
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY EVALUATION
(WHO/ILO/UNEP)
Evaluation
ANTIDOTES FOR POISONING BY
METALS AND METALLOIDS
Completed August 2004
Edited by Nicola Bates MSc, MA: National Poisons Information Service (London Centre), UK
Peer-review by Dr P Dargan3, Dr L Murray4 & Dr B Groszek5
©World Health Organization 2004
All rights reserved. Publications of the World Health Organization can be obtained from Marketing and Dissemination, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 2476; fax: +41 22 791 4857; email: ). Requests for permission to reproduce or translate WHO publications — whether for sale or for noncommercial distribution — should be addressed to Publications, at the above address (fax: +41 22 791 4806; email: permissions@who.int).
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement.
The mention of specific companies or of certain manufacturers' products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
The World Health Organization does not warrant that the information contained in this publication is complete and correct and shall not be liable for any damages incurred as a result of its use.
Deferoxamine (desferrioxamine B) is derived from ferrioxamine B, a sideramine isolated in 1960 from Streptomyces pilosus. It has a high binding affinity for trivalent iron which can be exploited to clinically remove excess iron from blood and tissue (Keberle, 1964).
Currently, deferoxamine is used for the treatment of acute iron poisoning and iron-overload anaemias, such as thalassaemia major, as well as aluminium poisoning associated with chronic renal dialysis (Banner and Tong, 1986; Day & Ackrill, 1993).
Acute iron intoxication is often described as progressing through four stages (Proudfoot et al., 1986; McGuigan, 1996).
The first of these is the gastrointestinal phase which may begin within minutes and last for several hours. This phase is due to the corrosive effects of iron and results in abdominal pain, vomiting and diarrhoea with fluid and electrolyte losses and in severe cases, gastrointestinal bleeding with shock and metabolic acidosis. The second stage is a quiescent period where the patient may appear to be relatively stable. In the third stage acidosis and shock occur which may lead to cardiovascular collapse. This may begin within hours but may be delayed as long as 24 hours in some cases. The fourth stage is characterised by the after effects of mucosal corrosion such as stricture formation and pyloric stenosis. These can occur even in patients treated with deferoxamine (Saviuc et al., 1992). Hepatic necrosis has been reported and normally occurs within the first 48 hours. It is the most common cause of death after shock.
In acute or chronic iron poisoning the goal of treatment is the removal of toxic iron from the body by chelation in order to avoid acute organ damage. Early treatment prior to distribution of iron into tissue and the reticulo-endothelial system has long been thought to be most effective (Whitten et al, 1966).
Deferoxamine has intrinsic toxicity (Bentur et al., 1991). Subacute effects consisting of nephrotoxicity, ototoxicity and retinal toxicity have been reported. These effects generally appear following long-term administration for chronic iron overload, however nephrotoxicity has been reported in patients treated for acute iron poisoning (Hershko and Weatherall, 1988; Koren et al., 1989). Rapid infusion of a large dose has been associated with histamine-induced hypotension (Whitten et al., 1965; Barker et al., 1974) and acute renal insufficiency (Freedman et al., 1989; Koren et al., 1989). Prolonged infusion has been associated with pulmonary toxicity (Tenenbein et al., 1992; Adamson et al., 1993).
|
International non-proprietary name: |
deferoxamine |
|
Synonyms: |
desferroxamine, desferrioxamine |
|
IUPAC name: |
N-(5-C3-L (5 aminopentyl) hydroxycarbamoyl)-propionamido)pentyl)-3(5-(N-hydroxyacetoamido)-pentyl)carbamoyl)-proprionhydroxamic acid |
|
CAS No: |
70-51-9 (deferoxamine) |
|
138-14-7 (deferoxamine mesylate) |
|
|
Chemical formula: |
C25H48O8N6 |
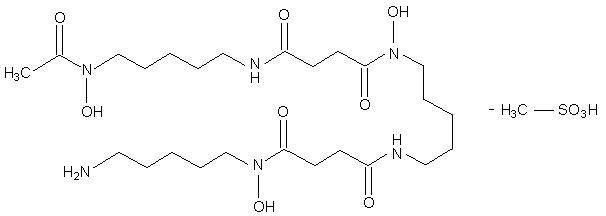
Figure 1: Deferoxamine mesylate
Relative molecular mass: 560.71 (656.8 as mesylate)
|
Conversion: |
1 mmol |
= |
560.71 mg |
|
1 g |
= |
1.78 mmol |
|
|
1 mmol/L |
= |
0.56 g/L |
|
|
1 g/L |
= |
1.78 mmol/L |
Manufacturers: Novartis Pharma AG (previously Ciba Geigy), Basel, Switzerland
|
Melting point: |
138-140°C |
|
Solubility in vehicle of administration: |
water at 20°C, 1.2% |
|
Optical properties: |
not applicable |
|
pH: |
3.5 - 5.5 in water as a 10% solution |
|
pKa: |
not applicable |
|
Stability in light: |
not applicable |
|
Thermal Stability: |
In water, stable for 1 day at room temperature (23°C or below). |
|
Refractive index: |
not applicable |
|
Loss of weight on drying: |
not applicable |
|
Proprietary names: |
Desferal®, Desferin®. |
Currently available commercially produced deferoxamine is a derivative of the iron-bearing metabolite, ferrioxamine B, from Streptomyces pilosus (Keberle, 1964). Ferrioxamine is isolated and iron is then removed by chemical means. The resulting deferoxamine is then exhaustively purified.
De novo organic synthesis was attempted in 1962 leading to a 6% yield (Prelog and Walser, 1962). Since then, a de novo synthesis has been developed leading to a greater overall yield with improved purity. The synthesis is very expensive.
Deferoxamine mesylate is available in vials of 500 mg or 2 g, as sterile powder for preparing injections or for oral use. The sterile content of a sealed container should be dissolved in the requisite amount of water for injection to form a clear pale yellow solution. The preparation should be used immediately and cloudy or discoloured solutions should be discarded.
As a measure of quality control, a deferoxamine solution can be tested by adding ferrous or ferric iron at a neutral pH and measuring absorbance at 430 nm compared to a known standard.
Identification of deferoxamine and ferrioxamine in solution or plasma is based on spectrophotometric analyses and the absorption of ferrioxamine at 430 nm. In solution, ferrioxamine can be measured directly. Deferoxamine is measured as ferrioxamine after addition of iron at low pH (Pippard and Stray, 1982). In plasma, the same assay can be used after deproteinisation with trichloroacetic acid.
Deferoxamine can be measured by atomic absorption spectroscopy (Allain et al., 1986) or by a high performance liquid chromatography (HPLC) technique which can separate deferoxamine and its metabolites, as well as the iron and aluminium chelate (Jenny and Peter, 1988).
Metals and metalloids can be analysed by sensitive standard methods, such as atomic absorption spectroscopy (AAS) or inductively coupled plasma-atomic emission spectroscopy (ICP-AES).
The shelf-life of deferoxamine is 4 years stored in an airtight container protected from light and heat. Vials of powder should be stored in a cool place (below 15°C) (Ciba-Geigy, 1994).
Deferoxamine appears only to chelate free iron (as the Fe3+ ion), and iron stored in the form of ferritin and haemosiderin, and to a very limited extent, transferrin; the iron of cytochromes and haemoglobin is unaffected (Keberle, 1964; Lipschitz et al., 1971; Hershko & Weatherall, 1988). One mole of deferoxamine binds one mole of iron to form the deferoxamine-iron complex, ferrioxamine. Almost all of the ferrioxamine formed is then eliminated by the kidneys within few hours, as demonstrated in studies in dogs, and the remainder is believed to be metabolised (Keberle, 1964). As such, the neutralisation of free iron by complexation and the renal excretion of the complexed iron reduces the amount of toxic iron available. This, in combination with the slower physiological movement of iron into storage compartments and the stabilisation of iron complexed to proteins, leads to an overall reduction of serum iron and decreased body burden (Bothwell et al., 1979). Ferrioxamine has a significantly lower volume of distribution (Keberle, 1964; Peters et al., 1966), is considered to be of lower toxicity than deferoxamine (Bentur et al., 1991) and complexation is important even without increased elimination of iron via ferrioxamine in the urine. Ferrioxamine can, however, be lethal if administered in the absence of free iron (Whitten et al., 1965; Adamson et al., 1993).
The failure of other chelators (e.g. diethylenetriaminepentaacetate; DTPA), which are excluded from the intracellular space, to aid in the prevention of iron-related morbidity, argues for a role for deferoxamine other than the quantitative removal of iron from serum (Whitten et al., 1965). An in vitro study of rat heart ventricular muscle cells in culture suggests a protective, in addition to a therapeutic, effect of deferoxamine. When these cells were exposed to iron concentrations of 20, 40 and 80 µg/mL (reflecting plasma concentrations in fatal intoxications) they developed arrhythmias and depression of contractility similar to that observed in terminal iron poisoning. The provision of deferoxamine was found to not only prevent the oxidative toxicity of iron but to reverse iron-induced tissue damage (Link et al., 1989). This may suggest that in both acute and chronic iron overload it is the provision of excess iron binding capacity (chelation) which prevents toxic iron damage, rather than the removal of iron as chelate.
Recently several authors have focused on the role of deferoxamine as an antioxidant and free-radical scavenger in non-iron intoxications (Hoffer et al., 1992; Van der Wal et al., 1992; Burkitt et al., 1993; Meulenbelt et al., 1993). The practical significance of this ability of deferoxamine in acute iron or other poisonings remains unproven. There is evidence that the mitochondrial respiration enzymes are the main target of acute iron toxicity (Link et al., 1998).
The high stability of the deferoxamine-aluminium complex (aluminoxamine) led to the use deferoxamine in dialysis-related aluminium toxicity syndromes (Day & Ackrill, 1993). Sudden decreases of aluminium concentrations do not occur because the excretion of deferoxamine is mainly renal. Aluminium appears to have some toxicity even bound to deferoxamine since the reduction of metal toxicity seems to depend on removal of the chelate complex by ambulatory peritoneal dialysis, haemodialysis, haemofiltration or haemoperfusion in patients with chronic renal failure (Allain et al., 1988; McCarthy et al., 1988). The possibility exists that the translocation of aluminium from bone may actually increase cerebral symptoms in a fashion analogous to the use of other chelators in other types of heavy metal poisoning. Our understanding of this phenomenon is limited.
Many studies have demonstrated that over relatively long intervals (weeks to months) the use of deferoxamine after dialysis at a dose not exceeding 40 mg/kg has led to a reduction in the severity of aluminium toxicity (encephalopathy, bone disease and anaemia). This improvement correlates approximately with concentrations of aluminium measured directly in tissues (bone) but not with serum concentrations (Day & Ackrill, 1993).
Deferoxamine produces its dynamic effect by the removal of excess inorganic iron. Inorganic iron is directly toxic to rabbit myocardium exposed to increasing amounts of iron leading to loss of contractile function (Artman et al., 1984). By binding to iron, deferoxamine may cause clinical improvement even before the iron is eliminated from the body. Deferoxamine also reverses iron-induced dysfunction of isolated rat heart muscle (Link et al., 1989).
Oral administration of deferoxamine in animals receiving oral lethal doses of iron demonstrates an interesting intraspecies difference. In rodents, deferoxamine had no effect on plasma iron concentrations (Dean and Krenzelok, 1987). In swine, significant differences in plasma iron concentrations were observed (Dean et al., 1988). One could argue that the pig model more closely resembles the human situation because of the greater similarity to human gastrointestinal iron handling. In one set of canine experiments, the deferoxamine-iron complex was absorbed directly as plasma iron concentrations rose rapidly after intragastric instillation of the ferrioxamine complex, and time to death was shorter than with iron alone (Whitten et al., 1965).
In a subsequent experiment by the same group, plasma iron concentrations rose less than in control dogs when intragastric and intravenous deferoxamine were given (Whitten et al., 1966). The presence of intravenous deferoxamine complicates the data analysis, but it is unlikely that the decrease in concentrations occurred as a result of the rapid clearance of ferrioxamine alone. Another study in dogs, however, revealed a survival advantage to dogs treated simultaneously with oral iron and oral deferoxamine (Brunner et al., 1965). In view of the difficulty in interpreting these data it is not surprising that the value of administration of intragastric deferoxamine in humans is controversial (Banner and Tong, 1986).
In a study in swine, 10 pigs were given elemental iron (60 mg/kg) + deferoxamine (1 g/kg) orally, whereas 13 pigs served as controls and were given iron only (Dean et al., 1996). Although deferoxamine reduced some peak concentrations, the total absorption of iron was unchanged.
Few adequate animal studies have been performed to mimic the actual conditions for which deferoxamine is used in aluminium toxicity. In most studies, animals are acutely loaded with aluminium, thus creating toxicity that may not relate to humans undergoing chronic dialysis. These studies yield conflicting results as to the value of deferoxamine as an antidote. In one study examining subacute aluminium loading in rabbits, deferroxamine treatment appeared to reduce tissue aluminium concentrations (Melograna & Yokel, 1984).
The dog has been the principle species used to examine the pharmacokinetics of deferoxamine (as opposed to the rodent models of chronic iron overload for chemical studies). Studies in the dog revealed a significant difference in the handling of the chelated and unchelated forms of deferoxamine (Peters et al., 1966). Ferrioxamine was distributed only to the extracellular space and excreted directly into the urine by glomerular filtration (clearance ferrioxamine/clearance inulin = 1.0). There was a small component of tubular reabsorption. In contrast, deferoxamine was distributed into total body water and is renally excreted by glomerular filtration with an easily overwhelmed tubular reabsorption mechanism.
In nephrectomised dogs, deferoxamine underwent hepatic metabolism with a plasma half-life of 76 ± 10 minutes. Ferrioxamine was not metabolised and plasma concentrations fell very slowly. Some metabolites of deferoxamine, most notably the product of oxidative deamination, also chelate iron, and thus the antidotal effect of the drug appears unaffected by hepatic metabolism (Peters et al., 1966).
When dogs were injected with tritrium-labelled deferoxamine, radioactivity was highest in the bile and brain, intermediate in the spleen, kidney and plasma, and lowest in the heart and lung (Hershko and Weatherall, 1988). In bile, deferoxamine exists primarily as metabolites. Ferrioxamine in bile is most likely derived from the deferoxamine chelation of iron in the hepatocyte, except in the case of nephrectomised dogs (and humans).
Deferoxamine may further complicate the picture by directly altering renal function to increase fractional excretion of sodium and decrease insulin clearance in the dog even with volume loading (Koren et al., 1989).
The toxicity of deferoxamine has recently been extensively reviewed by Bentur et al. (1991).
See table 1 for information on the acute toxicity of deferoxamine.
Table 1: The acute LD50 values of deferoxamine (Ciba Laboratories, 1964).
|
Species |
Route |
||
|
Intravenous LD50 |
Subcutaneous LD50 |
Oral LD50 |
|
|
Mouse |
340 mg/kg |
1600 mg/kg |
>3000 mg/kg |
|
Rat |
520 mg/kg |
>1000 mg/kg |
>1000 mg/kg |
|
Rabbit |
600 mg/kg |
- |
- |
Studies in dogs showed that deferoxamine administration was associated with depressed cardiac function and hypotension (Whitten et al., 1966). A reversible acute decrease in glomerular filtration rate and renal plasma flow, accompanied by disturbances in electrolyte excretion was demonstrated in dogs given deferoxamine. The mechanism of this nephrotoxic effect is not clear (Bentur et al., 1991).
Pulmonary effects have been observed with prolonged infusion of deferoxamine, but it has been argued that these effects may be to be due to iron toxicity, rather than deferoxamine itself (Shannon, 1992).
Pulmonary complications (ARDS, adult respiratory distress syndrome) associated with prolonged infusion of deferoxamine has been observed in humans (Tenenbein et al., 1992), and the same group later studied evidence of pulmonary toxicity in iron-poisoned mice (Adamson et al., 1993). Mice treated with one oral dose of iron followed by multiple doses of deferoxamine, or with ferrioxamine (the chelated complex) alone, did not develop lung lesions, even at doses which were fatal. To potentiate any possible free radical reaction some mice were also treated with oxygen (75-80% over 4 days). Ten of 12 mice receiving oxygen therapy in addition to 0.75 mg elemental iron and then deferoxamine (10 mg, 4 times/day for 4 days) died suddenly. Autopsy and electron microscopy of the lungs revealed changes as seen in ARDS. Furthermore, a free radical reaction product was demonstrated at the alveolar surface indicating that free radical formation was associated with the damages seen. Such changes were not seen in controls given deferoxamine or iron, plus hyperoxia. This study appears to confirm that the lung damage observed clinically was related to the prolonged use of deferoxamine. Although a free radical mechanism behind this toxicity may be possible, it remains unproven.
No evidence of a mutagenic potential has been observed in vitro with deferoxamine. Long-term carcinogenicity studies have not been performed. In rabbits exposed in utero deferoxamine caused skeletal malformations. However, these teratogenic effects were observed at doses which were toxic to the mother. In mice and rats deferoxamine appears to be free of teratogenic activity (Ciba-Geigy, 1994).
Deferoxamine is rapidly absorbed after intramuscular or subcutaneous administration, but only poorly absorbed from the gastrointestinal tract in the presence of intact mucosa (Ciba-Geigy, 1994).
Peak plasma concentrations of deferoxamine of 15.5 µmol/L (8.7 µg/mL) were measured 30 minutes after an intramuscular injection of 10 mg/kg (Allain et al., 1987). One hour after injection the peak concentration of ferrioxamine was 3.7 µmol/L (2.3 µg/mL).
Deferoxamine has a volume of distribution of 0.8-1.35 L/kg (Keberle 1964; Lee et al., 1993-4).
Less than 10% of deferoxamine is bound to serum proteins in vitro (Ciba-Geigy, 1994).
Normally, deferoxamine and ferrioxamine are excreted primarily by the kidneys after a wide range of dosages (Keberle, 1964). Biliary excretion of the chelate ferrioxamine only occurs when iron is chelated in the hepatocyte or when renal clearance is exceeded (Keberle, 1964; Octave et al., 1983). Deferoxamine distributes throughout total body water and is excreted by glomerular filtration and saturable tubular secretion. Ferrioxamine is excluded from the intracellular space and plasma concentrations are excreted entirely by the kidneys in patients without compromised renal function. In the nephrectomised human, injected labelled ferrioxamine appears in the faeces (Wolfe and Fosburg, personal communication 1989).
Both deferoxamine and ferrioxamine have a biphasic elimination pattern in healthy volunteers; for deferoxamine the half life is 1 hour, and for ferrioxamine 2.4 hours, in the first rapid phase (Allain et al., 1987). In the second slow phase the half-life is 6 hours for both. Within six hours of injection, 22% of the dose appears in the urine as deferoxamine and 1% as ferrioxamine.
In three healthy volunteers given 1 g of deferoxamine intravenously over one hour, the elimination was very rapid with a total plasma clearance of deferoxamine of 790-1090 mL/minute; no half-life could be calculated. The urinary excretion of ferrioxamine is very low (less than 1% of the dose). Deferoxamine excretion in urine in the first 24 hours was between 11 and 19%, with most of the drug excreted within the first 3 hours (Ciba-Geigy, 1987).
Deferoxamine is mainly metabolised in the plasma and hepatic metabolism is minimal. A number of metabolites have been isolated but not characterised (Lehmann & Heinrich, 1989; Kruck et al., 1993; Kraemer & Breithaupt, 1998). Animal studies have shown that some metabolites also chelate iron (Peters et al., 1966).
Seven volunteers were given 5 mg/kg elemental iron in a control phase and again in an experimental phase followed by a single equimolar dose of oral buffered deferoxamine solution (Jackson et al., 1995). No influence on iron absorption was demonstrated. In another controlled study, a premixed 1:3 (weight/weight) deferoxamine/activated charcoal slurry significantly reduced the gastrointestinal absorption of ferrous sulphate in adult volunteers (Gomez et al., 1997).
In reviewing the English language literature on deferoxamine the lack of controlled trials is striking. However, given the ample non-human literature regarding acute iron intoxication and the inherent difficulties in constructing comparative trials of an infrequent intoxication, this is not surprising. Although discussed in this section, the papers identified are primarily descriptive in nature and are not clinical trials in the real meaning of the term.
Whitten et al. (1965) reported a prospective open trial of deferoxamine in a group of 12 children aged 13-30 months. Serum iron as measured spectrophotometrically ranged from 53 to 141 µmol/L (300 to 792 µg/dL). Time after ingestion to assessment varied from 1.5 to 20 hours. Patients received intravenous and oral deferoxamine in a controlled manner. Three children received 800-1500 mg of deferoxamine over 15 minutes i.v. and two of these experienced severe hypotension including a 15 minute loss of blood pressure. The excretion of iron complex in the urine in these children was small with 25 mg/24 hours being the maximal rate in a child with an initial serum iron of 141 µmol/L (792 µg/dL). The only patient in this series with significant presenting symptoms (coma) remained comatose after treatment and had a poor neurological outcome. The authors concluded that the good outcome of their patients could not be related to deferoxamine as the iron excretion was limited compared to the amount ingested. It could, however, be argued that the assessment of deferoxamine efficacy by measuring renal excretion of iron may be difficult since the amount of iron absorbed, even in acute overdose, may not be more than the 10% observed following therapeutic ingestion. Furthermore, the process of chelation per se may be difficult to assess by the renal excretion of iron, especially in patients with hypotension and impaired renal function. This manuscript is important because of the observation regarding severe hypotension with rapid administration of deferoxamine in two patients.
McEnery & Greengard (1966) reported a prospective series of 20 children treated with deferoxamine over a two year period. Serum iron concentration on admission ranged from 11 to 296 µmol/L (64 to 1653 µg/dL). Deferoxamine therapy was started with oral doses in 16 patients and combinations of intravenous and intramuscular therapy in 17 of the 20. Nine patients received rapid infusions of i.v. deferoxamine without apparent sequelae. All patients survived and no morbidity was ascribed to the treatment. The lack of a control group and the relatively low serum iron concentrations in many patients makes it difficult to interpret this study particularly with respect to the efficacy of oral deferoxamine therapy.
Leikin et al. (1967) reported a randomised prospective clinical trial of oral, oral + intravenous and no chelation therapy in a group of 22 children aged from 1 to 6 years of age. Children with a history of ingestion and supranormal serum iron determinations on presentation were included in the trial. Serum irons were determined by colorimetric assay and ranged from 44 to 143 µmol/L (250 to 800 µg/dL). Assessment included estimation of quantity ingested, symptoms, time to treatment, initial serum iron, total urinary iron excretion and length of hospital stay. There were no fatalities in this series and the only serious morbidity was shock in a patient who inadvertently received 1000 mg of deferoxamine over 10 minutes. In the opinion of the authors the randomly assigned groups were comparable. The only appreciable benefit of therapy was an increase in urinary excretion which increased from 7.5 µg/h to 68.9 µg/h for oral therapy alone to 260.4 µg/h for oral + intravenous treatment. Once again there was no demonstrable benefit and one serious adverse event leading the authors to conclude that chelation was not predictably beneficial in a mild iron intoxication.
Barr & Fraser (1968) reported an open trial of 16 children with iron poisoning who were treated with deferoxamine (n=14) and DTPA (n=2). Serum iron concentrations at admission ranged from 69 to 404 µmol/L (390 to 2,260 µg/dL) in the deferoxamine treated group. The clinical symptoms varied widely but apparently correlated closely with serum iron concentration. A concentration greater than 179 µmol/L (1000 µg/dL) was associated with cardiovascular collapse, severe gastroenteritis, coma and death. This case series had no comparative group. The deferoxamine dose varied from 0 to 5 g orally and from 0.4 to 4 g parenterally with no consistent pattern. The authors concluded that deferoxamine was safe and biochemically effective but offered no comparative data, particularly to support the value of oral therapy.
Fischer et al (1971) reviewed 27 children treated for iron ingestion over a 10 year period. Serum iron ranged from 8 to 126 µmol/L (50 to 705 µg/dL). Nine patients received i.v. deferoxamine in doses ranging from 840 mg to 2000 mg over 12 hours. Three of the 27 patients received oral deferoxamine (7.5 to 8 g) in addition to their i.v therapy. No mortality was observed and the only morbidity was ascribed to the combination of 500 mg i.m. and 100 mg/h infusion of deferoxamine which produced hypotension. The authors correctly concluded that deferoxamine should be reserved for those patients in whom iron toxicity is likely, because of the risk of adverse effects from deferoxamine therapy.
Westlin (1971) reviewed the American clinical trials of deferoxamine undertaken by the manufacturer. Data on 474 patients were collected from multiple centres. One important contribution of this study was the finding that a serum iron less than 89 µmol/L (<500 µg/dL) had 6% incidence of serious signs and symptoms whereas an iron of greater than 179 µmol/L (1000 µg/dL) was associated with a 62% incidence of shock and coma. The deferoxamine dosage regimen was not controlled and included both oral, i.m. and i.v. administration at a variety of doses. There were 5 deaths in this series, attributable to very high iron concentrations and/or co-ingested toxins. Thirteen patients developed hypotension attributed to rapid administration of deferoxamine. Eight others had histaminergic symptoms also related to deferoxamine. The author correctly concluded that deferoxamine is an ‘adjunct to, and not a substitute for' supportive care. The incidence of hypotension was notable enough to prompt caution on the rate of i.v. administration.
McEnery (1971) presented a three year review of 55 patients treated with nasogastric deferoxamine (5 g) and i.v. deferoxamine (15 mg/kg/h). No adverse events related to deferoxamine were recorded in this series. Seventeen the patients excreted more than 5 mg of iron in the urine giving it a vin rose appearance and this correlated with the serum iron generally being greater than 89 µmol/L (500 µg/dL). The author inappropriately concluded that this series with 100% survival supported the approach of combined oral and parenteral deferoxamine although no comparative data of any type was presented.
Shnaps & Tirosh (1981) reported the survival of five patients with acute iron overdose managed with a combination of oral and i.v. deferoxamine, but included no control or detailed case reports. No conclusion can be drawn from this report.
Yaqoob et al. (1993) reported on the efficacy of deferoxamine to reverse resistance to recombinant human erythropoietin, due to aluminium overload, present in 9 of their 17 patients receiving erythropoietin for uraemic anaemia (haemoglobin <7 g/dL). Aluminium toxicity was diagnosed by a positive deferoxamine test and bone biopsy. Seven out of the 8 patients without aluminium toxicity responded to erythropoietin therapy. In the group with aluminium toxicity, only the 4 patients pretreated with deferoxamine responded, whereas the 5 patients not given deferoxamine were non-responders. All subjects in the latter group became responders when pretreated with deferoxamine. Resistance to erythropoietin therapy due to aluminium toxicity may therefore be reversed by low dose deferoxamine therapy in transfusion-dependent haemodialysis patients.
Isolated case reports on the use of deferoxamine have appeared in the literature and do not always contribute to our understanding of the actions or safety of this compound. Some reports however are worthy of note. The first two case reports are reviewed for historical reasons as the earliest demonstrations of deferoxamine use.
Henderson et al. (1963) reported of the use of deferoxamine in a 14 month old child following an ingestion of ferrous gluconate. The initial serum iron concentration at least 4.5 hours after ingestion was 456 µmol/L (2,550 µg/dL) and the child was tachycardic and acidotic. Both oral and intravenous deferoxamine were administered with the i.v. dose given as 800 mg in 20 mL of water. Urinary excretion was reported at 25 mg of iron over 3 days. At 24 hours after admission the child suffered 5 generalised seizures and had a residual hemiparesis that resolved over a period of months. The authors attributed the prompt improvement in the child's clinical status to deferoxamine administration, although other supportive therapies were instituted simultaneously.
Santos & Pisciotta (1964) reported the treatment of a 14 month old child with deferoxamine for acute iron poisoning. The initial serum iron concentration approximately 1 hour after ingestion was 102 µmol/L (575 µg/dL) and the child was treated with oral, intravenous and intramuscular deferoxamine. Urinary excretion of iron was 4.6 mg over 6 days. The authors suggest that the good outcome in this case was due in part to deferoxamine.
Peck et al. (1982) reported continuous deferoxamine administration in a 19 year old using an infusion of 15 mg/kg/h up to a total of 37.1 g over a 52 hour period. The patient continued to excrete a vin rose urine over this time period but recovered without sequelae from either toxin or treatment. This exceeds the generally recommended 6 g/24 hour dosage regimen for deferoxamine.
More recently, Sipahi et al. (2002) reported using deferoxamine to treat two cases of symptomatic iron poisoning in children aged 60 and 14 months with serum iron concentrations of 332 µg/dL (60 µmol/L) and 302 µg/dL (54 µmol/L), respectively. Both did well despite ingesting 88 mg/kg and 100 mg/kg of elemental iron respectively, although the need for antidotal therapy in such cases could be questioned.
In a retrospective study of 32 patients with moderately severe iron poisoning (serum iron concentrations of 300-500 µg/dL; 53-92 µmol/L), conservative management without deferoxamine therapy was sufficient (Bosse, 1995). However, both early intravenous deferoxamine therapy (at a rate of 15 mg/kg/hour) and gastrotomy did not prevent the death of a 17 month old child who presented with haemorrhagic emesis, coagulopathy, shock, acidosis, respiratory failure, and an initial blood iron concentration of 18,570 µg/dL (3,305 µmol/L) (Perrone et al., 2000).
Various indicators have been used to assess iron toxicity, but they are generally unreliable. In the past, a serum iron concentration in excess of the total iron-binding capacity (TIBC) has been considered as an indication for deferoxamine therapy in acute iron poisoning. Traditionally the presence of radiopacities on abdominal X-rays, leucocytosis, hyperglycaemia, metabolic acidosis and diarrhoea were thought to be associated with severe poisoning and high serum iron concentrations, but this is not the case (Palatnick & Tenenbein, 1996). Provocative chelation with deferoxamine has also been espoused as a measure of severity of iron poisoning.
Chyka & Butler (1993) in their review of 128 cases of acute iron poisoning in children found that 65 patients had a serum iron and a TIBC taken simultaneously allowing the authors to test the predictive value of these and other parameters, in the assessment of the severity of poisoning. They were unable to confirm that vomiting, diarrhoea, leucocytosis, hyperglycaemia and radiopacities were associated with a serum iron concentration in excess of 300 µg/dL (54 µmol/L); only one observation, coma, was associated (p <0.02) with a serum iron concentration greater than 500 µg/dL (90 µmol/dL). Coma, radiopacities, leucocytosis, and an elevated anion gap were concurrently present (predictive value positive = 100%) when the serum iron concentration was greater than 500 µg/dL; moreover, they were absent collectively (predictive value negative = 95%) when the serum iron concentration was below 500 µg/dL. Individually, these features had a low positive predictive value, but the absence of any one of these variables was likely to be associated with serum concentrations less than 500 µg/dL (predictive value negative >93%). The ratio of serum iron concentration to the TIBC was not associated with symptoms of systemic iron toxicity or the presence of vin rose urine after parenteral deferoxamine administration.
The fact that a serum iron concentration in excess of TIBC may not identify patients with serious iron poisoning (Chyka & Butler, 1993; Siff et al., 1999), may be partly related to the unsatisfactory low precision found for this analysis (variation coefficient 16%) and the observation that the in vitro addition of iron to test serum samples produced a relative increase in the TIBC (Tenenbein & Yatscoff, 1991). Toxicity is seen in patients in whom it has been determined that the TIBC is greater than the serum iron concentration (Schauben et al., 1990; Tenenbein & Yatscoff, 1991). The presence of deferoxamine itself makes the measurement of TIBC inaccurate (Siff et al., 1991). As such, the TIBC should not be used in the decision for the initiation of deferoxamine therapy in acute iron poisoning, and high TIBC values occasionally seen in such patients should not be considered as providing a protective effect.
Provocative chelation with deferoxamine has not proven clinically useful in the diagnosis of iron poisoning. Administration of a single dose of deferoxamine does indicate free unbound iron in the plasma if the urine subsequently turns pinkish in colour (vin rose urine). However it has been previously demonstrated that the urine can contain considerable ferrioxamine without any detectable colour change (Eisen et al., 1988), therefore provocative chelation as a diagnostic strategy has limited precision and usefulness.
A serum iron concentration taken at 4 hours post-ingestion is generally the best guide to severity and indicator of the need to treat with deferoxamine. Westlin (1971) reviewed 474 patients with iron poisoning and patients with a serum iron concentration less than 89 µmol/L (<500 µg/dL) had 6% incidence of serious toxicity whereas those with a serum iron greater than 179 µmol/L (1000 µg/dL) had a 62% incidence of shock and coma. Based on this and other clinical data, normal practice is to treat all patients with a serum iron concentration greater than 90 µmol/L(500 µg/dL) at 4 hours with deferoxamine. While a serum iron concentration obtained 4 hours post-exposure may be preferred, serum iron concentrations obtained anywhere between 3 to 6 hours after exposure are serviceable in predicting the clinical severity to be anticipated from iron poisoning.
The criteria for stopping deferoxamine therapy in the acutely iron poisoned patient are not precise, recommendations being to stop chelation when the vin rose colour of the urine disappears or 24 hours later (Robotham & Leitman, 1980). A urinary iron assay has been developed for this assessment and may be useful, especially in centres which regularly treat these patients, however further work is required to validate this assay and assess its clinical applicability (Yatscoff et al., 1991).
The problem of iron intoxication in pregnancy has been the subject of several reviews (McElhatton et al., 1991; Lacoste et al., 1992; Tran et al., 2000) and case reports (Rayburn et al., 1983; Blanc et al., 1984; Schauben et al., 1990; Turk et al., 1993).
In a review of iron overdose in 49 pregnant women the dose ingested was known in 48 patients and in 28 (60%) exceeded 20 mg/kg of elemental iron. The serum iron concentration was measured in 36 women, and 20 patients had a concentration greater than 60 µmol/L (338 µg/dL). Of the 49 pregnancies 43 resulted in live babies; there were 2 spontaneous abortions and 4 elective terminations. Three babies were premature and 3 had malformations. A total of 25 women (51%) were treated with deferoxamine, 2 of these had babies with malformations. However, all babies with malformations were born to mothers who had taken the overdose after the first trimester, so the abnormalities cannot be directly associated with the overdose or the treatment. The authors recommended that pregnancy should not be considered a contraindication to deferoxamine therapy in iron poisoning (McElhatton et al., 1991).
Rayburn et al. (1983) reported an iron overdose during the third trimester of pregnancy showing that iron is not specifically sequestered in the fetus. The peak serum iron concentration in the mother was 247 µmol/L (1,380 µg/dL). Fourteen hours later a viable preterm infant was born with a serum iron of 21 µmol/L (121 µg/dL). Deferoxamine was administered to the mother prior to the delivery. Of note is that the follow up iron concentrations in the infant showed the rapid onset of iron deficiency, which was treated with oral iron. Both mother and infant did well.
In the fifteenth week of gestation a 15 year old ingested 50 x 300 mg ferrous sulphate tablets (approximately 30 to 50 mg/kg elemental iron). The patient was treated with i.m. deferoxamine (total 3 g) without difficulty. The serum iron concentration between 4 and 5 hours post-ingestion (but not available until 24 hours later) was 324 µg/dL (58 µmol/L). The patient recovered without sequelae and later delivered a healthy term infant (Blanc et al., 1984).
Schauben et al (1990) report an 18 year old in week 25 of gestation who was treated with 2 g i.m. deferoxamine after ingesting 45 mg/kg elemental iron. Abdominal X rays showed an iron bezoar distal to the pylorus and whole bowel irrigation was performed using a polyethylene glycol solution. Serum iron concentrations were 289, 415 and 384 µg/dL (51, 74 and 68 µmol/L) at 2, 3 and 4 hours post-ingestion, respectively. She made a complete recovery and delivered a healthy infant at 39 weeks term.
Turk et al. (1993) reported ingestion of a large dose of iron (3.9 g of elemental iron; 36 mg/kg or 64 mg/kg calculated on lean body weight) in an adult patient in week 26 of gestation. The serum iron concentration about 2 hours after ingestion was 91 µmol/L (507 µg/dL). She was treated with 10.2 g of deferoxamine i.v. over 14 hours and whole bowel irrigation for 12 hours. The patient survived without sequelae and no adverse effects were reported in the fetus.
Severe hypotension was associated with rapid infusion of deferoxamine in two children given 80-150 mg/kg over 15 minutes (Whitten et al., 1965). Hypotension from rapid infusion of deferoxamine has also been demonstrated in other patients (Leikin et al., 1967; Fisher et al., 1971; Westlin, 1971; Dickerhoff, 1987). Nephrotoxicity may also occur and can develop without hypotension (Koren et al., 1989).
Tenenbein et al. (1992) reported fatal lung injury in four patients, aged 19-26 years, who received deferoxamine infusions of 15 mg/kg/hour for 65-92 hours. Respiratory distress developed after 32-72 hours. Their suggestion that the ARDS seen in these patients was a consequence of free-radical generation was supported by their later study in mice (Adamson et al., 1993). Severe ARDS developed in a 3.5 year old girl who was treated for iron poisoning with deferoxamine at 15 mg/kg/hour for 20 hours and then 5 mg/kg/hour for 14 hours (Ioannides & Panisello, 2000). Respiratory distress may, however, occur in severe iron poisoning (Shannon, 1992). It is generally recommended that a deferoxamine infusion should not be given for more than 24 hours, although prolonged infusions have been tolerated by several patients (Peck et al., 1982; Shannon, 1992).
While not generally occurring in the setting of acute iron intoxication, it is nevertheless important to review the accumulated knowledge concerning the toxic effects of chronic deferoxamine therapy.
Infectious complications of iron overdose and of treatment with deferoxamine are areas of controversy.
Melby et al. (1982) and Mofenson et al. (1987), reported children with ingestion of iron and subsequent deferoxamine therapy who developed septicaemia with Yersinia enterocolitica. It is unclear whether these cases represent compromise of the gastrointestinal tract from iron-induced enteritis, promotion of growth by the propensity for Yersinia enterocolitica to use iron when present in high concentrations or deferoxamine acting as a specific siderophore promoting the growth of this organism (Brock et al. 1988).
Seifert et al. (1987) found a general correlation between iron overload states and bacterial infections in patients receiving regular haemodialysis. Deferoxamine seemed to have no influence on the incidence of bacteraemia in the 39 study patients compared to a control group of 193 patients on haemodialysis, but not receiving deferoxamine. Fungal infections have also been reported in association with chronic iron overload treated with deferoxamine. Boelaert et al. (1988) and Rex et al. (1988) reported cases of fungal infections in a total of four patients. Similarly, Koren et al. (1988) reported a similar saprophytic infection, Pneumocystis carinii, in a patient, without evidence of HIV infection, with transfusional iron overload receiving deferoxamine.
Both visual toxicity (decreased acuity, night blindness, cataract formation, retinal degeneration) and ototoxicity attributable to deferoxamine have been described in patients with thalassaemia receiving repeated infusions of the drug (Davies et al., 1983; Olivieri et al., 1986; Bentur et al., 1991; Howland, 1996).
Reports of neurotoxicity began to emerge after approximately 20 years of deferoxamine use for iron chelation in patients receiving repeated blood transfusions (Freedman et al., 1988). Davies et al. (1983) and Lakhanpal et al. (1984) reported on patients receiving high dose therapy for chronic iron overload. These patients were found to have retrobulbar optic neuropathy with central scotomas and colour vision abnormalities. Most patients improved following discontinuation of therapy. Orton et al. (1985) reported ocular and auditory toxicity from chronic deferoxamine therapy. While the visual changes were partially reversible, both of the patients reported had permanent sensorineural hearing loss.
Blake et al. (1985) and Pall et al. (1986) suggested the possibility that these acute neurotoxic effects of deferoxamine may be related to chelation of copper. To further add to the confusion, Dickerhoff (1987) reported hypotension, transient blindness and aphasia in a patient who received a sudden bolus of deferoxamine. After restoration of intravascular volume, the symptoms and signs reversed and were attributed to a vasoregulatory phenomenon. Of particular concern to toxicologists is a report by Pengloan et al. (1987) describing two patients who received 10 mg/kg/h infusions of deferoxamine for four hours for the diagnosis of aluminium overload during chronic dialysis. These patients both had an acute reversible deterioration of vision. A similar case was reported by Bene (1989) in a patient receiving 1.8 g of deferoxamine over a two hour period to evaluate aluminium accumulation.
Although now considered a rare idiopathic effect, cataract formation has been reported in patients receiving deferoxamine (Bloomfield et al., 1978). More recently, as a result of higher infusion doses (75-235 mg/kg/day), note has been made of a retinopathy resembling tapetoretinal dystrophy (Davies et al., 1983; Olivieri et al., 1986; Hershko & Weatherall, 1988).
The most frequent adverse effects of deferoxamine are local reactions at the site of infusion or injection, such as pain, swelling, induration, erythema, burning, pruritus, wheals and rash, occasionally accompanied by fever, chills and malaise (Ciba-Geigy, 1994).
Hypotension is the most frequently reported adverse effect of deferoxamine. It appears to be due to a direct effect on mast cells resulting in histamine release, and is most frequent with rapid administration. True IgE mediated anaphylaxis has been reported (Athanasion et al., 1977), but this appears to be infrequent. An isolated case report of thrombocytopenia associated with intravenous deferoxamine is unclear as to aetiology (Walker et al., 1985). Another study of chronic toxicity by De Virgiliis et al. (1988) has demonstrated that excessive deferoxamine therapy in children resulted in poor longitudinal growth. Batey et al. (1979) reported a single case of acute renal failure associated with chronic deferoxamine therapy in a 14 year old treated for chronic iron overload following treatment for thalassaemia. The patient died from hyperkalaemia with renal pathology consistent with a proliferative glomerulonephritis.
Renal toxicity has been reported in a few cases of high-dose deferoxamine therapy (180 mg/kg/day); this may have significance for high-dose deferoxamine treatment in acute iron poisoning. The patient reported by Freedman et al. (1989) had mildly elevated creatinine concentrations, which returned to normal when therapy was discontinued. Koren et al. (1989) reported three patients with acute impairment of renal function from deferoxamine, one of whom was treated for acute iron poisoning.
The effectiveness of deferoxamine treatment in iron poisoning is dependent on an adequate urine output in order to ensure that the iron-deferoxamine complex (ferrioxamine) is excreted. If oliguria or anuria develops, peritoneal dialysis, haemodialysis, or haemofiltration may be necessary (Ciba-Geigy, 1994). The extent to which the dose of deferoxamine should be reduced in renal failure has not been studied.
Iron poisoning in pregnant women can result in spontaneous abortion, preterm delivery, or, in rare instances, maternal and/or perinatal death. Deferoxamine has been given to pregnant women without adverse effect, although its efficacy in pregnancy has not been studied in controlled clinical trials.
An 18 year old pregnant woman ingested 45mg/kg of elemental iron at 25 weeks gestation and was treated with whole bowel irrigation and i.m. deferoxamine (2 g), with subsequent complete recovery and delivery of an apparently normal male infant at term (Schauben et al., 1990). A 27 year old woman ingested 24 mg/kg of elemental iron in a suicide attempt at 27 weeks gestation (Tran et al., 1998). Deferoxamine treatment was performed without any complications. Tran and associates (2000) performed a meta-analysis of 61 published cases of intentional iron poisoning in pregnancy, including 29 cases in which deferoxamine was given. The authors observe that deferoxamine was withheld from 47% of eligible symptomatic patients despite the lack of evidence of adverse fetal effects of the drug. They recommend the use of deferoxamine in pregnant women with serious acute iron poisoning.
Experimental studies have shown that deferoxamine may counteract the toxic effects of free iron. It is thought to act by provision of excess iron binding capacity (chelation), and its effects are not necessarily dependent on the subsequent excretion of iron. Studies in animals give evidence of reduced serum iron concentration associated with deferoxamine administration, although conflicting data exists. No clinical studies have demonstrated any beneficial effect of deferoxamine on morbidity or mortality in subjects with acute iron poisoning. However, in some case reports the use of deferoxamine has been associated with a favourable outcome. As such, supportive care is the cornerstone of treatment for the most severe cases of iron poisoning with deferoxamine as an important adjunct.
In chronic aluminium toxicity (that is, patients on haemodialysis) there are still too few reports to evaluate a possible role of deferoxamine therapy.
Deferoxamine is indicated for the management of acute iron intoxication as an important adjunct to supportive care.
In considering triage of iron intoxications, it is important to remember that a 300 mg ferrous sulphate tablet contains 60 mg of elemental iron. Other preparations such as paediatric multivitamins usually contain only 4 to 20 mg of elemental iron per tablet. It is generally recommended that ingestions in excess of 60 mg/kg of elemental iron be considered for intensive gut decontamination and monitoring of the serum iron concentration. Ingestions of elemental iron in the 30-60 mg/kg range may still warrant decontamination, but are less likely to need further intervention. Obviously, in some cases the history may be unavailable or misleading and adjunctive support such as abdominal radiographs and examination of vomitus or gastric contents may be helpful in determining appropriate management. The final arbiter of any decision to discharge a patient should be the clinical condition of the patient, in conjunction with the serum iron concentration at 4-6 hours post-ingestion.
There is little controversy that serum iron concentrations above 89 µmol/L (>500 µg/dL) and/or patients with serious clinical manifestations of iron toxicity (e.g. coma, gastrointestinal haemorrhage) warrant treatment. Similarly patients with serum concentrations less than 53 µmol/L (300 µg/dL) generally do not show significant urinary excretion of iron during chelation (i.e. they do not display vin rose urine). Controversy exists over the patient with a serum iron between 53 µmol/L (300 µg/dL) and 90 µmol/L (500 µg/dL). In these patients, and if serum concentrations are not available, a history of a large ingestion and clinical signs of severity should dictate therapy (see section 13.1).
The serum iron concentration is unreliable in deferoxamine-treated patients and declines in both chelated and non-chelated patients due to redistribution. The decision to terminate chelation therapy is a clinical judgement made on the basis of the patient's condition and clearing of the vin rose colouration of urine (if present).
At the current time an intravenous infusion of 15 mg/kg/h of deferoxamine is recommended for acute iron intoxication. Since bolus deferoxamine may release histamine, a controlled infusion rate is critical to avoid adverse effects. A small degree of histamine release in a hypovolaemic patient may compromise the circulation and so maintenance of intravascular volume is recommended prior to administration of deferoxamine. This may also prevent deferoxamine-induced nephrotoxicity which can occur without hypotension. This dose of deferoxamine is empirical and has not been validated, however it is widely recommended by many authors.
The total intravenous dose of deferoxamine should generally not exceed 80 mg/kg/24 hours. There have, however, been reports of administration of much higher doses with no adverse effects, for example 425 mg/kg over 24 hours in one report of patients with thalassaemia (Propper et al., 1976).
The intramuscular route is discouraged and should be avoided in shock states. However, this route may be recommended in certain transport situations or where accurate delivery by infusion pump cannot be guaranteed. Doses ranging from 20 to 100 mg/kg every 4 to 8 hours i.m. have been recommended with an upper limit of 6 g/24 hours.
The subcutaneous dosing for chronic iron and aluminium overload (usually at 20-40 mg/kg/day) is not evaluated in this monograph.
Supportive care remains the mainstay of treatment for iron intoxication. Initial management should focus upon support of the airway, respiration and cardiac function.
Fluid resuscitation needs may be tremendous (up to 100 ml/kg) and it is clear that intravascular monitoring such as central venous pressure or Swan-Ganz catheterisation is an important adjunct to the management of critically ill patients with iron intoxication. Evidence of negative inotropic and chronotropic effects of iron in overdose supports the use of catecholamines in hypotension or low perfusion states unresponsive to fluid therapy (Vernon et al., 1989). Iron intoxication results in acidosis directly from liberation of an unbuffered hydrogen ion when ferrous iron is converted to ferric iron and indirectly from the effects of hypoperfusion; it should be corrected.
Gut decontamination may be undertaken considering the following points. Ipecacuanha syrup and gastric lavage may be appropriate choices, if given early, because iron poisoning may involve large tablets in various states of decomposition. The use of various alkalinising agents in attempts to maintain the iron in an insoluble state has been suggested and studied in vitro and in vivo (Czajka et al., 1981). Dilute sodium bicarbonate solution may be a reasonable approach, as may the use of antacids or H2 antagonists to achieve the same effect. Phosphate used for the same purpose has been demonstrated to produce severe systemic toxicity and is no longer recommended. Given the direct damage to the gastric mucosa by iron, protection of the stomach by decreasing gastric acid secretion with H2 antagonists and antacids seems prudent. However, there are no compelling data to support the efficacy of any of these agents at controlling tablet dissolution and absorption rate in a clinical setting.
Tenenbein (1987) has reported a series of patients treated with whole bowel irrigation and this may be useful in selected patients. In several anecdotal cases (Peterson and Fifield, 1980), gastrotomy for removal of tablets has been suggested, if a large number of tablets are visible in one location on abdominal radiograph.
Activated charcoal has low maximal binding for iron and therefore has limited value for the large amounts that may be present in the gastrointestinal tract. While deferoxamine has a relatively good absorptive quality for charcoal, the relative amounts of iron that can be bound to deferoxamine make the combination of charcoal and deferoxamine a relatively ineffective approach to binding iron in the gastrointestinal tract. Unless other agents have also been ingested, there is probably no role for charcoal in iron intoxication.
In a number of the more serious cases of iron intoxication, necrosis of the gastrointestinal tract (involving areas from the stomach to the ileocecal junction) has been reported. Ferrioxamine may promote the growth of organisms such as Yersinia enterocolitica and this may contribute to bacterial sepsis. These complications should be considered in any patient who deteriorates suddenly.
Some sources continue to recommend the administration of oral deferoxamine to form ferrioxamine in the stomach, although efficacy remains unproven. Controversial evidence is available that ferrioxamine is absorbed and is toxic although less toxic than the iron absorbed uncomplexed. The recommended dose of 2-10 grams is very expensive, and this use may decrease the availability of deferoxamine for parenteral use in areas where supplies are limited. At present the oral use of deferoxamine cannot be recommended.
Infusion rates of deferoxamine above the currently recommended 15 mg/kg/h may be tolerated in most patients. There are no large series or carefully acquired animal data regarding the risk of higher infusion rates in patients with acute iron poisoning.
The use of oral ascorbate (200 mg/day in divided doses) has been studied in patients with chronic iron excess being treated with deferoxamine; these studies have shown an increase in urinary iron excretion with the addition of ascorbate (Hussain et al., 1977; Pippard et al., 1982). There have been no studies that have assessed the potential role for ascorbate in acute iron poisoning.
Newer iron-chelating agents, such as the oral drug deferiprone (Berkovitch et al., 2000) and NaHBED ligand (Bergeron, 2002), may be promising alternatives to deferoxamine. However, they require further experimental studies before they can be recommended. N,N'-Bis(2-hydroxybenzylethylenediamine-N,N'-diacetic acid (HBED), a synthetic hexadentate ligand, forms a high-affinity, selective bond to iron and can be administered intravenously as a monosodium salt. Unlike deferoxamine it does not produce hypotension and in monkeys induced iron clearance 2-3 times greater than that produced by an equimolar dose of deferoxamine (Bergeron, 1998; Bergeron, 1999).
Chemical modification of deferoxamine, by conjugating the drug to polysaccharides such as dextran and hydroxylethyl starch, has been investigated as a means of improving its efficacy. This creates a high molecular weight drug that retains the chelant efficacy of deferoxamine while being retained in the vascular compartment for a prolonged period of time. Consequently larger doses and higher blood concentrations of the drug can be achieved, with greater chelating capacitance and a lower risk of such adverse effects as hypotension (Dragsten et al., 2000).
A detailed animal study to evaluate the efficacy and safety of oral deferoxamine in acute iron poisoning is critical because of the continued use of this approach. The risk that this may worsen outcome, for example by increasing absorption or altering bowel flora, mandate clarification of this issue.
Animal data followed by collaborative studies in iron intoxicated patients may help to define the range of safe doses of infused deferoxamine in patients with dramatically elevated serum iron concentrations. Animal data may demonstrate the benefit of pretreatment with antihistamine drugs in avoiding the cardiovascular effects of deferoxamine. Further animal and human studies of the safety and efficacy of high molecular weight deferoxamine chelants in the context of acute iron poisoning are also warranted.
Some recent work has demonstrated that the use of adjunctive techniques such as charcoal haemoperfusion and dialysis (Chang and Barre, 1983; Molitoris et al., 1988; Weiss et al., 1989), in some cases with deferoxamine bound to polymers outside the dialysis cartridge (Ambrus et al., 1987), may be methods of removing the iron complex from the circulation in patients with chronic iron and aluminium overload. The potential usefulness of this approach in acute iron intoxication has been supported in an animal model (Banner et al., 1989).
The criteria for cessation of deferoxamine therapy are not well defined, however a preliminary study has shown that urine free iron:creatinine ratio could be used as a guide (Yatscoff et al., 1991). Further work is required to validate the assay that was used in this study and to assess the iron:creatinine cut off at which deferoxamine can be stopped.
The relatively extensive use of deferoxamine over several years for acute iron poisoning has revealed few severe side effects, if used as recommended by the manufacturer.
In patients with acute iron poisoning hypotension may develop if deferoxamine is infused rapidly (>15 mg/kg/h); renal failure may also occur. Use of deferoxamine for more than 24 hours may be associated with severe pulmonary complications (ARDS) as shown in some iron-poisoned patients and supported by one experimental study in mice. Infectious complications, especially septicaemia with Yersinia enterocolitica, has been seen in some patients but may not be related to use of deferoxamine.
Chronic deferoxamine therapy in iron-overloaded patients has been associated with ocular and auditory toxicity and renal impairment.
None, except for previous anaphylaxis to deferoxamine, which has been reported but appears to be very uncommon.
In renal failure the dose must be reduced accordingly. Haemodialysis may be necessary to remove the ferrioxamine.
Deferoxamine is indicated for the management of acute iron intoxication where the total body burden is great, symptoms and signs severe, or the serum iron concentration is greater than 90 µmol/L (500 µg/dL) at 4-6 hours after ingestion. The indications for the use of deferoxamine following an acute ingestion include:
Deferoxamine may be given by subcutaneous, intramuscular or, preferably, by intravenous routes of administration.
Intravenous administration represents the most consistent mode of drug delivery but must be carefully controlled to deliver no more than 15 mg/kg/h as a continuous infusion, in order to avoid hypotension. This dose should be reduced as soon as the clinical situation permits, usually after 4 to 6 hours so that the total dose does not exceed 80 mg/kg in any 24 hour period. A 10% Desferal® solution (5 mL of sterile water into 500 mg of deferoxamine, then shake well) can be further diluted with isotonic saline, glucose or Ringer's lactate.
Intramuscular therapy may be indicated in doses of 20-100 mg/kg for treatment where intravenous therapy may be difficult to control adequately, such as during patient transport. No more than 80 mg/kg per 24 hour period should be given as for the intravenous route. The manufacturer recommends the following intramuscular dose to normotensive patients: A single dose of 2 g for an adult and 1 g for a child. The subcutaneous route is not recommended in acutely poisoned patients.
The decision to terminate deferoxamine treatment is essentially based on the clinical condition of the patient, supported by the disappearance of the vin rose colour of urine which is often, but not always, seen. The fact that severe pulmonary toxicity may develop after more than 24 hours of therapy must also be taken into account.
There are no specific contraindications to the use of deferoxamine in acute iron intoxication except for a history of prior hypersensitivity. Control of the infusion rate to avoid rapid administration is required to prevent hypotension. An adequate output of urine is essential to ensure that the iron-deferoxamine complex (ferrioxamine) is excreted. In the presence of acute renal failure, dialysis or haemofiltration will be required to remove this complex.
Hypotension is well documented as an adverse effect of rapid infusion of deferoxamine. However, it has never been seen when the drug is given at a rate less than 1 mg/kg/minute - a rate that rarely needs be exceeded.
The possibility that renal function may deteriorate during therapy should be considered and monitored. Pulmonary toxicity can occur with prolonged infusions.
Formation of the iron-deferoxamine complex changes the spectral characteristics of deferoxamine and forms an orange-red colour that when present in urine has been described as vin rose. This colouration of urine indicates excretion of chelated iron and is not an adverse event.
Local reactions at site of infusion or injection are common ranging from pain and erythema to wheals and rash, occasionally accompanied by fever, chills and malaise.
In chronic iron intoxication, auditory and ocular function should be monitored periodically to avoid toxicity, which is usually reversible.
The US Food and Drug Administration has categorised this drug as a Category C product (that is, risk cannot be ruled out). Skeletal abnormalities have occurred in the offspring of exposed mice and rabbits, but not rats. However, single case reports of treatment for acute iron intoxication in pregnant humans have not demonstrated any adverse effects.
Vials of powder should be stored in a cool place (below 15°C). This drug is manufactured as a lyophilised powder. After reconstitution it is stable at room temperature in the dark for up to one day.
Adamson IY, Sienko A & Tenenbein M (1993) Pulmonary toxicity of deferoxamine in iron-poisoned mice. Toxicol Appl Pharmacol, 120: 13-19.
Allain P, Mauras Y, Chaleil D, Simon P, Ang KS & Cam G (1987) Pharmacokinetics and renal elimination of desferrioxamine and ferrioxamine in healthy patients and in patients with haemochromatosis. Br J Clin Pharmacol, 24: 207-212.
Allain P, Leblondel G & Mauras Y (1988) Effect of aluminum and deferoxamine on biliary iron elimination in the rat. Proc Soc Exp Biol Med, 188(4): 471-473.
Allain P, Mauras Y, Beaudeau G & Hingouet P (1986) Indirect microscale method for the determination of desferrioxamine and its aluminum and iron chelated forms in biological samples by atomic absorption spectrometery with electrothermal atomisation. Analyst, 111: 531-533.
Ambrus CM, Anthone S, Deshpande G, Horvath C & Kalghatgi K (1987) Extracorporeal removal of iron with immobilized desferrioxamine. ASAIO Trans, 33: 749-753.
Artman M, Olson RD, Boucek RJ Jr, Ghishan FK & Boerth RC (1984) Acute effects of iron on contractile function in isolated rabbit myocardium. Dev Pharmacol Ther, 7: 50-60.
Athanasion A, Shepp MA & Necheles TF (1977) Anaphylactic reaction to desferrioxamine [letter]. Lancet, 2(8038): 616.
Banner W Jr & Tong TG (1986) Iron poisoning. Pediatr Clin North Am, 33: 393-409.
Banner W Jr, Vernon DD, Ward RM, Sweeley J & Dean M (1989) Continuous arteriovenous hemofiltration in experimental iron intoxication. Crit Care Med, 17(11): 1187-1190.
Barker G, Brown T & Hosking C (1974) Acute iron poisoning - clinical features and management. Anaesth Intensive Care, 4: 345-350.
Barr DG & Fraser DK (1968) Acute iron poisoning in children: role of chelating agents. Br Med J, 1:737-41.
Batey R, Scott J, Jain S & Sherlock S (1979) Acute renal insufficiency occurring during intravenous desferrioxamine therapy. Scand J Haematol, 22(3): 277-279.
Bene C, Manzler A, Bene D & Kranias G (1989) Irreversible ocular toxicity from single "challenge" dose of deferoxamine. Clin Nephrol, 31(1): 45-48.
Bentur Y, McGuigan M & Koren G (1991) Deferoxamine. New toxicities for an old drug. Drug Saf, 6: 37-46.
Bergeron RJ, Wiegand J & Brittenham GM (1998) HBED: a potential alternative to deferoxamine for iron-chelating therapy. Blood, 91: 1446-1452.
Bergeron RJ, Wieland J & Brittenham GM (1999) HBED: the continuing development of a potential alternative to deferoxamine for iron-chelating therapy. Blood, 93: 370-375.
Bergeron R J, Wiegand J & Brittenham GM (2002) HBED ligand: preclinical studies of a potential alternative to deferoxamine for treatment of chronic iron overload and acute iron poisoning. Blood, 99 (8): 3019-3026.
Berkovitch M, Livne A, Lushkov G, Segal M, Talmor C, Bentur Y, Klein J & Koren G (2000) The efficacy of oral deferiprone in acute iron poisoning. Am J Emerg Med, 18(1): 36-40.
Blake DR, Winyard P, Lunec J, Williams A, Good PA, Crewes SJ, Gutteridge JM, Rowley D, Halliwell B, Cornish A & Hider RC (1985) Cerebral and ocular toxicity induced by desferrioxamine. Q J Med, 56: 345-355.
Blanc P, Hryhorczuk D & Danel I (1984) Deferoxamine treatment of acute iron intoxication in pregnancy. Obstet Gynecol, 64 (3 Suppl): 12S-14S.
Bloomfield S, Markenson AL, Miller DR & Peterson CM (1978) Lens opacities in thalassemia. J Pediatr Ophthalmol Strabismus, 15:154-156.
Boelaert JR, van Roost GF, Vergauwe PL, Verbanck JJ, de Vroey C & Segaert MF (1988) The role of desferrioxamine in dialysis associated mucormycosis: report of three cases and review of the literature. Clin Nephrol, 29: 261-266.
Bosse GM (1995) Conservative management of patients with moderately elevated serum iron levels. J Toxicol Clin Toxicol 33: 135-140.
Bothwell T, Charleton R, Cook J & Finch C (1979) Iron metabolism in man. Oxford, Blackwell Scientific.
Brock JH, Liceaga J & Kontoghiorghes GJ (1988) The effect of synthetic iron chelators on bacterial growth in human serum. FEMS Microbiol Immunol, 1(1): 55-60.
Brunner H, Hedwall H, Keberle H & Schmid K (1965) The influence of desferrioxamine on an acute systemic poisoning with iron (II) in a dog (German). Helv Physiol Pharmacol Acta, 23: C81.
Burkitt MJ, Kadiiska MB, Hanna PM, Jordan SJ & Mason RP (1993) Electron spin resonance spin-trapping investigation into the effects of paraquat and deferoxamine on hydroxyl radical generation during acute iron poisoning. Mol Pharmacol, 43: 257-263.
Chang TM & Barre P (1983) Effect of desferrioxamine on removal of aluminum and iron by coated charcoal haemoperfusion and haemodialysis. Lancet, 2(8358): 1051-1053.
Chyka PA & Butler AY (1993) Assessment of acute iron poisoning by laboratory and clinical observations. Am J Emerg Med, 11: 99-103.
Chyka PA, Butler AY & Holley JE (1996) Serum iron concentrations and symptoms of acute iron poisoning in children. Pharmacotherapy, 16: 1053-1058.
Ciba-Laboratories (1964) Desferal®. A specific iron chelating agent. Ciba Laboratories Ltd, Horsham, Sussex, UK.
Ciba-Geigy (1987) Report B 76/1987. Basle, Switzerland, p34.
Ciba-Geigy (1994) Desferal. Basic drug information. Basle, Switzerland, p24.
Czajka PA, Konrad JD & Duffy JP (1981) Brief clinical and laboratory observations. J Pediatr, 98: 491-494.
Davies SC, Marcus RE, Hungerford JL, Miller MH, Arden GB & Huehns ER (1983) Ocular toxicity of high dose intravenous desferrioxamine. Lancet, 2(8343): 181-184.
Day JP & Ackrill P (1993) The chemistry of deferoxamine chelation for aluminium overload in renal dialysis patients. Ther Drug Monit, 15: 598-601.
Dean B & Krenzelok E (1987) In vivo effectiveness of oral complexation agents in the management of iron poisoning. Clin Toxicol, 25: 221-230.
Dean BS, Oehme FW, Krenzelok EP, Griffith GR, Hines RH (1996) Iron complexation with oral deferoxamine in a swine model. Vet Hum Toxicol, 38: 96-98.
De Virgiliis S, Congia M, Frau F, Argiolu F, Diana G, Cucca F, Varsi A, Sanna G, Podda G, Fodde M, Pirastu GF & Cao A (1988) Deferoxamine induced growth retardation in patients with thalassemia major. J Pediatr, 113(4): 661-669.
Dickerhoff R (1987) Acute aphasia and loss of vision with desferrioxamine overdose. Am J Pediatr Hematol Oncol, 9:287-288.
Dragsten PR, Hallaway PE, Hanson GJ, Berger AE, Bernard B & Hedlund BE (2000) First human studies with a high molecular weight iron chelator. J Lab Clin Med, 135(1): 57-65.
Eisen TF, Lacouture PG & Woolf A (1988) Visual detection of ferrioxamine color changes in urine. Vet Human Toxicol, 30: 369.
Fischer DS, Parkman R & Finch SC (1971) Acute iron poisoning in children. The problem of appropriate therapy. J Am Med Assoc, 218(8): 1179-1184.
Freedman MH, Boyden M, Taylor M & Skarf B (1988) Neurotoxicity associated with deferoxamine therapy. Toxicology, 49: 283-290.
Freedman M, Benton Y & Koren G (1989) Complications of deferroxamine therapy. In: Lucarelli G ed. Bone marrow transplantation in thalassemia and related problems. New York, Alan R. Liss.
Gomez HF, McClafferty HH, Flory D, Brent J & Dart RC (1997) Prevention of gastrointestinal iron absorption by chelation from an orally administered premixed deferoxamine/charcoal slurry. Ann Emerg Med, 30: 587-592.
Henderson F, Vietti TJ & Brown EB (1963) Desferrioxamine in the treatment of acute toxic reaction to ferrous gluconate. J Am Med Assoc, 186: 1139-1141.
Hershko C & Weatherall D (1988) Iron chelating therapy. Crit Rev Clin Lab Sci, 26: 303-346.
Hoffer E, Zonis Z, Tabak A & Taitelman U (1992) The administration of deferoxamine to paraquat-intoxicated rats. Vet Hum Toxicol, 34: 300-303.
Howland MA (1996) Risks of parenteral deferoxamine for acute iron poisoning. J Toxicol Clin Toxicol, 34 (5): 491-497.
Hussain MA, Green N, Flynn DM & Hoffbrand AV (1977) Effect of dose, time and ascorbate on iron excretion after subcutaneous deferoxamine. Lancet, 1: 977-979.
Ioannides AS & Panisello JM (2000) Acute respiratory distress syndrome in children with acute iron poisoning: the role of intravenous desferrioxamine. Eur J Pediatr, 159(3): 158-159.
Jackson TW, Ling LJ & Washington V (1995) The effect of oral deferoxamine on iron absorption in humans. J Toxicol Clin Toxicol, 33: 325-329.
Jenny H & Peter H (1988) Determination of desferrioxamine B, by high performance liquid chromatography using a polymer column. J Chromatogr, 438(2): 433-37.
Keberle H (1964) The biochemistry of deferroxamine and its relation to iron metabolism. Ann NY Acad Sci, 119: 758-768.
Koren G, Bentur Y, Strong D, Harvey E, Klein J, Baumal R, Speilberg S & Freedman M (1989) Deferoxamine therapy. Am J Dis Child, 143:1077-1080.
Koren PA, Slapak CA, Rosenwasser LJ & Miller KB (1988) Pneumocystis carinii pneumonia as a complication of desferrioxamine therapy. Br J Haematol, 70(3): 383-384.
Kraemer HJ & Breithaupt H (1998) Quantification of desferrioxamine, ferrioxamine and aluminoxamine by post-column derivatization high-performance liquid chromatography. Non-linear calibration resulting from second-order reaction kinetics. J Chromatogr B Biomed Sci Appl, 710: 191-204.
Kruck TP, Fisher EA & McLachlan DR (1993) A predictor for side effects in patients with Alzheimer's disease treated with deferoxamine mesylate. Clin Pharmacol Ther, 53(1): 30-37.
Lacoste H, Goyert GL, Goldman LS, Wright DJ & Schwartz DB (1992) Acute iron intoxication in pregnancy: case report and review of the literature. Obstet Gynecol, 80: 500-501.
Lakhanpal V, Schocket SS & Jiji R (1984) Deferoxamine (Desferal) induced toxic retinal pigmentary degeneration and presumed optic neuropathy. Ophthalmology, 91(5): 443-451.
Lee P, Mohamed N, Marshall L, Abeysinghe RD, Hider RC, Porter JB & Singh S. (1993) Intravenous infusion pharmacokinetics of desferrioxamine in thalassaemic patients. Drug Metab Dispos, 21: 640-644.
Lehmann WD & Heinrich HC (1990) Ferrioxamine and its hexadentate iron-chelating metabolites in human post-desferal urine studied by high-performance liquid chromatography and fast atom bombardment mass spectrometry. Anal Biochem, 184(2): 219-227.
Leikin S, Vossough P & Mochir-Fatemi F (1967) Chelation therapy in acute iron poisoning. J Pediatr, 71(3): 425-430.
Link G, Athias P, Grynberg A, Pinson A & Hershko C (1989) Effect of iron loading on transmembrane potential, contraction, and automaticity of rat ventricular muscle cells in culture. J Lab Clin Med, 113(1): 103-111.
Link G, Saada A, Pinson A, Konijn AM & Hershko C (1998) Mitochondrial respiratory enzymes are a major target of iron toxicity in rat heart cells. J Lab Clin Med, 131: 466-474.
Lipschitz DA, Dugard J, Simon MO, Bothwell TH & Charlton RW (1971) The site of action of desferrioxamine. Br J Haematol, 20: 395-404.
McCarthy JT, Milliner DS, Schmidt DF, Schniepp BJ, Kurtz SB & Johnson WJ (1988) Deferoxamine and coated charcoal hemoperfusion to remove aluminum in dialysis patients. Kidney Int, 34(6): 804-808.
McElhatton PR, Roberts JC & Sullivan FM (1991) The consequences of iron overdose and its treatment with desferrioxamine in pregnancy. Hum Exp Toxicol, 10(4): 251-259.
McEnery JT (1971) Hospital management of acute iron ingestion. Clin Toxicol, 4: 603-613.
McEnery JT & Greengard J (1966) Treatment of acute iron ingestion with deferoxamine in 20 children. J Pediatr, 68: 773-779.
McGuigan MA (1996) Acute iron poisoning. Pediatr Ann, 25: 33-38.
Melby K, Slördahl S, Gutteberg TJ & Nordby SA (1982) Septicaemia due to Yersinia enterocolitica after oral overdoses of iron. Br Med J, 285: 467.
Melograna JM & Yokel RA (1984) Effects of subchronic desferrioxamine infusion on aluminium toxicity in rabbits. Res Commun Chem Path Pharmacol, 44: 411-422.
Meulenbelt J, Dormans JA, van-Bree L, Rombout PJ & Sangster B (1993) Deferoxamine treatment reduces histological evidence of lung damage in rats after acute nitrogen dioxide (NO2) intoxication. Hum Exp Toxicol, 12: 389-395.
Mofenson HC, Caraccio TR & Sharieff N (1987) Iron sepsis: Yersinia enterocolitica septicemia possibly caused by an overdose of iron. N Engl J Med, 316: 1092-1093.
Molitoris BA, Alfrey AC, Alfrey PS & Miller NL (1988) Rapid removal of DFO chelated aluminum during hemodialysis using polysulfone dialyzers. Kidney Int, 34(1): 98-101.
Octave JN, Schneider YJ, Crichton RR & Trouet A (1983) Iron mobilization from cultured hepatocytes: effect of deferroxamine B. Biochem Pharmacol, 32: 3413-3418.
Olivieri NF, Buncic JR, Chew E, Gallant T, Harrison RV, Keenan N, Logan W, Mitchell D, Ricci G, Skarf B, Taylor M & Freedman MH (1986) Visual and auditory neurotoxicity in patients receiving subcutaneous deferoxamine infusions. N Engl J Med, 314: 869-873.
Orton R B, de Veber LL & Sulh HM (1985) Ocular and auditory toxicity of long term, high dose subcutaneous deferoxamine therapy. Can J Ophthalmol, 20: 153-156.
Palatnick W & Tenenbein M (1996) Leukocytosis, hyperglycemia, vomiting, and positive X-rays are not indicators of severity of iron overdose in adults. Am J Emerg Med, 14: 454-455.
Pall H, Blake DR, Good PA, Winyard P & Williams AC (1986) Copper chelation and the neuro-ophthalmic toxicity of desferrioxamine [letter]. Lancet, 2: 1279.
Peck MG, Rogers JF & Rivenbark JF (1982) Use of high doses of deferoxamine (Desferal) in an adult patient with acute iron overdosage. J Toxicol Clin Toxicol. 19(8): 865-869.
Pengloan J, Dantal J, Rossazza C, Abazza M & Nivet H (1987) Ocular toxicity after a single intravenous dose of desferrioxamine in 2 hemodialyzed patients [letter]. Nephron, 46: 211-212.
Perrone J, Lawless ST, Quintana E & De Roos F (2000) Fatal iron poisoning associated with massive iron level (18,570 µg/dL) [abstract]. J Toxicol Clin Toxicol, 38: 551.
Peters G, Keberle H, Schmid K & Brunner H (1966) Distribution and renal excretion of desferrioxamine and ferrioxamine in the dog and in the rat. Biochem Pharmacol, 15(1): 93-109.
Peterson CD & Fifield GC (1980) Emergency gastrotomy for acute iron poisoning. Ann Emerg Med, 9: 262-264.
Pippard M & Stray S (1982) Simple assay for urinary iron after deferroxamine therapy. Am J Clin Pathol, 77: 324-327.
Pippard MJ, Callendar ST & Finch CA (1982) Ferrioxamine excretion in iron-loaded man. Blood, 60: 288-294.
Prelog V & Walser A (1962) De novo synthesis of desferrioxamine B. Helv Chim Acta 45: 631.
Propper RD, Shurin SB & Nathan DG (1976) Reassessment of the use of desferrioxamine B in iron overload. N Engl J Med, 294(26): 1421-1423.
Proudfoot AT, Simpson D & Dyson EH (1986) Management of acute iron poisoning. Med Toxicol, 1: 83-100.
Rayburn WF, Donn SM & Wulf ME (1983) Iron overdose during pregnancy: successful therapy with deferoxamine. Am J Obstet Gynecol, 147(6): 717-718.
Rex JH, Ginsberg AM, Fries LF, Pass HI & Kwon-Chung KJ (1988) Cunninghamella bertholletiae infection associated with deferoxamine therapy. Rev Infect Dis, 10(6): 1187-94.
Robotham JL & Lietman PS (1980) Acute iron poisoning. A review. Am J Dis Child, 134: 875-879.
Santos AS & Pisciotta AV (1964) Acute iron intoxication. Treatment with desferrioxamine (Ba-29837). Am J Dis Child, 107: 424-427.
Saviuc P, Joannard A, Mazingue SD, Danel V & Dyon JF (1992) Intoxication par le perchlorure de fer compliquee d'une stenose gastrique. Pediatrie Bucur, 47: 37-39.
Schauben J L, Augenstein W L, Cox J & Sato R (1990) Iron poisoning: report of three cases and a review of therapeutic intervention. J Emerg Med, 8: 309-319.
Seifert A, von Herrath D & Schaefer K (1987) Iron overload, but not treatment with desferrioxamine favours the development of septicemia in patients on maintenance hemodialysis. Q J Med, 65(248): 1015-1024.
Shannon M (1992) Deferoxamine in acute iron poisoning. Lancet, 339: 1601.
Shnaps Y & Tirosh M (1981) Iron poisoning [letter]. Am J Dis Child, 135(1): 83.
Siff JE, Meldon SW & Tomassoni AJ (1999) Usefulness of the total iron binding capacity in the evaluation and treatment of acute iron overdose. Ann Emerg Med, 33(1): 73-76.
Sipahi T, Karakurt C, Bakirtas A & Tavil B (2002) Acute iron ingestion. Indian J Pediatr, 69(11): 947-949.
Tenenbein M (1987) Whole bowel irrigation in iron poisoning. J Pediatr, 3: 142-145.
Tenenbein M & Yatscoff RW (1991) The total iron-binding capacity in iron poisoning. Is it useful? Am J Dis Child, 145: 437-439.
Tenenbein M, Kowalski S, Sienko A, Bowden D & Adamson IYR (1992) Pulmonary toxic effects of continuous deferoxamine administration in acute iron poisoning. Lancet, 339: 699-701.
Tran T, Wax JR, Steinfeld JD & Ingardia CJ (1998) Acute intentional iron overdose in pregnancy. Obstet Gynecol, 92:678-680.
Tran T, Wax JR, Philput C, Steinfeld JD & Ingardia CJ (2000) Intentional iron overload in pregnancy – management and outcome. J Emerg Med, 18 (2): 225-228.
Turk J, Aks S, Ampuero F & Hryhorczuk DO (1993) Successful therapy of iron intoxication in pregnancy with intravenous deferoxamine and whole bowel irrigation. Vet Hum Toxicol 35: 441-444.
Van der Wal NA, Smith LL, van Oirschot JF & van Asbeck BS (1992) Effect of iron chelators on paraquat toxicity in rats and alveolar type cells. Am Rev Respir Dis, 145: 180-186.
Vernon DD, Banner W & Dean JM (1989) Hemodynamic effects of experimental iron poisoning. Ann Emerg Med, 18: 863-866.
Walker JA, Sherman RA & Eisinger RP (1985) Thrombocytopenia associated with intravenous desferrioxamine. Am J Kidney Dis, 6: 254-256.
Weiss LG, Danielson BG, Fellstrom B & Wikstrom B (1989) Aluminum removal with hemodialysis, hemofiltration and charcoal hemoperfusion in uremic patients after desferrioxamine infusion. A comparison of efficiency. Nephron, 51(3): 325-329.
Westlin W (1971) Desferrioxamine in the treatment of acute iron poisoning. Clin Toxicol, 4: 597-602.
Whitten CF, Chen YC & Gibson GW (1966) Studies in acute iron poisoning. II. Further observations on desferrioxamine in the treatment of acute experimental iron poisoning. Pediatrics, 38: 102-110.
Whitten CF, Gibson GW, Good MH, Goodwin JF & Brough AJ (1965) Studies in acute iron poisoning. I. Desferrioxamine in the treatment of acute iron poisoning: clinical observations, experimental studies, and theoretical considerations. Pediatrics, 36: 322-335.
Yaqoob M, Ahmad R, McClelland P, Shivakumar KA, Sallomi DF, Fahal IH, Roberts NB & Helliwell T (1993) Resistance to recombinant human erythropoietin due to aluminium overload and its reversal by low dose deferoxamine therapy. Postgrad Med J, 69: 124-128.
Yatscoff RW, Wayne EA & Tenenbein M (1991) An objective criterion for the cessation of deferoxamine therapy in the acutely iron poisoned patient. J Toxicol Clin Toxicol, 29: 1-10.
ENDNOTES:
See Also: Toxicological Abbreviations