
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 163
CHLOROFORM
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First draft prepared by Dr. J. de Fouw
National Institute of Public Health and
Environmental Protection, Bilthoven,
Netherlands.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1994
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Chloroform.
(Environmental health criteria ; 163)
1.Chloroform - adverse effects
I.Series
ISBN 92 4 157163 2 (NLM Classification: QV 81)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1994
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR CHLOROFORM
1. SUMMARY
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Sampling and analysis in air
2.4.1.1 Direct measurement
2.4.1.2 Adsorption-liquid desorption
2.4.1.3 Adsorption-thermal desorption
2.4.1.4 Cold trap-heating
2.4.2. Sampling and analysis in water
2.4.3. Sampling and analysis in biological samples
2.4.3.1 Blood and tissues
2.4.3.2 Urine
2.4.3.3 Fish
2.4.4. Sampling and analysis in soil gas
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production
3.2.1.1 Direct production levels and processes
3.2.1.2 Indirect production
3.2.1.3 Emissions from direct production and
use
3.2.1.4 Emissions from indirect production
3.2.2. Uses
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Transport
4.1.2. Distribution
4.1.3. Removal from the atmosphere
4.2. Biotic degradation
4.3. Bioaccumulation
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Ambient air
5.1.2. Indoor air
5.1.3. Water
5.1.3.1 Sea water
5.1.3.2 Rivers and lakes
5.1.3.3 Rain water
5.1.3.4 Waste water
5.1.3.5 Ground water
5.1.3.6 Drinking-water
5.1.4. Soil
5.1.5. Foodstuffs
5.2. General population exposure
5.2.1. Outdoor air
5.2.2. Indoor air
5.2.3. Drinking-water
5.2.4. Foodstuffs
5.3. Occupational exposure during manufacture, formulation or
use
6. KINETICS IN LABORATORY ANIMALS AND HUMANS
6.1. Pharmacokinetics
6.1.1. Absorption
6.1.1.1 Oral
6.1.1.2 Dermal
6.1.1.3 Inhalation
6.1.2. Distribution
6.1.3. Elimination and fate
6.1.4. Physiologically based pharmacokinetic modelling
for chloroform
6.2. Biotransformation and covalent binding of metabolites
6.3. Human studies
6.3.1. Uptake
6.3.1.1 Oral
6.3.1.2 Dermal
6.3.1.3 Inhalation
6.3.2. Distribution
6.3.3. Elimination
6.3.4. Biotransformation
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Lethality
7.1.2. Non-lethal effects
7.1.2.1 Oral exposure
7.1.2.2 Subcutaneous and intraperitoneal
exposure
7.1.2.3 Inhalation exposure
7.1.2.4 Dermal exposure
7.2. Short-term exposure
7.2.1. Oral exposure
7.2.1.1 Mice
7.2.1.2 Rats
7.2.2. Inhalation exposure
7.3. Long-term exposure
7.4. Skin and eye irritation
7.5. Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1. Reproduction
7.5.2. Embryotoxicity and teratogenicity
7.5.2.1 Oral exposure
7.5.2.2 Inhalation exposure
7.6. Mutagenicity and related end-points
7.7. Carcinogenicity
7.7.1. Mice
7.7.2. Rats
7.7.3. Dogs
7.7.4. Studies on initiating-promoting activity
7.7.4.1 Mice
7.7.4.2 Rats
7.8. In vitro studies
7.9. Factors modifying toxicity; toxicity of metabolites
8. EFFECTS ON HUMANS
8.1. Acute non-lethal effects
8.2. Epidemiology
8.2.1. Occupational exposure
8.2.2. General exposure
8.3. Abuse and addiction
9. EFFECTS ON OTHER ORGANISMS IN THE
LABORATORY AND FIELD
9.1. Freshwater organisms
9.1.1. Short-term toxicity
9.1.2. Long-term toxicity
9.2. Marine organisms
10. EVALUATION OF HUMAN HEALTH RISKS AND
EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health risks
10.1.1. Exposure
10.1.2. Health effects
10.1.3. Approaches to risk assessment
10.1.3.1 Non-neoplastic effects
10.1.3.2 Neoplastic effects
10.2. Evaluation of effects in the environment
11. FURTHER RESEARCH
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
REFERENCES
RESUME
RESUMEN
WHO TASK GROUP ON ENVIRONMENTAL HEALTH
CRITERIA FOR CHLOROFORM
Members
Dr M.W. Anders, Department of Pharmacology, University of Rochester,
Rochester, New York, USA
Dr D.Anderson, British Industrial Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United
Kingdom
Dr R.J. Bull, Washington State University, College of Pharmacy,
Pullman, Washington, USA
Dr C.D. Carrington, Food and Drug Administration, Washington DC, USA
Dr M. Crookes, Environment Section, Building Research Establishment,
Garston, Watford, United Kingdom
Dr E. Elovaara, Institute of Occupational Health, Department of
Industrial Hygiene and Toxicology, Helsinki, Finland
Dr J. de Fouw, Toxicology Advisory Centre, National Institute of
Public Health and Environmental Protection (RIVM), Bilthoven,
the Netherlands (Rapporteur)
Dr M.E. Meek, Environmental Health Directorate, Health Protection
Branch, Health and Welfare, Ottawa, Canada (Chairperson)
Dr R. Pegram, Environmental Toxicology Division, Health Effects
Research Laboratory, US Environmental Protection Agency,
Research Triangle Park, North Carolina, USA
Dr S.A. Soliman, Department of Pesticide Chemistry, College of
Agriculture and Veterinary Medicine, King Saud
University-Al-Qasseem, Bureidah, Saudi Arabia (Vice-Chairman)
Dr L. Vittozzi, Istituto Superiore di Sanità, Laboratorio di
Tossicologia, Comparata ed Ecotossicologia, Rome, Italy
(Vice-Chairman)
Dr P.P. Yao, Institute of Occupational Medicine, Chinese Academy of
Preventive Medicine, Beijing, China
Representatives of other Organizations
Dr B. Butterworth, International Life Sciences Institute, Risk
Science Institute, Washington DC, USA
Secretariat
Dr B.H. Chen, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Dr P.G. Jenkins, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland
Dr C. Partensky, International Agency for Research on Cancer, Lyon,
France
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the
criteria monographs as accurately as possible without unduly
delaying their publication. In the interest of all users of the
Environmental Health Criteria monographs, readers are kindly
requested to communicate any errors that may have occurred to the
Director of the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland, in order that they may be
included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Case
postale 356, 1219 Châtelaine, Geneva, Switzerland (Telephone No.
9799111).
* * *
This publication was made possible by grant number 5 U01
ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial
support from the European Commission.
ENVIRONMENTAL HEALTH CRITERIA FOR CHLOROFORM
A WHO Task Group on Environmental Health Criteria for
Chloroform met in Geneva from 15 to 19 November 1993. Dr B.H Chen,
IPCS, welcomed the participants on behalf of the Director, IPCS, and
the three IPCS cooperating organizations (UNEP/ILO/WHO). The Task
Group reviewed and revised the draft document and made an evaluation
of risks for human health and the environment from exposure to
chloroform.
The first draft was prepared by Dr J. de Fouw of the National
Institute of Public Health and Environmental Protection (RIVM),
Bilthoven, Netherlands. The second draft was also prepared by Dr
J.de Fouw incorporating comments received following the circulation
of the first draft to the IPCS Contact Points for Environmental
Health Criteria monographs. Dr M.E. Meek (Health and Welfare,
Canada) made a considerable contribution to the preparation of the
final text.
Dr B.H. Chen and Dr P.G. Jenkins, both members of the IPCS
Central Unit, were responsible for the overall scientific content
and technical editing, respectively.
The efforts of all who helped in the preparation and
finalization of the monograph are gratefully acknowledged.
ABBREVIATIONS
ALAT alanine aminotransferase
ASAT aspartate aminotransferase
Brdu bromodeoxyuridine
DENA diethylnitrosamine
ENU ethylnitrosourea
GGTase gamma-glutamyl transpeptidase
LI labelling index
NOAEL no-observed-adverse-effect level
NOEC no-observed-effect concentration
NOEL no-observed-effect level
NOLC no-observed-lethal concentration
PBPK physiologically based pharmacokinetics
SCE sister-chromatid exchange
SGPT serum glutamine-pyruvate transaminase
UDS unscheduled DNA synthesis
1. SUMMARY
Chloroform is a clear, colourless, volatile liquid with a
characteristic odour and a burning, sweet taste. It is degraded
photochemically, is not flammable and is soluble in most organic
solvents. However, its solubility in water is limited. Phosgene and
hydrochloric acid may be formed by chemical degradation.
Chloroform is used in pesticide formulations, as a solvent and
chemical intermediate. Its use as an anaesthetic and in proprietary
medicines is banned in some countries. The commercial production
amounted to 440 000 tonnes in 1987. Significant amounts of
chloroform are also produced in the chlorination of water and the
bleaching of paper pulp.
There are several analytical methods for the analysis of
chloroform in air, water and biological materials. The majority of
these methods are based on direct column injection, adsorption on
activated adsorbent or condensation in a cool trap, then desorption
or evaporation by solvent extraction or heating and subsequent gas
chromatographic analysis.
It is assumed that most chloroform present in water is
ultimately transferred to air, due to its volatility. Chloroform has
a residence time in the atmosphere of several months and is removed
from the atmosphere through chemical transformation. It is resistant
to biodegradation by aerobic microbial populations of soils and
aquifers subsisting on endogenous substrates or supplemented with
acetate. Biodegradation may occur under anaerobic conditions. The
bioconcentration in freshwater fish is low. Depuration is rapid.
Based on estimates of mean exposure from various media, the
general population is exposed to chloroform principally in food,
drinking-water and indoor air in approximately equivalent amounts.
The estimated intake from outdoor air is considerably less. The
total estimated mean intake is approximately 2 µg/kg body weight per
day. Available data also indicate that water use in homes
contributes considerably to levels of chloroform in indoor air and
to total exposure. For some individuals living in dwellings supplied
with tap water containing relatively high concentrations of
chloroform, estimated total intakes are up to 10 µg/kg body weight
per day.
Chloroform is well absorbed in animals and humans after oral
administration but the absorption kinetics are dependent upon the
vehicle of delivery. After inhalation exposure in humans, 60-80% of
the inhaled quantity is absorbed. The primary factors affecting the
absorption kinetics of chloroform following inhalation are its
concentration and species-specific metabolic capacities. It is
readily absorbed through the skin of humans and animals and
significant dermal absorption of chloroform from water while
showering has been demonstrated. Hydration of the skin appears to
accelerate absorption of chloroform.
Chloroform distributes throughout the whole body. Highest
tissue levels are reached in the fat, blood, liver, kidneys, lungs
and nervous system. Distribution is dependent on exposure route;
extrahepatic tissues receive a higher dose from inhaled or dermally
absorbed chloroform than from ingested chloroform. Placental
transfer of chloroform has been demonstrated in several animal
species and humans. Chloroform is eliminated primarily as exhaled
carbon dioxide. Unmetabolized chloroform is retained longer in fat
than in any other tissue.
The oxidative biotransformation of chloroform is catalysed by
cytochrome P-450 to produce trichloromethanol. Loss of HCl from
trichloromethanol produces phosgene as a reactive intermediate.
Phosgene may be detoxified by reaction with water to produce carbon
dioxide or with thiols including glutathione or cysteine to produce
adducts. The reaction of phosgene with tissue proteins is associated
with cell damage and death. Little binding of chloroform metabolites
to DNA is observed. Chloroform also undergoes P-450-catalysed
reductive biotransformation to produce the dichloromethyl radical,
which becomes covalently bound to tissue lipids. A role for
reductive biotransformation in the cytotoxicity of chloroform has
not been established.
In animals and humans exposed to chloroform, carbon dioxide and
unchanged chloroform are eliminated in the expired air. The fraction
of the dose eliminated as carbon dioxide varies with the dose and
the species. The rate of biotransformation to carbon dioxide is
higher in rodent (hamster, mouse, rat) hepatic and renal microsomes
than in human hepatic and renal microsomes. Also, chloroform is
biotransformed more rapidly in mouse than in rat renal microsomes.
The liver is the target organ for acute toxicity in rats and
several strains of mice. Liver damage is characterized mainly by
early fatty infiltration and balloon cells, progressing to
centrilobular necrosis and then massive necrosis. The kidney is the
target organ in male mice of other more sensitive strains. The
kidney damage starts with hydropic degeneration and progresses to
necrosis of the proximal tubules. Significant renal toxicity has not
been observed in female mice of any strain.
Acute toxicity varies depending upon the strain, sex and
vehicle. In mice the oral LD50 values range from 36 to 1366 mg
chloroform/kg body weight, whereas for rats, they range from 450 to
2000 mg chloroform/kg body weight. After a single inhalation
exposure of 4 h, liver toxicity was observed in mice and rats at
chloroform levels of 490 and 1410 mg/m3, respectively.
The most universally observed toxic effect of chloroform is
damage to the liver. The severity of these effects per unit dose
administered depends on the species, vehicle and the method by which
the chloroform is administered. The lowest dose at which liver
damage has been observed is 15 mg/kg body weight per day
administered to beagle dogs in a toothpaste base over a period of
7.5 years. Effects at lower doses were not examined. Somewhat higher
doses are required to produce hepatotoxic effects in other species.
Although duration of exposure varied in these studies, the
no-observed-adverse-effect levels ranged between 15 and 125 mg/kg
body weight per day.
Effects in the kidney have been observed in male mice of
sensitive strains and in the F-344 rat. Severe effects have been
observed in a particularly sensitive strain of male mice at doses as
low as 36 mg/kg body weight per day.
Daily 6 h inhalation of chloroform for 7 consecutive days
induced atrophy of Bowman's glands and new bone growth in the nasal
turbinates of F-344 rats. The no-observed-effect level (NOEL) for
these effects was 14.7 mg/m3 (3 ppm). The significance of these
effects is being further investigated in longer-term studies.
Chloroform induced hepatic tumours in mice when administered by
gavage in corn oil at doses in the range of 138 to 477 mg/kg body
weight per day. However, when similar doses were administered in
drinking-water, there was no effect of chloroform on the yield of
hepatic tumours in mice. Moreover, when chloroform was administered
in drinking-water as a promoter in initiation/promotion studies, it
actually appeared to inhibit the development of diethylnitrosamine-
initiated liver tumours in mice. Thus, the vehicle utilized and/or
the method in which chloroform is administered is an important
variable in its induction of hepatic tumours in mice.
Chloroform induced kidney tumours in rats at doses of 90 to 200
mg/kg body weight per day in corn oil by gavage. However, in this
species, results were similar when the chemical was administered in
the drinking-water, indicating that the response is not entirely
dependent on the vehicle used.
The carcinogenic effects of chloroform on the liver and kidney
of rodents appear to be closely related to cytotoxic and cell
replicative effects observed in the target organs. The effects on
cell replication were found to parallel the modifications of
carcinogenic responses to chloroform that were induced by vehicle
and mode of administration. The weight of the available evidence
indicates that chloroform has little, if any, capability to induce
gene mutation or other types of direct damage to DNA. Moreover,
chloroform does not appear capable of initiating hepatic tumours in
mice or of inducing unscheduled DNA synthesis in vivo. On the
other hand, hepatic tumours can be efficiently promoted by
chloroform when it is administered in an oil vehicle. Consequently,
it is likely that, in the case of prolonged administration of
chloroform, cytotoxicity followed by cell proliferation is the most
important cause for the development of liver and kidney tumours in
rodents.
There are some limited data to suggest that chloroform is toxic
to the fetus, but only at doses that are maternally toxic.
In general, chloroform elicits the same symptoms of toxicity in
humans as in animals. In humans, anaesthesia may result in death due
to respiratory and cardiac arrhythmias and failure. Renal tubular
necrosis and renal dysfunction have also been observed in humans.
The lowest levels at which liver toxicity due to occupational
exposure to chloroform has been reported are in the range of 80 to
160 mg/m3 (with an exposure period of less than 4 months) in one
study and in the range of 10 to 1000 mg/m3 (with exposure periods
of 1 to 4 years) in another study. The mean lethal oral dose for an
adult is estimated to be about 45 g, but large interindividual
differences in susceptibility occur. There is some weight of
evidence for an association between exposure to disinfection
by-products in drinking-water and colorectal and bladder cancer in
some epidemiological studies. However, these studies are compromised
by inadequate account of potential confounding factors and other
weaknesses. The evidence for the carcinogenicity of chlorinated
drinking-water in humans is inadequate. In addition, the
disinfection by-products cannot be attributed to chloroform per se.
Chloroform is toxic to the embryo-larval stages of some
amphibian and fish species. The lowest reported LC50 is 0.3
mg/litre for the embryo-larval stages of Hyla crucifer. Chloroform
is less toxic to fish and Daphnia magna. The LC50 values for
several species of fish are in the range of 18 to 191 mg/litre.
There is little difference in sensitivity between freshwater and
marine fish. The lowest reported LC50 for Daphnia magna is 29
mg/litre. Chloroform is of low toxicity to algae and other
microorganisms.
The Task Group concluded that the available data are sufficient
to develop a tolerable daily intake (TDI) for non-neoplastic effects
and risk-specific intakes for carcinogenic effects of chloroform on
the basis of studies in animal species; the value will serve as
guidance in the development of exposure limits by appropriate
authorities. However, it is cautioned that where local circumstances
require that a choice must be made between meeting microbiological
limits or limits for disinfection by-products such as chloroform,
the microbiological quality must always take precedence. Efficient
disinfection must never be compromised.
Based on the study by Heywood et al. (1979) in which slight
hepatotoxicity (increases in hepatic serum enzymes and fatty cysts)
was observed in beagle dogs ingesting 15 mg/kg body weight per day
in toothpaste for 7.5 years, and incorporating an uncertainty factor
of 1000 (x10 for interspecies variation, x10 for intraspecies
variation and x10 for use of an effect level rather than a no-effect
level and a subchronic study), a TDI of 15 µg/kg body weight per day
is obtained.
Based on the available mechanistic data, the approach
considered most appropriate for provision of guidance based on mouse
liver tumours is division of a no-effect level for cell
proliferation by an uncertainty factor. Based on the NOEL for
cytolethality and cell proliferation in B6C3F1 mice of 10 mg/kg
body weight per day, following administration in corn oil for 3
weeks in the study of Larson et al. (1994a) and incorporating an
uncertainty factor of 1000 (x10 for interspecies variation, x10 for
intraspecies variation and x10 for severity of effect, i.e.
carcinogenicity, and less-than-chronic study), a TDI of 10 µg/kg
body weight per day is obtained.
It is recognized that the kidney tumours in rats may similarly
be associated with cell lethality and proliferation. However, since
data on cell proliferation are not available in the strain where
tumours were observed and identified information on cell
proliferation and lethality are short-term (one single gavage and
7-day inhalation exposure), it is considered premature to deviate
from the default model (i.e. linearized multistage) as a basis for
estimation of lifetime cancer risk. The total daily intake
considered to be associated with a 10-5 excess lifetime risk,
based on the induction of renal tumours (adenomas and
adenocarcinomas) in male rats in the study by Jorgenson et al.
(1985), is 8.2 µg/kg body weight per day.
Levels of chloroform in surface waters are generally low and
would not be expected to present a hazard to aquatic organisms.
However, higher levels of chloroform in surface water resulting from
industrial discharges or spills may be hazardous to the
embryo-larval stages of some aquatic species.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1 Identity
Chemical formula: CHCl3
Chemical structure:
H
'
Cl - C - Cl
'
Cl
Common name: chloroform
Common synonyms: trichloromethane, methane trichloride,
trichloroform, methyl trichloride,
methenyl trichloride
CAS chemical name: chloroform
CAS registry number: 67-66-3
RTECS registry number: FS 9100000
2.2 Physical and chemical properties
The most important physical properties of chloroform (IARC,
1979; Windholz, 1983) are given in Table 1.
Chloroform is a clear, colourless, very volatile liquid with a
characteristic odour and a burning sweet taste. It is not flammable;
however, the substance may be oxidized by strong oxidizing agents
with the formation of phosgene and chlorine gas. Pure chloroform is
light-sensitive. Reagent grade chloroform therefore usually contains
0.75% ethanol as a stabilizer to avoid photochemical transformation
to phosgene and hydrogen chloride (IARC, 1979; Budavari, 1989). In
the absence of light this reaction may be catalysed by iron. By the
application of stabilizers, such as methanol or ethanol, the
auto-oxidation may be prevented since the phosgene is fixed as
carbon dioxide dimethyl (or ethyl) ester. Chloroform stabilized with
0.006% amylenes is now available. This is important for toxicology
studies to avoid contamination with by-products that might be formed
by reaction with ethanol. The substance is soluble in most organic
solvents, such as alcohol, benzene, ether, petroleum ether, carbon
tetrachloride, oils and carbon disulfide. Its solubility in water is
limited.
Table 1. Physical properties of chloroform
Colour colourless
Relative molecular mass 119.38
Boiling point at 101.3 kPa 61.3 °C
Melting point -63.2 °C
Relative density (20 °C) 1.484
Refraction index (Nd 20) 1.4467
Heat capacity (20 °C) 0.979 kJ/kg °C
Critical temperature 263.4 °C
Critical pressure 5.45 MPa
Critical density 500 kg/m3
Auto-ignition temperature > 1000 °C
Solubility of chloroform in water (25 °C) 7.5-9.3 g/litre
Heat of combustion 373 kJ/mol
Evaporation heat at standard
boiling point 247 kJ/kg
Vapour density (101.3 kPa, 0 °C) 4.36 kg/m3
Vapour pressure (0 °C) 8.13 kPa
Vapour pressure (20 °C) 21.28 kPa
Stability air- and light-
sensitive, breaks down
to phosgene, HCl and
chlorine
log Kow (octanol/water partition
coefficient) 1.97
Chloroform produces a hydrate, CHCl3.17H2O, which
decomposes at 1.6 °C and 8 kPa. In contact with water, at normal
temperatures in the absence of oxygen, chloroform remains stable. It
is stable at temperatures up to 290 °C. Heating it in the presence
of a diluted caustic solution leads to the formation of formic acid.
The pyrolysis of chloroform vapour at temperatures above 450 °C
produces tetrachloroethane, hydrochloric acid and various
chlorinated hydrocarbons. In the presence of potassium amalgam or
hot copper, acetylene is formed. The reaction with primary amines in
an alkaline environment is known as the isonitrile reaction;
aromatic hydroxyaldehydes are formed in the presence of phenolates
(Reimer-Tiemann reaction). In the Friedel-Crafts reaction,
chloroform and benzene produce triphenyl methane. Chlorination of
the compound produces tetrachloromethane; bromination of chloroform
vapour at 225-275 °C produces CCl2Br2 and CClBr3. Chloroform
reacts with aluminium bromide to form bromoform (CHBr3).
Fluoroform (CHF3) is produced in the reaction with hydrogen
fluoride in the presence of a metallic fluoride as a catalyst.
Iodoform (CHI3) is produced by allowing chloroform to react with
ethyl iodide in the presence of aluminium chloride. Unstabilized
chloroform reacts with aluminium, zinc and iron. Chloroform mixed
with methanolic sodium hydroxide or acetone, in the presence of a
base, gives a violent reaction.
2.3 Conversion factors
1 mg chloroform/m3 air = 0.204 ppm at 25 °C and 101.3 kPa
(760 mmHg)
1 ppm = 4.9 mg chloroform/m3 air
2.4 Analytical methods
Many analytical methods for the determination of chloroform
residues in air, water and biological samples have been reported.
Table 2 summarizes some of the procedures used in the literature for
sampling and determining chloroform in different media. The
detection limits are included in Table 2. Although all of these
methods were developed to detect chloroform at very low levels, some
of them can be used only in cases where chloroform is present at
relatively high levels.
Since chloroform is very volatile, care must be taken while
sampling and handling samples to prevent any chloroform from being
lost during such procedures. In this case, accuracy depends very
much on the repeatability of the method being used. All but one of
the methods given in Table 2 use gas chromatographic techniques with
electron capture detection (ECD), flame ionisation detection (FID),
photo-ionisation detection (PID) or mass spectrometry (MS) for
Table 2. Sampling and analysis of chloroform
Medium Sample method Analytical method Detection limit Sample size Comments Reference
Air aspiration velocity of MIRAN-infrared 300 µg/m3 can be used only when Lioy & Lioy
28 litres/min, trajectory spectrometer CHCl3 is presented at (1983)
of 20 m high levels
Air direct injection GC with a 0.5 µg/m3 5 ml injected method involves the use of Lasa et al.
coulometric ECD a continuously operating (1979)
automatic GC monitor
Air direct injection, GC with two > 0.4 µg/m3 8 ml injected efficiency followed from Lillian &
calibration gas used for ECDs installed (estimated) signal ratios of the Singh (1974)
reliability serially two ECDs
Air AIRSCAN/PHOTOVAC GC-PID 0.5 µg/m3 0.05-1 ml portable machine, suitable Leveson et
direct injection for field monitoring al. (1981)
Air adsorption on activated GC-ECD approximately 1 m3/24 h in 1984 the draft standard NNI (1984)
charcoal, desorption 0.1 µg/m3 NVN 2794 needed to be
with CS2 tested for usefulness
Air adsorption on Porapak-N, GC-ECD 1 µg/m3 20 litres advantage of methanol is the Van Tassel et
desorption with 1-2 ml absence of a background al. (1981)
methanol signal in the ECD
Air adsorption on Porapak-N, GC-ECD estimated to 0.3-3 litres confirmation of results by Russell &
thermal desorption at be 0.05 µg/m3 use of GC-MS Shadoff (1977)
200 °C
Air adsorption on GC-ECD-FID two approximately 1-3 litres Heil et al.
Chromosorb-102, thermal detectors 0.06 µg/m3 (1979)
desorption at 150 °C positioned in
parallel
Table 2 (contd)
Medium Sample method Analytical method Detection limit Sample size Comments Reference
Air adsorption on Tenax, GC-FID 0.08 µg/m3 2 ml injected Kebbekus &
sample rate 10-15 ml/min, GC-MS Bozzelli (1982)
thermal desorption and
cryofocusing
Air adsorption on Tenax-GC, GC-MS 0.2 µg/m3 20 litres Krost et al.
cooled with liquid (1982)
nitrogen, thermal
desorption at 270 °C
Air adsorption on activated GC-FID with 0.15 mg up to 30 these two types of detection Morele et
coal, desorption with TCEP, detector litres can be appeared to complement al. (1989)
CS2, using Chromosorbsen sitivity sampled each other
methylcyclohexane as IS column
adsorption on activated GC-ECD with 5% 2 µg is
coal, desorption with CV17, Chromosorb minimum
ethanol, using column quantifiable
trichloroethylene as IS value
Air collection on charcoal, GC-FID 0.01 mg per up to 15 suitable for simultaneous US NIOSH
desorption with CS2 using sample litres can be analysis of two or more (1984)
n-undecane as IS estimated sampled substances
Air cold trap, heating the GC-ECD 0.01 µg/m3 30 ml in air samples were taken Harsch &
cold trap cold trap in the stratosphere Cronn (1978)
Air injection into cold trap, GC-MS (SIM) 0.03 µg/m3 100 ml in Cronn &
heating the cold trap cold trap Harsch (1979)
Table 2 (contd)
Medium Sample method Analytical method Detection limit Sample size Comments Reference
Air cold trap after desication GC-PID-ECD-FID, 0.005 µg/m3 1 litre during the process the Rudolph &
with magnesium 3 detectors column is kept at -103 °C Jebsen (1983)
perchlorate, heating the placed (cryofocusing)
cold trap to 257 °C sequentially
Breath collection on Tenax GC GC-MS 0.11 µg/m3 suitable for quantitative Pellizzari
cartridge, thermal analysis, one sample in et al.
desorption 1.5 h (1985b)
Water headspace, CH2Br2 was headspace GC-ECD 0.02 µg/litre 500 µl suitable for routine Herzfeld et
used as IS injected analysis over a wide range al. (1989)
of differently composed
river waters
Water pentane extraction GC-ECD using 1 µg/litre 100 ml suitable for routine Oliver (1983)
2 mm x 4 mm i.d. extracted with measurements in
column backed with 10 ml pentane, drinking-water
Squalane on 24 litres of
Chromosorb P extract used
for injection
Water liquid-liquid extraction GC with a Hall 0.10 µg/litre 3 µl injected suitable for routine Mehran et al.
with pentane electrolyte analyses (1984)
conductivity
detector,
Tenax-GC column
Water direct aqueous injection GC-ECD with a 0.02 µg/litre 2 µl injected suitable for analyses of Grob (1984)
of sample into GC fused silica halocarbons in the 0.01-10
capillary column ppb range
Table 2 (contd)
Medium Sample method Analytical method Detection limit Sample size Comments Reference
Water direct aqueous injection GC-ECD with a 0.1 µl/litre 1 µl injected easy, fast and reliable Temmerman &
of sample into GC methyl-silicone technique for everyday Quaghebeur
fused silica quality control (1990)
capillary column
Aqueous diethyl ether extraction GC-MS with a < 1 µg/litre 200 ml suitable for water and Meier et al.
with 25 µg fused silica and recovery extracted, homogenized environmental (1985)
p-bromofluorobenzene capillary column efficiency of extract samples
as IS 0.85 concentrated
to 1 ml, 2 µl
injected
Blood headspace, magnesium headspace 0.0225 µg/litre 200 µl suitable for direct Aggazzotti
sulfate heptahydrate and GC-ECD, with (2.5 times injected measurements of CHCl3 et al.
n-octyl alcohol were Chromosorb standard (1987)
added to the plasma W AW column deviation)
Blood passing inert gas over GC-MS 3 µg/litre 1-10 ml suitable for quantitative Pellizzari
warmed blood sample, analysis of CHCl3 in et al.
collection on Tenax-GC, blood (1985a)
thermal desorption
Blood diethyl ether extraction GC-MS with a qualitative (no 1-5 ml, suitable for identification Mink et al.
plasma (1:1) with 3 different fused silica detection limit extract of CHCl3 in biological (1983)
and internal standards added capillary column was given) concentrated samples
stomach to the concentrated to 1 ml of
contents extract of which 2µl
is injected
Table 2 (contd)
Medium Sample method Analytical method Detection limit Sample size Comments Reference
Tissue maceration in water, GC-MS 6 µg/kg 5 g suitable for semi- Pellizzari
collection on Tenax-GC, quantitative analysis of et al.
thermal desorption chloroform in tissues (1985a)
Urine pentane extraction GC-ECD < 1 µg/litre 2 µl of convenient and sensitive Youssefi
extract means for determining et al.
injected light halogenated (1978)
hydrocarbons
Fish extraction with pentane GC-ECD with a 1 µg/kg in 2 µl extraction efficiency of Baumann
and isopropanol, fused silica fresh injected 67% Ofstad et
bromotrichloromethane capillary column material al. (1981)
used as IS
Abbreviations:
ECD = electron capture detector; FID = flame ionisation detector; GC = gas chromatography; IS = internal standard;
MS = mass spectrometry; PID = photo-ionisation detector; SIM = selected ion monitoring
measuring chloroform residues. Only the first method listed depends
on the use of a MIRAN-infrared spectrometer. The sensitivity of this
method is very poor.
2.4.1 Sampling and analysis in air
The methods reported in Table 2 for sampling and analysis of
chloroform levels in air can be grouped into four different
categories.
2.4.1.1 Direct measurement
In this type of procedure, air is aspirated or injected
directly into the measuring instrument without pretreatment.
Although these methods are simple, they can be used only when
chloroform is present in the air at relatively high levels (e.g.,
urban source areas, see section 5.1.1).
2.4.1.2 Adsorption-liquid desorption
Air samples analysed for their chloroform levels are conducted
through an activated adsorbing agent (e.g., charcoal or Porapak-N).
The adsorbed chloroform is then desorbed with an appropriate solvent
(e.g., carbon disulfide or methanol) and subsequently passed through
the gas chromatograph (GC) for measurement.
2.4.1.3 Adsorption-thermal desorption
In this technique, air samples are also passed through an
activated absorbing agent (e.g., Tenax-GC, Porapak-Q, Porapak-N or
carbon molecular sieve). The adsorbed chloroform is then thermally
desorbed and driven into the GC column for determination.
2.4.1.4 Cold trap-heating
In this type of procedure, air samples are injected into a cold
trap (liquid nitrogen or liquid oxygen are used for cooling). The
trap is then heated while transferring its chloroform content into
the packed column of a GC for measurement.
2.4.2 Sampling and analysis in water
Several methods of sampling and analysing water for chloroform
content are included in Table 2. In some of these methods, water
samples are directly injected into a wide bore or fused silica
capillary column to which an ECD is attached. In some other water
analysis procedures mentioned in Table 2, the chloroform in the
water samples is first extracted by means of a non-polar,
non-halogenated solvent (e.g., n-pentane). Samples of the obtained
extracts are then injected into the GC for determining chloroform.
In another procedure, referred to as "close-loop-stripping analysis"
(CLSA), chloroform is removed from the water sample by purging it
with a large volume of a gas (e.g., nitrogen); the gas is then
passed through an adsorption tube and subsequently analysed by
GC-MS. Using this latter method, a million-fold concentration can be
achieved, so that chloroform can be quantified even at very low
levels. A headspace GC technique with ECD has also been used for
measuring chloroform levels in water samples (see Table 2).
2.4.3 Sampling and analysis in biological samples
2.4.3.1 Blood and tissues
Several procedures for determining chloroform in blood and
tissue samples are presented in Table 2. A headspace GC technique
has been used for direct measurement of chloroform in plasma
obtained from subjects exposed to low levels in air (Aggazzotti et
al., 1987). The second procedure (Kroneld, 1985) depends on
liquid-liquid extraction of chloroform from blood samples and
subsequent injection of the extract into a GC system for
quantification. In the method of Pellizzari et al. (1985a),
chloroform is evaporated by passing an inert gas over a warmed
plasma or macerated tissue sample with adsorption of the vapour on a
Tenax GC column, and is then recovered by thermal desorption and
analysed by GC-MS.
2.4.3.2 Urine
Youssefi et al. (1978) measured chloroform concentration in
urine using pentane extraction and GC-ECD analysis.
2.4.3.3 Fish
The procedure of Baumann Ofstad et al. (1981) for determining
chloroform in fish samples is based on extraction by n-pentane and
subsequent analysis of the extracts by GC/ECD. It has been reported
that the sensitivity of this method is greatly affected by the fat
content of the fish samples.
2.4.4 Sampling and analysis in soil gas
Kerfoot (1987) determined the level of chloroform in soil gas
samples in order to use the results as an indication of ground water
contamination by this pollutant. In the procedure used, a 75-ml soil
gas sample was drawn from a depth of 1.3 m by means of a sampling
probe. The chloroform content of the subsample was directly measured
in the field using an on-site GC-ECD. The detection limit for
chloroform in soil gas by this method was reported to be 5 parts per
billion by volume.
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Information on the natural occurrence of chloroform has not
been identified.
3.2 Anthropogenic sources
3.2.1 Production
3.2.1.1 Direct production levels and processes
Chloroform was prepared, almost simultaneously in 1831, by the
action of alkali on chloral (Liebig) and by treating bleaching
powder with ethanol or acetone (Soubeirain) (Hardie, 1964). It is
currently manufactured in the USA by hydrochlorination of methanol
or by chlorination of methane. All chloroform production in Japan
and western Europe is by chlorination of methane (IARC, 1979). It
can also be manufactured by oxychlorination of methane (ECDIN,
1992).
In the years 1984-1987, the worldwide production of chloroform
increased from 360 to 440 kilotonnes (see Table 3).
3.2.1.2 Indirect production
An important contribution to the total emission of chloroform
is made through its formation from other substances. In particular
the reaction of chlorine with organic compounds may produce
substantial quantities of chloroform. With respect to the formation
of chloroform in the aquatic medium, it may be assumed that the
quantities produced are ultimately emitted totally to the
atmosphere.
The following sources are known to contribute to the formation
and emission of chloroform:
* Paper bleaching with chlorine (US EPA, 1984; Rosenberg et al.,
1991).
* Chlorination of drinking-water (US EPA, 1984).
* Chlorination of swimming pool water (Bätjer et al., 1980). A
study on emissions in indoor public swimming pools in Bremen
(Germany) revealed that an average of 10 g chloroform may be
produced daily.
* Chlorination of cooling water. The quantity of chloroform
formed depends on a vast range of factors, such as acidity and
the concentration of organic materials.
Table 3. Chloroform production and production capacity expressed in
kilotonnes over a period of 15 years (1973-1988)
Country Year Production Capacity
USA 1975 118 -
1980 160 -
1984 179 -
1985 - 200
1986 191 -
1987 204 -
1988 - 218
Japan 1984 46 -
1985 - 55
1987 55 -
1988 - 60
Italy 1973 13 -
1988 - 55
France 1973 14 -
1987 45 -
1988 - 55
Federal Republic of Germany 1973 22 -
Netherlands 1973 8 -
Belgium 1973 15 -
European Economic Community 1979 80 -
1980 95 -
1982 - 155
1984 130 -
1985 - 160
1987 150 -
1988 - 200
World 1984 360 -
1987 440 -
1988 - 500
From: ECDIN (1992)
* Chlorination of waste water.
* Exhaust emissions from traffic. The exhaust fumes of vehicles
have been demonstrated to contain chloroform; this originates
from the decomposition of 1,2-dichloroethane, which is added
to petrol as a lead scavenger (US EPA, 1984). Rem et al. (1982)
estimated the amount of chloroform to be 1% of the amount of
1,2-dichloroethane added.
* Decomposition of trichloroethene in the atmosphere. At high
concentrations (1 ppm) in the presence of light and NO2, 1%
was estimated to be converted (Appleby et al., 1976). US EPA
(1984) estimates this emission to be 780 tonnes/year in the
USA.
* Decomposition of 1,1,1-trichloroethane has also been suggested
as a source (van der Heijden et al., 1986).
Appleby et al. (1976) found that, at relatively high
concentrations (1 ppm), trichloroethene may yield about 1%
chloroform under the influence of light and NOx. The estimated
production of chloroform from trichloroethene is, at most, about 3 x
106 kg/year; in reality the value is likely to be lower.
A possible source of chloroform (van der Heijden et al., 1986)
is its production from 1,1,1-trichloroethane via the photolysis of
the formed chloral. The increase of chloroform levels in the
southern hemisphere since 1974 (from 3 to 11 ppt), is in accordance
with the increase in the levels of 1,1,1-trichloroethane during the
same period (from 25 to 116 ppt).
3.2.1.3 Emissions from direct production and use
Almost all of the emissions arise from production, storage,
transit and use.
Estimations of emission factors for the production of
chloroform range from 0.51 kg chloroform/tonne chloroform
(controlled) to 3.35 kg chloroform/tonne chloroform (uncontrolled)
(US EPA, 1984). The Federal Office of the Environment (1981)
published a higher emission factor of 18 kg chloroform/tonne
chloroform.
With respect to emissions of chloroform in the production of
chlorodifluoromethane, emission factors ranging from 0.077-0.33 kg
chloroform/tonne chlorodifluoromethane (controlled) to 0.59-2.5 kg
chloroform/tonne chlorodifluoromethane (uncontrolled) have been
reported (US EPA, 1984). The Federal Office of the Environment
(1981) reported an emission factor of 8 kg chloroform/tonne
chlorodifluoromethane.
3.2.1.4 Emissions from indirect production
Significant losses of chloroform can also be expected from
indirect production of chloroform during the chlorination of water
and paper pulp. Data on the magnitude of such emissions have not
been identified.
3.2.2 Uses
In the period 1980-1987, the use of chloroform increased in the
USA from 170 to 200 kilotonnes and in the EEC from 90 to 110
kilotonnes. The use in Japan was 70 kilotonnes in 1987 (ECDIN,
1992). Chloroform is used in pesticide formulations, in the
production of other chemicals, and as a solvent. More than 80% of
the produced chloroform is used for the production of
chlorodifluoromethane (ECDIN, 1992). This use is likely to decrease
in the future due to planned phase-out under the Copenhagen
Amendment to the Montreal Protocol (1992). Chloroform was formally
used as an anaesthetic (IARC, 1979).
In many countries the use of chloroform is banned as an
ingredient (active or inactive) in human drug and cosmetic products
(US FDA, 1976). However any drug product containing chloroform in
residual amounts, resulting from its use as a processing solvent in
manufacture or as a by-product from the synthesis of an ingredient,
is not considered to contain chloroform as an ingredient (US FDA,
1976). Chloroform is registered for use in the USA as an
insecticidal fumigant for stored barley, corn, oats, popcorn, rice,
rye, sorghum and wheat (US EPA, 1971).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Transport
Owing to its relatively high volatility, chloroform is
preferentially transferred from surface water to air. The
experimental half-life of chloroform in water (1 ppm solution with a
depth of 6.5 cm at 25 °C) was found to be 18.5 to 25.7 min in a
volatilization study by Dilling (1977). In the case of ground
waters, however, exchange with the atmosphere may not take place as
readily (Uchrin & Mangels, 1986).
4.1.2 Distribution
Adsorption - desorption
Uchrin & Mangels (1986) described the sorptive behaviour of
chloroform to solids from the Cohansey (90% sand, 8% silt, 2% clay,
4.4% organic matter) and Potomac-Raritan-Magothy (70.4% sand, 24%
silt, 5.6% clay, 2.2% organic matter) aquifer systems, located in
the southern New Jersey coastal plain. The fact that chloroform
showed a greater tendency to adsorb to the Cohansey material than to
the Potomac-RM material might be explained by the difference in
organic matter content. The organic carbon normalized partition
coefficient Koc was calculated by Uchrin & Mangels (1986) in two
ways and appeared to be 57.5 or 70.8. These values are in agreement
with the Koc values of 86.7 and 63.4 obtained for Cohansey and
Potomac-RM aquifer solids, respectively. Results from the
consecutive desorption experiments suggest that the sorption
processes in the systems used are not completely reversible.
4.1.3 Removal from the atmosphere
Since no data on the removal rate of chloroform through
deposition are available, the values are based on estimates and
calculations. These values, however, differ widely. The estimated
half-lives range from 92 to 900 years for wet deposition and from 20
days to 22 years for dry deposition.
The calculated half-lives for chloroform degradation are
reported to be approximately 100 to 180 days. Reaction with hydroxyl
radicals is likely to be the only mechanism for the decomposition of
chloroform in the atmosphere (van der Heijden et al., 1986). Cox et
al. (1976) determined the relative rate constant for chloroform in
comparison with methane in smog chamber studies to be K = 270
ppm-1 min-1. However, it is known that the decomposition of
chlorinated hydrocarbons may lead to intermediary products that can
accelerate the decomposition process. Dimitriades et al. (1983)
noted that, in a smog chamber, tetrachloroethene is degraded more
rapidly than might be expected on the basis of the reaction rate
constant. Another drawback of the method of Cox et al. (1976) is the
false assumption that the decomposition of hydrocarbons always leads
to a transformation of two NO molecules for each carbohydrate
molecule transformed. The absolute rate constants determined by
Howard Carleton & Evenson (1976) and by Davis et al. (1976) are in
agreement with each other, and are K(OH) = 170 ± 20 ppm-1
min-1 and K(OH) = 160 ± 10 ppm-1 min-1, respectively. Based
on these rate constants of 170 and 160 ppm-1 min-1, a half-life
of approximately 60 days can be calculated for the decomposition of
chloroform in the atmosphere, assuming a 12-h daytime average
hydroxyl radical concentration of 2 x 10-15 mol/litre (Lyman et
al., 1982).
When chloroform is irradiated in the presence of chlorine, a
rapid reaction takes place, resulting in the formation of radicals.
At later stages the trichloromethyl radical may also be formed from
the reaction of CHCl3 with the hydroxyl radical. The
trichloromethyl radical subsequently reacts with oxygen to form the
trichloromethyl peroxyl radical, which ultimately leads to the
formation of phosgene (Spence et al., 1976). This is a possible
mechanism for the formation of phosgene in ambient air from
chlorination.
4.2 Biotic degradation
Strand & Shippert (1986) reported that chloroform is resistant
to biodegradation by aerobic microbial communities of soils and
aquifers subsisting on endogenous substrates or supplemented with
acetate (Wilson et al., 1981; Bouwer & McCarty 1983). Strand &
Shippert (1986) used Indianola sandy loam to study the oxidation of
chloroform to carbon dioxide in natural gas-enriched soils. It
appeared that some chloroform was oxidized by soils that were
exposed to cylinder air only, but that the rate in natural
gas-enriched soils was four times higher. Chloroform oxidation rates
increased with increasing chloroform concentrations up to 5 µg/g
soil (see Table 4). Chloroform oxidation continued up to 31 days but
was inhibited by acetylene and higher concentrations of methane,
indicating that methane-oxidizing bacteria may catalyse chloroform
oxidation.
Bouwer et al. (1981) found significant degradation of
chloroform in seeded cultures, relative to controls, at initial
concentrations of 16 and 34 µg/litre. At a high initial chloroform
concentration of 157 µg/litre, degradation was less evident,
although there was a gradual reduction in chloroform concentration
relative to the sterile controls. The anaerobic degradation appeared
to be the result of biological action, although a combination of
chemical and biological mechanisms is also possible.
Table 4. Effect of chloroform concentration on chloroform oxidation
Applied chloroform concentration Chloroform oxidized
(µg/g soil) (ng/5 g soil)a
0.02 2.8 ± 1.3
0.11 8.9 ± 7.7
0.55 3.2 ± 7.7
1.09 11.1 ± 3.6
5.47 20.7 ± 9.6
a Measured during an 8-day incubation in 5 g of aerobic soil
acclimated to natural gas
Chloroform can be degraded by reductive dehalogenation under
anaerobic conditions. It can be reduced by pure cultures of the
methanogen Methanobacterium thermoautotrophicum or the
sulfate-reducing bacterium Desulfobacterium autotrophicum (Egli et
al., 1987). In anaerobic sediments, chloroform is probably degraded
to carbon dioxide via a carbene mechanism (Bouwer & McCarty, 1983).
Van Beelen & Van Keulen (1990) studied the degradation of
radiolabelled chloroform under natural conditions in microcosm
experiments. In these experiments, the degradation was monitored by
the appearance of radiolabelled carbon dioxide rather than by the
disappearance of chloroform. This has the advantage that sorption,
which can also lead to disappearance of chloroform, does not
interfere with the measurements. At a concentration of 4 µg
chloroform/litre, the degradation followed first-order kinetics,
with half-lives of 12 days at 10 °C and 2.6 days at 20 °C. At a
concentration of 400 µg chloroform/litre, the degradation rate
increased with time. After 63 days, the final percentages of label
in carbon dioxide and chloroform happened to be similar to the
values of the 4-µg/litre experiment. At the other time intervals the
percentages of formed carbon dioxide were lower at the higher
chloroform concentration. Evidently the degradation rate of
chloroform at 400 µg/litre increases with time due to adaptation of
the bacteria in the sediment.
4.3 Bioaccumulation
Anderson & Lusty (1980) determined bioaccumulation in four
species of fish (Salmo gairdneri, Lepomis macrochirus, Micropterus
salmoides and Ictalurus punctatus). The bioaccumulation factor (on
a fresh weight basis) appeared to be maximal in Salmo gairdneri
(approximately 10). Depuration was complete in this species within
48 h. A similar value of 6 (whole body; fresh weight) in Lepomis
macrochirus was reported by Veith et al. (1978).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Ambient air
An overview of the concentrations of chloroform measured in
areas far from anthropogenic sources is presented in Table 5.
Table 5. Reported concentrations of chloroform in remote areas
(From: van der Heijden et al., 1986).
Northern hemisphere Southern hemisphere
Locality Year Level Locality Year Level
(µgm3) (µgm3)
Cork, Ireland 1974 0.133 Cape Town 1974 < 0.015
Pacific Ocean 1976 0.044 South Africa 1977 < 0.015
(N.W.)
California 1976 0.085 Pacific Ocean 1981 0.105
30-40°S, 138-146°E
California 1977 0.100 South Pole 1981 0.08
Kansas 1978 0.08 Australia 1981 0.110
Marshall Islands 1981 0.130 Samoa 1981 0.110
Cape Meares, Oregon 1981 0.225 Eastern Pacific 1981 0.055
0-40°S
Pt Barrow, Alaska 1981 0.195
Hawaii 1981 0.160
Eastern Pacific 1981 0.105
0-40°N
Chloroform levels in urban centres may be elevated in
comparison with concentrations in remote areas. As in the case of
other countries, levels in ambient air in remote areas of the USA
range from 0.1 to 0.25 µg/m3. In urban and source-dominated areas,
concentrations are 0.3-9.9 µg/m3 and 4.1-110 µg/m3, respectively
(ATSDR, 1991). The population-weighted mean concentration of
chloroform at 17 urban sites sampled across Canada in 1989 was 0.2
µg/m3 (Environment Canada, 1991).
Su & Goldberg (1976) reported chloroform levels of 1-15 µg/m3
in Japanese and European cities. Hourly average concentrations of
chloroform in the Netherlands, determined during 1979-1981, were
generally 0.15 µg/m3 or less (estimated detection limit), the
maximum value being 10 µg/m3 (Den Hartog, 1980, 1981). Average
concentrations of chloroform during 1990 in four German cities
(Berlin, Tübingen, Freudenstadt and Leipzig) ranged from 0.26 to 0.9
µg/m3; the maximum value was 30 µg/m3 detected in Tübingen
(Toxicology and Environmental Health Institute of Munich Technical
University, 1992).
5.1.2 Indoor air
In a study conducted by the US EPA, volatile organic compounds
including chloroform were determined in breath, breathing zone air,
fixed outdoor air, drinking-water and some foodstuffs of populations
in the USA (Wallace, 1987). The observed increase in the median
concentration of indoor versus outdoor air (approximately 85%) was
considered to be consistent with assumptions concerning daily water
use and likely release of chloroform from water into air (Wallace,
1987). Based on a survey conducted in 1981 in the Federal Republic
of Germany, Bauer (1981) reported that levels of chloroform may be
higher in kitchens where foodstuffs and water are heated.
Taketomo & Grimsrud (1977) reported average indoor air
concentrations of chloroform to be 0.3 µg/m3 in a family house and
1.0-3.4 µg/m3 in an apartment in Montana, USA, compared to 0.2
µg/m3 in outdoor air. In a nationwide survey of 757 randomly
selected one-family houses in Canada sampled over a 10-month period
in 1991, the mean level of chloroform in indoor air was 4.1 µg/m3;
the maximum value was 69 µg/m3 (Otson et al., 1992). Ullrich
(1982) reported comparable concentrations in indoor air (1-3
µg/m3) in Germany, although data on outdoor air levels in the
vicinity were not presented. Taketomo & Grimsrud (1977) reported
indoor air chloroform concentrations of between 2 and 10 µg/m3 in
buildings other than residences, e.g., restaurants and shops.
Higher levels of chloroform occur in the air of enclosed
swimming pools, resulting from water chlorination with sodium
hypochlorite and subsequent release to air. Over a period of eleven
months, the levels of chloroform directly above the water surface in
indoor public swimming pools in Bremen, Germany, ranged from 10 to
380 µg/m3, with an average of about 100 µg/m3 (Bätjer et al.,
1980; Lahl et al., 1981a). Ullrich (1982) reported a similar mean
value in four public swimming pools in Germany. Chloroform levels in
the air of enclosed swimming pools are a function of several factors
such as the degree of ventilation, the level of chlorination, water
temperature, the degree of mixing at the water surface, and the
quantity of organic precursors present (Lahl et al., 1981a).
5.1.3 Water
5.1.3.1 Sea water
The maximum concentration of chloroform determined in a survey
of bay water at 172 locations was 1 µg/litre (Pearson & McConnell,
1975). Reported levels in the open ocean (east Pacific) and off the
coast of California were 0.015 µg/litre and 0.009-0.012 µg/litre,
respectively (Su & Goldberg, 1976).
5.1.3.2 Rivers and lakes
Concentrations of chloroform in surface water vary, depending
upon the proximity to industrial sources. Concentrations of up to
394 µg/litre have been reported in rivers in highly industrial
cities (Ewing et al., 1977; Pellizzari et al., 1979). Levels in
areas not affected heavily by industrial sources ranged from trace
to 22 µg/litre (Ohio River Valley Water Sanitation Commission, 1980,
1982). Concentrations in river water in Germany and Switzerland
ranged from about 0.01 to 30 µg/litre (Reynolds & Harrison, 1982).
Average concentrations of chloroform detected in 1989 in German
rivers ranged from 0.131 to 3.17 µg/litre, with a maximum level of
5.1 µg/litre detected in the River Main (Toxicology and
Environmental Health Institute of Munich Technical University,
1992).
5.1.3.3 Rain water
Kawamura & Kaplan (1983) measured 0.25 µg chloroform/litre in
Los Angeles rain water samples taken in the spring of 1982.
5.1.3.4 Waste water
Based on two to four samplings at each of 37 plants (22
branches of industry), Van Luin & Van Starkenburg (1984) detected
chloroform mainly in the waste water of flavouring and
pharmaceutical industries at concentrations of 300 and 16 µg/litre,
respectively. Concentrations were lower in the waste water of
slaughter-houses, laundries, and textile, rubber and dye industries.
In waste-water discharges from the treatment of sewage and
industrial wastes in the USA, chloroform was detected at
concentrations ranging from 7.1 to 12.1 µg/litre (Europ-Cost, 1976).
5.1.3.5 Ground water
Concentrations of chloroform in ground water vary widely,
depending principally on proximity to hazardous waste sites (ATSDR,
1993). Chloroform was detected at levels ranging from 11 to 866
µg/litre in samples from 5 out of 6 monitoring wells drilled 64 m
apart in a direction perpendicular to the northward flow of ground
water at a contaminated site in Pittman, Nevada, USA (the depth of
unconfined ground water was 2 to 4 m at this selected site)
(Kerfoot, 1987). In a survey of potentially contaminated sites
conducted by the US EPA, chloroform was detected at 45% of the
sites. The median and maximum concentrations were 1.5 and 300
µg/litre, respectively (Westrick et al., 1989). In 8 out of 29 deep
wells in the Netherlands sampled at least twice since 1980 at
several depths (± 10 and 25 m below ground level), chloroform was
detected (limit of detection, 0.1 µg/litre) (Van der Heijden et al.,
1986).
5.1.3.6 Drinking-water
Chloroform can be formed from naturally occurring organic
compounds during the chlorination of drinking-water with the rate
and degree of formation being a function primarily of the
concentrations of chlorine and humic acid, temperature and pH.
Levels vary seasonally, the concentrations generally being greater
in summer than winter.
Stander (1980) detected chloroform in 16 out of 20 tap water
samples from the USA and western Europe. The highest concentration
was 60 µg/litre.
In a national survey of 450 community water supplies in the USA
sampled in 1978, chloroform was detected in 94% of surface water
supplies and 34% of ground-water supplies. Median concentrations in
surface and ground-water supplies were 60 µg/litre and less than the
detection limit (0.5 µg/litre), respectively (Brass et al., 1981).
Finished drinking-water collected in 1988 from 35 sources in the
USA, of which 10 were located in California, sampled in all four
seasons (spring, summer and autumn in 1988 and winter in 1989),
contained median concentrations of chloroform ranging from 9.6 to 15
µg/litre. The overall median for all four seasons was 14 µg/litre
(Krasner et al., 1989). In a survey conducted in the USA between
October 1987 and March 1989, the mean concentration in finished
water for surface water systems serving more than 10 000 people was
38.9 µg/litre (90th percentile, 74.4 µg/litre). The comparable mean
value in the distribution system was 58.7 µg/litre (US EPA, 1992).
In a national survey of the water supplies of 70 communities in
Canada conducted during the winter of 1976/1977, concentrations of
chloroform in treated water of the distribution system 0.8 km from
the treatment plant averaged 22.7 µg/litre (Williams et al., 1980).
Concentrations at 10 different locations in southern Ontario sampled
in the early 1980s were 4.5 to 60 µg/litre in water leaving the
treatment plant and 7.1 to 63 µg/litre one mile from the plant
(Oliver, 1983).
Chloroform levels in drinking-water in 100 German cities
sampled in 1977 ranged from < 0.1 to 14.2 µg/litre and averaged 1.3
µg/litre. Concentrations were similar in other surveys conducted in
Germany in the late 1970s and early 1980s (Lahl et al., 1981a).
Concentrations of chloroform in chlorinated samples of Rhine river
water were 9 µg/litre, compared to 0.1 µg/litre in untreated water
from the river (Zoeteman et al., 1982)
In Japan, chloroform was detected at concentrations of 18 and
36 µg/litre in drinking-water (Kajino, 1977).
5.1.4 Soil
No data on concentrations of chloroform in uncontaminated soil
have been identified. Chloroform has been detected, however, in 9.9%
of hazardous waste sites in the USA; the median concentration was
12.5 µg/kg (ATSDR, 1993).
5.1.5 Foodstuffs
Chloroform has been detected in several foodstuffs, in
particular in decaffeinated coffee (20 µg/kg), olive oil (28 µg/kg),
pork (10 µg/kg) and sausages (17 µg/kg). Occasionally,
concentrations were higher: up to 80 µg/kg in coffee and 90 µg/kg in
sausages. Levels of 1 to 10 µg/kg have been detected in flour
products, potatoes, cod liver oil, margarine, lard, fish, mussels
and milk; levels in most foodstuffs, however, were less than 1 µg/kg
(Bauer, 1981).
Daft (1988) reported that chloroform was detected in about 90
of 300 samples in a market-basket survey of 231 "table ready"
foodstuffs (prepared and cooked as normally served) in the USA, most
often in fat-containing samples. In a later account, it was reported
that 2 to 830 µg chloroform/kg food was detected in 68% of 549
samples of foodstuffs obtained in a market-basket survey, grouped as
fat, non-fat, grain-based and non-grain-based (average of 71 µg/kg)
(Daft, 1989).
Entz et al. (1982) did not detect chloroform in composite
samples of meat/fish/poultry or in composite samples of oil/fat in
39 different foods in the USA, although it should be noted that the
quantification limits were higher (18 to 28 µg/kg) than those in the
studies described above. However, the authors did detect chloroform
at a concentration of 17 µg/litre in the composite of dairy foods.
Concentrations of chloroform in soft drinks range from 3 to 50
µg/litre, with levels for cola being at the upper end of the range
(Abdel-Rahman, 1982; Entz et al., 1982; Wallace et al., 1984).
5.2 General population exposure
Based on estimates of mean exposure from various media, the
general population is exposed to chloroform principally in food
(approximately 1 µg/kg body weight per day), drinking-water
(approximately 0.5 µg/kg body weight per day) and indoor air (0.3 to
1 µg/kg body weight per day) in approximately equivalent amounts.
Estimated intake from outdoor air is considerably less (0.01 µg/kg
body weight per day). For some individuals living in dwellings
supplied with tap water containing relatively high concentrations of
chloroform, exposures may be as high as 10 µg/kg body weight per
day.
5.2.1 Outdoor air
Based on a daily inhalation volume for adults of 22 m3, a
mean body weight for males and females of 64 kg, the assumption that
4 out of 24 h are spent outdoors (WHO, in press), and the mean
levels of chloroform in ambient air in cities presented in section
5.1.1 (0.2 µg/m3), mean intake of chloroform from ambient air for
the general population is estimated to be 0.01 µg/kg body weight per
day.
5.2.2 Indoor air
Based on a daily inhalation volume for adults of 22 m3, a
mean body weight for males and females of 64 kg, the assumption that
20 out of 24 h are spent indoors (WHO, in press), and the mean
levels of chloroform in indoor air presented in section 5.1.2 (1 to
4 µg/m3), mean intake of chloroform from indoor air for the
general population is estimated to be 0.3 to 1.2 µg/kg body weight
per day.
Aggazzotti et al. (1990) determined levels of chloroform in
samples of plasma of swimmers and visitors taken "a few minutes
after" exposure at indoor swimming pools with water chloroform
concentrations of 16.9-47 µg/litre. Concentrations of chloroform in
the plasma of all 127 subjects who attended the pools averaged 0.82
µg/litre and ranged from 0.1 to 3 µg/litre, whereas in the plasma
samples of 40 nonexposed subjects, chloroform was not detected
(limit of quantification, 0.1 µg/litre). The mean level of
chloroform in the plasma was significantly higher in swimmers who
breathed under stress for a long time directly at the surface of the
water (training for competitions).
Individuals may be exposed to elevated concentrations of
chloroform (from chlorinated tap water) during showering (Jo et al.,
1990a,b).
After showering for 10 min in water containing 5 to 36 µg
chloroform/litre, the concentrations of chloroform in the breath of
six individuals ranged from 6.0 to 21 µg/m3, while none was
detected (detection limit 0.86 µg/m3) in any of the samples of
breath collected prior to a shower (Jo et al., 1990b). Based on
assumptions of an absorption efficiency from the respiratory tract
of 0.77, a breathing rate of 0.014 m3/min for a 70-kg adult, a
shower air concentration of 157 µg chloroform/m3 and a ratio of
body burden resulting from dermal exposure to that of inhalation
exposure of 0.93, the authors estimated that the average intake of
chloroform (inhalation and dermal absorption) was 0.5 µg/kg body
weight per shower for a person weighing 70 kg.
Based on a review of relevant estimates, Maxwell et al. (1991)
concluded that the ratio of the dose of chloroform received over a
lifetime from inhalation to that received from ingestion of
drinking-water is probably in the range of 0.6-1.5 but could be as
high as 5.7. The ratio of the dose received dermally compared to
that received orally over a lifetime from drinking-water was
considered to be approximately 0.3 but could be as high as 1.8.
5.2.3 Drinking-water
Based on a daily volume of ingestion for adults of 1.4 litres
and a mean body weight for males and females of 64 kg (WHO, in
press), and the mean levels of chloroform presented in section 5.1.3
(generally < 20 µg/litre), estimated mean intake of chloroform from
drinking-water for the general population is less than 0.5 µg/kg
body weight per day. As discussed by Bauer (1981), actual levels of
exposure may be less than those estimated on the basis of mean
levels in drinking-water since most of the chloroform would be
expelled from drinking-water that is heated before consumption (tea,
coffee, soups, sauces). For example, approximately 96% of the total
volatile halogenated hydrocarbon fraction was eliminated in water
boiling for 5 min, whereas 50-90% was eliminated upon heating at
70-90 °C (Bauer, 1981). It should be noted, however, that owing to
the wide variations in concentrations of chloroform in water
supplies, intake from drinking-water could be considerably greater
than estimated here for some segments of the general population.
5.2.4 Foodstuffs
Based on a daily volume of ingestion of solid foodstuffs for
reference adults of 1.536 kg and a mean body weight for males and
females of 64 kg (WHO, in press), and the mean level and percentage
detection of chloroform in foodstuffs in a market-basket survey
reported by Daft (1989) (section 5.1.5), estimated daily intake of
chloroform from foodstuffs is approximately 1 µg/kg body weight per
day.
5.3 Occupational exposure during manufacture, formulation or use
Workers may be exposed to chloroform during, for example, the
production of chloroform itself, the synthesis of substances derived
from chloroform (for example chlorodifluoromethane), the use of
chloroform as a solvent in bleaching of paper, and in sewage
treatment facilities. Based on a national survey conducted from 1981
to 1983, NIOSH estimated that approximately 96 000 workers in the
USA are potentially exposed to chloroform (ATSDR, 1993).
Chloroform is used as a solvent both industrially and in the
laboratory; several studies on concentrations in laboratories have
been published. Taketomo & Grimsrud (1977) reported levels of
2.3-8.6 mg/m3 in three laboratories in Montana, USA. In an office
situated in the same building but distant from the laboratories,
levels were similar; this was attributed to transfer through the
air-conditioning system. Levels found by NIOSH in laboratories
ranged from 0.5 to 24.9 mg/m3 (Salisbury, 1982). Time-weighted (4
h) average levels during laboratory practicals were 0-375 mg/m3
(Hertlein, 1980).
Some data on exposure of workers at sewage treatment facilities
and at indoor pools and spas have also been reported. Lurker et al.
(1983) reported a maximum level of 0.02 mg/m3 in sewage treatment
facilities. Maintenance workers, attendants and life guards at
indoor pools and spas were exposed to 0.025 and 0.075 mg/m3,
respectively (Armstrong & Golden, 1986; Benoit & Jackson, 1987).
Generally low levels of chloroform were detected by Rosenberg
et al. (1991) in a softwood and hardwood kraft pulp mill. Chloroform
levels ranged from 50 to 290 µg/m3 and from 220 to 5400 µg/m3 in
the softwood and the hardwood bleaching plants, respectively.
Chloroform has been and still is often used in dentistry as one
of the ingredients of root canal sealers or as a solvent. The
results of a study by Allard & Andersson (1992) showed that a dental
team could be exposed to quite high concentrations, ranging from 2.2
to 19.1 mg/m3.
6. KINETICS IN LABORATORY ANIMALS AND HUMANS
6.1 Pharmacokinetics
6.1.1 Absorption
6.1.1.1 Oral
Chloroform is well absorbed after oral administration. After
intragastric administration of chloroform (75 mg/kg body weight) in
water or vegetable oil to male Wistar rats, peak blood
concentrations were observed in about 6 min, but blood
concentrations were higher (39.3 versus 5.9 µg/ml) with water than
with olive oil as the vehicle (Withey et al., 1983). The area under
the blood concentration-time course curve (AUC) after chloroform
administration in water was 8.7 times greater than the AUC derived
from vegetable oil delivery.
Corley et al. (1990) used the data of Withey et al. (1983) to
compute gavage absorption rate constants, which were 0.6 h-1 and
5.0 h-1 for corn oil and water, respectively.
6.1.1.2 Dermal
Chloroform is absorbed through the intact skin. Most studies
have examined the systemic appearance of chloroform (or its
appearance in expired air) to quantify absorption. Tsuruta (1975)
estimated an absorption rate of 329 nmol/min per cm2 of skin
surface for pure chloroform in mice, but this study did not correct
for metabolism. Morgan et al. (1991) measured blood chloroform
levels in male F-344 rats during 24-h dermal exposures of a shaved
region of the back to pure chloroform or to aqueous chloroform
solutions. The blood chloroform level peaked at 51 mg/litre after
exposure to the pure chemical for 4 to 8 h, and remained about
constant for the duration of the exposure period. More rapid
absorption rates were observed during exposure to the aqueous
solutions, which resulted in peak blood chloroform levels after
about 2 h. The authors attributed this difference to hydration of
the skin. Bogen et al. (1992) applied aqueous solutions of
[14C]-chloroform to most of the body surface of hairless
guinea-pigs and obtained a permeability coefficient of 0.13 ml/cm2
per h. This study recovered metabolites as well as expired
chloroform to measure absorption.
Indirect evidence of chloroform absorption was obtained by
observation of damage to kidney tubules in rabbits treated with 1, 2
or 4 g chloroform/kg applied under an impermeable plastic cuff held
tightly to the belly of rabbits for 24 h (Torkelson et al., 1976).
6.1.1.3 Inhalation
Lehmann & Hasegawa (1910) exposed rabbits to chloroform vapour
concentrations of around 20, 54 or 80 g/m3. About 35% of the
inhaled dose was retained during the first hour of the exposure
period. The fraction retained declined progressively after longer
periods of exposure (5 to 10% after 4 h; 2% after 8 h). In dogs
exposed to 73.2 g chloroform/m3, a steady-state blood
concentration of 354 mg chloroform/litre was reached within 2 h (Von
Oettingen et al., 1950).
Corley et al. (1990) developed a pharmacokinetic model for
chloroform (see section 6.1.4), which was based on inhalation
studies in a closed-atmosphere chamber (concentrations of 490-24 500
mg/m3; 100-5000 ppm). Given the same chloroform concentration
(4900 mg/m3; 1000 ppm), uptake over 6 h in male B6C3F1 mice
(total body weight = 450 g) was much more rapid and complete than in
male F-344 rats (total body weight = 690 g). This difference is due
primarily to the higher rate of chloroform metabolism in mice.
6.1.2 Distribution
Cohen & Hood (1969) performed autoradiography studies in male
NMRI mice after inhalation or intravenous injection of anaesthetic
doses of chloroform and found high levels of radioactivity in fat
and liver. Following a 10-min inhalation exposure, the tissue:blood
ratios at 0, 15 and 120 min post-exposure were 1.56, 2.10 and 6.7
for the liver and 6.42, 9.25 and 7.18 for fat, respectively. The
increase in radioactivity in the liver was attributed to the
accumulation of non-volatile, ether-extractable products. Other
tissues (blood, brain, muscle, lung and kidney) contained lesser and
more uniform amounts of radioactivity. Two hours after intravenous
injection of [14C]-chloroform, non-volitive radioactivity in the
liver accounted for 2% of the total dose.
Bergman (1984) studied the distribution of [14C]-chloroform
in mice after inhalation of 5 µl of [14C]-chloroform (reported
dose: 280 mg/kg) during 10 min. Whole-body autoradiography,
immediately after exposure and 2 h thereafter, showed high
concentrations of radioactivity in fat, blood, lungs, liver,
kidneys, spinal cord and nerves, meninges and cerebellar cortex.
After heating and extraction of the sections, it appeared that
non-volatile radioactivity was bound in the bronchi, nasal mucosa,
liver, kidneys, salivary glands and in the duodenal contents. High
levels of volatile or extractable radioactivity were found in
testes, preputial gland and epididymis.
Danielsson et al. (1986) observed tissue binding in gestational
C57BL mice and their fetuses after inhalation of very low
concentrations of [14C]-chloroform for 10 min, and in 4-day-old
C57BL mice after intraperitoneal injection of 0.4 µmoles of
[14C]-chloroform, respectively. The animals were killed 0, 1, 4
and 24 h after exposure. Low temperature autoradiograms, as well as
scintillation spectrometry, showed a high uptake of radioactivity
(volatile and non-volatile) directly after inhalation, especially in
the respiratory epithelium and liver, fat, lung, brain and segments
of tubuli in the renal cortex. Tissue-bound (non-volatile)
radioactivity was found in the respiratory tract, centrilobular
regions of the liver, salivary glands, and the conjunctiva of the
eye. Volatile radioactivity was no longer present 24 h after
exposure and the non-volatile activity had decreased with time in
all organs measured. Accumulation of non-volatile metabolites was
also found in the fetal respiratory tract.
The placental transport of chloroform was first demonstrated by
Nicloux (1906) in guinea-pigs. Danielsson et al. (1986) reported
that chloroform was transported to the conceptus at all stages of
gestation in mice. Non-volatile metabolites of chloroform
accumulated in the conceptus with time, especially in the amniotic
fluid at mid-gestation. The fetal uptake of chloroform was low,
which, according to the authors, was attributable to the low fat
content in the fetus. An accumulation of non-extractable metabolites
was found in the fetal respiratory tract in late gestation.
Withey & Karpinski (1985) exposed Sprague-Dawley rats on the
17th day of pregnancy to a series of different concentrations of
chloroform (111 to 1984 ppm; 544 to 9722 mg/m3) for 5 h.
Chloroform distribution did not appear to be related to fetal
position in the uterine horn. There was a highly significant
inter-litter variation in fetal concentration, and additional tests
showed that the maternal chloroform concentration accounted for only
part of the variation. However, the fetal and maternal blood
concentrations were linear functions of the administered dose, with
a fetal/maternal ratio of 0.316.
A sex difference in tissue-bound radioactivity in mice given
[14C]-chloroform was reported by Taylor et al. (1974).
Autoradiographic studies showed that the renal cortex of male CF/LP,
CBA and C57BL mice accumulated more label than the renal cortex of
female mice of the same strains. Treatment with testosterone
resulted in an increase in tissue binding in the females and
castration reduced the binding in the males (Taylor et al., 1974).
Sex differences in renal binding were not found in the rat or monkey
(Brown et al., 1974b).
6.1.3 Elimination and fate
The results of a pharmacokinetic study in male Wistar rats
indicated that the elimination of chloroform after intravenous
administration (jugular vein) at dose levels of 3, 6, 9, 12 or 15
mg/kg body weight followed a three-compartment model. Chloroform
was eliminated at a slower rate from fat (half-life of 106 min) than
from any other tissue examined. The elimination rates from all
tissues, except fat, were similar to those derived from blood
analysis (Whithey & Collins, 1980). The elimination half-lives for
the water and vegetable oil vehicles were 46 and 39 min,
respectively.
Various studies on the elimination of chloroform have been
reported (Paul & Rubinstein, 1963; Van Dyke et al., 1964; Lavigne &
Marchand, 1974). Corley et al. (1990) exposed B6C3F1 mice and
Osborne-Mendel rats to a range of chloroform concentrations for 6 h
and measured the radioactivity in exhaled air, urine, faeces,
carcass and skin and in the cage wash (Table 6). The fraction of the
dose exhaled as unchanged chloroform increased with increasing
exposure concentration in both mice and rats. [14C]-CO2 was the
major metabolite exhaled. The data indicate partial metabolic
saturation at the higher doses studied.
Brown et al. (1974b) administered [14C]-chloroform (60 mg/kg
body weight) to mice, rats and squirrel monkeys by the oral route.
The radioactivity was measured in the exhaled air, urine, faeces and
carcasses up to 48 h after dosing. The recovery percentages (of the
dose) are listed in Table 7.
About 50% of the radioactivity in the urine of the mouse and
the rat consisted of [14C]-urea and [14C]-bicarbonate.
Auto-radiography revealed biliary excretion of radioactivity in the
monkey. A high concentration of radioactivity in the bile was
present as unchanged chloroform.
The excreted quantities of chloroform and carbon dioxide in the
rat, as reported by Brown et al. (1974b), correspond to those
reported by Reynolds et al. (1984), who found that after oral doses
of 12 or 36 mg chloroform/kg body weight to the rat, about 70% of
the dose was excreted as carbon dioxide and 12% as chloroform in the
24 h following oral administration.
6.1.4 Physiologically based pharmacokinetic modelling for
chloroform
Corley et al. (1990) developed a physiologically based
pharmacokinetic model (PBPK) for mice, rats and humans that
incorporated literature values for physiological parameters, tissue
partition coefficients and metabolic constants. The metabolic
constants were derived from results of rodent in vivo gas-uptake
studies and in vitro metabolic studies with rodent and human (n=9)
microsomes. The tissue:air partition coefficients were determined by
a vial-equilibration technique with tissue homogenates.
Macromolecular binding constants, which define the fraction of the
total metabolites that bind covalently to proteins, were estimated
Table 6. Radioactivity (mg eq/kg body weight) in B6C3F1 mice and
Osborne-Mendel rats during and up to 48 h after 6-h
exposures to [14C]-chloroform (From: Corley et al., 1990)
Concentration Exhaled Exhaled Urine Faeces Residuea
(ppm) 14C- 14C-CO2
chloroform
Mice
10 0.03 7.22 0.95 0.05 0.19
89 0.47 70.35 7.46 1.24 2.32
366 23.03 217.85 21.24 3.84 9.68
Rats
93 0.76 31.84 3.34 0.40 1.09
356 16.15 54.85 6.53 0.81 2.18
1041 78.27 89.04 11.83 1.16 3.95
a Residues comprising total 14C-label present in carcass, skin
and cage wash at the end of post-exposure collection period
Table 7. Percentage recovery of radioactivity after
[14C]-chloroform administration
(From: Brown et al., 1974b)
Species In breath In faeces In carcassa
and urine
chloroform CO2
Mouse 5.2-7.1 84-87 2.1-3.0 1.2-2.3
Rat 20 67 8 NA
Monkey 79 18 2 NA
a NA = not analysed
from in vivo binding data obtained following inhalation exposures
to radiolabelled chloroform. The model parameters that were derived
for the three species by Corley et al. (1990) are presented in Table
8.
Table 8. Parameters used in the physiologically based
pharmacokinetic model for chloroforma
Mouse Rat Human
Partition coefficients
Blood/air 21.3 20.8 7.43
Liver/air 19.1 21.1 17.0
Kidney/air 11.0 11.0 11.0
Fat/air 242 203 280
Rapidly perfused/air 19.1 21.1 17.0
Slowly perfused/air 13.0 13.9 12.0
Metabolic and macromolecular binding constants
VmaxC (mg/h per kg) 22.8 6.8 15.7
Km (mg/litre) 0.352 0.543 0.448
fMMBb (h-1), liver 0.003 0.00104 0.00202
fMMBb (h-1), kidney 0.010 0.0086 0.00931
a From: Corley et al. (1990)
b MMB = macromolecular binding of reactive metabolites;
fMMB = fraction of MMB of particular organ
The blood:air partition coefficients for rodents were
approximately three times greater than for humans. Metabolism was
described by a single saturable pathway for each species, but in
mice, equations accounting for enzyme loss had to be incorporated.
The VmaxC values reflect the greater metabolic capacity of the
mouse compared to the rat, which has been shown in numerous studies.
The model generated predictions consistent with experimental data
for target organ-specific protein binding in rodents as well as
total chloroform metabolized and total exhaled chloroform in both
rodents and humans. Predictions of protein binding suggest a
relative sensitivity ranking for the three species as follows: mouse
> rat > humans, assuming that equivalent levels of binding produce
equivalent toxicities in target tissues (Corley et al., 1990).
Blancato & Chiu (1993) used the PBPK model of Corley et al.
(1990) to predict the relative contributions of different exposure
routes to target tissue doses of chloroform in humans. Tissue
macromolecular binding was predicted as a dose surrogate. With
respect to liver dose, a 10-min shower was predicted to contribute
about 25% of the total dose, with 57% from drinking-water. Showering
was predicted to account for more than 53% of the total dose to the
kidney, while drinking-water was estimated to contribute only 7% of
the dose. This difference was attributed to the absence of a
first-pass effect with dermal absorption and inhalation exposures.
Gearhart et al. (1993) recently described an additional PBPK
model for chloroform in B6C3F1 mice. This model accounts for
decreases in body temperature associated with exposure to high
chloroform concentrations. The authors contend that the inclusion of
an enzyme loss equation for mice in the model of Corley et al.
(1990) was inappropriate and that the incorporation of temperature
corrections greatly improved the overall fit of gas uptake data. The
authors also obtained better model simulations of gas-uptake data by
including a first-order rate constant, which is consistent with in
vitro work demonstrating multiple pathways of chloroform
biotransformation (Pohl, 1979; Testai et al., 1990).
6.2 Biotransformation and covalent binding of metabolites
Chloroform may undergo both oxidative and reductive
biotransformation (Fig. 1). The oxygenation of chloroform is
catalysed by cytochrome P450 and produces trichloromethanol.
Elimination of HCl from trichloromethanol gives phosgene as a
reactive intermediate (Mansuy et al., 1977; Pohl et al., 1977).
There is considerable evidence available to support this
reaction mechanism for the formation of phosgene in the
biotransformation of chloroform: the biotransformation of chloroform
to phosgene requires NADPH and oxygen. The phosgene formed in the
biotransformation of chloroform can be trapped by reaction with
cysteine to give 2-oxothiazolidine-4-carboxylic acid, and the
biotransformation of [14C]-chloroform in the presence of cysteine
gives [14C]-2-oxothiazolidine-4-carboxylic acid. When the
biotransformation of chloroform was studied in the presence of
[18O]-dioxygen or [35S]-cysteine, [2-18O]- and
[1-35S]-2-oxothiazolidine-4-carboxylic acid, respectively, are
formed. Deutero-chloroform is biotransformed more slowly than
chloroform (Mansuy et al., 1977; Pohl et al., 1977, 1979, 1980; Pohl
& Krishna, 1978). Moreover, when [36Cl]-chloroform,
[3H]-chloroform, or [14C]-chloroform were incubated with liver
microsomes from phenobarbital-treated Sprague-Dawley rats, only
label from [14C]-chloroform became covalently bound to proteins
(Pohl et al., 1980).
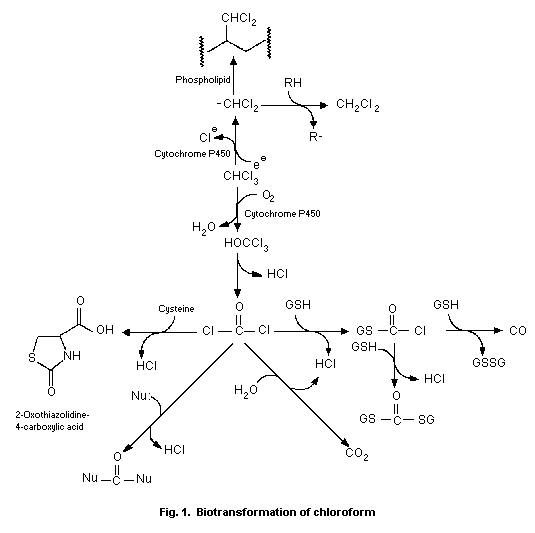 Phosgene reacts rapidly with water to give CO2 and HCl as
products, which explains the formation of CO2 as a metabolite of
chloroform (Fry et al., 1972; Brown et al., 1974b). Phosgene may
also react with tissue nucleophiles to form covalently bound
products (Uehleke & Werner, 1975). Cysteine blocks the covalent
binding of [14C]-chloroform-derived radioactivity, which supports
a role for phosgene in the formation of covalent adducts from
chloroform (Pohl et al., 1977, 1980). Alternatively, phosgene may
react with glutathione to form S-(chlorocarbonyl)glutathione; this
intermediate may react with glutathione to give diglutathionyl
dithiocarbonate (Pohl et al., 1981) or to give glutathione disulfide
and carbon monoxide as minor products (Ahmed et al., 1977).
The reductive biotransformation of chloroform is also catalysed
by cytochromes P450 (Testai & Vittozzi, 1986) (Fig. 1). Reduction of
chloroform gives rise to the dichloromethyl radical, which has been
identified by spin trapping and ESR (Tomasi et al., 1985). No
evidence for the formation of the dichloromethyl carbanion has been
presented, whereas the formation of chlorocarbene has been ruled out
(Wolf et al., 1977). The dichloromethyl radical may react
preferentially with the fatty acid skeleton of phospholipids to give
covalently bound adducts (De Biasi et al., 1992).
The balance between the oxidative and reductive
biotransformation of chloroform depends on several factors,
including oxygen and chloroform concentrations, animal species,
strain, enzyme induction, and the site of metabolism. Oxidative
metabolism is favoured at low (< 0.1 mM) chloroform concentrations
(Testai et al., 1990, 1991). Under these conditions, the oxygenation
of chloroform is catalysed by cytochrome P450 2E1 (Brady et al.,
1989; Guengerich et al., 1991), and covalent binding of chloroform
metabolites to proteins and lipids in incubation mixtures containing
mouse (B6C3F1 or C57BL/6J) liver microsomes is higher than in
incubation mixtures containing rat (Osborne-Mendel or
Sprague-Dawley) liver microsomes (Testai et al., 1991).
Chloroform reduction is increased at high substrate
concentrations (Testai et al., 1990), but oxidative metabolism is
quantitatively more important. In incubation mixtures containing 5
mM chloroform, both oxygenation and reduction of chloroform depend
on the oxygen tension in the incubation flask. Chloroform reduction
is particularly evident with microsomes from B6C3F1 mice and
Osborne-Mendel rats. At high chloroform concentrations (approx. 5
mM), the oxygenation of chloroform may be catalysed by cytochrome
P450 2B1, as suggested by the induction of the metabolism due to
pretreatment by phenobarbital (Branchflower et al., 1983; Testai &
Vittozzi, 1986; Nakajima et al., 1991). Phenobarbital or
ß-naphthoflavone pretreatment of Sprague-Dawley rats also stimulates
the formation of reduced intermediates of chloroform (Testai &
Vittozzi, 1986). Levels of the in vitro covalent binding of
[14C]-chloroform metabolites to proteins were higher with hepatic
microsomes from rabbits and human biopsies than with hepatic
microsomes from rats or mice (Uehleke & Werner, 1975).
The in vitro formation of dichloromethane as a stable
end-product of chloroform metabolism was addressed in early studies.
Dichloromethane was detected in mouse liver slices incubated with
chloroform (Butler, 1961), but not in slices or subcellular
fractions of rat liver incubated with chloroform (Paul & Rubinstein,
1963; Rubinstein & Kanics, 1964). These discrepancies, however, may
have been due to the incubation conditions employed in these early
studies. in vivo results with rats, dogs, mice and human
volunteers exposed to chloroform consistently indicated no
expiration of dichloromethane (Butler, 1961; Paul & Rubinstein,
1963; Fry et al., 1972; Brown et al., 1974b).
Interspecies differences in the oxidative metabolism of
chloroform have been found in vivo. After a [14C]-chloroform
dose of 60 mg/kg body weight, 85%, 66% and 18% was excreted as
[14C]-CO2 in C57BL, CF/PL and CBA mice, Sprague-Dawley rats, and
squirrel monkeys, respectively. Expiration of 14C accounted for
the elimination of most of the remaining dose (recoveries of 93-98%)
(Brown et al., 1974b). Mink et al. (1986) and Corley et al. (1990)
also showed that chloroform is metabolized in the mouse to a greater
extent than in the rat. Corley et al. (1990) demonstrated that the
covalent binding of [14C]-chloroform metabolites to liver and
kidney proteins in vivo was higher in B6C3F1 mice than in
Osborne-Mendel rats.
In several strains of mice given [14C]-chloroform, more
binding occurred in the kidney tissue of males than in that of
females (Ilett et al., 1973; Taylor et al., 1974). Male DBA mice
accumulate twice as much radioactivity in their kidneys as do male
C57BL mice. This strain difference shows intermediate or
multifactorial heredity (Hill et al., 1975).
Differences in binding were associated with variations in
toxicity (Hill et al., 1975; Clemens et al., 1979). The
nephrotoxicity of chloroform in male mice of susceptible strains
(see chapter 7) is most probably related to in situ renal
metabolic activation of chloroform (Zaleska-Rutczynska & Krus, 1973;
Hill, 1978; Clemens et al., 1979; Smith & Hook, 1983; Smith et al.,
1984). Indeed the overall biotransformation of chloroform in both
sexes is equal, whereas males exhibit more extensive formation of
renal tissue-bound metabolites than females (Taylor et al., 1974;
Smith & Hook, 1984). Smith et al. (1985) observed little chloroform
metabolism in rat (male, Fischer-344) renal cortical microsomes.
Additional studies, however, have demonstrated chloroform-induced
cytolethality and regenerative cell damage in male, Fischer-344 rat
kidney (Larson et al., 1993). Culliford & Hewitt (1957) reported
that females became more susceptible after pretreatment with
androgens, and the sensitivity of the males was reduced after
castration.
In the rat and mouse, chloroform biotransformation occurs
mainly in the liver, but other tissues also show metabolic activity.
After oral administration of chloroform to mice, maximum covalent
binding in the liver was observed after 3 h, whereas in the kidney,
maximum binding was found after 6 to 12 h. Binding appears to be
dose dependent up to doses of 3 mmol/kg body weight. At higher
doses, a plateau is reached (Ilett et al., 1973). Löfberg & Tjälve
(1986) studied the extra-hepatic metabolism of [14C]-chloroform in
Sprague-Dawley rats. Autoradiography was used to localize
metabolites in freeze-dried, extracted tissues to distinguish
between total and bound radioactivity. in vitro autoradiography,
in which tissue slices were incubated with [14C]-chloroform and
then examined autoradiographically, showed the capacity of several
tissues to metabolize [14C]-chloroform: liver, kidney cortex,
mucosa of the bronchial tree, tracheal mucosa, olfactory and
respiratory nasal mucosa, Bowman's glands in the olfactory lamina
propria mucosae, Steno's gland (the lateral nasal gland), mucosa of
the oesophagus, larynx, tongue, gingiva, cheek, naso-pharyngeal
duct, pharynx and the soft palate. Furthermore, autoradiographic
studies showed that a correlation exists between the ability of the
tissues to retain metabolites in vivo and the ability of these
tissues to metabolize chloroform in vitro.
The distribution of the covalent binding of 14C to DNA or RNA
after an intraperitoneal injection of [14C]-chloroform to Balb/c
mice or to Wistar rats shows several differences from the
distribution of the covalent binding to tissue proteins (Colacci et
al., 1991). The highest levels of covalent binding to DNA were
observed in mouse kidney (3.17 nmol/g DNA) and lung (3.65 nmol/g
DNA). In mice, binding to liver DNA (0.83 nmol/g DNA) was lower than
binding to stomach DNA (1.52 nmol/g DNA).
Differences in DNA binding of chloroform metabolites among rat
organs have been found to be limited, and the absolute values were
lower than those seen in mouse organs. In mice, RNA binding levels
were high in both the liver and kidney; in rats, they were higher in
the kidney than in other organs. In mice, protein binding was
highest in the liver (47.5 nmol/g), whereas in rats it was high in
both the liver (27.4 nmol/g) and kidney (30.7 nmol/g). In
incubations containing low (< 0.1 mM) chloroform concentrations and
human liver microsomes, little formation of reactive metabolites was
seen (Vittozzi et al., 1991). Detectable covalent binding was
observed in microsomes from some samples of colonic and ileal mucosa
of human patients but not of male Sprague-Dawley rats (Testai et
al., 1991).
Glutathione (GSH) is an important factor controlling the
binding of chloroform metabolites to proteins and lipids. In in
vitro studies, physiological concentrations of GSH (2-5 mM)
strongly reduced the covalent binding of chloroform metabolites to
proteins (Sipes et al., 1977; Cresteil et al., 1979; Smith & Hook,
1984). In later studies, 3 mM GSH blocked the covalent binding of
chloroform metabolites to proteins and to phospholipid polar heads,
whereas covalent binding to the phospholipid fatty acyl chains (due
to the radical metabolite) was only slightly affected (Testai &
Vittozzi, 1986; Testai et al., 1990, 1991; De Biasi et al., 1992).
Pretreatment of rats with diethylmaleate or buthionine sulfoximine
(BSO) increased the binding of administered [14C]-chloroform to
proteins (Stevens & Anders, 1981). Pretreatment of
phenobarbital-induced Sprague-Dawley rats with cysteine decreased
the covalent binding (Stevens & Anders, 1981b). Toxic effects
paralleled the covalent binding levels after these pretreatments
(Stevens & Anders, 1981a).
6.3 Human studies
6.3.1 Uptake
6.3.1.1 Oral
When Fry et al. (1972) dosed eight volunteers with 0.5 g
chloroform in olive oil in capsules, approximately 50% of the oral
dose was metabolized to carbon dioxide. Maximal blood levels of 1 to
5 ng chloroform/litre were achieved after 1.5 h. In two of the
subjects, the decline in blood levels could be described by a
two-compartment model with a half-life of 13 min for the initial
phase and a half-life of 90 min for the second phase. Chiou (1975)
reanalysed the data obtained from the two subjects mentioned above
and calculated an apparent volume of distribution of approximately
160 litres. The author estimated the hepatic first-pass effect to be
about 32% and the pulmonary first-pass effect to be 16%. Hence after
a single oral dose of 0.5 g chloroform, about 52% of the dose may be
available to the system. Pulmonary and metabolic clearances of 0.7
and 0.6 litre/min, respectively, gave a total body clearance of 1.3
litre/min.
6.3.1.2 Dermal
Jo et al. (1990a) studied the relative contributions of dermal
and pulmonary uptake of chloroform in individuals taking showers.
Post-exposure exhaled air concentrations of chloroform were measured
to estimate chloroform uptake and were 6 to 21 µg/m3 for normal
showers and 2.4 to 10 µg/m3 for inhalation-only exposure. The
difference between normal and inhalation-only exposure was
significant, and the authors concluded that the contribution of
dermal exposure was approximately equivalent to inhalation exposure.
Chinery & Gleason (1993) modified an existing PBPK model to
predict the exhaled air concentration of chloroform in individuals
exposed to the chemical while showering. Calibration of the model
with measured exhaled air concentrations of chloroform in
individuals exposed while showering either with or without dermal
absorption generated an expected value for skin-blood partitioning
of 1.2. This assumes a degree of transfer of chloroform from shower
water into shower air of 61%. The stratum corneum permeability
coefficient for chloroform was estimated to be within a range of
0.16-0.36 cm/h, and the expected value was 0.2 cm/h. The estimated
ratio of dermally:inhaled absorbed doses while showering ranged
between 0.6 and 2.2 and the expected value was 0.75.
6.3.1.3 Inhalation
The inhalation uptake of chloroform in humans was studied by
Lehmann & Hasegawa in 1910. More recently, Morgan et al. (1970)
measured the absorption of chloroform after a single inhalation
exposure to approximately 5 mg of [38Cl]-chloroform. About 80% of
the chloroform was absorbed under these conditions.
Prolonged inhalation of anaesthetic concentrations (about 50 g
chloroform/m3 air) gave rise to blood chloroform concentrations of
about 100 mg/litre (Smith et al., 1973).
The relative contribution of inhalation to chloroform uptake
during showering has been determined (see section 6.3.1.2, Jo et
al., 1990a).
6.3.2 Distribution
Corley et al. (1990) determined partition coefficients for
human tissues (see Table 8).
McConnell et al. (1975) analysed chloroform levels in
postmortem tissue from eight persons (four males and four females,
48 to 82 years old) living in non-industrial areas of the United
Kingdom. The chloroform levels (µg/kg wet tissue weight) observed
were: body fat, 5-68 (mean = 51); liver, 1-10 (mean = 7.2); kidney,
2-5; and brain, 2-4.
Phillips & Birchard (1991) reported on a nationwide survey of
the general population by the US EPA's National Human Adipose Tissue
Survey. Several hundred fat samples were pooled into 46 composite
samples by age and geographic region and were analysed. Chloroform
was detected at levels ranging from 5 to 580 ng/g in 29 of the
composite samples.
Dowty et al. (1976) detected chloroform in human maternal and
placental cord blood. Erickson et al. (1981) found chloroform,
supposedly originating from environmental exposure, present in
mother's milk (concentration not specified). Chloroform was
identified, but not quantified, in mother's milk samples collected
from 49 lactating women living in the vicinity of chemical
manufacturing plants or industrial user facilities in Pennsylvania,
New Jersey and Louisiana, USA (Pellizzari et al., 1982).
6.3.3 Elimination
Human volunteers given oral doses of 500 mg chloroform
eliminated on average 50% of the dose as CO2 and 40% as unchanged
chloroform during 8 h after dosing (Fry et al., 1972). The amount of
expired chloroform varied from 18 to 66%, depending on the obesity
of individuals.
After administration of 100, 250 or 1000 mg, the authors
recovered 0, 12 and 65% of the dose in the expired air,
respectively. After administration of [14C]-chloroform to a man
and a woman, approximately 50% of the dose was found in the exhaled
air (as CO2) in 7.5 h after dosing. Virtually no chloroform was
excreted by the kidneys (Fry et al., 1972).
After inhalation of chloroform (concentrations of 21 or 35 g
chloroform/m3), Lehmann & Hasegawa (1910) found little pulmonary
excretion, i.e. approximately 2% of the absorbed quantity within 30
min after the exposure. A pulmonary excretion of 10% of the body
content during the first hour after exposure was reported by Morgan
et al. (1970).
6.3.4 Biotransformation
Human cytochrome P450 2E1 catalyses the oxygenation of
chloroform (Guengerich et al., 1991).
Corley et al. (1990) quantified CO2 production from
incubations of human liver microsomes with 0.049-0.058 mM
chloroform. The average activity of samples from nine individuals
was 8.15 ± 0.02 pmol chloroform oxidized/min per mg protein. These
data were correlated with rodent in vitro and in vivo conversion
rates to estimate human in vivo metabolic rate constants (see
Table 8).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Lethality
The LD50 values of chloroform for mice and rats are given in
Tables 9 and 10, respectively. Chloroform-induced death is usually
due to liver damage, with the exception of male mice of very
sensitive strains, whose death is caused by kidney damage. The
higher susceptibility to chloroform acute toxicity in these strains
of mice (such as DBA, C3H, C3Hf, CBA, Balb/c, C3H/He), with respect
to other strains, is genetically controlled. An absolute sex-related
difference with respect to kidney damage, but not to liver damage,
has been described in mice: female mice do not develop renal
lesions. This is independent of the strain. Some influence of age on
chloroform acute toxicity in rats has also been described (Kimura et
al., 1971).
For the rat, the LD50 values ranged from 450 to 2000 mg
chloroform/kg body weight, and in this species no sex difference in
susceptibility was found (Kimura et al., 1971; Chu et al., 1980).
For OF1 female mice, a LC50 value of 6150 mg chloroform/m3 (6
h exposure) was reported by Gradiski et al. (1978).
A dose of 3070 mg chloroform/kg body weight in mineral oil to
rats resulted in death due to CNS depression within minutes, and a
dose of 980 mg chloroform/kg body weight resulted in hepatic
centrilobular necrosis (Reynolds & Yee, 1967). When administered to
newborn rats, chloroform was lethal at oral doses of 1500 mg/kg body
weight; smaller doses were not administered (Kimura et al., 1971).
7.1.2 Non-lethal effects
7.1.2.1 Oral exposure
Chloroform is a potent anaesthetic. Anaesthesia may result from
oral administration of chloroform; this was established by Bowman et
al. (1978) in the ICR mouse with a dose of 500 mg chloroform/kg body
weight in aqueous emulsion. The ED50 (50% of animals showing
effect at this dose level) in mice for acute neurological effects
(ataxia, incoordination and anaesthesia) was 484 mg chloroform/kg
body weight (Balster & Borzelleca, 1982).
After oral administration of chloroform in olive oil to Swiss
mice (both sexes), Jones et al. (1958) found the median narcotic
dose to be 350 mg/kg body weight and the median hepatotoxic dose to
be 35 mg/kg body weight. At this dose level, the liver showed
centrilobular fatty infiltration and at 350 mg/kg body weight
centrilobular necrosis was found.
Table 9. Representative LD50 values (mg chloroform/kg body weight) for mice
Sex/strain Route Vehicle Observed LD50 Reference
period
Male
C3H/tif oral sesame oil 15 days 36 Pericin & Thomann (1979)
DBA/2/j oral sesame oil 15 days 101 Pericin & Thomann (1979)
Tif:MAGf oral sesame oil 15 days 213 Pericin & Thomann (1979)
A/J oral sesame oil 15 days 253 Pericin & Thomann (1979)
Tif:MF2f oral sesame oil 15 days 336 Pericin & Thomann (1979)
C57BL/6J oral sesame oil 15 days 460 Pericin & Thomann (1979)
Princeton subcutaneous peanut oil 10 days 696 Plaa et al. (1958)
Swiss albino subcutaneous olive oil 10 days 3245 Kutob & Plaa (1962a)
Female
C3H/tif oral sesame oil 15 days 353 Pericin & Thomann (1979)
DBA/2/j oral sesame oil 15 days 679 Pericin & Thomann (1979)
A/J oral sesame oil 15 days 774 Pericin & Thomann (1979)
C57BL/6J oral sesame oil 15 days 820 Pericin & Thomann (1979)
Tif:MF2f oral sesame oil 15 days 1126 Pericin & Thomann (1979)
Tif:MAGf oral sesame oil 15 days 1366 Pericin & Thomann (1979)
OF1 intraperitoneal olive oil 14 days 880 Gradiski et al. (1974)
Table 10. Representative LD50 values (mg chloroform/kg body weight) for rats
Sex/strain Route Vehicle Observed LD50 Reference
period
Male
Sprague-Dawley oral none unknown 450 Kimura et al. (1971)
(14 days old)
unknown oral none unknown 1200 Kimura et al. (1971)
(older adults)
Sprague-Dawaley oral none 14 days 908 Chu et al. (1980)
Sprague-Dawley oral none 14 days 2000 Torkelson et al. (1976)
Female
Sprague-Dawley oral none unknown 450 Kimura et al. (1971)
(14 days old)
Sprague-Dawley oral none 14 days 1117 Chu et al. (1980)
Sprague-Dawley intraperitoneal peanut oil 24 h 1379 Lundberg et al. (1986)
14 days 894
Hill (1978) investigated strain and sex differences in
chloroform-induced toxicity in mice. Male mice of three strains
(DBA/2J, B6D2F1/J, and C57BL/6J) were given single oral doses of
chloroform in oil. No clear difference in hepatotoxicity between
strains was observed; centrilobular necrosis occurred at doses
greater than 250 mg/kg body weight in all three strains. In
contrast, there were differences between species in renal toxicity.
Doses of 89 mg/kg body weight caused glucosuria and/or proteinuria
in half of the DBA/2J animals, while doses of 119 and 163 mg/kg body
weight were required to produce these effects in half the B6D2F1/J
and C57BL/6J mice, respectively.
In male CFLP Swiss mice, Moore et al. (1982) found neither
histological changes in the liver or kidney nor biochemical changes
in plasma 4 days after oral administration of 17 mg chloroform/kg
body weight in corn oil. Administration of 66 mg chloroform/kg body
weight caused slight hepatotoxicity and a more severe
nephrotoxicity.
Chu et al. (1980, 1982a) observed piloerection, sedation,
flaccid muscle tone, ataxia, prostration and dacryorrhoea after
administration of chloroform to rats. Food intake in the males was
reduced. Histological and biochemical examination revealed effects
on liver, kidneys and red and white blood cells. Upon histological
examination, no lesions were found in other tissues with chloroform
doses up to 2100 mg/kg body weight. In this study the lowest
administered dose was 546 mg chloroform/kg body weight, a level at
which toxic effects were still found.
Reitz et al. (1982) determined the cellular regeneration (as
3H-thymidine uptake in DNA) 48 h after administration of
chloroform to male B6C3F1 mice and male Osborne-Mendel rats. In
the mice, 3H-thymidine uptake was significantly increased in
kidneys at a dose level of 60 mg chloroform/kg body weight and in
kidneys and liver at 240 mg chloroform/kg body weight. In the rats,
only a slight increase in 3H-thymidine uptake in liver and kidneys
was found at a dose level of 180 mg chloroform/kg body weight.
Torkelson et al. (1976) reported dose-related liver and kidney
changes in adult rats at dose levels as low as 250 mg chloroform/kg
body weight. Tyson et al. (1983) found an elevation of serum
aminotransferase levels in rats at dose levels above 200 mg/kg body
weight in oil.
One study examined the organ-specific toxicity of acute doses
of chloroform (Larson et al., 1993). Male F-344 rats were given
chloroform by gavage in corn oil at doses of 0, 34, 180 or 477 mg/kg
body weight and necropsied 24 h later. Additional rats were given a
single dose of 180 mg chloroform/kg and administered
bromodeoxyuridine (BrdU) 2 h prior to necropsy at 0.5, 1, 2, 4, and
8 days after chloroform treatment to label cells in S-phase. The
kidneys of male rats administered 34, 180 and 477 mg chloroform/kg
exhibited mild to severe proximal tubular necrosis in a
dose-dependent manner. A 20-fold increase in the labelling index
(LI, % of nuclei in S-phase) in the proximal tubule cells was
observed 2 days after treatment at a dose of 180 mg/kg body weight.
The livers of the male rats exhibited only slight to moderate
multifocal centrilobular necrosis at 180 and 477 mg/kg body weight.
A 10-fold increase in the LI was observed in the liver of male rats
given 477 mg/kg body weight, but no increase was observed at 180
mg/kg body weight (Larson et al., 1993).
Female B6C3F1 mice were given chloroform by gavage (0, 34,
238 or 477 mg/kg body weight) and necropsied 24 h after treatment.
Additional mice were given a single dose of 350 mg chloroform/kg
body weight, labelled with BrdU, and necropsied 0.5, 1, 2, 4, and 8
days after treatment. Female mice developed a dose-dependent
centrilobular hepatic necrosis at 238 and 477 mg/kg body weight. No
renal lesions were observed in female mice at any dose. A peak
increase in LI of 38-fold was observed in hepatocytes in the livers
of female mice 2 days after treatment with 350 mg chloroform/kg, but
the increase in LI observed in the kidneys was only 2-fold (Larson
et al., 1993). These data indicate that acute chloroform-induced
cytolethality leads to increased cell proliferation and that the
organ-specific pattern of toxicity is the same as the organ-specific
pattern of tumour formation (see NCI, 1976a,b, and section 7.7.1).
Groger & Grey (1979) intubated Colworth Wistar rats (6 of each
sex per group) daily with chloroform in peanut oil (0 to 50 mg/kg
body weight) for periods of 1, 5 or 10 days. There were changes in
the activity of several liver enzymes, the toxicological
significance of which is unclear.
Balster & Borzelleca (1982) administered chloroform in water to
male ICR mice (8-12/group) and examined their performances in a
battery of neurobehavioural tests (several exposure periods and
several dose levels). The only effect observed was a reduced
achievement in an operant behaviour test after dosing with 100 and
400 mg chloroform/kg body weight in water for 60 days. At the
chloroform level of 400 mg/kg body weight, about half the treated
animals died. No adverse effects on behaviour were observed after 90
days of dosing with 31 mg chloroform/kg body weight in water.
7.1.2.2 Subcutaneous and intraperitoneal exposure
A sex difference in toxicity was found after subcutaneous and
intraperitoneal administration of chloroform to mice. In males, the
kidney appeared to be more susceptible than in females, in which the
liver was found to be the target organ. Smith et al. (1983) exposed
male and female mice of the ICR strain to chloroform doses of 75 to
1500 mg/kg body weight (subcutaneous and intraperitoneal).
Hepatotoxicity was dose-related in both sexes from 375 mg
chloroform/kg body weight upwards. After the subcutaneous
administration of 375 mg chloroform/kg body weight, an increase in
the serum alanine aminotransferase (ALAT) and a decrease in the
liver non-protein sulfhydryl groups (NPSH) were observed.
Histological examinations showed centrilobular swelling of the liver
and necrosis of the hepatocytes in both sexes. At 24 h after
intraperitoneal exposure to 375 mg chloroform/kg body weight, renal
toxicity was observed in males but not in females. A decrease in the
renal NPSH concentration of about 60% in males and 20% in females
was found. The concentration in females, but not in males, returned
to normal within 24 h post-dosing. Histological examination of male
kidneys showed proximal tubular lesions with pyknotic nuclei and
loss of reticular cytoplasmic structure, necrosis of the cells of
the proximal tubuli and occlusion of the tubular lumens with hyaline
casts (Smith et al., 1983).
Skrzypinska et al. (1991) administered chloroform
intraperitoneally to Balb/c mice as a single dose ranging from 12.5
to 100% of the approximate lethal dose. At different time periods
after administration, mice were sacrificed. Serum glutamine-pyruvate
transaminase (SGPT) and sorbitol dehydrogenase (SDH), as well as
glutathione (GSH) and malondialdehyde (MDA) levels in the liver,
were determined. Increased SGPT and SDH levels were found for all
doses exceeding one eighth of the approximate lethal dose. The
depletion of GSH level was kept within 40% for all doses. A 2- to
4-fold increase of hepatic MDA level was found. The depletion of
hepatic GSH, and to some extent the increase of SGPT and SDH,
occurred in a biphasic fashion. Dose-effect functions for these
biochemical alterations could only be constructed for the second
delayed phase of action. It is postulated that the hepatotoxicity of
chloroform is mainly dependent on radical formation in the course of
biotransformation.
Plaa & Larson (1965) observed renal toxicity after an
intraperitoneal dose as low as 48 mg chloroform/kg body weight in
male Swiss mice. The authors reported that chloroform was the most
potent nephrotoxic agent of 14 short-chain chlorinated hydrocarbons
in male mice.
Ahmadizadeh et al. (1984) found an increase in the relative
kidney weight after intraperitoneal administration of chloroform in
peanut oil (150 mg/kg body weight) to male DBA mice, but not after
chloroform administration to DBA female mice or to male or female
mice of the C57BL strain.
Hepatic toxicity, which is the predominant effect in most
species, was found after a parenteral dose of 450 and 150 mg
chloroform/kg body weight in the rat and the guinea-pig,
respectively (Klaassen & Plaa, 1969; Divincenzo & Krasavage, 1974).
Detection of lipoperoxidation in the liver of PB-induced rats
exposed to chloroform has been reported by several authors (Klaassen
& Plaa, 1969; Brown, 1972; Brown et al., 1974a; Masuda et al.,
1980).
An increased bile duct/pancreatic fluid flow and a changed
composition of this fluid were observed after an intraperitoneal
dose of 1500 mg chloroform/kg body weight to rats (Harms et al.,
1976; Hamada & Peterson, 1977).
A single, liver-damaging intraperitoneal dose of chloroform led
to a maximal glutathione depletion in the liver of PB-pretreated
rats shortly after dosing (1-2 h) but not in saline-treated rats.
However, the maximal histopathological findings (centrilobular
necrosis) occurred much later (after about 24 h) (Docks & Krishna,
1976).
In dogs, liver toxicity has been found after intraperitoneal
administration of chloroform. The ED50 for an increased serum ALAT
activity via this route appears to be 300 mg chloroform/kg body
weight. At near-ED50 doses, chloroform caused centrilobular
vacuolization and centrilobular and subcapsular necrosis. The ED50
for renal dysfunction in the dog appears to be 645 mg chloroform/kg
body weight (Klaassen & Plaa, 1967).
Bai et al. (1992) evaluated the suitability of nine different
serum bile acids (SBA) as markers of chloroform exposure in rats.
Increases in specific SBA levels were observed following three daily
intraperitoneal administrations of chloroform at doses as low as 0.1
mmol/kg body weight. The effects on SBA levels were detectable at
much lower doses than were effects on histopathological indices or
on levels of alanine aminotransferase, aspartatetransaminase,
alkaline phosphatase, bilirubin or total bile acid.
Chloroform doses as low as 45 mg/kg body weight reduced the
microsomal Ca++/Mg++-ATP-ase activity (liver microsome calcium
pump) in rats (Moore, 1980).
7.1.2.3 Inhalation exposure
After exposure to chloroform vapour, the same pattern of
toxicity in mice was observed as after oral, intraperitoneal or
subcutaneous administration (Deringer et al., 1953; Hewitt, 1956).
Deringer et al. (1953) found necrosis in the proximal and distal
convoluted tubules, hyaline casts in the convoluted tubules and
collecting ducts, calcification of the cortex, and death after
exposure of male C3H mice to chloroform concentrations of 3400 to
5400 mg/m3 for 1 to 3 h; anaesthesia was not observed.
Kylin et al. (1963) exposed female albino mice of an undefined
strain to chloroform vapour for 4 h and reported hepatotoxic
effects. At 24 h after exposure to chloroform concentrations of 490
mg/m3 or more, a concentration-related fatty infiltration was
observed. From 980 mg chloroform/m3 upwards, necrosis of liver
cells and a rise in the serum ornithine carbamoyltransferase level
were seen. In mice, rabbits, guinea-pigs and cats, anaesthesia was
induced by exposure to chloroform concentrations in the range of 10
to 100 g/m3 for periods of 30 min to a few hours. In rabbits and
guinea-pigs such exposures can cause death (review by Lehmann &
Flury, 1943).
As with other anaesthetics, prolonged anaesthesia with
chloroform may result in respiratory depression, cardiac arrhythmia
and finally in cardiac arrest. Heart failure is probably due to
increased sensitivity of the heart muscle to adrenaline (Von
Oettingen et al., 1950; Von Oettingen, 1964). Exposure of rabbits to
224 mg/m3 for 1 min led to decreased diastolic pressure, reduction
of the stroke volume, blood pressure and cardiac output, and an
increase in the peripheral vascular resistance. The cardiac effects
were probably not due to respiratory effects, as blood oxygen and
carbon dioxide tension and pH were not significantly changed (Taylor
et al., 1976).
Exposure of rats to a chloroform concentration of 49 g/m3 for
5 h resulted in respiratory acidosis. Liver cells showed swollen
rough endoplasmic reticulum with a loss of ribosomes, mitochondrial
lesions, and cistern-like dilatation of tubular areas of the smooth
endoplasmic reticulum. An accumulation of fat droplets and reduced
amino acid incorporation into protein were also found in liver cells
(Scholler, 1966, 1967).
Brondeau et al. (1983) found increased serum activities of
glutamate dehydrogenase and sorbitol dehydrogenase after a single 4
h exposure of male rats to a chloroform concentration of 1410
mg/m3. The effects were dose-related and at the highest
concentrations tested (4600 and 5250 mg/m3) serum aspartate
aminotransferase levels (ASAT) were also increased.
7.1.2.4 Dermal exposure
Single application of 1 or 4 g chloroform/kg body weight for 24
h to the belly of rabbits, under an impermeable plastic cuff,
resulted in extensive necrosis and weight loss at both levels. The
kidneys of all animals showed dose-related degenerative changes in
the tubules. Livers were not grossly affected (see also section 7.4)
(Torkelson et al., 1976).
7.2 Short-term exposure
7.2.1 Oral exposure
7.2.1.1 Mice
Condie et al. (1983) dosed male CD1 mice daily with 0, 37, 74
and 148 mg chloroform/kg body weight in corn oil for 14 days.
Histological changes turned out to be the most sensitive indicators
of liver and kidney toxicity. Dose-related effects were observed at
dose levels from 37 mg/kg body weight upwards. Kidneys showed
intra-tubular mineralization, epithelial hyperplasia and cytomegaly.
Livers showed centrilobular cytoplasmic pallor, marked cell
proliferation and focal inflammation. After 14 days the body weight
in the highest dose group was reduced.
Female and male CD1 mice (7-12 animals of each sex per group)
were administered daily 0, 50, 125 and 250 mg/kg body weight in
water by gavage for 14 and 90 days (Munson et al., 1982). Many
histological and biochemical parameters were examined. After 14
days, the most important effects were a dose-related decrease in the
number of antibody-forming cells (as IgM response to sheep red blood
cells) in both sexes (> 50 mg/kg body weight) and an increase in
the liver weight of males at doses > 125 mg/kg body weight and of
females at the highest dose level. The serum ASAT level was
increased in males and females at the highest dose level and serum
ALAT was increased in females at the highest dose level. After 90
days, a depression in the number of antibody-forming cells was found
at the highest dose level in both sexes. In females at the highest
dose level, a decrease in cell-mediated type hypersensitivity was
observed. Liver weight was increased after 90 days of exposure to
doses > 50 mg chloroform per kg body weight in the females and at
250 mg chloroform/kg body weight in the males. After 90 days of
exposure, the animals showed a tolerance against a challenging dose
of 1000 mg chloroform/kg body weight. The kidneys and livers of all
dosed animals showed histological changes. In the kidneys these
changes included small intertubular collections of chronic
inflammatory cells, whereas in the liver they included generalized
hydropic degeneration of hepatocytes and occasional small focal
collections of lymphocytes. In females, small amounts of
extravasated bile were occasionally noted in the sinusoidal Kupffer
cells.
Jorgenson & Rushbrook (1980) administered chloroform to female
B6C3F1 mice for 90 days in the drinking-water at concentrations of
0, 200, 400, 600, 900, 1800 and 2700 mg/litre (measured daily
chloroform doses of 0, 34, 66, 92, 132, 263 and 400 mg/kg body
weight, respectively). In the first week of the experiment some mice
in the higher dose groups died of dehydration due to reduced
drinking. Depression of the central nervous system occurred in the
animals receiving chloroform and was concentration-related. The only
treatment-related histopathological findings consisted of a mild
adaptive and transitory fatty change in the livers of animals dosed
with 66 mg chloroform/kg body weight or more and a mild lymphoid
atrophy of the spleen at the higher dose levels.
There is evidence that the vehicle in which chloroform is
administered significantly affects its toxicity. Bull et al. (1986)
found that chloroform administered by gavage in corn oil was
significantly more hepatotoxic than equivalent doses administered in
an aqueous emulsion (2% Emulphor(R), polyoxyethylated vegetable
oil, GAF Corp.). Doses of 0, 130 and 270 mg/kg were administered to
male and female B6C3F1 mice for 90 days. Liver body weight ratios
were significantly higher in all dose groups and in both sexes when
chloroform was administered in corn oil. The SGPT level was
significantly elevated in both sexes at the high dose level of
chloroform administered in corn oil, but not in those treated with
the same dose in Emulphor. Mice treated at all levels of chloroform
in corn oil showed evidence of extensive vacuolation and those
treated with the high dose in corn oil showed extensive disruption
of hepatic architecture including cirrhosis. No such pathological
changes were observed in any of the animals treated with chloroform
in 2% Emulphor.
One study contrasted the toxic responses of chloroform
administered by gavage in corn oil or given ad libitum in the
drinking-water (Larson et al., 1994a). Female B6C3F1 mice were
administered oral doses (0, 3, 10, 34, 90, 238, or 477 mg/kg per
day) of chloroform dissolved in corn oil for 4 days or for 5
days/week for 3 weeks, or were continually exposed to chloroform in
the drinking-water at concentrations of 0, 60, 200, 400, 900 or 1800
mg/litre for 4 days or 3 weeks, at which time they were necropsied.
BrdU was delivered via osmotic pumps implanted 3.5 days prior to
necropsy. Cell proliferation was evaluated as a BrdU labelling index
(LI) in histological tissue sections. Dose-dependent changes
included centrilobular necrosis and markedly elevated LI in mice
given chloroform in corn oil at 238 or 477 mg/kg, the average daily
doses that produced tumours in the gavage cancer bioassay (NCI,
1976a,b). The no-observed-effect level (NOEL) for histopathological
changes was 10 mg/kg body weight per day and for induced cell
proliferation 34 mg/kg body weight per day. Chloroform given in the
drinking-water did not increase the hepatic LI after either 4 days
or 3 weeks in any of the dose groups, nor were any microscopic
alterations observed in the liver, even though the cumulative daily
amount of chloroform ingested in the 1800-mg/litre exposure group
was 329 mg/kg body weight per day (Larson et al., 1994a). Thus, the
authors concluded that liver detoxification mechanisms are
overwhelmed when chloroform is given as a single bolus dose, but the
liver can detoxify the same daily dose if it is given in small
amounts resulting from sips of water throughout the day. The authors
also concluded that the sustained increase in LI in the livers of
mice administered hepatocarcinogenic doses of chloroform in corn
oil, but not in the case of chloroform in drinking-water, supports
the hypothesis that chloroform-induced mouse liver cancer is
secondary to events associated with induced cytolethality and cell
proliferation (see also NCI, 1976a,b and section 7.7).
7.2.1.2 Rats
Chu et al. (1982a) exposed male weanling Sprague-Dawley rats to
chloroform via drinking-water for 28 days. The following chloroform
exposure doses were calculated: 0, 0.13, 1.3 and 11 mg/rat per day
(0, 0.7, 7.4 and 63 mg/kg body weight, respectively). The only
treatment-related effect observed was a decrease in the neutrophils
in the 11-mg group. In a 90-day study by Chu et al. (1982b) male and
female Sprague-Dawley rats were exposed to chloroform via
drinking-water at dose levels of 0, 0.17, 1.3, 12 and 40 mg/rat per
day for males and 0, 0.12, 1.3, 9.5 and 29 mg/rat per day for
females; this was followed by 90 days of recovery. Water and food
intake were reduced in the highest dose group. At the 40-mg level a
higher incidence of spontaneous death occurred. Histological
examination showed mild liver and thyroid lesions, especially in the
highest dose group. Livers of both males and females showed: an
increase in cytoplasmic homogeneity; density of the hepatocytes in
the periportal area; mid-zonal and centrilobular increase in
cytoplasmic volume; vacuolation due to fatty infiltration and
occasional nucleic vesiculation; and hyperplasia of biliary
epithelial cells. Thyroid lesions consisted of a reduction in
follicular size and colloid density, increase in epithelial cell
height and occasional collapse of follicles. Liver and thyroid
lesions diminished in severity during the 90 days recovery period.
Jorgenson & Rushbrook (1980) administered chloroform in the
drinking-water to male Osborne-Mendel rats for 90 days at
concentrations of 0, 200, 400, 600, 900 and 1800 mg/litre
(calculated to be 0, 20, 38, 57, 81 and 160 mg chloroform/kg body
weight, respectively). A concentration-related central nervous
system depression was seen. Body weights in the 160-mg group were
reduced throughout the study. Biochemical investigations of serum
showed no important deviations from control values other than a
dose-related increase in cholesterol at dose levels of 38 mg
chloroform/kg body weight or more after 60 days and a decrease in
triglycerides in the highest dose group from 30 days onwards. After
90 days of administration, however, these parameters were affected
in the two highest dose groups only. No dose-related
histopathological changes were reported.
7.2.2 Inhalation exposure
The severity of liver injury due to inhaled chloroform is not
only influenced by the administered concentration but also by the
shape of the exposure profile. This was observed by Plummer et al.
(1990), who exposed male black-hooded Wistar rats (36 per group) for
4 weeks to chloroform vapour as a constant concentration (245
mg/m3; 24 h/day; 7 days a week) or as repeated concentrations
(1387 mg/m3; 6 h/day; 5 days a week), with a similar total
exposure (154 g/m3-hours) for the two ways of exposure (levels
were monitored). Hepatic injury appeared to be more severe in the
continuously exposed group, in which microvesicular fatty change was
the most prominent feature, while focal necrosis was a minor
feature. Livers of the animals receiving repeated exposures showed
only minor injuries in the form of scattered hepatocytes containing
small fat droplets and a few foci of liver cell necrosis.
Torkelson et al. (1976) exposed male and female rats (10-12 of
each sex per group), rabbits (2-3 of each sex per group) and
guinea-pigs (8-12 of each sex per group) to concentrations of 0,
110, 230 and 410 mg chloroform/m3 air for 7 h/day, 5 days/week,
during 6 months. In the male and female rats, relative kidney weight
was increased at all exposure levels. In the males, at all levels,
kidneys showed cloudy swelling of the tubular epithelium and the
livers showed lobular granular degeneration with focal necrosis. At
the higher exposure levels the effects became more pronounced. The
effects observed in the males exposed to 110 mg chloroform/m3
disappeared within 6 weeks after exposure. At 410 mg
chloroform/m3, death, due to interstitial pneumonitis, occurred in
the males. No effects were seen in the male rats after 1, 2 or 4 h
of exposure to 110 mg chloroform/m3 (same schedule of exposure).
The results obtained after exposure of rabbits and guinea-pigs were
inconsistent because of low numbers of animals and/or the absence of
dose-effect relationships.
The toxicity of one-week exposures to inhaled chloroform has
been investigated in female B6C3F1 mice and in male F-344 rats
(Larson et al., 1994b; Méry et al., 1994). Rodents were exposed to
chloroform vapour at concentrations of 0, 4.9, 14.7, 49, 147, 490 or
1470 mg/m3 (0, 1, 3, 10, 30, 100 or 300 ppm) for 7 consecutive
days and necropsied on day 8. Cell proliferation was quantified as
the percentage of cells in S-phase (BrdU labelling index) measured
by immunohistochemical detection of BrdU-labelled nuclei. Mice
exposed to 490 or 1470 mg/m3 exhibited centrilobular hepatocyte
necrosis and severe vacuolar degeneration of mid-zonal and
periportal hepatocytes, while exposure to 49 or 147 mg/m3 resulted
in mild to moderate vacuolar changes in centrilobular hepatocytes.
Slight, dose-related increases in the hepatocyte LI were observed
for exposure concentrations of 4.9-14.7 mg/m3, while the LI was
increased more than 30-fold in the 490- and 1470-mg/m3 groups. The
kidneys of mice were affected only at 1470 mg/m3 exposure, with
approximately half of the proximal tubules lined by regenerating
epithelium and an 8-fold increase in the LI of tubule cells compared
with controls (Larson et al., 1994b).
In rats, mild centrilobular vacuolation was observed only in
the livers of animals exposed to 1470 mg/m3. The hepatocyte LI in
rats was increased only at 490 and 1470 mg/m3 (3-fold and 7-fold
over control, respectively). The kidneys of the male rats were
affected only at 1470 mg/m3. About 25 to 50% of the proximal
tubules were lined by regenerating epithelium in this exposure
group, while the LI for tubule cells was increased 2-fold over
controls (Larson et al., 1994b).
In the nasal passages of rats, chloroform concentrations of 49
mg/m3 or more induced histopathological changes that exhibited
clear concentration-related severity. Chloroform-induced changes
included increased epithelial mucosubstances in the respiratory
epithelium of the nasopharyngeal meatus, primarily in the rats. A
complex set of responses was seen in specific regions of the ethmoid
turbinates, predominantly in the rats. These lesions in the ethmoid
region, which involved all of the endo- and ectoturbinates, were
most severe peripherally and generally spared the tissue adjacent to
the medial airways. These changes were characterized by atrophy of
Bowman's glands, increased numbers of vimentin-positive cells in the
periosteum, new bone formation and increased number of periosteal
cells in S-phase as determined by BrdU incorporation. Additional
changes were site-specific loss of mucosubstances and loss of
immunocyto-chemical staining of acini and ducts of Bowman's glands
for P450-2E1 and pancytokeratin, and loss of P450-2E1 immunostaining
of the olfactory epithelium. The only change noted in the mice was
increased periosteal cell proliferation without new bone growth
(Méry et al., 1994).
7.3 Long-term exposure
In a carcinogenicity bioassay, female B6C3F1 mice were
exposed to 0, 200, 400, 900 or 1800 mg chloroform/litre
drinking-water (number of animals: 430, 430, 150, 50 and 50,
respectively) for a period of two years (Jorgenson et al., 1982)
(see also section 7.7.1). These concentrations (monitored by
analysis) correspond to time-weighted average daily chloroform doses
of 0, 34, 65, 130 and 263 mg/kg body weight (Jorgenson et al.,
1985). Matched controls (50 females) received an amount of water
without chloroform equal to that consumed by the 1800-mg/litre
group. Additional mice were used for intermediate biochemical and
histopathological examination. Early mortality in the high-dose
group was observed. After 3 months, livers of animals exposed to
chloroform concentrations of 65 mg/kg body weight or more showed a
higher fat content than those of the controls (as examined by
chemical techniques). After 6 months, liver fat content was
increased in all exposed groups. Data on organ weights were not
provided.
In a carcinogenicity bioassay, male Osborne-Mendel rats were
exposed to 0, 200, 400, 900 or 1800 mg chloroform/litre
drinking-water (number of animals: 330, 330, 150, 50 and 50,
respectively) for a period of two years (Jorgenson et al., 1982)
(see section 7.7.2). These concentrations (monitored by analysis)
correspond to time-weighted average daily chloroform doses of 0, 19,
38, 81 and 160 mg/kg body weight (Jorgenson et al., 1985). Matched
controls received an amount of water without chloroform equal to
that consumed by the 1800-mg/litre group. Additional rats were used
for intermediate biochemical and histopathological examination. The
survival was indirectly proportional to the dose levels.
Concentration-related decreases in water uptake and growth were
seen. The latter effects were also observed in the matched controls,
and thus may be attributed to the reduced intake of water.
Biochemical examination of blood after 6, 12 and 18 months showed
that chlorine, potassium, total iron and albumin levels and the
albumin/globulin ratio tended to be increased after chloroform
treatment, whereas levels of cholesterol, triglycerides and lactate
dehydrogenase were decreased in all treated groups. These deviations
were also observed in the matched controls, but the decreases in
serum triglycerides and cholesterol levels were more severe at the
two highest dose levels than in the matched control group. Data on
organ weights were not provided.
Beagle dogs were given chloroform in a toothpaste base in
gelatin capsules, 6 days/week for 7.5 years (Heywood et al., 1979).
The doses were 15 and 30 mg/kg and there were eight male and eight
female dogs in each dose group. Dogs given the high dose began to
show significant increases in SGPT levels at 6 weeks of treatment.
At the low dose level, significant increases were observed at 34
weeks and after. Similar effects were not observed in the vehicle
control (16 dogs of each sex) or untreated control (eight dogs of
each sex) groups. "Fatty cysts" of the liver were observed in both
dose groups at the end of this study (see section 7.7.3).
7.4 Skin and eye irritation
Adequate data on the skin irritation potential of chloroform
has not been identified. Torkelson et al. (1976) applied liquid
chloroform to the rabbit ear and found slight hyperaemia and
exfoliation after one to four treatments (period between application
and observation not specified). More frequent application did not
increase the severity of the injuries. A 24-h application of
chloroform on a cotton pad on the belly of rabbits produced slight
hyperaemia, moderate necrosis and eschar formation. Chloroform
delayed healing of mechanically damaged skin on the application
site.
Application of chloroform droplets in the rabbit eye caused a
transient slight irritation of the conjunctiva and corneal injury. A
purulent exudate occurred for 2 or more days after the treatment
(Torkelson et al., 1976).
Duprat et al. (1976) applied undiluted chloroform into the eyes
of six New Zealand white rabbits. It produced severe eye irritation,
with mydriasis and keratitis in all rabbits. Translucent zones in
the cornea were observed in four animals and a purulent haemorrhagic
discharge was also reported (number of rabbits unknown). The effects
had disappeared 2-3 weeks after application, except for one rabbit
that still showed corneal opacity after 3 weeks.
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1 Reproduction
Borzelleca & Carchman (1982) studied the reproductive toxicity
of chloroform in a three-generation experiment with ICR mice. They
administered the chemical (0.1% Emulphor in deionised water) via
drinking-water (in closed bottles) to males (10/group) and females
(30/group), at concentrations of 0, 0.1, 1 and 5 mg/ml, from 5 weeks
before F0 mating throughout the entire study until sacrifice of
the F2b pups. Death occurred among the males and females of the
highest dose group, and body weights in this group were reduced. At
1 mg/ml, the body weights of F1b females were also reduced.
Dose-related hepatotoxicity was found in the F0 and F1b animals
(symptoms varying from "slight yellow-grey colouring" in the lowest
dose group to "grey to black discolouration" with large nodules
(> 3 mm) upon and within the liver in the highest dose group).
The treatment resulted in reduced fertility, litter size, gestation
index and viability index in all F1 and F2 generations,
statistically significant at 5 mg/ml. No evidence for a teratogenic
potential was obtained.
7.5.2 Embryotoxicity and teratogenicity
Chloroform has not been found to be teratogenic but has been
shown to induce fetotoxic effects.
7.5.2.1 Oral exposure
No evidence for a teratogenic effect of chloroform was obtained
in a three-generation study with ICR mice (Borzelleca & Carchman,
1982).
In a study by Thompson et al. (1974), female Sprague-Dawley
rats (25/group) were intubated with chloroform in corn oil (0, 10,
25 and 63 mg/kg body weight) twice daily on days 6-15 of gestation.
A reduced body weight gain and anorexia were seen in the dams of the
two higher dose groups. Tissues from two dams of each dose group
were microscopically examined and fatty changes were observed in the
livers of both females at 63 mg/kg and in one female at 25 mg/kg.
Other signs of maternal toxicity were not found at these dose
levels. The fetuses of the 63-mg/kg groups had a smaller weight at
delivery than those of the control group. The incidence of bilateral
extralumbar ribs was significantly increased among the fetal
population of the 63-mg/kg dose group. Other minor visceral and
skeletal abnormalities were seen, but not at significantly elevated
levels. In the same study female Dutch-Belted rabbits (15/group)
were dosed orally with chloroform in corn oil (0, 20, 35 and 50
mg/kg body weight) once daily during days 6-18 of gestation.
Administration of chloroform produced a decrease in fetal body
weight and incomplete ossification of skeletal elements (skull
bones) in the 20- and 50-mg/kg dose groups. At the highest dose
level the dams showed decreased weight gain. Signs of embryotoxicity
and teratogenicity were not observed.
Ruddick et al. (1983) gave pregnant Sprague-Dawley rats
(15/group) chloroform in corn oil (0, 100, 200 and 400 mg
chloroform/kg body weight) daily by gavage from days 6 to 15 of
gestation. All doses caused reduced weight gain in the dams and
increased liver weight. At the highest dose level, there was an
increase in the kidney weight of the dams. Haematological
examinations showed dose-dependent reductions in haemoglobin and
haematocrit (14% maximally, both parameters). In the highest dose
group, the red blood cell count was also reduced.
According to Burkhalter & Balster (1979), oral administration
of chloroform to mice from 3 weeks before mating until the end of
the lactating period (in both sexes the dose was 31 mg/kg body
weight) did not result in retardation of the development of
responses to a battery of neurobehavioural tests in the pups.
7.5.2.2 Inhalation exposure
Schwetz et al. (1974) reported effects on pregnancy and on the
incidence of fetal malformations in Sprague-Dawley rats exposed to
chloroform concentrations of 147, 490 and 1470 mg/m3 (30, 100 and
300 ppm) for 7 h/day during days 6-15 of gestation (analysis 3 times
daily showed concentrations of 147, 466 and 1426 mg/m3; 30, 95 and
291 ppm, respectively). The two highest concentrations were toxic to
the dams (anorexia and reduced weight gain, increases in relative
and absolute liver weight). There was a dose-dependent decrease in
the pregnancy percentage (100% in the control group versus 15% in
the 1426-mg/m3 group) and in the number of living fetuses per
litter. An increase was observed in the percentage of
post-implantation losses (resorptions) in the highest dose group,
and a dose-dependent increase was seen in the percentage of litters
with resorptions (from 57% in the control group to 100% at the
highest concentration). At all exposure levels, fetuses showed
growth retardation and minor skeletal aberrations (delayed
ossification of skull and sternebrae). Exposure to 147 mg/m3
caused minor embryo- and fetotoxicity, and concentrations of 466 and
1426 mg/m3 in the inhaled air were embryo- and fetotoxic to the
rat. At the higher levels, subcutaneous oedema and other unspecified
fetal soft tissue anomalies were also observed.
Murray et al. (1979) exposed pregnant CF1 mice (35-40/group)
to 0 and 490 mg/m3 (0 and 100 ppm) for 7 h each day throughout
days 1-7, 6-15 or 8-15 of gestation. The ability of females to
maintain pregnancy was significantly decreased after exposure to
chloroform during days 1-7 or 6-15 of gestation (44 and 43% in the
treated groups versus 74 and 91% in the respective control groups).
The dosed animals consumed slightly less food than the control
animals, resulting in reduced body weight gain. Absolute and
relative liver weights were increased in the groups exposed during
days 6-15 and 8-15. After exposure during days 6-15, ALAT levels
were significantly increased in pregnant and non-pregnant animals,
the pregnant animals showing the smaller increase. Among the
controls, no difference in ALAT activity was observed. An increase
in total litter resorptions was observed after exposure through days
8-15. Mean fetal body weight and crown-rump length were decreased
significantly if the dams had been exposed through days 1-7 or 6-15
of pregnancy. In the exposed groups an increased number of fetuses
with delayed ossification of skull bones and sternebrae was
observed, especially in the days 1-7 and 8-15 exposed groups. The
incidence of cleft palates significantly increased in fetuses from
dams exposed to 490 mg/m3 through days 8-15 of gestation for 7 h
each day.
Published information for embryotoxicity and teratogenicity of
chloroform in rat, mouse and rabbit by oral and inhalation exposure
are summarized in Table 11.
7.6 Mutagenicity and related end-points
Very many genotoxicity assays have been conducted with
chloroform and the data currently available are summarized in Tables
12 and 13. Some of these reports are from a large collaborative
study comparing intra-laboratory variations in testing methodology
(De Serres & Ashby, 1981).
Two problems potentially compromise the interpretation of
mutagenicity data on chloroform. First, there is a possibility that
ethyl and diethylcarbonate, produced by reaction of phosgene with
ethanol that is routinely added to U.S.P (US Pharmacopoeia)
chloroform, could generate false positive results. Secondly, testing
of chloroform must be done in a sealed system because of its
volatility, and so studies that did not take this factor into
account could give false negative results.
In data presented in Table 12, three separate studies using the
Ames assay were conducted under sealed conditions to assure
chloroform retention. All three studies yielded negative test
results.
In not all studies was it reported whether an in vitro assay
was performed in a sealed chamber to prevent chloroform evaporation
(Table 12). However, dimethylsulfoxide (DMSO) was often used as a
solvent, thus increasing retention in the media. Furthermore, even
in the case of an unsealed chamber, chloroform would be expected to
stay in the media for a period of hours, and very high doses (up to
10 mg/plate) were often used.
Chloroform has been tested by a number of authors in validated
bacterial systems with Salmonella typhimurium and Escherichia
coli and showed to be negative both with and without metabolic
activation. Only in one uncommon test with Photobacterium
phosphorum was a positive effect found (Wecher & Scher, 1982).
Table 11. Embryotoxicity, fetotoxicity and teratogenicity produced in animals by exposure to chloroform
Species Dose Gestational days Route of Result Reference
administered administration
Rat 30, 100, 300 ppm 6-15 inhalation embryotoxic Schwetz et al. (1974)
(7 h/day) fetotoxic
Rat 20, 50, 126 mg/kg per day 6-15 oral fetotoxic Thompson et al. (1974)
Rat 100, 200, 400 mg/kg per day 6-15 oral fetotoxic Ruddick et al. (1983)
Mouse 100 ppm 1-7, inhalation embryotoxic Murray et al. (1979)
6-15, fetotoxic
8-15
(7 h/day)
Rabbit 20, 35, 50 mg/kg per day 6-18 oral fetotoxic Thompson et al. (1974)
Table 12. Mutagenicity studies with chloroform
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Bacterial systems
Salmonella TA1535 base-pair substitution 14C-labelled compound + ra i.m., - Uehleke et al.
typhimurium TA1538 frame-shift mutation tested; no further i.n.r. (1976, 1977)
details reported
S. typhimurium TA1535 base-pair substitution 5 mM tested; incubation + m PB - Uehleke et al.
TA1538 frame-shift mutation in closed containers (1976, 1977)
(survival > 80%)
S. typhimurium TA98 frame-shift mutation suspension test and - i.n.r. - Simmon et al.
TA1537 frame-shift mutation vapour test; concentration + r - (1977)
TA1538 frame-shift mutation in suspension test was 10%
TA100 base-pair substitution v/v, no further details
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation up to 3600 µg/plate; - - Gocke et al.
TA1537 frame-shift mutation incubation in air-tight + r PCB - (1981)
TA1538 frame-shift mutation desiccators
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation test conditions not - - Trueman (1981)
TA1537 frame-shift mutation reported + r PCB -
TA1538 frame-shift mutation
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation test conditions not - - Ichinotsubo
TA100 base-pair substitution reported + - et al. (1981b)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. typhimurium TA98 frame-shift mutation 0.5, 1.0, 5.0, 100, - - Venitt & Crofton-
TA100 base-pair substitution 200, 500 µg/plate + r PCB - Sleigh (1981)
S. typhimurium TA98 frame-shift mutation microtitre fluctuation - - Gatehouse
TA1537 frame-shift mutation test; 1, 5, 10 µg/ml + r PCB - (1981)
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation fluctuation test; 1-500 - ± Hubbard et
TA100 base-pair substitution µg/ml (not specified) + r - al. (1981)
S. typhimurium TA98 frame-shift mutation solvent DMSO; no further - - Baker &
TA1537 frame-shift mutation details + r PCB - Bonin (1981)
TA1538 frame-shift mutation
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation test conditions not - ± Garner et al.
TA1537 frame-shift mutation reported + r PB - (1981)
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation 50, 100, 200, 1000, - - MacDonald
TA1537 frame-shift mutation 2000, 5000 µg/plate + r PCB - (1981)
TA100 base-pair substitution
S. typhimurium TA98 frame-shift mutation solvents DMSO; no further - - Nagao &
TA1537 frame-shift mutation details + r PCB - Takahashi
TA100 base-pair substitution (1981)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. typhimurium TA98 frame-shift mutation 0.1, 1.0, 10, 100, 500, - - Rowland &
TA1537 frame-shift mutation 2000 µg/plate; solvent + r PCB - Severn (1981)
TA1538 frame-shift mutation DMSO
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA1535 base-pair substitution 10, 100, 1000, 10 000 - - Richold &
TA1537 frame-shift mutation µg/plate; solvent + r PCB - Jones (1981)
TA1538 frame-shift mutation DMSO
S. typhimurium TA98 frame-shift mutation test conditions not - - Simmon &
TA1537 frame-shift mutation reported + r PCB - Shepherd
TA1538 frame-shift mutation (1981)
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation solvent DMSO or water; - - Brooks &
TA1537 frame-shift mutation 0.2, 2, 20, 200, 2000 + r PCB - Dean (1981)
TA1538 frame-shift mutation µg/plate
TA100 base-pair substitution
TA1535 base-pair substitution
TA92 interstrand DNA
crosslinks
S. typhimurium TA98 frame-shift mutation 10, 100, 1000, 10 000 - - Van Abbé et
TA1537 frame-shift mutation µg/plate + r PCB - al. (1982)
TA1538 frame-shift mutation + m PCB -
TA100 base-pair substitution
S. typhimurium TA1535 base-pair substitution vapour test; exposure - - Van Abbé et
TA1538 frame-shift mutation for 2, 4, 6 or 8 h + r PCB - al. (1982)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. typhimurium TM 677 forward mutation to solvent DMSO; up to + r PCB - Skopek et al.
azaguanine resistance 300 µg/ml (1981)
Escherichia coli WP2 uvrA reversion to trp+ 10, 100, 1000 µg/plate + r PCB - Gatehouse (1981)
E. coli WP2 uvrA reversion to trp+ test conditions not + r PCB - Matsushima et
reported al. (1981)
E. coli WP2p reversion to trp+ solvent acetone; 0.1, 1, + r - Kirkland et
WP2 uvrA 10, 100, 1000, 10 000 - PCB - al. (1981)
µg/plate
E. coli WP2p reversion to trp+ 0.5, 1.0, 5, 10, 50, - - Venitt &
WP2 uvr-p 100, 200, 500 µg/plate + r PCB - Crofton-Sleigh
(1981)
E. coli K12 base-pair substitution 14C-labelled compound + ra i.m. - Greim et al.
(not specified) tested; no further details (1977)
reported
Photobacterium PPL- reversion to normal disc-diffusion assay; no - + Wecher &
phosphoreum light emission further details reported Scher (1982)
Non-mammalian eukaryotic systems
Allium cepa chromosomal aberrations solvent: DMSO; 0, 250, 500, + Cortés et al.
1000, 1500, 2500, 5000, (1985)
10 000 µg/ml
Saccharomyces D7 mitotic gene conversion no details reported + n.r. i.n.r. - Zimmermann &
cerevisiae at trp 5 locus Scheel (1981)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. cerevisiae D7 mitotic gene conversion 21, 41, 54 mM; incubation - + Callen et al.
at trp 5; mitotic in screw-capped glass (1980)
recombination at ade 2, tubes
reversion at ilv 1 loci
S. cerevisiae D6 mitotic aneuploidy agar added + r PCB - Parry & Sharp
(1981)
S. cerevisiae D6 mitotic aneuploidy direct incubation in + r PCB ± Parry & Sharp
plastic bottles, 25, 50, (1981)
100 µg/ml; idem in glass + r PCB -
bottles
S. cerevisiae JD1 mitotic gene conversion up to 1000 µg/ml; + r PCB ± Sharp & Parry
at trp 5 locus and his incubation in plastic (1981a)
5 polaron containers
S. cerevisiae JD1 idem as above idem as above, only + r PCB - Sharp & Parry
incubation in glass (1981a)
containers
S. cerevisiae D4 mitotic gene conversion 0.33, 1.0, 3.33, 100, - - Jagannath et
at ade 2 and trp 5 loci 333.3 µg/plate + r PCB - al. (1981)
S. cerevisiae T1 mitotic crossing over 100, 1000 µg/plate - - Kassinova et
T2 at ade 2 + r PCB - al. (1981)
S. cerevisiae XV 185-14 reversion at his 1, solvent DMSO; 111, - - Mehta & Von
C (haploid) hom 3, and arg 4 loci 1111 µg/ml + n.r. i.n.r. - Borstel (1981)
Schizosaccharomyces P1 forward mutation at ade 5, 7.5, 10 µg/ml - - Loprieno
myces pombe 1, 3, 4, 5 and 9 loci + r PCB (+) (1981)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Aspergillus 35 forward mutation 0.5% (survival 26%) ? - Gualandi
nidulans (haploid) (induction of methionine (1984)
suppressors)
A. nidulans P1 somatic segregation 0.5% (survival 16.5%) ? - Gualandi
(crossing-over and non- (1984)
disjunction)
Drosophila Berlin K sex-linked recessive Basc-test; 24 mM; adult - Gocke et al.
melanogaster wild and lethal test feeding (1981)
Basc
D. melanogaster Berlin K sex-linked recessive Basc-test; solvent DMSO; - Vogel et al.
wild and lethal test 0.1, 0.2% treated at (1981)
y mei 9a 25 °C for 3 days with
mei-41 D5 standard feeding
technique
In vitro mammalian systems
Chinese hamster V79 forward mutation to 1, 1.5, 2, 2.5% - - Sturrock
8-azaguanine resistance (1977)
Human lymphocytes chromosome breakage solvent acetone; 50, 100, + r PCB - Kirkland et
200, 400 µg/ml al. (1981)
In vivo mammalian systems
Mouse CD1 micronuclei in intraperitoneal, 0.015, - Tsuchimoto &
polychromatic 0.03, 0.06 ml/kg body Matter (1981)
erythrocytes of bone weight at 0 and 24 h
marrow
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Mouse NMRI micronuclei in intraperitoneal, 238, 476, - Gocke et al.
polychromatic 952 mg/kg body weight at (1981)
erythrocytes of bone 0 and 24 h
marrow
Mouse B6C3F1 micronuclei in intraperitoneal, about - Salamone et
polychromatic 0.088 ml/kg body weight al. (1981)
erythrocytes of bone at 0 and 24 h or at
marrow 0 h only
Mouse ? micronuclei in route not reported; 100, (+) Agustin &
polychromatic 200, 400, 600, 700, 800, Lim-Sylianco
erythrocytes of bone 900 mg/kg body weight (1978)
marrow
Rat Long-Evans chromosomal aberrations intraperitoneal, + Fujie et al.
in bone marrow 1.2-119.4 mg/kg body (1990)
6-597 mg/kg body weight weight; oral,
Host-mediated assays
S. typhimurium TA1535 base-pair substitution test conditions not - Agustin & Lim-
reported Sylianco (1978)
S. typhimurium TA1537 frame-shift mutation test conditions not + Agustin & Lim-
reported Sylianco (1978)
Table 12 (contd)
a + = with metabolic activation b PB = phenobarbital c + = positive
- = without metabolic activation PCB = polychlorinated biphenyls (+) = weakly positive
m = mouse i.m. = intact microsomes added ± = equivocal; study cannot be evaluated
r = rat i.n.r. = inducer not reported - = negative
ra = rabbit
n.r. = species not reported
? = not reported if metabolic
action was used
Table 13. Indicator studies with chloroform
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Bacterial systems
Escherichia coli WP2, WP67 DNA damage (growth test concentrations not - - Tweats (1981)
uvrA pol inhibition) specified; no further + r PCB -
A & CM 871 details
uvrA lexA
recA
E. coli WP2, WP67 DNA damage test conditions not - - Green (1981)
uvrA pol reported
A & CM 871
uvrA lexA
recA
E. coli W3110 DNA damage liquid suspension test; - - Rosenkranz et
(polA+), 25 mg/ml; solvent DMSO + r PCB (+) al. (1981)
P3478 or water
(POLA1-)
E. coli JC 2921 rec DNA damage test conditions not - + - Ichinotsubo
JC 9238 rec reported + + et al.
JC 8471 rec (1981a)
JC 5519 rec
JC 7689 rec
JC 7623 rec
E. coli 56-161 induction of prophage 0.5, 5 mg/ml + r PCB - Thomson (1981)
env A lambda in lysogenic -
C 600 E. coli
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Bacillus subtilis H17 rec+ DNA damage not further maximum concentration: + r - Kada (1981)
M45 rec- specified 20 µl/plate + yf -
+ jc -
Non-mammalian eukaryotic systems
Allium cepa SCE solvent DMSO; 0, 250, 500, ± Cortés et al.
1000, 1500, 2500, 5000, (1985)
10 000 µg/ml
Saccharomyces T4 DNA repair solvent DMSO; 0.1, 1.0% - - Kassinova et
cerevisiae T5 al. (1981)
S. cerevisiae 197/2d DNA repair 100, 300, 600, 750 µg/ml; - ± Sharp & Parry
rad 3, incubation in plastic + ± (1981b)
rad 18, bottles
rad 52,
trp 2
In vitro mammalian systems
Chinese hamster ovary SCE 0.7% (after exposure, 78% + r PCB - White et al.
of dose remained) (1979)
Chinese hamster ovary SCE 0.01, 0.1 µg/ml; solvent + r PCB - Perry & Thomson
DMSO (1981)
Chinese hamster ovary SCE 0.001, 0.01, 0.1 mM - + d Athanasiou &
Kyrtopoulos
(1981)
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Rat erythroblast SCE only 1.0 mM tested + (+) Fujie et al.
(1993)
Syrian hamster embryo adenovirus 0.12, 0.25, 0.50, 1.0, - + Hatch et al.
transformation 2.0 ml/sealed chamber (1983)
(4.6 litre)
Baby hamster kidney cell transformation test conditions not - (+) Daniel & Dehnel
reported + r - (1981)
Baby hamster kidney cell transformation 0.25, 2.5, 25, 250 µl/ml - - Styles (1979,
1981)
Rat primary UDS 0.00084-8.4 mM - - Althaus et al.
hepatocytes (1982)
Mouse (B6C3F1) primary UDS 0.01-10 mM - - Larson et al.
hepatocyte (1994c)
Human primary UDS 4 cases 0.01-1.0 mM - - Butterworth
hepatocyte et al. (1989)
Human lymphocytes SCE 0.016-50 mM - + Morimoto &
Koizumi (1983)
Human lymphocytes SCE solvent acetone; 25, 50, + r PCB - Kirkland et
75, 100, 200, 400 µg/ml al. (1981)
Human lymphocytes UDS 0.1, 1.0, 10 mM - - Perocco et
+ r PB - al. (1983)
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Human lymphocytes UDS 2.5, 5, 10 µg/ml - - Perocco &
+ r PB - Prodi (1981)
Human Hela cells UDS 0.1-100 µg/ml; solvent - - Martin &
DMSO +r PB - McDermid (1981)
In vivo mammalian systems
Mouse JCR/SJ SCE in bone marrow oral: 25, 50, 100, + Morimoto &
cells 250 mg/kg body weight Koizumi (1983)
per day for 5 days
Mouse B6C3F1 DNA repair in liver oral: 240 mg/kg body - Reitz et al.
weight (1982)
Mouse C57BL sperm abnormalities vapour exposure: 0.04, + Land et al.
x C3H 0.08%; 4 h/day for 5 days (1981)
Mouse CBA x sperm abnormalities intraperitoneal: 0.025, - Topham (1980,
BALB/C 0.05, 0.075, 0.1, 0.25 mg/kg 1981)
body weight per day for
5 days; vehicle corn oil
Rat F-344 UDS in hepatocytes oral: 40, 400 mg/kg body - Mirsalis et
weight, single dose, al. (1982)
vehicle corn oil
Mouse B6C3F1 UDS in hepatocytes 238, 477 mg/kg body weight, - Larson et al.
single dose, corn oil (1994c)
vehicle
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Rat (neonatal) liver and DNA damage (3H elution) vehicle corn oil; 200-400 - Petzold &
kidney cells mg/kg body weight Swenberg (1978)
a + = with metabolic activation; - = without metabolic activation; r = rat; yf = Yellowtail fish; jc = Japanese clam
b PB = phenobarbital; PCB = polychlorinated biphenyls
c + = positive; (+) = weakly positive; ± = equivocal; study cannot be evaluated; - = negative;
d In this test chromosome aberrations were also reported to occur; no details on this finding were reported.
The majority of studies with non-mammalian eukaryotic systems
(yeasts and other fungi) were negative. Positive results were
obtained with Saccharomyces cerevisiae D7, but only at the highest
concentration tested, at which there was a marked toxic effect
(Callen et al., 1980). It should be noted that this strain of yeast
contains an endogenous cytochrome P450-dependent monooxygenase
system. In Schizosaccharomyces pombe an indication for a mutagenic
effect was observed (Loprieno, 1981). The inconsistent results with
Saccharomyces cerevisiae D6 and JD 1 were probably due to
inadequate test conditions (exposure in plastic rather than glass
containers) and therefore it can be considered that chloroform was
non-mutagenic in these tests (Parry & Sharp, 1981; Sharp & Parry,
1981a). In two sex-linked recessive lethal tests with Drosophila
melanogaster, no mutagenic activity was observed (Gocke et al.,
1981; Vogel et al., 1981).
Chloroform did not induce gene mutations in V79 Chinese hamster
cells (Sturrock, 1977), or chromosomal aberrations in human
lymphocytes in vitro (Kirkland et al., 1981).
In vivo mammalian testing comprised four micronucleus tests
in mice, three of which gave a negative result (Tsuchimoto & Matter,
1981; Gocke et al.,1981; Salamone et al., 1981). The fourth
micronucleus test was reported to have given a weakly positive
result (Agustin & Lim-Sylianco, 1978). The same authors found a
positive effect in the mouse host-mediated assay with Salmonella
typhimurium TA1537 but not with TA1535.
Indicator studies showed that chloroform induces
sister-chromatid exchange (SCE) in hamster and human cells in vitro
in the absence of metabolic activation, and in mice in vivo
(Athanasiou & Kyrtopoulos, 1981; Morimoto & Koizumi, 1983). Positive
or weakly positive results were reported in two tests on DNA damage
and DNA repair with Escherichia coli and Saccharomyces cerevisiae
(Sharp & Parry, 1981b; Rosenkranz et al., 1981; Ichinotsubo et al.,
1981a).
The ability of chloroform to induce unscheduled DNA synthesis
(UDS) was examined in the in vitro and in vivo hepatocyte DNA
repair assays for the most sensitive site for tumour formation, the
female mouse liver (NCI, 1976a,b). In the in vitro assay, primary
hepatocyte cultures from female B6C3F1 mice were incubated with
concentrations from 0.01 to 10 mM chloroform in the presence of
3H-thymidine. UDS was assessed by quantitative autoradiography. No
induction of DNA repair was observed at any concentration. In the
in vivo assay, animals were treated by gavage with chloroform in
corn oil (238 and 477 mg/kg body weight). Primary hepatocyte
cultures were prepared 2 and 12 h later, incubated with
3H-thymidine and assessed for induction of UDS. No DNA repair
activity was seen at either dose or at either time point. These
negative results in the target organ are consistent with the
suggestion that neither chloroform nor its metabolites react
directly with DNA in vivo.
The ability of chloroform to induce DNA repair was examined in
freshly prepared primary cultures of human hepatocytes from
discarded surgical material. No activity was seen in cultures from
four different individuals at concentrations as high as 1 mM
chloroform (Butterworth et al., 1989).
Given the large number of sensitive assays to which chloroform
has been submitted, it is noteworthy that the reported positive
responses are so few. Furthermore, these few positive responses were
randomly distributed amongst the various assays with no apparent
pattern or clustering for any test system. Taken together, the
weight of evidence indicates that neither chloroform not its
metabolites would appear to interact directly with DNA or possess
genotoxic activity. The conclusion is consistent with the lack of
initiating activity of chloroform (see section 7.7.4).
7.7 Carcinogenicity
7.7.1 Mice
In a National Cancer Institute carcinogenicity study, B6C3F1
mice received USP grade chloroform stabilized with ethanol (0.5-1%)
in corn oil 5 times a week by gavage (NCI, 1976a,b). Dosing was
stopped after 78 weeks and the animals were sacrificed after 92
weeks. There were 20 animals per sex in the control group and 50
animals per sex in the dosed groups. The dose levels changed after
18 weeks, resulting in time-weighted average dose levels of 138
(low) and 277 (high) mg chloroform/kg body weight for male mice and
238 (low dose) and 477 (high dose) mg chloroform/kg body weight for
female mice. Administration of the highest dose of chloroform
reduced survival in the female mice. Causes of death were related to
the observed liver tumours, pulmonary inflammation and cardiac
thrombosis. This latter lesion was not observed in either the
control or the low-dose group. Dose-related increased frequencies of
hepatocellular carcinomas were found, the incidences being 1/18,
18/50 and 44/45 at 0, 138 and 277 mg chloroform/kg body weight in
the males and 0/20, 36/45 and 39/41 at 0, 238 and 477 mg chloroform
per kg body weight in the females, respectively. Mice presented
clinical signs of illness, i.e. a reduced food intake and an untidy
appearance, but clear information on non-neoplastic lesions was not
provided. There is evidence that tumour formation may have been
secondary to induced cytolethality and regenerative cell
proliferation (see Larson et al., 1994a, and section 7.2.1.1).
Jorgenson et al. (1985) exposed female B6C3F1 mice to
chloroform in their drinking-water for a period of two years. The
concentrations were 0, 200, 400, 900 and 1800 mg/litre, and the
numbers of animals were 430, 430, 150, 50 and 50 per group,
respectively. Time-weighted average daily doses were 0, 34, 65, 130
and 263 mg/kg body weight. Additional matched controls (50 animals)
received the same quantity of drinking-water (without chloroform) as
was consumed by the animals in the highest dose groups. Initially,
25% of the animals in the two highest dose groups died, but later on
the death rate was more or less equal to that in the control group.
No treatment-related effects on either liver or total tumour
incidence were observed. Lack of tumour formation is consistent with
the lack of induced liver necrosis or regenerative hepatocyte cell
proliferation when chloroform is administered in the drinking-water
(see Larson et al., 1994a and section 7.2.1.1).
The difference between the results obtained in the NCI study
(1976a,b) and the Jorgenson et al. (1985) study is probably related
to the manner in which the compound was administered. When given in
the drinking-water, only small amounts of chloroform reach the
liver, corresponding to each sip taken. Apparently, these small
doses and delivery rates can be metabolized, detoxified and
eliminated without liver damage (Larson et al., 1994a). When similar
daily amounts are given as a single bolus dose in corn oil, it is
probable that the high rate of delivery to the liver results in the
production of toxic metabolites that overwhelm detoxification
mechanisms, resulting in cell death and regenerative cell
proliferation (Larson et al., 1994a). The choice of vehicle may also
contribute to the observed difference in toxicity (Bull et al.,
1986) (see also section 7.2.1.1).
Roe et al. (1979) administered daily chloroform (British
Pharmacopoeia quality) in a toothpaste base (vehicle) to ICI mice
(control group 104 animals per sex, dose groups 52 animals per sex)
by gavage, 6 days a week for 80 weeks, followed by a 16-week
observation period. The dose levels were 0 (controls), 17 and 60
mg/kg body weight. Mice that died during the first 15 weeks of the
experiment were replaced by animals from a reserve group (which were
probably also dosed, although this was not specified). The control
toothpaste did not contain eucalyptol and peppermint oil, whereas
the toothpaste containing chloroform did contain these substances.
Treatment with chloroform resulted in slightly increased survival,
especially in the males. The most common cause of death was
respiratory failure. A slightly increased incidence of fatty
degeneration was observed among the chloroform-treated animals.
Total tumour incidence was increased in the male mice (20/37 and
21/38 at 17 and 60 mg/kg body weight, respectively, versus 20/72 in
the controls). Renal tumours (3 hypernephromas and 5 cortical
adenomas) were reported in 8 out of 38 males of the high-dose group.
In a second experiment by Roe et al. (1979), the influence of
peppermint oil, eucalyptol and chloroform was determined separately.
In this study, male ICI mice received 60 mg chloroform/kg body
weight daily, in the same way as in the study reported above. The
vehicle control (toothpaste without chloroform, eucalyptol and
peppermint oil) and dose groups consisted of 260 and 52 male
animals, respectively (the groups receiving a dose of peppermint or
eucalyptol also consisted of 52 animals). Again, the survival in the
chloroform-dosed group was better than in the control group. Total
tumour incidence was lower in the chloroform-treated group (30/49
versus 170/240 in the controls). However, administration of
chloroform resulted in a kidney tumour frequency (hypernephromas and
adenomas) of 9/49, compared with a control value of 6/240.
In a third study by Roe et al. (1979), 60 mg chloroform/kg body
weight in toothpaste (containing eucalyptol and peppermint oil) was
administered daily to male mice (52 per group) of the ICI, CBA,
C57BL and the CF1 strain for a period of 80 weeks. The chemical
was also administered in arachis oil to male mice of the ICI strain.
Each strain had its own control group. Terminal sacrifice was at 93,
97-99, 104 and 104 weeks for the CF1, ICI, C57BL and CBA strains,
respectively. In this study, a treatment-related increase in the
survival was found in all strains tested, except for the CF1
strain. Treatment with chloroform resulted in a higher incidence of
renal changes in the CBA and CF1 strains but not in the C57BL
strain. The cause of death in all four strains was renal neoplasia
in combination with respiratory and renal disease. In the C57BL, CBA
and CF1 strains no changes in tumour frequencies were observed. In
the ICI mice, after treatment with chloroform in either the
toothpaste vehicle or arachis oil, an increase in the incidence of
malignant kidney tumours was found (3/47 versus 0/49 in the
controls, toothpaste vehicle; 9/48 versus 0/50 in the controls,
arachis oil vehicle).
Though full results are not yet available, an additional
carcinogenesis bioassay in which mice were exposed to chloroform by
inhalation is under way (Matsushima, personal communication, 1993).
7.7.2 Rats
In a National Cancer Institute carcinogenicity study,
Osborne-Mendel rats received USP grade chloroform stabilized with
ethanol (0.5-1%) in corn oil 5 times a week by gavage (NCI,
1976a,b). Dosing was stopped after 78 weeks and the animals were
sacrificed after 111 weeks. There were 20 animals per sex in the
control group and 50 animals per sex in the dosed groups. The dose
levels changed after 23 weeks, resulting in time-weighted average
dose levels of 90 (low dose) and 180 (high dose) mg chloroform/kg
body weight for males and 100 (low) and 200 (high) mg chloroform/kg
body weight for females. Administration of chloroform reduced
survival in male and female rats in all dose groups. A clear
pathological reason for this effect in the rats was not given. In
male rats, dose-related increased frequencies of kidney epithelial
tumours were observed (incidences: 0/19, 4/50 and 12/50 at 0, 90 and
180 mg chloroform/kg body weight, respectively). In the females a
non-significant increase in the frequency of thyroid tumours was
found (incidences: 1/19, 8/49 and 10/46 at 0, 100 and 200 mg
chloroform/kg body weight, respectively). Rats presented clinical
signs of illness, i.e. a reduced food intake and an untidy
appearance. However, clear information on non-neoplastic lesions was
not provided.
Reuber (1979) re-evaluated the histological sections of the NCI
study (1976a,b) and reported the same neoplastic lesions as the NCI.
In addition, he noted that chloroform-dosed female rats developed
liver lesions that were not seen in the control females (i.e.
cholangiofibromas 0/20, 1/39 and 3/39; cholangiocarcinomas 0/20,
2/39 and 8/39; hyperplastic nodules 1/20, 7/39 and 12/39; and
hepatocellular carcinomas 0/20, 2/39 and 2/39, for the control, low-
and high-dose groups, respectively).
Jorgenson et al. (1985) exposed male Osborne-Mendel rats via
drinking-water to 0, 200, 400, 900 and 1800 mg chloroform/litre for
a period of two years. Time-weighted average daily chloroform doses
were 0, 19, 38, 81 and 160 mg/kg body weight and the numbers of
animals were 330, 330, 150, 50 and 50 per group, respectively.
Additional matched controls (50 animals) received the same quantity
of drinking-water (without chloroform) as was consumed by the
animals in the highest dose groups. As a probable consequence of
reduced drinking and reduced body weights, death rate was reduced
with increasing chloroform dosage and in the matched control group.
The only dose-related effect was an increase in renal tubular cell
adenomas and adenocarcinomas. The incidence for all kidney tumours
was 5/301, 1/50, 6/313, 7/148, 3/48 and 7/50 for control, matched
control and the 19, 38, 81 and 160 mg/kg groups, respectively. From
38 mg/kg body weight upwards the increase in the frequency of all
kidney tumours was statistically significant.
In an inadequately reported study, Tumasonis et al. (1985)
exposed male and female Wistar rats to 0 or 2900 mg chloroform per
litre drinking-water during the lifetime of the animals. Animal
numbers were 26 and 22 in the male and female control groups and 32
and 45 in the male and female treated groups, respectively. The
experiment started with weanlings. After 72 weeks, the
drinking-water chloroform concentrations were reduced because of an
increased intake of water by exposed animals. However, daily intakes
of chloroform varied considerably and so the time-weighted average
daily doses were estimated roughly from a figure in the report. They
appeared to be around 180 mg/kg body weight in the males and around
240 mg/kg body weight in the females. Body weights were decreased
and life-span was increased in the exposed animals. A severe hepatic
adenofibrosis (cholangiofibrosis) was observed in the exposed
animals. Ten out of the 40 females examined showed hepatic
hyperplastic nodules (none did in the control group). In the males
no increase in the incidence of neoplastic nodules was found.
Although full results are not yet available, an additional
carcinogenesis bioassay in which rats were exposed to chloroform by
inhalation is under way (Matsushima, personal communication, 1993).
7.7.3 Dogs
Heywood et al. (1979) administered chloroform to Beagle dogs at
dose levels of 0, 15 and 30 mg/kg body weight (6 days/week) in
toothpaste in a gelatin capsule for a period of 7.5 years. Sacrifice
followed after an observation period of 19 to 23 weeks, during which
the chloroform treatment was withdrawn. The control group consisted
of 16 animals of each sex and the dose groups of 8 animals of each
sex. There were no treatment-related increases in tumours.
7.7.4 Studies on initiating-promoting activity
7.7.4.1 Mice
One week after a single intraperitoneal administration of
ethylnitrosourea (0, 5 or 20 mg/kg body weight) to 15 days old CD1
Swiss mice (both sexes), Pereira et al. (1985) exposed the animals
to chloroform via drinking-water at concentrations of 0 or 1800
mg/litre until they were 51 weeks of age, after which the animals
were sacrificed. The chloroform treatment did not affect the liver
or lung tumour frequency in the females and the lung tumour
frequency in the males. However, the liver tumour frequency in the
males appeared to be reduced after the treatment.
Capel et al. (1979) administered chloroform as a drinking-water
solution (estimated daily doses of 0, 0.15 or 15 mg/kg body weight)
to male mice either from 14 days before or from 14 days before to 14
days after intraperitoneal injection with Ehrlich ascites cells (TO
strain), subcutaneous injection with B16 melanoma cells (C57BL
strain) or intramuscular injection with Lewis lung carcinoma cells
(C57BL strain). Chloroform treatment enhanced the growth of Ehrlich
ascites cells (measured as intraperitoneal tumour cell DNA) at the
high dose level. In comparison with the controls, more animals
receiving chloroform at both dose levels had organs invaded with B16
melanoma cells. Lewis lung tumour growth, measured as primary tumour
size or pulmonary metastases, was not significantly enhanced at
low-dose chloroform treatment, but after treatment with the high
dose the number of pulmonary metastases and tumour size were
markedly increased.
In a two-stage (initiation/promotion) treatment protocol,
Klaunig et al. (1986) studied the effect on liver tumour incidence
in male B6C3F1 mice (35/group) after continuous treatment with 600
and 1800 mg chloroform/litre drinking-water for 52 weeks to
determine if chloroform expresses its hepatocarcinogenicity through
tumour promotion mechanisms. Two groups received 600 and 1800 mg
chloroform/litre drinking-water containing diethylnitrosamine (DENA;
10 mg/litre) during the first 4 weeks of exposure. Two other groups
received 600 and 1800 mg chloroform/litre drinking-water without
DENA. The DENA groups constituted the initiated groups. One
initiated and one non-initiated control group were included.
Chloroform did not affect the incidence of liver or lung tumours by
itself, and even inhibited liver and lung tumorigenesis in the
DENA-initiated mice, compared with DENA treatment alone.
7.7.4.2 Rats
Deml & Oesterle (1985, 1987) studied the ability of chloroform
to promote the development of liver tumours. Female Sprague-Dawley
rats were initiated for liver tumours by administration of a single
dose of 8 mg dimethyl nitrosamine/kg body weight. This was followed
by administering chloroform (25, 100, 200 and 400 mg/kg body weight)
in an olive oil vehicle twice weekly for 11 consecutive weeks. There
was a dose-related increase of ATPase-negative, gamma-glutamyl
transpeptidase (GGTase)-positive and glycogen-storing foci of cells
within the liver. For example, ATPase-deficient foci were increased
from approximately 2-fold to 5-fold by doses of 100 and 400 mg/kg,
respectively. These data demonstrate that chloroform in an oil
vehicle will probably promote development of hepatic tumours in
rats.
Herren-Freund & Pereira (1986) evaluated the ability of
chloroform to act as an initiator, promoter and co-carcinogen in
B6C3F1 mice and male Sprague-Dawley rats. In rats, the initiator
was administered 18-24 h following a two-thirds partial hepatectomy.
Diethylnitrosamine (0.5 mmol/kg body weight) was used as the
positive control for initiation and phenobarbital (500 mg/litre
drinking-water) was used as the positive control for promotion.
Ethylnitrosourea (ENU) was the positive control for initiator in
15-day-old mice and phenobarbital (500 mg/litre drinking-water) was
used as the positive control for promotion. Chloroform was
administered as a single dose of 180 and 360 mg/kg body weight as an
initiator (no vehicle) in the rat and 1800 mg/litre drinking-water
for 48 weeks as a promoter. There was no evidence that chloroform
was able to act as an initiator in rats. Moreover, it did not act as
a tumour promoter in either mice or rats, but actually decreased the
numbers of hepatic tumours induced in neonatal mice by ENU.
Concurrent administration of chloroform and DENA to the rat had no
significant effect on foci or tumour development in rats. These data
further suggest that the corn oil vehicle is important to the
hepatocarcinogenic effects of chloroform.
In a previous experiment, Pereira et al. (1982) had examined
the effect of chloroform as an initiator and promoter. Chloroform
was administered at 180 mg/kg body weight in a single dose as an
initiator and 180 mg/kg body weight twice a week for 53 days as a
promoter. In this case, tricaprylin was the vehicle. Chloroform had
no activity as an initiator. There was a small, but statistically
significant, increase in the numbers of GGTase-positive foci in the
promotion study.
Although chloroform is an established rodent carcinogen,
several studies have shown that chloroform administered in impolar
solvents also has anti-cancer properties as it inhibits tumour
growth in mouse liver and in the gastrointestinal tract of the rat
(Pereira et al., 1985; Daniel et al., 1989).
Chloroform administered in drinking-water (0, 900 and 1800
mg/litre) to Fischer-344 rats significantly decreased
gastrointestinal (GI) tumours that were initiated by a single 200
mg/kg dose of dimethyl hydrazine (DMH) (Daniel et al., 1989). GI
tumour incidence was 14/39 in animals treated with DMH alone and
5/39 and 5/40 in the groups in which DMH treatment was followed by
900 and 1800 mg chloroform/litre, respectively, for 39 weeks.
Chloroform also inhibits the propensity for three
gastrointestinal tract carcinogens, benzo (a)pyrene (BAP),
1,2-dimethylhydrazine (DMH) and methylnitrosourea (MNU), to induce
nuclear anomalies in the proximal colon of B6C3F1 mice (Daniel et
al., 1991). These authors found that in mice pre-adapted to 1800 mg
chloroform/litre drinking-water for 30 days prior to the carcinogen
administration the level of nuclear anomalies induced in the
proximal colon was reduced by four-fold for BAP and two-fold for
both MNU and DMH. In the duodenum, chloroform was effective at
inhibiting unclear anomalies only for MNU.
Reddy et al. (1992) demonstrated that chloroform inhibits the
development of diethylnitrosamine-initiated, phenobarbital-promoted
gamma-glutamyl transpeptidase and placental form
glutathione- S-transferase-positive foci in the liver of male
Fischer-344 rats. They suggested that chloroform exerts its focal
inhibitory effect by selectively killing the putative initiated
cells.
The lack of initiating activity in these initiation-promotion
assays supports the conclusion that chloroform is non-genotoxic
(section 7.6), and also indicates that the carcinogenic action of
chloroform is attributable to a non-genotoxic/cytotoxic mode of
action (sections 7.2.1.1 and 7.7). Interestingly, more of the above
studies reported that chloroform inhibited the growth or formation
of precancerous or cancerous cells than those that reported that
chloroform had promotional activity.
7.8 In vitro studies
In vitro studies frequently provide insight into how
chemicals induce cytotoxic effects. However, at high concentrations
(e.g., 5 mM and above), the solvent effects of chloroform on cell
membranes complicate the interpretation of these experiments. The
preparations that have been studied are precision-cut slices taken
from the liver, primary hepatocytes suspensions and cultures.
Azri-Meehan et al. (1992) studied the cytotoxic effects of
chloroform in liver slices taken from phenobarbital-treated rats. No
comparison was made with non-induced animals. Concentrations in the
range of 0.5 to 1.6 mM induced loss of intracellular potassium and
glutathione. Reduced mitochondrial function (measured as decreases
in dye reduction) was observed in the same dose range. A
concentration of 0.2 mM had no effect.
Glende & Recknagel (1992) examined the ability of a number of
chlorinated hydrocarbons to activate phospholipase A2, presumably
through damage to calcium-binding sites in the endoplasmic
reticulum. At doses that induce 30 to 70% release of cellular
lactate dehydrogenase (i.e. 9.8 mM), chloroform did activate
phospholipase A2. This concentration is similar to that necessary to
destroy the calcium-binding capacity of the endoplasmic reticulum.
O'Hara et al. (1991) examined the effects of chloroform on the
viability of hepatocytes in suspension (measured by potassium
retention). These hepatocytes were isolated from control
phenobarbital-treated rats. The minimum concentration required to
produce an effect on potassium retention decreased from 10 mM in
control hepatocytes to 1 mM in hepatocytes obtained from induced
animals.
A number of studies of chloroform cytotoxicity in suspensions
of rat hepatocytes have been reported (Stacey, 1987). However, the
very high nominal concentrations of chloroform that were apparently
necessary to produce significant effects (i.e. 30 and 60 mM) raise
considerable questions as to their relevance to in vivo hepatic
toxicity.
An innovative approach has been developed for incubating
hepatocyte suspensions with the chemical of interest, followed by
observation of the cytotoxic response after placing the treated
cells into culture (Kedderis et al., 1993a). Such cytotoxicity was
observed when hepatocyte suspensions derived from B6C3F1 mice were
incubated with concentrations of chloroform between 1.3 and 3.8 mM.
These concentrations were consistent with peak liver concentrations
expected with the high doses of chloroform utilized in the
assessment of chloroform carcinogenicity in mice (NCI, 1976a,b), as
predicted by the Corley et al. (1990) pharmacokinetic model
(Kedderis et al., 1993b). The cytotoxicity of chloroform was
potentiated by pretreating the mice with acetone to induce
cytochrome P450 2E1.
Although there have been substantial advances in the study of
in vitro chloroform toxicity, the applicability of the results
that are available to date to estimate hazards in humans remains to
be established.
7.9 Factors modifying toxicity; toxicity of metabolites
The in vivo toxicity of chloroform is modified by a range of
factors. The rate of its biotransformation is a significant
determinant of its toxicity. Hence, factors that increase or
decrease chloroform biotransformation may alter the intensity of
chloroform-induced toxicity. The activities of the cytochrome P450
isoforms that catalyse the biotransformation of chloroform differ
among species and between sexes of experimental animals. Moreover
the activities of the enzymes that metabolize chloroform may be
increased or decreased by exposure to chemicals, and exposure to
chloroform itself may alter chloroform metabolism.
In addition to differences in the rates of chloroform
bioactivation, treatments that alter susceptibility are also
important determinants of chloroform-induced toxicity. Cellular
glutathione concentrations are an important determinant of
susceptibility, and perturbations of glutathione homeostasis may
affect markedly the toxicity of chloroform. Finally, for some of the
treatments that alter chloroform toxicity discussed in this section,
the mechanistic basis of these interactions is not well understood.
Brown et al. (1974a) reported that inhalation exposure of
phenobarbital-treated male Sprague-Dawley rats to chloroform at
doses of 2.45 or 4.9 g/m3 (500 or 1000 ppm) for 2 h produced
marked centrilobular necrosis that was accompanied by decreased
hepatic glutathione concentrations. in vitro studies showed that
glutathione reduced the covalent binding of [14C]-chloroform
metabolites to microsomal protein.
Docks & Krishna (1976) observed that administration of
chloroform decreased hepatic glutathione concentrations in
phenobarbital-treated rats (male, Sprague-Dawley), but not in
control animals 1 to 2 h after administration, and caused liver
necrosis. Administration of isopropanol or acetone, which increased
the covalent binding of chloroform metabolites (Sipes et al., 1973),
did not alter hepatic glutathione concentrations.
Starvation and carbohydrate restriction increase the in vivo
metabolism of chloroform and its hepato- and nephrotoxicity in rats
(Nakajima & Sato, 1979; McMartin et al., 1981; Nakajima et al.,
1982). In contrast, protein deficiency does not alter chloroform
toxicity (McLean, 1970).
Several authors have demonstrated that administration of
alcohols, including ethanol (Kutob & Plaa, 1962b; Sato et al., 1980,
1981), or ketones increases chloroform metabolism and
hepatotoxicity. An extension of these studies to include a range of
alcohols showed that methanol, ethanol, isopropanol, tert-butanol,
pentanol, hexanol, octanol and decanol all potentiate
chloroform-induced liver injury and lower the LD50 of chloroform
in male Sprague-Dawley rats (Ray & Mehendale, 1990). Aliphatic
ketones, including acetone, 2-butanone, 2-pentanone, 2-hexanone,
2,5-hexanedione, 2-heptanone and methyl isobutyl ketone, also
increase chloroform-induced hepatotoxicity (Hewitt et al., 1990;
Vézina et al., 1990), but treatment with 2-hexanone does not
increase chloroform-dependent lipid peroxidation either in vivo or
in vitro (Cowlen et al., 1984a,b). The potentiating effect of
alcohols and ketones in chloroform-induced hepatotoxicity is
attributed to an increase in the activity of the cytochromes P450
that metabolize chloroform (Koop et al., 1982; Ryan et al., 1986;
Brady et al., 1989; Vézina et al., 1990).
Harris et al. (1982) evaluated, by the intraperitoneal route,
the toxicity of chloroform (0.2 ml/kg body weight) and carbon
tetrachloride (0.1 ml/kg body weight) given alone or together to
male rats. At these doses, neither chloroform nor carbon
tetrachloride produced toxicity, but increases in SGPT activity and
hepatic triglyceride and calcium concentrations were seen when both
compounds were given together. Ikatsu & Nakajima (1992) showed that
a single inhalation exposure to 490 mg/m3 (100 ppm) chloroform for
8 h resulted in mid-zonal hepatotoxicity. In ethanol-treated rats
exposed to both chloroform (50 ppm) and carbon tetrachloride (10
ppm), liver necrosis and elevated plasma GOT/GPT activities were
observed. These findings indicate that the toxicity of chloroform is
elevated in the presence of carbon tetrachloride. O'Hara et al.
(1991) studied the effect of chloroform and carbon tetrachloride in
rat hepatocytes and demonstrated that the combined toxicity of both
compounds was greater than additive.
The pesticide kepone (chlordecone), but not its non-ketonic
analogue mirex, increases chloroform-induced hepato- and
nephrotoxicity (Hewitt et al., 1979, 1982; Iijima et al., 1983). In
Mongolian gerbils, which are susceptible to chloroform-induced
toxicity (200 or 500 µl/kg body weight, intraperitoneal), treatment
with phenobarbital or chlordecone decreased the hepatotoxicity of
chloroform (Ebel et al., 1987); in contrast, rats given 50 to 500
µl/kg body weight chloroform (intraperitoneally) showed little
hepatotoxicity, but toxicity was increased after treatment with
phenobarbital or chlordecone.
The drinking-water contaminants dichloroacetic acid (DCA) and
trichloroacetic acid (TCA) potentiate chloroform toxicity (Davis,
1992). Male and female rats (Sprague-Dawley) were orally treated
with 0.92 or 2.45 mmol/kg body weight DCA or TCA and were given 0.75
mg/kg body weight chloroform (intraperitoneally) 3 h later.
Increases in plasma ALAT activities were observed in female, but not
in male rats 24 and 48 h after giving DCA; in contrast, plasma ALAT
activities were increased 24, but not 48 h, after giving TCA in both
male and female rats. DCA administration increased blood urea
nitrogen concentrations in female rats, but produced little effect
in male rats, whereas TCA administration produced an effect only in
female rats 48 h after treatment. The mechanism of the effect of DCA
and TCA was not elaborated.
Monochloroacetic acid (MCA) given by gavage to male (188 mg/kg
body weight) or female (94 mg/kg body weight) Sprague-Dawley rats
one hour before giving chloroform (520 mg/kg body weight)
intraperitoneally increased chloroform-induced hepatotoxicity in
male rats, but had little effect in female rats (Davis & Berndt,
1992). Treatment with MCA alone decreased glomerular filtration
rates in female rats. The mechanism by which MCA potentiated
chloroform toxicity was not elucidated.
Temporal variations in chloroform-induced hepatotoxicity have
been observed in rats (Lavigne et al., 1983). Male Sprague-Dawley
rats were given chloroform (0.5 ml/kg body weight) intraperitoneally
at 9:00, 13:00, 17:00, 21:00 or 03:00 h and were killed 4 h after
treatment. Hepatotoxicity, as assessed by serum GPT, GOT and LDH
activities, was minimal and maximal at 09:00 h and 21:00 h,
respectively, whereas glucose-6-phosphatase activity was decreased
at 03:00 h and 13:00 h. When rats were starved for 16 h before
giving chloroform at 09:00 h, toxicity was increased substantially.
Charbonneau et al. (1991) studied the effect of acetone
treatment on the toxicity with a range of binary mixtures of
haloalkanes in rats. An increased hepatotoxic response was observed
with binary mixtures containing chloroform, carbon tetrachloride,
1,1,2-trichloroethane or 1,1-dichloroethylene.
In vitamin-A-deficient rats, serum ALAT and particularly ASAT
activities were increased after intraperitoneal administration of
chloroform, compared to control rats (Savoure et al., 1992).
8. EFFECTS ON HUMANS
8.1 Acute non-lethal effects
Chloroform is irritating to mucous membranes, producing
gastroenteritis with persistent nausea and vomiting. Symptoms
following ingestion of chloroform are similar to those following
inhalation (van der Heijden et al., 1986).
Cases of severe intoxication after suicidal attempts, with the
same pattern of symptoms as after anaesthetical use, have been
reported by Schröder (1965). There are considerable inter-individual
differences in susceptibility. Some persons presented serious
illness after an oral dose of 7.5 g of chloroform, whereas others
survived a dose of 270 g chloroform. The mean lethal dose for an
adult is estimated to be about 45 g (Winslow & Gerstner, 1978).
Rao et al. (1993) successfully managed acute toxicity from
chloroform in a 33-year-old white woman who attempted suicide by
injecting 0.5 ml of chloroform, and then drank half a cup the next
morning. Plasma chloroform levels, measured by headspace GC,
declined rapidly. Sequential measurement of biomarkers in serum for
liver cell necrosis, liver function and liver regeneration indicated
the presence of initial liver damage followed by recovery. The
authors suggested that, in addition to biomarkers for liver
necrosis, serial determinations of markers for liver regeneration
provide objective evidence for recovery from chloroform poisoning.
It has been reported that chloroform can cause severe toxic
effects in humans exposed to 9960 mg/m3 (2000 ppm) for 60 min,
symptoms of illness at 2490 mg/m3 (500 ppm) and can cause
discomfort at levels below 249 mg/m3 (50 ppm) (Verschueren, 1983).
Most data on the controlled exposure of man to chloroform have
resulted from its clinical use as an anaesthetic. This use of
chloroform was described as early as 1847 (Simpson, 1847). Induction
of anaesthesia may result from inhalation of chloroform vapours at a
concentration of 24 to 73 g/m3 air. For maintenance of
anaesthesia, concentrations in the range of 12 to 48 g/m3 are
required. As with animals, chloroform anaesthesia may result in
death in humans due to respiratory and cardiac arrhythmias and
failure. Because of the relatively high frequency of "late
chloroform poisoning" (liver toxicity), its use as anaesthetic has
been abandoned.
Other effects related to chloroform inhalation are: increase in
the rate and depth of respiration during induction and light
anaesthesia, minute volume decrease in deep anaesthesia,
hypothermia, depletion of adrenal adrenaline content, hypotension,
depression of gastrointestinal tract motility, respiratory acidosis,
hyperglycaemia, ketosis, constriction of the spleen, increase in the
number of leucocytes (especially polymorphonuclear cells), a
decrease in clotting time and an increase in prothrombin time. The
characteristics and severity of the effects depend on depth and
duration of anaesthesia (Adriani, 1970).
The cardiac effects might be secondary and due to hypoxia,
caused by depression of respiratory activity. No studies have been
found in which this problem has been investigated in man (e.g., by
forced respiration), but Taylor et al. (1976) obtained indications
that chloroform itself produces cardiovascular disturbances in
rabbits (viz. disturbances in left ventricular functioning and an
increase in peripheral resistance; see section 7.1.2.3) after
exposure to 244 mg/m3 for 1 min.
In man, as well as in animals, renal tubular necrosis and renal
dysfunction (anuria, proteinuria, uraemia, increase in blood urea
nitrogen) have been observed (Kluwe, 1981). Recovering from
chloroform anaesthesia, some patients may show the symptoms of a
delayed chloroform poisoning several days later. Prostration,
protracted nausea, vomiting, jaundice and coma due to hepatic
dysfunction are observed. The patient may die within 5 days after
anaesthesia. At autopsy, degeneration and necrosis of liver tissue
have been found (Goodman & Gilman, 1970). In general the symptoms
appear to be similar to those observed in animals.
According to Oettel (1936) and Winslow & Gerstner (1978),
exposure to concentrated chloroform vapours causes a stinging
sensation in the eye. Splashing of the liquid into the eye evokes
burning, pain and redness of the conjunctival tissue. Occasional
injury of the corneal epithelium will recover fully within a few
days. Dermal contact with chloroform causes chemical dermatitis
(symptoms: irritation, reddening, blistering and burns).
8.2 Epidemiology
8.2.1 Occupational exposure
Challen et al. (1958) reported the effects of exposure of
workers (mostly female) to chloroform vapour in a factory during
manufacture of lozenges containing the chemical. Eight workers (four
working full-time and four half-time) were exposed to chloroform
concentrations of 375 to 1330 mg/m3, with a peak concentration of
5680 mg chloroform/m3, for periods of 3 to 10 years. The symptoms
reported were lassitude, thirst, gastrointestinal distress, frequent
and scalding urination, lack of concentration, depression and
irritability. The management stated that some of the employees had
been noticed staggering about at work. Nine other workers (one
full-time, eight half-time), who were exposed to chloroform
concentrations of 110 to 350 mg/m3 for 10 to 24 months, suffered
from the same complaints as stated above, but to a lesser degree.
Several liver function tests did not reveal signs of liver toxicity,
but these tests were not very sensitive.
Bomski et al. (1967) investigated the occurrence of hepatitis
in a chemical factory in relation to the occurrence of this disease
in the city where the factory was located. The 68 workers were
exposed to occupational chloroform concentrations of 10 to 1000
mg/m3 for 1 to 4 years. In this group of employees, a higher
frequency of hepatitis was found than in the city inhabitants.
Seventeen workers showed hepatomegaly and in three of them hepatitis
was observed. Ten workers showed splenomegaly, but the cause of the
splenomegaly was not discussed.
The finding of a high frequency of hepatitis among
occupationally chloroform-exposed workers, as compared to that in
the city inhabitants, is supported by a recent report on a
16-year-old patient who attempted suicide by ingesting chloroform.
This led to the development of toxic hepatitis (Hakim et al., 1992).
In a study by Phoon et al. (1975), the air in the workroom of
13 persons with jaundice originally diagnosed as having viral
hepatitis was analysed for chloroform. The chloroform concentration
in the workroom appeared to be more than 1950 mg/m3. The period of
exposure was less than 6 months. Because no worker had a history of
fever and there was no relation to past medical history, it was
concluded that the original diagnosis must have been wrong and
should have been toxic jaundice. Five of the people with jaundice
and four other colleagues had blood chloroform levels in the range
of 1 to 2.9 mg/litre.
In another factory 18 cases of what seemed to be hepatitis B
were reported (Phoon et al., 1983). Investigation of the
occupational environment revealed a constant exposure to chloroform,
with concentrations in the range of 80 to 160 mg/m3. The exposure
period of these workers was less than 4 months and the conclusion
was drawn that these were cases of toxic jaundice related to
chloroform exposure, because no infection with the hepatitis B virus
could be established.
A historical mortality study was carried out by Linde & Mesnick
(1979). They investigated the cause of death of white male
anaesthesiologists, who were occupationally exposed to chloroform
vapours (extent of exposure was unknown). The death certificates
used were of persons, who were presumed to be exposed during the
1880-1890 period and who died during the 1930-1946 period.
Comparison of their death certificates with those of several control
groups did not exclude a possible association between cancer and
chloroform exposure.
8.2.2 General exposure
There have been numerous reports over the last 15 years which
have evaluated the relationship between chlorinated water and the
incidence of cancer. Chloroform is but one of many by-products
produced by reaction of chlorine with naturally occurring material
in source waters (Bull & Kapfler, 1991). Many of these studies noted
increased risk of cancer which at least partially fulfilled criteria
for causality (e.g., consistency, specificity and temporal
relationships).
IARC (1991) reviewed the available studies and concluded the
strongest evidence of increased risk related to exposure to
chlorinated surface water relative to unchlorinated ground water for
the incidence of cancer of the urinary bladder. However, the
weight-of-the-evidence evaluation by IARC concluded that there is
inadequate evidence for the carcinogenicity of chlorinated
drinking-water in humans.
Morris et al. (1992) conducted a meta-analysis which attempted
to integrate quantitatively the results of previously published
studies in which individual exposures were evaluated (i.e. case
control and cohort studies). The authors identified increased rates
of bladder and colo-rectal cancer in individuals exposed to
chlorinated surface water, which appeared to exhibit a dose-related
trend. Although this study was confounded by substantial differences
in exposure variables that occur in different water supplies, higher
risk rates were estimated when the analysis was restricted to those
studies which were judged to have the highest quality exposure
assessments. Because of the confounding of these results by chlorine
residual levels and a multiplicity of other chemicals which are
animal carcinogens and mutagens, none of the drinking-water studies
specifically implicate chloroform as a human carcinogen.
Kramer et al. (1992) studied the association between exposure
to trihalomethanes in the water supply and adverse reproductive
outcomes in the state of Iowa (USA). Estimations of chloroform
exposure were based on municipal water surveys. After adjustment for
maternal age, parity, prenatal care, marital status, education and
maternal smoking, an increased risk for intrauterine growth
retardation (abnormally low birth weight) was associated with
chloroform concentrations above 10 µg/litre. Limitations of the
study involve the ascertainment and classification of exposures to
trihalomethanes (such as fluctuation of levels and exposure at
individual level) and the influence of potential confounding
influences of unmeasured contaminants.
8.3 Abuse and addiction
Exposure to chloroform may result in euphoria and therefore
people expose themselves to chloroform by drinking the liquid or
sniffing the vapours (Storms, 1973). Addiction to chloroform and
chloroform-containing cough syrups has been reported by Heilbrunn et
al. (1945) and Conlon (1963). According to Heilbrunn et al. (1945),
addicts tolerated very high daily doses and presented neurological
symptoms and degenerative changes in the brains.
After an intravenous injection of 7.5 g of chloroform, a
patient showed signs of pulmonary malfunction and haemolysis. In
this case, kidney or liver toxicity was not reported (Timms et al.,
1975).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Freshwater organisms
The data on the toxicity of chloroform to several freshwater
organisms are listed in Table 14.
Due to the volatility of chloroform, caution must be exercised
in interpreting the test results, particularly those in open static
systems where no chemical analysis of the actual concentration was
carried out.
9.1.1 Short-term toxicity
The chemical is of low toxicity to unicellular plants and other
microorganisms (concentration range of initial population growth
inhibition: 125 to > 3200 mg/litre). Chloroform is moderately toxic
to Daphnia magna (LC50 = 29 mg/litre).
The LC50 values for several species of fish are in the range
of to 191 mg/litre. However, initial toxicity may occur at lower
levels: the no-observed-lethal concentrations (NOLCs) for Salmo
gairdneri and Lepomis macrochirus appear to be 8 and 3 mg/litre,
respectively. At lower concentrations (< 13 mg/litre), Salmo
gairdneri shows loss of equilibrium, slow operculum movement and
narcosis (Anderson & Lusty, 1980).
In Gasterosteus aculeatus, chloroform produced anaesthesia
which could be maintained for at least 90 min at concentrations of
210 mg/litre. Exposure to concentrations higher than 300 mg/litre
resulted in decreased oxygen consumption and death (Jones, 1947),
whereas concentrations lower than 120 mg/litre excited the animals
and gave rise to considerable higher oxygen uptake.
Chloroform is considerably more toxic to the juvenile stages of
several species of amphibians. In a continuous-flow system, Birge et
al. (1980) tested the toxicity of chloroform to embryo-larval stages
of several species of amphibians after exposure for 7-9 days (Table
14). Hyla crucifer appeared to be the most susceptible species. An
effect was found on the hatching rate of the embryos, which declined
from 97% at 8 µg/litre to 4% at 7340 µg/litre. In addition there was
some evidence of teratic larvae. During the 4 days post-hatching the
LC50 declined from 760 to 270 µg/litre. The other species tested
were less affected and only Rana pipiens showed a high
teratogenicity frequency in the offspring (100% at 27 mg/litre at
18% hatching rate).
Table 14. Chloroform toxicity to water organisms
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Short-term toxicity
Bacteria
Pseudomonas 25 acc.d Bringmann & S - 16 h initial reduction of 125 Bringmann &
putida Kühn (1977) cell multiplication Kühn (1977)
Pseudomonas 25 acc.d Bringmann S - 16 h initial change of 125 Bringmann (1973)
fluorescens (1973) culture turbidity
Algae
Microcystis 27 acc.d Bringmann S - 192 h initial reduction of 185 Bringmann (1975)
aeruginosa (1975) cell multiplication
Scenedesmus 25 acc.d Bringmann & S - 192 h initial reduction of 1100 Bringmann &
quadricauda Kühn (1977) cell multiplication Kühn (1977)
Haematococcus 20 acc.d Tümpling S - 4 h 10% reduction of 440 Knie et al. (1983)
pluvialis (1972) oxygen production
Protozoans
Entosiphon 25 Bringmann (1978) S - 72 h initial reduction of > 6560 Bringmann (1978)
sulcatum cell multiplication
Uronema 25 Bringmann & S - 20 h initial reduction of > 6560 Bringmann &
parduczi Kühn (1980) cell multiplication Kühn (1980)
Chilomonas 20 Bringmann et al. S - 48 h initial reduction of > 3200 Bringmann et al.
paramaecium (1980) cell multiplication (1980)
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Crustaceans
Daphnia magna 22 reconstituted well S - 48 h LC50 29 LeBlanc (1980)
water, pH 7,
hardness 173 mg
CaCO3/litre
Daphnia magna 19.8-20.9 lake water, pH S - 48 h LC50 65.7 Gersich et al.
8.0, hardness (1986)
157 mg CaCO3/litre
Daphnia magna 23 distilled water S - 48 h LC50 78.9 Abernethy et al.
(1986)
Fish
Cyprinus carpio 26 filtered well water S A until LC50 97 Mattice et al.
(mixed gametes) hatching (1981)
(3-5 days)
Pimephales 25 carbon filtered S - 96 h LC50 129 Mayes et al.
promelas lake water, pH (1983)
(10-15 days) 7.6-8.3, hardness
125 mg CaCO3/litre
(30-35 days) 22 idem S - 96 h LC50 171
(60-100 days) 22 idem S - 96 h LC50 103
Brachydanio 20 dechlorinated CF - 48 h LC50 100 Slooff (1979)
rerio tap water, pH 8,
hardness 10 d.H.
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Salmo 20 dechlorinated CF - 48 h initial reduction 20 Slooff (1979)
gairdneri tap water, pH 8, of respiration
hardness 10 d.H. frequency
Leuciscus 20 acc.d Mann (1975) S - 48 h LC50 162-191 Juhnke &
idus melanotus Lüdemann (1978)
Carassius 5 aerated tap water S - 1 h EC50 97-167 Cherkin &
auratus (anaesthesia) Catchpool (1964)
20 aerated tap water S - 1 h EC50 167
(anaesthesia)
Salmo 19 aerated river water CF A 96 h LC50 18 Anderson & Lusty
gairdneri NOLC 8 (1980)
Leopomis 19 aerated river water CF A 96 h LC50 18 Anderson & Lusty
macrochirus NOLC 3 (1980)
Micropterus 19 aerated river water CF A 96 h LC50 51 Anderson & Lusty
salmoides NOLC 39 (1980)
Ictalurus 19 aerated river water CF A 96 h LC50 75 Anderson & Lusty
punctatus NOLC 68 (1980)
Amphibians
Hyla crucifer 20.5 acc.d Birge et al. CF A until 4 LC50 0.3 Birge et al. (1980)
(eggs; 2 to (1979); pH 7.6, days after
6h post- hardness 107 mg hatching
spawning) CaCO3/litre or death
(7 days in
total)
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Amphibians (contd)
20.5 idem CF A idem NOLC 0.009
Rana pipiens 20.5 idem CF A idem (9 LC50 4.2 Birge et al. (1980)
(eggs; 30 min days in
after total)
fertilization) 20.5 idem CF A idem NOLC 0.16
Rana palustris 21.5 acc.d Birge et al. CF A idem (8 LC50 20.6 Birge et al. (1980)
(eggs; 2 to (1979); pH 7.6, days in
6 h post- hardness 104 mg total)
spawning) CaCO3/litre
21.5 idem CF A idem NOLC 0.33
Bufo fowleri 21.5 idem CF A idem (7 LC50 35.1 Birge et al. (1980)
(eggs; 2 to days in
6 h post- total)
spawning) 21.5 idem CF A idem NOLC 0.33
Long-term toxicity
Fish
Poecilia 22 Alabaster & Abram Sb - 14 days LC50 102 Könemann (1981)
reticulata (1964)
Salmo 13.5 ± 1 acc.d Birge et al. CF A until 4 LC50 2.0 Birge et al. (1979)
gairdneri (1979); pH 7.3, days after
(eggs; 20 min hardness 48 mg hatching
after CaCO3/litre or death
fertilization (27 days
totally)
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
13.5 ± 1 idem CF A idem NOLC 0.004
13.5 ± 1 idem, hardness 210 CF A idem LC50 1.24
mg CaCO3/litre
13.5 ± 1 idem CF A idem NOLC 0.003
a S = static, CF = continuous flow;
b static conditions but test water changed every 24 h
c A = concentration of test compound analysed during assay; - = no data
d acc. = according to the medium described in these references
9.1.2 Long-term toxicity
Birge et al. (1979) tested the toxicity of chloroform for
embryo-larval stages of Salmo gairdneri (Table 14) after 27 days.
The chemical was especially toxic for the unhatched embryos (LC50
is about 2 mg/litre), but did not cause death in the larvae at
concentrations up to 10.6 mg/litre. The occurrence of teratic
survivors in the hatched population increased from 3% at 56 µg/litre
to 40% at 10 mg/litre.
9.2 Marine organisms
The acute toxicity of chloroform to Artemia salina was tested
by Robinson et al. (1965). The observed effect was anaesthesia and
the EC50 value was 68 mg/litre after 10 h of exposure in
artificial sea water in closed containers under static conditions.
The 50% immobilization concentration (IC50) of chloroform for
Artemia salina nauplii, subjected to salinity stress, was
determined in a static study using artificial sea water by Foster &
Tullis (1985). The toxicity test began 30 h after hatching had
commenced and lasted for 24 h. The IC50 was 37 mg/litre.
Stewart et al. (1979) tested the acute toxicity of chloroform
to larvae of Crassostrea virginica. Chemical analysis showed a
rapid decline of chloroform concentrations in the sea-water medium.
The estimated LC50 was 1 mg/litre.
Pearson & McConnell (1975) tested the acute toxicity to
Limanda limanda in a continuous-flow system containing natural sea
water and obtained an LC50 of 28 mg/litre.
Cowgill et al. (1989) determined the sensitivity of the marine
diatom Skeletonema costatum to chloroform after exposure for 5
days under static conditions. The EC50 values calculated were 477
mg/litre and 437 mg/litre based on total cell count and total cell
volume, respectively. The NOEC was 216 mg/litre.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Based on estimates of mean exposure from various media, the
general population is exposed to chloroform principally in food
(approximately 1 µg/kg body weight per day), drinking-water
(approximately 0.5 µg/kg body weight per day) and indoor air (0.3 to
1 µg/kg body weight per day). Estimated intake from outdoor air is
considerably less (0.01 µg/kg body weight per day). The total
estimated mean intake for the general population is approximately 2
µg/kg body weight per day. Available data also indicate that water
use in homes contributes considerably to levels of chloroform in
indoor air and to total exposure. For some individuals living in
dwellings supplied with tap water containing relatively high
concentrations of chloroform, estimated total intakes are up to 10
µg/kg body weight per day.
Workers may be exposed to chloroform during, for example, the
production of chloroform itself, the synthesis of substances derived
from chloroform (for example, chlorodifluoromethane), and the use of
chloroform as a solvent, and also as a consequence of its formation
in paper bleaching and sewage treatment facilities. For example,
based on a national survey conducted from 1981 to 1983, NIOSH
estimated that approximately 96 000 workers in the USA are
potentially exposed to chloroform.
10.1.2 Health effects
The most important effects of chloroform are those on the liver
and kidney. These effects are associated with the metabolism of
chloroform to the reactive intermediate, phosgene. There are
substantial interspecies and sex differences in the rates at which
chloroform is metabolized. Data also indicate that reductive
metabolism differs among species.
The most universally observed toxic effect of chloroform is
damage to the liver. The severity of these effects per unit dose
administered depends on the species, the vehicle and the method by
which the chloroform is administered. The lowest dose at which liver
damage has been observed is 15 mg/kg body weight per day,
administered to beagle dogs in a toothpaste base over a period of
7.5 years. Effects at lower doses were not examined. Somewhat higher
doses are required to produce hepatotoxic effects in other species.
Although duration of exposure varied in these studies,
no-observed-adverse-effect levels (NOAELs) ranged between 15 and 125
mg/kg body weight per day.
Effects on the kidney have been observed in male mice of
sensitive strains and in F-344 rats. Severe effects have been
observed in a particularly sensitive strain of male mice at doses as
low as 36 mg/kg body weight per day.
Daily 6-h inhalation of chloroform for 7 days consecutively
induced atrophy of Bowman's glands and new bone growth in the nasal
turbinates of F-344 rats. The NOEL for these effects was 14.7
mg/m3 (3 ppm). The significance of these effects is being further
investigated in longer term studies.
The weight of the available evidence indicates that chloroform
has little, if any, capability to induce gene mutation, chromosomal
damage and DNA repair. There is some evidence of low-level binding
to DNA, however. Chloroform does not appear capable of inducing
unscheduled DNA synthesis in vivo.
Chloroform induced hepatic tumours in mice when administered by
gavage in corn oil. However, when similar doses were administered in
drinking-water to mice, hepatic tumours were not induced.
The carcinogenic effects of chloroform on the mouse liver
appear to be closely related to cytotoxic and cell replicative
effects. The effects on cell replication paralleled variations in
carcinogenic responses to chloroform due to vehicle and method of
administration. It is of interest, in this regard, that chloroform
administered in drinking-water was incapable of promoting, but
rather inhibited, the development of liver tumours in mice.
Chloroform does not appear capable of initiating liver tumours or
inducing unscheduled DNA synthesis in the mouse liver. It would
appear, therefore, that cytotoxicity followed by cell replication
with prolonged administration of chloroform is associated with the
development of liver tumours in mice.
Chloroform induced kidney tumours in rats when administered by
gavage in corn oil. However, results for this species were similar
when the chemical was administered in the drinking-water.
Experiments in F-344 rats have indicated that chloroform could
cause damage and increase cell replication in the kidney at doses
similar to those that induce renal tumours in Osborne-Mendel rats.
These effects are produced by both oral (one single gavage) and
7-day inhalation exposure. While these results are suggestive of an
association, it is difficult to associate with any certainty the
carcinogenic response with the toxic and replicative effects.
Indeed, toxicity studies are short term and involve a rat strain
that is unusually sensitive to the nephrotoxic effects of
chloroform. This strain is different from that in which tumours were
observed.
There are some limited data to suggest that chloroform is toxic
to the fetus, but only at doses that are maternally toxic.
10.1.3 Approaches to risk assessment
The following guidance is provided as a potential basis for the
derivation of exposure limits by relevant authorities. By allocation
of the tolerable and risk-specific intakes presented below based,
for example, on the proportion of total intakes originating from
each environmental medium presented in chapter 5, limits for
exposure in drinking-water, food and air could be developed by local
authorities (WHO, in press). However, local authorities may also
wish to take into account local variations in the proportions of
exposure from various media or factors such as cost, ease and
effectiveness of control in order to develop risk management
strategies appropriate for local circumstances. However, the
ultimate objective should be reduction of total exposure from all
sources to levels below the tolerable maximum intake and
risk-specific intakes presented below. Moderate to short-term
excedence of limits based on the guidance presented below does not
necessarily imply significant risk to health and relevant public
health authorities should be contacted before taking remedial
action.
Moreover, disinfection is unquestionably the most important
step in the treatment of water for public supply. The paramount
importance of microbiological quality requires some flexibility in
the derivation of limits for exposure to chloroform in
drinking-water. Where local circumstances require that a choice must
be made between meeting microbiological limits or limits for
disinfection byproducts, the microbiological quality must always
take precedence. Efficient disinfection must never be compromised.
10.1.3.1 Non-neoplastic effects
The Task Group concluded that the data available are sufficient
to develop a tolerable intake for non-neoplastic effects of
chloroform on the basis of effects in animal species.
The lowest effect level in long-term studies in animal species
is that reported by Heywood et al. (1979) where slight
hepatotoxicity (increases in hepatic serum enzymes and fatty cysts)
was observed in beagle dogs that ingested 15 mg/kg body weight per
day in toothpaste for 7.5 years. Liver fat content was also
increased in B6C3F1 mice that ingested 34 mg/kg body weight per
day in drinking-water for 2 years (Jorgenson et al., 1985). On the
basis of these data, a tolerable daily intake (TDI) can be derived
as follows:
15 mg/kg body
weight per day
TDI = ----------------- = 0.015 mg/kg body weight per day
1000 (15 µg/kg body weight per day)
where:
* 15 mg/kg body weight per day is the lowest-identified-effect
level (slight hepatotoxicity in the study on beagle dogs by
Heywood et al., 1979);
* 1000 is the uncertainty factor (x 10 for interspecies
variation, x 10 for intraspecies variation and x 10 for use of
an effect level rather than a no-effect level).
This value is likely to be conservative. It should be noted
that no effects have been observed in adequate studies on other
species exposed to higher doses administered in other vehicles.
10.1.3.2 Neoplastic effects
The Task Group concluded that the carcinogenic effects of
chloroform should also be considered in the development of limits of
exposure.
a) Liver tumours in female B6C3F1 mice
Based on the available mechanistic data, the approach
considered most appropriate for provision of guidance based on mouse
liver tumours is division of a no-effect level for cell
proliferation by an uncertainty factor. The NOEL for cytolethality
and cell proliferation in B6C3F1 mice was 10 mg/kg body weight per
day following administration in corn oil for 3 weeks (Larson et al.,
1994a).
On the basis of these data, a tolerable daily intake is derived
as follows:
10 mg/kg body
weight per day
TDI = ------------------ = 0.01 mg/kg body weight per day
1000 (10 µg/kg body weight per day)
where:
* 10 mg/kg body weight per day is the NOEL for cytolethality and
cell proliferation in B6C3F1 mice observed in the short-term
study of Larson et al. (1994a);
* 1000 is the uncertainty factor (x10 for interspecies variation,
x10 for intraspecies variation and x 10 for severity of effect
(i.e. carcinogenicity) and less-than-chronic study).
b) Kidney tumours in male Osborne-Mendel rats
Since data on cell proliferation are not available for the
strain in which tumours were observed (Osborne-Mendel rats) and
identified information on cell proliferation and lethality are short
term (one single gavage and a 7-day inhalation exposure in F-344
rats), it was considered premature to deviate from the default model
(i.e. linearized multistage) as a basis for estimation of lifetime
cancer risk.
Based on the induction of renal tumours (adenomas and
adenocarcinomas) in male rats in the study by Jorgenson et al.
(1985), the total daily intake considered to be associated with a
10-5 excess lifetime risk, calculated on the basis of the Global
82 version of the linearized multistage model, is 0.0082 mg/kg body
weight per day (8.2 µg/kg body weight per day). A body surface area
correction was not incorporated due to the fact that chloroform is
an indirect-acting carcinogen and that the rate of metabolism is
similar in rodents and man.
10.2 Evaluation of effects in the environment
Chloroform may be released into the environment during its
production, storage, transport and use. Significant amounts of
chloroform may also enter the environment as a consequence of its
formation during some chlorination processes (e.g., chlorination of
water, paper bleaching).
Chloroform is expected to volatilize readily from surface water
and the surface of soils. It is also expected to be highly mobile in
soils and may reach ground water.
Chloroform has a residence time of several months in the
atmosphere and can therefore be transported over long distances from
the point of emission. Degradation by reaction with hydroxyl
radicals is likely to be the only significant mechanism for
decomposition of chloroform in the atmosphere. A half-life of around
60 days has been estimated for this process.
Chloroform appears to be resistant to biodegradation under
aerobic conditions but is degraded under certain anaerobic
conditions.
Chloroform is toxic to the embryo-larval stages of some
amphibian and fish species. The lowest reported LC50 is 0.3
mg/litre (4- or 7-day exposure) for the embryo-larval stages of
Hyla crucifer. It is less toxic to fish and Daphnia magna. The
LC50 values for several species of fish are in the range of 18 to
191 mg/litre. There is little difference in sensitivity between
freshwater and marine fish. The lowest reported LC50 for Daphnia
magna is 29 mg/litre (48-h exposure). Chloroform is of low
toxicity to algae and other microorganisms.
Levels of chloroform in surface water are generally low and
would not be expected to present a hazard to aquatic organisms.
However, higher levels of chloroform in surface water resulting from
industrial discharges or spills may be hazardous to the
embryo-larval stages of some aquatic species.
11. FURTHER RESEARCH
A number of further studies is considered to be necessary:
* A study of compensatory cell regeneration in the liver and
kidney of the Osborne-Mendel rat
* Determination of reactive metabolite formation in situ
* Studies on the mechanism of the species-specific
carcinogenicity of chloroform including a) the identification
of the intermediate/metabolite responsible for the
carcinogenicity of chloroform and b) its mode of action
* An inhalation carcinogenicity bioassay
* Further validation of PBPK models for chloroform with
interspecies variations, including humans and dogs
* Further studies concerning the progression of nasal lesions in
the rat
* Additional long-term toxicity tests in aquatic organisms
* in vitro cytotoxicity/metabolism studies with human tissues
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
The International Agency for Research on Cancer evaluated
chloroform in 1978 (IARC, 1979) and re-evaluated it in 1987 (IARC,
1987). The conclusions were that there is inadequate evidence for
the carcinogenicity of chloroform in humans but sufficient evidence
for its carcinogenicity in experimental animals. The overall
evaluation was that chloroform is possibly carcinogenic to humans
(Group 2B).
Chlorinated drinking-water was evaluated in 1990 (IARC, 1991)
and the overall evaluation was that chlorinated drinking-water is
not classifiable as to its carcinogenicity to humans (Group 3).
Studies with chlorinated drinking-water gave no evidence for
carcinogenicity of chloroform in humans (Group 3) (IARC, 1991).
A drinking-water guideline value of 200 µg/litre for an excess
lifetime cancer risk of 10-5 has been recommended for chloroform
by the World Health Organization (WHO, 1993).
REFERENCES
Abdel-Rahman HS (1982) The presence of trihalomethanes in soft
drinks. J Appl Toxicol, 2: 165-166.
Abernethy S, Bobra AM, Shiu WY, Wells PG, & Mackay D (1986) Acute
lethal toxicity of hydrocarbons and chlorinated hydrocarbons to two
planktonic crustaceans: the key role of organism-water partitioning.
Aquat Toxicol, 8: 163-174.
Adriani J (1970) The pharmacology of anaesthetic drugs. Springfield,
Illinois, Charles C. Thomas, pp 57-60.
Aggazzotti G, Predieri G, Fantuzzi G, & Benedetti A (1987) Headspace
gas chromatographic analysis for determining low levels of
chloroform in human plasma. J Chromatogr, 416: 125-130.
Aggazzotti G, Fantuzzi G, Tartoni PL, & Predieri G (1990) Plasma
chloroform concentrations in swimmers using indoor swimming pools.
Arch Environ Health, 45(3): 175-179.
Agustin JS & Lim-Sylianco CY (1978) Mutagenic and clastogenic
effects of chloroform. Bull Phil Biochem Sci, 1: 17-23.
Ahmadizadeh M, Echt R, Kuo C-H, & Hook JB (1984) Sex and strain
differences in mouse kidney: Bowman's capsule morphology and
susceptibility to chloroform. Toxicol Lett, 20: 161-172.
Ahmed AF, Kubic VL, & Anders MW (1977) Metabolism of haloforms to
carbon monoxide: I. in vitro studies. Drug Metab Dispos, 5:
198-204.
Allard U & Andersson L (1992) Exposure of dental personnel to
chloroform in root filling procedures. Endod Dent Traumatol, 8:
155-159.
Althaus FR, Lawrence SD, Sattler GL, Longfellow DG, & Pitot HC
(1982) Chemical quantification of unscheduled DNA synthesis in
cultured hepatocytes as an assay for the rapid screening of
potential chemical carcinogens. Cancer Res, 42: 3010-3015.
Anderson DR & Lusty EW (1980) Acute toxicity and bioaccumulation of
chloroform to 4 species of freshwater fish. Richland, WA, Battelle
Pacific North West Laboratory (NUREG/CR-0893).
Appleby A, Kazazis J, Lillian D, & Singh HB (1976) Atmospheric
formation of chloroform from trichloroethylene. J Environ Sci
Health, A11: 711-715.
Armstrong DW & Golden T (1986) Determination of distribution and
concentration of trihalomethanes in aquatic recreational and
therapeutic facilities by electron capture GC. LC-GC, 4: 652-655.
Athanasiou K & Kyrtopoulos SA (1981) Induction of sister chromatid
exchange by non-mutagenic carcinogens. NATO Adv Study Inst Ser A,
40: 557-562.
ATSDR (1993) Toxicological profile for chloroform (update). Atlanta,
Georgia, Agency for Toxic Substances and Disease Registry.
Azri-Meehan S, Mata MP, Gandafi AJ, & Brendel K (1992) The
hepatotoxicity of chloroform in precision-cut rat liver slices.
Toxicology, 73: 239-250.
Bai C-L, Canfield PJ, & Stacey NH (1992) Individual serum bile acids
as early indicators of carbon tetrachloride- and chloroform-induced
liver injury. Toxicology, 75: 221-234.
Baker SU & Bonin AM (1981) Study of 42 coded compounds with the
Salmonella/mammalian microsome assay. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 249-260 (Progress in Mutation Research,
Volume 1).
Balster RL & Borzelleca JF (1982) Behavioural toxicity of
trihalomethane contaminants of drinking water in mice. Environ
Health Perspect, 46: 127-136.
Bätjer K, Cetinkaya M, Düszeln v J, Gabel B, Lahl U, Stachel B, &
Thiemann W (1980) Chloroform emission into urban atmosphere.
Chemosphere, 9: 311-316.
Bauer U (1981) [Human exposure to environmental chemicals -
Investigations on volatile organic halogenated compounds in water,
air, food and human tissues. III. Communication: Results of
investigations.] Zbl Bakt Hyg, 1. Abt Orig, B174: 200-237 [in
German].
Baumann OE, Drangsholt H, & Carlberg GE (1981) Analysis of volatile
halogenated organic compounds in fish. Sci Total Environ, 20:
205-215.
Benoit FM & Jackson R (1987) Trihalomethane formation in whirlpool
spas. Water Res, 2: 353-357.
Bergman K (1984) Application and results of whole-body
autoradiography in distribution studies of organic solvents. Crit
Rev Toxicol, 12: 59-119.
Birge WJ (1980) Effects of organic compounds on amphibian
reproduction. Lexington, University of Kentucky, Water Resources
Research Institute (Research Report No. 121; PB80-147523).
Birge WJ, Black JA & Bruser DM (1979) Toxicity of organic chemicals
to embryo-larval stages of fish. Washington, DC, Environmental
Protection Agency (EPA-560/11-79-007; PB80-101637).
Blancato JN & Chiu (1994) Use of pharmacokinetic models to estimate
internal doses from exposure. In: Wang R ed. Water contamination and
health. New York, Marcel Dekker, Inc.
Bogen KT, Colston BW, & Machicao LK (1992) Dermal absorption of
dilute aqueous chloroform, trichloroethylene, and
tetrachloroethylene in hairless guinea pigs. Fundam Appl Toxicol,
18: 30-39.
Bomski HA, Sobdewska A, & Strakowski A (1967) [Toxic damage of the
liver by chloroform in chemical industry workers.] Int Arch
Gewerbepathol Gewerbehyg, 24: 127-134 [in German]
Borzelleca JF & Carchman RA (1982) Effect of selected organic
drinking water contaminants on male reproduction. Research Triangle
Park, North Carolina, US Environmental Protection Agency (EPA
600-/1-82-009; PB82-259847).
Bouwer EJ & McCarty PL (1983) Transformation of 1- and 2-carbon
halogenated aliphatic organic compounds under methanogenic
conditions. Appl Environ Microbiol, 45: 1286-1294.
Bouwer EJ, Rittmann BE, & McCarty PL (1981) Anaerobic degradation of
halogenated 1-and 2-carbon organic compounds. Environ Sci Technol,
15: 596-599.
Bowman FJ, Borzelleca JF, & Munson AE (1978) The toxicity of some
halomethanes in mice. Toxicol Appl Pharmacol, 44: 213-215.
Brady JF, Li D, Ishizaki H, Lee M, Ning SM, Xiao F, & Yang CS (1989)
Induction of cytochromes P450IIE1 and P450IIB1 by secondary ketones
and the role of P450IIE1 in chloroform metabolism. Toxicol Appl
Pharmacol, 100: 342-349.
Branchflower RV, Schulick RD, George JW, & Pohl LR (1983) Comparison
of the effects of methyl n-butyl ketone and phenobarbital on rat
liver cytochrome P-450 and the metabolism of chloroform to phosgene.
Toxicol Appl Pharmacol, 71: 414-421.
Brass JH, Feige MA, & Halloran T (1977) The national organic
monitoring survey: sampling and analyses of purgeable organic
compounds. In: Drinking water quality enhancement source protection,
pp 393-416.
Bringmann G (1973) [Determination of the harmful biological effect
of water pollutants from the inhibition of glucose assimilation in
the bacterium Pseudomonas fluorescens.] Gesund-Ing, 94: 366-369
(in German).
Bringmann G (1975) [Determination of the harmful biological effect
of water pollutants from the inhibition of cell reproduction in the
blue alga Microcystis.] Gesund-Ing, 96: 238-241 (in German).
Bringmann G (1978) [Determination of the harmful biological effect
of water pollutants on protozoa. I. Bacteriophagic flagellates.] Z
Wasser-Abwasser Forsch, 11: 210-215 (in German).
Bringmann G & Kühn R (1977) [Threshold values for the harmful effect
of water pollutants on bacteria (Pseudomonas putida) and green
algae (Scenedesmus quadricauda) in the cell reproduction
inhibition test.] Z Wasser-Abwasser Forsch, 10: 87-97 (in German).
Bringmann G & Kühn R (1980) [Determination of the harmful biological
effect of water pollutants on protozoa. II. Bacteriophagic
ciliates.] Z Wasser-Abwasser Forsch, 13: 26-31 (in German).
Bringmann G, Kühn R, & Winter A (1980) [Determination of the harmful
biological effect of water pollutants on protozoa. III. Saprozoic
flagellates.] Z Wasser-Abwasser Forsch, 13: 170-173 (in German).
Brondeau MT, Bonnet P, Guenier JP, & De Ceaurriz J (1983) Short-term
inhalation test for evaluating industrial hepatotoxicants in rats.
Toxicol Lett, 19: 139-146.
Brooks TM & Dean BJ (1981) Mutagenic activity of 42 coded compounds
in the Salmonella/microsome assay with preincubation. In: De Serres
FJ & Ashby J ed. Evaluation of short-term tests for carcinogens:
Report of the international collaborative study. Amsterdam, Oxford,
New York, Elsevier North/Holland, pp 261-270 (Progress in Mutation
Research, Volume 1).
Brown BR Jr (1972) Hepatic microsomal lipoperoxidation and
inhalation anaesthetics: A biochemical and morphologic study in the
rat. Anesthesiology, 36: 458-465.
Brown BR, Sipes IG, & Sagalyn AM (1974a) Mechanisms of acute hepatic
toxicity: chloroform, halothane and glutathione. Anesthesiology 41:
554-561.
Brown DM, Langley PF, Smith D, & Taylor DC (1974b) Metabolism of
chloroform. I. The role of [14C]-chloroform by different species.
Xenobiotica, 4: 151-163.
Budavari S ed. (1989) The Merck index: an encyclopedia of chemicals,
drugs and biologicals, 11th ed. Rahway, New Jersey, Merck and Co.
Bull RJ & Kopfler FC (1991) Health Effects of disinfectants and
disinfection by-products. Denver, Colorado, AWWA Research Foundation
and American Water Works Association, pp 14, 99-104.
Bull RJ, Brown JM, Meierhenry EA, Jorgenson TA, Robinson M, & Stober
JA (1986) Enhancement of the hepatotoxicity of CHCl3 in B6C3F1
mice by corn oil: Implications for CHCl3 carcinogenesis. Environ
Health Perspect, 69: 49-58.
Burkhalter JE & Balster RL (1979) Behavioral teratology evaluation
of trichloromethane in mice. Neurobehav Toxicol, 1: 199-205.
Butler TC (1961) Reduction of carbon tetrachloride in vivo and
reduction of carbon tetrachloride and chloroform in vitro by
tissues and tissue constituents. J Pharmacol Exp Ther, 134: 311-319.
Butterworth BE, Smith-Oliver T, Earle L, Loury DJ, White RD,
Doolittle DJ, Working PK, Cattley RC, Jirtle R, Michalopoulos G, &
Strom S (1989) Use of primary cultures of human hepatocytes in
toxicology studies. Cancer Res, 49: 1075-1084.
Callen DF, Wolf CR, & Philpot RM (1980) Cytochrome P-450 mediated
genetic activity and cytotoxicity of seven halogenated aliphatic
hydrocarbons in Saccharomyces cerevisiae. Mutat Res, 77: 55-63.
Capel ID, Dorrell HM, Jenner M, Pinnock MH, & Williams DC (1979) The
effect of chloroform ingestion on the growth of murine tumours. Eur
J Cancer, 15: 1485-1490.
Challen PJR, Hickish DE, & Bedford J (1958) Chronic chloroform
intoxication. Br J Ind Med, 15: 243-249.
Charbonneau M, Greselin E, Brodeur J, & Plaa GL (1991) Influence of
acetone on the severity of the liver injury induced by haloalkane
mixtures. Can J Physiol Pharmacol, 69(12): 1901-1907.
Cherkin A & Catchpool JF (1964) Temperature dependence of anesthesia
in goldfish. Science, 144: 1460-1462.
Chinery RL & Gleason AK (1993) A compartmental model for prediction
of breath concentration and absorbed dose of chloroform after
exposure while showering. Risk Anal, 13(1): 51-62.
Chiou WL (1975) Quantitation of hepatic and pulmonary first-pass
effect and its implications in pharmacokinetic study. I.
Pharmacokinetics of chloroform in man. J Pharmacokinet Biopharm, 3:
193-201.
Chu I, Secours V, Marino I, & Villeneuve DC (1980) The acute
toxicity of four trihalomethanes in male and female rats. Toxicol
Appl Pharmacol, 52: 351-353.
Chu I, Villeneuve DC, Secours VE, & Becking GC (1982a) Toxicity of
trihalomethanes. I. The acute and subacute toxicity of chloroform,
bromodichloromethane, chlorodibromomethane and bromoform in rats. J
Environ Sci Health, B17: 205-224.
Chu I, Villeneuve DC, Secours VE, & Becking GC (1982b) Toxicity of
trihalomethanes. II. Reversibility of toxicological changes produced
by chloroform, bromodichloromethane, chlorodibromomethane and
bromoform in rats. J Environ Sci Health, B17: 225-240.
Clemens TL, Hill RN, Bullock LP, Johnson WD, Sultatos LG, & Vessel
ES (1979) Chloroform toxicity in the mouse: Role of genetic factors
and steroids. Toxicol Appl Pharmacol, 48: 117-130.
Cohen EN & Hood N (1969) Application of low-temperature
autoradiography to studies of the uptake and metabolism of volatile
anaesthetic in the mouse. Anesthesiology, 30: 306-314.
Colacci A, Bartoli S, Bonora B, Guidotti L, Lattanzi G, Mazzullo M,
Niero A, Perocco P, Silingardi P, & Grilli S (1991) Chloroform
bioactivation leading to nucleic acids binding. Tumori, 77(4):
285-290.
Condie LW, Smallwood CL, & Laurie RD (1983) Comparative renal and
hepatotoxicity of halomethanes: bromodichloromethane, bromoform,
chloroform, dibromochloromethane and methylene chloride. Drug Chem
Toxicol, 6: 563-578.
Conlon MF (1963) Addiction to chlorodyne. Br Med J, 2: 1177-1178.
Corley RA, Mendrala AL, Smith FA, Staats DA, Gargas ML, Conolly RB,
Andersen ME, & Reitz RH (1990) Development of a physiologically
based pharmacokinetic model for chloroform. Toxicol Appl Pharmacol,
103: 512-527.
Cortés F, Mateos S, & Escalza P (1985) C-Mitosis, chromosomal
aberrations and sister chromatid exchanges induced by chloroform in
root-tip cells of Allium cepa. Cytobios, 44: 231-237.
Cowgill UM, Milazzo DP, & Landenberger BD (1989) Toxicity of nine
benchmark chemicals to Skeletonema costatum, a marine diatom.
Environ Toxicol Chem, 8: 451-455.
Cowlen MS, Hewitt WR, & Schroeder F (1984a) 2-Hexanone potentiation
of (14C)chloroform hepatotoxicity: covalent interaction of a
reactive intermediate with rat liver phospholipids. Toxicol Appl
Pharmacol, 73: 478-491.
Cowlen MS, Hewitt WR, & Schroeder F (1984b) Mechanism in 2-hexanone
potentiation of chloroform hepatotoxicity. Toxicol Lett, 22:
293-299.
Cox RA, Derwent RG, Eggleton AEJ, & Lovelock JE (1976) Photochemical
oxidation of halocarbons in the troposphere. Atmos Environ, 10:
305-308.
Cresteil TL, Beaune Ph, Leroux JP, Lange M, & Mansuy D (1979)
Biotransformation of chloroform by rat and human liver microsomes;
in vitro effect on some enzyme activities and mechanisms of
irreversible binding to macromolecules. Chem-Biol Interact, 24:
153-165.
Cronn DR & Harsch DE (1979) Determination of atmospheric halocarbon
concentration by gas chromatographic-mass spectrometry. Anal Lett,
12: 1489-1496.
Culliford D & Hewitt HB (1957) The influence of sex hormone status
on the susceptibility of mice to chloroform-induced necrosis of
renal tubules. J Endocrinol, 14: 381-393.
Daft JA (1988) Fumigant contamination during large-scale food
sampling for analysis. Arch Environ Contam Toxicol, 17: 177-181.
Daft JA (1989) Determination of fumigants and related chemicals in
fatty and non-fatty foods. J Agric Food Chem, 37: 560-564.
Daniel MR & Dehnel JM (1981) Cell transformation test with baby
hamster kidney cells. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford New York, Elsevier
North/Holland, pp 626-637 (Progress in Mutation Research, Volume 1).
Daniel FB, De Angelo AB, Stober JA, Pereira MA, & Olson GR (1989)
Chloroform inhibition of 1,2-dimethylhydrazine-induced
gastrointestinal tract tumours in the Fischer 344 rat. Fundam Appl
Toxicol, 13: 40-45.
Daniel FB, Reddy TV, Stober JA, & Olson GR (1991) Site specific
modulation of carcinogen-induced gastrointestinal tract nuclear
anomalies in B6C3F1 mice by chloroform. Anticancer Res, 11:
665-670.
Danielsson BRG, Ghantous H, & Dencker L (1986) Distribution of
chloroform and methyl chloroform and their metabolites in pregnant
mice. Biol Res Pregnancy, 7: 77-83.
Davis ME (1992) Dichloroacetic acid and trichloroacetic acid
increase chloroform toxicity. J Toxicol Environ Health, 37: 139-148.
Davis ME & Berndt WO (1992) Sex differences in monochloroacetate
pretreatment effects on chloroform toxicity in rats. Fundam Appl
Toxicol, 18: 66-71.
Davis DD, Machado G, Conaway B, Oh Y, & Watson R (1976) A
temperature dependent kinetics study of the reaction of OH with
CH3Cl, CH2Cl2, CHCl3 and CH3Br. J Chem Phys, 65:
1268-1274.
De Biasi A, Sbraccia M, Keizer J, Testai E, & Vittozzi L (1992) The
regioselective binding of CHCl3 reactive intermediates to
microsomal phospholipids. Chem-Biol Interact, 85: 229-242.
Deml E & Oesterle D (1985) Dose-dependent promoting activity of
chloroform in rat liver foci bioassay. Cancer Letters, 29: 59-63.
Deml E & Oesterle D (1987) Dose-response of promotion by
polychlorinated biphenyls and chloroform in rat liver foci bioassay.
Arch Toxicol, 60: 209-211.
Den Hartog JC (1980, 1981) [Analysis of gaseous organic components
in the outdoor air.] Rijswijk, The Netherlands, TNO, Prins Maurits
Laboratory (Reports PML-TNO Nos 1980-182, 1981-149, 1981-155,
1981-158( (in Dutch).
Deringer MK, Dunn TB, & Heston WE (1953) Results of strain C3H mice
to chloroform. Proc Soc Exp Biol Med, 83: 474-479.
De Serres FJ & Ashby J ed. (1981) Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland (Progress in
Mutation Research, Volume 1).
Dilling WL (1977) Interphase transfer process. II. Evaporation rates
of chloromethanes, ethanes, ethylenes, propanes, and propylenes from
dilute aqueous solutions. Comparisons with theoretical predictions.
Environ Sci Technol, 11: 405-409.
Dimitriades B, Gay BW Jr, Arnts RR, & Seila RL (1983) Photochemical
reactivity of perchloroethylene: a new appraisal. J Air Pollut
Control Assoc, 33: 575.
Divincenzo GD & Krasavage WJ (1974) Serum ornithine carbamyl
transferase as a liver response test for exposure to organic
solvents. Am Ind Hyg Assoc J, 35: 21-29.
Docks EL & Krishna G (1976) The role of glutathione in chloroform
induced hepatotoxicity. Exp Mol Pathol, 24: 13-22.
Dowty BJ, Laseter JL, & Storer J (1976) The transplacental migration
and accumulation in blood of volatile organic constituents. Pediatr
Res, 10: 696-701.
Duprat P, Delsaut L, & Gradiski D (1976) Pouvoir irritant des
principaux chlorés aliphatiques sur la peau et les muqueuses
oculaires du lapin. Eur J Toxicol Environ Hyg, 9: 171-177.
Ebel RE, Barlow RL, & McGrath EA (1987) Chloroform hepatotoxicity in
the Mongolian gerbil. Fundam Appl Toxicol, 8: 207-216.
ECDIN (1992) On-line search in the environmental chemicals data and
information network. ECDIN/copyright JRC-CEC/ISPRA.
Egli C, Scholtz R, Cook AM, & Leisinger T (1987) Anaerobic
dechlorination of tetrachloromethane and 1,2-dichloroethane to
degradable products by pure cultures of Desulfobacterium sp. and
Methanobacterium sp. FEMS Microbiol Lett, 43: 257-261.
Entz RC, Thomas KW, & Diachenko GW (1982) Residues of volatile
halocarbons in foods using headspace gas chromatography. J Agric
Food Chem, 30: 846-849.
Environment Canada (1986) Ambient air concentration of volatile
organic compounds in Toronto and Montreal. Ottawa, Environment
Canada, Pollution Measurement Division (Unpublished report).
Environment Canada (1992) Volatile organic compounds measurements in
Canadian urban and rural areas: 1989-1991. Ottawa, Environment
Canada, Technology Development Branch (Internal report 92-75.16).
Erickson MD (1981) Acquisition and chemical analysis of mother's
milk for selected toxic substances. Gov Rep Announc Index US, 5096
(PB81-231029).
Europ-Cost (1976) A comprehensive list of polluting substances which
have been identified in various fresh waters, effluent discharges,
aquatic animals and plants, and bottom sediments, 2nd ed.
Luxembourg, Commission of the European Communities, p 31
(EUCO/MDU/73/76, XII/476/76).
Ewing BB, Chian ESK, & Cook JC (1977) Monitoring to detect
previously unrecognized pollutants in surface waters. Appendix:
Organic analysis data. Washington, DC, US Environmental Protection
Agency (EPA/560/6-77-015).
Federal Office of the Environment (1981) [Clean air '81:
development, situation, trends.] Berlin, Federal Office of the
Environment (BUA) (in German).
Foster GD & Tullis RE (1985) Quantitative structure-toxicity
relationships with osmotically stressed Artemia salina nauplii.
Environ Pollut, A38: 273-281.
Fry J, Taylor R, & Hathway DE (1972) Pulmonary elimination of
chloroform and its metabolite in man. Arch Int Pharmacodyn Ther,
196: 98-111.
Fujie K, Aoki T, & Wada M (1990) Acute and subacute cytogenetic
effects of the trihalomethanes on rat bone marrow cells in vivo.
Mutat Res, 242: 111-119.
Fujie K, Aoki T, & Mae S (1993) Sister-chromatid exchanges induced
by trihalomethanes in rat erythroblastic cells. Mutat Res, 300:
241-246.
Garner RC, Welch A, & Pickering C (1981) Mutagenic activity of 42
coded compounds in the Salmonella/microsome assay. In: De Serres FJ
& Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 280-284 (Progress in Mutation
Research, Volume 1).
Gatehouse D (1981) Mutagenic activity of 42 coded compounds in the
"microtiter" fluctuation test. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 376-386 (Progress in Mutation Research,
Volume 1).
Gearhart JM, Seckel C, & Vinegar A (1993) in vivo metabolism of
chloroform in B6C3F1, mice determined by the method of gas uptake:
the effects of body temperature on tissue partition coefficients and
metabolism. Toxicol Appl Pharmacol, 119: 258-266.
Gersich FM, Blanchard FA, Applegath SL, & Park CN (1986) The
precision of Daphnid ( Daphnia magna Straus, 1820) static acute
toxicity tests. Arch Environ Contam Toxicol, 15: 741-749.
Glende EA & Recknagel RO (1992) Phosphodipase A2 activation and cell
injury in isolated rat hepatocytes exposed to bromotrichloromethane,
chloroform, and 1,1-dichloroethylene as compared to effects of
carbon tetrachloride. Toxicol Appl Pharmacol, 113: 159-162.
Gocke E, King M-T, Eckhardt K, & Wild D (1981) Mutagenicity of
cosmetics ingredients licensed by the European Communities. Mutat
Res, 90: 91-109.
Goodman LS & Gilman AG (1970) The pharmacological basis of
therapeutics. New York, McMillan Co.
Gradiski D, Magadur JL, Baillot M, Danière MC, & Schuh MB (1974)
Toxicité comparée des principaux solvants chlorés aliphatiques. J
Eur Toxicol, 7: 247-254.
Gradiski D, Bonnet P, Raoult G, & Magadur JL (1978) Toxicité aiguë
comparée par inhalation des principaux solvants aliphatiques
chlorés. Arch Mal Prof Méd Trav Sécur Soc, 39: 249-257.
Green MHL (1981) A differential killing test using an improved
repair-deficient strain of Escherichia coli. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 183-194 (Progress in Mutation
Research, Volume 1).
Greim H, Bimboe SD, Egert G, Göggelmann W, & Krämer M (1977)
Mutagenicity and chromosomal aberrations as an analytical tool for
in vitro detection of mammalian enzyme-mediated formation of
reactive metabolites. Arch Toxicol, 39: 159-169.
Grob K (1984) Further development of direct aqueous injection with
electron-capture detection in gas chromatography. J Chromatogr, 299:
1-11.
Groger WKL & Grey TF (1979) Effect of chloroform on the activities
of liver enzymes in rats. Toxicology, 14: 23-28.
Gualandi G (1984) Genotoxicity of the free-radical producers CCl4
and lipoperoxidation in Aspergillus nidulans. Mutat Res, 136:
109-114.
Guengerich FP, Kim DH, & Iwasaki M (1991) Role of human cytochrome
P450 II E1 in the oxidation of suspected carcinogens. Chem Res
Toxicol, 4: 160-179.
Hakim A, Jain AK, & Jain R (1992) Chloroform ingestion causing toxic
hepatitis. J Assoc Phys India, 40: 477.
Hamada N & Peterson RE (1977) Effect of chlorinated aliphatic
hydrocarbons on excretion of protein and electrolytes by rat
pancreas. Toxicol Appl Pharmacol, 39: 185-194.
Hardie DWF (1964) Chlorocarbons and chlorohydrocarbons. Chloroform.
In: Kirk RE & Othmer DF ed. Encyclopedia of chemical technology, 2nd
ed. Vol 5, pp 119-127.
Harms MS, Peterson RE, Fujimoto JM, & Erwin CP (1976) Increased
"bile duct-pancreatic fluid" flow in chlorinated hydrocarbon-treated
rats. Toxicol Appl Pharmacol, 35: 41-49.
Harris RN, Ratnayake JH, Garry VF, & Anders MW (1982) Interactive
hepatotoxicity of chloroform and carbon tetrachloride. Toxicol Appl
Pharmacol, 63: 281-289.
Harsch DE & Cronn DR (1978) Low-pressure sample-transfer technique
for analysis of stratospheric air samples. J Chromatogr Sci, 16:
363-367.
Hatch GC, Mamay DD, Ayer ML, Casto BC, & Nesnow S (1983) Chemical
enhancement of viral transformation in Syrian hamster embryo cells
by gaseous and volatile chlorinated methanes and ethanes. Cancer
Res, 43: 1945-1950.
Heil E, Oeser H, Hatz R, & Kelker H (1979) [Gas-chromatographic
determination of traces of C1- and C2- fluoro- and
chlorohydrocarbons in air.] Fresenius Z Anal Chem, 297: 357-364 (in
German).
Heilbrunn G, Liebert E, & Szanto PB (1945) Chronic chloroform
poisoning. Arch Neurol Psychiatr, 53: 68-72.
Herren-Freund SL & Pereira MA (1986) Carcinogenicity of by-products
of disinfection in mouse and in rat liver. Environ Health Perspect,
69: 59-66.
Hertlein F (1980) Monitoring airborne contaminants in chemical
laboratories. ACS Symp Ser, 120: 215-230.
Herzfeld D, Van der Gun KD, & Louw R (1989) Quantitative
determination of volatile organochlorine compounds in water by
GC-headspace analysis with dibromomethane as an internal standard.
Chemosphere, 18: 1425-1430.
Hewitt HB (1956) Renal necrosis in mice after accidental exposure to
chloroform. Br J Exp Pathol, 37: 32-39.
Hewitt WR, Miyajima H, Côté MG, & Plaa GL (1979) Acute alteration of
chloroform-induced hepato- and nephrotoxicity by mirex and kepone.
Toxicol Appl Pharmacol, 48: 509-527.
Hewitt WR, Brown EM, Côté MG, Plaa GL, & Miyajima H (1982)
Alteration of chloroform-induced nephrotoxicity by exogenous
ketones. In: Porter G ed. Nephrotoxic mechanisms of drugs and
environmental toxins. New York, London, Plenum, Press, pp 357-365.
Hewitt LA, Palmason C, Masson S, & Plaa GL (1990) Evidence for the
involvement of organelles in the mechanism of ketone-potentiated
chloroform-induced hepatotoxicity. Liver, 10: 35-48.
Heywood R, Sortwell RJ, Noel PRB, Street AE, Prentice DE, Roe FJC,
Wardsworth PF, Worden AN, & Van Abbé NJ (1979) Safety evaluation of
toothpaste containing chloroform. III. Long-term study in beagle
dogs. J Environ Pathol Toxicol, 2: 835-851.
Hill RN (1978) Differential toxicity of chloroform in the mouse. Ann
NY Acad Sci, 298: 170-175.
Hill RN, Clemens TL, Liu DK, Vesell ES, & Johnson WD (1975) Genetic
control of chloroform toxicity in mice. Science, 190: 159-160.
Howard Carleton J & Evenson KM (1976) Rate constants for the
reactions of OH with CH4 and fluorine, chlorine and bromine
substituted methanes at 296 K. J Chem Phys, 64: 197.
Hubbard SA, Green MHL, Bridges BA, Wain AJ, & Bridges JW (1981)
Fluctuation test with S9 and hepatocyte activation. In: De Serres
FJ & Ashby J ed. Evaluation of short-term tests for carcinogens:
Report of the international collaborative study. Amsterdam, Oxford,
New York, Elsevier North/Holland, pp 361-370 (Progress in Mutation
Research, Volume 1).
IARC (1979) Chloroform. In: Some halogenated hydrocarbons. Lyon,
International Agency for Research on Cancer, pp 401-427 (IARC
Monographs on the Evaluation of the Carcinogenic Risk of Chemicals
to Humans, Volume 20).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs volumes 1 to 42. Lyon, International Agency for
Research on Cancer, pp 152-154 (IARC Monographs on the Evaluation of
the Carcinogenic Risk of Chemicals to Humans, Supplement 7).
IARC (1991) Chlorinated drinking-water. In: Chlorinated
drinking-water; chlorination by-products; some other halogenated
compounds; cobalt and cobalt compounds. Lyon, International Agency
for Research on Cancer, pp 45-141 (IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans, Volume 52).
Ichinotsubo D, Mower H, & Mandel M (1981a) Testing of a series of
paired compounds (carcinogen and non-carcinogenic structural analog)
by DNA repair-deficient E. coli strains. In: De Serres FJ & Ashby
J ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 195-198 (Progress in Mutation Research,
Volume 1).
Ichinotsubo D, Mower H, & Mandel M (1981b) Mutagen testing of a
series of paired compounds with the Ames Salmonella testing system.
In: De Serres FJ & Ashby J eds. Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 298-301
(Progress in Mutation Research, Volume 1).
Iijima M, Côté MG, & Plaa GL (1983) A semiquantitative morphologic
assessment of chlordecone-potentiated chloroform hepatotoxicity.
Toxicol Lett, 17: 307-314.
Ikatsu H & Nakajima T (1992) Hepatotoxic interaction between carbon
tetrachloride and chloroform in ethanol treated rats. Arch Toxicol,
66: 580-586.
Ilett KF, Reid WD, Sipes IG, & Krishna G (1973) Chloroform toxicity
in mice: correlation of renal and hepatic necrosis with covalent
binding of metabolites to tissue macromolecules. Exp Mol Pathol, 19:
215-229.
Jagannath DR, Vultaggio DM, & Brusick DJ (1981) Genetic activity of
42 coded compounds in the mitotic gene conversion assay using
Saccharomyces cerevisiae strain D4. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 456-467 (Progress in Mutation Research,
Volume 1).
Jo WK, Weisel CP, & Lioy PJ (1990a) Routes of chloroform exposure
and body burden from showering with chlorinated tap water. Risk
Anal, 10: 575-580.
Jo WK, Weisel CP, & Lioy PJ (1990b) Chloroform exposure and the
health risk associated with multiple uses of chlorinated tap water.
Risk Anal, 10: 581-585.
Jones JRE (1947) The oxygen consumption of Gasterosteus aculeatus
in toxic solutions. Exp Biol, 23: 298-311.
Jones WM, Margolis G, & Stephen CR (1958) Hepatotoxicity of
inhalation anaesthetic drugs. Anesthesiology, 19: 715-723.
Jorgenson TA, Meierhenry EF, Rushbrook CJ, Bull RJ, & Robinson M
(1985) Carcinogenicity of chloroform in drinking water to male
Osborne-Mendel rats and female B6C3F1 mice. Fundam Appl Toxicol,
5: 760-769.
Jorgenson TA & Rushbrook CJ (1980) Effects of chloroform in the
drinking water of rats and mice. Ninety-day subacute toxicity study.
Washington, DC, US Environmental Protection Agency
(EPA-600/1-80-030; NTIS PB80-219108).
Jorgenson TA, Rushbrook CJ, & Jones DCL (1982) Dose-response study
of chloroform carcinogenesis in the mouse and rat: status report.
Environ Health Perspect, 46: 141-149.
Juhnke I & Lüdemann D (1978) [Results of the testing of 200 chemical
compounds for acute toxicity for fish by means of the golden orfe
test.] Z Wasser-Abwasser Forsch, 11: 161-164 (in German).
Kada T (1981) The DNA-damaging activity of 42 coded compounds in the
Rec-assay. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp
175-182 (Progress in Mutation Research, Volume 1).
Kajino M (1977) [Formation of chloroform during chlorination of
drinking water.] Chem Abstr, 88, 27631K (in Japanese).
Kassinova GV, Kovaltsova SV, Marfin SV, & Zakharov IA (1981)
Activity of 40 coded compounds in differential inhibition and
mitotic crossing-over assays in yeast. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 434-455 (Progress in Mutation Research,
Volume 1).
Kawamura K & Kaplan IR (1983) Notes: Organic compounds in the
rainwater of Los Angeles. Environ Sci Technol, 17: 497-501.
Kebbekus BB & Bozzelli JW (1982) Determination of selected toxic
organic vapours in air by adsorbent trapping and capillary GC. J
Environ Sci Health, A17: 713-723.
Kedderis GL, Held SD, Pearson AC, & Carfagns MA (1993a) Isolated
mouse hepatocytes as an in vitro model for the in vivo
metabolism and cytolethality of chloroform (CF). Toxicologist, 13:
198.
Kedderis GL, Carfagna MA, Held SD, Batra R, Murphy JE, & Gargas ML
(1993b) Kinetic analysis of furan biotransformation by F-344 rats
in vivo and in vitro. Toxicol Appl Pharmacol, 123: 274-282.
Kerfoot HB (1987) Shallow-probe soil-gas sampling for indication of
ground-water contamination by chloroform. Int J Environ Anal Chem,
30: 167-181.
Kimura ET, Ebert DM, & Dodge PW (1971) Acute toxicity and limits of
solvent residue for sixteen organic solvents. Toxicol Appl
Pharmacol, 19: 699-704.
Kirkland DJ, Smith KL, & Van Abbé NJ (1981) Failure of chloroform to
induce chromosome damage or sister-chromatid exchange in cultured
human lymphocytes and failure to induce reversion in Escherichia
coli. Food Cosmet Toxicol, 19: 651-656.
Klaassen CD & Plaa GL (1967) Relative effects of various chlorinated
hydrocarbons on liver and kidney in dogs. Toxicol Appl Pharmacol,
10: 119-131.
Klaassen CD & Plaa GL (1969) Comparison of the biochemical
alterations elicited in livers from rats treated with carbon
tetrachloride, chloroform, 1,1,2-trichloroethane and
1,1,1-trichloroethane. Biochem Pharmacol, 18: 2019-2027.
Klaunig JE, Ruch RJ, & Pereira MA (1986) Carcinogenicity of
chlorinated methane and ethane compounds administered in drinking
water to mice. Environ Health Perspect, 69: 89-95.
Kluwe WM (1981) The nephrotoxicity of low molecular weight
halogenated alkane solvents, pesticides and chemical intermediates.
In: Hook JB ed. Toxicology of the kidney. New York, Raven Press Ltd,
pp 179-226.
Knie J, Hälke A, Juhnke I, & Schiller W (1983) [Results of studies
on chemical substances with four biotests.] Dtsch Gewässerkd Mitt,
27: 77-79 (in German).
Könemann H (1981) Quantitative structure-activity relationships in
fish toxicity studies. Part 1: Relationships for 50 industrial
pollutants. Toxicology, 19: 209-211.
Koop DR, Morgan ET, Tarr GE, & Coon MJ (1982) Purification and
characterization of a unique isozyme of cytochrome P450 from liver
microsomes of ethanol-treated rabbits. J Biol Chem, 257: 8472-8480.
Kramer MD, Lynch CF, Isacson P, & Hanson JW (1992) The association
of waterborne chloroform with intrauterine growth retardation.
Epidemiology, 3: 407-413.
Krasner SW, McGuire MJ, Jacangelo JG, Patania NL, Reagan KM, & Aieta
EM (1989) The occurrence of disinfection by-products in U.S.
drinking water. J Am Water Works Assoc, 81: 41-53.
Kroneld R (1985) Recovery and reproducibility in determination of
volatile halocarbons in water and blood. Bull Environ Contam
Toxicol, 34: 486-496.
Krost KJ, Pellizzari ED, Walburn SG, & Hubbard SA (1982) Collection
and analysis of hazardous organic emissions. Anal Chem, 54: 810-817.
Kutob SD & Plaa GL (1962a) A procedure of estimating the hepatotoxic
potential of certain industrial solvents. Toxicol Appl Pharmacol, 4:
354-361.
Kutob SD & Plaa GL (1962b) The effect of acute ethanol intoxication
on chloroform-induced liver damage. J Pharmacol Exp Teratol, 135:
245-251.
Kylin B, Reichard H, Sümegi I, & Yllner S (1963) Hepatotoxicity of
inhaled trichloroethylene, tetrachloroethylene and chloroform.
Single exposure. Acta Pharmacol Toxicol, 20: 16-26.
Lahl U, Bätjer K, Düszeln v J, Gabel B, Stachel B, & Thiemann W
(1981a) Distribution and balance of volatile halogenated
hydrocarbons in the water and air of covered swimming pools using
chlorine for water disinfection. Water Res, 15: 803-814.
Land PC, Owen EL, & Linde HW (1981) Morphologic changes in mouse
spermatozoa after exposure to inhalational anaesthetics during early
spermatogenesis. Anesthesiology, 54: 53-56.
Larson JL, Wolf DC, & Butterworth BE (1993) The acute hepatotoxicity
and nephrotoxic effects of chloroform in male F-344 rats and female
B6C3F1 mice. Fundam Appl Toxicol 20: 302-315.
Larson JL, Wolf DC, & Butterworth BE (1994a) Induced cytotoxicity
and cell proliferation in the hepatocarcinogenicity of chloroform in
female B6C3F1 mice. Comparison of administration by gavage in corn
oil vs ad libitum in drinking water. Fundam Appl Toxicol, 22:
90-102.
Larson JL, Wolf DC, Morgan KT, Méry S, & Butterworth BE, (1994b) The
toxicity of one week exposures to inhaled chloroform in female
B6C3F1 mice and male F-344 rats. Fundam Appl Toxicol, 22: 431-446.
Larson JL, Sprankle CS, & Butterworth BE (1994c) Lack of
chloroform-induced DNA repair in vitro and in vivo in
hepatocytes of female B6C3F1 mice. Environ Mol Mutagen, 23:
132-136.
Lasa J & Rosiek J (1979) [Detection of halogen compounds in air with
a colometric ECD.] Chem Anal, 24: 819-826 (in Polish).
Lavigne JG & Marchand C (1974) The role of metabolism in chloroform
hepatocyto-toxicity. Toxicol Appl Pharmacol, 29: 312-326.
Lavigne JG, Belanger PM, Dore F, & Labrecque G (1983) Temporal
variations in chloroform-induced hepatotoxicity in rats. Toxicology,
26: 267-273.
LeBlanc GA (1980) Acute toxicity of priority pollutants to water
flea (Daphnia magna). Bull Environ Contam Toxicol, 24: 684-691.
Lehmann KB & Flury FF (1943) Chlorinated hydrocarbons. In: Lehmann
KB & Flury FF ed. Toxicology and hygiene of industrial solvents.
Baltimore, Maryland, Williams & Wilkins Co, pp 138-145, 191-196.
Lehmann KB & Hasegawa (1910) [Study on the absorption of chlorinated
hydrocarbons from the air by animal and man.] Arch Hyg, 72: 327-342
(in German).
Leveson R & Barker N (1981) Airscan: an ultrasensitive trace air
impurity analyzer for use in toxic aviation environments (AGARD Conf
Proc AGARD-CP-309, B15/1-B15/12).
Lillian D & Singh HB (1974) Absolute determination of atmospheric
halocarbons by gas phase coulometry. Anal Chem, 46: 1060-1063.
Linde HW & Mesnick PS (1979) Causes of death of anaesthesiologists
from the chloroform era. Washington, DC, US Environmental Protection
Agency (EPA 600/1-179-043; NTIS PB80-125172).
Lioy PJ & Lioy MJ ed. (1983) Air sampling instruments for evaluation
of atmospheric contaminants, 6th ed. Cincinnati, Ohio, American
Conference of Governmental Industrial Hygienists, pp V/85-V/86.
Löfberg B & Tjälve H (1986) Tracing tissues with
chloroform-metabolizing capacity in rats. Toxicology, 39: 13-35.
Loprieno N (1981) Screening of coded carcinogenic/non carcinogenic
chemicals by a forward-mutation system with the yeast
Schizosaccharomyces pombe. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 424-425 (Progress in Mutation Research,
Volume 1).
Lundberg I, Ekdahl M, Kronevi T, Lidums V, & Lundberg S (1986)
Relative hepatotoxicity of some industrial solvents after
intraperitoneal injection or inhalation exposure in rats. Environ
Res, 40: 411-420.
Lurker PA, Clark CS, Elia VJ, Gartside PS, & Kinman RN (1983) Worker
exposure to chlorinated organic compounds from the activated sludge
wastewater treatment process. Am Ind Hyg Assoc J, 44: 109-112.
Lyman WJ, Reehl WF, & Rosenblatt DH (1982) Handbook of chemical
property estimation methods. New York, McGraw-Hill Book Company.
McConnell G, Ferguson DM, & Pearson CR (1975) Chlorinated
hydrocarbons and the environment. Endeavor, 34: 13-18.
MacDonald DJ (1981) Salmonella/microsome tests on 42 coded
chemicals. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp
285-297 (Progress in Mutation Research, Volume 1).
McLean AEM (1970) The effect of protein deficiency and microsomal
enzyme induction by DDT and phenobarbitone on the acute toxicity of
chloroform and a pyrrolizidine alkaloid, retrorsine. Br J Exp
Pathol, 511: 317-321.
McMartin DN, O'Connor JA, & Kaminsky LS (1981) Effects of
differential changes in rat hepatic and renal cytochrome P-450
concentrations on hepatotoxicity and nephrotoxicity of chloroform.
Res Commun Chem Pathol Pharmacol, 31: 99-110.
Mansuy D, Beaune P, Cresteil T, Lange M, & Leroux JP (1977) Evidence
for phosgene formation during liver microsomal oxidation of
chloroform. Biochem Biophys Res Commun, 79: 513-517.
Martin CN & McDermid AC (1981) Testing of 42 coded compounds for
their ability to induce unscheduled DNA repair synthesis in HeLa
cells. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 533-537
(Progress in Mutation Research, Volume 1).
Masuda Y, Yano I, & Murano T (1980) Comparative studies on the
hepatotoxic actions of chloroform and related halogenomethanes in
normal and phenobarbital-pretreated animals. J Pharmacobio-Dyn, 3:
53-64
Matsushima T, Takamoto Y, Shirai A, Sawamura M, & Sugimura T (1981)
Reverse mutation test on 42 coded compounds with the E. coli WP2
system. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp
387-395 (Progress in Mutation Research, Volume 1).
Mattice JS, Tsai SC, & Burch MB (1981) Toxicity of trihalomethanes
to common carp embryos. Trans Am Fish Soc, 110: 261-269.
Maxwell MI, Burmaster DE, & Ozonoff D (1991) Trihalomethanes.
Maximum contaminant levels: the significance of inhalation, dermal
exposures to chloroform in household water. Regul Toxicol Pharmacol,
14: 297-312.
Mayes MA, Alexander HC, & Dill DC (1983) A study to assess the
influence of age on the response of Fathead minnows in static acute
toxicity tests. Bull Environ Contam Toxicol, 31: 139-147.
Mehran MF, Slifker RA, & Cooper WJ (1984) A simplified liquid-liquid
extraction method for analysis of trihalomethanes in drinking water.
J Chromatogr Sci, 22: 241-243.
Mehta RD & Von Borstel RV (1981) Mutagenic activity of 42 encoded
compounds in the haploid yeast reversion assay, strain XV185-14C.
In: De Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 414-423
(Progress in Mutation Research, Volume 1).
Meier JR, Ringhand HP, Coleman WE, Munch JW, Streicher RP, Kaylor
WH, & Schenck KM (1985) Identification of mutagenic compounds formed
during chlorination of humic acid. Mutat Res, 157: 111-122.
Méry S, Larson JL, Butterworth BE, Wolf DC, Harden R, & Morgan KT
(1994) Nasal toxicity of chloroform in male F-344 rats and female
B6C3F1 mice following a one week inhalation exposure. Toxicol Appl
Pharmacol, 125: 214-227.
Mink FL, Coleman WE, Munch JW, Kaylor WH, & Ringhand HP (1983) in
vivo formation of halogenated reaction products following peroral
sodium hypochlorite. Bull Environ Contam Toxicol, 30: 394-399.
Mink FL, Brown TJ, & Rickabaugh J (1986) Absorption, distribution,
and excretion of 14C-trihalomethanes in mice and rats. Bull
Environ Contam Toxicol, 37: 752-758.
Mirsalis JC, Tyson CK, & Butterworth BE (1982) Detection of
genotoxic carcinogens in the in vivo-in vitro hepatocyte DNA
repair assay. Environ Mutagen, 4: 553-562.
Moore L (1980) Inhibition of liver-microsome calcium pump by in
vivo administration of CCl4, CHCl3 and 1,1-dichloroethylene.
Biochem Pharmacol, 29: 2505-2511.
Moore DH, Chasseaud LF, Majeed SK, Prentice DE, Roe FJC, & Van Abbé
NJ (1982) The effect of dose and vehicle on early tissue damage and
regenerative activity after chloroform administration to mice. Food
Chem Toxicol, 20: 951-954.
Morele Y, Lefevre C, Ferrari P, Guenier JP, & Muller J (1989)
Sampling and analysis of airborne chlorinated methanes - comparison
of FID and ECD. Chromatographia, 28: 617-619.
Morgan A, Black A, & Belcher DR (1970) The excretion in breath of
some aliphatic halogenated hydrocarbons following administration by
inhalation. Ann Occup Hyg, 13: 219-233.
Morgan DL, Cooper SW, Carlock DL, Sykora JJ, Sutton B, Mattie DR, &
McDougal JN (1991) Dermal absorption of neat and aqueous volatile
organic chemicals in the Fischer-344 rat. Environ Res, 55: 51-63.
Morimoto K & Koizumi A (1983) Trihalomethanes induce sister
chromatid exchanges in human lymphocytes in vitro and mouse bone
marrow cells in vivo. Environ Res, 32: 72-79.
Morris RD, Audet AM, Angelillo IF, Chalmers TC, & Mosteller F (1992)
Chlorination, chlorination by-products and cancer. A meta-analysis.
Am J Public Health, 82: 955.
Munson AE, Sain LE, Sanders VM, Kauffmann BM, White KL, Page DG,
Barnes DW, & Borzelleca JF (1982) Toxicology of organic drinking
water contaminants: trichloromethane, bromodichloromethane,
dibromomethane and tribromomethane. Environ Health Perspect, 46:
117-126.
Murray FJ, Schwetz BA, McBride JF, & Staples RE (1979) Toxicity of
inhaled chloroform in pregnant mice and their offspring. Toxicol
Appl Pharmacol, 50: 515-522.
Nagao M & Takahashi Y (1981) Mutagenic activity of 42 coded
compounds in the Salmonella/microsome assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 302-313 (Progress in Mutation
Research, Volume 1).
Nakajima T & Sato A (1979) Enhanced activity of liver
drug-metabolising enzymes for aromatic and chlorinated hydrocarbons
following food deprivation. Toxicol Appl Pharmacol, 50: 549-556.
Nakajima T, Koyama Y, & Sato A (1982) Dietary modification of
metabolism and toxicity of chemical substances with specific
reference to carbohydrate. Biochem Pharmacol, 31: 1005-1011.
Nakajima T, Elovaara E, Park SS, Gelboin HV, & Vainio H (1991)
Immunochemical detection of cytochrome P450 isozymes induced in rat
liver by n-hexane, 2-hexanone and acetonyl acetone. Arch Toxicol,
65: 542-547.
National Cancer Institute (1976a) Carcinogenesis bioassay of
chloroform. Bethesda, Maryland, National Cancer Institute (NTIS
PB-264018/AS).
National Cancer Institute (1976b) Report on carcinogenesis bioassay
of chloroform. Bethesda, Maryland, National Cancer Institute (NTIS
PB-264-018).
Nicloux MM (1906) Passage du chloroforme de la mère au foetus. C R
Soc Biol, 60: 373-375.
NNI (Dutch Standardisation Institute) (1984) [Air quality - Outdoor
air. Determination of chlorinated hydrocarbons - Coal
adsorption/liquid desorption/GC-method.] Dutch Standardisation
Institute (Standard NVN 2794) (in Dutch).
Oettel H (1936) [Effects of organic fluids on the skin.] Arch Exp
Pathol Pharmacol, 183: 641-696 (in German).
O'Hara TM, Sheppard MA, Clarke EC, Borzelleca JF, Gennings C, &
Condie LW (1991) A CCl4/CHCl3 interaction study in isolated
hepatocytes: non-induced and phenobarbital-pretreated cells. J Appl
Toxicol, 11: 147-154.
Ohio River Valley Water Sanitation Commission (1980) Assessment of
water quality conditions. Ohio River mainstream 1978-79. Cincinnati,
Ohio, Ohio River Valley Water Sanitation Commission.
Ohio River Valley Water Sanitation Commission (1982) Assessment of
water quality conditions Ohio River mainstream 1980-81. Cincinnati,
Ohio, Ohio River Valley Water Sanitation Commission.
Oliver BG (1983) Dihaloacetonitriles in drinking water. Algae and
fulvic acid as precursors. Environ Sci Technol, 17: 80-83.
Otson R (1990) A Health and Welfare Canada program to develop
personal exposure monitors for airborne organics at µg/m3.
Proceedings of the 1990 EPA/A & WWMA International Symposium on
Measurement of Toxic and Related Air Pollutants (VIP-17). Pittsburg,
Pennsylvania, Air & Waste Water Management Association, pp 483-488.
Otson R, Fellin P, & Whitmore R (1992) A national pilot study of
airborne VOCs in residences - design and progress. Presented at the
1992 EPA/A & WWMA Symposium on Measurement of Toxic and Related Air
Pollutants, Durham, NC, 4-8 May. Pittsburg, Pennsylvania, Air &
Waste Water Management Association.
Parry JM & Sharp DC (1981) Induction of mitotic aneuploidy in the
yeast strain D6 by 42 coded compounds. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 468-480 (Progress in Mutation Research,
Volume 1).
Paul BB & Rubinstein D (1963) Metabolism of carbon tetrachloride and
chloroform by the rat. J Pharmacol Exp Ther, 141: 141-148.
Pearson CR & McConnell G (1975) Chlorinated C1 and C2
hydrocarbons in the marine environment. Proc R Soc Lond, B189:
305-322.
Pellizzari ED, Erickson MD, & Zweidinger (1979) Formulation of
preliminary assessment of halogenated organic compounds in man and
environmental media. Research Triangle Park, North Carolina, US
Environmental Protection Agency (EPA 560/13-79-006).
Pellizzari ED, Hartwell TD, Harris BSH, Waddel RD, Whitaker DA, &
Erickson MD (1982) Purgeable organic compounds in mothers' milk.
Bull Environ Contam Toxicol, 28: 322-328.
Pellizzari ED, Sheldon LS, & Bursey JT (1985a) Method 25: GC/MS
determination of volatile halocarbons in blood and tissue. Environ
Carcinog Sel Methods Anal, 7: 435-444.
Pellizzari ED, Zweidinger RA, & Sheldon LS (1985b) Method 24: GC/MS
determination of volatile hydrocarbons in breath samples. Environ
Carcinog Sel Methods Anal, 7: 413-431.
Pereira MA, Lin L-HC, Lippitt HM, & Herren SL (1982) Trihalomethanes
as initiators and promoters of carcinogenesis. Environ Health
Perspect, 46: 151-156.
Pereira MA, Knutsen GL, & Herren-Freund SL (1985) Effect of
subsequent treatment of chloroform or phenobarbital on the incidence
of liver and lung tumours, initiated by ethylnitrosourea in 15 days
old mice. Carcinogenesis, 6: 203-207.
Pericin C & Thomann P (1979) Comparison of the acute toxicity of
clioquinol, histamine and chloroform in different strains of mice.
Arch Toxicol, 2(Suppl): 371-373.
Perocco P & Prodi G (1981) DNA damage by haloalkanes in human
lymphocytes cultured in vitro. Cancer Lett, 13: 213-218.
Perocco P, Bolognesi S, & Alberghini W (1983) Toxic activity of
seventeen industrial solvents and halogenated compounds on human
lymphocytes cultured in vitro. Toxicol Lett, 16: 69-75.
Perry PE & Thomson EJ (1981) Evaluation of the sister chromatid
exchange method in mammalian cells as a screening system for
carcinogens. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp.
560-569 (Progress in Mutation Research, Volume 1).
Petzold GL & Swenberg JA (1978) Detection of DNA damage induced in
vivo following exposure of rats to carcinogens. Cancer Res, 38:
1589-1594.
Phillips LJ & Birchard GF (1991) Regional variations in human toxic
exposure in the USA: An analysis based on the national human adipose
tissue survey. Arch Environ Contam Toxicol, 21: 159-168.
Phoon WH, Liang OK, & Kee CP (1975) An epidemiological study of an
outbreak of jaundice in a factory. Ann Acad Med Singap, 4: 396-399.
Phoon WH, Goh KT, Lee LT, Tan KT, & Kwok SF (1983) Toxic jaundice
from occupational exposure to chloroform. Med J Malays, 38: 31-34.
Plaa GL & Larson RE (1965) Relative nephrotoxicity properties of
chlorinated methane, ethane and ethylene derivatives in mice.
Toxicol Appl Pharmacol, 7: 34-44.
Plaa GL, Evans EA, & Hine CH (1958) Relative hepatotoxicity of seven
halogenated hydrocarbons. J Pharmacol Exp Ther, 123: 224-229.
Plummer JL, Hall P de la M, Ilsley AH, Jenner MA, & Cousins MJ
(1990) Influence of enzyme induction and exposure profile on liver
injury due to chlorinated hydrocarbon inhalation. Pharmacol Toxicol,
67: 329-335.
Pohl LR (1979) Biochemical toxicology of chloroform. Rev Biochem
Toxicol, 1: 79-107.
Pohl LR & Krishna G (1978) Deuterium isotope effect in bioactivation
and hepatotoxicity of chloroform. Life Sci, 23: 1067-1072.
Pohl LR, Brooshan B, Whittaker NF, & Krishna G (1977) Phosgene: A
metabolite of chloroform. Biochem Biophys Res Commun, 79: 684-691.
Pohl LR, George JW, Martin JL, & Krishna G (1979) Deuterium isotope
effect in in vivo bioactivation of chloroform to phosgene. Biochem
Pharmacol, 28: 561-563.
Pohl LR, Martin JL, & George JW (1980) Mechanism of metabolic
activation of chloroform by rat liver microsomes. Biochem Pharmacol,
29: 3271-3276.
Pohl LR, Branchflower RV, Highet RJ, Martin JL, Nunn DS, Monks TJ,
George JW, & Hinson JA (1981) The formation of diglutathionyl
dithiocarbonate as a metabolite of chloroform, bromotrichloromethane
and carbon tetrachloride. Drug Metab Dispos, 9: 334-339.
Rao KN, Virji MA, Moraca MA, Diven WF, Martin TG, & Schneider SM
(1993) Role of serum markers for liver function and regeneration in
the management of chloroform poisoning. J Anal Toxicol, 17: 99-102.
Ray SD & Mehendale HM (1990) Potentiation of CCl4 and CHCl3
hepatotoxicity and lethality by various alcohols. Fundam Appl
Toxicol, 15: 429-440.
Reddy TV, Daniel FB, Lin EL, Stober JA, & Olson GR (1992) Chloroform
inhibits the development of diethylnitrosamine-initiated,
phenobarbital-promoted gamma-glutamyltranspeptidase and placental
form glutathione S-transferase positive foci in rat liver.
Carcinogenesis, 13: 1325-1330.
Reitz RH, Fox TR, & Quast JF (1982) Mechanistic considerations for
carcinogenic risk estimation: chloroform. Environ Health Perspect,
46: 163-168.
Rem RM, Anderson ME, Daletsky SA, Misenheimer DC, & Rollius HF
(1982) Chloroform materials balance (EPA Draft Report 68-02-3168).
Reuber MD (1979) Carcinogenicity of chloroform. Environ Health
Perspect, 31: 171-182.
Reynolds ES & Yee AG (1967) Liver parenchymal cell injury. V.
Relationships between patterns of chloromethane-C14 incorporation
into constituents of liver in vivo and cellular injury. Lab
Invest, 16: 591-603.
Reynolds LF & Harrison DW (1982) Studies of discharges of: benzene,
chloroform and carbon tetrachloride into the aquatic environment and
the best technical means for reduction of water pollution from such
discharges. Brixham, Devon, ICI Brixham Laboratory (Report No.
BL/A/2198).
Reynolds ES, Treinen RJ, Farrish HH, & Moslen MT (1984) Metabolism
of [14C] carbon tetrachloride to exhaled, excreted and bound
metabolites. Biochem Pharmacol 21: 3363-3374.
Richold M & Jones M (1981) Mutagenic activity of 42 coded compounds
in the Salmonella/microsome assay. In: De Serres FJ & Ashby J ed.
Evaluation on short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 314-322 (Progress in Mutation Research,
Volume 1).
Robinson AB, Manly KF, Anthony MP, Catchpool JF, & Pauling L (1965)
Anesthesia of Artemia larvae. Method for quantitative study.
Science, 149: 1255-1258.
Roe FJC, Palmer AK, Worden AN, & Van Abbé NJ (1979) Safety
evaluation of toothpaste containing chloroform. 1. Long-term studies
in mice. J Environ Pathol Toxicol, 2: 799-819.
Rosenberg C, Nylund L, Aalto T, Kontsas H, Norppa H, Jäppinen P, &
Vainio H (1991) Volatile organohalogen compounds from the bleaching
of pulp - Occurrence and genotoxic potential in the work
environment. Chemosphere, 23: 1617-1628.
Rosenkranz HS, Hyman J, & Leifer Z (1981) DNA polymerase deficient
assay. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 210-218
(Progress in Mutation Research, Volume 1).
Rowland I & Severn B (1981) Mutagenicity of carcinogens and
non-carcinogens in the Salmonella/microsome test. In: De Serres FJ &
Ashby J eds. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 323-332 (Progress in Mutation
Research, Volume 1).
Rubinstein D & Kanics L (1964) The conversion of carbon
tetrachloride and chloroform to carbon dioxide by rat liver
homogenates. Can J Biochem, 42: 1577-1585.
Ruddick JV, Villeneuve DC, & Chu I (1983) A teratological assessment
of four trihalomethanes in the rat. J Environ Sci Health, B18:
333-349.
Rudolph J & Jebsen C (1983) The use of photoionization,
flameionization and electron capture detectors in series for the
determination of low molecular weight trace components in the non
urban atmosphere. Int J Environ Anal Chem, 13: 129-139.
Russell JW & Shadoff LA (1977) The sampling and determination of
halocarbons in ambient air using concentration on porous polymer. J
Chromatogr, 132: 375-384.
Ryan DE, Koop DR, Thomas PE, Coon MJ, & Levin W (1986) Evidence that
isoniazid and ethanol induce the same microsomal cytochrome P-450 in
rat liver, an isozyme homologous to rabbit cytochrome P-450 isozyme
3a. Arch Biochem Biophys, 246: 633-644.
Salamone MF, Heddle JA, & Katz M (1981) Mutagenic activity of 41
compounds in the in vivo micronucleus assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 686-697 (Progress in Mutation
Research, Volume 1).
Salisbury S (1982) Health hazard evaluation. Charleston Laboratory
(Report No. HETA-81-359-1058).
Sato A, Nakajima T, & Koyama Y (1980) Effects of chronic ethanol
consumption on hepatic metabolism of aromatic and chlorinated
hydrocarbons in rats. Br J Ind Med, 37: 382-386.
Sato A, Najajiwa T, & Koyama Y (1981) Dose-related effect of a
single dose of ethanol on the metabolism in rat liver of some
aromatic and chlorinated hydrocarbons. Toxicol Appl Pharmacol, 60:
8-15.
Savoure N, Maudet M, & Nicol M (1992) Toxicité du chloroforme et
statut vitaminique A chez le rat. J Toxicol Clin Exp, 12: 97-108.
Scholler KL (1966) [Electron microscope studies on rat liver cells
after anaesthesia with various inhalational anaesthetics.]
Anaesthesist, 15: 145-148 (in German).
Scholler KL (1967) [The effect of halothane and chloroform on the
fine structure and protein synthesis of rat's liver.] Experientia
(Basel), 23: 652-655 (in German).
Schröder HG (1965) Acute and delayed chloroform poisoning. A case
report. Br J Anaesth, 37: 971-975.
Schwetz BA, Leong BKJ, & Gehring PJ (1974) Embryo- and fetotoxicity
of inhaled chloroform in rats. Toxicol Appl Pharmacol, 28: 442-451.
Sharp DC & Parry JM (1981a) Induction of mitotic gene conversion by
41 coded compounds using the yeast culture JD1. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 491-501 (Progress in Mutation
Research, Volume 1).
Sharp DC & Parry JM (1981b) Use of repair-deficient strains of yeast
to assay the activity of 40 coded compounds. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 502-516 (Progress in Mutation
Research, Volume 1).
Simmon VF & Shepherd GH (1981) Mutagenic activity of 42 coded
compounds in the Salmonella/microsome assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 333-342 (Progress in Mutation
Research, Volume 1).
Simmon VF, Kauhanen K, & Tardiff RG (1977) Mutagenic activity of
chemicals identified in drinking water. In: Scott D ed. Progress in
genetic toxicology. Amsterdam, Oxford, New York, Elsevier/North
Holland Biomedical Press, pp 249-258.
Simpson JY (1847) On a new anaesthetic agent, more efficient than
sulfuric ether. Lancet, 2: 549-550.
Sipes IG, Stripp B, Krishna G, Maling HM, & Gillette JR (1973)
Enhanced hepatic microsomal activity by pretreatment of rats with
acetone or isopropanol. Proc Soc Exp Biol Med, 142: 237-240.
Sipes IG, Krishna G, & Gillette JR (1977) Bioactivation of carbon
tetrachloride, chloroform and bromotrichloromethane: role of
cytochrome P-450. Life Sci, 20: 1541-1548.
Skopek TS, Andon BM, Kaden DA, & Thilly WG (1981) Mutagenic activity
of 42 coded compounds using 8-azaguanine resistance as a genetic
marker in Salmonella typhimurium. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 371-375 (Progress in Mutation Research,
Volume 1).
Skrzypinska GM, Piotrowski JK, & Koralewska J (1991) The hepatotoxic
action of chloroform: short-time dynamics of biochemical alterations
and dose-effect relationships. Pol J Occup Med, 4(1): 77-84.
Slooff W (1979) Detection limits of a biological monitoring system
based on fish respiration. Bull Environ Contam Toxicol, 23: 517-523.
Smith JH & Hook JB (1983) Mechanism of chloroform nephrotoxicity.
II. in vitro evidence for renal metabolism of chloroform in mice.
Toxicol Appl Pharmacol, 70: 480-485.
Smith JH & Hook JB (1984) Mechanism of chloroform nephrotoxicity.
III. Renal and hepatic microsomal metabolism of chloroform in mice.
Toxicol Appl Pharmacol, 73: 511-524.
Smith AA, Volpitto PP, Gramling ZW, DeVore MB, & Glassman AB (1973)
Chloroform, haloethane and regional anesthesia: A comparative study.
Anesth Analg, 52: 1-11.
Smith JH, Maita K, Sleight SD, & Hook JB (1983) Mechanism of
nephrotoxicity. I. Time course of chloroform toxicity in male and
female mice. Toxicol Appl Pharmacol, 70: 467-479.
Smith JH, Maita K, Sleight SD, & Hook JB (1984) Effect of sex
hormones status on chloroform nephrotoxicity and renal mixed
function oxidase in mice. Toxicology, 30: 305-316.
Smith JH, Hewitt WR, & Hook JB (1985) Role of intrarenal
biotransformation in chloroform-induced nephrotoxicity in rats.
Toxicol Appl Pharmacol, 79: 166-174.
Spence JW, Philip LH, Hanst PL, & Gray BW (1976) Atmospheric
oxidation of methyl chloride, methylene chloride and chloroform. J
Air Pollut Control Assoc, 26: 994.
Stacey NH (1987) Assessment of the toxicity of chemical mixtures
with isolated rat hepatocytes: cadmium and chloroform. Fundam Appl
Toxicol, 9: 616-622.
Stander GJ (1980) Micro-organic compounds in the water environment
and their impact on the quality of potable water supplies. Water S
Afr, 6: 1-14.
Stevens JL & Anders MW (1981a) Effect of cysteine, diethylmaleate
and phenobarbital treatments on the hepatotoxicity of [1H]- and
[2H]-chloroform. Chem-Biol Interact, 37: 207-217.
Stevens JL & Anders MW (1981b) Metabolism of haloforms to carbon
monoxide. IV. Studies on the reaction mechanism in vivo. Chem-Biol
Interact, 37: 365-374.
Stewart ME, Blogoslawski WJ, Hsu RY, & Helz GR (1979) By-products of
oxidative biocides: toxicity to oyster larvae. Mar Pollut Bull, 10:
166-169.
Storms WW (1973) Chloroform parties. J Am Med Assoc, 255: 160.
Strand SE & Shippert L (1986) Oxidation of chloroform in an aerobic
soil exposed to natural gas. Appl Environ Microbiol, 52: 203-205.
Sturrock J (1977) Lack of mutagenic effect of halothane or
chloroform on cultured cells using the azaguanine test system. Br J
Anaesth, 49: 207-210.
Styles JA (1979) Cell transformation assays. In: Paget GE ed. Topics
in toxicology - Mutagens in sub-mammalian systems: Status and
significance. Baltimore, Maryland, University Park Press, pp
147-161.
Styles JA (1981) Activity of 42 coded compounds in the BHK-21 cell
transformation test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 638-646 (Progress in Mutation Research, Volume 1).
Su C & Goldberg E (1976) Environmental concentration and fluxes of
some halocarbons. Mar Pollut Transf, 353-373.
Taketomo AP & Grimsrud E (1977) An analysis of halocarbons in the
air of several working and living environments. Proc Mont Acad Sci,
37: 128-134.
Taylor DC, Brown DM, Keeble R, & Langley PF (1974) Metabolism of
chloroform. II. A sex difference in the metabolism of (14C)
chloroform in mice. Xenobiotica, 4: 165-174.
Taylor GJ, Drew RT, Lores EM, & Clemmer TA (1976) Cardiac depression
by haloalkane propellants, solvents and inhalation anaesthetics in
rabbits. Toxicol Appl Pharmacol, 38: 379-387.
Temmerman IFMM & Quaghebeur DJM (1990) Analysis of trihalomethanes
by direct aqueous injection (THM-DAI). Short communications. J High
Res Chromatogr, 13: 379-381.
Testai E & Vittozzi L (1986) Biochemical alterations elicited in rat
liver microsomes by oxidation and reduction products of chloroform
metabolism. Chem-Biol Interact, 59: 157-171.
Testai E, Di Marzio S, & Vittozzi L (1990) Multiple activation of
chloroform in hepatic microsomes from uninduced B6C3F1 mice.
Toxicol Appl Pharmacol, 104: 496-503.
Testai, E, Keizer J, Pacifici GM, & Vittozzi L (1991) Chloroform
bioactivation by microsomes from colonic and ileal mucosa of rat and
man. Toxicol Lett, 57: 19-27.
Thompson DJ, Warner SD, & Robinson VB (1974) Teratology studies on
orally administered chloroform in the rat and rabbit. Toxicol Appl
Pharmacol, 29: 348-357.
Thomson JA (1981) Mutagenic activity of 42 coded compounds in the
Lambda induction assay. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 224-235 (Progress in Mutation Research, Volume 1).
Timms RM & Moser KM (1975) Toxicity secondary to intravenously
administered chloroform in humans. Arch Intern Med, 135: 1601-1603.
Tomasi A, Albano E, Biasi F, Slater TF, Vannini V, & Dianzani MU
(1985) Activation of chloroform and related trihalomethanes to free
radical intermediates in isolated hepatocytes and in the rat in
vivo as detected by the ESR-spin trapping technique. Chem-Biol
Interact, 55: 303-316.
Topham JC (1980) Do induced sperm-head abnormalities in mice
specifically identify mammalian mutagens rather than carcinogens?
Mutat Res, 74: 379-387.
Topham JC (1981) Evaluation of some chemicals by the sperm
morphology assay. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 718-720 (Progress in Mutation Research, Volume 1).
Torkelson TR, Oyen F, & Rowe VK (1976) The toxicity of chloroform as
determined by single and repeated exposure of laboratory animals. Am
Ind Hyg Assoc J, 37: 697-705.
Toxicology and Environmental Health Institute of Munich Technical
University (1992) [Recommendations on chloroform/emission data. BUA
plenary session in Merseburg, 24-25 August 1992.] Berlin, Federal
Office of the Environment (BUA) (in German).
Trueman RW (1981) Activity of 42 coded compounds in the Salmonella
reverse mutation test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 343-350 (Progress in Mutation Research, Volume 1).
Tsuchimoto T & Matter BE (1981) Activity of coded compounds in the
micronucleus test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 705-711 (Progress in Mutation Research, Volume 1).
Tsuruta H (1975) Percutaneous absorption of organic solvents. Ind
Health, 13: 227-236.
Tumasonis CF, McMartin DN, & Bush B (1985) Lifetime toxicity of
chloroform and bromodichloromethane when administered over a
lifetime in rats. Ecotoxicol Environ Saf, 9: 233-240.
Tweats DJ (1981) Activity of 42 coded compounds in a differential
killing test using Escherichia coli strains WP2, WP67 (uvra po1A),
and CM871 (uvrA AlexA recA). In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 199-209 (Progress in Mutation Research,
Volume 1).
Tyson CA, Hawk-Prather K, Story DL, & Gould DH (1983) Correlations
of in vitro and in vivo hepatotoxicity for five haloalkanes.
Toxicol Appl Pharmacol, 70: 289-302.
Uchrin CG & Mangels G (1986) Chloroform sorption to New Jersey
coastal plain ground water aquifer solids. Environ Toxicol Chem, 5:
339-343.
Uehleke H & Werner T (1975) A comparative study on the irreversible
binding of labelled halothane, trichlorofluoromethane, chloroform
and carbon tetrachloride to hepatic protein and lipids in vitro
and in vivo. Arch Toxicol, 34: 289-308.
Uehleke H, Greim H, Krämer M, & Werner (1976) Covalent binding of
haloalkanes to liver constituents but absence of mutagenicity on
bacteria in metabolising test system. Mutat Res, 38: 114.
Uehleke H, Werner T, Greim H, & Krämer M (1977) Metabolic activation
of haloethanes and tests in vitro for mutagenicity. Xenobiotica,
7: 393-400.
Ullrich D (1982) [Organic halogen compounds in the air of some
Berlin indoor swimming pools.] WoBoLu-Berlin, 1: 50-52 (in German).
US EPA (1971) Compendium of registered pesticides. Washington, DC,
US Environmental Protection Agency, p III-C-19.
US EPA (1984) Locating and estimating air emissions from sources of
chloroform. US Washington, DC, Environmental Protection Agency
(Report PB-84-200617).
US EPA (1992) Technical Support Division (TSD). Disinfection
by-products field study data base. Washington, DC, US Environmental
Protection Agency, Office of Ground Water and Drinking Water,
Technical Support Division.
US FDA (US Food and Drug Administration (1976) Chloroform as an
ingredient of human drug and cosmetic products. Fed Reg, 41:
26842-26845.
US NIOSH (1984) Manual of analytical methods - Volume 1: Method
1003. Cincinnati, Ohio, National Institute for Occupational Safety
and Health.
US NIOSH (1989) National occupational exposure survey (NOES) as of
March 29, 1989. Cincinnati, Ohio, National Institute for
Occupational Safety and Health.
Van Abbé NJ, Green TJ, Jones E, Richold M, & Roe FJC (1982)
Bacterial mutagenicity studies on chloroform in vitro. Food Cosmet
Toxicol, 20: 557-561.
Van Beelen P & Van Keulen F (1990) The kinetics of the degradation
of chloroform and benzene in anaerobic sediment from the river
Rhine. Hydrobiol Bull, 24: 13-21.
Van der Heijden CA, Speijers GJA, Ros JPM, Huldy HJ, Besemer AC,
Lanting RW, Maas RJM, Heijna-Merkus E, Bergshoeff G, Gerlofsma A,
Mennes WC, Van der Most PFJ, De Vrijer FL, Janssen PCJM, Knaap AGAC,
Huijgen C, Duiser JA, & De Jong P (1986) Criteria document on
chloroform. Bilthoven The Netherlands, National Institute of Public
Health and Environmental Protection (Report No. 738513004).
Van Dyke RA, Chenoweth MB, & Van Poznak A (1964) Metabolism of
volatile anaesthetics-I. Conversion in vivo of several
anaesthetics to 14CO2 and chloride. Biochem Pharmacol, 13:
1239-1247.
Van Luin AB & Van Starkenburg W (1984) Hazardous substances in waste
water. Water Sci Tech, 17: 843-853.
Van Tassel S, Amalfitano N, & Narang RS (1981) Determination of
arenes and volatile halo-organic compounds in air at microgram per
cubic meter levels by gas chromatography. Anal Chem, 53: 2130-2135.
Veith GD, Macek KJ, Petrocelli SR, & Carroll J (1978) An evaluation
of using partition coefficients and water solubility to estimate
bioconcentration factors for organic chemicals in fish. In: Eaton
JG, Parrish PR, & Hendricks AC ed. Aquatic toxicology. Proceedings
of the Third Annual Symposium on Aquatic Toxicology. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 116-129
(STP 707).
Venitt S & Crofton-Sleigh C (1981) Mutagenicity of 42 coded
compounds in a bacterial assay using Escherichia coli and
Salmonella typhimurium. In: De Serres FJ & Ashby J ed. Evaluation
of short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 351-360 (Progress in Mutation Research, Volume 1).
Verschueren U (1983) Handbook of environmental data on organic
chemicals, 2nd ed. New York, Van Nostrand Reinhold Company, pp
367-369.
Vézina M, Kobusch AB, Du Souich P, Greselin E, & Plaa GL (1990)
Potentiation of chloroform-induced hepatotoxicity by methyl isobutyl
ketone and two metabolites. Can J Physiol Pharmacol, 68: 1055-1061.
Vittozzi L, Testai E, & De Biasi A (1991) Multiple bioactivation of
chloroform: a comparison between man and experimental animals. Adv
Exp Med Biol, 283: 665-667.
Vogel E, Blijleven WGH, Kortselius MJH, & Zijlstra JA (1981)
Mutagenic activity of 17 coded compounds in the sex-linked recessive
lethal test in Drosophila melanogaster. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 660-665 (Progress in Mutation Research,
Volume 1).
Von Oettingen WF (1964) The halogenated hydrocarbons of industrial
and toxicological importance. Amsterdam, Oxford, New York, Elsevier
Science Publishers, pp 77-108.
Von Oettingen WF, Powell CC, Sharpless NE, Alford WC, & Pecoza LJ
(1950) Comparative studies of the toxicity and pharmacodynamic
action of chlorinated methanes with special reference to their
physical and chemical action. Arch Int Pharmacodyn, 81: 17-34.
Wallace LA (1987) The total exposure assessment methodology (TEAM)
study: Summary and analysis. Volume I (EPA/600/6 - 87/002a).
Wallace LA, Pellizzari E, Hartwell T, Rosenzweig M, Erickson M,
Sparacino, C & Zelon H (1984) Personal exposure to volatile organic
compounds. I. Direct measurements in breathing-zone air, drinking
water, food and exhaled breath. Environ Res, 35: 293-319.
Wecher RA & Scher S (1982) Bioassay procedures for identifying
genotoxic agents using light emitting bacteria as indicator
organisms. In: Seno M & Pazzagli M ed. Luminescent assays:
perspectives in endocrinology and clinical chemistry. New York,
Raven Press Ltd, pp 109-113.
Westrick JJ, Mello JW, & Thomas RF (1989) The groundwater supply
survey. J Am Water Works Assoc, 76: 52-59.
White AE, Takehisa S, Eger EI, Wolff S, & Stevens WC (1979) Sister
chromatid exchanges induced by inhaled anaesthetics. Anesthesiology,
50: 426-430.
WHO (1993) Guidelines for drinking-water quality, 2nd ed. Volume 1:
Recommendations. Geneva, World Health Organization.
WHO (in press) IPCS Environmental Health Criteria 170: Assessing
human health risks of chemicals: Derivation of guidance values for
health-based exposure limits. Geneva, World Health Organization.
Williams DT, Olson R, Bothwell PD, Murphy KI, & Robertson JL (1980)
Trihalomethane levels in Canadian drinking water. Environ Sci Res,
16: 503-512.
Wilson JT, Enfield CG, Dunlap WJ, Cosby RL, Foster DA, & Baskin LB
(1981) Transport and fate of selected organic pollutants in a sandy
soil. J Environ Qual, 10: 501-506.
Windholz M ed. (1983) The Merck index: an encyclopedia of chemicals,
drugs, and biologicals, 10th ed. Rahway, New Jersey, Merck & Co.,
Inc.
Winslow SG & Gerstner HB (1978) Health aspects of chloroform. A
review. Drug Chem Toxicol, 1: 259-275.
Withey JR & Collins BT (1980) Chlorinated aliphatic hydrocarbons
used in the foods industry: the comparative pharmacokinetics of
methylene chloride, 1,2-dichloroethane, chloroform and
trichloroethylene after i.v. administration in the rat. J Environ
Pathol Toxicol, 3: 313-332.
Withey JR & Karpinski K (1985) The fetal distribution of some
aliphatic chlorinated hydrocarbons in the rat after vapour phase
exposure. Biol Res Pregnancy, 6: 79-88.
Withey JR, Collins BT, & Collins PG (1983) Effect of vehicle on the
pharmacokinetics and uptake of four halogenated hydrocarbons by the
gastrointestinal tract of the rat. J Appl Toxicol, 3: 249-253.
Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, & Ullrich V (1977)
The reduction of polyhalogenated methanes by liver microsomal
cytochrome P-450. Mol Pharmacol, 13: 698-705.
Youssefi M, Faust SD, & Zenchelsky ST (1978) Rapid determination of
light halogenated hydrocarbons in urine. Clin Chem, 24: 1109-1111.
Zaleska-Rutczynska Z & Krus S (1973) Genetic aspects of the high and
low sensitivity to chloroform of male mice of various inbred strains
and their crosses. Pol Med Sci Hist Bull Abstr, 1: 245-250.
Zimmermann FK & Scheel I (1981) Induction of mitotic gene conversion
in strain D7 of Saccharomyces cerevisiae by 42 coded chemicals.
In: De Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 481-490
(Progress in Mutation Research, Volume 1).
Zoeteman BCJ, Hrubec J, de Greef E, & Kool HJ (1982) Mutagenic
activity associated with by-products of drinking water disinfection
by chlorine, chlorine dioxide, ozone and UV-irradiation. Environ
Health Perspect, 46: 197-205.
RESUME
Le chloroforme se présente sous la forme d'un liquide volatil,
limpide et incolore, à l'odeur caractéristique et au goût âcre et
douceâtre. Il peut être décomposé par voie photochimique, il n'est
pas inflammable et il est soluble dans la plupart des solvants
organiques. Toutefois sa solubilité dans l'eau est limitée. Lors de
la décomposition chimique, il peut y avoir formation de phosgène et
d'acide chlorhydrique.
Le chloroforme s'emploie dans certaines formulations de
pesticides, comme solvant et comme intermédiaire dans la fabrication
de certains dérivés. Son utilisation comme anesthésique ou dans des
spécialités pharmaceutiques est interdite dans un certain nombre de
pays. La production de chloroforme à des fins commerciales a atteint
440 000 tonnes en 1987. Du chloroforme se forme également en
quantités appréciables lors de la chloration de l'eau et du
blanchiment de la pâte à papier.
L'analyse de l'air, de l'eau et d'échantillons biologiques pour
la recherche et le dosage du chloroforme peut s'effectuer selon
plusieurs méthodes. La majorité d'entre elles consiste en une
injection directe sur colonne, une adsorption sur un adsorbant
activé ou une condensation dans un piège froid; on procède ensuite à
une désorption ou à une extraction par un solvant qui est ensuite
chassé avant analyse finale par chromatographie en phase gazeuse.
On pense que la majeure partie du chloroforme présent dans
l'eau finit par passer dans l'air, en raison de la volatilité de ce
composé. Le temps de séjour du chloroforme dans l'atmosphère est de
plusieurs mois et il en est éliminé après transformation chimique.
Il résiste à la biodégradation aérobie par les bactéries du sol et
des nappes phréatiques qui se développent sur des substrats
endogènes ou en présence d'un supplément d'acétate. Il peut y avoir
biodégradation en anaérobiose. La bioconcentration est faible chez
les poissons d'eau douce. La dépuration est rapide.
D'après l'estimation de l'exposition moyenne due aux divers
milieux, on pense que la population générale est principalement
exposée au chloroforme par l'intermédiaire de la nourriture, de
l'eau de boisson et de l'air intérieur, dans des proportions à peu
près égales. L'absorption estimative à partir de l'air intérieur est
cependant beaucoup moindre. L'absorption moyenne totale estimative
est d'environ 2 µg/kg de poids corporel, par jour. Les données
disponibles indiquent également que l'utilisation domestique de
l'eau contribue de façon très importante à la concentration du
chloroforme dans l'air intérieur et par voie de conséquence à
l'exposition totale. Pour certaines personnes qui vivent dans des
habitations où l'eau de distribution renferme des concentrations
relativement élevées de chloroforme, on estime que l'absorption
totale peut aller jusqu'à 10 µg/kg de poids corporel et par jour.
Une fois administré par voie orale, le chloroforme est bien
résorbé chez l'animal et l'homme, mais la cinétique d'absorption
dépend du véhicule. Chez l'homme, après exposition par la voie
respiratoire, 60 à 80% de la dose inhalée sont absorbés. Les
principaux facteurs qui agissent sur la cinétique d'absorption du
chloroforme après inhalation sont la concentration ainsi que la
capacité de métabolisation, qui dépend de l'espèce. Chez l'homme et
l'animal, le chloroforme est rapidement résorbé par la peau et l'on
a montré qu'il pouvait être également absorbé par voie percutanée
dans une proportion importante à partir de l'eau lors d'une douche.
Il semble que l'hydratation de l'épiderme accélère la résorption du
chloroforme.
Le chloroforme se répartit dans l'ensemble de l'organisme.
C'est dans les graisses, le sang, le foie, les reins, les poumons et
le système nerveux que l'on trouve les plus fortes concentrations
tissulaires. La répartition du chloroforme dépend de la voie
d'exposition; la dose est plus forte dans les tissus
extra-hépatiques après inhalation ou absorption percutanée qu'après
ingestion. On a montré que chez plusieurs espèces animales et chez
l'homme, le chloroforme pouvait traverser la barrière placentaire.
Il s'élimine essentiellement dans l'air expiré sous forme de dioxyde
de carbone. Non métabolisé, il demeure plus longtemps dans les
graisses que dans les autres tissus.
La biotransformation oxydative du chloroforme en
trichlorométhanol est catalysée par le cytochrome P-450. Le
trichlorométhanol produit, par élimination d'HCl, un intermédiaire
réactif, le phosgène. Le phosgène peut être détoxifié en dioxyde de
carbone par réaction avec l'eau ou en divers adduits par réaction
avec des thiols, notamment le glutathion ou la cystéine. La réaction
du phosgène sur les protéines tissulaires entraîne des lésions
cellulaires et la mort. La liaison des métabolites du chloroforme à
l'ADN est limitée. Le chloroforme peut également subir une
biotransformation réductrice catalysée par le P-450, qui donne
naissance au radical dichlorométhyl, lequel se fixe ensuite par
liaison covalente aux lipides tissulaires. On n'a pas déterminé si
cette biotransformation réductrice jouait également un rôle dans la
cytotoxicité du chloroforme.
Chez l'animal et l'homme exposés à du chloroforme, le
chloroforme est éliminé d'une part sous forme de dioxyde de carbone
et d'autre part sous forme inchangée. La fraction de la dose qui est
éliminée sous forme de dioxyde de carbone varie avec cette dose et
l'espèce en cause. La vitesse de biotransformation en dioxyde de
carbone est plus élevée dans les microsomes hépatiques et rénaux des
rongeurs (hamster, souris, rat) que dans ceux de l'homme. La
biotransformation du chloroforme est également plus rapide dans les
microsomes rénaux des souris que dans ceux des rats.
En ce qui concerne la toxicité aiguë, c'est le foie qui est
l'organe-cible chez le rat et plusieurs souches de souris. Les
lésions hépatiques se caractérisent essentiellement, au début, par
une infiltration graisseuse et une ballonisation des cellules, qui
évoluent vers une nécrose centrilobulaire, puis une nécrose massive.
Le rein est l'organe-cible chez les souris mâles appartenant à des
souches plus sensibles. Au niveau du rein, les lésions débutent par
une dégénérescence hydropigène qui évolue vers la nécrose des
tubules proximaux. On n'a pas observé de toxicité rénale importante
chez les femelles d'aucune souche de souris.
La toxicité aiguë varie en fonction de la souche, du sexe et du
véhicule. Chez la souris, la DL50 par voie orale varie de 36 à
1366 mg/kg de poids corporel alors que chez le rat, elle peut aller
de 450 à 2000 mg de chloroforme par kg de poids corporel. Après une
seule exposition de 4 heures par voie respiratoire, on a observé des
effets toxiques sur le foie chez la souris et le rat à des
concentrations de chloroforme respectivement égales à 490 et 1410
mg/m3.
Ce sont les lésions du foie qui sont l'effet toxique du
chloroforme le plus universellement observé. La gravité de ces
effets par dose unitaire administrée dépend de l'espèce, du véhicule
et du mode d'administration du chloroforme. La dose la plus faible à
laquelle on ait observé ces lésions est de 15 mg/kg de poids
corporel et par jour, administrée à des chiens "beagle" dans une
base de pâte dentifrice, pendant une période de 7,5 années. On n'a
pas recherché s'il y avait des effets à des doses plus faibles. Chez
les autres espèces, les doses nécessaires pour produire des effets
hépatotoxiques sont un peu plus élevées. Bien qu'au cours de ces
différentes études, la durée d'exposition ait été variable, on a pu
fixer la concentration sans effets nocifs observables à 15-125 mg/kg
de poids corporel et par jour.
Les effets au niveau du rein ont été observés chez des mâles
appartenant à des souches sensibles de souris ainsi que chez des
rats F-344. Ces effets étaient graves chez les mâles appartenant à
une souche de souris particulièrement sensible, à des doses ne
dépassant pas 36 mg/kg de poids corporel et par jour.
Chez des rats F-344 à qui l'on avait fait inhaler du
chloroforme 7 jours de suite, tous les jours pendant 6 heures, on a
observé une atrophie des glandes de Bowman ainsi que la présence
d'os néoformés dans les cornets du nez. La dose sans effets
observables correspondante se situait à 14,7 mg/m3 (3 ppm). Des
études à long terme se poursuivent afin d'évaluer la portée de ces
effets.
On a constaté l'apparition de tumeurs hépatiques chez des
souris à qui l'on avait administré par gavage des doses quotidiennes
de chloroforme dans de l'huile de maïs, à raison de 138 à 477 mg/kg
de poids corporel. Toutefois, lorsque des doses analogues étaient
administrées dans l'eau de boisson, le chloroforme était sans
influence sur la proportion des tumeurs hépatiques qui se formaient
chez ces souris. De plus, lors d'études sur le caractère promoteur
éventuel de ce composé, on a observé, qu'administré dans l'eau de
boisson, le chloroforme avait en fait une action inhibitrice sur la
formation de tumeurs du foie provoquées chez la souris avec de la
diéthylnitrosamine comme initiateur. Le véhicule utilisé ou la
manière d'administrer le chloroforme conditionne donc de façon
importante son pouvoir tumoro-inducteur au niveau du foie chez la
souris.
Le chloroforme a produit des tumeurs rénales chez des rats qui
en avaient reçu quotidiennement par gavage, dans de l'huile de maïs,
des doses allant de 90 à 200 mg/kg de poids corporel. Toutefois,
chez cette espèce, les résultats se sont révélés analogues lorsque
le produit était administré dans l'eau de boisson, ce qui indique
que les effets ne dépendent pas entièrement du véhicule utilisé.
Il semble que les effets cancérogènes du chloroforme sur le
foie et le rein des rongeurs soient étroitement liés à son action
cytotoxique ainsi qu'aux effets que ce composé exerce sur la
réplication cellulaire dans les organes-cibles. On a constaté que
ces derniers effets suivaient de près les modifications de la
réponse cancérogène au chloroforme en fonction du type de véhicule
et du mode d'administration. A la lumière des données disponibles,
il semble que le chloroforme ne soit guère capable d'induire des
mutations géniques ou d'autres types de lésions directes de l'ADN.
En outre, le chloroforme ne semble pas non plus capable de jouer le
rôle d'initiateur tumoral au niveau du foie chez la souris ni
d'induire une synthèse non programmée de l'ADN in vivo. En
revanche, lorsqu'il est administré dans un véhicule huileux, le
chloroforme peut se révéler un promoteur efficace des tumeurs
hépatiques. Par conséquent, il est probable que, lors de
l'administration prolongée de chloroforme, la cytotoxicité de ce
composé et la prolifération cellulaire qu'il détermine sont les
causes les plus importantes de la formation de tumeurs hépatiques et
rénales chez les rongeurs.
On dispose de quelques données limitées selon lesquelles le
chloroforme serait toxique pour le foetus, mais uniquement à des
doses auxquelles il est également toxique pour la mère.
En général, le chloroforme détermine les mêmes symptômes
toxiques chez l'homme que chez l'animal. Chez l'homme, l'anesthésie
peut entraîner la mort par suite d'arythmie et d'insuffisance
respiratoire et cardiaque. On a également observé chez l'homme une
nécrose des tubules rénaux et une insuffisance rénale. Les doses les
plus faibles pour lesquelles des cas de toxicité hépatique due à une
exposition professionnelle au chloroforme ont fait l'objet de
rapports, se situaient dans les limites de 80 à 160 mg/m3 (durée
d'exposition de moins de 4 mois) selon une étude et allaient de 10 à
1000 mg/m3 (durée d'exposition: 1 à 4 ans) selon une autre étude.
On estime que la dose mortelle moyenne par voie orale pour un adulte
est d'environ 45 g, mais on note d'importantes différences de
sensibilité selon les individus. On est fondé à croire, selon
certaines études épidémiologiques, qu'il existe une association
entre l'exposition aux sous-produits des désinfectants présents dans
l'eau de boisson et les cancers colorectaux ou vésicaux. Cependant,
ces études souffrent de la présence de facteurs de confusion, entre
autres faiblesses. Les preuves avancées à l'appui de la
cancérogénicité pour l'homme de l'eau de boisson chlorée, sont
insuffisantes. En outre, la présence de sous-produits des
désinfectants utilisés ne peut être attribuée au chloroforme
lui-même.
Le chloroforme est toxique pour les stages embryo-larvaires de
certaines espèces d'amphibiens et de poissons. La CL50 la plus
faible dont il ait été fait état, se situait à 0,3 mg/litre pour les
stades embryo-larvaires de Hyla crucifer. Le chloroforme est moins
toxique pour les poissons et pour la daphnie Daphnia magna. Pour
plusieurs espèces de poissons, les valeurs de la CL50 se situent
dans les limites de 18 à 191 mg/litre. Il n'y a guère de différences
de sensibilité entre les poissons d'eau douce et les poissons de
mer. En ce qui concerne Daphnia magna, la valeur la plus faible de
la CL50 qui ait été signalée, était de 29 mg/litre. Le chloroforme
est peu toxique pour les algues et autres microorganismes.
Le Groupe de travail a estimé que les données disponibles
étaient suffisantes pour établir une dose journalière tolérable
(DJT) pour les effets non cancérogènes du chloroforme, ainsi qu'une
dose spécifiquement liée au risque d'effets cancérogènes, sur la
base des études effectuées chez l'animal; les valeurs ainsi fixées
serviront de guide pour l'établissement de limites d'exposition par
les autorités compétentes. Cependant, il est rappelé que lorsque les
conditions locales imposent un choix entre le respect des limites
microbiologiques ou celles qui concernent la présence de
sous-produits de désinfection tels que le chloroforme, c'est la
qualité microbiologique qui doit toujours l'emporter. Il ne faut
jamais transiger sur l'efficacité de la désinfection.
En se fondant sur l'étude de Heywood et al. (1979) et en
introduisant un facteur d'incertitude de 1000 (x10 pour les
variations interspécifiques, x10 pour les variations
intraspécifiques et x10 pour l'utilisation d'une dose avec effet
plutôt que d'une dose sans effet lors d'une étude subchronique), on
obtient une DJT de 15 µg/kg de poids corporel; il faut rappeler que
cette étude avait révélé l'existence d'une légère hépatotoxicité (à
savoir une augmentation des enzymes hépatiques sériques et des
kystes graisseux) chez des chiens "beagle" à qui l'on avait fait
ingérer pendant 7,5 ans, une pâte dentifrice contenant du
chloroforme à la dose de 15 mg/kg de poids corporel et par jour.
En se fondant sur ce que l'on sait du mécanisme de ces
phénomènes, la méthode que l'on juge la mieux adaptée pour obtenir
une valeur-guide consiste à diviser la valeur de la concentration
sans effet observable sur la prolifération cellulaire par un certain
facteur d'incertitude. C'est ainsi que si l'on utilise la valeur de
la dose sans effets observables obtenue par Larson et al. (1993b)
pour la cytoléthalité et la prolifération cellulaire chez des souris
B6C3F1 qui avaient reçu pendant 3 semaines, dans de l'huile de
maïs, une dose quotidienne de chloroforme équivalant à 10 mg/kg de
poids corporel, et en introduisant un facteur d'incertitude de 1000
(x10 pour les variations interspécifiques, x10 pour les variations
intraspécifiques et x10 pour la gravité de l'effet, c'est-à-dire la
cancérogénicité et parce qu'il s'agit d'une étude subchronique), on
obtient une DJT de 10 µg/kg de poids corporel.
On admet que les tumeurs rénales observées chez le rat peuvent
également être liées à l'action létale du chloroforme sur les
cellules et à ses effets sur leur prolifération. Cependant, étant
donné que l'on ne possède pas de données sur la prolifération
cellulaire chez les souches où l'on a observé des tumeurs et qu'en
outre, ce que l'on peut savoir de cet effet et de l'effet létal du
chloroforme sur les cellules n'a été observé qu'à court terme (un
seul gavage et une exposition par voie respiratoire de 7 jours), on
estime qu'il est prématuré de s'écarter du modèle par défaut
(c'est-à-dire multistade linéarisé) pour l'estimation du risque de
cancer sur la durée de vie. D'après l'étude de Jorgenson et al.
(1985) qui portait sur l'induction de tumeurs rénales (adénomes et
adénocarcinomes), on a fixé à 8,2 µg/kg de poids corporel et par
jour, la dose quotidienne totale jugée capable de produire un excès
de risque de 10-5 sur toute la durée de la vie.
La concentration de chloroforme dans les eaux de surface est
généralement faible et ne semble pas présenter de danger pour les
organismes aquatiques. Toutefois, la décharge ou le déversement de
produits industriels pourrait entraîner la présence de
concentrations plus élevées de chloroforme dans ces eaux et les
rendre dangereuses pour les stades embryo-larvaires de certaines
espèces aquatiques.
RESUMEN
El cloroformo es un líquido transparente, incoloro y volátil,
con un olor característico y un sabor dulce ardiente. Se degrada
fotoquímicamente, no es inflamable y es soluble en la mayor parte de
los disolventes orgánicos. Sin embargo, su solubilidad en agua es
limitada. Por degradación química del mismo pueden formarse fosgeno
y ácido hidroclorhídrico.
El cloroformo se utiliza en la formulación de plaguicidas, como
disolvente y como intermedio químico. Su utilización como anestésico
y en especialidades farmacéuticas está prohibida en algunos países.
La producción comercial ascendió a 440 000 toneladas en 1987.
También se producen cantidades apreciables de cloroformo en la
cloración del agua y en el blanqueado de la pasta papelera.
Existen varios métodos analíticos para determinar la presencia
de cloroformo en el aire, el agua y los materiales biológicos. La
mayor parte de esos métodos se basan en la inyección directa en
columna, la adsorción en adsorbentes activados o la condensación en
una cámara fría y posteriormente la desorción o evaporación mediante
la extracción por disolventes o el calentamiento y el subsiguiente
análisis por cromatografía de gases.
Se supone que la mayor parte del cloroformo presente en el agua
se transfiere finalmente al aire debido a su volatilidad. El
cloroformo tiene un tiempo de residencia en la atmósfera de varios
meses y desaparece de la misma por transformación química. Es
resistente a la biodegradación por la población microbiana aeróbica
de los suelos y de las capas acuíferas que viven en subestratos
endógenos o con el suplemento de acetato. La biodegradación es
posible en condiciones anaeróbicas. La bioconcentración en los peces
de agua dulce es baja. La depuración es rápida.
Las estimaciones de la exposición media calculadas a partir de
diversos medios indican que la población en general está expuesta al
cloroformo principalmente a través de los alimentos, el agua de
bebida y el aire de los interiores en cantidades aproximadamente
equivalentes. La inhalación estimada por conducto del aire exterior
es considerablemente menor. La ingesta media estimada total es de
aproximadamente 2 µg/kg de peso corporal por día. Los datos
disponibles también indican que el agua de uso doméstico contribuye
considerablemente a los niveles de cloroformo en el aire de los
interiores y a la exposición total. La ingesta total estimada de
algunos individuos que viven en lugares con un abastecimiento de
agua corriente con concentraciones relativamente elevadas de
cloroformo asciende a 10 µg/kg de peso corporal por día.
Los animales y los seres humanos absorben bien el cloroformo
después de la administración por vía oral, pero la cinética de la
absorción depende del vehículo suministrado. Tras la exposición por
inhalación, los seres humanos absorben del 60 al 80% de la cantidad
inhalada. Los factores principales que afectan a la cinética de la
absorción del cloroformo después de la inhalación son su
concentración y la capacidad metabólica específica de la especie.
Los seres humanos y los animales lo absorben fácilmente a través de
la piel y se ha demostrado que durante la ducha la absorción dérmica
del cloroformo del agua es apreciable. La hidratación de la piel
parece acelerar la absorción de cloroformo.
El cloroformo se distribuye en todo el cuerpo. Los niveles
tisulares más elevados se alcanzan en el tejido adiposo, la sangre,
el hígado, los riñones, los pulmones y el sistema nervioso. La
distribución depende de la vía de exposición; los tejidos
extrahepáticos reciben una dosis más elevada del cloroformo inhalado
o absorbido por la piel que del cloroformo ingerido. Se ha
demostrado que en varias especies animales y en el ser humano el
cloroformo se transfiere a través de la placenta. El cloroformo se
elimina principalmente como dióxido de carbono exhalado. El
cloroformo no metabolizado se mantiene más tiempo en el tejido
adiposo que en cualquier otro tejido.
El citocromo P-450 cataliza la biotransformación oxidativa del
cloroformo en triclorometanol. La pérdida de HCl del triclorometanol
produce fosgeno como reactivo intermedio. El fosgeno puede
destoxificarse por reacción con el agua produciendo dióxido de
carbono o con tioles, inclusive con glutatión o cisteína,
produciendo aductos. La reacción del fosgeno con proteínas tisulares
está asociada con daño y necrosis celulares. Se observa un escaso
enlace de los metabolitos del cloroformo con el ADN. El cloroformo
también es objeto de una biotransformación reductiva catalizada por
el P-450 que produce radicales de diclorometilo y éstos contraen
enlaces covalentes con los lípidos tisulares. No se ha determinado
el papel de la biotransformación reductiva en la citotoxicidad del
cloroformo.
Los animales y los seres humanos expuestos al cloroformo
eliminan con el aire espirado el dióxido de carbono y el cloroformo
que no se ha transformado. La fracción de dosis eliminada como
dióxido de carbono varía según la dosis y la especie. La tasa de
biotransformación en dióxido de carbono es más elevada en los
microsomas hepáticos y renales de roedores (hámster, ratón, rata)
que en los microsomas hepáticos y renales humanos. Además, el
cloroformo se biotransforma más rápidamente en los microsomas
renales del ratón que en los de la rata.
El hígado es el órgano vulnerable a la toxicidad aguda en las
ratas y en varias estirpes de ratones. La lesión hepática se
caracteriza principalmente por una infiltración grasa temprana y
células con forma de globo y evoluciona hacia la necrosis
centrilobular seguida de necrosis general. El riñón es el órgano
vulnerable en los ratones macho de otras estirpes más sensibles. La
lesión renal comienza con una degeneración hidrópica que avanza
hacia la necrosis de los tubos proximales. No se ha observado una
toxicidad renal apreciable en las ratas hembra de ninguna estirpe.
La toxicidad aguda varía según la raza, el sexo y el vehículo.
En el ratón, la DL50 por vía oral oscila entre 36 y 1366 mg de
cloroformo/kg de peso corporal, mientras que en las ratas oscila
entre 450 y 2000 mg de cloroformo/kg de peso corporal. Después de
una sola exposición de cuatro horas por inhalación, se observó
toxicidad hepática en ratones y ratas cuando el nivel de cloroformo
alcanzaba, respectivamente, 490 y 1410 mg/m3.
Los efectos tóxicos del cloroformo más generales observados
consisten en lesiones hepáticas. La gravedad de esos efectos por
unidad de dosis administrada depende de la especie, del vehículo de
administración y del método por el cual se haya administrado el
cloroformo. La dosis más baja causante de lesión hepática observada
es de 15 mg/kg de peso corporal por día, administrada a perros
pachones en una base de pasta dentífrica durante un periodo de 7,5
años. No se han examinado efectos con dosis más bajas. Se necesitan
dosis algo más elevadas para producir efectos hepatotóxicos en otras
especies. En esos estudios, aunque la duración de la exposición
variaba, los niveles sin efectos adversos observados oscilaban entre
15 y 125 mg/kg de peso corporal por día.
Se han observado efectos en el riñón de ratones macho de
estirpes sensibles y en la rata F-344. Se han observado efectos
graves en una estirpe especialmente sensible de ratones macho con
dosis de sólo 36 mg/kg de peso corporal por día.
La inhalación de cloroformo seis horas por día durante siete
días consecutivos produjo atrofia de las glándulas de Bowman y
neoplasia ósea en la concha nasal de ratas F-344. El nivel en que no
se observaron esos efectos fue de 14,7 mg/m3 (3 ppm). La
importancia de dichos efectos se está investigando más a fondo en
estudios de larga duración.
El cloroformo administrado por sonda en un vehículo de aceite
de maíz en dosis de 138 a 477 mg/kg de peso corporal por día indujo
tumores hepáticos en ratones. Sin embargo, dosis semejantes de
cloroformo administradas en el agua de bebida no produjeron tumores
hepáticos en ratones. Por otra parte, en estudios de
iniciación/promoción, el cloroformo administrado en el agua de
bebida como promotor parecía inhibir el desarrollo de tumores
hepáticos iniciados por dietilnitrosamina en ratones. Así pues, el
vehículo y/o el método de administración del cloroformo es una
variable importante en relación con la inducción de tumores
hepáticos en el ratón.
El cloroformo administrado por sonda en aceite de maíz en dosis
de 90 a 200 mg/kg de peso corporal por día indujo tumores renales en
ratas. Sin embargo, en esa especie se observaron efectos semejantes
tras la administración de cloroformo en el agua de bebida, lo que
indica que la reacción no depende exclusivamente del vehículo
utilizado.
Los efectos carcinogénicos del cloroformo en el hígado y los
riñones de roedores parecen estar estrechamente relacionados con
efectos citotóxicos y de replicación celular observados en los
órganos vulnerables. Se ha observado que los efectos en la
replicación celular eran paralelos a las modificaciones de las
respuestas carcinogénicas al cloroformo inducidas por el vehículo y
por la modalidad de administración. Las observaciones realizadas
indican que el cloroformo tiene poca o ninguna capacidad para
inducir mutaciones genéticas o daños directos de otro tipo en el
ADN. Por otra parte, el cloroformo no parece poder iniciar tumores
hepáticos en ratones ni de inducir síntesis imprevistas de ADN in
vivo. Por otra parte, el cloroformo puede promover la neoplasia
hepática cuando se administra en un vehículo oleoso. Por
consiguiente, es probable que, tras la administración prolongada de
cloroformo, la citotoxicidad seguida de proliferación celular sea la
causa más importante del desarrollo de tumores hepáticos y renales
en los roedores.
Algunos datos limitados sugieren que el cloroformo es tóxico
para el feto, pero sólo en dosis tóxicas para la madre.
En general, el cloroformo provoca en el ser humano los mismos
síntomas de toxicidad que en los animales. En el ser humano, la
anestesia puede causar la muerte por arritmia e insuficiencia
respiratoria y cardíaca. En el ser humano también se ha observado
necrosis de los tubos renales y disfunción renal. Los niveles más
bajos en los que se haya comunicado toxicidad hepática debida a la
exposición ocupacional al cloroformo se sitúan entre 80 y 160
mg/m3 (con un periodo de exposición inferior a cuatro meses) en un
estudio y entre 10 y 1000 mg/m3 (con periodos de exposición de uno
a cuatro años) en otro estudio. La dosis letal media de un adulto se
estima en unos 45 g, pero hay grandes diferencias de vulnerabilidad
de un individuo a otro. En algunos estudios epidemiológicos hay
ciertas indicaciones de que existe una asociación entre la
exposición a los subproductos de la desinfección del agua de bebida
y el cáncer colorrectal y de vejiga. Sin embargo, hay factores
confusos y otras insuficiencias que ponen en entredicho esos
estudios. Las pruebas de la carcinogenicidad del agua de bebida
clorada en el ser humano son insuficientes. Además, los subproductos
de la desinfección no pueden atribuirse al cloroformo por sí solo.
El cloroformo es tóxico en las fases embriolarvales de algunas
especies de anfibios y de peces. La CL50 más baja comunicada es de
0,3 mg/litro en las fases embriolarvales de Hyla crucifer. El
cloroformo es menos tóxico para los peces y para Daphnia magna. La
CL50 de varias especies de peces se halla entre 18 y 191 mg/litro.
Hay pocas diferencias de sensibilidad entre los peces de agua dulce
y salada. La CL50 más baja comunicada en Daphnia magna es de 29
mg/litro. El cloroformo es poco tóxico para las algas y otros
microorganismos.
El Grupo Especial llegó a la conclusión de que los datos
disponibles son suficientes para fijar una ingesta diaria tolerable
sin efectos neoplásicos e ingestas con riesgos carcinogénicos
específicos del cloroformo sobre la base de los estudios realizados
en especies animales; las dosis servirán como orientación para que
las autoridades competentes fijen límites de exposición. Sin
embargo, se advierte que, cuando las circunstancias locales exijan
optar entre el cumplimiento de límites microbiológicos y el de
límites para subproductos de la desinfección tales como el
cloroformo, debe siempre prevalecer la calidad microbiológica.
Nunca debe comprometerse una desinfección eficaz.
Sobre la base del estudio de Heywood et al. (1979) en el cual
se observó una ligera hepatotoxicidad (aumento de las enzimas del
suero hepático y quistes grasos) en perros pachones que habían
ingerido 15 mg/kg de peso corporal por día en pasta dentífrica
durante 7,5 años, incorporando un factor de incertidumbre de 1000
(x10 para la variación entre especies, x10 para la variación dentro
de la especie y x10 para utilizar un nivel con efectos en lugar de
sin efectos y un estudio subcrónico), se obtiene una ingesta diaria
tolerable (IDT) de 15 µg/kg de peso corporal por día.
En función de los datos disponibles sobre los mecanismos
determinantes, el método que se considera más apropiado para
establecer orientaciones fundadas en los tumores hepáticos de
ratones es dividir un nivel sin efectos de proliferación celular por
un factor de incertidumbre. A partir del nivel sin efectos
observados de citoletalidad y proliferación celular en ratones
B6C3F1, de 10 mg/kg de peso corporal por día administrados en
aceite de maíz durante tres semanas, comunicado en el estudio de
Larson et al. (1993b), incorporando un factor de incertidumbre de
1000 (x10 para la variación entre especies, x10 para la variación
dentro de la especie y x10 para la gravedad del efecto, es decir,
carcinogenicidad, y estudio subcrónico) se obtiene una IDT de 10
µg/kg de peso corporal por día.
Se reconoce que los tumores renales en ratas también pueden
estar asociados con letalidad y proliferación celular. Sin embargo,
dado que no se dispone de datos sobre proliferación celular en la
estirpe en la que se observaron tumores y la información sobre
proliferación y letalidad celulares es de corto plazo (una sola
sonda y exposición por inhalación durante siete días), se considera
prematuro alejarse del modelo establecido por defecto (es decir,
fases múltiples linearizadas) como base para estimar el riesgo de
cáncer durante una vida. La ingesta diaria total que se considera
asociada con un riesgo excesivo de 10-5 durante una vida, sobre la
base de la inducción de tumores renales (adenomas y adenocarcinomas)
en ratas macho en el estudio de Jorgenson et al. (1985), es de 8,2
µg/kg de peso corporal por día.
Los niveles de cloroformo en las aguas superficiales son
generalmente bajos y no se prevé que constituyan un peligro para los
organismos acuáticos. Sin embargo, niveles más elevados de
cloroformo en las aguas superficiales como consecuencia de las
descargas o los derrames industriales tal vez sean peligrosos en las
fases embriolarvales de algunas especies acuáticas.
Phosgene reacts rapidly with water to give CO2 and HCl as
products, which explains the formation of CO2 as a metabolite of
chloroform (Fry et al., 1972; Brown et al., 1974b). Phosgene may
also react with tissue nucleophiles to form covalently bound
products (Uehleke & Werner, 1975). Cysteine blocks the covalent
binding of [14C]-chloroform-derived radioactivity, which supports
a role for phosgene in the formation of covalent adducts from
chloroform (Pohl et al., 1977, 1980). Alternatively, phosgene may
react with glutathione to form S-(chlorocarbonyl)glutathione; this
intermediate may react with glutathione to give diglutathionyl
dithiocarbonate (Pohl et al., 1981) or to give glutathione disulfide
and carbon monoxide as minor products (Ahmed et al., 1977).
The reductive biotransformation of chloroform is also catalysed
by cytochromes P450 (Testai & Vittozzi, 1986) (Fig. 1). Reduction of
chloroform gives rise to the dichloromethyl radical, which has been
identified by spin trapping and ESR (Tomasi et al., 1985). No
evidence for the formation of the dichloromethyl carbanion has been
presented, whereas the formation of chlorocarbene has been ruled out
(Wolf et al., 1977). The dichloromethyl radical may react
preferentially with the fatty acid skeleton of phospholipids to give
covalently bound adducts (De Biasi et al., 1992).
The balance between the oxidative and reductive
biotransformation of chloroform depends on several factors,
including oxygen and chloroform concentrations, animal species,
strain, enzyme induction, and the site of metabolism. Oxidative
metabolism is favoured at low (< 0.1 mM) chloroform concentrations
(Testai et al., 1990, 1991). Under these conditions, the oxygenation
of chloroform is catalysed by cytochrome P450 2E1 (Brady et al.,
1989; Guengerich et al., 1991), and covalent binding of chloroform
metabolites to proteins and lipids in incubation mixtures containing
mouse (B6C3F1 or C57BL/6J) liver microsomes is higher than in
incubation mixtures containing rat (Osborne-Mendel or
Sprague-Dawley) liver microsomes (Testai et al., 1991).
Chloroform reduction is increased at high substrate
concentrations (Testai et al., 1990), but oxidative metabolism is
quantitatively more important. In incubation mixtures containing 5
mM chloroform, both oxygenation and reduction of chloroform depend
on the oxygen tension in the incubation flask. Chloroform reduction
is particularly evident with microsomes from B6C3F1 mice and
Osborne-Mendel rats. At high chloroform concentrations (approx. 5
mM), the oxygenation of chloroform may be catalysed by cytochrome
P450 2B1, as suggested by the induction of the metabolism due to
pretreatment by phenobarbital (Branchflower et al., 1983; Testai &
Vittozzi, 1986; Nakajima et al., 1991). Phenobarbital or
ß-naphthoflavone pretreatment of Sprague-Dawley rats also stimulates
the formation of reduced intermediates of chloroform (Testai &
Vittozzi, 1986). Levels of the in vitro covalent binding of
[14C]-chloroform metabolites to proteins were higher with hepatic
microsomes from rabbits and human biopsies than with hepatic
microsomes from rats or mice (Uehleke & Werner, 1975).
The in vitro formation of dichloromethane as a stable
end-product of chloroform metabolism was addressed in early studies.
Dichloromethane was detected in mouse liver slices incubated with
chloroform (Butler, 1961), but not in slices or subcellular
fractions of rat liver incubated with chloroform (Paul & Rubinstein,
1963; Rubinstein & Kanics, 1964). These discrepancies, however, may
have been due to the incubation conditions employed in these early
studies. in vivo results with rats, dogs, mice and human
volunteers exposed to chloroform consistently indicated no
expiration of dichloromethane (Butler, 1961; Paul & Rubinstein,
1963; Fry et al., 1972; Brown et al., 1974b).
Interspecies differences in the oxidative metabolism of
chloroform have been found in vivo. After a [14C]-chloroform
dose of 60 mg/kg body weight, 85%, 66% and 18% was excreted as
[14C]-CO2 in C57BL, CF/PL and CBA mice, Sprague-Dawley rats, and
squirrel monkeys, respectively. Expiration of 14C accounted for
the elimination of most of the remaining dose (recoveries of 93-98%)
(Brown et al., 1974b). Mink et al. (1986) and Corley et al. (1990)
also showed that chloroform is metabolized in the mouse to a greater
extent than in the rat. Corley et al. (1990) demonstrated that the
covalent binding of [14C]-chloroform metabolites to liver and
kidney proteins in vivo was higher in B6C3F1 mice than in
Osborne-Mendel rats.
In several strains of mice given [14C]-chloroform, more
binding occurred in the kidney tissue of males than in that of
females (Ilett et al., 1973; Taylor et al., 1974). Male DBA mice
accumulate twice as much radioactivity in their kidneys as do male
C57BL mice. This strain difference shows intermediate or
multifactorial heredity (Hill et al., 1975).
Differences in binding were associated with variations in
toxicity (Hill et al., 1975; Clemens et al., 1979). The
nephrotoxicity of chloroform in male mice of susceptible strains
(see chapter 7) is most probably related to in situ renal
metabolic activation of chloroform (Zaleska-Rutczynska & Krus, 1973;
Hill, 1978; Clemens et al., 1979; Smith & Hook, 1983; Smith et al.,
1984). Indeed the overall biotransformation of chloroform in both
sexes is equal, whereas males exhibit more extensive formation of
renal tissue-bound metabolites than females (Taylor et al., 1974;
Smith & Hook, 1984). Smith et al. (1985) observed little chloroform
metabolism in rat (male, Fischer-344) renal cortical microsomes.
Additional studies, however, have demonstrated chloroform-induced
cytolethality and regenerative cell damage in male, Fischer-344 rat
kidney (Larson et al., 1993). Culliford & Hewitt (1957) reported
that females became more susceptible after pretreatment with
androgens, and the sensitivity of the males was reduced after
castration.
In the rat and mouse, chloroform biotransformation occurs
mainly in the liver, but other tissues also show metabolic activity.
After oral administration of chloroform to mice, maximum covalent
binding in the liver was observed after 3 h, whereas in the kidney,
maximum binding was found after 6 to 12 h. Binding appears to be
dose dependent up to doses of 3 mmol/kg body weight. At higher
doses, a plateau is reached (Ilett et al., 1973). Löfberg & Tjälve
(1986) studied the extra-hepatic metabolism of [14C]-chloroform in
Sprague-Dawley rats. Autoradiography was used to localize
metabolites in freeze-dried, extracted tissues to distinguish
between total and bound radioactivity. in vitro autoradiography,
in which tissue slices were incubated with [14C]-chloroform and
then examined autoradiographically, showed the capacity of several
tissues to metabolize [14C]-chloroform: liver, kidney cortex,
mucosa of the bronchial tree, tracheal mucosa, olfactory and
respiratory nasal mucosa, Bowman's glands in the olfactory lamina
propria mucosae, Steno's gland (the lateral nasal gland), mucosa of
the oesophagus, larynx, tongue, gingiva, cheek, naso-pharyngeal
duct, pharynx and the soft palate. Furthermore, autoradiographic
studies showed that a correlation exists between the ability of the
tissues to retain metabolites in vivo and the ability of these
tissues to metabolize chloroform in vitro.
The distribution of the covalent binding of 14C to DNA or RNA
after an intraperitoneal injection of [14C]-chloroform to Balb/c
mice or to Wistar rats shows several differences from the
distribution of the covalent binding to tissue proteins (Colacci et
al., 1991). The highest levels of covalent binding to DNA were
observed in mouse kidney (3.17 nmol/g DNA) and lung (3.65 nmol/g
DNA). In mice, binding to liver DNA (0.83 nmol/g DNA) was lower than
binding to stomach DNA (1.52 nmol/g DNA).
Differences in DNA binding of chloroform metabolites among rat
organs have been found to be limited, and the absolute values were
lower than those seen in mouse organs. In mice, RNA binding levels
were high in both the liver and kidney; in rats, they were higher in
the kidney than in other organs. In mice, protein binding was
highest in the liver (47.5 nmol/g), whereas in rats it was high in
both the liver (27.4 nmol/g) and kidney (30.7 nmol/g). In
incubations containing low (< 0.1 mM) chloroform concentrations and
human liver microsomes, little formation of reactive metabolites was
seen (Vittozzi et al., 1991). Detectable covalent binding was
observed in microsomes from some samples of colonic and ileal mucosa
of human patients but not of male Sprague-Dawley rats (Testai et
al., 1991).
Glutathione (GSH) is an important factor controlling the
binding of chloroform metabolites to proteins and lipids. In in
vitro studies, physiological concentrations of GSH (2-5 mM)
strongly reduced the covalent binding of chloroform metabolites to
proteins (Sipes et al., 1977; Cresteil et al., 1979; Smith & Hook,
1984). In later studies, 3 mM GSH blocked the covalent binding of
chloroform metabolites to proteins and to phospholipid polar heads,
whereas covalent binding to the phospholipid fatty acyl chains (due
to the radical metabolite) was only slightly affected (Testai &
Vittozzi, 1986; Testai et al., 1990, 1991; De Biasi et al., 1992).
Pretreatment of rats with diethylmaleate or buthionine sulfoximine
(BSO) increased the binding of administered [14C]-chloroform to
proteins (Stevens & Anders, 1981). Pretreatment of
phenobarbital-induced Sprague-Dawley rats with cysteine decreased
the covalent binding (Stevens & Anders, 1981b). Toxic effects
paralleled the covalent binding levels after these pretreatments
(Stevens & Anders, 1981a).
6.3 Human studies
6.3.1 Uptake
6.3.1.1 Oral
When Fry et al. (1972) dosed eight volunteers with 0.5 g
chloroform in olive oil in capsules, approximately 50% of the oral
dose was metabolized to carbon dioxide. Maximal blood levels of 1 to
5 ng chloroform/litre were achieved after 1.5 h. In two of the
subjects, the decline in blood levels could be described by a
two-compartment model with a half-life of 13 min for the initial
phase and a half-life of 90 min for the second phase. Chiou (1975)
reanalysed the data obtained from the two subjects mentioned above
and calculated an apparent volume of distribution of approximately
160 litres. The author estimated the hepatic first-pass effect to be
about 32% and the pulmonary first-pass effect to be 16%. Hence after
a single oral dose of 0.5 g chloroform, about 52% of the dose may be
available to the system. Pulmonary and metabolic clearances of 0.7
and 0.6 litre/min, respectively, gave a total body clearance of 1.3
litre/min.
6.3.1.2 Dermal
Jo et al. (1990a) studied the relative contributions of dermal
and pulmonary uptake of chloroform in individuals taking showers.
Post-exposure exhaled air concentrations of chloroform were measured
to estimate chloroform uptake and were 6 to 21 µg/m3 for normal
showers and 2.4 to 10 µg/m3 for inhalation-only exposure. The
difference between normal and inhalation-only exposure was
significant, and the authors concluded that the contribution of
dermal exposure was approximately equivalent to inhalation exposure.
Chinery & Gleason (1993) modified an existing PBPK model to
predict the exhaled air concentration of chloroform in individuals
exposed to the chemical while showering. Calibration of the model
with measured exhaled air concentrations of chloroform in
individuals exposed while showering either with or without dermal
absorption generated an expected value for skin-blood partitioning
of 1.2. This assumes a degree of transfer of chloroform from shower
water into shower air of 61%. The stratum corneum permeability
coefficient for chloroform was estimated to be within a range of
0.16-0.36 cm/h, and the expected value was 0.2 cm/h. The estimated
ratio of dermally:inhaled absorbed doses while showering ranged
between 0.6 and 2.2 and the expected value was 0.75.
6.3.1.3 Inhalation
The inhalation uptake of chloroform in humans was studied by
Lehmann & Hasegawa in 1910. More recently, Morgan et al. (1970)
measured the absorption of chloroform after a single inhalation
exposure to approximately 5 mg of [38Cl]-chloroform. About 80% of
the chloroform was absorbed under these conditions.
Prolonged inhalation of anaesthetic concentrations (about 50 g
chloroform/m3 air) gave rise to blood chloroform concentrations of
about 100 mg/litre (Smith et al., 1973).
The relative contribution of inhalation to chloroform uptake
during showering has been determined (see section 6.3.1.2, Jo et
al., 1990a).
6.3.2 Distribution
Corley et al. (1990) determined partition coefficients for
human tissues (see Table 8).
McConnell et al. (1975) analysed chloroform levels in
postmortem tissue from eight persons (four males and four females,
48 to 82 years old) living in non-industrial areas of the United
Kingdom. The chloroform levels (µg/kg wet tissue weight) observed
were: body fat, 5-68 (mean = 51); liver, 1-10 (mean = 7.2); kidney,
2-5; and brain, 2-4.
Phillips & Birchard (1991) reported on a nationwide survey of
the general population by the US EPA's National Human Adipose Tissue
Survey. Several hundred fat samples were pooled into 46 composite
samples by age and geographic region and were analysed. Chloroform
was detected at levels ranging from 5 to 580 ng/g in 29 of the
composite samples.
Dowty et al. (1976) detected chloroform in human maternal and
placental cord blood. Erickson et al. (1981) found chloroform,
supposedly originating from environmental exposure, present in
mother's milk (concentration not specified). Chloroform was
identified, but not quantified, in mother's milk samples collected
from 49 lactating women living in the vicinity of chemical
manufacturing plants or industrial user facilities in Pennsylvania,
New Jersey and Louisiana, USA (Pellizzari et al., 1982).
6.3.3 Elimination
Human volunteers given oral doses of 500 mg chloroform
eliminated on average 50% of the dose as CO2 and 40% as unchanged
chloroform during 8 h after dosing (Fry et al., 1972). The amount of
expired chloroform varied from 18 to 66%, depending on the obesity
of individuals.
After administration of 100, 250 or 1000 mg, the authors
recovered 0, 12 and 65% of the dose in the expired air,
respectively. After administration of [14C]-chloroform to a man
and a woman, approximately 50% of the dose was found in the exhaled
air (as CO2) in 7.5 h after dosing. Virtually no chloroform was
excreted by the kidneys (Fry et al., 1972).
After inhalation of chloroform (concentrations of 21 or 35 g
chloroform/m3), Lehmann & Hasegawa (1910) found little pulmonary
excretion, i.e. approximately 2% of the absorbed quantity within 30
min after the exposure. A pulmonary excretion of 10% of the body
content during the first hour after exposure was reported by Morgan
et al. (1970).
6.3.4 Biotransformation
Human cytochrome P450 2E1 catalyses the oxygenation of
chloroform (Guengerich et al., 1991).
Corley et al. (1990) quantified CO2 production from
incubations of human liver microsomes with 0.049-0.058 mM
chloroform. The average activity of samples from nine individuals
was 8.15 ± 0.02 pmol chloroform oxidized/min per mg protein. These
data were correlated with rodent in vitro and in vivo conversion
rates to estimate human in vivo metabolic rate constants (see
Table 8).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Lethality
The LD50 values of chloroform for mice and rats are given in
Tables 9 and 10, respectively. Chloroform-induced death is usually
due to liver damage, with the exception of male mice of very
sensitive strains, whose death is caused by kidney damage. The
higher susceptibility to chloroform acute toxicity in these strains
of mice (such as DBA, C3H, C3Hf, CBA, Balb/c, C3H/He), with respect
to other strains, is genetically controlled. An absolute sex-related
difference with respect to kidney damage, but not to liver damage,
has been described in mice: female mice do not develop renal
lesions. This is independent of the strain. Some influence of age on
chloroform acute toxicity in rats has also been described (Kimura et
al., 1971).
For the rat, the LD50 values ranged from 450 to 2000 mg
chloroform/kg body weight, and in this species no sex difference in
susceptibility was found (Kimura et al., 1971; Chu et al., 1980).
For OF1 female mice, a LC50 value of 6150 mg chloroform/m3 (6
h exposure) was reported by Gradiski et al. (1978).
A dose of 3070 mg chloroform/kg body weight in mineral oil to
rats resulted in death due to CNS depression within minutes, and a
dose of 980 mg chloroform/kg body weight resulted in hepatic
centrilobular necrosis (Reynolds & Yee, 1967). When administered to
newborn rats, chloroform was lethal at oral doses of 1500 mg/kg body
weight; smaller doses were not administered (Kimura et al., 1971).
7.1.2 Non-lethal effects
7.1.2.1 Oral exposure
Chloroform is a potent anaesthetic. Anaesthesia may result from
oral administration of chloroform; this was established by Bowman et
al. (1978) in the ICR mouse with a dose of 500 mg chloroform/kg body
weight in aqueous emulsion. The ED50 (50% of animals showing
effect at this dose level) in mice for acute neurological effects
(ataxia, incoordination and anaesthesia) was 484 mg chloroform/kg
body weight (Balster & Borzelleca, 1982).
After oral administration of chloroform in olive oil to Swiss
mice (both sexes), Jones et al. (1958) found the median narcotic
dose to be 350 mg/kg body weight and the median hepatotoxic dose to
be 35 mg/kg body weight. At this dose level, the liver showed
centrilobular fatty infiltration and at 350 mg/kg body weight
centrilobular necrosis was found.
Table 9. Representative LD50 values (mg chloroform/kg body weight) for mice
Sex/strain Route Vehicle Observed LD50 Reference
period
Male
C3H/tif oral sesame oil 15 days 36 Pericin & Thomann (1979)
DBA/2/j oral sesame oil 15 days 101 Pericin & Thomann (1979)
Tif:MAGf oral sesame oil 15 days 213 Pericin & Thomann (1979)
A/J oral sesame oil 15 days 253 Pericin & Thomann (1979)
Tif:MF2f oral sesame oil 15 days 336 Pericin & Thomann (1979)
C57BL/6J oral sesame oil 15 days 460 Pericin & Thomann (1979)
Princeton subcutaneous peanut oil 10 days 696 Plaa et al. (1958)
Swiss albino subcutaneous olive oil 10 days 3245 Kutob & Plaa (1962a)
Female
C3H/tif oral sesame oil 15 days 353 Pericin & Thomann (1979)
DBA/2/j oral sesame oil 15 days 679 Pericin & Thomann (1979)
A/J oral sesame oil 15 days 774 Pericin & Thomann (1979)
C57BL/6J oral sesame oil 15 days 820 Pericin & Thomann (1979)
Tif:MF2f oral sesame oil 15 days 1126 Pericin & Thomann (1979)
Tif:MAGf oral sesame oil 15 days 1366 Pericin & Thomann (1979)
OF1 intraperitoneal olive oil 14 days 880 Gradiski et al. (1974)
Table 10. Representative LD50 values (mg chloroform/kg body weight) for rats
Sex/strain Route Vehicle Observed LD50 Reference
period
Male
Sprague-Dawley oral none unknown 450 Kimura et al. (1971)
(14 days old)
unknown oral none unknown 1200 Kimura et al. (1971)
(older adults)
Sprague-Dawaley oral none 14 days 908 Chu et al. (1980)
Sprague-Dawley oral none 14 days 2000 Torkelson et al. (1976)
Female
Sprague-Dawley oral none unknown 450 Kimura et al. (1971)
(14 days old)
Sprague-Dawley oral none 14 days 1117 Chu et al. (1980)
Sprague-Dawley intraperitoneal peanut oil 24 h 1379 Lundberg et al. (1986)
14 days 894
Hill (1978) investigated strain and sex differences in
chloroform-induced toxicity in mice. Male mice of three strains
(DBA/2J, B6D2F1/J, and C57BL/6J) were given single oral doses of
chloroform in oil. No clear difference in hepatotoxicity between
strains was observed; centrilobular necrosis occurred at doses
greater than 250 mg/kg body weight in all three strains. In
contrast, there were differences between species in renal toxicity.
Doses of 89 mg/kg body weight caused glucosuria and/or proteinuria
in half of the DBA/2J animals, while doses of 119 and 163 mg/kg body
weight were required to produce these effects in half the B6D2F1/J
and C57BL/6J mice, respectively.
In male CFLP Swiss mice, Moore et al. (1982) found neither
histological changes in the liver or kidney nor biochemical changes
in plasma 4 days after oral administration of 17 mg chloroform/kg
body weight in corn oil. Administration of 66 mg chloroform/kg body
weight caused slight hepatotoxicity and a more severe
nephrotoxicity.
Chu et al. (1980, 1982a) observed piloerection, sedation,
flaccid muscle tone, ataxia, prostration and dacryorrhoea after
administration of chloroform to rats. Food intake in the males was
reduced. Histological and biochemical examination revealed effects
on liver, kidneys and red and white blood cells. Upon histological
examination, no lesions were found in other tissues with chloroform
doses up to 2100 mg/kg body weight. In this study the lowest
administered dose was 546 mg chloroform/kg body weight, a level at
which toxic effects were still found.
Reitz et al. (1982) determined the cellular regeneration (as
3H-thymidine uptake in DNA) 48 h after administration of
chloroform to male B6C3F1 mice and male Osborne-Mendel rats. In
the mice, 3H-thymidine uptake was significantly increased in
kidneys at a dose level of 60 mg chloroform/kg body weight and in
kidneys and liver at 240 mg chloroform/kg body weight. In the rats,
only a slight increase in 3H-thymidine uptake in liver and kidneys
was found at a dose level of 180 mg chloroform/kg body weight.
Torkelson et al. (1976) reported dose-related liver and kidney
changes in adult rats at dose levels as low as 250 mg chloroform/kg
body weight. Tyson et al. (1983) found an elevation of serum
aminotransferase levels in rats at dose levels above 200 mg/kg body
weight in oil.
One study examined the organ-specific toxicity of acute doses
of chloroform (Larson et al., 1993). Male F-344 rats were given
chloroform by gavage in corn oil at doses of 0, 34, 180 or 477 mg/kg
body weight and necropsied 24 h later. Additional rats were given a
single dose of 180 mg chloroform/kg and administered
bromodeoxyuridine (BrdU) 2 h prior to necropsy at 0.5, 1, 2, 4, and
8 days after chloroform treatment to label cells in S-phase. The
kidneys of male rats administered 34, 180 and 477 mg chloroform/kg
exhibited mild to severe proximal tubular necrosis in a
dose-dependent manner. A 20-fold increase in the labelling index
(LI, % of nuclei in S-phase) in the proximal tubule cells was
observed 2 days after treatment at a dose of 180 mg/kg body weight.
The livers of the male rats exhibited only slight to moderate
multifocal centrilobular necrosis at 180 and 477 mg/kg body weight.
A 10-fold increase in the LI was observed in the liver of male rats
given 477 mg/kg body weight, but no increase was observed at 180
mg/kg body weight (Larson et al., 1993).
Female B6C3F1 mice were given chloroform by gavage (0, 34,
238 or 477 mg/kg body weight) and necropsied 24 h after treatment.
Additional mice were given a single dose of 350 mg chloroform/kg
body weight, labelled with BrdU, and necropsied 0.5, 1, 2, 4, and 8
days after treatment. Female mice developed a dose-dependent
centrilobular hepatic necrosis at 238 and 477 mg/kg body weight. No
renal lesions were observed in female mice at any dose. A peak
increase in LI of 38-fold was observed in hepatocytes in the livers
of female mice 2 days after treatment with 350 mg chloroform/kg, but
the increase in LI observed in the kidneys was only 2-fold (Larson
et al., 1993). These data indicate that acute chloroform-induced
cytolethality leads to increased cell proliferation and that the
organ-specific pattern of toxicity is the same as the organ-specific
pattern of tumour formation (see NCI, 1976a,b, and section 7.7.1).
Groger & Grey (1979) intubated Colworth Wistar rats (6 of each
sex per group) daily with chloroform in peanut oil (0 to 50 mg/kg
body weight) for periods of 1, 5 or 10 days. There were changes in
the activity of several liver enzymes, the toxicological
significance of which is unclear.
Balster & Borzelleca (1982) administered chloroform in water to
male ICR mice (8-12/group) and examined their performances in a
battery of neurobehavioural tests (several exposure periods and
several dose levels). The only effect observed was a reduced
achievement in an operant behaviour test after dosing with 100 and
400 mg chloroform/kg body weight in water for 60 days. At the
chloroform level of 400 mg/kg body weight, about half the treated
animals died. No adverse effects on behaviour were observed after 90
days of dosing with 31 mg chloroform/kg body weight in water.
7.1.2.2 Subcutaneous and intraperitoneal exposure
A sex difference in toxicity was found after subcutaneous and
intraperitoneal administration of chloroform to mice. In males, the
kidney appeared to be more susceptible than in females, in which the
liver was found to be the target organ. Smith et al. (1983) exposed
male and female mice of the ICR strain to chloroform doses of 75 to
1500 mg/kg body weight (subcutaneous and intraperitoneal).
Hepatotoxicity was dose-related in both sexes from 375 mg
chloroform/kg body weight upwards. After the subcutaneous
administration of 375 mg chloroform/kg body weight, an increase in
the serum alanine aminotransferase (ALAT) and a decrease in the
liver non-protein sulfhydryl groups (NPSH) were observed.
Histological examinations showed centrilobular swelling of the liver
and necrosis of the hepatocytes in both sexes. At 24 h after
intraperitoneal exposure to 375 mg chloroform/kg body weight, renal
toxicity was observed in males but not in females. A decrease in the
renal NPSH concentration of about 60% in males and 20% in females
was found. The concentration in females, but not in males, returned
to normal within 24 h post-dosing. Histological examination of male
kidneys showed proximal tubular lesions with pyknotic nuclei and
loss of reticular cytoplasmic structure, necrosis of the cells of
the proximal tubuli and occlusion of the tubular lumens with hyaline
casts (Smith et al., 1983).
Skrzypinska et al. (1991) administered chloroform
intraperitoneally to Balb/c mice as a single dose ranging from 12.5
to 100% of the approximate lethal dose. At different time periods
after administration, mice were sacrificed. Serum glutamine-pyruvate
transaminase (SGPT) and sorbitol dehydrogenase (SDH), as well as
glutathione (GSH) and malondialdehyde (MDA) levels in the liver,
were determined. Increased SGPT and SDH levels were found for all
doses exceeding one eighth of the approximate lethal dose. The
depletion of GSH level was kept within 40% for all doses. A 2- to
4-fold increase of hepatic MDA level was found. The depletion of
hepatic GSH, and to some extent the increase of SGPT and SDH,
occurred in a biphasic fashion. Dose-effect functions for these
biochemical alterations could only be constructed for the second
delayed phase of action. It is postulated that the hepatotoxicity of
chloroform is mainly dependent on radical formation in the course of
biotransformation.
Plaa & Larson (1965) observed renal toxicity after an
intraperitoneal dose as low as 48 mg chloroform/kg body weight in
male Swiss mice. The authors reported that chloroform was the most
potent nephrotoxic agent of 14 short-chain chlorinated hydrocarbons
in male mice.
Ahmadizadeh et al. (1984) found an increase in the relative
kidney weight after intraperitoneal administration of chloroform in
peanut oil (150 mg/kg body weight) to male DBA mice, but not after
chloroform administration to DBA female mice or to male or female
mice of the C57BL strain.
Hepatic toxicity, which is the predominant effect in most
species, was found after a parenteral dose of 450 and 150 mg
chloroform/kg body weight in the rat and the guinea-pig,
respectively (Klaassen & Plaa, 1969; Divincenzo & Krasavage, 1974).
Detection of lipoperoxidation in the liver of PB-induced rats
exposed to chloroform has been reported by several authors (Klaassen
& Plaa, 1969; Brown, 1972; Brown et al., 1974a; Masuda et al.,
1980).
An increased bile duct/pancreatic fluid flow and a changed
composition of this fluid were observed after an intraperitoneal
dose of 1500 mg chloroform/kg body weight to rats (Harms et al.,
1976; Hamada & Peterson, 1977).
A single, liver-damaging intraperitoneal dose of chloroform led
to a maximal glutathione depletion in the liver of PB-pretreated
rats shortly after dosing (1-2 h) but not in saline-treated rats.
However, the maximal histopathological findings (centrilobular
necrosis) occurred much later (after about 24 h) (Docks & Krishna,
1976).
In dogs, liver toxicity has been found after intraperitoneal
administration of chloroform. The ED50 for an increased serum ALAT
activity via this route appears to be 300 mg chloroform/kg body
weight. At near-ED50 doses, chloroform caused centrilobular
vacuolization and centrilobular and subcapsular necrosis. The ED50
for renal dysfunction in the dog appears to be 645 mg chloroform/kg
body weight (Klaassen & Plaa, 1967).
Bai et al. (1992) evaluated the suitability of nine different
serum bile acids (SBA) as markers of chloroform exposure in rats.
Increases in specific SBA levels were observed following three daily
intraperitoneal administrations of chloroform at doses as low as 0.1
mmol/kg body weight. The effects on SBA levels were detectable at
much lower doses than were effects on histopathological indices or
on levels of alanine aminotransferase, aspartatetransaminase,
alkaline phosphatase, bilirubin or total bile acid.
Chloroform doses as low as 45 mg/kg body weight reduced the
microsomal Ca++/Mg++-ATP-ase activity (liver microsome calcium
pump) in rats (Moore, 1980).
7.1.2.3 Inhalation exposure
After exposure to chloroform vapour, the same pattern of
toxicity in mice was observed as after oral, intraperitoneal or
subcutaneous administration (Deringer et al., 1953; Hewitt, 1956).
Deringer et al. (1953) found necrosis in the proximal and distal
convoluted tubules, hyaline casts in the convoluted tubules and
collecting ducts, calcification of the cortex, and death after
exposure of male C3H mice to chloroform concentrations of 3400 to
5400 mg/m3 for 1 to 3 h; anaesthesia was not observed.
Kylin et al. (1963) exposed female albino mice of an undefined
strain to chloroform vapour for 4 h and reported hepatotoxic
effects. At 24 h after exposure to chloroform concentrations of 490
mg/m3 or more, a concentration-related fatty infiltration was
observed. From 980 mg chloroform/m3 upwards, necrosis of liver
cells and a rise in the serum ornithine carbamoyltransferase level
were seen. In mice, rabbits, guinea-pigs and cats, anaesthesia was
induced by exposure to chloroform concentrations in the range of 10
to 100 g/m3 for periods of 30 min to a few hours. In rabbits and
guinea-pigs such exposures can cause death (review by Lehmann &
Flury, 1943).
As with other anaesthetics, prolonged anaesthesia with
chloroform may result in respiratory depression, cardiac arrhythmia
and finally in cardiac arrest. Heart failure is probably due to
increased sensitivity of the heart muscle to adrenaline (Von
Oettingen et al., 1950; Von Oettingen, 1964). Exposure of rabbits to
224 mg/m3 for 1 min led to decreased diastolic pressure, reduction
of the stroke volume, blood pressure and cardiac output, and an
increase in the peripheral vascular resistance. The cardiac effects
were probably not due to respiratory effects, as blood oxygen and
carbon dioxide tension and pH were not significantly changed (Taylor
et al., 1976).
Exposure of rats to a chloroform concentration of 49 g/m3 for
5 h resulted in respiratory acidosis. Liver cells showed swollen
rough endoplasmic reticulum with a loss of ribosomes, mitochondrial
lesions, and cistern-like dilatation of tubular areas of the smooth
endoplasmic reticulum. An accumulation of fat droplets and reduced
amino acid incorporation into protein were also found in liver cells
(Scholler, 1966, 1967).
Brondeau et al. (1983) found increased serum activities of
glutamate dehydrogenase and sorbitol dehydrogenase after a single 4
h exposure of male rats to a chloroform concentration of 1410
mg/m3. The effects were dose-related and at the highest
concentrations tested (4600 and 5250 mg/m3) serum aspartate
aminotransferase levels (ASAT) were also increased.
7.1.2.4 Dermal exposure
Single application of 1 or 4 g chloroform/kg body weight for 24
h to the belly of rabbits, under an impermeable plastic cuff,
resulted in extensive necrosis and weight loss at both levels. The
kidneys of all animals showed dose-related degenerative changes in
the tubules. Livers were not grossly affected (see also section 7.4)
(Torkelson et al., 1976).
7.2 Short-term exposure
7.2.1 Oral exposure
7.2.1.1 Mice
Condie et al. (1983) dosed male CD1 mice daily with 0, 37, 74
and 148 mg chloroform/kg body weight in corn oil for 14 days.
Histological changes turned out to be the most sensitive indicators
of liver and kidney toxicity. Dose-related effects were observed at
dose levels from 37 mg/kg body weight upwards. Kidneys showed
intra-tubular mineralization, epithelial hyperplasia and cytomegaly.
Livers showed centrilobular cytoplasmic pallor, marked cell
proliferation and focal inflammation. After 14 days the body weight
in the highest dose group was reduced.
Female and male CD1 mice (7-12 animals of each sex per group)
were administered daily 0, 50, 125 and 250 mg/kg body weight in
water by gavage for 14 and 90 days (Munson et al., 1982). Many
histological and biochemical parameters were examined. After 14
days, the most important effects were a dose-related decrease in the
number of antibody-forming cells (as IgM response to sheep red blood
cells) in both sexes (> 50 mg/kg body weight) and an increase in
the liver weight of males at doses > 125 mg/kg body weight and of
females at the highest dose level. The serum ASAT level was
increased in males and females at the highest dose level and serum
ALAT was increased in females at the highest dose level. After 90
days, a depression in the number of antibody-forming cells was found
at the highest dose level in both sexes. In females at the highest
dose level, a decrease in cell-mediated type hypersensitivity was
observed. Liver weight was increased after 90 days of exposure to
doses > 50 mg chloroform per kg body weight in the females and at
250 mg chloroform/kg body weight in the males. After 90 days of
exposure, the animals showed a tolerance against a challenging dose
of 1000 mg chloroform/kg body weight. The kidneys and livers of all
dosed animals showed histological changes. In the kidneys these
changes included small intertubular collections of chronic
inflammatory cells, whereas in the liver they included generalized
hydropic degeneration of hepatocytes and occasional small focal
collections of lymphocytes. In females, small amounts of
extravasated bile were occasionally noted in the sinusoidal Kupffer
cells.
Jorgenson & Rushbrook (1980) administered chloroform to female
B6C3F1 mice for 90 days in the drinking-water at concentrations of
0, 200, 400, 600, 900, 1800 and 2700 mg/litre (measured daily
chloroform doses of 0, 34, 66, 92, 132, 263 and 400 mg/kg body
weight, respectively). In the first week of the experiment some mice
in the higher dose groups died of dehydration due to reduced
drinking. Depression of the central nervous system occurred in the
animals receiving chloroform and was concentration-related. The only
treatment-related histopathological findings consisted of a mild
adaptive and transitory fatty change in the livers of animals dosed
with 66 mg chloroform/kg body weight or more and a mild lymphoid
atrophy of the spleen at the higher dose levels.
There is evidence that the vehicle in which chloroform is
administered significantly affects its toxicity. Bull et al. (1986)
found that chloroform administered by gavage in corn oil was
significantly more hepatotoxic than equivalent doses administered in
an aqueous emulsion (2% Emulphor(R), polyoxyethylated vegetable
oil, GAF Corp.). Doses of 0, 130 and 270 mg/kg were administered to
male and female B6C3F1 mice for 90 days. Liver body weight ratios
were significantly higher in all dose groups and in both sexes when
chloroform was administered in corn oil. The SGPT level was
significantly elevated in both sexes at the high dose level of
chloroform administered in corn oil, but not in those treated with
the same dose in Emulphor. Mice treated at all levels of chloroform
in corn oil showed evidence of extensive vacuolation and those
treated with the high dose in corn oil showed extensive disruption
of hepatic architecture including cirrhosis. No such pathological
changes were observed in any of the animals treated with chloroform
in 2% Emulphor.
One study contrasted the toxic responses of chloroform
administered by gavage in corn oil or given ad libitum in the
drinking-water (Larson et al., 1994a). Female B6C3F1 mice were
administered oral doses (0, 3, 10, 34, 90, 238, or 477 mg/kg per
day) of chloroform dissolved in corn oil for 4 days or for 5
days/week for 3 weeks, or were continually exposed to chloroform in
the drinking-water at concentrations of 0, 60, 200, 400, 900 or 1800
mg/litre for 4 days or 3 weeks, at which time they were necropsied.
BrdU was delivered via osmotic pumps implanted 3.5 days prior to
necropsy. Cell proliferation was evaluated as a BrdU labelling index
(LI) in histological tissue sections. Dose-dependent changes
included centrilobular necrosis and markedly elevated LI in mice
given chloroform in corn oil at 238 or 477 mg/kg, the average daily
doses that produced tumours in the gavage cancer bioassay (NCI,
1976a,b). The no-observed-effect level (NOEL) for histopathological
changes was 10 mg/kg body weight per day and for induced cell
proliferation 34 mg/kg body weight per day. Chloroform given in the
drinking-water did not increase the hepatic LI after either 4 days
or 3 weeks in any of the dose groups, nor were any microscopic
alterations observed in the liver, even though the cumulative daily
amount of chloroform ingested in the 1800-mg/litre exposure group
was 329 mg/kg body weight per day (Larson et al., 1994a). Thus, the
authors concluded that liver detoxification mechanisms are
overwhelmed when chloroform is given as a single bolus dose, but the
liver can detoxify the same daily dose if it is given in small
amounts resulting from sips of water throughout the day. The authors
also concluded that the sustained increase in LI in the livers of
mice administered hepatocarcinogenic doses of chloroform in corn
oil, but not in the case of chloroform in drinking-water, supports
the hypothesis that chloroform-induced mouse liver cancer is
secondary to events associated with induced cytolethality and cell
proliferation (see also NCI, 1976a,b and section 7.7).
7.2.1.2 Rats
Chu et al. (1982a) exposed male weanling Sprague-Dawley rats to
chloroform via drinking-water for 28 days. The following chloroform
exposure doses were calculated: 0, 0.13, 1.3 and 11 mg/rat per day
(0, 0.7, 7.4 and 63 mg/kg body weight, respectively). The only
treatment-related effect observed was a decrease in the neutrophils
in the 11-mg group. In a 90-day study by Chu et al. (1982b) male and
female Sprague-Dawley rats were exposed to chloroform via
drinking-water at dose levels of 0, 0.17, 1.3, 12 and 40 mg/rat per
day for males and 0, 0.12, 1.3, 9.5 and 29 mg/rat per day for
females; this was followed by 90 days of recovery. Water and food
intake were reduced in the highest dose group. At the 40-mg level a
higher incidence of spontaneous death occurred. Histological
examination showed mild liver and thyroid lesions, especially in the
highest dose group. Livers of both males and females showed: an
increase in cytoplasmic homogeneity; density of the hepatocytes in
the periportal area; mid-zonal and centrilobular increase in
cytoplasmic volume; vacuolation due to fatty infiltration and
occasional nucleic vesiculation; and hyperplasia of biliary
epithelial cells. Thyroid lesions consisted of a reduction in
follicular size and colloid density, increase in epithelial cell
height and occasional collapse of follicles. Liver and thyroid
lesions diminished in severity during the 90 days recovery period.
Jorgenson & Rushbrook (1980) administered chloroform in the
drinking-water to male Osborne-Mendel rats for 90 days at
concentrations of 0, 200, 400, 600, 900 and 1800 mg/litre
(calculated to be 0, 20, 38, 57, 81 and 160 mg chloroform/kg body
weight, respectively). A concentration-related central nervous
system depression was seen. Body weights in the 160-mg group were
reduced throughout the study. Biochemical investigations of serum
showed no important deviations from control values other than a
dose-related increase in cholesterol at dose levels of 38 mg
chloroform/kg body weight or more after 60 days and a decrease in
triglycerides in the highest dose group from 30 days onwards. After
90 days of administration, however, these parameters were affected
in the two highest dose groups only. No dose-related
histopathological changes were reported.
7.2.2 Inhalation exposure
The severity of liver injury due to inhaled chloroform is not
only influenced by the administered concentration but also by the
shape of the exposure profile. This was observed by Plummer et al.
(1990), who exposed male black-hooded Wistar rats (36 per group) for
4 weeks to chloroform vapour as a constant concentration (245
mg/m3; 24 h/day; 7 days a week) or as repeated concentrations
(1387 mg/m3; 6 h/day; 5 days a week), with a similar total
exposure (154 g/m3-hours) for the two ways of exposure (levels
were monitored). Hepatic injury appeared to be more severe in the
continuously exposed group, in which microvesicular fatty change was
the most prominent feature, while focal necrosis was a minor
feature. Livers of the animals receiving repeated exposures showed
only minor injuries in the form of scattered hepatocytes containing
small fat droplets and a few foci of liver cell necrosis.
Torkelson et al. (1976) exposed male and female rats (10-12 of
each sex per group), rabbits (2-3 of each sex per group) and
guinea-pigs (8-12 of each sex per group) to concentrations of 0,
110, 230 and 410 mg chloroform/m3 air for 7 h/day, 5 days/week,
during 6 months. In the male and female rats, relative kidney weight
was increased at all exposure levels. In the males, at all levels,
kidneys showed cloudy swelling of the tubular epithelium and the
livers showed lobular granular degeneration with focal necrosis. At
the higher exposure levels the effects became more pronounced. The
effects observed in the males exposed to 110 mg chloroform/m3
disappeared within 6 weeks after exposure. At 410 mg
chloroform/m3, death, due to interstitial pneumonitis, occurred in
the males. No effects were seen in the male rats after 1, 2 or 4 h
of exposure to 110 mg chloroform/m3 (same schedule of exposure).
The results obtained after exposure of rabbits and guinea-pigs were
inconsistent because of low numbers of animals and/or the absence of
dose-effect relationships.
The toxicity of one-week exposures to inhaled chloroform has
been investigated in female B6C3F1 mice and in male F-344 rats
(Larson et al., 1994b; Méry et al., 1994). Rodents were exposed to
chloroform vapour at concentrations of 0, 4.9, 14.7, 49, 147, 490 or
1470 mg/m3 (0, 1, 3, 10, 30, 100 or 300 ppm) for 7 consecutive
days and necropsied on day 8. Cell proliferation was quantified as
the percentage of cells in S-phase (BrdU labelling index) measured
by immunohistochemical detection of BrdU-labelled nuclei. Mice
exposed to 490 or 1470 mg/m3 exhibited centrilobular hepatocyte
necrosis and severe vacuolar degeneration of mid-zonal and
periportal hepatocytes, while exposure to 49 or 147 mg/m3 resulted
in mild to moderate vacuolar changes in centrilobular hepatocytes.
Slight, dose-related increases in the hepatocyte LI were observed
for exposure concentrations of 4.9-14.7 mg/m3, while the LI was
increased more than 30-fold in the 490- and 1470-mg/m3 groups. The
kidneys of mice were affected only at 1470 mg/m3 exposure, with
approximately half of the proximal tubules lined by regenerating
epithelium and an 8-fold increase in the LI of tubule cells compared
with controls (Larson et al., 1994b).
In rats, mild centrilobular vacuolation was observed only in
the livers of animals exposed to 1470 mg/m3. The hepatocyte LI in
rats was increased only at 490 and 1470 mg/m3 (3-fold and 7-fold
over control, respectively). The kidneys of the male rats were
affected only at 1470 mg/m3. About 25 to 50% of the proximal
tubules were lined by regenerating epithelium in this exposure
group, while the LI for tubule cells was increased 2-fold over
controls (Larson et al., 1994b).
In the nasal passages of rats, chloroform concentrations of 49
mg/m3 or more induced histopathological changes that exhibited
clear concentration-related severity. Chloroform-induced changes
included increased epithelial mucosubstances in the respiratory
epithelium of the nasopharyngeal meatus, primarily in the rats. A
complex set of responses was seen in specific regions of the ethmoid
turbinates, predominantly in the rats. These lesions in the ethmoid
region, which involved all of the endo- and ectoturbinates, were
most severe peripherally and generally spared the tissue adjacent to
the medial airways. These changes were characterized by atrophy of
Bowman's glands, increased numbers of vimentin-positive cells in the
periosteum, new bone formation and increased number of periosteal
cells in S-phase as determined by BrdU incorporation. Additional
changes were site-specific loss of mucosubstances and loss of
immunocyto-chemical staining of acini and ducts of Bowman's glands
for P450-2E1 and pancytokeratin, and loss of P450-2E1 immunostaining
of the olfactory epithelium. The only change noted in the mice was
increased periosteal cell proliferation without new bone growth
(Méry et al., 1994).
7.3 Long-term exposure
In a carcinogenicity bioassay, female B6C3F1 mice were
exposed to 0, 200, 400, 900 or 1800 mg chloroform/litre
drinking-water (number of animals: 430, 430, 150, 50 and 50,
respectively) for a period of two years (Jorgenson et al., 1982)
(see also section 7.7.1). These concentrations (monitored by
analysis) correspond to time-weighted average daily chloroform doses
of 0, 34, 65, 130 and 263 mg/kg body weight (Jorgenson et al.,
1985). Matched controls (50 females) received an amount of water
without chloroform equal to that consumed by the 1800-mg/litre
group. Additional mice were used for intermediate biochemical and
histopathological examination. Early mortality in the high-dose
group was observed. After 3 months, livers of animals exposed to
chloroform concentrations of 65 mg/kg body weight or more showed a
higher fat content than those of the controls (as examined by
chemical techniques). After 6 months, liver fat content was
increased in all exposed groups. Data on organ weights were not
provided.
In a carcinogenicity bioassay, male Osborne-Mendel rats were
exposed to 0, 200, 400, 900 or 1800 mg chloroform/litre
drinking-water (number of animals: 330, 330, 150, 50 and 50,
respectively) for a period of two years (Jorgenson et al., 1982)
(see section 7.7.2). These concentrations (monitored by analysis)
correspond to time-weighted average daily chloroform doses of 0, 19,
38, 81 and 160 mg/kg body weight (Jorgenson et al., 1985). Matched
controls received an amount of water without chloroform equal to
that consumed by the 1800-mg/litre group. Additional rats were used
for intermediate biochemical and histopathological examination. The
survival was indirectly proportional to the dose levels.
Concentration-related decreases in water uptake and growth were
seen. The latter effects were also observed in the matched controls,
and thus may be attributed to the reduced intake of water.
Biochemical examination of blood after 6, 12 and 18 months showed
that chlorine, potassium, total iron and albumin levels and the
albumin/globulin ratio tended to be increased after chloroform
treatment, whereas levels of cholesterol, triglycerides and lactate
dehydrogenase were decreased in all treated groups. These deviations
were also observed in the matched controls, but the decreases in
serum triglycerides and cholesterol levels were more severe at the
two highest dose levels than in the matched control group. Data on
organ weights were not provided.
Beagle dogs were given chloroform in a toothpaste base in
gelatin capsules, 6 days/week for 7.5 years (Heywood et al., 1979).
The doses were 15 and 30 mg/kg and there were eight male and eight
female dogs in each dose group. Dogs given the high dose began to
show significant increases in SGPT levels at 6 weeks of treatment.
At the low dose level, significant increases were observed at 34
weeks and after. Similar effects were not observed in the vehicle
control (16 dogs of each sex) or untreated control (eight dogs of
each sex) groups. "Fatty cysts" of the liver were observed in both
dose groups at the end of this study (see section 7.7.3).
7.4 Skin and eye irritation
Adequate data on the skin irritation potential of chloroform
has not been identified. Torkelson et al. (1976) applied liquid
chloroform to the rabbit ear and found slight hyperaemia and
exfoliation after one to four treatments (period between application
and observation not specified). More frequent application did not
increase the severity of the injuries. A 24-h application of
chloroform on a cotton pad on the belly of rabbits produced slight
hyperaemia, moderate necrosis and eschar formation. Chloroform
delayed healing of mechanically damaged skin on the application
site.
Application of chloroform droplets in the rabbit eye caused a
transient slight irritation of the conjunctiva and corneal injury. A
purulent exudate occurred for 2 or more days after the treatment
(Torkelson et al., 1976).
Duprat et al. (1976) applied undiluted chloroform into the eyes
of six New Zealand white rabbits. It produced severe eye irritation,
with mydriasis and keratitis in all rabbits. Translucent zones in
the cornea were observed in four animals and a purulent haemorrhagic
discharge was also reported (number of rabbits unknown). The effects
had disappeared 2-3 weeks after application, except for one rabbit
that still showed corneal opacity after 3 weeks.
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1 Reproduction
Borzelleca & Carchman (1982) studied the reproductive toxicity
of chloroform in a three-generation experiment with ICR mice. They
administered the chemical (0.1% Emulphor in deionised water) via
drinking-water (in closed bottles) to males (10/group) and females
(30/group), at concentrations of 0, 0.1, 1 and 5 mg/ml, from 5 weeks
before F0 mating throughout the entire study until sacrifice of
the F2b pups. Death occurred among the males and females of the
highest dose group, and body weights in this group were reduced. At
1 mg/ml, the body weights of F1b females were also reduced.
Dose-related hepatotoxicity was found in the F0 and F1b animals
(symptoms varying from "slight yellow-grey colouring" in the lowest
dose group to "grey to black discolouration" with large nodules
(> 3 mm) upon and within the liver in the highest dose group).
The treatment resulted in reduced fertility, litter size, gestation
index and viability index in all F1 and F2 generations,
statistically significant at 5 mg/ml. No evidence for a teratogenic
potential was obtained.
7.5.2 Embryotoxicity and teratogenicity
Chloroform has not been found to be teratogenic but has been
shown to induce fetotoxic effects.
7.5.2.1 Oral exposure
No evidence for a teratogenic effect of chloroform was obtained
in a three-generation study with ICR mice (Borzelleca & Carchman,
1982).
In a study by Thompson et al. (1974), female Sprague-Dawley
rats (25/group) were intubated with chloroform in corn oil (0, 10,
25 and 63 mg/kg body weight) twice daily on days 6-15 of gestation.
A reduced body weight gain and anorexia were seen in the dams of the
two higher dose groups. Tissues from two dams of each dose group
were microscopically examined and fatty changes were observed in the
livers of both females at 63 mg/kg and in one female at 25 mg/kg.
Other signs of maternal toxicity were not found at these dose
levels. The fetuses of the 63-mg/kg groups had a smaller weight at
delivery than those of the control group. The incidence of bilateral
extralumbar ribs was significantly increased among the fetal
population of the 63-mg/kg dose group. Other minor visceral and
skeletal abnormalities were seen, but not at significantly elevated
levels. In the same study female Dutch-Belted rabbits (15/group)
were dosed orally with chloroform in corn oil (0, 20, 35 and 50
mg/kg body weight) once daily during days 6-18 of gestation.
Administration of chloroform produced a decrease in fetal body
weight and incomplete ossification of skeletal elements (skull
bones) in the 20- and 50-mg/kg dose groups. At the highest dose
level the dams showed decreased weight gain. Signs of embryotoxicity
and teratogenicity were not observed.
Ruddick et al. (1983) gave pregnant Sprague-Dawley rats
(15/group) chloroform in corn oil (0, 100, 200 and 400 mg
chloroform/kg body weight) daily by gavage from days 6 to 15 of
gestation. All doses caused reduced weight gain in the dams and
increased liver weight. At the highest dose level, there was an
increase in the kidney weight of the dams. Haematological
examinations showed dose-dependent reductions in haemoglobin and
haematocrit (14% maximally, both parameters). In the highest dose
group, the red blood cell count was also reduced.
According to Burkhalter & Balster (1979), oral administration
of chloroform to mice from 3 weeks before mating until the end of
the lactating period (in both sexes the dose was 31 mg/kg body
weight) did not result in retardation of the development of
responses to a battery of neurobehavioural tests in the pups.
7.5.2.2 Inhalation exposure
Schwetz et al. (1974) reported effects on pregnancy and on the
incidence of fetal malformations in Sprague-Dawley rats exposed to
chloroform concentrations of 147, 490 and 1470 mg/m3 (30, 100 and
300 ppm) for 7 h/day during days 6-15 of gestation (analysis 3 times
daily showed concentrations of 147, 466 and 1426 mg/m3; 30, 95 and
291 ppm, respectively). The two highest concentrations were toxic to
the dams (anorexia and reduced weight gain, increases in relative
and absolute liver weight). There was a dose-dependent decrease in
the pregnancy percentage (100% in the control group versus 15% in
the 1426-mg/m3 group) and in the number of living fetuses per
litter. An increase was observed in the percentage of
post-implantation losses (resorptions) in the highest dose group,
and a dose-dependent increase was seen in the percentage of litters
with resorptions (from 57% in the control group to 100% at the
highest concentration). At all exposure levels, fetuses showed
growth retardation and minor skeletal aberrations (delayed
ossification of skull and sternebrae). Exposure to 147 mg/m3
caused minor embryo- and fetotoxicity, and concentrations of 466 and
1426 mg/m3 in the inhaled air were embryo- and fetotoxic to the
rat. At the higher levels, subcutaneous oedema and other unspecified
fetal soft tissue anomalies were also observed.
Murray et al. (1979) exposed pregnant CF1 mice (35-40/group)
to 0 and 490 mg/m3 (0 and 100 ppm) for 7 h each day throughout
days 1-7, 6-15 or 8-15 of gestation. The ability of females to
maintain pregnancy was significantly decreased after exposure to
chloroform during days 1-7 or 6-15 of gestation (44 and 43% in the
treated groups versus 74 and 91% in the respective control groups).
The dosed animals consumed slightly less food than the control
animals, resulting in reduced body weight gain. Absolute and
relative liver weights were increased in the groups exposed during
days 6-15 and 8-15. After exposure during days 6-15, ALAT levels
were significantly increased in pregnant and non-pregnant animals,
the pregnant animals showing the smaller increase. Among the
controls, no difference in ALAT activity was observed. An increase
in total litter resorptions was observed after exposure through days
8-15. Mean fetal body weight and crown-rump length were decreased
significantly if the dams had been exposed through days 1-7 or 6-15
of pregnancy. In the exposed groups an increased number of fetuses
with delayed ossification of skull bones and sternebrae was
observed, especially in the days 1-7 and 8-15 exposed groups. The
incidence of cleft palates significantly increased in fetuses from
dams exposed to 490 mg/m3 through days 8-15 of gestation for 7 h
each day.
Published information for embryotoxicity and teratogenicity of
chloroform in rat, mouse and rabbit by oral and inhalation exposure
are summarized in Table 11.
7.6 Mutagenicity and related end-points
Very many genotoxicity assays have been conducted with
chloroform and the data currently available are summarized in Tables
12 and 13. Some of these reports are from a large collaborative
study comparing intra-laboratory variations in testing methodology
(De Serres & Ashby, 1981).
Two problems potentially compromise the interpretation of
mutagenicity data on chloroform. First, there is a possibility that
ethyl and diethylcarbonate, produced by reaction of phosgene with
ethanol that is routinely added to U.S.P (US Pharmacopoeia)
chloroform, could generate false positive results. Secondly, testing
of chloroform must be done in a sealed system because of its
volatility, and so studies that did not take this factor into
account could give false negative results.
In data presented in Table 12, three separate studies using the
Ames assay were conducted under sealed conditions to assure
chloroform retention. All three studies yielded negative test
results.
In not all studies was it reported whether an in vitro assay
was performed in a sealed chamber to prevent chloroform evaporation
(Table 12). However, dimethylsulfoxide (DMSO) was often used as a
solvent, thus increasing retention in the media. Furthermore, even
in the case of an unsealed chamber, chloroform would be expected to
stay in the media for a period of hours, and very high doses (up to
10 mg/plate) were often used.
Chloroform has been tested by a number of authors in validated
bacterial systems with Salmonella typhimurium and Escherichia
coli and showed to be negative both with and without metabolic
activation. Only in one uncommon test with Photobacterium
phosphorum was a positive effect found (Wecher & Scher, 1982).
Table 11. Embryotoxicity, fetotoxicity and teratogenicity produced in animals by exposure to chloroform
Species Dose Gestational days Route of Result Reference
administered administration
Rat 30, 100, 300 ppm 6-15 inhalation embryotoxic Schwetz et al. (1974)
(7 h/day) fetotoxic
Rat 20, 50, 126 mg/kg per day 6-15 oral fetotoxic Thompson et al. (1974)
Rat 100, 200, 400 mg/kg per day 6-15 oral fetotoxic Ruddick et al. (1983)
Mouse 100 ppm 1-7, inhalation embryotoxic Murray et al. (1979)
6-15, fetotoxic
8-15
(7 h/day)
Rabbit 20, 35, 50 mg/kg per day 6-18 oral fetotoxic Thompson et al. (1974)
Table 12. Mutagenicity studies with chloroform
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Bacterial systems
Salmonella TA1535 base-pair substitution 14C-labelled compound + ra i.m., - Uehleke et al.
typhimurium TA1538 frame-shift mutation tested; no further i.n.r. (1976, 1977)
details reported
S. typhimurium TA1535 base-pair substitution 5 mM tested; incubation + m PB - Uehleke et al.
TA1538 frame-shift mutation in closed containers (1976, 1977)
(survival > 80%)
S. typhimurium TA98 frame-shift mutation suspension test and - i.n.r. - Simmon et al.
TA1537 frame-shift mutation vapour test; concentration + r - (1977)
TA1538 frame-shift mutation in suspension test was 10%
TA100 base-pair substitution v/v, no further details
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation up to 3600 µg/plate; - - Gocke et al.
TA1537 frame-shift mutation incubation in air-tight + r PCB - (1981)
TA1538 frame-shift mutation desiccators
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation test conditions not - - Trueman (1981)
TA1537 frame-shift mutation reported + r PCB -
TA1538 frame-shift mutation
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation test conditions not - - Ichinotsubo
TA100 base-pair substitution reported + - et al. (1981b)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. typhimurium TA98 frame-shift mutation 0.5, 1.0, 5.0, 100, - - Venitt & Crofton-
TA100 base-pair substitution 200, 500 µg/plate + r PCB - Sleigh (1981)
S. typhimurium TA98 frame-shift mutation microtitre fluctuation - - Gatehouse
TA1537 frame-shift mutation test; 1, 5, 10 µg/ml + r PCB - (1981)
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation fluctuation test; 1-500 - ± Hubbard et
TA100 base-pair substitution µg/ml (not specified) + r - al. (1981)
S. typhimurium TA98 frame-shift mutation solvent DMSO; no further - - Baker &
TA1537 frame-shift mutation details + r PCB - Bonin (1981)
TA1538 frame-shift mutation
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation test conditions not - ± Garner et al.
TA1537 frame-shift mutation reported + r PB - (1981)
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation 50, 100, 200, 1000, - - MacDonald
TA1537 frame-shift mutation 2000, 5000 µg/plate + r PCB - (1981)
TA100 base-pair substitution
S. typhimurium TA98 frame-shift mutation solvents DMSO; no further - - Nagao &
TA1537 frame-shift mutation details + r PCB - Takahashi
TA100 base-pair substitution (1981)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. typhimurium TA98 frame-shift mutation 0.1, 1.0, 10, 100, 500, - - Rowland &
TA1537 frame-shift mutation 2000 µg/plate; solvent + r PCB - Severn (1981)
TA1538 frame-shift mutation DMSO
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA1535 base-pair substitution 10, 100, 1000, 10 000 - - Richold &
TA1537 frame-shift mutation µg/plate; solvent + r PCB - Jones (1981)
TA1538 frame-shift mutation DMSO
S. typhimurium TA98 frame-shift mutation test conditions not - - Simmon &
TA1537 frame-shift mutation reported + r PCB - Shepherd
TA1538 frame-shift mutation (1981)
TA100 base-pair substitution
TA1535 base-pair substitution
S. typhimurium TA98 frame-shift mutation solvent DMSO or water; - - Brooks &
TA1537 frame-shift mutation 0.2, 2, 20, 200, 2000 + r PCB - Dean (1981)
TA1538 frame-shift mutation µg/plate
TA100 base-pair substitution
TA1535 base-pair substitution
TA92 interstrand DNA
crosslinks
S. typhimurium TA98 frame-shift mutation 10, 100, 1000, 10 000 - - Van Abbé et
TA1537 frame-shift mutation µg/plate + r PCB - al. (1982)
TA1538 frame-shift mutation + m PCB -
TA100 base-pair substitution
S. typhimurium TA1535 base-pair substitution vapour test; exposure - - Van Abbé et
TA1538 frame-shift mutation for 2, 4, 6 or 8 h + r PCB - al. (1982)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. typhimurium TM 677 forward mutation to solvent DMSO; up to + r PCB - Skopek et al.
azaguanine resistance 300 µg/ml (1981)
Escherichia coli WP2 uvrA reversion to trp+ 10, 100, 1000 µg/plate + r PCB - Gatehouse (1981)
E. coli WP2 uvrA reversion to trp+ test conditions not + r PCB - Matsushima et
reported al. (1981)
E. coli WP2p reversion to trp+ solvent acetone; 0.1, 1, + r - Kirkland et
WP2 uvrA 10, 100, 1000, 10 000 - PCB - al. (1981)
µg/plate
E. coli WP2p reversion to trp+ 0.5, 1.0, 5, 10, 50, - - Venitt &
WP2 uvr-p 100, 200, 500 µg/plate + r PCB - Crofton-Sleigh
(1981)
E. coli K12 base-pair substitution 14C-labelled compound + ra i.m. - Greim et al.
(not specified) tested; no further details (1977)
reported
Photobacterium PPL- reversion to normal disc-diffusion assay; no - + Wecher &
phosphoreum light emission further details reported Scher (1982)
Non-mammalian eukaryotic systems
Allium cepa chromosomal aberrations solvent: DMSO; 0, 250, 500, + Cortés et al.
1000, 1500, 2500, 5000, (1985)
10 000 µg/ml
Saccharomyces D7 mitotic gene conversion no details reported + n.r. i.n.r. - Zimmermann &
cerevisiae at trp 5 locus Scheel (1981)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
S. cerevisiae D7 mitotic gene conversion 21, 41, 54 mM; incubation - + Callen et al.
at trp 5; mitotic in screw-capped glass (1980)
recombination at ade 2, tubes
reversion at ilv 1 loci
S. cerevisiae D6 mitotic aneuploidy agar added + r PCB - Parry & Sharp
(1981)
S. cerevisiae D6 mitotic aneuploidy direct incubation in + r PCB ± Parry & Sharp
plastic bottles, 25, 50, (1981)
100 µg/ml; idem in glass + r PCB -
bottles
S. cerevisiae JD1 mitotic gene conversion up to 1000 µg/ml; + r PCB ± Sharp & Parry
at trp 5 locus and his incubation in plastic (1981a)
5 polaron containers
S. cerevisiae JD1 idem as above idem as above, only + r PCB - Sharp & Parry
incubation in glass (1981a)
containers
S. cerevisiae D4 mitotic gene conversion 0.33, 1.0, 3.33, 100, - - Jagannath et
at ade 2 and trp 5 loci 333.3 µg/plate + r PCB - al. (1981)
S. cerevisiae T1 mitotic crossing over 100, 1000 µg/plate - - Kassinova et
T2 at ade 2 + r PCB - al. (1981)
S. cerevisiae XV 185-14 reversion at his 1, solvent DMSO; 111, - - Mehta & Von
C (haploid) hom 3, and arg 4 loci 1111 µg/ml + n.r. i.n.r. - Borstel (1981)
Schizosaccharomyces P1 forward mutation at ade 5, 7.5, 10 µg/ml - - Loprieno
myces pombe 1, 3, 4, 5 and 9 loci + r PCB (+) (1981)
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Aspergillus 35 forward mutation 0.5% (survival 26%) ? - Gualandi
nidulans (haploid) (induction of methionine (1984)
suppressors)
A. nidulans P1 somatic segregation 0.5% (survival 16.5%) ? - Gualandi
(crossing-over and non- (1984)
disjunction)
Drosophila Berlin K sex-linked recessive Basc-test; 24 mM; adult - Gocke et al.
melanogaster wild and lethal test feeding (1981)
Basc
D. melanogaster Berlin K sex-linked recessive Basc-test; solvent DMSO; - Vogel et al.
wild and lethal test 0.1, 0.2% treated at (1981)
y mei 9a 25 °C for 3 days with
mei-41 D5 standard feeding
technique
In vitro mammalian systems
Chinese hamster V79 forward mutation to 1, 1.5, 2, 2.5% - - Sturrock
8-azaguanine resistance (1977)
Human lymphocytes chromosome breakage solvent acetone; 50, 100, + r PCB - Kirkland et
200, 400 µg/ml al. (1981)
In vivo mammalian systems
Mouse CD1 micronuclei in intraperitoneal, 0.015, - Tsuchimoto &
polychromatic 0.03, 0.06 ml/kg body Matter (1981)
erythrocytes of bone weight at 0 and 24 h
marrow
Table 12 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Mouse NMRI micronuclei in intraperitoneal, 238, 476, - Gocke et al.
polychromatic 952 mg/kg body weight at (1981)
erythrocytes of bone 0 and 24 h
marrow
Mouse B6C3F1 micronuclei in intraperitoneal, about - Salamone et
polychromatic 0.088 ml/kg body weight al. (1981)
erythrocytes of bone at 0 and 24 h or at
marrow 0 h only
Mouse ? micronuclei in route not reported; 100, (+) Agustin &
polychromatic 200, 400, 600, 700, 800, Lim-Sylianco
erythrocytes of bone 900 mg/kg body weight (1978)
marrow
Rat Long-Evans chromosomal aberrations intraperitoneal, + Fujie et al.
in bone marrow 1.2-119.4 mg/kg body (1990)
6-597 mg/kg body weight weight; oral,
Host-mediated assays
S. typhimurium TA1535 base-pair substitution test conditions not - Agustin & Lim-
reported Sylianco (1978)
S. typhimurium TA1537 frame-shift mutation test conditions not + Agustin & Lim-
reported Sylianco (1978)
Table 12 (contd)
a + = with metabolic activation b PB = phenobarbital c + = positive
- = without metabolic activation PCB = polychlorinated biphenyls (+) = weakly positive
m = mouse i.m. = intact microsomes added ± = equivocal; study cannot be evaluated
r = rat i.n.r. = inducer not reported - = negative
ra = rabbit
n.r. = species not reported
? = not reported if metabolic
action was used
Table 13. Indicator studies with chloroform
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Bacterial systems
Escherichia coli WP2, WP67 DNA damage (growth test concentrations not - - Tweats (1981)
uvrA pol inhibition) specified; no further + r PCB -
A & CM 871 details
uvrA lexA
recA
E. coli WP2, WP67 DNA damage test conditions not - - Green (1981)
uvrA pol reported
A & CM 871
uvrA lexA
recA
E. coli W3110 DNA damage liquid suspension test; - - Rosenkranz et
(polA+), 25 mg/ml; solvent DMSO + r PCB (+) al. (1981)
P3478 or water
(POLA1-)
E. coli JC 2921 rec DNA damage test conditions not - + - Ichinotsubo
JC 9238 rec reported + + et al.
JC 8471 rec (1981a)
JC 5519 rec
JC 7689 rec
JC 7623 rec
E. coli 56-161 induction of prophage 0.5, 5 mg/ml + r PCB - Thomson (1981)
env A lambda in lysogenic -
C 600 E. coli
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Bacillus subtilis H17 rec+ DNA damage not further maximum concentration: + r - Kada (1981)
M45 rec- specified 20 µl/plate + yf -
+ jc -
Non-mammalian eukaryotic systems
Allium cepa SCE solvent DMSO; 0, 250, 500, ± Cortés et al.
1000, 1500, 2500, 5000, (1985)
10 000 µg/ml
Saccharomyces T4 DNA repair solvent DMSO; 0.1, 1.0% - - Kassinova et
cerevisiae T5 al. (1981)
S. cerevisiae 197/2d DNA repair 100, 300, 600, 750 µg/ml; - ± Sharp & Parry
rad 3, incubation in plastic + ± (1981b)
rad 18, bottles
rad 52,
trp 2
In vitro mammalian systems
Chinese hamster ovary SCE 0.7% (after exposure, 78% + r PCB - White et al.
of dose remained) (1979)
Chinese hamster ovary SCE 0.01, 0.1 µg/ml; solvent + r PCB - Perry & Thomson
DMSO (1981)
Chinese hamster ovary SCE 0.001, 0.01, 0.1 mM - + d Athanasiou &
Kyrtopoulos
(1981)
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Rat erythroblast SCE only 1.0 mM tested + (+) Fujie et al.
(1993)
Syrian hamster embryo adenovirus 0.12, 0.25, 0.50, 1.0, - + Hatch et al.
transformation 2.0 ml/sealed chamber (1983)
(4.6 litre)
Baby hamster kidney cell transformation test conditions not - (+) Daniel & Dehnel
reported + r - (1981)
Baby hamster kidney cell transformation 0.25, 2.5, 25, 250 µl/ml - - Styles (1979,
1981)
Rat primary UDS 0.00084-8.4 mM - - Althaus et al.
hepatocytes (1982)
Mouse (B6C3F1) primary UDS 0.01-10 mM - - Larson et al.
hepatocyte (1994c)
Human primary UDS 4 cases 0.01-1.0 mM - - Butterworth
hepatocyte et al. (1989)
Human lymphocytes SCE 0.016-50 mM - + Morimoto &
Koizumi (1983)
Human lymphocytes SCE solvent acetone; 25, 50, + r PCB - Kirkland et
75, 100, 200, 400 µg/ml al. (1981)
Human lymphocytes UDS 0.1, 1.0, 10 mM - - Perocco et
+ r PB - al. (1983)
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Human lymphocytes UDS 2.5, 5, 10 µg/ml - - Perocco &
+ r PB - Prodi (1981)
Human Hela cells UDS 0.1-100 µg/ml; solvent - - Martin &
DMSO +r PB - McDermid (1981)
In vivo mammalian systems
Mouse JCR/SJ SCE in bone marrow oral: 25, 50, 100, + Morimoto &
cells 250 mg/kg body weight Koizumi (1983)
per day for 5 days
Mouse B6C3F1 DNA repair in liver oral: 240 mg/kg body - Reitz et al.
weight (1982)
Mouse C57BL sperm abnormalities vapour exposure: 0.04, + Land et al.
x C3H 0.08%; 4 h/day for 5 days (1981)
Mouse CBA x sperm abnormalities intraperitoneal: 0.025, - Topham (1980,
BALB/C 0.05, 0.075, 0.1, 0.25 mg/kg 1981)
body weight per day for
5 days; vehicle corn oil
Rat F-344 UDS in hepatocytes oral: 40, 400 mg/kg body - Mirsalis et
weight, single dose, al. (1982)
vehicle corn oil
Mouse B6C3F1 UDS in hepatocytes 238, 477 mg/kg body weight, - Larson et al.
single dose, corn oil (1994c)
vehicle
Table 13 (contd)
Species Strain/cells Measured end-point Test conditions Activationa Inducerb Resultc Reference
Rat (neonatal) liver and DNA damage (3H elution) vehicle corn oil; 200-400 - Petzold &
kidney cells mg/kg body weight Swenberg (1978)
a + = with metabolic activation; - = without metabolic activation; r = rat; yf = Yellowtail fish; jc = Japanese clam
b PB = phenobarbital; PCB = polychlorinated biphenyls
c + = positive; (+) = weakly positive; ± = equivocal; study cannot be evaluated; - = negative;
d In this test chromosome aberrations were also reported to occur; no details on this finding were reported.
The majority of studies with non-mammalian eukaryotic systems
(yeasts and other fungi) were negative. Positive results were
obtained with Saccharomyces cerevisiae D7, but only at the highest
concentration tested, at which there was a marked toxic effect
(Callen et al., 1980). It should be noted that this strain of yeast
contains an endogenous cytochrome P450-dependent monooxygenase
system. In Schizosaccharomyces pombe an indication for a mutagenic
effect was observed (Loprieno, 1981). The inconsistent results with
Saccharomyces cerevisiae D6 and JD 1 were probably due to
inadequate test conditions (exposure in plastic rather than glass
containers) and therefore it can be considered that chloroform was
non-mutagenic in these tests (Parry & Sharp, 1981; Sharp & Parry,
1981a). In two sex-linked recessive lethal tests with Drosophila
melanogaster, no mutagenic activity was observed (Gocke et al.,
1981; Vogel et al., 1981).
Chloroform did not induce gene mutations in V79 Chinese hamster
cells (Sturrock, 1977), or chromosomal aberrations in human
lymphocytes in vitro (Kirkland et al., 1981).
In vivo mammalian testing comprised four micronucleus tests
in mice, three of which gave a negative result (Tsuchimoto & Matter,
1981; Gocke et al.,1981; Salamone et al., 1981). The fourth
micronucleus test was reported to have given a weakly positive
result (Agustin & Lim-Sylianco, 1978). The same authors found a
positive effect in the mouse host-mediated assay with Salmonella
typhimurium TA1537 but not with TA1535.
Indicator studies showed that chloroform induces
sister-chromatid exchange (SCE) in hamster and human cells in vitro
in the absence of metabolic activation, and in mice in vivo
(Athanasiou & Kyrtopoulos, 1981; Morimoto & Koizumi, 1983). Positive
or weakly positive results were reported in two tests on DNA damage
and DNA repair with Escherichia coli and Saccharomyces cerevisiae
(Sharp & Parry, 1981b; Rosenkranz et al., 1981; Ichinotsubo et al.,
1981a).
The ability of chloroform to induce unscheduled DNA synthesis
(UDS) was examined in the in vitro and in vivo hepatocyte DNA
repair assays for the most sensitive site for tumour formation, the
female mouse liver (NCI, 1976a,b). In the in vitro assay, primary
hepatocyte cultures from female B6C3F1 mice were incubated with
concentrations from 0.01 to 10 mM chloroform in the presence of
3H-thymidine. UDS was assessed by quantitative autoradiography. No
induction of DNA repair was observed at any concentration. In the
in vivo assay, animals were treated by gavage with chloroform in
corn oil (238 and 477 mg/kg body weight). Primary hepatocyte
cultures were prepared 2 and 12 h later, incubated with
3H-thymidine and assessed for induction of UDS. No DNA repair
activity was seen at either dose or at either time point. These
negative results in the target organ are consistent with the
suggestion that neither chloroform nor its metabolites react
directly with DNA in vivo.
The ability of chloroform to induce DNA repair was examined in
freshly prepared primary cultures of human hepatocytes from
discarded surgical material. No activity was seen in cultures from
four different individuals at concentrations as high as 1 mM
chloroform (Butterworth et al., 1989).
Given the large number of sensitive assays to which chloroform
has been submitted, it is noteworthy that the reported positive
responses are so few. Furthermore, these few positive responses were
randomly distributed amongst the various assays with no apparent
pattern or clustering for any test system. Taken together, the
weight of evidence indicates that neither chloroform not its
metabolites would appear to interact directly with DNA or possess
genotoxic activity. The conclusion is consistent with the lack of
initiating activity of chloroform (see section 7.7.4).
7.7 Carcinogenicity
7.7.1 Mice
In a National Cancer Institute carcinogenicity study, B6C3F1
mice received USP grade chloroform stabilized with ethanol (0.5-1%)
in corn oil 5 times a week by gavage (NCI, 1976a,b). Dosing was
stopped after 78 weeks and the animals were sacrificed after 92
weeks. There were 20 animals per sex in the control group and 50
animals per sex in the dosed groups. The dose levels changed after
18 weeks, resulting in time-weighted average dose levels of 138
(low) and 277 (high) mg chloroform/kg body weight for male mice and
238 (low dose) and 477 (high dose) mg chloroform/kg body weight for
female mice. Administration of the highest dose of chloroform
reduced survival in the female mice. Causes of death were related to
the observed liver tumours, pulmonary inflammation and cardiac
thrombosis. This latter lesion was not observed in either the
control or the low-dose group. Dose-related increased frequencies of
hepatocellular carcinomas were found, the incidences being 1/18,
18/50 and 44/45 at 0, 138 and 277 mg chloroform/kg body weight in
the males and 0/20, 36/45 and 39/41 at 0, 238 and 477 mg chloroform
per kg body weight in the females, respectively. Mice presented
clinical signs of illness, i.e. a reduced food intake and an untidy
appearance, but clear information on non-neoplastic lesions was not
provided. There is evidence that tumour formation may have been
secondary to induced cytolethality and regenerative cell
proliferation (see Larson et al., 1994a, and section 7.2.1.1).
Jorgenson et al. (1985) exposed female B6C3F1 mice to
chloroform in their drinking-water for a period of two years. The
concentrations were 0, 200, 400, 900 and 1800 mg/litre, and the
numbers of animals were 430, 430, 150, 50 and 50 per group,
respectively. Time-weighted average daily doses were 0, 34, 65, 130
and 263 mg/kg body weight. Additional matched controls (50 animals)
received the same quantity of drinking-water (without chloroform) as
was consumed by the animals in the highest dose groups. Initially,
25% of the animals in the two highest dose groups died, but later on
the death rate was more or less equal to that in the control group.
No treatment-related effects on either liver or total tumour
incidence were observed. Lack of tumour formation is consistent with
the lack of induced liver necrosis or regenerative hepatocyte cell
proliferation when chloroform is administered in the drinking-water
(see Larson et al., 1994a and section 7.2.1.1).
The difference between the results obtained in the NCI study
(1976a,b) and the Jorgenson et al. (1985) study is probably related
to the manner in which the compound was administered. When given in
the drinking-water, only small amounts of chloroform reach the
liver, corresponding to each sip taken. Apparently, these small
doses and delivery rates can be metabolized, detoxified and
eliminated without liver damage (Larson et al., 1994a). When similar
daily amounts are given as a single bolus dose in corn oil, it is
probable that the high rate of delivery to the liver results in the
production of toxic metabolites that overwhelm detoxification
mechanisms, resulting in cell death and regenerative cell
proliferation (Larson et al., 1994a). The choice of vehicle may also
contribute to the observed difference in toxicity (Bull et al.,
1986) (see also section 7.2.1.1).
Roe et al. (1979) administered daily chloroform (British
Pharmacopoeia quality) in a toothpaste base (vehicle) to ICI mice
(control group 104 animals per sex, dose groups 52 animals per sex)
by gavage, 6 days a week for 80 weeks, followed by a 16-week
observation period. The dose levels were 0 (controls), 17 and 60
mg/kg body weight. Mice that died during the first 15 weeks of the
experiment were replaced by animals from a reserve group (which were
probably also dosed, although this was not specified). The control
toothpaste did not contain eucalyptol and peppermint oil, whereas
the toothpaste containing chloroform did contain these substances.
Treatment with chloroform resulted in slightly increased survival,
especially in the males. The most common cause of death was
respiratory failure. A slightly increased incidence of fatty
degeneration was observed among the chloroform-treated animals.
Total tumour incidence was increased in the male mice (20/37 and
21/38 at 17 and 60 mg/kg body weight, respectively, versus 20/72 in
the controls). Renal tumours (3 hypernephromas and 5 cortical
adenomas) were reported in 8 out of 38 males of the high-dose group.
In a second experiment by Roe et al. (1979), the influence of
peppermint oil, eucalyptol and chloroform was determined separately.
In this study, male ICI mice received 60 mg chloroform/kg body
weight daily, in the same way as in the study reported above. The
vehicle control (toothpaste without chloroform, eucalyptol and
peppermint oil) and dose groups consisted of 260 and 52 male
animals, respectively (the groups receiving a dose of peppermint or
eucalyptol also consisted of 52 animals). Again, the survival in the
chloroform-dosed group was better than in the control group. Total
tumour incidence was lower in the chloroform-treated group (30/49
versus 170/240 in the controls). However, administration of
chloroform resulted in a kidney tumour frequency (hypernephromas and
adenomas) of 9/49, compared with a control value of 6/240.
In a third study by Roe et al. (1979), 60 mg chloroform/kg body
weight in toothpaste (containing eucalyptol and peppermint oil) was
administered daily to male mice (52 per group) of the ICI, CBA,
C57BL and the CF1 strain for a period of 80 weeks. The chemical
was also administered in arachis oil to male mice of the ICI strain.
Each strain had its own control group. Terminal sacrifice was at 93,
97-99, 104 and 104 weeks for the CF1, ICI, C57BL and CBA strains,
respectively. In this study, a treatment-related increase in the
survival was found in all strains tested, except for the CF1
strain. Treatment with chloroform resulted in a higher incidence of
renal changes in the CBA and CF1 strains but not in the C57BL
strain. The cause of death in all four strains was renal neoplasia
in combination with respiratory and renal disease. In the C57BL, CBA
and CF1 strains no changes in tumour frequencies were observed. In
the ICI mice, after treatment with chloroform in either the
toothpaste vehicle or arachis oil, an increase in the incidence of
malignant kidney tumours was found (3/47 versus 0/49 in the
controls, toothpaste vehicle; 9/48 versus 0/50 in the controls,
arachis oil vehicle).
Though full results are not yet available, an additional
carcinogenesis bioassay in which mice were exposed to chloroform by
inhalation is under way (Matsushima, personal communication, 1993).
7.7.2 Rats
In a National Cancer Institute carcinogenicity study,
Osborne-Mendel rats received USP grade chloroform stabilized with
ethanol (0.5-1%) in corn oil 5 times a week by gavage (NCI,
1976a,b). Dosing was stopped after 78 weeks and the animals were
sacrificed after 111 weeks. There were 20 animals per sex in the
control group and 50 animals per sex in the dosed groups. The dose
levels changed after 23 weeks, resulting in time-weighted average
dose levels of 90 (low dose) and 180 (high dose) mg chloroform/kg
body weight for males and 100 (low) and 200 (high) mg chloroform/kg
body weight for females. Administration of chloroform reduced
survival in male and female rats in all dose groups. A clear
pathological reason for this effect in the rats was not given. In
male rats, dose-related increased frequencies of kidney epithelial
tumours were observed (incidences: 0/19, 4/50 and 12/50 at 0, 90 and
180 mg chloroform/kg body weight, respectively). In the females a
non-significant increase in the frequency of thyroid tumours was
found (incidences: 1/19, 8/49 and 10/46 at 0, 100 and 200 mg
chloroform/kg body weight, respectively). Rats presented clinical
signs of illness, i.e. a reduced food intake and an untidy
appearance. However, clear information on non-neoplastic lesions was
not provided.
Reuber (1979) re-evaluated the histological sections of the NCI
study (1976a,b) and reported the same neoplastic lesions as the NCI.
In addition, he noted that chloroform-dosed female rats developed
liver lesions that were not seen in the control females (i.e.
cholangiofibromas 0/20, 1/39 and 3/39; cholangiocarcinomas 0/20,
2/39 and 8/39; hyperplastic nodules 1/20, 7/39 and 12/39; and
hepatocellular carcinomas 0/20, 2/39 and 2/39, for the control, low-
and high-dose groups, respectively).
Jorgenson et al. (1985) exposed male Osborne-Mendel rats via
drinking-water to 0, 200, 400, 900 and 1800 mg chloroform/litre for
a period of two years. Time-weighted average daily chloroform doses
were 0, 19, 38, 81 and 160 mg/kg body weight and the numbers of
animals were 330, 330, 150, 50 and 50 per group, respectively.
Additional matched controls (50 animals) received the same quantity
of drinking-water (without chloroform) as was consumed by the
animals in the highest dose groups. As a probable consequence of
reduced drinking and reduced body weights, death rate was reduced
with increasing chloroform dosage and in the matched control group.
The only dose-related effect was an increase in renal tubular cell
adenomas and adenocarcinomas. The incidence for all kidney tumours
was 5/301, 1/50, 6/313, 7/148, 3/48 and 7/50 for control, matched
control and the 19, 38, 81 and 160 mg/kg groups, respectively. From
38 mg/kg body weight upwards the increase in the frequency of all
kidney tumours was statistically significant.
In an inadequately reported study, Tumasonis et al. (1985)
exposed male and female Wistar rats to 0 or 2900 mg chloroform per
litre drinking-water during the lifetime of the animals. Animal
numbers were 26 and 22 in the male and female control groups and 32
and 45 in the male and female treated groups, respectively. The
experiment started with weanlings. After 72 weeks, the
drinking-water chloroform concentrations were reduced because of an
increased intake of water by exposed animals. However, daily intakes
of chloroform varied considerably and so the time-weighted average
daily doses were estimated roughly from a figure in the report. They
appeared to be around 180 mg/kg body weight in the males and around
240 mg/kg body weight in the females. Body weights were decreased
and life-span was increased in the exposed animals. A severe hepatic
adenofibrosis (cholangiofibrosis) was observed in the exposed
animals. Ten out of the 40 females examined showed hepatic
hyperplastic nodules (none did in the control group). In the males
no increase in the incidence of neoplastic nodules was found.
Although full results are not yet available, an additional
carcinogenesis bioassay in which rats were exposed to chloroform by
inhalation is under way (Matsushima, personal communication, 1993).
7.7.3 Dogs
Heywood et al. (1979) administered chloroform to Beagle dogs at
dose levels of 0, 15 and 30 mg/kg body weight (6 days/week) in
toothpaste in a gelatin capsule for a period of 7.5 years. Sacrifice
followed after an observation period of 19 to 23 weeks, during which
the chloroform treatment was withdrawn. The control group consisted
of 16 animals of each sex and the dose groups of 8 animals of each
sex. There were no treatment-related increases in tumours.
7.7.4 Studies on initiating-promoting activity
7.7.4.1 Mice
One week after a single intraperitoneal administration of
ethylnitrosourea (0, 5 or 20 mg/kg body weight) to 15 days old CD1
Swiss mice (both sexes), Pereira et al. (1985) exposed the animals
to chloroform via drinking-water at concentrations of 0 or 1800
mg/litre until they were 51 weeks of age, after which the animals
were sacrificed. The chloroform treatment did not affect the liver
or lung tumour frequency in the females and the lung tumour
frequency in the males. However, the liver tumour frequency in the
males appeared to be reduced after the treatment.
Capel et al. (1979) administered chloroform as a drinking-water
solution (estimated daily doses of 0, 0.15 or 15 mg/kg body weight)
to male mice either from 14 days before or from 14 days before to 14
days after intraperitoneal injection with Ehrlich ascites cells (TO
strain), subcutaneous injection with B16 melanoma cells (C57BL
strain) or intramuscular injection with Lewis lung carcinoma cells
(C57BL strain). Chloroform treatment enhanced the growth of Ehrlich
ascites cells (measured as intraperitoneal tumour cell DNA) at the
high dose level. In comparison with the controls, more animals
receiving chloroform at both dose levels had organs invaded with B16
melanoma cells. Lewis lung tumour growth, measured as primary tumour
size or pulmonary metastases, was not significantly enhanced at
low-dose chloroform treatment, but after treatment with the high
dose the number of pulmonary metastases and tumour size were
markedly increased.
In a two-stage (initiation/promotion) treatment protocol,
Klaunig et al. (1986) studied the effect on liver tumour incidence
in male B6C3F1 mice (35/group) after continuous treatment with 600
and 1800 mg chloroform/litre drinking-water for 52 weeks to
determine if chloroform expresses its hepatocarcinogenicity through
tumour promotion mechanisms. Two groups received 600 and 1800 mg
chloroform/litre drinking-water containing diethylnitrosamine (DENA;
10 mg/litre) during the first 4 weeks of exposure. Two other groups
received 600 and 1800 mg chloroform/litre drinking-water without
DENA. The DENA groups constituted the initiated groups. One
initiated and one non-initiated control group were included.
Chloroform did not affect the incidence of liver or lung tumours by
itself, and even inhibited liver and lung tumorigenesis in the
DENA-initiated mice, compared with DENA treatment alone.
7.7.4.2 Rats
Deml & Oesterle (1985, 1987) studied the ability of chloroform
to promote the development of liver tumours. Female Sprague-Dawley
rats were initiated for liver tumours by administration of a single
dose of 8 mg dimethyl nitrosamine/kg body weight. This was followed
by administering chloroform (25, 100, 200 and 400 mg/kg body weight)
in an olive oil vehicle twice weekly for 11 consecutive weeks. There
was a dose-related increase of ATPase-negative, gamma-glutamyl
transpeptidase (GGTase)-positive and glycogen-storing foci of cells
within the liver. For example, ATPase-deficient foci were increased
from approximately 2-fold to 5-fold by doses of 100 and 400 mg/kg,
respectively. These data demonstrate that chloroform in an oil
vehicle will probably promote development of hepatic tumours in
rats.
Herren-Freund & Pereira (1986) evaluated the ability of
chloroform to act as an initiator, promoter and co-carcinogen in
B6C3F1 mice and male Sprague-Dawley rats. In rats, the initiator
was administered 18-24 h following a two-thirds partial hepatectomy.
Diethylnitrosamine (0.5 mmol/kg body weight) was used as the
positive control for initiation and phenobarbital (500 mg/litre
drinking-water) was used as the positive control for promotion.
Ethylnitrosourea (ENU) was the positive control for initiator in
15-day-old mice and phenobarbital (500 mg/litre drinking-water) was
used as the positive control for promotion. Chloroform was
administered as a single dose of 180 and 360 mg/kg body weight as an
initiator (no vehicle) in the rat and 1800 mg/litre drinking-water
for 48 weeks as a promoter. There was no evidence that chloroform
was able to act as an initiator in rats. Moreover, it did not act as
a tumour promoter in either mice or rats, but actually decreased the
numbers of hepatic tumours induced in neonatal mice by ENU.
Concurrent administration of chloroform and DENA to the rat had no
significant effect on foci or tumour development in rats. These data
further suggest that the corn oil vehicle is important to the
hepatocarcinogenic effects of chloroform.
In a previous experiment, Pereira et al. (1982) had examined
the effect of chloroform as an initiator and promoter. Chloroform
was administered at 180 mg/kg body weight in a single dose as an
initiator and 180 mg/kg body weight twice a week for 53 days as a
promoter. In this case, tricaprylin was the vehicle. Chloroform had
no activity as an initiator. There was a small, but statistically
significant, increase in the numbers of GGTase-positive foci in the
promotion study.
Although chloroform is an established rodent carcinogen,
several studies have shown that chloroform administered in impolar
solvents also has anti-cancer properties as it inhibits tumour
growth in mouse liver and in the gastrointestinal tract of the rat
(Pereira et al., 1985; Daniel et al., 1989).
Chloroform administered in drinking-water (0, 900 and 1800
mg/litre) to Fischer-344 rats significantly decreased
gastrointestinal (GI) tumours that were initiated by a single 200
mg/kg dose of dimethyl hydrazine (DMH) (Daniel et al., 1989). GI
tumour incidence was 14/39 in animals treated with DMH alone and
5/39 and 5/40 in the groups in which DMH treatment was followed by
900 and 1800 mg chloroform/litre, respectively, for 39 weeks.
Chloroform also inhibits the propensity for three
gastrointestinal tract carcinogens, benzo (a)pyrene (BAP),
1,2-dimethylhydrazine (DMH) and methylnitrosourea (MNU), to induce
nuclear anomalies in the proximal colon of B6C3F1 mice (Daniel et
al., 1991). These authors found that in mice pre-adapted to 1800 mg
chloroform/litre drinking-water for 30 days prior to the carcinogen
administration the level of nuclear anomalies induced in the
proximal colon was reduced by four-fold for BAP and two-fold for
both MNU and DMH. In the duodenum, chloroform was effective at
inhibiting unclear anomalies only for MNU.
Reddy et al. (1992) demonstrated that chloroform inhibits the
development of diethylnitrosamine-initiated, phenobarbital-promoted
gamma-glutamyl transpeptidase and placental form
glutathione- S-transferase-positive foci in the liver of male
Fischer-344 rats. They suggested that chloroform exerts its focal
inhibitory effect by selectively killing the putative initiated
cells.
The lack of initiating activity in these initiation-promotion
assays supports the conclusion that chloroform is non-genotoxic
(section 7.6), and also indicates that the carcinogenic action of
chloroform is attributable to a non-genotoxic/cytotoxic mode of
action (sections 7.2.1.1 and 7.7). Interestingly, more of the above
studies reported that chloroform inhibited the growth or formation
of precancerous or cancerous cells than those that reported that
chloroform had promotional activity.
7.8 In vitro studies
In vitro studies frequently provide insight into how
chemicals induce cytotoxic effects. However, at high concentrations
(e.g., 5 mM and above), the solvent effects of chloroform on cell
membranes complicate the interpretation of these experiments. The
preparations that have been studied are precision-cut slices taken
from the liver, primary hepatocytes suspensions and cultures.
Azri-Meehan et al. (1992) studied the cytotoxic effects of
chloroform in liver slices taken from phenobarbital-treated rats. No
comparison was made with non-induced animals. Concentrations in the
range of 0.5 to 1.6 mM induced loss of intracellular potassium and
glutathione. Reduced mitochondrial function (measured as decreases
in dye reduction) was observed in the same dose range. A
concentration of 0.2 mM had no effect.
Glende & Recknagel (1992) examined the ability of a number of
chlorinated hydrocarbons to activate phospholipase A2, presumably
through damage to calcium-binding sites in the endoplasmic
reticulum. At doses that induce 30 to 70% release of cellular
lactate dehydrogenase (i.e. 9.8 mM), chloroform did activate
phospholipase A2. This concentration is similar to that necessary to
destroy the calcium-binding capacity of the endoplasmic reticulum.
O'Hara et al. (1991) examined the effects of chloroform on the
viability of hepatocytes in suspension (measured by potassium
retention). These hepatocytes were isolated from control
phenobarbital-treated rats. The minimum concentration required to
produce an effect on potassium retention decreased from 10 mM in
control hepatocytes to 1 mM in hepatocytes obtained from induced
animals.
A number of studies of chloroform cytotoxicity in suspensions
of rat hepatocytes have been reported (Stacey, 1987). However, the
very high nominal concentrations of chloroform that were apparently
necessary to produce significant effects (i.e. 30 and 60 mM) raise
considerable questions as to their relevance to in vivo hepatic
toxicity.
An innovative approach has been developed for incubating
hepatocyte suspensions with the chemical of interest, followed by
observation of the cytotoxic response after placing the treated
cells into culture (Kedderis et al., 1993a). Such cytotoxicity was
observed when hepatocyte suspensions derived from B6C3F1 mice were
incubated with concentrations of chloroform between 1.3 and 3.8 mM.
These concentrations were consistent with peak liver concentrations
expected with the high doses of chloroform utilized in the
assessment of chloroform carcinogenicity in mice (NCI, 1976a,b), as
predicted by the Corley et al. (1990) pharmacokinetic model
(Kedderis et al., 1993b). The cytotoxicity of chloroform was
potentiated by pretreating the mice with acetone to induce
cytochrome P450 2E1.
Although there have been substantial advances in the study of
in vitro chloroform toxicity, the applicability of the results
that are available to date to estimate hazards in humans remains to
be established.
7.9 Factors modifying toxicity; toxicity of metabolites
The in vivo toxicity of chloroform is modified by a range of
factors. The rate of its biotransformation is a significant
determinant of its toxicity. Hence, factors that increase or
decrease chloroform biotransformation may alter the intensity of
chloroform-induced toxicity. The activities of the cytochrome P450
isoforms that catalyse the biotransformation of chloroform differ
among species and between sexes of experimental animals. Moreover
the activities of the enzymes that metabolize chloroform may be
increased or decreased by exposure to chemicals, and exposure to
chloroform itself may alter chloroform metabolism.
In addition to differences in the rates of chloroform
bioactivation, treatments that alter susceptibility are also
important determinants of chloroform-induced toxicity. Cellular
glutathione concentrations are an important determinant of
susceptibility, and perturbations of glutathione homeostasis may
affect markedly the toxicity of chloroform. Finally, for some of the
treatments that alter chloroform toxicity discussed in this section,
the mechanistic basis of these interactions is not well understood.
Brown et al. (1974a) reported that inhalation exposure of
phenobarbital-treated male Sprague-Dawley rats to chloroform at
doses of 2.45 or 4.9 g/m3 (500 or 1000 ppm) for 2 h produced
marked centrilobular necrosis that was accompanied by decreased
hepatic glutathione concentrations. in vitro studies showed that
glutathione reduced the covalent binding of [14C]-chloroform
metabolites to microsomal protein.
Docks & Krishna (1976) observed that administration of
chloroform decreased hepatic glutathione concentrations in
phenobarbital-treated rats (male, Sprague-Dawley), but not in
control animals 1 to 2 h after administration, and caused liver
necrosis. Administration of isopropanol or acetone, which increased
the covalent binding of chloroform metabolites (Sipes et al., 1973),
did not alter hepatic glutathione concentrations.
Starvation and carbohydrate restriction increase the in vivo
metabolism of chloroform and its hepato- and nephrotoxicity in rats
(Nakajima & Sato, 1979; McMartin et al., 1981; Nakajima et al.,
1982). In contrast, protein deficiency does not alter chloroform
toxicity (McLean, 1970).
Several authors have demonstrated that administration of
alcohols, including ethanol (Kutob & Plaa, 1962b; Sato et al., 1980,
1981), or ketones increases chloroform metabolism and
hepatotoxicity. An extension of these studies to include a range of
alcohols showed that methanol, ethanol, isopropanol, tert-butanol,
pentanol, hexanol, octanol and decanol all potentiate
chloroform-induced liver injury and lower the LD50 of chloroform
in male Sprague-Dawley rats (Ray & Mehendale, 1990). Aliphatic
ketones, including acetone, 2-butanone, 2-pentanone, 2-hexanone,
2,5-hexanedione, 2-heptanone and methyl isobutyl ketone, also
increase chloroform-induced hepatotoxicity (Hewitt et al., 1990;
Vézina et al., 1990), but treatment with 2-hexanone does not
increase chloroform-dependent lipid peroxidation either in vivo or
in vitro (Cowlen et al., 1984a,b). The potentiating effect of
alcohols and ketones in chloroform-induced hepatotoxicity is
attributed to an increase in the activity of the cytochromes P450
that metabolize chloroform (Koop et al., 1982; Ryan et al., 1986;
Brady et al., 1989; Vézina et al., 1990).
Harris et al. (1982) evaluated, by the intraperitoneal route,
the toxicity of chloroform (0.2 ml/kg body weight) and carbon
tetrachloride (0.1 ml/kg body weight) given alone or together to
male rats. At these doses, neither chloroform nor carbon
tetrachloride produced toxicity, but increases in SGPT activity and
hepatic triglyceride and calcium concentrations were seen when both
compounds were given together. Ikatsu & Nakajima (1992) showed that
a single inhalation exposure to 490 mg/m3 (100 ppm) chloroform for
8 h resulted in mid-zonal hepatotoxicity. In ethanol-treated rats
exposed to both chloroform (50 ppm) and carbon tetrachloride (10
ppm), liver necrosis and elevated plasma GOT/GPT activities were
observed. These findings indicate that the toxicity of chloroform is
elevated in the presence of carbon tetrachloride. O'Hara et al.
(1991) studied the effect of chloroform and carbon tetrachloride in
rat hepatocytes and demonstrated that the combined toxicity of both
compounds was greater than additive.
The pesticide kepone (chlordecone), but not its non-ketonic
analogue mirex, increases chloroform-induced hepato- and
nephrotoxicity (Hewitt et al., 1979, 1982; Iijima et al., 1983). In
Mongolian gerbils, which are susceptible to chloroform-induced
toxicity (200 or 500 µl/kg body weight, intraperitoneal), treatment
with phenobarbital or chlordecone decreased the hepatotoxicity of
chloroform (Ebel et al., 1987); in contrast, rats given 50 to 500
µl/kg body weight chloroform (intraperitoneally) showed little
hepatotoxicity, but toxicity was increased after treatment with
phenobarbital or chlordecone.
The drinking-water contaminants dichloroacetic acid (DCA) and
trichloroacetic acid (TCA) potentiate chloroform toxicity (Davis,
1992). Male and female rats (Sprague-Dawley) were orally treated
with 0.92 or 2.45 mmol/kg body weight DCA or TCA and were given 0.75
mg/kg body weight chloroform (intraperitoneally) 3 h later.
Increases in plasma ALAT activities were observed in female, but not
in male rats 24 and 48 h after giving DCA; in contrast, plasma ALAT
activities were increased 24, but not 48 h, after giving TCA in both
male and female rats. DCA administration increased blood urea
nitrogen concentrations in female rats, but produced little effect
in male rats, whereas TCA administration produced an effect only in
female rats 48 h after treatment. The mechanism of the effect of DCA
and TCA was not elaborated.
Monochloroacetic acid (MCA) given by gavage to male (188 mg/kg
body weight) or female (94 mg/kg body weight) Sprague-Dawley rats
one hour before giving chloroform (520 mg/kg body weight)
intraperitoneally increased chloroform-induced hepatotoxicity in
male rats, but had little effect in female rats (Davis & Berndt,
1992). Treatment with MCA alone decreased glomerular filtration
rates in female rats. The mechanism by which MCA potentiated
chloroform toxicity was not elucidated.
Temporal variations in chloroform-induced hepatotoxicity have
been observed in rats (Lavigne et al., 1983). Male Sprague-Dawley
rats were given chloroform (0.5 ml/kg body weight) intraperitoneally
at 9:00, 13:00, 17:00, 21:00 or 03:00 h and were killed 4 h after
treatment. Hepatotoxicity, as assessed by serum GPT, GOT and LDH
activities, was minimal and maximal at 09:00 h and 21:00 h,
respectively, whereas glucose-6-phosphatase activity was decreased
at 03:00 h and 13:00 h. When rats were starved for 16 h before
giving chloroform at 09:00 h, toxicity was increased substantially.
Charbonneau et al. (1991) studied the effect of acetone
treatment on the toxicity with a range of binary mixtures of
haloalkanes in rats. An increased hepatotoxic response was observed
with binary mixtures containing chloroform, carbon tetrachloride,
1,1,2-trichloroethane or 1,1-dichloroethylene.
In vitamin-A-deficient rats, serum ALAT and particularly ASAT
activities were increased after intraperitoneal administration of
chloroform, compared to control rats (Savoure et al., 1992).
8. EFFECTS ON HUMANS
8.1 Acute non-lethal effects
Chloroform is irritating to mucous membranes, producing
gastroenteritis with persistent nausea and vomiting. Symptoms
following ingestion of chloroform are similar to those following
inhalation (van der Heijden et al., 1986).
Cases of severe intoxication after suicidal attempts, with the
same pattern of symptoms as after anaesthetical use, have been
reported by Schröder (1965). There are considerable inter-individual
differences in susceptibility. Some persons presented serious
illness after an oral dose of 7.5 g of chloroform, whereas others
survived a dose of 270 g chloroform. The mean lethal dose for an
adult is estimated to be about 45 g (Winslow & Gerstner, 1978).
Rao et al. (1993) successfully managed acute toxicity from
chloroform in a 33-year-old white woman who attempted suicide by
injecting 0.5 ml of chloroform, and then drank half a cup the next
morning. Plasma chloroform levels, measured by headspace GC,
declined rapidly. Sequential measurement of biomarkers in serum for
liver cell necrosis, liver function and liver regeneration indicated
the presence of initial liver damage followed by recovery. The
authors suggested that, in addition to biomarkers for liver
necrosis, serial determinations of markers for liver regeneration
provide objective evidence for recovery from chloroform poisoning.
It has been reported that chloroform can cause severe toxic
effects in humans exposed to 9960 mg/m3 (2000 ppm) for 60 min,
symptoms of illness at 2490 mg/m3 (500 ppm) and can cause
discomfort at levels below 249 mg/m3 (50 ppm) (Verschueren, 1983).
Most data on the controlled exposure of man to chloroform have
resulted from its clinical use as an anaesthetic. This use of
chloroform was described as early as 1847 (Simpson, 1847). Induction
of anaesthesia may result from inhalation of chloroform vapours at a
concentration of 24 to 73 g/m3 air. For maintenance of
anaesthesia, concentrations in the range of 12 to 48 g/m3 are
required. As with animals, chloroform anaesthesia may result in
death in humans due to respiratory and cardiac arrhythmias and
failure. Because of the relatively high frequency of "late
chloroform poisoning" (liver toxicity), its use as anaesthetic has
been abandoned.
Other effects related to chloroform inhalation are: increase in
the rate and depth of respiration during induction and light
anaesthesia, minute volume decrease in deep anaesthesia,
hypothermia, depletion of adrenal adrenaline content, hypotension,
depression of gastrointestinal tract motility, respiratory acidosis,
hyperglycaemia, ketosis, constriction of the spleen, increase in the
number of leucocytes (especially polymorphonuclear cells), a
decrease in clotting time and an increase in prothrombin time. The
characteristics and severity of the effects depend on depth and
duration of anaesthesia (Adriani, 1970).
The cardiac effects might be secondary and due to hypoxia,
caused by depression of respiratory activity. No studies have been
found in which this problem has been investigated in man (e.g., by
forced respiration), but Taylor et al. (1976) obtained indications
that chloroform itself produces cardiovascular disturbances in
rabbits (viz. disturbances in left ventricular functioning and an
increase in peripheral resistance; see section 7.1.2.3) after
exposure to 244 mg/m3 for 1 min.
In man, as well as in animals, renal tubular necrosis and renal
dysfunction (anuria, proteinuria, uraemia, increase in blood urea
nitrogen) have been observed (Kluwe, 1981). Recovering from
chloroform anaesthesia, some patients may show the symptoms of a
delayed chloroform poisoning several days later. Prostration,
protracted nausea, vomiting, jaundice and coma due to hepatic
dysfunction are observed. The patient may die within 5 days after
anaesthesia. At autopsy, degeneration and necrosis of liver tissue
have been found (Goodman & Gilman, 1970). In general the symptoms
appear to be similar to those observed in animals.
According to Oettel (1936) and Winslow & Gerstner (1978),
exposure to concentrated chloroform vapours causes a stinging
sensation in the eye. Splashing of the liquid into the eye evokes
burning, pain and redness of the conjunctival tissue. Occasional
injury of the corneal epithelium will recover fully within a few
days. Dermal contact with chloroform causes chemical dermatitis
(symptoms: irritation, reddening, blistering and burns).
8.2 Epidemiology
8.2.1 Occupational exposure
Challen et al. (1958) reported the effects of exposure of
workers (mostly female) to chloroform vapour in a factory during
manufacture of lozenges containing the chemical. Eight workers (four
working full-time and four half-time) were exposed to chloroform
concentrations of 375 to 1330 mg/m3, with a peak concentration of
5680 mg chloroform/m3, for periods of 3 to 10 years. The symptoms
reported were lassitude, thirst, gastrointestinal distress, frequent
and scalding urination, lack of concentration, depression and
irritability. The management stated that some of the employees had
been noticed staggering about at work. Nine other workers (one
full-time, eight half-time), who were exposed to chloroform
concentrations of 110 to 350 mg/m3 for 10 to 24 months, suffered
from the same complaints as stated above, but to a lesser degree.
Several liver function tests did not reveal signs of liver toxicity,
but these tests were not very sensitive.
Bomski et al. (1967) investigated the occurrence of hepatitis
in a chemical factory in relation to the occurrence of this disease
in the city where the factory was located. The 68 workers were
exposed to occupational chloroform concentrations of 10 to 1000
mg/m3 for 1 to 4 years. In this group of employees, a higher
frequency of hepatitis was found than in the city inhabitants.
Seventeen workers showed hepatomegaly and in three of them hepatitis
was observed. Ten workers showed splenomegaly, but the cause of the
splenomegaly was not discussed.
The finding of a high frequency of hepatitis among
occupationally chloroform-exposed workers, as compared to that in
the city inhabitants, is supported by a recent report on a
16-year-old patient who attempted suicide by ingesting chloroform.
This led to the development of toxic hepatitis (Hakim et al., 1992).
In a study by Phoon et al. (1975), the air in the workroom of
13 persons with jaundice originally diagnosed as having viral
hepatitis was analysed for chloroform. The chloroform concentration
in the workroom appeared to be more than 1950 mg/m3. The period of
exposure was less than 6 months. Because no worker had a history of
fever and there was no relation to past medical history, it was
concluded that the original diagnosis must have been wrong and
should have been toxic jaundice. Five of the people with jaundice
and four other colleagues had blood chloroform levels in the range
of 1 to 2.9 mg/litre.
In another factory 18 cases of what seemed to be hepatitis B
were reported (Phoon et al., 1983). Investigation of the
occupational environment revealed a constant exposure to chloroform,
with concentrations in the range of 80 to 160 mg/m3. The exposure
period of these workers was less than 4 months and the conclusion
was drawn that these were cases of toxic jaundice related to
chloroform exposure, because no infection with the hepatitis B virus
could be established.
A historical mortality study was carried out by Linde & Mesnick
(1979). They investigated the cause of death of white male
anaesthesiologists, who were occupationally exposed to chloroform
vapours (extent of exposure was unknown). The death certificates
used were of persons, who were presumed to be exposed during the
1880-1890 period and who died during the 1930-1946 period.
Comparison of their death certificates with those of several control
groups did not exclude a possible association between cancer and
chloroform exposure.
8.2.2 General exposure
There have been numerous reports over the last 15 years which
have evaluated the relationship between chlorinated water and the
incidence of cancer. Chloroform is but one of many by-products
produced by reaction of chlorine with naturally occurring material
in source waters (Bull & Kapfler, 1991). Many of these studies noted
increased risk of cancer which at least partially fulfilled criteria
for causality (e.g., consistency, specificity and temporal
relationships).
IARC (1991) reviewed the available studies and concluded the
strongest evidence of increased risk related to exposure to
chlorinated surface water relative to unchlorinated ground water for
the incidence of cancer of the urinary bladder. However, the
weight-of-the-evidence evaluation by IARC concluded that there is
inadequate evidence for the carcinogenicity of chlorinated
drinking-water in humans.
Morris et al. (1992) conducted a meta-analysis which attempted
to integrate quantitatively the results of previously published
studies in which individual exposures were evaluated (i.e. case
control and cohort studies). The authors identified increased rates
of bladder and colo-rectal cancer in individuals exposed to
chlorinated surface water, which appeared to exhibit a dose-related
trend. Although this study was confounded by substantial differences
in exposure variables that occur in different water supplies, higher
risk rates were estimated when the analysis was restricted to those
studies which were judged to have the highest quality exposure
assessments. Because of the confounding of these results by chlorine
residual levels and a multiplicity of other chemicals which are
animal carcinogens and mutagens, none of the drinking-water studies
specifically implicate chloroform as a human carcinogen.
Kramer et al. (1992) studied the association between exposure
to trihalomethanes in the water supply and adverse reproductive
outcomes in the state of Iowa (USA). Estimations of chloroform
exposure were based on municipal water surveys. After adjustment for
maternal age, parity, prenatal care, marital status, education and
maternal smoking, an increased risk for intrauterine growth
retardation (abnormally low birth weight) was associated with
chloroform concentrations above 10 µg/litre. Limitations of the
study involve the ascertainment and classification of exposures to
trihalomethanes (such as fluctuation of levels and exposure at
individual level) and the influence of potential confounding
influences of unmeasured contaminants.
8.3 Abuse and addiction
Exposure to chloroform may result in euphoria and therefore
people expose themselves to chloroform by drinking the liquid or
sniffing the vapours (Storms, 1973). Addiction to chloroform and
chloroform-containing cough syrups has been reported by Heilbrunn et
al. (1945) and Conlon (1963). According to Heilbrunn et al. (1945),
addicts tolerated very high daily doses and presented neurological
symptoms and degenerative changes in the brains.
After an intravenous injection of 7.5 g of chloroform, a
patient showed signs of pulmonary malfunction and haemolysis. In
this case, kidney or liver toxicity was not reported (Timms et al.,
1975).
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Freshwater organisms
The data on the toxicity of chloroform to several freshwater
organisms are listed in Table 14.
Due to the volatility of chloroform, caution must be exercised
in interpreting the test results, particularly those in open static
systems where no chemical analysis of the actual concentration was
carried out.
9.1.1 Short-term toxicity
The chemical is of low toxicity to unicellular plants and other
microorganisms (concentration range of initial population growth
inhibition: 125 to > 3200 mg/litre). Chloroform is moderately toxic
to Daphnia magna (LC50 = 29 mg/litre).
The LC50 values for several species of fish are in the range
of to 191 mg/litre. However, initial toxicity may occur at lower
levels: the no-observed-lethal concentrations (NOLCs) for Salmo
gairdneri and Lepomis macrochirus appear to be 8 and 3 mg/litre,
respectively. At lower concentrations (< 13 mg/litre), Salmo
gairdneri shows loss of equilibrium, slow operculum movement and
narcosis (Anderson & Lusty, 1980).
In Gasterosteus aculeatus, chloroform produced anaesthesia
which could be maintained for at least 90 min at concentrations of
210 mg/litre. Exposure to concentrations higher than 300 mg/litre
resulted in decreased oxygen consumption and death (Jones, 1947),
whereas concentrations lower than 120 mg/litre excited the animals
and gave rise to considerable higher oxygen uptake.
Chloroform is considerably more toxic to the juvenile stages of
several species of amphibians. In a continuous-flow system, Birge et
al. (1980) tested the toxicity of chloroform to embryo-larval stages
of several species of amphibians after exposure for 7-9 days (Table
14). Hyla crucifer appeared to be the most susceptible species. An
effect was found on the hatching rate of the embryos, which declined
from 97% at 8 µg/litre to 4% at 7340 µg/litre. In addition there was
some evidence of teratic larvae. During the 4 days post-hatching the
LC50 declined from 760 to 270 µg/litre. The other species tested
were less affected and only Rana pipiens showed a high
teratogenicity frequency in the offspring (100% at 27 mg/litre at
18% hatching rate).
Table 14. Chloroform toxicity to water organisms
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Short-term toxicity
Bacteria
Pseudomonas 25 acc.d Bringmann & S - 16 h initial reduction of 125 Bringmann &
putida Kühn (1977) cell multiplication Kühn (1977)
Pseudomonas 25 acc.d Bringmann S - 16 h initial change of 125 Bringmann (1973)
fluorescens (1973) culture turbidity
Algae
Microcystis 27 acc.d Bringmann S - 192 h initial reduction of 185 Bringmann (1975)
aeruginosa (1975) cell multiplication
Scenedesmus 25 acc.d Bringmann & S - 192 h initial reduction of 1100 Bringmann &
quadricauda Kühn (1977) cell multiplication Kühn (1977)
Haematococcus 20 acc.d Tümpling S - 4 h 10% reduction of 440 Knie et al. (1983)
pluvialis (1972) oxygen production
Protozoans
Entosiphon 25 Bringmann (1978) S - 72 h initial reduction of > 6560 Bringmann (1978)
sulcatum cell multiplication
Uronema 25 Bringmann & S - 20 h initial reduction of > 6560 Bringmann &
parduczi Kühn (1980) cell multiplication Kühn (1980)
Chilomonas 20 Bringmann et al. S - 48 h initial reduction of > 3200 Bringmann et al.
paramaecium (1980) cell multiplication (1980)
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Crustaceans
Daphnia magna 22 reconstituted well S - 48 h LC50 29 LeBlanc (1980)
water, pH 7,
hardness 173 mg
CaCO3/litre
Daphnia magna 19.8-20.9 lake water, pH S - 48 h LC50 65.7 Gersich et al.
8.0, hardness (1986)
157 mg CaCO3/litre
Daphnia magna 23 distilled water S - 48 h LC50 78.9 Abernethy et al.
(1986)
Fish
Cyprinus carpio 26 filtered well water S A until LC50 97 Mattice et al.
(mixed gametes) hatching (1981)
(3-5 days)
Pimephales 25 carbon filtered S - 96 h LC50 129 Mayes et al.
promelas lake water, pH (1983)
(10-15 days) 7.6-8.3, hardness
125 mg CaCO3/litre
(30-35 days) 22 idem S - 96 h LC50 171
(60-100 days) 22 idem S - 96 h LC50 103
Brachydanio 20 dechlorinated CF - 48 h LC50 100 Slooff (1979)
rerio tap water, pH 8,
hardness 10 d.H.
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Salmo 20 dechlorinated CF - 48 h initial reduction 20 Slooff (1979)
gairdneri tap water, pH 8, of respiration
hardness 10 d.H. frequency
Leuciscus 20 acc.d Mann (1975) S - 48 h LC50 162-191 Juhnke &
idus melanotus Lüdemann (1978)
Carassius 5 aerated tap water S - 1 h EC50 97-167 Cherkin &
auratus (anaesthesia) Catchpool (1964)
20 aerated tap water S - 1 h EC50 167
(anaesthesia)
Salmo 19 aerated river water CF A 96 h LC50 18 Anderson & Lusty
gairdneri NOLC 8 (1980)
Leopomis 19 aerated river water CF A 96 h LC50 18 Anderson & Lusty
macrochirus NOLC 3 (1980)
Micropterus 19 aerated river water CF A 96 h LC50 51 Anderson & Lusty
salmoides NOLC 39 (1980)
Ictalurus 19 aerated river water CF A 96 h LC50 75 Anderson & Lusty
punctatus NOLC 68 (1980)
Amphibians
Hyla crucifer 20.5 acc.d Birge et al. CF A until 4 LC50 0.3 Birge et al. (1980)
(eggs; 2 to (1979); pH 7.6, days after
6h post- hardness 107 mg hatching
spawning) CaCO3/litre or death
(7 days in
total)
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
Amphibians (contd)
20.5 idem CF A idem NOLC 0.009
Rana pipiens 20.5 idem CF A idem (9 LC50 4.2 Birge et al. (1980)
(eggs; 30 min days in
after total)
fertilization) 20.5 idem CF A idem NOLC 0.16
Rana palustris 21.5 acc.d Birge et al. CF A idem (8 LC50 20.6 Birge et al. (1980)
(eggs; 2 to (1979); pH 7.6, days in
6 h post- hardness 104 mg total)
spawning) CaCO3/litre
21.5 idem CF A idem NOLC 0.33
Bufo fowleri 21.5 idem CF A idem (7 LC50 35.1 Birge et al. (1980)
(eggs; 2 to days in
6 h post- total)
spawning) 21.5 idem CF A idem NOLC 0.33
Long-term toxicity
Fish
Poecilia 22 Alabaster & Abram Sb - 14 days LC50 102 Könemann (1981)
reticulata (1964)
Salmo 13.5 ± 1 acc.d Birge et al. CF A until 4 LC50 2.0 Birge et al. (1979)
gairdneri (1979); pH 7.3, days after
(eggs; 20 min hardness 48 mg hatching
after CaCO3/litre or death
fertilization (27 days
totally)
Table 14 (contd)
Organism Temperature Medium Stat/ Analysisc Exposure Parameter Concentration Reference
(°C) flowa duration (mg/litre)
13.5 ± 1 idem CF A idem NOLC 0.004
13.5 ± 1 idem, hardness 210 CF A idem LC50 1.24
mg CaCO3/litre
13.5 ± 1 idem CF A idem NOLC 0.003
a S = static, CF = continuous flow;
b static conditions but test water changed every 24 h
c A = concentration of test compound analysed during assay; - = no data
d acc. = according to the medium described in these references
9.1.2 Long-term toxicity
Birge et al. (1979) tested the toxicity of chloroform for
embryo-larval stages of Salmo gairdneri (Table 14) after 27 days.
The chemical was especially toxic for the unhatched embryos (LC50
is about 2 mg/litre), but did not cause death in the larvae at
concentrations up to 10.6 mg/litre. The occurrence of teratic
survivors in the hatched population increased from 3% at 56 µg/litre
to 40% at 10 mg/litre.
9.2 Marine organisms
The acute toxicity of chloroform to Artemia salina was tested
by Robinson et al. (1965). The observed effect was anaesthesia and
the EC50 value was 68 mg/litre after 10 h of exposure in
artificial sea water in closed containers under static conditions.
The 50% immobilization concentration (IC50) of chloroform for
Artemia salina nauplii, subjected to salinity stress, was
determined in a static study using artificial sea water by Foster &
Tullis (1985). The toxicity test began 30 h after hatching had
commenced and lasted for 24 h. The IC50 was 37 mg/litre.
Stewart et al. (1979) tested the acute toxicity of chloroform
to larvae of Crassostrea virginica. Chemical analysis showed a
rapid decline of chloroform concentrations in the sea-water medium.
The estimated LC50 was 1 mg/litre.
Pearson & McConnell (1975) tested the acute toxicity to
Limanda limanda in a continuous-flow system containing natural sea
water and obtained an LC50 of 28 mg/litre.
Cowgill et al. (1989) determined the sensitivity of the marine
diatom Skeletonema costatum to chloroform after exposure for 5
days under static conditions. The EC50 values calculated were 477
mg/litre and 437 mg/litre based on total cell count and total cell
volume, respectively. The NOEC was 216 mg/litre.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health risks
10.1.1 Exposure
Based on estimates of mean exposure from various media, the
general population is exposed to chloroform principally in food
(approximately 1 µg/kg body weight per day), drinking-water
(approximately 0.5 µg/kg body weight per day) and indoor air (0.3 to
1 µg/kg body weight per day). Estimated intake from outdoor air is
considerably less (0.01 µg/kg body weight per day). The total
estimated mean intake for the general population is approximately 2
µg/kg body weight per day. Available data also indicate that water
use in homes contributes considerably to levels of chloroform in
indoor air and to total exposure. For some individuals living in
dwellings supplied with tap water containing relatively high
concentrations of chloroform, estimated total intakes are up to 10
µg/kg body weight per day.
Workers may be exposed to chloroform during, for example, the
production of chloroform itself, the synthesis of substances derived
from chloroform (for example, chlorodifluoromethane), and the use of
chloroform as a solvent, and also as a consequence of its formation
in paper bleaching and sewage treatment facilities. For example,
based on a national survey conducted from 1981 to 1983, NIOSH
estimated that approximately 96 000 workers in the USA are
potentially exposed to chloroform.
10.1.2 Health effects
The most important effects of chloroform are those on the liver
and kidney. These effects are associated with the metabolism of
chloroform to the reactive intermediate, phosgene. There are
substantial interspecies and sex differences in the rates at which
chloroform is metabolized. Data also indicate that reductive
metabolism differs among species.
The most universally observed toxic effect of chloroform is
damage to the liver. The severity of these effects per unit dose
administered depends on the species, the vehicle and the method by
which the chloroform is administered. The lowest dose at which liver
damage has been observed is 15 mg/kg body weight per day,
administered to beagle dogs in a toothpaste base over a period of
7.5 years. Effects at lower doses were not examined. Somewhat higher
doses are required to produce hepatotoxic effects in other species.
Although duration of exposure varied in these studies,
no-observed-adverse-effect levels (NOAELs) ranged between 15 and 125
mg/kg body weight per day.
Effects on the kidney have been observed in male mice of
sensitive strains and in F-344 rats. Severe effects have been
observed in a particularly sensitive strain of male mice at doses as
low as 36 mg/kg body weight per day.
Daily 6-h inhalation of chloroform for 7 days consecutively
induced atrophy of Bowman's glands and new bone growth in the nasal
turbinates of F-344 rats. The NOEL for these effects was 14.7
mg/m3 (3 ppm). The significance of these effects is being further
investigated in longer term studies.
The weight of the available evidence indicates that chloroform
has little, if any, capability to induce gene mutation, chromosomal
damage and DNA repair. There is some evidence of low-level binding
to DNA, however. Chloroform does not appear capable of inducing
unscheduled DNA synthesis in vivo.
Chloroform induced hepatic tumours in mice when administered by
gavage in corn oil. However, when similar doses were administered in
drinking-water to mice, hepatic tumours were not induced.
The carcinogenic effects of chloroform on the mouse liver
appear to be closely related to cytotoxic and cell replicative
effects. The effects on cell replication paralleled variations in
carcinogenic responses to chloroform due to vehicle and method of
administration. It is of interest, in this regard, that chloroform
administered in drinking-water was incapable of promoting, but
rather inhibited, the development of liver tumours in mice.
Chloroform does not appear capable of initiating liver tumours or
inducing unscheduled DNA synthesis in the mouse liver. It would
appear, therefore, that cytotoxicity followed by cell replication
with prolonged administration of chloroform is associated with the
development of liver tumours in mice.
Chloroform induced kidney tumours in rats when administered by
gavage in corn oil. However, results for this species were similar
when the chemical was administered in the drinking-water.
Experiments in F-344 rats have indicated that chloroform could
cause damage and increase cell replication in the kidney at doses
similar to those that induce renal tumours in Osborne-Mendel rats.
These effects are produced by both oral (one single gavage) and
7-day inhalation exposure. While these results are suggestive of an
association, it is difficult to associate with any certainty the
carcinogenic response with the toxic and replicative effects.
Indeed, toxicity studies are short term and involve a rat strain
that is unusually sensitive to the nephrotoxic effects of
chloroform. This strain is different from that in which tumours were
observed.
There are some limited data to suggest that chloroform is toxic
to the fetus, but only at doses that are maternally toxic.
10.1.3 Approaches to risk assessment
The following guidance is provided as a potential basis for the
derivation of exposure limits by relevant authorities. By allocation
of the tolerable and risk-specific intakes presented below based,
for example, on the proportion of total intakes originating from
each environmental medium presented in chapter 5, limits for
exposure in drinking-water, food and air could be developed by local
authorities (WHO, in press). However, local authorities may also
wish to take into account local variations in the proportions of
exposure from various media or factors such as cost, ease and
effectiveness of control in order to develop risk management
strategies appropriate for local circumstances. However, the
ultimate objective should be reduction of total exposure from all
sources to levels below the tolerable maximum intake and
risk-specific intakes presented below. Moderate to short-term
excedence of limits based on the guidance presented below does not
necessarily imply significant risk to health and relevant public
health authorities should be contacted before taking remedial
action.
Moreover, disinfection is unquestionably the most important
step in the treatment of water for public supply. The paramount
importance of microbiological quality requires some flexibility in
the derivation of limits for exposure to chloroform in
drinking-water. Where local circumstances require that a choice must
be made between meeting microbiological limits or limits for
disinfection byproducts, the microbiological quality must always
take precedence. Efficient disinfection must never be compromised.
10.1.3.1 Non-neoplastic effects
The Task Group concluded that the data available are sufficient
to develop a tolerable intake for non-neoplastic effects of
chloroform on the basis of effects in animal species.
The lowest effect level in long-term studies in animal species
is that reported by Heywood et al. (1979) where slight
hepatotoxicity (increases in hepatic serum enzymes and fatty cysts)
was observed in beagle dogs that ingested 15 mg/kg body weight per
day in toothpaste for 7.5 years. Liver fat content was also
increased in B6C3F1 mice that ingested 34 mg/kg body weight per
day in drinking-water for 2 years (Jorgenson et al., 1985). On the
basis of these data, a tolerable daily intake (TDI) can be derived
as follows:
15 mg/kg body
weight per day
TDI = ----------------- = 0.015 mg/kg body weight per day
1000 (15 µg/kg body weight per day)
where:
* 15 mg/kg body weight per day is the lowest-identified-effect
level (slight hepatotoxicity in the study on beagle dogs by
Heywood et al., 1979);
* 1000 is the uncertainty factor (x 10 for interspecies
variation, x 10 for intraspecies variation and x 10 for use of
an effect level rather than a no-effect level).
This value is likely to be conservative. It should be noted
that no effects have been observed in adequate studies on other
species exposed to higher doses administered in other vehicles.
10.1.3.2 Neoplastic effects
The Task Group concluded that the carcinogenic effects of
chloroform should also be considered in the development of limits of
exposure.
a) Liver tumours in female B6C3F1 mice
Based on the available mechanistic data, the approach
considered most appropriate for provision of guidance based on mouse
liver tumours is division of a no-effect level for cell
proliferation by an uncertainty factor. The NOEL for cytolethality
and cell proliferation in B6C3F1 mice was 10 mg/kg body weight per
day following administration in corn oil for 3 weeks (Larson et al.,
1994a).
On the basis of these data, a tolerable daily intake is derived
as follows:
10 mg/kg body
weight per day
TDI = ------------------ = 0.01 mg/kg body weight per day
1000 (10 µg/kg body weight per day)
where:
* 10 mg/kg body weight per day is the NOEL for cytolethality and
cell proliferation in B6C3F1 mice observed in the short-term
study of Larson et al. (1994a);
* 1000 is the uncertainty factor (x10 for interspecies variation,
x10 for intraspecies variation and x 10 for severity of effect
(i.e. carcinogenicity) and less-than-chronic study).
b) Kidney tumours in male Osborne-Mendel rats
Since data on cell proliferation are not available for the
strain in which tumours were observed (Osborne-Mendel rats) and
identified information on cell proliferation and lethality are short
term (one single gavage and a 7-day inhalation exposure in F-344
rats), it was considered premature to deviate from the default model
(i.e. linearized multistage) as a basis for estimation of lifetime
cancer risk.
Based on the induction of renal tumours (adenomas and
adenocarcinomas) in male rats in the study by Jorgenson et al.
(1985), the total daily intake considered to be associated with a
10-5 excess lifetime risk, calculated on the basis of the Global
82 version of the linearized multistage model, is 0.0082 mg/kg body
weight per day (8.2 µg/kg body weight per day). A body surface area
correction was not incorporated due to the fact that chloroform is
an indirect-acting carcinogen and that the rate of metabolism is
similar in rodents and man.
10.2 Evaluation of effects in the environment
Chloroform may be released into the environment during its
production, storage, transport and use. Significant amounts of
chloroform may also enter the environment as a consequence of its
formation during some chlorination processes (e.g., chlorination of
water, paper bleaching).
Chloroform is expected to volatilize readily from surface water
and the surface of soils. It is also expected to be highly mobile in
soils and may reach ground water.
Chloroform has a residence time of several months in the
atmosphere and can therefore be transported over long distances from
the point of emission. Degradation by reaction with hydroxyl
radicals is likely to be the only significant mechanism for
decomposition of chloroform in the atmosphere. A half-life of around
60 days has been estimated for this process.
Chloroform appears to be resistant to biodegradation under
aerobic conditions but is degraded under certain anaerobic
conditions.
Chloroform is toxic to the embryo-larval stages of some
amphibian and fish species. The lowest reported LC50 is 0.3
mg/litre (4- or 7-day exposure) for the embryo-larval stages of
Hyla crucifer. It is less toxic to fish and Daphnia magna. The
LC50 values for several species of fish are in the range of 18 to
191 mg/litre. There is little difference in sensitivity between
freshwater and marine fish. The lowest reported LC50 for Daphnia
magna is 29 mg/litre (48-h exposure). Chloroform is of low
toxicity to algae and other microorganisms.
Levels of chloroform in surface water are generally low and
would not be expected to present a hazard to aquatic organisms.
However, higher levels of chloroform in surface water resulting from
industrial discharges or spills may be hazardous to the
embryo-larval stages of some aquatic species.
11. FURTHER RESEARCH
A number of further studies is considered to be necessary:
* A study of compensatory cell regeneration in the liver and
kidney of the Osborne-Mendel rat
* Determination of reactive metabolite formation in situ
* Studies on the mechanism of the species-specific
carcinogenicity of chloroform including a) the identification
of the intermediate/metabolite responsible for the
carcinogenicity of chloroform and b) its mode of action
* An inhalation carcinogenicity bioassay
* Further validation of PBPK models for chloroform with
interspecies variations, including humans and dogs
* Further studies concerning the progression of nasal lesions in
the rat
* Additional long-term toxicity tests in aquatic organisms
* in vitro cytotoxicity/metabolism studies with human tissues
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
The International Agency for Research on Cancer evaluated
chloroform in 1978 (IARC, 1979) and re-evaluated it in 1987 (IARC,
1987). The conclusions were that there is inadequate evidence for
the carcinogenicity of chloroform in humans but sufficient evidence
for its carcinogenicity in experimental animals. The overall
evaluation was that chloroform is possibly carcinogenic to humans
(Group 2B).
Chlorinated drinking-water was evaluated in 1990 (IARC, 1991)
and the overall evaluation was that chlorinated drinking-water is
not classifiable as to its carcinogenicity to humans (Group 3).
Studies with chlorinated drinking-water gave no evidence for
carcinogenicity of chloroform in humans (Group 3) (IARC, 1991).
A drinking-water guideline value of 200 µg/litre for an excess
lifetime cancer risk of 10-5 has been recommended for chloroform
by the World Health Organization (WHO, 1993).
REFERENCES
Abdel-Rahman HS (1982) The presence of trihalomethanes in soft
drinks. J Appl Toxicol, 2: 165-166.
Abernethy S, Bobra AM, Shiu WY, Wells PG, & Mackay D (1986) Acute
lethal toxicity of hydrocarbons and chlorinated hydrocarbons to two
planktonic crustaceans: the key role of organism-water partitioning.
Aquat Toxicol, 8: 163-174.
Adriani J (1970) The pharmacology of anaesthetic drugs. Springfield,
Illinois, Charles C. Thomas, pp 57-60.
Aggazzotti G, Predieri G, Fantuzzi G, & Benedetti A (1987) Headspace
gas chromatographic analysis for determining low levels of
chloroform in human plasma. J Chromatogr, 416: 125-130.
Aggazzotti G, Fantuzzi G, Tartoni PL, & Predieri G (1990) Plasma
chloroform concentrations in swimmers using indoor swimming pools.
Arch Environ Health, 45(3): 175-179.
Agustin JS & Lim-Sylianco CY (1978) Mutagenic and clastogenic
effects of chloroform. Bull Phil Biochem Sci, 1: 17-23.
Ahmadizadeh M, Echt R, Kuo C-H, & Hook JB (1984) Sex and strain
differences in mouse kidney: Bowman's capsule morphology and
susceptibility to chloroform. Toxicol Lett, 20: 161-172.
Ahmed AF, Kubic VL, & Anders MW (1977) Metabolism of haloforms to
carbon monoxide: I. in vitro studies. Drug Metab Dispos, 5:
198-204.
Allard U & Andersson L (1992) Exposure of dental personnel to
chloroform in root filling procedures. Endod Dent Traumatol, 8:
155-159.
Althaus FR, Lawrence SD, Sattler GL, Longfellow DG, & Pitot HC
(1982) Chemical quantification of unscheduled DNA synthesis in
cultured hepatocytes as an assay for the rapid screening of
potential chemical carcinogens. Cancer Res, 42: 3010-3015.
Anderson DR & Lusty EW (1980) Acute toxicity and bioaccumulation of
chloroform to 4 species of freshwater fish. Richland, WA, Battelle
Pacific North West Laboratory (NUREG/CR-0893).
Appleby A, Kazazis J, Lillian D, & Singh HB (1976) Atmospheric
formation of chloroform from trichloroethylene. J Environ Sci
Health, A11: 711-715.
Armstrong DW & Golden T (1986) Determination of distribution and
concentration of trihalomethanes in aquatic recreational and
therapeutic facilities by electron capture GC. LC-GC, 4: 652-655.
Athanasiou K & Kyrtopoulos SA (1981) Induction of sister chromatid
exchange by non-mutagenic carcinogens. NATO Adv Study Inst Ser A,
40: 557-562.
ATSDR (1993) Toxicological profile for chloroform (update). Atlanta,
Georgia, Agency for Toxic Substances and Disease Registry.
Azri-Meehan S, Mata MP, Gandafi AJ, & Brendel K (1992) The
hepatotoxicity of chloroform in precision-cut rat liver slices.
Toxicology, 73: 239-250.
Bai C-L, Canfield PJ, & Stacey NH (1992) Individual serum bile acids
as early indicators of carbon tetrachloride- and chloroform-induced
liver injury. Toxicology, 75: 221-234.
Baker SU & Bonin AM (1981) Study of 42 coded compounds with the
Salmonella/mammalian microsome assay. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 249-260 (Progress in Mutation Research,
Volume 1).
Balster RL & Borzelleca JF (1982) Behavioural toxicity of
trihalomethane contaminants of drinking water in mice. Environ
Health Perspect, 46: 127-136.
Bätjer K, Cetinkaya M, Düszeln v J, Gabel B, Lahl U, Stachel B, &
Thiemann W (1980) Chloroform emission into urban atmosphere.
Chemosphere, 9: 311-316.
Bauer U (1981) [Human exposure to environmental chemicals -
Investigations on volatile organic halogenated compounds in water,
air, food and human tissues. III. Communication: Results of
investigations.] Zbl Bakt Hyg, 1. Abt Orig, B174: 200-237 [in
German].
Baumann OE, Drangsholt H, & Carlberg GE (1981) Analysis of volatile
halogenated organic compounds in fish. Sci Total Environ, 20:
205-215.
Benoit FM & Jackson R (1987) Trihalomethane formation in whirlpool
spas. Water Res, 2: 353-357.
Bergman K (1984) Application and results of whole-body
autoradiography in distribution studies of organic solvents. Crit
Rev Toxicol, 12: 59-119.
Birge WJ (1980) Effects of organic compounds on amphibian
reproduction. Lexington, University of Kentucky, Water Resources
Research Institute (Research Report No. 121; PB80-147523).
Birge WJ, Black JA & Bruser DM (1979) Toxicity of organic chemicals
to embryo-larval stages of fish. Washington, DC, Environmental
Protection Agency (EPA-560/11-79-007; PB80-101637).
Blancato JN & Chiu (1994) Use of pharmacokinetic models to estimate
internal doses from exposure. In: Wang R ed. Water contamination and
health. New York, Marcel Dekker, Inc.
Bogen KT, Colston BW, & Machicao LK (1992) Dermal absorption of
dilute aqueous chloroform, trichloroethylene, and
tetrachloroethylene in hairless guinea pigs. Fundam Appl Toxicol,
18: 30-39.
Bomski HA, Sobdewska A, & Strakowski A (1967) [Toxic damage of the
liver by chloroform in chemical industry workers.] Int Arch
Gewerbepathol Gewerbehyg, 24: 127-134 [in German]
Borzelleca JF & Carchman RA (1982) Effect of selected organic
drinking water contaminants on male reproduction. Research Triangle
Park, North Carolina, US Environmental Protection Agency (EPA
600-/1-82-009; PB82-259847).
Bouwer EJ & McCarty PL (1983) Transformation of 1- and 2-carbon
halogenated aliphatic organic compounds under methanogenic
conditions. Appl Environ Microbiol, 45: 1286-1294.
Bouwer EJ, Rittmann BE, & McCarty PL (1981) Anaerobic degradation of
halogenated 1-and 2-carbon organic compounds. Environ Sci Technol,
15: 596-599.
Bowman FJ, Borzelleca JF, & Munson AE (1978) The toxicity of some
halomethanes in mice. Toxicol Appl Pharmacol, 44: 213-215.
Brady JF, Li D, Ishizaki H, Lee M, Ning SM, Xiao F, & Yang CS (1989)
Induction of cytochromes P450IIE1 and P450IIB1 by secondary ketones
and the role of P450IIE1 in chloroform metabolism. Toxicol Appl
Pharmacol, 100: 342-349.
Branchflower RV, Schulick RD, George JW, & Pohl LR (1983) Comparison
of the effects of methyl n-butyl ketone and phenobarbital on rat
liver cytochrome P-450 and the metabolism of chloroform to phosgene.
Toxicol Appl Pharmacol, 71: 414-421.
Brass JH, Feige MA, & Halloran T (1977) The national organic
monitoring survey: sampling and analyses of purgeable organic
compounds. In: Drinking water quality enhancement source protection,
pp 393-416.
Bringmann G (1973) [Determination of the harmful biological effect
of water pollutants from the inhibition of glucose assimilation in
the bacterium Pseudomonas fluorescens.] Gesund-Ing, 94: 366-369
(in German).
Bringmann G (1975) [Determination of the harmful biological effect
of water pollutants from the inhibition of cell reproduction in the
blue alga Microcystis.] Gesund-Ing, 96: 238-241 (in German).
Bringmann G (1978) [Determination of the harmful biological effect
of water pollutants on protozoa. I. Bacteriophagic flagellates.] Z
Wasser-Abwasser Forsch, 11: 210-215 (in German).
Bringmann G & Kühn R (1977) [Threshold values for the harmful effect
of water pollutants on bacteria (Pseudomonas putida) and green
algae (Scenedesmus quadricauda) in the cell reproduction
inhibition test.] Z Wasser-Abwasser Forsch, 10: 87-97 (in German).
Bringmann G & Kühn R (1980) [Determination of the harmful biological
effect of water pollutants on protozoa. II. Bacteriophagic
ciliates.] Z Wasser-Abwasser Forsch, 13: 26-31 (in German).
Bringmann G, Kühn R, & Winter A (1980) [Determination of the harmful
biological effect of water pollutants on protozoa. III. Saprozoic
flagellates.] Z Wasser-Abwasser Forsch, 13: 170-173 (in German).
Brondeau MT, Bonnet P, Guenier JP, & De Ceaurriz J (1983) Short-term
inhalation test for evaluating industrial hepatotoxicants in rats.
Toxicol Lett, 19: 139-146.
Brooks TM & Dean BJ (1981) Mutagenic activity of 42 coded compounds
in the Salmonella/microsome assay with preincubation. In: De Serres
FJ & Ashby J ed. Evaluation of short-term tests for carcinogens:
Report of the international collaborative study. Amsterdam, Oxford,
New York, Elsevier North/Holland, pp 261-270 (Progress in Mutation
Research, Volume 1).
Brown BR Jr (1972) Hepatic microsomal lipoperoxidation and
inhalation anaesthetics: A biochemical and morphologic study in the
rat. Anesthesiology, 36: 458-465.
Brown BR, Sipes IG, & Sagalyn AM (1974a) Mechanisms of acute hepatic
toxicity: chloroform, halothane and glutathione. Anesthesiology 41:
554-561.
Brown DM, Langley PF, Smith D, & Taylor DC (1974b) Metabolism of
chloroform. I. The role of [14C]-chloroform by different species.
Xenobiotica, 4: 151-163.
Budavari S ed. (1989) The Merck index: an encyclopedia of chemicals,
drugs and biologicals, 11th ed. Rahway, New Jersey, Merck and Co.
Bull RJ & Kopfler FC (1991) Health Effects of disinfectants and
disinfection by-products. Denver, Colorado, AWWA Research Foundation
and American Water Works Association, pp 14, 99-104.
Bull RJ, Brown JM, Meierhenry EA, Jorgenson TA, Robinson M, & Stober
JA (1986) Enhancement of the hepatotoxicity of CHCl3 in B6C3F1
mice by corn oil: Implications for CHCl3 carcinogenesis. Environ
Health Perspect, 69: 49-58.
Burkhalter JE & Balster RL (1979) Behavioral teratology evaluation
of trichloromethane in mice. Neurobehav Toxicol, 1: 199-205.
Butler TC (1961) Reduction of carbon tetrachloride in vivo and
reduction of carbon tetrachloride and chloroform in vitro by
tissues and tissue constituents. J Pharmacol Exp Ther, 134: 311-319.
Butterworth BE, Smith-Oliver T, Earle L, Loury DJ, White RD,
Doolittle DJ, Working PK, Cattley RC, Jirtle R, Michalopoulos G, &
Strom S (1989) Use of primary cultures of human hepatocytes in
toxicology studies. Cancer Res, 49: 1075-1084.
Callen DF, Wolf CR, & Philpot RM (1980) Cytochrome P-450 mediated
genetic activity and cytotoxicity of seven halogenated aliphatic
hydrocarbons in Saccharomyces cerevisiae. Mutat Res, 77: 55-63.
Capel ID, Dorrell HM, Jenner M, Pinnock MH, & Williams DC (1979) The
effect of chloroform ingestion on the growth of murine tumours. Eur
J Cancer, 15: 1485-1490.
Challen PJR, Hickish DE, & Bedford J (1958) Chronic chloroform
intoxication. Br J Ind Med, 15: 243-249.
Charbonneau M, Greselin E, Brodeur J, & Plaa GL (1991) Influence of
acetone on the severity of the liver injury induced by haloalkane
mixtures. Can J Physiol Pharmacol, 69(12): 1901-1907.
Cherkin A & Catchpool JF (1964) Temperature dependence of anesthesia
in goldfish. Science, 144: 1460-1462.
Chinery RL & Gleason AK (1993) A compartmental model for prediction
of breath concentration and absorbed dose of chloroform after
exposure while showering. Risk Anal, 13(1): 51-62.
Chiou WL (1975) Quantitation of hepatic and pulmonary first-pass
effect and its implications in pharmacokinetic study. I.
Pharmacokinetics of chloroform in man. J Pharmacokinet Biopharm, 3:
193-201.
Chu I, Secours V, Marino I, & Villeneuve DC (1980) The acute
toxicity of four trihalomethanes in male and female rats. Toxicol
Appl Pharmacol, 52: 351-353.
Chu I, Villeneuve DC, Secours VE, & Becking GC (1982a) Toxicity of
trihalomethanes. I. The acute and subacute toxicity of chloroform,
bromodichloromethane, chlorodibromomethane and bromoform in rats. J
Environ Sci Health, B17: 205-224.
Chu I, Villeneuve DC, Secours VE, & Becking GC (1982b) Toxicity of
trihalomethanes. II. Reversibility of toxicological changes produced
by chloroform, bromodichloromethane, chlorodibromomethane and
bromoform in rats. J Environ Sci Health, B17: 225-240.
Clemens TL, Hill RN, Bullock LP, Johnson WD, Sultatos LG, & Vessel
ES (1979) Chloroform toxicity in the mouse: Role of genetic factors
and steroids. Toxicol Appl Pharmacol, 48: 117-130.
Cohen EN & Hood N (1969) Application of low-temperature
autoradiography to studies of the uptake and metabolism of volatile
anaesthetic in the mouse. Anesthesiology, 30: 306-314.
Colacci A, Bartoli S, Bonora B, Guidotti L, Lattanzi G, Mazzullo M,
Niero A, Perocco P, Silingardi P, & Grilli S (1991) Chloroform
bioactivation leading to nucleic acids binding. Tumori, 77(4):
285-290.
Condie LW, Smallwood CL, & Laurie RD (1983) Comparative renal and
hepatotoxicity of halomethanes: bromodichloromethane, bromoform,
chloroform, dibromochloromethane and methylene chloride. Drug Chem
Toxicol, 6: 563-578.
Conlon MF (1963) Addiction to chlorodyne. Br Med J, 2: 1177-1178.
Corley RA, Mendrala AL, Smith FA, Staats DA, Gargas ML, Conolly RB,
Andersen ME, & Reitz RH (1990) Development of a physiologically
based pharmacokinetic model for chloroform. Toxicol Appl Pharmacol,
103: 512-527.
Cortés F, Mateos S, & Escalza P (1985) C-Mitosis, chromosomal
aberrations and sister chromatid exchanges induced by chloroform in
root-tip cells of Allium cepa. Cytobios, 44: 231-237.
Cowgill UM, Milazzo DP, & Landenberger BD (1989) Toxicity of nine
benchmark chemicals to Skeletonema costatum, a marine diatom.
Environ Toxicol Chem, 8: 451-455.
Cowlen MS, Hewitt WR, & Schroeder F (1984a) 2-Hexanone potentiation
of (14C)chloroform hepatotoxicity: covalent interaction of a
reactive intermediate with rat liver phospholipids. Toxicol Appl
Pharmacol, 73: 478-491.
Cowlen MS, Hewitt WR, & Schroeder F (1984b) Mechanism in 2-hexanone
potentiation of chloroform hepatotoxicity. Toxicol Lett, 22:
293-299.
Cox RA, Derwent RG, Eggleton AEJ, & Lovelock JE (1976) Photochemical
oxidation of halocarbons in the troposphere. Atmos Environ, 10:
305-308.
Cresteil TL, Beaune Ph, Leroux JP, Lange M, & Mansuy D (1979)
Biotransformation of chloroform by rat and human liver microsomes;
in vitro effect on some enzyme activities and mechanisms of
irreversible binding to macromolecules. Chem-Biol Interact, 24:
153-165.
Cronn DR & Harsch DE (1979) Determination of atmospheric halocarbon
concentration by gas chromatographic-mass spectrometry. Anal Lett,
12: 1489-1496.
Culliford D & Hewitt HB (1957) The influence of sex hormone status
on the susceptibility of mice to chloroform-induced necrosis of
renal tubules. J Endocrinol, 14: 381-393.
Daft JA (1988) Fumigant contamination during large-scale food
sampling for analysis. Arch Environ Contam Toxicol, 17: 177-181.
Daft JA (1989) Determination of fumigants and related chemicals in
fatty and non-fatty foods. J Agric Food Chem, 37: 560-564.
Daniel MR & Dehnel JM (1981) Cell transformation test with baby
hamster kidney cells. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford New York, Elsevier
North/Holland, pp 626-637 (Progress in Mutation Research, Volume 1).
Daniel FB, De Angelo AB, Stober JA, Pereira MA, & Olson GR (1989)
Chloroform inhibition of 1,2-dimethylhydrazine-induced
gastrointestinal tract tumours in the Fischer 344 rat. Fundam Appl
Toxicol, 13: 40-45.
Daniel FB, Reddy TV, Stober JA, & Olson GR (1991) Site specific
modulation of carcinogen-induced gastrointestinal tract nuclear
anomalies in B6C3F1 mice by chloroform. Anticancer Res, 11:
665-670.
Danielsson BRG, Ghantous H, & Dencker L (1986) Distribution of
chloroform and methyl chloroform and their metabolites in pregnant
mice. Biol Res Pregnancy, 7: 77-83.
Davis ME (1992) Dichloroacetic acid and trichloroacetic acid
increase chloroform toxicity. J Toxicol Environ Health, 37: 139-148.
Davis ME & Berndt WO (1992) Sex differences in monochloroacetate
pretreatment effects on chloroform toxicity in rats. Fundam Appl
Toxicol, 18: 66-71.
Davis DD, Machado G, Conaway B, Oh Y, & Watson R (1976) A
temperature dependent kinetics study of the reaction of OH with
CH3Cl, CH2Cl2, CHCl3 and CH3Br. J Chem Phys, 65:
1268-1274.
De Biasi A, Sbraccia M, Keizer J, Testai E, & Vittozzi L (1992) The
regioselective binding of CHCl3 reactive intermediates to
microsomal phospholipids. Chem-Biol Interact, 85: 229-242.
Deml E & Oesterle D (1985) Dose-dependent promoting activity of
chloroform in rat liver foci bioassay. Cancer Letters, 29: 59-63.
Deml E & Oesterle D (1987) Dose-response of promotion by
polychlorinated biphenyls and chloroform in rat liver foci bioassay.
Arch Toxicol, 60: 209-211.
Den Hartog JC (1980, 1981) [Analysis of gaseous organic components
in the outdoor air.] Rijswijk, The Netherlands, TNO, Prins Maurits
Laboratory (Reports PML-TNO Nos 1980-182, 1981-149, 1981-155,
1981-158( (in Dutch).
Deringer MK, Dunn TB, & Heston WE (1953) Results of strain C3H mice
to chloroform. Proc Soc Exp Biol Med, 83: 474-479.
De Serres FJ & Ashby J ed. (1981) Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland (Progress in
Mutation Research, Volume 1).
Dilling WL (1977) Interphase transfer process. II. Evaporation rates
of chloromethanes, ethanes, ethylenes, propanes, and propylenes from
dilute aqueous solutions. Comparisons with theoretical predictions.
Environ Sci Technol, 11: 405-409.
Dimitriades B, Gay BW Jr, Arnts RR, & Seila RL (1983) Photochemical
reactivity of perchloroethylene: a new appraisal. J Air Pollut
Control Assoc, 33: 575.
Divincenzo GD & Krasavage WJ (1974) Serum ornithine carbamyl
transferase as a liver response test for exposure to organic
solvents. Am Ind Hyg Assoc J, 35: 21-29.
Docks EL & Krishna G (1976) The role of glutathione in chloroform
induced hepatotoxicity. Exp Mol Pathol, 24: 13-22.
Dowty BJ, Laseter JL, & Storer J (1976) The transplacental migration
and accumulation in blood of volatile organic constituents. Pediatr
Res, 10: 696-701.
Duprat P, Delsaut L, & Gradiski D (1976) Pouvoir irritant des
principaux chlorés aliphatiques sur la peau et les muqueuses
oculaires du lapin. Eur J Toxicol Environ Hyg, 9: 171-177.
Ebel RE, Barlow RL, & McGrath EA (1987) Chloroform hepatotoxicity in
the Mongolian gerbil. Fundam Appl Toxicol, 8: 207-216.
ECDIN (1992) On-line search in the environmental chemicals data and
information network. ECDIN/copyright JRC-CEC/ISPRA.
Egli C, Scholtz R, Cook AM, & Leisinger T (1987) Anaerobic
dechlorination of tetrachloromethane and 1,2-dichloroethane to
degradable products by pure cultures of Desulfobacterium sp. and
Methanobacterium sp. FEMS Microbiol Lett, 43: 257-261.
Entz RC, Thomas KW, & Diachenko GW (1982) Residues of volatile
halocarbons in foods using headspace gas chromatography. J Agric
Food Chem, 30: 846-849.
Environment Canada (1986) Ambient air concentration of volatile
organic compounds in Toronto and Montreal. Ottawa, Environment
Canada, Pollution Measurement Division (Unpublished report).
Environment Canada (1992) Volatile organic compounds measurements in
Canadian urban and rural areas: 1989-1991. Ottawa, Environment
Canada, Technology Development Branch (Internal report 92-75.16).
Erickson MD (1981) Acquisition and chemical analysis of mother's
milk for selected toxic substances. Gov Rep Announc Index US, 5096
(PB81-231029).
Europ-Cost (1976) A comprehensive list of polluting substances which
have been identified in various fresh waters, effluent discharges,
aquatic animals and plants, and bottom sediments, 2nd ed.
Luxembourg, Commission of the European Communities, p 31
(EUCO/MDU/73/76, XII/476/76).
Ewing BB, Chian ESK, & Cook JC (1977) Monitoring to detect
previously unrecognized pollutants in surface waters. Appendix:
Organic analysis data. Washington, DC, US Environmental Protection
Agency (EPA/560/6-77-015).
Federal Office of the Environment (1981) [Clean air '81:
development, situation, trends.] Berlin, Federal Office of the
Environment (BUA) (in German).
Foster GD & Tullis RE (1985) Quantitative structure-toxicity
relationships with osmotically stressed Artemia salina nauplii.
Environ Pollut, A38: 273-281.
Fry J, Taylor R, & Hathway DE (1972) Pulmonary elimination of
chloroform and its metabolite in man. Arch Int Pharmacodyn Ther,
196: 98-111.
Fujie K, Aoki T, & Wada M (1990) Acute and subacute cytogenetic
effects of the trihalomethanes on rat bone marrow cells in vivo.
Mutat Res, 242: 111-119.
Fujie K, Aoki T, & Mae S (1993) Sister-chromatid exchanges induced
by trihalomethanes in rat erythroblastic cells. Mutat Res, 300:
241-246.
Garner RC, Welch A, & Pickering C (1981) Mutagenic activity of 42
coded compounds in the Salmonella/microsome assay. In: De Serres FJ
& Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 280-284 (Progress in Mutation
Research, Volume 1).
Gatehouse D (1981) Mutagenic activity of 42 coded compounds in the
"microtiter" fluctuation test. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 376-386 (Progress in Mutation Research,
Volume 1).
Gearhart JM, Seckel C, & Vinegar A (1993) in vivo metabolism of
chloroform in B6C3F1, mice determined by the method of gas uptake:
the effects of body temperature on tissue partition coefficients and
metabolism. Toxicol Appl Pharmacol, 119: 258-266.
Gersich FM, Blanchard FA, Applegath SL, & Park CN (1986) The
precision of Daphnid ( Daphnia magna Straus, 1820) static acute
toxicity tests. Arch Environ Contam Toxicol, 15: 741-749.
Glende EA & Recknagel RO (1992) Phosphodipase A2 activation and cell
injury in isolated rat hepatocytes exposed to bromotrichloromethane,
chloroform, and 1,1-dichloroethylene as compared to effects of
carbon tetrachloride. Toxicol Appl Pharmacol, 113: 159-162.
Gocke E, King M-T, Eckhardt K, & Wild D (1981) Mutagenicity of
cosmetics ingredients licensed by the European Communities. Mutat
Res, 90: 91-109.
Goodman LS & Gilman AG (1970) The pharmacological basis of
therapeutics. New York, McMillan Co.
Gradiski D, Magadur JL, Baillot M, Danière MC, & Schuh MB (1974)
Toxicité comparée des principaux solvants chlorés aliphatiques. J
Eur Toxicol, 7: 247-254.
Gradiski D, Bonnet P, Raoult G, & Magadur JL (1978) Toxicité aiguë
comparée par inhalation des principaux solvants aliphatiques
chlorés. Arch Mal Prof Méd Trav Sécur Soc, 39: 249-257.
Green MHL (1981) A differential killing test using an improved
repair-deficient strain of Escherichia coli. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 183-194 (Progress in Mutation
Research, Volume 1).
Greim H, Bimboe SD, Egert G, Göggelmann W, & Krämer M (1977)
Mutagenicity and chromosomal aberrations as an analytical tool for
in vitro detection of mammalian enzyme-mediated formation of
reactive metabolites. Arch Toxicol, 39: 159-169.
Grob K (1984) Further development of direct aqueous injection with
electron-capture detection in gas chromatography. J Chromatogr, 299:
1-11.
Groger WKL & Grey TF (1979) Effect of chloroform on the activities
of liver enzymes in rats. Toxicology, 14: 23-28.
Gualandi G (1984) Genotoxicity of the free-radical producers CCl4
and lipoperoxidation in Aspergillus nidulans. Mutat Res, 136:
109-114.
Guengerich FP, Kim DH, & Iwasaki M (1991) Role of human cytochrome
P450 II E1 in the oxidation of suspected carcinogens. Chem Res
Toxicol, 4: 160-179.
Hakim A, Jain AK, & Jain R (1992) Chloroform ingestion causing toxic
hepatitis. J Assoc Phys India, 40: 477.
Hamada N & Peterson RE (1977) Effect of chlorinated aliphatic
hydrocarbons on excretion of protein and electrolytes by rat
pancreas. Toxicol Appl Pharmacol, 39: 185-194.
Hardie DWF (1964) Chlorocarbons and chlorohydrocarbons. Chloroform.
In: Kirk RE & Othmer DF ed. Encyclopedia of chemical technology, 2nd
ed. Vol 5, pp 119-127.
Harms MS, Peterson RE, Fujimoto JM, & Erwin CP (1976) Increased
"bile duct-pancreatic fluid" flow in chlorinated hydrocarbon-treated
rats. Toxicol Appl Pharmacol, 35: 41-49.
Harris RN, Ratnayake JH, Garry VF, & Anders MW (1982) Interactive
hepatotoxicity of chloroform and carbon tetrachloride. Toxicol Appl
Pharmacol, 63: 281-289.
Harsch DE & Cronn DR (1978) Low-pressure sample-transfer technique
for analysis of stratospheric air samples. J Chromatogr Sci, 16:
363-367.
Hatch GC, Mamay DD, Ayer ML, Casto BC, & Nesnow S (1983) Chemical
enhancement of viral transformation in Syrian hamster embryo cells
by gaseous and volatile chlorinated methanes and ethanes. Cancer
Res, 43: 1945-1950.
Heil E, Oeser H, Hatz R, & Kelker H (1979) [Gas-chromatographic
determination of traces of C1- and C2- fluoro- and
chlorohydrocarbons in air.] Fresenius Z Anal Chem, 297: 357-364 (in
German).
Heilbrunn G, Liebert E, & Szanto PB (1945) Chronic chloroform
poisoning. Arch Neurol Psychiatr, 53: 68-72.
Herren-Freund SL & Pereira MA (1986) Carcinogenicity of by-products
of disinfection in mouse and in rat liver. Environ Health Perspect,
69: 59-66.
Hertlein F (1980) Monitoring airborne contaminants in chemical
laboratories. ACS Symp Ser, 120: 215-230.
Herzfeld D, Van der Gun KD, & Louw R (1989) Quantitative
determination of volatile organochlorine compounds in water by
GC-headspace analysis with dibromomethane as an internal standard.
Chemosphere, 18: 1425-1430.
Hewitt HB (1956) Renal necrosis in mice after accidental exposure to
chloroform. Br J Exp Pathol, 37: 32-39.
Hewitt WR, Miyajima H, Côté MG, & Plaa GL (1979) Acute alteration of
chloroform-induced hepato- and nephrotoxicity by mirex and kepone.
Toxicol Appl Pharmacol, 48: 509-527.
Hewitt WR, Brown EM, Côté MG, Plaa GL, & Miyajima H (1982)
Alteration of chloroform-induced nephrotoxicity by exogenous
ketones. In: Porter G ed. Nephrotoxic mechanisms of drugs and
environmental toxins. New York, London, Plenum, Press, pp 357-365.
Hewitt LA, Palmason C, Masson S, & Plaa GL (1990) Evidence for the
involvement of organelles in the mechanism of ketone-potentiated
chloroform-induced hepatotoxicity. Liver, 10: 35-48.
Heywood R, Sortwell RJ, Noel PRB, Street AE, Prentice DE, Roe FJC,
Wardsworth PF, Worden AN, & Van Abbé NJ (1979) Safety evaluation of
toothpaste containing chloroform. III. Long-term study in beagle
dogs. J Environ Pathol Toxicol, 2: 835-851.
Hill RN (1978) Differential toxicity of chloroform in the mouse. Ann
NY Acad Sci, 298: 170-175.
Hill RN, Clemens TL, Liu DK, Vesell ES, & Johnson WD (1975) Genetic
control of chloroform toxicity in mice. Science, 190: 159-160.
Howard Carleton J & Evenson KM (1976) Rate constants for the
reactions of OH with CH4 and fluorine, chlorine and bromine
substituted methanes at 296 K. J Chem Phys, 64: 197.
Hubbard SA, Green MHL, Bridges BA, Wain AJ, & Bridges JW (1981)
Fluctuation test with S9 and hepatocyte activation. In: De Serres
FJ & Ashby J ed. Evaluation of short-term tests for carcinogens:
Report of the international collaborative study. Amsterdam, Oxford,
New York, Elsevier North/Holland, pp 361-370 (Progress in Mutation
Research, Volume 1).
IARC (1979) Chloroform. In: Some halogenated hydrocarbons. Lyon,
International Agency for Research on Cancer, pp 401-427 (IARC
Monographs on the Evaluation of the Carcinogenic Risk of Chemicals
to Humans, Volume 20).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC monographs volumes 1 to 42. Lyon, International Agency for
Research on Cancer, pp 152-154 (IARC Monographs on the Evaluation of
the Carcinogenic Risk of Chemicals to Humans, Supplement 7).
IARC (1991) Chlorinated drinking-water. In: Chlorinated
drinking-water; chlorination by-products; some other halogenated
compounds; cobalt and cobalt compounds. Lyon, International Agency
for Research on Cancer, pp 45-141 (IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans, Volume 52).
Ichinotsubo D, Mower H, & Mandel M (1981a) Testing of a series of
paired compounds (carcinogen and non-carcinogenic structural analog)
by DNA repair-deficient E. coli strains. In: De Serres FJ & Ashby
J ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 195-198 (Progress in Mutation Research,
Volume 1).
Ichinotsubo D, Mower H, & Mandel M (1981b) Mutagen testing of a
series of paired compounds with the Ames Salmonella testing system.
In: De Serres FJ & Ashby J eds. Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 298-301
(Progress in Mutation Research, Volume 1).
Iijima M, Côté MG, & Plaa GL (1983) A semiquantitative morphologic
assessment of chlordecone-potentiated chloroform hepatotoxicity.
Toxicol Lett, 17: 307-314.
Ikatsu H & Nakajima T (1992) Hepatotoxic interaction between carbon
tetrachloride and chloroform in ethanol treated rats. Arch Toxicol,
66: 580-586.
Ilett KF, Reid WD, Sipes IG, & Krishna G (1973) Chloroform toxicity
in mice: correlation of renal and hepatic necrosis with covalent
binding of metabolites to tissue macromolecules. Exp Mol Pathol, 19:
215-229.
Jagannath DR, Vultaggio DM, & Brusick DJ (1981) Genetic activity of
42 coded compounds in the mitotic gene conversion assay using
Saccharomyces cerevisiae strain D4. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 456-467 (Progress in Mutation Research,
Volume 1).
Jo WK, Weisel CP, & Lioy PJ (1990a) Routes of chloroform exposure
and body burden from showering with chlorinated tap water. Risk
Anal, 10: 575-580.
Jo WK, Weisel CP, & Lioy PJ (1990b) Chloroform exposure and the
health risk associated with multiple uses of chlorinated tap water.
Risk Anal, 10: 581-585.
Jones JRE (1947) The oxygen consumption of Gasterosteus aculeatus
in toxic solutions. Exp Biol, 23: 298-311.
Jones WM, Margolis G, & Stephen CR (1958) Hepatotoxicity of
inhalation anaesthetic drugs. Anesthesiology, 19: 715-723.
Jorgenson TA, Meierhenry EF, Rushbrook CJ, Bull RJ, & Robinson M
(1985) Carcinogenicity of chloroform in drinking water to male
Osborne-Mendel rats and female B6C3F1 mice. Fundam Appl Toxicol,
5: 760-769.
Jorgenson TA & Rushbrook CJ (1980) Effects of chloroform in the
drinking water of rats and mice. Ninety-day subacute toxicity study.
Washington, DC, US Environmental Protection Agency
(EPA-600/1-80-030; NTIS PB80-219108).
Jorgenson TA, Rushbrook CJ, & Jones DCL (1982) Dose-response study
of chloroform carcinogenesis in the mouse and rat: status report.
Environ Health Perspect, 46: 141-149.
Juhnke I & Lüdemann D (1978) [Results of the testing of 200 chemical
compounds for acute toxicity for fish by means of the golden orfe
test.] Z Wasser-Abwasser Forsch, 11: 161-164 (in German).
Kada T (1981) The DNA-damaging activity of 42 coded compounds in the
Rec-assay. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp
175-182 (Progress in Mutation Research, Volume 1).
Kajino M (1977) [Formation of chloroform during chlorination of
drinking water.] Chem Abstr, 88, 27631K (in Japanese).
Kassinova GV, Kovaltsova SV, Marfin SV, & Zakharov IA (1981)
Activity of 40 coded compounds in differential inhibition and
mitotic crossing-over assays in yeast. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 434-455 (Progress in Mutation Research,
Volume 1).
Kawamura K & Kaplan IR (1983) Notes: Organic compounds in the
rainwater of Los Angeles. Environ Sci Technol, 17: 497-501.
Kebbekus BB & Bozzelli JW (1982) Determination of selected toxic
organic vapours in air by adsorbent trapping and capillary GC. J
Environ Sci Health, A17: 713-723.
Kedderis GL, Held SD, Pearson AC, & Carfagns MA (1993a) Isolated
mouse hepatocytes as an in vitro model for the in vivo
metabolism and cytolethality of chloroform (CF). Toxicologist, 13:
198.
Kedderis GL, Carfagna MA, Held SD, Batra R, Murphy JE, & Gargas ML
(1993b) Kinetic analysis of furan biotransformation by F-344 rats
in vivo and in vitro. Toxicol Appl Pharmacol, 123: 274-282.
Kerfoot HB (1987) Shallow-probe soil-gas sampling for indication of
ground-water contamination by chloroform. Int J Environ Anal Chem,
30: 167-181.
Kimura ET, Ebert DM, & Dodge PW (1971) Acute toxicity and limits of
solvent residue for sixteen organic solvents. Toxicol Appl
Pharmacol, 19: 699-704.
Kirkland DJ, Smith KL, & Van Abbé NJ (1981) Failure of chloroform to
induce chromosome damage or sister-chromatid exchange in cultured
human lymphocytes and failure to induce reversion in Escherichia
coli. Food Cosmet Toxicol, 19: 651-656.
Klaassen CD & Plaa GL (1967) Relative effects of various chlorinated
hydrocarbons on liver and kidney in dogs. Toxicol Appl Pharmacol,
10: 119-131.
Klaassen CD & Plaa GL (1969) Comparison of the biochemical
alterations elicited in livers from rats treated with carbon
tetrachloride, chloroform, 1,1,2-trichloroethane and
1,1,1-trichloroethane. Biochem Pharmacol, 18: 2019-2027.
Klaunig JE, Ruch RJ, & Pereira MA (1986) Carcinogenicity of
chlorinated methane and ethane compounds administered in drinking
water to mice. Environ Health Perspect, 69: 89-95.
Kluwe WM (1981) The nephrotoxicity of low molecular weight
halogenated alkane solvents, pesticides and chemical intermediates.
In: Hook JB ed. Toxicology of the kidney. New York, Raven Press Ltd,
pp 179-226.
Knie J, Hälke A, Juhnke I, & Schiller W (1983) [Results of studies
on chemical substances with four biotests.] Dtsch Gewässerkd Mitt,
27: 77-79 (in German).
Könemann H (1981) Quantitative structure-activity relationships in
fish toxicity studies. Part 1: Relationships for 50 industrial
pollutants. Toxicology, 19: 209-211.
Koop DR, Morgan ET, Tarr GE, & Coon MJ (1982) Purification and
characterization of a unique isozyme of cytochrome P450 from liver
microsomes of ethanol-treated rabbits. J Biol Chem, 257: 8472-8480.
Kramer MD, Lynch CF, Isacson P, & Hanson JW (1992) The association
of waterborne chloroform with intrauterine growth retardation.
Epidemiology, 3: 407-413.
Krasner SW, McGuire MJ, Jacangelo JG, Patania NL, Reagan KM, & Aieta
EM (1989) The occurrence of disinfection by-products in U.S.
drinking water. J Am Water Works Assoc, 81: 41-53.
Kroneld R (1985) Recovery and reproducibility in determination of
volatile halocarbons in water and blood. Bull Environ Contam
Toxicol, 34: 486-496.
Krost KJ, Pellizzari ED, Walburn SG, & Hubbard SA (1982) Collection
and analysis of hazardous organic emissions. Anal Chem, 54: 810-817.
Kutob SD & Plaa GL (1962a) A procedure of estimating the hepatotoxic
potential of certain industrial solvents. Toxicol Appl Pharmacol, 4:
354-361.
Kutob SD & Plaa GL (1962b) The effect of acute ethanol intoxication
on chloroform-induced liver damage. J Pharmacol Exp Teratol, 135:
245-251.
Kylin B, Reichard H, Sümegi I, & Yllner S (1963) Hepatotoxicity of
inhaled trichloroethylene, tetrachloroethylene and chloroform.
Single exposure. Acta Pharmacol Toxicol, 20: 16-26.
Lahl U, Bätjer K, Düszeln v J, Gabel B, Stachel B, & Thiemann W
(1981a) Distribution and balance of volatile halogenated
hydrocarbons in the water and air of covered swimming pools using
chlorine for water disinfection. Water Res, 15: 803-814.
Land PC, Owen EL, & Linde HW (1981) Morphologic changes in mouse
spermatozoa after exposure to inhalational anaesthetics during early
spermatogenesis. Anesthesiology, 54: 53-56.
Larson JL, Wolf DC, & Butterworth BE (1993) The acute hepatotoxicity
and nephrotoxic effects of chloroform in male F-344 rats and female
B6C3F1 mice. Fundam Appl Toxicol 20: 302-315.
Larson JL, Wolf DC, & Butterworth BE (1994a) Induced cytotoxicity
and cell proliferation in the hepatocarcinogenicity of chloroform in
female B6C3F1 mice. Comparison of administration by gavage in corn
oil vs ad libitum in drinking water. Fundam Appl Toxicol, 22:
90-102.
Larson JL, Wolf DC, Morgan KT, Méry S, & Butterworth BE, (1994b) The
toxicity of one week exposures to inhaled chloroform in female
B6C3F1 mice and male F-344 rats. Fundam Appl Toxicol, 22: 431-446.
Larson JL, Sprankle CS, & Butterworth BE (1994c) Lack of
chloroform-induced DNA repair in vitro and in vivo in
hepatocytes of female B6C3F1 mice. Environ Mol Mutagen, 23:
132-136.
Lasa J & Rosiek J (1979) [Detection of halogen compounds in air with
a colometric ECD.] Chem Anal, 24: 819-826 (in Polish).
Lavigne JG & Marchand C (1974) The role of metabolism in chloroform
hepatocyto-toxicity. Toxicol Appl Pharmacol, 29: 312-326.
Lavigne JG, Belanger PM, Dore F, & Labrecque G (1983) Temporal
variations in chloroform-induced hepatotoxicity in rats. Toxicology,
26: 267-273.
LeBlanc GA (1980) Acute toxicity of priority pollutants to water
flea (Daphnia magna). Bull Environ Contam Toxicol, 24: 684-691.
Lehmann KB & Flury FF (1943) Chlorinated hydrocarbons. In: Lehmann
KB & Flury FF ed. Toxicology and hygiene of industrial solvents.
Baltimore, Maryland, Williams & Wilkins Co, pp 138-145, 191-196.
Lehmann KB & Hasegawa (1910) [Study on the absorption of chlorinated
hydrocarbons from the air by animal and man.] Arch Hyg, 72: 327-342
(in German).
Leveson R & Barker N (1981) Airscan: an ultrasensitive trace air
impurity analyzer for use in toxic aviation environments (AGARD Conf
Proc AGARD-CP-309, B15/1-B15/12).
Lillian D & Singh HB (1974) Absolute determination of atmospheric
halocarbons by gas phase coulometry. Anal Chem, 46: 1060-1063.
Linde HW & Mesnick PS (1979) Causes of death of anaesthesiologists
from the chloroform era. Washington, DC, US Environmental Protection
Agency (EPA 600/1-179-043; NTIS PB80-125172).
Lioy PJ & Lioy MJ ed. (1983) Air sampling instruments for evaluation
of atmospheric contaminants, 6th ed. Cincinnati, Ohio, American
Conference of Governmental Industrial Hygienists, pp V/85-V/86.
Löfberg B & Tjälve H (1986) Tracing tissues with
chloroform-metabolizing capacity in rats. Toxicology, 39: 13-35.
Loprieno N (1981) Screening of coded carcinogenic/non carcinogenic
chemicals by a forward-mutation system with the yeast
Schizosaccharomyces pombe. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 424-425 (Progress in Mutation Research,
Volume 1).
Lundberg I, Ekdahl M, Kronevi T, Lidums V, & Lundberg S (1986)
Relative hepatotoxicity of some industrial solvents after
intraperitoneal injection or inhalation exposure in rats. Environ
Res, 40: 411-420.
Lurker PA, Clark CS, Elia VJ, Gartside PS, & Kinman RN (1983) Worker
exposure to chlorinated organic compounds from the activated sludge
wastewater treatment process. Am Ind Hyg Assoc J, 44: 109-112.
Lyman WJ, Reehl WF, & Rosenblatt DH (1982) Handbook of chemical
property estimation methods. New York, McGraw-Hill Book Company.
McConnell G, Ferguson DM, & Pearson CR (1975) Chlorinated
hydrocarbons and the environment. Endeavor, 34: 13-18.
MacDonald DJ (1981) Salmonella/microsome tests on 42 coded
chemicals. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp
285-297 (Progress in Mutation Research, Volume 1).
McLean AEM (1970) The effect of protein deficiency and microsomal
enzyme induction by DDT and phenobarbitone on the acute toxicity of
chloroform and a pyrrolizidine alkaloid, retrorsine. Br J Exp
Pathol, 511: 317-321.
McMartin DN, O'Connor JA, & Kaminsky LS (1981) Effects of
differential changes in rat hepatic and renal cytochrome P-450
concentrations on hepatotoxicity and nephrotoxicity of chloroform.
Res Commun Chem Pathol Pharmacol, 31: 99-110.
Mansuy D, Beaune P, Cresteil T, Lange M, & Leroux JP (1977) Evidence
for phosgene formation during liver microsomal oxidation of
chloroform. Biochem Biophys Res Commun, 79: 513-517.
Martin CN & McDermid AC (1981) Testing of 42 coded compounds for
their ability to induce unscheduled DNA repair synthesis in HeLa
cells. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 533-537
(Progress in Mutation Research, Volume 1).
Masuda Y, Yano I, & Murano T (1980) Comparative studies on the
hepatotoxic actions of chloroform and related halogenomethanes in
normal and phenobarbital-pretreated animals. J Pharmacobio-Dyn, 3:
53-64
Matsushima T, Takamoto Y, Shirai A, Sawamura M, & Sugimura T (1981)
Reverse mutation test on 42 coded compounds with the E. coli WP2
system. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp
387-395 (Progress in Mutation Research, Volume 1).
Mattice JS, Tsai SC, & Burch MB (1981) Toxicity of trihalomethanes
to common carp embryos. Trans Am Fish Soc, 110: 261-269.
Maxwell MI, Burmaster DE, & Ozonoff D (1991) Trihalomethanes.
Maximum contaminant levels: the significance of inhalation, dermal
exposures to chloroform in household water. Regul Toxicol Pharmacol,
14: 297-312.
Mayes MA, Alexander HC, & Dill DC (1983) A study to assess the
influence of age on the response of Fathead minnows in static acute
toxicity tests. Bull Environ Contam Toxicol, 31: 139-147.
Mehran MF, Slifker RA, & Cooper WJ (1984) A simplified liquid-liquid
extraction method for analysis of trihalomethanes in drinking water.
J Chromatogr Sci, 22: 241-243.
Mehta RD & Von Borstel RV (1981) Mutagenic activity of 42 encoded
compounds in the haploid yeast reversion assay, strain XV185-14C.
In: De Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 414-423
(Progress in Mutation Research, Volume 1).
Meier JR, Ringhand HP, Coleman WE, Munch JW, Streicher RP, Kaylor
WH, & Schenck KM (1985) Identification of mutagenic compounds formed
during chlorination of humic acid. Mutat Res, 157: 111-122.
Méry S, Larson JL, Butterworth BE, Wolf DC, Harden R, & Morgan KT
(1994) Nasal toxicity of chloroform in male F-344 rats and female
B6C3F1 mice following a one week inhalation exposure. Toxicol Appl
Pharmacol, 125: 214-227.
Mink FL, Coleman WE, Munch JW, Kaylor WH, & Ringhand HP (1983) in
vivo formation of halogenated reaction products following peroral
sodium hypochlorite. Bull Environ Contam Toxicol, 30: 394-399.
Mink FL, Brown TJ, & Rickabaugh J (1986) Absorption, distribution,
and excretion of 14C-trihalomethanes in mice and rats. Bull
Environ Contam Toxicol, 37: 752-758.
Mirsalis JC, Tyson CK, & Butterworth BE (1982) Detection of
genotoxic carcinogens in the in vivo-in vitro hepatocyte DNA
repair assay. Environ Mutagen, 4: 553-562.
Moore L (1980) Inhibition of liver-microsome calcium pump by in
vivo administration of CCl4, CHCl3 and 1,1-dichloroethylene.
Biochem Pharmacol, 29: 2505-2511.
Moore DH, Chasseaud LF, Majeed SK, Prentice DE, Roe FJC, & Van Abbé
NJ (1982) The effect of dose and vehicle on early tissue damage and
regenerative activity after chloroform administration to mice. Food
Chem Toxicol, 20: 951-954.
Morele Y, Lefevre C, Ferrari P, Guenier JP, & Muller J (1989)
Sampling and analysis of airborne chlorinated methanes - comparison
of FID and ECD. Chromatographia, 28: 617-619.
Morgan A, Black A, & Belcher DR (1970) The excretion in breath of
some aliphatic halogenated hydrocarbons following administration by
inhalation. Ann Occup Hyg, 13: 219-233.
Morgan DL, Cooper SW, Carlock DL, Sykora JJ, Sutton B, Mattie DR, &
McDougal JN (1991) Dermal absorption of neat and aqueous volatile
organic chemicals in the Fischer-344 rat. Environ Res, 55: 51-63.
Morimoto K & Koizumi A (1983) Trihalomethanes induce sister
chromatid exchanges in human lymphocytes in vitro and mouse bone
marrow cells in vivo. Environ Res, 32: 72-79.
Morris RD, Audet AM, Angelillo IF, Chalmers TC, & Mosteller F (1992)
Chlorination, chlorination by-products and cancer. A meta-analysis.
Am J Public Health, 82: 955.
Munson AE, Sain LE, Sanders VM, Kauffmann BM, White KL, Page DG,
Barnes DW, & Borzelleca JF (1982) Toxicology of organic drinking
water contaminants: trichloromethane, bromodichloromethane,
dibromomethane and tribromomethane. Environ Health Perspect, 46:
117-126.
Murray FJ, Schwetz BA, McBride JF, & Staples RE (1979) Toxicity of
inhaled chloroform in pregnant mice and their offspring. Toxicol
Appl Pharmacol, 50: 515-522.
Nagao M & Takahashi Y (1981) Mutagenic activity of 42 coded
compounds in the Salmonella/microsome assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 302-313 (Progress in Mutation
Research, Volume 1).
Nakajima T & Sato A (1979) Enhanced activity of liver
drug-metabolising enzymes for aromatic and chlorinated hydrocarbons
following food deprivation. Toxicol Appl Pharmacol, 50: 549-556.
Nakajima T, Koyama Y, & Sato A (1982) Dietary modification of
metabolism and toxicity of chemical substances with specific
reference to carbohydrate. Biochem Pharmacol, 31: 1005-1011.
Nakajima T, Elovaara E, Park SS, Gelboin HV, & Vainio H (1991)
Immunochemical detection of cytochrome P450 isozymes induced in rat
liver by n-hexane, 2-hexanone and acetonyl acetone. Arch Toxicol,
65: 542-547.
National Cancer Institute (1976a) Carcinogenesis bioassay of
chloroform. Bethesda, Maryland, National Cancer Institute (NTIS
PB-264018/AS).
National Cancer Institute (1976b) Report on carcinogenesis bioassay
of chloroform. Bethesda, Maryland, National Cancer Institute (NTIS
PB-264-018).
Nicloux MM (1906) Passage du chloroforme de la mère au foetus. C R
Soc Biol, 60: 373-375.
NNI (Dutch Standardisation Institute) (1984) [Air quality - Outdoor
air. Determination of chlorinated hydrocarbons - Coal
adsorption/liquid desorption/GC-method.] Dutch Standardisation
Institute (Standard NVN 2794) (in Dutch).
Oettel H (1936) [Effects of organic fluids on the skin.] Arch Exp
Pathol Pharmacol, 183: 641-696 (in German).
O'Hara TM, Sheppard MA, Clarke EC, Borzelleca JF, Gennings C, &
Condie LW (1991) A CCl4/CHCl3 interaction study in isolated
hepatocytes: non-induced and phenobarbital-pretreated cells. J Appl
Toxicol, 11: 147-154.
Ohio River Valley Water Sanitation Commission (1980) Assessment of
water quality conditions. Ohio River mainstream 1978-79. Cincinnati,
Ohio, Ohio River Valley Water Sanitation Commission.
Ohio River Valley Water Sanitation Commission (1982) Assessment of
water quality conditions Ohio River mainstream 1980-81. Cincinnati,
Ohio, Ohio River Valley Water Sanitation Commission.
Oliver BG (1983) Dihaloacetonitriles in drinking water. Algae and
fulvic acid as precursors. Environ Sci Technol, 17: 80-83.
Otson R (1990) A Health and Welfare Canada program to develop
personal exposure monitors for airborne organics at µg/m3.
Proceedings of the 1990 EPA/A & WWMA International Symposium on
Measurement of Toxic and Related Air Pollutants (VIP-17). Pittsburg,
Pennsylvania, Air & Waste Water Management Association, pp 483-488.
Otson R, Fellin P, & Whitmore R (1992) A national pilot study of
airborne VOCs in residences - design and progress. Presented at the
1992 EPA/A & WWMA Symposium on Measurement of Toxic and Related Air
Pollutants, Durham, NC, 4-8 May. Pittsburg, Pennsylvania, Air &
Waste Water Management Association.
Parry JM & Sharp DC (1981) Induction of mitotic aneuploidy in the
yeast strain D6 by 42 coded compounds. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 468-480 (Progress in Mutation Research,
Volume 1).
Paul BB & Rubinstein D (1963) Metabolism of carbon tetrachloride and
chloroform by the rat. J Pharmacol Exp Ther, 141: 141-148.
Pearson CR & McConnell G (1975) Chlorinated C1 and C2
hydrocarbons in the marine environment. Proc R Soc Lond, B189:
305-322.
Pellizzari ED, Erickson MD, & Zweidinger (1979) Formulation of
preliminary assessment of halogenated organic compounds in man and
environmental media. Research Triangle Park, North Carolina, US
Environmental Protection Agency (EPA 560/13-79-006).
Pellizzari ED, Hartwell TD, Harris BSH, Waddel RD, Whitaker DA, &
Erickson MD (1982) Purgeable organic compounds in mothers' milk.
Bull Environ Contam Toxicol, 28: 322-328.
Pellizzari ED, Sheldon LS, & Bursey JT (1985a) Method 25: GC/MS
determination of volatile halocarbons in blood and tissue. Environ
Carcinog Sel Methods Anal, 7: 435-444.
Pellizzari ED, Zweidinger RA, & Sheldon LS (1985b) Method 24: GC/MS
determination of volatile hydrocarbons in breath samples. Environ
Carcinog Sel Methods Anal, 7: 413-431.
Pereira MA, Lin L-HC, Lippitt HM, & Herren SL (1982) Trihalomethanes
as initiators and promoters of carcinogenesis. Environ Health
Perspect, 46: 151-156.
Pereira MA, Knutsen GL, & Herren-Freund SL (1985) Effect of
subsequent treatment of chloroform or phenobarbital on the incidence
of liver and lung tumours, initiated by ethylnitrosourea in 15 days
old mice. Carcinogenesis, 6: 203-207.
Pericin C & Thomann P (1979) Comparison of the acute toxicity of
clioquinol, histamine and chloroform in different strains of mice.
Arch Toxicol, 2(Suppl): 371-373.
Perocco P & Prodi G (1981) DNA damage by haloalkanes in human
lymphocytes cultured in vitro. Cancer Lett, 13: 213-218.
Perocco P, Bolognesi S, & Alberghini W (1983) Toxic activity of
seventeen industrial solvents and halogenated compounds on human
lymphocytes cultured in vitro. Toxicol Lett, 16: 69-75.
Perry PE & Thomson EJ (1981) Evaluation of the sister chromatid
exchange method in mammalian cells as a screening system for
carcinogens. In: De Serres FJ & Ashby J ed. Evaluation of short-term
tests for carcinogens: Report of the international collaborative
study. Amsterdam, Oxford, New York, Elsevier North/Holland, pp.
560-569 (Progress in Mutation Research, Volume 1).
Petzold GL & Swenberg JA (1978) Detection of DNA damage induced in
vivo following exposure of rats to carcinogens. Cancer Res, 38:
1589-1594.
Phillips LJ & Birchard GF (1991) Regional variations in human toxic
exposure in the USA: An analysis based on the national human adipose
tissue survey. Arch Environ Contam Toxicol, 21: 159-168.
Phoon WH, Liang OK, & Kee CP (1975) An epidemiological study of an
outbreak of jaundice in a factory. Ann Acad Med Singap, 4: 396-399.
Phoon WH, Goh KT, Lee LT, Tan KT, & Kwok SF (1983) Toxic jaundice
from occupational exposure to chloroform. Med J Malays, 38: 31-34.
Plaa GL & Larson RE (1965) Relative nephrotoxicity properties of
chlorinated methane, ethane and ethylene derivatives in mice.
Toxicol Appl Pharmacol, 7: 34-44.
Plaa GL, Evans EA, & Hine CH (1958) Relative hepatotoxicity of seven
halogenated hydrocarbons. J Pharmacol Exp Ther, 123: 224-229.
Plummer JL, Hall P de la M, Ilsley AH, Jenner MA, & Cousins MJ
(1990) Influence of enzyme induction and exposure profile on liver
injury due to chlorinated hydrocarbon inhalation. Pharmacol Toxicol,
67: 329-335.
Pohl LR (1979) Biochemical toxicology of chloroform. Rev Biochem
Toxicol, 1: 79-107.
Pohl LR & Krishna G (1978) Deuterium isotope effect in bioactivation
and hepatotoxicity of chloroform. Life Sci, 23: 1067-1072.
Pohl LR, Brooshan B, Whittaker NF, & Krishna G (1977) Phosgene: A
metabolite of chloroform. Biochem Biophys Res Commun, 79: 684-691.
Pohl LR, George JW, Martin JL, & Krishna G (1979) Deuterium isotope
effect in in vivo bioactivation of chloroform to phosgene. Biochem
Pharmacol, 28: 561-563.
Pohl LR, Martin JL, & George JW (1980) Mechanism of metabolic
activation of chloroform by rat liver microsomes. Biochem Pharmacol,
29: 3271-3276.
Pohl LR, Branchflower RV, Highet RJ, Martin JL, Nunn DS, Monks TJ,
George JW, & Hinson JA (1981) The formation of diglutathionyl
dithiocarbonate as a metabolite of chloroform, bromotrichloromethane
and carbon tetrachloride. Drug Metab Dispos, 9: 334-339.
Rao KN, Virji MA, Moraca MA, Diven WF, Martin TG, & Schneider SM
(1993) Role of serum markers for liver function and regeneration in
the management of chloroform poisoning. J Anal Toxicol, 17: 99-102.
Ray SD & Mehendale HM (1990) Potentiation of CCl4 and CHCl3
hepatotoxicity and lethality by various alcohols. Fundam Appl
Toxicol, 15: 429-440.
Reddy TV, Daniel FB, Lin EL, Stober JA, & Olson GR (1992) Chloroform
inhibits the development of diethylnitrosamine-initiated,
phenobarbital-promoted gamma-glutamyltranspeptidase and placental
form glutathione S-transferase positive foci in rat liver.
Carcinogenesis, 13: 1325-1330.
Reitz RH, Fox TR, & Quast JF (1982) Mechanistic considerations for
carcinogenic risk estimation: chloroform. Environ Health Perspect,
46: 163-168.
Rem RM, Anderson ME, Daletsky SA, Misenheimer DC, & Rollius HF
(1982) Chloroform materials balance (EPA Draft Report 68-02-3168).
Reuber MD (1979) Carcinogenicity of chloroform. Environ Health
Perspect, 31: 171-182.
Reynolds ES & Yee AG (1967) Liver parenchymal cell injury. V.
Relationships between patterns of chloromethane-C14 incorporation
into constituents of liver in vivo and cellular injury. Lab
Invest, 16: 591-603.
Reynolds LF & Harrison DW (1982) Studies of discharges of: benzene,
chloroform and carbon tetrachloride into the aquatic environment and
the best technical means for reduction of water pollution from such
discharges. Brixham, Devon, ICI Brixham Laboratory (Report No.
BL/A/2198).
Reynolds ES, Treinen RJ, Farrish HH, & Moslen MT (1984) Metabolism
of [14C] carbon tetrachloride to exhaled, excreted and bound
metabolites. Biochem Pharmacol 21: 3363-3374.
Richold M & Jones M (1981) Mutagenic activity of 42 coded compounds
in the Salmonella/microsome assay. In: De Serres FJ & Ashby J ed.
Evaluation on short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 314-322 (Progress in Mutation Research,
Volume 1).
Robinson AB, Manly KF, Anthony MP, Catchpool JF, & Pauling L (1965)
Anesthesia of Artemia larvae. Method for quantitative study.
Science, 149: 1255-1258.
Roe FJC, Palmer AK, Worden AN, & Van Abbé NJ (1979) Safety
evaluation of toothpaste containing chloroform. 1. Long-term studies
in mice. J Environ Pathol Toxicol, 2: 799-819.
Rosenberg C, Nylund L, Aalto T, Kontsas H, Norppa H, Jäppinen P, &
Vainio H (1991) Volatile organohalogen compounds from the bleaching
of pulp - Occurrence and genotoxic potential in the work
environment. Chemosphere, 23: 1617-1628.
Rosenkranz HS, Hyman J, & Leifer Z (1981) DNA polymerase deficient
assay. In: De Serres FJ & Ashby J ed. Evaluation of short-term tests
for carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 210-218
(Progress in Mutation Research, Volume 1).
Rowland I & Severn B (1981) Mutagenicity of carcinogens and
non-carcinogens in the Salmonella/microsome test. In: De Serres FJ &
Ashby J eds. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 323-332 (Progress in Mutation
Research, Volume 1).
Rubinstein D & Kanics L (1964) The conversion of carbon
tetrachloride and chloroform to carbon dioxide by rat liver
homogenates. Can J Biochem, 42: 1577-1585.
Ruddick JV, Villeneuve DC, & Chu I (1983) A teratological assessment
of four trihalomethanes in the rat. J Environ Sci Health, B18:
333-349.
Rudolph J & Jebsen C (1983) The use of photoionization,
flameionization and electron capture detectors in series for the
determination of low molecular weight trace components in the non
urban atmosphere. Int J Environ Anal Chem, 13: 129-139.
Russell JW & Shadoff LA (1977) The sampling and determination of
halocarbons in ambient air using concentration on porous polymer. J
Chromatogr, 132: 375-384.
Ryan DE, Koop DR, Thomas PE, Coon MJ, & Levin W (1986) Evidence that
isoniazid and ethanol induce the same microsomal cytochrome P-450 in
rat liver, an isozyme homologous to rabbit cytochrome P-450 isozyme
3a. Arch Biochem Biophys, 246: 633-644.
Salamone MF, Heddle JA, & Katz M (1981) Mutagenic activity of 41
compounds in the in vivo micronucleus assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 686-697 (Progress in Mutation
Research, Volume 1).
Salisbury S (1982) Health hazard evaluation. Charleston Laboratory
(Report No. HETA-81-359-1058).
Sato A, Nakajima T, & Koyama Y (1980) Effects of chronic ethanol
consumption on hepatic metabolism of aromatic and chlorinated
hydrocarbons in rats. Br J Ind Med, 37: 382-386.
Sato A, Najajiwa T, & Koyama Y (1981) Dose-related effect of a
single dose of ethanol on the metabolism in rat liver of some
aromatic and chlorinated hydrocarbons. Toxicol Appl Pharmacol, 60:
8-15.
Savoure N, Maudet M, & Nicol M (1992) Toxicité du chloroforme et
statut vitaminique A chez le rat. J Toxicol Clin Exp, 12: 97-108.
Scholler KL (1966) [Electron microscope studies on rat liver cells
after anaesthesia with various inhalational anaesthetics.]
Anaesthesist, 15: 145-148 (in German).
Scholler KL (1967) [The effect of halothane and chloroform on the
fine structure and protein synthesis of rat's liver.] Experientia
(Basel), 23: 652-655 (in German).
Schröder HG (1965) Acute and delayed chloroform poisoning. A case
report. Br J Anaesth, 37: 971-975.
Schwetz BA, Leong BKJ, & Gehring PJ (1974) Embryo- and fetotoxicity
of inhaled chloroform in rats. Toxicol Appl Pharmacol, 28: 442-451.
Sharp DC & Parry JM (1981a) Induction of mitotic gene conversion by
41 coded compounds using the yeast culture JD1. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 491-501 (Progress in Mutation
Research, Volume 1).
Sharp DC & Parry JM (1981b) Use of repair-deficient strains of yeast
to assay the activity of 40 coded compounds. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 502-516 (Progress in Mutation
Research, Volume 1).
Simmon VF & Shepherd GH (1981) Mutagenic activity of 42 coded
compounds in the Salmonella/microsome assay. In: De Serres FJ &
Ashby J ed. Evaluation of short-term tests for carcinogens: Report
of the international collaborative study. Amsterdam, Oxford, New
York, Elsevier North/Holland, pp 333-342 (Progress in Mutation
Research, Volume 1).
Simmon VF, Kauhanen K, & Tardiff RG (1977) Mutagenic activity of
chemicals identified in drinking water. In: Scott D ed. Progress in
genetic toxicology. Amsterdam, Oxford, New York, Elsevier/North
Holland Biomedical Press, pp 249-258.
Simpson JY (1847) On a new anaesthetic agent, more efficient than
sulfuric ether. Lancet, 2: 549-550.
Sipes IG, Stripp B, Krishna G, Maling HM, & Gillette JR (1973)
Enhanced hepatic microsomal activity by pretreatment of rats with
acetone or isopropanol. Proc Soc Exp Biol Med, 142: 237-240.
Sipes IG, Krishna G, & Gillette JR (1977) Bioactivation of carbon
tetrachloride, chloroform and bromotrichloromethane: role of
cytochrome P-450. Life Sci, 20: 1541-1548.
Skopek TS, Andon BM, Kaden DA, & Thilly WG (1981) Mutagenic activity
of 42 coded compounds using 8-azaguanine resistance as a genetic
marker in Salmonella typhimurium. In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 371-375 (Progress in Mutation Research,
Volume 1).
Skrzypinska GM, Piotrowski JK, & Koralewska J (1991) The hepatotoxic
action of chloroform: short-time dynamics of biochemical alterations
and dose-effect relationships. Pol J Occup Med, 4(1): 77-84.
Slooff W (1979) Detection limits of a biological monitoring system
based on fish respiration. Bull Environ Contam Toxicol, 23: 517-523.
Smith JH & Hook JB (1983) Mechanism of chloroform nephrotoxicity.
II. in vitro evidence for renal metabolism of chloroform in mice.
Toxicol Appl Pharmacol, 70: 480-485.
Smith JH & Hook JB (1984) Mechanism of chloroform nephrotoxicity.
III. Renal and hepatic microsomal metabolism of chloroform in mice.
Toxicol Appl Pharmacol, 73: 511-524.
Smith AA, Volpitto PP, Gramling ZW, DeVore MB, & Glassman AB (1973)
Chloroform, haloethane and regional anesthesia: A comparative study.
Anesth Analg, 52: 1-11.
Smith JH, Maita K, Sleight SD, & Hook JB (1983) Mechanism of
nephrotoxicity. I. Time course of chloroform toxicity in male and
female mice. Toxicol Appl Pharmacol, 70: 467-479.
Smith JH, Maita K, Sleight SD, & Hook JB (1984) Effect of sex
hormones status on chloroform nephrotoxicity and renal mixed
function oxidase in mice. Toxicology, 30: 305-316.
Smith JH, Hewitt WR, & Hook JB (1985) Role of intrarenal
biotransformation in chloroform-induced nephrotoxicity in rats.
Toxicol Appl Pharmacol, 79: 166-174.
Spence JW, Philip LH, Hanst PL, & Gray BW (1976) Atmospheric
oxidation of methyl chloride, methylene chloride and chloroform. J
Air Pollut Control Assoc, 26: 994.
Stacey NH (1987) Assessment of the toxicity of chemical mixtures
with isolated rat hepatocytes: cadmium and chloroform. Fundam Appl
Toxicol, 9: 616-622.
Stander GJ (1980) Micro-organic compounds in the water environment
and their impact on the quality of potable water supplies. Water S
Afr, 6: 1-14.
Stevens JL & Anders MW (1981a) Effect of cysteine, diethylmaleate
and phenobarbital treatments on the hepatotoxicity of [1H]- and
[2H]-chloroform. Chem-Biol Interact, 37: 207-217.
Stevens JL & Anders MW (1981b) Metabolism of haloforms to carbon
monoxide. IV. Studies on the reaction mechanism in vivo. Chem-Biol
Interact, 37: 365-374.
Stewart ME, Blogoslawski WJ, Hsu RY, & Helz GR (1979) By-products of
oxidative biocides: toxicity to oyster larvae. Mar Pollut Bull, 10:
166-169.
Storms WW (1973) Chloroform parties. J Am Med Assoc, 255: 160.
Strand SE & Shippert L (1986) Oxidation of chloroform in an aerobic
soil exposed to natural gas. Appl Environ Microbiol, 52: 203-205.
Sturrock J (1977) Lack of mutagenic effect of halothane or
chloroform on cultured cells using the azaguanine test system. Br J
Anaesth, 49: 207-210.
Styles JA (1979) Cell transformation assays. In: Paget GE ed. Topics
in toxicology - Mutagens in sub-mammalian systems: Status and
significance. Baltimore, Maryland, University Park Press, pp
147-161.
Styles JA (1981) Activity of 42 coded compounds in the BHK-21 cell
transformation test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 638-646 (Progress in Mutation Research, Volume 1).
Su C & Goldberg E (1976) Environmental concentration and fluxes of
some halocarbons. Mar Pollut Transf, 353-373.
Taketomo AP & Grimsrud E (1977) An analysis of halocarbons in the
air of several working and living environments. Proc Mont Acad Sci,
37: 128-134.
Taylor DC, Brown DM, Keeble R, & Langley PF (1974) Metabolism of
chloroform. II. A sex difference in the metabolism of (14C)
chloroform in mice. Xenobiotica, 4: 165-174.
Taylor GJ, Drew RT, Lores EM, & Clemmer TA (1976) Cardiac depression
by haloalkane propellants, solvents and inhalation anaesthetics in
rabbits. Toxicol Appl Pharmacol, 38: 379-387.
Temmerman IFMM & Quaghebeur DJM (1990) Analysis of trihalomethanes
by direct aqueous injection (THM-DAI). Short communications. J High
Res Chromatogr, 13: 379-381.
Testai E & Vittozzi L (1986) Biochemical alterations elicited in rat
liver microsomes by oxidation and reduction products of chloroform
metabolism. Chem-Biol Interact, 59: 157-171.
Testai E, Di Marzio S, & Vittozzi L (1990) Multiple activation of
chloroform in hepatic microsomes from uninduced B6C3F1 mice.
Toxicol Appl Pharmacol, 104: 496-503.
Testai, E, Keizer J, Pacifici GM, & Vittozzi L (1991) Chloroform
bioactivation by microsomes from colonic and ileal mucosa of rat and
man. Toxicol Lett, 57: 19-27.
Thompson DJ, Warner SD, & Robinson VB (1974) Teratology studies on
orally administered chloroform in the rat and rabbit. Toxicol Appl
Pharmacol, 29: 348-357.
Thomson JA (1981) Mutagenic activity of 42 coded compounds in the
Lambda induction assay. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 224-235 (Progress in Mutation Research, Volume 1).
Timms RM & Moser KM (1975) Toxicity secondary to intravenously
administered chloroform in humans. Arch Intern Med, 135: 1601-1603.
Tomasi A, Albano E, Biasi F, Slater TF, Vannini V, & Dianzani MU
(1985) Activation of chloroform and related trihalomethanes to free
radical intermediates in isolated hepatocytes and in the rat in
vivo as detected by the ESR-spin trapping technique. Chem-Biol
Interact, 55: 303-316.
Topham JC (1980) Do induced sperm-head abnormalities in mice
specifically identify mammalian mutagens rather than carcinogens?
Mutat Res, 74: 379-387.
Topham JC (1981) Evaluation of some chemicals by the sperm
morphology assay. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 718-720 (Progress in Mutation Research, Volume 1).
Torkelson TR, Oyen F, & Rowe VK (1976) The toxicity of chloroform as
determined by single and repeated exposure of laboratory animals. Am
Ind Hyg Assoc J, 37: 697-705.
Toxicology and Environmental Health Institute of Munich Technical
University (1992) [Recommendations on chloroform/emission data. BUA
plenary session in Merseburg, 24-25 August 1992.] Berlin, Federal
Office of the Environment (BUA) (in German).
Trueman RW (1981) Activity of 42 coded compounds in the Salmonella
reverse mutation test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 343-350 (Progress in Mutation Research, Volume 1).
Tsuchimoto T & Matter BE (1981) Activity of coded compounds in the
micronucleus test. In: De Serres FJ & Ashby J ed. Evaluation of
short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 705-711 (Progress in Mutation Research, Volume 1).
Tsuruta H (1975) Percutaneous absorption of organic solvents. Ind
Health, 13: 227-236.
Tumasonis CF, McMartin DN, & Bush B (1985) Lifetime toxicity of
chloroform and bromodichloromethane when administered over a
lifetime in rats. Ecotoxicol Environ Saf, 9: 233-240.
Tweats DJ (1981) Activity of 42 coded compounds in a differential
killing test using Escherichia coli strains WP2, WP67 (uvra po1A),
and CM871 (uvrA AlexA recA). In: De Serres FJ & Ashby J ed.
Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 199-209 (Progress in Mutation Research,
Volume 1).
Tyson CA, Hawk-Prather K, Story DL, & Gould DH (1983) Correlations
of in vitro and in vivo hepatotoxicity for five haloalkanes.
Toxicol Appl Pharmacol, 70: 289-302.
Uchrin CG & Mangels G (1986) Chloroform sorption to New Jersey
coastal plain ground water aquifer solids. Environ Toxicol Chem, 5:
339-343.
Uehleke H & Werner T (1975) A comparative study on the irreversible
binding of labelled halothane, trichlorofluoromethane, chloroform
and carbon tetrachloride to hepatic protein and lipids in vitro
and in vivo. Arch Toxicol, 34: 289-308.
Uehleke H, Greim H, Krämer M, & Werner (1976) Covalent binding of
haloalkanes to liver constituents but absence of mutagenicity on
bacteria in metabolising test system. Mutat Res, 38: 114.
Uehleke H, Werner T, Greim H, & Krämer M (1977) Metabolic activation
of haloethanes and tests in vitro for mutagenicity. Xenobiotica,
7: 393-400.
Ullrich D (1982) [Organic halogen compounds in the air of some
Berlin indoor swimming pools.] WoBoLu-Berlin, 1: 50-52 (in German).
US EPA (1971) Compendium of registered pesticides. Washington, DC,
US Environmental Protection Agency, p III-C-19.
US EPA (1984) Locating and estimating air emissions from sources of
chloroform. US Washington, DC, Environmental Protection Agency
(Report PB-84-200617).
US EPA (1992) Technical Support Division (TSD). Disinfection
by-products field study data base. Washington, DC, US Environmental
Protection Agency, Office of Ground Water and Drinking Water,
Technical Support Division.
US FDA (US Food and Drug Administration (1976) Chloroform as an
ingredient of human drug and cosmetic products. Fed Reg, 41:
26842-26845.
US NIOSH (1984) Manual of analytical methods - Volume 1: Method
1003. Cincinnati, Ohio, National Institute for Occupational Safety
and Health.
US NIOSH (1989) National occupational exposure survey (NOES) as of
March 29, 1989. Cincinnati, Ohio, National Institute for
Occupational Safety and Health.
Van Abbé NJ, Green TJ, Jones E, Richold M, & Roe FJC (1982)
Bacterial mutagenicity studies on chloroform in vitro. Food Cosmet
Toxicol, 20: 557-561.
Van Beelen P & Van Keulen F (1990) The kinetics of the degradation
of chloroform and benzene in anaerobic sediment from the river
Rhine. Hydrobiol Bull, 24: 13-21.
Van der Heijden CA, Speijers GJA, Ros JPM, Huldy HJ, Besemer AC,
Lanting RW, Maas RJM, Heijna-Merkus E, Bergshoeff G, Gerlofsma A,
Mennes WC, Van der Most PFJ, De Vrijer FL, Janssen PCJM, Knaap AGAC,
Huijgen C, Duiser JA, & De Jong P (1986) Criteria document on
chloroform. Bilthoven The Netherlands, National Institute of Public
Health and Environmental Protection (Report No. 738513004).
Van Dyke RA, Chenoweth MB, & Van Poznak A (1964) Metabolism of
volatile anaesthetics-I. Conversion in vivo of several
anaesthetics to 14CO2 and chloride. Biochem Pharmacol, 13:
1239-1247.
Van Luin AB & Van Starkenburg W (1984) Hazardous substances in waste
water. Water Sci Tech, 17: 843-853.
Van Tassel S, Amalfitano N, & Narang RS (1981) Determination of
arenes and volatile halo-organic compounds in air at microgram per
cubic meter levels by gas chromatography. Anal Chem, 53: 2130-2135.
Veith GD, Macek KJ, Petrocelli SR, & Carroll J (1978) An evaluation
of using partition coefficients and water solubility to estimate
bioconcentration factors for organic chemicals in fish. In: Eaton
JG, Parrish PR, & Hendricks AC ed. Aquatic toxicology. Proceedings
of the Third Annual Symposium on Aquatic Toxicology. Philadelphia,
Pennsylvania, American Society for Testing and Materials, pp 116-129
(STP 707).
Venitt S & Crofton-Sleigh C (1981) Mutagenicity of 42 coded
compounds in a bacterial assay using Escherichia coli and
Salmonella typhimurium. In: De Serres FJ & Ashby J ed. Evaluation
of short-term tests for carcinogens: Report of the international
collaborative study. Amsterdam, Oxford, New York, Elsevier
North/Holland, pp 351-360 (Progress in Mutation Research, Volume 1).
Verschueren U (1983) Handbook of environmental data on organic
chemicals, 2nd ed. New York, Van Nostrand Reinhold Company, pp
367-369.
Vézina M, Kobusch AB, Du Souich P, Greselin E, & Plaa GL (1990)
Potentiation of chloroform-induced hepatotoxicity by methyl isobutyl
ketone and two metabolites. Can J Physiol Pharmacol, 68: 1055-1061.
Vittozzi L, Testai E, & De Biasi A (1991) Multiple bioactivation of
chloroform: a comparison between man and experimental animals. Adv
Exp Med Biol, 283: 665-667.
Vogel E, Blijleven WGH, Kortselius MJH, & Zijlstra JA (1981)
Mutagenic activity of 17 coded compounds in the sex-linked recessive
lethal test in Drosophila melanogaster. In: De Serres FJ & Ashby J
ed. Evaluation of short-term tests for carcinogens: Report of the
international collaborative study. Amsterdam, Oxford, New York,
Elsevier North/Holland, pp 660-665 (Progress in Mutation Research,
Volume 1).
Von Oettingen WF (1964) The halogenated hydrocarbons of industrial
and toxicological importance. Amsterdam, Oxford, New York, Elsevier
Science Publishers, pp 77-108.
Von Oettingen WF, Powell CC, Sharpless NE, Alford WC, & Pecoza LJ
(1950) Comparative studies of the toxicity and pharmacodynamic
action of chlorinated methanes with special reference to their
physical and chemical action. Arch Int Pharmacodyn, 81: 17-34.
Wallace LA (1987) The total exposure assessment methodology (TEAM)
study: Summary and analysis. Volume I (EPA/600/6 - 87/002a).
Wallace LA, Pellizzari E, Hartwell T, Rosenzweig M, Erickson M,
Sparacino, C & Zelon H (1984) Personal exposure to volatile organic
compounds. I. Direct measurements in breathing-zone air, drinking
water, food and exhaled breath. Environ Res, 35: 293-319.
Wecher RA & Scher S (1982) Bioassay procedures for identifying
genotoxic agents using light emitting bacteria as indicator
organisms. In: Seno M & Pazzagli M ed. Luminescent assays:
perspectives in endocrinology and clinical chemistry. New York,
Raven Press Ltd, pp 109-113.
Westrick JJ, Mello JW, & Thomas RF (1989) The groundwater supply
survey. J Am Water Works Assoc, 76: 52-59.
White AE, Takehisa S, Eger EI, Wolff S, & Stevens WC (1979) Sister
chromatid exchanges induced by inhaled anaesthetics. Anesthesiology,
50: 426-430.
WHO (1993) Guidelines for drinking-water quality, 2nd ed. Volume 1:
Recommendations. Geneva, World Health Organization.
WHO (in press) IPCS Environmental Health Criteria 170: Assessing
human health risks of chemicals: Derivation of guidance values for
health-based exposure limits. Geneva, World Health Organization.
Williams DT, Olson R, Bothwell PD, Murphy KI, & Robertson JL (1980)
Trihalomethane levels in Canadian drinking water. Environ Sci Res,
16: 503-512.
Wilson JT, Enfield CG, Dunlap WJ, Cosby RL, Foster DA, & Baskin LB
(1981) Transport and fate of selected organic pollutants in a sandy
soil. J Environ Qual, 10: 501-506.
Windholz M ed. (1983) The Merck index: an encyclopedia of chemicals,
drugs, and biologicals, 10th ed. Rahway, New Jersey, Merck & Co.,
Inc.
Winslow SG & Gerstner HB (1978) Health aspects of chloroform. A
review. Drug Chem Toxicol, 1: 259-275.
Withey JR & Collins BT (1980) Chlorinated aliphatic hydrocarbons
used in the foods industry: the comparative pharmacokinetics of
methylene chloride, 1,2-dichloroethane, chloroform and
trichloroethylene after i.v. administration in the rat. J Environ
Pathol Toxicol, 3: 313-332.
Withey JR & Karpinski K (1985) The fetal distribution of some
aliphatic chlorinated hydrocarbons in the rat after vapour phase
exposure. Biol Res Pregnancy, 6: 79-88.
Withey JR, Collins BT, & Collins PG (1983) Effect of vehicle on the
pharmacokinetics and uptake of four halogenated hydrocarbons by the
gastrointestinal tract of the rat. J Appl Toxicol, 3: 249-253.
Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, & Ullrich V (1977)
The reduction of polyhalogenated methanes by liver microsomal
cytochrome P-450. Mol Pharmacol, 13: 698-705.
Youssefi M, Faust SD, & Zenchelsky ST (1978) Rapid determination of
light halogenated hydrocarbons in urine. Clin Chem, 24: 1109-1111.
Zaleska-Rutczynska Z & Krus S (1973) Genetic aspects of the high and
low sensitivity to chloroform of male mice of various inbred strains
and their crosses. Pol Med Sci Hist Bull Abstr, 1: 245-250.
Zimmermann FK & Scheel I (1981) Induction of mitotic gene conversion
in strain D7 of Saccharomyces cerevisiae by 42 coded chemicals.
In: De Serres FJ & Ashby J ed. Evaluation of short-term tests for
carcinogens: Report of the international collaborative study.
Amsterdam, Oxford, New York, Elsevier North/Holland, pp 481-490
(Progress in Mutation Research, Volume 1).
Zoeteman BCJ, Hrubec J, de Greef E, & Kool HJ (1982) Mutagenic
activity associated with by-products of drinking water disinfection
by chlorine, chlorine dioxide, ozone and UV-irradiation. Environ
Health Perspect, 46: 197-205.
RESUME
Le chloroforme se présente sous la forme d'un liquide volatil,
limpide et incolore, à l'odeur caractéristique et au goût âcre et
douceâtre. Il peut être décomposé par voie photochimique, il n'est
pas inflammable et il est soluble dans la plupart des solvants
organiques. Toutefois sa solubilité dans l'eau est limitée. Lors de
la décomposition chimique, il peut y avoir formation de phosgène et
d'acide chlorhydrique.
Le chloroforme s'emploie dans certaines formulations de
pesticides, comme solvant et comme intermédiaire dans la fabrication
de certains dérivés. Son utilisation comme anesthésique ou dans des
spécialités pharmaceutiques est interdite dans un certain nombre de
pays. La production de chloroforme à des fins commerciales a atteint
440 000 tonnes en 1987. Du chloroforme se forme également en
quantités appréciables lors de la chloration de l'eau et du
blanchiment de la pâte à papier.
L'analyse de l'air, de l'eau et d'échantillons biologiques pour
la recherche et le dosage du chloroforme peut s'effectuer selon
plusieurs méthodes. La majorité d'entre elles consiste en une
injection directe sur colonne, une adsorption sur un adsorbant
activé ou une condensation dans un piège froid; on procède ensuite à
une désorption ou à une extraction par un solvant qui est ensuite
chassé avant analyse finale par chromatographie en phase gazeuse.
On pense que la majeure partie du chloroforme présent dans
l'eau finit par passer dans l'air, en raison de la volatilité de ce
composé. Le temps de séjour du chloroforme dans l'atmosphère est de
plusieurs mois et il en est éliminé après transformation chimique.
Il résiste à la biodégradation aérobie par les bactéries du sol et
des nappes phréatiques qui se développent sur des substrats
endogènes ou en présence d'un supplément d'acétate. Il peut y avoir
biodégradation en anaérobiose. La bioconcentration est faible chez
les poissons d'eau douce. La dépuration est rapide.
D'après l'estimation de l'exposition moyenne due aux divers
milieux, on pense que la population générale est principalement
exposée au chloroforme par l'intermédiaire de la nourriture, de
l'eau de boisson et de l'air intérieur, dans des proportions à peu
près égales. L'absorption estimative à partir de l'air intérieur est
cependant beaucoup moindre. L'absorption moyenne totale estimative
est d'environ 2 µg/kg de poids corporel, par jour. Les données
disponibles indiquent également que l'utilisation domestique de
l'eau contribue de façon très importante à la concentration du
chloroforme dans l'air intérieur et par voie de conséquence à
l'exposition totale. Pour certaines personnes qui vivent dans des
habitations où l'eau de distribution renferme des concentrations
relativement élevées de chloroforme, on estime que l'absorption
totale peut aller jusqu'à 10 µg/kg de poids corporel et par jour.
Une fois administré par voie orale, le chloroforme est bien
résorbé chez l'animal et l'homme, mais la cinétique d'absorption
dépend du véhicule. Chez l'homme, après exposition par la voie
respiratoire, 60 à 80% de la dose inhalée sont absorbés. Les
principaux facteurs qui agissent sur la cinétique d'absorption du
chloroforme après inhalation sont la concentration ainsi que la
capacité de métabolisation, qui dépend de l'espèce. Chez l'homme et
l'animal, le chloroforme est rapidement résorbé par la peau et l'on
a montré qu'il pouvait être également absorbé par voie percutanée
dans une proportion importante à partir de l'eau lors d'une douche.
Il semble que l'hydratation de l'épiderme accélère la résorption du
chloroforme.
Le chloroforme se répartit dans l'ensemble de l'organisme.
C'est dans les graisses, le sang, le foie, les reins, les poumons et
le système nerveux que l'on trouve les plus fortes concentrations
tissulaires. La répartition du chloroforme dépend de la voie
d'exposition; la dose est plus forte dans les tissus
extra-hépatiques après inhalation ou absorption percutanée qu'après
ingestion. On a montré que chez plusieurs espèces animales et chez
l'homme, le chloroforme pouvait traverser la barrière placentaire.
Il s'élimine essentiellement dans l'air expiré sous forme de dioxyde
de carbone. Non métabolisé, il demeure plus longtemps dans les
graisses que dans les autres tissus.
La biotransformation oxydative du chloroforme en
trichlorométhanol est catalysée par le cytochrome P-450. Le
trichlorométhanol produit, par élimination d'HCl, un intermédiaire
réactif, le phosgène. Le phosgène peut être détoxifié en dioxyde de
carbone par réaction avec l'eau ou en divers adduits par réaction
avec des thiols, notamment le glutathion ou la cystéine. La réaction
du phosgène sur les protéines tissulaires entraîne des lésions
cellulaires et la mort. La liaison des métabolites du chloroforme à
l'ADN est limitée. Le chloroforme peut également subir une
biotransformation réductrice catalysée par le P-450, qui donne
naissance au radical dichlorométhyl, lequel se fixe ensuite par
liaison covalente aux lipides tissulaires. On n'a pas déterminé si
cette biotransformation réductrice jouait également un rôle dans la
cytotoxicité du chloroforme.
Chez l'animal et l'homme exposés à du chloroforme, le
chloroforme est éliminé d'une part sous forme de dioxyde de carbone
et d'autre part sous forme inchangée. La fraction de la dose qui est
éliminée sous forme de dioxyde de carbone varie avec cette dose et
l'espèce en cause. La vitesse de biotransformation en dioxyde de
carbone est plus élevée dans les microsomes hépatiques et rénaux des
rongeurs (hamster, souris, rat) que dans ceux de l'homme. La
biotransformation du chloroforme est également plus rapide dans les
microsomes rénaux des souris que dans ceux des rats.
En ce qui concerne la toxicité aiguë, c'est le foie qui est
l'organe-cible chez le rat et plusieurs souches de souris. Les
lésions hépatiques se caractérisent essentiellement, au début, par
une infiltration graisseuse et une ballonisation des cellules, qui
évoluent vers une nécrose centrilobulaire, puis une nécrose massive.
Le rein est l'organe-cible chez les souris mâles appartenant à des
souches plus sensibles. Au niveau du rein, les lésions débutent par
une dégénérescence hydropigène qui évolue vers la nécrose des
tubules proximaux. On n'a pas observé de toxicité rénale importante
chez les femelles d'aucune souche de souris.
La toxicité aiguë varie en fonction de la souche, du sexe et du
véhicule. Chez la souris, la DL50 par voie orale varie de 36 à
1366 mg/kg de poids corporel alors que chez le rat, elle peut aller
de 450 à 2000 mg de chloroforme par kg de poids corporel. Après une
seule exposition de 4 heures par voie respiratoire, on a observé des
effets toxiques sur le foie chez la souris et le rat à des
concentrations de chloroforme respectivement égales à 490 et 1410
mg/m3.
Ce sont les lésions du foie qui sont l'effet toxique du
chloroforme le plus universellement observé. La gravité de ces
effets par dose unitaire administrée dépend de l'espèce, du véhicule
et du mode d'administration du chloroforme. La dose la plus faible à
laquelle on ait observé ces lésions est de 15 mg/kg de poids
corporel et par jour, administrée à des chiens "beagle" dans une
base de pâte dentifrice, pendant une période de 7,5 années. On n'a
pas recherché s'il y avait des effets à des doses plus faibles. Chez
les autres espèces, les doses nécessaires pour produire des effets
hépatotoxiques sont un peu plus élevées. Bien qu'au cours de ces
différentes études, la durée d'exposition ait été variable, on a pu
fixer la concentration sans effets nocifs observables à 15-125 mg/kg
de poids corporel et par jour.
Les effets au niveau du rein ont été observés chez des mâles
appartenant à des souches sensibles de souris ainsi que chez des
rats F-344. Ces effets étaient graves chez les mâles appartenant à
une souche de souris particulièrement sensible, à des doses ne
dépassant pas 36 mg/kg de poids corporel et par jour.
Chez des rats F-344 à qui l'on avait fait inhaler du
chloroforme 7 jours de suite, tous les jours pendant 6 heures, on a
observé une atrophie des glandes de Bowman ainsi que la présence
d'os néoformés dans les cornets du nez. La dose sans effets
observables correspondante se situait à 14,7 mg/m3 (3 ppm). Des
études à long terme se poursuivent afin d'évaluer la portée de ces
effets.
On a constaté l'apparition de tumeurs hépatiques chez des
souris à qui l'on avait administré par gavage des doses quotidiennes
de chloroforme dans de l'huile de maïs, à raison de 138 à 477 mg/kg
de poids corporel. Toutefois, lorsque des doses analogues étaient
administrées dans l'eau de boisson, le chloroforme était sans
influence sur la proportion des tumeurs hépatiques qui se formaient
chez ces souris. De plus, lors d'études sur le caractère promoteur
éventuel de ce composé, on a observé, qu'administré dans l'eau de
boisson, le chloroforme avait en fait une action inhibitrice sur la
formation de tumeurs du foie provoquées chez la souris avec de la
diéthylnitrosamine comme initiateur. Le véhicule utilisé ou la
manière d'administrer le chloroforme conditionne donc de façon
importante son pouvoir tumoro-inducteur au niveau du foie chez la
souris.
Le chloroforme a produit des tumeurs rénales chez des rats qui
en avaient reçu quotidiennement par gavage, dans de l'huile de maïs,
des doses allant de 90 à 200 mg/kg de poids corporel. Toutefois,
chez cette espèce, les résultats se sont révélés analogues lorsque
le produit était administré dans l'eau de boisson, ce qui indique
que les effets ne dépendent pas entièrement du véhicule utilisé.
Il semble que les effets cancérogènes du chloroforme sur le
foie et le rein des rongeurs soient étroitement liés à son action
cytotoxique ainsi qu'aux effets que ce composé exerce sur la
réplication cellulaire dans les organes-cibles. On a constaté que
ces derniers effets suivaient de près les modifications de la
réponse cancérogène au chloroforme en fonction du type de véhicule
et du mode d'administration. A la lumière des données disponibles,
il semble que le chloroforme ne soit guère capable d'induire des
mutations géniques ou d'autres types de lésions directes de l'ADN.
En outre, le chloroforme ne semble pas non plus capable de jouer le
rôle d'initiateur tumoral au niveau du foie chez la souris ni
d'induire une synthèse non programmée de l'ADN in vivo. En
revanche, lorsqu'il est administré dans un véhicule huileux, le
chloroforme peut se révéler un promoteur efficace des tumeurs
hépatiques. Par conséquent, il est probable que, lors de
l'administration prolongée de chloroforme, la cytotoxicité de ce
composé et la prolifération cellulaire qu'il détermine sont les
causes les plus importantes de la formation de tumeurs hépatiques et
rénales chez les rongeurs.
On dispose de quelques données limitées selon lesquelles le
chloroforme serait toxique pour le foetus, mais uniquement à des
doses auxquelles il est également toxique pour la mère.
En général, le chloroforme détermine les mêmes symptômes
toxiques chez l'homme que chez l'animal. Chez l'homme, l'anesthésie
peut entraîner la mort par suite d'arythmie et d'insuffisance
respiratoire et cardiaque. On a également observé chez l'homme une
nécrose des tubules rénaux et une insuffisance rénale. Les doses les
plus faibles pour lesquelles des cas de toxicité hépatique due à une
exposition professionnelle au chloroforme ont fait l'objet de
rapports, se situaient dans les limites de 80 à 160 mg/m3 (durée
d'exposition de moins de 4 mois) selon une étude et allaient de 10 à
1000 mg/m3 (durée d'exposition: 1 à 4 ans) selon une autre étude.
On estime que la dose mortelle moyenne par voie orale pour un adulte
est d'environ 45 g, mais on note d'importantes différences de
sensibilité selon les individus. On est fondé à croire, selon
certaines études épidémiologiques, qu'il existe une association
entre l'exposition aux sous-produits des désinfectants présents dans
l'eau de boisson et les cancers colorectaux ou vésicaux. Cependant,
ces études souffrent de la présence de facteurs de confusion, entre
autres faiblesses. Les preuves avancées à l'appui de la
cancérogénicité pour l'homme de l'eau de boisson chlorée, sont
insuffisantes. En outre, la présence de sous-produits des
désinfectants utilisés ne peut être attribuée au chloroforme
lui-même.
Le chloroforme est toxique pour les stages embryo-larvaires de
certaines espèces d'amphibiens et de poissons. La CL50 la plus
faible dont il ait été fait état, se situait à 0,3 mg/litre pour les
stades embryo-larvaires de Hyla crucifer. Le chloroforme est moins
toxique pour les poissons et pour la daphnie Daphnia magna. Pour
plusieurs espèces de poissons, les valeurs de la CL50 se situent
dans les limites de 18 à 191 mg/litre. Il n'y a guère de différences
de sensibilité entre les poissons d'eau douce et les poissons de
mer. En ce qui concerne Daphnia magna, la valeur la plus faible de
la CL50 qui ait été signalée, était de 29 mg/litre. Le chloroforme
est peu toxique pour les algues et autres microorganismes.
Le Groupe de travail a estimé que les données disponibles
étaient suffisantes pour établir une dose journalière tolérable
(DJT) pour les effets non cancérogènes du chloroforme, ainsi qu'une
dose spécifiquement liée au risque d'effets cancérogènes, sur la
base des études effectuées chez l'animal; les valeurs ainsi fixées
serviront de guide pour l'établissement de limites d'exposition par
les autorités compétentes. Cependant, il est rappelé que lorsque les
conditions locales imposent un choix entre le respect des limites
microbiologiques ou celles qui concernent la présence de
sous-produits de désinfection tels que le chloroforme, c'est la
qualité microbiologique qui doit toujours l'emporter. Il ne faut
jamais transiger sur l'efficacité de la désinfection.
En se fondant sur l'étude de Heywood et al. (1979) et en
introduisant un facteur d'incertitude de 1000 (x10 pour les
variations interspécifiques, x10 pour les variations
intraspécifiques et x10 pour l'utilisation d'une dose avec effet
plutôt que d'une dose sans effet lors d'une étude subchronique), on
obtient une DJT de 15 µg/kg de poids corporel; il faut rappeler que
cette étude avait révélé l'existence d'une légère hépatotoxicité (à
savoir une augmentation des enzymes hépatiques sériques et des
kystes graisseux) chez des chiens "beagle" à qui l'on avait fait
ingérer pendant 7,5 ans, une pâte dentifrice contenant du
chloroforme à la dose de 15 mg/kg de poids corporel et par jour.
En se fondant sur ce que l'on sait du mécanisme de ces
phénomènes, la méthode que l'on juge la mieux adaptée pour obtenir
une valeur-guide consiste à diviser la valeur de la concentration
sans effet observable sur la prolifération cellulaire par un certain
facteur d'incertitude. C'est ainsi que si l'on utilise la valeur de
la dose sans effets observables obtenue par Larson et al. (1993b)
pour la cytoléthalité et la prolifération cellulaire chez des souris
B6C3F1 qui avaient reçu pendant 3 semaines, dans de l'huile de
maïs, une dose quotidienne de chloroforme équivalant à 10 mg/kg de
poids corporel, et en introduisant un facteur d'incertitude de 1000
(x10 pour les variations interspécifiques, x10 pour les variations
intraspécifiques et x10 pour la gravité de l'effet, c'est-à-dire la
cancérogénicité et parce qu'il s'agit d'une étude subchronique), on
obtient une DJT de 10 µg/kg de poids corporel.
On admet que les tumeurs rénales observées chez le rat peuvent
également être liées à l'action létale du chloroforme sur les
cellules et à ses effets sur leur prolifération. Cependant, étant
donné que l'on ne possède pas de données sur la prolifération
cellulaire chez les souches où l'on a observé des tumeurs et qu'en
outre, ce que l'on peut savoir de cet effet et de l'effet létal du
chloroforme sur les cellules n'a été observé qu'à court terme (un
seul gavage et une exposition par voie respiratoire de 7 jours), on
estime qu'il est prématuré de s'écarter du modèle par défaut
(c'est-à-dire multistade linéarisé) pour l'estimation du risque de
cancer sur la durée de vie. D'après l'étude de Jorgenson et al.
(1985) qui portait sur l'induction de tumeurs rénales (adénomes et
adénocarcinomes), on a fixé à 8,2 µg/kg de poids corporel et par
jour, la dose quotidienne totale jugée capable de produire un excès
de risque de 10-5 sur toute la durée de la vie.
La concentration de chloroforme dans les eaux de surface est
généralement faible et ne semble pas présenter de danger pour les
organismes aquatiques. Toutefois, la décharge ou le déversement de
produits industriels pourrait entraîner la présence de
concentrations plus élevées de chloroforme dans ces eaux et les
rendre dangereuses pour les stades embryo-larvaires de certaines
espèces aquatiques.
RESUMEN
El cloroformo es un líquido transparente, incoloro y volátil,
con un olor característico y un sabor dulce ardiente. Se degrada
fotoquímicamente, no es inflamable y es soluble en la mayor parte de
los disolventes orgánicos. Sin embargo, su solubilidad en agua es
limitada. Por degradación química del mismo pueden formarse fosgeno
y ácido hidroclorhídrico.
El cloroformo se utiliza en la formulación de plaguicidas, como
disolvente y como intermedio químico. Su utilización como anestésico
y en especialidades farmacéuticas está prohibida en algunos países.
La producción comercial ascendió a 440 000 toneladas en 1987.
También se producen cantidades apreciables de cloroformo en la
cloración del agua y en el blanqueado de la pasta papelera.
Existen varios métodos analíticos para determinar la presencia
de cloroformo en el aire, el agua y los materiales biológicos. La
mayor parte de esos métodos se basan en la inyección directa en
columna, la adsorción en adsorbentes activados o la condensación en
una cámara fría y posteriormente la desorción o evaporación mediante
la extracción por disolventes o el calentamiento y el subsiguiente
análisis por cromatografía de gases.
Se supone que la mayor parte del cloroformo presente en el agua
se transfiere finalmente al aire debido a su volatilidad. El
cloroformo tiene un tiempo de residencia en la atmósfera de varios
meses y desaparece de la misma por transformación química. Es
resistente a la biodegradación por la población microbiana aeróbica
de los suelos y de las capas acuíferas que viven en subestratos
endógenos o con el suplemento de acetato. La biodegradación es
posible en condiciones anaeróbicas. La bioconcentración en los peces
de agua dulce es baja. La depuración es rápida.
Las estimaciones de la exposición media calculadas a partir de
diversos medios indican que la población en general está expuesta al
cloroformo principalmente a través de los alimentos, el agua de
bebida y el aire de los interiores en cantidades aproximadamente
equivalentes. La inhalación estimada por conducto del aire exterior
es considerablemente menor. La ingesta media estimada total es de
aproximadamente 2 µg/kg de peso corporal por día. Los datos
disponibles también indican que el agua de uso doméstico contribuye
considerablemente a los niveles de cloroformo en el aire de los
interiores y a la exposición total. La ingesta total estimada de
algunos individuos que viven en lugares con un abastecimiento de
agua corriente con concentraciones relativamente elevadas de
cloroformo asciende a 10 µg/kg de peso corporal por día.
Los animales y los seres humanos absorben bien el cloroformo
después de la administración por vía oral, pero la cinética de la
absorción depende del vehículo suministrado. Tras la exposición por
inhalación, los seres humanos absorben del 60 al 80% de la cantidad
inhalada. Los factores principales que afectan a la cinética de la
absorción del cloroformo después de la inhalación son su
concentración y la capacidad metabólica específica de la especie.
Los seres humanos y los animales lo absorben fácilmente a través de
la piel y se ha demostrado que durante la ducha la absorción dérmica
del cloroformo del agua es apreciable. La hidratación de la piel
parece acelerar la absorción de cloroformo.
El cloroformo se distribuye en todo el cuerpo. Los niveles
tisulares más elevados se alcanzan en el tejido adiposo, la sangre,
el hígado, los riñones, los pulmones y el sistema nervioso. La
distribución depende de la vía de exposición; los tejidos
extrahepáticos reciben una dosis más elevada del cloroformo inhalado
o absorbido por la piel que del cloroformo ingerido. Se ha
demostrado que en varias especies animales y en el ser humano el
cloroformo se transfiere a través de la placenta. El cloroformo se
elimina principalmente como dióxido de carbono exhalado. El
cloroformo no metabolizado se mantiene más tiempo en el tejido
adiposo que en cualquier otro tejido.
El citocromo P-450 cataliza la biotransformación oxidativa del
cloroformo en triclorometanol. La pérdida de HCl del triclorometanol
produce fosgeno como reactivo intermedio. El fosgeno puede
destoxificarse por reacción con el agua produciendo dióxido de
carbono o con tioles, inclusive con glutatión o cisteína,
produciendo aductos. La reacción del fosgeno con proteínas tisulares
está asociada con daño y necrosis celulares. Se observa un escaso
enlace de los metabolitos del cloroformo con el ADN. El cloroformo
también es objeto de una biotransformación reductiva catalizada por
el P-450 que produce radicales de diclorometilo y éstos contraen
enlaces covalentes con los lípidos tisulares. No se ha determinado
el papel de la biotransformación reductiva en la citotoxicidad del
cloroformo.
Los animales y los seres humanos expuestos al cloroformo
eliminan con el aire espirado el dióxido de carbono y el cloroformo
que no se ha transformado. La fracción de dosis eliminada como
dióxido de carbono varía según la dosis y la especie. La tasa de
biotransformación en dióxido de carbono es más elevada en los
microsomas hepáticos y renales de roedores (hámster, ratón, rata)
que en los microsomas hepáticos y renales humanos. Además, el
cloroformo se biotransforma más rápidamente en los microsomas
renales del ratón que en los de la rata.
El hígado es el órgano vulnerable a la toxicidad aguda en las
ratas y en varias estirpes de ratones. La lesión hepática se
caracteriza principalmente por una infiltración grasa temprana y
células con forma de globo y evoluciona hacia la necrosis
centrilobular seguida de necrosis general. El riñón es el órgano
vulnerable en los ratones macho de otras estirpes más sensibles. La
lesión renal comienza con una degeneración hidrópica que avanza
hacia la necrosis de los tubos proximales. No se ha observado una
toxicidad renal apreciable en las ratas hembra de ninguna estirpe.
La toxicidad aguda varía según la raza, el sexo y el vehículo.
En el ratón, la DL50 por vía oral oscila entre 36 y 1366 mg de
cloroformo/kg de peso corporal, mientras que en las ratas oscila
entre 450 y 2000 mg de cloroformo/kg de peso corporal. Después de
una sola exposición de cuatro horas por inhalación, se observó
toxicidad hepática en ratones y ratas cuando el nivel de cloroformo
alcanzaba, respectivamente, 490 y 1410 mg/m3.
Los efectos tóxicos del cloroformo más generales observados
consisten en lesiones hepáticas. La gravedad de esos efectos por
unidad de dosis administrada depende de la especie, del vehículo de
administración y del método por el cual se haya administrado el
cloroformo. La dosis más baja causante de lesión hepática observada
es de 15 mg/kg de peso corporal por día, administrada a perros
pachones en una base de pasta dentífrica durante un periodo de 7,5
años. No se han examinado efectos con dosis más bajas. Se necesitan
dosis algo más elevadas para producir efectos hepatotóxicos en otras
especies. En esos estudios, aunque la duración de la exposición
variaba, los niveles sin efectos adversos observados oscilaban entre
15 y 125 mg/kg de peso corporal por día.
Se han observado efectos en el riñón de ratones macho de
estirpes sensibles y en la rata F-344. Se han observado efectos
graves en una estirpe especialmente sensible de ratones macho con
dosis de sólo 36 mg/kg de peso corporal por día.
La inhalación de cloroformo seis horas por día durante siete
días consecutivos produjo atrofia de las glándulas de Bowman y
neoplasia ósea en la concha nasal de ratas F-344. El nivel en que no
se observaron esos efectos fue de 14,7 mg/m3 (3 ppm). La
importancia de dichos efectos se está investigando más a fondo en
estudios de larga duración.
El cloroformo administrado por sonda en un vehículo de aceite
de maíz en dosis de 138 a 477 mg/kg de peso corporal por día indujo
tumores hepáticos en ratones. Sin embargo, dosis semejantes de
cloroformo administradas en el agua de bebida no produjeron tumores
hepáticos en ratones. Por otra parte, en estudios de
iniciación/promoción, el cloroformo administrado en el agua de
bebida como promotor parecía inhibir el desarrollo de tumores
hepáticos iniciados por dietilnitrosamina en ratones. Así pues, el
vehículo y/o el método de administración del cloroformo es una
variable importante en relación con la inducción de tumores
hepáticos en el ratón.
El cloroformo administrado por sonda en aceite de maíz en dosis
de 90 a 200 mg/kg de peso corporal por día indujo tumores renales en
ratas. Sin embargo, en esa especie se observaron efectos semejantes
tras la administración de cloroformo en el agua de bebida, lo que
indica que la reacción no depende exclusivamente del vehículo
utilizado.
Los efectos carcinogénicos del cloroformo en el hígado y los
riñones de roedores parecen estar estrechamente relacionados con
efectos citotóxicos y de replicación celular observados en los
órganos vulnerables. Se ha observado que los efectos en la
replicación celular eran paralelos a las modificaciones de las
respuestas carcinogénicas al cloroformo inducidas por el vehículo y
por la modalidad de administración. Las observaciones realizadas
indican que el cloroformo tiene poca o ninguna capacidad para
inducir mutaciones genéticas o daños directos de otro tipo en el
ADN. Por otra parte, el cloroformo no parece poder iniciar tumores
hepáticos en ratones ni de inducir síntesis imprevistas de ADN in
vivo. Por otra parte, el cloroformo puede promover la neoplasia
hepática cuando se administra en un vehículo oleoso. Por
consiguiente, es probable que, tras la administración prolongada de
cloroformo, la citotoxicidad seguida de proliferación celular sea la
causa más importante del desarrollo de tumores hepáticos y renales
en los roedores.
Algunos datos limitados sugieren que el cloroformo es tóxico
para el feto, pero sólo en dosis tóxicas para la madre.
En general, el cloroformo provoca en el ser humano los mismos
síntomas de toxicidad que en los animales. En el ser humano, la
anestesia puede causar la muerte por arritmia e insuficiencia
respiratoria y cardíaca. En el ser humano también se ha observado
necrosis de los tubos renales y disfunción renal. Los niveles más
bajos en los que se haya comunicado toxicidad hepática debida a la
exposición ocupacional al cloroformo se sitúan entre 80 y 160
mg/m3 (con un periodo de exposición inferior a cuatro meses) en un
estudio y entre 10 y 1000 mg/m3 (con periodos de exposición de uno
a cuatro años) en otro estudio. La dosis letal media de un adulto se
estima en unos 45 g, pero hay grandes diferencias de vulnerabilidad
de un individuo a otro. En algunos estudios epidemiológicos hay
ciertas indicaciones de que existe una asociación entre la
exposición a los subproductos de la desinfección del agua de bebida
y el cáncer colorrectal y de vejiga. Sin embargo, hay factores
confusos y otras insuficiencias que ponen en entredicho esos
estudios. Las pruebas de la carcinogenicidad del agua de bebida
clorada en el ser humano son insuficientes. Además, los subproductos
de la desinfección no pueden atribuirse al cloroformo por sí solo.
El cloroformo es tóxico en las fases embriolarvales de algunas
especies de anfibios y de peces. La CL50 más baja comunicada es de
0,3 mg/litro en las fases embriolarvales de Hyla crucifer. El
cloroformo es menos tóxico para los peces y para Daphnia magna. La
CL50 de varias especies de peces se halla entre 18 y 191 mg/litro.
Hay pocas diferencias de sensibilidad entre los peces de agua dulce
y salada. La CL50 más baja comunicada en Daphnia magna es de 29
mg/litro. El cloroformo es poco tóxico para las algas y otros
microorganismos.
El Grupo Especial llegó a la conclusión de que los datos
disponibles son suficientes para fijar una ingesta diaria tolerable
sin efectos neoplásicos e ingestas con riesgos carcinogénicos
específicos del cloroformo sobre la base de los estudios realizados
en especies animales; las dosis servirán como orientación para que
las autoridades competentes fijen límites de exposición. Sin
embargo, se advierte que, cuando las circunstancias locales exijan
optar entre el cumplimiento de límites microbiológicos y el de
límites para subproductos de la desinfección tales como el
cloroformo, debe siempre prevalecer la calidad microbiológica.
Nunca debe comprometerse una desinfección eficaz.
Sobre la base del estudio de Heywood et al. (1979) en el cual
se observó una ligera hepatotoxicidad (aumento de las enzimas del
suero hepático y quistes grasos) en perros pachones que habían
ingerido 15 mg/kg de peso corporal por día en pasta dentífrica
durante 7,5 años, incorporando un factor de incertidumbre de 1000
(x10 para la variación entre especies, x10 para la variación dentro
de la especie y x10 para utilizar un nivel con efectos en lugar de
sin efectos y un estudio subcrónico), se obtiene una ingesta diaria
tolerable (IDT) de 15 µg/kg de peso corporal por día.
En función de los datos disponibles sobre los mecanismos
determinantes, el método que se considera más apropiado para
establecer orientaciones fundadas en los tumores hepáticos de
ratones es dividir un nivel sin efectos de proliferación celular por
un factor de incertidumbre. A partir del nivel sin efectos
observados de citoletalidad y proliferación celular en ratones
B6C3F1, de 10 mg/kg de peso corporal por día administrados en
aceite de maíz durante tres semanas, comunicado en el estudio de
Larson et al. (1993b), incorporando un factor de incertidumbre de
1000 (x10 para la variación entre especies, x10 para la variación
dentro de la especie y x10 para la gravedad del efecto, es decir,
carcinogenicidad, y estudio subcrónico) se obtiene una IDT de 10
µg/kg de peso corporal por día.
Se reconoce que los tumores renales en ratas también pueden
estar asociados con letalidad y proliferación celular. Sin embargo,
dado que no se dispone de datos sobre proliferación celular en la
estirpe en la que se observaron tumores y la información sobre
proliferación y letalidad celulares es de corto plazo (una sola
sonda y exposición por inhalación durante siete días), se considera
prematuro alejarse del modelo establecido por defecto (es decir,
fases múltiples linearizadas) como base para estimar el riesgo de
cáncer durante una vida. La ingesta diaria total que se considera
asociada con un riesgo excesivo de 10-5 durante una vida, sobre la
base de la inducción de tumores renales (adenomas y adenocarcinomas)
en ratas macho en el estudio de Jorgenson et al. (1985), es de 8,2
µg/kg de peso corporal por día.
Los niveles de cloroformo en las aguas superficiales son
generalmente bajos y no se prevé que constituyan un peligro para los
organismos acuáticos. Sin embargo, niveles más elevados de
cloroformo en las aguas superficiales como consecuencia de las
descargas o los derrames industriales tal vez sean peligrosos en las
fases embriolarvales de algunas especies acuáticas.