
LEVAMISOLE
1. EXPLANATION
Levamisole is a broad spectrum anthelminthic drug widely used to
control internal parasites in livestock. It is the levoenantiomer of
tetramisole. Optical resolution of d,l- tetramisole into its dextro-
and levoisomers revealed the levo form to have more anthelminthic
activity; thus this isomer is used as the anthelminthic. Levamisole
can be administered orally or parenterally, and in cattle dermally.
Depending on the parasite and host animal the therapeutic dose levels
vary between 5 and 40 mg/kg body weight.
Levamisole is used in human medicine as an anthelminthic in a
single dose form at 2.5 mg/kg. It is also used for a variety of other
indications including adjuvant therapy in cancer treatment. It is
anticipated this use will expand due to results in recent clinical
trials.
Levamisole has not been evaluated previously by the Joint FAO/WHO
Expert Committee on Food Additives. The structure of levamisole is
shown in Figure 1.
 2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Absorption, distribution, and excretion
Distribution of 3H-levamisole was studied in Charles River CD
cobs rats and C3H black mice. The rodents were given a single oral
dose of 2.5 mg/kg and serially sacrificed at 0.5, 1, 4, 8, 16, 24, 48,
72, 96 hours and 8 days. Distribution was evaluated using whole body
sagittal sections. The highest activity was present 30 minutes after
dosing in both species. The liver concentrated the highest activity in
both species, with trace amounts remaining in liver 4 days after
dosing. A high level of activity was found in bile in the mouse. A
significant binding to melanin was also suggested in the mouse (Benard
et al., 1980).
Radiolabelled levamisole was administered orally to rats to study
absorption, distribution, and elimination of the drug. Groups of 5
rats were administered a single bolus of 15 mg/kg levamisole via
gavage. Rats were then housed in individual metabolism cages for the
duration of the study. Four different radiotracers were used: 14C
in the 8 position was primarily used, while 14C in the 6 position,
35S, and 3H on the para position of the phenyl ring were used to a
lesser extent. The radiolabel was monitored for 8 days following dose
administration.
Radioactivity was rapidly absorbed, distributed throughout the
body, and eliminated via urine and faeces. An estimated 91% of the
dose was recovered during the 8 days. The other 9% was attributed to
experimental error rather than loss to unidentified pathways.
Approximately 40% of the dose was excreted in the urine within 12
hours; however, further renal elimination was limited, with only about
48% of the total dose excreted via this pathway. About 41% of the
dose was eliminated in faeces, primarily during the initial 48 hours.
About 0.9% of the dose was retained in the tissues after 8 days. The
highest tissue levels were found in liver and kidney. At 48 hours 0.8
and 0.57 ppm were found in liver and kidney respectively (Gatterdam
et al., 1966 and Boyd et al., 1973).
Total radioactivity and drug concentrations were determined in
plasma, organs and excreta of male Sprague-Dawley rats given a single
oral or intramuscular dose of 7.5 mg/kg of 3H-levamisole. Rats were
sacrificed and tissues collected at 1, 4, 8, and 24 hours after
dosing. Excreta were collected at 4, 8, 12, 24, 32, 48, 56 and 72
hours. A separate group of rats was used for bile duct cannulation.
Only 45% of plasma radioactivity at one hour was levamisole.
During the initial 24 hours following administration the intestinal
tract contained the largest fraction of the dose regardless of route
of administration. Most of the radioactivity was excreted in urine
(68-78%) rather than the faeces (17-33%) during the initial 72 hours.
More than 50% was excreted in urine during the first 12 hours. Only
6.3-8.5% of the urine fraction was levamisole, while 5.8 - 8.0% was
the 4-hydroxylevamisole metabolite. Biliary excretion was 26% for i.m
administration but 13% for oral administration (Galtier et al.,
1983).
The absorption of levamisole was studied in 3 New Zealand white
rabbits after a single dose of 10 mg/kg given by gavage. Peak plasma
levels of 0.25 ppm were obtained at 30 minutes, while plasma half-life
was 425 minutes. At 4 hours plasma levels were <0.01 ppm (Michiels
et al., 1978).
2.1.2 Biotransformation
Metabolites from administration of levamisole to rats were
evaluated using TLC. Over 50 distinct metabolites were determined;
however, the qualitative pattern in urine, faeces, and tissue extracts
was nearly the same. Evaluation of metabolites suggests there are
four initial metabolic processes, each giving rise to a family of
metabolites (Figure 2). The most important quantitatively is (1) the
oxidative introduction of a double bond into the imidazole ring,
accompanied by or followed by oxidation of the sulfur to sulfoxide and
introduction of a hydroxy group in the para position of the phenyl
ring. The second most important pathway (2) is the hydrolysis of the
thiazolidine ring to an oxoimidazole metabolite. The third (3)
pathway is formation of p-hydroxytetramisole and its subsequent
conjugation with glucuronic acid. The least important pathway is (4)
the hydrolysis of the thiazole ring to yield the mercaptoethyl
intermediate and subsequent oxidation to sulphoxide and sulphone. The
metabolic pattern suggests these are not the only pathways involved,
with about 20% of the metabolites remaining unidentified. The
metabolic scheme depicted in Figure 2, with the various pathways, has
been proposed based on this information (Gatterdam et al., 1966
and Boyd et al., 1973).
2.1.3. Pharmacokinetics
Three healthy male volunteers each received a single oral dose of
150 mg 3H-levamisole. Peak plasma levels of levamisole were reached
2-4 hours after dosing. Parent drug represented 40% of total
reactivity. Total radioactivity and unchanged levamisole were
measured in plasma, urine and faeces for 72 hours post-dosing. The
half-life of levamisole was 4 hours while the half life for total
reactivity was 16 hours. The primary route of excretion was via urine
- 70% - while only 3-5% appeared in the faeces. About 4% of the
excreted material was parent compound compared with the remainder
being metabolites (Heykants, 1976). A more recent study is in
agreement with this data (Kouassi et al., 1986).
2.2 Toxicological studies
2.2.1 Acute toxicity studies
The results of acute toxicity studies with levamisole are shown
in Table 1.
2.2.2 Short-term studies
2.2.2.1 Rats
Sixty Alderley Park albino rats were divided into groups of 10
rats/sex/dose and received 0, 50 or 100 mg/kg body weight daily via
gavage for 30 days. Clinical signs, weight, haematology, and
urinalysis were monitored at intervals during the study. At sacrifice,
gross necropsy was performed and organ weights were determined.
Tissues were examined histopathologically. A moderate reduction in
weight gain occurred in treated groups. An increase in liver and
kidney weights of treated rats also occurred (Davey, 1968a).
Six to eight week old Wistar rats were divided into 4 groups of
10 rats/sex/dose and received target doses of 0, 10, 40, or 160 mg/kg
b.w./day levamisole in the feed at rates of 0, 100, 400, or 1600 ppm
daily for 13 weeks. The following parameters were monitored:
survival, behavior and appearance, food consumption (weekly), body
weight (weekly), haematology (terminal), clinical chemistry
(terminal), urinalysis (terminal), organ weight, and gross pathology
and histopathology. Survival and clinical signs were not effected by
treatment. Food consumption was decreased in the 160 mg/kg b.w./day
group, while weight gains were reduced in all female treatment groups
and the 40 and 160 mg/kg b.w./day male treatment groups.
Haematology, clinical chemistry, and urinalysis of treated groups
were normal except for elevated urine pH. Organ weights were in
general reduced in the 160 mg/kg b.w./day group. No treatment-related
histopathologic changes were observed (Marsboom et al., 1969).
2.2.2.2 Dogs
Levamisole was administered to 10 beagle dogs orally at the rate
of 15 mg/kg b.w./day in three divided doses of 5 mg/kg body weight.
Drug related vomiting was observed in two dogs (Desplenter, 1983).
2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Absorption, distribution, and excretion
Distribution of 3H-levamisole was studied in Charles River CD
cobs rats and C3H black mice. The rodents were given a single oral
dose of 2.5 mg/kg and serially sacrificed at 0.5, 1, 4, 8, 16, 24, 48,
72, 96 hours and 8 days. Distribution was evaluated using whole body
sagittal sections. The highest activity was present 30 minutes after
dosing in both species. The liver concentrated the highest activity in
both species, with trace amounts remaining in liver 4 days after
dosing. A high level of activity was found in bile in the mouse. A
significant binding to melanin was also suggested in the mouse (Benard
et al., 1980).
Radiolabelled levamisole was administered orally to rats to study
absorption, distribution, and elimination of the drug. Groups of 5
rats were administered a single bolus of 15 mg/kg levamisole via
gavage. Rats were then housed in individual metabolism cages for the
duration of the study. Four different radiotracers were used: 14C
in the 8 position was primarily used, while 14C in the 6 position,
35S, and 3H on the para position of the phenyl ring were used to a
lesser extent. The radiolabel was monitored for 8 days following dose
administration.
Radioactivity was rapidly absorbed, distributed throughout the
body, and eliminated via urine and faeces. An estimated 91% of the
dose was recovered during the 8 days. The other 9% was attributed to
experimental error rather than loss to unidentified pathways.
Approximately 40% of the dose was excreted in the urine within 12
hours; however, further renal elimination was limited, with only about
48% of the total dose excreted via this pathway. About 41% of the
dose was eliminated in faeces, primarily during the initial 48 hours.
About 0.9% of the dose was retained in the tissues after 8 days. The
highest tissue levels were found in liver and kidney. At 48 hours 0.8
and 0.57 ppm were found in liver and kidney respectively (Gatterdam
et al., 1966 and Boyd et al., 1973).
Total radioactivity and drug concentrations were determined in
plasma, organs and excreta of male Sprague-Dawley rats given a single
oral or intramuscular dose of 7.5 mg/kg of 3H-levamisole. Rats were
sacrificed and tissues collected at 1, 4, 8, and 24 hours after
dosing. Excreta were collected at 4, 8, 12, 24, 32, 48, 56 and 72
hours. A separate group of rats was used for bile duct cannulation.
Only 45% of plasma radioactivity at one hour was levamisole.
During the initial 24 hours following administration the intestinal
tract contained the largest fraction of the dose regardless of route
of administration. Most of the radioactivity was excreted in urine
(68-78%) rather than the faeces (17-33%) during the initial 72 hours.
More than 50% was excreted in urine during the first 12 hours. Only
6.3-8.5% of the urine fraction was levamisole, while 5.8 - 8.0% was
the 4-hydroxylevamisole metabolite. Biliary excretion was 26% for i.m
administration but 13% for oral administration (Galtier et al.,
1983).
The absorption of levamisole was studied in 3 New Zealand white
rabbits after a single dose of 10 mg/kg given by gavage. Peak plasma
levels of 0.25 ppm were obtained at 30 minutes, while plasma half-life
was 425 minutes. At 4 hours plasma levels were <0.01 ppm (Michiels
et al., 1978).
2.1.2 Biotransformation
Metabolites from administration of levamisole to rats were
evaluated using TLC. Over 50 distinct metabolites were determined;
however, the qualitative pattern in urine, faeces, and tissue extracts
was nearly the same. Evaluation of metabolites suggests there are
four initial metabolic processes, each giving rise to a family of
metabolites (Figure 2). The most important quantitatively is (1) the
oxidative introduction of a double bond into the imidazole ring,
accompanied by or followed by oxidation of the sulfur to sulfoxide and
introduction of a hydroxy group in the para position of the phenyl
ring. The second most important pathway (2) is the hydrolysis of the
thiazolidine ring to an oxoimidazole metabolite. The third (3)
pathway is formation of p-hydroxytetramisole and its subsequent
conjugation with glucuronic acid. The least important pathway is (4)
the hydrolysis of the thiazole ring to yield the mercaptoethyl
intermediate and subsequent oxidation to sulphoxide and sulphone. The
metabolic pattern suggests these are not the only pathways involved,
with about 20% of the metabolites remaining unidentified. The
metabolic scheme depicted in Figure 2, with the various pathways, has
been proposed based on this information (Gatterdam et al., 1966
and Boyd et al., 1973).
2.1.3. Pharmacokinetics
Three healthy male volunteers each received a single oral dose of
150 mg 3H-levamisole. Peak plasma levels of levamisole were reached
2-4 hours after dosing. Parent drug represented 40% of total
reactivity. Total radioactivity and unchanged levamisole were
measured in plasma, urine and faeces for 72 hours post-dosing. The
half-life of levamisole was 4 hours while the half life for total
reactivity was 16 hours. The primary route of excretion was via urine
- 70% - while only 3-5% appeared in the faeces. About 4% of the
excreted material was parent compound compared with the remainder
being metabolites (Heykants, 1976). A more recent study is in
agreement with this data (Kouassi et al., 1986).
2.2 Toxicological studies
2.2.1 Acute toxicity studies
The results of acute toxicity studies with levamisole are shown
in Table 1.
2.2.2 Short-term studies
2.2.2.1 Rats
Sixty Alderley Park albino rats were divided into groups of 10
rats/sex/dose and received 0, 50 or 100 mg/kg body weight daily via
gavage for 30 days. Clinical signs, weight, haematology, and
urinalysis were monitored at intervals during the study. At sacrifice,
gross necropsy was performed and organ weights were determined.
Tissues were examined histopathologically. A moderate reduction in
weight gain occurred in treated groups. An increase in liver and
kidney weights of treated rats also occurred (Davey, 1968a).
Six to eight week old Wistar rats were divided into 4 groups of
10 rats/sex/dose and received target doses of 0, 10, 40, or 160 mg/kg
b.w./day levamisole in the feed at rates of 0, 100, 400, or 1600 ppm
daily for 13 weeks. The following parameters were monitored:
survival, behavior and appearance, food consumption (weekly), body
weight (weekly), haematology (terminal), clinical chemistry
(terminal), urinalysis (terminal), organ weight, and gross pathology
and histopathology. Survival and clinical signs were not effected by
treatment. Food consumption was decreased in the 160 mg/kg b.w./day
group, while weight gains were reduced in all female treatment groups
and the 40 and 160 mg/kg b.w./day male treatment groups.
Haematology, clinical chemistry, and urinalysis of treated groups
were normal except for elevated urine pH. Organ weights were in
general reduced in the 160 mg/kg b.w./day group. No treatment-related
histopathologic changes were observed (Marsboom et al., 1969).
2.2.2.2 Dogs
Levamisole was administered to 10 beagle dogs orally at the rate
of 15 mg/kg b.w./day in three divided doses of 5 mg/kg body weight.
Drug related vomiting was observed in two dogs (Desplenter, 1983).
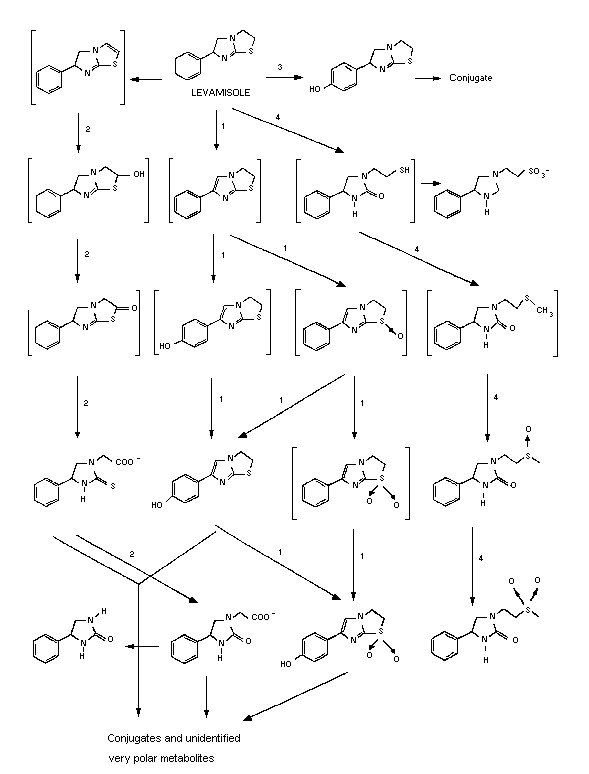 Table 1: Results of acute toxicity studies with levamisole
Species Sex Route LD50 Reference
(mg/kg)
Mouse M&F oral 205-285 Davey, undated a
Niemegeers, 1975
M&F i.v. 20-28 Davey, undated a
Niemegeers, 1975
M&F s.c. 102-121 Davey, undated a
Niemegeers, 1975
Rat M&F oral 458-1095 Davey, undated a
Niemegeers, 1975
M&F 286-566 Wallwork &
James, 1975
M i.v. 17-28 Davey, undated a
Niemegeers, 1975
M&F s.c. 81-89 Davey, undated a
Niemegeers, 1975
dermal 252 Wallwork & James,
1975
Rabbit M oral 458 Davey, undated a
F i.v. 25 Davey, undated a
Swine M&F oral in no Wang, 1970a
feed at lethality
40 mg/kg
A one month oral study with levamisole was conducted in beagles.
Twenty-four dogs were divided into 3 groups containing 4 dogs/sex.
Dogs received 0, 10, or 20 mg/kg b.w./day. Clinical signs included
ataxia and convulsions; one death occurred in the high dose group.
Treated groups lost weight during the study. Haematologic and
clinical chemistry parameters were evaluated but no treatment-related
effects occurred. Gross necropsy was performed on all dogs and organ
weights were obtained. Histopathologic evaluation was generally
unremarkable although some lymphoid cuffing occurred in the cerebral
vessels of high dose dogs (Davey, 1968b).
Groups of beagle dogs (2/sex) received 0, 1.5, 3, or 6 mg/kg
b.w./day of levamisole daily for 90 days. Dogs were up to two years
of age at study initiation. Parameters evaluated included: clinical
signs, food intake, body weight, haematology, clinical chemistry,
organ weights and histopathology (6 mg/kg b.w./day group only was
sacrificed). Special emphasis was placed on evaluation of possible
haemolytic effects of levamisole. Dogs were distributed into dose-
groups based on predetermined susceptibility to erythrocyte
haemolysis. Methaemoglobin and erythroblastosis were monitored. No
treatment-related effects were observed (Huchison et al., 1967).
The effect of levamisole treatment for one year in dogs was
evaluated. Four groups of 8-10 month old beagles containing 3
dogs/sex/group were administered levamisole orally in gelatin capsules
daily six times/week for 12 months. Dogs received 20, 5, or 1.25
mg/kg b.w./day levamisole or 250 mg lactose. Dogs were acclimated and
baseline parameter values obtained during a 6 week period prior to
dosing. The following parameters were evaluated: Clinical signs
including ophthalmoscopy (0, 6, and 12 months), ECG and blood pressure
(monthly), food consumption, body weight (weekly), haematology, and
clinical chemistry and urinalysis (monthly). Gross pathology and
organ weights were monitored at necropsy, and histopathologic
examination was performed on a standard array of tissues.
During the 8th week all 20 mg/kg b.w./day dogs and one 5 mg/kg
b.w./day female dog experienced severe treatment induced haemolytic
anaemia. Decreases in haematocrit, haemoglobin, and RBC count
occurred with increases in erythoroblasts and immature granulocytes.
These dogs were removed from the study. Their haematology parameters
returned to normal about 2 weeks after dosing was stopped, but anaemia
returned when treatment was reinstituted. An in vitro serum RBC
agglutinizing factor which required at least 100 µg/ml of levamisole
was demonstrated. No other treatment-related effects were found in
this study (Marsboom et al., 1975).
The effect of levamisole administered daily as a pour on was
studied in dogs. Twenty-four beagles were divided into 4 groups of
3/sex and received 0, 2.5, 10, or 40 mg/kg b.w. daily as a 20%
solution applied to the back. The following parameters were
evaluated: clinical signs, ophthalmoscopic examination, body weight,
heart rate and ECG, blood pressure, haematology, clinical chemistry,
urinalysis, and at necropsy organ weights, gross pathology and
histopathology. Significant weight loss occurred in the 40 mg/kg
b.w./day group. No other treatment-related effects were observed in
this study (Verstraeten et al., 1983).
A series of clinical reports on levamisole-associated haemolysis
in dogs were evaluated. All reports were related to long-term use of
levamisole for dirofilaria treatment. The dose regimens generally
were altered at two week intervals with peak doses of 10-24 mg/kg
b.w./day and low doses of 2-3 mg/kg b.w./day. Anaemia developed at 3-
6 weeks of treatment (Maes & Marsboom, 1988; Atwell et al., 1981).
2.2.2.3 Pigs
Levamisole-HCl was administered to pigs at the rate of 0, 8, 24,
and 40 mg/kg body weight in feed or drinking water as a single
treatment. Five pigs/sex/dose were treated with each route of
administration. The treatment was repeated one week later. More
severe signs were observed in the drinking water group. Salivation
and vomiting were dose-related, while tremor, tachypnoea, transient
recumbency and 10% mortality were observed in the 40 mg/kg body weight
group. Salivation and vomiting only were observed in the 40 mg/kg
body weight feed-route pigs. Some suggestion of hepatic toxicity was
found upon histopathologic evaluation. The authors concluded that,
although some toxicity occurred, the 40 mg/kg body weight feed-
treatment and 24 mg/kg body treatment water-treatment were safe for
swine based on reversibility of clinical signs and hepatic lesions
(Wang, 1970b).
2.2.2.4 Primates
A 23 kg male baboon was treated dermally with 10, 20, and 40
mg/kg body weight levamisole with intervals of 10 and 4 days between
treatments. Effects including minor excitement were observed
following the 40 mg per kg body weight dose (Desplenter, 1980).
2.2.3 Long-term/carcinogenicity studies
2.2.3.1 Mice
An oral carcinogenicity bioassay of levamisole was conducted
using albino Swiss mice. Four hundred 7 to 8 week old mice were
divided into 4 groups containing 50 mice/sex. Following an
acclimatization period of one week, levamisole was provided at the
rate of 0, 12.5, 50, and 200 ppm in drinking water for 18 months to
provide target doses of 0, 5, 20, and 80 mg/kg b.w./day based on
consumption of 100 ml/week/mouse. The test article was stored at room
temperature in well-closed amber bottles. Solutions were prepared
fresh weekly. The animals were initially housed 25/cage but after 7
months were housed individually to prevent fighting. Clinical
examinations were performed daily except Sundays, but were not
reported. Gross necropsy was performed on all mice that died or were
sacrificed due to moribund condition as well as at study termination.
Lung, liver, pancreas, kidney, spleen, testis, epididymis, ovary,
uterus, mammary gland, adrenals, hypophysis, lymph nodes, and any
other indicated tissue were examined histopathologically. Survival
rates at the end of the study were: 0 ppm - 32%M/22%F, 12.5 ppm -
20%M/40%F, 50 ppm - 18%M/40%F, and 200 ppm - 20%M/38%F. However the
survival rate at 12-14 months was reduced considerably from that
expected for a useful carcinogenicity bioassay: less than 50% in all
male treatment groups and about 40% in the female control group. No
treatment-related gross pathology was reported. No treatment-related
effects were seen with respect to number of tumour-bearing mice or
incidence of various tumour types. However, appropriate tissues
available for examination were fewer than expected for a
carcinogenicity bioassay (Vandenberghe et al., 1980).
2.2.3.2 Rats
The chronic toxicity of levamisole was examined in rats. Six to
eight week old Wistar rats were divided into 4 groups of 20/sex and
received target doses of 40, 10, 2.5 and 0 mg/kg b.w. levamisole daily
provided in the feed at rates of 800, 200, 50, or 0 ppm. Diets were
prepared weekly. An interim sacrifice of 10 rats/sex was performed at
12 months with the final sacrifice at 18 months. The following
parameters were evaluated: Clinical signs (daily), ophthalmoscopic
exam (terminal sacrifice), food consumption (weekly), body weight
(weekly), and haematology, clinical chemistry, and urinalysis
(terminal sacrifice). Gross necropsy was performed at sacrifice.
Organ weights were obtained for heart, spleen, thymus, liver, kidney,
pancreas, adrenals, thyroid, brain, genital organs, and lung.
Histopathology was performed on tissue from the above organs and the
following array of tissues: Lymph nodes, urinary bladder, alimentary
tract tissues, parathyroid, hypophysis, diaphragm, striated muscle,
tongue, teeth, skin, bone marrow, and eye. No treatment related
clinical signs or mortality occurred. Mean food consumption was
reduced in both sexes of the high dose group. Weight gains were
significantly decreased for both sexes of the high dose group
throughout the study. The middle dose group weight gain was generally
reduced throughout the study but was significant only for the 12 month
females. Clinical chemistry parameters which were altered included
marginal increases in alkaline phosphatase in high dose females, and
bilirubin in 12 month high dose males and females. In the high dose
group the absolute weights of organs generally were decreased, while
the relative weights were increased. Histopathologic treatment-
related changes were limited to a degeneration of the germinal
epithelium of the testes and a mild chronic stimulation of the liver
seen only in the high-dose group (Marsboom et al., 1972).
The tumourigenic potential of levamisole was studied in Wistar
rats. Four hundred 3 month old rats were divided into 4 groups of
50/sex and following a one week acclimatization period were provided
levamisole in the diet at rates of 0, 50, 200, or 800 ppm to provide
target doses of 0, 2.5, 10, or 40 mg/kg b.w./day. Test diet was
prepared every two weeks. Animals were examined daily but clinical
signs were not reported. A full necropsy was performed on all
surviving animals at 24 month study termination and all animals which
died or were sacrificed due to moribund condition during the study.
The following tissues were examined histopathologically: Lung, liver,
pancreas, kidney, spleen, testis, epididymis, ovary, mammary gland,
adrenals, thyroids, parathyroids, hypophysis, lymph nodes, and any
other indicated tissue. Survival rates for the various groups were:
0 ppm - 4%M/10%F, 50 ppm - 2%M/8%F, 200 ppm - 2%M/20%F, and 800 ppm -
6%M/14%F. Survival rates for all male study groups were well below
50% by 16 months. No treatment-related effects were seen with respect
to number of tumour-bearing rats or incidence of various tumour types.
However, appropriate tissues available for examination were fewer than
expected for a carcinogenicity bioassay (Marsboom & Herin, 1980).
2.2.4 Reproduction studies
2.2.4.1 Rats
The effect of levamisole administered prior to breeding for 60
days in males and 14 days in females on the reproductive performance
of rats was studied. To accomplish the study 320 sexually mature
Wistar rats were divided into groups of 20 rats/sex and received
levamisole target doses of 1.25, 5, 20, or 80 mg/kg b.w./day in the
feed at rates of 25, 100, 400, or 1600 ppm for the prescribed duration
of treatment. Fresh diets were prepared weekly. Treated rats were
mated with nontreated rats. No control matings were performed with
untreated animals. Matings were validated by daily vaginal washings
for sperm. Female weights were determined at insemination and weekly
thereafter. Half the pregnant females were sacrificed on gestation
day 13 and the uterus was examined for number and distribution of
embryos, and resorptions as well as unusual uterine condition. The
remaining females were sacrificed on gestation day 22. The number and
distribution of fetuses were recorded and fetuses were examined
externally. Fetuses were processed and examined for internal
abnormalities via clearing and alizarin red staining of bones and
serial sectioning as well as X-ray analysis. No treatment-related
effects on male or female fertility or offspring normality were
reported in this study (Marsboom, 1969a).
The potential toxicity of levamisole administered during the
peri- and post-natal periods was evaluated in female Wistar rats.
Five groups of 20 three month old rats received 0, 25, 100, 400, and
1600 ppm levamisole in the diet from gestation day 16 through the 3
week lactation period to provide target doses of 0, 1.25, 5, 20, and
80 mg/kg b.w./day. Female weights were recorded on gestation days 1,
7, 14, and 21 post parturition, and on lactation days 14 and 21. Live
and dead pups were examined, counted, and weighed a few hours after
parturition. Females not delivering by gestation day 24 were
sacrificed, necropsied, and uteruses evaluated for distribution of
placental sites, resorptions and fetuses. Surviving pups were weighed
on lactation days 14 and 21. There was a decrease in weight gain for
the 1600 ppm dams. Additionally an increased incidence of stillborn
pups, reduced birth weight, reduced lactational weight gains, and
decreased 3-week survival rate were recorded for the 1600 ppm pups
(Marsboom, 1969b).
The potential embryotoxic and teratogenic effects of levamisole
were evaluated during the gestation periods of three consecutive
generations of treated pregnant rats. Eighty virgin 3-4 month old
rats were divided into 4 groups of 20 rats each and provided
levamisole at the rate of 0, 100, 400, and 1600 ppm in the diet,
equivalent to 0, 5, 20 and 80 mg/kg body weight/day on gestation days
6-15. Prior to and after the period of organogenesis the rats were
provided the control diet. At the age of 3 months 80 virgin females
were selected from those born to treated mothers of the prior
generation and mated with 40 non-littermate males also born to treated
dams from the preceding generation. The males were treated in the
same manner as their dams. The succeeding generation was established
in the same manner except that dams were sacrificed on gestation day
22 for evaluation of teratogenic and embryotoxic parameters. Females
were weighed on the 1st, 7th, 14th and 21st days of gestation, while
pups were weighed shortly after parturition and on lactation days 4,
14, and 21. If parturition had not occurred by gestation day 24 rats
were sacrificed, necropsied and distribution of fetuses and
resorptions were noted. The litters from the 3rd generation were
examined for distribution of live and dead fetuses and resorptions.
All fetuses were examined for external abnormalities, radiographic
examination was carried out, and fetuses were processed for soft
tissue (1/3) and skeletal (2/3) examination. No treatment-related
effects were recorded on dams or fetuses (Marsboom, 1974).
2.2.5 Special studies on embryotoxicity/teratogenicity
2.2.5.1 Rats
The potential embryotoxicity or teratogenicity of levamisole was
studied in Wistar rats. Sexually mature female Wistar rats were mated
with fertile males and received target doses of 0, 1.25, 5, 20, or 80
mg/kg b.w./day levamisole based on 25, 100, 400, and 1600 ppm
levamisole in the diet on gestation days 6-15. Mating was established
by vaginal plug. Weights of females were obtained at mating and
weekly thereafter. Food consumption was recorded individually during
gestation. Females were sacrificed on gestation day 22 and the
following litter and fetal parameters obtained: Number and
distribution of live and dead fetuses and resorptions within
abnormalities (external - all fetuses, skeletal - 2/3 and soft
tissue - 1/3). At 1600 ppm the incidence of resorptions increased
slightly. No other parameters were effected (Marsboom, 1972).
A teratology study with levamisole at doses of 0, 10, 50, and 100
mg/kg body weight/day was carried out on rats. Female rats were
divided into 4 groups of 20 rats and received levamisole on gestation
days 0-20 by mouth. Half the females were sacrificed on day 20,
litters were examined grossly, fetuses cleared and stained with
alizarin red and examined for skeletal effects. The remaining females
littered and reared their litters to lactation day 21. Maternal
weight gain was reduced in a dose-related manner. No effect on litter
parameters, embryotoxicity or post natal growth was observed (Davey,
undated c).
2.2.5.2 Rabbits
The teratogenic potential of levamisole was studied in rabbits.
Sixty mature female New Zealand rabbits were divided into 3 groups of
20 rabbits each. Rabbits were inseminated artificially due to low
pregnancy rate in the rabbit colony. Does received 0, 10 or 40 mg/kg
b.w./day levamisole by gavage on gestation days 6-18. Does were
observed daily for toxicity and weights were recorded at insemination,
gestation days 6-18, and at sacrifice. Rabbits were sacrificed on
gestation day 28, and necropsied. Fetuses were examined grossly and
then processed for soft tissue examination (1/3) or skeletal
examination (2/3). Survival of does was not affected by treatment.
There was a marked reduction in mean weight gain in the 40 mg/kg
b.w./day group compared with controls (250 vs 432, respectively). The
incidence of resorptions and dead fetuses was doubled in the 40 mg/kg
b.w./day group vs controls (10 vs 5), while the incidence of litters
with abnormalities was 4 in the 40 mg/kg b.w./day group vs 0 in the
control and 10 mg/kg b.w/day groups. However, this was within the
historical control range. No treatment-related effects from
levamisole occurred in this study (Marsboom, 1972).
Nineteen female Dutch rabbits were divided into 3 groups of 6-7
rabbits each to evaluate the potential teratogenic effect of
levamisole. Rabbits received 0, 25, or 75 mg/kg b.w. orally
throughout the gestation period. Does were sacrificed on gestation
day 28 and litter parameters evaluated. After examination the fetuses
were processed for skeletal examination. No treatment-related effects
were reported (Davey, undated c).
2.2.5.3 Pigs
Twenty pregnant sows received 10 mg/kg body weight levamisole 4
times at 2-day intervals intramuscularly. A group of 20 untreated
sows served as controls. The sows were selected to have produced one
previous litter of >7 pigs. The sows were divided into 4 groups of
5 each and received treatment on gestation days 8-14, 16-22, 24-30, or
32-38. Parturition data, length of gestation, litter size, sex,
weight, suckling behavior and external abnormalities were evaluated.
No treatment-related embryotoxic or teratogenic effects occurred in
this study (Veys & Dony, 1985).
2.2.6 Special studies on genotoxicity
The results of genotoxicity studies with levamisole are
summarized in Table 2.
2.3 Observations in humans
The safety of levamisole use in humans has recently been
reviewed. Levamisole has been used in humans for the following
indications: anthelminthic, rheumatoid arthritis, inflammatory
disease, infectious disease, and cancer therapy. Aside from its use
for parasitic infection, these uses appear to be based on the
immunomodulating properties of levamisole. Although a number of side
effects have been reported, the most important are haematologic,
including reversible leukopenia, agranulocytosis, and
thrombocytopenia. A low incidence of agranulocytosis appears to occur
in all continuous dose regimens ranging from 50 to 200 mg daily and
with 3 days treatment every other week to continuous therapy.
Haematologic effects were seen in nearly 5% of rheumatic patients but
only 0.2% of infectious patients. Based on his review of the data,
the author concluded that agranulocytosis was dose-related in onset
(Reyntjens, 1989).
A survey of published literature on levamisole-associated
agranulocytosis identified 110 citations. These reports occurred
between 1973 and 1984, and, based on volume of levamisole uses,
agranulocytosis correlated closely with human use of levamisole for
immunotherapy but not veterinary use of levamisole (Veys, 1989).
Numerous clinical reports of levamisole side effects are in the
literature. Two reports are reviewed for illustrative purposes:
Observations were made on a single cancer patient who was given
only levamisole at the rate of 2.5 mg/kg body weight for 2 consecutive
days per week for 13 weeks. The patient developed an agranulocytosis
and therapy was discontinued. His total white cell count was 1100
cells/mm3. Bone-marrow was described as hypercellular with relative
erythroid hyperplasia and virtually no myelopoiesis (0.5% myeloblasts,
no mature neutrophils, 2% eosinophils, 0.5% basophils, 63%
lymphocytes, 5% plasma cells, 0.5% monocytes, 23.5% erythroid forms,
and 5% reticulum cells). Megakargocytes were present. Seven days
later the patient's bone marrow was hypocellular but demonstrated
myelopoiesis: 12% myeloblasts, 15% promyelocytes, 2% myelocytes, 8%
metamyelocytes; and 1% neutrophils. The red cell series was reduced
and megakaryocytes were markedly reduced. A transient but severe
thrombocytopenia (without bleeding) to 30,000 cells/mm3 also occurred
during the first week.
Table 2: Genotoxicity of levamisole
Test system Test Object Concentration Results Reference
Chromosomal Human from humans positive Berger
aberrations lymphocytes treated with et al.,
150 mg 1980
levamisole
(in vivo) or
250 µg/ml
(in vitro)
Sister Humanadded lymphocyte positive Berger
Chromatid lymphocytes cultures from et al.,
Exchange untreated 1980
volunteers
Ames test TA 1535 10-10,000 µg/ml negative Richold
(+/- S9) TA 1537 et al.,
TA 98 1979
TA 100
Chromosomal Cultured +S9: 5000 µg/ml negative Enninga,
aberrations human -S9: 1000 µg/ml 1988
(+/- S9) lymphocytes
Micronucleus Mouse 3-75 mg/kg negative Ikegami,
test erythrocytesorally et al., 1981
Dominant Male and Single oral negative Marsboom,
lethal test female dose: 10, 40 1977
mice or 160 mg/kg
b.w.
The marrow recovered in 10-14 days with circulating neutrophils
rebounding to raised levels. The immunologic basis for these
levamisole effects were explored using the leuco agglutination test
and Clq deviation test for circulating immune complexes. Levamisole-
dependent leuco agglutinations were present in the patient's serum in
high titre (1/512) during the acute phase. During the first week the
patient's serum induced some spontaneous leuco agglutination; however,
after one week added levamisole was required for leuco agglutination.
Circulating Clq-reactive materials reached a peak seven days after
onset of neutropenia coincident with the peak in leuco agglutination
titre (Parkinson et al., 1977).
A clinical report of a 52 year old female patient with a history
of current seronegative rheumatoid arthritis, erythema nodosum in
childhood, and angioneurotic edema at age 35 to 37 years was reviewed.
She received 50 mg levamisole once weekly. After each dose she
experienced a mild flu-like syndrome. Following the sixth dose this
syndrome was more severe and mouth ulcers developed. This syndrome
became more pronounced following the seventh dose. She had a
polymorph count of 225 per mm3 6 days later. Bone marrow examination
showed increased promyelocytes but maturation arrest at the myelocyte
stage. Erythropoiesis was normal. No sideroblasts were present. A
dose of 10 mg levamisole was given 2 months later. No clinical side
effects were noted but a polymorph decrease of 31% occurred at 24
hours post dose. A dose of 25 mg of levamisole two weeks later
produced clinical signs and a 43% reduction of polymorphs at 32 hours.
Twenty leucocyte lines were tested for agglutination with 19/20
yielding negative results. Cytotoxicity tests were also negative
(Felix-Davies, 1978).
In addition to individual case studies and clinical reports, some
early epidemiologic reports or reviews of levamisole-associated
haematologic adverse effects were available. Adverse reactions in 267
of 6217 patients on levamisole were analyzed via a questionnaire.
Significant reactions were agranulocytosis (2.3%), skin rash and
febrile illness. These occurred primarily in patients (particularly
females) with rheumatoid arthritis. Agranulocytosis was spontaneously
reversible (Symoens et al., 1978). A low incidence of leukopenia
and agranulocytosis (0.4%) was reported after use of levamisole
adjuvant therapy in 203 neoplastic patients (Colizza et al.,
1981). Forty-six controlled studies (2635 patients) of adjuvant
levamisole treatment in cancer were reviewed with respect to efficacy
and incidence of agranulocytosis. More patients receiving levamisole
for 2 consecutive days every week developed agranulocytosis (3.1%)
than those receiving it for 3 days every other week (0.1%). Dose
regimens were in the 5 -200 mg/kg range (Amery & Butterworth, 1983).
The pathogenesis of levamisole-associated agranulocytosis has
been studied. Based on levamisole's immunomodulating properties,
these efforts have focused on immunologic parameters. Serum from 10
neutropenic patients and 10 normal patients receiving levamisole was
evaluated. All 10 neutropenic patient serums were strongly
granulocytotoxic. IgM granulocytotoxic antibodies were identified.
None of the control patient sera were granulocytotoxic. A variety of
other immunologic parameters were evaluated. Tests for a hapten
mechanism were negative (Thompson et al., 1980). Three
neutropenic patients receiving levamisole were found to have
complement-dependent granulocytotoxic antibodies. The agranulocytosis
appeared to be related to auto-antibodies since serum
granulocytotoxicity correlated closely with agranulocytosis (Drew
et. al., 1980). More recently the possible similar aetiologies of
levamisole-induced agranulocytosis in humans and haemolytic anaemia in
dogs were reviewed. The authors concluded the evidence is compatible
with agranulocytosis in man and haemolytic anaemia in dogs being
different clinical manifestations of the same immuno-allergic reaction
(Maes and Marsboom, 1988).
A recent review of proposed immunomodulating activity etiologies
concluded, however, that none of the presently proposed hypotheses
adequately explains levamisole therapeutic activity (Van Wauwe &
Janssen, 1989).
3. COMMENTS
The Committee considered results from studies on metabolism,
short-term studies, studies on carcinogenicity, effects on
reproduction and development and mutagenicity, and from clinical
reports in humans.
The absorption, distribution, excretion, and biotransformation of
levamisole were studied primarily in rats. The results showed that
the drug is rapidly absorbed and metabolized, with only 45% of the
radioactivity in the plasma being accounted for by the parent compound
after one hour. More than 90% of the radioactivity was excreted in
the urine and faeces within 8 days. Over 50 metabolites were
identified from these studies and four major metabolic pathways have
been proposed. Following oral administration of levamisole to humans,
the half-life was estimated to be 4 hours, while the half-life for
total radioactivity was 16 hours. Most of the radioactivity was
excreted in the urine, with 4% as unchanged levamisole and the
remainder as metabolites.
In a 13-week study in rats, in which levamisole was administered
in the diet, reduced weight gains were noted at doses of 40 and 160
mg/kg b.w./day in both sexes. This effect was also observed at 10
mg/kg b.w./day in female rats. In a 90-day study in dogs, no drug-
related effects were noted at doses of up to 6 mg/kg b.w./day.
Groups of three dogs of each sex were dosed for one year (six
days per week) by oral administration of the compound in capsules.
During the eighth week, all six dogs that received levamisole at 20
mg/kg b.w./day and one dosed at 5 mg/kg b.w./day developed severe
haemolytic anaemia. The haematocrit level and total erythrocyte
counts were decreased while the number of erythroblasts and immature
granulocytes were increased. Treatment was discontinued in these
affected dogs, and their haematological parameters returned to normal
about two weeks later. However, anaemia developed again when
treatment was resumed. A levamisole-dependent erythrocyte-
agglutinating factor was demonstrated in vitro in the serum of these
dogs. No other treatment-related effects were found. The no-
observed-effect level was 1.25 mg/kg b.w./day in this study.
The Committee noted the deficiencies in the study, including the
low number of animals per group, completion of the study with only two
treated groups and uncertainty regarding the mechanism for levamisole-
induced haemolysis, which appeared to have an immunological basis.
In a chronic toxicity study in rats in which levamisole was
administered in the diet for 12 or 18 months, a significant reduction
in weight gain was observed at 80 mg/kg b.w./day in both sexes. This
effect occurred to a lesser degree at 20 mg/kg b.w./day. In general,
absolute organ weights were generally decreased, while relative organ
weights were increased at 80 mg/kg b.w./day. Other changes observed
in this treatment group included slight increases in alkaline
phosphatase (in females) and bilirubin (at 12 months), and
degeneration of the germinal epithelium of the testis. The no-
observed-effect level in this study was 20 mg/kg b.w./day.
In an 18-month carcinogenicity study in mice in which levamisole
was administered in drinking-water, no treatment-related effects were
evident with respect to the number of tumour-bearing mice or the
incidence of various types of tumor. Dose levels of up to 80 mg/kg
b.w./day were used in this study, but the Committee noted the lack of
toxicity data provided to support selection of these doses. There was
no evidence for a carcinogenic effect in female mice. However, the
survival rate of male mice beyond 12-15 months was poor. In addition,
the Committee noted that a high percentage of the male mice had not
been examined microscopically, which further reduced the sensitivity
of the study. The Committee concluded that this study did not fully
assess the carcinogenic potential of levamisole in male mice.
In a 24-month carcinogenicity study in rats in which levamisole
was administered in feed at doses up to the equivalent of 40 mg/kg
b.w./day, no treatment-related effects with respect to the number of
tumour-bearing rats or incidence of various tumour types were
reported. There was no evidence of a carcinogenic effect in female
rats and survival was not affected by treatment. However, taking into
account low survival rate beyond 18 months, the Committee concluded
that this study did not fully assess the carcinogenic potential of
levamisole in male rats.
In three reproduction studies in rats in which levamisole was
administered in the diet, there was decreased maternal weight gain,
increased incidence of still birth, reduced birth weight, and reduced
weight gain by the pups during suckling, and decreased survival at 3
weeks at 80 mg/kg b.w./day. The no-observed-effect level for effects
on reproduction was 20 mg/kg b.w./day.
Levamisole was further evaluated for effects on development in
studies in which it was administered to rats and rabbits during all or
part of gestation. In rats the incidence of resorptions was increased
slightly after oral administration at 80 mg/kg b.w./day, which was the
highest dose tested. In the rabbit, the highest dose of 40 mg/kg
b.w./day caused a marked reduction in maternal weight gain, and an
increase in the incidence of fetal death and abnormalities. The no-
observed-effect levels for these studies were 20 mg/kg b.w./day for
the rat and 10 mg/kg b.w./day for the rabbit.
The genotoxic potential of levamisole was investigated in a
number of test systems. Positive results were reported in the
chromosomal aberration test and the sister chromatid exchange test
using human lymphocytes. Negative results were reported in a series
of Ames tests, a further chromosomal aberration test, a mouse
micronucleus test, and a dominant lethal assay.
The Committee considered data from numerous clinical reports on
the use of levamisole in human therapy. Adverse effects were rare,
the most important being agranulocytosis and neutropenia. Although
these haematological disorders were generally reversible, certain
factors, such as the concurrent use of other drugs, made the
interpretation of occasional fatalities extremely difficult.
Fatalities were usually associated with concurrent infection before
agranulocytosis had been recognized. The Committee noted that the
increase in the number of IgM antibodies observed in some patients
suggested an immunological basis for the granulocytopenia; this effect
was not associated with the single dose of 2.5 mg per kg of body
weight used for the treatment of human parasites. Thrombocytopenia
and haemolytic anaemia have also been reported in patients being
treated with levamisole.
The Committee noted that there were large numbers of published
reports concerning the induction of granulocytopenia or
agranulocytosis by levamisole, primarily in patients being treated for
rheumatoid arthritis or cancer. However, the data were inadequate for
the purposes of establishing a no-observed-effect level for induction
of these haematological effects. The results of a survey showed a
0.1% incidence of agranulocytosis in patients receiving levamisole at
50-200 mg for three days every other week. However, agranulocytosis
and thrombocytopenia have been reported in a cancer patient treated
only with levamisole at 2.5 mg per kg of body weight on two
consecutive days each week for 13 weeks. Although the bone marrow was
initially depressed, the patient's immunological indicators returned
to normal within two weeks of the cessation of treatment. The
immunological basis for this condition was further supported by the
presence of levamisole-dependent leukoagglutinins correlating with the
acute phase of granulocytopenia. A report of a patient with a history
of prior treatment with levamisole who developed mild granulocytopenia
following the administration of a single challenge dose of
approximately 0.2 mg per kg of body weight, suggested that this dose
may be near the no-observed-effect level in humans.
The Committee considered that levamisole-induced haemolysis in
dogs and agranulocytosis in humans may have a similar immunological
basis. However, there was insufficient evidence to confirm or exclude
this possibility. It was noted that thrombocytopenia and haemolytic
anaemia have been observed in both dogs and human patients treated
with levamisole. However, neither the data from case reports of human
therapeutic use nor those from the study in dogs were considered
adequate for the purpose of establishing a full ADI. In the former
instance, most of the patients were suffering from cancer or diseases
associated with underlying autoimmune disorder, and had been exposed
to a variety of therapeutic regimens before receiving levamisole;
furthermore, there was no information about the threshold dose at
which agranulocytosis occurred. In the study in dogs, which used an
inadequate number of animals, haemolysis rather than agranulocytosis
was the end-point, and there was uncertainty whether the pathogenesis
of this effect was identical to that responsible for the
agranulocytosis in humans.
4. EVALUATION
A temporary ADI of 0-0.003 mg/kg of body weight was established
for levamisole, based on a no-observed-effect level of 1.25 mg/kg
b.w./day for the induction of haemolysis in dogs, and a safety factor
of 500, which the Committee selected for this compound after taking
into consideration the uncertainties regarding the relevance of the
dog model and the fact that no threshold could be established from the
human data.
The Committee noted that the lowest level of levamisole reported
to be associated with even mild neutropenia in a compromised (ill)
human patient was at least 60 times the temporary ADI. The Committee
assumed that residues other than levamisole have the same potential
toxicity as the parent drug.
The Committee requires the following by 1994:
1. A comprehensive assessment of the incidence of granulocytopenia
and agranulocytosis, thrombocytopenia and haemolytic anaemia in
humans receiving levamisole, together with dose-response
information.
2. The results of studies that demonstrate that the mechanisms for
the production of haemolytic anaemia in dogs and neutropenia or
agranulocytosis in humans are related phenomena, such as
experiments in which the specificity of the antibody response and
of the target cells is studied.
3. A comparison of the metabolites of levamisole produced in humans,
laboratory animals, and food-producing animals; if differences in
major metabolites are demonstrated, evidence should be obtained
on the potential of the metabolites that are produced in food-
producing animals to induce haemoatological effects.
5. REFERENCES
AMERY, W., & BUTTERWORTH, B. (1983). Review: Commentary on the Dosage
Regimen of Levamisole in Cancer: Is it Related to Efficacy and Safety.
Int. J. Immunopharm., 5, 1, 1-9.
ATWELL, R.B., THORNTON, J.R., & ODLUM, J. (1981). Inspected Drug
Induced Thrombocytopenia associated with Levamisole Therapy in a Dog.
Australian Vet., J. 57, 74-81.
BENARD, P., BRUNET, C., CAZIN, M., BRAUN, J.P., BURGAT-SACAZE, V., &
RICO, A.G. (1980). Whole Body Distribution of 3H Levamisole in Rats
and Mice. Mechanisms of Toxicity and Hazard Evaluation.
Elsevier/North-Holland Biomedical Press.
BERGER, R., BERHEIM, A., FEINGOLD, J., & ANDRIEU, J.M. (1980). The
Effect of Levamisole on Human Chromosomes. Path. Biol., 28, 323-
324.
BOYD, J.E., BULLOCK, M.W., CHAMPAGNE, D.A., GATTERDAM, P.E., MORICI,
I.J., PLAISTED, P.H., SPICER, L.D., WAYNE, R.S., & ZULALIAN, J.
(1973). Metabolism of 1-tetramisole in Rats. Unpublished; Submitted
to WHO by Janssen Pharmaceutica.
COLIZZA, S., BAGOLAN, P., & DI PAOLA, M. (1981). Side Effects to
Levamisole Given to Neoplastic Patients as Adjuvant to Surgery: A new
Case of Agranulocytosis. J. Surg. Oncol., 16, 259-64.
DAVEY, D.G. (1968a). One Month Toxicity Test in Dogs. Unpublished.
Imperial Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (1968b). One Month Toxicity Test in Rats. Unpublished.
Imperial Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (undated a). Acute Toxicity. Unpublished. Imperial
Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (undated b). Teratogenic Test: Rabbits. Unpublished,
Imperial Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (undated c). Teratogenic Studies. Unpublished, Imperial
Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DESPLENTER, L. (1980). Tolerance of Levamisole Pour-on Formulation in
a Baboon. Unpublished; submitted to WHO by Janssen Pharmaceutica.
DESPLENTER, L. (1983). Levamisole - Induced Vomiting in Dogs.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
DREW, S., CARTER, B., NATHANON, D., & TERASAKI, P. (1980). Levamisole-
associated Neutropenia and Autoimmune Granulocytotoxins. Annals
Rheumatic Diseases, 39, 59-63.
ENNINGA, I.C. (1988). Evaluation of the Ability of R12564
(levamisole) to Induce Chromosomal Aberrations in Cultured Peripheral
Human Lymphcytes. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
FELIX-DAVIES, (1978). Neutropenia on Low Dosage Levamisole Therapy
(Discussion). In: Proceedings of International Symposium on
Levamisole in Rheumatoid Arthritis.; University of Antwerp, Belgium,
June 23, 1978; J. Rheumatol; 5 (Suppl. 4), 93-95.
GALTIER, P., COCHE, Y., & ALVINERIE, M. (1983). Tissue distribution
and Elimination of [3H]levamisole in the Rat After Oral and
Intramuscular Administration. Xenobiotica, 13, 7, 407-413.
GATTERDAM, P.E., CHAMPAGNE, D.A., & BOYD, J.E. (1966). Tetramisole:
Preliminary Elimination Studies of the Absorption, Metabolism, and
Excretion in Rats. Unpublished, American Cyanamid. Submitted to WHO
by Janssen Pharmaceutica.
HUCHISON, E.B., MCNERY, J.,M. RIBELIN, WE., & LEVINSKAS, G.J. (1967).
1-Tetramisole: Ninety-Day Repeated Feeding to Dogs. Rpt. No. 67-118.
Unpublished, American Cyanamid. Submitted to WHO by Janssen
Pharmaceutica.
HEYKANTS, J. (1976). Clinical Research Reports. Original NDA.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
IKEGAMI, J., NISHIKAWA, T., HARA,T., MIYAZAKI, E., & OHGURO, T.
(1981). Oral Micronucleus Test in Male Mouse. Unpublished, Kyowa
Hakko Kogya Co., Ltd. Submitted to WHO by Janssen Pharmaceutica.
KOUASSI, E., Caille, G., Lery, L., Lariviere, L., & Vezina, M. (1986).
Novel Assay and Pharmacokinetics of Levamisole and P-hydroxylevamisole
in Human Plasma and Urine. Bio Pharm. Drug Dispos., 7, 71-89.
MAES, L., & MARSBOOM, R. (1988). Granulocytopenia as a Side-effect of
Prolonged Therapy with Levamisole in Man - Is there a Relationship
with the Wide-spread Use of Levamisole in Veterinary Medicine?
Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R. (1969a). Effects of Oral R12564 (levamisole) on Male and
Female fertility. Rpt. No. 239. Unpublished; submitted to WHO by
Janssen Pharmaceutica.
MARSBOOM, R. (1969b). Effects of R12546 (levamisole) in Rats After
Oral Administration During the Peri and Postnatal Period. Rpt. No.
238. Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R. (1972). Potential of Oral R12564 (levamisole) for
Embryotoxicity and Teratogenic Effects in Rats. Rpt. No. 237.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R. (1974). Three-generation Reproduction Study in Rats with
Oral R12546 (levamisole). Rpt. No. 537. Unpublished; submitted to WHO
by Janssen Pharmaceutica.
MARSBOOM, R. (1977). Dominant Lethal Test in Male Mice - Single Oral
Dose. Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R., & HERIN, V. (1980). Oral Carcinogenicity Study in
Albino Wistar Rats. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
MARSBOOM, R., HERIN, V., VANDESTEENE, R., & VAN BELLE, H. (1969).
Thirteen Week Feeding Study of Levamisole-HCl in Rats,. Unpublished;
submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R., VANDESTEENE, R. & PARDOEL, L. (1972). Eighteen Month
Feeding Study in Rats with Levamisole. Rpt. No. 154. Unpublished;
submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R., HERIN, V., VANDESTEENE, R. & PARDOEL, L. (1975). Oral
Toxicity Study in Beagle Dogs Repeated Dosage for 12 Months.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
MICHIELS, M., WOESTENBORGHS, R., & HEYKANTS, J. (1978). Absorption
and Plasma Levels of Oral Levamisole in the Rabbit. Unpublished;
submitted to WHO by Janssen Pharmaceutica.
NIEMEGEERS, C.M. (1975). The Acute Intravenous, Subcutaneous, and
Oral Toxicity of Levamisole in Mice and Rats. Unpublished; submitted
to WHO by Janssen Pharmaceutica.
PARKINSON, D.R., JERRY, L.M., SHIBATA, H.K., LEWIS, M.G., CARO, P.O.,
CAPEK, A., MANSELL, P.W., & MARQUIS, G. (1977). Complications of
Cancer Immunotherapy with Levamisole. Lancet, 1, 1129-1132.
REYNTJENS. (1989). Clinical Expert Report on Use of Levamisole for
Treatment of Dukes C Colorectal Cancer in Man. Unpublished; submitted
to WHO by Janssen Pharmaceutica.
RICHOLD, M. JONES, E., & PROUDLOCK, R. (1979). Ames Metabolic
Activation Test to Assess the Potential Mutagenic Effect of
Levamisole. Unpublished, Huntingdon Research Center. Submitted to
WHO by Janssen Pharmaceutica.
SYMOENS, J. VEYS, E., MIELANTS, M., & PINALS, R. (1978). Adverse
Reactions to Levamisole. Cancer Treatment Reports, 62, 11, 1721-30.
THOMPSON, J., HERBICK, J., KLASSEN, C. SEVERSON, C., VELIN,V.,
BLASCHKE, J., SILVERMAN, M., & VOGEL, C. (1980). Studies on
Levamisole - Induced Agranulocytosis. Blood, 56, 3, 388-396.
VANDENBERGHE, J., MARSBOOM, R., & HERIN, V. (1980). Oral
Carcinogenicity Study in Albino Swiss Mice. Unpublished; submitted to
WHO by Janssen Pharmaceutica.
VAN WAUWE, J, & JANSSEN, P. (1989). The Cardinal Question About
Levamisole: How Does It Work? Unpublished; submitted to WHO by
Janssen Pharmaceutica.
VERSTRAETEN, A., HERIN, V., & MARSBOOM, R. (1983). Subacute Toxicity
Study in Beagle Dogs Repeated Dosage for 3 Months Pour On.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
VEYS, P., & DONY, J. (1985). Embryotoxicity and Teratogenicity Study
with Levamisole in Swine. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
VEYS, P. (1989). Literature Retrieval of Reports and Publications on
Levamisole-related Granulocytopenia. Unpublished; submitted to WHO by
Janssen Pharmaceutica.
WALLWORK, L.M. & JAMES, J.A. (1975). Rat Acute Oral and Dermal
Toxcicity Using a "Pour-on" Formulation. Unpublished, Wellcome
Research Laboratories. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
WANG, G.T. (1970a). Safety study of Levamisole in Swine.
Unpublished, American Cyanamide. Submitted to WHO by Janssen
Pharamaceutica.
WANG, G.T. (1970b). Safety study of Levamisole in Swine.
Unpublished, American Cyanamide. Submitted to WHO by Janssen
Pharamaceutica.
Table 1: Results of acute toxicity studies with levamisole
Species Sex Route LD50 Reference
(mg/kg)
Mouse M&F oral 205-285 Davey, undated a
Niemegeers, 1975
M&F i.v. 20-28 Davey, undated a
Niemegeers, 1975
M&F s.c. 102-121 Davey, undated a
Niemegeers, 1975
Rat M&F oral 458-1095 Davey, undated a
Niemegeers, 1975
M&F 286-566 Wallwork &
James, 1975
M i.v. 17-28 Davey, undated a
Niemegeers, 1975
M&F s.c. 81-89 Davey, undated a
Niemegeers, 1975
dermal 252 Wallwork & James,
1975
Rabbit M oral 458 Davey, undated a
F i.v. 25 Davey, undated a
Swine M&F oral in no Wang, 1970a
feed at lethality
40 mg/kg
A one month oral study with levamisole was conducted in beagles.
Twenty-four dogs were divided into 3 groups containing 4 dogs/sex.
Dogs received 0, 10, or 20 mg/kg b.w./day. Clinical signs included
ataxia and convulsions; one death occurred in the high dose group.
Treated groups lost weight during the study. Haematologic and
clinical chemistry parameters were evaluated but no treatment-related
effects occurred. Gross necropsy was performed on all dogs and organ
weights were obtained. Histopathologic evaluation was generally
unremarkable although some lymphoid cuffing occurred in the cerebral
vessels of high dose dogs (Davey, 1968b).
Groups of beagle dogs (2/sex) received 0, 1.5, 3, or 6 mg/kg
b.w./day of levamisole daily for 90 days. Dogs were up to two years
of age at study initiation. Parameters evaluated included: clinical
signs, food intake, body weight, haematology, clinical chemistry,
organ weights and histopathology (6 mg/kg b.w./day group only was
sacrificed). Special emphasis was placed on evaluation of possible
haemolytic effects of levamisole. Dogs were distributed into dose-
groups based on predetermined susceptibility to erythrocyte
haemolysis. Methaemoglobin and erythroblastosis were monitored. No
treatment-related effects were observed (Huchison et al., 1967).
The effect of levamisole treatment for one year in dogs was
evaluated. Four groups of 8-10 month old beagles containing 3
dogs/sex/group were administered levamisole orally in gelatin capsules
daily six times/week for 12 months. Dogs received 20, 5, or 1.25
mg/kg b.w./day levamisole or 250 mg lactose. Dogs were acclimated and
baseline parameter values obtained during a 6 week period prior to
dosing. The following parameters were evaluated: Clinical signs
including ophthalmoscopy (0, 6, and 12 months), ECG and blood pressure
(monthly), food consumption, body weight (weekly), haematology, and
clinical chemistry and urinalysis (monthly). Gross pathology and
organ weights were monitored at necropsy, and histopathologic
examination was performed on a standard array of tissues.
During the 8th week all 20 mg/kg b.w./day dogs and one 5 mg/kg
b.w./day female dog experienced severe treatment induced haemolytic
anaemia. Decreases in haematocrit, haemoglobin, and RBC count
occurred with increases in erythoroblasts and immature granulocytes.
These dogs were removed from the study. Their haematology parameters
returned to normal about 2 weeks after dosing was stopped, but anaemia
returned when treatment was reinstituted. An in vitro serum RBC
agglutinizing factor which required at least 100 µg/ml of levamisole
was demonstrated. No other treatment-related effects were found in
this study (Marsboom et al., 1975).
The effect of levamisole administered daily as a pour on was
studied in dogs. Twenty-four beagles were divided into 4 groups of
3/sex and received 0, 2.5, 10, or 40 mg/kg b.w. daily as a 20%
solution applied to the back. The following parameters were
evaluated: clinical signs, ophthalmoscopic examination, body weight,
heart rate and ECG, blood pressure, haematology, clinical chemistry,
urinalysis, and at necropsy organ weights, gross pathology and
histopathology. Significant weight loss occurred in the 40 mg/kg
b.w./day group. No other treatment-related effects were observed in
this study (Verstraeten et al., 1983).
A series of clinical reports on levamisole-associated haemolysis
in dogs were evaluated. All reports were related to long-term use of
levamisole for dirofilaria treatment. The dose regimens generally
were altered at two week intervals with peak doses of 10-24 mg/kg
b.w./day and low doses of 2-3 mg/kg b.w./day. Anaemia developed at 3-
6 weeks of treatment (Maes & Marsboom, 1988; Atwell et al., 1981).
2.2.2.3 Pigs
Levamisole-HCl was administered to pigs at the rate of 0, 8, 24,
and 40 mg/kg body weight in feed or drinking water as a single
treatment. Five pigs/sex/dose were treated with each route of
administration. The treatment was repeated one week later. More
severe signs were observed in the drinking water group. Salivation
and vomiting were dose-related, while tremor, tachypnoea, transient
recumbency and 10% mortality were observed in the 40 mg/kg body weight
group. Salivation and vomiting only were observed in the 40 mg/kg
body weight feed-route pigs. Some suggestion of hepatic toxicity was
found upon histopathologic evaluation. The authors concluded that,
although some toxicity occurred, the 40 mg/kg body weight feed-
treatment and 24 mg/kg body treatment water-treatment were safe for
swine based on reversibility of clinical signs and hepatic lesions
(Wang, 1970b).
2.2.2.4 Primates
A 23 kg male baboon was treated dermally with 10, 20, and 40
mg/kg body weight levamisole with intervals of 10 and 4 days between
treatments. Effects including minor excitement were observed
following the 40 mg per kg body weight dose (Desplenter, 1980).
2.2.3 Long-term/carcinogenicity studies
2.2.3.1 Mice
An oral carcinogenicity bioassay of levamisole was conducted
using albino Swiss mice. Four hundred 7 to 8 week old mice were
divided into 4 groups containing 50 mice/sex. Following an
acclimatization period of one week, levamisole was provided at the
rate of 0, 12.5, 50, and 200 ppm in drinking water for 18 months to
provide target doses of 0, 5, 20, and 80 mg/kg b.w./day based on
consumption of 100 ml/week/mouse. The test article was stored at room
temperature in well-closed amber bottles. Solutions were prepared
fresh weekly. The animals were initially housed 25/cage but after 7
months were housed individually to prevent fighting. Clinical
examinations were performed daily except Sundays, but were not
reported. Gross necropsy was performed on all mice that died or were
sacrificed due to moribund condition as well as at study termination.
Lung, liver, pancreas, kidney, spleen, testis, epididymis, ovary,
uterus, mammary gland, adrenals, hypophysis, lymph nodes, and any
other indicated tissue were examined histopathologically. Survival
rates at the end of the study were: 0 ppm - 32%M/22%F, 12.5 ppm -
20%M/40%F, 50 ppm - 18%M/40%F, and 200 ppm - 20%M/38%F. However the
survival rate at 12-14 months was reduced considerably from that
expected for a useful carcinogenicity bioassay: less than 50% in all
male treatment groups and about 40% in the female control group. No
treatment-related gross pathology was reported. No treatment-related
effects were seen with respect to number of tumour-bearing mice or
incidence of various tumour types. However, appropriate tissues
available for examination were fewer than expected for a
carcinogenicity bioassay (Vandenberghe et al., 1980).
2.2.3.2 Rats
The chronic toxicity of levamisole was examined in rats. Six to
eight week old Wistar rats were divided into 4 groups of 20/sex and
received target doses of 40, 10, 2.5 and 0 mg/kg b.w. levamisole daily
provided in the feed at rates of 800, 200, 50, or 0 ppm. Diets were
prepared weekly. An interim sacrifice of 10 rats/sex was performed at
12 months with the final sacrifice at 18 months. The following
parameters were evaluated: Clinical signs (daily), ophthalmoscopic
exam (terminal sacrifice), food consumption (weekly), body weight
(weekly), and haematology, clinical chemistry, and urinalysis
(terminal sacrifice). Gross necropsy was performed at sacrifice.
Organ weights were obtained for heart, spleen, thymus, liver, kidney,
pancreas, adrenals, thyroid, brain, genital organs, and lung.
Histopathology was performed on tissue from the above organs and the
following array of tissues: Lymph nodes, urinary bladder, alimentary
tract tissues, parathyroid, hypophysis, diaphragm, striated muscle,
tongue, teeth, skin, bone marrow, and eye. No treatment related
clinical signs or mortality occurred. Mean food consumption was
reduced in both sexes of the high dose group. Weight gains were
significantly decreased for both sexes of the high dose group
throughout the study. The middle dose group weight gain was generally
reduced throughout the study but was significant only for the 12 month
females. Clinical chemistry parameters which were altered included
marginal increases in alkaline phosphatase in high dose females, and
bilirubin in 12 month high dose males and females. In the high dose
group the absolute weights of organs generally were decreased, while
the relative weights were increased. Histopathologic treatment-
related changes were limited to a degeneration of the germinal
epithelium of the testes and a mild chronic stimulation of the liver
seen only in the high-dose group (Marsboom et al., 1972).
The tumourigenic potential of levamisole was studied in Wistar
rats. Four hundred 3 month old rats were divided into 4 groups of
50/sex and following a one week acclimatization period were provided
levamisole in the diet at rates of 0, 50, 200, or 800 ppm to provide
target doses of 0, 2.5, 10, or 40 mg/kg b.w./day. Test diet was
prepared every two weeks. Animals were examined daily but clinical
signs were not reported. A full necropsy was performed on all
surviving animals at 24 month study termination and all animals which
died or were sacrificed due to moribund condition during the study.
The following tissues were examined histopathologically: Lung, liver,
pancreas, kidney, spleen, testis, epididymis, ovary, mammary gland,
adrenals, thyroids, parathyroids, hypophysis, lymph nodes, and any
other indicated tissue. Survival rates for the various groups were:
0 ppm - 4%M/10%F, 50 ppm - 2%M/8%F, 200 ppm - 2%M/20%F, and 800 ppm -
6%M/14%F. Survival rates for all male study groups were well below
50% by 16 months. No treatment-related effects were seen with respect
to number of tumour-bearing rats or incidence of various tumour types.
However, appropriate tissues available for examination were fewer than
expected for a carcinogenicity bioassay (Marsboom & Herin, 1980).
2.2.4 Reproduction studies
2.2.4.1 Rats
The effect of levamisole administered prior to breeding for 60
days in males and 14 days in females on the reproductive performance
of rats was studied. To accomplish the study 320 sexually mature
Wistar rats were divided into groups of 20 rats/sex and received
levamisole target doses of 1.25, 5, 20, or 80 mg/kg b.w./day in the
feed at rates of 25, 100, 400, or 1600 ppm for the prescribed duration
of treatment. Fresh diets were prepared weekly. Treated rats were
mated with nontreated rats. No control matings were performed with
untreated animals. Matings were validated by daily vaginal washings
for sperm. Female weights were determined at insemination and weekly
thereafter. Half the pregnant females were sacrificed on gestation
day 13 and the uterus was examined for number and distribution of
embryos, and resorptions as well as unusual uterine condition. The
remaining females were sacrificed on gestation day 22. The number and
distribution of fetuses were recorded and fetuses were examined
externally. Fetuses were processed and examined for internal
abnormalities via clearing and alizarin red staining of bones and
serial sectioning as well as X-ray analysis. No treatment-related
effects on male or female fertility or offspring normality were
reported in this study (Marsboom, 1969a).
The potential toxicity of levamisole administered during the
peri- and post-natal periods was evaluated in female Wistar rats.
Five groups of 20 three month old rats received 0, 25, 100, 400, and
1600 ppm levamisole in the diet from gestation day 16 through the 3
week lactation period to provide target doses of 0, 1.25, 5, 20, and
80 mg/kg b.w./day. Female weights were recorded on gestation days 1,
7, 14, and 21 post parturition, and on lactation days 14 and 21. Live
and dead pups were examined, counted, and weighed a few hours after
parturition. Females not delivering by gestation day 24 were
sacrificed, necropsied, and uteruses evaluated for distribution of
placental sites, resorptions and fetuses. Surviving pups were weighed
on lactation days 14 and 21. There was a decrease in weight gain for
the 1600 ppm dams. Additionally an increased incidence of stillborn
pups, reduced birth weight, reduced lactational weight gains, and
decreased 3-week survival rate were recorded for the 1600 ppm pups
(Marsboom, 1969b).
The potential embryotoxic and teratogenic effects of levamisole
were evaluated during the gestation periods of three consecutive
generations of treated pregnant rats. Eighty virgin 3-4 month old
rats were divided into 4 groups of 20 rats each and provided
levamisole at the rate of 0, 100, 400, and 1600 ppm in the diet,
equivalent to 0, 5, 20 and 80 mg/kg body weight/day on gestation days
6-15. Prior to and after the period of organogenesis the rats were
provided the control diet. At the age of 3 months 80 virgin females
were selected from those born to treated mothers of the prior
generation and mated with 40 non-littermate males also born to treated
dams from the preceding generation. The males were treated in the
same manner as their dams. The succeeding generation was established
in the same manner except that dams were sacrificed on gestation day
22 for evaluation of teratogenic and embryotoxic parameters. Females
were weighed on the 1st, 7th, 14th and 21st days of gestation, while
pups were weighed shortly after parturition and on lactation days 4,
14, and 21. If parturition had not occurred by gestation day 24 rats
were sacrificed, necropsied and distribution of fetuses and
resorptions were noted. The litters from the 3rd generation were
examined for distribution of live and dead fetuses and resorptions.
All fetuses were examined for external abnormalities, radiographic
examination was carried out, and fetuses were processed for soft
tissue (1/3) and skeletal (2/3) examination. No treatment-related
effects were recorded on dams or fetuses (Marsboom, 1974).
2.2.5 Special studies on embryotoxicity/teratogenicity
2.2.5.1 Rats
The potential embryotoxicity or teratogenicity of levamisole was
studied in Wistar rats. Sexually mature female Wistar rats were mated
with fertile males and received target doses of 0, 1.25, 5, 20, or 80
mg/kg b.w./day levamisole based on 25, 100, 400, and 1600 ppm
levamisole in the diet on gestation days 6-15. Mating was established
by vaginal plug. Weights of females were obtained at mating and
weekly thereafter. Food consumption was recorded individually during
gestation. Females were sacrificed on gestation day 22 and the
following litter and fetal parameters obtained: Number and
distribution of live and dead fetuses and resorptions within
abnormalities (external - all fetuses, skeletal - 2/3 and soft
tissue - 1/3). At 1600 ppm the incidence of resorptions increased
slightly. No other parameters were effected (Marsboom, 1972).
A teratology study with levamisole at doses of 0, 10, 50, and 100
mg/kg body weight/day was carried out on rats. Female rats were
divided into 4 groups of 20 rats and received levamisole on gestation
days 0-20 by mouth. Half the females were sacrificed on day 20,
litters were examined grossly, fetuses cleared and stained with
alizarin red and examined for skeletal effects. The remaining females
littered and reared their litters to lactation day 21. Maternal
weight gain was reduced in a dose-related manner. No effect on litter
parameters, embryotoxicity or post natal growth was observed (Davey,
undated c).
2.2.5.2 Rabbits
The teratogenic potential of levamisole was studied in rabbits.
Sixty mature female New Zealand rabbits were divided into 3 groups of
20 rabbits each. Rabbits were inseminated artificially due to low
pregnancy rate in the rabbit colony. Does received 0, 10 or 40 mg/kg
b.w./day levamisole by gavage on gestation days 6-18. Does were
observed daily for toxicity and weights were recorded at insemination,
gestation days 6-18, and at sacrifice. Rabbits were sacrificed on
gestation day 28, and necropsied. Fetuses were examined grossly and
then processed for soft tissue examination (1/3) or skeletal
examination (2/3). Survival of does was not affected by treatment.
There was a marked reduction in mean weight gain in the 40 mg/kg
b.w./day group compared with controls (250 vs 432, respectively). The
incidence of resorptions and dead fetuses was doubled in the 40 mg/kg
b.w./day group vs controls (10 vs 5), while the incidence of litters
with abnormalities was 4 in the 40 mg/kg b.w./day group vs 0 in the
control and 10 mg/kg b.w/day groups. However, this was within the
historical control range. No treatment-related effects from
levamisole occurred in this study (Marsboom, 1972).
Nineteen female Dutch rabbits were divided into 3 groups of 6-7
rabbits each to evaluate the potential teratogenic effect of
levamisole. Rabbits received 0, 25, or 75 mg/kg b.w. orally
throughout the gestation period. Does were sacrificed on gestation
day 28 and litter parameters evaluated. After examination the fetuses
were processed for skeletal examination. No treatment-related effects
were reported (Davey, undated c).
2.2.5.3 Pigs
Twenty pregnant sows received 10 mg/kg body weight levamisole 4
times at 2-day intervals intramuscularly. A group of 20 untreated
sows served as controls. The sows were selected to have produced one
previous litter of >7 pigs. The sows were divided into 4 groups of
5 each and received treatment on gestation days 8-14, 16-22, 24-30, or
32-38. Parturition data, length of gestation, litter size, sex,
weight, suckling behavior and external abnormalities were evaluated.
No treatment-related embryotoxic or teratogenic effects occurred in
this study (Veys & Dony, 1985).
2.2.6 Special studies on genotoxicity
The results of genotoxicity studies with levamisole are
summarized in Table 2.
2.3 Observations in humans
The safety of levamisole use in humans has recently been
reviewed. Levamisole has been used in humans for the following
indications: anthelminthic, rheumatoid arthritis, inflammatory
disease, infectious disease, and cancer therapy. Aside from its use
for parasitic infection, these uses appear to be based on the
immunomodulating properties of levamisole. Although a number of side
effects have been reported, the most important are haematologic,
including reversible leukopenia, agranulocytosis, and
thrombocytopenia. A low incidence of agranulocytosis appears to occur
in all continuous dose regimens ranging from 50 to 200 mg daily and
with 3 days treatment every other week to continuous therapy.
Haematologic effects were seen in nearly 5% of rheumatic patients but
only 0.2% of infectious patients. Based on his review of the data,
the author concluded that agranulocytosis was dose-related in onset
(Reyntjens, 1989).
A survey of published literature on levamisole-associated
agranulocytosis identified 110 citations. These reports occurred
between 1973 and 1984, and, based on volume of levamisole uses,
agranulocytosis correlated closely with human use of levamisole for
immunotherapy but not veterinary use of levamisole (Veys, 1989).
Numerous clinical reports of levamisole side effects are in the
literature. Two reports are reviewed for illustrative purposes:
Observations were made on a single cancer patient who was given
only levamisole at the rate of 2.5 mg/kg body weight for 2 consecutive
days per week for 13 weeks. The patient developed an agranulocytosis
and therapy was discontinued. His total white cell count was 1100
cells/mm3. Bone-marrow was described as hypercellular with relative
erythroid hyperplasia and virtually no myelopoiesis (0.5% myeloblasts,
no mature neutrophils, 2% eosinophils, 0.5% basophils, 63%
lymphocytes, 5% plasma cells, 0.5% monocytes, 23.5% erythroid forms,
and 5% reticulum cells). Megakargocytes were present. Seven days
later the patient's bone marrow was hypocellular but demonstrated
myelopoiesis: 12% myeloblasts, 15% promyelocytes, 2% myelocytes, 8%
metamyelocytes; and 1% neutrophils. The red cell series was reduced
and megakaryocytes were markedly reduced. A transient but severe
thrombocytopenia (without bleeding) to 30,000 cells/mm3 also occurred
during the first week.
Table 2: Genotoxicity of levamisole
Test system Test Object Concentration Results Reference
Chromosomal Human from humans positive Berger
aberrations lymphocytes treated with et al.,
150 mg 1980
levamisole
(in vivo) or
250 µg/ml
(in vitro)
Sister Humanadded lymphocyte positive Berger
Chromatid lymphocytes cultures from et al.,
Exchange untreated 1980
volunteers
Ames test TA 1535 10-10,000 µg/ml negative Richold
(+/- S9) TA 1537 et al.,
TA 98 1979
TA 100
Chromosomal Cultured +S9: 5000 µg/ml negative Enninga,
aberrations human -S9: 1000 µg/ml 1988
(+/- S9) lymphocytes
Micronucleus Mouse 3-75 mg/kg negative Ikegami,
test erythrocytesorally et al., 1981
Dominant Male and Single oral negative Marsboom,
lethal test female dose: 10, 40 1977
mice or 160 mg/kg
b.w.
The marrow recovered in 10-14 days with circulating neutrophils
rebounding to raised levels. The immunologic basis for these
levamisole effects were explored using the leuco agglutination test
and Clq deviation test for circulating immune complexes. Levamisole-
dependent leuco agglutinations were present in the patient's serum in
high titre (1/512) during the acute phase. During the first week the
patient's serum induced some spontaneous leuco agglutination; however,
after one week added levamisole was required for leuco agglutination.
Circulating Clq-reactive materials reached a peak seven days after
onset of neutropenia coincident with the peak in leuco agglutination
titre (Parkinson et al., 1977).
A clinical report of a 52 year old female patient with a history
of current seronegative rheumatoid arthritis, erythema nodosum in
childhood, and angioneurotic edema at age 35 to 37 years was reviewed.
She received 50 mg levamisole once weekly. After each dose she
experienced a mild flu-like syndrome. Following the sixth dose this
syndrome was more severe and mouth ulcers developed. This syndrome
became more pronounced following the seventh dose. She had a
polymorph count of 225 per mm3 6 days later. Bone marrow examination
showed increased promyelocytes but maturation arrest at the myelocyte
stage. Erythropoiesis was normal. No sideroblasts were present. A
dose of 10 mg levamisole was given 2 months later. No clinical side
effects were noted but a polymorph decrease of 31% occurred at 24
hours post dose. A dose of 25 mg of levamisole two weeks later
produced clinical signs and a 43% reduction of polymorphs at 32 hours.
Twenty leucocyte lines were tested for agglutination with 19/20
yielding negative results. Cytotoxicity tests were also negative
(Felix-Davies, 1978).
In addition to individual case studies and clinical reports, some
early epidemiologic reports or reviews of levamisole-associated
haematologic adverse effects were available. Adverse reactions in 267
of 6217 patients on levamisole were analyzed via a questionnaire.
Significant reactions were agranulocytosis (2.3%), skin rash and
febrile illness. These occurred primarily in patients (particularly
females) with rheumatoid arthritis. Agranulocytosis was spontaneously
reversible (Symoens et al., 1978). A low incidence of leukopenia
and agranulocytosis (0.4%) was reported after use of levamisole
adjuvant therapy in 203 neoplastic patients (Colizza et al.,
1981). Forty-six controlled studies (2635 patients) of adjuvant
levamisole treatment in cancer were reviewed with respect to efficacy
and incidence of agranulocytosis. More patients receiving levamisole
for 2 consecutive days every week developed agranulocytosis (3.1%)
than those receiving it for 3 days every other week (0.1%). Dose
regimens were in the 5 -200 mg/kg range (Amery & Butterworth, 1983).
The pathogenesis of levamisole-associated agranulocytosis has
been studied. Based on levamisole's immunomodulating properties,
these efforts have focused on immunologic parameters. Serum from 10
neutropenic patients and 10 normal patients receiving levamisole was
evaluated. All 10 neutropenic patient serums were strongly
granulocytotoxic. IgM granulocytotoxic antibodies were identified.
None of the control patient sera were granulocytotoxic. A variety of
other immunologic parameters were evaluated. Tests for a hapten
mechanism were negative (Thompson et al., 1980). Three
neutropenic patients receiving levamisole were found to have
complement-dependent granulocytotoxic antibodies. The agranulocytosis
appeared to be related to auto-antibodies since serum
granulocytotoxicity correlated closely with agranulocytosis (Drew
et. al., 1980). More recently the possible similar aetiologies of
levamisole-induced agranulocytosis in humans and haemolytic anaemia in
dogs were reviewed. The authors concluded the evidence is compatible
with agranulocytosis in man and haemolytic anaemia in dogs being
different clinical manifestations of the same immuno-allergic reaction
(Maes and Marsboom, 1988).
A recent review of proposed immunomodulating activity etiologies
concluded, however, that none of the presently proposed hypotheses
adequately explains levamisole therapeutic activity (Van Wauwe &
Janssen, 1989).
3. COMMENTS
The Committee considered results from studies on metabolism,
short-term studies, studies on carcinogenicity, effects on
reproduction and development and mutagenicity, and from clinical
reports in humans.
The absorption, distribution, excretion, and biotransformation of
levamisole were studied primarily in rats. The results showed that
the drug is rapidly absorbed and metabolized, with only 45% of the
radioactivity in the plasma being accounted for by the parent compound
after one hour. More than 90% of the radioactivity was excreted in
the urine and faeces within 8 days. Over 50 metabolites were
identified from these studies and four major metabolic pathways have
been proposed. Following oral administration of levamisole to humans,
the half-life was estimated to be 4 hours, while the half-life for
total radioactivity was 16 hours. Most of the radioactivity was
excreted in the urine, with 4% as unchanged levamisole and the
remainder as metabolites.
In a 13-week study in rats, in which levamisole was administered
in the diet, reduced weight gains were noted at doses of 40 and 160
mg/kg b.w./day in both sexes. This effect was also observed at 10
mg/kg b.w./day in female rats. In a 90-day study in dogs, no drug-
related effects were noted at doses of up to 6 mg/kg b.w./day.
Groups of three dogs of each sex were dosed for one year (six
days per week) by oral administration of the compound in capsules.
During the eighth week, all six dogs that received levamisole at 20
mg/kg b.w./day and one dosed at 5 mg/kg b.w./day developed severe
haemolytic anaemia. The haematocrit level and total erythrocyte
counts were decreased while the number of erythroblasts and immature
granulocytes were increased. Treatment was discontinued in these
affected dogs, and their haematological parameters returned to normal
about two weeks later. However, anaemia developed again when
treatment was resumed. A levamisole-dependent erythrocyte-
agglutinating factor was demonstrated in vitro in the serum of these
dogs. No other treatment-related effects were found. The no-
observed-effect level was 1.25 mg/kg b.w./day in this study.
The Committee noted the deficiencies in the study, including the
low number of animals per group, completion of the study with only two
treated groups and uncertainty regarding the mechanism for levamisole-
induced haemolysis, which appeared to have an immunological basis.
In a chronic toxicity study in rats in which levamisole was
administered in the diet for 12 or 18 months, a significant reduction
in weight gain was observed at 80 mg/kg b.w./day in both sexes. This
effect occurred to a lesser degree at 20 mg/kg b.w./day. In general,
absolute organ weights were generally decreased, while relative organ
weights were increased at 80 mg/kg b.w./day. Other changes observed
in this treatment group included slight increases in alkaline
phosphatase (in females) and bilirubin (at 12 months), and
degeneration of the germinal epithelium of the testis. The no-
observed-effect level in this study was 20 mg/kg b.w./day.
In an 18-month carcinogenicity study in mice in which levamisole
was administered in drinking-water, no treatment-related effects were
evident with respect to the number of tumour-bearing mice or the
incidence of various types of tumor. Dose levels of up to 80 mg/kg
b.w./day were used in this study, but the Committee noted the lack of
toxicity data provided to support selection of these doses. There was
no evidence for a carcinogenic effect in female mice. However, the
survival rate of male mice beyond 12-15 months was poor. In addition,
the Committee noted that a high percentage of the male mice had not
been examined microscopically, which further reduced the sensitivity
of the study. The Committee concluded that this study did not fully
assess the carcinogenic potential of levamisole in male mice.
In a 24-month carcinogenicity study in rats in which levamisole
was administered in feed at doses up to the equivalent of 40 mg/kg
b.w./day, no treatment-related effects with respect to the number of
tumour-bearing rats or incidence of various tumour types were
reported. There was no evidence of a carcinogenic effect in female
rats and survival was not affected by treatment. However, taking into
account low survival rate beyond 18 months, the Committee concluded
that this study did not fully assess the carcinogenic potential of
levamisole in male rats.
In three reproduction studies in rats in which levamisole was
administered in the diet, there was decreased maternal weight gain,
increased incidence of still birth, reduced birth weight, and reduced
weight gain by the pups during suckling, and decreased survival at 3
weeks at 80 mg/kg b.w./day. The no-observed-effect level for effects
on reproduction was 20 mg/kg b.w./day.
Levamisole was further evaluated for effects on development in
studies in which it was administered to rats and rabbits during all or
part of gestation. In rats the incidence of resorptions was increased
slightly after oral administration at 80 mg/kg b.w./day, which was the
highest dose tested. In the rabbit, the highest dose of 40 mg/kg
b.w./day caused a marked reduction in maternal weight gain, and an
increase in the incidence of fetal death and abnormalities. The no-
observed-effect levels for these studies were 20 mg/kg b.w./day for
the rat and 10 mg/kg b.w./day for the rabbit.
The genotoxic potential of levamisole was investigated in a
number of test systems. Positive results were reported in the
chromosomal aberration test and the sister chromatid exchange test
using human lymphocytes. Negative results were reported in a series
of Ames tests, a further chromosomal aberration test, a mouse
micronucleus test, and a dominant lethal assay.
The Committee considered data from numerous clinical reports on
the use of levamisole in human therapy. Adverse effects were rare,
the most important being agranulocytosis and neutropenia. Although
these haematological disorders were generally reversible, certain
factors, such as the concurrent use of other drugs, made the
interpretation of occasional fatalities extremely difficult.
Fatalities were usually associated with concurrent infection before
agranulocytosis had been recognized. The Committee noted that the
increase in the number of IgM antibodies observed in some patients
suggested an immunological basis for the granulocytopenia; this effect
was not associated with the single dose of 2.5 mg per kg of body
weight used for the treatment of human parasites. Thrombocytopenia
and haemolytic anaemia have also been reported in patients being
treated with levamisole.
The Committee noted that there were large numbers of published
reports concerning the induction of granulocytopenia or
agranulocytosis by levamisole, primarily in patients being treated for
rheumatoid arthritis or cancer. However, the data were inadequate for
the purposes of establishing a no-observed-effect level for induction
of these haematological effects. The results of a survey showed a
0.1% incidence of agranulocytosis in patients receiving levamisole at
50-200 mg for three days every other week. However, agranulocytosis
and thrombocytopenia have been reported in a cancer patient treated
only with levamisole at 2.5 mg per kg of body weight on two
consecutive days each week for 13 weeks. Although the bone marrow was
initially depressed, the patient's immunological indicators returned
to normal within two weeks of the cessation of treatment. The
immunological basis for this condition was further supported by the
presence of levamisole-dependent leukoagglutinins correlating with the
acute phase of granulocytopenia. A report of a patient with a history
of prior treatment with levamisole who developed mild granulocytopenia
following the administration of a single challenge dose of
approximately 0.2 mg per kg of body weight, suggested that this dose
may be near the no-observed-effect level in humans.
The Committee considered that levamisole-induced haemolysis in
dogs and agranulocytosis in humans may have a similar immunological
basis. However, there was insufficient evidence to confirm or exclude
this possibility. It was noted that thrombocytopenia and haemolytic
anaemia have been observed in both dogs and human patients treated
with levamisole. However, neither the data from case reports of human
therapeutic use nor those from the study in dogs were considered
adequate for the purpose of establishing a full ADI. In the former
instance, most of the patients were suffering from cancer or diseases
associated with underlying autoimmune disorder, and had been exposed
to a variety of therapeutic regimens before receiving levamisole;
furthermore, there was no information about the threshold dose at
which agranulocytosis occurred. In the study in dogs, which used an
inadequate number of animals, haemolysis rather than agranulocytosis
was the end-point, and there was uncertainty whether the pathogenesis
of this effect was identical to that responsible for the
agranulocytosis in humans.
4. EVALUATION
A temporary ADI of 0-0.003 mg/kg of body weight was established
for levamisole, based on a no-observed-effect level of 1.25 mg/kg
b.w./day for the induction of haemolysis in dogs, and a safety factor
of 500, which the Committee selected for this compound after taking
into consideration the uncertainties regarding the relevance of the
dog model and the fact that no threshold could be established from the
human data.
The Committee noted that the lowest level of levamisole reported
to be associated with even mild neutropenia in a compromised (ill)
human patient was at least 60 times the temporary ADI. The Committee
assumed that residues other than levamisole have the same potential
toxicity as the parent drug.
The Committee requires the following by 1994:
1. A comprehensive assessment of the incidence of granulocytopenia
and agranulocytosis, thrombocytopenia and haemolytic anaemia in
humans receiving levamisole, together with dose-response
information.
2. The results of studies that demonstrate that the mechanisms for
the production of haemolytic anaemia in dogs and neutropenia or
agranulocytosis in humans are related phenomena, such as
experiments in which the specificity of the antibody response and
of the target cells is studied.
3. A comparison of the metabolites of levamisole produced in humans,
laboratory animals, and food-producing animals; if differences in
major metabolites are demonstrated, evidence should be obtained
on the potential of the metabolites that are produced in food-
producing animals to induce haemoatological effects.
5. REFERENCES
AMERY, W., & BUTTERWORTH, B. (1983). Review: Commentary on the Dosage
Regimen of Levamisole in Cancer: Is it Related to Efficacy and Safety.
Int. J. Immunopharm., 5, 1, 1-9.
ATWELL, R.B., THORNTON, J.R., & ODLUM, J. (1981). Inspected Drug
Induced Thrombocytopenia associated with Levamisole Therapy in a Dog.
Australian Vet., J. 57, 74-81.
BENARD, P., BRUNET, C., CAZIN, M., BRAUN, J.P., BURGAT-SACAZE, V., &
RICO, A.G. (1980). Whole Body Distribution of 3H Levamisole in Rats
and Mice. Mechanisms of Toxicity and Hazard Evaluation.
Elsevier/North-Holland Biomedical Press.
BERGER, R., BERHEIM, A., FEINGOLD, J., & ANDRIEU, J.M. (1980). The
Effect of Levamisole on Human Chromosomes. Path. Biol., 28, 323-
324.
BOYD, J.E., BULLOCK, M.W., CHAMPAGNE, D.A., GATTERDAM, P.E., MORICI,
I.J., PLAISTED, P.H., SPICER, L.D., WAYNE, R.S., & ZULALIAN, J.
(1973). Metabolism of 1-tetramisole in Rats. Unpublished; Submitted
to WHO by Janssen Pharmaceutica.
COLIZZA, S., BAGOLAN, P., & DI PAOLA, M. (1981). Side Effects to
Levamisole Given to Neoplastic Patients as Adjuvant to Surgery: A new
Case of Agranulocytosis. J. Surg. Oncol., 16, 259-64.
DAVEY, D.G. (1968a). One Month Toxicity Test in Dogs. Unpublished.
Imperial Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (1968b). One Month Toxicity Test in Rats. Unpublished.
Imperial Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (undated a). Acute Toxicity. Unpublished. Imperial
Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (undated b). Teratogenic Test: Rabbits. Unpublished,
Imperial Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DAVEY, D.G. (undated c). Teratogenic Studies. Unpublished, Imperial
Chemical Industries Limited. Submitted to WHO by Janssen
Pharmaceutica.
DESPLENTER, L. (1980). Tolerance of Levamisole Pour-on Formulation in
a Baboon. Unpublished; submitted to WHO by Janssen Pharmaceutica.
DESPLENTER, L. (1983). Levamisole - Induced Vomiting in Dogs.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
DREW, S., CARTER, B., NATHANON, D., & TERASAKI, P. (1980). Levamisole-
associated Neutropenia and Autoimmune Granulocytotoxins. Annals
Rheumatic Diseases, 39, 59-63.
ENNINGA, I.C. (1988). Evaluation of the Ability of R12564
(levamisole) to Induce Chromosomal Aberrations in Cultured Peripheral
Human Lymphcytes. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
FELIX-DAVIES, (1978). Neutropenia on Low Dosage Levamisole Therapy
(Discussion). In: Proceedings of International Symposium on
Levamisole in Rheumatoid Arthritis.; University of Antwerp, Belgium,
June 23, 1978; J. Rheumatol; 5 (Suppl. 4), 93-95.
GALTIER, P., COCHE, Y., & ALVINERIE, M. (1983). Tissue distribution
and Elimination of [3H]levamisole in the Rat After Oral and
Intramuscular Administration. Xenobiotica, 13, 7, 407-413.
GATTERDAM, P.E., CHAMPAGNE, D.A., & BOYD, J.E. (1966). Tetramisole:
Preliminary Elimination Studies of the Absorption, Metabolism, and
Excretion in Rats. Unpublished, American Cyanamid. Submitted to WHO
by Janssen Pharmaceutica.
HUCHISON, E.B., MCNERY, J.,M. RIBELIN, WE., & LEVINSKAS, G.J. (1967).
1-Tetramisole: Ninety-Day Repeated Feeding to Dogs. Rpt. No. 67-118.
Unpublished, American Cyanamid. Submitted to WHO by Janssen
Pharmaceutica.
HEYKANTS, J. (1976). Clinical Research Reports. Original NDA.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
IKEGAMI, J., NISHIKAWA, T., HARA,T., MIYAZAKI, E., & OHGURO, T.
(1981). Oral Micronucleus Test in Male Mouse. Unpublished, Kyowa
Hakko Kogya Co., Ltd. Submitted to WHO by Janssen Pharmaceutica.
KOUASSI, E., Caille, G., Lery, L., Lariviere, L., & Vezina, M. (1986).
Novel Assay and Pharmacokinetics of Levamisole and P-hydroxylevamisole
in Human Plasma and Urine. Bio Pharm. Drug Dispos., 7, 71-89.
MAES, L., & MARSBOOM, R. (1988). Granulocytopenia as a Side-effect of
Prolonged Therapy with Levamisole in Man - Is there a Relationship
with the Wide-spread Use of Levamisole in Veterinary Medicine?
Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R. (1969a). Effects of Oral R12564 (levamisole) on Male and
Female fertility. Rpt. No. 239. Unpublished; submitted to WHO by
Janssen Pharmaceutica.
MARSBOOM, R. (1969b). Effects of R12546 (levamisole) in Rats After
Oral Administration During the Peri and Postnatal Period. Rpt. No.
238. Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R. (1972). Potential of Oral R12564 (levamisole) for
Embryotoxicity and Teratogenic Effects in Rats. Rpt. No. 237.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R. (1974). Three-generation Reproduction Study in Rats with
Oral R12546 (levamisole). Rpt. No. 537. Unpublished; submitted to WHO
by Janssen Pharmaceutica.
MARSBOOM, R. (1977). Dominant Lethal Test in Male Mice - Single Oral
Dose. Unpublished; submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R., & HERIN, V. (1980). Oral Carcinogenicity Study in
Albino Wistar Rats. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
MARSBOOM, R., HERIN, V., VANDESTEENE, R., & VAN BELLE, H. (1969).
Thirteen Week Feeding Study of Levamisole-HCl in Rats,. Unpublished;
submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R., VANDESTEENE, R. & PARDOEL, L. (1972). Eighteen Month
Feeding Study in Rats with Levamisole. Rpt. No. 154. Unpublished;
submitted to WHO by Janssen Pharmaceutica.
MARSBOOM, R., HERIN, V., VANDESTEENE, R. & PARDOEL, L. (1975). Oral
Toxicity Study in Beagle Dogs Repeated Dosage for 12 Months.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
MICHIELS, M., WOESTENBORGHS, R., & HEYKANTS, J. (1978). Absorption
and Plasma Levels of Oral Levamisole in the Rabbit. Unpublished;
submitted to WHO by Janssen Pharmaceutica.
NIEMEGEERS, C.M. (1975). The Acute Intravenous, Subcutaneous, and
Oral Toxicity of Levamisole in Mice and Rats. Unpublished; submitted
to WHO by Janssen Pharmaceutica.
PARKINSON, D.R., JERRY, L.M., SHIBATA, H.K., LEWIS, M.G., CARO, P.O.,
CAPEK, A., MANSELL, P.W., & MARQUIS, G. (1977). Complications of
Cancer Immunotherapy with Levamisole. Lancet, 1, 1129-1132.
REYNTJENS. (1989). Clinical Expert Report on Use of Levamisole for
Treatment of Dukes C Colorectal Cancer in Man. Unpublished; submitted
to WHO by Janssen Pharmaceutica.
RICHOLD, M. JONES, E., & PROUDLOCK, R. (1979). Ames Metabolic
Activation Test to Assess the Potential Mutagenic Effect of
Levamisole. Unpublished, Huntingdon Research Center. Submitted to
WHO by Janssen Pharmaceutica.
SYMOENS, J. VEYS, E., MIELANTS, M., & PINALS, R. (1978). Adverse
Reactions to Levamisole. Cancer Treatment Reports, 62, 11, 1721-30.
THOMPSON, J., HERBICK, J., KLASSEN, C. SEVERSON, C., VELIN,V.,
BLASCHKE, J., SILVERMAN, M., & VOGEL, C. (1980). Studies on
Levamisole - Induced Agranulocytosis. Blood, 56, 3, 388-396.
VANDENBERGHE, J., MARSBOOM, R., & HERIN, V. (1980). Oral
Carcinogenicity Study in Albino Swiss Mice. Unpublished; submitted to
WHO by Janssen Pharmaceutica.
VAN WAUWE, J, & JANSSEN, P. (1989). The Cardinal Question About
Levamisole: How Does It Work? Unpublished; submitted to WHO by
Janssen Pharmaceutica.
VERSTRAETEN, A., HERIN, V., & MARSBOOM, R. (1983). Subacute Toxicity
Study in Beagle Dogs Repeated Dosage for 3 Months Pour On.
Unpublished; submitted to WHO by Janssen Pharmaceutica.
VEYS, P., & DONY, J. (1985). Embryotoxicity and Teratogenicity Study
with Levamisole in Swine. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
VEYS, P. (1989). Literature Retrieval of Reports and Publications on
Levamisole-related Granulocytopenia. Unpublished; submitted to WHO by
Janssen Pharmaceutica.
WALLWORK, L.M. & JAMES, J.A. (1975). Rat Acute Oral and Dermal
Toxcicity Using a "Pour-on" Formulation. Unpublished, Wellcome
Research Laboratories. Unpublished; submitted to WHO by Janssen
Pharmaceutica.
WANG, G.T. (1970a). Safety study of Levamisole in Swine.
Unpublished, American Cyanamide. Submitted to WHO by Janssen
Pharamaceutica.
WANG, G.T. (1970b). Safety study of Levamisole in Swine.
Unpublished, American Cyanamide. Submitted to WHO by Janssen
Pharamaceutica.