According to the authors (Bruckner et al.,
1986), a NOAEL of 1 mg/kg body weight of carbon tetrachloride could be
established.
Allis et al. (1990) administered carbon tetrachloride in corn oil
to male Fischer-344 rats by gavage at dose levels of 0, 20 or 40 mg/kg
body weight for 12 weeks. At both dose levels ALAT, ASAT and LDH
levels were elevated and hepatic CYP protein concentrations were
reduced. Histopathology showed cirrhotic livers, vacuolar degeneration
and hepatocellular necrosis at both dose levels, but this was more
severe in the higher dose group. At day 8 and 15 after exposure, all
serum indicators and CYP protein concentration had returned to normal
levels. In both dose groups, hepatocellular necrosis disappeared by
day 8 and vacuolar degeneration decreased in severity but was still
present at day 15. Cirrhosis persisted in the high-dose group,
although it was less severe. Furthermore the relative liver weight in
animals receiving 40 mg/kg body weight remained elevated.
Several tests on renal function were conducted on adult male
Fischer-344 rats treated for 15 days with carbon tetrachloride in corn
oil at doses of 50, 150, 450 or 1350 mg/kg body weight. At 1350 mg/kg
body weight, kidney injury could be observed, as indicated by
haematuria, enzymeuria and decreases in kidney weight and serum
glucose concentration. At 450 mg/kg body weight, a lower body weight
and a reduction in the maximum urine-concentrating ability were
observed (Kluwe, 1981).
c) Dogs
Young adult Beagle dogs of the Alderly Park strain (6/sex) that
received carbon tetrachloride (in gelatin capsules) as a daily dose of
0.05 ml/kg body weight (80 mg/kg body weight) for 28 days, showed
elevated ALAT and ornithine carbamyl transferase activities, whereas
no effects were observed on ASAT and alkaline phosphatase. Furthermore
there were changes in the livers of all dogs characterized
histologically by fatty vacuolation of centrilobular cells. In 3 of
the 12 dogs, the vacuolation also occurred mid-zonally and
periportally. There was some evidence of individual cell necrosis, and
in some cases the sinusoids were mildly congested. No changes in
plasma enzyme activities and no histological changes in the liver were
observed when three females received a daily dose of 0.02 ml/kg body
weight (32 mg/kg body weight) of carbon tetrachloride for 8 weeks.
(Litchfield & Gartland, 1974).
7.2.2 Inhalation exposure
a) Mice
As reported in a translated, extensive summary, BDF1 mice
(10/sex/group) were exposed (whole-body) to atmospheres of 0, 10, 30,
90, 270 or 810 ppm carbon tetrachloride (0, 64, 192, 577, 1731 or 5192
mg/m3, respectively) for 6 h a day, 5 days a week for 13 weeks. Mice
were observed daily for clinical signs, behavioural changes and
mortality and were weighed each week. Urinalysis, haematology, blood
chemistry and microscopy were performed at the scheduled end of the
experiment. No compound-related deaths occurred. Body weight gain was
depressed in males at 30 ppm or more. Slight, but statistically
significant, changes in haematology were observed in males at 810 ppm
(decreased Hb and increased MPV) and in females at the two highest
dose levels (decreased Hb, Ht and RBC). Increased liver enzymes in
blood were observed in both sexes at the three highest dose levels.
Urinalysis showed a decrease in pH at the highest dose level in
females only. Microscopic examination showed slight to moderate
dose-related changes in the liver including cytological alterations,
even at the lowest dose level in males. At higher dose levels there
were more severe changes described as collapse, deposit of ceroid,
proliferative ducts, increase in mitosis, pleomorphism and foci (Japan
Bioassay Research Centre, 1998). A NOAEL cannot be established on the
basis of these results.
b) Rats
Exposure (whole body) of Sprague-Dawley rats (IFFA CREDO; 8
males/group) to a carbon tetrachloride atmosphere of 3308 mg/m3 (516
ppm) for 6 h a day for 2 or 4 consecutive days resulted in increased
serum activities of glutamate dehydrogenase, ASAT, ALAT and SDH after
4 days exposure. After 2 days only the SDH level was significantly
increased (Brondeau et al., 1983).
When male hooded rats were exposed (whole body) 8 h a day for 12
days to carbon tetrachloride levels of 68 or 680 ppm (436 or 4360
mg/m3) increased ASAT levels were found at low- and high-dose levels
after 4 and 2 days, respectively. At both doses the level of liver
lipids reached a maximum after 8 days, the level being related to the
dose. Additional exposure resulted in a decrease (Kanics & Rubinstein,
1968).
An increased liver triglyceride content was also reported by
Shimizu et al. (1973) who exposed (whole body) female Sprague-Dawley
rats to 10, 50 and 100 ppm of carbon tetrachloride vapour (64, 320 and
641 mg/m3) for 3 h a day for 6 to 8 weeks. Exposures to 320 and 641
mg/m3 resulted in striking increases in the hepatic trigly cerides to
a maximum during the first 3 weeks. Afterward this level was nearly
maintained in both groups. At 64 mg/m3 the rise in triglycerides was
minimal and was maintained for 2 weeks.
David et al. (1981) compared the serum enzyme activities and
liver lesions in rats exposed to various concentration-time
combinations. After four exposures to 50 ppm (320 mg/m3) for 6 h per
day, the enzyme activities were significantly increased by 50 to 70%,
and steatosis and hydropic changes were found in the liver. The
changes were significantly more intensive in rats exposed to 250 ppm
(1600 mg/m3) for 72 min per day, not withstanding that the
concentration-time product was equal. The same was true for two-fold
concentrations and 18 exposures.
Bogers et al. (1987) performed 4-week studies in male Wistar rats
by exposing (whole body) them to 63 or 80 ppm (404 or 513 mg/m3)
carbon tetrachloride in three different concentration profiles: 1)
continuous exposure of 6 h/day for 5 days/week; 2) exposure of 2 × 3
h/day (1.5 h interruption) for 5 days/week; and 3) peak loads of 382
ppm (2450 mg/m3) for 5 min (4 peaks for 3 h) with and without the
1.5-h interruption. The interruption of the daily 6-h exposures did
not result in less severe but rather in slightly more severe
hepatotoxic effects, such as changes in enzyme levels, fat
accumulation, increased relative liver weight, lower microsomal
protein content and hydropic degeneration of liver cells. Peak loads
did not affect the severity of the hepatotoxic effects.
Plummer et al. (1990) exposed (whole body) male black-hooded
Wistar rats (36/group) for 4 weeks to carbon tetrachloride both
continuously (24 h per day, 7 days per week) to 32 ppm (205 mg/m3) or
intermittently (6 h per day, 5 days per week) to 176 ppm (1128
mg/m3). The concentration-time products were similar for both groups.
The-carbon tetrachloride-induced hepatotoxicity appeared to be similar
in the two exposure profiles. However, when rats received the
enzyme-inducing agents phenobarbitone or 1,3-butanediol during the
study via their drinking-water, the liver injury appeared to be
exacerbated in 1,3-butanediol-treated rats, especially in the
intermittent exposure profile.
Groups of male Sprague Dawley rats were exposed (whole body) to
100 ppm (641 mg/m3) of [14C]carbon tetrachloride for either 8 or
11.5 h/day for periods of 1 to 10 days, and examined with and without
recovery in a tissue distribution study (see section 6.1.3 and 6.1.4).
The only significant difference between rats exposed to the two
different schedules was the serum SDH activity, which was almost
always significantly greater for rats exposed to the 11.5 h/day
schedule than for the comparable groups exposed to the 8 h/day
schedule, except when measured after a recovery period. (Paustenbach
et al., 1986b).
F-344 rats (10/sex/group) were exposed (whole-body) in a 13-week
inhalation study to 10, 30, 90, 270 or 810 ppm carbon tetrachloride
(64.1, 192.3, 576.9, 1730.7 or 5192.1 mg/m3, respectively) for 6 h a
day, 5 days a week. Control groups were included. Animals were
observed for clinical signs, behavioural changes and mortality once a
day, and they were weighed once a week. Urinalysis was performed at
the end of the dosing period, and haematology, blood biochemistry and
microscopy were performed at the scheduled sacrifice. At 810 ppm the
body weight gain was depressed in both sexes. Haematological changes
were observed at 90 ppm or more in both sexes, and at 30 ppm in
females. Increased liver enzymes in blood and urinalysis changes were
observed in males at 270 ppm or more and in females at 90 ppm or more.
Increased creatine phosphokinase (CPK) was seen at 30 ppm in females.
Microscopic examination showed slight to marked changes in the liver
described as fatty change, cytological alterations, deposition of
ceroid, proliferative ducts, increase in mitosis, pleomorphism,
cirrhosis and foci. Furthermore, vacuolic change of tubule, hyaline
degeneration of glomerulus and protein cast of the kidney were noted
at the two highest dose levels (Japan Bioassay Research Centre, 1998).
An NOAEL could not be established on the basis of these results.
c) Comparisons between species
Adams et al. (1952) exposed (whole body) rats, guinea-pigs,
albino rabbits and rhesus monkeys to various concentrations of carbon
tetrachloride in air. Histological data were not reported for the
control groups; these groups were, however, used for comparison with
dose groups. Dose-related effects were seen in Wistar rats
(15/sex/group) after exposure to carbon tetrachloride at
concentrations of 5, 10, 25, 50, 100, 200 and 400 ppm (32, 63, 160,
320, 630, 1282 and 2520 mg/m3) for 7 h a day, 5 days a week, during
approximately 5.5-6.5 months. Increased liver weight, increased liver
fat content (especially neutral fat and esterified cholesterol) and
fatty degeneration of the liver were observed after 2 to 3 weeks of
exposure to concentrations of 63 mg/m3 or more. Cirrhotic livers were
found from 630 mg/m3 upwards. At a concentration of 320 mg/m3,
kidney tubular epithelium was affected and death rate seemed to be
increased, especially in the males. At the two highest exposure levels
testicular weights were decreased. At the 32 mg/m3 level, no adverse
effects were seen. After exposure for 5 days a week during 13 weeks to
2520 mg/m3 for 3 min a day or to 630 mg/m3 for 18 min a day, no
effects could be observed. Similar results were obtained in
guinea-pigs from 63 mg/m3 upwards. Rabbits developed slight to
moderate fatty degeneration and cirrhosis of the liver at 160 mg/m3
or more; there was no reported effect at 63 mg/m3. Rhesus monkeys
developed slight-to-moderate fatty degeneration of the liver 630
mg/m3 (2 monkeys); there was no reported effect at 320 mg/m3 (2
monkeys).
Prendergast et al. (1967) studied the effects of continuous and
repeated exposure to carbon tetrachloride vapour in rats, guinea-pigs,
New Zealand rabbits, beagle dogs and squirrel monkeys (see Table 8).
After repeated exposure to 515 mg/m3, all species showed pulmonary
interstitial fibrosis or pneumonitis. Mottled livers were seen in all
species except in the dog. Histological examination revealed fatty
changes in the livers of all species. In addition, fibrosis, bile duct
proliferation, hepatocyte degeneration and regeneration, focal
inflammatory infiltration and portal cirrhosis were observed in the
guinea-pigs. After continuous exposure to 61 mg/m3 all species showed
growth retardation and all squirrel monkeys showed alopecia and
emaciation. Histopathological examination showed liver changes similar
to those reported after repeated exposures. After continuous exposure
to 6.1 mg/m3 carbon tetrachloride in 61 mg/m3 n-octane (as a
carrier) no visible signs of toxicity were noted in any of the species
and no animals died. At termination, all species except the rat showed
less body weight gain than the controls. Histopathological examination
revealed non-specific inflammatory changes in the lungs of all species
and in the liver, kidney and heart of several animals, but no specific
pathological changes attributable to the exposure were noted.
7.2.3 Intraperitoneal exposure
Biochemical and morphological characterization of
carbon-tetrachloride-induced lung fibrosis were investigated in rats
after intraperitoneal administration of 1.0 ml/kg body weight (1600
mg/kg in paraffin oil) twice a week for 2 or 5 weeks, and examined
after the end of the exposure. The third group (5 rats) was treated
for 2 weeks and examined after 3 weeks of recovery. Acute haemorrhagic
interstitial pneumonia resulted from the 2 week exposure, while
chronic interstitial pneumonia was observed in rats exposed for 5
weeks and in the third group after 3 week of recovery (Pääkkö et al.,
1996).
7.3 Long-term exposure
In a carcinogenicity study described in section 7.7 (Reuber &
Glover, 1970), young male rats were administered 1.3 ml/kg by
subcutaneous injection twice a week. Severe cirrhosis was observed in
all (16/16) Sprague-Dawley rats by 5 to 16 weeks (the time of death of
the animals) and in 13/17 Black rats by 7 to 18 weeks. In Wistar rats,
6/12 rats developed moderate and 6/12 severe cirrhosis by 17-68 weeks,
while the cirrhosis was mild in 2/13, moderate in 7 and severe in 4
Osborne Mendel rats by 10-105 weeks; in Japanese rats, the cirrhosis
was mild in 9/15, moderate in 5 and severe in one rat by 8 to 78
weeks.
Alumot et al. (1976) exposed rats (strain unknown) to carbon
tetrachloride in feed at measured levels of 0, 80 and 200 mg/kg feed
for 2 years. The highest concentration corresponded to a daily dose of
10 to 18 mg/kg body weight. Because of chronic respiratory disease in
all animals beginning at 14 months, which resulted in increased
mortality, the results reported upon necropsy at 2 years were
inadequate for a health risk evaluation.
Muños Torres et al. (1988) administered carbon tetrachloride to
female Wistar rats (150 g initial body weight) as weekly
intraperitoneal injections at a dose of 0.2 ml in mineral oil for 46
weeks. The hepatic lesions were macroscopically and microscopically
evaluated after 8, 16, 22, 30 and 46 injections. After 8 injections,
changes in the hepatic architecture due to an increase in the
collagenous component accompanied by formation of fibrous bridges were
seen. After 46 injections a clearly established cirrhosis with nodules
of different sizes was seen.
Table 8. Mortality in animals exposed to carbon tetrachloride
(from Prendergast at al., 1967).
Concentration Type of Ratc Guinea-pig Rabbit Dog Monkey
(mg/m3) studyb (Hartley) (New Zealand) (Beagle) (Squirrel)
515 R 0/15 3/15 0/3 0/2 1/3
61 C 0/15 3/15 0/2 0/2 0/3
6.1a C 0/15 0/15 0/3 0/2 0/3
a in 61 mg/m3 n-octane (as a carrier)
b R = 30 exposures, 8 h/day, 5 days/week for 6 weeks; C = continuous 90-day exposure.
c Long-Evans or Sprague-Dawley rats
BDF1 mice (50/sex/group) were exposed (whole-body) in a 2-year
inhalation study to 0, 5, 25 or 125 ppm carbon tetrachloride (0,
32.05, 160.25 or 801.25 mg/m3) for 6 h a day, 5 days a week. Animals
were observed for clinical signs, behavioural changes and mortality
once a day, and they were weighed once a week for the first 13 weeks
and every 4 weeks thereafter. Urinalysis was performed at the end of
the dosing period, and haematology, blood biochemistry and microscopy
were performed at the scheduled sacrifice. No compound-related effects
were observed at 5 ppm in female mice. The results from male mice
could not be evaluated due to anomalous control group liver enzyme
data. A significant decrease in survival was observed at 25 and 125
ppm. Liver tumours were the main cause of death at the highest dose
level. Body weight gain was depressed at 25 and 125 ppm. Changes in
haematology, blood biochemistry including liver enzymes, and
urinalysis were observed at 25 ppm or more. Microscopic examination
showed changes of the liver (deposit of ceroid, cyst and
degeneration), the kidney (protein cast) and the spleen (increased
deposit of haemosiderin at 25 ppm and increased extramedullary
haematopoiesis at 125 ppm) at the two highest dose levels in male
mice. In female mice changes in the liver included deposit of ceroid,
thrombus, necrosis, degeneration and cyst at 25 and 125 ppm. At 25 ppm
an increased deposit of haemosiderin of the spleen was observed while
at 125 ppm deposit of ceroid of the ovary was seen (Japan Bioassay
Research Centre, 1998). A NOAEL could not be established on the basis
of these results.
F-344 rats (50/sex/group) were exposed (whole-body) in a 2-year
inhalation study to 0, 5, 25 or 125 ppm carbon tetrachloride (0,
32.05, 160.25 or 801.25 mg/m3, respectively) for 6 h a day, 5 days a
week. Animals were observed for clinical signs, behavioural changes
and mortality once a day. They were weighed once a week for the first
13 weeks and every 4 weeks thereafter. Urinalysis was performed at the
end of the dosing period, and haematology, blood biochemistry and
microscopy were performed at the scheduled sacrifice. A significant
decrease in survival was observed at 125 ppm, with liver tumours
and/or chronic nephropathy being the main cause of death. Body weight
gain was depressed at 25 and 125 ppm. Changes in haematology, blood
biochemistry including liver enzymes, and urinalysis were observed at
25 ppm and even at 5 ppm for the nitrate and protein level in the
urine of the rats. At the 125 ppm level, only one male and three
female rats survived, so no statistical test was performed.
Microscopic examination showed changes of the liver (including fatty
change, deposit of ceroid, fibrosis, granulation and cirrhosis) at the
two highest dose levels in both sexes. An increased deposit of
haemosiderin in the spleen was observed in males at all dose levels.
An eosinophilic change of the nasal cavity was observed in females at
all dose levels and in males at 25 and 125 ppm. A chronic nephropathy
(progressive glomerulonephrosis) developed in females at 25 ppm and in
both sexes at 125 ppm. At 125 ppm deposit of ceroid and granulation of
the lymph node were observed in both sexes (Japan Bioassay Research
Centre, 1998). A NOAEL could not be established on the basis of these
results.
7.4 Irritation
7.4.1 Skin irritation
Epicutaneous administration of 1 ml of carbon tetrachloride has
been demonstrated to induce degenerative changes in the epidermis 15
min to 16 h after application (Kronevi et al., 1979; see section
7.1.2.4).
Moderate dermal irritation was observed after application of 0.5
ml carbon tetrachloride (under occlusion) onto the shaven skin of
rabbits (only abraded skin) and male Hartley guinea-pigs (normal and
abraded skin) (Roudabush et al., 1965).
In a study conducted according to Draize protocol, 0.5 ml carbon
tetrachloride was applied under an occlusive dressing for 24 h to the
intact and abraded skin of rabbits. Irritation was assessed at 24 and
72 h. Carbon tetrachloride was classified as a "medium" skin irritant.
Histopathology of skin samples taken from the application site on day
3 after exposure confirmed the irritant reaction (Duprat et al.,
1976).
Undiluted carbon tetrachloride (10 µl) was applied to the open
skin of guinea-pigs 3 times daily for 3 days. A skin reaction (no
further details provided) was observed on day 2 and an average score
described as "redness" was seen on day 4 (Anderson et al., 1988).
Wahlberg (1984a) rubbed 0.1 ml (159 mg) of carbon tetrachloride
into the skin of rabbits and guinea-pigs for ten consecutive days and
observed oedema and erythema.
7.4.2 Eye irritation
In a study conducted according to Draize protocol, 0.1 ml of
carbon tetrachloride caused a mild irritant response in rabbits. The
response was evident at 24, 48 and 72 h after exposure and recovery
was complete by day 14 (Duprat et al., 1976).
7.5 Toxicity to the reproductive system, embryotoxicity, teratogenicity
7.5.1 Reproduction
Groups of six male rats received a single intraperitoneal
injection of coconut oil or carbon tetrachloride in coconut oil (1:1
mixture) as 3 ml/kg rat weight (2378 mg/kg body weight) After 15 days,
a significant increase in the weight of the pituitary and a decrease
in the weights of the testes and seminal vesicles were observed.
Histological examination showed testicular atrophy and some
abnormality in the process of spermatogenesis in the experimental
animals (Chatterjee, 1966).
In a study of similar design with female rats, effects on the
reproductive system were seen 10 days after dosing. The effects
reported were: inhibition of estrous rhythm, reduction in ovarian and
uterine weights and vascularization, an increase in adrenal weight and
a marked reduction in pituitary gonadotrophin potency (Chatterjee
(1968).
Kalla & Bansal (1975) injected male rats with a mixture of 3
ml/kg body weight carbon tetrachloride in coconut oil (1:1, v/v) (2378
mg carbon tetrachloride/kg body weight) through an intraperitoneal
route for 10, 15 or 20 consecutive days. All dosing periods resulted
in decreased weights of testicles and accessory sex organs and
impairments in spermatogenesis. Dosing for 20 days resulted in an
entire deterioration of testicular tissue accompanied by an absence of
spermatids. The study was not reported adequately; number and strain
of rats were not reported.
7.5.2 Embryotoxicity and teratogenicity
The available data suggest that the fetus is not preferentially
sensitive to carbon tetrachloride, and effects of carbon tetrachloride
on fetal development and post-natal survival are likely to be
secondary to maternal toxicity.
7.5.2.1 Oral exposure
When carbon tetrachloride was administered by gavage to F-344
rats on gestation days 6-15 at 0, 25, 50 and 75 mg/kg per day in
either corn oil or in an aqueous vehicle containing 10% Emulphor(R),
it was more maternally toxic when administered in corn oil,
particularly at the highest dose. Full litter resorption (FLR)
occurred at 50 and 75 mg/kg with both vehicles. At 75 mg/kg, dams
receiving carbon tetrachloride in corn oil had a significantly higher
rate of FLR (67%) than those given the aqueous vehicle counterpart
(8%) (Narotsky et al., 1997a). Ammonium sulfide staining was used to
detect the resorption sites (Narotsky et al., 1997b).
Thiersch (1971) dosed pregnant rats with carbon tetrachloride (in
corn oil) at a level of 1000 mg/kg body weight on days 7, 7 and 8, 11,
or 11 and 12 of gestation. No malformations in the offspring were
reported, but the litters of the dams that had received two doses
showed more resorptions than the litters of those receiving one dose.
Clear information on a control group was not provided.
Hamlin et al. (1993) examined the effect on B6D2F1 mice of oral
administration of carbon tetrachloride (in corn oil) at concentrations
of 82.6 or 826.3 mg/kg body weight for five consecutive days beginning
on day 1, 6 or 11 of gestation. No effects were seen on maternal or
various neonatal parameters such as weight and crown rump length. No
malformations were detected in any pup on day 1 post-partum and the
pups developed normally.
7.5.2.2 Inhalation exposure
When Schwetz et al. (1974) exposed pregnant Sprague-Dawley rats
to measured carbon tetrachloride concentrations of 334 or 1004 ppm
(214 or 6435 mg/m3) for 7 h a day on days 6 to 15 of gestation, the
dams showed a dose-related decrease in food consumption (and body
weight gain). Signs of hepatotoxicity (increased ALAT activity) were
observed at both dose levels, but were not dose-related. Fetal body
weight and crown-rump length were significantly decreased. No
anomalies were seen upon gross examination of the fetuses. In both
exposure groups the incidence of fetuses with subcutaneous oedema was
increased but was statistically significant only in the lower dose
group. The incidence of sternebral anomalies (bipartite and delayed
ossification) was significantly increased in the fetuses of rats
exposed to the higher dose.
In an inhalation study by Gilman (1971), exposure of pregnant
rats to carbon tetrachloride at 1575 mg/m3 for 8 h a day on days 10
to 15 of gestation decreased the lactation index (83% compared to 98%
in the controls) and the viability index (83% as compared to 99% in
the controls).
7.6 Mutagenicity
The data from genotoxicity assays conducted with carbon
tetrachloride are summarized in Table 9. Since carbon tetrachloride is
a volatile compound that partitions preferentially in the hydrophobic
phase, the conditions adopted for in vitro experiments are important
to the outcome, but these conditions are often not reported in
sufficient detail. Carbon tetrachloride was not mutagenic to
Salmonella typhimurium in a large number of studies. It did,
however, induce DNA damage and mutations in single studies with
Escherichia coli. In fungi it induced intrachromosomal and mitotic
recombination. However, it did not induce aneuploidy in one study on
the yeast Saccharomyces cerevisiae, although aneuploidy was induced
in another single study with Aspergillus nidulans. In the only study
with Drosophila melanogaster, sex-linked recessive lethal mutations
were not induced by carbon tetrachloride.
In mammalian in vitro assays, carbon tetrachloride induced cell
transformation in a single study with Syrian hamster cells and
centromere-positive-staining micronuclei in human cell lines
expressing cDNAs for CYP1A2, CYP2A6, CYP3A4, epoxide hydrolase or
CYP2E1. The AHH-1 cell line constitutively expressing CYP1A1 showed no
increase in either total micronucleus frequency or centromere-staining
micronucleus frequency. There is little evidence for the induction
in vitro of DNA damage, unscheduled DNA synthesis, sister-chromatid
exchange or chromosomal aberrations.
In mammalian in vivo tests, carbon tetrachloride did induce DNA
strand breakage in one study but not in four others and did not
induce: a) unscheduled DNA synthesis in rat hepatocytes; b)
micronuclei in mouse hepatocytes, bone marrow cells or peripheral
blood erythrocytes; c) chromosomal aberrations in mouse bone marrow;
or d) aneuploidy in mouse hepatocytes. Binding of carbon tetra
chloride to liver cell DNA has been observed in rats, mice and Syrian
hamsters treated in vivo. There has been a report of a reduction in
I-compounds (species- and tissue-specific DNA adducts) in mouse liver.
The only clear evidence for genotoxicity comes from a number of
fungal cell experiments involving mutation and recombinational events.
Effects in mammalian cells indicate damage during cytokinesis. This
type of damage could result from interactions with proteins, rather
than DNA, e.g., of the trichloromethyl radical, and could be induced
secondarily to the toxicity of carbon tetrachloride (McGregor & Lang,
1996). Thus, no carbon-tetrachloride-DNA adduct identification has
been made, while the polar adducts observed in Syrian hamster liver
DNA appear to be derived from lipid peroxidation products (Wang &
Liehr, 1995). Consequently, strand-breakage and aneuploidy could arise
from the effects of lipid peroxidation products rather than carbon
tetrachloride or its metabolites; linoleic acid hydroperoxide, for
example, can induce single-strand breaks in DNA of cultured
fibroblasts (Nakayama et al., 1986). No resolved DNA damage has been
observed in vivo. It is concluded that although carbon tetrachloride
has some effects upon genetic material and these could be due to a
direct effect of carbon tetrachloride, no supporting evidence is
available; the effects are explicable in terms of nuclear protein or
DNA damage induced secondarily to carbon tetrachloride toxicity.
7.7 Carcinogenicity
7.7.1 Mice
After administration of 0.1 ml of a 40% solution of carbon
tetrachloride in olive oil (64 mg/mouse) by stomach tube to male C3H
mice, female C mice, and male and female A and Y mice 2 or 3 times a
week for 8 to 16 weeks (23 to 58 treatments), hepatomas developed in
126/143 (88%), 34/41 (83%), 63/64 (98%) and 9/15 (60%) of the C3H, C,
A and Y mice, respectively. No concurrent control data were reported.
Historical control data indicated the following incidence of hepatomas
(%) at one year or more: male C3H, 27%; female C, 0%; male and female
A, 1.5%; male and female Y, 1.6% (Edwards, 1941; Edwards & Dalton,
1942). In 34 of 73 male and female mice of the L strain that received
0.04 ml carbon tetrachloride per treatment by stomach tube 2 or 3
times a week for 46 treatments and were killed 3 to 3.5 months
following treatment, hepatomas were found that were similar to those
found in the other strains (Edwards et al., 1942).
Table 9. Mutagenicity studies with carbon tetrachloride
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
Bacterial systems
S. typhimurium TA 100 base-pair substitution not reported + rat PCB - McCann et al.,
TA 1535 1975
S. typhimurium G 46 base-pair substitution not reported + mouse i.n.r. - Kraemer et al.,
1974
S. typhimurium TA 1950 base-pair substitution concentrations: unknown - Braun &
G 46 10, 20 and 40 mg/ml Schöneich, 1975
S. typhimurium TA 1535 base-pair substitution test performed in + rabbit i.n.r. - Uehleke et al., 1977
TA 1538 frameshift mutation desiccators i.m.
S. typhimurium TA 100 base-pair substitution test performed in + rabbit PCB - Simmon et al., 1977
TA 1535 desiccators
TA 98 frameshift mutation
TA 1537
TA 1538
S. typhimurium TA 1535 base-pair substitution concentration: 8 mM, + mouse PB - Uehleke et al., 1977
TA 1538 frameshift mutation incubation in closed i.m.
containers
(survival > 90%)
S. typhimurium TA 100 base-pair substitution test performed in -/+ i.n.r. - Simmon & Tardiff,
TA 1535 desiccators sp. n.r. 1978
S. typhimurium TA 100 base-pair substitution concentrations: 4, 5.7, - - Barber et al.,
TA 1535 7, 10.2, 12.3, 18.4 1981
TA 98 frameshift mutation µmoles/plate; test for + rat PCB -
TA 1537 volatile liquids
TA 1538
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
S. typhimurium TA1535/pSK1002 SOS induction open + - Nakamura et al.,
1987
S. typhimurium TA1535/pSK 1002 SOS induction open + - Brams et al., 1987
S. typhimurium TA1535/pSK 1002 forward mutation open + - Roldàn-Arjona
et al., 1991
S. typhimurium TA1535/pSK 1002 forward mutation open + - Roldàn-Arjona &
Pueyo, 1993
S. typhimurium TA100 reverse mutation open + - Zeiger et al., 1988
TA1535
TA1537
TA97
TA98
S. typhimurium TA100 reverse mutation open + - Brams et al., 1987
TA98
TA97
S. typhimurium TA 100 base-pair substitution concentration: below - - De Flora, 1981;
TA 1535 toxicity limit De Flora et al.,
TA 98 frameshift mutation 1984
TA 1537
TA 1538
E. coli K 12 gene mutations not reported + mouse i.n.r. - Kraemer et al., 1974
i.m.
E. coli K 12 gene mutations not reported + rabbit i.n.r. - Uehleke et al., 1976
i.m.
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
E. coli uvrA back-mutation to trp concentration: + mouse i.n.r. - Norpoth et al., 1980
0.01% v/v
E. coli WP2 uvrA reverse mutation 2.5% atmosphere + (+) Norpoth et al., 1980
Non-mammalian eukaryotic systems
A. Nidulans unknown somatic segregation spot test technique n.r. - Bignami, 1977
(crossing-over and
non-disjunction)
A. Nidulans unknown induction of 8-aza- spot test technique n.r. - Bignami, 1977
guanine resistance
A. Nidulans 35 (haploid) forward mutation concentration: n.r. (+) Gualandi, 1984
1 (diploid) somatic segregation 0.5% v/v n.r. +
A. Nidulans aneuploidy - + Benigni et al., 1993
S. cerevisiae D7 gene conversion at concentration: 21, 28, - + Callen et al., 1980
trps-locus; mitotic 34 mM; test performed
recombination at ade-2; in screw-capped glass
gene conversion at ilv-1 tubes
S. cerevisiae AGY31DEL intrachromosomal - + Schiestl et al.,
recombination 1989
S. cerevisiae AGY31DEL intrachromosomal - + Galli & Schiestl,
recombination 1995
S. cerevisiae D61.M mitotic chromosome - - Whittaker et al.,
loss 1989
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
S. cerevisiae mitotic recombination - + Galli & Schiestl,
1996
Drosophila SLRL mutation feeding - Foureman et al.,
melanogaster 1994
Drosophila SLRL mutation injection - Foureman et al.,
melanogaster 1994
Chinese hamster V79 aneuploidy 2500 µg/ml - + Önfelt, 1987
lung
Cell line AHH1 (expressing aneuploidy 10 mM - - Doherty et al., 1996
CYP1A1) (centromere staining)
Cell line MCL-5 (cDNAs for aneuploidy 2 mM - + Doherty et al., 1996
CYP1A2, CYP2A6, (centromere staining)
CYP3A4, CYP2E1
and epoxide
hydrolase)
Cell line h2E1 (cDNAs for aneuploidy 2 mM - + Doherty et al., 1996
CYP2E1) (centromere staining)
Cell line AHH1 (expressing micronucleus 10 mM - - Doherty et al., 1996
CYP1A1)
Cell line MCL-5 (cDNAs for micronucleus 2 mM - + Doherty et al., 1996
CYP1A2, CYP2A6,
CYP3A4, CYP2E1
and epoxide
hydrolase
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
Cell line h2E1 (cDNAs for micronucleus 2 mM - + Doherty et al., 1996
CYP2E1)
In vitro mammalian systems
Chinese hamster ovary cells anaphase analysis concentration: 5 µl/ml - (+) Coutino, 1979
for chromosomal
rearrangements
Chinese hamster ovary cells chromosomal highest concentration: + rat PCB - Loveday et al., 1990
aberrations + SCE 3000 µg/ml
- -
Rat liver epithelium metaphase analysis concentration: 0.005, cells - Dean &
for chromosomal 0.01, 0.02 µl/ml in posses Hodson-Walker,
abnormalities sealed flasks; toxicity intrinsic 1979
observed activity
Human lymphocyte chromosomal concentration: + sp.n.r. i.n.r - Garry et al., 1990
aberrations + SCE 3.8-76 µg/ml
Host-mediated assays
S. typhimurium TA 1950 base-pair substitution mice NMRI; - Braun & Schöneich,
subcutaneous 1975
4 ml/kg body weight
S. typhimurium TA 1950 base-pair substitution mice CBA*C57Bl/6; - Shapiro & Fonshtein,
subcutaneous 20% 1979
solution in sunflower
oil
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
Syrian hamster cell transformation 3 µg/ml - (+) Amacher & Zelljadt,
embryo cells (clonal assay) 1983
Rat hepatocytes UDS 100 mg/kg × 1 p.o. or - Doolittle et al.,
in vivo 14 p.o. 1987
Rat hepatocytes SCE and chromosomal 1600 mg/kg × 1 p.o. - Sawada et al., 1991
in vivo aberrations and
micronucleus
Mouse bone chromosomal 8000 mg/kg × 1 - Lil'p, 1983
marrow in vivo aberrations
Mouse bone marrow micronucleus 2000 mg/kg and - Suzuki et al., 1997
and peripheral 3000 mg/kg
erythrocytes
in vivo
Mouse liver binding to DNA + Diaz-Gomez & Castro,
in vitro 1980a
Mouse, rat, binding to DNA + Castro et al., 1989
Syrian hamster
liver in vivo
Mouse liver ICR I-compound reduction 1600 mg/kg × 1 p.o. + Nath et al., 1990
in vivo (32P-post-labelling)
Syrian hamster binding to DNA + Wang & Liehr, 1995
liver and kidney
in vivo
a S. typhimurium = Salmonella typhimurium; A. Nidulans = Aspergillus nidulans; S. cerevisiae = Saccharomyces cerevisiae
b SCE = sister chromatid exchange; UDS = unscheduled DNA synthesis
c + = with metabolic activation; - = without metabolic activation; sp.n.r. = species not reported; n.r. = not reported whether metabolic
activation was used
d i.n.r. = inducer not reported; i.m. = intact microsomes added; PCB = polychlorinated biphenyls; PB = phenobarbital
e - = negative; + = positive; (+) = weakly positive
Table 10. Indicator tests with carbon tetrachloride
Species Strain Measured end-point Test conditions Activationa Inducationb Resultsc Reference
Bacterial systems
Escherichia coli WP2 (repair DNA repair variation 1: liquid - + De Flora et al.,
proficient WP67 micro method 1984
(uvrA polA-)
CM871 (uvrAa, + rat PCB +
recA-, lexA-)
variation 2: 24 h - +
preincubation and
plating out + rat PCB -
In vitro mammalian cells
Chinese hamster ovary cells SCE concentration: 0.001, - +d Athanasiou &
0.1, 1 mM; solvent Kyrtopoulos, 1981
DMSO
Chinese hamster ovary cells SCE -S9: toxic at 1490 + rat PCB - Loveday et al.,
µg/ml; +S9: 2930 1990
µg/ml highest dose
tested
Human lymphocyte SCE concentration: + sp.n.r. i.n.r. - Garry et al.,
3.8-76 µg/ml - - 1990
Human lymphocyte UDS concentration: 2.5, - PB - Perocco &
5 µg/ml; solvent + rat - Prodi, 1981
DMSO
Rat hepatocyte DNA single strand concentration: 0.03, - + Sina et al., 1983
breaks 0.3, 3 mM
Table 10. (Continued)
Species Strain Measured end-point Test conditions Activationa Inducationb Resultsc Reference
In vivo mammalian systems
Mouse NMRI DNA single strand 2.5 mg/kg body - Schwartz et al.,
breaks weight; single oral 1979
dose, undiluted or in
corn oil
Mouse CBA*BALB/c sperm head 0.1, 0.25, 0.5, 1.0, - Topham, 1980
abnormalities 1.5 mg/kg body
weight (i.p.) for 5
days; solvent corn oil
Mouse CDI DNA damage in 3H- 0.02-0.1 ml/kg body + Gans & Korson,
1984 labelled liver weight; single oral
cells dose in corn oil
Rat Wistar UDS in liver cells 4000 mg/kg body ee Craddock &
weight; single oral Henderson, 1978
dose, 2 and 17 h
exposure
Rat Fischer-344 UDS in liver cells 10 or 100 mg/kg - Mirsalis &
body weight; single Butterworth, 1980
oral dose in corn oil,
2 h exposure
Rat Wistar DNA damage single oral dose of - Stewart, 1981
(hepatectomized) 200-800 mg/kg body
weight in corn oil,
3 weeks after
hepatectomy
Table 10. (Continued)
Species Strain Measured end-point Test conditions Activationa Inducationb Resultsc Reference
Rat Fischer-344 DNA single strand 400 mg/kg body - Bermudez et al.,
breaks weight; single oral 1982
dose in corn oil
Rat Fischer-344 UDS in liver cells 40 or 400 mg/kg - Mirsalis et al.,
body weight; single 1982
oral dose up to 48 h
of exposure
Rat Sprague-Dawley DNA damage 200 mg/kg body - Brambilla et al.,
weight; single i.p. 1983
dose
a + = with metabolic activation; - = without metabolic activation; sp.n.r.= species not reported
b PCB = polychlorinated biphenyls; PB = phenobarbital; i.n.r.; = inducer not reported
c - = negative; + = positive; e = equivocal
d chromosome aberrations occurred, but no details on this finding were reported
e increase in DNA associated with tissue regeneration, but no increase in unscheduled DNA synthesis
Eschenbrenner & Miller (1944) administered 30 doses (each 0.005
ml) of 32%, 16%, 8%, 4% and 2% solutions of carbon tetrachloride in
olive oil (2540, 1270, 635, 318 or 160 mg/kg body weight) at 1- to
5-day intervals to mice of the A strain. Control group animals were
administered olive oil. All animals were examined for hepatomas 150
days after the first dose. Hepatomas were found in 33/60, 32/60,
25/60, 23/60 and 23/60 mice of the 32, 16, 8, 4 and 2% dose groups. A
variation in the numerical incidence of hepatomas at a given time was
observed to be related both to the total amount administered and to
the interval elapsing between successive doses. The incidence of
hepatomas increased with the interval between dosing from 1 to 4 days.
Weisburger (1977) reported that, in B6C3F1 mice dosed by gavage
with carbon tetrachloride in corn oil at levels of 1250 or 2500 mg/kg
body weight 5 days/week for 78 weeks and killed at 90 weeks, hepatomas
were found in 47/48 males and 43/45 females receiving the higher dose
and in 49/49 males and 40/40 females receiving the lower dose. Control
incidences of hepatomas were 3/18 in males and 1/18 in females. An
increase in adrenal tumours was also reported in the treated mice.
Groups of 50 male and 50 female BDF1 mice, 6 weeks of age, were
exposed by whole-body inhalation to 0, 5, 25 or 125 ppm (0, 32, 160 or
800 mg/m3) carbon tetrachloride (purity > 99.8%) for 6 h per day on
5 days a week for 104 weeks. Survival of the mid- and high-dose groups
in both sexes (35/50, 36/50, 25/50 and 1/50 males; 26/50, 24/49, 10/50
and 1/49 females) was decreased due to liver tumours. Significantly
increased incidences of hepatocellular adenomas (9/50, 10/50, 27/50
and 16/50 males; 2/50, 8/49, 17/50 and 5/49 females) were observed in
mid- and high-dose males (p < 0.01, Chi-square test) and in low- and
mid-dose females (low dose, p < 0.05; mid-dose, p < 0.01),
hepatocellular carcinomas (17/50, 12/50, 44/50 and 47/50 males; 2/50,
1/49, 33/50 and 48/49 females) in mid- and high-dose males and females
(p < 0.01) and pheochromocytomas of the adrenal gland (0/50, 0/50,
16/50 and 31/50 males; 0/50, 0/49,0/50 and 22/49 females) in mid- and
high-dose males (p < 0.01) and high-dose females (p < 0.01) (Nagano
et al., 1998).
7.7.2 Rats
In a study reported by Reuber & Glover (1970) male rats of the
Osborne-Mendel, Japanese, Wistar, Black and Sprague-Dawley strains
received twice a week a subcutaneous injection (1.3 ml/kg body weight)
of a 50% solution of carbon tetrachloride in corn oil (1036 mg/kg body
weight). The Japanese rats survived for 47 weeks and Osborne-Mendel
rats for 44 weeks. Wistar rats lived for 33 weeks and Black and
Sprague-Dawley rats lived only for 11 and 33 weeks, respectively.
Hepatocellular carcinomas developed in 8/13 Osborne-Mendel rats and in
12/15 Japanese rats, and hyperplastic nodules were also found in these
strains. The hepatocellular carcinomas found in 4/12 Wistar rats were
smaller. Nodules and hyperplasia were also observed in Wistar rats, as
well as moderate to severe cirrhosis. The Black rats and
Sprague-Dawley rats died with severe cirrhosis before they developed
carcinomas. No hepatocellular carcinomas developed in the controls of
any strain.
Weisburger (1977) reported increases in the incidence of
neoplastic nodules and hepatocellular carcinomas after oral
administration of carbon tetrachloride in corn oil to Osborne-Mendel
rats at doses of 47 and 94 mg/kg body weight (males) and 80 and 160
mg/kg body weight (females) for 78 weeks. The rats were killed at 110
weeks. The incidences of hepatocellular carcinomas in the controls
were 0/20 in males and 1/20 in females, and the incidences of liver
neoplastic nodules were 0/20 in males and 1/20 in females.
Groups of 50 male and 50 female F-344 rats, 6 weeks of age, were
exposed by whole-body inhalation to 0, 5, 25 or 125 ppm (0, 32, 160,
800 mg/m3) carbon tetrachloride (purity > 99.8%) for 6 h per day on
5 days a week for 104 weeks. Survival of the high-dose groups in both
sexes (22/50, 29/50, 19/50 and 3/50 males; 39/50, 43/50, 39/50 and
1/50 females) was decreased due to liver tumours and chronic
nephropathy (progressive glomerulonephrosis). There were significantly
increased incidences of hepatocellular adenomas (0/50, 1/50, 1/50 and
21/50 males; 0/50, 0/50, 0/50 and 40/50 females) and hepatocellular
carcinomas (1/50, 0/50, 0/50 and 32/50 males; 0/50, 0/50, 3/50 and
15/50 females) in high-dose rats of each sex (p < 0.01, Chi-square
test) (Nagano et al., 1998).
7.8 Special studies
7.8.1 Immunotoxicity
Carbon tetrachloride treatment of B6C3F1 female mice resulted in
marked suppression of both humoral and cell-mediated immune functions.
Humoral immunity, as measured by the T-dependent antibody response to
sheep red blood cells (SRBC), proved to be the most sensitive
indicator of carbon-tetrachloride-induced immunotoxicity. Carbon
tetrachloride was immunotoxic in female B6C3F1 mice at all doses
tested (500-5000 mg/kg body weight) and there were no significant
differences in the magnitude of immunosuppression between oral and
intraperitoneal routes of exposure. There was no dose-response
relationship with respect to SRBC antibody responses; all dose levels
resulted in approximately 50% suppression of the control response. To
determine whether a dose-response relationship could be attained,
female mice were treated at lower carbon tetrachloride levels (25, 50
and 100 mg/kg body weight) for 30 consecutive days. It appeared that
doses as low as 50 mg/kg body weight produced the maximum attainable
inhibition of SRBC response (50%); 25 mg/kg body weight caused a 20%
reduction (Kaminsky et al., 1989; 1990).
Delaney & Kaminski (1994) studied the immunomodulatory activity
of serum isolated from carbon tetrachloride-treated B6C3F1 mice on
T-cell-independent humoral immune responses. The results of the study
suggested that carbon tetrachloride has bifurcating immunological
effects. Exposure to carbon tetrachloride appears to suppress
T-cell-dependent immune responses but enhance the activity of B-cells.
Both effects appear to be mediated by blood-borne factors. Incubation
of sera from carbon-tetrachloride-treated mice with neutralizing
monoclonal antibodies toward transforming growth factor ß1 reversed
the immunosuppression, indicating that TGF ß1 at least in part
mediates the immunosuppression induced by carbon tetrachloride.
In contrast to the results found in mice by Kaminsky et al.
(1989, 1990), Smialowicz et al. (1991) found no consistent alterations
in humoral or cell-mediated immune function in male Fischer-344 rats
at dosages that clearly resulted in body weight decreases and
hepatotoxicity. However, the dose levels used in this study (up to 40
mg/kg body weight) were much lower than those reported by Kaminsky et
al. (1989, 1990).
Mice (A/PhJ) were administered carbon tetrachloride (about 300
mg/kg per day intraperitoneally in olive oil) for 2, 7, 14 and 23
days. A variety of immunological parameters were evaluated.
Morphological examination by light microscopy revealed significant
activation of lymphoid tissues in T-cell-dependent areas and only
slight activation in B-cell-dependent areas (Jirova et al., 1996).
Thymus weights (after exposure for 2, 14 and 23 days) and spleen
weights (after exposure for 2 and 23 days) decreased significantly
when weights at 23 days were compared to those of controls. The
response of SRBC was permanently suppressed from the beginning of
exposure.
7.8.2 Influence of oxygen levels
It is known that under normal atmospheric conditions carbon
tetrachloride initiates lipid peroxidation in mice and rat liver
microsomes and in mice and rats in vivo (Sagai & Tappel, 1979;
Kornbrust & Mavis, 1980; Gee et al., 1981; Lee et al., 1982). At
reduced oxygen concentrations the carbon-tetrachloride-induced lipid
peroxidation is greatly enhanced in in vivo experiments and in
microsomal preparations, but in these in vitro systems, the process
is entirely blocked under anoxic conditions (Kieczka & Kappus, 1980;
Noll & De Groot, 1984).
Covalent binding to RNA and DNA was enhanced in the absence of
oxygen when male rat (Sprague-Dawley) hepatocytes were treated with
carbon tetrachloride (Cunningham et al., 1981). DiRenzo et al. (1984)
observed that the enhanced covalent binding to protein or lipid,
caused by carbon tetrachloride in cultured male rat (Sprague-Dawley)
hepatocytes, at decreased oxygen tension was most evident for the
binding to lipids.
Shen et al. (1982) studied the effect of oxygen concentrations on
carbon-tetrachloride-induced hepatotoxicity in male Long-Evans rats
exposed to differing oxygen concentrations combined with different
carbon tetrachloride concentrations. In this in vivo experiment,
carbon tetrachloride appeared to be more toxic when oxygen
concentrations were reduced, as shown by increased ALAT activity, more
severe centrilobular necrosis, and increased covalent binding to
hepatic microsomal lipids and proteins.
An increased metabolism of carbon tetrachloride under hypoxic
conditions, and consequently an aggravated hepatotoxicity, was
reported in male Wistar rats by Siegers et al. (1985). In agreement
with the reports of Kieczka & Kappus (1980) and Noll & De Groot
(1984), a more pronounced lipid peroxidation was observed, as shown by
exhaled ethane, than in animals kept under normal oxygen conditions
and treated with the same dose of carbon tetrachloride.
Male Sprague-Dawley rats were used in a study by Burk et al.
(1988) to examine the effect of hyperbaric oxygen on carbon
tetrachloride metabolism by different isoenzymes of cytochrome P-450.
The authors concluded that under low oxygen tensions the rate of
carbon tetrachloride metabolism depended largely on the amount of
cytochrome P-450 present, while under higher oxygen tensions the major
determinant was the type of cytochrome P-450.
7.9 Factors modifying toxicity
7.9.1 Dosing vehicles
Several studies have demonstrated that
carbon-tetrachloride-induced hepatotoxicity, like absorption (see
section 6.1.1.1), can be influenced by the dosing vehicle.
In order to evaluate the effect of vehicle on the hepatotoxicity
in mice, Condie et al. (1986) treated male and female CD-1 mice with
oral doses of carbon tetrachloride (0, 1.2, 12 or 120 mg/kg body
weight) in either corn oil or 1% Tween-60 vehicle, 5 times/week for 90
days. Differences between the vehicles were observed in mice at the 12
mg/kg dose level. Hepatomegaly and more fat accumulation were observed
when carbon tetrachloride was administered in corn oil. At the highest
dose level the usage of corn oil as vehicle caused a greater
hepatotoxic effect, as shown by necrosis and fatty infiltration in the
mice. The data indicated that the NOAEL for hepatotoxic effects after
carbon tetrachloride exposure in corn oil was 1.2 mg/kg body weight,
while the NOAEL for the Tween-60 groups was 12 mg/kg body weight.
When pregnant F-344 rats were dosed by gavage with carbon
tetrachloride in corn oil or an aqueous vehicle containing 10%
Emulphor during gestation, corn oil was associated with a full litter
resorption (FLR) rate of 67%, compared to 8% in those dosed with
Emulphor (Narotsky et al., 1997a). Further details regarding this
study are described in section 7.5.2.1.
Koporec et al. (1995) determined the vehicle effects on the
subchronic toxicity of carbon tetrachloride in male Sprague-Dawley
rats. Carbon tetrachloride was administered at dose levels of 0, 25 or
100 mg/kg body weight by gavage in either corn oil or a 1% Emulphor
aqueous emulsion 5 days/week for 13 weeks. It was concluded that there
was no difference in the subchronic hepatotoxicity of carbon
tetrachloride in rats when given in corn oil or as an aqueous
emulsion. This result contrasts with the result found in mice
described by Condie et al. (1986), and with the result of the study in
male Sprague-Dawley rats by Kim et al. (1990b), who reported that the
hepatotoxicity in male Sprague-Dawley rats was less pronounced at each
dose level when corn oil was used as a vehicle. Administration of
undiluted carbon tetrachloride or of an aqueous emulsion produced
comparable toxicity.
Szende et al. (1994) dosed male F-344 rats with carbon
tetrachloride (0.2 ml/kg body weight) dissolved in various oils
(sunflower, corn, fish or olive oil) by gastric intubation 3
times/week for 8 weeks. The increase of collagen fibres in the liver
was only 2-4% when olive oil was used as a vehicle, instead of the
6-8% increase when the other oils were used.
7.9.2 Diet
In male NMRI mice, fasted for 24 h before receiving an
intraperitoneal injection (0.1 ml/kg body weight) of carbon
tetrachloride (159.4 mg/kg body weight) in olive oil, higher hepatic
carbon tetrachloride and chloroform levels were found (Pentz &
Strubelt, 1983).
Fed male Sprague-Dawley rats appeared to be more resistant to the
toxic action of carbon tetrachloride than rats starved overnight.
Contrary to findings in mice, the carbon tetrachloride concentrations
were similar in the livers of fed and fasted rats (Díaz Gómez et al.,
1975b).
Male Wistar rats were fed various test diets in order to assess
the nutritional effects on the liver mixed-function oxidases (MFO) and
consequently on the carbon tetrachloride metabolism. The MFO activity
increased almost linearly with decreasing food intake. Furthermore it
was shown that a diet deficient in carbohydrate enhanced the
metabolism and thus the toxic action of carbon tetrachloride,
irrespective of the protein or fat content of the diet (Nakajima et
al., 1982; Sato & Nakajima, 1985).
Cervinková et al. (1987) investigated the effect of long-term
intake of high or low protein diet on liver repair processes after the
administration of carbon tetrachloride. Rats were fed for 21 days on a
low-protein diet (LPD), a standard diet (SD) and a high-protein diet
(HPD) and were then given a single intraperitoneal injection (0.75
ml/kg body weight) of carbon tetrachloride (calculated to be 1196
mg/kg body weight). The HPD was found to increase sensitivity to
carbon tetrachloride, but it also promoted liver repair processes. The
LPD raised liver resistance to carbon tetrachloride, but the
development of liver repair activity differed from the process after
the SD and HPD, since polyploidy of the hepatocytes predominated and
there was also an increase in the number of binuclear hepatocytes.
Cell hypertrophy was expressed less in rats fed on the LPD. As far as
liver repair was concerned, the HPD showed no explicit advantage over
the SD.
7.9.3 Alcohol
Several studies have demonstrated that ethanol, methanol and
other alcohols potentiate the hepatic toxicity of carbon tetrachloride
(Traiger & Plaa, 1971; Cantilena et al., 1979; Harris & Anders, 1980;
Ray & Mehendale, 1990; Simko et al., 1992). Dietary ethanol (2 g/80 ml
liquid diet for 3 weeks) potentiated carbon tetrachloride (inhalation
exposure to 10 ppm (64.1 mg/m3) for 8 h) hepatotoxicity, measured by
serum aminotransferases and liver malonaldehyde concentrations, in
male Wistar rats. Potentiation did not occur upon exposure to 5 ppm
(32 mg/m3) for 8 h (Ikatsu et al., 1991; Ikatsu & Nakajima, 1992).
Only a minor potentiating effect on weight gain, but no effect on
carbon-tetrachloride-induced hepatotoxicity was observed, when rats
were simultaneously treated with ethanol (æ 0.5 ml/kg) and carbon
tetrachloride (20 mg/kg) by gavage for 14 days (Berman et al., 1992).
Micronodular cirrhosis was observed in all treated rats after 10 weeks
of inhalation exposure to carbon tetrachloride (513 mg/m3, 6 h/day, 5
days/week) when the animals were simultaneously given ethanol as a
part of a liquid diet, while no animal treated with either ethanol or
carbon tetrachloride alone developed cirrhosis (Hall et al., 1991).
Similar cirrhosis was also observed in Porton rats treated with carbon
tetrachloride and ethanol (Hall et al., 1994). Inhalation exposure to
methanol (10 000 ppm for 6 h) increased the liver toxicity of carbon
tetrachloride (a single dose of 0.075 ml/kg after 24 h) (Simmons et
al., 1995). Similar exposure to methanol also increased the toxicity
of inhaled carbon tetrachloride (100, 250 to 1000 ppm (641, 1602 to
6410 mg/m3) for 6 h at 26-27 h after the beginning of the methanol
exposure). This potentiation subsided when the interval between
methanol and carbon tetrachloride exposures was increased by 24 h
(Evans & Simmons, 1996). Malonaldehyde generation induced by carbon
tetrachloride in vitro was enhanced by prior exposure of the rats to
methanol (10 000 ppm for 6 h); this enhancement coincided with an
increased microsomal activity of para-nitrophenol hydroxy lase, used
as a marker of cytochrome P-450 2E1; inhibition of CYP 2E1 by allyl
sulfone abolished the carbon-tetrachloride-induced lipid peroxidation
(Allis et al., 1996).
Sato & Nakajima (1985) reported that the metabolism of carbon
tetrachloride in the rat was enhanced by pretreatment with ethanol. It
was found that the increase in carbon tetrachloride hepatotoxicity was
related to the degree of the enhancement. Similar observations were
made by Sato et al. (1980), Teschke et al. (1984), Strubelt (1984) and
Reinke et al. (1988).
In a study by Wang et al. (1997), before exposure to carbon
tetrachloride, rats were kept either on an ethanol-containing (2 g/80
ml per rat per day) liquid diet for 3 weeks, to obtain a maximal
induction of the alcohol-inducible CYP 2E1 isoenzyme, or on a liquid
diet with no alcohol. Both groups were exposed to carbon tetrachloride
by inhalation (0, 320 or 3205 mg/m3 for 6 h), or by oral or
intraperitoneal administration (0, 0.105 or 1.675 mmol/kg).
Ethanol-pretreatment increased significantly the metabolism of carbon
tetrachloride as indicated by the carbon tetrachloride concentrations
in blood samples. Plasma ALAT and ASAT levels, assayed 24 h after
carbon tetrachloride treatment, were highly significantly increased in
all carbon tetrachloride-dosed rats pretreated with ethanol, whereas
in control diet groups only a slight elevation of transaminases was
observed after the high-dose carbon tetrachloride treatment.
Shibayama (1988) compared the hepatotoxicity of carbon
tetrachloride (intraperitoneal administration in olive oil) in male
Wistar rats fed a standard diet and 5 or 20% ethanol solution with the
hepatotoxicity of carbon tetrachloride in control rats, which received
water instead of ethanol for a period of 1 to 100 weeks. The results
indicated that the effect of ethanol on the hepatotoxicity is
dependent on the daily amount of alcohol intake and is not affected by
the duration of the alcohol consumption. Kniepert et al. (1990),
however, reported that an increased duration (30 or 52 weeks instead
of 1 or 10) of ethanol pretreatments (10% in drinking water) caused a
decrease in ethanol potentiation of carbon-tetrachloride-induced
toxicity in male Wistar rats.
Ray & Mehendale (1990) studied the effect caused by a single dose
of carbon tetrachloride after pretreatment with various homologous
alcohols in male Sprague-Dawley rats. A combination of the alcohols
methanol, ethanol, isopropanol and decanol with carbon tetrachloride
potentiated liver injury but did not affect lethality. A combination
of t-butanol, pentanol, hexanol and octanol potentiated liver injury
and decreased animal survival significantly. Eicosanol potentiated
neither liver injury nor lethality.
Hall et al. (1994) observed that chronic administration of
alcohol and 'low-dose' carbon tetrachloride vapour caused cirrhosis in
all male Porton rats receiving this treatment for 5-7 weeks. A
possible mechanism for the interaction between alcohol and carbon
tetrachloride is the alcohol-dependent induction of cytochrome P-450
2E1, resulting in enhanced production of toxic metabolites of carbon
tetrachloride. These in turn are responsible for the initiation of
lipid peroxidation and impaired conjugation of carbon tetrachloride
metabolites with glutathione.
To determine the dose-response relationships in the production of
hepatic fibrosis and cirrhosis, the livers of male Porton rats (4
animals/group) were examined after combined exposure to carbon
tetrachloride and alcohol (Plummer et al., 1994). Carbon tetrachloride
was administered by inhalation 6 h/night for 5 nights/week at
concentrations of 10, 20 or 40 ppm (actual values of 60, 120.1 or
240.1 mg/m3, respectively). Ethanol was administered orally at levels
of 75, 150, or 300 kcal/litre liquid diet, leading to mean daily
intakes of 2.29, 4.61 and 8.16 g ethanol/kg body weight, respectively.
It was proposed to continue administration of alcohol and carbon
tetrachloride until animals became cirrhotic, as diagnosed by liver
biopsy, or for a maximum of 20 weeks. However, the alcohol consumption
declined gradually, approximately to half that at the beginning.
Results of the study show that both alcohol and carbon tetrachloride
contribute to the liver injury in a dose-related manner. All four rats
that received the high dose of both carbon tetrachloride and alcohol,
and one of four rats that received the medium alcohol and high-dose
carbon tetrachloride treatments, showed liver cirrhosis after 10 weeks
of exposure. Two of four rats that received the low alcohol in
combination with the high-dose of carbon tetrachloride showed
cirrhosis after 20 weeks. Although cirrhosis was observed only at the
highest carbon tetrachloride dose, some degree of hepatic fibrosis was
observed in all treated rats in a dose-related manner.
Daniluk et al. (1994) reported that acute liver injury due to
intraperitoneal carbon tetrachloride administration combined with a
long-term alcohol consumption may act synergistically in depressing
interferon production in C3H/He mice.
Strain differences in response to carbon tetrachloride have been
described for both mice (see section 7.1.2.3) and rats (see section
7.3).
7.9.4 Enhancement of carbon tetrachloride-induced hepatotoxicity by various compounds
According to Klingensmith et al. (1983) the LD50 of carbon
tetrachloride after oral administration dropped by a factor of 14
after pretreatment with chlordecone. The potentiation of carbon
tetrachloride toxicity by chlordecone appears to be related to a
chlordecone-dependent increase in the biotransformation rate of carbon
tetrachloride and a chlordecone-dependent reduction of the
liver-regenerating capacity (Mehendale, 1984).
Mehendale (1989) described a mechanism for the potentiation of
carbon tetrachloride hepatotoxicity by chlordecone. The mechanism
underlying the highly unusual amplification of carbon tetrachloride
toxicity relates to the suppression of the initial hepatocellular
regeneration, ordinarily stimulated by low doses of carbon
tetrachloride.
Pretreatment with phenobarbital enhanced the metabolism of carbon
tetrachloride in rats, and consequently the toxicity (Bechtold et al.,
1982; Fander et al., 1982; Sato & Nakajima, 1985), whereas
pretreatment with PCBs or 3-methylcholanthrene for a few days hardly
influenced the carbon tetrachloride metabolism and carbon
tetrachloride toxicity (Sato & Nakajima, 1985). Prolonged pretreatment
with hexachlorobenzene, PBBs or PCBs, however, made male rats
considerably more susceptible to the toxic effects of carbon
tetrachloride (Kluwe et al., 1982).
The reaction of the liver to carbon tetrachloride was studied in
the adaptive stage of organic solvent poisoning. Rats were pretreated
daily with benzene, toluene, xylene, phenobarbital or oil for 4 days.
On day 4, carbon tetrachloride was given orally and 24 h later the
rats were killed. Histological, histochemical and electron microscopic
examination revealed a potentiating interaction between the solvents
and carbon tetrachloride, similar to the potentiation of carbon
tetrachloride toxicity by simultaneous phenobarbital administration.
Centrilobular necrosis caused by carbon tetrachloride became confluent
and turned submassive in the livers of pretreated animals, and it
appeared that in the rats treated with solvent and carbon
tetrachloride, the amount of damaged area was twice that induced by
carbon tetrachloride alone (Tátrai et al., 1979).
Qazi & Alam (1988) reported that phenobarbitone treatment in
drinking-water together with carbon tetrachloride (by intraperitoneal
injection) induced severe liver cirrhosis with marked proliferation of
the bile ducts in female rats. Females receiving only carbon
tetrachloride showed moderate cirrhosis whereas the females receiving
only phenobarbitone remained healthy, showing only an increased liver
weight.
ElSisi et al. (1993a,b) studied the effect of retinol (vitamin A)
on carbon-tetrachloride-induced hepatoxicity in a time-response and a
dose-response study. In the time-response study, male Sprague-Dawley
rats were given 75 mg/kg body weight retinol for 1 or 3 days, or 1,2,3
or 5 weeks. In the dose-response study, retinol was given at daily
doses of 30 to 75 mg/kg body weight for 3 weeks. At 24 h after the
last dose of retinol, 0.15 ml carbon tetrachloride/kg body weight (239
mg/kg body weight) in corn oil was given by intraperitoneal injection
and another 24 h later the animals were killed. All treatment
durations with retinol, except 1 day, resulted in equivalent
potentiation of carbon tetrachloride hepatotoxicity. All rats
pretreated with retinol and subsequent administration of carbon
tetrachloride had more extensive liver injury than those given carbon
tetrachloride alone. As the daily dose of retinol increased, so did
the degree of potentiation of carbon tetrachloride hepatotoxicity.
Pretreatment with ketonic or ketogenic compounds (e.g., hexane,
acetone, isopropanol) potentiated the liver injury in Sprague-Dawley
rats produced by an intraperitoneal carbon tetrachloride injection
(Charbonneau et al., 1985).
The hepatotoxicity of the liver to carbon tetrachloride is
considerably enhanced in alloxan-diabetic rats. This potentiation
effect can be reversed by an additional insulin treatment before the
carbon tetrachloride administration (Villarruel et al., 1982).
Intraperitoneal treatment of male Sprague-Dawley rats with
pyrazole increased the sensitivity of these rats to carbon
tetrachloride hepatotoxicity as assessed by significant loss of
cytochrome P-450 and increases in ASAT and ALAT levels. As stated by
the author (Ebel, 1989), these observations are consistent with the
hypothesis that only certain forms of P-450 (and in this case the
'alcohol-inducible form') are capable of activation of hepatotoxins
and potentiate the toxicity.
Imidazole and pyrazole, inducers of CYP 2E1, caused 3- to 25-fold
enhanced rates of carbon-tetrachloride-induced lipid perioxidation
(and chloroform production from carbon tetrachloride); the increase
was directly related to the amount of this cytochrome in the
microsomes (Johansson & Ingelman-Sundberg, 1985).
Acetone, methyl ethylketone and methyl isobutylketone (6.8
mmol/kg body weight for 3 days) increased the hepatotoxicity of carbon
tetrachloride (Raymond & Plaa, 1995a); this enhanced toxicity was
coincident with an increased microsomal aniline hydroxylase activity
(Raymond & Plaa, 1995b). In addition to the effect on cytochrome
P-450, acetone, but not the other ketones, increased basal canalicular
membrane fluidity, as measured by fluorescence polarization of
1,6-diphenyl-1,3,5-hexatriene or
1,4-(trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene (Raymond &
Plaa, 1996).
Treatment of male athymic nude rats, male and female
Sprague-Dawley rats, and male Fischer-344 rats with retinol (75
mg/kg/day for 7 days) greatly enhanced the hepatotoxicity of carbon
tetrachloride in F-344 rats (0.2 or 0.1 mg/kg i.p.), while it
protected Balb/C, C3H/HeJ, athymic nude and Swiss-Webster mice against
carbon tetrachloride hepatotoxicity (0.0125 to 0.02 ml/kg,
respectively) (Hooser et al., 1994). In male Sprague-Dawley rats
retinol (> 100 000 IU/kg per day for 3 weeks or 250 000 IU/kg per
day for > 1 week) greatly increased the hepatotoxicity of carbon
tetrachloride (0.15 ml/kg intraperitoneally) (ElSisi et al., 1993a).
There was a simultaneous six- to eight-fold increase in the amount of
exhaled ethane and a less than two-fold increase in covalent binding
to liver proteins in rats treated with retinol (250 000 IU or 75 mg/kg
per day for 1 week) and carbon tetrachloride (0.15 ml/kg), in
comparison with rats treated with carbon tetrachloride alone. However,
there was no increase in the exhaled 14CO2, exhaled organics or
metabolites excreted in the urine, or covalent binding to hepatic
lipids from 14C-carbon tetrachloride. Aminobenzotriazole (50 mg/kg
intraperitoneally, 2 h before carbon tetrachloride), an inhibitor of
cytochrome P-450, blocked the retinol-induced potentiation of the
hepatotoxicity of carbon tetrachloride (ElSisi et al., 1993b). A
single dose of retinol (> 75 mg/kg orally) 24 h before carbon
tetrachloride also very significantly potentiated carbon tetrachloride
hepatotoxicity (Badger et al., 1996). While the total cytochrome P-450
content of the liver was not affected by the retinol treatment, the
concentration (Western blot analysis) and activity (aniline
hydroxylase) of CYP 2E1 were both elevated. Isolated hepatocytes from
retinol-treated rats also exhibited enhanced susceptibility to carbon
tetrachloride (Badger et al., 1996).
Concomitant administration of a single, non-hepatotoxic dose of 6
mmol dichloromethane (DCM)/kg and 308 mg carbon tetrachloride
intraperitoneally potentiated carbon tetrachloride-induced
hepatotoxicity, as measured by SDH and ALAT. When a radiolabelled
tracer dose of carbon tetrachloride was included in the treatment, DCM
was found to significantly increase the covalent binding of
[14C]-carbon tetrachloride metabolites to microsomal lipids. However,
DCM did not affect lipid peroxidation induced by carbon tetrachloride
(Kim, 1997).
Potentiation of carbon tetrachloride hepatotoxicity was studied
after inhalation exposure at concentrations of 0, 5 or 10 ppm (0, 160
or 320 mg/m3) for 8 h and simultaneous exposure to chloroform (0, 25
or 50 ppm for 8 h). While carbon tetrachloride exposure had no effect
on plasma ALAT or ASAT levels, co-exposure to chloroform resulted in a
slight increase of the transaminase activity in blood (Ikatsu &
Nakajima, 1992).
7.9.5 Reduction of carbon tetrachloride-induced hepatotoxicity by various compounds
Vitamin E reduces the carbon-tetrachloride-induced lipid
peroxidation in rat liver and kidney slices (Gavino et al., 1984), but
in an in vivo experiment in rats only limited protection by vitamin
E against this process was seen (Gee et al., 1981). A protective
action of vitamin E against liver cell membrane damage in Wistar rats
was reported in several studies (Ozeki et al., 1982; Martínez-Calva et
al., 1984). This protective action could be reinforced by
co-administration of vitamin E and riboflavin tetrabutyrate (Miyazawa
et al., 1984). When male Wistar rats were given vitamin E 15 h before
carbon tetrachloride administration, a partial or complete protection
against the necrogenic effect of carbon tetrachloride was induced,
depending on the concentration of carbon tetrachloride used.
Furthermore, the vitamin supplementation prevented the
carbon-tetrachloride-induced increase in total hepatic calcium content
(Biasi et al., 1991).
Protection, apparent as a decrease in mortality, less pronounced
histological damage and lower serum aminotransferase levels was
afforded by intravenous administration of alpha-tocopherol as a suspen
sion or in liposomes, which are accumulated in Kupffer cells (Yao et
al., 1994; Liu et al., 1995). Where incorporated into liposomes, other
antioxidants, such as butylated hydroxytoluene and ascorbic acid
palmitate, also protected mice against carbon tetrachloride toxicity
(Yao et al., 1994).
Preventive effects on hepatotoxicity in rats were also reported
for vitamin D3 and vitamin C by Fander et al. (1982) and Ademuyiwa et
al. (1994), respectively.
In contrast to the results of ElSisi et al. (1993a,b), who
reported a potentiation in hepatotoxicity when retinol was given to
rats prior to carbon tetrachloride administration, Rosengren et al.
(1995) observed a protective effect on the liver when retinol was
given to mice prior to carbon tetrachloride administration.
Studies reported by Bishayee & Chatterjee (1993) and Mandal et
al. (1993) indicated a possible hepatoprotective role of carrot
(Daucus carota) aqueous extract and Mikania cordata root extract
in male Swiss mice. The increased lipid peroxidation and decreased
glutathione levels, resulting from carbon tetrachloride treatment,
were significantly reversed in a dose-related way after pretreatment
of the mice with the carrot extract. According to authors this
protective role of carrot extract could be attributed to its
antioxidant properties.
Pretreatment of rats with SKF-525 A (inhibitor of
drug-metabolizing enzymes), cysteine (in particular in combination
with tryptophan) or reduced glutathione decreased the
carbon-tetrachloride-induced hepatotoxicity (Bechtold et al., 1982; de
Ferreyra et al., 1983; Gorla et al., 1983).
Administration of
6,6'-methylene-bis(2,2,4-trimethyl-1,2-dihydroquinoline),
tert-butyl-4-hydroxyanisole, oltipraz or anethol dithiolthione
protects mice against the acute toxic effects of carbon tetrachloride
(Fehér et al., 1982; Toncsev et al., 1982; Ansher et al., 1983;
Benson, 1993). A preventive effect was also observed when
diethyldithiocarbamate and carbon disulfide were administered to mice.
Diethyldithiocarbamate was most effective when given orally, while the
action of carbon disulfide was less dependent on the route of
administration (Masuda & Nakayama, 1982).
Rao & Mehendale (1989) reported that administration of fructose
1,6-diphosphate decreased the toxicity of carbon tetrachloride in rats
(as shown by a 50-70% decrease in the activity of serum
transaminases). This decrease was accompanied by elevated activities
of enzymes involved in the polyamine metabolism (important for
hepatocellular regeneration and recovery).
It appears that nicotinamide administered to male Sprague-Dawley
rats at late stages of carbon tetrachloride poisoning (e.g., 6 or 10 h
after the hepatotoxin) significantly prevents the liver necrogenic
effects of carbon tetrachloride at 24 h. A study by de Ferreyra et al.
(1994) did not reveal any relevant effect when nicotinamide was given
30 min before the hepatotoxin.
Simultaneous treatment of carbon-tetrachloride-intoxicated rats
with zinc (227 mg/litre in drinking-water) resulted in improved serum
and liver enzyme levels and attenuated histological abnormalities as
well as NADPH-dependent lipid peroxidation (Dhawan & Goel, 1994).
Studies of Kaminski et al. (1989, 1990) demonstrated that carbon
tetrachloride administration in mice results in a marked suppression
of humoral and cell-mediated immune functions. Ahn & Kim (1993)
observed that PMC (diphenyl dimethyl dicarboxylate) had a significant
preventive effect on carbon-tetrachloride-induced immunotoxic status
in ICR mice that were immunized and challenged with sheep red blood
cells (SRBC) and were subsequently given PMC (3 or 6 mg/kg body
weight; oral administration) once a day for 28 days in combination
with carbon tetrachloride (1 ml/kg body weight, 25%, oral
administration) twice a week, 2 h after PMC administration.
Gadolinium chloride (10 mg/kg), given intravenously 24 h prior to
an intragastric dose of carbon tetrachloride (4 g/kg), nearly
completely protected rats from hepatic necrosis, as measured by serum
ASAT levels and trypan blue exclusion, without an effect on CYP 2E1
(Edwards et al., 1993). This was interpreted to indicate a role of
Kupffer cells in carbon-tetrachloride-induced hepatic damage, since
gadolinium chloride at this concentration strongly inhibits Kupffer
cell phagocytosis (Hustzik et al., 1980). Similar dosage of gadolinium
chloride was, however, also reported to decrease the total amount of
hepatic cytochrome P-450 in rats, as well as the activity of aniline
para-hydroxylase (Badger et al., 1997). In support of the role of
Kupffer cells in carbon-tetrachloride-induced hepatic damage, it was
reported that gadolinium chloride (10 mg/kg given intravenously 24 h
before carbon tetrachloride administration) prevented and methyl
palmitate, another Kupffer cell inhibitor, attenuated the periportal
oedema observed using proton magnetic imaging 1-2 h after carbon
tetrachloride administration (0.8 ml/kg given intraperitoneally)
(Towner et al., 1994). In vivo pin trapping using
alpha-phenyl- N-tert- butylnitrone and a subsequent electron
paramagnetic resonance study of the liver indicated that gadolinium
chloride did not affect the generation of trichloromethyl radicals
from carbon tetrachloride (Towner et al., 1994). Gadolinium chloride
(10 mg/kg given intravenously), methyl palmitate,
polyethylene-glycol-coupled superoxide dismutase and
polyethylene-glycol-coupled catalase protected Sprague-Dawley rats
against retinol-induced potentiation of carbon tetrachloride
hepatotoxicity, both after a single dose and daily dosing for seven
days of retinol (ElSisi et al., 1993a; Sauer & Sipes, 1995; Badger et
al., 1996). Dietary alpha-tocopherol (250 mg/kg diet) partly protected
male Wistar rats against carbon-tetrachloride-induced (0.15 ml 3 times
a week for 5 weeks) hepatic damage (Parola et al., 1992). In an acute
experiment, alpha-tocopheryl hemisuccinate (0.19 mmol, approx. or
equivalent 100 mg/kg by gavage) afforded a partial protection against
the hepatotoxicity of carbon tetrachloride (1.0 g/kg) administered 18
h later (Tirmenstein et al., 1997).
7.10 Mode of action
Recknagel & Glende (1973) suggested that carbon tetrachloride
toxicity requires cleavage of the carbon-chlorine bond and that the
cleavage takes place after binding of carbon tetrachloride to
cytochrome P-450 apoprotein in the mixed-function oxidase system
located in the hepatocellular endoplasmatic reticulum. However,
cytochrome P-450 is encased in lipid, and peroxidative decomposition
of the lipid is initiated by the free radicals formed by the cleavage.
Owing to the decomposition of the lipid and the attack on protein
functional groups by lipid peroxides, the structure and function of
the endoplasmatic reticulum is destroyed.
The carbon-tetrachloride-induced destruction of microsomal
cytochrome P-450 in vitro (De Groot & Haas, 1980, 1981) and
in vivo (Pasquali-Ronchetti et al., 1980; Shen et al., 1982)
inhibits the further biotransformation of carbon tetrachloride, the
generation of radicals and the concomitant peroxidation of endoplasmic
reticular lipids.
Burk et al. (1983) proposed a theory in which the toxic action of
carbon tetrachloride in vivo is dependent on either trichloromethyl
or trichloromethylperoxide radical formation, which is controlled by
the oxygen status of the liver cell (peripheral or centrilobular),
leading to damage to tissue macromolecules. This theory has been
supported by mechanistical arguments as summed up by Slater (1982) and
Dianzani (1984), who proposed the trichloromethyl radical as the main
covalently binding agent (haloalkylation) and the
trichloromethylperoxide radical as the main
lipid-peroxidation-inducing agent.
Oral dosage of carbon tetrachloride (2.5 ml/kg) decreased the
ATP-dependent calcium uptake of liver microsomes within 30 min in
Sprague-Dawley rats (Moore et al., 1976). The cytosolic concentration
of Ca2+ increased 100-fold in hepatocytes exposed to carbon
tetrachloride (about 1 mmol/litre), and this was paralleled by an
inhibition of the endoplasmic reticulum Ca-Mg ATPase (Long & Moore,
1986). The inhibition of the ATPase by carbon tetrachloride exposure
has been confirmed in several studies (Srivastava et al., 1990), and
has led to the hypothesis that this is the specific mechanism by which
radical intermediates from carbon tetrachloride lead to cell death.
Calcium chelating agents, Calcion and alizarin sodium sulfonate,
administered 6 or 10 h after a necrogenic intraperitoneal dose of
carbon tetrachloride (1 ml/kg), markedly decreased the necrotizing
effect of carbon tetrachloride on the liver, and decreased the hepatic
calcium concentration but did not affect carbon-tetrachloride-induced
lipid peroxidation in vitro, or lipid accumulation in the liver (de
Ferreyra et al., 1989, 1992). Carbon tetrachloride (0.01-0.12
mmol/litre) induced a complete release of calcium from calcium-loaded
microsomes in the presence of NADPH; this release was blocked by
adding the spin trapping agent, phenyl- tert- butylnitrone (PBN)
after a lag period that was dependent on the concentration of carbon
tetrachloride. The lag period was shortened using microsomes from
pyrazole-treated rats, which showed an elevated activity for
para-nitrophenol oxidation, and lengthened in the presence of the
CYP-450 2E1 inhibitor, methylpyrazole, or an anti-CYP-450 2E1
antibody. Calcium release was practically complete at concentrations
of carbon tetrachloride that had no effect on the Ca-Mg ATPase
activity. Ruthenium red, a specific ryanodine receptor inhibitor,
completely blocked the carbon-tetrachloride-induced calcium release at
a concentration (0.02 mmol/litre) that had no effect on
para-nitrophenol hydroxylation or formation of PBN-carbon
tetrachloride adducts (Stoyanovsky & Cederbaum, 1996). These results
support the notions that the hepatotoxicity of carbon tetrachloride
requires metabolism to the *CCl3 radical and is mediated by calcium
release from intracellular stores, most likely from the
ryanodine-sensitive calcium store.
Brattin et al. (1984) stated that the disturbance of the
intracellular Ca2+ balance cannot be regarded as an intracellular
"toxic messenger". Recknagel (1983), however, suggested that an early
disturbance in hepatocellular Ca2+ homoeostasis may be involved in
the pathological changes elicited by carbon tetrachloride (Recknagel,
1983). In addition, other authors suggest that the depression of the
Ca2+- sequestration capacity of the endoplasmic reticulum
(microsomes), resulting in a rise in the concentration of Ca2+ in the
cytosol, is an important factor in carbon-tetrachloride-induced
hepatotoxicity (Waller et al., 1983; Srivastava et al., 1990;
Yamamoto, 1990; Glende & Recknagel, 1991). Mehendale (1990, 1991)
showed that hepatic microsomes from carbon-tetrachloride-treated rats
accumulated progressively greater concentrations of Ca2+ in response
to the rise of Ca2+ levels in cytosol.
It is well known that lactic acid plays an important role in
hepatic fibrogenesis. Ayub-Ayala et al. (1993) determined the
relationship between short-term carbon tetrachloride administration
and the rise in lactic acid levels, before the appearance of any signs
of hepatic cirrhosis. After intraperitoneal administration of one
single and three consecutive carbon tetrachloride doses of 2.0 ml/kg
body weight (1:1 dilution in mineral oil) to Sprague-Dawley rats, the
blood lactic acid levels were raised, whereas the administration of
mineral oil did not increase them. Since carbon tetrachloride
increases lactic acid levels prior to cirrhosis development, the
authors suggested that chronic presence of lactic acid is one of the
factors in hepatic fibrogenesis caused by carbon tetrachloride.
Castro et al. (1990) stated that the understanding of the
mechanism of the liver carcinogenic effects of carbon tetrachloride
might be of relevance because there are reasons to believe that carbon
tetrachloride might be one of those carcinogens of a non-genotoxic
nature. The authors showed that liver nuclei from three species tested
(rat, hamster and mouse) were able to promote a lipid peroxidation
process in the presence of carbon tetrachloride, and that NADPH was
only required in part for carbon-tetrachloride-induced lipid
peroxidation in the case of the mice. There was no correlation between
the intensity of carbon-tetrachloride-induced lipid peroxidation,
either in liver nuclear or liver slices preparations, in the three
species tested and their carcinogenic response to carbon
tetrachloride. These results suggest that lipid peroxidation is not
determinant or rate-limiting in the process of liver cancer induction
by carbon tetrachloride, but does not exclude its participation in
given stages of the overall process of cancer development, as is
actually believed to occur during chemical carcinogen insult.
Carbon tetrachloride induced a hepatic cell proliferation,
increasing the frequency of cells in S-phase from < 1% in control
animals to around 10% in B6C3F1 mice (100 mg/kg by gavage 48 h before
sacrifice) (Mirsalis et al., 1985). In rats a similar increase was
observed after a dose of 400 mg/kg (Mirsalis et al., 1985), and the
increase was around 30% in CD mice (50 mg/kg) (Doolittle et al.,
1987). In male Fischer-344 rats, the frequency of S-phase cells was
elevated in one study to 30% 24 h after the only dose tested, 0.4
ml/rat (Cunningham & Matthews, 1991). In another study with 400 mg/kg
carbon tetrachloride, the frequency was increased to 3% in fed and to
15% in fasting F-344 rats (Asakura et al., 1994). In yet another study
with Fischer rats, an increase to 5% was observed 24 h after an
intraperitoneal dose of 400 mg/kg carbon tetrachloride (Mirsalis et
al., 1985). An even lower response, approximately 2%, was observed in
Tif:RAIf rats (400 mg/kg) (Puri & Müller, 1989). An increase in DNA
synthesis was observed 48 h (and the number of ras transcripts was
elevated 36-48 h) after an intragastric dose (2.5 ml/kg) of carbon
tetrachloride in Sprague-Dawley rats (Goyette et al., 1983). A rapid
transient increase in c- fos and c- jun mRNA (1-2 h post-treatment)
was also observed in the liver of male Sprague-Dawley rats after a
single dose of 160 mg/kg carbon tetrachloride (Zawaski et al., 1993).
An increase in the c- fos, c- jun and c- myc nRNA was also observed
in male Wistar rats after a single dose of carbon tetrachloride (2
ml/kg intragastrically) (Coni et al., 1990, 1993). In rat liver,
ras and myc proteins were observed by immunohistochemical
techniques. Their concentrations peaked in periportal areas 32 h after
dosing with carbon tetrachloride (2.5 ml/kg), and staining throughout
the lobule peaked 96 h after the carbon tetrachloride dose (Richmond
et al., 1992). The sequence of fos, myc and Ha- ras mRNA
expression, followed by hepatocyte proliferation, was also observed in
F-344 rats after a single intraperitoneal dose of 2000 mg/kg carbon
tetrachloride by gavage (Goldsworthy et al., 1994). Injection of a
polyclonal antiserum to murine tumour necrosis factor alpha one hour
before a challenge with carbon tetrachloride (0.1 ml/kg) blocked the
increase in c- fos and c- jun mRNA expression and the subsequent
increase of S-phase cells, while at the same time prolonging the
elevation of serum ALAT, ASAT and sorbitol dehydrogenase (SDH) in
female B6C3F1 mice. When recombinant TNF alpha was injected to mice,
rapid expression of c- jun and c- fos proto-oncogene mRNA was
observed, thus supporting the notion that TNF alpha has a role in the
hepatocellular regeneration after carbon tetrachloride administration
(Bruccoleri et al., 1997). This idea was originally put forward after
the demonstration of increased expression of TNF alpha following an
administration of a hepatotoxic dose of carbon tetrachloride (Czaja et
al., 1989). On the other hand, injection of soluble TNF alpha receptor
preparation to rats had a protective effect against a higher dose of
carbon tetrachloride (0.5 ml/kg), reducing the mortality, serum
aminotransferase levels and the extent of histological liver damage
(Czaja et al., 1995).
8. EFFECTS ON HUMANS
8.1 Controlled studies
8.1.1 Inhalation
Six healthy male volunteers were exposed (three times 4 weeks
apart) to carbon tetrachloride vapour at concentrations of 49 ppm (314
mg/m3) for 70 min, 11 ppm (70.5 mg/m3) for 180 min and 10 ppm (64.1
mg/m3) for 180 min. At the high concentration level, all subjects
smelled a sweetish odour. None of the volunteers reported irritation,
nausea, lightheadedness or disturbance in coordination. No increase in
ASAT activity was observed, but a decrease in serum iron concentration
was observed in two out of four subjects at the highest concentration.
No effects were observed at the lower concentrations. Carbon
tetrachloride was detected in exhaled breath at all three exposure
levels (Stewart et al., 1961).
8.1.2 Dermal
Daily treatments for 10 days with carbon tetrachloride did not
cause any increase in skin-fold thickness or erythema on the volar
surface of the forearms of a healthy human volunteer. However, no
occlusion was used, so it is probable that the chemical evaporated
after administration (Wahlberg, 1984a).
When an excess of carbon tetrachloride was applied in a glass
ring to the volar surface of the forearms of a healthy man for 5 min
there was an immediate increase in blood flow. A spontaneous transient
whitening of the skin was observed after 5 min and after 10 to 20 min
a slight, transient erythema appeared (Wahlberg, 1984b).
Immersion of the thumbs of three volunteers for 30 min in carbon
tetrachloride caused mild erythema that disappeared in 1 to 2 h after
exposure. The volunteers reported a burning sensation in the thumbs,
which subsided within 10 min after the immersion (Stewart & Dodd,
1964).
8.2 Case reports
Cases of poisoning with carbon tetrachloride have resulted from
the accidental or suicidal ingestion of carbon tetrachloride, but the
majority resulted from the inhalation of carbon tetrachloride vapour;
the concentration of the saturated vapour at ambient temperature can
reach 800 000 mg/m3. Carbon tetrachloride appears to be toxic to the
liver and kidney. The clinical picture of carbon tetrachloride
poisoning is characterized, independent of the route of intake, in the
first 24 h with gastrointestinal and neurological symptoms, such as
nausea, headache, dizziness, vomiting, diarrhoea and dyspnoea. Liver
damage appears, at the earliest, after 24 h. In serious cases, ascites
and hepatic coma develop, often accompanied by haemorrhages. Kidney
damage is detected later, in 1 to 6 days, but often only 2-3 weeks
following the poisoning (Zimmerman, 1978; Kluwe, 1981; Monster &
Zielhuis, 1983).
Von Oettingen (1964) reviewed the literature on acute carbon
tetrachloride intoxication in humans. He concluded that exposure to
10-80 ppm (64.1 to 512.8 mg/m3) for 3-4 h has no adverse effects. At
higher concentrations nausea, vomiting, headache, rapid pulse, rapid
respiration, sleepiness, dizziness, unconsciousness and immediate
death can occur even after only 10-30 min of exposure.
Bagnasco et al. (1978) reported a case in which a 22-year-old man
ingested 355 ml carbon tetrachloride and an equal amount of water to
commit suicide. His liver function deteriorated over the first 24 h,
but gradually within the following 3 to 4 days the patient improved.
According to the authors fatal cases have been reported with as little
as 1.5 ml carbon tetrachloride, whereas some patients have been known
to survive after swallowing more than 100 ml.
Smetana (1939) described two cases of carbon tetrachloride
poisoning. One person, who drank an unknown quantity of carbon
tetrachloride, died. Apart from the general symptoms, jaundice, anuria
and malaise were observed. Aside from lesions in the liver there was
clinical evidence of functional damage of the kidneys, recognized by
the presence of albumin in the urine, oliguria, nitrogen retention,
oedema and acute hypertension. The patients had a history of
alcoholism.
Norwood et al. (1950) reported three cases of severe intoxication
arising from use of carbon tetrachloride, two by inhalation and one by
drinking. The two people who inhaled carbon tetrachloride died, and
nephrosis was found to have occurred. All three had a history of heavy
drinking.
Tracey & Sherlock (1968) described a 59-year-old man, with a
history of moderate alcohol consumption, who was exposed to carbon
tetrachloride vapour. Five days later, he developed nausea, vomiting
and diarrhoea, followed by jaundice and acute renal failure. The liver
was found to be enlarged. He recovered uneventfully and liver
functions returned to normal.
McDermott & Hardy (1963) reported three cases of liver cirrhosis
in which repeated exposure to carbon tetrachloride vapour occurred
over a number of years. One of the cases involved mixed solvent
exposure. There was no evidence of significant alcohol intake for any
of the patients.
Ruprah et al. (1985) reported details of 19 patients poisoned
with carbon tetrachloride during the period 1981-1984. Eight of these
patients were known to have ingested other substances, including
phenothiazine and benzodiazepine tranquillizers, trichloroethane and
trichloroethanol. Carbon tetrachloride exposure was by inhalation (4
cases) or ingestion (15 cases). In each case the diagnosis was
confirmed by laboratory analysis of blood specimens. The age of the
patients ranged from 3 to 79 years and the whole-blood concentrations
at the time of hospital admission varied from 0.1 to 31.5 mg/litre.
However, actual doses and exposure concentrations were not known and
are difficult to estimate. In none of these cases was the intoxication
associated with occupational use or exposure. The commonest symptoms
found in these patients were vomiting, abdominal pain, diarrhoea,
dizziness, headache and coma. There were no fatalities.
Norwood et al. (1950) reported 51 very mild and 4 mild
intoxications among industrial workers using carbon tetrachloride. The
4 mildly intoxicated patients showed the general symptoms of carbon
tetrachloride intoxication. No data on exposure levels were given.
Kazantzis & Bomford (1960) examined a group of 17 factory
workers, exposed to carbon tetrachloride atmospheric concentrations of
45 to 100 ppm (288-641 mg/m3). During periods of 24 months to 1 week
before the examination, 12 out of 17 workers had experienced one or
more of the following symptoms: nausea, anorexia, vomiting,
flatulence, epigastric discomfort or distention, depressive symptoms,
headache or giddiness. After taking measures to reduce carbon
tetrachloride evaporation, these symptoms disappeared and follow-up
for 6 months revealed no recurrences.
Fourteen workers in an isopropyl alcohol packaging plant became
ill after exposure to unspecified carbon tetrachloride levels (Folland
et al., 1976). The illness was characterized by the gradual onset of
nausea, vomiting, weakness, headache and abdominal pain. In three
heavily exposed subjects renal failure developed. Air concentrations
of isopropanol in the plant were measured 12 and 9 months before and 2
months after the exposure and were about 400 ppm (2564 mg/m3).
Brugnone et al. (1983) investigated 40 workers occupationally
exposed to carbon tetrachloride vapour. After several measurements it
turned out that the alveolar carbon tetrachloride concentration
corresponded to about 53% of the environmental concentration measured
in the breathing zone (mean 3.5 ± 5.9 mg/m3). Among the 40 workers,
two suffered accidental carbon tetrachloride intoxication with acute
renal impairment.
Manno et al. (1996) reported that five workers were exposed to
carbon tetrachloride vapour for 2 h and two for 6 h following fire
accidents. Carbon tetrachloride was present in the fire-extinguishing
liquid. Symptoms of carbon tetrachloride poisoning (diarrhoea, nausea,
vomiting, fever and liver and kidney impairment) developed only in two
heavily drinking workers who consumed, respectively, about 120 and 250
g ethanol daily. The other workers, consuming less than 50 g ethanol
per day, did not develop symptoms. Exposure data were not available.
8.3 Epidemiology
8.3.1 Non-cancer epidemiology
In a mortality study in a metal fabrication plant (Teta & Ott,
1988) slight increase in mortality from liver cirrhosis was observed.
The highest increase (SMR 2.7) was found in workers potentially
exposed to carbon tetrachloride before the use of this solvent was
discontinued. The authors consider carbon tetrachloride exposure as a
possible contributing risk factor for the cirrhosis findings. However,
exposure data for carbon tetrachloride, data on other exposures and
alcohol consumption were not available, which limit the ability to
draw conclusions regarding carbon tetrachloride.
Volunteers from three plants were divided into four groups on the
basis of estimated exposure to carbon tetrachloride: none (n=262), low
(1 ppm (6.4 mg/m3) or less, n=40), medium (1-4 ppm (6.4-25.6 mg/m3),
n=54) and high (more than 4 ppm (25.6 mg/m3), n=61). The alcohol
consumption was at the same level in all groups. ALAT, ASAT, alkaline
phosphatase, gamma-glutamyltransferase, glutamate dehydrogenase, and
other biochemical and haematological variables were determined. The
percentages of values above the normal range were 2.7% in the
non-exposed group and 7.8% in combined exposed groups for ALAT, and 3%
and 10.9% for gamma-glutamyltransferase, respectively (both differences
statistically significant). The low exposure group did not differ
significantly in any enzymatic activity test from the non-exposed
group (Tomenson et al., 1995).
8.3.2 Cancer epidemiology
A number of epidemiological studies (e.g., cohort mortality,
retrospective cohorts, and case-control) have examined potential
cause-effect relationships between carbon tetrachloride exposure and
incidence of cancer. Because these studies are all characterized by
mixed exposures and a lack of carbon tetrachloride exposure data, any
contribution from carbon tetrachloride cannot be reliably identified.
Thus, information from these studies is not useful for quantitative
health risk evaluation.
Ott et al. (1985) conducted a cohort mortality study of 1919 men
employed for one or more years between 1940 and 1969 at a chemical
manufacturing facility in the USA. This cohort included 226 workers
assigned to a unit that produced chlorinated methanes (methyl
chloride, dichloromethane, chloroform, and carbon tetrachloride) and,
recently, perchloroethylene. Exposure levels were not reported. The
follow-up period was from 1940 to 1979 and follow-up was 94% complete.
Expected numbers of cancer deaths were based on US white male cancer
rates for the full cohort; the expected numbers in the full cohort
were used for sub-cohort analyses. There were 42 deaths, including
nine cancers, three of which were pancreatic cancers. The standardized
mortality ratios for all deaths and for all cancers were not elevated.
Blair et al. (1990) performed a study to examine the risk of
cancer and other causes of death among a cohort of 5365 members of a
dry cleaners' union in the USA. The cohort consisted of people who
were union members for one or more years before 1978 and had been
employed in dry cleaning establishments. Carbon tetrachloride was used
extensively in dry cleaning between 1930 and 1960, although other
solvents, such as white spirit (Stoddard solvent), were also widely
used. The exposure assessment classified members by level of exposure
to solvents, but not by type of solvent. The mean year at entry into
the cohort was 1956. Follow-up was from 1948 to 1978 and was 88%
complete. There were 294 cancer deaths, including a significant excess
of oesophageal cancer. Non-significant excesses of several other
cancers were found, but only the risk of lymphatic and haematopoietic
cancers appeared to be related to the level of solvent exposure.
Blair et al. (in press) performed a retrospective cohort
mortality study of 14 457 workers employed for at least one year
between 1952 and 1956 at an aircraft maintenance facility in the USA.
Among this cohort were 6737 workers who had been exposed to carbon
tetrachloride (Stewart et al., 1991). Among women, exposure to carbon
tetrachloride was associated with an increased risk of non-Hodgkin's
lymphoma and multiple myeloma, but among men the corresponding risks
were lower. No association was observed with breast cancer and no
other site-specific results for carbon tetrachloride were presented.
Exposure levels for carbon tetrachloride were not reported, and
overlapping exposure to other solvents limits the ability to draw
conclusions regarding carbon tetrachloride.
A nested case-control study within a cohort of rubber workers in
the USA was performed to examine the relationship between exposure to
24 solvents (levels of exposure not reported) and the risk of cancer
(Checkoway et al., 1984; Wilcosky et al., 1984). The cohort consisted
of 6678 male rubber workers who were either active or retired between
1964 and 1973. The cases comprised all persons with fatal stomach
cancer (n=30), respiratory system cancer (n=101), prostrate cancer
(n=33), lymphosarcoma (n=9) and lymphocytic leukaemia (n=10). The
control group was a 20% age-stratified random sample of the cohort
(n=1350). Although an association was observed between exposure for
one or more years to carbon tetrachloride and lymphocytic leukaemia
and lymphosarcoma after adjusting for year of birth, overlapping
exposures limit the ability to draw conclusions regarding carbon
tetrachloride.
Bond et al. (1986) conducted a nested case-control study of lung
cancer among a cohort of 19 608 white male chemical workers in the
USA. They were employed for one or more years between 1940 and 1980 at
a large facility that produced chlorinated solvents, plastics,
chlorine, caustic soda, ethylene, styrene, epoxy, latex, magnesium
metal, chlor-nitrogen agricultural chemicals and glycols. The cases
were 308 lung cancer deaths that occurred among cohort members between
1940 and 1981. Two control groups, one consisting of other deaths
(n=308) and the other a "living" series (n=97), were matched for race,
year of birth, and year of hire. No association was observed between
having been exposed (levels not reported) to carbon tetrachloride
("ever" versus "never") and lung cancer.
Linet et al. (1987) performed an analysis to compare two
different methods for determining occupational exposure in a
population-based case-control study of chronic lymphocytic leukaemia.
No association between chronic lymphocytic leukaemia and carbon
tetrachloride was observed in either set of analyses.
Heineman et al. (1994) performed a case-control study to examine
the relationship between occupational exposure to six chlorinated
aliphatic hydrocarbons and risk of astrocytic brain cancer. The study
was conducted in three areas of the USA, and 300 cases and 320
controls were included in the analysis. Exposure was assessed using a
semi-quantitative job exposure matrix developed for the study (Gómez
et al., 1994), and probability of exposure, duration of exposure,
average intensity and cumulative exposure were examined. There were
137 cases and 123 controls classified as having been exposed at some
time. There was an association between the incidence of astrocytic
brain cancer and chlorinated solvent exposure, but not specifically
carbon tetrachloride.
Cantor et al. (1995) performed a case-control study to examine
the relationship between occupational exposure and female breast
cancer mortality in 24 states in the USA. Probability and level of
workplace exposure to 31 chemical and physical agents were estimated
using a job exposure matrix. No association was found with probability
of exposure to carbon tetrachloride. After adjustment for age and
socioeconomic status, a slightly but significantly elevated risk was
observed at the highest exposure level among white women but not among
black women. However, the designation of the usual occupation from
death certificates in combination with a job-exposure matrix may be a
poor indicator of exposure to carbon tetrachloride.
Holly et al. (1996) performed a case-control study of intraocular
melanoma to examine the role of chemical exposure. Cases were white
male patients referred to the Ocular Oncology Unit at the University
of California San Francisco between 1978 and 1987. Two white males
matched on age and geographic area were selected for each case using
random digit dialling. A total of 221 cases and 447 control (93% and
85% participation rates, respectively) were interviewed for the study.
Although an association with exposure ("ever" versus "never") to
"carbon tetrachloride and other cleaning fluids" was observed, the
potential for recall bias for exposure history and the lack of
characterization of the exposure atmospheres precludes the ability to
draw conclusions regarding carbon tetrachloride alone.
In a case-control study carried out in Montreal, the
investigators estimated the associations between 293 workplace
substances and several types of cancer (Siemiatycki, 1991). Carbon
tetrachloride was one of the substances. About 4% of the study
subjects had been exposed to carbon tetrachloride at some time. Among
the main occupations for which carbon tetrachloride was attributed in
this study were fire fighters, machinists and electricians. For most
types of cancer examined (oesophagus, stomach, colon, pancreas,
prostrate, kidney, skin melanoma, non-Hodgkin's lymphoma), there was
no indication of an excess risk. There were, however, elevated risks
for rectal cancer and, in the population subgroup of French-Canadians,
bladder cancer.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
Owing to the volatility of carbon tetrachloride, care must be
taken in interpreting test results, particularly those in open static
systems where no chemical analysis of the actual concentration was
carried out.
9.1 Toxicity to microorganisms
Carbon tetrachloride appeared to be of low toxicity to several
microorganisms (see Table 11).
During studies to determine the toxicity threshold, an initial
reduction of cell multiplication or change in culture turbidity was
seen at 30 mg/litre in aerobic bacteria, but an IC50 as low as 6.4
mg/litre was found for methanobacteria. The toxicity threshold for
protozoa was much higher (> 300 mg/litre).
Walton et al. (1989) studied the effect of carbon tetrachloride
on the microbial respiration of two slightly acidic soil types, a
Captina silt loam (1.49% organic carbon) from Roane County, Tennessee,
USA, and a sandy loam (0.66% organic carbon) from Stone County,
Mississippi, USA. Carbon tetrachloride was applied at a rate of 1000
µg/g soil (dry weight) and microbial respiration, measured as CO2
efflux, was monitored at 24-h intervals over a 6-day period. Carbon
tetrachloride had no effect on the respiration of the silt loam. The
CO2 efflux of the sandy loam decreased relative to the control soil
but recovered within the 6-day exposure period.
9.2 Aquatic toxicity
9.2.1 Algae
Data in Table 11 show the toxicity of carbon tetrachloride to
algae to be low.
9.2.2 Invertebrates
The acute toxicity values for carbon tetrachloride in
Daphnia magna (Table 12) range from 28 to > 770 mg/litre.
Carbon tetrachloride had no effect on the embryonic development
of sea urchin (Paracentrotus lividus) eggs at concentrations up to
the saturated solution concentration (Congiu et al., 1984).
9.2.3 Vertebrates
Acute toxicity data for fish are summarized in Table 12. The
acute LC50 values for fish range from 13 to 472 mg/litre for the
Golden orfe (Leusiscus idus melanotus).
Table 11. Carbon tetrachloride toxicity to bacteria, protozoa and algae
Organism Test conditions End-point Nominal Reference
concentration
(mg/litre)
Bacteria
Pseudomonas fluorescens 16 h, 25 °C, static 3% reduction of turbidity 30 Bringmann, 1973
threshold, log phase
Pseudomonas putida 16 h, 25 °C, static 3% reduction of turbidity 30 Bringmann & Kühn, 1977a
threshold, log phase
Nitromonas sp. 24 h, 25 °C, static IC50, 50% reduction in NH3 51 Blum & Speece, 1991
consumption
Methanogens 24 h, 35 °C, static IC50, 50% reduction in gas 6.4 Blum & Speece, 1991
production
Aerobic heterotrophs. 24 h, 35 °C, static IC50, 50% reduction in oxygen 130 Blum & Speece, 1991
uptake
Protozoa
Bacteriovorous flagellate 72 h, 25 °C, static 5% reduction cell count >770 Bringmann, 1978
(Entosiphon sulcatum)
Bacteriovorous flagellate 20 h, 25 °C, static 5% reduction in cell count >616 Bringmann & Kühn, 1980
(Uronema parduczi)
Saprozoic flagellate 48 h, 20 °C, static 5% reduction in cell count >300 Bringmann et al., 1980
(Chilomonas paramecium)
Table 11. (Continued)
Organism Test conditions End-point Nominal Reference
concentration
(mg/litre)
Algae
Blue-green alga 192 h, 27 °C, static 1% reduction of turbidity 105 Bringmann, 1975
(Microcystis aeroginosa) threshold
Green alga 192 h, 27 °C, static 3% reduction of turbidity >600 Bringmann & Kühn, 1977a
(Scenedesmus quadricauda) threshold
Haematococcus pluvialis 4 h, 20 °C, static EC10, 10% reduction in >136 Knie et al., 1983
oxygen uptake
Table 12. Carbon tetrachloride toxicity to invertebrates and fish
Organism Test conditions Parameter Concentration Reference
(mg/litre)
Invertebrates
Daphnia magna 21-23 °C reconstituted static 48 h LC50 35 nominal LeBlanc, 1980
well water; pH 7.4-9.4; 24 h LC50 35 nominal
hardness 173 mg
CaCO3/litre
Daphnia magna 20-22 °C dechlorinated static 24 h LC50 >770 Bringmann & Kühn,
tap water; pH 7.6; 1977b
hardness
173 mg CaCO3
Daphnia magna static 24 h LC50 28 Knie et al., 1983
Fish
Freshwater
Guppy 22 °C hardness 25 mg static 336 h LC50 67 Könemann, 1981
(Poecilia reticulata) CaCO3/litre renewal
Golden orfe 20 °C static 48 h LC50 95 nominala Juhnke &
(Leuciscus idus melanotus) 472 nominala Lüdemann, 1978
Golden orfe static 48 h LC50 13 Knie et al., 1983
(Leuciscus idus melanotus)
Bluegill sunfish 23 °C well water; static 96 h LC50 125 nominal Dawson et al.,
(Lepomis macrochirus) pH 7.6-7.9; hardness 1975/77
55 mg CaCO3/litre
Table 12. (Continued)
Organism Test conditions Parameter Concentration Reference
(mg/litre)
Bluegill sunfish 21-23 °C pH 6.7-6.8; static 96 h LC50 27 nominal Buccafusco et al.,
(Lepomis macrochirus) hardness 32-48 1981
mg CaCO3/litre
Fathead minnow - - flow - LC50 43.1 measured US EPA, 1984b
(Pimephales promelas)
Fathead minnow 21.7 °C pH 6.8; hardness flow 96 h LC50 41.4 measured National Library of
(Pimephales promelas) 49.2 mg CaCO3/litre Medicine, 1997
Marine
Dab - natural seawater flow 96 h LC50 50 measured Pearson &
(Limanda limanda) McConnell, 1975
Tidewater silverside 20 °C saltwater; static 96 h LC50 150 nominal Dawson et al.,
(Menidia beryllina) pH 7.6-7.9; hardness 1975/77
55 mg CaCO3/litre
a The authors tested 200 selected chemicals with the golden orfe test under comparable conditions in two different
laboratories and found LC50 values of 95 mg/litre (Juhnke) and 472 mg/litre (Lüdemann), respectively, for carbon
tetrachloride
Rainbow trout (Oncorhynchus mykiss) were exposed to carbon
tetrachloride concentrations of between 1 and 80 mg/litre for up to
336 h under semi-static conditions (water was renewed every 48 h). No
mortality was observed and no significant changes in enzyme activity
were found (Statham et al., 1978).
Toxicity data for embryo-larval stages of fish and amphibians are
given in Table 13. Carbon tetrachloride is considerably more toxic to
the embryo-larval stages of several species of fish and amphibians
than it is to the adults (Birge, 1980; Black et al., 1982). The common
bullfrog (Rana catesbeiana) was the most susceptible species. At 60
µg/litre the incidence of teratic larvae was 1%, rising to 17% at 7.8
mg/litre. A more striking effect was found in the hatchability of the
embryos, which declined from 92% at 60 µg/litre to 23% at 7.8 mg/litre
(Birge, 1980).
9.3 Terrestrial toxicity
9.3.1 Earthworms
Red earthworms (Eisenia foetida) were exposed to carbon
tetrachloride via filter paper in glass vials. An LC50 of 160 µg/cm2
was found (Neuhauser et al., 1985).
Table 13. Carbon tetrachloride toxicity to embryo-larval stages of fish and amphibians
Organism Test conditions Exposure Parameter Measured Reference
period concentration
(days) (mg/litre)
Fish
Rainbow trout 13 °C pH 9.2; hardness flow 27a LC50 1.97 Black et al., 1982
(Oncorhynchus mykiss) 104 mg CaCO3/litre
Fathead minnow 20 °C pH 6.4; hardness flow 9b LC50 4.0 Black et al., 1982
(Pimephales promelas) 96 mg CaCO3/litre
Amphibians
Bullfrog 21 °C pH 8; hardness flow 8b LC50 0.9 Birge, 1980
(Rana catesbeiana) 108 mg CaCO3/litre
Pickerel frog 22 °C pH 7.7; hardness flow 8b LC50 2.4 Birge, 1980
(Rana palustris) 104 mg CaCO3/litre
Fowler's toad 22 °C pH 7.7; hardness flow 7b LC50 2.8 Birge, 1980
(Bufo fowleri) 104 mg CaCO3/litre
European common frog 19 °C pH 7.7; hardness flow 9a LC50 1.2 Black et al., 1982
(Rana temporaria) 96 mg CaCO3/litre
Leopard frog 19 °C pH 7.7; hardness flow 9a LC50 1.6 Black et al., 1982
(Rana pipiens) 96 mg CaCO3/litre
African clawed toad 19 °C pH 7.7; hardness flow 6a LC50 22.4 Black et al., 1982
(Xenopus laevis) 96 mg CaCO3/litre
Northwestern salamander 19 °C pH 7.7; hardness flow 9.5a LC50 1.98 Black et al., 1982
(Ambystoma gracile) 96 mg CaCO3/litre
a The organisms were exposed from fertilization until 4 days after hatching
b The organisms were exposed from 2-8 h post spawning to 4 days after hatching
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Carbon tetrachloride can be detected ubiquitously in the
environment, mostly in the air due to its volatility and high vapour
pressure. Furthermore it is found in foodstuffs and drinking-water.
Based on the estimates of mean exposure from various media, as
reported in chapter 5, the general population may be exposed via air
at a concentration of 0.5-1.0 µg/m3 (retention calculated to be
0.068-0.136 µg/kg body weight per day, assuming 40% retention, ATSDR,
1994), and via drinking-water at levels of 0.1-3 µg/litre (calculated
to be 0.003-0.094 µg/kg body weight). Intake via foodstuffs is
estimated to be very small (0.13 µg/day) (Yoshida, 1993), but could
have been larger in the past in individuals who consumed fumigated or
otherwise contaminated foods. These are presumably no longer on the
market since this use of carbon tetrachloride has ceased. This means
for the general population an estimated maximum daily carbon
tetrachloride intake of about 0.23 µg/kg body weight, assuming (IPCS,
1994):
- a body weight of 64 kg;
- an inhalation volume of 22 m3/day;
- a water consumption of 2 litres/day;
- a food consumption of 1.536 kg/day.
According to estimates made by the ATSDR and in Japan and Germany
the general population is considered to be exposed to carbon
tetrachloride via ingestion and inhalation leading to an average daily
intake of 0.1 to 0.27 µg/kg body weight.
Workers involved in the production or use of carbon tetrachloride
are likely to be exposed to higher levels than the general population.
Based on a national survey conducted from 1981 to 1983, NIOSH
estimated that 58 208 workers were potentially exposed to carbon
tetrachloride in the USA during that period.
Exposure to higher levels of carbon tetrachloride could occur as
a result of accidental spillage or near hazardous waste sites
contaminated with carbon tetrachloride.
10.1.2 Health effects
Acute symptoms after human exposure to carbon tetrachloride are
characterized by gastrointestinal and neurological symptoms, such as
nausea, vomiting, headache, dizziness and dyspnoea. Liver damage
appears after 24 h or more. Kidney damage is evident often only 2 to 3
weeks following the poisoning. Short-term and long-term exposure to
low concentrations of carbon tetrachloride can also produce hepatic
and renal damage. The toxicity of carbon tetrachloride is associated
with the formation of reactive metabolites, the principal enzyme
involved being CYP 2E1. The severity of the effects on the liver
depends on a number of factors, such as species, susceptibility, route
and mode of exposure, diet or co-exposure to other compounds, in
particular, ethanol. How these factors affect the CNS and kidney
responses is not known. No adequate long-term oral study on laboratory
animals, suitable for quantitative health risk evaluation of carbon
tetrachloride, was available (section 7.3).
In a 12-week oral study on rats (5 days/week), a NOAEL of 1 mg/kg
body weight was reported. The LOAEL reported in this study was 10
mg/kg body weight, showing a slight, but significant increase in SDH
activity and mild hepatic centrilobular vacuolization (Bruckner et
al., 1986). A NOAEL of 1.2 mg/kg body weight was reported in a 90-day
oral study on mice (5 days/week). On the basis of hepato toxicity, the
LOAEL was 12 mg/kg body weight (Condie et al., 1986).
When rats were exposed to carbon tetrachloride by inhalation for
approximately 6 months, 5 days/week, 7 h/day, the NOAEL was 32 mg/m3.
The LOAEL, based on changes in the liver morphology, was 63 mg/m3
(Adams et al., 1952). In a 90-day study on rats, a NOAEL of 6.1 mg/m3
was found after continuous exposure to carbon tetrachloride
(Prendergast, 1967).
An exposure level of 32 mg/m3 (the lowest concentration studied)
in a 2-year inhalation study on rats caused marginal effects (Japan
Bioassay Research Centre, 1998).
In experiments with mice and rats, carbon tetrachloride proved to
be capable of inducing hepatomas and hepatocellular carcinomas. The
doses inducing hepatic tumours were higher than those inducing cell
toxicity. It is likely that the carcinogenicity of carbon
tetrachloride is secondary to its hepatotoxic effects.
There is little evidence to suggest that carbon tetrachloride is
genotoxic.
Based on the weight of evidence it can be concluded that the
hepatic tumours are induced by an indirect mechanism and that a
tolerable daily intake or concentration can be derived.
The available data suggest that carbon tetrachloride can induce
embryotoxic and embryolethal effects, but only at doses that are
maternally toxic. Carbon tetrachloride is not teratogenic in rats and
mice.
10.1.3 Approaches to health risk assessment
There is little evidence to suggest that carbon tetrachloride is
genotoxic. A quantitative risk assessment for threshold effects (IPCS,
1994), which includes the effects of non-genotoxic carcinogens, was
therefore adopted.
10.1.3.1 Calculation of a TDI based on oral data
Calculations of tolerable daily intake (TDI) were based on the
12-week oral study on rats (Bruckner et al., 1986) and the 90-day oral
study on mice (Condie et al., 1986), where NOAEL values of 1 mg/kg
body weight and 1.2 mg/kg body weight were identified, respectively.
a) Rat
1 mg/kg body weight × (5/7)
TDI = 500 = 1.42 µg/kg body weight
where:
* 1 mg/kg body weight is the NOAEL in the 12-week oral study on
rats
* (5/7) is the conversion from 5 days/week of dosing to 7 days/week
* 500 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 10 for a less-than-long-term
study; a modifying factor of 0.5 was applied because this was a
bolus study).
b) Mouse
TDI = 1.2 mg/kg body weight × (5/7) = 1.72 µg/kg body weight
500
where:
* 1.2 mg/kg body weight is the NOAEL in the 90-day oral study on
mice
* (5/7) is the conversion from 5 days/week of dosing to 7 days/week
* 500 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 10 for a less-than-long-term
study; a modifying factor of 0.5 was applied because this was a
bolus study).
10.1.3.2 Calculation of a tolerable concentration based on inhalation
data
Calculations of tolerable concentrations (TC) were based on: (a)
the 90-day study of Prendegast (1967) where a NOAEL of 6.1 mg/m3 was
identified for continuous exposure; (b) the 6-month study of Adams et
al. (1952) where a NOAEL of 32 mg/m3 was found; and (c) the 2-year
inhalation study by Japan Bioassay Research Centre (1998) where a
LOAEL with a marginal adverse effect was 32 mg/m3.
a)
TC = 6.1 mg/m3 = 6.1 µg/m3
1000
where:
* 6.1 mg/m3 is the NOAEL in the 90-day inhalation study on rats
* 1000 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation; and 10 for a less-than-long-term
study)
b)
TC = 32 mg/m3 × (7/24) × (5/7) = 6.7 µg/m3
1000
where:
* 32 mg/m3 is the NOAEL in the 6-month inhalation study on rats
* (7/24) × (5/7) is the conversion from 7 h/day and 5 days/week to
continuous exposure.
* 1000 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 10 for a less-than-long-term
study).
c)
TC = 32 mg/m3 × (6/24) × (5/7) = 11.4 µg/m3
500
* 32 mg/m3 is the LOAEL in the 2-year inhalation study on rats
* (6/24) × (5/7) is the conversion from 6 h/day and 5 days/week to
continuous exposure
* 500 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 5 for use of a marginal effect rather
than a no-observed-effect level).
It is noted that the end-point on which the LOAEL is based in the
recent 2-year inhalation bioassay on rats (i.e. proteinuria) was not
investigated in the studies of Adams et al. (1952) and Prendergast
(1967).
10.1.3.3 Summary of the results of risk assessment
TDI
Oral studies 1.42 µg/kg body weight
1.72 µg/kg body weight
TC TDI (calculated from the TC)
Inhalation studies 6.1 µg/m3 0.85 µg/kg body weight
6.7 µg/m3 0.92 µg/kg body weight
11.4 µg/m3 1.56 µg/kg body weight
10.1.3.4 Conclusions based on exposure and health risk assessment
On the basis of exposure data presented in chapter 5, an
approximate upper-limit estimate of the daily intake of carbon
tetrachloride for long-term exposure of the general population can be
made for prevailing normal exposure and for a "worst case scenario".
The following concentration ranges are considered:
* ambient air, 0.5-1.0 µg/m3 (worst case 6 µg/m3 );
* indoor air in dwellings, 0.6-2.0 µg/m3 (worst case 9 µg/m3);
* drinking-water, 0.0002-2.3 µg/litre (worst case 16 µg/litre; the
abnormally high value of 39.5 µg/litre reported in Spain was not
considered);
* foodstuffs (particularly table-ready foods) 0.1-6.0 µg/kg (worst
case 31 µg/kg).
The daily estimates are summarized in Table 14.
As is seen from Table 14, the upper limit of human daily intake
under prevailing conditions is estimated to be 0.2 µg/kg body weight,
well below the lowest tolerable daily intake (0.85 µg/kg body weight)
presented in section 10.1.3.3. This leads to the conclusion that the
currently prevailing exposure of the general population to carbon
tetrachloride from all sources is unlikely to cause excessive intake
of the chemical.
The hypothetical worst case scenario of exposure may bring about
a daily intake of 2.5 µg/kg body weight, more than ten times the daily
intake under currently prevailing conditions, and three times the
lowest tolerable intake. This would indicate a need for caution.
However, conditions similar to those of the worst case scenario are
very unlikely to occur in future, due to the expected fall in the use
of carbon tetrachloride as a consequence of the Amended Montreal
Protocol.
10.2 Evaluation of effects on the environment
Carbon tetrachloride may be released into the environment during
its production, storage, transport and use. Owing to its volatility,
most of the substance emitted into the environment can be found in the
air. The residence time of carbon tetrachloride in the atmosphere is
long, and it can therefore be transported over long distances from the
point of emission. The main degradation site of carbon tetrachloride
is the stratosphere where it is photolytically degraded by UV
radiation. Carbon tetrachloride contributes both to ozone depletion
and to global warming.
Carbon tetrachloride is, in general, resistant to aerobic biode
gradation, but less so to anaerobic. Acclimation increases
biodegradation rates. Although the octanol-water partition coefficient
indicates a moderate potential for bioaccumulation, the short lifetime
in tissues reduces this tendency.
Table 14. Daily intake of carbon tetrachloride for long-term exposure of the general population
Prevailing upper limits Worst case scenario
Concentration Daily intake Concentration Daily intake
Air
1 µg/m3 1 µg/m3 × 22 m3 × 0.4a = 8.8 µg 9 µg/m3 9 µg/m3 × 22 m3 × 0.4a = 79.2 µg
Water
0.1 µg/litre 0.1 µg/litre × 2 litres = 0.2 µg 16 µg/litre 16 µg/litres × 2 litres = 32 µg
Food
3 µg/kg 3 µg/kg × 1.5 kg = 4.5 µg 31 µg/kg 31 µg/kg × 1.5 kg = 46.5 µg
Total daily intake 13.5 µg 157 µg
Total daily intake per kg 0.2 µg 2.5 µg
body weight
a The value of 0.4 derives from the 40% retention reported by ATSDR (1994)
Carbon tetrachloride is of low toxicity to the algae and
microorganisms tested; the lowest toxic concentration of carbon
tetrachloride reported was for methanogenic bacteria (IC50 = 6.4
mg/litre). In aquatic invertebrates, LC50 values range from 28
mg/litre to over 770 mg/litre.
The lowest acutely toxic concentration found for freshwater fish
was an LC50 of 13 mg/litre for the golden orfe
(Leuciscus idus melanotus). The lowest LC50 for a marine species
was 50 mg/litre for the dab (Limanda limanda).
Carbon tetrachloride is toxic to embryo-larval stages of fish and
of amphibians. The most sensitive species tested was the common
bullfrog (Rana catesbeiana) with an LC50 of 0.92 mg/litre for the
period from fertilization to 4 days post-hatching.
Comparing the LC50 value for the most sensitive aquatic species
(0.9 mg/litre) with typical levels of carbon tetrachloride in water
(< 1.0 µg/litre) gives a ratio of > 900. Therefore, the general risk
to aquatic organisms is low. However, carbon tetrachloride may present
a risk to embryo-larval stages of aquatic organisms at, or near, sites
of industrial discharges or spills, where much higher levels have been
reported.
11. FURTHER RESEARCH
a) Physiologically based pharmacokinetic models for carbon tetra
chloride should be further developed in order to improve their
use in defining target organ doses in human exposure conditions.
b) Since there is a lack of epidemiological data in those countries
where carbon tetrachloride is still used, epidemiological studies
of exposed populations would be useful.
c) No further research topics are recommended in view of the
phase-out of the production and use of carbon tetrachloride as a
result of the Montreal Protocol on Substances that Deplete the
Ozone Layer.
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
A drinking-water guideline value of 2 µg/litre has been
recommended for carbon tetrachloride by the World Health Organization
(WHO, 1993), based on a risk assessment approach for non-genotoxic
carcinogens. A NOAEL of 1 mg/kg body weight and an uncertainty factor
of 1000 were adopted for calculations.
The International Agency for Research on Cancer evaluated carbon
tetrachloride in 1978 (IARC, 1979), and re-evaluated it in 1987 (IARC,
1987) and 1998 (IARC, in press). The conclusions from the most recent
evaluation were that there is inadequate evidence for carcinogenicity
of carbon tetrachloride in humans but sufficient evidence for its
carcinogenicity in experimental animals. The overall evaluation was
that carbon tetrachloride is possibly carcinogenic to humans (Group
2B).
REFERENCES
Adams EM, Spencer HC, Rowe VK, McCollister DD, & Irish DD (1952) Vapor
toxicity of carbon tetrachloride determined by experiments on
laboratory animals. Arch Ind Hyg Occup Med, 6: 50-66.
Ademuyiwa O, Adesanya O, & Ajuwon OR (1994) Vitamin C in CCl4
hepatotoxicity -- A preliminary report. Hum Exp Toxicol, 13: 107-109.
Ahmadizadeh M, Echt R, Heusner WW, Ross LM, & Roth RA (1990) Effect of
carbon tetrachloride on hamster tracheal epithelial cells. J Toxicol
Environ Health, 30: 273-285.
Ahn YK & Kim JH (1993) Preventive effects of diphenyl dimethyl
dicarboxylate on the immunotoxicity of carbon tetrachloride in ICR
mice. J Toxicol Sci, 18: 185-195.
Ahr HJ, King LJ, Nastainczyk W, & Ullrich V (1980) The mechanism of
chloroform and carbon monoxide formation from carbon tetrachloride by
microsomal cytochrome P-450. Biochem Pharmacol, 29: 2855-2861.
Akahori A, Masui M, Kagawa K, Enomoto M, & Saito M (1983) Time course
of biochemical and histological alterations following a single feeding
of carbon tetrachloride to mice. Jpn J Exp Med, 53: 199-209.
Allis JW, Ward TR, Seely JC, & Simmons JE (1990) Assessment of hepatic
indicators of subchronic carbon tetrachloride injury and recovery in
rats. Fundam Appl Toxicol, 15: 558-570.
Allis JW, Brown BL, Simmons JE, Hatch GE, McDonald A, & House DE
(1996) Methanol potentiation of carbon tetrachloride hepatotoxicity:
The central role of cytochrome P450. Toxicology, 112: 131-140.
Alumot E, Nachtomi E, Mandel E, & Holstein P (1976) Tolerance and
acceptable daily intake of chlorinated fumigants in the rat diet. Food
Cosmet Toxicol, 14: 105-110.
Amacher DE & Zelljadt I (1983) The morphological transformation of
Syrian hamster embryo cells by chemicals reportedly nonmutagenic to
Salmonella typhimurium. Carcinogenesis, 4: 291-295.
Anders MW & Jakobson J (1985) Biotransformation of halogenated
solvents. Scand J Work Environ Health, 11(suppl 1): 23-32.
Anderson C, Sundberg K, & Groth O (1988) Animal model for assessment
of skin irritancy. Contact Dermatitis, 15: 143-151.
Anderson TA, Beauchamp JJ, & Walton BT (1991) Organic chemicals in the
environment. Fate of volatile and semivolatile organic chemicals in
soil: Abiotic versus biotic losses. J Environ Qual. 20: 420-424.
Ansari GA, Treinen Moslen M, & Reynolds ES (1982) Evidence for
in vivo covalent binding of CCl3 derived from CCl4 to cholesterol
of rat liver. Biochem Pharmacol, 31: 3509-3510.
Ansher SS, Dolan P, & Bueding E (1983) Chemoprotective effects of two
dithiolthiones and of butylhydroxyanisole against carbon tetrachloride
and acetaminophen toxicity. Hepatology, 3: 932-935.
Asakura S, Sawada S, Daimon H, Fukuda T, Ogura K, Yamatsu K, &
Furihata C (1994) Effects of dietary restriction on induction of
unscheduled DNA synthesis (UDS) and replicative synthesis (RDS) in rat
liver. Mutat Res, 322: 257-264.
Athanasiou K & Kyrtopoulos Sa (1981) Induction of sister chromatid
exchange by nonmutagenic carcinogens. NATO Adv Study Inst Ser A Life
Sci, 41: 557-562.
ATSDR (1994) Toxicological profile for carbon tetrachloride (Update).
Atlanta, Georgia, US Department of Health and Human Services, Agency
for Toxic Substances and Disease Registry, 229 pp (TP-93/02).
Ausloos PJ, Rebbert RE, & Glasgow L (1977) Photodecomposition of
chloromethanes adsorbed on silica surfaces. J Res Natl Stand, 82: 1-8.
Ayub-Ayala M, Flores-Alvarado LJ, Bueno Topete MR, Ortiz GG,
Gudiño-Cabrera G, & Ortuño-Sahagun D (1993) Effect of short-term
carbon tetrachloride administration on blood lactic acid levels. Gen
Pharmacol, 24(3): 627-630.
Badger DA, Sauer JM, Hoglen NC, Jolley CS, & Sipes IG (1996) The role
of inflammatory cells and cytochrome P450 in the potentiation of
CCl4-induced injury by a single dose of retinol. Toxicol Appl
Pharmacol, 141: 507-519.
Badger DA, Kuester RK, Sauer JM, & Sipes IG (1997) Gadolinium chloride
reduces cytochrome P450: Relevance to chemical-induced hepatotoxicity.
Toxicology, 121: 143-153.
Bagnasco FM, Stringer B, & Muslim AM (1978) Carbon tetrachloride
poisoning: radiographic findings. NY State J Med, 78: 646-647.
Barber ED, Donish WH, & Mueller KR (1981) A procedure for the
quantitative measurement of the mutagenicity of volatile liquids in
the Ames Salmonella/microsome assay. Mutat Res, 90: 31-48.
Barrows ME, Petrocelli SR, Macek KJ, & Carroll JJ (1980)
Bioconcentration and elimination of selected water pollutants by
bluegill sunfish. In: Haque R ed. Dynamics, exposure and hazard
assessment of toxic chemicals. Ann Arbor, Michigan, Ann Arbor Science
Publishers, Inc.
Bauer U (1981) [Human exposure to environmental chemicals
-- Investigations on volatile organic halogenated compounds in water,
air, food and human tissues. III. Communication: Results of
investigations.] Zent.bl Bakteriol Hyg I Abt Orig B, 174: 200-237 (in
German).
Baumann Ofstad E, Drangsholt H, & Carlberg GE (1981) Analysis of
volatile halogenated organic compounds in fish. Sci Total Environ, 20:
205-215.
Bechtold MM, Gee DL, Bruenner U, & Tappel AL (1982) Carbon
tetrachloride-mediated expiration of pentane and chloroform by the
intact rat: The effects of pretreatment with diethyl maleate, SKF-525A
and phenobarbital. Toxicol Lett, 11: 165-171.
Benedetti A, Fulceri R, Ferrali M, Ciccoli L, Esterbauer H, & Comporti
M (1982) Detection of carbonyl formation in phospholipids of liver
microsomes in carbon tetrachloride- and bromotrichloromethane-poisoned
rats. Biochim Biophys Acta, 712(3): 628-638.
Benigni R, Andreoli C, Conti L, Tafani P, Cotta Ramusino M, Carere A,
& Crebelli R (1993) Quantitative structure-activity relationship
models correctly predict the toxic and aneuploidizing properties of
six halogenated methanes in Aspergillus nidulans. Mutagenesis, 8:
301-305.
Benson AB (1993) Oltipraz: a laboratory and clinical review. J Cell
Biochem, 17(suppl F): 278-291.
Bergman K (1984) Application and results of whole-body radiography in
distribution studies of organic solvents. Crit Rev Toxicol, 12: 59-119
Berman E, House DE, Allis JW, & Simmons JE (1992) Hepatotoxic
interactions of ethanol with allyl alcohol or carbon tetrachloride in
rats. J Toxicol Environ Health, 37: 161-176.
Bermudez E, Mirsalis JC, & Eales HC (1982) Detection of DNA damage in
primary cultures of rat hepatocytes following in vivo and
in vitro exposure to genotoxic agents. Environ Mutagen, 4: 667-679.
Bhathal PS, Rose NR, Mackay IR, & Whittingham S (1983) Strain
differences in mice in carbon tetrachloride-induced liver injury. Br J
Exp Pathol, 64: 524-533.
Biasi F, Albano E, Chiarpotto E, Corongiu FP, Pronzato MA, Marinari
UM, Parola M, Dianzani MU, & Poli G (1991) In vivo and in
vitro evidence concerning the role of lipid peroxidation in the
mechanism of hepatocyte death due to carbon tetrachloride. Cell
Biochem Funct, 9: 111-118.
Bignami M (1977) [Relationship between mutagenic activity and chemical
structure of certain pesticides tested with Aspergillus nidulans. ]
Atti Asoc Genet Ital, 22: 143-149 (in Italian).
Birge WJ (1980) Effects of organic compounds on amphibian
reproduction. Lexington, Kentucky, University of Kentucky, Water
Resources Research Institute (Research report No. 121; PB80-147523).
Bishayee A & Chatterjee M (1993) Carrot aqueous extract protection
against hepatic oxidative stress and lipid peroxidation induced by
acute carbon tetrachloride intoxication in mice. Fitoterapia, 64(3):
261-265.
BIT-SC (Bureau international technique-Solvants chlorés) (1976)
Standardization of methods for the determination of volatile
chlorinated hydrocarbons in air and water by GC. Anal Chim Acta, 82:
1-17.
Black JA, Birge WJ, McDonnell WE, Westerman AG, Ramey BA, & Bruser DM
(1982) The aquatic toxicity of organic compounds to embryo-larval
stages of fish and amphibians. Springfield, Virginia, US Department of
Commerce (PB82-224601).
Blair A, Hartge P, Stewart PA, McAdams M, & Lubin J (in press)
Mortality and cancer incidence of workers at an aircraft maintenance
facility exposed to trichloroethylene and other organic solvents and
chemicals: Extended follow-up. Occup Environ Med.
Blair A, Stuart PA, Tolbert PE, Grauman D, Moran FX, Vaught J, &
Reyner J (1990) Cancer and other causes of death among a cohort of dry
cleaners. Br J Ind Med, 47: 162-168.
Blum DJW & Speece RE (1991) A database of chemical toxicity to
environmental bacteria and its use in interspecies comparisons and
correlations. Res J Water Pollut Control Fed, 63: 198-207.
Bogers M, Appelman LM, Feron VJ, Beems RB, & Notten WRF (1987) Effects
of the exposure profile on the inhalation toxicity of carbon
tetrachloride in male rats. J Appl Toxicol, 7: 185-191.
Bond GG, Flores GH, Shellenberger RJ, Cartmille JB, Fishbeck WA, &
Cook RR (1986) Nested-case control study of lung cancer among chemical
workers. Am J Epidemiol, 124: 53-66.
Bouwer EJ & McCarty PL (1983a) Transformations of 1- and 2-carbon
halogenated aliphatic organic compounds under methanogenic conditions.
Appl Environ Microbiol, 45: 1286-1294.
Bouwer EJ & McCarty PL (1983b) Transformations of halogenated organic
compounds under denitrification conditions. Appl Environ Microbiol,
45: 1295-1299.
Boyd MR, Statham CN, & Longo NS (1980) The pulmonary Clara cell as
target for toxic chemicals requiring metabolic activation; studies
with carbon tetrachloride. J Pharmacol Exp Ther, 212: 109-114.
Bozzelli JW & Kebbekus BB (1982) A study of some aromatic and
halocarbon vapors in the ambient atmosphere of New Jersey. J Environ
Sci Health, A17: 693-711.
Brambilla G, Carlo P, Finollo R, Bignone FA, Ledda A, & Cajelli E
(1983) Viscometric detection of liver DNA fragmentation in rats
treated with minimal doses of chemical carcinogens. Cancer Res, 43:
202-209.
Brams A, Buchet JP, Crutzen MC, De Meester C, Lowerys R, & Leonard A
(1987) A comparative study, with 40 chemicals, of the efficiency of
the Salmonella assay and the SOS chromotest (kit procedure). Toxicol
Lett, 38: 123-133.
Brattin WJ, Pencil SD, Waller RL, Glende EA, & Recknagel RO (1984)
Assessment of the role of calcium ion in halocarbon hepatotoxicity.
Environ Health Perspect, 57: 321-323.
Braun R & Schöneich J (1975) The influence of ethanol and carbon
tetrachloride in the host-mediated assay with
Salmonella typhimurium. Mutat Res, 31: 191-194.
Bringmann G (1973) [Determination of biological hazards of water
polluting substances from the inhibition of glucose assimilation of
the bacterium Pseudomonas fluorescens. ] Gesundh-Ing, 94(12):
366-369(in German).
Bringmann G (1975) [Determination of the biological damage of
water-endangering compounds by means of the inhibition of cell
proliferation in the blue alga Microcystis.] Gesundh-Ing, 96(9):
238-241 (in German).
Bringmann G (1978) [Determination of biological adverse action of
water pollutants against protozoa: I. Bacteriological flagellates.] Z
Wasser-Abwasser Forsch, 11: 210-215 (in German).
Bringmann G & Kühn R (1977a) [Limit values for adverse action of water
pollutants against bacteria (Pseudomonas putida) and green algae
(Scenedesmus quadricauda) in the cell proliferation inhibition
test.] Z Wasser-Abwasser Forsch, 10: 87-97 (in German).
Bringmann G & Kühn R (1977b) [Results of adverse action of water
pollutants against Daphnia magna. ] Z Wasser-Abwasser Forsch, 10:
161-166 (in German).
Bringmann G & Kühn R (1980) [Determination of biological adverse
action of water pollutants against protozoa: II. Bacteriological
ciliates.] Z Wasser-Abwasser Forsch, 13: 26-31.
Bringmann G, Kühn R, & Winter A (1980) [Determination of biological
adverse action of water pollutants against protozoa: III. Saprozoic
flagellates.] Z Wasser-Abwasser Forsch, 13: 170-173 (in German).
Brondeau MT, Bonnet P, Guenier JP, & De Ceaurriz J (1983) Short-term
inhalation test for evaluating industrial hepatotoxicants in rats.
Toxicol Lett, 19: 139-146.
Brown RHA, Cape JN, & Farmer JG (1998) Partitioning of chlorinated
solvents between pine needles and air. Chemosphere, 36: 1799-1810.
Bruccoleri A, Ballucci R, Germolec DR, Blackshear P, Simeonova P,
Thurman RG, & Luster MI (1997) Induction of early-immediate genes by
tumor necrosis factor A contribute to liver repair following
chemical-induced hepatotoxicity. Hepatology, 25: 133-141.
Bruckner JV, MacKenzie WF, Muralidhara S, Luthra R, Kyle GM, & Acosta
D (1986) Oral toxicity of carbon tetrachloride: Acute, subacute, and
subchronic studies in rats. Fundam Appl Toxicol, 6: 16-34.
Brugnone F, Apostoli P, Perbellini L, Silvestri R, & Cocheo V (1983)
Monitoring of occupational exposure to low concentration of carbon
tetrachloride. Dev Toxicol Environ Sci, 11: 575-578.
BUA (1990) [Tetrachloromethane. Report of the German Chemical
Society-Advisory Committee on Existing Chemicals of Environmental
Relevance.] Stuttgart, S. Hirzel Wissenschaftliche Verlagsgesellschaft
(BUA Report 45) (in German).
Buccafusco RJ, Ells SJ, & LeBlanc GA (1981) Acute toxicity of priority
pollutants to Bluegill (Lepomis macrochirus). Bull Environ Toxicol,
26: 446-452.
Burk RF, Patel K, & Lane JM (1983) Reduced glutathione protection
against rat liver microsomal injury by carbon tetrachloride:
Dependence on O2. Biochem J, 215: 441-445.
Burk RF, Lane JM, & Patel K (1984) Relationship of oxygen and
glutathione in protection against carbon tetrachloride-induced hepatic
microsomal lipid peroxidation and covalent binding in the rat. J Clin
Invest, 74: 1996-2001.
Burk RF, Hill KE, & Lane JM (1988) Inhibition of CCl4 metabolism by
oxygen varies between isoenzymes of cytochrome P-450. Biochem Biophys
Res Commun, 152: 1463-1467.
Burston MW, Nazari MN, Bishop PK, & Lerner DN (1993) Pollution of
ground water in the Coventry region (UK) by chlorinated hydrocarbon
solvents. J Hydrol, 149: 137-161.
Callen DF, Wolf CR, & Philpot RM (1980) Cytochrome P-450 mediated
genetic activity and cytotoxicity of seven halogenated aliphatic
hydrocarbons in Saccharomyces cerevisiae. Mutat Res, 77: 55-63.
Cambon-Gros C, Deltour P, Boigegrain RA, Fernandez Y, & Mitjavila S
(1986) Short communications: Radical activation of carbon
tetrachloride in foetal and maternal rat liver microsomes. Biochem
Pharmacol, 35: 2041-2044.
Cantilena LR, Cagen SZ, & Klaassen CD (1979) Methanol potentiation of
carbon tetrachloride-induced hepatotoxicity. Proc Soc Exp Biol Med,
162: 90-95.
Cantor KP, Stuart PA, Brinton LA, & Dosemeci M (1995) Occupational
exposures and female breast cancer mortality in the United States. J
Occup Environ Med, 37: 336-348.
Castro GD , Díaz-Gomez MI, & Castro JA (1989) Species differences in
the interaction between CCl4 reactive metabolites and liver DNA or
nuclear protein fractions. Carcinogenesis, 10: 289-294.
Castro GD, Díaz-Gomez MI, & Castro JA (1990) Biotransformation of
carbon tetrachloride and lipid peroxidation promotion by liver nuclear
preparations from different animal species. Cancer Lett ,53: 9-15.
Cervinková Z, Bgatova NP, Shorina TG, Holecek M, Subrtová D,
Vosvrelová H, Shkurupy VA, & Simek J (1987) Structural and functional
changes after the administration of tetrachloromethane in the liver of
rats fed on diets with different protein contents. Physiol Bohemoslov,
36: 349-359.
Charbonneau M, Iijima M, Cote MG, & Plaa GL (1985) Temporal analysis
of rat liver injury following potentiation of carbon tetrachloride
hepatotoxicity with ketonic or ketogenic compounds. Toxicology, 35(2):
95-112.
Chatterjee A (1966) Testicular degeneration in rats by carbon
tetrachloride intoxication. Experientia(Basel), 226: 395-396.
Chatterjee A (1968) Effect of CCl4 on gonadal physiology in female
rats. Acta Anat, 71: 82-86.
Checkoway H, Wilcosky T, Wolf P, & Tyroler H (1984) An evaluation of
the associations of leukemia and rubber industry solvent exposures. Am
J Ind Med, 5: 239-249.
Chen WJ, Chi EY, & Smuckler EA (1977) Carbon tetrachloride-induced
changes in mixed function oxidases and microsomal cytochromes in the
rat lung. Lab Invest, 36: 388-394.
Clark CS (1981) Evaluation of the health risk associated with the
treatment and disposal of municipal wastewater and sludge. Washington,
DC, US Environmental Protection Agency (EPA-600/1-80-030).
Clarke H, Egan DA, Hefferman M, Doyle S, Byrne C, Kilty C, & Ryan MP
(1997) Alpha-glutathione S-transferase (alpha-GST) release, an early
indicator of carbon tetrachloride hepatotoxicity in the rat. Hum Exp
Toxicol, 16: 154-157.
Clyne MAA & Holt PM (1978) Reaction kinetics involving ground X2Pi
and excited A2Sigma+ hydroxyl radicals. J Chem Soc Faraday Trans
II, 75: 569-581.
Cochran JW, Yates MV, & Henson JM (1988) A modified fuge-and-trap gas
chromatography method for analysis of volatile halocarbons in
microbiological degradation studies. J Microbiol Methods, 8: 347-354.
Comporti M, Benedetti A, Ferrali M, & Fulceri R (1984) Reactive
aldehydes (4-hydroxyalkenals) originating from the peroxidation of
liver microsomal lipids: biological effects and evidence for their
binding to microsomal protein in CCl4 or BrCCl3 intoxication. Front
Gastrointest Res, 8: 46-62.
Condie LW, Laurie RD, Mills T, Robinson M, & Bercz JP (1986) Effect of
gavage vehicle on hepatotoxicity of carbon tetrachloride in CD-1 mice:
corn oil versus Tween-60 aqueous emulsion. Fundam Appl Toxicol, 7:
199-206.
Congiu AM, Calendi E, & Ugazio G (1984) Effects of metal ions and
CCl4 on sea urchin embryo (Paracentrotus lividus). Res Commun Chem
Pathol Pharmacol ,43: 317-323.
Coni P, Pichiri-Coni G, Ledda-Columbano GM, Rao PM, Sarma DSR, &
Columbano A (1990) Liver hyperplasia is not necessarily associated
with increased expression of c-fos and c-myc RNA. Carcinogenesis, 11:
835-839.
Coni P, Simbula G, de Prati AC, Menegazzi M, Suzuki H, Sarma DSR,
Ledda-Columbano GM, & Columbano A (1993) Differences in the
steady-state levels of c-fos, c-jun and c-myc messenger RNA during
mitogen-induced liver growth and compensatory regeneration.
Hepatology, 17: 1109-1116.
Coutant RW & Scott DR (1982) Applicability of passive dosimeters for
ambient air monitoring of toxic organic compounds. Environ Sci
Technol, 16: 410-413.
Coutino RR (1979) Analysis of anaphase in cell culture: an adequate
test system for the distinction between compounds which selectively
alter the chromosome structure or the mitotic apparatus. Environ
Health Perspect, 31: 131-136.
Cox RA, Derwent RG, Eggleston AEJ, & Lovelock JE (1976) Photochemical
oxidation of halocarbons in the troposphere. Atmos Environ, 10:
305-308.
Craddock VM & Henderson AR (1978) De novo and repair replication of
DNA in liver or carcinogen-treated animals. Cancer Res, 38: 2135-2143.
Crescentini G, Mangani F, Mastrogiacomo AR, & Bruner F (1981)
Calibration method for the gas chromatographic analysis of halocarbons
in atmospheric samples using permeation tubes and an electron-capture
detector. J Chromatogr, 204: 445-451.
Crescentini G, Mangani F, Mastrogiacomo AR, Cappiello A, & Bruner F
(1983) Fast determination of some halocarbons in the atmosphere by gas
chromatography-high-resolution mass spectrometry. J Chromatogr, 280:
146-151.
Cronn DR & Harsch DE (1979) Determination of atmospheric halocarbons
concentrations by gas chromatographic-mass spectrometry. Anal Lett,
12B(4): 1489-1496.
Cronn DR, Rasmussen RA, Robinson E, & Harsch DE (1977) Halogenated
compound identification and measurement in the troposphere and lower
stratosphere. J Geophys Res, 82: 5935-5944.
Cunningham ML & Matthews HB (1991) Relationship of
hepatocarcinogenicity and hepatocellular proliferation induced by
mutagenic noncarcinogens vs carcinogens. II. 1- vs 2-nitropropane.
Toxicol Appl Pharmacol 110: 505-513.
Cunningham ML, Gandolfi AJ, Brendel K, & Sipes IG (1981) Covalent
binding of halogenated volatile solvents to subcellular macromolecules
in hepatocytes. Life Sci, 29: 1207-1212.
Cupitt LT (1980) Fate of toxic and hazardous materials in the
environment. Washington, DC, US Environmental Protection Agency (EPA
600/53-80-084).
Czaja MJ, Flanders KC, Biempica I, Klein C, Zern MA, & Weiner FR
(1989) Expression of tumor necrosis factor alpha and transforming
factor beta1 in acute liver injury. Growth Factors, 1: 219-226.
Czaja MJ, Xu J, & Alt E (1995) Prevention of carbon
tetrachloride-induced rat liver injury by soluble tumor necrosis
factor receptor. Gastroenterology, 108: 1849-1854.
Daft JL (1988) Rapid determination of fumigant and industrial chemical
residues in food. J Assoc Off Anal Chem, 71: 748-760.
Daft JL (1989) Determination of fumigants and related chemicals in
fatty and nonfatty foods. J Agric Food Chem 37: 560-564.
Daft JL (1991) Fumigants and related chemicals in foods. Review of
residue findings, contamination sources, and analytical methods. Sci
Total Environ, 100: 501-518.
Dai Y & Cederbaum AI (1995) Inactivation and degradation of human
cytochrome P4502E1 by CCl4 in a transfected HepG2 cell line.
J Pharmacol Exp Ther, 275: 1614-1622.
Daniluk J, Chibowski D, Szuster-Ciesielska A, & Kandefer-Szerszen M
(1994) Effect of carbon tetrachloride intoxication and ethanol
ingestion on interferon production in mice. Arch Immunol Ther Exp
(Warsz), 42(4): 325-330.
David A, Frantik E, Holusa R, & Nováková O (1981) Role of time and
concentration on carbon tetrachloride toxicity in rats. Int Arch Occup
Environ Health, 48: 49-60.
Dawson GW, Jennings AL, Drozdowski D, & Rider E (1975/77) The acute
toxicity of 47 industrial chemicals to fresh and saltwater fishes.
J Hazard Mater, 1: 303-318.
Dean BJ & Hodson-Walker G (1979) An in vitro chromosome assay using
cultured rat-liver cells. Mutat Res, 64: 329-337.
Deer HM, McJilton CE, & Harein PK (1987) Respiratory exposure of grain
inspection workers to carbon tetrachloride fumigant. Am Ind Hyg Assoc
J, 48: 586-593.
De Ferreyra EC, De Fenos OM, & Castro JA (1983) Tryptophan
potentiation of the late cysteine preventive effects in carbon
tetrachloride-induced necrosis. Res Commun Chem Pathol Pharmacol, 40:
515-518.
De Ferreyra EC, Villarruel MC, Bernacchi AS, Fernandez G, de Fenos OM,
& Castro JA (1989) Late preventive effects against carbon
tetrachloride-induced liver necrosis of the calcium chelating agent
Calcion. Arch Toxicol, 63: 450-455.
De Ferreyra EC, Villarruel MC, Bernacchi AD, de Fenos OM, & Castro JA
(1992) Prevention of carbon tetrachloride-induced liver necrosis by
the chelator alizarin sodium sulfonate. Exp Mol Pathol, 56: 197-207.
De Ferreyra EC, Bernacchi AS, San Martín MF, Castro GD, & Castro JA
(1994) Nicotinamide late protective effects against carbon
tetrachloride-induced liver necrosis. Exp Mol Pathol, 60: 214-223.
De Flora S (1981) Study of 106 organic and inorganic compounds in the
Salmonella/microsome test. Carcinogenesis, 2: 283-298.
De Flora S, Zanacchi P, Camoirano A, Bennicelli C, & Badolati GS
(1984) Genotoxic activity and potency of 135 compounds in the Ames
reversion test and in a bacterial DNA-repair test. Mutat Res, 133:
161-198.
De Groot H & Haas W (1980) O2-independent damage of cytochrome P-450
by CCl4 metabolites in hepatic microsomes. Febs Lett, 115: 253-256.
De Groot H & Haas W (1981) Self-catalysed, O2-independent
inactivation of NADPH- or dithionite- reduced microsomal cytochrome
P-450 by carbon tetrachloride. Biochem Pharmacol, 30: 2343-2347.
Delaney B & Kaminski NE (1994) Induction of serum borne
immunomodulatory factors in B6C3F1 mice by carbon tetrachloride.
Toxicology, 88: 201-212.
Dent JG & Graichen EM (1982) Effect of hepatocarcinogens on epoxide
hydrolase and other xenobiotic metabolizing enzymes. Carcinogenesis,
3: 733-738.
De Vries JW, Larson PA, Bowers RH, Keating JA, Broge JM, Wehling PS,
Patel HH, & Zurawski JW (1985) Improved codistillation method for
determination of carbon tetrachloride, ethylene-dichloride and
ethylene-dibromide in grain and grain-based products. J Assoc Off Anal
Chem, 68(4): 759-762.
De Zwart LL, Venhorst J, Groot M, Commandeur JN, Hermanns RC, Meerman
JH, Fan Baar BL, & Vermeulen NP (1997) Simultaneous determination of
eight lipid peroxidation degradation products in urine of rats treated
with carbon tetrachloride using gas chromatography with
electron-capture detection. J Chromatogr Biomed Appl, B694: 277-287.
Dhawan DK & Goel A (1994) Protective role of zinc on rat liver
function in long-term toxicity induced by carbon tetrachloride.
J Trace Elem Exp Med, 7: 1-9.
Dianzani MU (1984) Lipid peroxidation and haloalkylation: two distinct
mechanisms for carbon tetrachloride-induced liver damage. Int Congr
Ser Exerpta Med, 632: 39-50.
Díaz Gómez MI & Castro JA (1980a) Covalent binding of carbon
tetrachloride metabolites to liver nuclear DNA, protein and lipids.
Toxicol Appl Pharmacol, 56: 199-206.
Díaz Gómez MI & Castro JA (1980b) Nuclear activation of carbon
tetrachloride and chloroform. Res Commun Pathol Pharmacol, 27:
191-194.
Díaz Gómez MI, de Castro CR, D'Acosta N, de Fenos OM, de Ferreyra EC,
& Castro JA (1975a) Species differences in carbon
tetrachloride-induced hepatotoxicity: the role of CCl4 activation and
of lipid peroxidation. Toxicol Appl Pharmacol, 34: 102-114.
Díaz Gómez MI, de Castro CR, de Ferreyra EC, D'Acosta N, de Fenos OM,
& Castro JA (1975b) Mechanistic studies on carbon tetrachloride
hepatotoxicity in fasted and fed rats. Toxicol Appl Pharmacol, 32:
101-108.
Dickson AG & Riley JP (1976) The distribution of short-chain
halogenated aliphatic hydrocarbons in some marine organisms. Mar
Pollut Bull, 7: 167-169.
Dilling WL, Tefertiller NB, & Kallos GJ (1975). Evaporation rates and
reactivities of methylene chloride, chloroform, 1,1,1-trichloroethane,
trichloroethylene, tetrachloroethylene and other chlorinated compounds
in dilute aqueous solutions. Environ Sci Technol, 9(9): 833-838.
Dingell JV & Heimberg M (1968) The effects of aliphatic halogenated
hydrocarbons on hepatic drug metabolism. Biochem Pharmacol, 17:
1269-1278.
DiRenzo AB, Gandolfi AJ, Sipes IG, Brendel K, & Byard JL (1984) Effect
of O2 tension on the bioactivation and metabolism of aliphatic
halides by primary rat-hepatocyte cultures. Xenobiotica, 14: 521-525.
Divincenzo GD & Krasavage WJ (1974) Serum ornithine carbamyl
transferase as a liver response test for exposure to organic solvents.
Am Ind Hyg Assoc J, 35: 21-29.
Doherty AT, Ellard S, Parry EM, & Parry JM (1996) An investigation
into the activation and deactivation of chlorinated hydrocarbons to
genotoxins in metabolically competent human cells. Mutagenesis, 11:
247-274.
Doolittle DJ, Muller G, & Scribner HE (1987) Relationship between
hepatotoxicity and induction of replicative DNA synthesis following
single or multiple doses of carbon tetrachloride. J Toxicol Environ
Health, 22: 63-78.
Doong RA & Wu SC (1995) Substrate effects on the enhanced
biotransformation of polychlorinated hydrocarbons under anaerobic
condition. Chemosphere, 30: 1499-1511.
Doong RA & Wu SC (1996) Effect of substrate concentration on the
biotransformation of carbon tetrachloride and 1,1,1-trichloroethane
under anaerobic condition. Water Res, 30: 577-586.
Doong RA, Wu SC, & Chen TF (1996) Anaerobic biotransformation of
polychlorinated methane and ethene under various redox conditions.
Chemosphere, 32: 377-390.
Duffy CC, McCallister DL, & Renken RR (1997) Carbon tetrachloride
retention by modern and buried soil A horizons. J Environ Qual, 26:
1123-1127.
Duprat P, Delsart L, & Gradiski P (1976) Irritant potency of the
principle aliphatic chloride solvents on the skin and ocular mucous
membranes of rabbits. Eur J Toxicol, 9: 171-177.
Ebel RE (1989) Pyrazole treatment of rats potentiates CCl4-but not
CHCl3-hepatotoxicity. Biochem Biophys Res Commun, 161: 615-618.
ECDIN (1992) On-line search in the environmental chemicals data and
information network. ECDIN/copyright Joint Research Centre/European
Commission, Ispra.
Edwards JE (1941) Hepatomas in mice induced with carbon tetrachloride.
J Natl Cancer Inst, 2: 197-199.
Edwards JE & Dalton AJ (1942) Induction of cirrhosis of the liver and
of hepatomas in mice with carbon tetrachloride. J Natl Cancer Inst, 3:
19-41.
Edwards JE, Heston WE, & Dalton AJ (1942) Induction of the carbon
tetrachloride hepatoma in strain L mice. J Natl Cancer Inst, 3:
297-301.
Edwards PR, Campbell I, & Milne GS (1982a) The impact of
chloromethanes on the environment - Part 1: The atmospheric chlorine
cycle. Chem Ind, 21 August: 574-578.
Edwards PR, Campbell I, & Milne GS (1982b) The impact of
chloromethanes on the environment, part 3: Chloroform and carbon
tetrachloride. Chem Ind, 18 September: 714-718.
Edwards MJ, Keller BJ, Kauffman FC, & Thurman RG (1993) The
involvement of Kupfer cells in carbon tetrachloride toxicity. Toxicol
Appl Pharmacol, 119: 275-279.
Egli C, Scholtz R, Cook AM, & Leisinger T (1987) Anaerobic
dechlorination of tetrachloromethane and 1,2-dichloroethane to
degradable products by pure cultures of Desulfobacterium sp. and
Methanobacterium sp. FEMS Microbiol Lett, 43: 257-261.
Eichelberger JW, Bellar TA, Donnelly JP, & Budde WL (1990)
Determination of volatile organics in drinking water with US EPA
method 524.2 and the ion trap detector. J Chromatogr Sci, 28: 460-467.
Elias L (1977) In-situ quantitation of background halofluorocarbon
levels. NBS Special Publication (USA), vol 464, pp 435-438.
ElSisi AE, Hall P, Sim W-LW, Earnest DL, & Sipes IG (1993a)
Characterization of vitamin A potentiation of carbon
tetrachloride-induced liver injury. Toxicol Appl Pharmacol, 119:
280-288.
ElSisi AE, Earnest DL, & Sipes IG (1993b) Vitamin A potentiation of
carbon tetrachloride hepatotoxicity: role of liver macrophages and
active oxygen species. Toxicol Appl Pharmacol, 119: 295-301.
Ember LR, Layman PL, Lepkowski W, & Zurer PS (1986) The changing
atmosphere. Chem Eng News, 24 November: 14-64.
Entz RC & Hollifield HC (1982) Headspace gas chromatographic analysis
of foods for volatile halocarbons. J Agric Food Chem, 30: 84-88.
Entz RC, Thomas KW & Diachenko GW (1982) Residues of volatile
halocarbons in foods using headspace gas chromatography. J Agric Food
Chem, 30: 846-849.
Erickson MD (1981) Acquisition and chemical analysis of mother's milk
for selected toxic substances. Gov Rep Announc Index US, 81: 5096
(Publication PB81-231029).
Eschenbrenner AB & Miller EB (1944) Studies on hepatomas: I. Size and
spacing of multiple doses in the induction of carbon tetrachloride
hepatomas. J Natl Cancer Inst, 4: 385-388.
Evans MV & Simmons JE (1996) Physiologically based pharmacokinetic
estimated metabolic constants and hepatotoxicity of carbon
tetrachloride after methanol pretreatment in rats. Toxicol Appl
Pharmacol, 140: 245-253.
Evans MV, Crank WD, Yang HM, & Simmons JE (1994) Applications of
sensitivity analysis to a physiologically based pharmacokinetic model
for carbon tetrachloride in rats. Toxicol Appl Pharmacol, 128: 36-44.
Fander U, Haas W, & Kröner H (1982) The damage of the hepatic mixed
functional oxygenase system by CCl4: Significance of incorporation of
14CCl4 metabolites in vivo. Exp Mol Pathol, 36: 34-43.
Fanelli SL & Castro JA (1995) Covalent binding of carbon tetrachloride
reactive metabolites to liver microsomal and nuclear lipid and
phospholipid classes from Sprague-Dawley and Osborne Mendel male rats.
Teratog Carcinog Mutagen, 15: 155-166.
Fehér J, Bar-Pollák ZS, Sréter L, Fehér E, & Toncsev H (1982)
Biochemical markers in carbon tetrachloride- and galactosamine-induced
acute liver injuries: The effects of dihydroquinoline-type
antioxidants. Br J Exp Pathol, 63: 394-400.
Fernández G, Villarruel MC, De Toranzo EGD, & Castro JA (1982)
Covalent binding of carbon tetrachloride metabolites to the heme
moiety of cytochrome P-450 and its degradation products. Res Commun
Chem Pathol Pharmacol, 35: 283-290.
Fogelqvist E (1985) Carbon tetrachloride, tetrachloroethylene,
1,1,1-trichloroethane and bromoform in arctic seawater. J Geophys Res,
90: 9181-9193.
Folland DS, Schaffner W, Ginn HE, Crofford OB, & McMurray DR (1976)
Carbon tetrachloride toxicity potentiated by isopropyl alcohol. J Am
Med Assoc, 236: 1853-1856.
Foureman P, Mason JM, Valencia R, & Zimmering S (1994) Chemical
mutagenesis testing in Drosophila: X. Results of 70 coded chemicals
tested for the National Toxicology Program. Environ Mol Mutagen, 23:
208-227.
Fowler JSL (1969) Carbon tetrachloride metabolism in the rabbit. Br J
Pharmacol 37: 733-737.
Frank H & Frank W (1990) Concentrations of airborne C1 - and
C2 - halocarbons in forest areas in West Germany: results of
three campaigns in 1986, 1987 and 1989. Atmos Environ, 24A: 1735-1739.
Frank H & Frank W (1986) Photochemical activation of chloroethenes
leading to destruction of photosynthetic pigments. Experientia
(Basel), 42: 1267-1269.
Frantik E & Benes V (1984) Central nervous effect and blood level
regressions on exposure time paralleled in solvents (toluene, carbon
tetrachloride and chloroform. Activ Nerv Sup (Praha), 26: 131-133.
Fraser P & Derek N (1994) Halocarbons, nitrous oxide, methane and
carbon monoxide -- the GAGE Program. In: Dick C & Fras J ed. Baseline
1991. Washington, DC, Bureau of Meteorology (CSIRO), pp 70-81.
Fraser P, Penkett S, Gunson M, Weiss R, & Rowland FS (1994)
Measurements. In: Kaye J, Penkett S, & Ormond F ed. Concentrations,
lifetimes and trends of CFCs, halons and related species. Washington,
DC, NASA, pp 1/1-1/68 (Report No.1339).
Freiria-Gandara MJ, Lorenzo-Ferreira RA, Alvarez-Deveja A, & Bermejo F
(1992) Occurrence of halogenated hydrocarbons in the water supply of
different cities of Galicia (Spain). Environ Technol, 13: 437-447.
Fujii T (1977) The determination of traces of organohalogen compounds
in aqueous solution by direct injection gas chromatography -- mass
spectrometry and single ion detection. Anal Chim Acta, 92: 117-122.
Gäb S, Schmitzer J, Turner WV, & Korte F (1980) Mineralization of
CCl4 and CCl2F2 on solid surfaces. Z Naturforsch, 35B: 946-952.
Galbally IE (1976) Man-made carbon tetrachloride in the atmosphere.
Science, 193: 573-576.
Galli A & Schiestl RH (1995) Salmonella test positive and negative
carcinogens show different effects on intrachromosomal recombination
in G2 cells cycle arrested yeast cells. Carcinogenesis, 16: 659-663.
Galli A & Schiestl RH (1996) Effects of Salmonella assay negative
and positive carcinogens on intrachromosomal recombination in
G1-arrested yeast cells. Mutat Res, 370: 209-221.
Gans JH & Korson R (1984) Liver nuclear DNA synthesis in mice
following carbon tetrachloride administration or partial hepatectomy.
Proc Soc Exp Biol Med, 175: 237-242.
Gargas ML, Andersen ME, & Clewell HJ III (1986) A physiologically
based simulation approach for determining metabolic constants from gas
uptake data. Toxicol Appl Pharmacol, 86: 341-352.
Garry VF, Nelson RL, Griffith J, & Harkins M (1990) Preparations for
human study of pesticide applicators: Sister chromatid exchanges and
chromosome aberrations in cultured human lymphocytes exposed to
selected chemicals. Teratog Carcinog Mutagen, 10: 21-29.
Gavino VC, Dillard CJ, & Tappel AL (1984) Release of ethane and
pentane from rat tissue slices: Effect of vitamin E, halogenated
hydrocarbons and iron overload. Arch Biochem Biophys, 233: 741-747.
Gee DL, Bechtold MM, & Tappel AL (1981) Carbon tetrachloride-induced
lipid peroxidation: simultaneous in vivo measurements of pentane and
chloroform exhaled by the rat. Toxicol Lett, 8: 299-306.
Gehring PJ (1968) Hepatotoxic potency of various chlorinated
hydrocarbons vapours relative to their narcotic and lethal potencies
in mice. Toxicol Appl Pharmacol, 13: 287-298.
Geyer H, Politzki G, & Freitag D(1984) Prediction of ecotoxicological
behaviour of chemicals: Relationship between n-octanol/water
partition coefficient and bioaccumulation of organic chemicals by alga
Chlorella. Chemosphere, 13: 269-284.
Giger W, Molnar-Kubica E, & Wakeham S (1978) Volatile chlorinated
hydrocarbons in ground and lake waters. In: Aquatic pollution
-- Transformation of biological effects. Oxford, New York, Pergamon
Press, pp 101-123 (Pergamon Series on Environmental Science).
Gilli G, Scursatone E, Bono R, Natale P, & Grosa M (1990) An overview
of atmospheric pollution in Italy before the use of new gasoline. Sci
Total Environ, 93: 51-56.
Gilman MR (1971) A preliminary study of the teratogenic effects of
inhaled carbon tetrachloride and ethyl alcohol consumption in the rat.
Philadelphia, Pennsylvania, Drexel University (Dissertation).
Glende EA & Recknagel RO (1991) An indirect method demonstrating that
CCl4-dependent hepatocyte injury is linked to a rise in intracellular
calcium ion concentration. Res Commun Chem Pathol Pharmacol, 73:
41-52.
Goldsworthy TL, Goldsworthy SM, Sprankle CS , & Butterworth BE (1994)
Expression of myc, fos and Ha-ras associated with chemically induced
cell proliferation in the rat liver. Cell Prolif, 27: 269-278.
Gómez MR, Cocco P, Dosemeci M, & Stewart PA (1994) Occupational
exposure to chlorinated aliphatic hydrocarbons: Job exposure matrix.
Am J Ind Med, 26: 171-183.
Gorla N, de Ferreyra EC, Villarruel MC, de Fenos OM, & Castro JA
(1983) Studies on the mechanism of glutathione prevention of carbon
tetrachloride-induced liver injury. Br J Exp Pathol, 64: 388-395.
Goyette M, Petropulos CJ, Shank PR, & Fausto N (1983) Expression of
cellular oncogene during liver regeneration. Science, 219: 510-512.
Gradiski D, Magadur J-L, Baillot M, Danière MC, & Schuh MB (1974)
Toxicité comparée des principaux solvants chlorés aliphatiques. J Eur
Toxicol, 7: 247-254.
Gradiski D, Bonnet P, Raoult G, & Magadur JL (1978) Toxicité aiguë
comparée par inhalation des principaux solvants aliphatiques chlorés.
Arch Mal Prof Méd Trav Sécur Soc, 39: 249-257.
Grob K (1984) Further development of direct aqueous injection with
electron-capture detection in gas chromatography. J Chromatogr, 299:
1-11.
Gruebele A, Zawaski K, Kapalan D, & Novak RF (1996) Cytochrome
P4502E1- and cytochrome P4502B1/2B2-catalysed carbon tetrachloride
metabolism. Drug Metab Dispos, 24: 15-22.
Gualandi G (1984) Genotoxicity of the free-radical producers CCl4 and
lipoperoxide in Aspergillus nidulans. Mutat Res, 136: 109-114.
Guicherit R & Schulting FL (1985) The occurrence of organic chemicals
in the atmosphere of the Netherlands. Sci Total Environ, 43: 193-219.
Hall PM, Plummer JL, Ilsley AH, & Cousins MJ (1991) Hepatic fibrosis
and cirrhosis after chronic administration of alcohol and "low-dose"
carbon tetrachloride vapour in the rat. Hepatology, 13: 815-819.
Hall PM, Plummer JL, Ilsley AH, Ahern MJ, Cmielewski PL, & Williams RA
(1994) The pathology of liver injury induced by the chronic
administration of alcohol and "low dose" carbon tetrachloride in
Porton rats. J Gastroenterol Hepatol, 9: 250-256.
Hamlin GP, Kholkute SD, & Dukelow WR (1993) Toxicology of maternally
ingested carbon tetrachloride (CCl4) on embryonal and fetal
development and in vitro fertilization in mice. Zool Sci, 10:
111-116.
Harris RN & Anders MW (1980) Effect of fasting, diethyl maleate, and
alcohols on carbon tetrachloride-induced hepatotoxicity. Toxicol Appl
Pharmacol, 56: 191-198.
Harsch DE & Cronn DR (1978) Low-pressure sample-transfer technique for
analysis of stratospheric air samples. J Chromatogr Sci ,16(8):
363-367.
Hayes JR, Condie LW, & Borzelleca JF (1986) Acute, 14-day repeated
dosing, and 90-day subchronic toxicity studies of carbon tetrachloride
in CD-1 mice. Fundam Appl Toxicol, 7: 454-463.
Heikes DL (1987) Purge and trap method for determination of volatile
hydrocarbons and carbon disulfide in table ready foods. J Assoc Off
Anal Chem, 70: 215-226.
Heil E, Oeser H, Hatz R, & Kelker H (1979) [Gas-chromatographic
determination of traces of C1- and C2-fluoro- and chlorohydrocarbons
in air.] Fresenius Z Anal Chem, 297(5): 357-364 (in German).
Heineman EF, Cocco P, Gómez MR, Dosemeci M, Stewart PA, Hayes RB, Zahm
SH, Thomas TL, & Blair A (1994) Occupational exposure to chlorinated
aliphatic hydrocarbons and risk of astrocytic brain cancer. Am J Ind
Med, 26: 155-169.
Herzfeld D, Van der Gun KD, & Louw R (1989) Quantitative determination
of volatile organochlorine compounds in water by GC-headspace analysis
with dibromomethane as an internal standard. Chemosphere, 18(7/8):
1425-1430.
Hollinger MA (1982) Biochemical evidence for pulmonary endothelial
cell injury after carbon tetrachloride administration in mice. J
Pharmacol Exp Ther, 222: 641-644.
Holly EA, Aston DA, Ahn DK, & Smith AH (1996) Intraocular melanoma
linked to occupations and chemical exposures. Epidemiology, 7: 55-61.
Honma J (1990) Effects of trichloroethylene, 1,1,1-trichloroethane and
carbon tetrachloride on plasma fibroproteins of rats. Ind Health, 28:
159-174.
Hooser SB, Rosengren RJ, Hill DA, Mobley SA, & Sipes IG (1994) Vitamin
A modulation of xenobiotic-induced hepatotoxicity in rodents. Environ
Health Perspect, 102(suppl 9): 39-43.
Howard PH ed. (1990) Handbook of environmental fate and exposure data
for organic chemicals: Volume II. Solvents. Chelsea, Michigan, Lewish
Publishers, Inc., pp 85-91.
Howard Carleton J & Evenson P (1976) Rate constants for the reactions
of OH with CH4 and fluorine, chlorine and bromine substituted
methanes at 296 K. J Chem Phys, 64: 197.
Howard PH, Boethling RS, & Jarvis WF ed. (1991) Handbook of
environmental degradation rates. Chelsea, Michigan, Lewis Publishers,
Inc., pp 34-35.
Huiskamp R, Van der Heijden CA, Speijers GJA, Ros JPM, Huldy HJ,
Besemer AC, Lanting RW, Maas RJM, Heijna-Merkus E, Bergshoeff G,
Gerlofsma A, Mennes WC, Van der Most PFJ, De Vrijer FL, Janssen PCJM,
Knaap AGAC, Huijgen C, Duiser JA, & De Jong P (1986) Integrated
criteria document: Carbon tetrachloride -- Air effects. Bilthoven, The
Netherlands, National Institute of Public Health and Environmental
Protection (Report No. 738513005).
Hustzik E, Lazar G, & Parducz A (1980) Electron microscopic study of
Kupfer-cell phagocytosis blockade induced by gadolinium chloride. Br J
Exp Pathol, 61: 624-630.
IARC (1979) Some halogenated hydrocarbons. Lyon, International Agency
for Research on Cancer, pp 377-399 (IARC Monographs on the Evaluation
of the Carcinogenic Risk of Chemicals to Humans, Volume 20).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs, volumes 1 to 42. Lyon, International Agency for
Research on Cancer, pp 143-144 (IARC Monographs on the Evaluation of
Carcinogenic Risks to Humans, Supplement 7).
IARC (in press) Part one: Re-evaluation of some organic chemicals.
Lyon, International Agency for Research on Cancer (IARC Monographs on
the Evaluation of Carcinogenic Risks to Humans, Volume 71).
Ikatsu H & Nakajima T (1992) Hepatotoxic interaction between carbon
tetrachloride and chloroform in ethanol treated rats. Arch Toxicol,
66: 580-586.
Ikatsu H, Okino T, & Nakajima T (1991) Ethanol and food deprivation
induced enhancement of hepatotoxicity in rats given carbon
tetrachloride at low concentration. Br J Ind Med, 48: 636-642.
Inoko M, Tsuchiya M, & Matsuno T (1984) Determination of lower
halogenated hydrocarbons by the water-xylene extraction method.
Environ Pollut, B7: 129-140.
IPCC (WMO/UNEP Intergovernmental Panel on Climate Change) (1990) In:
Houghton JT, Jenkins GJ & Ephraums JJ ed. Climate change: The IPCC
scientific assessment. Cambridge, United Kingdom, Cambridge University
Press, 356 pp.
IPCC (WMO/UNEP Intergovernmental Panel on Climate Change) (1995) In:
Houghton JT, Meira Filho LG, & Bruce J ed. Climate change 1994:
Radiative forcing of climate change and an evaluation of the IPCC IS92
emission scenarios. Cambridge, United Kingdom, Cambridge University
Press, 337 pp.
IPCS (1994) Environmental health criteria 170: Assessing human health
risks of chemicals: Derivation of guidance values for health-based
exposure limits. Geneva, World Health Organization
Isaksen ISA & Stordal F (1981) The influence of man on the ozone
layer, readjusting the estimates. Ambio, 10: 9-17.
Isidorov VA, Zenkerich IG, & Ioffe BV (1990) Volatile organic
compounds in solfataric gases. J Atmos Chem, 10: 329-340.
Jaeger RJ, Conolly RB, & Murphy SD (1975) Short term inhalation
toxicity of halogenated hydrocarbons. Arch Environ Health, 30: 26-31.
Jakobson I, Wahlberg JE, Holmberg B, & Johansson G (1982) Uptake via
the blood and elimination of 10 organic solvents following
epicutaneous exposure of anesthetized guinea pigs. Toxicol Appl
Pharmacol, 63: 181-187.
Japan Bioassay Research Centre (1998)Thirteen-week and two-year
inhalation studies on F-344 rats and BDF1 mice (Studies Nos 0020,
0021, 0043 and 0044). Kanagawa, Japan Industrial Safety and Health
Association, Japan Bioassay Research Centre (Unpublished report to the
Ministry of Labour).
Jeffers PM, Brenner C, & Wolfe NL (1996) Hydrolysis of carbon
tetrachloride. Environ Toxicol Chem, 15: 1064-1065.
Jin G & Englarde AJ (1996) Redox potential as a controlling factor in
an enhancing carbon tetrachloride biodegradation. Water Sci Technol,
34: 59-66.
Jirova D, Sperlingova I, Halaskova M, Bendova H, & Dabrowska L (1996)
Immunotoxic effects of carbon tetrachloride -- the effect on
morphology and function of the immune system in mice. Centr Eur J
Public Health, 4: 16-20.
Johansson I & Ingelman-Sundberg M (1985) Carbon tetrachloride-induced
lipid peroxidation dependent on an ethanol-inducible form of rabbit
liver microsomal cytochrome P450. FEBS Lett, 183: 265-269.
Juhnke I & Lüdemann D (1978) [Results from a study of 200 chemical
compounds on the acute fish toxicity test with Golden Orfe.] Z
Wasser-Abwasser Forsch, 11: 161-164 (in German).
Jury WA, Spencer WF, & Farmer WJ (1984) Behavior assessment model for
trace organics in soil: III. Application of screening model. J Environ
Qual, 13: S73-S79.
Kaiser KLE, Comba ME, & Hunealt H (1983) Volatile halogen contaminants
in the Niagara River and Lake Ontario. J Great Lakes Res, 9: 212-223.
Kalla NR & Bansal MP (1975) Effect of carbon tetrachloride on gonadal
physiology in male rats. Acta Anat, 91: 380-385.
Kaminski NE, Jordan SD, & Holsapple MP (1989) Suppression of humoral
and cell-mediated immune responses by carbon tetrachloride. Fundam
Appl Toxicol, 12: 117-128.
Kaminski NE, Barnes DW, Jordan SD, & Holsapple MP (1990) The role of
metabolism in carbon tetrachloride-mediated immunosuppression:
in vivo studies. Toxicol Appl Pharmacol, 102: 9-20.
Kanaghinis T, Avgerinos A, Scliros P, Kalantzis N, Hatzioannou J,
Nikolopoulou P, Anagnostou D, Katsas A, Demopoulos J, Rekoumis G, &
Stathakos D (1982) Plasma lipoprotein pattern in relation to liver
histology after toxic hepatitis and experimental biliary obstruction
in rabbits. Am J Gastroenterol, 77: 512-522.
Kanics L & Rubinstein D (1968) The effect of chronic carbon
tetrachloride inhalation on the secretion of lipid by rat liver.
Biochem Pharmacol, 17: 1959-1967.
Kazantzis G & Bomford RR (1960) Dyspepsia due to inhalation of carbon
tetrachloride vapour. Lancet, 1: 360-362.
Kenaga EE (1980) Predicted bioconcentration factors and soil
adsorption coefficients of pesticides and other chemicals. Ecotoxicol
Environ Saf, 4: 26-38.
Kennedy GL, Ferenz RL, & Burgess BA (1986) Estimation of acute oral
toxicity in rats by determination of the approximate lethal dose
rather than the LD50. J Appl Toxicol, 6: 145-148.
Kieczka H & Kappus H (1980) Oxygen dependence of CCl4-induced lipid
peroxidation in vitro and in vivo. Toxicol Lett, 5: 191-196.
Kim YC (1997) Dichloromethane potentiation of carbon tetrachloride
hepatotoxicity in rats. Fundam Appl Toxicol, 35: 138-141.
Kim HJ, Bruckner JV, Dallas CE, & Gallo JM (1990a) Effect of dosing
vehicles on the pharmacokinetics of orally administered carbon
tetrachloride in rats. Toxicol Appl Pharmacol, 102: 50-60.
Kim HJ, Odend'hal S, & Bruckner JV (1990b) Effect of oral dosing
vehicles on the acute hepatotoxicity of carbon tetrachloride in rats.
Toxicol Appl Pharmacol, 102: 34-49.
Klaassen CD & Plaa GL (1967a) Susceptibility of male and female mice
to the nephrotoxic and hepatotoxic properties of chlorinated
hydrocarbons. Proc Soc Exp Biol Med, 124: 1163-1166.
Klaassen CD & Plaa GL (1967b) Relative effects of various chlorinated
hydrocarbons on liver and kidney function in dogs. Toxicol Appl
Pharmacol, 10: 119-131.
Klaassen CD & Plaa GL (1969) Comparison of the biochemical alterations
elicited in livers from rats treated with carbon tetrachloride,
chloroform, 1,1,2-trichloroethane and 1,1,1-trichloroethane. Biochem
Pharmacol, 18: 2019-2027.
Klecka GM & Gonsior SJ (1984) Reductive dichlorination of chlorinated
methanes and ethanes by reduced iron (II) porphyrins. Chemosphere, 13:
391-402.
Klingensmith JS, Lockard V, & Mehendale HM (1983) Acute hepatotoxicity
and lethality of CCl4 in chlordecone-pretreated rats. Exp Mol Pathol,
39: 1-10.
Kluwe WM (1981) Renal function tests as indicators of kidney injury in
subacute toxicity studies. Toxicol Appl Pharmacol, 57: 414-424.
Kluwe WM, Hook JB, & Bernstein J (1982) Synergistic toxicity of carbon
tetrachloride and several aromatic organohalide compounds. Toxicology,
23: 321-336.
Knie J, Hälke A, Juhnke I, & Schiller W (1983) [Results of studies on
chemical substances with four biotests.] Dtsch Gewässerkd Mitt, 27:
77-79 (in German).
Kniepert E, Siegemund A, & Görisch V (1990) Influence of ethanol
pretreatment of differing duration on toxic effects of carbon
tetrachloride in rats. Biomed Biochem Acta, 49: 1097-1102.
Könemann H (1981) Quantitative structure-activity relationships in
fish toxicity studies. Part 1: Relationship for 50 industrial
pollutants. Toxicology, 19: 209-221.
Koporec KP, Kim HJ, MacKenzie WF, & Bruckner JV (1995) Effect of oral
dosing vehicles on the subchronic hepatotoxicity of carbon
tetrachloride in the rat. J Toxicol Environ Health, 44: 13-27.
Kornbrust DJ & Mavis RD (1980) Microsomal lipid peroxidation: II.
Stimulation by carbon tetrachloride. Mol Pharmacol, 17: 408-414.
Korsrud GO, Grice HC, & McLaughlan JM (1972) Sensitivity of several
serum enzymes in detecting carbon tetrachloride-induced liver damage
in rats. Toxicol Appl Pharmacol, 22: 474-483.
Kraemer M, Bimboes D, & Greim H (1974) Use of S. typhimurium and
E. coli to detect chemical mutagens. Naunyn-Schmiedeberg's Arch
Pharmacol, 284: 46R.
Kroneld R (1985) Recovery and reproducibility in determination of
volatile halocarbons in water and blood. Bull Environ Contam Toxicol,
34: 486-496.
Kroneld RC (1989) Volatile pollutants in the environment and human
tissues. Bull Environ Contam Toxicol, 42: 873-877.
Kronevi T, Wahlberg J, & Holmberg B (1979) Histopathology of skin,
liver and kidney after epicutaneous administration of five industrial
solvents to guinea pigs. Environ Res, 19: 56-69.
Krost KJ, Pellizzari ED, Walburn SG, & Hubbard SA (1982) Collection
and analysis of hazardous organic emissions. Anal Chem, 54: 810-817.
Kubic VL & Anders MW (1980) Metabolism of carbon tetrachloride to
phosgene. Life Sci, 26: 2151-2155.
Kutob SD & Plaa GL (1962) A procedure for estimating the hepatotoxic
potential of certain industrial solvents. Toxicol Appl Pharmacol, 4:
354-361.
Lahl U (1983) [Analysis and evaluation of potential adverse effects of
selected volatile halogenated hydrocarbons in the human environment.]
Bremen, Germany, University of Bremen, pp 52-91 (Dissertation) (in
German).
Lahl U, Cetinkaya M, Düszeln J V, Stachel B, Thiemann W, Gabel B,
Kozicki R, & Podbielski A (1981) Health risks from volatile
halogenated hydrocarbons ? Sci Total Environ, 20: 171-189.
Lai EK, McCay PB, Noguchi T, & Fong K-L (1979) In vivo spin-trapping
of trichloromethyl radicals from CCl4. Biochem Pharmacol, 28:
2231-2235.
Larson RE & Plaa GL (1965) A correlation of the effects of cervical
cordotomy, hypothermia and catecholamines on carbon
tetrachloride-induced hepatic necrosis. J Pharmacol Exp Ther, 147:
103-111.
Lasa J, Rosiek J, & Wcislak L (1979) [Detection of halogen compounds
in air with coulometric ECD.] Chem Anal, 24: 819-826 (in Polish).
LeBlanc GA (1980) Acute toxicity of priority pollutants to water flea
(Daphnia magna). Bull Environ Contam Toxicol, 24: 684-691.
Lee PY, McCay PB, & Hornbrook KR (1982) Evidence for carbon
tetrachloride-induced lipid peroxidation in mouse liver. Biochem
Pharmacol, 31: 405-409.
Lehmann KB & Schmidt-Kehl L (1936) [The thirteen most important
chlorinated aliphatic hydrocarbons from the standpoint of industrial
hygiene.] Arch Hyg, 116: 131-268 (in German).
Lillian D & Singh HB (1974) Absolute determination of atmospheric
halocarbons by gas phase coulometry. Anal Chem, 46: 1060-1063.
Lillian D, Singh HB, Appleby A, Lobban L, Arnts R, Gumpert R, Hague R,
Toomey J, Kazazis J, Antell M, Hansen D, & Scott B (1975) Atmospheric
fates of halogenated compounds. Environ Sci Technol, 9: 1042-1048.
Lil'p IG (1983) Instability of the chromosomes in 101/H and C57BI/6
mice during aging. Soviet Genet, 18: 1467-1472.
Linet MS, Stuart WF, Van Natta ML, McCaffrey LD, & Szklo M (1987)
Comparison of methods for determining occupational exposure in a
case-control interview study of chronic lymphocytic leukemia. J Occup
Med, 29: 136-141.
Link B, Durk H, Thiel D, & Frank H (1984) Binding of trichloromethyl
radicals to lipids of the hepatic endoplasmic reticulum during
tetrachloromethane metabolism. Biochem J, 233(3): 577-586.
Lioy PJ & Lioy MJ ed. (1983) Air sampling instruments for evaluation
of atmospheric contaminants, 6th ed. Cincinnati, Ohio, American
Conference of Governmental Industrial Hygienist, pp V/85-V/86.
Litchfield MH & Gartland CJ (1974) Plasma enzyme activity and
hepatocellular changes in the beagle dog after single or repeated
administration of carbon tetrachloride. Toxicol Appl Pharmacol, 30:
117-128.
Liu SL, Esposti SD, Yao T, Diehl AM, & Zern MA (1995) Vitamin E
therapy of acute CCl4-induced hepatic injury in mice is associated
with inhibition of nuclear factor kappa B binding. Hepatology, 22:
1474-1481.
Long RM & Moore L (1986) Elevated cytosolic calcium in rat hepatocytes
exposed to carbon tetrachloride. J Pharmacol Exp Ther, 238: 186-191.
Loveday KS, Anderson BE, Resnick MA, & Zeiger E (1990) Chromosome
aberration and sister chromatid exchange tests in Chinese hamster
ovary cells in vitro: V. Results with 46 chemicals. Environ Mol
Mutagen, 16: 272-303.
Lowrey K, Glende EA, & Recknagel RO (1981) Rapid depression of rat
liver microsomal calcium pump activity after administration of carbon
tetrachloride or bromotrichloromethane and lack of effect after
ethanol. Toxicol Appl Pharmacol, 59: 389-394.
Lundberg I, Ekdahl M, Kronevi T, Lidums V, & Lundberg S (1986)
Relative hepatotoxicity of some industrial solvents after
intraperitoneal injection or inhalation exposure in rats. Environ Res,
40: 411-420.
Lurker PA, Clark CS, Elia VJ, Gartside PS, & Kinman RN (1983) Worker
exposure to chlorinated organic compounds from the activated-sludge
wastewater treatment process. Am Ind Hyg Assoc J, 44: 109-122.
Lyman WJ, Reehl WF, & Rosenblatt DH (1982) Handbook of chemical
property estimation methods. New York, McGraw-Hill Book Company.
Mabey W & Mill T (1978) Critical review of hydrolysis of organic
compounds in water under environmental conditions. J Phys Chem Ref
Data, 7(2): 383-415.
McCann J, Choi E, Yamasaki E, & Ames BN (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of
300 chemicals. Proc Natl Acad Sci (USA), 72: 5135-5139.
McCay PB, Lai EK, Poyer JL, DuBose CM, & Janzen EG (1984) Oxygen- and
carbon-centered free radical formation during carbon tetrachloride
metabolism. Observations of lipid radicals in vivo and in vitro. J
Biol Chem, 259: 2135-2143.
McCollister DD, Beamer WH, Atchison GJ, & Spencer HC (1951) The
absorption, distribution and elimination of radioactive carbon
tetrachloride by monkeys upon exposure to low vapor concentrations. J
Pharmacol Exp Ther, 102: 112-124.
McConnell G, Ferguson DM, & Pearson CR (1975) Chlorinated hydrocarbons
in the environment. Endeavour, 34: 13-18.
McDermott WV & Hardy HL (1963) Cirrhosis of the liver following
chronic exposure to carbon tetrachloride. J Occup Med, 5: 249-251.
McGregor D & Lang M (1996) Carbon tetrachloride: genetic effects and
other modes of action. Mutat Res, 366: 181-195.
McKone TE (1987) Human exposure to volatile organic compounds in
household tap water: the indoor inhalation pathway. Environ Sci
Technol, 21: 1194-1201.
Magos L, Snowden R, White INH, Butler WH, & Tuffery AA (1982) Isotoxic
oral and inhalation exposure of carbon tetrachloride in Porton-Wistar
and Fischer rats. J Appl Toxicol, 2: 238-240.
Makide Y & Yokohata A (1983) Ultratrace analysis of CCl2F2, CCl3F,
CCl2FCClF2, CH3CCl3, CCl4, CHCl=CCl2 and CCl2=CCl2 in the
atmosphere. J Trace Microprobe Tech, 1: 265-292.
Makide Y, Tominaga T, & Rowland S (1979) Gas chromatographic analysis
of halogenated hydrocarbons in air over Japan. Chem Lett, 1979:
355-358.
Makide Y, Kanai Y, & Tominaga T (1980) Background atmospheric
concentrations of halogenated hydrocarbons in Japan. Bull Chem Soc
Jpn, 53: 2681-2682.
Mandal PK, Bishayee A, & Chatterjee M (1993) Short communication:
Stimulation of tissue repair by Mikania cordata root extract in
carbon tetrachloride-induced liver injury in mice. Phytother Res, 7:
103-105.
Mann H (1975) [The Golden Orfe test: A German proposal to test the
effects of chemical compounds in fish.] Vom Wasser, 44: 1-13 (in
German).
Manno M, Rezzadore M, Grossi M, & Sbrana C (1996) Potentiation of
occupational carbon tetrachloride toxicity by ethanol abuse. Hum Exp
Toxicol, 15: 294-300.
Marchand C, McLean S, & Plaa GL (1970) The effect of SKF 525A on the
distribution of carbon tetrachloride in rats. J Pharmacol Exp Ther,
174: 232-238.
Martínez-Calva I, Campos-Apáez A, Rosales-Vega E, & Mourelle M (1984)
Vitamin E improves membrane lipid alterations induced by CCl4
intoxication. J Appl Toxicol, 4: 270-272.
Masuda Y & Nakayama N (1982) Protective effect of
diethyldithiocarbamate and carbon disulfide against liver injury
induced by various hepatotoxic agents. Biochem Pharmacol, 31:
2713-2725.
Mehendale HM (1984) Potentiation of halomethane hepatotoxicity:
Chlordecone and carbon tetrachloride. Fundam Appl Toxicol, 4: 295-308.
Mehendale HM (1989) Mechanism of lethal interaction of chlordecone and
CCl4 at non-toxic doses. Toxicol Lett, 49: 215-241.
Mehendale HM (1990) Potentiation of halomethane hepatotoxicity by
chlordecone: a hypothesis for the mechanism. Med Hypotheses, 33:
289-299.
Mehendale HM (1991) Commentary: Role of hepatocellular regeneration
and hepatolobular healing in the final outcome of liver injury. A
two-stage model of toxicity. Biochem Pharmacol, 42: 1155-1162.
Mico BA & Pohl LR (1983) Reductive oxygenation of carbon
tetrachloride. Trichloromethylperoxyl radical as a possible
intermediate in the conversion of carbon tetrachloride to
electrophilic chlorine. Arch Biochem Biophys, 225: 596-609.
Mirsalis JC & Butterworth BE (1980) Detection of unscheduled DNA
synthesis in hepatocytes isolated from rats treated with genotoxic
agents: An in vivo-in vitro assay for potential carcinogens and
mutagens. Carcinogenesis, 1: 621-625.
Mirsalis JC, Tyson CK, & Butterworth BE (1982) Detection of genotoxic
carcinogens in the in vivo- in vitro hepatocyte DNA repair assay.
Environ Mutagen, 4: 553-562.
Mirsalis JC, Tyson CK, Loh EN, Steinmetz KL, Bakke JP, Hamilton CM,
Spak DK, & Spalding JW (1985) Induction of hepatic cell proliferation
and unscheduled DNA synthesis in mouse hepatocytes following
in vivo treatment. Carcinogenesis, 6: 1521-1524.
Miyazawa T, Tsuchiya K, & Kaneda T (1984) Riboflavin tetrabutyrate: an
antioxidative synergist of alpha-tocopherol as estimated by hepatic
chemiluminescence. Nutr Rep Int, 29: 157-165.
Molina MJ & Rowland FS (1974) Predicted present stratospheric
abundances of chlorine species for photodissociation of carbon
tetrachloride. Geophys Res Lett, 1: 309-312.
Monster AC & Zielhuis RL (1983) Chlorinated hydrocarbon solvents. In:
Human biological monitoring of industrial chemical series. Ispra,
Italy, European Commission, Joint Research Centre, pp 63-65 (EUR 8476
EN).
Moore L, Davenport GR, & Landon EJ (1976) Calcium uptake of a rat
liver microsomal subcellular fraction in response to
in vivo administration of carbon tetrachloride. J Biol Chem, 25:
1197-1201.
Morele Y, Lefevre C, Ferrari P, Guenier JP, & Muller J (1989) Sampling
and analysis of airborne chlorinated methanes -- comparison of FID and
ECD. Chromatographia, 28: 617-619.
Morgan A, Black A, & Belcher DR (1970) The excretion in breath of some
aliphatic halogenated hydrocarbons following administration by
inhalation. Ann Occup Hyg, 13: 219-233.
Muños Torres E, Paz Bouza JI, Abad Hernandez MM, Alonso Martin MJ, &
Lopez Bravo A (1988) Experimental carbon tetrachloride-induced
cirrhosis of the liver. Int J Tissue React, X: 245-251.
Murphy SD & Malley S (1969) Effect of carbon tetrachloride on
induction of liver enzymes by acute stress or corticosterone. Toxicol
Appl Pharmacol, 15: 117-130.
Nagano K, Nishizava T, Yamamoto S, & Matsushima T (1998) Inhalation
carcinogenesis studies of six halogenated hydrocarbons in rats and
mice. In: Chiyotani K, Hosoda Y, & Aizawa Y ed. Advances in the
prevention of occupational respiratory diseases. Amsterdam, Oxford,
New York, Elsevier Science Publishers, pp 741-746.
Nakajima T, Koyama Y, & Sato A (1982) Dietary modification of
metabolism and toxicity of chemical substances with special reference
to carbohydrate. Biochem Pharmacol, 31: 1005-1011.
Nakamura SI, Oda Y, Shimada T, Oki I, & Sugimoto K (1987) SOS-inducing
activity of chemical carcinogens and mutagens in
Salmonella typhimurium TA1535/pSK1002: examination with 151
chemicals. Mutat Res, 192: 239-246.
Nakata R, Tsukamoto I, Miyoshi M, & Kojo S (1985) Liver regeneration
after carbon tetrachloride intoxication in the rat. Biochem Pharmacol,
34: 586-588.
Nakayama T, Kaneko M, & Kodama M (1986) Detection of DNA damage in
cultured human fibroblasts induced by methyl linoleate hydroperoxide.
Agric Biol Chem, 50: 261-262.
Narotsky MG, Pegram RA, & Kavlock RJ (1997a) Effect of dosing vehicle
on the developmental toxicity of bromodichloromethane and carbon
tetrachloride in rats. Fundam Appl Toxicol, 40: 30-36.
Narotsky MG, Brownie CF, & Kavlock RJ (1997b) Critical period of
carbon tetrachloride-induced pregnancy loss in Fischer-344 rats, with
insights into the detection of resorption sites by ammonium sulfide
staining. Teratology, 56: 252-261.
Nath RG, Li DH, & Randerath K (1990) Acute and long-term effects of
carbon tetrachloride on DNA modifications (I-compounds) in male mouse
liver. Chem-Biol Interact, 76: 343-357.
National Library of Medicine (1992) Hazardous substances data bank.
Bethesda, Maryland, National Library of Medicine, National Toxicology
Information Programme.
National Library of Medicine (1997) Hazardous substances data bank.
Bethesda, Maryland, National Library of Medicine, National Toxicology
Information Programme.
Neely WB, Branson DR, & Blau GE (1974) Partition coefficient to
measure bioconcentration potential of organic chemicals in fish.
Environ Sci Technol, 8: 1113-1115.
Neuhauser EF, Loehr RC, Malecki MR, Milligan DL, & Durkin PR (1985)
The toxicity of selected organic chemicals to the earthworm
Eisenia foetida. J Environ Qual, 14: 383-388.
NIOSH (1977) Manual of analytical methods -- Part 2: Standards
completion program validated methods 3:S314. Cincinnati, Ohio,
National Institute of Occupational Safety and Health, pp 1-9.
NIOSH (1984) Manual of analytical methods: Method 1003. Cincinnati,
Ohio, National Institute of Occupational Safety and Health, pp 1-9.
Noguchi T, Fong K-L, Lai EK, Olson L, & McCay PB (1982a) Selective
early loss of polypeptides in liver microsomes of CCl4-treated rats.
Relationship to cytochrome P-450 content. Biochem Pharmacol, 31:
609-614.
Noguchi T, Fong K-L, Lai EK, Alexander SS, King MM, Olson L, Poyer JL,
& McCay PB (1982b) Specificity of a phenobarbital-induced cytochrome
P-450 for metabolism of carbon tetrachloride to the trichloromethyl
radical. Biochem Pharmacol, 31: 615-624.
Noll T & De Groot H (1984) The critical steady-state hypoxic
conditions in carbon tetrachloride-induced lipid peroxidation in rat
liver microsomes. Biochim Biophys Acta, 795: 356-362.
Norpoth KH, Reisch KA, & Heinecke A (1980) Biostatistics of Ames-test
data. In: Norpoth KH & Garmer RC ed. Short-term systems for detecting
carcinogens. Berlin, Springer-Verlag, pp 312-323.
Norwood WD, Fuqua PA, & Scudder BC (1950) Carbon tetrachloride
poisoning. Arch Ind Hyg Occup Med, 1: 90-100.
Öenfelt A (1987) Spindle disturbances in mammalian cells - III.
Toxicity, c-mitosis and aneuploidy with 22 different compounds:
Specific and unspecific mechanisms. Mutat Res, 182: 135-154.
Oldenhuis R, Vink RJLM, Janssen DB, & Witholt B (1989) Degradation of
chlorinated aliphatic hydrocarbons by
Methylosinus trichosporium OB3b expressing soluble methane
monooxygenase. Appl Environ Microbiol, 55: 2819-2826.
Oraumbo IF & Van Duuren BL (1989) Evidence for the covalent
interaction of carbon tetrachloride with mouse liver chromatin DNA
in vivo. J Environ Pathol Toxicol Oncol, 9: 13-18.
Ott MG, Carlo GL, Steinberg S, & Bond GG (1985) Mortality among
employees engaged in chemical manufacturing and related activities. Am
J Epidemiol, 122: 311-322.
Ozeki T, Iwaki K, Taoka Y, Yamashina A, Ouchi K, & Kan M (1982) The
effects of vitamin E on the indicator enzymes of organelle membranes
in the injured liver. Gastroenterol Jpn, 17: 441-446.
Pääkkö P, Anttila S, Sormunen R, Ala-Kokko L, Peura R, Ferrans VJ, &
Ryhanen L (1996) Biochemical and morphological characterization of
carbon tetrachloride-induced lung fibrosis in rats. Arch Toxicol, 70:
540-552.
Packer JE, Slater TF, & Willson RL (1978) Reactions of the carbon
tetrachloride-related peroxy free radical (CCl3O2.) with amino
acids: Pulse radiolysis evidence. Life Sci, 23: 2617-2620.
Page DA & Carlson GP (1994) The role of the intestinal tract in the
elimination of carbon tetrachloride. Toxicol Appl Pharmacol, 124:
268-274.
Parola M, Leonarduzzi G, Biasi F, Albano E, Biocca ME, Poli G, &
Dianzani M (1992) Vitamin E dietary supplementation protects against
carbon tetrachloride-induced chronic liver damage and cirrhosis.
Hepatology, 16: 1014-1021.
Parsons F, Barrio Lage G, & Rice R (1985) Biotransformation of
chlorinated organic solvents in static microcosms. Environ Toxicol
Chem, 4: 739-742.
Pasquali-Ronchetti I, Bini A, Botti B, De Alojsio G, Fornieri C, &
Vannini V (1980) Ultrastructural and biochemical changes by
progressive lipid peroxidation on isolated microsomes and rat liver
endoplasmic reticulum. Lab Invest, 42: 457-468.
Paul BB & Rubinstein D (1963) Metabolism of carbon tetrachloride and
chloroform by the rat. J Pharmacol Exp Ther, 141: 141-148.
Paustenbach DJ, Carlson GP, Christian JE, & Born GS (1986a) A
comparative study of the pharmacokinetics of carbon tetrachloride in
the rat following repeated inhalation exposures of 8 and 11.5-hr/day.
Fundam Appl Toxicol, 6: 484-497.
Paustenbach DJ, Christian JE, Carlson GP, & Born GS (1986b) The effect
of an 11.5-hr/day exposure schedule on the distribution and toxicity
of inhaled carbon tetrachloride in the rat. Fundam Appl Toxicol, 6:
472-483.
Paustenbach DJ, Clewell HJ, Gargas ML, & Andersen ME (1988) A
physiologically based pharmacokinetic model for inhaled carbon
tetrachloride. Toxicol Appl Pharmacol, 96: 191-211.
Pearson CR & McConnell G (1975) Chlorinated C1 and C2 hydrocarbons
in the marine environment. Proc R Soc Lond B, 189: 305-332.
Pellizzari ED, Sheldon LS, & Bursey JT (1985) Method 25: GC/MS
determination of volatile halocarbons in blood and tissue. Environ
Carcinog Sel Methods Anal, 7: 435-444.
Pentz R & Strubelt O (1983) Fasting increases the concentration of
carbon tetrachloride and of its metabolite chloroform in the liver of
mice. Toxicol Lett, 16: 231-234.
Peoples AJ, Pfaffenberger CD, Shafik TM, & Enos HF (1979)
Determination of volatile purgeable halogenated hydrocarbons in human
adipose tissue and blood serum. Bull Environ Contam Toxicol, 23:
244-249.
Perocco P & Prodi G (1981) DNA Damage by haloalkanes in human
lymphocytes cultured in vitro. Cancer Lett, 13: 213-218.
Pierotti D, Rasmussen RA, & Dalluge R (1980) Measurements of N2O,
CF2Cl2, CFCl3, CH3CCl3, CCl4, CH3Cl in the troposphere and
lower stratosphere over North America. J Geomagn Geoelectr, 32:
181-205.
Plummer JL, Hall PD, Ilsley AH, Jenner MA, & Cousins MJ (1990)
Influence of enzyme induction and exposure profile on liver injury due
to chlorinated hydrocarbon inhalation. Pharmacol Toxicol, 67: 329-335.
Plummer JL, Hall PD, Ilsley AH, Cmielewski PL, Ahern MJ, & Williams RA
(1994) Dose-response relationships in hepatic injury produced by
alcohol and carbon tetrachloride. Alcohol Clin Exp Res, 18(6):
1523-1526.
Pohl LR, Branchflower RV, Highet RJ, Martin JL, Nunn DS, Monks TJ,
George JW, & Hinson JA (1981) The formation of diglutathionyl
dithiocarbonate as a metabolite of chloroform, bromotrichloromethane,
and carbon tetrachloride. Drug Metab Dispos, 9: 334-339.
Pohl LR, Schulick RD, Highet RJ, & George JW (1984)
Reductive-oxygenation mechanism of metabolism of carbon tetrachloride
to phosgene by cytochrome P-450. Mol Pharmacol, 25: 318-321.
Poyer JL, Floyd RA, McCay PB, Janzen EG, & Davis ER (1978)
Spin-trapping of the trichloromethyl radical produced during enzymic
NADPH oxidation in the presence of carbon tetrachloride or
bromotrichloromethane. Biochem Biophys Acta, 539: 402-409.
Poyer JL, McCay PB, Lai EK, Janzen EG, & Davis ER (1980) Confirmation
of assignment of the trichloromethyl radical spin adduct detected by
spin trapping during 13C-carbon tetrachloride metabolism
in vitro and in vivo. Biochem Biophys Res Commun, 94: 1154-1160.
Prendergast JA, Jones RA, Jenkins LJ, & Siegel J (1967) Effects on
experimental animals of long-term inhalation of trichloroethylene,
carbon tetrachloride, 1,1,1-trichloroethane, dichlorodifluoromethane,
and 1,1-dichloroethylene. Toxicol Appl Pharmacol, 10: 270-289.
Prinn RG, Simmonds PG, Rasmussen RA, Rosen RD, Alyea FN, Cardelino CA,
Crawford AJ, Cunnold DM, Fraser PJ, & Lovelock JE (1983) The
atmospheric lifetime experiment. I. Introduction, instrumentation, and
overview. J Geophys Res, 88: 8353-8367.
Pritchard DJ, Wright MG, Sulsh S, & Butler WH (1987) The assessment of
chemically induced liver injury in rats. J Appl Toxicol, 7: 229-236.
Puri EC & Müller D (1989) Testing of hydralizine in
in vivo-in vitro hepatocyte assays for UDS and stimulation of
replicative DNA synthesis. Mutat Res, 218: 13-19.
Qazi FM & Alam SM (1988) Potentiating effects of phenobarbitone on the
induction of cirrhosis in rats by carbon tetrachloride. J Pak Med
Assoc, 38(11): 296-299.
Rao SB & Mehendale HM (1989) Protective role of fructose
1,6-biphosphate during CCl4 hepatotoxicity in rats. Biochem J, 262:
721-725.
Rasmussen RA & Khalil MAK (1983) Natural and anthropogenic trace gases
in the lower troposphere of the Arctic. Chemosphere, 12: 371-375.
Rasmussen RA & Khalil MAK (1986) Atmospheric trace gases: Trends and
distributions over the last decade. Science, 232: 1623-1624.
Rasmussen RA, Khalil MAK, & Dalluge RW (1981) Atmospheric trace gases
in Antarctica. Science, 211: 285-287.
Raucy JL, Kraner JC, & Lasker JM (1993) Bioactivation of halogenated
hydrocarbons by cytochrome P4502E1. Crit Rev Toxicol, 23: 1-20.
Ray SD & Mehendale HM (1990) Potentiation of CCl4 and CHCl3
hepatotoxicity and lethality by various alcohols. Fundam Appl
Toxicol, 15: 429-440.
Raymond P & Plaa GL (1995a) Ketone potentiation of haloalkane-induced
hepato- and nephrotoxicity: I. Dose-response relationships. J Toxicol
Environ Health, 45: 465-480.
Raymond P & Plaa GL (1995b) Ketone potentiation of haloalkene-induced
hepato- and nephrotoxicity: II. Implication of monooxygenases. J
Toxicol Environ Health, 46: 317-328.
Raymond P & Plaa GL (1996) Ketone potentiation of haloalkene-induced
hepatotoxicity: CCl4 and ketone treatment on hepatic membrane
integrity. J Toxicol Environ Health, 49: 285-300.
Raymond P & Plaa GL (1997) Effect of dosing vehicle on the
hepatotoxicity of CCl4 and nephrotoxicity of CHCl3 in rats. J
Toxicol Environ Health, 51: 463-476.
Recknagel RO (1983) A new direction in the study of carbon
tetrachloride hepatotoxicity. Life Sci, 33: 401-408.
Recknagel RO & Glende EA Jr (1973) Carbon tetrachloride
hepatotoxicity: An example of lethal cleavage. CRC Crit Rev Toxicol,
2: 263-297.
Reiner O, Athanassopoulos S, Hellmer KH, Murray RE, & Uehleke H (1972)
[Formation of chloroform from carbon tetrachloride in liver
microsomes, lipid peroxidation and destruction of cytochrome P-450.]
Arch Toxikol, 29(3): 219-233 (in German).
Reinke LA, Lai EK, & McCay PB (1988) Ethanol feeding stimulates
trichloromethyl radical formation from carbon tetrachloride in liver.
Xenobiotica, 18: 1311-1318.
Reuber MD & Glover EL (1970) Cirrhosis and carcinoma of the liver in
male rats given subcutaneous carbon tetrachloride. J Natl Cancer Inst,
44: 419-427.
Reynolds LF & Harrison DW (1982) Study of discharge of benzene,
chloroform and carbon tetrachloride into the aquatic environment and
the best technical means for the reduction of water pollution from
such discharges -- Final report. Brixham, United Kingdom, ICI
Laboratory (Report No. BL/A/2189).
Reynolds LF, Harrison DW, & Carter L (1982) Contract No. ENV/223/74-EN
-- Third revision. Luxembourg, Commission of the European Communities,
Environment and Consumer Protection Service, pp 103-107.
Reynolds ES, Treinen RJ, Farrish HH, & Treinen Moslen M (1984)
Metabolism of [14C]carbon tetrachloride to exhaled, excreted and
bound metabolites. Biochem Pharmacol, 33: 3363-3374.
Richmond RE, DeAngelo AB, & Daniel FB (1992) Immunohistochemical
detection of ras and myc oncogene expression in regenerating rat
liver. Toxicol Lett, 60: 119-129.
Rivett MO, Lerner DN, Lloyd JW, & Clark L (1990a) Organic
contamination of the Birmingham aquifer, UK. J Hydrol, 113: 307-323.
Rivett MO, Lerner DN, & Lloyd JW (1990b) Chlorinated solvents in the
UK aquifers. J Inst Water Environ Manage, 4: 242-250.
Rogers HR, Crathorne B, & Watts CD (1992) Sources and fate of organic
contaminants in the Mersey Estuary: volatile organohalogen compounds.
Mar Pollut Bull, 24: 82-91.
Roldan-Arjona T & Pueyo C (1993) Mutagenic and lethal effects of
halogenated methanes in the Ara test of Salmonella typhimurium:
quantitative relationship with chemical reactivity. Mutagenesis, 8:
127-131.
Roldan-Arjona T, Garcia-Pedrajas MD, Luque-Romero FL, Hera C, & Pueyo
C (1991) An association between mutagenicity of the Ara test of
Salmonella typhimurium and carcinogenicity in rodents for 16
halogenated aliphatic hydrocarbons. Mutagenesis, 6: 199-205.
Rosen ME, Pankow JF, Gibs J, & Imbrigiotta TE (1992) Comparison of
downhole and surface sampling for the determination of volatile
organic compounds (VOC) in ground water. Ground Water Monit Rev, 12:
126-133.
Rosengren RJ, Sauer JM, Hooser SB, & Sipes IG (1995) The interactions
between retinol and five different hepatotoxicants in the Swiss
Webster mouse. Fundam Appl Toxicol, 25: 281-292.
Roudabush RL, Terhaar CJ, Fassett DW, & Dziuba SP (1965) Comparative
acute effects of some chemicals on the skin of rabbits and guinea
pigs. Toxicol Appl Pharmacol, 7: 559-565.
Rowland FS (1985) Methane and chlorocarbons in the earth's atmosphere.
Orig Life, 15: 279-297.
Rubinstein D & Kanics L (1964) The conversion of carbon tetrachloride
and chloroform to carbon dioxide by rat liver homogenates. Can J
Biochem, 42: 1577-1585.
Rudolph J & Jebsen C (1983) The use of photoionization, flame
ionization and electron capture detectors in series for the
determination of low molecular weight trace components in the
non-urban atmosphere. Intern. J Environ Anal Chem, 13: 129-139.
Ruprah M, Mant TGK & Flanagan RJ (1985) Acute carbon tetrachloride
poisoning in 19 patients: implications for diagnosis and treatment.
Lancet, May 4: 1027-1029.
Russell JW & Shadoff LA (1977) The sampling and determination of
halocarbons in ambient air using concentration on porous polymer. J
Chromatogr, 134(2):375-384.
Sagai M & Tappel AL (1979) Lipid peroxidation induced by some
halomethanes as measured by in vivo pentane production in the rat.
Toxicol Appl Pharmacol, 49: 283-291.
Sanzgiri UY, Kim HJ, Muralidhara S, Edallas E & Bruckner JV (1995)
Effect of route and pattern of exposure on the pharmacokinetics and
acute hepatotoxicity of carbon tetrachloride. Tox Appl Pharmacol 134:
148-154.
Sanzgiri UY & Bruckner JV (1997) Effect of Emulphor, an emulsifier, on
the pharmacokinetics and hepatotoxicity of oral carbon tetrachloride
in the rat. Fundam Appl Toxicol, 36: 54-61.
Sato A & Nakajima T (1985) Enhanced metabolism of volatile
hydrocarbons in rat liver following food deprivation, restricted
carbohydrate intake, and administration of ethanol, phenobarbital,
polychlorinated biphenyl and 3-methylcholanthrene: a comparative
study. Xenobiotica, 15: 67-75.
Sato A, Nakajima T, & Koyama Y (1980) Effects of chronic ethanol
consumption on hepatic metabolism of aromatic and chlorinated
hydrocarbons in rats. Br J Ind Med, 37: 382-286.
Sauer JM & Sipes IG (1995) Modulation of chemical-induced lung and
liver toxicity by all-trans-retinol in the male Sprague-Dawley rat.
Toxicology, 105: 237-249.
Sawada S, Yamanaka T, Yamatsu K, Furihata C, & Matsushima T (1991)
Chromosome aberrations, micronuclei and sister-chromatid exchanges
(SCEs) in rat liver induced in vivo by hepatocarcinogens including
heterocyclic amines. Mutat Res, 251: 59-69.
Schiestl RH, Gietz RD, Mehta RD, & Hastings PJ (1989) Carcinogens
induce intrachromosomal recombination in yeast. Carcinogenesis, 10:
1445-1455.
Schwarz M, Hummel J, Appel KE, Rickert R, & Kunz W (1979) DNA damage
induced in vivo evaluated with a non-radioactive alkaline elution
technique. Cancer Lett, 6: 221-226.
Schwetz BA, Leong BKJ, & Gehring PJ (1974) Embryo- and fetotoxicity of
inhaled carbon tetrachloride, 1,1-dichloroethane and methyl ethyl
ketone in rats. Toxicol Appl Pharmacol, 28: 452-464.
Shah JJ & Heyerdahl EK (1988) National ambient volatile organic
compounds (VOCs) data base update. Research Triangle Park, North
Carolina, US Environmental Protection Agency (PB88-195631).
Shah H, Hartman SP, & Weinhouse S (1979) Formation of carbonyl
chloride in carbon tetrachloride metabolism by rat liver
in vitro. Cancer Res, 39: 3942-3947.
Shapiro AA & Fonshtein LM (1979) Study of the mutagenic action of
cyclophosphane on bacteria in host-mediated assay. Biol Bull Acad Sci
(USSR), 6: 310-315.
Shen ES, Garry VF, & Anders MW (1982) Effect of hypoxia and carbon
tetrachloride hepatotoxicity. Biochem Pharmacol, 31: 3783-3793.
Shertzer HG, Reitman FA, & Tabor MW (1988) Influence of diet on the
expression of hepatotoxicity from carbon tetrachloride in ICR mice.
Drug-Nutr Interact, 5: 275-282.
Shibayama Y (1988) Hepatotoxicity of carbon tetrachloride after
chronic ethanol consumption. Exp Mol Pathol, 49: 234-242.
Shimizu Y, Nagase C, & Kawai K (1973) Accumulation and toxicity of
carbon tetrachloride after repeated inhalation in rats. Ind Health,
11: 48-54.
Shinozuka H (1971) Unusual membrane alterations in the rough
endoplasmic reticulum of rat liver cells after carbon tetrachloride
injury. Exp Cell Res, 64: 380-386.
Siegers C-P, Horn W, & Younes M (1985) Effect of hypoxia on the
metabolism and hepatotoxicity of carbon tetrachloride and vinylidene
chloride in rats. Acta Pharmacol Toxicol, 56: 81-86.
Siemiatycki J ed. (1991) Risk factors for cancer in the work place.
Boca Raton, Florida, CRC Press, 310 pp.
Simko V, Michael S, Katz J, Oberstein E, & Popescu A (1992) Protective
effect of oral acetylcysteine against the hepatorenal toxicity of
carbon tetrachloride potentiated by ethyl alcohol. Alcohol Clin Exp
Res, 16: 759-799.
Simmon VF & Tardiff RG (1978) Mutagenic activity of halogenated
compounds found in chlorinated drinking waters. In: Proceedings of the
Conference on Water Chlorination Environmental Impact and Health
Effects, vol 2, pp 417-431.
Simmon VF, Kavhanen K, & Tardiff RG (1977) Mutagenic activity of
chemicals identified in drinking waters. In: Scott D, Bridges BA, &
Sobesl FH ed. Progress in genetic toxicology. Amsterdam, Oxford, New
York, Elsevier/North-Holland Biochemical Press, pp 249-258.
Simmonds PG & Kerns E (1979) Direct aqueous injection gas
chromatography for the analysis of trace organics in water. J
Chromatogr, 186: 785-794.
Simmonds PG, Alyea FN, Cardelino CA, Crawford AJ, Cunnold DM, Lane BC,
Lovelock JE, Prinn RG, & Rasmussen RA (1983) The atmospheric lifetime
experiment: 6. Results for carbon tetrachloride based on 3 years data.
J Geophys Res, 88: 8427-8441.
Simmonds PG, Cunnold DM, Alyea FN, Cardelino CA, Crauford AJ, Prinn
RG, Fraser PJ, Rasmussen RA, & Rosen RD (1988) Carbon tetrachloride
lifetimes and emissions determined from daily global measurements
during 1978-1985. J Atmos Chem, 7: 35-58.
Simmons JE, McDonald A, Seely JC, & Sey YM (1995) Potentiation of
carbon tetrachloride hepatotoxicity by inhaled methanol: Time course
of injury and recovery. J Toxicol Environ Health, 46: 203-216.
Sina JF, Bean CL, Dysart GR, Taylor VI, & Bradley MO (1983) Evaluation
of the alkaline elution/rat/hepatocyte assay as a predictor of
carcinogenic/mutagenic potential. Mutat Res, 113: 357-391.
Singh HB, Lillian D, Appleby A, & Lobban L (1975) Atmospheric
formation of carbon tetrachloride from tetrachloroethylene. Environ
Lett, 10: 253-256.
Singh HB, Fowler DP, & Peyton TO (1976) Atmospheric carbon
tetrachloride: another man-made pollutant. Science, 192: 1231-1234.
Singh HB, Salas L, Shigeishi H, & Crawford A (1977) Urban -- nonurban
relationships of halocarbons, SF6, N2O and other atmospheric trace
constituents. Atmos Environ, 11: 819-828.
Singh HB, Salas LJ, Shigeishi H, & Scribner E (1979) Atmospheric
halocarbons, hydrocarbons, and sulfur hexafluoride: global
distributions, sources and sinks. Science, 203: 899-903.
Singh HB, Salas LJ, Smith AJ, & Shigeishi H (1980) Measurements of
some potentially hazardous organic chemicals in urban environments.
Atmos Environ, 15: 601-612.
Singh HB, Salas LJ, & Stiles RE (1982) Distribution of selected
gaseous organic mutagens and suspect carcinogens in ambient air.
Environ Sci Technol, 16: 872-880.
Singh HB, Salas LJ, & Stiles RE (1983) Selected man-made halogenated
chemicals in the air and oceanic environment. J Geophys Res, 88:
3675-3683.
Sipes IG, Krishna G, & Gillette JR (1977) Bioactivation of carbon
tetrachloride, chloroform and bromotrichloromethane: role of
cytochrome P-450. Life Sci, 20: 1541-1548.
Slater TF (1982) Activation of carbon tetrachloride: chemical
principles and biological significance. In: McBrien DCH & Slater TF
ed. Free radicals, lipid peroxidation and cancer. New York, London,
San Francisco, Academic Press.
Smejkalová J, Simek J, Rouchal J, & Dvorácková I (1985) The time
course of biochemical and histological changes following carbon
tetrachloride-induced liver damage in rats of both sexes. Physiol
Bohemoslov, 34: 494-501.
Smetana H (1939) Nephrosis due to carbon tetrachloride. Arch Intern
Med, 63: 760-777.
Smialowicz RJ, Simmons JE, Luebke RW, & Allis JW (1991)
Immunotoxicologic assessment of subacute exposure of rats to carbon
tetrachloride with comparison to hepatotoxicity and nephrotoxicity.
Fundam Appl Toxicol, 17: 186-196.
Smyth HF, Weil CS, West JS, & Carpenter CP (1970) An exploration of
joint toxic action: II. Equitoxic versus equivolume mixtures. Toxicol
Appl Pharmacol, 17: 498-503.
Srivastava SP, Chen NQ, & Holtzman JL (1990) The
in vitro NADPH-dependent inhibition by CCl4 of the ATP-dependent
calcium uptake of hepatic microsomes from male rats. J Biol Chem, 265:
8392-8399.
Staples CA, Werner AF, & Hoogheem TJ (1985) Assessment of priority
pollutant concentrations in the United States using STORET database.
Environ Toxicol Chem, 4: 131-142.
Statham CN, Croft WA, & Lech JJ (1978) Uptake, distribution and
effects of carbon tetrachloride in rainbow trout
(Salmo gairdneri). Toxicol Appl Pharmacol, 45: 131-140.
Stewart BW (1981) Generation and persistence of carcinogens-induced
repair intermediates in rat liver DNA in vivo. Cancer Res, 41:
3238-3243.
Stewart RD & Dodd HC (1964) Absorption of carbon tetrachloride,
trichloroethylene, tetrachloroethylene, methylene chloride, and
1,1,1,-trichloroethane through the human skin. Am Ind Hyg Assoc J, 25:
439-446.
Stewart RD, Arbor A, Gay HH, Erley DS, Hake CL, & Peterson JE (1961)
Human exposure to carbon tetrachloride vapor. J Occup Med, 3: 586-590.
Stewart PA, Lee JS, Marano DE, Spirtas R, Forbes CD, & Blair A (1991)
Retrospective cohort mortality study of workers at an aircraft
maintenance facility: II. Exposures and their assessment. Br J Ind
Med, 48: 531-537.
Stoyanovsky DA & Cederbaum AI (1996) Thiol oxidation and cytochrome
P450-dependent metabolism of CCl4 triggers Ca2+ release from liver
microsomes. Biochemistry, 35: 15839-15845.
Striker GE, Smuckler EA, Kohnen PW, & Nagle RB (1968) Structural and
functional changes in rat kidney during CCl4 intoxication. Am J
Pathol, 53: 769-789.
Strubelt O (1984) Alcohol potentiation of liver injury. Fundam Appl
Toxicol, 4: 144-151.
Su C & Goldberg E (1976) Environmental concentrations and fluxes of
some halocarbons. Mar Pollut Transf, 14: 353-374.
Suffet IH, Gibs J, Coyle JA, Chrobak RS, & Yohe TL (1985) Applying
analytical techniques to solve ground water problems. J Am Water Works
Assoc, 77: 65-72.
Sugama Y, Kubo M, Nonaka T, Nozawa T, & Tsuchihasi Y (1995)
[Measurement of atmospheric concentration of low boiling point organic
chlorine compounds and CFCs.] Tochigi-ken Kogai Kenkyusho Nenpo, 19:
66-75 (in Japanese with English abstract).
Suzuki H, Hirano N, Watanabe C, & Tarumoto Y (1997) Carbon
tetrachloride does not induce micronucleus in either mouse bone marrow
or peripheral blood. Mutat Res, 394: 77-80.
Svirbely JL, Highman B, Alford WC, & Von Oettingen WF (1947) The
toxicity and narcotic action of monochloro-monobromomethane with
special reference to inorganic and volatile bromide in blood, urine
and brain. J Ind Hyg Toxicol, 29: 382-389.
Szende B, Timár F, & Hargitai B (1994) Olive oil decreases liver
damage in rats caused by carbon tetrachloride (CCl4). Exp Toxicol
Pathol, 46: 355-359.
Tabak HH, Quave SA, Mashni CI, & Barth EF (1981) Biodegradability
studies with organic priority pollutant compounds. J Water Pollut
Control Fed, 53: 1503-1518.
Taketomo AP & Grimsrud E (1977) An analysis of halocarbons in the air
of several working and living environments. Proc Mont Acad Sci, 37:
128-134.
Tátrai E, Hudák A, & Ungváry G (1979) Simultaneous effect on the rat
liver of benzene, toluene, xylene and CCl4. Acta Physiol Acad Sci
Hung, 53: 261 (abstract).
Teschke R, Vierke W, & Goldermann L (1983) Carbon tetrachloride
(CCl4) levels and serum activities of liver enzymes following acute
CCl4 intoxication. Toxicol Lett, 17: 175-180.
Teschke R, Vierke W, & Gellert J (1984) Effect of ethanol on carbon
tetrachloride levels and hepatotoxicity after acute carbon
tetrachloride poisoning. Arch Toxicol, 56: 78-82.
Teta MJ & Ott MG (1988) A mortality study of a research, engineering,
and metal fabrication facility in Western New York State. Am J
Epidemiol, 127: 540-551.
Thiersch JB (1971) Differential effect of compounds on rat litter and
mother. In: Tuchmann-Duplesis H ed. Malformations congénitales des
mammifères. Paris, Masson & Co., pp 99-100.
Tierney DJ, Haas AL, & Koop DR (1992) Degradation of cytochrome
P4502E1: selective loss after labilization of the enzyme. Arch Biochem
Biophys, 293: 9-16.
Tirmenstein MA, Leraas TL, & Fariss MW (1997) Alpha-tocopheryl
hemisuccinate administration increases rat liver subcellular
alpha-tocopherol levels and protects against carbon
tetrachloride-induced hepatotoxicity. Toxicol Lett, 92: 67-77.
Tjälve H & Löfberg B (1983) Extrahepatic sites of metabolism of carbon
tetrachloride in rats. Chem-Biol Interact, 46: 299-316.
Tomasi A, Albano E, Lott KAK, & Slater TF (1980) Spin trapping of free
radical products of CC14 activation using pulse radiolysis and high
energy radiation procedures. FEBS Lett, 122: 303-306.
Tomenson JA, Baron CE, O'Sullivan JJ, Edwards JC, Stonard MD, Walker
RJ, & Fearnley DM (1995) Hepatic function in workers occupationally
exposed to carbon tetrachloride. Occup Environ Med, 52: 508-514.
Toncsev H, Pollák ZS, Kiss A, Sréter L, & Fehér J (1982) Acute carbon
tetrachloride-induced lysosomal membrane damage and the membrane
protecting effect of a new dihydroquinoline-type antioxidant. Int J
Tissue React, 4: 325-330.
Topham JC (1980) Do sperm-head abnormalities in mice specifically
identify mammalian mutagens rather than carcinogens? Mutat Res, 74:
379-387.
Towner RA, Reinke LA, Janzen EG, & Yamashiro S (1994)
In vivo magnetic resonance imaging study of Kupfer cell involvement
in CCl4-induced hepatotoxicity in rats. Can J Physiol Pharmacol, 72:
441-446.
Tracey JP & Sherlock P (1968) Hepatoma following carbon tetrachloride
poisoning. NY State J Med, 68: 2202-2204.
Traiger GJ & Plaa GL (1971) Differences in the potentiation of carbon
tetrachloride in rats by ethanol and isopropanol pretreatment. Toxicol
Appl Pharmacol, 20: 105-112.
Tsuruta H (1975) Percutaneous absorption of organic solvents: I.
Comparative study of the in vivo percutaneous absorption of
chlorinated solvents in mice. Ind Health, 13: 227-236.
Uehleke H, Hellmer KH, & Tabarelli S (1973) Binding of 14C-carbon
tetrachloride to microsomal proteins in vitro and formation of
CHCl3 by reduced liver microsomes. Xenobiotica, 3: 1-11.
Uehleke H, Greim H, Krämer M, & Werner T (1976) Covalent binding of
haloalkanes to liver constituents but absence of mutagenicity on
bacteria in a metabolizing test system. Mutat Res, 38: 114.
Uehleke H, Werner T, Greim H, & Krämer M (1977) Metabolic activation
of haloalkanes and tests in vitro for mutagenicity. Xenobiotica, 7:
393-400.
Uemitsu N (1986) Inhalation pharmacokinetics of carbon tetrachloride
in rats based on arterial blood: inhaled air concentration ratios.
Toxicol Appl Pharmacol, 83: 20-29.
Uemitsu N, Minobe Y, & Nakayoshi H (1985) Concentration-time-response
relationship under conditions of single inhalation of carbon
tetrachloride. Toxicol Appl Pharmacol, 77: 260-266.
Ukai H, Inui S, Takada S, Dendo J, Ogawa J, Isobe K, Ashida T, Tamura
M, Tabuki K, & Ikeda M (1997) Types of organic solvents used in
small- to medium-scale industries in Japan; a nationwide field survey.
Int Arch Occup Environ Health, 70: 385-392.
Ullrich D (1982) [Organohalogen compounds in the air of some indoor
swimming pools in Berlin.] WaBoLu-Berlin, 1: 50-52 (in German).
UNEP (1989) Technical progress on protecting the ozone layer: Report
of the Technology Review Panel. Nairobi, Kenya, United Nations
Environment Programme.
UNEP (1996) The 1987 Montreal protocol on substances that deplete the
ozone layer as adjusted and amended by the Second, Fourth and Seventh
Meeting of the Parties. In: Handbook for the international treaties
for the protection of the ozone layer, 4th ed. Nairobi, Kenya, United
Nations Environment Programme, pp 18-39.
Uryvaeva IV & Delone GV (1995) An improved method of mouse liver
micronucleus analysis: An application to age-related genetic
alteration and polyploidy study. Mutat Res, 334(1): 71-80.
US EPA (1984a) Locating and estimating air emissions from sources of
carbon tetrachloride. Washington, DC, US Environmental Protection
Agency (Report No. 450/4-84-0067 b).
US EPA (1984b) Health assessment document for carbon tetrachloride.
Washington, DC, US Environmental Protection Agency (Report No.
600/8-82-001F).
US EPA (1991) Toxics in the community: National and local
perspectives. Washington, DC, US Environmental Protection Agency,
Office of Toxic Substances (EPA 560/4-91-014).
Vannelli T, Logan M, Arciero DM, & Hooper AB (1990) Degradation of
halogenated aliphatic compounds by the ammonia-oxidising bacterium
Nitrosomonas europaea. Appl Environ Microbiol, 56: 1169-1171.
Van Rensburg JFJ, Van Huyssteen JJ, & Hassett AJ (1978) A
semi-automated technique for the routine analysis of volatile
organohalogens in water purification process. Water Res, 12: 127-131.
Van Tassel S, Amalfitano N, & Narang RS (1981) Determination of arenes
and volatile haloorganic compounds in air at microgram per cubic meter
levels by gas chromatography. Anal Chem, 53: 2130-2135.
Van Zoest R & Van Eck GTM (1991) Occurrence and behaviour of several
groups of organic pollutants in the Scheldt estuary. Sci Total
Environ, 103: 57-71.
Veith GD (1978) An evaluation of using partition coefficients and
water solubility to estimate bioconcentration factors for organic
chemicals in fish. In: Aquatic toxicology: Proceedings of the Third
Annual Symposium on Aquatic Toxicology. Philadelphia, Pennsylvania,
American Society for Testing and Materials, pp 116-129 (ASTM STP 707).
Veng-Pedersen P, Paustenbach DJ, Carlson GP, & Suarez L (1987) A
linear systems approach to analyzing the pharmacokinetics of carbon
tetrachloride in the rat following repeated exposures of 8 and 11.5
h/day. Arch Toxicol, 60: 355-364.
Versar Inc. (1979) Water related environmental fate of 129 priority
pollutants. Washington, DC, US Environmental Protection Agency, vol 2,
chapter 41 (EPA 440/4-79-029B).
Villarruel M del C, de Toranzo EGD, & Castro JA (1977) Carbon
tetrachloride activation, lipid peroxidation and the mixed function
oxygenase activity of various rat tissues. Toxicol Appl Pharmacol, 41:
337-344.
Villarruel M del C, Fernández G, De Ferreyra EC, De Fenos OM, & Castro
JA (1982) Studies on the mechanism of alloxan-diabetus potentiation of
carbon tetrachloride-induced liver necrosis. Br J Exp Pathol, 63:
388-393.
Von Düszeln J & Thiemann W (1985) Volatile chlorinated hydrocarbons in
a coastal urban atmosphere. Sci Total Environ, 41: 187-194.
Von Oettingen WF (1964) The halogenated hydrocarbons of industrial and
toxicological importance. In: Browning E ed. Elsevier monographs on
toxic agents. Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp 107-170.
Von Oettingen WF, Powell CC, Sharpless NE, Alford WC, & Pecora LJ
(1950) Comparative studies of the toxicity and pharmacodynamic action
of chlorinated methanes with special reference to their physical and
chemical characteristics. Ach Int Pharmacodyn, 81: 17-34.
Wahlberg JE (1984a) Edema-inducing effects of solvents following
topical administration. Dermatosen Beruf Umw, 32: 91-94.
Wahlberg JE (1984b) Erythema-inducing effects of solvents following
epicutaneous administration to man -- Studied by laser Doppler
flowmetry. Scand J Work Environ Health, 10: 159-162.
Wahlberg JE & Boman A (1979) Comparative percutaneous toxicity of ten
industrial solvents in the guinea pig. Scand J Work Environ Health, 5:
345-351.
Wallace LA (1986) Personal exposures, indoor and outdoor air
concentrations and exhaled breath concentrations of selected volatile
organic compounds measured for 600 residents of New Jersey, North
Dakota, North Carolina and California. Toxicol Environ Chem, 12:
215-236.
Waller RL, Glende EA, & Recknagel RO (1983) Carbon tetrachloride and
bromotrichloromethane toxicity. Biochem Pharmacol, 32: 1613-1617.
Walton BT, Anderson TA, Hendricks MS, & Talmage SS (1989)
Physicochemical properties as predictors of organic chemical effects
on soil microbial respiration. Environ Toxicol Chem, 8: 53-63.
Walton BT, Hendricks MS, Anderson TA, Griest WH, Merriweather R,
Beauchamp JJ, & Francis CW (1992) Soil sorption of volatile and
semivolatile organic compounds in a mixture. J Environ Qual, 21:
552-558.
Wang MY & Liehr JG (1995) Lipid hydroperoxidase-induced endogenous DNA
adducts in hamsters: possible mechanism of lipid
hydroperoxide-mediated carcinogenesis. Arch Biochem Biophys, 316:
38-46.
Wang PY, Kaneko T, Tsukada H, Nakano M, & Sato A (1997) Dose- and
route-dependent alterations in metabolism and toxicity of chemical
compounds in ethanol-treated rats: difference between highly
(chloroform) and poorly (carbon tetrachloride) metabolized hepatotoxic
compounds. Toxicol Appl Pharmacol, 142: 13-21.
Watanabe A, Shiota T, Takei N, Fujiwara M, & Nagashima H (1986) Blood
to brain transfer of carbon tetrachloride and lipoperoxidation in rat
brain. Res Commun Chem Pathol Pharmacol, 51: 137-140.
Weisburger EK (1977) Carcinogenicity studies on halogenated
hydrocarbons. Environ Health Perspect, 21: 7-16.
Westrick JJ, Mello JW, & Thomas RF (1984) The groundwater supply
survey. J Am Water Works Assoc, 76: 52-59.
Whittaker SG, Zimmermann FK, Dicus B, Piegorsch WW, Fogel S, & Resnick
MA (1989) Detection of induced mitotic chromosome loss in
Saccharomyces cerevisiae -- an interlaboratory study. Mutat Res,
224: 31-78.
WHO (1993) Guidelines for drinking-water quality -- Volume 1:
Recommendations. Geneva, World Health Organization, pp 57-58.
Wilcosky TC, Checkoway H, Marshall EG, & Tyroler HA (1984) Cancer
mortality and solvent exposures in the rubber industry. J Am Ind Hyg
Assoc, 45: 809-811.
Withey JR, Collins BT, & Collins PG (1983) Effect of vehicle on the
pharmacokinetics and uptake of four halogenated hydrocarbons from the
gastrointestinal tract of the rat. J Appl Toxicol, 3: 249-253.
WMO (1991) Global ozone research and monitoring project. Scientific
assessment of ozone depletion: 1991. Geneva, World Meteorological
Organization (Report No. 25).
Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, & Ullrich V (1977)
The reduction of polyhalogenated methanes by liver microsomal
cytochrome P450. Mol Pharmacol, 13: 698-705.
Wong LCK & DiStefano V (1966) Rapid accumulation of renal fat in cats
after single inhalations of carbon tetrachloride. Toxicol Appl
Pharmacol, 9: 485-494.
Yamamoto HA (1990) Relation of Ca++ accumulation and lipid
peroxidation with CCl4-induced toxicity in the rat liver. Pharmacol
Toxicol, 66: 213-216.
Yao T, Degli Esposito S, Huang L, Arnon R, Spangenberger A, & Zern MA
(1994) Inhibition of carbon tetrachloride-induced liver injury by
liposomes containing vitamin E. Am J Physiol, 267: G476-G484.
Yoshida K (1993) Preliminary exposure assessment of volatile
chlorinated hydrocarbons in Japan. Chemosphere, 27: 621-630.
Youssefi M, Faust SD, & Zenchelsky ST (1978) Rapid determination of
light halogenated hydrocarbons in urine. Clin Chem, 24(7): 1109-1111.
Zawaski K, Gruebele A, Kaplan D, Reddy S, Mortensen A, & Novak RF
(1993) Evidence for enhanced expression c-fos, c-jun, and the
Ca2+-activated neutral protease in rat liver following carbon
tetrachloride administration. Biochem Biophys Res Commun, 197:
585-590.
Zeiger E, Anderson B, Haworth S, Lawlor T, & Mortelmans K (1988)
Salmonella mutagenicity tests: IV. Results from the testing of 300
chemicals. Environ Mol Mutagen, 12 (suppl 11): 1-157.
Zimmerman HJ (1978) Syndromes of environmental hepatotoxicity. In:
Zimmerman HJ ed. Hepatotoxicity -- the adverse effect of drugs and
other chemicals on the liver. New York, Appleton-Century-Crofts, pp
297-302.
Zoeteman BCJ, Harmsen K, Linders JBHJ, Morra CFH, & Slooff W (1980)
Persistent organic pollutants in river water and ground water of the
Netherlands. Chemosphere, 9: 231-249.
RÉSUMÉ
Le tétrachlorure de carbone est un liquide limpide, incolore et
volatil qui dégage une odeur douceâtre caractéristique. Il est
miscible à la plupart des solvants aliphatiques et il est lui-même un
solvant. Il est peu soluble dans l'eau. Le tétrachlorure de carbone
n'est pas inflammable et il est stable à l'air et à la lumière. En se
décomposant, il peut donner naissance à du phosgène, à du dioxyde de
carbone et à de l'acide chlorhydrique.
La présence de tétrachlorure de carbone dans l'environnement est
vraisemblablement presque exclusivement d'origine humaine. La majeure
partie du tétrachlorure de carbone produit sert à la préparation de
chlorofluorocarbures (CFC) et autres hydrocarbures chlorés. En 1987,
la production mondiale de tétrachlorure de carbone a été de 960 000
tonnes. Toutefois, depuis que le Protocole de Montréal relatif aux
substances qui appauvrissent la couche d'ozone (1987) et ses
amendements de 1990 et de 1992, a établi un calendrier pour l'abandon
progressif de la production et de la consommation de tétrachlorure de
carbone, la production a reculé et continuera à le faire.
On a mis au point plusieurs méthodes suffisamment sensibles et
précises pour la recherche et le dosage du tétrachlorure de carbone
dans l'air, l'eau et les milieux biologiques. La plupart d'entre elles
sont basées, soit sur l'injection directe de l'échantillon dans un
chromatographe en phase gazeuse, soit sur une adsorption sur charbon
actif, suivie d'une désorption ou d'une évaporation puis d'une
détection par chromatographie en phase gazeuse.
La presque totalité du tétrachlorure de carbone libéré dans
l'environnement finira tôt ou tard dans l'atmosphère en raison de sa
grande volatilité. Comme sa durée de séjour dans l'atmosphère est
longue, il est largement distribué. Au cours de la période 1980-1990,
la concentration atmosphérique du tétrachlorure de carbone était de
l'ordre de 0,5-1,0 µg/m3. Les estimations de sa durée de séjour
atmosphériques sont variables mais on pense que la valeur la plus
raisonnable est de 45 à 50 ans. Le tétrachlorure de carbone contribue
à la fois à la destruction de la couche d'ozone et au réchauffement du
climat. Il est généralement résistant à la biodégradation aérobie,
mais moins à la biodégradation anaérobie. La vitesse de biodégradation
peut s'accroître par suite d'un processus d'acclimatation. Bien que le
coefficient de partage octanol-eau indique un potentiel de
bioaccumulation moyen, cette tendance est réduite par la brièveté de
la demi-vie tissulaire.
Dans l'eau, on fait état de concentrations inférieures à 10
ng/litre pour les océans et de moins de 1 µg/litre pour les eaux
douces, mais de valeurs beaucoup plus fortes à proximité des sites de
décharge. On a mesuré des valeurs allant jusqu'à 60 µg/kg dans des
denrées alimentaires qui avaient été traitées avec du tétrachlorure de
carbone, mais cette pratique a cessé.
C'est essentiellement par l'intermédiaire de l'air que la
population dans son ensemble est exposée au tétrachlorure de carbone.
Si l'on se base sur les concentrations relevées dans l'air ambiant,
les denrées alimentaires et l'eau de boisson, on peut estimer à
environ 1 µg/kg de poids corporel la dose de tétrachlorure de carbone
absorbée. Cette estimation est probablement un peu forte à l'heure
actuelle, du fait qu'on n'utilise plus de tétrachlorure de carbone
pour la fumigation des céréales et que les concentrations annoncées
pour les aliments et utilisées pour ce calcul, correspondaient tout
particulièrement aux matières grasses et aux produits à base de
céréales. D'autres sources font état de valeurs comprises entre 0,1 et
0,27 µg/kg p.c. pour l'exposition journalière de la population
générale. Une exposition plus importante au tétrachlorure de carbone
peut se produire sur le lieu de travail en cas de déversement
accidentel.
Le tétrachlorure de carbone est bien résorbé au niveau des voies
digestives et respiratoires de l'Homme et des animaux. Il peut
également y avoir absorption percutanée du produit liquide, mais dans
le cas de la vapeur, cette absorption est lente.
Le tétrachlorure de carbone se répartit dans tout l'organisme,
mais se concentre surtout dans le foie, le cerveau, les reins, les
muscles, les tissus adipeux et le sang. Le composé initial s'élimine
principalement dans l'air expiré et, en proportion minime, dans
l'urine et les matières fécales.
La première étape de la biotransformation du tétrachlorure de
carbone est catalysée par les enzymes du cytochrome P-450 et aboutit à
la formation d'un radical réactif, le radical trichlorométhyl. La voie
de biotransformation la plus importante conduisant à l'élimination de
ce radical consiste dans une oxydation en un radical encore plus
réactif, le radical trichlorométhylperoxyl, qui peut réagir à son tour
pour donner du phosgène. Le phosgène peut être détoxifié par réaction
avec l'eau pour donner du dioxyde de carbone ou par réaction avec le
glutathion ou la cystéine. En anaérobiose, il y a formation de
chloroforme et de dichlorocarbène.
Les intermédiaires métaboliques du tétrachlorure de carbone
peuvent former des liaisons covalentes avec des macromolécules et
provoquer la peroxydation des lipides.
L'action toxique du tétrachlorure de carbone a pour organes
cibles le foie et le rein. La gravité des effets hépatiques dépend
d'un certain nombre de facteurs tels que la sensibilité de l'espèce,
la voie et le mode d'exposition, le régime alimentaire et une
exposition concomitante éventuelle à d'autres substances, notamment
l'éthanol. En outre, il semble qu'un traitement préalable par divers
composés, comme le phénobarbital ou la vitamine A, accroisse
l'hépatotoxicité du tétrachlorure de carbone, alors que d'autres, au
contraire, la réduisent, comme la vitamine E.
Après application sur l'épiderme de lapins et de cobayes, on a
constaté une irritation modérée et une réaction également modérée a
été observée après instillation dans l'oeil du lapin.
La DL50 la plus faible (2391 mg/kg p.c. sur une période de 14
jours) a été obtenue à l'issue d'une étude sur des chiens qui
recevaient le composé par voie intrapéritonéale. Chez le rat, on a
obtenu des valeurs comprises entre 2821 et 10 054 mg/kg p.c.
Lors d'une étude de 12 semaines sur des rats comportant
l'administration du produit par la voie buccale 5 jours par semaine,
on a obtenu une dose sans effet nocif observable (NOAEL) de 1 mg/kg
p.c. La dose la plus faible produisant un effet nocif observable
(LOAEL) était de 10 mg/kg p.c., les effets observés étant une
augmentation légère, mais significative, de l'activité de la
sorbitol-déshydrogénase et une vacuolisation modérée des hépatocytes
centrilobulaires. Une NOAEL similaire de 1,2 mg/kg p.c. (5 jours par
semaine) a été obtenue chez des souris lors d'une étude de 90 jours
avec administration buccale; dans la même étude, la LOAEL
(hépatotoxicité) a été trouvée égale à 12 mg/kg p.c.
En exposant des rats à du tétrachlorure de carbone par la voie
respiratoire pendant environ 6 mois, 5 jours par semaine, 7 heures par
jour, on a obtenu une NOAEL de 32 mg/m3 LOAEL, basée sur des
anomalies de la morphologie hépatique, a été trouvée égale à 63
mg/m3. Dans une autre étude de 90 jours sur des rats, on a obtenu une
NOAEL de 6,1 mg/m3 après exposition continue à du tétrachlorure de
carbone. Lors d'une étude d'inhalation de 2 ans portant également sur
des rats, la concentration la plus faible étudiée (32 mg/m3) a
provoqué des effets marginaux.
La seule étude toxicologique à long terme dont on dispose a
consisté à faire ingérer du tétrachlorure de carbone à des rats
pendant 2 ans, aux doses respectives de 0, 80 et 200 mg de produit par
kg de nourriture. En raison d'une affection respiratoire chronique qui
a touché tous les animaux à partir du 14ème mois et a provoqué une
augmentation de la mortalité, les résultats de l'autopsie effectuée au
bout de deux ans ne peuvent pas être utilisés pour une évaluation du
risque sanitaire.
Les études d'inhalation effectuées sur des rats et des souris ont
permis de conclure que le tétrachlorure de carbone peut avoir des
effets embryotoxiques pouvant aller jusqu'à la mort de l'embryon.
Toutefois ces effets ne se manifestent qu'aux doses toxiques pour les
femelles gravides. Le tétrachlorure de carbone n'est pas tératogène.
De nombreuses études de génotoxicité ont été effectuées sur le
tétrachlorure de carbone. Sur la base des données disponibles, on peut
considérer que ce composé n'est pas génotoxique.
Le tétrachlorure de carbone provoque l'apparition d'hépatomes et
de carcinomes hépatocellulaires chez le rat et la souris. Les doses
qui entraînent la formation de tumeurs hépatiques sont supérieures aux
doses cytotoxiques.
Chez l'Homme, les manifestations aiguës qui surviennent après
exposition au tétrachlorure de carbone sont indépendants du mode
d'absorption et se caractérisent par des symptômes gastrointestinaux
et neurologiques tels que nausées, vomissements, céphalées,
étourdissements, dyspnée qui finissent par aboutir à la mort. Des
lésions hépatiques apparaissent au bout de 24 h ou davantage. Les
lésions rénales ne se manifestent souvent que 2 ou 3 semaines après
l'intoxication.
Les études épidémiologiques n'ont pas permis d'établir
l'existence d'une association entre l'exposition au tétrachlorure de
carbone et un accroissement du risque de mortalité, de cancer ou
d'affection hépatique. Certains travaux incitent à penser qu'il
pourrait y avoir augmentation du risque de lymphome non Hodgkinien
et,selon une étude particulière, du risque de mortalité et de cirrhose
du foie. Il faut cependant préciser que toutes ces études ne portaient
pas spécifiquement sur l'exposition au tétrachlorure de carbone et
qu'il n'y avait pas, en tout cas, de corrélations statistiques fortes.
En général, le tétrachlorure de carbone se révèle peu toxique
pour les bactéries, les protozoaires et les algues. La concentration
toxique la plus faible a été mesurée chez les bactéries méthanogènes
(CI50 = 6,4 mg/litre). Pour les invertébrés aquatiques, les valeurs
de la Cl50 aiguë varient de 28 à > 770 mg/litre. Dans le cas des
poissons d'eau douce, c'est chez l'orfe (Leuciscus idus
melanotus) que l'on a trouvé la valeur la plus faible de la CL50
aiguë, avec 13 mg/litre. Chez les espèces marines, c'est la limande
(Limanda limanda) qui présente la plus faible valeur de la CL50,
avec 50 mg/litre. Chez les poissons et les amphibiens, le
tétrachlorure de carbone se révèle plus toxique pour les stades
embryo-larvaires que pour les adultes. La grenouille-taureau commune
(Rana catesbeiara) est l'espèce la plus sensible, avec une CL50 de
0,92 mg/litre (de la fécondation à 4 jours après l'éclosion).
Les données disponibles montrent que le mécanisme de formation
des tumeurs hépatiques n'est pas de nature génotoxique et il est donc
admissible de fixer une dose journalière tolérable par ingestion (TDI)
et une concentration journalière tolérable dans l'air (TC).
En s'appuyant sur l'étude de Bruckner et al. (1986) qui ont
déterminé une dose sans effet nocif observable (NOAEL) de 1 mg/kg p.c.
lors d'une étude de 12 semaines sur des rats auxquels on avait fait
ingérer du tétrachlorure de carbone, et en utilisant un facteur de
conversion de 5/7 pour la dose journalière et un coefficient
d'incertitude de 500 (100 pour les variations inter- et
intraspécifiques, 10 pour la durée de l'étude plus un facteur de 0,5
pour tenir compte du fait que l'on avait utilisé des boulettes), on
parvient à une TDI de 1,42 µg/kg de poids corporel.
En s'appuyant sur une étude d'inhalation de 90 jours pratiquée
sur des rats (Prendergast et al., 1967), qui a permis d'aboutir à une
NOAEL de 6,1 mg/m3 on a calculé une TC de 6,1 g/m3 en utilisant les
coefficients de 7/24 et de 5/7 pour passer à une exposition en continu
et un coefficient d'incertitude de 1000 (100 pour les variations
inter- et intraspécifiques et 10 pour la durée de l'étude). Cette TC
correspond à une TDI de 0,85 µg/kg de poids corporel.
En comparant la limite supérieure de la dose journalière absorbée
par l'Homme, c'est-à-dire 0,2 µg/kg p.c., à la valeur la plus faible
de la TDI, soit 0,85 µg/kg p.c., on peut conclure que l'exposition
actuelle de la population générale au tétrachlorure de carbone de
toutes origines a peu de chances de causer une absorption excessive de
ce composé.
En règle générale, les organismes aquatiques ne courent guère de
risque imputable au tétrachlorure de carbone. Toutefois, il peut y
avoir un danger pour les stades embryo-larvaires sur les sites de
décharge ou de déversement de produits industriels ou à proximité de
ces sites.
RESUMEN
El tetracloruro de carbono es un líquido volátil transparente,
incoloro, con un olor dulce característico. Es miscible con la mayor
parte de los disolventes alifáticos y tiene a su vez propiedades
disolventes. La solubilidad en agua es baja. El tetracloruro de
carbono no es inflamable y se mantiene estable en presencia del aire y
de la luz. Su descomposición puede producir fosgeno, anhídrido
carbónico y ácido clorhídrico.
La fuente de tetracloruro de carbono en el medio ambiente con
toda probabilidad tiene un origen casi exclusivamente antropogénico.
La mayor parte del tetracloruro de carbono producido se emplea en la
fabricación de clorofluorocarbonos y otros hidrocarburos clorados. La
producción mundial ascendió en 1987 a 960 000 toneladas. Sin embargo,
desde que en el Protocolo de Montreal relativo a las sustancias que
agotan la capa de ozono (1987) y en sus enmiendas (1990 y 1992) se
estableció un calendario para la reducción progresiva de la producción
y consumo del tetracloruro de carbono, su fabricación ha disminuido y
seguirá descendiendo.
Se han elaborado varios métodos analíticos suficientemente
sensibles y precisos para la determinación del tetracloruro de carbono
en muestras de aire, agua y biológicas. La mayoría de estos métodos se
basan en la inyección directa en un cromatógrafo de gases o la
adsorción en carbón activado, seguida de la desorción o la evaporación
y la posterior detección por cromatografía de gases.
Casi todo el tetracloruro de carbono que se libera en el medio
ambiente estará en último término presente en la atmósfera, debido a
su volatilidad. Dado que su tiempo de permanencia en la atmósfera es
prolongado, tiene una distribución muy amplia. Durante el período
1980-1990, las concentraciones atmosféricas fueron de alrededor de
0,5-1 µg/m3. Las estimaciones de su permanencia en la atmósfera son
variables, pero se aceptan como los valores más razonables los 45-50
años. El tetracloruro de carbono contribuye tanto a la reducción del
ozono como al calentamiento mundial. Es en general resistente a la
biodegradación aerobia, pero menos a la anaerobia. La aclimatación
aumenta la velocidad de biodegradación. Aunque el coeficiente de
reparto octanol/agua indica un potencial de bioacumulación moderado,
el breve período de permanencia en los tejidos reduce esta tendencia.
En el agua, se han notificado concentraciones inferiores a 10
ng/litro en los océanos y generalmente inferiores a 1 µg/litro en el
agua dulce, pero con valores mucho más elevados cerca de los lugares
de liberación. Se han registrado concentraciones de hasta 60 µg/kg en
alimentos elaborados con tetracloruro de carbono, pero esta práctica
se ha suprimido.
La población general está expuesta al tetracloruro de carbono
fundamentalmente a través del aire. A partir de las concentraciones
notificadas en el aire ambiente, los productos alimenticios y el agua
potable, se ha estimado que la ingesta diaria de tetracloruro de
carbono es de alrededor de 1 µg/kg de peso corporal. En la actualidad
esta estimación es probablemente demasiado elevada, porque se ha
suprimido el uso del tetracloruro de carbono como fumigante de los
cereales y los valores notificados para los alimentos y utilizados en
el cálculo fueron fundamentalmente los obtenidos en alimentos a base
de grasas y cereales. En otras partes se han descrito valores de 0,1 a
0,27 µg/kg de peso corporal para la exposición diaria de la población
general. Se puede producir una exposición a concentraciones más
elevadas de tetracloruro de carbono en el lugar de trabajo debido a un
derrame accidental.
El tetracloruro de carbono se absorbe bien de los tractos
gastrointestinal y respiratorio en los animales y en el ser humano. Es
posible la absorción cutánea de tetracloruro de carbono líquido, pero
la absorción cutánea del vapor es lenta.
El tetracloruro de carbono se distribuye por todo el organismo,
alcanzando las concentraciones más altas en el hígado, el cerebro, el
riñón, los músculos, la grasa y la sangre. El compuesto original se
elimina fundamentalmente en el aire exhalado, mientras que se excretan
cantidades mínimas en las heces y la orina.
En la biotransformación del tetracloruro de carbono, el primer
paso es la catálisis por enzimas del citocromo P-450 para formar un
radical reactivo, el triclorometilo. La biotransformación oxidativa es
la ruta más importante en la eliminación del radical, produciendo otro
radical incluso más reactivo, el triclorometilperoxilo, que puede
reaccionar de nuevo para formar fosgeno. Éste se puede destoxificar
mediante la reacción con el agua para producir anhídrido carbónico, o
con el glutatión o la cisteína. En condiciones anaerobias se forma
cloroformo y diclorocarbeno.
Se produce la unión a macromoléculas mediante enlaces covalentes
y la peroxidación de lípidos a través de intermediarios metabólicos
del tetracloruro de carbono.
El hígado y el riñón son los órganos destinatarios de la
toxicidad de este compuesto. La gravedad de los efectos hepáticos
depende de diversos factores, como la susceptibilidad de la especie,
la ruta y el modo de exposición, la alimentación o la exposición
simultánea a otros compuestos, en particular el etanol. Además, parece
que el tratamiento previo con diversos compuestos, como el
fenobarbital y la vitamina A, aumenta la hepatotoxicidad, mientras que
otros compuestos, como la vitamina E, reducen la acción hepatotóxica
del tetracloruro de carbono.
Tras la aplicación cutánea se ha observado una irritación
moderada en la piel de conejos y cobayas, y se puso de manifiesto una
reacción leve después de aplicar el compuesto en el ojo del conejo.
La DL50 más baja, de 2391 mg/kg de peso corporal (período de 14
días), se notificó en un estudio realizado con perros mediante
administración intraperitoneal. En ratas, los valores de la DL50
oscilaron entre 2821 y 10 054 mg/kg de peso corporal.
En un estudio de administración por vía oral a ratas durante 12
semanas (5 días/semana), la concentración sin efectos adversos
observados (NOAEL) fue de 1 mg/kg de peso corporal. La concentración
más baja sin efectos adversos observados (LOAEL) notificada en este
estudio fue de 10 mg/kg de peso corporal, registrándose un aumento
ligero, pero significativo, de la actividad de la sorbitol
deshidrogenasa y una ligera vacuolación centrilobular hepática. En un
estudio de 90 días por vía oral realizado en ratones, se encontró una
NOAEL semejante de 1,2 mg/kg de peso corporal (5 días/semana) con una
LOAEL de 12 mg/kg de peso corporal cuando se produjo hepatotoxicidad.
Cuando se expusieron ratas a tetracloruro de carbono mediante
inhalación durante unos seis meses, cinco días a la semana, siete
horas diarias, se notificó una NOAEL de 32 mg/m3. Se informó de una
LOAEL, basada en cambios en la morfología del hígado, de 63 mg/m3. En
otro estudio de 90 días en ratas se encontró una NOAEL de 6,1 mg/m3
tras una exposición continua a tetracloruro de carbono. El nivel de
exposición más bajo, de 32 mg/m3 (la concentración más baja
estudiada), en un estudio de inhalación en ratas de dos año produjo
efectos marginales.
El único estudio de toxicidad prolongada por vía oral fue uno de
dos años realizado en ratas expuestas a 0, 80 y 200 mg de tetracloruro
de carbono/kg de alimentos. Debido a una enfermedad respiratoria
crónica que contrajeron todos los animales a partir del 14° mes y que
provocó un aumento de la mortalidad, los resultados notificados de la
necropsia a los dos años son insuficientes para evaluar el riesgo para
la salud.
Se llegó a la conclusión de que el tetracloruro de carbono puede
inducir efectos embriotóxicos y embrioletales, pero sólo a dosis
tóxicas para la madre, como se observó en los estudios de inhalación
realizados en ratas y ratones. El tetracloruro de carbono no es
teratogénico.
Se han realizado numerosas valoraciones de la genotoxicidad del
tetracloruro de carbono. Tomando como base los datos disponibles, se
puede considerar que este producto es un compuesto no genotóxico.
El tetracloruro de carbono induce la formación de hepatomas y
carcinomas hepatocelulares en ratones y ratas. Las dosis que inducen
la formación de tumores hepáticos son más elevadas que las que
producen toxicidad celular.
En el ser humano, los síntomas agudos tras la exposición a
tetracloruro de carbono son independientes de la ruta de ingestión y
se caracterizan por síntomas gastrointestinales y neurológicos, como
náuseas, vómitos, dolor de cabeza, desvanecimiento, disnea y la
muerte. Después de las 24 horas o más aparecen lesiones hepáticas. Son
evidentes los trastornos renales con frecuencia sólo dos o tres
semanas después de la intoxicación.
Los estudios epidemiológicos no han establecido una asociación
entre la exposición al tetracloruro de carbono y el aumento del riesgo
de mortalidad, neoplasia o enfermedad hepática. Algunos estudios han
indicado una asociación con un aumento del riesgo de linfoma
no-Hodgkin y, en un estudio, con la mortalidad y la cirrosis hepática.
Sin embargo, no en todos estos estudios se señaló la exposición
específica al tetracloruro de carbono y las asociaciones estadísticas
no fueron convincentes.
En general, el tetracloruro de carbono parece tener una toxicidad
baja para las bacterias, los protozoos y las algas; la concentración
tóxica más baja notificada para las bacterias metanogénicas
correspondió a una CI50 de 6,4 mg/litro. Para los invertebrados
acuáticos, los valores de la CL50 aguda fueron de 28 a >770
mg/litro. En los peces de agua dulce el valor más bajos de la CL50
aguda, de 13 g/litro, se encontró en el cachuelo dorado
(Leuciscus idus melanotus), y para las especies marinas se notificó
un valor de la CL50 de 50 mg/litro para la limanda
(Limanda limanda). El tetracloruro de carbono parece ser más tóxico
para las fases embrionaria y larvaria de los peces y anfibios que para
los adultos. La rana toro común (Rana catesbeiara) es la especie más
susceptible, con una CL50 de 0,92 mg/litro (desde la fecundación
hasta los cuatro días después de la eclosión).
Los datos disponibles indican que la inducción de tumores
hepáticos se debe a un mecanismo no genotóxico, por lo que parece
aceptable el establecimiento de una ingesta diaria tolerable (IDT) y
de una concentración diaria tolerable (CDT) en el aire para el
tetracloruro de carbono.
Tomando como base el estudio de Bruckner et al. (1986), en el
cual se observó una NOAEL de 1 mg/kg de peso corporal en un estudio de
12 semanas con administración por vía oral a ratas, e incorporando un
factor de conversión de 5/7 para la dosificación diaria y aplicando un
factor de incertidumbre de 500 (100 por la variación interespecífica e
intraespecífica, 10 por la duración del estudio y un factor de
modificación de 0,5 porque se trataba de un estudio de bolo), se
obtiene una IDT de 1,42 µg/kg de peso corporal.
Al comparar el límite superior estimado de la ingesta diaria
predominante de 0,2 µg/kg de peso corporal con el valor más bajo de la
IDT (0,85 µg/kg de peso corporal), se puede llegar a la conclusión de
que la exposición predominante en la actualidad de la población
general al tetracloruro de carbono procedente de todas las fuentes es
poco probable que dé lugar a una ingesta excesiva de la sustancia
química.
En general, el riesgo del tetracloruro de carbono para los
organismos acuáticos es bajo. Sin embargo, puede presentar un riesgo
para las fases embrionaria y larvaria en lugares de vertidos o escapes
industriales o en zonas próximas a ellos.
Condensation of phosgene with cysteine leads to the formation of
2-oxothiazolidine-4-carboxylic acid (Shah et al., 1979; Kubic &
Anders, 1980). The condensation of phosgene with glutathione (GSH),
resulting in diglutathionyl dithiocarbonate, has been demonstrated by
Pohl et al. (1981) in in vivo experiments.
Formation of chloroform and CCl2-carbene occur under
O2-deficient circumstances (Reiner et al., 1972; Shah et al., 1979;
Pohl et al., 1984). Under in vivo conditions CCl2-carbene is of
minor importance as an intermediate. Castro et al. (1990) examined the
biotransformation of carbon tetrachloride to chloroform by liver
nuclear preparations of three different species: C3H mice,
Sprague-Dawley rats and Syrian golden hamsters. All species were able
to transform carbon tetrachloride to chloroform. This ability was not
NADPH dependent and proceeded to an equal extent under nitrogen and
air. The relative transforming intensity was mice > hamsters > rats
under anaerobic and hamsters >> mice > rats under aerobic
conditions, respectively. More detailed experiments with preparations
of C3H mice suggested the presence of enzymatic and non-enzymatic
pathways of carbon tetrachloride transformation, as revealed by their
heat susceptibility and the inhibitory effects of EDTA.
Many studies on covalent binding of carbon tetrachloride
metabolites to tissue macromolecules have been carried out, but most
of them have measured only radioactivity and not identified the adduct
formed. Many studies ignored the radioactive impurities present in the
carbon tetrachloride used as well as the possible incorporation of
14C-radioactivity via carbon dioxide riginating from carbon
tetrachloride. For these reasons, the binding of carbon tetrachloride
should be considered as an association of radioactivity, unless
further information is provided.
According to Shertzer et al. (1988), the active radical metabolic
intermediate of carbon tetrachloride may covalently bind to
macromolecules, produce lipid peroxidation, and result in the loss of
intrahepatic calcium homoeostasis.
Cambon-Gros et al. (1986) showed that the fetal rat liver during
the last days of pregnancy, as well as the mother liver, can
metabolize carbon tetrachloride into a free radical: CCl3*. This
radical may bind covalently to the microsomal membranes and cause the
destruction of cytochrome P-450 as well as the inhibition of one of
the main microsomal activities, the ability to store Ca2+. Contrary
to the situation in adults, these radicals do not provoke a membrane
phospholipid peroxidation in the fetus. The animals used in the study
were nulliparous pregnant female Sprague-Dawley rats (twentieth day of
gestation).
Tjälve & Löfberg (1983) showed that covalent binding (probably
due to metabolic activation) occurred in many tissues of exposed rats
(liver, kidney cortex, mucosae of the respiratory tract, the mouth
cavity and the oesophagus).
The non-extractable part of non-volatile radioactivity in liver
and kidney may indicate covalent binding to tissue components
(Bergman, 1984). Association of radioactivity to tissue components
also appeared to occur in the testis, the uterine and vaginal mucosa
and in the nasal mucosa.
A similar pattern of association with tissue components to that
observed by Bergman (1984) has been found by Tjälve & Löfberg (1983)
after intravenous and intraperitoneal administration, indicating that
the tissue binding in the nasal mucosa is not specific to the route of
administration.
Díaz Gómez et al. (1975a) investigated the relations between
liver carbon tetrachloride levels, lipid peroxidation, the covalent
binding to liver lipids and hepatic centrilobular necrosis after
in vivo adminis tration of equimolar doses of carbon tetrachloride
in different animal species. The results support the hypothesis that
carbon tetrachloride-induced lipid peroxidation is not the only
mechanism of its toxic action. In fact, there seems to be a better
correlation between irreversible association with tissue components
and carbon tetrachloride toxicity than between lipid peroxidation and
carbon tetrachloride toxicity.
According to Villarruel et al. (1977) association of carbon
tetrachloride metabolites with lipids occurs mostly in the liver and
kidney cortex and medulla. Ansari et al. (1982) demonstrated the
binding of trichloromethyl radicals originating from carbon
tetrachloride to cholesterol. Binding to membrane lipids, eventually
leading to cross-linking, has been demonstrated by Link et al. (1984).
Association of carbon tetrachloride derivatives with
macromolecules in vitro has been found mainly in microsomal systems,
but binding to lipids and proteins also occurs in purified nuclear
preparations (Díaz Gómez & Castro, 1980b).
The covalent binding of carbon tetrachloride reactive metabolites
to different nuclear and microsomal lipids was studied by Fanelli &
Castro (1995) in male Sprague Dawley and Osborne Mendel rats, strains
with a marked difference in the carcinogenic response to carbon
tetrachloride, the Sprague-Dawley being non-susceptible and the
Osborne-Mendel being responsive. The intensity of covalent binding to
microsomal lipids in vivo and in vitro was higher in the Osborne
Mendel rats. Most of the covalent binding of carbon tetrachloride
reactive metabolites in both rat strains occurs in the phospholipid
and in the cholesterol/cholesterol ester fractions. The covalent
binding to phospholipids is higher in the Sprague Dawley strain, while
binding to cholesterol and cholesterol ester is more intense in the
Osborne Mendel rat.
After administration of [14C]carbon tetrachloride to rats and
mice (9 µmol/kg body weight), the quantities of label associated with
DNA at 6 h post-dosing were 0.52 and 0.72 pmol/mg DNA, respectively.
In this study binding also occurred to nuclear proteins and lipids,
especially to phospholipids (diphosphatidylglycerol) and diglycerides
(Díaz Gómez & Castro, 1980a).
The results of the study of Oraumbo & Van Duuren (1989) indicated
that under aerobic incubation conditions, carbon tetrachloride is
metabolized to one or more electrophilic metabolites, which bind
covalently to chromatin DNA in a dose- and time-dependent manner. In
this study chromatin was isolated from male B6C3F1 hybrid mice and
incubated with [14C]carbon tetrachloride in the presence of hepatic
microsomes from the same animals and a NADPH-regenerating system. The
study was carried out with various carbon tetrachloride concentrations
and incubation times.
6.3 Human studies
6.3.1 Uptake
6.3.1.1 Dermal
Immersion of a thumb in liquid carbon tetrachloride for 30 min
produced a maximum alveolar carbon tetrachloride concentration of
about 3.8 mg/m3 air 30 min after the end of exposure (Stewart & Dodd,
1964). The concentration declined with a half-life of about 2.5 h.
6.3.1.2 Inhalation
Lehmann & Schmidt-Kehl (1936) reported that 60% of the quantity
of carbon tetrachloride inhaled was retained in an experiment
involving a 30-min exposure of a volunteer to 4200 mg/m3.
6.3.2 Elimination
The pulmonary excretion of 33% of the absorbed quantity of
[38Cl] carbon tetrachloride occurred during the first hour after a
single breath by a volunteer (Morgan et al., 1970). Erickson (1981)
found carbon tetrachloride in mother's milk (concentration and
exposure not specified).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Lethality
LD50 values of carbon tetrachloride for various mammalian
species are represented in Table 7.
For female OF1 mice a LC50 of 7176 ppm (45 998 mg/m3) was
reported after a 6-h inhalation exposure to carbon tetrachloride (14
days observation period) (Gradiski et al., 1978). Svirbely et al.
(1947) reported a LC50 of 50 000 mg/m3 after a 7-h inhalation
exposure (8-h observation period) for male and female Swiss mice.
A 100% death rate of Wistar rats due to anaesthesia was observed
by Adams et al. (1952) after inhalation exposure to carbon
tetrachloride at concentrations of 121 600 mg/m3 for 2.2 h or 46 700
mg/m3 for 8 h.
Roudabush et al. (1965) reported that the acute dermal LD50
values of carbon tetrachloride for rabbits and guinea-pigs were in
excess of 15 g/kg body weight. Data on mortality and observation
period were not given.
Wahlberg & Boman (1979) applied carbon tetrachloride in
quantities of 800 and 3200 mg (approximately 2100 and 8500 mg/kg body
weight) to the skin of guinea-pigs. After 21 days 5/20 and 13/20 of
the animals had died in the 800 mg and 3200 mg groups, respectively.
7.1.2 Non-lethal effects
7.1.2.1 Oral exposure
Effects on the liver with changes in several enzyme levels are
reported to be the major effects resulting from an acute oral exposure
to carbon tetrachloride. In addition to the effects on the liver,
effects have also been reported in other organs such as lungs and
kidneys.
a) Mice
Akahori et al. (1983) examined the biochemical alterations in
liver and blood, and the histological findings in the liver of female
C57BL/6J mice after a single oral administration of carbon
tetrachloride in liquid paraffin at doses of 0.02, 0.5 or 1.5 ml/kg
body weight (calculated to be 32, 797 or 2391 mg/kg body weight) at
various times from 15 to 327 h after administration. The biochemical
changes in the liver (decreases in protein, glucose, phospholipids,
DNA and RNA concentrations; increases in triglycerides, glycogen, and
Table 7. LD50 values (mg/kg body weight) for mammals
Species/strain Sex Route Vehicle Observed period LD50a Reference
Mice
Swiss Webster male intraperitoneal corn oil 24 h 4144 Klaassen & Plaa (1967a)
female intraperitoneal corn oil 24 h 4463 Klaassen & Plaa (1967a)
Strain and sex
unknown oral unknown not reported 12 100-14 400 IARC (1979)
Swiss Webster female intraperitoneal corn oil 24 h 4676 Gehring (1968)
OF1 (SPF) female intraperitoneal olive oil 14 days 3350 Gradiski et al. (1974)
Rats
Wistar female oral unknown 14 days 2821 Smyth et al. (1970)
Sprague-Dawley male intraperitoneal corn oil 48 h 4463 Klingensmith et al. (1983)
Sprague-Dawley male intraperitoneal corn oil 24 h 3029 Klaassen & Plaa (1969)
Sprague-Dawley female intraperitoneal peanut oil 24 h 6603 Lundberg et al. (1986)
14 days 2824 Lundberg et al. (1986)
Charles River male oral corn oil 14 days 10 054 Kennedy et al. (1986)
Dogs
Mongrel male/female intraperitoneal corn oil 24 h 2391 Klaassen & Plaa (1967b)
a Most of the values are calculated values because they were reported as ml/kg body weight
free and esterified cholesterol concentrations) and in blood (an
increase in serum aspartate aminotransferase (ASAT) activity and free
and esterified cholesterol concentration; a decrease in glucose
concentration) were generally dose-related but occurred more slowly in
the highest dose groups. The biochemical alterations were reflected in
the histological findings in the liver (centrilobular necrosis in the
low-dose and a mild midzonal necrosis in the mid- and high-dose
groups). These histological findings occurred later in the high-dose
group.
A dose-related increase in the serum angiotensin converting
enzyme level, indicative of pulmonary endothelial cell injury, was
reported by Hollinger (1982), who administered carbon tetrachloride in
vegetable oil at doses of 0.1 to 2.8 ml/kg body weight (159 to 4463
mg/kg body weight) to male Swiss-Webster mice.
Boyd et al. (1980) found morphological effects on the pulmonary
Clara cells of mice, including severe dilations of endoplasmic
reticulum and occasional cellular necrosis, after administration of
2.5 ml/kg body weight (3985 mg/kg body weight) of carbon tetrachloride
in sesame oil. Oral doses of less than 1 ml/kg body weight (1594 mg/kg
body weight) failed to produce visible pulmonary lesions.
b) Rats
Murphy & Malley (1969) reported dose-related increases in liver
and serum alanine aminotransferase (ALAT), liver tyrosine transaminase
and alkaline phosphatase activities after a single oral dose of 0.5 to
2 ml/kg body weight (797 to 3188 mg/kg body weight) of undiluted
carbon tetrachloride in male Holtzman rats.
In a study by Korsrud et al. (1972), fasted male Wistar rats
received carbon tetrachloride at a dose ranging from 0 to 2.5 ml/kg
body weight (0 to 3985 mg/kg body weight) in corn oil. The rats were
killed 18 h later. At a dose of 0.0125 ml/kg body weight (19.9 mg/kg),
there was histopathological evidence of toxic effects on the liver. At
0.025 ml/kg body weight (39.9 mg/kg), liver fat and weight, serum urea
and the activities of sorbitol dehydrogenase, fructose-l-P-aldolase,
isocitrate dehydrogenase, ALAT and aspartate aminotransferase (ASAT)
were increased.
According to Teschke et al. (1984), liver enzymes such as ALAT,
ASAT and glutamate dehydrogenase measured in serum reached maximal
activities 12-48 h following a single intragastric dose of carbon
tetrachloride (1.5 ml/kg body weight) to female Wistar rats.
A maximum increase of ASAT and ALAT after 48 h was reported by
Nakata et al. (1985) after administration of 5 ml/kg body weight (7970
mg/kg body weight) of carbon tetrachloride in corn oil to male Wistar
rats. Regeneration of the liver was characterized by a normalization
of the ASAT and ALAT levels and an increase in hepatic thymidylate
synthetase and thymidine kinase levels, two enzymes that are believed
to be indicative of tissue regeneration.
A single oral bolus of carbon tetrachloride (17.5 or 179 mg/kg)
to male Sprague-Dawley rats induced a dose-dependent increase in serum
sorbitol dehydrogenase and ALAT activities, and a decrease in the
hepatic cytochrome P-450 content and glucose-6-phosphatase activity.
When the same dose was given as a gastric infusion for 2 h, or by
inhalation exposure, the effects were much smaller (Sanzgiri et al.,
1995). No statistically significant difference was observed in the
toxicity of carbon tetrachloride administered orally in either corn
oil, Emulphor, or Tween-85 (Raymond & Plaa, 1997).
Significant increases in alpha-GSH, a cytosolic enzyme of short
half- life found in high concentration throughout the liver, were
detected 2 h after gavage dosing of male Sprague-Dawley rats (Clarke
et al., 1997). It was concluded that alpha-GST is a more sensitive and
accurate measure of carbon tetrachloride hepatotoxicity than ASAT.
Lowrey et al. (1981) reported a dose-dependent decreased capacity
of rat liver microsomes to sequester calcium 5 min after the
administration of carbon tetrachloride to fasted male Sprague-Dawley
rats at doses ranging from 0.025 to 5 ml/kg body weight (40 to 7970
mg/kg body weight). At 10 min after a carbon tetrachloride dose of 2.5
ml/kg body weight (3985 mg/kg body weight), the microsomal calcium
uptake was reduced to 15% of the control levels.
Chen et al. (1977) observed marked decreases in cytochrome P-450
content and P-450-related N-demethylation of dimethylaniline in the
microsomes of the lungs of male Sprague-Dawley rats after an oral
carbon tetrachloride dose in mineral oil of 2.5 ml/kg body weight
(3985 mg/kg body weight).
Shinozuka (1971) found alterations of the rough endoplasmic
reticulum membranes of rat hepatic cells, such as detachment of
ribosomes, narrowing of cisternal spaces, fusion of membranes and
eventual collapse, within 30 min after an administration of 5 ml/kg
body weight (7970 mg/kg body weight) of carbon tetrachloride in
mineral oil to Wistar rats.
Boyd et al. (1980) found lesions in the lungs of male
Sprague-Dawley rats that were similar to the lesions found in mice
(enlarged pulmonary Clara cells with dilations of the endoplasmic
reticulum, occasional cellular necrosis) after carbon tetrachloride
administration at doses of 2.4, 3.2 or 4.5 ml/kg body weight (3825,
5100 or 7173 mg/kg body weight) in sesame oil. These lesions, however,
were less pronounced and less frequent than in mice.
Striker et al. (1968) observed reversible lesions limited to the
proximal tubules in the kidneys of male Sprague-Dawley rats after
administration of 0.25 ml/kg body weight (400 mg/kg body weight) of
carbon tetrachloride in mineral oil. The earliest morphological change
was seen in the mitochondria, followed by cellular swelling, loss of
basilar interdigitations and swollen microvilli. Proliferation of the
smooth endoplasmic reticulum occurred later. Serum parameters, such as
creatinine, blood urea nitrogen and bilirubin, temporarily increased.
Furthermore, a decrease in the ability to preserve sodium ions and
water was observed, accompanied by a reduction of succinate
dehydrogenase activity.
Rats administered carbon tetrachloride in corn oil as a single
intraperitoneal dose had significantly prolonged clotting times that
appeared prior to liver necrosis (Pritchard et al., 1987).
c) Rabbits
Rabbits (strain unspecified), given an intragastric dose of 0.15
ml/kg body weight of a 33% carbon tetrachloride solution in liquid
paraffin (equivalent to 239 mg/kg body weight) showed an abnormal
electrophoretic lipoprotein pattern. This correlated with the degree
of liver injury as measured by ASAT and ALAT activities and plasma
lipid levels (Kanaghinis et al., 1982).
d) Monkeys
Centrilobular hepatocellular necrosis was observed in two out of
four monkeys 24 h after administration of a single oral dose of carbon
tetrachloride (1590 mg/kg).
e) Dogs
Dogs were administered single oral doses of 159, 318 and 477
mg/kg. Increased serum ALAT and ASAT activities were observed at 318
mg/kg or more.
7.1.2.2 Inhalation exposure
a) Mice
Boyd et al. (1980) exposed male Swiss mice to carbon
tetrachloride concentrations of 0.46 or 0.92 mmol/litre air for 1 h,
1.84 mmol/litre air for 12 min, and 3.68 mmol/litre air for 2 min
(70 750, 141 500, 283 000 and 566 000 mg/m3 air). All exposures
produced marked Clara cell lesions, similar to those caused by oral
exposure, and hepatic necrosis.
b) Rats
Brondeau et al. (1983) exposed male Sprague-Dawley rats (IFFA
CREDO; 8 males/group) to carbon tetrachloride at concentrations of
259, 531, 967 and 1459 ppm (1660, 3404, 6198 and 9352 mg/m3 air) for
4 h and examined the ASAT, ALAT, SDH and glutamate dehydrogenase
activities in the serum 24 h post-exposure. At the lowest exposure
level only the glutamate dehydrogenase activity was increased, whereas
at the higher exposure levels increases were observed in all enzyme
activities. Similar increases in serum activities of liver enzymes
have been reported in other rat strains (Magos et al., 1982
(Porton-Wistar and Fischer rats); Jaeger et al., 1975 (Holtzman rats);
Siegers et al., 1985 (Wistar rats)).
When male Sprague-Dawley rats were exposed to carbon
tetrachloride under conditions of various combinations of
concentration (; 1350 to 6900 ppm) and exposure time, it appeared that
the concentration had more influence on the hepatotoxicity than the
exposure time (Uemitsu et al., 1985).
Chen et al. (1977) observed a decrease in the cytochrome P-450
content and P-450-related demethylation of dimethylaniline in the
microsomes of lungs of male Sprague-Dawley rats exposed for 30 min to
air containing 4.38% carbon tetrachloride (280 758 mg/m3).
Morphological analysis of the lungs revealed focal changes in
pulmonary architecture consisting of alveolar collapse, septal
thickening and atypical type II pneumocyte configuration.
c) Cats
Wong & DiStefano (1966) exposed cats to carbon tetrachloride at a
concentration of 10 000 ppm (64 100 mg/m3) for 15, 30, 60 and 240
min. After 15 min the renal lipid content reached maximal levels. An
increase of kidney weight occurred within 60 min and was maintained
throughout the 24-h observation period. The total lipid content of the
liver had increased significantly at 3, 12 and 24 h after the 4-h
exposure period. Increased liver weight was observed 24 h after the
withdrawal of carbon tetrachloride. According to the authors, the
early increases in both the weight and fat content of the kidney
suggests that the renal changes precede the liver damage.
7.1.2.3 Subcutaneous and intraperitoneal exposure
a) Mice
A subcutaneous dose of 28 mg/kg body weight of carbon
tetrachloride in olive oil to male Swiss mice (10/group) appeared to
be the ED50 for causing prolongation of pentobarbital-induced
sleeping time. Histological examination showed changes in the liver
after administration of 77 mg/kg body weight. (Kutob & Plaa, 1962).
Intraperitoneal administration of carbon tetrachloride in corn
oil induced an elevation of the ALAT activity at calculated dose
levels of 11.2 to 15.9 mg/kg body weight in female Swiss mice and at
14.4 to 15.9 mg/kg body weight in male Swiss mice (Klaassen & Plaa,
1967a).
Bhathal et al. (1983) reported striking differences in the degree
of hepatic cell injury among four different strains of mice upon
histological examination after subcutaneous injection of 0.3 ml/mouse
of olive oil containing 5, 10 and 20% carbon tetrachloride. The SJL/J
strain appeared to be the least susceptible and the BALB/c strain the
most susceptible one. The hepatic lesions in the C3H and C57BL/6
strains were intermediate.
b) Rats
The study of Smejkalová et al. (1985) showed the existence of sex
differences in the sensitivity of the liver to carbon tetrachloride,
including a difference in the rate and quality of liver regeneration.
It appeared that in male Wistar rats the biochemical changes occurred
earlier (as early as 6 h after intraperitoneal administration of 1200
mg/kg body weight) and persisted longer (reaching a maximum after 12 h
and persisting for more than 72 h) than in females. In females these
changes reached a maximum after 24 h, and after 72 h the levels were
identical to the control values. Whereas in females the liver
regeneration started sooner than in males and led to complete healing
of the liver tissue, the regeneration in males started more slowly and
healing followed a different course, showing the development of
fibrosis.
Carbon tetrachloride dissolved in olive oil was injected
intraperitoneally to male Fischer rats at doses of 30 to 1000 mg/kg
body weight. Free and esterified cholesterol, triglycerides,
phospholipids and total lipids in plasma were reduced in a
dose-dependent manner. Cholesterol, triglyceride, phospholipid and
total lipid concentrations in the plasma were significantly lower in
rats given 30 mg/kg body weight than in control rats (p < 0.01)
(Honma, 1990).
When male Wistar rats received carbon tetrachloride as an
intraperitoneal injection of 16 or 96 mg/kg body weight in olive oil,
the calcium ion (Ca2+) content of liver microsomes was significantly
increased by 20% in rats treated with 96 mg/kg body weight. The
mitochondrial Ca2+ content was increased in both the 16 and 96 mg/kg
body weight group (600% and 1100%, respectively, 3 h after
administration) (Yamamoto, 1990).
c) Guinea-pigs
Divincenzo & Krasavage (1974) administered intraperitoneally 5,
25, 50, 75 or 150 mg/kg body weight to guinea-pigs. At 25 mg/kg or
more, increased ornithine decarboxylase activity in the serum was
found, an effect that was reflected by histological changes in the
liver.
d) Hamsters
Carbon tetrachloride produced injury to ciliated and non-ciliated
tracheal cells (swollen, loss of staining capacity, diluted nuclei) of
adult male Syrian golden hamsters that received carbon tetrachloride
intraperitoneally at a dose of 2.5 ml/kg body weight (3985 mg/kg body
weight). Groups of three hamsters were killed 1, 4, 12 or 24 h after
treatment. The number of damaged cells increased markedly after 1 h in
the lower trachea, but not until after 4 h in the upper trachea. By 24
h the number of injured cells approached normal values. Effects were
consistent within each group (Ahmadizadeh et al., 1990).
e) Dogs
When five mongrel dogs were given carbon tetrachloride at
different intraperitoneal doses, increases in ALAT were observed. The
calculated ED50 24-h after exposure was 32 mg/kg (Klaassen & Plaa,
1967b).
De Zwart et al. (1997) have identified eight urinary degradation
products of carbon tetrachloride-induced lipid peroxidation as
potentially useful biomarkers of in vivo hepatocellular damage. Male
Wistar rats were injected intraperitoneally with single doses of 38,
77 and 154 mg/kg body weight and the following substances were
identified in urine 12 to 48 h later: formaldehyde, acetaldehyde,
acetone, propanol, butanol, pentanal, hexanal and malondialdehyde. A
dose-dependent increase in histological and clinical chemistry
evidence of hepatocellular damage, along with these degradation
products, was observed. Increases in urinary concentrations of all
eight products were statistically significant at doses of 77 and 154
mg/kg. At 38 mg/kg, acetaldehyde and propanol were the only urinary
markers to exhibit a statistically significant increase.
7.1.2.4 Dermal exposure
The histopathology of the skin, liver, and kidney in the
guinea-pig (weighing 440 to 570 g) was studied by Kronevi et al.
(1979) at 15 min and 1, 4 and 16 h after occlusive epicutaneous
administration of 1 ml of carbon tetrachloride. After 15 min, some
degenerative changes in the epidermis, such a moderate karyopyknosis,
marked spongiosis and perinuclear oedema was observed. These changes
became more obvious with time, and at 16 h a slight karyolysis also
was seen. A junctional separation and cellular infiltration in the
dermis was observed after 4 and 16 h. Carbon tetrachloride exposure
caused hepatic centrilobular hydropic changes and, in addition, a
tendency to necrotic lesions after 16 h. Kidney histology was normal
for all exposed guinea-pigs.
7.2 Short-term exposure
7.2.1 Oral exposure
a) Mice
Hayes et al. (1986) administered carbon tetrachloride in corn oil
to CD-1 ICR mice (20/sex/group) for 14 consecutive days at dose levels
of 0, 625, 1250 or 2500 mg/kg body weight and for 90 consecutive days
at dose levels of 0, 12, 120, 540 or 1200 mg/kg body weight. No
compound-related deaths were seen in the 90-day study whereas, in the
14-day study, 6, 8 and 12 males and 0, 1 and 2 females died within 2-4
days in the 625, 1250 and 2500 mg/kg body weight groups, respectively.
Dose-dependent effects in the 14-day study consisted of decreased
fibrinogen and lymphocyte levels, increased LDH, ALAT and ASAT levels,
increased absolute and relative liver weights in both sexes, and
decreased lung, thymus and kidney weights in males. In the 90-day
study LDH, ASAT, ALAT and AP, cholesterol and bilirubin levels in the
blood were increased in a dose-dependent manner while blood glucose
levels were decreased at all dose levels. In both sexes and in all
90-day dose groups absolute and relative liver, spleen and thymus
weights were increased and liver damage was observed.
The results of a 90-day oral study in CD-1 mice by Condie et al.
(1986), indicated that the no-observed-adverse-effect level (NOAEL)
for hepatotoxic effects after administration of carbon tetrachloride
in corn oil was 1.2 mg/kg body weight (see also section 7.9).
b) Rats
Four groups of weanling rats (six males and six females per
group) were fed diet containing 0, 150, 225 or 520 mg/kg; estimated
daily doses were 0, 7-13, 13-24, 21-27 mg/kg body weight. The fat
content of liver was increased significantly in two higher dose groups
in both males (exposed for 6 weeks) and females (exposed for 5 weeks);
the difference compared to the control group was 50 to 200% (Alumot et
al., 1976).
Carbon tetrachloride treatment for 5 consecutive days at a dose
level of 400 mg/kg body weight in corn oil to male Fischer-344 rats
(CD F/CrlBr) increased the relative liver weight, decreased the CYP
enzyme concentration and activity in the liver, and increased the ALAT
levels (Dent & Graichen, 1982).
Bruckner et al. (1986) administered carbon tetrachloride in corn
oil to male Sprague-Dawley rats (5/group) for 5 consecutive days for
12 weeks, then 2 days without dosing followed by another 4 consecutive
days of dosing. Groups of rats weighing 300 to 350 g received 0, 20,
40 or 80 mg/kg body weight, whereas groups of rats weighing 200 to 250
g received 0, 20, 80 or 160 mg/kg body weight. In rats weighing
300-350 g, 20 mg/kg body weight caused vacuolization of hepatocytes
adjacent to the central vein of most liver lobules. The other dose
levels in this group produced comparable increases in SDH and ALAT
activities, and vacuolization of 25 to 35% of the hepatocytes in each
liver lobule. Carbon tetrachloride appeared to be more toxic to the
rats weighing 200-250 g with regard to necrotic cells, which were
rarely seen in livers of the 300-350 g rats, while in each 200-250 g
rat given 80 mg/kg body weight necrosis was observed.
Groups of 15-16 male Sprague-Dawley rats weighing 200 to 250 g
were given carbon tetrachloride in corn oil at doses of 0, 1, 10 and
33 mg/kg body weight for 5 days a week. The rats were dosed during the
dark part of their light cycle. After 12 weeks, 7 to 9 rats/group were
killed. The remaining animals were killed 13 days after exposure. At
10 mg/kg body weight a slightly but significantly increased SDH
activity and mild hepatic centrilobular vacuolization was seen.
Administration of 33 mg/kg body weight caused elevated serum levels of
SDH, ornithine-carbamyl transferase (OCT) and ALAT, which returned to
normal during the recovery period except for the OCT. Histopathology
of the livers of the 33 mg/kg body weight group revealed cirrhosis,
characterized by bile duct proliferation, fibrosis, lobular
distortion, parenchymal regeneration, hyperplastic nodules and
single-cell necrosis. According to the authors (Bruckner et al.,
1986), a NOAEL of 1 mg/kg body weight of carbon tetrachloride could be
established.
Allis et al. (1990) administered carbon tetrachloride in corn oil
to male Fischer-344 rats by gavage at dose levels of 0, 20 or 40 mg/kg
body weight for 12 weeks. At both dose levels ALAT, ASAT and LDH
levels were elevated and hepatic CYP protein concentrations were
reduced. Histopathology showed cirrhotic livers, vacuolar degeneration
and hepatocellular necrosis at both dose levels, but this was more
severe in the higher dose group. At day 8 and 15 after exposure, all
serum indicators and CYP protein concentration had returned to normal
levels. In both dose groups, hepatocellular necrosis disappeared by
day 8 and vacuolar degeneration decreased in severity but was still
present at day 15. Cirrhosis persisted in the high-dose group,
although it was less severe. Furthermore the relative liver weight in
animals receiving 40 mg/kg body weight remained elevated.
Several tests on renal function were conducted on adult male
Fischer-344 rats treated for 15 days with carbon tetrachloride in corn
oil at doses of 50, 150, 450 or 1350 mg/kg body weight. At 1350 mg/kg
body weight, kidney injury could be observed, as indicated by
haematuria, enzymeuria and decreases in kidney weight and serum
glucose concentration. At 450 mg/kg body weight, a lower body weight
and a reduction in the maximum urine-concentrating ability were
observed (Kluwe, 1981).
c) Dogs
Young adult Beagle dogs of the Alderly Park strain (6/sex) that
received carbon tetrachloride (in gelatin capsules) as a daily dose of
0.05 ml/kg body weight (80 mg/kg body weight) for 28 days, showed
elevated ALAT and ornithine carbamyl transferase activities, whereas
no effects were observed on ASAT and alkaline phosphatase. Furthermore
there were changes in the livers of all dogs characterized
histologically by fatty vacuolation of centrilobular cells. In 3 of
the 12 dogs, the vacuolation also occurred mid-zonally and
periportally. There was some evidence of individual cell necrosis, and
in some cases the sinusoids were mildly congested. No changes in
plasma enzyme activities and no histological changes in the liver were
observed when three females received a daily dose of 0.02 ml/kg body
weight (32 mg/kg body weight) of carbon tetrachloride for 8 weeks.
(Litchfield & Gartland, 1974).
7.2.2 Inhalation exposure
a) Mice
As reported in a translated, extensive summary, BDF1 mice
(10/sex/group) were exposed (whole-body) to atmospheres of 0, 10, 30,
90, 270 or 810 ppm carbon tetrachloride (0, 64, 192, 577, 1731 or 5192
mg/m3, respectively) for 6 h a day, 5 days a week for 13 weeks. Mice
were observed daily for clinical signs, behavioural changes and
mortality and were weighed each week. Urinalysis, haematology, blood
chemistry and microscopy were performed at the scheduled end of the
experiment. No compound-related deaths occurred. Body weight gain was
depressed in males at 30 ppm or more. Slight, but statistically
significant, changes in haematology were observed in males at 810 ppm
(decreased Hb and increased MPV) and in females at the two highest
dose levels (decreased Hb, Ht and RBC). Increased liver enzymes in
blood were observed in both sexes at the three highest dose levels.
Urinalysis showed a decrease in pH at the highest dose level in
females only. Microscopic examination showed slight to moderate
dose-related changes in the liver including cytological alterations,
even at the lowest dose level in males. At higher dose levels there
were more severe changes described as collapse, deposit of ceroid,
proliferative ducts, increase in mitosis, pleomorphism and foci (Japan
Bioassay Research Centre, 1998). A NOAEL cannot be established on the
basis of these results.
b) Rats
Exposure (whole body) of Sprague-Dawley rats (IFFA CREDO; 8
males/group) to a carbon tetrachloride atmosphere of 3308 mg/m3 (516
ppm) for 6 h a day for 2 or 4 consecutive days resulted in increased
serum activities of glutamate dehydrogenase, ASAT, ALAT and SDH after
4 days exposure. After 2 days only the SDH level was significantly
increased (Brondeau et al., 1983).
When male hooded rats were exposed (whole body) 8 h a day for 12
days to carbon tetrachloride levels of 68 or 680 ppm (436 or 4360
mg/m3) increased ASAT levels were found at low- and high-dose levels
after 4 and 2 days, respectively. At both doses the level of liver
lipids reached a maximum after 8 days, the level being related to the
dose. Additional exposure resulted in a decrease (Kanics & Rubinstein,
1968).
An increased liver triglyceride content was also reported by
Shimizu et al. (1973) who exposed (whole body) female Sprague-Dawley
rats to 10, 50 and 100 ppm of carbon tetrachloride vapour (64, 320 and
641 mg/m3) for 3 h a day for 6 to 8 weeks. Exposures to 320 and 641
mg/m3 resulted in striking increases in the hepatic trigly cerides to
a maximum during the first 3 weeks. Afterward this level was nearly
maintained in both groups. At 64 mg/m3 the rise in triglycerides was
minimal and was maintained for 2 weeks.
David et al. (1981) compared the serum enzyme activities and
liver lesions in rats exposed to various concentration-time
combinations. After four exposures to 50 ppm (320 mg/m3) for 6 h per
day, the enzyme activities were significantly increased by 50 to 70%,
and steatosis and hydropic changes were found in the liver. The
changes were significantly more intensive in rats exposed to 250 ppm
(1600 mg/m3) for 72 min per day, not withstanding that the
concentration-time product was equal. The same was true for two-fold
concentrations and 18 exposures.
Bogers et al. (1987) performed 4-week studies in male Wistar rats
by exposing (whole body) them to 63 or 80 ppm (404 or 513 mg/m3)
carbon tetrachloride in three different concentration profiles: 1)
continuous exposure of 6 h/day for 5 days/week; 2) exposure of 2 × 3
h/day (1.5 h interruption) for 5 days/week; and 3) peak loads of 382
ppm (2450 mg/m3) for 5 min (4 peaks for 3 h) with and without the
1.5-h interruption. The interruption of the daily 6-h exposures did
not result in less severe but rather in slightly more severe
hepatotoxic effects, such as changes in enzyme levels, fat
accumulation, increased relative liver weight, lower microsomal
protein content and hydropic degeneration of liver cells. Peak loads
did not affect the severity of the hepatotoxic effects.
Plummer et al. (1990) exposed (whole body) male black-hooded
Wistar rats (36/group) for 4 weeks to carbon tetrachloride both
continuously (24 h per day, 7 days per week) to 32 ppm (205 mg/m3) or
intermittently (6 h per day, 5 days per week) to 176 ppm (1128
mg/m3). The concentration-time products were similar for both groups.
The-carbon tetrachloride-induced hepatotoxicity appeared to be similar
in the two exposure profiles. However, when rats received the
enzyme-inducing agents phenobarbitone or 1,3-butanediol during the
study via their drinking-water, the liver injury appeared to be
exacerbated in 1,3-butanediol-treated rats, especially in the
intermittent exposure profile.
Groups of male Sprague Dawley rats were exposed (whole body) to
100 ppm (641 mg/m3) of [14C]carbon tetrachloride for either 8 or
11.5 h/day for periods of 1 to 10 days, and examined with and without
recovery in a tissue distribution study (see section 6.1.3 and 6.1.4).
The only significant difference between rats exposed to the two
different schedules was the serum SDH activity, which was almost
always significantly greater for rats exposed to the 11.5 h/day
schedule than for the comparable groups exposed to the 8 h/day
schedule, except when measured after a recovery period. (Paustenbach
et al., 1986b).
F-344 rats (10/sex/group) were exposed (whole-body) in a 13-week
inhalation study to 10, 30, 90, 270 or 810 ppm carbon tetrachloride
(64.1, 192.3, 576.9, 1730.7 or 5192.1 mg/m3, respectively) for 6 h a
day, 5 days a week. Control groups were included. Animals were
observed for clinical signs, behavioural changes and mortality once a
day, and they were weighed once a week. Urinalysis was performed at
the end of the dosing period, and haematology, blood biochemistry and
microscopy were performed at the scheduled sacrifice. At 810 ppm the
body weight gain was depressed in both sexes. Haematological changes
were observed at 90 ppm or more in both sexes, and at 30 ppm in
females. Increased liver enzymes in blood and urinalysis changes were
observed in males at 270 ppm or more and in females at 90 ppm or more.
Increased creatine phosphokinase (CPK) was seen at 30 ppm in females.
Microscopic examination showed slight to marked changes in the liver
described as fatty change, cytological alterations, deposition of
ceroid, proliferative ducts, increase in mitosis, pleomorphism,
cirrhosis and foci. Furthermore, vacuolic change of tubule, hyaline
degeneration of glomerulus and protein cast of the kidney were noted
at the two highest dose levels (Japan Bioassay Research Centre, 1998).
An NOAEL could not be established on the basis of these results.
c) Comparisons between species
Adams et al. (1952) exposed (whole body) rats, guinea-pigs,
albino rabbits and rhesus monkeys to various concentrations of carbon
tetrachloride in air. Histological data were not reported for the
control groups; these groups were, however, used for comparison with
dose groups. Dose-related effects were seen in Wistar rats
(15/sex/group) after exposure to carbon tetrachloride at
concentrations of 5, 10, 25, 50, 100, 200 and 400 ppm (32, 63, 160,
320, 630, 1282 and 2520 mg/m3) for 7 h a day, 5 days a week, during
approximately 5.5-6.5 months. Increased liver weight, increased liver
fat content (especially neutral fat and esterified cholesterol) and
fatty degeneration of the liver were observed after 2 to 3 weeks of
exposure to concentrations of 63 mg/m3 or more. Cirrhotic livers were
found from 630 mg/m3 upwards. At a concentration of 320 mg/m3,
kidney tubular epithelium was affected and death rate seemed to be
increased, especially in the males. At the two highest exposure levels
testicular weights were decreased. At the 32 mg/m3 level, no adverse
effects were seen. After exposure for 5 days a week during 13 weeks to
2520 mg/m3 for 3 min a day or to 630 mg/m3 for 18 min a day, no
effects could be observed. Similar results were obtained in
guinea-pigs from 63 mg/m3 upwards. Rabbits developed slight to
moderate fatty degeneration and cirrhosis of the liver at 160 mg/m3
or more; there was no reported effect at 63 mg/m3. Rhesus monkeys
developed slight-to-moderate fatty degeneration of the liver 630
mg/m3 (2 monkeys); there was no reported effect at 320 mg/m3 (2
monkeys).
Prendergast et al. (1967) studied the effects of continuous and
repeated exposure to carbon tetrachloride vapour in rats, guinea-pigs,
New Zealand rabbits, beagle dogs and squirrel monkeys (see Table 8).
After repeated exposure to 515 mg/m3, all species showed pulmonary
interstitial fibrosis or pneumonitis. Mottled livers were seen in all
species except in the dog. Histological examination revealed fatty
changes in the livers of all species. In addition, fibrosis, bile duct
proliferation, hepatocyte degeneration and regeneration, focal
inflammatory infiltration and portal cirrhosis were observed in the
guinea-pigs. After continuous exposure to 61 mg/m3 all species showed
growth retardation and all squirrel monkeys showed alopecia and
emaciation. Histopathological examination showed liver changes similar
to those reported after repeated exposures. After continuous exposure
to 6.1 mg/m3 carbon tetrachloride in 61 mg/m3 n-octane (as a
carrier) no visible signs of toxicity were noted in any of the species
and no animals died. At termination, all species except the rat showed
less body weight gain than the controls. Histopathological examination
revealed non-specific inflammatory changes in the lungs of all species
and in the liver, kidney and heart of several animals, but no specific
pathological changes attributable to the exposure were noted.
7.2.3 Intraperitoneal exposure
Biochemical and morphological characterization of
carbon-tetrachloride-induced lung fibrosis were investigated in rats
after intraperitoneal administration of 1.0 ml/kg body weight (1600
mg/kg in paraffin oil) twice a week for 2 or 5 weeks, and examined
after the end of the exposure. The third group (5 rats) was treated
for 2 weeks and examined after 3 weeks of recovery. Acute haemorrhagic
interstitial pneumonia resulted from the 2 week exposure, while
chronic interstitial pneumonia was observed in rats exposed for 5
weeks and in the third group after 3 week of recovery (Pääkkö et al.,
1996).
7.3 Long-term exposure
In a carcinogenicity study described in section 7.7 (Reuber &
Glover, 1970), young male rats were administered 1.3 ml/kg by
subcutaneous injection twice a week. Severe cirrhosis was observed in
all (16/16) Sprague-Dawley rats by 5 to 16 weeks (the time of death of
the animals) and in 13/17 Black rats by 7 to 18 weeks. In Wistar rats,
6/12 rats developed moderate and 6/12 severe cirrhosis by 17-68 weeks,
while the cirrhosis was mild in 2/13, moderate in 7 and severe in 4
Osborne Mendel rats by 10-105 weeks; in Japanese rats, the cirrhosis
was mild in 9/15, moderate in 5 and severe in one rat by 8 to 78
weeks.
Alumot et al. (1976) exposed rats (strain unknown) to carbon
tetrachloride in feed at measured levels of 0, 80 and 200 mg/kg feed
for 2 years. The highest concentration corresponded to a daily dose of
10 to 18 mg/kg body weight. Because of chronic respiratory disease in
all animals beginning at 14 months, which resulted in increased
mortality, the results reported upon necropsy at 2 years were
inadequate for a health risk evaluation.
Muños Torres et al. (1988) administered carbon tetrachloride to
female Wistar rats (150 g initial body weight) as weekly
intraperitoneal injections at a dose of 0.2 ml in mineral oil for 46
weeks. The hepatic lesions were macroscopically and microscopically
evaluated after 8, 16, 22, 30 and 46 injections. After 8 injections,
changes in the hepatic architecture due to an increase in the
collagenous component accompanied by formation of fibrous bridges were
seen. After 46 injections a clearly established cirrhosis with nodules
of different sizes was seen.
Table 8. Mortality in animals exposed to carbon tetrachloride
(from Prendergast at al., 1967).
Concentration Type of Ratc Guinea-pig Rabbit Dog Monkey
(mg/m3) studyb (Hartley) (New Zealand) (Beagle) (Squirrel)
515 R 0/15 3/15 0/3 0/2 1/3
61 C 0/15 3/15 0/2 0/2 0/3
6.1a C 0/15 0/15 0/3 0/2 0/3
a in 61 mg/m3 n-octane (as a carrier)
b R = 30 exposures, 8 h/day, 5 days/week for 6 weeks; C = continuous 90-day exposure.
c Long-Evans or Sprague-Dawley rats
BDF1 mice (50/sex/group) were exposed (whole-body) in a 2-year
inhalation study to 0, 5, 25 or 125 ppm carbon tetrachloride (0,
32.05, 160.25 or 801.25 mg/m3) for 6 h a day, 5 days a week. Animals
were observed for clinical signs, behavioural changes and mortality
once a day, and they were weighed once a week for the first 13 weeks
and every 4 weeks thereafter. Urinalysis was performed at the end of
the dosing period, and haematology, blood biochemistry and microscopy
were performed at the scheduled sacrifice. No compound-related effects
were observed at 5 ppm in female mice. The results from male mice
could not be evaluated due to anomalous control group liver enzyme
data. A significant decrease in survival was observed at 25 and 125
ppm. Liver tumours were the main cause of death at the highest dose
level. Body weight gain was depressed at 25 and 125 ppm. Changes in
haematology, blood biochemistry including liver enzymes, and
urinalysis were observed at 25 ppm or more. Microscopic examination
showed changes of the liver (deposit of ceroid, cyst and
degeneration), the kidney (protein cast) and the spleen (increased
deposit of haemosiderin at 25 ppm and increased extramedullary
haematopoiesis at 125 ppm) at the two highest dose levels in male
mice. In female mice changes in the liver included deposit of ceroid,
thrombus, necrosis, degeneration and cyst at 25 and 125 ppm. At 25 ppm
an increased deposit of haemosiderin of the spleen was observed while
at 125 ppm deposit of ceroid of the ovary was seen (Japan Bioassay
Research Centre, 1998). A NOAEL could not be established on the basis
of these results.
F-344 rats (50/sex/group) were exposed (whole-body) in a 2-year
inhalation study to 0, 5, 25 or 125 ppm carbon tetrachloride (0,
32.05, 160.25 or 801.25 mg/m3, respectively) for 6 h a day, 5 days a
week. Animals were observed for clinical signs, behavioural changes
and mortality once a day. They were weighed once a week for the first
13 weeks and every 4 weeks thereafter. Urinalysis was performed at the
end of the dosing period, and haematology, blood biochemistry and
microscopy were performed at the scheduled sacrifice. A significant
decrease in survival was observed at 125 ppm, with liver tumours
and/or chronic nephropathy being the main cause of death. Body weight
gain was depressed at 25 and 125 ppm. Changes in haematology, blood
biochemistry including liver enzymes, and urinalysis were observed at
25 ppm and even at 5 ppm for the nitrate and protein level in the
urine of the rats. At the 125 ppm level, only one male and three
female rats survived, so no statistical test was performed.
Microscopic examination showed changes of the liver (including fatty
change, deposit of ceroid, fibrosis, granulation and cirrhosis) at the
two highest dose levels in both sexes. An increased deposit of
haemosiderin in the spleen was observed in males at all dose levels.
An eosinophilic change of the nasal cavity was observed in females at
all dose levels and in males at 25 and 125 ppm. A chronic nephropathy
(progressive glomerulonephrosis) developed in females at 25 ppm and in
both sexes at 125 ppm. At 125 ppm deposit of ceroid and granulation of
the lymph node were observed in both sexes (Japan Bioassay Research
Centre, 1998). A NOAEL could not be established on the basis of these
results.
7.4 Irritation
7.4.1 Skin irritation
Epicutaneous administration of 1 ml of carbon tetrachloride has
been demonstrated to induce degenerative changes in the epidermis 15
min to 16 h after application (Kronevi et al., 1979; see section
7.1.2.4).
Moderate dermal irritation was observed after application of 0.5
ml carbon tetrachloride (under occlusion) onto the shaven skin of
rabbits (only abraded skin) and male Hartley guinea-pigs (normal and
abraded skin) (Roudabush et al., 1965).
In a study conducted according to Draize protocol, 0.5 ml carbon
tetrachloride was applied under an occlusive dressing for 24 h to the
intact and abraded skin of rabbits. Irritation was assessed at 24 and
72 h. Carbon tetrachloride was classified as a "medium" skin irritant.
Histopathology of skin samples taken from the application site on day
3 after exposure confirmed the irritant reaction (Duprat et al.,
1976).
Undiluted carbon tetrachloride (10 µl) was applied to the open
skin of guinea-pigs 3 times daily for 3 days. A skin reaction (no
further details provided) was observed on day 2 and an average score
described as "redness" was seen on day 4 (Anderson et al., 1988).
Wahlberg (1984a) rubbed 0.1 ml (159 mg) of carbon tetrachloride
into the skin of rabbits and guinea-pigs for ten consecutive days and
observed oedema and erythema.
7.4.2 Eye irritation
In a study conducted according to Draize protocol, 0.1 ml of
carbon tetrachloride caused a mild irritant response in rabbits. The
response was evident at 24, 48 and 72 h after exposure and recovery
was complete by day 14 (Duprat et al., 1976).
7.5 Toxicity to the reproductive system, embryotoxicity, teratogenicity
7.5.1 Reproduction
Groups of six male rats received a single intraperitoneal
injection of coconut oil or carbon tetrachloride in coconut oil (1:1
mixture) as 3 ml/kg rat weight (2378 mg/kg body weight) After 15 days,
a significant increase in the weight of the pituitary and a decrease
in the weights of the testes and seminal vesicles were observed.
Histological examination showed testicular atrophy and some
abnormality in the process of spermatogenesis in the experimental
animals (Chatterjee, 1966).
In a study of similar design with female rats, effects on the
reproductive system were seen 10 days after dosing. The effects
reported were: inhibition of estrous rhythm, reduction in ovarian and
uterine weights and vascularization, an increase in adrenal weight and
a marked reduction in pituitary gonadotrophin potency (Chatterjee
(1968).
Kalla & Bansal (1975) injected male rats with a mixture of 3
ml/kg body weight carbon tetrachloride in coconut oil (1:1, v/v) (2378
mg carbon tetrachloride/kg body weight) through an intraperitoneal
route for 10, 15 or 20 consecutive days. All dosing periods resulted
in decreased weights of testicles and accessory sex organs and
impairments in spermatogenesis. Dosing for 20 days resulted in an
entire deterioration of testicular tissue accompanied by an absence of
spermatids. The study was not reported adequately; number and strain
of rats were not reported.
7.5.2 Embryotoxicity and teratogenicity
The available data suggest that the fetus is not preferentially
sensitive to carbon tetrachloride, and effects of carbon tetrachloride
on fetal development and post-natal survival are likely to be
secondary to maternal toxicity.
7.5.2.1 Oral exposure
When carbon tetrachloride was administered by gavage to F-344
rats on gestation days 6-15 at 0, 25, 50 and 75 mg/kg per day in
either corn oil or in an aqueous vehicle containing 10% Emulphor(R),
it was more maternally toxic when administered in corn oil,
particularly at the highest dose. Full litter resorption (FLR)
occurred at 50 and 75 mg/kg with both vehicles. At 75 mg/kg, dams
receiving carbon tetrachloride in corn oil had a significantly higher
rate of FLR (67%) than those given the aqueous vehicle counterpart
(8%) (Narotsky et al., 1997a). Ammonium sulfide staining was used to
detect the resorption sites (Narotsky et al., 1997b).
Thiersch (1971) dosed pregnant rats with carbon tetrachloride (in
corn oil) at a level of 1000 mg/kg body weight on days 7, 7 and 8, 11,
or 11 and 12 of gestation. No malformations in the offspring were
reported, but the litters of the dams that had received two doses
showed more resorptions than the litters of those receiving one dose.
Clear information on a control group was not provided.
Hamlin et al. (1993) examined the effect on B6D2F1 mice of oral
administration of carbon tetrachloride (in corn oil) at concentrations
of 82.6 or 826.3 mg/kg body weight for five consecutive days beginning
on day 1, 6 or 11 of gestation. No effects were seen on maternal or
various neonatal parameters such as weight and crown rump length. No
malformations were detected in any pup on day 1 post-partum and the
pups developed normally.
7.5.2.2 Inhalation exposure
When Schwetz et al. (1974) exposed pregnant Sprague-Dawley rats
to measured carbon tetrachloride concentrations of 334 or 1004 ppm
(214 or 6435 mg/m3) for 7 h a day on days 6 to 15 of gestation, the
dams showed a dose-related decrease in food consumption (and body
weight gain). Signs of hepatotoxicity (increased ALAT activity) were
observed at both dose levels, but were not dose-related. Fetal body
weight and crown-rump length were significantly decreased. No
anomalies were seen upon gross examination of the fetuses. In both
exposure groups the incidence of fetuses with subcutaneous oedema was
increased but was statistically significant only in the lower dose
group. The incidence of sternebral anomalies (bipartite and delayed
ossification) was significantly increased in the fetuses of rats
exposed to the higher dose.
In an inhalation study by Gilman (1971), exposure of pregnant
rats to carbon tetrachloride at 1575 mg/m3 for 8 h a day on days 10
to 15 of gestation decreased the lactation index (83% compared to 98%
in the controls) and the viability index (83% as compared to 99% in
the controls).
7.6 Mutagenicity
The data from genotoxicity assays conducted with carbon
tetrachloride are summarized in Table 9. Since carbon tetrachloride is
a volatile compound that partitions preferentially in the hydrophobic
phase, the conditions adopted for in vitro experiments are important
to the outcome, but these conditions are often not reported in
sufficient detail. Carbon tetrachloride was not mutagenic to
Salmonella typhimurium in a large number of studies. It did,
however, induce DNA damage and mutations in single studies with
Escherichia coli. In fungi it induced intrachromosomal and mitotic
recombination. However, it did not induce aneuploidy in one study on
the yeast Saccharomyces cerevisiae, although aneuploidy was induced
in another single study with Aspergillus nidulans. In the only study
with Drosophila melanogaster, sex-linked recessive lethal mutations
were not induced by carbon tetrachloride.
In mammalian in vitro assays, carbon tetrachloride induced cell
transformation in a single study with Syrian hamster cells and
centromere-positive-staining micronuclei in human cell lines
expressing cDNAs for CYP1A2, CYP2A6, CYP3A4, epoxide hydrolase or
CYP2E1. The AHH-1 cell line constitutively expressing CYP1A1 showed no
increase in either total micronucleus frequency or centromere-staining
micronucleus frequency. There is little evidence for the induction
in vitro of DNA damage, unscheduled DNA synthesis, sister-chromatid
exchange or chromosomal aberrations.
In mammalian in vivo tests, carbon tetrachloride did induce DNA
strand breakage in one study but not in four others and did not
induce: a) unscheduled DNA synthesis in rat hepatocytes; b)
micronuclei in mouse hepatocytes, bone marrow cells or peripheral
blood erythrocytes; c) chromosomal aberrations in mouse bone marrow;
or d) aneuploidy in mouse hepatocytes. Binding of carbon tetra
chloride to liver cell DNA has been observed in rats, mice and Syrian
hamsters treated in vivo. There has been a report of a reduction in
I-compounds (species- and tissue-specific DNA adducts) in mouse liver.
The only clear evidence for genotoxicity comes from a number of
fungal cell experiments involving mutation and recombinational events.
Effects in mammalian cells indicate damage during cytokinesis. This
type of damage could result from interactions with proteins, rather
than DNA, e.g., of the trichloromethyl radical, and could be induced
secondarily to the toxicity of carbon tetrachloride (McGregor & Lang,
1996). Thus, no carbon-tetrachloride-DNA adduct identification has
been made, while the polar adducts observed in Syrian hamster liver
DNA appear to be derived from lipid peroxidation products (Wang &
Liehr, 1995). Consequently, strand-breakage and aneuploidy could arise
from the effects of lipid peroxidation products rather than carbon
tetrachloride or its metabolites; linoleic acid hydroperoxide, for
example, can induce single-strand breaks in DNA of cultured
fibroblasts (Nakayama et al., 1986). No resolved DNA damage has been
observed in vivo. It is concluded that although carbon tetrachloride
has some effects upon genetic material and these could be due to a
direct effect of carbon tetrachloride, no supporting evidence is
available; the effects are explicable in terms of nuclear protein or
DNA damage induced secondarily to carbon tetrachloride toxicity.
7.7 Carcinogenicity
7.7.1 Mice
After administration of 0.1 ml of a 40% solution of carbon
tetrachloride in olive oil (64 mg/mouse) by stomach tube to male C3H
mice, female C mice, and male and female A and Y mice 2 or 3 times a
week for 8 to 16 weeks (23 to 58 treatments), hepatomas developed in
126/143 (88%), 34/41 (83%), 63/64 (98%) and 9/15 (60%) of the C3H, C,
A and Y mice, respectively. No concurrent control data were reported.
Historical control data indicated the following incidence of hepatomas
(%) at one year or more: male C3H, 27%; female C, 0%; male and female
A, 1.5%; male and female Y, 1.6% (Edwards, 1941; Edwards & Dalton,
1942). In 34 of 73 male and female mice of the L strain that received
0.04 ml carbon tetrachloride per treatment by stomach tube 2 or 3
times a week for 46 treatments and were killed 3 to 3.5 months
following treatment, hepatomas were found that were similar to those
found in the other strains (Edwards et al., 1942).
Table 9. Mutagenicity studies with carbon tetrachloride
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
Bacterial systems
S. typhimurium TA 100 base-pair substitution not reported + rat PCB - McCann et al.,
TA 1535 1975
S. typhimurium G 46 base-pair substitution not reported + mouse i.n.r. - Kraemer et al.,
1974
S. typhimurium TA 1950 base-pair substitution concentrations: unknown - Braun &
G 46 10, 20 and 40 mg/ml Schöneich, 1975
S. typhimurium TA 1535 base-pair substitution test performed in + rabbit i.n.r. - Uehleke et al., 1977
TA 1538 frameshift mutation desiccators i.m.
S. typhimurium TA 100 base-pair substitution test performed in + rabbit PCB - Simmon et al., 1977
TA 1535 desiccators
TA 98 frameshift mutation
TA 1537
TA 1538
S. typhimurium TA 1535 base-pair substitution concentration: 8 mM, + mouse PB - Uehleke et al., 1977
TA 1538 frameshift mutation incubation in closed i.m.
containers
(survival > 90%)
S. typhimurium TA 100 base-pair substitution test performed in -/+ i.n.r. - Simmon & Tardiff,
TA 1535 desiccators sp. n.r. 1978
S. typhimurium TA 100 base-pair substitution concentrations: 4, 5.7, - - Barber et al.,
TA 1535 7, 10.2, 12.3, 18.4 1981
TA 98 frameshift mutation µmoles/plate; test for + rat PCB -
TA 1537 volatile liquids
TA 1538
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
S. typhimurium TA1535/pSK1002 SOS induction open + - Nakamura et al.,
1987
S. typhimurium TA1535/pSK 1002 SOS induction open + - Brams et al., 1987
S. typhimurium TA1535/pSK 1002 forward mutation open + - Roldàn-Arjona
et al., 1991
S. typhimurium TA1535/pSK 1002 forward mutation open + - Roldàn-Arjona &
Pueyo, 1993
S. typhimurium TA100 reverse mutation open + - Zeiger et al., 1988
TA1535
TA1537
TA97
TA98
S. typhimurium TA100 reverse mutation open + - Brams et al., 1987
TA98
TA97
S. typhimurium TA 100 base-pair substitution concentration: below - - De Flora, 1981;
TA 1535 toxicity limit De Flora et al.,
TA 98 frameshift mutation 1984
TA 1537
TA 1538
E. coli K 12 gene mutations not reported + mouse i.n.r. - Kraemer et al., 1974
i.m.
E. coli K 12 gene mutations not reported + rabbit i.n.r. - Uehleke et al., 1976
i.m.
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
E. coli uvrA back-mutation to trp concentration: + mouse i.n.r. - Norpoth et al., 1980
0.01% v/v
E. coli WP2 uvrA reverse mutation 2.5% atmosphere + (+) Norpoth et al., 1980
Non-mammalian eukaryotic systems
A. Nidulans unknown somatic segregation spot test technique n.r. - Bignami, 1977
(crossing-over and
non-disjunction)
A. Nidulans unknown induction of 8-aza- spot test technique n.r. - Bignami, 1977
guanine resistance
A. Nidulans 35 (haploid) forward mutation concentration: n.r. (+) Gualandi, 1984
1 (diploid) somatic segregation 0.5% v/v n.r. +
A. Nidulans aneuploidy - + Benigni et al., 1993
S. cerevisiae D7 gene conversion at concentration: 21, 28, - + Callen et al., 1980
trps-locus; mitotic 34 mM; test performed
recombination at ade-2; in screw-capped glass
gene conversion at ilv-1 tubes
S. cerevisiae AGY31DEL intrachromosomal - + Schiestl et al.,
recombination 1989
S. cerevisiae AGY31DEL intrachromosomal - + Galli & Schiestl,
recombination 1995
S. cerevisiae D61.M mitotic chromosome - - Whittaker et al.,
loss 1989
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
S. cerevisiae mitotic recombination - + Galli & Schiestl,
1996
Drosophila SLRL mutation feeding - Foureman et al.,
melanogaster 1994
Drosophila SLRL mutation injection - Foureman et al.,
melanogaster 1994
Chinese hamster V79 aneuploidy 2500 µg/ml - + Önfelt, 1987
lung
Cell line AHH1 (expressing aneuploidy 10 mM - - Doherty et al., 1996
CYP1A1) (centromere staining)
Cell line MCL-5 (cDNAs for aneuploidy 2 mM - + Doherty et al., 1996
CYP1A2, CYP2A6, (centromere staining)
CYP3A4, CYP2E1
and epoxide
hydrolase)
Cell line h2E1 (cDNAs for aneuploidy 2 mM - + Doherty et al., 1996
CYP2E1) (centromere staining)
Cell line AHH1 (expressing micronucleus 10 mM - - Doherty et al., 1996
CYP1A1)
Cell line MCL-5 (cDNAs for micronucleus 2 mM - + Doherty et al., 1996
CYP1A2, CYP2A6,
CYP3A4, CYP2E1
and epoxide
hydrolase
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
Cell line h2E1 (cDNAs for micronucleus 2 mM - + Doherty et al., 1996
CYP2E1)
In vitro mammalian systems
Chinese hamster ovary cells anaphase analysis concentration: 5 µl/ml - (+) Coutino, 1979
for chromosomal
rearrangements
Chinese hamster ovary cells chromosomal highest concentration: + rat PCB - Loveday et al., 1990
aberrations + SCE 3000 µg/ml
- -
Rat liver epithelium metaphase analysis concentration: 0.005, cells - Dean &
for chromosomal 0.01, 0.02 µl/ml in posses Hodson-Walker,
abnormalities sealed flasks; toxicity intrinsic 1979
observed activity
Human lymphocyte chromosomal concentration: + sp.n.r. i.n.r - Garry et al., 1990
aberrations + SCE 3.8-76 µg/ml
Host-mediated assays
S. typhimurium TA 1950 base-pair substitution mice NMRI; - Braun & Schöneich,
subcutaneous 1975
4 ml/kg body weight
S. typhimurium TA 1950 base-pair substitution mice CBA*C57Bl/6; - Shapiro & Fonshtein,
subcutaneous 20% 1979
solution in sunflower
oil
Table 9. (Continued)
Speciesa Strain Measured end-pointb Test conditions Activationc Inductiond Resultse Reference
Syrian hamster cell transformation 3 µg/ml - (+) Amacher & Zelljadt,
embryo cells (clonal assay) 1983
Rat hepatocytes UDS 100 mg/kg × 1 p.o. or - Doolittle et al.,
in vivo 14 p.o. 1987
Rat hepatocytes SCE and chromosomal 1600 mg/kg × 1 p.o. - Sawada et al., 1991
in vivo aberrations and
micronucleus
Mouse bone chromosomal 8000 mg/kg × 1 - Lil'p, 1983
marrow in vivo aberrations
Mouse bone marrow micronucleus 2000 mg/kg and - Suzuki et al., 1997
and peripheral 3000 mg/kg
erythrocytes
in vivo
Mouse liver binding to DNA + Diaz-Gomez & Castro,
in vitro 1980a
Mouse, rat, binding to DNA + Castro et al., 1989
Syrian hamster
liver in vivo
Mouse liver ICR I-compound reduction 1600 mg/kg × 1 p.o. + Nath et al., 1990
in vivo (32P-post-labelling)
Syrian hamster binding to DNA + Wang & Liehr, 1995
liver and kidney
in vivo
a S. typhimurium = Salmonella typhimurium; A. Nidulans = Aspergillus nidulans; S. cerevisiae = Saccharomyces cerevisiae
b SCE = sister chromatid exchange; UDS = unscheduled DNA synthesis
c + = with metabolic activation; - = without metabolic activation; sp.n.r. = species not reported; n.r. = not reported whether metabolic
activation was used
d i.n.r. = inducer not reported; i.m. = intact microsomes added; PCB = polychlorinated biphenyls; PB = phenobarbital
e - = negative; + = positive; (+) = weakly positive
Table 10. Indicator tests with carbon tetrachloride
Species Strain Measured end-point Test conditions Activationa Inducationb Resultsc Reference
Bacterial systems
Escherichia coli WP2 (repair DNA repair variation 1: liquid - + De Flora et al.,
proficient WP67 micro method 1984
(uvrA polA-)
CM871 (uvrAa, + rat PCB +
recA-, lexA-)
variation 2: 24 h - +
preincubation and
plating out + rat PCB -
In vitro mammalian cells
Chinese hamster ovary cells SCE concentration: 0.001, - +d Athanasiou &
0.1, 1 mM; solvent Kyrtopoulos, 1981
DMSO
Chinese hamster ovary cells SCE -S9: toxic at 1490 + rat PCB - Loveday et al.,
µg/ml; +S9: 2930 1990
µg/ml highest dose
tested
Human lymphocyte SCE concentration: + sp.n.r. i.n.r. - Garry et al.,
3.8-76 µg/ml - - 1990
Human lymphocyte UDS concentration: 2.5, - PB - Perocco &
5 µg/ml; solvent + rat - Prodi, 1981
DMSO
Rat hepatocyte DNA single strand concentration: 0.03, - + Sina et al., 1983
breaks 0.3, 3 mM
Table 10. (Continued)
Species Strain Measured end-point Test conditions Activationa Inducationb Resultsc Reference
In vivo mammalian systems
Mouse NMRI DNA single strand 2.5 mg/kg body - Schwartz et al.,
breaks weight; single oral 1979
dose, undiluted or in
corn oil
Mouse CBA*BALB/c sperm head 0.1, 0.25, 0.5, 1.0, - Topham, 1980
abnormalities 1.5 mg/kg body
weight (i.p.) for 5
days; solvent corn oil
Mouse CDI DNA damage in 3H- 0.02-0.1 ml/kg body + Gans & Korson,
1984 labelled liver weight; single oral
cells dose in corn oil
Rat Wistar UDS in liver cells 4000 mg/kg body ee Craddock &
weight; single oral Henderson, 1978
dose, 2 and 17 h
exposure
Rat Fischer-344 UDS in liver cells 10 or 100 mg/kg - Mirsalis &
body weight; single Butterworth, 1980
oral dose in corn oil,
2 h exposure
Rat Wistar DNA damage single oral dose of - Stewart, 1981
(hepatectomized) 200-800 mg/kg body
weight in corn oil,
3 weeks after
hepatectomy
Table 10. (Continued)
Species Strain Measured end-point Test conditions Activationa Inducationb Resultsc Reference
Rat Fischer-344 DNA single strand 400 mg/kg body - Bermudez et al.,
breaks weight; single oral 1982
dose in corn oil
Rat Fischer-344 UDS in liver cells 40 or 400 mg/kg - Mirsalis et al.,
body weight; single 1982
oral dose up to 48 h
of exposure
Rat Sprague-Dawley DNA damage 200 mg/kg body - Brambilla et al.,
weight; single i.p. 1983
dose
a + = with metabolic activation; - = without metabolic activation; sp.n.r.= species not reported
b PCB = polychlorinated biphenyls; PB = phenobarbital; i.n.r.; = inducer not reported
c - = negative; + = positive; e = equivocal
d chromosome aberrations occurred, but no details on this finding were reported
e increase in DNA associated with tissue regeneration, but no increase in unscheduled DNA synthesis
Eschenbrenner & Miller (1944) administered 30 doses (each 0.005
ml) of 32%, 16%, 8%, 4% and 2% solutions of carbon tetrachloride in
olive oil (2540, 1270, 635, 318 or 160 mg/kg body weight) at 1- to
5-day intervals to mice of the A strain. Control group animals were
administered olive oil. All animals were examined for hepatomas 150
days after the first dose. Hepatomas were found in 33/60, 32/60,
25/60, 23/60 and 23/60 mice of the 32, 16, 8, 4 and 2% dose groups. A
variation in the numerical incidence of hepatomas at a given time was
observed to be related both to the total amount administered and to
the interval elapsing between successive doses. The incidence of
hepatomas increased with the interval between dosing from 1 to 4 days.
Weisburger (1977) reported that, in B6C3F1 mice dosed by gavage
with carbon tetrachloride in corn oil at levels of 1250 or 2500 mg/kg
body weight 5 days/week for 78 weeks and killed at 90 weeks, hepatomas
were found in 47/48 males and 43/45 females receiving the higher dose
and in 49/49 males and 40/40 females receiving the lower dose. Control
incidences of hepatomas were 3/18 in males and 1/18 in females. An
increase in adrenal tumours was also reported in the treated mice.
Groups of 50 male and 50 female BDF1 mice, 6 weeks of age, were
exposed by whole-body inhalation to 0, 5, 25 or 125 ppm (0, 32, 160 or
800 mg/m3) carbon tetrachloride (purity > 99.8%) for 6 h per day on
5 days a week for 104 weeks. Survival of the mid- and high-dose groups
in both sexes (35/50, 36/50, 25/50 and 1/50 males; 26/50, 24/49, 10/50
and 1/49 females) was decreased due to liver tumours. Significantly
increased incidences of hepatocellular adenomas (9/50, 10/50, 27/50
and 16/50 males; 2/50, 8/49, 17/50 and 5/49 females) were observed in
mid- and high-dose males (p < 0.01, Chi-square test) and in low- and
mid-dose females (low dose, p < 0.05; mid-dose, p < 0.01),
hepatocellular carcinomas (17/50, 12/50, 44/50 and 47/50 males; 2/50,
1/49, 33/50 and 48/49 females) in mid- and high-dose males and females
(p < 0.01) and pheochromocytomas of the adrenal gland (0/50, 0/50,
16/50 and 31/50 males; 0/50, 0/49,0/50 and 22/49 females) in mid- and
high-dose males (p < 0.01) and high-dose females (p < 0.01) (Nagano
et al., 1998).
7.7.2 Rats
In a study reported by Reuber & Glover (1970) male rats of the
Osborne-Mendel, Japanese, Wistar, Black and Sprague-Dawley strains
received twice a week a subcutaneous injection (1.3 ml/kg body weight)
of a 50% solution of carbon tetrachloride in corn oil (1036 mg/kg body
weight). The Japanese rats survived for 47 weeks and Osborne-Mendel
rats for 44 weeks. Wistar rats lived for 33 weeks and Black and
Sprague-Dawley rats lived only for 11 and 33 weeks, respectively.
Hepatocellular carcinomas developed in 8/13 Osborne-Mendel rats and in
12/15 Japanese rats, and hyperplastic nodules were also found in these
strains. The hepatocellular carcinomas found in 4/12 Wistar rats were
smaller. Nodules and hyperplasia were also observed in Wistar rats, as
well as moderate to severe cirrhosis. The Black rats and
Sprague-Dawley rats died with severe cirrhosis before they developed
carcinomas. No hepatocellular carcinomas developed in the controls of
any strain.
Weisburger (1977) reported increases in the incidence of
neoplastic nodules and hepatocellular carcinomas after oral
administration of carbon tetrachloride in corn oil to Osborne-Mendel
rats at doses of 47 and 94 mg/kg body weight (males) and 80 and 160
mg/kg body weight (females) for 78 weeks. The rats were killed at 110
weeks. The incidences of hepatocellular carcinomas in the controls
were 0/20 in males and 1/20 in females, and the incidences of liver
neoplastic nodules were 0/20 in males and 1/20 in females.
Groups of 50 male and 50 female F-344 rats, 6 weeks of age, were
exposed by whole-body inhalation to 0, 5, 25 or 125 ppm (0, 32, 160,
800 mg/m3) carbon tetrachloride (purity > 99.8%) for 6 h per day on
5 days a week for 104 weeks. Survival of the high-dose groups in both
sexes (22/50, 29/50, 19/50 and 3/50 males; 39/50, 43/50, 39/50 and
1/50 females) was decreased due to liver tumours and chronic
nephropathy (progressive glomerulonephrosis). There were significantly
increased incidences of hepatocellular adenomas (0/50, 1/50, 1/50 and
21/50 males; 0/50, 0/50, 0/50 and 40/50 females) and hepatocellular
carcinomas (1/50, 0/50, 0/50 and 32/50 males; 0/50, 0/50, 3/50 and
15/50 females) in high-dose rats of each sex (p < 0.01, Chi-square
test) (Nagano et al., 1998).
7.8 Special studies
7.8.1 Immunotoxicity
Carbon tetrachloride treatment of B6C3F1 female mice resulted in
marked suppression of both humoral and cell-mediated immune functions.
Humoral immunity, as measured by the T-dependent antibody response to
sheep red blood cells (SRBC), proved to be the most sensitive
indicator of carbon-tetrachloride-induced immunotoxicity. Carbon
tetrachloride was immunotoxic in female B6C3F1 mice at all doses
tested (500-5000 mg/kg body weight) and there were no significant
differences in the magnitude of immunosuppression between oral and
intraperitoneal routes of exposure. There was no dose-response
relationship with respect to SRBC antibody responses; all dose levels
resulted in approximately 50% suppression of the control response. To
determine whether a dose-response relationship could be attained,
female mice were treated at lower carbon tetrachloride levels (25, 50
and 100 mg/kg body weight) for 30 consecutive days. It appeared that
doses as low as 50 mg/kg body weight produced the maximum attainable
inhibition of SRBC response (50%); 25 mg/kg body weight caused a 20%
reduction (Kaminsky et al., 1989; 1990).
Delaney & Kaminski (1994) studied the immunomodulatory activity
of serum isolated from carbon tetrachloride-treated B6C3F1 mice on
T-cell-independent humoral immune responses. The results of the study
suggested that carbon tetrachloride has bifurcating immunological
effects. Exposure to carbon tetrachloride appears to suppress
T-cell-dependent immune responses but enhance the activity of B-cells.
Both effects appear to be mediated by blood-borne factors. Incubation
of sera from carbon-tetrachloride-treated mice with neutralizing
monoclonal antibodies toward transforming growth factor ß1 reversed
the immunosuppression, indicating that TGF ß1 at least in part
mediates the immunosuppression induced by carbon tetrachloride.
In contrast to the results found in mice by Kaminsky et al.
(1989, 1990), Smialowicz et al. (1991) found no consistent alterations
in humoral or cell-mediated immune function in male Fischer-344 rats
at dosages that clearly resulted in body weight decreases and
hepatotoxicity. However, the dose levels used in this study (up to 40
mg/kg body weight) were much lower than those reported by Kaminsky et
al. (1989, 1990).
Mice (A/PhJ) were administered carbon tetrachloride (about 300
mg/kg per day intraperitoneally in olive oil) for 2, 7, 14 and 23
days. A variety of immunological parameters were evaluated.
Morphological examination by light microscopy revealed significant
activation of lymphoid tissues in T-cell-dependent areas and only
slight activation in B-cell-dependent areas (Jirova et al., 1996).
Thymus weights (after exposure for 2, 14 and 23 days) and spleen
weights (after exposure for 2 and 23 days) decreased significantly
when weights at 23 days were compared to those of controls. The
response of SRBC was permanently suppressed from the beginning of
exposure.
7.8.2 Influence of oxygen levels
It is known that under normal atmospheric conditions carbon
tetrachloride initiates lipid peroxidation in mice and rat liver
microsomes and in mice and rats in vivo (Sagai & Tappel, 1979;
Kornbrust & Mavis, 1980; Gee et al., 1981; Lee et al., 1982). At
reduced oxygen concentrations the carbon-tetrachloride-induced lipid
peroxidation is greatly enhanced in in vivo experiments and in
microsomal preparations, but in these in vitro systems, the process
is entirely blocked under anoxic conditions (Kieczka & Kappus, 1980;
Noll & De Groot, 1984).
Covalent binding to RNA and DNA was enhanced in the absence of
oxygen when male rat (Sprague-Dawley) hepatocytes were treated with
carbon tetrachloride (Cunningham et al., 1981). DiRenzo et al. (1984)
observed that the enhanced covalent binding to protein or lipid,
caused by carbon tetrachloride in cultured male rat (Sprague-Dawley)
hepatocytes, at decreased oxygen tension was most evident for the
binding to lipids.
Shen et al. (1982) studied the effect of oxygen concentrations on
carbon-tetrachloride-induced hepatotoxicity in male Long-Evans rats
exposed to differing oxygen concentrations combined with different
carbon tetrachloride concentrations. In this in vivo experiment,
carbon tetrachloride appeared to be more toxic when oxygen
concentrations were reduced, as shown by increased ALAT activity, more
severe centrilobular necrosis, and increased covalent binding to
hepatic microsomal lipids and proteins.
An increased metabolism of carbon tetrachloride under hypoxic
conditions, and consequently an aggravated hepatotoxicity, was
reported in male Wistar rats by Siegers et al. (1985). In agreement
with the reports of Kieczka & Kappus (1980) and Noll & De Groot
(1984), a more pronounced lipid peroxidation was observed, as shown by
exhaled ethane, than in animals kept under normal oxygen conditions
and treated with the same dose of carbon tetrachloride.
Male Sprague-Dawley rats were used in a study by Burk et al.
(1988) to examine the effect of hyperbaric oxygen on carbon
tetrachloride metabolism by different isoenzymes of cytochrome P-450.
The authors concluded that under low oxygen tensions the rate of
carbon tetrachloride metabolism depended largely on the amount of
cytochrome P-450 present, while under higher oxygen tensions the major
determinant was the type of cytochrome P-450.
7.9 Factors modifying toxicity
7.9.1 Dosing vehicles
Several studies have demonstrated that
carbon-tetrachloride-induced hepatotoxicity, like absorption (see
section 6.1.1.1), can be influenced by the dosing vehicle.
In order to evaluate the effect of vehicle on the hepatotoxicity
in mice, Condie et al. (1986) treated male and female CD-1 mice with
oral doses of carbon tetrachloride (0, 1.2, 12 or 120 mg/kg body
weight) in either corn oil or 1% Tween-60 vehicle, 5 times/week for 90
days. Differences between the vehicles were observed in mice at the 12
mg/kg dose level. Hepatomegaly and more fat accumulation were observed
when carbon tetrachloride was administered in corn oil. At the highest
dose level the usage of corn oil as vehicle caused a greater
hepatotoxic effect, as shown by necrosis and fatty infiltration in the
mice. The data indicated that the NOAEL for hepatotoxic effects after
carbon tetrachloride exposure in corn oil was 1.2 mg/kg body weight,
while the NOAEL for the Tween-60 groups was 12 mg/kg body weight.
When pregnant F-344 rats were dosed by gavage with carbon
tetrachloride in corn oil or an aqueous vehicle containing 10%
Emulphor during gestation, corn oil was associated with a full litter
resorption (FLR) rate of 67%, compared to 8% in those dosed with
Emulphor (Narotsky et al., 1997a). Further details regarding this
study are described in section 7.5.2.1.
Koporec et al. (1995) determined the vehicle effects on the
subchronic toxicity of carbon tetrachloride in male Sprague-Dawley
rats. Carbon tetrachloride was administered at dose levels of 0, 25 or
100 mg/kg body weight by gavage in either corn oil or a 1% Emulphor
aqueous emulsion 5 days/week for 13 weeks. It was concluded that there
was no difference in the subchronic hepatotoxicity of carbon
tetrachloride in rats when given in corn oil or as an aqueous
emulsion. This result contrasts with the result found in mice
described by Condie et al. (1986), and with the result of the study in
male Sprague-Dawley rats by Kim et al. (1990b), who reported that the
hepatotoxicity in male Sprague-Dawley rats was less pronounced at each
dose level when corn oil was used as a vehicle. Administration of
undiluted carbon tetrachloride or of an aqueous emulsion produced
comparable toxicity.
Szende et al. (1994) dosed male F-344 rats with carbon
tetrachloride (0.2 ml/kg body weight) dissolved in various oils
(sunflower, corn, fish or olive oil) by gastric intubation 3
times/week for 8 weeks. The increase of collagen fibres in the liver
was only 2-4% when olive oil was used as a vehicle, instead of the
6-8% increase when the other oils were used.
7.9.2 Diet
In male NMRI mice, fasted for 24 h before receiving an
intraperitoneal injection (0.1 ml/kg body weight) of carbon
tetrachloride (159.4 mg/kg body weight) in olive oil, higher hepatic
carbon tetrachloride and chloroform levels were found (Pentz &
Strubelt, 1983).
Fed male Sprague-Dawley rats appeared to be more resistant to the
toxic action of carbon tetrachloride than rats starved overnight.
Contrary to findings in mice, the carbon tetrachloride concentrations
were similar in the livers of fed and fasted rats (Díaz Gómez et al.,
1975b).
Male Wistar rats were fed various test diets in order to assess
the nutritional effects on the liver mixed-function oxidases (MFO) and
consequently on the carbon tetrachloride metabolism. The MFO activity
increased almost linearly with decreasing food intake. Furthermore it
was shown that a diet deficient in carbohydrate enhanced the
metabolism and thus the toxic action of carbon tetrachloride,
irrespective of the protein or fat content of the diet (Nakajima et
al., 1982; Sato & Nakajima, 1985).
Cervinková et al. (1987) investigated the effect of long-term
intake of high or low protein diet on liver repair processes after the
administration of carbon tetrachloride. Rats were fed for 21 days on a
low-protein diet (LPD), a standard diet (SD) and a high-protein diet
(HPD) and were then given a single intraperitoneal injection (0.75
ml/kg body weight) of carbon tetrachloride (calculated to be 1196
mg/kg body weight). The HPD was found to increase sensitivity to
carbon tetrachloride, but it also promoted liver repair processes. The
LPD raised liver resistance to carbon tetrachloride, but the
development of liver repair activity differed from the process after
the SD and HPD, since polyploidy of the hepatocytes predominated and
there was also an increase in the number of binuclear hepatocytes.
Cell hypertrophy was expressed less in rats fed on the LPD. As far as
liver repair was concerned, the HPD showed no explicit advantage over
the SD.
7.9.3 Alcohol
Several studies have demonstrated that ethanol, methanol and
other alcohols potentiate the hepatic toxicity of carbon tetrachloride
(Traiger & Plaa, 1971; Cantilena et al., 1979; Harris & Anders, 1980;
Ray & Mehendale, 1990; Simko et al., 1992). Dietary ethanol (2 g/80 ml
liquid diet for 3 weeks) potentiated carbon tetrachloride (inhalation
exposure to 10 ppm (64.1 mg/m3) for 8 h) hepatotoxicity, measured by
serum aminotransferases and liver malonaldehyde concentrations, in
male Wistar rats. Potentiation did not occur upon exposure to 5 ppm
(32 mg/m3) for 8 h (Ikatsu et al., 1991; Ikatsu & Nakajima, 1992).
Only a minor potentiating effect on weight gain, but no effect on
carbon-tetrachloride-induced hepatotoxicity was observed, when rats
were simultaneously treated with ethanol (æ 0.5 ml/kg) and carbon
tetrachloride (20 mg/kg) by gavage for 14 days (Berman et al., 1992).
Micronodular cirrhosis was observed in all treated rats after 10 weeks
of inhalation exposure to carbon tetrachloride (513 mg/m3, 6 h/day, 5
days/week) when the animals were simultaneously given ethanol as a
part of a liquid diet, while no animal treated with either ethanol or
carbon tetrachloride alone developed cirrhosis (Hall et al., 1991).
Similar cirrhosis was also observed in Porton rats treated with carbon
tetrachloride and ethanol (Hall et al., 1994). Inhalation exposure to
methanol (10 000 ppm for 6 h) increased the liver toxicity of carbon
tetrachloride (a single dose of 0.075 ml/kg after 24 h) (Simmons et
al., 1995). Similar exposure to methanol also increased the toxicity
of inhaled carbon tetrachloride (100, 250 to 1000 ppm (641, 1602 to
6410 mg/m3) for 6 h at 26-27 h after the beginning of the methanol
exposure). This potentiation subsided when the interval between
methanol and carbon tetrachloride exposures was increased by 24 h
(Evans & Simmons, 1996). Malonaldehyde generation induced by carbon
tetrachloride in vitro was enhanced by prior exposure of the rats to
methanol (10 000 ppm for 6 h); this enhancement coincided with an
increased microsomal activity of para-nitrophenol hydroxy lase, used
as a marker of cytochrome P-450 2E1; inhibition of CYP 2E1 by allyl
sulfone abolished the carbon-tetrachloride-induced lipid peroxidation
(Allis et al., 1996).
Sato & Nakajima (1985) reported that the metabolism of carbon
tetrachloride in the rat was enhanced by pretreatment with ethanol. It
was found that the increase in carbon tetrachloride hepatotoxicity was
related to the degree of the enhancement. Similar observations were
made by Sato et al. (1980), Teschke et al. (1984), Strubelt (1984) and
Reinke et al. (1988).
In a study by Wang et al. (1997), before exposure to carbon
tetrachloride, rats were kept either on an ethanol-containing (2 g/80
ml per rat per day) liquid diet for 3 weeks, to obtain a maximal
induction of the alcohol-inducible CYP 2E1 isoenzyme, or on a liquid
diet with no alcohol. Both groups were exposed to carbon tetrachloride
by inhalation (0, 320 or 3205 mg/m3 for 6 h), or by oral or
intraperitoneal administration (0, 0.105 or 1.675 mmol/kg).
Ethanol-pretreatment increased significantly the metabolism of carbon
tetrachloride as indicated by the carbon tetrachloride concentrations
in blood samples. Plasma ALAT and ASAT levels, assayed 24 h after
carbon tetrachloride treatment, were highly significantly increased in
all carbon tetrachloride-dosed rats pretreated with ethanol, whereas
in control diet groups only a slight elevation of transaminases was
observed after the high-dose carbon tetrachloride treatment.
Shibayama (1988) compared the hepatotoxicity of carbon
tetrachloride (intraperitoneal administration in olive oil) in male
Wistar rats fed a standard diet and 5 or 20% ethanol solution with the
hepatotoxicity of carbon tetrachloride in control rats, which received
water instead of ethanol for a period of 1 to 100 weeks. The results
indicated that the effect of ethanol on the hepatotoxicity is
dependent on the daily amount of alcohol intake and is not affected by
the duration of the alcohol consumption. Kniepert et al. (1990),
however, reported that an increased duration (30 or 52 weeks instead
of 1 or 10) of ethanol pretreatments (10% in drinking water) caused a
decrease in ethanol potentiation of carbon-tetrachloride-induced
toxicity in male Wistar rats.
Ray & Mehendale (1990) studied the effect caused by a single dose
of carbon tetrachloride after pretreatment with various homologous
alcohols in male Sprague-Dawley rats. A combination of the alcohols
methanol, ethanol, isopropanol and decanol with carbon tetrachloride
potentiated liver injury but did not affect lethality. A combination
of t-butanol, pentanol, hexanol and octanol potentiated liver injury
and decreased animal survival significantly. Eicosanol potentiated
neither liver injury nor lethality.
Hall et al. (1994) observed that chronic administration of
alcohol and 'low-dose' carbon tetrachloride vapour caused cirrhosis in
all male Porton rats receiving this treatment for 5-7 weeks. A
possible mechanism for the interaction between alcohol and carbon
tetrachloride is the alcohol-dependent induction of cytochrome P-450
2E1, resulting in enhanced production of toxic metabolites of carbon
tetrachloride. These in turn are responsible for the initiation of
lipid peroxidation and impaired conjugation of carbon tetrachloride
metabolites with glutathione.
To determine the dose-response relationships in the production of
hepatic fibrosis and cirrhosis, the livers of male Porton rats (4
animals/group) were examined after combined exposure to carbon
tetrachloride and alcohol (Plummer et al., 1994). Carbon tetrachloride
was administered by inhalation 6 h/night for 5 nights/week at
concentrations of 10, 20 or 40 ppm (actual values of 60, 120.1 or
240.1 mg/m3, respectively). Ethanol was administered orally at levels
of 75, 150, or 300 kcal/litre liquid diet, leading to mean daily
intakes of 2.29, 4.61 and 8.16 g ethanol/kg body weight, respectively.
It was proposed to continue administration of alcohol and carbon
tetrachloride until animals became cirrhotic, as diagnosed by liver
biopsy, or for a maximum of 20 weeks. However, the alcohol consumption
declined gradually, approximately to half that at the beginning.
Results of the study show that both alcohol and carbon tetrachloride
contribute to the liver injury in a dose-related manner. All four rats
that received the high dose of both carbon tetrachloride and alcohol,
and one of four rats that received the medium alcohol and high-dose
carbon tetrachloride treatments, showed liver cirrhosis after 10 weeks
of exposure. Two of four rats that received the low alcohol in
combination with the high-dose of carbon tetrachloride showed
cirrhosis after 20 weeks. Although cirrhosis was observed only at the
highest carbon tetrachloride dose, some degree of hepatic fibrosis was
observed in all treated rats in a dose-related manner.
Daniluk et al. (1994) reported that acute liver injury due to
intraperitoneal carbon tetrachloride administration combined with a
long-term alcohol consumption may act synergistically in depressing
interferon production in C3H/He mice.
Strain differences in response to carbon tetrachloride have been
described for both mice (see section 7.1.2.3) and rats (see section
7.3).
7.9.4 Enhancement of carbon tetrachloride-induced hepatotoxicity by various compounds
According to Klingensmith et al. (1983) the LD50 of carbon
tetrachloride after oral administration dropped by a factor of 14
after pretreatment with chlordecone. The potentiation of carbon
tetrachloride toxicity by chlordecone appears to be related to a
chlordecone-dependent increase in the biotransformation rate of carbon
tetrachloride and a chlordecone-dependent reduction of the
liver-regenerating capacity (Mehendale, 1984).
Mehendale (1989) described a mechanism for the potentiation of
carbon tetrachloride hepatotoxicity by chlordecone. The mechanism
underlying the highly unusual amplification of carbon tetrachloride
toxicity relates to the suppression of the initial hepatocellular
regeneration, ordinarily stimulated by low doses of carbon
tetrachloride.
Pretreatment with phenobarbital enhanced the metabolism of carbon
tetrachloride in rats, and consequently the toxicity (Bechtold et al.,
1982; Fander et al., 1982; Sato & Nakajima, 1985), whereas
pretreatment with PCBs or 3-methylcholanthrene for a few days hardly
influenced the carbon tetrachloride metabolism and carbon
tetrachloride toxicity (Sato & Nakajima, 1985). Prolonged pretreatment
with hexachlorobenzene, PBBs or PCBs, however, made male rats
considerably more susceptible to the toxic effects of carbon
tetrachloride (Kluwe et al., 1982).
The reaction of the liver to carbon tetrachloride was studied in
the adaptive stage of organic solvent poisoning. Rats were pretreated
daily with benzene, toluene, xylene, phenobarbital or oil for 4 days.
On day 4, carbon tetrachloride was given orally and 24 h later the
rats were killed. Histological, histochemical and electron microscopic
examination revealed a potentiating interaction between the solvents
and carbon tetrachloride, similar to the potentiation of carbon
tetrachloride toxicity by simultaneous phenobarbital administration.
Centrilobular necrosis caused by carbon tetrachloride became confluent
and turned submassive in the livers of pretreated animals, and it
appeared that in the rats treated with solvent and carbon
tetrachloride, the amount of damaged area was twice that induced by
carbon tetrachloride alone (Tátrai et al., 1979).
Qazi & Alam (1988) reported that phenobarbitone treatment in
drinking-water together with carbon tetrachloride (by intraperitoneal
injection) induced severe liver cirrhosis with marked proliferation of
the bile ducts in female rats. Females receiving only carbon
tetrachloride showed moderate cirrhosis whereas the females receiving
only phenobarbitone remained healthy, showing only an increased liver
weight.
ElSisi et al. (1993a,b) studied the effect of retinol (vitamin A)
on carbon-tetrachloride-induced hepatoxicity in a time-response and a
dose-response study. In the time-response study, male Sprague-Dawley
rats were given 75 mg/kg body weight retinol for 1 or 3 days, or 1,2,3
or 5 weeks. In the dose-response study, retinol was given at daily
doses of 30 to 75 mg/kg body weight for 3 weeks. At 24 h after the
last dose of retinol, 0.15 ml carbon tetrachloride/kg body weight (239
mg/kg body weight) in corn oil was given by intraperitoneal injection
and another 24 h later the animals were killed. All treatment
durations with retinol, except 1 day, resulted in equivalent
potentiation of carbon tetrachloride hepatotoxicity. All rats
pretreated with retinol and subsequent administration of carbon
tetrachloride had more extensive liver injury than those given carbon
tetrachloride alone. As the daily dose of retinol increased, so did
the degree of potentiation of carbon tetrachloride hepatotoxicity.
Pretreatment with ketonic or ketogenic compounds (e.g., hexane,
acetone, isopropanol) potentiated the liver injury in Sprague-Dawley
rats produced by an intraperitoneal carbon tetrachloride injection
(Charbonneau et al., 1985).
The hepatotoxicity of the liver to carbon tetrachloride is
considerably enhanced in alloxan-diabetic rats. This potentiation
effect can be reversed by an additional insulin treatment before the
carbon tetrachloride administration (Villarruel et al., 1982).
Intraperitoneal treatment of male Sprague-Dawley rats with
pyrazole increased the sensitivity of these rats to carbon
tetrachloride hepatotoxicity as assessed by significant loss of
cytochrome P-450 and increases in ASAT and ALAT levels. As stated by
the author (Ebel, 1989), these observations are consistent with the
hypothesis that only certain forms of P-450 (and in this case the
'alcohol-inducible form') are capable of activation of hepatotoxins
and potentiate the toxicity.
Imidazole and pyrazole, inducers of CYP 2E1, caused 3- to 25-fold
enhanced rates of carbon-tetrachloride-induced lipid perioxidation
(and chloroform production from carbon tetrachloride); the increase
was directly related to the amount of this cytochrome in the
microsomes (Johansson & Ingelman-Sundberg, 1985).
Acetone, methyl ethylketone and methyl isobutylketone (6.8
mmol/kg body weight for 3 days) increased the hepatotoxicity of carbon
tetrachloride (Raymond & Plaa, 1995a); this enhanced toxicity was
coincident with an increased microsomal aniline hydroxylase activity
(Raymond & Plaa, 1995b). In addition to the effect on cytochrome
P-450, acetone, but not the other ketones, increased basal canalicular
membrane fluidity, as measured by fluorescence polarization of
1,6-diphenyl-1,3,5-hexatriene or
1,4-(trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene (Raymond &
Plaa, 1996).
Treatment of male athymic nude rats, male and female
Sprague-Dawley rats, and male Fischer-344 rats with retinol (75
mg/kg/day for 7 days) greatly enhanced the hepatotoxicity of carbon
tetrachloride in F-344 rats (0.2 or 0.1 mg/kg i.p.), while it
protected Balb/C, C3H/HeJ, athymic nude and Swiss-Webster mice against
carbon tetrachloride hepatotoxicity (0.0125 to 0.02 ml/kg,
respectively) (Hooser et al., 1994). In male Sprague-Dawley rats
retinol (> 100 000 IU/kg per day for 3 weeks or 250 000 IU/kg per
day for > 1 week) greatly increased the hepatotoxicity of carbon
tetrachloride (0.15 ml/kg intraperitoneally) (ElSisi et al., 1993a).
There was a simultaneous six- to eight-fold increase in the amount of
exhaled ethane and a less than two-fold increase in covalent binding
to liver proteins in rats treated with retinol (250 000 IU or 75 mg/kg
per day for 1 week) and carbon tetrachloride (0.15 ml/kg), in
comparison with rats treated with carbon tetrachloride alone. However,
there was no increase in the exhaled 14CO2, exhaled organics or
metabolites excreted in the urine, or covalent binding to hepatic
lipids from 14C-carbon tetrachloride. Aminobenzotriazole (50 mg/kg
intraperitoneally, 2 h before carbon tetrachloride), an inhibitor of
cytochrome P-450, blocked the retinol-induced potentiation of the
hepatotoxicity of carbon tetrachloride (ElSisi et al., 1993b). A
single dose of retinol (> 75 mg/kg orally) 24 h before carbon
tetrachloride also very significantly potentiated carbon tetrachloride
hepatotoxicity (Badger et al., 1996). While the total cytochrome P-450
content of the liver was not affected by the retinol treatment, the
concentration (Western blot analysis) and activity (aniline
hydroxylase) of CYP 2E1 were both elevated. Isolated hepatocytes from
retinol-treated rats also exhibited enhanced susceptibility to carbon
tetrachloride (Badger et al., 1996).
Concomitant administration of a single, non-hepatotoxic dose of 6
mmol dichloromethane (DCM)/kg and 308 mg carbon tetrachloride
intraperitoneally potentiated carbon tetrachloride-induced
hepatotoxicity, as measured by SDH and ALAT. When a radiolabelled
tracer dose of carbon tetrachloride was included in the treatment, DCM
was found to significantly increase the covalent binding of
[14C]-carbon tetrachloride metabolites to microsomal lipids. However,
DCM did not affect lipid peroxidation induced by carbon tetrachloride
(Kim, 1997).
Potentiation of carbon tetrachloride hepatotoxicity was studied
after inhalation exposure at concentrations of 0, 5 or 10 ppm (0, 160
or 320 mg/m3) for 8 h and simultaneous exposure to chloroform (0, 25
or 50 ppm for 8 h). While carbon tetrachloride exposure had no effect
on plasma ALAT or ASAT levels, co-exposure to chloroform resulted in a
slight increase of the transaminase activity in blood (Ikatsu &
Nakajima, 1992).
7.9.5 Reduction of carbon tetrachloride-induced hepatotoxicity by various compounds
Vitamin E reduces the carbon-tetrachloride-induced lipid
peroxidation in rat liver and kidney slices (Gavino et al., 1984), but
in an in vivo experiment in rats only limited protection by vitamin
E against this process was seen (Gee et al., 1981). A protective
action of vitamin E against liver cell membrane damage in Wistar rats
was reported in several studies (Ozeki et al., 1982; Martínez-Calva et
al., 1984). This protective action could be reinforced by
co-administration of vitamin E and riboflavin tetrabutyrate (Miyazawa
et al., 1984). When male Wistar rats were given vitamin E 15 h before
carbon tetrachloride administration, a partial or complete protection
against the necrogenic effect of carbon tetrachloride was induced,
depending on the concentration of carbon tetrachloride used.
Furthermore, the vitamin supplementation prevented the
carbon-tetrachloride-induced increase in total hepatic calcium content
(Biasi et al., 1991).
Protection, apparent as a decrease in mortality, less pronounced
histological damage and lower serum aminotransferase levels was
afforded by intravenous administration of alpha-tocopherol as a suspen
sion or in liposomes, which are accumulated in Kupffer cells (Yao et
al., 1994; Liu et al., 1995). Where incorporated into liposomes, other
antioxidants, such as butylated hydroxytoluene and ascorbic acid
palmitate, also protected mice against carbon tetrachloride toxicity
(Yao et al., 1994).
Preventive effects on hepatotoxicity in rats were also reported
for vitamin D3 and vitamin C by Fander et al. (1982) and Ademuyiwa et
al. (1994), respectively.
In contrast to the results of ElSisi et al. (1993a,b), who
reported a potentiation in hepatotoxicity when retinol was given to
rats prior to carbon tetrachloride administration, Rosengren et al.
(1995) observed a protective effect on the liver when retinol was
given to mice prior to carbon tetrachloride administration.
Studies reported by Bishayee & Chatterjee (1993) and Mandal et
al. (1993) indicated a possible hepatoprotective role of carrot
(Daucus carota) aqueous extract and Mikania cordata root extract
in male Swiss mice. The increased lipid peroxidation and decreased
glutathione levels, resulting from carbon tetrachloride treatment,
were significantly reversed in a dose-related way after pretreatment
of the mice with the carrot extract. According to authors this
protective role of carrot extract could be attributed to its
antioxidant properties.
Pretreatment of rats with SKF-525 A (inhibitor of
drug-metabolizing enzymes), cysteine (in particular in combination
with tryptophan) or reduced glutathione decreased the
carbon-tetrachloride-induced hepatotoxicity (Bechtold et al., 1982; de
Ferreyra et al., 1983; Gorla et al., 1983).
Administration of
6,6'-methylene-bis(2,2,4-trimethyl-1,2-dihydroquinoline),
tert-butyl-4-hydroxyanisole, oltipraz or anethol dithiolthione
protects mice against the acute toxic effects of carbon tetrachloride
(Fehér et al., 1982; Toncsev et al., 1982; Ansher et al., 1983;
Benson, 1993). A preventive effect was also observed when
diethyldithiocarbamate and carbon disulfide were administered to mice.
Diethyldithiocarbamate was most effective when given orally, while the
action of carbon disulfide was less dependent on the route of
administration (Masuda & Nakayama, 1982).
Rao & Mehendale (1989) reported that administration of fructose
1,6-diphosphate decreased the toxicity of carbon tetrachloride in rats
(as shown by a 50-70% decrease in the activity of serum
transaminases). This decrease was accompanied by elevated activities
of enzymes involved in the polyamine metabolism (important for
hepatocellular regeneration and recovery).
It appears that nicotinamide administered to male Sprague-Dawley
rats at late stages of carbon tetrachloride poisoning (e.g., 6 or 10 h
after the hepatotoxin) significantly prevents the liver necrogenic
effects of carbon tetrachloride at 24 h. A study by de Ferreyra et al.
(1994) did not reveal any relevant effect when nicotinamide was given
30 min before the hepatotoxin.
Simultaneous treatment of carbon-tetrachloride-intoxicated rats
with zinc (227 mg/litre in drinking-water) resulted in improved serum
and liver enzyme levels and attenuated histological abnormalities as
well as NADPH-dependent lipid peroxidation (Dhawan & Goel, 1994).
Studies of Kaminski et al. (1989, 1990) demonstrated that carbon
tetrachloride administration in mice results in a marked suppression
of humoral and cell-mediated immune functions. Ahn & Kim (1993)
observed that PMC (diphenyl dimethyl dicarboxylate) had a significant
preventive effect on carbon-tetrachloride-induced immunotoxic status
in ICR mice that were immunized and challenged with sheep red blood
cells (SRBC) and were subsequently given PMC (3 or 6 mg/kg body
weight; oral administration) once a day for 28 days in combination
with carbon tetrachloride (1 ml/kg body weight, 25%, oral
administration) twice a week, 2 h after PMC administration.
Gadolinium chloride (10 mg/kg), given intravenously 24 h prior to
an intragastric dose of carbon tetrachloride (4 g/kg), nearly
completely protected rats from hepatic necrosis, as measured by serum
ASAT levels and trypan blue exclusion, without an effect on CYP 2E1
(Edwards et al., 1993). This was interpreted to indicate a role of
Kupffer cells in carbon-tetrachloride-induced hepatic damage, since
gadolinium chloride at this concentration strongly inhibits Kupffer
cell phagocytosis (Hustzik et al., 1980). Similar dosage of gadolinium
chloride was, however, also reported to decrease the total amount of
hepatic cytochrome P-450 in rats, as well as the activity of aniline
para-hydroxylase (Badger et al., 1997). In support of the role of
Kupffer cells in carbon-tetrachloride-induced hepatic damage, it was
reported that gadolinium chloride (10 mg/kg given intravenously 24 h
before carbon tetrachloride administration) prevented and methyl
palmitate, another Kupffer cell inhibitor, attenuated the periportal
oedema observed using proton magnetic imaging 1-2 h after carbon
tetrachloride administration (0.8 ml/kg given intraperitoneally)
(Towner et al., 1994). In vivo pin trapping using
alpha-phenyl- N-tert- butylnitrone and a subsequent electron
paramagnetic resonance study of the liver indicated that gadolinium
chloride did not affect the generation of trichloromethyl radicals
from carbon tetrachloride (Towner et al., 1994). Gadolinium chloride
(10 mg/kg given intravenously), methyl palmitate,
polyethylene-glycol-coupled superoxide dismutase and
polyethylene-glycol-coupled catalase protected Sprague-Dawley rats
against retinol-induced potentiation of carbon tetrachloride
hepatotoxicity, both after a single dose and daily dosing for seven
days of retinol (ElSisi et al., 1993a; Sauer & Sipes, 1995; Badger et
al., 1996). Dietary alpha-tocopherol (250 mg/kg diet) partly protected
male Wistar rats against carbon-tetrachloride-induced (0.15 ml 3 times
a week for 5 weeks) hepatic damage (Parola et al., 1992). In an acute
experiment, alpha-tocopheryl hemisuccinate (0.19 mmol, approx. or
equivalent 100 mg/kg by gavage) afforded a partial protection against
the hepatotoxicity of carbon tetrachloride (1.0 g/kg) administered 18
h later (Tirmenstein et al., 1997).
7.10 Mode of action
Recknagel & Glende (1973) suggested that carbon tetrachloride
toxicity requires cleavage of the carbon-chlorine bond and that the
cleavage takes place after binding of carbon tetrachloride to
cytochrome P-450 apoprotein in the mixed-function oxidase system
located in the hepatocellular endoplasmatic reticulum. However,
cytochrome P-450 is encased in lipid, and peroxidative decomposition
of the lipid is initiated by the free radicals formed by the cleavage.
Owing to the decomposition of the lipid and the attack on protein
functional groups by lipid peroxides, the structure and function of
the endoplasmatic reticulum is destroyed.
The carbon-tetrachloride-induced destruction of microsomal
cytochrome P-450 in vitro (De Groot & Haas, 1980, 1981) and
in vivo (Pasquali-Ronchetti et al., 1980; Shen et al., 1982)
inhibits the further biotransformation of carbon tetrachloride, the
generation of radicals and the concomitant peroxidation of endoplasmic
reticular lipids.
Burk et al. (1983) proposed a theory in which the toxic action of
carbon tetrachloride in vivo is dependent on either trichloromethyl
or trichloromethylperoxide radical formation, which is controlled by
the oxygen status of the liver cell (peripheral or centrilobular),
leading to damage to tissue macromolecules. This theory has been
supported by mechanistical arguments as summed up by Slater (1982) and
Dianzani (1984), who proposed the trichloromethyl radical as the main
covalently binding agent (haloalkylation) and the
trichloromethylperoxide radical as the main
lipid-peroxidation-inducing agent.
Oral dosage of carbon tetrachloride (2.5 ml/kg) decreased the
ATP-dependent calcium uptake of liver microsomes within 30 min in
Sprague-Dawley rats (Moore et al., 1976). The cytosolic concentration
of Ca2+ increased 100-fold in hepatocytes exposed to carbon
tetrachloride (about 1 mmol/litre), and this was paralleled by an
inhibition of the endoplasmic reticulum Ca-Mg ATPase (Long & Moore,
1986). The inhibition of the ATPase by carbon tetrachloride exposure
has been confirmed in several studies (Srivastava et al., 1990), and
has led to the hypothesis that this is the specific mechanism by which
radical intermediates from carbon tetrachloride lead to cell death.
Calcium chelating agents, Calcion and alizarin sodium sulfonate,
administered 6 or 10 h after a necrogenic intraperitoneal dose of
carbon tetrachloride (1 ml/kg), markedly decreased the necrotizing
effect of carbon tetrachloride on the liver, and decreased the hepatic
calcium concentration but did not affect carbon-tetrachloride-induced
lipid peroxidation in vitro, or lipid accumulation in the liver (de
Ferreyra et al., 1989, 1992). Carbon tetrachloride (0.01-0.12
mmol/litre) induced a complete release of calcium from calcium-loaded
microsomes in the presence of NADPH; this release was blocked by
adding the spin trapping agent, phenyl- tert- butylnitrone (PBN)
after a lag period that was dependent on the concentration of carbon
tetrachloride. The lag period was shortened using microsomes from
pyrazole-treated rats, which showed an elevated activity for
para-nitrophenol oxidation, and lengthened in the presence of the
CYP-450 2E1 inhibitor, methylpyrazole, or an anti-CYP-450 2E1
antibody. Calcium release was practically complete at concentrations
of carbon tetrachloride that had no effect on the Ca-Mg ATPase
activity. Ruthenium red, a specific ryanodine receptor inhibitor,
completely blocked the carbon-tetrachloride-induced calcium release at
a concentration (0.02 mmol/litre) that had no effect on
para-nitrophenol hydroxylation or formation of PBN-carbon
tetrachloride adducts (Stoyanovsky & Cederbaum, 1996). These results
support the notions that the hepatotoxicity of carbon tetrachloride
requires metabolism to the *CCl3 radical and is mediated by calcium
release from intracellular stores, most likely from the
ryanodine-sensitive calcium store.
Brattin et al. (1984) stated that the disturbance of the
intracellular Ca2+ balance cannot be regarded as an intracellular
"toxic messenger". Recknagel (1983), however, suggested that an early
disturbance in hepatocellular Ca2+ homoeostasis may be involved in
the pathological changes elicited by carbon tetrachloride (Recknagel,
1983). In addition, other authors suggest that the depression of the
Ca2+- sequestration capacity of the endoplasmic reticulum
(microsomes), resulting in a rise in the concentration of Ca2+ in the
cytosol, is an important factor in carbon-tetrachloride-induced
hepatotoxicity (Waller et al., 1983; Srivastava et al., 1990;
Yamamoto, 1990; Glende & Recknagel, 1991). Mehendale (1990, 1991)
showed that hepatic microsomes from carbon-tetrachloride-treated rats
accumulated progressively greater concentrations of Ca2+ in response
to the rise of Ca2+ levels in cytosol.
It is well known that lactic acid plays an important role in
hepatic fibrogenesis. Ayub-Ayala et al. (1993) determined the
relationship between short-term carbon tetrachloride administration
and the rise in lactic acid levels, before the appearance of any signs
of hepatic cirrhosis. After intraperitoneal administration of one
single and three consecutive carbon tetrachloride doses of 2.0 ml/kg
body weight (1:1 dilution in mineral oil) to Sprague-Dawley rats, the
blood lactic acid levels were raised, whereas the administration of
mineral oil did not increase them. Since carbon tetrachloride
increases lactic acid levels prior to cirrhosis development, the
authors suggested that chronic presence of lactic acid is one of the
factors in hepatic fibrogenesis caused by carbon tetrachloride.
Castro et al. (1990) stated that the understanding of the
mechanism of the liver carcinogenic effects of carbon tetrachloride
might be of relevance because there are reasons to believe that carbon
tetrachloride might be one of those carcinogens of a non-genotoxic
nature. The authors showed that liver nuclei from three species tested
(rat, hamster and mouse) were able to promote a lipid peroxidation
process in the presence of carbon tetrachloride, and that NADPH was
only required in part for carbon-tetrachloride-induced lipid
peroxidation in the case of the mice. There was no correlation between
the intensity of carbon-tetrachloride-induced lipid peroxidation,
either in liver nuclear or liver slices preparations, in the three
species tested and their carcinogenic response to carbon
tetrachloride. These results suggest that lipid peroxidation is not
determinant or rate-limiting in the process of liver cancer induction
by carbon tetrachloride, but does not exclude its participation in
given stages of the overall process of cancer development, as is
actually believed to occur during chemical carcinogen insult.
Carbon tetrachloride induced a hepatic cell proliferation,
increasing the frequency of cells in S-phase from < 1% in control
animals to around 10% in B6C3F1 mice (100 mg/kg by gavage 48 h before
sacrifice) (Mirsalis et al., 1985). In rats a similar increase was
observed after a dose of 400 mg/kg (Mirsalis et al., 1985), and the
increase was around 30% in CD mice (50 mg/kg) (Doolittle et al.,
1987). In male Fischer-344 rats, the frequency of S-phase cells was
elevated in one study to 30% 24 h after the only dose tested, 0.4
ml/rat (Cunningham & Matthews, 1991). In another study with 400 mg/kg
carbon tetrachloride, the frequency was increased to 3% in fed and to
15% in fasting F-344 rats (Asakura et al., 1994). In yet another study
with Fischer rats, an increase to 5% was observed 24 h after an
intraperitoneal dose of 400 mg/kg carbon tetrachloride (Mirsalis et
al., 1985). An even lower response, approximately 2%, was observed in
Tif:RAIf rats (400 mg/kg) (Puri & Müller, 1989). An increase in DNA
synthesis was observed 48 h (and the number of ras transcripts was
elevated 36-48 h) after an intragastric dose (2.5 ml/kg) of carbon
tetrachloride in Sprague-Dawley rats (Goyette et al., 1983). A rapid
transient increase in c- fos and c- jun mRNA (1-2 h post-treatment)
was also observed in the liver of male Sprague-Dawley rats after a
single dose of 160 mg/kg carbon tetrachloride (Zawaski et al., 1993).
An increase in the c- fos, c- jun and c- myc nRNA was also observed
in male Wistar rats after a single dose of carbon tetrachloride (2
ml/kg intragastrically) (Coni et al., 1990, 1993). In rat liver,
ras and myc proteins were observed by immunohistochemical
techniques. Their concentrations peaked in periportal areas 32 h after
dosing with carbon tetrachloride (2.5 ml/kg), and staining throughout
the lobule peaked 96 h after the carbon tetrachloride dose (Richmond
et al., 1992). The sequence of fos, myc and Ha- ras mRNA
expression, followed by hepatocyte proliferation, was also observed in
F-344 rats after a single intraperitoneal dose of 2000 mg/kg carbon
tetrachloride by gavage (Goldsworthy et al., 1994). Injection of a
polyclonal antiserum to murine tumour necrosis factor alpha one hour
before a challenge with carbon tetrachloride (0.1 ml/kg) blocked the
increase in c- fos and c- jun mRNA expression and the subsequent
increase of S-phase cells, while at the same time prolonging the
elevation of serum ALAT, ASAT and sorbitol dehydrogenase (SDH) in
female B6C3F1 mice. When recombinant TNF alpha was injected to mice,
rapid expression of c- jun and c- fos proto-oncogene mRNA was
observed, thus supporting the notion that TNF alpha has a role in the
hepatocellular regeneration after carbon tetrachloride administration
(Bruccoleri et al., 1997). This idea was originally put forward after
the demonstration of increased expression of TNF alpha following an
administration of a hepatotoxic dose of carbon tetrachloride (Czaja et
al., 1989). On the other hand, injection of soluble TNF alpha receptor
preparation to rats had a protective effect against a higher dose of
carbon tetrachloride (0.5 ml/kg), reducing the mortality, serum
aminotransferase levels and the extent of histological liver damage
(Czaja et al., 1995).
8. EFFECTS ON HUMANS
8.1 Controlled studies
8.1.1 Inhalation
Six healthy male volunteers were exposed (three times 4 weeks
apart) to carbon tetrachloride vapour at concentrations of 49 ppm (314
mg/m3) for 70 min, 11 ppm (70.5 mg/m3) for 180 min and 10 ppm (64.1
mg/m3) for 180 min. At the high concentration level, all subjects
smelled a sweetish odour. None of the volunteers reported irritation,
nausea, lightheadedness or disturbance in coordination. No increase in
ASAT activity was observed, but a decrease in serum iron concentration
was observed in two out of four subjects at the highest concentration.
No effects were observed at the lower concentrations. Carbon
tetrachloride was detected in exhaled breath at all three exposure
levels (Stewart et al., 1961).
8.1.2 Dermal
Daily treatments for 10 days with carbon tetrachloride did not
cause any increase in skin-fold thickness or erythema on the volar
surface of the forearms of a healthy human volunteer. However, no
occlusion was used, so it is probable that the chemical evaporated
after administration (Wahlberg, 1984a).
When an excess of carbon tetrachloride was applied in a glass
ring to the volar surface of the forearms of a healthy man for 5 min
there was an immediate increase in blood flow. A spontaneous transient
whitening of the skin was observed after 5 min and after 10 to 20 min
a slight, transient erythema appeared (Wahlberg, 1984b).
Immersion of the thumbs of three volunteers for 30 min in carbon
tetrachloride caused mild erythema that disappeared in 1 to 2 h after
exposure. The volunteers reported a burning sensation in the thumbs,
which subsided within 10 min after the immersion (Stewart & Dodd,
1964).
8.2 Case reports
Cases of poisoning with carbon tetrachloride have resulted from
the accidental or suicidal ingestion of carbon tetrachloride, but the
majority resulted from the inhalation of carbon tetrachloride vapour;
the concentration of the saturated vapour at ambient temperature can
reach 800 000 mg/m3. Carbon tetrachloride appears to be toxic to the
liver and kidney. The clinical picture of carbon tetrachloride
poisoning is characterized, independent of the route of intake, in the
first 24 h with gastrointestinal and neurological symptoms, such as
nausea, headache, dizziness, vomiting, diarrhoea and dyspnoea. Liver
damage appears, at the earliest, after 24 h. In serious cases, ascites
and hepatic coma develop, often accompanied by haemorrhages. Kidney
damage is detected later, in 1 to 6 days, but often only 2-3 weeks
following the poisoning (Zimmerman, 1978; Kluwe, 1981; Monster &
Zielhuis, 1983).
Von Oettingen (1964) reviewed the literature on acute carbon
tetrachloride intoxication in humans. He concluded that exposure to
10-80 ppm (64.1 to 512.8 mg/m3) for 3-4 h has no adverse effects. At
higher concentrations nausea, vomiting, headache, rapid pulse, rapid
respiration, sleepiness, dizziness, unconsciousness and immediate
death can occur even after only 10-30 min of exposure.
Bagnasco et al. (1978) reported a case in which a 22-year-old man
ingested 355 ml carbon tetrachloride and an equal amount of water to
commit suicide. His liver function deteriorated over the first 24 h,
but gradually within the following 3 to 4 days the patient improved.
According to the authors fatal cases have been reported with as little
as 1.5 ml carbon tetrachloride, whereas some patients have been known
to survive after swallowing more than 100 ml.
Smetana (1939) described two cases of carbon tetrachloride
poisoning. One person, who drank an unknown quantity of carbon
tetrachloride, died. Apart from the general symptoms, jaundice, anuria
and malaise were observed. Aside from lesions in the liver there was
clinical evidence of functional damage of the kidneys, recognized by
the presence of albumin in the urine, oliguria, nitrogen retention,
oedema and acute hypertension. The patients had a history of
alcoholism.
Norwood et al. (1950) reported three cases of severe intoxication
arising from use of carbon tetrachloride, two by inhalation and one by
drinking. The two people who inhaled carbon tetrachloride died, and
nephrosis was found to have occurred. All three had a history of heavy
drinking.
Tracey & Sherlock (1968) described a 59-year-old man, with a
history of moderate alcohol consumption, who was exposed to carbon
tetrachloride vapour. Five days later, he developed nausea, vomiting
and diarrhoea, followed by jaundice and acute renal failure. The liver
was found to be enlarged. He recovered uneventfully and liver
functions returned to normal.
McDermott & Hardy (1963) reported three cases of liver cirrhosis
in which repeated exposure to carbon tetrachloride vapour occurred
over a number of years. One of the cases involved mixed solvent
exposure. There was no evidence of significant alcohol intake for any
of the patients.
Ruprah et al. (1985) reported details of 19 patients poisoned
with carbon tetrachloride during the period 1981-1984. Eight of these
patients were known to have ingested other substances, including
phenothiazine and benzodiazepine tranquillizers, trichloroethane and
trichloroethanol. Carbon tetrachloride exposure was by inhalation (4
cases) or ingestion (15 cases). In each case the diagnosis was
confirmed by laboratory analysis of blood specimens. The age of the
patients ranged from 3 to 79 years and the whole-blood concentrations
at the time of hospital admission varied from 0.1 to 31.5 mg/litre.
However, actual doses and exposure concentrations were not known and
are difficult to estimate. In none of these cases was the intoxication
associated with occupational use or exposure. The commonest symptoms
found in these patients were vomiting, abdominal pain, diarrhoea,
dizziness, headache and coma. There were no fatalities.
Norwood et al. (1950) reported 51 very mild and 4 mild
intoxications among industrial workers using carbon tetrachloride. The
4 mildly intoxicated patients showed the general symptoms of carbon
tetrachloride intoxication. No data on exposure levels were given.
Kazantzis & Bomford (1960) examined a group of 17 factory
workers, exposed to carbon tetrachloride atmospheric concentrations of
45 to 100 ppm (288-641 mg/m3). During periods of 24 months to 1 week
before the examination, 12 out of 17 workers had experienced one or
more of the following symptoms: nausea, anorexia, vomiting,
flatulence, epigastric discomfort or distention, depressive symptoms,
headache or giddiness. After taking measures to reduce carbon
tetrachloride evaporation, these symptoms disappeared and follow-up
for 6 months revealed no recurrences.
Fourteen workers in an isopropyl alcohol packaging plant became
ill after exposure to unspecified carbon tetrachloride levels (Folland
et al., 1976). The illness was characterized by the gradual onset of
nausea, vomiting, weakness, headache and abdominal pain. In three
heavily exposed subjects renal failure developed. Air concentrations
of isopropanol in the plant were measured 12 and 9 months before and 2
months after the exposure and were about 400 ppm (2564 mg/m3).
Brugnone et al. (1983) investigated 40 workers occupationally
exposed to carbon tetrachloride vapour. After several measurements it
turned out that the alveolar carbon tetrachloride concentration
corresponded to about 53% of the environmental concentration measured
in the breathing zone (mean 3.5 ± 5.9 mg/m3). Among the 40 workers,
two suffered accidental carbon tetrachloride intoxication with acute
renal impairment.
Manno et al. (1996) reported that five workers were exposed to
carbon tetrachloride vapour for 2 h and two for 6 h following fire
accidents. Carbon tetrachloride was present in the fire-extinguishing
liquid. Symptoms of carbon tetrachloride poisoning (diarrhoea, nausea,
vomiting, fever and liver and kidney impairment) developed only in two
heavily drinking workers who consumed, respectively, about 120 and 250
g ethanol daily. The other workers, consuming less than 50 g ethanol
per day, did not develop symptoms. Exposure data were not available.
8.3 Epidemiology
8.3.1 Non-cancer epidemiology
In a mortality study in a metal fabrication plant (Teta & Ott,
1988) slight increase in mortality from liver cirrhosis was observed.
The highest increase (SMR 2.7) was found in workers potentially
exposed to carbon tetrachloride before the use of this solvent was
discontinued. The authors consider carbon tetrachloride exposure as a
possible contributing risk factor for the cirrhosis findings. However,
exposure data for carbon tetrachloride, data on other exposures and
alcohol consumption were not available, which limit the ability to
draw conclusions regarding carbon tetrachloride.
Volunteers from three plants were divided into four groups on the
basis of estimated exposure to carbon tetrachloride: none (n=262), low
(1 ppm (6.4 mg/m3) or less, n=40), medium (1-4 ppm (6.4-25.6 mg/m3),
n=54) and high (more than 4 ppm (25.6 mg/m3), n=61). The alcohol
consumption was at the same level in all groups. ALAT, ASAT, alkaline
phosphatase, gamma-glutamyltransferase, glutamate dehydrogenase, and
other biochemical and haematological variables were determined. The
percentages of values above the normal range were 2.7% in the
non-exposed group and 7.8% in combined exposed groups for ALAT, and 3%
and 10.9% for gamma-glutamyltransferase, respectively (both differences
statistically significant). The low exposure group did not differ
significantly in any enzymatic activity test from the non-exposed
group (Tomenson et al., 1995).
8.3.2 Cancer epidemiology
A number of epidemiological studies (e.g., cohort mortality,
retrospective cohorts, and case-control) have examined potential
cause-effect relationships between carbon tetrachloride exposure and
incidence of cancer. Because these studies are all characterized by
mixed exposures and a lack of carbon tetrachloride exposure data, any
contribution from carbon tetrachloride cannot be reliably identified.
Thus, information from these studies is not useful for quantitative
health risk evaluation.
Ott et al. (1985) conducted a cohort mortality study of 1919 men
employed for one or more years between 1940 and 1969 at a chemical
manufacturing facility in the USA. This cohort included 226 workers
assigned to a unit that produced chlorinated methanes (methyl
chloride, dichloromethane, chloroform, and carbon tetrachloride) and,
recently, perchloroethylene. Exposure levels were not reported. The
follow-up period was from 1940 to 1979 and follow-up was 94% complete.
Expected numbers of cancer deaths were based on US white male cancer
rates for the full cohort; the expected numbers in the full cohort
were used for sub-cohort analyses. There were 42 deaths, including
nine cancers, three of which were pancreatic cancers. The standardized
mortality ratios for all deaths and for all cancers were not elevated.
Blair et al. (1990) performed a study to examine the risk of
cancer and other causes of death among a cohort of 5365 members of a
dry cleaners' union in the USA. The cohort consisted of people who
were union members for one or more years before 1978 and had been
employed in dry cleaning establishments. Carbon tetrachloride was used
extensively in dry cleaning between 1930 and 1960, although other
solvents, such as white spirit (Stoddard solvent), were also widely
used. The exposure assessment classified members by level of exposure
to solvents, but not by type of solvent. The mean year at entry into
the cohort was 1956. Follow-up was from 1948 to 1978 and was 88%
complete. There were 294 cancer deaths, including a significant excess
of oesophageal cancer. Non-significant excesses of several other
cancers were found, but only the risk of lymphatic and haematopoietic
cancers appeared to be related to the level of solvent exposure.
Blair et al. (in press) performed a retrospective cohort
mortality study of 14 457 workers employed for at least one year
between 1952 and 1956 at an aircraft maintenance facility in the USA.
Among this cohort were 6737 workers who had been exposed to carbon
tetrachloride (Stewart et al., 1991). Among women, exposure to carbon
tetrachloride was associated with an increased risk of non-Hodgkin's
lymphoma and multiple myeloma, but among men the corresponding risks
were lower. No association was observed with breast cancer and no
other site-specific results for carbon tetrachloride were presented.
Exposure levels for carbon tetrachloride were not reported, and
overlapping exposure to other solvents limits the ability to draw
conclusions regarding carbon tetrachloride.
A nested case-control study within a cohort of rubber workers in
the USA was performed to examine the relationship between exposure to
24 solvents (levels of exposure not reported) and the risk of cancer
(Checkoway et al., 1984; Wilcosky et al., 1984). The cohort consisted
of 6678 male rubber workers who were either active or retired between
1964 and 1973. The cases comprised all persons with fatal stomach
cancer (n=30), respiratory system cancer (n=101), prostrate cancer
(n=33), lymphosarcoma (n=9) and lymphocytic leukaemia (n=10). The
control group was a 20% age-stratified random sample of the cohort
(n=1350). Although an association was observed between exposure for
one or more years to carbon tetrachloride and lymphocytic leukaemia
and lymphosarcoma after adjusting for year of birth, overlapping
exposures limit the ability to draw conclusions regarding carbon
tetrachloride.
Bond et al. (1986) conducted a nested case-control study of lung
cancer among a cohort of 19 608 white male chemical workers in the
USA. They were employed for one or more years between 1940 and 1980 at
a large facility that produced chlorinated solvents, plastics,
chlorine, caustic soda, ethylene, styrene, epoxy, latex, magnesium
metal, chlor-nitrogen agricultural chemicals and glycols. The cases
were 308 lung cancer deaths that occurred among cohort members between
1940 and 1981. Two control groups, one consisting of other deaths
(n=308) and the other a "living" series (n=97), were matched for race,
year of birth, and year of hire. No association was observed between
having been exposed (levels not reported) to carbon tetrachloride
("ever" versus "never") and lung cancer.
Linet et al. (1987) performed an analysis to compare two
different methods for determining occupational exposure in a
population-based case-control study of chronic lymphocytic leukaemia.
No association between chronic lymphocytic leukaemia and carbon
tetrachloride was observed in either set of analyses.
Heineman et al. (1994) performed a case-control study to examine
the relationship between occupational exposure to six chlorinated
aliphatic hydrocarbons and risk of astrocytic brain cancer. The study
was conducted in three areas of the USA, and 300 cases and 320
controls were included in the analysis. Exposure was assessed using a
semi-quantitative job exposure matrix developed for the study (Gómez
et al., 1994), and probability of exposure, duration of exposure,
average intensity and cumulative exposure were examined. There were
137 cases and 123 controls classified as having been exposed at some
time. There was an association between the incidence of astrocytic
brain cancer and chlorinated solvent exposure, but not specifically
carbon tetrachloride.
Cantor et al. (1995) performed a case-control study to examine
the relationship between occupational exposure and female breast
cancer mortality in 24 states in the USA. Probability and level of
workplace exposure to 31 chemical and physical agents were estimated
using a job exposure matrix. No association was found with probability
of exposure to carbon tetrachloride. After adjustment for age and
socioeconomic status, a slightly but significantly elevated risk was
observed at the highest exposure level among white women but not among
black women. However, the designation of the usual occupation from
death certificates in combination with a job-exposure matrix may be a
poor indicator of exposure to carbon tetrachloride.
Holly et al. (1996) performed a case-control study of intraocular
melanoma to examine the role of chemical exposure. Cases were white
male patients referred to the Ocular Oncology Unit at the University
of California San Francisco between 1978 and 1987. Two white males
matched on age and geographic area were selected for each case using
random digit dialling. A total of 221 cases and 447 control (93% and
85% participation rates, respectively) were interviewed for the study.
Although an association with exposure ("ever" versus "never") to
"carbon tetrachloride and other cleaning fluids" was observed, the
potential for recall bias for exposure history and the lack of
characterization of the exposure atmospheres precludes the ability to
draw conclusions regarding carbon tetrachloride alone.
In a case-control study carried out in Montreal, the
investigators estimated the associations between 293 workplace
substances and several types of cancer (Siemiatycki, 1991). Carbon
tetrachloride was one of the substances. About 4% of the study
subjects had been exposed to carbon tetrachloride at some time. Among
the main occupations for which carbon tetrachloride was attributed in
this study were fire fighters, machinists and electricians. For most
types of cancer examined (oesophagus, stomach, colon, pancreas,
prostrate, kidney, skin melanoma, non-Hodgkin's lymphoma), there was
no indication of an excess risk. There were, however, elevated risks
for rectal cancer and, in the population subgroup of French-Canadians,
bladder cancer.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
Owing to the volatility of carbon tetrachloride, care must be
taken in interpreting test results, particularly those in open static
systems where no chemical analysis of the actual concentration was
carried out.
9.1 Toxicity to microorganisms
Carbon tetrachloride appeared to be of low toxicity to several
microorganisms (see Table 11).
During studies to determine the toxicity threshold, an initial
reduction of cell multiplication or change in culture turbidity was
seen at 30 mg/litre in aerobic bacteria, but an IC50 as low as 6.4
mg/litre was found for methanobacteria. The toxicity threshold for
protozoa was much higher (> 300 mg/litre).
Walton et al. (1989) studied the effect of carbon tetrachloride
on the microbial respiration of two slightly acidic soil types, a
Captina silt loam (1.49% organic carbon) from Roane County, Tennessee,
USA, and a sandy loam (0.66% organic carbon) from Stone County,
Mississippi, USA. Carbon tetrachloride was applied at a rate of 1000
µg/g soil (dry weight) and microbial respiration, measured as CO2
efflux, was monitored at 24-h intervals over a 6-day period. Carbon
tetrachloride had no effect on the respiration of the silt loam. The
CO2 efflux of the sandy loam decreased relative to the control soil
but recovered within the 6-day exposure period.
9.2 Aquatic toxicity
9.2.1 Algae
Data in Table 11 show the toxicity of carbon tetrachloride to
algae to be low.
9.2.2 Invertebrates
The acute toxicity values for carbon tetrachloride in
Daphnia magna (Table 12) range from 28 to > 770 mg/litre.
Carbon tetrachloride had no effect on the embryonic development
of sea urchin (Paracentrotus lividus) eggs at concentrations up to
the saturated solution concentration (Congiu et al., 1984).
9.2.3 Vertebrates
Acute toxicity data for fish are summarized in Table 12. The
acute LC50 values for fish range from 13 to 472 mg/litre for the
Golden orfe (Leusiscus idus melanotus).
Table 11. Carbon tetrachloride toxicity to bacteria, protozoa and algae
Organism Test conditions End-point Nominal Reference
concentration
(mg/litre)
Bacteria
Pseudomonas fluorescens 16 h, 25 °C, static 3% reduction of turbidity 30 Bringmann, 1973
threshold, log phase
Pseudomonas putida 16 h, 25 °C, static 3% reduction of turbidity 30 Bringmann & Kühn, 1977a
threshold, log phase
Nitromonas sp. 24 h, 25 °C, static IC50, 50% reduction in NH3 51 Blum & Speece, 1991
consumption
Methanogens 24 h, 35 °C, static IC50, 50% reduction in gas 6.4 Blum & Speece, 1991
production
Aerobic heterotrophs. 24 h, 35 °C, static IC50, 50% reduction in oxygen 130 Blum & Speece, 1991
uptake
Protozoa
Bacteriovorous flagellate 72 h, 25 °C, static 5% reduction cell count >770 Bringmann, 1978
(Entosiphon sulcatum)
Bacteriovorous flagellate 20 h, 25 °C, static 5% reduction in cell count >616 Bringmann & Kühn, 1980
(Uronema parduczi)
Saprozoic flagellate 48 h, 20 °C, static 5% reduction in cell count >300 Bringmann et al., 1980
(Chilomonas paramecium)
Table 11. (Continued)
Organism Test conditions End-point Nominal Reference
concentration
(mg/litre)
Algae
Blue-green alga 192 h, 27 °C, static 1% reduction of turbidity 105 Bringmann, 1975
(Microcystis aeroginosa) threshold
Green alga 192 h, 27 °C, static 3% reduction of turbidity >600 Bringmann & Kühn, 1977a
(Scenedesmus quadricauda) threshold
Haematococcus pluvialis 4 h, 20 °C, static EC10, 10% reduction in >136 Knie et al., 1983
oxygen uptake
Table 12. Carbon tetrachloride toxicity to invertebrates and fish
Organism Test conditions Parameter Concentration Reference
(mg/litre)
Invertebrates
Daphnia magna 21-23 °C reconstituted static 48 h LC50 35 nominal LeBlanc, 1980
well water; pH 7.4-9.4; 24 h LC50 35 nominal
hardness 173 mg
CaCO3/litre
Daphnia magna 20-22 °C dechlorinated static 24 h LC50 >770 Bringmann & Kühn,
tap water; pH 7.6; 1977b
hardness
173 mg CaCO3
Daphnia magna static 24 h LC50 28 Knie et al., 1983
Fish
Freshwater
Guppy 22 °C hardness 25 mg static 336 h LC50 67 Könemann, 1981
(Poecilia reticulata) CaCO3/litre renewal
Golden orfe 20 °C static 48 h LC50 95 nominala Juhnke &
(Leuciscus idus melanotus) 472 nominala Lüdemann, 1978
Golden orfe static 48 h LC50 13 Knie et al., 1983
(Leuciscus idus melanotus)
Bluegill sunfish 23 °C well water; static 96 h LC50 125 nominal Dawson et al.,
(Lepomis macrochirus) pH 7.6-7.9; hardness 1975/77
55 mg CaCO3/litre
Table 12. (Continued)
Organism Test conditions Parameter Concentration Reference
(mg/litre)
Bluegill sunfish 21-23 °C pH 6.7-6.8; static 96 h LC50 27 nominal Buccafusco et al.,
(Lepomis macrochirus) hardness 32-48 1981
mg CaCO3/litre
Fathead minnow - - flow - LC50 43.1 measured US EPA, 1984b
(Pimephales promelas)
Fathead minnow 21.7 °C pH 6.8; hardness flow 96 h LC50 41.4 measured National Library of
(Pimephales promelas) 49.2 mg CaCO3/litre Medicine, 1997
Marine
Dab - natural seawater flow 96 h LC50 50 measured Pearson &
(Limanda limanda) McConnell, 1975
Tidewater silverside 20 °C saltwater; static 96 h LC50 150 nominal Dawson et al.,
(Menidia beryllina) pH 7.6-7.9; hardness 1975/77
55 mg CaCO3/litre
a The authors tested 200 selected chemicals with the golden orfe test under comparable conditions in two different
laboratories and found LC50 values of 95 mg/litre (Juhnke) and 472 mg/litre (Lüdemann), respectively, for carbon
tetrachloride
Rainbow trout (Oncorhynchus mykiss) were exposed to carbon
tetrachloride concentrations of between 1 and 80 mg/litre for up to
336 h under semi-static conditions (water was renewed every 48 h). No
mortality was observed and no significant changes in enzyme activity
were found (Statham et al., 1978).
Toxicity data for embryo-larval stages of fish and amphibians are
given in Table 13. Carbon tetrachloride is considerably more toxic to
the embryo-larval stages of several species of fish and amphibians
than it is to the adults (Birge, 1980; Black et al., 1982). The common
bullfrog (Rana catesbeiana) was the most susceptible species. At 60
µg/litre the incidence of teratic larvae was 1%, rising to 17% at 7.8
mg/litre. A more striking effect was found in the hatchability of the
embryos, which declined from 92% at 60 µg/litre to 23% at 7.8 mg/litre
(Birge, 1980).
9.3 Terrestrial toxicity
9.3.1 Earthworms
Red earthworms (Eisenia foetida) were exposed to carbon
tetrachloride via filter paper in glass vials. An LC50 of 160 µg/cm2
was found (Neuhauser et al., 1985).
Table 13. Carbon tetrachloride toxicity to embryo-larval stages of fish and amphibians
Organism Test conditions Exposure Parameter Measured Reference
period concentration
(days) (mg/litre)
Fish
Rainbow trout 13 °C pH 9.2; hardness flow 27a LC50 1.97 Black et al., 1982
(Oncorhynchus mykiss) 104 mg CaCO3/litre
Fathead minnow 20 °C pH 6.4; hardness flow 9b LC50 4.0 Black et al., 1982
(Pimephales promelas) 96 mg CaCO3/litre
Amphibians
Bullfrog 21 °C pH 8; hardness flow 8b LC50 0.9 Birge, 1980
(Rana catesbeiana) 108 mg CaCO3/litre
Pickerel frog 22 °C pH 7.7; hardness flow 8b LC50 2.4 Birge, 1980
(Rana palustris) 104 mg CaCO3/litre
Fowler's toad 22 °C pH 7.7; hardness flow 7b LC50 2.8 Birge, 1980
(Bufo fowleri) 104 mg CaCO3/litre
European common frog 19 °C pH 7.7; hardness flow 9a LC50 1.2 Black et al., 1982
(Rana temporaria) 96 mg CaCO3/litre
Leopard frog 19 °C pH 7.7; hardness flow 9a LC50 1.6 Black et al., 1982
(Rana pipiens) 96 mg CaCO3/litre
African clawed toad 19 °C pH 7.7; hardness flow 6a LC50 22.4 Black et al., 1982
(Xenopus laevis) 96 mg CaCO3/litre
Northwestern salamander 19 °C pH 7.7; hardness flow 9.5a LC50 1.98 Black et al., 1982
(Ambystoma gracile) 96 mg CaCO3/litre
a The organisms were exposed from fertilization until 4 days after hatching
b The organisms were exposed from 2-8 h post spawning to 4 days after hatching
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Carbon tetrachloride can be detected ubiquitously in the
environment, mostly in the air due to its volatility and high vapour
pressure. Furthermore it is found in foodstuffs and drinking-water.
Based on the estimates of mean exposure from various media, as
reported in chapter 5, the general population may be exposed via air
at a concentration of 0.5-1.0 µg/m3 (retention calculated to be
0.068-0.136 µg/kg body weight per day, assuming 40% retention, ATSDR,
1994), and via drinking-water at levels of 0.1-3 µg/litre (calculated
to be 0.003-0.094 µg/kg body weight). Intake via foodstuffs is
estimated to be very small (0.13 µg/day) (Yoshida, 1993), but could
have been larger in the past in individuals who consumed fumigated or
otherwise contaminated foods. These are presumably no longer on the
market since this use of carbon tetrachloride has ceased. This means
for the general population an estimated maximum daily carbon
tetrachloride intake of about 0.23 µg/kg body weight, assuming (IPCS,
1994):
- a body weight of 64 kg;
- an inhalation volume of 22 m3/day;
- a water consumption of 2 litres/day;
- a food consumption of 1.536 kg/day.
According to estimates made by the ATSDR and in Japan and Germany
the general population is considered to be exposed to carbon
tetrachloride via ingestion and inhalation leading to an average daily
intake of 0.1 to 0.27 µg/kg body weight.
Workers involved in the production or use of carbon tetrachloride
are likely to be exposed to higher levels than the general population.
Based on a national survey conducted from 1981 to 1983, NIOSH
estimated that 58 208 workers were potentially exposed to carbon
tetrachloride in the USA during that period.
Exposure to higher levels of carbon tetrachloride could occur as
a result of accidental spillage or near hazardous waste sites
contaminated with carbon tetrachloride.
10.1.2 Health effects
Acute symptoms after human exposure to carbon tetrachloride are
characterized by gastrointestinal and neurological symptoms, such as
nausea, vomiting, headache, dizziness and dyspnoea. Liver damage
appears after 24 h or more. Kidney damage is evident often only 2 to 3
weeks following the poisoning. Short-term and long-term exposure to
low concentrations of carbon tetrachloride can also produce hepatic
and renal damage. The toxicity of carbon tetrachloride is associated
with the formation of reactive metabolites, the principal enzyme
involved being CYP 2E1. The severity of the effects on the liver
depends on a number of factors, such as species, susceptibility, route
and mode of exposure, diet or co-exposure to other compounds, in
particular, ethanol. How these factors affect the CNS and kidney
responses is not known. No adequate long-term oral study on laboratory
animals, suitable for quantitative health risk evaluation of carbon
tetrachloride, was available (section 7.3).
In a 12-week oral study on rats (5 days/week), a NOAEL of 1 mg/kg
body weight was reported. The LOAEL reported in this study was 10
mg/kg body weight, showing a slight, but significant increase in SDH
activity and mild hepatic centrilobular vacuolization (Bruckner et
al., 1986). A NOAEL of 1.2 mg/kg body weight was reported in a 90-day
oral study on mice (5 days/week). On the basis of hepato toxicity, the
LOAEL was 12 mg/kg body weight (Condie et al., 1986).
When rats were exposed to carbon tetrachloride by inhalation for
approximately 6 months, 5 days/week, 7 h/day, the NOAEL was 32 mg/m3.
The LOAEL, based on changes in the liver morphology, was 63 mg/m3
(Adams et al., 1952). In a 90-day study on rats, a NOAEL of 6.1 mg/m3
was found after continuous exposure to carbon tetrachloride
(Prendergast, 1967).
An exposure level of 32 mg/m3 (the lowest concentration studied)
in a 2-year inhalation study on rats caused marginal effects (Japan
Bioassay Research Centre, 1998).
In experiments with mice and rats, carbon tetrachloride proved to
be capable of inducing hepatomas and hepatocellular carcinomas. The
doses inducing hepatic tumours were higher than those inducing cell
toxicity. It is likely that the carcinogenicity of carbon
tetrachloride is secondary to its hepatotoxic effects.
There is little evidence to suggest that carbon tetrachloride is
genotoxic.
Based on the weight of evidence it can be concluded that the
hepatic tumours are induced by an indirect mechanism and that a
tolerable daily intake or concentration can be derived.
The available data suggest that carbon tetrachloride can induce
embryotoxic and embryolethal effects, but only at doses that are
maternally toxic. Carbon tetrachloride is not teratogenic in rats and
mice.
10.1.3 Approaches to health risk assessment
There is little evidence to suggest that carbon tetrachloride is
genotoxic. A quantitative risk assessment for threshold effects (IPCS,
1994), which includes the effects of non-genotoxic carcinogens, was
therefore adopted.
10.1.3.1 Calculation of a TDI based on oral data
Calculations of tolerable daily intake (TDI) were based on the
12-week oral study on rats (Bruckner et al., 1986) and the 90-day oral
study on mice (Condie et al., 1986), where NOAEL values of 1 mg/kg
body weight and 1.2 mg/kg body weight were identified, respectively.
a) Rat
1 mg/kg body weight × (5/7)
TDI = 500 = 1.42 µg/kg body weight
where:
* 1 mg/kg body weight is the NOAEL in the 12-week oral study on
rats
* (5/7) is the conversion from 5 days/week of dosing to 7 days/week
* 500 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 10 for a less-than-long-term
study; a modifying factor of 0.5 was applied because this was a
bolus study).
b) Mouse
TDI = 1.2 mg/kg body weight × (5/7) = 1.72 µg/kg body weight
500
where:
* 1.2 mg/kg body weight is the NOAEL in the 90-day oral study on
mice
* (5/7) is the conversion from 5 days/week of dosing to 7 days/week
* 500 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 10 for a less-than-long-term
study; a modifying factor of 0.5 was applied because this was a
bolus study).
10.1.3.2 Calculation of a tolerable concentration based on inhalation
data
Calculations of tolerable concentrations (TC) were based on: (a)
the 90-day study of Prendegast (1967) where a NOAEL of 6.1 mg/m3 was
identified for continuous exposure; (b) the 6-month study of Adams et
al. (1952) where a NOAEL of 32 mg/m3 was found; and (c) the 2-year
inhalation study by Japan Bioassay Research Centre (1998) where a
LOAEL with a marginal adverse effect was 32 mg/m3.
a)
TC = 6.1 mg/m3 = 6.1 µg/m3
1000
where:
* 6.1 mg/m3 is the NOAEL in the 90-day inhalation study on rats
* 1000 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation; and 10 for a less-than-long-term
study)
b)
TC = 32 mg/m3 × (7/24) × (5/7) = 6.7 µg/m3
1000
where:
* 32 mg/m3 is the NOAEL in the 6-month inhalation study on rats
* (7/24) × (5/7) is the conversion from 7 h/day and 5 days/week to
continuous exposure.
* 1000 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 10 for a less-than-long-term
study).
c)
TC = 32 mg/m3 × (6/24) × (5/7) = 11.4 µg/m3
500
* 32 mg/m3 is the LOAEL in the 2-year inhalation study on rats
* (6/24) × (5/7) is the conversion from 6 h/day and 5 days/week to
continuous exposure
* 500 is the uncertainty factor (10 for interspecies variation, 10
for intraspecies variation and 5 for use of a marginal effect rather
than a no-observed-effect level).
It is noted that the end-point on which the LOAEL is based in the
recent 2-year inhalation bioassay on rats (i.e. proteinuria) was not
investigated in the studies of Adams et al. (1952) and Prendergast
(1967).
10.1.3.3 Summary of the results of risk assessment
TDI
Oral studies 1.42 µg/kg body weight
1.72 µg/kg body weight
TC TDI (calculated from the TC)
Inhalation studies 6.1 µg/m3 0.85 µg/kg body weight
6.7 µg/m3 0.92 µg/kg body weight
11.4 µg/m3 1.56 µg/kg body weight
10.1.3.4 Conclusions based on exposure and health risk assessment
On the basis of exposure data presented in chapter 5, an
approximate upper-limit estimate of the daily intake of carbon
tetrachloride for long-term exposure of the general population can be
made for prevailing normal exposure and for a "worst case scenario".
The following concentration ranges are considered:
* ambient air, 0.5-1.0 µg/m3 (worst case 6 µg/m3 );
* indoor air in dwellings, 0.6-2.0 µg/m3 (worst case 9 µg/m3);
* drinking-water, 0.0002-2.3 µg/litre (worst case 16 µg/litre; the
abnormally high value of 39.5 µg/litre reported in Spain was not
considered);
* foodstuffs (particularly table-ready foods) 0.1-6.0 µg/kg (worst
case 31 µg/kg).
The daily estimates are summarized in Table 14.
As is seen from Table 14, the upper limit of human daily intake
under prevailing conditions is estimated to be 0.2 µg/kg body weight,
well below the lowest tolerable daily intake (0.85 µg/kg body weight)
presented in section 10.1.3.3. This leads to the conclusion that the
currently prevailing exposure of the general population to carbon
tetrachloride from all sources is unlikely to cause excessive intake
of the chemical.
The hypothetical worst case scenario of exposure may bring about
a daily intake of 2.5 µg/kg body weight, more than ten times the daily
intake under currently prevailing conditions, and three times the
lowest tolerable intake. This would indicate a need for caution.
However, conditions similar to those of the worst case scenario are
very unlikely to occur in future, due to the expected fall in the use
of carbon tetrachloride as a consequence of the Amended Montreal
Protocol.
10.2 Evaluation of effects on the environment
Carbon tetrachloride may be released into the environment during
its production, storage, transport and use. Owing to its volatility,
most of the substance emitted into the environment can be found in the
air. The residence time of carbon tetrachloride in the atmosphere is
long, and it can therefore be transported over long distances from the
point of emission. The main degradation site of carbon tetrachloride
is the stratosphere where it is photolytically degraded by UV
radiation. Carbon tetrachloride contributes both to ozone depletion
and to global warming.
Carbon tetrachloride is, in general, resistant to aerobic biode
gradation, but less so to anaerobic. Acclimation increases
biodegradation rates. Although the octanol-water partition coefficient
indicates a moderate potential for bioaccumulation, the short lifetime
in tissues reduces this tendency.
Table 14. Daily intake of carbon tetrachloride for long-term exposure of the general population
Prevailing upper limits Worst case scenario
Concentration Daily intake Concentration Daily intake
Air
1 µg/m3 1 µg/m3 × 22 m3 × 0.4a = 8.8 µg 9 µg/m3 9 µg/m3 × 22 m3 × 0.4a = 79.2 µg
Water
0.1 µg/litre 0.1 µg/litre × 2 litres = 0.2 µg 16 µg/litre 16 µg/litres × 2 litres = 32 µg
Food
3 µg/kg 3 µg/kg × 1.5 kg = 4.5 µg 31 µg/kg 31 µg/kg × 1.5 kg = 46.5 µg
Total daily intake 13.5 µg 157 µg
Total daily intake per kg 0.2 µg 2.5 µg
body weight
a The value of 0.4 derives from the 40% retention reported by ATSDR (1994)
Carbon tetrachloride is of low toxicity to the algae and
microorganisms tested; the lowest toxic concentration of carbon
tetrachloride reported was for methanogenic bacteria (IC50 = 6.4
mg/litre). In aquatic invertebrates, LC50 values range from 28
mg/litre to over 770 mg/litre.
The lowest acutely toxic concentration found for freshwater fish
was an LC50 of 13 mg/litre for the golden orfe
(Leuciscus idus melanotus). The lowest LC50 for a marine species
was 50 mg/litre for the dab (Limanda limanda).
Carbon tetrachloride is toxic to embryo-larval stages of fish and
of amphibians. The most sensitive species tested was the common
bullfrog (Rana catesbeiana) with an LC50 of 0.92 mg/litre for the
period from fertilization to 4 days post-hatching.
Comparing the LC50 value for the most sensitive aquatic species
(0.9 mg/litre) with typical levels of carbon tetrachloride in water
(< 1.0 µg/litre) gives a ratio of > 900. Therefore, the general risk
to aquatic organisms is low. However, carbon tetrachloride may present
a risk to embryo-larval stages of aquatic organisms at, or near, sites
of industrial discharges or spills, where much higher levels have been
reported.
11. FURTHER RESEARCH
a) Physiologically based pharmacokinetic models for carbon tetra
chloride should be further developed in order to improve their
use in defining target organ doses in human exposure conditions.
b) Since there is a lack of epidemiological data in those countries
where carbon tetrachloride is still used, epidemiological studies
of exposed populations would be useful.
c) No further research topics are recommended in view of the
phase-out of the production and use of carbon tetrachloride as a
result of the Montreal Protocol on Substances that Deplete the
Ozone Layer.
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
A drinking-water guideline value of 2 µg/litre has been
recommended for carbon tetrachloride by the World Health Organization
(WHO, 1993), based on a risk assessment approach for non-genotoxic
carcinogens. A NOAEL of 1 mg/kg body weight and an uncertainty factor
of 1000 were adopted for calculations.
The International Agency for Research on Cancer evaluated carbon
tetrachloride in 1978 (IARC, 1979), and re-evaluated it in 1987 (IARC,
1987) and 1998 (IARC, in press). The conclusions from the most recent
evaluation were that there is inadequate evidence for carcinogenicity
of carbon tetrachloride in humans but sufficient evidence for its
carcinogenicity in experimental animals. The overall evaluation was
that carbon tetrachloride is possibly carcinogenic to humans (Group
2B).
REFERENCES
Adams EM, Spencer HC, Rowe VK, McCollister DD, & Irish DD (1952) Vapor
toxicity of carbon tetrachloride determined by experiments on
laboratory animals. Arch Ind Hyg Occup Med, 6: 50-66.
Ademuyiwa O, Adesanya O, & Ajuwon OR (1994) Vitamin C in CCl4
hepatotoxicity -- A preliminary report. Hum Exp Toxicol, 13: 107-109.
Ahmadizadeh M, Echt R, Heusner WW, Ross LM, & Roth RA (1990) Effect of
carbon tetrachloride on hamster tracheal epithelial cells. J Toxicol
Environ Health, 30: 273-285.
Ahn YK & Kim JH (1993) Preventive effects of diphenyl dimethyl
dicarboxylate on the immunotoxicity of carbon tetrachloride in ICR
mice. J Toxicol Sci, 18: 185-195.
Ahr HJ, King LJ, Nastainczyk W, & Ullrich V (1980) The mechanism of
chloroform and carbon monoxide formation from carbon tetrachloride by
microsomal cytochrome P-450. Biochem Pharmacol, 29: 2855-2861.
Akahori A, Masui M, Kagawa K, Enomoto M, & Saito M (1983) Time course
of biochemical and histological alterations following a single feeding
of carbon tetrachloride to mice. Jpn J Exp Med, 53: 199-209.
Allis JW, Ward TR, Seely JC, & Simmons JE (1990) Assessment of hepatic
indicators of subchronic carbon tetrachloride injury and recovery in
rats. Fundam Appl Toxicol, 15: 558-570.
Allis JW, Brown BL, Simmons JE, Hatch GE, McDonald A, & House DE
(1996) Methanol potentiation of carbon tetrachloride hepatotoxicity:
The central role of cytochrome P450. Toxicology, 112: 131-140.
Alumot E, Nachtomi E, Mandel E, & Holstein P (1976) Tolerance and
acceptable daily intake of chlorinated fumigants in the rat diet. Food
Cosmet Toxicol, 14: 105-110.
Amacher DE & Zelljadt I (1983) The morphological transformation of
Syrian hamster embryo cells by chemicals reportedly nonmutagenic to
Salmonella typhimurium. Carcinogenesis, 4: 291-295.
Anders MW & Jakobson J (1985) Biotransformation of halogenated
solvents. Scand J Work Environ Health, 11(suppl 1): 23-32.
Anderson C, Sundberg K, & Groth O (1988) Animal model for assessment
of skin irritancy. Contact Dermatitis, 15: 143-151.
Anderson TA, Beauchamp JJ, & Walton BT (1991) Organic chemicals in the
environment. Fate of volatile and semivolatile organic chemicals in
soil: Abiotic versus biotic losses. J Environ Qual. 20: 420-424.
Ansari GA, Treinen Moslen M, & Reynolds ES (1982) Evidence for
in vivo covalent binding of CCl3 derived from CCl4 to cholesterol
of rat liver. Biochem Pharmacol, 31: 3509-3510.
Ansher SS, Dolan P, & Bueding E (1983) Chemoprotective effects of two
dithiolthiones and of butylhydroxyanisole against carbon tetrachloride
and acetaminophen toxicity. Hepatology, 3: 932-935.
Asakura S, Sawada S, Daimon H, Fukuda T, Ogura K, Yamatsu K, &
Furihata C (1994) Effects of dietary restriction on induction of
unscheduled DNA synthesis (UDS) and replicative synthesis (RDS) in rat
liver. Mutat Res, 322: 257-264.
Athanasiou K & Kyrtopoulos Sa (1981) Induction of sister chromatid
exchange by nonmutagenic carcinogens. NATO Adv Study Inst Ser A Life
Sci, 41: 557-562.
ATSDR (1994) Toxicological profile for carbon tetrachloride (Update).
Atlanta, Georgia, US Department of Health and Human Services, Agency
for Toxic Substances and Disease Registry, 229 pp (TP-93/02).
Ausloos PJ, Rebbert RE, & Glasgow L (1977) Photodecomposition of
chloromethanes adsorbed on silica surfaces. J Res Natl Stand, 82: 1-8.
Ayub-Ayala M, Flores-Alvarado LJ, Bueno Topete MR, Ortiz GG,
Gudiño-Cabrera G, & Ortuño-Sahagun D (1993) Effect of short-term
carbon tetrachloride administration on blood lactic acid levels. Gen
Pharmacol, 24(3): 627-630.
Badger DA, Sauer JM, Hoglen NC, Jolley CS, & Sipes IG (1996) The role
of inflammatory cells and cytochrome P450 in the potentiation of
CCl4-induced injury by a single dose of retinol. Toxicol Appl
Pharmacol, 141: 507-519.
Badger DA, Kuester RK, Sauer JM, & Sipes IG (1997) Gadolinium chloride
reduces cytochrome P450: Relevance to chemical-induced hepatotoxicity.
Toxicology, 121: 143-153.
Bagnasco FM, Stringer B, & Muslim AM (1978) Carbon tetrachloride
poisoning: radiographic findings. NY State J Med, 78: 646-647.
Barber ED, Donish WH, & Mueller KR (1981) A procedure for the
quantitative measurement of the mutagenicity of volatile liquids in
the Ames Salmonella/microsome assay. Mutat Res, 90: 31-48.
Barrows ME, Petrocelli SR, Macek KJ, & Carroll JJ (1980)
Bioconcentration and elimination of selected water pollutants by
bluegill sunfish. In: Haque R ed. Dynamics, exposure and hazard
assessment of toxic chemicals. Ann Arbor, Michigan, Ann Arbor Science
Publishers, Inc.
Bauer U (1981) [Human exposure to environmental chemicals
-- Investigations on volatile organic halogenated compounds in water,
air, food and human tissues. III. Communication: Results of
investigations.] Zent.bl Bakteriol Hyg I Abt Orig B, 174: 200-237 (in
German).
Baumann Ofstad E, Drangsholt H, & Carlberg GE (1981) Analysis of
volatile halogenated organic compounds in fish. Sci Total Environ, 20:
205-215.
Bechtold MM, Gee DL, Bruenner U, & Tappel AL (1982) Carbon
tetrachloride-mediated expiration of pentane and chloroform by the
intact rat: The effects of pretreatment with diethyl maleate, SKF-525A
and phenobarbital. Toxicol Lett, 11: 165-171.
Benedetti A, Fulceri R, Ferrali M, Ciccoli L, Esterbauer H, & Comporti
M (1982) Detection of carbonyl formation in phospholipids of liver
microsomes in carbon tetrachloride- and bromotrichloromethane-poisoned
rats. Biochim Biophys Acta, 712(3): 628-638.
Benigni R, Andreoli C, Conti L, Tafani P, Cotta Ramusino M, Carere A,
& Crebelli R (1993) Quantitative structure-activity relationship
models correctly predict the toxic and aneuploidizing properties of
six halogenated methanes in Aspergillus nidulans. Mutagenesis, 8:
301-305.
Benson AB (1993) Oltipraz: a laboratory and clinical review. J Cell
Biochem, 17(suppl F): 278-291.
Bergman K (1984) Application and results of whole-body radiography in
distribution studies of organic solvents. Crit Rev Toxicol, 12: 59-119
Berman E, House DE, Allis JW, & Simmons JE (1992) Hepatotoxic
interactions of ethanol with allyl alcohol or carbon tetrachloride in
rats. J Toxicol Environ Health, 37: 161-176.
Bermudez E, Mirsalis JC, & Eales HC (1982) Detection of DNA damage in
primary cultures of rat hepatocytes following in vivo and
in vitro exposure to genotoxic agents. Environ Mutagen, 4: 667-679.
Bhathal PS, Rose NR, Mackay IR, & Whittingham S (1983) Strain
differences in mice in carbon tetrachloride-induced liver injury. Br J
Exp Pathol, 64: 524-533.
Biasi F, Albano E, Chiarpotto E, Corongiu FP, Pronzato MA, Marinari
UM, Parola M, Dianzani MU, & Poli G (1991) In vivo and in
vitro evidence concerning the role of lipid peroxidation in the
mechanism of hepatocyte death due to carbon tetrachloride. Cell
Biochem Funct, 9: 111-118.
Bignami M (1977) [Relationship between mutagenic activity and chemical
structure of certain pesticides tested with Aspergillus nidulans. ]
Atti Asoc Genet Ital, 22: 143-149 (in Italian).
Birge WJ (1980) Effects of organic compounds on amphibian
reproduction. Lexington, Kentucky, University of Kentucky, Water
Resources Research Institute (Research report No. 121; PB80-147523).
Bishayee A & Chatterjee M (1993) Carrot aqueous extract protection
against hepatic oxidative stress and lipid peroxidation induced by
acute carbon tetrachloride intoxication in mice. Fitoterapia, 64(3):
261-265.
BIT-SC (Bureau international technique-Solvants chlorés) (1976)
Standardization of methods for the determination of volatile
chlorinated hydrocarbons in air and water by GC. Anal Chim Acta, 82:
1-17.
Black JA, Birge WJ, McDonnell WE, Westerman AG, Ramey BA, & Bruser DM
(1982) The aquatic toxicity of organic compounds to embryo-larval
stages of fish and amphibians. Springfield, Virginia, US Department of
Commerce (PB82-224601).
Blair A, Hartge P, Stewart PA, McAdams M, & Lubin J (in press)
Mortality and cancer incidence of workers at an aircraft maintenance
facility exposed to trichloroethylene and other organic solvents and
chemicals: Extended follow-up. Occup Environ Med.
Blair A, Stuart PA, Tolbert PE, Grauman D, Moran FX, Vaught J, &
Reyner J (1990) Cancer and other causes of death among a cohort of dry
cleaners. Br J Ind Med, 47: 162-168.
Blum DJW & Speece RE (1991) A database of chemical toxicity to
environmental bacteria and its use in interspecies comparisons and
correlations. Res J Water Pollut Control Fed, 63: 198-207.
Bogers M, Appelman LM, Feron VJ, Beems RB, & Notten WRF (1987) Effects
of the exposure profile on the inhalation toxicity of carbon
tetrachloride in male rats. J Appl Toxicol, 7: 185-191.
Bond GG, Flores GH, Shellenberger RJ, Cartmille JB, Fishbeck WA, &
Cook RR (1986) Nested-case control study of lung cancer among chemical
workers. Am J Epidemiol, 124: 53-66.
Bouwer EJ & McCarty PL (1983a) Transformations of 1- and 2-carbon
halogenated aliphatic organic compounds under methanogenic conditions.
Appl Environ Microbiol, 45: 1286-1294.
Bouwer EJ & McCarty PL (1983b) Transformations of halogenated organic
compounds under denitrification conditions. Appl Environ Microbiol,
45: 1295-1299.
Boyd MR, Statham CN, & Longo NS (1980) The pulmonary Clara cell as
target for toxic chemicals requiring metabolic activation; studies
with carbon tetrachloride. J Pharmacol Exp Ther, 212: 109-114.
Bozzelli JW & Kebbekus BB (1982) A study of some aromatic and
halocarbon vapors in the ambient atmosphere of New Jersey. J Environ
Sci Health, A17: 693-711.
Brambilla G, Carlo P, Finollo R, Bignone FA, Ledda A, & Cajelli E
(1983) Viscometric detection of liver DNA fragmentation in rats
treated with minimal doses of chemical carcinogens. Cancer Res, 43:
202-209.
Brams A, Buchet JP, Crutzen MC, De Meester C, Lowerys R, & Leonard A
(1987) A comparative study, with 40 chemicals, of the efficiency of
the Salmonella assay and the SOS chromotest (kit procedure). Toxicol
Lett, 38: 123-133.
Brattin WJ, Pencil SD, Waller RL, Glende EA, & Recknagel RO (1984)
Assessment of the role of calcium ion in halocarbon hepatotoxicity.
Environ Health Perspect, 57: 321-323.
Braun R & Schöneich J (1975) The influence of ethanol and carbon
tetrachloride in the host-mediated assay with
Salmonella typhimurium. Mutat Res, 31: 191-194.
Bringmann G (1973) [Determination of biological hazards of water
polluting substances from the inhibition of glucose assimilation of
the bacterium Pseudomonas fluorescens. ] Gesundh-Ing, 94(12):
366-369(in German).
Bringmann G (1975) [Determination of the biological damage of
water-endangering compounds by means of the inhibition of cell
proliferation in the blue alga Microcystis.] Gesundh-Ing, 96(9):
238-241 (in German).
Bringmann G (1978) [Determination of biological adverse action of
water pollutants against protozoa: I. Bacteriological flagellates.] Z
Wasser-Abwasser Forsch, 11: 210-215 (in German).
Bringmann G & Kühn R (1977a) [Limit values for adverse action of water
pollutants against bacteria (Pseudomonas putida) and green algae
(Scenedesmus quadricauda) in the cell proliferation inhibition
test.] Z Wasser-Abwasser Forsch, 10: 87-97 (in German).
Bringmann G & Kühn R (1977b) [Results of adverse action of water
pollutants against Daphnia magna. ] Z Wasser-Abwasser Forsch, 10:
161-166 (in German).
Bringmann G & Kühn R (1980) [Determination of biological adverse
action of water pollutants against protozoa: II. Bacteriological
ciliates.] Z Wasser-Abwasser Forsch, 13: 26-31.
Bringmann G, Kühn R, & Winter A (1980) [Determination of biological
adverse action of water pollutants against protozoa: III. Saprozoic
flagellates.] Z Wasser-Abwasser Forsch, 13: 170-173 (in German).
Brondeau MT, Bonnet P, Guenier JP, & De Ceaurriz J (1983) Short-term
inhalation test for evaluating industrial hepatotoxicants in rats.
Toxicol Lett, 19: 139-146.
Brown RHA, Cape JN, & Farmer JG (1998) Partitioning of chlorinated
solvents between pine needles and air. Chemosphere, 36: 1799-1810.
Bruccoleri A, Ballucci R, Germolec DR, Blackshear P, Simeonova P,
Thurman RG, & Luster MI (1997) Induction of early-immediate genes by
tumor necrosis factor A contribute to liver repair following
chemical-induced hepatotoxicity. Hepatology, 25: 133-141.
Bruckner JV, MacKenzie WF, Muralidhara S, Luthra R, Kyle GM, & Acosta
D (1986) Oral toxicity of carbon tetrachloride: Acute, subacute, and
subchronic studies in rats. Fundam Appl Toxicol, 6: 16-34.
Brugnone F, Apostoli P, Perbellini L, Silvestri R, & Cocheo V (1983)
Monitoring of occupational exposure to low concentration of carbon
tetrachloride. Dev Toxicol Environ Sci, 11: 575-578.
BUA (1990) [Tetrachloromethane. Report of the German Chemical
Society-Advisory Committee on Existing Chemicals of Environmental
Relevance.] Stuttgart, S. Hirzel Wissenschaftliche Verlagsgesellschaft
(BUA Report 45) (in German).
Buccafusco RJ, Ells SJ, & LeBlanc GA (1981) Acute toxicity of priority
pollutants to Bluegill (Lepomis macrochirus). Bull Environ Toxicol,
26: 446-452.
Burk RF, Patel K, & Lane JM (1983) Reduced glutathione protection
against rat liver microsomal injury by carbon tetrachloride:
Dependence on O2. Biochem J, 215: 441-445.
Burk RF, Lane JM, & Patel K (1984) Relationship of oxygen and
glutathione in protection against carbon tetrachloride-induced hepatic
microsomal lipid peroxidation and covalent binding in the rat. J Clin
Invest, 74: 1996-2001.
Burk RF, Hill KE, & Lane JM (1988) Inhibition of CCl4 metabolism by
oxygen varies between isoenzymes of cytochrome P-450. Biochem Biophys
Res Commun, 152: 1463-1467.
Burston MW, Nazari MN, Bishop PK, & Lerner DN (1993) Pollution of
ground water in the Coventry region (UK) by chlorinated hydrocarbon
solvents. J Hydrol, 149: 137-161.
Callen DF, Wolf CR, & Philpot RM (1980) Cytochrome P-450 mediated
genetic activity and cytotoxicity of seven halogenated aliphatic
hydrocarbons in Saccharomyces cerevisiae. Mutat Res, 77: 55-63.
Cambon-Gros C, Deltour P, Boigegrain RA, Fernandez Y, & Mitjavila S
(1986) Short communications: Radical activation of carbon
tetrachloride in foetal and maternal rat liver microsomes. Biochem
Pharmacol, 35: 2041-2044.
Cantilena LR, Cagen SZ, & Klaassen CD (1979) Methanol potentiation of
carbon tetrachloride-induced hepatotoxicity. Proc Soc Exp Biol Med,
162: 90-95.
Cantor KP, Stuart PA, Brinton LA, & Dosemeci M (1995) Occupational
exposures and female breast cancer mortality in the United States. J
Occup Environ Med, 37: 336-348.
Castro GD , Díaz-Gomez MI, & Castro JA (1989) Species differences in
the interaction between CCl4 reactive metabolites and liver DNA or
nuclear protein fractions. Carcinogenesis, 10: 289-294.
Castro GD, Díaz-Gomez MI, & Castro JA (1990) Biotransformation of
carbon tetrachloride and lipid peroxidation promotion by liver nuclear
preparations from different animal species. Cancer Lett ,53: 9-15.
Cervinková Z, Bgatova NP, Shorina TG, Holecek M, Subrtová D,
Vosvrelová H, Shkurupy VA, & Simek J (1987) Structural and functional
changes after the administration of tetrachloromethane in the liver of
rats fed on diets with different protein contents. Physiol Bohemoslov,
36: 349-359.
Charbonneau M, Iijima M, Cote MG, & Plaa GL (1985) Temporal analysis
of rat liver injury following potentiation of carbon tetrachloride
hepatotoxicity with ketonic or ketogenic compounds. Toxicology, 35(2):
95-112.
Chatterjee A (1966) Testicular degeneration in rats by carbon
tetrachloride intoxication. Experientia(Basel), 226: 395-396.
Chatterjee A (1968) Effect of CCl4 on gonadal physiology in female
rats. Acta Anat, 71: 82-86.
Checkoway H, Wilcosky T, Wolf P, & Tyroler H (1984) An evaluation of
the associations of leukemia and rubber industry solvent exposures. Am
J Ind Med, 5: 239-249.
Chen WJ, Chi EY, & Smuckler EA (1977) Carbon tetrachloride-induced
changes in mixed function oxidases and microsomal cytochromes in the
rat lung. Lab Invest, 36: 388-394.
Clark CS (1981) Evaluation of the health risk associated with the
treatment and disposal of municipal wastewater and sludge. Washington,
DC, US Environmental Protection Agency (EPA-600/1-80-030).
Clarke H, Egan DA, Hefferman M, Doyle S, Byrne C, Kilty C, & Ryan MP
(1997) Alpha-glutathione S-transferase (alpha-GST) release, an early
indicator of carbon tetrachloride hepatotoxicity in the rat. Hum Exp
Toxicol, 16: 154-157.
Clyne MAA & Holt PM (1978) Reaction kinetics involving ground X2Pi
and excited A2Sigma+ hydroxyl radicals. J Chem Soc Faraday Trans
II, 75: 569-581.
Cochran JW, Yates MV, & Henson JM (1988) A modified fuge-and-trap gas
chromatography method for analysis of volatile halocarbons in
microbiological degradation studies. J Microbiol Methods, 8: 347-354.
Comporti M, Benedetti A, Ferrali M, & Fulceri R (1984) Reactive
aldehydes (4-hydroxyalkenals) originating from the peroxidation of
liver microsomal lipids: biological effects and evidence for their
binding to microsomal protein in CCl4 or BrCCl3 intoxication. Front
Gastrointest Res, 8: 46-62.
Condie LW, Laurie RD, Mills T, Robinson M, & Bercz JP (1986) Effect of
gavage vehicle on hepatotoxicity of carbon tetrachloride in CD-1 mice:
corn oil versus Tween-60 aqueous emulsion. Fundam Appl Toxicol, 7:
199-206.
Congiu AM, Calendi E, & Ugazio G (1984) Effects of metal ions and
CCl4 on sea urchin embryo (Paracentrotus lividus). Res Commun Chem
Pathol Pharmacol ,43: 317-323.
Coni P, Pichiri-Coni G, Ledda-Columbano GM, Rao PM, Sarma DSR, &
Columbano A (1990) Liver hyperplasia is not necessarily associated
with increased expression of c-fos and c-myc RNA. Carcinogenesis, 11:
835-839.
Coni P, Simbula G, de Prati AC, Menegazzi M, Suzuki H, Sarma DSR,
Ledda-Columbano GM, & Columbano A (1993) Differences in the
steady-state levels of c-fos, c-jun and c-myc messenger RNA during
mitogen-induced liver growth and compensatory regeneration.
Hepatology, 17: 1109-1116.
Coutant RW & Scott DR (1982) Applicability of passive dosimeters for
ambient air monitoring of toxic organic compounds. Environ Sci
Technol, 16: 410-413.
Coutino RR (1979) Analysis of anaphase in cell culture: an adequate
test system for the distinction between compounds which selectively
alter the chromosome structure or the mitotic apparatus. Environ
Health Perspect, 31: 131-136.
Cox RA, Derwent RG, Eggleston AEJ, & Lovelock JE (1976) Photochemical
oxidation of halocarbons in the troposphere. Atmos Environ, 10:
305-308.
Craddock VM & Henderson AR (1978) De novo and repair replication of
DNA in liver or carcinogen-treated animals. Cancer Res, 38: 2135-2143.
Crescentini G, Mangani F, Mastrogiacomo AR, & Bruner F (1981)
Calibration method for the gas chromatographic analysis of halocarbons
in atmospheric samples using permeation tubes and an electron-capture
detector. J Chromatogr, 204: 445-451.
Crescentini G, Mangani F, Mastrogiacomo AR, Cappiello A, & Bruner F
(1983) Fast determination of some halocarbons in the atmosphere by gas
chromatography-high-resolution mass spectrometry. J Chromatogr, 280:
146-151.
Cronn DR & Harsch DE (1979) Determination of atmospheric halocarbons
concentrations by gas chromatographic-mass spectrometry. Anal Lett,
12B(4): 1489-1496.
Cronn DR, Rasmussen RA, Robinson E, & Harsch DE (1977) Halogenated
compound identification and measurement in the troposphere and lower
stratosphere. J Geophys Res, 82: 5935-5944.
Cunningham ML & Matthews HB (1991) Relationship of
hepatocarcinogenicity and hepatocellular proliferation induced by
mutagenic noncarcinogens vs carcinogens. II. 1- vs 2-nitropropane.
Toxicol Appl Pharmacol 110: 505-513.
Cunningham ML, Gandolfi AJ, Brendel K, & Sipes IG (1981) Covalent
binding of halogenated volatile solvents to subcellular macromolecules
in hepatocytes. Life Sci, 29: 1207-1212.
Cupitt LT (1980) Fate of toxic and hazardous materials in the
environment. Washington, DC, US Environmental Protection Agency (EPA
600/53-80-084).
Czaja MJ, Flanders KC, Biempica I, Klein C, Zern MA, & Weiner FR
(1989) Expression of tumor necrosis factor alpha and transforming
factor beta1 in acute liver injury. Growth Factors, 1: 219-226.
Czaja MJ, Xu J, & Alt E (1995) Prevention of carbon
tetrachloride-induced rat liver injury by soluble tumor necrosis
factor receptor. Gastroenterology, 108: 1849-1854.
Daft JL (1988) Rapid determination of fumigant and industrial chemical
residues in food. J Assoc Off Anal Chem, 71: 748-760.
Daft JL (1989) Determination of fumigants and related chemicals in
fatty and nonfatty foods. J Agric Food Chem 37: 560-564.
Daft JL (1991) Fumigants and related chemicals in foods. Review of
residue findings, contamination sources, and analytical methods. Sci
Total Environ, 100: 501-518.
Dai Y & Cederbaum AI (1995) Inactivation and degradation of human
cytochrome P4502E1 by CCl4 in a transfected HepG2 cell line.
J Pharmacol Exp Ther, 275: 1614-1622.
Daniluk J, Chibowski D, Szuster-Ciesielska A, & Kandefer-Szerszen M
(1994) Effect of carbon tetrachloride intoxication and ethanol
ingestion on interferon production in mice. Arch Immunol Ther Exp
(Warsz), 42(4): 325-330.
David A, Frantik E, Holusa R, & Nováková O (1981) Role of time and
concentration on carbon tetrachloride toxicity in rats. Int Arch Occup
Environ Health, 48: 49-60.
Dawson GW, Jennings AL, Drozdowski D, & Rider E (1975/77) The acute
toxicity of 47 industrial chemicals to fresh and saltwater fishes.
J Hazard Mater, 1: 303-318.
Dean BJ & Hodson-Walker G (1979) An in vitro chromosome assay using
cultured rat-liver cells. Mutat Res, 64: 329-337.
Deer HM, McJilton CE, & Harein PK (1987) Respiratory exposure of grain
inspection workers to carbon tetrachloride fumigant. Am Ind Hyg Assoc
J, 48: 586-593.
De Ferreyra EC, De Fenos OM, & Castro JA (1983) Tryptophan
potentiation of the late cysteine preventive effects in carbon
tetrachloride-induced necrosis. Res Commun Chem Pathol Pharmacol, 40:
515-518.
De Ferreyra EC, Villarruel MC, Bernacchi AS, Fernandez G, de Fenos OM,
& Castro JA (1989) Late preventive effects against carbon
tetrachloride-induced liver necrosis of the calcium chelating agent
Calcion. Arch Toxicol, 63: 450-455.
De Ferreyra EC, Villarruel MC, Bernacchi AD, de Fenos OM, & Castro JA
(1992) Prevention of carbon tetrachloride-induced liver necrosis by
the chelator alizarin sodium sulfonate. Exp Mol Pathol, 56: 197-207.
De Ferreyra EC, Bernacchi AS, San Martín MF, Castro GD, & Castro JA
(1994) Nicotinamide late protective effects against carbon
tetrachloride-induced liver necrosis. Exp Mol Pathol, 60: 214-223.
De Flora S (1981) Study of 106 organic and inorganic compounds in the
Salmonella/microsome test. Carcinogenesis, 2: 283-298.
De Flora S, Zanacchi P, Camoirano A, Bennicelli C, & Badolati GS
(1984) Genotoxic activity and potency of 135 compounds in the Ames
reversion test and in a bacterial DNA-repair test. Mutat Res, 133:
161-198.
De Groot H & Haas W (1980) O2-independent damage of cytochrome P-450
by CCl4 metabolites in hepatic microsomes. Febs Lett, 115: 253-256.
De Groot H & Haas W (1981) Self-catalysed, O2-independent
inactivation of NADPH- or dithionite- reduced microsomal cytochrome
P-450 by carbon tetrachloride. Biochem Pharmacol, 30: 2343-2347.
Delaney B & Kaminski NE (1994) Induction of serum borne
immunomodulatory factors in B6C3F1 mice by carbon tetrachloride.
Toxicology, 88: 201-212.
Dent JG & Graichen EM (1982) Effect of hepatocarcinogens on epoxide
hydrolase and other xenobiotic metabolizing enzymes. Carcinogenesis,
3: 733-738.
De Vries JW, Larson PA, Bowers RH, Keating JA, Broge JM, Wehling PS,
Patel HH, & Zurawski JW (1985) Improved codistillation method for
determination of carbon tetrachloride, ethylene-dichloride and
ethylene-dibromide in grain and grain-based products. J Assoc Off Anal
Chem, 68(4): 759-762.
De Zwart LL, Venhorst J, Groot M, Commandeur JN, Hermanns RC, Meerman
JH, Fan Baar BL, & Vermeulen NP (1997) Simultaneous determination of
eight lipid peroxidation degradation products in urine of rats treated
with carbon tetrachloride using gas chromatography with
electron-capture detection. J Chromatogr Biomed Appl, B694: 277-287.
Dhawan DK & Goel A (1994) Protective role of zinc on rat liver
function in long-term toxicity induced by carbon tetrachloride.
J Trace Elem Exp Med, 7: 1-9.
Dianzani MU (1984) Lipid peroxidation and haloalkylation: two distinct
mechanisms for carbon tetrachloride-induced liver damage. Int Congr
Ser Exerpta Med, 632: 39-50.
Díaz Gómez MI & Castro JA (1980a) Covalent binding of carbon
tetrachloride metabolites to liver nuclear DNA, protein and lipids.
Toxicol Appl Pharmacol, 56: 199-206.
Díaz Gómez MI & Castro JA (1980b) Nuclear activation of carbon
tetrachloride and chloroform. Res Commun Pathol Pharmacol, 27:
191-194.
Díaz Gómez MI, de Castro CR, D'Acosta N, de Fenos OM, de Ferreyra EC,
& Castro JA (1975a) Species differences in carbon
tetrachloride-induced hepatotoxicity: the role of CCl4 activation and
of lipid peroxidation. Toxicol Appl Pharmacol, 34: 102-114.
Díaz Gómez MI, de Castro CR, de Ferreyra EC, D'Acosta N, de Fenos OM,
& Castro JA (1975b) Mechanistic studies on carbon tetrachloride
hepatotoxicity in fasted and fed rats. Toxicol Appl Pharmacol, 32:
101-108.
Dickson AG & Riley JP (1976) The distribution of short-chain
halogenated aliphatic hydrocarbons in some marine organisms. Mar
Pollut Bull, 7: 167-169.
Dilling WL, Tefertiller NB, & Kallos GJ (1975). Evaporation rates and
reactivities of methylene chloride, chloroform, 1,1,1-trichloroethane,
trichloroethylene, tetrachloroethylene and other chlorinated compounds
in dilute aqueous solutions. Environ Sci Technol, 9(9): 833-838.
Dingell JV & Heimberg M (1968) The effects of aliphatic halogenated
hydrocarbons on hepatic drug metabolism. Biochem Pharmacol, 17:
1269-1278.
DiRenzo AB, Gandolfi AJ, Sipes IG, Brendel K, & Byard JL (1984) Effect
of O2 tension on the bioactivation and metabolism of aliphatic
halides by primary rat-hepatocyte cultures. Xenobiotica, 14: 521-525.
Divincenzo GD & Krasavage WJ (1974) Serum ornithine carbamyl
transferase as a liver response test for exposure to organic solvents.
Am Ind Hyg Assoc J, 35: 21-29.
Doherty AT, Ellard S, Parry EM, & Parry JM (1996) An investigation
into the activation and deactivation of chlorinated hydrocarbons to
genotoxins in metabolically competent human cells. Mutagenesis, 11:
247-274.
Doolittle DJ, Muller G, & Scribner HE (1987) Relationship between
hepatotoxicity and induction of replicative DNA synthesis following
single or multiple doses of carbon tetrachloride. J Toxicol Environ
Health, 22: 63-78.
Doong RA & Wu SC (1995) Substrate effects on the enhanced
biotransformation of polychlorinated hydrocarbons under anaerobic
condition. Chemosphere, 30: 1499-1511.
Doong RA & Wu SC (1996) Effect of substrate concentration on the
biotransformation of carbon tetrachloride and 1,1,1-trichloroethane
under anaerobic condition. Water Res, 30: 577-586.
Doong RA, Wu SC, & Chen TF (1996) Anaerobic biotransformation of
polychlorinated methane and ethene under various redox conditions.
Chemosphere, 32: 377-390.
Duffy CC, McCallister DL, & Renken RR (1997) Carbon tetrachloride
retention by modern and buried soil A horizons. J Environ Qual, 26:
1123-1127.
Duprat P, Delsart L, & Gradiski P (1976) Irritant potency of the
principle aliphatic chloride solvents on the skin and ocular mucous
membranes of rabbits. Eur J Toxicol, 9: 171-177.
Ebel RE (1989) Pyrazole treatment of rats potentiates CCl4-but not
CHCl3-hepatotoxicity. Biochem Biophys Res Commun, 161: 615-618.
ECDIN (1992) On-line search in the environmental chemicals data and
information network. ECDIN/copyright Joint Research Centre/European
Commission, Ispra.
Edwards JE (1941) Hepatomas in mice induced with carbon tetrachloride.
J Natl Cancer Inst, 2: 197-199.
Edwards JE & Dalton AJ (1942) Induction of cirrhosis of the liver and
of hepatomas in mice with carbon tetrachloride. J Natl Cancer Inst, 3:
19-41.
Edwards JE, Heston WE, & Dalton AJ (1942) Induction of the carbon
tetrachloride hepatoma in strain L mice. J Natl Cancer Inst, 3:
297-301.
Edwards PR, Campbell I, & Milne GS (1982a) The impact of
chloromethanes on the environment - Part 1: The atmospheric chlorine
cycle. Chem Ind, 21 August: 574-578.
Edwards PR, Campbell I, & Milne GS (1982b) The impact of
chloromethanes on the environment, part 3: Chloroform and carbon
tetrachloride. Chem Ind, 18 September: 714-718.
Edwards MJ, Keller BJ, Kauffman FC, & Thurman RG (1993) The
involvement of Kupfer cells in carbon tetrachloride toxicity. Toxicol
Appl Pharmacol, 119: 275-279.
Egli C, Scholtz R, Cook AM, & Leisinger T (1987) Anaerobic
dechlorination of tetrachloromethane and 1,2-dichloroethane to
degradable products by pure cultures of Desulfobacterium sp. and
Methanobacterium sp. FEMS Microbiol Lett, 43: 257-261.
Eichelberger JW, Bellar TA, Donnelly JP, & Budde WL (1990)
Determination of volatile organics in drinking water with US EPA
method 524.2 and the ion trap detector. J Chromatogr Sci, 28: 460-467.
Elias L (1977) In-situ quantitation of background halofluorocarbon
levels. NBS Special Publication (USA), vol 464, pp 435-438.
ElSisi AE, Hall P, Sim W-LW, Earnest DL, & Sipes IG (1993a)
Characterization of vitamin A potentiation of carbon
tetrachloride-induced liver injury. Toxicol Appl Pharmacol, 119:
280-288.
ElSisi AE, Earnest DL, & Sipes IG (1993b) Vitamin A potentiation of
carbon tetrachloride hepatotoxicity: role of liver macrophages and
active oxygen species. Toxicol Appl Pharmacol, 119: 295-301.
Ember LR, Layman PL, Lepkowski W, & Zurer PS (1986) The changing
atmosphere. Chem Eng News, 24 November: 14-64.
Entz RC & Hollifield HC (1982) Headspace gas chromatographic analysis
of foods for volatile halocarbons. J Agric Food Chem, 30: 84-88.
Entz RC, Thomas KW & Diachenko GW (1982) Residues of volatile
halocarbons in foods using headspace gas chromatography. J Agric Food
Chem, 30: 846-849.
Erickson MD (1981) Acquisition and chemical analysis of mother's milk
for selected toxic substances. Gov Rep Announc Index US, 81: 5096
(Publication PB81-231029).
Eschenbrenner AB & Miller EB (1944) Studies on hepatomas: I. Size and
spacing of multiple doses in the induction of carbon tetrachloride
hepatomas. J Natl Cancer Inst, 4: 385-388.
Evans MV & Simmons JE (1996) Physiologically based pharmacokinetic
estimated metabolic constants and hepatotoxicity of carbon
tetrachloride after methanol pretreatment in rats. Toxicol Appl
Pharmacol, 140: 245-253.
Evans MV, Crank WD, Yang HM, & Simmons JE (1994) Applications of
sensitivity analysis to a physiologically based pharmacokinetic model
for carbon tetrachloride in rats. Toxicol Appl Pharmacol, 128: 36-44.
Fander U, Haas W, & Kröner H (1982) The damage of the hepatic mixed
functional oxygenase system by CCl4: Significance of incorporation of
14CCl4 metabolites in vivo. Exp Mol Pathol, 36: 34-43.
Fanelli SL & Castro JA (1995) Covalent binding of carbon tetrachloride
reactive metabolites to liver microsomal and nuclear lipid and
phospholipid classes from Sprague-Dawley and Osborne Mendel male rats.
Teratog Carcinog Mutagen, 15: 155-166.
Fehér J, Bar-Pollák ZS, Sréter L, Fehér E, & Toncsev H (1982)
Biochemical markers in carbon tetrachloride- and galactosamine-induced
acute liver injuries: The effects of dihydroquinoline-type
antioxidants. Br J Exp Pathol, 63: 394-400.
Fernández G, Villarruel MC, De Toranzo EGD, & Castro JA (1982)
Covalent binding of carbon tetrachloride metabolites to the heme
moiety of cytochrome P-450 and its degradation products. Res Commun
Chem Pathol Pharmacol, 35: 283-290.
Fogelqvist E (1985) Carbon tetrachloride, tetrachloroethylene,
1,1,1-trichloroethane and bromoform in arctic seawater. J Geophys Res,
90: 9181-9193.
Folland DS, Schaffner W, Ginn HE, Crofford OB, & McMurray DR (1976)
Carbon tetrachloride toxicity potentiated by isopropyl alcohol. J Am
Med Assoc, 236: 1853-1856.
Foureman P, Mason JM, Valencia R, & Zimmering S (1994) Chemical
mutagenesis testing in Drosophila: X. Results of 70 coded chemicals
tested for the National Toxicology Program. Environ Mol Mutagen, 23:
208-227.
Fowler JSL (1969) Carbon tetrachloride metabolism in the rabbit. Br J
Pharmacol 37: 733-737.
Frank H & Frank W (1990) Concentrations of airborne C1 - and
C2 - halocarbons in forest areas in West Germany: results of
three campaigns in 1986, 1987 and 1989. Atmos Environ, 24A: 1735-1739.
Frank H & Frank W (1986) Photochemical activation of chloroethenes
leading to destruction of photosynthetic pigments. Experientia
(Basel), 42: 1267-1269.
Frantik E & Benes V (1984) Central nervous effect and blood level
regressions on exposure time paralleled in solvents (toluene, carbon
tetrachloride and chloroform. Activ Nerv Sup (Praha), 26: 131-133.
Fraser P & Derek N (1994) Halocarbons, nitrous oxide, methane and
carbon monoxide -- the GAGE Program. In: Dick C & Fras J ed. Baseline
1991. Washington, DC, Bureau of Meteorology (CSIRO), pp 70-81.
Fraser P, Penkett S, Gunson M, Weiss R, & Rowland FS (1994)
Measurements. In: Kaye J, Penkett S, & Ormond F ed. Concentrations,
lifetimes and trends of CFCs, halons and related species. Washington,
DC, NASA, pp 1/1-1/68 (Report No.1339).
Freiria-Gandara MJ, Lorenzo-Ferreira RA, Alvarez-Deveja A, & Bermejo F
(1992) Occurrence of halogenated hydrocarbons in the water supply of
different cities of Galicia (Spain). Environ Technol, 13: 437-447.
Fujii T (1977) The determination of traces of organohalogen compounds
in aqueous solution by direct injection gas chromatography -- mass
spectrometry and single ion detection. Anal Chim Acta, 92: 117-122.
Gäb S, Schmitzer J, Turner WV, & Korte F (1980) Mineralization of
CCl4 and CCl2F2 on solid surfaces. Z Naturforsch, 35B: 946-952.
Galbally IE (1976) Man-made carbon tetrachloride in the atmosphere.
Science, 193: 573-576.
Galli A & Schiestl RH (1995) Salmonella test positive and negative
carcinogens show different effects on intrachromosomal recombination
in G2 cells cycle arrested yeast cells. Carcinogenesis, 16: 659-663.
Galli A & Schiestl RH (1996) Effects of Salmonella assay negative
and positive carcinogens on intrachromosomal recombination in
G1-arrested yeast cells. Mutat Res, 370: 209-221.
Gans JH & Korson R (1984) Liver nuclear DNA synthesis in mice
following carbon tetrachloride administration or partial hepatectomy.
Proc Soc Exp Biol Med, 175: 237-242.
Gargas ML, Andersen ME, & Clewell HJ III (1986) A physiologically
based simulation approach for determining metabolic constants from gas
uptake data. Toxicol Appl Pharmacol, 86: 341-352.
Garry VF, Nelson RL, Griffith J, & Harkins M (1990) Preparations for
human study of pesticide applicators: Sister chromatid exchanges and
chromosome aberrations in cultured human lymphocytes exposed to
selected chemicals. Teratog Carcinog Mutagen, 10: 21-29.
Gavino VC, Dillard CJ, & Tappel AL (1984) Release of ethane and
pentane from rat tissue slices: Effect of vitamin E, halogenated
hydrocarbons and iron overload. Arch Biochem Biophys, 233: 741-747.
Gee DL, Bechtold MM, & Tappel AL (1981) Carbon tetrachloride-induced
lipid peroxidation: simultaneous in vivo measurements of pentane and
chloroform exhaled by the rat. Toxicol Lett, 8: 299-306.
Gehring PJ (1968) Hepatotoxic potency of various chlorinated
hydrocarbons vapours relative to their narcotic and lethal potencies
in mice. Toxicol Appl Pharmacol, 13: 287-298.
Geyer H, Politzki G, & Freitag D(1984) Prediction of ecotoxicological
behaviour of chemicals: Relationship between n-octanol/water
partition coefficient and bioaccumulation of organic chemicals by alga
Chlorella. Chemosphere, 13: 269-284.
Giger W, Molnar-Kubica E, & Wakeham S (1978) Volatile chlorinated
hydrocarbons in ground and lake waters. In: Aquatic pollution
-- Transformation of biological effects. Oxford, New York, Pergamon
Press, pp 101-123 (Pergamon Series on Environmental Science).
Gilli G, Scursatone E, Bono R, Natale P, & Grosa M (1990) An overview
of atmospheric pollution in Italy before the use of new gasoline. Sci
Total Environ, 93: 51-56.
Gilman MR (1971) A preliminary study of the teratogenic effects of
inhaled carbon tetrachloride and ethyl alcohol consumption in the rat.
Philadelphia, Pennsylvania, Drexel University (Dissertation).
Glende EA & Recknagel RO (1991) An indirect method demonstrating that
CCl4-dependent hepatocyte injury is linked to a rise in intracellular
calcium ion concentration. Res Commun Chem Pathol Pharmacol, 73:
41-52.
Goldsworthy TL, Goldsworthy SM, Sprankle CS , & Butterworth BE (1994)
Expression of myc, fos and Ha-ras associated with chemically induced
cell proliferation in the rat liver. Cell Prolif, 27: 269-278.
Gómez MR, Cocco P, Dosemeci M, & Stewart PA (1994) Occupational
exposure to chlorinated aliphatic hydrocarbons: Job exposure matrix.
Am J Ind Med, 26: 171-183.
Gorla N, de Ferreyra EC, Villarruel MC, de Fenos OM, & Castro JA
(1983) Studies on the mechanism of glutathione prevention of carbon
tetrachloride-induced liver injury. Br J Exp Pathol, 64: 388-395.
Goyette M, Petropulos CJ, Shank PR, & Fausto N (1983) Expression of
cellular oncogene during liver regeneration. Science, 219: 510-512.
Gradiski D, Magadur J-L, Baillot M, Danière MC, & Schuh MB (1974)
Toxicité comparée des principaux solvants chlorés aliphatiques. J Eur
Toxicol, 7: 247-254.
Gradiski D, Bonnet P, Raoult G, & Magadur JL (1978) Toxicité aiguë
comparée par inhalation des principaux solvants aliphatiques chlorés.
Arch Mal Prof Méd Trav Sécur Soc, 39: 249-257.
Grob K (1984) Further development of direct aqueous injection with
electron-capture detection in gas chromatography. J Chromatogr, 299:
1-11.
Gruebele A, Zawaski K, Kapalan D, & Novak RF (1996) Cytochrome
P4502E1- and cytochrome P4502B1/2B2-catalysed carbon tetrachloride
metabolism. Drug Metab Dispos, 24: 15-22.
Gualandi G (1984) Genotoxicity of the free-radical producers CCl4 and
lipoperoxide in Aspergillus nidulans. Mutat Res, 136: 109-114.
Guicherit R & Schulting FL (1985) The occurrence of organic chemicals
in the atmosphere of the Netherlands. Sci Total Environ, 43: 193-219.
Hall PM, Plummer JL, Ilsley AH, & Cousins MJ (1991) Hepatic fibrosis
and cirrhosis after chronic administration of alcohol and "low-dose"
carbon tetrachloride vapour in the rat. Hepatology, 13: 815-819.
Hall PM, Plummer JL, Ilsley AH, Ahern MJ, Cmielewski PL, & Williams RA
(1994) The pathology of liver injury induced by the chronic
administration of alcohol and "low dose" carbon tetrachloride in
Porton rats. J Gastroenterol Hepatol, 9: 250-256.
Hamlin GP, Kholkute SD, & Dukelow WR (1993) Toxicology of maternally
ingested carbon tetrachloride (CCl4) on embryonal and fetal
development and in vitro fertilization in mice. Zool Sci, 10:
111-116.
Harris RN & Anders MW (1980) Effect of fasting, diethyl maleate, and
alcohols on carbon tetrachloride-induced hepatotoxicity. Toxicol Appl
Pharmacol, 56: 191-198.
Harsch DE & Cronn DR (1978) Low-pressure sample-transfer technique for
analysis of stratospheric air samples. J Chromatogr Sci ,16(8):
363-367.
Hayes JR, Condie LW, & Borzelleca JF (1986) Acute, 14-day repeated
dosing, and 90-day subchronic toxicity studies of carbon tetrachloride
in CD-1 mice. Fundam Appl Toxicol, 7: 454-463.
Heikes DL (1987) Purge and trap method for determination of volatile
hydrocarbons and carbon disulfide in table ready foods. J Assoc Off
Anal Chem, 70: 215-226.
Heil E, Oeser H, Hatz R, & Kelker H (1979) [Gas-chromatographic
determination of traces of C1- and C2-fluoro- and chlorohydrocarbons
in air.] Fresenius Z Anal Chem, 297(5): 357-364 (in German).
Heineman EF, Cocco P, Gómez MR, Dosemeci M, Stewart PA, Hayes RB, Zahm
SH, Thomas TL, & Blair A (1994) Occupational exposure to chlorinated
aliphatic hydrocarbons and risk of astrocytic brain cancer. Am J Ind
Med, 26: 155-169.
Herzfeld D, Van der Gun KD, & Louw R (1989) Quantitative determination
of volatile organochlorine compounds in water by GC-headspace analysis
with dibromomethane as an internal standard. Chemosphere, 18(7/8):
1425-1430.
Hollinger MA (1982) Biochemical evidence for pulmonary endothelial
cell injury after carbon tetrachloride administration in mice. J
Pharmacol Exp Ther, 222: 641-644.
Holly EA, Aston DA, Ahn DK, & Smith AH (1996) Intraocular melanoma
linked to occupations and chemical exposures. Epidemiology, 7: 55-61.
Honma J (1990) Effects of trichloroethylene, 1,1,1-trichloroethane and
carbon tetrachloride on plasma fibroproteins of rats. Ind Health, 28:
159-174.
Hooser SB, Rosengren RJ, Hill DA, Mobley SA, & Sipes IG (1994) Vitamin
A modulation of xenobiotic-induced hepatotoxicity in rodents. Environ
Health Perspect, 102(suppl 9): 39-43.
Howard PH ed. (1990) Handbook of environmental fate and exposure data
for organic chemicals: Volume II. Solvents. Chelsea, Michigan, Lewish
Publishers, Inc., pp 85-91.
Howard Carleton J & Evenson P (1976) Rate constants for the reactions
of OH with CH4 and fluorine, chlorine and bromine substituted
methanes at 296 K. J Chem Phys, 64: 197.
Howard PH, Boethling RS, & Jarvis WF ed. (1991) Handbook of
environmental degradation rates. Chelsea, Michigan, Lewis Publishers,
Inc., pp 34-35.
Huiskamp R, Van der Heijden CA, Speijers GJA, Ros JPM, Huldy HJ,
Besemer AC, Lanting RW, Maas RJM, Heijna-Merkus E, Bergshoeff G,
Gerlofsma A, Mennes WC, Van der Most PFJ, De Vrijer FL, Janssen PCJM,
Knaap AGAC, Huijgen C, Duiser JA, & De Jong P (1986) Integrated
criteria document: Carbon tetrachloride -- Air effects. Bilthoven, The
Netherlands, National Institute of Public Health and Environmental
Protection (Report No. 738513005).
Hustzik E, Lazar G, & Parducz A (1980) Electron microscopic study of
Kupfer-cell phagocytosis blockade induced by gadolinium chloride. Br J
Exp Pathol, 61: 624-630.
IARC (1979) Some halogenated hydrocarbons. Lyon, International Agency
for Research on Cancer, pp 377-399 (IARC Monographs on the Evaluation
of the Carcinogenic Risk of Chemicals to Humans, Volume 20).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs, volumes 1 to 42. Lyon, International Agency for
Research on Cancer, pp 143-144 (IARC Monographs on the Evaluation of
Carcinogenic Risks to Humans, Supplement 7).
IARC (in press) Part one: Re-evaluation of some organic chemicals.
Lyon, International Agency for Research on Cancer (IARC Monographs on
the Evaluation of Carcinogenic Risks to Humans, Volume 71).
Ikatsu H & Nakajima T (1992) Hepatotoxic interaction between carbon
tetrachloride and chloroform in ethanol treated rats. Arch Toxicol,
66: 580-586.
Ikatsu H, Okino T, & Nakajima T (1991) Ethanol and food deprivation
induced enhancement of hepatotoxicity in rats given carbon
tetrachloride at low concentration. Br J Ind Med, 48: 636-642.
Inoko M, Tsuchiya M, & Matsuno T (1984) Determination of lower
halogenated hydrocarbons by the water-xylene extraction method.
Environ Pollut, B7: 129-140.
IPCC (WMO/UNEP Intergovernmental Panel on Climate Change) (1990) In:
Houghton JT, Jenkins GJ & Ephraums JJ ed. Climate change: The IPCC
scientific assessment. Cambridge, United Kingdom, Cambridge University
Press, 356 pp.
IPCC (WMO/UNEP Intergovernmental Panel on Climate Change) (1995) In:
Houghton JT, Meira Filho LG, & Bruce J ed. Climate change 1994:
Radiative forcing of climate change and an evaluation of the IPCC IS92
emission scenarios. Cambridge, United Kingdom, Cambridge University
Press, 337 pp.
IPCS (1994) Environmental health criteria 170: Assessing human health
risks of chemicals: Derivation of guidance values for health-based
exposure limits. Geneva, World Health Organization
Isaksen ISA & Stordal F (1981) The influence of man on the ozone
layer, readjusting the estimates. Ambio, 10: 9-17.
Isidorov VA, Zenkerich IG, & Ioffe BV (1990) Volatile organic
compounds in solfataric gases. J Atmos Chem, 10: 329-340.
Jaeger RJ, Conolly RB, & Murphy SD (1975) Short term inhalation
toxicity of halogenated hydrocarbons. Arch Environ Health, 30: 26-31.
Jakobson I, Wahlberg JE, Holmberg B, & Johansson G (1982) Uptake via
the blood and elimination of 10 organic solvents following
epicutaneous exposure of anesthetized guinea pigs. Toxicol Appl
Pharmacol, 63: 181-187.
Japan Bioassay Research Centre (1998)Thirteen-week and two-year
inhalation studies on F-344 rats and BDF1 mice (Studies Nos 0020,
0021, 0043 and 0044). Kanagawa, Japan Industrial Safety and Health
Association, Japan Bioassay Research Centre (Unpublished report to the
Ministry of Labour).
Jeffers PM, Brenner C, & Wolfe NL (1996) Hydrolysis of carbon
tetrachloride. Environ Toxicol Chem, 15: 1064-1065.
Jin G & Englarde AJ (1996) Redox potential as a controlling factor in
an enhancing carbon tetrachloride biodegradation. Water Sci Technol,
34: 59-66.
Jirova D, Sperlingova I, Halaskova M, Bendova H, & Dabrowska L (1996)
Immunotoxic effects of carbon tetrachloride -- the effect on
morphology and function of the immune system in mice. Centr Eur J
Public Health, 4: 16-20.
Johansson I & Ingelman-Sundberg M (1985) Carbon tetrachloride-induced
lipid peroxidation dependent on an ethanol-inducible form of rabbit
liver microsomal cytochrome P450. FEBS Lett, 183: 265-269.
Juhnke I & Lüdemann D (1978) [Results from a study of 200 chemical
compounds on the acute fish toxicity test with Golden Orfe.] Z
Wasser-Abwasser Forsch, 11: 161-164 (in German).
Jury WA, Spencer WF, & Farmer WJ (1984) Behavior assessment model for
trace organics in soil: III. Application of screening model. J Environ
Qual, 13: S73-S79.
Kaiser KLE, Comba ME, & Hunealt H (1983) Volatile halogen contaminants
in the Niagara River and Lake Ontario. J Great Lakes Res, 9: 212-223.
Kalla NR & Bansal MP (1975) Effect of carbon tetrachloride on gonadal
physiology in male rats. Acta Anat, 91: 380-385.
Kaminski NE, Jordan SD, & Holsapple MP (1989) Suppression of humoral
and cell-mediated immune responses by carbon tetrachloride. Fundam
Appl Toxicol, 12: 117-128.
Kaminski NE, Barnes DW, Jordan SD, & Holsapple MP (1990) The role of
metabolism in carbon tetrachloride-mediated immunosuppression:
in vivo studies. Toxicol Appl Pharmacol, 102: 9-20.
Kanaghinis T, Avgerinos A, Scliros P, Kalantzis N, Hatzioannou J,
Nikolopoulou P, Anagnostou D, Katsas A, Demopoulos J, Rekoumis G, &
Stathakos D (1982) Plasma lipoprotein pattern in relation to liver
histology after toxic hepatitis and experimental biliary obstruction
in rabbits. Am J Gastroenterol, 77: 512-522.
Kanics L & Rubinstein D (1968) The effect of chronic carbon
tetrachloride inhalation on the secretion of lipid by rat liver.
Biochem Pharmacol, 17: 1959-1967.
Kazantzis G & Bomford RR (1960) Dyspepsia due to inhalation of carbon
tetrachloride vapour. Lancet, 1: 360-362.
Kenaga EE (1980) Predicted bioconcentration factors and soil
adsorption coefficients of pesticides and other chemicals. Ecotoxicol
Environ Saf, 4: 26-38.
Kennedy GL, Ferenz RL, & Burgess BA (1986) Estimation of acute oral
toxicity in rats by determination of the approximate lethal dose
rather than the LD50. J Appl Toxicol, 6: 145-148.
Kieczka H & Kappus H (1980) Oxygen dependence of CCl4-induced lipid
peroxidation in vitro and in vivo. Toxicol Lett, 5: 191-196.
Kim YC (1997) Dichloromethane potentiation of carbon tetrachloride
hepatotoxicity in rats. Fundam Appl Toxicol, 35: 138-141.
Kim HJ, Bruckner JV, Dallas CE, & Gallo JM (1990a) Effect of dosing
vehicles on the pharmacokinetics of orally administered carbon
tetrachloride in rats. Toxicol Appl Pharmacol, 102: 50-60.
Kim HJ, Odend'hal S, & Bruckner JV (1990b) Effect of oral dosing
vehicles on the acute hepatotoxicity of carbon tetrachloride in rats.
Toxicol Appl Pharmacol, 102: 34-49.
Klaassen CD & Plaa GL (1967a) Susceptibility of male and female mice
to the nephrotoxic and hepatotoxic properties of chlorinated
hydrocarbons. Proc Soc Exp Biol Med, 124: 1163-1166.
Klaassen CD & Plaa GL (1967b) Relative effects of various chlorinated
hydrocarbons on liver and kidney function in dogs. Toxicol Appl
Pharmacol, 10: 119-131.
Klaassen CD & Plaa GL (1969) Comparison of the biochemical alterations
elicited in livers from rats treated with carbon tetrachloride,
chloroform, 1,1,2-trichloroethane and 1,1,1-trichloroethane. Biochem
Pharmacol, 18: 2019-2027.
Klecka GM & Gonsior SJ (1984) Reductive dichlorination of chlorinated
methanes and ethanes by reduced iron (II) porphyrins. Chemosphere, 13:
391-402.
Klingensmith JS, Lockard V, & Mehendale HM (1983) Acute hepatotoxicity
and lethality of CCl4 in chlordecone-pretreated rats. Exp Mol Pathol,
39: 1-10.
Kluwe WM (1981) Renal function tests as indicators of kidney injury in
subacute toxicity studies. Toxicol Appl Pharmacol, 57: 414-424.
Kluwe WM, Hook JB, & Bernstein J (1982) Synergistic toxicity of carbon
tetrachloride and several aromatic organohalide compounds. Toxicology,
23: 321-336.
Knie J, Hälke A, Juhnke I, & Schiller W (1983) [Results of studies on
chemical substances with four biotests.] Dtsch Gewässerkd Mitt, 27:
77-79 (in German).
Kniepert E, Siegemund A, & Görisch V (1990) Influence of ethanol
pretreatment of differing duration on toxic effects of carbon
tetrachloride in rats. Biomed Biochem Acta, 49: 1097-1102.
Könemann H (1981) Quantitative structure-activity relationships in
fish toxicity studies. Part 1: Relationship for 50 industrial
pollutants. Toxicology, 19: 209-221.
Koporec KP, Kim HJ, MacKenzie WF, & Bruckner JV (1995) Effect of oral
dosing vehicles on the subchronic hepatotoxicity of carbon
tetrachloride in the rat. J Toxicol Environ Health, 44: 13-27.
Kornbrust DJ & Mavis RD (1980) Microsomal lipid peroxidation: II.
Stimulation by carbon tetrachloride. Mol Pharmacol, 17: 408-414.
Korsrud GO, Grice HC, & McLaughlan JM (1972) Sensitivity of several
serum enzymes in detecting carbon tetrachloride-induced liver damage
in rats. Toxicol Appl Pharmacol, 22: 474-483.
Kraemer M, Bimboes D, & Greim H (1974) Use of S. typhimurium and
E. coli to detect chemical mutagens. Naunyn-Schmiedeberg's Arch
Pharmacol, 284: 46R.
Kroneld R (1985) Recovery and reproducibility in determination of
volatile halocarbons in water and blood. Bull Environ Contam Toxicol,
34: 486-496.
Kroneld RC (1989) Volatile pollutants in the environment and human
tissues. Bull Environ Contam Toxicol, 42: 873-877.
Kronevi T, Wahlberg J, & Holmberg B (1979) Histopathology of skin,
liver and kidney after epicutaneous administration of five industrial
solvents to guinea pigs. Environ Res, 19: 56-69.
Krost KJ, Pellizzari ED, Walburn SG, & Hubbard SA (1982) Collection
and analysis of hazardous organic emissions. Anal Chem, 54: 810-817.
Kubic VL & Anders MW (1980) Metabolism of carbon tetrachloride to
phosgene. Life Sci, 26: 2151-2155.
Kutob SD & Plaa GL (1962) A procedure for estimating the hepatotoxic
potential of certain industrial solvents. Toxicol Appl Pharmacol, 4:
354-361.
Lahl U (1983) [Analysis and evaluation of potential adverse effects of
selected volatile halogenated hydrocarbons in the human environment.]
Bremen, Germany, University of Bremen, pp 52-91 (Dissertation) (in
German).
Lahl U, Cetinkaya M, Düszeln J V, Stachel B, Thiemann W, Gabel B,
Kozicki R, & Podbielski A (1981) Health risks from volatile
halogenated hydrocarbons ? Sci Total Environ, 20: 171-189.
Lai EK, McCay PB, Noguchi T, & Fong K-L (1979) In vivo spin-trapping
of trichloromethyl radicals from CCl4. Biochem Pharmacol, 28:
2231-2235.
Larson RE & Plaa GL (1965) A correlation of the effects of cervical
cordotomy, hypothermia and catecholamines on carbon
tetrachloride-induced hepatic necrosis. J Pharmacol Exp Ther, 147:
103-111.
Lasa J, Rosiek J, & Wcislak L (1979) [Detection of halogen compounds
in air with coulometric ECD.] Chem Anal, 24: 819-826 (in Polish).
LeBlanc GA (1980) Acute toxicity of priority pollutants to water flea
(Daphnia magna). Bull Environ Contam Toxicol, 24: 684-691.
Lee PY, McCay PB, & Hornbrook KR (1982) Evidence for carbon
tetrachloride-induced lipid peroxidation in mouse liver. Biochem
Pharmacol, 31: 405-409.
Lehmann KB & Schmidt-Kehl L (1936) [The thirteen most important
chlorinated aliphatic hydrocarbons from the standpoint of industrial
hygiene.] Arch Hyg, 116: 131-268 (in German).
Lillian D & Singh HB (1974) Absolute determination of atmospheric
halocarbons by gas phase coulometry. Anal Chem, 46: 1060-1063.
Lillian D, Singh HB, Appleby A, Lobban L, Arnts R, Gumpert R, Hague R,
Toomey J, Kazazis J, Antell M, Hansen D, & Scott B (1975) Atmospheric
fates of halogenated compounds. Environ Sci Technol, 9: 1042-1048.
Lil'p IG (1983) Instability of the chromosomes in 101/H and C57BI/6
mice during aging. Soviet Genet, 18: 1467-1472.
Linet MS, Stuart WF, Van Natta ML, McCaffrey LD, & Szklo M (1987)
Comparison of methods for determining occupational exposure in a
case-control interview study of chronic lymphocytic leukemia. J Occup
Med, 29: 136-141.
Link B, Durk H, Thiel D, & Frank H (1984) Binding of trichloromethyl
radicals to lipids of the hepatic endoplasmic reticulum during
tetrachloromethane metabolism. Biochem J, 233(3): 577-586.
Lioy PJ & Lioy MJ ed. (1983) Air sampling instruments for evaluation
of atmospheric contaminants, 6th ed. Cincinnati, Ohio, American
Conference of Governmental Industrial Hygienist, pp V/85-V/86.
Litchfield MH & Gartland CJ (1974) Plasma enzyme activity and
hepatocellular changes in the beagle dog after single or repeated
administration of carbon tetrachloride. Toxicol Appl Pharmacol, 30:
117-128.
Liu SL, Esposti SD, Yao T, Diehl AM, & Zern MA (1995) Vitamin E
therapy of acute CCl4-induced hepatic injury in mice is associated
with inhibition of nuclear factor kappa B binding. Hepatology, 22:
1474-1481.
Long RM & Moore L (1986) Elevated cytosolic calcium in rat hepatocytes
exposed to carbon tetrachloride. J Pharmacol Exp Ther, 238: 186-191.
Loveday KS, Anderson BE, Resnick MA, & Zeiger E (1990) Chromosome
aberration and sister chromatid exchange tests in Chinese hamster
ovary cells in vitro: V. Results with 46 chemicals. Environ Mol
Mutagen, 16: 272-303.
Lowrey K, Glende EA, & Recknagel RO (1981) Rapid depression of rat
liver microsomal calcium pump activity after administration of carbon
tetrachloride or bromotrichloromethane and lack of effect after
ethanol. Toxicol Appl Pharmacol, 59: 389-394.
Lundberg I, Ekdahl M, Kronevi T, Lidums V, & Lundberg S (1986)
Relative hepatotoxicity of some industrial solvents after
intraperitoneal injection or inhalation exposure in rats. Environ Res,
40: 411-420.
Lurker PA, Clark CS, Elia VJ, Gartside PS, & Kinman RN (1983) Worker
exposure to chlorinated organic compounds from the activated-sludge
wastewater treatment process. Am Ind Hyg Assoc J, 44: 109-122.
Lyman WJ, Reehl WF, & Rosenblatt DH (1982) Handbook of chemical
property estimation methods. New York, McGraw-Hill Book Company.
Mabey W & Mill T (1978) Critical review of hydrolysis of organic
compounds in water under environmental conditions. J Phys Chem Ref
Data, 7(2): 383-415.
McCann J, Choi E, Yamasaki E, & Ames BN (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of
300 chemicals. Proc Natl Acad Sci (USA), 72: 5135-5139.
McCay PB, Lai EK, Poyer JL, DuBose CM, & Janzen EG (1984) Oxygen- and
carbon-centered free radical formation during carbon tetrachloride
metabolism. Observations of lipid radicals in vivo and in vitro. J
Biol Chem, 259: 2135-2143.
McCollister DD, Beamer WH, Atchison GJ, & Spencer HC (1951) The
absorption, distribution and elimination of radioactive carbon
tetrachloride by monkeys upon exposure to low vapor concentrations. J
Pharmacol Exp Ther, 102: 112-124.
McConnell G, Ferguson DM, & Pearson CR (1975) Chlorinated hydrocarbons
in the environment. Endeavour, 34: 13-18.
McDermott WV & Hardy HL (1963) Cirrhosis of the liver following
chronic exposure to carbon tetrachloride. J Occup Med, 5: 249-251.
McGregor D & Lang M (1996) Carbon tetrachloride: genetic effects and
other modes of action. Mutat Res, 366: 181-195.
McKone TE (1987) Human exposure to volatile organic compounds in
household tap water: the indoor inhalation pathway. Environ Sci
Technol, 21: 1194-1201.
Magos L, Snowden R, White INH, Butler WH, & Tuffery AA (1982) Isotoxic
oral and inhalation exposure of carbon tetrachloride in Porton-Wistar
and Fischer rats. J Appl Toxicol, 2: 238-240.
Makide Y & Yokohata A (1983) Ultratrace analysis of CCl2F2, CCl3F,
CCl2FCClF2, CH3CCl3, CCl4, CHCl=CCl2 and CCl2=CCl2 in the
atmosphere. J Trace Microprobe Tech, 1: 265-292.
Makide Y, Tominaga T, & Rowland S (1979) Gas chromatographic analysis
of halogenated hydrocarbons in air over Japan. Chem Lett, 1979:
355-358.
Makide Y, Kanai Y, & Tominaga T (1980) Background atmospheric
concentrations of halogenated hydrocarbons in Japan. Bull Chem Soc
Jpn, 53: 2681-2682.
Mandal PK, Bishayee A, & Chatterjee M (1993) Short communication:
Stimulation of tissue repair by Mikania cordata root extract in
carbon tetrachloride-induced liver injury in mice. Phytother Res, 7:
103-105.
Mann H (1975) [The Golden Orfe test: A German proposal to test the
effects of chemical compounds in fish.] Vom Wasser, 44: 1-13 (in
German).
Manno M, Rezzadore M, Grossi M, & Sbrana C (1996) Potentiation of
occupational carbon tetrachloride toxicity by ethanol abuse. Hum Exp
Toxicol, 15: 294-300.
Marchand C, McLean S, & Plaa GL (1970) The effect of SKF 525A on the
distribution of carbon tetrachloride in rats. J Pharmacol Exp Ther,
174: 232-238.
Martínez-Calva I, Campos-Apáez A, Rosales-Vega E, & Mourelle M (1984)
Vitamin E improves membrane lipid alterations induced by CCl4
intoxication. J Appl Toxicol, 4: 270-272.
Masuda Y & Nakayama N (1982) Protective effect of
diethyldithiocarbamate and carbon disulfide against liver injury
induced by various hepatotoxic agents. Biochem Pharmacol, 31:
2713-2725.
Mehendale HM (1984) Potentiation of halomethane hepatotoxicity:
Chlordecone and carbon tetrachloride. Fundam Appl Toxicol, 4: 295-308.
Mehendale HM (1989) Mechanism of lethal interaction of chlordecone and
CCl4 at non-toxic doses. Toxicol Lett, 49: 215-241.
Mehendale HM (1990) Potentiation of halomethane hepatotoxicity by
chlordecone: a hypothesis for the mechanism. Med Hypotheses, 33:
289-299.
Mehendale HM (1991) Commentary: Role of hepatocellular regeneration
and hepatolobular healing in the final outcome of liver injury. A
two-stage model of toxicity. Biochem Pharmacol, 42: 1155-1162.
Mico BA & Pohl LR (1983) Reductive oxygenation of carbon
tetrachloride. Trichloromethylperoxyl radical as a possible
intermediate in the conversion of carbon tetrachloride to
electrophilic chlorine. Arch Biochem Biophys, 225: 596-609.
Mirsalis JC & Butterworth BE (1980) Detection of unscheduled DNA
synthesis in hepatocytes isolated from rats treated with genotoxic
agents: An in vivo-in vitro assay for potential carcinogens and
mutagens. Carcinogenesis, 1: 621-625.
Mirsalis JC, Tyson CK, & Butterworth BE (1982) Detection of genotoxic
carcinogens in the in vivo- in vitro hepatocyte DNA repair assay.
Environ Mutagen, 4: 553-562.
Mirsalis JC, Tyson CK, Loh EN, Steinmetz KL, Bakke JP, Hamilton CM,
Spak DK, & Spalding JW (1985) Induction of hepatic cell proliferation
and unscheduled DNA synthesis in mouse hepatocytes following
in vivo treatment. Carcinogenesis, 6: 1521-1524.
Miyazawa T, Tsuchiya K, & Kaneda T (1984) Riboflavin tetrabutyrate: an
antioxidative synergist of alpha-tocopherol as estimated by hepatic
chemiluminescence. Nutr Rep Int, 29: 157-165.
Molina MJ & Rowland FS (1974) Predicted present stratospheric
abundances of chlorine species for photodissociation of carbon
tetrachloride. Geophys Res Lett, 1: 309-312.
Monster AC & Zielhuis RL (1983) Chlorinated hydrocarbon solvents. In:
Human biological monitoring of industrial chemical series. Ispra,
Italy, European Commission, Joint Research Centre, pp 63-65 (EUR 8476
EN).
Moore L, Davenport GR, & Landon EJ (1976) Calcium uptake of a rat
liver microsomal subcellular fraction in response to
in vivo administration of carbon tetrachloride. J Biol Chem, 25:
1197-1201.
Morele Y, Lefevre C, Ferrari P, Guenier JP, & Muller J (1989) Sampling
and analysis of airborne chlorinated methanes -- comparison of FID and
ECD. Chromatographia, 28: 617-619.
Morgan A, Black A, & Belcher DR (1970) The excretion in breath of some
aliphatic halogenated hydrocarbons following administration by
inhalation. Ann Occup Hyg, 13: 219-233.
Muños Torres E, Paz Bouza JI, Abad Hernandez MM, Alonso Martin MJ, &
Lopez Bravo A (1988) Experimental carbon tetrachloride-induced
cirrhosis of the liver. Int J Tissue React, X: 245-251.
Murphy SD & Malley S (1969) Effect of carbon tetrachloride on
induction of liver enzymes by acute stress or corticosterone. Toxicol
Appl Pharmacol, 15: 117-130.
Nagano K, Nishizava T, Yamamoto S, & Matsushima T (1998) Inhalation
carcinogenesis studies of six halogenated hydrocarbons in rats and
mice. In: Chiyotani K, Hosoda Y, & Aizawa Y ed. Advances in the
prevention of occupational respiratory diseases. Amsterdam, Oxford,
New York, Elsevier Science Publishers, pp 741-746.
Nakajima T, Koyama Y, & Sato A (1982) Dietary modification of
metabolism and toxicity of chemical substances with special reference
to carbohydrate. Biochem Pharmacol, 31: 1005-1011.
Nakamura SI, Oda Y, Shimada T, Oki I, & Sugimoto K (1987) SOS-inducing
activity of chemical carcinogens and mutagens in
Salmonella typhimurium TA1535/pSK1002: examination with 151
chemicals. Mutat Res, 192: 239-246.
Nakata R, Tsukamoto I, Miyoshi M, & Kojo S (1985) Liver regeneration
after carbon tetrachloride intoxication in the rat. Biochem Pharmacol,
34: 586-588.
Nakayama T, Kaneko M, & Kodama M (1986) Detection of DNA damage in
cultured human fibroblasts induced by methyl linoleate hydroperoxide.
Agric Biol Chem, 50: 261-262.
Narotsky MG, Pegram RA, & Kavlock RJ (1997a) Effect of dosing vehicle
on the developmental toxicity of bromodichloromethane and carbon
tetrachloride in rats. Fundam Appl Toxicol, 40: 30-36.
Narotsky MG, Brownie CF, & Kavlock RJ (1997b) Critical period of
carbon tetrachloride-induced pregnancy loss in Fischer-344 rats, with
insights into the detection of resorption sites by ammonium sulfide
staining. Teratology, 56: 252-261.
Nath RG, Li DH, & Randerath K (1990) Acute and long-term effects of
carbon tetrachloride on DNA modifications (I-compounds) in male mouse
liver. Chem-Biol Interact, 76: 343-357.
National Library of Medicine (1992) Hazardous substances data bank.
Bethesda, Maryland, National Library of Medicine, National Toxicology
Information Programme.
National Library of Medicine (1997) Hazardous substances data bank.
Bethesda, Maryland, National Library of Medicine, National Toxicology
Information Programme.
Neely WB, Branson DR, & Blau GE (1974) Partition coefficient to
measure bioconcentration potential of organic chemicals in fish.
Environ Sci Technol, 8: 1113-1115.
Neuhauser EF, Loehr RC, Malecki MR, Milligan DL, & Durkin PR (1985)
The toxicity of selected organic chemicals to the earthworm
Eisenia foetida. J Environ Qual, 14: 383-388.
NIOSH (1977) Manual of analytical methods -- Part 2: Standards
completion program validated methods 3:S314. Cincinnati, Ohio,
National Institute of Occupational Safety and Health, pp 1-9.
NIOSH (1984) Manual of analytical methods: Method 1003. Cincinnati,
Ohio, National Institute of Occupational Safety and Health, pp 1-9.
Noguchi T, Fong K-L, Lai EK, Olson L, & McCay PB (1982a) Selective
early loss of polypeptides in liver microsomes of CCl4-treated rats.
Relationship to cytochrome P-450 content. Biochem Pharmacol, 31:
609-614.
Noguchi T, Fong K-L, Lai EK, Alexander SS, King MM, Olson L, Poyer JL,
& McCay PB (1982b) Specificity of a phenobarbital-induced cytochrome
P-450 for metabolism of carbon tetrachloride to the trichloromethyl
radical. Biochem Pharmacol, 31: 615-624.
Noll T & De Groot H (1984) The critical steady-state hypoxic
conditions in carbon tetrachloride-induced lipid peroxidation in rat
liver microsomes. Biochim Biophys Acta, 795: 356-362.
Norpoth KH, Reisch KA, & Heinecke A (1980) Biostatistics of Ames-test
data. In: Norpoth KH & Garmer RC ed. Short-term systems for detecting
carcinogens. Berlin, Springer-Verlag, pp 312-323.
Norwood WD, Fuqua PA, & Scudder BC (1950) Carbon tetrachloride
poisoning. Arch Ind Hyg Occup Med, 1: 90-100.
Öenfelt A (1987) Spindle disturbances in mammalian cells - III.
Toxicity, c-mitosis and aneuploidy with 22 different compounds:
Specific and unspecific mechanisms. Mutat Res, 182: 135-154.
Oldenhuis R, Vink RJLM, Janssen DB, & Witholt B (1989) Degradation of
chlorinated aliphatic hydrocarbons by
Methylosinus trichosporium OB3b expressing soluble methane
monooxygenase. Appl Environ Microbiol, 55: 2819-2826.
Oraumbo IF & Van Duuren BL (1989) Evidence for the covalent
interaction of carbon tetrachloride with mouse liver chromatin DNA
in vivo. J Environ Pathol Toxicol Oncol, 9: 13-18.
Ott MG, Carlo GL, Steinberg S, & Bond GG (1985) Mortality among
employees engaged in chemical manufacturing and related activities. Am
J Epidemiol, 122: 311-322.
Ozeki T, Iwaki K, Taoka Y, Yamashina A, Ouchi K, & Kan M (1982) The
effects of vitamin E on the indicator enzymes of organelle membranes
in the injured liver. Gastroenterol Jpn, 17: 441-446.
Pääkkö P, Anttila S, Sormunen R, Ala-Kokko L, Peura R, Ferrans VJ, &
Ryhanen L (1996) Biochemical and morphological characterization of
carbon tetrachloride-induced lung fibrosis in rats. Arch Toxicol, 70:
540-552.
Packer JE, Slater TF, & Willson RL (1978) Reactions of the carbon
tetrachloride-related peroxy free radical (CCl3O2.) with amino
acids: Pulse radiolysis evidence. Life Sci, 23: 2617-2620.
Page DA & Carlson GP (1994) The role of the intestinal tract in the
elimination of carbon tetrachloride. Toxicol Appl Pharmacol, 124:
268-274.
Parola M, Leonarduzzi G, Biasi F, Albano E, Biocca ME, Poli G, &
Dianzani M (1992) Vitamin E dietary supplementation protects against
carbon tetrachloride-induced chronic liver damage and cirrhosis.
Hepatology, 16: 1014-1021.
Parsons F, Barrio Lage G, & Rice R (1985) Biotransformation of
chlorinated organic solvents in static microcosms. Environ Toxicol
Chem, 4: 739-742.
Pasquali-Ronchetti I, Bini A, Botti B, De Alojsio G, Fornieri C, &
Vannini V (1980) Ultrastructural and biochemical changes by
progressive lipid peroxidation on isolated microsomes and rat liver
endoplasmic reticulum. Lab Invest, 42: 457-468.
Paul BB & Rubinstein D (1963) Metabolism of carbon tetrachloride and
chloroform by the rat. J Pharmacol Exp Ther, 141: 141-148.
Paustenbach DJ, Carlson GP, Christian JE, & Born GS (1986a) A
comparative study of the pharmacokinetics of carbon tetrachloride in
the rat following repeated inhalation exposures of 8 and 11.5-hr/day.
Fundam Appl Toxicol, 6: 484-497.
Paustenbach DJ, Christian JE, Carlson GP, & Born GS (1986b) The effect
of an 11.5-hr/day exposure schedule on the distribution and toxicity
of inhaled carbon tetrachloride in the rat. Fundam Appl Toxicol, 6:
472-483.
Paustenbach DJ, Clewell HJ, Gargas ML, & Andersen ME (1988) A
physiologically based pharmacokinetic model for inhaled carbon
tetrachloride. Toxicol Appl Pharmacol, 96: 191-211.
Pearson CR & McConnell G (1975) Chlorinated C1 and C2 hydrocarbons
in the marine environment. Proc R Soc Lond B, 189: 305-332.
Pellizzari ED, Sheldon LS, & Bursey JT (1985) Method 25: GC/MS
determination of volatile halocarbons in blood and tissue. Environ
Carcinog Sel Methods Anal, 7: 435-444.
Pentz R & Strubelt O (1983) Fasting increases the concentration of
carbon tetrachloride and of its metabolite chloroform in the liver of
mice. Toxicol Lett, 16: 231-234.
Peoples AJ, Pfaffenberger CD, Shafik TM, & Enos HF (1979)
Determination of volatile purgeable halogenated hydrocarbons in human
adipose tissue and blood serum. Bull Environ Contam Toxicol, 23:
244-249.
Perocco P & Prodi G (1981) DNA Damage by haloalkanes in human
lymphocytes cultured in vitro. Cancer Lett, 13: 213-218.
Pierotti D, Rasmussen RA, & Dalluge R (1980) Measurements of N2O,
CF2Cl2, CFCl3, CH3CCl3, CCl4, CH3Cl in the troposphere and
lower stratosphere over North America. J Geomagn Geoelectr, 32:
181-205.
Plummer JL, Hall PD, Ilsley AH, Jenner MA, & Cousins MJ (1990)
Influence of enzyme induction and exposure profile on liver injury due
to chlorinated hydrocarbon inhalation. Pharmacol Toxicol, 67: 329-335.
Plummer JL, Hall PD, Ilsley AH, Cmielewski PL, Ahern MJ, & Williams RA
(1994) Dose-response relationships in hepatic injury produced by
alcohol and carbon tetrachloride. Alcohol Clin Exp Res, 18(6):
1523-1526.
Pohl LR, Branchflower RV, Highet RJ, Martin JL, Nunn DS, Monks TJ,
George JW, & Hinson JA (1981) The formation of diglutathionyl
dithiocarbonate as a metabolite of chloroform, bromotrichloromethane,
and carbon tetrachloride. Drug Metab Dispos, 9: 334-339.
Pohl LR, Schulick RD, Highet RJ, & George JW (1984)
Reductive-oxygenation mechanism of metabolism of carbon tetrachloride
to phosgene by cytochrome P-450. Mol Pharmacol, 25: 318-321.
Poyer JL, Floyd RA, McCay PB, Janzen EG, & Davis ER (1978)
Spin-trapping of the trichloromethyl radical produced during enzymic
NADPH oxidation in the presence of carbon tetrachloride or
bromotrichloromethane. Biochem Biophys Acta, 539: 402-409.
Poyer JL, McCay PB, Lai EK, Janzen EG, & Davis ER (1980) Confirmation
of assignment of the trichloromethyl radical spin adduct detected by
spin trapping during 13C-carbon tetrachloride metabolism
in vitro and in vivo. Biochem Biophys Res Commun, 94: 1154-1160.
Prendergast JA, Jones RA, Jenkins LJ, & Siegel J (1967) Effects on
experimental animals of long-term inhalation of trichloroethylene,
carbon tetrachloride, 1,1,1-trichloroethane, dichlorodifluoromethane,
and 1,1-dichloroethylene. Toxicol Appl Pharmacol, 10: 270-289.
Prinn RG, Simmonds PG, Rasmussen RA, Rosen RD, Alyea FN, Cardelino CA,
Crawford AJ, Cunnold DM, Fraser PJ, & Lovelock JE (1983) The
atmospheric lifetime experiment. I. Introduction, instrumentation, and
overview. J Geophys Res, 88: 8353-8367.
Pritchard DJ, Wright MG, Sulsh S, & Butler WH (1987) The assessment of
chemically induced liver injury in rats. J Appl Toxicol, 7: 229-236.
Puri EC & Müller D (1989) Testing of hydralizine in
in vivo-in vitro hepatocyte assays for UDS and stimulation of
replicative DNA synthesis. Mutat Res, 218: 13-19.
Qazi FM & Alam SM (1988) Potentiating effects of phenobarbitone on the
induction of cirrhosis in rats by carbon tetrachloride. J Pak Med
Assoc, 38(11): 296-299.
Rao SB & Mehendale HM (1989) Protective role of fructose
1,6-biphosphate during CCl4 hepatotoxicity in rats. Biochem J, 262:
721-725.
Rasmussen RA & Khalil MAK (1983) Natural and anthropogenic trace gases
in the lower troposphere of the Arctic. Chemosphere, 12: 371-375.
Rasmussen RA & Khalil MAK (1986) Atmospheric trace gases: Trends and
distributions over the last decade. Science, 232: 1623-1624.
Rasmussen RA, Khalil MAK, & Dalluge RW (1981) Atmospheric trace gases
in Antarctica. Science, 211: 285-287.
Raucy JL, Kraner JC, & Lasker JM (1993) Bioactivation of halogenated
hydrocarbons by cytochrome P4502E1. Crit Rev Toxicol, 23: 1-20.
Ray SD & Mehendale HM (1990) Potentiation of CCl4 and CHCl3
hepatotoxicity and lethality by various alcohols. Fundam Appl
Toxicol, 15: 429-440.
Raymond P & Plaa GL (1995a) Ketone potentiation of haloalkane-induced
hepato- and nephrotoxicity: I. Dose-response relationships. J Toxicol
Environ Health, 45: 465-480.
Raymond P & Plaa GL (1995b) Ketone potentiation of haloalkene-induced
hepato- and nephrotoxicity: II. Implication of monooxygenases. J
Toxicol Environ Health, 46: 317-328.
Raymond P & Plaa GL (1996) Ketone potentiation of haloalkene-induced
hepatotoxicity: CCl4 and ketone treatment on hepatic membrane
integrity. J Toxicol Environ Health, 49: 285-300.
Raymond P & Plaa GL (1997) Effect of dosing vehicle on the
hepatotoxicity of CCl4 and nephrotoxicity of CHCl3 in rats. J
Toxicol Environ Health, 51: 463-476.
Recknagel RO (1983) A new direction in the study of carbon
tetrachloride hepatotoxicity. Life Sci, 33: 401-408.
Recknagel RO & Glende EA Jr (1973) Carbon tetrachloride
hepatotoxicity: An example of lethal cleavage. CRC Crit Rev Toxicol,
2: 263-297.
Reiner O, Athanassopoulos S, Hellmer KH, Murray RE, & Uehleke H (1972)
[Formation of chloroform from carbon tetrachloride in liver
microsomes, lipid peroxidation and destruction of cytochrome P-450.]
Arch Toxikol, 29(3): 219-233 (in German).
Reinke LA, Lai EK, & McCay PB (1988) Ethanol feeding stimulates
trichloromethyl radical formation from carbon tetrachloride in liver.
Xenobiotica, 18: 1311-1318.
Reuber MD & Glover EL (1970) Cirrhosis and carcinoma of the liver in
male rats given subcutaneous carbon tetrachloride. J Natl Cancer Inst,
44: 419-427.
Reynolds LF & Harrison DW (1982) Study of discharge of benzene,
chloroform and carbon tetrachloride into the aquatic environment and
the best technical means for the reduction of water pollution from
such discharges -- Final report. Brixham, United Kingdom, ICI
Laboratory (Report No. BL/A/2189).
Reynolds LF, Harrison DW, & Carter L (1982) Contract No. ENV/223/74-EN
-- Third revision. Luxembourg, Commission of the European Communities,
Environment and Consumer Protection Service, pp 103-107.
Reynolds ES, Treinen RJ, Farrish HH, & Treinen Moslen M (1984)
Metabolism of [14C]carbon tetrachloride to exhaled, excreted and
bound metabolites. Biochem Pharmacol, 33: 3363-3374.
Richmond RE, DeAngelo AB, & Daniel FB (1992) Immunohistochemical
detection of ras and myc oncogene expression in regenerating rat
liver. Toxicol Lett, 60: 119-129.
Rivett MO, Lerner DN, Lloyd JW, & Clark L (1990a) Organic
contamination of the Birmingham aquifer, UK. J Hydrol, 113: 307-323.
Rivett MO, Lerner DN, & Lloyd JW (1990b) Chlorinated solvents in the
UK aquifers. J Inst Water Environ Manage, 4: 242-250.
Rogers HR, Crathorne B, & Watts CD (1992) Sources and fate of organic
contaminants in the Mersey Estuary: volatile organohalogen compounds.
Mar Pollut Bull, 24: 82-91.
Roldan-Arjona T & Pueyo C (1993) Mutagenic and lethal effects of
halogenated methanes in the Ara test of Salmonella typhimurium:
quantitative relationship with chemical reactivity. Mutagenesis, 8:
127-131.
Roldan-Arjona T, Garcia-Pedrajas MD, Luque-Romero FL, Hera C, & Pueyo
C (1991) An association between mutagenicity of the Ara test of
Salmonella typhimurium and carcinogenicity in rodents for 16
halogenated aliphatic hydrocarbons. Mutagenesis, 6: 199-205.
Rosen ME, Pankow JF, Gibs J, & Imbrigiotta TE (1992) Comparison of
downhole and surface sampling for the determination of volatile
organic compounds (VOC) in ground water. Ground Water Monit Rev, 12:
126-133.
Rosengren RJ, Sauer JM, Hooser SB, & Sipes IG (1995) The interactions
between retinol and five different hepatotoxicants in the Swiss
Webster mouse. Fundam Appl Toxicol, 25: 281-292.
Roudabush RL, Terhaar CJ, Fassett DW, & Dziuba SP (1965) Comparative
acute effects of some chemicals on the skin of rabbits and guinea
pigs. Toxicol Appl Pharmacol, 7: 559-565.
Rowland FS (1985) Methane and chlorocarbons in the earth's atmosphere.
Orig Life, 15: 279-297.
Rubinstein D & Kanics L (1964) The conversion of carbon tetrachloride
and chloroform to carbon dioxide by rat liver homogenates. Can J
Biochem, 42: 1577-1585.
Rudolph J & Jebsen C (1983) The use of photoionization, flame
ionization and electron capture detectors in series for the
determination of low molecular weight trace components in the
non-urban atmosphere. Intern. J Environ Anal Chem, 13: 129-139.
Ruprah M, Mant TGK & Flanagan RJ (1985) Acute carbon tetrachloride
poisoning in 19 patients: implications for diagnosis and treatment.
Lancet, May 4: 1027-1029.
Russell JW & Shadoff LA (1977) The sampling and determination of
halocarbons in ambient air using concentration on porous polymer. J
Chromatogr, 134(2):375-384.
Sagai M & Tappel AL (1979) Lipid peroxidation induced by some
halomethanes as measured by in vivo pentane production in the rat.
Toxicol Appl Pharmacol, 49: 283-291.
Sanzgiri UY, Kim HJ, Muralidhara S, Edallas E & Bruckner JV (1995)
Effect of route and pattern of exposure on the pharmacokinetics and
acute hepatotoxicity of carbon tetrachloride. Tox Appl Pharmacol 134:
148-154.
Sanzgiri UY & Bruckner JV (1997) Effect of Emulphor, an emulsifier, on
the pharmacokinetics and hepatotoxicity of oral carbon tetrachloride
in the rat. Fundam Appl Toxicol, 36: 54-61.
Sato A & Nakajima T (1985) Enhanced metabolism of volatile
hydrocarbons in rat liver following food deprivation, restricted
carbohydrate intake, and administration of ethanol, phenobarbital,
polychlorinated biphenyl and 3-methylcholanthrene: a comparative
study. Xenobiotica, 15: 67-75.
Sato A, Nakajima T, & Koyama Y (1980) Effects of chronic ethanol
consumption on hepatic metabolism of aromatic and chlorinated
hydrocarbons in rats. Br J Ind Med, 37: 382-286.
Sauer JM & Sipes IG (1995) Modulation of chemical-induced lung and
liver toxicity by all-trans-retinol in the male Sprague-Dawley rat.
Toxicology, 105: 237-249.
Sawada S, Yamanaka T, Yamatsu K, Furihata C, & Matsushima T (1991)
Chromosome aberrations, micronuclei and sister-chromatid exchanges
(SCEs) in rat liver induced in vivo by hepatocarcinogens including
heterocyclic amines. Mutat Res, 251: 59-69.
Schiestl RH, Gietz RD, Mehta RD, & Hastings PJ (1989) Carcinogens
induce intrachromosomal recombination in yeast. Carcinogenesis, 10:
1445-1455.
Schwarz M, Hummel J, Appel KE, Rickert R, & Kunz W (1979) DNA damage
induced in vivo evaluated with a non-radioactive alkaline elution
technique. Cancer Lett, 6: 221-226.
Schwetz BA, Leong BKJ, & Gehring PJ (1974) Embryo- and fetotoxicity of
inhaled carbon tetrachloride, 1,1-dichloroethane and methyl ethyl
ketone in rats. Toxicol Appl Pharmacol, 28: 452-464.
Shah JJ & Heyerdahl EK (1988) National ambient volatile organic
compounds (VOCs) data base update. Research Triangle Park, North
Carolina, US Environmental Protection Agency (PB88-195631).
Shah H, Hartman SP, & Weinhouse S (1979) Formation of carbonyl
chloride in carbon tetrachloride metabolism by rat liver
in vitro. Cancer Res, 39: 3942-3947.
Shapiro AA & Fonshtein LM (1979) Study of the mutagenic action of
cyclophosphane on bacteria in host-mediated assay. Biol Bull Acad Sci
(USSR), 6: 310-315.
Shen ES, Garry VF, & Anders MW (1982) Effect of hypoxia and carbon
tetrachloride hepatotoxicity. Biochem Pharmacol, 31: 3783-3793.
Shertzer HG, Reitman FA, & Tabor MW (1988) Influence of diet on the
expression of hepatotoxicity from carbon tetrachloride in ICR mice.
Drug-Nutr Interact, 5: 275-282.
Shibayama Y (1988) Hepatotoxicity of carbon tetrachloride after
chronic ethanol consumption. Exp Mol Pathol, 49: 234-242.
Shimizu Y, Nagase C, & Kawai K (1973) Accumulation and toxicity of
carbon tetrachloride after repeated inhalation in rats. Ind Health,
11: 48-54.
Shinozuka H (1971) Unusual membrane alterations in the rough
endoplasmic reticulum of rat liver cells after carbon tetrachloride
injury. Exp Cell Res, 64: 380-386.
Siegers C-P, Horn W, & Younes M (1985) Effect of hypoxia on the
metabolism and hepatotoxicity of carbon tetrachloride and vinylidene
chloride in rats. Acta Pharmacol Toxicol, 56: 81-86.
Siemiatycki J ed. (1991) Risk factors for cancer in the work place.
Boca Raton, Florida, CRC Press, 310 pp.
Simko V, Michael S, Katz J, Oberstein E, & Popescu A (1992) Protective
effect of oral acetylcysteine against the hepatorenal toxicity of
carbon tetrachloride potentiated by ethyl alcohol. Alcohol Clin Exp
Res, 16: 759-799.
Simmon VF & Tardiff RG (1978) Mutagenic activity of halogenated
compounds found in chlorinated drinking waters. In: Proceedings of the
Conference on Water Chlorination Environmental Impact and Health
Effects, vol 2, pp 417-431.
Simmon VF, Kavhanen K, & Tardiff RG (1977) Mutagenic activity of
chemicals identified in drinking waters. In: Scott D, Bridges BA, &
Sobesl FH ed. Progress in genetic toxicology. Amsterdam, Oxford, New
York, Elsevier/North-Holland Biochemical Press, pp 249-258.
Simmonds PG & Kerns E (1979) Direct aqueous injection gas
chromatography for the analysis of trace organics in water. J
Chromatogr, 186: 785-794.
Simmonds PG, Alyea FN, Cardelino CA, Crawford AJ, Cunnold DM, Lane BC,
Lovelock JE, Prinn RG, & Rasmussen RA (1983) The atmospheric lifetime
experiment: 6. Results for carbon tetrachloride based on 3 years data.
J Geophys Res, 88: 8427-8441.
Simmonds PG, Cunnold DM, Alyea FN, Cardelino CA, Crauford AJ, Prinn
RG, Fraser PJ, Rasmussen RA, & Rosen RD (1988) Carbon tetrachloride
lifetimes and emissions determined from daily global measurements
during 1978-1985. J Atmos Chem, 7: 35-58.
Simmons JE, McDonald A, Seely JC, & Sey YM (1995) Potentiation of
carbon tetrachloride hepatotoxicity by inhaled methanol: Time course
of injury and recovery. J Toxicol Environ Health, 46: 203-216.
Sina JF, Bean CL, Dysart GR, Taylor VI, & Bradley MO (1983) Evaluation
of the alkaline elution/rat/hepatocyte assay as a predictor of
carcinogenic/mutagenic potential. Mutat Res, 113: 357-391.
Singh HB, Lillian D, Appleby A, & Lobban L (1975) Atmospheric
formation of carbon tetrachloride from tetrachloroethylene. Environ
Lett, 10: 253-256.
Singh HB, Fowler DP, & Peyton TO (1976) Atmospheric carbon
tetrachloride: another man-made pollutant. Science, 192: 1231-1234.
Singh HB, Salas L, Shigeishi H, & Crawford A (1977) Urban -- nonurban
relationships of halocarbons, SF6, N2O and other atmospheric trace
constituents. Atmos Environ, 11: 819-828.
Singh HB, Salas LJ, Shigeishi H, & Scribner E (1979) Atmospheric
halocarbons, hydrocarbons, and sulfur hexafluoride: global
distributions, sources and sinks. Science, 203: 899-903.
Singh HB, Salas LJ, Smith AJ, & Shigeishi H (1980) Measurements of
some potentially hazardous organic chemicals in urban environments.
Atmos Environ, 15: 601-612.
Singh HB, Salas LJ, & Stiles RE (1982) Distribution of selected
gaseous organic mutagens and suspect carcinogens in ambient air.
Environ Sci Technol, 16: 872-880.
Singh HB, Salas LJ, & Stiles RE (1983) Selected man-made halogenated
chemicals in the air and oceanic environment. J Geophys Res, 88:
3675-3683.
Sipes IG, Krishna G, & Gillette JR (1977) Bioactivation of carbon
tetrachloride, chloroform and bromotrichloromethane: role of
cytochrome P-450. Life Sci, 20: 1541-1548.
Slater TF (1982) Activation of carbon tetrachloride: chemical
principles and biological significance. In: McBrien DCH & Slater TF
ed. Free radicals, lipid peroxidation and cancer. New York, London,
San Francisco, Academic Press.
Smejkalová J, Simek J, Rouchal J, & Dvorácková I (1985) The time
course of biochemical and histological changes following carbon
tetrachloride-induced liver damage in rats of both sexes. Physiol
Bohemoslov, 34: 494-501.
Smetana H (1939) Nephrosis due to carbon tetrachloride. Arch Intern
Med, 63: 760-777.
Smialowicz RJ, Simmons JE, Luebke RW, & Allis JW (1991)
Immunotoxicologic assessment of subacute exposure of rats to carbon
tetrachloride with comparison to hepatotoxicity and nephrotoxicity.
Fundam Appl Toxicol, 17: 186-196.
Smyth HF, Weil CS, West JS, & Carpenter CP (1970) An exploration of
joint toxic action: II. Equitoxic versus equivolume mixtures. Toxicol
Appl Pharmacol, 17: 498-503.
Srivastava SP, Chen NQ, & Holtzman JL (1990) The
in vitro NADPH-dependent inhibition by CCl4 of the ATP-dependent
calcium uptake of hepatic microsomes from male rats. J Biol Chem, 265:
8392-8399.
Staples CA, Werner AF, & Hoogheem TJ (1985) Assessment of priority
pollutant concentrations in the United States using STORET database.
Environ Toxicol Chem, 4: 131-142.
Statham CN, Croft WA, & Lech JJ (1978) Uptake, distribution and
effects of carbon tetrachloride in rainbow trout
(Salmo gairdneri). Toxicol Appl Pharmacol, 45: 131-140.
Stewart BW (1981) Generation and persistence of carcinogens-induced
repair intermediates in rat liver DNA in vivo. Cancer Res, 41:
3238-3243.
Stewart RD & Dodd HC (1964) Absorption of carbon tetrachloride,
trichloroethylene, tetrachloroethylene, methylene chloride, and
1,1,1,-trichloroethane through the human skin. Am Ind Hyg Assoc J, 25:
439-446.
Stewart RD, Arbor A, Gay HH, Erley DS, Hake CL, & Peterson JE (1961)
Human exposure to carbon tetrachloride vapor. J Occup Med, 3: 586-590.
Stewart PA, Lee JS, Marano DE, Spirtas R, Forbes CD, & Blair A (1991)
Retrospective cohort mortality study of workers at an aircraft
maintenance facility: II. Exposures and their assessment. Br J Ind
Med, 48: 531-537.
Stoyanovsky DA & Cederbaum AI (1996) Thiol oxidation and cytochrome
P450-dependent metabolism of CCl4 triggers Ca2+ release from liver
microsomes. Biochemistry, 35: 15839-15845.
Striker GE, Smuckler EA, Kohnen PW, & Nagle RB (1968) Structural and
functional changes in rat kidney during CCl4 intoxication. Am J
Pathol, 53: 769-789.
Strubelt O (1984) Alcohol potentiation of liver injury. Fundam Appl
Toxicol, 4: 144-151.
Su C & Goldberg E (1976) Environmental concentrations and fluxes of
some halocarbons. Mar Pollut Transf, 14: 353-374.
Suffet IH, Gibs J, Coyle JA, Chrobak RS, & Yohe TL (1985) Applying
analytical techniques to solve ground water problems. J Am Water Works
Assoc, 77: 65-72.
Sugama Y, Kubo M, Nonaka T, Nozawa T, & Tsuchihasi Y (1995)
[Measurement of atmospheric concentration of low boiling point organic
chlorine compounds and CFCs.] Tochigi-ken Kogai Kenkyusho Nenpo, 19:
66-75 (in Japanese with English abstract).
Suzuki H, Hirano N, Watanabe C, & Tarumoto Y (1997) Carbon
tetrachloride does not induce micronucleus in either mouse bone marrow
or peripheral blood. Mutat Res, 394: 77-80.
Svirbely JL, Highman B, Alford WC, & Von Oettingen WF (1947) The
toxicity and narcotic action of monochloro-monobromomethane with
special reference to inorganic and volatile bromide in blood, urine
and brain. J Ind Hyg Toxicol, 29: 382-389.
Szende B, Timár F, & Hargitai B (1994) Olive oil decreases liver
damage in rats caused by carbon tetrachloride (CCl4). Exp Toxicol
Pathol, 46: 355-359.
Tabak HH, Quave SA, Mashni CI, & Barth EF (1981) Biodegradability
studies with organic priority pollutant compounds. J Water Pollut
Control Fed, 53: 1503-1518.
Taketomo AP & Grimsrud E (1977) An analysis of halocarbons in the air
of several working and living environments. Proc Mont Acad Sci, 37:
128-134.
Tátrai E, Hudák A, & Ungváry G (1979) Simultaneous effect on the rat
liver of benzene, toluene, xylene and CCl4. Acta Physiol Acad Sci
Hung, 53: 261 (abstract).
Teschke R, Vierke W, & Goldermann L (1983) Carbon tetrachloride
(CCl4) levels and serum activities of liver enzymes following acute
CCl4 intoxication. Toxicol Lett, 17: 175-180.
Teschke R, Vierke W, & Gellert J (1984) Effect of ethanol on carbon
tetrachloride levels and hepatotoxicity after acute carbon
tetrachloride poisoning. Arch Toxicol, 56: 78-82.
Teta MJ & Ott MG (1988) A mortality study of a research, engineering,
and metal fabrication facility in Western New York State. Am J
Epidemiol, 127: 540-551.
Thiersch JB (1971) Differential effect of compounds on rat litter and
mother. In: Tuchmann-Duplesis H ed. Malformations congénitales des
mammifères. Paris, Masson & Co., pp 99-100.
Tierney DJ, Haas AL, & Koop DR (1992) Degradation of cytochrome
P4502E1: selective loss after labilization of the enzyme. Arch Biochem
Biophys, 293: 9-16.
Tirmenstein MA, Leraas TL, & Fariss MW (1997) Alpha-tocopheryl
hemisuccinate administration increases rat liver subcellular
alpha-tocopherol levels and protects against carbon
tetrachloride-induced hepatotoxicity. Toxicol Lett, 92: 67-77.
Tjälve H & Löfberg B (1983) Extrahepatic sites of metabolism of carbon
tetrachloride in rats. Chem-Biol Interact, 46: 299-316.
Tomasi A, Albano E, Lott KAK, & Slater TF (1980) Spin trapping of free
radical products of CC14 activation using pulse radiolysis and high
energy radiation procedures. FEBS Lett, 122: 303-306.
Tomenson JA, Baron CE, O'Sullivan JJ, Edwards JC, Stonard MD, Walker
RJ, & Fearnley DM (1995) Hepatic function in workers occupationally
exposed to carbon tetrachloride. Occup Environ Med, 52: 508-514.
Toncsev H, Pollák ZS, Kiss A, Sréter L, & Fehér J (1982) Acute carbon
tetrachloride-induced lysosomal membrane damage and the membrane
protecting effect of a new dihydroquinoline-type antioxidant. Int J
Tissue React, 4: 325-330.
Topham JC (1980) Do sperm-head abnormalities in mice specifically
identify mammalian mutagens rather than carcinogens? Mutat Res, 74:
379-387.
Towner RA, Reinke LA, Janzen EG, & Yamashiro S (1994)
In vivo magnetic resonance imaging study of Kupfer cell involvement
in CCl4-induced hepatotoxicity in rats. Can J Physiol Pharmacol, 72:
441-446.
Tracey JP & Sherlock P (1968) Hepatoma following carbon tetrachloride
poisoning. NY State J Med, 68: 2202-2204.
Traiger GJ & Plaa GL (1971) Differences in the potentiation of carbon
tetrachloride in rats by ethanol and isopropanol pretreatment. Toxicol
Appl Pharmacol, 20: 105-112.
Tsuruta H (1975) Percutaneous absorption of organic solvents: I.
Comparative study of the in vivo percutaneous absorption of
chlorinated solvents in mice. Ind Health, 13: 227-236.
Uehleke H, Hellmer KH, & Tabarelli S (1973) Binding of 14C-carbon
tetrachloride to microsomal proteins in vitro and formation of
CHCl3 by reduced liver microsomes. Xenobiotica, 3: 1-11.
Uehleke H, Greim H, Krämer M, & Werner T (1976) Covalent binding of
haloalkanes to liver constituents but absence of mutagenicity on
bacteria in a metabolizing test system. Mutat Res, 38: 114.
Uehleke H, Werner T, Greim H, & Krämer M (1977) Metabolic activation
of haloalkanes and tests in vitro for mutagenicity. Xenobiotica, 7:
393-400.
Uemitsu N (1986) Inhalation pharmacokinetics of carbon tetrachloride
in rats based on arterial blood: inhaled air concentration ratios.
Toxicol Appl Pharmacol, 83: 20-29.
Uemitsu N, Minobe Y, & Nakayoshi H (1985) Concentration-time-response
relationship under conditions of single inhalation of carbon
tetrachloride. Toxicol Appl Pharmacol, 77: 260-266.
Ukai H, Inui S, Takada S, Dendo J, Ogawa J, Isobe K, Ashida T, Tamura
M, Tabuki K, & Ikeda M (1997) Types of organic solvents used in
small- to medium-scale industries in Japan; a nationwide field survey.
Int Arch Occup Environ Health, 70: 385-392.
Ullrich D (1982) [Organohalogen compounds in the air of some indoor
swimming pools in Berlin.] WaBoLu-Berlin, 1: 50-52 (in German).
UNEP (1989) Technical progress on protecting the ozone layer: Report
of the Technology Review Panel. Nairobi, Kenya, United Nations
Environment Programme.
UNEP (1996) The 1987 Montreal protocol on substances that deplete the
ozone layer as adjusted and amended by the Second, Fourth and Seventh
Meeting of the Parties. In: Handbook for the international treaties
for the protection of the ozone layer, 4th ed. Nairobi, Kenya, United
Nations Environment Programme, pp 18-39.
Uryvaeva IV & Delone GV (1995) An improved method of mouse liver
micronucleus analysis: An application to age-related genetic
alteration and polyploidy study. Mutat Res, 334(1): 71-80.
US EPA (1984a) Locating and estimating air emissions from sources of
carbon tetrachloride. Washington, DC, US Environmental Protection
Agency (Report No. 450/4-84-0067 b).
US EPA (1984b) Health assessment document for carbon tetrachloride.
Washington, DC, US Environmental Protection Agency (Report No.
600/8-82-001F).
US EPA (1991) Toxics in the community: National and local
perspectives. Washington, DC, US Environmental Protection Agency,
Office of Toxic Substances (EPA 560/4-91-014).
Vannelli T, Logan M, Arciero DM, & Hooper AB (1990) Degradation of
halogenated aliphatic compounds by the ammonia-oxidising bacterium
Nitrosomonas europaea. Appl Environ Microbiol, 56: 1169-1171.
Van Rensburg JFJ, Van Huyssteen JJ, & Hassett AJ (1978) A
semi-automated technique for the routine analysis of volatile
organohalogens in water purification process. Water Res, 12: 127-131.
Van Tassel S, Amalfitano N, & Narang RS (1981) Determination of arenes
and volatile haloorganic compounds in air at microgram per cubic meter
levels by gas chromatography. Anal Chem, 53: 2130-2135.
Van Zoest R & Van Eck GTM (1991) Occurrence and behaviour of several
groups of organic pollutants in the Scheldt estuary. Sci Total
Environ, 103: 57-71.
Veith GD (1978) An evaluation of using partition coefficients and
water solubility to estimate bioconcentration factors for organic
chemicals in fish. In: Aquatic toxicology: Proceedings of the Third
Annual Symposium on Aquatic Toxicology. Philadelphia, Pennsylvania,
American Society for Testing and Materials, pp 116-129 (ASTM STP 707).
Veng-Pedersen P, Paustenbach DJ, Carlson GP, & Suarez L (1987) A
linear systems approach to analyzing the pharmacokinetics of carbon
tetrachloride in the rat following repeated exposures of 8 and 11.5
h/day. Arch Toxicol, 60: 355-364.
Versar Inc. (1979) Water related environmental fate of 129 priority
pollutants. Washington, DC, US Environmental Protection Agency, vol 2,
chapter 41 (EPA 440/4-79-029B).
Villarruel M del C, de Toranzo EGD, & Castro JA (1977) Carbon
tetrachloride activation, lipid peroxidation and the mixed function
oxygenase activity of various rat tissues. Toxicol Appl Pharmacol, 41:
337-344.
Villarruel M del C, Fernández G, De Ferreyra EC, De Fenos OM, & Castro
JA (1982) Studies on the mechanism of alloxan-diabetus potentiation of
carbon tetrachloride-induced liver necrosis. Br J Exp Pathol, 63:
388-393.
Von Düszeln J & Thiemann W (1985) Volatile chlorinated hydrocarbons in
a coastal urban atmosphere. Sci Total Environ, 41: 187-194.
Von Oettingen WF (1964) The halogenated hydrocarbons of industrial and
toxicological importance. In: Browning E ed. Elsevier monographs on
toxic agents. Amsterdam, Oxford, New York, Elsevier Science
Publishers, pp 107-170.
Von Oettingen WF, Powell CC, Sharpless NE, Alford WC, & Pecora LJ
(1950) Comparative studies of the toxicity and pharmacodynamic action
of chlorinated methanes with special reference to their physical and
chemical characteristics. Ach Int Pharmacodyn, 81: 17-34.
Wahlberg JE (1984a) Edema-inducing effects of solvents following
topical administration. Dermatosen Beruf Umw, 32: 91-94.
Wahlberg JE (1984b) Erythema-inducing effects of solvents following
epicutaneous administration to man -- Studied by laser Doppler
flowmetry. Scand J Work Environ Health, 10: 159-162.
Wahlberg JE & Boman A (1979) Comparative percutaneous toxicity of ten
industrial solvents in the guinea pig. Scand J Work Environ Health, 5:
345-351.
Wallace LA (1986) Personal exposures, indoor and outdoor air
concentrations and exhaled breath concentrations of selected volatile
organic compounds measured for 600 residents of New Jersey, North
Dakota, North Carolina and California. Toxicol Environ Chem, 12:
215-236.
Waller RL, Glende EA, & Recknagel RO (1983) Carbon tetrachloride and
bromotrichloromethane toxicity. Biochem Pharmacol, 32: 1613-1617.
Walton BT, Anderson TA, Hendricks MS, & Talmage SS (1989)
Physicochemical properties as predictors of organic chemical effects
on soil microbial respiration. Environ Toxicol Chem, 8: 53-63.
Walton BT, Hendricks MS, Anderson TA, Griest WH, Merriweather R,
Beauchamp JJ, & Francis CW (1992) Soil sorption of volatile and
semivolatile organic compounds in a mixture. J Environ Qual, 21:
552-558.
Wang MY & Liehr JG (1995) Lipid hydroperoxidase-induced endogenous DNA
adducts in hamsters: possible mechanism of lipid
hydroperoxide-mediated carcinogenesis. Arch Biochem Biophys, 316:
38-46.
Wang PY, Kaneko T, Tsukada H, Nakano M, & Sato A (1997) Dose- and
route-dependent alterations in metabolism and toxicity of chemical
compounds in ethanol-treated rats: difference between highly
(chloroform) and poorly (carbon tetrachloride) metabolized hepatotoxic
compounds. Toxicol Appl Pharmacol, 142: 13-21.
Watanabe A, Shiota T, Takei N, Fujiwara M, & Nagashima H (1986) Blood
to brain transfer of carbon tetrachloride and lipoperoxidation in rat
brain. Res Commun Chem Pathol Pharmacol, 51: 137-140.
Weisburger EK (1977) Carcinogenicity studies on halogenated
hydrocarbons. Environ Health Perspect, 21: 7-16.
Westrick JJ, Mello JW, & Thomas RF (1984) The groundwater supply
survey. J Am Water Works Assoc, 76: 52-59.
Whittaker SG, Zimmermann FK, Dicus B, Piegorsch WW, Fogel S, & Resnick
MA (1989) Detection of induced mitotic chromosome loss in
Saccharomyces cerevisiae -- an interlaboratory study. Mutat Res,
224: 31-78.
WHO (1993) Guidelines for drinking-water quality -- Volume 1:
Recommendations. Geneva, World Health Organization, pp 57-58.
Wilcosky TC, Checkoway H, Marshall EG, & Tyroler HA (1984) Cancer
mortality and solvent exposures in the rubber industry. J Am Ind Hyg
Assoc, 45: 809-811.
Withey JR, Collins BT, & Collins PG (1983) Effect of vehicle on the
pharmacokinetics and uptake of four halogenated hydrocarbons from the
gastrointestinal tract of the rat. J Appl Toxicol, 3: 249-253.
WMO (1991) Global ozone research and monitoring project. Scientific
assessment of ozone depletion: 1991. Geneva, World Meteorological
Organization (Report No. 25).
Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, & Ullrich V (1977)
The reduction of polyhalogenated methanes by liver microsomal
cytochrome P450. Mol Pharmacol, 13: 698-705.
Wong LCK & DiStefano V (1966) Rapid accumulation of renal fat in cats
after single inhalations of carbon tetrachloride. Toxicol Appl
Pharmacol, 9: 485-494.
Yamamoto HA (1990) Relation of Ca++ accumulation and lipid
peroxidation with CCl4-induced toxicity in the rat liver. Pharmacol
Toxicol, 66: 213-216.
Yao T, Degli Esposito S, Huang L, Arnon R, Spangenberger A, & Zern MA
(1994) Inhibition of carbon tetrachloride-induced liver injury by
liposomes containing vitamin E. Am J Physiol, 267: G476-G484.
Yoshida K (1993) Preliminary exposure assessment of volatile
chlorinated hydrocarbons in Japan. Chemosphere, 27: 621-630.
Youssefi M, Faust SD, & Zenchelsky ST (1978) Rapid determination of
light halogenated hydrocarbons in urine. Clin Chem, 24(7): 1109-1111.
Zawaski K, Gruebele A, Kaplan D, Reddy S, Mortensen A, & Novak RF
(1993) Evidence for enhanced expression c-fos, c-jun, and the
Ca2+-activated neutral protease in rat liver following carbon
tetrachloride administration. Biochem Biophys Res Commun, 197:
585-590.
Zeiger E, Anderson B, Haworth S, Lawlor T, & Mortelmans K (1988)
Salmonella mutagenicity tests: IV. Results from the testing of 300
chemicals. Environ Mol Mutagen, 12 (suppl 11): 1-157.
Zimmerman HJ (1978) Syndromes of environmental hepatotoxicity. In:
Zimmerman HJ ed. Hepatotoxicity -- the adverse effect of drugs and
other chemicals on the liver. New York, Appleton-Century-Crofts, pp
297-302.
Zoeteman BCJ, Harmsen K, Linders JBHJ, Morra CFH, & Slooff W (1980)
Persistent organic pollutants in river water and ground water of the
Netherlands. Chemosphere, 9: 231-249.
RÉSUMÉ
Le tétrachlorure de carbone est un liquide limpide, incolore et
volatil qui dégage une odeur douceâtre caractéristique. Il est
miscible à la plupart des solvants aliphatiques et il est lui-même un
solvant. Il est peu soluble dans l'eau. Le tétrachlorure de carbone
n'est pas inflammable et il est stable à l'air et à la lumière. En se
décomposant, il peut donner naissance à du phosgène, à du dioxyde de
carbone et à de l'acide chlorhydrique.
La présence de tétrachlorure de carbone dans l'environnement est
vraisemblablement presque exclusivement d'origine humaine. La majeure
partie du tétrachlorure de carbone produit sert à la préparation de
chlorofluorocarbures (CFC) et autres hydrocarbures chlorés. En 1987,
la production mondiale de tétrachlorure de carbone a été de 960 000
tonnes. Toutefois, depuis que le Protocole de Montréal relatif aux
substances qui appauvrissent la couche d'ozone (1987) et ses
amendements de 1990 et de 1992, a établi un calendrier pour l'abandon
progressif de la production et de la consommation de tétrachlorure de
carbone, la production a reculé et continuera à le faire.
On a mis au point plusieurs méthodes suffisamment sensibles et
précises pour la recherche et le dosage du tétrachlorure de carbone
dans l'air, l'eau et les milieux biologiques. La plupart d'entre elles
sont basées, soit sur l'injection directe de l'échantillon dans un
chromatographe en phase gazeuse, soit sur une adsorption sur charbon
actif, suivie d'une désorption ou d'une évaporation puis d'une
détection par chromatographie en phase gazeuse.
La presque totalité du tétrachlorure de carbone libéré dans
l'environnement finira tôt ou tard dans l'atmosphère en raison de sa
grande volatilité. Comme sa durée de séjour dans l'atmosphère est
longue, il est largement distribué. Au cours de la période 1980-1990,
la concentration atmosphérique du tétrachlorure de carbone était de
l'ordre de 0,5-1,0 µg/m3. Les estimations de sa durée de séjour
atmosphériques sont variables mais on pense que la valeur la plus
raisonnable est de 45 à 50 ans. Le tétrachlorure de carbone contribue
à la fois à la destruction de la couche d'ozone et au réchauffement du
climat. Il est généralement résistant à la biodégradation aérobie,
mais moins à la biodégradation anaérobie. La vitesse de biodégradation
peut s'accroître par suite d'un processus d'acclimatation. Bien que le
coefficient de partage octanol-eau indique un potentiel de
bioaccumulation moyen, cette tendance est réduite par la brièveté de
la demi-vie tissulaire.
Dans l'eau, on fait état de concentrations inférieures à 10
ng/litre pour les océans et de moins de 1 µg/litre pour les eaux
douces, mais de valeurs beaucoup plus fortes à proximité des sites de
décharge. On a mesuré des valeurs allant jusqu'à 60 µg/kg dans des
denrées alimentaires qui avaient été traitées avec du tétrachlorure de
carbone, mais cette pratique a cessé.
C'est essentiellement par l'intermédiaire de l'air que la
population dans son ensemble est exposée au tétrachlorure de carbone.
Si l'on se base sur les concentrations relevées dans l'air ambiant,
les denrées alimentaires et l'eau de boisson, on peut estimer à
environ 1 µg/kg de poids corporel la dose de tétrachlorure de carbone
absorbée. Cette estimation est probablement un peu forte à l'heure
actuelle, du fait qu'on n'utilise plus de tétrachlorure de carbone
pour la fumigation des céréales et que les concentrations annoncées
pour les aliments et utilisées pour ce calcul, correspondaient tout
particulièrement aux matières grasses et aux produits à base de
céréales. D'autres sources font état de valeurs comprises entre 0,1 et
0,27 µg/kg p.c. pour l'exposition journalière de la population
générale. Une exposition plus importante au tétrachlorure de carbone
peut se produire sur le lieu de travail en cas de déversement
accidentel.
Le tétrachlorure de carbone est bien résorbé au niveau des voies
digestives et respiratoires de l'Homme et des animaux. Il peut
également y avoir absorption percutanée du produit liquide, mais dans
le cas de la vapeur, cette absorption est lente.
Le tétrachlorure de carbone se répartit dans tout l'organisme,
mais se concentre surtout dans le foie, le cerveau, les reins, les
muscles, les tissus adipeux et le sang. Le composé initial s'élimine
principalement dans l'air expiré et, en proportion minime, dans
l'urine et les matières fécales.
La première étape de la biotransformation du tétrachlorure de
carbone est catalysée par les enzymes du cytochrome P-450 et aboutit à
la formation d'un radical réactif, le radical trichlorométhyl. La voie
de biotransformation la plus importante conduisant à l'élimination de
ce radical consiste dans une oxydation en un radical encore plus
réactif, le radical trichlorométhylperoxyl, qui peut réagir à son tour
pour donner du phosgène. Le phosgène peut être détoxifié par réaction
avec l'eau pour donner du dioxyde de carbone ou par réaction avec le
glutathion ou la cystéine. En anaérobiose, il y a formation de
chloroforme et de dichlorocarbène.
Les intermédiaires métaboliques du tétrachlorure de carbone
peuvent former des liaisons covalentes avec des macromolécules et
provoquer la peroxydation des lipides.
L'action toxique du tétrachlorure de carbone a pour organes
cibles le foie et le rein. La gravité des effets hépatiques dépend
d'un certain nombre de facteurs tels que la sensibilité de l'espèce,
la voie et le mode d'exposition, le régime alimentaire et une
exposition concomitante éventuelle à d'autres substances, notamment
l'éthanol. En outre, il semble qu'un traitement préalable par divers
composés, comme le phénobarbital ou la vitamine A, accroisse
l'hépatotoxicité du tétrachlorure de carbone, alors que d'autres, au
contraire, la réduisent, comme la vitamine E.
Après application sur l'épiderme de lapins et de cobayes, on a
constaté une irritation modérée et une réaction également modérée a
été observée après instillation dans l'oeil du lapin.
La DL50 la plus faible (2391 mg/kg p.c. sur une période de 14
jours) a été obtenue à l'issue d'une étude sur des chiens qui
recevaient le composé par voie intrapéritonéale. Chez le rat, on a
obtenu des valeurs comprises entre 2821 et 10 054 mg/kg p.c.
Lors d'une étude de 12 semaines sur des rats comportant
l'administration du produit par la voie buccale 5 jours par semaine,
on a obtenu une dose sans effet nocif observable (NOAEL) de 1 mg/kg
p.c. La dose la plus faible produisant un effet nocif observable
(LOAEL) était de 10 mg/kg p.c., les effets observés étant une
augmentation légère, mais significative, de l'activité de la
sorbitol-déshydrogénase et une vacuolisation modérée des hépatocytes
centrilobulaires. Une NOAEL similaire de 1,2 mg/kg p.c. (5 jours par
semaine) a été obtenue chez des souris lors d'une étude de 90 jours
avec administration buccale; dans la même étude, la LOAEL
(hépatotoxicité) a été trouvée égale à 12 mg/kg p.c.
En exposant des rats à du tétrachlorure de carbone par la voie
respiratoire pendant environ 6 mois, 5 jours par semaine, 7 heures par
jour, on a obtenu une NOAEL de 32 mg/m3 LOAEL, basée sur des
anomalies de la morphologie hépatique, a été trouvée égale à 63
mg/m3. Dans une autre étude de 90 jours sur des rats, on a obtenu une
NOAEL de 6,1 mg/m3 après exposition continue à du tétrachlorure de
carbone. Lors d'une étude d'inhalation de 2 ans portant également sur
des rats, la concentration la plus faible étudiée (32 mg/m3) a
provoqué des effets marginaux.
La seule étude toxicologique à long terme dont on dispose a
consisté à faire ingérer du tétrachlorure de carbone à des rats
pendant 2 ans, aux doses respectives de 0, 80 et 200 mg de produit par
kg de nourriture. En raison d'une affection respiratoire chronique qui
a touché tous les animaux à partir du 14ème mois et a provoqué une
augmentation de la mortalité, les résultats de l'autopsie effectuée au
bout de deux ans ne peuvent pas être utilisés pour une évaluation du
risque sanitaire.
Les études d'inhalation effectuées sur des rats et des souris ont
permis de conclure que le tétrachlorure de carbone peut avoir des
effets embryotoxiques pouvant aller jusqu'à la mort de l'embryon.
Toutefois ces effets ne se manifestent qu'aux doses toxiques pour les
femelles gravides. Le tétrachlorure de carbone n'est pas tératogène.
De nombreuses études de génotoxicité ont été effectuées sur le
tétrachlorure de carbone. Sur la base des données disponibles, on peut
considérer que ce composé n'est pas génotoxique.
Le tétrachlorure de carbone provoque l'apparition d'hépatomes et
de carcinomes hépatocellulaires chez le rat et la souris. Les doses
qui entraînent la formation de tumeurs hépatiques sont supérieures aux
doses cytotoxiques.
Chez l'Homme, les manifestations aiguës qui surviennent après
exposition au tétrachlorure de carbone sont indépendants du mode
d'absorption et se caractérisent par des symptômes gastrointestinaux
et neurologiques tels que nausées, vomissements, céphalées,
étourdissements, dyspnée qui finissent par aboutir à la mort. Des
lésions hépatiques apparaissent au bout de 24 h ou davantage. Les
lésions rénales ne se manifestent souvent que 2 ou 3 semaines après
l'intoxication.
Les études épidémiologiques n'ont pas permis d'établir
l'existence d'une association entre l'exposition au tétrachlorure de
carbone et un accroissement du risque de mortalité, de cancer ou
d'affection hépatique. Certains travaux incitent à penser qu'il
pourrait y avoir augmentation du risque de lymphome non Hodgkinien
et,selon une étude particulière, du risque de mortalité et de cirrhose
du foie. Il faut cependant préciser que toutes ces études ne portaient
pas spécifiquement sur l'exposition au tétrachlorure de carbone et
qu'il n'y avait pas, en tout cas, de corrélations statistiques fortes.
En général, le tétrachlorure de carbone se révèle peu toxique
pour les bactéries, les protozoaires et les algues. La concentration
toxique la plus faible a été mesurée chez les bactéries méthanogènes
(CI50 = 6,4 mg/litre). Pour les invertébrés aquatiques, les valeurs
de la Cl50 aiguë varient de 28 à > 770 mg/litre. Dans le cas des
poissons d'eau douce, c'est chez l'orfe (Leuciscus idus
melanotus) que l'on a trouvé la valeur la plus faible de la CL50
aiguë, avec 13 mg/litre. Chez les espèces marines, c'est la limande
(Limanda limanda) qui présente la plus faible valeur de la CL50,
avec 50 mg/litre. Chez les poissons et les amphibiens, le
tétrachlorure de carbone se révèle plus toxique pour les stades
embryo-larvaires que pour les adultes. La grenouille-taureau commune
(Rana catesbeiara) est l'espèce la plus sensible, avec une CL50 de
0,92 mg/litre (de la fécondation à 4 jours après l'éclosion).
Les données disponibles montrent que le mécanisme de formation
des tumeurs hépatiques n'est pas de nature génotoxique et il est donc
admissible de fixer une dose journalière tolérable par ingestion (TDI)
et une concentration journalière tolérable dans l'air (TC).
En s'appuyant sur l'étude de Bruckner et al. (1986) qui ont
déterminé une dose sans effet nocif observable (NOAEL) de 1 mg/kg p.c.
lors d'une étude de 12 semaines sur des rats auxquels on avait fait
ingérer du tétrachlorure de carbone, et en utilisant un facteur de
conversion de 5/7 pour la dose journalière et un coefficient
d'incertitude de 500 (100 pour les variations inter- et
intraspécifiques, 10 pour la durée de l'étude plus un facteur de 0,5
pour tenir compte du fait que l'on avait utilisé des boulettes), on
parvient à une TDI de 1,42 µg/kg de poids corporel.
En s'appuyant sur une étude d'inhalation de 90 jours pratiquée
sur des rats (Prendergast et al., 1967), qui a permis d'aboutir à une
NOAEL de 6,1 mg/m3 on a calculé une TC de 6,1 g/m3 en utilisant les
coefficients de 7/24 et de 5/7 pour passer à une exposition en continu
et un coefficient d'incertitude de 1000 (100 pour les variations
inter- et intraspécifiques et 10 pour la durée de l'étude). Cette TC
correspond à une TDI de 0,85 µg/kg de poids corporel.
En comparant la limite supérieure de la dose journalière absorbée
par l'Homme, c'est-à-dire 0,2 µg/kg p.c., à la valeur la plus faible
de la TDI, soit 0,85 µg/kg p.c., on peut conclure que l'exposition
actuelle de la population générale au tétrachlorure de carbone de
toutes origines a peu de chances de causer une absorption excessive de
ce composé.
En règle générale, les organismes aquatiques ne courent guère de
risque imputable au tétrachlorure de carbone. Toutefois, il peut y
avoir un danger pour les stades embryo-larvaires sur les sites de
décharge ou de déversement de produits industriels ou à proximité de
ces sites.
RESUMEN
El tetracloruro de carbono es un líquido volátil transparente,
incoloro, con un olor dulce característico. Es miscible con la mayor
parte de los disolventes alifáticos y tiene a su vez propiedades
disolventes. La solubilidad en agua es baja. El tetracloruro de
carbono no es inflamable y se mantiene estable en presencia del aire y
de la luz. Su descomposición puede producir fosgeno, anhídrido
carbónico y ácido clorhídrico.
La fuente de tetracloruro de carbono en el medio ambiente con
toda probabilidad tiene un origen casi exclusivamente antropogénico.
La mayor parte del tetracloruro de carbono producido se emplea en la
fabricación de clorofluorocarbonos y otros hidrocarburos clorados. La
producción mundial ascendió en 1987 a 960 000 toneladas. Sin embargo,
desde que en el Protocolo de Montreal relativo a las sustancias que
agotan la capa de ozono (1987) y en sus enmiendas (1990 y 1992) se
estableció un calendario para la reducción progresiva de la producción
y consumo del tetracloruro de carbono, su fabricación ha disminuido y
seguirá descendiendo.
Se han elaborado varios métodos analíticos suficientemente
sensibles y precisos para la determinación del tetracloruro de carbono
en muestras de aire, agua y biológicas. La mayoría de estos métodos se
basan en la inyección directa en un cromatógrafo de gases o la
adsorción en carbón activado, seguida de la desorción o la evaporación
y la posterior detección por cromatografía de gases.
Casi todo el tetracloruro de carbono que se libera en el medio
ambiente estará en último término presente en la atmósfera, debido a
su volatilidad. Dado que su tiempo de permanencia en la atmósfera es
prolongado, tiene una distribución muy amplia. Durante el período
1980-1990, las concentraciones atmosféricas fueron de alrededor de
0,5-1 µg/m3. Las estimaciones de su permanencia en la atmósfera son
variables, pero se aceptan como los valores más razonables los 45-50
años. El tetracloruro de carbono contribuye tanto a la reducción del
ozono como al calentamiento mundial. Es en general resistente a la
biodegradación aerobia, pero menos a la anaerobia. La aclimatación
aumenta la velocidad de biodegradación. Aunque el coeficiente de
reparto octanol/agua indica un potencial de bioacumulación moderado,
el breve período de permanencia en los tejidos reduce esta tendencia.
En el agua, se han notificado concentraciones inferiores a 10
ng/litro en los océanos y generalmente inferiores a 1 µg/litro en el
agua dulce, pero con valores mucho más elevados cerca de los lugares
de liberación. Se han registrado concentraciones de hasta 60 µg/kg en
alimentos elaborados con tetracloruro de carbono, pero esta práctica
se ha suprimido.
La población general está expuesta al tetracloruro de carbono
fundamentalmente a través del aire. A partir de las concentraciones
notificadas en el aire ambiente, los productos alimenticios y el agua
potable, se ha estimado que la ingesta diaria de tetracloruro de
carbono es de alrededor de 1 µg/kg de peso corporal. En la actualidad
esta estimación es probablemente demasiado elevada, porque se ha
suprimido el uso del tetracloruro de carbono como fumigante de los
cereales y los valores notificados para los alimentos y utilizados en
el cálculo fueron fundamentalmente los obtenidos en alimentos a base
de grasas y cereales. En otras partes se han descrito valores de 0,1 a
0,27 µg/kg de peso corporal para la exposición diaria de la población
general. Se puede producir una exposición a concentraciones más
elevadas de tetracloruro de carbono en el lugar de trabajo debido a un
derrame accidental.
El tetracloruro de carbono se absorbe bien de los tractos
gastrointestinal y respiratorio en los animales y en el ser humano. Es
posible la absorción cutánea de tetracloruro de carbono líquido, pero
la absorción cutánea del vapor es lenta.
El tetracloruro de carbono se distribuye por todo el organismo,
alcanzando las concentraciones más altas en el hígado, el cerebro, el
riñón, los músculos, la grasa y la sangre. El compuesto original se
elimina fundamentalmente en el aire exhalado, mientras que se excretan
cantidades mínimas en las heces y la orina.
En la biotransformación del tetracloruro de carbono, el primer
paso es la catálisis por enzimas del citocromo P-450 para formar un
radical reactivo, el triclorometilo. La biotransformación oxidativa es
la ruta más importante en la eliminación del radical, produciendo otro
radical incluso más reactivo, el triclorometilperoxilo, que puede
reaccionar de nuevo para formar fosgeno. Éste se puede destoxificar
mediante la reacción con el agua para producir anhídrido carbónico, o
con el glutatión o la cisteína. En condiciones anaerobias se forma
cloroformo y diclorocarbeno.
Se produce la unión a macromoléculas mediante enlaces covalentes
y la peroxidación de lípidos a través de intermediarios metabólicos
del tetracloruro de carbono.
El hígado y el riñón son los órganos destinatarios de la
toxicidad de este compuesto. La gravedad de los efectos hepáticos
depende de diversos factores, como la susceptibilidad de la especie,
la ruta y el modo de exposición, la alimentación o la exposición
simultánea a otros compuestos, en particular el etanol. Además, parece
que el tratamiento previo con diversos compuestos, como el
fenobarbital y la vitamina A, aumenta la hepatotoxicidad, mientras que
otros compuestos, como la vitamina E, reducen la acción hepatotóxica
del tetracloruro de carbono.
Tras la aplicación cutánea se ha observado una irritación
moderada en la piel de conejos y cobayas, y se puso de manifiesto una
reacción leve después de aplicar el compuesto en el ojo del conejo.
La DL50 más baja, de 2391 mg/kg de peso corporal (período de 14
días), se notificó en un estudio realizado con perros mediante
administración intraperitoneal. En ratas, los valores de la DL50
oscilaron entre 2821 y 10 054 mg/kg de peso corporal.
En un estudio de administración por vía oral a ratas durante 12
semanas (5 días/semana), la concentración sin efectos adversos
observados (NOAEL) fue de 1 mg/kg de peso corporal. La concentración
más baja sin efectos adversos observados (LOAEL) notificada en este
estudio fue de 10 mg/kg de peso corporal, registrándose un aumento
ligero, pero significativo, de la actividad de la sorbitol
deshidrogenasa y una ligera vacuolación centrilobular hepática. En un
estudio de 90 días por vía oral realizado en ratones, se encontró una
NOAEL semejante de 1,2 mg/kg de peso corporal (5 días/semana) con una
LOAEL de 12 mg/kg de peso corporal cuando se produjo hepatotoxicidad.
Cuando se expusieron ratas a tetracloruro de carbono mediante
inhalación durante unos seis meses, cinco días a la semana, siete
horas diarias, se notificó una NOAEL de 32 mg/m3. Se informó de una
LOAEL, basada en cambios en la morfología del hígado, de 63 mg/m3. En
otro estudio de 90 días en ratas se encontró una NOAEL de 6,1 mg/m3
tras una exposición continua a tetracloruro de carbono. El nivel de
exposición más bajo, de 32 mg/m3 (la concentración más baja
estudiada), en un estudio de inhalación en ratas de dos año produjo
efectos marginales.
El único estudio de toxicidad prolongada por vía oral fue uno de
dos años realizado en ratas expuestas a 0, 80 y 200 mg de tetracloruro
de carbono/kg de alimentos. Debido a una enfermedad respiratoria
crónica que contrajeron todos los animales a partir del 14° mes y que
provocó un aumento de la mortalidad, los resultados notificados de la
necropsia a los dos años son insuficientes para evaluar el riesgo para
la salud.
Se llegó a la conclusión de que el tetracloruro de carbono puede
inducir efectos embriotóxicos y embrioletales, pero sólo a dosis
tóxicas para la madre, como se observó en los estudios de inhalación
realizados en ratas y ratones. El tetracloruro de carbono no es
teratogénico.
Se han realizado numerosas valoraciones de la genotoxicidad del
tetracloruro de carbono. Tomando como base los datos disponibles, se
puede considerar que este producto es un compuesto no genotóxico.
El tetracloruro de carbono induce la formación de hepatomas y
carcinomas hepatocelulares en ratones y ratas. Las dosis que inducen
la formación de tumores hepáticos son más elevadas que las que
producen toxicidad celular.
En el ser humano, los síntomas agudos tras la exposición a
tetracloruro de carbono son independientes de la ruta de ingestión y
se caracterizan por síntomas gastrointestinales y neurológicos, como
náuseas, vómitos, dolor de cabeza, desvanecimiento, disnea y la
muerte. Después de las 24 horas o más aparecen lesiones hepáticas. Son
evidentes los trastornos renales con frecuencia sólo dos o tres
semanas después de la intoxicación.
Los estudios epidemiológicos no han establecido una asociación
entre la exposición al tetracloruro de carbono y el aumento del riesgo
de mortalidad, neoplasia o enfermedad hepática. Algunos estudios han
indicado una asociación con un aumento del riesgo de linfoma
no-Hodgkin y, en un estudio, con la mortalidad y la cirrosis hepática.
Sin embargo, no en todos estos estudios se señaló la exposición
específica al tetracloruro de carbono y las asociaciones estadísticas
no fueron convincentes.
En general, el tetracloruro de carbono parece tener una toxicidad
baja para las bacterias, los protozoos y las algas; la concentración
tóxica más baja notificada para las bacterias metanogénicas
correspondió a una CI50 de 6,4 mg/litro. Para los invertebrados
acuáticos, los valores de la CL50 aguda fueron de 28 a >770
mg/litro. En los peces de agua dulce el valor más bajos de la CL50
aguda, de 13 g/litro, se encontró en el cachuelo dorado
(Leuciscus idus melanotus), y para las especies marinas se notificó
un valor de la CL50 de 50 mg/litro para la limanda
(Limanda limanda). El tetracloruro de carbono parece ser más tóxico
para las fases embrionaria y larvaria de los peces y anfibios que para
los adultos. La rana toro común (Rana catesbeiara) es la especie más
susceptible, con una CL50 de 0,92 mg/litro (desde la fecundación
hasta los cuatro días después de la eclosión).
Los datos disponibles indican que la inducción de tumores
hepáticos se debe a un mecanismo no genotóxico, por lo que parece
aceptable el establecimiento de una ingesta diaria tolerable (IDT) y
de una concentración diaria tolerable (CDT) en el aire para el
tetracloruro de carbono.
Tomando como base el estudio de Bruckner et al. (1986), en el
cual se observó una NOAEL de 1 mg/kg de peso corporal en un estudio de
12 semanas con administración por vía oral a ratas, e incorporando un
factor de conversión de 5/7 para la dosificación diaria y aplicando un
factor de incertidumbre de 500 (100 por la variación interespecífica e
intraespecífica, 10 por la duración del estudio y un factor de
modificación de 0,5 porque se trataba de un estudio de bolo), se
obtiene una IDT de 1,42 µg/kg de peso corporal.
Al comparar el límite superior estimado de la ingesta diaria
predominante de 0,2 µg/kg de peso corporal con el valor más bajo de la
IDT (0,85 µg/kg de peso corporal), se puede llegar a la conclusión de
que la exposición predominante en la actualidad de la población
general al tetracloruro de carbono procedente de todas las fuentes es
poco probable que dé lugar a una ingesta excesiva de la sustancia
química.
En general, el riesgo del tetracloruro de carbono para los
organismos acuáticos es bajo. Sin embargo, puede presentar un riesgo
para las fases embrionaria y larvaria en lugares de vertidos o escapes
industriales o en zonas próximas a ellos.