
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 141
QUALITY MANAGEMENT FOR CHEMICAL SAFETY TESTING
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1992
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Quality management for chemical safety testing.
(Environmental health criteria ; 141)
1.Hazardous substances - toxicity 2.Laboratories - standards
3.Quality control 4.Toxicology - methods
I.Series
ISBN 92 4 157141 1 (NLM Classification: QV 602)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1992
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
QUALITY MANAGEMENT FOR CHEMICAL SAFETY TESTING
INTRODUCTION
1. GENERAL QUALITY MANAGEMENT APPROACH
FOR QUALITY ASSURANCE
1.1. Organization and personnel
1.1.1. Introduction
1.1.2. Organization
1.1.3. Test facility management
1.1.4. Study director
1.1.5. Support personnel
1.1.6. Quality assurance function
1.1.7. Personnel selection and development
1.1.8. Orientation and training of new personnel
1.2. Quality assurance programme
1.2.1. Introduction
1.2.2. Quality assurance and quality control
1.2.3. Organization and personnel
1.2.4. Inspections and audits
1.2.5. Records and reports
1.2.6. Quality assurance SOPs
1.3. Facilities and equipment
1.3.1. Introduction
1.3.2. Facilities for handling test, control
and reference substances
1.3.3. Field study facilities
1.3.4. Equipment
1.4. Study plan
1.4.1. Introduction
1.4.2. Study plan preparation
1.4.3. Format and content of study plan
1.4.4. Use of study plan
1.5. Standard operating procedures
1.5.1. Introduction
1.5.2. Format and content of SOPs
1.5.3. Preparation of SOPs
1.5.4. Typical SOPs
1.5.5. Use and availability of SOPs
1.5.6. Adequacy of SOPs
1.5.7. Maintenance of SOPs
1.6. Test, control and reference substances
1.6.1. Introduction
1.6.2. Test, control and reference substance
characterization
1.6.3. Handling
1.6.4. Storage
1.6.5. Distribution
1.6.6. Mixtures of substances with vehicles
(carriers)
1.6.7. Stability
1.6.8. Labelling
1.6.9. Facilities and equipment
1.7. Quality control
1.7.1. Introduction
1.7.2. Level of quality control
1.7.3. Pre-analytical quality control
1.7.4. Analytical quality control
1.7.5. Statistical considerations
1.7.6. Analytical performance evaluation
1.8. Documentation and record keeping
1.8.1. Introduction
1.8.2. Manual data records
1.8.3. Computer data records
1.8.4. Indirect computer data records
1.9. Final report
1.9.1. Introduction
1.9.2. Contents
1.9.3. Indexing
1.10. Archiving and retention of data
1.10.1. Introduction
1.10.2. Facilities
1.10.3. Responsibilities for an archive
1.10.4. SOPs for archiving
1.10.5. Receiving, indexing and identification
1.10.6. Filing and storage
1.10.7. Access and security
1.10.8. Retrieval of data and control of access
1.10.9. Retention of information
2. QUALITY MANAGEMENT APPLIED TO TOXICITY
STUDIES
2.1. Introduction
2.2. Procedural requirements
2.3. Phases of animal use
2.4. Obtaining animals
2.5. Shipping and receipt of animals
2.6. Animal care facilities
2.7. Animal husbandry supply facilities
2.8. Facilities for handling test, control and
reference substances
2.9. Pre-study evaluation of animals
2.10. Allocation of animals to a study
2.11. Exposure of animals to a test or control substance
2.12. Control of laboratory environment
2.13. Evaluation of in-life animal responses to test
and control substances
2.14. Removal of animals from a study
2.15. Transfer of animal tissues and specimens to
archives
3. QUALITY MANAGEMENT APPLIED TO HUMAN
AND ENVIRONMENTAL MONITORING STUDIES
3.1. Introduction
3.2. Procedural requirements
3.3. Selection of sampling strategies and study design
3.4. Sampling procedures and documentation
3.5. Handling of samples
3.6. Analytical performance evaluation
3.7. The regression method
3.8. Practical application of the regression method
3.9. Other analytical performance evaluation programmes
3.10. Analytical performance criteria
3.11. Quality control samples
REFERENCES
APPENDIX I
WHO TASK GROUP ON QUALITY MANAGEMENT FOR CHEMICAL SAFETY TESTING
Members
Professor W. Almeida, Department of Preventive Medicine,
State University of Campinas, São Paulo, Brazil
Professor E.A. Bababumni, Department of Biochemistry,
University of Ibadan, Ibadan, Nigeria
Dr A.W. Choudhry, Kenya Medical Research Institute, Nairobi,
Kenya (Chairman)
Dr C. Morris, International Chemical Consultants, Alexandria,
Virginia, USA
Dr M. Ruchirawat, Chulabhorn Research Institute, Bangkok,
Thailand (Vice-Chairman)
Dr A. Strik, National Institute of Public Health and
Environmental Protection, Bilthoven, The Netherlands
(Rapporteur)
Dr K. Kanagalingam, Office of Compliance Monitoring,
United States Environmental Protection Agency,
Washington, DC, USA
Professor M. Vahter, Institute of Environmental Medicine,
Karolinska Institute, Stockholm, Sweden
Dr Z. Xing-Quan, Institute of Environmental Health
Monitoring, Beijing, China
Mr R. Zisa, Office of Compliance Monitoring, United States
Environmental Protection Agency, Washington, DC, USA
Representatives of other organizations
Dr R.F.M. Herber, Coronel Laboratory, University of
Amsterdam, Amsterdam, The Netherlands, representing the
International Union of Pure and Applied Chemistry
(IUPAC)
Professor D. de Wied, Rudolf Magnus Institute of
Pharmacology, University of Utrecht, Utrecht, The
Netherlands, representing the International Union of
Pharmacology (IUPHAR)
Dr H. Könemann, Public Health Inspectorate, Ministry of
Welfare, Health and Cultural Affairs, Rijswijk, The
Netherlands, representing the Organisation for Economic
Co-operation and Development (OECD)
Dr R. Länge, Schering A.G., Berlin, Germany, representing the
European Chemical Industry Ecology and Toxicology Centre
(ECETOC)
Secretariat
Dr E.M. Smith, International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland ( Secretary)
Dr D.T. Mage, Prevention of Environmental Pollution, Division
of Environmental Health, World Health Organization,
Geneva, Switzerland
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are kindly requested to communicate any
errors that may have occurred to the Director of the International
Programme on Chemical Safety, World Health Organization, Geneva,
Switzerland, in order that they may be included in corrigenda.
QUALITY MANAGEMENT FOR CHEMICAL SAFETY TESTING
The WHO Task Group on Quality Management for Chemical Safety
Testing met in Bilthoven, The Netherlands, from 28 May to 1 June 1990.
Dr E. Smith welcomed the participants on behalf of the heads of the
three IPCS cooperating organizations (UNEP/ILO/WHO). The Task Group
reviewed and revised the draft monograph and extended its scope.
The first draft of the part of the monograph dealing with quality
assurance of toxicological studies was prepared by Mr E.A. Brisson (US
Food and Drug Agency, Rockville, Maryland, USA) and the part dealing
with quality control in human health monitoring was prepared by
Professor M. Vahter (Karolinska Institute, Stockholm, Sweden) with
contributions from Professor R. Herber (Coronel Laboratory, University
of Amsterdam, The Netherlands). Additional text on quality assurance
for environmental monitoring was prepared by Dr R. Länge (Schering AG,
Berlin, Germany). Dr H. Könemann (Ministry of Welfare, Health and
Cultural Affairs, Rijswijk, The Netherlands) reviewed and integrated
the text. Finally, Dr Könemann, Professor R. Herber (Coronel
Laboratory, University of Amsterdam, The Netherlands) and Professor M.
Vahter acted as an ad hoc editorial group and gave valuable
assistance in the preparation of the final text. Dr E.M. Smith and Dr
P.G. Jenkins, both members of the IPCS Central Unit, were responsible
for the overall scientific content and technical editing,
respectively, of this monograph.
Support for the meeting was provided by The Netherlands National
Institute of Public Health and Environmental Protection (RIVM).
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
EQC External Quality Control
HEAL Human Exposure Assessment Locations
IQC Internal Quality Control
MAD Maximum Allowable Deviation
MRBIS Mean Running Bias Index Score
MRVIS Mean Running Variance Index Score
NEQUAS National External Quality Assessment Scheme (UK)
QA Quality Assurance
QAP Quality Assurance Programme
QC Quality Control
QCP Quality Control Programme
SOP Standard Operating Procedure
INTRODUCTION
Chemical safety is a world priority. Considerable effort is being
devoted by governments and industries to ensure that the manufacture
and use of chemicals will not have an adverse effect on human health
or the environment. Many governments have introduced laws,
regulations, and guidelines designed to prevent human health risks and
environmental degradation.
Concerns about the potential of chemicals to have adverse effects
on human health and the natural environment has led the World Health
Organization (WHO), together with the International Labour
Organisation (ILO) and the United Nations Environment Programme
(UNEP), to cooperate actively in the International Programme on
Chemical Safety (IPCS). The objectives of the IPCS include the
evaluation of the effects of chemicals on human health and the
environment and the development of methodology and testing methods in
order to produce internationally comparable results. The importance of
the quality of data in achieving these objectives is self-evident.
The need for quality of data generated in laboratory and field
toxicological studies is parallelled by the need for quality of
analytical data on tissue concentrations, environmental exposure, and
exposure surveillance and monitoring studies. It is important to
realize that chemical risk assessment utilizes not only data from
studies carried out for regulatory purposes, such as notification, but
also data from many types of research studies, both pure and applied.
In the process of risk assessment, biological dose-effect and
dose-response data are integrated with analytical data. Thus, overall
quality management of data generation and application of quality
assurance and quality control are crucial. Data quality is inherent
in the IPCS evaluations of the risk to human health and the
environment of chemicals, published as Environmental Health Criteria
(EHC) monographs. The relevance of quality assurance and quality
control for the generation of sound data is a key element in the
monographs dealing with principles and methods for the evaluation of
toxicity, e.g., the monograph on Principles of Toxicokinetic Studies
(WHO, 1986a).
The validity and usefulness of the results from experimental
studies, whether these relate to basic research or tests carried out
to meet regulatory requirements, are critically dependent on the way
in which they are designed, managed and performed. In risk
assessment, the quantity and quality of data are both important.
Limited or inadequate data, even where there are no doubts on their
quality, cannot result in a balanced evaluation, and the overall
conclusions on risks to human health and the environment are
inevitably limited. An extensive data base that is of poor quality
also gives rise to poor assessments and, probably, erroneous
conclusions.
The quality of data is considered to be an objective matter. Data
should be meaningful and reliable for use in the assessment of the
safety of chemicals. Of course, quality of data depends to a large
extent on the quality of the scientists and other individuals involved
in the production of these data, but many aspects of quality can be
verified, measured or assessed objectively, and therefore, also
improved systematically.
Promoting quality is a management responsibility. Quality
management is a broad approach, using all possible tools to carry this
responsibility. Studies are complex activities involving people,
facilities, equipment, test systems, chemicals and materials, often
over a considerable period of time. Therefore activities must be
carefully coordinated so that specific events occur when scheduled and
in the way intended.
In the field of safety testing of chemicals and preparations,
quality management was introduced in the 1970s after the discovery of
some cases of fraudulent tests and increasing concern over the
careless way in which many tests supporting the registration of drugs
were being carried out. The United States Food and Drug
Administration responded to these concerns by developing regulations
on Good Laboratory Practices (Lepore, 1979; FDA, 1987a,b). The
philosophy and many of the requirements were based on the quality
management systems developed for industrial production, such as that
of military equipment. Experience had already been gained in applying
similar principles, e.g., the application of Good Manufacturing
Practices to the production of pharmaceutical products.
The International Organization for Standardization in ISO 8402
(ISO, 1986a) defines quality as "the totality of features and
characteristics of a product or service that bear on its ability to
satisfy stated or implied needs", quality management as "that aspect
of the overall management function that determines and implements the
quality policy", and quality policy as the "overall quality intentions
and direction of an organization as regards quality, as formally
expressed by top management". Quality management focuses on the
organizational process. It is directly relevant to the conditions
under which laboratory studies are planned, performed, monitored,
recorded, reported and archived. Quality management principles have
been laid down in national legislation and in documents from
international organizations. Major examples are the Principles of
Good Laboratory Practice produced by the Organisation for Economic
Co-operation and Development (OECD, 1982), publications of the
International Standardization Organization, such as ISO 8402 (ISO,
1986a) and ISO 9000 and 9004 (ISO, 1987a,b) and quality assurance
principles for analytical laboratories of the Association of Official
Analytical Chemists (AOAC, 1984). The philosophy followed in these
various documents is broadly similar and has served as guidance for
developing this monograph.
A prerequisite for producing data of good quality is the
availability of adequate facilities, equipment, and personnel who are
both well educated and well trained. Studies need to be adequately
designed and planned. Routine laboratory procedures need to be well
defined, so that they can be carried out in the best possible way and
in a consistent and reproducible manner. Other key elements in
performing and reporting studies are the test, control and reference
substances, the documentation and record keeping, the final report
and, lastly, the archiving and retention of data. This monograph
provides guidance for these activities.
An important feature of quality is that it should be verifiable
and indeed be verified. For that purpose the quality assurance
approach has been developed (Burck, 1979). Quality assurance includes
independent study monitoring that assures laboratory management and
users of data that facilities, equipment, personnel, methods,
practices, records and controls conform to accepted quality management
principles. An effective quality assurance system provides confidence
that a study report meets the pre-established quality standards of
accuracy, integrity, completeness and clarity. Quality assurance
should be integrated within the entire study process to ensure that
the results are valid and that the final report accurately reflects
these results. This monograph therefore deals with the organization
of a quality assurance programme.
An important quality assurance tool is known as quality control.
Quality control is the quality assessment of quantitative, routine
laboratory determinations. Many techniques are used for this purpose.
Quality control plays a major role in monitoring studies, but in
addition, it is frequently applied in toxicity studies, for instance,
to assess the quality of biochemical and haematological data.
The application of modern quality management approaches to the
two fields of safety assessment of chemicals is crucial. These fields
are a) quality management of laboratory toxicity studies and b)
studies to monitor the extent of exposure and effects and the presence
of chemicals in man and the environment.
Quality has its price. Considerable investment, especially in
terms of human resources, has to be made in a laboratory before a
quality management system is operational. Furthermore, it requires
continued effort to keep the system functioning. On the other hand,
quality assurance is a major help for management in organizing the
work, planning it well, ensuring continuity and increasing
productivity. In addition, an efficient quality assurance system
reduces considerably the risk that erroneous data will lead to
non-acceptance of studies and rejection of reports or the need to
repeat studies.
The following guidance is not directly based on legal precepts
but follows the same philosophy, and it describes quality assurance
considerations involved in the production of reproducible and reliable
test data. It addresses basic elements of a quality management
programme for chemical safety testing with the aim of promoting the
quality assurance concept in test facilities throughout the world and
of facilitating the global sharing of useful information on chemicals.
It is not intended to deal with the details of quality management of
toxicity and ecotoxicity testing and chemical analysis, but chapter 2
describes quality assurance as applied to biological testing in some
detail because this is an area that is not familiar to all scientists
working in this field. Even though certain countries may not have
extensive testing facilities, the national and international
acceptability of their research data can be greatly enhanced by
utilizing these basic quality assurance and management principles. In
addition they can apply the principles of quality assurance and
quality control to assure the quality and acceptability of safety data
presented to them for risk assessment purposes.
1. GENERAL QUALITY MANAGEMENT APPROACH FOR QUALITY ASSURANCE
1.1 Organization and personnel
1.1.1 Introduction
To ensure that studies are valid and properly conducted, an
organizational structure should be established that will best serve
the needs of the testing facility and will employ an adequate number
of personnel that are well qualified by education, training and/or
experience to perform their assigned tasks.
1.1.2 Organization
Regardless of the specific organizational structure, facility
personnel include test facility management, study directors, quality
assurance programme personnel and support personnel. The
responsibilities for successfully running a laboratory are shared
among these groups.
The typical composition and responsibilities of each of these
groups are discussed below.
1.1.3 Test facility management
In general, duties that are more administrative than scientific
are the direct responsibility of management. The primary duty of
management is to ensure that all studies are properly planned,
conducted and reported, and that sufficient staff and resources are
made available for the successful conduct of studies. Before any
studies are started, management must make various administrative
decisions concerning the nature of the studies that will be conducted.
Management must decide which controls will be in place to ensure the
successful conduct and completion of studies, and decide the
organizational coordination within the test facility to carry out the
studies. It must establish a quality assurance programme that assures
compliance with the study plan test facility procedures, and, where
necessary, government regulations.
Properly trained personnel are crucial to the conduct of a
quality study. It is management's responsibility to see that staff,
both professional and technical, receive adequate training to ensure
that they are competent to carry out their duties. Management must
make available to all personnel the opportunity for external and
on-the-job training to maintain and improve necessary laboratory
skills.
Management must implement organizational and personnel directing
and coordinating mechanisms that will adequately serve the facility.
It must also be certain that an adequate number of qualified employees
are available who clearly understand the functions they are to
perform, as well as any health and safety precautions that need to be
taken during the conduct of a study.
In addition, management must apply proper managerial skills in
dealing with the staff in order to promote a high level of job
performance. Positive feedback to the personnel for recognition of
good work performance should be practised continually by management.
All personnel should view their work as a positive contribution to a
high quality end product, the final study report.
The most fundamental decisions of management concern the types
and numbers of studies that will be conducted at the testing facility.
Taking into account the experience of personnel both within the test
facility and outside, such as consultants, management must take
decisions on a number of aspects, e.g., what type of test systems will
be maintained, whether studies will be of a short-term or long-term
nature or a combination of both, whether other external test
facilities will be required to conduct certain portions of a study,
and how many studies can be properly handled considering the
personnel, equipment and facilities available in the test facility.
Management must establish procedural controls that will assure
that all study activities will be properly conducted. Typically,
these include the requirement that written Standard Operating
Procedures (SOPs) exist for all routine functions conducted within the
test facility or by other external facilities, and that the test
facility management is kept informed of study progress or set-backs.
The designation of job responsibilities and the assignment of
these responsibilities to the various entities within the organization
should be documented. Management must appoint a study director to be
responsible for overall study management and the single point of study
control. A qualified person must be designated to assume the study
director's responsibilities on a temporary basis in his absence. If
a study director needs to be replaced on a permanent basis, this
should be done promptly.
Management delegates much of its responsibilities and authorities
to the appropriate divisions and personnel within the organization.
Each division must be held accountable to laboratory management to
accomplish its assigned tasks. This requires open communication
between the various organizational personnel and laboratory
management.
1.1.4 Study director
Experience has shown that unless responsibility for the proper
conduct of a study is assigned to one person, there is a potential for
personnel to receive conflicting instructions, which can result in a
poor study. Therefore, it is recommended practice, before a study is
initiated, for management to designate one individual who will serve
as study director and be considered the chief scientist in charge of
a study. It is essential for the study director to have a strong
scientific background, as well as proven managerial abilities,
strengths in communication and problem solving, and the ability to
organize the day-to-day and long-term objectives of a study.
Specific responsibilities of a study director include assuring
that:
* the study plan is agreed;
* the procedures specified in a study plan are followed by
personnel engaged in the conduct of that study;
* all revisions to a study plan are brought to the attention of
appropriate personnel;
* test systems are appropriate for the study;
* personnel involved in the study clearly understand their
functions and are qualified to perform them;
* data are accurately and promptly recorded and verified;
* health hazards associated with the test system are recognized and
controlled, including hazards to personnel as well as to the
integrity and quality of the test system;
* studies are conducted in a manner that is safe for personnel;
* all raw data, samples and specimens are archived promptly at
the completion of the study, as appropriate;
* the reported results of the study accurately reflect the raw
data;
* unforeseen circumstances that may affect the quality and
integrity of a study are noted, corrected and documented.
The study director must use the advice, education, experience and
assistance of other scientists participating in the study.
Concerning the study director's workload, both the types and
lengths of the studies have a significant impact on the number of
studies for which a single study director may be responsible. The
maximum number of studies assigned to one study director should be
very carefully assessed by management.
1.1.5 Support personnel
A variety of specialists is required to conduct a study
adequately and to provide the administrative support to ensure its
proper conduct. In addition to clerical and administrative support to
staff, each study requires the participation of such specialists as
toxicologists, biochemists, clinical pathologists, veterinarians,
chemists, histologists, statisticians, equipment and maintenance
specialists, computer specialists, and animal caretakers. The
personnel needs of each facility will be guided by the particular
studies to be conducted.
1.1.6 Quality assurance function
In many laboratories the quality assurance function is run by
quality assurance programme (QAP) staff carrying out quality assurance
duties on a full-time basis. The primary functions of quality
assurance personnel are to conduct facility inspections and data
audits. Through inspections they ensure, in general, that studies are
conducted in accordance with the quality assurance principles; and in
particular, that the study plans and SOPs are followed. Through their
data audit function they assure that the final reports of studies are
verifiable from the raw data, and that these data were collected,
recorded and maintained according the quality assurance principles.
QAP staff or those exercising a quality assurance function should
have a scientific or technical background. They should be well versed
in:
* basic management skills;
* communication skills;
* understanding of intra-organizational relationships and
functions;
* good record keeping;
* negotiation skills.
Generally, quality assurance personnel are drawn from experienced
study personnel and are qualified for their quality assurance
responsibilities through formal training courses and on-the-job
training exercises. It is often helpful for the test facility to
structure the on-the-job training so that personnel are certified
proficient as they successfully accomplish each duty listed in the job
description. For example, when laboratory staff demonstrate that they
can review a study plan for adherence to acceptable laboratory
practices, this would be indicated in their personal records.
1.1.7 Personnel selection and development
All personnel engaged in the conduct of a study should have
sufficient education, experience and training to perform their
assigned duties. The careful and systematic recruitment and
employment of laboratory personnel should be considered an important
management function. The job requirements should be delineated in
written job descriptions established for each position available at
the laboratory. Job descriptions should specify the duties and
responsibilities, the reporting relationships (i.e. to whom the
employee should routinely report the study status and unexpected
emergencies) and the reporting requirements expected from the
employee.
1.1.8 Orientation and training of new personnel
The need for adherence to the principles of quality assurance and
to safety procedures, personal sanitation, and clothing and health
restrictions should be discussed with each new employee. Copies of
the SOPs defining the laboratory's personnel health and safety
regulations and copies of relevant SOPs for their areas of
responsibility should also be provided to employees. Before beginning
any work, employees should be instructed by their immediate supervisor
on their particular job duties, responsibilities and working
conditions. Employees should be required to review SOPs and quality
assurance manuals pertinent to their specific jobs.
This orientation process is followed by a period of on-the-job
training under careful and direct supervision. The amount of training
required for each function will be determined by the nature of the job
function. When a supervisor believes that an employee can
proficiently perform a specific job function, the employee should be
authorized to perform such functions independently. Documentation of
this proficiency and authorization should be maintained by the
appropriate department (e.g., personnel department and/or division
where the employee is directly employed). Supervisors should hold
additional training sessions as often as needed to emphasize a
procedure or explain a change in a procedure. These sessions should
also be appropriately documented.
The laboratory should have documentation that explains how
employees should be oriented and trained for their new positions, as
well as how their training will be documented.
1.2 Quality assurance programme
1.2.1 Introduction
All testing facilities (e.g., laboratories, field operations),
regardless of size, that generate data for assessing the impact of
chemicals (e.g., industrial chemicals, pesticides or pharmaceuticals)
on human health or the environment should have an efficient management
system. The establishment of an independent quality assurance
programme (QAP) is an essential mechanism for accomplishing these
goals.
A QAP does not necessarily require a separate organizational
entity consisting of personnel permanently assigned to this task.
However, it is critical that an organizational separation exist
between quality assurance inspection and study personnel if a
laboratory or field operation is to expect an impartial analysis of
the accomplishments and operations encountered in the conduct of a
study.
1.2.2 Quality assurance and quality control
It is important, at the outset, to distinguish between the
related concepts of quality assurance and quality control. Quality
control is a valuable quality assurance tool. Assessing study quality
periodically is an essential aspect of quality assurance. Thus, the
quality of an analytical measurement may be validated, for example, by
comparing analytical results against a known standard taking into
consideration the sensitivity, accuracy, precision, calibration and
maintenance of the analytical equipment. These measurements would be
part of a quality control system.
1.2.3 Organization and personnel
Management assures adherence to the principles of good laboratory
management practices, authorizes study plans and standard operating
procedures, and corrects operational defects that are reported by the
quality assurance programme staff. A quality assurance programme must
be established by management so that impartial inspections can be
conducted and observed defects can be corrected promptly. Most
laboratories and field operations generating data for regulatory
purposes have a quality assurance programme unit as a distinct
organizational entity reporting to management. If scientists carrying
out testing are also to conduct quality assurance inspections and
audits, they should have no scientific or personal involvement in the
studies being quality assured and must be impartial.
It is important for a quality assurance programme to be
implemented by highly motivated, qualified, and trained individuals
who possess sufficient knowledge of experimental procedures to permit
an adequate impartial assessment.
1.2.4 Inspections and audits
General inspection of facilities and critical activities and
auditing final reports are very important tasks in a quality assurance
programme. The purpose of inspection is to verify that the study is
being conducted in accordance with the study plan, the SOPs (see
chapter 4) and the applicable principles of good laboratory management
practices. The goal is to detect and correct systematic or
unintentional flaws in the study process before the flaws have an
adverse impact on the quality of the study. It is very important to
assure that a study is conducted as directed by the study plan.
Auditing has two purposes. The first is to confirm that the results
presented in the final report accurately reflect the raw data that
were collected during the study and the second is to highlight any
circumstances that would adversely affect the study. SOPs should be
developed to cover procedures of study conduct and for the audit of
the final report. The QAP must compare these audit findings against
the study plan and the supporting raw data to ensure that the report
accurately reflects the results of the study in accordance with the
study plan. The SOP should also indicate which raw data are to be
reviewed during the audit and how they are to be reviewed.
Prior to initiating an inspection of the facilities and
equipment, the designated quality assurance person should be familiar
with the experimental operation to be inspected. This requires a
review of the study plan and the SOPs that relate to the laboratory or
field operations. Personnel files and equipment logs should be
reviewed as well as the report of the previous inspection of the same
operation. These inspections should be conducted in such a manner as
to minimize any disruption of normal operations. The quality
assurance person should explain the purpose of the inspection to
laboratory or field staff. Any deviations should immediately be made
known to the responsible personnel. The kinds of items to be examined
include reagent preparation and labelling, personnel files, presence
of authorized SOPs, equipment logs, recorded data entries,
environmental controls, storage for specimens and test substances, and
general cleanliness and orderliness of the laboratory or field
operation areas.
Quality assurance staff may interview laboratory or field
personnel whenever information on a study-related process is needed.
Most importantly, quality assurance staff must witness the actual
operations to assure that they are being conducted in accordance with
the study plan and SOPs. It is not possible to estimate how much time
should be spent in observing each operation, because this depends on
the complexity of the operation. In any event, enough of the
operation should be observed to conclude that the operations are in
control and are being conducted in accordance with the SOPs.
Large amounts of data are often collected during studies, which
are summarized in the final report. It is important, therefore, to
assure that the summarized data in the final reports are accurate and
complete. When large amounts of raw data are to be audited, several
approaches can be used to select data points to evaluate. One
approach is a pragmatic sampling plan which is based on the experience
of the quality assurance staff with certain operational units. For
example, activities associated with receipt and handling of the test
or reference substances generally result in very few recording errors,
and it may be necessary to sample only 10-20% of the recorded
information. In contrast, the recording of test system observation
may frequently be a problem; 75-100% of these observations should be
audited and the activity inspected on a regular basis. Another
approach used is a random sampling plan. These plans are developed
with the aid of statisticians and they provide a statistical
confidence level for verifying the data in the final report. In
accordance with these plans, a certain fraction of the data is
examined, and if this fraction is found to be accurate, it is assumed
that the whole report is accurate. If the fraction examined is shown
to contain errors, then a larger number of data entries is examined,
and so on. One should take into account, however, that errors may not
occur at random and that statistical sampling plans may give
misleading results. Auditing final reports occurs prior to the
archiving of the study records. The quality assurance auditor
assembles the raw data and all other study records for the study to be
audited. All of these records are read for completeness and those
that contain raw data are checked against the data in the final report
in accordance with a random sampling plan. It is important to locate
missing records and it is equally important to confirm the presence of
all specimens and reserve samples of test and reference substances.
All errors in the final report should be brought to the attention of
the study director for resolution.
1.2.5 Records and reports
As with other activities in the laboratory or field operations,
it is important that timely and accurate reports on quality assurance
inspections are made and careful records kept.
There are three kinds of reports:
a) A routine, periodic report to management and to the study
directors of all quality assurance work that was conducted
during a defined period (e.g., one month). This report
should give an overall summary of the inspections and audits
that were conducted. It should highlight emerging problem
areas, if any, and should forecast projected quality
assurance activities.
b) A report made to the study director and to management
whenever significant problems are found that can have an
adverse effect on a study's quality. The report should
describe completely the observed deficiencies.
c) The report that accompanies the final study report. This
report lists the dates and phases of inspection conducted in
the course of the study, and the dates when the inspectional
findings were reported to management.
It is necessary for a quality assurance programme to keep copies
of the following records: the master schedule sheet of all studies
being conducted by the facility; study plans; periodic reports to
management; inspection and audit reports; an index of quality
assurance records and a compendium of all the test facility SOPs; and
quality assurance programme SOPs.
1.2.6 Quality assurance standard operating procedures (SOPs)
In order to accomplish its purpose, a quality assurance programme
must continually review study records for ongoing and completed
studies, and inspect ongoing studies and all related facilities and
equipment. This requires a number of SOPs specifically for quality
assurance in order to describe the methods for accomplishing these
tasks.
The content of the SOPs will vary depending on the organization,
but generally the following areas should be covered: structure and
organization of the quality assurance programme; to whom and how the
quality assurance staff report findings; procedures for study
inspection; procedures for auditing the final report; scheduling of
inspection and audits; and procedures for indexing and maintaining
records.
An SOP describes the organization of the quality assurance
programme and how it relates to the organizational structure at the
testing facility. This is important for a number of reasons. It
should include the following issues. Firstly, a quality assurance
programme must provide reports directly to management to be effective.
The SOP should describe the reporting procedure. It should also
address the internal organization of a quality assurance programme.
Secondly, a quality assurance programme must be composed of an
adequate number of trained individuals to carry out its
responsibilities, and the required qualifications for each position
must be known. The SOP should include procedures for training
personnel so that the personnel meet the stated qualifications.
The SOP needs to include how often reports are made and the
format in which the reports are to be prepared. Internal reporting
within the quality assurance programme should be in writing. There
are two important aspects of reporting that should be addressed in the
SOP: the manner in which reported deficiencies are corrected, and the
manner in which any corrections are to be documented.
The SOP covers procedures for the in-process inspection of
critical laboratory operations and their scheduling. It is necessary
for the quality assurance personnel to visit the various areas
regularly and to observe ongoing study-related activities to assure
compliance with the study plan and relevant SOPs.
1.3 Facilities and equipment
1.3.1 Introduction
A test site must have suitable facilities and equipment to ensure
the proper conduct of studies. These must be of adequate design,
construction, capacity and location for their intended purpose. In
addition, they must be properly used, maintained, and cleaned.
Equipment used for generation, measurement or assessment of data must
be calibrated and/or standardized with maintenance of appropriate
records.
1.3.2 Facilities for handling test, control and reference substances
The facilities must allow separation of areas involved in the
storage, handling and distribution of test, control and reference
substances. This is necessary to prevent contamination of facilities,
equipment, personnel and test systems, as well as to prevent confusion
of substances. To accomplish this, there should be separate areas
for:
a) receipt and storage of test, control and reference substances;
b) mixing of test, control and reference substances with a carrier
(e.g., feed);
c) storage of the test, control and reference substance mixtures.
In addition, storage areas for the test, control and reference
substances should be separate from areas housing the test systems.
The storage facilities should be adequate to preserve the stability of
the test, control and reference substances. For example, adequate
refrigerator or freezer storage space should be available for test,
control and reference substances requiring low temperature storage.
There needs to be separate space for the performance of the
routine and specialized procedures required by study plans. This
would include specialized areas for performing activities such as
necropsy, histology, X-rays, and analytical and clinical chemistry.
The extent of space needed and the degree of separation required
will vary widely depending upon the nature and amount of work being
conducted. Larger laboratories may require a complete array of
facilities capable of carrying out every phase of a study, whereas
smaller laboratories may hire contract testing laboratories to perform
some phases of the study that they do not routinely perform, such as
histopathological slide preparation, clinical chemistry tests, or
analytical chemistry for test substance characterization. In either
case, laboratory operations must be performed under conditions that
provide an adequate degree of separation to preclude confusion and
interference between areas performing different aspects of the study.
1.3.3 Field study facilities
Adequate facilities should be available for the receipt, handling
and storage of chemicals, such as herbicides, pesticides, and
fertilizers, necessary for the conduct of good agricultural practices.
These areas should be separated from the areas containing the test,
control and reference substances. Adequate space should also be
available for processing soil and plant residue samples and there
should be adequate refrigerator and freezer space for interim storage
before shipment to analytical residue laboratories.
1.3.4 Equipment
Along with adequate buildings, a test facility or field operation
must have equipment of appropriate design and adequate capacity to
function according to the study plan. This applies especially to
equipment used in the generation, application, measurement or
assessment of data. It also includes equipment used for laboratory
environmental control, such as refrigerators, freezers, and air
conditioning for test system areas and test substance preparation and
storage areas. All equipment should be suitably located for ease of
operation, inspection, cleaning, maintenance and safety.
Proper maintenance and calibration of equipment is a fundamental
quality assurance management practice. Equipment should be inspected,
cleaned and maintained. In addition, equipment used for the
generation, application, measurement or assessment of data should be
tested, calibrated and/or standardized.
A testing facility should have SOPs that describe in sufficient
detail the methods, materials and schedules to be used in the routine
operation, inspection, cleaning, maintenance, testing, calibration
and/or standardization of equipment, and there should be appropriate
documentation. These procedures should specify remedial action to be
taken in the event of failure or malfunction and should designate the
person responsible for performing each operation. Copies of these
SOPs should be readily available to personnel in the vicinity of the
equipment. In order to assure that all equipment associated with a
study was operating properly at the time of its use, SOPs must be
followed and records maintained for all equipment inspection, testing,
calibrating, standardizing and maintenance. The records should
contain the date the operation was performed and, in the case of
maintenance operation or repairs, the type of function or malfunction,
how and when it was observed, and, as appropriate, any remedial action
taken.
1.4 Study plan
1.4.1 Introduction
A clearly written, comprehensive study plan is an essential
element of all chemical safety studies. A study plan should state the
objectives, schedules and all methods for the conduct of a study. The
plan, in combination with the SOPs, should provide the complete
specifications and instructions for the execution of the study.
There is a practical difference between the role of the SOPs and
the role of the study plan. The SOPs describe how specific routine
operations are to be conducted. The study plan contains the design
specifications for a single study or a group of related studies. Its
role is to describe what the study objectives are and what methods are
to be employed to achieve the stated objectives. In short, the plan
describes what is to be done and when, while the SOPs address how
specific study events are to be accomplished.
As the design specification for a study, the plan has an
important quality assurance function: it serves as the reference for
measuring study performance. If the plan of a well-designed study is
followed, the study objectives should be achieved. However, if the
plan is inadequate or is not followed, the intended objectives of the
study may not be met. In this case, the study plan cannot serve as a
suitable reference against which the quality of the study can be
assessed.
The study plan is a useful management tool; it helps the long-
term planning of, for instance, workload, manpower implications and
the necessary facilities and equipment.
Test facilities should have SOPs covering the preparation and
approval of study plans to help assure an orderly, complete and
consistent review process. This is especially important in the
preparation of the study plan, because errors or omissions can
jeopardize the entire study.
Generally, SOPs for study plans should address the persons who
will be responsible for preparation and review, how the preparation
process begins and ends, what elements of format and content should be
included in the plan and to whom it is to be distributed.
1.4.2 Study plan preparation
The study director has the primary responsibility for preparing
the study plan and is generally responsible for designing the plan
scientifically to assure that the study will achieve its intended
purpose. Senior scientists who will participate in various aspects of
the study should be involved in the preparation and/or review of the
plan. This may include writing portions of the plan that involve heir
area of expertise. For example, the pathologist for the study may
prepare the pathology section of the plan. Involvement of other
scientists may include reviewing what the study director has prepared,
correcting errors and omissions, and commenting on its adequacy.
Technical personnel should be involved in plan preparation to ensure
that the functions specified in the plan can be performed.
Management is also involved in the preparation and review of the
study plan since it is its responsibility to assure that personnel,
resources, facilities, equipment, materials and methodologies are
available as scheduled in the plan. It is also useful for the study
plan to be reviewed from the quality assurance perspective to assure
that it contains the basic elements required in the applicable SOP.
For studies conducted under contract, the sponsor of a study
should also be involved in the preparation and review of the study
plan and must approve it prior to the initiation of the study. This
is necessary to permit a clear understanding between the study
director and the sponsor of the requirements and objectives of the
study.
1.4.3 Format and content of study plan
The specific contents of each study plan will vary according to
the objectives of a given study and the particular methods and
procedures that will be employed. However, there are basic elements
common to most chemical safety testing studies that should be included
in the study plan:
a) A descriptive title and a statement of the purpose of the study.
Stating the title and purpose of the study in the study plan
clearly establishes the study objectives, and the study
performance can later be assessed to ensure that the stated
objectives were achieved.
b) The identification of the test, reference and control substances
by name, chemical abstract number or code number. If code
numbers are used to identify the test substance, such numbers
should be unique to this substance.
c) The test system, i.e. any human, animal, plant or microbial
system, as well as other cellular, subcellular, chemical or
physical systems, or a combination thereof, which is used for the
test or control substance. The selection of the appropriate test
system is an important feature of study design, and should be
described fully. The failure to do so could result in the
inadvertent use of a test system other than that intended by the
study director or the sponsor.
d) The specific procedure to be used for the identification of
components of the test system. This most commonly refers to the
method of identifying test animals within their respective test
groups. Proper identification of the test system is necessary to
prevent confusion of animals, samples and/or specimens.
e) A description of the experimental design, including the methods
for the control of bias.
f) The type and frequency of tests, analyses, and measurements to be
made. The plan should specify which parameters are to be
measured and how often such determinations are to be made.
g) The records to be maintained. Because written records and
electronically captured data are the tangible results of any
study, the study plan should identify which records are to be
kept. Many test facilities have developed SOPs that describe the
identity, use, and retention of study-related records. In
identifying records to be maintained, reference to the SOPs is
appropriate.
h) A statement of the proposed plan for choosing the appropriate
methods for statistical analyses of the data set. Although the
study plan may assume that a particular test is applicable, a
statistical test may show that it is not appropriate and that
another test (e.g. nonparametric) must be chosen.
The above elements are not the only items that can be addressed
in the study plan. For example, in the case where a study is modelled
on an established procedure published in the scientific literature,
the study plan may include literature references. The study plan may
also include such items as the proposed starting and completion dates
of the study and the name and address of the testing facility.
The study plan should be written in clear, concise language with
adequate reference to the SOPs involved in the application of the test
methods described. The pages should be numbered consecutively and the
total number of pages should also be given. The body of the plan may
be divided into clearly identified sections for ease of reference.
1.4.4 Use of study plan
A study plan must be used properly to achieve valid study
results. Adherence to the study plan requirements, documenting
adherence, and controlling and documenting any deviations or changes
to the study should be monitored by the study director.
Although all personnel associated with the study refer to the
study plan, the individuals most involved on a daily basis are the
personnel performing study operations and the study director.
Personnel use the plan to guide the performance of their duties. The
study director uses it as a means of informing study personnel of the
study requirements. This individual has the overall responsibility
for assuring that the personnel are aware of the current study plan
requirements and that the study is carried out as directed by the
study plan.
Once the study has started, a quality assurance programme also
uses the study plan, e.g., to determine that no deviations from
approved plans or SOPs are made without proper authorization and to
audit the final report. To accomplish this, a quality assurance
programme must maintain a copy of all study plans and current
amendments. Quality assurance staff use these to compare actual
practices against those specified in the plan for a given study. The
plan often contains a schedule of study events that is useful to a
quality assurance programme in scheduling its inspection of a study.
In this regard, the quality assurance unit must verify that raw data
are available to document that the approved study plan was followed.
To be used effectively, the study plan should be available,
familiar to personnel and in a convenient format. However, the
copying and distribution of the study plan should be controlled to
prevent unauthorized copying and distribution. A copy of the study
plan should be located in the immediate area of the test facility
where a study event is being performed.
A final requirement for the effective use of the study plan is
that it must be current. Although a well-designed and well-prepared
plan should require little change after the study has begun, changes
will almost always be required as the study progresses. The reasons
for the planned and unplanned changes are many. For example, after
the study has begun, an unusual or unexpected toxic effect may be
observed that requires additional tests or changes in the frequency of
observations. The study plan should be updated to reflect these
changes in the form of appendices. The study director must approve,
by means of a dated signature, the change or revision and document the
reason for the change. In addition to dating, it is also desirable to
number the amendments sequentially so that the most recent amendments
can be quickly identified and study personnel can be sure they have
received all amendments. Also, if study plan amendments are
themselves revised in subsequent amendments, the current amendment
should reference the superseded amendment.
Complete records are important for documenting adherence to the
study plan. These include proper recording of study data and of
records related to study performance. When the plan directs the
performance of a study event, records should be kept to document that
the study plan was followed.
1.5 Standard operating procedures
1.5.1 Introduction
Quality assurance involves the development and use of standard
operating procedures (SOPs). SOPs are written procedures which
describe how to perform certain routine laboratory tests or
activities, normally not specified in detail in study plans or test
guidelines. SOPs set forth in detail how specific routine laboratory
operations are to be carried out and complement the study plan by
describing all routine study methods. SOPs are not described in
detail in this monograph. There are no universal SOPs, although those
detailing the same operation will have many close similarities from
laboratory to laboratory. The act of developing SOPs, taking account
of the operation and how its conduct is directly influenced by
laboratory organization and structure, is a key function of laboratory
management.
The importance of effective SOPs in conducting a study must be
emphasized. SOPs are management directives designed to ensure that
all personnel associated with a laboratory study will be familiar with
and use the same procedures. If different individuals perform
important study functions, such as dosing animals, preparing
solutions, archiving documents or receiving test articles, these
operations should be performed in the same manner. By standardizing
procedures for the conduct of studies, SOPs have a valuable quality
assurance function. They prevent the introduction of possible errors
in the generation, collection and reporting of data.
The development of SOPs includes the following aspects: who
should prepare, review and authorize SOPs; which laboratory operations
require SOPs; and what information the SOPs should contain. The
nature of the laboratory work being done and the training and
experience of the laboratory personnel at a particular facility will
determine exactly how extensive the content needs to be.
1.5.2 Format and content of standard operating procedures
SOPs should be easy to use. They should be identified by a
descriptive title and a number. The individual pages of an SOP should
be numbered, and each page should note the total number of pages
contained in each individual procedure. This practice is especially
valuable where SOPs are loosely bound, because it enables individuals
using the procedure to assure themselves that pages are not missing.
In addition to the descriptive title an SOP should have a short
statement of its specific purpose or objective. This should be
presented under a separate heading at the very beginning of the SOP.
A section listing cross-references to related SOPs or other literature
is another format element. For example, an SOP for the operation of
a piece of laboratory equipment should reference maintenance and use
of the equipment on the basis of the manufacturer's literature. An
SOP for determination of animal body weights or organ weights may
reference the SOP covering calibration and use of the scales to
measure the weights. The appropriate use of cross-references can
reduce the SOPs volume by eliminating redundancy and can assure that
users are aware of all other important procedures related to the
operation that they are performing.
SOPs should have a section listing all materials and equipment
required for performance of the indicated operation. This is
especially true for highly technical laboratory procedures where the
unavailability of required equipment or omission of required materials
may result in the improper performance of the procedure.
The exact content of SOPs will vary accordingly to the specific
procedures with which they are concerned. SOPs concerning the use of
laboratory equipment should describe the methods, materials and
schedules to be used for the routine inspection, cleaning,
maintenance, testing, calibration and/or standardization, and the way
to document these operations. They should also specify remedial
action to be taken in the event of failure or malfunction, and should
designate who is responsible for the performance of each of the
operations mentioned above. Depending upon the complexity of the
equipment, the SOP should contain sufficient detail to permit trained
laboratory personnel to operate the instrument. For example, the use
of equipment logbooks or forms should be described completely. SOPs
should provide procedures for handling emergency situations, such as
the spillage of chemicals or fires.
1.5.3 Preparation of standard operating procedures
Laboratory management is primarily responsible for the
development of adequate SOPs.
Scientists, laboratory technicians and quality assurance staff
play a significant role in the preparation and review of SOPs. They
should be written by scientists and laboratory personnel who actually
perform the studies and should be approved by management. SOPs that
are prepared by a single department should be reviewed centrally by
someone outside that department. This will assure that newly prepared
SOPs are not in conflict with existing SOPs of other departments.
Central review may be performed by a quality assurance programme or by
representatives from each of the departments in the laboratory (i.e.
a standard operating procedures review committee).
1.5.4 Typical standard operating procedures
It is important to identify which specific routine operations
require SOPs. Although specific examples of operations requiring SOPs
exist, there is no single list that includes all operations that
require these procedures. Different laboratories will require the
existence of different SOPs depending upon such considerations as the
nature of the work, the kinds of equipment and facilities, and the
qualifications and training of personnel. Laboratory management
should decide which SOPs are required to assure the quality of the
laboratory's work. At any rate, there should be SOPs to cover those
areas and operations that are routinely involved in the conduct of a
study and that involve or impact on the generation, collection or
reporting of data.
To assure proper preparation and maintenance of SOPs, the
laboratory should have an SOP covering the format, initial
preparation, review, and approval of SOPs, changes, continuing review,
update, and distribution. The SOPs should also identify who is
responsible for each of the steps in the preparation process. The SOP
covering changes to these procedures should identify what types of
changes will be made (i.e. minor, major, etc.), who will be
responsible for making changes, and the documentation required to make
changes.
In a toxicological laboratory, the following SOPs are typically
needed:
* preparation, review and distribution of SOPs;
* receipt, identification, labelling, handling, sampling, usage and
storage of test and reference substances;
* maintenance, cleaning and calibration of measuring apparatus
and environmental control equipment;
* preparation of reagents and dosing formulations;
* record-keeping, reporting, storage and retrieval of records and
reports;
* data collection;
* preparation and environmental control of areas housing the test
systems;
* receipt, transfer, location, characterization, identification and
care of test systems;
* handling of the test systems before, during and at the
termination of the study;
* disposal of materials;
* use of pest control and cleaning agents;
* quality assurance programme operations;
* safety and emergency procedures.
1.5.5 Use and availability of standard operating procedures
SOPs are used by laboratory management, scientific and technical
personnel, a quality assurance programme, and other interested parties
for several different purposes. Management uses SOPs as a means of
directing and instructing personnel in procedures that are in
accordance with the requirements of quality assurance. Scientific and
technical personnel use the SOPs directly in the performance of their
duties and as a means of training new personnel in the conduct of
study operations. A quality assurance programme uses SOPs both
directly and indirectly: directly to carry out its operations and
indirectly to monitor the operations of every other laboratory
section. Part of quality assurance inspections and reviews must
include a comparison of the procedures that are being used against
those specified in the SOPs to determine if these are being used and
are adequate. Since it is the role of a quality assurance programme
to assure management that the relevant SOPs exist and are being
followed, it is desirable for a quality assurance programme to
maintain a current copy of all of the laboratory's SOPs so they can be
available for ready reference. The SOPs may also used by parties
outside the laboratory, such as inspectors from a regulatory
authority, and during study audits.
Every laboratory should have a formal procedure for training
employees in the use of SOPs, and there should be documentation
verifying the completion of such training. Some laboratories
accomplish this by making the laboratory supervisor responsible for
providing new personnel with copies of the SOPs and reviewing them
with the employee. The employee is then supervised over a period of
time to ensure that the relevant SOPs are fully understood and the
procedures are being followed correctly. This may be documented in
the employee's training file by a record signed by the laboratory
supervisor indicating the procedures in which the employee is
proficient. Some laboratories also require employees to sign a
statement that they have received and read the SOPs pertinent to their
responsibilities. Of course, employees should be aware of all SOPs
that may relate to them, not just the ones they use daily in their own
limited area. For example, a laboratory animal technician collecting
specimens of blood for the haematology laboratory needs to be aware of
the haematology laboratory's SOPs with respect to the labelling,
storage and processing of specimens.
Management should identify who is responsible for distributing
new or revised SOPs and who is responsible for removing obsolete
procedures from use. It should also establish a standard distribution
pattern to assure that all involved personnel and departments receive
copies of updated SOPs and properly remove obsolete copies. In
addition, it should address procedures to prevent unauthorized copying
of SOPs. Some laboratories have approached this by the use of an
official stamp or other mark on each page of the SOPs that will not
photocopy. Thus, when there is a quality assurance inspection, any
copies of the SOPs not bearing this mark will be recognized as
unauthorized copies and can be removed from use.
Copies of the SOPs should be located in the immediate area where
they are to be used, preferably at each work station. For example,
SOPs covering the calibration and use of a piece of equipment should
be located near the equipment, those covering procedures performed in
animal rooms should be located in the animal rooms, and those covering
the performance of necropsy or preparation of tissues for
histopathological evaluation should be located in the laboratory
performing these operations.
Copies of all the SOPs should be kept together in a manual, and,
depending upon the size of the laboratory, there may be one or more
such manuals. In laboratories that have SOPs divided into individual
manuals by department or operation, a general index listing the
various department's manuals is often useful. In order for laboratory
personnel to refer easily to those SOPs that apply to the operations
for which they are responsible, a table of contents for each
individual manual is also useful. In smaller laboratories, where all
the SOPs are contained in one manual, a single table of contents is
adequate.
A complete set of all current SOPs should be kept by a quality
assurance programme and by management.
1.5.6 Adequacy of standard operating procedures
The first evaluation of the adequacy of an SOP is made during
preparation and review. Once an SOP has been developed and approved,
its adequacy still needs to be continually evaluated, because of the
changing nature of laboratory operations. The continuous evaluation
of the adequacy of an SOP is the responsibility of the laboratory
personnel using it and of quality assurance programme personnel.
During its inspections of laboratory operations, the quality assurance
personnel should examine SOPs and compare them against actual
operations. If there are discrepancies, the existing SOP may be
inadequate and revision warranted. Also, if there are continuing
problems in particular operations, this could be an indication of an
inadequate SOP. Obsolete SOPs should be kept and not discarded since
they may be necessary to reconstruct certain aspects of completed
studies.
1.5.7 Maintenance of standard operating procedures
The responsibility for maintaining currently active SOPs is
shared by laboratory management, technical and scientific personnel,
and a quality assurance programme. Management is responsible for
reviewing and authorizing new SOPs or significant changes to existing
ones that are required to keep them current. Technical and scientific
personnel who use the SOPs daily are primarily responsible for
identifying those that have become obsolete. These individuals should
identify required changes and seek appropriate action from their
supervisors to update the SOP. Laboratory personnel, especially the
first line supervisors, are also responsible for seeing that current
SOPs are physically maintained in the laboratory and available for
use, although a quality assurance programme is primarily responsible
for verifying that SOPs in use are current, approved, adequate, and
available.
The SOPs covering continuing review and update should indicate
how often SOPs will be reviewed and who is responsible for review and
update. There are two approaches to continuing review. Some
laboratories have no programme for regular review of SOPs but simply
rely on the users to initiate updating as needed. In this case, the
SOP covering review and update of SOPs would not specify how often
they are reviewed but should still address how the frequency of review
and updating is established. Other facilities have a programme that
requires a regular scheduled review of all SOPs regardless of apparent
need. Either approach is acceptable provided it assures that the
procedures are maintained up-to-date.
Maintenance of historical files of SOPs should be a central
responsibility. This could be part of a quality assurance programme
or be located in the unit responsible for maintaining data archives.
Alternatively, the various departments could be responsible for their
respective SOPs. The historical files of SOPs should include all
revisions of the SOPs and the dates of the revisions.
If historical files of SOPs are not maintained by a central unit
but are instead kept by each laboratory department, the individual
within each department responsible for maintenance of the historical
file should be identified. Also, because there will be a number of
different individuals involved, there should be a uniform
laboratory-wide index to facilitate the retrieval of historical SOPs
from each department by the quality assurance programme or other
personnel outside of the specific department. The location(s) of
historical files should be identified in SOPs covering their
maintenance.
1.6 Test, control and reference substances
1.6.1 Introduction
The term "test substance" refers to a chemical or a mixture which
is under investigation for the purpose of evaluating its effects on
the test system. A test substance can be any class of product, e.g.,
drugs, biological products, food additives and pesticides. Control
and reference substances are chemicals that are administered to the
test system as a positive or negative reference for the purpose of
establishing a basis for comparison with the test substance. For
example, in a mutagenicity assay such as the Ames test, a known
mutagen is used as a means of comparing the response of the test
system to the test substance. The known mutagen is the reference
substance. The test, control and reference substances and the test
system are the major components of a nonclinical safety study. Unless
the laboratory has adequate procedures, facilities and equipment for
proper characterization and handling of these substances and their
mixtures, a valid safety assessment of the test substance cannot be
made.
There should be adequate procedures covering the characterization
of test, control and reference substances, the handling and
distribution of test and reference substances, and the preparation and
handling of mixtures of substances with carriers such as feed.
Characterization includes determining the identity, strength, purity
and composition or any other characteristics that will appropriately
define the test or reference substances. Stability determination is
considered to be a part of the characterization process. The
importance of this process in the study is paramount because it is
essential to know what is being tested before a meaningful evaluation
can be made of study findings.
Proper handling of test, control and reference substances is also
required to preclude the possibility of contamination, deterioration,
damage or misidentification during the testing process. Proper
handling includes adequate study records that document the receipt and
use of the test and reference substances to establish their
administration, in known quantities, to the specified test system.
Laboratories should have the necessary equipment and facilities
for handling test and reference substances. They should have separate
areas for receipt and storage of test and reference substances, mixing
of test and reference substances with carriers, storage of test and
reference substance mixtures, and for carrying out experiments, in
order to prevent contamination and mix-ups. Laboratories should also
have necessary equipment to test, prepare, store and administer test
and reference substances. Equipment must be maintained, cleaned,
calibrated and inspected to assure adequate performance.
1.6.2 Test, control and reference substance characterization
The identity, strength, purity and composition or other
characteristics (such as impurities) that will appropriately define
the test, control or reference substance should be determined and
documented prior to study initiation. Methods of synthesis,
manufacture or derivation of the test and reference substances should
be documented by the sponsor or the testing facility. It is necessary
in any study to characterize fully discrete quantities of these
substances. To do this, specific batches and lots must be obtained
and used in the test. The sponsor or the laboratory conducting the
test must have information such as laboratory analytical reports
and/or batch records documenting the manufacturing and testing of
these materials to show that they conform to the specified standards
of identity, strength and purity.
The test, control and reference substance containers must carry
identifying information. This is facilitated by the use of a batch
number. A batch is a specific quantity of substance that has a common
origin and defined physical/chemical characteristics. For a study,
the same batch of test substance should be used for the entire study,
after being appropriately characterized. If more than one batch is
used, each should be fully characterized. Complete documentation must
be maintained. It is not a preferred practice to use different
batches of the same substance without prior characterization.
When the test, control and reference substances are received for
the test, the name of the substances and the lot number must be
recorded on all study records relating to any analysis of the
substances. For example, chromatographs from analysis of the test
substance for purity or identity should include the chemical or code
number identifying the compound analysed and the specific batch or lot
number.
When marketed products are used as reference substances, full
characterization testing is not usually necessary because these
products can be characterized by their labelling. However, the
laboratory should have procedures for ensuring adequate label review
and procedures for accepting shipments of reference substances. Also,
label information such as lot numbers, expiration dates, and the name
of the manufacturer should be documented in the study records so that
the substances used in the study can be traced. Again, one batch
should be used for the entire study, if possible.
The stability of each test, control or reference substance should
be determined by the testing facility or by the sponsor before
initiation of a study. It is necessary to know the stability of the
test and reference substances to ensure that the test system is
exposed to substances of adequate potency and concentration over the
full course of the study.
Although laboratory management should assure that the stability
has been adequately determined, the sponsor may assume this
responsibility under the same conditions as specified above for
characterization of the substances; namely, this assumption of
responsibility should be described in the protocol and/or contractual
agreement.
If the stability of the test, control and reference substances
cannot be determined before initiation of a study, schedules should be
established and followed to provide for periodic re-analysis of each
batch. For test substances whose stability cannot be determined prior
to study, SOPs or the study plan should specify procedures and
schedules for sampling and testing the substance for stability. The
study plan can be amended as the results of the stability test become
known.
There must be ample storage facilities for any stability samples
retained. The storage facilities must provide the same environmental
conditions as those facilities used to store the test and reference
substances during the study. The storage conditions, i.e.
temperature, humidity and other conditions as required, must be
documented.
Each storage container for a test, control or reference substance
must be labelled by name, chemical abstract number or code number,
batch number and expiry date (if any). Where appropriate, storage
conditions necessary to maintain the identity, strength, purity and
composition as well as safety information, if available, of the test
or reference substances must be indicated. If safety information is
not available, this should also be noted on the label. The laboratory
should have procedures for labelling the test substances. These SOPs
should require that this information be clearly indicated on the
label. They should also address who will be responsible for labelling
the containers and/or verifying the label of any containers received
by the laboratory. The SOPs should also address how labels will be
applied. It is generally unacceptable to place the only identifying
label on the lid of the container. This practice may lead to
confusion between containers when lids are removed during use. SOPs
regarding labelling may include procedures for assuring that labels
will remain affixed and legible during conditions of storage and use.
For example, test substance containers may be exposed to moisture or
solvents that could alter the labelling.
Storage containers should be assigned to a particular test
substance for the duration of the study. This is necessary to
preclude the possibility of cross-contamination, which may occur when
the same container is used subsequently to store a different test
substance. It is good practice to dispose of empty test substance
containers after a study has been completed rather than attempt to
decontaminate and reuse them.
Retention of samples of test, control and reference substances is
useful should a question arise regarding the quality or identity of
the test substances actually administered to the animals.
1.6.3 Handling
A laboratory should have adequate procedures for a system of
handling the test, control and reference substances to ensure that
there is proper storage, that distribution is made in a manner
designed to preclude the possibility of contamination, deterioration
or damage, that proper identification is maintained throughout the
distribution process, and that receipt and distribution of each batch
is documented. These procedures should include a description of the
records to be maintained in order to document that procedures are
being followed.
1.6.4 Storage
Handling procedures should include proper storage. Storage
requirements vary with the specific test, control and reference
substances used. For example, some test substances may require
storage at temperatures below 0 °C, while others are stable at room
temperature. Some may require storage in the dark or in special
containers and cabinets due to high volatility. In all cases, the
laboratory must have procedures that will require each test or
reference substance to be evaluated upon receipt for special storage
and handling conditions, and to assure that these conditions are met.
For example, there may be a standard form that is filled out upon
receipt of a substance that requires the recipient to record any
special storage conditions required, to ensure that these conditions
are recorded on the label, and that the substances are stored
accordingly. Laboratory procedures for receipt of the test and
reference substances should also require the person receiving them to
determine, if possible, the storage conditions of the substances
during shipment and at the time of receipt. For example, if the
substances are to remain frozen until used, they should be inspected
upon receipt for thawing and this should be documented in the study
records. The storage requirements for the test, control and reference
substances should generally be provided by the sponsor or manufacturer
responsible for initially providing the substances.
Procedures for ensuring proper storage need, at a minimum, for
required storage conditions to be specified in the labelling and study
records, necessary facilities to be identified and available to meet
required storage conditions, storage conditions during transport and
use to be monitored and documented, and deviations from the required
conditions to be investigated and documented. Where deviations occur,
the study director should determine their impact on the study and
record in the final report any that may seriously affect the study.
1.6.5 Distribution
Procedures covering test, control and reference substance
handling should ensure that distribution is made in a manner designed
to preclude the possibility of contamination, deterioration or damage.
Such procedures might include the types of containers used to
transport articles to the laboratory areas where they are to be used
and the designation of specific storage areas. Laboratories dealing
with large numbers of test substances should have SOPs identifying
where the substances are to be stored. Procedures should also
identify where and how containers of test and reference substances are
to be opened for dispensing. This usually takes place in an area
specially designed for this purpose with an isolated environment.
Many laboratories have a specially designated room or laboratory bench
top under a laminar air flow to prevent the possibility of
cross-contamination of equipment and facilities, as well as to protect
employees from potentially hazardous test substances. Special gowning
and decontamination procedures are generally required for the test
substance dispensing areas and are included in the SOPs for test and
reference substance handling. Adherence to these procedures must be
documented. Such documentation will include records of cleaning the
areas between use for different compounds.
A systematic process for receipt and distribution of test,
control and reference substances should be fully described in
laboratory SOPs for test, control and reference substance handling.
Ideally, there should be a centralized procedure responsible for
receiving and logging in test, control and reference substances. This
should ensure that proper identification is made initially and then
maintained throughout the distribution process.
Documentation should specifically include the date and quantity
of each batch distributed or returned. An inventory record is often
used to record the receipt of all test and reference substances.
Entries include the name or identifying number of the substance
received, the batch or lot number, the date of receipt, the amount
received, and the signature of the individual who received the
substance and completed the record. Many laboratories record the
weight of the container and its contents as received. When a portion
of the test substance is subsequently dispensed, the amount is weighed
and recorded in the inventory record. The record should also reflect
to whom and for what purpose the substance was dispensed. As an
additional quality assurance measure, many laboratories not only weigh
out the amount to be dispensed, but also weigh and record the
remaining substance in the bulk container. This provides a continual
inventory of the test substance and allows detection of any dispensing
or record-keeping errors that may indicate a potential misdosing
problem.
It is clear that there should be procedures covering every aspect
of the handling of test, control and reference substances to ensure
proper storage, distribution, identification and accountability.
Records should be maintained to document that these procedures were
actually followed. All study records pertaining to use, testing and
distribution of the test or reference substances should be directly
traceable to the specific lot or batch of materials issued, and
accountability records should document that the quantities and lot
number used were consistent with study plan requirements. Periodic
checks should be made that the amount of test substance in the
inventory matches with the amount used in the study.
1.6.6 Mixtures of substances with vehicles (Carriers)
A vehicle (or carrier) is the material with which the test,
control or reference substance may be mixed for administration in the
test system. It can be feed, water, solvents and/or excipients,
depending on the form of dosage and route of administration. A common
example of a carrier is the rodent feed used to mix a test substance
for a chronic feeding study. However, the phrase "mixture of
substance and vehicle" also refers to solutions and suspensions, e.g.,
a solution of test substance in distilled water.
A laboratory should have procedures for the preparation,
analysis, storage, labelling, distribution and return or disposal of
mixtures of substances with vehicles. The procedures should
accomplish the same goals of maintaining proper identity and ensuring
proper storage and use as those procedures already discussed for the
test and reference substances alone. In addition to the concerns
already mentioned, mixtures of substances with vehicles present
special problems. For example, there must be procedures to assure
that the mixtures used are uniformly mixed, stable, and provide the
proper concentration of the test or reference substance. To
accomplish this for each test, control or reference substance mixture,
appropriate analytical methods should be used to determine the
uniformity of the mixture and to determine, periodically, the
concentration of the test or reference substance in the mixture. The
standard of uniformity should be determined before the start of the
study.
Each time a batch of substance carrier mixture is made, it should
be recorded that the batch was properly formulated and mixed according
to procedure. For liquid preparations, uniformity determinations need
not be made on true solutions, but should be made for suspensions. In
laboratories that perform numerous short-term assays (e.g.
mutagenicity studies) by using liquid dosage forms, it may not be
practical to analyse dosing solutions from every assay. In this case,
it is imperative for the laboratory to have well-documented dilution
and dosing procedures and SOPs for determining and documenting
solubility of the test and reference substances.
1.6.7 Stability
In addition to analyses for uniformity and concentration, tests
should be conducted to determine the stability of the test, control
and reference substances in the substance carrier mixture. If the
stability cannot be determined before the study is initiated, the
study plan should provide for periodic re-analysis of the test and
reference substances in mixtures. The laboratory must determine the
stability of the mixtures over their periods of use. This would
include the period between the day the batch is prepared and the day
the last portion of the batch is used. Stability should be determined
under actual conditions of storage and use. Once the stability of a
given concentration of a test substance carrier mixture is
substantiated for one batch, no further stability testing is necessary
for each subsequent batch of that concentration. However, periodic
re-analysis to determine concentration must be carried out, as
discussed previously.
A sound practice related to stability testing is the use of
expiry dates. Where any of the components for the test or reference
substance carrier mixture has an expiry date, that date should be
clearly shown on the container. If more than one component has an
expiry date, the earliest date should be shown. This requirement is
necessary to assure that outdated or unstable mixtures are not used.
1.6.8 Labelling
The importance of proper labelling cannot be overemphasized.
This is especially true for the labelling of test, control or
reference substances, as well as for their mixtures. Misapplied or
inadequate labelling can lead to major problems that can invalidate
study results. For this reason, every laboratory should have SOPs for
labelling test, control or reference substances and their mixtures
with carriers. These SOPs should address how labelling will be
applied and what its content will be.
Labelling for a mixture container should include the expiry date
of the mixture; the use of expiry dates is intended to preclude the
use of mixtures that may have deteriorated. Procedures should exist
to assure that outdated or deteriorated batches of test and reference
substance mixtures are not used.
1.6.9 Facilities and equipment
There should be a separate area for the receipt and storage of
test, control and reference substances. Laboratories may have a
centrally located facility for this purpose. These areas are usually
supplied with storage cabinets, freezers, refrigerators, balances and
related equipment required for the orderly receipt, storage and
distribution of the substances received. These areas are sometimes
equipped with special environmental equipment, such as laminar air
flow and biohazardous hoods, depending on the extent to which these
areas may be used for opening and dispensing test and reference
substances for use in the mixing areas. If containers of test and
reference substances are opened in the receiving/storage area, SOPs
should prescribe the necessary precautions to be taken to avoid
contamination of the area and equipment with the various substances.
Employee safety precautions in handling chemicals are essential.
These should include decontamination procedures for spills and other
emergencies, as well as procedures for routine cleaning. The nature
and extent of the facilities and their use will dictate what
procedures and equipment will be required. The area should be
physically secured and only authorized personnel permitted to store or
remove materials. In smaller laboratories, there may not be room for
a separate facility for this purpose. In this case, test and
reference substances may be stored in areas used for other purposes;
however, the same general requirements apply as stated above. In
particular the area, such as a cabinet, should be isolated as much as
possible from other areas and activities and should be made physically
secure with authorized access only. It should not be used for any
other purpose. Limited access to test substances is required to
prevent unauthorized use and mix-ups. Accountability records should be
maintained, documenting receipt and dispersal of test and reference
substances. Such documentation for all substances is often maintained
in the receipt and storage area. The entry or removal of substances
from the designated areas should be documented.
In addition to separate areas for receipt and storage of test,
control and reference substances, the laboratory should provide an
isolated area for preparing and mixing these substances with carriers.
The extent and nature of these facilities will depend upon the scope
and type of the laboratory. Larger laboratories may have elaborate
equipment, facilities and procedures for mixing test, control and
reference substances with carriers. In a typical laboratory, engaged
routinely in conducting safety studies, there will usually be a
separate area or areas where test substances are stored and weighed.
The pure substances are then transferred to the separate mixing and
dosage preparation areas.
Another major consideration in the design of a mixing area is
cleaning and decontamination. The area should be easily cleanable.
The walls, ceiling, and floor should be smooth and impervious to
cleaning agents used. Cleaning and decontamination design
considerations also apply to personnel who work in the area.
There must be specific SOPs to cover the cleaning and/or
decontamination of equipment. This is especially important for
equipment, such as mixers, that may come in direct contact with test
substances. In specifying cleaning agents, the SOP should take into
account the solubility of materials to be removed from the equipment
and also the removal of any cleaning agent residues. In studies where
the test material may present unusual equipment cleaning problems, the
procedures for cleaning may be included in the study plan. It is also
very important for the cleaning of important equipment to be monitored
and documented.
Once the test, control or reference substances have been mixed or
otherwise prepared, they should be moved to a separate area designated
for the storage of these mixtures.
SOPs should also deal with the disposal and/or reuse of
containers used to store test and reference substances and their
mixtures. In laboratories where containers are reused to store these
substances, there should be strict references and documentation
covering decontamination and labelling. Procedures for final disposal
of hazardous waste should be defined by national or local governmental
regulations. Laboratories should ensure that they conform to these
procedures.
1.7 Quality control
1.7.1 Introduction
Quality control is applied to routine laboratory biological,
chemical and physical analyses in order to assure reliability and
comparability of test data. It involves statistical approaches
designed to demonstrate the constancy or variability, and precision of
analytical data.
When the concentration of a chemical is determined repeatedly in
the same laboratory a certain scatter is always seen. The scatter is
due to variation in the analytical step as well as to changes that may
take place during the collection, preparation and storage of the
samples.
Quality control depends, among other factors, on the type of
compounds involved. A major issue is "to keep the compound as it is"
and it is reasonable to make a distinction between inorganic and
organic compounds. Since over ten million organic compounds exist, a
further distinction must be made between simple organic compounds,
such as solvents and metabolites, more complex compounds that may have
a low vapour pressure, such as some pesticides, compounds that are
capable of being adsorbed, and very complex biological compounds such
as proteins and enzymes.
In the case of inorganic compounds, contamination of the test
substance with the same compound from an external source is a major
concern. Some elements are volatile, e.g., elemental mercury, and
special care is needed to avoid loss of analyte. Certain elements are
readily reduced to a different oxidation state even within the
container used. Examples are the reduction from Cr(VI) to Cr(III) and
the reduction from Hg(II) via Hg(I) to elemental mercury, which
disappears from the test material. There may be effects due to
compounds already present in the test substance, to compounds added
for preservation or to wall effects of the container. The latter
category can be separated into chemical effects, e.g., reduction and
oxidation, and physical effects, e.g., adsorption to the wall or
stopper, and precipitation/ coprecipitation of the analyte alone or
together with other compounds.
In the case of simple organic compounds, a well-sealed container
is needed to prevent vaporization loss. The container or stopper can
be responsible for adsorption or leakage. Reduction as well as
oxidation of metabolites in biological material may occur.
Ultraviolet light, as occurs in sunlight, may decompose organic
compounds by photochemical reactions, and the infrared radiation in
sunlight may cause thermal degradation of organic compounds. Thermal
degradation may also occur due to other infrared sources such as heat
radiators or laboratory apparatus. Another factor which may lead to
thermal degradation is the temperature of the environment of the test
substance from sampling until final analysis within the apparatus.
In the case of more complex compounds with a low vapour pressure,
such as some pesticides, vaporization of the analyte is unlikely.
Certain pesticides may undergo reduction or oxidation. Photochemical
and thermal degradation may occur in this class of compounds.
Proteins and enzymes are extremely vulnerable to changes in pH
and redox potential, heat and exposure to ultraviolet light.
Moreover, as many proteins can bind easily to heavy metals, they need
special care.
Another important item is the concentration of the compound of
interest. Concentrations of compounds in biological materials range
in general from parts per million, e.g., essential elements in plant
and animal, to parts per trillion for very toxic compounds such as
dioxins. This enormous range of concentrations is also seen in the
soil, surface water, indoor air, and, to a lesser extent, in sea
water, drinking-water and outdoor air. As precision is often
inversely related to concentration, it is evident that the
determination of sub-ppm concentrations of compounds is always
problematic, regardless of the method of choice.
Analytical variation may be divided into two major categories:
accuracy and precision. Accuracy refers to the agreement between the
measure and the true amounts of the analyte, and precision refers to
the random variability or reproducibility of the method.
In order to assure the reliability and comparability of the test
data, an extensive quality assurance programme should be implemented.
This should cover the sampling and sample handling (preanalytical
quality control), as well as the analytical procedures (analytical
quality control). The purpose of quality assurance is to identify
different types of errors that may invalidate the test data and to
make sure that the total of all errors is below certain established
limits.
1.7.2 Level of quality control
A basic question in quality control is how accurate and precise
the different measurements need to be in order to provide reliable
exposure assessments. This decision may be based on prior information
on the inherent variability in the quantity being measured, and this
is likely to differ for each pollutant and medium. Existing
information from measurements already conducted in the locality in
question, or in similar areas or groups of people, can be used to
provide a guide. The variability can then be assessed as the relative
standard deviation, expressing the standard deviation as a percentage
of the mean, assuming that a normal distribution basis is adequate for
the present purpose.
An important issue in biological test substances is the
biological variability, which can be separated into the
intra-individual variability, i.e. the variability within an
individual, and the inter-individual variability, i.e. the variability
between individuals. The variability may be expressed as a relative
standard deviation. The allowed standard deviation for a
determination used in clinical and environmental chemistry is half the
standard deviation of the inter-individual variability at a maximum,
or, alternatively half of the sampling error (Youden, 1967).
As another example of how to deal with precision, the procedure
in the UNEP/WHO (1984) and WHO (1986b) studies may be used. Within
these studies a relative standard deviation of 20% was set
arbitrarily.
The error in precision within a laboratory is thought to be of
the order of one-half to two-thirds that of the inter-laboratory
error.
1.7.3 Pre-analytical quality control
It is essential to ensure correct sampling, i.e. that the
collected air, particles, water, food or other samples really
represent the whole sampling period and the subjects concerned, and
that contamination of the samples is avoided. It is important for the
field personnel and the laboratory staff to be properly trained.
Detailed guidelines for the sampling procedures should be prepared
prior to the sample collection and discussed with all the people
concerned.
In the case of many pollutants, it is necessary to check their
content in containers and other equipment used for sampling and
storage before sampling in order to avoid contamination.
Audit procedures are useful to control the reliability of the
sample collection, transport and storage. The major steps to be
audited during sampling would be:
* (preventative) maintenance
* calibration of sampling and analytical measurement equipment
* procedural control checks
* tick-off chart
* cleanliness during sampling, sample transport and storage
* sample deterioration, temperature control and stabilizers
* time lapse before analysis
* data recording, calculations and record keeping
Sampling should be representative, and the number of samples
should be adequate. Sampling can be random or selective, depending on
the goal of the study. If sampling should be random, all subsequent
steps should also be carried out in a randomized manner. For example,
if an "exposed" population of animals or humans is compared with a
reference group, sampling should be performed randomly. Also the
pre-analytical steps, such as the distribution of the samples in the
containers and the time lapse before determination, should be
performed carefully to avoid any systematic influence.
Although sampling errors are often the greatest source of
variability, especially in environmental measurements, these errors
are mostly beyond the control of the laboratory, unless the laboratory
is also responsible for the sampling operation (Horwitz, 1989).
Regardless of who collects the samples, this source of variability
must be considered individually for each lot or population to be
examined. The magnitude of this source of variability is assessed by
taking a number of random samples from the population and examining
them individually (see also section 1.7.5).
In occupational toxicology and sometimes in environmental health,
the timing of specimen collection is important. Some industrial
chemicals have a long biological half-life in various body
compartments, and the time of sampling is not critical (Herber &
Schaller, 1986). For other chemicals the timing of sampling is
critical because after exposure the compound and/or metabolites may be
rapidly eliminated from the organism.
In the case of urine samples, 24-h or 6-h urine samples are
preferable to spot samples. Preservation against bacterial growth and
fungi can be achieved by adding a solution of sodium azide.
Determination of enzymes should generally be performed within 24 h.
However, some stable enzymes may be determined after a longer time
interval, but preferably within a week when stored at 4 °C.
In the case of blood samples, clotting is a major source of
errors and this should be avoided at all costs. Clotted blood samples
cannot be used for analysis and must be rejected. Many urine samples
have a precipitate but generally this can be redissolved by acids
without influencing the determination, by carefully heating up to 37
°C, or by ultrasonic treatment. Another possibility is to remove the
precipitate by adequate centrifuging. When a precipitate forms during
cooling, care must be taken that the samples are stirred so that any
deposits are dissolved or evenly distributed. All pre-analytical
procedures must be checked comprehensively before starting routine
determinations.
Storage of samples is optimal at -80 °C. At this temperature
samples may be stored for years. A more practical temperature is -20
°C. At this temperature proteins can be stored for months. Inorganic
compounds maybe stored at +4 °C, provided that no vaporization takes
place.
1.7.4 Analytical quality control
Validation of analytical procedures with respect to accuracy and
sensitivity should be accomplished by appropriate quality control
studies. The accuracy of the methods used by the different
laboratories should preferably be established by an external quality
control programme, in which the reference values of the quality
control samples are unknown to the laboratories. If this is not
possible, comparisons with reference methods or the analysis of
certified reference samples may be used. It is also important to
establish the reproducibility of the routine analytical procedures
used. Acceptable limits of variation for control samples should be
set primarily by considering the data quality requirement rather than
the analytical characteristics of the procedure. The stringency of
the limits depends largely on the purpose for which the test data are
intended.
In case of random sampling, the complete analytical procedures
should be randomized. Thus, destructions or extractions must be
performed for an approximately equal number of "exposed" and "control"
test specimens within the same run. This applies to instrumental
techniques including spectrophotometry, chromatography and atomic
absorption spectrometry. Calibration and reference specimens must be
distributed randomly between the test specimens.
A major item is the influence of the matrix and, especially in
urine, the matrix variability; an example is the variable salt
concentration in urine which may cause problems in the determination
of metals.
1.7.5 Statistical considerations
According to Taylor (1988a), it is essential to define the degree
of uncertainty of the data. This is necessary in order to provide
evidence that samples are "representative" for the environment or
population to be sampled. Two factors influence the uncertainty of
the data, i.e. measurement uncertainty and sample uncertainty. The
uncertainty of the measurements can be estimated by replicate
measurements on a sufficient number of samples. Taylor proposed that
the measurement uncertainty should not exceed one-third of the total
uncertainty tolerance.
The sample uncertainty is based on the variability of the
population or environment to be sampled. Sampling strategies based on
statistical considerations address this factor. During sampling a
number of items can represent components of the sampling uncertainty.
These are, for instance, systematic components like the properties of
sampling equipment and the variability within the actual sampling
process. All these components add up to the total sampling
uncertainty. It is practical to define acceptable total uncertainty,
and then estimate the different components contributing to this
uncertainty.
Smith et al. (1988) stated that quality assurance procedures of
sampling data require the definition of the precision, the bias, the
representativeness, the completeness and the comparability. Precision
in this case is the agreement of individual measurements of the same
property under the same conditions. It is best expressed in terms of
standard deviations. In environmental sampling, it means that the
total variance of the measurements is the result of the precision of
the analytical measurements and the precision in sampling. The
analytical precision is obtained by determining the standard deviation
of individual samples, whereas the precision of the sampling as such
is determined by analysing a number of replicated field samples. The
presentation of results from environmental measurements should include
regression equation coefficients, when appropriate, means and standard
deviations, and ideally a graph containing the actual data points, the
best-fit curve and confidence intervals, in order to provide
information for a quality assessment.
Bias is the degree of agreement of a measurement or an average of
measurements having an accepted reference or true value. It is
usually expressed as the difference between these two values or as a
percentage of the true or reference value. Bias is frequently
expressed as recovery (100% bias) in environmental monitoring
programmes, and can be estimated by using spiked field samples for the
bias in the field sampling phase, and by the repeated analysis of
field samples or reference material for the analytical phase.
Representativeness is the degree to which data represent
precisely and accurately a characteristic of a population or
environmental condition. This can be assured by the use of
appropriate sampling techniques.
Completeness is a measure of the amount of valid data obtained
from a measurement system compared to the amount that was expected to
be obtained to fulfil the objectives. Various conditions, e.g.,
meteorological conditions, accidents, during sampling and analysis
could result in the incompleteness of data, particularly during field
sampling. In any case there should be an evaluation of the influence
of missing or lost data on the overall result of a study.
Finally, comparability expresses the confidence with which one
data set can be compared to another. For this purpose, there should
be descriptions of the comparabilities of sites elected for sampling,
the calculation and statistical analysis, and the sampling and
analysis protocol.
1.7.6 Analytical performance evaluation
There are various ways of performing analytical quality
evaluation. Since it is impossible to produce errorless analytical
data or to obtain a measure of the accuracy, it is important to
estimate the limits of uncertainty of produced data.
Some general statistical approaches to evaluating the quality of
chemical measurements have recently been reviewed (Taylor, 1987,
1988b). A well-established procedure for comparing the analytical
performance of several laboratories is the round-robin test using a
Youden plot (Youden & Steiner, 1975). Two similar samples, designated
X and Y, are sent to each collaborating laboratory for analysis. The
results provide a pair of coordinates which are plotted in a diagram.
If random errors predominate, the results will fall within a circle
with the center showing the "true" value. Due to systematic errors
most of the observations are usually found along a 45° axis.
Evaluation of the regression line of reported versus reference
values for a set of quality control samples is a useful method of
guarding against systematic errors in the likely concentration range
(Vahter, 1982; UNEP/WHO, 1984; Friberg, 1988). Further discussion of
this method is given in section 3.7.
1.8 Documentation and record keeping
1.8.1 Introduction
Any study report must be capable of being validated before it can
be fully relied upon for accuracy and completeness of findings and
before any scientific conclusions can be derived from it. This means
that the information and conclusions stated in the report must be
fully supported by raw data documented in the laboratory records
covering the conduct of the study. It requires the existence of a
complete data trail from the initiation of the study to the time when
the last data point is recorded. The data trail should be detailed
enough to allow an independent party to trace every aspect of the
study. Validation of a study by an independent party is sometimes
necessary to assure that all the provisions of the study plan and SOPs
were followed.
"Raw data" covers all of the original observations of the study.
For example, the term "raw data" may mean any laboratory worksheets,
records, memoranda or notes (or exact copies of these) that are the
result of original observations and activities of a study and that are
necessary for the reconstruction and evaluation of the final report.
Exact copies of raw data may be valid, provided that they are verified
as accurate by the dated signature of the person making the copies.
Other examples of raw data include photographs, microfilm or
microfiche copies, computer printouts, magnetic media, including
dictated observations, and recorded data from automated instruments.
There are three common processes for capturing data. These
processes include manual recording of data, direct computer entry of
data, and entry of written data into a computer.
1.8.2 Manual data records
Many testing facilities find it useful to have standard data
collection forms for recording items such as health status
information, body weights, body length, clinical observations, test
system care, necropsy, clinical chemistry, haematology, histological
processing, and environmental condition. These forms should be
designed with care and should be readily available to personnel
conducting study functions. Procedures for proper use of data
collection forms should be described in SOPs or the study plan. When
these forms are used during the course of the study, they should be
identified by study number, when applicable, test substance and
species, and they should carry entry points not only for the collected
data but also for dates and initials of the person or persons entering
the data.
All data should be recorded in indelible ink, preferably black,
and corrections in the data should be made so as not to obscure the
original entry. The use of white-out, correction tape, erasers,
overwriting or any other means to obscure the original entry is not
acceptable. Changes made in records of original observations should
ordinarily be made by the individual responsible for the original
entry. All corrections should be initialled and dated at the time the
correction is made and a justification should be given in the record.
The record should be corrected in a timely manner. Some laboratories
use codes or abbreviations to indicate common reasons for corrections.
Such codes should be defined in the record or the associated SOP and
should be used consistently by all employees making corrections.
Standard data collection forms should be designed so that they
are unambiguous and so that they require a notation to indicate
whether an activity has been performed or an observation has been
made. Leaving data entry spaces blank is an unacceptable means of
documenting an observation. For example, a standard form to document
the collection of tissue at necropsy may list all possible tissues and
provide a space to document the collection of specific tissues. If a
technician leaves a blank space to record that no tissue was taken, an
individual reviewing the record can never be sure if the tissue was,
indeed, not taken or the technician merely forgot to fill in the space
when the tissue was collected. Similarly, a line drawn through all
data entry spaces on a record can be ambiguous, unless its meaning is
clearly defined in the record or SOP and is understood by all
individuals completing the record.
A study notebook in which narrative notes may be kept of
significant events, such as animal deaths, changes in batches of test
substances, clinical examinations, disease outbreaks and animal
sacrifices, supplements the standard data collection forms. Actual
data elements required by the study plan for the test system can be
recorded in the study notebook.
1.8.3 Computer data records
Although there is still a need for written records to document
important daily events, computers may now be used to save time and
labour in data collection, validation and report generation. Direct
entry of data may be accomplished by use of keyboards, optical bar
code readers, touch screens, optical scan sheets, and direct digital
input from laboratory instruments or sensors. In order to capture and
correlate effectively diverse data from multiple sources, most
laboratories collecting data in this manner use a distributed data
processing system, where remote terminals and microcomputers collect
data, which are later transmitted to the central computer for
permanent storage and/or processing.
In systems such as this, data should be verified at the time of
collection prior to transfer to the central computer. This can be
done by review of the terminal screen at the time of data entry and/or
review of data print-outs. As with data collected manually,
computer-entered data must include the identity of the individual
making the observation and the date the observation was made. Any
correction made later at the local level require that the change be
verified in the central computer to check that it has been recorded
correctly. Experience has revealed cases where legitimate corrections
were made at a local level but not submitted to the central computer
or included in the final report. The same principles apply for
correcting computer-collected data as for paper data entries, i.e. the
original entry must not be lost, and the change must be documented to
show who made the correction, and when and why the correction was
made.
A computer system must be designed to meet the requirements of
quality assurance. It must be tested prior to initial use and
whenever significant changes are made to the computer hardware or
software. The testing process must include a documented review of the
system design, development, and acceptance measurements to assure
proper system performance. This should be done prior to testing by
laboratory personnel. Testing often involves the checking of a
previously validated array of hand-collected data against the same
data collected concurrently and reported by computer for one or more
studies. For each study, the acceptance testing needs to cover each
of the data points the system is designed to collect and process.
For the purposes of retaining raw data, the magnetic media
containing the raw data (i.e. disc, tape) and/or a verified printout
should be kept. However, manufacturers of magnetic media advise that
data retention is not guaranteed beyond a limited time, and, if discs
are erased, revised periodically or reused, some errors in the discs
can arise. Thus, a paper print-out of the data for data review and
storage as original data is essential. However, any print-out
retained as original raw data must be verified. This means it must be
reviewed for accuracy and this review must be documented by the
signature and date of the individual(s) performing it. Proper
verification would include review by the supervisor and/or study
director and quality assurance personnel.
Any computerized data collection system must include SOPs to
describe operations, specification, security, validation and
maintenance of both the local data acquisition systems and the central
computer system. These documents should be in the appropriate work
stations and should contain such information as how the data are to be
entered and what remedial action is to be taken in the event of system
failure.
1.8.4 Indirect computer data records
The principles outlined above also apply to manual data that are
subsequently entered into a computer. In addition, evidence must
exist that the manual data were correctly and completely entered.
Frequently, this is a quality assurance programme function. The
extent to which the "raw data" are compared with those of the initial
paper print-out (or screen display) needs to be defined in SOPs.
1.9 Final report
1.9.1 Introduction
The final report is the end-product of a carefully planned and
conducted study. The report must be well organized and reflect
accurately all the experimental data. It must contain a detailed
account of the study including, where relevant, unexpected deviations
in the controls, study plan and environmental conditions.
1.9.2 Contents
The final report should include the following information:
a) a descriptive title;
b) clearly defined objective(s) and study plan;
c) an informative summary of the results of the study;
d) an identification of the test and reference substances by
chemical name, code or chemical abstracts number;
e) a description of the characterization of the test and
reference substance including purity, stability and
homogeneity;
f) name and address of the test facility, and the name and
address of any facility that may have performed parts of the
study;
g) the name of the study director and any other principal
scientists who contributed reports included in the final
report;
h) the dates on which the study was initiated and completed;
i) a description of the methods, procedures and materials used,
highlighting any changes in the study plan;
j) all information and data called for in the study plan,
including "outliers";
k) a description of the test system, and the procedure used for
identification;
l) exposure conditions;
m) an accurate presentation of results, including calculations,
and a description of the statistical methods used to analyse
the data;
n) an evaluation and discussion of test results and any
conclusions drawn from the results;
o) the dated signature of the study director.
1.9.3 Indexing
An index is essential for each volume of data comprising the
final report. Appropriate references to appended tables and figures
are necessary to facilitate the peer review of the validity of the
conclusions drawn from the study. Appendices containing graphs,
tables and statistical evaluations of all the data and observations
are as important as the narrative report. A glossary of terms and
abbreviations used in the report should also be included, since this
will facilitate an understanding of the findings of the report.
1.10 Archiving and retention of data
1.10.1 Introduction
Records, specimens and reports constitute lasting proof of the
validity of a study; facilities and procedures for their archiving
should be available. The following basic points should be observed:
* all raw data, documentation, study plans, appropriate specimens
and final reports generated as a result of a study should be
retained;
* there should be archives for the orderly storage and retrieval of
all raw data, documentation (e.g., training and qualification
records), equipment maintenance, master schedule of studies and
quality assurance reports, study plans, specimens, and interim
and final reports;
* material retained or referred to in the archives should be
indexed by test substance, date of study, test system and nature
of study;
* an individual should be responsible for the archives;
* only authorized personnel should be permitted to enter the
archives.
1.10.2 Facilities
Storage facilities required for archiving data and specimens must
be adequate to preserve specimen quality and to control access. The
storage conditions in the archives should be monitored to prevent
accelerated deterioration of data and/or specimens. Archives should
be designed to have a controlled access area, i.e. with access limited
by doors that lock, and the area should be isolated from operational
laboratory areas but still conveniently accessible. Environmental
conditions should avoid extremes of temperature and humidity. For
example, tissue blocks should be kept cool and dry enough to prevent
melting, sticking or mould growth. Paper records should be protected
from fire, water, rodents, etc. Environmental conditions in the
archives should be monitored to assure that appropriate conditions are
maintained. Finally, proper storage cabinets are needed to organize
and store data and specimens for easy retrieval.
1.10.3 Responsibilities for an archive
Normally a testing facility has the responsibility for providing
an archive facility and authorized archivist. However, when there is
a sponsor/contractor relationship involved, there can be problems over
the allocation of responsibility for archiving material between the
laboratory and the sponsor. During the course of the study, the
original data remains with the test facility conducting the study.
When the study has been completed, there are several ways that data
may be archived, depending on the contract between the sponsor and the
test facility. Firstly, the test facility may archive the data.
Secondly, it may transfer all archival material (e.g., data, slides,
tissues) to the sponsor for archiving. In this case, there should be
a record of transfer, and documentation identifying the storage
location should be maintained in the contract facility's archive. The
third option is for the test facility to send the originals to the
sponsor and maintain copies in its archive.
1.10.4 Standard operating procedures for archiving
There must be SOPs for each activity performed in operating an
archive. Some of the relevant SOPs are:
* receiving, indexing and identification
* filing and storage
* access to archives
* security
* data retrieval
* retention of material
* removal and return of records and raw data (as appropriate)
1.10.5 Receiving, indexing and identification
Generally, the central archive of a test facility is utilized for
the paper and computer data from a study. This includes all raw data
in notebooks, forms and/or computer print-outs, approved study plans,
amendments, and final reports generated. These materials should be
received and accepted by an authorized person (the designated
archivist or his replacement).
Prior to accepting the data, the archivist should be given an
index, signed by the study director, of the submitted data listing the
items being archived. When the items have been checked against the
index and the archivist is satisfied that the package is complete, the
index should be signed and dated by the archivist. After acceptance,
the total study package should be archived using a classification
system (e.g., a unique number).
1.10.6 Filing and storage
Material files should be filed and cross-referenced for easy
access and retrieval of data. Index cards are often used for
cross-referencing, but data are increasingly being indexed on
computers.
Archived material should be paginated and indexed by test
substance, date of study, test system and nature of study. Other
titles used for cross-referencing are unique study numbers, the study
title and the name of the study director.
In the case of specimens, slides, blocks, tissues, test
substances and other materials that are not in the central archive
with the raw data and final report, there should be a method for
indicating where these articles are archived. One method is the use
of a form to be filled out by the person maintaining the items. For
instance, if the teratologist maintains an archive for fetal specimens
and the pathologist maintains a separate archive for wet tissue,
blocks and slides, they should complete and sign a form certifying the
location and storage of the specimens. This information would be
maintained in the central archive with the other paperwork generated
as a result of the study.
1.10.7 Access and security
There should be control of access to any archive where data,
specimens and/or slides from studies are stored. The door to the
archive should be locked at all times when it is unattended. Only the
archivist should have keys for access to the archive records.
Ideally, the archive should be designed with a separate area for
people to wait while the archivist is retrieving the requested data in
order to help maintain the limited access environment.
1.10.8 Retrieval of data and control of access
Although stored data and specimens should be easily identified
and retrievable, their removal from the archive should be discouraged.
For the retrieval of data specimens, there must be a standard
operating procedure. The approval required may vary, but in many
testing facilities, the authority to release data is given to the
archivist. For records management and to maintain a tracking system,
a "Data and Specimen Request Form" is useful because it becomes a
permanent record for the study file and helps make the person
requesting data aware of the need to ensure the security and integrity
of the items removed from the archive.
The title of the study, name of the borrower, purpose,
organization and items requested are entered on the form. When the
request is completed, the archivist will sign and date the form and
the borrower will sign, indicating that the items were received and
that the security and integrity of the data will be ensured. This
form remains in the archive.
When the borrower returns the data, the archivist must check the
items returned against those listed on the request form. If all items
are present and the archivist is satisfied that the integrity of the
study has not been compromised, the form is signed by both the
borrower and archivist and becomes a permanent record for the study
file.
1.10.9 Retention of information
The length of time that study records must be retained will vary,
this depending mainly on legal considerations. It is the
responsibility of test facility management and/or the study sponsor to
ensure that records, specimens and reports are retained for the
required time.
For archived material, such as wet specimens, plates for
mutagenicity testing, test and reference substances, haematology
slides and histochemical and other specimens that are relatively
fragile and that vary in stability and quality during storage, the
retention period can be only as long as the period during which the
material is of a quality that affords a valid and meaningful
evaluation. Management should develop an SOP establishing the
retention time for these types of specimens.
2. QUALITY MANAGEMENT APPLIED TO TOXICITY STUDIES
2.1 Introduction
Laboratory animals are the test system for many studies. In
order to provide meaningful safety data, the animals must be properly
selected and cared for to ensure that any response observed in the
animals is caused by controlled exposure to the test, control or
reference substances and not by uncontrolled variables such as disease
and adverse environmental conditions. Appropriate laboratory practice
needed to assure proper care and use of animals includes providing
proper facilities and employing necessary procedures to eliminate or
control factors that could interfere with the response of animals to
the test and reference substances. The care, housing and treatment of
animals used in research and testing is regulated by law in many
countries.
Animal care facilities can comprise a large portion of the
physical plant of a laboratory because sufficient animal rooms or
areas are needed to ensure separation of species or test systems, and
provide for quarantine requirements and routine or specialized housing
of animals.
2.2 Procedural requirements
* There should be SOPs for housing, environmental conditions,
feeding, handling and care of animals.
* Animals recently received from outside sources should be
appropriately isolated and their health status should be
evaluated before they are used in a study.
* At the beginning of a study, animals should be free of diseases
that might interfere with the purpose or conduct of the study.
* Animals that become diseased while under study should be
appropriately isolated to prevent infection of other animals.
* Such animals may be treated if treatment is authorized and
documented, and if the treatment will not interfere with the
interpretation of the test.
* Animals should be identified individually as appropriate (e.g.,
tattoo, ear tag) to assure that they can be identified with their
respective dose regimens and the in-life and postmortem
observations. Their individual housing units (e.g., cages)
should be labelled with all the information needed to identify
specifically each animal within the unit.
* Animals of different species and/or projects should be
appropriately separated to preclude inadvertent exposure to test
or reference substances, disease transmission or other
uncontrolled stress to the animals.
* Animal cages, racks and accessory equipment should be cleaned at
appropriate intervals.
* Food and water used for animals should be analysed periodically
for potential interfering contaminants, as specified in the study
plan.
* Bedding used in animal cages and pens should not interfere with
the purpose or conduct of the study, and should be changed as
needed to keep animals clean and dry.
* The use of any pest control materials should be approved by the
veterinarian/study director and be documented. Cleaning and pest
control materials that interfere with the study should not be
used.
2.3 Phases of animal use
The procedures listed in section 2.2 must be applied in the
various phases of animal use, which include:
* obtaining animals
* shipment and receipt of animals
* pre-study evaluation of animals
* allocation of animals to the study
* exposure of animals to the test substance
* evaluation of in-life animal response to test and reference
substances
* removal of animals from a study
* transfer of animal tissues and specimens to archives
2.4 Obtaining animals
Some laboratories obtain their animals from commercial breeders,
while others maintain their own breeding colonies. In either case,
the laboratory should have SOPs for animal procurement. These SOPs
should address the source of supply as well as the responsibility for
initiating and authorizing animal orders.
With respect to the source of animals, the SOP should
specifically identify acceptable suppliers for each of the different
species of animals used at the laboratory. Ideally, the list of
suppliers should be developed by a laboratory veterinarian or other
animal health care professional with responsibility for directing the
laboratory's overall animal health programme. Animal suppliers should
be evaluated for their ability to provide healthy animals of known
genetic and health background. The laboratory should keep records of
its animal health evaluations or should ask to review such records at
the animal supplier in order to establish which suppliers can provide
suitable animals consistently. Some laboratories require that their
veterinarian or health care professional inspect the facilities of
animal suppliers as part of the evaluation of the supplier's
suitability. Once a list of acceptable suppliers has been developed,
only animals from this source should be used. This practice, coupled
with an effective programme at the laboratory for evaluating the
health status of incoming animals, will help assure the quality of
incoming animals.
If animals are from the laboratory's own breeding colony, there
should be SOPs for the maintenance and testing of the colony to assure
the production of animals of consistent quality. Breeding colonies
should be kept separate from the areas housing animals being tested.
This helps to preclude inadvertent exposure of the breeding colony to
test substances and disease. Records of health examinations,
treatments and breeding should be kept for review by a health care
professional responsible for assessing the suitability of animals for
use in tests.
In addition to addressing the source of supply, the SOPs for
ordering animals should define the person(s) with responsibility for
ordering animals, the information required to order animals, and the
documentation to be maintained. A single individual or department
should be assigned responsibility for ordering animals. It is good
practice to have a single authority for ordering animals to assure
that the animals are obtained only from an acceptable source. This is
often the animal husbandry department, which is headed by the
laboratory's veterinarian. However, the individual or department
charged with ordering animals cannot assume full responsibility for
this function alone. The study director is responsible for assuring
that the test system (i.e. the laboratory animals) used is the one
specified in the study plan. One means of ensuring this is to require
that the study director provides a written request to the animal
husbandry department specifying the species, strain and number of
animals required for a given study.
The SOP for ordering animals should specify the information to be
included in the request. Normally, this will be the same as the
information on the test system in the study plan. For example, when
requesting animals, the study director should specify at least the
following: the species, strain or substrain; number and sex of
animals; weight range and/or age of animals; and any special
requirements such as timed-pregnant animals or surgically altered
animals. The request should also state the proposed starting date of
the study so that animals will be available on time and within the
specified age/weight range. The number of animals ordered should also
take into account the loss of animals due to shipping. The written
request for animals should be signed by the study director indicating
that the authorization and animal specifications conform to the study
plan. The written request for animals should be retained with the raw
data to document the involvement of the study director in this phase
of the study. The SOP for ordering animals should also specify what
documentation is to be retained. In addition to the request for
animals, some laboratories retain purchase orders, shipping tickets
and/or other records to document the date the order was made, that the
order conformed to the request for animals and that it was placed with
an acceptable animal supplier.
2.5 Shipping and receipt of animals
When animals are shipped from an outside source, the receiving
laboratory may not have much influence over the handling of animals
during shipment. Most reputable animal suppliers will select the
shipper and provide adequate housing, feed and water to accommodate
the animals' needs during shipment. However, the testing laboratory
should be familiar with the supplier's normal means of shipment and
inform the supplier of any special shipping requirements. For
example, a supplier may normally make deliveries to a laboratory on
certain days of the week. However, if for some reason, there will be
no one at the laboratory to receive the animals on the normal day, the
supplier/shipper should be advised. Otherwise, animals may be left
unattended in an unsuitable environment.
In laboratories that breed their own animals, transport of the
animals from the breeding colony to the areas where the animals are
received and used should be defined in an SOP for transfer of animals.
The SOP should include a description of the housing to be used during
transport as well as the precautions to assure that animals are
properly fed and watered from the time they leave the breeding
facilities until the time they arrive at their point of use. The
procedures should clearly identify the individuals responsible for
transfer of the animals and should provide for direct communication
between the breeding colony and the department that is to receive the
animals. The SOP should include any procedures required to preclude
unnecessary stress to the animal during transport. Animals must not
be allowed to remain in an uncontrolled or a stressful environment any
longer than necessary. It is also important that animals transported
in-house be clearly identified on their housing as to their origin and
destination in order to assure proper routing and use.
Unlike shipping, the receipt of animals ordered by the laboratory
from its own breeding colony is under its full control. The
laboratory should have SOPs covering the receipt of animals, which
define, at a minimum, who will be responsible for receiving animals,
where the animals are to be received, which tests should be done
immediately upon receipt, and what documentation should be kept.
At the time of receipt, animals are usually counted and assigned
temporary numbers or other means of individual identification. They
are placed in standard housing units for transfer to the area where
their health status is to be checked (i.e. quarantine). There should
be an SOP that covers identification and housing of animals during
this pre-study evaluation period. Animals should be identified so
that observations of their health can be clearly documented for the
individual animal.
Documentation of animal receipt is important information and
should be kept with the other study records. It should cover the
following details: where the animals were shipped from; the date and
time they were received at the laboratory; who received the shipment;
the general condition of the shipment, including a description of the
number of containers; and the number of live, dead and moribund
animals in each container. The assignment of animals to cages for
transfer to the pre-study evaluation area should be documented and
should include the actual number of animals transferred. Any other
information obtained for each animal during the receipt phase should
also be recorded. For example, animals may be weighed or, in the case
of some large animals, vaccinated or otherwise treated. This
treatment should be recorded. It is useful to retain a copy of the
shipping tickets and animal container labels in the study records in
order to document the receipt of the animals.
2.6 Animal care facilities
Depending on the nature of a laboratory's work, animal care
facilities may comprise a large portion of the physical plant. The
laboratory should have a sufficient number of animal rooms or areas to
assure proper separation of species or test systems, isolation of
individual projects, quarantine of animals, and routine or specialized
housing of animals.
The need for separation of species should be evaluated by a
veterinarian or animal health professional to assess where separation
by species is required to prevent disease or stress in the animals.
Although not ideal, it is possible to house different species together
where it has been determined that such housing arrangements will not
potentially affect the health of the animals or otherwise adversely
affect the study. This is not a desirable practice, however, and
should be avoided.
Individual projects should be isolated to avoid potential
confusion and cross-contamination of animals. As a general rule,
animals being used in separate projects should be housed in different
rooms. This is especially true in feeding studies where environmental
contamination may be present due to dust from handling dosed feed and
spillage of feed by animals. Although it may be permissible to house
two different projects together under some conditions (e.g., same test
substance, same species), it is generally inadvisable to house
projects together that use different test substances. In any event,
precautions should be taken to minimize or eliminate exposure of
laboratory personnel to the test substance.
Facilities for quarantine or isolation of animals are essential.
Laboratories should have space available to isolate animals
effectively, when necessary, to avoid adverse effects on the conduct
of studies. For example, many laboratories quarantine incoming
animals in the room where the study will be conducted. They are held
in that room until their health status is evaluated and then they are
placed on study in the same room. Thus, they are effectively
quarantined from other animals at the laboratory, even though there is
not an animal room strictly dedicated for quarantine.
Areas for routine or specialized housing are required. Routine
housing of animals may include the maintenance of a breeding colony or
the housing of animals in a routine study, such as a chronic feeding
study. Specialized housing should be available where required. For
example, studies requiring collection of metabolism or behavioral data
may require special caging or environmental controls. Also, animals
used in inhalation studies may be housed in special inhalation
chambers. The laboratory management should assure that these and all
other facilities are available as required.
In addition to the animal facilities discussed above, a
toxicological laboratory should have a number of animal rooms or areas
separate from these to ensure isolation of studies being done with
test systems or test and control substances known to be biohazardous,
including volatile substances, aerosols, radioactive materials, and
infectious agents. These areas should include facilities and
equipment for disposal of waste and other contaminated material from
this area, and special equipment or clothing to protect laboratory
personnel.
Separate areas should also be provided, where appropriate, for
the diagnoses, treatment and control of laboratory animal diseases.
These areas should provide effective isolation for the housing of
animals either known or suspected of being diseased or of being
carriers of disease from other animals. The extent of, and the need
for, these facilities will depend upon the testing laboratory's policy
regarding the handling of diseased animals. Many laboratories using
rodents have a policy of immediately removing and discarding any
animal that develops disease. Such a policy would essentially
eliminate the need for any facilities dedicated to diagnosis and
treatment of disease in these animals. However, when primates, dogs
or other larger animals are used, they frequently are treated for
disease. In this case, special areas should be provided for such
diagnosis, treatment and isolation. The extent to which diseased
animals need to be isolated to prevent adverse effects on other test
animals will require the judgment of the laboratory veterinarian or
animal health care professional. However, should isolation be
required, facilities must be available. The need for dedicated
treatment facilities will also vary depending on the nature of the
treatment. Treatment that is highly stressful to the animal or which
includes aseptic procedures, such as surgery, should not be performed
in the animal room. On the other hand, oral administration of
medication and monitoring of body temperature are examples of
diagnostic and treatment procedures that may be carried out in the
animal room.
Adequate facilities for animal care must include facilities for
collection and disposal of animal waste. In testing facilities that
do not dispose of the waste directly, there must be facilities for
safe sanitary storage of waste until it can be removed from the
laboratory for disposal. Disposal facilities should be designed and
operated to minimize vermin infestation, odours, disease hazards and
environmental contamination. Facilities for storage and disposal of
waste should be adequately separated from animal areas to preclude
contamination of the animals, and schedules must be established,
followed and documented to assure the timely performance of waste
removal activities (e.g., animal cage changing).
A schedule for routine maintenance of buildings should be
established. Although many laboratories have such schedules for
equipment, many do not have building maintenance schedules. The need
for such schedules is most easily illustrated in the area of animal
facilities. These facilities receive much wear from both animals and
personnel. Walls, floors and ceilings may crack, making these
surfaces uncleanable or exposing underlying surfaces that may produce
dust. Paint may also crumble. Light fixtures and doors wear out.
All of these conditions may have an adverse effect on the animals
housed in the facility. It is difficult to repair some of these
conditions after a study is underway without adversely affecting the
animals' environment. Consequently, maintenance should be scheduled
to eliminate the need for repairs while studies are underway. This is
especially true for rooms used to house animals in chronic studies.
Of course, consideration must always be given to the potential impact
on the study of chemical agents used in building maintenance and
cleaning. Similarly, attention must be paid to the possible
environmental contamination of the building during construction,
renovation or repair of laboratory facilities, e.g., the use of
certain plastics that leach out toxic chemicals or asbestos used in
ceilings or to protect pipes. A schedule of routine preventive
maintenance for buildings will ensure proper cleaning and maintenance
of all areas.
2.7 Animal husbandry supply facilities
Storage areas should be supplied, as needed, for feed, bedding,
supplies and equipment related to animal care and use. Storage areas
for feed and bedding should be separated from areas housing the test
systems and should be protected against infestation or contamination.
There should also be facilities, as needed, for the preservation of
perishable supplies, e.g., certain feeds requiring refrigeration.
Most large testing facilities maintain central storage areas for
bulk storage of feed and bedding. In the case of feed, the individual
responsible for running the bulk feed storage facility will dispense
small lots of feed to the various animal areas for short-term storage
and immediate use. These small lots are often stored in separate
rooms in the immediate animal area under the responsibility of animal
husbandry personnel working in the area. Feed may then be further
dispensed to relatively small storage containers in the actual animal
rooms. Feed is dispensed from these containers into feeders for the
animals. Despite the difference in size and location of feed storage
facilities at the testing laboratory, they must be protected against
infestation or contamination. This is generally not a problem where
feed is stored in designated areas outside of the animal rooms. In
these designated areas, feeds are bagged and stored to facilitate
stock rotation and to preclude contamination with cleaning agents.
The chance of vermin infestation is also reduced. However, if feed is
to be stored in animal rooms, special precautions must be taken.
Firstly, only small amounts of feed should be permitted to be stored
in the room, i.e. amounts of feed necessary for two weeks or less.
Secondly, the feed must be stored in sealed vermin-proof containers
that can protect feed from contamination during cleaning of the room.
Thirdly, the feed must be clearly identified and stored in an area
separate from the animal housing unit and designated for no other
purpose. Finally, there should be procedures in place to assure the
proper rotation of feed. Often, new feed is dumped into a container
in the room without emptying it of the previous feed. This practice,
if continued, may lead to contamination of the feed with mould and
accompanying mycotoxins.
There should be facilities for the storage and handling of
bedding so as to prevent contamination and vermin infestation. Some
laboratories store substantial amounts of bedding in the area where
cages are cleaned. This bedding is sometimes placed in the cages
prior to their transport to the animal room. In this case, the
facilities must be adequate to protect the bedding from contamination
by equipment cleaning agents and dirty equipment brought to this area
for cleaning.
Finally, animal supply facilities should provide areas for the
storage of clean and dirty equipment, as well as facilities for
cleaning it. Such equipment would include cages, racks, water
bottles, feeders, and similar items. Some facilities have corridors
and rooms dedicated strictly to either clean or dirty equipment. The
laboratory and flow of work are designed to prevent the mingling
and/or mixing of clean and dirty equipment. Although designated
clean/dirty facilities are ideal, an adequate degree of separation can
be maintained by scheduling work so that corridors and cleaning areas
are not occupied by clean and dirty equipment at the same time.
2.8 Facilities for handling test, control and reference substances
The facilities must allow separation of areas involved in the
storage, handling and distribution of test, control and reference
substances and of mixtures containing them. This is necessary to
prevent contamination of facilities, equipment, personnel and test
systems, as well as to prevent confusion of substances. To accomplish
this, there should be separate areas for:
a) receipt and storage of test, control and reference substances;
b) mixing of test, control and reference substances with a carrier
(e.g., feed);
c) storage of the test, control and reference substance mixtures.
In addition, storage areas for the test and control substances
and test and control mixtures should be separate from areas housing
the test systems. The storage facilities should be adequate to
preserve the stability of the substance and mixtures. For example,
adequate refrigerator or freezer storage space should be available for
substances requiring low temperature storage.
2.9 Pre-study evaluation of animals
A fundamental laboratory practice is to place all newly received
animals in quarantine until their health status has been evaluated.
The procedures for doing this vary from laboratory to laboratory.
Some maintain animal colonies under barrier conditions with extensive
programmes for the evaluation of animals entering the barrier. These
include thorough screening for bacterial, viral and fungal pathogens.
Sentinel animals may also be maintained in the room housing the
animals under test. These sentinel animals are evaluated in the same
manner as the animals sampled during the pre-study health evaluation.
Whatever pre-study evaluation is employed, a laboratory should
have SOPs to assure that the animals' health status is fully evaluated
prior to the test. This evaluation may be performed by the laboratory
veterinarian, animal care employees or the study director. These
individuals should have documented training and experience that
qualifies them to recognize symptoms and to diagnose disease problems
for the animal species involved.
A laboratory should have facilities that can adequately separate
newly received animals from those already housed, and a designated
quarantine area is desirable. Laboratories may quarantine newly
received animals in the room where they will be housed during the
study. This is acceptable provided that employees follow adequate
procedures to prevent cross-exposure of other animal rooms with
equipment, waste or other materials from the rooms where the animals
are housed for pre-study evaluation. The use of the same room for
pre-study health evaluation and conduct of the study also permits the
acclimatization of animals to the actual environmental conditions in
the room prior to the beginning of the study. The facilities required
for housing animals during quarantine are generally the same as for
housing animals being tested. There should be an adequate number of
cages of appropriate design, construction and size for the species to
be housed. There should also be facilities for the removal and
disposal of animal wastes, and adequate environmental controls for
temperature, humidity and lighting. Technical guidance regarding
facilities, equipment and proper animal husbandry procedures may form
part of government regulations for animal welfare.
SOPs for the pre-study health evaluation of animals should define
the parameters to be monitored for each species during the pre-study
evaluation period and the frequency of observations. For example, the
SOP might require rodents to be observed daily for mortality and
clinical signs of illness such as rough hair coat, diarrhoea, laboured
breathing and weight loss. A very important SOP topic is the length
of the pre-study evaluation period. Animals should be isolated long
enough to provide reasonable assurance that the most common diseases
affecting that species will have ample time to manifest themselves
clinically. This time varies from species to species and should be
based on acceptable veterinary medical practice. The SOP should
identify who will make the observations, define the circumstances
under which a veterinarian or the study director should be called, and
what, if any, treatment is to be given to the animals. Any signs of
illness observed or treatment rendered should be documented in study
records as described in the SOP or study plan. Records of clinical
signs of illness should include entries specifically identifying
individual animal(s) involved, with a detailed description of the
clinical features, the date and time of onset and duration. The
records should identify who made the observation.
Records of treatment should include the name of any medications
used and the complete details of their administration, including the
date of each administration and the amount given. Records should
clearly indicate when treatment began and ended for each animal and
who authorized treatment; the laboratory veterinarian, in consultation
with the study director, should authorize all treatment. These basic
requirements apply whether treatment is administered during the
pre-study evaluation period or during the study itself. In any case,
no treatment should be administered that will interfere with the
purpose or conduct of the study. All records of animal observations
and treatment made during the pre-study evaluation period should be
retained with the study records.
The SOP covering pre-study evaluation must reference the SOP for
housing, feeding, handling and care of the animals. It should specify
the types and sizes of cages to be used, feeds and feeding schedules,
bedding schedule and procedures for changing and cleaning cages and
equipment, procedures for watering animals, and procedures for
monitoring and documenting environmental conditions such as lighting,
temperature and humidity. The SOP should specify acceptable ranges
for these parameters and remedial action to be taken when readings are
outside these ranges. Like the SOP for treatment of animals, the SOPs
for the housing, feeding, handling and care of animals is general and
required for all animals whether the animals are in quarantine, on
test or in the breeding colony.
2.10 Allocation of animals to a study
Most laboratories document the suitability of the test animals at
the end of the quarantine period by means of a form signed by the
veterinarian in charge of quarantine, which authorizes the release of
the animals for use in the study. The study director is notified and
the animals are transferred to the room where the study will be
conducted if they are not already housed there. At this point,
animals are ready for allocation to the study.
Three important events occur during the allocation to study
phase. These are:
* Animals are evaluated to determine if they meet study plan
requirements such as age, weight and physiological condition.
* Animals are permanently identified so that they can be
specifically related to any data or specimens generated during
the test. Animal housing units are also identified so that all
information needed to identify each animal within the housing
unit is located on that unit.
* Animals are allocated to the respective test groups in a manner
that precludes bias (i.e. they are randomized).
Related procedures and documentation should exist to assure that
each one of these vital study functions is properly performed. In the
animal evaluation, some overlap may occur with the quarantine phase of
the study. For example, it may be necessary to use animals that have
specific blood chemistry values. To establish these values, several
measurements may be required over a period of weeks. Blood samples
may have already been drawn and analysed for several weeks prior to
release of animals from quarantine. Now, however, the results of
those blood analysis must be compared against the study plan
requirements, and the study director must decide which animals qualify
for study. Also, weight requirements and other parameters that must
be met at the time of study initiation should now be evaluated for
conformance to the study plan. These are some examples of pre-study
evaluations that are made during the allocation phase of the study.
They are dictated by the study plan, and the study director has the
final say as to whether the criteria are met. The most important
record-keeping requirement at this point includes all records of
laboratory reports and other observations made to determine if the
animals conform to study plan requirements. There should also be a
record of the individual reviewing this documentation and authorizing
the animals for use in the study.
At this point, animals should be individually identified in such
a way that they can be traced for any in-life or postmortem
observation. This is often done by the use of tattoos or ear notches.
Whatever method is chosen, it should be thoroughly described in the
SOP for identifying animals. The method selected should not interfere
with the study, should not separate from the animal, and should not be
obliterated over the course of the study. It should also be easily
read by animal technicians and all other laboratory personnel handling
animals. It is especially important to provide adequate instruction
to animal technicians on the proper reading of the identification code
used. For example, a system of ear notches may be used to represent
animal numbers.
Such a code should be fully explained in an SOP maintained in the
animal room for ready reference. The SOP should also cover how
numbers are assigned for a given study to assure their traceability.
Study records must very clearly document the assignment of permanent
animal numbers in such a way that they can be accurately
cross-referenced with any temporary number assigned during quarantine.
This is necessary to maintain data integrity for a given animal from
receipt to disposal.
In addition to identifying individual animals, housing units
should be marked to indicate which animals are housed in the unit.
Most laboratories utilize cage cards or labels for this purpose. The
cage identification should include the number, sex, dose group and
study number for the animals housed in the cage. This practice helps
assure that the animals are selected from and returned to their proper
cage during cage-changing operations and during removal of the animals
for required study observations.
The allocation of animals to their assigned test groups is
another important study event. Once animals have been appropriately
evaluated and approved for use on the study, they must be divided into
treatment groups. In a typical study, there are four treatment groups
of animals for each sex: control, low-dose, mid-dose and high-dose.
Additional groups may be required as dictated by the study plan.
Animals should be placed in these groups in a manner that is designed
to eliminate bias. This is accomplished by using a procedure that
randomly assigns animals to a given treatment group. The procedure
for randomizing animals should be described in a written SOP or the
study plan. The allocation of animals using the specified procedure
should be fully documented.
In addition to randomizing animals, many laboratories employ
procedures to eliminate bias in the housing of animals. This is
necessary because, even in the best animal facilities, environmental
conditions within the room will vary. For example, there may be areas
of lower air circulation or uneven heating and cooling.
The microenvironment of the animal cages in a single rack housing
multiple cages may also vary. For example, animals in higher cages
may be exposed to more intense light and higher temperatures than
animals in lower cages. To overcome this, most laboratories have
established SOPs for the placement and rotation of animal housing
units within a rack and racks within a room. Commonly, in rodent
studies, cages housing animals of a single dose group are placed
together in a structured fashion to facilitate easy identification of
the group during observation, feeding and dosing. The cages are then
moved systematically as a group through all locations within the rack
during the course of the study. The racks themselves are
systematically moved through all locations in the room. This assures
uniform exposure of the test animals to the full range of
environmental variations within the room. The moves are often
performed with the same frequency as other animal room procedures,
such as cage changing. The moves should be documented by recording
the date of the move. Laboratories may use diagrams of racks and
rooms to document the movements and specific location of racks and
cages.
2.11 Exposure of animals to a test or control substance
There are two major concerns during this phase of the study.
Firstly, known quantities of defined substances should be administered
to specifically identified test animals over a given period of time.
Secondly, test animals must not be exposed to uncontrolled stress that
may affect their response to the test substance. Both of these
concerns can be met by appropriate care and use of laboratory animals.
The administration of substances should be carried out in
accordance with the SOP and/or the study plan requirements to assure
proper treatment of the test animals. A laboratory should have SOPs
to cover fully every aspect of receipt, testing, preparation,
distribution and use of test and control substances. Laboratory
procedures should ensure that properly defined and known quantities of
correctly labelled test and control substances are delivered to the
laboratory personnel for administering to the test animals. Personnel
should have their SOPs and study plan readily available to assure
proper administration of the materials. The study plan should specify
the route of administration and the dosage levels. However, there
should also be SOPs to cover the technical aspects of administering
the substances, as well as specific procedures to prevent confusion of
animals or test and control substances during administration.
Examples of SOPs for dosing animals might include instructions on
how to insert a stomach tube for administration of materials by oral
gavage or how to administer substances intravenously. Examples of
SOPs designed to prevent confusion would include procedures that
address the order in which the animals are to be dosed, how many
animals can be removed for dosing at one time, and procedures for
verifying animal and test substance identification. The SOP or study
plan directions for the administration of test substances should
provide directions on how to document the administration of the test
and reference substances. This is crucial to the subsequent
validation of study findings. Such documentation usually consists of
a dosing record kept by means of a standard form. Documentation
should include the following information: the name, lot number and
expiration date of the test and reference substances, as applicable;
the exact quantity of substance administered; the specific identity of
the individual animal receiving it; the date, time and duration, as
applicable, of administration; and the names of the individuals
administering the dose. The individuals involved should sign or
initial the record. During test and reference substance
administration, the fact that each animal was dosed and the specific
weight or volume of dose that each animal received must be recorded.
If the test substances must be prepared or manipulated by technicians,
this should also be documented. It is equally important to document
positively that control animals were treated according to study plan
requirements.
2.12 Control of laboratory environment
The second major concern during the dosing phase is to prevent
the exposure of test animals to uncontrolled stress. This is
important throughout the in-life portion of the study. Uncontrolled
stress can be caused by numerous factors: these include inadequately
controlled animal room environment; disease; contaminants in feed,
bedding and water; exposure to cleaning agents, pesticides and
uncontrolled test substances; poor husbandry practices; improper
handling; and inadequate facilities.
To prevent interference from environmental factors, the
laboratory must have animal facilities that will provide a controlled
environment appropriate for the species housed. This generally means
that temperature, humidity, lighting, ventilation, noise and personnel
activity in the animal rooms should be monitored and/or controlled.
The monitoring and control activities should be recorded.
Laboratories housing rodents should have the capability to
control temperature and humidity. There should be SOPs to define
acceptable ranges for environmental parameters and to describe actions
to be taken if acceptable ranges are exceeded. Some laboratories
continuously monitor and record the temperature and humidity (by means
of hygrothermographs). The recording charts from such devices should
be regularly checked and retained as study records. The charts should
be initialled and dated by the individual checking them and there
should be explanations for any reading that is abnormal. Such
readings should be brought to the attention of the study director so
that their impact on the study can be assessed.
Although they are desirable, hygrothermographs or other elaborate
temperature and humidity recording devices are not absolutely
essential. Many laboratories monitor these parameters with
nonautomatic instruments on a twice daily basis to coincide with other
activities that are being performed. All that is required is an
accurate thermometer and a hygrometer or sling psychrometer. If these
are used there should be SOPs for their use and the collected records
should be reviewed and retained as raw data. If temperature is to be
observed only once a day, it is desirable to have a thermometer that
will show the minimum and maximum temperatures reached over the 24-h
period. It is also good practice to have any temperature monitoring
device periodically calibrated against an official standard to assure
accuracy. This also applies to thermocouples or other electronic
sensors used to monitor temperature and humidity.
Other environmental factors that affect animals include lighting,
noise and ventilation. Lighting is often controlled by the use of
timers to cycle lights on and off on a regular schedule. For small
animals, it is common practice to provide equal periods of light and
dark over a 24-h period (i.e. 12 h on, 12 h off). Lighting cycles
should be monitored periodically to assure that they meet study plan
requirements. Such monitoring should be recorded. This sometimes
presents a problem and may be overlooked when the light cycles occur
after normal working hours. To overcome this problem, some
laboratories have SOPs that direct the security or building
maintenance personnel to monitor and document the light cycles after
working hours. This is an acceptable practice, provided accurate
documentation is maintained. It is also good practice to have animal
room doors identified with the name and telephone number of the study
director or other responsible individual so that they may be contacted
should a problem with the animal room be detected after working hours.
Noise in animal facilities is generally controlled by facility
design and proper placement of animals. In most laboratories, noise
usually comes from three sources: animals, equipment and personnel.
To control animal noise, noisy animals such as primates and dogs
should be housed separately from other animals. Their housing should
have doors and windows designed to reduce noise transmission between
rooms and provide for arrangement of animals to accommodate their
social behaviour. For example, animals housed so that they can see
each other are often quieter. Limiting the entrance of unnecessary
personnel into these areas will greatly reduce noise levels.
The most stressful noise probably originates from equipment and
personnel. For example, cage washing operations often generate sharp
irregular and/or continuous noise. Noise from these sources can be
controlled by locating these areas out of hearing range of animal
housing areas. Employees slamming doors, moving racks and cages, and
playing radios can also create uncontrolled stress in the animals.
Ventilation of animal rooms should always be controlled and
monitored. Proper ventilation with clear, filtered air is necessary
to maintain acceptable temperature and humidity levels and to remove
offensive odours or other airborne contaminants. SOPs for animal care
and housing should specify acceptable air exchange rates for animal
rooms. These should be monitored and documented periodically.
Another factor that may produce uncontrolled stress in test
animals is disease. In cases where animals become diseased during a
study, the disease may be treated. However, such treatment must be
fully authorized and documented as discussed in section 2.6, and the
study director must determine that treatment will not interfere with
the purpose or conduct of the study.
Contaminants in feed, water and bedding can adversely affect the
animals. To preclude this, feed and water used for animals should be
analysed periodically to ensure that contaminants known to be capable
of interfering with the study, and reasonably expected to be present
in such feed and water, are not present at levels above those
specified in the study plan. Documentation of such analysis should be
maintained as raw data. The study director should identify any such
contaminants in the study plan. Likewise, bedding should be free of
any interfering contaminants or naturally occurring constituents that
could interfere with the test. For example, pine and cedar shavings
may contain naturally occurring aromatic hydrocarbons that can affect
the liver metabolism of laboratory animals.
Other potential contaminants may be introduced into the animals'
environment in the form of cleaning agents, pesticides and test or
control substances from different studies. There should be SOPs
covering the identification, use and monitoring of these agents. For
example, the facility should have an SOP detailing the cleaning agents
acceptable for use in animal facilities or on equipment coming into
contact with animals. This would include detergents and sanitizers
for cleaning floors, walls, ceilings and counter tops in animal rooms,
as well as these same agents used to clean cages, pans, water bottles,
racks and feed mixers. The SOP for cleaning must also include
procedures for documenting that cleaning was performed and that the
agents used were in accordance with the SOP. A similar SOP should
exist for any pest control materials that are used. The SOP should
identify which chemical pesticides are approved for use, the method of
application and the specific locations where they can be used. The
SOP should include the provision that responsible laboratory personnel
should accompany any contract pest control personnel when they apply
pesticides to assure that they are applied in accordance with the SOP.
This should be documented in writing. Many laboratories forbid the
use of any pesticides and instead rely on mechanical traps and
barriers. The use of these should also be described in the SOP.
Other sources of uncontrolled stress to animals include
laboratory personnel and poor husbandry practices. There should be an
SOP regarding the admittance of personnel to the animal facilities;
this should identify which employees are permitted in the animal rooms
and under what circumstances. It should also describe any special
health or safety precautions necessary to protect employees and
animals. For example, the SOP should describe any protective clothing
to be worn or decontamination procedures to be followed by personnel
entering or leaving animal areas. Many laboratories have procedures
designed to control disease and environmental contamination of the
animal colony by prohibiting movement of employees between different
animal facilities or laboratories. SOPs should also exclude from
direct contact with animals an employee with an illness that may
adversely affect animal health. Such employees should be reassigned
until their health recovers.
Poor husbandry practices can be avoided by the use of adequate
SOPs covering housing, feeding, handling and care of animals. This
should include procedures to assure that animal cages, racks and
accessory equipment are cleaned and sanitized at appropriate
intervals. SOPs should also specify minimum acceptable cage sizes and
room capacities to prevent animals being overcrowded. Every activity
performed in the animal room during a study should be documented. To
do this, some laboratories keep room activity logs. These records are
maintained in the animal rooms during a study. Each time anyone
enters the room they must sign the log documenting who they are, when
they entered the room, what activity they were performing, and when
they finished and left the room. This is a good practice for
controlling and monitoring the level of activity in an animal room.
Excessive activity can increase stress in the animals.
These animal care and use procedures are not all inclusive but
they do cover the major factors. The procedures apply not only during
the exposure of the animals to the test substances, but also to every
phase of studies involving the maintenance and use of animals in
laboratories.
2.13 Evaluation of in-life animal responses to test and control
substances
This phase of animal use covers the period from the first
administration of the test substances until the animals are submitted
for postmortem evaluation. It is during this phase that crucial data
are obtained by observing the response of test animals to exposure of
test and reference substances.
In a typical chronic rodent study, animals are observed as
specified in the study plan and/or SOPs. Examples of observations to
be made include the following: general physical examination; daily
observations for mortality and toxicological or pharmacological
effects, body weight and feed consumption measurements; palpation for
tissue masses; and special procedures such as ophthalmoscopy. The
study plan must specify the frequency of these observations. Also,
each of these procedures should be covered by the study plan or an
SOP; SOPs often complement the study plan by providing specific
guidance. For example, the study plan may simply state that animals
are to be observed for toxicological effects. The SOP may define such
effects as lethargy, hyperactivity or convulsions.
SOPs can standardize observations by prescribing the use of
standard terminology and methods. For example, in palpating animals
for tissue masses, the SOP should require that the location and size
of masses be reported. It should define the limits of specific
location description, such as dorsal front right, to distinguish
masses in this area from adjoining areas. The SOP should also require
that masses reported in a previous observation be positively accounted
for during each subsequent observation session from the time they
appear until the time they regress or are observed at postmortem
observation. SOPs should also cover how and where observations will
be recorded.
Proper identification of animals is important because all
observations made must be related to specific animals. There should
be SOPs that assure verification of the accuracy of animal
identification during dosing, observation and transfer of animals.
All documentation should clearly identify who made an observation,
when it was made and to which animal it pertained.
An important consideration is the training of personnel. It is
crucial to have employees who are trained and experienced in working
with animals to make in-life observations. All personnel should have
documentation of their training and experience. Complete familiarity
with a species and its characteristics is essential to detect the
often subtle clinical changes that may be indicative of toxic effects
of the test substances.
2.14 Removal of animals from a study
Animals may be removed from a study because they die while the
study is in progress, because they become sick or because they are
sacrificed at the times specified in the study plan. In these cases,
it is important that proper identification of the animals and related
specimens is maintained and that the animals are submitted promptly
for postmortem evaluation to preclude the loss of data due to
autolytic changes in tissue. This is accomplished by requiring
multiple daily checks for mortality and morbidity and specifying
after-hours procedures for storage of dead animals.
Documentation should be maintained for unscheduled removal of a
dead or moribund animal and include, at a minimum, the identity of the
animal, the date and time of removal, the reason for removal, and the
identity of the individual who removed the animal. For moribund
animals, the reason for removal should include specific observations
of animal behaviour and physical condition. The study records should
clearly indicate whether the animal was sacrificed or found dead.
Animals that are killed accidentally during the study should be fully
reported.
The same general requirements, with respect to the required
documentation, apply to scheduled deaths. An important requirement in
the scheduled sacrifice of animals is that documentation should be
maintained to demonstrate the method of sacrifice used. This may be
specified in the study plan or SOP, but should include a method that
will not interfere with postmortem evaluation of animal tissues and
specimens.
Postmortem evaluation includes gross necropsy, histological
preparation of tissues and organs, and histopathological evaluation.
A major concern is integrity of animal identification. There should
be procedures covering the identification of animals and their
respective tissues and specimens from the time they are submitted for
postmortem evaluation until they are sent to storage.
There should always be positive identification attached to or
accompanying each animal. Many laboratories have a form that
accompanies the animal to necropsy, identifying it by number, sex,
dose group and study. For animals that are individually numbered with
ear tags, tattoos or other permanent identification, SOPs generally
require the necropsy personnel to compare the animal received against
the form to confirm proper submission. Animals are then necropsied
and tissues, organs and specimens are collected and processed as
directed by the study plan. Each of the items collected should be
identified in a manner that relates it back to the animal it came
from. This is often accomplished by placing unique identifying
information on containers used in the storage or processing of these
tissues, organs or specimens. A unique sequential accession number
can be assigned to containers that relate to the animal, or the unique
animal identification number may be utilized. Whatever method is
used, it must be traceable back to a specific animal in a specific
test. The procedures used must be fully described in the SOP. Not
only must tissues be identified, but all documentation for the
receipt, preparation and evaluation of tissues must be identified to
relate them to a specific animal from a specific study. The records
of gross necropsy findings, microscopic findings and in-life
observations must account for all gross observations. For example, if
an animal technician reports four tissue masses during the last
in-life observation period of an animal prior to submission for
necropsy, the necropsy records must confirm the presence or absence of
each of the masses reported.
To achieve this, the last in-life observations must be available
to the individual making necropsy observations. Each mass accounted
for at necropsy should be uniquely reported and identified in the
gross necropsy findings for the animals. Trimming and tissue
processing records must also account for masses collected at necropsy
for processing. Finally, necropsy findings should be provided to the
pathologist making microscopic evaluation of the tissues. Records of
this evaluation, likewise, must account for all tissues, masses and
lesions reported. To facilitate tissue accountability for animals
with multiple masses, number or letter designations should be assigned
to each mass at necropsy. The method used to assure identification
and accountability of animals and tissues during this phase of the
study must be fully described in the SOP.
2.15 Transfer of animal tissues and specimens to archives
Safety studies generally produce large amounts of data and
specimens. To accommodate their storage and retrieval, a laboratory
should have space designated as an archive for all raw data and
specimens from completed studies. This area should have access
limited to authorized personnel only. Although most laboratories have
a centrally located archive to support the whole testing facility,
some have a number of archives. For example, some support
departments, such as histology and chemistry, may maintain space for
storage of data and specimens they generated or analysed. This is
acceptable, provided access to these materials is limited to
authorized personnel and the data and specimens are properly
identified and indexed as to their location. The best situation,
however, is to have archive facilities available to provide central
storage of all data and specimens. These should be under the control
of a designated individual.
After evaluation of animal tissues and specimens, the final phase
is the transfer of these materials to the archives. At this point,
the gross remains of animals are placed in sealed containers while the
tissues are contained in blocks and slides. There should be an SOP to
assure that all these items are properly identified, accounted for,
inventoried and placed in the archives. It is the study director's
responsibility to assure that all specimens are transferred to the
archives at the conclusion of the study.
3. QUALITY MANAGEMENT APPLIED TO HUMAN AND ENVIRONMENTAL MONITORING
STUDIES
3.1 Introduction
The manufacture and use of chemicals can lead to the deliberate
or unintentional exposure of natural ecosystems to potentially
hazardous substances. In this case, humans may be affected by contact
with the chemicals through the consumption of food and drinking-water,
and air inhalation. The natural environment can change its structures
due to effects on the complex processes governing the functions of
ecosystems.
In recent years, the awareness of risks of adverse health effects
due to exposure to various environmental factors has increased
substantially. In many countries major health problems and nuisances
are still related to particulate matter, sulfur dioxide and
pesticides. In the more developed industrialized countries the
interest has shifted to effects such as cancer, allergy and disturbed
function of the central nervous system, caused by exposure to
pollutants such as hydrocarbons, nitrogen oxides and toxic metals. In
the future, interest is likely to focus on environmental health
effects, e.g., effects on the nervous and immune systems, that are
more difficult to detect and often appear after long periods of
exposure to relatively low doses of specific environmental factors or
to combinations of different factors.
One aim for continuous health surveillance is to follow the
development of morbidity and mortality patterns within various
population groups. This requires access to extensive descriptive
data. In order to relate observed health effects to certain
environmental factors, reliable exposure data are needed. It is
important that observed changes in the health effect pattern are
validated on a local level. Many observed changes have been shown to
be caused by artefacts.
The function of an ecosystem is determined by the physical and
chemical processes, as well as by the relationships and interactions
of the living organisms within the system. With the overall aim of
the safe manufacture and use of chemicals, several objectives are
covered by monitoring studies in natural environments. These can be
described, for instance, as the monitoring of the levels of chemicals
in various compartments within ecosystems, i.e. a descriptive study
of situations and trends. Furthermore, programmes monitoring the fate
and effects of chemical substances in the field are carried out in
order to compare the results with existing information from similar
studies carried out in the laboratory, e.g., the investigation of
exposure-response relationships for risk assessment. In addition,
monitoring of contamination levels in a selected environment is
performed in order to evaluate compliance with specific environmental
quality criteria or targets, for instance, from a regulatory point of
view.
Consequently, in environmental monitoring studies of natural
ecosystems, it is predominantly the concentrations of chemicals,
transformation processes and products which are investigated, as well
as accumulation and ecological effects in wildlife, and the abundance
and distribution of species. For this purpose, environmental
monitoring studies are carried out in different compartments of
various ecosystems, e.g., sediments and soils, air and water, and
terrestrial and aquatic biota.
The incorporation of quality management approaches in
environmental monitoring studies should ensure that the resulting data
are reliable and reconstructible. Quality management should cover all
phases of an environmental monitoring study, i.e. the planning of the
study, selection and preparation of sampling equipment, sampling
procedures, analyses, measurements and observations, and the
reporting.
Reliable exposure data are essential for the establishment of
dose-response relationships for toxic effects of environmental
pollutants or other chemicals in human subjects, in epidemiological
studies and for the assessment of risks of adverse health effects upon
contact with such substances. Traditionally, pollution monitoring has
been concerned primarily with the movement of pollutants through the
environment and the concentrations in various environmental media such
as air, food and water. Such monitoring provides little information
on the amounts of pollutants actually coming in contact with people.
Direct measurements of human exposure levels may involve
determination of the chemical under study in air, food and water,
preferably through personal monitoring, or determination of the
chemical or its metabolites in tissues or body fluids (biological
monitoring). Ideally, the concentration of a pollutant, or its
metabolites, in an indicator medium will give information on the
degree of exposure, the dose at the critical organ and the risk of
adverse health effects. Some information concerning the type and
degree of exposure may also be obtained from determinations of
pollutant concentrations in micro-environments combined with studies
of human activity patterns.
The World Health Organization (WHO) and United Nations
Environment Programme (UNEP) have been involved in human exposure
monitoring since 1977, when global studies on the biological
monitoring of lead and cadmium (Braux et al., 1979; Vahter, 1982;
Friberg & Vahter, 1983; Bruaux & Svartengren, 1985; Vahter & Slorach,
1989) and organochlorine compounds (Slorach & Vaz, 1983) were
initiated. On the basis of this work the WHO health-related
programme, which involves monitoring of pollutants in air, water and
food, has included a new component, the Human Exposure Assessment
Locations (HEAL) programme. Initially, the HEAL programme has focused
on methods for exposure monitoring, including methods for quality
assurance. General principles and procedures for the development of
quality assurance in relation to exposure monitoring have been
prepared (UNEP/WHO, 1984; WHO, 1986b). A comprehensive document on
quality assurance in biological monitoring of metals has been
published (Friberg, 1988).
3.2 Procedural requirements
A crucial point in exposure monitoring is to ensure correct
sampling, e.g., that the collected air particles, food, blood and
other samples really cover a representative period and that the
correct sampling procedures have been used. As in other types of
studies, it is important that the field personnel and the laboratory
staff are properly trained. In many exposure monitoring activities
the subjects under study collect the samples themselves, e.g.,
duplicate diets. In this case it is necessary to train the subjects
properly, and to have staff available for assistance or advice during
the entire sampling period. A study plan and detailed SOPs for the
sampling procedures should be prepared and discussed with all the
people concerned. It is also important to motivate the subjects and to
give them adequate information about the aim and the design of the
project before starting collecting the samples.
The SOPs should cover all aspects of the monitoring exercise,
particularly the sampling procedure, use of equipment and materials,
transport and processing of samples, and analytical and observation
methods. For example, a description of the equipment to be used for
field sampling should take into account that outdoor application
requires robust constructions, simple handling and easy-to-read
instructions. Careful transportation is necessary to maintain the
functioning of equipment. Furthermore, calibration procedures should
be established which appropriately consider that the performance of
many instruments, not particularly designed for outdoor use, may alter
under field conditions; the laboratory calibrations may not be valid.
The SOP should consequently define how the functions of the
instruments for field measurements are to be tested.
The correct, unambiguous and non-erodible labelling of samples
and specimens is particularly essential, since the samples taken in
the field are usually transported and analysed or measured at other
locations and at a later date. Thus the conditions under which the
samples have to be transported and stored must be established in an
SOP. The correct recovery of a chemical from an environmental sample,
for instance, is very much dependent on its chemical and biological
stability in the medium. Considerable care has, therefore, to be
taken to ensure that the content and often the structure of a sample
remain unchanged until it is analysed, or until observations or
measurements have been carried out.
In the study plan, the personnel responsible for different parts
of the study should be clearly identified, since the field work may be
carried out by different institutions to those that carry out the
laboratory analyses and measurements. Therefore, the study plan is
the most important document for providing information to all
participants on all aspects of the study, including sampling dates,
amounts and character of samples.
Deviations from the study plan, particularly concerning changes
in the selected sampling stations, sampled populations and sampling
tissues, necessitate a detailed amendment to the study plan. This is
of great importance, since in many monitoring studies sampling occurs
on a regular basis, and the data obtained may be worthless if the
preset sampling scheme is not followed. Therefore, changes need
authorization of the study director. Acceptable levels of deviations
in respect of time or space of sampling should also be stated.
If a change in a given parameter or the impact of a certain
process has to be monitored over time or space, a control site or
population often has to be selected. The selection of the control
must ensure that conditions are similar to those of the site under
investigation but that the influencing factor is lacking. In
contrast, background sites (or populations) that are not affected by
human activities (Black, 1988) can be treated as "blanks" in
environmental monitoring programmes.
3.3 Selection of sampling strategies and study design
The study plan for a monitoring study must state specifically the
selected sampling area, sampling station or population to be sampled,
medium to be sampled, frequency and time of sampling, numbers of
samples and the parameters for evaluation. For instance, when the
contamination level in an aquatic ecosystem is monitored over a long
period, samples from the water column, sediments and living organisms
are commonly taken on a regular basis. The number of samples is
selected according to the size and character of the investigation
zone. The volume of the sampled medium is chosen according to the
expected concentration of the chemical and the sensitivity of the
analytical method. The wildlife species to be sampled should be
representative for the biota and known to accumulate the substance of
concern sufficiently.
Different objectives of environmental monitoring programmes
require various kinds of sampling techniques. In general, it has to
be decided if random or non-random sampling should be applied to
achieve the objectives.
For example, if the fate and effect of a contaminant distributed
by a diffuse source has to be monitored, a random sampling technique
is appropriate. On the other hand, if the dilution of an industrial
effluent by natural surface waters is monitored, the sampling stations
need to be selected according to various factors, e.g. water flow,
while sampling times may be randomly chosen.
In cases where exposure data for a group of individuals are used
for estimating exposure in the population, the selection of
individuals for monitoring is a critical step. The sample of
individuals has to be selected so that it will provide valid
inferences for the target population. Thus, the selected individuals
must be representative of the population under study. Quality
assurance related to selection of study groups and individuals is
described in the guidance provided by WHO on sample selection and data
analysis for HEAL studies (WHO, 1992).
Three major random sampling techniques exist; these are commonly
described as "stratified random sampling", "simple random sampling",
and "systematic sampling" (Cochran, 1963; Kelley, 1976; Sokal & Rohlf,
1981). All three techniques have a common purpose, namely that the
samples are representative for the population or compartment to be
sampled.
In simple random sampling the selection process is totally,
unconditionally random. The disadvantage of this method lies in the
possibility that the samples may unintentionally be clumped together,
while other parts of the population or compartment may not be sampled
at all. This may be particularly unfavourable when samples show a
large variation.
Distribution problems can be solved by dividing the population or
compartment into either equal segments, where the investigator selects
the interval systematically and in which segment a sample is to be
taken, or into segments that are unequal in size or number ("strata"),
and where at least one sample is taken in each segment. The first
situation (systematic sampling) is unfavourable in cases where, during
repeated sampling, the functions to be monitored vary, perhaps
periodically. In the second case (stratified random sampling), some
information on functions that are monitored must be available before
the study starts and has to be used in designing the strata. Within
each stratum the samples are selected randomly. The sampling size can
be adjusted for each segment of the population or compartment.
Thus, the quality and value of the data obtained by the
monitoring exercise is governed to a great extent by the selected
sampling technique. For example, if the effects of the so-called acid
rain on forest ecosystems should be monitored, simple random sampling
might not lead to valid results, since the effects might vary
considerably between, for instance, deciduous and coniferous forest
systems.
All these aspects demonstrate the extent to which a well-
designed study influences the quality of the results.
3.4 Sampling procedures and documentation
The parameters to be measured or observed must be specified by
SOPs and/or the study plan. Where and by which means the samples are
taken and how measurements and observations are carried out should be
described. For instance, if concentrations of a substance are
monitored in natural aquatic ecosystems, it is necessary to establish
the volume of water to be sampled, from which depth the samples must
be taken, and which device should be used. For blood samples the time
of sampling can be of great importance.
For collecting human samples, new syringes and containers should
preferably be used, but for environmental samples this may not always
be feasible. In any event, clean sampling devices and containers
should always be used. In order to avoid contamination of samples,
the necessary cleaning procedures for sampling devices and containers
must be specified. During sample collection the sampling devices can
contribute to chemical contamination due to the use of improper or
unstable material as well as improper cleaning procedures. The SOPs
should state clearly the material to be used for sampling and storage
of samples, as well as the cleaning procedures. The
cross-contamination of biological material can also occur, e.g., a net
sample with planktonic organisms, where organisms from a previous
sample are introduced into another because the net was not properly
rinsed. In this example, if the species composition is monitored,
biased results can be expected. Thorough rinsing would avoid this.
In the case of chemical contamination, cleaning procedures such as
acid washing may be useful.
Containers and chemicals may be potential sources of
contamination, for instance with trace metals. Special cleaning
procedures are needed to minimize this problem.
3.5 Handling of samples
The samples obtained during a study are normally shipped from
the field into the laboratory where they are to be evaluated. Often
there is a considerable delay between sampling and arrival at the
laboratory where the samples and specimens are to be stored.
For many pollutants there is a great risk of introducing errors
during sampling, sample handling and chemical analysis. The risk of
contamination of the sample during sampling and sample handling is
particularly great for samples with low concentrations of pollutants
ubiquitous in the environment or present in materials and tools coming
into contact with the sample. For example, a blood sample of 1 ml
normally contains as little as 0.1-0.5 ng cadmium, and it is obvious
that considerable measures have to be taken in order to avoid
contamination. Tobacco smoke often contains cadmium at levels that
may seriously contaminate the blood samples. Therefore, all sample
handling must be carried out in rooms where smoking is prohibited, and
preferably by non-smokers.
Concentration changes during storage may be due to precipitation
or evaporation of the analyte or to evaporation of the solvent.
Adsorption of the analyte to the container may also occur. Changes in
the sample matrix, e.g., clotting of blood, may change the
concentration of the chemical to be measured. Often the samples have
to undergo a series of preparation steps before the determination of
the concentration of a pollutant is carried out. Mistakes in the
dilution, weight determination and calculations may appear. Chemicals
added to the samples may be contaminated with the substance under
study.
The use of blanks is the most common analytical tool for
controlling contamination. Blanks can be used in the laboratory in
the form of instrument blanks, calibration blanks and reagent blanks.
In the field, matched-matrix blanks (determination of contamination
during sample collection, handling, storage and transport) are used to
simulate the sample matrix and are carried through all processes.
Blanks of the media used in sampling (like filters, nets, traps and
containers) and of the sampling devices (by collecting the rinsing
media) should also be used.
Appropriate provision must be made and measures taken in order to
keep the samples under the conditions required to maintain the
character of the samples. The stability of the chemical to be
analysed, in terms of, for instance, physical, chemical or biological
degradation, must be known. If there are special storage conditions
required, adherence to these conditions must be controlled and
documented.
One means of controlling the stability of the samples for
chemical analysis is the use of spiked samples. These samples contain
the same matrix as the environmental sample but without the respective
analyte, which is then added in a known quantity. The spike samples
are then handled and stored in the same way and under the same
conditions as the environmental samples and are concomitantly
analysed. It should be noted that the integrity of volatile fractions
in samples is particularly difficult to maintain.
The stability of biological material is typically ensured by the
use of preservation materials or processes, but proper and appropriate
preservation methods must be used so that very fragile structures,
such as single cells in plankton samples, are not damaged or
destroyed. This is necessary to ensure that measurements or
observations of the samples are representative.
3.6 Analytical performance evaluation
The main objective of the analytical performance evaluation is to
assess the accuracy of data. This evaluation should be published
along with the data so that users can assess the quality. The
following techniques are available for analytical performance
evaluation:
* external quality control programmes;
* comparison with results using an independent technique;
* comparison with results of a reference laboratory;
* analysis of commercial standard reference materials;
* analysis of spiked samples prepared at the laboratory.
Ideally, the analytical performance evaluation should be
coordinated by an external quality assurance coordinating centre or by
the quality assurance unit of the laboratory. The coordinators
provide the laboratory with external quality control (EQC) samples,
the concentrations of the substance being unknown to the analyst. The
EQC samples should be analysed together with the collected samples,
and the results should be evaluated by the quality assurance
personnel.
If it is not possible to have the analytical performance
evaluated by an external quality assurance personnel, duplicate
samples should be analysed using other analytical techniques,
preferably at a reference laboratory. If other techniques are not
available, duplicate samples should be analysed at a reference
laboratory.
Since the certified concentrations of standard reference
materials are published, and this is known to the analyst, analysis of
such samples alone is often not enough for independent evaluation of
the accuracy of the resulting data. Correct results for the internal
quality control (IQC) samples do not always guarantee accurate results
for the EQC samples. Also, standard reference materials are often
only available at one or two concentrations. The quality control
samples should cover the range of concentrations likely to be found in
the monitoring samples. Good analytical performance at one
concentration is no guarantee of good performance at other
concentrations.
The analytical performance evaluation should be carried out at
the time of the analysis of the monitoring samples. Reference to
previous participation in interlaboratory comparison programmes cannot
be used for evaluating the accuracy of the data produced.
3.7 The regression method
Unfortunately it is not possible to measure of the accuracy of
data produced, but the limits of uncertainty can be estimated.
Principles for analytical quality assessment in human exposure
monitoring, giving limits for the uncertainty, have been recommended
by WHO (1986b). The method is based on the quality control programmes
developed in the UNEP Biological Monitoring of Lead and Cadmium
(Vahter, 1982; Friberg & Vahter, 1983; Vahter & Slorach, 1990).
Basically, it involves the analysis of sets of quality control samples
and evaluation of the results using linear regression analysis. In
the WHO/UNEP Human Exposure Assessment Locations (HEAL) monitoring
programme for lead and cadmium, a quality control set consisted of 3-6
external EQC samples, the metal concentrations of which were not known
to the laboratories, as well as 1-2 IQC samples, the metal
concentrations of which were given. The IQC samples are used for the
analyst's own control of the analytical conditions, while the EQC
samples are used for the analytical performance evaluation. One or
more sets of quality control samples were analysed together with the
monitoring samples, and the results were evaluated by a coordinating
centre.
The regression method, i.e. the evaluation of the regression line
of reported versus "true" values for a set of quality control samples
analysed together with the monitoring samples, is a useful method of
guarding against systematic errors in the whole range of
concentrations likely to occur (Vahter, 1982; UNEP/WHO, 1984; Friberg,
1988). The regression line represents the average analytical
performance.
In order to obtain limits for the uncertainty of the data
produced, the maximum allowable deviation (MAD) of the empirical
regression line from the ideal line y = x is defined. The MAD
criteria have to be decided separately for each pollutant and for each
medium. In the UNEP/WHO HEAL project on lead and cadmium, the MAD was
generally set to ± (5-10% ± sigma), where sigma was the estimated
error of the method, based on several quality control runs.
Since the regression line, based on the results of a set of
quality control samples, has an operating error, the decision on
acceptance or rejection of the regression line must be based on
statistical criteria, i.e. the probability of making right or wrong
decisions. A laboratory's results may be erroneously rejected when in
fact the laboratory performance is satisfactory, or erroneously
accepted when the performance is bad. Table 1 illustrates the
different decisions that can be made on the basis of the results of
the quality control analyses, and the associated probabilities
(UNEP/WHO, 1984).
Table 1. Decision-making on the basis of quality control resultsa
True condition Decision
Acceptance Rejection
Methodology is correct decision wrong decision
satisfactory (1-alpha) Type I error (alpha)
Methodology is not wrong decision correct decision
satisfactory Type II error (ß) Power (1-ß)
a From: UNEP/WHO (1984)
In the HEAL project, a total power of 90% was employed, which
means that the probability of accepting an unsatisfactory performance
(the true regression line falling outside the MAD interval) was not
more than 10%. For acceptance of data the empirical regression lines
had to fall not only inside the MAD interval, but also inside an
acceptance interval.
Fig. 1 shows an example of a regression line of the results of
six quality control samples, the MAD interval (solid lines) and the
acceptance interval (broken lines). The distance between the MAD
lines and the acceptance lines is 1.645 times the operating error,
sigmað, calculated according to the formula:
sigma2ð = sigma2y/x (1 + d2 )
n (n-1).sigma2x
where
n = number of observations
d = difference between x value and x mean
sigmax = standard deviation of x values
sigmay/x = error of method or residual deviation (estimated from
previous analyses)
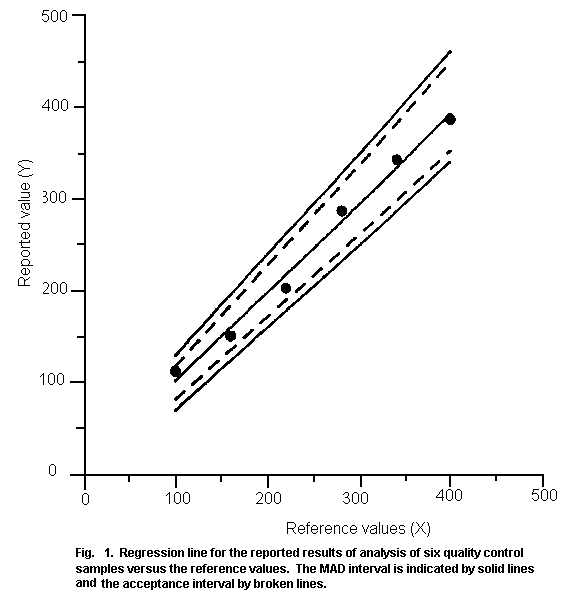 It is obvious from the formula that the acceptance interval (AI)
lines will get closer to the MAD lines with increasing numbers of data
points (quality control samples). Also, a smaller error of method
will decrease the difference between the MAD lines and the AI lines.
The random error of method may be calculated from the results of
each quality control set. However, with quality control sets
consisting of only 4-6 samples, the empirical error of method may be
largely influenced by one or two occasional gross errors. Therefore,
it is good practice to estimate the error of method based on previous
experience. It is nevertheless important to calculate the empirical
error of method, since it may serve as a supplementary guidance in the
evaluation of the analytical performance. When the current random
error of method deviates too greatly from the estimated one, this is
an indicator of bad performance, and the evaluation should not be
based on the estimated error of method.
For many types of chemical measurements, the error variance tends
to vary with the true concentration. Variance-stabilizing
transformations, e.g., logarithmic ( z = ln x ) or square-root
( z = x ) transformation, of the data may make the error variance
independent of the true concentration (Starks, 1989). In the HEAL
nitrogen dioxide project, where many quality control samples were
used, the quality control results were considered satisfactory if the
regression line, the 90% confidence interval and all data points were
inside the MAD interval (Matsushita & Tanabe, 1991).
The regression method may give valuable information concerning
the type of error. For example, a regression line parallel to the
ideal line ( y = x ) indicates an absolute error caused by, for
instance, a false blank value. A slope deviating from 1.0 indicates
a constant relative error due to, for instance, incorrect standards or
errors in the concentrations of the standards.
3.8 Practical application of the regression method
The regression method for analytical performance evaluation was
developed for use in a WHO study on the assessment of human exposure
to lead and cadmium through biological monitoring. Since then, the
method has been used in a number of studies, involving both metals
(Lind et al., 1987, 1988a,b; Zheng & Ji, 1987; Vahter & Slorach, 1990)
and nitrogen dioxide (Matsushita & Tanabe, 1991).
3.9 Other analytical performance evaluation programmes
There are several other methods for external analytical
performance assessment (ISO, 1986b; Horwitz, 1988). Some examples are
given below. Most of these methods are descriptive and do not give
acceptability limits for possible errors.
The WHO European Regional Study on Health Effects of Exposure to
Cadmium, coordinated by the Coronal Laboratory for Occupational and
Environmental Health, was specially developed for countries
participating in the United Nations Development Programme. A quality
control programme was developed for the determination of lead and
cadmium in blood, as well as cadmium, ß2-microglobulin and retinol
binding protein in urine (Herber, 1990). In the metal programme,
30-40 laboratories each analysed six samples of human blood and six
samples of human urine. The samples contained cadmium and lead
nitrate at levels up to about 20 µg/litre for cadmium and up to 800
µg/litre for lead.
Regarding the proteins in urine, there were only six or seven
participating laboratories, and another, more simple, evaluation
procedure was used. Urine from patients with kidney disease was
diluted with urine with normal physiological protein concentrations.
The acceptance criteria were ± 20% of the median for albumin and ± 40%
of the median for ß2-microglobulin and retinol binding protein.
The Guildford Trace Element Quality Assessment Scheme,
coordinated by Robens Institute of Industrial and Environmental Health
and Safety and St. Luke's Hospital, Guildford, United Kingdom, is an
external quality assessment scheme for trace elements in human
biological fluids (Taylor et al., 1985). More than 100 laboratories
from over 15 countries participate in the scheme. The programme
includes several combinations of analytes and biological samples.
Every month during a six-month cycle, three specimens of each matrix
are sent to the participants. Monthly reports include consensus mean,
standard deviation, relative standard deviation, a histogram of
distribution and a tabulation of the results. At the end of the
six-month cycle, the 18 results for each sample-matrix analyte are
summarized and the analytical performance is assessed based on the
proximity to the consensus mean, the difference between results
analysed on two occasions and the recovery of added analyte. A
"performance score" is calculated for each individual laboratory.
Targets or markers of satisfactory performance have been established
based on what is necessary for clinical purposes and what can be
achieved with available analytical techniques.
The Guildford external quality assessment (EQA) programme for
aluminium in serum has been in operation since 1981 (Taylor, 1988).
Samples of horse serum, supplemented with known amounts of aluminium,
are prepared for a series of 6-monthly cycles. During a cycle, nine
different specimens are distributed in duplicate, three samples each
month. The results are evaluated as described above. The programme
has shown poor performance in large numbers of laboratories and
excellent performance in a few laboratories. In an attempt to explain
this, an evaluation of instrumentation and methodologies was carried
out. It showed that the quality of the results was not influenced by,
for example, specific features of equipment or instrumentation but
rather by good, careful analysts, who were able to ensure that
everything was properly set up and that contamination was avoided.
The National External Quality Assessment Scheme (NEQAS),
coordinated by the Wolfson Research Laboratory in Birmingham, is
another external quality assessment programme in the United Kingdom
(Bullock & Wilde, 1985). Samples are distributed to the participating
laboratories for analysis, and based on the results obtained, a
variance index (VI) is calculated. The VI is the difference between
the result obtained and a trimmed mean, divided by a chosen relative
standard deviation for the analyte and expressed as a percentage. A
Mean Running Variance Index Score (MRVIS) and a Mean Running Bias
Index Score (MRBIS) are also calculated. It is believed that a MRVIS
below 33 should be the target for the participants, and that any MRVIS
over 66 should stimulate investigation of the laboratory's method.
3.10 Analytical performance criteria
The limits of the uncertainty in the produced data should be
based on the accuracy requirements for the monitoring data and what
can be achieved with available analytical techniques. The accuracy
and precision of routine and reference methods should be evaluated in
order to determine the feasibility of the proposed criteria.
Often the criteria for the analytical performance are determined
by the sensitivity and accuracy of the analytical method. In the
WHO/UNEP monitoring project on cadmium and lead in blood, the limits
for the MAD lines for lead in blood were set to
y = x ± (0.1 x + 20). This means that if the average
concentration of lead in blood in a group of people was found to be,
for example, 100 µg/litre, the criteria guaranteed with a probability
of 90% that the true average concentration was somewhere between 70
and 130 µg/litre. If the average blood lead concentration found was
50 µg/litre, the criteria guaranteed that the true average was between
25 and 75 µg/litre. With such a great "uncertainty" in the monitoring
data, it is of course difficult to detect differences in lead exposure
between various groups in the general population.
The experience from the quality control activities in the HEAL
pilot project on lead and cadmium shows that, for lead concentrations
in blood (µg/litre), experienced laboratories can meet the MAD
criteria y = x ± (0.05 x + 10), which guarantees that an obtained
mean blood lead concentration of 50 µg/litre lies with 90% probability
between 37.5 and 62.5 µg/litre. For concentrations of cadmium in
blood, well-experienced and well-equipped laboratories met the MAD
criteria y = x ± (0.05 x + 0.2), which guarantees that an
obtained mean blood cadmium concentration of, for example, 0.5
µg/litre lies with 90% probability between 0.28 and 0.72 µg/litre.
3.11 Quality control samples
The analytical procedures and performance may vary considerably
between various types of samples. Good analytical performance for a
pollutant in one type of medium is no guarantee of good performance
with other media. Thus, it is important for quality control samples
to have a matrix similar to that of the monitoring samples.
As already mentioned above, the quality control sample should
cover the range of concentrations likely to be found in the monitoring
samples. It must be emphasized that good analytical performance at
one concentration is no guarantee for good performance at other
concentrations.
Various types of reference samples are commercially available
(Muntau et al., 1983; Belliardo & Wagstaffe, 1988; Klich & Caliste,
1988; Okamoto, 1988; Parr et. al., 1988; Rasberry, 1988). However,
many of the commercially available reference materials are certified
for a limited number of substances, and usually only for one or two
different concentrations. Commercially available reference materials
are suitable for internal quality control purposes but cannot, as a
rule, be used for published data.
If possible, specimens of the samples should finally be archived
in order to prove again the validity of an environmental monitoring
study, if it should be necessary. This may be appropriate where the
samples are still integral after evaluation.
Biological tissue is typically contained in slides or blocks,
while specimens are stored in containers with appropriate fixation.
Aliquots from the various media for chemical analysis may be stored
after appropriate preservation, carrying a label with an expiry date
based on stability analysis. All written documentation including the
raw data is also transferred to the archive at the conclusion of the
study.
REFERENCES
AOAC (1984) Quality assurance principles for analytical laboratories.
Washington, DC, Association of Official Analytical Chemists, 224 pp.
Belliardo JJ & Wagstaff PJ (1988) BCR reference materials for food and
agricultural analysis: An overview. Fresenius Z Anal Chem, 332(6):
533-538.
Black SC (1988) Defining control sites and blank sample needs. In:
Keith LH ed. Principles in environmental samples. Washington, DC,
American Chemical Society, pp 109-118.
Bruaux P & Svartengren M ed. (1985) Global Environmental Monitoring
System (GEMS). Assessment of human exposure to lead: Comparison
between Belgium, Malta, Mexico and Sweden. Stockholm, National Swedish
Institute of Environmental Medicine and Department of Environmental
Hygiene, Karolinska Institute, and Brussels, Institute of Hygiene and
Epidemiology, 57 pp (Prepared for United Nations Environment Programme
and World Health Organization).
Bruaux P, Claeys-Thoreau F, Ducoffre G, & Lafontaine A (1979)
Exposition saturnine de la population bruxelloise. Choix des groupes
de population. Choix des indicateurs. Premiers résultats. Arch Belg
Méd Soc, 37: 488-501.
Bullock DG & Wilde CE (1985) External quality assessment of urinary
pregnancy oestrogen assay: further experience in the United Kingdom.
Ann Clin Biochem, 22: 273-282.
Burck K (1979) The role of quality assurance in good laboratory
practices. Clin Toxicol, 15: 627-640.
Cochran WG (1963) Sampling techniques. New York, John Wiley & Sons, 13
pp.
FDA (1987a) Good laboratory practice regulations. Fed Reg, 52(172):
33768 (21 CFR Part 58) (Docket No. 83N-0142).
FDA (1987b) Part 58 - Good laboratory practice for non-clinical
laboratory studies. Code Fed Regul, 21(Ch1): 228-242.
Friberg L (1988) Quality assurance. In: Clarkson TW, Friberg L,
Nordberg GF, & Sager PR ed. Biological monitoring of toxic metals. New
York, Plenum Press, pp 103-126.
Friberg L & Vahter M (1983) Assessment of exposure to lead and cadmium
through biological monitoring. Results of a UNEP/WHO global study.
Environ Res, 30: 95-128.
Herber RFM (1990) The World Health Organization study on health
effects of exposure to cadmium. Toxicol Environ Chem, 27: 1-10.
Herber RFM & Schaller KH (1986) Analytical variability of biological
parameters of exposure and early effects. In: Notten WRF, Herber RFM,
Hunter WJ, Monster AC, & Zielhuis RL ed. Health surveillance of
individual workers exposed to chemical agents. Berlin, Springer
Verlag, pp 102-109.
Horwitz W (1988) Protocol for the design, conduct and interpretation
of collaborative studies: IUPAC Workshop on the Harmonization of
Collaborative Analytical Studies, Geneva, Switzerland, 4-5 May 1987.
Pure Appl Chem, 60: 855-864.
Horwitz W (1989) Harmonization of methodology: international
perspective. In: Cowgill UM & Williams LR ed. Aquatic toxicology and
hazard assessment. Philadelphia, American Society for Testing and
Materials, pp 5-10 (Special Technical Publication No. 1027).
ISO (1986a) International Standard 8402. Quality - Vocabulary. Geneva,
International Organization for Standardization, 15 pp.
ISO (1986b) International Standard 5725: Precision of test methods -
Determination of repeatability and reproducibility for a standard test
method by inter-laboratory tests: 2nd edition. Geneva, International
Organization for Standardization, 49 pp.
ISO (1987a) International Standard 9000. Quality management and
quality assurance standards: guidelines for selection and use. Geneva,
International Organization for Standardization, 6 pp.
ISO (1987b) International Standard 9004. Quality management and
quality system elements - Guidelines. Geneva, International
Organization for Standardization, 19 pp.
Kelley JC (1976) Sampling the sea In: Cushing DH & Walsh JJ ed. The
ecology of the seas. Oxford, Blackwell Scientific Publications, pp
361-387.
Klich H & Caliste JP (1988) COMAR - database for certified reference
materials. Fresenius Z Anal Chem, 332(6): 552-555.
Lepore PD (1979) FDA's good laboratory practice regulations. Pharm
Technol, 3: 71-74.
Lind B, Elinder C-G, Friberg L, Nilsson B, Svartengren M, & Vahter M
(1987) Quality control in the analysis of lead and cadmium in blood.
Fresenius Z Anal Chem, 326: 647-655.
Lind B, Vahter M, Rahnster B, & Bjors U (1988a) Quality control
samples for the determination of lead and cadmium in blood, feces, air
filters and dust. Fresenius Z Anal Chem, 332(6): 741-743.
Lind B, Bigras L, Cernichiari E, Clarkson TW, Friberg L, Hellman M,
Kennedy P, Kirkbride J, Kjellstrom T, & Ohlin B (1988b) Quality
control of analyses of mercury in hair. Fresenius Z Anal Chem, 332(6):
620-622.
Matsushita H & Tanabe K (1991) Earthwatch - Global Environmental
Monitoring System (GEMS). Exposure monitoring of nitrogen dioxide. An
international pilot study within the WHO/UNEP Human Exposure
Assessment Location (HEAL) Programme. Nairobi, United Nations
Environment Programme, 109 pp.
Muntau H, Schramel P, Brätter P, & Knapp G (1983) Trace element
investigations on some new biological test materials. In: Brätter P &
Schramel P ed. Trace element - Analytical chemistry in medicine and
biology. Berlin, New York, Walter de Gruyter & Co., vol 2, pp 819-831.
OECD (1982) Good laboratory practice in the testing of chemicals.
Paris, Organisation for Economic Co-operation and Development, 62 pp.
Okamoto K (1988) Biological reference materials from the National
Institute of Environmental Studies (Japan). Fresenius Z Anal Chem,
332(6): 524-527.
Parr RM, Schelenz R, & Ballestra S (1988) IAEA biological reference
materials. Fresenius Z Anal Chem, 332(6): 518-523.
Rasberry SD (1988) NBS activities in biological reference materials.
Fresenius Z Anal Chem, 332(6): 528-532.
Slorach SA & Vaz R ed. (1983) Global Environmental Monitoring System
(GEMS). Assessment of human exposure to selected organochlorine
compounds through biological monitoring. Uppsala, Swedish National
Food Administration, 134 pp (Prepared for United Nations Environment
Programme and the World Health Organization).
Smith F, Kulkarni S, Myers LE, & Messner MJ (1988) Evaluating and
presenting quality assurance sampling data. In: Keith LH ed.
Principles in environmental samples. Washington, DC, American Chemical
Society, pp 157-168.
Sokal RR & Rohlf FJ (1981) Biometry: The principles and practice of
statistics in biological research, 2nd ed. New York, Oxford, W.H.
Freeman, 859 pp.
Starks TN (1989) Global Environmental Monitoring System (GEMS).
Statistical guidelines for quality assurance in human exposure
assessment location studies. Geneva, World Health Organization, 48 pp
(Document PEP/89.9).
Taylor A (1988) Reference materials for measurement of aluminium in
biological samples. Fresenius Z Anal Chem, 332(6): 616-619.
Taylor JK (1987) Quality assurance of chemical measurements. Chelsea,
Michigan, Lewis Publishers, 328 pp.
Taylor JK (1988a) Defining the accuracy, precision and confidence
limits of sample data. In: Keith LH ed. Principles in environmental
samples. Washington, DC, American Chemical Society, pp 101-108.
Taylor JK (1988b) The role of statistics in quality assurance.
Fresenius Z Anal Chem, 332(6): 722-725.
Taylor A, Starkey BJ, & Walker AW (1985) Determination of aluminium in
serum: findings of an external quality assessment scheme. Ann Clin
Biochem, 22: 351-358.
UNEP/WHO (1984) Principles and procedures for quality assurance in
environmental pollution exposure monitoring. Geneva, World Health
Organization, 31 pp (EFP/HEAL/84.4).
Vahter M (1982) Global Environmental Monitoring System (GEMS).
Assessment of human exposure to lead and cadmium through biological
monitoring. Stockholm, National Swedish Institute of Environmental
Medicine and Department of Environmental Hygiene, Karolinska
Institute, 136 pp (Prepared for United Nations Environment Programme
and World Health Organization).
Vahter M & Slorach S ed. (1989) Global Environmental Monitoring System
(GEMS). Exposure monitoring of lead and cadmium: An international
pilot study within the UNEP/WHO Human Exposure Assessment Location
(HEAL) Programme. Nairobi, United Nations Environment Programme, 82
pp.
Vahter M & Slorach S ed. (1990) Global Environmental Monitoring System
(GEMS). Exposure monitoring of lead and cadmium: An international
pilot study within the WHO/UNEP Human Exposure Assessment Location
(HEAL) Programme. Nairobi, United Nations Environment Programme, 82
pp.
WHO (1986a) Environmental Health Criteria 57: Principles of
toxicokinetic studies. Geneva, World Health Organization, pp 20-24.
WHO (1986b) UNEP/WHO Global Environmental Monitoring System (GEMS).
Human Exposure Assessment Location (HEAL) Project: Guidelines for
integrated air, water, food and biological exposure monitoring.
Geneva, World Health Organization, 389 pp (Document PEP/86.6).
WHO (1992) Earthwatch - Global Environmental Monitoring System (GEMS).
Guidance on survey design for Human Exposure Assessment Locations
(HEAL) Studies. Geneva, World Health Organization (Document
WHO/PEP/92.6).
Youden WJ (1967) The role of statistics in regulatory work. J Assoc
Off Anal Chem, 50: 1007-1013.
Youden WJ & Steiner EH (1975) Statistical manual of the Association of
Official Analytical Chemists. Washington, DC, Association of Official
Analytical Chemists, 88 pp.
Zheng XQ & Ji RD (1987) Assessment of lead contamination of the
general population through blood lead levels. Environ Monit Assess, 9:
169-177.
APPENDIX I
GLOSSARY
Batch: A specific quantity or lot of a test or reference substance
produced during a defined cycle of manufacture, in such a way that it
could be expected to be of a uniform character and should be
designated as such.
Carrier (vehicle): Any agent that serves as a vehicle used to mix,
disperse, or solubilize the test or reference substance to facilitate
the administration to the test system.
Calibration: Assigning values to the output of an instrument or
other device. Calibration can apply to instruments, glassware or any
other measuring device.
Calibration verification (calibration check): Determining if the
output of an instrument or device is within some established
tolerance.
Code number: A unique number assigned to each packaged unit of the
test substance.
Final report: A comprehensive report that includes all information
relative to the evaluation of the results of a specific nonclinical
laboratory study.
Laboratory technician: A laboratory worker who performs an operation
covered by an SOP, under the supervision of a professional scientist.
Master schedule sheet: A document that contains essential
information on completed and in-progress studies in the laboratory,
i.e. a list of studies, initiation and completion dates, test systems,
route of administration and name of study director.
Quality assurance programme (QAP): An internal control system of
inspections and study audits designed to ascertain that a study is in
compliance with defined guidance principles. It assures laboratory
management that facilities, equipment, personnel, methods, practices,
records and controls conform with these principles.
Quality assurance unit (QAU): Any person or organizational element,
designated by testing facility management to perform inspections of
laboratory operations and study audits relating to quality assurance
of studies.
Quality control system: A system used to assess product quality
whenever the quality attributes of the final product can be expressed
as discretely measured parameters.
Raw data: All original laboratory records and documentation, or
verified copies thereof, which are the result of the original
observations and activities in a study.
Sample: Any quantity of the test or reference substance.
Specimen: Any material derived from a test system for examination,
analysis or storage.
Sponsor: A person or entity who commissions and/or supports a study.
A testing facility can also be a sponsor if it both initiates and
actually conducts the study.
Standardization: Determining the response of an instrument to known
quantities of a test agent according to a specific method; accurately
determining the concentration of a solution.
Standardization verification (standardization check): Determining if
the response of an instrument to a particular standard is within some
established tolerance (e.g., verifying that a previously prepared
standard curve (or other procedure) is still valid).
Standard operating procedure (SOP): A written procedure that
describes how to perform certain routine laboratory tests or
activities that are normally not specified in detail in study plans or
test guidelines.
Study: An experiment or set of experiments in which a test substance
is examined to obtain data on its properties and/or its safety with
respect to human health and the environment.
Study audit: A comparison of the raw data and associated records
with the interim or final report in order to determine whether the raw
data was accurately reported, and whether testing was carried out in
accordance with the study plan and SOPs to obtain additional
information not provided in the report, and to establish whether
practices were employed in the development of data that would impair
their validity.
Study director: The individual responsible for the overall conduct
of the study.
Study plan: A protocol which defines the entire scope of the study.
Test facility (testing facility): The persons, premises and
operational unit(s) that are necessary for conducting the study.
Test facility management: Consists of the executive level of
management charged with the ultimate responsibility of the test
facility and studies.
Test substance: A chemical substance or a mixture that is under
investigation.
Test system: Any human, other animal, plant, microbial, as well as
other cellular, subcellular, chemical or physical system or
combination of these, that is exposed to a test, control or reference
substance.
Validation: Establishing documented evidence that provides a high
degree of assurance that the intended use of such things as a
procedure, test system, test substance or control substance is
accomplished.
Validation procedure(s): A written procedure stating how validation
will be conducted. Validation procedures should exist for all
elements of the test system and should specify the procedures and
tests that will be conducted and the data to be collected. Examples
include the validation procedure used for animal supply vendors,
facility equipment, test substances control substances, and SOPs.
It is obvious from the formula that the acceptance interval (AI)
lines will get closer to the MAD lines with increasing numbers of data
points (quality control samples). Also, a smaller error of method
will decrease the difference between the MAD lines and the AI lines.
The random error of method may be calculated from the results of
each quality control set. However, with quality control sets
consisting of only 4-6 samples, the empirical error of method may be
largely influenced by one or two occasional gross errors. Therefore,
it is good practice to estimate the error of method based on previous
experience. It is nevertheless important to calculate the empirical
error of method, since it may serve as a supplementary guidance in the
evaluation of the analytical performance. When the current random
error of method deviates too greatly from the estimated one, this is
an indicator of bad performance, and the evaluation should not be
based on the estimated error of method.
For many types of chemical measurements, the error variance tends
to vary with the true concentration. Variance-stabilizing
transformations, e.g., logarithmic ( z = ln x ) or square-root
( z = x ) transformation, of the data may make the error variance
independent of the true concentration (Starks, 1989). In the HEAL
nitrogen dioxide project, where many quality control samples were
used, the quality control results were considered satisfactory if the
regression line, the 90% confidence interval and all data points were
inside the MAD interval (Matsushita & Tanabe, 1991).
The regression method may give valuable information concerning
the type of error. For example, a regression line parallel to the
ideal line ( y = x ) indicates an absolute error caused by, for
instance, a false blank value. A slope deviating from 1.0 indicates
a constant relative error due to, for instance, incorrect standards or
errors in the concentrations of the standards.
3.8 Practical application of the regression method
The regression method for analytical performance evaluation was
developed for use in a WHO study on the assessment of human exposure
to lead and cadmium through biological monitoring. Since then, the
method has been used in a number of studies, involving both metals
(Lind et al., 1987, 1988a,b; Zheng & Ji, 1987; Vahter & Slorach, 1990)
and nitrogen dioxide (Matsushita & Tanabe, 1991).
3.9 Other analytical performance evaluation programmes
There are several other methods for external analytical
performance assessment (ISO, 1986b; Horwitz, 1988). Some examples are
given below. Most of these methods are descriptive and do not give
acceptability limits for possible errors.
The WHO European Regional Study on Health Effects of Exposure to
Cadmium, coordinated by the Coronal Laboratory for Occupational and
Environmental Health, was specially developed for countries
participating in the United Nations Development Programme. A quality
control programme was developed for the determination of lead and
cadmium in blood, as well as cadmium, ß2-microglobulin and retinol
binding protein in urine (Herber, 1990). In the metal programme,
30-40 laboratories each analysed six samples of human blood and six
samples of human urine. The samples contained cadmium and lead
nitrate at levels up to about 20 µg/litre for cadmium and up to 800
µg/litre for lead.
Regarding the proteins in urine, there were only six or seven
participating laboratories, and another, more simple, evaluation
procedure was used. Urine from patients with kidney disease was
diluted with urine with normal physiological protein concentrations.
The acceptance criteria were ± 20% of the median for albumin and ± 40%
of the median for ß2-microglobulin and retinol binding protein.
The Guildford Trace Element Quality Assessment Scheme,
coordinated by Robens Institute of Industrial and Environmental Health
and Safety and St. Luke's Hospital, Guildford, United Kingdom, is an
external quality assessment scheme for trace elements in human
biological fluids (Taylor et al., 1985). More than 100 laboratories
from over 15 countries participate in the scheme. The programme
includes several combinations of analytes and biological samples.
Every month during a six-month cycle, three specimens of each matrix
are sent to the participants. Monthly reports include consensus mean,
standard deviation, relative standard deviation, a histogram of
distribution and a tabulation of the results. At the end of the
six-month cycle, the 18 results for each sample-matrix analyte are
summarized and the analytical performance is assessed based on the
proximity to the consensus mean, the difference between results
analysed on two occasions and the recovery of added analyte. A
"performance score" is calculated for each individual laboratory.
Targets or markers of satisfactory performance have been established
based on what is necessary for clinical purposes and what can be
achieved with available analytical techniques.
The Guildford external quality assessment (EQA) programme for
aluminium in serum has been in operation since 1981 (Taylor, 1988).
Samples of horse serum, supplemented with known amounts of aluminium,
are prepared for a series of 6-monthly cycles. During a cycle, nine
different specimens are distributed in duplicate, three samples each
month. The results are evaluated as described above. The programme
has shown poor performance in large numbers of laboratories and
excellent performance in a few laboratories. In an attempt to explain
this, an evaluation of instrumentation and methodologies was carried
out. It showed that the quality of the results was not influenced by,
for example, specific features of equipment or instrumentation but
rather by good, careful analysts, who were able to ensure that
everything was properly set up and that contamination was avoided.
The National External Quality Assessment Scheme (NEQAS),
coordinated by the Wolfson Research Laboratory in Birmingham, is
another external quality assessment programme in the United Kingdom
(Bullock & Wilde, 1985). Samples are distributed to the participating
laboratories for analysis, and based on the results obtained, a
variance index (VI) is calculated. The VI is the difference between
the result obtained and a trimmed mean, divided by a chosen relative
standard deviation for the analyte and expressed as a percentage. A
Mean Running Variance Index Score (MRVIS) and a Mean Running Bias
Index Score (MRBIS) are also calculated. It is believed that a MRVIS
below 33 should be the target for the participants, and that any MRVIS
over 66 should stimulate investigation of the laboratory's method.
3.10 Analytical performance criteria
The limits of the uncertainty in the produced data should be
based on the accuracy requirements for the monitoring data and what
can be achieved with available analytical techniques. The accuracy
and precision of routine and reference methods should be evaluated in
order to determine the feasibility of the proposed criteria.
Often the criteria for the analytical performance are determined
by the sensitivity and accuracy of the analytical method. In the
WHO/UNEP monitoring project on cadmium and lead in blood, the limits
for the MAD lines for lead in blood were set to
y = x ± (0.1 x + 20). This means that if the average
concentration of lead in blood in a group of people was found to be,
for example, 100 µg/litre, the criteria guaranteed with a probability
of 90% that the true average concentration was somewhere between 70
and 130 µg/litre. If the average blood lead concentration found was
50 µg/litre, the criteria guaranteed that the true average was between
25 and 75 µg/litre. With such a great "uncertainty" in the monitoring
data, it is of course difficult to detect differences in lead exposure
between various groups in the general population.
The experience from the quality control activities in the HEAL
pilot project on lead and cadmium shows that, for lead concentrations
in blood (µg/litre), experienced laboratories can meet the MAD
criteria y = x ± (0.05 x + 10), which guarantees that an obtained
mean blood lead concentration of 50 µg/litre lies with 90% probability
between 37.5 and 62.5 µg/litre. For concentrations of cadmium in
blood, well-experienced and well-equipped laboratories met the MAD
criteria y = x ± (0.05 x + 0.2), which guarantees that an
obtained mean blood cadmium concentration of, for example, 0.5
µg/litre lies with 90% probability between 0.28 and 0.72 µg/litre.
3.11 Quality control samples
The analytical procedures and performance may vary considerably
between various types of samples. Good analytical performance for a
pollutant in one type of medium is no guarantee of good performance
with other media. Thus, it is important for quality control samples
to have a matrix similar to that of the monitoring samples.
As already mentioned above, the quality control sample should
cover the range of concentrations likely to be found in the monitoring
samples. It must be emphasized that good analytical performance at
one concentration is no guarantee for good performance at other
concentrations.
Various types of reference samples are commercially available
(Muntau et al., 1983; Belliardo & Wagstaffe, 1988; Klich & Caliste,
1988; Okamoto, 1988; Parr et. al., 1988; Rasberry, 1988). However,
many of the commercially available reference materials are certified
for a limited number of substances, and usually only for one or two
different concentrations. Commercially available reference materials
are suitable for internal quality control purposes but cannot, as a
rule, be used for published data.
If possible, specimens of the samples should finally be archived
in order to prove again the validity of an environmental monitoring
study, if it should be necessary. This may be appropriate where the
samples are still integral after evaluation.
Biological tissue is typically contained in slides or blocks,
while specimens are stored in containers with appropriate fixation.
Aliquots from the various media for chemical analysis may be stored
after appropriate preservation, carrying a label with an expiry date
based on stability analysis. All written documentation including the
raw data is also transferred to the archive at the conclusion of the
study.
REFERENCES
AOAC (1984) Quality assurance principles for analytical laboratories.
Washington, DC, Association of Official Analytical Chemists, 224 pp.
Belliardo JJ & Wagstaff PJ (1988) BCR reference materials for food and
agricultural analysis: An overview. Fresenius Z Anal Chem, 332(6):
533-538.
Black SC (1988) Defining control sites and blank sample needs. In:
Keith LH ed. Principles in environmental samples. Washington, DC,
American Chemical Society, pp 109-118.
Bruaux P & Svartengren M ed. (1985) Global Environmental Monitoring
System (GEMS). Assessment of human exposure to lead: Comparison
between Belgium, Malta, Mexico and Sweden. Stockholm, National Swedish
Institute of Environmental Medicine and Department of Environmental
Hygiene, Karolinska Institute, and Brussels, Institute of Hygiene and
Epidemiology, 57 pp (Prepared for United Nations Environment Programme
and World Health Organization).
Bruaux P, Claeys-Thoreau F, Ducoffre G, & Lafontaine A (1979)
Exposition saturnine de la population bruxelloise. Choix des groupes
de population. Choix des indicateurs. Premiers résultats. Arch Belg
Méd Soc, 37: 488-501.
Bullock DG & Wilde CE (1985) External quality assessment of urinary
pregnancy oestrogen assay: further experience in the United Kingdom.
Ann Clin Biochem, 22: 273-282.
Burck K (1979) The role of quality assurance in good laboratory
practices. Clin Toxicol, 15: 627-640.
Cochran WG (1963) Sampling techniques. New York, John Wiley & Sons, 13
pp.
FDA (1987a) Good laboratory practice regulations. Fed Reg, 52(172):
33768 (21 CFR Part 58) (Docket No. 83N-0142).
FDA (1987b) Part 58 - Good laboratory practice for non-clinical
laboratory studies. Code Fed Regul, 21(Ch1): 228-242.
Friberg L (1988) Quality assurance. In: Clarkson TW, Friberg L,
Nordberg GF, & Sager PR ed. Biological monitoring of toxic metals. New
York, Plenum Press, pp 103-126.
Friberg L & Vahter M (1983) Assessment of exposure to lead and cadmium
through biological monitoring. Results of a UNEP/WHO global study.
Environ Res, 30: 95-128.
Herber RFM (1990) The World Health Organization study on health
effects of exposure to cadmium. Toxicol Environ Chem, 27: 1-10.
Herber RFM & Schaller KH (1986) Analytical variability of biological
parameters of exposure and early effects. In: Notten WRF, Herber RFM,
Hunter WJ, Monster AC, & Zielhuis RL ed. Health surveillance of
individual workers exposed to chemical agents. Berlin, Springer
Verlag, pp 102-109.
Horwitz W (1988) Protocol for the design, conduct and interpretation
of collaborative studies: IUPAC Workshop on the Harmonization of
Collaborative Analytical Studies, Geneva, Switzerland, 4-5 May 1987.
Pure Appl Chem, 60: 855-864.
Horwitz W (1989) Harmonization of methodology: international
perspective. In: Cowgill UM & Williams LR ed. Aquatic toxicology and
hazard assessment. Philadelphia, American Society for Testing and
Materials, pp 5-10 (Special Technical Publication No. 1027).
ISO (1986a) International Standard 8402. Quality - Vocabulary. Geneva,
International Organization for Standardization, 15 pp.
ISO (1986b) International Standard 5725: Precision of test methods -
Determination of repeatability and reproducibility for a standard test
method by inter-laboratory tests: 2nd edition. Geneva, International
Organization for Standardization, 49 pp.
ISO (1987a) International Standard 9000. Quality management and
quality assurance standards: guidelines for selection and use. Geneva,
International Organization for Standardization, 6 pp.
ISO (1987b) International Standard 9004. Quality management and
quality system elements - Guidelines. Geneva, International
Organization for Standardization, 19 pp.
Kelley JC (1976) Sampling the sea In: Cushing DH & Walsh JJ ed. The
ecology of the seas. Oxford, Blackwell Scientific Publications, pp
361-387.
Klich H & Caliste JP (1988) COMAR - database for certified reference
materials. Fresenius Z Anal Chem, 332(6): 552-555.
Lepore PD (1979) FDA's good laboratory practice regulations. Pharm
Technol, 3: 71-74.
Lind B, Elinder C-G, Friberg L, Nilsson B, Svartengren M, & Vahter M
(1987) Quality control in the analysis of lead and cadmium in blood.
Fresenius Z Anal Chem, 326: 647-655.
Lind B, Vahter M, Rahnster B, & Bjors U (1988a) Quality control
samples for the determination of lead and cadmium in blood, feces, air
filters and dust. Fresenius Z Anal Chem, 332(6): 741-743.
Lind B, Bigras L, Cernichiari E, Clarkson TW, Friberg L, Hellman M,
Kennedy P, Kirkbride J, Kjellstrom T, & Ohlin B (1988b) Quality
control of analyses of mercury in hair. Fresenius Z Anal Chem, 332(6):
620-622.
Matsushita H & Tanabe K (1991) Earthwatch - Global Environmental
Monitoring System (GEMS). Exposure monitoring of nitrogen dioxide. An
international pilot study within the WHO/UNEP Human Exposure
Assessment Location (HEAL) Programme. Nairobi, United Nations
Environment Programme, 109 pp.
Muntau H, Schramel P, Brätter P, & Knapp G (1983) Trace element
investigations on some new biological test materials. In: Brätter P &
Schramel P ed. Trace element - Analytical chemistry in medicine and
biology. Berlin, New York, Walter de Gruyter & Co., vol 2, pp 819-831.
OECD (1982) Good laboratory practice in the testing of chemicals.
Paris, Organisation for Economic Co-operation and Development, 62 pp.
Okamoto K (1988) Biological reference materials from the National
Institute of Environmental Studies (Japan). Fresenius Z Anal Chem,
332(6): 524-527.
Parr RM, Schelenz R, & Ballestra S (1988) IAEA biological reference
materials. Fresenius Z Anal Chem, 332(6): 518-523.
Rasberry SD (1988) NBS activities in biological reference materials.
Fresenius Z Anal Chem, 332(6): 528-532.
Slorach SA & Vaz R ed. (1983) Global Environmental Monitoring System
(GEMS). Assessment of human exposure to selected organochlorine
compounds through biological monitoring. Uppsala, Swedish National
Food Administration, 134 pp (Prepared for United Nations Environment
Programme and the World Health Organization).
Smith F, Kulkarni S, Myers LE, & Messner MJ (1988) Evaluating and
presenting quality assurance sampling data. In: Keith LH ed.
Principles in environmental samples. Washington, DC, American Chemical
Society, pp 157-168.
Sokal RR & Rohlf FJ (1981) Biometry: The principles and practice of
statistics in biological research, 2nd ed. New York, Oxford, W.H.
Freeman, 859 pp.
Starks TN (1989) Global Environmental Monitoring System (GEMS).
Statistical guidelines for quality assurance in human exposure
assessment location studies. Geneva, World Health Organization, 48 pp
(Document PEP/89.9).
Taylor A (1988) Reference materials for measurement of aluminium in
biological samples. Fresenius Z Anal Chem, 332(6): 616-619.
Taylor JK (1987) Quality assurance of chemical measurements. Chelsea,
Michigan, Lewis Publishers, 328 pp.
Taylor JK (1988a) Defining the accuracy, precision and confidence
limits of sample data. In: Keith LH ed. Principles in environmental
samples. Washington, DC, American Chemical Society, pp 101-108.
Taylor JK (1988b) The role of statistics in quality assurance.
Fresenius Z Anal Chem, 332(6): 722-725.
Taylor A, Starkey BJ, & Walker AW (1985) Determination of aluminium in
serum: findings of an external quality assessment scheme. Ann Clin
Biochem, 22: 351-358.
UNEP/WHO (1984) Principles and procedures for quality assurance in
environmental pollution exposure monitoring. Geneva, World Health
Organization, 31 pp (EFP/HEAL/84.4).
Vahter M (1982) Global Environmental Monitoring System (GEMS).
Assessment of human exposure to lead and cadmium through biological
monitoring. Stockholm, National Swedish Institute of Environmental
Medicine and Department of Environmental Hygiene, Karolinska
Institute, 136 pp (Prepared for United Nations Environment Programme
and World Health Organization).
Vahter M & Slorach S ed. (1989) Global Environmental Monitoring System
(GEMS). Exposure monitoring of lead and cadmium: An international
pilot study within the UNEP/WHO Human Exposure Assessment Location
(HEAL) Programme. Nairobi, United Nations Environment Programme, 82
pp.
Vahter M & Slorach S ed. (1990) Global Environmental Monitoring System
(GEMS). Exposure monitoring of lead and cadmium: An international
pilot study within the WHO/UNEP Human Exposure Assessment Location
(HEAL) Programme. Nairobi, United Nations Environment Programme, 82
pp.
WHO (1986a) Environmental Health Criteria 57: Principles of
toxicokinetic studies. Geneva, World Health Organization, pp 20-24.
WHO (1986b) UNEP/WHO Global Environmental Monitoring System (GEMS).
Human Exposure Assessment Location (HEAL) Project: Guidelines for
integrated air, water, food and biological exposure monitoring.
Geneva, World Health Organization, 389 pp (Document PEP/86.6).
WHO (1992) Earthwatch - Global Environmental Monitoring System (GEMS).
Guidance on survey design for Human Exposure Assessment Locations
(HEAL) Studies. Geneva, World Health Organization (Document
WHO/PEP/92.6).
Youden WJ (1967) The role of statistics in regulatory work. J Assoc
Off Anal Chem, 50: 1007-1013.
Youden WJ & Steiner EH (1975) Statistical manual of the Association of
Official Analytical Chemists. Washington, DC, Association of Official
Analytical Chemists, 88 pp.
Zheng XQ & Ji RD (1987) Assessment of lead contamination of the
general population through blood lead levels. Environ Monit Assess, 9:
169-177.
APPENDIX I
GLOSSARY
Batch: A specific quantity or lot of a test or reference substance
produced during a defined cycle of manufacture, in such a way that it
could be expected to be of a uniform character and should be
designated as such.
Carrier (vehicle): Any agent that serves as a vehicle used to mix,
disperse, or solubilize the test or reference substance to facilitate
the administration to the test system.
Calibration: Assigning values to the output of an instrument or
other device. Calibration can apply to instruments, glassware or any
other measuring device.
Calibration verification (calibration check): Determining if the
output of an instrument or device is within some established
tolerance.
Code number: A unique number assigned to each packaged unit of the
test substance.
Final report: A comprehensive report that includes all information
relative to the evaluation of the results of a specific nonclinical
laboratory study.
Laboratory technician: A laboratory worker who performs an operation
covered by an SOP, under the supervision of a professional scientist.
Master schedule sheet: A document that contains essential
information on completed and in-progress studies in the laboratory,
i.e. a list of studies, initiation and completion dates, test systems,
route of administration and name of study director.
Quality assurance programme (QAP): An internal control system of
inspections and study audits designed to ascertain that a study is in
compliance with defined guidance principles. It assures laboratory
management that facilities, equipment, personnel, methods, practices,
records and controls conform with these principles.
Quality assurance unit (QAU): Any person or organizational element,
designated by testing facility management to perform inspections of
laboratory operations and study audits relating to quality assurance
of studies.
Quality control system: A system used to assess product quality
whenever the quality attributes of the final product can be expressed
as discretely measured parameters.
Raw data: All original laboratory records and documentation, or
verified copies thereof, which are the result of the original
observations and activities in a study.
Sample: Any quantity of the test or reference substance.
Specimen: Any material derived from a test system for examination,
analysis or storage.
Sponsor: A person or entity who commissions and/or supports a study.
A testing facility can also be a sponsor if it both initiates and
actually conducts the study.
Standardization: Determining the response of an instrument to known
quantities of a test agent according to a specific method; accurately
determining the concentration of a solution.
Standardization verification (standardization check): Determining if
the response of an instrument to a particular standard is within some
established tolerance (e.g., verifying that a previously prepared
standard curve (or other procedure) is still valid).
Standard operating procedure (SOP): A written procedure that
describes how to perform certain routine laboratory tests or
activities that are normally not specified in detail in study plans or
test guidelines.
Study: An experiment or set of experiments in which a test substance
is examined to obtain data on its properties and/or its safety with
respect to human health and the environment.
Study audit: A comparison of the raw data and associated records
with the interim or final report in order to determine whether the raw
data was accurately reported, and whether testing was carried out in
accordance with the study plan and SOPs to obtain additional
information not provided in the report, and to establish whether
practices were employed in the development of data that would impair
their validity.
Study director: The individual responsible for the overall conduct
of the study.
Study plan: A protocol which defines the entire scope of the study.
Test facility (testing facility): The persons, premises and
operational unit(s) that are necessary for conducting the study.
Test facility management: Consists of the executive level of
management charged with the ultimate responsibility of the test
facility and studies.
Test substance: A chemical substance or a mixture that is under
investigation.
Test system: Any human, other animal, plant, microbial, as well as
other cellular, subcellular, chemical or physical system or
combination of these, that is exposed to a test, control or reference
substance.
Validation: Establishing documented evidence that provides a high
degree of assurance that the intended use of such things as a
procedure, test system, test substance or control substance is
accomplished.
Validation procedure(s): A written procedure stating how validation
will be conducted. Validation procedures should exist for all
elements of the test system and should specify the procedures and
tests that will be conducted and the data to be collected. Examples
include the validation procedure used for animal supply vendors,
facility equipment, test substances control substances, and SOPs.