
IPCS/CEC EVALUATION OF ANTIDOTES SERIES
VOLUME 1
NALOXONE, FLUMAZENIL AND DANTROLENE AS ANTIDOTES
IPCS/CEC Evaluation of Antidotes Series
IPCS International Programme on Chemical Safety
CEC Commission of the European Communities
Volume 1 Naloxone, flumazenil and dantrolene as antidotes
Volume 2 Antidotes for poisoning by cyanide
This important new series will provide definitive and authoritative
guidance on the use of antidotes to treat poisoning. The
International Programme on Chemical Safety (IPCS) and the Commission
of the European Communities (CEC) (ILO/UNEP/WHO) have jointly
undertaken a major programme to evaluate antidotes used clinically
in the treatment of poisoning. The aim of this programme has been
to identify and evaluate for the first time in a scientific and
rigorous way the efficacy and use of a wide range of antidotes.
This series will therefore summarise and assess, on an antidote by
antidote basis, their clinical use, mode of action and efficacy. The
aim has been to provide an authoritative consensus statement which
will greatly assist in the selection and administration of an
appropriate antidote. This scientific assessment is complemented by
detailed clinical information on routes of administration,
contraindications, precautions and so on. The series will therefore
collate a wealth of useful information which will be of immense
practical use to clinical toxicologists and all those involved in the
treatment and management of poisoining.
Scientific Editors
T.J. MEREDITH
Department of Health, London, United Kingdom
D. JACOBSEN
Ulleval University Hospital, Oslo, Norway
J.A. HAINES
International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
J-C. BERGER
Health and Safety Directorate,
Commission of the European Communities, Luxembourg
EUR 14797 EN
Published by Cambridge University Press on behalf of the World Health
Organization and of the Commission of the European Communities
CAMBRIDGE UNIVERSITY PRESS
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned.
Neither the Commission of the European Communities nor any person
acting on behalf of the Commission is responsible for the use which
might be made of the information contained in this report.
(c) World Health Organization, Geneva, 1993 and
ECSC-EEC-EAEC, Brussels-Luxembourg, 1993
First published 1993
Publication No. EUR 14797 EN of the Commission of the European
Communities, Dissemination of Scientific and Technical Knowledge
Unit, Directorate-General Information Technologies and Industries,
and Telecommunications, Luxembourg
ISBN 0 521 45459 X hardback
CONTENTS
PREFACE
ABBREVIATIONS
1. INTRODUCTION TO THE SERIES
2. NALOXONE
2.1. Introduction
2.2. Name and chemical formula
2.3. Physico-chemical properties
2.4. Pharmaceutical formulation and synthesis
2.5. Analytical methods
2.5.1. Quality control
2.5.2. Identification
2.5.3. Quantification of the antidote
2.5.4. Analysis of toxic agents
2.6. Shelf-life
2.7. General properties
2.8. Animal studies
2.8.1. Pharmacodynamics
2.8.2. Pharmacokinetics
2.8.3. Toxicology
2.9. Volunteer studies
2.9.1. Pharmacokinetics
2.9.2. Pharmacodynamics
2.9.3. Effects of high doses of naloxone
2.10. Clinical studies - clinical trials
2.10.1. Effects in therapeutic use of opioids
2.10.2. Effects in acute opioid poisoning
2.11. Clinical studies - case reports
2.11.1. Naloxone in clonidine poisoning
2.12. Summary of evaluation
2.12.1. Indications
2.12.2. Advised routes and dose
2.12.3. Other consequential or supportive therapy
2.12.4. Areas where there is insufficient information
to make recommendations
2.12.5. Proposals for further studies
2.12.6. Adverse effects
2.12.7. Restrictions of use
2.13. Model information sheet
2.13.1. Uses
2.13.2. Dosage and route
2.13.3. Precautions/contraindications
2.13.4. Adverse effects
2.13.5. Use in pregnancy and lactation
2.13.6. Storage
2.14. References
3. FLUMAZENIL
3.1. Introduction
3.2. Name and chemical formula of antidote
3.3. Physico-chemical properties
3.4. Pharmaceutical formulation and synthesis
3.5. Analytical methods
3.5.1. Identification of the antidote
3.5.1.1 Infrared spectroscopy
3.5.1.2 Ultraviolet absorption
3.5.1.3 Thin-layer chromatography
3.5.2. Quantification of the antidote in biological
samples
3.5.3. Analysis of the toxic agent in biological
samples
3.6. Shelf-life
3.7. General properties
3.8. Animal studies
3.8.1. Pharmacodynamics
3.8.2. Pharmacokinetics
3.8.2.1 Absorption
3.8.2.2 Distribution
3.8.2.3 Elimination
3.8.3. Toxicology
3.8.3.1 Acute toxicity
3.8.3.2 Subacute toxicity
3.8.3.3 Chronic toxicity
3.8.3.4 Embryotoxicity
3.8.3.5 Mutagenicity
3.9. Volunteer studies
3.9.1. Pharmacodynamics
3.9.1.1 BZD antagonist effect
3.9.1.2 Intrinsic effects
3.9.2. Pharmacokinetics
3.9.2.1 Absorption
3.9.2.2 Distribution
3.9.2.3 Elimination
3.9.3. Tolerance of flumazenil
3.9.4. Other studies
3.10. Clinical studies - clinical trials
3.10.1. Anaesthesiology
3.10.1.1 General anaesthesia
3.10.1.2 Conscious sedation
3.10.2. Benzodiazepine overdose or intoxication
3.11. Clinical studies - case reports
3.12. Summary of evaluation
3.12.1. Indications
3.12.2. Dosage and route
3.12.3. Other consequential or supportive therapy
3.12.4. Areas where there is insufficient information to
make recommendations
3.12.5. Proposals for further study
3.12.6. Adverse effects
3.12.7. Restrictions of use
3.13. Model information sheet
3.13.1. Uses
3.13.2. Dosage and route
3.13.3. Precautions/contraindications
3.13.3.1 Pharmaceutical precautions
3.13.3.2 Other precautions
3.13.4. Adverse effects
3.13.5. Use in pregnancy and lactation
3.13.6. Storage
3.13.7. Special risk groups
3.14. References
4. DANTROLENE SODIUM
4.1. Introduction
4.2. Name and chemical formula of antidote
4.3. Physico-chemical properties
4.4. Pharmaceutical formulation and synthesis
4.5. Analytical methods
4.5.1. Identification and quantification of dantrolene
sodium and its formulation
4.5.2. Quantification of dantrolene in body fluids
4.5.2.1 Spectrofluorimetry
4.5.2.2 High-performance liquid chromatography
4.6. Shelf life
4.7. General properties
4.8. Animal studies
4.8.1. Pharmacodynamics
4.8.1.1 Effect on skeletal muscle
4.8.1.2 Effects on other tissues
4.8.1.3 Studies in malignant hyperthermia-
susceptible pigs
4.8.2. Pharmacokinetics
4.8.3. Toxicology
4.8.3.1 Acute toxicity
4.8.3.2 Subacute toxicity
4.8.3.3 Chronic toxicity
4.8.3.4 Teratogenicity
4.9. Volunteer studies
4.9.1. Administration and plasma concentrations
4.9.2. Distribution
4.9.2.1 Distribution to the fetus and
newborn baby
4.9.3. Elimination
4.9.4. Human in vitro pharmacodynamics
4.10. Clinical studies - clinical trials
4.11. Clinical studies - case reports
4.11.1. Use in malignant hyperthermia
4.11.1.1 Prophylaxis of malignant hyperthermia
4.11.1.2 Prophylaxis of malignant hyperthermia
during pregnancy
4.11.2. Use in neuroleptic malignant syndrome
4.11.3. Use in other drug-induced hyperthermia
4.12. Summary of evaluation
4.12.1. Indications
4.12.1.1 Treatment of malignant hyperthermia
4.12.1.2 Treatment of neuroleptic malignant
syndrome
4.12.1.3 Treatment of hyperthermia induced by
muscle rigidity in poisoning
4.12.2. Advised routes and doses
4.12.2.1 Treatment of severe drug-induced
hyperthermia, including malignant
hyperthermia
4.12.2.2 Prophylaxis of malignant hyperthermia
prior to anaesthesia in susceptible
patients
4.12.3. Other consequential or supportive therapy
4.12.4. Controversial issues and areas of insufficient
information
4.12.5. Proposals for further studies
4.12.6. Adverse effects
4.12.6.1 Hepatoxicity
4.12.6.2 Interaction with calcium antagonists
4.12.7. Restrictions for use
4.13. Model information sheet
4.13.1. Uses as an antidote
4.13.2. Dosage and route
4.13.3. Precautions and contraindications
4.13.4. Pharmaceutical incompatibilities and drug
interactions
4.13.5. Adverse effects
4.13.6. Use in pregnancy and lactation
4.13.7. Storage
4.14. References
APPENDIX I List of antidotes
APPENDIX II Principles for the evaluation of antidotes
APPENDIX III Proforma for monographs on antidotes for
specific toxic agents
WORKING GROUP ON VOLUME 1, EVALUATION OF ANTIDOTES
Members
Dr D.N. Bateman, Department of Clinical Pharmacology, University of
Newcastle, Newcastle-upon Tyne, United Kingdom
Professor C. Bismuth, Hôpital Fernand Widal, Paris, France
Dr R.E. Ferner, West Midlands Poisons Unit, Dudley Road Hospital,
Birmingham, United Kingdom (Joint Rapporteur)
Dr T.J. Meredith, Department of Health, London, United Kingdom
Dr H. Persson, Poison Information Centre, Karolinska Sjukhuset,
Stockholm, Sweden (Joint Chairman)
Professor L. Prescott, Scottish Poison Information Service, The Royal
Infirmary, Edinburgh, Scotland (Joint Chairman)
Dr M.-L. Ruggerone, Ospedale Niguarda, Centro Antiveleni, Milan, Italy
Dr H. Smet, Centre Belge Anti-Poisons, Brussels, Belgium
Dr U. Taitelman, National Poisons Information Centre, Rambam Medical
Centre, Haifa, Israel
Dr W. Temple, National Toxicology Group, Otago University Medical
School, Dunedin, New Zealand (Joint Rapporteur)
Professor A.N.P. van Heijst, Bosch en Duin, The Netherlands
Dr G. Volans, Poisons Unit, New Cross Hospital, London, United Kingdom
Dr E. Wickstrom, National Poison Centre, Oslo, Norway
Observer
Dr G. Olibet, Centro Antiveleni, Milan, Italy
Secretariat
Dr J.-C. Berger, Health and Safety Directorate, Commission of the
European Communities, Luxembourg
Dr J.A. Haines, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr M. ten Ham, Pharmaceuticals Programme, World Health Organization,
Geneva, Switzerland
PREFACE
At a joint meeting of the World Federation of Associations of
Clinical Toxicology and Poison Control Centres, the International
Programme on Chemical Safety (IPCS), and the Commission of the
European Communities (CEC), held at the headquarters of the World
Health Organization in October 1985, the evaluation of antidotes used
in the treatment of poisonings was identified as a priority area for
international collaboration. During 1986, the IPCS and CEC undertook
the preparatory phase of a joint project on this subject. For the
purpose of the project an antidote was defined as a therapeutic
substance used to counteract the toxic action(s) of a specified
xenobiotic. Antidotes, as well as other agents used to prevent the
absorption of poisons, to enhance their elimination and to treat their
effects on body functions, were listed and preliminarily classified
according to the urgency of treatment and efficacy in practice. With
respect to efficacy in practice, they were classified as: (1) those
generally accepted as useful; (2) those widely used and considered
promising but not yet universally accepted as useful and requiring
further research concerning their efficacy and/or their indications
for use; and (3) those of questionable usefulness. Additionally,
certain antidotes or agents used for specific purposes were considered
to correspond to the WHO criteria for essential drugs (see Criteria
for the Selection of Essential Drugs, WHO Technical Report Series 722,
Geneva, 1985).
A methodology for the principles of evaluating antidotes and
agents used in the treatment of poisonings and a proforma for
preparing monographs on antidotes for specific toxins were drafted
(Appendices II and III respectively).
Monographs are being prepared, using the proforma, for those
antidotes and agents provisionally classified in category 1 as regards
efficacy in practice. For those classified in categories 2 and 3,
where there are insufficient data or controversy regarding efficacy in
practice, it was agreed that further study was necessary.
Accordingly, several were selected for initial review and evaluation,
among which were naloxone as an antagonist for opioids, flumazenil as
a benzodiazepine antagonist and dantrolene for malignant hyperthermia.
The review and evaluation of these antidotes was initiated at a
joint meeting of the IPCS and the CEC, organized by the Northern
Poisons Unit and held at the Medical School of the University of
Newcastle-upon-Tyne, United Kingdom, 13-17 March 1989. In preparation
for this meeting, monographs were drafted, using the proforma, on
naloxone by Dr D.N. Bateman, on flumazenil by Dr A. Brovard and
Professor C. Bismuth and on dantrolene by Dr H. Smet and Professor C.
Bismuth. The draft document on naloxone was reviewed by a working
group consisting of Professor L.F. Prescott (Chairman), Dr W. Temple
(Rapporteur), Dr D. Bateman, Dr M. Ten Ham, Dr A.N.P. van Heijst, Dr
G.N. Volans and Dr E. Wickstrom. The draft documents on flumazenil
and dantrolene were reviewed by a working group consisting of Dr H.
Persson (Chairman), Dr R.E. Ferner (Rapporteur), Dr J.-C. Berger,
Professor C. Bismuth, Dr G. Olibet, Dr M.-L. Ruggerone, Dr H. Smet and
Dr U. Taitelman.
Following the meeting further drafting work was undertaken by the
authors, with the assistance of Drs R.E. Ferner, B. Britt (Department
of Anaesthesia, Faculty of Medicine, University of Toronto, Canada),
and T. Fagerlund (Institute of Medical Genetics, University of Oslo,
Norway) in the redrafting of the dantrolene monograph. Draft texts
were further revised by the series editors (Dr T.J. Meredith, Dr D.
Jacobsen, Dr J.A. Haines, and Dr J.-C. Berger), who also prepared an
introduction to the series. This introduction summarizes the results
of the preparatory phase and indicates the volumes currently planned
for this series. The efforts of all who helped in the preparation and
finalization of this volume are gratefully acknowledged.
ABBREVIATIONS
BZD benzodiazepine
CAT computer-assisted tomography
CNS central nervous system
GABA gamma-aminobutyric acid
GLC gas-liquid chromatography
HIV human immunodeficiency virus
HPLC high-performance liquid chromatography
LSD lysergic acid diethylamide
RIA radio-immunoassay
TLC thin-layer chromatography
UV ultraviolet
1. INTRODUCTION TO THE SERIES
Antidotes play a vital role in the treatment of poisoned
patients. Good supportive care, directed particularly at the cardiac
and respiratory systems, and the use of elimination techniques when
indicated, enable the majority of poisoned patients to make a full
recovery. However, in certain circumstances the use of antidotes can
be life-saving, and in other circumstances the use of antidotes may
reduce morbidity as well as medical and other resources required in
the care of a patient. In areas remote from hospital care, and
particularly in developing countries where facilities for supportive
care outside hospital are often limited, the availability of certain
antidotes is even more essential for the successful treatment of a
poisoned patient.
However, there remains controversy about the clinical efficacy
and indications for use of many of the antidotes conventionally
employed in the treatment of poisoning. There is also sometimes
difficulty in obtaining antidotes in an emergency situation,
particularly if the substance in question is not available as a
pharmaceutical preparation.
The need for an international evaluation of the clinical efficacy
of antidotes and other substances used in the treatment of poisoning
was first recognized at a joint meeting of the World Federation of
Associations of Clinical Toxicology Centres and Poisons Control
Centres, the International Programme on Chemical Safety (IPCS) and the
Commission of the European Communities (CEC), held at WHO
headquarters, Geneva, 6-9 October 1985. At the same time, the need to
encourage the more widespread availability of those antidotes that are
effective was also recognized. As a result, a joint IPCS/CEC project
was subsequently initiated to address these problems.
In a preparatory phase of the project, an antidote was defined
for working purposes as a therapeutic substance used to counteract the
toxic action(s) of a specified xenobiotic. A preliminary list of
antidotes for review, as well as of other agents used to prevent the
absorption of poisons, to enhance their elimination and to treat their
effects on body functions, was established. For the purposes of the
review process, antidotes and other substances were classified
according to the urgency with which treatment with the antidote was
thought on current evidence to be required and the (currently judged)
clinical efficacy of the antidote in practice. Those corresponding to
the WHO concept of an essential drug were designated as such. Some
have already been incorporated into the WHO list of essential
drugsa. Antidotes and similar substances for veterinary use were
a WHO (1988) Use of Essential Drugs. Model list of essential drugs
(fifth list). Third Report of the WHO Expert Committee. WHO
Technical Report Series 770, Geneva World Health Organization.
also listed. A methodology on the principles for evaluation of
antidotes and other agents used in the treatment of poisonings was
developed and this has subsequently been used as a framework for
drafting monographs on specific antidotes. The list of antidotes and
other agents established as a result of the preparatory phase and the
preliminary classification is given in Appendix I. The principles for
evaluation are detailed in Appendix II.
Early during the course of the preparatory phase, it became
apparent that the availability of antidotes differed from one country
to another. Problems of availability fell into three interrelated
categories, namely:
* scientific, technical and economic aspects;
* regulatory and administrative requirements;
* geospatial and time considerations.
Problems of availability of antidotes used in the treatment of
poisonings were therefore examined by an IPCS/CEC Working Group,
hosted by the Norwegian National Poisons Information Centre and held
in Oslo, 20-22 June 1988. The record of this meeting is given in
ICS/88.44. In preparation for this meeting, a preliminary survey was
undertaken of selected poisons control centres in order to identify
more precisely the practical difficulties encountered in obtaining
antidotes. The survey showed that, in general, poisons centres in
industrialized countries had few problems in obtaining most antidotes,
although lack of suitable preparations/importers/manufacturers
together with administrative difficulties did hinder access to certain
antidotes. In contrast, centres in developing countries reported many
problems in obtaining even those antidotes that are readily available
elsewhere.
A report was prepared by the IPCS/CEC Working Group setting out
the problems associated with the availability of antidotes and
suggesting ways in which the availability of antidotes might be
ensured for the treatment of poisoned individuals. In due course, it
is intended that this report will be brought to the attention of all
relevant national drug regulatory and importation authorities,
pharmaceutical manufacturers, distributors of pharmaceutical
materials, and all poisons control centres. The IPCS Guidelines for
Poisons Control summarize the problems and issues of availability
identified by the Working Groupb.
b WHO (in press) - Guidelines for Poisons Control, Part II, section
6 Geneva, World Health Organization
Aspects of the evaluation of antidotes
The development and evaluation of substances to counteract the
toxic action(s) of a xenobiotic is principally a task for the
scientific community, particularly those working in experimental
pharmacology, toxicology and clinical medicine. The efficacy of a
substance intended for use as an antidote must first be demonstrated
in an appropriate animal model. The next step, demonstration of
efficacy in humans, it is often more difficult because there is rarely
an opportunity for controlled clinical trials. Even if a substance
is shown to be effective as an antidote, the potential intrinsic
toxicity of the substance also needs to be considered prior to its
more widespread use, and, as with all drugs, the possibility of an
adverse drug reaction should be considered. A clinician is more
likely to be prepared to use a relatively "non-toxic" antidote (even
one whose efficacy has still to be established with certainty) than
one with intrinsic toxicity. An antidote which is potentially toxic
should only be used if it is therapeutically effective and the
indication for use is clear. Although possible long-term adverse
effects and chronic toxicity need to be considered, they are usually
of less consequence than for an ordinary pharmaceutical agent because
treatment with an antidote is rarely required more than once in any
particular individual. A final consideration in the use of an
antidote is that increased toxicity should not result from
mobilization of the toxin from tissue stores or from changes in tissue
distribution.
The concept of relative "efficacy" of antidotes
It is important that clinicians employing antidotes in the
treatment of poisoned patients recognize that the clinical "efficacy"
of antidotes varies considerably. On the one hand there are antidotes
whose clinical effect is both rapid and dramatic. Examples would be
naloxone or flumazenil, which act as very specific competitive
antagonists at opioid and benzodiazepines receptors, respectively.
On the other hand, there are antidotes that are able to counter
only some of the toxic effects of a particular compound; if the dose
of the compound in question is sufficiently high then the patient is
likely to die despite the use of an antidote. Chelating agents
provide good examples of antidotes that fall into this category of
efficacy. Nevertheless, chelating agents have a valuable role to play
in the treatment of heavy metal poisoning, and many are recommended
for this purpose in volume V of this series.
Some agents are loosely termed antidotes even though they may
have little or no true antidotal effect; they may nonetheless form
valuable adjuncts to treatment. Diazepam, used in the treatment of
organophosphate poisoning (volume IV), is one such example.
Provisional list of volumes in the IPCS/CEC antidotes series
It is intended that the IPCS/CEC series of monographs on
antidotes will cover all antidotes that are commonly employed - or
which have been proposed for use - in the treatment of human
poisoning. Once this aim has been achieved, it is intended that the
volumes will be periodically updated in order to meet the needs of
health care professionals. At present, the proposed volumes for this
series include:
Volume 2
Evaluation of antidotes for cyanide poisoning:
* oxygen
* sodium thiosulfate
* hydroxocobalamin
* dicobalt edetate
* amyl nitrite
* sodium nitrite
* 4-dimethylaminophenol
* antidotes to methaemoglobin-forming agents (methylene blue,
toluidine blue)
* analytical methods for cyanide alone and in combination with
cyanide antidotes
Volume 3
Evaluation of antidotes for paracetamol poisoning
* overview
* N-acetylcysteine
* methionine
Volume 4
Evaluation of antidotes for organophosphate poisoning
* overview
* atropine
* diazepam
* obidoxime
* pralidoxime
Volume 5
Evaluation of chelating agents for heavy metal poisoning
* overview
* deferoxamine
* prussian blue
* trientine
* calcium disodium edetate
* DTPA
* DMPS
* DMSA
* dimercaprol
* penicillamine and N-acetyl penicillamine
Volume 6
Antidotes for methanol and ethylene glycol poisoning.
Volume 7
Antidotes for amatoxin, gyrometrine and isoniazid poisoning
Volume 8
Evaluation of the various pharmaceutical substances used for enhanced
elimination and prevention of absorption.
Further volumes are planned for:
* General antidotes and sorbents
* Antidotes based on immunotoxicology
International evaluation process
Experts are requested by the IPCS to prepare draft monographs on
specific antidotes or agents, or on specific aspects associated with
their therapeutic use. Original literature references must be used
according to the criteria established for Environmental Health
Criteria documents. In order to ensure that monographs are written
according to agreed standards, a common format has been established
following the methodology on principles for evaluation of antidotes
(Appendix II) and the guidelines to authors (Appendix III). The
series editors examine the drafts to ensure that they conform to the
standard format and are of acceptable quality for peer review. For
certain volumes a guest editor is also appointed. The IPCS sends the
drafts to selected experts for comment and for possible additional
information. A working group of authors and experts in the field is
then convened by the IPCS and CEC. The task of this group is to:
(i) examine the literature referred to in the monographs for its
relevance, including case data experience;
(ii) identify any gaps in knowledge or scientific unknowns;
(iii) make an evaluation of the clinical efficacy of the antidote
for a particular poisoning or pathological condition resulting
from the poisoning;
(iv) provide guidance on the treatment regimens, under various
conditions of use of the antidote, including, where
appropriate, field and primary health care use, advise on the
accompanying supportive care, and give particular attention to
paediatric doses, contraindications and special
considerations.
Following the working group meeting further drafting may need to
be undertaken by the original author in consultation with the series
and guest editors. An overview chapter summarizing the issues and
giving the evaluation of a series of antidotes for specific types of
poisoning cases is drafted by the editors or invited experts. The
IPCS and CEC may convene a further editorial meeting to finalize the
monographs for a particular volume and to approve the overview
chapter. The volume is then processed by the WHO editor for
publication by Cambridge University Press.
2. NALOXONE
2.1 Introduction
Naloxone is an opioid antagonist acting at all three types of
opioid receptors. It appears devoid of agonist activity (Martin,
1976). Naloxone is indicated in the treatment of opiate poisoning.
Although naloxone has also been reported to be of benefit as an
antidote in benzodiazepine (BZD) poisoning (Bell, 1975), other workers
failed to demonstrate an effect in a double-blind study of
diazepam-induced sedation (Christensen & Huttel, 1979). However,
Jordan (1980) demonstrated some reversal of diazepam-induced
respiratory depression by naloxone. Thus there is a need for further
controlled studies, particularly in cases of poisoning.
Naloxone has also been claimed to have an effect on
ethanol-induced central nervous system (CNS) depression, and in one
study appeared to cause an improvement in 20% of treated cases
(Jefferys et al., 1980). However, this finding has not been confirmed
by other workers (Handal et al., 1983; Nuotto et al., 1984).
The possible beneficial effects of naloxone in non-opiate
poisoning probably reflect the involvement of endogenous opioids in
the depressant action of some non-opioid drugs (McNicholas & Martin,
1984).
2.2 Name and Chemical Formula
Naloxone
6-Allylnoroxymorphone
17-Allyl-6-deoxy-7,8-dihydro-14-hydroxy-6-oxo-17-normorphone
Empirical formula: C19 H21 NO4
Relative molecular mass: 327
CAS number: 465-65-6
Trade names: Narcan, Nalone, Narcanti (Du Pont Pharmaceuticals)
Naloxone is available for clinical use as the hydrochloride salt,
which may be anhydrous (CAS-357-08-4) or contain 2 molecules of water
of hydration (CAS 51481-60-8). The relative molecular mass of the
free base is 327.37 and of the anhydrous salt 363.84.
Conversion table: 1 g = 3.1 mmol
1 mmol = 327.4 mg
1 mg/ml = 3.1 mmol/l
1 mmol/l = 0.33 mg/ml
The molecular structure of naloxone hydrochloride is shown below.
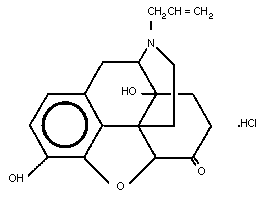 2.3 Physico-chemical Properties
Naloxone hydrochloride has a melting range of 200-205 °C. It is
soluble in water, dilute acids and strong alkalis, and is slightly
soluble in alcohol but practically insoluble in ether. Aqueous
solutions are acidic (pH 3 to 4.5) (United States Pharmacopeia, 1980)
and an 8.08% solution in water is isotonic with serum (Hassan et al.,
1985). A 25% solution of naloxone hydrochloride rotates light between
-170 and -181. Naloxone crystals from ethyl acetate have a specific
optical rotation at 20 °C ([alpha]D20; 9.3 g/l chloroform) of
-194.5 ° (Windholz, 1983).
Naloxone has a pKa (20 °C) values for the nitrogen and phenolic
H groupings of 7.94 and 9.44, respectively (Kaufman et al., 1975).
On drying at 105 °C, the anhydrous form loses not more than 0.5%
and the hydrated form not more than 11% of its weight.
The solution for injection is made up in water and should be
protected from light. Naloxone can be diluted in 0.9% saline or 5%
dextrose and should then be used within 24 h. It should not be mixed
in solutions containing metasulfite, metabisulfite, or long-chain or
high relative molecular mass anions, or in those with an alkaline pH.
2.4 Pharmaceutical Formulation and Synthesis
Three synthetic routes for the production of naloxone have been
reported (Hassan et al., 1985). Oxymorphone is a starting point for
two of the synthetic processes and 14-hydroxycodeinone for the third.
Noroxymorphone hydrochloride is a potential impurity from the
manufacturing process.
2.5 Analytical Methods
2.5.1 Quality control
Naloxone hydrochloride can be assayed by gas chromatography with
flame ionization detection (United States Pharmacopeia, 1980).
2.5.2 Identification
About 150 mg of the unknown substance is dissolved in 25 ml of
water and a few drops of 6N ammonium hydroxide are added. Three 5-ml
portions of chloroform are used for extraction and the extract is
filtered. The filtrate is collected, evaporated to dryness using a
steam bath, and dried at 105 °C for one hour. The infrared absorption
spectrum of a 1-in-50 solution of the residue obtained in chloroform
will have maxima at the same wavelengths as those of a similar
solution of naloxone reference standard.
The addition of one drop of ferric chloride solution to 1 ml of
a 1-in-100 solution of naloxone hydrochoride results in a clear
purplish-blue colour.
2.5.3 Quantification of the antidote
Assay methods for naloxone in biological fluids employing
gas-liquid chromatography (GLC) (Meffin & Smith, 1980),
radio-immunoassay (RIA) (Berkowitz et al., 1975; Hahn et al., 1983)
and high-performance liquid chromatography (HPLC) (Asali, 1983; Terry
et al., 1984) have all been reported. The GLC method involves
derivatization and the specific antibody for the RIA is not widely
available. The HPLC methods reported appear sensitive and
reproducible, and are therefore probably the methods of choice.
2.5.4 Analysis of toxic agents
In the majority of cases in which naloxone is used as an
antidote, there is no way of measuring the level of the opioid poison.
Present assay techniques for many opiates are difficult, and RIA
suffers from lack of specificity in many cases. Some opiates, e.g.,
morphine, also appear to have active metabolites (Bodd et al., 1990).
The most widely used method for opioid detection is RIA of urine.
2.6 Shelf-life
The shelf-life of naloxone for intravenous injection in temperate
countries is 3 years and has a similar length in tropical countries.
2.7 General Properties
Naloxone is a specific opioid antagonist (Martin, 1976) and it is
for this reason that it is used in the treatment of poisoning. There
are reports that it may reverse the central effects of ethanol and BZD
poisoning in man. However, these are experimental uses that remain
unproven, and any observed effects probably reflect the involvement of
endogenous opioids in the nonspecific depressant action of those
agents (McNicholas & Martin, 1984).
2.8 Animal Studies
2.8.1 Pharmacodynamics
Naloxone is a competitive antagonist at opiate receptors, and
appears to be effective at all three types of receptor (mu, kappa and
sigma) (Martin, 1976). It does not produce habituation in animal or
human models of opiate tolerance and appears to be free of agonist
activity in most laboratory test models (Jasinski, 1967; McNicholas &
Martin, 1984). It produces a parallel shift in the in vitro dose-
response effects of pure agonist opioids, such as morphine, and
partial agonists, such as pentazocine (Smits & Takemori, 1970),
buprenorphine and dextropropoxyphene.
Since the range and relative quantities of opioid receptors vary
in different animal tissues, a range of concentrations of naloxone is
required to antagonize opioid effects in different test systems.
Confusion has arisen as to whether naloxone is a pure antagonist.
This is because some opioid receptors act as modulators and enhance
nociceptive stimuli. Thus, in some animal models naloxone appears to
possess agonist effects, but this is in fact incorrect (Sawynok et
al., 1979). Naloxone has also been observed in some experiments to
antagonize the antinociceptive effects of some non-opiate drugs.
Again it seems likely that this reflects an involvement of opioid
receptors in the mechanism of action of these drugs (Sawynok et al.,
1979). However, in a recent study in rats, Kotlinska & Langwinski
(1990) failed to find any evidence for the participation of the opioid
system in the mediation of acute ethanol effects in rats.
Naloxone has been reported to either decrease or have no
influence on barbiturate-induced anaesthesia. This paradox may be a
result of the dose-response relationship of the effects of naloxone,
which at high doses may have a potentiating effect (Sawynok et al.,
1979). Naloxone has some activity as a GABA antagonist and may thus
have convulsant activity. However, this is likely to be at much
higher concentrations that those encountered clinically (Dingledine et
al., 1978), since in mice a dose of 100 mg/kg was required to produce
convulsions.
Naloxone has also been shown to have a number of biochemical
effects in the rat, including inhibition of lipolysis and a subsequent
increase in circulating free tryptophan (Badawy et al., 1983).
2.8.2 Pharmacokinetics
Naloxone appears to be readily absorbed after oral administration
but undergoes extensive first-pass hepatic metabolism, which results
in a very low bioavailability (Misra, 1978). Studies of the
pharmacokinetics of intravenous naloxone have been performed in a
variety of animal species including the rat, rabbit and dog. Many of
these studies are based on radio-immunoassay of naloxone.
The serum concentration of naloxone found 5 min after injection
was similar (5 mg/kg) in the rat and the dog (Ngai et al., 1976; Pace
et al., 1979). The half-life of the parent drug in the rat (30 min)
was approximately half that in the dog (71 min).
Ngai et al. (1976) also examined the brain:serum ratio of
naloxone and found this to vary in the rat between 2.7:1 and 4.6:1.
Intravenously administered naloxone acts rapidly on the brain. The
brain:serum ratio was higher, however, when the naloxone was
administered subcutaneously. These workers also studied, in a
parallel group of animals, the distribution of morphine and noted that
the brain:serum ration was 1:10.
The initial distribution of naloxone may account for the rapid
onset of its reversal of opiate effects when it is given
intravenously. The major metabolite of naloxone is the glucuronide.
Naloxone-3-glucuronide has been found, for example, in the rabbit
(Fujimoto, 1969). A conjugated 6-hydroxy product of naloxone,
N-allyl-14-hydroxy-7,8-dihydronormorphine-3-glucuronide was
identified in the chicken by Fujimoto (1969); this conjugate was also
identified in the rabbit by Weinstein et al. (1974) but only in small
amounts.
The relatively short action of naloxone appears to result from
the ease with which it enters the brain after intravenous dosing and
the subsequent rapid redistribution, elimination and consequent fall
in brain naloxone levels (Berkowitz, 1976).
Hydroxylated metabolites of naloxone appear to possess narcotic
antagonist activities, but their potencies are much weaker than the
parent compound. Thus they are unlikely to be of significance in view
of the small amounts produced (Fujimoto et al., 1975).
The distribution of naloxone has not been found to be altered by
a 25-fold range of morphine concentration in the rat (Fishman et al.,
1975).
2.8.3 Toxicology
Acute toxicity studies with naloxone have been performed in mice,
rats and dogs. The LD50 for intravenous administration was 150
mg/kg in mice, 109 mg/kg in rats and 80 mg/kg in dogs (Social Welfare
Board, 1976). For 24-h-old rats the LD50 was 260 mg/kg when given
subcutaneously (Blumberg et al., 1966). The maximum nontoxic
subcutaneous dose in rats was found to be of the order of 50 mg/kg
(Blumberg et al., 1966). This dose was tolerated for 24 days, whereas
200 mg/kg resulted in tremor, convulsions and salivation.
Daily doses of 0.2 mg/kg given intravenously to dogs for 16 days
and 5 mg/kg given subcutaneously to monkeys for 30 days caused no
toxicity. However, a subcutaneous dose of 20 mg/kg resulted in
lethargy and tremor in monkeys.
No teratogenic effects were observed in mice, rats or rabbits
when naloxone was given parenterally over the period of organogenesis
(Social Welfare Board, 1976). No studies on mutagenicity have been
published.
2.9 Volunteer Studies
Studies of the pharmacokinetics and pharmacodynamics of naloxone
have been performed in volunteers.
2.9.1 Pharmacokinetics
Using an RIA assay, the pharmacokinetics of naloxone were found
to fit a two-compartment model, with a rapid distribution phase and a
slower elimination phase, having a half-life of 64 min (Ngai et al.,
1976). More recent studies using HPLC to assay naloxone suggest that
the apparent volume of distribution, half-life and clearance all show
differences within groups of normal volunteers. Thus Aitkenhead et
al. (1984) reported a mean apparent volume of distribution at steady
state of 3.65 l/kg (range 1.43-7.05 l/kg) and a mean half-life of
151.2 min (range 47.1-313.2 min). Using an HPLC assay, Goldfrank et
al. (1986) found less variability in patients (half-life 28-55 min).
The kinetics of naloxone in infants appear similar to those in
adults (Stile et al., 1984).
Orally administered radiolabelled naloxone undergoes extensive
first-pass metabolism in normal subjects (Fishman et al., 1973).
After intravenous administration, most (70%) of the radioactivity was
recovered in urine, the major part of which was conjugated as the
glucuronide. In addition other metabolites were found in small
quantities, i.e. the glucuronide conjugates of 7,8-dihydro-14-hydroxy-
normorphine, and N-allyl-7,8-dihydro-14-hydroxy-normorphine
(Weinstein et al., 1971).
As a consequence of the high hepatic clearance of naloxone and
relatively weak agonist activity of its metabolites, it is unlikely
that dose adjustments would be necessary in cases of renal failure.
Naloxone is only 54% protein-bound in adult plasma (61.5% in fetal
plasma), and this binding is not concentration-dependent over the
range 9 ng/ml to 2.5 µg/ml (Asali & Brown, 1984). Thus protein-
binding interactions seem unlikely.
The elimination of naloxone might be altered in patients with
liver disease, but no studies appear to have been performed.
2.9.2 Pharmacodynamics
Studies have been conducted on the duration of action and potency
of naloxone in reversing respiratory depression induced by morphine
(intravenous doses of 5 mg plus 10 mg) in volunteers (Kaufman et al.,
1981). The effect of naloxone against this therapeutic dose of
morphine reached a peak at around 30 min, which was equatable with the
probable peak in brain concentration. It should be noted that the
times of onset and peak effect of naloxone differed. The duration of
action of naloxone appeared to be about 1.5 h in this experimental
model.
Johnstone (1974) examined the effects of an infusion of naloxone
in volunteers who had received 2 mg/kg morphine intravenously and been
anaesthetized for 5 h. Intravenous naloxone given to these volunteers
at a rate of 40 µg/kg over a 10-h period reversed the central
depressant effects of morphine on respiratory function (measured by
CO2 responsiveness) and higher functions (assessed by a vigilance
test). No tachyphalaxis to the effects of naloxone was observed over
this period (Johnstone et al., 1974).
It has been suggested that ethanol may exert some of its effects
via the endogenous opiate system, as illustrated by the study by
Jeffferys et al. (1980) and Jeffcoate et al. (1979) where naloxone was
found to antagonize some of the ethanol effects. However, these
findings could not be confirmed by Handal et al. (1983) or Nuotto et
al. (1984). In the latter study, the effect of naloxone on ethanol-
induced impairment of psychomotor performance was first studied in two
placebo-controlled, double-blind, cross-over trials in 17 healthy male
volunteers. The main conclusion was that naloxone (intravenous doses
of 0.4 plus 2 mg) had no significant antagonizing effects on the
impairment induced by ethanol (1.5 g/kg). However, a slight but
significant effect on ethanol-induced nystagmus was noted. A placebo-
controlled, double-blind study was subsequently conducted on male
alcoholics admitted for acute ethanol intoxication (the mean blood
ethanol level was 2.9 g/l (64 mmol/l)). In this case, neither naloxone
(intravenous doses of 0.4 plus 2 mg; n=11) nor saline (n=7) had any
effect, as judged from a clinical inebriation test (Nuotto et al.,
1984).
2.9.3 Effects of high doses of naloxone
Naloxone has been administered to healthy volunteers at dose
levels of 0.3-4 mg/kg. These high dose levels produced dose-dependent
dysphasia and memory impairment. In addition, increases in blood
pressure and respiratory rate were noted, together with increases in
cortisol and growth hormone levels (Cohen et al., 1983). These
findings have been used to support the hypothesis that endogenous
opioids play a normal regulatory physiological role, but obviously
have potential therapeutic implications if large doses of naloxone are
used to treat poisoned patients.
2.10 Clinical Studies - Clinical Trials
Naloxone has been investigated in clinical studies on both
patients who have received a therapeutic dose of an opiate (see
section 2.9) and those who have been poisoned with opiates. Since
naloxone is a competitive antagonist, the dose required to reverse the
clinical effects of a specific opiate will depend on the dose of the
opiate, its duration of action, and its pharmacological properties,
particularly whether it has partial agonist activity or shows
selectivity at one type of opioid receptor subgroup (Martin, 1976).
2.10.1 Effects in therapeutic use of opioids
An alternative method of studying the response to naloxone was
reported by Drummond et al. (1977). They studied patients who had been
anaesthetized and had received the synthetic opiate fentanyl.
Naloxone produced a dose-dependent increase in respiratory function
(measured as minute volume or respiratory rate) with intravenous doses
of 0.1, 0.2 and 0.4 mg.
Hatano et al. (1975) reported an open study on 80 patients
undergoing a variety of surgical procedures including cardiopulmonary
bypass. Premedication included pethidine (meperidine) and induction
was achieved with pentazocine and diazepam. The doses of pentazocine
in males were 2 mg/kg and females 1.5 mg/kg, and those of diazepam
were 0.4 and 0.3 mg/kg, respectively. The authors used a stepwise
increment of naloxone (0.2-mg intravenous boluses) to achieve reversal
of the opiate effect of pentazocine at the end of the operative
procedure and noted a stepwise reversal of the opiate effects in their
patients as the opiate dose was increased (the average total dose
given was 2.5 mg/kg body weight).
The duration of action of naloxone in reversing the effects of
morphine (5 or 10 mg, intramuscular) in patients recovering from
surgery is relatively short (Longnecker et al., 1973). The authors
suggested that the use of a combination of intravenous and
intramuscular naloxone might be an appropriate regimen in the post-
operative situation; this has also been suggested for the treatment
of acute overdoses in heroin addicts (see sections 2.12.2 & 2.13.2).
2.10.2 Effects in acute opioid poisoning
Two important studies have demonstrated the efficacy of naloxone
in reversing opiate poisoning. Evans et al. (1973) reported a study
in which naloxone (0.4-1.2 mg, intravenous) resulted in recovery of
consciousness within 1-2 min in nine patients with a history of opiate
ingestion. This was associated with improvement in respiratory
function in the six patients in whom this could be measured with
minute volume and respiratory rate. The opiates taken by these
patients were reported as dipipanone (3), pethidine (2),
dihydrocodeine (2), pentazocine (1) and heroin (1). In contrast, none
of 13 patients overdosed with a variety of other central nervous
system depressants showed improvement after having been given a total
intravenous dose of 1.2 mg naloxone. This rapid and clear benefit of
therapy was also reported by Buchner et al. (1972), who studied the
effects of naloxone (0.005 to 0.01 mg/kg) in 10 children with
methadone poisoning. Although they did not study a control group,
they did confirm the presence of methadone in biological fluids in
some of their patients. These authors stress the importance of an
adequate period of observation for patients poisoned with long-acting
opiates and the necessity of repeated doses of naloxone.
Since the onset of the effects of naloxone is so rapid, it has
proved relatively easy to confirm its effectiveness in opiate
poisoning at restoring consciousness and improving respiration.
Further extensive clinical trials in opiate poisoning have, therefore,
not been performed.
Henry & Volans (1984) have stressed the importance of classifying
drugs correctly as opioids. A list of opioids is a useful reminder
(Table 1) that agents such as loperamide and diphenoxylate may produce
significant systemic toxicity in overdose.
One particular aspect of naloxone use that requires consideration
is that of the most appropriate dosage regimen. Early human studies
confirmed that the duration of action of naloxone was shorter than
might have been expected from its plasma half-life (Berkowitz et al.,
1975). The long duration of action of some opiates is also a factor
in the need to repeat the initial dose of naloxone in poisoned
patients (Gober et al., 1979). As an alternative to repetitive
dosing, several research workers have suggested that intravenous
loading doses followed by a steady-state infusion of the drug would be
appropriate both in children (Gourlay & Coulthard, 1983; Tenenbein,
1984) and in adults (Bradberry & Raebel, 1981; Goldfrank et al., 1986)
suffering opiate poisoning. These regimens have appeared safe and
effective in clinical use, but do not obviate the need for close
monitoring during treatment of respiratory function, conscious level
and cardiovascular function. It is important to remember that some
synthetic opioids, e.g., dextropropoxyphene, have been reported to
produce toxic effects at high doses, which are not reversible by
naloxone (Barraclough & Lowe, 1982). These effects may be due to a
direct action of dextropropoxyphene on cardiac cell membranes.
Table 1. Alphabetical list of opioid drugsa
Alletorphine Levorphanol
Alphaprodine Loperamide
Anileridine Meptazinol
Azidomorphine Methadone
Bezitramide Metofoline
Buprenorphine Morphine
Butorphanol Nalbuphine
Codeine Norpipanone
Dextromoramide Opium
Dextropropoxyphene Oxycodone
Diamorphine (Heroin) Oxymorphone
Difenoxin Papaveretum
Dihydrocodeine Pentazocine
Diphenoxylate Pethidine (Meperidine)
Dipipanone Phenadoxone
Ethoheptazine Phenazocine
Ethylmorphine Phenoperidine
Etorphine Piminodine
Fentanyl Piritramide
Hydrocodone Thebacon
Hydromorphone Tilidate
Ketobemidone Tramadol
Levomethadyl Trimeperidine
a From Martindale (1982). Some of these drugs may be marketed as
part of a combination preparation.
2.11 Clinical Studies - Case Reports
Individual published case reports have confirmed efficacy for the
majority of opiates (Handal et al., 1983). In patients who are
narcotic addicts, naloxone may precipitate features of acute opiate
withdrawal. Doses of up to 20 mg naloxone have been used in children
without associated adverse effects (Handal et al., 1983).
If patients with acute renal failure are given morphine over
several days for various reasons (e.g., for sedation while on a
respirator), opioid toxicity may occur due to accumulation of the
active metabolite morphine-6-glucuronide, which is renally excreted
(Bodd et al., 1990). In such cases, the opioid toxicity may last for
up to two weeks after the cessation of morphine therapy, and the
patient will need naloxone infusion in order to avoid respiratory
depression.
2.11.1 Naloxone in clonidine poisoning
Clonidine hydrochloride is a central and peripheral
alpha-adrenergic antagonist that is still used in the treatment of
hypertension. It has also been suggested for the treatment of opiate
withdrawal (Gold et al., 1980). The mechanism for this effect and for
the claimed effect of naloxone in some cases of clonidine poisoning
(North et al., 1981; Kulig et al., 1982) is not clear, but the
involvement of endogenous opioids has been suggested. However, the
effect of naloxone in clonidine poisoning could not be confirmed by
Banner et al. (1983). In a retrospective study of 47 consecutive
children admitted for clonidine poisoning (Wiley et al., 1990), only
3 out of the 19 given naloxone showed a temporary response. One child
had an episode of severe hypertension associated with naloxone
administration (0.1 mg/kg). Thus, there is no clear documentation for
the beneficial effect of naloxone in clonidine poisoning.
2.12 Summary of Evaluation
2.12.1 Indications
Naloxone has been reported to significantly antagonize acute
opioid toxicity and opioid effects within anaesthesia. Its high
therapeutic index and possible beneficial effect in other poisonings
allow for diagnostic use in critically ill patients when opioid
poisoning may be a differential diagnosis.
2.12.2 Advised routes and dose
In patients with definite opiate poisoning, naloxone should be
given by the intravenous route until an improvement in conscious level
and respiration is observed. This may involve the administration of
several milligrams of naloxone if partial opioid agonists are given,
but 0.8-1.2 mg is usually sufficient in morphine or heroin poisonings.
It is important to stress that a pharmacologically active dose of
naloxone in opiate poisoning may be more than that normally
recommended in anaesthetic practice.
In patients with suspected opiate poisoning, an intravenous
injection of up to 2 mg naloxone should be administered and the
patient's response closely monitored. If there is improvement in
conscious level, respiratory rate or cardiovascular parameters,
further doses of naloxone should be administered. The effect of
naloxone should be visible within 1 to 2 min after administration.
Once a patient has regained consciousness, it is necessary to
continue to monitor respiration and cardiovascular status at regular
intervals. In the patient who has taken a large opiate overdose or an
overdose of a long-acting opiate, it may be necessary to repeat dosing
with naloxone. This may be conveniently done by establishing an
intravenous infusion of naloxone. A guide to the required dosage has
been suggested by Goldfrank et al. (1986). From studies of the
pharmacokinetics of naloxone in patients suffering opiate poisoning,
they calculated that an hourly infusion of two-thirds of the dose
required initially to reverse the effects of the opiate would maintain
naloxone levels at approximately those present 30 min after the
initial bolus administration.
Another approach to opioid poisoning that may sometimes be
usefully employed in addicts is to give 0.8-1.2 mg naloxone
intramuscularly before awakening the patient with an intravenous
naloxone dose of 0.4-0.8 mg (higher doses are rarely needed) (personal
communication by D. Jacobsen, 1991). This has been shown to be a
useful practical approach, since many addicts leave the hospital
immediately following the effect of the intravenous dose. Since
naloxone has a shorter duration of action than the opiate, patients
are commonly readmitted within one hour with miosis, coma and impaired
respiration. This approach to treatment, however, requires adequate
ventilatary support for the patient because of the short delay before
the intravenous dose is given.
Naloxone may also be given as a continuous intravenous infusion
(about 0.5 mg/h in isotonic saline) to counteract effects of morphine
metabolites in patients with acute renal failure (Bodd et al., 1990).
2.12.3 Other consequential or supportive therapy
Since many of these patients suffer from impaired respiration or
respiratory arrest, it is extremely important to give oxygen and to
support ventilation immediately while waiting for naloxone to be
available for injection. If ventilation is under control and cyanosis
is regressing, one should consider giving an intramuscular dose of
naloxone before the intravenous dose (see section 2.12.2).
Pulmonary congestion or oedema is occasionally seen in opioid
(heroin) poisoning. It is usually transient and responds to supportive
therapy (oxygen and ventilation support) and naloxone.
2.12.4 Areas where there is insufficient information to make
recommendations
There are anecdotal reports of beneficial effect of naloxone in
other types of acute poisoning, e.g., with ethanol or clonidine. In
the case of ethanol, these results have not been confirmed in well-
controlled studies on volunteers or in intoxicated patients (Nuotto et
al., 1984). The claimed effect in clonidine poisoning has also been
challenged (Wiley et al., 1990). There are insufficient data to
recommend the use of naloxone in poisonings other than those involving
opioids.
2.12.5 Proposals for further studies
Studies of the effect of naloxone in other acute poisonings
should be encouraged. It could, however, be argued that enough studies
have been performed on the use of naloxone in ethanol intoxication to
rule out a possible beneficial effect. On the other hand, there is
certainly a lack of controlled studies on the possible effect of
naloxone in clonidine poisoning.
If effects of naloxone are observed in patients assumed to have
been poisoned by non-opioids, urine specimens should be collected and
analysed by RIA for presence of opioids. Otherwise such "case
reports" are of little value.
2.12.6 Adverse effects
Naloxone possesses a high therapeutic index, but it may provoke
withdrawal signs and symptoms, e.g., seizures, in (heroin) addicts.
Other adverse reactions, as described below, are very rarely seen.
Cardiac arrhythmias and, in particular, ventricular fibrillation
have resulted from rapid reversal of opiate effects with naloxone.
Such events may be a particular problem in patients who have recently
undergone surgery or those habituated to opiates (Cuss et al., 1984).
These reactions may result from a release of sympathetic transmitters,
since a rise in blood pressure and tachycardia have also been
demonstrated.
Some cases of pulmonary oedema following naloxone use in
anaesthetic practice have been reported, but it is unclear in this
situation which is the responsible agent: the anaesthetic, the opiate
or the antagonist (Partridge & Ward, 1986).
2.12.7 Restrictions of use
The fear of provoking withdrawal signs and symptoms should not
hinder use of naloxone in those who need it clinically.
2.13 Model Information Sheet
2.13.1 Uses
Naloxone is indicated in the management of opiate poisoning, both
definite and suspected. Opiate poisoning should be considered in
comatose patients with impaired respiration. Miosis is an unreliable
sign and is not required for a diagnosis of opioid poisoning. The
high wide therapeutic index of naloxone allows its use when a
diagnosis of opioid poisoning is uncertain.
2.13.2 Dosage and route
Since naloxone is a competitive antagonist of opiate poisoning,
there can be no absolute guidelines on dosage. Naloxone should be
given intravenously, in successive doses of 0.4 to 2.0 mg, until the
desired response has been obtained. It should be noted that to
reverse the effects of partial agonists/antagonists, e.g.,
pentazocine, buprenorphine and dextropropoxyphene, much larger doses
may be required, and it may prove impossible to reverse the effects of
buprenorphine.
Failure to respond to a total dose of 10 mg usually indicates: a)
that poisoning is not due to opiates; b) that poisoning is due to a
partial agonist/antagonist; or c) that hypoxic brain damage has
occurred. It should be noted that dextropropoxyphene has been reported
to produce cardiac toxicity that is not reversible by naloxone
administration.
The duration of action of naloxone is short; careful monitoring
is required and repeated doses may be necessary. The alternative is
an intravenous infusion of naloxone. The use of an hourly infusion of
two-thirds of the dose of naloxone required to resuscitate the patient
has been reported to be effective, but dosage should be always
titrated to the individual patient.
Another alternative, which may be appropriate for opiate addicts,
is to give naloxone (0.8-1.2 mg) intramuscularly before waking the
patient with an intravenous dose of 0.4-0.8 mg. However, adequate
ventilatory support must be given. The patient then has a "depot" of
antidote in case he/she departs soon after the initial treatment (as
many addicts do).
The dose given to children should be reduced according to body
weight (0.01 mg/kg initially).
2.13.3 Precautions/contraindications
Naloxone may induce symptoms and signs of acute opiate withdrawal
in addicts. If seizures occur they are best controlled with diazepam
(10-30 mg, intravenously). No dosage alterations seem necessary in
the case of changes in renal function. The dose in children should be
adjusted on a body-weight basis to that used in adults.
Appropriate protective precautions need to be taken by hospital
staff in the case of opiate addicts, bearing in mind the risk of
infection from blood-borne diseases such as hepatitis B and human
immunodeficiency virus (HIV).
2.13.4 Adverse effects
Naloxone has a very high therapeutic index and adverse effects
are rarely seen. Ventricular arrhythmias including ventricular
fibrillation have been reported following rapid reversal of severe
opiate intoxication. This may be avoided if oxygen and adequate
ventilatory support are also given. The management of withdrawal
symptoms in addicts is discussed in section 2.13.3.
2.13.5 Use in pregnancy and lactation
Naloxone is not teratogenic in animals, but no relevant human
data exist. Naloxone treatment does not appear to be a
contraindication to breast feeding, although the opiate poisoning
being treated may itself be a contraindication.
2.13.6 Storage
Naloxone for injection should be stored protected from light.
Its shelf-life is 3 years.
2.14 References
Aitkenhead AR, Derbyshire DR, Pinnock CA, Achola K, & Smith G (1984)
Pharmacokinetics of intravenous naloxone in healthy volunteers.
Anaesthesiology, 61: A381.
Asali LA (1983) Determination of naloxone in blood by high
performance liquid chromatography. J Chromatogr, 278: 329-335.
Asali LA & Brown KF (1984) Naloxone protein binding in adult and
fetal plasma. Eur J Clin Pharmacol, 27: 459-464.
Badawy AA-R, Evans M, Punjani NF, & Morgan CJ (1983) Does naloxone
always act as an opiate antagonist? Life Sci, 33(Suppl 1): 739-742.
Banner W, Lund ME, & Clawson L (1983) Failure of naloxone to reverse
clonidine toxic effect. Am J Dis Child, 137: 1170-1171.
Barraclough CJ & Lowe RA (1982) Failure of naloxone to reverse the
cardiotoxicity of Distalgesic overdose. Postgrad Med J, 58: 667-668.
Bell EF (1975) The use of naloxone in the treatment of diazepam
poisoning. J Paediatr, 87: 803-804.
Berkowitz BA, Ngai SH, Hempstead J, & Spector S (1975) Disposition of
naloxone: Use of a new radio-immunoassay. J Pharm Exp Ther, 195(2):
499-504.
Berkowitz BA (1976) The relationship of pharmacokinetics to
pharmacological activity: Morphine, methadone and naloxone. Clin
Pharmacokinet, 1: 219-230.
Blumberg H, Wernick T, Dayton HB, Hansen RE, & Rapaport DN (1966)
Toxicological studies on the narcotic antagonist naloxone. Toxicol
Appl Pharmacol, 8: 335.
Bodd E, Jacobsen D, Lund E, Ripel A, Morland J, & Wiik-Larsen E
(1990) Morphine-6-glucuronide might mediate the prolonged opioid
effect of morphine in acute renal failure. Hum Exp Toxicol, 9:
317-321.
Bradberry JC & Raebel MA (1981) Continuous infusion of naloxone in
the treatment of narcotic overdose. Drug Intell Clin Pharm, 15:
945-950.
Buchner LH, Cimino JA, Raybin HW, & Stewart B (1972) Naloxone
reversal of methadone poisoning. NY State J Med, 72: 2305-2309.
Christensen KN & Huttel M (1979) Naloxone does not antagonise
diazepam-induced sedation. Anaesthesiology, 51: 187.
Cohen MR, Cohen RM, Pickar D, Weingartner H, & Murphy DL (1983) High
dose naloxone infusions in normals. Dose-dependent behavioural,
hormonal and physiological responses. Arch Gen Psychiatry, 40:
613-619.
Cuss FM, Colaço CB, & Baron JH (1984) Cardiac arrest after reversal
of effects of opiates with naloxone. Br Med J, 288: 363-364.
Dingledine R, Inversen LL, & Breuker E (1978) Naloxone as a GABA
antagonist: evidence from iontophoretic, receptor binding and
convulsant studies. Eur J Pharmacol, 47: 19-27.
Drummond GB, Davie IT, & Scott DB (1977) Naloxone: dose-dependent
antagonism of respiratory depression by fentanyl in anaesthetised
patients. Br J Anaesth, 49: 151-154.
Evans LEJ, Roscoe P, Swainson CP, & Prescott LF (1973) Treatment of
drug overdose with naloxone, a specific narcotic antagonist. Lancet,
1: 452.
Fishman J, Roffwarg H, & Hellman, L. (1973) Disposition of naloxone
- 7,8,3H in normal and narcotic-dependent men. J Pharmacol Exp Ther,
187: 575-580.
Fishman J, Hahn EF, & Norton BI (1975) Comparative in vivo
distribution of opiate agonists and antagonists by means of double
isotope techniques. Life Sci, 17: 1119-1126.
Fujimoto JM (1969) Isolation of two different glucuronide metabolites
of naloxone from the urine of rabbit and chicken. J Pharmacol Exp
Ther, 168: 180-186.
Fujimoto JM, Roerig S, Wang RIH, Chatterjie N, & Intrirrisi CE (1975)
Narcotic antagonist activity of several metabolites of naloxone and
naltrexone tested in morphine dependent mice (38558). Proc Soc Exp
Biol Med, 148: 443-448.
Gober AE, Kearns GL, Yorkel RA, & Danziger L (1979) Repeated naloxone
administration for morphine overdose in a 1 month old infant.
Paediatrics, 63: 606-608.
Gold MS, Pottash AC, Sweeney DR, & Kleber HD (1980) Opiate withdrawal
using clonidine. A safe, effective, and rapid nonopiate treatment. J
Am Med Assoc, 243: 343-346.
Goldfrank L, Weisman RS, Errick JK, & Lo MW (1986) A dosing nomogram
for continuous infusion intravenous naloxone. Ann Emergency Med, 15:
566-570.
Gourlay GK & Coulthard K (1983) The role of naloxone infusions in the
treatment of overdoses of long half-life narcotic agonists.
Application to nor-methadone. Br. J Clin Pharmacol., 15: 269-271.
Handal KA, Schauben JL, & Salamone FR (1983) Naloxone. Ann Intern
Med, 12: 438-445.
Hahn EF, Lahita R, Kreek J, Duma C, & Intrurrisi CE (1983) Naloxone
radio-immunoassay: an improved antiserum. J Pharm Pharmacol, 35:
833-836.
Hassan MMA, Mohammed ME, & Mian MS (1985) Naloxone hydrochloride.
Anal Profiles Drug Subst, 14: 453-489.
Hatano S, Keane DM, Wade MA, & Sadove MS (1975) Naloxone reversal for
anaesthetic dosages of pentazocine. Anaesth Rev, 2: 11-15.
Henry J & Volans G (1984) ABC of Poisoning: Analgesics: Opioids. Br
Med J, 289: 990-993.
Jasinski DR, Martin WR, & Haertzen CA (1967) The human pharmacology
and abuse potential of N-allyl noroxymorphone (naloxone). J Pharm Exp
Ther, 157: 420-426.
Jeffcoate WJ, Herbert M, Cullen MH, Hastings AG, & Walder CP (1979)
Prevention of effects of alcohol intoxication by naloxone. Lancet, 2:
1157-1159.
Jefferys DB, Flanagan RJ, & Volans GN (1980) Reversal of ethanol-
induced coma with naloxone. Lancet, 1: 308-309.
Johnstone RE, Jobes DR, Kennell EM, Behar MG, & Smith TC (1974)
Reversal of morphine anaesthesia with naloxone. Anaesthesiology, 41:
361-367.
Jordan C (1980) Respiratory depression following diazepam: reversal
with high-dose naloxone. Anaesthesiology, 53: 293-298.
Kaufman RD, Gabathuler ML, & Bellville JW (1981) Potency, duration of
action and pA2 in man of intravenous naloxone measured by reversal of
morphine-depressed respiration. J Pharmacol Exp Ther, 219: 156-162.
Kaufman JJ, Semo NM, & Koski WS (1975) Microelectrometric titration
measurements of the pKa's and partition and drug distribution
coefficients of narcotics and narcotic antagonists and their pH and
temperature dependence. J Med Chem, 18: 647-655.
Kotlinska J & Langwinski R (1990) The lack of effect of opioid
agonists and antagonists on some acute effects of ethanol. Pol J
Pharmacol Pharm, 42: 129-135.
Kulig K, Duffy J, & Rumack BH (1982) Naloxone for treatment of
clonidine overdose. J Am Med Assoc, 247: 1697.
Longnecker DD, Grazis PA, & Eggors WWN (1973) Naloxone for antagonism
of morphine-induced respiratory depression. Anaesth Analg, 53:
447-452.
McNicholas LF & Martin WR (1984) New and experimental therapeutic
roles for naloxone and related opioid antagonists. Drugs, 27: 98-93.
Martin WR (1976) Naloxone. Ann Intern Med, 85: 765-768.
Martindale (1982) In: Reznolds JEF ed. The extra pharmacopoeia, 28th
ed. London, Pharmaceutical Press.
Meffin PF & Smith KJ (1980) Gas chromatographic analysis of naloxone
in biological fluids. J Chromatogr, 183: 352-356.
Misra AL (1978) Metabolism of opiates. In: Factors affecting the
action of narcotics. New York, Raven Press, pp 297-343.
Ngai SH, Berkowitz BA, Yang JC, Hampstead J, & Spector S (1976)
Pharmacokinetics of naloxone in rats and in man: Basis for its potency
and short duration of action. Anaesthesiology, 44: 398-401.
North DS, Wieland MJ, & Peterson CD (1981) Naloxone administration in
clonidine overdosage. Ann Emergency Med, 10: 397.
Nuotto E, Palva ES, & Seppala T (1984) Naloxone-ethanol interaction in
experimental and clinical situations. Acta Pharmacol Toxicol, 54:
278-284.
Pace NL, Parrish RG, Lieberman MM, Wong KC, & Blatnick RA (1979)
Pharmacokinetics of naloxone and naltrexone in the dog. J Pharmacol
Exp Ther, 208: 254-256.
Partridge BL & Ward CF (1986) Pulmonary edema following low-dose
naloxone administration. Anesthesiology, 65: 709-710.
Sawynok J, Pinsky C, & Labella FS (1979) Mini review on the
specificity of naloxone as an opiate antagonist. Life Sci, 25:
1621-1632.
Smits SE & Takemori AE (1970) Quantitative studies on the antagonism
by naloxone of some narcotic and narcotic-antagonist analgesics. Br
J Pharmacol, 39: 627-638.
Social Welfare Board (1976) Nalone (naloxon). Uppsala, Sweden, Social
Welfare Board, Pharmaceuticals Department, pp 7-9.
Stile IL, Fort M, Marotta F, Wurzburger R, Hiatt IM, & Hegyi T (1984)
Pharmacokinetics of naloxone in premature infants. Paediatr Res,
18(392): 161A.
Tenenbein M (1984) Continuous naloxone infusion for opiate poisoning
in infancy. J Pediatr, 105: 645-647.
Terry MD, Hisayasu GH, Kern JW, & Cohen JL (1984) High performance
liquid chromatographic analysis of naloxone in human serum. J
Chromatogr, 311: 213-217.
United States Pharmacopeia (1980) 20th ed. Rockville, Maryland,
United States Pharmacopeial Convention, Inc.
Weinstein SH, Pfeffer M, & Schor JM (1974) Metabolism and
pharmacokinetics of naloxone. Adv Biochem Psychopharmacol, 8:
525-535.
Weinstein SH, Pfeffer M, Schor JM, Indindoli L, & Mintz M (1971)
Metabolites of naloxone in human urine. J Pharm Sci, 60: 1567-1568.
Wiley JF, Wiley CC, Torrey SB, & Henretig FM (1990) Clonidine
poisoning in young children. J Pediatr, 116: 654-658.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs and biologicals, 10th ed. Rahway, New Jersey, Merck and Co,
Inc.
3. FLUMAZENIL
3.1 Introduction
Acute poisoning is currently one of the main causes of hospital
admission in developed countries. Benzodiazepines (BZDs) are the most
commonly used drugs throughout the world and their abuse may be
responsible for the impairment of memory and for dependence. An acute
overdose can result in long-lasting coma, which is generally treated
with supportive measures. Flumazenil, an imidazobenzo-diazepine
(AnexateTM), has been shown to reverse the sedative, anti-
convulsant, and muscle-relaxant effects of BDZs. It has no convulsive
action in itself and its use has therefore been proposed to counteract
benzodiazepine action in anaesthetics, clinical toxicology and
intensive care.
3.2 Name and Chemical Formula of Antidote
* Flumazenil AnexateR (Roche Laboratories)
* Ethyl-8-fluoro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5-a][1,4]
benzo-diazepine-3-carboxylate
* Empirical formula: C15H14O3N3F
* Relative molecular mass: 303.3
* Therapeutic class : Imidazobenzodiazepine
* CAS number: 78 755-81-4
* Conversions:1 mmol = 303.3 mg
1 g = 3.3 mmol
µmol/l = 3.3 x µg/ml
µg/ml = 0.3 x µmol/l
3.3 Physico-chemical Properties
Physico-chemical properties of flumazenil are given in Table 1.
Flumazenil remains stable when exposed to light and when stored
for 2 years at 35° C. The loss of weight on drying is up to 1%.
3.4 Pharmaceutical Formulation and Synthesis
No information is available on the routes of synthesis and
manufacture.
Flumazenil is supplied for parenteral administration in vials
containing 5 or 10 ml aqueous solution (0.1 mg/ml). It is available
for oral administration as tablets of 10, 20 or 30 mg.
Table 1. Physico-chemical properties of flumazenila
Melting point 198-202 °C
Solubility in water < 1 g/l
Solubility in organic solvents (g/l)
chloroform < 250
methanol < 17
ethyl acetate < 3
diethyl ether < 1
Solubility at various pH values (g/l)
(in aqueous buffered solution at 37° C)
pH 1.2 3
pH 5.3 0.7
pH 7.5 0.6
Acidity (10% aqueous solution) 4.5-7.5
pKa (in weak base) 1.7
a Personal communication from Roche Laboratories (1988)
3.5 Analytical Methods
3.5.1 Identification of the antidote
Information on the identification of flumazenil was provided by
Roche Laboratories (personal communication, 1988).
3.5.1.1 Infrared spectroscopy
The infrared spectrum (625-4000 cm-1) of a sample (a 1:300
solid dispersion in potassium bromide) is compared qualitatively with
that of a reference substance.
3.5.1.2 Ultraviolet absorption
A portion (85-95 mg) of the sample is dissolved in approximately
100 ml of ethanol and diluted to 150 ml with ethanol (solution 1).
This solution is then diluted ten-fold with ethanol to give solution
2, which is further diluted ten-fold with ethanol to give solution 3.
The position and absorbance of solution 3 is measured
spectrophotometrically at the maximum (245 nm) and minimum (228 nm)
wavelengths, against ethanol, in quartz cells.
3.5.1.3 Thin-layer chromatography
The TLC details are as follows:
* layer: Silica gel 60 F254
* mobile phase: Chloroform/ethanol (90/10 v/v)
* sample solution: 10 ml of the ampoule solution is extracted
with 1 ml of chloroform
* standard solution: 5 mg of flumazenil is dissolved in 5 ml
of chloroform saturated with water
* front distance: 12 cm
* migration time approx: 30 min
* detection: the plate is dried in a current of warm air for
5 min, and examined under shortwave light. Decreasing
fluorescence due to flumazenil occurs at 254 nm
(ultraviolet region). When the plate is sprayed with
Dragendorff's reagent, flumazenil appears as an orange
spot. The Rf value is approximately 0.5.
3.5.2 Quantification of the antidote in biological samples
The determination of flumazenil in plasma by gas-liquid
chromatography (GLC) with nitrogen phosphorus detection is a sensitive
and specific method, the detection limit being 3 ng/ml (Abernethy et
al., 1983). An ethyl acetate extraction (neutral pH) of 0.1-3 ml
plasma is used for sample preparation. When methylclonazepam is used
as an internal standard, the graph is linear for plasma concentrations
up to 200 ng/ml. The retention time for flumazenil is 3.96 min.
High-performance liquid chromatography (HPLC) with UV detection
at 254 nm is a sensitive method for determination in urine or plasma,
the detection limit being about 10 ng/ml (Timm & Zell, 1983; Bun et
al., 1989). When the n-propyl ester analogue is used as an internal
standard, the graph is linear for plasma concentrations up to 320
ng/ml.
3.5.3 Analysis of the toxic agent in biological samples
Three major methods for the quantitative analysis of BZDs in
plasma or serum are used:
* HPLC with UV detection at 246 nm (detection limits are 5-50 µg/ml
of serum) (Rocher, 1984);
* immunoenzymology by the EMIT method for a semiquantitative
determination (metabolites also measured) of diazepam levels,
completed by a chromatographic method (sensitivity from 0.3 to 2
µg/ml) (Rocher, 1984);
* gas-liquid chromatography (Pellerin, 1986).
3.6 Shelf-life
Vials ready for use are stable at room temperature (15-25° C) for
three years.
3.7 General Properties
Flumazenil has been shown to block all the typical BZD effects
(anticonvulsive, sedative, anxiolytic, muscle relaxant, and amnesic).
It acts as a potent BZD-specific antagonist by competing at the
central synaptic gamma-aminobutyric acid (GABA) receptor sites in a
dose-dependent manner, but does not seem to antagonise BZD effects at
peripheral GABA-ergic (renal, cardiac, etc.) receptor sites (Mohler et
al., 1981). It possesses agonist properties and has a specific, but
discreet, anticonvulsive effect without inducing drowsiness or muscle
relaxation (Abernethy et al., 1983; Timm & Zell, 1983; Haaefely, 1983;
Rocher, 1984; Scollo-Lavizzari, 1984; personal communication by Roche
Laboratories, 1988). In addition, it antagonizes the sedative effects
of other compounds that act through GABA receptors, such as zopiclone
(Mohler et al., 1981).
3.8 Animal Studiesc
3.8.1 Pharmacodynamics
Flumazenil has been tested for its ability to induce withdrawal
signs in animals pretreated with benzodiazepine; the signs included
emesis, tremors, rigidity and clonic convulsions.
c Personal communication by Roche Laboratories to the IPCS, 1988
Rats that had been pretreated with an oral dose (10 or 100 mg/kg)
of diazepam for 12 days were administered flumazenil (10 mg/kg)
intravenously. Signs were very mild even at 100 mg/kg.
Cats were pretreated intraperitoneally for 16 days with either a
10-mg/kg dose of lorazepam twice daily or a 1-mg/kg dose of triazolam
once daily. Flumazenil (100 mg/kg) was then administered
intraperitoneally either immediately or 1.5, 6, 12, 48, and 60 h after
the last dose. Symptoms such as rigidity, vocalization and tachypnoea
lasted 30 min, whereas others such as hypersalivation lasted 2 h.
Flumazenil (1 to 15 mg/kg) was administered intragastrically to
rats that had been pretreated with daily diazepam doses of 113 mg/kg
for about 6 months. Abstinence syndromes increased with increasing
dose of flumazenil and reached a plateau.
The intragastric administration of flumazenil (15 mg/kg per day)
to cats pretreated with flurazepam (5 mg/kg per day) for 35 days led
to withdrawal symptoms (increasing muscle tone, tremors, piloerection,
mydriasis, and hypersalivation) 24 h after the last dose of
flurazepam. No convulsions were observed.
Intramuscular administration of flumazenil (5 mg/kg) to squirrel
monkeys and baboons, pretreated with oral doses of lorazepam,
triazolam (3 mg/kg per day), oxazepam (40 or 80 mg/kg per day) or
diazepam (8-20 mg/kg per day), produced withdrawal signs. However, no
withdrawal signs were precipitated by flumazenil in monkeys treated
with oral midazolam (30 mg/kg) or in barbital-dependent rhesus monkeys
(the length of pretreatment with BZD was not specified).
The severity of withdrawal signs resulting from the blocking of
BZD receptors by flumazenil depends on the species tested, the dose of
BDZ used to develop physiological dependence, and the duration of
treatment.
3.8.2 Pharmacokinetics
3.8.2.1 Absorption
A single dose of flumazenil (125 mg/kg) in a carboxymethyl
cellulose suspension produced a maximum plasma concentration in rats
of 9.9 µg/ml after 20 min. The bioavailability was 0.55. In rabbits,
the maximum concentration 90 min after a single dose of flumazenil
(150 mg/kg) was 15 µg/ml. The bioavailability was 0.60.
3.8.2.2 Distribution
When total radioactivity was measured in rats 0.5, 7, 24, 96, and
192 h after an intravenous dose of 14C-labelled flumazenil (2
mg/kg), the highest level was found at 0.5 h in the kidney, liver and
intestine. None was found at 192 h. The volume of distribution
ranged from 0.71 to 1.87 l/kg.
3.8.2.3 Elimination
Studies on rats given an oral dose (50 mg/kg) of 14C-labelled
flumazenil and on dogs given an intravenous dose of 4 mg/kg showed
three main inactive metabolites:
* Ro 15-3890 acid and major metabolite (72% in the rat, 30-60% in
the dog);
* Ro 15-4965 hydroxyethyl derivative (3% in the rat);
* Ro 15-6877 N-demethyl derivative (1% in the rat, 1-13% in the
dog).
Table 2 presents elimination data in three different species. In
these species, 90% of the intravenously or orally administered
flumazenil was eliminated, mainly as metabolites, within 48 h. One
third was eliminated in the faeces and two-thirds in the urine.
Table 2. Elimination of flumazenil in the rat, rabbit and dog
Species Dose Total plasma T´
clearance (min)
(ml/min per kg)
Rat 2 mg/kg 114 7.4
Rabbit 0.5 mg/kg 24 34
Dog 5 mg 21 48
3.8.3 Toxicology
3.8.3.1 Acute toxicity
a) Intravenous administration to rats and mice
An aqueous solution of 0.1 mg flumazenil/ml was used and was
administered at a dose of 2.5 mg/kg to mice and 1 mg/kg to rats. No
abnormal clinical signs and no deaths occurred. LD50 values were
not determined; these doses (50 to 250 times higher than the clinical
doses) were well tolerated in the two species.
A flumazenil solution with a concentration of 50 mg/ml was
subsequently used and the LD50 values given in Table 3 were obtained
(95% confidence intervals). Deaths occurred 30 min after the
injection, preceded by rigidity and clonic convulsions.
Table 3. Intravenous LD50 values (mg/kg) for the mouse and rat
Species Male Female
Mouse 143-198 145-175
Rat 85-167 112-231
b) Intravenous administration to the dog
The administration of daily doses of 0.01 to 0.03 mg/kg was well
tolerated and no deaths were observed. LD50 values were not
determined; the doses (15 to 30 times higher than the clinical doses)
were again well tolerated.
c) LD50 values (mg/kg) for the rat, mouse and rabbit
When flumazenil was administered orally to rats, mice and rabbits
(Table 4), deaths were observed within three days, associated with
decreased motor activity, catatonic state and tremors.
Table 4. LD50 values (mg/kg) for the rat, mouse and rabbit
Species Male Female
Mouse 2500 1300
Rat 4200 4200
Rabbit 2000 2000
3.8.3.2 Subacute toxicity
Systemic tolerance was good in both rats and dogs administered
flumazenil intravenously at dosages up to 10 mg/kg per day for 4
weeks.
3.8.3.3 Chronic toxicity
In 13-week studies using an oral aqueous solution of flumazenil,
very good tolerance was shown by rats at dosages of 0.5, 25 and 125
mg/kg per day and by dogs at 0.5, 20, and 80 mg/kg per day. No
haematological, biochemical or gross pathological abnormalities were
observed.
3.8.3.4 Embryotoxicity
Studies on rats (between the 7th and 16th day of gestation) and
rabbits (between the 7th and 19th day of gestation) revealed no signs
of embryotoxicity at dosages of 15, 50, and 150 mg/kg per day.
3.8.3.5 Mutagenicity
Flumazenil was not mutagenic in the Ames test or micronucleus
test, or in tests using Saccharomyces cerevisiae or Chinese hamster
V79 cells.
3.9 Volunteer Studies
3.9.1 Pharmacodynamics
3.9.1.1 BZD antagonist effect
Efficacy studies were performed on 125 healthy volunteers with
oral doses of flumazenil up to 20 mg, the aim being to antagonize the
effects of diazepam, flunitrazepam and midazolam on the CNS (Darragh,
1981; Lupolover, 1983). These studies demonstrated the antagonist
effect of flumazenil, which rapidly abolished the hypnotic-sedative
BZD effects. Other studies used meclonazepam (Darragh et al., 1981),
diazepam (Darragh et al., 1982), flunitrazepam (Gaillard & Blois,
1983) and midazolam (Forster et al., 1983). In studies by Ziegler &
Schalch (1983) and Lauven et al. (1985), flumazenil was administered
to subjects during continuous midazolam infusion after the attainment
of a pharmacokinetic and pharmacodynamic steady state, at which point
subjects were deeply asleep. The degree and duration of the effect of
flumazenil depended on the BZD dose, the antagonist dose and the time
that had elapsed since the BZD was given. In the study by Ziegler &
Schalch (1983), baseline levels of vigilance and orientation were
reached within 1 min. Lauven et al. (1985) used higher midazolam and
flumazenil dosages and his patients awoke within 28 to 48 seconds. No
signs of BZD withdrawal effects were seen in short-term studies (one
single dose) on healthy volunteers given flumazenil to antagonize BZDs
(Amrein, 1987).
The efficacy of flumazenil in antagonizing the effects of
midazolam was also clearly demonstrated in the double-blind placebo-
controlled study by Rouiller et al. (1987).
3.9.1.2 Intrinsic effects
Most studies on healthy human volunteers have shown little or no
intrinsic effect of flumazenil when administered alone. The mild
sedation reported by Amrein (1987) occurred after the administration
of oral doses greater than 100 mg.
Scollo-Lavizzari (1984) observed some anticonvulsant effects in
epileptic patients. Decreased amplitude of auditory evoked potentials
has also been described (Laurian et al., 1984; Schoepf et al., 1984).
Mild, nonspecific effects such as increased alertness may occur after
the administration of doses very much higher than those used
clinically (Laurian et al., 1987).
3.9.2 Pharmacokinetics
3.9.2.1 Absorption
Following oral administration of a 200-mg dose of flumazenil, the
highest plasma concentration (Cmax) ranged from 147 to 349 µg/l and
was reached within 20 to 45 min. The mean bioavailability of the
tablets used was about 17% and the inter-individual variability was
7-29% (Pellerin, 1986).
3.9.2.2 Distribution
The proportion of flumazenil bound to plasma proteins is 50%
(two-thirds of which is bound to albumin). Values for the mean
steady-state volume of distribution of 0.95 l/kg (personal
communication by Roche Laboratories, 1988) and 1.23 l/kg (Roncari et
al., 1986) have been determined.
3.9.2.3 Elimination
Ninety-nine per cent of the flumazenil administered is
metabolized by the liver, and 1% is excreted unchanged in the urine.
Mean total blood clearance, for which values of 59 l/h (Pellerin,
1986) and 72 l/h (Roncari et al., 1986) have been determined, is
essentially due to the hepatic clearance. The apparent plasma half-
life in healthy volunteers has been reported to be 53-58 min (Roncari
et al., 1986; personal communication by Roche Laboratories, 1988).
3.9.3 Tolerance of flumazenil
In the study by Rouiller et al. (1987), no objective agonist
effects or biological toxicity of flumazenil could be demonstrated in
six healthy volunteers.
3.9.4 Other studies
There is evidence that central nervous system effects of ethanol
are mediated through the GABA system. For this reason, the effect of
flumazenil on psychometric performance was studied in eight healthy
volunteers with stable blood ethanol levels of 1.6 g/l (35 mmol/l)
under a placebo-controlled double-blind design (Clausen et al., 1990).
Flumazenil did not improve psychomotor functions in these ethanol-
intoxicated subjects, which is in agreement with experience in
clinical toxicology.
3.10 Clinical Studies - Clinical Trials
Flumazenil was first used clinically in patients with iatrogenic
benzodiazepine overdose due to mechanical ventilation or status
epilepticus (Scollo-Lavizzari, 1983).
Clinical studies can be grouped under the headings
anaesthesiology and toxicology (Amrein, 1986).
3.10.1 Anaesthesiology
3.10.1.1 General anaesthesia
Three placebo-controlled studies have been conducted in patients
who were given flunitrazepam for general anaesthesia.
Jensen et al. (1985) reported that a 0.3-mg to 0.7-mg dose of
flumazenil awoke all patients within 5 min, compared with only 35% of
the patients in the placebo-treated group (P < 0.001 for sedation,
orientation and amnesia).
In a study of 60 patients, Tolksdorf et al. (1986) found that
patients treated with flumazenil were less sedated than placebo-
treated patients (P < 0.05) following flunitrazepam sedation (from 5
min to 1 h after the administration of flumazenil), better orientated
at 15 min, and less amnesic. Ellmauer et al. (1986) reported similar
results in 57 patients given a 0.1- to 1-mg dose of flunitrazepam (P
< 0.005).
No significant difference was observed after 2 h between the
placebo-treated and flumazenil-treated patients in any of the three
studies described in this section.
Midazolam effects were reversed by flumazenil in an open study
including 18 intracranial surgery patients (Chiolero et al., 1988).
3.10.1.2Conscious sedation
In a 74-patient open study (Geller et al., 1986) and a 40-patient
placebo-controlled study (Knudsen et al., 1986), in which either
midazolam or diazepam was used, there was a significant difference
between flumazenil- and placebo-treated patients. In the former
study, patients were awakened by a 0.1- to 0.6-mg dose of flumazenil
within 1 to 2 min. In the study by Knudsen et al. (1986), 80% of the
flumazenil-treated patients were awake 5 min after receiving the dose
compared with 50% in the placebo group (P < 0.05).
3.10.2 Benzodiazepine overdose or intoxication
Three different studies have indicated that flumazenil may be an
effective tool for the management of intoxication (either intentional
or iatrogenic) with BZD in the presence or absence of other agents.
Owing to its safety and specificity, flumazenil could be used in the
initial treatment of poisoning and coma of unknown origin. In a study
by Hofer & Scollo-Lavizzari (1985) based on 13 patients, a 1.5-to 10-
mg dose of flumazenil administered intravenously at a rate of 1.5 to
2.5 mg/min reversed the CNS depression induced by various BZDs within
1 to 2 min.
Geller et al. (1985) treated 34 patients (23 cases of intentional
drug intoxication and 11 of iatrogenic BZD overdose) by means of
intravenous injections of 0.1 mg flumazenil every 30 seconds until the
patient regained consciousness. The treatment proved to be extremely
effective, providing reversal effects lasting up to 2 h.
Bismuth et al. (1985) treated patients for BZD overdose in a
double-blind randomized study, injecting a single dose of either
flumazenil or placebo. Two of the 20 placebo patients awoke
partially, compared with 17 of the 20 flumazenil-treated patients (one
experienced seizures interrupting the study). In a second open study
(Bismuth et al., 1986) based on 37 patients, 6 showed no response to
doses of flumazenil ranging from 5 to 9.5 mg (mixed intoxication), 11
showed partial awakening (no possible written response) at a dose of
2.1 ± 1.6 mg (mixed intoxication), and 20 were completely awakened by
a dose of 1.4 ± 0.7 mg. The awakening was only temporary and return
to coma occurred after an interval of 15 min to 5 h. Permanent
recovery occurred in a patient suffering intoxication due to
triazolam, a BZD with a short half-life, after a single administration
of flumazenil.
More recent placebo-controlled double-blind studies have
confirmed the beneficial effect of flumazenil in cases of BZD
poisoning (Aarseth et al., 1988; Ritz et al., 1990).
3.11 Clinical Studies - Case Reports
The many controlled clinical studies of the effect of flumazenil
limit the need for information from case reports. In the clinical
studies reported, there have been few adverse effects associated with
the use of flumazenil. There have, however, been case reports of
seizures followed by ventricular tachycardia associated with the use
of flumazenil in combined poisonings with cyclic antidepressants and
BZD (Bismuth et al., 1985).
In one report, death was claimed to have been associated with
flumazenil administration in an old, obese and anaemic woman who had
been sedated with midazolam (4 mg, intravenous) prior to gastroscopy
(Lim, 1989). During the investigation she suffered cardiac arrest;
flumazenil was given promptly and she recovered temporarily, but then
gradually deteriorated and died 16 h later. According to Birch &
Miller (1990), the death of this patient was probably not related to
flumazenil administration.
Recently, successful treatment was achieved by administering
flumazenil as an intravenous bolus (0.02 mg/kg) and then as a
maintenance dose of 0.05 mg/kg per h to a newborn baby with recurrent
apnoea due to BZDs taken by his mother (Richard et al., 1991).
The benefit from the diagnostic use of flumazenil in coma of
unknown origin has been reported in two recent cases (Burkhart &
Kulig, 1990). When flumazenil is used with caution in such
situations, time may be saved and further expensive diagnostic
procedures, e.g., cerebral computerized tomographic (CT) scan,
avoided.
3.12 Summary of Evaluation
Flumazenil appears to be a antagonist to BZDs and other GABA-
ergic agents. This antagonism, following intravenous injection, has
been reported to be sensitive in cases of intoxication resulting
solely from BZDs (the reversal of BZD effects being observed with
doses of less than 2 mg), rapid in onset (within 2 min), and short-
lived (effects last for less than 30 min).
3.12.1 Indications
In controlled clinical trials, flumazenil significantly
antagonizes BZD-induced coma arising from anaesthesia or acute
overdose. However, the use of flumazenil has not been shown to reduce
mortality or sequelae in such cases. As the mortality in pure BZD
poisoning is extremely low, studies with mortality as end-point are
impractical since a reasonable level of statistical significance could
probably never be obtained. However, in cases of mixed intoxication,
especially with ethanol and triazolam/flunitrazepam, the use of
flumazenil may be life-saving due to the poten-tiation of BZD toxicity
by ethanol. Given this situation, it is obvious that the routine use
of flumazenil in BZD poisoning is not indicated and that
recommendations for its use in clinical toxicology must be based on
pragmatic considerations made by clinicians experienced in treating
these patients. Flumazenil is a relatively expensive drug and this
may also influence its use, especially in areas with limited
resources.
The use of flumazenil in BZD poisoning should, therefore, only be
advocated in situations with complications, which are rarely seen
except in cases of mixed ingestion. Although not of life-saving
significance, it also seems reasonable to advocate the use of
flumazenil if intubations (before gastric lavage) and mechanical
ventilation can thereby be avoided (see section 3.13.1). The proposed
uses of flumazenil within acute medicine and anaesthesia are listed in
section 3.13.1. Acute poisoning is always an important differential
diagnosis in cases of coma in children and young adults. The
diagnostic use of flumazenil in such cases can be justified by its
high therapeutic index and the fact that this may limit the use of
other diagnostic procedures such as cerebral CT scan, clinical
chemistry analyses and even lumbar puncture.
3.12.2 Dosage and route
Flumazenil is available for intravenous and oral administration.
The need for the latter formulation may be questioned in view of the
fact that drugs should generally be given intravenously in the
emergency situation and the bioavailability is low and variable. Thus
the intravenous route is preferable. Doses need to be adjusted
according to individual clinical response, bearing in mind the very
high therapeutic index of flumazenil.
a) In anaesthetics and in intensive care, doses of 0.2-0.5 mg should
be used to reduce sedation and doses of 0.5-1 mg to abolish other
BZD effects (Amrein, 1987).
b) In cases of BZD overdosage, single doses of 0.3-1 mg can be given
and repeated as necessary. If there is no clinical response to
2 mg flumazenil given over a period of 5-10 minutes, diagnoses
other than BZD poisoning are likely. It is also possible to
administer a continuous infusion (0.3-1 mg/h) of flumazenil
(diluted in 0.9% sodium chloride solution or 5% glucose solution)
in patients relapsing into a coma and/or respiratory depression
following an initial effect of flumazenil injection.
c) In children, experience is limited and dosage regimens less well
documented (Lheureux & Askenasi, 1988; Wood et al., 1987). It
is suggested that intravenous doses of 0.1 mg should be given
once per minute until the child is awake. It may be necessary to
give a subsequent continuous intravenous infusion at a rate of
0.1 to 0.2 mg/h.
3.12.3 Other consequential or supportive therapy
Treatment with flumazenil requires continuous intensive
observation. After the administration of a single dose of flumazenil,
the patient must be observed for at least 2 h to be certain that BZD-
induced complications will not recur. The termination of continuous
infusion requires intensive care monitoring.
3.12.4 Areas where there is insufficient information to make
recommendations
There is insufficient information to make recommendations in the
case of hepatic encephalopathy (indication is based on the hypothesis
that hepatic encephalopathy is associated with increased GABA-mediated
inhibitory neurotransmission).
3.12.5 Proposals for further study
The use and dosages of flumazenil in children require further
study. Indications for utilization of oral preparations need to be
clarified. The use in coma of unknown origin merits further studies.
3.12.6 Adverse effects
The most frequent adverse effects have been reviewed by Amrein
(1987). When flumazenil is used in anaesthesia, the main adverse
effects that have been reported are nausea and vomiting (placebo:
7.5%; < 1 mg flumazenil: 12.1%; 1-10 mg flumazenil: 24.5%). Other
adverse effects, which have been reported in less than 5% of cases,
are tremor, involuntary movements, dizziness, agitation, discomfort,
tears, anxiety, and a sensation of cold.
Minor effects occur when flumazenil is used in intensive care,
where agitation is the commonest adverse effect (10%). When it is
administered to patients showing BZD habituation, the following
features occur: anxiety, tenseness, fear, agitation, confusion,
convulsions (Marchant & Wray, 1989) and myoclonic seizures. Their
frequency and intensity depend on the degree of dependency and they
are believed to be related to some sort of BZD abstinence syndrome.
When administered rapidly, flumazenil can cause hypertension,
tachycardia and acute anxiety. This equivalent of an "exercise test"
was observed with the 1 mg/ml solution, which is no longer used.
3.12.7 Restrictions of use
In certain circumstances, BZD antagonism by flumazenil may be
harmful:
a) An acute withdrawal syndrome can occur in patients showing BZD
habituation following therapy or abuse.
b) Convulsions can occur in cases of mixed drug overdosage where BZD
has been taken with a drug liable to cause convulsions (such as
a tricyclic antidepressive agent).
c) Convulsions can be induced in patients treated with BZD for
seizure disorders or in patients who for years have been using
BZD for sleep disturbances.
There are other limitations to the use of flumazenil.
a) It has a short-lived effect and repeated injection or continuous
infusion is often necessary unless a short-acting BZD (e.g.,
triazolam) has been ingested.
b) In cases of mixed drug overdosage, the patient may remain
unresponsive when other drugs are contributing to the coma.
c) The treatment costs are high and supportive treatment may be
cheaper.
3.13 Model Information Sheet
3.13.1 Uses
Flumazenil is a specific antagonist of the effects of BZD at
central GABA-ergic receptors.
Within the domains of intensive care and anaesthesia, flumazenil
may be valuable in the following circumstances:
a) to diagnose BZD-induced unconsciousness in patients presenting
coma of unknown origin;
b) to terminate long-term BZD-induced sedation in the intensive care
unit (e.g., weaning from ventilatory support);
c) to reduce BZD-induced sedation or to counteract paradoxical
anxiety reactions to BZD in anaesthesia;
d) to antagonise BZD-induced sedation after short diagnostic
procedures where a long-acting BZD has been used.
Flumazenil may be justified in the following situations in cases
of BZD poisoning:
a) to facilitate gastric lavage and avoid intubation in comatose
patients;
b) to treat complications in severe cases of mixed poisoning where
BZD is thought to be one of the major toxic agents;
c) to avoid the need for mechanical ventilation in cases where there
is respiratory depression.
The routine use of flumazenil for the treatment of BZD overdosage
is not recommended.
3.13.2 Dosage and route
The intravenous route of administration is recommended when
flumazenil is given as a BZD antagonist. Doses need to be adjusted
according to individual clinical response and the following are
recommended.
a) In anaesthetics and in intensive care (adults), doses of 0.2-0.5
mg should be used to reduce sedation and doses of 0.5-1 mg to
abolish BZD effects.
b) In cases of BZD overdosage (adults), single doses of 0.3-1 mg can
be given and repeated as necessary. The absence of clinical
response to 2 mg flumazenil within 5-10 min indicates that BZD
poisoning is not the major cause of coma and other complications.
It is also possible to administer a continuous infusion (0.3 to
1 mg/h) of flumazenil (diluted in 0.9% sodium chloride solution
or 5% glucose solution) following an initial response to
flumazenil.
c) In children, it is suggested that intravenous doses of 0.1 mg
should be given once per minute until the child is awake. It may
be necessary to give a subsequent continuous intravenous infusion
at a rate of 0.1 to 0.2 mg/h.
3.13.3 Precautions/contraindications
3.13.3.1 Pharmaceutical precautions
Solutions of flumazenil should be stored at +4 °C. No other drug
should be injected or infused with the flumazenil, which should be
made up in 0.9% sodium chloride or 5% glucose (dextrose) solution.
3.13.3.2 Other precautions
Treatment with flumazenil requires continuous intensive
observation. After the administration of a single dose of flumazenil,
the patient must be observed for at least 2 h to be certain that
BZD-induced complications will not recur. The termination of
continuous infusion requires intensive care monitoring.
Note that in cases of mixed drug overdosage, the patient may
remain unresponsive where other drugs are contributing to the coma.
BZD antagonism by flumazenil may in certain circumstances be
harmful.
a) An acute withdrawal syndrome can occur in patients showing BZD
habituation following therapy or abuse.
b) Convulsions can occur in cases of mixed drug overdosage where BZD
has been taken with a drug liable to cause convulsions (such as
a tricyclic antidepressive agent).
c) Convulsions can be induced in patients treated with BZD for
seizure disorders.
The three above-mentioned situations may be considered relative
contraindications to its use; flumazenil should only be used when it
is strongly indicated. In these situations, it should be given more
slowly than usual (e.g., 0.3 mg intravenously over 3 min, a 3-min
pause, then a further 0.3 mg at the same rate, and so on).
3.13.4 Adverse effects
Various effects have been reported when flumazenil is used in
anaesthesia. These include (in order of decreasing frequency):
nausea, vomiting, tremor, involuntary movements, dizziness, agitation,
discomfort, tears, anxiety, and sensation of cold.
Similar effects occur when flumazenil is used in intensive care,
agitation being the commonest adverse effect. When flumazenil is
administered to patients showing BZD habituation, the following can
occur: anxiety, tenseness, fear, agitation, confusion, convulsions and
myoclonic seizures.
When administered rapidly, flumazenil can cause hypertension,
tachycardia and acute anxiety.
3.13.5 Use in pregnancy and lactation
Even though animal studies showed no embryotoxicity or
teratogenicity at high doses, flumazenil, like any new drug, should be
avoided at the beginning of pregnancy. However, in life-threatening
situations its possible risk to the fetus is probably far outweighed
by its beneficial effects. Isolated administration of flumazenil
during lactation is not contraindicated.
3.13.6 Storage
No special storage conditions are required.
3.13.7 Special risk groups
Acute withdrawal syndrome can occur in patients showing BZD
habituation following therapy or abuse (see section 3.13.3).
3.14 References
Aarseth HP, Bredesen JE, Grynne B, Lyngdal PT, Storstein L, & WiiK-
Larsen E (1988) Benzodiazepine-receptor antagonist, a clinical double
blind study. J Toxicol Clin Toxicol, 26: 283-292.
Abernethy DR, Arend R, Lauven P, & Greenblatt D (1983) Determination
of Ro 15-1788, a benzodiazepine antagonist in human plasma, by gas-
liquid chromatography with nitrogen phosphorus detection.
Pharmacology, 26(5): 285-289.
Amrein R (1986) Clinical documentation on flumazenil. Basel,
Switzerland, Hoffmann-Laroche Laboratory (Unpublished report).
Amrein R, Leishman B, Bentzinger C, & Roncari G (1987) Flumazenil in
benzodiazepine antagonism. Med Toxicol, 2: 411-429.
Bismuth C, Baud FJ, Fournier PE, & Mellerio F (1985) Antagoniste
Ro-15-1788 dans l'intoxication volontaire par benzodiazépines. Valeur
diagnostique et thérapeutique. Etude randomisée sur 40 cas. J Toxicol
Clin Exp, 5: 403-404.
Bismuth C, Baud F, Fournier E, & Mellerio F (1986) Antagoniste Ro
15-1788 dans l'intoxication volontaire par les benzodiazépines.
Valeur diagnostique et thérapeutique. Etude ouverte sur 40 cas. Ann
Méd Interne, 137(4): 363-364.
Birch BRP & Miller RA (1990) Death after flumazenil? Br Med J, 300:
467-468.
Bun H, Duplan V, Crevat-Pisano P, Liurens M, & Durand A (1989) Rapid
determination of the benzodiazepine antagonist flumazenil, Ro 15-1788,
by high performance liquid chromatography. Biomed Chromatogr, 3:
269-271.
Burkhart KK & Kulig KW (1990) The diagnostic utility of flumazenil in
coma of unknown etiology. Ann Emerg Med, 19: 319-321.
Chiolero R, Ravussin P, Chassot PG, Neff R, & Freeman J (1988) Ro
15-1788 for rapid recovery after craniotomy. Ann Fr Anesth Réanim, 7:
17-21.
Clausen TG, Wolff J, Carl P, & Theilgaard A (1990) The effect of the
benzodiazepine antagonist, flumazenil, on psychometric performance in
acute ethanol intoxication in man. Eur J Clin Pharmacol, 38: 233-236.
Darragh A (1981) A study of efficacy of Ro 15-1788 (600mg) as an
antagonist of the sedative effects of Ro 11-3128 in healthy
volunteers. Basel, Switzerland, Hoffmann-Laroche Laboratory
(Unpublished report).
Darragh A, Lambe R, Scully M, Brick I, O'Boyle C, & Dowrie WW (1981)
Investigation in man of the efficacy of a benzodiazepine antagonist,
Ro 15-1788. Lancet, 2(8236): 8-10.
Darragh A, O'Boyle C, & Cambe RI (1982) Antagonist of the central
effects of diazepam in man by Ro 15-1788, a new benzodiazepine
antagonist. Irish J Med Sci, 151(3): 90.
Ellmauer S, Muller H, & Christen M (1986) [Clinical investigations on
the efficacy and safety of the benzodiazepine antagonist Ro 15-1788.]
Beitr Anaesthesiol Intensivmed, 17: 78 (in German).
Forster A, Rouiller M, Morel D, & Gemperle M (1983) Double-blind
randomized study evaluating a specific benzodiazepine antagonist.
Anaestesiology, 59(3): A375.
Gaillard JM & Blois R (1983) Effect of the benzodiazepine antagonist
Ro 15-1788 on flunitrazepam-induced sleep changes. Br J Clin
Pharmacol, 15: 529-536.
Geller E & Niv M (1985) Ro 15-1788, a benzodiazepine antagonist in
the treatment of 34 intoxicated patients. Anaesthesiology, 61(1): 35.
Geller E, Chernilas J, Halpern P, Niv D, & Miller HI (1986)
Hemodynamics following reversal of benzodiazepine sedation with Ro
15-1788 in cardiac patients. Anaesthesiology, 65: A49 (Abstract).
Haaefely W (1983) Antagonist of benzodiazepine. Encéphale, 9:
143B-150B.
Hofer P & Scollo-Lavizzari G (1985) First clinical investigation of
the benzodiazepine antagonist Ro 15-1788 in comatose patients. Arch
Intern Med, 145(4): 663-664.
Jensen S, Kirkegaard L, & Andersen BN (1985) Benzodiazepine
antagonist Ro 15-1788: randomized clinical investigation of Ro 15-1788
in reversing the central effects of flunitrazepam. Acta Anaesthesiol
Scand, 29(Suppl): 89.
Knudsen L & Kirkegaard L (1986) Benzodiazepine antagonist Ro 15-1788:
antagonism of diazepam sedation in out-patients undergoing
gastroscopy. Beitr Anaesthesiol Intensivmed, 17: 268.
Laurian S, Gaillard JM, & Le PK (1984) Dose effects of the
benzodiazepine antagonist Ro 15-1788 on the auditory evoked
potentials. In: Abstracts of the 14th Congress of the Collegium
Internationale Neuropsychopharmacologicum, Firenze, Italy, 19-23 June
1984. Fidia Research Biomedical Information (Abstract No. 33).
Laurian S, Despland PA, Gaillard JM, Grasset F, & Le PK (1987)
Psychotropic drugs and EEG organization. Int J Neurosci, 32(12): B370.
Lupolover R (1983) Evaluation of the efficacy and safety (tolerance)
of Ro 15-1788 intravenously administered in normal healthy volunteers.
Basel, Switzerland, Hoffmann-Laroche Laboratory (Unpublished report).
Lauven PM, Schwilden H, & Stoeckel H (1985) The effects of a
benzodiazepine antagonist Ro 15-1788 in the presence of stable
concentrations of midazolam. Anesthesiology, 63: 61-64.
Lheureux P & Askenasi R (1988) Specific treatment of benzodiazepine
overdose. Hum Toxicol, 7: 165-170.
Lim AG (1989) Death after flumazenil. Br Med J, 299: 858-859.
Marchant B & Wray R (1989) Flumazenil causing convulsions and
ventricular tachycardia. Br Med J, 299: 860.
Mohler H, Pieri L, Polc P, Bonetti EP, Cumiu R, Schaffner R, & Haefely
W (1981) Selective antagonists of benzodiazepines. Nature(Lond), 290:
514-516.
Pellerin F (1986) Pharmaceutical documentation on flumazenil. Basel,
Switzerland, Hoffmann-Laroche Laboratory (Unpublished report).
Richard P, Autret E, Bardol J, Soyez C, Barbier P, Jonville AP, &
Ramponi N (1991) The use of flumazenil in a neonate. J Toxicol Clin
Toxicol, 29: 137-140.
Ritz R, Zuber M, Elsasser S, & Scollo-Lavizzari G (1990) Use of
flumazenil in intoxicated patients with coma. A double-blind placebo-
controlled study in ICU. Intensive Care Med, 16: 242-247.
Rocher JP (1984) Benzodiazepines. Lyon Pharm, 35(3): 149-167.
Roncari G, Ziegler QH, & Guentert TW (1986) Pharmacokinetics of the
new benzodiazepin antagonist Ro 15-1788 in man following intravenous
and oral administration. Br J Clin Pharmacol, 22: 421-428.
Rouiller M, Forster A, & Gemperle M (1987) Double blind randomized
study evaluating a specific benzodiazepine antagonist. Ann Fr Anesth
Réanim, 6: 1-6.
Scoepf J, Laurian S, Le PK, & Gaillard JM (1984) Intrinsic activity of
the benzodiazepine antagonist Ro-1788 in man: an electrophysiological
investigation. Pharmacopsychiatry, 17: 79-83.
Scollo-Lavizzari G (1984) The anticonvulsant effect of the
benzodiazepine antagonist, Ro 15-1788: an EEG study in 4 cases. Eur
Neurol, 23: 1-6.
Timm U & Zell M (1983) Determination of the benzodiazepine antagonist
Ro 15-1788 in plasma by high performance liquid chromatography with UV
detection. Arzneimittelforschung, 33(1): 358-362.
Tolksdorf W, Pirwitz A, & Pfeiffer D (1986) [Effects and side-
effects of the benzodiazepine antagonist Ro 15-1788 following mixed
anaesthesia in association with flunitrazepam, compared with a
placebo: a double-blind study.] Beitr Anaesthesiol Intensivmed, 35:
121 (in German).
Wood C, Bismuth C, Oriot D, Devictor D, & Juault G (1987)
Réversibilité d'un coma par benzodiazépine par perfusion de flumazenil
chez un enfant. Presse Méd, 16: 1483.
Ziegler WH & Schalch E (1983) Antagonism of benzodiazepine-induced
sedation in man. In: In sleep. Proceedings of the 6th European
Congress of Sleep Research, Zurich, 1982. Basel, Karger, pp 427-429.
4. DANTROLENE SODIUM
4.1 Introduction
Dantrolene sodium, a hydantoin-furan derivative, causes skeletal
muscle relaxation by preventing calcium flux across the sarcoplasmic
reticulum. It was first synthesized in 1967 and the initial use was
to treat muscle spasm (Dykes, 1975). More recently dantrolene has
been used successfully in the treatment of malignant
hyperpyrexia/hyperthermia and neuroleptic malignant syndrome,
hypercatabolic syndromes that have previously been associated with
high mortality rates.
Malignant hyperthermia results from a genetic susceptibility to
certain anaesthetic agents (autosomal dominant myopathy), and is
usually fatal unless appropriate treatment is given. However,
provided the diagnosis is made early and treatment with dantrolene and
necessary supportive measures is given at once, there is rapid
resolution of the hyperthermia. Signs of malignant hyperthermia may
occur from minutes to 1-2 h after the induction of anaesthesia and are
thought to result from an acute influx of calcium into the muscle
cytoplasm from the sarcoplasmic reticulum, which results in abnormal
muscle contraction, hypermetabolism, hyperthermia and muscle damage.
In patients with a family history or previous episodes of
malignant hyperthermia, prophylactic treatment with dantrolene prior
to anaesthesia has been advocated (Shime et al., 1988). The
prophylactic use of dantrolene with susceptible patients is, however,
controversial. Those opposing this approach advocate the agreed
safety precautions and the availability of dantrolene in order to
intervene promptly if any crisis occurs (Harrison, 1988; Hackl et al.,
1990). At present, most anaesthesiologists do not use dantrolene
prophylactically in these patients.
The predisposition for malignant hyperthermia is probably
heterogenous and not only linked to the CRC (calcium release channel)
gene on chromosome 19 (Fagerlund et al., 1992). It is therefore
premature to use DNA markers flanking this gene as the major test to
diagnose susceptibility to the malignant hyperthermia syndrome,
although this has been suggested (Healy et al., 1991). Predisposition
to the disease is still best determined through a halothane- and
caffeine-induced contracture test on a skeletal muscle biopsy
(MacLennan et al., 1990).
Neuroleptic malignant syndrome occurs in 0.2-1% of patients
taking neuroleptics, especially when haloperidol and the depot
fluphenazines are used (Pope et al., 1986). One component of the
syndrome appears to be sustained extrapyramidal rigidity causing
hyperthermia and muscle necrosis (rhabdomyolysis). The syndrome may
last for 10-14 days or 4 weeks after cessation of oral or depot
neuroleptics, respectively.
Dantrolene has also been used successfully in the treatment of a
few cases of heat-stroke (Lydiatt & Hill, 1981), which has many
similarities to malignant hyperthermia.
This monograph will restrict itself to the use of dantrolene in
the treatment of drug-induced hypercatabolic syndromes.
4.2 Name and Chemical Formula of Antidote
Dantrolene sodium
IUPAC name: 1-[[5-( p-nitrophenyl)
furfurylidene]amino]hydantoin sodium
hydrate
Empirical formula: C14H9N4NaO5 (anhydrous salt)
Relative molecular mass: 336 (anhydrous salt)
The hydrated salt contains approximately 15% water (3´ moles) and
has a molecular weight of 399.
CAS numbers: 7261-97-4 Dantrolene
14663-23-1 Dantrolene sodium,
anhydrous
24868-20-0 Dantrolene sodium salt,
hemiheptahydrate
(Martindale, 1989)
Molecular structure:
2.3 Physico-chemical Properties
Naloxone hydrochloride has a melting range of 200-205 °C. It is
soluble in water, dilute acids and strong alkalis, and is slightly
soluble in alcohol but practically insoluble in ether. Aqueous
solutions are acidic (pH 3 to 4.5) (United States Pharmacopeia, 1980)
and an 8.08% solution in water is isotonic with serum (Hassan et al.,
1985). A 25% solution of naloxone hydrochloride rotates light between
-170 and -181. Naloxone crystals from ethyl acetate have a specific
optical rotation at 20 °C ([alpha]D20; 9.3 g/l chloroform) of
-194.5 ° (Windholz, 1983).
Naloxone has a pKa (20 °C) values for the nitrogen and phenolic
H groupings of 7.94 and 9.44, respectively (Kaufman et al., 1975).
On drying at 105 °C, the anhydrous form loses not more than 0.5%
and the hydrated form not more than 11% of its weight.
The solution for injection is made up in water and should be
protected from light. Naloxone can be diluted in 0.9% saline or 5%
dextrose and should then be used within 24 h. It should not be mixed
in solutions containing metasulfite, metabisulfite, or long-chain or
high relative molecular mass anions, or in those with an alkaline pH.
2.4 Pharmaceutical Formulation and Synthesis
Three synthetic routes for the production of naloxone have been
reported (Hassan et al., 1985). Oxymorphone is a starting point for
two of the synthetic processes and 14-hydroxycodeinone for the third.
Noroxymorphone hydrochloride is a potential impurity from the
manufacturing process.
2.5 Analytical Methods
2.5.1 Quality control
Naloxone hydrochloride can be assayed by gas chromatography with
flame ionization detection (United States Pharmacopeia, 1980).
2.5.2 Identification
About 150 mg of the unknown substance is dissolved in 25 ml of
water and a few drops of 6N ammonium hydroxide are added. Three 5-ml
portions of chloroform are used for extraction and the extract is
filtered. The filtrate is collected, evaporated to dryness using a
steam bath, and dried at 105 °C for one hour. The infrared absorption
spectrum of a 1-in-50 solution of the residue obtained in chloroform
will have maxima at the same wavelengths as those of a similar
solution of naloxone reference standard.
The addition of one drop of ferric chloride solution to 1 ml of
a 1-in-100 solution of naloxone hydrochoride results in a clear
purplish-blue colour.
2.5.3 Quantification of the antidote
Assay methods for naloxone in biological fluids employing
gas-liquid chromatography (GLC) (Meffin & Smith, 1980),
radio-immunoassay (RIA) (Berkowitz et al., 1975; Hahn et al., 1983)
and high-performance liquid chromatography (HPLC) (Asali, 1983; Terry
et al., 1984) have all been reported. The GLC method involves
derivatization and the specific antibody for the RIA is not widely
available. The HPLC methods reported appear sensitive and
reproducible, and are therefore probably the methods of choice.
2.5.4 Analysis of toxic agents
In the majority of cases in which naloxone is used as an
antidote, there is no way of measuring the level of the opioid poison.
Present assay techniques for many opiates are difficult, and RIA
suffers from lack of specificity in many cases. Some opiates, e.g.,
morphine, also appear to have active metabolites (Bodd et al., 1990).
The most widely used method for opioid detection is RIA of urine.
2.6 Shelf-life
The shelf-life of naloxone for intravenous injection in temperate
countries is 3 years and has a similar length in tropical countries.
2.7 General Properties
Naloxone is a specific opioid antagonist (Martin, 1976) and it is
for this reason that it is used in the treatment of poisoning. There
are reports that it may reverse the central effects of ethanol and BZD
poisoning in man. However, these are experimental uses that remain
unproven, and any observed effects probably reflect the involvement of
endogenous opioids in the nonspecific depressant action of those
agents (McNicholas & Martin, 1984).
2.8 Animal Studies
2.8.1 Pharmacodynamics
Naloxone is a competitive antagonist at opiate receptors, and
appears to be effective at all three types of receptor (mu, kappa and
sigma) (Martin, 1976). It does not produce habituation in animal or
human models of opiate tolerance and appears to be free of agonist
activity in most laboratory test models (Jasinski, 1967; McNicholas &
Martin, 1984). It produces a parallel shift in the in vitro dose-
response effects of pure agonist opioids, such as morphine, and
partial agonists, such as pentazocine (Smits & Takemori, 1970),
buprenorphine and dextropropoxyphene.
Since the range and relative quantities of opioid receptors vary
in different animal tissues, a range of concentrations of naloxone is
required to antagonize opioid effects in different test systems.
Confusion has arisen as to whether naloxone is a pure antagonist.
This is because some opioid receptors act as modulators and enhance
nociceptive stimuli. Thus, in some animal models naloxone appears to
possess agonist effects, but this is in fact incorrect (Sawynok et
al., 1979). Naloxone has also been observed in some experiments to
antagonize the antinociceptive effects of some non-opiate drugs.
Again it seems likely that this reflects an involvement of opioid
receptors in the mechanism of action of these drugs (Sawynok et al.,
1979). However, in a recent study in rats, Kotlinska & Langwinski
(1990) failed to find any evidence for the participation of the opioid
system in the mediation of acute ethanol effects in rats.
Naloxone has been reported to either decrease or have no
influence on barbiturate-induced anaesthesia. This paradox may be a
result of the dose-response relationship of the effects of naloxone,
which at high doses may have a potentiating effect (Sawynok et al.,
1979). Naloxone has some activity as a GABA antagonist and may thus
have convulsant activity. However, this is likely to be at much
higher concentrations that those encountered clinically (Dingledine et
al., 1978), since in mice a dose of 100 mg/kg was required to produce
convulsions.
Naloxone has also been shown to have a number of biochemical
effects in the rat, including inhibition of lipolysis and a subsequent
increase in circulating free tryptophan (Badawy et al., 1983).
2.8.2 Pharmacokinetics
Naloxone appears to be readily absorbed after oral administration
but undergoes extensive first-pass hepatic metabolism, which results
in a very low bioavailability (Misra, 1978). Studies of the
pharmacokinetics of intravenous naloxone have been performed in a
variety of animal species including the rat, rabbit and dog. Many of
these studies are based on radio-immunoassay of naloxone.
The serum concentration of naloxone found 5 min after injection
was similar (5 mg/kg) in the rat and the dog (Ngai et al., 1976; Pace
et al., 1979). The half-life of the parent drug in the rat (30 min)
was approximately half that in the dog (71 min).
Ngai et al. (1976) also examined the brain:serum ratio of
naloxone and found this to vary in the rat between 2.7:1 and 4.6:1.
Intravenously administered naloxone acts rapidly on the brain. The
brain:serum ratio was higher, however, when the naloxone was
administered subcutaneously. These workers also studied, in a
parallel group of animals, the distribution of morphine and noted that
the brain:serum ration was 1:10.
The initial distribution of naloxone may account for the rapid
onset of its reversal of opiate effects when it is given
intravenously. The major metabolite of naloxone is the glucuronide.
Naloxone-3-glucuronide has been found, for example, in the rabbit
(Fujimoto, 1969). A conjugated 6-hydroxy product of naloxone,
N-allyl-14-hydroxy-7,8-dihydronormorphine-3-glucuronide was
identified in the chicken by Fujimoto (1969); this conjugate was also
identified in the rabbit by Weinstein et al. (1974) but only in small
amounts.
The relatively short action of naloxone appears to result from
the ease with which it enters the brain after intravenous dosing and
the subsequent rapid redistribution, elimination and consequent fall
in brain naloxone levels (Berkowitz, 1976).
Hydroxylated metabolites of naloxone appear to possess narcotic
antagonist activities, but their potencies are much weaker than the
parent compound. Thus they are unlikely to be of significance in view
of the small amounts produced (Fujimoto et al., 1975).
The distribution of naloxone has not been found to be altered by
a 25-fold range of morphine concentration in the rat (Fishman et al.,
1975).
2.8.3 Toxicology
Acute toxicity studies with naloxone have been performed in mice,
rats and dogs. The LD50 for intravenous administration was 150
mg/kg in mice, 109 mg/kg in rats and 80 mg/kg in dogs (Social Welfare
Board, 1976). For 24-h-old rats the LD50 was 260 mg/kg when given
subcutaneously (Blumberg et al., 1966). The maximum nontoxic
subcutaneous dose in rats was found to be of the order of 50 mg/kg
(Blumberg et al., 1966). This dose was tolerated for 24 days, whereas
200 mg/kg resulted in tremor, convulsions and salivation.
Daily doses of 0.2 mg/kg given intravenously to dogs for 16 days
and 5 mg/kg given subcutaneously to monkeys for 30 days caused no
toxicity. However, a subcutaneous dose of 20 mg/kg resulted in
lethargy and tremor in monkeys.
No teratogenic effects were observed in mice, rats or rabbits
when naloxone was given parenterally over the period of organogenesis
(Social Welfare Board, 1976). No studies on mutagenicity have been
published.
2.9 Volunteer Studies
Studies of the pharmacokinetics and pharmacodynamics of naloxone
have been performed in volunteers.
2.9.1 Pharmacokinetics
Using an RIA assay, the pharmacokinetics of naloxone were found
to fit a two-compartment model, with a rapid distribution phase and a
slower elimination phase, having a half-life of 64 min (Ngai et al.,
1976). More recent studies using HPLC to assay naloxone suggest that
the apparent volume of distribution, half-life and clearance all show
differences within groups of normal volunteers. Thus Aitkenhead et
al. (1984) reported a mean apparent volume of distribution at steady
state of 3.65 l/kg (range 1.43-7.05 l/kg) and a mean half-life of
151.2 min (range 47.1-313.2 min). Using an HPLC assay, Goldfrank et
al. (1986) found less variability in patients (half-life 28-55 min).
The kinetics of naloxone in infants appear similar to those in
adults (Stile et al., 1984).
Orally administered radiolabelled naloxone undergoes extensive
first-pass metabolism in normal subjects (Fishman et al., 1973).
After intravenous administration, most (70%) of the radioactivity was
recovered in urine, the major part of which was conjugated as the
glucuronide. In addition other metabolites were found in small
quantities, i.e. the glucuronide conjugates of 7,8-dihydro-14-hydroxy-
normorphine, and N-allyl-7,8-dihydro-14-hydroxy-normorphine
(Weinstein et al., 1971).
As a consequence of the high hepatic clearance of naloxone and
relatively weak agonist activity of its metabolites, it is unlikely
that dose adjustments would be necessary in cases of renal failure.
Naloxone is only 54% protein-bound in adult plasma (61.5% in fetal
plasma), and this binding is not concentration-dependent over the
range 9 ng/ml to 2.5 µg/ml (Asali & Brown, 1984). Thus protein-
binding interactions seem unlikely.
The elimination of naloxone might be altered in patients with
liver disease, but no studies appear to have been performed.
2.9.2 Pharmacodynamics
Studies have been conducted on the duration of action and potency
of naloxone in reversing respiratory depression induced by morphine
(intravenous doses of 5 mg plus 10 mg) in volunteers (Kaufman et al.,
1981). The effect of naloxone against this therapeutic dose of
morphine reached a peak at around 30 min, which was equatable with the
probable peak in brain concentration. It should be noted that the
times of onset and peak effect of naloxone differed. The duration of
action of naloxone appeared to be about 1.5 h in this experimental
model.
Johnstone (1974) examined the effects of an infusion of naloxone
in volunteers who had received 2 mg/kg morphine intravenously and been
anaesthetized for 5 h. Intravenous naloxone given to these volunteers
at a rate of 40 µg/kg over a 10-h period reversed the central
depressant effects of morphine on respiratory function (measured by
CO2 responsiveness) and higher functions (assessed by a vigilance
test). No tachyphalaxis to the effects of naloxone was observed over
this period (Johnstone et al., 1974).
It has been suggested that ethanol may exert some of its effects
via the endogenous opiate system, as illustrated by the study by
Jeffferys et al. (1980) and Jeffcoate et al. (1979) where naloxone was
found to antagonize some of the ethanol effects. However, these
findings could not be confirmed by Handal et al. (1983) or Nuotto et
al. (1984). In the latter study, the effect of naloxone on ethanol-
induced impairment of psychomotor performance was first studied in two
placebo-controlled, double-blind, cross-over trials in 17 healthy male
volunteers. The main conclusion was that naloxone (intravenous doses
of 0.4 plus 2 mg) had no significant antagonizing effects on the
impairment induced by ethanol (1.5 g/kg). However, a slight but
significant effect on ethanol-induced nystagmus was noted. A placebo-
controlled, double-blind study was subsequently conducted on male
alcoholics admitted for acute ethanol intoxication (the mean blood
ethanol level was 2.9 g/l (64 mmol/l)). In this case, neither naloxone
(intravenous doses of 0.4 plus 2 mg; n=11) nor saline (n=7) had any
effect, as judged from a clinical inebriation test (Nuotto et al.,
1984).
2.9.3 Effects of high doses of naloxone
Naloxone has been administered to healthy volunteers at dose
levels of 0.3-4 mg/kg. These high dose levels produced dose-dependent
dysphasia and memory impairment. In addition, increases in blood
pressure and respiratory rate were noted, together with increases in
cortisol and growth hormone levels (Cohen et al., 1983). These
findings have been used to support the hypothesis that endogenous
opioids play a normal regulatory physiological role, but obviously
have potential therapeutic implications if large doses of naloxone are
used to treat poisoned patients.
2.10 Clinical Studies - Clinical Trials
Naloxone has been investigated in clinical studies on both
patients who have received a therapeutic dose of an opiate (see
section 2.9) and those who have been poisoned with opiates. Since
naloxone is a competitive antagonist, the dose required to reverse the
clinical effects of a specific opiate will depend on the dose of the
opiate, its duration of action, and its pharmacological properties,
particularly whether it has partial agonist activity or shows
selectivity at one type of opioid receptor subgroup (Martin, 1976).
2.10.1 Effects in therapeutic use of opioids
An alternative method of studying the response to naloxone was
reported by Drummond et al. (1977). They studied patients who had been
anaesthetized and had received the synthetic opiate fentanyl.
Naloxone produced a dose-dependent increase in respiratory function
(measured as minute volume or respiratory rate) with intravenous doses
of 0.1, 0.2 and 0.4 mg.
Hatano et al. (1975) reported an open study on 80 patients
undergoing a variety of surgical procedures including cardiopulmonary
bypass. Premedication included pethidine (meperidine) and induction
was achieved with pentazocine and diazepam. The doses of pentazocine
in males were 2 mg/kg and females 1.5 mg/kg, and those of diazepam
were 0.4 and 0.3 mg/kg, respectively. The authors used a stepwise
increment of naloxone (0.2-mg intravenous boluses) to achieve reversal
of the opiate effect of pentazocine at the end of the operative
procedure and noted a stepwise reversal of the opiate effects in their
patients as the opiate dose was increased (the average total dose
given was 2.5 mg/kg body weight).
The duration of action of naloxone in reversing the effects of
morphine (5 or 10 mg, intramuscular) in patients recovering from
surgery is relatively short (Longnecker et al., 1973). The authors
suggested that the use of a combination of intravenous and
intramuscular naloxone might be an appropriate regimen in the post-
operative situation; this has also been suggested for the treatment
of acute overdoses in heroin addicts (see sections 2.12.2 & 2.13.2).
2.10.2 Effects in acute opioid poisoning
Two important studies have demonstrated the efficacy of naloxone
in reversing opiate poisoning. Evans et al. (1973) reported a study
in which naloxone (0.4-1.2 mg, intravenous) resulted in recovery of
consciousness within 1-2 min in nine patients with a history of opiate
ingestion. This was associated with improvement in respiratory
function in the six patients in whom this could be measured with
minute volume and respiratory rate. The opiates taken by these
patients were reported as dipipanone (3), pethidine (2),
dihydrocodeine (2), pentazocine (1) and heroin (1). In contrast, none
of 13 patients overdosed with a variety of other central nervous
system depressants showed improvement after having been given a total
intravenous dose of 1.2 mg naloxone. This rapid and clear benefit of
therapy was also reported by Buchner et al. (1972), who studied the
effects of naloxone (0.005 to 0.01 mg/kg) in 10 children with
methadone poisoning. Although they did not study a control group,
they did confirm the presence of methadone in biological fluids in
some of their patients. These authors stress the importance of an
adequate period of observation for patients poisoned with long-acting
opiates and the necessity of repeated doses of naloxone.
Since the onset of the effects of naloxone is so rapid, it has
proved relatively easy to confirm its effectiveness in opiate
poisoning at restoring consciousness and improving respiration.
Further extensive clinical trials in opiate poisoning have, therefore,
not been performed.
Henry & Volans (1984) have stressed the importance of classifying
drugs correctly as opioids. A list of opioids is a useful reminder
(Table 1) that agents such as loperamide and diphenoxylate may produce
significant systemic toxicity in overdose.
One particular aspect of naloxone use that requires consideration
is that of the most appropriate dosage regimen. Early human studies
confirmed that the duration of action of naloxone was shorter than
might have been expected from its plasma half-life (Berkowitz et al.,
1975). The long duration of action of some opiates is also a factor
in the need to repeat the initial dose of naloxone in poisoned
patients (Gober et al., 1979). As an alternative to repetitive
dosing, several research workers have suggested that intravenous
loading doses followed by a steady-state infusion of the drug would be
appropriate both in children (Gourlay & Coulthard, 1983; Tenenbein,
1984) and in adults (Bradberry & Raebel, 1981; Goldfrank et al., 1986)
suffering opiate poisoning. These regimens have appeared safe and
effective in clinical use, but do not obviate the need for close
monitoring during treatment of respiratory function, conscious level
and cardiovascular function. It is important to remember that some
synthetic opioids, e.g., dextropropoxyphene, have been reported to
produce toxic effects at high doses, which are not reversible by
naloxone (Barraclough & Lowe, 1982). These effects may be due to a
direct action of dextropropoxyphene on cardiac cell membranes.
Table 1. Alphabetical list of opioid drugsa
Alletorphine Levorphanol
Alphaprodine Loperamide
Anileridine Meptazinol
Azidomorphine Methadone
Bezitramide Metofoline
Buprenorphine Morphine
Butorphanol Nalbuphine
Codeine Norpipanone
Dextromoramide Opium
Dextropropoxyphene Oxycodone
Diamorphine (Heroin) Oxymorphone
Difenoxin Papaveretum
Dihydrocodeine Pentazocine
Diphenoxylate Pethidine (Meperidine)
Dipipanone Phenadoxone
Ethoheptazine Phenazocine
Ethylmorphine Phenoperidine
Etorphine Piminodine
Fentanyl Piritramide
Hydrocodone Thebacon
Hydromorphone Tilidate
Ketobemidone Tramadol
Levomethadyl Trimeperidine
a From Martindale (1982). Some of these drugs may be marketed as
part of a combination preparation.
2.11 Clinical Studies - Case Reports
Individual published case reports have confirmed efficacy for the
majority of opiates (Handal et al., 1983). In patients who are
narcotic addicts, naloxone may precipitate features of acute opiate
withdrawal. Doses of up to 20 mg naloxone have been used in children
without associated adverse effects (Handal et al., 1983).
If patients with acute renal failure are given morphine over
several days for various reasons (e.g., for sedation while on a
respirator), opioid toxicity may occur due to accumulation of the
active metabolite morphine-6-glucuronide, which is renally excreted
(Bodd et al., 1990). In such cases, the opioid toxicity may last for
up to two weeks after the cessation of morphine therapy, and the
patient will need naloxone infusion in order to avoid respiratory
depression.
2.11.1 Naloxone in clonidine poisoning
Clonidine hydrochloride is a central and peripheral
alpha-adrenergic antagonist that is still used in the treatment of
hypertension. It has also been suggested for the treatment of opiate
withdrawal (Gold et al., 1980). The mechanism for this effect and for
the claimed effect of naloxone in some cases of clonidine poisoning
(North et al., 1981; Kulig et al., 1982) is not clear, but the
involvement of endogenous opioids has been suggested. However, the
effect of naloxone in clonidine poisoning could not be confirmed by
Banner et al. (1983). In a retrospective study of 47 consecutive
children admitted for clonidine poisoning (Wiley et al., 1990), only
3 out of the 19 given naloxone showed a temporary response. One child
had an episode of severe hypertension associated with naloxone
administration (0.1 mg/kg). Thus, there is no clear documentation for
the beneficial effect of naloxone in clonidine poisoning.
2.12 Summary of Evaluation
2.12.1 Indications
Naloxone has been reported to significantly antagonize acute
opioid toxicity and opioid effects within anaesthesia. Its high
therapeutic index and possible beneficial effect in other poisonings
allow for diagnostic use in critically ill patients when opioid
poisoning may be a differential diagnosis.
2.12.2 Advised routes and dose
In patients with definite opiate poisoning, naloxone should be
given by the intravenous route until an improvement in conscious level
and respiration is observed. This may involve the administration of
several milligrams of naloxone if partial opioid agonists are given,
but 0.8-1.2 mg is usually sufficient in morphine or heroin poisonings.
It is important to stress that a pharmacologically active dose of
naloxone in opiate poisoning may be more than that normally
recommended in anaesthetic practice.
In patients with suspected opiate poisoning, an intravenous
injection of up to 2 mg naloxone should be administered and the
patient's response closely monitored. If there is improvement in
conscious level, respiratory rate or cardiovascular parameters,
further doses of naloxone should be administered. The effect of
naloxone should be visible within 1 to 2 min after administration.
Once a patient has regained consciousness, it is necessary to
continue to monitor respiration and cardiovascular status at regular
intervals. In the patient who has taken a large opiate overdose or an
overdose of a long-acting opiate, it may be necessary to repeat dosing
with naloxone. This may be conveniently done by establishing an
intravenous infusion of naloxone. A guide to the required dosage has
been suggested by Goldfrank et al. (1986). From studies of the
pharmacokinetics of naloxone in patients suffering opiate poisoning,
they calculated that an hourly infusion of two-thirds of the dose
required initially to reverse the effects of the opiate would maintain
naloxone levels at approximately those present 30 min after the
initial bolus administration.
Another approach to opioid poisoning that may sometimes be
usefully employed in addicts is to give 0.8-1.2 mg naloxone
intramuscularly before awakening the patient with an intravenous
naloxone dose of 0.4-0.8 mg (higher doses are rarely needed) (personal
communication by D. Jacobsen, 1991). This has been shown to be a
useful practical approach, since many addicts leave the hospital
immediately following the effect of the intravenous dose. Since
naloxone has a shorter duration of action than the opiate, patients
are commonly readmitted within one hour with miosis, coma and impaired
respiration. This approach to treatment, however, requires adequate
ventilatary support for the patient because of the short delay before
the intravenous dose is given.
Naloxone may also be given as a continuous intravenous infusion
(about 0.5 mg/h in isotonic saline) to counteract effects of morphine
metabolites in patients with acute renal failure (Bodd et al., 1990).
2.12.3 Other consequential or supportive therapy
Since many of these patients suffer from impaired respiration or
respiratory arrest, it is extremely important to give oxygen and to
support ventilation immediately while waiting for naloxone to be
available for injection. If ventilation is under control and cyanosis
is regressing, one should consider giving an intramuscular dose of
naloxone before the intravenous dose (see section 2.12.2).
Pulmonary congestion or oedema is occasionally seen in opioid
(heroin) poisoning. It is usually transient and responds to supportive
therapy (oxygen and ventilation support) and naloxone.
2.12.4 Areas where there is insufficient information to make
recommendations
There are anecdotal reports of beneficial effect of naloxone in
other types of acute poisoning, e.g., with ethanol or clonidine. In
the case of ethanol, these results have not been confirmed in well-
controlled studies on volunteers or in intoxicated patients (Nuotto et
al., 1984). The claimed effect in clonidine poisoning has also been
challenged (Wiley et al., 1990). There are insufficient data to
recommend the use of naloxone in poisonings other than those involving
opioids.
2.12.5 Proposals for further studies
Studies of the effect of naloxone in other acute poisonings
should be encouraged. It could, however, be argued that enough studies
have been performed on the use of naloxone in ethanol intoxication to
rule out a possible beneficial effect. On the other hand, there is
certainly a lack of controlled studies on the possible effect of
naloxone in clonidine poisoning.
If effects of naloxone are observed in patients assumed to have
been poisoned by non-opioids, urine specimens should be collected and
analysed by RIA for presence of opioids. Otherwise such "case
reports" are of little value.
2.12.6 Adverse effects
Naloxone possesses a high therapeutic index, but it may provoke
withdrawal signs and symptoms, e.g., seizures, in (heroin) addicts.
Other adverse reactions, as described below, are very rarely seen.
Cardiac arrhythmias and, in particular, ventricular fibrillation
have resulted from rapid reversal of opiate effects with naloxone.
Such events may be a particular problem in patients who have recently
undergone surgery or those habituated to opiates (Cuss et al., 1984).
These reactions may result from a release of sympathetic transmitters,
since a rise in blood pressure and tachycardia have also been
demonstrated.
Some cases of pulmonary oedema following naloxone use in
anaesthetic practice have been reported, but it is unclear in this
situation which is the responsible agent: the anaesthetic, the opiate
or the antagonist (Partridge & Ward, 1986).
2.12.7 Restrictions of use
The fear of provoking withdrawal signs and symptoms should not
hinder use of naloxone in those who need it clinically.
2.13 Model Information Sheet
2.13.1 Uses
Naloxone is indicated in the management of opiate poisoning, both
definite and suspected. Opiate poisoning should be considered in
comatose patients with impaired respiration. Miosis is an unreliable
sign and is not required for a diagnosis of opioid poisoning. The
high wide therapeutic index of naloxone allows its use when a
diagnosis of opioid poisoning is uncertain.
2.13.2 Dosage and route
Since naloxone is a competitive antagonist of opiate poisoning,
there can be no absolute guidelines on dosage. Naloxone should be
given intravenously, in successive doses of 0.4 to 2.0 mg, until the
desired response has been obtained. It should be noted that to
reverse the effects of partial agonists/antagonists, e.g.,
pentazocine, buprenorphine and dextropropoxyphene, much larger doses
may be required, and it may prove impossible to reverse the effects of
buprenorphine.
Failure to respond to a total dose of 10 mg usually indicates: a)
that poisoning is not due to opiates; b) that poisoning is due to a
partial agonist/antagonist; or c) that hypoxic brain damage has
occurred. It should be noted that dextropropoxyphene has been reported
to produce cardiac toxicity that is not reversible by naloxone
administration.
The duration of action of naloxone is short; careful monitoring
is required and repeated doses may be necessary. The alternative is
an intravenous infusion of naloxone. The use of an hourly infusion of
two-thirds of the dose of naloxone required to resuscitate the patient
has been reported to be effective, but dosage should be always
titrated to the individual patient.
Another alternative, which may be appropriate for opiate addicts,
is to give naloxone (0.8-1.2 mg) intramuscularly before waking the
patient with an intravenous dose of 0.4-0.8 mg. However, adequate
ventilatory support must be given. The patient then has a "depot" of
antidote in case he/she departs soon after the initial treatment (as
many addicts do).
The dose given to children should be reduced according to body
weight (0.01 mg/kg initially).
2.13.3 Precautions/contraindications
Naloxone may induce symptoms and signs of acute opiate withdrawal
in addicts. If seizures occur they are best controlled with diazepam
(10-30 mg, intravenously). No dosage alterations seem necessary in
the case of changes in renal function. The dose in children should be
adjusted on a body-weight basis to that used in adults.
Appropriate protective precautions need to be taken by hospital
staff in the case of opiate addicts, bearing in mind the risk of
infection from blood-borne diseases such as hepatitis B and human
immunodeficiency virus (HIV).
2.13.4 Adverse effects
Naloxone has a very high therapeutic index and adverse effects
are rarely seen. Ventricular arrhythmias including ventricular
fibrillation have been reported following rapid reversal of severe
opiate intoxication. This may be avoided if oxygen and adequate
ventilatory support are also given. The management of withdrawal
symptoms in addicts is discussed in section 2.13.3.
2.13.5 Use in pregnancy and lactation
Naloxone is not teratogenic in animals, but no relevant human
data exist. Naloxone treatment does not appear to be a
contraindication to breast feeding, although the opiate poisoning
being treated may itself be a contraindication.
2.13.6 Storage
Naloxone for injection should be stored protected from light.
Its shelf-life is 3 years.
2.14 References
Aitkenhead AR, Derbyshire DR, Pinnock CA, Achola K, & Smith G (1984)
Pharmacokinetics of intravenous naloxone in healthy volunteers.
Anaesthesiology, 61: A381.
Asali LA (1983) Determination of naloxone in blood by high
performance liquid chromatography. J Chromatogr, 278: 329-335.
Asali LA & Brown KF (1984) Naloxone protein binding in adult and
fetal plasma. Eur J Clin Pharmacol, 27: 459-464.
Badawy AA-R, Evans M, Punjani NF, & Morgan CJ (1983) Does naloxone
always act as an opiate antagonist? Life Sci, 33(Suppl 1): 739-742.
Banner W, Lund ME, & Clawson L (1983) Failure of naloxone to reverse
clonidine toxic effect. Am J Dis Child, 137: 1170-1171.
Barraclough CJ & Lowe RA (1982) Failure of naloxone to reverse the
cardiotoxicity of Distalgesic overdose. Postgrad Med J, 58: 667-668.
Bell EF (1975) The use of naloxone in the treatment of diazepam
poisoning. J Paediatr, 87: 803-804.
Berkowitz BA, Ngai SH, Hempstead J, & Spector S (1975) Disposition of
naloxone: Use of a new radio-immunoassay. J Pharm Exp Ther, 195(2):
499-504.
Berkowitz BA (1976) The relationship of pharmacokinetics to
pharmacological activity: Morphine, methadone and naloxone. Clin
Pharmacokinet, 1: 219-230.
Blumberg H, Wernick T, Dayton HB, Hansen RE, & Rapaport DN (1966)
Toxicological studies on the narcotic antagonist naloxone. Toxicol
Appl Pharmacol, 8: 335.
Bodd E, Jacobsen D, Lund E, Ripel A, Morland J, & Wiik-Larsen E
(1990) Morphine-6-glucuronide might mediate the prolonged opioid
effect of morphine in acute renal failure. Hum Exp Toxicol, 9:
317-321.
Bradberry JC & Raebel MA (1981) Continuous infusion of naloxone in
the treatment of narcotic overdose. Drug Intell Clin Pharm, 15:
945-950.
Buchner LH, Cimino JA, Raybin HW, & Stewart B (1972) Naloxone
reversal of methadone poisoning. NY State J Med, 72: 2305-2309.
Christensen KN & Huttel M (1979) Naloxone does not antagonise
diazepam-induced sedation. Anaesthesiology, 51: 187.
Cohen MR, Cohen RM, Pickar D, Weingartner H, & Murphy DL (1983) High
dose naloxone infusions in normals. Dose-dependent behavioural,
hormonal and physiological responses. Arch Gen Psychiatry, 40:
613-619.
Cuss FM, Colaço CB, & Baron JH (1984) Cardiac arrest after reversal
of effects of opiates with naloxone. Br Med J, 288: 363-364.
Dingledine R, Inversen LL, & Breuker E (1978) Naloxone as a GABA
antagonist: evidence from iontophoretic, receptor binding and
convulsant studies. Eur J Pharmacol, 47: 19-27.
Drummond GB, Davie IT, & Scott DB (1977) Naloxone: dose-dependent
antagonism of respiratory depression by fentanyl in anaesthetised
patients. Br J Anaesth, 49: 151-154.
Evans LEJ, Roscoe P, Swainson CP, & Prescott LF (1973) Treatment of
drug overdose with naloxone, a specific narcotic antagonist. Lancet,
1: 452.
Fishman J, Roffwarg H, & Hellman, L. (1973) Disposition of naloxone
- 7,8,3H in normal and narcotic-dependent men. J Pharmacol Exp Ther,
187: 575-580.
Fishman J, Hahn EF, & Norton BI (1975) Comparative in vivo
distribution of opiate agonists and antagonists by means of double
isotope techniques. Life Sci, 17: 1119-1126.
Fujimoto JM (1969) Isolation of two different glucuronide metabolites
of naloxone from the urine of rabbit and chicken. J Pharmacol Exp
Ther, 168: 180-186.
Fujimoto JM, Roerig S, Wang RIH, Chatterjie N, & Intrirrisi CE (1975)
Narcotic antagonist activity of several metabolites of naloxone and
naltrexone tested in morphine dependent mice (38558). Proc Soc Exp
Biol Med, 148: 443-448.
Gober AE, Kearns GL, Yorkel RA, & Danziger L (1979) Repeated naloxone
administration for morphine overdose in a 1 month old infant.
Paediatrics, 63: 606-608.
Gold MS, Pottash AC, Sweeney DR, & Kleber HD (1980) Opiate withdrawal
using clonidine. A safe, effective, and rapid nonopiate treatment. J
Am Med Assoc, 243: 343-346.
Goldfrank L, Weisman RS, Errick JK, & Lo MW (1986) A dosing nomogram
for continuous infusion intravenous naloxone. Ann Emergency Med, 15:
566-570.
Gourlay GK & Coulthard K (1983) The role of naloxone infusions in the
treatment of overdoses of long half-life narcotic agonists.
Application to nor-methadone. Br. J Clin Pharmacol., 15: 269-271.
Handal KA, Schauben JL, & Salamone FR (1983) Naloxone. Ann Intern
Med, 12: 438-445.
Hahn EF, Lahita R, Kreek J, Duma C, & Intrurrisi CE (1983) Naloxone
radio-immunoassay: an improved antiserum. J Pharm Pharmacol, 35:
833-836.
Hassan MMA, Mohammed ME, & Mian MS (1985) Naloxone hydrochloride.
Anal Profiles Drug Subst, 14: 453-489.
Hatano S, Keane DM, Wade MA, & Sadove MS (1975) Naloxone reversal for
anaesthetic dosages of pentazocine. Anaesth Rev, 2: 11-15.
Henry J & Volans G (1984) ABC of Poisoning: Analgesics: Opioids. Br
Med J, 289: 990-993.
Jasinski DR, Martin WR, & Haertzen CA (1967) The human pharmacology
and abuse potential of N-allyl noroxymorphone (naloxone). J Pharm Exp
Ther, 157: 420-426.
Jeffcoate WJ, Herbert M, Cullen MH, Hastings AG, & Walder CP (1979)
Prevention of effects of alcohol intoxication by naloxone. Lancet, 2:
1157-1159.
Jefferys DB, Flanagan RJ, & Volans GN (1980) Reversal of ethanol-
induced coma with naloxone. Lancet, 1: 308-309.
Johnstone RE, Jobes DR, Kennell EM, Behar MG, & Smith TC (1974)
Reversal of morphine anaesthesia with naloxone. Anaesthesiology, 41:
361-367.
Jordan C (1980) Respiratory depression following diazepam: reversal
with high-dose naloxone. Anaesthesiology, 53: 293-298.
Kaufman RD, Gabathuler ML, & Bellville JW (1981) Potency, duration of
action and pA2 in man of intravenous naloxone measured by reversal of
morphine-depressed respiration. J Pharmacol Exp Ther, 219: 156-162.
Kaufman JJ, Semo NM, & Koski WS (1975) Microelectrometric titration
measurements of the pKa's and partition and drug distribution
coefficients of narcotics and narcotic antagonists and their pH and
temperature dependence. J Med Chem, 18: 647-655.
Kotlinska J & Langwinski R (1990) The lack of effect of opioid
agonists and antagonists on some acute effects of ethanol. Pol J
Pharmacol Pharm, 42: 129-135.
Kulig K, Duffy J, & Rumack BH (1982) Naloxone for treatment of
clonidine overdose. J Am Med Assoc, 247: 1697.
Longnecker DD, Grazis PA, & Eggors WWN (1973) Naloxone for antagonism
of morphine-induced respiratory depression. Anaesth Analg, 53:
447-452.
McNicholas LF & Martin WR (1984) New and experimental therapeutic
roles for naloxone and related opioid antagonists. Drugs, 27: 98-93.
Martin WR (1976) Naloxone. Ann Intern Med, 85: 765-768.
Martindale (1982) In: Reznolds JEF ed. The extra pharmacopoeia, 28th
ed. London, Pharmaceutical Press.
Meffin PF & Smith KJ (1980) Gas chromatographic analysis of naloxone
in biological fluids. J Chromatogr, 183: 352-356.
Misra AL (1978) Metabolism of opiates. In: Factors affecting the
action of narcotics. New York, Raven Press, pp 297-343.
Ngai SH, Berkowitz BA, Yang JC, Hampstead J, & Spector S (1976)
Pharmacokinetics of naloxone in rats and in man: Basis for its potency
and short duration of action. Anaesthesiology, 44: 398-401.
North DS, Wieland MJ, & Peterson CD (1981) Naloxone administration in
clonidine overdosage. Ann Emergency Med, 10: 397.
Nuotto E, Palva ES, & Seppala T (1984) Naloxone-ethanol interaction in
experimental and clinical situations. Acta Pharmacol Toxicol, 54:
278-284.
Pace NL, Parrish RG, Lieberman MM, Wong KC, & Blatnick RA (1979)
Pharmacokinetics of naloxone and naltrexone in the dog. J Pharmacol
Exp Ther, 208: 254-256.
Partridge BL & Ward CF (1986) Pulmonary edema following low-dose
naloxone administration. Anesthesiology, 65: 709-710.
Sawynok J, Pinsky C, & Labella FS (1979) Mini review on the
specificity of naloxone as an opiate antagonist. Life Sci, 25:
1621-1632.
Smits SE & Takemori AE (1970) Quantitative studies on the antagonism
by naloxone of some narcotic and narcotic-antagonist analgesics. Br
J Pharmacol, 39: 627-638.
Social Welfare Board (1976) Nalone (naloxon). Uppsala, Sweden, Social
Welfare Board, Pharmaceuticals Department, pp 7-9.
Stile IL, Fort M, Marotta F, Wurzburger R, Hiatt IM, & Hegyi T (1984)
Pharmacokinetics of naloxone in premature infants. Paediatr Res,
18(392): 161A.
Tenenbein M (1984) Continuous naloxone infusion for opiate poisoning
in infancy. J Pediatr, 105: 645-647.
Terry MD, Hisayasu GH, Kern JW, & Cohen JL (1984) High performance
liquid chromatographic analysis of naloxone in human serum. J
Chromatogr, 311: 213-217.
United States Pharmacopeia (1980) 20th ed. Rockville, Maryland,
United States Pharmacopeial Convention, Inc.
Weinstein SH, Pfeffer M, & Schor JM (1974) Metabolism and
pharmacokinetics of naloxone. Adv Biochem Psychopharmacol, 8:
525-535.
Weinstein SH, Pfeffer M, Schor JM, Indindoli L, & Mintz M (1971)
Metabolites of naloxone in human urine. J Pharm Sci, 60: 1567-1568.
Wiley JF, Wiley CC, Torrey SB, & Henretig FM (1990) Clonidine
poisoning in young children. J Pediatr, 116: 654-658.
Windholz M ed. (1983) The Merck index: An encyclopedia of chemicals,
drugs and biologicals, 10th ed. Rahway, New Jersey, Merck and Co,
Inc.
3. FLUMAZENIL
3.1 Introduction
Acute poisoning is currently one of the main causes of hospital
admission in developed countries. Benzodiazepines (BZDs) are the most
commonly used drugs throughout the world and their abuse may be
responsible for the impairment of memory and for dependence. An acute
overdose can result in long-lasting coma, which is generally treated
with supportive measures. Flumazenil, an imidazobenzo-diazepine
(AnexateTM), has been shown to reverse the sedative, anti-
convulsant, and muscle-relaxant effects of BDZs. It has no convulsive
action in itself and its use has therefore been proposed to counteract
benzodiazepine action in anaesthetics, clinical toxicology and
intensive care.
3.2 Name and Chemical Formula of Antidote
* Flumazenil AnexateR (Roche Laboratories)
* Ethyl-8-fluoro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[1,5-a][1,4]
benzo-diazepine-3-carboxylate
* Empirical formula: C15H14O3N3F
* Relative molecular mass: 303.3
* Therapeutic class : Imidazobenzodiazepine
* CAS number: 78 755-81-4
* Conversions:1 mmol = 303.3 mg
1 g = 3.3 mmol
µmol/l = 3.3 x µg/ml
µg/ml = 0.3 x µmol/l
3.3 Physico-chemical Properties
Physico-chemical properties of flumazenil are given in Table 1.
Flumazenil remains stable when exposed to light and when stored
for 2 years at 35° C. The loss of weight on drying is up to 1%.
3.4 Pharmaceutical Formulation and Synthesis
No information is available on the routes of synthesis and
manufacture.
Flumazenil is supplied for parenteral administration in vials
containing 5 or 10 ml aqueous solution (0.1 mg/ml). It is available
for oral administration as tablets of 10, 20 or 30 mg.
Table 1. Physico-chemical properties of flumazenila
Melting point 198-202 °C
Solubility in water < 1 g/l
Solubility in organic solvents (g/l)
chloroform < 250
methanol < 17
ethyl acetate < 3
diethyl ether < 1
Solubility at various pH values (g/l)
(in aqueous buffered solution at 37° C)
pH 1.2 3
pH 5.3 0.7
pH 7.5 0.6
Acidity (10% aqueous solution) 4.5-7.5
pKa (in weak base) 1.7
a Personal communication from Roche Laboratories (1988)
3.5 Analytical Methods
3.5.1 Identification of the antidote
Information on the identification of flumazenil was provided by
Roche Laboratories (personal communication, 1988).
3.5.1.1 Infrared spectroscopy
The infrared spectrum (625-4000 cm-1) of a sample (a 1:300
solid dispersion in potassium bromide) is compared qualitatively with
that of a reference substance.
3.5.1.2 Ultraviolet absorption
A portion (85-95 mg) of the sample is dissolved in approximately
100 ml of ethanol and diluted to 150 ml with ethanol (solution 1).
This solution is then diluted ten-fold with ethanol to give solution
2, which is further diluted ten-fold with ethanol to give solution 3.
The position and absorbance of solution 3 is measured
spectrophotometrically at the maximum (245 nm) and minimum (228 nm)
wavelengths, against ethanol, in quartz cells.
3.5.1.3 Thin-layer chromatography
The TLC details are as follows:
* layer: Silica gel 60 F254
* mobile phase: Chloroform/ethanol (90/10 v/v)
* sample solution: 10 ml of the ampoule solution is extracted
with 1 ml of chloroform
* standard solution: 5 mg of flumazenil is dissolved in 5 ml
of chloroform saturated with water
* front distance: 12 cm
* migration time approx: 30 min
* detection: the plate is dried in a current of warm air for
5 min, and examined under shortwave light. Decreasing
fluorescence due to flumazenil occurs at 254 nm
(ultraviolet region). When the plate is sprayed with
Dragendorff's reagent, flumazenil appears as an orange
spot. The Rf value is approximately 0.5.
3.5.2 Quantification of the antidote in biological samples
The determination of flumazenil in plasma by gas-liquid
chromatography (GLC) with nitrogen phosphorus detection is a sensitive
and specific method, the detection limit being 3 ng/ml (Abernethy et
al., 1983). An ethyl acetate extraction (neutral pH) of 0.1-3 ml
plasma is used for sample preparation. When methylclonazepam is used
as an internal standard, the graph is linear for plasma concentrations
up to 200 ng/ml. The retention time for flumazenil is 3.96 min.
High-performance liquid chromatography (HPLC) with UV detection
at 254 nm is a sensitive method for determination in urine or plasma,
the detection limit being about 10 ng/ml (Timm & Zell, 1983; Bun et
al., 1989). When the n-propyl ester analogue is used as an internal
standard, the graph is linear for plasma concentrations up to 320
ng/ml.
3.5.3 Analysis of the toxic agent in biological samples
Three major methods for the quantitative analysis of BZDs in
plasma or serum are used:
* HPLC with UV detection at 246 nm (detection limits are 5-50 µg/ml
of serum) (Rocher, 1984);
* immunoenzymology by the EMIT method for a semiquantitative
determination (metabolites also measured) of diazepam levels,
completed by a chromatographic method (sensitivity from 0.3 to 2
µg/ml) (Rocher, 1984);
* gas-liquid chromatography (Pellerin, 1986).
3.6 Shelf-life
Vials ready for use are stable at room temperature (15-25° C) for
three years.
3.7 General Properties
Flumazenil has been shown to block all the typical BZD effects
(anticonvulsive, sedative, anxiolytic, muscle relaxant, and amnesic).
It acts as a potent BZD-specific antagonist by competing at the
central synaptic gamma-aminobutyric acid (GABA) receptor sites in a
dose-dependent manner, but does not seem to antagonise BZD effects at
peripheral GABA-ergic (renal, cardiac, etc.) receptor sites (Mohler et
al., 1981). It possesses agonist properties and has a specific, but
discreet, anticonvulsive effect without inducing drowsiness or muscle
relaxation (Abernethy et al., 1983; Timm & Zell, 1983; Haaefely, 1983;
Rocher, 1984; Scollo-Lavizzari, 1984; personal communication by Roche
Laboratories, 1988). In addition, it antagonizes the sedative effects
of other compounds that act through GABA receptors, such as zopiclone
(Mohler et al., 1981).
3.8 Animal Studiesc
3.8.1 Pharmacodynamics
Flumazenil has been tested for its ability to induce withdrawal
signs in animals pretreated with benzodiazepine; the signs included
emesis, tremors, rigidity and clonic convulsions.
c Personal communication by Roche Laboratories to the IPCS, 1988
Rats that had been pretreated with an oral dose (10 or 100 mg/kg)
of diazepam for 12 days were administered flumazenil (10 mg/kg)
intravenously. Signs were very mild even at 100 mg/kg.
Cats were pretreated intraperitoneally for 16 days with either a
10-mg/kg dose of lorazepam twice daily or a 1-mg/kg dose of triazolam
once daily. Flumazenil (100 mg/kg) was then administered
intraperitoneally either immediately or 1.5, 6, 12, 48, and 60 h after
the last dose. Symptoms such as rigidity, vocalization and tachypnoea
lasted 30 min, whereas others such as hypersalivation lasted 2 h.
Flumazenil (1 to 15 mg/kg) was administered intragastrically to
rats that had been pretreated with daily diazepam doses of 113 mg/kg
for about 6 months. Abstinence syndromes increased with increasing
dose of flumazenil and reached a plateau.
The intragastric administration of flumazenil (15 mg/kg per day)
to cats pretreated with flurazepam (5 mg/kg per day) for 35 days led
to withdrawal symptoms (increasing muscle tone, tremors, piloerection,
mydriasis, and hypersalivation) 24 h after the last dose of
flurazepam. No convulsions were observed.
Intramuscular administration of flumazenil (5 mg/kg) to squirrel
monkeys and baboons, pretreated with oral doses of lorazepam,
triazolam (3 mg/kg per day), oxazepam (40 or 80 mg/kg per day) or
diazepam (8-20 mg/kg per day), produced withdrawal signs. However, no
withdrawal signs were precipitated by flumazenil in monkeys treated
with oral midazolam (30 mg/kg) or in barbital-dependent rhesus monkeys
(the length of pretreatment with BZD was not specified).
The severity of withdrawal signs resulting from the blocking of
BZD receptors by flumazenil depends on the species tested, the dose of
BDZ used to develop physiological dependence, and the duration of
treatment.
3.8.2 Pharmacokinetics
3.8.2.1 Absorption
A single dose of flumazenil (125 mg/kg) in a carboxymethyl
cellulose suspension produced a maximum plasma concentration in rats
of 9.9 µg/ml after 20 min. The bioavailability was 0.55. In rabbits,
the maximum concentration 90 min after a single dose of flumazenil
(150 mg/kg) was 15 µg/ml. The bioavailability was 0.60.
3.8.2.2 Distribution
When total radioactivity was measured in rats 0.5, 7, 24, 96, and
192 h after an intravenous dose of 14C-labelled flumazenil (2
mg/kg), the highest level was found at 0.5 h in the kidney, liver and
intestine. None was found at 192 h. The volume of distribution
ranged from 0.71 to 1.87 l/kg.
3.8.2.3 Elimination
Studies on rats given an oral dose (50 mg/kg) of 14C-labelled
flumazenil and on dogs given an intravenous dose of 4 mg/kg showed
three main inactive metabolites:
* Ro 15-3890 acid and major metabolite (72% in the rat, 30-60% in
the dog);
* Ro 15-4965 hydroxyethyl derivative (3% in the rat);
* Ro 15-6877 N-demethyl derivative (1% in the rat, 1-13% in the
dog).
Table 2 presents elimination data in three different species. In
these species, 90% of the intravenously or orally administered
flumazenil was eliminated, mainly as metabolites, within 48 h. One
third was eliminated in the faeces and two-thirds in the urine.
Table 2. Elimination of flumazenil in the rat, rabbit and dog
Species Dose Total plasma T´
clearance (min)
(ml/min per kg)
Rat 2 mg/kg 114 7.4
Rabbit 0.5 mg/kg 24 34
Dog 5 mg 21 48
3.8.3 Toxicology
3.8.3.1 Acute toxicity
a) Intravenous administration to rats and mice
An aqueous solution of 0.1 mg flumazenil/ml was used and was
administered at a dose of 2.5 mg/kg to mice and 1 mg/kg to rats. No
abnormal clinical signs and no deaths occurred. LD50 values were
not determined; these doses (50 to 250 times higher than the clinical
doses) were well tolerated in the two species.
A flumazenil solution with a concentration of 50 mg/ml was
subsequently used and the LD50 values given in Table 3 were obtained
(95% confidence intervals). Deaths occurred 30 min after the
injection, preceded by rigidity and clonic convulsions.
Table 3. Intravenous LD50 values (mg/kg) for the mouse and rat
Species Male Female
Mouse 143-198 145-175
Rat 85-167 112-231
b) Intravenous administration to the dog
The administration of daily doses of 0.01 to 0.03 mg/kg was well
tolerated and no deaths were observed. LD50 values were not
determined; the doses (15 to 30 times higher than the clinical doses)
were again well tolerated.
c) LD50 values (mg/kg) for the rat, mouse and rabbit
When flumazenil was administered orally to rats, mice and rabbits
(Table 4), deaths were observed within three days, associated with
decreased motor activity, catatonic state and tremors.
Table 4. LD50 values (mg/kg) for the rat, mouse and rabbit
Species Male Female
Mouse 2500 1300
Rat 4200 4200
Rabbit 2000 2000
3.8.3.2 Subacute toxicity
Systemic tolerance was good in both rats and dogs administered
flumazenil intravenously at dosages up to 10 mg/kg per day for 4
weeks.
3.8.3.3 Chronic toxicity
In 13-week studies using an oral aqueous solution of flumazenil,
very good tolerance was shown by rats at dosages of 0.5, 25 and 125
mg/kg per day and by dogs at 0.5, 20, and 80 mg/kg per day. No
haematological, biochemical or gross pathological abnormalities were
observed.
3.8.3.4 Embryotoxicity
Studies on rats (between the 7th and 16th day of gestation) and
rabbits (between the 7th and 19th day of gestation) revealed no signs
of embryotoxicity at dosages of 15, 50, and 150 mg/kg per day.
3.8.3.5 Mutagenicity
Flumazenil was not mutagenic in the Ames test or micronucleus
test, or in tests using Saccharomyces cerevisiae or Chinese hamster
V79 cells.
3.9 Volunteer Studies
3.9.1 Pharmacodynamics
3.9.1.1 BZD antagonist effect
Efficacy studies were performed on 125 healthy volunteers with
oral doses of flumazenil up to 20 mg, the aim being to antagonize the
effects of diazepam, flunitrazepam and midazolam on the CNS (Darragh,
1981; Lupolover, 1983). These studies demonstrated the antagonist
effect of flumazenil, which rapidly abolished the hypnotic-sedative
BZD effects. Other studies used meclonazepam (Darragh et al., 1981),
diazepam (Darragh et al., 1982), flunitrazepam (Gaillard & Blois,
1983) and midazolam (Forster et al., 1983). In studies by Ziegler &
Schalch (1983) and Lauven et al. (1985), flumazenil was administered
to subjects during continuous midazolam infusion after the attainment
of a pharmacokinetic and pharmacodynamic steady state, at which point
subjects were deeply asleep. The degree and duration of the effect of
flumazenil depended on the BZD dose, the antagonist dose and the time
that had elapsed since the BZD was given. In the study by Ziegler &
Schalch (1983), baseline levels of vigilance and orientation were
reached within 1 min. Lauven et al. (1985) used higher midazolam and
flumazenil dosages and his patients awoke within 28 to 48 seconds. No
signs of BZD withdrawal effects were seen in short-term studies (one
single dose) on healthy volunteers given flumazenil to antagonize BZDs
(Amrein, 1987).
The efficacy of flumazenil in antagonizing the effects of
midazolam was also clearly demonstrated in the double-blind placebo-
controlled study by Rouiller et al. (1987).
3.9.1.2 Intrinsic effects
Most studies on healthy human volunteers have shown little or no
intrinsic effect of flumazenil when administered alone. The mild
sedation reported by Amrein (1987) occurred after the administration
of oral doses greater than 100 mg.
Scollo-Lavizzari (1984) observed some anticonvulsant effects in
epileptic patients. Decreased amplitude of auditory evoked potentials
has also been described (Laurian et al., 1984; Schoepf et al., 1984).
Mild, nonspecific effects such as increased alertness may occur after
the administration of doses very much higher than those used
clinically (Laurian et al., 1987).
3.9.2 Pharmacokinetics
3.9.2.1 Absorption
Following oral administration of a 200-mg dose of flumazenil, the
highest plasma concentration (Cmax) ranged from 147 to 349 µg/l and
was reached within 20 to 45 min. The mean bioavailability of the
tablets used was about 17% and the inter-individual variability was
7-29% (Pellerin, 1986).
3.9.2.2 Distribution
The proportion of flumazenil bound to plasma proteins is 50%
(two-thirds of which is bound to albumin). Values for the mean
steady-state volume of distribution of 0.95 l/kg (personal
communication by Roche Laboratories, 1988) and 1.23 l/kg (Roncari et
al., 1986) have been determined.
3.9.2.3 Elimination
Ninety-nine per cent of the flumazenil administered is
metabolized by the liver, and 1% is excreted unchanged in the urine.
Mean total blood clearance, for which values of 59 l/h (Pellerin,
1986) and 72 l/h (Roncari et al., 1986) have been determined, is
essentially due to the hepatic clearance. The apparent plasma half-
life in healthy volunteers has been reported to be 53-58 min (Roncari
et al., 1986; personal communication by Roche Laboratories, 1988).
3.9.3 Tolerance of flumazenil
In the study by Rouiller et al. (1987), no objective agonist
effects or biological toxicity of flumazenil could be demonstrated in
six healthy volunteers.
3.9.4 Other studies
There is evidence that central nervous system effects of ethanol
are mediated through the GABA system. For this reason, the effect of
flumazenil on psychometric performance was studied in eight healthy
volunteers with stable blood ethanol levels of 1.6 g/l (35 mmol/l)
under a placebo-controlled double-blind design (Clausen et al., 1990).
Flumazenil did not improve psychomotor functions in these ethanol-
intoxicated subjects, which is in agreement with experience in
clinical toxicology.
3.10 Clinical Studies - Clinical Trials
Flumazenil was first used clinically in patients with iatrogenic
benzodiazepine overdose due to mechanical ventilation or status
epilepticus (Scollo-Lavizzari, 1983).
Clinical studies can be grouped under the headings
anaesthesiology and toxicology (Amrein, 1986).
3.10.1 Anaesthesiology
3.10.1.1 General anaesthesia
Three placebo-controlled studies have been conducted in patients
who were given flunitrazepam for general anaesthesia.
Jensen et al. (1985) reported that a 0.3-mg to 0.7-mg dose of
flumazenil awoke all patients within 5 min, compared with only 35% of
the patients in the placebo-treated group (P < 0.001 for sedation,
orientation and amnesia).
In a study of 60 patients, Tolksdorf et al. (1986) found that
patients treated with flumazenil were less sedated than placebo-
treated patients (P < 0.05) following flunitrazepam sedation (from 5
min to 1 h after the administration of flumazenil), better orientated
at 15 min, and less amnesic. Ellmauer et al. (1986) reported similar
results in 57 patients given a 0.1- to 1-mg dose of flunitrazepam (P
< 0.005).
No significant difference was observed after 2 h between the
placebo-treated and flumazenil-treated patients in any of the three
studies described in this section.
Midazolam effects were reversed by flumazenil in an open study
including 18 intracranial surgery patients (Chiolero et al., 1988).
3.10.1.2Conscious sedation
In a 74-patient open study (Geller et al., 1986) and a 40-patient
placebo-controlled study (Knudsen et al., 1986), in which either
midazolam or diazepam was used, there was a significant difference
between flumazenil- and placebo-treated patients. In the former
study, patients were awakened by a 0.1- to 0.6-mg dose of flumazenil
within 1 to 2 min. In the study by Knudsen et al. (1986), 80% of the
flumazenil-treated patients were awake 5 min after receiving the dose
compared with 50% in the placebo group (P < 0.05).
3.10.2 Benzodiazepine overdose or intoxication
Three different studies have indicated that flumazenil may be an
effective tool for the management of intoxication (either intentional
or iatrogenic) with BZD in the presence or absence of other agents.
Owing to its safety and specificity, flumazenil could be used in the
initial treatment of poisoning and coma of unknown origin. In a study
by Hofer & Scollo-Lavizzari (1985) based on 13 patients, a 1.5-to 10-
mg dose of flumazenil administered intravenously at a rate of 1.5 to
2.5 mg/min reversed the CNS depression induced by various BZDs within
1 to 2 min.
Geller et al. (1985) treated 34 patients (23 cases of intentional
drug intoxication and 11 of iatrogenic BZD overdose) by means of
intravenous injections of 0.1 mg flumazenil every 30 seconds until the
patient regained consciousness. The treatment proved to be extremely
effective, providing reversal effects lasting up to 2 h.
Bismuth et al. (1985) treated patients for BZD overdose in a
double-blind randomized study, injecting a single dose of either
flumazenil or placebo. Two of the 20 placebo patients awoke
partially, compared with 17 of the 20 flumazenil-treated patients (one
experienced seizures interrupting the study). In a second open study
(Bismuth et al., 1986) based on 37 patients, 6 showed no response to
doses of flumazenil ranging from 5 to 9.5 mg (mixed intoxication), 11
showed partial awakening (no possible written response) at a dose of
2.1 ± 1.6 mg (mixed intoxication), and 20 were completely awakened by
a dose of 1.4 ± 0.7 mg. The awakening was only temporary and return
to coma occurred after an interval of 15 min to 5 h. Permanent
recovery occurred in a patient suffering intoxication due to
triazolam, a BZD with a short half-life, after a single administration
of flumazenil.
More recent placebo-controlled double-blind studies have
confirmed the beneficial effect of flumazenil in cases of BZD
poisoning (Aarseth et al., 1988; Ritz et al., 1990).
3.11 Clinical Studies - Case Reports
The many controlled clinical studies of the effect of flumazenil
limit the need for information from case reports. In the clinical
studies reported, there have been few adverse effects associated with
the use of flumazenil. There have, however, been case reports of
seizures followed by ventricular tachycardia associated with the use
of flumazenil in combined poisonings with cyclic antidepressants and
BZD (Bismuth et al., 1985).
In one report, death was claimed to have been associated with
flumazenil administration in an old, obese and anaemic woman who had
been sedated with midazolam (4 mg, intravenous) prior to gastroscopy
(Lim, 1989). During the investigation she suffered cardiac arrest;
flumazenil was given promptly and she recovered temporarily, but then
gradually deteriorated and died 16 h later. According to Birch &
Miller (1990), the death of this patient was probably not related to
flumazenil administration.
Recently, successful treatment was achieved by administering
flumazenil as an intravenous bolus (0.02 mg/kg) and then as a
maintenance dose of 0.05 mg/kg per h to a newborn baby with recurrent
apnoea due to BZDs taken by his mother (Richard et al., 1991).
The benefit from the diagnostic use of flumazenil in coma of
unknown origin has been reported in two recent cases (Burkhart &
Kulig, 1990). When flumazenil is used with caution in such
situations, time may be saved and further expensive diagnostic
procedures, e.g., cerebral computerized tomographic (CT) scan,
avoided.
3.12 Summary of Evaluation
Flumazenil appears to be a antagonist to BZDs and other GABA-
ergic agents. This antagonism, following intravenous injection, has
been reported to be sensitive in cases of intoxication resulting
solely from BZDs (the reversal of BZD effects being observed with
doses of less than 2 mg), rapid in onset (within 2 min), and short-
lived (effects last for less than 30 min).
3.12.1 Indications
In controlled clinical trials, flumazenil significantly
antagonizes BZD-induced coma arising from anaesthesia or acute
overdose. However, the use of flumazenil has not been shown to reduce
mortality or sequelae in such cases. As the mortality in pure BZD
poisoning is extremely low, studies with mortality as end-point are
impractical since a reasonable level of statistical significance could
probably never be obtained. However, in cases of mixed intoxication,
especially with ethanol and triazolam/flunitrazepam, the use of
flumazenil may be life-saving due to the poten-tiation of BZD toxicity
by ethanol. Given this situation, it is obvious that the routine use
of flumazenil in BZD poisoning is not indicated and that
recommendations for its use in clinical toxicology must be based on
pragmatic considerations made by clinicians experienced in treating
these patients. Flumazenil is a relatively expensive drug and this
may also influence its use, especially in areas with limited
resources.
The use of flumazenil in BZD poisoning should, therefore, only be
advocated in situations with complications, which are rarely seen
except in cases of mixed ingestion. Although not of life-saving
significance, it also seems reasonable to advocate the use of
flumazenil if intubations (before gastric lavage) and mechanical
ventilation can thereby be avoided (see section 3.13.1). The proposed
uses of flumazenil within acute medicine and anaesthesia are listed in
section 3.13.1. Acute poisoning is always an important differential
diagnosis in cases of coma in children and young adults. The
diagnostic use of flumazenil in such cases can be justified by its
high therapeutic index and the fact that this may limit the use of
other diagnostic procedures such as cerebral CT scan, clinical
chemistry analyses and even lumbar puncture.
3.12.2 Dosage and route
Flumazenil is available for intravenous and oral administration.
The need for the latter formulation may be questioned in view of the
fact that drugs should generally be given intravenously in the
emergency situation and the bioavailability is low and variable. Thus
the intravenous route is preferable. Doses need to be adjusted
according to individual clinical response, bearing in mind the very
high therapeutic index of flumazenil.
a) In anaesthetics and in intensive care, doses of 0.2-0.5 mg should
be used to reduce sedation and doses of 0.5-1 mg to abolish other
BZD effects (Amrein, 1987).
b) In cases of BZD overdosage, single doses of 0.3-1 mg can be given
and repeated as necessary. If there is no clinical response to
2 mg flumazenil given over a period of 5-10 minutes, diagnoses
other than BZD poisoning are likely. It is also possible to
administer a continuous infusion (0.3-1 mg/h) of flumazenil
(diluted in 0.9% sodium chloride solution or 5% glucose solution)
in patients relapsing into a coma and/or respiratory depression
following an initial effect of flumazenil injection.
c) In children, experience is limited and dosage regimens less well
documented (Lheureux & Askenasi, 1988; Wood et al., 1987). It
is suggested that intravenous doses of 0.1 mg should be given
once per minute until the child is awake. It may be necessary to
give a subsequent continuous intravenous infusion at a rate of
0.1 to 0.2 mg/h.
3.12.3 Other consequential or supportive therapy
Treatment with flumazenil requires continuous intensive
observation. After the administration of a single dose of flumazenil,
the patient must be observed for at least 2 h to be certain that BZD-
induced complications will not recur. The termination of continuous
infusion requires intensive care monitoring.
3.12.4 Areas where there is insufficient information to make
recommendations
There is insufficient information to make recommendations in the
case of hepatic encephalopathy (indication is based on the hypothesis
that hepatic encephalopathy is associated with increased GABA-mediated
inhibitory neurotransmission).
3.12.5 Proposals for further study
The use and dosages of flumazenil in children require further
study. Indications for utilization of oral preparations need to be
clarified. The use in coma of unknown origin merits further studies.
3.12.6 Adverse effects
The most frequent adverse effects have been reviewed by Amrein
(1987). When flumazenil is used in anaesthesia, the main adverse
effects that have been reported are nausea and vomiting (placebo:
7.5%; < 1 mg flumazenil: 12.1%; 1-10 mg flumazenil: 24.5%). Other
adverse effects, which have been reported in less than 5% of cases,
are tremor, involuntary movements, dizziness, agitation, discomfort,
tears, anxiety, and a sensation of cold.
Minor effects occur when flumazenil is used in intensive care,
where agitation is the commonest adverse effect (10%). When it is
administered to patients showing BZD habituation, the following
features occur: anxiety, tenseness, fear, agitation, confusion,
convulsions (Marchant & Wray, 1989) and myoclonic seizures. Their
frequency and intensity depend on the degree of dependency and they
are believed to be related to some sort of BZD abstinence syndrome.
When administered rapidly, flumazenil can cause hypertension,
tachycardia and acute anxiety. This equivalent of an "exercise test"
was observed with the 1 mg/ml solution, which is no longer used.
3.12.7 Restrictions of use
In certain circumstances, BZD antagonism by flumazenil may be
harmful:
a) An acute withdrawal syndrome can occur in patients showing BZD
habituation following therapy or abuse.
b) Convulsions can occur in cases of mixed drug overdosage where BZD
has been taken with a drug liable to cause convulsions (such as
a tricyclic antidepressive agent).
c) Convulsions can be induced in patients treated with BZD for
seizure disorders or in patients who for years have been using
BZD for sleep disturbances.
There are other limitations to the use of flumazenil.
a) It has a short-lived effect and repeated injection or continuous
infusion is often necessary unless a short-acting BZD (e.g.,
triazolam) has been ingested.
b) In cases of mixed drug overdosage, the patient may remain
unresponsive when other drugs are contributing to the coma.
c) The treatment costs are high and supportive treatment may be
cheaper.
3.13 Model Information Sheet
3.13.1 Uses
Flumazenil is a specific antagonist of the effects of BZD at
central GABA-ergic receptors.
Within the domains of intensive care and anaesthesia, flumazenil
may be valuable in the following circumstances:
a) to diagnose BZD-induced unconsciousness in patients presenting
coma of unknown origin;
b) to terminate long-term BZD-induced sedation in the intensive care
unit (e.g., weaning from ventilatory support);
c) to reduce BZD-induced sedation or to counteract paradoxical
anxiety reactions to BZD in anaesthesia;
d) to antagonise BZD-induced sedation after short diagnostic
procedures where a long-acting BZD has been used.
Flumazenil may be justified in the following situations in cases
of BZD poisoning:
a) to facilitate gastric lavage and avoid intubation in comatose
patients;
b) to treat complications in severe cases of mixed poisoning where
BZD is thought to be one of the major toxic agents;
c) to avoid the need for mechanical ventilation in cases where there
is respiratory depression.
The routine use of flumazenil for the treatment of BZD overdosage
is not recommended.
3.13.2 Dosage and route
The intravenous route of administration is recommended when
flumazenil is given as a BZD antagonist. Doses need to be adjusted
according to individual clinical response and the following are
recommended.
a) In anaesthetics and in intensive care (adults), doses of 0.2-0.5
mg should be used to reduce sedation and doses of 0.5-1 mg to
abolish BZD effects.
b) In cases of BZD overdosage (adults), single doses of 0.3-1 mg can
be given and repeated as necessary. The absence of clinical
response to 2 mg flumazenil within 5-10 min indicates that BZD
poisoning is not the major cause of coma and other complications.
It is also possible to administer a continuous infusion (0.3 to
1 mg/h) of flumazenil (diluted in 0.9% sodium chloride solution
or 5% glucose solution) following an initial response to
flumazenil.
c) In children, it is suggested that intravenous doses of 0.1 mg
should be given once per minute until the child is awake. It may
be necessary to give a subsequent continuous intravenous infusion
at a rate of 0.1 to 0.2 mg/h.
3.13.3 Precautions/contraindications
3.13.3.1 Pharmaceutical precautions
Solutions of flumazenil should be stored at +4 °C. No other drug
should be injected or infused with the flumazenil, which should be
made up in 0.9% sodium chloride or 5% glucose (dextrose) solution.
3.13.3.2 Other precautions
Treatment with flumazenil requires continuous intensive
observation. After the administration of a single dose of flumazenil,
the patient must be observed for at least 2 h to be certain that
BZD-induced complications will not recur. The termination of
continuous infusion requires intensive care monitoring.
Note that in cases of mixed drug overdosage, the patient may
remain unresponsive where other drugs are contributing to the coma.
BZD antagonism by flumazenil may in certain circumstances be
harmful.
a) An acute withdrawal syndrome can occur in patients showing BZD
habituation following therapy or abuse.
b) Convulsions can occur in cases of mixed drug overdosage where BZD
has been taken with a drug liable to cause convulsions (such as
a tricyclic antidepressive agent).
c) Convulsions can be induced in patients treated with BZD for
seizure disorders.
The three above-mentioned situations may be considered relative
contraindications to its use; flumazenil should only be used when it
is strongly indicated. In these situations, it should be given more
slowly than usual (e.g., 0.3 mg intravenously over 3 min, a 3-min
pause, then a further 0.3 mg at the same rate, and so on).
3.13.4 Adverse effects
Various effects have been reported when flumazenil is used in
anaesthesia. These include (in order of decreasing frequency):
nausea, vomiting, tremor, involuntary movements, dizziness, agitation,
discomfort, tears, anxiety, and sensation of cold.
Similar effects occur when flumazenil is used in intensive care,
agitation being the commonest adverse effect. When flumazenil is
administered to patients showing BZD habituation, the following can
occur: anxiety, tenseness, fear, agitation, confusion, convulsions and
myoclonic seizures.
When administered rapidly, flumazenil can cause hypertension,
tachycardia and acute anxiety.
3.13.5 Use in pregnancy and lactation
Even though animal studies showed no embryotoxicity or
teratogenicity at high doses, flumazenil, like any new drug, should be
avoided at the beginning of pregnancy. However, in life-threatening
situations its possible risk to the fetus is probably far outweighed
by its beneficial effects. Isolated administration of flumazenil
during lactation is not contraindicated.
3.13.6 Storage
No special storage conditions are required.
3.13.7 Special risk groups
Acute withdrawal syndrome can occur in patients showing BZD
habituation following therapy or abuse (see section 3.13.3).
3.14 References
Aarseth HP, Bredesen JE, Grynne B, Lyngdal PT, Storstein L, & WiiK-
Larsen E (1988) Benzodiazepine-receptor antagonist, a clinical double
blind study. J Toxicol Clin Toxicol, 26: 283-292.
Abernethy DR, Arend R, Lauven P, & Greenblatt D (1983) Determination
of Ro 15-1788, a benzodiazepine antagonist in human plasma, by gas-
liquid chromatography with nitrogen phosphorus detection.
Pharmacology, 26(5): 285-289.
Amrein R (1986) Clinical documentation on flumazenil. Basel,
Switzerland, Hoffmann-Laroche Laboratory (Unpublished report).
Amrein R, Leishman B, Bentzinger C, & Roncari G (1987) Flumazenil in
benzodiazepine antagonism. Med Toxicol, 2: 411-429.
Bismuth C, Baud FJ, Fournier PE, & Mellerio F (1985) Antagoniste
Ro-15-1788 dans l'intoxication volontaire par benzodiazépines. Valeur
diagnostique et thérapeutique. Etude randomisée sur 40 cas. J Toxicol
Clin Exp, 5: 403-404.
Bismuth C, Baud F, Fournier E, & Mellerio F (1986) Antagoniste Ro
15-1788 dans l'intoxication volontaire par les benzodiazépines.
Valeur diagnostique et thérapeutique. Etude ouverte sur 40 cas. Ann
Méd Interne, 137(4): 363-364.
Birch BRP & Miller RA (1990) Death after flumazenil? Br Med J, 300:
467-468.
Bun H, Duplan V, Crevat-Pisano P, Liurens M, & Durand A (1989) Rapid
determination of the benzodiazepine antagonist flumazenil, Ro 15-1788,
by high performance liquid chromatography. Biomed Chromatogr, 3:
269-271.
Burkhart KK & Kulig KW (1990) The diagnostic utility of flumazenil in
coma of unknown etiology. Ann Emerg Med, 19: 319-321.
Chiolero R, Ravussin P, Chassot PG, Neff R, & Freeman J (1988) Ro
15-1788 for rapid recovery after craniotomy. Ann Fr Anesth Réanim, 7:
17-21.
Clausen TG, Wolff J, Carl P, & Theilgaard A (1990) The effect of the
benzodiazepine antagonist, flumazenil, on psychometric performance in
acute ethanol intoxication in man. Eur J Clin Pharmacol, 38: 233-236.
Darragh A (1981) A study of efficacy of Ro 15-1788 (600mg) as an
antagonist of the sedative effects of Ro 11-3128 in healthy
volunteers. Basel, Switzerland, Hoffmann-Laroche Laboratory
(Unpublished report).
Darragh A, Lambe R, Scully M, Brick I, O'Boyle C, & Dowrie WW (1981)
Investigation in man of the efficacy of a benzodiazepine antagonist,
Ro 15-1788. Lancet, 2(8236): 8-10.
Darragh A, O'Boyle C, & Cambe RI (1982) Antagonist of the central
effects of diazepam in man by Ro 15-1788, a new benzodiazepine
antagonist. Irish J Med Sci, 151(3): 90.
Ellmauer S, Muller H, & Christen M (1986) [Clinical investigations on
the efficacy and safety of the benzodiazepine antagonist Ro 15-1788.]
Beitr Anaesthesiol Intensivmed, 17: 78 (in German).
Forster A, Rouiller M, Morel D, & Gemperle M (1983) Double-blind
randomized study evaluating a specific benzodiazepine antagonist.
Anaestesiology, 59(3): A375.
Gaillard JM & Blois R (1983) Effect of the benzodiazepine antagonist
Ro 15-1788 on flunitrazepam-induced sleep changes. Br J Clin
Pharmacol, 15: 529-536.
Geller E & Niv M (1985) Ro 15-1788, a benzodiazepine antagonist in
the treatment of 34 intoxicated patients. Anaesthesiology, 61(1): 35.
Geller E, Chernilas J, Halpern P, Niv D, & Miller HI (1986)
Hemodynamics following reversal of benzodiazepine sedation with Ro
15-1788 in cardiac patients. Anaesthesiology, 65: A49 (Abstract).
Haaefely W (1983) Antagonist of benzodiazepine. Encéphale, 9:
143B-150B.
Hofer P & Scollo-Lavizzari G (1985) First clinical investigation of
the benzodiazepine antagonist Ro 15-1788 in comatose patients. Arch
Intern Med, 145(4): 663-664.
Jensen S, Kirkegaard L, & Andersen BN (1985) Benzodiazepine
antagonist Ro 15-1788: randomized clinical investigation of Ro 15-1788
in reversing the central effects of flunitrazepam. Acta Anaesthesiol
Scand, 29(Suppl): 89.
Knudsen L & Kirkegaard L (1986) Benzodiazepine antagonist Ro 15-1788:
antagonism of diazepam sedation in out-patients undergoing
gastroscopy. Beitr Anaesthesiol Intensivmed, 17: 268.
Laurian S, Gaillard JM, & Le PK (1984) Dose effects of the
benzodiazepine antagonist Ro 15-1788 on the auditory evoked
potentials. In: Abstracts of the 14th Congress of the Collegium
Internationale Neuropsychopharmacologicum, Firenze, Italy, 19-23 June
1984. Fidia Research Biomedical Information (Abstract No. 33).
Laurian S, Despland PA, Gaillard JM, Grasset F, & Le PK (1987)
Psychotropic drugs and EEG organization. Int J Neurosci, 32(12): B370.
Lupolover R (1983) Evaluation of the efficacy and safety (tolerance)
of Ro 15-1788 intravenously administered in normal healthy volunteers.
Basel, Switzerland, Hoffmann-Laroche Laboratory (Unpublished report).
Lauven PM, Schwilden H, & Stoeckel H (1985) The effects of a
benzodiazepine antagonist Ro 15-1788 in the presence of stable
concentrations of midazolam. Anesthesiology, 63: 61-64.
Lheureux P & Askenasi R (1988) Specific treatment of benzodiazepine
overdose. Hum Toxicol, 7: 165-170.
Lim AG (1989) Death after flumazenil. Br Med J, 299: 858-859.
Marchant B & Wray R (1989) Flumazenil causing convulsions and
ventricular tachycardia. Br Med J, 299: 860.
Mohler H, Pieri L, Polc P, Bonetti EP, Cumiu R, Schaffner R, & Haefely
W (1981) Selective antagonists of benzodiazepines. Nature(Lond), 290:
514-516.
Pellerin F (1986) Pharmaceutical documentation on flumazenil. Basel,
Switzerland, Hoffmann-Laroche Laboratory (Unpublished report).
Richard P, Autret E, Bardol J, Soyez C, Barbier P, Jonville AP, &
Ramponi N (1991) The use of flumazenil in a neonate. J Toxicol Clin
Toxicol, 29: 137-140.
Ritz R, Zuber M, Elsasser S, & Scollo-Lavizzari G (1990) Use of
flumazenil in intoxicated patients with coma. A double-blind placebo-
controlled study in ICU. Intensive Care Med, 16: 242-247.
Rocher JP (1984) Benzodiazepines. Lyon Pharm, 35(3): 149-167.
Roncari G, Ziegler QH, & Guentert TW (1986) Pharmacokinetics of the
new benzodiazepin antagonist Ro 15-1788 in man following intravenous
and oral administration. Br J Clin Pharmacol, 22: 421-428.
Rouiller M, Forster A, & Gemperle M (1987) Double blind randomized
study evaluating a specific benzodiazepine antagonist. Ann Fr Anesth
Réanim, 6: 1-6.
Scoepf J, Laurian S, Le PK, & Gaillard JM (1984) Intrinsic activity of
the benzodiazepine antagonist Ro-1788 in man: an electrophysiological
investigation. Pharmacopsychiatry, 17: 79-83.
Scollo-Lavizzari G (1984) The anticonvulsant effect of the
benzodiazepine antagonist, Ro 15-1788: an EEG study in 4 cases. Eur
Neurol, 23: 1-6.
Timm U & Zell M (1983) Determination of the benzodiazepine antagonist
Ro 15-1788 in plasma by high performance liquid chromatography with UV
detection. Arzneimittelforschung, 33(1): 358-362.
Tolksdorf W, Pirwitz A, & Pfeiffer D (1986) [Effects and side-
effects of the benzodiazepine antagonist Ro 15-1788 following mixed
anaesthesia in association with flunitrazepam, compared with a
placebo: a double-blind study.] Beitr Anaesthesiol Intensivmed, 35:
121 (in German).
Wood C, Bismuth C, Oriot D, Devictor D, & Juault G (1987)
Réversibilité d'un coma par benzodiazépine par perfusion de flumazenil
chez un enfant. Presse Méd, 16: 1483.
Ziegler WH & Schalch E (1983) Antagonism of benzodiazepine-induced
sedation in man. In: In sleep. Proceedings of the 6th European
Congress of Sleep Research, Zurich, 1982. Basel, Karger, pp 427-429.
4. DANTROLENE SODIUM
4.1 Introduction
Dantrolene sodium, a hydantoin-furan derivative, causes skeletal
muscle relaxation by preventing calcium flux across the sarcoplasmic
reticulum. It was first synthesized in 1967 and the initial use was
to treat muscle spasm (Dykes, 1975). More recently dantrolene has
been used successfully in the treatment of malignant
hyperpyrexia/hyperthermia and neuroleptic malignant syndrome,
hypercatabolic syndromes that have previously been associated with
high mortality rates.
Malignant hyperthermia results from a genetic susceptibility to
certain anaesthetic agents (autosomal dominant myopathy), and is
usually fatal unless appropriate treatment is given. However,
provided the diagnosis is made early and treatment with dantrolene and
necessary supportive measures is given at once, there is rapid
resolution of the hyperthermia. Signs of malignant hyperthermia may
occur from minutes to 1-2 h after the induction of anaesthesia and are
thought to result from an acute influx of calcium into the muscle
cytoplasm from the sarcoplasmic reticulum, which results in abnormal
muscle contraction, hypermetabolism, hyperthermia and muscle damage.
In patients with a family history or previous episodes of
malignant hyperthermia, prophylactic treatment with dantrolene prior
to anaesthesia has been advocated (Shime et al., 1988). The
prophylactic use of dantrolene with susceptible patients is, however,
controversial. Those opposing this approach advocate the agreed
safety precautions and the availability of dantrolene in order to
intervene promptly if any crisis occurs (Harrison, 1988; Hackl et al.,
1990). At present, most anaesthesiologists do not use dantrolene
prophylactically in these patients.
The predisposition for malignant hyperthermia is probably
heterogenous and not only linked to the CRC (calcium release channel)
gene on chromosome 19 (Fagerlund et al., 1992). It is therefore
premature to use DNA markers flanking this gene as the major test to
diagnose susceptibility to the malignant hyperthermia syndrome,
although this has been suggested (Healy et al., 1991). Predisposition
to the disease is still best determined through a halothane- and
caffeine-induced contracture test on a skeletal muscle biopsy
(MacLennan et al., 1990).
Neuroleptic malignant syndrome occurs in 0.2-1% of patients
taking neuroleptics, especially when haloperidol and the depot
fluphenazines are used (Pope et al., 1986). One component of the
syndrome appears to be sustained extrapyramidal rigidity causing
hyperthermia and muscle necrosis (rhabdomyolysis). The syndrome may
last for 10-14 days or 4 weeks after cessation of oral or depot
neuroleptics, respectively.
Dantrolene has also been used successfully in the treatment of a
few cases of heat-stroke (Lydiatt & Hill, 1981), which has many
similarities to malignant hyperthermia.
This monograph will restrict itself to the use of dantrolene in
the treatment of drug-induced hypercatabolic syndromes.
4.2 Name and Chemical Formula of Antidote
Dantrolene sodium
IUPAC name: 1-[[5-( p-nitrophenyl)
furfurylidene]amino]hydantoin sodium
hydrate
Empirical formula: C14H9N4NaO5 (anhydrous salt)
Relative molecular mass: 336 (anhydrous salt)
The hydrated salt contains approximately 15% water (3´ moles) and
has a molecular weight of 399.
CAS numbers: 7261-97-4 Dantrolene
14663-23-1 Dantrolene sodium,
anhydrous
24868-20-0 Dantrolene sodium salt,
hemiheptahydrate
(Martindale, 1989)
Molecular structure:
 Proprietary names: DantriumR (Norwich Eaton UK,
Australia, Belgium, Canada, France,
Netherlands, New Zealand, South Africa,
USA) (Martindale, 1989).
DanleneR (SIT, Italy)
DantamacrinR (Röhm Pharma, D-6108
Weiterstadt);
Boehringer Mannheim, Switzerland)
DantralenR (Lafarquim, Spain)
DantriumR (Norwich Eaton, Norwich NY
13185, USA;
Formenti, I-20149 Milan, Italy; Smith
Kline & French, USA; Yamanouchi, Japan)
DantrixR (SIT, I-27035 Mede, Italy)
(Martindale, 1989;
Index Nominum, 1990)
4.3 Physico-chemical Properties
Dantrolene sodium is an orange, odorless powder with a melting
point of 279-280 °C. It is slightly soluble in water, but it
hydrolyses and precipitates the extremely insoluble (< 1 mg/l) free
acid, dantrolene. This hydrolysis may be prevented to some extent by
the addition of small amounts of sodium hydroxide, but this procedure
is complicated by the precipitation of dantrolene sodium by the common
ion effect. The amount remaining in solution is a function of the
total ionic strength.
The approximate solubility data given in Table 1 were obtained at
room temperature.
Table 1. Solubility of Dantrolene sodium in various solvents
Solvent Solubility (g/l)
Propylene glycol 40
Glycerine 25
Polyethylene glycol 400 80
Dimethylformamide 15a
Dimethylacetamide 15a
0.2% Morpholine in water 0.2
1% Morpholine in water 0.5
2% Morpholine in water 0.9
Chloroform < 20 (70 g/l for dantrolene)
Acetone 20-25
a complex formed
Dantrolene (the free acid of dantrolene sodium) is a weak acid
with a pKa of about 7.5. However, the extremely low solubility of the
free acid prevents an accurate determination of its pKa (personal
communication from A.W. Castellion, Norwich Easton, to the IPCS).
Dantrolene sodium solutions should be protected from light.
Dantrolene sodium capsules and powder for injection should be kept at
a temperature below 40 °C, preferably between 15 and 30 °C, and the
capsules should be stored in well-closed containers. Following
reconstitution with 60 ml of sterile water for injection, dantrolene
sodium injection solution is stable for 6 h when stored at 15-30 °C
and protected from light.
The excipients used with dantrolene are mannitol and sodium
hydroxide
4.4 Pharmaceutical Formulation and Synthesis
Dantrolene sodium is available as capsules of 25 mg, 50 mg and
100 mg.
It is also available as an orange powder for preparing injections
in vials of 20 mg, with 3 g mannitol and sodium hydroxide. The powder
is dissolved by the addition of 60 ml of water, producing a highly
irritant solution with a pH of about 9.5.
Snyder et al. (1967) described the synthesis of a series of 1-
[(5-arylfurfurylidene)amino]hydantoins from the appropriate
aryldiazonium chlorides and 2-furaldehyde, coupled in aqueous acetone
with cupric chloride as the catalyst. The intermediate aldehydes were
condensed directly with 1-aminohydantoin hydrochloride to give the
aminohydantoin derivatives.
4.5 Analytical Methods
4.5.1 Identification and quantification of dantrolene sodium and its
formulation
The thin-layer chromatography details are as follows:
* layer: silica gel G, 250 µm thick
* mobile phase: chloroform:acetone (4:1), retention factor: 19;
ethyl acetate:methanol:strong ammonia solution
(85:10:5), retention factor: 0.9;
ethyl acetate, retention factor: 36 (Moffat,
1986)
An alkaline solution of dantrolene sodium gives a peak in the
ultraviolet spectrum at 314 nm (Moffat, 1986). In the infrared
spectrum, principal peaks occur at the following wavenumbers: 1600,
1225, 1510, 850, 1713, 1108 (dantrolene sodium, KBR disc) (Moffat,
1986).
Saxena et al. (1977) reported a method for the determination of
dantrolene sodium in a dosage form by converting the drug to its free
acid in acidic media (pH 2.5-4.0) using 2N HCl. This is followed by
extraction into a 1-butanol-chloroform mixture and quantification by
high-performance liquid chromatography (HPLC) using carbon
tetrachloride:dimethylformamide (90:10) as mobile phase. The peaks
are detected at 375 nm.
4.5.2 Quantification of dantrolene in body fluids
4.5.2.1 Spectrofluorimetry
This is a rapid and sensitive method for the quantitative
determination of dantrolene in plasma, blood and urine, and consists
of a direct extraction of dantrolene into a nitropropane-heptane (1:1)
solvent mixture. The excitation peak is at 395 nm and the
fluorescence peak at 530 nm (Hollifield & Conklin, 1968). The
sensitivity is 100 ng/ml in plasma, blood and urine, and 400 ng/ml in
bile and tissue (Moffat, 1986)
4.5.2.2 High-performance liquid chromatography
Dantrolene and its metabolites 5-hydroxydantrolene and
nitroreduced acetylated dantrolene (F490) are detected by a reversed-
phase HPLC method after a preliminary extraction step into a
chloroform-butanol mixture for plasma samples. The detection limit is
20 ng/l (Wuis et al., 1983).
Lalande et al. (1988) have reported an alternative method for the
determination of dantrolene and its reduced and oxidized metabolites
in plasma.
4.6 Shelf Life
The following instructions for storage conditions are recommended
by the manufacturer:
Dosage form Storage requirements Shelf-life
As supplied below 30 °C 3 years
Reconstituted 15-30 °C 6 h when protected from
light
4.7 General Properties
Dantrolene is a peripheral striated muscle relaxant agent which
most probably acts by preventing calcium flux across the sarcoplasmic
reticulum, thereby reducing the intracellular free calcium
concentration (Lopez et al., 1987). Its site of action is in the
muscle itself, beyond the motor end-plate. Although this mechanism of
action seems logical, other authors stress that we are still a long
way from understanding the finer details of its mode of action and,
correspondingly, those of the pathogenesis of malignant hyperthermia
(Harrison, 1988). The genetic defect in susceptible individuals is
considered to reside in the calcium release channel (CRC) of the
sarcoplasmic reticulum of skeletal muscle (MacLennan et al., 1990).
The effects of dantrolene on other calcium-dependant systems seem
negligible at doses in current use (Hall et al., 1982).
4.8 Animal Studies
4.8.1 Pharmacodynamics
4.8.1.1 Effect on skeletal muscle
Dantrolene inhibits the development of tension in animal muscle
preparations in vitro caused by caffeine or by depolarization. The
effect of dantrolene can be antagonized by raising the extracellular
calcium concentration (Ellis & Wessells, 1977; Yamamoto et al., 1977;
Anderson et al., 1978; Nelson, 1983).
Dantrolene also inhibits the contractile response to ryanodine,
which is known to cause release of calcium from skeletal muscle
sarcoplasmic reticulum (Fairhust et al., 1980).
Dantrolene, therefore, probably inhibits skeletal muscle
contraction by preventing release of Ca2+ from sarcoplasmic
reticulum. The results obtained by Ohta et al. (1990), using skinned
skeletal muscle from guinea-pigs, suggested that dantrolene acts as a
selective inhibitor of the calcium-induced Ca2+ release mechanism
without having an effect on the Ca2+ pump of the sarcoplasmic
reticulum or on the contractile machinery of skeletal muscle.
Dantrolene also diminishes exercise-induced muscle damage in the
rat, as judged from creatine kinase isoenzyme patterns in plasma
before and after a 2-h run on a treadmill (Amelink et al., 1990).
4.8.1.2 Effects on other tissues
The effects of dantrolene on cardiac muscle, smooth muscle, the
nervous system and endocrine glands have been studied in animal
experiments, and the data have been reviewed by Ward et al. (1986).
These data are not of clinical importance.
4.8.1.3 Studies in malignant hyperthermia-susceptible pigs
Certain breeds of pig exhibit a stress-related syndrome very
similar to that of anaesthetic-induced malignant hyperthermia in
humans, and they are a useful investigational model for malignant
hyperthermia.
Dantrolene inhibits contractures of skeletal muscle preparations
induced by halothane, caffeine, suxamethonium, and potassium chloride
and thymol (Okumura et al., 1980; Sullivan & Denborough, 1981). It
also prevents the accumulation of myocytoplasmic calcium in the
mitochondria of pigs susceptible to malignant hyperthermia
(Stadhouders et al., 1984) and decreases the release of calcium from
the sarcoplasmic reticulum of muscle from susceptible pigs (Ohnishi et
al., 1983). The reduction of the free intracellular calcium
concentration is probably the mechanism of action of dantrolene in
these animals (Lopez et al., 1987). It has been suggested that
dantrolene may act at the initial stage of excitation (Miyamoto &
Racker, 1982).
Dantrolene has been demonstrated to be effective in treating
malignant hyperthermia induced by halothane or suxamethonium
anaesthesia in susceptible pigs (Gronert et al., 1976; Harrison, 1977;
Kerr et al., 1978; Hall et al., 1982). It is also effective when
given prophylactically before the induction of anaesthesia (Harrison,
1977; Kerr et al., 1978).
4.8.2 Pharmacokinetics
The pharmacokinetics of dantrolene have been studied in human
volunteers and this limits the need for animal data in this area.
Following a single oral dose of 5 mg/kg in dogs and 1 mg/kg in
rats, 20% and 29%, respectively, is absorbed (Fournier, 1982).
4.8.3 Toxicology
4.8.3.1 Acute toxicity
The oral LD50 has been estimated to be 29 g/kg in newborn rats.
No deaths occurred after the oral administration of 8 g/kg to young
adult rats, mice, hamsters and rabbits (Fournier, 1982).
The LD50 after the intraperitoneal injection of dantrolene was
780 mg/kg in rats (Fournier, 1982) and 1400 mg/kg in mice (Ellis &
Carpenter, 1974). The intravenous LD50 was > 40 mg/kg in rats and
> 80 mg/kg in mice (Fournier, 1982).
4.8.3.2 Subacute toxicity
Four out of 20 mice given dantrolene (84 mg/kg per day) by mouth
for one month developed hepatic steatosis. Renal tubular damage,
crystal deposition in the renal tract, and hepatocellular necrosis
were seen in rats after they had received oral doses of 60 to 500
mg/kg per day for one month (Fournier, 1982). Intraperitoneal doses
of 50 mg/kg per day lowered the level of serum glucocorticoids over 5
days (Francis & Hamrick, 1980). No anatomical or histological changes
were seen after one month with oral doses of up to 500 mg/kg per day
in dogs or 90 mg/kg per day in monkeys, although animals given high
doses developed anorexia, hypotonia and weight loss (Fournier, 1982).
4.8.3.3 Chronic toxicity
Rats receiving oral dantrolene doses of 15 mg/kg per day or more
showed a reduction in weight gain, hepatocellular degeneration,
crystalluria and keratitis with corneal opacities. Females developed
mammary tumours, with a statistically significant increase in the
incidence of breast adenocarcinoma in those receiving 60 mg/kg per day
(Fournier, 1982).
Dogs showed no effects when treated with dantrolene (15 mg/kg per
day) for 12 months. However, higher doses caused a reduction in
weight gain, and at 60 mg/kg per day impaired hepatocellular function,
anaemia and crystalluria were apparent. One case of intrahepatic
cholestasis occurred (Fournier, 1982).
4.8.3.4 Teratogenicity
There was no evidence of teratogenic effects from dantrolene when
doses of up to 45 mg/kg per day were given to rats, rabbits or monkeys
(Pinder et al., 1977).
Using the maternal-fetal sheep model in nine pregnant ewes, Craft
et al. (1988) found an equilibrium between maternal and fetal plasma
dantrolene concentrations 5 min after dosing. The fetal level of
dantrolene was about 10% of that of the mother. It was concluded that
the administration of intravenous dantrolene (1.2 or 2.4 mg/kg) had no
clinically significant adverse effect on mother or fetus in the sheep
model.
4.9 Volunteer Studies
Included in this section are studies involving healthy volunteers
and patients undergoing surgery from whom informed consent was
obtained.
4.9.1 Administration and plasma concentrations
Peak plasma dantrolene concentrations of 0.5 to 0.95 mg/l
occurred 4 to 8 h after 50 mg dantrolene was administered orally to
six healthy volunteers. The corresponding peak 5-hydroxydantrolene
concentration was 0.11 to 0.3 mg/l after 6 to 8 h (Katogi et al.,
1982). In the study of Allen et al. (1988), a total oral dose of 5 mg
dantrolene/kg was given to ten malignant hyperthermia-susceptible
patients prior to anaesthesia. All subjects had plasma dantrolene
levels above 2.8 mg/l, indicating a high bioavailability of
dantrolene.
In six patients, who had previously suffered from suspected or
proven malignant hyperthermia and who received prophylactic
intravenous dantrolene (2.5 mg/kg) before anaesthesia, peak blood
concentrations of 4.3 to 6.5 mg/l were found (Flewellen & Nelson
1985). Lerman et al. (1989) infused dantrolene (2.4 mg/kg) over 10
min in 10 children susceptible to malignant hyperthermia. Mean
dantrolene blood concentrations 1 min and 1 h after infusion were 6.03
mg/l (SD ± 0.93) and 3.56 (SD ± 0.49) mg/l, respectively. The blood
concentrations of the metabolite 5-hydroxydantrolene rose to a maximum
of 0.6 mg/l at 8 h and then fell with an apparent half-life of 9 h.
Plasma dantrolene concentrations during prolonged treatment are
similar to those found after single oral dosing (Vallner et al., 1979;
Meyler et al. 1981). Metabolite concentrations may be relatively
elevated after prolonged (> 2 months) administration (Vallner et al.
1979).
4.9.2 Distribution
In the 10 children studied by Lerman et al. (1989), the apparent
volume of distribution for dantrolene was 0.54 l/kg (SD ± 0.08). In
vitro studies show that dantrolene interacts with human serum
albumin at a minimum of two sites on the protein (Vallner et al.,
1976), but exact figures for the degree of protein binding have not
been reported.
4.9.2.1 Distribution to the fetus and newborn baby
The oral administration of dantrolene (total doses of 250 and 600
mg) to two pregnant patients thought to be susceptible to malignant
hyperthermia resulted in a fetal/maternal serum concentration ratio of
approximately 0.4, thus indicating that this agent reaches the
placenta in appreciable concentrations (Morison 1983). In a study of
20 pregnant women susceptible to malignant hyperthermia, the mean
maternal predelivery dantrolene level was 0.99 ± 0.5 mg/l and the mean
neonatal cord blood dantrolene level 0.68 ± 0.3 mg/l (Shime et al.,
1988).
4.9.3 Elimination
The major metabolite of dantrolene in humans is
5-hydroxydantrolene, produced by hepatic microsomal oxidation,
although minor metabolites also exist (Lietman et al., 1974; Ellis &
Wessels, 1978). According to in vitro data, this major metabolite
is half as potent as the parent drug, whereas the other metabolites
appear to be inactive (Ellis & Wessels, 1978). When dantrolene is
administered orally, 15-25% of the dose is excreted renally,
predominantly as 5-hydroxydantrolene but with small amounts of reduced
acetylated dantrolene and unchanged drug (Lietman et al., 1974).
The elimination half-life of orally administered dantrolene in
humans is probably between 6 and 9 h, although values of 3 to 22 h
have been observed (Lietman et al., 1974; Dykes 1975; Meyler et al.,
1979, 1981; Wuis et al., 1983; Allen et al., 1988). The elimination
half-life after intravenous dosing was reported to be 12 h in both
healthy volunteers (Flewellen et al., 1983) and patients known to
suffer from malignant hyperthermia (Flewellen & Nelson, 1985). This
is in accordance with the value of 10 h (SD ± 2.6) reported in
children (Lerman et al., 1989). In the study by Shime et al. (1988)
in neonatals, the dantrolene half-life was 20 h.
The median elimination half-life for 5-hydroxydantrolene, the
major metabolite of dantrolene, was found to be 15.5 h (range, 8.1 to
29.4 h) in healthy volunteers (Meyler et al., 1979), whereas it was 9
h (SD ± 2.5) in the 10 children studied by Lerman et al. (1989).
4.9.4 Human in vitro pharmacodynamics
Dantrolene inhibits the contraction of isolated human skeletal
muscle induced by halothane or suxamethonium (succinylcholine) (Nelson
& Denborough 1977; Hallsall & Ellis 1979; Fletcher & Rosenberg 1985).
4.10 Clinical Studies - Clinical Trials
The rarity of malignant hyperthermia, the variability of its
clinical manifestations and the range of adjunctive therapy have
precluded controlled trials or comparative studies of dantrolene in
its treatment.
4.11 Clinical Studies - Case Reports
This section presents several case reports where the use of
dantrolene is associated with a favourable outcome. Even if some of
the case reports (or series of such reports) may seem convincing,
regarding the efficacy of dantrolene, one should still bear in mind
the lack of controlled studies.
4.11.1 Use in malignant hyperthermia
Ward et al. (1986) and Harrison (1988) have reviewed the
published case reports. In children, an initial intravenous dose of
1 mg/kg has usually been effective if given within 2 h of the onset of
signs and symptoms, although a boy of 11 years died in spite of
receiving dantrolene (1 mg/kg) about 2 h after the onset of malignant
hyperthermia (Desparmet et al., 1983). Doses of up to 3.6 mg/kg have
been given to patients who subsequently recovered (Maruta et al.,
1980). Experienced clinicians prefer a more aggressive dosing of 1
mg/kg per min up to a total of 10 mg/kg or even more (personal
communications by B.A. Britt and T. Fagerlund to the IPCS, 1992). The
time factor is critical, since dantrolene is often ineffective if
treatment is delayed for 2 h after the onset of signs in children
(personal communication by B.A. Britt to the IPCS, 1992).
In adults, dantrolene appears uniformly effective if given within
6 h of the precipitating anaesthetic agent, but death may supervene if
treatment is delayed beyond this (Kolb et al., 1982; personal
communication by B.A. Britt to the IPCS, 1992). Death occurred in one
case despite a massive initial intravenous dose (Mathieu et al., 1979)
and in two others despite prolonged oral and intravenous therapy over
10 to 21 days (Kolb et al., 1982). Presumably irreversible changes
took place before treatment was started. In the study by Kolb et al.
(1982), intravenous dantrolene (a total dose of 1-7 mg/kg) lead to the
successful treatment of three patients with probable and eight
patients with unequivocal malignant hyperthermia.
4.11.1.1 Prophylaxis of malignant hyperthermia
Where a family history of malignant hyperthermia has been
particularly strong or where a patient has had previous confirmed or
suspected episodes of anaesthetic-induced malignant hyperthermia, the
prophylactic administration of dantrolene has been discussed, along
with the avoidance of "trigger" agents (Ward et al., 1986). An oral
dosage of 4 to 8 mg/kg per day, given in three or four divided doses
for 1 or 2 days with the last dose being administered 3 to 4 h prior
to anaesthesia, has been recommended (Ward et al., 1986).
Alternatively, a single intravenous dose of 2.5 mg/kg has been
proposed, to be given just prior to surgery (Flewellen et al., 1983).
Most of the patients treated in this manner have undergone
uneventful surgery although in three cases postoperative tachycardia,
increased blood pressure, and respiratory and metabolic acidosis
occurred without hyperthermia or muscle rigidity. These patients were
all successfully treated with either further intravenous dantrolene
(Fitzgibbons, 1981; Ruhland & Hinkle, 1984) or by correcting the
metabolic acidosis (de Pinna, 1978).
There is, however, no consensus on this prophylactic use of
dantrolene (Harrison, 1988) although most experienced clinicians are
now in favour of using no prophylactic treatment (personal
communication by B.A Britt and T. Fagerlund to the IPCS, 1992).
Recently, a series of 30 general anaesthesias in 24 patients
susceptible to malignant hyperthermia was reported. In all patients,
malignant hyperthermia susceptibility was confirmed by in vitro
testing of skeletal muscles obtained from a biopsy of the quadriceps
femoris muscle, and four patients had experienced previous episodes of
malignant hyperthermia during anaesthesia. No clinical or biochemical
features of malignant hyperthermia were observed (Hackl et al., 1990).
These authors concluded that safe anaesthesia for these patients could
easily be provided simply by avoiding trigger agents and carefully
preparing the monitoring equipment and anaesthesia machine.
Dantrolene should be reserved for the treatment of malignant
hyperthermia.
4.11.1.2 Prophylaxis of malignant hyperthermia during pregnancy
In a prospective study of 32 pregnant women known to be
susceptible to malignant hyperthermia, 17 received oral dantrolene
sodium beginning 5 days prior to planned delivery, while the other 15
did not receive prophylactic dantrolene. Malignant hyperthermia
developed immediately postpartum in one mother and her baby who had
not received prophylactic dantrolene, but both mother and baby
responded rapidly to intravenous dantrolene. None of the other
pregnancies was complicated and all fetal examinations and neonatal
neurological assessments were normal (Ward et al., 1986).
In a series of 20 malignant hyperthermia-susceptible pregnant
patients, dantrolene was given orally for 5 days before and 3 days
after delivery (in three cases, delivery was by Caesarean section)
(Shime et al., 1988). No signs of malignant hyperthermia or adverse
effects of dantrolene were seen in this non-controlled study.
There have been several other case reports of successful use of
prophylactic intravenous dantrolene in pregnant women who were
susceptible to this condition and who underwent anaesthesia during
birth (Cupryn et al., 1984; Glassenberg & Cohen, 1984) and of the
reversal of established malignant hyperthermia with intravenous
dantrolene in both mothers and their neonates (Sewall et al., 1980;
Lips et al., 1982). Weingarten et al. (1987) reported a case of
postpartum uterine atony in a patient who received dantrolene during
a Caesarian section.
Although there are several non-controlled reports of the
successful use of dantrolene prior to planned delivery, there is no
established consensus on this use.
4.11.2 Use in neuroleptic malignant syndrome
In the neuroleptic malignant syndrome, thermogenesis is
ultimately due to tonic contraction of skeletal muscles. Thus, the
use of dantrolene may be helpful by relaxing skeletal muscle.
Since the initial observations (Bismuth et al., 1982; Boles et
al., 1982), there have been many case reports of the use of dantrolene
in the neuroleptic malignant syndrome (Ward et al., 1986; Harrison
1988) with no consensus concerning dosage. Single intravenous bolus
doses (less than 3 to 10 mg/kg) and repeated oral doses (25 to 600
mg/day) have been used, often in combination with other drugs and
supportive treatment. Dantrolene apparently reduces the pyrexia,
usually within 12 h of intravenous therapy and over a somewhat longer
period with oral dantrolene therapy. Most patients also improve
clinically, although this occurs at a later stage. In some cases
withdrawal of oral dantrolene therapy has been associated with a
deterioration of the patient's condition and an increase in body
temperature.
4.11.3 Use in other drug-induced hyperthermia
Hyperthermia due to or associated with increased muscular
rigidity can occur in poisoning with strychnine (Gordon & Richards,
1979), phencyclidine (Jan et al., 1978) or Cicuta species (water
hemlock) (Starreveld & Hope, 1975). In cases of poisoning with these
agents, the direct action of dantrolene on muscle activity is likely
to be of benefit in controlling the hyperpyrexia. It would be logical
to give intravenous dantrolene as in cases of malignant hyperthermia,
provided it was combined with standard supportive measures such as
external cooling. There is, however, no experimental verification of
this view, nor any report of a relevant clinical case.
Amphetamines (Jordan & Hampson, 1960; Kendrick et al., 1977;
Ginsberg et al., 1970), monoamine oxidase inhibitors (Mirchandani &
Reich, 1985), lysergic acid diethylamide (Friedman & Hirsch, 1971;
Mercieca & Brown, 1984), and cocaine (Roberts et al., 1984) all cause
hyperthermia, which is due at least in part to motor overactivity with
or without convulsions. Where hyperthermia does not respond to
sedation with a major tranquillizer, and where paralysis and
artificial ventilation are inappropriate or ineffective, dantrolene
should be of benefit, but documentation is still lacking.
A case of overdosage with phenelzine, a monoamine oxidase
inhibitor, was treated successfully with intravenous dantrolene after
conventional therapy had failed (Kaplan et al. 1986), as was a case of
carbon monoxide poisoning with hyperthermia and rigidity (Holter &
Schellens, 1988). In a fatal case of theophylline poisoning with
rhabdomyolysis, dantrolene administration was claimed to be useful in
controlling the hypermetabolic state (Parr & Willatts, 1991).
Hyperthermia in poisoning with salicylates, dinitrophenol and the
herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is due to uncoupling
of oxidative phosphorylation. Tricyclic antidepressives and other
anticholinergic drugs reduce sweating and so impair heat loss.
Dantrolene would not be expected to have a role in the treatment of
hyperpyrexia induced by these mechanisms.
4.12 Summary of Evaluation
4.12.1 Indications
4.12.1.1 Treatment of malignant hyperthermia
When combined with standard cooling and supportive therapy,
dantrolene has proved effective in rapidly reversing clinical symptoms
in anaesthetic-induced malignant hyperthermia, as judged from clinical
case reports. Failure of dantrolene to prevent the often fatal
consequences of malignant hyperthermia is usually a consequence of
late diagnosis and therapy.
4.12.1.2 Treatment of neuroleptic malignant syndrome
Dantrolene may be helpful as an adjunct to supportive therapy in
neuroleptic malignant syndrome induced by drugs such as dopamine
antagonists, particularly the major tranquillizers. There is no
report of a controlled trial in this rare illness, and no study
comparing dantrolene with bromocriptine, which has also been used in
its management.
4.12.1.3 Treatment of hyperthermia induced by muscle rigidity in
poisoning
Though documentation is lacking, there are good theoretical
grounds for using dantrolene to treat hyperthermia due to poisoning
with strychnine, phencyclidine or Cicuta species (water hemlock). It
might also be useful in the management of hyperthermia in patients
poisoned with amphetamines, cocaine, lysergic acid diethylamine (LSD)
or monoamine oxidase inhibitors, where hyperthermia is associated with
motor overactivity.
4.12.2 Advised routes and doses
4.12.2.1 Treatment of severe drug-induced hyperthermia, including
malignant hyperthermia
Dantrolene (2.5 mg/kg) should be given intravenously as soon as
possible. A response to the treatment should be apparent within
minutes; if not, up to 10 mg/kg may be given again every 15 min until
there is an effect. Subsequent to intravenous therapy, dantrolene (4
mg/kg per day in divided doses for 48 h) can be given orally to
prevent recurrence. The patient should stay in the intensive care
unit for at least 24 h after the hyperthermia reaction has subsided
(personal communication by T. Fagerlund to the IPCS, 1992). The
proposed dosage regimen for dantrolene is not clearly defined (see
section 4.12.4).
In cases of neuroleptic malignant syndrome, the use of dantrolene
and dopamine agonists (amantadine and bromocriptine) has been
described (Boles et al., 1982; Goulon et al., 1983) and has produced
promising results.
4.12.2.2 Prophylaxis of malignant hyperthermia prior to anaesthesia
in susceptible patients
Triggering anaesthetic agents have to be avoided in patients
known to be susceptible to malignant hyperthermia. Such agents are
depolarizing muscle relaxants (succinylcholine), halogenated
inhalational anaesthetics (e.g., halothane), haloperidol and
promethazine. Regional anaesthetic techniques should be preferred if
possible.
Oral dantrolene (4-8 mg/kg per day) administered for 1-2 days
prior to anaesthesia may prevent malignant hyperthermia in known
susceptible individuals. Alternatively, intravenous dantrolene (2.5
mg/kg) can be given just prior to anaesthesia (Flewellen et al.,
1983). Oral dantrolene given 5 days before and 3 days after delivery
has been associated with favourable outcome in pregnant patients
susceptible to malignant hyperthermia (Shime et al., 1988). Such
prophylaxis with dantrolene is controversial and not generally
recommended.
4.12.3 Other consequential or supportive therapy
It is very important for the administration of dantrolene to be
accompanied by the use of aggressive supportive therapy, including
central cooling techniques, an inspired oxygen concentration enriched
up to 100%, and correction of metabolic acidosis. Serum potassium
concentrations should be monitored closely.
Further supportive therapy should be directed towards
complications such as respiratory acidosis, cardiac arrhythmias, and
instability of blood pressure. Special attention must be given to the
development of rhabdomyolysis leading to elevated serum muscle enzymes
(creatine kinase, aspartate transaminase, aldolase). If creatine
kinase activities are above 10 000 U/l and/or meat-coloured urine is
present, together with areas of swollen and tender skeletal muscles,
the patient may develop acute renal failure as muscle debris is
precipitated in the kidneys (myoglobinuria). Associated electrolyte
disturbances are hyperkalaemia, hypocalcaemia and hyperphosphataemia.
Acute renal failure of this nature has been reported to be prevented,
even at creatine kinase activities above 70 000 U/l, when prompt
forced alkaline diuresis treatment has been instituted in order to
prevent debris deposition in the kidneys (personal communication by D.
Jacobsen to the IPCS, 1992). Urine output should exceed 2 ml/kg per
h in these patients.
4.12.4 Controversial issues and areas of insufficient information
The prophylactic use of dantrolene prior to anaesthesia in
susceptible patients should be considered controversial and an
international consensus has yet to be established (Ward et al., 1986;
Shime et al., 1988; Harrison, 1988; Hackl et al., 1990). This use of
dantrolene is not generally recommended (personal communications by T.
Fagerlund and B.A. Britt to the IPCS, 1992).
The dose regimen of dantrolene is not clearly defined. The
initial dose recommended in the USA is generally 1 mg/kg per min
(personal communication by B.A. Britt to the IPCS, 1992), whereas the
European recommendation is usually an initial dose of 2.5 mg/kg
(personal communication by T. Fagerlund to the IPCS, 1992). There is,
however, a consensus on the maximum dose of 10 mg/kg per 15 min.
Furthermore, there is no clear overall maximum dose of dantrolene
during the first hours of treatment. This may be important, as
malignant hyperthermia could be incorrectly diagnosed in places with
limited laboratory facilities. In such situations, one should not
continue to give dantrolene in doses of 10 mg/kg per 15 min.
Therefore, if more than 20 mg/kg has been given during the first 30
min without any effect, the diagnosis of malignant hyperthermia should
certainly be questioned (personal communication by T. Fagerlund to the
IPCS, 1992). Another reason for lack of effect could be that
dantrolene therapy has been initiated too late, since the time factor
is critical.
It is not clear whether doses higher than 10 mg/kg per day add
any beneficial effects concerning the prognosis. However, clinicians
with experience in this field do recommend higher doses for critically
ill patients who do not respond to the initial dose (personal
communications by T. Fagerlund and B.A. Britt to the IPCS, 1992).
The use of external cooling techniques with these patients has
been generally recommended for years. However, this treatment is now
being questioned, since dantrolene treatment alone lowers temperatures
to the normal range. External cooling also prevents heat loss by
inducing peripheral vasoconstriction and increases heat production by
stimulating shivering and non-shivering thermogenesis (personal
communication by B.A. Britt to the IPCS, 1992).
4.12.5 Proposals for further studies
The effect of dantrolene in established drug-induced hyperthermia
has not been documented from a scientific point of view. However, the
effect, as indicated in several case reports (and series of such
reports), is so convincing that it may be difficult to perform a
controlled double-blind study due to ethical considerations.
The prophylactic use of dantrolene prior to anaesthesia in
susceptible patients may warrant a controlled study. Due to the
relatively small number of patients, this should preferably be done
under a multicenter design.
In cases of neuroleptic malignant syndrome, a treatment protocol
using dantrolene versus dantrolene/bromocriptine may be warranted
(Boles et al., 1982; Goulon et al., 1983).
The optimum dose regimen of dantrolene is not clearly defined and
warrants further study.
4.12.6 Adverse effects
Dantrolene sodium solution is highly alkaline (pH 9.6) and
extravasation during intravenous injection may cause tissue necrosis
(Harrison, 1988). Due to this risk of severe thrombophlebitis,
intravenous administration should preferably be given through a
central venous catheter. Apart from this, dantrolene appears to be
well tolerated for the short-term use discussed in this monograph
(Kolb et al., 1982, Shime et al., 1988).
Several side effects have been reported in patients receiving
chronic dantrolene treatment for spasticity. Muscular weakness,
drowsiness, dizziness and general malaise commonly occur and
gastrointestinal disturbances are seen less frequently. Rare adverse
effects have included hallucinations (Andrews et al., 1975),
exacerbation of respiratory depression (Rivera et al., 1975),
pleuropericardial reaction (Petusevsky et al., 1979; Miller & Haas
1984), lymphocytic lymphoma (Wan & Tucker, 1980), and leucopenia
(Greenspun & Pacho, 1981). The most serious reaction in long-term use
is hepatotoxicity.
4.12.6.1 Hepatotoxicity
Hepatotoxicity may occur when patients are treated with
dantrolene, as indicated in a review by Chan (1990). In general,
therapy was given for at least two months before injury became
evident. In 107 adult cases of dantrolene hepatotoxicity, 40 (37%)
received 200 mg/day or less; 48 (45%) received up to 400 mg/day, and
the highest dose given was 1600 mg/day (Chan, 1990). Biochemical
abnormalities of liver function were observed in about 1.8% of
patients being given long-term dantrolene treatment and fatal
hepatitis occurred in 0.3%. The risk of hepatic injury was greatest
in females, in patients receiving doses over 300 mg/day, and in those
treated for over 60 days (Utili et al., 1977). The major histological
pathology was subacute hepatic necrosis or chronic active hepatitis,
although cholestasis has been reported.
4.12.6.2 Interaction with calcium antagonists
The combination of calcium antagonists and dantrolene may result
in systemic hyperkalaemia and even cardiovascular collapse, and should
therefore be avoided (Yoganathan, 1988).
4.12.7 Restrictions for use
When dantrolene was used prophylactically 5 days before delivery
to twenty pregnant women, no adverse effect on the fetus or newborn
infant could be detected (Shime et al., 1988). A possible teratogenic
effect could, however, not be ruled out from these data.
Reconstituted dantrolene should not be used if more than 6 h has
passed since it was made up or if the solution has not been protected
from light.
4.13 Model Information Sheet
The current theory is that dantrolene relaxes peripheral striated
muscle by preventing Ca2+ flux across the sarcoplasmic reticulum and
thereby reducing the free intracellular calcium concentration. Since
malignant hyperthermia and other drug-induced hyperthermias are
considered to be a paroxysmal hypercatabolic reaction of these
muscles, the hyperpyrexia is counteracted by the effect of dantrolene.
When the doses recommended in this monograph are used, dantrolene has
no significant effect on other calcium-dependent systems.
4.13.1 Uses as an antidote
Use of dantrolene is indicated in:
a) the treatment of malignant hyperthermia induced in susceptible
individuals by anaesthetic agents or skeletal muscle relaxants;
b) the treatment of malignant neuroleptic syndrome;
c) the treatment of hyperpyrexia due to poisoning with strychnine,
Cicuta species (water hemlock) or phencyclidine, and perhaps also
hyperpyrexia due to poisoning with amphetamines, cocaine,
lysergic acid diethylamine (LSD), theophylline or monoamine
oxidase inhibitors.
The prophylactic use of dantrolene prior to anaesthesia to
susceptible patients should be considered controversial and is not
generally recommended.
4.13.2 Dosage and route
Whenever indicated, dantrolene treatment must be started without
delay. Dantrolene is often ineffective if treatment is delayed for 2
h after the onset of signs in children, or 6 h after the onset of
signs in adults.
In the treatment of severe drug-induced hyperthermia, including
acute malignant hyperthermia induced by anaesthetic agents, dantrolene
must be given intravenously as soon as the diagnosis is made. The
initial dose is 1-2.5 mg/kg given intravenously (1 mg/kg per min), and
the effectiveness of the treatment should be monitored closely. A
dose of up to 10 mg/kg may be given again every 15 min (1 mg/kg per
min) if signs and symptoms do not subside. Doses greater than 10
mg/kg are rarely needed.
The recurrence of symptoms should be treated in the same way. To
prevent this, dantrolene can be given orally in divided doses at a
total dosage of 4 mg/kg per day.
Provided adequate supportive therapy (section 12.3) is given,
dantrolene treatment can usually be stopped within 2 days in cases of
malignant hyperthermia. Oral treatment may be given for up to 10-14
days in cases of malignant neuroleptic syndrome, or even longer if
depot neuroleptics have precipitated the syndrome.
In the controversial prophylaxis of malignant hyperthermia in
susceptible individuals, dantrolene may either be given orally at a
dosage of 4-8 mg/kg per day for 1 to 2 days prior to anaesthesia, or
intravenously as a single dose of 2.5 mg/kg 1-2 h prior to
anaesthesia. This approach is, however, not generally recommended.
4.13.3 Precautions and contraindications
Solutions of dantrolene should be protected from light.
Solutions of dantrolene are strongly alkaline (pH 9.5).
Extravasation may cause tissue necrosis. The dantrolene solution
should therefore be injected via a fast-flowing intravenous infusion
into a large vein, or preferably through a central venous catheter.
Hepatic failure should be considered as a relative
contraindication and there should be a firm diagnosis for the use of
dantrolene is this situation (e.g., no prophylactic use).
4.13.4 Pharmaceutical incompatibilities and drug interactions
Dantrolene sodium powder should only be diluted with sterile
water for injection, and is incompatible with other infusion fluids.
The combination of dantrolene and calcium antagonists should be
avoided, as severe hyperkalaemia and myocardial depression may occur.
Treatment with calcium antagonists should therefore be discontinued
if dantrolene is given or may be given.
4.13.5 Adverse effects
The major adverse effect is hepatic toxicity, which may be fatal,
although this is unlikely in the acute setting discussed here. Few
side effects have been reported at the dose levels recommended in this
monograph.
In chronic treatment (muscle rigidity), dantrolene may also cause
muscle weakness, drowsiness, dizziness and malaises. Hallucinations,
exacerbation of respiratory depression, pleuroperitonitis, lymphocytic
lymphoma and leucopenia have been reported rarely.
When given intravenously, dantrolene solution is highly irritant.
Extravasation may cause tissue necrosis.
4.13.6 Use in pregnancy and lactation
There is no evidence that dantrolene is harmful when given
prophylactically to predisposed mothers before delivery. No adverse
effects on the fetus or newborn infant have been reported. Dantrolene
crosses the placenta (the fetal:maternal concentration ratio is 0.4:1)
and is excreted in breast milk. It would be prudent to avoid breast
feeding if dantrolene were being taken for prophylaxis or treatment.
4.13.7 Storage
Dantrolene powder and capsules are stable at temperatures below
40 °C. The capsules should be stored in well-sealed containers.
Following reconstitution with sterile water, dantrolene sodium
injection solution is stable for only 6 h at room temperature.
Solutions should be protected from light.
4.14 References
Allen GC, Cattran CB, Peterson RG, & Lalande M (1988) Plasma levels
of dantrolene following oral administration in malignant hyperthermia-
susceptible patients. Anaesthesiology, 69: 900-904.
Amelink GJH, Van der Kallen CJH, Wokke JHJ, & Bar PRD (1990)
Dantrolene sodium diminishes exercise-induced muscle damage in the
rat. Eur J Pharmacol, 179: 187-192.
Anderson IL, Lipicky RJ, & Jones EW (1978) Dantrolene sodium in
porcine malignant hyperthermia: Studies on isolated muscle strips. In:
Adrete JA & Britt BA ed. Second International Symposium on Malignant
Hyperthermia, Denver, Colorado, 1-3 April 1977. New York, Grune &
Stratton, pp 509-534.
Andrews LG, Muzumdar AS, & Pinkerton AC (1975) Hallucinations
associated with dantrolene sodium therapy. Can Med Assoc J, 112: 148.
Bismuth C, Elkharrat D, Rohan-Chabot P, & Conso F (1982) Theoretical
indication of dantrolene in neuroleptic malignant syndrome. Efficacy
in three cases. Vet Hum Toxicol, 24(Suppl 1): 52-55.
Boles JM, Lecam B, Mialon P, Pennec Y, & Garre M (1982) Hyperthermie
maligne des neuroleptiques: guérison rapide par le dantrolène. Nouv
Presse Méd, 11: 674.
Chan CH (1990) Dantrolene sodium and hepatic injury. Neurology, 40:
1427-1432.
Craft JB, Goldberg NH, Lim M, Landsberger E, Mazel P, Abramson FP,
Stolte AL, Braswell ME, & Farina JP (1988) Cardiovascular effects and
placental passage of dantrolene in the maternal-fetal sheep model.
Anaesthesiology, 68: 68-72.
Cupryn JP, Kennedy A, & Byrick RJ (1984) Malignant hyperthermia in
pregnancy. Am J Obstet Gynecol, 150: 327-328.
De Pinna GA (1978) A case of osteogenesis imperfecta, malignant
hyperthermia susceptible - anesthetic management. In: Aldrete JA &
Britt BA ed. Malignant hyperthermia. New York, Grune & Stratton, pp
409-418.
Desparmet J, Josse E, Bavoux F, Laguenie G, & Saint-Maurice C (1983)
Fatal malignant hyperthermia in an eleven-year old child. Cah
Anesthésiol, 31: 303-305.
Dykes HHM (1975) Evaluation of a muscle relaxant: Dantrolene sodium
(Dantrium). J Am Med Assoc, 231: 862-864.
Ellis KO & Carpenter JF (1974) A comparative study of dantrolene
sodium and other skeletal muscle relaxants with the Straub tail mouse.
Neuropharmacology, 13: 211-214.
Ellis KO & Wessels FL (1977) Dantrolene sodium effects on caffeine
contractures in frog sartorius muscle. In Wassermann RH ed. Calcium-
binding proteins and calcium function: Proceedings of the 2nd
International Symposium on Calcium-Binding Proteins and Calcium
Function in Health and Disease, Cornell University, Ithaca, New York,
5-9 June 1977. Amsterdam, Elsevier North-Holland, pp 273-274.
Ellis KO & Wessels FL (1978) Muscle relaxant properties of the
identified metabolites of dantrolene. Naunyn-Schmiedeberg's Arch
Pharmacol, 301: 237-240.
Fagerlund T, Islander G, Ranklew E, Harbitz I, Hauge JG, Mokleby E, &
Berg K (1992) Genetic recombination between malignant hyperthermia
and calcium release in skeletal muscle. Clin Genet, 41: 270-272.
Fairhurst AS, Hamamoto V, & Macri J (1980) Modification of ryanodine
toxicity by dantrolene and halothane in a model of malignant
hyperthermia. Anaesthesiology, 53: 199-204.
Fitzgibbons DC (1981) Malignant hyperthermia following preoperative
oral administration of dantrolene. Anaesthesiology, 54: 73-75.
Fletcher JE & Rosenberg H (1985) In vitro interaction between
halothane and succinylcholine in human skeletal muscle: implications
for malignant hyperthermia and masseter muscle rigidity.
Anaesthesiology, 63: 190-194.
Flewellen EH & Nelson TE (1985) Intravenous dantrolene
pharmacokinetics in malignant hyperthermia suspect patients.
Anaesthesiology, 63(Suppl 3A): 300.
Flewellen EH, Nelson TE, Jones WP, Arens JF, & Wagner DL (1983)
Dantrolene dose response in awake man: implications for management of
malignant hyperthermia. Anaesthesiology, 59: 275-280.
Fournier P (1982) Dossier toxicologique, pharmacologique,
pharmacocinétique du Dantrium IV. Dantrium et hyperthermie maligne.
Lyon, Laboratoire Oberval (Unpublished internal report).
Francis KT & Hamrick ME (1980) Effects of dantrolene on adrenal
cortical function. Biochem Pharmacol, 29(12): 1669-1672.
Friedman SA & Hirsch SE (1971) Extreme hyperthermia after LSD
ingestion. J Am Med Assoc, 217: 1549-1550.
Ginsberg MD, Hertzman M, & Schmidt-Nowara WW (1970) Amphetamine
intoxication with coagulopathy hyperthermia and reversible renal
failure. Ann Intern Med, 75: 81-85.
Glassenberg R & Cohen H (1984) Intravenous dantrolene in a pregnant
malignant hyperthermia susceptible patient. Anesthesiology, 61(Suppl
3): 404.
Gordon AM & Richards DW (1979) Strychnine intoxication. J Am Coll
Emergency Physicians, 8: 520-522.
Goulon M, De Rohan-Chabot P, Elkharrat D, Gajdos PH, Bismuth C, &
Conso F (1983) Beneficial effect of dantrolene in the treatment of
neuroleptic malignant syndrome: a report of two cases. Neurology, 33:
516-518.
Greenspun B & Pacho A (1981) Leukopenia with dantrolene sodium. Arch
Phys Med Rehabil, 62: 521.
Gronert GA, Milde JH, & Theye RA (1976) Dantrolene in porcine
malignant hyperthermia. Anaesthesiology, 44: 488-495.
Hackl W, Mauritz W, Winkler M, Sporn P, & Sternberetner K (1990)
Anaesthesia in malignant hyperthermia-susceptible patients without
dantrolene prophylaxis: a report of 30 cases. Acta Anaesthesiol Scand,
34: 534-537.
Hall GM, Lucke JN, Orchard C, Lovell R, & Lister D (1982) Effect of
dantrolene on leg metabolism in porcine malignant hyperthermia.
Ananesthesia, 37: 1167-1170.
Hallsall PJ & Ellis FR (1979) A screening test for the malignant
hyperpyrexia phenotype using suxamethonium-induced contracture of
muscle treated with caffeine, and its inhibition by dantrolene. Br J
Anaesthesiol, 51: 753-756.
Harrison G (1977) The control and prevention of malignant hyperthermia
in MHS pigs. Some experimental observations. In: Anaesthesiology.
Amsterdam, Exerpta Medica, pp 452-454.
Harrison GG (1988) Malignant hyperthermia. Dantrolene dynamics and
kinetics. Br J Anaesthesiol, 60: 279-286.
Healy JMS, Heffron JJA, Lehane M, Bradley DG, Johnson K, & McCarthy TV
(1991) Diagnosis of susceptibility to malignant hyperthermia with
flanking DNA markers. Br Med J, 303: 1225-1228.
Hollifield RD & Conklin JD (1968) A spectrophotofluorometric procedure
for the determination of dantrolene in blood and urine. Arch Int
Pharmacodyn, 174: 333-341.
Holter MT & Schellens LAM (1988) Dantrolene sodium for treatment of
carbon monoxide poisoning. Br Med J, 296: 1772-1773.
Index Nominum (1990) International drug directory. Zurich, Medpharm.
Jan KM, Dorsey S, & Bernstein A (1978) Hot hog: hyperthermia from
phencyclidine. New Engl J Med, 299: 722.
Jordan SC & Hampson F (1960) Amphetamine poisoning associated with
hyperpyrexia. Br Med J, 2: 844.
Kaplan RF, Feinglass NG, Webster W, & Mudra S (1986) Phenelzine
overdose treated with dantrolene sodium. J Am Med Assoc, 255: 642-644.
Katogi Y, Tamaki N, & Adachi M (1982) Simultaneous determination of
dantrolene and its metabolite, 5-hydroxydantrolene, in human plasma by
high-performance liquid chromatography. J Chromatogr, 228: 404-408.
Kendrick WC, Hull AR, & Knochel JP (1977) Rhabdomyolysis and shock
after intravenous amphetamine administration. Ann Intern Med, 86:
381-387.
Kerr DD, Wingard DW, & Gatz EE (1978) Prevention of porcine malignant
hyperthermia by oral dantrolene. In: Aldrete JA & Britt BA ed. Second
International Symposium on Malignant Hyperthermia, Denver, Colorado,
1-3 April 1977. New York, Grune & Stratton, pp 499-507.
Kolb ME, Horne ML, & Martz R (1982) Dantrolene in human malignant
hyperthermia. Anesthesiology, 56: 254-262.
Lalande M, Mills P, & Peterson RG (1988) Determination of dantrolene
and its reduced and oxidized metabolites in plasma by high-performance
liquid chromatography. J Chromatogr, 430: 187-191.
Lerman J, McLeod ME, & Strong HA (1989) Pharmacokinetics of dantrolene
in children. Anaesthesiology, 70: 625-629.
Lietman PS, Haslam RHA, & Walcher JR (1974) Pharmacology of dantrolene
sodium in children. Arch Phys Med Rehabil, 55: 388-392.
Lips FJ, Newland M, & Dutton G (1982) Malignant hyperthermia
triggered by cyclopropane during caesarian section. Anesthesiology,
56: 144-146.
Lopez JR, Allen P, Alamo L, Ryan JF, Jones DE, & Sreter F (1987)
Dantrolene prevents the malignant hyperthermic syndrome by reducing
free intracellular calcium concentration in skeletal muscle of
susceptible swine. Cell calcium, 8: 385-396.
Lydiatt JS & Hill GE (1981) Treatment of heat stroke with dantrolene.
J Am Med Assoc, 246: 41-42.
MacLennan DH, Duff C, Zorzato F, Fujii J, Phillips M, Korneluk RG,
Frodis W, Britt BA, & Worton RG (1990) Ryanodine receptor gene is a
candidate for predisposition to malignant hyperthermia. Nature(Lond),
343: 559-561.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, Pharmaceutical Press, p 1896.
Maruta H, Soga T, Okutsu Y, & Asari H (1980) Malignant hyperthermia:
a case report. Hiroshima J Anaesth, 16(Suppl): 35-39.
Mathieu A, Bogosian AJ, Ryan JF, Crone RK, & Crocker D (1979)
Recrudescence after survival of an initial episode of malignant
hyperthermia. Anaesthesiology, 51: 454-455.
Mercieca J & Brown EA (1984) Acute renal failure due to rhabdomyolysis
associated with use of a straight jacket in lysergide intoxication. Br
Med J, 288: 1949-1950.
Meyler WJ, Mols-Thurkow HW, & Wesseling H (1979) Relationship between
plasma concentration and effect of dantrolene sodium in man. Eur J
Clin Pharmacol, 16: 203-209.
Meyler WJ, Bakker H, Kok JJ, Agoston S, & Wesseling H (1981) The
effect of dantrolene sodium in relation to blood levels in spastic
patients after prolonged administration. J Neurol Neurosurg
Psychiatry, 44: 334-339.
Miller DH & Haas LF (1984) Pneumonitis, pleural effusion and
pericarditis follwoing treatment with dantrolene. J Neurol Neurosurg
Psychiatry, 47: 553-554.
Mirchandani H & Reich LE (1985) Fatal malignant hyperthermia as a
result of the ingestion of tranylcypromine (Parnate) combined with
white wine and cheese. J Forensic Sci, 30: 217-230.
Miyamoto H & Racker E (1982) Mechanism of calcium release from
skeleteal sarcoplasmic reticulum. J Membrane Biol, 66: 193-201.
Moffat AC (1986) Clarke's isolation and identification of drugs in
pharmaceuticals, body fluids and post mortem materials, 2nd ed.
London, The Pharmaceutical Press, p 508.
Morison DH (1983) Placental transfer of dantrolene. Anaesthesiology,
59: 265.
Nelson TE (1983) Dantrolene does not block abnormal calcium efflux
from a putative calcium channel of MH sarcoplasmic reticulum
(abstract). Anaesthesiology, 59: A225.
Nelson TE & Denborough MA (1977) Studies on normal skeletal muscle in
the relation to the pathopharmacology of malignant hyperpyrexia. Clin
Exp Pharmacol Physiol, 4: 315-322.
Ohnishi ST, Taylor S, & Gronert GA (1983) Calcium-induced calcium
release from sarcoplasmic reticulum of pigs susceptible to malignant
hyperthermia. The effect of halothane and dantrolene. FEBS Lett, 261:
103-107.
Ohta T, Ito S, & Ohga A (1990) Inhibitory action of dantrolene on
Ca-induced Ca2+ release from sarcoplasmic reticulum in guinea pig
skeletal muscle. Eur J Pharmacol, 178: 11-19.
Okumura F, Crocker BD, & Denborough MA (1980) Site of the muscle cell
abnormality in swine susceptible to malignant hyperpyrexia. Br J
Anaesthesiol, 52: 377-383.
Parr MJA & Willatts SM (1991) Fatal theophylline poisoning with
rhabdomyolysis. Anaesthesia, 46: 557-559.
Petusevsky ML, Ealing LJ, Rocklin RE, Snider GL, Merliss AD, Moses JM,
& Dorman SA (1979) Pleuropericardial reaction to treatment with
dantrolene. J Am Med Assoc, 242: 2772.
Pinder RM, Brogden RN, Speight TM, & Avery GS (1977) Dantrolene
sodium: A review of its pharmacological properties and therapeutic
efficacy in spasticity. Drugs, 13: 3-23.
Pope HG, Cole JO, Choras PT, & Fulwiler CE (1986) Apparent neuroleptic
malignant syndrome with clozapine and lithium. J Nerv Ment Dis, 174:
493-495.
Rivera VM, Breitbach WB, & Swanke L (1975) Dantrolene in amyotrophic
lateral scierosis. J Am Med Assoc, 233: 863-864.
Roberts JR, Quattrocchi E, & Howland MA (1984) Severe hyperthermia
secondary to intravenous drug abuse. Am J Emergency Med, 2: 373.
Ruhland G & Hinkle AJ (1984) Malignant hyperthermia after oral and
intravenous pretreatment with dantrolene in a patient susceptible to
malignant hyperthermia. Anaesthesiology, 60: 159-160.
Saxena SJ, Honigberg IL, Stewart JT, & Vallner JJ (1977) Liquid
chromatography in pharmaceutical analysis VI: determination of
dantrolene sodium in a dosage form. J Pharm Sci, 66: 286-288.
Sewall K, Flaverdew RMM, & Bromberger P (1980) Severe muscular
rigidity at birth; malignant hyperthermia syndrom? Can Anaesth Soc J,
27: 279-282.
Shime J, Gare D, Andrews J, & Britt B (1988) Dantrolene in pregnancy:
Lack of adverse effects on the fetus and newborn infant. Am J Obstet
Gynecol, 159: 831-834.
Snyder HR, Davis CS, Bickerton RK, & Halliday RP (1967) 1-((5-
arylfurfurylidene) amino)hydantoins. A new class of muscle relaxants.
J Med Chem, 10: 807-810.
Stadhouders AM, Viering WAL, Verburg MP, Ruitenbeek W, & Sengers RCA
(1984) In vivo induced malignant hyperthermia in pigs. III.
Localization of calcium in skeletal muscle mitochondria by means of
electron-microscopy and microprobe analysis. Acta Anaesthesiol Scand,
28: 14-26.
Starreveld E & Hope CE (1975) Cicutoxin poisoning (Water hemlock).
Neurology, 25: 730-735.
Sullivan JS & Denborough MA (1981) Temperature dependence of muscle
function in malignant hyperpyrexia-susceptible swine. Br J
Anaesthesiol, 53: 1217-1222.
Utili R, Boitnott JK, & Zimmerman HJ (1977) Dantrolene associated
hepatic injury: incidence and character. Gastroenterology, 72:
610-616.
Vallner JJ, Sternson LA, & Parsons DL (1976) Interaction of dantrolene
sodium with human serum albumin. J Pharm Sci, 675: 873-877.
Vallner JJ, Honigberg IL, Stewart JT, & Peyton AB (1979) Dantrolene
and metabolite levels in six patients on chronic therapy. Curr Ther
Res, 25: 79-91.
Wan HH & Tucker JS (1980) Dantrolene and lymphocytic lymphoma.
Postgrad Med J, 56: 261.
Ward A, Chaffman MO, & Sorkin EM (1986) Dantrolene (review). Drugs,
32: 130-168.
Weingarten AE, Korsh JI, Neuman GG, & Stern SB, (1987) Postpartum
uterine atony after intravenous dantrolene. Anesthesiol Analg, 66:
269-270.
Wuis EW, Grutters ACLM, Vree TB, & Van der Kleyn E (1983) Simultaneous
determination of dantrolene and its metabolites, 5-hydroxydantrolene
and nitro-reduced acetylated dantrolene in plasma and urine of man and
dog by HPLC. J Chromatogr, 231: 401-409.
Yamamoto T, Suzuki A, & Hotta K (1977) Dissociation of excitation and
contraction in skeletal muscle induced by denterium oxide and
dantrolene-sodium. Jpn J Physiol, 27: 95-109.
Yoganathan T, Casthely, PA, & Lamprou M (1988) Dantrolene-induced
hyperkalemia in a patient treated with diliazem and metrprolol. J
Cardiothorac Anesth, 2: 363-364.
APPENDIX 1
List of Antidotes
Group 1
Antidote Main indication Other possible
of pathological applications
condition
ACETYLCYSTEINE PARACETAMOL ORGANOCHLORINE
SOLVENTS, AMANITIN
AMYL NITRITE CYANIDE HYDROGEN SULFIDE
ASCORBIC ACID ORGANIC PEROXIDES
(OSMIUM)
ATROPINE CHOLINERGIC SYNDROME
AURINTRICARBOXYLIC BERYLLIUM
ACID (ATA)
BENZYLPENICILLIN AMANITINES
BETA-AMINOPROPIONITRILE CAUSTICS
CALCIUM CHLORIDE OR HF, FLUORIDES, CALCIUM
OTHER CALCIUM SALTS OXALATES ANTAGONISTS
DANTROLENE MALIGNANT MALIGNANT NEUROLEPTIC
HYPERTHERMIA SYNDROME
DEFEROXAMINE IRON, ALUMINIUM PARAQUAT
Antidote Main indication Other possible
of pathological applications
condition
DIAZEPAM CHLOROQUINE
DICOBALT EDETATE CYANIDE
DIGOXIN SPECIFIC DIGOXIN/DIGITOXIN,
ANTIBODY FRAGMENTS DIGITALIS GLYCOSIDES
DIMERCAPROL ARSENIC COPPER, GOLD, MERCURY
(INORGANIC), LEAD
ENCEPHALOPATHY
4-DIMETHYLAMINOPHENOL CYANIDE HYDROGEN SULFIDE
(4-DMAP)
ETHANOL METHANOL, ETHYLENE ALKOXYSILANES
GLYCOL, GLYCOL ETHERS
FLUMAZENIL BENZODIAZEPINES
FOLINIC ACID FOLINIC ACID ANTAGONISTS
GLUCAGON BETA-BLOCKERS
GLUCOSE INSULIN
GUANIDINE BOTULISM
HYDROXOCOBALAMIN CYANIDE
Antidote Main indication Other possible
of pathological applications
condition
ISOPRENALINE BETA-BLOCKERS
METHIONINE PARACETAMOL
4-METHYLPYRAZOLE ETHYLENE GLYCOL, COPRIN & DISULFIRAM
METHANOL
METHYLTHIONINIUM METHAEMOGLOBINAEMIA
CHLORIDE
(METHYLENE BLUE)
N-ACETYL PENICILLAMINE MERCURY
NALOXONE OPIATES
NEOSTIGMINE NEUROMUSCULAR BLOCK
(CURARE TYPE) PERIPHERAL
ANTI-CHOLINERGIC
POISONING
OXIMES ORGANOPHOSPHATES
OXYGEN CYANIDE, CARBON MONOXIDE,
HYDROGEN SULFIDE
OXYGEN-HYPERBARIC CARBON MONOXIDE CYANIDE, HYDROGEN
SULFIDE, CARBON
TETRACHLORIDE
Antidote Main indication Other possible
of pathological applications
condition
PENICILLAMINE COPPER GOLD, LEAD,
MERCURY
PENTETIC ACID (DTPA) RADIOACTIVE METALS
PHENTOLAMINE ALPHA-ADRENERGIC
POISONING
PHYSOSTIGMINE CENTRAL ANTI-CHOLINERGIC CENTRAL ANTI-CHOLINERGIC
SYNDROME FROM ATROPINE SYNDROME FROM
& DERIVATIVES OTHER DRUGS
PHYTOMENADIONE COUMARIN DERIVATIVES
(VITAMIN K)
POTASSIUM HEXACYANOFERRAT THALLIUM
(PRUSSIAN BLUE C177520)
PRENALTEROL BETA-BLOCKERS
PROPRANOLOL BETA-ADRENERGIC
POISONING
PROTAMINE SULFATE HEPARIN
PYRIDOXINE ISONIAZID ETHYLENE GLYCOL,
GYROMETRINE,
HYDRAZINES
Antidote Main indication Other possible
of pathological applications
condition
SILIBININ AMANITINE
SODIUM NITRITE CYANIDE HYDROGEN SULFIDE
SODIUM NITROPRUSSIDE ERGOTISM
SODIUM SALICYLATE BERYLLIUM
SODIUM THIOSULFATE CYANIDE BROMATE, CHLORATE,
IODINE
SUCCIMER (DMSA) LEAD, MERCURY
TOCOPHEROL CARBON MONOXIDE OXYGEN TOXICITY
TOLONIUM CHLORIDE METHAEMOGLOBINAEMIA
(TOLUIDINE BLUE)
TRIENTINE (TRIETHYLENE COPPER
TETRAMINE)
UNITHIOL (DMPS) ARSENIC COPPER, NICKEL,
LEAD, CADMIUM,
MERCURY (METHYL
AND INORGANIC)
Group 2
Agents used to prevent the absorption of poisons,
to enhance their elimination or to treat
symptomatically their effects on body functions
A. Emetics
APOMORPHINE
IPECACUANHA
B. Cathartics and solutions used for whole gut lavage
MAGNESIUM CITRATE
MAGNESIUM SULFATE
MANNITOL
SODIUM SULFATE
SORBITOL
WHOLE GUT LAVAGE FLUIDS
C. Agents to modify urinary pH
AMMONIUM CHLORIDE
ARGININE HYDROCHLORIDE
HYDROCHLORIC ACID (O.1 N)
SODIUM BICARBONATE
D. Agents to prevent absorption of toxic substances in the
gastrointestinal tract
ACTIVATED CHARCOAL (FOR MOST POISONINGS)
DIGITALIS, COUMARIN, KEPONE
FULLERS EARTH PARAQUAT, DIQUAT, POTASSIUM,
COPPER, FERROCYANIDE
SIMETHICONE FOAMING DETERGENTS
SODIUM BICARBONATE IRON, MERCURY,
ORGANOPHOSPHATES
SODIUM SULFATE LEAD, BISMUTH, BARIUM
STARCH IODINE
E. Agents to prevent absorption and/or damage in the skin
CALCIUM GLUCONATE HYDROFLUORIC ACID
GEL
MACROGOL 400 PHENOL
Group 3
Other useful therapeutic agents for
the treatment of poisoning
Below are listed certain therapeutic agents which are not
antidotes according to the accepted definition, but which through
their importance and sometimes specific role in the treatment of
poisonings, border on the concept of "antidotes".
In practice, these agents are used very often in cases of
poisoning and in other medical circumstances. The usefulness of these
agents is in general well established, most of them are considered
essential drugs, and they should be available for immediate use.
Agents Indications - symptoms arising from poisoning
BENZTROPINE dystonia
CHLORPROMAZINE hallucinatory and psychotics states
CORTICOSTEROIDS acute allergic reactions, largyngeal oedema,
(systemic/tropical/bronchoconstriction)
DIAZEPAM convulsions, excitation, anxiety, muscular
hypertonia
DIPHENHYDRAMINE dystonia
DOBUTAMINE myocardial depression
DOPAMINE myocardial depression, vascular relaxation
EPINEPHRINE (ADRENALINE) anaphylactic shock, cardiac arrest
FUROSEMIDE fluid retention, left ventricular failure
GLUCOSE hypoglycaemia
HALOPERIDOL hallucinatory and psychotic states
HEPARIN hypercoagulability states
LIDOCAINE ventricular arrhythmias
MANNITOL (IV) cerebral oedema, fluid retention
Agents Indications - symptoms arising from poisoning
OXYGEN hypoxia
PANCURONIUM muscular rigidity, convulsions
PROMETHAZINE allergic reactions
SALBUTAMOL bronchoconstriction (systemic/inhaled)
SODIUM BICARBONATE acidosis, some cardiac disturbances (e.g.,
TCA poisonings)
Group 4
Antidotes and related agents considered obsolete
Antidote Indicated for
ASCORBIC ACID Methaemoglobinaemia
CYCLOPHOSPHAMIDE Gold-Paraquat
CYSTEAMINE Paracetamol
DIETHYLDITHIOCARBAMATE Thallium
FRUCTOSE Ethanol
LEVALLORPHAN Opiates
NALORPHINE Opiates
POTASSIUM PERMANGANATE Fluorides
SULFADILIDINE Amanitine
TANNINS Alkaloids
THIOCTIC ACID Amanitine
TOCOPHEROL (Vitamin E) Paraquat
UNIVERSAL ANTIDOTE Ingested poisons
COPPER SULFATE as an emetic
SODIUM CHLORIDE as an emetic
CASTOR OIL as a cathartic
ACETAZOLAMIDE as a urinary pH modifier
APPENDIX 2
PRINCIPLES FOR EVALUATION OF ANTIDOTES
For many antidotes, the data on both efficacy and the optimum
methods of use are inadequate. Furthermore, there are problems in
both the pre-clinical and clinical aspects of antidote use, as well as
variations in licensing requirements between different countries.
Consequently, the IPCS and the CEC decided to evaluate antidotes and
other agents used in the treatment of poisoning cases. For this
purpose, a methodology has been developed for evaluation of antidotes
based on the principles concerned with the pharmaceutical properties
of the substance, its toxicity, as derived from animal and other
experiments, and the clinical studies in man, which demonstrate its
efficacy as an antidote and its safety in clinical use. All aspects
of these principles, as described below, may not be applicable to each
antidote and agent, but they provide the basis for assessment of those
in current use and under development.
1. Pharmaceutical Properties
As is true of any material to be administered to man, clear
guidelines on the pharmaceutical properties of an antidote are
required (reference should be made to the criteria as formulated in
generally accepted National or Regional Pharmacopoeias). The
information on an antidote should, therefore, include its chemical
formula and physical properties, such as melting point, solubility in
appropriate vehicles for administration, optical properties, acidity,
stability in light, and thermal stability. Particular account must be
taken of the wide range of temperatures to which such compounds may be
exposed in clinical situations (-30 to +40 °C). For liquids, the
refractive index and specific gravity should be considered, and, for
solids, their loss of weight on drying may be important.
To ensure purity and uniformity of the antidote preparation, the
route of synthesis, manufacturing process, and excipients need to be
considered and may have to be specified. Similarly, analytical
methods for the quality control or identification of the antidote
should be established.
Storage requirements and limitations need to be considered and
the shelf-life is very important for compounds that may be held for
long periods, particularly under tropical conditions. Evaluations of
shelf-life will need to be revised as further information on the
effects of storage under various conditions becomes available.
Mention should be made of any incompatibility with other
pharmaceuticals or food.
2. Pre-clinical Studies
Pre-clinical studies include those undertaken in vitro, in
laboratory animals, and in man (human volunteer studies). In the case
of antidotes, unlike other pharmaceutical products, studies of this
nature may first be indicated following the clinical use of postulated
antidotes or as part of a re-evaluation procedure, in particular where
comparison of the efficacy of antidotes is indicated.
2.1 In vitro studies
The type of in vitro study will vary depending on the antidote
being evaluated. Standardized studies of properties such as
adsorption capacity, chelating activity, anticaustic activity,
neutralization (e.g., for acids or alkalis), biochemical actions, and
pharmacological properties may be appropriate for the range of
different kinds of antidote.
Isolated cell, tissue culture, and isolated organ techniques may
be particularly relevant and in vitro studies should enable
subsequent studies in animals and man to be targeted more
specifically.
2.2 In vivo studies
2.2.1 Pharmacodynamic studies
Pharmacodynamic studies are aimed at assessing the mode of action
and efficacy of an antidote and its use in any particular type of
poisoning. Studies should be performed in relevant animal species and
should include investigations of both the pharmacological activity of
the antidote alone and the efficacy against the toxic agent when this
is administered in appropriate dosages. Ideally studies should be
conducted on two unrelated species that show qualitatively similar
response to the toxin in humans, and the efficacy against two
appropriate doses of the toxic agent (moderate and high toxicity)
should be evaluated. If animals are anaesthetized for such studies,
then it is important that consideration be given to the effects of any
anaesthetic agent, and, ideally, two different types of anaesthetic
should be used in parallel experiments.
The toxic compound should be administered by a clinically
relevant route and, if possible, a dose-response relationship for the
antidote established. Opportunity should be taken to study the
pharmacokinetics of the toxic agent and antidote during these studies,
particularly examining interactions in the distribution and clearance
of antidote and toxic compound.
2.2.2 Kinetic studies
Studies of the absorption, distribution, and elimination of an
antidote should, where possible, include monitoring of its metabolism
and also include a study of the effects of dose on its kinetics.
Doses used should be relevant to the likely clinical situation.
Kinetic studies should take into account the likely route of use of
the antidote (e.g., oral, parenteral, or topical) and should, ideally,
include the effect of the antidote on the kinetics of the toxic
substance against which it is used. Studies on the influence of
single or multiple organ failure on the elimination and metabolism of
the antidote or antidote complex may also be relevant.
2.2.3 Toxicological studies
Ideally, toxicological studies should be performed on species
that show similarity to humans as regards the kinetics and metabolism
of the antidote. Acute toxicological studies on two (unrelated)
species would be appropriate. The extent of toxicity evaluation would
depend on the proposed use of the antidote, and, for the situations in
which repeated doses of an antidote are necessary, chronic toxicity
studies need to be undertaken. Consideration should also be given to
the general approach to acute toxicity studies, bearing in mind the
doubts about the usefulness of LD50 values. Depending on likely
use, teratogenicity, mutagenicity, and carcinogenicity testing may be
deemed necessary.
2.3 Human volunteer studies
Human volunteer studies with antidotes present specific problems
and should be carried out in accordance with the Declaration of
Helsinki and the Council for the International Organizations of
Medical Sciences (CIOMS) Proposed International Guidelines for
Biomedical Research involving Human Subjectsa. The ethical
implications require particular attention. However, volunteer studies
may be important and useful for evaluating the human pharmacology of
certain antidotes. On special occasions, it may also be possible to
study interaction with a potentially toxic substance in volunteers,
subject to full and appropriate ethical review. Such investigations
may be particularly relevant for antidotes that are likely to be
widely used. They may also aid in choosing the appropriate dosage
regimen of an antidote for clinical use. In some circumstances
studies on patients with a particular disease or of particular age
groups may be indicated (e.g., renal, cardiac, or hepatic impairment,
and the elderly). In the rare situation in which pharmacogenetic
differences due to polymorphism of metabolism of the antidote is
important, such human volunteer studies will obviously be valuable.
a Proposed International Guidelines for Biomedical research
Involving human Subjects, 1982, Geneva, World Health
Organization.
3. Clinical Studies
By their very nature, poisonings of whatever sort are
unpredictable, and it is often difficult to establish the exact dose
of toxic compound to which a patient has been exposed. Clinical
studies of antidotes are therefore inherently more difficult than
studies of the effects of pharmaceutical preparations in many other
conditions, since definition of the severity of the poisoning is often
a problem.
The precise combination of approaches used in clinical studies of
an individual antidote needs to be carefully tailored. More than one
antidote may sometimes be needed, and, in these circumstances, an
evaluation of the combined treatment approach should be given.
3.1 Literature evaluation
Published work on antidotes is frequently in the form of case
reports, which are by their nature uncontrolled. Such studies are an
important source of data and can provide information on both the
effect of an antidote in the clinical setting and the likely pattern
of toxicity associated with the poison. They may thus prevent
needless duplication of studies. A principal role for a continued
evaluation of the literature is to identify specific areas for future
work and areas of ignorance.
Case reports are often not well documented and it is rarely
possible to make strict comparisons between published cases undertaken
at different centres. The human toxicological scientific basis could
be greatly enhanced by the establishment of a comparable basis for
case data collection and reporting (see below).
3.2 General approach to clinical studies
Double-blind controlled studies are the ideal in most areas of
therapeutics. In the assessment of antidotes, however, particularly
for rarer poisonings and those with a high morbidity or mortality,
such an approach presents major ethical and technical problems. Thus,
it may not be possible to obtain the consent of the patient or his
relatives and an individual clinician is unlikely to see a sufficient
number of patients to make a formal study worthwhile. The proper
construction of a control group may also raise ethical dilemmas.
Thus, a prospective approach to the study of antidotes should be
considered in parallel with the critical retrospective analysis of
existing patient data. Such studies may be best carried out on a
multi-centre or multi-national basis. This retrospective analysis,
often of unpublished data, could perhaps be organized centrally.
Retrospective data will provide a useful baseline from which to mount
prospective studies.
3.3 Methodology
Since case records are a major data source in antidote studies,
it is essential that records are kept carefully in a standardized
format. Case records should, where possible, include a detailed
personal history to cover occupational exposure, toxic agents,
relevant hobbies, previous medical history, and concurrent medication
including non-prescribed medicines. Physical examination should pay
particular attention to specific signs of intoxication and their
physiological consequences. Careful measurement and monitoring of
physiological changes is essential; in particular, careful
documentation of changes, such as the level of consciousness, should
be established. A scoring system for monitoring changes in the level
of consciousness and an agreed coma scale for grouping patients into
classes of comparable severity should be adopted where possible.
Detailed consecutive records of clinical progress in an individual
patient are important. Accurate recording of the time of the
intoxication, of its presentation to treatment centres, and of
initiation of treatment are particularly important.
The clinical examination should be supported by appropriate
analytical data on samples of body fluid such as blood, urine, and
gastric aspirate. The collection, handling, and analysis of samples
needs to be standardized and guidelines for individual toxic compounds
laid down. Accurate records of the time of clinical examination and
toxicological sampling are necessary and, where possible, the two
should be concurrent and continue throughout the clinical course of
the poisoning. This may enable a relationship between the two to be
established, as well as providing careful documentation of the effect
of an antidote. The routine saving of multiple biological samples
from a patient suffering from intoxication is therefore necessary.
It should be stressed that objective measures are preferable to
clinical impressions and that the opportunity should be taken at all
stages of management of intoxications to quantify physiological
parameters. Where possible, accepted standardized prognostic factors
should be utilized both to determine the appropriate use of the
antidote and to evaluate its effect.
3.4 Controlled studies
In the past, controlled studies have provided the basis for
important advances in the management of poisonings. As indicated in
section 3.2, there are ethical consideration that need to be
considered carefully. Furthermore, such studies need to be carefully
tailored to the particular antidote and poison under investigation.
The general poisons made in section 3.3 will also apply.
Careful consideration needs to be given to the establishment of
a control group. This should ideally be a parallel and comparable
group of patients but may be retrospective. Establishment of a
parallel control group may be easier in the situation where two active
treatments are being compared. It should be remembered, however, that
there may be a risk attached to using an antidote, and the use of a
control group that receives full supportive therapy may enable a more
rapid decision on the efficacy of an antidote to be obtained.
In controlled studies, an opportunity may arise to study the
toxicity and kinetics of the antidote; it should be grasped whenever
possible.
The period of follow-up needs to be determined, and end-points
must be clearly established and defined. Statistical considerations
are important and need to be fully incorporated at the time of trial
design.
4. Centralized Record System
It is suggested that a central pooling of the experience of
various treatment centres, particularly with respect to rarer
antidotes, would be a great advantage. An international network of
IPCS-designed clinical centres could provide a mechanism for this
purpose. This pool of data would be used as a basis for the
international evaluation of antidotes but would remain confidential
and would not be published without the consent of the individual
clinician concerned. Data submitted to such a pool would remain the
property of the investigator, who would be at liberty to publish it
separately.
Poison control centres appear to be in a unique position to
collect data on antidotes for rarer poisoning and to ensure that
appropriate clinical and toxicological data are collected. This
process might be facilitated if a poison control centre acted as a
base for the supply of antidotes.
The amount of information required from individual patients
entered on the case record system would have to be carefully planned
and the collection and reporting of the data coordinated.
The establishment of such a centralized case record system would
also be likely to stimulate further international collaboration in the
evaluation of antidotes by controlled studies and could lead to
recommendations for further areas of research.
APPENDIX 3
PROFORMA FOR MONOGRAPHS ON ANTIDOTES FOR SPECIFIC TOXIC AGENTS
Guidelines to Authors
These guidelines provide a unified format for monographs written
on individual antidotes used in the management of poisoning using
existing published (and unpublished) literature. For a number of
antidotes currently used clinically some of the information suggested
will be missing: these gaps in knowledge should be stated. For those
antidotes currently under in development, this proforma is designed to
give an idea of the sort of information that would be required for
evaluation and worldwide acceptance for use. Suggested section
headings and an outline guide to contents are given below.
1. Introduction
This should be brief (usually 200-400 words) and include:
- indications for antidote use
- rationale for the choice of the antidote, mentioning areas where
there are doubts about efficacy
- an indication of specific groups at risk from treatment with the
antidote
2. Name and Chemical Formula of Antidote
International non-proprietary name (when available); CAS number
(Chemical Abstracts Service); IUPAC name (International Union of Pure
and Applied Chemistry); manufacturer and commercial names, formula
(include figure of structure); relative molecular mass; specification
of chemical salts used; conversion table from mass units to SI units.
3. Physico-chemical Properties
- melting point, boiling point
- solubility in vehicles for administrations
- optical properties
- acidity
- pKa
- stability in light
- thermal stability/flammability (including vehicle if antidote
usually in solution or suspension)
- for liquids, refractive index and specific gravity
- for solids, loss of weight on drying
- the excipients and pharmaceutical aids
- pharmaceutical incompatibilities
4. Pharmaceutical Formulation and Synthesis
4.1 Routes of synthesis (brief details only)
4.2 Manufacturing processes (indicate, where known, possible
contaminants)
4.3 Presentation and formulation
5. Analytical Methods
In this section, the passive voice should be used, e.g., "the
solution is mixed to dissolve the reagents". Vocative instructions
should not be used, e.g., "dissolve 0.5 g in 20 ml of water". To
include:
5.1 Quality control procedures for the antidote and/or its
formulation
5.2 Methods for identification of the antidote
5.3 Methods for analysis of the antidote in biological samples
5.4 Analysis of the toxic agent in biological samples
referring to the preferred assay techniques
6. Shelf-life
Attention should be given to specific conditions of temperature
and humidity, including tropical conditions, and instructions on
storage conditions.
7. General Properties
This section should be particularly tailored to the antidote in
question and include:
- information on the mode of antidotal activity of the compound
(e.g., chelating agent, receptor antagonist)
- other relevant biochemical and pharmacological profiles (e.g.,
anticholinergic, antiadrenergic properties) as demonstrated in
vivo and in vitro
This section can be subdivided as needed.
8. Animal Studies
It is important to exclude human data from this section. Even if
a paper presents both human and animal data, the human data should be
given in the clinical sections 9, 10 or 11, as appropriate. An
attempt should be made to evaluate the statistical significance of the
results. As a rule of thumb, this term should imply a level of
statistical significance at the 5% level (P <0.05). If other
parameters are used, they should be given. If both positive and
negative results have been reported, possible reasons for such
differences may be briefly discussed (routes of administration, time
before giving the antidote, dose, etc.).
8.1 Pharmacodynamics
This section should include data on efficacy, examining the
protective effects against moderate and high doses of the toxin. The
efficacy parameter end-point should be defined before the data are
discussed, e.g., is it reduced mortality or increased excretion of the
toxic agent that has been studied? Data on the dose-response
relationship of the antidote and on the pharmacokinetics of any
interaction between antidote and toxic compound should be sought. Both
the toxic agent and the antidote should be given by the clinically
relevant route(s). Information should also be sought on the time after
the administration of the toxic agent at which the antidote is likely
to be of any clinical benefit.
8.2 Pharmacokinetics
Studies on the bioavailability, half-life and clearance of the
antidote by the relevant route of administration; studies on the dose
dependency of the pharmacokinetics relevant to the doses used in the
clinical settings and details of the metabolism of the compound. If
there are data on more than one species, these should be included.
8.3 Toxicology
Details of acute, subacute or chronic toxicity, depending on the
likely clinical use of the compound, should be discussed. Such
studies should use appropriate routes of administration. Where
available, details of mutagenicity and teratogenicity testing would be
of interest. LD50 values for different species and different routes
of administration should be presented.
9. Volunteer Studies
Information on the pharmacokinetics of the antidote in man and
possible effects of disease on the handling of the antidote should be
given, where available. Studies on the interaction with the toxic
agent given at subtoxic doses may also be available. All human data
based on volunteer studies should be included in this section.
10. Clinical Studies - Clinical Trials
Literature evaluation of clinical trials. For each trial the
following information should be given:
(a) evidence of poisoning, i.e. inclusion criteria
(i) definition of assay techniques to measure the poison and
its effects
(ii) physiological changes of poisoning recorded; accepted
methods
(b) number of patients studied
(c) was there a control group? type, i.e. parallel or retrospective;
appropriate, i.e. were the treated and control groups
comparable (e.g., age, sex, time of presentation, severity of
poisoning)?
(d) details of antidote used
(i) dose and route
(ii) time between exposure to the toxin and administration of
antidote
(iii) evidence of any toxic effect of the antidote
(e) outcome assessment
(i) clinical
(ii) biochemical
(iii) end-points - death, pathological damage
(f) overall view of likelihood of benefit due to antidote
It may sometimes be difficult to define what is a clinical trial.
Retrospective studies and use of historical controls should normally
not be considered as clinical trials and may then be described under
section 11.
11. Clinical Studies - Case Reports
Many authors of case reports claim a favourable outcome "due to
the use of the antidote". In most cases, however, the best conclusion
drawn from such studies is that the use of the antidote was associated
with a favourable outcome and nothing more. Some case reports are
thoroughly performed with measurement of half-lives in different body
compartments and amount of toxic agent eliminated by different
elimination techniques. When referring to such studies, the end-point
parameter, e.g., reduction in plasma half-life, should be evaluated
critically. There may be too few plasma levels over a too short a
time period to allow for any conclusion.
Other important items to evaluate are:
- evidence of poisoning (clinical, pathological);
- was an appropriate assay/measurement technique used?
- dose and route of antidote
- evidence of outcome
- evidence of toxicity or side-effects of antidote
It should be borne in mind that most poisoned patients will
recover with intensive supportive care alone. If such cases have been
reported, a brief mention may be appropriate.
12. Summary of Evaluation
This section is frequently misunderstood by authors writing their
first draft in this series. Section 12 is not to be a copy of section
13. In section 12 the author should try to evaluate or summarize
the data, previously presented in the monograph, as a basis for the
recommendations under the following subheadings:
12.1 Indications
12.2 Advised routes and dose
12.3 Other consequential or supportive therapy
12.4 Controversial issues and areas of use where there is
insufficient information to make recommendations
12.5 Proposals for further studies
12.6 Adverse effects
12.7 Restrictions for use
13. Model Information Sheet
This should be a concise user's manual for this antidote in the
emergency situation. This section may, for example, be faxed to the
clinician treating the patient. No references or abbreviations should
be given since all the information given should have been discussed in
previous sections.
13.1 Uses
13.2 Dosage and route
13.3 Precautions/contraindications
13.4 Pharmaceutical incompatibilities and drug interactions
13.5 Adverse effects
13.6 Use in pregnancy and lactation
13.7 Storage
14. References
References should be given in text as (McMartin et al., 1980) or
(Henry & Volans, 1984). Several references to the same statement are
placed in chronological order.
In the reference list, the names of all authors and their
initials must be given. Only initial letters are capitalized. Titles
of articles in languages other than English, except those in French,
should be translated into English. Translated titles appear in square
brackets with the original language in parentheses.
Periodicals
Names of journals should be abbreviated according to the ISDS
(International Serials Data System) List of Serial Title Word
Abbreviations (a condensed list is obtainable from the RO) or
otherwise given in full. The initial letter of each abbreviation is
capitalized. The volume number is indicated in bold print and is
followed by the issue number (if any) in parentheses. First and last
page numbers must be given.
McMartin KE, Ambre JJ & Tephly TR (1980) Methanol poisoning in human
subjects. Am J Med, 68: 414-418.
Henry J & Volans G (1984) ABC of poisoning. Br Med J, 289: 990-993.
Conference proceedings
The following elements are necessary: name(s) and initial(s) of
author(s); the year of publication; title of paper; the word "In:" the
editors of the proceedings; the full title of the conference (not
abbreviated); the place and date of the conference; the place of
publication; the publisher; the volume number (if any) and the page
numbers.
Example
Wassermann M (1984) L'étude de la toxicologie des pesticides en
climat tropical In: Smith JH ed. Proceedings of the 14th
International Congress of Occupational Health, Madrid, 16-21 May
1983. Amsterdam, Excerpta Medica, vol 3, 1728-1733.
Books
Windholz M ed. (1983) The Merck Index: an encyclopedia of
chemicals, drugs, and biologicals, 10th ed. Rahway, New Jersey, Merck
and Co., Inc.
Reference to a chapter in a book should be given as follows:
Weinstein L (1974) Pathogenic properties of invading
microorganisms. In: Sodeman WA Jr ed. Pathologic physiology:
mechanisms of disease. New York, Academic Press, pp 457-472.
Agency report
US EPA (1984) Mercury health effects update: health issue
assessment. Washington, DC, US Environmental Protection Agency
(EPA-60018-84-019F).
Order of entries in the list
The following rules are applied:
a) Several papers by different authors with the same surname
are listed alphabetically according to their initials.
b) Several papers by one author are listed chronologically.
c) Several papers by the author plus a co-author are listed
alphabetically.
d) Several papers by the author plus two or more coauthors are
listed chronologically.
Example
Smith DE (1985)
Smith JH (1983)
Smith JH (1984)
Smith JH & Barns MP (1986)
Smith JH & Jones TD (1979)
Smith JH, Jones TD, & Barnes MP (1981)
Smith JH, Barnes MP, & Jones TD (1983)
Normally one should not refer to abstracts or works published as
supplements. Under special circumstances, e.g., little has been
published and the person has great experience with the topic, personal
communications may be inserted in the text as follows: (Vale JA,
personal communication).
15. Author(s) Name, Address
16. Additional Information
e.g., availability of supply.
Proprietary names: DantriumR (Norwich Eaton UK,
Australia, Belgium, Canada, France,
Netherlands, New Zealand, South Africa,
USA) (Martindale, 1989).
DanleneR (SIT, Italy)
DantamacrinR (Röhm Pharma, D-6108
Weiterstadt);
Boehringer Mannheim, Switzerland)
DantralenR (Lafarquim, Spain)
DantriumR (Norwich Eaton, Norwich NY
13185, USA;
Formenti, I-20149 Milan, Italy; Smith
Kline & French, USA; Yamanouchi, Japan)
DantrixR (SIT, I-27035 Mede, Italy)
(Martindale, 1989;
Index Nominum, 1990)
4.3 Physico-chemical Properties
Dantrolene sodium is an orange, odorless powder with a melting
point of 279-280 °C. It is slightly soluble in water, but it
hydrolyses and precipitates the extremely insoluble (< 1 mg/l) free
acid, dantrolene. This hydrolysis may be prevented to some extent by
the addition of small amounts of sodium hydroxide, but this procedure
is complicated by the precipitation of dantrolene sodium by the common
ion effect. The amount remaining in solution is a function of the
total ionic strength.
The approximate solubility data given in Table 1 were obtained at
room temperature.
Table 1. Solubility of Dantrolene sodium in various solvents
Solvent Solubility (g/l)
Propylene glycol 40
Glycerine 25
Polyethylene glycol 400 80
Dimethylformamide 15a
Dimethylacetamide 15a
0.2% Morpholine in water 0.2
1% Morpholine in water 0.5
2% Morpholine in water 0.9
Chloroform < 20 (70 g/l for dantrolene)
Acetone 20-25
a complex formed
Dantrolene (the free acid of dantrolene sodium) is a weak acid
with a pKa of about 7.5. However, the extremely low solubility of the
free acid prevents an accurate determination of its pKa (personal
communication from A.W. Castellion, Norwich Easton, to the IPCS).
Dantrolene sodium solutions should be protected from light.
Dantrolene sodium capsules and powder for injection should be kept at
a temperature below 40 °C, preferably between 15 and 30 °C, and the
capsules should be stored in well-closed containers. Following
reconstitution with 60 ml of sterile water for injection, dantrolene
sodium injection solution is stable for 6 h when stored at 15-30 °C
and protected from light.
The excipients used with dantrolene are mannitol and sodium
hydroxide
4.4 Pharmaceutical Formulation and Synthesis
Dantrolene sodium is available as capsules of 25 mg, 50 mg and
100 mg.
It is also available as an orange powder for preparing injections
in vials of 20 mg, with 3 g mannitol and sodium hydroxide. The powder
is dissolved by the addition of 60 ml of water, producing a highly
irritant solution with a pH of about 9.5.
Snyder et al. (1967) described the synthesis of a series of 1-
[(5-arylfurfurylidene)amino]hydantoins from the appropriate
aryldiazonium chlorides and 2-furaldehyde, coupled in aqueous acetone
with cupric chloride as the catalyst. The intermediate aldehydes were
condensed directly with 1-aminohydantoin hydrochloride to give the
aminohydantoin derivatives.
4.5 Analytical Methods
4.5.1 Identification and quantification of dantrolene sodium and its
formulation
The thin-layer chromatography details are as follows:
* layer: silica gel G, 250 µm thick
* mobile phase: chloroform:acetone (4:1), retention factor: 19;
ethyl acetate:methanol:strong ammonia solution
(85:10:5), retention factor: 0.9;
ethyl acetate, retention factor: 36 (Moffat,
1986)
An alkaline solution of dantrolene sodium gives a peak in the
ultraviolet spectrum at 314 nm (Moffat, 1986). In the infrared
spectrum, principal peaks occur at the following wavenumbers: 1600,
1225, 1510, 850, 1713, 1108 (dantrolene sodium, KBR disc) (Moffat,
1986).
Saxena et al. (1977) reported a method for the determination of
dantrolene sodium in a dosage form by converting the drug to its free
acid in acidic media (pH 2.5-4.0) using 2N HCl. This is followed by
extraction into a 1-butanol-chloroform mixture and quantification by
high-performance liquid chromatography (HPLC) using carbon
tetrachloride:dimethylformamide (90:10) as mobile phase. The peaks
are detected at 375 nm.
4.5.2 Quantification of dantrolene in body fluids
4.5.2.1 Spectrofluorimetry
This is a rapid and sensitive method for the quantitative
determination of dantrolene in plasma, blood and urine, and consists
of a direct extraction of dantrolene into a nitropropane-heptane (1:1)
solvent mixture. The excitation peak is at 395 nm and the
fluorescence peak at 530 nm (Hollifield & Conklin, 1968). The
sensitivity is 100 ng/ml in plasma, blood and urine, and 400 ng/ml in
bile and tissue (Moffat, 1986)
4.5.2.2 High-performance liquid chromatography
Dantrolene and its metabolites 5-hydroxydantrolene and
nitroreduced acetylated dantrolene (F490) are detected by a reversed-
phase HPLC method after a preliminary extraction step into a
chloroform-butanol mixture for plasma samples. The detection limit is
20 ng/l (Wuis et al., 1983).
Lalande et al. (1988) have reported an alternative method for the
determination of dantrolene and its reduced and oxidized metabolites
in plasma.
4.6 Shelf Life
The following instructions for storage conditions are recommended
by the manufacturer:
Dosage form Storage requirements Shelf-life
As supplied below 30 °C 3 years
Reconstituted 15-30 °C 6 h when protected from
light
4.7 General Properties
Dantrolene is a peripheral striated muscle relaxant agent which
most probably acts by preventing calcium flux across the sarcoplasmic
reticulum, thereby reducing the intracellular free calcium
concentration (Lopez et al., 1987). Its site of action is in the
muscle itself, beyond the motor end-plate. Although this mechanism of
action seems logical, other authors stress that we are still a long
way from understanding the finer details of its mode of action and,
correspondingly, those of the pathogenesis of malignant hyperthermia
(Harrison, 1988). The genetic defect in susceptible individuals is
considered to reside in the calcium release channel (CRC) of the
sarcoplasmic reticulum of skeletal muscle (MacLennan et al., 1990).
The effects of dantrolene on other calcium-dependant systems seem
negligible at doses in current use (Hall et al., 1982).
4.8 Animal Studies
4.8.1 Pharmacodynamics
4.8.1.1 Effect on skeletal muscle
Dantrolene inhibits the development of tension in animal muscle
preparations in vitro caused by caffeine or by depolarization. The
effect of dantrolene can be antagonized by raising the extracellular
calcium concentration (Ellis & Wessells, 1977; Yamamoto et al., 1977;
Anderson et al., 1978; Nelson, 1983).
Dantrolene also inhibits the contractile response to ryanodine,
which is known to cause release of calcium from skeletal muscle
sarcoplasmic reticulum (Fairhust et al., 1980).
Dantrolene, therefore, probably inhibits skeletal muscle
contraction by preventing release of Ca2+ from sarcoplasmic
reticulum. The results obtained by Ohta et al. (1990), using skinned
skeletal muscle from guinea-pigs, suggested that dantrolene acts as a
selective inhibitor of the calcium-induced Ca2+ release mechanism
without having an effect on the Ca2+ pump of the sarcoplasmic
reticulum or on the contractile machinery of skeletal muscle.
Dantrolene also diminishes exercise-induced muscle damage in the
rat, as judged from creatine kinase isoenzyme patterns in plasma
before and after a 2-h run on a treadmill (Amelink et al., 1990).
4.8.1.2 Effects on other tissues
The effects of dantrolene on cardiac muscle, smooth muscle, the
nervous system and endocrine glands have been studied in animal
experiments, and the data have been reviewed by Ward et al. (1986).
These data are not of clinical importance.
4.8.1.3 Studies in malignant hyperthermia-susceptible pigs
Certain breeds of pig exhibit a stress-related syndrome very
similar to that of anaesthetic-induced malignant hyperthermia in
humans, and they are a useful investigational model for malignant
hyperthermia.
Dantrolene inhibits contractures of skeletal muscle preparations
induced by halothane, caffeine, suxamethonium, and potassium chloride
and thymol (Okumura et al., 1980; Sullivan & Denborough, 1981). It
also prevents the accumulation of myocytoplasmic calcium in the
mitochondria of pigs susceptible to malignant hyperthermia
(Stadhouders et al., 1984) and decreases the release of calcium from
the sarcoplasmic reticulum of muscle from susceptible pigs (Ohnishi et
al., 1983). The reduction of the free intracellular calcium
concentration is probably the mechanism of action of dantrolene in
these animals (Lopez et al., 1987). It has been suggested that
dantrolene may act at the initial stage of excitation (Miyamoto &
Racker, 1982).
Dantrolene has been demonstrated to be effective in treating
malignant hyperthermia induced by halothane or suxamethonium
anaesthesia in susceptible pigs (Gronert et al., 1976; Harrison, 1977;
Kerr et al., 1978; Hall et al., 1982). It is also effective when
given prophylactically before the induction of anaesthesia (Harrison,
1977; Kerr et al., 1978).
4.8.2 Pharmacokinetics
The pharmacokinetics of dantrolene have been studied in human
volunteers and this limits the need for animal data in this area.
Following a single oral dose of 5 mg/kg in dogs and 1 mg/kg in
rats, 20% and 29%, respectively, is absorbed (Fournier, 1982).
4.8.3 Toxicology
4.8.3.1 Acute toxicity
The oral LD50 has been estimated to be 29 g/kg in newborn rats.
No deaths occurred after the oral administration of 8 g/kg to young
adult rats, mice, hamsters and rabbits (Fournier, 1982).
The LD50 after the intraperitoneal injection of dantrolene was
780 mg/kg in rats (Fournier, 1982) and 1400 mg/kg in mice (Ellis &
Carpenter, 1974). The intravenous LD50 was > 40 mg/kg in rats and
> 80 mg/kg in mice (Fournier, 1982).
4.8.3.2 Subacute toxicity
Four out of 20 mice given dantrolene (84 mg/kg per day) by mouth
for one month developed hepatic steatosis. Renal tubular damage,
crystal deposition in the renal tract, and hepatocellular necrosis
were seen in rats after they had received oral doses of 60 to 500
mg/kg per day for one month (Fournier, 1982). Intraperitoneal doses
of 50 mg/kg per day lowered the level of serum glucocorticoids over 5
days (Francis & Hamrick, 1980). No anatomical or histological changes
were seen after one month with oral doses of up to 500 mg/kg per day
in dogs or 90 mg/kg per day in monkeys, although animals given high
doses developed anorexia, hypotonia and weight loss (Fournier, 1982).
4.8.3.3 Chronic toxicity
Rats receiving oral dantrolene doses of 15 mg/kg per day or more
showed a reduction in weight gain, hepatocellular degeneration,
crystalluria and keratitis with corneal opacities. Females developed
mammary tumours, with a statistically significant increase in the
incidence of breast adenocarcinoma in those receiving 60 mg/kg per day
(Fournier, 1982).
Dogs showed no effects when treated with dantrolene (15 mg/kg per
day) for 12 months. However, higher doses caused a reduction in
weight gain, and at 60 mg/kg per day impaired hepatocellular function,
anaemia and crystalluria were apparent. One case of intrahepatic
cholestasis occurred (Fournier, 1982).
4.8.3.4 Teratogenicity
There was no evidence of teratogenic effects from dantrolene when
doses of up to 45 mg/kg per day were given to rats, rabbits or monkeys
(Pinder et al., 1977).
Using the maternal-fetal sheep model in nine pregnant ewes, Craft
et al. (1988) found an equilibrium between maternal and fetal plasma
dantrolene concentrations 5 min after dosing. The fetal level of
dantrolene was about 10% of that of the mother. It was concluded that
the administration of intravenous dantrolene (1.2 or 2.4 mg/kg) had no
clinically significant adverse effect on mother or fetus in the sheep
model.
4.9 Volunteer Studies
Included in this section are studies involving healthy volunteers
and patients undergoing surgery from whom informed consent was
obtained.
4.9.1 Administration and plasma concentrations
Peak plasma dantrolene concentrations of 0.5 to 0.95 mg/l
occurred 4 to 8 h after 50 mg dantrolene was administered orally to
six healthy volunteers. The corresponding peak 5-hydroxydantrolene
concentration was 0.11 to 0.3 mg/l after 6 to 8 h (Katogi et al.,
1982). In the study of Allen et al. (1988), a total oral dose of 5 mg
dantrolene/kg was given to ten malignant hyperthermia-susceptible
patients prior to anaesthesia. All subjects had plasma dantrolene
levels above 2.8 mg/l, indicating a high bioavailability of
dantrolene.
In six patients, who had previously suffered from suspected or
proven malignant hyperthermia and who received prophylactic
intravenous dantrolene (2.5 mg/kg) before anaesthesia, peak blood
concentrations of 4.3 to 6.5 mg/l were found (Flewellen & Nelson
1985). Lerman et al. (1989) infused dantrolene (2.4 mg/kg) over 10
min in 10 children susceptible to malignant hyperthermia. Mean
dantrolene blood concentrations 1 min and 1 h after infusion were 6.03
mg/l (SD ± 0.93) and 3.56 (SD ± 0.49) mg/l, respectively. The blood
concentrations of the metabolite 5-hydroxydantrolene rose to a maximum
of 0.6 mg/l at 8 h and then fell with an apparent half-life of 9 h.
Plasma dantrolene concentrations during prolonged treatment are
similar to those found after single oral dosing (Vallner et al., 1979;
Meyler et al. 1981). Metabolite concentrations may be relatively
elevated after prolonged (> 2 months) administration (Vallner et al.
1979).
4.9.2 Distribution
In the 10 children studied by Lerman et al. (1989), the apparent
volume of distribution for dantrolene was 0.54 l/kg (SD ± 0.08). In
vitro studies show that dantrolene interacts with human serum
albumin at a minimum of two sites on the protein (Vallner et al.,
1976), but exact figures for the degree of protein binding have not
been reported.
4.9.2.1 Distribution to the fetus and newborn baby
The oral administration of dantrolene (total doses of 250 and 600
mg) to two pregnant patients thought to be susceptible to malignant
hyperthermia resulted in a fetal/maternal serum concentration ratio of
approximately 0.4, thus indicating that this agent reaches the
placenta in appreciable concentrations (Morison 1983). In a study of
20 pregnant women susceptible to malignant hyperthermia, the mean
maternal predelivery dantrolene level was 0.99 ± 0.5 mg/l and the mean
neonatal cord blood dantrolene level 0.68 ± 0.3 mg/l (Shime et al.,
1988).
4.9.3 Elimination
The major metabolite of dantrolene in humans is
5-hydroxydantrolene, produced by hepatic microsomal oxidation,
although minor metabolites also exist (Lietman et al., 1974; Ellis &
Wessels, 1978). According to in vitro data, this major metabolite
is half as potent as the parent drug, whereas the other metabolites
appear to be inactive (Ellis & Wessels, 1978). When dantrolene is
administered orally, 15-25% of the dose is excreted renally,
predominantly as 5-hydroxydantrolene but with small amounts of reduced
acetylated dantrolene and unchanged drug (Lietman et al., 1974).
The elimination half-life of orally administered dantrolene in
humans is probably between 6 and 9 h, although values of 3 to 22 h
have been observed (Lietman et al., 1974; Dykes 1975; Meyler et al.,
1979, 1981; Wuis et al., 1983; Allen et al., 1988). The elimination
half-life after intravenous dosing was reported to be 12 h in both
healthy volunteers (Flewellen et al., 1983) and patients known to
suffer from malignant hyperthermia (Flewellen & Nelson, 1985). This
is in accordance with the value of 10 h (SD ± 2.6) reported in
children (Lerman et al., 1989). In the study by Shime et al. (1988)
in neonatals, the dantrolene half-life was 20 h.
The median elimination half-life for 5-hydroxydantrolene, the
major metabolite of dantrolene, was found to be 15.5 h (range, 8.1 to
29.4 h) in healthy volunteers (Meyler et al., 1979), whereas it was 9
h (SD ± 2.5) in the 10 children studied by Lerman et al. (1989).
4.9.4 Human in vitro pharmacodynamics
Dantrolene inhibits the contraction of isolated human skeletal
muscle induced by halothane or suxamethonium (succinylcholine) (Nelson
& Denborough 1977; Hallsall & Ellis 1979; Fletcher & Rosenberg 1985).
4.10 Clinical Studies - Clinical Trials
The rarity of malignant hyperthermia, the variability of its
clinical manifestations and the range of adjunctive therapy have
precluded controlled trials or comparative studies of dantrolene in
its treatment.
4.11 Clinical Studies - Case Reports
This section presents several case reports where the use of
dantrolene is associated with a favourable outcome. Even if some of
the case reports (or series of such reports) may seem convincing,
regarding the efficacy of dantrolene, one should still bear in mind
the lack of controlled studies.
4.11.1 Use in malignant hyperthermia
Ward et al. (1986) and Harrison (1988) have reviewed the
published case reports. In children, an initial intravenous dose of
1 mg/kg has usually been effective if given within 2 h of the onset of
signs and symptoms, although a boy of 11 years died in spite of
receiving dantrolene (1 mg/kg) about 2 h after the onset of malignant
hyperthermia (Desparmet et al., 1983). Doses of up to 3.6 mg/kg have
been given to patients who subsequently recovered (Maruta et al.,
1980). Experienced clinicians prefer a more aggressive dosing of 1
mg/kg per min up to a total of 10 mg/kg or even more (personal
communications by B.A. Britt and T. Fagerlund to the IPCS, 1992). The
time factor is critical, since dantrolene is often ineffective if
treatment is delayed for 2 h after the onset of signs in children
(personal communication by B.A. Britt to the IPCS, 1992).
In adults, dantrolene appears uniformly effective if given within
6 h of the precipitating anaesthetic agent, but death may supervene if
treatment is delayed beyond this (Kolb et al., 1982; personal
communication by B.A. Britt to the IPCS, 1992). Death occurred in one
case despite a massive initial intravenous dose (Mathieu et al., 1979)
and in two others despite prolonged oral and intravenous therapy over
10 to 21 days (Kolb et al., 1982). Presumably irreversible changes
took place before treatment was started. In the study by Kolb et al.
(1982), intravenous dantrolene (a total dose of 1-7 mg/kg) lead to the
successful treatment of three patients with probable and eight
patients with unequivocal malignant hyperthermia.
4.11.1.1 Prophylaxis of malignant hyperthermia
Where a family history of malignant hyperthermia has been
particularly strong or where a patient has had previous confirmed or
suspected episodes of anaesthetic-induced malignant hyperthermia, the
prophylactic administration of dantrolene has been discussed, along
with the avoidance of "trigger" agents (Ward et al., 1986). An oral
dosage of 4 to 8 mg/kg per day, given in three or four divided doses
for 1 or 2 days with the last dose being administered 3 to 4 h prior
to anaesthesia, has been recommended (Ward et al., 1986).
Alternatively, a single intravenous dose of 2.5 mg/kg has been
proposed, to be given just prior to surgery (Flewellen et al., 1983).
Most of the patients treated in this manner have undergone
uneventful surgery although in three cases postoperative tachycardia,
increased blood pressure, and respiratory and metabolic acidosis
occurred without hyperthermia or muscle rigidity. These patients were
all successfully treated with either further intravenous dantrolene
(Fitzgibbons, 1981; Ruhland & Hinkle, 1984) or by correcting the
metabolic acidosis (de Pinna, 1978).
There is, however, no consensus on this prophylactic use of
dantrolene (Harrison, 1988) although most experienced clinicians are
now in favour of using no prophylactic treatment (personal
communication by B.A Britt and T. Fagerlund to the IPCS, 1992).
Recently, a series of 30 general anaesthesias in 24 patients
susceptible to malignant hyperthermia was reported. In all patients,
malignant hyperthermia susceptibility was confirmed by in vitro
testing of skeletal muscles obtained from a biopsy of the quadriceps
femoris muscle, and four patients had experienced previous episodes of
malignant hyperthermia during anaesthesia. No clinical or biochemical
features of malignant hyperthermia were observed (Hackl et al., 1990).
These authors concluded that safe anaesthesia for these patients could
easily be provided simply by avoiding trigger agents and carefully
preparing the monitoring equipment and anaesthesia machine.
Dantrolene should be reserved for the treatment of malignant
hyperthermia.
4.11.1.2 Prophylaxis of malignant hyperthermia during pregnancy
In a prospective study of 32 pregnant women known to be
susceptible to malignant hyperthermia, 17 received oral dantrolene
sodium beginning 5 days prior to planned delivery, while the other 15
did not receive prophylactic dantrolene. Malignant hyperthermia
developed immediately postpartum in one mother and her baby who had
not received prophylactic dantrolene, but both mother and baby
responded rapidly to intravenous dantrolene. None of the other
pregnancies was complicated and all fetal examinations and neonatal
neurological assessments were normal (Ward et al., 1986).
In a series of 20 malignant hyperthermia-susceptible pregnant
patients, dantrolene was given orally for 5 days before and 3 days
after delivery (in three cases, delivery was by Caesarean section)
(Shime et al., 1988). No signs of malignant hyperthermia or adverse
effects of dantrolene were seen in this non-controlled study.
There have been several other case reports of successful use of
prophylactic intravenous dantrolene in pregnant women who were
susceptible to this condition and who underwent anaesthesia during
birth (Cupryn et al., 1984; Glassenberg & Cohen, 1984) and of the
reversal of established malignant hyperthermia with intravenous
dantrolene in both mothers and their neonates (Sewall et al., 1980;
Lips et al., 1982). Weingarten et al. (1987) reported a case of
postpartum uterine atony in a patient who received dantrolene during
a Caesarian section.
Although there are several non-controlled reports of the
successful use of dantrolene prior to planned delivery, there is no
established consensus on this use.
4.11.2 Use in neuroleptic malignant syndrome
In the neuroleptic malignant syndrome, thermogenesis is
ultimately due to tonic contraction of skeletal muscles. Thus, the
use of dantrolene may be helpful by relaxing skeletal muscle.
Since the initial observations (Bismuth et al., 1982; Boles et
al., 1982), there have been many case reports of the use of dantrolene
in the neuroleptic malignant syndrome (Ward et al., 1986; Harrison
1988) with no consensus concerning dosage. Single intravenous bolus
doses (less than 3 to 10 mg/kg) and repeated oral doses (25 to 600
mg/day) have been used, often in combination with other drugs and
supportive treatment. Dantrolene apparently reduces the pyrexia,
usually within 12 h of intravenous therapy and over a somewhat longer
period with oral dantrolene therapy. Most patients also improve
clinically, although this occurs at a later stage. In some cases
withdrawal of oral dantrolene therapy has been associated with a
deterioration of the patient's condition and an increase in body
temperature.
4.11.3 Use in other drug-induced hyperthermia
Hyperthermia due to or associated with increased muscular
rigidity can occur in poisoning with strychnine (Gordon & Richards,
1979), phencyclidine (Jan et al., 1978) or Cicuta species (water
hemlock) (Starreveld & Hope, 1975). In cases of poisoning with these
agents, the direct action of dantrolene on muscle activity is likely
to be of benefit in controlling the hyperpyrexia. It would be logical
to give intravenous dantrolene as in cases of malignant hyperthermia,
provided it was combined with standard supportive measures such as
external cooling. There is, however, no experimental verification of
this view, nor any report of a relevant clinical case.
Amphetamines (Jordan & Hampson, 1960; Kendrick et al., 1977;
Ginsberg et al., 1970), monoamine oxidase inhibitors (Mirchandani &
Reich, 1985), lysergic acid diethylamide (Friedman & Hirsch, 1971;
Mercieca & Brown, 1984), and cocaine (Roberts et al., 1984) all cause
hyperthermia, which is due at least in part to motor overactivity with
or without convulsions. Where hyperthermia does not respond to
sedation with a major tranquillizer, and where paralysis and
artificial ventilation are inappropriate or ineffective, dantrolene
should be of benefit, but documentation is still lacking.
A case of overdosage with phenelzine, a monoamine oxidase
inhibitor, was treated successfully with intravenous dantrolene after
conventional therapy had failed (Kaplan et al. 1986), as was a case of
carbon monoxide poisoning with hyperthermia and rigidity (Holter &
Schellens, 1988). In a fatal case of theophylline poisoning with
rhabdomyolysis, dantrolene administration was claimed to be useful in
controlling the hypermetabolic state (Parr & Willatts, 1991).
Hyperthermia in poisoning with salicylates, dinitrophenol and the
herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is due to uncoupling
of oxidative phosphorylation. Tricyclic antidepressives and other
anticholinergic drugs reduce sweating and so impair heat loss.
Dantrolene would not be expected to have a role in the treatment of
hyperpyrexia induced by these mechanisms.
4.12 Summary of Evaluation
4.12.1 Indications
4.12.1.1 Treatment of malignant hyperthermia
When combined with standard cooling and supportive therapy,
dantrolene has proved effective in rapidly reversing clinical symptoms
in anaesthetic-induced malignant hyperthermia, as judged from clinical
case reports. Failure of dantrolene to prevent the often fatal
consequences of malignant hyperthermia is usually a consequence of
late diagnosis and therapy.
4.12.1.2 Treatment of neuroleptic malignant syndrome
Dantrolene may be helpful as an adjunct to supportive therapy in
neuroleptic malignant syndrome induced by drugs such as dopamine
antagonists, particularly the major tranquillizers. There is no
report of a controlled trial in this rare illness, and no study
comparing dantrolene with bromocriptine, which has also been used in
its management.
4.12.1.3 Treatment of hyperthermia induced by muscle rigidity in
poisoning
Though documentation is lacking, there are good theoretical
grounds for using dantrolene to treat hyperthermia due to poisoning
with strychnine, phencyclidine or Cicuta species (water hemlock). It
might also be useful in the management of hyperthermia in patients
poisoned with amphetamines, cocaine, lysergic acid diethylamine (LSD)
or monoamine oxidase inhibitors, where hyperthermia is associated with
motor overactivity.
4.12.2 Advised routes and doses
4.12.2.1 Treatment of severe drug-induced hyperthermia, including
malignant hyperthermia
Dantrolene (2.5 mg/kg) should be given intravenously as soon as
possible. A response to the treatment should be apparent within
minutes; if not, up to 10 mg/kg may be given again every 15 min until
there is an effect. Subsequent to intravenous therapy, dantrolene (4
mg/kg per day in divided doses for 48 h) can be given orally to
prevent recurrence. The patient should stay in the intensive care
unit for at least 24 h after the hyperthermia reaction has subsided
(personal communication by T. Fagerlund to the IPCS, 1992). The
proposed dosage regimen for dantrolene is not clearly defined (see
section 4.12.4).
In cases of neuroleptic malignant syndrome, the use of dantrolene
and dopamine agonists (amantadine and bromocriptine) has been
described (Boles et al., 1982; Goulon et al., 1983) and has produced
promising results.
4.12.2.2 Prophylaxis of malignant hyperthermia prior to anaesthesia
in susceptible patients
Triggering anaesthetic agents have to be avoided in patients
known to be susceptible to malignant hyperthermia. Such agents are
depolarizing muscle relaxants (succinylcholine), halogenated
inhalational anaesthetics (e.g., halothane), haloperidol and
promethazine. Regional anaesthetic techniques should be preferred if
possible.
Oral dantrolene (4-8 mg/kg per day) administered for 1-2 days
prior to anaesthesia may prevent malignant hyperthermia in known
susceptible individuals. Alternatively, intravenous dantrolene (2.5
mg/kg) can be given just prior to anaesthesia (Flewellen et al.,
1983). Oral dantrolene given 5 days before and 3 days after delivery
has been associated with favourable outcome in pregnant patients
susceptible to malignant hyperthermia (Shime et al., 1988). Such
prophylaxis with dantrolene is controversial and not generally
recommended.
4.12.3 Other consequential or supportive therapy
It is very important for the administration of dantrolene to be
accompanied by the use of aggressive supportive therapy, including
central cooling techniques, an inspired oxygen concentration enriched
up to 100%, and correction of metabolic acidosis. Serum potassium
concentrations should be monitored closely.
Further supportive therapy should be directed towards
complications such as respiratory acidosis, cardiac arrhythmias, and
instability of blood pressure. Special attention must be given to the
development of rhabdomyolysis leading to elevated serum muscle enzymes
(creatine kinase, aspartate transaminase, aldolase). If creatine
kinase activities are above 10 000 U/l and/or meat-coloured urine is
present, together with areas of swollen and tender skeletal muscles,
the patient may develop acute renal failure as muscle debris is
precipitated in the kidneys (myoglobinuria). Associated electrolyte
disturbances are hyperkalaemia, hypocalcaemia and hyperphosphataemia.
Acute renal failure of this nature has been reported to be prevented,
even at creatine kinase activities above 70 000 U/l, when prompt
forced alkaline diuresis treatment has been instituted in order to
prevent debris deposition in the kidneys (personal communication by D.
Jacobsen to the IPCS, 1992). Urine output should exceed 2 ml/kg per
h in these patients.
4.12.4 Controversial issues and areas of insufficient information
The prophylactic use of dantrolene prior to anaesthesia in
susceptible patients should be considered controversial and an
international consensus has yet to be established (Ward et al., 1986;
Shime et al., 1988; Harrison, 1988; Hackl et al., 1990). This use of
dantrolene is not generally recommended (personal communications by T.
Fagerlund and B.A. Britt to the IPCS, 1992).
The dose regimen of dantrolene is not clearly defined. The
initial dose recommended in the USA is generally 1 mg/kg per min
(personal communication by B.A. Britt to the IPCS, 1992), whereas the
European recommendation is usually an initial dose of 2.5 mg/kg
(personal communication by T. Fagerlund to the IPCS, 1992). There is,
however, a consensus on the maximum dose of 10 mg/kg per 15 min.
Furthermore, there is no clear overall maximum dose of dantrolene
during the first hours of treatment. This may be important, as
malignant hyperthermia could be incorrectly diagnosed in places with
limited laboratory facilities. In such situations, one should not
continue to give dantrolene in doses of 10 mg/kg per 15 min.
Therefore, if more than 20 mg/kg has been given during the first 30
min without any effect, the diagnosis of malignant hyperthermia should
certainly be questioned (personal communication by T. Fagerlund to the
IPCS, 1992). Another reason for lack of effect could be that
dantrolene therapy has been initiated too late, since the time factor
is critical.
It is not clear whether doses higher than 10 mg/kg per day add
any beneficial effects concerning the prognosis. However, clinicians
with experience in this field do recommend higher doses for critically
ill patients who do not respond to the initial dose (personal
communications by T. Fagerlund and B.A. Britt to the IPCS, 1992).
The use of external cooling techniques with these patients has
been generally recommended for years. However, this treatment is now
being questioned, since dantrolene treatment alone lowers temperatures
to the normal range. External cooling also prevents heat loss by
inducing peripheral vasoconstriction and increases heat production by
stimulating shivering and non-shivering thermogenesis (personal
communication by B.A. Britt to the IPCS, 1992).
4.12.5 Proposals for further studies
The effect of dantrolene in established drug-induced hyperthermia
has not been documented from a scientific point of view. However, the
effect, as indicated in several case reports (and series of such
reports), is so convincing that it may be difficult to perform a
controlled double-blind study due to ethical considerations.
The prophylactic use of dantrolene prior to anaesthesia in
susceptible patients may warrant a controlled study. Due to the
relatively small number of patients, this should preferably be done
under a multicenter design.
In cases of neuroleptic malignant syndrome, a treatment protocol
using dantrolene versus dantrolene/bromocriptine may be warranted
(Boles et al., 1982; Goulon et al., 1983).
The optimum dose regimen of dantrolene is not clearly defined and
warrants further study.
4.12.6 Adverse effects
Dantrolene sodium solution is highly alkaline (pH 9.6) and
extravasation during intravenous injection may cause tissue necrosis
(Harrison, 1988). Due to this risk of severe thrombophlebitis,
intravenous administration should preferably be given through a
central venous catheter. Apart from this, dantrolene appears to be
well tolerated for the short-term use discussed in this monograph
(Kolb et al., 1982, Shime et al., 1988).
Several side effects have been reported in patients receiving
chronic dantrolene treatment for spasticity. Muscular weakness,
drowsiness, dizziness and general malaise commonly occur and
gastrointestinal disturbances are seen less frequently. Rare adverse
effects have included hallucinations (Andrews et al., 1975),
exacerbation of respiratory depression (Rivera et al., 1975),
pleuropericardial reaction (Petusevsky et al., 1979; Miller & Haas
1984), lymphocytic lymphoma (Wan & Tucker, 1980), and leucopenia
(Greenspun & Pacho, 1981). The most serious reaction in long-term use
is hepatotoxicity.
4.12.6.1 Hepatotoxicity
Hepatotoxicity may occur when patients are treated with
dantrolene, as indicated in a review by Chan (1990). In general,
therapy was given for at least two months before injury became
evident. In 107 adult cases of dantrolene hepatotoxicity, 40 (37%)
received 200 mg/day or less; 48 (45%) received up to 400 mg/day, and
the highest dose given was 1600 mg/day (Chan, 1990). Biochemical
abnormalities of liver function were observed in about 1.8% of
patients being given long-term dantrolene treatment and fatal
hepatitis occurred in 0.3%. The risk of hepatic injury was greatest
in females, in patients receiving doses over 300 mg/day, and in those
treated for over 60 days (Utili et al., 1977). The major histological
pathology was subacute hepatic necrosis or chronic active hepatitis,
although cholestasis has been reported.
4.12.6.2 Interaction with calcium antagonists
The combination of calcium antagonists and dantrolene may result
in systemic hyperkalaemia and even cardiovascular collapse, and should
therefore be avoided (Yoganathan, 1988).
4.12.7 Restrictions for use
When dantrolene was used prophylactically 5 days before delivery
to twenty pregnant women, no adverse effect on the fetus or newborn
infant could be detected (Shime et al., 1988). A possible teratogenic
effect could, however, not be ruled out from these data.
Reconstituted dantrolene should not be used if more than 6 h has
passed since it was made up or if the solution has not been protected
from light.
4.13 Model Information Sheet
The current theory is that dantrolene relaxes peripheral striated
muscle by preventing Ca2+ flux across the sarcoplasmic reticulum and
thereby reducing the free intracellular calcium concentration. Since
malignant hyperthermia and other drug-induced hyperthermias are
considered to be a paroxysmal hypercatabolic reaction of these
muscles, the hyperpyrexia is counteracted by the effect of dantrolene.
When the doses recommended in this monograph are used, dantrolene has
no significant effect on other calcium-dependent systems.
4.13.1 Uses as an antidote
Use of dantrolene is indicated in:
a) the treatment of malignant hyperthermia induced in susceptible
individuals by anaesthetic agents or skeletal muscle relaxants;
b) the treatment of malignant neuroleptic syndrome;
c) the treatment of hyperpyrexia due to poisoning with strychnine,
Cicuta species (water hemlock) or phencyclidine, and perhaps also
hyperpyrexia due to poisoning with amphetamines, cocaine,
lysergic acid diethylamine (LSD), theophylline or monoamine
oxidase inhibitors.
The prophylactic use of dantrolene prior to anaesthesia to
susceptible patients should be considered controversial and is not
generally recommended.
4.13.2 Dosage and route
Whenever indicated, dantrolene treatment must be started without
delay. Dantrolene is often ineffective if treatment is delayed for 2
h after the onset of signs in children, or 6 h after the onset of
signs in adults.
In the treatment of severe drug-induced hyperthermia, including
acute malignant hyperthermia induced by anaesthetic agents, dantrolene
must be given intravenously as soon as the diagnosis is made. The
initial dose is 1-2.5 mg/kg given intravenously (1 mg/kg per min), and
the effectiveness of the treatment should be monitored closely. A
dose of up to 10 mg/kg may be given again every 15 min (1 mg/kg per
min) if signs and symptoms do not subside. Doses greater than 10
mg/kg are rarely needed.
The recurrence of symptoms should be treated in the same way. To
prevent this, dantrolene can be given orally in divided doses at a
total dosage of 4 mg/kg per day.
Provided adequate supportive therapy (section 12.3) is given,
dantrolene treatment can usually be stopped within 2 days in cases of
malignant hyperthermia. Oral treatment may be given for up to 10-14
days in cases of malignant neuroleptic syndrome, or even longer if
depot neuroleptics have precipitated the syndrome.
In the controversial prophylaxis of malignant hyperthermia in
susceptible individuals, dantrolene may either be given orally at a
dosage of 4-8 mg/kg per day for 1 to 2 days prior to anaesthesia, or
intravenously as a single dose of 2.5 mg/kg 1-2 h prior to
anaesthesia. This approach is, however, not generally recommended.
4.13.3 Precautions and contraindications
Solutions of dantrolene should be protected from light.
Solutions of dantrolene are strongly alkaline (pH 9.5).
Extravasation may cause tissue necrosis. The dantrolene solution
should therefore be injected via a fast-flowing intravenous infusion
into a large vein, or preferably through a central venous catheter.
Hepatic failure should be considered as a relative
contraindication and there should be a firm diagnosis for the use of
dantrolene is this situation (e.g., no prophylactic use).
4.13.4 Pharmaceutical incompatibilities and drug interactions
Dantrolene sodium powder should only be diluted with sterile
water for injection, and is incompatible with other infusion fluids.
The combination of dantrolene and calcium antagonists should be
avoided, as severe hyperkalaemia and myocardial depression may occur.
Treatment with calcium antagonists should therefore be discontinued
if dantrolene is given or may be given.
4.13.5 Adverse effects
The major adverse effect is hepatic toxicity, which may be fatal,
although this is unlikely in the acute setting discussed here. Few
side effects have been reported at the dose levels recommended in this
monograph.
In chronic treatment (muscle rigidity), dantrolene may also cause
muscle weakness, drowsiness, dizziness and malaises. Hallucinations,
exacerbation of respiratory depression, pleuroperitonitis, lymphocytic
lymphoma and leucopenia have been reported rarely.
When given intravenously, dantrolene solution is highly irritant.
Extravasation may cause tissue necrosis.
4.13.6 Use in pregnancy and lactation
There is no evidence that dantrolene is harmful when given
prophylactically to predisposed mothers before delivery. No adverse
effects on the fetus or newborn infant have been reported. Dantrolene
crosses the placenta (the fetal:maternal concentration ratio is 0.4:1)
and is excreted in breast milk. It would be prudent to avoid breast
feeding if dantrolene were being taken for prophylaxis or treatment.
4.13.7 Storage
Dantrolene powder and capsules are stable at temperatures below
40 °C. The capsules should be stored in well-sealed containers.
Following reconstitution with sterile water, dantrolene sodium
injection solution is stable for only 6 h at room temperature.
Solutions should be protected from light.
4.14 References
Allen GC, Cattran CB, Peterson RG, & Lalande M (1988) Plasma levels
of dantrolene following oral administration in malignant hyperthermia-
susceptible patients. Anaesthesiology, 69: 900-904.
Amelink GJH, Van der Kallen CJH, Wokke JHJ, & Bar PRD (1990)
Dantrolene sodium diminishes exercise-induced muscle damage in the
rat. Eur J Pharmacol, 179: 187-192.
Anderson IL, Lipicky RJ, & Jones EW (1978) Dantrolene sodium in
porcine malignant hyperthermia: Studies on isolated muscle strips. In:
Adrete JA & Britt BA ed. Second International Symposium on Malignant
Hyperthermia, Denver, Colorado, 1-3 April 1977. New York, Grune &
Stratton, pp 509-534.
Andrews LG, Muzumdar AS, & Pinkerton AC (1975) Hallucinations
associated with dantrolene sodium therapy. Can Med Assoc J, 112: 148.
Bismuth C, Elkharrat D, Rohan-Chabot P, & Conso F (1982) Theoretical
indication of dantrolene in neuroleptic malignant syndrome. Efficacy
in three cases. Vet Hum Toxicol, 24(Suppl 1): 52-55.
Boles JM, Lecam B, Mialon P, Pennec Y, & Garre M (1982) Hyperthermie
maligne des neuroleptiques: guérison rapide par le dantrolène. Nouv
Presse Méd, 11: 674.
Chan CH (1990) Dantrolene sodium and hepatic injury. Neurology, 40:
1427-1432.
Craft JB, Goldberg NH, Lim M, Landsberger E, Mazel P, Abramson FP,
Stolte AL, Braswell ME, & Farina JP (1988) Cardiovascular effects and
placental passage of dantrolene in the maternal-fetal sheep model.
Anaesthesiology, 68: 68-72.
Cupryn JP, Kennedy A, & Byrick RJ (1984) Malignant hyperthermia in
pregnancy. Am J Obstet Gynecol, 150: 327-328.
De Pinna GA (1978) A case of osteogenesis imperfecta, malignant
hyperthermia susceptible - anesthetic management. In: Aldrete JA &
Britt BA ed. Malignant hyperthermia. New York, Grune & Stratton, pp
409-418.
Desparmet J, Josse E, Bavoux F, Laguenie G, & Saint-Maurice C (1983)
Fatal malignant hyperthermia in an eleven-year old child. Cah
Anesthésiol, 31: 303-305.
Dykes HHM (1975) Evaluation of a muscle relaxant: Dantrolene sodium
(Dantrium). J Am Med Assoc, 231: 862-864.
Ellis KO & Carpenter JF (1974) A comparative study of dantrolene
sodium and other skeletal muscle relaxants with the Straub tail mouse.
Neuropharmacology, 13: 211-214.
Ellis KO & Wessels FL (1977) Dantrolene sodium effects on caffeine
contractures in frog sartorius muscle. In Wassermann RH ed. Calcium-
binding proteins and calcium function: Proceedings of the 2nd
International Symposium on Calcium-Binding Proteins and Calcium
Function in Health and Disease, Cornell University, Ithaca, New York,
5-9 June 1977. Amsterdam, Elsevier North-Holland, pp 273-274.
Ellis KO & Wessels FL (1978) Muscle relaxant properties of the
identified metabolites of dantrolene. Naunyn-Schmiedeberg's Arch
Pharmacol, 301: 237-240.
Fagerlund T, Islander G, Ranklew E, Harbitz I, Hauge JG, Mokleby E, &
Berg K (1992) Genetic recombination between malignant hyperthermia
and calcium release in skeletal muscle. Clin Genet, 41: 270-272.
Fairhurst AS, Hamamoto V, & Macri J (1980) Modification of ryanodine
toxicity by dantrolene and halothane in a model of malignant
hyperthermia. Anaesthesiology, 53: 199-204.
Fitzgibbons DC (1981) Malignant hyperthermia following preoperative
oral administration of dantrolene. Anaesthesiology, 54: 73-75.
Fletcher JE & Rosenberg H (1985) In vitro interaction between
halothane and succinylcholine in human skeletal muscle: implications
for malignant hyperthermia and masseter muscle rigidity.
Anaesthesiology, 63: 190-194.
Flewellen EH & Nelson TE (1985) Intravenous dantrolene
pharmacokinetics in malignant hyperthermia suspect patients.
Anaesthesiology, 63(Suppl 3A): 300.
Flewellen EH, Nelson TE, Jones WP, Arens JF, & Wagner DL (1983)
Dantrolene dose response in awake man: implications for management of
malignant hyperthermia. Anaesthesiology, 59: 275-280.
Fournier P (1982) Dossier toxicologique, pharmacologique,
pharmacocinétique du Dantrium IV. Dantrium et hyperthermie maligne.
Lyon, Laboratoire Oberval (Unpublished internal report).
Francis KT & Hamrick ME (1980) Effects of dantrolene on adrenal
cortical function. Biochem Pharmacol, 29(12): 1669-1672.
Friedman SA & Hirsch SE (1971) Extreme hyperthermia after LSD
ingestion. J Am Med Assoc, 217: 1549-1550.
Ginsberg MD, Hertzman M, & Schmidt-Nowara WW (1970) Amphetamine
intoxication with coagulopathy hyperthermia and reversible renal
failure. Ann Intern Med, 75: 81-85.
Glassenberg R & Cohen H (1984) Intravenous dantrolene in a pregnant
malignant hyperthermia susceptible patient. Anesthesiology, 61(Suppl
3): 404.
Gordon AM & Richards DW (1979) Strychnine intoxication. J Am Coll
Emergency Physicians, 8: 520-522.
Goulon M, De Rohan-Chabot P, Elkharrat D, Gajdos PH, Bismuth C, &
Conso F (1983) Beneficial effect of dantrolene in the treatment of
neuroleptic malignant syndrome: a report of two cases. Neurology, 33:
516-518.
Greenspun B & Pacho A (1981) Leukopenia with dantrolene sodium. Arch
Phys Med Rehabil, 62: 521.
Gronert GA, Milde JH, & Theye RA (1976) Dantrolene in porcine
malignant hyperthermia. Anaesthesiology, 44: 488-495.
Hackl W, Mauritz W, Winkler M, Sporn P, & Sternberetner K (1990)
Anaesthesia in malignant hyperthermia-susceptible patients without
dantrolene prophylaxis: a report of 30 cases. Acta Anaesthesiol Scand,
34: 534-537.
Hall GM, Lucke JN, Orchard C, Lovell R, & Lister D (1982) Effect of
dantrolene on leg metabolism in porcine malignant hyperthermia.
Ananesthesia, 37: 1167-1170.
Hallsall PJ & Ellis FR (1979) A screening test for the malignant
hyperpyrexia phenotype using suxamethonium-induced contracture of
muscle treated with caffeine, and its inhibition by dantrolene. Br J
Anaesthesiol, 51: 753-756.
Harrison G (1977) The control and prevention of malignant hyperthermia
in MHS pigs. Some experimental observations. In: Anaesthesiology.
Amsterdam, Exerpta Medica, pp 452-454.
Harrison GG (1988) Malignant hyperthermia. Dantrolene dynamics and
kinetics. Br J Anaesthesiol, 60: 279-286.
Healy JMS, Heffron JJA, Lehane M, Bradley DG, Johnson K, & McCarthy TV
(1991) Diagnosis of susceptibility to malignant hyperthermia with
flanking DNA markers. Br Med J, 303: 1225-1228.
Hollifield RD & Conklin JD (1968) A spectrophotofluorometric procedure
for the determination of dantrolene in blood and urine. Arch Int
Pharmacodyn, 174: 333-341.
Holter MT & Schellens LAM (1988) Dantrolene sodium for treatment of
carbon monoxide poisoning. Br Med J, 296: 1772-1773.
Index Nominum (1990) International drug directory. Zurich, Medpharm.
Jan KM, Dorsey S, & Bernstein A (1978) Hot hog: hyperthermia from
phencyclidine. New Engl J Med, 299: 722.
Jordan SC & Hampson F (1960) Amphetamine poisoning associated with
hyperpyrexia. Br Med J, 2: 844.
Kaplan RF, Feinglass NG, Webster W, & Mudra S (1986) Phenelzine
overdose treated with dantrolene sodium. J Am Med Assoc, 255: 642-644.
Katogi Y, Tamaki N, & Adachi M (1982) Simultaneous determination of
dantrolene and its metabolite, 5-hydroxydantrolene, in human plasma by
high-performance liquid chromatography. J Chromatogr, 228: 404-408.
Kendrick WC, Hull AR, & Knochel JP (1977) Rhabdomyolysis and shock
after intravenous amphetamine administration. Ann Intern Med, 86:
381-387.
Kerr DD, Wingard DW, & Gatz EE (1978) Prevention of porcine malignant
hyperthermia by oral dantrolene. In: Aldrete JA & Britt BA ed. Second
International Symposium on Malignant Hyperthermia, Denver, Colorado,
1-3 April 1977. New York, Grune & Stratton, pp 499-507.
Kolb ME, Horne ML, & Martz R (1982) Dantrolene in human malignant
hyperthermia. Anesthesiology, 56: 254-262.
Lalande M, Mills P, & Peterson RG (1988) Determination of dantrolene
and its reduced and oxidized metabolites in plasma by high-performance
liquid chromatography. J Chromatogr, 430: 187-191.
Lerman J, McLeod ME, & Strong HA (1989) Pharmacokinetics of dantrolene
in children. Anaesthesiology, 70: 625-629.
Lietman PS, Haslam RHA, & Walcher JR (1974) Pharmacology of dantrolene
sodium in children. Arch Phys Med Rehabil, 55: 388-392.
Lips FJ, Newland M, & Dutton G (1982) Malignant hyperthermia
triggered by cyclopropane during caesarian section. Anesthesiology,
56: 144-146.
Lopez JR, Allen P, Alamo L, Ryan JF, Jones DE, & Sreter F (1987)
Dantrolene prevents the malignant hyperthermic syndrome by reducing
free intracellular calcium concentration in skeletal muscle of
susceptible swine. Cell calcium, 8: 385-396.
Lydiatt JS & Hill GE (1981) Treatment of heat stroke with dantrolene.
J Am Med Assoc, 246: 41-42.
MacLennan DH, Duff C, Zorzato F, Fujii J, Phillips M, Korneluk RG,
Frodis W, Britt BA, & Worton RG (1990) Ryanodine receptor gene is a
candidate for predisposition to malignant hyperthermia. Nature(Lond),
343: 559-561.
Martindale (1989) In: Reynolds JEF ed. The extra pharmacopoeia, 29th
ed. London, Pharmaceutical Press, p 1896.
Maruta H, Soga T, Okutsu Y, & Asari H (1980) Malignant hyperthermia:
a case report. Hiroshima J Anaesth, 16(Suppl): 35-39.
Mathieu A, Bogosian AJ, Ryan JF, Crone RK, & Crocker D (1979)
Recrudescence after survival of an initial episode of malignant
hyperthermia. Anaesthesiology, 51: 454-455.
Mercieca J & Brown EA (1984) Acute renal failure due to rhabdomyolysis
associated with use of a straight jacket in lysergide intoxication. Br
Med J, 288: 1949-1950.
Meyler WJ, Mols-Thurkow HW, & Wesseling H (1979) Relationship between
plasma concentration and effect of dantrolene sodium in man. Eur J
Clin Pharmacol, 16: 203-209.
Meyler WJ, Bakker H, Kok JJ, Agoston S, & Wesseling H (1981) The
effect of dantrolene sodium in relation to blood levels in spastic
patients after prolonged administration. J Neurol Neurosurg
Psychiatry, 44: 334-339.
Miller DH & Haas LF (1984) Pneumonitis, pleural effusion and
pericarditis follwoing treatment with dantrolene. J Neurol Neurosurg
Psychiatry, 47: 553-554.
Mirchandani H & Reich LE (1985) Fatal malignant hyperthermia as a
result of the ingestion of tranylcypromine (Parnate) combined with
white wine and cheese. J Forensic Sci, 30: 217-230.
Miyamoto H & Racker E (1982) Mechanism of calcium release from
skeleteal sarcoplasmic reticulum. J Membrane Biol, 66: 193-201.
Moffat AC (1986) Clarke's isolation and identification of drugs in
pharmaceuticals, body fluids and post mortem materials, 2nd ed.
London, The Pharmaceutical Press, p 508.
Morison DH (1983) Placental transfer of dantrolene. Anaesthesiology,
59: 265.
Nelson TE (1983) Dantrolene does not block abnormal calcium efflux
from a putative calcium channel of MH sarcoplasmic reticulum
(abstract). Anaesthesiology, 59: A225.
Nelson TE & Denborough MA (1977) Studies on normal skeletal muscle in
the relation to the pathopharmacology of malignant hyperpyrexia. Clin
Exp Pharmacol Physiol, 4: 315-322.
Ohnishi ST, Taylor S, & Gronert GA (1983) Calcium-induced calcium
release from sarcoplasmic reticulum of pigs susceptible to malignant
hyperthermia. The effect of halothane and dantrolene. FEBS Lett, 261:
103-107.
Ohta T, Ito S, & Ohga A (1990) Inhibitory action of dantrolene on
Ca-induced Ca2+ release from sarcoplasmic reticulum in guinea pig
skeletal muscle. Eur J Pharmacol, 178: 11-19.
Okumura F, Crocker BD, & Denborough MA (1980) Site of the muscle cell
abnormality in swine susceptible to malignant hyperpyrexia. Br J
Anaesthesiol, 52: 377-383.
Parr MJA & Willatts SM (1991) Fatal theophylline poisoning with
rhabdomyolysis. Anaesthesia, 46: 557-559.
Petusevsky ML, Ealing LJ, Rocklin RE, Snider GL, Merliss AD, Moses JM,
& Dorman SA (1979) Pleuropericardial reaction to treatment with
dantrolene. J Am Med Assoc, 242: 2772.
Pinder RM, Brogden RN, Speight TM, & Avery GS (1977) Dantrolene
sodium: A review of its pharmacological properties and therapeutic
efficacy in spasticity. Drugs, 13: 3-23.
Pope HG, Cole JO, Choras PT, & Fulwiler CE (1986) Apparent neuroleptic
malignant syndrome with clozapine and lithium. J Nerv Ment Dis, 174:
493-495.
Rivera VM, Breitbach WB, & Swanke L (1975) Dantrolene in amyotrophic
lateral scierosis. J Am Med Assoc, 233: 863-864.
Roberts JR, Quattrocchi E, & Howland MA (1984) Severe hyperthermia
secondary to intravenous drug abuse. Am J Emergency Med, 2: 373.
Ruhland G & Hinkle AJ (1984) Malignant hyperthermia after oral and
intravenous pretreatment with dantrolene in a patient susceptible to
malignant hyperthermia. Anaesthesiology, 60: 159-160.
Saxena SJ, Honigberg IL, Stewart JT, & Vallner JJ (1977) Liquid
chromatography in pharmaceutical analysis VI: determination of
dantrolene sodium in a dosage form. J Pharm Sci, 66: 286-288.
Sewall K, Flaverdew RMM, & Bromberger P (1980) Severe muscular
rigidity at birth; malignant hyperthermia syndrom? Can Anaesth Soc J,
27: 279-282.
Shime J, Gare D, Andrews J, & Britt B (1988) Dantrolene in pregnancy:
Lack of adverse effects on the fetus and newborn infant. Am J Obstet
Gynecol, 159: 831-834.
Snyder HR, Davis CS, Bickerton RK, & Halliday RP (1967) 1-((5-
arylfurfurylidene) amino)hydantoins. A new class of muscle relaxants.
J Med Chem, 10: 807-810.
Stadhouders AM, Viering WAL, Verburg MP, Ruitenbeek W, & Sengers RCA
(1984) In vivo induced malignant hyperthermia in pigs. III.
Localization of calcium in skeletal muscle mitochondria by means of
electron-microscopy and microprobe analysis. Acta Anaesthesiol Scand,
28: 14-26.
Starreveld E & Hope CE (1975) Cicutoxin poisoning (Water hemlock).
Neurology, 25: 730-735.
Sullivan JS & Denborough MA (1981) Temperature dependence of muscle
function in malignant hyperpyrexia-susceptible swine. Br J
Anaesthesiol, 53: 1217-1222.
Utili R, Boitnott JK, & Zimmerman HJ (1977) Dantrolene associated
hepatic injury: incidence and character. Gastroenterology, 72:
610-616.
Vallner JJ, Sternson LA, & Parsons DL (1976) Interaction of dantrolene
sodium with human serum albumin. J Pharm Sci, 675: 873-877.
Vallner JJ, Honigberg IL, Stewart JT, & Peyton AB (1979) Dantrolene
and metabolite levels in six patients on chronic therapy. Curr Ther
Res, 25: 79-91.
Wan HH & Tucker JS (1980) Dantrolene and lymphocytic lymphoma.
Postgrad Med J, 56: 261.
Ward A, Chaffman MO, & Sorkin EM (1986) Dantrolene (review). Drugs,
32: 130-168.
Weingarten AE, Korsh JI, Neuman GG, & Stern SB, (1987) Postpartum
uterine atony after intravenous dantrolene. Anesthesiol Analg, 66:
269-270.
Wuis EW, Grutters ACLM, Vree TB, & Van der Kleyn E (1983) Simultaneous
determination of dantrolene and its metabolites, 5-hydroxydantrolene
and nitro-reduced acetylated dantrolene in plasma and urine of man and
dog by HPLC. J Chromatogr, 231: 401-409.
Yamamoto T, Suzuki A, & Hotta K (1977) Dissociation of excitation and
contraction in skeletal muscle induced by denterium oxide and
dantrolene-sodium. Jpn J Physiol, 27: 95-109.
Yoganathan T, Casthely, PA, & Lamprou M (1988) Dantrolene-induced
hyperkalemia in a patient treated with diliazem and metrprolol. J
Cardiothorac Anesth, 2: 363-364.
APPENDIX 1
List of Antidotes
Group 1
Antidote Main indication Other possible
of pathological applications
condition
ACETYLCYSTEINE PARACETAMOL ORGANOCHLORINE
SOLVENTS, AMANITIN
AMYL NITRITE CYANIDE HYDROGEN SULFIDE
ASCORBIC ACID ORGANIC PEROXIDES
(OSMIUM)
ATROPINE CHOLINERGIC SYNDROME
AURINTRICARBOXYLIC BERYLLIUM
ACID (ATA)
BENZYLPENICILLIN AMANITINES
BETA-AMINOPROPIONITRILE CAUSTICS
CALCIUM CHLORIDE OR HF, FLUORIDES, CALCIUM
OTHER CALCIUM SALTS OXALATES ANTAGONISTS
DANTROLENE MALIGNANT MALIGNANT NEUROLEPTIC
HYPERTHERMIA SYNDROME
DEFEROXAMINE IRON, ALUMINIUM PARAQUAT
Antidote Main indication Other possible
of pathological applications
condition
DIAZEPAM CHLOROQUINE
DICOBALT EDETATE CYANIDE
DIGOXIN SPECIFIC DIGOXIN/DIGITOXIN,
ANTIBODY FRAGMENTS DIGITALIS GLYCOSIDES
DIMERCAPROL ARSENIC COPPER, GOLD, MERCURY
(INORGANIC), LEAD
ENCEPHALOPATHY
4-DIMETHYLAMINOPHENOL CYANIDE HYDROGEN SULFIDE
(4-DMAP)
ETHANOL METHANOL, ETHYLENE ALKOXYSILANES
GLYCOL, GLYCOL ETHERS
FLUMAZENIL BENZODIAZEPINES
FOLINIC ACID FOLINIC ACID ANTAGONISTS
GLUCAGON BETA-BLOCKERS
GLUCOSE INSULIN
GUANIDINE BOTULISM
HYDROXOCOBALAMIN CYANIDE
Antidote Main indication Other possible
of pathological applications
condition
ISOPRENALINE BETA-BLOCKERS
METHIONINE PARACETAMOL
4-METHYLPYRAZOLE ETHYLENE GLYCOL, COPRIN & DISULFIRAM
METHANOL
METHYLTHIONINIUM METHAEMOGLOBINAEMIA
CHLORIDE
(METHYLENE BLUE)
N-ACETYL PENICILLAMINE MERCURY
NALOXONE OPIATES
NEOSTIGMINE NEUROMUSCULAR BLOCK
(CURARE TYPE) PERIPHERAL
ANTI-CHOLINERGIC
POISONING
OXIMES ORGANOPHOSPHATES
OXYGEN CYANIDE, CARBON MONOXIDE,
HYDROGEN SULFIDE
OXYGEN-HYPERBARIC CARBON MONOXIDE CYANIDE, HYDROGEN
SULFIDE, CARBON
TETRACHLORIDE
Antidote Main indication Other possible
of pathological applications
condition
PENICILLAMINE COPPER GOLD, LEAD,
MERCURY
PENTETIC ACID (DTPA) RADIOACTIVE METALS
PHENTOLAMINE ALPHA-ADRENERGIC
POISONING
PHYSOSTIGMINE CENTRAL ANTI-CHOLINERGIC CENTRAL ANTI-CHOLINERGIC
SYNDROME FROM ATROPINE SYNDROME FROM
& DERIVATIVES OTHER DRUGS
PHYTOMENADIONE COUMARIN DERIVATIVES
(VITAMIN K)
POTASSIUM HEXACYANOFERRAT THALLIUM
(PRUSSIAN BLUE C177520)
PRENALTEROL BETA-BLOCKERS
PROPRANOLOL BETA-ADRENERGIC
POISONING
PROTAMINE SULFATE HEPARIN
PYRIDOXINE ISONIAZID ETHYLENE GLYCOL,
GYROMETRINE,
HYDRAZINES
Antidote Main indication Other possible
of pathological applications
condition
SILIBININ AMANITINE
SODIUM NITRITE CYANIDE HYDROGEN SULFIDE
SODIUM NITROPRUSSIDE ERGOTISM
SODIUM SALICYLATE BERYLLIUM
SODIUM THIOSULFATE CYANIDE BROMATE, CHLORATE,
IODINE
SUCCIMER (DMSA) LEAD, MERCURY
TOCOPHEROL CARBON MONOXIDE OXYGEN TOXICITY
TOLONIUM CHLORIDE METHAEMOGLOBINAEMIA
(TOLUIDINE BLUE)
TRIENTINE (TRIETHYLENE COPPER
TETRAMINE)
UNITHIOL (DMPS) ARSENIC COPPER, NICKEL,
LEAD, CADMIUM,
MERCURY (METHYL
AND INORGANIC)
Group 2
Agents used to prevent the absorption of poisons,
to enhance their elimination or to treat
symptomatically their effects on body functions
A. Emetics
APOMORPHINE
IPECACUANHA
B. Cathartics and solutions used for whole gut lavage
MAGNESIUM CITRATE
MAGNESIUM SULFATE
MANNITOL
SODIUM SULFATE
SORBITOL
WHOLE GUT LAVAGE FLUIDS
C. Agents to modify urinary pH
AMMONIUM CHLORIDE
ARGININE HYDROCHLORIDE
HYDROCHLORIC ACID (O.1 N)
SODIUM BICARBONATE
D. Agents to prevent absorption of toxic substances in the
gastrointestinal tract
ACTIVATED CHARCOAL (FOR MOST POISONINGS)
DIGITALIS, COUMARIN, KEPONE
FULLERS EARTH PARAQUAT, DIQUAT, POTASSIUM,
COPPER, FERROCYANIDE
SIMETHICONE FOAMING DETERGENTS
SODIUM BICARBONATE IRON, MERCURY,
ORGANOPHOSPHATES
SODIUM SULFATE LEAD, BISMUTH, BARIUM
STARCH IODINE
E. Agents to prevent absorption and/or damage in the skin
CALCIUM GLUCONATE HYDROFLUORIC ACID
GEL
MACROGOL 400 PHENOL
Group 3
Other useful therapeutic agents for
the treatment of poisoning
Below are listed certain therapeutic agents which are not
antidotes according to the accepted definition, but which through
their importance and sometimes specific role in the treatment of
poisonings, border on the concept of "antidotes".
In practice, these agents are used very often in cases of
poisoning and in other medical circumstances. The usefulness of these
agents is in general well established, most of them are considered
essential drugs, and they should be available for immediate use.
Agents Indications - symptoms arising from poisoning
BENZTROPINE dystonia
CHLORPROMAZINE hallucinatory and psychotics states
CORTICOSTEROIDS acute allergic reactions, largyngeal oedema,
(systemic/tropical/bronchoconstriction)
DIAZEPAM convulsions, excitation, anxiety, muscular
hypertonia
DIPHENHYDRAMINE dystonia
DOBUTAMINE myocardial depression
DOPAMINE myocardial depression, vascular relaxation
EPINEPHRINE (ADRENALINE) anaphylactic shock, cardiac arrest
FUROSEMIDE fluid retention, left ventricular failure
GLUCOSE hypoglycaemia
HALOPERIDOL hallucinatory and psychotic states
HEPARIN hypercoagulability states
LIDOCAINE ventricular arrhythmias
MANNITOL (IV) cerebral oedema, fluid retention
Agents Indications - symptoms arising from poisoning
OXYGEN hypoxia
PANCURONIUM muscular rigidity, convulsions
PROMETHAZINE allergic reactions
SALBUTAMOL bronchoconstriction (systemic/inhaled)
SODIUM BICARBONATE acidosis, some cardiac disturbances (e.g.,
TCA poisonings)
Group 4
Antidotes and related agents considered obsolete
Antidote Indicated for
ASCORBIC ACID Methaemoglobinaemia
CYCLOPHOSPHAMIDE Gold-Paraquat
CYSTEAMINE Paracetamol
DIETHYLDITHIOCARBAMATE Thallium
FRUCTOSE Ethanol
LEVALLORPHAN Opiates
NALORPHINE Opiates
POTASSIUM PERMANGANATE Fluorides
SULFADILIDINE Amanitine
TANNINS Alkaloids
THIOCTIC ACID Amanitine
TOCOPHEROL (Vitamin E) Paraquat
UNIVERSAL ANTIDOTE Ingested poisons
COPPER SULFATE as an emetic
SODIUM CHLORIDE as an emetic
CASTOR OIL as a cathartic
ACETAZOLAMIDE as a urinary pH modifier
APPENDIX 2
PRINCIPLES FOR EVALUATION OF ANTIDOTES
For many antidotes, the data on both efficacy and the optimum
methods of use are inadequate. Furthermore, there are problems in
both the pre-clinical and clinical aspects of antidote use, as well as
variations in licensing requirements between different countries.
Consequently, the IPCS and the CEC decided to evaluate antidotes and
other agents used in the treatment of poisoning cases. For this
purpose, a methodology has been developed for evaluation of antidotes
based on the principles concerned with the pharmaceutical properties
of the substance, its toxicity, as derived from animal and other
experiments, and the clinical studies in man, which demonstrate its
efficacy as an antidote and its safety in clinical use. All aspects
of these principles, as described below, may not be applicable to each
antidote and agent, but they provide the basis for assessment of those
in current use and under development.
1. Pharmaceutical Properties
As is true of any material to be administered to man, clear
guidelines on the pharmaceutical properties of an antidote are
required (reference should be made to the criteria as formulated in
generally accepted National or Regional Pharmacopoeias). The
information on an antidote should, therefore, include its chemical
formula and physical properties, such as melting point, solubility in
appropriate vehicles for administration, optical properties, acidity,
stability in light, and thermal stability. Particular account must be
taken of the wide range of temperatures to which such compounds may be
exposed in clinical situations (-30 to +40 °C). For liquids, the
refractive index and specific gravity should be considered, and, for
solids, their loss of weight on drying may be important.
To ensure purity and uniformity of the antidote preparation, the
route of synthesis, manufacturing process, and excipients need to be
considered and may have to be specified. Similarly, analytical
methods for the quality control or identification of the antidote
should be established.
Storage requirements and limitations need to be considered and
the shelf-life is very important for compounds that may be held for
long periods, particularly under tropical conditions. Evaluations of
shelf-life will need to be revised as further information on the
effects of storage under various conditions becomes available.
Mention should be made of any incompatibility with other
pharmaceuticals or food.
2. Pre-clinical Studies
Pre-clinical studies include those undertaken in vitro, in
laboratory animals, and in man (human volunteer studies). In the case
of antidotes, unlike other pharmaceutical products, studies of this
nature may first be indicated following the clinical use of postulated
antidotes or as part of a re-evaluation procedure, in particular where
comparison of the efficacy of antidotes is indicated.
2.1 In vitro studies
The type of in vitro study will vary depending on the antidote
being evaluated. Standardized studies of properties such as
adsorption capacity, chelating activity, anticaustic activity,
neutralization (e.g., for acids or alkalis), biochemical actions, and
pharmacological properties may be appropriate for the range of
different kinds of antidote.
Isolated cell, tissue culture, and isolated organ techniques may
be particularly relevant and in vitro studies should enable
subsequent studies in animals and man to be targeted more
specifically.
2.2 In vivo studies
2.2.1 Pharmacodynamic studies
Pharmacodynamic studies are aimed at assessing the mode of action
and efficacy of an antidote and its use in any particular type of
poisoning. Studies should be performed in relevant animal species and
should include investigations of both the pharmacological activity of
the antidote alone and the efficacy against the toxic agent when this
is administered in appropriate dosages. Ideally studies should be
conducted on two unrelated species that show qualitatively similar
response to the toxin in humans, and the efficacy against two
appropriate doses of the toxic agent (moderate and high toxicity)
should be evaluated. If animals are anaesthetized for such studies,
then it is important that consideration be given to the effects of any
anaesthetic agent, and, ideally, two different types of anaesthetic
should be used in parallel experiments.
The toxic compound should be administered by a clinically
relevant route and, if possible, a dose-response relationship for the
antidote established. Opportunity should be taken to study the
pharmacokinetics of the toxic agent and antidote during these studies,
particularly examining interactions in the distribution and clearance
of antidote and toxic compound.
2.2.2 Kinetic studies
Studies of the absorption, distribution, and elimination of an
antidote should, where possible, include monitoring of its metabolism
and also include a study of the effects of dose on its kinetics.
Doses used should be relevant to the likely clinical situation.
Kinetic studies should take into account the likely route of use of
the antidote (e.g., oral, parenteral, or topical) and should, ideally,
include the effect of the antidote on the kinetics of the toxic
substance against which it is used. Studies on the influence of
single or multiple organ failure on the elimination and metabolism of
the antidote or antidote complex may also be relevant.
2.2.3 Toxicological studies
Ideally, toxicological studies should be performed on species
that show similarity to humans as regards the kinetics and metabolism
of the antidote. Acute toxicological studies on two (unrelated)
species would be appropriate. The extent of toxicity evaluation would
depend on the proposed use of the antidote, and, for the situations in
which repeated doses of an antidote are necessary, chronic toxicity
studies need to be undertaken. Consideration should also be given to
the general approach to acute toxicity studies, bearing in mind the
doubts about the usefulness of LD50 values. Depending on likely
use, teratogenicity, mutagenicity, and carcinogenicity testing may be
deemed necessary.
2.3 Human volunteer studies
Human volunteer studies with antidotes present specific problems
and should be carried out in accordance with the Declaration of
Helsinki and the Council for the International Organizations of
Medical Sciences (CIOMS) Proposed International Guidelines for
Biomedical Research involving Human Subjectsa. The ethical
implications require particular attention. However, volunteer studies
may be important and useful for evaluating the human pharmacology of
certain antidotes. On special occasions, it may also be possible to
study interaction with a potentially toxic substance in volunteers,
subject to full and appropriate ethical review. Such investigations
may be particularly relevant for antidotes that are likely to be
widely used. They may also aid in choosing the appropriate dosage
regimen of an antidote for clinical use. In some circumstances
studies on patients with a particular disease or of particular age
groups may be indicated (e.g., renal, cardiac, or hepatic impairment,
and the elderly). In the rare situation in which pharmacogenetic
differences due to polymorphism of metabolism of the antidote is
important, such human volunteer studies will obviously be valuable.
a Proposed International Guidelines for Biomedical research
Involving human Subjects, 1982, Geneva, World Health
Organization.
3. Clinical Studies
By their very nature, poisonings of whatever sort are
unpredictable, and it is often difficult to establish the exact dose
of toxic compound to which a patient has been exposed. Clinical
studies of antidotes are therefore inherently more difficult than
studies of the effects of pharmaceutical preparations in many other
conditions, since definition of the severity of the poisoning is often
a problem.
The precise combination of approaches used in clinical studies of
an individual antidote needs to be carefully tailored. More than one
antidote may sometimes be needed, and, in these circumstances, an
evaluation of the combined treatment approach should be given.
3.1 Literature evaluation
Published work on antidotes is frequently in the form of case
reports, which are by their nature uncontrolled. Such studies are an
important source of data and can provide information on both the
effect of an antidote in the clinical setting and the likely pattern
of toxicity associated with the poison. They may thus prevent
needless duplication of studies. A principal role for a continued
evaluation of the literature is to identify specific areas for future
work and areas of ignorance.
Case reports are often not well documented and it is rarely
possible to make strict comparisons between published cases undertaken
at different centres. The human toxicological scientific basis could
be greatly enhanced by the establishment of a comparable basis for
case data collection and reporting (see below).
3.2 General approach to clinical studies
Double-blind controlled studies are the ideal in most areas of
therapeutics. In the assessment of antidotes, however, particularly
for rarer poisonings and those with a high morbidity or mortality,
such an approach presents major ethical and technical problems. Thus,
it may not be possible to obtain the consent of the patient or his
relatives and an individual clinician is unlikely to see a sufficient
number of patients to make a formal study worthwhile. The proper
construction of a control group may also raise ethical dilemmas.
Thus, a prospective approach to the study of antidotes should be
considered in parallel with the critical retrospective analysis of
existing patient data. Such studies may be best carried out on a
multi-centre or multi-national basis. This retrospective analysis,
often of unpublished data, could perhaps be organized centrally.
Retrospective data will provide a useful baseline from which to mount
prospective studies.
3.3 Methodology
Since case records are a major data source in antidote studies,
it is essential that records are kept carefully in a standardized
format. Case records should, where possible, include a detailed
personal history to cover occupational exposure, toxic agents,
relevant hobbies, previous medical history, and concurrent medication
including non-prescribed medicines. Physical examination should pay
particular attention to specific signs of intoxication and their
physiological consequences. Careful measurement and monitoring of
physiological changes is essential; in particular, careful
documentation of changes, such as the level of consciousness, should
be established. A scoring system for monitoring changes in the level
of consciousness and an agreed coma scale for grouping patients into
classes of comparable severity should be adopted where possible.
Detailed consecutive records of clinical progress in an individual
patient are important. Accurate recording of the time of the
intoxication, of its presentation to treatment centres, and of
initiation of treatment are particularly important.
The clinical examination should be supported by appropriate
analytical data on samples of body fluid such as blood, urine, and
gastric aspirate. The collection, handling, and analysis of samples
needs to be standardized and guidelines for individual toxic compounds
laid down. Accurate records of the time of clinical examination and
toxicological sampling are necessary and, where possible, the two
should be concurrent and continue throughout the clinical course of
the poisoning. This may enable a relationship between the two to be
established, as well as providing careful documentation of the effect
of an antidote. The routine saving of multiple biological samples
from a patient suffering from intoxication is therefore necessary.
It should be stressed that objective measures are preferable to
clinical impressions and that the opportunity should be taken at all
stages of management of intoxications to quantify physiological
parameters. Where possible, accepted standardized prognostic factors
should be utilized both to determine the appropriate use of the
antidote and to evaluate its effect.
3.4 Controlled studies
In the past, controlled studies have provided the basis for
important advances in the management of poisonings. As indicated in
section 3.2, there are ethical consideration that need to be
considered carefully. Furthermore, such studies need to be carefully
tailored to the particular antidote and poison under investigation.
The general poisons made in section 3.3 will also apply.
Careful consideration needs to be given to the establishment of
a control group. This should ideally be a parallel and comparable
group of patients but may be retrospective. Establishment of a
parallel control group may be easier in the situation where two active
treatments are being compared. It should be remembered, however, that
there may be a risk attached to using an antidote, and the use of a
control group that receives full supportive therapy may enable a more
rapid decision on the efficacy of an antidote to be obtained.
In controlled studies, an opportunity may arise to study the
toxicity and kinetics of the antidote; it should be grasped whenever
possible.
The period of follow-up needs to be determined, and end-points
must be clearly established and defined. Statistical considerations
are important and need to be fully incorporated at the time of trial
design.
4. Centralized Record System
It is suggested that a central pooling of the experience of
various treatment centres, particularly with respect to rarer
antidotes, would be a great advantage. An international network of
IPCS-designed clinical centres could provide a mechanism for this
purpose. This pool of data would be used as a basis for the
international evaluation of antidotes but would remain confidential
and would not be published without the consent of the individual
clinician concerned. Data submitted to such a pool would remain the
property of the investigator, who would be at liberty to publish it
separately.
Poison control centres appear to be in a unique position to
collect data on antidotes for rarer poisoning and to ensure that
appropriate clinical and toxicological data are collected. This
process might be facilitated if a poison control centre acted as a
base for the supply of antidotes.
The amount of information required from individual patients
entered on the case record system would have to be carefully planned
and the collection and reporting of the data coordinated.
The establishment of such a centralized case record system would
also be likely to stimulate further international collaboration in the
evaluation of antidotes by controlled studies and could lead to
recommendations for further areas of research.
APPENDIX 3
PROFORMA FOR MONOGRAPHS ON ANTIDOTES FOR SPECIFIC TOXIC AGENTS
Guidelines to Authors
These guidelines provide a unified format for monographs written
on individual antidotes used in the management of poisoning using
existing published (and unpublished) literature. For a number of
antidotes currently used clinically some of the information suggested
will be missing: these gaps in knowledge should be stated. For those
antidotes currently under in development, this proforma is designed to
give an idea of the sort of information that would be required for
evaluation and worldwide acceptance for use. Suggested section
headings and an outline guide to contents are given below.
1. Introduction
This should be brief (usually 200-400 words) and include:
- indications for antidote use
- rationale for the choice of the antidote, mentioning areas where
there are doubts about efficacy
- an indication of specific groups at risk from treatment with the
antidote
2. Name and Chemical Formula of Antidote
International non-proprietary name (when available); CAS number
(Chemical Abstracts Service); IUPAC name (International Union of Pure
and Applied Chemistry); manufacturer and commercial names, formula
(include figure of structure); relative molecular mass; specification
of chemical salts used; conversion table from mass units to SI units.
3. Physico-chemical Properties
- melting point, boiling point
- solubility in vehicles for administrations
- optical properties
- acidity
- pKa
- stability in light
- thermal stability/flammability (including vehicle if antidote
usually in solution or suspension)
- for liquids, refractive index and specific gravity
- for solids, loss of weight on drying
- the excipients and pharmaceutical aids
- pharmaceutical incompatibilities
4. Pharmaceutical Formulation and Synthesis
4.1 Routes of synthesis (brief details only)
4.2 Manufacturing processes (indicate, where known, possible
contaminants)
4.3 Presentation and formulation
5. Analytical Methods
In this section, the passive voice should be used, e.g., "the
solution is mixed to dissolve the reagents". Vocative instructions
should not be used, e.g., "dissolve 0.5 g in 20 ml of water". To
include:
5.1 Quality control procedures for the antidote and/or its
formulation
5.2 Methods for identification of the antidote
5.3 Methods for analysis of the antidote in biological samples
5.4 Analysis of the toxic agent in biological samples
referring to the preferred assay techniques
6. Shelf-life
Attention should be given to specific conditions of temperature
and humidity, including tropical conditions, and instructions on
storage conditions.
7. General Properties
This section should be particularly tailored to the antidote in
question and include:
- information on the mode of antidotal activity of the compound
(e.g., chelating agent, receptor antagonist)
- other relevant biochemical and pharmacological profiles (e.g.,
anticholinergic, antiadrenergic properties) as demonstrated in
vivo and in vitro
This section can be subdivided as needed.
8. Animal Studies
It is important to exclude human data from this section. Even if
a paper presents both human and animal data, the human data should be
given in the clinical sections 9, 10 or 11, as appropriate. An
attempt should be made to evaluate the statistical significance of the
results. As a rule of thumb, this term should imply a level of
statistical significance at the 5% level (P <0.05). If other
parameters are used, they should be given. If both positive and
negative results have been reported, possible reasons for such
differences may be briefly discussed (routes of administration, time
before giving the antidote, dose, etc.).
8.1 Pharmacodynamics
This section should include data on efficacy, examining the
protective effects against moderate and high doses of the toxin. The
efficacy parameter end-point should be defined before the data are
discussed, e.g., is it reduced mortality or increased excretion of the
toxic agent that has been studied? Data on the dose-response
relationship of the antidote and on the pharmacokinetics of any
interaction between antidote and toxic compound should be sought. Both
the toxic agent and the antidote should be given by the clinically
relevant route(s). Information should also be sought on the time after
the administration of the toxic agent at which the antidote is likely
to be of any clinical benefit.
8.2 Pharmacokinetics
Studies on the bioavailability, half-life and clearance of the
antidote by the relevant route of administration; studies on the dose
dependency of the pharmacokinetics relevant to the doses used in the
clinical settings and details of the metabolism of the compound. If
there are data on more than one species, these should be included.
8.3 Toxicology
Details of acute, subacute or chronic toxicity, depending on the
likely clinical use of the compound, should be discussed. Such
studies should use appropriate routes of administration. Where
available, details of mutagenicity and teratogenicity testing would be
of interest. LD50 values for different species and different routes
of administration should be presented.
9. Volunteer Studies
Information on the pharmacokinetics of the antidote in man and
possible effects of disease on the handling of the antidote should be
given, where available. Studies on the interaction with the toxic
agent given at subtoxic doses may also be available. All human data
based on volunteer studies should be included in this section.
10. Clinical Studies - Clinical Trials
Literature evaluation of clinical trials. For each trial the
following information should be given:
(a) evidence of poisoning, i.e. inclusion criteria
(i) definition of assay techniques to measure the poison and
its effects
(ii) physiological changes of poisoning recorded; accepted
methods
(b) number of patients studied
(c) was there a control group? type, i.e. parallel or retrospective;
appropriate, i.e. were the treated and control groups
comparable (e.g., age, sex, time of presentation, severity of
poisoning)?
(d) details of antidote used
(i) dose and route
(ii) time between exposure to the toxin and administration of
antidote
(iii) evidence of any toxic effect of the antidote
(e) outcome assessment
(i) clinical
(ii) biochemical
(iii) end-points - death, pathological damage
(f) overall view of likelihood of benefit due to antidote
It may sometimes be difficult to define what is a clinical trial.
Retrospective studies and use of historical controls should normally
not be considered as clinical trials and may then be described under
section 11.
11. Clinical Studies - Case Reports
Many authors of case reports claim a favourable outcome "due to
the use of the antidote". In most cases, however, the best conclusion
drawn from such studies is that the use of the antidote was associated
with a favourable outcome and nothing more. Some case reports are
thoroughly performed with measurement of half-lives in different body
compartments and amount of toxic agent eliminated by different
elimination techniques. When referring to such studies, the end-point
parameter, e.g., reduction in plasma half-life, should be evaluated
critically. There may be too few plasma levels over a too short a
time period to allow for any conclusion.
Other important items to evaluate are:
- evidence of poisoning (clinical, pathological);
- was an appropriate assay/measurement technique used?
- dose and route of antidote
- evidence of outcome
- evidence of toxicity or side-effects of antidote
It should be borne in mind that most poisoned patients will
recover with intensive supportive care alone. If such cases have been
reported, a brief mention may be appropriate.
12. Summary of Evaluation
This section is frequently misunderstood by authors writing their
first draft in this series. Section 12 is not to be a copy of section
13. In section 12 the author should try to evaluate or summarize
the data, previously presented in the monograph, as a basis for the
recommendations under the following subheadings:
12.1 Indications
12.2 Advised routes and dose
12.3 Other consequential or supportive therapy
12.4 Controversial issues and areas of use where there is
insufficient information to make recommendations
12.5 Proposals for further studies
12.6 Adverse effects
12.7 Restrictions for use
13. Model Information Sheet
This should be a concise user's manual for this antidote in the
emergency situation. This section may, for example, be faxed to the
clinician treating the patient. No references or abbreviations should
be given since all the information given should have been discussed in
previous sections.
13.1 Uses
13.2 Dosage and route
13.3 Precautions/contraindications
13.4 Pharmaceutical incompatibilities and drug interactions
13.5 Adverse effects
13.6 Use in pregnancy and lactation
13.7 Storage
14. References
References should be given in text as (McMartin et al., 1980) or
(Henry & Volans, 1984). Several references to the same statement are
placed in chronological order.
In the reference list, the names of all authors and their
initials must be given. Only initial letters are capitalized. Titles
of articles in languages other than English, except those in French,
should be translated into English. Translated titles appear in square
brackets with the original language in parentheses.
Periodicals
Names of journals should be abbreviated according to the ISDS
(International Serials Data System) List of Serial Title Word
Abbreviations (a condensed list is obtainable from the RO) or
otherwise given in full. The initial letter of each abbreviation is
capitalized. The volume number is indicated in bold print and is
followed by the issue number (if any) in parentheses. First and last
page numbers must be given.
McMartin KE, Ambre JJ & Tephly TR (1980) Methanol poisoning in human
subjects. Am J Med, 68: 414-418.
Henry J & Volans G (1984) ABC of poisoning. Br Med J, 289: 990-993.
Conference proceedings
The following elements are necessary: name(s) and initial(s) of
author(s); the year of publication; title of paper; the word "In:" the
editors of the proceedings; the full title of the conference (not
abbreviated); the place and date of the conference; the place of
publication; the publisher; the volume number (if any) and the page
numbers.
Example
Wassermann M (1984) L'étude de la toxicologie des pesticides en
climat tropical In: Smith JH ed. Proceedings of the 14th
International Congress of Occupational Health, Madrid, 16-21 May
1983. Amsterdam, Excerpta Medica, vol 3, 1728-1733.
Books
Windholz M ed. (1983) The Merck Index: an encyclopedia of
chemicals, drugs, and biologicals, 10th ed. Rahway, New Jersey, Merck
and Co., Inc.
Reference to a chapter in a book should be given as follows:
Weinstein L (1974) Pathogenic properties of invading
microorganisms. In: Sodeman WA Jr ed. Pathologic physiology:
mechanisms of disease. New York, Academic Press, pp 457-472.
Agency report
US EPA (1984) Mercury health effects update: health issue
assessment. Washington, DC, US Environmental Protection Agency
(EPA-60018-84-019F).
Order of entries in the list
The following rules are applied:
a) Several papers by different authors with the same surname
are listed alphabetically according to their initials.
b) Several papers by one author are listed chronologically.
c) Several papers by the author plus a co-author are listed
alphabetically.
d) Several papers by the author plus two or more coauthors are
listed chronologically.
Example
Smith DE (1985)
Smith JH (1983)
Smith JH (1984)
Smith JH & Barns MP (1986)
Smith JH & Jones TD (1979)
Smith JH, Jones TD, & Barnes MP (1981)
Smith JH, Barnes MP, & Jones TD (1983)
Normally one should not refer to abstracts or works published as
supplements. Under special circumstances, e.g., little has been
published and the person has great experience with the topic, personal
communications may be inserted in the text as follows: (Vale JA,
personal communication).
15. Author(s) Name, Address
16. Additional Information
e.g., availability of supply.